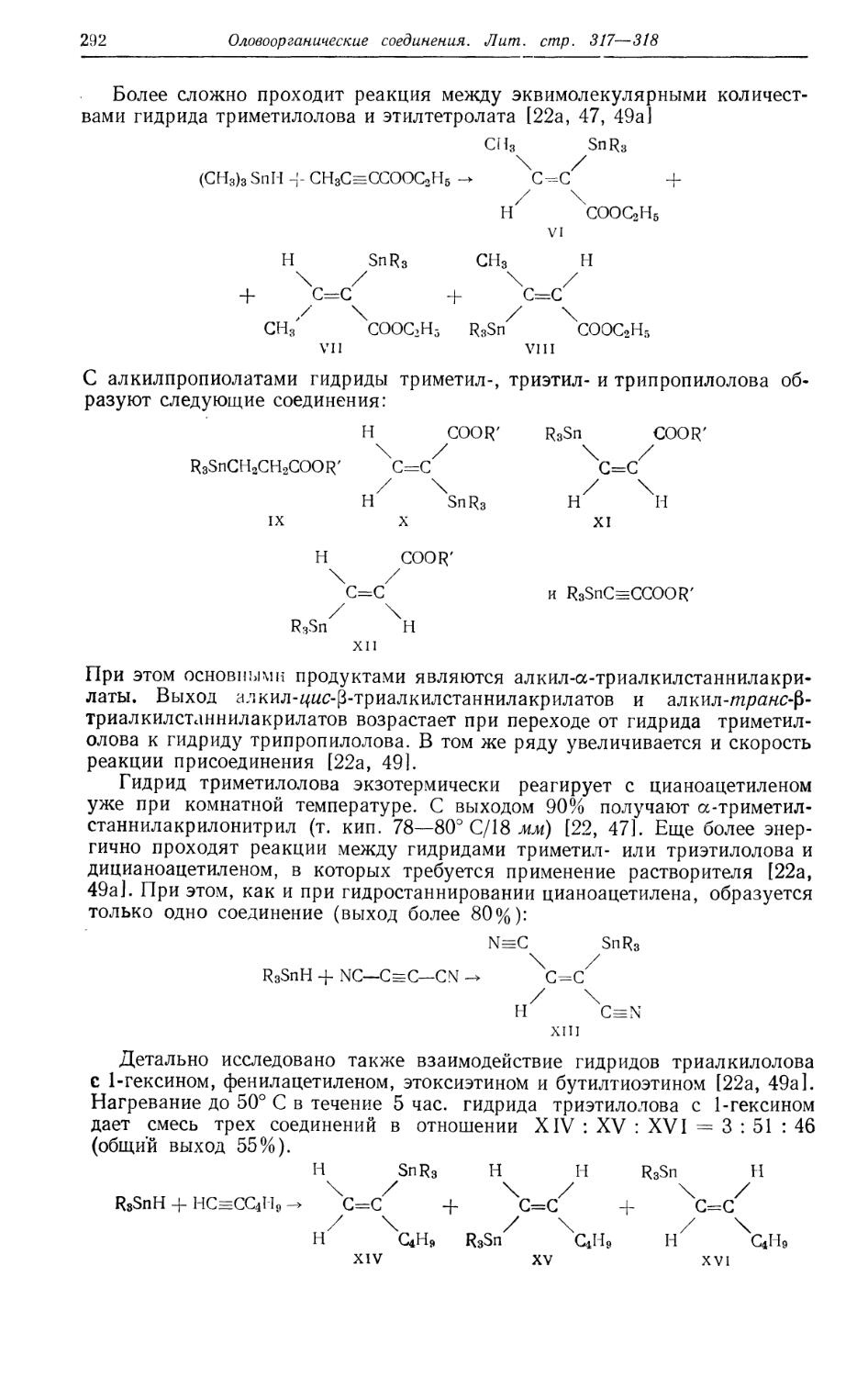
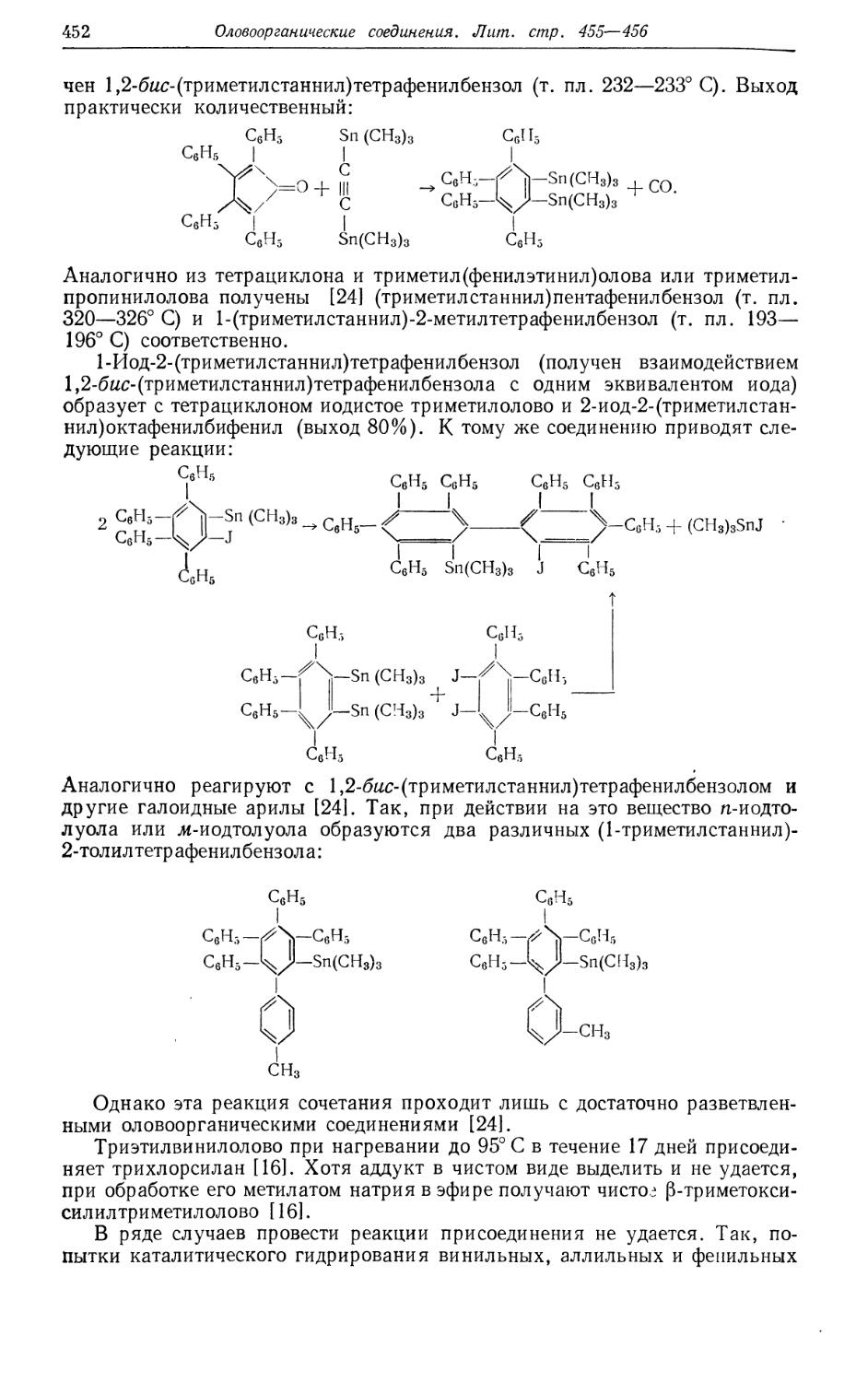
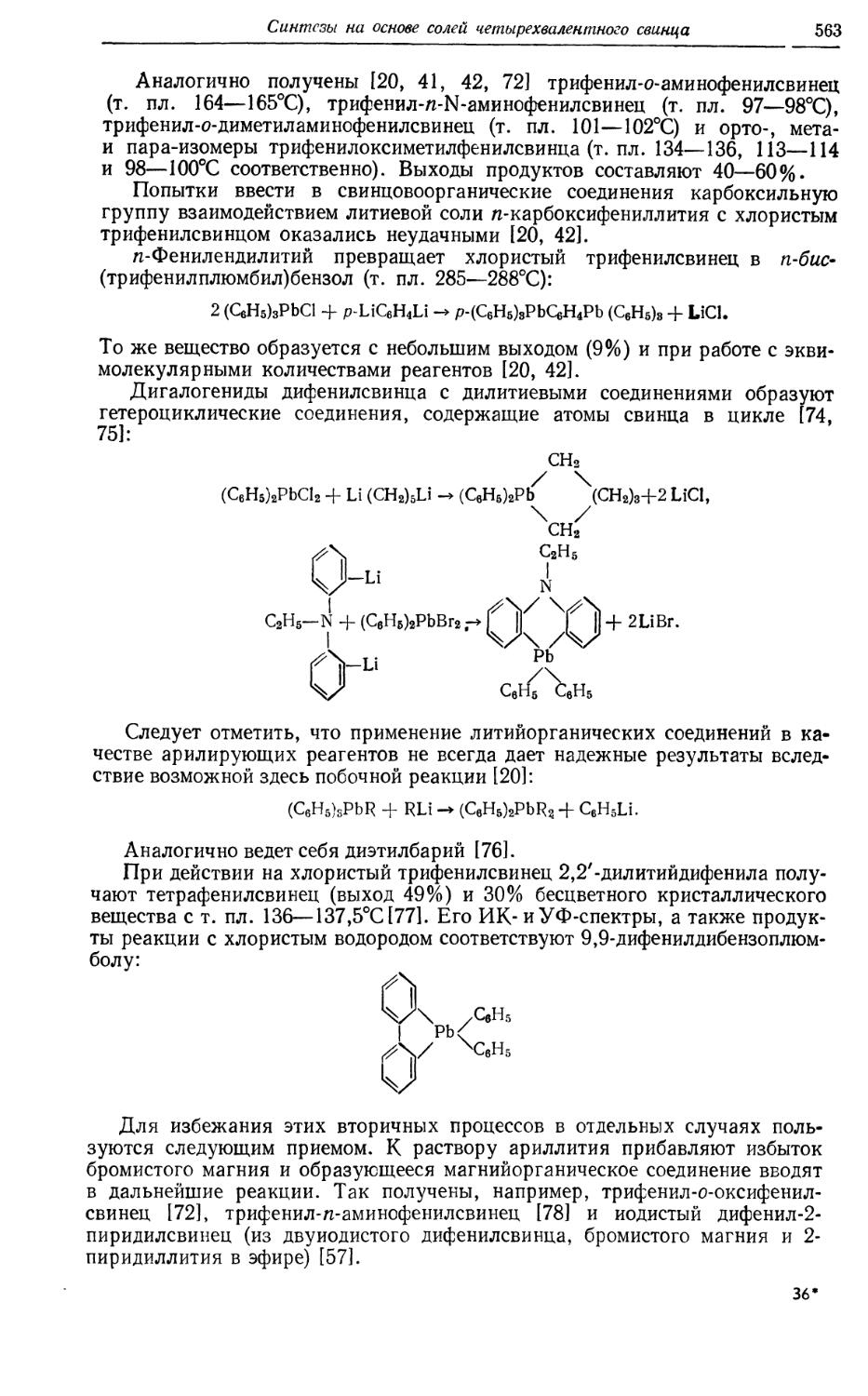
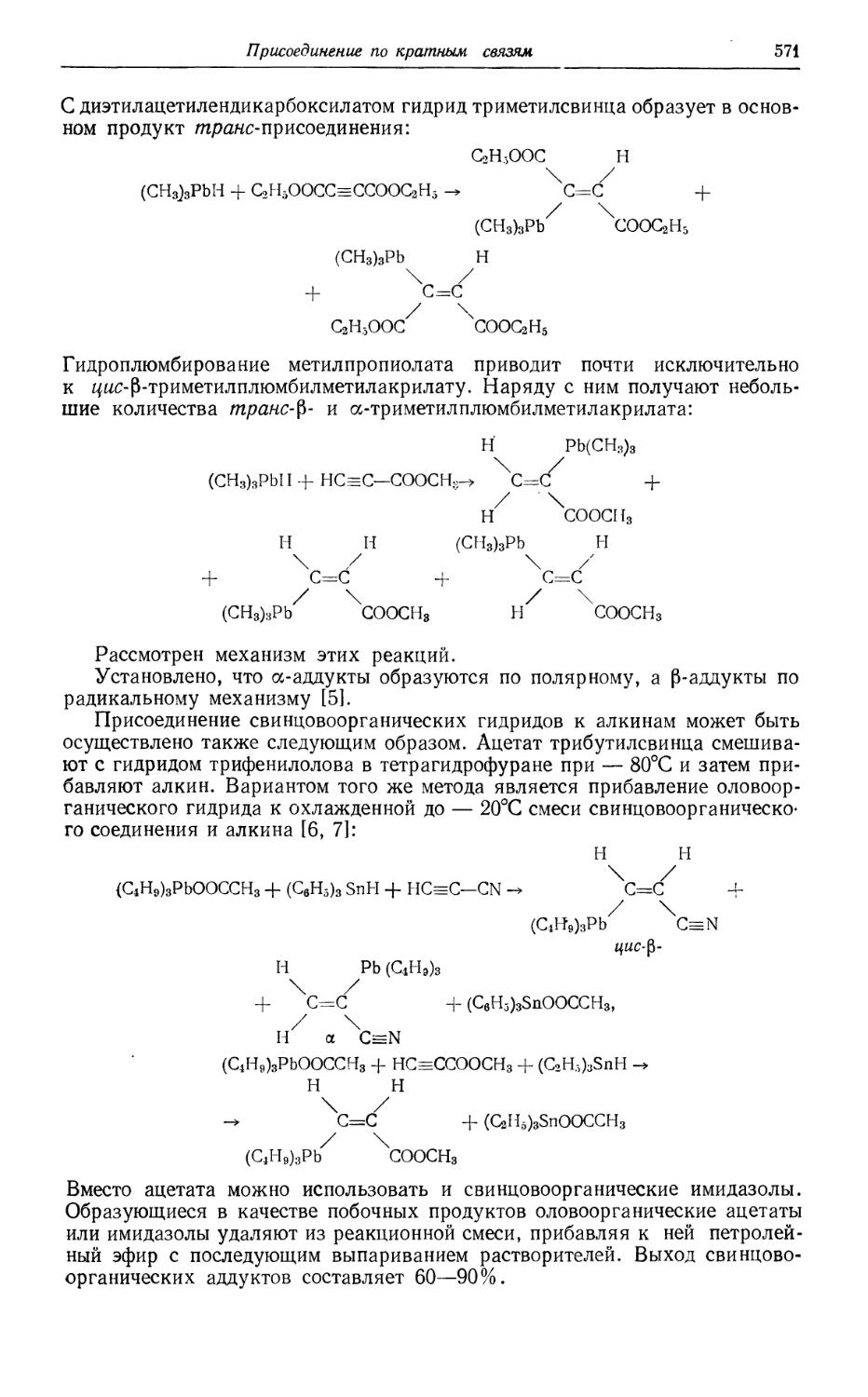
/
























































































































































































































































































































































































































































































































































































































































































































Автор: Несмеянов А.Н.
Теги: производство органических веществ органические соединения неорганическая химия химия
Год: 1968
Текст
МЕТОДЫ
ЭЛЕМЕНТО-
ОРГАНИЧЕСКОЙ
ХИМИИ
е п ' i
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ
ЭЛЕМЕНТООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Под общей редакцией
А. Н. НЕСМЕЯНОВА и К. А. КОЧЕШКОВА
МЕТОДЫ
ЭЛЕМЕНТО-
ОРГАНИЧЕСКОЙ
ХИМИИ
ИЗДАТЕЛЬСТВО «НАУКА»
Москва • 1 968
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ
ЭЛЕМЕНТООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
К. А. КОЧЕШКОВ, Н. Н. ЗЕМАЯНСКИЙ,
Я. Я. ШЕВЕРАЯНА, Е. М. ПАНОВ
МЕТОДЫ
ЭЛЕМЕНТО-
ОРГАНИЧЕСКОЙ
ХИМИИ
ГЕРМАНИИ
ОЛОВО
СВИНЕЦ
ИЗДАТЕЛЬСТВО «НАУКА»
Москва• 1 968
УДК 661.718:546.289 + 546.81
Методы элементоорганической химии. Германий, олово, свинец. Кочешков К. А.,
Землянский Н. Н., Шевердина Н. И., Панов Е. М. Изд-во «Наука», 1968 г.
Монография посвящена методам синтеза и реакциям металлоорганических
соединений германия, олова и свинца, имеющим большое научное и
практическое значение и интенсивно разрабатываемым в последние годы. Обобщен
обширный материал, охватывающий с максимально возможной полнотой
многочисленные работы зарубежных и отечественных исследователей, в том
числе работы авторов монографии. Наряду с ранее известными рассматриваются
новые классы германий-,олово- и свинцовоорганических соединений —
органические гидриды этих металлов и соединения, содержащие два и более атомов
металла в молекуле. Описываются физико -химические свойства и методы
анализа указанных типов металлоорганических соединений, рассматриваются
основные области их практического использования.
Книга рассчитана на химиков: работников научно-исследовательских
учреждений и заводских лабораторий, а также преподавателей и студентов
зузов.
Таблиц 10. Иллюстраций 3. Библ. назв. 4522.
Ответственный редактор
академик А. Н. НЕСМЕЯНОВ
2-5-3
283-68(1)
ПРЕДИСЛОВИЕ
Настоящая книга из серии монографий «Методы элементоорганической
химии» посвящена металлоорганическим соединениям германия, олова и
свинца.
В последние 2—3 десятилетия химия оловоорганических соединений
развивалась весьма интенсивно, несколько менее — химия германийорга-
нических соединений и по сравнению с производными этих металлов
довольно малое место занимают исследования в области свинцовоорганических
соединений. Во всяком случае за 20 лет, прошедших со времени выхода
монографии «Синтетические методы в области металлоорганических
соединений элементов IV группы», написанной одним из авторов настоящей
книги *, объем материала возрос примерно в 4—5 раз. Возник целый ряд
совершенно новых разделов в области синтеза, реакций, а также и в области
практического применения этих металлоорганических соединений. Таким
образом, необходимо было создавать книгу по существу заново.
Для всех трех рассматриваемых элементов известны соединения
следующих основных типов: R4M, R3MX, R2MX2 и R6M2, где R —
алифатический, ароматический или гетероциклический радикал; X — галоид,
остаток минеральной, органической кислоты или другая функциональная
группа (например OR, SR, NR2 и т. п.). Подробно изучены также германий-
и оловоорганические соединения типа RMX3. Соответствующие
производные свинца длительное время не были известны, и только в последние годы
разработаны методы синтеза триацилатов арилсвинца — ArPb(OOCR)3.
В отношении алифатических свинцовоорганических соединений этого типа
в литературе имеются противоречивые данные.
По методам синтеза и свойствам среди других классов выделяются
металл оорганические гидриды и соединения, содержащие два или более
атомов металла в молекуле.
Систематические исследования германий- и оловоорганических гидридов
начаты только в последние годы. Теперь это большой и разнообразный
материал. Основные типы органических гидридов: R3MH, R2MH2, R2MHX,
RMH3; органические гидриды свинца типа R3PbH и R2PbH2 известны пока
лишь в алифатическом ряду, не описаны и R2PbHX и RPbH3. Различие в
развитии химии указанных металлов связано, вероятно, с неодинаковой
устойчивостью органических производных: Ge > Sn > Pb.
Относительная реакционная способность отвечает тому же ряду.
В области полиметаллических соединений наиболее подробно изучены
соединения типа R3GeM, R3SnM и R3PbM (M — щелочной металл), для
олова известны также соединения со связями Sn—Cd, а для германия —
соединения с рядом элементов II—IV групп.
* К. А. Кочешков. Синтетические методы в области металлоорганических
соединений элементов IV группы. М.—Л., Изд-во АН СССР, 1947.
•6
Предисловие
Значительный интерес представляют соединения германия, олова и
свинца с карбонилами переходных металлов, например R3GeRe(CO)5,
R3SnMn(CO)5, R3PbCo(CO)5 и др. Обращает на себя внимание
устойчивость связи металл — металл в этих соединениях к действию электрофиль-
ных реагентов.
Соединения, отвечающие формально низшей валентности
рассматриваемых металлов, существуют, вероятно, лишь в полимерной форме,
определенные данные имеются только в отношении (R2Sn),2.
Переходя к сравнительной характеристике методов синтеза германий-,
олово- и свинцовоорганических соединений, необходимо отметить, что
исходными веществами обычно являются галоидные соли четырехвалентного
олова и германия или двухвалентного свинца (четыреххлористый свинец,
как известно, неустойчив и обладает окислительными свойствами). В
качестве алкилирующих (или арилирующих) агентов чаще всего применяют
магний-, литий-, натрий- и цинкорганические соединения. В последнее
время для этой цели начали использоваться и алюминийорганические
соединения. Сюда же примыкают реакции дигалогенидов германия и олова с ди-
арилртутью, приводящие к Ar2SnX2 или Ar2GeJ2. Использование в
реакциях с диарилртутьютетраацилатов свинца дало возможность получить
новый класс свинцовоорганических соединений—ArPb(OOCR)3.
Прямой синтез из галоидного алкила и металла разработан
неравномерно. Так, взаимодействие германия (обычно применяется смесь германий—
медь) и хлористых алкилов ограничено соединениями метильного и этиль-
ного ряда. Для оловоорганических соединений реакции прямого синтеза
с участием хлористых алкилов пока разработаны на ограниченном числе
примеров: получение бензильных производных из порошка олова и
хлористого бензила в растворе, метальных производных из хлористого метила
и окиси олова или некоторых алкильных производных из хлористых
алкилов и олова в присутствии третичных аминов. Реакции между
йодистыми алкилами (или смеси их с бромистыми алкилами) и оловом в
присутствии катализаторов проходят достаточно хорошо. Под действием
у-лучей (Со60) порошок олова реагирует с любыми бромистыми алкилами
с образованием с хорошими выходами соединений типа R2SnX2. Для
свинцовоорганических соединений реакции прямого синтеза пока не изучены.
Однако в промышленном синтезе тетраэтилсвинца широко применяют
сплавы свинца с натрием и хлористый этил.
В синтезе металллоорганических соединений с различными
заместителями в алифатическом радикале у атома металла важное значение имеют
реакции присоединения гидридов германия, олова или свинца к алкенам
и алкинам.
Метод наиболее подробно изучен в ряду германийорганических
соединений, где можно исходить как из неорганических, так и из органических
гидридов. Гидриды германия типа HGeX3 присоединяются к олефинам уже
при комнатной температуре и в отсутствие катализаторов. С германийорга-
ническими гидридами реакция, как правило, проходит в присутствии
катализаторов (например H2PtCl6).
Что касается оловоорганических гидридов, то во многих случаях они
присоединяются к олефинам при простом нагревании смеси. Часто реакцию
проводят в присутствии каталитических количеств азодиизобутиронитрила.
Свинцовоорганические гидриды в этом плане остаются мало
изученными. Согласно имеющимся немногочисленным данным, они присоединяются
к алкенам и алкинам в гораздо более мягких условиях, чем гидриды олова.
Необходимо отметить, что к достаточно полярной кратной связи могут
присоединяться и другие соединения германия, олова и свинца. Так, ал-
кокситриалкилгерманий в присутствии спиртового раствора йодной ртути
Предисловие
7
присоединяется к кетену. Алкоксисоединения олова и станниламины с ке-
теном реагируют экзотермически. Энергично присоединяются к кетену и
гидроокиси или ацилаты триарилсвинца. Однако этокситриметилсвинец в
этих условиях с кетеном не реагирует.
Германий-, олово- и свинцовоорганические соединения могут быть
получены при действии алифатических диазосоединений на галоидные соли
этих металлов, а также действием на органические гидриды германия и
■олова производных' диазоуксусного эфира. Использование солей диазония
также позволяет синтезировать металлоорганические соединения германия,
олова и свинца.
По своим химическим свойствам металлоорганические соединения
германия, олова и свинца имеют много общего, хотя есть и существенные
различия. Прежде всего необходимо отметить, что увеличение межатомных
расстояний и связанное с этим ослабление внутримолекулярных связей
приводит к постепенному уменьшению прочности связи металл — углерод при
переходе от германия к олову и свинцу. Относительная устойчивость
этой связи изменяется в следующем порядке: Ge>»Sn»Pb.
В этом отношении значительный интерес представляют реакции
перехода органических радикалов от олова или свинца к германию или от
свинца к олову, например при нагревании тетраалкильных соединений с гало-
генидами соответствующих металлов.
При действии галоидов или галоидоводородов на соединения типа R4M
в случае германийорганических соединений наблюдается отщепление
только одного радикала. Дигалогениды диалкилгермания получают в
присутствии катализаторов типа хлористого алюминия. Деалкилирование олово-
органических соединений проходит при температурах, близких к
комнатным, или при нагревании смеси до 50—60° С. При этом отщепляются один,
два, или все четыре органических радикала. Со свинцовоорганическими
соединениями аналогичные реакции проходят уже при низких
температурах.
Реакция деалкилирования галоидными солями одноименных металлов,
столь четко протекающая у оловоорганических соединений, в случае
германийорганических соединений проходит более сложно, приводя к смеси
продуктов различной степени алкилирования, причем второй радикал
отщепляется только в присутствии катализатора. У свинцовоорганических
соединений реакции этого типа остаются мало изученными.
Германий-, олово- и свинцовоорганические галогениды легко гидроли-
зуются водными растворами щелочи и аммиака с образованием
гидроокисей, окисей или соответствующих кислот. Однако для соединений германия
и олова более характерно образование полимерных и олигомерных окисей,
в то время как дигалогенидам диалкилсвинца присуще, по-видимому,
образование дигидроокисей диалкилсвинца.
Что касается реакций, связанных с преобразованием функциональных
групп у атома металла, то они довольно широко изучены в ряду германий-
и оловоорганических соединений, менее — у свинцовоорганических
соединений. Существенных различий в этих реакциях для указанных
металлов не наблюдается.
Наоборот, реакции, связанные с преобразованием в радикале (стоящем
у атома металла), более характерны для германийорганических соединений
в связи с большей прочностью связи Ge—С (например присоединение
галоидов и галоидоводородов к винильным и аллильным соединениям
германия). Активные группы (CN, галоид, COOR) в радикале металлооргани-
ческого соединения не только германия, но и олова и свинца могут
вступать в обычные реакции, характерные для этих групп без разрыва связи
С—М.
8
Предисловие
Что касается практического применения рассматриваемых металлоор-
ганических соединений, то в настоящее время оловоорганические
соединения используются как стабилизаторы полимеров (например поливинил-
хлорида), как катализаторы различных процессов (конденсация,
полимеризация), а также в качестве физиологически активных веществ. Германий-
органические соединения пока практического применения не нашли.
Классическое применение тетраэтилсвинца — в качестве добавки к моторному
топливу как антидетонатора до сих пор сохраняет свое значение.
Многочисленные работы, посвященные синтезу полимерных материалов
с атомами германия, олова и свинца (по аналогии с кремнийорганическими
полимерами), пока еще далеки от завершения.
Следует упомянуть о токсичности оловоорганических и свинцовоорга-
нических соединений (особенно последних). Токсичность германийоргани-
ческих соединений в настоящее время изучена не достаточно.
При написании данной книги использовались соответствующие разделы
в монографии Кочешкова [1], Губена [2], Краузе и Гроссе [3], Рунге [4],
Шмидта [5], Гилмана [6], Рохова, Херда и Льюиса [7], а также
монографий Коатса [8, 9], Лукевица и Воронкова [10, 11], Брилкиной и
Шушунова [12]. Книги и обзорные статьи, касающиеся в отдельности германия,
олова или свинца, приведены во введениях к каждой части настоящей
книги.
В настоящей книге оригинальные работы использованы до 1
октября 1967 г. (считая дату по реферативному журналу «Химия»).
ЛИТЕРАТУРА
1. Кочешков К А., Синтетические методы в области металлоорганических
соединений элементов IV группы. М.— Л., Изд-во АН СССР, 1947.
2. Но u be nJ., Die Methoden der organischen Chemie, Bd. 4. Leipzig, VerlagG. Thieme,.
1924.
3. KrauseE., GrosseA., Die Chemie der metallorganischen Verbindungen. Berlin,.
Borntrager, 1937.
4. R u n g e F., Organometallverbindungen, Teil I. Stuttgart, WissenschaftlicheVerlaggesell-
schaft, 1932 (русск. перевод: М.—Л., ОНТИ, 1937).
5. Schmidt J., Organometallverbindungen, Teil II. Stuttgart, Wissenschaftliche Verlag-
• gesellschaft, 1934 (русск. перевод: Л., ОНТИ, 1937).
6. G i 1 m a n H., Organic chemistry. An advanced treatise, vol.1, New York, J. Wiiley, 1943.
7. R о с h о w E. G., H u r d D. Т., L e w i s R. N., The chemistry of organometallic
compounds, New York, J. Wiiley, 1957 (русск. перевод: М., ИЛ, 1963).
8. С о a t e s G. E., Organometallic compounds. London, Methuen, 1956.
9. С о a t e s G. E., Organometallic compounds, 2 ed. London, Methuen. 1960.
10. Л у к е в и ц Э. Я., В о р о н к о в М. Г., Гидросилилирование, гидрогермилирование
и гидростаннилирование. Рига, Изд-во АН ЛатвССР, 1964.
11. L u k e v i t s E. Y., V о г о п к о v M. G., Organic insertion reactions of group IV
elements. Consultants Bureau—New York, 1966.
12. Брилкина Т. Г., Ш у ш у н о в В. А., Реакции металлоорганических соединений
с кислородом и перекисями. М., «Наука», 1966.
ГЕРМАНИИОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ВВЕДЕНИЕ
Химия германийорганических соединений начала развиваться в
последнее десятилетие, а особенно интенсивно в последние два — три года. Первый
обзор Джонсона [1], изданный в 1951 г., содержал лишь около ста литера-
турных ссылок, включая и статьи, посвященные неорганическим
соединениям германия. В настоящее время число работ превышает несколько
сотен.
В области германийорганических соединений известны соединения
типов R4Ge, R6Ge2, R4-nGeXn где R — ароматический, алифатический,
гетероциклический радикал; X — галоид, кислород, гидроксил,
различные ацилаты, алкокси-, амино-, меркаптогруппы и др. Радикалы в
германийорганических соединениях могут быть одинаковыми и разными.
В последние годы.интенсивно развивается область химии
органических гидридов германия различной степени алкилирования, таких как
R3GeH, R2GeH2, RGeH3, а также R2Ge (X)H, RGe (X2)H, RGe (X)H2 (где
X — галоид, чаще всего хлор).
Много внимания уделяется соединениям типа R3GeM. В 1965 г.
получены также соединения типа R2GeM2.
Как правило, германийорганические соединения — это вещества,
устойчивые на воздухе; в алифатическом ряду — жидкости, в ароматическом —
твердые вещества. Органические гидриды германия менее стабильны в
присутствии воздуха, работу с ними проводят в атмосфере инертного газа.
С точки зрения стабильности органические гидриды германия занимают
промежуточное место между соответствующими гидридами олова и кремния
(последние наиболее устойчивы).
Что касается физиологической активности германийорганических
соединений, то на основании имеющихся данных можно, видимо, сделать
заключение, что они значительно менее токсичны, чем соответствующие
соединения олова и свинца [2].
Одним из главнейших методов синтеза германийорганических
соединений является действие магнийорганических соединений на галоидные
соли четырехвалентного германия или галогениды алкилгермания
4RMgX + GeX4 -* R4Ge + 4MgX2.
В последнем случае можно получать и соединения с различными
радикалами.
В ароматическом ряду для получения соединений типа R4Ge удобно
применять и литийорганические соединения.
Магний- и литийорганический синтезы иногда применяют и для синтеза
соединений типа R4_„GeX„, однако этот метод не является характерным
в связи с возникающими трудностями разделения образующих галогенидов
алкилгермания и наличием других более удобных методов их получения.
10 Германийорганичвские соединения. Лит. стр. 16—18
Синтез с участием металлического натрия (по аналогии с реакцией Вюр-
ца) дает низкие выходы, однако он имеет некоторое значение для получения
соединений, имеющих в молекуле, кроме германия, еще другой металл,
например:
Na
(СНз)з GeCl + (СНз)з МСН = СНС1 -* (CH3)3GeCH= CHM (СН8)3.
(M = Sn, Si, Pb).
Применение цинкорганических соединений в синтезе
германийорганических соединений (R4Ge) является полезным только при получении малых
количеств особо чистых веществ. Однако применение растворов
цинкорганических соединений может иметь ряд преимуществ перед гриньяровским
синтезом (это направление еще мало разработано).
С помощью алюминийорганических соединений германийорганические
соединения (R4Ge) получены только в отдельных случаях.
Описанные выше методы приводят к соединениям типа R4Ge. Для
синтеза германийорганических соединений типа R4_rt GeX„ (где R — в
основном СН3 или С2Н5, п = 2 и 3) на первое место необходимо поставить
прямой метод синтеза. В алифатическом ряду он имеет преимущественное
значение перед другими методами.
RCl + Ge/Cu-^R4_„GeCl„
(я = 2 и 3).
Особенностью прямого метода синтеза германийорганических соединений
по сравнению с кремнийорганическими соединениями является то, что в
случае германия не образуются соединения типа RGeHCl2, структурные
аналоги которых RSiHCl2 часто преобладают при взаимодействии RC1 с
кремнемедными сплавами.
Для синтеза соединений типа R4_nGeXn, где п = 3, имеет некоторое
значение действие йодистых алкилов на соли двухвалентного германия
C2H5J + GeJ2 -* C2H5GeJ3.
Представляет интерес открытая недавно своеобразная реакция иодбен-
зола с четырехбромистым или четыреххлористым германием, приводящая
к труднодоступному иными путями ArGeJ3.
Действие ртутноорганических соединений на соли четырехвалентного
германия приводит к получению соединений низшей степени арилирования
и может быть представлено реакцией
Ar2Hg + GeCl* -* ArGeCl3 + ArHgCl.
Интересно отметить, что реакция восстановления с участием солей
двухвалентного германия дает возможность получать соединения более высокой
степени арилирования
Ar2Hg + GeJ2 -> Ar2GeJ2 + Hg.
В последнее время область германийорганических соединений
обогащена рядом ценных реакций. Сложные эфиры кислот, имеющие в
молекуле атом германия, хорошо получаются по реакции
/Ох О
2R3GeJ + Hg CH2C - 2R3GeCH2C + HgJ2.
V OR'Л OR'
Сложные эфиры триалкилстаннилуксусной кислоты при реакции с
четыреххлористым германием также образуют эфиры алкилгермилуксусной
Введение
11
КИСЛОТЫ
— >Cl3GeCH2COOR
-^-- Cl2Ge (CH2COOR)2
-^-->ClGe(GH2GOOR)3
-i!f~> Ge (CH2COOR)4
Большое значение в синтезе функциональнозамещенных германийорга-
нических соединений в настоящее время приобрело присоединение
различных органических и неорганических гидридов германия к кратным связям.
Например, гибрид трех хлор исто го германия легко присоединяется
практически к любым непредельным соединениям при комнатной температуре
в отсутствие катализаторов. При более высокой температуре (140—150° С)
также реагируют с непредельными соединениями галоидгидриды алкил-
германия (катализатор — H2PtCl6). В обоих случаях присоединение
распространено на алкены, алкины, диеновые соединения, ряд непредельных
соединений: галоидные алкенилы, спирты, альдегиды, кетоны, карбоновые кислоты
и их производные, а также ряд металлоорганических соединений, имеющих
непредельные радикалы в молекуле. GeJ2 присоединяется легко к
ацетиленовым соединениям, образуя шестичленные циклические соединения,
имеющие атомы германия в цикле.
Следует отметить присоединение алкоксисоединений германия к кете-
ну, идущее в двояком направлении (I) и (II).
R3GeOR + СН2 = С =р -► R3GeCH2COOR. (I)
Реакция (I) идет только в присутствии йодной ртути (спиртовый раствор),
■без катализатора образуются эфиры |3-триалкилгермилоксивинилуксусной
кислоты
|R3GeOR + 2СН2 =С-=0->СН2 = С- CH2COOR
Диазометан при низкой температуре реагирует с четыреххлористым
германием, образуя с хорошим выходом треххлористый хлорметилгерманий.
GsGU + CH2N2 -* Cl3GeCH2Cl + N3.
В присутствии порошка меди диазоуксусный эфир, диазоацетон и диа-
зоацетофенон реагируют с гидридами алкилгермания с образованием
функциональнозамещенных в а-положении по отношению к атому германия
германийорганических соединений (хороший выход)
R3GeH + N2CH2COOC2H5 -> R3GeGH2GOOC2H5 + N2.}
Что касается ароматического ряда, то если разложению подвергнуть
«борфториды арилдиазониев в присутствии GeCl4, то образуются германий-
органические соединения низшей степени арилирования ArGeCl3.
Многочисленные другие реакции в настоящей книге сведены в основном
к четырем типам. Реакции деалкилирования, реакции гидролиза, реакции,
связанные с изменением функциональных групп у атома германия, и
реакции, связанные с изменением в радикале германийорганических
соединений.
Реакция деалкилирования у германийорганических соединений
происходит гораздо труднее, чем в случае оловоорганических соединений,
-{-R.tSnCH,COOR
UeU4 _R3SnCl
12
Германийорганические соединения. Лит. стр. 16—18
и чаще всего приводит к получению смеси органических галогенидов германия
различной степени алкилирования, разделение которых представляет ряд
экспериментальных трудностей вследствие близких температур кипения.
В качестве деалкилирующих агентов чаще всего используют галоиды (бром,
иод), галоидоводородные кислоты, галоидные соли германия (GeCl4, GeBr4),
в отдельных случаях применяют и галоидные алкилы. При деалкилирова-
нии галоидами, как правило, отщепляется один радикал, второй радикал
отщепляется в присутствии катализаторов типа хлористого алюминия. Из
галоидоводородных кислот наиболее удобным деалкилирующим агентом
является фтористый водород, отщепляющий только один радикал (далее
реакция не идет).
Реакция деалкилирования с помощью GeX4, так широко применяемая
в ряду оловоорганических соединений, для германийорганических
соединений идет труднее. Реакция требует применения катализаторов —
хлористого алюминия или алюминийорганических соединений. Соединения типа
RGeX3 методом деалкилирования получаются плохо, их лучше
синтезировать другими способами, например прямым методом.
Гидролиз органических галогенидов германия чаще всего проводят
водным раствором щелочи. По сравнению с кремнийорганическими
соединениями гидролиз германийорганических соединений происходит труднее и
неполно:
2R3GeX ■£. R3GeOH ^ (R3Ge)2 О,
R2GeX2 ^ [R2Ge (OH)2] •£. R2GeO,
2RGeX3 ;± [RGe (OH)3] ^ 2RGeOOH ^ (RGeOfeO.
Гидролиз галогенидов алкилгермания приводит к гидроокисям (чаще
к гексаалкилдигерманоксанам), окисям и соответствующим германиевым
кислотам или их ангидридам. Окиси диалкил- и диарилгермания, так же
как и окиси диалкилолова, склонны к полимеризации и существуют обычно
в виде различных олигомеров.
Реакции, связанные с изменением функциональных групп у атома
германия, чрезвычайно многочисленны. Гидроокиси, гексаалкилдигерманок-
саны, алкилгерманиевые кислоты и их ангидриды легко реагируют с
органическими и неорганическими кислотами, заменяя кислород на остаток
кислоты.
Органические галогениды германия легко обменивают галоид на
остаток кислоты при действии серебряных (реже калиевых) солей. В обмен
вступают и другие германийорганические производные. Обменные
реакции для различных германийорганических производных изображаются
(Андерсон) в виде следующего ряда, характеризующего направление
замещения (идет всегда вправо):
J -* S -> Br -* CN -> NGS, CI -> NGO -+ OCOR -> F.
Интересно отметить, что в реакции обмена вступают и различные
германийорганические ацилаты, алкокси- и аминосоединения.
Для германийорганических соединений характерно большое число
реакций, связанных с преобразованием органического радикала у атома
германия, а именно: присоединение к непредельным алифатическим
радикалам, многочисленные реакции подвижных атомов галоида в радикале-
германийорганических соединений, реакции алкиновой группы
германийорганических соединений с магнийорганическими соединениями и
гидридами металлоорганических соединений IV группы.
Широко развитая в последнее время область химии
германийорганических гидридов имеет большое значение в синтезе германийорганических
соединений с различными функциональными группами. Методы синтеза.
Введение
13
органических гидридов германия основаны на восстановлении различных
галогенидов алкил- и арилгермания, реже — алкоксисоединений или
окисей. В качестве восстановителей наиболее часто применяют литийалюми-
нийгидрид, натрийборгидрид, иногда гидрид лития.
Получение органических галоидгидридов германия основано на
частичном галоидировании уже готовых гидридов, или частичном восстановлении
,дигалогенидов диалкилгермания с помощью моногидридов три ал кил герма-
пи я
R2GeX2 + R3GeH -» R2Ge(X)H + R3GeX.
Удобным методом получения галоидгидридов является также действие га-
.лоидных алкилов на гидриды диалкилгермания,
R2GeH3 + R'X -> R2GeH(X) + R'H.
Германийорганические гидриды окисляются труднее, чем
соответствующие оловоорганические гидриды, но легче, чем кремнийорганические
гидриды. При окислении кислородом воздуха все типы гидридов R3GeH,
R2GeH2 и RGeH3 образуют соответствующие кислородные соединения.
В присутствии порошка меди органические гидриды германия
реагируют с соединениями, имеющими подвижный водород, такими как вода,
спирты, гликоли, фенолы, органические кислоты с образованием связи
•Ge—О и выделением водорода
R3GeH + R'OH -* R3GeOR' + Н2.
В случае альдегидоз и кетоноз происходит присоединение к двойной связи
.карбонильной группы
R R
/ /
R3GeH + О = С -* R3GeOCH.
\ \
R R
Интересно отметить, что непредельные органические кислоты могут
реагировать с органическими гидридами германия двояко. В присутствии
лорошка меди реакция происходит аналогично насыщенным кислотам по
карбонильной группе
Си
СН2 = СНСОЭН + R3GeH -> СН2 = CHCOOGeR3 + Н2.
В присутствии платинохлористоводородной кислоты (а в отдельных
случаях и без катализатора) германийорганические гидриды присоединяются
по кратной связи
H2PtCl,
СН2 = СНСООН + RsGeH .-* R8GeCH2CH2COOH.
В противоположность органическим гидридам кремния, которые не
реагируют с галоидными алкилами, гидриды алкилгермания реагируют с
большой легкостью с галоидными алкилами в отсутствие катализаторов; по
реакционной способности их можно сравнить с оловоорганическими
гидридами. Обмен водород — галоид происходит экзотермически и почти
количественно.
R3GeH + R'X -* R3GeX + R'H,
RGeH3 + 3R'X -> RGeX3 + 3R'H.
С галоидными арилами органические гидриды германия не реагируют.
14
Германийорганшеские соединения. Лит. стр. 16—18
В последние годы в химии германийорганических соединений большое
место стали занимать соединения, содержащие два (или более) металла.
Это соединения, в которых либо имеется непосредственная связь между
германием и другим металлом, либо германий и металл разделены
углеродным мостиком.
Синтез германийорганических соединений, в которых германий связан
с другим металлом через углеродный мостик, может быть осуществлен с
помощью магний- и натрийорганических соединений
R3MC6H4MgX + R3GeX -* R3MC6H4GeR3 + MgX2,
R3MC = CMgX + R3GeX -> R3MC = CGeR3 + MgX2,
Na
RsMCH = CHX + R3GeX — R3MCH = CHGeR3 + NaX
(M = Si, Sn, Pb).
или присоединением гидридов металлоорганических соединений IV
группы периодической системы к металлоорганическим соединениям, имеющим
алкиновую группу:
R3GeC = СН + RsMH -> R3GeCH = CHMR3,
R3MC = СН + R3GeH -» R3MCH = CHGeR3
(M = Si, Sn).
Для синтеза соединений RgGeM^Rn-x (где М — иной металл, чем германий;
п — валентность металла; R — ароматический, алифатический радикал)
существует два пути: действие органических гидридов германия на
различные металлоорганические соединения, или действие германийорганических
соединений типа R3GeM (где М — Li, Na) на галогениды алкильных или
арильных органических производных других металлов. Соединения типа
R3GeM (где М — Na, К, Li) могут быть получены и другими путями.
Из соединений типа RgGeM^R^-! наиболее исследованы соединения с
участием щелочных металлов, а именно типы R3GeM и менее — типы
R2GeM2 (где М — Li, Na, К)- В последнее время круг металлов,
связанных с Ge, расширен на металлы второй (Zn, Cd, Hg), третьей (Т1),
четвертой (Si, Sn, Pb) и в малой степени пятой (Bi) группы периодической
системы. Наконец, описаны своеобразные биметаллические соединения типа
Ar3GeM(CO)5 (где М — Re, Мп).
Соединения типа R3GeM (где М — Li, Na, К) не были выделены в
чистом виде, а были получены лишь в растворах. чНо эти растворы, очевидно,
благодаря наличию в них щелочного металла очень реакционноспособны; в
литературе описаны их многочисленные реакции. Из других
биметаллических германийорганических соединений наиболее изучены соединения
Ge с другими металлоорганическими соединениями IV группы
периодической системы (Si, Sn, Pb). Соединения со связью Ge — Hg, Ge—Cd, Ge—Bi,
Ge—Tl, например типа R3GeCdGeR3, а также с карбонилами переходных
металлов (Re, Mn) получены лишь в последнее время; изучение их свойств
только начинается.
Практическое применение германийорганических соединений в
настоящее время развито очень мало. Непредельные соединения, имеющие атом
германия в цепи, исследовались в реакции полимеризации и сополимери-
зации. Пока полученные полимеры и сополимеры не представили особого
интереса. До сих пор не получены германийорганические полимеры с
достаточно большим молекулярным весом.
Германийорганические соединения исследовались в качестве
катализаторов полимеризации этилена [3], а также а-моноолефинов [4].
Исследовалось также влияние на термораспад поливинилхлорида добавок тетраэтил-
германия [5].
Введение
15
Номенклатура германийорганических соединений
Формула
германий-
органического
соединения
RGeH3
R2GeH3
R3GeH
R^GeHCl
RGeX3
R2GeX2
R3GeX
R4Ge
RGeOOH
R2GeO
R3GeOGeR3
RaGeOH
R3GeOR
R.GeOOCR
R3GeSR
R3GeNR2
R3GeGeR3
RsGeLi
Название,
принятое в настоящей
книге
Тригидрид
алкилгермания
Дигидрид
диалкилгермания
Гидрид триалкил-
германия
Хлоргидрид
диалкилгермания
Трехгалоидный
алкилгерманий
Двугалоидный
ди алкилгерманий
Галоидный три-
алкилгерманий
Тетраалкилгер-
маний
Алкилгермание-
вая кислота
Окись
диалкилгермания
Гексаалкилдигер-
маноксан
Гидроокись три-
алкилгермания
Алкокситриалкил-
германий
Ацилат триалкил-
германия
Алкилмеркаптид
триалкилгерма-
(Диалкиламино)-
триалкилгсрма-
ний
Гексаалкилдигер-
ман
Триалкилгерма-
ниллитий
Другое,
встречающееся в русской
литературе, название
Алкилгерман
Диалкилгерман
Триалкилгерман
—
Алкилтригалоген-
герман
Диалкилгалогенгер-
ман
Триалкилгалогенгер-
ман
Тетраалкилгерман
—
—
—
—
Триалкилалкокси-
герман
—
—
—
"
Английское
название
Alkylgermane
Dialkylgerman
Trialkylgerman,
Trialkylgermanium hydride
Dialkylchlorogermane
Alkylgermaniiim
trihalid
Dialkylgermanium
dihalid
Trialkylgerman fum
halid
Germanium tetraalkyl,
Tetraalkylgermane
Alkylgermanicacid
Dialkylgermanium-
oxyd
Trialkylgermanium
oxyde
Trialkylgermanol
Trialkylalkoxygerman
Try alky lgermanium
carboxylate
Alkylmercaptides i
trialkylgermanium
-
Hexaalkyldigermane
Lithium trialkylger-
manide
Немецкое название
-
—
Trialkylgerman
—
Alkylgermanium
trihalogenid
Dialkylgermanium-
dihalogenid
Trialkylgerman ium
halogenid
Germaniumtetraalkyi
Alkylgermanosaure
Dialky lgermanium
oxyd
Trialkylgermanium
oxyd
Trialkylgermanium.
hydroxyd
—
Hexaalkyldigerman,
digermanium hexaal-
kyl
Большое число работ посвящено исследованию физических свойств
различных германийорганических соединений. Рассмотрение их не входит в
задачу настоящего издания. Однако можно отметить отдельные
оригинальные статьи, посвященные различным физико-химическим исследованиям
германийорганических соединений. Описаны инфракрасные спектры
поглощения [6—45], Раман-спектры [7, 18, 37, 44, 46—52], спектры ядерного
магнитного резонанса [35, 43, 53—68], ультрафиолетовые спектры [69—
73], квадрупольный резонанс [74]. Определялась энергия связи Ge—С
[75—77], проведен расчет частот и форм нормальных колебаний связи С^С
10
Германийорганшеские соединения
в винильных производных германия [39], а также шестичленного цикла,
содержащего два атома германия [78]. Выполнены рентгенографические
исследования [79] и электронографические исследования [80], масс-спектро-
метрические [81, 82] и магнетито-химические исследования [83—86].
Определялись дипольные моменты ряда соединений [13, 87—91]. Определялся
молекулярный вес (C2H5)4Ge [92] и растворимость тетраэтил- и тетрафенил-
германия [92, 93]. Вычислялась молекулярная рефракция ряда германий-
органических соединений [94]. Вычислялись и определялись температура
кипения некоторых гидридов германия и моногидридов алкилгермания
[95]. Измерялась электропроводность хлористого трифенилгермания в
жидком безводном хлористом водороде [96].
Различные физико-химические исследования ряда соединений,
особенно ИК-спектры, приводятся в статьях, посвященных синтезу и реакциям
германийорганических соединений.
Химия германийорганических соединений освещена в нескольких
обзорных статьях. Это статьи Джонсона [1], Куэйна и сотр. [97], Рикенса и
сотр. [2]. Химии германийорганических гидридов посвящена обзорная
статья Сатже [98]; имеется несколько обзорных статей, посвященных
отдельным вопросам химии германийорганических соединений [99—109]. В 1967 г.
издана монография Миронова и Гар [ПО].
ЛИТЕРАТУРА
1. JohnsonO. H., Chem. Revs., 48, 259 (1951).
2. Ингам Р., РозенбергС, Г и л м а н Г., РикенсФ., Оловоорганические
и германийорганические соединения. М., ИЛ, 1962, стр. 186.
3. Ray R.J., S i s t r u n k Т. О., Пат. США 2868772 (1959); РЖХим., 1961, 8П139.
4. ShearerN.H, С о о v e r H. W., Пат. США 2925409 (1960); РЖХим., 1962, 4П177.
5. Попова 3. В., Тихова Н. В., Вязанкин Н. С, Высокомолекулярные
соединения. Сб. «Химические свойства и модификация полимеров». М., «Наука», 1964,
стр. 175.
6. YoungC. W., К о е h 1 е г J. S., М с k i n n е у D. S., J. Am. Chem. Soc, 69, 1410
(1947).
7. L i p p i n с о t t E. R., Т о b i n M. С, J. Am. Chem. Soc, 75, 4141 (1953).
8. К г о s s R. D., F a s s e 1 V. A., J. Am. Chem. Soc, 77, 5858 (1955).
9. Steingros s W., Zeil W., J. Organometal. Chem., 6, 464 (1966).
10. Б а р ч у к о в А. И., П р о х о р о в А. М., Оптика и спектроскопия, 4, 799 (1958).
11. No I tes J. G., H enry M. С, J a nsse n M. J., Chem. and Ind., 1959, 298.
12. Батуев М. И., Пономаренко В. А., Матвеева А. Д., Взенкова
Г. Я., Изв. АН СССР, ОХН, 1959, 2226.
13. LaurieV. W., J. Chem. Phys., 30, 1210 (1959).
14. W e s t R., В a n e у R. H., J. Phys. Chem., 64 822 (1960).
15. Полякова А. М., Чума ев с кий Н. А., ДАН СССР, 130, 1037 (1960).
16. С т е р л и н Р. Н., Д у б о в С. С, Ж. Всесоюзн. хим., общ. им. Д. И. Менделеева,
7, 117 (1962).
17. ЛопатинБ.Ф., Я к о в л е в И. П., Изв. АН СССР, серия физич., 1962, 1288.
18. Ч у м а е в с к и й Н. А., Оптика и спектроскопия, 8, 68 (1962).
19. Е г о р о в Ю. П., К и р е й Г. Г., Л е й т е с Л. А., М и р о н о в В. Ф.,
Петров А. Д., Изв. АН СССР, ОХН, 1962, 1880.
20. Вышинский Н. Н., Александров Ю. А., Рудневский Н. К., Изв.
АН СССР, серия физич,, 1962, 1285.
21. GoldfarbT. D., SujishiS., Internat. Sympos. Molec. Struct, and Spectrosc,
Tokyo, 1962; РЖХим., 1983, 17Б77.
22. Л е й те с Л. А., Д у л о в а В. Г., В о л ь п и н М. Е., Изв. АН СССР, ОХН, 1963,
731.
23. Н е п г у М. С, N о 1 t e s J. G., J. Am. Chem. Soc, 82, 555 (1960).
24. S r i v a s t a v a T. V., OnyszchukM. Canad., J. Chem., 41, 1244 (1963).
25. G r i f f i t h s J. E., J. Chem. Phys., 38, 2879 (1963).
26. Burger H., Sawodny W„ Ynorgan, and Nucl. Chem. Letters, 2, 209 (1963).
27. Gross R. J., GlocklingF., J. Organometal. Chem., 3, 146 (1963).
28. Ойкав a X., J. Chem. Soc. Japan, Pure Chem. Sec, 84, 5 Al (1963); РЖХим.,
1964, 96151.
Введение
17
29. О и к а в а X., J. Chem. Sec. Japan. Pure Chem., Sec, 84, 1 (1963); РЖХим., 1964,
9Б138.
30. Маркова С. В., Оптика и спектроскопия. Сборник статей, т. 2. М.—Л., Изд-во
АН СССР, 1963, стр. 173.
31. Sac her R. E. Lemmon D. II., Miller F. A., Spectrochim. Acta, 23A, 1169
(1957).
32. Вышинский Н. Н., Труды по химии и хим. технологии (Горький), 1963, 18.
33. Е г о р о в Ю. П., К и р е й Г. Г., ЖОХ, 34, 3615 (1964).
34. Гастилович Е. А., Шигорин Д. Н., Комаров Н. В., Труды Комиссии
по спектроскопии АН СССР, вып. 1, 70 (1964).
35. S с h е г е г О. J., S с h m i d t M., J. Organometal. Chem., 1, 490 (1964).
36. Ma p ко в а С. В., Оптика и спектроскопия, 16, 776 (1964).
37. О б р е и м о в И. В., Ч у м а е в с к и й Н. А., Ж. структурн. хим., 5, 59 (1964).
38. О б р е и м о в И. В., Ч у м а е в с к и й Н. А., Ж. структурн. хим., 5, 137 (1964).
39. Л и в ш и ц Б. Л., Ч у м а е в с к и й Н. А., Ж- структурн. хим., 5, 140 (1964).
40. Л е й т е с Л. А., Г а р Т. К., М и р о н о в В. Ф., ДАН СССР, 158, 400 (1964).
41. Е г о р о в Ю. П., Теорет. и экспер. химия, 1, 30 (1965).
42. Солодовников С. П., Чернышев Е. А., Труды Совещания по физическим
методам исследования органических соединений и химических процессов. Фрунзе,
«Илим» 1964, стр. 196.
43. Р о t t е г Р. Е., Р г a t t L., W i 1 k i n s о n G., J. Chem. Soc, 1964, 524.
44. A r о u s J. R., D u r i g J. R., Spectrochim. Acta, 20, 219 (1964).
45. П е т у x о в В. А., Миронов В. И., К р а в ч е н к о А. Л., Изв. АН СССР,
серия хим., 1966, 156.
46. S i e b e r t H., Z. anorg. allgem. Chem., 263, 82 (1950).
47. S i e b e r t H., Z. anorg. allgem. Chem., 268, 177 (1952).
48. L i p p i nco t t E. R., M e г с i e r P., TobinM.C, J. Phys. Chem., 57, 939
(1953).
49. Wa t e r s D. N., W о о d w а г d L. A., P г о с. Roy. Soc, 246, 119 (1958).
50. В г о w n M. P., С a r t m e 1 1 E., F о w 1 e s G. W. A., J. Chem. Soc, 1960, 506.
51. ЕгоровЮ. П., Л е й т е с Л. А., К р а в ц о в а И. Д., Миронов В. Ф., Изв.
АН СССР, серия хим., 1963, 1114.
52. Е г о р о в Ю. П., Изв. АН СССР, ОХН, 1957, 124.
53. В г о w n M. P., W e b s t e r D. Е., J. Phys. Chem., 64, 698 (1960).
54. N а г a s i m h a n Р. Т., R о g e r s M. Т., J. Am. Chem. Soc, 82, 5983 (1960).
55. Coyle T. D., Stafford S. L., Stone F. G. A., Spectrochim. Acta, 17, 968
(1961).
56. E г о p о ч к ii ii A. H., Хидекель М. Л., Пономаре и ко В. А.,
Зуева Г. Я., С в и р е ж е в а С. С, РазуваевГ. А., Изв. АН СССР, серия хим.,
1983, 1865.
57. С a w 1 е у S., D a n у 1 u k S. S. Canad. J. Chem., 41, 1850 (1963).
58. М а с к а у К. М., W a t t R., J. Organometal. Chem., 6, 336 (1966).
59. С a w 1 e v S., D a n у 1 и к S. S., J. Chem. Phys., 38, 285 (1963).
60. Schmidbaur H., Ber., 97, 1639 (1964).
61. Б и р ю к о в И. П., С а ф и н И. А., В о р о н к о в М. Г., Изв. АН ЛатвССР,
серия хим., 1964, 181.
62. Егорочкин А. Н., Хидекель М. Л., П о н о м а р е н к о В. А., 3 у е-
в а Г. Я., Р а з у в а е в Г. А. И з в. АН СССР, серия хим., 1964, 373.
63. Егорочкин А. Н., X и д е к е л ь М. Л., Р а з у в а е в Г. А., М и р о н о в В. Ф.,
Кравченко А. Л., Изв. АН СССР, серия хим., 1964, 1312.
64. К е 1 е n G. P., Verdonck L, Vondel Van der D., Bull. Soc. chim. belg.,
73, 733 (1964).
65. Петровская Л.И., Б у р л а ч е н к о Г. С, Ф е д и н Э. И., Б а у к о в Ю. И.,
Л у цен ко И. Ф.,Ж. структурн. хим., 6, 781 (1965).
66. S m i t h G. W., J. Chem. Phys., 42, 4229 (1965).
67. H e ф е д о в О. М., Колесников СП., Ш е й ч е н к о В. И., Шейн-
керЮ. Н., ДАН СССР, 162, 589 (1965).
68. Купрневич В. А., Егоров Ю. П., Д я д ю ш а Г. Г., ДАН АН УССР,
1964, 508; РЖХпм., 1964, 18г)62.
69. М а г г о t J., M a i r e J.— С, С a s s a n J., Compt. rend., 260, 3931 (1960).
70. L a P a g 1 i a S. R., J. Molec. Spectrosc, 7, 427 (1961).
71. Drenth W„ Janssen M.J., van der Ker kG.J. M.,Vl iegen tha r t J. A.,
J. Organometall. Chem., 2, 265 (1964).
72. Hague D. N., PrinceR.D., J. Chem. Soc, 1965, 4690.
73. Петухов В. А., Миронов В. Ф., Шорыгин П. П., Изв. АН СССР, серия
хим., 1964, 2203.
74. Л е й т е с Л. А., Изв. АН СССР, серия хим., 1963, 1527.
75. HugginsM. L., J. Am. Chem. Soc, 75, 4123 (1953).
18
Германийорганические соединения
76. Рабиновичи. Б., Тельной В. И., К а р я к и н Н. В., Р а з у в а е в Г. А.,
ДАН СССР, 149, 324 (1963).
77. В i 1 1 s J. L., С о t t о n F. A., J. Phys. Chem., 68, 806 (1964).
78. Г а л ь п е р и н Е. Г., М а я н ц Л. С, Ж. структурн. хим., 6, 785 (1965).
79. К а г n a t a k R., С h a n d г a S. P., J. chim. phys. el phys. chim. biol., 62, 883 (1965).
80. В и л к о в Л. В., М а с т р ю к о в В. С, Ж. структурн. хим., 6, 811 (1965).
81. Хмельницкий Р. А., Полякова А. А., Петров А. А., Медве-
д е в Ф. А., С т а д н и ч у к М. Д., ЖОХ, 35, 773 (1965).
82. Г о р о х о в Л. Н., Ж. структурн. хим., 6, 766 (1965).
83. A b е 1 Е. W., В u s h R. P., J e n k i n s C. R., Z о b e 1 Т., Trans. Faraday Soc,
60, 1214 (1964).
84. L a b a r r e J.— FM Mazerolles P., Compt. rend., 254, 3998 (1962).
85. LesbreM., Mazerolles P.Joigt D.; Compt. rend., 240, 622 (1955).
86. MazerollesP, Voigt D., Compt. rend., 240, 2144 (1955).
87. К а р ц е в Г. Н., С ы р к и н Я. К., М и р о н о в В. М., Изв. АН СССР, ОХН, 1960,
948.
88. Карцев Г. Н., С ы р к и н Я. К., Кравченко А. Л., М и р о н о в В. Ф.,
Ж. структурн. хим., 5, 639 (1964).
89. Барчуков А. И., Петров Ю. Н., Оптика и спектроскопия, 11, 129 (1961).
90. К а р ц е в Г. Н., Сыркин Я. К., К р а в ч е н к о А. Л., М и р о н о в В. Ф.,
Ж. структурн. хим., 5, 492 (1964).
91. О s t h о I I R. С, Rochow E.G.,J. Am. Chem. Soc, 74, 845 (1952).
92. Strohmeier W., HumpenerK., Miltenberger K., SeifertF.,
Z. fur Elektrochemie, 63, 537 (1959).
93. Strohmeier W., Miltenberger K., Ber., 91, 1357 (1958).
94. Christopher P. M.,J. Chem. and Engng. Data, 10, 44 (1965).
95. E n g 1 i s h W. D., J. Am. Chem. Soc, 74, 2927 (1952).
96. P e а с h M. E., Wadd ington T.C., J. Chem. Soc, 1961, 1238.
97. Q u a n e D., В о t t e 1 R. S., Chem. Revs., 63, 403 (1963).
98. Satge J., Ann. de chim., 6, 519 (1961).
99. WittenbergD., GilmanH., Quart. Rev., 13, 116 (1959).
100. Gilraan H., Bull. Soc. chim. France, 1963, 1356.
101. LesbreM., Chim. mod., 8, 3 (1963); РЖХим., 1963, 17Ж259.
102. GlocklingR, Quart. Rev., 20, 45 (1966).
103. В я з а н к и н Н. С, К р у г л а я О. А., Усп. хим., 35, 1388 (1966).
104. Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Усп. хим., 35, 1430 (1966).
105. A i t k e n I. D., SheldonR., S t a p 1 e t о n G. В., Brit. Plast., 34, 662 (1961).
106. Б р а у н Д. В., Усп. хим., 31, 769 (1962).
107. M a r 1 e t t E. M., Ann. N.— Y. Acad. Sci., 125, 12 (1965).
108. H e n г у M. C, Davidson W.E., Ann. N.— Y. Acad. Sci., 125, 172 (1965).
109. SchmidbaurH., Angew. Chem., 77, 206 (1965).
110. Миронов В.Ф.,Гар Т. К., Органические соединения германия. М., «Наука», 1967-
Глава 1
ПРЯМОЙ МЕТОД СИНТЕЗА
ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ ИЗ ГАЛОИДНЫХ
АЛКИЛОВ И МЕТАЛЛИЧЕСКОГО ГЕРМАНИЯ
Рохов впервые в 1947 г. показал возможность получения галоидных ал-
кил- или арилгерманийорганических соединений реакцией прямого
синтеза из металлического германия (в присутствии порошка меди) и
галоидных алкилов или арилов при температуре 300—360° С [1—8] аналогично
приготовлению метилхлорсиланов.
300—360° С
RC1 + Ge / Си > R„GeCl4_„
(л = 1, 2).
В прямом синтезе Рохов применял хлористый метил [1], этил [1, 2, 4, 51,
пропил [3], фенил [1], а также бромистый метил [11. В случае R = СН3
и С2Н5 образуются R2GeCl2 и RGeCl3 в соотношении 2:1.
В дальнейшем Петров и сотр. [9] показали, что если реакцию вести при
более высокой температуре (—550° С), то содержание двуххлористых ди-
алкилгерманийорганических соединений резко снижается. Двухлористый
диэтилгерманий вообще не был обнаружен, был получен лишь
треххлористый этил германий, а двухлористый диметил германий образовывался
примерно в таких же количествах, как и треххлористый метилгерманий. Те же
авторы впервые в прямом синтезе использовали хлористый аллил и метал-
лил и получили с хорошими выходами треххлористый аллил- и металлил-
германий.
При дальнейшем исследовании реакции прямого синтеза было показано
[10, 111, что в случае хлористого или бромистого метила соотношение
между образующимися соединениями (CH3)2GeX2 (I) и CH3GeX3 (II) при
данной температуре зависит в значительной степени от содержания меди в
смеси (или сплаве) германия и меди. Относительный выход (II) тем больше
(особенно при использовании бромистого метила), чем выше процент меди
в сплаве или в механической смеси с германием. Например, 50%-ный сплав
германий — медь приводит к получению смеси (CH3)2GeBr2 : CH3GeBr3 --=
= 1 : 2,5. Если же применять сплав германия, содержащий 20% меди, то
соотношение между (I) и (II) становится равным 1 : 0,7. Оно меняется
и в ходе процесса. Так, в случае хлористого метила в одном из опытов
(Ge : Си =- 4 : 1) это соотношение было равно 4,5 : 1 в начале реакции;
2,4 : 1 в середине реакции и 1,4 : 1 в конце реакции. Полученные результаты
легко объясняются увеличивающимся относительным содержанием меди в
сплаве по мере вступления германия в реакцию.
В результате изучения реакции прямого синтеза [11] было
установлено, что повышение температуры и процентного содержания меди в сплаве
(или смеси) с германием способствуют образованию соединений типа RGeX3
и снижению выхода соединений типа R2GeX2. Условия опытов могут быть
2*
20 Германийорганичгскис создинения. Лит. стр. 21
подобраны таким образом, что будет образовываться практически один
лишь RGeX3.
В дальнейших исследованиях были установлены наиболее оптимальные
условия для получения двухлористого диметил- [Па] и диэтилгермания
[116]. При получении прямым синтезом хлористого триметилгермания
рекомендуется в контактную массу добавлять от 0,5 до 2,5% порошка
алюминия [Ив]. Хлористый пропил реагирует в условиях прямого синтеза
плохо [Ив].
Необходимо подчеркнуть существенную особенность прямого синтеза
германийорганических соединений по сравнению с кремнийорганическими
соединениями. В случае германия ни одному из авторов не удалось
выделить соединения типа RGeHCU, структурные аналоги которых RSiHCl2
являются очень часто преобладающими продуктами при взаимодействии
RC1 с кремнемедным сплавом.
Синтез германийорганических соединений прямым путем был
использован как препаративный метод рядом авторов [12, 13, 13а, 136]. Для
приготовления относительно больших количеств треххлористого метил
германия, как было найдено, реакцию нужно проводить в отсутствие меди. При
этом в интервале температур 500—520° С получается оптимальный выход,
равный 70% [136].
В противоположность олову металлический германий не реагирует с
хлористым бензилом с образованием (C6H5CH2)nGeX4-n. Вместо этого
были выделены лишь полибензилы [14].
Миронов и сотр. [15] показали, что при взаимодействии дихлорметана
с металлическим германием при 350° С в присутствии порошка меди
реакция протекает по уравнению
С12
Ge
/ \
Ge/Cu . H2C 9На
СН2С12 > СИзйеСЬ (27%) + Cl3GeCH2GeCl3 (23%) + (19%)
Cl2Ge GeCl2
Н2
При нагревании (300—400° С) йодистого метила с металлическим
германием в присутствии гюро'лка меди или без него образуется смесь
CH3GeJ3, (CH3)2GeJo, (CH3)3GeJ, а также в некоторых случаях примесь
GeJ4 [161.
Изучение взаимодействия германиевой губки с йодистым этилом в
проточной системе [17] показало, что до 280° С образуется C2H5GeJ3, при 350° С—
GeJ4, а при 320° С — C2H5GeJ3 и GeJ4. Добавление меди оказывает
каталитическое действие — при 260° С выход C2H5GeJ3 повышается, однако при
280° С добавление меди не увеличивает выхода C2H5GeJ3.
Реакция между хлористым этилом и смесью металлического германия и меди [2]. 32,7 г
восстановленного медного порошка и 186 г порошка чистого металлического германия
(40 меш) помещают в стеклянную трубку диаметром 20 мм и длиной 450 мм между двумя
слоями стеклянной ваты. Трубка с одной стороны соединена с обратным холодильником,
охлаждаемым водой. Часть трубки, содержащую смесь германий — медь, нагревают в
обычной трубчатой печи, через трубку медленно пропускают ток хлористого этила. К
моменту, когда температура нагрева достигает 356° С, на стенках холодильника начинает
конденсироваться жидкость. При скорости пропускания хлористого этила 150 мл в 1 мин. при
температуре 317° С в течение 49 час. собирают 687 г жидкости (главную часть жидкости
собирают за первые 16 час). Полученную жидкость фракционируют. Она состоит из непрореа-
гировавшего хлористого этила, 61,5 г треххлористого этилгермания и 91,1 г двухлористого
диэтилгермания.
Прямой метод синтеза германийорганических соединений
21
Получение треххлористгоо аллилгермания [9]. Через стеклянную трубку диаметром
20лш, заполненную смесью 25 г мелкораздробленного германия и 10 г медных опилок и
помещенную в трубчатую электропечь, пропускают хлористый аллил. Пропускание 300 г
хлористого аллила проводят при 340° С +10° со скоростью 60 капель в 1 мин. Собирают 170 г
конденсата, перегонкой его на колонке выделяют 85 г треххлористого аллилгермания;
т. кип. 153,8°С/743,5лл; d|° 1,5274; п2£ 1,4928.
При использовании в аналогичных условиях хлористого металл ил а по
лучают треххлористый металлилгерманий (24%); т. кип. 42°С/7 мм.
ЛИТЕРАТУРА
1. RochowE.G., J.Am. Chem. Soc, 69, 1729 (1947); 70, 436 (1948).
2. R о с h о w E. G., J. Am. Chem. Soc, 72, 198 (1950).
3. RochowE.G., DidtschenkoR., West R.C., J. Am. Chem. Soc, 73,
5486 (1951).
4. R о с h о w E. G., R о с h о w Т. G., J. Phys. Colloid chem., 55, 9 (1951).
5. OsthoffR. C, R о с h о w E. G., J. Am. Chem. Soc, 74, 845 (1952).
6. British Thomson-Houston Co., Ltd. Англ. пат. 626398 (1949); С. А., 44, 2548a (1950).
7. RochowE.G., Пат. США 2444270 (1948); С. А., 42 7318в (1948).
8. R о с h о w E. G, П а т. США 2451871 (1948); С. А., 43 2631е (1949).
9. П е т р о в А. Д., М и р о н о в В. Ф., Д о л г и й И. Е., Изв. АН СССР, ОХН,
1956, 1146.
10. П о н о м а р е н к о В. А., В з е н к о в а Г. Я., Изв. АН СССР, ОХН, 1957, 994.
11. Миронов В. Ф., ПономаренкоВ. А., Взенкова Г. Я.,Долгий И.Е.
Петров А. Д., Химия и практическое применение кремнийорганических
соединений, вып. 1. Л., изд. БЦТИ, 1958, стр. 192.
Па. Зуева Г. Я., Погорелов А. Г., Писаренко В. И., Снегова А. Д..
Пономаренко В. А., Изв. АН СССР, серия неорг. матер., 2, 1359 (1966).
116. 3 у е в а Г. Я., Л у к ь я н к и н а Н.В., Кечина А. Г., Пономаренко В.А.,
Изв. АН СССР, серия хим., 1966, 1843.
Ив. Зуева Г. Я., Лукьянкина Н. В., Пономаренко В. А., Изв. АН
СССР, серия хим., 1967, 192.
12. Н е ф е д о в О. М., М а н а к о в М. Н., П е т р о в А. Д., ДАН СССР. 147, 1376
(1962).
13. V a n d е V о n d е 1 D. F., J. Organometal. Chem., 3, 400 (1965).
13а. Moedritzer К., J. Organometal. Chem., 6, 282 (1966).
136. WieberM., F г о h n i n g С. D., S с h m i d t M., J. Organometal. Chem., 6,427
(1966).
14. G г о h n H., P a u d e r t R., Z. chem., 3, 89 (1963).
15. M и р о н о в В. Ф., Г а р Т. К., Изв. АН СССР, серия хим., 1964, 1887.
16. Zablotna R., Akerman K.,SzuchnikA., Bull. Acad. Sci. Ser. sci. chim ,
12, 695 (1964); РЖХим., 1965, 23Ж328.
17. Zablotna R., Akerman K-, S z u с h n i k A., Bull. Acad. Sci., Ser. sci
chim., 13, 527 П967); РЖХим., 1966, 19Ж323.
Глава II
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ ГАЛОИДНЫХ АЛКИЛОВ НА СОЛИ ГЕРМАНИЯ
ИЛИ ИХ КОМПЛЕКСЫ
Действие галоидных алкилов на соли двухвалентного германия
приводит к образованию с хорошими выходами трехгалоидных германииалкилов
или арилов. При действии двуиодистого германия на йодистый этил [1]
протекает следующая реакция:
C2H5J + GeJ2 -> C2H5GeJ3.
Выход трехиодистого этилгермания равен 86%, т. кип. 281° С/755 мм.
Получение трехиодистого метилгермания [2]. Юг двуиодистого германия помещают в
толстостенную ампулу из стекла пирекс (рис. 1) и соединяют с помощью большого крана с
вакуумной системой, в которую включена ампула с йодистым метилом. Ампула, содержащая
двуиодистыи германий, имеет боковой отвод, в который может быть отогнан и удален по
Вакуум
Рис. 1. Прибор для получения трехиодистого метилгермания
окончании реакции избыток йодистого метила. По удалении воздуха из системы ампулу
помещают в охладительную смесь, и при комнатной температуре в нее перегоняют 2,1 мл
йодистого метила (4,8 г, 10% избыток). Ампулу запаивают и нагревают при 110° С в течение
24 час.(в начале реакции в ампуле создается большое давление и может произойти взрыв, если
ее стенки недостаточно прочны). Во время реакции желтые кристаллы двуиодистого германия
постепенно исчезают и образуется светло-желтая жидкость. Если иодид содержит примесь
окиси, она остается нерастворенной. При охлаждении продукт затвердевает, за
исключением случая, когда применяют большой избыток йодистого метила. По охлаждении
реакционной смеси боковую ампулу помещают в охладительную смесь, перегоняют непро-
реагировавший йодистый метил и ампулу отпаивают. После этого продукт реакции
подвергают соответствующей очистке путем перегонки. Выход близок к количественному.
Синтез действием галоидных алкилов на соли германия
23
Трехиодистый метилгерманий представляет лимонно-желтые кристаллы, т. пл. 48,5° С.
Он термически стабилен и может быть перегнан без заметного разложения; т. кип.
232° С/752° мм. Растворимость в органических растворителях позволяет перекристаллизовать
его из петролейного эфира при охлаждении до —33° С насыщенного при +50° С раствора.
Вещество возгоняется в вакууме при 45° С в большие светло-желтые кристаллы.
Лесбр с сотр. [3] подробно исследовал реакцию двуиодистого германия
с галоидными алкилами и арилами и нашел, что в ходе этих реакций
германий переходит из двухвалентного в четырехвалентное состояние.
Так, при 40° С в запаянной ампуле двуиодистый германий реагирует
с йодистым бутилом, образуя трехиодистый бутилгерманий; т. кип. 119—
121 °С/1 мм. При взаимодействии с иодбензолом при 160° С образуется
трехиодистый фенилгерманий. Йодистый метилен и двуиодистый германий
дают в запаянной ампуле при 80° С трехиодистый иодметилгерманий:
GeJ2 + JCH2J -> J3GeCH2J.
С 1,2-дииодэтаном реакция протекает более сложно. Иодметиловый эфир
количественно реагирует на холоду с двуиодистым германием
GeJ2 + JCH2OCH3 -> J3GeCH?OCH3.
Этиловый эфир иодуксусной кислоты при 90° С образует этиловый эфир
трииодгермилуксусной кислоты
GeJ2 + JCH2COOC2H5 -> J3GeCH2COOC2H5.
Двуиодистый германий реагирует также и с бромистыми производными.
При нагревании с бромистым бутилом реакция завершается за 5 дней при
150° С и приводит к образованию смеси продуктов, которые, будучи
обработаны бромистым бутилмагнием, дают триэтшгбутилгерманий. По
мнению авторов, реакция протекает по уравнению
GeJ2 + QH9Br-> Br3GeQH9 + Br2JGeQHf + J3GeQH9 -*
C3H5MgBr
> (C2H5)3GeQH9-«.
Действие двуиодистого германия на 1,4-дибромбутан ведет к получению
смеси поли галоидных производных
GeJ2 + Br (CH2)4 Br -* X3Ge (CH2)4 Br
(Х = Br или J).
Интересно отметить работы по получению перфторированных моноал-
кильных производных германийорганических соединений [4, 5].
Получение трехиодистого трифторметилгермания [4]. 59,7 г (0,183 моля) двуиодистого
германия помещают в автоклав из нержавеющей стали и в него конденсируют 112 г (0,57
моля) йодистого трифторметила. Автоклав нагревают в течение 10 дней при 130—135° С,
полученный летучий продукт фракционируют. Получают 84,2 г (0,43 моля) непрореагировав-
шего йодистого трифторметила и небольшое количество (0,3 г) бесцветной жидкости, по
анализу являющейся двуиодистым бис-(трифторметил)германием (CF3)2GeJ2. Желтое масло,
оставшееся в автоклаве, отделяют от непрореагировавшего двуиодистого германия отгонкой
в высоком вакууме, а последующей повторной перегонкой получают 40 г (0,078 молей)
трехиодистого трифторметилгермания с т. кип. 40—-42° С /10""3 мм и т. пл. 8,4° С; п2£ 1,6571.
Своеобразный метод, предложенный для получения
германийорганических соединений типа RGcX3, основан на реакции
RJ + CsGenCl3 -* CsJ + RGe1 VC13.
По этому методу был получен треххлористый этил- и фенилгерманий, а
при действии на йодистый метилен (нагревание в запаянной трубке при
24
Г ерманийорганичсские соединения. Лит. стр. 25
200° С в течение 30 час.) с выходом 80% образуется СН2 (GeG3L [6]. С
хлорбензолом реакция на идет.
Миронов с сотр. [7, 81 нашел, что при кипячении четыреххлористого
или четырехбромистого германия с галоидными арилами в присутствии
порошка меди образуются производные трехгалоидною арилгермания
Си
ArJ + GeX4 -» ArGeXs (80%).
Скорость протекания реакции зависит главным образом от природы GeX4
и в меньшей степени от природы галоидного арил а. Например, четырех-
бромистый германий гораздо быстрее (4 часа) реагирует с иодбеизолом,
чем четыреххлористый германий (12 час). Однако в первом случае наряду
с трехбромистым фенилгерманием образуются заметные количества дву-
бромистого дифенилгермания
Си
C6H5J + GeBr4 -* CGH>GeBr3 (51%) + (CeH5)3 GeBr2 (28%).
С другими йодистыми арилами реакция протекает успешно только при
использовании четырехбромистого германия. Длительное кипячение (13
час.) смеси четыреххлористого германия, меди и а-ноднафталина или ^-иод-
толуола не приводит к образованию в заметных количествах
соответствующих германийорганических соединений. Если эту реакцию проводить с
четырехбромистым германием, то после 1 — 1,5-часового кипячения с
высокими выходами образуются соответствующие соединения.
Алифатические иодиды в реакцию с четырехбромистым германием
вступают плохо. Йодистый бутил и йодистый аллил образуют с
четырехбромистым германием трехбромистый бутил- и аллилгерманий с выходами 25%.
Замена меди в этой реакции на другие металлы — цинк, железо,
натрий — или иодбензола на бромбензол, так же как и замена четыреххлор-
стого германия на треххлористый метилгерманий, не приводит к успеху.
Авторы считают, учитывая доступность исходных веществ и простоту
проведения реакций, что этот новый путь синтеза треххлористого и трех-
бромистого фенилгермания в настоящее время является лучшим из
известных методов.
Получение треххлористого фенилгермания [7]. В колбу емкостью 50 мл, снабженную
обратным холодильником, помещают 21 2 иодбензола, 24,6 2 четыреххлористого германия и
20 г порошка меди. После 12-часового кипячения содержимое колбы отфильтровывают от
твердого остатка, который промывают изопентаном. При перегонке фильтрата получают 5 г
иодбензола, 6 г четыреххлористого германия и 16,5 г треххлористого фенилгермания;
т. кип. 118—119°{ С/24 мм; df 1, 5972; п2£ 1,5540; выход 62,7% (80% на прореагировавший
четыреххлористый германий).
Аналогично из иодбензола и четырехбромистого германия получают
трехбромистый фенилгерманий; т. кип. 160—161° С/24 мм; d\° 2,2641; п2£
1,6330; выход 51%. Кроме того, выделяют 3 г (28%) двубромистого дифе-
нилгермания; т. кип. 208—210°С/24лш; п2£ 1,6380; 2—3-кратная перегонка
полученных германийорганических соединений над медным порошком
полностью освобождает их от следов иода.
Аналогично получены трехбромистый а-нафтилгерманий (т. пл. 52,5—
53,6°С, выход 78%), трехбромистый ж-толилгерманий (т. кип. 186—187°
С/37 мм, выход 86,6%), трехбромистый ж-фторфенилгерманий (т. кип. 151 —
152—С/23 мм, выход 74%), трехбромистый а-тиенилгерманий (т. кип. 152—
153°С/23 мм, выход 41%), трехбромистый ж-иодфенил германий (т. пл.
73—74°С/выход 19,2%), трехбромистый фенилгерманий (т. кип. 160—
16ГС/24 мм, выход 51%) [8].
Синтез действием галоидных аякилов на соли германия 25-
ЛИТЕРАТУРА
1. F 1 о о d E. A., J. Am. Chem. Sec, 55, 4935 (1933).
2. F 1 о о d E. A., Godfrev K.L., Foster L.S., Inorg. Syntheses, 3, 64 (1950),
3. LesbreM., Mazerol les P., Manuel G, Compt. rend., 257, 2303 (1963)..
4. С 1 a r к H. C, W i 1 1 i s С J., J. Am. Chem. Soc, 84, 898 (1962).
5. ClarkH.C, WillisCJ., Proc. Chem. Soc, 1960, 282.
6. T с h а к i r i a n A., Lewinsohn M.t Compt. Rend., 201, 835 (1935).
7 M и р о и о в В. Ф., Ф е д о т о в Н. С, ЖОХ, 34, 4122 (1964).
8. М и р о к о в В. Ф., Федотов Н. С, ЖОХ, 36, 556 (1966).
Глава HI
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ МАГНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ТИПА R4Ge
Магнийорганический синтез в получении германийорганических
соединений является одним из самых важных методов, особенно при
получении соединений жирного ряда.
Реакцию обычно проводят в эфирной среде, применяя четыреххлори-
стый или четырехбромистый германий. В некоторых случаях эфир заменяют
на более высококипящие растворители, например бензол, ксилол или
высшие эфиры, особенно при получении тетраметилгермания, так как его
температура кипения близка к температуре кипения эфира. Первый
представитель гомологического ряда — тетраметилгерманий был получен
авторами [1—13] действием йодистого метилмагния на четыреххлористый
германий. Реже применяют бромистый и хлористый .метилмагний. По данным
Липпинкотта с сотр. [4], при проведении реакции в эфире выход не
превышает 10% по сравнению с 40%, получаемыми в ди-я-бутиловом эфире.
Большую трудность представляет отделение тетраметилгермания от
эфира и йодистого метила. Для этого применяются различные приемы. Так,
Юнг и сотр. [3], осуществив реакцию в ди-я-бутиловом эфире, встряхивали
реакционную смесь с концентрированной серной кислотой и после
трехкратной промывки водой перегоняли из колбы Кляйзена. Для отделения
от примеси йодистого метила [5J реакционную смесь нагревали с пиридином
в течение часа в запаянной трубке при 80° С. Миронов и Кравченко [6]
проводили реакцию в ди-я-пропиловом эфире, реакционную смесь
перегоняли на колонке примерно в 20 теоретических тарелок и достигали выхода
81%.
Получение тетраметилгермания [7]. В 300 мл сухого ди-я-бутилового эфира
суспендируют 40 г магниевых стружек и нагревают в трехгорлой колбе, снабженной мешалкой, обратным
холодильником, капельной воронкой и термометром. Для инициирования реакции
добавляют 1 мл йодистого метила и несколько кристалликов иода. Колбу помещают в
охлаждающую баню и прибавляют 200 г бромистого метила, растворенного в 150 мл ди-я-бутилового
эфира, с таким расчетом, чтобы температура не поднималась выше 40° С. По окончании
прибавления бромистого метила реакционную смесь нагревают при 40° С до полного
растворения металлического магния. Затем реакционную смесь охлаждают до —15° С и по каплям
прибавляют раствор 48 г четыреххлористого германия в 50 мл ди-«-бутилового эфира. По
окончании прибавления температуру медленно поднимают до 80° С и нагревают при этой
температуре в течение 2 час. После охлаждения прибавляют по каплям воду для разложения
избытка реактива Гриньяра. Отгоняют реакционную смесь до тех пор, пока температура
паров не достигнет температуры кипения ди-я-бутилового эфира. Это соответствует примерно
100 мл. Полученный отгон сушат над хлористым кальцием и фракционируют. Получают 20 г
тетраметилгермания с т. кип. 43—44° С. Реакционную смесь, оставшуюся в трехгорлой
колбе, фильтруют. Разгонка фильтрата дает дополнительно еще 4 г тетраметилгермания. Выход
составляет 80%. Масс-спектрометрический анализ полученного продукта указывает, что
примесь бромистого и йодистого метила в нем не превышает 2%.
Синтез с помощью магнийорганических соединений
27
В отношении остальных членов гомологического ряда-применение
эфира в качестве растворителя не препятствует выделению германийорганиче-
ских соединений. Так, тетраэтилгерманий был получен с выходом 91% при
действии бромистого этилмагния в эфире на четырехбромистый германий
[13—17]. Аналогично был получен с выходом 75% .тетра-^-пропилгерма-
ний (т. кип. 225°С/746жж) [13, 16] и тетра-я-бутилгерманий (т. кип. 153—
155°С/18лш); п2о 1,4575 [13, 18—21]. Однако выходы полученных тетраал-
кильныхсоединений германия в работах различных авторов сильно
колеблются и в отдельных случаях не превышают 58,5% [20]. Андерсон [21]
указывает, что при получении тетра-я-бутилгермания только 74% германия
переходит в тетрабутилгерманий, а 20% —в бромистый трибутилгерманий.
Проведение реакции в растворителе, кипящем выше 100° С, способствует
повышению выхода.
При переходе к высшим членам гомологического ряда выходы
получающихся тетраалкильных соединений германия значительно снижаются,
даже при замене эфира на ксилол и длительном нагревании реакционной
смеси. Так, тетра-я-додецилгерманий был получен с выходом 47%, тетра-я-
тетрадецилгерманий — 37,6%, тетра-я-гексадецилгерманий — 36,0% и тет-
ра-я-октадецил германий — 24,0%.
Получение тетра-#-тетрадецилгермания [22]. Эфирный раствор 0?33 моля бромистого
миристилмагния, приготовленный из 96,5 г (0,348 моля) бромистого миристила и 8,46 г
(0,348 г-атома) магния, помещают в колбу на 500 мл, снабжедную мешалкой, обратным
холодильником, трубкой для ввода азота и капельной воронкой. При перемешивании
добавляют 12,5 г (0,058 моля) четырех хлор и стог о германия, растворенного в 80 мл сухого
ксилола. После того как прибавление закончено, добавляют 80 мл сухого ксилола, а большую
часть эфира отгоняют. Оставшийся ксилольный раствор нагревают при 100° С в течение трех
дней и затем гидролизуют 10%-ным раствором соляной кислоты. Небольшое количество
эфира добавляют для полного растворения осадка. Органический слой отделяют и
концентрируют его на водяной бане при небольшом вакууме. От полученного остатка отгоняют высшие
углеводороды, образовавшиеся при гидролизе реактива Гриньяра, др температуры 105° С/
/0,03 мм. Германийорганические соединения подвергают молекулярной перегонке при
температуре бани 360—370°Си давлении 0,0005 мм. Получают 18,85 г (37,6%) тетра-«-тетраде-
цилгермания.
Французские авторы [23, 24] получили для физико-химических
исследований ряд тетраалкильных соединений германия (С = -1—8) по реакции
Гриньяра, однако выходов авторы не указывают.
В литературе имеются различные данные по синтезу, тетраалкильных
соединений германия изо-строения. Тетраизоамилгерманий был получен
с выходом 87%, т. кип. 163—164°С/10лш [16]. Аналогично Лесбр с сотр.
,[25] получили тетраизобутилгерманий с выходом 70—80%.
Однако Андерсону [26, 27] не удалось получить тетраизопропилгерма-
ний при действии избытка галоидного изопропилмагния на четыреххло-
ристый или четырехбромистый германий. Образовывались лишь частично
замещенные германийорганические соединения.
Мазеролл [28] при реакции четыреххлористого гер-мания с бромистым
изопропилмагнием выделил лишь гидрид триизопропилгермания с выходом
36%, тетраизопропилгерманий получен не был. Авторы объясняют
аномальное течение реакции стерическими препятствиями и протекающей,
очевидно, восстановительной конденсацией (см. стр. 31).
Тетрабензилгерманий, т. пл. 107—108° С [29—31] был получен почти
с количественным выходом, однако из 2-бромэтилбензола соответствующее
тетразамещенное германийорганическое соединение получено с выходом
лишь 21,6% [22].
Получение тетрабензилгермания [30]. Хлористый бензилмагний получают из 65 г
хлористого бензила, 15 г магния в 330 мл абс. эфира. К полученному раствору реактива
Гриньяра прибавляют 13 2 четыреххлористого германия в 100 мл толуола. Нагревают с обратным
холодильником в течение 8 час, после гидролиза и обычной обработки получают 24—2~> г
28 Г ерманийорганически° соединения. Лит. стр. 43—46
тетрабензилгермания; т. кип. 107—110° С. Результаты получают аналогичные, если
реактив Гриньяра отфильтровывают от избытка металлического магния и еслл избыток
магния не отфильтровывают.
Сейферт [32] в 1957 г. впервые получил тетравинилгерманий действием
бромистого винилмагния на четыреххлористый германий в тетрагидро-
фуране с еыходсм 35,4% наряду с гексаЕинилдигермгнсм (см. стр. 32).
Мазеролл с сотр. [33, 34], не приЕодя эксперимента, упоминают о
получении тетрааллилгермания. Тетраперфторвинилгерманий был получен
действием йодистого или бромистого перфторвинилмагния на
четыреххлористый германий в тетрагидрсфуране [36].
Интересно отметить получение пслнозамещенных германийорганических
соединений, имеющих атом германия в иикле [35] при действии
четыреххлористого германия на димагниеЕые соединения дибромбутана и дибром-
пентана. Шварц и Рейнхарт [37] при реакции четыреххлористого германия
и димагниевого производного 1,5-дибромпентана получили двухлористый
пентаметиленгерманий лишь с очень незначительным выходом. Мазеролл
[36] указывает, что эта реакция ведет к получению сложной смеси, однако
восстановление с помсилю литийа^юминийгидрила полученных в виде
примеси частично замещенных соединений дает возможность выделить
циклические производные германия.
Получение германо-5-спиро-(4,4)-нонана [35]
СН2
сп3
В трехгорлую колбу (3 л), снабженную капельной воронкой, мешалкой и обратным
холодильником, закрытым осушающей трубкой, помещают раствор 70 г (0,32 моля)
четыреххлористого германия в 1,5 л абс. эфира. В капельную воронку помещают реактив Гриньяра,
полученный из 24 г (1 г-атом) магния и 95 г (0,44 моля) 1,4-дибромбутана в 500 мл эфира
(два слоя), и прибавляют при перемешивании к раствору четыреххлористого германия. При
этом наблюдают выпадение галоидной соли магния. По окончании прибавления (1 час.)
реакционную смесь нагревают в течение 5 час. при непрерывном перемешивании,
фильтруют в токе азота через стеклянный фильтр, фильтрат перегоняют. Вязкий остаток постепенно
нагревают в вакууме на парафиновой бане до 200° С. Собирают 47,1 г продукта,
перегоняющегося между 65 и 105° С/15 мм. После восстановления сырого продукта с помощью 12 г
литийалюминийгидрида (см. стр. 124) отгоняют 7,93 г дигидрида циклотетраметиленгерма-
ния. Выход составляет 18,6% (при 188—189° С) германо-5-спиро-(4,4)-нонана; d2^ 1,1837; п2^
1,5118. ИК-спектр этого продукта не содержит полосы Ge—Н.
Необходимо отметить, что в идентичных условиях четыреххлористый
кремний дает другие результаты: выход двухлористого циклического
производного более еысокий, но, по-видимому, спиро-соединений не
образуется [38]. При аналогичной реакции четыреххлористого германия с
соответствующим производным 1,5-дибромпентана получают германо-6-спиро-
(5,5)-ундекан, однако недостаточно чистый. После длительного
встряхивания с концентрированной серной кислотой, промывки раствором поташа,
водой и сушки над хлористым кальцием получают чистый спиран; т. кип.
108—109° С/17 мм; df 1,1400, п^ 1,5060. В ИК-спектре отсутствует
полоса Ge—Н.
Что касается ароматических германийорганических соединений, то
получение их представляет ряд экспериментальных трудностей и требует
точного соблюдения условий проведения процесса, так как в ряде случаев
продукты реакции получаются загрязненными примесью
германийорганических галогенидов, отделение которых представляет большие
трудности. Другой примесью яеляются гексаарилдигерманы (см. стр. 32). По
более ранним данным, даже применение £6 молей магнийорганического сое-
СН2 — CH2MgBr
+ GeCl4 -
СН2 —CH2MgBr
СН2 —СП2 СН2 —
/ \
СН2 — Cri2 СН2 —
Синтез с помощью магнийорганшеских соединений 29
динения на 1 моль галоидной соли германия дает выход тетрафенил германия
40%, при 8 молях на 1 моль — всего 22°о [16, 39, 40], а при действии 5
молей реактива Гриньяра на 1 моль четырехбромистого германия тетрафенил-
германий не был получен вовсе (вместо него выделен только бромистый
трифенилгерманий и менее фенилированные продукты [38]). Уоррель,
заменив эфир на толуол, повысил температуру реакции и достиг выхода тет-
рафенилгермания 80% [41]. В дальнейшем тетрафенил германий был
получен рядом авторов [42—47] в основном по методике, данной Джонсоном с
сотр. [42]. Авторы считают, что основным условием для получения
хорошего выхода тетрафенилгермания является немедленное удаление эфира из
реакционной сме и, так как несоблюдение этого условия приводит к
образованию большего количества гексафенилдигермана.
Получение тетрафенилгермания [44]. В трехгорлую колбу, снабженную обратным
холодильником, капельной воронкой и механической мешалкой, помещают 24,3 г (1 г-атом)
стружек магния и 500 мл абс. эфира. Через капельную воронку по каплям добавляют 157 г
бромбензола (1 моль), реакционную смесь кипятят в течение часа. Затем по каплям
прибавляют 21,4 г (0,1 моля) четыреххлористого германия [48] в 250 мл толуола, а эфир путем
отгонки по возможности быстро заменяют на толуол. После кипячения с обратным
холодильником в течение свыше час. избыток бромистого фенилмагния разлагают 50%-ной уксусной
кислотой. Рекомендуют также разложение разбавленной соляной кислотой. (1 объем
12 М кислоты на 10 объемов воды). При этом не происходит образования твердых
продуктов. Раствор нагревают до кипения, горячий толуольный слой отделяют. По
охлаждении толуольного раствора выделяют 26,4—28 г (67—72%) тетрафенилгермания, т. пл. от
225 до 228° С.
