/











































Теги: химия химическая промышленность химические реакции
Год: 1961




Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СОВЕТА МИНИСТРОВ СССР
ПО ХИМИИ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 3
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
МОСКВА—1961
СОДЕРЖАНИЕ
Антразо. А. М. Лукин, Г. С. Петрова ..................... 5
Антрахас. А. М. Лукан, Г. С. Петрова ............. 8
N-Бензоилфенилгидроксиламин. А. М Лукан, Е. Я. Яровенко . . 11
Биссалицилаль-этилендиамин. А. М. Лукан, Г. Б. Заварахина . 14
Биурет. А М. Лукан, Е. Я. Яровенко...................... 15
а, а'-Диангримид. Е. Я. Яровенко ........................17
Дикетон. Л. М Моисеева, И. М. Кузнецова................. 19
Мурексид. А. М. Лукин, И. Д Калинина.................... 21
Нафтазарин. А. М. Лукин, Е. Я. Яровенко............• . . 23
Нингидрин. Г. И. Кошелева............................... 26
Родизонат натрия. В. Г. Брудзъ, Л. И. Гричева, Д. А. Драпкина 29
Салицилаль-о-аминофенол. А. М. Лукин, |£. Д. Осетрова\ ... 31
Тиооксин и 8,8'-дихинолилдисульфид. И. Шевчук, Э. Лукша . ... 33
1,1-Гидразиндиуксусная кислота. Р. П. Ластовский, В. Я. Т, мкина,
Е. И, Миронова....................................... 39
З-Оксиэтилиминодиуксусная кислота. Р. П. Ластовский, В. Я. Тем-
кина, Е. И. Миронова .................... 40
Этилендиаминтетраацетат свинца. Р.П. Ластовский, В. Я. Темкана,
И. А. Селиверстова................................... 41
2,5-Дннитрофенол. Г. И. Кошелева, Г. Д. Глебова......... 42
Нитразиновый желтый. Г. Н. Кошелева, Г. Д. Глебова...... 44
Пропиловый красный. Г. Н. Кошелева..................... 46
Метакрезоловый пурпуровый. Г. Н. Кошелева, Е. С. Фомина ... 48
Ксиленоловый синий. Г. Н. Кошелева...................... 50
Бромкрезоловый зеленый. Г. Н. Кошелева.................. 52
Бромфеноловый синий Г. Н. Кошелева ..................... 54
Бромфеноловый красный. Г. Н. Кошелева................... 56
Бромксиленоловый синий. Г. Н. Кошелева.................. 58
Хлорфеноловый красный. Г. Н. Кошелева................... 60
Этиловый синий. Г. Н. Кошелева, Г. Д. Глебова.........> . 61
4-Этоксиакридон. Г. Н. Кошелева, Е. С. Фомина........... 64
2,3-Днциангидрохинон. Г. Н. Кошелева.................... 66
Диоксифталимид. Г. Н. Кошелева.......................... 68
Метилвиологендихлорид. Г. Н. Кошелева, Г. Н. Налецкая .... 69
Эухризин ЗР. Е. Я. Яровенко, Г. Н. Кошелева............. 72
Люцигенин. Е. Я. Яровенко, Г. Н. Кошелева............... 73
10,10'-Диметил-9,9'-биакриден. Е. Я. Яровенко, Г. Н. Кошелева . . 77
Лофин. Е. Я- Яровенко, Г. Н. Кошелева ............. 79
Пирокатехиновый фиолетовый. Е. Я. Яровенко, Г. Н. Кошелева . 81
Пирокатехинфталеин. Е. Я. Яровенко, Г. Н. Кошелева.......83
о-Сульфобензойной кислоты моноаммонийная соль. Е. Я. Яровенко,
Г. Н. Кошелева ....................... 84
о Сульфобензойной кислоты ангидрид. Е. Я. Яровенко, Г. Н. Ко-
шелева ............................................ 85
3
АНТРАЗО
Антрахинон-( 1-азо-Г)-4л-диметиланилин хлоргидрат
А. М. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
/ \__n = N- / х
°=С>=°
c22h1802n3ci
N(CHS).3.HC1
м. в. 391,84
Антразо применяется для количественного определения
Sniv, Pb и Те [1, 2, 3].
Свободное основание этого реактива, в числе многих дру-
гих азокрасителей антрахинонового ряда, впервые было опи-
сано в 1921 г. Гаттерманом [4]; в 1940 г. В. И. Кузнецов [1]
описал впервые синтез его хлоргидрата. Однако оба автора,
приводя условия сочетания а-диазоантрахинона с диметилани-
лином, по-видимому, не занимались исследованием этой реак-
ции.
Наша работа [5] привела к установлению оптимальных ус-
ловий процессов диазотирования и сочетания.
Проводя реакцию диазотирования а-аминоантрахинона в
солянокислой среде, удалось повысить выход диазосоедине-
ния до 95% теории, считая на амин. Далее, при проведении
реакции сочетания с применением 10 молей ацетата натрия на
1 моль диазоантрахинона был получен реактив, по составу
близкий к химически чистому, с выходом также -~95% теории,
считая на 100%-ный а-аминоантрахинон. Таким образом, со-
четание в наших условиях идет количественно.
Синтез хлоргидрата антрахинон-( 1-азо-Г)-4'-диметиланилина
/~\-NH2 0^1
O = f ^=O+NaNO2+2HCl—0=:^ ^=O+NaCl+2H2O
^~^-n2ci
О=/ ^ = 0+/“\-N(CH3),-
^~^-N=N-/“~\-N(CH8)2+HCl
—<-0=/ ^. = 0
/ ; — N=N - —N(CH3)S
O=\ = O J-HC1 —
- N = N - \ - N(CHg)2 • HC1
->0 = / x — О
Характеристика основного сырья
а-Аминоантрахинон, 95—96%-ный, техн,
Диметиланилин хлоргидрат, ч., ГОСТ 3444—51.
Натрий уксуснокислый трехводный, ч.д.а., ГОСТ 199—52.
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
Растворяют 23,5 г (0,1 М) 95%-ного а-аминоантрахинона
при нагревании на водяной бане в 125 .ил концентрированной
серной кислоты. Полученный раствор выливают в 1 л холод-
ной воды, 'а выпавший осадок отфильтровывают и промывают
6
водой до нейтральной реакции промывных вод. Полученную
пасту смешивают с 60 мл концентрированной соляной кислоты
и 445 мл воды.
Образовавшуюся суспензию, охлажденную до 10—11°, диа-
зотируют 10%-ным водным раствором нитрита натрия (7,24 г
ЫаМОг) до положительной пробы на йодкрахмальную бу-
мажку. В процессе диазотирования диазосоединение выпадает
в форме пластинчатых кристаллов (см. примечание). По окон-
чании диазотирования к реакционной массе приливают 450 мл
воды и при размешивании массу осторожно подогревают на
водяной бане до полного растворения кристаллов диазосоеди-
нения (температура реакционной массы не должна превы-
шать 25—30°). Полученный раствор быстро отфильтровывают,
а фильтрат тотчас же используют для сочетания, прибавляя
по каплям при 10—12° к раствору 16,7 г хлоргидрата диме-
тиланилина и 136 г ацетата натрия в 300 мл воды. После за-
грузки диазосоединения массу размешивают 30 минут и вы-
держивают 3—4 часа. Полученную после фильтрования фио-
летовую пасту переносят в круглодонную колбу, добавляют
90 г безводного углекислого натрия и 500 мл воды. Из полу-
ченной суспензии избыточный диметиланилин отгоняют с па-
ром, а оставшийся после отгонки красный осадок отфильтро-
вывают и промывают дистиллированной водой до нейтраль-
ной реакции промывных вод. Пасту замешивают с 20 мл кон-
центрированной соляной кислоты при нагревании на водяной
бане, а затем высушивают при 100° до постоянного веса.
Получают 37,5 г антразо (95% теории, считая на 100%-ный
а-аминоантрахинон).
Анализ основания.
Найдено %: N 11,36; 11,34.
С^НпОгИз. Вычислено %: N 11,83.
Примечание. Все работы, связанные с раствором диазоантрахи-
нона, необходимо производить в затемненном месте. Следует особенно
остерегаться солнечных лучен.
ЛИТЕРАТУРА
1. В. И. Кузнецов, Ж. прикл. химии, 13. 769 и 1724 (1940).
2. В. И. Кузнецов, Ж. прикл. химии, 16, 328 (1943).
3. В. И. Кузнецов, Ж. аналит. химии, 1, 259 (1946).
4. L. Gattermann, Н. Rolfes, Liebigs Ann. Chem., 425, 135 (1921).
5. A. M. Лукин, Г. С. Петрова, Труды ИРЕА, вып. 21, стр. 77, Госхим-
издат, 1956.
АНТРАХАС
1-Антрахинонарсоновая кислота
А. М. ЛУКИН, Г. С. ПЕТРОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
О
ссо
YY ,
C14H9O6As О AsO3H2 м. в. 332,12
1-Антрахинонарсоновая кислота применяется как реактив
для осаждения малых количеств олова [1], а также для опре-
деления некоторых других катионов.
По литературным данным, 1-антрахинонарсоновую кислоту
получают по реакции Барта из диазотированного а-амино-
антрахинона и мышьяковистого ангидрида в щелочной среде
[1, 2, 3] (см. примечание).
В результате изучения оптимальных условий всех стадий
синтеза реактива удалось значительно усовершенствовать ме-
тодику его получения [4].
Синтез 1-антрахинонарсоновой кислоты
О NH2
-f- NaNOa Н- H2SO4
О n2-hso4
|+NaHSO4+2HaO
О
AssO3+4NaOH----> 2Na2HAsO3+H2O
О N2-HSO4 О AsO8HNa
II I II I
I I I | + Na2HAsO3— I I I I +NaHSO4 + N,
О AsOjHNa
О AsO8H2
4-NaNO8
Характеристика основного сырья
а-Аминоантрахинон, 95%-ный техн.
Мышьяковистый ангидрид, х. ч., 98%-ный, ГОСТ 1973—43.
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
В фарфоровый стакан емкостью 200 мл, снабженный рам-
ной мешалкой, термометром и капельной воронкой, вносят
23,5 г (0,1 М) технического а-аминоантрахинона, медленно
добавляют 40 мл концентрированной серной кислоты, после
чего осторожно размешивают образовавшийся сульфат а-ами-
поантрахинона, охлаждая разогревшуюся реакционную массу
до комнатной температуры.
Затем к массе постепенно добавляют при размешивании
120 мл дистиллированной воды, при этом температура реак-
ционной массы поднимается до 60°. Выпавшую обильную
красную массу а-аминоантрахинона охлаждают до 10° и при
энергичном размешивании прибавляют к ней по каплям в
течение 1,5 часов 40%-ный водный раствор нитрита натрия до
положительной пробы на йодкрахмальную бумажку. По окон-
чании диазотирования реакционную массу охлаждают до тем-
пературы от 0 до +2°, выдерживают при этой температуре
30 минут, затем отфильтровывают осадок диазосоединения и
промывают его 50 мл ледяной дистиллированной воды.
Одновременно с диазотированием готовят в фарфоровом
стакане раствор, содержащий 13,2 г (0,13 Л4) мышьяковистого
ангидрида, 8 г едкого натра, 8 г безводного углекислого нат-
рия в 700 мл дистиллированной воды. Растворение мышьяко-
вистого ангидрида проводят при нагревании на водяной бане
до 90°.
К охлажденному раствору полученного арсенита натрия
прибавляют раствор 4 г медного купороса в 40 мл воды. Смесь
выливают в 2-литровый фарфоровый стакан, помещенный в
баню и снабженный рамной мешалкой, термометром и стек-
лянной трубкой с диаметром не более 25 мм, конец которой
погружен в реакционную массу. При 10° и энергичном переме-
шивании прибавляют через трубку суспензию диазосоедине-
9
ния, приготовленную взмучиванием влажного осадка сульфа-
та диазония в 350 мл воды. При добавлении диазосоединения
наблюдается сильное пенообразоваиие, для уничтожения ко-
торого изредка по каплям прибавляют изоамиловый спирт.
Суспензию диазосоединения прибавляют в 5—6 приемов в те-
чение 15—20 минут, следя за тем, чтобы среда была все время
слабощелочной и температура не поднималась выше 16е.
О наличии слабощелочной среды судят по слабо-розовой
окраске фенолфталеиновой бумажки. Обычно в конце прили-
вания суспензии диазосоединения добавляют смесь 6 г едкого
натра и 6 г соды, растворенную в 40 мл воды. В этих условиях
конечное значение pH реакционной массы равно 8,8—9 (по
универсальной индикаторной бумаге).
По окончании прибавления суспензии реакционную массу
размешивают 1 час и оставляют на 8—10 часов. Затем к ре-
акционной массе прибавляют 15 г едкого натра, 40 а активи-
рованного древесного угля, нагревают до 90° и выдерживают
при этой температуре 30 минут, далее отфильтровывают и к
теплому фильтрату осторожно прибавляют 80 мл азотной кис-
лоты уд. в. 1,35 до сильно кислой реакции по бумажке конго;
полученную суспензию нагревают до 90—95° и выдерживают
при этой температуре 10—15 минут; охлаждают до 10—15°; от-
фильтровывают желтый осадок 1-антрахинонарсоновой кисло-
ты, тщательно отжимают и промывают дистиллированной во-
дой до нейтральной реакции промывных вод. Сушат при 80—
90°. Получают 19,6 а 1-антрахиноиарсоновой кислоты (59%
теории, считая на 100%-ный а-аминоантрахинон).
Найдено %: As (связанного) 23,05.
Найдено %: As (свободного) 0,05.
C14H9O5AS. Вычислено %: As 22,56.
Примечание. В литературе [1, 2] отмечается ядовитость 1-антра-
хинонарсоновой кислоты, во много раз превышающая ядовитость мышья-
ковых соединений бензольного ряда.
ЛИТЕРАТУРА
1. В. И. Кузнецов, Заводск. лаборатория, 11, 263 (1945).
2. L. Benda, J. prakt. Chem.. 95, 74 (1917).
3. Stelkopf, Schmidt, Ber., 61. 675 (1928).
4. A. M. Лукин, Г. С. Петрова, Ж. общ. химии, 27, 2171 (1957).
V-БЕ НЗОИЛ ФЕНИЛ ГИДРОКСИЛ АМИН
Л. М. ЛУКИН, Е. Я. ЯРОВЕНКО
(ВНИИ химических реактивов и особо чистых химических веществ)
ОН о
Ci3HuO2N м. в. 213,08
Бензоилфенилгидроксиламин (БФГА) предложен как реак-
тив для количественного определения весовым путем ряда ка-
тионов [1], в том числе меди, железа, алюминия и титана [2],
циркония [3], скандия [4], ниобия [5, 6], тантала [7] и др.
По литературным данным, бензоилфенилгидроксиламин
образуется под влиянием света из бензальдегида и нитробен-
зола [8], а также при взаимодействии фенилгидроксиламина с
бензойным ангидридом [9] или с бензоилхлоридом [1, 2].
Мы проверили последний метод и уточнили условия неко-
торых его стадий. Исходный фенилгидроксиламин был полу-
чен путем восстановления нитробензола цинковой пылью [10].
Синтез бензоилфенилгидроксиламина
1) ^-NO24-2Zn-ЬЗН.О-*
\ -NHOH 4-2 Zn(OH)2
________. ___ NaHCO,
2а) / у-NHOH + / ^-СОС1-►
— / \-N-CO-(^ у + НС1
ОН
-- --- NaHCO,
26) \ -CO-N-/ / + х \~COC1-----►
ОН
- / X—N-CO-/ /4-НС1
о
I _____
со-\ /
(Побочный процесс)
11
3) / \_N-CO-/ \ 4- NH4OH
ОН
— / N— СО-\ 4- Н2О
onh4
4) 2^ \-N-CO-^ \ H2SO4 -
ONH4
-> 2(~У~N~co-/ у + (NH4)2SO4
OH
Характеристика основного сырья
Аммиак водный, ГОСТ 9—40.
Бензоил хлористый, ч., ТУ МХП 92—51.
Нитробензол, ч., ГОСТ 5846—51.
Цинковая пыль, 85%-ная.
Условия получения
Получение фенилгидроксиламииа
К раствору 50 г хлористого аммония в 1600 мл воды в
двухлитровом стакане добавляют, при размешивании, 100 г
нитробензола, а затем маленькими порциями 130 г цинковой
пыли в течение 15—20 минут. Реакция восстановления сопро-
вождается повышением температуры реакционной массы до
60—65°. В том случае, если повышения температуры не наб-
людается, реакционную смесь необходимо осторожно подо-
греть до указанной температуры. Реакционную массу вы-
держивают при температуре 60—65° в течение 10—
15 минут. Горячую реакционную массу фильтруют для удале-
ния гидроокиси цинка, которую затем промывают 200 мл горя-
чей воды. Фильтрат и промывные воды насыщают хлористым
натрием (500 г; необходимо следить за тем, чтобы хлористый
натрий целиком растворился); охлаждают до 0° и выдержива-
ют при этой температуре в течение 0,5 часа.
Выпавший фенилгидроксиламин быстро отфильтровывают,
хорошо отжимают и тотчас же используют для конденсации
с хлористым бензоилом (см. примечание 1).
Вес сырого продукта 118 г (93% теории, принимая во вни-
мание, что влажность его составляет 30%).
12
Получение бензоилфенилгидроксиламина
В трехлитровой колбе растворяют 67,5 г сырого, хорошо
отжатого фенилгидроксиламина в 2 .« теплой воды (40°); по-
лученный раствор отфильтровывают. Фильтрат, охлажденный
до 10°, подщелачивают бикарбонатом натрия до слабощелоч-
ной реакции на лакмус и, при энергичном размешивании, к
нему по каплям добавляют 78 г бензоилхлорида (см. приме-
чание 2).
После добавления всего количества хлористого бензоила
реакционную массу выдерживают при размешивании в течение
1,5 часа. Далее, выпавшую смесь моно- и ди-бензоильных про-
изводных отфильтровывают, выделенный осадок тщательно
растирают с 50 мл 10%-ного раствора бикарбоната натрия,
фильтруют, после чего осадок промывают водой до удаления
запаха хлористого бензола, монобензоильное производное от-
деляют от дибензоильного обработкой смеси водным раствором
аммиака (100 мл), в котором растворяется только монобен-
зоилфенилгидроксиламин. Массу отфильтровывают, а фильт-
рат приливают при энергичном размешивании к 200 мл раз-
бавленной серной кислоты (1 :4). Полученные белые кристал-
лы бензоилфенилгидроксиламина отфильтровывают и промы-
вают холодной водой до отсутствия кислой реакции в промыв-
ных водах по бумажке конго.
Вес технического бензоилфенилгидроксиламина равен 14 г
(27% теории), т. пл. 120—121°.
Полученный продукт перекристаллизовывают из этилово-
го спирта. Вес перекристаллизованного продукта 8,4 г, что со-
ставляет 16% теории, считая на фенилгидроксиламин. Пере-
кристаллизованное вещество имеет т. пл. 121 —122°, что соот-
ветствует литературным данным.
Пр имечания. 1. При хранении сырой фенилгидроксиламин легко
окисляется.
2. При добавлении хлористого бензоила необходимо поддерживать в
реакционной массе щелочную среду, что достигается параллельным прибав-
лением бикарбоната натрия (50 г).
ЛИТЕРАТУРА
1. Е. Bamberger, Вег., 52, 1116(1919).
2. S. Shome, The Analyst, 75, 27 (1950).
3. И. П. Алимарин, Цзе Юнь-Сян, Ж. апалит. химии, 14, 574 (1959).
4. И. П. Алимарин, Цзе Юнь-Сян, Заводск. лаборатория, 25, 1435
(1959).
5. А. К. Majumdar, А. /(. Mukherjel, Anal. Chim. Acta, 21, 245 (1959).
6. И. П. Алимарин, О. М. Петрухин, Докл. АН СССР, 136. 1073 (1961).
7. R. Ц7. Mosbier, J. Е. Schwarberg, Anal. Chetn. 29, 947 (1957).
8. Ciamictan Silber, Gazzetta Chimica Italiana, 36, II, 190; Beilst., 15, 8.
9. E. Beckmann, J. Pract. chem., [2]. 56, 87 (1897).
10. «Синтезы органических препаратов» сб. 1, стр. 432, ИЛ, 1949.
БИССАЛ ИЦИЛАЛЬ-ЭТИЛ ЕНДИАМИН
А. М. ЛУКИН, Г. Б. ЗАВАРИХИНА
(ВНИИ химических реактивов и особо чистых химических веществ)
он но
/ \-CH=N-CH,-CH2-N=CH-/ \
\- / /
C16H16OaN8
м. в. 268,32
Биссалицилаль-этилендиамин предложен как реактив в ос-
новном для люминесцентного определения магния в неводных
средах; это определение характеризуется весьма высокой чув-
ствительностью и избирательностью [1].
Указанное вещество имеет только один метод получения —
конденсацию салицилового альдегида с этилендиамином, осу-
ществляемую в разных средах [2, 3, 4]. Этот метод синтеза
был нами проверен, причем наилучшие результаты, после не-
которых изменений по сравнению с данными литературы, по-
лучены при конденсации в этиловом спирте. В установленных
условиях продукт конденсации образуется даже с лучшим вы-
ходом и при применении дает большую чувствительность ре-
акции с магнием, чем это приводят авторы метода определе-
ния магния, получавшие реактив в бензоле.
Синтез соединения
О-ОН
2 +H2N-CH2-CH2—NH2 —
'х/!-сон
он но
CH=N—СНа-СН8-N=CH— / 4- 2НаО
Характеристика основного сырья
Салициловый альдегид, ч.д.а. ТУ 27—1865.
Этилендиамин, ч., ВТУ-П 327—58, 70%-ный водный раст-
вор.
Условия получения
К раствору 2,4 г салицилового альдегида (0,02 Л4) в 10 мл
этилового спирта добавляют при комнатной температуре 0,8 г
70%-ного раствора этилендиамина (0,009 М). По мере добав-
14
Ления амина в реакционной массе образуются лимонно-жел-
тые кристаллы продукта конденсации; одновременно наблю-
дается небольшой саморазогрев. Массу выдерживают при раз-
мешивании 30—40 минут, и после того как температура вновь
станет комнатной, осадок отфильтровывают, промывают на
фильтре 5 мл этилового спирта и сушат при 60—70°. Вес полу-
ченного вещества 2,4 г, что составляет ~ 93% теоретического
выхода.
Т. пл. полученного продукта 123,5—124°. По данным лите-
ратуры, вещество имеет т. пл. 124—125° [3, 4].
ЛИТЕРАТУРА
1. Ch. Е. White, F. Cuttitta, Analyt. Chem., 31, 2083 (1959).
2. A. T. Mason, Ber„ 20, 267 (1887).
3. А. П. Терентьев, E. Г. Рухадзе, Вести. Моск, ун-та, 4, № 2, 89,
(1949).
4. D. С. Freeman и Ch. Е. White, J. Amer. Chem., Soc. 78, 2678 (1956).
БИУРЕТ
Амид аллофановой кислоты
А. М. ЛУКИН, Е. Я. ЯРОВЕНКО
(ВНИИ химических реактивов и особо чистых химических веществ)
CaHjOjNg
,CONH2
hn/
XCONH,
м. в. 103,08
Биурет находит применение в аналитической практике как
эталон сравнения для обнаружения —СО—NH— группы (би-
уретовой реакции).
По литературным данным, биурет получают путем , нагре-
вания одной мочевины при температуре 150—170° [1—5] или
вместе с РС13 при 100° [6]; при пропускании в расплавленную
мочевину паров HCN [7], хлора [8] или высушенного хлористо-
го водорода [9]; при конденсации мочевины с фумаровой кис-
лотой [10].
15
В основу нижеописанной прописи был положен метод син-
теза из мочевины нагреванием ее при пониженном давлении
[2]. Полученные результаты подтвердили данные патента, од-
новременно были уточнены некоторые детали процесса.
Синтез биурета
,NH2 CONH2
2ОС( ------->HNf +NH3
XNH2 xCONHs
Характеристика основного сырья
Мочевина, ч., ГОСТ 2081—43.
Условия получения
В круглодонную колбу емкостью 1 литр, соединенную с ва-
куумной системой, загружают 100 г (1,6 Af) мочевины и вы-
держивают ее при атмосферном давлении на масляной бане,
нагретой до 145°, до полного расплавления. Затем в колбе
устанавливают вакуум не более 50 мм остаточного давления
(см, примечание 1). После этого мочевину нагревают при тем-
пературе (в массе) 160—169° в течение 30—45 минут, далее
поднимают температуру до 169—174° и выдерживают при этой
температуре еще 40—50 минут. По окончании этого времени
вакуум выключают, а реакционную смесь еще горячей выли-
вают в 380 мл горячей же воды, раствор нагревают до кипе-
ния и фильтруют горячим. Фильтрат охлаждают до 0—' +5° и
выдерживают при этой температуре 6—8 часов, при этом вы-
падают белые кристаллы биурета. Их отфильтровывают и пе-
рекристаллизовывают из 380 мл воды. Высушивают в вакуум-
эксикаторе над хлористым кальцием. Получают 77 г (около
90% теории). Вещество имеет т. пл. 190° (см. примечание 2).
Примечания. 1. Необходимо точно держать вакуум около 50 мм
остаточного давления (не более) и тщательно соблюдать температурные
условия, в противном случае получается продукт, имеющий температуру
плавления значительно выше температуры плавления биурета.
2. По данным литературы, т. пл. биурета равна 188° [12], 190° [9], 192°
[13], 192,5—193° [Ю], 196—197° [И].
ЛИТЕРАТУРА
1, Е. Wiedemann, Liebigs Ann. Chem., 68, 325 (1832).
2. U. Harmon, Пат. США 2145 392 (1939).
3. A. Sonn, Герм. пат. 726 290 (1942).
4. К ОНп, Пат. США 2.370065 (1945).
5. U. Werner, J. Gray, J. Chem. Soc., 24, 111 (1946).
6. Weith, Ber„ 10, 1743 (1877).
7. C. Finckh, Liebigs Ann. Chem., 124, 336 (1862).
16
8. J. Thiele, E. Uhlfllder, Liebigs Ann. Chem., 303, 95 (1898),
9. A. Hoffman, Ber., 4, 255 (1871).
10. P. Friedlander. Lieb'gs Ann. Chem., 229, 240 (1885).
11. Z. Jerzmanow dia—Sienklewlezowa, Roczn. chem., 15, 510 (1937).
12. J. W,gierJauregg, Liebigs Ann. Chem., 561, 87 (Ь48).
13. Bougault, Leboucg, Bull. Soc. chim. France [4], 47, 594 (1930).
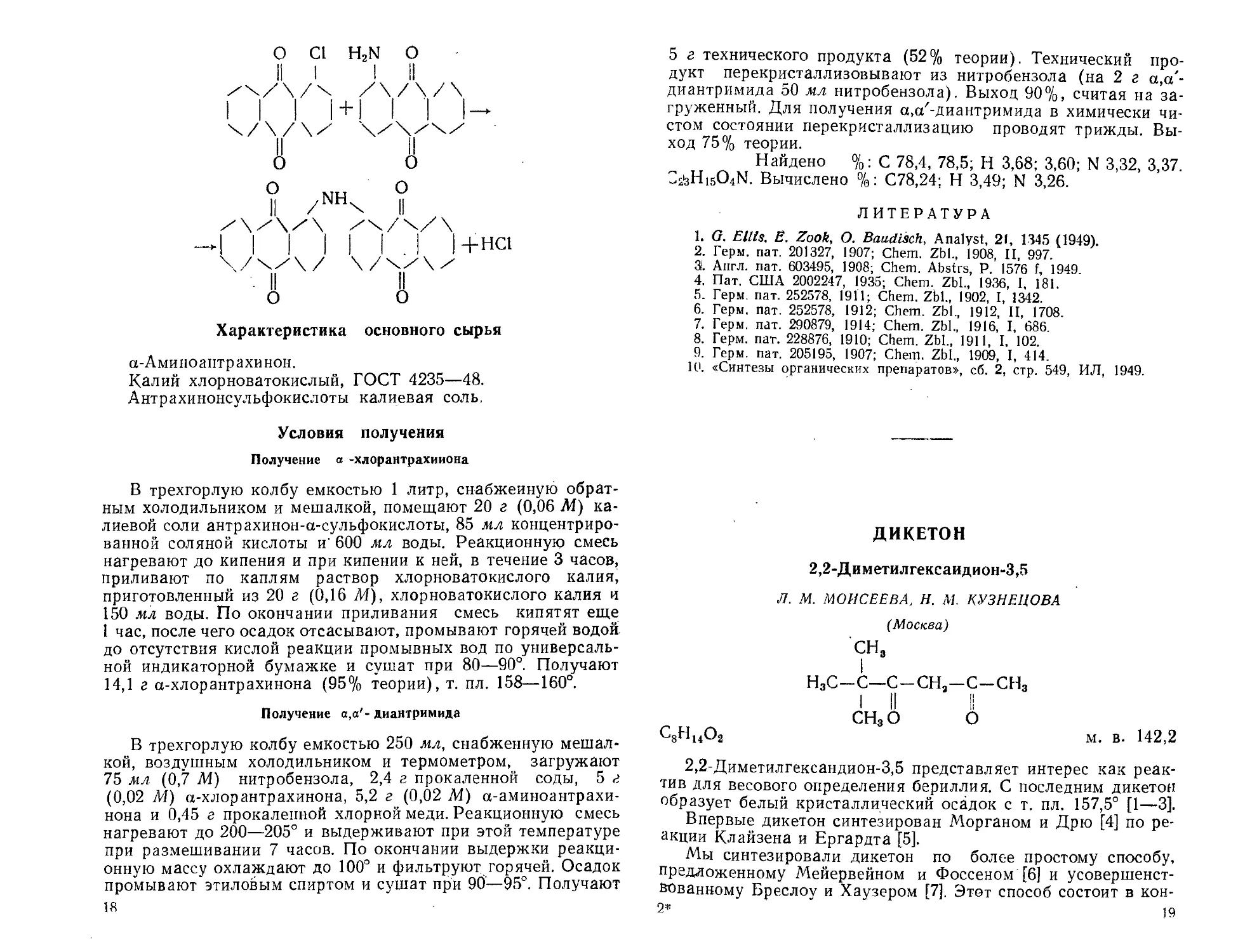
а,а-ДИАНТРИМИД
Диантрахинониламин
E. Я- ЯРОВЕНКО
(ВНИИ химических реактивов и особо чистых химических веществ)
C28H15O4N
м. в. 429,41
а.а'-Диантримид применяется как реактив для микроопре-
деления бора [1].
По литературным данным, а.а'-диантримид получают из
а-аминоантрахинона и а-хлорантрахииона [2, 3], или из 1-хлор-
2-фталимидоантрахинона и 1-хлорантрахинона [4]. а-Хлоран-
трахипон получают из а-аминоантрахинона [5], из а-нитроан-
трахинона [6], из а-оксиантрахинона [7], из антрацен-а-сульфо-
кислоты [8] или из антрахинон-а-сульфокислоты [9, 10].
Нами а,а'-диантримид был получен из а-аминоантрахино- •
на и а-хлорантрахинона, последний получали из калиевой со-
ли антрахинон-а-сульфокислоты [10]. Уточнены условия синте-
за и получен образец в химически чистом состоянии.
Синтез а.а'-диантримида
О SO3K
КС1ОГ) НС1
О С1
Зак. 973
Характеристика основного сырья
а-Амипоаптрахинон.
Калий хлорноватокислый, ГОСТ 4235—48.
Антрахинонсульфокислоты калиевая соль.
Условия получения
Получение а -хлорантрахииона
В трехгорлую колбу емкостью 1 литр, снабженную обрат-
ным холодильником и мешалкой, помещают 20 г (0,06 М) ка-
лиевой соли антрахинон-а-сульфокислоты, 85 мл концентриро-
ванной соляной кислоты и' 600 мл воды. Реакционную смесь
нагревают до кипения и при кипении к ней, в течение 3 часов,
приливают по каплям раствор хлорноватокислого калия,
приготовленный из 20 г (0,16 М), хлорноватокислого калия и
150 мл воды. По окончании приливания смесь кипятят еще
1 час, после чего осадок отсасывают, промывают горячей водой
до отсутствия кислой реакции промывных вод по универсаль-
ной индикаторной бумажке и сушат при 80—90°. Получают
14,1 г а-хлорантрахинона (95% теории), т. пл. 158—160°.
Получение а,а'- диантримида
В трехгорлую колбу емкостью 250 мл, снабженную мешал-
кой, воздушным холодильником и термометром, загружают
75 мл (0,7 М) нитробензола, 2,4 г прокаленной соды, 5 г
(0,02 М) а-хлорантрахинона, 5,2 г (0,02 М) а-аминоантрахи-
нона и 0,45 г прокаленной хлорной меди. Реакционную смесь
нагревают до 200—205° и выдерживают при этой температуре
при размешивании 7 часов. По окончании выдержки реакци-
онную массу охлаждают до 100° и фильтруют горячей. Осадок
промывают этиловым спиртом и сушат при 90—95°. Получают
18
5 г технического продукта (52% теории). Технический про-
дукт перекристаллизовывают из нитробензола (на 2 а а,а'-
диантримида 50 мл нитробензола). Выход 90%, считая на за-
груженный. Для получения а.а'-диантримида в химически чи-
стом состоянии перекристаллизацию проводят трижды. Вы-
ход 75% теории.
Найдено %: С 78,4, 78,5; Н 3,68; 3,60; N 3,32, 3,37.
C^Hi5O4N. Вычислено %: С78.24; Н 3,49; N 3,26.
ЛИТЕРАТУРА
1. G. Ellis. Е. Zook, О. Batidisch, Analyst, 21, 1345 (1949).
2. Герм. пат. 201327, 1907; Chem. Zbl., 1908, II, 997.
3) . Англ. пат. 603495, 1908; Chem. Abstrs, Р. 1576 f, 1949.
4. Пат. США 2002247, 1935; Chem. Zbl., 1936, I, 181.
5. Герм. пат. 252578, 1911; Chem. Zbl., 1902, I, 1342.
6. Герм. пат. 252578, 1912; Chem. Zbl., 1912, II, 1708.
7. Герм. пат. 290879, 1914; Chem. Zbl., 1916, I, 686.
8. Герм. пат. 228876, 1910; Chem. Zbl., 1911, I, 102.
9. Герм. пат. 205195, 1907; Chem. Zbl., 1909, I, 414.
10. «Синтезы органических препаратов», сб. 2, стр. 549, ИЛ, 1949.
ДИКЕТОН
2,2-Диметилгексаидион-3,5
Л. М. МОИСЕЕВА, И. М. КУЗНЕЦОВА
(Москва)
сн8
Н3С-С—С-СН3-С-СН3
I II II
СН3О о
С8НиОа м. в. 142,2
2,2 -Диметилгександион-3,5 представляет интерес как реак-
тив для весового определения бериллия. С последним дикетон
образует белый кристаллический осадок с т. пл. 157,5° [1—3].
Впервые дикетон синтезирован Морганом и Дрю [4] по ре-
акции Клайзена и Ергардта [5].
Мы синтезировали дикетон по более простому способу,
предложенному Мейервейном и Фоссеном [6] и усовершенст-
вованному Бреслоу и Хаузером [7]. Этот способ состоит в кон-
2* 19
денсации уксусного ангидрида с кетонами в присутствии фто-
ристого бора в качестве катализатора. Конденсацией уксусно-
го ангидрида с пинаколином данный дикетон получен впервые.
Синтез дикетона
СНз
I bf8
HjC—-С - С-СН8 + (СН3СО)2О-------->
СНз О
СНз
Н3С-С- С-СНа-С-СН3 + СН3СООН
I II II
СНз О О
Характеристика основного сырья
Пинаколин, ч., ГОСТ 2621—51.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Фтористый бор (получение описано в примечании 1).
Условия получения
В трехгорлую круглодонную колбу, емкостью 1 л, снабжен-
ную мешалкой с ртутным затвором, трубкой для введения
фтористого бора и хлоркальциевой трубкой, помещают 150 г
высушенного над СаС1л и перегнанного пинаколина и 210 г
перегнанного над пятиокисью фосфора уксусного ангидрида.
Конец трубки для введения фтористого бора должен находить-
ся при этом на расстоянии 1 см от поверхности жидкости.
Колбу помещают в лед, включают мешалку (см. примечание 2)
и начинают пропускать фтористый бор до полного насыщения
реакционной смеси (см. примечание 3). Затем реакционную
смесь выливают в 13%-ный раствор уксуснокислого натрия,
из расчета 1 л раствора на каждый моль загруженного пина-
колина. Дикетон отгоняют с паром (см. примечание 4). Ди-
стиллат нейтрализуют 10%-ным раствором едкого натра до
pH 4—5 по универсальному индикатору, и дикетон экстраги-
руют эфиром. Эфирный экстракт высушивают хлористым
кальцием или сернокислым натрием и, после отгонки эфира,
полученное вещество фракционируют, отбирая фракцию, ки-
пящую при 169—172°.
Выход дикетона 89 г (41% теории). Температура кипения
170—17Г, температура плавления медной соли 190° [1].
Пр имечаиия. 1. Для получения фтористого бора поступают сле-
дующим образом. В колбу Вюрца емкостью 2 л помещают 600 г аммония
20
борфтористоводородного (чистого), 102 г борного ангидрида (чистого) и
800 мл 5—6%-ного олеума или столько же концентрированной серной кис-
лоты. Колбу закрывают резиновой пробкой (корковые пробки быстро раз-
рушаются фтористым бором) н реакционную смесь нагревают. Температу-
ру нагревания регулируют в зависимости от скорости выделения фтористо-
го бора. Слишком сильное нагревание в начале процесса может вызвать
выбрасывание пробки и реакционной смеси. Нагревание продолжают до
прекращения выделения газа. Перед введением фтористого бора в реак-
ционную колбу его очищают от HF и SiF4, пропуская через насыщенный
раствор борного ангидрида в олеуме (или серной кислоте).
2. Применять для охлаждения реакционной смеси снег с солью нельзя:
это ускоряет процесс самокондеисации уксусного ангидрида с образовани-
ем ацетилацетоиа.
3. О насыщении реакционной смеси фтористым бором свидетельствует
обильное выделение белых паров из хлоркальциевой трубки. Скорость тока
фтористого бора устанавливается такой, чтобы насыщение закончилось за
4—5 часов.
4. Окончание отгонки определяют по отсутствию красного окрашива-
ния дистиллата при добавлении к нему раствора FeCls.
ЛИТЕРАТУРА
1. Е. С. Пржевальский, Л. М. Моисеева, Вести. Моск, ун-та № 1, 203
(1959).
2. Е. С. Пржевальский, Л. М. Моисеева, Ж. аналит. химии, 15, 117
(1960).
3. Л. М. Моисеева Н. М. Кузнецова, И. И. Пальшина, Ж- аналит. хи-
мии, 15, 561 (1960).
4. О. Т Morgan. Н. D. К- Drew. J. Chem. Soc 121, 937 (1919).