Тетрафенилгерманий — белое кристаллическое вещество, т. пл. 235,7° С. Он хорошо
растворим в хлороформе, умеренно — в четыреххлористом углероде, сероуглероде и ацетоне.
Хорошо растворим в горячем бензоле и толуоле (удобные растворители для
перекристаллизации), плохо растворим в спирте, эфире и петролейном эфире.
Глоклинг с сотр. [30] считает, что наилучшим методом получения
тетрафенилгермания, дающим наиболее устойчивый выход, является
проведение реакции в тетрагидрофуране.
Получение тетрафенилгермания [30]. Раствор Юг четыреххлористого германия в 60 мл
тетра гидрофура на прибавляют в течение 10 мин. к профильтрованному раствору бромистого
фенилмагния, приготовленному из 88 г бромбензола и 15,6 г магния в 180 мл тетрагидро-
•фурана. После кипячения в течение 18 час. сырой тетрафенилгерманий, не прибегая к
гидролизу, отделяют фильтрованием, промывают разбавленной уксусной кислотой и
кристаллизуют из толуола; получают 15,3 г (85%). Т. пл. 237—238° С. При этом методе гексафенил-
дигерман не образуется.
Описано получение ряда полнозамещенных ароматических германий-
органических соединений по методике, приведенной Симонсом с сотр. 140].
Быпи получены тегра-о-толил-[40, 49], тетра-ж-толил-[40], тетра- п-тол ил -
[16, 40, 501 и тетра-л-бифзнил германий [161.
Глоклинг с сотр. [301 указывает, что получение орто-, мета- и пара-то-
лильных производных тетраарилгермания не происходит так просто, как
описывает Симоне 140]. Например, тетра-д-толилгерманий образуется с
выходом 52% только при применении раствора бромистого /г-толилмагния,
отфильтрованного от избытка металлического магния. В противном случае
основным продуктом реакции является гекса-л-толилдигерман и гидрид три-
/7-толил германия.
Реакция четыреххлористого германия и брэмистого #-толилмагния [30]. Раствор
бромистого я-толилмагния, полученный из 137 г я-бромтолуола и 30 г магния в 500 мл абс.
эфира, разделяют на две равные части (а) и (б ). К части (а), содержащей избыток
металлического магния, прикапывают раствор 10 г четыреххлористого германия в 100 мл толуола и
кипятят с обратным холодильником 2 часа. После гидролиза коричневого раствора
получают 3,4 г гекса-я-толилдигермана; т. пл. 345° С после кристаллизации из толуола. При
перегонке коричневого маслоообразного остатка выделяют 4,5 г гидрида три-я-толилгер-
мания (образуется за счет гидролиза промежуточно образовавшегося R3GeMgX см.
стр. 149); т. кип. 160° С/Ю~3 мм; т. пл. 81° С (кристаллизация из петролейного эфира,
кипящего в пределах 40—60° С). Из остатка после перегонки получают некоторое
количество тетра-я-толилгермания, т. пл. 224—225° С. Часть (б) эфирного раствора бромистого
30
Германийорганические соединения. Лит. стр. 43—46
л-толилмагния тщательно отфильтровывают от избытка металлического магния, дальше
поступают аналогично описанному выше. После гидролиза желтого раствора получают 10,6 г
(52%) тетра-я-толилгермания; т. пл. 222—225° С. Из маточного раствора выделяют
бромистый три-я-толилгерманий (т. пл. 128—129° С) и хлористый три-я-толилгерманий (т. пл.
121° С).
Для орто- и мета-изомеров реакция протекает аналогично. Так, при
реакции четыреххлористого германия с бромистым ж-толилмагнием в
присутствии избытка металлического магния образуется 21,5% гекса-ж-толилди-
германа, гидрид три-ж-толилгермания и лишь следы тетра-ж-толилгермания.
В случае проведения реакции в отсутствие избытка металлического магния
получают 52% тетра-ж-толилгермания и в виде примеси 6% гекса-ж-то-
лилдигермана.
Тетра-о-толилгерманий не был получен совсем даже при тщательной
очистке бромистого о-толилмагния от избытка металлического магния.
Единственными продуктами реакции были гекса-о-толилдигерман и
бромистый три-о-тол ил германий.
Полнозамещенные германийорганические соединения нафтильного ряда
с помощью реактива Гриньяра получены не были [51].
В гетероциклическом ряду получен тетра-а-тиенилгерманий с выходом.
72%. Т. пл. 149—150° С [52].
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ТИПА R3GeX, R2GeX2 и RGeX3
При действии магнийорганических соединений на соли
четырехвалентного германия наряду с R4Ge часто получается смесь германийорганических
соединений класса R3GeX, R2GeX2 и RGeX3, особенно в ароматическом
ряду. Однако эта реакция не является методом синтеза указанных германий-
органических соединений в связи с возникающими трудностями разделения
смеси германийержанических соединений различной степени алкилирова-
ния, а также благодаря наличию других более удобных приемов их
получения (см. стр. 19,22). В литературе имеются лишь отдельные примеры
применения магнийорганических соединений для этой цели. Шмидт с сотр.
[53] указывает, что при попытке получить йодистый триметилгерманий
при действии йодистого метилмагния на четыреххлористый германий в
отношении 3 : 1 йодистого триметилгермания не образуется. Захаркин с
сотр. [54, 55] получил хлористый три-я-бутилгерманий с выходом 60% при
действии смеси хлористого бутила и четыреххлористого германия на
металлический магний без растворителя. При синтезе тетра-я-бутилгермания
действием реактива Гриньяра бромистый трибутил германий образуется в
количестве до 20% [21]. Бромистый три-я-гексилгерманий может быть
получен при помощи реактива Гриньяра, однако продукт недостаточно чист.
Получение бромистого три-и-гексилгермания [56]. К 19,4 мл (0,17 моля)
четыреххлористого германия прибавляют 0,57 моля бромистого я-гексилмагния, полученного из 14,6 г
магния и 84,4 мл бромистого я-гексила. Полученную суспензию фильтруют, фильтрат
перегоняют. Сначала отгоняется непрореагировавший бромистый я-гексил и растворитель, а
затем — 28 г (40%) бромистого три-я-гексил германия с т. кип. 143—145° С/0,5 мм.
Дополнительно получают еще 15,4 г жидкости с т. кип. 145—160° С/0,5 мм, являющейся смесью
бромистого три-я-гексилгермания и тетра-я-гексилгермания. Бромистый три-я-гексилгер-
маний также был получен из четыреххлористого германия и 3,3 эквивалента бромистого
я-гексилмагния, однако продукт не был очищен от примеси двубромистого ди-я-гексил-
германия. Выходы авторы не указывают [56].
Лесбр и Сатже [57] сообщают (без приведения эксперимента), что им
удалось получить RGeCl3(R — бутил, амил, гексил, гептил и октил) с
выходами, превышающими 50%, при действии магнийорганических
соединений на избыток четыреххлористого германия.
Рохов [581 при реакции 1,5 эквивалентов хлористого я-пропилмагния с-
1 эквивалентом четыреххлористого германия получил треххлористый.
Синтез с помощью магнийорганических соединений
31
я-пропилгерманий с т. кип. 167° С/767 мм\п2о 1,4780; п23 1,4718. Выход
продукта не указан.
Что касается частично замещенных германийорганических соединений
изостроения, то, как уже указывалось выше (см. стр. 27), в ряде случаев
они образуются вместо тетразамещенных германийорганических
соединений. Особенно это отмечено в изопропильном ряду. Так, Рохов [58]
приготовил при действии эквивалентного количества хлористого
изопропилмагния на четыреххлористый германий в эфире треххлористый изопро-
пилгерманий; т. кип. 164,5° С/765 мм; n2S 1,4760; п2о 1,4700.
Андерсон [26] указывает, что в отличие от бромистого я-пропилмагния
избыток 1алоидного изопропилмагния реагирует с четырехбромистым
германием с образованием (вследствие, очевидно, стерического эффекта)
только частично замещенных продуктов — (t-C3H7)2GeBr2, (j-C3H7)3GeBr и
твердого полимера (t-C3H7Ge)n.
Получение хлористого триизопропилгермания [27]. К 67 г четыреххлористого германия
медленно прибавляют 1,1 л 1,3 М раствора хлористого изопропилмагния, через 2 часа после
окончания прибавления реакционную смесь разлагают водой. Для удаления магниевых солей
органический слой промывают водой, затем встряхивают с избытком едкого натра и
экстрагируют гексаном, который затем отгоняют. Перегонка при 9 мм дает 15 г гидроокиси
триизопропилгермания; т. кип. 90—92° С /9 мм. При дальнейшей перегонке (при 1 мм) получают
10 г смеси, кипящей при 126° С, и окончательно 24 г довольно чистого тримера окиси диизо-
пропилгермания [(i-C3H7)2GeO]3, гидроокись триизопропилгермания, затем с помощью НС1
переводят в хлористый триизопропилгерманий.
Мазеролл [36], а ранее Шварц с сотр. [37] описали получение двухло-
ристого циклотетраметиленгермания. Реакция четыреххлористого
германия с магнийорганическим производным 1,4-дибромбутана может
протекать согласно уравнениям:
BrMgCH2 — СН2 СН2 - СН2
1) GeCl4 +
2MgBrCl +
GeC
BrMgCH2 — CH2 CH2 — СН.
2) ОеСЦ + 2
BrMgCH2 — СН2 СН2 — СН2 СН2 — СИ2
\ /
Ge
/ \
BrMgCH2 - СН2 СН2 — СН2 СН2 — СН2
+ 4MgBrCl,
3) GeCl4 + BrMg (CH2)4 MgBr + GeCl4 -* 2 MgBrCl + Cl3Ge (CH2)4 GeCl3.
Все эти продукты реакции и были выделены после соответствующей
обработки реакционной смеси.
В случае циклогексильных и ряда ароматических производных даже
при действии большого избытка реактива Гриньяра образуются лишь три-
замещенные производные германия. Например, вместо тетрациклогексил-
германия [59] был выделен бромистый трициклогексилгерманий.
Получение трициклогексилгерманола [20]. В трехгорлую колбу, снабженную обратным
холодильником, мешалкой и капельной воронкой, помещают 163- г бромистого циклогексила,
300 мл абс. эфира и 24,5 г магниевой стружки. По окончании реакции добавляют раствор
21,4 г четыреххлористого германия в 200 мл абс. бензола. Кипятят 2 часа и затем перегоняют
с водяным паром. Твердый остаток отфильтровывают, хорошо отжимают, растворяют в
спирте и осаждают трициклогексилгерманол путем выливания спиртового раствора в воду.
Перекристаллизацией из петролейного эфира получают 10 г белых кристаллов с т. пл. 171 —
173° С; выход 29,6% оттеорет., считая на четыреххлористый германий.
По данным Веста [51], тетра-а-нафтилгерманий не может быть получен
прямой реакцией бромистого а-нафтилмагния и четыреххлористого
германия (это не удается даже при применении литийорганических соединений),
в результате реакции получается лишь бромистый три-а-нафтилгерманий..
-32 Гсрманийорганиiecxue соединения. Лит. стр. 43—46
Получение бромистого три-а-нафтилгермания [51]. Реакцию проводят под азотом в
трехгорлой колбе, снабженной капельной воронкой, обратным холодильником и трубкой
для ввода азота. К реактиву Гриньяра, полученному из 50 г (0,24 моля) а-бромнафталина
и 7 2 магния в 250 мл эфира, прибавляют в течение 20 мин. раствор 14 г (0,36 моля) четырех-
бромистого германия в 100 мл бензола. Смесь кипятят 6 час. с обратным холодильником,
затем разлагают льдом и соляной кислотой. Органический слой отделяют, быстро
промывают водой, фильтруют и сушат над хлористым кальцием. Раствор концентрируют, под конец
нагревая до 130° С при 3 мм для удаления нафталина. Коричневый резиноподобный остаток
переносят в теплый этилацетат, фильтруют через активированный уголь и оставляют
стоять в течение месяца в холодильнике. За это время 4 раза выделившиеся кристаллы отделяют
фильтрованием и получают всего 7,3 г (38%) сырого бромистого три-а-нафтилгермания,
т. пл. 230—238° С. Несколько кристаллизации из этилацетата поднимают температуру до
242—253° С. Чистый продукт представляет бесцветные призмы.
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ТИПА R3Ge — GeR3
Морган с сотр. [39] при действии четырехбромистого германия на
бромистый фенилмагний ошибочно констатировал отсутствие образования гек-
сафенилдигермана. В дальнейшем Джонсон и Гаррис [42] впервые
показали, что при продолжительном кипячении галоидной соли германия с
реактивом Гриньяра образуются некоторые ароматиче:кие дигерманы. Однако,
если необходимо, реакция образования дигерманов может быть полностью
подавлена путем повышения температуры нагревания, что приводит к Ar4Ge.
Получение гексафенилдигермана [60]
4 GeCl4 + 14CeH3MgBr -» 2 (СбН5)3 GeGe (СсН5)з + 7MgBr2 + 7MgCl2 + 2QH.C1.
Раствор 181 г (1 моль) бромистого фенилмагния в 500 мл абс. эфира помещают в трех-
горлую колбу, снабженную механической мешалкой, обратным холодильником и капельной
воронкой. К этому раствору по каплям добавляют 21,4 г четыреххлористого германия (0,1
моля) в 225 мл толуола. Нагревают с обратным холодильником в течение 3 час, добавляют
20(Ьотолуола и отгонкой доводят объем реакционной смеси до 600 мл. 'Избыток реактива
Гриньяра разлагают 50%-ным раствором уксусной кислоты или 200 мл AM соляной кислоты,
водный слой отделяют с помощью резиновой груши. Органический слой с оставшимся в нем
осадком промывают 3 раза горячей водой, отделяя водный слой тем же приемом. Толуоль-
ный слой нагревают до кипения для растворения тетрафенилгермания, образовавшегося в
качестве побочного продукта. Гексафенилдигерман в горячем толуоле не растворяется, его
отделяют путем горячего фильтрования. Небольшое дополнительное количество
гексафенилдигермана получают доведением фильтрата до малого объема с последующим горячим
фильтрованием. При этом получают практически чистый гексафенилдигерман; т. пл. 325—327° С.
Все же в нем содержится некоторое количество тетрафенилгермания. Окончательная
очистка достигается фракционированной возгонкой. Тетрафенилгерманий возгоняется при
210° С/1 мм. Возгонка должна проводиться очень осторожно, так как гексафенилдигерман сам
возгоняется в тех же условиях при 275° С. После удаления тетрафенилгермания
гексафенилдигерман возгоняют и затем перекристаллизовывают из хлороформа. Получают
мелкокристаллическое вещество с т. пл. 330—331°. Выход 18 г (59%). Гексафенилдигерман очень
слабо растворим в обычных органических растворителях и нерастворим в жидком аммиаке.
i
Гилман с сотр. [14] при синтезе тетрафенилгермания с помощью
реактива Гриньяра выделил гексафенилдигерман с выходом 8%. Гексабензил-
дигерман был получен с выходом 11,4% [31].
Сейферт [31] при взаимодействии бромистого винилмагния с четырех-
хлористым германием в тетрагидрофуране выделил 31%
тетравинилгермания и 25% гексавинилдигермана. Автор объясняет механизм реакции
■ следующей схемой:
GeCl4 + 2СН2 = CHMgBr.-т* GeCi2 + CH2 = СН - СН = СН2 + MgCl2 + MgBr*,
GeCl2 + 2СН2 == CHMgBr -» (СН2 = СН)2 Ge + MgCl2 + MgBr2,
(CH2 = CH)2 Ge + СН2 = CHMgBr -» (СН2 = СН)3 GeMgBr,
Синтез с помощью магнийорганических соединений
33
(СН2 = СН)3 GeMgBr + (СН2 = СН)3 GeCl -> (СН2 = СН)3 GeGe (СН = СН2)3 +
+ V2MgCl2 + V2MgBr2,
т. е. четыреххлористый германий восстанавливается до двухвалентного
германия с образованием промежуточного нестойкого дивинил германия с
последующим превращением в магниевое производное (СН2 = CH)3GeMgBr
Сейферт считает, что такое течение реакции объясняется более высокой
электроотрицательностью германия по сравнению, например, с кремнием
и оловом. Обычно при действии бромистого винилмагния на
четыреххлористый кремний и олово образуются полнозамещенные винильные
производные.
Глоклинг с сотр. [30] при повторении реакции получения гексафенил-
дигермана [60] столкнулся с фактом, когда даже при соблюдении
совершенно идентичных условий выходы колеблются от 0 до 60%. Предполагается,
что это происходит от присутствия в реакционной смеси избыточного
количества мелко диспергированного металлического магния, который не
всегда удается удалить декантацией или грубой фильтрацией. Гексаарилдигер-
маны образуются, когда реакция арилирования образовавшегося R3GeX
затруднена стерическими препятствиями или реакционная способность
соответствующего RMgX низка. Авторы исследовали реакцию четырех-
хлористого германия в фенильном, о-, м- и n-толильных рядах при избытке
металлического магния и в его отсутствие. Избыток мелко
диспергированного металлического магния всегда приводит к образованию
соответствующих дигерманов. Однако, очевидно, вследствие большой реакционной
способности хлористого бензилмагния образование соответствующего дигер-
мана не наблюдалось как в присутствии металлического магния, так и в
отсутствие. Авторы предлагают следующую схему реакции:
1. GeX4+RMgX-»R8GeX,
2. R3GeX + RMgX -» R4Ge,
3. R3GeX + Mg-* RsGeMgX,
4. RsGeMgX + R3GeX -> R6Ge2,
5. R3GeMgX + H20 -» R3GeH,
6. R3GeX + RMgX -> R3GeMgX + RX.
Они подтверждают ее исследованием промежуточных продуктов
реакции.
Каков бы ни был механизм реакции, представляемый различными
авторами, вполне вероятным является образование в процессе реакции
соединения со связью Ge—Mg. R3GeMgX (о получении этого соединения см.
стр. 149) легко реагирует с молекулой R3GeX, давая R3Ge—ReR3 и MgX2.
О реакции бромистого n-толилмагния и четыреххлористого германия в
отсутствие и при избытке металлического магния см. стр. 29.
Интересно отметить опубликованные в последнее время исследования
по получению смешанных алкильных производных дигерманов [61]. Было
приготовлено несколько несимметричных гексаалкилдигерманов формулы
Ge2(CH3)jcRe-jc (R = QH5, п-С3Н7 и i-C3H7) с выходом от 7 до 35% при
реакции четыреххлористого германия со смесью различных
магнийорганических соединений. Были синтезированы диэтилтетраметил-, триэтилтри-
метил-, тетраметилди-я-пропил- и диизопропилтетраметилдигерманы. С
помощью газожидкостной хроматографии были определены соотношения
изомеров для некоторых из этих соединений; проведен элементарный
анализ и исследованы ИК-спектры.
3 Заказ JM& 207
34
Германийорганшеские соединения. Лит. стр. 43—46
СИНТЕЗ НЕСИММЕТРИЧНЫХ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МАГНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА ГЕРМАНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Для германия известно большое количество полнозамещенных германий-
органических соединений, имеющих различные радикалы при атоме
германия, как, например, следующие: RgGeR1, RaGeRa1, R2GeR!Rn.
Соединения типа I^GeRHR111!?™ в литературе пока не описаны, но возможность
их существования не исключена. Обычным путем синтеза упомянутых выше
германийорганических соединений является действие реактива Гриньяра
на соответствующие галоидопроизводные германийорганических
соединений.
R3GeX + R'MgX -> R3GeR' + MgX2,
R2GeX2 + 2R'MgX -* RaGeR^ + 2MgX2,
RGeX3+ 3R'MgX -» RGeRg+ 3MgX2.
Для получения германийорганических соединений с тремя различными
радикалами исходят из смешанного несимметричного германийорганиче-
ского соединения:
R2R'GeX + R"MgX -> R2R'GeR" + MgX2.
Исходные R2R'GeX получают последовательным деалкилированием
полнозамещенных германийорганических соединений при определенном
подборе радикалов в соответствии с прочностью их связи с германием (см.
главу о деалкилировании).
Синтез несимметричных германийорганических соединений, несмотря
на многочисленные примеры, довольно однотипен. Выбор соответствующего
реактива Гриньяра и германийорганичского соединения, входящего в
реакцию, зависит от легкости приготовления последнего. Так, для
получения R3GeR', где R = С2Н5, С4Н9 или С6Н5 и другие радикалы, исходят
обычно из соответствующего R3GeX. Однако, например, в случае
необходимости синтеза (CH3)3GeR, очевидно, в связи с малой доступностью тет-
раметилгермания и его галоидных производных, чаще исходят из RGeX3 и
действуют на него йодистым метил магнием. Ввиду ряда синтетических
особенностей образования несимметричных германийорганических
соединений, имеющих в молекуле непредельные радикалы, и возможности их
применения в получении полимерных материалов они рассматриваются
отдельно.
В литературе имеются отдельные примеры применения бромистого три-
метилгермания в реакциях с реактивами Гриньяра, полученными из
бромистого изобутила [62], я-дибромбензола [63] или хлористого я-хлорбен-
зила [64]. Образующийся при этом триметилизобутилгерманий имеет т. кип.
121° С, триметил-я-бромфенилгерманий — 118° С/20 мм, а триметил-я-
хлорбензилгерманий — 112° С/14 мм.
Бромистый триэтилгерманий, так же как и хлористый триэтилгерманий,
применяют для подобных синтезов довольно часто.
Гилман и сотр. [14], действуя на хлористый триэтилгерманий
хлористым бензилмагнием и магнийорганическим производным хлористого этил-
бензола, получили германийорганические соединения с выходом 79%.
Иборн с сотр. [65, 66] получил ряд несимметричных
германийорганических соединений, использовав бромистый триэтилгерманий.
Получение арилтриэтилгермания через реактив Гриньяра [65]. Раствор 0,05 моля
бромистого триэтилгерманий в 100 мл эфира прибавляют к реактиву Гриньяра,
приготовленному из 0,07 моля бромистого арила и 0,075 г-атома магния в 100 мл эфира. Реакционную
Синтез с помощью магнийорганических соединений
35
смесь кипятят 2—12 час, разлагают разбавленной соляной кислотой, отделяют эфирный
слой, водный слой промывают эфиром. Эфирные вытяжки сушат над сернокислым натрием
и фракционируют.
Были получены несимметричные триэтиларильные соединения
германия с радикалами: я-толил (выход 65%, т. кип. 136° С/16 мм, n2S 1,5134);
я-изопропилбензол (выход 70%, т. кип. 136° С/77 мм; n2S 1,5095); п-трет-
бутилбензол (выход 85%, т. кип. 125°С/2 мм); я-фторбензол (выход 75%,
т. кип. 12ГС/15,5 мм); ж-хлорбензол (выход 60%, т. кип. 132—133°С/8 мм);
я-хлорбензол (выход 60%, т. кип. 130°С/6 мм). Однако триэтилфени л
германий содержал примесь дифенила; авторам не удалось его очистить даже
путем многократной перегонки. Аналогично был получен триэтил-я-меток-
Сифенилгерманий [67], выход 79%; т. кип. 14ГС/10 мм, 118°С/3 мм; п2£
1,5217.
Лесбр и Мазеролл [68] действовали на R3GeX (R = С2Н5, С4Н9, С6Н5;
X = Вг) магнийорганическими производными циклопентадиена, флуорена
и индена. Авторы, впрочем, не приводят методики эксперимента и выходов,
даны лишь температуры кипения или плавления, показатели преломления,
плотности и анализы.
Те же Лесбр и Мазеролл [68], исследуя реакцию иода с различными
тетраалкильными производными германия (см. стр. 82), получили ряд
йодистых солей триалкилгермания и использовали их для получения
несимметричных германийорганических соединений. Так, из йодистого три-
этилгермания были получены метилтриэтилгерманий (т. кип. 135°С/760 мм;
af 0,9906; п*о 1,4332); я-пропилтриэтилгерманий (т. кип. 73—74°С/20 мм,
af0,9810; п2о 1,4460); я-бутилтриэтилгерманий (т. кип. 91—92°С/20 мм,
af0,971; n2£ 1,4483); я-гептилтриэтилгерманий (т. кип. 104—105°С/20 мм,
a24°0,9625; /z2D° 1,4495).
Йодистые соединения метилгермания различной степени алкилирования
использовались также для получения несимметричных
германийорганических соединений с помощью реактива Гриньяра. Был получен диметилди-
я-пропилгерманий (т. кип. 141° С, выход 75%) и триметил-я-пропил-
германий (т. кип. 99,5° С, выход 73%) [69].
Интересный метод получения несимметричных германийорганических
соединений состоит в одновременном прибавлении двух различных
растворов магнийорганических соединений к эфирному раствору четыреххлори-
стого германия. Таким образом была получена смесь бензилтрибутилгер-
мания (10,2%), дибензилдибутилгермания (46%) и трибензилбутилгермания
16% [31].
Мезеролл [70, 71] нашел общий метод приготовления со -галоидирован-
ных производных германия, исходя из хлоргидринов гликоля. Хлоргид-
рины с С4 и С5 получались очень легко, исходя из тетрагидрофурана или
тетрагидропирана [72]. Пираниловые производные со-хлоргидринов легко
дают в среде тетрагидрофурана соответствующие магнийорганические
производные, которые реагируют обычным образом с галогенидами
германийорганических соединений. Продукт реакции нагревают в присутствии
фосфорной кислоты и получают германийорганическиесоединения с гидрок-
сильной группой (выход 85%). Методику эксперимента автор не приводит.
О
/ О (СН,)„ MgCl + RsGeX - R3Ge (CH^-O-l^ -f MgClX
О О
R3Ge (CH,)n-0-l J -5^-* R3Ge(CH2)„OH +
yS нагрев
3*
36 Германийорганжеские соединения. Лит. стр. 43—46
Таким образом былиполученытриэтилбутанолгерманий (C2H5)3Ge (CH2)4OH,
т. кип. 134° С/13 мм, ,4° 1,0572; п2о 1,4688 и триэтилгептанолгерманий
(C2H5)3Ge(CH2)7OH, т. кип. 145°С/17 мм, df 1,0454; n*S 1,4700.
В ароматическом ряду для синтеза несимметричных германийоргани-
ческих соединений еще в ранних работах [18, 29] применялся бромистый
трифенилгерманий (получение трифенил->г-толилгермания, трифенилэтил-
германия), а также бромистый трибензилгерманий. Необходимо отметить
работы Гилмана с сотр. [73—75]. Трифенил-я-октадецилгерманий был
получен ими с выходом 30%, т. пл. 79,5—80,5° С [76], а трифенилциклогек-
силгерманий — с выходом 31%, т. пл. 143—146° С.
Получение я-трифенилгермилфенилдифенилметана [73]
(СбН5)з GeBr + (СбН5)2 CH—/^4-MgBr ~* (С«Н^3 Ge—/^-СН(СвНв)2.
Реактив Гриньяра приготовляют из 10 г (0,031 моля) я-бромфенилдифенилметана и
0,96 г (0,04 г-атома) магния в 100 мл эфира. Реакцию инициируют прибавлением 0,2 мл
бромистого этила, реакционную смесь нагревают с обратным холодильником в течение 24
час. Титрование аликвотной части реакционной смеси показывает выход реактива Гриньяра,
равный 62%. К 2,85 г бромистого трифенил германия в 10 мл эфира прибавляют 50 мл
реактива Гриньяра, реакционную смесь нагревают 24 часа. Эфир отгоняют, а остаток при
перемешивании нагревают при 100° С 2 часа, затем добавляют 50 мл бензола и перемешивают в
течение ночи. Реакционную смесь гидролизуют водой и разбавленной кислотой, бензольный
раствор сушат, бензол отгоняют в вакууме. К оставшемуся резиноподобному остатку
добавляют 20 мл петролейного эфира (т. кип. 60—70° С), который осаждает 2,3 г (57%) я-трифенил-
гермилфенилдифенилметана т. пл. 207—209° С (после перекристаллизации из смеси бензола
и ггетролейного эфира т. пл. до 208—210° С). Из петролейного эфира получают
дополнительно некоторое количество вещества; общий выход 65%.
Манулкин с сотр. [76] получил ряд несимметричных ароматических
германийорганических соединений общей формулы (C6H5)3GeC6H5X, где
X — изопропил-(выход 48,6%), изобутил- (45,3%), изоамил-(47%), я-гек-
сил- (62,7%), я-гептил- (50,8%), я-октил- (60,6%), я-нонил- (52,9%), я-децил-
(48,5%), циклогексил- (47,4%). Он синтезировал также отолилтрифенил-
германий (45%) и (С6Н5)зОеС6Н4ОС2Н5->г (58,3%). Аналогично г^ыли
синтезированы три-я-пропилфенил германий (72,8%) трибутилфенилгерманий
(64,8%), триизобутилфенилгерманий (63,4%), три-я-амилфенилгерманий
(75%), триизоамилфенилгерманий (65,9%), три-я-гептилфенил германий
(64%), три-я-октилфенил германий (61,4%), три-я-нонилфенилгерманий
(67,1%) и три-я-децил фенил германий (52,5%) [76а].
Действие магнийорганических соединений на R2GeX2 изучалось в
сравнительно немногих работах.
Флуд [77] впервые синтезировал дифенилдиэтилгерманий; т. кип.
316° С/760 мм. Исходя из двуиодистого диэтилгермания [68], был получен
диметилдиэтилгерманий (т. кип. 108—109° С/760 мм, df 0,9885; nb° 1,4516)
и диэтилди-я-бутилгерманий (т. кип. 109—110° С/14 мм, df 0,9547;
п2о 1,4516); выход не указан. Теми же авторами [67] получен дифенилди-
инденилгерманий, т. пл. 144—145° С.
Продолжая работы по исследованию циклических
германийорганических соединений, Мазеролл [35, 78] получил дигалоидные производные
циклотетраметиленгермания и циклопентаметиленгермания и показал, что
к соответствующим диалкильным и диарильным производным можно
подойти двумя путями:
1. Действием дигалогенидов алкилгермания, имеющих готовый цикл в
молекуле, на магнийорганические соединения:
Синтез с помощью магнийорганических соединений
37
СНг—СН2 Br CH2—СН2 СгН]
\ /
Ge +2C2H5MgBr-
/ \
5
Ч /
Ge + 2MgBr2,
/ \
СНг—СН2 Br CH2—CH2 G2H5
СН2—GH2 C1 СНа—СН2 С6Н5
/ \ / / \ /
СН2 Ge + 2C6H5MgBr-* СН2 Ge + 2MgBrCl.
\ / \ \ / \
СНа—GH2 C1 СН2—СНа СбНб
2. Действием дигалогенида диалкилгермания на димагнийорганическое
производное. В эфире эта реакция не идет, однако в тетрагидрофуране
проходит достаточно энергично
СНа—СНа С2Н5
I \ /
(С2Н5)2 GeBr2 + BrMg (CH2)4 MgBr -> Ge + 2MgBr.
I / \
СНа—СНа С3Н5
По мнению авторов, первый метод более удобен для получения чистых
продуктов.
Получение диэтилциклотетраметиленгермания [35]. К раствору бромистого этилмагния,
приготовленному из 8 г (0,333дмоля) магния, прибавляют 13,15 г (0,045 моля) чистого дву-
бромистого циклотетраметиленгермания. После отгонки большей части эфира реакционную
смесь нагревают в течение часа на водяной бане; по охлаждении гидролизуют водой. После
обычной обработки получают 7,35 г диэтилциклотет-раметиленгермания. Выход 86%;
т. кип. 173—174° С/750 мм; df 1,0761; п2£ 1,4725.
Если взять в качестве исходных соединений двубромистый диэтилгер-
маний и магнийорганическое производное 1,4-дибромбутана, то выход
равен 41%. В случае синтеза диэтилциклопентаметиленгермания (т. кип.
116—117 С/72 мм, df 1,0663; п\° 1,4742) выход равен 87%, если в качестве
исходного продукта берут двубромистый циклопентаметиленгерманий, и
59%, если исходят из двубромистого диэтилгермания. Аналогично получают
дифенилциклотетраметиленгерманий (выход 83%, т. кип. 115° С/0,2 мм;
df 1,2189, п2о 1,5971) и дифенилциклопентаметиленгерманий (выход 79%,
т. кип. 150° С/1 мм; df 1,2061; n2S 1,5932). При действии димагнийорга-
нического производного 1,4-дибромбутана на двубромистый
циклопентаметиленгерманий с выходом 45% был получен германо-5-спиро-(4,5)-декан
[36] (т. кип. 101° С/23 мм; df 1,1474; n2D° 1,5058).
Манулкин с сотр. [79, 80] получил ряд несимметричных германийорга-
нических соединений, исходя из двубромистого дифенилгермания.
Получение дифенилди-(я-метоксифенил)германия [79]. Приготовляют удвоенное
количество (по сравнению с теорией) реактива Гриньяра из 0,75 г магния и 5,79 г я-броманизола
в 30 мл абс. эфира. К теплому раствору при перемешивании порциями добавляют 3 г (СбНб)2
GeBr2, при этом наблюдается вскипание эфира. Для завершения реакции реакционную смесь
нагревают 2 часа на водяной бане. Эфир отгоняют и, заменив 30 мл толуола,
продолжают нагревание на масляной бане при ПО—120° С в течение 2 час. По охлаждении
разлагают холодной водой и 1%-ным раствором соляной кислоты. Толуольный слой отделяют,
водный дважды обрабатывают эфиром. Эфирный слой соединяют с толуольным, после
удаления растворителя остаток перегоняют с паром. Перегоняющуюся густую вязкую массу
отделяют от воды, растворяют в эфире и сушат хлористым кальцием. После отгонки
растворителя твердый осадок обрабатывают смесью эфира со спиртом, после чего выделяют
кристаллы. Получают 1,54 г дифенилди-(/г-метоксифенил)германия с т. пл. 120—121° С, выход
43,6%.
Аналогично получены дифенилди-о-толилгерманий (т. пл. 165—166° С,
выход 48,7%), дифенилди-(/г-этилфенил)германий (т. пл. 111—112° С, выход
39,2%)\ дифенилди(изопропилфенил)германий (т. пл. 127—128° С, выход
38
Германийорганические соединения. Лит. стр. 43—46
32,5%), дифенилди-(/г-этоксифенил)германий (т. пл. ПО—111° С, выход
33,3%), дифенилди-(п-феноксифенил)германий (т. пл. 129—130° С, выход
24,2%), дифенилди-(п-бромфенил)германий (т. пл. 129—130° С, выход 23%),
дифенилди-я-пропилгерманий (т. кип. 173—175° С/4 мм, выход 61,9%)
дифенилдиизобутилгерманий (т. кип. 160—162° С/4 мм, выход 56%),
дифенилдиизоамилгерманий (т. кип. 187—189° С/4,им, выход 67%), дифе-
нилди-я-гексилгерманий (т. кип. 203—205° С/4 мм, выход 66%), дифенил-
ди-я-гептилгерманий (т. кип. 215—217° С/3 мм, выход 65%), дифенилди-я-
октилгерманий (т. кип. 225—226° С/3 мм, выход 75%), дифенилди-я-но-
нилгерманий (т. кип. 229—230° С/3 мм, $ыход 50%), дифенилди-я-децил-
германий (т. кип. 234—235° С/6 мм, выход 84,1%). Также получены
некоторые производныедифенилгермания с непредельными радикалами [80].
Пономаренко с сотр. [81] при действии бромистого метилмагния на двух-
хлористый диметилгерманий получил хлористый триметилгерманий (выход
18%).
Бауер с сотр. [82] впервые показал, что при действии йодистого метил-
магния и йодистого этилмагния на трехиодистый фенилгерманий образуется
фенилтриметилгерманий (т. кип. 182—183°С) и фенилтриэтилгерманий (т.
кип. 116—117° С/13 мм). Получен также и n-толилтриэтилгерманий (т.
кип. 125—126° С/12 мм).
Лесбр с сотр. [68, 83] из йодистых алкильных производных германия с
помощью реактива Гриньяра получил ряд несимметричных германийорга-
нических соединений: этилтриметилгерманий (т. кип. 79—80° С/760 мм\
df 0,9843; n2S 1,4090); этилтри-я-бутилгерманий (т. кип. 148—149°
С/7мм; df 0,9274; п2£ 1,4565), а также бутилтриэтилгерманий [83].
Миронов и Гар [84], действуя бромистым метилмагнием на треххлори-
стый трет-бутилгерманий, синтезировали с выходом 76% mpem-бутилтри-
метилгерманий; т. кип. 111° С/758 мм.
Поскольку галоид, связанный с германием, более реакционноспособен,
чем галоид, связанный с углеродом, возникает возможность получать
германийорганические соединения с галоидом в радикале [75, 84а, 85, 86].
Получение хлорметилтриметилгермания [84а]. К 3 молям бромистого метилмагния в 1 л
эфира прибавляют смесь 58,8 г (0,258 моля) треххлористого хлорметилгермания и двухло-
ристого дихлорметилгермания, разбавленную равным по объему количеством эфира, с такой
скоростью, чтобы эфир энергично кипел. По окончании прибавления реакционную смесь
нагревают 14 час, охлаждают до 0° С и добавляют 400 мл насыщенного раствора
хлористого аммония. Органический слой отделяют. К водному слою добавляют разбавленную
соляную кислоту; выделившийся при этом небольшой органический слой прибавляют к
основному. После сушки и удаления растворителей перегонкой получают 49,7 г
хлорметилтриметилгермания с т. кип. 113—114°С/761, Ъмм, df 1,189; n2gl, 4389; выход 79,8%.
В литературе известны несколько представителей несимметричных гер-
манийорганических соединений с тремя различными радикалами; этил-
изопропилдифенил германий, этилизопропилди-п-толилгерманий, этил-
фенилди-п-толилгерманий [53]. Указанные соединения получены
последовательным расщеплением и алкилированием соответствующих германийор-
ганических соединений.
Описаны и отдельные представители элементоорганических соединений,
имеющих в молекуле различные элементы, разделенные углеродным
мостиком [6, 62].
Получение /г-триметилгермилтриметилсилилбензола (СНз)зОеСбН481(СНз)з [6]. К
реактиву Гриньяра, полученному из 25,5 г триметил-я-бромфенилкремния, 42,43 г магния
добавляют 19,7 г бромистого триметилгермания. После 10 час. кипячения содержимое колбы
разлагают водой, эфирный слой отделяют, водный слой экстрагируют эфиром. Эфирный
экстракт сушат над сернокислым натрием; при перегонке получают 26 г вещества с т. кип.
104—107° С/7 мм после перекристаллизации из гексана т. пл. 98° С (выход 89%).
Синтез с помощью магнииорганических соединений 39
Аналогично получают и (CH3)3GeCH2Si(CH3)3, т. кип. 139° С/752 мм;
df 0,9541; /ig 1,4329 (90,5%).
Синтез непредельных несимметрических германийорганических
соединений может быть осуществлен двумя путями [87, 88]: обычный путь —
действие реактива Гриньяра на галогениды германийорганических
соединений, имеющих в молекуле непредельные радикалы, и действие магний-
органических соединений непредельного ряда на галогениды
германийорганических соединений. Открытие Норманом [89] простого метода
синтеза непредельных магнииорганических соединений в тетрагидрофуране
дало возможность синтезировать разнообразные германийорганические
соединения непредельного ряда. Так, Петровым и сотр. [90] были
получены при действии хлористого метилмагния на треххлористый аллилгерма-
ний в эфире с выходом 68,1 % аллилтриметил германий (т. кип. 101° С/764 мм,
df 0,9952; п2о 1,4333), с выходом 52,3% — аллилтриэтилгерманий (т. кип,
190,5°ьС/747 мм; df 1,0004; п2£ 1,4594), с выходом 42,5% — металлилтри-
метилгерманий (т. кип. 121° С/733 мм; df 0,9908; п™ 1,4416).
С другой стороны, из магниевого производного бромистого аллила те же
авторы синтезировали диаллилдиметилгерманий с выходом 42% (т. кип.
146° С/735 мм; df 1,0337; n2S 1,4645) и триаллилметилгерманий с выходом
45% (т. кип. 190,5° С/747 мм; df 1,0222; п2£> 1,4867).
Сейферт [32], использовав реактив Нормана, впервые приготовил ряд
несимметричных винильных производных германия, в дальнейшем их
синтез был осуществлен и рядом других авторов.
Получение диэтилдивинилгермания [32]. К 0,5 моля бромистого винилмагния в 200 мл
тетрагидрофурана прибавляют 18 г (0,089 моля) двухлористого диэтилгермания,
растворенного в равном по объему количестве тетрагидрофурана, с таким расчетом, чтобы все время
было энергичное кипение реакционной смеси. Затем реакционную смесь нагревают с
обратным холодильником в течение 20 час. и гидролизуют 75 мл насыщенного водного раствора
хлористого аммония. Органический слой декантируют, водный слой промывают эфиром,
эфирные слои добавляют к органическому слою. После отгонки растворителя и
последующей перегонки получают 11,5 г (70%) диэтилдивинилгермания; т. кип. 59—60° С/28,5 мм;
п% 1,4540.
Аналогично диэтилдивинилгерманий был синтезирован и другими
авторами [91, 92]. Этилтривинилгерманий получен [32, 92] с выходом 50%
<т. кип. 55—57° С/28 мм; п23 1,4605); а триэтилвинилгерманий [33,93, 94] —
с выходом 35,5% (т. кип. 151—152° С/748 мм и 40° С/4 мм; п2£ 1,4518).
Мазеролл [33] и Лабарри, Мазеролл [94] синтезировали три-я-бутил-
винилгерманий (т. кип. 108—109° С/2 мм; df 0,9479; п™ 1,4598) и три-я-
бутилаллилгерманий (т. кип. 116—117° С/2 мм, df 0,9629, n2S 1,4664).
Несимметричные германийорганические соединения, имеющие метальные и
винильные или аллильные радикалы, были получены Петровым с сотр. [95].
Наметкин с сотр. аналогично получил триметилвинилгерманий, однако
он значительно был загрязнен примесью тетрагидрофурана вследствие
близких температур кипения [95а].
Ряд германийорганических соединений был получен из бромистого три-
алкилгермания и различных непредельных магнииорганических
соединений в условиях реакции Нормана [956].
Нолтэс впервые использовал бромистый трифенилгерманий в реакции
с бромистым винилмагнием [47].
Получение трифенилвинилгермания [47]. Бромистый трифенилгерманий (11,7 г; 0,03
моля) растворяют в 30 мл бензола и прибавляют по каплям к 0,07 моля бромистого винил-
магния в 70 мл тетрагидрофурана; после нагревания в течение часа смесь гидролизуют
обычным путем. При испарении органического слоя остается масло, которое растворяют в 96%-
ном спирте. После охлаждения получают 8,3 г (84%) бесцветных кристаллов, т. пл. 62—64° С.
40
Германийорганические соединения. Лит. стр. 43—46
Аналогично были получены дифенилдивинилгерманий (67%), т. кип.
111—113° С/0,003 мм; дифенилдиаллилгерманий (77%), т. кип. 117—120°
С/0,007 мм и трифенилаллилгерманий (59%), т. пл. 90—91° С [75, 76].
При действии бромистым аллилмагнием на бромистый фенилциклотетра-
метиленгерманий был получен с выходом 82% аллилфенилциклотетраме-
тиленгерманий [78].
Сейферт [96], без приведения эксперимента, упоминает о получении по
гриньяровскому синтезу трифенилперфторвинил германия. Исследование
Либрика и Рамсдена [97] по получению хлористого n-винилфенил магния
дали развитие большому числу работ по получению полнозамещенных
металл оорганических соединений кремния, германия, олова и свинца, у
которых один из радикалов является остатком стирола. Нолтэс с сотр. [98]
впервые получил при действии хлористого n-винилфенилмагния на
бромистый трифенилгерманий и двубромистый дифенилгерманий
соответствующие моно- и дизамещенные производные стирола по реакции
(QH5)4_„ GeBr„ n-C1MgCeH4CH=CH2-> (CH.)^ Ge (C6H4CH=CH2)n
тетрагидрофуран
(л=1 и 2).
Аналогичная реакция была проведена и другими авторами [99].
Получение трифенил-я-стирилгермания [98]. К раствору 0,1 моля хлористого я-винил-
фенилмагния в 50 мл тетрагидрофурана, приготовленного согласно методике, разработанной
Либрик и Рамсден [97], по каплям прибавляют 28,8 г (0,075 моля) бромистого
трифенилгермания в 100 мл тетрагидрофурана. Реакционную смесь нагревают 3 часа при 50°С, после
прибавления 0,25 г n-mp^m-бутилкатехола разлагают холодным раствором хлористого аммония.
Органический слой отделяют, водный слой дважды экстрагируют эфиром. Соединенные вы
тяжки сушат над сульфатом магния, растворитель отгоняют в вакууме. Остаток (32 г)
растворяют в 100 мл горячего петролейного эфира; после отгонки последнего до объема 40 мл
и охлаждения до 0° С получают 20,3 г (67%) бесцветных кристаллов, т. пл. 114—117° С;
после перекристаллизации из спирта т. пл. 116—117° С. В горячем петролейном эфире
остается нерастворимым 4,2 г вещества с т. пл. 178—181° С, которое представляет собой гекса-
фенилдигерманоксан.
Аналогично синтезированы дифенилди-п-стирил германий (т. пл. 139—
140° С, выход 62%), триметил-п-стирилгерманий (т. кип. 39—41° С/0,05 лии;
df 1,1060, п2о 1,5400; 71%), триметил-я-а-метилстирилгерманий (т. кип.
55—57° С/0,3 мм; df 1,0936; п™ 1,5344; 57%) [100], триэтил-п-стирилгер-
маний [93, 101] и триэтил-а-метилстирилгерманий [102].
Исходя из реактива Гриньяра, полученного из Р-бромстирола и
йодистого триметилгермания, с выходом 46,4% получен
Р-стирилтриметилгерманий [102а].
Для германия, так же как и для кремния, олова и свинца,
синтезировано большое количество несимметричных германийорганических
соединений, у которых один или несколько радикалов имеют тройные
углерод-углеродные связи. Присутствие тройной связи значительно ослабляет связь
Ge—С. Как и для германийорганических соединений, имеющих радикал с
двойной связью, этот эффект ярко выражен тогда, когда кратная связь
находится в Р-положении по отношению к германию.
Первая попытка получения германийорганических соединений типа
R3Ge—С = СН была сделана Хартманом и сотр. [103], которые действовали
магнийорганическими производными ацетилена на галоидный ал кил
германий R3GeX. Однако при этом были получены лишь ацетиленовые
производные, содержащие два атома германия:
(QHs^Ge—C=C—Ge (QHgfo (т. кип. 50°С/14 мм)\
(QHufeGe—CeeC—Ge (C6Hn)3 (т. пл. 158° С);
(C6H5)3Ge—C=C—Ge (СбН5)3 (т. пл. 127° С).
Методику эксперимента авторы не приводят.
Синтез с помощью магнийорганических соединений
41
Коршак с сотр. [104] при действии на димагниевое производное
ацетилена германийорганических соединений класса R2GeX2 получил элементо-
органические полимеры.
Мазеролл [105], позжеЛабари Мазеролл [94], а также и другие авторы
[106, 107] показали, что наиболее удобным методом синтеза монозамещен-
ных ацетиленовых производных тетраалкилгермания является действие
галогенида алкилгермания на чистые мономагнииорганические производные
ацетилена, полученные в тетрагидрофуране
HteCH + C2H5MgBr тетрагИДР°ФураН-> HfeCMgBr + С2Н6,
R3GeBr + HC=CMgBr -> R3Ge-G-GH + MgBr2.
Аналогично реакция протекает и с производными ацетилена
R'CeeCH + C2H5MgBr -* R'teCMgBr + С2Н6,
R3GeX + R'CEECMgBr -» R3GefeCR' + MgBrX.
Для получения пропаргильных соединений германия бромистый про-
паргилмагний получают при температуре ниже 20° С, что позволяет
осуществить дальнейший синтез с выходом 77%.
Получение три-я-бутилэтинилгермания [105]. В колбу емкостью 500 мл, снабженную
обратным холодильником, мешалкой, капельной воронкой и трубкой, погруженной до дна
колбы, помещают 100 мл сухого тетра гидрофура на, который насыщают ацетиленом.
Продолжая пропускание ацетилена, по каплям и при перемешивании прибавляют раствор
бромистого этил магния (из 6 г магния)*в тетрагидрофуране. При этом наблюдают выделение
этана и сильное вспенивание жидкости. После перемешивания в течение 20 мин. при
комнатной температуре реакционную смесь охлаждают до 0° С, в течение 15 мин. прибавляют 25 г
бромистого три-«-бутилгермания. После перемешивания в течение 15 час. реакционную
смесь гидролизуют и обрабатывают обычным способом. Получают 14,6 г ацетиленового
производного, т. кип. 99—100° С/1,7 мм; п2£ 1,4580.
Аналогично был получен триэтилэтинилгерманий (т. кип. 70—71°
C/G5MM,df 1,0241; n2S 1,4485) и триметилэтинилгерманий (выход 76%,
т. кип. 72,5° С/760 мм; df 1,0391; n2D° 1,4180 [61).
Получение триэтилпропаргилгермания [105]. В колбу (500 мл) со шлифами, снабженную
капельной воронкой, мешалкой с ртутным затвором, обратным холодильником, термометром
и вводом сухого азота, помещают 12 г магния, немного сухого эфира и несколько
миллилитров бромистого пропаргила. Начало реакции инициируется добавлением хлористой ртути и
легким подогреванием. Затем прибавляют 60 г бромистого пропаргила, разбавленного 5
объемами эфира. Реакционную смесь при перемешивании охлаждают до температуры ниже
20° С и при этой температуре перемешивают в течение 2 час. Полученное магнийорганиче-
ское соединение декантируют, охлаждают до 0° С и прибавляют небольшими порциями
30,3 г бромистого триэтилгермания. По окончании прибавления реакционную смесь
перемешивают 10 час. при 20° С, затем избыток магнийорганического соединения гидролизуют
горячей дистиллированной водой, органический слой обрабатывают обычным способом.
Получают 19,5 г (77%) триэтилпропаргилгермания, т. кип. 76—77° С/18 мм, df 1,0326;
n2g 1,4669.
Аналогично получены три-я-бутилпентенил-1 -германий (выход 69%;
т. кип. 116—117° С/0,5 мм; df 0,9425; n2S 1,4645),
три-я-бутилгексинил-1-германий (выход 71%; т. кип. 132° С/0,7 мм; df 0,9381; n2D° 1,4648.
В ряде работ использованы и другие производные ацетилена.
Например, большое число германийорганических соединений XC6H4C=CGeR3
было получено с хорошими выходами при реакции бромистого
триэтилгермания с XC6H4C=CMgBr [1081.
Получение триэтил-я-метоксифенилэтинилгермания [108]. Бромистый триэтилгерманий
(0,025 моля) в тетрагидрофуране (15мл) прибавляютк бромистому
л-метоксифенилэтинилмагнию, приготовленному при реакции л-метоксифенилацетилена (0,025 моля) с бромистым
этилмагнием (0,025 моля) в тетрагидрофуране (25 мл). Смесь кипятят 10 мин., охлаждают
и разлагают насыщенным раствором хлористого аммония. После обработки получают с
выходом 90% триэтил-я-метоксифенилэтини л германий.
42
Германийорганические соединения. Лит. стр. 43—46
Таблица 1
Германийорганические соединения ряда фенилацетилена[108]
Формула соединения
(С2Н5)зОеС=ССбН4СНз-о
(C2H5bGeCEECC6H4CH3-m
(С2Н5)зОеС=ССбН4С(СН3)з-р
(С2Н5)зОеС=ССбНз(СНз)2-2,3
(С2Н5)заеС~СС6Н2(СНз)з-2,4,6
(C3H5)3GeC=CC6H4OGH3-o
(C2H5)3GeC=CC6H4OCH3-m
(QHsJsGeC^CQP^OCHg-p
(C2H5)3GeC-CC6H4Cl-o
(C2H5)sGeC5ECC6H4Cl-m
(C2H5)3GeC=CQH4Cl-p
(C2H5)3GeC=CC6H4Br-o
(C2H5)3GeC=CC6H4Br-w
(C2H5)3GeC=CC6H4Br-p
(C2H5)3GeC=CC6H4F-p
(C2H5)3GeC=CC6H4J-p
(C2H5)3GeC-CC6H4CF3-/r2
Выход,
%
80
82
88
80
92
84
86
90
88
95
86
78
85
86
90
78
90
Т. кип., °С/мм
рт. ст.
119/19
115/1,2
147/1,9
132/1,8
128/0,8
138/2,0
140/2,0
135/1,2
138/2,8
128/1,2
118/0,9
138/1,5
116/0,3
139/1,5
106/1,6
160/1,8
112/2,6
25
nD
1,5330
1,5336
1,5290
1,5291
1,5330
1,5300
1,5390
1,5450
1,5451
1,5430
1,5466
1,5570
1,5580
1,5612
1,5188
1,5882
1,4898
Полученные в работе [108] соединения приведены в табл. 1.
По аналогичной методике были получены 1-триэтилгерманилбутен-З-
ин-1 (C2H5)3GeC=C—СН=СН2 (выход 72%; т. кип. 62—63° С/5 мм;
df 1,0333; п2о 1,4850), 1-триэтилгерманил-3-метилбутен-3-ин-1 (C2H5)3Ge—С=
ее С—С(СН3)=СН2 (выход 75%; т. кип. 70—71° С/5 мм; df 1,0047; n2D°
1,4808), 1-триэтилгерманилпентен-3-ин-1 (C2H5)3Ge—Ce=C—СН=СН—СН3
(выход 80%; т. кип. 78—79° С/5 мм; df 1,0173; n2D° 1,4900 [109], а также
триметилвинилэтинилгерманий (выход 50%; т. кип. 133—134° С/55 мм;
df 1,0711, n2S 1,4655 [6]).
Двухлористый диэтил- и двухлористый дифенилгерманий были с
успехом использованы в реакции с магнийорганическими производными
ацетилена [110] для получения новых германийорганических производных
формулы R2Ge(C=CR')2 (R = С2Н5 и С6Н5; R'= я-С3Н7, п-С4Н9 и С6Н5).
В аналогичной реакции был использован и двубромистый дифенилгерма-
ний [111].
Манулкин и сотр. [112] в реакции бромистого трифенилгермания и дву-
бромистого дифенилгермания использовали реактив Нормана и получили
ряд соединений типа (C6H5)3GeR и (C6H5)2GeR2 (где R—я-пропенил и
изобутенил), а также соединения типа (C6H5)2GeC6H4X (где X — аллил,
изопропенил, изобутенил) и (C6H5)2Ge(C6H4X)2 (где X — ал кил, изопропенил и
изобутенил).
Кремнийорганические соединения ацетиленового ряда способны давать
соответствующие магнийорганические производные. Используя это,
Миронов и Кравченко [6] осуществили синтез металлоорганических
соединений с двумя различными металлами в молекуле.
Получение триметилсилилтриметилгерманилацетилена (CH3)3GeC=CSi(CH3)3 [6].
К реактиву Гриньяра, полученному из 2,5 г магния и И г бромистого этила в 150 мл эфира,
добавляют 10 г триметилэтинилкремния. Реакционную смесь оставляют при комнатной тем-
Синтез с помощью магнийорганических соединений 43
пературе на 24 часа и затем добавляют 20 г бромистого триметилгермания. После 20 час.
кипячения и обычной обработки разгонкой на колонке выделяют 19 г вещества; т. кип. 150—
151° С/750 мм; т. пл. 25° С; df 0,9642; п2* 1,4405.
Шихеев и сотр. [113—124] изучали взаимодействие галоидных солей
элементоорганических соединений IV группы (кремния, германия) с
реактивом Иоцича.
Реакции протекают по уравнениям
OMgBr
(Alk)a С + R3GeBr -> R3GeC=C-COH (Alk)2, (I)
\
CEEECMgBr
OMgBr
2 (Alk)2C + R2GeBr2 -» R2Ge [C=C—СОН (Alk)2]2, (II)
\
C=CMgBr
OMgBr
3 (Alk)aC + RGeBr3 -> RGe [C=C-COH (Alk)2]3. (Ill)
\
CEECMgBr
Синтез (2-метилбутин-3-ол-2)-4-триметилгермана [113]. К реактиву Иоцича, приготов-
ленномуиз 0,5моля бромистого зтилмагнияи21 г (0,25 моля)диметилэтинилкарбинолав
присутствии Си2С12 и 1 г HgCl2 при охлаждении до —10° С и перемешивании постепенно
прибавляют 50 г (0,25 моля) эфирного раствора бромистого триметилгермания. Реакционную смесь
нагревают на водяной бане в течение 8 час, затем обрабатывают 5%-ной соляной кислотой,
эфирный слой промывают раствором бикарбоната натрия, водой и сушат над сернокислым
натрием. Эфир отгоняют и остаток перегоняют в вакууме. После двукратной перегонки
выделяют 22,6 г вещества (45%), т. кип. 72—73° С/7 мм, т. пл. 30° С. Продукт хорошо
растворим в эфире и других органических растворителях.
По этой методике авторами синтезировано большое число соединений.
Указанными авторами [120] описан синтез некоторых представителей
германийорганических одноатомных диацетиленовых спиртов с
изолированными тройными связями при взаимодействии реактива Иоцича и
германийорганических соединений ацетиленового ряда
OMgBr
R2C + (С2Н5)з GeC=CC (CH8) (C2H5) C1 -*
\
C=CMgBr
-» (QH5)3 GeC=C-C (СН3) (С2Н5) C=C-CR2OH.
ЛИТЕРАТУРА
1. DennisLM., HanceF. Е., J. Phys. Chem., 30, 1055 (1926).
2. DennisL.M., P a t n о d e W. I. J. Am Chem. Soc, 52, 2779 (1930).
3. YoungCW., Koehler J.S., McKinney D.S., J. Am. Chem. Soc, 69,
1410 (1947).
4. Lippincott E. R., TobinM. C, J. Am. Chem. Soc, 75, 4141 (1953).
5. WatersD.N., W о о d w а г d L. A., Proc Roy. Soc, 246, 119 (1958).
6. M и р о н о в В. Ф., К р а в ч е н к о А. Л., Изв. АН СССР, серия хим., 1965, 1026.
7. Van de V о n d e 1 D. R, J. Organometal. Chem., 3, 400 (1965).
8. RochowE.G., J. Am. Chem. Soc, 70, 1801 (1948).
9. S i b e r t H., Z. anorg. allgem. Chem., 263, 82 (1950).
10. Г л а дштей н Б. М., Ро дэ В. В., С о б о р о в с к и й Л. 3., ЖОХ, 29, 2155
(1959).
44
Германийорганшеские соединения
11. A b е 1 Е. W., N i с к 1 е s s G., Р о 1 1 а г d F. H., Proc. Chem. Soc, 1960, 288.
12. Z a b 1 о t n a R., Bull. Acad, polon. sci., Ser. Sci. chim., 12, 475 (1964).
13. S e m 1 у e n J. A., WalkerG.R., В 1 о f e 1 d R. E., P h i 1 1 i p s C. S. G.,
J. Chem. Soc, 1964, 4948.
14. GilmanH., HughesM.B., GerowCW., J. Org. Chem., 24, 352 (1959).
15. KrausC. A., F 1 о о d E. A., J. Am. Chem. Soc, 54, 1635 (1932).
16. TabernD.L, Orndorf f W. K., Dennis.L.M., J. Am. Chem. Soc, 47,
2039 (1925).
17. Strohmeier W., HumpenerK., MiltenbergerK., Seifert F.,
Z. fur Elekrochemie, 63, 537 (1959).
18. Orndorf! W. K., TabernD.L, DennisL.M., J. Am. Chem. Soc, 49,
2512 (1927).
19. G i 1 m a n H., LeeperR.W., J. Org. Chem., 16, 466 (1951).
20. Колесников Г. С., Давыдова С. Л., КлиментоваН. В., Высоко-
молек. соед., 4, 1098 (1962).
21. A n d e r s о n H. H., J. Am. Chem., Soc, 73, 5800 (1951).
22. Fuc h s R., Moo re L., MilesD., GilmanH., J. Org. Chem., 21, 1113
(1956).
23. LesbreM., Mazerol les P., VoigtD., Compt. rend., 240, 622 (1955).
24. M a z e г о 1 1 e s P., VoigtD., Compt. rend., 240, 2144 (1955).
25. LesbreM., M a z e г о 1 1 e s P., Compt. rend., 246, 1708 (1958).
26. AndersonH.H., J. Am. Chem. Soc, 75, 814 (1953).
27. AndersonH.H., J. Am. Chem. Soc, 78, 1692 (1956).
28. M a z e г о 1 1 e s P., Dissertation, Toulouse, 1959.
29. В a u e r H., Burschkies K., Ber., 67, 1041 (1934).
30. Glockling R, H о о t о n К. A., J. Chem. Soc, 1962, 3509.
31. С г о s s R. J., Glockling F., J., Chem. Soc, 1964, 4125.
32. Sey f er t h D., J. Am Chem. Soc, 79, 2738 (1957).
33. M a z e г о 1 1 e s P., LesbreM., Compt. rend., 248, 2018 (1959).
34. Labarre J.F., M a z e г о 1 1 e s P., Compt. rend., 254, 3998 (1962).
35. M a z e г о 1 1 e s P., Bull. Sec chim. France, 1962, 1907.
36. С т е р л и н Р. Н., Дубов С. С, Ли Вей-чан, Вахомчик Л. П.,
Кнунянц И. Л., Ж. Всесоюзн. хим., общ. им. Д. И. Менделеева, 6, ПО (1961).
37. S с h w a r z R., R e i n h a r d t W., Ber., 65, 1743 (1932).
38. West R., J. Am. Chem. Soc, 76, 6012 (1954).
39. M о r g a n G. Т., DrewH.D. K., J. Chem. Soc, 127, 1760 (1925).
40. SimonsJ.K., W a g n e r E. C, Muller J.H., J. Am. Chem. Soc, 55, 3705
(1933).
41. W о r r a 1 D. E., J. Am. Chem. Soc, 62, 3267 (1940).
42. JohnsonO.H., HarrisD.M., J. Am. Chem. Soc, 72, 5564 (1950).
43. KrossR.D., F a s s e 1 V. A., J. Am. Chem. Soc, 77, 5858 (1955).
44. Harris D. M., Neberga 1 1 W. H., J о n s о п О. Н., Inorg. Syntheses, 5, 70
(1957).
45. StrohmeierW., MiltenbergerK., Ber., 91, 1357 (1958).
46. N о 1 t e s J. G., H e n г у M. C, J a n s s e n M. J., Chem. and Ind., 1959, 298.
47. H e n г у M. C, NoltesJ.G., J. Am. Chem. Soc, 82, 555 (1960).
48. F о s t e r L. S., DrenanJ.W., W i 1 1 i s t о n A. F., Inorg. Syntheses, 2, 109
(1946).
49. S i m о n s J. K., J. Am. Chem. Soc, 57, 1299 (1935).
50. SchwarzR., LewinsonM., Ber., 64, 2352 (1931).
51. West R., J. Am. Chem. Soc, 74, 4363 (1952).
52. KrauseE., RenwanzG, Ber., 65, 777 (1932).
53. S с h m' i d t M., R u i d i s с h I., Z. anorg. allgem. Chem., 311, 331 (1961).
54. 3 a x a p к и н Л. И., Охлобыстин О.Ю., Струнин Б. Н., ДАН СССР,
144, 1299 (1962).
55. ЗахаринЛ. И., Охлобыстин О. Ю., Струнин Б. Н., Изв. АН СССР,
ОХН, 1961, 2254.
56. F u с h s R., GilmanH., J. Org. Chem., 23, 911 (1958).
57. LesbreM., SatgeJ., Bull. Soc chem. France, 1959, 789.
58. RochowE.G., Didtschenko R., WestR.C, J. Am. Chem. Soc, 73,
5486 (1951).
59. BauerH., Burschkies K., Ber., 65, 956 (1932).
60. HarrisD.M., Nebergall W. H., JohnsonO.H., Inorg. Syntheses, 5,
72 (1957).
61. S e m 1 у e n J. A., W a 1 k e r G. R., P h i 1 1 i p s С S. G., J. Chem. Soc, 1965,
1197.
62. SchmidbaurH., W a 1 d m a n n S., Ber., 97, 3381 (1964).
63. С h a t t J., W i 1 1 i a m s A. A., J. Chem. Soc, 1954, 4403.
Синтез с помощью магнийорганических соединений
45
64. BottR. W., E a born С, Р a n d е К. С, S w a d d 1 е Т. W., J. Chem. Soc,
1962, 1217.
65. Е a b о г п С, Р a n d е К. С, J. Chem., Soc, 1960, 3200.
66. Е a b о г п С, Р a n d е К. С, J. Chem. Soc, 1960, 1566.
67. Lesbre M., M a z е г о 1 1 е s P., Compt. rend., 255, 544 (1962).
68. LesbreM., Mazerolles P., Compt. rend., 246, 1708 (1958).
•69. ZablotnaR., AkermanK., SzuchnikA., Bull. Acad, polon. sci. Ser.
sci. chim., 12, 695 (1964); РЖХим., 1965, 23Ж328.
70. M a z e г о 1 1 e s P., Compt., rend., 257, 1481 (1963).
71. Mazerolles P., Bull. Soc. chem. France, 1965, 464.
72. F u s a r i S. A., Greenlee K. W., В г о w n J., J. Am. Oil Chem. Soc, 28,
416 (1951).
73. В г о о к A. G., GilmanH., J. Am. Chem. Soc, 76, 77 (1954).
74. FuchsR., GilmanH., J. Org. Chem., 22, 1009 (1957).
75. GilmanH., GerowCW., J. Am. Chem. Soc, 79, 342 (1957).
76. M а н у л к и н 3. М. К у ч к а р е в А. Б., С а р а н к и н а С. А., ДАН СССР,
149, 318 (1963).
76 а. С а р а н к и н а С. А., Манул кин З.М., Кучка рев А. Б., Ж О X, 37,
217 (1967).
77. F 1 о о d E. A., J. Am. Chem. Soc, 54, 1663 (1932).
78. М a z е г о 1 1 е s P., D u b а с J., Compt. rend., 257, 1103 (1963).
79. С а р а н к и н а С. А., МанулкинЗ.М, ЖОХ, 35, 845 (1965).
£0. Саранкина С. А., Манулкин 3. М., Кучкаров А. Б., Докл. АН
УзССР, 1965, 35; РЖХим., 1966, ЗЖ415.
8\. П о н о м а р е н к о В. А., Взенкона Г. Я., Изв. АН СССР, ОХН, 1957, 994.
82. Bauer H., В u r s с h k i e s К., Вег., 66, 1156 (1933).
83. L е s b г е М., М a z е г о 1 1 е s P., ManyelG., Compt. rend., 257, 2303 (1963).
а МироновВ.Ф., ГарТ. К., Изв. АН СССР, серия хим., 1965, 291.
84 a. S е у f e r t h D., Roch ow E.G., J. Am. Chem. Soc, 77, 907 (1965).
85. M и р о н о в В. Ф., Д ж у р и н с к а я Н. Г., Изв. АН СССР, ОХН, 1963, 75.
86. L e s b r e M., S a t g ё J., MassolM., Сотр. rend., 257, 2665 (1963).
87. Р е t г о v A. D., MironovV.R, Angew. Chem., 73, 59 (1961).
88. В г a u n D., Angew. Chem., 73, 197 (1961).
89. N о r m a n t H.. Bull. Soc. chim. France, 5, 728 (1957).
90. П е т т.. о в А. Д., Миронов В. Ф., Долгий И. Е., Изв. АН СССР, ОХН.
1956, 1146.
91. МироновВ. Ф„ Петров А.Д., Максимова Н. Г., Изв. АН СССР,
ОХН, 1959, 1954.
92. П е т р о в А. Д., МироновВ.Ф., Изв. АН СССР, ОХН, 1957, 1491.
93. Ко л ее н и ко в Г. С, Д а в ы д о в а С. Л., ЖОХ, 29, 2042(1959).
94. Labarre J.— F., M a z е г о 1 1 е s P., Compt. rend., 254, 3998 (1962).
'95. Миронов В. Ф., Пономаренко В. Я., Взенкова Г. Я.,
Долгий И. Е., П е т р о в А. Д., Химия и практическое применение кремнийорганических
соединений, вып. 1. Л., изд. БЦТИ, 1958, стр. 192.
95а. Наметкин Н. С, Дургарьян С. Г., Тихонова Л. И., ДАН СССР,
72, 615 (1967).
956. ТурсунбаевГ. Л., Манулкин 3. М., ЖОХ, 37, 219 (1967).
96. S е у f e r t h D., В г a n d 1 е К., R a a b G., Angew. Chem., 72, 77 (1960).
97. L e e b r i с k J. R., Rarasden H.E, J. Org. chem., 23, 935 (1958).
98. Noltes J.G., BuddingH. A., van der К e r k G. J. M.,« Rec. Trav. Chem.,
79, 408 (1969).
99. Чернышев Е. А., Петров А. Д., Краснова Т. Л., Сб. «Синтез и
свойства мономеров». М., «Наука», 1964, стр. 103.
100. N о 1 t e s J. G., В u d d i n g H. A., van der К e r k G. J. M., Rec. trav. Chem.,
79, 1076 (1960).
101. П е т р о в А. Д., ЧернышевЕ.А, Краснова Т.Л., ДАН СССР, 140,
837 (1961).
102. Полякова А. М., Коршак В. В., Тамбовцева Е. С, Высоко-
молек. соед., 3, 662 (1961).
102а. Наметкин Н. С, Дургарьян С. Г., Тихонова Л. И., ДАН СССР,
172, 687 (1967).
103. Hartmann H., A h r e n s J. U., Angew. Chem., 70, 75 (1958).
104. К о р ш а к В. В., С л а д к о в А. М., Л у н е в а Л. К., Изв. АН СССР, ОХН,
1962, 2251.
105. Mazerolles P., Bull. Soc. chim. France, 1960, 856.
106. J о n e s E. R. H., ScattebolL, W h i t i n g M. C, J. Org. Chem., 21, 4765
(1956).
107. H e с м е я н о в А. Н., Борисов ДАН СССР, 174, 96 (1967).
46
Германийорганические соединения
108. EabornC, W а 1 t о п D. R. M., Organometal. Chem., 2, 95, (1964).
109. Ста дни чу к М. Д., Петров А. А., ЖОХ, 35, 451 (1965).
ПО. I b e k w e S. D., Newlands M. J., J. Chem. Soc, 1965, 4608.
111. Сл ад ко в A.M., Л у н е в а Л. К., ЖОХ, 36, 553 (1966).
112. С а р а н к и н а С. А., М а н у л к и н 3. М., Узб. хим. ж., 1965, 30; РЖХим.,
1966, 12Ж389.
113. Ш и х и е в И. А., Ш о с т а к о в с к и й М. Ф., Комаров Н. В., А с л а-
н о в И. А., ЖОХ, 29, 1549 (1959).
114. ШихиевИ. А., ШостаковскийМ. Ф.,Комаров Н. В., Асланов И.,
Авт. свид. СССР 117493 (1959); РЖХим., 1960, 39634П; Бюлл. изобрет., №2 (1953).
115. Ю с у ф о в Б. Г., Ш и х и е в И. А., А с л а н о в И. А., Авт. свид. СССР 140426
(1961); РЖХим., 1962, 10Л87; Бюлл. изобрет., № 16 (1961).
116. Ш и х иев И. А., Асланов И. А., Ю с у ф о в Б. Г., ЖОХ, 31,3647(1961).
117. Ш и х и е в И. А., А с л а н о в И. А., Ю с у ф о в Б. Г., ЖОХ, 32, 3148 (1962).
118. Шихиев И. А., Асланов И. А., ЖОХ, 34,397(1964).
119. Шихиев И. А., Г у с е й н з а д е Б. М., МехмандароваН. Т., Асла-
н о в И. А., ЖОХ, 34, 394 (1964).
120. ШихиевИ. А., Асланов И. А., МехмандароваН. Т., ЖОХ, 35, 459
(1965).
121. Ш и х и е в И. А., Гусейнзаде Б. М., А б д у л а е в а Н. Д., Докл. АН
АзербССР, 20, 13 (1964); РЖХим., 1965, 23Ж326.
122. Ш и х и е в И. А., А с л а н о в И. А., ЖОХ, 35, 459 (1965).
123. ШихиевИ. А., Асланов И. А., МехмандароваН. Т., ЖОХ, 36,
1297 (1966).
124. Ш и х и е в И. А., А б д у л л а е в Н. Д., ЖОХ, 35, 1348 (1965).
Глава IV
СИНТЕЗ ГЕРМАНИИОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ МЕТДЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЛИТИЯ, НАТРИЯ, ЦИНКА, РТУТИ, АЛЮМИНИЯ, ОЛОВА
И СВИНЦА
СИНТЕЗ ГЕРМАНИИОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Джонсон с сотр. [1] указывает, что применение литийорганических
соединений в синтезе германииорганических соединений имеет ряд преимуществ
по сравнению с магнииорганическим синтезом; однако эта область развита
очень мало — имеются только отдельные работы.
Шмидт и Руйдиш [2] при алкилировании двухлористого диметилгер-
мания метиллитием (отношение 1 : 1) получили хлористый триметилгер-
маний с выходом 30—35% и побочно тетраметилгерманий в количестве 25%.
При применении йодистого метилмагния выход был равен лишь 25%. Ал-
килирование двухлористого метилхлорметилгермания с помощью метил-
лития привело к получению хлористого диметилхлорметилгермания
наряду с триметилхлорметилгерманием [2а].
Гилман и сотр. [3] при попытке синтезировать тетраэтилгерманий с
помощью этиллития добились лишь 12%-ного выхода. Наряду с этим был
получен гексаэтилдигерман (6—8%) и большое количество неидентифици-
рованных, вероятно, полимерных материалов. В случае я-пропиллития
была выделена смесь только моно-, дву- и треххлористого я-пропилгерма-
ния (выход последнего 16%) [4]. Тетрациклогексил германий с помощью
литийорганических соединений не образуется даже при применении 100%
избытка циклогексиллития, при этом получают только хлористый трицик-
логексилгерманий с выходом 70% [1]. Были получены тетра-2-этилгексил-
германий (27%), тетра-2-циклогексилэтилгерманий (5%), тетра-я-октилгер-
маний (21,3%) и тетра-я-децилгерманий (18%) [5].
Несмеянов с сотр. [6] синтезировал тетра-^с-пропенилгерманий (28%)
и тетра-траяс-пропенилгерманий (28%), исходя из ^с-пропениллития и
соответственно /яраяс-пропениллития и четыреххлористогогермания.
4CH3CH=CHLi + GeCl4 -> (СН3СН=СН)й Ge + 4L1C1.]
Изопропениллитий с четыреххлористым германием также образовывал
тетраизопропенилгерманий с выходом 36%. 1;
Получение тетра-^#с-пропенилгермания [6]. К цш>пропениллитию [7], полученному
из 30 г бромистого ^r/c-пропенила и 3 г лития в 200 мл сухого эфира, медленно прибавляют
7 г четыреххлористого германия в 5 мл эфира при 0—5° С. После 2 час. перемешивания
реакционную смесь обрабатывают 200 мл насыщенного раствора хлористого аммония при
температуре 5—10° С. Эфирный слой отделяют, водный экстрагируют 50 мл эфира. Соединенный
вместе эфирный раствор сушат хлористым кальцием. После отгонки 3/4 объема растворителя
реакционную смесь обрабатывают сухим аммиаком, образовавшийся осадок
отфильтровывают, от фильтрата отгоняют эфир, остаток перегоняют в вакууме.
48
Германийорганические соединения. Лит. стр. 57—59
Тетра-^с-пропенилгерманий получают в количестве 2,16 г (28%);
т. кип: 77,5° — 78,5е С/4 мм; df 1,0347; /iS 1,5040.
Из германийорганических соединений ароматического ряда с хорошими
выходами получены тетрафенилгерманий (выход 90%) [1], тетрафенил-
этинилгерманий (выход 60%) [8], однако выход тетра-2-фурилгермания
(т. пл. 99—100°С) составил всего 32% [9]. Имеется указание и о
возможности получения тетраперфторфенилгермания [10, 11], а также
(С6Н5СеееС)4 Ge [12] и (BrCeH4C=C)4Ge[13].
Получение тетрафенилэтинилгермания (C6H5C=C)4Ge[8l. К 10,8 г фенилацетиленида
лития в 150 мл абс. эфира в атмосфере сухого азота медленно прикапывают 6 г четыреххло-
ристого германия в 75 мл абс. бензола. После нагревания в течение чгса реакционную смесь
фильтруют от выделившихся неорганических солей, фильтрат разлагают раствором
хлористого аммония. После сушки органического слоя над сульфатом натрия растворитель
отгоняют, а полученную зелено-желтую кристаллическую массу несколько раз кристаллизуют
из смеси бензола и петролейного эфира, выход 60%, т. пл. 187—188° С.
В литературе имеется сообщение о получении полнозамещенного
германийорганического соединения с атомами кремния в молекуле
[(CH3)3SiC6H4]4Ge (выход не указан) [14].
Имеется значительное число примеров получения несимметричных
германийорганических соединений с помощью литийорганических соединений.
В ряде случаев применение литийорганических соединений дает ряд
преимуществ перед магнийорганическим синтезом. Чаще всего речь идет о
введении одной алкильнои или арильнои группы, т. е. о получении соединений
типа R3GeR' и реже типа R2GeR'2. Так, пропенилтриметилгерманий был
получен с выходом 75% при реакции пропениллития и хлористого триме-
тилгермания [15].
Получение пропенилтриметилгермания [15]. В трехгорлую колбу емкостью 200 мл,
снабженную магнитной мешалкой, обратным холодильником и капельной воронкой, помещают
70 мл абс. эфира и 1,12 г (0,16 г-атома) мелко нарезанной проволоки металлического лития
(литий содержит 1% натрия). Прибавляют по каплям 10 г (0,083 моля) 1-бромпропена (смесь
изомеров) в 15 мл эфира с таким расчетом, чтобы эфир энергично кипел. К полученному ли-
тийорганическому соединению прибавляют 10,2 г (0,067 моля) хлористого триметилгермания,
реакционную смесь кипятят с обратным холодильником в течение 30 мин., затем гидролизуют
насыщенным раствором хлористого аммония. После разгонки сухого органического слоя
получают 7,9 г(75%) пропенилтриметилгермания; т. кип. 100—103° С.Продукт содержит 70%
цис- и 30% транс- из ом ер а, которые разделяют с помощью газовой хроматографии.
Несмеянов с сотр. [6] из бромистого триэтилгермания и
соответствующих производных пропениллития получил цис- и траяс-изомеры триэтил-
пропенилгермания с выходом 77 и 61 %, а также триэтилпропенилгерманий
(выход 66%).
Наметкин и сотр. [14а] получили триметилвинилгерманий (исходя из
виниллития и йодистого триметилгермания), выход равен 52%.
Аналогично был получен и изопропенилтриметилгерманий [146].
Фенилацетиленид и я-толилацетиленид лития были использованы для
синтеза несимметричных германийорганических соединений (исходя из
хлористого триэтилгермания); выходы составляли соответственно 92 и 84%
[161.
Иборн и Панде [17, 18] получили с выходом 80—90% ряд соединений
формулы XC6H4Ge (С2Н5)3, где X = /-С4Н9, р-С6Н5, o-(CH3)2N, p-(CH3)2N,
2,4,6-(СН3)3С6Н2, о-СН30, т-СН30, р-С6НбО, /?-(CH3)3SiCH2-, p-Br, p-J
(исходя из литийорганических соединений формулы XC6H4Li и хлористого
триэтилгермания). Хлористый трициклогексилгерманий также был
успешно применен для синтеза соединений формулы XC6H4Ge (C6Hn)3 [17].
Интересно отметить получение несимметричных германийорганических
соединений, имеющих два атома германия в молекуле [18а], с помощью литийор-
Синтез с помощью металлоорганических соединений
49
ганических соединений по реакции
BrC6H4Ge (С2Н5)з + Li -> LiC6H4Ge (C2H5)3,
(С2Н5)з GeCl + LiC6H4Ge (С2Н5)з -> (QHsb GeC6H4Ge (C2H5)3.
В синтезе несимметричных германийорганических соединений
использовался галоидный трифенилгерманий. Можно упомянуть о его реакции с
октиллитием [19], с аллиллитием [20] и рядом литийорганических
соединений— производных бензола [21, 22].
Получение /г-диметиламинофенилтрифенилгермания [21]. Приготовляют раствор /г-ди-
метиламинофениллития из 15 г (0,075 моля) /г-бромдиметиланилина и 14 г (10,2-г-атома)
лития в 100 мл эфира (атмосфера азота, кипячение 1/2 часа) и прибавляют 24,2 г (0,063 моля)
бромистого трифенилгермания, растворенного в бензоле. Реакционную смесь кипятят в
течение 2 час, эфир отгоняют и заменяют бензолом. Полученный раствор кипятят с обратным
холодильником в течение ночи и осторожно разлагают водой. Органический слой отделяют,
после отгонки бензола получают/г-диметиламинофенилтрифенилгерманий (79%),т. пл. 140—
141° С.
Действие литий- или дилитийорганических соединений на дигалоидные
производные алкил- или арилгерманийорганических соединений также
приводит к получению полнозамещенных несиммметричных
германийорганических соединений.
При действии фениллития на двухлористый диклопентаметиленгерма-
ний был получен циклопентаметилендифенилгерманий, т. кип. 125—
128° С/0,08 мм, п2о 1,5873 [23], а синтез пятичленных гетероциклических
соединений, содержащих германий в качестве гетероатома, был осуществлен
при действии дилитийтетрафенилбутадиена на соединение типаR2GeX2 [24].
Гилман и сотр. [25] описывают реакцию N-этилдилитийдифениламина с
двухлористым дифенилгерманием и четыреххлористым германием.
Получение 5-этил-5,10-дигидро-10,10-дифенилфеназагермина [25]
2п-С4Н9Ы
QH5—N
У
К раствору 1М-этил-2,2'-дилитийдифениламина, приготовленного из 5,33 г (0,015 моля)
1Ч-этил-2,2'-дибромдифениламина и 0,03 моля я-бутиллития в 50 мл эфира, прибавляют
4,46 г (0,015 моля) двухлористого дифенилгермания в 50 мл эфира, реакционную смесь
кипятят с обратным холодильником 16 час, затем добавляют 50 мл сухого толуола, эфир
отгоняют и толуольный раствор кипятят 2 часа с обратным холодильником. Реакционную
смесь гидролизуют 50 мл воды, органический слой отделяют и сушат сернокислым
натрием, растворитель отгоняют в вакууме без нагревания. Остаток перекристаллизовывают
дважды из петролейного эфира (т. кип. 60—75е С), получают 3,47 г (52,4%) бесцветных
кристаллов, т. пл. 120—121° С.
Получение 5,5'-диэтил-10, Ю'-спиро-бйС-(5, Ю-дигидрофеназогермина) [25]
QH5
50
Германийорганические соединения. Лит. стр. 57—59
К раствору 0,03 моля М-этил-2,2'-дилитийдифениламина в 150 мл эфира прибавляют
3,21 г (0,015 моля) четыреххлористого германия в 50 мл эфира; реакционную смесь кипятят
с обратным холодильником в течение 16 час. Затем добавляют 100 ms сухого толуола, эфир
отгоняют и толуольную суспензию нагревают в течение 4 час. Iiocjie гидролиза
добавляют эфир, органический слой отделяют и сушат. Растворитель отгоняют в вакууме без
нагревания, и остаток кристаллизуют из этилацетата. Получают 1,76 г бесцветного осадка,
т. пл. 205—211° С. После перекристаллизации из этилацетата получают 1,26 г (18,1%)
бесцветных игл, т. пл. 212—214° С.
Петров и сотр. [26] действовали литийорганическими соединениями ди-
фенила и я-дифенилметана на двухлористый диметилгерманий.
Описано [27] взаимодействие метилития с гексаметилдигерманоксаном,
приводящее к образованию тетраметилгермания.
В отдельных случаях применяют не литийорганические соединения, а
проводят реакцию между галоидными алкилами и галогенидами германий-
органических соединений в присутствии металлического лития (по типу
реакции Вюрца) [28, 29].
Несимметричные гексаалкилдигерманы были получены с выходом от 7
до 35% при реакции четыреххлористого германия со смесью двух ал кил
литиевых соединений [30].
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ НАТРИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
При нагревании галоидных алкилов или арилов с четыреххлористым
германием в присутствии металлического натрия, вероятно, через стадию
промежуточного образования натрийорганических соединений
получаются полнозамещенные германийорганические соединения
4RX + GeCU + 8Na -» R4Ge + 4NaCl + 4NaX.
По этому методу были синтезированы тетраэтил-, тетрапропил-, тетраизо-
амил-, тетрафенил- и тетра-я-толилгерманий [31], а также тетра-Р-стирил-
германий (исходя из Р-бромстирола). Однако выход продуктов достигает
лишь 17,5% [8]. Метод широкого развития не получил.
Позже натрийорганические соединения применялись для получения
несимметричных германийорганических соединений, а также соединений,
имеющих два атома металла в молекуле.
Как правило, реакцию проводят без специального выделения
натрийорганических соединений, хотя в отдельных случаях к этому и прибегают.
Так, а-стирилтриметилгерманий был синтезирован при нагревании
йодистого триметилгермания и а-бромстирола с металлическим натрием [146].
Получение а-стирилтриметилгермания [146]. К 5,4г (0,235 г-атома) мелко
диспергированного натрия в 90 мл эфира приливают раствор 22,75 г (0,0932 моля) йодистого
триметилгермания в 10 мл эфира и спустя 15 мин. медленно прикапывают в течение часа раствор 15,68 г
(0,08 молей) а-бромстирола в 10 мл эфира. Температура не должна повышаться выше 45° С.
После окончания кипения эфира реакционную смесь перемешивают 5 час. при комнатной
температуре, фильтруют, промывая остаток на фильтре эфиром. К эфирному слою
добавляют 25 мл метанола и 0,8 г гидрохинона. Спустя 30 мин. растворитель отгоняют, а остаток
перегоняют в вакууме. Получают 8,2 г а-стирилтриметилгермания. Выход 36%. Попытка
получать триметилвинилгерманий (исходя из йодистого триметилгермания, бромистого
винила и металлического натрия) дала выход конечного продукта, равный лишь 27% [14а].
Несимметричные германийорганические соединения синтезированы при
нагревании бромистого триэтилгерманйя с хлористыми арилами формулы
XArCl, где X = о-СН, (выход 74%), т-СН3 (63%), р-С2Н5 (65%), о-С6Н4
(55%) [17].
Синтез с помощью металлоорганшеских соединений
51
Получение арилтриэтилгерманиев [17]. Смесь хлористого арила (0,05 моля) и бромистого
триэтилгермания (0,05 моля) прибавляют осторожно к энергично перемешиваемой
суспензии металлического натрия (0,11 г-атома) в кипящем толуоле (ПО мл). После кипячения
в течение часа реакционную смесь фильтруют и фильтрат подвергают фракционированной
перегонке.
Из 1- и 2-бромнафталина аналогично были приготовлены триэтил-1-
нафтилгерманий (25%, т. кип. 153° С/4 мм) и триэтил-2-нафтилгерманий
(64%, т. кип. 151° С/3—4 мм).
С помощью ацетиленида натрия был осуществлен синтез ряда ацетиле-
нидов германийорганических соединений [31а]. Так, был получен тетра-
этинилгерманий (34%), дифенилдиэтинилгерманий (56%), ди-я-бутилдиэти-
нилгерманий (57%) и трифенилэтинилгерманий (13,7%).
Получение ди-#-бутилдиэтинилгермания [31а]. Суспензию из 17 г (350 ммолей)
свежеприготовленного моноацетил ени да натрия в 150 мл сухого тетрагидрофурана помещают в
трехгорлую колбу (500 мл)^соединенную с обратным холодильником, капельной воронкой и
магнитной мешалкой, и добавляют 40 г (155 ммолей) двухлористого дибутилгермания. 1/3
двухлористого дибутилгермания прибавляют сразу при энергичном перемешивании, кипятят
реакционную смесь 15 мин; затем оставшийся двухлористый дибутилгерманий прибавляют
в течение 20 мин. Бесцветный раствор оставляют стоять в течение 20 мин. Раствор за это
время становится красно-коричневым. Когда температура реакционной смеси достигнет
комнатной, ее фильтруют, а фильтрат удаляют в вакууме. Остаток растворяют в 500 мл гексана,
затем фильтруют, растворитель опять удаляют. Оставшуюся жидкость перегоняют в вакууме.
Выход 21 г (57%); т. кип. 55° С/0,5 мм.
Мазеролл и сотр. [316] упоминают без приведения эксперимента о
получении соответствующих этинильных производных германия при действии
ацетиленида натрия на со-бромпроизводные германия в присутствии гек-
саметилфосфотр иамида.
ГМФТ
(QHsb Ge (CH2)4 Br + HC~CNa > (QHsb Ge (CH2)4 C=CH+ NaBr.
Выход достигает 60%, но продукт содержит небольшое количество алле-
нового изомера.
При действии на бромистый трифенилгерманий в тетрагидрофуране ди-
натрийдиацетиленида получают (C6H5)3Ge—CeeeC—С=е С—Ge(C6H5)3;
т. пл. 294° С [32]. Реакция с ацетиленидом натрия приводит к соединению
(C6H5)3GeC=CH [33].
Гилман и сотр. [34, 35] исследовали реакцию бромистого трифенилгер-
мания с натриевыми производными дифенил- и трифенилметанового ряда
и получили соответствующие германийорганические соединения.
Бромистый трифенилгерманий реагирует с динатриевым производным
ферроцена в кипящем пентане с образованием трифенилгермилферроцена
(10%) Fe(C5H5)[C5H4Ge(C6H5)3] и 1,Г-^^-(трифенилгермил)ферроцена
Fe[C6H4Ge(C6H5)3]2 [36].
Циклические дигерманы получают при действии металлического натрия
на бромистый фенилциклотетраметиленгерманий, выход 80% [37].
СН2-СН2 С6Н5 СН2-СН2 СН2—СН2
ч /
Ge +2 Na«
/ \
Ge-—Ge 4-2NaBr.
/I I \ I
CH3—CH2 Br iH2—CH3 СбНб СвН5СН2—CHa
Дибутилциклотриметиленгерманий был получен с выходом 75% при
действии металлического натрия в кипящем толуоле на хлористый
дибутил хлорпропил германий [38]
СН2
/ \
(С4Н9)2 Ge (C1) СзН6С1 + 2Na -* (C4H9)2Ge CH2 + 2NaCl.
СН2
4*
52 Германийорганичсские соединения. Лит. стр. 57—59
Соответствующее этильное производное было получено лишь с выходом
35%. Дибутилциклотриметиленгерманий был получен также при
взаимодействии металлического натрия с эквимолекулярной смесью двухлористо-
го дибутилгермания и 1,3-дихлорпропана в ксилоле [38].
сн2
/ \
(С4Н9)2 GeCb + 4Na + С1СН2СН2СН2С1 -* (QH9)2 Ge CH2 + 4NaCl.
CH2
Авторы исследовали устойчивость кольца и нашли, что по сравнению с
5- и 6-членными циклами полученные соединения менее устойчивы и легко
раскрывают циклы при действии брома, литийалюминийгидрида и галоидо-
водородных кислот.
Метод получения германийорганических соединений с помощью натрий-
органических соединений дает возможность получить и металлоорганиче-
ские соединения, имеющие два атома металла в молекуле, причем связь
между металлами может быть или непосредственная М—М или через тот
или иной углеродный мостик [17, 18, 18а]. Так, при действии на
бромистый триэтилгерманий BrC6H4OSi(CH3)3 в присутствии металлического
натрия образуется (CH3)3SiOC6H4Ge (C2H5)3; выход 42% [16]. Аналогично
получают триметил-4-триметилсилилбензилгерманий (CH3)3SiC6H4CH2Ge(CH3)3
с выходом 80% [39].
Миронов и сотр. [28, 40, 41] при нагревании с металлическим натрием
галоидопроизводных германийорганических соединений или галоидопро-
изводных германий- и кремнийорганических соединений получили
соединения, имеющие или два атома германия, или атомы германия и кремния в
молекуле.
При действии на Р-хлорвинилтриметилгерманий хлористого триметил-
германия в присутствии металлического натрия реакция идет по схеме
Nt
(СНз)з GeCH=CHCl + CIGe (СН3)з ► (СН3)3 GeCH=CHGe (CH3)3.
Аналогичная реакция проведена с ClSi(CH3)3 [3]. При реакции а-хлорви-
нилтриметилгермания с (CH3)3GeCl образуется 1,1-бас-(триметилгермил)-
этилен [40]
Ge(CH3)3
СН2=С
Ge(CH3)3
Получение триметилгермил-2-триметилсилилэтилена [28]
(СН3)3 GeCl + (СН3)з SiCH=CHCl —- (СН3)3 SiCH=CHGe (CH3)3.
9 г натрия расплавляют в 60 мл кипящего толуола и с помощью быстро вращающейся
мешалки мелко дробят. После охлаждения толуол сливают и заменяют 250 мл эфира. Затем
.добавляют 25 & хлористого три метил германия, 2 мл (CH3)3SiCH =СНС1 и через несколько
^яинут — 0,5 мл этйлацетата. Начинается реакция с равномерным кипением эфира, которую
поддерживают добавление** остальных 25 г (CH3)3SiCH=CHCl в течение 2 час. Содержимое
колбы оставляют на ночь и отфильтровывают от хлористого натрия, промывая свежим
эфиром. После отгонки эфира остаток разгоняют на колонке. Получают 16 г три метил гер мил-2-
тримегилсилилэтилена; выход 56%; т. кип. 158° С/760 мм; df 0,9557; /z^l,4473. Кроме этого
выделяют 10 г гексаметилдигермана.
Синтез с помощью металлоорганических соединений
53
Аналогично получают триметилгермил-1-триметилсилилэтилен с
выходом 61,5%.
(CH3)3Ge
\
С=СН2.
(CH3)3Si
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ЦИНКОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Впервые тетраэтилгерманий был получен через цинкорганические
соединения Винклером [42] при действии диэтилцинка на четыреххлористый
германий
2(С2Н5)2 Zn + GeCl4 -* (С2Н5)4 Ge + 2ZnCl2.
В дальнейшем Деннис с сотр. [43] повторил эту реакцию и нашел, что
если в качестве катализатора прибавить небольшое количество хлористого
цинка и тетраэтилгермания, реакция протекает более ровно и дает хорошие
выходы.
Метод вряд ли имеет большое практическое значение в связи с
необходимостью предварительного получения индивидуальных алифатических
цинкорганических соединений, однако, например, синтез, малых количеств
особо чистого тетраметилгермания с помощью диметилцинка [44] является
удобным.
Больший интерес представляет действие растворов цинкорганических
соединений на галоидные соли четырехвалентного германия. Этот метод
дал возможность получить ряд полнозамещенных ароматических
соединений германия с лучшим выходом, чем по гриньяровскому синтезу [45, 46].
Получение тетрафенилгермания [46]. К реактиву Гриньяра (из 14>2 г магния, 76,4 г
бромбензола в 200 мл эфира) прибавляют 33,2 г безводного хлористого цинка, растворенного
в 100 мл эфира. Реакционную смесь при перемешивании охлаждают в течение часа и затем
нагревают 1 час на водяной бане, прибавляют 100 мл сухого толуола, затем отгоняют эфир.
Оставшуюся смесь охлаждают до 0° С и при энергичном перемешивании прибавляют 14,9 г
четырехбромистого германия, растворенного в толуоле, после чего нагревают на масляной
бане в течение 16 час.(в атмосфере азота). Реакционную смесь охлаждают и разлагают 200 мл
2N серной кислоты. Толуольный слой отделяют, высушивают, толуол отгоняют в вакууме,
полученный продукт кристаллизуют из бензола; т. пл. 235° С; выход 95,4 %.
Аналогичным образом получают тетра-я-толилгерманий (65%), тетра-ж-
толилгерманий (75%) и тетра-о-толилгерманий (30%) [45].
Растворы цинкорганических соединений были применены и для
получения несимметричных германийорганических соединений. При действии
раствора хлористого n-толилцинка на бромистый три-ж-толилгерманий был
получен три-ж-толил-я-толилгерманий и аналогично три-л-толил-о-толил-
германий [47]. В последнее время появилась американская патентная
работа по получению винилфенильных соединений германия с помощью
растворов цинкорганических соединений [48].
О реакции цинкорганических соединений с солями двухвалентного
германия в литературе имеется лишь одна работа Джекобса [49], который,
действуя на двуиодистый германий ди-я-бутил- и диизобутилцинком,
выделил германийорганические соединения типа R2GeJ2, в частности,
двуиодистый ди-я-бутил германий.
54 Германийорганические соединения. Лит. стр. 57—59
СИНТЕЗ ГЕРМАНИИОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Действие ароматических ртутноорганических соединений на соли
четырехвалентного германия приводит к образованию германииорганических
соединений низшей степени арилирования и может быть представлено
реакцией
(СюН7)2 Hg + GeCl4 -> CioH7GeCl3 + Ci0H7HgCl.
Так был получен треххлористый а-нафтилгерманий [50], который
выделялся не непосредственно, а действием едкого натра переводился в ангидрид
а-нафтилгерманиевой кислоты.
Аналогично выделены и ангидриды фенил-, п-толш- и бензил
германиевой кислот [51], а также треххлористый винил германий [52].
В литературе имеется указание о получении трехбромистого перфтор-
фенилгермания при действии четырехбромистого германия на метилпер-
фторфенилртуть [53].
Использование солей двухвалентного германия, в частности, в реакции
с алифатическими ртутноорганическими соединениями не дало достаточно
удовлетворительных результатов [48].
Макаровой [54] и Макаровой и Несмеяновым с сотр. [55] показано, что
кипячение в течение 15 мин. эквимолекулярных количеств диарилртути и
двуиодистого германия в толуоле дает с высокими выходами диарилиро-
ванные германийорганические соединения с примесью три- и моноарилиро-
ванных продуктов. Авторы полагают, что в основном имеет место
следующий процесс:
Ar2Hg + GeJ3 -> Ar2GeJ3 + Hg.
Ar3GeJ образуется далее вследствие обмена
Ar2Hg + Ar2GeJ2 -► Ar3G3j + ArHgJ,
a ArGeJ3 получается в очень незначительных количествах по реакции
ArHgJ + G3J2 -* ArGeJ3 + Hg.
Реакция была проведена на примере взаимодействия .двуиодистого
германия с дифенил-, ди-о-толил-, ди-ж-толил-, ди-я-толил-, ди-о-хлорфенил-,
ди-^-хлорфенил-, ди-п-хлорфенил-, ди-о-бромфенил-, ди-л-бромфенил-, ди-
д-анизил-, ди-о-фенетил-, ди-я-фенетил- и ди-|3-нафтилртутью. Выходы
диарилированных замещенных германия от 40 до 70%.