5. L. Claisen, E. F. Ehrhardt, Ber. 22, 1009 (1889).
6. H. Meerwein, D. Vossen, J. pract. Chem, 141, 149 (1934).
7. D. S. Breslow, C. R. Hauser, J. Amer. Chem. Soc., 62, 2385 (1940).
МУРЕКСИД
Пурпурат аммония
A. M. ЛУКИН, И. Д. КАЛИНИНА
(ВНИИ химических реактивов и особо чистых химических веществ)
О о
II II
HN—С С—NH
II II
О=С С —N=CJC=O
I .11 II
HN-C C-NH
ONH4 О
C8H8OeNe м. в. 284, 19
21
Мурексид применяется как реактив для определения неко-
торых катионов, в особенности кальция [1]. В отличие от
большинства описанных в литературе методов получения му-
рексида [2, 3, 4] лишь метод синтеза Пилоти и Финка из алло-
ксантина [5] позволяет получать чистый реактив, что необхо-
димо для удовлетворительного определения кальция. Однако
при увеличении масштаба синтеза, хотя бы в 3—4 раза по
сравнению с данными статьи [5], реактив и по этому методу
получается загрязненным.
Нами было установлено, что образование примесей при
синтезе мурексида по [5] в укрупненном масштабе объясняется
гидролитическим расщеплением мурексида. Отсюда вытекает,
что увеличение в реакционной массе концентрации иона NH4+
по сравнению с данными [5] должно предотвратить указанное
расщепление мурексида. Экспериментальная проверка под-
твердила правильность этого предположения [6] и в настоя-
щее время по приведенной ниже методике нашей химической
промышленностью выпущено значительное количество мурек-
сида.
Синтез мурексида
NH—С=О О=С - NH
О=С НС--О---СОН C=O+2NH3
II II
NH—С=О О=С - NH
[CHsCOONH41
[(nh4),co,j
NH - C=O
----у O=c C- N
I . II
NH-C
ONH4
O=C-NH
I I
= C C=o+2HSO
I I
O=C- NH
Характеристика основного сырья
Аллоксантин, ч., ТУ 694—52. .
Аммоний углекислый, ч., ГОСТ 3770—47.
Аммоний уксуснокислый, ч„ ГОСТ 3117—46.
Условия получения
В раствор 145 г ацетата аммония в 90 мл воды при 70° вно-
сят 11 г карбоната аммония и тотчас же при энергичном пе-
22
ремешивании приливают горячий (80°) раствор 18 а аллоксан-
тина в 540 мл воды. Реакционную массу перемешивают 3—
5 минут при 80—90°. В течение этого времени происходит вы-
падение мурексида в виде блестящих бордово-красных кри-
сталлов с зеленоватым блеском (примечание 1), образовав-
шийся осадок быстро отфильтровывают (примечание 2) и про-
мывают 20 мл спирта. Сушат при 100—110°. Получают 10,8 г
вещества (70% теории).
Найдено %: С 33,91, 33,81; Н 3,18, 3,30.
С8Н8О6Нб Вычислено %: С 33,82; Н 2, 84.
Примечания. I. Контроль синтеза осуществляют при помощи
микроскопа; выпадающие цветные кристаллы должны быть однородными;
появление слабо окрашенных частиц свидетельствует о начале разложения.
2. Качество мурексида значительно ухудшается при длительном на-
хождении выделившегося осадка в реакционной массе, в связи с чем сле-
дует как можно быстрее проводить фильтрование.
ЛИТЕРАТУРА
1. G. Schwarzenbach, W. Biedertnan, F. Bangerter, Helv. chim. acta,
29, 811 (1946).
2. N. Prout, Ann. chim. phys., (2) 11, 48 (1819).
3. F. Wdhler, Liebigs Ann. Chem., 26, 319 (1838).
4. W. Gregory, Liebigs Ann. Chem., 33, 334 (1840).
5. O. PiMy, K. Flnckh, Liebigs Ann, Chem., 333, 22 (1904).
6. A. M. Лукин, И. Д. Калинина, «Вещества высокой степени чистоты
и реактивы», сб. статей ИРЕА, вып. 23, стр. 63, Госхимиздат, .1959.
НАФТАЗАРИН
5,8-Диокси, 1,4-нафтохинон
А. М. ЛУКИН, Е. Я. Я РОВЕН КО .
(ВНИИ химических реактивов и особо чистых химических веществ)
но о
С10НвО4
м. в. 190,10
23
Нафтазарин был предложен как реактив для определения
некоторых катионов, в частности бериллия [1, 2], но в настоя-
щее время широкого применения не имеет.
По л,итературным данным, нафтазарин получают из 1,5-ди-
нитронафталина нагреванием его или с дымящей серной кис-
лотой [3] или с одной частью серы и 10 частями 40 %-кого
олеума [4] или с раствором серы в 40%-ном олеуме [5]; из
1,8-динитронафталина нагреванием его с серной кислотой и
каким-либо восстановителем [6] или электрохимическим вос-
становлением [7] из 1, 2, 5, 8-тетранитронафталина нагрева-
нием с дымящей серной кислотой в присутствии олова [8].
Данные литературы о получении нафтазарина из 1,5-дини-
тронафталина и из 1,8-динитронафталина свидетельствуют о
возможности его получения из технического динитронафтали-
на, представляющего смесь двух вышеназванных изомеров.
Применяя технический продукт, мы действительно получи-
ли нафтазарин в состоянии, близком к химически чистому.
Для осуществления синтеза были использованы условия, ука-
занные в методике [5] с некоторыми изменениями.
Синтез нафтазарина
Химизм образования нафтазарина из нйтронафталинов до
сего времени не выяснен. Имеются различные толкования
этого процесса [9, 10]. Приводим лишь две из предложенных
схем.
NO, NHOH ОН NHa
NO2 ОН NO О NOH
24
о он
\/\/
II I
о он
Характеристика основного сырья
Динитронафталин технический, ОСТ/НКТП-2500.
Моногидрат.
Олеум 35%, ГОСТ 2184—43.
Сера порошковая, фармацевтическая.
Условия получения
Приготовление раствора серы в олеуме
В плоскодонную колбу емкостью 600 мл загружают 225 г
35%-ного олеума и к нему небольшими порциями, при разме-
шивании, добавляют 22,5 г порошка серы, с такой скоростью,
чтобы реакционная масса не разогревалась выше 85°. Обра-
зуется синий раствор.
Получение иафтазарина
В круглодонную трехгорлую колбу на 1,5 л, снабженную
термометром и мешалкой, загружают 45 г технического дини-
тронафталина, растертого в порошок, и 900 г моногидрата и
добавляют в течение двух часов, при размешивании, раствор
серы так, чтобы температура реакционной массы не поднима-
лась выше 40—45° (см. примечание). Затем реакционную мас-
су осторожно, медленно'выливают в трехлитровый сосуд, со-
держащий 2 500 г льда. Массу фильтруют, фильтрат и промы-
вные воды (250 мл) нагревают до кипения, при этом окра-
ска раствора переходит из фиолетовой в красную. При охлаж-
дении выпадает осадок, который отфильтровывают, промыва-
ют водой до исчезновения кислой реакции по бумажке конго
и сушат на воздухе. Выход технического нафтазарина 7,8 г
(20% теории).
Технический нафтазарин перекристаллизовывают из авиа-
ционного бензина с т. кип. 80—110° (на 1 г нафтазарина берут
50 мл бензина).
Найдено %: С 63,12; 63,06; Н 3,23, 3, 36.
СюН6О4. Вычислено %: С 63,12; Н 3,16.
Примечание. Проба иа полноту реакции: 1—2 капли реакционной
массы разбавляют водой, появившаяся при этом синяя окраска подтверж-
дает полноту реакции; красная окраска показывает, что реакция не дошла
до конца.
25
ЛИТЕРАТУРА
1. Т. Toribara, A. Underwood, Anal. Chem, 21, 1352 (1949).
2. Г. Г. Каранович, Труды ИРЕА, вып. 21, стр 43 (1956).
3. De Aqular А. А., А. Bayer, Вег, 4, 253 (1871).
4. Герм, пат., 71386 (1893); 77330 (1894).
5. Р. Мелау, Г. Т. Бухерер, .Практическое руководство по химии
красящих веществ', стр. 259 (1927).
6. Герм, пат., 76922 (1894).
7. Герм, пат., 79406 (1895).
8. W. Will, Вег., 28, 2234 (1895).
9. Вицингер, «Органические красители», стр. LIX, 1936.
10. Q. Flere—David, „Kunstliche Organische Farbstoffe“, стр. 568, 1926.
НИНГИДРИН
Трикетогидринден гидрат
Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
| СО-Н2О
ч/xcoz
С9Н4О3-Н2О
м. в. 178,15
Нингидрин применяется для качественного и количествен-
ного определения аминокислот в самых разнообразных слу-
чаях, например: в бумажной хроматографии, при обнаружи-
вании ракового заболевания, при определении индикана в мо-
че, при определении туберкулеза, при обнаруживании загни-
вания рыбы, в судебной медицине при обнаруживании отпечат-
ков пальцев и т. д.
Нингидрин получают взаимодействием а- или [3-гидриндо-
на с n-нитрозодиметиланилином с последующим разложени-
ем разбавленной кислотой полученного соединения [1]; окисле-
нием индандиона [2] или его изонитрозопроизводного [3] селе-
нистой кислотой, или нитрованием индандиона, бромировани-
ем полученного нитроиндандиона и разложением бромпроиз-
водного [4].
Индандион, в свою очередь, получают или из диэтилового
[2] или диметилового [5] эфиров фталевой кислоты конденса-
26
цией их с этилацетатом в присутствии металлического натрия
и омылением полученного промежуточного продукта, иЛй из/
фталилуксусной кислоты обработкой ее метилатом натрия [6].
Нами проверен и уточнен способ получения нингидрина из
индандиона окислением его селенистой кислотой. Индандион
получали из диэтилфталата.
/Х/соос2н5
| | H-Na+CH3COOC2H5----->
х/хСООС2Н5
/Х/СО
---► I I CNaCOC)C2H5+2CaHBOH
соч
CNaCOOC2H6+H2O
СОХ
H,SO4
/ \/СО\
I | СН2+СО2+СаНБОН
\ \ сох
/Х/СО\
I I CH2-|-H2SeOs
х /XCOZ
--->| СО • H2O+H2O-|-Se
х/ \ со7
Характеристика основного сырья
Диэтилфталат, ч., ТУ МХП 2966—54.
Натрий металлический, ГОСТ 3273—55.
Этилацетат, ч„ ОСТ НКПП 528.
Селенистая кислота, ч., ЦМТУ 1304—46.
Диоксан, ч„ ВТУ МХП 3111—52.
Условия получения
Получение нидаидиома
В трехгорлую трехлитровую круглодонную колбу, снаб-
женную обратным холодильником, капельной воронкой и ме-
ханической мешалкой, загружают 500 мл ксилола (см. приме-
чание 1), 100 г мелко нарезанного металлического натрия и
500 г диэтилфталата. Нагревают содержимое колбы до 80° и
при этой температуре и при размешивании начинают прибав-
лять по каплям смесь из 10 мл абсолютного этилового спирта
и 400 г этилацетата. Скорость прибавления смеси регулируют
27
таким образом, чтобы содержимое колбы равномерно кипело.
Обычно на это требуется 1,5 часа. Температура реакционной
смеси постепенно поднимается и достигает 120—130°, и при
этой температуре ее выдерживают 6 часов. Затем массу охла-
ждают до комнатной температуры и выливают на смесь 250 г
льда и 750 мл воды. (см. примечание 2). Выпавший желтый
кристаллический продукт отсасывают, хорошо отжимают и
сушат при 60—70°. Получают 380—400 г натриевого соедине-
ния эфира дикетогидринденкарбоновой кислоты.
В 8-литровый фарфоровый бачок с механической ме-
шалкой загружают 380—400 г полученного натриевого соеди-
нения и 4,5 л горячей (80—90°) воды. Перемешивают и к ра-
створу добавляют тонкой струйкой 250 мл серной кислоты
(1:3). При этом выделяется углекислый газ и выпадает ин-
дандион. Перемешивают еще 45 минут, проверяют реакцию
раствора по бумажке конго (реакция должна быть кислой).
Охлаждают смесь до +10’ добавлением внутрь кусочков льда,
фильтруют с отсасыванием и промывают индандиои ледяной
водой до отсутствия кислой реакции по конго.
Индандион сушат при 50—60°. Выход 190 г (80% теории).
Т. пл. 122—127°.
Получение нингидрина
В 5-литровую трехгорлую круглодонную колбу, снабжен-
ную обратным холодильником и механической мешалкой, за-
гружают 2,4 л диоксана, 30 мл воды, 138 г селенистой кислоты
и нагревают при перемешивании до 60°. К полученному раство-
ру добавляют 150 г индандиона и кипятят 6 часов. Затем ре-
акционную смесь охлаждают до 50°, фильтруют с отсасывани-
ем от металлического селена и отгоняют от маточника 1800 мл
диоксана (см. примечание 3). К остатку добавляют 1 л дистил-
лированной воды, кипятят 10 минут, охлаждают до комнат-
ной температуры и фильтруют от смолы. От маточника отго-
няют 1050 мл воды в смеси с диоксаном. Остаток еще раз
фильтруют, упаривают на водяной бане до объема 350 мл и
охлаждают до +4°. Выделившийся нингидрин отсасывают и
сушат при 50—60°.
Выход технического продукта 90 г.
Его перекристаллизовывают с углем из трехкратного ко-
личества воды и получают 50 г продукта. Маточник от пере-
кристаллизации упаривают, выделяют нингидрин, перекри-
сталлизовывают его еще раз. Всего получают 60 г продукта
(33% теории).
Качество продукта проверяют по ВТУ МХП 3188—52.
Примечания. 1. Все применяемое сырье должно быть тщательно
высушено.
2. Если реакция полностью не закончилась, то возможно наличие остат-
ков металлического натрия в реакционной смеси.
28
3. Отогнанный диоксаи может быть после перегонки употреблен для
следующего синтеза.
ЛИТЕРАТУРА
1. Ruheman. J. Chem. Soc., 97, 1445 (1910); 99, 799 (1911).
2. Teeters, Shriner, J. Amer. Chem. Soc. 55, 3026 (1933);
Кошелева, Труды ИРЕА, вып. 20, стр. 251, Госхимиздат, 1951.
3. Egaslra, J. Chem. Soc. Japan, 75, 308 (1954).
4. Gustowski, Przem. chem., 11, 694 (1951);
Wanag, Ber., 69. 1069 (1936);
Wanag, Lode, Ber., 71, 1267 (1938);
Christensen, Wang, Davies, Harris, Analyt. Chem., 21, 1573 (1949).
5. Ванач, Галвииь, 1 ирзитис, Изв. АН Латв. ССР, № 4, 131 (1954);
РЖХим, 1956, реф. № 10673.
6. Dominguez, J. Chem. Educ., 30, 624 (1953).
РОДИЗОНАТ НАТРИЯ
В. Г. БРУДЗЬ, л. Ц. ГРАЧЕВА, Д. А. ДРАПКИНА
(ВНИИ химических реактивов и особо чистых химических веществ)
О
II
0= х \ONa
O='4J.'ONa
CeHeNa2
м. в. 214,05
Родизонат натрия в промышленности получают из хлор-
анила через нитранилат, диаминотетраоксибензол и трихино-
ил. Процесс протекает в четыре стадии и требует применения
больших количеств ацетона. Общая продолжительность синте-
за около 42 часов. В литературе описан метод получения ро-
дизоната натрия конденсацией глиоксаля в водном растворе
сульфита и карбоната натрия [1]. Автором указано, что выход
соли родизоновой кислоты приближается к теоретическому.
Однако, при проверке этого метода нами и другими автора-
ми [2] выход родизоната натрия не превышал 5—10% теории.
В более поздней работе [3] роднзоновую кислоту предложено
получать из глиоксаля через тетраоксибензохинон и трихиноил
С общим выходом около 9% теории.
29
В описанный метод [1] получения родизоната натрия нами
внесены следующие изменения: 1) в качестве исходного сырья
применен глиоксальсульфат [4], 2) использован цианистый ка-
лий как катализатор реакции конденсации глиоксаля. Приме-
нение цианистого калия позволило повысить выход, родизона-
та натрия до 16—18% теории. Общая продолжительность син-
теза родизоната натрия по разработанному нами методу со-
ставляет 20 часов, включая стадию получения глиоксальсуль-
фата.
Синтез родизоната натрия
Н
OaS( | ^SO24-2NaaCO3
\о-с-ох
н
н
с=о
---| 2Na2SO44" 2СОа
с—о
I
1
н
н о
з С = ° °*___________, O = /\,ONa
। NasCO3, NaaSO3, KCN О=Д J.'oNa
c=o и
Характеристика основного сырья
Глиоксальсульфат, ч., РТУ 557—60.
Сода кальцинированная техн., ГОСТ 5100—49.
Сульфит натрия кристаллический, ГОСТ 903—41.
Цианистый калий, ГОСТ 8465—57.
Условия получения
Получение раствора глиоксаля. В 460 мл воды
постепенно вносят цри размешивании и температуре 70—75°
115 г глиоксальсульфата. Раствор охлаждают до 30—35°, ней-
трализуют содой до:pH 5—6 и отфильтровывают. Получают
около 550 мл раствора глиоксаля....
30
Получение родизоната натрия. В раствор, при-
готовленный из 500 мл воды, 100 г кальцинированной соды,
63 г кристаллического сульфита натрия, при размешивании и
температуре 30° вносят 2,0 г цианистого калия. Затем добав-
ляют раствор глиоксаля. После внесения всего раствора гли-
оксаля реакционную массу выдерживают при 50° и размеши-
вании в течение 2 часов, далее охлаждают до 18—20° и от-
фильтровывают. Осадок родизоната натрия, тщательно отжа-
тый, промывают сначала 15 мл 10%-ного раствора ацетата
натрия и затем 2 раза (по 15 мл) спиртом. Сушат при 60°.
Выход 5,9—6,5 а (16—17,5% теории). Качество препарата со-
ответствует техническим требованиям ВТУ 2892—53.
ЛИТЕРАТУРА
1. В. Homolke, Вег., 54, 1393 (1921); герм. пат. ЗШ46 (1921).
2. «Реактивы и препараты лабораторного назначения» под редакцией
В. В. Лонгинова, стр. 259, ГОНТИ, 1938.
3. В. Eistert, Q. Bock, Angew. Chemie, 70, 19, 595 (1958).
4. В. Г. Брудзь, Д. А. Драпкина, И. С. Маркович, «Химические реакти-
вы и препараты», сб. статей ИРЕА, вып. 24, стр. 98, Госхимиздат, 1961.
САЛИЦИЛАЛЬ- О-АМИНОФЕНОЛ
А. М. ЛУКИН,\~Ё. Д. ОСЕТРОВ А |
(ВНИИ химических реактивов и особо чистых химических веществ)
ОН но
\—/ н \_/
ClsHuOaN м. в. 213,23
Салицилаль-о-аминофенол был предложен Хольцбехером
[1] для открытия и определения алюминия. Метод основан на!
возникновении люминесценции под ультрафиолетовыми луча-
ми при прибавлении раствора салицилаль-о-аминофенола к
водному раствору соли алюминия. После дополнительного ис-
следования этого метода во ВНИИ химических реактивов [2]
были найдены оптимальные условия определения и достигну-
та чувствительность реакции, в 20 раз большая указанной
Хольцбехером. В результате этих работ салицилаль-о-амино-
фенол можно считать реактивом для количественного люми-
31
несцентного определения алюминия с высокой степенью чувст-
вительности (0,0025 у А1 в 5 мл раствора).
Салицилаль-о-аминофенол, по данным литературы, полу-
чают конденсацией салицилового альдегида с о-аминофено-
лом в 50%-ной уксусной кислоте [3]. Данный метод был нами
проверен [4], причем в условия синтеза были внесены лишь не-
значительные изменения и уточнения. Полученное вещество
пригодно для люминесцентного определения алюминия с ука-
занной выше чувствительностью.
Синтез салицилаль- о -аминофенола
ОН НО
Характеристика основного сырья
Салициловый альдегид, ч.д.а., СТ 27—1865.
о-Аминофенол, ТУ МХП 1979—49.
Условия получения
В трехгорлой колбе (с термометром, мешалкой и обратным
холодильником) нагревают на водяной бане суспензию 21,85 г
о-аминофенола (0,2 М) в 200 мл 50%-ной уксусной кислоты
при размешивании до температуры 68—71° и выдерживают
0,5 часа при этой температуре. В течение этого времени пер-
воначальная белая суспензия переходит в розовато-желтую.
Затем к реакционной массе добавляют 24,45 г (0,2 М)
(21,2 мл) салицилового альдегида. По мере внесения альдеги-
да суспензия тотчас же становится оранжевато-красной. Да-
лее, массу размешивают при той же температуре еще один
час, после чего быстро охлаждают (снегом или льдом) и раз-
мешивают еще один час. После этого массу отфильтровывают,
тщательно отсасывают под вакуумом и промывают водой до
получения бесцветного фильтрата. Промытую и отжатую па-
сту сушат при температуре 90—100°. Вес полученного вещест-
ва 38,5 г, т. пл. 185—187° (т. пл., по данным литературы [5],
равна 185°).
Найдено %: N 6,45; 6,38.
С13НцО£М. Вычислено %: N 6,57.
32
Таким образом, вещество получается в состоянии, близком
к химически чистому, и выход его составляет 90% теории.
ЛИТЕРАТУРА
1. Holzbecher, Zavis, Chetn. listy. 47, 5, 680 (1953).
2. E. А. Божевольнов, Труды ИРЕА, вып. 22, стр. 70, Госхимиздат, 1958.
3. Е. Haegele, Вег., 25, 2753 (1892).
4. А. М. Лукин, |Е. Д, Осетр ова\, Труды ИРЕА, вып. 21, стр. 3, Гос-
химиздат, 1956.
5. Е. Haegele, Вег., 26, 394 (1893).
тиооксин
(8-меркаптохинолин)
И 8,8-ДИХИНОЛ ИЛДИСУЛЬФ^ИД
И. ШЕВЧУК, Э. ЛУКША
(Завод химреактивов и Институт Химии АН Латв. ССР)
8-Меркаптохинолин и его дисульфид (8,8'-дихинолилди-
сульфид), как установлено работами Банковского и его сот-
рудников, обладают ценными качествами как аналитические
реактивы.
Метод синтеза тиооксина (8-меркаптохинолина) впервые
описан Эдингером [1], упрощен Банковским с сотрудниками
[2, 3]. Нами [4] (в экспериментальной работе принимал участие
Иванов И.. М.) разработанный метод синтеза значительно со-
кращен и имеет ряд преимуществ перед ранее разработан-
ными.
Синтез и очистка натриевой соли 8-меркаптохинолина
HaSO4
3 Зак. 973
I N
SOgH
NaOH
I N
SO9Na
33
PC1S
SnCl2
—*
| | |-HaSnCle
Y nz
SH
HaSnCl6
NaOH
|ХЧ|/Х|-ЗН2°
I N
SNa
[Z YYh2O
Y V
SNa
HC1
|/X|/4|'2H»0
\/\/
I N
SH
|Z Y X|-2H2O
Y Y
SH
C2H6ONa
•3H2O
Характеристика основного сырья
Хинолин, ч„ ТУ МХП 93—47.
Олеум 65%, х.ч., ВТУ МХП 1477—53.
Кислота серная, х.ч., ГОСТ 4204—48.
Едкий натр, х.ч., ГОСТ 4328—48.
Фосфор пятихлористый, ч.д.а.
Аммиак водный, 25%, ГОСТ 3760—47.
Кислота соляная, х.ч., ГОСТ 3118—46.
Кислота соляная, реактивная.
Олово двуххлористое, ч.д.а., ГОСТ 36—40.
Эфир диэтиловый, наркозный.
Спирт этиловый, ректификат.
Натрий металлический, ч.
Натрий виннокислый, средний, ч.д.а, ГОСТ 3656—51.
34
Условия получения тйооксина
Получение хииолин-8-сульфокислоты и ее натриевой соли
50 г хинолина в течение 10 минут приливают при переме-
шивании к 40 г серной кислоты (1,84). Охлажденный твердый
сульфат хинолина при перемешивании небольшими порциями
в течение 1,5—2 часов вносят в 200 г олеума (50—55%). на-
литого в трехгорлую колбу емкостью 250 мл, снабженную при-
шлифованным обратным холодильником, через который про-
пущена стеклянная мешалка (в этой операции можно ограни-
читься перемешиванием вручную). Колба устанавливается в
ледяную баню. Во второе горло колбы вставляется термометр.
Через третье горло вводится сульфат хинолина. Во время
внесения сульфата хинолина температура смеси не должна
превышать 100°.
После внесения сульфата хинолина колбу устанавливают
в масляную баню и выдерживают при 160° в течение 4 часов.
Охлажденное содержимое колбы выливают на 0,5 кг льда и
через 2 часа отфильтровывают выпавшую сульфокислоту, ко-
торую промывают на фильтре двумя порциями воды по 10 мл
и отжимают.
Полученную хинолин-8-сульфокислоту помещают в фарфо-
ровую чашку диаметром 20—25 см и при перемешивании при-
ливают небольшими порциями 20%-ный раствор едкого нат-
ра до нейтральной реакции. Раствор натриевой соли осто-
рожно выпаривают почти досуха и высушивают при 160° в
течение 8—10 часов. Сухую соль тщательно измельчают в
фарфоровой ступке до мелкого порошка. Получают 51—56 г
натриевой соли. Для дальнейшего синтеза важна степень из-
мельчения натриевой соли. Она не должна содержать на
ощупь твердых крупинок.
Получение хинолии-8-сульфохлорида
Полученную натриевую соль путем встряхивания смешива-
ют в колбе емкостью 0,5 л с закрытой пробкой с равным по
весу количеством пятихлористого фосфора, который предвари-
тельно измельчен до порошка. Смесь переносят в стакан ем-
костью 150—200 мл, который погружают приблизительно до
половины в масляную баню и нагревают. При температуре
80—100° происходит разжижение смеси. При постоянном пе-
ремешивании смеси температуру бани поднимают до 140—145°
и выдерживают до начала загустевания, что достигается в
течение 40—60 минут. Стакан вынимают из бани и немедлен-
но все содержимое переносят в большую фарфоровую ступку
или чашку и распределяют равномерным слоем. После начала
схватывания массу соскабливают, одновременно измельчая и
перемешивая. Затем в ступку наливают 300—400 мл воды со
льдом и продолжают измельчение под водой в течение 10 ми-
3* 35
нут. Полученный сульфохлорид на фильтре промывают двумя
порциями воды по 100 мл и отсасывают. Выход 63—65 г влаж-
ного сульфохлорида. Измельчение реакционной массы следует
производить еще при полужидком состоянии, так как после
затвердевания она очень трудно измельчается.
Получение хлороловянной соли 8-меркаптохииолина
Сульфохлорид немедленно подвергают дальнейшей обра-
ботке во избежание разложения его. 160 г хлористого олова
растворяют в 400 мл концентрированной соляной кислоты.
63—65 г сульфохлорида растворяют в 250 мл концентрирован-
ной соляной кислоты и немедленно приливают тонкой струей
полученный раствор к заранее приготовленному раствору хло-
ристого олова при постоянном перемешивании. Смесь разогре-
вается, и выпадает лимонно-желтый кристаллический осадок
хлороловянной соли 8-меркаптохинолина. После охлаждения
соль отфильтровывают, промывают соляной кислотой (1:1),
затем 1—2 раза водой и тщательно отжимают. Получают 93—
100 г влажной соли.
Получение натриевой соли 8-меркаптохинолииа
Растирают в ступке 100 г хлороловянной соли и 100 мл
воды до получения однородной пасты и переносят небольши-
ми порциями дистиллированной воды в стакан емкостью 2 л.
Общий объем израсходованной воды не должен превышать
400 мл. В суспензию вносят 92,6 г среднего тартрата натрия и
55,6 г едкого натра и перемешивают стеклянной палочкой до
растворения тартрата и едкого натра. Смесь при этом разо-
гревается и цвет переходит из оранжево-желтого в серовато-
желтый. Комочки неразложившейся хлороловянной соли сле-
дует размять стеклянной палочкой и для завершения разло-
жения смесь нагреть до кипения. Охлажденную до 15° смесь
фильтруют, осадок промывают 100 мл раствора, содержащего
5 г едкого натра и 15 г среднего тартрата натрия, и отжимают.
Фильтрат отбрасывают.
Получение дигидрата 8-меркаптохииолииа
Сырую натриевую соль переносят в стакан емкостью 2 л и
растворяют при перемешивании в 1 л дистиллированной воды.
Буровато-желтый раствор доводят соляной кислотой осторож-
но, при постоянном перемешивании, до pH 5,5—6. Начиная с
pH 8 выпадает ярко-красный дигидрат 8-меркаптохинолина, и
по мере добавления кислоты смесь густеет от выпадающего
осадка. Образовавшийся дигидрат оставляют на ночь. На сле-
дующее утро к дигидрату приливают перегнанный аммиак до
образования красновато-оранжевого (йо не желтого!) раство-
ра. Полученный раствор фильтруют через самый плотный
двойной фильтр. Прозрачный фильтрат немедленно, переносят
36
в стакан (2 л) и тотчас Доводят особо чистой соляной кисло-
той до pH 5,5. Выпавший при этом красный осадок дигидрата
8-меркаптохинолина фильтруют через беззольный фильтр,
промывают 50 мл бидистиллированной воды и тщательно от-
жимают с хорошим отсасыванием.
Получение чистой натриевой соли 8-меркаптохииолииа
Полученные 50 г влажного дигидрата переносят в кониче-
скую литровую колбу из термостойкого стекла, приливают
220 мл этанола и легким нагреванием растворяют. К раствору
дигидрата медленно приливают раствор этилата натрия (4 г
чистого металлического натрия в 400 мл этанола) до перехода
окраски раствора из темно-красной в чисто-желтую, затем еще
5 мл раствора этилата. Некоторое количество этилата может
остаться неиспользованным. Колбу с образовавшимся желтым
раствором и желтыми кристаллами нагревают до кипения. Ес-
ли кристаллы при этом не растворились, добавляют необходи-
мое количество этанола и снова нагревают до кипения и воз-
можно быстро фильтруют через беззольный фильтр. Фильтрат
быстро переносят в стакан и оставляют для кристаллизации.
После полного осаждения лимонно-желтые кристаллы натри-
евой соли 8-меркаптохинолина отфильтровывают, промывают
100 мл диэтилового эфира, переносят на фильтровальную бу-
магу и сушат на воздухе 20—30 минут. Выход 28—30 г, или
30—32% теории, считая на хинолин.
Синтез 8,8'дихинолилдисульфида
I N
SH
Характеристика основного сырья
Пергидроль, ос. ч., ГОСТ 177—41.
Хлороформ, х.ч., ч.д.а., ГОСТ 3160—51.
Спирт этиловый, ректификат или спирт метиловый, синте-
тический, ч.д.а., ВТУ МХП 2220—50.
о-Ксилол, ВТУ ЛАХП 2940—51.
Гипофосфит натрия, ч.д.а., ГОСТ 200—41.
Едкий натр, х. ч., ГОСТ 4328—48.
Условия получения 8,8'-дихинолилдисульфида
Получение 8,8'-дихинолилдисульфида из аммиачного раствора
8-меркаптохинолина
В чистый стакан емкостью 2 л помещают не содержащий
тяжелых металлов аммиачный раствор 8-меркаптохинолина
37
(см. получение дигидрата 8-меркаптохинолина) и небольшими
порциями (по 0,5—2 мл) осторожно, при постоянном переме-
шивании приливают перекись водорода (10—30%) до исчезно-
вения желтой окраски раствора (не более!). Выпавший белый
творожистый осадок дисульфида тщательно промывают на
фильтре дистиллированной водой, отсасывают и сушат при
120—130° в течение 6 часов.
Для получения дисульфида из очищенной натриевой соли
8-меркаптохинолина 25 г соли растворяют в 1,5—2 г дистилли-
рованной воды и дальше поступают, как описано выше.
Очистка 8,8'-дихинолилдисульфида
25 г полученного сухого дисульфида в литровой конической
колбе заливают 500 мл хлороформа и при встряхивании раст-
воряют до исчезновения кусков. Раствор фильтруют через
фильтр средней плотности в коническую колбу емкостью
3 литра. К желтоватому фильтрату добавляют 2 л этанола или
метанола. Через 2—3 часа выпавший дисульфид отфильтровы-
вают на нутче. Температура плавления чистого 8,8'-дихино-
лилдисульфида по Эдингеру [1] 206°. Выход 68—70% кристал-
лического дисульфйда от взятого количества некристаллизо-
ванного продукта.
Для перекристаллизации из ксилола 25 г сухого дисульфи-
да растворяют в 400 мл кипящего ксилола. Раствор фильтру-
ют по возможности горячим. Фильтрат переносят в коническую
колбу на 0,5 л и оставляют на 2—3 часа в холодильном шкафу
для кристаллизации. Кристаллы отфильтровывают и промы-
вают двумя порциями (по 50 мл) этанола. Выход — 75—80%
от взятого количества. Продукт для аналитических целей ме-
нее пригоден, чем полученный по первому способу.
Восстановление 8,8'-дихинолилдисульфида
25 г 8,8'-дихинолилдисульфида, 12,5 г NaH2PO2-H2O, 38 мл
конц. соляной кислоты и 12 мл дистиллированной воды поме-
щают в трехлитровую коническую колбу и кипятят на элек-
троплитке в течение 2—3 минут. Охлажденную смесь разбав-
ляют дистиллированной водой до 2 л и 20%-ным раствором
едкого натра осторожно, при постоянном перемешивании, ней-
трализуют кислоту до pH 5,0—5,5. Полученный дигидрат 8-
меркаптохинолина превращают в натриевую соль 8-меркапто-
хинолина, как описано выше. Выход 20,5—22 г чистой натрие-
вой соли 8-меркаптохинолина (55—60% теории, считая на ди-
сульфид).
ЛИТЕРАТУРА
1. A. Edinger., Вег., 41, 937 (1908).
2. JO. А. Банковский, Изв. АН Латв. ССР, 1952, № 12, 127.
38
3. Ю. А. Банковский, А. Ф. Иевиныи и Э. А. Лукша Ж. общ. химии,
28, 2273, (1958).
4. И. Шевчук, Э. Лукша. Изв. АН Латв. ССР, 1961, К» 2, 127.
1,1- ГИДРАЗИНДИУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИИ, В. Я. ТЕМКИНА, Е. И. МИРОНОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
CHgCOOH
h2nn^
' СН3СООН
C4H8O4N4 м. в. 148,12
1,1-Гидразиндиуксусная кислота может быть получена вза-
имодействием гидразингидрата с монохлоруксусной кислотой
в среде абсолютного спирта [1].
При проверке описанного способа нами внесены измене-
ния, существенно упрощающие проведение реакции.
Синтез соединения
H2NNH2+2ClCH2COOH4-2NaOH —
. СН2СООН
--->H2NN( 4-2NaCl+2HaO
х СН2СООН
Характеристика основного сырья
Гидразингидрат, ч., ОСТ 10955—40.
Условия получения
Монохлоруксусная кислота, техническая, 1 сорт, ОСТ 40052.
В трехгорлую колбу, снабженную обратным холодильни-
ком, термометром и мешалкой, помещают 80 г (0,85 М) моно-
хлоруксусной кислоты и нейтрализуют ее 20%-ным раствором
едкого натра с такой скоростью, чтобы температура не превы-
шала 40°. К полученному раствору, нагретому до 60°, посте-
пенно при перемешивании прибавляют раствор 21 г (0,42 М)
гидразингидрата в 140 мл 20%-ного раствора едкого натра,
следя за тем, чтобы температура не поднималась выше 80°.
Реакционную смесь выдерживают при температуре 70—80° в
39
течение 6 часов, поддерживая pH 9—10 прибавлением щелочи.
После охлаждения раствор фильтруют, фильтрат подкисляют
концентрированной соляной кислотой. Выпавший осадок про-
мывают дистиллированной водой и спиртом. Выход 27 г (45%
теории). Содержание основного вещества 98% (определено ме-
тодом потенциометрического титрования).
Найдено %: N18.8, 19,1.
C4H8O4N2. Вычислено %: N 18,9.
ЛИТЕРАТУРА
1. Т. Curtius, L, Hussong, J. Prakt. Chem., (2), 83, 271 (1911).
Р-ОКСИЭТИЛИМИНОДИУКСУСНАЯ КИСЛОТА
P. П. ЛАСТОВСКИЙ, В. Я. ТЕМКИНА, Е. И. МИРОНОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
СН2СООН
HOCHjCH2-N^
СН2СООН
C6HnO5N м. в. 177,16
По литературным данным, ₽-оксиэтилиминодиуксусная
кислота может быть получена взаимодействием этаноламина с
этиловым эфиром монобромуксусной кислоты в среде хлоро-
форма [1], взаимодействием этаноламина с монохлоруксусной
кислотой в водной среде [2]. Выход продукта в работах не
приведен.
Нами проверен второй метод получения комплексона и
внесены некоторые уточнения.
Синтез соединения
HOCH2CH,NH2+2ClCH2COOH+2NaOH —>
CHjCOOH
—>HOCH2CH2N^ +2NaCl+2H2O
'CH2COOH
Характеристика основного сырья
Этаноламин, ч., ВТУ 632—52.
Монохлоруксусная кислота, техническая, 1 сорт, ОСТ 40052.
40
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, термометром и мешалкой, помешают 105 г (1,1 Л4) моно-
хлоруксусной кислоты, нейтрализуют ее 20%-ным едким нат-
ром, нагревают до 60° и добавляют, при перемешивании, 30 а
(0,5 М) этаноламина с такой скоростью, чтобы температура
внутри колбы не поднималась выше 80°. Реакционную массу
выдерживают при 70—80° в течение 4 часов, поддерживая pH
9—10 прибавлением 20%-ного едкого натра. Охлажденный
раствор фильтруют и фильтрат подкисляют концентрирован-
ной соляной кислотой до pH 2. Выпавший осадок отфильтро-
вывают, промывают дистиллированной водой и затем спир-
том.
Выход комплексона 45 г (55% теории). Содержание основ-
ного вещества 98% (определено методом потенциометрическо-
го титрования).
Найдено %: N 8,0, 8,1.
CeHnOgN. Вычислено %: N 7,9.
ЛИТЕРАТУРА
1. А. И. Киприянов, Укр. хим. ж., 4, 239 (1929).
2. О. Schwarzenbach, G. Anderegg, IF. Schneider, H. Senn, Helv. Chim.
Acta, 38, 1166 (1955).
ЭТИЛЕНДИАМИНТЕТРААЦЕТАТ СВИНЦА
P. IL ЛАСТОВСКИЙ, В. Я. ТЕМКИНА, И. А. СЕЛИВЕРСТОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
H,[Pb(C10HJ2NaO8)]
CioHi4Na08Pb м. в. 497,44
По литературным данным, этилендиаминтетраацетат свин-
ца получают взаимодействием эквивалентных количеств гидро-
окиси свинца и двунатриевой соли этилеидиаминтетрауксус-
ной кислоты с последующим осаждением хлорной кислотой и
ацетоном [1]. Условия синтеза и выделения комплексоната не
приведены.