Попытка синтезировать полнозамещенные германийорганические
соединения длительным кипячением в ксилолеиодистоготри-л*-толилгермания
и ди-^-тол ил ртути оказалась неудачной.
. Взаимодействие ди-/г-толилртути с двуиодистым германием [55]. 3,3 г (0,008 моля)
(p-CH3C6H4)2Hg и 3,3 г (0,01 моля) GeJ2 в 30 мл сухого толуола кипятят в течение 15 мин.
В образовавшемся зеленовато-сером осадке, промытом 2 раза толуолом, обнаруживают
металлическую ртуть, Hg2J2» HgJ и непрореагировавший GeJ2. От толуольного фильтрата
растворитель отгоняют в небольшом вакууме, к остатку приливают 20 мл спирта и 15 мл 15%-ного
раствора едкого натра. Смесь кипятят на водяной бане с одновременной отгонкой спирта
около часа. Остаток разбавляют водой, образовавшиеся желтоватые кристаллы тщательно отмы
вают водой, а затем растворяют в бензоле (полное растворение). Из бензольного раствора
получают 1,72 г (79% от теорет.) бесцветных игольчатых кристаллов; т. пл. 220—223° С.
После кристаллизации из смеси петролейного эфира с бензолом выделяют кристаллы
Kp-CHgQH^GeOla с т. пл. 221,5—222,5° С, а из щелочного фильтрата при подкислении
получают 0,21 г (p-CHgCe^GeO^O в виде слегка желтоватого порошка; выход 33% от теорет.
Большой интерес в области синтеза германииорганических соединений,
а также соответствующих производных олова и кремния через ртутноорга-
Синтез с помощью металлоорганических соединений- - 55
нические соединения представляют работы Луценко с сотр. [56—59].
Авторы предложили новый метод получения сложных эфиров, содержащих
атомы германия, олова или кремния в а-положении, исходя из а-меркури-
рованных сложных эфиров и элементоорганических производных Si, Ge и
Sn. Наиболее удобными для получения германийорганических сложных
эфиров оказались соединения типа R3GeJ (гидриды триалкилгермания
взаимодействуют с эфирами меркур-бш>уксусной кислоты менее энергично, чем
соответствующие оловоорганические] гидриды, и выходы при этом ниже).
2R3GeJ+ Hg CH2C -+ R3GeCH2C + HgJ2.
у° v
\
OR'-
\ /°
-> RsGeCH2C
/ \
'% OR'
При добавлении йодистого триалкилгермания к суспензии эфира мер-
кур-бас-уксусной кислоты в инертном растворителе реакционная смесь
разогревается и появляется красный осадок йодной ртути. В препаративных
целях удобнее проводить реакцию, добавляя йодистый триалкилгерманий
к раствору ртутного эфира и пиридина (взятых в соотношении 1 : 2) в
инертном растворителе. При этом комплекс йодной ртути с пиридином HgJ2-
•2C5H5N выпадает в осадок и может быть легко отделен фильтрованием.
Германийорганические сложные эфиры выделяют из фильтрата разгонкой.
Авторы получили ряд германийорганических сложных эфиров формулы
R3GeCH2COOR', где R = С2Н5, /г-С3Н7 и /г-С4Н9; R' - СН3, С2Н5,
я-С3Н7.
Взаимодействие йодистого трипропилгермания с метиловым эфиром меркур-б"дс-уксус-
ной кислоты [58]. К раствору 17,5 г метилового эфира меркур-б«£-уксусной кислоты и 6 мл
пиридина в петролейном эфире прибавляют по каплям 33 г йодистого трипропилгермания,
кипятят 30 мин. и фильтруют. При разгонке получают 19,5 г метилового эфира трипропил-
гермилуксусной кислоты с т. кип. 81,5—84° С/1,5 мм\ d|° 1,0518; п2£ 1,4580; выход 71%.
В дальнейшем исследовалось действие йодистого трипропилгермания
на иодмеркурацетон и было показано, что реакция с галогенидами алкил-
германия протекала без переноса реакционного центра — происходило
образование только продуктов со связью Ge—С [59]
JHgCH2COCH8 + (C3H7)3GeJ -► (С3Н7)3 GeCHaCOCH3 + HgJ2.
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛЮМИНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Германийорганические соединения с помощью алюминийорганических
соединений получены только на отдельных примерах, но метод, возможно,
имеет известные перспективы для своего развития по крайней мере в
алифатическом ряду.
В английском патенте [60] впервые упоминается о получении тетра-
этилгермания действием четыреххлористого германия на триэтилалюминий
в присутствии хлористого натрия; выход 88,7%. Реакция протекает по
уравнению
4(С3НБ)зА1 + 3GeCl4-* 3(C2H5)4Ge + 4А1С13.
Получение тетраэтилгермания [61]. К 18,0; г триэтилалюминия добавляют при
перемешивании в атмосфере азота 25,6 г свежеперегнанного над ртутью четыреххллористого
германия с такой скоростью, чтобы температура смеси поддерживалась на уровне 80—90° С.
По окончании прибавления реакционную смесь нагревают в течение б час. при 120—130° С,
разбавляют эфиром и разлагают избытком 20%-ного едкого натра. Эфирный слой промывают
разбавленной уксусной кислотой и сушат хлористым кальцием. Выход 15,9 г (72,9%).
Аналогично получают тетраизобутилгерманий.
56
Германийорганические соединения. Лит. стр. 57— 59
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ОЛОВО- И СВИНЦОВ00РГАНИ4ЕСКИХ СОЕДИНЕНИЙ
Эфиры карбоновых кислот, содержащие атом германия в а-положении к
эфирной группе, как уже описывалось ранее, могут быть получены диазо-
методом (действие гидридов ал кил германия на диазоуксусный эфир, см.
стр. 75) или реакцией йодистого алкилгерманияс эфирами меркур-бис-ук-
сусной кислоты (см. стр. 55).
' Луценкоисотр. [62,63] изучили возможность использования в реакции
с галогенидами германия и алкилгермания легко доступных эфиров триал-
килстаннилуксусной кислоты [64]. Оказалось, что эфиры триалкилстан-
нилуксусной кислоты реагируют с галогенидами германия, причем в
некоторых случаях можно с успехом испбльзовать и хлориды — самые
доступные галогениды германия, которые, как правило, с эфирами меркур-
бш>уксусной кислоты не реагируют.
В зависимости от соотношения исходных реагентов можно заменить
в четыреххлористом германии один, два, три или все четыре атома хлора на
органический радикал
1:1
>Cl3GeCH2COOCH3
■f R3SnCH2COOCH3
GeCl4 —
-R3SnX
— Cl2Ge (СНзСООСНзЬ
-J— CiGe (CH2COOCH3)3
1 :4
>Ge(CH2COOCH3)4
Выходы эфиров гермилуксусной кислоты составляют 75—85%. Также
легко реагирует с эфирами триалкилстаннилуксусной кислоты и хлористый
триалкоксигермаций (хорошие выходы).
+ (C4H,)8SnCH2COOCH3
(CH30)3GeCl ,- » ч с Г| *(CH30)3GeCH2COOCH3.
Что касается хлористого триалкилгермания, то он оказался
значительно менее реакционноспособным. После нагревания в течение 4 час. на
кипящей водяной бане хлористого триэтилгермания с метиловым эфиром
трибутилстаннилуксусной кислоты было выделено только 27% метилового
эфира триэтилгермилуксусной кислоты. Хороший выход был получен при
использовании более активного йодистого триэтилгермания. При
взаимодействии йодистого триэтилгермания с трибутилстанн ил ацетоном с
выходом 92% получен триэтилгермилацетон [58]; т. кип. 85,5—87° С/6,5 мм.
Реакция четыреххлористого германия с метиловым эфиром триалкилстаннилуксусной
кислоты [.62, 63].
а) Получение метилового эфира трихлоргермилуксусной кислоты. 101,6 а метилового
эфира трибутилстаннилуксусной кислоты прибавляют по каплям к 80 г четыреххлористого
германия (соотношение 1:1), реационную смесь нагревают 4 часа при 60° С. После отгонки
избытка четыреххлористого германия остаток фракционируют. После повторной перегонки
фракции (88—125° С/9 мм) получают 46,1 г метилового эфира трихлоргермилуксусной
кислоты; т. кип. 70—71° С/6,5 мм; выход 87% на прореагировавший
четыреххлористый-германий.
б) Получение метилового эфира дихлоргермил-бис-уксусной кислоты. 11,1 г
четыреххлористого германия и 29 г метилового эфира триэтилстаннияуксусной кислоты (соотношение
1:2} нагревают на кипящей водяной бане 5 час. После фракционирования получают 25 г
(99%) хлористого триэтилолова и 11,2 г (75% от теорет.) метилового эфира дихлоргермил-
бис-уксусной кислоты; т. кип. 106—110° С/2 мм; df 1,5430; п2£ 1, 4870.
в) Получение метилового эфира хлоргермилтриуксусной кислоты. 11 г четыреххлористого
германия и 70 г метилового эфира триэтилстаннилуксусной кислоты (соотношение 1:3)
нагревают 8 час. при 130° С. При фракционированной разгонке получают 59 г хлористого три-
Синтез с помощью металлоорганических соединений
57
атилолова (98%) и 21,2 г метилового эфира хлоргермилтриуксусной кислоты; т. кип. 131 —
133° С/1 лш (77%).
г) Получение метилового эфира гермилтетрауксусной кислоты. Смесь 20 г четыреххло-
ристого германия и 146 г метилового эфира триэтилстаннилуксусной кислоты (соотношение
1:4) нагревают 2 часа на кипящей водяной бане и 6 час. при 130° С. При фракционированной
перегонке получают 25 г метилового эфира гермилтетрауксусной кислоты; т. кип. 125—
12б°С/0,045 мм. Выход 77% в расчете на вступивший в реакцию четырех хлор истый германий.
Реакция йодистого триэтилгермания с метиловым эфиром трибутилстаннилуксусной
кислоты [62, 63]. 10,2 г метилового эфира трибутилстаннилуксусной кислоты и 7,5 г йодистого
триэтилгермания нагревают в течение 2 час. на кипящей водяной бане. При фракционировании
получают5,1 г(85%)кметилового эфира триэтилгермилуксусной кислоты; т. кип. 80—82°С/7лш.
Реакция хлористого триметоксигермания с метиловым эфиром трибутилстаннилуксусной
кислоты [62, 63]. 12 г метилового эфира трибутилстаннилуксусной кислоты и 5,4 г
хлористого , триметоксигермания нагревают 5 час. на кипящей водяной бане и 2,5 часа при
130—140°С. При фракционировании выделяют 4,3 г (67%) метилового эфира триметокси-
гермилуксусной кислоты; т. кип. 61—62° С/1 мм\ df 1,3596; п2£ 1,4400.
Реакция йодистого триэтилгермания с трибутилстаннилацетоном [59]. 20 г трибутил-
станнилацетона и 12 г йодистого триэтилгермания нагревают 2 часа при 50° С.
Фракционированной перегонкой выделяют 8,4 г триэтилгермилацетона; т. кип. 85,5—87° С/6,5 мм
(92%).
Различие скоростей реакции деалкилирования оловоорганических и
германийорганических соединений было использовано для получения с
количественными выходами до этого времени трудно доступных C4H9GeCl3.
Было найдено [65], что реакция тетрабутилолова с четыреххлористым
германием и тетрабутилгермания с четыреххлористым оловом при комнатной
температуре не идет, при 110° С малозаметна, при 210° С в обеих системах
(одна бутильная группа обменивается с атомом хлора) — согласно
уравнениям
(С4Н9)4 Sn + GeCU -> (QHgb SnCl + C4H9GeCl3 (*)
И
(QH9)4 Ge + SnCU -»(QH9) SnCl3 + (C4H9)3 GeCl. (2)
Реакция (1) заканчивается через час (выход количественный). Реакция
(2) требует более продолжительного времени для своего завершения (6
час. в запаянном сосуде). Выход треххлористого бутилолова по уравнению
(2) составил 92%. Было установлено, что обе реакции не имеют тенденции
к дальнейшему протеканию. Получить более, юдного моля соединения
C4H9GfeCl^^H.QH9SnCl3) из каждого моля (С4Н9)4М (применение избытка
МС14) не удалось.
Методом ядерного магнитного резонанса была исследована кинетика
реакции' обмена радикалов между тетраметилоловом и четыреххлористым
германием [66].
Миронов и сотр. [67] показали, что свинцовоорганические соединения
реагируют аналогично оловоорганическим соединениям с
четыреххлористым германием. Треххлористый этилгерманий образуется с выходом 90%
по реакции
(С2Н5)4 Pb + GeCU -» (C2H5)3 PbCl + C2H5GeCl3.
ЛИТЕРАТУРА
1. JohnsonO.H., N е b е г g a 1 1 W. H., J. Am. Chem. Soc, 71, 1720 (1949).
2. Schmidt M., R ui d isch J., Z. anorg. allg. Chem., 311, 331 (1961).
2a. W i e b e г М., FrohningCD., Z. Naturforsch., 21b, 492 (1966).
3. GilmanH., Hughes M. В., G e г о w С. W., J. Org. Chem., 24, 352 (1959).
4. JohnsonO.H., JonesL, J. Org. Chem., 17, 1172 (1952).
5. FuchsR., Moore L,, Miles D., GilmanH., J. • Org. Chem., 21, 1113
(1956).
58
Германийорганшеские соединения
6. Несмеянов А.Н., Б.о р и с о в А. Е., Но в и к о в а Н. В., ДАН СССР, 165,
333 (1965).
7. Несмеянов А. Н., Борисов А. Е., Новикова Н. В., Изв. АН СССР,
ОХН, 1959, 1216.
8. В i г г К. Н., Kraft D., Z. anorg. allg. Chem., 311, 235 (1961).
9. G i 1 m a n H., Leeper R.W., J. Org. Chem., 16, 466 (1951).
10. FentonD.E., MasseyA.G., Chem. and Ind., 1964, 2100.
11. TamborskiC, S о 1 о s k i J., Dec S. M., J. Organometal. Chem., 4, 446 (1965).
12. HartmannH., Meyer K., Naturwissenschaften, 52, 303 (1965).
13. HartmannH., AlssarM. E., Naturwissenschaften, 52, 304 (1965).
14. MeenR.E, GilmanH., J. Org. Chem., 22, 564 (1957).
14a. Н'аметкинН. С, ДургарьянСГ., Тихонова Л.И., ДАН СССР,
172, 615 (1967).
146. Наметкин Н. С, ДургарьянСГ., Тихонова Л. И., ДАН СССР,
172, 867 (1967).
15. S (у f e r t h D., Vaughan L.G., J. Organometal. Chem., 1, 138 (1963).
16. EabornC, WaltonD.R. M.,J. Organometal. Chem., 2, 95 (1964).
17. EabornC, Pan de K.C, J. Chem. Soc, 1960, 3200.
18. EabornC, Leyshon K.( Pande K.C, J. Chem. Soc, 1960, 3423.
18a. L e n s i n k A. J., N о 1 t e s J. G., В u d d i n g H. A., V a n d e r KerkG.J.M.,
Recuel trav. chim., 83, 844 (1964).
19. FuchsR., GilmanH., J. Org. Chem., 22, 1009 (1957).
20. S e у f e r t h D., Weiner M. A., J. Org. Chem., 24, 1395 (1959).
21. В en keser R. A., De BoerC. E., Robinson R. E„ SanveD.M.,
J. Am. Chem. Soc, 78, 682 (1956).
22. О и к а в a X., J. Chem. Soc Japan, Pure Chem. Sec, 84, 272 (1963); РЖХим., 1964,
7Ж305.
23. В a j e r F. J., P о s t H. W., J. Org. Chem., 27, 1422 (1962).
24. L e a v i t t F. C, ManuelT.A., JahnsonF., J. Am. Chem. Soc, 81, 3163
(1959).
25. GilmanH., Z u e с h E. A., J. Am. Chem. Soc, 82, 2522 (1960).
26. П е т р о в А. Д., Ч е л ь ц о в а М. А., К о м а р о в а С. Д., Изв. АН СССР,
серия хим., 1965, 550.
27. R u i d i s с h J., SchmidtM., Ber., 96, 821 (1963).
28. МироновВ.Ф., К р а в ч е н к о А. А., Изв. АН СССР, серия хим.; 1965, 1026.
29. Н е ф е д о в О. М., М а н а к о в М. Н., П е т р о в А. Д., ДАН СССР, 147, 1376
(1962). .
30. S e m 1 у е n J. A., W а 1 k e r G. R., Phillips С. S. G.t J. Chem. Soc, 1965.
1197.
31. T a b e r n D. L., О r n d о r f f W. К., D e n n i s L. M., J. Am. Chem. Soc, 47, 2039
(1925).
31a. W e 1 z e 1 D., M a I к о 1 m С H., J. Organometal. Chem., 5, 29 (1966).
316. MazerollesP,, LesbreM., MarreS., Compt. rend., 261, 4134 (1965).
32. H a r t m a n n H., Dietz E., Komorn iezytk K., R e i ss W.,
Naturwissenschaften, 48, 570 (1961).
33. HartmannH., В e e r m a n n С Пат. ФРГ 1062244 (1960); РЖХим., 1961,
24Л101.
34. В г о о к A. G., GilmanH., J. Am. Chem. Soc, 76, 77 (1954).
35. GilmanH., GerowC.W, J. Am. Chem. Soc, 79, 342 (1957).
36. S e у f e r t h D., H о f m a n n H. P., BurtonR., H e 1 1 i n g J. F., Inorg,
Chem., 1, 227 (1962).
37. MazerollesP., D u b а с J., Compt. rend., 257, 1103 (1963).
38. M a z e г о 1 1 e s P., LesbreM., D u b а с J., Compt. rend., 260, 2255 (1965).
39. В о t t R. W., EabornC, PandeK.C, Swaddle T.W., J. Chem. Soc,
1962, 1217.
40. M и р о н о в В. Ф., К р а в ч е н к о А. Л., П е т р о в А. Д., ДАН СССР, 155,
843 (1964).
41. МироновВ.Ф., Д ж у р и н с к а я Н. Г., Изв. АН СССР, ОХН, 1963, 75.
42. W i n к 1 е г С, J. prakt. Chem. [2], 36, 177 (1887).
43. D e n n i s L. M., HanceF.E., J. Am. Chem. Soc, 47, 370, (1925).
44. L e n g e 1 J. H., DibelerV.H., J. Am. Chem. Soc, 74, 2683 (1952).
45. SimonsJ.K., WagnerE.C, Muller J.H., J. Am. Chem. Soc, 55, 3705
(1933).
46. К r a u s С A., FosterL.S., J. Am. Chem. Soc, 49, 459 (1927).
47. S i m о n s J. K., J. Am. Chem. Soc, 57, 1299 (1935).
48. R a m s d e n H. E., Пат. США 31Q9851 (1963); РЖХим., 1965, 17Н158П.
49. JacobsG., Compt. rend., 238, 1825 (1954).
50. BauerH., Burschkies K., Ber., 65, 956 (1932).
Синтез с помощью металлоорганических соединений
59
51. О г п d о г f f W. К., TabernD. L, DennisLM., J. Am. Chem. Soc, 49,
2512 (1927).
52. BrinckmanF.E., S t о n e F. G. A., J. Inorg. Nucl. Chem., 11, 24 (1959).
53. Chambers R. D.f Cunningham J., Tetrahedron Letters, 1965, 2389.
54. E м е л ь я н о в а Л. И., М а к а р о в а Л. Г., Изв. АН СССР, ОХН, I960, 2067.
55. Емельянова Л. И., Виноградова В. Н., М а к а р о в а Л. Г.,
Несмеянов А. Н., Изв. АН СССР, ОХН, 1962, 53.
56. Б а у к о в Ю. И., Л у ц е н к о И. Ф., ЖОХ, 32, 2746 (1962).
57. Л у ц е н к о И. Ф., Б а у к о в Ю. И., X а с а п о в Б. Н., ЖОХ, 33, 2724 (1963).
58. Б а у к о в Ю. И., Б у р л а ч е н к о Г. С, Л у ц е н к о И. Ф., ЖОХ, 35, 1173
(1965).
59. Б а у к о в Ю. И., Бурлаченко Г. С, Бел а вин И. Ю., Л у ц е н-
к о И. Ф., ЖОХ, 36, 153 (1966).
60. Kali Chemie. Англ. пат. 820146 (1959); С. А., 54, 6550е (1960).
61. Заха рки н Л. И., Охлобыстин О.Ю., ЖОХ, 31, 3662 (1961).
62. Авдеева В. И., Бурлаченко Г. С, БауковЮ. И., Луценко И.Ф.,
ЖОХ, 36, 1679 (1966).
63. L u t s e n k о I. F., В а и к о v Ju. I., В и г 1 а с h e n к о G. S., J. Organometal.
Chem., 6, 496 (1966). J
64. Л у ц е н к о И. Ф., П о н о м а р е в С. В., ЖОХ, 31, 2025 (1961).
65. L и i j t e n J. G. A., R i j к e n s F., Rec. trav. Chem., 83, 857 (1964).
66. GrantD., van W a z e r J. R., J. Organometal. Chem., 4, 229 (1965).
67. МироновВ.Ф., К р а в ч е н к о А. Л., ДАН СССР, 158, 656 (1964).
Г лава V
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИСОЕДИНЕНИЕМ РАЗЛИЧНЫХ ГЕРМАНИЙОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ И СОЛЕЙ ГЕРМАНИЯ К КРАТНЫМ СВЯЗЯМ
Реакция присоединения различных органических и неорганических
гидридов германия к кратным сйязям имеет большое значение в синтезе
германийорганических соединений и может успешно конкурировать с
другими методами их получения. Гидрид треххлористого германия легко
присоединяется практически к любым непредельным соединениям при комнатной
температуре и в отсутствие катализатороЁ. Галоидгидриды алкилгермания
также реагируют с непредельными соединениями, однако лишь при
нагревании до 140—150° С. Для присоединения гидридов ал кил- или арил
германия в большинстве случаев требуется присутствие катализаторов типа
H2PtCl6. Присоединение может идти лишь при нагревании, выходы
невысокие.
Ниже рассматриваются реакции присоединения различных гидридов
по кратным связям, приводящие к образованию германийорганических
соединений. Иное течение реакции, не приводящее к образованию новых
германийорганических соединений, рассмотрено в главе «Синтез и реакции
германийорганических гидридов».
ПРИСОЕДИНЕНИЕ HGeX3 К НЕПРЕДЕЛЬНЫМ СОЕДИНЕНИЯМ
Впервые Рохов с сотр. [1] провел присоединение гидрида
треххлористого германия к гексену-1 при длительном кипячении в присутствии
перекиси бензоила и отметил образование треххлористого гексилгермания с
выходом 22%. В тех же условиях было осуществлено присоединение
HGeCl3 к девяти другим олефинам [2].
Пономаренко, Взенкова и Егоров [3] проводили присоединение гидрида
треххлористого германия к этилену в присутствии H2PtCl6 в автоклаве.
Треххлористый этил германий был получен в этих условиях с выходом
менее 25%.
Петров, Миронов и сотр. [4—15] показали, что гидрид треххлористого
германия без каких-либо катализаторов и инициаторов энергично
присоединяется в момент контакта компонентов (с выделением тепла) уже при
комнатной температуре практически к любым непредельным соединениям.
\ / II
Cl3GeH + С=С -> Cl3GeC-CH.
/ \ II
Авторы пришли к заключению, что HGeCl3 более реакционноспособен в
этой реакции, чем HSiCl3, присоединение которого происходит лишь в
присутствии катализаторов и в более жестких условиях.
При барботировании этилена через гидрид треххлористого германия
после разгонки жидкости получен треххлористый этилгерманий; т. кип.
Присоединение германийорганических соединений по кратным связям 61
141,5° С; выход 55% [4]. Аналогично при пропускании тока пропилена
через гидрид треххлористого германия разгонкой на колонке был выделен
треххлористый я-пропилгерманий; т. кип. 163,5° С, выход 83% [7].
Авторы провели присоединение гидрида треххлористого германия и к буте-
ну-1, получив треххлористый я-бутилгерманий (т. кип. 182° С, выход 75%
171), к октену-1 (выход 55%) [4], к стиролу (выход Cl3GeCH2CH2C6H5 30%;
т. кип. 132° С/10 мм) [7].
В своих первых сообщениях [4, 7] авторы отмечают, что при
пропускании тока ацетилена в HGeCl3 с выходом 90% образуется соединение
Cl3GeCH2CH2GeCl3; т. кип. 130—131° С/12 мм. Если же добавлять HGeCl3 к
насыщенному ацетиленом гексану, удается выделить, кроме того, и
треххлористый винилгерманий [13]:
HGeCl3
СНеееСН — > CH2=CHGeCl3 (13%) + Cl3GeCH2CH2GeCl3 (17%).
В среде эфира или с эфиратом гидрида треххлористого германия
реакция протекает иначе [16]. В этом случае в зависимости от порядка4
смешения компонентов получается или только полимер (добавление HGeCl3 к
насыщенному ацетиленом эфиру), или наряду с полимером образуется и
1,2-бис-(трихлоргермил)этилен, если ацетилен пропускать через эфират
HGeCl3
HGeCb
СН=СН — > Cl3GeCH=CHGeCl3 +
эфир
Г С1
I
-Ge—CH^CH-
, I
L C1
Авторы считают, что образование этих веществ связано, вероятно, с
наличием в реакционной среде таких частиц, как GeCl2, GeCl3 и других
образующихся по схеме [10, И]
HGeCb ^ Н+ + (GeCl3r ^ Н+ + С1~ + GeCl2.
Что касается диеновых соединений, то найдено, что в
противоположность трихлорсилану, который присоединяется исключительно в
положении 1—4, HGeCl3 присоединяется [10, И, 17, 18] по схеме
СН-2-СН
HGeCl3 /
СН2=СНСН=СН2 ► Cl3GeCH2CH=CHCH3 + Cl2Ge
\
(I) СН2—СН
(II)
Соотношение между соединениями (I) и (II) от опыта к опыту колеблется
в пределах от 1 : 3 до 1 : 9.
Изопрен с гидридом треххлористого германия реагирует с образованием
гетероциклического соединения
HGeCl3 СНз
СН2=С-СН=СН2 ► СН=С—СНз 1
| | 1 + Cl3GeCrt2-C=CH-CH3.
СН3 СН2 СН2
\ /
GeCl2
Соотношение продуктов равно 1 : 3.
62
Германийорганические соединения. Лит. стр. 72—73
Пиперилен в этих условиях образует исключительно линейные
продукты
HGeCl3
СН3СН=СНСН=СН2 ► CH3CH2CH=CHCH2GeCl3 + СН3СН=СН—CHGeCl3.
СН8
Структура полученных соединений доказывается спектральным анализом.
HGeClg энергично присоединяется к циклопентадиену с образованием
Cl3GeC5H7; т. кип. 87° С/11 мм, выход 58% [7].
Гидрид треххлористого германия удалось присоединить и к
непредельной ароматической связи нафталина [19].
Колесников и сотр. [19а] нагревали 0,05 моля нафталина с 0,05 моля
гидрида треххлористого германия в течение 10—15 мин. при ПО—130° С.
После метилирования реакционной смеси с помощью бромистого метилмаг-
ния в эфире они получили с выходом 52,5% 1,3-бис-(триметилгермил)-1,2,3,
4-тетрагидронафталин; т. кип. 100—120° С/0,2 мм.
HGeCl3
^Y^VGeCl.3 CH^BTr f \ Ч-Ое(еНз/.
^^^G'eeb -.*».. €мхС<ЗДаЬ
Аналогичные результаты были получены при действии HGeCl3 на
1-метилнафталин, диметилнафталин, антрацен, фенантрен и их производные.
Бензол, толуол и ксилол в этих условиях с гидридом треххлористого
германия не реагируют. При реакции с анизолом (нагревание, 8,5 час. при
110° С) после действия бромистого метилмагния выделена смесь изомеров
метокси-т/шс-(триметилгермил)циклогексана.
С фенетолом реакция проходит аналогично (нагревание 30 мин. при
110—130° С). Ранее те же авторы [196] наблюдали, что при нагревании
алкоксибензола с гидридом треххлористого германия в эфире реакции
не происходят.
Циклопропан и его производные обладают известной ненасыщенностью.
В связи с этим была изучена реакция гидрида треххлористого германия с
арил- и алкилзамещенными циклопропана [20]. Было найдено, что реакция
протекает по уравнению
R—СН
\
сн2
/I
+|HGeCl3-» Cl8GeCHCH2CHg.
СН2 R
Присоединение было проведено к фенилциклопропану, я-амилциклопропа-
ну, 1-метил-2-я-бутилциклопропану и производным норкарана. В отличие
от алкил- и арилциклопропанов сам циклопропан и его хлорпроизводные
не реагируют с гидридом треххлористого германия или его эфиратом при
температурах от —60 до 75° С.
С непредельными галоидными алкилами гидрид треххлористого
германия (за исключением алкильных производных) реагирует так же, как и с
непредельными углеводородами. Происходит присоединение по кратным
связям в отсутствие катализаторов и при комнатной температуре. При
барботировании хлористого винила через HGeCl3 присоединение протекает
с саморазогреванием, однако в результате реакции образуется не треххло-
ристый а-хлорэтилгерманий, а треххлористый р-хлорэтилгерманий, выход
32,5% [5]
С1СН=СН2 + HGeCl3 -* ClCH3-CH2GeCl3.
Присоединение германийорганических соединений по кратным связям 03
Реакция была проведена с производными этилена различной степени
хлорирования [5, 7, 9]. С /яраяс-дихлорэтиленом получен треххлористый а,|3-
дихлорэтилгерманий; выход 62%; т. кип. 88°С/12 мм. С несимметричным
дихлорэтиленом выход треххлористого Р,|3-дихлорэтилгер мания составил
62%; т. кип. 74° С/10 мм. Трихлорэтилен реагирует аналогично с
образованием треххлористого триэтилгермания (55,3%; т. кип. 92° С/9 мм), а тет-
рахлорэтилен образует с выходом 48% треххлористый тетрахлорэтилгер-
маний (т. кип. 123° С/25 мм).
Присоединение гидрида треххлористого германия к аллильным и ме-
таллильным галоидным производным протекает несколько отлично. Если
реакцию проводить обычным образом, прибавляя хлористый аллил к
HGeGl3, происходит присоединение по кратной связи, с выходом 77,4%
образуется треххлористый 7"хлоРпРопилгеРман™; т- кип- Ю5° С/20 мм.
С хлористым металлилом также образуется треххлористый у-хлоризобу-
тилгерманий; выход 58,3%; т., кип. 90—93° С/11 мм [4].
Было найдено, что при прибавлении HGeCl3 к бромистому аллилу
протекает экзотермичная реакция, а вместо ожидаемого треххлористого
у-бромпропилгермания образуется треххлористый аллилгерманий с
выходом 50% [6, 8, 14].
CH2=CHCH2Br+HGeCl3 >CH2=CHCH2GeCl3+HBr.
В дальнейшем авторы [8, 14] нашли, что с хлористым аллилом, если
реакцию проводить в присутствии эфира, также протекает не реакция
присоединения, а реакция конденсации. Таким образом, реакция с хлористым
аллилом может протекать двояко:
HGeCbl-2^-* CH2=CHCH3GeCl8 (26%) + НС1
> Cl3GeCH2CH2CH2Cl (52%)
CH2=CHCH2C1-
Хлористый металлил реагирует аналогично:
эфир
СН2=ССН2С1
* Cl3GeCH2—С=СН2 (60%) + НС1
HGeCl3| I
СНз
£н ' > Cl3GeCH2—CH—СН2С1 (58%).
СНз
Йодистый аллил и бромистый металлил реагируют исключительно по схеме
конденсации.
Что касается реакции HGeCl3 с хлористыми бромистым пропаргилом [14],
то с этими соединениями протекает реакция конденсации, однако
исключить параллельно идущую реакцию присоединения не удается. В
результате одновременного протекания обеих реакций образуется смесь
продуктов
НС=ССН2С1 52!PilCl3GeGH=CHCH2GeCl3 + (Cl3Ge)2CH—CH2CH2GeCl3.
Реакция присоединения гидрида треххлористого германия была
изучена и на ряде других непредельных функциональнозамещенных
органических соединений. Так, HGeCl3 присоединяется к аллилацетату, образуя
с выходом 40,8% Cl8GeCH2CH2CH2OOCCH3 (т. кип. 127,5° С/12 мм) [4],
и винилацетату [7], образуя с выходом 57% С1зОеСН2СН2ООССНз (т. кип.
97°С/15жж).Акрилонитрил [4] реагирует с образованием треххлористого р-ци-
анэтилгермания ClsGeCH2CH2CN с выходом 53%; т. кип. 135° С/22 мм.
Реакции HGeCl3 с германий- или кремнийорганическими соединениями,
имеющими в молекуле непредельные радикалы, происходят нормально пу-
64 Германийорганические соединения. Лит. стр. 72—73
тем присоединения по кратным связям. Присоединение было проведено к
треххлористому аллилкремнию, аллилгерманию, двухлористому метилви-
нилкремнию [8] и к ацетилениду триметилгермания [12].
Гидрид трехбромистого германия в химических реакциях с
непредельными соединениями ведет себя аналогично гидриду треххлористого
германия, однако он более склонен к реакциям конденсации, чем присоединения
[15, 21].
ПРИСОЕДИНЕНИЕ ГИДРИДОВ АЛКИЛ- И АРИЛГЕРМАНИЯ
К КРАТНЫМ СВЯЗЯМ
Гилман и сотр. [22, 23] впервые показали, что гидрид трифенилгер-
мания в присутствии перекиси бензоила или при ультрафиолетовом
облучении присоединяется к непредельным углеводородам. Имеются об этом
указания и в американском патенте [24].
Реакция гидрида трифенилгермания и октена-1.
а) В присутствии перекиси бензоила [22]. Смесь 21,4 г (0,07 моля) гидрида
трифенилгермания, 25 мл петролейного эфира (т. кип. 60—70° С), 1,7 мл (0,01 моля) октена-1 и 0,3 г
перекиси бензоила нагревают на водяной бане при 75° С в течение 24 час. При перегонке в
вакууме выделяют непрореагировавший гидрид трифенилгермания и 3,8 г (91%) вещества
с т. кип. 190—198° С/0,25 мм, т. пл. 57—67° С. Две перекристаллизации из спирта
поднимают т. пл. до 71—72° С. Смешанная точка плавления с трифенил-1-октилгерманием не дает
депрессии.
б) При ультрафиолетовом облучении. Смесь 0,045 моля гидрида трифенилгермания, 0,006
моля октена-1 и 25 мл гептана в кварцевой колбе облучают в течение 48 час. После перегонки
выделяют 2 г (80%) трифенил-1-октилгермания и небольшую примесь тетрафенилгермания.
Аналогично происходит присоединение гидрида трифенилгермания в
присутствии перекиси бензоила к октадецену-1 [23]; выход трифенил-я-
октадецилгермания равен 67%; т. пл. 75,5—76,5° С.
Присоединение гидрида трифенилгермания к циклогексену [22] в
присутствии перекиси бензоила или при ультрафиолетовом облучении также
имеет место, однако в продуктах реакции было найдено значительное
количество тетрафенилгермания, образовавшегося за счет диспропорциониро-
вания гидрида трифенилгермания. Попытка присоединить гидрид
трифенилгермания в присутствии перекиси бензоила к 1,1-дифенилэтилену
окончилась неудачно, в результате реакции были выделены неидентифицирован-
ные полимерные продукты.
Гидрид трифенилгермания может присоединяться к непредельным
углеводородам и в отсутствие катализаторов, однако выходы при этом
значительно ниже и требуется более продолжительное нагревание [25].
Присоединение гидрида трифенилгермания к я-октену [25]. Смесь 1,12 г (10 ммолей)
я-октена и 3,05 г (10 ммолей) гидрида трифенилгермания нагревают на масляной бане при
ПО—115° С в течение 15 дней. Реакционную смесь нагревают с гексаном, по охлаждении
выделяют трифенилоктилгерманий. После перекристаллизации из гексана и метанола т. пл.
62—66° С; выход 29%.
Авторы [25] провели также присоединение гидрида трифенилгермания
к циклогексену (39%) и к стиролу (40%).
Органические гидриды германия реагируют с ацетиленовыми
углеводородами. Присоединение гидрида трифенилгермания к ацетилену в
отсутствие катализаторов происходит с выходом лишь 12% (нагревание на
масляной бане в течение четырех дней при 135° С).
Лесбр и Сатже [26, 27] нашли, что в присутствии платинохлористово-
дородной кислоты в спиртовом 0,1 ./V растворе присоединение гидридов ал-
килгермания к тройной связи алкинов проходит с почти количественными
выходами. Реакция идет вопреки правилу Марковникова и является об-
Присоединение германийорганических соединений по кратным связям 65
щим методом приготовления германийорганических соединений,
содержащих замещенную винильную группу.
Реакция была проведена с гидридами триалкилгермания и ацетиленом,
фенилацетиленом, гексином-1 при эквимолекулярных соотношениях
реагентов в количествах порядка 0,03 и 0,04 моля в присутствии 0,1 мл 0,1 .V
раствора платинохлористоводороднои кислоты в этиловом или изопропило-
вом спирте в атмосфере азота.
При пропускании ацетилена [27] в гидрид триэтилгермания при 100° С
в присутствии спиртового раствора платинохлористоводороднои кислоты
присоединение происходит в две стадии:
(C2H5)3GeH + СНеееСН -> (C2H5)3GeCH=:CH2,
(C2H6)eGeCII=CH2 + (C2H5)3GeH -> (C2H5)3GeCH2CII2Ge(C2H5)3.
Образующийся вначале триэтилвинилгерманий присоединяет новую
молекулу гидрида с образованием бас-(триэтилгермил)- 1,2-этана; выход 75%,
т." кип. 274° С.
Гидрид трибутилгермания количественно присоединяется в
присутствии катализатора к фенилацетилену с образованием трибутил-Р-стирилгер-
мания
(QH9)3GeH + СН=СС6Н5 -» (QH9)gGeCFI=CHCeH3.
Аналогично присоединяется и гидрид триэтилгермания к гексину-1
(C2H5)3GeH + СН=С-(СН2)3СНз-> (C2H5)3GeGH^CH(CH2)3CH3.
Дигидрид дибутилгермания 126—28] присоединяется при 100° С к
молекуле фенилацетилена, образуя гидрид дибутилстирилгермания
(C4H9)2Ge(H)CH —СНС6Н5, который при продолжительном нагревании
образует полимер в виде желтого масла. Тригидрид гептилгермания [26, 27]
при температуре 200° С присоединяет три молекулы гептина-1, образуя
тетраалкилгерманий, содержащий три замещенных винильных группы
(C7H15)Ge[CH=CH(CH2)4CH3]3; т. кип. 174°С/0,4лш;^0,9194; п™ 1,4798.
Гидриды триалкилгермания в присутствии катализаторов легко
присоединяются и к непредельным спиртам. Так, гидрид трибутилгермания
при нагревании с аллиловым спиртом в присутствии амината платины с
выходом 57% образует трибутилгермил-З-пропанол-1. В присутствии
перекиси бензоила реакция гидрида трипропилгермания и аллилового спирта
проходит лишь на 14 %. Перекись бензоила в данном случае является менее
активным катализатором [27, 291.
Петров и Миронов с сотр. [7] также исследовали присоединение гидрида
триэтилгермания к аллиловому и пропаргиловому спирту (катализатор
Н.2Р(С16-6Н20).
Присоединение гидрида триэтилгермания в аллиловому спирту [7]. Смесь, состоящую
из 18 г аллилового спирта, 20 г (С2Н5)зОеН и двух капель 0,1 N раствора H2PtCl6-6H20
в изопропиловом спирте нагревают при 90° С в течение 4 час. После вакуумной разгонки
выделяют 24 г (C2H5)3GeCH2CH2CH2OH; т. кип. 89—90° С/3 м, выход 73%.
Аналогично получен и (C2H5)3GeCH==CHCH2OH; т. кип. 112—
113°С/12 мм, выход 37%.
Сатже [27] нашел, что гидрид трибутилгермания без катализатора
легко присоединяется к пропаргиловому спирту; реакция начинается при70°С,
выход трибутилгермил-3-пропен-2-ол-1 составляет 80% после 6 час.
нагревания. Аналогично с выходом90% реагирует и СН = С—С(ОН)(СН3)2«
Петров и сотр. [30] исследовали реакцию между гидридом
триэтилгермания и различными винилэтинилкарбинолами в присутствии в качестве
катализатора 0,1 N раствора H2PtCl6 в изопропиловом спирте. Было най-
5 Заказ № 207
66
Германийорганические соединения. Лит. стр. 72—73
дено, что реакция между (C2H5)3GeH и исследуемыми карбинолами
проходит более гладко, чем в случае (C2H5)3SiH, и выходы выше.
Реакция проходит по схеме
R' R'
R__C(OH)~CeeC-CH=--CH2 + (CaHsbGeH -> R-C-C=CH-CH=CH2
I I
OH Ge(C3H5)3
R = R' = CH3; R = CH3, R' = C2H5;
,CHo—CH3 yCH3—СН2ч
R-R'= < I и < >CH2
\CH3—CH2 XCH2—CHU/
В результате реакции были получены следующие соединения:
СН3ч
>С(ОН)—C[Ge(CaH6)3]=CH—СН=СНа
сн3/
(т. кип. 112° С/6 мм; df 1,0399; /$1,4954; выход 70%);
(СоН5)(СНз)С (ОН)—С [Ge(C2H5)3]=CH—СН=СН2
(т. кип. 98—100° С/3 мм; df 1,0430; n2D°l,4972; выход 79,6%);
СН2—СН2ч
| >C(OlI)C [Ge(C2H5)3]=CH—CH=CH2
СН2—СНо/
(т. кип. 116—118° С/1 мм; 4° 1,0788; л!> 1,5129; выход 82,5%);
СНа—СН2
/ \
СН2 С(ОН)—C[Ge(C2H3)3]=CH—СН=СН2
\ /
СН3—СН2
(т. кип. 114—116° С/1 мм; df 1,0777; /$1,5140; выход 86,9%).
Аналогичная реакция была проведена с диацетиленовыми гликолями
[31, 32], с первичными и вторичными винилацетиленовыми карбинолами [33]
и несимметричными у-гликолями [34].
Шихеев с сотр. [35] исследовал присоединение гидрида
трибутилгермания к пропилэтинилкарбинолу и метилэтилэтинилкарбинолу, а также
различных гидридов триалкилгермания к соединениям RCH(OH)C = СН
[36] и RC(CH3)(OH)C=CH [37] и другим подобным соединениям [37а].
Реакция между гидридом трифенил германия и 2-метил-З-бутин-З-олом
в отсутствие катализатора привела к получению продукта присоединения
(CeH6)3GeCH=CHC(OH)(CH8)2 (49%) [25], аналогично получен три-
фенил-(5-оксипентил)германий (C6H5)3Ge(CH2)4CH2OH; т. пл. 51—61° С.
Гидриды триалкилгермания в отсутствие катализаторов присоединяются
к виниловым эфирам с хорошими выходами [27]. Так, гидрид трибутил
германия и винилбутиловый эфир при нагревании в течение 8 час. при 130° С
с выходом 72% образуют бутоксиэтилтрибутилгерманий
(C4H9)4GeH + СН2=СНОС4Н9 -» (С4Н9)зСеСН2СН2ОС4Н9.
Аналогично при присоединении тригидрида гептилгермания к винилбути-
ловому эфиру с выходом 70% получают три-бутоксиэтил-гептилгерманий
C7H15GeH3 + СНг^СНОСдНэ -> C7H15Ge(CH2CH2OC4H9)3.
• Непредельные альдегиды при добавлении ингибиторов полимеризации
присоединяют гидриды алкилгермания в присутствии платиновых катали-
Присоединение германийорганических соединений по кратным связям 67
заторов [27, 29]. При этом получаются германийорганические производные
насыщенных альдегидов
(C4H9)3GeH + СН.—СНСНО -* (QH9)3GeCH2CH2CHO,
(C6Hu)2GeH2 + СН3СН=СНСНО -> (C5Hn)2Ge(H)--CH(CH3)—CHoCHO.
Реакция гидрида трибутилгермания с акролеином [27]. Смесь 9,55 г (0,039 моля) гидрида
трибутилгермания, 2,18 г (0,039 моля) акролеина и 0,2 г гидрохинона нагревают 10 час.
при 90° С. в присутствии амината платины. При вакуумной перегонке получают 7 г (60%)
3-(трибутилгермил)пропанола-1. После вторичной перегонки т. кип. 125—128° С; д^°1,0314;
я2^1,4630.
Дигидрид диамилгермания с кротоновым альдегидом при нагревании в
течение 18 час. при 100° С образует смесь монозамещенного продукта (44%;
т. кип. 139—140° С/10 мм\ df 0,9910; п™ 1,4585) и дизамещенного
продукта, выделить который в чистом виде не удалось.
Непредельные кетоны присоединяют органические гидриды германия
по кратным связям при нагревании в отсутствие катализаторов [27, 29].
В противоположность гидридам кремния присоединения по
карбонильной группе не наблюдается. Дигидриддибутилгермания и винилметилкетон
при нагревании в течение 10 час. в присутствии гидрохинона с выходом 30%
образуют дибутилкето-3-бутил германий (ft-C4H9)2GeHCH2CH2COCH3 и с
выходом 27% —дибутилди(кето-З-бутил)германий (ft-C4H9)2Ge(CH2CH2COCH3)2-
Гидрид трифенилгермания и метилвинилкетон с выходом 53% образует
трифенил-(2-ацетилэтил)германий; т. пл. 144—146° С [25]. Аллидамин [14,
27] присоединяет гидриды алкилгермэния только в присутствии платиновых
катализаторов после продолжительного нагревания
R3GeH + CH2=CH—CH2NH2-> RsGeCHsCHsCHsNH*.
В случае оловоорганических гидридов при аналогичной реакции
выделяется пропен. Выход трибутилгермил-З-амино-1-пропана [27] при
реакции гидрида трибутилгермания и аллиламина составил 74%; т. кип.
161° С/10 мм\ df 0,9816; пЬ° 1,4700. Акриловая кислота [27, 29] при
нагревании с гидридами триалкил- и диал кил германия без катализатора дает
продукты присоединения
(C2H5)3GeH + СН2=СНСООН -» (QH5)3GeCH2CH,COOH,
(C3H7)2GeH2 + CH2=CHCOOH -> (C3H7)2Ge(H)CH2CH2COOH.
При реакции гидрида триэтилгермания с акриловой кислотой (нагревание
8 час. на водяной бане при 140—150° С) выход (C2H5)3GeCH2CH2COOH
составляет 49%. При аналогичной реакции дигидрида
дипропилгермания с акриловой кислотой выход (C3H7)2Ge(H)CH2CH2COOH равен 30%,
дизамещенный продукт в чистом виде выделен не был.
Эфиры непредельных кислот [27, 29] легко без катализатора
присоединяют гидриды триалкилгермания, однако выходы низкие вследствие
идущих побочных реакций:
(C3H7)3GeH + СН2=СНСООСН3-> (C3H7)3GeCH2CH2COOCH3.
Нагревание эквимолекулярных количеств гидрида трипропилгермания
и метилакрилата при 100° С в течение 8 час. без катализатора приводит к
(a-C3H7)3GeCH2CH2COOCH3 (34%); т. кип. 150—154° С/18 мм; df 1,040;
/22D° 1,4596. Аналогично получен (n-C4H9)3GeCH2CH2COOC2H5 (34%); т. кип.
122—124° С/0,5 мм; df 1,0028; n2D° 1,4595; (n-C4H9)3GeCH2CH2COOC2H5
получен с выходом 40%.
5*
68 Германийорганические соединения. Лит. стр. 72—73
Описано присоединение гидрида три метил- и триэтилгермания к эфиру
винилуксусной кислоты [38].
Гидрид трифенилгермания присоединяется к метилакрилату, давая
(C6H5)3GeCH2CH2COOCH3 с выходом 50%. К винилацетату [25] по виниль-
ной группе присоединение происходит на 54% (нагревание 6 час. при 50—
60° С). К аллилацетату гидрид триэтилгермания присоединяется на 50%
[8].
В литературе [25] имеется один пример присоединения гидрида
трифенилгермания к акриламиду. Авторами выделен линейный продукт
присоединения (без катализатора), однако выход не указан.
Непредельные нитрилы [25, 29] реагируют с гидридами алкилгермания
различно в зависимости от строения алкена. Акрилонитрил присоединяет
гидриды без катализатора при нагревании эквимолекулярной смеси
реагентов. К аллилнитрилу присоединение происходит лишь в присутствии
катализаторов, так как реакционная способность двойной связи аллилнит-
рила значительно ниже, чем акрилонитрила. В случае присоединения к ак-
рилонитрилу гидрида трибутилгермания и трипропилгермания выходы
соответствующих продуктов присоединения составляют 42 и 24%. Реакция
ссложняется сопутствующей реакцией полимеризации.
К аллилнитрилу был присоединен гидрид триизоамилгермания [27].
После кипячения в течение 20 час. в присутствии амината платины был
выделен триизоамилгермил-З-циано-1-пропан (^-С5Н11)зОеСН2СН2СН2СЫ с
выходом 50%; т. кип. 128° С/0,6 мм\ dfO.9635; n^ 1,4653.
Дигидриды диалкилгермания присоединяются к акрилонитрилу с
образованием монозамещенного соединения, содержащего нитрильную
группу и связь Ge—Н с выходом 21%. Гидрид трифенилгермания и
акрилонитрил легко образуют продукт присоединения [25] с выходом 83%, т. пл.
126—129 С.
В случае реакции с меркаптанами Сатже [27] не наблюдал реакции
восстановления функциональной группы, например при действии аллилмер-
каптана на гидрид трибутилгермания, но тем не менее продукт
присоединения получался с низким выходом (30%) даже в присутствии платиновых
катализаторов
R3GeH -J- CH2=CH—CH2SH -> R3GeCH2CH2CH2SH.
Что касается присоединения гидридов триалкилгермания к металлоор-
ганическим соединениям, содержащим винильные или аллильные радикалы,
то присоединение гидрида триэтилгермания к диметиловому эфиру аллил-
борной кислоты происходит с большой легкостью в отсутствие
катализатора [39] при ПО—190" С и приводит к получению диметилового эфира 3-
(триэтилгермил)пропилборной кислоты с выходом 77,8%
(C2H5)3GeH + СН3=СН—СН2В(ОСН8)а -> (С2Н5)зСе(СН2)зВ(ОСНз)2.
С еще большей легкостью идет присоединение гидрида
трифенилгермания, выход около 100%. Присоединение гидрида три-я-бутилгермания к
эфирам алкенилборных кислот исследовалось и другими аторами [40].
Сатже [27, 411 наблюдал, что после нескольких часов нагревания в при-
сутствии амината платины гидрид триэтилгермания количественно
присоединяется к триэтилвинилгерманию и к триэтилаллилгерманию
(C2H5)3GeCH=:CH2 + (C,H5)3GeH - (CzH^GeCH^CHaGefCaHsb,
(C2H5)3GeCH2CH = Crf2 + (C2H3)8GeH -> (C2H5)3GsGH2GH2CH2Ge(C2H5)3.
Попытка присоединить гидрид трифенилгермания к трифенилаллилолову
в присутствии перекиси бензоила окончилась неудачей [42]. В противо-
Присоединение германийорганических соединений по кратным связям 69
положность этому присоединение того же гидрида к трифенилаллил-
кремнию под влиянием перекиси бензоила прошло на 74% [43].
Миронов и сотр. [14] присоединили гидрид триэтилгермания к
CH2=CHCCOSi(CH3)3, присоединение прошло на 58%.
Гидрид три-я-бутилгермания реагирует с двух направлениях с три-
этилэтинилкремнием [44]; при этом образуется 1-(триэтилсилил)-2-(три-н-
бутилгермил)этилен по уравнению
(n-QH9)3GeH + (C2H5)3SiC=CH^ (/2-C4H9)3GeCH=CHSi(C2H5)3
и 1,2-бш>(три-я-бутилгермил)этилен. Образование последнего соединения
происходит за счет обмена этинильной группы, связанной с кремнием, на
атом Еодорода, соединенный с германием. При этом образовавшийся три-я-
бутилэтинилгерманий реагирует с гидридом три-я-бутилгермания по
уравнению
(rc-C4H9)3GeH + (GsHsfoSiteCH -* (n C4H9)3GeC-CH + (C2H5bSiH,
(/z-C4H9)3GeH + (n-C4H9)3GeC=CH -* (/i-C4H9)3GeCH=CHGe(C4Ho-«)8.
ПРИСОЕДИНЕНИЕ R2GeHX К КРАТНЫМ СВЯЗЯМ
Галоидгидриды алкилгермания присоединяются к различным
ненасыщенным углерод-углеродным связям без катализаторов с хорошими
выходами. Присутствие галоида в молекуле гидрида значительно "увеличивает
реакционную способность связи Ge—Н.
Лесбр и Сатже с сотр. [41] нашли, что хлор- и бромгидриды
диэтилгермания и хлор-, бром- и иодгидриды дибутилгермания легко присоединяются
к ненасыщенным углеводородам. Реакция обычно проводилась в запаянных
ампулах при температуре около 140° С. Полученные производные имели
линейную структуру:
R2GeHX + CH2=CHR' -> R2Ge(X)CH2CH2R\
При нагревании в запаянной ампуле эквимолекулярной смеси хлоргидрида
дибутилгермания и октена-1 продукт присоединения был выделен с выходом
80%. В тех же экспериментальных условиях бромгидрид дибутилгермания
и иодгидрид дибутилгермания присоединяются с выходом 85 и 90%. При
150° С присоединение хлоргидрида к двойной связи циклогексена приводит
к хлористому дибутилциклогексилгерманию с выходом 70%
(QH9)o(Cl)GeH + СН=СН(СН2)4-> (C4H9)2(C1)GeCH—СН*(СН2)4.
I [ ! I
Присоединение галоидгидридов диалкилгермания к диеновым
углеводородам происходит всегда в положение 1,4 [45]. Так, хлор гидрид и бром-
гидрид диэтил германия присоединяются к бутадиену в положение 1,4.
Реакцию проводят в автоклаве при температуре 110° С
(QH5)2Ge(X)H + Н2С=СН—СН=СН2-> (С2Н5)2(Х)СеСН2СН=СНСН3.
Еыхсд X = С1 — 35%, X - Вг — 49%.
Аналогично хлсргидриды диэтил- и дибутилгермания присоединяются к
диьс1Ил-2,2-бутэд:иену-1,3.
При пропускании ацетилена в течение 8 час. вхлоргидрид диэтил
германия при 1СС—110° С образуется (с еыходом 50%) 1,2-быс-(диэтилхлоргер-
\ил)зтгн. Регкьия прохс^ит в дие стадии:
(С2Н5)2(С1) GeH + СН=СП -» (С2Н5)2(С1) GeCH=CH2,
(С2Н5)о(С1) GeCH=CH3 + (C3H3)2(Cl)GeH-> (C2H5)2Ge(Cl)—СН2СН2—Ge(Cl)(C2H5)«.
70
Германийорганические соединения. Лит, стр. 72—73
Фенилацетилен реагирует с хлоргидридом дибутил германия с выходом
75%. Аналогично реагирует и гексин-1, выход количественный [46].
В противоположность алкилгалоидсиланам, присоединение которых к
хлористому аллилу трудно достигнуть даже в присутствии платиновых
катализаторов (так как преобладает реакция расщепления), галоидгидриды
алкилгермания при температуре 150° С присоединяются к хлористому аллилу
без всякого катализатора. Так, хлор гидрид диэтил- и дибутилгермания
присоединяются с выходом около 90% [41]
R2(Cl)GeH + СН2=СНСН2С1 -» R2(Cl)GeCH2CH2CH2Cl.
Бромистый аллил и бромгидрид диэтил германия в тех же
экспериментальных условиях реагируют согласно уравнению [41J:
(C2H5)2(Br)GeH + CH2=:CH—CH2Br -> (C2H5)2GeBr2 + СН2=СН—СН3.
Хлористый металлил ведет себя аналогично хлористому аллилу. Выход
продукта присоединения равен 80% [46].
Хлоргидрид дибутилгермания после 20 час. кипячения при 150° С без
катализатора присоединяется к аллиловому спирту с выходом 65%. Ранее
было отмечено (стр. 65), что аллиловый спирт реагирует с гидридом три-
бутилгермания только в присутствии платиновых' катализаторов и не
реагирует с аналогичными силанами.
Винилбутиловый эфир присоединяет хлоргидрид дибутилгермания при
140° С, выход 80% [41]. Метакролеин [47] присоединяет хлоргидрид
дибутилгермания лишь в присутствии платинохлористоводородной кислоты и
гидрохинона как ингибитора полимеризации. Присоединение идет всегда
в положение 1,2; выход 40%.
(C4H9)2Ge(Cl)H + СН2=С(СН3)—СНО -» (C4H9)2Ge(Cl)—CH2—СН(СН3)СНО.
Диэтилацеталь акролеина присоединяет хлоргидрид дибутилгермания с
выходом 70%:
(QH9)2(Cl)GeH + СН2=СН—СН(ОС>Н5)2 -> (QHe)2(Cl) GeCH2CH2CH(OC2H5)2.
Винилметилкетон [47] присоединяет хлоргидрид диэтилгермания.
После 4 час. кипячения диэтил хлор гермилэтилметилкетон был выделен с
выходом 70%
(C2H5)2(Cl)GeH + СН2=СНСОСН3 -» (С2Н5)2 (Cl)GeCH2CH2COCH3.
В этом случае заметна разница в реакционной способности хлоргидрида
диэтилгермания и гидрида триэтилгермания; последний в тех же
экспериментальных условиях с трудом присоединяется к винилметилкетону
(см. стр. 67).
Сложные эфиры ненасыщенных кислот присоединяют галоидгидрид ди-
алкилгермания с хорошими выходами без катализатора при нагревании до
130—140° С в запаянных ампулах. Исследовалось [47] присоединение
хлоргидрида дибутилгермания к метилакрилату (выход 65%), метилме-
такрилату (65%), аллилацетату (80%). Хлоргидрид диэтилгермания
исследовался в реакции присоединения к этилакрилату (выход 70%), винилаце-
тату, изопропенилацетату; выходы количественные. Во всех случаях были
получены линейные продукты без заметного протекания побочных реакций
Ненасыщенные нитрилы присоединяют галоидгидриды диалкилгерма-
ния при повышенной температуре без катализатора [41, 46]. Описано
присоединение хлоргидрида дибутилгермания к акрилнитрилу (выход 70%
температура реакции 100° С)
(С4Н9)2(С1) GeH + СН2=СН—CN -» (C4H9)2(Cl)GeCH2CH2CN
и присоединение хлоргидрида диэтилгермания к аллилнитрилу при
температуре 150° С.
Присоединение германийорганических соединений по кратным связям 71
ПРИСОЕДИНЕНИЕ АЛКОКСИСОЕДИНЕНИЙ ГЕРМАНИЯ К КЕТЕНУ
Луценко и сотр. [48, 49], продолжая исследование реакции кетенас
органическими алкоксисоединениями элементов IV группы, обратили
внимание на различие их химического поведения в зависимости от природы
металла. Органические алкоксисоединения олова реагируют с кетеном
легко и с разогреванием, давая с высокими выходами эфирыстаннилуксусной
кислоты (см. стр. 311), в то время как соответствующие алкоксисоединения
кремния в этих условиях с кетеном не взаимодействуют. Что касается
производных триалкилалкоксигермания, то они реагируют более вяло, чем
соответствующие оловоорганические соединения, и дают конечные
продукты иного строения. Так, кетен реагирует с треххлористымалкоксигерма-
нием по схеме
CbGeOR + СН2=С=:0-* Cl3GeCH2COOR
с образованием эфиров трихлоргермилуксусной кислоты. Был
синтезирован метиловый эфир трихлоргермилуксусной кислоты (выход 62%; т. кип.
68—70° С/7 мм; df\f6775; По 1,4835) и этиловый эфир
трихлоргермилуксусной кислоты (61,5%; т. кип. 49—50° С/1,5 мм; По 1,4750). После насы-
шения кетеном раствора триалкилалкоксигермания из реакционной смеси
с выходом 70% был выделен эфир Р-триалкилгермилоксивинилуксусной
кислоты, образовавшейся по уравнению
(C2H5)3GeOR 2GH2=G=Ci CH2=C—CH2COOR
I
OGe(C2H5)3
При пропускании кетена в спиртовый раствор триалкилалкоксигермания,
куда предварительно добавлено некоторое количество йодной ртути, в
качестве единственного продукта реакции был получен эфир триалкилгер-
милуксусной кислоты с выходом 92%; совершенно не было получено эфира
Р-триалкилгермилоксивинилуксусной кислоты
сн2=с=о
(C2H5)3GeOCH3 -тгт-"* (QH5)3GeCH2COOCH3.
HgJ2
Авторы приводят свои соображения о механизме реакции.
Реакция триэтилметоксигермания с кетеном [49]. Триэтилметоксигерманий был
получен при реакции йодистого триэтилгермания с трибутилметоксисловом; выход 97%;
т. кип. 56—57716 мм; of 1,0696; п™ 1,4376.
а) Через раствор 3,4 г триэтилметоксигермания пропускают кетен при температуре
реакционной смеси — 5 -. 10° С. При перегонке выделяют 0,9 г исходного
триэтилметоксигермания и 2,5 г метилового эфира (З-триэтилгермилоксивинилуксусной кислоты; т. кип.
81—83° С/1 мм; df 1,1272; п™ 1,4615.
б) При пропускании избытка кетена через раствор 5,2 г триэтилметоксигермания и 0,4 г
йодной ртути в 6 мл метилового спирта после перегонки получают 5,8 г (92% от теорет.)
метилового эфира триэтилгермилуксусной кислоты; т. кип. 50—52° С/1,5 мм.
ПРИСОЕДИНЕНИЕ ДВУИОДИСТОГО ГЕРМАНИЯ
К ПРОИЗВОДНЫМ АЦЕТИЛЕНА И БУТАДИЕНА
В ряде работ Кирсановым и Вольпиным с сотр. [50—56] была показана
возможность присоединения двуиодистого германия к толану или ацетилену.
Были выделены и с помощью физико-химических и чисто химических методов
подробно исследованы продукты этой реакции. Авторы рассматривали
полученные соединения как производные трехчленных циклов с участием
в цикле атома германия. Позже Джонсон и сотр. [57, 58] установили, что
здесь имеет место образование не трехчленных, а шестичленных циклов.
72
Германийорганические соединения
Мазеролл и сотр. [59] показали, что двуиодистый германий
количественно реагирует с изопреном и 2,3-диметилбутадиеном с образованием
соответствующих циклических германийорганических соединений.
GeJo -;- СН2=С— С=СН2
I !
СНз СН3
GeJo + СН3=С—СН=СН
1
СНз
J4 /Clb—С
- >G< !!
У ХСН,-С
-СНз
—СНз
J СН-—С—СЧ£
\ <у II
/ \ II
j c:io—сч
Реакция двуиодистого германия с изопреном [59]. В трубку «пирекс» в токе азота
помещают 15 г (0,046 моля) двуиодистого германия и 3,40 г (0,05 моля) изопрена. Трубку
запаивают и затем нагревают 3 часа при 60° С. После нагревания в трубке остается желтая
жидкость, чрезвычайно чувствительная к воздуху и разлагающаяся при перегонке; d%° 2,319;
л^ 1,6515; анализ неочищенного продукта показал, что полученное вещество соответствует
дииод-1,1-германо-1-метил-3-циклопентену-3.
Реакция двуиодистого германия с 2,3-диметилбутадиеном [59]. В колбу на \00 мл
в токе азота вносят 20 г (0,061 моля) порошка двуиодистого германия и 5,1 г (0,062 моля)
2,3-диметилбутадиена. При перемешивании реакционной смеси начинается экзотермическая
реакция, йодистый германий постепенно исчезает. По окончании экзотермической реакции
реакционную смесь нагревают до 110° С. Получают желто-оранжевую жидкость, которая
кристаллизуется при 0° С. Вещество не удается перегнать вследствие разложения, а также
перекристаллизовать из пентана. По анализу полученное вещество соответствует дииодиду
1,1-германо-1-диметил-3,4-циклопентену-3; т. пл. 32—34° С; d2® 2,224; п2£ 1,654.
Для доказательства строения полученных соединений авторы провели
ряд физико-химических исследований, а также химических реакций, в
результате которых получили их производные.
ЛИТЕРАТУРА
1. F i s h e r. L.f W e s t R. С, RochowE.G., J. Am. Chem. Soc, 76, 5878 (1954).
2. Riemschneider R., M e n g e К., К 1 a n g P., Z. Naturforsch., lib, 115
(1956).
3. П о н о м о р е н к о В. А., В з е н к о в а Г. Я., Е г о р о в Ю. П., ДАН СССР,
122, 405 (1958).
4. П е т р о в А. Д., М и р о н о в В. Ф., Д ж у р и н с к а я Н. Г., ДАН СССР, 128.
302 (1959).
5. Ми р о и о в В. Ф., Д ж у р и н с к а я Н. Г., П ет р о в А. Д., ДАН СССР, 131,
98 (1960).
6. Миронов В. Ф., ДжуринскаяН. Г., Петров А. Д., Изв. АН СССР,
ОХН, 1960, 2066.
7. ДжуринскаяН. Г. Миронов В. Ф., П е т р о в А. Д., ДАН СССР, 138,
1107 (1961).
8. Миронов В. Ф., Д ж у р и н с к а я Н. Г., Г а р Т. К., П е т р о в А. Д., Изв.
АН СССР, ОХН, 1962, 460.
9. М и р о н о в В. Ф., Д ж у р и н с к а я Н. Г., Изв. АН СССР, ОХН, 1963, 75.
10. М и р о н о в В. Ф., Г а р Т. К., Изв. АН СССР, ОХН, 1963, 578.
11. Миронов В.Ф., ГарТ. К., ДАН СССР, 152, 1111 (1963).
12. МироновВ.Ф., Кравченко А.Л., Петров А.Д., ДАН СССР, 155, 843
(1964).
13. Миронов В. Ф., ГарТ. К., Изв. АН СССР, серия хим., 1964, 1515.
14. МироновВ.Ф., ГарТ. К., Изв. АН СССР, серия хим., 1965, 291.
15. Миронов В. Ф., ГарТ. К., Изв. АН СССР, серия хим., 1965, 755.
16. Нефедов О. М., К о л е с н и к о в С. П., Изв. АН СССР, серия хим., 1963, 2068.
17. Н е ф е д о в О. М., Колесников С. П., X а ч а т у р о в А. С, Пет-
ровА. Д., ДАН СССР, 154, 1389 (1964).
Присоединение германийорганических соединений по кратным связям 73
18. МироновВ.Ф., Г а р Т. К., Изв. АН СССР, серия хим., 1966, 482.
19. Колесникове. П., Нефедов О. М., Ж. Всесоюзн. хим. общ. им. Д. И.
Менделеева, 10, 478 (1965).
19а. Колесникове. П., Нефедов О. М., Ш е й ч е н к о В. И., Изв. АН СССР,
серия хим., 1966, 443; Angew. Chem., 76, 498 (1964).
196. К о 1 е s n i k о v S. P., Nefedov O.M., Angew. Chem., 77, 345 (1965).
20. H e ф е д о в О. М., К о л е с н и к о в С. П., Новицкая Н. Н, Изв. АН СССР,
серия хим., 1965, 5791.
21. Г а р Т. К., Миронов В. Ф., Изв. АН СССР, серия хим., 1965, 855.
22. G i 1 m a n H., G е г о w С. W., J. Am. Chem. Soc, 79, 342 (1957).
23. F u с h s R., GilmanH, J. Org. Chem., 22, 1009 (1957).
24. R о h a M. E., В e e a r s W. L., Пат. США 2977381 (1961); РЖХим., 1962, 6Л100.
25. H e n г у М. С, DowneyM.R, J. Org. Chem., 26, 2299 (1961).
26. LesbreM., SatgeJ., Compt. rend., 250, 2220 (1960).
27. S atge J. Ann. de chim., 6, 519 (1961).
28. N о 1 1 e s J. G., van Kerk G. J.M., Rec. trav. Chem., 81, 24 (1962).
29. LesbreM, SatgeJ., Compt., rend., 247, 471 (1958).
30. Гвердцители И. М., Г у н ц а д з е Т. П., Петров А. Д., ДАН СССР, 153,
107 (1963).
31. Гвердцители И. М., Б у а ч и д з е М. А., ДАН СССР, 158, 147 (1964).
32. Гвердцители И. М., Б у а ч и д з е М. А., Сообщ. АН ГрузССР, 37, 323
(1965); РЖХим., 1965, 22Ж253.
33. Гвердцители И. М., Г у н ц а д з е Т. П., П е т р о в А. Д., ДАН СССР,
157, 607 (1964).
34. Гвердцители И. М., Б у а ч и д з е М. А., Сообщ. АН ГрузССР, 37, 59 (1965);
РЖХим., 1965, 21Ж278.
35. Ш и х и е в И. А., А б д у л л а е в Н. Д., А л и е в М. И., ЖОХ, 36, 942 (1966).
36. Ш и х и е в И. А., А с л а н о в И. А., М е х м а н д а р о в а Н. Т., Верди-
е в а С. Ш., Азерб. хим. ж., 1965, 42; РЖХим., 1966, 6Ж360.
37. Ш и х и е в И. А, А с л а н о в И. А., М е х м а н д а р о в а Н. Т., ЖОХ, 36, 1295
(1966).
37а. А л и - 3 а д е И. Г., Ш и х е е в а М. И., С а л и м о в М. А., А б д у л л а е в Н. Д.
Ш и х е е в И. А., ДАН СССР, 173, 89 (1967).
38. R ij kens F., lanssen M. J., Drenth W., van der К e r k G. J. M., J. Organo-
metal. Chem., 2, 347 (1964).
39. M и x а й л о в Б. М., Б у б н о в Ю. Н., К и с е л е в В. Г., Изв. АН СССР, серия
хим., 1965, 68.
40. Braun J., Compt. rend., 250, 218 (1965).
41. L e s b r e M., S a t g с J., M a s s о 1 M., Compt. rend., 256, 1548 (1963).
42. GilmanH., E i s с h J., J. Org. Chem., 20, 763 (19Г>Г>).
43. M e e n R. H., Oilman H., J. Org. Chem., 22, 684 (1957).
44. Несмеянов А.Н., Б о р и с о в А. Е., ДАН СССР, 174, 96 (1967).
45. S a t g ё J., LesbreM., Bull. Soc. Chim. France, 1965, 2578.
46. LesbreM., SatgeJ., MassolM, Compt. rend., 257, 2665 (1963).
47. Lesbre M., SatgeJ., MassolM., Compt. rend., 258, 2842 (1964).
48. Б у р л а ч е н к о Г. С, А в д е е в а В. И., Б а у к о в Ю. И., Л у ц е н к о И. Ф.,
ЖОХ, 35, 1881 (1965).
49. L u t senko I. F., В a u k о v Yu. I., Burlachenko G. S., J. Organometal,
Chem., 6, 4)6 (l'J66>.
50. В о л ь п и н М. Е., Курса нов Д. Н., Изв. АН СССР, ОХН, 1960, 1903.
51. Вольпин М. Е., К у р с а н о в Д. Н., ЖОХ, 32, 1455 (1960).
52. Vo 1 р i n М. Е., Koreshkov Yu. D, Dulova V.G, KursanovD. П.,
Tetrahedron, 18, 107 (1962).
53. К у р с а н о в Д. Н., В о л ь п и н М. Е, Ж. Всесоюзн. хим. общ. им. Д. И.
Менделеева, 7, 282 (1962).
54. ВольпинМ.Е., Дулова В. Г., К у р с а н о в Д. Н., Изв. АН СССР, ОХН,
19(53, 727.
55. Л е й т е с Л. А., Д у л о в а В. Г., Вольпин М. Е., Изв. АН СССР, ОХН,
1963, 731.
56. В о л ь п и н М. Е., С т р у ч к о в Ю. Т., В и л к о в Л. В., М а с т р ю к о в В. С,
Дулова В. Г., Курсанов Д. Н.. Изв. АН СССР, серия хим., 1963, 2067.
57. Johnson F.Gohlke R. S., Tetrahedron Letters, 26, 1291 (1962).
58. Johnson F., GohlkeR. S., NasutavicusW. A., J. Organometal.
Chem., 3, 233 (1965).
59. M a z e г о 1 1 e s P., ManuelG, Bull. Soc. chim. Frans, 1966, 327.
Г лава VI
ДИАЗОМЕТОД СИНТЕЗА
ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛИФАТИЧЕСКИХ ДИАЗОСОЕДИНЕНИЙ
Диазометан при низкой температуре реагирует с четыреххлористым
германием, образуя с выходом 93,7% треххлористый хлорметилгерманий [1]
GeCl4 + CH2N3 -» Cl3GeCH2Cl + Na.
В качестве примеси образуется незначительное количество двухлористого
бш>хлорметилгермания.
Получение треххлористого хлорметилгермания [1]. В трехгорлую колбу (1л),
снабженную механической мешалкой, термометром для низких температур, капельной воронкой и
осушающей трубкой, помещают 28 г (0,12 моля) четыреххлористого германия в 130 мл
сухого эфира, 0,5 г медного порошка, затем реакционную смесь охлаждают до —60° С. При
энергичном перемешивании медленно прибавляют холодный раствор 0,128 моля диазометана
в 250 мл сухого эфира. Выделение азота начинается немедленно. После окончания
прибавления реакционную смесь перемешивают в течение 2 час. при температуре—60н 70° С
(при непрерывном пропускании тока сухого азота), затем дают реакционной смеси нагреться
до комнатной температуры. Перемешивание продолжают при комнатной температуре в
течение 2 час. Эфир отгоняют на колонке. Остаток перегоняют в вакууме. Получают 7,2 г
непрореагировавшего четыреххлористого германия и 20,8 г треххлористого
хлорметилгермания (т. кип. 60—65° С/35 мм; df 1,833; я^ 1,4989; выход 97,3%, считая да
прореагировавший четырехлористый германий). При перегонке более высококипящего остатка выделяют
некоторое количество двухлористого бис- хлорметилгермания ст. кип. 90° С/21 мм; п2р 1,5176.
При изучении реакции между диазометаном и треххлористым фенил-
германием, двухлористым дифенилгерманием и хлористым трифенилгерма-
нием [2] было найдено, что последний не реагирует с диазометаном. Однако
при действии избытка диазометана на треххлористый фенилгерманий
(соотношение 3 : 1) в присутствии медного порошка почти с количественным
выходом образуется хлористый фенил-бш>хлорметилгерманий. При
молярном соотношении реагентов 1:1, даже в отсутствие порошка меди,
образуется двухлористый фенил хлорметилгерманий. Фенилтрихлорметил германий
не был получен даже при использовании большого избытка диазометана.
Реакция двухлористого дифенилгермания с диазометаном приводит к
получению хлористого дифенилхлорметилгермания с выходом 26%; т. кип.
100° С/0,0004 мм\ пЬ° 1,5990.
Реакция треххлористого фенилгермания с диазометаном [2]. Эфирный раствор 19,2 г
(0,075 моля) треххлористого фенилгермания смешивают на холоду с 300 мл (0,25 моля)
раствора диазометана, затем добавляют небольшое количество медной пыли; по окончании
реакции отфильтровывают медь и образовавшийся побочно полиметилен. Эфир отгоняют.
После перегонки остатка получают 18 г хлористогофенил-бис-хлорметилгермания ст. кип.
93° С/0,1 мм; п2£ 1,5723; выход 86%.
Диазометод синтеза германийорган ических соединений
75
При действии 32,7 мл (0,025 моля) эфирного раствора диазометана на 64 г (0,025 моля)
треххлористого фенилгермания получают 4,7 г двухлористого фенилхлорметилгермания
с т. кип. 136° С/10 мм; п2р 1,5658; выход 70%. Реакция заканчивается в течение часа без
прибавления медного порошка.
При действии диазометана на гидриды алкилгермания в присутствии
медного порошка или при ультрафиолетовом облучении с очень небольшими
выходами происходит метилирование соответствующих гидридов.
Исследовалось действие диазометана на гидрид триэтил-, три-#-пропил- и три-я-бу-
тилгермания. Выходы соответственно 9; 5 и 2%. Крамер с сотр. [3] считал,
что диазометан не реагирует с гидридом трифенил германия. Однако Сатже
с сотр. [За] нашел, что тригидрид фенилгермания после обработки 50%-
ным избытком диазометана в эфирной среде при облучении УФ в течение
6 час. образует C6H5GeH2CH3 с выходом 59% и C6H5GeH(CH3)2 с выходом
14%. Одновременно тригидрид фенилгермания (21%) выделен без
изменения. При действии диазометана на дигидрид дифенил германия образуется
смесь дифенилдиметилгермания и гидрида дифенилметилгермания [За, 4).
Интересно отметить работы Сатже с сотр. [5,6], а также и других
авторов [7], которые показали, что при реакции германийорганических гидридов
с некоторыми диазопроизводными образуются германийорганические
соединения, имеющие функциональную группу в а-положении по отношению к
атому германия.
Так, в присутствии порошка меди диазоуксусный эфир, диазоацетон и
диазоацетофенон в эфирном или бензольном растворе реагируют с
гидридами алкилгермания с выделением азота
R3GeH + N2CHCOOC2H5 -* R8GeCHaCOOC2H5 + N2,
R3GeH + N2CHCOCH3-> R3GeCH2COCH3 + N2.
При реакции дигидридов алкилгермания с диазоуксусным эфиром
реакция идет по схеме
R2GeH2+ NaCHGOOQaHs -» R2Ge(H) CH2COOC2H5 + N2.
Получение триэтил(карбэтоксиметил)германия [6]
(C2H5)3GeH + NaCHCOOCaHs -* (C2H5)3GeCHCOOC2H5 + N2.
К 6,8 г (0,042 моля) гидрида триэтилгермания прикапывают при перемешивании раствор
7,18 г (0,063 моля) диазоуксусного эфира. В присутствии порошка меди при 60° С
начинается выделение газа, продолжающееся в течение часа. Затем реакционную смесь
нагревают в течение 3 час. При перегонке получают 1,4 г непрореагировавшего гидрида и 3,8 г
(37%) светло-желтой жидкости с едким запахом, которую вновь перегоняют при
атмосферном давлении (гонится без разложения)., Получают (G>H5)3GeCHaCOOCoH5; т. кип. 222—
224° С.
При аналогичной реакции из гидрида трибутилгермания и диазоук-
усного эфира при 60° С в растворе толуола получен трибутил(карб-
этоксиметил)германий с т. кип. 160—165° С/10 мм; df 1,0271; п% 1,4590;
выход 40%.
Триамил(карбэтоксиметил)германий (^-C5H11)3GeCH2COOC2H5 (т. кип.
188° С/14 мм) получен с выходом 35%.
Получение гидрида дибутил(карбэтоксиметил)германия [6]
(C4H9)2GeH2 + N3CHCOOCaH3 -» (C4H9)2Ge(H)CH2COOCaII;) -; N2.
Смесь 8,5 г (0,045 молей) дигидрида дибутилгермания и 16 г (0,140 моля) диазоуксусного
эфира в 30 мл бензола нагревают при 100° С в присутствии порошка меди в течение 8 час.
При перегонке получают две фракции сырых продуктов: 3,5 г монозамещенного
производного (т. кип. 130—135° С/12 мм) и 4,8 г дизамещенного производного (т. кип. 190° С/12 мм).
Повторная перегонка первого продукта дает моноэфир, содержащий связь германий — во-
76 Германийорганичсскис соединения
дород; т. кип. 132—134° С/12 мм; df 1,0521; п2£ 1,4540; выход 28%. Из второй фракции
чистый продукт выделен не был.
Получение триэтилгермилацетона [6]
(C2H5)3GeH + N2CHCOCH3-* (С2Н5)зОеСН2СОСН3 + N2.
8 г (0,05 моля) гидрида триэтилгермания добавляют к 8 г (0,095 моля) диазоацетона в
15 мл эфира. Реакцию ведут при температуре 30° С в присутствии порошка меди.
Реакционную смесь перемешивают в течение 8 час. Получают 1,2 г непрореагировавшего гидрида три-
бутилгермания и 3,2 г желтой подвижной жидкости с сильным запахом; т. кип. 110° С/20 мм;
d^° 1,0763; п2£ 1,4604; выход 30%. Производное с 2,4-динитрофенилгидразином имеет
т. пл. 105° С.
Аналогично реагирует гидрид триэтилгермания с диазоацетофеноном.
Выход триэтилгермилметилфенилацетона равен 31%; т. кип. 135° С/3 мм.
Соответствующий 2,4-динитрофенилгидразон имеет т. пл. 218—220 ° С.
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ СОЛЕЙ ДИАЗОНИЯ
По поводу применения ароматических диазосоединений в синтезе гер-
манийорганических соединений Несмеянов с сотр. [8] сообщает, что четы-
реххлористый германий не образует двойных солей с хлоридами арил-
диазониев.
При разложении борфторидов арилдиазония порошками металлов
(лучше всего цинковой пылью в среде ацетона в присутствии четыреххлористо-
го германия) были получены моноарилированные соединения германия.
Соединений германия с большей степенью арилирования в этих условиях не
образуется. Производные треххлористого арилгермания выделялись и
анализировались в виде ангидридов арилгерманиевых кислот. Ангидрид фе-
нилгерманиевой кислоты получен с'выходом 28%, для других арилов —
р-СН3ОС6Н4, р-С2Н5ОС6Н4, /?-BrCGH4, р-С1С6Н4 — выходы были ниже.
Разложение борфторида фенилдиазония в присутствии четыреххлористого германия в
ацетоне цинковой пылью [8]. Реакцию проводят в длинном узком стакане, снабженном
мешалкой и термометром. В охлажденную до —8° С взвесь (17,4 г цинковой пыли в растворе
25,1 г четыреххлористого германия в 80 мл сухого ацетона) при энергично работающей
мешалке вносят небольшими порциями в течение 20 мин. 50 г борфторида фенилдиазония.
Прибавление проводят с такой скоростью, чтобы температура не повышалась выше + 5° С.
Реакционную смесь перемешивают при охлаждении в течение 5 час. На следующий день в
реакционную смесь добавляют 40 мл сухого ацетона, перемешивание продолжают до полного
разложения соли диазония (отрицательная реакция сР-нафтолом). Реакционную смесь
обрабатывают 20%-ным раствором едкого натра. Образовавшийся осадок через 3 часа
отфильтровывают и промывают ацетоном. Из осадка 20%-ным раствором едкого натра при нагревании
и последующем подкислении концентрированной соляной кислотой извлекают основное
количество ангидрида фенилгерманиевой кислоты (5,58 г, т. е. 27,3% от теорет.). Для анализа
ангидрид фенилгерманиевой кислоты очищают многократным переосаждением соляной
кислотой из 10%-ного раствора едкого натра при нагревании с последующей промывкой
пиридином, водой, спиртом, эфиром.
ЛИТЕРАТУРА
1. S е у f е г t h D., R о с h о w E. G., J. Am. Chem. Soc, 77, 907 (1955).
2. KramerK., Wright A. N.. Ber., 96, 1877 (1963).
3. К г a m e r K. A. W., Wright A. N.. J. Chem. Soc, 1963, 3604.
3a. Satgc J., R i v i e г е P., Bull. Soc. Chim. France, 1966, 1773.
4. К г a m e r K. A. W., Wright A. N.. Angew. Chem., 74, 468 (1962).
5. LesbreM., Satgt'J., Compt. Rend., 247, 471 (1958).
6. S a t g ё J., Ann. de chim., 6, 519 (1961).
7. RijkensF., JanssenM. J., DrenthW., van der Kerk G. J. M.,
J. Organometal. Chem., 2, 347 (1964).
8. Несмеянов А, Н., Емельянова А. И„ М а к а р о в а Л. Г., ДАН СССР,
122, 403 (1958).
Глава VII
СИНТЕЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЗАМЕНОЙ ВОДОРОДНОГО АТОМА НА АТОМ ГЕРМАНИЯ
(германирование)
Установление непосредственной связи между углеродом и германием
при прямом действии неорганических соединений германия на
углеводороды или их диалкиламинопроизводные описано только в отдельных случаях.
Так, утверждается, что при действии четыреххлористого германия на N-
диметилаиилин, моноэтиланилии и другие соединения, имеющие
подвижный водород в пара-положении, образуются германийорганические
соединения, которые выделяют в виде ангидридов соответствующих
германиевых кислот* (без указания выходов).
Получение ангидрида /г-мэнометилачинофенилгерманиевой кислоты *. 25 г свежепере-
гнапного монометиланилина нагревают 36 час. при 100° С. в автоклаве с 10,7 г
четыреххлористого германия. Реакционную смесь разбавляют соляной кислотой, нейтрализуют щелочью
и удаляют монометиланилин перегонкой с паром. Из отфильтрованного щелочного раствора
при насыщении его углекислым газом выделяют ангидрид в виде бесцветной
кристаллической пыли, легко растворимой в щелочи и минеральных кислотах.
Аналогично получают ангидриды я-диэтиламинофенил- и я-диметил-
аминофенилгерманиевых кислот.
* В а и е г Н., В и г s с h k i e s К., Вег., 65, 956 (1932).
Глава VIII
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ГЕРМАНИЯ
THnA(R3Ge)„, (R2Ge)rt, (RGe)„
Все описанные в литературе попытки получения соединений типа R3Ge,
R2Ge, RGe приводили к их полимерам.
Так, Краус и Браун [1] при синтезе органических соединений
двухвалентного германия в кипящем ксилоле по уравнению
(C6H5)2GeCl2 -f 2Na -» (C6H5)2Ge + 2NaCl
получили, судя по молекулярному весу, тетрамер.
Саммерс [2] сообщил о выделении дифенилгермания при реакции дву-
иодистого германия с фениллитием без приведения данных о молекулярном
весе. Попытки получить алкильные производные двувалентного германия
взаимодействием двуиодистого германия с диэтил- и ди-я-бутилртутью,
этил- и #-бутиллитием, ди-я-бутил- и диизобутилцинком оказались
неудачными [3]. Единственным производным германия, которое удалось
выделить, был продукт состава (C4H9)4Ge2J2, по предположению авторов
(C4H9)2JGe — GeJ (C4H9)2. Гилман и Героу [4] предположили, что трифенил-
гермиллитий диссоциирует на дифенилгерманий и фениллитий, однако им
не удалось получить никаких доказательств образования органических
соединений двухвалентного германия.
В результате попытки восстановления треххлористого фенилгермания
металлическим калием был получен полимерный продукт состава (C6H6Ge)6,
окончательная структура полученного соединения не была установлена
[5]. Дальнейшие попытки восстановления треххлористого фенилгермания
металлическим калием или амальгамой лития не дали хорошо
идентифицированных продуктов [6]. Попытка получить соединения типа R2Ge по
аналогии с соединениями олова путем пиролиза дигидрида
дифенилгермания при температуре 280—340° [7] оказалась неудачной. В результате
реакции были получены металлический германий, водород, бензол и тетра-
фенилгерманий. Авторы предполагают, что реакция протекает через стадию*
промежуточного образования дифенилгермания
(CGH5)2GeH2 -* Н2 + СбНб + (C6H5)2Ge,
(C6H5)2Ge -* (C6H5)4Ge + Ge.
Были сделаны попытки получить органические производные
двухвалентного германия [7] при действии на GeJ2 фениллития в эфире, бромистого фе-
нилмагния в тетрагидрофуране, трифенилалюминия в тетрагидрофуране
и толуоле и бромистого мезитилмагния в тетрагидрофуране. В результате-
проведенных исследований показано, что при реакции GeJ2 с фениллитием
при —25° С образуется темно-красное нерастворимое в эфире масло, из*
Соединения германия типа (R3Ge)rt, (R2Ge/z) и (RGe)^ 79
которого было выделено оранжевое разлагающееся на воздухе соединение;
ему была приписана формула (C6H5)2Ge2J; авторы считают, что полученный
продукт не является однородным, а представляет собой смесь иодидовС6Н5ОеJ
и (C6H5)2Ge-GeJ. При реакции избытка фениллития с GeJ2, например в
соотношении 2:1,былполучентетрафенилгерманий, красно-коричневый
аморфный полимер фенилгермания (C6H5Ge)10; главным продуктом реакции был
желтый аморфный полимер с молекулярным весом от 800 до 1700, который
являлся смесью фенил- и дифенил германия. При применении большого избытк а
фениллития был также получен тетрафенилгерманий, гидрид трифенил-
германия [последний образуется в результате гидролиза (C6H5)3GeLi] и
полимер фенилгермания, который, очевидно, является смесью полимеров
(R3Ge)n, (R2Ge)n и (RGe)n. Аналогичные продукты были получены и при
действии на GeJ2 реагентов C6H5MgBr, (C6H5)3A1 и (CH3)3C6H2MgBr.
Авторы делали попытку доказать структуру образующихся соединений
бромированием полученных полимеров, действием бромистого этилмагния
на продукты бромирования и дальнейшим хроматографическим анализом
полученных продуктов.
ЛИТЕРАТУРА
1. К г a u s С. A., BrownC.L, *J. Am. Chem. Soc, 52, 4031 (1930).
2. S u m m e r s L., Jowa State Coll. J. Sci., 26, 292 (1952).
3. J а с о b s G. Compt. rend., 238, 1825 (1954).
4. G i 1 m a n H., G e г о w С. W., J. Org. Chem., 23, 1582 (1958).
5. S с h w a r z R., Schmeisser M., Ber., 69, 579 (1936).
6. MetlesicsW., ZeissH., J. Am. Chem. Soc, 82, 3321 (1960).
7. Glockl ing R, H о о t о n К. A., J. Chem. Soc, 1963, 1849.
Глава IX
ДЕАЛКИЛИРОВАНИЕ
ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Деалкилирование полнозамещенных германийорганических соединений
с целью получения R3GeX, R2GeX2 и RGeX3 довольно широко применяется
в химии германийорганических соединений. В качестве деалкилирующих
агентов чаще всего используют галоиды (бром, иод), галоидоводородные
кислоты, галоидные соли германия (GeCl4, GeBr4). В последнее время
наблюдалась реакция деалкилирования и с помощью галоидных алкилов.
Однако реакции деалкилирования германийорганических соединений протекают
значительно сложнее и труднее, чем, например, в случае оловоорганиче-
ских соединений.
Для германийорганических соединений характерно, что реакция
деалкилирования обычно приводит к трудно разделяемым смесям галогенидов
алкилгермания различной степени алкилирования. Без катализатора в
алифатическом ряду деалкилирование можно проводить, например, при
действии брома и фтористого водорода лишь с отрывом одного радикала.
В ароматическом ряду при действии брома (без катализатора) можно
получить и соединения типа Ar2GeX2. В качестве катализаторов обычно
применяют галоидные соли алюминия, в отдельных случаях алюминийорганиче-
ские соединения. Применение солей других металлов возможно, но выходы
при этом ниже. Легче всего отщепляются радикалы, содержащие тройную
связь, затем аллильные и арильные; наиболее сложно протекают реакции
с алифатическими соединениями. Винильные соединения германия более
склонны к реакциям присоединения, чем к реакциям расщепления (см.
стр. 115).
При кипячении полнозамещенных германийорганических соединений
с бромом в различных растворителях довольно легко происходит замена
одного из радикалов на бром. Второй радикал заменяется гораздо
труднее, как правило, в присутствии катализаторов типа бромистого алюминия,
особенно в алифатическом ряду. Например, при кипячении тетраметилгер-
мания с бромом в бромистом пропиле легко получается бромистый триме-
тилгерманий. При дальнейшем действии брома на бромистый триметил-
германий в присутствии каталитических количеств бромистого алюминия
получен двубромистый диметилгерманий [1].
Получение бромистого триметилгермания [1]. К 200 г тетраметилгермания в 100 мл
бромистого пропила добавляют частями 256 г брома. После 20 час. кипячения температура
смеси поднимается с 47 до 78° С. Ректификацией на колонке получают 290 г бромистого
триметилгермания (98%); т. кип. 113,5 С/735 мм; df 1,5604; п^ 1,4672.
Получение двубромистого диметилгермания [1]. К 20 г бромистого триметилгермания
добавляют 0,1 г А1Вг3 и 16,5 г брома. После кипячения в течение часа реакционную смесь
перегоняют на колонке. Получают 21 г двубромистого диметилгермания (79%); т. кип.
152,5° СПЩмм, df 2,1145; п™ 1,5280.
Деалкил ирован ие
81
Бромистый триэтилгерманий был получен при бромировании тетраэтил-
германия без растворителя [2, 3] или в бромистом этиле [4], выходы близки
к количественным. Бромистые трипропил- [5], трибутил- [6—8] и трибензил-
германий [9] получены аналогично.
Действие брома на ароматические полнозамещенные германийорганиче-
ские соединения, например на тетрафенилгерманий [8, 10—19] или другие
Ar4Ge [20], с целью получения Ar3GeBr проведено рядом авторов. Второй
ароматический радикал заменяется на галоид гораздо труднее. Вообще
метод не является удобным в препаративном отношении, так как приводит
к смеси галоидопроизводных германия разной степени алкилирования,
которые трудно расфракционировать. Одним из приемов разделения
является предварительное восстановление их с помощью литийалюминийгидрида
или перевод в окиси при действии щелочи с последующей разгонкой.
Получение бромистого трифенилгермания [15]. Раствор 58,4 г тетрафенилгермания
(0,15 моля) в 400 мл 1,2-дибромэтана помещают в трехгорлую колбу объемом 1 л,
снабженную холодильником, капельной воронкой и механической мешалкой. Раствор нагревают
до кипения и прикапывают 25,6 г (0,16 моля) брома. Реакционную смесь кипятят в течение
получаса. Растворитель и небольшое количество непрореатировавшего брома отгоняют в
вакууме. Получают около 50 мл остатка, из которого бромистый трифенилгерманий при
охлаждении выделяется в виде кристаллов. Сырой продукт перегоняют в вакууме и затем
дважды кристаллизуют из ледяной уксусной кислоты. Выход от 44 до 48 г (от 75 до 82%),
т. пл. 136—137° С. Бромистый трифенилгерманий легко растворим в обычных органических
растворителях, таких, как хлороформ, четыреххлористый углерод, бензол, толуол;
медленно гидролизуется водой и более быстро-— щелочами. В результате гидролиза образуется не
гидроокись, а гексафенилдигерманоксан.
Получение двубромистого дифенилгермания [14] действием брома на
тетрафенилгерманий с последующим восстановлением литийалюминийгид-
ридом приведено на стр. 126.
Действие брома на несимметричные германийорганические соединения
в литературе изучено на отдельных примерах. При бромировании трифе-
нил-я-толилгермания в четыреххлористом углероде на свету происходит
замена водорода метильной группы на бром. Бромирование в темноте
приводит к разрыву связи Ge — С. Изучение кинетики действия брома на
(C6H5)3Ge С6Н4Х позволило авторам [21] в порядке возрастания скорости
реакции расположить заместители X в следующий ряд:
p-(CH3)2N > р-ОСНз > о-ОСНз > р-СНз > о-СН3> ™-СН3.
В несимметричных полнозамещенных германийорганических соединениях,
имеющих алифатические и ароматические радикалы, в первую очередь
отщепляются ароматические.
Мазеролл [22—24], испытывая прочность цикла с участием атома
германия, показал, что бром реагирует на холоду с дифенилциклотетрамети-
ленгерманием с отрывом двух фенильных радикалов. В тех же
экспериментальных условиях бром не реагирует с диэтилциклотетраметиленгерма-
нием. Напротив, в присутствии бромистого алюминия реакция происходит
немедленно по схеме
СН2—Cri24 AIRr,
| >Ge(C2H5)2 + Br _-L (C2H5)2GeBr2+ C4H8.
CH3—CH2/
При действии брома на я-пропилфенилциклотетраметиленгерманий легко
образуется бромистый я-пропилциклотетраметиленгерманий [24].
При реакции брома с диэтилциклотриметиленгерманием [231 уже на
холоду происходит разрыв связи Ge — С
/СН2ч
(CoH5)2Ge( >СН2 + Br2-* (C2H5)2Ge(Br)CH2CH2CH2Br.
хсн2/
6 Заказ Ш 207
82 Германийорганические соединения. Лит. стр. 87—88
В ненасыщенных германийорганических соединениях связь Ge — С
в случае винильных радикалов более прочная по сравнению с аллильными
и другими ненасыщенными радикалами. При действии брома (при—80° С)
происходит присоединение брома по двойной связи к винилтрибутилгерма-
нию. При реакции с аллилтрибутилгерманием даже при такой низкой
температуре бром отщепляет аллильный радикал [25]. С триэтилциклопента-
диенилгерманием бром в растворе бромистого этила реагирует по уравнению
[26]
(СоЬУзСеСзНз + Вг-^ (С2Н5)зОеВг + полимерный остаток.
Аналогично действует бром и на трибутил-|3-стирилгерманий [27], а также
и на соединения, имеющие ацетиленовые радикалы [28—30]. При реакции
с гексаалкил- и гексаарилдигерманием бром количественно разрывает связь
Ge —Ge [3, 31].
Действие иода на тетраалкильные соединения германия
исследовалось не раз [6, 32, 33], однако лишь Лесбр и сотр. [34] разработали
количественный метод получения иодидов алкилгермания при действии иода на
R4Ge в присутствии трехиодистого алюминия. Например, при действии
эквимолекулярного количества иода на тетраметил- и тетраэтилгерманий
реакция идет по схеме
R4Ge + J2 -* R3GeJ + R J. (1)
Однако для германийорганических соединений, начиная от тетрапропил-
германия, процесс идет иначе:
2R4Ge + J3-> 2RsGeJ + R — R. (2)
Кроме углеводорода типа R—R, в этой реакции были выделены и
низшие алкилы. Реакция (2) не имеет места в случае органических соединений
олова.
По Мазероллу [34], в известных условиях иод может последовательно
разрывать две и даже три связи Ge—С (исследовано на примере тетраэтил-
германия). По методу Мазеролла был получен другими авторами йодистый
триметилгерманий [35] и йодистый трибутилгерманий [7]. Иод, так же как
и бром, реагирует с триэтилаллилгерманием без катализатора при 20° С>
отрывая алл ильный радикал [25].
Из всех галоидоводородов наиболее удобным реагентом для реакции
деалкилирования является фтористый водород. Однако даже при действии
избытка фтористого водорода не удается отщепить более одной алкильной
группы. Метод позволяет синтезировать фтористый триалкилгерманий в
чистом виде без примеси ди- и трифторпроизводных [36], что делает его
удобным в препаративном отношении.