Нами найдены оптимальные условия проведения реакции и
осаждения этилендиаминтетраацетата свинца.
Синтез соединения
Na2[H2(C10H12NaO8)]+Pb(OH)2—> Na2[Pb(C10H12N2O8)]4-2H2O
Naa[Pb(CrtH12N208)]+2HG104—>H2[Pb(C10H12N2O8)]-|-2NaClO4
41
Характеристика основного сырья
Трилон Б, ч., РУ-819-53.
Гидрат окиси свинца, свежеприготовленный.
Хлорная кислота, уд. в. 1,53, ТУ МХП № ОРУ 87—57.
Условия получения
В трехгорлой колбе, снабженной обратным холодильником,
мешалкой и термометром, растворяют при 70°—80° 18,6 г
(0,05 М) трилона Б в 100 мл дистиллированной воды. К раст-
вору добавляют 12,06 г (0,05 М) свежеприготовленной гидро-
окиси свинца (см. примечание 1) и выдерживают реакцион-
ную массу до полного растворения гидроокиси. Охлажденный
до комнатной температуры раствор (pH 3,5—4,0%) фильтру-
ют. К раствору добавляют 50 мл ацетона и подкисляют хлор-
ной кислотой до pH 1,5, после чего высаживают комплексонат
прибавлением 100 мл ацетона (см. примечание 2).
Выпавший белый кристаллический осадок отфильтровыва-
ют, промывают ацетоном и сушат при 80°.
Выход 21 г (84,4% теории).
Найдено %: РЬ 41,58.
CioHuNaOsPb. Вычислено %: РЬ 41,65.
Примечания. 1. Для получения гидроокиси свинца к горячему ра.
створу 100 г уксуснокислого свинца (ч.д.а.) пря перемешивании добавляют
40 мл NH<OH, проверяя полноту осаждения. Осадок промывают горячей
водой до отсутствия запаха уксусной кяслоты и сушат при 100° [2].
Выход 52,8 г гидроокиси (87% теории).
2. Наиболее сложным моментом данного синтеза является выделение
комплексоната в кислотной форме. Следует строго соблюдать порядок при-
бавления реагентов я pH среды, в противном случае может выпасть этп-
лендяаминтетрауксусная кислота.
ЛИТЕРАТУРА
1. Л. Saito. К. Suga, Nuovo Cimento, IL, X, 11, 600 (1959).
2. Ю. В. Карякин, \И. И. Ангелов |, .Чистые химические реактивы",
стр. 471, Госхимиздат, 1955.
2,5-ДИНИТРОФЕНОЛ
у-Динитрофенол
Г. И. КОШЕЛЕВА, Г. Д. ГЛЕБОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
ОН
|/V|-NO2
C6H4O5Na OaN/X/ м. в. 184,15
42
у-Динитрофенол является кислотно-основным индикатором
с переходом окраски от бесцветной к желтой в интервале pH
4,0—5,4.
Применяется при определении концентрации водородных
ионов по Михаэлису в бактериологии и при титровании ела
бых кислот и оснований в спиртовых растворах.
у-Динитрофенол обычно получают нитрованием л-нитрофе-
нола [1] с выходом 15% теоретического. Наряду с 2,5-динитро-
фенолом, при этом получают значительные количества 2,3- и
3,4-динитрофенолов. Отделение от изомеров проводят путем
перекристаллизации из спирта, бензола или уксусной кислоты.
Другие методы получения [2] продукта по недоступности сырья
или малому выходу практического значения не имеют.
Нами предложено [3] получать у-динитрофенол окислением
5-нитро-2-аминофенола кислотой Каро, что позволило значи-
тельно увеличить выход продукта и избежать сложного разде-
ления изомеров перекристаллизацией.
ОН ОН
' /NH, /I /NOа
| | (NHJ2S2O8+H2SO4
O2N/X/ O2N/X/
Характеристика основного сырья
5-Нитро-2-аминофенол, 99,7%, ВТУ завода.
Серная кислота, ч., ГОСТ 4204—48.
Персульфат аммония, ч., ГОСТ 3766—47.
Условия получения
Растирают в фарфоровой ступке 52,9 г (0,3 Л4) 5-нитро-2-
аминофенола в 300 мл концентрированной серной кислоты до
получения прозрачного раствора.
В 0,5 л фарфоровый стакан загружают 350 г персульфата
аммония и 500 мл воды, перемешивают механической мешал-
кой до растворения и к раствору добавляют по каплям раст-
вор 5-нитро-2-аминофенола с такой скоростью, чтобы темпера-
тура реакционной смеси не поднималась выше 30°. Затем пе-
ремешивают еще 4,5 часа при 29—32° и оставляют до следую-
щего дня.
Выделившийся у-динитрофенол, отсасывают, промывают
4%-ным раствором соляной кислоты до получения светло-жел-
тых промывных вод, затем водой до отсутствия кислой реак-
ции по бумажке конго и сушат при .50—60°. Получают 35—
39. г технического продукта. Его перекристаллизовывают с ак-
тивированным углем из 700 мл 50%-ного спирта. '
43
Выход у-динитрофенола с т. пл, 104—107,5° составляет
29 г (48% теории).
Химически чистый образец получают при 2-кратной пере-
кристаллизации технического продукта из 10-кратного количе-
ства спирта.
Найдено %: N 15,32; 15,56.
C6H4O5N2. Вычислено %: N 15,21.
. ЛИТЕРАТУРА
1. А. Bantlin, Вег., 8, 21 (1875); 11. 2108 (1878);
R. Henrigues, Liebigs Ann. Chem., 215, 324 (1882).
2. F. Reverdin. Ber., 39, 2691 (1906);
T. Slgiwlck, V. Aldous, J. Chem. Soc., 119, 1002 (1921);
M -И. Богданов, Анилинокрасочная пром-сть, 3, 133 (1933).
3. Г. Кошелева, Г. Глебова, Д. Фрайштат, Авт. свнд. СССР 111826,
2.08.57.
НИТРАЗИНОВЫЙ ЖЕЛТЫЙ
Дельта, динатриевая соль 2,4-динитробензол-азо-1-нафтол-3,6-
дисульфокислоты
Г. Н. КОШЕЛЕВА, Г. Д. ГЛЕБОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
^leHeOnNiNaaSj
м. в. 542,39
Нитразиновый желтый является кислотно-основным инди-
катором с переходом окраски от желтой к синей в интервале
pH 6,2—7,6. Применяется при титровании сильных кислот и
оснований, для приготовления индикаторной бумаги и для ана-
лиза молока.
Нитразиновый желтый получают сочетанием диазотирован-
ного 2.4-динитроанилина с 1-нафтол-3,6-дисульфокислотой [1].
Нами был проверен и уточнен метод получения, описанный
в литературе.
44
20tN-^ NH2+2NaNO24-2HsSO4 —>
П
NOa
Характеристика основного сырья
2,4-Дииитроанилин, ВТУ РУ 860-53.
Нитрит натрия, ч., ГОСТ 4197—48.
Кислота серная, ч., ГОСТ 4204—48.
1-Нафтол-3,6-дисульфокислота, ВТУ завода.
Условия получения
В фарфоровый стакан с мешалкой загружают 50 мл кон-
центрированной серной кислоты и при размешивании посте-
пенно порциями, при температуре не выше 10° вносят 4,5 г
(0,064 М) хорошо растертого нитрита натрия. Затем для пол-
ного растворения смесь нагревают до 50°. Полученную нитро-
зилсерную кислоту охлаждают до 10° и к ней порциями при
перемешивании при температуре 10—15° добавляют 13 г
(0,071 Л4) динитроанилина. Затем перемешивают еще час и
реакционный раствор выливают на 320 г мелко толченного
льда. Диазораствор быстро фильтруют через охлажденную
воронку от небольшого количества непрореагировавшего 2,4-
динитроанилина.
В фарфоровый стакан на 2 л с мешалкой загружают раст-
вор 20 г (0,058 М) натриевой соли 1 -нафтол-3,6-дисульфокис-
лоты в 1000 мл воды и 96 а уксуснокислого аммония. Пускают
в ход мешалку, охлаждают раствор до 10° и приливают про-
фильтрованный диазораствор. Краситель выпадает в виде
красно-коричневого осадка.
45
Его отсасывают и сушат при 30° до постоянного веса. Вы-
ход индикатора с содержанием основного вещества не менее
80% (спектрофотометрически) составляет 25—26 г (69—71%
теории).
Для получения химически чистого образца технический
продукт ДваждЬьперекристаллизовывают из воды в соотноше-
нии 1: 10 и дважды Из 20%-ной уксусной кислоты в соотно-
шении 1:12.
Найдено %: N 11,27, 10,96.
C16H10N4O11SS'. Вычислено %: N 11,25.
ЛИТЕРАТУРА
1. Вассерман, Заводск. лаборатория, 868 (1934);
Wenker, Industr. and Eng. Chem., 26, 350 (1934).
ПРОПИЛОВЫЙ КРАСНЫЙ
ЬДипропиланилин^-азо-Г-бензол-г'-карбоновая кислота
Г. И. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
/Х/СООН
\ х \ N=N - / _ ) -N(CsH7)2
С19Н2зО21^з м. в. 325,42
Пропиловый красный применяют в качестве кнслотнооснов-
ного индикатора для титрований в водных и неводных раство-
рах. По индикаторным свойствам он близок к метиловому
красному, но имеет более глубокую и интенсивную окраску.
Интервал перехода окраски от красной к желтой при pH
4,6—6,4.
Получают пропиловый красный диазотированием антрани-
ловой кислоты и сочетанием полученного диазония с дипро-
пиланилином в солянокислой среде [1].
Полученный продукт очищают перекристаллизацией из
смеси ацетон — метанол (1:1) [2].
Нами предложено проводить сочетание в спиртовой среде,
что увеличивает выход готового продукта в три раза, значи-
тельно сокращает время сочетания и устраняет необходимость
очистки.
46
соон
I I 4-NaNO2+HCl —>
XXXNHa
"\/C°X
—> | | O+NaCl-}-2H2O
X/'xn2/
Характеристика основного сырья
Антраниловая кислота, ч„ ТУ МХП 1713—50.
Нитрит натрия, ч., ГОСТ 4197—48.
Дипропиланилин, ч., ВТУ завода.
Условия получения
В фарфоровый стакан с мешалкой, погруженный в баню сс
льдом, загружают 41 г (0,3 Л1) антраниловой кислоты, 21,5 г
(0,31 М) нитрита натрия и 100 мл воды. Перемешивают и пос-
ле растворения при температуре 4-2-г- +5° к раствору по кап-
лям добавляют концентрированную соляную кислоту в тече-
ние 1,5 часов, затем дают 30-минутную выдержку и проверяют
окончание диазотирования по йодкрахмальной бумажке и бу-
мажке конго. После этого добавляют около 120 г мелко рас-
тертого уксуснокислого натрия, проверяют отсутствие кислой
реакции раствора по бумажке конго (не должно быть синего
окрашивания бумажки).
Полученный совершенно прозрачный желтый раствор диа-
зония (см. примечание 1) добавляют при перемешивании К
раствору 46 г (0,25 М дипропиланилина в 500 мл спирта с та-
кой скоростью, чтобы температура при сочетании не превыша-
ла + 5° (наружное охлаждение льдом). Затем реакционную
смесь медленно, в течение часа нагревают до 30° и оставляют
при комнатной температуре до следующего дня.
Выделившийся индикатор отсасывают, промывают неболь-
шим количеством спирта и сушат при 70—80°.
47
Выход индикатора с содержанием основного вещества 90%
(спектрофотометрически) составляет 52—бо г (62—71% тео-
рии), т. пл. 169—174°.
Для получения химически чистого образца технический
пропиловый красный перекристаллизовывают из 17-кратного
количества смеси метанол — ацетон. Продукт имеет т. пл.
175—176°.
Найдено %: N 12,93, 12,75.
C19H23O2N3. Вычислено %: N 12,90.
Примечание. Если раствор диазония получен мутным, то его сле-
дует профильтровать на холоду (температура не выше -4-5°!).
ЛИТЕРАТУРА
1. Lubs, Clark, J. Wash. Acad, Sei., 5, 609 (1915).
2. Slotta, Franke, Ber., 66, 104 (1933).
МЕТАКРЕЗОЛОВЫЙ ПУРПУРОВЫЙ
Метакрезолсульфофталеин
Г. Н. КОШЕЛЕВА, Е. С. ФОМИНА
(ВНИИ химических реактивов и особо чистых химических веществ)
CjiHjgOjS
м. в. 382,44
Метакрезоловый пурпуровый является кислотно-основным
индикатором с переходом окрасок от красной к желтой при pH
1,2—2,8 и от желтой к пурпуровой при pH 7,4—9,0. Применя-
ется для определения концентрации водородных ионов, для
неводных титрований, для составления смешанных индикато-
ров и как сырье для получения-бромкрезолового зеленого.
48
Метакрезоловый пурпуровый получают конденсацией
м-крезола с о-сульфобензойной кислотой, ее ангидридом или
хлорангидридом в присутствии хлористого цинка [1], хлороки-
си фосфора или смеси хлористый цинк — хлорокись фосфора
[2]. Все методы, описанные в литературе, обеспечивают лишь
небольшие выхода и рекомендуют сложный способ выделения
продукта [3].
Нами была уточнена методика конденсации в присутствии
смеси хлорокись фосфора — хлористый цинк и разработаны
простые условия выделения продукта.
СООН
SO3H
Характеристика основного сырья
о-Сульфобензойная кислота, ч., ВТУ МХП 2833—53.'
-и-Крезол, ч., ТУ МХП 2687 51.
Хлорокись фосфора, ТУ ГХП от 8/V—41.
Цинк хлористый, ч., ГОСТ 4529—48.
«-Бутиловый спирт, ч., ГОСТ 6006—51.
Условия получения
В трехгорлую круглодонную колбу, снабженную обрат-
ным холодильником и мешалкой, загружают 285 а (1,4 М) без-
водной о-сульфобензойной кислоты (см. примечание 1), 300 мл
.«-крезола (80%), 120 г плавленного хлористого цинка (см. при-
мечание 2) и 120 мл хлорокиси фосфора и нагревают при пе-
ремешивании до 95—100°. Начинается реакция, и температура
смеси самопроизвольно поднимается до ПО—115°. При этой
температуре дают выдержку 45 минут. Затем охлаждают
смесь до 100° и осторожно добавляют к ней 300 мл горячей
воды (см. примечание 3).
Полученный раствор выливают в 5 л холодной воды и
оставляют до следующего дня. Выпавший, иногда смолистый,
осадок отделяют от воды и смешивают с 350 мл «-бутилово-
го спирта. Кипятят 10 минут и фильтруют с отсасыванием че-
рез нагретую воронку. Осадок дважды промывают 150 мл
«-бутилового спирта и сушат при 105°.
Выход индикатора 80 г (13—15% теории) с содержанием
основного вещества 70—80% (спектрофотометрически). .
4 Зак. 973 491
Для получения химически'чистого образца растворяют
технический продукт в 7%-ном растворе бикарбоната натрия,
осаждают соляной кислотой, фильтруют, отмывают от хлор-
иона и сушат. Затем дважды обрабатывают 10-кратным коли-
чеством кипящего этилового спирта, как указано выше для
бутилового спирта.
Найдено %: С 66,5, 66,2; Н 4,67, 4,65.
C21H18O5S. Вычислено %: С 65,95; Н 4,74.
Примечания. 1. Содержание основного вещества в лг-крезоле
должно быть не ниже 80%.
Содержание основного вещества в о-сульфобензойной кислоте (титро-
ванием 0,1 н. раствором едкого натра) должно быть не ниже 98%, а со-
держание сульфатов (титрованием хлористым барием по родизонату нат-
рия) — не выше 1 % >
2. Хлористый цинк необходимо предварительно обезвоживать при 260’
в течение 2 часов и по охлаждении быстро растереть.
3. Если реакция прошла не полностью, то остается хлорокись фосфо-
ра и при добавлении воды возможна бурная реакция С выбросом реакци-
онной смеси.
ЛИТЕРАТУРА
1. Cohen, Public Health Reports, 38, 199 (1923); 41, 3061 (1926);
Orndorf, Purdy, J. Amer. Chem. Soc., 48, 2212 (1926).
2. Михайлов, Кошелева, Авт. свид. СССР 104125, 31.08.56.
3. Lemlnger. Chem. prumysl., 5, 7 (1955).
КСИЛЕНОЛОВЫЙ СИНИЙ
Ксиленолсульфофталеин
Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
сн3 СН3
\/\с^\/
1 I 1
СН3 \ (Д13
I I -SO3H
C23H2SO5S м. в. 410,50
Ксиленоловый синий является кислотно-основным индика-
тором с переходом окраски от красной к желтой в интервале
pH 1,2—2,8 и от желтой к синей при pH 8,0—9,6. РекомеНду-
50
ется также в качестве флуоресцирующего индикатора для
работы в ультрафиолетовом свете [1].
В литературе описан способ получения ксиленолового си-
него конденсацией ангидрида или хлорангидрида о-сульфо-
бензойной кислоты с n-ксиленолом в присутствии хлористого
цинка [2].
Нами предложен способ конденсации о-сульфобензойной
кислоты с n-ксиленолом в присутствии смеси: хлористый
цинк — хлорокись фосфора [3], что дает увеличение выхода
продукта и улучшает его качество.
-СООН
-SO8H
Характеристика основного сырья
о-Сульфобензойная кислота, ч., ВТУ МХП 2833—53.
n-Ксиленол, т. пл. 69—71°.
Хлорокись фосфора, ТУ ГХП от 8/V—41.
Цинк хлористый, ч., ГОСТ 4529—48.
Условия получения
В трехгорлую круглодонную колбу с мешалкой, обратным
холодильником и термометром загружают 68,5 г (0,34 М) без-
водной о-сульфобензойной кислоты (cto. примечание 1), рас-
плавляют ее и прибавляют к ней при перемешивании 21 г мел-
корастертого хлористого цинка (см. примечание 2) и 84 г
(0,68 М) n-ксиленола. Реакционную смесь нагревают до 130°,
хорошо перемешивают и охлаждают до 90-?-95°. При этой тем-
пературе быстро приливают 21 мл хлорокиси фосфора и дают
2-часовую выдержку при температуре 100—105°. Затем добав-
ляют еще 12 мл хлорокиси фосфора и выдерживают еЩе 0,5
часа.
Масса постепенно густеет и окрашивается в темно-фиолето-
вый цвет. По окончании выдержки ее растворяют в 660 мл на-
сыщенного раствора соды и от раствора отгоняют с паром не
вступивший в реакцию n-ксиленол (около 15—20 г). Остаток
в перегонной колбе фильтруют от небольшого количества смо-
листых-примесей и маточник подкисляют 10%-ной соляной
кислотой. Выпадает осадок ксиленолового синего. Его отфиль-
4* 51
тровывают с отсасыванием, промывают небольшим количест-
вом воды и сушат.
Выход индикатора составляет 92—98 г (79—83% теорети-
ческого). Качество продукта проверяют по ВТУ МГ УХП
№ 104—58.
Примечания. 1. о-Сульфобензойную кислоту обезвоживают, нагре-
вая ее в течение 2 часов при 140°.
2. Хлористый цинк необходимо предварительно обезвоживать при 260е
в течение 2 часов и по охлаждении быстро растереть.
ЛИТЕРАТУРА
1. Czebelledy, Site. Magyar Jydquszeresztud Tarsas Eztesitoje, 14, 409
(1938); Chem. Lbl, 2, 1819 (1938).
2. A. Cohen, Biochem. J., 16, 31 (1922).
3. Г. И. Кошелева, Ж. прикл. химии, 28, 307 (1955).
БРОМКРЕЗОЛОВЫЙ ЗЕЛЕНЫЙ
Тетрабром- м -крезолсульфофталеин
Г. и. кошелеза
(ВНИИ химических реактивов и особо чистых химических веществ)
Вт Вг
i I
НО-/Х.
Вг~\/хс/'х/~Вг
СН3 L СН3
। |-SO8H
CslHleO6Br4S м. в. 700,09
Бромкрезоловый зеленый является кислотно-основным ин-
дикатором с переходом окраски от желтой к синей в интерва-
ле pH 3,8—5,4. Он применяется для определения концентра-
ции водородных ионов, для производства индикаторной бу-
маги, для приготовления смешанных индикаторов, в бактерио-
логии, в качестве адсорбционного индикатора при определе-
нии тиоцианата.
52
Бромкрезоловый зеленый получают бромированием бро-
мом /«-крезолового пурпурового в среде ледяной уксусной кис-
лоты [1], или бромированием бромом реакционной массы, по-
лученной после конденсации /«-крезола с ангидридом о-суль-
фобензойной кислоты и содержащей м-крезояовый пурпуро-
вый [2].
Нами предложен [3] способ бромирования /«-крезолового
пурпурового в водной среде раствором гипобромита натрия,
что значительно упрощает процесс синтеза.
НО-
/\
N II 4- 4NaBrO + 4НС1
\ /\Гх7\/
। 7 1
СН8 J. СНз
Характеристика основного сырья
/«-Крезоловый пурпуровый, инд., ВТУ МХП 4090—55.
Соляная кислота, ч., ГОСТ 3118—46.
Бром, ч., ГОСТ 4109—48.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
Растворяют 10 г (0,036 М) метакрезолового пурпурового
(60%) при перемешивании в'400 мл 0,1 н. раствора соды, до-
бавляют к раствору 50 мл раствора гипобромита натрия (при-
мечание) и выдерживают 30 минут. Затем к реакционному ра-
створу добавляют 100 г льда и 75 мл соляной кислоты (1:1).
Перемешивают 30 минут, отфильтровывают индикатор, промы-
вают его (трщраза. по 30 мл) соляной кислотой (1:1) и сушат
при .60°.
Выход составляет 10,5 г (76% теоретического).
Для получения химически чистого образца 5 г индикатора
растворяют при нагревании в смеси 20 мл уксусной кислоты и
53
46 мл ацетона;, .кипятят е активированным углем, фильтруют и
отгоняют от маточника ацетон/ Вып^вшцй индикатор; отфиль-
тровывают, промывают уксусной кислотой и сушат при 120°.
Найдено %: С 36,42, 36,19; Н 2,17, 2,10.
C2IHI6O5Br4S. Вычислено %: С 36,13, Н:2,02.
Примечание. Раствор гипобромита иатрия должен содержать
23 г активного брома и Т г свободной щелочи в 100 мл. Его получают ра-
створением 50 мл брома в 620 мл 13'%,-ного раствора едкого натра при тем-
пературе не выше +5°
ЛИТЕРАТУРА
1. Cohen. Public Health Reports, 41, 3061 (1926),
Orndorff Purdy, J. Amer. Chem. Soc., 48, 2212 (1926).
2. Sirokman, Otvos, Acta phys. et chem. Szeged, 4, 73 (1958); РЖХим
№ 38603 (1959).
3. Кошелева, Зимакоьа, Петровская, Ефимова, Авт. свид. СССР 127254,
18.07.59.
БРОМФЕНОЛОВЫЙ СИНИЙ
Тетрабромфенолсульфоф|талеии
Г. Н. КОШЕЛЕВА
(ВНИИ химических пеактивов и особо чистых химических веществ)
CigtijoOgBr^S
м.: в. 670,02
Бромфеноловый синий является кислотно-Основным инди-
катором с переходом окраски от желтой к пурпурной в; интер-
вале pH 3,0—4,6. Применяется при определении концентра-
ции водородных ионов, при бумажной хроматографии и т. п.
Индикатор получают бромированием фенолового красного
бромом в среде ледяной уксусной кислоты [1].
54
Нами предложен способ бромирования фенолового крас-
ного в водной среде раствором гипрбромита натрия [2], ч+о
значительно упрощает процесс синтеза.
Н°\/ \ /X
। Ч1 ГТ
\/\сА/ +4NaBrO-f-4HCI------------->
|/Xj -SO8H
Вг fef
н0\Д /\^°'
I i г
----> Br/' / XC//' /''Br -|-4NaCI4-4H2O
i
|/Xp-SO3H
Характеристика основного сырья
Феноловый красный, инд., ГОСТ 4599—51.
Соляная кислота, ч„ ГОСТ 3118—46.
Бром, ч., ГОСТ 4109—48.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
Растворяют 10 г (0,028 Л4) фенолового красного при пере-
мешивании в 500 мл 0,1 н. раствора едкого натра и добавляют
93 мл раствора гипобромита натрия (см. примечание). Пере-
мешивают 30 минут, добавляют 70 мл соляной кислоты (1 :1)
и выдерживают при перемешивании еще 30 минут.
Выпавший индикатор отсасывают, промывают три раза (по
20 мл) соляной кислотой (1 : 1) и сушат при 60°.
Выход составляет 17,3 г (91% теории).
Для получения химически чистого образца 5 г индикатора
растворяют при нагреваний в смеси 20 мл уксусной кислоты й
30 мл ацетона, кипятят с активированным углем, фильтруют
и от маточника отгоняют ацетон. Выпавший индикатор отфиль-
тровывают, промывают,уксусной кислото|й и сушат при 120°.
Найдено typ: С 34,22 , 34,08; ?Н 1,20, 1,15.
CigHioOsBnS. Вычислено %: С 34,04; Н 1,50.
Примечание. Раствор гипобромита должен содержать 23 г актив-
ного брома и 1 г свободной щелочи в 100' мл. Его !получают растворением
55.
50 мл брома в 62О’ л1.г 13%-ного раствора едкого натра при температуре
не, выше +5°.
ЛИТЕРАТУРА
1. White, Acree, J. Amer. Chem. Soc., 41, 1205 (1919).
2. Кошелева, Зпмакова, Петровская, Ефимова, Авт. свид. СССР 127254,
18.07.59.
БРОМФЕНОЛОВЫЙ КРАСНЫЙ
Дибромфенолсульфофталеин
Г., И. КОШЕЛЕВА '
(ВНИИ химических реактивов и особо чистых химических веществ)
Вг Вг
НОХ/!Х
I '''j— SO3H
QgHiaOBraS м. в. 512,21
Бромфеноловый красный является кислотно-основным ин-
дикатором с переходом окрасок от желтой к красной при pH
5,4—7,0. Он применяется при определении концентрации водо-
родных ионов и при определении pH молока.
Бромфеноловый красный получают бромированием фено-
лрвого красного бромом [1], конденсацией о-бромфенола с ан-
гидридом о-сульфобензойной кислоты [2]. В последнем случае
описано выделение продукта экстракцией «-бутиловым спир-
том [3].
Нами предложен способ получения бромфенолового крас-
ного бромированием фенолового красного раствором гипобро-
мита натрия [4], что значительно упрощает процесс синтеза.
НОч ,. /\^°
J 4-2NaBrO4-2HCl------------
С*
I
j/4j- SOsH
56
Характеристика основного сырья
Феноловый красный,, инд., ГОСТ 4599—51.
Натр едкий, ч., ГОСТ 4328—48.
Бром, ч., ГОСТ 4109—48.
Соляная кислота, ч., ГОСТ 4599—51.
Условия получения
, В фарфоровый стакан с мешалкой и термометром загружа-
ют 25 мл 0,1 н. раствора едкого натра, 175 мл воды и 10 г
(0,028 Л4) фенолового красного, пускают в ход мешалку и
цосле растворения фенолового красного добавляют 32,5 мл
раствора гипобромита натрия (примечание 1). После часового
перемешивания добавляют в реакционную массу 50 г льда
(смесь охлаждается до +5°) и затем приливают 37,5 мл кон-
центрированной соляной кислоты до полноты выделения ин-
дикатора. Перемешивают еще час и оставляют до следующего
дня.
Осадок индикатора отфильтровывают, сушат при 60—70°.
Выход продукта с содержанием основного вещества не ме-
нее 85% (спектрофотрметрически) составляет 12 г (84%
теории).
Пр имеча ние. Применяют раствор гипобромита с содержанием
19 г активного брома и 1 г свободной щелочи в 100 мл. Гипобромит ука-
занной концентрации получают (при растворении 42 мл брома в 16%-ном
растворе едкого натра.
ЛИТЕРАТУРА
1. Sohon, Am. Chem. J. 20,f 284 (1898).
2. Cohen, Public Health Reports, 38, 199 (1923); 41. 3061 (1926).
3. Leminger, Chem. pruumysl, 5, 7 (1955);
Leminger, Чешек, пат/ 83863 от 25/V11I-1951.
4. Кошелева, Зи макова, Петровская, Ефимова, Авт. спид. СССР 127254,
18.07.59. '
БРОМКСИЛЕНОЛОВЫЙ СИНИЙ
Дибромксилеиолсульфофталеии ;'
Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
CssHjoOgBrjS
м. в. 568,31
Бромксиленоловый синий является кислотно-основным ин-
дикатором с переходом окраски от желтой к синей при pH
6,0—7,6.
Применяется в ацидиметрии, при определении кислотности
молока, как составная часть смешанных индикаторов.
Бромксиленоловый синий получают бромированием ксиле-
нолового синего [1] бромом в спиртовой среде.
Нами был проверен и уточнен указанный метод получения.
СН, СН3
58
Характеристика основного сырья
Ксиленоловый синий, инд., ВТУ УХП 75—58.
Бром, ч., ГОСТ 4109—48.
( Спирт этиловый, ректификат, ГОСТ 5962—51.
Условия получения
В трехгорлую круглодонную колбу с обратным холодиль-
ником, мешалкой и термометром загружают 26 г (0,061 Л1)
ксиленолового синего и 104 мл этилового спирта. Пускают в
ход мешалку, нагревают реакционную смесь до 40°, прибав-
ляют к ней 7 мл (0,136М) брома и выдерживают при 50—55г
в течение часа. За это'й^емя бромирование заканчивается (см.
примечание).
По окончании реакции смесь охлаждают до 10°, фильтру-
ет . осадок промывают 10 мл спирта и сушат при 50—60°..
Выход бромксиленолового синего составляет 26—28 г
,(Т5—81 % теории).
Качество продукта проверяют по РТУ № 223—57. ,
, Примечание. Проба на конец реакции: каплю реакционной смеси
растворяют .в 5 мл спирта; в две пробирки наливают по 5 мл буферных
смесей: в 1-то пробирку с pH 7,8, во 2-ю с pH 9,8 и добавляют в обе про
бйркй!по Капле раствора реакционной смеси; если бромирование закончено,
то окраска обоих растворов должна быть одинакова. В противном civile
идгревают реакционную смесь еще 0,5 часа и повторяют пробу.
ЛИТЕРАТУРА
i„fi. Cohen, Biochem. .1. 17 535 (1923).
ХЛОРФЕНОЛОВЫЙ КРАСНЫЙ
Дихлорфенолсульфофталеин
Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
С1 С1
н°х/\ /1../°
Д/SOgH
CI9H12O6Ci2S ' м. в. 423,29
Хлорфеноловый красный является кислотно-основным ин-
дикатором с переходом окраски от желтой к красной при pH
4,8—6,8. Он применяется при определении концентрации водо-
родных ионов, для определения культур бактерий, для прояв-
ления бумажных хроматограмм органических кислот, для ана-
лиза качества молока и для приготовления бумаги для анали-
за молока.
Хлорфеноловый красный получают конденсацией о-хлор-1
фенола с ангидридом о-сульфобензойной кислоты [1]. По это-
му способу выделение продукта осуществляется очень сложно.
Нами предложен способ получения индикатора хлориро-
ванием фенолового красного раствором гипохлорита натрия [2].
что значительно увеличивает выход и упрощает процесс синтеза.
НО / . VO
\/\ Х\//
\У^Г//-^/ 4~2NaC10-f-2HCl------->
I
|Х"|- SO3H
Cl Cl
но\/\ Л/°
-----* \ / \г.^\ / 4-2NaC14-2HsO
( SOsH
60
Характеристика основного сырья
Феноловый красный, инд., ГОСТ 4599—51.
Соляная кислота, ч., ГОСТ 3118—46.
Бром, ч., ГОСТ 4109—48.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
В фарфоровый стакан с мешалкой и термометром загружа-
ют 500 мл воды, 16 г борной кислоты и 10 г (0,028 Л4) феноло-
вого красного. Перемешивают и к раствору добавляют 88 мл
раствора гипохлорита натрия (см. примечание 1). Перемеши-
вают 10 минут, охлаждают до 10° и подкисляют концентриро-
ванной соляной кислотой до полноты осаждения (~ 150 мл).
Осадок отфильтровывают и сушат при 60—70°.
Выход индикатора с содержанием основного вещества не
ниже 80% (спектрофотометрически) 9,4 г (84% теории).
Примечание. Применяют раствор гипохлорита с содержанием
5,2 г активного хлора и 0,9 г свободной щелочи в 100 мл.
ЛИТЕРАТУРА
1. В. Cohen, Public Health Reports, 41, 3061 (1926);
О
Lemtnger, Chem. prumysl, 5, 7 (1955);
Leminger, Чешек, пат. 33863 от 25/V11I-1951.
2. Кошелева, Зимакова, Грушецкий, Ефимова, Авт. свид. СССР 127264,
29.07.59.
ЭТИЛОВЫЙ СИНИЙ
Этил-бис-(2,4-дииитрофеиилацетат)
Г. Н. КОШЕЛЕВА, Г. Д. ГЛЕБОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
NO,
,СНСООС2Н,
°SN ’\ _/ z
I
* no2
Ci»H12O10N4 m. b. 420,30
Этил-бис-(2,4-динитрофенилацетат) является кислотно-ос-
новным индикатором с изменением окраски от бесцветной до
61
синей в интервале pH ,7,0—9,4. Применяется, при титровании
темно-окрашенных масел, определении кислотного числа и
числа омыления масел.
Этил-бис-(2,4-динитрофенилацетат) получают из динитро-
бромбензола и этилового эфира натрийацетоуксусной кислоты
[1], при взаимодействии натриймалонового эфира с динитро-
бромбензолом [2] или динитрохлорбензолом [3], а также при
нитровании нитрующей смесью дифенилуксусной кислоты [4].
Нами проверен и уточнен второй из перечисленных мето-
дов.
/COOCsHs ^СООС2Н5
СН2 +2C2H6ONa—> CNa2 +2С2НВОН
ХСООС2Н6 ХСООС2Н5
7СООС2Н5 ।
/ —NO
CNa2 + 2| |
СООС2Н5
NO,
СООС2Н6
I
C-COOC2H5+2NaCl
I
|/Xj-NO2|/X'|-NO2
X/ ' \/
NO3
no2
СООС2Н5
Ч-.’- ......
С-СООС2Н, +НаО----
NO» NO,
Np2
?СН -G0OC2H6+CO24-C2HSOH
Характеристика основного сырья
Малоновый эфир, ч., ВТУ МХП 4162—54.
Этилат натрия, техн., 16% ВТУ завода.
Динитрохлорбензол техн., ГОСТ 625—41.
Спирт этиловый, ГОСТ 5962—51.
Условия получения
В трехгорлую круглодонную колбу, снабженную мешалкой,
капельной воронкой и обратным холодильником с хлоркаль-
циевой трубкой на конце, загружают 248 мл (0,5 М) 16%-но-
го спиртового раствора этилата натрия. Включают мешалку,
прикапывают в течение 10—15 минут 40,5 г (0,25 Л4) малоно-
вого эфира и перемешивают один час.
К образовавшейся густой массе прибавляют по каплям в
течение 0,5 часа горячий (50—60°) раствор 101,3 г (0,5 М)
1,2,4-динитрохлорбензола в 100 мл абсолютного спирта. Полу-
ченную смесь кипятят 7 часов и оставляют до следующего дня,
а затем выливают в 500 мл воды. Образовавшуюся темно-бу-
рую реакционную массу подкисляют концентрированной сер-
ной кислотой (~5 мл) до кислой реакции по бумажке конго.
При этом отделяется тяжелая смолистая масса. Через час
верхний слой декантируют, а смолистую массу дважды про-
мывают декантацией теплой водой, а затем теплым 60° спиртом
до получения твердого осадка. Отсасывают и промывают на
фильтре спиртом, сушат при температуре ~ 100°.
Выход индикатора светло-желтого или светло-серого цвета
с содержанием основного вещества не менее 90% (ванадато-
метрически) 24 г (21% теории).
Химически чистый образец получают при перекристаллиза-
ции технического продукта из 10-кратного количества бензо-
ла, а затем из 13-кратного количества ледяной уксусной кис-
лоты.
Найдено %: N 13,25; 13,26.
C16H12O10N4. Вычислено %: N 13,33.
ЛИТЕРАТУРА
1. Heckman, Liebigs Ann Chem., 220, 137 (1883).
2. Richter, Ber., 21, 2470 (1888).
3. Fhenel, Amstutz, Industr. and Eng. Chem., Anal, ed., 16,53(1944).
4. Werner, Ber., 39, 1290 (1896).
4-ЭТО КСИ АКР ИДО Н
Г.. Н. КОШЕЛЕВА. Е. С. ФОМИНА
(ВНИИ химических реактивов и особо чистых химических веществ)
c18h13osn
м. в. 239,28
4-Этоксиакридон является флуоресцентным кислотно-ос-
новным индикатором с переходом цвета флуоресценции в уль-
трафиолетовом свете от зеленого до синего в интервале pH
1,2—3,0.
4-Этоксиакридон получают внутримолекулярной конденса-
цией 2'-этоксидифениламинокарбоновой кислоты — (2) в при-
сутствии серной кислоты [1] с выходом 48% теоретического.
2'-Этоксидифениламинокарбоновую кислоту получают кон-
денсацией о-хлорбензойной кислоты с о-фенетидииом.
Нами проверен и уточнен способ получения 2'-этоксидифе-
ниламинокарбоновой кислоты—(2) и предложен способ полу-
чения из нее 4-этоксиакридона конденсацией в присутствии
хлорокиси фосфора [2], что дает значительное увеличение вы-
хода. В качестве промежуточного продукта при этом образует-
ся ранее не описанный в литературе мезохлоракридин, выде-
ленный нами в химически чистом состоянии.
/Х/СООН zNHa ХСООН
fi +ГТ ‘ -if _ +нс1
/хХ! "' ОС^Н- 'X/XNH-/
Г
осгн6
С1
СООН /\
3| (' --f-POClH----->3f I ( I+H3PO44-3HSO
ос2н5
ос2н5
64
Cl
( YYY«°
V Y
OC2H8
OC2H6/
Характеристика основного сырья
о-Хлорбензойная кислота, ч., ВТУ 4134—54.
о-Фенетидин, ч., ВТУ МХП 4176—54.
Медь металлическая в порошке, ГОСТ 1124—41.
Спирт изоамиловый, ч., ОСТ НКТП 7670—621.
Хлорокись фосфора, ТУ ГХП от 8/V—41.
Анилин, ч., ОСТ 10157—39.
Условия получения
Получение 2'-этоксидифениламинокарбоновой кислоты — (2).