Получение фтористого триметилгермания [36]. В медный цилиндр, охлажденный
твердой углекислотой и снабженный медным обратным холодильником, помешают 13 г тетраме-
тилгермания и быстро приливают 10 г фтористого водорода. Реакция начинается при
температуре —10 -? 15° С и почти полностью заканчивается при 20—25° С. Для окончания
реакции реакционную смесь нагревают 2—3 часа на водяной бане при 50—55° С. Окончание
реакции определяют по прекращению выделения метана, собираемого в газометр. Для
удаления избытка фтористого водорода реакционную смесь обрабатывают безводным
фтористым калием, декантируют и перегоняют. Выход фтористого триметилгермания 9,6 г (71%);
т. кип. 77—78° С/755 мм; df 1,2114; п™ 1,3855.
Аналогично получают фтористый триэтилгерманий; т. кип. 147—
148° С/744 мм, df 1,1527; п™ 1,4206; выход 71%.
Мазеролл показал [23], что галоидоводородные кислоты реагируют
количественно с диалкилциклотриметиленгерманием, разрывая связь
Ge — С в цикле
Деалкил ирован ие
83
СН2
(С4Н9)2 Ge СНа + ИХ -* (QH9)2 Ge (X) CH2CH2CII3.
CH2
Остальные галоидоводороды реагируют с R4Ge лишь в присутг ЛШИ г-а-
лоидных солей алюминия. Применяя газообразный хлористыг L ВОДОрод
в присутствии хлористого алюминия, получают хлористый триу ,етилгерма-
ний (95%) [37]. Газообразный бромистый водород также лип- ib B присут.
ствии бромистого алюминия [38] отрывает метальную группу о /Т тетраметил-
германия и от ряда тетраарильных соединений германия [39]. Авторы
указывают, что радикалы можно расположить в следующий А ряд по степени
трудности их отщепления: n-толил, ж-толил, фенил, бензи л>
Концентрированная соляная кислота на холоду не г действует на тетра-
метилгерманий [33]. Бромистоводородная кислота прт л ~80° С
присоединяется к винильному радикалу три-н-бутилвинилг ермапия [25]. В тех
же условиях при действии на три-я-бутилаллилгерм'аНий
бромистоводородная кислота отщепляет аллильный радикал с образ ованием бромистого три-
и-бутилгермания и пропана.
Иборн и Панд в ряде работ изучили кинетику реакции расщепления
различных германийорганических соединений при действии водного или
спиртового раствора хлорной кислоты [40—-45] или спиртового раствора
щелочи [46, 47]. Действие щелочи на перфтортетравинил германий
исследовали Кнунянц и сотр. [48].
Сильные органические кислоты, например хлоруксусная кислота, не
реагируют с насыщенными тетраалкильными соединениями германия,
однако легко отщепляют ненасыщенные радикалы. Например, при действии
дихлоруксусной кислоты на аллилфенилциклотетраметилеигерманий легко
идет следующая реакция (выход 88%) [24]:
СН2 — СНг СбН^
\ /
Ge + СНС12СООН ->
/ \
СИ-. —СНо СН2СН=СН2
сн2-сн2 с6п5
\ /
Ge +CH3CH=CII3.
/ \
СНа - СН2 ООССНС12
Аналогично происходит отщепление радикалов, имеющих тройные
ненасыщенные связи [28].
В литературе описан один пример [49] действия 70%-ного раствора
азотной кислоты в уксусном ангидриде на (C2H5)3GeC6H4Ge(C2H5)3.
Реакция при этом идет следующим образом:
jHNOg
(С2Н5)з GeC6H4Ge(C2H3)3 > (С2НГ)): GeCGH4N02.
Реакция была проведена с мета- и пара-изомерами.
Концентрированная серная кислота на холоду реагирует с диэтил-
циклотетраметиленгерманием, раскрывая кольцо и разрывая связь Ge—С
122].
Смесь концентрированной азотной и серной кислот полностью сжигает
молекулу германийорганического соединения; метод применяют для
анализа германийорганических соединений (см. стр. 162).
Описывается действие сернистого газа на />(C2H5)3GeCoH4Ge(C2H5)3 в
четыреххлористом углероде с образованием (74%) /г"-триэтилгермилбензол-
сульфокислоты [50].
6*
84
Германийорганические соединения. Лит. стр. 87—88
О деалкилировании с помощью солей германия в литературе известен
лишь пример образования хлористого трифенилгермания в очень жестких
условиях (нагревание при 350° С в течение 36 час.) из четыреххлористого
германия и тетрафенилгермания [20, 51]. Обычно переход алкильных групп
от R4Ge KGeCl4 без катализатора не имеет места [3, 52].
Мазеролл и сотр. [53] впервые сообщили, что реакция
перераспределения радикалов, приводящая к образованию соединений типа R2GeX2, на
примере двубромистого диэтил- и дибутилгермания происходит с выходом
соответственно 85 и 73% при нагревании в запаянных трубках тетраалкиль-
ных соединений германия с четырехбромистым германием в присутствии
больших количеств бромистого алюминия. Аналогично получают и дву-
бромистый дифенилгерманий [26].
Ван-дер-Керк с сотр. [52, 54] подробно изучил реакцию деалкилирования
германийорганических соединений галоидными солями германия. Авторы
показали, что,при нагревании в запаянных трубках при температуре 200° С
четыреххлористого германия и тетрабутилгермания в присутствии
хлористого алюминия легко происходит реакция перераспределения радикалов
с образованием хлористого трибутилгермания и двухлористого
дибутилгермания [52]. Треххлористый бутилгерманий образуется, но с низким
выходом. В дальнейшем ими была показана возможность образования и трех-
хлористого бутилгермания [54]. Было обнаружено, что образующийся
в процессе реакции треххлористый бутилгерманий и двухлористый дибу-
тилгерманий легче алкилируются тетрабутилгерманием, чем четыреххлори-
стый германий. Таким образом, в первую очередь в процессе реакции
образуется хлористый трибутилгерманий, который в свою очередь выступает как
алкилирующий агент по отношению к менее алкилированным продуктам.
Реакция может быть представлена следующими схемами, подтвержденными
на многочисленных примерах.
(С4Н9)4 Ge + GeCU -* (QH9)3 GeCl + QH9GeCl3 (медленно),
(С4Н9)4 Ge + C4H9GeCl3 -* (С4Н9)3 GeCl + (С4Н9)2 GeCl2 (быстро),
(С4Н9)4 Ge + (QH9)2 GeCl2 -> 2 (C4H9)3 GeCl (быстро);
3 (C4H9)4 Ge + GeCl4 -> 4(C4H9)3 GeCl,
(C4H9)3 GeCl + GeCl4 -> (QHgb GeCl2 + (CiH9) G2CI3 (медленно),
(QH9)3 GeCl + (QHb) GeCl3 -> 2 (C4H9)2 GeCl2 (быстро);
2(QH9)3 GeCl + GeCl4 -> 3(QH9)2 GeCl3.
Специальным исследованием показано, что при приготовлении трех-
хлористого бутилгермания из тетрабутилгермания и четыреххлористого
германия даже применение избытка последнего не предотвращает
образования дихлорида. Удлинение времени реакции незначительно повышает
выход C4H9GeCl3. Повышение температуры вызывает побочные реакции.
Например, через 6 час. при 250° С выход треххлористого бутилгермания равен
45%. По мнению авторов [54], оптимальными условиями при приготовлении
треххлористого бутилгермания являются высокая температура и короткий
период проведения реакции.
Хлористый алюминий в качестве катализатора реакции
деалкилирования тетраалкильных соединений германия применялся довольно широко.
Так, были исследованы реакции GeCl4 с тетраметил-, тетраэтил-, тетрапро-
пил-, тетраамил-, тетрагексил- и тетрагептилгерманием, а также и с тетра-
фенилгерманием. Реакция между четыреххлористым германием и
тетрабутилгерманием заканчивается через 5 час. при 200° С. Для завершения
реакции с тетраметил-, тетраэтил-, и тетрапропилгерманием требуется
более продолжительное время. Деалкилирование тетрафенилгермания
происходит быстрее и при более низкой температуре. Уже при 120° С выход
Деалкилирование
85
хлорида и дихлорида достигает 80%, если исходить из GeCl4 и (C6H5)4Ge или
(C6H5)3GeCl. Даже образование треххлористого фенил германия полностью
заканчивается через 6 час. при 125 ° С.
Авторы считают [54], что каталитическая активность хлористого
алюминия основывается на промежуточном образовании галогенидов алкил-
алюминия, являющихся переносчиками алкильных радикалов. В ряде
экспериментов хлористый алюминий был заменен тризтилалюминием, три-
бутилалюминием, хлористым диэтилалюминием или двухлористым бутил-
алюминием, причем выходы были равны выходам при условии применения
хлористого алюминия. Некоторые другие соединения, такие как безводный
хлористый цинк, хлористый бериллий, хлористый бор, треххлористый
фосфор, хлорокись фосфора, пятихлористый фосфор или смесь двух
последних соединений, обладают более слабым каталитическим действием.
При деалкилировании при атмосферном давлении верхний
температурный предел лимитируется температурой кипения четыреххлористого
германия (83° С).
Так как реакция деалкилирования фенильных производных германия
происходит при .более низкой температуре, то ее с успехом можно вести в
открытых сосудах. В случае бутильных производных, где требуется более
высокая температура, применяют следующий прием: смесь тетрабутилгер-
мания и хлористого алюминия нагревают до 200° С, а четыреххлористый
германий прибавляют лишь постепенно, поддерживая температуру
реакционной смеси 195—200° С (образование моно- и дихлорида).
Авторы пришли к выводу, что реакция деалкилирования при
атмосферном давлении в присутствии хлористого алюминия или алкильных
производных алюминия может быть применена для получения всех трех типов
фенильных производных германия и для получения соединений типа R3GeCl
и R2GeCl2 (где R — С3Н7 и другие высшие алкильные радикалы),
приготовление же (C2H5)3GeCl и (С2Н5)2 GeCl2 может быть проведено лишь в
запаянных сосудах. Для получения производных метилгермания предпочтительно
применять другие методы, например прямой метод (см. стр. 19).
Реакция деалкилирования тетрабутилгермания четыреххлористым германием [52].
Для достижения максимального превращения тетрабутилгермания и четыреххлористого
германия в продукты с промежуточной степенью алкилирования необходима температура
около 150° С и присутствие хлористого алюминия. В последующей работе [54] авторы
указывают оптимальную температуру нагревания реакционной смеси 200° С и
продолжительность нагревания 4—5 час.
Если исходные реагенты берут точно в стехиометрических соотношениях, образуются
галогениды бутилгермания с более низкой степенью алкилирования, чем можно ожидать,
поэтому для предотвращения образования других хлоридов бутилгермания рекомендуется
применять избыток 10—20% тетрабутилгермания по сравнению с расчетным количеством.
Таким образом можно получить хлористый трибутилгерманий или двухлористый б^тилгер-
маний с выходом выше 80%. Для обработки реакционной смеси применяют следующую
схему:
1) Реакционную смесь подвергают перегонке в вакууме на эффективной колонке, при
этом выделяют четыреххлористый германий (охлажденная ловушка), треххлористый бутил-
германий и двухлористый дибутилгерманий. 2) Остаток, содержащий тетрабутилгерманий и
хлористый трибутилгерманий, гидролпзуют водным растЕором едкого натра для пеусвода
хлористого трибутилгермания в гексабутилдигерманоксан. 3) После экстрагирования пстро-
лейным эфиром проводят перегонку для выделения непрореагироЕавшего
тетрабутилгермания. 4) Остаток, содержащий гексабутилдигерманоксан, обрабатывают соляной кислотой;
образовавшийся хлористый трибутилгерманий экстра1ируют петролейным эфиром и
перегоняют.
Получение хлористого трибутилгермания [54а]. 35,5 г (0,166 моля) четыреххлористого
германия прибавляют по каплям в течение 90 мин. к смеси 159 г (0,528 молей)
тетрабутилгермания и 12,8 г (98 ммолей) треххлористого алюминия, нагретой до 200° С. По окончании
прибавления реакционную смесь нагревают в течение 15 мин., затем промывают несколько раз
200 мл 12 N соляной кислоты и экстрагируют петролейным эфиром. Органические слои
соединяют и сушат над хлористым кальцием. Растворитель отгоняют и остаток фракционируют.
Получают 164 г хлористого трибутилгермания; т. кип. 131 —133° С/17 мм\ п2£ 1,4650;
выход 84,6%.
S6
Германийорганические соединения. Лит. стр. 87—88
Получение двухлористого дибутилгермания [54а]. 8,4 г (39,2 ммоля) четыреххлористого
германия прибавляют по каплям в течение 2 час. к смеси 13,9 г (46,2 ммоля)
тетрабутилгермания и 1 г (7,5 ммоля) хлористого алюминия при температуре 200° С. По окончании
прибавления нагревание продолжают в течение 3 час. При перегонке в вакууме собирают
основную фракцию при т. кип. 98—106° С/10 мм; п^ 1,4730. Получают 15,6 г двухлористого
дибутилгермания; выход 70,9%.
Получение треххлористого бутилгермания [54а]. Смесь 21,4 г (0,1 моля)
четыреххлористого германия, 6 г (0,02 моля) тетрабутил германия и 1 г (7,5 ммоля) хлористого алюминия
нагревают в трубке Кариуса в течение 5 час. при 200° С. Реакционную смесь перегоняют в
вакууме. При этом получают 14,7 г четыреххлористого германия, 4 г треххлористого бутил-
германия (15,6%) и 4,7 г двухлористого дибутилгермания (15,2%). Треххлористый бутил-
германий — бесцветная жидкость с острым запахом (т. кип. 69°С/12 мм\ d20 1,4496; 72^1,4761);
легко гидролизуется водным раствором едкого натра с образованием натриевой соли бутил"
германиевой кислоты.
Получение хлористого триамилгермания и гексаамилдигерманоксана [54]. 250 г (0,7
моля) тетраамилгермания и 5,6 г (0,04 моля) возогнанного хлористого алюминия нагревают до
200° С. В течение 3 час. прибавляют 45,5 г (0,21 моля) четыреххлористого германия, при этом
температура реакционной смеси колеблется между 200 и 210° С. Отекание с обратного
холодильника прекращается после 13 час. дополнительного нагревания. Реакционную смесь
промывают концентрированной соляной кислотой, масляный слой отделяют. Водный слой
экстрагируют петролейным эфиром, полученный экстракт соединяют с масляным слоем и сушат.
Небольшой образец реакционной смеси осторожно перегоняют и получают чистый
хлористый триамилгерманий; т. кип. НО—114° С/0,3 мм; п2£ 1,4656. Боле низкокипящая фракция
имеет более высокое содержание хлора, чем это соответствует содержанию хлора в хлористом
триамилгерманий. После отгонки этой части (от главной массы) получают 230,9 г (78,8%)
практически чистого хлористого триамилгермания; п~^ 1,4690. Хлористый триамилгерманий
гидролизуют при кипячении 30%-ным водным раствором едкого натра. После экстракции
петролейным эфиром, сушки и перегонки получают 180,5 г чистого
гексаамилдигерманоксана; т. кип. 156—158°С/0,06 мм; п2р 1,4656; выход 67,3%, считая на исходный тетраамил-
германий.
Получение хлористого тригептилгермания [54]. Тетрагептилгерманий (37,6 г, 0,08 моля)
и возогнанный хлористый алюминий (0,7 г, 0,005 моля) нагревают до 210° С. Через 15 мин.
прибавляют по каплям 5,35 г (0,025 моля) четыреххлористого германия, при этом
температуру реакционной смеси поднимают до 210° С. Отекание с холодильника прекращается через
30 мин. Температуру реакционной смеси поддерживают в течение часа при 210° С и затем
охлаждают. Перегонку реакционной смеси проводят в высоком вакууме, при этом в
охлаждающей ловушке собирают лишь 0,4 г жидкости (п2^ 1,3920). Остаток промывают
концентрированной соляной кислотой, водный слой экстрагируют петролейным эфиром,
полученный экстракт соединяют с масляным слоем, сушат и перегоняют. Получают следующие
фракции. Фракция I: 1,8 г; т. кип. 120—150° С/0,12 мм; п2£ 1,4697; фракция II: 29,7 г; т. кип.
152° С/0,12 мм; п™ 1,4671.
Получение хлористого трифенилгермания [54а]. Смесь 28,4 г (75 ммолей) тетрафенилгер-
мания, 0,9 г (6,8 ммолей) хлористого алюминия и 5 г (23 ммоля) четыреххлористого германия
нагревают в течение 4 час. при 120° С. Реакционная смесь становится гомогенной и застывает
при охлаждении и кристаллизации из петролейного эфира. Получают 25,6 г хлористого
трифенилгермания с т. пл. 114° С. Из маточного раствора выделяют еще 3,9 г вещества с т. пл.
109—110° С; выход 83,7°/0.
Получение двухлористого дифенилгермания [54а]. Смесь 16,4 г (A3 ммоля) тетрафенил-
германия, 1 г (7,5 ммоля) хлористого алюминия, 8,4 г (39,2 ммоля) четыреххлористого
германия нагревают при 120° С в течение 4 час. Реакционная смесь остается жидкой при
комнатной температуре, ее промывают концентрированной соляной кислотой и экстрагируют
бензолом. Полученную бензольную вытяжку сушат и перегоняют. Получают 18 г фракции
ст. кип. 118—125° С/0,5 мм; п2£ 1,6003; выход 80,5%. Чистый двухлористый дифенилгер-
маний имеет т. кип. ПО—120° С/0,15 мм; п2£ 1,5975.
Получение треххлористого фенилгермания [54а]. Смесь 13,75 г (64,1 ммоля)
четыреххлористого германия, Юг (26,3 ммоля) тетрафенилгермания и 0,7г (5,3 ммоля) хлористого
алюминия нагревают в трубке Кариуса в течение 6 час. при 125° С. Реакционную смесь для
удаления хлористого алюминия промывают концентрированной соляной кислотой и затем
экстрагируют бензолом. После сушки бензольный слой фракционируют. Получают 17,3 г
(74 8%) треххлористого фенилгермания; т. кип. 104° С/15 мм; п2^ 1,5540. В реакционной
смеси присутствует небольшое количество (1,1 г) непрореагировавшего четыреххлористого
германия. Из остатка после перегонки выделяют 4 г двухлористого дифенилгермания.
Деалкилирован иг
87
В литературе описано также деалкилирование тетраперфторфенилгер-
мания с помощью четыреххлористого германия [55]. Хлористый трипер-
фторфенилгерманий имеет т. пл. 103—104° С. Галоидные соли других
металлов в качестве деалкилирующих агентов исследованы мало.
Миронов с сотр. [56] нашел, что германийорганические соединения,
содержащие атом кремния или второй атом германия, претерпевают реакцию,
ранее открытую для кремнийорганических соединений:
Г СН3 -1
А1Вг3 |
(CH3)3Ge(CH2)„Ge(CH3)3 > (CH3)4Ge + - (CH2)ftGe-
L СНз Хг
Германийорганические соединения более склонны к этой реакции.
Сулема не реагирует с тетравинилгермаиием [57]. При действии трех-
хлористого галлия на тетраметил германий с выходом 91% был получен
двухлористый метилгаллий [58]
(СН3)4 Ge + GaCl3 -> (CH3)3 GeCl + CH3GaCl2.
С треххлористым индием реакция проходит при 220° С, но образующийся
двухлористый метилиндий не стабилен [58].
При нагревании тетрабутилгермания с элементарной серой (8 час, 230°С)
[59] было показано, что сера деалкилирует тетрабутилгерманий с
образованием дибутилсульфида и тримера дибутилгерманийсульфида
Ge (C4H9)2
S S
3 (С4Н9)4 Ge + 6 S -> I I +3 (QH9)3 S.
(C4H9)2Ge Ge(C4II9)2
При реакции тетрафенилгермания с серой (8 час, 270° С) было выделено
некоторое количество дифен ил сульфида и металлический германий.
Имеются указания, что тетравинилгерманий деалкилируется
щелочными металлами Na, К, Li, Rb, Cs с образованием, например, виниллития на
90?о [60J. Разуваев и сотр. [61] показали, что при действии хлористого изо-
пропила на тетраэтилгерманий в присутствии хлористого алюминия с
выходом 60% образуется хлористый триэтилгерманий. Аналогичная реакция была
проведена на примере тетрабутилгермания с хлористым и бромистым изоами-
лом. Выход хлористого w-бутилгермания равен 55%, бромистого я-бутил-
германия — 67% [62].
Миронов и сотр. [1] позже на примере тетраметилгермания отметили,
что реакция деалкилирования может происходить при действии галоидных
алкилав (А1Вг3). С бромистым изопропилом выход бромистого триметил-
германия равен 92%. Бромистые ал килы могут быть заменены на хлористые.
В этой реакции может быть использован и триметилфенилгерманий.
ЛИТЕРАТУРА
1. Миронов В. Ф., К р а в ч е н к о А. Л., Изв. АН СССР, серия хим., 1965, 1026.
2. AndersonH.H., J. Am. Chem. Soc, 72, 2089 (1950).
3. К r a u s С A., F 1 о о d E. A., J. Am. Chem. Soc, 54, 1635 (1932).
4. Shackelford J. M., DeSchmertzing H.fHeuther С. Н., Р о d a 11 H.,
J. Org. Chem., 28, 1700 (1963).
5. A n d e г s о n H. H., J. Am. Chem. Soc, 73, 5440 (1951).
6. G i 1 m a n H., LeeperR.W., J. Org. Chem., 16, 466 (1951).
7. AndersonH.H., J. Am. Chem. Soc, 73, 5800 (1951).
88
Германийорганические соединения
8. Колесников Г. С, Давыдова С. Л., Климентова Н. В. Высо-
комолек. соед., 4, 1098 (1962).
9. Cross R. J., GlocklingF., J. Chem. Soc, 1964, 4125.
10. Orndorff W. K., TabernD.L, Dennis L.M., J. Am. Chem. Soc, 49,
2512 (1927).
И. К r a u s C. A., BrownCL, J. Am. Chem. Soc, 52, 3690 (1930).
12. К r a u s С A., FosterL.S., J. Am. Chem. Soc, 49, 457 (1927).
13. T h о m a s А. В., RochowE.G., J. Am. Chem. Soc, 79, 1843 (1957).
14. JohnsonO.H., HarrisD.M., Inorg. Syntheses, 5, 74 (1957).
15. JohnsonO.H., N e b e r g a 1 1 W. H., HarrisD.M., Inorg. Syntheses, 5, 76
(1957).
16. JohnsonO.H., S с h m a 1 1 E. A., J. Am. Chem. Soc, 80, 2931 (1958).
17. Metlesics W., ZeissH., J. Am. Chem. Soc, 82, 3324 (1960).
18. WestR., В a n e у R. H., PowellD.L, J. Am. Chem. Soc, 82, 6269 (1960).
19. BrookA.G., PeddleG. J., J. Am. Chem. Soc, 85, 1869 (1963).
20. S с h w a r z R., LewinsohnM., Ber., 64, 2352 (1931).
21. О и k а в a X., J. Chem. Soc. Japan, Pure Chem. Sec, 84, 510 (1963); РЖХим., 1964„
17Ж32.
22. M a z e г о 1 1 e s P., Bull. Soc. chim. France, 1962, 1907.
23. M a z e г о 1 1 e s P., LesbreM., D u b а с J., Compt. rend., 260, 2255 (1965).
24. M a z e г о 1 1 e s P., D u b а с J., Compt. rend., 257, 1103 (1963).
25. M a z e г о 1 1 e s P., LesbreM., Compt. rend., 248, 2018 (1959).
26. LesbreM., M a z e г о 1 1 e s P., Compt. rend., 255, 544 (1962).
27. LesbreM., S a t g ё J., Compt. rend., 250, 2220 (1960).
28. M a z e г о 1 1 e s P., Bull. Soc Chim. France, 1960, 856.
29. Freed man H. H., Пат. США 3090797 (1963); РЖХим., 1965, 8Н103П.
30. С а р а н к и н а С. А., М а н у л к и н 3. М., ЖОХ, 36, 1299 (1966).
31. Sem ly en J. A., W а 1 ker G. R., P h i 1 1 i ps С. S. G., J. Chem. Soc, 1965, 1197.
32. Tabern D. L, Orndorff W. K., DennisLM., J. Am. Chem. Soc, 47,
2039 (1925).
33. Dennis L.M., HanceF.E., J. Phys. Chem., 30, 1055 (1926).
34. LesbreM., M a z e г о 1 1 e s P., Compt. rend., 246, 1708 (1958).
35. S e у f e r t h D., К a h 1 e n N.. J. Org. Chem., 25, 809 (1960).
36. Гладштейн Б.М., РодэВ.В., СоборовскийЛ.З., ЖОХ, 29, 2155
(1959).
37. V а п d е V о п d е 1 D. F., J. Organometal. Chem., 3, 400 (1965).
38. D e n n i s L. M., P a t n о d e W. I., J. Am. Chem. Soc, 52, 2779 (1930).
39. S i m о n s J. K., J. Am. Chem. Soc, 57, 1299 (1935).
40. E a born C, PandeK.C. J. Chem. Soc, 1960, 1566.
41. EabornC, PandeK.C, J. Chem. Soc, 1961, 297.
42. Bott R. W., EabornC, PandeK.C, Swaddle T. W., J. Chem.
Soc, 1962, 1217.
43. Bo t t R. W. E a bo r n C, Wa 1 to n D. R. M., J. Organometal.Chem., 1,420(1964).
44. В о t t R. W., EabornC, GreaslevP.M, J. Chem. Soc, 1964, 4804.
45. В о t t R. W., EabornC, WaltonD.R. M., J. Organometal. Chem., 2, 154
(1964).
46. BottR.W., EabornC, SwaddleT.W., J. Chem. Soc, 1963, 2342.
47. E a b о r n C, WaltonD.R., J. Organometal. Chem., 4, 217 (1965).
48. С т е р л и н Р. Н., Д у б о в С С, Ли В а й-Ч а н„ В а х о м ч и к Л. П., К н у-
н я и ц И. Л., Ж. Всесоюзн. хим. общ. им. Д. И. Менделеева, 6, 110 (1961).
49. EabornC, Leyshon К., Р a n d е К. С, J. Chem. Soc, 1960, 3423.
50. В о t t R. W., E a b о r n С, H a s h i m о t о Т., J. Chem. Soc, 1963, 3906.
51. Schwarz R., Schmeisser E., Ber., 69, 579 (1936).
52. V a n d e г К e r k G. J. M., R i j k e n s F., J a n s s e n M. J., Rec Trav. Chim.,
81, 764 (1962).
53. M a z e г о 1 1 e s P., Bull. Soc chim. France, 1961, 1911.
54. R i j k e n s F., vander KerkG. J.M., Rec trav. chem., 83, 723 (1964).
54a. R i j k e n s F„ v a n d e г К e r k G. J. M., Investigations in the field of organoger-
manium chemistrv. Germanium research commitee, Nederland, 1964.
55. F e n t о n D. E., "M a s s e у A. G., Chem. and Ind., 1964, 2100.
56. M и р о н о в В. Ф., К р а в ч е и к о А. Л., Изв, АН СССР, серия хим., 1964, 768.
57. S е у f e r t h D., J. Org. Chem., 22, 478 (1957).
58. Schmidbaur H., FindeissW., Angew., 76, 752 (1964).
59. Schmidt M.t S с h u m a n n H., Z. annorg. allgem. Chem., 325, 130 (1963).
60. HoneycuttJ, Пат. США 3059036 (1962); РЖХим., 1964, 12H71.
61. P а з у в а е в Г. А., ВязанкинН. С, Дьячковская О. С, Киселе
в а И. Г., Д е р г у н о в Ю. И., ЖОХ, 31, 4056 (1961).
62. D а о - Н и у G i а о., Compt. rond., 260, 6937 (1965).
Глава X
ГИДРОЛИЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Галогениды алкил- и арилгермания легко гидролизуются водным
раствором щелочи с образованием в подавляющем большинстве случаев
соответствующих окисей или ангидридов кислот, так как промежуточно
образующиеся гидроокиси нестойки и легко отщепляют воду
2R3GeX •£. [R3GeOH]2 ^ (R3Ge)2 О,
R2GeX2 ;± [R2Ge (OH)2] ■£ R2GeO,
2RGeX3^ [RGe (OH)8] ^ 2RGeOOH ^ (RGeO)20.
Рохов с сотр. [1] сопоставил гидролиз галогенидов алкилгермания с
гидролизом аналогичных соединений других элементов IV группы.
Большинство галогенидов алкилгермания гидролизуются с трудом, к тому же
не полностью, поскольку имеет место равновесие. Аналогичные соединения
кремния гидролизуются легко и нацело. Рохов ошибочно считал, что в
неводных растворителях, таких как диметилформамид, ацетонитрил [2],
происходит образование окисей германийорганических соединений,
однако в дальнейшем было установлено [3, 4], что это явление, очевидно,
следует приписать наличию следов воды в растворителе [3]. Изучение
разложения двухлористого диметилгермания водой [5] показало, что это
соединение ведет себя, подобно сильной двухосновной кислоте, и гидролиз
протекает почти нацело, тогда как двухлористое диметилолово в тех же
условиях гидролизуется лишь на 10—45%. Рохов исследовал также гидролиз
водой и этильных соединений германия [6]. Гидролиз водой хлористого
диметилхлорметилгермания происходит по уравнению [6а]
СН3 СНз
I I
2 (CICHo) (CH3)2 GeCl + Ы20 -* С1СН2 - Ge - О - Ge - СН2С1 + 2НС1.
I I
СНз СНз
Как уже было упомянуто выше, в результате щелочного гидролиза
галогенидов типа R3GeX образуется (R3Ge)20. Образование гидроокисей
триизопропил- [7], трифенил- [8—12] и три-а-нафтилгермания [13] путем
гидролиза соответствующих галогенидов составляет исключение.
Стабильность гидроксильных групп в этих соединениях приписывают влиянию
стерических факторов. Гидроокись трифенилгермания была получена также
гидролизом бензольного раствора (C6H5)3GeONa [14].
Получение гидроокиси трифенилгермания [8]. Раствор 0,5 г едкого кали ъЪ мл воды и
50 мл спирта кипятят с обратным холодильником в течение 1 часа с 2 г (0,СС5 моля)
бромистого трифенилгермания. Полученный растЕср охлаждают и вылиЕают в 2С0 мл воды. Выпав-
90
Германийорганические согдингная. Лит. стр. 93—94
ший белый осадок (1,8 г) отсасывают, сушат и перекристаллизовывают из смеси (1:1)
хлороформа и петролейного эфира (т. кип. 60—70° С). Получают 1,4 г (84%) гидроокиси
трифенилгермания, т. пл. 127—131° С (после дальнейшей перекристаллизации т. пл. 133—134° С).
В работе [15] изучена кинетика гидролиза соединений R3GeCl (где
R = С6Н5, /?-CGH4CH3 и i-C3H7) в водных растворах эфира и ацетона
(определены константы равновесия реакции)
R3GeX + Н20 т1 R3GeOH + HC1.
Деннис с сотр. [16] впервые указал на существование гексаметилди-
германоксана без описания свойств. В дальнейшем при гидролизе
бромистого триметилгермания водным раствором едкого натра с последующей
экстракцией петролейным эфиром и перегонкой была получена бесцветная
жидкость с приятным запахом; т. кип. 138° С; п2* 1,4299 [17—19].
Аналогично получен гексаэтилдигерманоксан с выходом 97% [20, 21]; это
бесцветная жидкость с запахом камфоры, не застывает при охлаждении до—50° С,
т. кип. 253,9° С; растворяется в обычных органических растворителях в
частности в этиламине, не растворяется в воде и жидком аммиаке.
Подобным же образом были получены соединения типа (R3Ge)20, где R — я-про-
пил [21, 22], я-бутил- [21, 23, 24], я-амил-[25], я-гексил-[26]. Гексабен-
зилдигерманоксан был получен при кипячении бромистого трибензил-
германия со спиртовым раствором едкого кал^ [27].
По поводу получения гидроокиси трифенилгермания (см. выше) в более
ранних работах указывается, что при щелочном гидролизе бромистого
трифенилгермания образуется гексафенилдигерманоксан [12, 14, 28]. Это
же соединение было получено и при гидролизе водой трифенилгермил-
амина [29]
2(QH5)3GeNH2 + П20~* [(CGH5)3 Ge]2 О + 3NH3.
Кипячение в течение 10 час. бромистого три-я-толилгермания с 0,5 N
раствором едкого натра также приводит к гекса-я-толилдигерманоксану [30]
Гидролиз соединений типа R2GeX2 дает обычно окиси диалкилгермания,
которые склонны очень легко образовывать циклические олигомеры.
Число описанных окисей диалкилгермания сравнительно невелико. Строение их
наиболее подробно изучено на примере окиси диметилгермания [5, 19,
31—34]. При гидролизе двухлористого диметилгермания щелочью
образуется циклический тетрамер с т. пл. 92° С, легко растворимый в углеводородах.
Молекулярный вес его равен 470 (криоскопия — бензол), что отвечает
формуле [(CH3)2GeO]4 (мол. вес. 475). В отличие от соответствующих
соединений кремния он растворим в воде. При упаривании водного раствора вме*
сто тетрамера выпадает волокнистый полимер с т. пл. 132—133° С,
нерастворимый в бензоле и четыреххлористом углероде. Криоскопические
измерения в воде показывают, что в растворе находятся только мономерные
молекулы за счет деполимеризации [5]
[(СН3)2 GeO]4 + 4Н20 -» 4 (СН3)2 Ge (ОИ)2.
Тетрамер (т. пл. 92 °С) и полимер (т. пл. 133° С), нагретые до 210° С,
превращаются в нестабильный циклический тример, растворимый в
органических растворителях, легко переходящий в полимер (т. пл. 133° С).
При быстром охлаждении паров тримера до комнатной температуры тример
выделяется в виде жидкости. Взаимные переходы различных форм окиси
диметилгермания можно выразить следующим образом:
Гидролиз
91
перегонка
(СН3)2 GeCU + NaOH -* [(CH3)2 GeO]4 > [(СН8)2 GeO]3
t
н.о I
нагревание
I lidri'etsiiHwt; IX IX
(С1-18)3 Ge (ОН)2 с [(СН3)2 GeO]n смесь [(СН3)2 GeO]4 и [(СН3)2 GeO]„
На основании исследования ИК-спектров различным олигомерам окиси ди-
метилгермания придается следующее строение [191:
Ge (CH3)a
/ \
0 О
1 I
(СН3)2 Ge Ge (CH3)2
v
Гексаметилтригерманоксан
сн3
(СН3)2 Ge О Ge (CH8)3
О
(СН3)3 Ge О Ge (CH3)2
Октаметилтетрагерманоксан
_ Ge — О -
. I
L СН3 -in
Полидиметилгерманоксан
Получение тетрамерной окиси диметилгермания [33]. 21,8 г двухлористого диметилгер-
мания гидролизуют 9,85 г едкого натра в 10 мл воды, прибавляют 30 мл петролейного
эфира, реакционную смесь нагревают с обратным холодильником 4 часа. Органический слой
отделяют, щелочной слой экстрагируют 4 раза петролейным эфиром. После сушки над
сернокислым кальцием петролейный эфир отгоняют в атмосфере азота и получают 10,0 г (67%)
тетрамера с т. пл. 88—92° С. После перекристаллизации т. пл. 91—92° С.
Получение высокополимерной окиси диметилгермания [33]. Водный раствор 0,97 г
окиси диметилгермания в 2 мл воды оставляют испаряться при комнатной температуре в
течение 48 час. Получают вещество ст. пл. 132—133° С, не растворимое в бензоле, циклогек-
сане и четыреххлористом углероде, но легко растворимое в спирте и уксусной кислоте.
Получение трехмерной окиси диметилгермания [33]. Образец (0,2 г) тетрамера или
полимерной окиси диметилгермания (т. пл. 133° С) помещают в кварцевую трубку (диаметр 15 мм,
длина 200 мм), запаянную с одного конца. Трубку эвакуируют, запаивают и нагревают в
течение 24 час. при 210—220° С. При быстром охлаждении холодной водой получают
нестабильные кристаллы. Молекулярный вес (криоскопия, в бензоле) отвечает тримеру.
Окись диметилгермания была получена также гидролизом сульфида
диметилгермания в присутствии серной кислоты или перекиси водорода [35,
36]. Окиси диэтил- и ди-я-пропилгермания получены в виде трех олиго-
меров—три-,тетра- и полимеров. Как и в случае окиси диметилгермания, три-
мерная форма окиси диэтилгермания [37] и ди-я-пропилгермания [38]
неустойчивы и легко полимеризуются при хранении. Полимер при
нагревании вновь превращается в тример. Имеются указания о существовании
этих окисей и в виде тетрамеров [39—41]. Описывается также окись ди-я-
бутилгермания [42, 43] и окись диизопропилгермания, последняя в виде
соответствующего тримера [7, 13].
Из аналогичных соединений ароматического ряда в литературе описаны
полимерные окиси диарилгермания. При гидролизе двубромистого ди-
фенилгермания получена смесь продуктов с различной структурой и
молекулярным весом [28, 44, 45]. Длительное кипячение этой смеси с уксусной
кислотой приводит к образованию циклической тетрамерной окиси дифе-
нилгермания, а перегонкой тетрамера получают циклический тример.
Тример окиси дифенилгермания опять можно перевести в тетрамер, при
длительном кипячении с уксусной кислотой. В свою очередь тетрамер
переходит в полимер при нагревании с водным раствором спирта. Взаимные пере-
92
Германийорганшеские соединения. Лит. стр. 93—94
ходы различных форм окиси дифенилгермания можно представить следую*
щей схемой [45]:
(C6M5)2GeBr2 + NaOH-
l(C6H5)2GeO].
l(C6HJ2GeO}n
Т. пл 230-295° С
KC6H3)2GeO]4
Т. пл- 218°С
(CGII5)2GeBr,
KC6H5)2GeOJ3
Получение тетрамерной окиси дифенилгермания с т. пл. 218° С [45]. Юг (0,026 моля)
тетрафенилгермания в 70 мл кипящего 1,2-дибромэтана обрабатывают в течение нескольких
часов 10,5 г брома (0,066 моля, 27% избыток), растворенными в 20 мл 1,2-дибромэтана. По
окончании прибавления реакционную смесь нагревают с обратным холодильником 15 час,
растворитель удаляют в вакууме при нагревании на водяной бане. Остаток гидролизуют
водным раствором едкого кали и перегоняют с паром. Белый осадок после перегонки с паром
представляет окись дифенилгермания с т. пл. 230—295° С, легко растворимую в обычных
органических растворителях. После продолжительного нагревания с уксусной кислотой осадок
переходит в раствор. При медленном разбавлении его водой и охлаждении выпадают
кристаллы тетрамерной окиси дифенилгермания с т. пл. 215—218° С; выход 5,75 г (91%). После
кристаллизации из эфира т. пл. 219—221° С. При кипячении с водным раствором спирта
40:60 полученная тетрамерная форма окиси дифенилгермания переходит в полимерную окись
с т. пл. 295—298° С.
Получение тримерной окиси дифенилгермания с т. пл. 149° С [45]. 1,5 г окиси
дифенилгермания с т. пл. 290—295 °С нагревают в запаянной трубке при 100° С с 15 мл иодисто-
водородпой кислоты (уд. вес 1,7) в течение 30 мин. Трубку охлаждают, прибавляют эфир и
затем воду, отделяют эфирный слой, промывают его несколько раз водой, водным раствором
тиосульфата натрия и бикарбонатом натрия. Эфир удаляют и добавляют петролейный эфир
для отделения кристаллической окиси дифенилгермания с т. пл. 142—148° С. После
нескольких кристаллизации из смеси бензол — гексан, эфир — гексан получают чистую тример-
ную форму окиси дифенилгермания ст. пл. 147—149° С. Тримерная форма окиси
дифенилгермания может быть перегнана при 300° С (температура бани) и давлении 0,2 мм.
Получается бесцветная стеклообразная масса, которая кристаллизуется при перемешивании острой
палочкой (нанесение царапин).
В литературе описаны также окиси диарилгермания формулы Ar2GeO,
где Аг = С6Н5, о-, га-, /?-СН3СбН4, о, га-, /?-С1С6Н4, о-, /?-ВгС6Н4, /?-СН3ОС6Н4,
о-, р- C6H5OCGH4,a-C10H7 [46, 47].
Алкил- или арилгерманиевые кислоты и их ангидриды типа RGeOOH
и (RGgO)20 получаются при гидролизе соответствующих соединений RGeX3.
Однако литература по получению этих соединений незначительна и
относится в большинстве своем к более раннему периоду. Германийорганические
соединения описаны там в качестве промежуточной стадии при разделении
продуктов различной степени алкилирования. Описаны следующие алкил-
германиевые кислоты или их ангидриды: C2H5GeOOH [37, 48], C3H7GeOOH
Гидролиз
93
[49], (C2H5GeO)20 [50], (C3H7GeO)20 [41, 49] и (/i-C4H9GeO)aO [51].
Большинство веществ не было выделено в чистом виде.
Получение ангидрида я-бутилгерманиевой кислоты [51]. 7,9 г треххлористого w-бутил-
германия встряхивают дважды с 8 М раствором аммиака порциями по 24 г, добавляют 24 г
четыреххлористого углерода. Отделенный слой четыреххлористого углерода промывают
водой, сушат сернокислым натрием, затем осторожно испаряют растворитель и остаток сушат
в течение 3 час. при 140° С/1 мм. Полученный ангидрид бутил германиевой кислоты
[(/2-C4H9GeO)20]4 весит 4,83 г, выход 93%.
Что касается арилгерманиевых кислот и их ангидридов, то при
гидролизе трехбромистого фенилгермания был получен бесцветный аморфный
продукт, растворимый в щелочах [28, 52], которому было приписано строение
фенилгерманиевой кислоты; однако в чистом виде кислота не была выделена.
Ее ангидрид описан в виде белого аморфного осадка, не растворимого в
воде и органических растворителях, но растворимого в избытке щелочи
[28, 52—55]. Описаны и другие ангидриды арилгерманиевых кислот:
(/?-CH.,C6H4GeO)20 [52, 54], (/?-CH3OC6H4GeO)20, (/7-C2H5OC6H4GeO)20,
(/?-ClC6H4GeO)20, (/?-BrC6H4GeO)20 [55], (p-CH3NHC6H4GeO)20 [53],
[p-(CH3)2NC6H4GeO]20 [52, 53, 56], [/7-(C2H5)2NC6H4GeO]20 [53],
l(3-N02)(4-(CH8)2N) C6H3GeO]20 [56].
ЛИТЕРАТУРА
1. A 1 1 г e d A. L., Rochow E.G.,J. Inorg. Nucler. Chem, 5, 269 (1958).
2. IngoldK., Rocho w E. G., S eye ferthD., Smith A.C., West R., J. Am.
Chem. Soc, 74, 6306 (1952).
3. T h о m a s А. В., RochowE.G., J. Am. Chem. Soc, 79, 1843 (1957).
4. T h о m a s А. В., RochowE.G., J. Inorg. Nucl. Chem., 4, 205 (1957).
5. RochowE.G, A 1 1 r e d A. L., J. Am. Chem. Soc, 77, 4489 (1955).
6. RochowE.G, J. Am. Chem. Soc, 72, 198 (1950).
6a. W i e b e r M. F г о h n i n g С D. Z. Naturforsch, 21b, 492 (1966).
7. AndersonH.H, J. Am. Chem. Soc, 75, 814 (1953).
8. BrookA.G, GilmanH, J. Am. Chem. Soc, 76, 77 (1954).
9. WestR, Baney R.H, Powell D. L, J. Am. Chem. Soc, 82, 6269 (1960).
10. P i k e R. M, Rec trav. chim, 80, 885 (1961).
11. JohnsonO.H, S с h m a 1 1 E. A, J. Am. Chem. Soc, 80, 2931 (1958).
12. JohnsonO.H, Nebergall W.H, J.Am. Chem. Soc, 71, 1720 (1949).
13. West R, J. Am. Chem. Soc, 74, 4363 (1952).
14. KrausCA, FosterL.S., J. Am. Chem. Soc, 49, 457 (1927).
15. Chip per fie 1 d J. R, Prince R.H, J. Chem. Soc, 1963, 3567.
16. D e n n i s L. M, P a t n о d e W. I, J. Am. Chem. Soc, 52, 2779 (1930).
17. Schmidt M., R u i d i s с h I, Z. anorg. allgem. Chem, 311, 331 (1961).
18. G r i f f i t h s J. E, О n у s z с h u k M. Canad, J. Chem, 39, 339 (1961).
19. В г о w n M. P, О k a w a r a R, RochowE.G, Spectrochim. Acta, 16, 595
(1960).
20. К r a u s С A., F 1 о о d E. A, J. Am. Chem. Soc, 54, 1635 (1932).
21. AndersonH.H, J. Am. Chem. Soc, 73, 5439 (1951).
22. AndersonH.H, J. Am. Chem. Soc, 73, 5440 (1951).
23. AndersonH.H, J. Am. Chem. Soc, 73, 5800 (1951).
24. V a n d e r KerkG. J. M, R i j kens F, Janssen M.J, Rec trav. chim,
81, 764 (1962).
25. R i j k e n s F, van der Kerk G. J.M, Rec trav. chem, 83, 723 (1964).
26. FuchsR, GilmanH, J. Org. Chem, 23, 911 (1958).
27. BauerH, BurschkiesK., Ber, 67, 1041 (1934).
28. Morgan G. T, Drew H. D. K., J. Chem. Soc, 127, 1730 (1925).
29. К r a u s С A, WoosterC.B, J. Am. Chem. Soc, 52, 372 (1930).
30. SimonsJ. K., WagnerE.C, M u 1 1 e r J. H, J. Am. Chem. Soc, 55, 3705
(1933).
31. RochowE.G, SindlerB. M, J. Am. Chem. Soc, 72, 1218 (1950).
32. RochowE.G, RochowT.G, J. Phys. Colloid. Chem, 55, 9 (1951).
33. В г о w n M. P, R о с h о w E. G, J. Am. Chem. Soc, 82, 4166 (I960).
34. P о x о в Е, Сб. «Успехи неорганической и элементоорганической химии». М, ИЛ
1963, стр. 59.
94
Германийоргаиические соединения
35. RochowE. G., J. Am. Chem. Soc, 70, 1801 (1948).
36. British Thomson — Houston Co Ltd. Англ. пат. 654571 (1951); С. А., 46, 4561b (1952)..
37. F 1 о о d E. A., J. Am. Chem. Soc, 54, 1663 (1932).
38. A n d e r s о n H. H., J. Am. Chem. Soc, 74, 2370 (1952).
39. AndersonH.H., J. Am. Chem. Soc, 72, 2089 (1950).
40. AndersonH.H., J. Am. Chem. Soc, 72, 194 (1950).
41. JohnsonO.H., J ohes L. V., J. Org. Chem., 17, 1172 (1952).
42. Ma zero 1 1 es P., Bull. Soc chim. France, 1961, 1911.
43. AndersonH.H., J. Am. Chem. Soc, 83, 547 (Ш61).
44. К r a u s С. А., В г о w n С L., J. Am. Chem. Soc, 52, 3690 (1930).
45. Metlesic's W., Z e i s s H. J. Am. Chem. Soc, 82, 3324 (1960).
46. E м е л ь я н о в а Л. И., М а к а р о в а Л. Г., Изв. АН СССР, ОХН, 1960, 2067.
47. Емельянова Л. И., Виноградова В. Н., Макарова Л. Г., Н е-
с м е я н о в А. Н., Изв. АН СССР, ОХН, 1962, 53.
48. Т с h a k i r i a n A., LewinsohnM., Compt. rend., 201, 835 (1935).
49. J о n e s L. V., Dissertation Abstr., 13, 308 (1953).
50. F 1 о о d E. A., J., Am. Chem. Soc, 55, 4935 (1933).
51. A n d e r s о n H. H., J. Am. Chem. Soc, 82, 3016 (1960).
52. Orndorff W. K., TabernD.L, Dennis L. M., J. Am. Chem. Soc, 49,
2512 (1927).
53. В a u e г Н., В u r s с h к i e s K., Ber., 65, 956 (1932).
54. RothermundtM., В urschkies K., Z. Immunitatsforsch., 87, 445 (1936).
55. E м е л ь я н о в а Л. И., М а к а р о в а Л. Г., ДАН СССР, 122, 403 (1958).
56. В ur sch k ies K-, Вег., 69, 1143(1936).
Глава XI
РЕАКЦИИ ГЕРМАНИЙОРГДНИЧЕСКИХ СОЕДИНЕНИЙ,
СВЯЗАННЫЕ С ЛРЕСЪРАЗСВАИМЕМ ФУНКЦИОНАЛЬНЫХ ГРУПП
У АТОМА ГЕРМАНИЯ
РЕАКЦИИ ГЕРМАНИЙОРГДНИЧЕСКИХ ГИДРООКИСЕЙ,
ОКИСЕЙ И АНГИДРИДОВ ГЕРМАНИЙОРГАНИЧЕСКИХ КИСЛОТ
Германийорганические окиси типа (R3Ge)20, R2GeO и (RGeO)20 легко
реагируют с органическими и минеральными кислотами, заменяя
кислород на остаток кислоты. Легкость обмена, количественные выходы в
большинстве случаев и простота эксперимента дают возможность получать
таким путем различные новые германийорганические соединения.
Большинство реакций подобного типа описаны Андерсоном. Основные
уравнения реакции можно представить в следующем виде:
(R3Ge)2 О -f 2HX -» 2R8GeX + Н20,
R2GeO + 2НХ-> R2GeX2 + Н20,
(RGeO)2 О + 6НХ -> 2RGeX3 + ЗН20
(X — галоид, остаток минеральной или органической кислоты).
Действие галоидоводородных кислот иллюстрируется следующими
примерами.
Получение фтористого три-я-пропилгермания [1]. 5,8 г гекса-я-пропилдигерманоксана
встряхивают с 12 мл 48%-ной фтористоводородной кислоты в прозрачной пластмассовой
пробирке. При этом температура реакционной смеси поднимается на 10° С. Когда реакционная
смесь простояла 2 часа, ее экстрагируют 4 раза петролейным эфиром (т. кип. 30—60° С).
После сушки в течение ночи безводным сульфатом натрия и отгонки растворителя получают
6,05 г фтористого три-я-пропилгермания; т. кип. 203° С; т. пл. 27,5° С; п2^ 1,4340. После трех
перегонок продукт содержит 0,4% свободного фтористого водорода, очистку от которого
проводят перегонкой над фтористым натрием.
Аналогичным образом получают, например, хлористый три-я-пропил-
германий (еыход количественный; т. кип. 227° С; 98—99° С/11 мм; п2£ 1,4661),
бромистый три-я-пропилгерманий (93%; т. кип. 112—113° С/12 мм; ri2g
1,4832), йодистый три-я-пропилгерманий (85%; т. кип. 122—123° С/10 мм;
п2£ 1,5144), а также галоидные соли других алкильных производных Ge:
(C2H5)3GeX [2, 3], (i-C8H7)3GeX [4], (/2-C4H9)3GeX [5], (/2-C6H13)3GeX
[6], (C6Hn)3GeX [7], (C2H5CH2)3GeX [8]. В ароматическом ряду
галоидные соли триарилгермания получают тем же путем [9—151.
Что касается двухгалоидных соединений, то они количественно
образуются из окисей и галоидоводородных кислот.
Получение двухлористсго, двубрсмистого и двуиодистого лкизопрспилгермаьия [4, 16].
В каждом случае 4 г трехмерной окиси диизопропилгермания и 20 мл концентрированной
галоидоводородной кислоты нагревают до 85е С и сильно встряхивают в течение 5 мин.
После охлаждения в течение 2 час. двухлористый и дв>бромистый днизопропилгерманий
выделяют экстрагированием петролейным эфиром, сушкой над сульфатом натрия и отгонкой
растворителя. Двуиодистый днизопропилгерманий отделяют в делительной воронке. Выходы
близки к количественным.
9fi Германайорганические соединения. Лит. стр. 112—ИЗ
Аналогично описано получение двубромистого диэтилгермания [17],
двуфтористого, двухлористого, двубромистого и двуиодистого ди-я-про-
пил- [18] и диизопропилгермания [4], двухлористого ди-я-бутилгермания
[19], двухлористого циклотетраметиленгермания [20], двуиодистого дифе-
нилгермания [21].
Действие галоидоводородных кислот на ангидриды алкилгерманиевых
кислот в литературе описаны только на отдельных примерах. Так, при
действии 48%-ной бромистоводородной кислоты на ангидрид
я-бутилгерманиевой кислоты был получен трехбромистый я-бутилгерманий [22];
выход 67%. Описано также действие галоидоводородных кислот и на
ангидриды арилгерманиевых кислот [23, 24].
Обработкой концентрированной серной кислотой гексаэтилгерманок-
сана и окиси диэтилгермания были легко получены соответствующие
сульфаты [(C2H5)3Ge]2S04 и (C2H5)2GeS04, однако конечный продукт не был
достаточно очищен и всегда содержал примесь серной кислоты [25].
В литературе описан пример получения сульфида дифенилгермания при
действии на тример окиси дифенилгермания, подкисленного уксусной
кислотой, водного раствора сульфида натрия [26].
Шмидт с сотр. [27] показал, что при действии на гексаметилдигерманок-
сан различных окислов металлов и металлоидов идет следующая реакция:
3 [(СНз)з Ge]2 О + As303 -* 2 [(СН3)з GeO]3 AsO.
Аналогичная реакция идет с Se03 [27, 28], V205, Cr03, Re207 [29], Р4О10
[30].
В литературе описан один пример действия на гексаэтилдигерманоксан-
фенилмеркаптана с выходом 60% [31]; по другим данным, с выходом 94%
[32] был выделен фенилмеркаптид триэтилгермания.
Синильная кислота и изотиоцианистая реагируют с гексаалкилдигерма-
ноксанами и с окисями диалкилгермания аналогично галоидоводородным
кислотам. В литературе описаны производные синильной кислоты R3GeCN
(где R — этил, я-пропил) [33], производные изотиоциановой кислоты
R3GeNCS (R — этил, я-пропил, я-бутил [33] и изопропил [4]) и
R2Ge(NCS)2 (R—этил [33], изопропил [16], я-бутил [20]). Интересно
отметить, что изоциановая кислота не реагирует с гексаэтилдигерманокса-
ном даже при нагревании [33].
Органические кислоты при действии на германийорганические
гидроокиси или гексаалкилдигерманоксаны легко дают большое число
различных германийорганических ацилатов. Ацетаты впервые были приготовлены
при действии уксусного ангидрида на гидроокись трициклогексил- [34],
триэтил- [35], три-я-пропилгермания [36]. Подобным же образом
приготовлены и формиаты R3GeOOCH, где R — этил [35], пропил [36],
бутил [5].
Имеются примеры получения солей других кислот, например пропиона-
та и я-бутирата триэтилгермания [37], а также производных монохлор-,
дихлор-, трихлор-, монобром-, моноиодуксусной кислот, а- и |3-хлор-,
бром- и а-иодопропионовой кислоты (действие на гексаэтилдигерманоксан
[37]).
Взаимодействие гекса-я-пропилдигерманоксана с монохлор-, дихлор-,
монобром-и трифторуксуснои кислотой дает соответствующие ацилаты [36],
однако лишь производные трифторуксуснои кислоты являются достаточно
стабильными. Описаны также соответствующие галоидированные ацилаты
триизопропилгермания [38], атакжетрифторацетаттри-я-бутилгермания [5].
При реакции гексаэтилдигерманоксана с безводной меркаптоуксусной
кислотой HSCH2COOH был получен (C2H5)3Ge(SCH2COO)Ge(C2H5)3 [35].
Преобразование функциональных групп у атома германия 97
Получение формиата три-«-пропилгермания [36], При встряхивании 5 г гекса-«-про-
пилдигерманоксана и 3,3 г 90%-ной муравьиной кислоты температура реакционной смеси
поднимается на 7° С и образуется два слоя. Нижний слой составляет 0,5 мл. После
медленной отгонки воды и кислоты (2,7 мл) остаток перегоняют в вакууме. Получают б г формиата
три-я-пропилгермания; т. кип. 108—109° С/12 мм; df 1,094; п2£ 1,4505.
Получение монохлорацетата триэтилгермания [37]. 2,3 г гексаэтилдигерманокслна и 1,5 г
сухой монохлоруксусной кислоты нагревают при перемешивании в течение 10 мин. при
100° С и охлаждают до комнатной температуры. После центрифугирования большую часть
образовавшейся воды удаляют с помощью микропипетки. Остатки воды удаляют
сушкой в течение 3 час. над сульфатом натрия. Большую часть ацилата переносят в
перегонную колбу с помощью шприца. Сульфат натрия промывают 0,6 мл петролейного
эфира. После отгонки петролейного эфира ацилат перегоняют. Монохлор ацетат
триэтилгермания имеет т. кип. ИЗ—114° С/б мм', d^° 1,243; п2£ 1,4672; выход количественный.
Получение трифторацетата три-я-бутилгермания [5]. Смешивают 4 г чистого гекса-я-
бутилдигерманоксана и 4 г трифторуксусной кислоты, причем происходит сильное
разогревание. После осторожного нагревания большую часть воды удаляют центрифугированием.
Выход трифторацетата три-я-бутилгермания количественный. Однако продукт значительно
загрязнен трифторуксусной кислотой. После повторного нагревания дополнительно с 0,3 г
окиси в продукте остается 1% свободной трифторуксусной кислоты; трифторацетат три-я-
бутилгермания имеет т. кип. 147—149° С/19 мм; п2£ 1,419.
Интересно отметить, что при действии на гидроокись трифенилгерма-
ния трифенилгермилмуравьиной кислоты с выходом 84% [39] проходит
реакция по уравнению
(С6Н5)3 GeOH + (СбН5)з GeCOOH -* (СбН5)3 GeCOOGe (QH5)3 + Н20.
При действии на гексаметил- и гексаэтилдигерманоксан дифторангид-
рида метилфосфиновой кислоты получаются триалкилгерманоловые эфи-
ры метилфторфосфиновой кислоты [40].
Окиси диалкилгермания реагируют при нагревании с органическими
кислотами или их ангидридами, легко образуя соответствующие диаци-
латы. Были получены диформиат и диацетат диэтилгермания [35], диаце-
тат и димонохлорацетат ди-я-пропилгермания [18], диацетат
диметилгермания [41].
Получение диацетата ди-#-пропилгермания [18]. 3,5 г окиси ди-я-пропилгермания и
4,5 г уксусного ангидрида нагревают с обратным холодильником в течение 3 час. После
отгонки избытка уксусного ангидрида получают диацетат ди-я-пропилгермания; т. кип.
244,6 °С; т. пл. 35,6° С; выход 90%.
При нагревании окиси диэтил германия с фенилмеркаптаном (выход 94%)
был получен дифенилмеркаптид диэтил германия; т. кип. 153—154° С/0,15 мм;
п™ 1,6242 [32].
Алкилгерманиевые кислоты и их ангидриды не реагируют с уксусным
ангидридом [35, 41], для получения соединений типа RQe(OOCR)3
используются другие методы (см. стр. 100, 111).
При действии сероводорода на ангидриды арилгерманиевой кислоты в
кислом растворе были получены соответствующие сульфиды (RGeS2)S [7].
Гексаметилдигерманоксаны при действии металлоорганических соединений,
например метиллития в эфире или тетрагидрофуране, легко расщепляются
с образованием соответствующих германолятов [42, 43]
(CH3)3GeOGe (СН3)з + CH3Li -* (CH3)3 GeOLi + (CH3)4 Ge.
Интересно отметить, что если гидроокись трибутилгермания, окись ди-
бутил- и дифенилгермания нагревать в бензоле в присутствии /г-толуолсуль-
фокислоты как катализатора со спиртами, гликолями, ацетилацетоном и
замещенными аминами, то образуются соответствующие алкоксисоедине-
7 Заказ М 20"
1)8
Германийорганические соединения. Лит. стр. 112—113
кия [43а]:
R2GeO + 2R0H -» R2Ge (0R)2 + Н20
(R' = С4Н9; R = СН3, 0>Н5, С3Н7,
(R' = CeH3; R-C2H5,
[(С4Н9)3 Ge]2 0 + 2R0H -» 2C4H9GeOR + Н20
(R = C2H3,
0
, / \
RoGe0 + R (0Н)2 -> R2Ge R + Н20
\ /
0
(R=CH2CHa;
f-C3H7, s-C4H9, /-C4H9)
1-СзН7, /-QH9).
/-C3H7, /-C4H9).
R' = C4H9, C6H5)
[R = CHoCH (CH3); R' = QH9].
:4H9)3 Ge]2 0 + R (0H)2 -» [(QH9)3 GeO]2 R + H20
[R = CH2CH2; CH3CH (CH3)].
HOCH2 OCH2
R2GeO +
R2Ge
+ HoO
H2NCH2 HNCH2
(R' = C4H9; QH5).
Трибутоксибутилгерманий был приготовлен при нагревании
ангидрида бутилгерманиевой кислоты с бутиловым спиртом [436]
(C4H9GeO)2 О + 6С4Н9ОН -> 2C4H9Ge (ОС4Н9)3 + ЗН20.
Получение бутилтри-и-бутоксигермания [436]. 1,27 г ангидрида бутилгерманиевой
кислоты, 12 г я-бутилового спирта и 20 г бензола нагревают с обратным холодильником в
течение 6 час. Образующаяся в процессе реакции вода удаляется путем азеотропной
отгонки. Полученный бутил три-я-бутокси германий перегоняют в вакууме; т. кип. 111—
114° С/0,5 мм; nD 1,4310; выход 70%.
Окись дибутилгермания и гексабутилдигерманоксан в присутствии п-
толуолсульфокислоты реагируют и с тиоспиртами с образованием
соответствующих меркаптидов ал кил германия [43в1.
(С4Н9)2 GeO + 2QH9SH -> (QH9)2 Ge (SC4H9)2 + H20,
[(C4H9)3 Ge]2 О + 2QH9SH -» 2 (C4H9)3rGeSC4H9 + H20.
Рикенс и сотр. [43г, 43д] показали, что при нагревании гексаалкилдигер-
маноксанов с некоторыми гетероциклами типа имидазола, пиразола
образуются новые германийорганические соединения, имеющие связь Ge—N.
Получение N-триэтилгермилимидазола [43г]. Смесь Юг гексаэтилдигерманоксана и
4,1 г имидазола нагревают при 160° С в колбе, снабженной капельной воронкой и
нисходящим холодильником. Воду, образующуюся при реакции, отгоняют прибавлением
небольшого количества бензола и отгонкой азеотропной смеси. После перегонки реакционной смеси
получают 1,7 г N-триэтилгермилимидазола; выход 13%; т. кип. 155-—156° С/13 мм\ п2р 1,4947.
РЕАКЦИИ ОБМЕНА
ГЕРМАНИЙОРГАНИЧЕСКИХ ГАЛОИДОПРОИЗВОДНЫХ
При действии солей органических и неорганических кислот на
германийорганические галоидопроизводные легко происходит обмен галоида на
остаток кислоты. В ряде случаев метод более удобен и дает более чистые
продукты реакции, чем действие кислот на германийорганические гидроокиси,
окиси или алкилгерманиевые кислоты. Наиболее часто применяют
серебряные соли, в отдельных случаях и калиевые соли.
Преобразование функциональных групп у атсма германия 99
В отдельных случаях, впрочем, реакция идет в других направлениях.
Так, например, при кипячении спиртового раствора бромистого трифенил-
германия с азотнокислым серебром с количественным выходом был
получен гексафенилдигерманоксан [44] и соответственно гексабензил- [8], гек-
са-я-толил-, гекса-л*-толилдигерманоксаны [14], а также гидроокись три-о-
тол ил германия [4] и гидроокись трициклогексил германия [7, 34].
Однако если азотнокислое серебро действует на галогениды алкил-
германия в инертных апротонных растворителях, то легко образуются
соответствующие эфиры азотной кислоты [30].
Хлористый триметилгерманий реагирует при комнатной температуре
с сухим нитратом серебра в тетрагидрофуране по уравнению
(CH3)3GeCi + AgN03 -> (CH3)3 GeON02 + AgCl.
Аналогичная реакция происходит с серебряной солью ортофосфорной
кислоты [30]
3(СН3)3GeCl + Ag3P04-» [(CH8)3GeO]3P04 + 3AgCl,
серебряной солью ортомышьяковой кислоты [27]
3(СН3)з GeCl + Ag3As04 -> [(CH3)3 GeO]3 As04 + 3AgCl.
Шмидт и сотр. исследовали реакцию хлористого триметилгермания с
серебряными солями ряда кислот. При проведении реакции в инертных
растворителях были получены соответствующие эфиры кислот триметил-
германия. Реакция была проведена с Ag3V04, Ag2Cr04, AgRe04 [27], Ag2Se04
[27, 28], AgC104 [45].
При действии углекислого серебра на бромистый триэтилгерманий в
сухом бензоле был выделен лишь гексаэтилдигерманоксан [33]. Аналогичный
продукт реакции был получен и при действии углекислого серебра на
бромистый триметилгерманий [46].
Сернистое серебро и иодиды триалкилгермания реагируют по уравнению
(хорошие выходы) [16,32]
2R3GeJ + Ag2S -> (R3Ge)2 S + 2AgJ.
Серебряные соли циановой, изотиоциановой и изоциановой кислот
реагируют аналогично:
R3GeX + AgCN -»R3GeCN + AgX,
R2GeX2 + 2AgNCS -> R2Ge (NCS)2 + 2AgX,
RGeX3 + 3AgNCO -> RGe(NCO)3 + 3AgX.
Реакция проходит, как правило, без растворителя, реже ее проводят в
бензоле при кипячении в течение короткого времени.
Получение изоцианата три-н-бутилгермания [5]. Смесь 7 г чистого хлористого три-«-бу-
тилгермания и 12 г изоцианата серебра, 25 мл бензола и 2 мл нитрометана кипятят с
обратным холодильником; при этом получают с количественным выходом (rc-QHiihGeNCO;
т. кип. 109—110° С/2 мм; п™ 1,4595.
Аналогично был синтезирован изоцианат триэтилгермания, диизоциа-
нат диэтилгермания, триизоцианат этилгермания [3], изоцианат три-я-про-
пилгермания [1], изоцианат триизопропилгермания [38], диизоцианат ди-
изопропилгермания [16]. Действием изоцианового серебра на двуиодистый
ди-я-бутилгерманий с выходом 67% получен диизоцианат ди-я-бутилгерма-
ния [20]. Описаны также изоцианат триметилгермания [47] и изоцианат
трифенилгермания [48].
7*
100 Гернанийорганические соединения. Лит. стр. 112—113
Андерсон [4, 5, 16, 33, 41, 49] изучал реакцию обмена солей германий-
органических соединений с различными серебряными солями и показал,
что обмен может происходить в соответствии со следующим рядом и всегда
в направлении вправо:
J-»S-*Br-*CN-*NCS, Cl->NCO->0, OCOR-*F.
Андерсон провел ряд обменных реакций различных солей германийорга-
нических соединений и серебряных солей кислот, согласно табл. 2.
Т аблица 2
Обменные реакции германийорганических солей
Германийорганиче-
ская соль
[(C2H5)3Ge]20
(CaHebGeOCOCH.
(/-CsH7hGe(NCO)2
(fz-CsH7)3GeCN
(;-C3H7)3Ge(NCS)3
(/-C3H7)3GeBr
(/-C3H7)3GeBr
[(/-C3H7)2GeS]2
[(/-C3H7)2GeS]2
[(*'-C3H7)2GeS]2
(i-C3H7)3GeH2
Серебряная
соль
AgF
AgF
AgOCOC3H7-n
AgOCOCsHs
AgOCOCH3
AgCN
AgNGS
AgOCOCH3
AgCN !
AgBr
AgOCOCH3
Продукт реакции
(CaHsbGeF
(C2H5)3GeF
(/-C3H7)2Ge(OCOC3H7-/i)2
(w-C3H7)3GeOCOC2H5
(/-C3H7)2Ge(OCOGH3)2
(/-QH^sGeCN
(/-G3H7)3GeNCS
(/-C5H7)2Ge(OCOCH3)2
(i-C3H7)2Ge(CN)2
(i-QHihGeBrz
.(/-C3H7)3Ge(OCOCH3)2
Выход,
%
60
80
78
80
80
—
—
—
—
—
—
При нагревании галогенидов алкилгермания с серебряными солями
органических кислот%егко и с хорошими выходами получаются
соответствующие ацилаты.
Получение а-хлорпропионата триэтилгермания [37]. Раствор 2,83 г бромистого триэтил-
германия и 3,5 г серебряной соли а-хлорпропионовой кислоты в 8 мл четырех хлор и сто го
углерода кипятят с обратным холодильником в течение часа. Реакционную смесь
отфильтровывают от бромистого серебра. От фильтрата отгоняют растворитель, после перегонки в
вакууме получают 3,13 г а-хлорпропионата триэтилгермания. Выход близок к
количественному; т. кип. 73—75° С/1 мм; п™ 1,4619.
Аналогично с выходом 90% получен я-валерат триэтилгермания [38]
бензоат триэтилгермания [31]. Исходя из хлористого триизопропилгерма-
ния и соответствующих серебряных солей органических кислот, получены
ацетат, пропионат, я-бутират, я-валерат, монохлорацетат, Р-хлорпропио-
нат, а-хлорпропионат и бензоат триизопропилгермания [5].
Ги^ман и сотр. [50], действуя серебряной солью трифенилгермилуксус-
ной кислоты на бромистый трифенилгерманий, с выходом 53% получили
своеобразное соединение (СбН5)3 GeOOCGe(C6H5)3.
В литературе описано получение и диацилатов: диацетата, дипропиона-
та, ди-я-бутирата и дибензоата диизопропилгермания [16] действием
серебряных солей соответствующих органических кислот на двухлористый
диизопропилгерманий.
Получены также и триацетат, трипропионат, трия-бутират и три-я-ва-
лерат этилгермания [3]. Отметим применение формиата свинца в реакции
с трехиодистым этилгерманием [41].
Преобразование функциональных групп у атома германия
101
Флоринский [51] из серебряной соли метакриловой кислоты и
бромистого трифенилгермания получил метакрилат трифенилгермания (25,7%),
Калиевые соли некоторых непредельных кислот в присутствии
полухлористой меди были успешно использованы Колесниковым и сотр. [52—
55] для синтеза соответствующих ацилатов, использованных в дальнейшем
в исследовании германийорганических полимеров.
Получение метакрилата триэтилгермания [52, 53]. В трехгорлую кслбу, снабженную
обратным холодильником, мешалкой и капельной воронкой, помещают 5 г безводной
калиевой соли метакриловой кислоты, 60 мл абсолютного метанола и небольшое количество
полухлористой меди. К смеси медленно прибавляют 10 г бромистого триэтилгермания, нагревают
в течение часа и оставляют на ночь. Выпавший бромистый калий отфильтровывают, отгоняют
растворитель в вакууме (15 мм) при температуре не выше 50° С, остаток перегоняют в
вакууме. Получают метакрилат триэтилгермания; т. кип. 69—70° С/5 мм, п2£ 1,4569; выход
6 г (61%).
Аналогично получены метакрилаты трибутилгермания (т. кип. 130—
132° С/4 мм; п™ 1,4602), трифенилгермания (т. пл. 180°С), трициклогек-
силгермания (т. пл. 82—84° С), а также акрилат триэтилгермания (т. кип.
88—90°С/12 мм; п™ 1,4582), трибутилгермания (т. кип. 131° С/4 мм;
п™ 1,4609) и трифенилгермания (т. пл. 178—178,5° С).
Сообщение о существовании германийорганических перекисей в
литературе появилось сравнительно недавно [56—61].
Известны два типа германийорганических перекисей R3GeOOR' и
RgGeOOGeRg. Была сделана также попытка получения перекисей типа
R2Ge(OOR)2 [56].
Для получения перекисей первого типа применяют различные методы.
Например, органическими гидроперекисями действуют на гадогениды
триалкил- и триарилгермания в присутствии сухого аммиака или
органических оснований:
R3GeX + R'OOH -» R3GeOOR' + НХ.
Применение органических оснований, например эквивалентных
количеств пиридина или диметиламина, менее удобно, так как образующиеся
соли заметно растворимы в органических растворителях, применяемых в
реакции (эфир, пентан); это затрудняет очистку полученных продуктов.
Более удобно реакцию проводить в присутствии газообразного аммиака.
Аммиак участвует в реакции, образуя, например, трифениламиногерманий [11].
Обмен аминогруппы на алкилпероксигруппу происходит почти мгновенно
при смешении эквимолекулярных количеств реагентов при комнатной
температуре; после удаления растворителя в вакууме получают почти
аналитически чистую германийорганическую перекись
(CeHfi)8GeNH2 + R'OOH -> (C6H5)3GeOOR + NH8.
Получение (CeHebGeOOCfCHsbfCeHs) [61]. В раствор 2,5 г бромистого трифенилгер-
ния в 50 мл эфира пропускают газообразный аммиак до прекращения выделения бромистого
аммония, который затем отсасывают; к фильтрату прибавляют растЕор 1 г (СсН5)(СН3)2СООН
в 10 мл эфира. Поддерживают температуру реакционной смеси 20° С. После отгонки
растворителя в вакууме получают 2,3 г (СбН6)зСеССС(СН3)2 (СеН5); выход 78% ; т. пл. 106—108° С;
после перекристаллизации из бензина т. кип. 70—80° С.
Получение (С2Н5)зОеООС(СНз)з [61]. К раствору 4 г хлористого триэтилгермания в 50
мл эфира прикапывают раствор 2 г (СН3)3СООН в 20 мл эфира при температуре 20° С.
Пропускают ток газообразного аммиака с такой скоростью, чтобы выпадение хлористого
аммония закончилось в течение 20 мин. Осадок отсасывают, фильтрат испаряют в вакууме,
получают 2,8 г {С2Нб)зСеООС(СН3)з; выход 55%; т. кип. 78—79° С/14—15 мм; п^ 1,4368.
102 Германийорганические соединения. Лит. стр. 112—113
По этому методу были получены следующие германийорганические
перекиси:
(СвН5)з GeOOC (С6Н5)3 (54%), т. пл. 193 —195° С (из эфира);
(С6Н5)3GeOOQoHu (95%); т. пл 112—114°С (из бензина);
(С6Н5)3 GeOOQHeO (86%), т. пл. 159—161° С (из бензола и пентана).
Вторым удобным методом получения германийорганических перекисей
является взаимодействие хлористого или бромистого триэтил- или
трифенилгермания с натриевыми солями гидроперекисей
[ R3GeX + R'OONa -> R3GeOOR' -f NaX.
Получение (СбН5)3ОеООС(СН3)з [61]. К 0,4 г тонко измельченного амида натрия в 30 м*
пентана при энергичном перемешивании и комнатной температуре прикапывают раствор
0,6 г (СН3)3СООН в 30 мл пентана. Реакционную смесь перемешивают в течение часа.
К полученной суспензии натриевой соли гидроперекиси при перемешивании прибавляют
раствор 2,55 г бромистого трифенилгермания в 60 мл эфира при температуре 20° С;
перемешивание продолжают еще в течение часа. Выпавший осадок отсасывают, растворитель из
фильтрата удаляют в вакууме. Получают 2,4 г (92%) (СбН5)зСеООС(СНз)з; т- пл- 55—57° С
(из пентана.)
Действие органических гидроперекисей на алкоксисоединения триал-
килгермания также является хорошим методом получения
германийорганических перекисей
R3GeOR' + R"OOH _> R3GeOOR" + R'OH.
Однако обмена алкоксигруппы на пероксигруппу в случае ароматических
германийорганических соединений, например трифенилгермания, не
происходит.
Получение (CoH-^GeOOCXCHab [61]. Раствор 2,7 г метокситриэтилгермания в 20 мл
эфира и 1,3 г (СН3)3СООН в 10 мл эфира оставляют стоять короткое время при комнатной
температуре, затем нагревают с обратным холодильником, отгоняют в вакууме
растворитель и получают 2,6 г (80%) (С2Н5)зОеООС(СН3)з.
Германийорганические перекиси типа R3GeOOGeR3, в которых два атома
металла связаны перекисной группой, получают теми же методами, что и
соединения типа R3GeOOR', но вместо органических гидроперекисей
применяют перекись водорода, например при реакции галогенидов
трифенилгермания и перекиси водорода в присутствии аммиака. Метод
особенно удобен для получения перекиси бас-(трифенилгермания)
2 (СвН5)з GeNH2 + Н202 -> (QH5)3 GeOOGe(C6H5)3 + 2 NH3.
На перекисную группу можно заменить в подобной реакции и алко-
ксигруппу. Так, из метокситриэтилгермания и перекиси водорода с
выходом 77% была получена перекись бас-(триэтилгермания); т. кип. 56—
57° С/0,05 мм] п™ 1,4603.
Попытка получить трифенилтриметилсилилпероксигерманий (С6Н5)3-
GeOOSi(CH3)3 из бромистого трифенилгермания, хлористого триметил-
кремния и перекиси водорода в присутствии аммиака окончилась
неудачей, были выделены лишь перекись бас-(трифенилгермания) и перекись
бш>(триметил кремния).
Германийорганические перекиси в чистом виде бесцветны, термически
довольно устойчивы. Они могут храниться длительное время при комнатной
температуре и не взрываться, не чувствительны к трению и удару. Водой
легко гидролизуются с образованием исходной гидроперекиси или
перекиси водорода.
Преобразование функциональных групп у атома германия
103
Арильные производные гидролизуются легче, чем соответстующие ал-
кильные соединения. При пропускании хлористого водорода в растворы
германийорганических перекисей образуется хлористый триалкил- или
триарилгерманий и исходная гидроперекись.
Описан класс германийорганических алкоксисоеддаений, которые
образуются при реакции органических галогенидов гершетия с алксятуштами
щелочных металлов. Так, например, Вест и сотр. [62] впервые получили
метоксипроизводные метил германия. Метокситриметилгерманий имеет т. кип.
87—88° С/753 мм, п2* 1,401; диметоксидиметилгерманий — т. кип. 118—
118,5 ° С/763 мм, п2* 1,4093; триметоксиметилгерманий — т. кип. 136,5—
138° С/760 мм, п2* 1,4053. Метокситриметилгерманий был получен позже
и другими авторами [46]. Описаны также аллилокситриэтилгерманий [63],
метокси-а-нафтилфенилметилгерманий [64], метокситрифенилгерманий [39].
Получение триметоксиметилгермания [62]. Раствор 13,3 г (0,58 z-am<HtfSf Я&фйЩъ
125 мл метанола прибавляют по каплям к 34,6 г (0,178 ммоля) треххлористогЧУ
ffieffiAГермания. Реакционную смесь нагревают с обратным холодильником в течение 2 чйс. Выдавший
хлористый натрий отсасывают, фильтрат подвергают фракционированной перегонке.
Выход 21,9 г (66%); т. кип. 136,5—138° С/760 мм; п2* 1,4053.
Для получения алкильных производных алкоксигермания реакцию
иногда проводят, действуя на галогениды алкилгермания спиртом в
присутствии аммиака [65] по реакции
с н
R2GeCl2 + 2ROH + 2NH3 -^-* R2Ge (OR)2 + 2NH4Ct.
Таким образом были получены производные (C4H9)2Ge(OR)2, где R =
СН3, С2Н5, /г-С3Н7, /-С3НТ, /г-С4Н9, s-C4H9, *-С4Н9, ^-С4Не, /-С5НИ,
(СН3)(С3Н7) СН.
Получение диметоксидибутилгермания [65]. Медленно пропускают аммиак в смесь 8 г
двухлористого дибутилгермания, 15 г метанола и 30—50 мл бензола. Реакция
сопровождается вначале медленным выделением хлористого аммония. Через несколько минут
развивается довольно быстро экзотермическая реакция, выделение хлористого аммония ускоряется.
Когда реакционная смесь остынет, аммиак прекращают пропускать. После хранения в
течение ночи хлористый аммоний отсасывают и диметоксидибутилгерманий перегоняют в
вакууме; т. кип. ИЗ—116° С/13 мм\ выход 84%.
В случае получения алкоксисоединений, содержащих третичную алко-
ксигруппу, двухлористый дибутилгерманий по каплям при перемешивании
прибавляют к смеси пиридина (2 моля на 1 моль двухлористого
дибутилгермания) и третичного спирта в бензоле, затем пропускают аммиак.
Получение бутилтриэтоксигермания [436]. Сухой аммиак медленно пропускают в смесь
6,55 г треххлористого бутилгермания, 15 г этилового спирта и 60 г бензола. При этом
начинается образование хлористого аммония и разогревание реакционной смеси. Аммиак
пропускают до охлаждения реакционной смеси до комнатной температуры, после чего ее
оставляют стоять в течение ночи, хлористый аммоний отфильтровывают. От фильтрата отгоняют
спирт и бензол. Бутилтриэтоксигерманий перегоняют; т. кип. 90—92° С/8 мм; п2£ 1,4330;
выход 90%.
При действии на двухлористый диметилгерманий о-оксибензилового
спирта в присутствии триэтиламина было получено с количественным
выходом новое германиевое гетероциклическое соединение в виде
светло-желтых кристаллов, хорошо растворимых в органических растворителях,
имеющее т. кип. 118° С/10 мм [66]
СН2ОН СН2 - О
(f J + (СНз)о GeCl2 + 2(СвН3)з N -> fY I +
ОН О Ge(CH3)2
-i-2(C2H5)3N - НС1.
104 Германийорганические соединений. Лит. стр. 112—113
Галогениды трифенилгермания в присутствии бромистого магния [671
реагируют с окисью пропилена по реакции
MgBr2
(СбН5)3 GeX + СИ3 - СН - СН2 > (С6Н5)3 GeOCH2CHXCH3.
\ /
О
Галогениды германийорганических соединений реагируют легко с литий-
и натрийалкилсиланолятами, образуя соединения со звеномQe—О—Si.
Например [68], литийтриметилсиланолят при 10° С реагирует со стехиометри-
ческим количеством чистого хлористого триметилгермания, образуя с
выходом 70% гексаметилгерманосилоксан
(СН3)з GeCi + LiOSi (CH3)3 -» (СН3)3 GeOSi (CH8)8 + LiCl.
Двухлористый диметилгерманий реагирует аналогично. Если
молекулярные соотношения реагентов 1 : 2, то реакция протекает по уравнению
(СН3)2 GeCl2 + 2MOSi (СН3)3 -» [(СН8)8 SiO]2 Ge (CH3)2 + 2МС1
(M = Li, Na, К).
Если молекулярные соотношения реагентов 1:1, то уравнение реакции
будет выглядеть следующим образом:
(СН3)2 GeCl2 +'MOSi (СНС)8 -* (СН8)з SiOGe (СН3)2 CI + MC1.
Одновременно с синтезом соединений со звеном Qe—О—Si была
изучена прочность связей. Расщепление связей Ge—О—Si проводилось
хлористым алюминием [69], S03 и СЮ3 [70], а также РОС13 и Р203С14 [71]. Авторы
высказали соображения о механизме реакции.
При действии сероводорода на галогениды алкилгермания в
присутствии пиридина легко образуются соответствующие тиолы
(СбН5)3 GeBr + H2S + C5H5N -* (С6Н5)3 GeSH + C5H5N • HBr.
Тиолтрифенилгерманий — белое кристаллическое вещество,
устойчивое в сухой атмосфере. Во влажном воздухе легко выделяет сероводород
[72].
Получение тиолтрифенилгермания [72]. Юг бромистого трифенилгермания и 21 г
пиридина растворяют в 150 мл бензола. В прозрачный раствор при перемешивании магнитной
мешалкой пропускают сероводород в течение 1,5 час. Выделившийся осадок бромистоводо-
родной соли пиридина отсасывают, фильтрат испаряют досуха в вакууме при 25—30° С.
Получают 8,65 г (97%) белого кристаллического вещества. После перекписталлизации из
бензола получают чистый тиолтрифенилгерманий.
При окислении иодом тиолтрифенилгермания образуется бх/с-трифенил-
германийдисульфид
2 (С6Н5)3 GeSH + J2 -> (C6H5)3 GeSSGe (СбН5)3 + 2HJ.
При действии на германиевые тиолы галоидалкилов RX в пиридине с
хорошим выходом образуются алкилмеркаптиды трифенилгермания [72]
C5H6N
(C6H5)3GeSH + RX > (C6H5)3GeSR + HX.
Ё дальнейшем было найдено, что при действии меркаптанов в
присутствий третичных аминов на хлористый или бромистый трифенилгерманий
Преобразование функциональных групп у атома германия
105
легко получаются алкилмеркаптиды трифенилгермания [73, 74]:
(C6H5)3GeX + RSH > (C6H6)3GeSR + C5H5N . НХ.
C.H.N или (C2H,)3N
(R = (CH2)ioCH8l CH2CH2OH, CH2CH=CH2, СН2СООН, С(0)СН8,
СН2С(0)СН3, CH2C(0)NH2, СбН5, QF*, р-С1С6Н4,
2-СюН7, 17-(3-меркаптотестостерон).
Полученные меркаптиды термически и гидролитически устойчивы [73].
Сатже и сотр. [32] нашли, что Pb(SR/)2 реагирует в бензольной среде
с хлоридами алкилгермания с образованием алкилмеркаптидов триалкил-
и диалкилгермания с выходом в среднем 65%
2R3GeCl + Pb(SR')2 -* 2R3GeSR' + РЬС12,
R2GeCl2 + Pb (SR')2 -* R2Ge (SR')2 + PbCl2.
Были получены R3GeSR' (где R = C2H5, C4H9; R' = C2H5, QH9) и
R2Ge(SR/)2 (где R = C4H9, R'= C2H5). Хлоргидриддиэтилгерманий
реагировал аналогично
2 (C2H5)2Ge (CI) H + Pb (SC2H5)2 — (G2H5)2 Ge (H) (SC2H5) + PbCl2.
81%
Рохов [75—77] описал реакцию двухлористого диметилгермания с
сероводородом
(CH3)2GeCl2 + H2S -* (CH8)£GeS + 2НС1.
Получение сульфида диметилгермания [75]. 100 г двухлористого диметилгермания*
перемешивают в 1 л воды и пропускают сероводород в течение нескольких дней. Осадок
отсасывают, промывают водой, растворяют в 100 мл теплого спирта, высаживают добавлением
четырех объемов холодной воды. Смесь сильно охлаждают, насыщают сероводородом для
предотвращения гидролиза и затем отсасывают. Осадок промывают на фильтре водой,
содержащей сероводород, и сушат в эксикаторе. Получают 73,4 г (94,5%) сульфида
диметилгермания; т. пл. 55,5° С, т. кип. 302° С.
При действии на двугалоидные соединения дифенилгермания двух
молей меркаптана в присутствии третичных аминов было получено [731.
большое число дитиопроизводных дифенилгермания (С6Н5)2 Ge(SR)2, где R =
(СН2)10СН3, СН2СН=СН2, CH2C(0)NH2, С6Н5, /7-С1СбН4, /7-Ш2СбН4,
p-NH2CeH4, СбН5СН2, С(0)СбН5, а-С10Н7.
Этилентиогликоль, тиогликолевая кислота и 2-оксиэтантиол с
двубромистым дифенилгерманием легко образуют соединения со
своеобразными гетероциклами [731
н2с — сн3 о=с — сн2 н2с — сн2
S S
\ /
Ge(C6H5)2
О S
\ /
Ge (C6H5)2
О S
\ /
Ge(C6H5)2
Подобные гетероциклические соединения были получены и при реакции
двухлористого диметилгермания с различными тиогликолями или тиофе-
нолами [78—811.
Другим путем синтеза алкилмеркаптидов трифенилгермания является
действие галоидных алкилов на натрийтионолят трифенилгермания. Нат-
рийтионолят трифенилгермания может быть приготовлен и выделен с
хорошим выходом при прибавлении по каплям бензольного раствора
бромистого трифенилгермания к свежеприготовленной суспензии сульфида
натрия в этаноле [82]
rCeH5)3GeBr + Na2S -> (C6H5)3GeSNa + NaBr.
106
Германийорганические соединения. Лит. стр. 112—113
Чистая соль довольно гигроскопична, но в эксикаторе может
храниться больше месяца без изменения. Она растворима в воде, бензоле, спирте.
При нагревании ее с абс. спиртом образуется бш>(трифенилгерма-
ний)сульфид и сульфид натрия
С2Н5ОН
(C6H5)2GeSNa (C6H5)3GeSGe (CGH5)3 + Na2S.
Аналогично сульфид натрия действует и на двубромистый дифенилгер-
маний [72]
(C6H5)2GeBr2 + 2Na2S -* (C6H5)2Ge (SNa)2 + 2NaBr.
При реакции натрийтионолята трифенилгермания с галоидными алкилами
был выделен ряд алкилмеркаптидов трифенилгермания [82]
(CeH5)3GeSNa + RX ^ (CeH5)3GeSR
(R=CH3, П-С4Н9, С6Н5СН2, С(0)С6Н5, C(0)C6H4N02-p, Ge(C6H5)3, GH2SGH3).
В отличие от сульфида натрия селенид натрия при действии на двухлори-
стый диметилгерманий образует не соответствующие натрийселеноляты ди-
метилгермания, а циклический трехмерный селенид диметилгермания [83—
851:
3(CH3)2GeCl2 + 3Na2Se -> [(CH3)2GeSe]3 + 2NaCl.
Селенид диметил германия при 0° С реагирует с метиллитием в абс.
эфире по уравнению
[(CH3)2GeSe]3 + 3CH3Li -> 3 (CH3)3GeSeLi.
Литийтриметилгерманийселенид — вещество чрезвычайно легко гидро-
лизующееся влагой воздуха. Легко растворимо в эфире, реагирует с
соединениями типа R3MC1, где М = Si, Ge, Sri
(CH3)3GeSeLi + (СН3)3/ЧС1 -> (CH3)3GeSeM (CH3)3 + LiCl.
Реакция была проведена с хлористым триметилгерманием, а также с
кремнием и оловом [85].
При действии газообразного аммиака на бромистый трифенилгерманий
или другие галогениды трифенилгермания в органических растворителях
легко образуется трифенилгермиламин [9, 11]
(C6H5)3GeBr> 2NH3 -> (C6H5)3GeNH2 + NH4Br.
Полученные аминопроизводные легко гидролизуются даже влагой воздуха.
Другим путем для получения трифенилгермиламина является действие
амида калия на бромистый трифенилгерманий
(C6H5)3GeBr + KNH2 -> (C6H5)3GeNH2 + KBr,
(C6H5)3GeNH2 + KNH2 -> (GQH5)3GeNHK + NH3,
(C6H5)3GeNHK + NH4Br -> (G6H5)3GeNH2 + KBr + NH3.
При действии аммиака на дигалоидные соединения легко получаются
имины [17, 86]
(C6H5)2GeCl2 + 3NH3 -> (C6H5)2GeNH + 2NH4C1.
При действии жидкого аммиака на трехиодистыйэтилгерманий
образуется белый порошок, нерастворимый в жидком аммиаке [87]. Как
предполагают авторы, идет образование нитрида согласно уравнению
QHsGeJg + 4NH3 -> QH5GeN + 3NH4J.
Преобразование функциональных групп у атома германия
107
Андерсон [88] показал, что треххлористый этил германий реагирует с
диметил- и диэтиламином, образуя производные алкиламиногермания.
C2H5Ge[N(CH3)2]3HMeeTT.KHn.l05—107° С/34 л**;**» Ш9;С2Н5Ое[К(С2Н5)2]з—
т. кип. 117—118° С/12 мм\ df 1,108.
Двухлористый диметилгерманий и метиламин легко реагируют в эфире
с образованием тримера диметилгерманилметиламина [89] (выход 75%).
Это бесцветная легкоподвижная жидкость с т. кип. 80° С/2 мм, хорошо
растворимая в органических растворителях.
NCH3
/ \
(CH3)2Ge Ge (CH3)2
3(CH3)3GeCl2 + 9CH3NH2-
+ 6CH3NH2.HC1.
CH3N NCH3
\ /
Ge (CH3)2
Полученный тример количественно реагирует с метиллитием в эфирном
растворе, образуя литийтриметилгерманилметиламид
Li
[(CFI3)oGeNCH3]3 + 3LiCH3 -> 3 (CH3)3GeN
\
CH3
Литийтриметилгерманилметиламид очень реакционноспособен. Он
легко реагирует с хлористым триметилгерманием, оловом, кремнием, свинцом,
образуя с хорошим выходом новые соединения, имеющие связь Ge—N—М
Li
(CH3)3GeN + (СНз)8МС1 - (CH3)3GeN (СН3) М (СН3)3 + LiCl
\
СНз
(М = Si, Ge, Sn, Pb).
Было найдено, что хлористый триалкилгерманий с натрий-бас-(триме-
тилсилил)амидом в сухих органических растворителях реагирует с
хорошим выходом по уравнению
[(CH3)3Si]2NNa + R3GeCl -> [(CH3)3Si]2N—GeR3 + NaCl
(R=CH3, n-C4H9).
Двухлористый диметилгерманий реагирует с теми же соединениями, но
выход продуктов реакции составляет лишь 30—40% [90].
(CH3)aGeCla + 2 [(CH3)3Si]2NNa -* [(CH3)3Si]2N-Ge (CH3),-N [Si (CH3)3]2 + 2NaCl.
Сатже с сотр. [91] нашел, что при действии аминомагниевых и амино-
литиевых соединений на хлориды триалкилгермания в среде углеводородов
или тетрагидрофурана с хорошими выходами образуются соединения диал-
киламинотриал кил германия:
R3GeCl + RgNMgBr -> R^eNR^ + MgBrCl,
RsGeCl + RgNLi - R3GeNR2 + LiCl.
Большая реакционная способность связи Ge—N позволила
синтезировать при действии углекислого газа, сероуглерода, фенилизоцианатов,
108 Германийорганжсские соединения. Лит. стр. 112—113
фенилизотиоцианатов ряд новых германийорганических производных:
(C2H5)3GeN(QH5)2 + 0=С= О -* (C2H5)3Ge-0—С—N(C2H5)2,
90% ||
О
(C2H5)3GeN(CH3)2 + S=C=S -> (C2H5)3Ge—S-C—N(CH3)2,
90% ||
S
(C2H5)3GeN(C2H5)2 + CeH5N=C=0 -> (C2H5)3Ge-N-C-N(GH5)2,
80% | ||
QII5O
(C2H5)3GeNR2 + C6H6N=C=S -> (C2H5)bGe—N C-NR2
I I!
CGH5 S
(R = CH3 и С2Н5, выход 72 и 60%).
Полученные соединения охарактеризованы рядом химических реакций.
Хлористый трифенилгерманий при реакции с N-натрий или литийдифенил-
амином образуют соответствующие дифениламинопроизводные германия
[92]
(C6H5)2NLi (Na) + (CGH5)3GeCl -> (C6H5)2N-Ge (C6H5)3
(т. пл. 153,5—155° С).
Аналогично (C2H5)2NLi реагирует с дифеноксидиметилгерманием с
образованием (CH3)2Ge[N(C2H5)2]2 [93].
Рикенс и сотр. [43г, 43д] использовали галогениды триалкилгермания в
реакциях с различными Na-, К- или Li-производными гетероциклов типа
имидазола, пиразола, пиррола и некоторых других для получения ряда
новых соединений со связью Ge—N.
Получение N-трифенилгермилпиррола [43г]. К суспензии пиррола калия,
приготовленного из 5,9 г (0,08 молей) пиррола и 2,4 г (0,06 моля) металлического калия в 50 мл толуола,
прибавляют по каплям раствор 18,4 г (0,054 моля) хлористого трифенилгермания в толуоле.
Полученную фиолетово-красную реакционную смесь нагревают с обратным холодильником
в течение часа и фильтруют. Осадок промывают 100 мл бензола. После удаления большей
части растворителя и охлаждения получают белые кристаллы. После
перекристаллизации из бензола получают 8,8 г (44%) N-трифенилгермилпиррола; т. пл. 198—199° С;
дополнительно из фильтрата получают еще 43 г вещества (21,5%) с т. пл. 196—199° С.
Хлористый триметилгерманий и двухлористый диметилгерманий
в эфире реагируют с азидом натрия, образуя триметилгерманилазид и
диметилгерманилдиазид [94]
[(CH3)3GeCI + NaN3 -> (CH3)bGeN3 + NaCl,
(CH3)2 GeCl2 -f 2NaN3 -> (CH3)2 Ge (N3)2 + 2NaCl.
Оба полученных соединения — бесцветные легкоподвижные жидкости,
хорошо растворимые в органических растворителях, легко гидролизую-
щиеся водой с образованием соответствующих окисей.
Последующими исследованиями было показано, что
триметилгерманилазид может быть приготовлен в эфирно-водной среде в присутствии
соляной кислоты; для улучшения выхода требуется избыток аммиака
(CH3)8GeX + HN3 -> (CH3)3GeN3 + Н+ + X".
Полученный триметилгерманилазид очищают фракционированной
перегонкой; т. кип. 132—135° С [95].
Бромистый трифенилгерманий при реакции с азидом натрия образует
с выходом 59% азид трифенилгермания; т. пл. 102—103° С [96] или 107—
Преобразование функциональных групп у атома германия
109
107,5° С [97]. При реакции азидтрифенилгермания с трифенилфосфином
получены стабильные фосфоимины (СбН5)3ОеМ=Р(СбН5)з [96].
В американском патенте [98] приводится получение соединений
формулы Ri-n Qe (N = YR'R"R'")n, где R — алкил, арил, галоид, алкоксил,
амино-, алкилтио- или азидогруппа; Y — Р, As или Sb; R\ R", R"' — арил,
алкил, алкоксил, арилокси-, амино-, алкилтио- или арилтиогруппа; /г= 1—4.
Бромистый трифенилгерманий при нагревании в эфире с трифенилфос-
финметиленом образует [(C6H5)3PCH2Ge(C6H5)3]Br; т. пл. 121—122° С
[99].
При реакции бромистого триэтилгермания с (C6H5)2PLi в тетрагидрофу-
ране получен ((^Нб)/деР(<^Н5)2 [100].
Продолжая исследования в области карбонилов переходных металлов,
Несмеянов и Анисимов с сотр. [101—103] показали, что при реакции гало-
генидов арилгермания с карбонилами марганца и рения образуются
биметаллические соединения карбонилов переходных металлов.
С карбонилами марганца реакция протекает так:
(СвНв)4.„ОеВгд + /iNa.Vta (GO). -> (C3H5)4_„Ge [Мп (СО)5]а + /iNaBr
(/1 = 1 или 2).
По этой реакции при п = 1 авторы получили (C6H5)3Ge[Mn(CO)5] (I) с
выходом 61%; при п = 2 получены одновременно два продукта (C6H5)2Ge-
.[Мп(СО)5]2 (II) и (C6H5)2[(CO)5Mn]Ge-Ge[Mn(CO)5](C6H5)2 (III).