В двухлитровую круглодонную колбу, снабженную мешал-
кой, обратным холодильником и погруженную в масляную ба-
ню, загружают 150 г (0,95 М) о-хлорбензойной кислоты, 150 г
(1,1 М) свежеперегнанного о-фенетидина, 150 г (1,1 М) све-
жепрокаленного поташа, 20 г медного порошка и 1 л изо-
амилового спирта. Смесь кипятят в течение 3 часов. Затем ме-
няют обратный холодильник на нисходящий и отгоняют изо-
амиловый спирт с паром. При этом отгоняется примерно 1 л
спирта и 1,5—2 л воды. Оставшуюся в перегонной колбе смесь
фильтруют. На фильтре остается медь и небольшое количест-
во смолистых примесей. Маточник разбавляют водой до объе-
ма 5 л, нагревают до 70° и подкисляют соляной кислотой до
кислой реакции по конго (~200 мл). Выпадает 2/-этоксидифе-
ниламинокарбоновая кислота—(2). Ее отфильтровывают,
промывают горячей (~70°) водой до исчезновения кислой ре-
акции по конго 1,5—2 л воды) и сушат при 60—70°. Выход
составляет 196—200 г (81% теории), т. пл. 146—152°.'’
Технический продукт перекристаллизовывают из 7,5-крат-
ного количества спирта (с углем). Выход составляет 75% за-
груженного, т. пл. 155—158°.
Получение мезохлоракридина
В круглодонную колбу с обратным холодильником загру-
жают 50 г (0,2 М) 2/-этоксидиФениламинокарбоновой кисло-
ты— (2) и 200 мл хлорокиси фосфора. Нагревают до кипения
и кипятят полученный раствор в течение часа. Затем меняют
обратный холодильник па нисходящий и отгоняют 120 мл
хлорокиси фосфора. Остаток после отгонки выливают в 1 л
5 Зек. 973 ' &5
воды, фильтруют от незначительного количества смолистых
примесей и добавляют к фильтрату 10%-ный раствор едкого
натра до слабощелочной реакции по бриллиантовой желтой
бумажке. Выделившийся 4-этокси-9-хлоракридин отфильтро-
вывают, промывают водой и сушат.
Выход составляет 43,5 г (89% теории), т. пл. 124—125°.
Найдено %: С1 13,85; 13,52.
C15H12ONCI. Вычислено %: С1 13,7.
Получение 4-этоксиакридоиа
Растворяют 43,8 г (0,17 М) 4-этокси-9-хлоракридина в
750 мл воды и 23 мл концентрированной соляной кислоты. Ра-
створ нагревают при перемешивании в течение 1,5 часа. Вы-
павший осадок 4-этоксиакридона отфильтровывают, промыва-
ют 100 мл воды и сушат при 60—70°. Выход 4-этоксиакридоиа
составляет 36,5 г (79% теории). Его перекристаллизовывают
из 70 мл смеси анилина и ледяной уксусной кислоты (1 : 25 по
объему) с добавлением активированного угля.
Выход продукта после перекристаллизации 90% от загру-
Найдено %: N 5,96, 6,16.
C15H13O2N. Вычислено %: N 5,86.
ЛИТЕРАТУРА
1. Matsumura, J. Amer. Chem. Soc., 49, 810 (1927).
2. Г. H. Кошелева, Ж- общ. химии, 26, 2567 (1956).
2,3-ДИЦИАНГИДРОХИНОН
3,6-Диоксидинитрил фталевой кислоты
Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ
ОН
|/\-cn.2hsO
\/-CN
C8H4OaNa-2H2O ОН м. в. 196,17
Дициангидрохинон является кислотно-основным флуоре-
сцентным индикатором с переходом флуоресценции в ультра-
фиолетовом-свете от-синей к зеленой" в интервале pH 6,0—8,0.
66
Дициангидрохинон получают действием цианистого водоро-
да в момент выделения на хинон [1] или хлорхинон [2].
Нами проверен и уточнен первый из литературных методов.
О ОН
21 I+2HCN I I
\/ \/“CN
О ОН
он
*0
он
Характеристика основного сырья
Хинон, ч., ТУ МХП 3041—51.
Натрий цианистый, ч., ВТУ.
Серная кислота, ч., ГОСТ 4204—48.
Условия получения
-В четырехлитровую трехгорлую колбу (Всю работу необ-
ходимо проводить под хорошо действующей тягой, так как воз-
можно выделение цианистого водорода!) наливают 660 мл
воды и нагревают ее до 90°. Затем добавляют 55 г (0,51 М)
хинона, включают мешалку и после растворения хинона до-
бавляют 184 мл 24,5% серной кислоты. Понижают температу-
ру до 70° и из делительной воронки спускают в течение 2—3
минут раствор ПО г (1,7 М) цианистого калия в 180 мл воды.
При этом температура реакционного раствора поднимается
до 86°. Перемешивают 10 минут, охлаждают реакционную
смесь до 70° и добавляют из делительной воронки 164 мл
24,5% серной кислоты. Через 10 минут проверяют реакцию по
бумажке конго (реакция кислая) (см. примечание) и переса-
сывают реакционный раствор в 5-литровую колбу, в которой
находится 1350 г мелкотолченого льда. Выпавший дициангид-
рохинон отсасывают, промывают водой до отсутствия кислой
реакции по бумажке конго и сушат при 100—110°.
Выход безводного продукта с содержанием основного ве-
щества не менее 90% (спектрофотометрически по интенсивно-
сти люминесценции при 460 -ип) 30 г (60% теории).
Примечание. Если реакция не кислая, то добавляют еще некоторое
количество серной кислоты той же концентрации и снова проверяют
реакцию.
ЛИТЕРАТУРА
1. Helferlch, Вег., 54, 156, (1925);
Graves. Adams, J. Amer. Chem. Soc., 45, 2447 (1923);
Allen, Wilson, J. Amer. Chem Soc., 63, 1756 (1946);
Thiele. Qilnter, Liebigs Ann. Chem, 349, 59 (1906);
Pai, Guha, J. Indian, Chem. Soc., 11, 231 (1934).
-2.. Thiele, GtMer, Liebigs Ann. .Chem,, 349,-48 (1906).
5* 67
ДИОКСИФТАЛИМИД
4,7-Диокси-1,3-дикето-изоиндолии
Г. И. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
ОН
|/X|”C04>NH
^J-CO/
\
ОН
C8H6O4N м. в. 179,14
Диоксифталимид является кислотно-основным флуорес-
центным индикатором с переходом цвета . флуоресценции в
ультрафиолетовом свете от синего к зеленому в интервале pH
О—2,4 и от зеленого к желтому в интервале pH 6,0—8,0.
Диоксифталимид получают омылением дициангидрохино-
на [1] или метилового эфира гентизиновой кислоты [2].
Нами проверен и уточнен первый из способов получения.
ОН
+зн2о
H»SO*
он
<Х1--СО\
M_Co>H+NH‘OH
ОН
Характеристика основного сырья
Дицнангидрохинон, ВТУ завода.
Серная кислота, ч„ ГОСТ 4204—48.
Условия получения
В трехгорлую круглодонную колбу, помещенную в водяную
баню, загружают 25 г (0,15 М) двуводного дициангидрохино-
на, 100 мл концентрированной серной кислоты, включают ме-
шалку и греют 15 минут при температуре кипения бани. Затем
реакционную смесь выливают в стакан на смесь льда (100 г),
воды (400 мл) и угля (7 г) и перемешивают от руки 15 минут.
Уголь отфильтровывают, а маточник оставляют до следую-
щего дня.
Выпавший осадок диоксифталимида отфильтровывают, про-
мывают 250—300 мл 20%-ного раствора хлористого натрия и
68
затем 50 мл воды до отсутствия кислой реакции по бумажке
конго и сушат при 100—110° (см. примечание).
Выход продукта 10—11 г (39% теории).
Химически чистый образец получают двукратной перекри-
сталлизацией из 20-кратного количества воды.
Найдено %: С 53,21, 52,97; Н 3,01, 3,07; N 7,6, 7,76.
C8H5O4N. Вычислено %: С 53,64; Н 2,81; N 7,81.
Примечание. Качество диоксифталимида проверяют хроматогра-
фией на бумаге; в качестве растворителя применяют 0,3 н. раствор серни-
стой кислоты; хроматограмму высушивают и рассматривают в фильтрован,
ном ультрафиолетовом свете. Не должно быть прнмеси исходного дициан-
гидрохиноиа.
ЛИТЕРАТУРА
1. Helf erich, Ber., 54, 156 (1926).
2. Новохатка, Лазурьевский, Уч. зап. Кишиневск. ун-та. 14, 63 (1953).
МЕТИЛВИОЛОГЕНДИХЛОРИД
7У,А^-Диметил-4,4'-дипиридилийдихлорид
Г. Н. КОШЕЛЕВА, Г. Н. НАЛЕЦКАЯ
(ВНИИ химических реактивов и особо чистых химических веществ)
HSC-+N^ >N+-CHe
ci- х—7 —7 Cl- ’
C12H14NaCl9 m.b. 257,17
Метилвиологендихлорид является ценным окислительно-
восстановительным, индикатором для микробиологии: Eh =
—0,446.
По литературным данным [1, 2], метилвиологендихло-
рид можно получить метилированием 4,4'-дипиридила диме-
тилсульфатом с последующей обработкой метилвиологенди-
сульфата соляной кислотой.
Выход составляет всего 14%.
Нами был предложен способ получения метилвиологенди-
хлорида, который заключается в замене иода в М,М'-диме-
тил-4,4'-дипиридилийдийодиде на хлор, с помощью хлористой
ртути [3], что позволило упростить процесс синтеза и значи-
тельно повысить выход продукта.
69
Получение Метилвиологендийодида было осуществлено ме
тилированием 4,4'-дипиридила йодистым мерилом [4].
N.\ /N-I-2CH3J—*
->н,с-+ыЛ ^N+-CH8
J- Х— Z J-
HSC—+N^ ")-( /N+-CH3+2HgCl —>
J- — х —7 J-
Н3С—+n/ \n+—CH3+2HgJ
Cl- —7 Cl-
Характеристика основного сырья
4,4/-Дипиридил, ч., ВТУ РУ 661—52.
йодистый метил, ч., ТУ МХП 49—51.
Хлористая ртуть (каломель), ч., ГОСТ 3203—46.
Условия получения
Получение метилвиологендийодида
В трехгорлую колбу, снабженную механической мешалкой,
шариковым холодильником и термометром, помещают 10 г
(0,052 М) 4,4'-дипиридила в виде его дигидрата, 20 мл (46 г,
0,32 М) свежеперегнанного йодистого метила (т. кип. 41,5—
43,5о/760 мм) и 50 мл изопропилового спирта. Реакционную
массу кипятят в течение 3 часов (температура массы 55—58°),
после чего охлаждают до 20°. Выделившиеся оранжевые кри-
сталлы метилвиологендийодида отфильтровывают под ваку-
умом, промывают на фильтре 5—7 мл изопропилового спирта
и сушат на воздухе.
Выход сухого, технического 92%-ного метилвиологендийо-
дида 21,3 г (85,5% теории) (см. примечание 1).
Получение метилвиологендихлорида
В трехгорлую колбу, снабженную механической мешалкой,
обратным холодильником и термометром, помещают 20,8 г
92%-ного (0,043 М) метилвиологендийодида, 200 мл воды и
30,4 г (0,12 М) хлористой ртути.
Реакционную смесь нагревают при перемешивании в тече-
ние 2 часов при температуре 70°.
Конец реакции определяют по качественной пробе (см. при-
мечание 2). При наличии положительной пробы реакционную
смесь охлаждают до комнатной температуры, йодистую ртуть
отфильтровывают и промывают ее иа фильтре 10—15 мл воды.
70
От маточника отгоняют при атмосферном давлении 4/5
первоначального объема воды.
Остаток выпаривают досуха в фарфоровой чашке на во-
дяной бане. Технический продукт сушат до постоянного веса
в сушильном шкафу при 105°.
Выход сухого технического 91%-ного метилвиологенди-
хлорида 11,7 г (см. примечание 3) (94,2% теоретического в
пересчете на метилвиологендийодид и 81,5% теоретического в
пересчете на 4,4/-дипиридил).
Для очистки технический метилвиологендихлорид перекри-
сталлизовывают из этилового спирта. Выход продукта после
перекристаллизации составляет 77% от загруженного.
Чистый метилвиологендихлорид содержит не менее 95%
основного вещества.
Найдено %: N 11,14.
C12H14N2CI2. Вычислено %: N 10,89.
Примечания. 1. Содержание метилвиологендийодида определяют
методом роданидометрии.
2. К небольшому количеству прозрачного раствора, взятого из реак-
ционной колбы, предварительно отфильтрованного от солей ртути, добав-
ляют по каплям 0,1 н. раствор азотнокислой закисной ртути. Если при этом
образуется осадок желтого или желтоватого цвета (йодистая ртуть)
вследствие наличия в растворе пепрореагировавшего метилвнологендийоди-
да — значит реакция ие прошла до конца. Если же при добавлении раст-
вора азотнокислой закисной ртути к испытуемому раствору образуется
осадок белого цвета (хлористая ртуть) — значит реакция прошла пол-
ностью. Если проба отрицательна, продолжают нагревание еще в течение
1 часа и опять проверяют конец реакции.
# 3. Содержание метилвиологендийодида определяют меркурометриче-
ским способом.
ЛИТЕРАТУРА
1. L. Michaelis, Biochem. Z., 230, 564 ('932).
2. L. Michaelis, J. Amer. Chem. Soc., 55, 1481 (1933).
3. Г. H. Кошелева и Г. H. Налецкая, Авт. свид. СССР, 133544, 22.02.60.
4. А. Р. Phillips, /.Mentha, J. Amer. Chem. Soc., 77, 6394 (1955).
ЭУХРИЗИН ЗР
Основание акридинового оранжевого, основание
3,6-бисдиметиламииоакр идина
Е. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
Q17H19N3
(CHs)2N
-N(CH3)2
м. в. 265,36
Эухризин применяется как флуоресцентный индикатор с
переходом цвета флуоресценции от оранжевой к зеленой в ин-
тервале pH 8,4—10,4 [1—2].
Эухризин нами получался из цинковой соли акридинового
оранжевого (двойной соли акридинового оранжевого с хлори-
стым цинком) растворением ее в уксусной кислоте и последу-
ющим фракционированным осаждением щелочью. При этом
были уточнены условия синтеза и получен образец эухризина
в химически чистом состоянии.
N
(CH,),nY Y y4|-N(CH3), ,ZnCli СН^Са°°”-
Х//ХN
(CH3)SN--Y Y YN<CH.b +ZnCl2
\ / \ / х /
Характеристика основного сырья
Акридинового оранжевого
цинковая соль.
Промежуточный продукт прн полу-
чении акридинового оранжевого
Кислота уксусная, д., ГОСТ 61—51.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
В стакан емкостью 1 литр, снабженный мешалкой, загру-
жают 8 г (0,02 М) цинковой соли акридинового оранжевого и
270 мл 1 н. уксусной кислоты. Реакционную массу размеши-
вают при комнатной температуре до почти полного растворе-
ния и фильтруют от небольшого количества нерастворившего-
ся осадка. К фильтрату добавляют 120 мл 10%-ного раствора
едкого натра до pH 7 по бумажке «Рифан» и выпавший в не-
72
большом количестве темно-оранжевый осадок отфильтровыва-
ют. К фильтрату добавляют еще 20 мл 10%-ного раствора ед-
кого натра до сильно щелочной реакции, выпавший эухризин
отфильтровывают и промывают на фильтре 50 мл воды. Су-
шат при 80—90°. Получают 3 г (37,5% теории) технического
эухризина.
Для очистки 3 г технического эухризина растворяют при
кипении в 80 мл этилового спирта, в колбе емкостью 150 мл,
снабженной обратным холодильником, и кипятят в течение 20
минут. Раствор отфильтровывают и к фильтрату добавляют
120 мл воды для наиболее полного выделения эухризина. По-
лученный осадок светло-оранжевого цвета отфильтровывают
и промывают 20 мл воды. Сушат при 100°. Получают 1,5 г
эухризина, что составляет 50% от загруженного технического
эухризина.
Найдено . %: N 15,77, 15,83.
C17H19N3. Вычислено %: N 15,83.
ЛИТЕРАТУРА
1. К. Jensen, Z. Analyt. Chem., 94, 177 (1933).
2. М. ОёНЬёгё, Ann. Chlm. anal. Chim. appl, [3],
19, 290 (1937); Chem. Zbl., 1938, 1, 2758.
ЛЮЦИГЕНИН
А^ТУ'-Диметилбиакридилий нитрат
E. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
C28H2SN4O6
м. в. 510,49
Люцигенин находит применение как хемилюминесцентный
индикатор с зеленоватым свечением при pH 9,02—9,97 [1]. По
73
литературным данным, люцигенин получают по следующей
схеме: N-фенилантраниловая кислота -> 9-хлоракридин-9,9'-
биакридил—• бромистая соль 10,10/-диметил-9,9''-биакридилия
-> люцигенин.
9-Хлоракридин получают конденсацией N-фенил-антрани-
ловой кислоты с хлорокисью фосфора; 9,9'-биакридил — кон-
денсацией двух молекул хлоракридина в присутствии цинко-
вой пыли; бромистую соль 10,10'-диметилбиакридилия полу-
чают метилированием 9,9'-биакридила диметилсульфатом и,
наконец, люцигенин получают нагреванием бромистой соли
10,10'-диметилбиакридилия с азотной кислотой [2].
Нами люцигенин был получен по вышеприведенной схеме
из N-фенилантраниловой кислоты. Уточнены условия всех
стадий синтеза и получен образец люцигенина в химически
чистом состоянии.
NH . NH
1 । 1 J ----III
х/хсоонх/ \/\/х/
Cl
74
CHS
HNO3
Характеристика основного сырья
Анилин, ч., ГОСТ 5819—51.
Диметилсульфат, ВТУ У-59-51.
Калий бромистый, ч., ГОСТ 4160—48.
Кислота азотная, ч., ГОСТ 4461—48.
Кислота о-хлорбензойная, ч., ВТУ У—134—51.
Хлорокись фосфора, ВТУ У—166—51.
Условия получения
Получение 9-хлоракридина
В круглодонную колбу на 500 мл, снабженную обратным
холодильником, загружают 50,7 г (0,23 М) N-фенилантрани-
ловой кислоты и 253,5 г (1,6 М) хлорокиси фосфора. Смесь
выдерживают при кипении (на масляной бане) в течение 2 ча-
сов. Избыток хлорокиси фосфора отгоняют, а оставшуюся
массу выливают на лед и отфильтровывают. К фильтрату до-
бавляют 500 мл 12%-ного раствора аммиака до щелочной ре-
акции по бриллиантовой желтой бумажке (см. примечание 1).
Выпавший хлоракридин отфильтровывают, промывают водой
до отсутствия щелочной реакции в промывных водах по брил-
лиантовой желтой бумажке и сушат на воздухе. Получают
40,8 г технического продукта (80% теории), т. пл. 117—118°.
Получение 9,9'-биакридила
В круглодонную колбу емкостью 500 мл, снабженную ме-
шалкой и обратным холодильником, помещают 15 г (0,07 М)
9-хлоракридина, 3,0 г цинковой пыли и 380 мл метилового
спирта. Смесь кипятят при размешивании в течение 4 часов.
При этом вещество не растворяется, изменяется только его
цвет (желтеет). По охлаждений продукт отфильтровывают
%
и промывают на фильтре метиловым спиртом. Сушат на воз-
духе, получают 20 г технического Э.^-биакридила.
Для очистки 20 г технического 9,9'-биакридила растворяют
при кипении в 750 мл пиридина. Горячий раствор фильтруют,
на фильтре остается непрореагировавшая цинковая пыль. Из
фильтрата высаживают водой 9,9'-биакридил. Его отфильтро-
вывают и тщательно промывают несколько раз водой до ис-
чезновения запаха пиридина. Сушат на воздухе, получают
18 г 9,9'-биакридила (72% теории).