Выходы (II) и (III) зависят от температуры реакции; при —2°С преобладает
соединение (II), а соединение (III) выделено с выходом лишь 3,2%; однако
при 15° С получено 32% соединения (III) при незначительном увеличении
общего выхода [102]. Соединения типа R4-aGe[Re(CO)51n получены по
уравнению
R4_„G2Xn -J- nNaRe (СО)6 - R4_„Ge [Re (CO)5]„ + /iNaX
(R = СзН3; X = Gl, Br; n = 1 и 2).
Соединения (C6H5)3GeRe(CO)5 и (C6H5)2Ge[Re(CO)5]2 выделены с
выходами 87 и 60% соответственно в виде бесцветных кристаллов, стабильных на
воздухе [103]. Теми же авторами была осуществлена новая в ряду
производных карбонилов металлов реакция хлорпентакарбонила марганца с
гидридом треххлористого германия [101]
ClgGaH + С1Мп (СО)5 -> Cl3G3Mn (СО)5 + НС1.
Авторы изучили ряд химических превращений полученных соединений.
Описывается также реакция триметил(изо)цианогермания с пентакар-
бонилом железа [104]. В результате с хорошим выходом был получен
(CH3)3GeN=C-Fe(CO)4.
РЕАКЦИИ ОБМЕНА ГЕРМАНИЙОРГАНИЧЕСКИХ АЦИЛАТОВ,
АЛКОКСИ- И АИИНОСОЕДИНЕНИЙ
При нагревании ацилатов германийорганических соединений с
органическими кислотами, серной кислотой, алкилсульфокислотами,
меркаптанами более летучая кислота отгоняется и происходит замена
функциональной группы при атоме германия. Метод переацилирования может быть
широко использован для получения различных ацилатов, меркаптидов,
однако его ограничением является обязательный широкий интервал в
температурах кипения кислот
RsGaOOCR' + R"C03H -> R^eODCR" + R'GOOH.
110
Германийорганические соединения. Лит. стр. 112—113
Методом переацилирования были получены димонохлорацетат ди-я-
пропилгермания [18], димонохлорацетат и ди-Р-хлорпропионат диизопропил-
германия [16], Р-бромпропионат, а-бромпропионат, р-иодпропионат и циа-
ноацетат триэтилгермания [37], монохлорацетат, дихлорацетат и трихлор-
ацетаттри-я-пропилгермания [36], энантат три-я-бутилгермания [5], а
также о-аминобензоат триэтилгермания [31].
Интересно отметить, что этот метод может быть использован и для
получения соответствующих сульфатов. Так, были получены сульфат диизо-
пропилгермания [16], сульфат ди-я-пропилгермания [18], сульфат бис-
(три-я-пропил)германия [36] и сульфат <5#с-(триизопропил)германия [38].
Метод переацилирования используется также и для синтеза германий-
органических производных алкилсульфокислот [105].
Получение метилсульфоната трибутилгермания [105]. 5,7 г (15,9 моля) трифторацетата
трибутилгермания и 1,39 г (14,5 ммоля) метилсульфокислоты нагревают с обратным
холодильником в течение 10 мин. с одновременной отгонкой образовывающихся продуктов.
Вначале отгоняется 1,36 г (12,0 ммолей) трифторуксусной кислоты, 0,40, г избытка трифтораце-
тата трибутилгермания, затем 0,45 г сырого метилсульфоната трибутилгермания и,
наконец, 4,35 г чистого метилсульфоната трибутилгермания, т. кип. 150° С/1 мм\ выход 89%.
По аналогичной методике были получены метилсульфонат
триэтилгермания (т. кип. 116—117° С/1 мм), этилсульфонат триэтилгермания (т. кип.
125—126° С/1 мм), диметилсульфонат диизопропилгермания (т. кип. 180—
182° С/1 мм), метилсульфонаттри-я-бутилгермания (т. кип. 150—152°С/1 мм),
этилсульфонат три-я-бутилгермания (т. кип. 149—151° С/1 мм) [105].
Андерсон [105] провел большое число обменных реакций германийор-
ганических производных алкилсульфокислот с различными
неорганическими солями и металлоорганическими соединениями и показал, что алкил-
сульфогруппы легко обмениваются на другие атомы при простом
кипячении реагентов.
Метод применяется также и для получения арил;- и алкилмеркаптидов
триэтилгермания [31]
(C2H5)3GeOCOCH3 + HSC6H5 -> (QHebGeSCeHe + СН3СООН.
Был получен ряд новых ранее не описанных в литературе меркаптидов
типа (C2H5)3GeSR.
Сатже и сотр. [32] нашли, что при нагревании (100° С) метокситриалкил-
германия с малолетучими меркаптанами легко происходит замена метокси-
группы с образованием соответствующих алкил-или арилмеркаптидов три-
алкил германия
R3GeOCH3 + R'SH -> R3GeSR' + СН3ОН
(R=C4H9; R'=CeH6, /i-C8H17).
Аналогичная реакция была проведена с диметоксидиэтилгерманием и
фенилмеркаптаном, а также между диэтоксидибутилгерманием и додецил-
меркаптаном [43г]. При нагревании производных диалкиламинотриалкил-
германия с меркаптанами происходит количественная реакция с выделением
соответствующих аминов и образованием производных со связью Ge—S [32]
R3GeN:(CH3)2[+ R'SH -* R3GeSR' + (CH3)2NH.
Этот метод интересен тем, что позволяет выделить с высокими выходами ал-
килмеркаптиды триалкилгермания с вторичными и третичными радикалами,
получение которых другими путями затруднено.
Были получены я-октилмеркаптид триэтилгермания (т. кип. 161° С/7 мм;
п™ 1,4839; выход 89%), изопропилмеркаптид триэтилгермания (т. кип.
116° С/17 мм; п20 1,4886; выход 93%), mpem-бутилмеркаптид
трибутилгермания (т. кип. 169° С/18 мм; п™ 1,4835; выход 90%). Было найдено , что ди-
Преобразование функционалън ых групп у атсма германия
Hi
алкоксидибутилгерманий (C4H9)2Ge(OR)2 (R = С2Н5 и /-С3Н7) легко меняет
алкоксигруппу при реакции в бензоле с высшими спиртами [436, 1061:
(C4H9)2Ge (OR)2 + 2R'OjI (избыток)^1°4! (C4H9)2Ge (OR'\ + 2ROH
(R' = ^C4R9, .v-QH9l 1-С5Ни)г
Реакция в случае я-бутилового спирта заканчивается очень быстро.
С вторичным бутиловым спиртом реакция происходит более медленно и
полностью заканчивается лишь в присутствии /г-толуолсульфокислоты. С
третичными спиртами без катализатора (/г-толуолсульфокислота) реакция не
идет.
С гликолями реакция протекает с образованием циклических продуктов,
причем во всех случаях, кроме этиленгликоля, в присутствии л-толуол-
сульфокислоты как катализатора
О
(OS 9)2Ge(OR)2 + R' (ОН), —^ульфокислот^ g Q/ \, + 2ROn
\ /
О
(R^QH., или i-C3H7; R'=—СН2СН2—, —СН2СН2СН2—, — CII2CII2CH2CH2—,.
—СН2СН2СН (СНз)—, — СН (СН)3 СН (СНз)—, —CI IoCI 13ОСН3СНз—,
—СН (СНз) СН2С (СНз)2—)•
Аналогичная реакция была проведена с моноалкокситрибутилгерманием,
диалкоксидифенилгерманием и различными гликолями [107] в
присутствии /г-толуолсульфокислоты как катализатора.
Реакция между этокситри-я-бутилгерманием и этиленгликолем [107]. 1,55 г этокситри-я-
бутилгермания смешивают с 0,17 г этиленгликоля и 5 г бензола. Реакционную смесь
кипятят с обратным холодильником 4—5 час. в присутствии 0,02 г /г-толуолсульфокислоты как
катализатора. Во время нагревания отгоняется азеотропная смесь бензола и этилового
спирта. По окончании реакции катализатор разрушают добавлением нескольких капель триэтил-
амина. Бензол отгоняют при уменьшенном давлении, остаток перегоняют. (C4H9)3GeOCH2-
CH2OGe (С4Н9)3 имеет т. кип. 180° С/0,4 мм; это бесцветная, легко подвижная жидкость;
выход 84%.
В дальнейшем при изучении реакций алкоксисоединений германия было
найдено, что реакции обмена могут происходить с уксусной и бензойной
кислотами, а также с некоторыми оксикислотами [108]. Реакция протекает
в основном по следующим уравнениям:
OR'
/
R2Ge (OR')2 + R"COOH -> R2Ge + R'OH,
\
OOCR"
О
/ \
R2Ge (OR'b + HOR"COOH -* R2Ge R" +2R'OH,
\ /
ooc
(QHebGeOR' + R"COOH -» (C4H9)3GeOOCR" + R'OH
(R'—C4H9 или C6H5),
(C4H9)3 GeO
\
2 (C4H9)3 GeOR' + HOR"COOH -» R" + 2R'OH.
/
(C4H9)3 GeOOC
Реакция между этокситрибутилгерманием и бензойной кислотой в молярных
соотношениях 1:1 [108]. 1,23 г этокситрибутилгермания реагируют с 0,52 г бензойной кислоты в 20 г
бензола. Реакционную смесь нагревают 4 часа, при этом отгоняется азеотропная смесь
этилового спирта и бензола. Остаток бензола удаляют в вакууме. После перегонки получают
1,35 г продукта с т. кип. 160° С/0,5 мм\ выход 87%. По анализу полученный продукт
соответствует бензоату трибутилгермания.
112
Германийорганичвскиг соединения
ЛИТЕРАТУРА
1. AndersonH.H., J. Am. Chem. Soc, 73, 5440 (1951).
2. К r a u s С. A., F 1 о о d E. A., J. Am. Chem. Soc, 54, 1635 (1932).
3. A n d e r s о п H. H., J. Am. Chem. Soc, 71, 1799 (1949).
4. A n d e г so n H. H., J. Am. Chem. Soc, 75, 814 (1953).
5. AndersonH. H.,J. Am. Chem. Soc, 73, 5800 (1951).
6. FuchsR., GilmanH., J. Org. Chem., 23, 911 (1958).
7. BauerH., Burschkies K., Ber., 65, 956 (1932).
8. BauerH., Burschkies K., Ber., 67, 1041 (1934).
9. К г a u s С A., FosterLS., J. Am. Chem. Soc, 49, 457 (1927).
10. О r n d о r f f W. R., TabernD. L, DennisL.M., J. Am. Chem. Soc, 49,
2512 (1927).
11. К г a u s С A., W о о s t e г С. В., J. Am. Chem. Soc, 52, 372 (1930).
12. J о h n s о п О. H., S с h m a 1 1 E. A., J. Am. Chem. Soc, 80, 2931 (1958).
13. TabernD.L, О г п d о r f f W. R., DennisL.M, J. Am. Chem. Soc, 47,
^2039 (1925).
14. SimonsJ.K., WagnerE.C, M u i 1 e r J. H., J. Am. Chem. Soc, 55, 3705
(1933).
15. JS imonsJ.K., J. Am. Chem. Soc, 57, 1299 (1935).
16. A n d e r s о п H. H., J. Am. Chem. Soc, 78, 1692 (1956).
17. F 1 о о d E. A., J. Am. Chem. Soc, 54, 1663 (1932).
18. A n d e r s о п H. H., J. Am. Chem. Soc, 74, 2370 (1952).
19. AndersonH. H., J. Am. Chem. Soc, 83, 547 (1961).
20. M a z e г о 1 1 e s P., Bull. Soc chim. France, 1962, 1907.
21. JohnsonO.H., Harris D. M. J. Am. Chem. Soc, 72, 5564 (1950).
22. AndersonH.H. J. Am. Chem. Soc, 82, 3016 (1960).
23. TabernD.L, Orndorf f W. R., DennisL.M., J. Chem. Soc, 127, 1760
(1925).
24. BauerH., Burschkies K., Ber., 66, 1156 (1933).
25. AndersonH.H., J. Am. Chem., Soc, 72, 194 (1950).
26. R e i с h 1 e W. Т., J. Org. Chem., 26, 4634 (1961).
27. SchmidtM., R u i d i s с h I., Schmidbaur H., Ber., 94, 2451 (1961).
28. Schmidt M., Schmidbaur H., Ruidisch I., BornmannP., An-
gew. Chem., 73. 408 (1961).
29. S с h m i d t M., R u i d i s с h I., Angew. Chem., 73, 408 (1961).
30. SchmidtM., R u i d i s с h I., Ber., 95, 1454 (1962).
31 A n d e r s о п H. H., J. Org. Chem., 21, 869 (1956).
32. Satge J., Lesbre M., Bull. Soc. chim. France, 1965, 2578.
33. A n d e r s о п H. H., J. Am. Chem. Soc, 73, 5439 (1951).
34. JohnsonO.H, NebergallN.H., J. Am. Chem. Soc, 71, 1720 (1949).
35. A n d e r s о n H. H., J. Am. Chem. Soc, 72, 2089 (1950).
36. AndersonH.H., J. Am. Chem. Soc, 73, 5798 (1951).
37. AndersonH.H., J. Org. Chem., 20, 900 (1955).
38. AndersonH.H., J. Org. Chem., 20, 536 (1955).
39. Brook A.G, J.Am. Chem. Soc, 77, 4827 (1955).
40. СоборовскийЛ. 3., ГладштейнБ. М., КулюлинИ. П., Авт. свид.
СССР 168693 (1965); РЖХим, 1966, 10Н113П; Бюлл. изобрет., №5, 23 (1965).
41. A n d е г s о п Н. Н., J. Am. Chem. Soc, 74, 2371 (1952).
42. R u i d i s с h I., SchmidtM., Angew. Chem., 75, 575 (1963).
43. R u i d i s с h I., SchmidtM., Ber., 96, 821 (1963).
43a. MehrotraR.C, MathurS., J. Organometal. Chem:, 6, 11 (1966).
436. MehrotraR.C, MathurS., J. Organometal. Chem., 7, 233 (1967).
43b. MehrotraR.C, Gupta V.D., Sukhani D.,J. Organometal. Chem., 9,
263 (1967).
43r. RijkensF., Van der KerkG. J. M., Investigations in field of organoger-
manium chemistry. Germanium research commitee, Niderland, 1964.
43д. Rijkens F., JanssenM. J., Van der Kerk G. J. M., Rec trav.
chim., 84, 1597 (1965).
44. M о r g a n G. Т., DrewH.D. K-, J. Chem. Soc, 125, 1261 (1924).
45. R u i d i s с h I., SchmidtM., Z. Naturforsch., 18b, 508 (1963).
46. G r i f f i t h s J. E., OnyszchukM., Canad. J. Chem., 39, 339 (1961).
47. S e у f e r t h D., К a h 1 e n N., J. Org. Chem., 25, 80$ (960).
48. Beck W., SchuiererE., Ber., 97, 3517 (1964). *
49. A n d e r s о п H. H., J. Am. Chem. Soc, 75, 1576 (1953).
50. В г о о k A. G., G i 1 m a n H., J. Am. Chem. Soc, 76, 77 (1954).
51. Флоринский Ф. С, ЖОХ, 32, 1443 (1962).
52. Колесников Г. С, Давыдова С. Л., Климентова В. В., Высоко
мол. соед., 2, 563 (1960).
Преобразование функциональных групп у атома германия 113
53. Л е й т е с Л. А., Е г о р о в Ю. П., К о л е с н и к о в Г. С, Д а в ы д о в а С. Л.,
Изв. АН СССР, ОХН, 1961, 1976.
54. К о л е с н и к о в Г. С, Давыдова С. Л., К л и м е н т о в а Н. В., Сб.
«Синтез и свойства мономеров». М., «Наука», 1964, стр. 113.
55. Колесников Г. С, Давыдова С. Л., КлиментоваН. В., Высоко-
молек. соед., 4, 1098 (1962).
56. D a v i e s A. G., Н а 1 1 С. D., J. Chem. Soc, 1959, 3835.
57. D a v i e s A. G., H a 1 1 С. D., Chem. and Ind., 1958, 1695.
58. R i е с h e A., D a h 1 m a n n J., Angew. Chem., 71, 194 (1959).
59. RiecheA., Angew. Chem., 72, 36 (1960).
60. К а г n о j i t z k у V., Chemie et industrie, 85, 92 (1961).
61. R i e с h e A., D a h 1 m a n n J., Ann., 675, 19 (1964).
62. West R., Hunt H. R., W h i p p 1 e R. O., J. Am. Chem. Soc, 76, 310 (1954).
63. Satge J., Bull. Soc. chim. France, 1964, 630.
64. В г о о k A. G., P e d d 1 е G. J., J. Am. Chem. Soc, 85, 1870 (1963).
65. Mathur S., С h a n d г a G., Rai A.K., Mehro tra R.C., J. Organometal.
Chem., 4, 294 (1965).
66. WieberM., SchmidtM., J. Organometal. chem., 1, 93 (1963).
67. P i k e R. M., L a v i g n e A. A., Rec trav. chim., 82, 49 (1963).
68. Schmidbaur H., SchmidtM., Ber., 94, 1138 (1961).
69. SchmidbaurH., ScmidtM., Ber., 94, 1349 (1961).
70. S с h m i d b a u r H., SchmidtM., Ber., 94, 2137 (1961).
71. Schmidt M., SchmidbaurH., R u i d i s с h I., Angew. Chem., 74, 408
(1961).
72. H e n г у M. C, DavidsonW.E, Canad. J. Chem., 41, 1276 (1963).
73. DavidsonW.E., H i 1 1 s K., H e n г у M. C, J. Organometal. Chem., 3, 285
(1965).
74. H о о t о n К. A., A 1 1 r e d A. L., Inorg. Chem., 4, 671 (1965).
75. R о с h о w E. G. J. Am. Chem. Soc. 70, 1801 (1948).
76. В г о w n M. P., RochowE.G., Am. Chem. Soc, 82, 4166 (1960).
77. RochowE.G., A 1 1 r e d A. L., J. Am. Chem. Soc, 77, 4489 (1955).
78. WieberM., SchmidtM., J. Organometal. Chem., 1, 336 (1964).
79. WieberM., SchmidtM., Z. Naturforsch., 18b, 847 (1963).
80. WieberM., SchmidtM., Z. Naturforsch., 18b, 849 (1963).
81. W i e b e r M. S с h m i d t M., J. Organometal. Chem., 2, 129 (1964).
82. HenryM.C, DavidsonW.E, J. Org. Chem., 27, 2252 (1962).
83. SchmidtM., R u f H., Angew. Chem., 73, 64 (1961).
84. SchmidtM., R u f H., Inorg. and Nucl. Chem., 25, 557 (1963).
85. R u i d i s с h I., SchmidtM., J. Organometal. Chem., 1, 160 (1963).
86. К r a u s С. А., В г о w n С L., J. Am. Chem. Soc, 52, 3690 (1930).
87. F 1 о о d E. A., J. Am. Chem. Soc, 55, 4935 (1933).
88. AndersonH.H., J. Am. Chem. Soc, 74, 1421 (1952).
89. R u i d i s с h I., SchmidtM., Angew. Chem., 76, 686 (1964).
90. S с h e r e r O., S с h m i d t M., Angew. Chem., 75, 642 (1963).
91. S a t g ё J., LesbreM., В a n d e t M., Compt. rend., 259, 4733 (1964).
92. В a u m U.LehnW.L, Tamborski C, J. Org. Chem., 29, 1264 (1964).
93. Koster — Pelugmacher A., Termin E., Naturwissenshaften, 51, 554
(1964).
94. Ruidisch I. I., Schm i d t M., J. Organometal. Chem., 1, 493 (1964).
95. Thayer J.S., WestR., Inorg. Chem., 3, 889 (1964).
96. T h а у e r J. S., WestR., Inorg. Chem., 3, 406 (1964).
97. R e i с h 1 e W. T. Inorg. Chem., 3, 402 (1964).
98. WashburnR.M., В a 1 d w i n R. А., Пат. США 3112331 (1963); РЖХим., 1965,
16Н61П.
99. Seyierth D.,GrimS.O., J. Am. Chem. Soc, 83, 1610 (1961).
100. Glokling F., H о о t о n K. A., Proc Chem. Soc, 1963, 146.
101. Несмеянов А. Н., Анисимов К. Н., Колобова Н. Е., Антоно
в а А. В., Изв. АН СССР, серия хим., 1965, 1309.
102. Несмеянов А. Н., Анисимов К. Н., Колобова Н. Е.,
Антоно в а А. Б., Изв. АН СССР, серия хим., 1966, 160.
103. Несмеянов А. Н., Колобова Н. Е., Анисимов К. Н., Хандож-
к о В. Н., Изв. АН СССР, серия хим., 1966, 163.
104. S е у i e r t h D., К a h 1 е n N.. J. Am. Chem. Soc, 82, 1080 (1960).
105. AndersonH.H., Inorg. Chem., 3, 910 (1964).
106. MathurS., ChandraG., Rai A. K., Mehrotra R.C., J. Organometal.
Chem., 4, 371 (1965).
107. MehrotraR.C, MathurS., J. Organometal. Chem., 6, 427 (1966).
108. MathurS., MehrotraR.C, J. Organometal. Chem., 7, 227 (1967).
8 Заказ П 207
Г лава XI/
РЕАКЦИИ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
СВЯЗАННЫЕ С ИЗМЕНЕНИЯМИ В РАДИКАЛЕ
Для германийорганических соединений характерно большое число
реакций, связанных с преобразованием органического радикала при атоме
германия: присоединение к непредельному алифатическому радикалу,
реакции подвижного галоида в радикале, замещение водорода в радикале на
галоид, реакции ацетиленовых (или вообще алкиновых) радикалов с маг-
нийорганическими соединениями и др.
В германийорганических соединениях, содержащих непредельные
алифатические радикалы, связь германий — углерод обладает достаточной
прочностью. Следовательно, здесь возможно осуществить широкий круг
реакций присоединения по двойной или тройной связи с целью синтеза
германийорганических соединений с различными функциональными группами.
Заметим, что у оловоорганических соединений связь непредельного
алифатического радикала с оловом очень нестойка, и при действии водорода,
галоидов, галоидоводородных кислот и других реагентов происходит
реакция деалкилирования.
Мазеролл и Лесбр с сотр. [1] провели каталитическое гидрирование
различных типов этиленовых и ацетиленовых производных алкилгермания
на никелевом катализаторе в эфире при температуре от 1 до 20° С и
атмосферном давлении. Было установлено, что гидрирование ненасыщенной связи
ва-положении относительно атома германия проходит более медленно, чем
в Р-положении. Длина цепи других алифатических радикалов, связанных с
атомом германия, не влияет на скорость гидрирования ненасыщенных
соединений. Так, при гидрировании соединений типа R3GeC = CR' (R' =
= Н, СН2ОН, С3Н7, Ge(C2H5)3) было установлено, что в соединениях, где
R' = Н, гидрирование происходит легко, в соединениях, где R = СН2ОН,
С3Н7 или Ge (C2H5)3, даже в течение многих часов оно не является полным.
Соединение (C2H5)3Ge— С — С—СН2ОН было прогидрировано в триэтил-
гермил-З-пропанол-1 (С2Н5)3 GeCH2CH2CH2OH лишь при 130° С и давлении
120 атм. Частичное гидрирование тройной связи было проведено на
дезактивированном палладии. Таким образом был получен триэтилгермил-6-
гексадиен-1,4, при этом двойная связь 1,2 не затрагивалась
(C2H5)3GeCH2—С=С—СН2СН=СН2 + Н2~ (C2H5)3Ge—СН2—СН=СН—СП2—СП=СП2.
Авторы осуществили частичное гидрирование германийорганических
соединений, имеющих две тройные связи в цепи, оно было направлено
преимущественно на тройную связь, более удаленную от атома германия.
Гидрирование полнозамещенных германийорганических соединений, имеющих
ненасыщенные радикалы, проводилось неоднократно [2—8]. Описано
также каталитическое дегидрирование, например гетероциклов, имеющих
атом германия в кольце, на окисном алюмохромовом катализаторе [9].
Реакции, связанные с изменениями в радикале
115
В ряде работ описывается действие галоидов или галоидоводородов на
ненасыщенные германийорганические соединения. Например, винилтри-
алкилгерманий при —80° С присоединяет бром и бромистоводородную
кислоту [2, 10]
(л-С4Н9)3 GeCH=CH3 + Вг2 -» (л-С4Н9)3 GeCHBr—СП2Вг,
(я-С4Н9)3 GeCH—СН2 + НВг -> (я-С4Н9)3 GeCH2—СН2Вг.
Соединения типа R3Ge(CH2)nCH=CH2 также легко присоединяют
бром при низкой температуре [10а]. Отщепление бромистого водорода от
полученных соединений спиртовой щелочью или амидом натрия в жидком
аммиаке или ксилоле легко приводит к образованию соответствующих
ацетиленовых производных. В аналогичных экспериментальных условиях
удается присоединить бром и к ацетилениду триэтилгермания [2]. К аллил-
триалкилгерманию галоиды и галоидоводородные кислоты не
присоединяются. Даже при низкой температуре (—80° С) бром и бромистоводород-
ная кислота вызывают разрыв связи германий — углерод [10]
(я-С4Н9)3 GeCH3CH=CH2 + НВг -* (л-С4Н9)3 GeBr + СН3=СНСН3.
Действие брома, растворенного в бромистом этиле, при температуре 0° С на
(C2H5)3GeCH2CH=CHCH3 также приводит к разрыву связи Ge—С,
очевидно, ослабленной наличием двойной связи в ^-положении [81.
Петров и сотр. [11] проводили бромирование в хлороформе при
температуре —10 ~ 15° С 1-триэтилгерманийбутен-3-ина-1, 1-триэтил герма-
ний-З-метилбутен-З-ина-1 и 1-триэтилгерманийпентен-З-ина-1 и нашли, что
независимо от строения германийуглеводородов бром присоединяется в
положения 1,4 и 3,4, причем образующиеся аддукты претерпевают при
перегонке ацетилен—алленовую— 1,3-диеновую перегруппировку с образованием
1,3-диеновых дибромидов. Треххлористый металлилгерманий присоединяет
бромистый водород по кратной связи [12]:
Ci3Ge—СН2—С=СН2 + НВг -> Br3GeCH2CBr(CH3)2.
I
СНз
Треххлористый винил- и аллилгерманий не присоединяют бромистый
водород по кратным связям, происходит лишь замена хлора на бром [12].
ClsGeCH._CK=--CH2 + ЗНВг-* BrbCeCII2CH=CH2+3HC1.
Манулкин и сотр. [13] нашли, что брсм и хлористый водород легко
присоединяются к (C6H5)2Ge(C6H4C(CH3)=CH2-p)2
Из других реакций присоединения следует упомянуть о присоединении
меркаптанов. Так, производные винил- и аллилтриалкилгермания легко
реагируют по уравнениям [10]
(С2Н5)3 GeCH=CH2 + HSCH.COOH -» (QH5)3GeCHcCJI25.CII;:CCCH,
(СгН5)3 GeCH£CH=CH2 -{ Н£СН2СССН -> (С2Н.->), GeCn,CH,CII2SCH2CCOn,
(С2Н5)8 GeCH2CH=CH2 + HSCH2CH2CH -» (C2H.03OeCH2CHXH2SCH2CII2OH.
Однако присоединение C6H5SH идет лишь при нагревании и в присутствии
катализатора [10]
(CoII5)3GeCII«CH - CH2+ CeH5SH-* (CsHsJaGeCHoCHoCILiSCelb.
Миронов и сотр. [14] действовали тиоуксусной кислотой на аллилтри-
метилгерманий, при этом они наблюдали саморазогревание реакционной
смеси и реакцию присоединения
(СН3)з GeCH2CH=CH2 + HSCCCH3-> (CII3)3 GeCH2CH2CH2SCCCH3
(выход 94%, т. кип. 90—91° С/7 мм).
8*
116
Германийорганшеские соединения. Лит. стр. 120—121
Действием щелочи и акрилонитрила на образовавшийся продукт была
осуществлена следующая последовательность реакций!
NaOH CH2=CHCN
(CtfsfoGeCHaCHaCHaSOCCHs Г (СНз)з GeCH2CH3CH3SH —>
68% 45%
-> (СНз)з GeCH3CH2CH2SCH3CHaCN.
Мягкое окисление аллилтриэтилгермания (1%-ным раствором перман-
ганата калия в среде водного ацетона при 0° С) [10] идет как обычно с
образованием 1,2-диола
3(QH5)3GeCHaCH=CHa + 2КМпЭ4 + 4НаО -*
-* 3(C2H5)3GeCH2—СН (ОН)— СЧ2ОН + 2ЧпЭ2 + 2КОН.
В присутствии перекиси бензоила удается осуществить радикальное
присоединение к винилтриалкилгерманию четыреххлористого углерода,
хлороформа и этилового эфира бромуксусной кислоты [10]
(л-С4Н9)3 <ЗеСН=СН3 + ССЦ (СбН5СО°^ (п-С*Н9)3 GeCHClCH2CCl3,
(/г-С4Н9)3 GeCH=CHa + GHGi3 (CeH5COO)i (л-С4Н9)3 GeCH2CH2CCl3,
(С2Н5)3 GeCH=CH2 + СН2ВгСООС2Н5 (СбН5СО°^ (C2H5)3 GeGHBr (GH2)2 COOC2H5.
Родан легко присоединяется к двойным связям германииорганических
соединений [15—17]. Метод является более универсальным для оценки
непредельности, чем метод бромных чисел.
К винильным производным алкилгермания легко присоединяются
органические гидриды металлоорганических соединений IV группы. О
присоединении органических гидридов германия см. стр. 68, органических
гидридов олова [18—20] см. стр. 286.
Интересно отметить действие йодистого иодметилцинка на винил-
триметилгерманий [21] и аллилтриметилгерманий [22], приводящее к
образованию германийсодержащих углеводородов ряда циклопропана
(СН3)з GeCH=CHa JCH2ZniS(CH3)3 GeCH—CH2
GH2
(СН3)з GeCHaCH=CH3 JCH2Zni (CH3)3GeCH3CH — CH2
CH2
Получение (триметилгермилметил)циклопропана (СН3)з GeCH2CH—CH2 [22]. В колбу
СН2
снабженную обратным холодильником и мешалкой, помещают 53,6 г йодистого метилена»
0,15 г иода, 16,3 г «цинк-медной пары» и 165 мл эфира. Реакционную смесь нагревают в
течение 30 мин., прибавляют 34 г аллилтриметилгермания и нагревание продолжают в тече*
ние 30 мин. Реакционную смесь отфильтровывают, фильтрат промывают 5%-ным раствором
соляной кислоты (3 раза по 50 мл), 50 мл насыщенного раствора соды, 50 мл насыщенного
раствора хлористого натра, сушат над хлористым кальцием и после отгонки эфира
разгоняют на колонке. Получают (триметилгермилметил)циклопропан с выходом 32,3%; т. кип.
126° С/745 мм; df 1,0290 п2£ 1,4355;
Путями синтеза германииорганических соединений, имеющими галоид
в органическом радикале, являются уже ранее описанные реакции
присоединения органических гидридов германия к непредельным галоидным ал-
килам, например к хлористому аллилу (см. стр. 63, 70), и реакции галоидных
солей алкилгермания с диазометаном (см. стр. 74). Что касается
непосредственной замены водорода в органическом радикале германииорганических
Реакции, связанные с изменениями в радикале
117
соединений на галоид, то оно возможно при фотохимическом хлорировании
или при действии смеси хлористого сульфурила с перекисью бензоила. Далее
галоид может быть введен при действии галоидных солей фосфора на герма-
нийорганические соединения, имеющие в радикале гидроксильные группы.
Было найдено [23—25], что при фотохимическом хлорировании галоге-
нидов метилгермания с удалением продуктов взаимодействия из зоны
реакции с довольно хорошими выходами можно провести замену водорода
метильной группы на хлор. Так, при хлорировании треххлористого метил-
германия были выделены как треххлористый хлорметилгерманий
(Cl3GeCH2Cl — 18%), так и треххлористый дихлорметилгерманий
(Cl3GeCHCl2—30%). Из двухлористого диметилгермания с хорошим выходом
был получен двухлористый метилхлорметилгерманий. Исчерпывающее
хлорирование треххлористого метилгермания привело к получению
треххлористого трихлорметилгермания (Cl3GeCCl3).
Другим реагентом для хлорирования галогенидов алкилгермания
является смесь хлористого сульфурила с перекисью бензоила [15, 17, 26, 27].
Найдено, что при хлорировании треххлористого этилгермания хлористым
сульфурилом образуется примерно в 6—8 раз больше треххлористого р-
хлорэтилгермания, чем а-изомера. Что касается дегидрохлорирования, то
при действии хинолина на треххлористый р-хлорэтилгерманий имее!
место, помимо образования СН2 = CHGeCl3, получение значительных
количеств четыреххлористого германия (17%). При дегидрохлорировании того
же продукта хлористым алюминием получается 41,5% четыреххлористого
германия и всего лишь 14% треххлористого винил германия. Кстати
говоря, фта реакция совершенно иначе проходит у аналогичных кремнийоргани-
ческих соединений. На основании полученных экспериментальных данных
авторы делают предположение об относительной реакционной способности
некоторых металлоорганических соединений IV группы.
Получение треххлористых а-хлорэтил- и Р-хлорэтилгермания [24].В колбу (500 мл),
снабженную обратным холодильником, закрытым ловушкой с серной кислотой, помещают 133 г
треххлористого этилгермания, 112 г хлористого сульфурила и 0,5 г сухой свежеосажденной
метиловым спиртом из хлороформенного раствора перекиси бензоила. Подогревание
содержимого колбы вызывает энергичное выделение газов в течение 2 час. Добавление еще 0,3 г
перекиси бензоила вызывает выделение газов еще в течение часа. После перегонки на
колонке получают 28 г исходного С^\ЬС^С\Ъ с т. кип. от 70 до 100° С; 30 г CH3ClCHGeCl3 с т. кип.
164—168° С и 73 г ClCH2CH2GeCl3 с т. кип. 188° С; df 1,7587; ng l ,5094; выход монохлоридов
60% (84% на вступивший в реакцию C2H5GeCl3).
Мазеролл [28] действовал хлористым сульфурилом на
тетраэтилгерманий и получил триэтил-а-хлорэтилгерманий (C2H5)3GeCHClCH3.
Общим методом синтеза со -галоидированных органических
производных германия R3Ge(CH2)nX является действие при 0° С трехбромистого
фосфора на германийорганические соединения, имеющие в радикале
гидроксильные группы [29]
3R8Ge (CH2)n ОН + PEr3 -* 3RbCe (CH2)n Вг + Н3Р03.
В литературе не описаны методы непосредственного введения галоидов
в ароматические радикалы германийорганических соединений. Для этой
цели обычно действуют на галогениды алкил- или арилгермания магний-
или литийорганическими соединениями, содержащими галоид в
ароматическом радикале [30—32].
Получение триэтил-п-бромфенилгермания [30]. 24 г бромистого триэтилгермания в
тетра гидрофура не прибавляют к растЕору реактиЕа Гриньяра, приготовленному из 5 г
магния и 48 г я-дибромбензола в том же растворителе. Реакционную смесь нагревают 2
часа, охлаждают, разлагают раствором хлористого аммония, органический слой отделяют,
водный — экстрагируют эфиром, эфирный экстракт присоединяют к органическому слою,
сушат, отгоняют растворители и остаток перегоняют в вакууме. Получают 11,2 г (46,6%)
триэтил-л-бромфенилгермания; т. кип. 126—129° С/Б мм; п2£ 1,5560. Выделяют также 14,4%
исходного л-дибромбензола.
118 Германийорганические соединения. Лит. стр. 120—121
Сейфертс и сотр. [33], продолжая исследования карбонильных
соединений переходных металлов, нашли, что при нагревании (150° С) в
атмосфере инертного газа триметилфенилгермания с Сг(СО)6 в диметоксиэтане
образуется
(CH3)3G<
"*^ЧСг(СО)3
Это желтые кристаллы, устойчивые на воздухе, но быстро окисляющиеся
в растворе. Выход 34%; т. пл. 79—80° С; т. кип. 60—65° С/0,01 мм.
Галоид, находящийся в радикале германийорганических соединений,
обладает большой подвижностью и способен ко всем реакциям,
свойственным галоиду в галоидных ал кил ах.
Миронов и сотр. [34, 35] исследовали ряд реакций нуклеофильного
замещения хлорметилтриметилгермания и обнаружили, что эти реакции у
германия протекают быстрее, чем с аналогичными соединениями
кремния.
Были проведены реакции
(CH3)3GeCH2Cl + KSCN -* (СН3)3 GeCH3SCN (81%),
(CH3)3GeCH2Cl + HN(C2H5)2-> (CH3)3GeCH2N(C2H5)2 (73,5%),
(CH3)3 GeCH2Cl + KJ -* (СН3)з GeCH2J,
(СНз)з GeCM2Cl + P (OC2H5)3 -> (CH3)3 GeCH2P (OC2H5)2 (48%),
II
О
(СНз)з GeCHaCl + NaOCH3 -> (СН3)з GeGH2OCH3 (81 %),
(СН3)з GeCH2Cl + NaCH (СООСН3)2 -* (CH3)3 GeCH2CH (COOCH3)2 (51%),
/
(CH3)3 GeCH2Cl + CH2=CHC -* (CH3)3 GeCH2—ОС—CH=CH2 (55%),
\ II
OK О
СНз
(СНз)з GeCH2Gl + CH2=C—С02К -> (СН3)3 GeCH20-С—C=CH2 (46,5%).
I II
СНз О
Этими же авторами [35] был получен с выходом 87% триметилгермил-
метанол по реакции
сн,он
(СН3)з GeCH2Cl + К02ССН3 -> (СН3)3 GeCH2OOCCH3 —-* (CH3)3 GeCH3OH.
H2SO{
Получение метакрилометилтриметилгермания СН2=С (СНц) С02СН2 Ge (СН3)з [35]. Смесь
состоящую из 7 г ClCH2Ge (СН3)3 и 7 г калиевой соли метакриловой кислоты, 1 г порошка
меди, 0,2 г гидрохинона и 3 г этанола, нагревают в запаянной ампуле при 150° С в течение
5 час. Образовавшуюся смесь отфильтровывают, промывают эфиром. После отгонки эфира
и спирта жидкий остаток перегоняют в вакууме. Получают 5 г продукта; т. кип. 53,5—
54° С/8 мм; df 1,1104; n2g 1,4490; выход 46,5%.
При действии жидкого аммиака в автоклаве при 100° С на (С4Н9)3-
•GeCH2CH2CH2Cl была осуществлена замена хлора на аминогруппу [36].
со-Галоидированные органические производные германия типа R3Ge-
(СН2)ПХ [37] количественно реагируют с цианистым калием в спиртовой
среде
R3Ge (CH2)„ Br + KCN -> R3Ge (CH2)„ CM + KBr.
Реакции, связанные с изменениями в радикале
119
Каталитическое гидрирование этих нитрилов в присутствии никеля Ренея
осуществлялось в среде насыщенного спиртового раствора аммиака
R3Ge (CH2)AlCN + 2Н2 -» R3Ge (СН3)„ CH2NH2.
Гидратацией нитрилов были получены соответствующие кислоты
R3Ge (СН2)„ CN + КОН + Н20 -> R3Ge (CH2)„ COOK + N Н3.
Аналогичные реакции были проведены с соединениями типа
R2Ge[(CH2)nBr]2 [37].
Галоид в радикале германийорганических соединений способен
реагировать с металлическим магнием, образуя магнийорганические
соединения, которым свойственны все их обычные реакции.
Так, хлорметилтриметилгерманий при реакции с магнием образовывал
реактив Гриньяра, который был успешно использован в следующих
реакциях [35]:
,0
СО 2
CH3GeCH2MgCl
> (СН3)3 GeCH3C
\
у?° ОН
сн2=снсг w
\
н
СН2=СН—СН2Вг
* (СН3)3 GeCH2GH (ОН) СН=СН2 (43%)
-»(СН3)з GeCH2CH2CH=CH2 (68%)
Получение f-бутенилтриметилгермания СН2=СНСН2СН2 Ge (CHS)3 [35]. К гриньяровско-
му реактиву, полученному в 50 мл эфира "из 2,9 г магния и 20 г ClCH2Ge(CH3)3, при
охлаждении прибавляют 20 г бромистого аллила. Содержимое колбы оставляют стоять
в течение ночи. Затем эфир отгоняют, а твердый остаток нагревают на кипящей водяной
бане в течение 5 час. Отогнанный эфир возвращают в колбу, а реакционную смесь
обрабатывают водой. После сушки эфирного слоя над хлористым кальцием и перегонки на
колонке получают 14 г продукта, т. кип. 124—125° С/755,5 мм\ df 0,9907; n2g 1,4363; выход
68%.
Аналогичная реакция была проведена с сулемой [34].
(CH3)3GeCH2MgCl + HgCl2 - (CH3)3GeCH2HgCl (68%).
Мазеролл и сотр. [29, 36], имея германийорганические соединения типа
R3Ge(CH2)nBr, ввели их в реакцию с магнием и после обычных для магний-
органических соединений реакций получили ряд новых функциональноза-
мещенных германийорганических соединений. Так, (C2H5)3Ge(CH2)4MgBr
исследован в реакции с водой, бромистым триэтилгерманием, иодметиловым
эфиром, бромистым аллилом, пропионовым альдегидом, ацетоном, окисью
этилена [29]. Для (я-С4Н9)3 Ge(CH2)3MgX были проведены реакции
окисления и взаимодействия с окисью этилена [36].
Германийорганические соединения, имеющие в радикале другие
функциональные группы, помимо атома галоида, в реакциях исследованы очень
мало. Проведено восстановление (C4H9)2Ge (C1)CH2CH2CN и (C2H5)2Ge (C1)CH2-
CH2CH2CN литийалюминийгидридом в эфирной среде, а также действие
бромистого этилмагния на (C2H5)2Ge(Cl)CH2CH2CH2CN [36]. Проводилось
также восстановление литийалюминийгидридом эфиров триалкилгермилук-
сусной кислоты, приводящее к получению соответствующих спиртов [38—
40]. В отдельных случаях полученные спирты ацетилировались кетеном.
Несимметричные германийорганические соединения, имеющие в
качестве одного радикала ацетиленовую группу или другую алкиновую группу,
120
Германийорганические соединения
реагируют с магнийорганическими соединениями [2, 10а, 411 по уравнению
R3Ge-C=CH + R'MgX -> RsGeC=CMgX + R'H,
R3Ge—CH3—C=CH + R'MgX -* R3GeCH2C=CMgX.
Полученные производные оказались достаточно11 реакционноспособными.
С ними были осуществлены многочисленные синтезы с хорошими выходами,
например, были проведены реакции карбонизации, изучалось действие
галоидных алкилов, альдегидов и кетонов.
Интересно отметить реакцию, разработанную Несмеяновым с сотр. [42],
открывающую пути синтеза соединений, имеющих два атома металла в
молекуле. Авторы показали, что гидрид три-я-бутилолова легко
присоединяется к трибутилэтинилгерманию по уравнению
(С|Н,)3 GeC = СН + (С4Н9)3 SnH -> (C4H9)3GeCH=:CHSn (QH9).
Исследования продуктов реакции методами ИК-спектров и спектров
ЯМР показывают, что реакция идет в ^ас-положение с образованием транс-
изомеров этиленового ряда, возможно, через я-комплекс или
четырехчленное переходное состояние [42].
R3GeC=CH + R3SnH -* R3GeCfEECH -> R3GeC=CH
HSnR3 j I
H SnR3
RsGe H
-> G=C
/ \
H SnR3.
ЛИТЕРАТУРА
1. Ma zero 1 1 es P., LesbreM., DaoHuy-Giao., Compt/rend., 253, 673(1961)
2. MazerollesP., Bull. Soc. chim. France, 1960, 856.
3. MazerollesR, D u b а с J., Compt. rend., 257, 1103 (1963).
4. LesbreM., S a t g ё J., Compt. rend., 250, 2220 (1960).
5. Ф р е й д л и н Л. X., Ж у к о в а И. Ф., МироновВ.Ф., Изв. АН СССР, ОХН,
1960. 2258.
6. LesbreM., MazerollesP., Compt. rend., 255, 544 (1962).
7. ГвердцителиИ. М., ГунцадзеТ. П., Сообщ. АН ГрузССР, 36, 574 (1964);
РЖХим., 1965, 22Ж252.
8. S a t g ё J., MassolM., Compt. rend., 261, 170 (1965).
9. НефедовО.М., М а н а к о в М. Н., Изв. АН СССР, ОХН, 1963, 769.
10. М a z е г о 1 1 е s P., LesbreM., Compt. rend., 248, 2018 (1959).
10а. MazerollesP., LesbreM., MarreS., Compt. rend., 261, 4131 (1965).
11. С т а д н и ч у к М. Д., П е т р о в А. А., ЖОХ, 35, 700 (1965).
12. Миронов В.Ф., Д ж у р и н с к а я И. Г., П е т р о в А. Д., Изв. АН СССР,
ОХН, 1961, 2095.
13. С а р а н к и н а С. А., Манулкин З.М., ЖОХ, 36, 1299 (1966).
14. Д ж у р и н с к а я Н. Г., МироновВ.Ф., П е т р о в А. Д., ДАН СССР, 138,
1107(1961).
15. Р е t г о w A. D., M i г о п о w W. F., Bugorkowa A.A., Fette, Seifen, Anst-
richmittel, 62, 1107 (1960).
16. Б у г о р к о в а А. А., Миронов В. Ф., П е т р о в А. Д., Изв. АН СССР,
ОХН, 1960, 474.
17. Миронов В. Ф., По н о м а р е н к о В. А., В з ен ко в а Г. Я., Д о л г и й И. Е.,
Петров А. Д., Химия и практическое применение кремнийорганических
соединений, вып. 1. Л., изд. ЦБТИ, 1958, стр. 192.
18. N о 1 t е s J. G., van d е г К е г k G. J. M., Rec. trav. chem., 80, 623 (1961).
19. Н е п г у М. С, N о 1 t e s J. G., J. Am. Chem. Soc, 82, 561 (1960).
20. Н е п г у М. С, N о 1 t e s I. G., J. Am. Chem. Soc, 82, 588 (1960).
Реакции, связанные с изменениями в радикале
121
21 S е у f e r t h D., CohenH., Inorg. Chem., 1, 913 (1962).
22. Долгий И. E., Мещеряков А. П., Гайворонская Г. К., Изв. АН
СССР, ОХН, 1963, 1111.
23. П о н о м а р е н к о В. А., В з е н к о в а Г. Я., Изв. АН СССР, ОХН, 1957, 994.
24. МироновВ.Ф., Е г о р о в Ю. П., Петров А.Д., Изв. АН СССР, ОХН,
1959, 1400.
25. Миронов В. Ф., ПонаморенкоВ. А, Изв. АН СССР, ОХН, 1957, 199.
26. П е т р о в А. Д., МироновВ.Ф., Долгий И. Е., Изв. АН СССР, ОХН,
1956, 1146.
27. МироновВ.Ф., Д ж у р и н с к а я Н. Г., Изв. АН СССР, ОХН, 1963, 75.
28. М a z е г о 1 1 е s P., Dissertation, Toulouse, 1959.
29. М a z е г о 1 1 е s P., Compt. rend., 257, 1481 (1963).
30. К о л е с н и к о в Г. С, Д а в ы д о в а С. Л., ЖОХ, 29, 2042 (1959).
31. С h a 11 J., W i 1 1 i a m s A. A., J. Chem. Soc, 1954, 4403.
32. Benkeser R. A., De В о е г С E., Robinson R. E., SauveD. M.,
J. Am. Chem. Soc, 78, 682 (1956).
33. S e у f e r t h D., Alles ton D. L, Inorg. Chem., 2, 417 (1963).
34. M и р о н о в В. Ф., К р а в ч е н к о А. Л., Изв. АН СССР, серия хим., 1963, 1563.
35. М и р о н о в В. Ф., К р а в ч е н к о А. Л., П е т р о в А. Д., Изв. АН СССР, серия
хим., 1964, 1209.
36. L e s b r e M., S a t g ё J., MassolM., Compt. rend., 257, 2665 (1963).
37. M a z e г о 1 1 e s P., Bull. Soc. chim. France, 1965, 464.
38. БауковЮ.И., Б у р л а ч е н к о Г. С, Л у цен ко И. Ф., ЖОХ, 35, 1173
(1965).
39. Л у ц е н к о И. Ф., Б а у к о в Ю. И., X а с а п о в Б. Н., ЖОХ, 33, 2724 (1963).
40. Б а у к о в Ю. И., ЛуценкоИ.Ф. ЖОХ, 34, 3453 (1964).
41. М a z е г о 1 1 е s P., Compt. rend., 251, 2041 (1960).
42. Н е с м е я н о в А. Н., Б о р и с о в А. Е., ДАН СССР, 174, 96 (1967).
Глава XIИ
СИНТЕЗ И РЕАКЦИИ
ГЕРМАНИЙОРГАНИЧЕСКИХ ГИДРИДОВ
СИНТЕЗ ОРГАНИЧЕСКИХ ГИДРИДОВ ГЕРМАНИЯ
Германийорганические гидриды имеют большое значение в синтезе
германийорганических соединений с различными функциональными
группами. Методы получения органических гидридов германия, имеющие
синтетическое значение, основаны на восстановлении различных
органических галоидопроизводных германия, реже соответствующих алкоксисое-
динений или окисей (отдельные примеры).
В качестве восстановителей наиболее часто применяют литийалюми-
нийгидрид или натрийборгидрид. Может быть применен и гидрид лития.
(Применение амальгамы цинка и концентрированной соляной кислоты не
имеет существенного значения.) Восстановление проводят в эфирной среде,
реже применяют дибутиловый эфир или диоксан. Выходы полученных
гидридов часто близки к количественным.
Германийорганические гидриды разлагаются под влиянием влаги и
кислорода воздуха, поэтому их получают в атмосфере инертного газа.
Первый представитель этого класса соединений — гидрид трифенил-
германия был синтезирован в 1927 г. [1] при действии бромистого аммония
на трифенилгермилнатрий в среде жидкого аммиака
NH3 жидкий
(СбН5)з GeNa + NH4Br > (С6Н5)3 GeH + NaBr + NH3.
Аналогично был получен и гидрид триэтилгермания [2].
Тригидриды метил-, этил-, пропил- и изоамилгермания были
синтезированы действием H3GeNa на соответствующие бромистые алкилы [3, 4]
NaGeH3 + RBr -> RGeH3 + NaBr.
Авторы не указывают выходы полученных продуктов.
Гидрид диэтилизоамилгермания и дигидрид этилизоамилгермания
получались взаимодействием в среде этиламина (C2H5)(C5Hn)GeHLi
с йодистым этилом и (C2H5)GeH2Li с бромистым изоамилом [4].
Вест [5] в 1953 г. описал получение небольших количеств гидрида три-
фенилгермания и дигидрида диметилгермания при действии амальгамы
цинка и концентрированной соляной кислоты в эфирно-спиртовой среде на
бромистый трифенилгерманий и сульфид диметилгермания. При
восстановлении треххлористого метилгермания тригидрид не был получен.
Синтез и реакции германийорганических гидридов
123
Восстановление гидридом лития
Пономаренко и сотр. [6, 7] впервые применили гидрид и дейтерид
лития для получения гидридов и дейтеридов алкилгермания. Так, были
получены гидриды (а также дейтериды) метил- и этилгермания из
соответствующих хлористых или бромистых алкильных производных германия
Полученные соединения приведены в табл. 3.
Таблица 3
Органические тригидриды германия
Полученные
гидриды и дейтериды
CH3GeH3
CH3GeD3
(CH3)2GeH2
(CH3)2GeD2
(CH3)3GeH
(CH3)3GeD
(C2H5)GeH3
(C2H5)GeD3
(C2H5)2GeH2
(C2H5)2GeD2
(CH8)2(CaH5)GeH
(CH3)2(C2H5)GeD
Выход,
%
50,0
52,0
99,0
67,0
72,0
70,0
54,0
56,0
—
—
—
—
Т. кип., °С/мм
рт. ст.
-23,5/745
—23,5/752
6,5/744
6,5/745
26/755,5
26/758,5
11,5/743,5
11,3/748,5
72,5/740,5
71,5/743,5
62/755,5
60/737,4
d
4
—
—
—
1,0128
1,0207
—
—
1,0378
1,0525
1,0158
1,0262
20
nD
—
—
—
1,3890
1,3893
—
—
1,4208
1,4200
1,4090
1,4083
Восстановление натрийборгидридом
Восстановление галогенидов триалкилгермания с помощью
натрийборгидрида в тетрагидрофуране проходит с почти количественным выходом
[8]. Реакция протекает по уравнению
R3GeX + NaBH4 -» R3Ge (BH4) + NaX,
R3Ge (BH4) + ЗН20 -» R3GeH + В (OH)3 + ЗН2.
Дигалогениды диалкилгермания в тех же условиях дают дигидриды с
выходом около 70%.
При получении ди- и тригидридов алкилгермания применение в
качестве восстановителя литийалюминийгидрида дает лучшие результаты.
Получение гидрида трибутилгермания [8]. К 12,3 г (0,033 моля) йодистого трибутил-
германия в 100 мл тетрагидрофурана прибавляют 3 г (0,08 моля) натрийборгидрида в
100 мл тетрагидрофурана. Реакционную смесь кипятят 4 часа, охлаждают до 15° С и по
каплям прибавляют воду. Затем реакционную смесь оставляют на ночь. После отделения
органического слоя водный слой экстрагируют 3 раза 200 мл эфира. После разгонки на
большой колонке получают 7,4 г (91%) гидрида трибутилгермания; т. кип. 121° С/19 мм\
п2£ 1,4510.
Получение дигидрида дибутилгермания [8]. В аналогичных условиях к 22 г (0,05 моля)
двуиодистого дибутилгермания в 100 мл тетрагидрофурана прибавляют по каплям 11,8 г
(0,312 моля) натрийборгидрида в 200 мл того же растворителя. После кипячения в течение
4 час. и аналогичной обработки получают 6,6 г (70%) дигидрида дибутилгермания;
т. кип. 172° С; п2£ 1,4429
При действии натрийборгидрида в водной среде на соответствующие
бромиды были получены следующие гидриды метил германия: CH3GeH3
(выход 99,796), (CH3)2GeH2 (93,7%), (CH3)3GeH (95,3%), (CD3)GeH3 (95,6%)
и (CD3)2GeH2.(99,4%) [9].
124 Германийорганические соединения. Лит. стр. 142—143
Восстановление литийалюминийгидридом
Реакция получения органических гидридов германия восстановлением
с помощью литийалюминийгидрида может быть выражена следующим
уравнением:
4RnGeX4_n + (4-л) LiAlH4-> 4RnGeH4__n 4 (4-/i) LiX + (4-/i) AIX3,
где X — чаще всего хлор и бром, в редких случаях ■— гидроксильная
группа.
В 1947 г. впервые при действии литийалюминийгидрида на хлористый
и бромистый трициклогексилгерманий в среде эфира был получен гидрид
трициклогексилгермания [10]; позднее он был получен и другими авторами
[11]. Андерсон [12] успешно применил эту реакцию для получения гидрида
триэтилгермания.
Сатже [8] впервые получил ряд производных R3GeH (R — метил, этил,
изопропил, я-пропил, я-бутил, изоамил, я-амил, я-гексил, я-гептил и н-
октил) при взаимодействии германийорганических галогенидов с
литийалюминийгидридом в среде эфира или дибутилового эфира. Автор считает,
что если берутся точные стехиометрические соотношения, соответствующие
уравнению реакции, то большая часть галогенида возвращается в конце
реакции без изменения. Если берется избыток гидрида, то выходы
значительно улучшаются и достигают 90%.
Аналогично восстановлением соответствующего бромида [13] с выходом
90% был получен гидрид
СН2—CH2 C4H9
I \ /
Ge
Сгь—СН2 H
Гидрид трибензилгермания получен с выходом 85% [14].
Метод получения гидридов ароматических германийорганических
соединений мало отличается от синтеза аналогичных гидридов в
алифатическом ряду. Были синтезированы гидрид трифенилгермания [15, 16], три-а-
нафтилгермания [17]. Интересно отметить получение оптически активных
германийорганических соединений: гидрида метилфенил-а-нафтилгерма-
ния — (СН3)(С6Н5) (а-С10Н7) GeH [18] и этилфенил-а-нафтилгермания —
(C2H5)(C6H5)(a-C10H7)GeH [19].
Джонсон [15] получил гидрид трициклогексил германия с
количественным выходом при восстановлении трициклогексилгерманола в среде
эфира, а гидрид трифенилгермания — при восстановлении гексафенил-
дигерманоксана; выход не указан. Полученные моногидриды приведены
в табл. 4.
Получение гидрида триэтилгермания [12]. 76 г хлористого триэтилгермания в течение
30 мин. прибавляют к раствору 15 г литийалюминийгидрида в 50 мл сухого эфира;
реакционную смесь нагревают с обратным холодильником 2 часа. После осторожного разложения
водой эфирный слой сушат над сернокислым натрием и, отгоняя эфир, получают 49 г (79%)
(C,H5)sGeH; т. кип. 124° С; df 1,009; n2D° 1,4382.
Получение гидрида трициклогексилгермания [15]. Раствор 3 г трициклогексилгерманола
в сухом эфире прибавляют к большому избытку литийалюминийгидрида в эфире;
реакционную смесь кипятят с обратным холодильником 2 часа. Избыток литийалюминийгидрида
разлагают разбавленной серной кислотой, эфир органического слоя отгоняют в вакууме.
Маслообразный остаток гидрида закристаллизовывают охлаждением в бане со льдом т. пл*
24—25° С; выход количественный.
Получение гидрида трифенилгермания [16]
4 (С6Н5)з GeBr + LiAiH4 ~> 4 (С6Н5)3 GeH -f Li Br + AlBr8.
Синтез и реакции германийорганических гидридов
125
Таблица 4
Органические моногидриды германия
Полученные R3GeH
(CH8)3GeII
(C2H5)oGeH
(rc-C3H7)3GeH
(/-C3H7)3GeH
(n-C4H9)3GeH
(rc-QHnbGeH
(/-CsHiOsGell
(n-C6H13)3GeH
(Az-C7H16)3GeH
(«-C8H17)3GeH
(CH3)2(C2H5)GeH
(Az-C4H9)2(CH2=CHCH2)GeH
(C2H5)2(/-C5Hn)Gen
(CeH5)3GeH
(a-Ci0H7)3GeH
(C6H5CH2)3GeH
Т. кип./С/лл
рт. ст.
26/755,5
124/760
183/742
174/760
123/20
150/11
140/17
169—170/9
182/1,7
179—180/0,4
62/755,5
93/13
—
41-41,5*
249—250*
80—82*
to
nD
1,3890
1,4382
1,4441
1,4505
1,4508
1,4542
1,4517
1,4582
1,4600
1,4610
1,4090
1,4608
—
—
—
—
20 j
dt
1,0128
1,0090
0,9694
0,9770
0,9455
0,9310
0,9238
0,9228
0,9108
0,9061
1,0158
0,9702
—
—
—
—
Литература
[6-9]
[2,8,12]
[8]
[8]
[8,20]
[8,20]
[8,20]
[8]
[8]
[8]
[(3,7]
[8]
[4]
[16]
[47]
[14]
• Температура плавления.
В трехгорлую колбу (1 л), снабженную обратным холодильником, капельной воронкой
и мешалкой, помещают 400 мл эфира и 10 г (0,26 моля) литийалюминийгидрида. Колбу
предварительно наполняют сухим гелием, аргоном или азотом. По каплям прибавляют 32,5 г
{0,085 моля) бромистого трифенилгермания, реакционную смесь нагревают в течение часа.
Эфир отгоняют до объема реакционной смеси 100 мл, прибавляют 100 мл петролейного
эфира (т. кип. 60—70° С) и опять отгоняют до объема реакционной смеси 100 мл. Эту операцию
повторяют 4 раза, пока температура отгонки окончательно не покажет отсутствие эфира.
Оставшуюся суспензию фильтруют через воронку со стеклянным фильтром, твердый осадок
промывают несколько раз петролейным эфиром. Полученные фильтраты соединяют, пет-
ролейный эфир отгоняют, оставшийся гидрид трифенилгермания очищают перегонкой.
Выход 16,4—20 г (61—79%). Дальнейшую очистку производят кристаллизацией из
метанола; т. пл. 41,0—41,5° С.
Получение гидрида трифенилгермания восстановлением гексафенилдигерманоксана
[15]. Раствор 2 г гексафенилдигерманоксана в 25 мл сухого эфира прибавляют по каплям
к эфирному раствору, содержащему избыток литийалюминийгидрида, и кипятят 3 часа с
обратным холодильником в аппаратуре, исключающей влагу. Литийалюминийгидрид
разлагают разбавленной серной кислотой. Органический слой отгоняют, полученный
гидрид трифенилгермания кристаллизуется при охлаждении в бане со льдом (т. пл. 27° С
Органические дигидриды германия получают аналогично
моногидридам, исходя из двугалоидных соединений германия.
Наибольшее число дигидридов диалкилгермания было синтезировано
Сатже с сотр. [8, 21, 22]. Авторы получили R2GeH2 (R —этил, я-пропил,
я-бутил, я-амил, я-гексил, я-гептил, н-октил) восстановлением с помощью
литийалюминийгидрида соответствующих R2GeX2 в среде эфира или дибу-
тилового эфира. Однако в работах не приводится методика эксперимента
и выходы полученных соединений.
Дигидрид изопропилгермания был получен Андерсеном [23] с выходом
54%.
При восстановлении литийалюминийгидридом двухлористого цикло-
пентилгермания и двухлористого циклогексилгермания получены
соответствующие дигидриды. Выходы 18,6 и 20,2% [24].
126 Германийорганические соединения. Лит. стр. 142—143
Из ароматических производных германийорганических дигидридов
известен лишь один представитель — дигидрид дифенилгермания [25, 26].
Физические константы полученных дигидридов приведены в табл. 5.
Таблица 5
Органические дигидриды германия
Полученные RiGeHi
(CH3)2GeH2
(QH5)2GeH2
(/-C3H7)2GeH2
(rt-C3H7)2GeH2
(rt-C4H9)2GeH2
(rt-C3Hn)2GeH2
(rt-C6Hi3)2GeH2
(Vc7Hi5)2GeH2
(AZ-C8Hi7)2GeH2
сн2—сн2ч
1 >GeH2
,CH2—СН2ч
CH2< >GeH2
XCH2-CH/
(C6H5)2GeH2
Т. кип., °С/мм
рт. ст.
•'3,5/744
74/760
110—111/760
126—127/760
86/46
92/12
113-114/3
148/10
164—165/9
91—92
119—120
91/5
20
nD
1,4219
1,432
1,4340
1,4428
1,4478
1,4522
1,4543
1,4568
1,4838
1,4835
1,5921 1
20
dl
1,0390
0,982
1,0030
0,9782
0,9595
0,9484
0,9348
0,9274
1,2261
1,1846
—
Литература
[6]
[6,8]
[23]
[8]
[8]
[8]
[8]
[8]
[8]
[24]
[24]
[25,26]
Получение дигидрида диизопропилгермания [23]. 8,55 г двуиодистого диизобутилгерма-
ния и 2,2 г литийалюминийгидрида в 40 мл сухого эфира кипятят 2 часа с обратным
холодильником. К холодному раствору прибавляют по каплям воду. Эфирный раствор
встряхивают с разбавленной серной кислотой и после перегонки получают 1,8 г сырого, свободного
от галоида дигидрида диизопропилгермания. Выход 54%; т. кип. ПО—111° С; п2р 1, 432.
Получение дигидрида дифенилгермания [26]
(QH5)4 Ge + 2Вг2 -^ (С6Нб)2 GeBr2 + 2C6H5Br,
2 (С6Н5)2 GeBr2 + LiAlH4 -> 2 (С6Н5)2 GeH2 + Li Br + AlBr3.
В трехгорлую колбу (1 л), снабженную механической мешалкой, обратным
холодильником и капельной воронкой, помещают 52,5 г (0,135 моля) тетрафенилгермания в 300 мл
1,2-дибромэтана. При кипячении с обратным холодильником осторожно по каплям
приливают 46 г (0,288 моля) брома. Исчезновение видимых- паров брома имеет место при
продолжающемся кипячении приблизительно через 30 мин. после окончания прибавления брома.
1,2-Дибромэтан, бромбензол и следы брома отгоняют в вакууме (1 мм). Остаток растворяют
в 50 мл сухого эфира и переносят в капельную воронку, соединенную с второй трехгорлой
колбой (1 л). В эту колбу помещают 20 г (0,53 моля) литийалюминийгидрида в 450 мл
сухого эф#ра. Колбу наполняют сухим гелием, аргоном или азотом. Раствор бромистого арил-
германЯя прибавляют по каплям с таким расчетом, чтобы реакционная смесь слабо кипела.
По ок ончании прибавления кипячение продолжают еще 2 часа. Эфир отгоняют до объема
реакционной смеси 100 мл, прибавляют 100 мл петролейного эфира (т. кип. 60—70° С) и
отгоняют до 100 мл объема реакционной смеси. Операцию повторяют 4 раза, пока
температура отгона не покажет полного отсутствия эфира. Реакционную смесь фильтруют через
воронку со стеклянным фильтром в токе инертного газа. Остаток на фильтре промывают пет-
ролейным эфиром. Отгоняют от фильтрата петролейный эфир, остаток перегоняют в вакууме
при 1 мм. Ниже 93° С отгоняют несколько капель жидкости. При 93° С отгоняется
практически весь дигидрид дифенилгермания. При этом получают очень чистый продукт.
Образовавшийся моногидрид остается в колбе. Получают 17,5 г (55%) дигидрида дифенилгермания;
т. кип. 95° С/1 мм. Бесцветная жидкость устойчива при хранении (0° С), при комнатной
температуре менее стабильна.
Тригидриды алкилгермания в настоящее время известны лишь в
алифатическом ряду и синтезируются по методике, аналогичной получению
моно- и дигидридов.
Синтез и реакции германийорганических гидридов
127
Сатже и сотр. [8, 21, 22] получили ряд алифатических тригидридов ал-
килгермания RGeH3 (R — я-бутил, я-амил, я-гексил, я-гептил, я-октил).
Аналогично был получен и тригидрид я-пропилгермания [27]. Тригидрид
винилгермания синтезирован восстановлением треххлористого винилгер-
мания в растворе диметилового эфира диэтиленгликоля; проведение реак*
ции в дибутиловом эфире, по указанию авторов, дает худшие результаты
[28].
В литературе описано получение гидрида триметилгермания (65%
выход), дигидрида диметил германия и тригидрида метил германия (в
последних двух случаях выход количественный) при восстановлении
соответствующих хлоридов в растворе диоксана с помощью LiAlH (OC4H9-O3 [29].
Константы полученных тригидридов алкилгермания представлены в табл. 6,
Таблица 6
Органические тригидриды германия
Полученные
RGeH3
CH3GeH3
CH2=CHGeH3
C2H5GeH3
ra-C3H7GeH3
fl-C4H9GeH3
/i-CsHnGeHa
/2-C6Hi3GeH3
ft-C7Hi5GeH3
/i-C8H17GeH3
Т. кип., °С/мм
рт. ст.
—23,5/745
— 3,5/760
11,5/743,5
30/760
74/760
104—105/760
128—129/760
85/74
80/31
20
nD
—
—
—
1,4200
1,4302
1,4350
1,4390
1,4422
20
d*
—
—
—
1,0220
1,0138
0,9972
0,9819
0,9717
Как уже указывалось выше, основной метод получения органических
гидридов германия сводится к восстановлению соответствующих галоидо-
производных с помощью различных восстановителей. При применении
таких сильных восстановителей, как LiAlH4, NaBH4, LiH, ни одному из
авторов не удалось осуществить ступенчатое восстановление галоидопроизвод-
ных с целью выхода к соединениям типа R2GeHX и RGeH2X (где X —
галоид).
Основные пути синтеза этих соединений заключаются в частичном га-
лоидировании уже готовых гидридов; имеется пример восстановления дига-
логенида диалкилгермания с помощью гидрида триалкилгермания
(см. стр. 128).
Андерсон [30] впервые показал возможность ступенчатого
восстановления германийорганических гидридов и получения соединений типа
R2GeH(X) и RGeH2(X). При постепенном прибавлении рассчитанного
количества сулемы, бромной ртути или иода к тригидридам или дигидридам
бутилгермания были получены соответствующие галоидгидриды бутилгер-
мания.
Получение иоддигидрида бутилгермания [30]. К 6,2 г тригидрида «-бутилгермания
постепенно в течение 2 час. при 0° С прибавляют 9,4 г мелкоизмельченного иода. Реакционную
смесь подвергают фракционированной перегонке. Получают небольшое количество непрореа-
гировавшего тригидрида бутилгермания, 8,4 г (70%) fl-C4H9GeH2J и некоторое количество
высококипящего вещества. После двух перегонок ft-G4HpGeH2J имеет т. кип. 18Г С; d2?
1,776; п2£ 1,5412. Вещество обладает своеобразным острым запахом.
Получение бромдигидрида бутилгермания [30]. Смесь 7,7 г HgBr2 и 6,2 г /г-С^-ЬСЗеНз
нагревают в течение 5 час. при 31° С и затем 10 мин. кипятят с обратным холодильником.
После фракционированной перегонки получают 1,0 г непрореагировавшего ft-QHoGeH^
7,7 2 (70%) /z-C4HgGeH2Br и 0,5 г остатка (выделена металлическая ртуть).
128 Германийорганические соединения. Лит. стр. 142—143
В дальнейшем Андерсон [31] при постепенном прибавлении роданистой
и цианистой ртути к дигидриду дибутилгермания получил с выходом 74%
<n-QH9)2QeHCN и (n-C4H9)2GeHNCS.
В литературе имеется лишь одно указание [32] без приведения
экспериментальных подробностей о получении (хлор)дигидрида метилгермания
(выход 51%) и (хлор)дигидрида метилгермания (выход 49%) при действии
в газовой фазе хлористого водорода на гидрид метилгермания в
присутствии хлористого алюминия. При аналогичном действии хлористого
водорода на дигидрид диметилгермания выход хлоргидрида диметилгермания
равен лишь 29%.
Сатже и сотр. [8, 33] осуществили синтез галоидгидридов диалкилгер-
мания частичным восстановлением дигалогенидов диалкилгермания с
помощью моногидридов триалкилгермания
R2GeX2 + R3GeH -> R2GeHX + R3GeX
(Х=С1|и*Вг)
Реакция происходит в присутствии катализаторов — хлористого или
бромистого алюминия — почти количественно. Авторы указывают, что
им не удалось осуществить реакцию, имеющую место в случае кремния [34]
А1С13
(С2Н5)3 SiH + (С2Н5)2 SiCl2 > (С2Н5)3 SiCl + (СзН5)з SiHCl + (C2H5)2 SiH2.
Получение хлоргидрида диэтилгермания [8]. В колбу емкостью 25 мл в токе азота
помещают 8,3 г (0,041 моля) (C2H5)2GeCl2 и стехиометрическое количество 6,58 г гидрида триэтил-
германия. При перемешивании добавляют 0,2 г хлористого алюминия. В присутствии
катализатора начинается экзотермическая реакция и выделяется газ. Реакционную смесь при
перемешивании нагревают в течение 2 час. при 150° С. Перегонкой профильтрованной
реакционной смеси получают две основные фракции. Первая фракция с т. кип. 135—150° С
(5 г) после повторной перегонки дает бесцветную жидкость с сильным запахом (т. кип. 135—
140° С; n2Q 1,4586), которая соответствует хлоргидриду диэтилгермания с примесью
моногидрида триэтилгермания (выход 51%). Вторая фракция с т. кип. 150—175°С (п2£ 1,4619)
состоит главным образом из (C2H5)3GeCI.
В тех же условиях эквимолекулярная смесь (C2H5)2GeBr2 и (C2H5)3GeH в
присутствии А1Вг3 дает (C2H5)2GeHBr с выходом 30%. Бромгидрид не был
выделен в чистом виде. Восстановление (C2H5)2GeJ2 гидридом
триэтилгермания в присутствии A1J3 не удалось осуществить.
Гораздо более удобный метод синтеза галоидгидридов алкилгермания
типа R2GeHX состоит в частичном замещении водорода на галоид при
действии рассчитанного количества галоидного алкила по схеме [8, 33]
R2GeH2 + R'X -> RgGeHX + R'H.
Реакция была проведена с (С2Н5)2 GeH2 и (/x-C4H9)2GeH2 действием
бромистого бутила в количестве 70% от теорет. Выход R2GeHBr достигал 45%.
Йодистый пропил при реакции с (n-C4H9)2GeH2 и (C2H5)2GeH2
дал соответствующие иодгидриды с выходом 40%.
Отметим также реакцию дигидридов диалкилгермания с хлорметиловым
эфиром [8, 33], приводящую к образованию с выходами до 80% хлоргид-
ридов
R_2GeH2 + С1Н2СОСН3 -* R2GeHCl + СН3ОСН3.
Получение хлоргидрида дибутилгермания [8]. 7,43 г (0,039 моля) дигидрида дибутилгер-
мания смешивают с 2,02 г (0,025 моля, 64% от теорет.) хлорметилового эфира и реакционную
смесь нагревают до 59° С. При этой температуре начинается энергичная реакция. После
15 мин. кипячения на водяной бане кипение прекращается. Фракционированная перегонка
реакционной смеси дает 2,8 г (C4H9)2GeH2 (38%) и 4,4 г жидкости с т. кип. 116—122740 мм,
из которой при повторной перегонке выделяют чистый (С4Н9)2СеС1Н; т. кип. 115° С/30 мм\
dP® 1,110; п2р 1,4620. Выход в расчете на взятый хлорметиловый эфир равен 80%.
Синтез и реакции германийорганинеских гидридов
129
В аналогичных условиях, исходя из (C2H5)2GeH2, получен с выходом 80%
(C2H5)2GeClH; т. кип. 136° С; d2l 1,2409; пЕ 1,4572.
Хлоргидриды фенил- и дифенилгермания были синтезированы с
хорошими выходами при реакции тригидрида фенилгермания и дигидрида фе-
нилгермания с хлорметиловым эфиром в присутствии хлористого алюминия
[33а]. Тем же авторам удалось получить бромгидриды фенил- и
дифенилгермания при действии стехиометрических количеств N-бромсукцинимида
на соответствующие гидриды фенилгермания
CeH3GeH3 + (СН2СО)2 NBr -> C6H5 GeH2Br + (CH2CO)2 NH,
(СвН3)2 GeH2 + (CH2GO), NBr -* (СвИ5)2 GeHBr + (CH2GO)8 NH.
Миронов и сотр. [35] нашли интересный метод синтеза дих лор гидридов
алкилгермания при действии эфирата HGeCl3 на тетраметилолово. Реакция
протекает по уравнению
эфир
(СН8)4 Sn + HGeCb > HGeCl2CH3 (80%) + (СН8)з SnCl (70%).
Аналогичным путем могут быть получены и другие дихлоргидриды
алкилгермания. Однако в этих случаях наряду с RGeClaH образуются замерные
количества треххлористого алкилгермания и, кроме того, выделяется
водород и соответствующий углеводород:
эфир
(CoH^Sn + HGeCb > HGeCl2C2H5 (45o/0)-f C2H5GeCl3 (31o/0) + (C2H5)3SnCl + Ha+CaHe,
(QH5)i Sn + HGeCb -> HGeCl2QH5 (~ 5%) + C6H5GeGl3 (45%).
Замена тетраэтилолова на тетраэтилсвинец также приводит к получению
этих же соединений в несколько иных соотношениях
(С2Н5)4 Pb + HGeCl3-* HGeCb (CaH6) (60%) + C2H5GeCl3 (20%).
В табл. 7 приведены свойства некоторых галоидгидридов алкилгермания.
Таблица 7
Органические галоидгидриды алкилгермания
Полученные
галоидгидриды
CH3GeH2Cl
CH3GeHCl2
(CH3)2GeHCl
(CaH5)2GeHCl
(C2H5)2GeHBr
(C2H,)2GeHJ
/2-C4H9GeH2Cl
/z-C4H9GeH2Br
/z-C4H9GeH2J
{/z-CH9)2GeHCl
(/i-QH9)2GsHBr
(/i-CjH9)2GeHJ
(/i-QH9)2GeHNCS
(«-C4H9)2GeHCN
Т. кип., °С[мм
рт. ст.
70,9
113,2
89,4
136
153
70/23
140
159
181
219
234
249
Разложение
243 с
разложением
20
nD
—
—
1,4572
1,4888
1,5382
1,4598
1,4910
1,5412
1,4618
1,4832
1,5148
1,5097
1,4527
f—
—
—
1,2409
1,5340
1,7717
1,246
1,536
1,776
1,107
1,305
1,470
1,123
1,050
РЕАКЦИИ ОРГАНИЧЕСКИХ ГИДРИДОВ ГЕРМАНИЯ
Реакции с кислородом, серой, селеном, теллуром
Гидриды алкилгермания окисляются труднее, чем соответствующие
гидриды алкилолова, но легче, чем гидриды алкилкремния. Три типа
гидридов R3GeH, R2GeH2 и RGeH3 медленно окисляются воздухом, образуя окиси
9 Заказ № 207
130 Германийорганические соединения. Лит. стр. 142—143
(R3Ge)20, R2GeO и (RGeO)20. Окиси, как правило, получаются в виде
циклических полимеров.
Так, при окислении тригидрида октилгермания быстро преобразуется
в жидкость с большой вязкостью, по анализу отвечающую составу (п-
C8Hi7GeO)20. Концентрированная соляная кислота легко превращает ее
в трихлорид n-C8H17GeCl3 [8]. Перманганат калия в ацетоне частично
превращает гидрид триэтилгермания в окись [(C2H6)3Ge]20, выход 28% [12].
Окисление гидрида трифенилгермания описано Джонсоном [15].
Мазеролл [24] исследовал окисление дигидрида циклотетраметилен-
германия и нашел, что при контакте с воздухом он легко превращается в
твердую полимерную окись
СН2—СН2
сн2—сн2
GeH2 + ЗОо -
СН2—СН2
1_сн2—сн2
/
GeO
4- ЗН20.
J3
Окисление дигидрида циклопентаметиленгермания происходит более
медленно, в несколько этапов. В первый момент имеет место окисление одной
связи Ge—Н, ИК-спектр промежуточного производного содержит при
2024 смгл характерную полосу связи Ge—Н и слабую полосу Ge—ОН, в то
н
время как полоса Ge отсутствует. При дальнейшем окислении вторая
ХН
связь Ge—Н окисляется в свою очередь и получается кристаллическая
циклическая окись, ИК-спектр которой не дает никаких следов
характеристических полос Ge—Н и Ge—ОН.
Следовательно, окисление дигидрида циклопентаметиленгермания можно
представить в виде уравнений
сн2-
/
н2
\
СН2-
сн
/
сн2
\
-СНз
\
GeH
/
-сн2
2—СН2
\
i + V
н
/
Ge
у
\
2о2
н
\
сн2-
/
-*сн2
\
СН2-
-СНа
\
н
/
Ge
/
-сн2
СН2—СН2
/
Ge
/
\
\
сн2
/
\
он
+ н2о
о2
СН2—СН2
О
СН2—СН2
сн2
о
СН2 СН2
I I
сн2 сн2
\/
—Ge
-О
СНг—СН2
-* СИЯ
/
Ge
Ge
\ /
СН2—СНо
о-
Ge^
/\
сн2 сн2
СН2 СН3
\ /
сн2
/
СН2—СН2
СН2—СН2
/
СНЯ
о
Синтез и реакции германийорганических гидридов
131
Была показана возможность синтеза [(C2H6)3Ge]2M, где М = S, Se, Те,
при нагревании элементарной серы, селена и теллура с гидридом триэтил-
германия [36, 37]. Реакция проводилась в эвакуированных запаянных
ампулах.
Реакции с соединениями,
содержащими подвижный водород
Гидриды алкилгермания в присутствии медного порошка реагируют с
водой и органическими соединениями, содержащими подвижный водород,
такими, как спирты, гликоли, фенолы, органические кислоты, с образованием
связи Ge—О [38]. Применение порошка меди, восстановленной водородом,
дает лучшие выходы [39].
Вода количественно реагирует с гидридом триэтилгермания в
присутствии медного порошка при температуре около 100° С [38]
2 (С2Н5)3 GeH + Н20 -> (С2Н5)3 GeOGe (QHeb + 2Н2.
Та же реакция, но более медленная, наблюдалась с дигидридом дибутил-
германия
(QH9)2GeH2 + Н20 -»(С4Н9)2 GeO + 2Н2.
Первичные и вторичные спирты при 95° С реагируют по уравнению
R3GeH + R'CH2OH -* R8GeOCH2R' + П2.
С метиловым и этиловым спиртом реакция на обычном порошке меди
не идет, по мнению авторов, из-за их низкой температуры кипения [38]. На
медном порошке, восстановленном водородом, метанол и этанол реагируют
с гидридом триэтил- и трибутилгермания при комнатной температуре с
почти количественными выходами [39]. Так же количественно протекает
реакция и с другими спиртами (п-С3Н7ОН, п-С4Н9ОН, я-С6Н13ОН и
С6Н5СН2ОН).
Эквимолекулярная смесь дигидрида дибутилгермания и я-гептилового
спирта при 150° С с выходом 40% дает гидрид дибутилгептоксигермания
[38, 41]
(GftHe)a GeH2 + л-С7Н15ОН -» (С4Н9)2 Ge (Н) ОС7Н15-л + Н2.
Изопропиловый спирт реагирует с гидридами триалкилгермания в
присутствии восстановленного медного порошка при 70° С. Та же реакция идет
при 150° С в присутствии обычного порошка меди [39]
(С2Н5)з GeH + (СН3)2 СНОН -* (С2Н5)3 GeOCH (СН3)2 + Н3.
Циклогексанол и метилгексилкарбинол, введенные в реакцию с гидридом
трибутилгермания при 150° С с выходом 80%, образуют соответственно
(n-C4H9)3GeOC6Hn (т. кип. 180° С/16 мм\ d204 0,9963; /ig 1,4680) и (n-QH9)3-
GeOCH(CH3)(CH2)6CH3 (т. кип. 180° С/12 мм, d204 0,9452; /ig 1, 4540),'[41].
Дигидрид дибутилгермания и циклогексанол при 160° С приводят к
образованию с выходом 65% дибутилдициклогексоксигермания (п-С4Н9)20е(ОСбНи)2
(т. кип. 149° С/0,5 мм; df 1,0468; /ig l, 4772) [38].
Реакция третичного бутилового спирта с гидридами протекает очень
медленно при 85° С [39]
Си (восст.)
(CaHsbGeH + (СН3)3С(ОН) ► (С2Н5)3 GeOC(CH3)3 + Н2
9*
132 Германий органические соединения. Лит. стр. 142—143
В присутствии восстановленного порошка меди аллиловыи спирт реагирует
с гидридом триэтилгермания не по реакции присоединения (см. стр. 65),
а происходит обычная реакция дегидроконденсации [39]
Си (восст.)
(СзНзЬ GeH + СН2=СНСН2ОН > (С2Н5)3 GeOCH2CH=:CH2 -f H2.
Аналогично реагирует металлиловый, кротоновый и фурфуриловый
спирты [39].
Ацетиленовые спирты в присутствии восстановленного медного порошка
реагируют с довольно плохими выходами. Реакция октин-З-ола-1 с
гидридом трибутилолова на восстановленном порошке меди проходит лишь на
25%
Си (восст.)
(n-QHe)3 GeH + СН3 (СН2)3 С=С—СН2СН2(Х1 >
-+ (/г-С4Н9)з Ge—О—СН2СП2С=С— (СН2)8С118+ Н2.
Этиленгликоль и бутандиол-1,4 при кипячении с обратным
холодильником с гидридом триэтилгермания дают б&с-(триэтилгермокси)-1,2-этан
(C2H5)3Ge~0—(CH2)20-Ge(C2H5)3 (т. кип. 162° С/11 мм; d*l 1,1490; n2D°
1,464) и бис-(триэтилгермокси)-1,4-бутан (С2Н5)3 GeO(CH2)4OGe(C2H5)3
(т. кип. 151° С/1 мм; df 1,1191, п2£ 1,4650; выход 82 и 90%).
2 (СаНз)з GeH + НО (СН2)лОН -* (С2Н5)з GeO- (CH2)n-OGe (С2Н5)з + 2Н2.
Фенолы реагируют аналогично при температуре выше 80° С
(QHsb GeH + QH5OH -» (С2Н5)3 GeO-QH, + 112
(выход 90%).
Гидрохинон с выходом 88% образует 1,4-био(триэтилгермокси)бензол;
т. кип. 157° С/0,35 мм; п™ 1,5103
Интересно отметить, что спирты, имеющие атом германия в алкильной
группе, реагируют с гидридами в присутствии медного порошка,
восстановленного водородом, аналогично простым спиртам [39]. Так, 3-(триэтил-
гермил)пропанол-1 (продукт присоединения гидрида триэтилгермания к
аллиловому спирту) при реакции с гидридом триэтилгермания образует
триэтилгермил-З-триэтилгермокси-1-пропан [39]
Си (восст.)
(СвН5)3 GeCH2CH2CH2OH + (СзН.ЬGeH * (GjHgbGeCH2CH2CH2OGe «VЪ)3 + Н2.
Аналогично реагирует 3-(триэтилгермил)пропин-3-ол-1. Выход конечного
продукта 20%
Си (восст.)
(С2Н5)0 GeH + (C2H5)3Ge-C-C-CH2OH >
-> (С2Н5)з Gc—C=C—CH2OGe (C2H5)3 + Н2.
Меркаптаны [39, 40] реагируют аналогично спиртам (в качестве
катализатора применяют платину на асбесте). Гидрид триэтилгермания и бу-
тилмеркаптан после нагревания с обратным холодильником в течение 72 час.
с выходом 75% взаимодействуют по уравнению
(С2Н5)3 GeH + C4H9SH -> (С2Н5)3 Ge—S—C4H, -f H2.
Органические кислоты реагируют с гидридами алкилгермания различно.
В отсутствие катализаторов ледяная уксусная и муравьиная кислоты не
Синтез и реакции германийорганшеских гидридов
133
реагируют с гидридом триэтилгермания [8, 12]. Однако Сатже [38]
указывает, что в присутствии порошка меди имеет место количественная реакция
(С4Н9)3 GeH + СН3СООН -> (QH9)3 GeOOCCH3 + Н2.
Аналогично акриловая кислота, которая в отсутствие катализатора
реагирует как ненасыщенная кислота с присоединением по двойной связи (см.
стр. 67), в присутствии порошка меди реагирует как насыщенная кислота
СН2=СН—СООН + (С2Н5)з GeH > CH2=CHCOOGe (QHsb + Н2.
60%
Более сильные органические кислоты, например фторированные
C3F7COOH и C2F5COOH, реагируют с гидридом триэтилгермания
количественно с образованием соответствующих сложных эфиров кислот [12]
(СаНзЬ GeH + C3F7GOOH -> (С2Н5)3 GeOOGC3F7 + Н2.
CFgCOOH, CHF2COOH и CH2FCOOH при аналогичной реакции дают
выходы эфиров соответственно 95, 60 и 20% [12]. Наоборот, трихлоруксус-
ная, трибромуксусная и моноиодуксусная кислоты восстанавливаются
(QH^GeH в уксусную кислоту. Выход 80—94% [12]. Реакция
восстановления происходит и при реакции трихлоруксуснои кислоты с тригидридом
гексилгермания при температуре 120° С [8, 31]
CeHnGeHs + CCI3COOH -> C6H5GeCl3 + CH3COOH.
Андерсон [12] объясняет различие действия фторуксусных кислот, с
одной стороны, и трихлор-, трибром- и иодуксусных кислот, с другой
стороны, различным значением энергии связей: для С—F 107 ккал/мол; С—Н
87 ккал/мол; С—С1 67 ккал/мол; С—Вг 54 ккал/мол; С—J 46 ккал/мол.
Ацетат ртути [12] и ацетат серебра [23] реагируют с образованием
эфиров.
Как уже упоминалось выше (см. стр. 66), с непредельными
альдегидами и кетонами в присутствии катализаторов идет присоединение по
двойной связи углерод — углерод. Полученные насыщенные производные не
содержат связи Ge—О.
Лесбр и Сатже [41] наблюдали реакцию конденсации гидридов алкил-
германия с карбонильной группой насыщенных альдегидов и кетонов
R3GeH + R'CHO -> R3GeOCH2R\
По мнению авторов, присоединение происходит по ионному механизму
R' R'
Ъ+ 5- 8- 8+/ /
R3Ge— Н + О—С -* RaGeO—CH
Выход при таких реакциях конденсации получается количественный,
особенно если он происходит в присутствии порошка меди. Так, например,
нагревание эквимолекулярной смеси гидрида триэтилгермания и гепта-
наля в течение 24 час. на масляной бане при температуре 150° С в
присутствии медного порошка дает с количественным выходом триэтилгептоксигер-
маний (C2H5)3GeO(CH2)6CH3; т. кип. 127° С/10 мм; d24° 0,9852; /t2D° 1,4460.
Бензальдегид конденсируется с гидридом триэтилгермания в
присутствии медного порошка после нагревания 40 час. при 150° С с образованием
триэтилбензоксигермания; выход 80%; т. кип. 150—156° С/18 мм;
d2l 1,1103; /г/? 1,5030. В тех же условиях этил бутил кетон и мети л гексил кетон
134
Германийорганические соединения. Лит. стр. 142—143
присоединяются к гидриду трибутилгермания с выходом в среднем 50% .
Полученные соединения
(С4Н9)3 GeOCH (СН3)з СН3 (я-С4Н9)3 GeOCH (СНо)б СН3
I и |
СН2СН3 СНз
(D (И)
имеют следующие константы: для (I) т. кип. 160° С/9 мм; df 0,9504; п2£
1,4532; для (II) т. кип. 180° С/12 мм; df 0,9452; az2d° l, 4540.
Циклогексанон присоединяется к (C2H5)3GeH количественно после
нагревания в течение 48 час. при 150° С в присутствии порошка меди.
Полученный (C2H5)3GeOC6Hn имеет т. кип. 134° С/18 мм; df 1,0612; п™ 1,4680.
При той же температуре, но без катализатора, гидрид трибутилгермания
с выходом 60% присоединяется к циклогексанону, давая (Ai-QH^gGeOCeHu*,
т.кип. 180° С/16 мм; df 0,9963; п2о 1,4680. Гидрид треххлористого германия с
ацетоном реагирует с образованием окиси мезитила (41а, 416).
Хлорангидриды кислот, реагируя с гидридами алкилгермания,
восстанавливаются до соответствующих альдегидов [8, 33]
R3GeH + R'COCl -» R3GeCl + R'CHO.
При нагревании гидрида трибутилгермания с хлористым бензоилом с
выходом 70% образуется хлористый трибутилгерманий и бензальдегид [8].
Реакции с галоидами, галоидводородными
и другими минеральными кислотами,
солями металлов и металлоидов
Гидриды алкилгермания энергично реагируют с галоидами [8] по
реакции
R3GeH + Х2 -* R3GeX + НХ,
RGeH3 + 3X2 -» RGeX3 + 3HX.
Обычно при действии брома или иода на гидриды алкилгермания в среде
бромистого этила, хлороформа, четыреххлористого углерода получают га-
логениды алкилгермания с количественным выходом [8, 15, 18, 24, 25, 27].
Андерсон [30] показал, что при действии иода (0°С) на избыток гидридов
алкилгермания типа R?GeH2 и RGeH3 идет частичное замещение водорода
и образуются соединения типа R2GeHX и RGeH2X (см. стр. 127).
В отношении действия галоидводородных кислот на гидриды
алкилгермания в литературе имеются несколько противоречивые данные. В
более ранней литературе имеются указания [1, 12], что гидриды
алкилгермания легко реагируют с галоидводородными кислотами с образованием
галоидных солей алкилгермания, однако Вест [5] получал гидрид трифе-
нил- и триметилгермания восстановлением соответствующих хлоридов или
сульфидов цинком в 12УУ соляной кислоте.
Сатже [8] указывает, что гидриды алкилгермания слабо реагируют с
концентрированными галоидводородными кислотами. После 4 час.
нагревания дигидрида дибутилгермания с 60%-ным раствором б ром истоводор
одной кислоты большая часть дигидрида остается без изменения. Безводная
соляная кислота реагирует с тригидридом я-амилгермания и дигидридом
дибутилгермания лишь в присутствии хлористого алюминия [8].
Амбергер [32] действовал газообразным хлористым водородом в
присутствии хлористого алюминия на тригидрид метилгермания и показал,
что в этой реакции образуется 51% (хлор)дигидрида метилгермания, 49%
Синтез и реакции германийорганических гидридов 135
(дихлор)гидрида метилгермания и лишь следы треххлористого метилгерма-
ния. При проведении аналогичной реакции с дигидридом диметилгермания
образуется 29% (хлор)гидрида диметилгермания и 71% двуххлористого
диметилгермания. Автор дает физико-химические константы полученных
соединений без приведения экспериментальных подробностей. Уравнение
реакции следующее:
+НС1 +НС1
GH8GeH8 >CH3GeH2Cl > CH3GeHCb>.
-н2 —н«
Что касается действия других минеральных кислот, то концентрированная
серная кислота медленно реагирует с гидридом триэтилгермания с
выделением водорода [12]
2 (C2H5)3GeH + H2S04 > [(C2H5)3 Ge]2 S04 + 2Н2.
70%
При действии серной кислоты на дигидрид ди-я-бутилгермания
количество выделившегося водорода (определение по методу Церевитинова)
равнялось 80% [8].
Дымящая азотная кислота бурно реагирует с гидридами алкилгерма-
ния. Тригидриды при соприкосновении взрываются. Смесь азотной и серной
кислоты превращает гидриды алкилгермания количественно в Ge02
(метод применяется для анализа германийорганических соединений, см.
стр. 162).
Бензолсульфокислота [8] при кипячении 6 час. в растворе толуола с
гидридом триэтилгермания реагирует с выделением водорода
толуол
(C2H5)3GeH+C6H5S03H > (СзВДз GeS03C6H5 + Ha.
70%
Борная кислота реагирует с гидридом триэтилгермания в присутствии
безводного хлористого никеля [8, 38].
В литературе имеются указания, что гидриды алкилгермания при
действии щелочей выделяют водород, превращаясь в соответствующие
кислородные соединения [8, 11, 42], причем наиболее быстро реакция протекает
с тригидридами.
Андерсон [12] изучил действие гидрида триэтилгермания на некоторые
соли переходных металлов. В этой реакции гидриды действуют в качестве
восстановителей. Гидрид триэтилгермания восстанавливает до металла соли
Pt, Pd, Au, Hg; соли Си11 восстанавливаются в соли Си1, TiIV в TiIU, Vv
в VIV и V111. Хлористый кадмий не восстанавливается.
При действии гидрида триэтилгермания на четырехх лор истый германий
[8, 33] реакция протекает в две стадии. При комнатной температуре
происходит образование двухлористого германия, а при дальнейшем
нагревании до 130° С выделяется металлический германий в виде зеркала.
Аналогично реагирует и хлористый алюминий [8, 33].
Реакция гидрида трибутилгермания с четыреххлористым углеродом
происходит с образованием на 90% хлористого трибутилгермания и
хлороформа.
Хлористый сульфурил энергично реагирует с гидридом
триэтилгермания при комнатной температуре. Галоидирование количественное [8, 33]
2 (С2Н5)3 GeH + SOaCl2 -» 2 (G>H5)3 GeCl + S02 + Ha.
136
Германийорганические соединения. Лит. стр. 142—143
Реакции с галоидными алкилами
В противоположность органическим гидридам кремния, которые не
реагируют с галоидными алкилами даже в присутствии катализаторов
(главным образом типа Фриделя — Крафтса), гидриды алкилгермания
реагируют с замечательной легкостью с галоидными алкилами в отсутствие
всяких катализаторов. Их реакционную способность можно сравнить с
соответствующими гидридами алкилолова, причем связь Ge—Н
превращается в Ge—X. Обмен водород—галоид происходит экзотермически и почти
количественно
R3GeH + R'X -> R3GeX + R'H,
RGeH3 + 3R'X -» RGeX3 + 3R'H.
Реакции гидридов алкилгермания с галоидными алкилами протекают по
механизму нуклеофильного замещения.
Йодистые и бромистые алкилы реагируют количественно с тремя типами
гидридов R3GeH, R2GeH2 и RGeH3 [8, 33].
Йодистый пропил медленно реагирует с гидридом трибутилгермания
с выделением пропана. При нагревании в течение 3 час. йодистый трибутил-
германий образуется с выходом 95%. В аналогичных условиях йодистый
пропил реагирует с гидридом триизобутилгермания. При реакции йодистого
пропила с дигидридом ди-я-амилгермания двуиодистый ди-я-амилгерма-
ний образуется с выходом 95% (нагревание в течение часа при 100° С). При
нагревании 60° С тригидрида я-гексилгермания с йодистым пропилом с
выходом 96% образуется трехиодистый я-гексилгерманий.
С бромистыми алкилами реакция протекает аналогично. Описано
взаимодействие гидрида трибутилгермания с бромистым гептилом (нагревание
4 часа при 150° С, выход (C4H9)3GeBr — 95%) и дигидрида дибутилгермания
с бромистым бутилом (кипячение при 100° С). Тригидрид я-гексилгер-
мания при 85° С реагирует с бромистыми алкилами количественно.
Мазеролл [14, 24] показал, что при действии бромистого и йодистого
бутила на дигидрид циклотетраметилен- и циклопентаметиленгермания с
количественным выходом образуются соответствующие двубромистые и дву-
иодистые производные.
С хлористыми алкилами выходы несколько ниже. Например, после 4 час.
нагревания гидрида трибутилгермания с избытком (30%) хлористого я-
гептила выход (C4H9)3GeCl равен 80%. Хлористый тритил (С6Н5)3СС1, в
котором галоид особенно подвижен, реагирует количественно с (C4H9)2GeH2.
О ступенчатых реакциях дигидридов диалкилгермания с галоидными
алкилами, приводящих к получению соединений типа R2GeHX, см. стр. 128.
Бромистый и хлористый аллил всегда реагируют с гидридами
алкилгермания с выделением пропена
(C4Hf)3GeH+CH2=CH—СН2Вг -> (C4Hf)3 GeBr + СН3—СН=СН2.
Даже в присутствии катализаторов никогда не наблюдалась реакция
присоединения по двойной связи [8, 33], что имеет место для гидрида треххло-
ристого германия [43] (см. стр. 63).
Что касается реакции гидридов алкилгермания с галоидными арилами, то
при нагревании в течение 6 час. дигидрида дибутилгермания с хлорбензолом
исходные реагенты были выделены без изменения. Иодбензол
восстанавливается в бензол с помощью дигидрида дибутилгермания с выходом 40%
при 175°С [8, 33].
Миронов и сотр. [44—47] нашли, что гидрид треххлористого германия
ведет себя несколько отлично от органических гидридов германия в
реакциях с галоидными алкилами. Было найдено, что первичный и вторичные
Синтез и реакции германийорганических гидридов
137
хлористые бутилы, а также хлористый я-гептил, бромистый изопропил,
хлористый я-пропил, ос-хлоруксусный эфир, хлористый триметилкрем-
ний, треххлористый хлорэтилкремний не реагируют с HGeCl3, однако
хлористый трет-бутил с хорошим выходом образует треххлористый трет-
бутилгерманий. Аналогично ведет себя и хлористый трифенилметан. В этих
реакциях необходимо присутствие эфира, в отсутствие эфира выход
продуктов конденсации снижается до 50 и 20% соответственно.
С хлористым бензилом в присутствии эфира с выходом 45% образуется
треххлористый бензилгерманий
?эфир
С6Н5СН2С1 + HGeCl3- > C6H5CH2GeCl3 + НС1.
Реакция с бромистым аллилом проходит при сливании реагентов, с выходом
50% образуется треххлористый аллилгерманий
СНа=СНСН2Вг + HGeCl3 -* CH2=CHCH3GeCl8 + HBr.
С хлористым аллилом без эфира происходит реакция присоединения (см.
стр. 63), в эфирной среде идет реакция конденсации с выходом 26%
эфир
СН2=СНСН2С1 + НСеС13 ► CH2=CHCH2GeCl8 + HC1.
Йодистый аллил и бромистый металл ил реагируют исключительно по
схеме конденсации. Хлористый металлил в присутствии эфира также с
выходом 60% образует
Cl3GeCH2—С=СН2
СН3
Гидрид трехбромистого германия еще в большей степени, чем гидрид трех-
хлористого германия, способен к реакциям конденсации при взаимодействии
с галоидными алкилами и алкенилами [48, 49].
Реакции с металлоорганическими соединениями
Гидриды алкил- и арилгермания реагируют очень активно со многими
металлоорганическими соединениями с образованием германийорганических
соединений со связью германий — металл. В некоторых более редких
случаях образуется связь германий — углерод.
Реакции с литийорганическими соединениями
Гидриды триарилгермания реагируют с литийорганическими
соединениями двояко. По данным Джонсона и Харрис [50], при прибавлении
гидрида трифенилгермания к избытку эфирного раствора фениллития
образуется тетрафенилгерманий с выходом 70%. По данным Гилмана с сотр.
[51, 52], при действии литийорганических соединений на гидрид
трифенилгермания образуется с хорошим выходом трифенилгермиллитий
(C6H5)3GeH + RLi -> (C6H5)3GeLi + RH.
Более подробно см. стр. 144.
Реакции с магнийорганическими соединениями
По данным Гилмана с сотр. [52], некоторые реактивы Гриньяра метал-
лируют гидриды трифенилгермания с образованием R3GeMgX.
Подробности см. стр. 149.
138 Гсрманийорганические соединения. Лит. стр. 142—143
Сатже [8] изучал реакцию гидридов алкилгермания с магнийоргани-
ческими соединениями в эфире, дибутиловом эфире и тетрагидрофуране.
Он отмечает большое различие в реакционной способности гидридов
алкилгермания и гидридов алкилкремния. По мнению автора, присутствие ал-
кильных электроположительных радикалов уменьшает
электроотрицательность группы R3Ge в гидридах триал кил германия. Реакция между
гидридами алкилгермания и магнийорганическими соединениями в эфире и
дибутиловом эфире практически не идет. В тетрагидрофуране, особенно при
продолжительном кипячении, имеет место реакция алкилирования,
характерная для гидридов алкилкремния
R2GeH2+R'MgX -> R2R'GeH + MgXH,
RGeH3 -f R'MgX -> RR'GeH2 + MgXH.
С бромистым амил-и бромистым бутилмагнием выходы очень низки, однако
хлористый аллил- и бензилмагний алкилируют гидриды алкилгермания с
лучшими выходами [8].
Реакции с ртутноорганическими соединениями
При реакции гидридов триалкилгермания с эфирами меркур-бш>уксус-
ной кислоты происходит образование сложных эфиров, содержащих атом
германия в а-положении по отношению к сложноэфирной группе [53]
(С2Н5)3 GeH + Hg (CH2COOCH3)2 -> (СзН5)3 GeCH2COOCH3 + Hg + CH3COOCH3.
Дигидриды диалкилгермания при взаимодействии с
эквимолекулярными количествами эфира меркур-бш>уксусной кислоты образуют эфиры ди-
алкилгерманилуксусных кислот с выходом 60% [54]
R2GeH2 + Hg (CH2COOCH3)2 -+ R2Ge (Н) СН2СООСН3 + Hg + СН3СООСН3.
При взаимодействии гидрида диалкилгермания с ртутным эфиром в
соотношении 1 : 2 в качестве основного продукта образуется эфир диалкил-
германил-бис-уксусной кислоты
R2GeH2 + 2Hg (CH2COOCH3)2 -> R2Ge (CH2COOCH3)2 + 2Hg + 2CH3COOCH3.
Если вместо гидридов берут галоидные соли алкилгермания, то реакция
происходит более энергично.
Хлоргидриды диалкилгермания реагируют с эфирами меркур-бис-ук-
сусной кислоты, при этом происходит замена гидридного атома водорода
на органический остаток [55]
R2Ge (С1) Н -|- Hg (СН2СООСН3)2 -> R2Ge (CI) CH2COOCH3 + Hg f- CH8COOCH8.
При реакции иодгидрида диалкилгермания с меркур-бш>уксусной
кислотой реагирует лишь галоид, а атом водорода в реакцию не вступает [55]
R2GeJH + Hg (CH2COOCH3)2 -> R2Ge (Н) СН2СООСН3 + JHgCH2COOCH3.
Ранее авторы изучали действие гидридов триалкилгермания на меркур-бш>
ацетальдегид [56]. Было найдено, что реакция протекает по уравнению
R3GeH + Hg | СН2С -> R3GeOCH=CH2 + Hg + CH3C
H/2 Н
Синтез и реакции германийорганических гидридов
139
Интересно отметить, что реакция моногидридов триалкилгермания с
ртутноорганическими соединениями типа R2Hg приводит к образованию
новой связи Ge—Hg.
Разуваев и сотр. [57] нашли, что при взаимодействии диэтилртути с
гидридом триэтилгермания в молярном соотношении 1 : 2 в отсутствие
кислорода воздуха при 100—120° С образуется бш;-(триэтилгермил) ртуть и
этан с выходами 66,5 и 96,8% соответственно
2 (С2Н5)3 GeH + (СзНвЬ Hg -> (C2H5)3 GeHgGe (C2H5)3 -f 2С2Нб.
бш>(Триэтилгермил)ртуть имеет т. кип. 118—120° С/1,5 мм; п2£ 1,5695.
Если на гидрид триэтилгермания действовать этил(триэтилсилил)ртутью
и этил(пентаэтилдисилил)ртутью [58], то при реакции выделяется этан;
процесс замещения этильной группы, связанной с ртутью, протекает очень
гладко в сравнительно мягких условиях (1—2 часа при 100° С)
(СаНб)з GeH + C2H5HgR -> C2H6 + (G2H5)3GeHgR
[R — (QHsbSi и (CaH5)3SiSi(C2H6)2].
Этим путем были синтезированы триэтилсилил(триэтилгермил)ртуть и
пентаэтилдисилил(триэтилгермил)ртуть — первые элементоорганические
соединения со связями Si—Hg—Ge и Si—Si—Hg—Ge. Действие на бис-
(триметилгермил)ртуть фенил-(бромдихлорметил)ртути привело лишь к
разрыву связи Ge—Hg [58a].
По данным Неймана с сотр. [59—61], при реакции дигидридов дифенил-
германия с диэтилртутью выделяется этан и образуются полимерные
соединения, имеющие связь Ge—Hg, которые при действии УФ-света или
нагревании легко выделяют металлическую ртуть с образованием октафенил-
циклотетрагермания и других полимеров
4 (СбН5)2 GeH2 -f- 4 (С2Н5)2 Hg -> A/n
С6Н5
I
—Ge—Hg
_ I
L C6H5
l-Hg
+ 8QH6
-Ge-
I
(66%)
Реакции с цинк- и кадмийорганическими соединениями
В результате реакции гидрида триэтилгермания с диэтилцинком (5 час.
при 125° С) образуется этан, цинк и тетраэтилгерманий с выходами
соответственно 94,3, 29,6 и 8,6%, а также выделен неперегоняющийся остаток
лимонно-желтого цвета, с которым авторы проделали некоторые реакции,
но состав его точно не установили [62].
140 Германийорганические соединения. Лит. стр. 142—143
Гидрид триэтилгермания в мягких условиях (3 часа при 80—85° С)
реагирует с диэтилкадмием с образованием бас-(триэтилгермил)кадмия и
этана. Выходы 78,6 и 90,3% [63]
2(QHe)3GeH + (QH6)3Cd -> 2С2Нб + (QH5)3GeCdGe(C2H5)3.
Полученное соединение не удалось выделить в достаточно чистом состоянии.
Полученный бш>(триэтилгермил)кадмий реагирует с бромистым этилом с
образованием бромистого кадмия и тетраэтилгермания. Реакция
заканчивается через 2 часа при 80° С [64].
бш>(Триэтилгермил)кадмий реагирует с уксусной кислотой, пропиловым
спиртом, гидридом триэтилолова, хлористым триэтилоловом с разрывом
связей Qe—Cd—Qe [64а]. При взаимодействии с четыреххлористым
углеродом б£/с-(триэтилгермил)кадмия (при обычной температуре) происходит
взрыв. Реакция поддается регулированию лишь при температурах ниже
—75° С. При этом с количественным выходом образуется хлористый
кадмий. Кроме того, авторам удалось изолировать хлористый триэтил германий
и бас-(триэтилгермил)дихлорметан. Выход составляет 41,0 и 55%
соответственно. Авторы исследуют механизм реакции [65].
Реакции с алюминий-, висмут- и таллийорганическими соединениями
В то время как гидрид триэтилолова вступает в реакцию водородного
обмена с гидридом диэтилалюминия [66], гидрид триэтилгермания даже при
60° С в эту реакцию не вступает.
Разуваев с сотр. [67], продолжая исследования в области биметаллоор-
ганических соединений, нашел, что при взаимодействии гидрида
триэтилгермания с триэтил висмутом без доступа воздуха образуются металлоорга-
нические соединения со связью Ge—Bi
(C2H5)3GeH + (С2Н5)3 Bi -> LC2H6 + [(QH^Ge]^ Bi (С,Н5)3_Л.
При нагревании до 140—145° С (п = 3) выход гарш>(триэтилгермил)висму-
та достигает 61,6%; т. кип. 163—168° С/2,5 мм. Легко окисляющаяся
жидкость лимонно-желтого цвета.
В более мягких условиях (7 час. 130—135° С) реакция идет таким
образом, что п = 2 даже тогда, когда гидрид берется в избытке. Выходы этана и
этил-бш>(триэтилгермил)висмута равны 100 и 86,3%. При нагревании экви-
молярных количеств реагентов (6 час. при 140—150° С) с низким выходом
(8,5%) образуется диэтил(триэтилгермил)висмут; т. кип. 132—136° С/2 мм.
В основном реакция идет с образованием соединения, где п = 3.
При реакции гидрида триэтилгермания с триэтилталлием впервые было
получено соединение со связью Ge—Т1 — т/жс-(триэтилгермил)таллий с
выходом 91% [68]. Соединение было изолировано в виде неперегоняющей-
ся жидкости темно-вишневого цвета.
Реакции с оловоорганическими соединениями
При реакции гидрида трифенилгермания со станниламинами
образуются соединения, имеющие связь Sn—Ge [69]. Реакция протекает по
уравнению
(СеН5)3 GeH + (QH9)3 SnN(C2H5)2 -» (С6Н5)3 Ge-Sn (C4H9)3 + HN(C2H5)2.
Выход83%; т. кип. 179° С/0,001 мм; т. пл. 23° С. Аналогичные реакции с
различными станниламинами исследовались и другими авторами [70, 71 ].
Было показано, что реакция гидридов трифенилгермания со станниламина-
Синтез и реакции германийорганических гидридов
141
ми проходит согласно следующим уравнениям:
(QHsbGeH + RaSnNfCsHsb -> R3Sn-Ge(C6H5)3 + (QH5)a NH
(R-C2H5,C6H5),
(CeH5)3GeH + (C2H5)2 Sn [N (СзНв)а]2 -> (CeH5)3Ge-Sn (0>Н5)3—N (C2H5)2 + (QH5)2 NH,
(QH5)3GeH + (C6H5)3Ge-Sn (QH5)2-N (QjH5)8 -*
-* (C6H5)3 Ge-Sn (C2H5)2—Ge (C6H5)3 + (QHs), NH,
3 (C6H5)3 GeH + (QH5) Sn [N (C2H5)2]3 -> QH5Sn [Ge (С6Н5)з]з + 3HN (C2H5)2
(выход 3%).
Реакции с диазопроизвсдными
Гидриды алкилгермания реагируют с диазопроизводными с образованием
функциональнозамещенных ва-положении по отношению к атому германия
германийорганических соединений [8] согласно уравнениям
R3GeH + N2CHCOOC2H5 -> R8GeCH3COOC2H5 + N2,
RnGeH + N2CHCOCH3 -> R3GeCH2COCH3 + N2,
RcGell -f N2CHCOCGH5 -> R8GeCH2COC0He + N2,
R2GeH2 + N2CHCOOC2H5 -* R2Ge(H)CH2COOG2H5.
Подробности реакции см. стр. 74.
Реакции с непредельными соединениями
Неорганические гидриды HGeCl3, гидриды алкил- и арилгермания, а
также галоидгидриды алкилгермания легко присоединяются к
непредельным органическим соединениям с образованием новых функционально
замещенных германийорганических соединений.
HGeCb + С=С -> Cl3GeC-CH ,
/ \ II
\ / II
R3GeH+C=C -> RsGeC—CH,
/ \ II
\ / II II
R3GeH2 + С=С -* R2Ge(H)C—CH -> R2Ge (С—СН)2,
/ \ II II
\ / II
R,GeHX +C=C -» R2Ge(X)C—CH.
/ \ II
Для HGeClg в большинстве случаев реакция протекает при комнатной
температуре в отсутствие катализаторов при простом смешении реагентов.
Галоидгидриды алкилгермания также легко присоединяются к
непредельным соединениям, но уже при повышенной температуре. Для присоединения
гидридов алкил- или арилгермания требуется присутствие катализаторов,
чаще это платинохлористоводородная кислота, реже применяется перекись
бензоила или УФ-облучение.
Подробности и примеры реакций см. в главе «Синтез
германийорганических соединений присоединением гидридов к кратным связям».
142
Германийорганические соединения
ЛИТЕРАТУРА
1. Kraus С. Ам Foster L. S., J. Am. Chem. Soc, 49, 457 (1927).
2. Kraus С. A., Flood E. A., J. Am. Chem. Soc, 54, 1635 (1932).
3. Teal G. K., Kraus С A., J. Am. Chem. Soc, 72, 4706 (1950).
4. Glarum S. N., Kraus C. A., J. Am. Chem. Soc, 72, 5398 (1950).
5. West R., J. Am. Chem. Soc 75, 6080(1953)
6. Понома ренко В. А., Взенкова Г. Я-, Егоров Ю. П., ДАН СССР, 122,
405 (1958).
7. Миронов В. Ф., Пономаренко В. А., Взенкова Г. Я-, Долгий
И. Е., Петров А. Д., Химия и практическое применение кремнийорганических
соединений, вып. 1. Л., изд. ЦБТИ, 1958, стр. 192.
8. S a t g ё J., Ann., de chim., 6, 519 (1961).
9. G r i f f i t h s J. E., Inorg. Chem., 2, 375 (1963).
10. F i n h о 1 t A. E., BondA.C, W i 1 z b а с h K. E., S с h 1 e s i n g e г Н. Т.,
J. Am. Chem. Soc, 69, 2692 (1947).
11. F u с h s R., G i 1 m a n H., J. Org. Chem., 23, 911 (1958)..
12. A n d e r s о n H. H., J. Am. Chem. Soc, 79, 326 (1957).
13. M a z e г о 1 1 e s P., D u b а с J., Compt. rend., 257, 1103 (1963).
14. CrossR.J., GlocklingF., J. Chem. Soc, 1964, 4125.
15. JohnsonO.H., N e b e r g a 1 1 W. H., J. Am. Chem. Soc, 71, 1720 (1949).
16. J о h n s о n O. H., Nebergall W. H., Harris D. M., Inorg. Syntheses, 5>
76 (1957).
17. West R., J. Am. Chem. Soc, 74, 4363 (1952).
18. В г о о k A. G., P e d d 1 e G. J., J. Am. Chem. Soc, 85, 1869 (1963).
19. В о t t R. W., E a b о r n C, V a r m a I. D., Chem. and Ind., 1963, 614.
20. LesbreM., SatgeJ., Compt. rend., 247 471 (1958).
21. Lesbre M., Satge J., Bull. Soc. chim. France, 1959, 789.
22. S a t g ё J., Mathis-NoelR., LesbreM., Compt. rend., 249, 131 (1959).
23. A n d e г s о n H. H., J. Am. Chem. Soc, 78, 1962 (1956).
24. M a z e г о 1 1 e s P., Bull. Soc. chim. France, 1962, 1907.
25. JohnsonO.H., Harris D. M., J. Am. Chem. Soc, 72, 5564 (1950).
26. JohnsonO.H., HarrisD.M., Inorg. Syntheses, 5, 74, (1957).
27. J о h n s о n O. H., J о n e s L. V., J. Org. Chem., 17, 1172 (1952).
28. BrinckmanF.E., S t о n e F. G. A., J. Inorg. Nucl. Chem., 11, 24 (1959).
29. Van de VondelD.F., J. Organometal. Chem., 3, 400 (1965).
30. AndersonH.H., J. Am. Chem. Soc, 82, 3016 (1960).
31. A n d e r s о n H. H., J. Am. Chem. Soc, 83, 547 (1961).
32. AmbergerE., Boeters H., Angew. Chem., 73, 114 (1961).
33. LesbreM., SatgeJ., Compt. rend., 252, 1974 (1961).
33a. SatgeJ., R i v i e r e P., Bull. Soc. Chim. France, 1966, 1775.
34. Whitmore F.C., P i e t r u s z a E. W., SommerL.H., J. Am. Chem.
Soc, 69, 2108 (1947).
35. M и р о н о в В. Ф., К р а в ч е н к о А. Л., ДАН СССР, 158, 656 (1964).
36. В я з а н к и н Н. С, Б о ч к а р е в М. Н., С а н и н а Л. П., ЖОХ, 36, 1154
(1966).
37. В я з а н к и н Н. С, Бочкарев М.Н., Санина Л.П., ЖОХ, 36, 166
(1966).
38. Lesbre M., Sa tge J., Compt. rend., 254, 4051 (1962).
39. Satge J., Bull. Soc. chim. France, 1964, 630.
40. SatgeJ., LesbreM., Bull. Soc. chim. France, 1965, 2578.
41. LesbreM., Satge J., Compt. rend., 254, 1453 (1962).
41a. ГарТД., МисоновВ.Ф, ЖОХ, 36, 1709 (1966).
416. Колесникове. П., Нефедов О. М., ЖОХ, 37, 746 (1967).
42. S с h о t t G., H a r z d о г f С, Z. anorg. allg. Chem., 307, 105 (1960).
43. П е т р о в А. Д., М и р о н о в В. Ф., Д ж у р и н с к а я Н. Г., ДАН СССР,
128, 302 (1959).
44. Миронов В. Ф., ДжуринскаяН. Г., Петров А. Д., Изв. АН СССР,
ОХН, 1960, 2066.
45. Миронов В. Ф., Д ж у р и н с к а я Н. Г., Г а р Т. К-, П е т р о в А. Д., Изв.
АН СССР, ОХН, 1962, 460.
46. М и р о н о в В. Ф., Г а р Т. К., Изв. АН СССР, серия хим., 1965, 291.
47. Миронов В. Ф., Д ж у р и н с к а я Н. Г., Г а р Т. К., Сб. «Синтез и свойства,
мономеров». М., «Наука», 1964, стр. 150.
48. Г а р Т. К., М и р о н о в В. Ф., Изв. АН СССР, серия хим., 1965, 855.
49. М и р о н о в В. Ф., Г а р Т. К., Изв. АН СССР, серия хим., 1965, 755.
50. J о h n s о п О. Н., Н а г г i s D. M., J. Am. Chem. Soc, 72, 5566 (1950).
51. G i 1 m a n H., GerowC.W., J. Am. Chem. Soc, 78, 5435 (1956).
Синтез и реакции германийорганических гидридов
143
52. G i 1 m a n Н., ZeuechE.A.. J. Org. Chem., 26, 3035 (1961).
53. Л у ц е н к о И. Ф., Б а у к о в Ю. И., X а с а п о в Б. И., ЖОХ, 33, 2724 (1963).
54. Б а у к о в Ю. И., Л у ц е н к о И. Ф., ЖОХ, 34, 3453 (1964).
55. Б а у к о в Ю. И., БелавинИ. Ю., Л у ц е н к о И. Ф., ЖОХ, 35, 1092
(1965).
56. Б а у к о в Ю. И., Л у ц е н к о И. Ф., ЖОХ, 32, 3838 (1962).
57. Вязанкин Н. С, РазуваевГ. А., Гладышев Е. Н., ДАН СССР,
151, 1326 (1963)
58. Вязанкин Н. С, РазуваевГ. А., Гладышев Е. Н., Гурикова
Т. Г., ДАН СССР, 155, 1108 (1964).
58а. S е у f е г t h D., С г о s s R. J., Р г о k a i В., J. Organometal. Chem., 7, P20 (1967).
59. N e u m a n n W. P., Angew. Chem., 75, 679 (1963).
60. NeuraannW.P, К u h 1 e i n K., Tetrahedron Letters., 1963, 1541.
61. Neumann W. P., Kuh lei п К., Ann., 683, 1(1965).
62. В я з а н к и н Н. С, Р а з у в а е в Г. А., Корнева С. П., Круглая О. А.,
Г а л и у л и н а Р. Ф., ДАН СССР, 158, 884 (1964).
63. Вязанкин Н. С, Разуваев Г. А., Бычков В. Т., ДАН СССР, 158,382 (1964).
64. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б ы ч к о в В. Т., ЖОХ, 35, 395 (1965).
64а. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б ы ч к о в В. Т., Изв. АН СССР,
серия хим., 1965, 1965.
65. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б ы ч к о в В. Т., 3 в е з д и н В. Л.,
Изв. АН СССР, серия хим., 1966, 562.
66. N е u m a n n W. P., SommerR, Angew. Chem., 75, 788 (1963).
67. К р у г л а я О. А., В я з а н к и н Н. С, Р а з у в а е в Г. А., ЖОХ, 35, 394
(1965).
68. В я з а н к и н Н. С, Митрофанова Е. В., Круглая О. А.,
Разуваев Г. А., ЖОХ, 36, 160 (1966).
69. S ommerR., Neumann W. P., Schneider В., Tetrahedron Letters,
1964, 3875.
70. С r e e m e r s H. M. J. C, NoltesJ.G., J. Organometal. chem., 7, 237 (1967).
71. С r e e m e r s H. M. J. C, Hydrostannolysis. A general method for establishing
tin-metal bond. Utrecht (Nederland), Schotanus and Jens Utrecht. New York, 1967.
Глава XIV
СИНТЕЗ И РЕАКЦИИ
ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
СОДЕРЖАЩИХ ДВА РАЗЛИЧНЫХ МЕТАЛЛА
Химия германийорганических соединений, содержащих в молекуле два
различных металла, получила в последние годы значительное развитие.
Германий в подобных соединениях может иметь непосредственную связь
Ge—М, или металлы могут быть разделены углеродным мостиком. В ряду
соединений, имеющих связь Ge—М, наиболее исследованы соединения с
участием щелочных металлов, а именно типы R3GeM и R2GeM2 (М = Li, Na, К).
В последнее время круг металлов, связанных с германием, расширен на
металлы второй (Zn, Cd), третьей (Т1), четвертой (Si, Sn, Pb) и в малой
степени пятой (Bi) групп периодической системы. Наконец, описаны
своеобразные биметаллические соединения типа Ar3GeM (СО)5 (где М = Re, Mn).
Синтез германийорганических соединений, в которых германий связан
с другим металлом через углеродный мостик, осуществлен с помощью маг-
нийорганических соединений (см. стр. 38)
R3MC6H4MgX + R3GeX -> I^MQAGeRs + MgX2,
RsMCsCMgX + RsGeX -> R3MC=CGaR8 + MgX3,
или через посредство натрийорганических соединений (см. стр. 52)
Na
R8MCH=CHC1 + R3GeX > RgMCH-CHGeRg + NaCl + NaX.
Для получения подобного рода биметаллических соединений могут быть
применены гидриды, например кремния и олова, согласно уравнению
(см. стр. 120)
R3GeC=CH + R3MH -* R3GeCH-CHMR3
(M = Si, Sn).
Эти же соединения могут быть получены и при иной комбинации реагентов
(см. стр.63, 69)
R3MC=CH > R3GeH -* R3MOT=CHGsR8
(М - Si, Sn).
СИНТЕЗ И РЕАКЦИИ СОЕДИНЕНИЙ
ТИПА RrtGeM4—я, ГДЕ M = Li, Na, К
Соединения R3GeM, где М = Li, Na, К, играют заметную роль в химии
германийорганических соединений и дают возможность получать ряд
новых типов германийорганических соединений.
Основные методы синтеза соединений типа R3GeM [1,2] следующие.
а) Действие щелочных металлов или сплавов щелочных металлов на
галоидсодержащие германийорганические соединения R3GeX (X— галоид).
Германииорганические соединения, содержащие два различных металла 145
б) Действие щелочных металлов на дигерманы или аналогичные
соединения, имеющие связь германия с другими металлами, например Ge—Si,
Ge — Sn и т. д.
в) Действие щелочных металлов на металлоорганические соединения
четырехвалентного германия, сопровождающееся отщеплением радикала.
г) Действие литийорганических соединений на гидриды алкилгермания.
Соединения типа R3GeM не были выделены в индивидуальном состоянии,
а получались лишь в растворах. Обычно это окрашенные растворы,
дающие положительную пробу Гилмана, что указывает на присутствие связи
Ge—М (см. ниже реакции R3GeM с кетонами).
Метод получения соединений типа R3GeM действием щелочных металлов
или их сплавов на галоидсодержащие германииорганические соединения
является наиболее общим.
В работах раннего периода при действии, например, на бромистый три-
фенилгерманий металлического натрия в кипящем ксилоле в качестве
единственного продукта реакции получали гексафенилдигерман [3—5] и
соответственно гекса-я-толилдигерман, гексабензилдигерман [6]. В
алифатическом ряду были получены аналогичные результаты. Так, при
действии металлического натрия в среде жидкого аммиака на бромистый три-
этилгерманий получался гексаэтилдигерман [7], а действие металлического
калия на бромистый триметилгерманий (без растворителя) приводило
к образованию гексаметилдигермана с выходом 74% [8]. Наконец,
действием металлического лития на хлористый или бромистый триэтилгерма-
ний в этиламине был синтезирован гексаэтилдигерман [7].
В последнее время на основе работ Гилмана с сотр. [9], применивших в
качестве растворителя тетрагидрофуран, с хорошим выходом удалось
получить соединения типа R3GeM. Так, при реакции бромистого трифенилгер-
мания с металлическим литием в тетрагидрофуране был получен трифенил-
гермиллитий с выходом 52,5%. При дальнейшей реакции с хлористым три-
этилкремнием было доказано образование трифенил(триэтилсилил)герма-
ния.
Тамборский и сотр. [10], исследуя реакцию между металлическим литием
и галоидными металлоорганическими соединениями IV группы
периодической системы, пришли к выводу, что реакция протекает через стадию
промежуточного образования соответствующих диметаллоорганических
соединений по уравнениям
(CGH5)3MCl-r2Li - (CeH3)3MLi-|-LiCl,
(CGH5)3 МЫ + (CeH5)3 MCI -> (СвНз)з M-MS(CeHB)3 + LiQ,
(СвН3)з М-М (СвНб)з + 2Li -* 2 (C6H5)3MLi.
(М — кремний, германий, олово, свинец).
Это основывается на следующих фактах.
1) При прибавлении прозрачного раствора хлорида в тетрагидрофуране
к литию спустя некоторое время появляется белый осадок, проба Гилмана,
характеризующая наличие связи М—М в этот момент отрицательна, однако
в дальнейшем осадок исчезает и образуется окрашенный раствор с
положительной пробой Гилмана.
2) В качестве побочного продукта обычно образуются небольшие
количества (С6Н5)3М —М(СвНБ)3.
3) Как известно (С6Н5)3М-М(С6Н5)3 легко расщепляются литием в
тетрагидрофуране, давая с прекрасным выходом соединения типа (СвН&),-
MLi.
Ю лаказ Xt 207
146 Германийорганические соединения. Лит. стр. 157—158
Получение трифенилгермиллития [9]. К перемешиваемой смеси 3,84 г (0,01 моля)
бромистого трифенилгермания и 0,69 г (0,1 г-атома) лития в течение часа прибавляют 50 мл
тетрагидрофурана. Через 15 мин. начинает выпадать белый осадок, а через 10 мин. раствор
принимает светло-желтый цвет. Перемешивание продолжают в течение 3 час. при
комнатной температуре, полученный красно-коричневый раствор фильтруют через стеклянную
вату. К фильтрату, имеющему положительную пробу Гилмана, для идентификации
трифенилгермиллития добавляют 1,5 г (0,01 моля) хлористого триэтилкремния. Полученную
реакционную смесь гидролизуют и экстрагируют эфиром. После удаления растворителя
получают 2,2 г (52,5%) трифенил(триэтилсилил)германия с т. пл. 95—96° С после
перекристаллизации из этанола.
Получение трифенилгермиллития [10]. Раствор 5,43 г (0,016 моля) хлористого
трифенилгермания в 17 мл тетрагидрофурана прибавляют в течение 4 мин. к перемешиваемой взвеси
0,62 г (0,08 г-атома) металлического лития в 16 мл тетрагидрофурана. Реакция слегка эк-
зотермична. Появляется белый осадок. При дальнейшем перемешивании в течение 3 час.
белый осадок исчезает и образуется темно-желтый раствор, дающий положительную пробу
Гилмана. После дополнительного перемешивания в течение 2,5 час. раствор фильтруют в
атмосфере азота через стеклянную вату. Выход полученного трифенилгермиллития
определяют прибавлением хлористого бензила и выделением бензилтрифенилгермания (60%).
В качестве побочного продукта реакции выделены следы гексафенилдигермана.
Об образовании соединений типа R3GeM при действии щелочных металлов
на производные дигермана указывается в ряде работ.
Так, при встряхивании гексафенилдигермана с металлическим натрием
в среде жидкого аммиака легко образуется трифенилгермилнатрий [4]
NH3 жидкий
(C6H5)3Ge-Ge(C6H5)3+2Na — > 2 (C6H5)3GeNa.
Металлический литий или калий в диэтиламине легко расщепляют гекса-
этилдигерман с образованием триэтилгермиллития
(С2Н5)3 Ge-Ge (С2Н5)3 + 2Li -> 2 (С2Н5)3 GeLi
или триэтилгермилкалия [7]
(С2Н5)з Ge~ Ge (С2Н5)з + 2К -> 2 (С2Н5)з GeK.
Гилмаы и сотр. [11, 12] подробно изучили расщепление
гексафенилдигермана щелочными металлами, показали большое влияние растворителей
на течение реакции и различие в реакционной способности
гексафенилдигермана и гексафенилдисилана. Например, гексафенилдисилан легко
расщепляется сплавом калий — натрий в эфире с образованием (C6H5)3SiK.
Реакция начинается через несколько минут и быстро полностью
заканчивается. Однако гексафенилдигерман не реагирует со сплавом калий —
натрий в эфире или бензоле (после реакции был выделен лишь непрореаги-
ровавший гексафенилдигерман). В случае проведения реакции в кипящем
дибутиловом эфире, ксилоле или диметиловом эфире этиленгликоля три-
фенилгермилкалий также не обнаружен (впрочем, и исходный
гексафенилдигерман не был выделен). При проведении реакции в тетрагидрофуране
авторы нашли, что сплав калий — натрий реагирует с растворителем.
Наилучшими условиями проведения реакции являются описываемые
в следующем примере.
Получение трифенилгермилкалия [11]. К 5 г (0,00823'моля) гексафенилдигермания в
10 мл сухого эфпра прибавляют 2 мл сплава натрий — калий 1:5. Смесь перемешивают 5 мин.
и прибавляют 20 капель тетрагидрофурана. Через2мин. последобавления тетрагидрофурана
раствор приобретает светло-коричневую окраску. После перемешивания в течение 30 мин.
добавляют 30 мл сухого эфира, реакционную смесь перемешивают в течение 27 час. Избыток
сплава амальгамируют. Суспензию трифенилгермилкалия в токе азота переводят в другую
реакционную колбу и добавляют к ней по каплям 2,5 г (0,0165 моля) хлористого
триэтилкремния; после обычной обработки получают 4,2 г (64,1%) триэтилсилилтрифенилгермания
с т. пл. 92,5—94,5° С. В случае замены тетрагидрофурана небольшими количествами бром-
бензола или тетрафенилгермания выходы триэтилсилилтрифенилгермания равны
соответственно 54,2 и 55,3%.
Германийорганические соединения, содержащие два различны^ металла 147
В отличие от сплава калий — натрий металлический литий расщепляет
гексафенилдигерман в диметиловом эфире этиленгликоля. Однако выход,
определенный по триэтилсилилтрифенилгерманию, не превышает 39,8%
[И].
При расщеплении германийорганических соединений, имеющих связь
германия с другими элементами IV группы периодической системы, также
образуются соответствующие соединения типа R3GeM. Так, при действии
сплава калий — натрий на трифенилметилтрифенилгерманий трифенил-
гермилкалий образуется с выходом 26% [12]. При реакции сплава калий —
натрий в эфире с трифенилсилилтрифенилгерманием (связь Ge—Si) трифенил-
гермилкарбоновая кислота после карбонизации получена с выходом 74%
[13].
Разуваев и сотр. [14, 14а] получили триэтилгермиллитий с выходом
более 90% реакцией бис-(триэтилгермил)ртути или т/шс-(триэтилгермил)-
таллия с литием в тетрагидрофуране в отсутствие кислорода воздуха
ТГФ
[(С2Н5)3 Ge]„M + п U > М + п (C2H5)3GeLi
M=Hg, n = 2) М=Т1, н=3).
Что касается метода получения этих соединений путем отщепления
радикалов, то трифенилгермилнатрий был получен при действии
металлического натрия на тетрафенилгермакий в жидком аммиаке
NH3 жидкий
(С6Н5)4 Ge + 2Na > (С6Н5)3 GeNa + NaNH2 + С6Н6.
Однако реакция протекает очень медленно и с плохим выходом вследствие,
как предполагают авторы, плохой растворимости тетрафенилгермания
в жидком аммиаке [4].
Позднее Гилман и сотр. [15] показали возможность получения трифенил-
гермиллития действием металлического лития на тетрафенилгерманий в
среде диметилового эфира этиленгликоля с выходом 70%. Метод не имеет общего
характера, так как попытки расщепить тетраэтилгерманий, тетрабутилгер-
маний, тетраоктилгерманий, тетрабензилгерманий и тетракис-(2-феяял-
этил)германий с помощью металлического лития в том же растворителе
оказались неудачными [16]. В случае действия на тетраэтилгерманий сплава
натрий — калий в различных растворителях авторами не были
выделены какие-либо определенные продукты. Возможно, имело место
образование триэтилгермилкалия и его последующая реакция с
растворителем [16].
При реакции несимметричных полнозамещенных германийорганических
соединений с металлическим литием в диметиловом эфире этиленгликоля
расщепление имеет место, в первую очередь отшепляется фенильная группа
[16].
Реакция гидридов алкилгермания со щелочными металлами (Na, Li) или
с литийорганическими соединениями в условиях, использовавшихся в
ранних работах [17, 18], протекала более сложно.
Так, Краус и сотр. [171 нашли, что тригидриды этил-,летил- и изо-
амилгермания реагируют со металлическим натрием в жидком аммиаке
или с литием в этиламине, давая RGeH2Na или соответственно RGeH2Li
(имеет место и побочная реакция, приводящая к выделению более чем
одного эквивалента водорода). Дигидрид этилизоамилгермания реагирует
с металлическим литием в этиламине количественное образованием С2Н5-
(/-C6Hu)GeHLi.
В отношении действия литийорганических соединений на гидриды три-
арилгермания Джонсон и Харрис [18] показали, что если прибавлять гид-
1Э*
148 Германийорганичсские соединения. Лит. стр. 157—158
рид трифенилгермания к избытку эфирного раствора фениллития
(кипячение с обратным холодильником в течение 12 час),то образуется с выходом
70% тетрафенилгерманий; при обратном порядке прибавления избытка
фениллития к кипящему раствору гидрида образуется с 50—60%-ным
выходом гексафенилдиперман (наряду с небольшим количеством тетрафенил-
германия). Авторы считают, что образование гексафенилдигермана идет
через промежуточное образование трифенилгермиллития по уравнению
(QH5)3GeLi + (CeH5)3G3H -> ^H^Gb-Gb (С3Ч3)з + LiH,
однако не приводят к этому никаких доказательств.
В дальнейшем Гилман с сотр. [19], а также и другие авторы [20]
показали, что при действии литийорганических соединений на гидрид
трифенилгермания образуется с хорошими выходами трифенилгермиллитий
наряду с небольшой примесью тетрафенилгермания. В данном случае
действие литийорганических соединений на гидриды триарилгермания
отличается от действия на соответствующие гидриды триарилкремния, так как
с последними образуются лишь полнозамещенные кремнийорганические
соединения.
Гилман и сотр. [19] указывают, что если фениллитий по каплям
прибавлять к эфирному раствору гидрида трифенилгермания и смесь кипятить
с обратным холодильником, образуется желтый раствор, из которого после
карбонизации можно выделить с выходом 86% соответствующую карбоно-
вую кислоту (и очень небольшое количество тетрафенилгермания)
1) С02
(G6H5)3GbH+ RLi - (C345)3G?Li -f RH --> (C345)3GbG334.
2) H>0
В тех же условиях бутиллитий дает количественный выход трифенил-
гермилкарбоновой кислоты. Метиллитий при комнатной температуре плохо
реагирует с гидридом трифенилгермания, но при кипячении с обратным
холодильником в течение 24 час. выход карбоновой кислоты достигает 70—
80% наряду с 16% трифенилметилгермания.
С целью уточнения условий образования гексафенилдигермана, как
это было отмечено выше, Гилман [19] специально исследовал действие
гидрида трифенилгермания на трифенилгермиллитий в диметиловом эфире
этиленгликоля, эфире и смеси обоих растворителей. Во всех этих случаях
гексафенилдигерман образовался лишь в незначительных количествах
(11—12%).
Таким образом, Гилман считает, что лучшим методом синтеза
трифенилгермиллития является действие бутиллития на гидрид трифенилгермания.
Реакция проходит количественно, быстро и без образования побочных
продуктов. Трифенилгермиллитий хорошо растворим в эфире и не реагирует с
растворителем; в реакционной смеси нет избытка металла, и дальнейшие
реакции могут проводиться без фильтрования в том же реакционном сосуде,
где была проведена и основная реакция.
Что касае#я соединений типа R2GeM2, то в литературе такие соединения
почти не описаны, однако сделано предположение [21, 22], что при действии
металлического лития на тетрабензилгерманий, возможно, происходит
отщепление второго бензильного радикала при реакции трибензилгермилли-
тия с образовавшимся в процессе реакции бензиллитием. Реакция
проводилась при 0° С в диметиловом эфире этиленгликоля.
Реакция между дигидридом дифенилгермания и бутиллитием, по
мнению авторов, также, очевидно, приводит к дилитиевому производному
Германийорганические соединения, содержащие два различных металла 149
(C6H5)2GeLi2, однако в смеси с другими производными
(C6H3)2GeH2H-C4H9Li
I
(QH5)2GeLi2 + (СбН5)2 GeLi (QH9) + (CGH5)2 Ge (С4Н9)2+ (C6H5)2 LiGe- GeLi (C6H5)2
J C2H5Br
(C6H5)2Ge (CsH5)2 + (C6H,)2 Ge(C4H9) (C2H5)+(C6H5)2 Ge (C4H9)2 +
2% 20% 12%
+ (C6H5)2 (C2H5) Ge-Ge (C2H5) (C6H5)2
28%
Попытка получения магниевого производного R3GeMgX действием
бромистого бутилмагния на гидрид трифенилгермания [19, 23] оказалась
неудачной. Отдельные авторы [24, 25] считают, что R3GeMgX образуется в
качестве промежуточного продукта реакции при получении гексаарилди-
германов через магнийорганические соединения (см. стр. 33).
Гилман и сотр. [23] нашли, однако, что некоторые реактивы Гринья-
ра замещают водород на MgX в гидриде трифенилгермания. Так, при
реакции гидрида трифенилгермания с хлористым аллилмагнием в кипящем
тетрагидрофуране образуется хлористый трифенилгермилмагний. Выход
полученного магниевого производного определялся хорошо известной
реакцией карбонизации
со2
(С6Н5)3 GeH + CH3=CHCH2MgCl -* (C6H5)3 GeMgCl > (CGH5)3GeCOOH.
н2о
Авторы установили кроме того, что полученное магниевое производное
расщепляет тетрагидрофуран и в продуктах реакции, после гидролиза всегда
находят 4-оксибутилтрифенилгерманий (соответствующее литиевое
производное менее активно по отношению к тетрагидрофурану [261).
ТГФ
(С6Н5), GeMgCl > (С6Н5)3 Ge (СН„)4 ОН.
Бромистый аллилмагний или бромистый фенилмагний ведут себя в
реакциях с гидридом трифенилгермания аналогично.
Отдельные авторы [27] наблюдали, что при реакции четыреххлористого
германия с магнийорганическими соединениями в ряде случаев образуется
примесь гидрида триалкилгермания. Проведенные исследования методом
ИК-спектроскопии показали, что в процессе реакции ни разу не
наблюдалось появления полосы Ge—Н. После гидролиза реакционной смеси в
продуктах реакции всегда наблюдалась полоса Ge—Н. При гидролизе
реакционной смеси тяжелой водой были получены соединения типа R3GeD.
Объяснение данному аномальному течению реакции авторы видят в
образовании промежуточных продуктов реакции со связью Ge—MgX, в
результате гидролиза которых и образуются гидриды.
РЕАКЦИИ СОЕДИНЕНИЙ ТИПА R3GeM
Растворы германийорганических соединений типа R3GeM являются
чрезвычайно реакционноспособными. Они энергично реагируют с влагой и
кислородом воздуха, поэтому работу с ними всегда проводят в атмосфере сухого
инертного газа. Наличие в этих соединениях щелочного металла
обусловливает их большую реакционную способность.
150 Германийорзанччзскчз создинзния. Лит. стр. 157—158
Реакции с кислородом, серой, селеном, теллуром
При окислении трифенилгермилнатрия в жидком аммиаке или бензоле
образуется натрийтрифепилгерманолят [4]
2 (С0Н3)8 GeNa + 02 -> 2(CeH5)3GeONa,
В последнее время з литературе [28, 28а] появилось описание действия
серы, селена и теллура на трифенилгермиллитий. Авторы считают, что
сера, селен и теллур, так же как и кислород, встают между германием и
литием, образуя соответствующие соединения по уравнениям
8(CGH5)3GeLi-I-S8 — 8 (C645)3GeSLi,
8 (C6HS)3 GeLi + Se8 -* 8 (C3H5)3 GeSeLi,
n (C6H5)S GeLi + Ten -> n (C6H5)3 GeTeLi.
Полученные сульфиды, селениды и теллуриды не выделялись из растворов,
а вводились в реакцию с хлористым бензоилом, бромистым трифепилгерма-
нием, хлористым трифенилоловом и хлористым трифенилсвинцом. Во всех
случаях были получены соединения типа R3GeXMR3 где R = С6Н5; X =
= S, Se, Те; М = Ge, Sn, Pb.
Реакция с углекислым гдзэм и карбэнильными соединениями
При действии углекислого газа на соединения типа R3GeM происходит
реакция карбонизации, характерная для металлоорганических соединений
щелочных металлов с образованием связи Ge—С
н2о
R3GeM+C02 > R3GeCODH.
Реакция часто употребляется для идентификации и определения выхода
полученных соединений R3GeM [12, 20, 21].
Что касается взаимодействия с альдегидами и кетонами, то было
найдено, что реакция с формальдегидом [29] в диметиловом эфире этиленгли-
коля происходит по уравнению
(CeH3)3GeLi + НСНО —--> (C845)3GeCri2OH.
Аналогично реагирует бензофенон с образованием трифенилгермилдифенил-
карбинола
(C6H5)3GeLi + (C3H-))2CO —1-» (C6H5)3GeC (ОН) (СвЧ5)2>
подобно тому, как это имеет место в случае щелочноорганических
соединений. Реакция трифенилгермиллития была проверена на ряде различных
кетонов [29а]. Выходы соответствующих карбинолов в большинстве
случаев получены от 50 до 80% . Дегидратация полученных карбинолов привела
к получению ряда германийорганических непредельных соединений.
Реакция соединений типа R3GeM (где М = Li, N, К) с кетоном Михлера часто
используется в качестве цветной реакции
При реакции трифенилгермиллития с (C6H5)3GeCOOCH3 и диэтилкарбо-
натом происходит декарбонилирование [15]
(C3H5)3GBLi + (G845)3GbG3X43 -> (C3H5)jG2-G2 (С3Ч3)3 +СЭ + LiOCH3,
2(CeH5)3G2Li+CD(OC2H5)2->(C345)3G2-G2(Ge45)3;+CD+ 2LOG2H5.
Германийорганические соединения, содержащие два различных металла 151
Реакция с углеводородами
Соединения типа R3GeM в отдельных случаях металлируют
ароматические углеводороды (замена водорода на щелочной металл). Так, Гилман
[30—32J указывает, что флуорен металлируется трифенилгермиллитием в
положение 9, образуя после карбонизации и гидролиза 9-флуоренилкар-
боновую кислоту.
В работе обсуждается механизм металлирования флуорена при помощи
(С6Н5)3 GeLi. Дибензофуран металлировать не удалось [30].
При реакции с непредельными углеводородами происходит
присоединение по двойной связи [14а, 33, 33а]. Трифенилгермиллитий и трифенил-
гермилкалий присоединяются по двойной связи к 1,1-дифенилэтилену с
образованием 1,1-дифенил-2-трифенилгермилэтана (выход 15%).
(С6Н5)3 GeM + (С6Н5)3С-СН2 — -> (С3И5)3 СНМ-СП2 Ge (CGH,)3
и не присоединяются, например, к траяс-стильбену. Трифенилгермиллитий
присоединяется к октадецену-1. С выходом 27,3% образуется трифенил-я-
октадецилгерманий. Аналогично получен триэтил-я-гексил германий
(выход, 8,6%) и триэтил-2-фенилэтилгерманий (выход 42,7%) [14а].
С бензальацетофеноном реакция протекает по уравнению
(CeH5)3GeLi + С6Ч5СН--СНСЭС6Н5 —--> С6Ч5—СН— CHU-CO—СвН5
I
Ог (С6Н3)з
(выход 26,4%).
Реакция с галоидными алкилами и арилами
Действие галоидных алкилов на соединения типа R3GeNa дает
возможность получать несимметричные германийорганические соединения.
При изучении реакции трифенилгермилнатрия с галоидными алкилами
в жидком аммиаке было найдено [5], что реакция протекает по уравнению
R4.nCXn + п NaGe (СбН5)3 - Р4_ЯС [Ge (СбН3)з]л + «NaX,
R — водород или углеродный радикал; X — галоид.
С моногалоидными соединениями реакция протекает почти
количественно. Так, при действии йодистого метила на трифенилгермилнатрий с
выходом 86% синтезирован метилтрифенил германий [5]. Аналогично получены
этилтрифенилгерманий, я-пропилтрифенилгерманий,
я-бутилтрифенилгерманий, я-амилтрифенилгерманий [34]. При действии бромистого этила на
триэтилгермилкалий получен тетраэтилгерманий [7], действием бромистого
этила на трибензилгермиллитий—трибензилэтилгерманий [21].
Действие хлористого бензила часто используется для количественного
определения выхода полученных соединений типа R3GeM [9,34]. Для
получения трифенил-2-фенилэтилгермания успешно был использован
трифенилгермиллитий и бромистый 2-фенилэтил; выход 60% [16]. Реакция
была проведена в диметиловом эфире этиленгликоля. По аналогичной
реакции получен я-децилтрифенилгерманий (70%), я-октадецилдифенил-2-фе-
нилэтилгерманий (17,6%) из я-октадецилдифенилгермиллития и
бромистого 2-фенилэтилена. Тот же продукт с выходом 67% был получен из дифенил-
2-фенилэтилгермиллития и бромистого октадецила [16].
С хлористым метиленом [5] основная реакция протекает по уравнению
2 (CGH5)3 GeNa + СН2С12 -» [(С6Н5)3 Ge]2 CH2 + 2NaCl.
152 Германийорганичсские соединения. Лит. стр. 157—158
В качестве примесей найдены метилтрифенилгерманий, окись трифенил-
германия и /яршхгрифенилгерманиламин [(C6H5)3Ge]3N.
Триметиленбромид и пентаметиленбромид [35] реагируют с трифенил-
гермилнатрием в жидком аммиаке с образованием б&с-трифенилгерманил-
пропана (60,5%) и бис-трифенилгерманилпентана (59,5%).
Вг (СН2)3 Вг + 2(СбН5)з GeNa -> (СбН5)з Ge (GH2)3 Ge (C6H5)3 + 2NaCL
При проведении реакции с хлороформом и четыреххлор истым углеродом
в эфире реакция протекает более сложно [5,35], главным продуктом
реакции является гексафенилдигерман.
Основным продуктом реакции между трифенилгермилнатрием и
галоидными арилами [5] в жидком аммиаке явилась окись трифенилгермания
н,о
Ск>Н7Вг + (СбН5)3 GeNa + NH3 — NaBr + d0H8 + (С6ЫГ))3 GeNH2 * [(СвН,)з Ge]3 О.
По сообщениям работ последних лет, действие галоидных алкилов на
соединения R3GeM применяется для установления выхода полученного
соединения R3GeM. Так, хлористый бензил прибавляют к раствору трифенилгер-
миллития в тетрагидрофуране, полученном или расщеплением гексафенил-
дигермана, или при действии металлического лития на хлористый трифенил-
германий [9].
Реакции с галоидными металлами, металлоидами
и металлоорганическими соединениями
Краус и сотр. [35] впервые исследовали действие треххлористого бора
на трифенилгермилнатрий в эфирном растворе. Реакция протекала очень
бурно, и авторам не удалось выделить строго идентифицированных
продуктов .
Трифенилгермиллитий с двуиодистым германием [36] реагирует
экзотермически, образуя темно-красный раствор, из которого после гидролиза
был выделен с выходом 36% [(C6H5)3Ge]3 GeH. Авторы предлагают
следующую схему протекания реакции:
(C6H5),GeLi
2(С6Н5)3 GeLi + Ge J2 -* [(С6Н5)3 Ge]2 Ge ►
н2о
-> [(G6H5)3 Ge]3 GeLi " * [(C6H5)3 Ge]3 GeH.
C,H9Li
При действии двухлористого свинца на,трифенилгермиллитий получен
[(C6H5)3Ge]4Pb с выходом 23%, однако он не был выделен в чистом виде
вследствие трудностей отделения от побочно образующегося (11%) гекса-
фенилдигермана. Аналогично при реакции трифенилгермиллития с четы-
реххлористым оловом [(С6Н5)3 Ge]4Sn также не был выделен в чистом виде
[37].
Трифенилгермилнатрий [38] и триэтилгермиллитий [14] реагируют
с трихлорсиланом по уравнению
3(С6Н5)3 GeNa + S1HCI3 -> [(CeH5)3 Ge]3 SiH + 3NaCl.
Однако аналогичная реакция с трибензилгермиллитием дала полимерный
материал, который не был идентифицирован [21].
Что касается действия отдельных элементоорганических соединений на
R3GeM, то можно отметить, например, что при действии хлористого дибу-
тилбора на трифенилгермилнатрий [39], хотя связь Ge—В и образуется,
но идениифицированных продуктов выделить не удалось. Действие три-
Германийорганичесше соединения, содержащие два различных металла 153
фенилбора на литиевые производные трифенилгермания приводит к
образованию комплекса, в котором впервые осуществлена непосредственная
связь Qe—В [40].
(СсН3)з GeLi + (С6Н5)3 В - Li [(CGH5)3 GeB (C6H5)3].
Полученный комплекс легко гидролизуется, однако его достаточная
устойчивость в метаноле создает возможность выполнить с ним различные
реакции. Подобно всем тетраалкилборанам полученный комплекс образует
нерастворимые соединения с различными неорганическими и органическими
катионами, например с тетраметиламмонием [(СНз)4М-[(С6Нб)зОеВ(СбН5)з].
Реакция между трифенилгермильными производными щелочных
металлов и трифенильными или триалкильными галогенидами элементов
IV А группы периодической системы изучена Гилманом и сотр. [11, 13, 41].
Было показано, что при действии на трифенилгермилкалий [11] или три-
фенилгермиллитий [7] хлористого триэтилкремния с количественным
выходом образуется триэтилсилилтрифенилгерманий. Аналогично при
действии на трибензилгермиллитий хлористого триметилкремния получен
трибензилгермилтриметилсилан [21 ].
В результате взаимодействия трифенилгермилкалия и хлористого три-
фенилкремния образуется трудноразделимая смесь, содержащая, как
предполагают авторы [13], следующие соединения:
(CaH5)3Ge-Ge(C6H5)3, (C6HJ3 Si-Si (C6H5)3 и (С6Н5)3 Ge-Si (C6H5)3.
Присутствие этих продуктов объясняется обменом галоид — металл
между реагентами и последующей конденсацией образующихся соединений.
Реакция триэтилгермиллития и двухлористого дифенилкремния была
изучена Разуваевым и сотр. [14]. Трифенилтриэтилдигерман был получен
при взаимодействии трифенилгермилнатрия с бромистым триэтилгерманием
[34] по реакции
(СвН5)8 GeNa + (C2H5)3 GeBr -* (СбН5)3 Ge-Ge (C2H5)3 + NaBr.
Трибензилтриэтилдигерман получен аналогично [21].
Краус и Фостер [4] провели аналогичную реакцию с бромистым три-
метилоловом и получили (C6H5)3Ge—Sn (СН3)3. Трифенилстаннилфенилгер-
маний был получен с выходом 60% (из трифенилгермилкалия и хлористого
трифенилолова) [41].
Сейферт с сотр. [42], изучая свойства перфторвинильных производных
кремнийорганических соединений, нашел, что при действии трифенилгер-
миллития на триэтилперфторвинилсилан в диметиловом эфире этиленгли-
коля с выходом 55% образуется (C2H5)3SiCF = CFGe(C6H5)3.
Было найдено, что при реакции триэтилгермиллития с гидридами
триэтилкремния, германия и олова [14] реакция протекает по уравнению
(С2Н5)3 GeLi + (СаН5)з МН -» (С2Н5)3 GeM (С2Н5)3 + LiH
(M = Si, Ge, Sn).
Реакция с аммиаком и азотсодержащими соединениями
Аммиак действует на соединения типа R3GeM, заменяя металл на
водород; аналогично реагирует и бромистый аммоний. При действии аммиака
и бромистого аммония на триэтилгермиллитий в диэтиламине был выделен
гидрид триэтилгермания [7].
(С2Н5)3 GeLi + NH3 -> (C2H5)3 GeH + LiNHa.
154 Г ерманийорганическиг соединения. Лит. стр. 157—158
Аналогично с количественным выходом образуется гидрид трифенилгерма-
ния при реакции трифенилгермилнатрия с бромистым аммонием в жидком
аммиаке [4].
Гилман и сотр. [43] изучили взаимодействие трифенилгермиллития с
азоксибензолом и азобензолом.
Авторы приводят следующую схему реакции:
О
II
(CoH-JaGeLi -f C6H5N=N—CeH5 -> [СвЧ3\-=ЫСз11-,] + (QH^GeOLi
Li H
I н.о |
[C0H5N=NQH5] + (C6H5)3GeLi -» CeH5N — NCeH5 --> CGH5 N-NC6H5
I I
Ge(CeH5)8 Ge(Ce4:>)3
Реакция приводит к получению замещенных гидразинов.
СИНТЕЗ И РЕАКЦИИ СОЕДИНЕНИЙ
ТИПОВ RsGeM^R^j и R3GeM(CO)5 (М == Re, Mn)
В настоящее время существуют два основных пути синтеза соединений
типа R3GeM/1R„-1 (M — иной металл, не германий, п — валентность
металла, R — ароматический, алифатический радикал). Это действие
органических гидридов германия на различные металлоорганические соединения
или действие германийорганических соединений типа R3GeM (М = Li, Na)
на галогениды алкильных или арильных органических производных
других металлов.
В качестве примеров реакции первого типа можно привести следующие.
С литийорганическими соединениями образуются соединения типа R3GeLi
[19, 23] (см. стр. 148).
R3GeH + RLi -> R3GeLi + RH.
С некоторыми магнийорганическими соединениями получены
соединения типа R3GeMgX [23, 44] (см. стр. 149).
Взаимодействие гидридов триалкилгермания с диэтилртутью протекает
по уравнению [45, 46].
2R8GeH + R2Hg -> R3GeHgGeR3.
В случае взаимодействия дигидрида дифенилгермания с диэтилртутью
образуются полимерные материалы со связью Ge—Hg [45—49].
Диэтилцинк, хотя и реагирует с гидридом триэтилгермания, однако
строго идентифицированных продуктов выделить не удалось [50], в то
время как с диэтилкадмием реакция протекает по схеме [51]
(C2H5)3GeH + (C2H5)2Cd -» (C2H5)3GeCdGe (С2П5)з.
С триэтилвисмутом образуются соединения со связью Ge—Bi [52].
Аналогично протекает реакция и с триэтилталлием [53]. т/шс-(Триэтил-
гермил)таллий получен с выходом 91%.
При нагревании гидрида трифенилгермания со станниламинами
получены соединения со связью Ge—Sn [54].
(C6H3)3GeH + (C4H„)8SnN (C2H5)2 -> (C6H3)3GeSn (QH9)3 + HN (C2H5)2.
В литературе описаны реакции соединений типа R3GeM (М = Li, Na,
К) с ВС13, GeJ2, PbCl2, SnCl4, а также с HSiCl3 [14, 21, 35—48], однако в
большинстве случаев выделить строго идентифицированные продукты
реакции авторам не удалось (см. стр. 152).
Германийорганические соединения, содержащие два различных металла 'Го
Реакции R3GeM (М, = Li, Na, К) с металлоорганическими соединениями
в литературе описаны на отдельных примерах [39, 40]. Наиболее широко
исследовалась реакция с галогенидами металлоорганических соединений
IV группы [4, 7, 11, 13, 14, 21, 411.
Своеобразный тип биметаллических соединений найден Несмеяновым и
сотр. [55, 56], которые показали, что при реакции галогенидов трифенил-
германйя с карбонилами марганца и рения образуются биметаллические
соединения карбонилов металлов.
(CGH3)4_rGe8r/z -f- /iNaMn (С ))3 - (C6H3)4_/zGe [Мп (СЭ)3]Л + "NaBr
(/2 = 1 ИЛИ 2)
(C6H3)4_„GeX„ + /zNaRe (CO)3 -> (CeH5)4-/lGe [Re (CO),'],, + nNaX
(Х=С1 и Вг; л = 1 и 2).
Была осуществлена также новая в ряду производных карбонилов металлов
реакция хлорпентакарбонила марганца с гидридом треххлористого герма
ния.
HGeCl3 + CIMn (СЭ)5 -> Cl3GeMn (CO)5 + НС1.
Свойства полиметаллических германийорганических соединений со
связью Ge—Hg, Ge—Cd, Ge—Bi, Ge—Tl были подробно изучены Разу-
ваевым с сотр. [45, 46, 50—53, 57, 58]. Все они легко окисляются, например,
кислородом воздуха
2 (C2H5)3GeHgGe (С2Н3)з + 02 -* 2 (C2H3)3GeOGe (С2Н3)3 + Hg.
Взаимодействие с водой протекает различно: *шс-(триэтилгермил)ртуть
не реагирует с водой даже при многочасовом контакте, бас-(триэтилгермил)-
кадмий медленно разлагается водой с выделением металлического кадмия,
окиси бис-(триэтил)германия и гидрида триэтилгермания; выходы
соответственно 100, 98,1 и 43,9%.
6ас-(Триэтилгермил)ртуть чувствительна к действию света и
разлагается количественно, выделяя ртуть и гексаэтилдигерман
hv
(C2H5)3GeHgGe (C2H5)3-* Hg + (C2H5)3GeGe (C2H3)3.
бш>(Триэтилгермил)ртуть, т/?ш>(триэтилгермил)висмут, этил-(6ас-
триэтилгермил)висмут и диэтил(триэтилгермил)-висмут являются
термически устойчивыми жидкостями и могут быть перегнаны при пониженном
давлении. В противоположность этому бас-(триэтилгермил)кадмий, а
также /ЯуРш:-(триэтилгермил)таллий нелетучи.
Ледяная уксусная кислота реагирует с бш>(триэтилгермил)ртутью и
/тгрш>(триэтилгермил)висмутом с выделением ацетата триэтилгермания,
гидрида триэтилгермания и металлической ртути или соответственно
висмута.
Описаны реакции с металлами. Так, т/?ш>(триэтилгермил)таллий
реагирует с металлической ртутью по уравнению
2 [{C2H3)3Ge]3Tl -f 3Hg -* ЗТ1 + 3 [(C2H5)3Ge]2Hg.
Выход бш>(триэтилгермил)ртути равен 77%.
При действии металлического лития и натрия на бас-(триэтилгермил)-
ртуть и 6ас-(триэтилгермил)кадмий образуются триэтилгермиллитий и три-
этилгермилнатрий.
Наконец, довольно подробно были изучены также реакции полученных
биметаллических соединений германия с галоидными алкилами и арилами.
156 Германийорганические соединения. Лит. стр. 157—158
Было показано, что бг/с-(триэтилгермил)кадмий реагирует уже при
комнатной температуре с галоидными алкилами по уравнению
;(QH5)tGeCdGe (QH5)3 + 2RBr -> 2 (C2H5)3GeR + CdBr2
(R=CaH5f C6H5CH2).
С бромбензолом реакция происходит лишь под влиянием УФ-света с
образованием бромистого триэтилгермания, дифенилкадмия и бромистого
кадмия.
Фотолиз бас-(триэтилгермил) ртути в среде а-бромнафталина, бромистого
бензила и бромистого этила описывается уравнением
C2H5)bGeHgGe (С2Н3)8 + 2RBr -* 2 (C2H5)3GeBr + R2Hg
(R = a-Ci0H7, C6H5CH2, C2H5).
С бромбензолом фотохимическая реакция протекает с образованием
бромистого триэтилгермания, дифенилртути и металлической ртути. С фтор-
бензолом гомолитические реакции расщепления связей С—F не
наблюдаются и реакция идет с количественным выделением [(C2H5)3Ge]20 и
металлической ртути.
Все соединения со связью Ge—Hg, Ge—Cd и Ge—Tl реагируют с 1,2-
дибромэтиленом при комнатной температуре.
Авторы подробно изучили поведение полученных соединений с
перекисью бензоила и некоторыми другими перекисями.
Что касается свойств биметаллических соединений германия с карбо-
нилами переходных металлов, то Несмеянов с сотр. [55, 56] показали, что
при действии брома на (С6Н5)3 GeMn (CO)5 и (C6H5)3GeRe(CO)5 (в смеси rert-
тан— бензол 1:1) при комнатной температуре в течение 2—3 час.
происходит разрыв связи Ge—Мп и Ge—Re с образованием соответствующих га-
лоидопроизводных
(C6H5)3GeMn (CO)o -f Br2 -> (C6H5)3GeBr + BrMn (СО)5,
(C6H3)8GeRe (СО)5 + Br2 -> (CeH5)3GeBr + BrRe (CO)5.
Однако при действии трех эквивалентов брома на (C6H5)3GeMn(CO)5 в ди-
бромэтане в течение 5 час. при постепенном повышении температуры от 20
до 130° С разрушается связь Ge—С
(C6H5)3GeMn (СО)з + 3Br2 -> Br.GeMn (GO)5 + ЗС6Н5Вг.
В случае (C6H5)3GeRe(CO)5 при нагревании в смеси гептан — бензол (30 мин.)
с бромом, взятым в количестве, рассчитанном на замещение у германия
одной, двух или трех фенильных групп, происходило образование Br3GeRe-
• (СО)5 с выходом до 80% и с возвращением части исходного продукта.
При действии хлора на (C6H5)3GeMn(CO)5 в четырехх лор истом углероде
цри температуре кипения растворителя в течение 30 мин. образовался
Cl3GeMn(CO)5.
Действие брома на (C6H5)2Ge[Re(CO)5]2 вызывает разрыв связей Ge—Re.
Хлористый водород при 0—5° С в четыреххлористом углероде с течение
30 мин. отщепляет от (C6H5)2Ge[Re(CO)5]2 группу Re(CO)5 с образованием
(C6H5)2GeClRe(CO)5. При реакции (C6H5)3GeMn (CO)5 в четыреххлористом
углероде в интервале температур—10—76° С с тем же галоидоводородом
количественно возвращался исходный продукт.
Авторы исследовали реакцию замещения карбонильных групп в
(C6H5)3GeMn(CO)5 и (С6Н5)3 GeRe(CO)5 на трифенилфесфин, трифениларсин
и трифенилстибин.
(C6H5)3GeMn (СО)5 + L -» (C6H5)3GeMn (CO)4L + CO,
(C645)GeRe (СЭ)5 + L -» (C6H5)3GeRe (CO)4L + СО
(L=(CGH5)3P, (C6H,)3 As, (C\R)LSb)
Германийорганические соединения, содержащие два различных металла 157
Таким образом, была показана возможность замещения СО-группы в
биметаллических фенилгерманиевых производных карбонила марганца и
рения на группу лиганда и получен ряд новых, не описанных в литературе,
соединений.
Получение (CeHsbGeReCCOJPCCeFbb; [55]. При нагревании до 200° С в течение 2 час.
1,6 г (C6H5)3GeRe (СО)5 и 0,66 г Р (С6Н5)3 выделено 2 г (92%) белого продукта с т. пл. 231 —
233° С.
Аналогично получены (C6H5)3GeRe(CO)4Sb (С6Н5)3, т. пл. 216—218° С;
(C6H5)3GeRe(CO}4AsiC6H6)3, т. пл. 225-227° C;(C6H5)3GeMn(CO)4P(C6H5)3,
т. пл. 249— 250°С; (C6H5)3GeM-(CO)4As(C6H5)3, т. пл. 229,5—231°С;
(CGH5)3 GeMn (CO)4Sb (СбН5)3, т. пл. 214,5—216,5° С.
ЛИТЕРАТУРУ!
l.GilmanH., Bull. Soc. chim. France, 1963, 1356.
2. Wittenberg D., Gilman H., Quart. Rev., 13, 116, (1959).
3. M о r g a n G. Т., DrewH.D. K-, J. Chem. Soc, 127, 1760 (1925).
4. К r a u s С A., Foster L.S., J. Am. Chem. Soc, 49, 457 (1927).
5. К r a u s C. A., Nutting H.S., J. Am. Chem. Soc, 54, 1622 (1932).
6. В a u e r H., Burschkies K., Ber., 67, 104 (1934).
7. К r a u s С A., F 1 о о d E. A., J. Am. Chem. Soc, 54, 1635 (1932).
8. В г о wn M. P., Fow 1 e s G. W. A., J. Chem. Soc, 1958, 2811.
9. G e о r g e M. V., Peterson D. J., Gilman H. J. Am. Chem. Soc, 82, 403
(1960).
10. Tamborski C, FordF. E., Lehn W. L., Moore G. J., S о 1 о s k i E.
J. Org. chem., 27, 619 (1962).
11. Gilman H., GerowC. W., J. Am. Chem. Soc, 77, 5509 (1955).
12. Brook A.G., GilmanH, J. Am. Chem. Soc, 76, 77 (1954).
13. GilmanH., GerowCW, J. Am. Chem. Soc, 78, 5823 (1956).
14. В я з а и к и н Н. С, Г л а д ы ш е в Е. Н., Разу ваев Г. А., Корне-
е в а С. П., ЖОХ, 36, 952 (1966).
14а. Vyazankin N. S., Razuvaev G. P., G 1 a d у s h e v E. N., К о г п е-
е v a S. P., J. Organometal. chem., 7, 357 (1966).
15. Gilman H., GerowC. W., J. Am. Chem. Soc, 77, 4675 (1955).
16. GilmanH, HughesM.B, GerowCW., J. Org. Chem., 24, 352 (1959).
17. GlarumS.N., К г a u s С A., J. Am. Chem. Soc, 72, 5398 (1950).
18. JohnsonO.H, HarrisD.M, J. Am. Chem. Soc, 72, 5566 (1950).
19. GilmanH., GerowC. W., J. Am. Chem. Soc. 78, 5435 (1956).
20. Brook A. G, P e d d 1 s G. J. D., J. Am. Chem. Soc, 85, 2338 (1963).
21. С г о s s R. J., GlocklingF., J. Chem. Soc, 1964, 4125.
22. GlocklingF., Quart. Rev., 20, 45 (1966).
23. GilmanH., ZeuechE.A, J. Org. Chem., 26, 3035 (1961).
24. Seyferth D., J. Am. Chem. Soc, 79, 2738 (1957).
25. Glockling F., Hooton K.A., J. Chem. Soc, 1962, 3509.
26. GilmanH., Car t 1 e dge F. K-, Sim S.—Y., J. Organometal. Chem., 4,
332 (1965).
27. Mendelsohn J.-C., MetrasF., V a 1 a d e J., Compt. rend. 261, 756
(1965).
28. SchumanH., Thorn K-— F., Schmidt M., J. Organometal. Chem., 4,
22 (1965).
28a. Schuraan H., Schmidt M., Angew. Chem., 77, 1049 (1965).
29. GilmanH., GerowCW., J. Am. Chem. Soc, 77, 5740 (1955).
29a. N i с h о 1 s о n D. A., A 1 1 r e d A. J., Inorg. Chem., 4, 1751 (1965).
30. G i 1 m a n H., GerowC. W., J. Org. Chem., 23, 1582 (1958).
31. GilmanH., MarrsO.L, T r e p k a W. J., D i e h 1 J. W., J. Org. Chem.
27, 1260 (1962).
32. Gilman G., Car t 1 edge F. K-, Organometal. Chem., 3, 255 (1965).
33. GilmanH., GerowCW., J. Am. Chem. Soc, 79, 342 (1957).
33a. ВязанкинН. С, ГладышевЕ. Н., КорнееваС. П., Разува-
e в Г. А., ЖОХ, 36, 2025 (1966).
34. Кг a us С. A., ShermanC.S., J. Am. Chem. Soc, 55, 4694 (1933).
35. Smith F. В., К г a u s С. A., J. Am. Chem. Soc, 74, J418 (1952).
36. G 1 о с k 1 i n g F., H о о t о n K. A., J. Chem. Soc, 1963, 1849.
158
Германийорганические соединения
37. W i 1 1 е m s e n s L. С, van der К е г к G. J. M., J. Organometal. Chem., 2y
260 (1964).
38. М i 1 1 i g a n J. G., К г a u s С. A., J. Am. Chem. Soc, 72, 5297 (1950).
39. Booth R.B., К r a u s С A., J. Am Chem. Soc., 74, 1415 (1952). '
40. S e у f e r t h D., R a a b G., GrimS.O., J. Org. chem., 26, 3034 (1961).
41. GilmanH., GerowCW., J. Org. chem., 22, 334 (1957).
42. S e у f e r t h D., W a d а Т., I n о г g. Chem., 1, 78 (1962).
43. G e о г g e M. V., T a 1 u к d a r P. Т., GerowC. W., G i 1 m a n H., J. Am.
Chem. Soc, 82, 4562 (1960).
44. Satge J., Ann. de chim., 6, 519 (1961) (франц.).
45. Вязанкин Н. С, Разуваев Г. А., Гладышев Е. Н., ДАН СССР,.
151, 1326 (1963).
46. ВязанкинН. С, Разуваев Г. А., ГладышевЕ. Н., Гурико-
ва Т. Г., ДАН СССР, 155, 1108 (1964).
47. N е u m a n n W. P., Angew. Chem., 75, 679 (1963).
48. N е u m a n n W. Р., К u h 1 e i n K., Tetrahedron Letters, 1963, 1541.
49. N e u m a n n W. P., К u h 1 e i n K., Ann., 683, 1 (1965).
50. Вязанкин Н. С, Р а з у в а е в Г. А., Корнева С. П., Круглая О. А.,
ГалиулинаР. Ф., ДАН СССР, 158, 884 (1964).
51. В я з а н к и н Н. С, Р а з у в а е в а Г. А., Б ы ч к о в В. Т., ДАН СССР, 158,
382 (1964).
52. К р у г л а я О. А., В я з а н к и н Н. С, Р а з у в а е в Г. А., ЖОХ, 35, 394
(1965).
53. В я з а н к и н Н. С, Митрофанова Е. В., К р у г л а я О. А.,
Разуваев Г. А., ЖОХ, 36, 160 (1966).
54. Sommer R., Neumann W. P., Schneider В., Tetrahedron Letters,
1964, 3875.
55. Несмеянов А. Н., К о л о б о в а Н. Е., А н и с и м о в Е. Н., X о н д о ж-
к о В. Н., Изв. АН СССР, серия хим., 1966, 163.
56. Несмеянов А. Н., А н и с и м о в К- Н., Колобова Н. Е.,
Антонова А. Б., Изв. АН СССР, серия хим., 1966. 160.
57. В я з а н к и н Н. С, Разуваев Г. А., Бычков В. Т., ЖОХ, 35, 395
(1965).
58. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б ы ч к г в R. Т., 3 в е з д и н В. А»,
Изв. АН СССР, серия хим., 1966, 562.
Глава XV
ПОЛИМЕРИЗАЦИЯ
ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В литературе описано несколько примеров полимеризации
непредельных германийорганических, соединений [1—3].
Изучалась полимеризация различных соединений типа RnGeR4^ (n =
= 1,2 или 3; R = СН3 или С2Н5; R' — винил или алкил) в присутствии
перекиси трет-бутша при давлении 6000 атм и температуре 120° С [4, 5].
Моноаллильные и моновинильные соединения дали маслянистые
низкомолекулярные полимеры; ди-, триаллильные и ди- и тривинильные
соединения легко полимеризовались в бесцветные стеклообразные материалы.
Были получены также сополимеры этих соединений со стиролом и метил-
мета к рил атом.
Полимеризация диметидиаллилгермания в присутствии катализаторов
Циглера, Натта привела к образованию полимеров низкомолекулярного
веса [6]. Авторы предлагают для полученного полимера циклическую
структуру
г-СНз -,
ХУ
Ge
L СИ3 СНз-1 п.
Триметилалл ил германий практически не полимеризуется [41.
Изучалась сополимеризация диаллильных соединений германия со стиролом
и метилметакрилатом в присутствии перекиси бензоила [7].
Попытка полимеризации тригидрида винилгермания привела к
получению белого порошка, разлагающегося при температуре около 273° С,
нерастворимого в органических растворителях [8].
Полимеризация металлоорганических производных стирола (Si, Ge, Sn)
исследовалась Колесниковым и сотр. [9] при использовании катализаторов
ионного типа. Ими было установлено, что эти катализаторы практически не
вызывают полимеризации металлоорганических производных стирола. В
дальнейшем удалось получить полимеры на основе германийорганических
производных стирола и а-метилстирола [10—12] в присутствии ъзо-бис-
изобутиронитрила как катализатора. Полимер, содержащий германий в
боковых цепях, был получен Колесниковым и сотр. [13—16] радикальной
полимеризацией метакрилата и акрилата триэтилгермания. Полученные
полимеры обладали плохими термомеханическими свойствами.
Ван-дер-Керк и сотр. [17] получили полимеры, содержащие элементы
IV группы периодической системы, реакцией присоединения дигидридов ди-
фен ил олова к соединениям типа (С6Н5)2М(С6Н4СН = СН2)2 (М — Ge, Sn,
160
Германийорганические соединения.
Pb). Авторы придают полученным полимерам следующую структуру:
QH5
I
-Sn—(СНа)з'
I
с6н5
с6н5
—(СП2)2—
Аналогичные соединения были получены и другими авторами [18].
Полимерные германийорганические соединения были получены при
взаимодействии двухлористого диметилгермания и стирола в тетрагидро-
фуране в присутствии металлического лития. После удаления непрореаги-
ровавших исходных продуктов была получена смола с содержанием
германия 25,16% [19—21].
В результате реакции диенов-1,3 и гидридов треххлористого германия
в среде простых эфиров были получены германиевые полимеры с
молекулярным весом от 1 000 до 2000 [21—24].
Коршак и сотр. [251 изучали возможность получения
высокомолекулярных соединений из дигидрида дифенилгермания в условиях реакции
полирекомбинации.
Описаны различные полигерманосилоксаны [26—28]. Эти соединения
получались присоединением соответствующих германосилоксанов к
ненасыщенным соединениям типа R2Ge (СН = СН2)2 и R2Ge(CH2CH = СН2)2
в присутствии платинохлористоводородной кислоты в качестве
катализатора. Полученным полимерам авторы придают следующую структуру:
R R' R' -
I I I
—(CH2)m—Ge— (СН2) — Si—О—Si—
I I I
R R" R' - ..
(т - 2, 3)
Полидиметилгерманосилоксаны были получены при полимеризации
низкомолекулярных полимеров, образующихся в результате совместного
гидролиза двухлористого диметилгермания и двухлористого диметилкрем-
ния. Полимер обладал свойствами, во многом напоминающими свойства
полидиметилсилоксанового каучука [29]. Имеется указание о получении
полидиметилгерманиевых соединений с галоидными концевыми группами
[30]. Недавно описан новый вид металлоорганических полиамидов [31],
полученных поликонденсацией соответствующих металоорганических
диаминов и дикарбоновых кислот, имеющих одинаковые или разные
металлы, например германий и кремний.
ЛИТЕРАТУРА
1. Б о н н о И., Усп. хим. 34, 850 (1965).
2. Т a n a k а Т., Engng. Mater., 12, 65 (1964); РЖХим., 1965, 2С206.
3. НепгуМ.С, DavidsonW.E., Ann. N. J. Acad. Sci., 125, 172 (1965); РЖХим
1966, 4C50.
4. К о р ш а к В. В., Полякова А.М
ДАН СССР, 112, 436 (1957).
5. К о р ш а к В. В., П о л я к о в а А. М.
Изв. АН СССР, ОХН, 1959, 178.
6. Колесников Г. С, Давыдова С.
комолек. соед., 1, 1493 (1959).
7. К о л е с н и к о в Г. С, Давыдова С. Л., Е р м о л а е в а Т. И., Ш м л с
в а Н. Д., Б ы х о в с к а я М. Б., Высокомолек. соед., 2, 567 (1960).
В г i n с k m a n F. Е., S t о n e F. G. A., J. Inorg. Nurl. Chem., 11, 24 (1959).
9. К о л е с н и к о в Г. С, Давыдова С. Л., ЖОХ, 29, 2042 (1959).
Ю. N о 1 t es J. G., BuddingH.A., van d e r KerkG.J.M., Rec. trav.
chem., 79, 1076 П960У
П е т р о в А. Д., М и р о и о в В. Ф.
М и р о н о в В. Ф., Петров А. Д.
Л., Ермолаева Т. И.,
Высо
Полимеризация
161
11. Полякова A.M., С а х а р о в а А. А., Ч е р н ы ш е в Е. А.,
Краснова Т. Л., К о р ш а к В. В., П е т р о в А. Д., Высокомолек. соед., 5, 353 (1963).
12. Полякова А. М., К о р ш а к В. В., Т а м б о в ц е в а Е. С, Высокомолек.
соед., 3, 662 (1961).
13. К о л е с н и к о в Г. С, Д а в ы д о в а С. Л., Климентова Н. В.,
Высокомолек. соед., 2, 563 (1960).
14. К о л е с н и к о в Г. С, Д а в ы д о в а С. Л., Климентова Н. В.,
Международный симпозиум по макромолекул яр ной химии, секция 1. М., 1960, стр. 156.
15. Кол есн и к о в Г. С, Г у р г е н и д з е Г. Т., Изв. АН СССР, ОХН, 1962,
1275.
16. К о л е с н и к о в Г. С, Д а в ы д о в а С. Л., К л и м е н т о в а Н. В., Сб.
«Синтез и свойства мономеров». М., «Наука», 1964 г., стр. ИЗ.
17. Noltes J. G., van der Ker k G. J.M., Rec. trav. chim., 80, 623 (1961).
18. Лунева Л. К., Сладков A.M., Коршак В. В., Высокомолек. соед., 7,
427 (1955).
19. Н ефе д о в О. М., М а н а к о в М. Н., Петров А. Д., ДАН СССР, 147,
1376 (1962).
20. N е f e d о w О. М., ManakowM. N., Ре trow A. D., Plaste und Kauts-
chuk, 10, 721 (1963).
21. Нефедов О. М., М а н а к о в М. Н., Ш и р я е в В. И., Авт. свид. СССР
157491 (1963); РЖХим., 1964, 13С202; Бюлл. изобрет., № 18, 57 (1963).
22. Н е'ф едовО. М., Колесникове. П., Высокомолек. соед., 7, 1857 (1965).
23. Н е ф е д о в О. М., К о л е с н и к о в С. П., Авт. свид. 161922 (1964); РДХим.,
1965, 14С273П; Бюлл. изобрет. СССР, № 8, 59 (1964).
24. Нефедов О. М., К о л е с н и к о в С. П., Авт. свид. СССР 161922 (1964);
Бюлл. изобрет., № 8, 59 (1964).
25. С о с и н С. Л., Коршак В. В., Алексеева В. П., Высокомолек. соед.,
6, 827 (1964).
26. Коршак В. В., П о л я к о в а А. М., В д о в и н В. М., Миронов В. Ф.,
П е т р о в А. Д., ДАН СССР, 128, 960 (1959).
27. Петров А. Д., 17 Международный конгресс по чистой и прикладной химии, Мюн-
хен, 1959.
28. Пол я ков a A.M., Ч у м а е в с к и й И. А., ДАН СССР, 130, 1037 (1960).
29. СтавицкийИ. К., Борисов С. Н., ПономаренкоВ. А.,
Свиридов а Н. Г., 3 у е в а Г. Д., Высокомолек. соед., 1, 1502 (1959).
30. М о е d r i t z е г К., Wazer J. R., Amer. Chem. Soc. Polymer. Preprints 6,
1140(1965); РЖХим., 1986, 12C178.
31. M a z e г о 1 1 e s P., Bull. Soc. chim. France, 1965, 464.
11 За саз М 207
Г лава XVI
АНАЛИЗ ГЕРМАНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Качественный пробой на присутствие германия служит появление серо-
белого налета на металлической (лучше платиновой пластинке) после
прокаливания образца в пламени горелки [1].
Для количественного определения чаще всего применяют обработку
навески германииорганического соединения смесью серной и азотной кислот
сначала на холоду, затем при нагревании прокаливают до постоянного
веса и определяют германий в виде двуокиси германия [2, 3]. В некоторых
случаях рекомендуют для разложения металлоорганического соединения
применять смесь серной кислоты и перекиси водорода [4]. Было показано
[5, 6], что для соединений, в которых атом германия связан с двумя или
более атомами галоида или кислорода, надежным способом разложения
является сплавление навески с едким кали или натром в никелевой бомбе
при нагревании до 700—750° С в течение 40—50 мин. Наиболее трудно
разлагаемыми являются тетраалкильные соединения германия, для
количественного разложения которых рекомендуют сплавление вещества с едким
натром и содой (1 : 1) в герметически закрытой никелевой бомбе при
температуре 920—940° С в течение 1,5—2 час. Этот метод применим также для
разложения германийорганических соединений любого строения, включая
полимеры. Ошибка метода + 0,4 абс.%.
В литературе описаны методы сожжения германийорганических
соединений с одновременным определением углерода и водорода [7] при
сжигании в присутствии W03, или определение углерода, водорода и германия
[8] в присутствии в качестве катализатора Сг203.
Галоид в германийорганических соединениях после минерализации
можно определять методом аргентометрии.
Для микроопределения алкоксильных групп в германийорганических
соединениях Климова и сотр. [9] рекомендуют видоизмененный метод Цей-
зеля — Фибека. Анализируемое вещество нагревают с концентрированной
иодистоводородной кислотой, образовавшийся йодистый ал кил отгоняют
и окисляют бромом в йодноватую кислоту, которую определяют иодометри-
чески. Чтобы избежать употребления иодистоводородной кислоты,
разлагающейся при хранении, авторы рекомендуют метод микроопределения
алкоксильных групп, в котором для разложения алкоксисоединений
применяют смесь йодистого калия с ортофосфорной кислотой (при нагревании
образуется иодистоводородная кислота).
Соединения алкокси- и меркаптоалкилгермания рекомендуют определять
методом ацетилирования или при помощи бромистоводородной кислоты
[10]. Непредельные связи германииорганического соединения определяют
методом родановых чисел [11 —13]. По мнению авторов, это является более
универсальным приемом оценки непредельности, чем метод бромных
чисел.
Анализ-
163
Для анализа германийорганических соединений в последнее время
начали разрабатываться хроматографические методы [14—19], но число
работ в этой области не очень велико.
ЛИТЕРАТУРА
1. Spialter L., BallesterM., Analyt. Chem., 34, 1183 (1962).
2. К r a u s С. A., BrownC.L, J. Am. Chem. Soc, 52, 3693 (1930).
3. SmithF.B., RrausCA, J. Am. Chem. Soc, 74, 1418 (1952).
4. Губен-Вейль. Методы органической химии, том II. Методы анализа (перевод с
немецкого). М., Госхимиздат, 1963, стр. 87.
5. В и та л и н а М. Д., Климова В. А., Ж- аналит. хим., 17, 1105 (1962).
6. Климова В. А., ВиталинаМ. Д., Ж- аналит. хим., 19, 1254 (1964).
7. Р i e t е г s H., Buis W.J., Microchem. J., 8, 326, 383 (1964); РЖХим., 1965,
18Г149.
8. Aranyine-HalmosT., Erdeyne-Schneer A., Magyar kem lapja.
20, 164 (1965) (венг.); РЖХим., 1965, 21Г197.
9. Климова В. А., Забродина К. С, Ш и т и к о в а Н. А., Изв. АН СССР,
серия хим., 1965, 178.
10. Magnuson J.A., KnaubE.W., Analyt. Chem., 37, 1607 (1965).
11. Б у г о р к о в а А. А., Миронов В. Ф., Петров А. Д., Изв. АН СССР,
ОХН, 1960, 474.
12. Petrow A. D., Mironow W. F., Bugorkowa A. A., Fette, Seifen, Ans-
trichmittel, 62, 1107 (1960).
13. Миронов В. Ф., Пономаренко В. А., Взенкова Г. Я-,
Долгий И. Е., П е т р о в А. Д., Химия и практическое применение кремнийоргани-
ческих соединений, вып. 1. Л., изд. ЦБТИ, 1958, стр. 192.
14. A b е 1 Е. W., N i с к 1 е s s G., Р о 1 1 а г d F. H., Proc. Chem. Soc, 1960, 288.
15. С н е г о в а А. Д., М а р к о в Л. К.» Пономаренко В. А., Ж- аналит.
хим., 19, 610 (1964).
16. S e m 1 у е n J. A., W а 1 k e r G. R., В 1 о f e I d R. E.f P h i 1 1 i p s С. S. G.,
J. Chem. Soc, 1964, 4948.
17. S e m 1 у e n J. A., Phil lips C.S. G., J. Chromatog., 18, 1 (1965).
18. S e m 1 у e n J. A., WalkerG.R., P h i 1 1 i p s С S. G., J. Chem. Soc, 1965,
1197.
19. P о 1 1 a r d F. H., NicklessG., UdenP.C, J. Chromatog., 19, 28 (1965).
11*
ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ВВЕДЕНИЕ
Оловоорганические соединения впервые получены в 1852 г. Левигом
при действии йодистого этила на сплав олова с натрием.
В дальнейшем химия оловоорганических соединений развивалась
довольно неравномерно. В период 1852—1900 гг. опубликовано ограниченное
количество статей, описывающих или действие сплавов олова на галоидные
алкилы, или простейшие реакции, касающиеся изменения функциональных
групп у атома олова. Позже в 1920—1926 гг. в основном немецкими
авторами изучались методы синтеза тетраалкильных и тетраарильных
производных олова с помощью органических соединений магния, ртути или цинка
и реакции деалкилирования оловоорганических соединений галоидами или
галоидоводородами.
С 1929 г. число работ значительно растет. Исследованиями советских
и немецких авторов^найдены различные реакции и новые классы
оловоорганических соединений.
В настоящее время область оловоорганических соединений развивается
весьма интенсивно. Если в 1959 г. в обзоре по этому вопросу [1] приведено
около 900 литературных ссылок, то в данной книге обработано свыше 2500
статей и патентов. \■!
Найден ряд новых классов веществ, среди которых необходимо отметить
соединения, содержащие различные металлы в молекуле, как разделенные
между собой метиленовыми мостиками, так и с прямой связью Sn—М (где
М = Li, Na, К, Mg, Zn, Cd, Si, Ge, Pb, Ti, Zr, Cr, Mo, W или другой металл).
Широко развиваются работы по синтезу и изучению свойств соединений
с различными функциональными группами в алифатическом радикале у
атома олова. Разработан новый метод синтеза ацетиленидоз олова.
Наконец, в последнее время внесен ценный вклад в изучение соединений со
связью Sn—N. Появляется большая группа работ, посвященных строению
оловоорганических соединений и механизму химических реакций с их
участием.
Оловоорганические соединения нашяи широкое применение в процессах
синтеза и переработки полимеров, например, в качестве катализаторов
различных процессоз, стабилизаторов полихлорвинила и т. д. Сюда
примыкают работы по исследованию полимерных материалоз на основе
оловоорганических соединений.
В свою очередь потребности практики выдзинули в качестве оснэзнэй
задачи разработку методов синтеза оловоорганических соединений в
промышленных масштабах.
Широко развиваются исследования по изучению физико-химических
свойств оловоорганических соединений. Ниже приводятся
литературные ссылки на специальные статьи по рефрактометрии и плотностям олово-
Введение 165
органических соединений [2—6], их спектрам поглощения в
ультрафиолетовой области [7—21], ИК-спектрам [18—84], спектрам комбинационного
рассеяния [23, 24, 31, 65, 66, 85—88], микроволновым спектрам [51, 89,
90]. Имеются работы по электронному [91—93] и ядерному [18, 64, 79,
81, 94—121] магнитному резонансу, эффекту Мессбауэра [122—138],
ядерному квадрупольному расщеплению [139], масс-спектрам [140], рентгено-
структурному анализу [141 —150], магнетохимическим исследованиям [84,
151—155]. Изучены также дипольные моменты [156—162] и
электропроводность [163—165] оловоорганических соединений. Измерены теплоты их
сгорания и образования [166—173]. Исследовано поведение различных
оловоорганических соединений в растворах [174—179]. В литературе имеются
также данные о возможности применения тетраметилолова в счетчиках
Гейгера— Мюллера [180]. Бомбардировкой пучком электронов паров тетра-
алкилолова получены [181] металлические пленки толщиной от 50 до 1500;А
и исследовано влияние условий разложения на качество этих пленок.
Наконец, имеются работы с использованием меченых атомов [182, 183]. Кроме
того, в ряде статей полученные соединения охарактеризованы с помощью
тех или иных физико-химических методов. (Поскольку данная книга
посвящена синтетическим методам элементоорганической химии, подробное
рассмотрение этих вопросов не входит в нашу задачу.)
Ниже указаны основные методы синтеза оловоорганических соединений.
1. Действие галоидных алкилов на металлическое олово. Большую
роль в этих реакциях игргют природа галоидного алкила и условия
проведения эксперимента. Нагревание до 130—180° С металлического
олова с йодистыми алкилами приводит к смеси соединений типа R3SnJ и
R2SnJ2. В случае низших йодистых алкилов применение катализаторов,
например магния (или цинка) и спирта (или тетрагидрофурана), не
оказывает существенного влияния на состав образующихся веществ.
Йодистый бутил и его высшие гомологи реагируют с; металлическим оловом
в присутствии указанных выше катализаторов с образованием дииодидов
диалкилолова.
Что касается бромистых алкилов, то при нагревании их с
металлическим оловом и небольшими количествами магния, йодистого алкила и спирта
образуется смесь оловоорганических бромидов различной степени алкили-
рования. Двубромистое дибутилолово получено при длительном
нагревании до 160° С бромистого бутила, металлического^олова и триэтиламина.
Метод не является, по-видимому, достаточно универсальным, так как уже
в случае бромистого этила он приводит к смеси бромистого триэтилолова и
двубромистого диэтилолова.
Чистые дибромиды диалкилолова удалось получить с высоким выходом
при облучении смеси бромистого алкила и олова у-лучами.
Хлористые алкилы реагируют с оловом без катализаторов только в очень
жестких условиях (при 300—350° С). Метод применен для синтеза двухло-
ристого диметилолова, но уже с хлористым бутилом он не приводит к цели.
Однако в присутствии трет-аминов и иода хлористый бутил реагирует с
оловом с образованием преимущественно хлористого трибутилолова или
двухлористого дибутилолова. То или иное направление реакции зависит
от условий проведения эксперимента.
Реакция между галоидными бензилами и металлическим оловом
проходит своеобразно. Нагреванием смеси в воде получают хлористое трибензил-
олово и двухлористое олово
н2о
ЗС6Н„СН2С1 + 2Sn _-> (C6H5CH2)3SnCl + SnCl2.
1 66 Оловоорганические соединения. Лит. стр. 175—180
В толуоле единственным продуктом той же реакции является двухлористое
дибензилолово свн6сн3
2С6Н,СН2С1 + Sn > (C6H5CH2)2SnCl2.
При замене олова на его сплавы с натрием могут быть получены
соединения типа R4Sn как в алифатическом, так и в ароматическом ряду. В этих
реакциях использованы йодистые, хлористые и бромистые алкилы. Метод
позволяет получать и оловоорганические моно- или дигалогениды. Менее
активны сплавы олова с медью или магнием. Например, реакция между
сплавом Sn—Си и йодистыми алкилами проходит достаточно легко, а с
бромистыми алкилами — только в присутствии йодистых алкилов.
Хлористые алкилы вообще не реагируют с этим сплавом.
2. Одним из давних и хорошо разработанных методов является действие
реактива Гриньяра на соли четырехвалентного олова. Метод широко
применяется для синтеза четырехзамещенных оловоорганических соединений
алифатического, ароматического и гетероциклического ряда.
SnX4 + 4RMgX -> R4Sn + 4MgX2.
Реакции обычно проводят с избытком реактива Гриньяра в среде эфира
или тетрагидрофурана. Разработаны варианты проведения этого синтеза и в
безэфирной среде.
При соблюдении стехиометрических количеств реагентов образуются
соединения типа R3SnX и R2SnX2:
SnX4 + 3RMgX -+ R3SnX + 3MgX2,
SnX4 + 2RMgX -> R2SnX2 + 2MgX2.
Метод использован также для синтеза тригалогенидов алкилолова.
Магнийорганические соединения применяются и при доалкилировании
оловоорганических галогенидов, как правило, с целью получения
соединений с различными органическими радикалами у атома олова:
R3SnX + R'MgX -> R3SnR' + MgX2,
R2S11X2 + 2R'MgX -* RaSnRg + MgX2.
Иногда в качестве алкилирующих или арилирующих агентов применяют
органические соединения лития, натрия, цинка или алюминия. Например,
литийорганические соединения используют в тех случаях, где
приготовление реактива Гриньяра связано с трудностями, или там, где необходим
реагент с повышенной реакционной способностью. Это относится прежде
всего к синтезу тетраарильных производных олова с заместителями в фениль-
ном ядре.
При получении оловоорганических ацетиленидов обычно исходят из
натрийорганических соединений. По-видимому, последние являются также
промежуточными продуктами и при синтезе тетраалкилолова реакцией
типа реакции Вюрца:
4RX + 8Na + SnCl4 -> R4Sn + 4NaCl + 4NaX.
Для синтеза тетраалкил- и тетраар ил олова и отчасти оловоорганических
галогенидов (особенно ароматического ряда) довольно широко
применяются алюминийорганические соединения:
3SnCl4 + 4R3A1 -> 3R4Sn + 4А1С13,
SnCl4 + R3AI -> R3SnX + AICI3.
Кроме того, в этих реакциях могут быть использованы R3A1X,
R3A12X3 (X — галоид) и R4A1M (M — щелочной металл).
3. Реакции металоорганических соединений с солями двухвалентного
олова могут проходить в двух направлениях. С магний- и литийорганичес-
Введение
167
кими соединениями образуются вещества полимерного характера со связью
олово — олово: с г. . опт . /п с ч . от _t
ЯпС12 + 2RLi -* (R2Sn)a + 2LiCl.
Другое направление основано на восстановлении ртутно- или таллийорга-
нических соединений
SnCI2 + R2Hg -> R2SnCl3 + Hg.
Этот метод создает возможность получения дигалогенидов диарилолова с
различными заместителями в ароматическом радикале.
4. Важная новая реакция получения оловоорганических соединений
с заместителями в алкильном радикале заключается в присоединении
оловоорганических гидридов к олефинам или алкинам:
R„SnH4_n + СН2= СНХ - RnSn (СН2СН2Х)4_Я,
R«SnH4-Az + СН=ССН2Х -> R„Sn (CH=CHCH3X)4_/l
(л = 1, 2, 3; Х=ОН, OR, OOCR, CN и др.).
Аналогичным образом ведут себя оловоорганические (галоид)гидриды,
например:
* к R2Sn (Н) С1 + СН2=СНХ -> R2Sn (СН2—СН2Х) CI.
Присоединение оловоорганических гидридов к олефинам в ряде случаев
проходит самопроизвольно, но обычно требуется нагревание смеси до 60—100° С
или применение катализаторов. Гидриды фенилолова более активны, чем
соответствующие алкилзамещенные соединения.