Получение бромистой соли
10,1 O'-ди м етил-9,9'-биакридил ия
В круглодонную колбу емкостью 200 мл, снабженную об-
ратным холодильником, загружают 7,7 г (0,02 Л1) 9,9'-биакри-
дила и 27 мл (0,21 Л1) сухого нейтрального диметилсульфата.
Реакционную смесь кипятят на масляной бане в течение 2 ча-
сов. По охлаждении к реакционной массе добавляют 60 мл
эфира, размешивают, затем эфирный раствор сливают с вы-
павшего полутвердого осадка. Осадок повторно обрабатыва-
ют эфиром, эфир сливают, а осадок при осторожном нагрева-
нии (примечание 2) растворяют в 100 мл воды. Раствор ки-
пятят с активированным углем в течение 15 минут, отфильтро-
вывают и к еще теплому маточнику добавляют горячий раст-
вор бромистого калия, приготовленный из 70 г (0,58 М) бро-
мистого калия и 100 мл воды. Выпавшие золотистые листочки
бромистой соли 10,10'-диметилбиакридилия отфильтровывают,
промывают водой и вновь растворяют в 100 мл воды при
~80°. Снова добавляют горячий раствор бромистого калия,
приготовленный таким же образом, выпавший бромид люци-
генина отфильтровывают, промывают водой и сушат при 70°.
Получают 9,3 г люцигенинбромида (80% теории).
Получение люцигенина
В стакане емкостью 500 мл, при ~80° растворяют 9,3 г
(0,02 Л1) бромида люцигенина в 372 мл 6%-ной азотной кис-
лоты. Образуется оранжево-желтый раствор, из которого по
охлаждении выпадают желтые листочки люцигенина. Получа-
ют 6,1 г технического продукта (70% теории).
Для очистки технический люцигенин перекристаллизовыва-
ют из 49-кратного количества воды. Получают желтовато-золо-
тистые кристаллы люцигенина в количестве 5,2 г (85%, считая
на технический продукт).
Найдено %: С 65,82, 65,80; Н 4,34, 4,33.
C28H22N4O6. Вычислено %: С 65, 81; Н 4,31.
76
Примечания. 1. Не следует оставлять на продолжительное время
необработанным раствор после конденсации N-фенилантраниловой кисло-
ты с хлорокисью фосфора.
2. Необходимо осторожное нагревание, так как удаляются остатки
эфира и диметилсульфата.
ЛИТЕРАТУРА
V . V
1. R. Parlzek, U. Moucka, Chem. listy, 48, (78) 4 (1954).
2. A. M. Григоровский «Исследования в области производных акриди-
на». Докторск. диссертация, 1951.
10,10'-ДИМЕТИЛ-9,9'-БИАКРИДЕН
Е. Я. ЯРОВЕНКО, Г: Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
СНз
м. в. 386,48
10,10''-Диметил-9,9''-биакриден является хемилюминесцент-
ным индикатором с свечением при pH 9—10 [1].
В литературе описано получение 10,10'-диметил-9,9'-биа-
кридена восстановлением бромида люцигенина в присутствии
скелетного никеля [2, 3].
Нами уточнены условия синтеза и получен образец в хими-
чески чистом состоянии.
77
НзС7 Br-
Характеристика основного сырья
Бромид люцигенина
Никель скелетный (катализатор)
Промежуточный продукт при по-
лучении люцигенина (см. стр. 76)
Условия получения
В круглодонную колбу емкостью 1 л, снабженную обрат-
ным холодильником и мешалкой, загружают 20 г бромида
люцигенина, 4 г пасты скелетного никеля и 500 мл метилового
спирта. Реакционную смесь кипятят при размешивании в те-
чение 3 часов. Охлаждают, осадок отфильтровывают и поме-
щают в круглодонную колбу емкостью 3,5 л, снабженную об-
ратным холодильником, прибавляют 3 л пиридина и кипятят
в течение 20 минут. Раствор горячим фильтруют, из фильтра-
та по охлаждении выпадают кристаллы 10,10'-диметил-9,9'-
биакридена, их отфильтровывают, промывают водой до уда-
ления запаха пиридина, сушат при 150°. Получают 10 г тех-
нического 10,10'-диметил-9,9'-биакриДена (71 % теории).
Для очистки технический продукт перекристаллизовывают
из 200-кратного количества пиридина. Выход 50% от загру-
женного.
Л ИТЕ.РАТУРА
1, Е. Я. Яровенко, Г. И. Кошелева. Авт. свид. СССР 116002, 17.02.58.
2. А. М. Григоровский «Исследование в области акридиновых произ-
водных», Докторск. диссертация, 1951.
3. «Синтезы органических препаратов», сб. 4, стр. 349, ИЛ.,’ 1953.
ЛОФИН
2,4,5-Трифенилиминазол
Е. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
HeC.-C-N
НвСв-С С.С,НВ
N
I
н
C81H)eNa м. в. 296,14
Лофин применяется как хемилюминесцентный индикатор с
белым свечением при pH 8,9—9,4 [1].
В литературе описано получение лофина взаимодействием
бензальдегида с аммиаком при последующем термическом раз-
ложении полученного гндробензамида [2—3].
Нами проверены и уточнены условия синтеза лофина и по-
лучен образец в химически чистом состоянии.
.О C6H6CH==N
ЗС6Н6С^ +2NH3--------> ^СН-СвН64-ЗНаО
ХН c6h6ch=n/
. H6Ce-C-N
свн6сн=н II II
хсн-с6н6—> нвс6-с С-С6НВ
CeH6CH = N'/ \/
N
н
Характеристика основного сырья
Аммиак водный, ч„ ГОСТ 9—57.
Бензойный альдегид, ч., ГОСТ 157—51.
Условия получения
Получение гидробензамида
В круглодонную колбу емкостью 150 мл, снабженную м<
шалкой с глицериновым затвором, загружают 63,6 г (0,6 Л
бензальдегида и 40 мл водного аммиака. Реакционную смес
перемешивают при комнатной температуре в течение 6 часов
затем оставляют без перемешивания еще на 18 часов.
79
При этом выпадает осадок, его отфильтровывают и промы-
вают водой до исчезновения в промывных водах щелочной ре-
акции по универсальной индикаторной бумажке. Сушат при
100°. Получают 54,7 г гидробензамида (91,7% теории), т. пл.
101—102°.
Получение лофина
27 г гидробензамида нагревают в круглодонной колбе ем-
костью 100 мл до 360°. При этом гидробензамид плавится и
превращается в желтую жидкость, которая по мере нагрева-
ния темнеет; по достижении 360° наступает бурная реакция с
выделением большого количества густых белых паров. Реак-
ционную массу при этой температуре выдерживают 5 минут и,
не давая охладиться, выливают в фарфоровую ступку. Зас-
тывшую массу растирают в порошок, примеси экстрагируют
эфиром на холоду (порциями по 30 мл) до бесцветной эфир-
ной вытяжки. Остаток растворяют в 100 мл уксусной кисло
ты, отфильтровывают от нерастворившихся примесей. Из филь-
трата добавлением воды высаживают технический лофин, его
отфильтровывают, промывают водой и сушат при 100°. Полу-
чают 23 г технического лофина (62% теории), т. пл. 273°.
Для очистки 2 г технического лофина растворяют в смеси
60 мл этилового спирта и 80 мл водного аммиака. Раствор
кипятят с обратным холодильником в течение 1 часа, отфиль-
тровывают, охлаждают и для полного выделения осаждают
лофин водой. Отфильтровывают, промывают водой до исчез-
новения щелочной реакции промывных вод по универсальной
индикаторной бумажке и сушат при 100°.
Получают 1,5 г лофина (75% от загруженного), т. пл.
274—275°.
Найдено %: N 9,42, 9,45.
C2iHi6N2- Вычислено %: N 9,45.
ЛИТЕРАТУРА
1. L. Ergey, J. Bizas, Analyt. Chim. Acta., 15, 4,3z2 (1956).
2. Br. Radztszewskl. Ber., 10, 70, (1877).
3. Br. Radztszewskl, Ber., 15, 1493 (1882).
ПИРОКАТЕХИНОВЫЙ ФИОЛЕТОВЫЙ
3,3'-4'-Триоксифуксон-2"-сульфокислота,
пирокатехинсульфофталеин
Е. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
cltH14o,s
м. в. 386,33
Пирокатехиновый фиолетовый является кислотно-основ-
ным индикатором с переходом окраски раствора при pH 0,2—
0,8 от розовой к оранжевой; pH 4,0—7,0 от желтой к зеленой;
pH 8,5—10,2 от фиолетовой к синей; pH 10,2—12,5 от синей к
зеленой [1]. Применяется, главным образом, как комплексо-
метрический индикатор, позволяющий, при определенных ус-
ловиях, титровать ряд катионов: Th, Bi, NT, Со, Мп, Zn, Mg,
Cd, Zr [2—7]. По литературным данным, пирокатехиновый фи-
олетовый получают конденсацией пирокатехина с симметрич-
ным дихлорангидридом о-сульфобензойной кислоты [8—9], с
ангидридом о-сульфобензойной кислоты [9—11]. Нами пирока-
техиновый фиолетовый получен конденсацией пирокатехина с
ангидридом о-сульфобензойной кислоты [11] (см. стр. 85).
При этом были уточнены условия синтеза и получен образен
индикатора в химически чистом состоянии.
ОН ОН ОН
I I '
2,/Х.-ОН <Х.-СО НО--< ,Х\-ОН
IJ + 'х J-so,/° —" U U +н,°
I О
/X I
I |-so,
6 Зак. 973
8!
Характеристика основного сырья
Ангидрид о-сульфобензойной кислоты, ч., ГОСТ 5829—51.
Пирокатехин, ч., ГОСТ 2792—51.
Условия получения
В колбу, погруженную в глицериновую баню и снабжен-
ную мешалкой и термометром, помещают 10 г (0,05 М) ангид-
рида о-сульфобёнзойной кислоты и 5,5 г (0,05 Л4) пирокатехи-
на. Смесь нагревают до 100—110° и выдерживают при этой
температуре и размешивании 1,5 часа. Образуется густая тем-
но-фиолетовая масса, которую при 100° (в этой же колбе) ра-
створяют при размешивании в 23 мл ледяной уксусной кисло-
ты. Выпавшие кристалл^ пирокатехинового фиолетового от-
фильтровывают и промывают 10 мл уксусной кислоты. Сушат
при 100°. Получают технический пирокат^хинрвый фиолетовый
в количестве 3,3 г (34% теории, считая на пирокатехин). Тех-
нический продукт перекристаллизовывают из 50-кратного ко-
личества ледяной уксусной кислоты. Выход 55% от загружен-
ного.
ЛИТЕРАТУРА
1. Ю. В. Карякин, «Кислотно-основные индикаторы», стр. 69, Госхим-
издат, 1951.
2. V. Suk, М. Maldt, Coll. Czech. Chem. Comm., 20, (Г), 150 (1954);
An. Abst., 2. 1764 (1955).
, 3. M Maldt, V. Suk O. Ryba, Chem. listy, 48, 2, 205 (1954).
4. M Maldt. Coll Czech. Chem. Comm., 6, 1156 (1954).
5. V. Suk. M. Maldt, O. Ryba, Chem. listy, 48, 535 (1954).
6. M. Maldt, V. Suk. A. JenLkova., Chem. listy, 48 (5) 665 (1954).
7. J. Mair, Chem. Abstrs, 14, 3607 (1920).
8. С. B. Wood, I. Amer. Chem. Soc., 52, 5463 (1930).
9. Чешек, пат. 84119, 1953.
10. V. Suk, M. NCldt, Chemist Analyst, 45, 2 (1956).
11. Z. Voddk. O. Leminger, Chem. listy, 48, 552 (1954).
П ИРОКАТЕХИ НФТАЛЕИ Н
Е. Я-Я РОВЕН КО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
но—// //-он
но-l \ * /он
1 i
//-со
GjoH14Oe м. в. 350,35
Пирокатехинфталеин представляет интерес как комплексо-
метрический индикатор. Проверен и уточнен метод его получе-
ния [1, 2]. Получен образец в химически чистом состоянии.
ОН
Характеристика основного сырья
Ангидрид фталевой кислоты, ч., ГОСТ 5869—51.
Пирокатехин, ч., ГОСТ 2792—51.
Хлористый цинк, ч., ГОСТ 4529—48.
: Условия получения
В трехгорлую колбу емкостью 250 мл помещают 22,2 г
(0,15 М) фталевого ангидрида и 11 г (0,1 М) пирокатехина.
Реакционную массу нагревают до расплавления и к ней, при
размешивании, по частяй й течёние 0,5 часа прибавляют
20,4 г (0,15 М) Хлористого цинка, предварительно обезвожен-
ного при 260° в течение,! часа. По добавлении всего хлористо-
го цинка: реакционную кассу нагревают до 145° и выдержива-
ют при этой температуре в течение 3 часов. По охлаждении
6* 83
реакционная масса затвердевает. Плав экстрагируют горячей
водой (порциями по 200 мл) до тех пор, пока проба водного
раствора не перестанет давать с едким натром синего окраши-
вания (~ 3 л). Раствор кипятят с активированным углем, от-
фильтровывают от него и оставляют на 12-—18 часов. Выпада-
ет маслянистое вещество, которое отделяют от водного слоя и
для освобождения от пирокатехина нагревают 30 минут с
100 мл бензола при 60—70°. По охлаждении осадок отфильтро-
вывают и сушат при 100°. Получают 20 г технического пиро-
катехинфталеина (57,3% теории, считая на пирокатехин).
Для очистки технический пирокатехинфталеин три раза пе-
рекристаллизовывают с углем из 13 частей воды.
Получают 8,2 г пирокатехинфталеина (23% теории, считая
на пирокатехин).
Найдено %: С 68,10; 68,10; Н 4,05; 3,95.
СгоНиОе. Вычислено %: С 68,3; Н 4,0.
ЛИТЕРАТУРА
1. /?. Meyer, Н. Pfotenhauer, Вег., 40, 1442 (1907).
2. R. Baeyer, Е. KochendOrfer, Вег., 22, 2196 (1889).
0-СУЛЬФОБЕНЗОЙНОЙ КИСЛОТЫ
МОНОАММОНИЙНАЯ СОЛЬ
Е. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
/ \-COOH
C,H9O5NS
м. в. 219,23
Моноаммонийная соль о-сульфобензойной кислоты приме-
няется для получения сульфофталеиновых индикаторов.
Ее обычно получают из имида о-сульфобензойной кисло-
ты [1—3].
Нами о-сульфобензойная кислота получена действием вод-
ного аммиака на раствор о-сульфобензойной кислоты.
СООН . ...
4-NH4OH
‘^y-SOaH.
.^Х-СООН
\ /'-SO3NH4
4-Н,0
84
Характеристика основного сырья
Аммиак водный, ч., ГОСТ 9—57.
о-Сульфобензойная кислота, ч., ВТУ МХП 2833—53.
Условия получения
В фарфоровой чашке емкостью 200 мл растворяют 100 г
(0,49 М) о-сульфобензойной кислоты в 125 мл воды, к раст-
вору прибавляют 32 мл водного аммиака (до pH 2—3 по уни-
версальной индикаторной бумажке). Раствор выпаривают на
водяной бане до объема 80—90 мл, охлаждают, выпавшие кри-
сталлы отфильтровывают и сушат на воздухе до постоянного
веса. Получают 81,1 г технической аммонийной соли о-суль-
фобензойной кислоты (74,8% теории).
Для очистки в стакан емкостью 200 мл, снабженный ме-
шалкой, загружают 70 г технической аммонийной соли о-суль-
фобензойной кислоты, 60 мл воды и 3,5 г активированного уг-
ля. Раствор при размешивании нагревают до 70°, выдерживают
при этой температуре 30 минут, фильтруют (горячим) и охла-
ждают. Выпавшие кристаллы фильтруют, промывают 10 мл
ледяной воды. Сушат на воздухе до постоянного веса. Полу-
чают 48,6 г аммонийной соли о-сульфобензойной кислоты с
содержанием азота не ниже 98% от вычисленного. Выход со-
ставляет 69,5% от взятой технической соли или 52,5% теории,
считая на о-сульфобензойную кислоту.
ЛИТЕРАТУРА
1. «Синтезы органических препаратов», сб. 1, стр. 275, ИЛ., 1949.
2. С. Fahlberg, R. Barge, Ber., 22, 755 (1889).
3. М. Remsen, Р. Zinn, J. Amer. Chem. Soc.. 11, 74, (1889).
о-СУЛЬФОБЕНЗОЙНОЙ КИСЛОТЫ АНГИДРИД
. E. Я. ЯРОВЕНКО, Г. Н. КОШЕЛЕВА
(ВНИИ химических реактивов и особо {чистых химических веществ)
C,H4SO4 м. в. 184,18
Ангидрид о-сульфобензойной кислоты применяется для по-
лучения сульфофталеиновых индикаторов.
85
По литературным данным, ангидрид о-сульфобензойной
кислоты может быть получен из кислой аммонийной соли о-
сульфобензойной кислоты [1], нагреванием о-сульфобензойной
кислоты [2], нагреванием ее с пятиокисью фосфора [3], воздей-
ствием на нее хлористым ацетилом [4, 5], нагреванием кислой
калиевой соли о-сульфобензойной кислоты с пятихлористым
фосфором [4] или с хлористым тионилом [6]. Нами ангидрид
о-сульфобензойной кислоты получен из о-сульфобензойной
кислоты и хлористого ацетила
i/Xj-COOH + СН3СООС1------►
!х /’-SOeH
\ СО \
I ;OLCH?COOH 4-HCI
/'-SO2/
Характеристика основного сырья
Ацетил хлористый, ч., ГОСТ 5829—51.
Кислота о-сульфобензойная, ТУ 2838—53.
Условия получения
В круглодонную колбу емкостью 150 мл, снабженную об-
ратным холодильником, загружают .48.г' (0,23 А1) безводной
о-сульфобензбйной кислоты (см. Примечание) и добавляют
72 г (0,91 М) хлористого ацетила. Раствор выдерживают 12—
18 часов при комнатной температуре, после чего реакционную
смесь нагревают на водяной бане в течение 2 часов при тем-
пературе бани 40—50° до полного растворения твердого осад-
ка. Охлаждают и выпавшие (через 12—18 часов) кристаллы
отфильтровывают и сушат при 100°. Получают 32 г техниче-
ского ангидрида о-сульфобензойной киблоты (73,5%' теории),
т. пл. ИЗ—115°.
Для очистки технйческйй ангидрид перекристаллизовыва-
ют из трехкратного количества бензола. Выход 80%, считая на
загруженный ангидрид о-сульфобензойной кислоты, т. пл.
122—124°.
Примечание. Обезвоживание о-сульфобензойной кислоты прово-
дят следующим образом: в предварительно взвешенный фарфоровый ста-
кан,1 погруженный в масляную баню, загружают о-сульфобснзойную кйс'ло^
ту, нагревают около двух часов при 140—150° (периодически взвешивая),
до прекращения уменьшения-веса, после чего кислоту выливают в фарфо-
ровую тарелку (размешивая при затвердевании) и измельчают..
86,
ЛИТЕРАТУРА
1. «Реактивы и препараты .лабораторного назначения». Под редакцией
В. В. Лонгинова, стр. 312, 1938.
2. A. Remsen, В. Dohme, J. Amer. Chem. Soc., 11, 343 (1889).
3. D. Fahlberg, N. Barge, Ber., 22, 757 (1889).
4. A. Cobb, J. Amer. Chem. Soc., 35, 499 (1906).
5. If. Flaschka, F. Sadek, Z. analyt. Chem., 150, 339 (1956).
6. M. Heitman, J. Amer. Chem. Soc., 34. 1594 (1912).