Скорость и направление реакций присоединения оловоорганических
гидридов к олефинам существенно зависят и от природы последних.
Наиболее легко проходит гидростаннирование эфиров акриловой и метакрило-
вой кислот, их нитрилов или амидов, а также металлоорганических
соединений с винильными радикалами. Более жесткие условия требуются в
случае незамещенных олефинов, непредельных спиртов и виниловых эфиров.
К галоидзамещенным олефинам, непредельным аминам или кислотам олово-
органические гидриды, как правило, не присоединяются. Здесь имеют
место следующие реакции:
R3SnH + СН2=СНСН2Вг -* R3SnBr + СН3=СНСН3,
_ с „ CH2=CHCH2NR2 песо
R3SnH -» R3SnSnR3,
R3SnH 4- СН3=СНСООН -* R3SnOOCCH==CH2 + Н2.
К алкинам оловоорганические гидриды присоединяются гораздо легче, чем
к алкенам.
Присоединение оловоорганических гидридов к алкенам и алкинам
приводит преимущественно к соединениям, замещенным у [J- или более
удаленного по отношению к атому олова углеродного атома. Общий метод синтеза
соединений с заместителями у а-углеродного атома основан на
присоединении алкоксисоединений олова или станниламинов к кетену или изопро-
пен ил ацетату:
J R3SnOR' + СН3=С^=0 -» R3SnCH2COOR\
R3SnNR2 + CH3=C=0 -» R3SnCH2CONR'2,
RsSnOR' + CH3C=-CH2 -> R3SnCH2COCH3 + CH3COOR'.
I
OOCCH3
Аналогичным образом ведут себя соответствующие диалкильные соединения
олова.
5. Дигалогениды диарилолова образуются при разложении двойных
диазониевых солей типа (ArN2Cl)2SnCl4 (реакция Несмеянова). Ароматнчес-
168 Оловоорганические соединения. Лит. стр. 175—180
кое ядро может содержать активные заместители, но в этом направлении
метод пока развит мало.
Что касается алифатического ряда, то при действии диазоалканов на
четыреххлористое олово или оловоорганические галогениды получают
соединения типа [ClCH(R)]nSnXw (п и т от 1 до 3):
R2SnCl2 + CH2N2 -* R2Sn (CH2Cl)2 + Na.
Алифатические диазосоединения легко реагируют и с гидридами олова
R3SnH + N2CHCOOC2H5 -* R3SnCH2COOC2H5 + N2.
К оловоорганическим соединениям приводят некоторые другие реакции.
Оловоорганические ацетилениды и другие соединения получают
нагреванием углеводородов с алкоксисоединеииями олова, станниламинами или
гексаалкилдистанноксанами:
R3SnOCH3 + НСее CR' -* R3SnC=CR' + CH3OH.
В основном принципиальный интерес представляют реакции металлоорга-
нических соединений с оловом, его сплавами или оловоорганическими
соединениями со связью олово — олово.
Электрохимические методы синтеза находятся еще в стадии начальной
разработки [184—194].
Переходя к характеристике превращений оловоорганических
соединений, необходимо отметить, что исходят обычно из соединений типа R4Sn.
Получение соединений низшей степени алкилирования широко было
осуществлено с помощью галогенидов олова (реакция Кочешкова):
3R4Sn + SnX4 -» 4R3SnX,
tf4Sn+ R2SnX2->2R3SnX,
R4Sn + SnX4 -> 2R2SnX2,
R3SnX + RSnX3 -> 2R2SnX2
(где R — алкил или арил),
Ar4Sn + 3SnX4 -> 4ArSnX3,
Ar2SnX2 + SnX4 -» 2ArSnX3.
Реакции обычно проводят путем нагревания до 180—200° С стехиомет-
рических количеств реагентов в течение 3—4 час. Галогениды моно- и ди-
винилолова образуются уже при обычной температуре.
Провести в этих условиях отщепление трех алифатических радикалов
не удается. Синтез тригалогенидов ал кил олова основан на следующих
реакциях:
AlkSnAr3 + 3SnX4 -* AlkSnX3 + 3ArSnX3
или
Alk4Sn + 2SnX4 -> 2AlkSnX3 + Alk2SnX2.
Оловоорганические соединения с различными радикалами у атома
олова превращают в соответствующие галогениды с помощью галоидов
R2SnR^ + Br2 -> R2R'SnBr + R'Br.
При этом наиболее легко от атома олова отщепляются ароматические и ви-
нильные группы, гораздо труднее — алифатические. Из ароматических
радикалов вначале отрывается более тяжелая группа (толил раньше
фенила). В алифатическом ряду имеет место обратная зависимость (прежде
всего на галоид заменяется метильная группа). Следует отметить, что
первый и второй радикалы отщепляются от тетраалкилолова при различной
температуре. Температурные условия отщепления третьего и четвертого
Введение
169
радикалов весьма близки, поэтому для синтеза соединений класса RSnX3
этот метод не применяется.
Что касается соединений, содержащих различные заместители в
алифатическом радикале, то галоидированиеа-замещенных оловоорганических
эфиров или нитрилов приводит к преимущественному отрыву замещенного
радикала. Однако если заместитель находится по отношению к атому олова
в Р- или еще более удаленном положении, то происходит отрыв
незамещенной группы:
(С4Н9)3 §nCH2CN + h -> (C4H9)3 SnJ + JCH2CN,
(CiHe)8 SnCH2CH2CN + Вга -> (С4Нв)2 (CNCH2CH2) SnBr + QH9Br.
При галоидировании тетраарилолова обычно образуется смесь продуктов
различной степени арилирования. Для получения чистых веществ
используют комплекс брома с пиридином, который обладает более мягкими де-
арилирующими свойствами, чем сам бром.
В качестве деарилирующих агентов можно применять также галоидово-
дороды. Этот метод используется в основном в тех случаях, когда молекула
оловоорганического соединения содержит в своем составе сложные
ароматические радикалы (фенантрил, бифенил и др.).
Тетраалкильные соединения олова довольно устойчивы к действию
органических кислот. Сильные кислоты, например трифторуксусная, отщепляют,
как правило, только один алифатический радикал. С более слабыми
кислотами реакция проходит в весьма незначительной степени. Отщепление двух
алифатических радикалов от атома олова не характерно и описано только
на отдельных примерах. Однако винильные группы и радикалы с
заместителями у а-углеродного атома отщепляются сравнительно легко. Тетрафе-
нилолово при нагревании до 100° С устойчиво к действию органических
кислот, но при более высокой температуре происходит отщепление всех
ароматических радикалов и образуются основные соли олова.
Иногда в качестве деалкилирующих агентов применяют соли металлов.
Несмотря на то, что эти реакции имеют ограниченное препаративное
значение, деалкилирующие свойства солей металлов изучены довольно подробно.
От тетраалкилолова они отщепляют не более двух алифатических радикалов.
В случае тетравинил- и тетраарилолова можно провести и полное
отщепление всех органических радикалов от атома олова. Метод используется для
синтеза органических соединений бора, таллия, германия, фосфора,
мышьяка, сурьмы и других элементоорганических соединений. Оловоорганические
соединения выступают в этих реакциях в качестве алкилирующих или ари-
лирующих агентов.
Деалкилирование и деарилирование соединений олова можно провести
также нагреванием R4Sn с серой, селеном или теллуром. Эти реакции
использованы, например, для синтеза сульфидов диалкил- и диарилолова.
Значительный интерес представляют работы по изучению термической
устойчивости оловоорганических соединений и их реакций с галоидными
алкилами под влиянием УФ-облучения или в присутствии перекисей.
Эта область широко исследуется в работах Разуваева и его школы.
Вторая группа реакций связана с преобразованием функциональных
групп у атома олова. Для синтеза оловоорганических ацилатов, меркап-
тидов, аминов, алкоксисоединений применяется два основных метода. По
первому из них оловоорганические галогениды последовательно
обрабатывают водным раствором щелочи, а затем соединениями с активным
атомом водорода. Галогениды R3SnX, R2SnX2 и RSnX3 при действии на них
водной щелочи или аммиака легко гидролизуются с образованием R3SnOSnR3,
(R2SnO)n и (RSnOOH)n. При проведении гидролиза в более мягких
условиях могут быть выделены и промежуточные продукты гидролиза, напри-
170
Оловоорганические соединения. Лит. стр. 175—180
мер, X(SnR20)nSnR2X (X — галоид или остаток органической кислоты).
Все они при обработке кислотами, меркаптанами и некоторыми другими
соединениями с достаточно подвижным атомом водорода превращаются
в соответствующие ацилаты, меркаптиды и др. Например:
R3SnO + 2R'COOH -» R2Sn (OOCR')2 + H20;
ClSnR2OSnR2Cl + 2R'COOH -> 2R2Sn(Cl) (OOCR') + H30.
Более общий метод синтеза различных производных олова заключается
в обмене оловоорганических галогенидов с солями (меркаптидами, алкого-
лятами, амидами и т. д.) натрия, калия или свинца:
R„SnCl4_„ + NaX - R„SnX4_„ + NaCl
(n = 1, 2, 3; X - F, J, OOCR, OR, SR, OOR, NR2 и др.).
При получении некоторых неустойчивых оловоорганических соединений
(например перекисей) в качестве исходных веществ применяют алкокси-
соединения олова или станниламины
R2Sn (OR')2 + 2 R"OOH -> R2Sn (OOR")2 + 2R'OH.
Интересно отметить, что различные оловоорганические соединения, такие
как алкоксисоединения олова, станниламины или гексаалкилдистанно-
ксаны, легко вступают в реакции присоединения к кетонам, сероуглероду,
арилизоцианатом, двуокиси углерода, трехокиси серы и другим
соединениям с полярной кратной связью. Например:
R3SnOR'+ R"N=C-0-* R3SnNR"COOR\
На направление химических реакций, каталитическую активность
структуру и свойства оловоорганических соединений существенное влияние
оказывает их стремление к образованию комплексов. Последние можно
условно разделить на три типа. К первому из них можно отнести комплексные
оловоорганические соединения с различного рода лигандами, имеющими
в своем составе гетероатомы с неподеленной парой электронов (азот,
кислород, серу и др.). Следует подчеркнуть, что акцепторные свойства атома
олова в значительной степени определяются характером окружающих его групп.
Так, склонность к комплексообразованию оловоорганических хлоридов
изменяется в следующем ряду:
C6H5SnCl3 > (С4Н9)2 SnCl3 > (QH5)2 SnCl2 > (С6Н5)3 SnCl > (С4Н9)3 SnCl.
Наряду с образованием комплексов с различного рода лигандами
оловоорганические соединения способны к внутримолекулярной координации
или межмолекулярной ассоциации. Такие типы координации возможны в
тех случаях, когда молекула оловоорганического соединения имеет в своем
составе электронодонорную группу (OR, OOCR, NR2 и др.).
Наконец, известны случаи, когда два различных оловоорганических
соединения могут образовать между собой комплексы за счет донорных групп
одного из компонентов и акцепторных способностей атома олова в другом.
Например, алкоксисоединения олова образуют с оловоорганическими гало-
генидами или ацилатами комплексы состава R2Sn (OR)2 • R2SnX2 (X —
галоид или остаток органической кислоты).
По методам синтеза и свойствам от оловоорганических соединений
других классов существенно отличаются оловоорганические гидриды и
соединения, содержащие два или более атомов металла в молекуле.
Оловоорганические гидриды обычно получают восстановлением
соответствующих галогенидов литийалюминийгидридом или гидридами диал-
Введение
171
килалюминия:
R„SnX4_n + L1AIH4 -* RnSnH4_n + LiX + А1Х3;
R„SnCl4_„ + R;A1H -» R„SnH4_„ + ^AIX
(я = 1, 2, 3).
Гидриды олова широко применяются для синтеза различных типов олово-
органических соединений. Помимо алкенов и алкинов (см. стр. 167), они
присоединяются к альдегидам и кетонам с образованием алкоксисоединений
олова:
R3SnH + R2CO-> R3SnOCHR2.
Присоединением оловоорганических гидридов к азометинам, азо- и нитро-
зосоединениям и арилизоцианатам получены соединения со связью Sn—N:
rt-CH8CeH4N=CHCeH5 + HSnR8^ /i-CH3CeH4N (SnR3)— GH2CeH5f
ArN=NAr + HSnR8 -» ArN (SnR3)—NHAr,
ArNO + HSnR3 -» ArNHOSnRg,
О
ArN-C-0 + R3SnH -» R3SnN (Ar) C-H.
Оловоорганические гидриды вступают также в реакции присоединения к
алкилизоцианатам и арилизотиоцианатам
R3SnH + AlkN-C-0 -» R3SnC (О) NHAlk,
R3SnH + ArN=C=S -» R3SnSCH = NAr.
Высокая подвижность связи олова с водородом в оловоорганических
гидридах позволяет широко использовать их в синтезе соединений со связью
олово — олово. Примером могут служить следующие реакции:
2R2SnH2 + 2R'COOH -» R'C003nR2SnR2OCR' + 2Н2,
nR2SnH2 > (R2Sn)„ + n H2,
2R3SnH + R2SnO -» R3SnSnR2SnR3 -f H20.
Взаимодействием оловоорганических гидридов с аминрпроизводными
германия, титана или циркония получены соединения со связью Sn—Ge,
Sn—Ti и Sn—Zr соответственно, например:
(C4H9)3 GeN (CH3)2 + (QH9)2 SnH2 -» (C4H9)3 GeSn (QH9)2 H + (CH3)2 NH.
Оловоорганические соединения использованы и для синтеза соединений со
связью Sn—Zn, Sn—Mg или Sn—Cd:
C2H5MgBr.2 (C2H5)2 О + (C6H5)3 SnH -» (С6Н5)з SnMgBr + C2H6.
Другая область применения оловоорганических гидридов основана на
их восстановительных свойствах. По сравнению с другими восстановителями
оловоорганические гидриды имеют некоторые преимущества, что позволяет
использовать их в препаративной органической химии, например при
восстановлении галоидных алкилов или арилов:
R3SnH + R'X ~> RsSnX + R'H.
172
Оловоорганические соединения. Лит, стр. 175—180
Альдегиды и кетоны гладко восстанавливаются оловоорганическимн
гидридами до соответствующих спиртов:
\ \
2R3SnH + C=0-»R3SnSnR8 + CHOH,
/ /
\ \
R2SnHa + C=0-*(R2Sn)/1 + CHOH,
/ /
\ \
RSnH3 + С=0 -> (RSn)„ + СНОН.
За последнее время все большее внимание исследователей привлекают
оловоорганические соединения, содержащие два и более атомов металла
в молекуле. Примером могут служить соединения типа R3SnM и R2SnM2
(где М — щелочной металл). Они образуются при действии щелочных
металлов на оловоорганические галогениды. По другому методу двухлористое
олово обрабатывают тремя молями ал кил- или ариллития:
SnCl2 + 3RLi -» RsSnLi + 2LiCl.
В индивидуальном состоянии литий- и натрийоловоорганические
соединения обычно не выделяют, а используют в виде растворов (окрашенные
растворы). Прибавлением к ним галоидных алкилов (или арилов) или га-
логенидов различных металлов получены соединения типа R3SnR' или
соответствующие биэлементоорганические производные олова:
2R3SnLi + RgSiCU -* RsSnSiRgSnRs + 2ЫС1,'
2РЬС12 + 4R3SnLi -» (R3Sn)4 Pb + Pb + 4LIC1,
3R3SnLi + SbCl3-* (R3Sn)3Sb + 3UC1.
Значительный интерес представляют соединения, в которых атом олова
связан с карбонилами молибдена, вольфрама, хрома и других металлов
VI—VIII групп периодической системы. Их получают по реакции
R3SnCl rf- NaM (CO)3 C5H5 -* R3SnM (CO)3 C5H5 + NaCl.
Оловоорганические соединения, в которых два атома металла разделены
углеводородными радикалами, получают реакцией Гриньяра
R'nMCH3CH3MgX + R3SnX -* R3SnCH2CH2MR^ -f MgX2
или присоединением оловоорганических гидридов к алкенильным или ал-
кинильным соединениям металлов. Второй метод позволил получить ряд
биметаллических производных этилена
R3SnH + RnMC=CH -> R3SnCH=CHMR/l
(M=Si, Ge, Sn, Sb).
Известен ряд реакций, приводящих к изменению в органическом
радикале у атома олова. Обычно исходят из оловоорганических соединений,
уже содержащих галоид, нитрильную или сложноэфирную группу в
органическом радикале. При этом действие электрофильных или нуклеофиль-
ных реагентов на оловоорганические соединения с заместителями у а-угле-
родного атома приводит к отщеплению замещенного радикала. Однако
если функциональные группы находятся у (3-углеродного атома или дальше,
то они легко вступают в характерные для них реакции, в то время как ос-
Введение
173
тальная часть молекулы остается незатронутой. Например:
LiAlH4
R3SnCH2CH2CN > R3SnCH2CH2CH2NH2;
LiAlH4
R3SnCH2CH2COOCH3 * R35nCH2CH2CH2OH;
RMgX
R3SnCH2CH2CN > R3SnCH2CH2COR.
Более сложно проходят реакции омыления оловоорганических эфиров
или нитрилов:
NaOH
R3SnCH2CH2CN > R3SnCH2CH2COONa,
HCI
R3SnGH2CH2COONa > [R3SnCH2CH2COOH] -* R2Sn+CH2CH2COCr.
Менее характерны реакции присоединения к винильным или аллильным
соединениям олова. Последние могут присоединять полисульфид водорода,
тиолуксусную кислоту, полигалоидметаны, а также вступают в реакции
типа реакции Дильса—Альдера с циклонами. Соединения, содержащие
тройную углерод-углеродную связь, присоединяют трифенилметильные
радикалы, диазометан и циклоны. Например:
QH5
|
П.-А-^Ч
Н-,Сб—\/
1
SnR3
1
С
=0+|||
с
1
SnR3
СвН3
|
H.Q-^-SnRs
H,Q—\/—SnR3
1
СвН3
Однако при действии галоидов или галоидоводородов на алкенильные или
алкинильные соединения олова наблюдаются только реакции деалкилиро-
вания. Также неудачными оказались попытки каталитического
гидрирования этих веществ.
Что касается реакций, приводящих к изменению в ароматическом
радикале у атома олова, то, например, окисление трифенил-д-оксиметилфенил-
олова приводит к соответствующей кислоте:
(С3Ч3)з SnC34iC i20Л + KMnDi -» (CGH3)3 SnC3H4GOOK -> (QH,)3 SnQHjCOOH.
Соединения, содержащие в ароматическом радикале аминогруппу, легко
вступают в реакции сочетания с солями диазония:
R3Sn~C=/~N (СНЗ)2 + X~C=/'~N2+> "^ ^SU-\^/~N (С113)2 + Н(+)
N-=N—^ у— X
Попытки непосредственного введения заместителей в ароматическое ядро
обычно не приводят к цели, а происходит отщепление ароматического
радикала от атома олова.
Хотя в задачу книги и не входит описание отдельных типов
оловоорганических соединений, мы считаем целесообразным поместить здесь краткую
их характеристику. В настоящее время известны следующие классы
соединений: R4Sn, R3SnX, R2SnX2, RSnX3, R3SnSnR3 и (R2Sn)n. Здесь R —
алифатический, ароматический или гетероциклический радикал; X —
остаток минеральной или органической кислоты, алкокси-, меркапто-,
амино- или другая функциональная группа. Кроме того, X может
представлять собой водород, щелочной металл или какую-либо элементоорганичес-
174
Оловоорганические соединения» ЛиШ. ctnp. 175—180
кую группу (R3Si, R3P, R3As и др.), связанную с оловом как
непосредственно, так и через гетероатом (О, S, Se и др.).
Номенклатура оловоорганических соединений
«С 1 , О) _
Ч Ли о «
>>° £ ° 5
s о f к
1оё2£
e§sS&
1
RSnH3
R2SnH2
RsSnH
R2SnHCl
RSnX3
R2SnX2
R3SnX
R4Sn
RSnOOH
R2SnO
R3SnOSnR3 1
R3SnOH
R3SnLi
R2SnLi2
R2Sn
R3SnSnR3
RsSnOR
R3SnOOCR
RsSnSR
R3SnNR2
Название, принятое
в настоящей книге
2
Тригидрид алкилоло-
ва
Дигидрид диалкил-
олова
Гидрид триалкилоло-
ва
(Хлор)гидрид диал-
килолова
Трехгалоидное ал-
килолово
Двугалоидное диал-
килолово
Галоидное триалкил-
олово
Тетраалкилолово
Алки летанноновая
кислота
Окись диалкилолова
Гексаалкилдистан-
ноксан
Гидроокись триал-
килолова
Триалкилстаннил-
литий
Диалкилстаннилен-
дилитий
Диалкилолово
Гексаалкилдистаннан
Алкокситриалкил-
олово
Ацилат триалкилоло-
ва
Алкилмеркаптид три-
алкилолова
(Диалкиламино)три-
алкилолово
Другое,
встречающееся в русской
литературе название
3
-
—
Триалкилстаннан
Диалкилхлорстаннан
Алкилтригалоген-
станнан, алкилолово-
тригалогенид
Диалкилдигалогсн-
станнан
Триалкилгалоген-
станнан
Тетраалкилстаннан
—
Диалкилстаннон
Окись триалкилоло-
ва, бис-(триалкило-
лово)окись
Триалкилстаннол
—
Дилитийдиалкилоло-
во, диалкилоловоди-
л итий
—
Триаллилалкокси-
станнан
. —
-
Диалкил(триалкил-
станнил)амин
Английское
название
1 4
Alkyltin trihydride,
Alkylstannane
Dialkyltin dihydride,
Dialkilstannane
Trialkyltin hydride,
Trialkylstannane
Dialkylchlorotin
hydride
Alkyltintrihalogenide
Dialkyltin dihalogenid
Trialkyltin ha'ogeni-
de, Trialkylchlorstan-
nane *
Tetraalkyltin
Alkylstannonic acid
Dialkyltin oxide
Bis(trialkyItin)oxide,
Hexaalkyldistan-
noxan, Trialkyltin
oxide
Trialkyltin hydroxide
Trialkyltinlithium,
Trialkylstannillithium
Dialkylstannylendilit-
hium
Dialkyltin
Hexaalkylditin, Hexa-
alkyldistannan
Trialkyltin alkoxide,
trialkylalkoxytin
Trialkyltin carboxyla-
te
Trialkyltin alkylmer-
captides
Trialkyl(dialkylami-
no)tin, (dialkylamino)
trialkylstannane
Немецкое
название
5
Alkylzinntrihydrid-
Dialkylzinndihyd-
rid
Trialkylzinnhydrid
Dialkylchloroztnn-
hydrid
Alkylzinntrihalo-
genid
Dialkylzinndihalo-
genid, Dialkyldi--
halogenstannan
Trialkylzinnhalo-
genidj
Tetraalkylzinn
Alkylstannonsaure-
Dialkylzinnoxid
Hexaalkyldistan-
noxane
Trialkilzinnhyd-
roxyde
Lithiumtrialkyls-
tannan, Trialkyl-
zinnlithium
Dilithiumdialkyl-
stannan
Zinndialkyl
Hexaalkyldistan-
nan
Trialkylzinnalko-
xid
—
-
Trfalkylzinndial-
kylamide
Тетраалкильные соединения олова — бесцветные жидкости, тетра-
арильные — твердые вещества. Все они устойчивы к действию воды и к
окислению. Однако введение электрофильных групп ка-углеродному атому
поляризует связь Sn—С, снижая тем самым ее устойчивость к действию
воды. Например, тетрафенилолово при нагревании с раствором щелочи
практически не изменяется, тогда как триэтилстаннилацетон с
саморазогреванием реагирует с водой, давая гидроокись триэтилолова и ацетон.
Галогениды триалкилолова (кроме фторидов) — жидкости с
характерным резким и неприятным запахом, галогениды триарилолова —
высокоплавкие кристаллы. Дигалогениды диалкил- и диарилолова относительно
низкоплавки, прекрасно растворимы в обычных органических
растворителях. Тригалогениды алкил- и арилолова являются жидкостями. Что ка-
Введение
175
сается оловоорганических фторидов, то они представляют собой
нерастворимые неплавкие порошки белого цвета.
Большинство оловоорганических соединений типа R3SnX, R2SnX2 и
RSnX3 не окисляются на воздухе. Их гидролитическая устойчивость
уменьшается с уменьшением числа органических радикалов у атома олова. Га-
логениды олова водой практически не гидролизуются. Напротив, многие
оловоорганические ацилаты, алкоксисоединения, перекиси.и амины
гидролизуются даже влагой воздуха. В этом отношении от соединений других
классов резко отличаются оловоорганические гидриды. Последние
инертны к действию воды, но легко окисляются кислородом воздуха.
Окиси бас-(триалкилолова), образующиеся при гидролизе соединений
типа R3SnX щелочью, представляют собой прозрачные жидкости, которые
легко очищаются перегонкой в вакууме. Гидроокиси триарилолова и окиси
бис- (триар ил олова) — кристаллические вещества, хорошо растворимые в
обычных органических растворителях. Напротив, окиси диалкил- и диарил-
олова — белые нерастворимые и неплавкие порошки, имеющие полимерный
характер. Ал кил- и арилстанноновые кислоты — также полимерные
продукты, но они растворяются в низших алифатических спиртах и
концентрированных щелочах.
Из соединений со связью Sn—М наиболее подробно изучены
соответствующие производные лития и натрия типа R3SnM и R2SnM2, а также вещества,
в основе которых лежит цепочка из атомов олова (например, R3SnSnR3>
(R2Sn)7l, R3Sn (SnR2)7lSnR3 и др.). Их характерной чертой является высокая
активность связи олово — олово к действию различных окислителей
(кислород, сера, галоиды, тетраацилаты свинца и др.). При нагревании гекса-
алкил(арил)дистаннанов или соединений типа (R2Sn)n они легко диспро-
порционируют с выделением R4Sn и металлического олова. В этой связи
обращает на себя внимание устойчивость связи олова с хромом,
молибденом, рением и другими металлами VI — VIII групп периодической
системы. Даже при действии на них галоидов или галоидоводородов эта связь
остается не затронутой, а происходит отщепление органического радикала
от атома олова.
Химии оловоорганических соединений посвящен ряд обзорных статей
[1, 195—201]. Кроме того, есть обзорные статьи по отдельным классам
оловоорганических соединений: гидридам [202—206], бис- и полиэлементоор-
ганическим соединениям олова [207—210], соединениям со связью Sn— N
[211, 211а], оловосилоксанам [212], оловоорганическим перекисям [213],
соединениям с различными заместителями в органическом радикале у
атома олова [214], а также оловоорганическим комплексам [215, 216] и
полимерным материалам,содержащим олово [217—223]. В статье Грожан с сотр. [2241
рассмотрен механизм и стереохимия нуклеофильного замещения у атома
олова.
ЛИТЕРАТУРА
1. Ингам Р., Розенберг С, ГилманГ., РикенсФ.,
Оловоорганические и германийорганические соединения. М., ИЛ, 1962.
2. VogelA. I., С г ess we 1 1 W. Т., Leicester J., J. Phys. Chem., 58, 174
(1954).
3. С u m m i n s R. A., D u n n P., Austral. J. Chem., 17, 411 (1964).
4. S а у r e R., J. Chem. and Engng. Data, 6, 560 (1961).
5. Л а п к и н И. И., Д v м л е р В. А., Уч. зап. Пермск. ун-та, 111, 190 (1964).
6. CresswellW. Т.," L e i с es t e r J., VogelA. I., Chem. a. Ind., 1953. 19.
7. MarrotJ., Ma ire J.— С, С a s s a n J., Compt. rend., 200, 3931 (1965).
8. П е т у x о в В. А., Миронов В. Ф., Шорыги н П.П., Изв. АН СССР, ОХН,
1964, 2203.
9. Hague D. N., Prince R.H., J. Chem. Soc, 1S65, 4690.
176
Оловоорганические соединения
10. Д у б о в С. С, С т р у к о в О. Г., Ж- Всесоюзн. хим. общ. им. Д. И. Менделеева,
8, 699 (1963).
11. Л а п к и н И. И., Д у мл ер В. А., Уч. зап. Пермск. ун-та, 111, 185 (1964).
12. D г е n t h W., J anssenM. J., van der К e r k G. J. M., J. Organometal.
Chem., 2, 265 (1964).
13. N e u m a n n W. P., KonigK., Ann., 677, 1 (1964).
14. BowdenK., BraudeE.A., J. Chem. Soc, 1952, 1068.
15. LaPagliaS. R., J. Molec Spectrosc, 7, 427 (1961).
16. G г i f f i t h s V. S., DerwischG.A.W., J. Molec. Spectrosc, 7, 233 (1961).
17. W a d a M., К a w a k a m i К., О k a w a r a R., J. Organometal. Chem., 4, 159
(1965).
18. DaveL. D., Evans D. F., Wilkinson G., J. Chem. Soc, 1959,3684.
19. Горшкова Г. Н., ЧубароваМ. А., СладковА. М., Лунева Л. Км
Касаточкин В. И., ЖФХ, 39, 2695 (1965).
20. Okawara R., J. Chem. Soc Japan, Pure Chem. Sec, 86, 543 (1965); РЖХим.,
1966, 8Б141.
21. A d d i s о n С. С, SimpsonW.B., W a 1 k e r A., J. Chem. Soc, 1964, 2360.
22. Youn'gCW., Kochler J. S., McKinney D.S., J.Am. Chem. Soc,
69, 1410 (1947).
23. L i p p i n с о t t E. P., TobinM.C, J. Am. Chem. Soc, 75, 4141 (1953).
24. D i 1 1 a r d C. R., LawsonJ. R., J. Opt. Soc America, 50, 1271 (1960).
25. Вышинский Н. Н., Рудневский Н.К., Оптика и спектроскопия, 10,
797 (1961).
26. Taimsalu P., WoodJ.L, Trans. Farad. Soc, 59, 1754 (1963).
27. CumminsR.A., Austral. J. Chem., 16, 985 (1963).
28. С u m m i n s R. A., Austral. J. Chem., 18, 985 (1965).
29. Kawasaki Y., T a n a k а Т., Okawara R., J. Organometal. Chem., 6,
97 (1966).
30. Mendelsohn J., MarchandA., ValadeJ., J. Organometal. Chem.,
6, 25 (1966).
31. Яковлева Т. В., Петров А. А., ЗавгороднийВ. С, Оптика и
спектроскопия, 12, 200 (1962).
32. Е г о р о в Ю. П., К и р е й Г. Г., ЖОХ, 34, 3615 (1964).
33. Ч у м а е в с к и й Н. А., ДАН СССР, 141, 168 (1961).
34. Schwartz W. Т., Р о s t H. W., J. Organometal. Chem., 2, 357 (1964).
35. О б р е и м о в И. В., Ч у м а е в с к и й Н. А., Ж- структурн. химии, 5, 59
(1964).
36. Ч у м а е в с к и й Н. А., Бор и'с о в А. Е., ДАН СССР, 161, 366 (1965).
37. 3 а в г о р о д н и й В. С, П е т р о в А. А., ДАН СССР, 143, 855 (1962).
38. ШигоринД. Н., ГастиловичЕ. А., Комаров Н. В., Труды
Комиссии по спектроскопии АН СССР, 1, 673 (1964).
39. ШостаковскийМ. Ф., Шергина Н. И., Голованова Н. И.,
Комаров Н. В., Б р одска я Э. И., М и с ю н а с В. К-> ЖОХ, 35, 1768
(1965).
40. Н е п г у М. С, N о 1 t e s J. G., J. Am. Chem. Soc. 82, 555 (1960).
41. Стерли н P. H., Дубов С. С, Ж- Всесоюзн. хим. общ. им. Д. И. Менделеева,
7, 117 (1962).
42. Гастилович Е. А., ШигоринД. Н., Комаров Н. В., Оптика и
спектроскопия, 16, 46 (1964).
43. О к к a b а X., J. Chem. Soc. Japan., Pure Chem. Sec, 84, 1 (1963); РЖХим., 1964,
9Б138.
44. G r i f f i t h s V. S., Der wish G. A. W., J. Molec. Spectrosc, 11, 81 (1963).
45. К a e s z H. D., S t о n e F. G. A., Spectrochim. acta, 1959, 360.
46. L о h m a n n D. H., J. Organometal. Chem., 4, 382 (1965).
47. T a i m s a 1 u P., WoodJ.L., Spectrochim. acta, 20, 1043 (1964).
48. Вышинский Н. H., Рудневский Н. К-, Сб. «Спектроскопия». М.,
«Наука», 1964, стр. 115.
49. Л и в ш и ц Б. Л., Ч у м а е в с к и й Н. А., Ж- структурн. химии, 5, 140 (1964).
50. Л е й т е с Л. А., П а в л о в а И. Д., Егоров Ю. П., Теорет. и эксперим.
химия, 1, 311 (1965).
51. В о л к о в В. Ф., Вышинский Н. Н., Рудневский Н. К., Изв. АН
СССР, серия физ., 26, 1282 (1962).
52. G r i f f i t h s V. S., Derwish G. A. W., J. Molec. Spectrosc, 9, 83 (1962).
53. С u m m i n s R. A., E v a n s J. V., Spectrochim. acta, 21, 1016 (1965).
54. Вышинский Н. Н., Александров Ю. А., Рудневский Н. К-,
Изв. СССР, серия физ., 26, 1285 (1962).
•55. В u t с h е г F. К., Gerrard W., М о о п е у Е. F., Rees R. G., W i 1 -
1 i s H. A., Spectrochim. acta, 20, 51 (1964).
Введение
177
56,/Lerndelsohn J'» March and A., ValadeJ., Compt rend., 261, 135
(1965).
57 К aw a s а к i Y., T a n а к а Т., OkawaraR, Bull. Chem. Soc. Japan.,
37, 903 (1964). F
58',^?Jh is"Noel R-. LesbreM., de Roch I. S. Compt. rend., 243, 257
(1956).
*л' £ \ [ eA s m a n n H'» Ulbricht K., Z. anorg. allgem. Chem., 328, 90 (1964).
60. E d gel 1 W. F., Ward С H., J. Moiec. Spectrosc., 8, 343 (1962).
61. Kawasaki Y., К a wa к am i К., Т ana к а Т., Bull. Chem. Soc. Japan,
38, 1102 (1965).
^F/iebeE., Kelker H., Z. analyt. Chem., 192, 267 (1963).
Ы. Ma lr e J.-C, Cass an J., Lepretre В., Mar rot J., Compt. rend..
2j60, 5290 (1965).
64'4M50nd°X M* L*' FlitcroftN-' KaeszH. D., J. Organometal. Chem.,
65. p i 1 1 a r d С R., M а у L., J. Molec. Spectrosc, 14, 250 (1964).
nocJ!pincott E,R'' MercierP., TobinM.C, J. Phys. Chem., 57, 939
(1953).
67. Domange L., Guy J., Ann. pharm. franc, 16, 161 (1958); С. А., 52, 19451
(1958).
68,/?^L?wn M- R> OkawaraR., RochowE.G, Spectrochim. acta, 16, 595
(1960).
69. В г о w n D. H., M о h a m m e d A., S h a r p D. W. A., Spectrochim. acta, 21, 1013
(1965).
70. В u t с h e r F. K., G e r r a r d W., M о о n e у E. F., R e e s R. G., Willis H. A.,
AndersonA., Gebbic H. A., J. Organometal. Chem., 1, 431 (1964).
11. Александров Ю. А., ВышинскийН. Н. Труды по химии и хим.
технологии (г. Горький), вып. 3, 1961, 656.
72. М а г с h a n d A., M e n d е 1 s о h n J., V а 1 a d e J., Compt. rend., 259, 1737 (1964).
7d. С 1 а г к Н. С, О'В г i e n R. J., Inorg. Chem., 2, 740 (1963).
li' £J с G г а d У м- М-> Т о b i a s R. S., J. Am Chem. Soc, 87, 1909 (1965).
ik n 3 * h a W a У B* J" W e b s { e r D* E" Proc* Chem' Soc>' 1963> 14*
/о. Вы шинский Н. Н. Труды по химии и хим. технологии (г. Горький), вып. 1,
19оЗ, 18.
77. Р о 1 1 е г R. С, J. Inorg. a. Nucl. Chem., 24, 593 (1962).
78. F r i t z H. P., F i s с h e r E. O., J. Chem. Soc, 1961, 547.
79. В г о w n T. L., M о r g a n G. L., Inorg. Chem., 2, 736 (1963).
80. С u m m i n s R. A., Austral. J. Chem., 18, 98 (1965).
Si. P о 1 t e r P. E., P r a t t L., W i 1 k i n s о n G., J. Chem. Soc, 1964, 524.
82. В a r t о с h а В., В r i n с k m a n F. E., KaeszH.D., S t о n e F. G. A., Proc
Chem. Soc, 1958, 116.
83. E г о р о в Ю. П., Кир ей Г. Г., Лейтес Л.А., Миронов В. Фм Пет-
Р о в А. Д., Изв. АН СССР, ОХН 1962, 1880.
84. Е г о р о в Ю. П., Теор и эксперим. химия, 1, 30 (1965).
85. S i e b е г t H., Z. anorg. allgem. Chem., 263, 82 (1950).
86. SavidanL, Bull. Soc. Chim. France, 1953, 411.
8/. К у п р и е в и ч В. А., Е г о р о в Ю. П., Д я д ю ш а Г. Г., ДАН УССР, 1964, 508.
88. Е г о р о в Ю. П., Л е й т е с Л. А., М и р о н о в В. Ф., Ж. структуры, химии, 2,
562 (1У61).
89. С a h i 1 1 Р., В u t с h e r S., J. Chem. Phys., 35, 2255 (1961).
90. L i d e D. R., J. Chem. Phys., 19, 1605 (1951).
91. N e u m a n n W. P., P e t e r s e n E., SommerR, Angew. Chem., 77, 622 (1965).
92. Солодовников С. П., ЧернышевЕ. А., Труды Совещания по физическим
методам исследования органических соединений и химических процессов, 1962. Фрунзе,
«Илим», 1964, стр. 196.
93. G о г g у W., М с С о г m i с С. G., J. Am. Chem. Soc, 78, 3243 (1956).
94. В u r k e J. J., L a u t e r b u г Р. С, J. Am. Chem. Soc, 83, 326 (1961).
95. E г о р о ч к и н А. Н., ХидекельМ.Л., Р а з у в а е в Г. А., Изв. АН СССР,
ОХН, 1966, 437.
96. Е г о р о ч к и н А. Н., В я з а н к и н Н. С, Р а з у в а е в Г. А., К р у г-
л а я О. А., Б о ч к а р е в М. Н., ДАН, СССР, 170, 333 (1966).
97. S m i t h G. W., J. Chem. Phys., 42, 4229 (1965).
98. В г о w n M. P., Webster D. E., J. Phys. Chem., 64, 698 (1960).
99. F r i t z H. P., KreiterCG., J. Organometal. Chem., 4, 313 (1965).
100. MooreD.W., H a p p e J. A., J. Phys. Chem., 65, 224 (1964).
101. M о e d r i t z e r K., V a n W a z e r J. R., Inorg. Chem., 3, 943 (1964).
102. M a s s e у A. G., R a n d a 1 1 E. W., S h a w D., Chem. a Ind., 1963, 1244.
103. ClarkH.C, К w о n J. Т., R e e v e s L. W., W e 1 1 s E. J. N.. Canad. J. Chem.,
41, 3005 (1963); Inorg. Chem., 3, 907 (1964).
12 Заказ jsft 207
178 Оловоорганические соединения
104. MaireJ.-CHeramertF., Bull. Soc. chem. France, 1963, 2785.
105. F 1 i t с г о f t N.. KaeszH.D., J. Am. Chem. Soc, 85, 1377 (1963).
106. A m b e r g e r E., Fritz H. P., KreiterC.G., К u 1 a M.-R., Ber.. 96, 3270
(1963).
107. S i m m о n i n M.- P., Compt. rend., 257, 1075 (1963).
108. KulaM.R., KreiterC.G., L о r b e r t h J., Ber., 97, 1294 (1964).
109. L о r b e r t h J., KulaM.R., Ber., 97, 3444 (1964).
110. Van der KelenG. P., VerdonckL, van deVondelD., Bull. Soc.
chim. belg., 73, 733 (1964).
111. Ku 1 aM. R., A m b er ger E., R u p pr ech t H., Ber., 98, 629 (1965).
112. L or b er t h J., KulaM.R., Ber., 98, 520 (1965).
113. К u 1 a M. R., A m b e r g e г Е., М а у е г К. К., Ber., 98, 634 (1965).
114. V e r d о n с к L., V a n der К e 1 e n G. P., Bull. Soc. Chim. belg., 74, 361 (1965).
115. P a t i 1 H. R. H., G r a h a m W. A. G., J. Am. Chem. Soc, 87, 673 (1965).
116. KloseG., Ann. Phys. (DDR), 10, 391 (1963); РЖХим., 1964, 5Б181.
117. VI a di mi r of f Т., M a 1 i h о w s к i E. R., J. Chem. Phys., 42, 440 (1965).
118. VerdonckL, van der KelenG. P., Ber. Bunsenges phys. Chem., 69, 478
(1965); РЖХим., 1966, 10Б274.
119. Coy 1 eT. D., St afford S. L., S t о n e F. G. A., Spectrochim. acta, 17, 96#
(1961).
120. Петровская Л. И., БурлаченкоГ. С, Федин Э. И., БауковЮ.И.,
ЛуценкоИ. Ф., Ж- структуры, химии, 6, 781 (1965).
121. Петров А. А., Л еб еде в В. Б., ЖОХ, 32, 657 (1962).
122. Н е г b е г R. Н., S t б с к 1 е г Н. A., Trans. N. Y. Acad. Sci, 26, 929 (1964).
123. G i b b Т. С, G г е е n w о о d N. N., J. Chem. Soc, 1966A, 43.
124. К а р а с е в А. Н., Колобова Н. Е., П о л а к Л. С, Ш п и н е л ь В. С, А н и-
с и м о в К- Н., Теор. и эксперим. химия, 2, 126(1966).
125. ГольданскийВ. И., БоршаговскийБ. В., Макаров Е. Ф., С т у-
• к а н Р. А., А н и с и м о в К-Н., Колобова Н. Е., СкрипкинВ. В. Тео-
рет.и эксперим. химия, 3, 478 (1967).
126. Гольданский В. И., Городи нскийГ.М., К а р я г и н С. В., Корыт-
к о Л. А., КрижанскийЛ. М., Макаров Е. Ф., С у з д а л е в И. П., X ра-
п о в В. В., ДАН СССР, 147, 127 (1962).
127. ГольданскийВ.И., Р о ч е в В. Я., X р а п о в В. В., ДАН СССР, 156, 909
(1964).
128. Брюханов В. А., ГольданскийВ. И., Деляг и н Н. Н., Корыт-
к о Л. А., М а к а р о в Е. Ф., С у з д а л е в И. П., Ш п и н е л ь В. С, Ж- эксперим.
итеор. физ., 43, 448(1962).
129. GolDanskiiV. I., Okhlobystin О. Yu., R о с h e v V. Ya., К h r a
р о v V. V., J. Organometal. Chem., 4, 160 (1965).
130. Александров А. Ю., Б р е г а д з е В. И., Г о л ь д а н с к и й В. И., 3 а-
х а р к и н Л. И., О х л о б ы с т и н О. Ю., X р а п о в В. В., ДАН СССР, 165, 593
(1965).
131. Н е г b е г R. Н., S t б с к 1 е г Н. A., R e i с hi e W. Т., J. Chem. Phys., 42, 2447
(1965).
132. Epstein L. M., Stranb D., Inorg. Ghem., 4, 1551 (1965); Her ber R. H,
Parisi G. J., Inorg. Ghem., 5, 769(1967).
133. Александров А. Ю., ДелягинН. Н., Митрофанов К. П., По-
л а к Л. С, Ш п и н е л ь В. С, ДАН СССР, 148, 126 (1963).
134. А л е к с а н д р о в А. Ю., ДелягинН.Н., Митрофанов К. П., По-
л а к Л. С, Ш п и н е л ь В. С, Ж. эксперим. и теор. физ., 43, 1242 (1962).
135. Александров А. Ю., ДорфманЯ. Г., ЛепендинаО. Л.,
Митрофанов К. П., Плотникова М. В., ПолакЛ. С, ТемкинА. Я.Дпи-
н е л ь В. С, ЖФХ, 38, 2190 (1964).
136. Александров А. Ю., Охлобыстин О. Ю., П о л а к Л. С, Ш п и-
н е л ь В. С, ДАН СССР, 157, 934 (1964).
137. Шпинель В. С, Александров А. Ю., Рясный Г. К., О х л о б ы с-
т и н О. Ю., Ж- эксперим. и теор. физ., 48, 69 (1965).
138. Н а у е s M. С, J. Inorg. Nucl. Chem., 26, 2306 (1964).
139. S w i g e r E. D., G г а у b e a 1 J. D., J. Am. Chem. Soc, 87, 1464 (1965).
140. D i b e 1 er V. H., J. Res. Natl. Bur. Standards, 49, 235 (1952).
141. M a 1 у J., T e p 1 у J., Chem. Zvesti, 7, 553 (1953).
142. T e p 1 у J., M a 1 у J., Chem. Zvesti, 7, 463 (1953).
143. W h e a t 1 e у P. J., J. Chem. Soc, 1961, 5027.
144. К i 1 b о u r n В. Т., Р о w e 1 1 H. M., Chem. a. Ind., 1964, 1578.
145. С 1 a r k H. С, ОЧВ r i e n R. J., T г о t t e r J., J. Chem. Soc, 1964, 2332.
146. MendelH., Acta crystalogr., 15, 9 (1962).
147. ИсмаилзадеИ. Г., Кристаллография, 3, 155 (1958).
Введение
179
148. ИсмаилзадеИ. Г., ЖФХ, 26, 1139 (1959).
149. Ж Д а н о в Г. С, И с м а и л з а д е И. Г., ДАН СССР, 68, 95 (1949).
150. Ж Д а н о в Г. С, И с м а и л з а д е И. Г., ЖФХ, 24, 1495 (1950).
151. V о i g t D., LesbreM., GallaisF., Compt. rend., 239, 1485 (1954).
152. К a d о m t z e f f I., Compt. rend., 230, 536 (1950).
153. К a d о m t z e f f I., Bull. Soc. Chim. France, 1949, D394.
154. A b e 1 E. W., BushR.P., JenkinsCR., Z о b e 1 Т., Trans. Farad. Soc, 60,
1214 (1964).
155. Д о р ф м а н Я. Г., ДАН СССР, 125, 765 (1959).
156. HuangH. H., HuiK.M., J. Organometal. Chem., 2, 288 (1964); 6, 504 (1966).
157. WeissE., Z. anorg. allgem. Chem., 287, 236 (1956).
158. StrohmeierW., von HobeD.,Z. Elektrochem., 64, 945 (1960).
159. Петров А. А., МингалеваК. С, ЗавгороднийВ. С, ЖОХ, 34, 533
(1964).
160. L о г b e r t h J., N б t h H., Ber., 98, 969 (1965).
161. ГольдштейнИ. П., Гурьянова Е. Н., ЗемлянскийН. Н., С ю т-
к и н а О. П., П а н о в Е. М., К о ч е ш к о в К. А., ДАН СССР, 175, 836 (1967).
162. М а 1 a t e s t a L., P i z z о t t i R., Gazz., 73, 344 (1943).
163. P e а с h M. E., W a d d i n g t о п Т. С, J. Chem. Soc, 1961, 1238.
164. T h о m a s А. В., R о с h о w E. G., J. Am. Chem. Soc, 79, 1843 (1957).
165. T h о m a s А. В., R о с h о w E. G., J. Inorg. Nucl. Chem., 4, 205 (1957).
166. M о r t i m e г С. Т., J. Chem. Educ, 35, 381 (1958).
167. DillardCR., McNeillE.H., SimmonsD.E, Y e 1 d e 1 1 J. В., J. Am.
Chem. Soc, 80, 3607 (1958).
168. P e d 1 e у J. В., S k i n n e r H. A., ChernickCL, Trans. Farad. Soc, 53, 1612
(1957).
169. P о р е Е. A., SkinnerH.A., Trans. Farad. Soc, 60, 1402 (1964).
170. T e л ь н о й В. И., Р а б и н о в и ч И. Б., ЖФХ, 39, 2076 (1965).
171. Баландин А. А., КлабуновскийЕ. И., Козина М. П.,
Ульянова С. Д., Изв. АН СССР, ОХН, 1958, 12.
172. Р е d 1 е у J. В., S k i n n e r H. A., Trans. Farad. Soc, 55, 544 (1959).
173. N a s h G. A., S к i n n e r H. A., S t а с к W. F., Trans. Farad. Soc, 61, 640 (1965).
174. HugelG., Kolloid-Z., 131, 4 (1953).
175. H e ф е д о в В. Д., В а р ш е в Н. А., ЖФХ, 2Ь, 981 (1954).
176. В a m b у п е к W., F r e i s e V., Z. phys. Chem., 7, 317 (1956).
177. Р г i n с е R. H., J. Chem. Soc, 1959, 1783.
178. J a n s s e n M. J., L u i j t e n J. G. A., Rec trav. chim., 82, 1008 (1963).
179. W e s t R., В a n e у R. H., P о w e 1 1 D. L., J. Am. Chem. Soc, 82, 6269 (1960).
180. В a m b у n e к W., F r e i s e V., Z. Naturforsch., 11a, 168 (1956).
181. В а к e r A. G., M о r r i s W. C, Rev. socient instrum., 32, 458 (1961); РЖХим., 1962,
1Б650.
182. С 1 a r i d g e R. F. С, M e r z E., R e i d e 1 H. J., Nucleonic, 7, 53 (1965).
183. НазароваЛ.М, ЖОХ, 31, 1119 (1961).
184. ЦиглерК,ЛемкульГ, Пат. СССР 132136 (1960); РЖХим., 1962, 9Л108.
185. Z i e g 1 е г К., Англ. пат. 814609 (1959); С. А., 53, 17733 (1959).
186. ZieglerK.,Lehmkuhl Н. Пат. ФРГ 1127900 (1962); РЖХим., 1964, ЗН103.
187. Z i e g 1 е г К. Англ. пат. 848364 (1960); С. А., 55, 5199 (1961).
188. KobetzP., PinkertonR. Пат. США 3028320 (1962); РЖХим., 1963, 15Н58.
189. R о е t h е 1 i В. Е., S i m p s о п I. В., Англ. пат. 797093 (1958); С. А., 53, 930(1959).
190. G i r a i t i s A. P. Пат. ФРГ 104617 (1958); С. А., 55, 383 (1961).
191. T о м и л о в А. П., К а а б а к Л. В., ЖПХ, 32, 2600 (1959).
192. К а а б а к Л. В., Т о м и л о в А. П., ЖОХ, 33, 2808 (1963).
193. Т о м и л о в А. П., С м и р н о в Ю. Д., В а р ш а в с к и й С. Л., ЖОХ, 35, 391
(1965).
194. К о р о т а е в с к и й К. Н., ЛысенкоЕ. Н., С м о л я н 3. С, М о н а с т ы р-
с к и й Л. М., А р м е н с к а я Л. В., ЖОХ, 36, 167 (1966).
195. Van der К е г k G. J. M., L u i j t e n J. G. A., N о 1 t e s J. G., Angew. Chem.,
70, 298 (1958).
196. I n g h a m R. K., RosenbergS. D., GilmanH., Chem. Revs., 60, 459 (1960).
197. P e t г о v A. D., M i г о n о v V. F., Angew. Chem., 73, 59 (1961).
198. Van der К e r k G. J. M., L и i j t e n J. G. A., van E g m о n d J. C, N ol -
t es J.G., Chimia, 16, 36(1962).
199. N e и m a n n W. P., Angew. Chem., 75, 225 (1963).
200. А з е р б а е в И. Н., К о ч к и н Д. А., Вестн. АН КазССР, 10, 18 (1963).
201. L u i j t e n J. G. A., Chem. end techn. rev., 20, 693 (1965); РЖХим., 1966, 5Ж358.
202. Neumann W. P., Angew. Chem., 76, 849 (1964).
203. KuivilaH., Advances in Organometallic chemistry, U 47 (1964).
204. S e v e s t r e J., Peinture, Pigment, vernis, 41, 278 (1965).
12*
180
Оловоорганические соединения
205. Van d е г К е г к G. J. М., N о 1 t e s J. G., Ann. N. Y. Acad. Sci., 125, 25 (1965).
206. ИмотоХидэдзи, Chemistry (Japan), 14, 860 (1959); РЖХим., 1961, 12Ж61.
207. В я з а н к и н Н. С, К р у г л а я О. А., Усп. хим., 35, 1388 (1966).
208. Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Усп. хим., 35, 1430 (1966).
209. W i t t e n b e r g D., G i 1 m a n H., Quart. Rev., 13, 116 (1959).
210. GilmanH., Bull. Soc. Chim. France, 1963, 1356.
211. J о n es K., L a p p er t M. F., Organometal. Chem. Rev., 1, 67 (1966).
211a. Lu i j t e n J. G. A., R i j kens F., van d er К er k E. J. M. Advance Organo-
metall. Ghem., vol. 3, New York —London, Acad. Press, 1965, 397.
212. SchmidbaurH., Angew. Chem., 77, 206 (1965).
213. К a r n о j i t z k у V., Chimie et industrie, 85, 92 (1961).
214. NoltesJ.G., van derKerkG. J. M., Functionally Substituted Organotin
Compounds, Tin Research Institute, Middlesex, England, 1958.
215. P о 1 1 e r R. C, J. Organometal. Chem., 3, 321 (1965).
216. CroattoU., В a r b i e r i R., Ricerca scient., 2A, 8, 441 (1965); РЖХим., 1966,
12B189.
217. MarkH.F, Chimia, 18, 270 (1964).
218. S p e n g 1 e r G., К о 1 1 M., Erdol und Kohle, 17, 107 (1964).
219. Aitkenl.D., SheldonR., S t a p 1 e t о n G. В., Brit. Plast., 34, 662 (1961).
220. Браун Д. В., Усп. хим., 31, 769 (1962).
221. BonnotJ., Rev. Inst, franc, petrole, 18, 284 (1963).
222. БонноИ, Усп. хим., 34, 850 (1965).
223. H e n г у М. С, D a v i d s о n W. E., Ann. N. Y. Acad. Sci., 125, 172 (1965).
224. GrosjeanM., GielenM., NasielskiJ., Ind. chim. beige, 28, 721 (1963).
Г лава I
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ ГАЛОИДНЫХ АЛКИЛОВ НА ОЛОВО,
ЕГО СПЛАВЫ ИЛИ СОЛИ ДВУХВАЛЕНТНОГО ОЛОВА
Метод получения оловоорганических соединений действием галоидных
алкилов на олово и его сплавы имел существенное значение в период
возникновения химии оловоорганических соединений. Однако в дальнейшем
этот путь не получил развития; только в последнее время интерес к прямому
методу значительно возрос в связи с тем большим значением, которое оло-
воорганические соединения приобрели в различных областях практического
применения.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ДЕЙСТВИИ ГАЛОИДНЫХ АЛКИЛОВ НА МЕТАЛЛ
Действие металлического олова на йодистые ал килы в некоторых
случаях на солнечном свету при обыкновенной температуре, а в иных случаях
при нагревании до 130—180° С приводит к смеси соединений типа R3SnJ
и R2SnJ2 [1—5].
В присутствии небольших количеств магния (или цинка) и спирта (или
тетрагидрофурана) указанная выше реакция проходит в более мягких
условиях [6—10]. Вероятно, роль этих добавок сводится к образованию
соединений типа ROMgJ, которые вместе с органическим растворителем
ускоряют реакцию [11]. Нагревание в автоклаве йодистого метила и фольги
олова в течение 2 час. в присутствии магния и метилового или я-бутилового
спирта приводит к смеси (CH3)3SnJ, (CH3)2 SnJ2 и CH3SnJ3 (выход 25—30,40—
46 и 20—27% соответственно) [6, 7]. Изменение соотношения реагентов,
количества добавок, температуры или продолжительности опыта не
оказывает существенного влияния на ход реакции [6, 12]. Аналогичным образом
из йодистого этила и олова получена [6, 12] смесь (С9Н5)35пЛ(выход 14—18%),
(C2H5)2SnJ2 (63—67%) и C2H5SnJ3 (3—6%).
С высшими йодистыми алкилами реакция проходит в основном в
сторону образования соединений типа R2SnJ2.
Получение двуиодистого ди-я-бутилолова [6]. Смесь 100 г фольги олова, 372 г йодистого
бутила, 0,3 г магния и 11,2 г бутилового спирта нагревают до 130—135° С при перемешивании
в течение 3 час. Смесь фильтруют и перегоняют в вакууме. Получают 381 г (95%)
двуиодистого ди-«-бутилолова ст. кип. 143—145° С/3 мм.
Следует отметить, что с увеличением длины алкильной группы выход
продукта уменьшается. Например, в случае двуиодистого ди-я-додецилоло-
ва он составляет только 59% [6].
Каталитическое влияние на реакцию йодистого бутила с оловом
оказывают, помимо магния, и другие металлы (литий, натрий, ртуть) [13], а
также свободный иод [14] в присутствии спиртов. Применение только одного
182 Оловоорганические соединения. Лит. стр. 190—192
компонента каталитической системы не оказывает удовлетворительного
действия. При замене ^-бутилового спирта на этоксиэтанол скорость реакции
заметно возрастает. Вероятно, в случае лития катализатором является
сольватированный йодистый литий или алкоголят лития. Их активность
зависит от электронодонорных свойств применяемых растворителей [131.
По-видимому, реакция йодистых алкилов с оловом может быть
распространена и на вещества с функциональными заместителями в алифатическом
радикале. Эммерт и Эллер [15] при действии олова на этиловый эфир иодук-
сусной кислоты получили соединение состава (C2H5OOCCH2)2SnJ2.
Бромистые алкилы реагируют с оловом при нагревании смеси реагентов
до 175—180° С с образованием соединений типа R2SnBr2. Вследствие
жестких условий реакции продукт загрязнен примесью значительных количеств
(до 25%) R3SnX. При неизбежном здесь применении запаянных сосудов
наблюдались сильные взрывы за счет накопления газообразных продуктов
[16]. Кроме того, реакция проходит лишь с низшими алкилами (метил,
этил) [17]. При нагревании до 180° С бромистого w-бутила и олова в течение
нескольких часов они не взаимодействуют между собой. Пары бромистого
w-бутила, пропущенные через расплавленное олово, дают также
отрицательный результат.
Реакция проходит легче и ее можно провести и с высшими бромистыми
алкилами, если к смеси прибавить небольшое количество магния, йодистого
ал кила и спирта [6, 18—20]. Однако в этом случае образуется смесь олово-
органических бромидов различной степени алкилирования. В присутствии
лития и w-бутилового спирта бромистый бутил с оловом не реагирует [13].
Каталитическое влияние на реакцию олова с бромистыми алкилами
оказывает также триэтиламин [20а]. При нагревании до 160° С в течение 6 час.
1 моля бромистого бутила и 0,1 моля триэтиламина с 0,33 г-атома порошка
олова в автоклаве при энергичном перемешивании получают 64,9% (в
расчете на металлическое олово) двубромистого дибутилолова. С бромистым
пропилом выход несколько ниже (55,3%). В случае бромистого этила
образуется смесь бромистого триэтилолова и двубромистого диэтилолова
[20а].
Кочешков и Шевердина с сотр. [21—26] изучили взаимодействие
галоидных алкилов с оловом в условиях облучения 7-лучами. Реакция между
бромистыми алкилами и оловом имеет место при обычной температуре,
достигая выхода 50 молекул на 100 эв (считая на галоидный алкил) [21]
Y
RBr + Sn чААЛ/V *-R2SnBr2
В отличие от реакции между RBr и оловом в условиях нагревания
( ~200° С) здесь имеет место образование лишь следов R3SnBr.
На примере хлористого я-пропила показано, что с хлористыми алкилами
в этих условиях реакция не идет. Йодистые алкилы реагируют легко,
выходы достигают 60 молекул на 100 эв [21 ].
Облучение смеси бромистого алкила и олова при умеренном нагревании
и перемешивании значительно увеличивает выход оловоорганического
соединения (до 180 молекул на 100 эв) и дает возможность провести реакцию
с высшими бромистыми алкилами (например бромистым #-гексилом,я-геп-
тилом, я-октилом или я-нонилом) [23, 24].
Позже взаимодействие бромистого бутила с оловом под влиянием у-ради-
ации исследовано и другими авторами [26а, 27]. Найдено, что существенное
влияние оказывают различные добавки. Например, введение в систему
небольшого количества бромистого водорода значительно сокращает
индукционный период и повышает выход двубромистого дибутилолова. По своей
Синтез действием галоидных алкилов на олово 183
эффективности исследованные добавки располагаются следующим образом:
HBr > ai-C4H9J > (n-C4H9)4Sn > SnBr2. С другой стороны, четырехброми-
стое олово и трихлорбромметан оказывают ингибирующее действие [27].
Что касается галоидопроизводных непредельного ряда, то бромистый
аллил легко реагирует с оловом при нагревании смеси реагентов в толуоле в
присутствии катализаторов (хлорной ртути или аминов) [28]. Вероятно,
роль хлорной ртути заключается в активировании поверхности олова
амальгамированием, тогда как амины способствуют поляризации связи С—Вг
в бромистом аллиле.
При одновременном прибавлении каломели и амина образуется
каталитически неактивный комплекс. Но при предварительной обработке порошка
олова хлорной ртутью в нагретом до кипения толуоле с последующим
прибавлением триэтиламина получают [28] двубромистое диаллилолово с
выходом 81,7%.
Существенное влияние на ход этой реакции оказывает также
присутствие небольших количеств воды. При работе в сухом толуоле и без
катализатора выход двубромистого диаллилолова составляет только 2%; он
повышается до 14% при применении влажного порошка олова. Напротив, в
присутствии хлорной ртути выход двубромистого диаллилолова снижается с 80%
(в сухом толуоле) до 44% (в толуоле, насыщенном водой).
Получение двубромистого диаллилолова [28]. К 17,8 г (0,15 моля) порошка олова в
150 мл толуола прибавляют 0,5 г (0,002 моля) хлорной ртути и при перемешивании смесь
нагревают до кипения в течение 30 мин. После охлаждения к ней добавляют 0,2 г (0,002
моля) триэтиламина, нагревают до кипения и при энергичном перемешивании прибавляют
по каплям 18,2 г бромистого аллила. После первоначального снижения точки кипения до
103° С она постепенно повышается (через 1,5 часа температура 111° С). Смесь фильтруют,
растворитель удаляют в вакууме и остаток перегоняют также в вакууме. Получают 22,3 г
(81,7%) двубромистого диаллилолова с т. кип. 77—79° С/2 мм; d^ 1,8640.
В аналогичных условиях взаимодействие первичного бромистого кротила
или 2-бромбутена-З с порошком олова приводит к смеси двубромистого ди-
кротилолова (выход 70%) и [СН2 - СН — СН (CH3)]2SnBr2 (— 6%).
Строение продуктов установлено по ИК-спектрам [28].
Своеобразная реакция проходит при нагревании олова с бромистым
метиленом [29, 301:
2 СН2Вг2 + 2 Sn -* 2 CH3SnBr3 + С.
Хлористый метилен с металлическим оловом, несмотря на 120-часовое
нагревание (185—220° С), почти не реагирует. С йодистым метиленом
образуется четырехиодистое олово [29, 30].
Получение двубромистого ди(карбэтсхсиметил)олова [31]. 5,93 г (0,05 г-атома)
порошка олова и 16,7 г (0,1 моля) этилового эфира бромуксусной4 кислоты нагревают до
кипения в течение 5,5 час. Выделившийся осадок промывают эфиром и перекристаллизовыва-
ют из бензола. Получают 3,5 г (15,5%) вещества с т. пл. 139° С.
Двубромистое ди-(2-этоксикарбонилэтил)олово получено [31]
нагреванием при 150° С в течение 20 час. 2,95 г олова с 20мл этилового эфираа-бром-
пропионовой кислоты. После завершения реакции избыток эфира отгоняют
и остаток перегоняют в вакууме. Собираютфракцию с т. кип. 82—85°С/1,5мм.
Аналогичным образом проходят реакции между порошком олова и
метиловым эфиром бромуксусной кислоты, фенацилбромидом,
бромэтилацетатом, 2-бромпиридином, 1-хлор-2-иодэтаном и другими замещенными га-
логенидами. Однако выделить образующиеся продукты в чистом виде
не удается [31].
Соединения типа (YC71H27l)2SnX2 (где X == CI, Br, J; Y = СООН,
COOR, CN, ОН, RCOO~, ROCH2 и др.; п — целое число) получены [32]
взаимодействием олова с веществами формулы XCnH27lY (1—3 часа при 60—
184
Оловоорганические соединения. Лит. стр. 190—192
160° С) в присутствии небольших количеств магния, йодистого бутила и
тетрагидрофурана или я-бутилового спирта.
Хлористые ал килы реагируют с оловом лишь в очень жестких условиях.
Двухлористое диметилолово получено [33—35] барботированием
хлористого метила через расплавленное олово, содержащее каталитические
количества цинка и меди при температуре 300—350° С. Основной продукт содержит
небольшую примесь хлористого триметилолова и треххлористого метилоло-
ва [34]. В качестве катализаторов этой реакции исследованы [34] другие
элементы, но лучшие результаты получены с добавкой меди.
Проведение реакции хлористого метила с фольгой олова в присутствии
магния, я-бутилового спирта или тетрагидрофурана и йодистого бутила
позволяет повысить выход двухлористого диметилолова до 92% [6,361.
Однако с другими хлористыми алкилами эта реакция проходит иначе.
Например, при нагревании хлористого бутила и олова в автоклаве в присутствии
небольшого количества магния, йодистого бутила и тетрагидрофурана
получают примерно 14% продукта, представляющего собой смесь
(C4H9)3SnCl, (C4H9)2SnCL и C4H9SnCl3. Незначительный выход объясняется
побочными процессами [6, 36]. В присутствии третичного амина и иода
реакция между хлористым бутилом и оловом легко проходит при
температуре 130—180° С [20а1. В зависимости от продолжительности реакции,
температуры и относительных количеств олова и триэтиламина
преимущественно образуется хлористое трибутилолово или двухлористое дибутилолово.
Нагревание смеси до 180° С в течение 6 час. приводит в основном к двухло-
ристому дибутилолову. Понижение температуры до 135° С и сокращение
продолжительности реакции до 1 часа благоприятствует образованию
хлористого трибутилолова. Этому способствует также увеличение
относительного количества металлического олова или триэтиламина. В предельном
случае, когда используется большое количество триэтиламина, образуется
только хлористое трибутилолово.
Реакция хлористого бутила с металлическим оловом в присутствии иода и триэтиламина
[20а]. Смесь 39,6 г (0,33 г-атома) порошкообразного олова, 92,6 г (1,0 моля) хлористого
бутила, 10,1 г (0,10 моля) триэтиламина и 5,1 г (0,02 моля) иода нагревают при 160° С в
стальном автоклаве, снабженном эффективной вращающейся мешалкой (1000 об/мин) в
течение 6 час. Порошок олова полностью вступает в реакцию. Продукт обрабатывают 200
мл воды и 15 мл 12 N соляной килоты для отделения оловоорганических галогенидов
от солянокислого триэтиламина, который не растворяется в эфире и не перегоняется. Оло-
воорганическое соединение трижды экстрагируют эфиром (порции по 200 мл). Объединенные
эфирные вытяжки упаривают, остаток перегоняют в вакууме. Получают 66,9 г продукта
с т. кип. 125—145° С/5 мм, состоящего из 48,4 г двухлористого дибутилолова и 18,5 г
хлористого трибутилолова.
Помимо триэтиламина, катализаторами реакции хлористого бутила с
оловом являются и другие основания, например пиридин, хинолин, анилин,
]\[,]\[-диметиланилин, пиперидин, этаноламин, трибутиламин, диэтиламин,
я-бутиламин, аммиак или трифенилфосфин. Во всех случаях вторым
компонентом каталитической системы является иод. При использовании пиридина
или хинолина с хорошим выходом получают чистое двухлористое
дибутилолово, тогда как в присутствии других первичных, вторичных или третичных
аминов, а также аммиака образуются смеси хлористого трибутилолова и
двухлористого дибутилолова.
Олово легко реагирует с бензилгалогенидами. Реакция может проходить
в нескольких направлениях. Состав образующихся продуктов определяется
условиями проведения эксперимента.
При нагревании порошка олова с хлористым или бромистым бензилом
без растворителя происходит выделение галоидоводорода и образуется дибен-
зил [37]. Оловоорганические соединения в смеси не обнаружены. При
размалывании на вибрационной мельнице металлического олова с йодистым,
бромистым или хлористым бензилом или м-замещенными галоидбензилами
Синтез действием галоидных алкилов на олово
185
образуются соответствующие дигалогениды дибензилолова. Эта реакция
подробно изучена Гроуном и Поудером [37—39]. Показано, что на выход
продуктов решающее влияние оказывают условия механической обработки
реагентов.
Целесообразнее проводить реакцию между оловом и бензилгалогенидами
в растворителях. При этом первоначальным продуктом реакции является
двухлористое дибензилолово [39а]. В полярных средах оно реагирует с
металлическим оловом с образованием хлористого трибензилолова, выход
которого увеличивается с повышением полярности растворителя. Так,
нагревание хлористого бензила с суспензией порошка олова в воде приводит к
хлористому трибензилолову [40—43]:
н2о
3 СвН5СН2С1 + 2 Sn (СбН5СН2)3 SnCl -f SnCl3.
Интересно отметить, что превращение двухлористого дибензилолова
в хлористое трибензилолово проходит и под влиянием других металлов:
кадмия, магния, цинка или алюминия. Если же олово заменить на титан,
медь, железо или двухлористое олово, то хлористого трибензилолова не
образуется [39а]. При проведении этой реакции важное значение имеет
достаточно хорошее перемешивание смеси. В противном случае возможны
значительные местные перегревы, в результате чего выход конечного
продукта снижается.
Получение хлористого трибензилолова [40]. К нагретой до кипения суспензии 17,8 г
(0,15 моля) порошка олова в 150 мл воды в течение 2 мин. при энергичном перемешивании
прибавляют 19,2 г (0,15 моля) хлористого бензила. Смесь кипятят при перемешивании 1,5
часа и отфильтровывают. Осадок, представляющий собой смесь хлористого трибензилолова
и непрореагировавшего олова, высушивают на воздухе и затем экстрагируют ацетоном в
аппарате Сокслета в течение 4 час. Выпаривание экстракта в вакууме дает 18,0 г хлористого
трибензилолова ст. пл. 142—144° С (из этилацетата). Сходным образом проходит реакция в
среде бутилового спирта.
Взаимодействие хлористого бензила с оловом в толуоле приводит к
двухлористому дибензилолову [40]
свн5сн3
2 СбН5СН2С1 + Sn > (CeH5CH2)3 SnCl,.
При замене толуола бензолом или лигроином образуется то же вещество, но
с более низким выходом.
Получение двухлористого дибензилолова [40]. 17,8 г (0,15 моля) порошка олова
растирают с 3 каплями воды и затем суспензируют в 150 мл толуола. К нагретой до кипения
суспензии при энергичном перемешивании прибавляют по каплям 19,2 г (0,15 моля) хлористого
бензила. Смесь кипятят в течение 3 час. и отфильтровывают. Осадок экстрагируют ацетоном
и экстракт выпаривают в вакууме. Получают 21,6 г сырого двухлористого дибензилолова.
Выпаривание толуольного раствора дает еще 3,0 г вещества. Для очистки его
кристаллизуют из этилацетата. Получают 22,4 г (80%) двухлористого дибензилолова с т. пл. 161 —
163° С.
Аналогичным образом проходят реакции с бензилгалогенидами,
содержащими заместители в бензольном кольце [40, 441: *
3 (СН3)2 СН—<Г==Ъ—СН2С1 + 2 Sn —-> Г (СН3)а СН—/^^-СнЛ SnCl + SnCIo,
2 (СН8)2 СН—/=\--СН2С1 + Sn 3-> Г(СН8)2 СН—<f=\—СИ2] SnCl2.
Как правило, наличие в бензольном кольце электронодонорных
заместителей, таких как алкил- или алкоксигруппы, снижает выход оловоорга-
нического соединения, если реакция проводится в воде, и несколько повы-
186
Оловоорганические соединения. Лит. стр. 190—192
шает его, если реакцию вести в толуоле. Введение в бензольное кольцо
атомов галоида значительно замедляет скорость образования оловооргани-
ческих соединений в воде, но выходы продуктов достаточно высокие [40].
о-Галоидбензилхлориды не реагируют с суспензией олова в толуоле.
С мета- и пара-замещенными бензилгалогенидами реакция проходит как
обььчно с образованием соединений типа R2SnX2 [40].
При обработке металлического олова ос-замещенными
бензилгалогенидами в воде или толуоле оловоорганические соединения не образуются;
с хорошим выходом получены продукты реакции Вюрца [45]. Например,
хлористый дифенилметил дает сшш-тетрафенилэтан с выходом 85%. В
случае а-фенилэтилхлорида получают смесь мезо- и 2,3-дифенилбутана с
выходом 82%. При проведении реакций в воде выходы продуктов конденсации
несколько ниже, чем в толуоле вследствие гидролиза [45].
Механо-химическая обработка бензотрихлорида и олова дает 1,1,2,2-те-
трахлор-1,2-дифенилэтан и двухлористое олово [37, 39]. Из Р-хлор- или
(3-бромэтилбензола получают 1,4-дифенилбутаны [37].
Галоидные арилы с металлическим оловом не реагируют [6, 7, 21, 28].
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ДЕЙСТВИИ ГАЛОИДНЫХ АЛКИЛОВ И АРИЛОВ НА СПЛАВЫ ОЛОВА
Действие галоидных алкилов или арилов на сплавы олова позволяет
получать производные олова как в алифатическом, так и в ароматическом
ряду.
Этот метод имел весьма важное значение в период возникновения химии
металлоорганических соединений. Впервые оловоорганические соединения
получены в 1852 г. Левигом [46] при действии йодистого этила на сплав
олова с натрием (I часть натрия и 6 частей олова). Из продуктов реакции им
выделено масло темного цвета, которое, по данным элементарного анализа,
соответствует диэтилолову — [(C2H5)2Sn]A- (ср. [47]).
Каур и др. [48] ближе изучили реакцию йодистых этилов со сплавом
олова с натрием. При действии йодистого этила на сплав олова с натрием,
содержащий 4 части олоза и 1 часть натрия, ими получено тетраэтилолово
(наряду с небольшим количеством гексаэтилдистаннана). В дальнейшем
Каур совместно с Демарсе [49, 50] показали, что в зависимости от
содержания натрия в сплаве можно получить оловоорганические соединения двух
типов. Так, при действии йодистых алкилов на сплав олова с малым
количеством натрия (5—6%) получаются соединения типа R2SnX2. При
применении сплава с большим количеством натрия (10%) образуются соединения
типа R3SnX.
Ладенбург [51], работая со сплавами, содержащими 14% натрия,
получил простейший представитель типа R4Sn — тетраметилолово. Позже эта
методика была воспроизведена Зибертом [52].
Леттс и Коли [53] нагреванием йодистого этила со сплавом олова,
цинка и меди получили тетраэтилолово с выходом 50%. Ими отмечено, что со
сплавом олова с медью в этих условиях реакция не идет.
Получение оловоорганических соединений действием йодистых алкилов
на сплавы олова с натрием описано и другими авторами [54—57]. Вместо
йодистых алкилов можно использовать и другие алкилгалогениды [58—62].
О кинетике реакции бромистого этила со сплавом NaSn и NaSn2 см. статью
Неймана и Шушунова [62].
Введение небольших количеств цинка в сплав олова с натрием
значительно повышает его активность [63—68]. Тетраэтил- [65—67], тегра-
пропил- [68] и другие тетраалкильные соединения олова [69] получены
взаимодействием соответствующих бромистых алкилов со сплавом Na—Sn—Zn.
Синтез действием галоидных алкилов на олово
187
Хлористый я-бутил легко реагирует со сплавом SnNa, содержащим 2%
цинка, или со сплавом состава Na0,92Ko,o8Sn при 150—180° С под давлением
с образованием смесей, содержащих 10—20% вес. тетрабутилолова и 80—
30% хлористого трибутилолова [64]. Сходным образом ведут себя
хлористый я-пропил и хлористый я-амил.
Если реакцию с хлористым бутилом проводить при более низкой
температуре или использовать бинарный сплав NaSn, то продукт содержит
значительные количества дибутилолова. Общую схему реакции можно
выразить следующим образом [64]:
2 NaSn + 2 RCl -> R2Sn + 2 NaCl + Sn,
R2Sn + RCl-^ R3SnCl,
2R2Sn-* R4Sn + Sn.
Выходы продуктов составляют 25—30% (в расчете на олово).
Помимо сплавов олова с натрием, для получения оловоорганических
юединений применяются сплавы олова и с другими металлами.
Захаркин, Охлобыстин [701 изучили взаимодействие сплава олова с
недью с различными бромистыми и йодистыми алкилами в присутствии
юльватирующих растворителей. Найдено, что реакция йодистых алкилов
с этим сплавом (4—5% меди) в значительной степени облегчается в
присутствии диметиловых эфиров этилен-и диэтиленгликоля. При замене же этих
4>иров близкокипящими углеводородами образуется лишь ничтожное
количество оловоорганических соединений.
Бромистые алкилы вступают в эту реакцию только в присутствии
примерно 10% соответствующего йодистого алкила.
Хлористые алкилы не реагируют с указанным сплавом даже в
присутствии диглима и йодистых алкилов [70].
По патентным данным [71], пропускание паров хлористого метила,
этила, пропила или бутила над твердым оловом, содержащим 40% меди, при
температуре 250—450° С. приводит к соединениям типа R2SnX2.
Нагревание при 150° С в автоклаве в течение 4—5 час. галоидных алкилов со
сплавом олова с магнием в присутствии катализаторов (третичных аминов или
смеси третичных аминов с хлорной ртутью) приводит к смеси
оловоорганических соединений различной степени алкилирования. Галоидные
алкилы с разветвленной цепью в этих условиях с оловом не реагируют [72].
Ван-дер-Керк и Люитен [17] подробно исследовали взаимодействие
бромистого и хлористого этила со сплавом Mg2Sn и предложили наиболее
вероятный механизм реакции. Ими показано, что взаимодействием сплава
с бромистым этилом можно, по желанию, получить тетраэтилолово или
бромистое триэтилолово.
При нагревании до 160 С бромистого этила и Mg2Sn в циклогексане в
течение 3 час. с хорошим выходом получают тетраэтилолово. Если реакцию
проводить с избытком галоидного алкила, то в качестве основного
продукта образуется бромистое триэтилолово [17] (ср. [73]). Растворитель имеет
важное значение в этих реакциях. Лучше всего применять примерно 40 мл
циклогексана на 7 г сплава. При замене циклогексана бензолом общий
выход снижается и продукт представляет собой смесь тетраэтилолова,
бромистого триэтилолова и двубромистого диэтилолова. Если реакцию
проводить в петролейном эфире, то выход продукта достаточно высокий и в
основном состоит из бромистого триэтилолова. Применение катализаторов —
ртути или хлорной ртути сокращает продолжительность опыта с 3 час. до
1 часа, но температура требуется та же.
Опыты с хлористым этилом (под давлением) показали, что реакция идет
только в присутствии катализатора (ртути или хлорной ртути) и избытка
188
Оловоорганические соединения. Лит. стр. 190—192
галоидного алкила. Продукт представляет собой смесь тетраэтилолова,
хлористого триэтилолова и двухлористого диэтилолова [17, 74].
Несмотря на хорошие выходы оловоорганических соединений (до 80%),
полностью превратить сплав Mg2Sn в тетраэтил олово не удается. Наряду с
основной реакцией
Mg2Sn + 4 QH5Br (CI) -* (С2Н5)4 Sn + 2 MgBr2 (Cla)
проходит дегидрогалоидирование бромистого (или хлористого) этила с
образованием этилена. В случае высших галоидных алкилов роль этой
побочной реакции возрастает, поэтому получить с хорошим выходом
другие тетраалкильные соединения олова этим способом не удается [17] (ср.
[75—77]). Следует отметить, что и в случае бромистого этила реакция
проходит только под давлением или при обычном давлении, но в присутствии
катализаторов (ртути или хлорной ртути). Бром, иод, двухлористое олово,
эфират трехфтористого бора, диэтиловый эфир и ацетальдегид почти
неактивны, так же как и добавки к сплаву меди или цинка. Этильные
соединения олова не образуются и при пропускании паров бромистого этила над
сплавом олова с магнием при температуре 200—350° С [17].
Действие галоидных арилов на сплавы олова изучено недостаточно
[78—80]. Полис [78] при нагревании в течение 30 час. бромбензола со
сплавом олово — натрий (23% Na) в присутствии небольшого количества этил-
ацетата получил первое оловоорганическое соединение ароматического
ряда — тетрафенилолово (выход не указан). Чемберс и Шерер [79],
изменив несколько условия реакции, работая со сплавом, содержащим 14% Na
и обрабатывая полученный продукт четыреххлористым углеродом,
получили тетрафенилолово с выходом 50%.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПРИ ДЕЙСТВИИ ГАЛОИДНЫХ АЛКИЛОВ
НА СОЕДИНЕНИЯ ДВУХВАЛЕНТНОГО ОЛОВА
Метод получения оловоорганических соединений действием галоидных
алкилов на окись двухвалентного олова, а также станнит калия имеет
ограниченное значение и, по-видимому, применим только для соединений
алифатического ряда. Выходы, как правило, остаются невысокими.
В реакциях с окисью олова применяют хлористые и бромистые ал килы.
Реакция протекает при температуре около 300 С. Иодиды в зтих условиях
недостаточно устойчивы. Галоидный алкил пропускают через трубки из
пирекса, содержащие окись двухвалентного олова в порошке, с добавлением
порошка меди или без него. Появляется желтое окрашивание, характерное
для соединений (R2Sn)rt. Реакция оканчивается на образовании
моногалоген ид а [81]:
3 SnO + 2 СН3С1 -* (СНз)2 Sn -|- SnOCl2 + Sn02,
(СНзЬ Sn + CH3CI -> (CI 13)з SnCl.
Получение хлористого триметилолова [81]. Хлористый метил пропускают через смесь
20 г окиси олова и 2 г окиси меди при 30С° С со скоростью 15 мл/мин. Образуется хлористое
триметилолово, которое после кристаллизации из бензола имеет т. пл. 37,5—39,5° С.
Бромистое триметилолово образуется аналогично. Йодистое
триметилолово выделено в весьма небольшом количестве.
Двухлористое диметилолово может быть получено взаимодействием
хлористого метила с двуокисью олова в присутствии (8% по весу) окиси меди.
Вероятно, двуокись олова восстанавливается хлористым метилом до окиси
Синтез действием галоидных алкилов на олово
189
олова и двухлористого олова, которые и реагируют с хлористым метилом
с образованием оловоорганических галогенидов. Двухлористое диметилоло-
во образуется в результате взаимодействия галогенидов различной степени
алкилирования (реакцию проводят при 300° С).
Андриановым с сотр. [82] изучено действие хлористого метила на окись
олова в присутствии губчатой меди. Реакция протекает с образованием
двухлористого диметилолова и треххлористого метилолова по схемам
2 SnO + 2 СН3С1 -* (СН3)2 SnCI2 + Sn02,
2 SnO + 3 СН3 CI -» CH3SnCl3 + Sn02 + C2H6.
Направление реакции существенно зависит от температуры. При 250° С
образующийся продукт содержит 98% двухлористого диметилолова и 2%
треххлористого метилолова, тогда как при 350° С — соответственно 75 и
25%.
Для получения треххлористого метилолова хлористый метил барботи-
руют в расплавленное безводное хлористое олово при 365° С. ]81]
CH3Ci + SnCl2 -> CHgSnCls.
В присутствии аминов с хлористым оловом реагируют и другие
хлористые алкилы [82а].
В случае алкилиодидов, как показали Пфейфер и Геллер [83, 84],
реакция протекает при более умеренных температурах
CH8J + SnJ2 -> CH3SnJ3.
Получение трехиодистого метилолова [84]. 3,5 г йодистого олова нагревают до 160° С
с двойным молекулярным количеством йодистого метила в запаянной трубке в течение 4 час.
После обработки сухим эфиром, фильтрования, отгонки эфира, перекристаллизации остатка
из лигроина получено 1,2 г трехиодистого метилолова, т. пл. 85° С.
Действие йодистых алкилов на активированный серебром цинк в
присутствии хлористого олова приводит к смеси оловоорганических моно- и
дииодидов [43, 85]:
н.,о
RJ + Zn (Ag) + SnCl2 * R3SnJ -j- R2SnJ3 + RH + RR + ZnJ2 + ZnCU
(R=az-CiH9, л-С8Н17, az-Ci6H33).
Вместо хлористого олова можно применять хлорное олово. Ояыты по
замене цинка магнием оказались безуспешными. Октальные производные
олова можно получать и без добавок серебра, но в этом случае их выход
заметно уменьшается.
Как показано Лесбром с сотр. [86], в реакциях с йодистыми ал килами
могут применяться и двойные соли двухвалентного олова:
RJ -\- MSnX3 -* MJ + R3nX3
(M = Rb, Cs, К; X —галоид).
Получение трехиодистого этилолова [86]. 5,29 г SnCl2-KCl (перед реакцией
комплексную соль SnCU- КС1- Н20 высушивают при 100° С), нагревают до 110° С в запаянной
трубке с избытком йодистого этила в течение 48 час. При последовавшей затем перегонке
собрана фракция 181—184,5° С/19 мм, отвечавшая по анализу трехиодистому этилолову.
Выход 37% •
Таким же образом были получены [861 CH3SnJ3 (продолжительность
реакции 48 час, выход 44%) и n-C3H7SnJ3 (25%).
оТрехиодистое бутилолово получено с выходом 25% нагреванием при
90° С KSnCl3 с избытком йодистого бутила в трубке Кариуса в течение
190
Оловоорганшеские соединения
72 час.; т. кип. 154° С/5 мм. Это светло-красная жидкость, которая
разлагается с выделением йодистого олова [31J.
Обращает на себя внимание тот факт, что во всех исследованных
случаях, за исключением йодистого изопропила, вместо ожидаемого RSnClgr
получаются иодиды RSnJ3. Авторы признают возможной
последовательность следующих реакций:
KSnCl3 + 2KJ -> SnJ2 -f 3 КС1,
HJ + SnJ2 -» RSnJ3.
По существу процесс сводится к упомянутому выше методу Пфейфера и
Геллера [84]. Однако в том или ином варианте реакция между солями
двухвалентного олова и галоидными алкилами заслуживает предпочтения
перед реакцией Мейера —Дрюса [87. 88], с использованием станнита калия.
Несмотря на относительно низкие выходы, метод получения оловоор-
ганических соединений с помощью галоидных алкилов и станнита кали*
до сих пор имеет некоторое значение для типа RSnX3 в алифатическом ряду:
OK R ОМ
/ \ /
RJ + Sn -» Sn -* RSnOOK.
\ / \
ОН J OK
Получение метилстанноновой кислоты [87, 89, 90]. Раствор 20 г хлористого олова
в 50 мл воды прибавляют при перемешивании к 100 мл 20%-ного едкого кали. После
добавления 14 г йодистого метила в 100 мл спирта смесь оставляют на 3 дня, часто встряхивая.
Спирт удаляют при 40° С, прибавляют разбавленную соляную кислоту до тех пор, пока
реакция не сделается кислой. Выпавший белый студенистый осадок отфильтровывают,
отмывают от хлоридов холодной водой и высушивают в вакууме над серной кислотой. Выход
13,5 г (на неочищенный продукт).
Получение этилстанноновой кислоты [91—93]. Раствор 30 г SnCl2-2 H20 в 100 мл
теплого спирта медленно вливают в холодный раствор 46 г едкого кали в 200 мл воды до тех пор,
пока образовавшийся осадок не перестанет растворяться при встряхивании. Через час
раствор отфильтровывают, добавляют 20 г бромистого этила, встряхивают около часа. Через
три дня спирт выпаривают на водяной бане, раствор слегка подкисляют разбавленной
соляной кислотой, выпавший белый осадок отсасывают, отмывают и высушивают на воздухе
или в эксикаторе над серной кислотой. Свежеосажденный продукт бесцветен, аморфен, но
после высушивания становится желтовато-коричневым и роговидным. Этилстанноновая
кислота не растворяется в обычных органических растворителях и в воде, но растворяется
в разбавленных кислотах, а свежеприготовленная — также и в водных щелочах.
Аналогичным образом получены я-пропил-, изопропил-, лаурил-,
винил- и аллилстанноновые кислоты 181, 91—93]. Эта реакция с
соединениями ароматического ряда приводит, по-видимому, к низким выходам [94Ц
ЛИТЕРАТУРА
1. F r a n k 1 a n d E., Ann., 85, 329 (1853).
2. С a h о u r s A., Ann., 114, 227 (1860).
3. CahoursA., Ann., 114, 354 (1860).
4. К а г a n t a s s i s Т., В a s i 1 e i a d о s К., Compt. rend., 205, 460 (1937).
5. KarantassisT., BasileiadosK., Compt. rend., 206, 842 (1938).
6. M a t s u d a S., M a t s u d a H., Bull. Chem. Soc. Japan, 35, 208 (1962).
7. MatsudaS., Matsuda H., J. Chem. Soc. Japan, Industr. Chem. Sec, 63, 114
(1960); РЖХим., 1961, 8Ж229; Японск. пат. 26194 (1964); РЖХим., 1967, 9Н97.
8. М а ц у д а С, Я т а н и Т., Японск. пат. 7810 (1960); РЖХим., 1962, 8Л99.
9. М а ц у д а С, М а ц у д а X., Японск. пат. 19115 (1963); РЖХим., 1965, 20Н74.
10. М а ц у д а С, М а ц у д а X., Японск. пат. 25664 (1963); РЖХим., 1965, 24Н54.
11. МацудаХ.,Накамура М., М а ц у д а С, J. Chem. Soc. Japan, Industr. Chem.
Sec, 64, 1948 (1961); РЖХим., 1963, 5Ж281.
12. M а ц у д a X., M а ц у д а С, J. Chem. Soc. Japan, Industr. Chem. Sec, 63, 1958 (I960);;
РЖХим., 1962, 15Ж295.
Синтез действием галоидных алкилов на олово
191
13. OaxesV., HuttonR. Е., J. Organometal. Chem., 3, 472 (1965). Англ. пат.
1021426 (19С(); РЖХим., 1967, 4Н136.
14. X о с а к а X., Японск. пат. 17669 (1963); РЖХим., 1965, 24Н55.
15. Е mm ег t В., Е 1 1 er W., Вег., 44, 2328 (1911).
16. G r u t t n e r G., К г a u s е Е., Вег., 50, 1549 (1917).
17. Van der Kerk G. J. M., Luijten J. G. A., J. appl. Chem., 4, 301 (1954).
18. МацудаХ., ХаясиД., МацудаС, J. Chem. Soc. Japan, Industr. Chem. Sec,
64, 1951 (1961); РЖХим., 1963, 5Ж282.
19. МацудаХ., МацудаС, J. Chem. Soc. Japan, Industr. Chem. Sec, 63, 1965 (1960);
РЖХим., 1962, 17Ж324.
20. M а ц у д а С, М а ц у д a X., Японск. пат. 19116 (1963); РЖХим., 1965, 20H75.
20a. S i s i d о К., К о z i m a S., T u z i Т., J. Organometal. Chem., 9, 109 (1967).
21. А б р а м о в а Л. В., Ш е в е р д и н а Н. И., К о ч е ш к о в К. А., ДАН СССР, 123,
681 (1958).
22. К о с h e s h k о v К. A., S h e v e r d i n a N. I., A b r a m о v a L. V., Ind. China.
Beige, Suppl., 2, 331 (1959).
23. Абрамова Л. В., ШевердинаН. И., Кочешков К-А., Авт. свид. СССР
136313 (1961); Бюлл. изобрет., № 5 (1961).
24. А б р а м о в а Л. В., Ш е в е р д и н а Н. И., К о ч е ш к о в К- А., Труды II
Всесоюзного совещания по радиационной химии. М., Изд-во АН СССР, 1962, стр. 394.
25. A b r a m о v a L. W., Vereschinsky I. W., К о с h e s h k о w К. А., М i-
rezkyW.J., Posdeev W. W., R jabuchi n J. S., ScheverdinaN. J.,
Industrial uses Large Radiao Sources, Vol. 1. Vienna, 1963, p. 83.
26. А в е р б у х Б. С, А б р а м о в а Л. В., Б р е г е р А. X., В а й н ш т е й н Б. И*,
Г о л ь д и н В. А., К о ч е ш к о в К. А., С ы р к у с Н. П., Шаляпин НД.,
Ш е в е р д и н а Н. И., ЖФХ, 38, 2445 (1964).
26а. Верещинский И. В., Поздеев В. В., Мирецкий В. Ю., Ива-
н о в А. Ю., Авт. свид. СССР 181104 (1966); Бюлл. изобрет., № 9, 24 (1966).
27. F е п t i m a n A. F., W у a n t R. E., J e f f г е у D. А., К i г с h e r J. F., J.
Organometal. Chem., 4, 302 (1965); 6, 645 (1966).
28. S i s i d о K., T a k e d a Y., J. Org. Chem., 26, 2301 (1961).
29. КочешковК.А, ЖРФХО, 60, 1191 (1928).
30. К о z e s с h k о w К. А., Вег., 61, 1659 (1928).
31. G i 1 m a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
32. M а ц у д а С, К и к к а в а С, J. Chem. Soc. Japan, Industr. Chem. Sec, 67, 852 (1964);
РЖХим., 1965, 4H52. 69,256, 646, 649 (1966); РЖХим., 1966, 23Ж354: 1967, 5Ж37 1—2.
33. SmithF. А., Пат. США 2625559 (1953); С. А., 47, 11224 (1953).
34. S m i t h A. C, R о с h о w E. G., J. Am. Chem. Soc, 75, 4103 (1953).
35. R о с h о w E. G., Пат. США 2679506 (1954); С. А., 49, 4705 (1955).
36. M а ц у д а X., T а н и г у т и X., M а ц у д а С, J. Chem. Soc Japan, Industr. Chem.
Sec, 64, 541 (1961); РЖХим., 1962, 2Л32.
37. G г о h n H., P a u d e r t R., Z. Chem., 3, 89 (1963).
38. GrohnH., P a u d e r t R., Chem. Techn., 12, 430 (1960).
39. G г о h n H., P a u d e r t R., W i s s Z., Techn. Hochschule Chem. Zeuna-Merseburg
3, 203 (1960—1961); РЖХим., 1963, 8Ж348.
39a. S i s i d о К., К о z i m a S., H a n a d а Т., J. Organometal. Chem., 9, 99 (1967).
40. S i s i d о К., T a k e d a G., J. Am. Chem. Soc, 83, 538 (1961).
41. ShishidoK., KinukawaJ, Японск. пат. 6626 (1953); С. А., 49, 9690 (1955).
42. Lui jt en J.G. A., van der К e r k G. J. M., J. appl. Chem., 11, 35 (1961).
43. Nosek J., Collect. Czechosl. Chem. Communs., 29, 597 (1964).
44. D a n e k O., Collect. Czechosl. Chem. Communs., 26, 2035 (1961).
45. Si si do K., Taked aG., Nozaki H., J. Org. Chem., 27,2411 (1962).
46. LowigC, Ann., 84, 308 (1852).
47. H a r a d а Т., J. Sci. Res. Inst. (Tokyo), 43, 31 (1948).
48. С a h о u r s A., Compt. rend., 35, 93 (1852).
49. С a h о u r s A., D e m a r s а у Е., Compt. rend., 88, 1112 (1879).
50. С a h о u r s A., DemarsayE, Compt. rend., 89, 68 (1879).
51. L a d e n b u r g A., Ann. (Spl.), 8, 55 (1872).
52. S i e b e r t H., Z. anorg. allgem. Chem., 263, 82 (1950).
53. L e t t s E. А., С о 1 1 i e N.. Proc Chem. Soc, 2, 166 (1886).
54. С a h о u r s A., Compt. rend., 76, 133 (1873).
55. G r i m m A., J. prakt. Chem., 62, 385 (1854).
56. L a d e n b u r g A., Ber., 3, 353 (1870).
57. W e r n e r A., P f e i f f e r P., Z. anorg. Chem., 17, 82 (1898).
58. К r a u s С A., CallisCC, Канадск. пат. 226140 (1926); С. А., 21, 917 (1927)..
59. К г a u s С А., С а 1 1 i s С. С, Пат. США 1639947 (1926); С А., 21, 3180 (1927).
60. Caste 1 R., GrasG., Trav. soc. pharm. Montpellier, 17, 205 (1957); С А.,.
52, 18990 (1958).
192
Оловоорганические соединения
61. В 1 i t z e r S. M., Z i e t z J. R., Пат. США 2852543 (1958); РЖХим., 1960, 93378
62. НейманМ.Б., Ш у ш у н о в В. А., ДАН СССР, 60, 1347 (1948).
63. Англ. пат. 736882 (1955); РЖХим., 1957, 42360.
64. Zi etz J. R., Blitzer S. M., Redman H. E., Robinson G. C,
J. Org. Chem., 22, 60 (1957).
65. Нага da Т., Sci. Papers Inst., Phys. Chem. Research (Tokyo), 35, 290 (1939);
С. А., 33, 5357 (1939).
66. G i 1 m a n H., ArntzenCE., J. Org. Chem., 15, 994 (1950).
67. ArntzenCE., Iowa State Coll. J. Sci., 18, 6 (1943); С. А., 38, 61 (1944).
68. H a r a d а Т., Repts. Sci. Res. Inst. (Japan), 24, 177 (1948); С. А., 45, 2356 (1951).
69. P о 1 к i n h о r n e H., TapleyC. G., Англ. пат. 736822 (1955); С. А., 50, 8725
(1956).
70. 3 a x a p к и н Л. И., О х л о б ы с т и н О. Ю., Изв. АН СССР, ОХН, 1963, 2202.
71. W e i n b е г g E. L., Пат. США 2679505 (1954); С. А., 49, 4705 (1955).
72. Кацумура Т. J. 6hem. Soc. Japan, Pure Chem. Sec, 83, 724 (1962); РЖХим.,
1963, 16Ж251.
73. I г e 1 a n d J., Англ. пат. 713727 (1954); С. А., 49, 12530 (1955).
74. Faulkner С. J., Пат. ФРГ 946447 (1956); С. А., 53, 7014 (1959).
75. V а п d е г К е г k G. J. M., Пат. ФРГ 946447 (1956); РЖХим., 1958, 2205.
76. Л а й н е Л. В., Ш о с т а к о в с к и й М. Ф., Котрелев В. Н., К а л и н и-
н а С. П., К у з н е ц о в а Г. И., Б о р и с о в а А. И., Авт. свид. СССР 137519
(1961); РЖХим., 1962, 4Л115.
77. Р о 1 k i п h о г п е Н., Т а р 1 е у С. G., Англ. пат. 761357 (1956); С. А., 51, 11382
(1957).
78. Р о 1 i s А., Вег., 22, 2915 (1889).
79. С h a m b e r s R. F., Scherer Р.С., J. Am. Chem. Soc, 48, 1054 (1926).
80. L e w i s F. В.,, Англ. пат. 854776 (1960); РЖХим., 1961, 18Л91.
81. S m i t h А. С, R о с h о w E. G., J. Am. Chem. Soc, 75, 4105 (1953).
82. Андрианов К. А., Васильева Т. В., Нудельман З.Н., Хана-
нашвили Л. М., К о ч е т к о в а А. С, Чередникова А. Г., ЖОХ, 32,
2307 (1962).
82а. Франц. пат. 1456268 (1967); Organomet. Сотр., 10, 138 (1967); Пат. США 3340283
(1967); Organomet. Сотр., 11, 207 (1967); Голл. пат. 14326 (1966); Organomet.
Сотр., 11, 25 (1967); Австр. пат. 256877 (1967); Organomet. Сотр., 11, 206
(1967); Англ. пат. 1064178(1967); 1074407 (1967); 1079641 (1967); Organomet. Сотр.,
10, 184(1967); И, 72 (1967); И, 103 (1967).
83. Р i e i f f e r P., Вег., 44, 1278 (1911).
84. Pf ei f f er P., H e 1 1 e r I., Ber., 37,4618(1904).
85. N о s e k J., Collect. Czechosl. Chem. Communs., 29, 3173 (1964).
86. Tchakirian A., Lesbre M., Lewinson M., Compt. rend., 202, 138
(1936).
87. Meyer G., Ber., 16,1439(1883).
88. DruceJ.G. F., J. Chem. Soc, 113, 715 (1918).
89. P f e i f f e r P., L e h n a r d t R., Ber., 36, 1054 (1903).
90. E b e r 1 у К. С, В е s t С. Е., Пат. США 2626955 (1953); С. А., 47, 11224 (1953).
91. Druce J. G. F., J. Chem. Soc, 119, 758 (1921).
92. DruceJ.G. F., Chem. Soc, 121, 1859 (1922).
93. S о 1 e r i о A., Gazz., 85, 61 (1955); РЖХим., 1955, 55176.
94. Lesbre M., Compt. rend., 202, 136 (1936).
Глава //
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА ДВУГАЛОИДНОЕ ОЛОВО
Для синтеза оловоорганических соединений соли двугалоидного олова
находят ограниченное применение. Реакция протекает в основном в двух
направлениях. С литий- и магнийорганическими соединениями образуются
соединения типа (R2Sn)n. С ртутно-, таллий- и свинцовоорганическими
соединениями происходит^выделение металлической ртути, хлористого таллия
или двухлористого свинца с образованием дихлоридов R2SnX2. Реакции,
приводящие к оловоорганическим соединениям других классов, для
хлористого олова менее характерны.
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ВЗАИМОДЕЙСТВИЕМ ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ДВУХЛОРИСТЫМ ОЛОВОМ
Взаимодействие хлористого олова с двумя эквивалентами литийорга-
нических соединений с хорошим выходом приводит к соединениям класса
(R2Sn)n. Это олигомеры со связями олово — олово
/xSnCl2 + 2n RLi -» (RsSn),, + 2ti LiCl.
Реакция протекает на холоду в обычных для литийорганических
соединений растворителях. Для метода характерны следующие побочные реакции:
в случае избытка литийорганического соединения происходит образование
триалкил- или триарилстанниллития
R2Sn+ RLi-* R3SnLi.
Эта реакция находит значительное применение для получения
соединений со связью олово—литий (см. стр. 497).
Соединения (R2Sn)n реагируют с галоидными алкилами (см. стр. 442),
присутствие которых возможно, если реакция образования
литийорганического соединения прошла неполностью
R3Sn+RX-*R3SnX.
Соединения (RgSn)^ выделены в достаточно чистом виде [1].
Образование веществ более сложного строения рассмотрено при аналогичной
реакции с помощью магнийорганических соединений (см. стр. 195).
Всю работу по синтезу и очистке (R2Sn)n ведут в инертном газе. На
воздухе они легко окисляются.
Получение ди-я-бутилолова [1]. При взаимодействии взвеси безводного хлористого олова
(0,25 моля) в смеси эфира с бензолом (1:4) при —10° С с «-бутиллитием (0,5 моля) в эфире
раствор окрашивается в темно-вишневый цвет. Реакционную массу разлагают небольшим
количеством воды (температура не выше 2° С). Выпавший осадок отфильтровывают, промы-
13 Заказ Kt 207
194
Оловоорганические соединения. Лит. стр. 203—204
вают горячим бензолом, фильтрат упаривают в вакууме. В остатке темно-красное масло с
небольшим количеством осадка. Добавляют гексан, еще раз фильтруют, гексан удаляют
в вакууме (ЫО""2 мм), а остальные примеси отгоняют на кипящей водяной бане в вакууме
5—7, 10~в мм. Получают 37 г (63,7%) темно-красного масла, представляющего собой чистое
ди-«-бутилолово с мол. весом 1745 (эбулиоскопия, в толуоле) и 1921 (криоскопия, фреон
112). Диэтилолово получают аналогично с выходом 40,8% в виде масла темно-вишневого
цвета с мол. весом 1633 и 1780.
Диэтил- и дибутилолово хорошо растворяются в гексане, бензоле,
толуоле, эфире, ацетоне и четыреххлористом углероде, плохо — в спирте
и ацетоне. Выше 230° С вещества разлагаются с выделением металла [1].
Получение дициклопентадиенилолова [2]. В трехгорлую колбу на 250 мл, снабженную
мешалкой, обратным холодильником и газоподводной трубкой, во встречном токе азота
быстро вводят 6 г (0,083 моля) циклопентадиениллития и раствор 8,6 г (0,045 моля)
безводного хлористого олова в 70 мл сухого диметилформамида. Смесь перемешивают в течение 1,5
час. на кипящей водяной бане. Колбу соединяют с водоструйным насосом, растворитель
отгоняют при 60° С до тех пор, пока вещество начнет возгоняться на стенки колбы.
Реакционную смесь оставляют на ночь, после чего тестообразную массу переносят в прибор для
сублимации. После двойной возгонки при 90—110° С в высоком вакууме получают бесцветный
порошок дициклопентадиенилолова. Выход 50—60%. Все операции проводят под азотом.
Дициклопентадиенилолово холодной водой не гидролизуется, но кис*
лотами циклопентадиенильная группа отщепляется [2].
Получение дифенилолова [3]. К суспензии 40 г (0,21 моля) хлористого олова в
небольшом количестве эфира добавляют при внешнем охлаждении и перемешивании в течение 4
час. 400 мл 1,05 N (0,42 моля) раствора фениллития. Образуется раствор темно-красного
цвета, реакция по фенолфталеину щелочная. Реакционную смесь при внешнем охлаждении
разлагают водой. Дифенилолово, которое частично выпадает в осадок, вновь переводят в
раствор нагретым бензолом. Раствор сливают с осадка, высушивают и при 40° С
концентрируют в вакууме до объема 150 мл. Упаренный раствор выливают в 700 мл спирта.
Выпадает золотисто-желтый осадок дифенилолова. Выход 42 г (73%).
В случае избытка фениллития в реакционной смеси появляется трифе-
нилстанниллитий [3, 4]. Гексаарилдистаннаны, если и образуются, то в
небольших количествах [5].
Известны модификации дифенилолова, отличающиеся по составу и
свойствам [3, 6]. Нейман и Кониг [4,7,8] нашли, что как дифенилолово, так и
другие соединения этого класса часто имеют более сложный состав, чем
(R2Sn)n. Наряду с группами R2Sn вещества содержат R3Sn и RSn. Многие
препараты окрашены в темный цвет.
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ВЗАИМОДЕЙСТВИЕМ РЕАКТИВА ГРИНЬЯРА
С ДВУХЛОРИСТЫМ ОЛОВОМ
Взаимодействие реактива Гриньяра с хлористым оловом [9—13] может
быть выражено следующим уравнением
2 RMgX + SnCl2 -* R2Sn + 2 MgXCl.
Однако реакция протекает сложно, часто с образованием неоднородных
продуктов. Получаемые соединения являются олигомерами [12, 14—17] со
связями олово — олово [18—20]. Это подтверждается рентгеноструктур-
ными исследованиями [18, 19] и изучением мессбауэровского спектра [20].
Так, дифенилолово в действительности является додекафенилциклогекса-
станнаном [(C6H5)2Sn]6 [19].
Высказывались сомнения в отношении идентичности препаратов,
описанных разными авторами [6—8, 21, 22].
По-видимому, в одних и тех же условиях синтеза можно получить
различные результаты [3, 10, 12]. Согласно исследованиям Неймана с сотр.
Синтез действием металлоорганических соединений на деугалоидное олово 195
[4, 22], в результате реакции образуются не только (R2Sn)n, но и
полимеры с открытой цепью, на концах которой находятся алкильные группы.
Цепь из атомов олова может быть разветвлена
R3Sn— (SnR2)rt—SnR3, R3Sn—SnR—(SnR2)„—SnR8.
SnR3
Соединения с разветвленной цепью не являются собственно диалкил(арил)
оловом. Установлено, что чем глубже окраска препарата, тем более
разветвлена полимерная цепь [4, 12].
Взаимодействие реактива Гриньяра с хлористым оловом лучше
исследовано в ароматическом ряду. Для реакции применяют сухое хлористое
олово (т. пл. 247° С, т. кип. 606° С) [3, 23]. В качестве растворителя
используют этиловый эфир, смесь эфира с бензолом и тетрагидрофуран.
Элементарный состав полученных соединений часто не удовлетворителен
вследствие загрязнений [9, 10, 12, 21, 22]. Побочной реакцией является
взаимодействие полученного продукта с невступившим в реакцию галоидным алки-
лом (см. стр. 441).
Избыток реактива Гриньяра, особенно при нагревании, способствует
реакции диспропорционирования с образованием дистаннанов (см. стр. 377),
Так, например, гексафенилдистаннан может быть получен нагреванием
хлористого олова с избытком бромистого фенилмагния [10]. Однако вторая
побочная реакция во многих случаях не имеет существенного значения.
Некоторые синтезы можно проводить с избытком реактива Гриньяра [9,
24].
Из соединений алифатического ряда наиболее подробно исследовано
«диэтилолово» — вязкая жидкость от желтого до ярко-красного цвета,
обесцвечивается кислородом воздуха, устойчиво к действию воды и
разбавленных кислот [13, 25, 26], в достаточно высоком вакууме может быть
перегнано [22] (при 25 мм около 150° С разлагается, не перегоняясь [13]).
Взаимодействие бромистого этилмагния с хлористым оловом [22]. К отфильтрованному
реактиву Гриньяра, полученному из 100 г (0,92 моля) бромистого этила и 60 г (2,5 моля)
магния в 800 мл эфира, прибавляют при перемешивании по частям 40 г (0,21 моля) сухого
хлористого олова. Реакционная смесь окрашивается в красный цвет. После
полуторачасового кипения содержимое колбы при охлаждении разлагают 500 мл разбавленной свободной
от воздуха соляной кислотой. Эфирный слой промывают раствором соды и водой, высушивают
хлористым кальцием и перегоняют в вакууме. Получают 31,0 г (83%) тяжелого красного
масла.
Полученный продукт не является однородным. Горячим спиртом может
быть извлечено желтое масло, содержащее группы (C2H5)2Sn — и (C2H5)3Sn—;
при обработке хлором (—60-; 70° С) дает 30 мол.% (C2H5)3SnCl
и 70 мол.% (C2H5)2SnCl2. Остаток — твердое красно-черное вещество
разветвленной структуры. С хлором дает наряду с моно- и дихлоридом три-
хлоридС2Н55пС13 122]. Таким образом, собственно диэтилолово [(C2H5)2Sn]n
является только одним из продуктов реакции.
Нейман [22] считает, что различное описание вещества [12, 27],
полученного с помощью реактива Гриньяра, объясняется частичной
изомеризацией с образованием разветвлений [22].
Реакция диспропорционирования первоначально образовавшегося ди-
алкилолова в дистаннан R3SnSnR3 с выделением металлического олова, по-
видимому, более характерна для соединений изостроения. Так, при
обработке галоидом продукта взаимодействия хлористого неопентилмагния с
хлористым оловом образуется преимущественно галоидное тринеопентил-
олово [28].
13»
196 Оловоорганические соединения. Лит. стр. 203—204
Получение дициклогексилолова [24]. Реактив Гриньяра, полученный из 40 г бромистого
циклогексила в 150 мл абсолютного эфира и 7 г магния, нагревают в течение 3 час. с
обратным холодильником на водяной бане (следить за тем, чтобы бромид полностью прореагировал).
Раствор сливают с непрореагировавшего магния и смешивают с 200 мл чистого бензола,
высушенного над натрием. Затем быстро маленькими порциями добавляют в токе азота при
энергичном перемешивании без охлаждения 9 г тонкого порошкообразного безводного
хлористого олова SnCl2 (предварительно перегнанного в токе хлористого водорода). Раствор
нагревается и окрашивается в интенсивный желто-коричневый цвет. После 2 час. нагревания
на водяной бане с перемешиванием (в атмосфере азота) реакционную смесь осторожно
разлагают 100 мл ледяной воды. Для лучшего разделения слоев еще раз нагревают на водяной
бане. Бензольно-эфирный слой темно-оранжево-красного цвета отделяют под азотом,
промывают 2 раза водой, просушивают коротким взбалтыванием с безводным сульфатом натрия,
фильтруют, концентрируют в токе азота до 10 мл и выливают при перемешивании в смесь из
180 мл безводного спирта и 20 мл сухого эфира. Дициклогексилолово выделяется в виде
светло-желтого пушистого порошка. Его растворяют в небольшом количестве бензола и
высаживают спиртом и эфиром. Если соединение содержит галоид, то его еще раз растворяют в
сухом бензоле, кипятят с бромистым циклогексилмагннем и опять обрабатывают указанным
выше путем. Отсасывание должно производиться в токе азота. Еще лучше избежать
фильтрования, осадок промыть декантацией. В этом случае оставшуюся кашицу выливают на
глиняную тарелку и помещают над пятиокисью фосфора в эксикатор, наполненный азотом.
После 2-часовой эвакуации вещество оставляют на короткое время под вакуумом в
свежезаряженном эксикаторе [24]. Образуется интенсивно желтый порошок, который при
нагревании до 130° С окрашивается в желто-оранжевый цвет, а при 176—178° С плавится, давая
рубиново-красный расплав. Температура разложения 260—285° С [24]. Твердое вещество
на воздухе окисляется крайне медленно (несколько месяцев), хотя быстро становится белым.
[24].
Дициклопентадиенилолово образуется с выходом 90% действием дицик-
лопентадиенилмагния на хлористое олово в ксилоле при 160° С [29].
Дифенилолово в виде соединения желтого цвета впервые описано Крау-
зе и Бекером [10].
Взаимодействие бромистого фенилмагния с хлористым оловом [10]. Реактив Гриньяра
приготавливают из 75 г бромбензола и небольшого избытка магния в 400 мл сухого эфира.
Нагревание в течение 10 час. и избыток магния необходимы для полного освобождения от
непрореагировавшего бромбензола. Затем постепенно при взбалтывании и охлаждении льдом
вводят 15 г (одну треть рассчитанного количества) растертого в мелкий порошок хлористого
олова (если прибавить большее количество хлористого олова, то получается продукт,
содержащий галоид). Эфир вскипает, раствор приобретает темно-красно-коричневый цвет, а
добавленное хлористое олово, поскольку оно не переходит в раствор, превращается в
золотисто-желтый аморфный порошок. Смесь часто и сильно взбалтывают в течение одного часа при
комнатной температуре. Реакцию завершают кипячением в течение 10 мин. на теплой
водяной бане с обратным холодильником. Более длительное кипячение или местный перегрев
на кипящей водяной бане ведет к выделению олова и образованию гексафенилдистаннана
(CeHsbSnSn (СбНб)3. Смесь осторожно разлагают маленькими кусочками льда при внешнем
охлаждении и экстрагируют бензолом. Бензольно-эфирный раствор золотисто-желтого
цвета высушивают и концентрируют в вакууме примерно до 70 мл (если сконцентрировать
сильнее, то соединение выпадает в виде темно-красной густой маслянистой массы).
Темно-красный раствор выливают при перемешивании в безводный спирт, затем выпавший осадок
промывают декантацией безводным спиртом, отсасывают и высушивают в вакууме над
пятиокисью фосфора. Получают 16 г (64%) светло-желтого блестящего порошка [10].
Рекомендуется перед внесением хлористого олова реакционную смесь разбавить чистым и высушенным
над натрием бензолом [28],
Синтез и очистку вещества производят в атмосфере азота. В сухом виде
оно более стабильно [10, 30].
Вещество не является однородным. Температуры плавления различных
препаратов значительно отличаются друг от друга. Содержание олова в
них переменное, обычно ниже вычисленного [31—34]. Известна плохо
растворимая модификация дифенилолова [30].
В своих обстоятельных работах Нейман и Кониг [4, 21] показали, что
взаимодействие хлористого олова с бромистым фенилмагнием, в
зависимости от условий эксперимента, приводит к соединениям циклического
строения или соединениям с группами R3Sn на концах цепи [4]:
[(QH5)2 Sn]^H (CeH5)3;Sn-[Sn (CeHsJ^-Sn (СвЩз.
Синтез действием металлоорганических соединений на двугалоидное олово 197
Обработкой вещества золотисто-желтого цвета при 20° С иодом они
получили 70—75 % двуиодистого дифенилолова и 25—30% йодистого трифенил-
олова и, таким образом, показали, что оно не является дифенилоловом, как
это предполагалось ранее [10], а состоит в основном из смеси веществ с
открытой цепью [4]. Последние не могут образоваться в результате
взаимодействия первично образовавшегося шестичленного цикла с реактивом
Гриньяра. Циклическое соединение в этих условиях устойчиво.
Предположение [31, 35], что в начале образуется мономерное дифенилолово, в
дальнейшем не подтвердилось [4]. Препарат, который быстро освобождается
от остатков растворителя, выдерживается при —70° С в вакууме, через час
после получения имеет окончательный молекулярный вес 1678 (в
приведенной формуле л =3—4) [4]. Продукты циклического строения [(С6Н5)28п]л
образуются, если в качестве растворителя применяют тетрагидрофуран и
раствор бромистого фенилмагния медленно прибавляют к раствору
эквивалентного количества хлористого олова. Наряду с переменным количеством
[(C6H5)2Sn]e получают желтый, плохо растворимый порошок, состав
которого отвечает формуле [(CeH^aSnln. Степень конденсации п изменяется в
зависимости от условий синтеза, но чаще всего равна 13—14. Группы
R3Sn или RSn в этом случае отсутствуют.
Получение высших олигомеров дифенилолова [4]. К раствору 5 г (0,0264 моля)
безводного хлористого олова в 65 мл сухого тетра гидрофура на прибавляют по каплям в течение
1 часа 30 мл 1,75 М раствора бромистого фенилмагния в тетрагидрофуране. Появляется
желтоватое помутнение. Приливают равный объем метанола и выпаривают до 60 мл. Получают
5,6 г светло-желтого, труднорастворимого в бензоле порошка, не содержащего
хлора и устойчивого на воздухе (п — 13—14). Мол. вес 3790, 3560. Из продукта экстрагируют
толуолом 3% [(C6H5)2SnIG.
В иных условиях (раствор хлористого олова вводят в реактив
Гриньяра) образуется соединение с группами R3Sn. В случае избытка магнийорга-
нического соединения образуется в значительных количествах (60%) гек-
сафенилдистаннан [4 ].
Высшие диарильные соединения олова изучены недостаточно. Довольно
подробно описаны ди-п-толилолово (т. пл. 119° С, выход 67%), ди-ога-
лоидбензилолово [36], ди-д-ксилилолово (т. пл. 157° С) и ди-а-нафтилолово
(при 196° С размягчается, т. пл. 200° С, выход 73%) [10]. Позже получено
ди-(9-фенантрил)олово [9] и ди-д-бифенилолово [5, 211. Образующиеся
продукты, по-видимому, не являются диарилоловом (Ar2Sn)n, они содержат
также группы Ar3Sn [21].
В тетрагидрофуране диарилолово (Ar2Sn)n образуется в более чистом
виде [4, 21]. Полученный продукт может быть выделен в виде двугалоид-
ного диарилолова. Двуиодистое ди-д-бифенилолово получено с выходом
74% [21].
Дифенантрилолово получают в смеси эфира с бензолом, так как зфи-
рат бромистого фенантрилмагния нерастворим в эфире [9].
Получение ди-(9-фенантрил)олова [9]. В хорошо перемешиваемую суспензию 7,0 г
(0,037 моля) тонкоизмельченного хлористого олова в 30 мл бензола добавляют по каплям в
течение часа 100 мл эфирно-бензольного раствора бромистого 9-фенантрилмагния (0,089
моля).
Золотисто-желтая окраска реакционной смеси постепенно переходит в ярко-красную.
Смесь перемешивают 2 часа при комнатной температуре, затем нагревают 2 часа на водяной
бане. Образуется суспензия красно-коричневого цвета. После охлаждения избыток реактива
Гриньяра осторожно разлагают не содержащим воздуха, охлажденным до нуля
концентрированным раствором хлористого аммония. Появляются два слоя: водный, в котором
суспензирована основная масса выпавшего осадка (основные соли магния и олова),и
ярко-красный эфирно-бензольный раствор дифенантрилолова. Фильтруют на воронке Бюхнера через
двойной фильтр вначале органический слой, а затем — водный. Фильтрат переносят в дели-
198 Олоеоорганические соединения. Лит, стр. 203—204
тельную воронку, слои разделяют. Эфирно-бензольный слой высушивают сульфатом натрия*
добавляют небольшое количество сухого бензола и концентрируют в вакууме до 50 мл
(более концентрированный раствор дает с этанолом маслянистый осадок). К полученному
темно-красному раствору при энергичном перемешивании прибавляют по каплям сухой этанол
или полностью очищенный от ненасыщенных соединений лигроин с т. кип. 70—80° С.
Выпавший осадок дифенантрилолова отсасывают, промывают этанолом и высушивают в вакууме
над пятиокисью фосфора. Все операции проводят под азотом. Воздух из применяемых
растворителей должен быть предварительно удален. Чистый сухой продукт на воздухе устойчив.
Выход 12,0 г (69% в расчете на взятое хлористое олово) [9]. Ди-(9-фенантрил)олово
растворяют в минимальном количестве сухого бензола и под азотом фильтруют в трех горл ую
колбу на 500 мл, снабженную мешалкой, обратным холодильником и капельной воронкой.
В раствор интенсивно-красного цвета добавляют по каплям вначале сухой уксусноэтиловый
эфир, а затем равный объем сухого этанола. Образовавшуюся суспензию перемешивают
полчаса при 30—40° С, осадок ди-(9-фенантрил)олова отсасывают и быстро переносят в
наполненный азотом ваккуум-эксикатор с Р205. Вещество оставляют на ночь, затем измельчают под
азотом и высушивают в течение двух дней при 50—60° С и давлении Ю-4 мм. Получают
желтый или желто-оранжевый порошок с т. пл. 179—180° С. Выход 50—80% от неочищенного
ди-(9-фенантрил)олова.
Ди-(9-фенантрил)олово может быть получено в тщательно очищенном от
перекисей тетрагидрофуране. На 0,1 моля 9-бромфенантрена берут 100—
150 мл тетрагидрофурана. Выход бромистого 9-фенантрилмагния составляет
91—94%. Экспериментальные условия немногим отличаются от описанных
выше. Выход ди-(9-фенантрил)олова равен 60,2% [9].
Чистое ди-(9-фенантрил)олово к окислителям сравнительно устойчиво,
после месячного хранения на воздухе остается практически без
изменения [9].
Дифенилолово при нагревании с избытком бромистого фенилмагния уже
при 100° С выделяет олово с образованием гексафенилдистаннана
3 (С6Н5)2 Sn -> (СбН5)з Sn-Sn (С8Н5)3 + Sn.
Получение гексафенилдистаннана [29]. В магнийорганическое соединение, полученное
из 156 г бромбензола и 24 г стружек магния в 400 мл сухого эфира, довольно быстро при
взбалтывании вносят 35 г безводного тонко размельченного хлористого олова таким образом,
чтобы кипящий эфир изолировал продукт реакции от внешнего воздуха (применение азота в
данном случае излишне). Полученный темно-коричневый раствор дифенилолова немедленно
(чтобы избежать окисления) энергично кипятят 3 часа на водяной бане с обратным
холодильником. Жидкость чернеет от выделившегося олова (иногда на стенках образуется темное
зеркало). Эфир отгоняют, остаток нагревают еще 1—2часа на кипящей водяной бане. Отогнанный
эфир приливают обратно к охлажденному остатку, соли магния разлагают осторожным
добавлением воды. Затем эфир полностью отгоняют или испаряют, осадок отсасывают,
высушивают при температуре не выше 100° С и экстрагируют бензолом. Т. пл. 237° С; выход 30 г,
Гексафенилдистаннан кристаллизуется с 2 молекулами бензола,
которые он теряет при хранении в вакуум-эксикаторе [26]. Указанный метод
приложим лишь для получения гексафенилдистанана, получить таким
путем соединения с радикалом тяжелее фенила не удается НО].
Вероятно, из-за стерических препятствий ди-(9-фенантрил)олово даже
в жестких условиях не диспропорционирует на металлическое олово и гек-
са-(9-фенантрил)дистаннан [5].
б#с-(Бифенил-2)-олово, образующееся при действии на хлористое олово
бромистого бифенил-2-магния или бифенил-2-лития, диспропорционирует.
Из продуктов реакции с низким выходом (5—6%) выделена низкоплавкая,
хорошо растворимая форма дистаннана [51. (О высоко плавкой форме этого
соединения см. стр. 504.)
Синтез действием металлоорганических соединений на двугалоидное олово 199
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ВЗАИМОДЕЙСТВИЕМ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ДВУХЛОРИСТЫМ ОЛОВОМ
В синтезе оловоорганических соединений ртутноорганические
соединения не имеют столь общего значения, как, например, реактив Гриньяра,
хотя с их помощью и могут быть получены основные типы
оловоорганических соединений R4Sn, R3SnX, R2SnX2.
Ртутноорганические соединения применяют в реакциях с SnCl2, SnBr2,
SnCl4 (см. стр. 260), Sn, SnNa, (R2Sn)n, (R3Sn)2 (см. стр. 269).
Наиболее разработанной является реакция с хлористым и бромистым
оловом, дающая возможность синтезировать оловоорганические
соединения типа R2SnX2 [37, 38].
R2Hg + SnX2 -* R2SnX2 + Hg.
Реакционная способность солей двухвалентного олова во многом зависит от
их растворимости. Обычно применяют хлористое или бромистое олово. В
некоторых случаях реакция с бромистым оловом идет быстрее, чем с
хлористым [39]. Йодистое олово по указанной схеме не реагирует, фтористое
и сернокислое олово реагируют крайне медленно [37].
Реакцию проводят обычно в спирте, ацетоне, петролейном эфире или
нагреванием тонкой смеси веществ без растворителя. В петролейном
эфире, несмотря на слабую растворимость в нем хлористого и бромистого
олова, реакция протекает достаточно легко [40].
В алифатическом ряду в реакциях с двугалоидным оловом находят
применение в основном ртутноорганические алкены. Соединения предельного
ряда вступают в реакцию крайне медленно, ртуть выделяется неполностью,
несмотря на многочасовое кипячение. В результате реакции обмена
(С2Н5)2 SnCl3 + (C2H5)2 Hg -* (С2Н5)з SnCl + C2H5HgCl
образуется смесь оловоорганических галогенидов различной степени алки-
лирования. Так, двухлористое диэтилолово получено с выходом 52% (т. пл.
83—83,5° С). Помимо того, выделены хлористое триэтилолово, хлористая
этилртуть, некоторое количество этана и металлическая ртуть [37, 38, 40].
Для синтеза оловоорганических алкенов типа R2SnX2 в качестве
растворителя лучше применять петролейный эфир. В этом случае образование
соединений типа (C2H50)2SnX2 исключается. Геометрическая конфигурация
изомеров при реакции сохраняется. Стереоизомерное двугалоидное дипро-
пенилолово выделено с выходом до 70—75%. В спирте или ацетоне пропе-
нильные соединения олова получить не удается [41, 42].
С диизопропенилртутью реакция протекает более сложно. В различных
растворителях образуется, помимо двубромистого диизопропенилолова,
тетраизопропенилолово, бромистая изопропенилртуть и металлическая
ртуть. Те же продукты получены при взаимодействии веществ без
растворителя [41—43].
Стереоизомерные хлориды ди-Р-хлорвинилолова образуются с хорошим
выходом в спиртовом растворе
(ClCH=CH)2Hg + SnCl2-> (С1СН=СН)2 SnCU + Hg,
траяс-траяс-Соединение реагирует заметно быстрее, чем его цис-цис-
изомер, но оба они реагируют легче, чем дифенилртуть. Реакция протекает
на холоду. Из твердого ртутного траяс-траяс-Р-хлорвинильного продукта
образуется твердое двухлористое траяс-траяс-ди-Р-хлорвинилолово, из
жидкой ^wc-ф/с-ди-Р-хлорвинилртути — жидкое двухлористое цис-цис-
ди-Р-хлорвинилолово. Оба вещества обладают типичным запахом оловоор-
200
Оловсорганические соединения. Лит. стр. 203—204
ганических соединений [44, 45]. Жидкий (С1СН = CH)2SnCl2 может быть
получен облучением ультрафиолетовым светом твердого стереоизомера [46].
Действие хлористого олова на /юра#с-/яра#с-ди-р-хлорвинилртуть [44]. 2 г (0,0063
моля) транс- транс- ди-|3-хлорвинилртути в Ъ мл сухого спирта, слегка подкисленного
сухим хлористым водородом, и 1,2 г (0,0063 моля) безводного хлористого олова в Ъ мл сухого
спирта сливают вместе. Реакция начинается уже на холоду. После нагревания в течение
10 мин. при 45—50° С и часового стояния при комнатной температуре происходит почти
количественное выделение мелкораздробленной ртути (1,2 г, 97%). После прекращения реакции
прозрачный раствор декантируют с выпавшей в осадок металлической ртути. Спирт отгоняют
возможно полно при 50—60° С (в вакууме водоструйного насоса), затем прибавляют 10 мл
петролейного эфира, с которым отгоняют остатки спирта. Содержимое колбы дважды пере-
кристаллизовывают из петролейного эфира. Получают 1,05 г (58%) белого кристаллического
вещества с т. пл. 77,5—78,5° С; при 135° С вещество в капилляре желтеет [44].
Дибензилртуть вступает в реакцию с двугалоидным оловом особенно
легко. Двухлористое дибензилолово (т. пл. 65° С) получено почти с коли-
чественным выходом, двубромистое дибензилолово (т. пл. 129°) — с выходом
88%. Следует применять свежеприготовленную дибензилртуть. С
хранившимся препаратом, даже после его перекристаллизации, выход ниже [37].
Двухлористое ди-|3-стирилолово получено с выходом 24% (т. пл. 116° С)
[47]. Взаимодействие /?2ра#£-а-меркур-б^£-стильбена с хлористым оловом в
ацетоне, спирте или без растворителя в расплаве при 240—260° С не
приводит к образованию оловоорганических соединений. С ртутноорганиче-
скими производными ^ш:-стильбена при комнатной температуре образуется
смесь дву хлор истого ди-а-стильбенилолова с треххлористым а-стильбенил-
оловом и ^wc-стильбеном. После гидролиза водным аммиаком
нерастворимая в органических растворителях окись ди-а-стильбенилолова может
быть отделена от а-стильбенилстанноновой кислоты (т. пл. 157—160° С).
При 70—80° С образуется только треххлористое а-стильбенилолово
(т. пл. 108—110° С) и 20—30% стильбена. Геометрическая конфигурация
стильбена в реакциях обмена сохраняется [48].
Синтез дигалогенидов диарилолова разработан наиболее широко. По
сравнению с реактивом Гриыьяра ртутноорганические соединения имеют то
основное преимущество, что позволяют синтезировать оловоорганические
соединения с активными заместителями в ароматическом ядре. В качестве
типичного в методическом отношении примера можно привести следующий.
Получение двухлористого дифенилолова [37, 38]. К 5 г дифенилртути и 4 г безводного
хлористого олова (избыток около 50%) прибавляют 80 мл сухого спирта. Реакция
начинается уже на холоду. Затем смесь нагревают до кипения в течение 30 мин. Выделившаяся ртуть
оседает в виде тяжелого порошка (2,73 г, 96%). Спирт (слитый с осадка) возможно полно
отгоняют, а к остатку прибавляют около 50 мл петролейного эфира, который при отгонке
увлекает за собой остатки спирта. Для извлечения образовавшегося оловоорганического
соединения остаток, состоящий из избытка хлористого олова и двухлористого дифенилолова,
обрабатывают несколько раз петролейным эфиром и раствор отфильтровывают. После
отгонки растворителя в колбе остается 3,7 г густого масла, нацело закристаллизовывающего-
ся после продолжительного стояния. Продукт перекристаллизовывают из лигроина; выход
80%; т. пл. 42° С [37, 38].
При взаимодействии дифенилртути с хлористым оловом в петролейном
эфире при различных температурных условиях образуется только
хлористое трифенилолово. Двухлористое дифенилолово в этом случае является
промежуточным продуктом [40].
Ди-я- и ди-0-толилртуть реагируют с хлористым оловом с образованием
только двухлористого ди-я-(т. пл. 49,5° С) и соответственно ди-о-толилолова
(т. пл. 49,5—50° С). С ди-а-нафтилртутью образуется двухлористое ди-а-
нафтилолово (т. пл. 136—137° С). Выходы достаточно высокие. Ароматические
соединения олова с заместителями в бензольном ядре типа НО или (CH3)2N
получить не удалось [40].
Синтез действием металлоорганических соединений на двугалоидное олово 201
Получение двухлористого ди-я-толилолова [40]. 3 г (0,0078 моля) ди-я-тол ил ртути и 3 г
(0,025 моля) хлористого олова в 20 мл лигроина (т. кип. 100—110° С) энергично
перемешивают при 90—95° С в течение 13 час. После охлаждения осадок отделяют, трижды
экстрагируют нагретым лигроином, затем метанолом, растворы соединяют вместе. После отгонки
растворителя жидкий остаток полностью закристаллизовывается. Выход 2,1 г (72%); т. пл.
40—43° С. Вещество перекристаллизовывают из петролейного эфира. Получают 1,85 г
двухлористого ди-л-толилолова с т. пл. 49,5° С. Металлической ртути выделено 1,51 г (96%).
Природа растворителя при реакции играет существенную роль. Все
ароматические соединения Ar2Hg в ацетоне или лигроине [40] реагируют
с образованием R2SnX2. В спиртовой среде ртутноорганические
соединения с боковой цепью в бензольном ядре и, в частности, а- и Р-нафтилртуть
претерпевают отщепление ароматического ядра в виде углеводорода [37,
49, 50]
R2Hg + SnX2 + С2Н5ОН -» 2 RH -f Hg + (C2H50)2 SnX3.
В большинстве случаев в сухом спирте обе реакции протекают
одновременно [37]. Поэтому при получении, например, дихлоридов ди-а-нафтил-
олова (87%), ди-Р-нафтилолова (66%), дибромидов ди-п-толилолова (88%),
ди-а-нафтилолова (60%) нужно работать в сухом ацетоне [37, 38] или пет-
ролейном эфире [40].
В сухом спирте получены: двухлористое ди-п-хлорфенилолово (т. пл.
86,5° С, 75%), двухлористое ди-я-бромфенилолово (т. пл. 103° С, 85%),
двухлоршгрэе ди-д-иодфенилолово (т. пл. 147° С, 75%), двубромистое ди-м-
хлорфенилолово (т. пл. 73° С, 92%), двубромистое ди-д-бромфенилолово
(т. пл. 82° С, выход близок к теорет.), двубромистое ди-п-иодфенилолово
(т. пл. 102° С, 70%). Во всех этих случаях можно вести реакцию и в
ацетоне, в котором, как и в спирте, хлористое и бромистое олово хорошо
растворимы.
Получение двубромистого ди-#-бромфенилолова [39]. 5,12 г (0,01 моля) ди-ос-бромфе-
нилртути и 4,2 г (0,015 моля) безводного бромистого олова кипятят в течение получаса в
30 мл сухого спирта. Выделившаяся ртуть весит 2 г (теоретическое количество). Густой
маслообразный остаток, полученный после обработки и отгонки петролейного эфира, через
один час нацело закристаллизовывается. Выход 5,7 г (96%). После перекристаллизации
из петролейного эфира т. пл. 82° С [39, 51].
Получение двухлористого ди-а-нафтилолова. 4,54 г (0,01 моля) ди-а-нафтилртути
и 2,85 г (0,015 моля) хлористого олова в 50 мл ацетона нагревают до кипения в течение
получаса. Количество выделившейся ртути близко к теоретическому. После отгонки ацетона
остаток обрабатывают петролейным эфиром, как обычно (см. выше). После
перекристаллизации из петролейного эфира получают двухлористое ди-а-нафтилолово ст. пл. 137—137,5° С;
выход 66% [37, 38, 40].
Дихлориды и дибромиды диарилолова легко гидролизуются, дииодиды
более устойчивы [41]. Примером аналогичной реакции без растворителя
может служить следующий.
Получение двухлористого ди-я-карбэтоксифенилолова (C2H5OOCC6H4)2SnCI2 [52]. 4 г
(0,021 моля) безводного хлористого олова расплавляют в колбе при 250—260° С (дана
температура бани) и к нему быстро прибавляют 5 г (0,01 моля) ди-п-карбэтоксифенилртути,
предварительно расплавленной в пробирке. Смесь перемешивают при этой температуре в
течение 5 мин., на дне колбы появляется металлическая ртуть. Реакционную смесь после
охлаждения обрабатывают петролейным эфиром (т. кип. 60—80° С). Из экстракта при
стоянии выпадает 3,5 г (73%) кристаллического осадка. Точка плавления один раз
перекристаллизованного двухлористого ди-п-карбэтоксифенилолова 83—84° С. Ртути выделяется
1,6 г (80%).
Ртутноорганические галогениды в реакциях с хлористым или бромистым
оловом применяются очень мало. Хлористая алкилртуть (RHgCl) по
отношению к двугалоидному олову чрезвычайно мало реакционноспособна
[37]. Хлористая бензилруть, хлористая фенилртуть и их аналоги вступа-
202 Оловоорганшеские соединения. Лит. стр. 203—204
ют в реакцию по уравнению
2RHgCl + 2 SnCl2 -* RsSnCls + 2 Hg + SnCl4.
Направление реакции зависит от ртутноорганического соединения и от
галоида [37]. Ароматические ртутноорганические бромиды реагируют сразу
по двум направлениям: аналогично хлоридам с образованием SnBr4 и,
помимо того, с выделением бромистой ртути
2 RHgBr + SnBr2 -> R2SnBr3 + Hg3Br2.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ ВЗАИМОДЕЙСТВИЕМ ТАЛЛИЙ-,
СВИНЦОВО- И ДРУГИХ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ДВУХЛОРИСТЫМ (ДВУБРОМИСТЫМ) ОЛОВОМ
Таллийорганические галогениды типа R2T1X находят применение в
основном для синтеза оловоорганических алкенов и оловоорганических
соединений ароматического ряда
(СН2=СН)2 Т1Х + SnX2 -* (СН2=СН)2 SnX2 + Т1Х.
Экспериментальные условия аналогичны описанным для двузамещен-
ных ртутноорганических соединений (см. стр. 199). Из бромистого диви-
нилталлия и бромистого олова в ацетоне при нагревании получают дву-
бромистое дивинилолово с т. кип. 47° С/2 мм; d*°2,1924; ng 1,5920. Дву-
хлористоедивинилолово (т. кип. 53—55° С/2 мм, dfl,7621; п2£ 1,5500)
образуется аналогично [53].
Хлористый ди-Р-хлорвинилталлий в сухом метиловом спирте реагирует
с хлористым оловом при комнатной температуре с образованием
кристаллического двухлористого ди-Р-хлорвинилолова. Продукт, вероятно, имеет
траяс-строение (т. пл. 76,5—78° С). Во всех исследованных случаях в
реакциях конфигурация хлорвинильного радикала остается неизменной
[54]. Хлористый ^с-фя>ди-|3-хлорвинилталлий реагирует с хлористым
оловом с образованием жидкого двухлористого цис- ^с-ди-|3-хлорвинилолова
(т. кип. 100—103° С/3 мм) [46]. Двубромистое дипропенилолово
синтезируют из бромистого дипропенилталлия и бромистого олова в бензоле при
50° С [41]. Оловоорганические соединения в отличие от галоидного таллия
растворимы в органических растворителях и легко могут быть от него
отделены.
Оловоорганические дигалогениды получены также нагреванием
исходных соединений без растворителя, выход от 50 до 85% [41].
Получение двубромистого диизопропенилолова [43]. 5,0 г (0,0136 моля) бромистого
диизопропенилталлия и 10 г (0,035 моля) бромистого олова тщательно растирают в тонкий
порошок и вносят в колбу Вюрца с низкоприпаянным отводом. При быстром нагревании
смеси до 200° С в приемник переходит 3,5 г (75%) жидкого неприятно пахнущего вещества,
которое перегоняют в вакууме. Получают 2,2 г двубромистого диизопропенилолова с т. кип.
100—110° С/10 мм [43].
Без растворителя, когда взаимодействие имеет место при сравнительно
высокой температуре, могут быть получены, по-видимому, только смеси
стереоизомеров [41 ].
Аналогично синтезированы двубромистое дипропенилолово (70%, т.
кип. 125° С/10 мм), двухлористые дифенилолово (69%, т. пл. 40—41° С)
и ди-д-толилолово (60%, т. пл. 49—49,5° С), а также двубромистое ди-а-
нафтилолово (50%, т. пл. 140,5—142° С) [43].
Свинцовоорганические соединения R2PbX2 вступают в реакцию с
хлористым оловом аналогично ртутно- или таллийорганическим соединениям
R2PbCl2 + SnCl2 -* R2SnCl2 + РЬС12.
Синтез действием металлоорганических соединений на двугалоидное олово 203
Реакцию проводят в сухом спирте или ацетоне. Двухлористое ди-Р-хлор-
винилолово образуется с выходом 67,1% уже при комнатной температуре
[55]. Обычно необходимо длительное нагревание.
Взаимодействие двухлористого диэтилсвинца с хлористым оловом [56]. 9 г дву
хлористого диэтил свинца и 5 г безводного хлористого олова в 90 мл сухого спирта, подкисленного
сухим хлористым водородом, кипятят с обратным холодильником в течение 15 час. После
охлаждения осадок отфильтровывают. Спирт из фильтрата отгоняют в вакууме досуха,
остатки спирта удаляют добавлением петролейного эфира с последующей отгонкой в
вакууме. Сухой остаток экстрагируют горячим сухим петролейным эфиром (70—90° С), эфир
испаряют. В остатке 3,3 г (51%) вещества с т. пл. 84° С (из петролейного эфира).
Двухлористое диметилолово получено аналогично с выходом 47% [56].
Дву хлор истый дифенилсвинец с безводным хлористым оловом образует
двухлористое дифенилолово, но с низким выходом [56]. Свинцовооргани-
ческие соединения класса RPbX3 в реакциях с солями двухвалентного
олова не исследованы. Четырехзамещенные свинцовоорганические
соединения, например тетрафенилсвинец, хлористым оловом деарилируются.
Реакция заканчивается образованием плохо растворимого (С6Н5)2РЬХ2
[57].
Из прочих металлоорганических соединений в реакциях с солями
двухвалентного олова применяют дициклопентадиенилнатрий для синтеза ди-
циклопентадиенилолова [58]. Борорганические соединения R3B и NaBR4?
которые приводят к R2Sn и Sn (BR4)2 [59—61], и алюминийорганические
соединения [62, 63] описаны очень кратко.
ЛИТЕРАТУРА
1. 3 е м л я н с к и й Н. Н., ПановЕ.М., К о ч е ш к о в К. А., ДАН СССР, 146,
1335 (1962).
2. F i s с h e r E. О., Grubert H., Z. Naturforsch., Ив, 423 (1956).
3. W i t t i g G., M e у e r F. J., LangeG., Ann., 571, 167 (1951).
4. N e u m a n n W. P., KonigK., Ann., 677, 1 (1964).
5. В a h r G., G e 1 i u s R., Ber., 91, 825 (1958).
6. К u i v i 1 a H. G., S a w у е г A., ArmourA.G, J. Org. Chem., 26, 1426
(1961).
7. N e u m a n n W. P., KonigK., Angew. Chem., Internat. Ed., 1, 212 (1962).
8. N e u m a n n W. P., KonigK., Angew. Chem., 74, 215 (1962).
9. В a h r G., GeliusR., Ber., 91, 829 (1958).
10. К r a u s e E., BeckerR., Ber., 53, 173, (1920).
11. KrauseE., RenwanzG, Ber., 60, 1582 (1927).
12. N e u m a n n W. P., Angew. chem., 75, 225 (1963).
13. P f e i f f e r P., Ber., 44, 1269 (1911).
И. Ингам Р., РозенбергС, ГильманГ., Р и к е н с Ф., Оловооргани-
ческие и германийорганические соединений. М., ИЛ, 1962.
15. Vander Kerk G. J. M., L u i j t e n J. G. A., Noltes J.G., Angew.
Chem., 70, 298 (1958).
16. F a r r a r W. V., S k i n n e r H. A., J. Organometal. Chem., 1, 434 (1964).
17. N e u m a n n W. P., Angew. Chem., Intern. Ed., 2, 173 (1963).
18. R i n d 1 e R. E., OlsonD.H., Inorg. Chem., 3, 596 (1964).
19. OlsonD.H., R i n d 1 e R. E., Inorg. Chem., 2, 1310 (1963).
20 Гол ь да иски й В. И., Рогов В. Я-, X р а п о в В. В., ДАН СССР, 151,
909 (1964).
21. N е u m a n W. P., KonigK., Ann., 677, 12 (1964).
22. NeumannW.P., P e d a i n J., Ann., 672, 34 (1964).
23. Б р а у е р Г., Руководство по препаративной неорганической химии. М., ИЛ,
1956, стр. 347.
24. К г a u s е Е., Р о h 1 a n d R., Ber., 57, 532 (1924).
25. П е т р о в А. А., 3 а в г о р о д н и й В. С, ЖОХ, 34, 2806 (1964).
26. KrauseE., Grosse A., «Die Chemie der metalloogranischen Verbindimgen».
Berlin, Gebr. Borutrager, 1937, S. 354, 356, 360.
204
Оловоорганические соединения
27. Coates G. E., Organo-metallic compounds, London — N. Y., John Willey, I960»
p. 200.
28. Zimmer H., Hechenb leikner J., HombergO. A., Danzi M.,
J. Org, Chem., 29, 2632 (1964).
29. R e i d A. F., W a i 1 e s P. C, Austral. J. Chem., 19, 309 (1966); РЖХим., 1966,
19Ж 307.
30. ChambersR.F., SchererP. C, J. Am. Chem. Soc, 48, 1054 (1926),
31. J e n s e n К. А., С 1 a u s о n-K ass N., Z. anorg. allgem., Chem., 250, 277 (1943).
32. В о e s с к e n J., Rutgers J.J., Rec. trav. chim., 42, 1017 (1923).
33. G i 1 m a n H., Car t ledge F. K-, Chem. and Ind., 1964, 1231.
34. К u i v i 1 a H. G., Beumel O.F., J. Am. chem. Soc, 80, 3250 (1958).
35. КрафтМ.Я., Бородина Г. М., ЖОХ, 32, 1665 (1962).
36. Bah r G., Zoche G., Ber., 88, 1450(1955).
37. К о ч е ш к о в К. А., Несмеянов А.Н., ЖРФХО, 62, 1795 (1930).
38. Nesmejanow A.N., К о с h e s с h к о w К. А., Вег., 63, 2496 (1930).
39. Н е с м е я н о в А. Н., К о ч е ш к о в К. А., ЖОХ, 1, 219 (1931).
40. Н е с м е я н о в А. Н., Борисов А. Е., Н о в и к о в а Н. В., О с и п о-
в а М. А., Изв. АН СССР, ОХН, 1959, 263.
41. Несмеянов А. Н., Борисов А. Е., Н о в и к о в а Н. В., Изв. АН СССР,
ОХН, 1959, 644.
42. Несмеянов А. Н., Борисов А.Е., Н о в и к о в а Н. В., Изв. АН СССР,
ОХН, 1959, 1216.
43. Н е с м е я н о в А. Н., Борисов А. Е., Новикова Н. В., Изв. АН СССР,
ОХН, 1959, 259.
44. Н е с м е я н о в А. Н., Борисов А. Е., Абрамова А. Н., Изв. АН СССР,
ОХН, 1946, 647.
45. Несмеянов А.Н., Борисов А.Е., ДАН СССР, 60, 67 (1948).
46. Несмеянов А. Н., Борисов А.Е., Шепелева Р. И., Изв. АН СССР,
ОХН, 1949, 582.
47. Н е с м е я н о в А. Н., Кудрявцева Т. А., Уч. зап. МГУ, 151, 57 (1951).
48. Несмеянов А. Н., Борисов А. Е., В о л ь к е н а у В. А., Изв. АН СССР,
ОХН, 1956, 162.
49. Несмеянов А.Н., Усп. хим., 3, 34 (1934).
50. Несмеянов А. Н., К о ч е ш к о в К. А., Уч. зап. МГУ, 3, 283 (1934).
51. Kozeschkow К- A., Nesmejanow A.N., Вег., 64, 628 (1931).
52. Э с к и н И. Т., Н е с м е я н о в А. Н., К о ч е ш к о в К. А., ЖОХ, 8, 35 (1938).
53. Несмеянов А. Н., Борисов А. Е., Савельева И. С, Голубе-
в а Е. И., Изв. АН СССР, ОХН, 1958, 1490.
54. Несмеянов А. Н., Ф р е й д л и н а Р. X., Кочетков А. Км Изв. АН
СССР, ОХН, 1948, 445.
55. Несмеянов А. Н., Ф рейдлина Р. Ф., К о ч е т к о в А. К., Изв.
АН СССР ОХН 1948 127
56. Кочетков АДм'фрейдлинаР.Х., Изв. АН СССР, ОХН, 1950, 203.
57. G о d d a r d А. Е., A s h 1 е у J. N.. Е v a n s R. В., J. Chem. Soc, 121, 978 (1922).
58. DaveL.D., E v a n s D. F., WilkinsonG., J. Chem. Soc, 1959, 3684.
59. R i d d 1 e J. M., Пат. США 2950302 (1960); РЖХим., 1961, 22Л71.
60. R i d d 1 e J. M., Пат. США 2950301 (1960); РЖХим., 1961, 23Л98.
61. К о b e t z P., Пат. США 3134796 (1964); РЖХим., 1965, 23H67.
62. S m i t h Т. D„ Nature, 199, 374 (1963).
63. M a n g h a n J. R., Пат. США 3095433 (1963); РЖХим., 1965, 23H67.
Глава HI
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА ПРОИЗВОДНЫЕ ЧЕТЫРЕХВАЛЕНТНОГО ОЛОВА
Действие металлоорганических соединений на соли четырехвалентного
олова или оловоорганические галогениды является наиболее
распространенным в лабораторных условиях методом синтеза оловоорганических
соединений. Вне зависимости от молярного соотношения реагентов реакция
обычно проходит в направлении образования четырехзамещенных
соединений олова. Применение этого метода для синтеза оловоорганических га-
логенидов не типично и описано лишь на отдельных примерах.
В качестве исходных соединений чаще всего используют легко
доступные галоидные соли олова или оловоорганические галогениды. Менее
характерно применение для этой цели окисей, алкоксисоединений или
сульфидов олова.
Из металлоорганических соединений наиболее широкое применение
нашли магнийорганические соединения. Соединения лития используют,
как правило, только тогда, когда приготовление реактива Гриньяра
связано с трудностями или там, где необходим реагент с повышенной
реакционной способностью.
В отдельных случаях, как, например, при получении оловоорганических
ацетиленидов, наиболее целесообразно исходить из натрийорганических
соединений. Из других металлоорганических соединений необходимо особо
отметить соединения алюминия.
Для получения оловоорганических соединений можно применять также
органические соединения цинка, таллия, ртути, свинца и некоторых
других металлов. Однако эти реакции не получили широкого
распространения.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ РЕАКТИВА ГРИНЬЯРА
Соли четырехвалентного олова (обычно галоидные соли) реагируют с
реактивом Гриньяра с образованием оловоорганических соединений. Для
получения простейших оловоорганических соединений типа R4Sn, которые
часто служат важным исходным материалом, действие магнииорганических
соединений является одним из самых удобных методов синтеза и
выражается уравнением
4 RMgCl + SnCU -* R4Sn + 4 MgCl2.
Для синтеза соединений R3SnCl и Я^пС^метод имеет ограниченное
применение, так как обычно при взаимодействии рассчитанных количеств
реагентов образуется трудноразделимая смесь продуктов с различной
степенью алкилирования.
206
Олоесорганшеские соединения. Лит. стр. 261—268
Взаимодействие хлорного олова со смесями магнийорганических
соединений находит применение, например, для синтеза диметилдиэтилолова
[1]
SnCl4 + 2 CH3MgBr + 2 C2H5MgBr -> (СН3)2 Sn (C2H5)2 «f 4 MgBr2.
Соединения со смешанными радикалами R3SnR', R2SnR'2 и R2R'SnX
получают действием R'MgX на оловоор ганические галогениды RgSnX и
R2SnX2.
Взаимодействие реактива Гриньяра с галогенидами олова
Реактив Гриньяра (как обычно в эфире) приготовляют из хлористых,,
бромистых и реже йодистых алкилов. Хлорное и бромное олово (как
неорганические компоненты) равноценны, но в лабораторных условиях удобно*
пользоваться бромным оловом в растворе бензола, толуола или эфира [2].
Алкоксисоединения Sn (OR)4 имеют ограниченное применение [3—5].
Хлорное олово образует с эфиром нерастворимый твердый эфират.
Помимо эфира, реакцию проводят также в дибутиловом эфире, тетра-
гидрофуране, реже — в углеводородах. Тетрагидрофуран как
растворитель имеет в известных случаях преимущество перед эфиром [6, 7]. Так,
ряд синтезов, например получение тетравинилолова, удается провести
только в тетрагидрофуране.
С помощью реактива Гриньяра получены оловоорганические
соединения с радикалами всех типов.
Тетраалкильные производные олова имеют особое
значение в качестве промежуточных продуктов. В алифатическом ряду —
это бесцветные жидкости, легко растворимые в органических
растворителях (начиная с тетрадодецилолова — твердые низкоплавкие вещества).
Соединения Ar4Sn — твердые кристаллические тела с высокой температурой
плавления.
Соединения этих типов устойчивы к действию воды и водного
раствора щелочи, воздуха и окислителей, но легко вступают в реакцию с
кислотами, галоидами, сулемой, хлорным оловом, переходя в соединения
с более низкой степенью алкилирования.
При синтезе необходимо применять избыток магнийорганического
соединения, так как иначе наряду с R4Sn образуется значительное количество
соединений типа R3SnX и R2SnX2 [6, 7]. Даже при избытке реактива
Гриньяра обычно остаются моногалогениды, которые трудно удалить путем
фракционирования. По-видимому, наиболее целесообразно, особенно при
большом количестве монохлорида, удалять его встряхиванием с водной
щелочью или водно-спиртовым раствором нейтрального фтористого калия.
В последнем случае выделяются труднорастворимые, хорошо
кристаллизующиеся фториды триалкилолова [8—10]. В случае малых количеств
монохлорида пропускают сухой аммиак в сухой эфирный раствор тетраалкилоло-
ва. При этом выделяется нерастворимый в эфире R3SnCl-2NH3 [11].
Предлагается разлагать избыток магнийорганического соединения
водой или раствором хлористого аммония на холоду. С соляной кислотой,
которая обычно применяется для этой цели, возможны реакции деалкили-
рования [5].
При синтезе оловоорганических соединений с более сложными
радикалами (вторичными, третичными) реакция заканчивается на образовании
моногалогенида. Побочной реакцией в этом случае является
восстановление оловоорганических соединений реактивом Гриньяра до R3Sn—SnR3 и
R2Sn (см. стр. 212).
Синтез на основе производных четырехвалентного олова
207
Получение тетраметилолова [12, 13]. В литровую круглодонную колбу, снабженную
мешалкой с ртутным затвором, обратным холодильником (защищать от влаги воздуха),
термометром и капельной воронкой, помещают 50 г (2,06 г-атома) стружек магния в 600 мл
дибутилового эфира. Приготовляют раствор 225 г (1,59 моля) свежеперегнанного йодистого
метила в равном объеме того же растворителя. После прибавления нескольких
кристалликов иода в колбу вводят при перемешивании 3—5 мл раствора йодистого метила. Реакция
начинается, как правило, легко, но иногда требуется нагревание. Остальное количество
йодистого метила прибавляют по каплям со скоростью, необходимой для умеренного кипения
реакционной среды, на что требуется около 3 час. В смесь после ее охлаждения до
комнатной температуры прибавляют по каплям в течение 2—2,5 час. 50—75 г (0,19—0,29) моля
безводного хлорного олова с такой скоростью, чтобы поддерживать умеренное кипение.
После введения хлорного олова реакционную смесь кипятят (85—95° С) еще час и оставляют
на несколько часов. Обратный холодильник заменяют нисходящим, сырой продукт отгоняют
из реакционной колбы. Получают смесь тетраметилолова и «-бутилового эфира с т. кип. 85—
95° С. Тетраметилолово выделяют перегонкой на колонке в 35—40 теоретических тарелок.
Основная его фракция перегоняется при 76,6 ° С /748 мм; выход 85—91% [12, 14].
Применение дибутилового эфира дает возможность повысить
температуру реакции и тем самым сократить время ее протекания. Однако дибу-
тиловый эфир как растворитель не получил пока еще достаточно широкого
применения в синтезе оловоорганических соединений. Синтез
тетраметилолова чаще проводят в этиловом эфире [15]. В этом случае выход
неустойчив и часто низок. Представляет затруднение отделение тетраметилолова
от большого количества эфира [16—18]. Достаточно эффективна колонка
высотой 75 см, диаметром 2 см, наполненная мелкими кусочками стеклянной
трубки [19]. Для удаления хлористого триметилолова, которое образуется
в значительных количествах, продукт промывают раствором
фтористого натрия, водой -и соляной кислотой [18]. Т. кип. 76,7° С;
77—77,5° С; d25l,2900, ng 1,4405 [10, 17, 20, 21]; 78,3° С/740 лш; az2J 1,439а
[18].
Имеется ряд достаточно подробных описаний синтеза [3, 5, 9, 22—28]
тетраметилолова. По-видимому, для завершения реакции необходима более
высокая температура, чем та, которую можно получить в этиловом эфире
при атмосферном давлении. Предложено, например, заканчивать синтез
следующим приемом: после введения хлорного олова в эфирный раствор
йодистого метилмагния реакционную смесь нагревают 4 часа на водяной
бане, затем всю жидкость отгоняют на масляной бане при 140° С. Тетра-
метилолово выделяют фракционированием дистиллята с выходом 70,4%;
т. кип. 75—77° С/720 мм (однако выход не стабилен) [22].
Тетраметилолово, полученное методом Гриньяра, может содержать
в виде примеси и йодистый метил. Его удаляют нагреванием с пиридином
в запаянной трубке в течение полутора часов при 80° С [29]. Свойства
тетраметилолова исследованы довольно подробно, приведены его
спектроскопические [16, 18], термохимические [17, 21, 23] и другие характеристики [30].
Этиловый эфир, как реакционная среда, находит широкое применение
в синтезе ближайших аналогов тетраметилолова. Для повышения
температуры в реакционной колбе растворитель отгоняют, но в отличие от
тетраметилолова более высококипящие оловоорганические соединения
остаются в реакционной колбе и могут прореагировать с избытком магнийорга-
нического соединения более полно. Тетраэтил-, тетрапропил-, тетрабутил-
олово в отличие от тетраметилолова образуется, как правило, с хорошим
выходом.
Получение тетраэтилолова [31]. В двухлитровую трехгорлую колбу, снабженную
обратным холодильником с широкой внутренней трубкой, мешалкой и капельной воронкой,
помещают 50 г (2,05 г-атома) мелких магниевых стружек. Приготовляют раствор 250 г
(175 мл\ 2,3 моля) бромистого этила в 500 мл абсолютного эфира. В капельную воронку
наливают 5 мл раствора, к нему добавляют 3 капли брома, после чего смесь прибавляют к
магнию. Сразу начинается реакция Гриньяра, для поддержания которой постепенно при-
208 Оловоорганические соединения. Лит. стр. 261—268
бавляют остальной эфирный раствор бромистого этила. После того как самопроизвольная
реакция закончится, смесь нагревают при перемешивании в течение 30 мин. так, чтобы
раствор кипел. Затем колбу охлаждают в бане со льдом и в течение 20 мин. при
перемешивании прибавляют 83 г (37 мл\ 0,32 моля) хлорного олова (капельная воронка не должна
содержать паров эфира, так как последние образуют с хлорным оловом твердый комплекс).
Смесь нагревают при температуре кипения еще 1 час. Обратный холодильник заменяют на
нисходящий, мешалку останавливают и нагревают колбу так, чтобы в течение 1,5 час. было
отогнано около 200 мл эфира. По мере удаления эфира температура в реакционной колбе
поднимается и достигает в центре реакционной смеси 60—65° С. Колбу опять охлаждают
в бане со льдом, отогнанный эфир возвращают в реакционную смесь и последнюю разлагают,
медленно прибавляя сначала 85 мл ледяной воды, а затем 400 мл охлажденной до 0° 10%-ной
соляной кислоты. Несколько минут смесь перемешивают, а затем содержимое колбы
переносят в делительную воронку. Эфирный слой отделяют, фильтруют через складчатый фильтр
и сушат хлористым кальцием. Эфир отгоняют, неочищенное тетраэтилолово перегоняют в
вакууме. Выход тетраэтилолова с т. кип. 63—65° С/12 мм составляет 67—72 г (89—96%);
(T-h 1,1916; n2g 1,4693—1,4699 [31], т. кип. 175—177° С [3—5, 19,122,|28,|32—351; 180—182° С
[36—38]. С трехкратными количествами реагентов получены сравнимые выходы [31].
Тетраэтилолово, полученное по данной прописи, содержит только следы
хлористого триэтилолова [22, 31]. Для отделения последнего, если это
необходимо, высушенный эфирный раствор обрабатывают сухим аммиаком и
образовавшийся осадок отфильтровывают [31]. Иногда [22, 25, 39]
эфирный раствор фракционируют над натрием. Следует иметь в виду, что
тетраэтилолово при температуре его кипения несколько реагирует с
металлическим натрием [40]. Значительное количество монохлорида удобнее
отделить встряхиванием органического слоя, выделенного после разложения
реакционной среды с 3jV едким натром [36, 41]. Примесь хлорида часто
удаляют также в виде фтористого триэтилолова [2, 10, 23, 29] (растворение в
метаноле и встряхивание с водным раствором фтористого калия [42]).
Тетраэтилолово из йодистого этила и хлорного олова может быть
получено в изооктане [42]. Тетра-я-пропил-[3, 5, 19, 28, 29, 31, 42, 43] и тет-
ра-я-бутилолово могут быть получены по той же методике и примерно с
теми же выходами [31, 35, 36]. Для завершения реакции следует отгонять
растворитель [41].
При синтезе тетра-я-пропил- и других высококипящих оловоорганиче-
ских соединений хлорное олово прибавляют иногда в растворе бензола
[33, 36, 44].
Тетра-я-бутилолово в настоящее время находит промышленное
применение (например является исходным в синтезе солей органических кислот
дибутилолова). Для его производства существенным является возможность
применения хлористого бутила и растворителя, менее огнеопасного, чем
диэтиловый эфир (например дибутилового эфира), или синтез в безэфирной
среде.
Хлорное олово легко вступает в реакцию с реактивом Гриньяра в
толуоле, ксилоле, петролейном эфире и других растворителях. Магнийоргани-
ческие соединения в этих случаях могут быть получены действием
галоидного алкила на магний в углеводородах при незначительной добавке эфира
в качестве катализатора [45—50а] или без него [47, 51, 52]. Для
инициирования реакции в отсутствие эфира необходимо нагревание до 80—100° С,
после чего она идет с выделением тепла. Особенно легко вступают в реакцию
высшие галоидные алкилы, начиная с бутила. Температуру реакции
поддерживают в интервале 80—150° С, регулируя скорость введения
галоидного алкила.
Оловоорганические соединения можно получать как в две стадии, так
и в одну. При двухстадийном варианте процесса магний в виде стружки или
опилок нагревают с небольшим количеством соответствующего галоидного
алкила до начала образования магнийорганического соединения (в виде
белого порошка), после чего добавляют либо сначала углеводород, а затем
Синтез на основе производных четырехвалентного олова
209
постепенно галоидный алкил, либо сразу их смесь, либо только галоидный
алкил, если реакция проводится без растворителя. К полученной
суспензии магнийорганического соединения добавляют затем хлорное олово,
причем вторая стадия процесса также, как правило, протекает экзотер-
мично.
В случае одностадийного процесса к магнию, начавшему реагировать с
галоидным алкилом, одновременно добавляют углеводород, галоидный
алкил и хлорное олово; возможно проведение реакции без растворителя [51].
Получение тетра-#-бутилолова [51]. К 48,6 г (2 моля) активированных иодом стружек
магния добавляют при перемешивании (в атмосфере азота) смесь 65,14 г (0,25 моля) хлорного
олова и 185,14 г (2 моля) хлористого я-бутила. Добавление ведут с такой скоростью, чтобы
температура реакционной смеси удерживалась на уровне 90—100° С, после чего реакционную
смесь нагревают до той же температуры в течение одного часа. Тетра-я-бутилолово отгоняют
непосредственно из|реакционной массы в вакууме. Выход 77г (90%); т. кип. 117°С/1 мм; d|°
1,0570; /г^° 1,4750.
Для получения оловоорганических соединений в значительных
количествах очень удобно применять дибутиловый эфир.
Получение тетра-я-бутилолова [53]. В четырехгорлую колбу, емкостью 5 л,
снабженную хорошо действующей мешалкой, термометром, обратным холодильником и капельной
воронкой, загружают 194 г (8 молей) металлического магния, 500 мл ди-я-бутилового
эфира и 120 г (135 мл) хлористого я-бутила. Для инициирования реакции вводят бром или
кристаллики иода и нагревают до 65—70° С. Через 20—30 мин. начинается энергичная реакция;
температура повышается до 100—110° С. Остальное количество хлористого я-бутила 700 мл
(620 г; всего берут 8 молей) растворяют в 1500 мл ди-я-бутилового эфира и прибавляют в
реакционную колбу с такой скоростью, чтобы температура смеси не превышала 70—80° С.
Мешалку включают тогда, когда прореагирует около половины магния. После введения
хлористого бутила реакционную смесь перемешивают еще 2 часа при той же температуре.
Содержимое колбы охлаждают ледяной водой до 10—15° С и, не снимая охлаждения,
прибавляют 232,1 мл (516,8 г, 1,98 моля) хлорного олова с такой скоростью, чтобы температура
не превышала 25—30° С. После введения хлорного олова смесь нагревают в течение 3 час.
при температуре 60—70° С. Разлагают ледяной водой (при наружном охлаждении льдом),
затем приливают 300 мл соляной кислоты (1:1). Органический слой отделяют, водный слой
экстрагируют ди-я-бутиловым эфиром, высушивают, растворитель отгоняют, остаток
перегоняют в вакууме. Получают 527,8 г (76,7%) тетра-я-бутилолова ст. кип. 128—130° С/3 мм
[53]; 175—176° С/40 мм; #°41,0559; п2£ 1,4735 [22]. Реакция заканчивается значительно
быстрее, если в дибутиловый эфир после начала образования магнийорганического соединения
вводят смесь хлористого или бромистого бутила и хлорного олова [54].
S этиловом эфире [22, 31] тетра-я-бутилолово образуется с выходом до
89—96% [28, 44, 55—58].
При получении высших членов ряда R4Sn выходы сильно уменьшаются
[59]. Даже если эфир отгоняют и заменяют кипящим ксилолом при
избытке реактива Гриньяра (18 молей на 1 моль хлорного олова), всегда остается
монохлорид, который полностью удалить не удается. Его отделяют в виде
монофторида [60]. Из соединений указанного ряда синтезированы тетра-я-
амилолово (т. кип. 181° С/10 мм), тетра-я-гексилолово (т. кип. 209°/10 мм)
[19, 28, 44, 59], тетра-я-гептилолово (т. кип. 239° С/10 мм) [44, 59]; тетра-
я-октилолово (т. кип. 268° С/10 мм) [44,59, 61]; тетра-я-додецилолово
(выход 45%; т. пл. 15—16° С—помутнение, 21° С—полная прозрачность;
п2о 1,4736); тетрагексадецилолово (выход76%, т. пл. 41,5—42,5° С) [62].
Получение тетра-я-тетрадецилолова [62]. К 0,087 моля бромистого
я-тетрадецилмагния в эфире прибавляют 5,5 г хлорного олова, смесь кипятят в течение часа и оставляют до
следующего дня. После этого реакционную смесь гидролизуют, эфир удаляют, из остатка
извлекают этилацетатом тетра-я-тетрадецилолово (12,7 г, 66%). Кристаллизуют его из того
же растворителя. Получают 8,6 г чистого продукта с т. пл. 33—34° С. Побочный продукт
октакозан (1,7 г) не растворяется в этилацетате.
14 Заказ Кя 207
210
Оловоорганические соединения. Лит. стр. 261—268
Для синтеза высших производных тетраалкилолова нормального
строения реакция Гриньяра не всегда дает надежные результаты. Соединения
нормального строения в некоторых случаях удобнее синтезировать
реакцией типа Вюрца (см. стр. 251). Наоборот, соединения R4Sn изостроения
методом Гриньяра удается получить и тогда, когда реакция Вюрца не идет
159]. Например, тетраизопропилолово [19, 41] получено с выходом 61%
163] и 85% [3,51; т. кип. 108—109° С/11 мм [41]; /$1,4749 [19, 22, 32, 64].
Реакция сопровождается образованием значительных количеств гексаизо-
иропилдистаннана [19]. Тетраизобутилолово [37, 64, 65] и тетраизоамил-
олово [8, 22] получены аналогично. Реакция между хлористым неопентил-
магнием и хлорным оловом приводит к смеси R3SnCl, R4Sn и R3SnSnR3,
в которой преобладает монохлорид (62%) [66]. Тетра-т/?ет-бутил- или
тетра-т/?ет-амилолово, по-видимому, получить не удается [4, 66, 67]. При
прибавлении хлорного олова к реактиву Гриньяра образуется т-трет-
алкилолово. Если меняют порядок смешения реагентов, получают дву-
хлористое ди-т/?ет-алкилолово [67].
Получение высших алифатических соединений R4Sn с разветвленной
цепью связано с трудностями. Являясь жидкостями, они имеют высокую
температуру кипения и не могут быть перегнаны без разложения.Обычно
ограничиваются отгонкой из сырого продукта образовавшихся
углеводородов. Выделенные таким образом продукты содержат значительные
количества монохлорида. Так, тетра-киС'(2-этил-Н'Гексш)олово содержит 1,4%
хлора [59]. (Тетраэтилолово, например, в близких условиях образуется с
выходом89—96% и содержит только следы хлористого триэтилолова [31].)
Получение тетра-(3,5,5-триметил-#-гексил)олова [59]. Для приготовления реактива
Гриньяра 360 г (2,2 моля) хлористого 3,5,5-триметил-я-гексила в 500 мл сухого эфира
прибавляют к 49 г (2 г-атома) магния в течение 1,5 час; реакционную смесь кипятят с
обратным холодильником еще 1,5 часа. К охлажденному раствору прибавляют при
перемешивании в течение 1,5 час. 87 г (39 мл, ]/з моля) хлорного олова. Реакционную смесь кипятят с
обратным холодильником 3 часа, затем растворитель отгоняют на кипящей водяной бане
в течение 2,5 час. Отогнанный эфир снова прибавляют к остатку и последний разлагают при
охлаждении прибавлением 600 мл 5%-ной соляной кислоты. Эфирный слой высушивают,
эфир удаляют. В остатке сырое тгт/?а/сг/с-(3,5,5-триметил-я-гексил)олово. Алканы удаляют
нагреванием при 240° С/0,8 мм. Остаток после фильтрования весит 139 г (66,5%). Он
содержит 1,3% хлора.
Для синтеза галогенидов R3SnCl, R2SnCl2 и RSnCl3 магнийорганические
соединения находят ограниченное применение. В этом случае обычно
образуется трудноразделимая смесь продуктов с различной степенью
замещения. В общем случае оловоорганические галогениды удобнее синтезировать
реакцией, обратной диспропорционированию или деалкилированием
соединений R4Sn (см. главу 8) и только в отдельных случаях прямым
действием магнийорганического соединения на хлорное олово.
. лучать моногалогениды R3SnX методом Гриньяра нецелесообразно,
так как образуется смесь галоидопроизводных с примесью R4Sn [33, 68—
71]. Индивидуальные соединения с достаточно хорошим выходом могут быть
выделены только в отдельных случаях. Йодистое триметилолово и двуио-
дистое диметилолово получены медленным прибавлением трех молей
йодистого метилмагния к одному молю йодного олова в эфире [72]. При
действии трех молей бромистого этилмагния на моль хлорного олова при 0° С
хлористое триэтилолово образуется с выходом 25,5% [73].
Если применяют магнийорганическое соединение в расчете на
исчерпывающее алкилирование, то образуются в основном два продукта R4Sn и
R3SnCl. Монохлорид может образовываться в значительных количествах
[74, 75], особенно в безэфирной среде. Так, при проведении реакций в
углеводородах с применением в качестве катализаторов алкоголятов ме-
Синтез на основе производных четырехвалентного олова 211
таллов получено 69,5% хлористого трибутилолова и 30,5% тетрабутилоло-
ва [48, 76].
Хлористое триэтил- и трибутилолово могут быть получены в толуоле.
Реактив Гриньяра и безводное хлорное олово применяют в отношении 4:1.
При применении бромистого бутила отпадает необходимость в абсолютном
эфире как каталитической добавке. При использовании хлористого бутила
добавление эфира необходимо, но в этом случае реакция протекает очень
медленно. Хлористый бутил вступает в реакцию очень быстро в
присутствии каталитических количеств бромистого бутила. Добавка СоС12
равноценна действию эфира; если применять сравнительно большие количества
(10%) СоС12, выход снижается [47].
Получение хлористого трибутилолова [47]. Смесь 50 г порошка магния и 2 г иода
нагревают при перемешивании до возгонки части иода, прибавляют 400 мл абс. толуола, 130 г
безводного хлорного олова и нагревают до 120° С; затем прибавляют последователько 10 г
бромистого бутила и 200 г хлористого бутила. После окончания самопроизвольной реакции
кипятят несколько часов, охлажденную массу выливают при охлаждении в смесь 1 л
концентрированной соляной кислоты и 1 л воды. Получают 144 г хлористого трибутилолова;
выход 88,3% (на хлорное олово), 72,26% (на хлористый бутил); т. кип. 145—147°
С/15 мм [47].
Оловоорганические галогениды, полученные реакцией Гриньяра,
иногда вместо фракционирования обрабатывают щелочью и разделяют
образовавшиеся окиси R2SnO, (R3Sn)20 и гидроокиси [77, 78]. Как правило,
продукты гидролиза значительно отличаются растворимостью и температурой
кипения. Соотношение реагентов подбирают таким образом, чтобы
побочные продукты реакции отделялись наиболее легко. Окиси R2SnO обычно
твердые, плохо растворимые соединения; (R3Sn)20 — жидкости с довольно
высокой температурой кипения или твердые, растворимые в органических
растворителях соединения.
Получение окиси <?ис-(три-я-пропилолова). Эфирный раствор, содержащий 3 л (4,5 моля)
хлористого я-пропилмагния, прибавляют в течение 3 час. при периодическом встряхивании
к 300 г (1,5 моля) хлорного олова в 500 мл сухого бензола, погруженного в баню с ледяной
водой. Реакционную смесь оставляют до утра, разлагают 10% -ной соляной кислотой,
органический слой отделяют и для превращения хлористого три-я-иропилолова в окись бис-
(три-я-пропилолова) встряхивают в течение 30 мин. с 400 млЗ N едкого натра.
Эфирно-бензольный слой отделяют, водный слой экстрагируют 400 мл гексана. Органический слой
высушивают сульфатом натрия, растворитель отгоняют при атмосферном давлении.
Дистилляцией получают 69 г окиси б^с-(три-я-пропилолова) с т. кип. 142—144° С/1 мм [77],
Аналогично образуется окись бш>(триэтилова) [78].
Алкилирование хлорного олова двумя молями хлористого бутилмагния
имеет значение для синтеза технически важного двухлористого дибутило-
лова. Реакцию проводят в дибутиловом эфире, хлорное олово вводят в маг-
нийорганическое соединение при температуре не выше 15° С. В качестве
побочного продукта возможно образование некоторого количества
хлористого трибутилолова. В этом случае содержание хлора в продукте будет
заниженным. Хлористое трибутилолово не отделяют, а переводят в дву-
хлористое дибутилолово по реакции диспропорционирования добавлением в
полученный продукт соответствующего количества хлорного олова с
последующим нагреванием (см. реакцию диспропорционирования, стр. 376)
[53].
Получение двухлористого ди-я-бутилолова [53]. Приготавливают раствор хлористого
«-бутилмагния в ди-я-бутиловом эфире (см. выше синтез тетрабутилолова, стр. 209);
содержание магнийорганического соединения в нем определяют ацидометрически (выход до 94%
от теорет.). К 751,2 г охлажденного до нуля реактива Гриньяра (0,3445 мг C4H9MgCl в 1 мл
раствора) в той же аппаратуре при 0° С постепенно прибавляют 842 г (387 мл) хлорного
олова так, чтобы температура не превышала 15° С. Затем реакционную смесь перемешивают еще
час при комнатной температуре и при наружном охлаждении водой со льдом осторожно
разлагают водой, а затем разбавленной (1:1) соляной кислотой. Масляный слой отделяют,
водный слой дважды экстрагируют дибутиловым эфиром, растворитель отгоняют. Остаток ди-
14*
212 Оловоорганические соединения. Лит. стр. 261—268
бутилового эфира удаляют под вакуумом (70—100 мм). Получают твердый продукт (найдено
хлора 23,73%, вычислено для двухлористого дибутилолова 23,38%) с т. кип. 130—135° С/3—
Б мм; выход 775,4 г (79,5%).
Двухлористое ди-я-бутилолово может быть синтезировано и без
растворителя в условиях, описанных для тетра-я-бутилолова (см. выше) [51, 52].
В безэфирной среде также может образоваться смесь веществ различной
степени замещения [46, 48] (см. стр. 210).
Двухлористое дилаурилолово получено действием бромистого лаурил-
магния на рассчитанное количество хлорного олова в эфире; т. пл. 19,4° С;
т. кип. 218° С с разложением [79].
Оловоорганические меркаптиды, применяемые в качестве
стабилизаторов, могут быть получены действием магнийорганических соединений на
(RS)4^ SnCl, [80].
С помощью реактива Гриньяра получены также соединения RSnCl3 в
алифатическом ряду. Приведенная методика, несмотря на низкий выход,
представляет интерес, так как вещества этого класса сравнительно мало
доступны другими методами (см. стр. 335).
Получение треххлористого #-бутилолова [81]. Раствор хлористого w-бутилмапшя,
приготовленный из 185 г (2 моля) хлористого бутила и 48,6 г (2 г-атома) магния в 500 мл
эфира прибавляют по каплям в течение 1,5 час. при постоянном перемешивании и охлаждении
к 234 мл (521 г, 2,0 моля) хлорного олова в 250 мл бензола. Реакционную смесь немедленно
после окончания прибавления разлагают 1200 мл воды. Органический слой отделяют,
фильтруют, водный слой экстрагируют 250 мл эфира. После высушивания безводным сульфатом
натрия растворитель удаляют дистилляцией. В остатке после первого фракционирования
получают 103 г сырого треххлористого я-бутилолова с т. кип. 111—115° С/12 мм и
содержанием хлора 35,7% . После фракционирования через колонку 50 см, наполненную
стеклянными спиралями, получают 74 г чистого треххлористого w-бутилолова с т. кип. 102—103 °С/
12 мм в виде бесцветной, слабо дымящейся на влажном воздухе жидкости. Выход 13% [81].
Основным продуктом реакции, даже при молярном соотношении
реагентов, является двухлористое дибутилолово [82].
Взаимодействие димагнийорганических соединений с хлорным оловом
приводит к высокомолекулярным соединениям с концевыми группами SnCl3
[83].
При получении методом Гриньяра соединений олова с третичной бу-
тильной или третичной амильной группами [67] вместо ожидаемого
соединения типа R4Sn получаются: 1) при действии хлорного олова на избыток
реактива Гриньяра соединения со связью олово — олово [67, 84—87]; 2)
при обратном введении реактивов (прибавление магнийорганического
соединения по каплям к бензольному раствору хлорного олова) соединения
типа R2SnX2 (X — галоид). Лишь при повторном действии реактива
Гриньяра на двухлористое диалкилолово удается ввести еще одну алкильную
группу и получить R3SnX. Однако эта реакция не проходит полностью.
Вероятная причина — наличие пространственных затруднений. Для получения
оловоорганических соединений с третичными радикалами лучше применять
хлористые алкилы. Бромистые и йодистые алкилы для этой цели не
годятся, так как при реакции с магнием они дают много побочных продуктов.
Получение двубромистогоди-/»р^/»-бутилолова[(СНз)зС]28пВг2[67]. В прозрачный
раствор, полученный из 28 г магния и 93 г хлористого mpem-бутила, добавляют по каплям 35 г
хлорного олова таким образом, чтобы пары кипящего эфира защищали реакционную смесь
от внешнего воздуха. Затем в смесь вводят по каплям воду; эфирный слой, окрашенный в
красновато-желтый цвет, декантируют в токе азота в колбу, наполненную азотом. Эфирный
раствор, содержащий главным образом дн-трет-бутилолово наряду с небольшим
количеством двухлористого ди-трега-бутилолова, сейчас же смешивают в токе азота с 16 г брома,
растворенного в эфире. Выход сырого двубромистого ди-тре/и-бутнлолова 39 г. С
целью очистки, а также для получения других солей эфирный раствор сырого дибромида
встряхиванием в делительной воронке с 15%-ным раствором едкого кали переводят в дигид-
Синтез на основе производных четырехвалентного олова
213
рооксид дн-трет-бутилолова. Это соединение выделяется в виде мелких кристаллов, которые
отсасывают, промывают водой и небольшим количеством эфира. Гидрооксид перекристалли-
зовывают из теплого эфира, в котором он трудно растворяется. Эфирный раствор (частично
в виде суспензии) чистого гидрооксида некоторое время сильно взбалтывают в делительной
воронке с 15% -ной бромистоводородной кислотой, отделяют от кислоты и высушивают
встряхиванием с некоторым количеством безводного бромистого кальция. Затем эфир отгоняют,
а оставшееся масло дистиллируют в вакууме. Жидкость бесцветна, т. кип. 128] С/14 мм.
Аналогично получены двубромистое ди-трет-амилолово, дигидрооксид ди-трст-амилолова
и двухлористое ди-треш-бутилолово [67].
Та же реакция, но проведенная в тетрагидрофуране, дает с хорошим
выходом двухлористое ди-/тг/?£/?г-бутилолово без образования красного
раствора «ди-т/?£т-дибутнлолова» [86].
Алифатические непредельные оловоорганические соединения стали
доступными лишь после работ Нормана [88, 89], который показал, что випиль-
ные галогениды в тетрагидрофуране вступают в реакцию с магнием с
образованием магнийорганических соединений. Были синтезированы олово-
органические соединения с винильной, перфторвинильной и другими
непредельными группами. В реакциях с тетрагалогенидами олова обычно
стремятся выйти к полнозамещенным соединениям, действуя избытком магний-
органического соединения
SnCl4 + 4 CH2=--CHMgCl -» (СН2 -=СН)4 Sn -f 4 MgCl2.
Получать продукты частичного алкилирования хлорного олова тем же
методом нецелесообразно. Здесь, как и в синтезе предельных оловоорга-
нических соединений, взаимодействие хлорного олова с рассчитанным
количеством магнииорганического соединения приводит к трудноразделимой
смеси веществ с различной степенью замещения галоида на вииильную
группу [90]. Хлористое тривинилолово, двухлористое дивинилолово и
другие продукты получают реакцией, обратной диспропорционированию'(см.
стр. 331), или деалкилированием тетравинилолова действием галоидов или
галоидоводородных кислот (см. стр. 333).
Получение тетравинилолова [90]. Приготовляют раствор 1212 г (19,4 моля) хлористого
винила в 3460 г (48 моля) тетрагидрофурана. В колбу емкостью 22 л помещают 394,0 г (16,2
г-атома) стружек магния и 100 мл приготовленного раствора, нагревают до кипения и для
инициирования реакции вводят 2,0 мл бромистого этила. После того как реакция начнется,
нагревание прекращают и остальное количество хлористого винила прибавляют с такой
скоростью, чтобы поддерживать температуру 48—56° С. Операция занимает 6 час.
Нагревание продолжают еще 3 часа. Затем к смеси прибавляют раствор 663,6 г (3,5 моля) хлорного
олова в 10,8 л пентана так, чтобы поддерживать кипение смеси. Операция занимает 4 часа.
Смесь кипятят с обратным холодильником еще 5 час.
После охлаждения реакционную смесь частично гидролизуют осторожным
прибавлением 2,8 л 10%-ного раствора соляной кислоты. Из органического слоя выпадает твердый
осадок хлористого магния. Органический слой декантируют в колбу емкостью 22 л.
Твердый остаток гидролизуют окончательно прибавлением 2 л воды. Образовавшийся
органический слой (около 300 мл) отделяют от водного и присоединяют к основному количеству.
Растворители — пентан и тетрагидрофуран быстро отгоняют на водяной бане при
атмосферном давлении. Остаток тщательно фракционируют в вакууме. В предварительной фракции
150 г тетрагидрофурана (т. кип. 25—30° С/100—120 мм). Затем отгоняют 642 г (62%)
тетравинилолова с т. кип. 55—577С 17 мм [90]; 67—70° С/28 мм [6,35, 91].
Тетравинилолово перегоняется без разложения при атмосферном
давлении в отсутствие ингибиторов полимеризации (некоторое разложение
наступает, если температура бани достигает 200° С); т. кип. 160—163° С/766 мм\
п% 1,4914 [6].
Тетраперфторвинилолово образуется при действии бромистого трифтор-
винилмагния на хлорное олово в тетрагидрофуране. Хлорное олово вводят
в реакцию в виде аддукта с тетрагидрофураном в избытке последнего.
Реакция протекает без затруднений. Тетраперфторвинилолово образуется с
выходом 31%, т. кип. 52—54° СУ19 мм. Оно более чувствительно к действию
21'i Оловоорганические соединения. Лит. стр 261—268
воздуха, влаги, кислот или оснований, чем тетравинилолово, но в их
отсутствие термически более устойчиво (в течение 6 час. при 150° С не
изменяется). В запаянной ампуле в присутствии кислорода через два дня
превращается в полимер, довольно устойчивый к окислению [92].
Тетраалл ил олово [43, 93, 94] образуется из хлорного олова в
бензольном растворе и бромистого аллилмагния. Это — бесцветная реакционноспо-
собная жидкость с т. кип. 87—88° С/4 мм\ п3£ 1,5324. По сравнению с
тетравинилоловом оно отличается малой стабильностью. Например,
нагреванием с этиловым спиртом при 130° С происходит полный распад
соединения с образованием пропилена и гидрата окиси олова.
Четырехзамещенные оловоорганические соединения алициклического
ряда могут быть получены обычным образом. Описаны тетрациклопро-
пилолово, т. кип. 67—68° С/0,3 мм [95]; тетрациклопентилолово, т. пл.
68—68,5° С [96]. Обычно образуется смесь продуктов
CeHuMgBr + SnCl4-* (QHnU Sn + (CeHii)3SnSn(C6H11)3.
Разделение кристаллизацией связано с большими потерями вещества.
Удобнее полученную смесь перевести в бромистое трициклогексилолово,
которое образуется как из тетрациклогексилолова, так и гексациклогек-
силдистаннана [67].
Получение тетрациклогексилолова [67]. Из 250 г бромистого циклогексила (или 182 г
хлористого циклогексила) приготовляют магнийорганическое соединение в эфире, сливают
его с остатка магния и разбавляют 300 мл чистого, перегнанного над натрием бензола.
В смесь при механическом перемешивании (охлаждение ледяной водой) вводят по каплям
раствор 75 г хлорного олова в 200 мл сухого бензола. Реакцию заканчивают после 3 час.
кипячения на водяной бане с обратным холодильником. Смесь охлаждают и разлагают
ледяной водой и 5%-ной соляной кислотой. Бензольно-эфирный слой отделяют, промывают
водой, высушивают над хлористым кальцием и концентрируют примерно до ЗБмл. По
охлаждении выделяются в большом количестве кристаллы, которые отсасывают и перекристалли-
зовывают из бензола с добавлением абсолютного спирта. Полученную смесь
тетрациклогексилолова с гексациклогексилдистаннаном бромируют в хлороформе или четыреххлористом
углероде (при охлаждении льдом) рассчитанным количеством брома. Растворитель отгоняют,
образовавшийся бромистый циклогексил удаляют нагреванием в вакууме (15 мм) на
кипящей водяной бане. Остаток промывают и перекристаллизовывают из небольшого
количества спирта. Полученное таким путем чистое бромистое трициклогексилолово дает после
вторичной обработки бромистым циклогексил магнием тетрациклогексилолово; т. пл. (после
кристаллизации из горячего бензола) 263—264° С.
Оловоорганические соединения, содержащие циклопентадиенильные
радикалы, изучены довольно подробно [97, 98].
Реакция между оловоорганическими галогенидами и бромистым 1-цик-
лопентадиенилмагнием протекает с заменой атомов галоида на 1-циклопен-
тадиенил. С хлорным оловом реакция протекает аналогично, но выделение
из реакционной смеси образовавшегося тетра-1-циклопентадиенилолова
представляет известные затруднения; оно легко разлагается.
Следует иметь в виду, что бромистый 1-циклопентадиенилмагний
реагирует с кетоном Михлера медленнее, чем обычные магнийорганические
соединения. Поэтому качественную пробу на присутствие бромистого 1-цик-
лопентадиенилмагния по обычно принятой методике следует проводить
при температуре около 30° С и увеличивать время взаимодействия до 3—10
мин. [97].
Свежеполученные оловоорганические соединения, содержащие
циклопентадиенильные. группы, в зависимости от числа циклопентадиенильных
групп имеют слабо-желтый или канареечный цвет. При хранении они
темнеют и, наконец, становятся ржаво-коричневыми. Скорость, с которой
изменяется цвет, зависит от числа циклопентадиенильных групп в молекуле
и ускоряется действием света и воздуха.
Синтез на основе производных четырехвалентного олова 215
Природа соединений, полученных при самопроизвольном разложении,
неизвестна. По-видимому, разложение является результатом окисления и
полимеризации. Тетра-1-циклопентадиенилолово пирофорно, бурно
реагирует с концентрированной азотной кислотой, которая применяется при
анализе полученного соединения.
Получение оловоорганических соединений с циклопентадиенильными
группами имеет свои особенности. Реакцию ведут в бензольно-эфирной
среде. Согласно применяемой методике, эфир и бензол берутся в таких
объемах, чтобы их смесь имела т. кип. 60° С. Бромистый 1-циклопентадиенил-
магний образуется с выходом 75—80% (по титрованию соляной кислотой)
[98].
Получение тетра-1 циклопентадиенилолова [98]. К 0,4 моля бромистого циклопентадие-
нилмагния в 415 мл смеси эфира с бензолом [99] прибавляют 13,05 г (0,05 моля) безводного
хлорного олова в 40 мл бензола. Смесь кипятят с обратным холодильником 24 часа и затем
концентрируют до состояния пасты. Последнюю обрабатывают 3 раза петролейным эфиром
(порциями по 200 мл; т. кип. 57—70° С) и полученный раствор отфильтровывают горячим.
Соединенные фильтраты концентрируют в вакууме до 100 мл и затем охлаждают сухим льдом
в ацетоне. Получают 12,5 г вещества с т. пл. 71—75° С (после высушивания в
вакуум-эксикаторе над парафинов в течение 3 час). Сырой продукт растворяют в минимальном количестве
горячего петролейного эфира (т. кип. 57—70е С) и отфильтровывают горячим. В остатке 2,24 г
аморфного твердого коричневого вещества. Фильтрат охлаждают сухим льдом в ацетоне.
Получают 8,70 г (41,5%) относительно чистого вещества с т. пл. 71—73° С.
Содержание олова в тетра-1 -циклопентадиенилолове ниже теоретической
величины; при хранении образца эта разница еще более увеличивается.
После высушивания вещества в вакуум-эксикаторе в течение 24 и 72 час.
найдено олово соответственно на 2,67 и 3,95% ниже вычисленного.
В приведенной методике избегают разложения реакционной смеси водой,
так как тетра-1-циклопентадиенилолово легко гидролизуется. Оловоорга-
ническое соединение отделяют от солей магния и бромистого циклопента-
диенилмагния экстракцией [98]. (Синтез оловоорганических соединений, в
которых циклопентадиенильные группы находятся наряду с другими
органическими радикалами, см. стр. 228.)
В ароматическом ряду реакция по уравнению
4 ArMgX + SnCU -> Ar4Sn + 4MgClX
является лучшим методом получения простейших тетраарильных
соединений олова. Например, тетрафенил-, тетра-о-толил-, тетра-ж-толил-, тетра-
n-толил- и тетрабензилолово могут быть получены в чистом виде, что не
всегда удается другими методами [100]. Оловоорганические соединения с
более сложными заместителями в ароматическом ядре с помощью реактива
Гриньяра образуются с плохим выходом, даже если заместители
индифферентны к раствору Гриньяра. Такие производные лучше получать с
помощью литийорганических соединений (см. стр. 242).
Примесями обычно являются продукты неполного замещения и
соединения со связью олово — олово.
Тетраарильные соединения олова — твердые высокоплавкие вещества,
часто плохо растворимые на холоду в обычных органических
растворителях.
Получение тетрафенилолова [101]. В эфирный раствор бромистого фенилмагния,
полученный из 175 г бромбензола и освобожденный декантацией от остатков магния, при
охлаждении водой и встряхивании вводят 50 г хлорного олова. После 2 час. кипячения с обратным
холодильником разлагают водой, 5% -ной соляной кислотой и, не разделяя слоев, эфир
отгоняют на водяной бане. Остаток отсасывают, промывают водой и спиртом и сушат на водяной бане.
Затем его смешивают с большим количеством бензола и кипятят с обратным холодильником.
Бензольный экстракт фильтруют горячим. По охлаждении из него выкристаллизовываются
белоснежные блестящие кристаллы тетрафенилолова. Экстракцию повторяют еще 2—3 ра.
216 Оловоорганические соединения. Лит. стр. 261—268
за, используя для этой цели маточник. Т. пл. 225° С [101], 229° С [32, 35, 37]. Вещество
относительно легко растворяется в хлороформе и горячем бензоле, плохо — в эфире, почти не
растворяется в спирте и не растворяется в воде.
Лучшие результаты получают, если применяют бромное олово и
заканчивают реакцию при более высокой температуре. Для этого к магнийорга-
ническому соединению в эфире прибавляют сухой толуол или бензол, эфир
отгоняют, к охлажденному раствору прибавляют бромное олово в
небольшом количестве толуола и нагревают на водяной бане около часа. На
следующий день реакционную смесь выливают в воду, подкисленную соляной
кислотой, и отсасывают. Получают тетрафеиилолово с выходом 70%.
После высушивания в вакууме т. пл. 225° С без перекристаллизации [102].
Готовый хлористый фенилмагний промышленного изготовления дает те же
результаты. Выход при взаимодействии его с хлорным оловом в толуоле
составляет 60—80%, в зависимости от качества исходного магиийоргаии-
ческого соединения [102].
Тетрафеиилолово с достаточно хорошими выходами может быть
получено и в безэфирной среде [49, 51, 52]. В технике применяют в основном
инертные высококипящие растворители, например, толуол, декалин,
иногда с каталитическими добавками эфира [45, 49, 50].
Получение тетрафенилолова в безэфирной среде [51, 52]. К 29,2 г (1, 2 моля)
активированных иодом стружек магния при перемешивании (в токе азота) добарляют раствор 172, 72 г
(1,1 моля) бромбензола в 200 мл декалина; температуру поддерживают в интервале 160—i70°
С. Затем в реакционную смесь постепенно приливают 39,08 г (0,15 моля) хлорного олова.
Смесь нагревают до 160—170° С 4 часа Из осадка выделяют 47 г (74%) тетрафенилолова,
т. пл. 225—225,5° С (из бензола) [51, 52, 103J.
Тетрафеиилолово получено в хлорбензоле с добавлением абс. эфира и
(C2H50)4Si. Для начала реакции применяют CH3J, содержащий 10% иода.
Так как при образовании реактива Гриньяра используется 30% магния,
то в данном случае необходимо брать его в избытке [47].
Оловоорганические соединения с атомами фтора в бензольном ядре
более доступны, чем фторсодержащие соединения алифатического ряда. Их
получают, например, действием бромистого я-фторфенил- [104, 105] или
перфторфенилмагния на хлорное олово. Тетрапентафторфенилолово
стабильно при температуре плавления (221 °С) [106—ПО].
Получение тетра-#-хлорфенилолова [111]. Реактив Гриньяра, приготовленный из п-
хлорбромбензола, кипятят 2 часа на водяной бане, сливают с остатка магния и к нему
добавляют по каплям 36 г (50% рассчитанного количества) хлорного олова. (Помимо
непосредственного прибавления хлорного олова, здесь с успехом был применен метод, описанный
ранее [74], а именно: к магнийорганическому соединению добавляют высушенный над
натрием бензол, к смеси прибавляют бензольный раствор хлорного олова). После 2 час.
нагревания на водяной бане смесь осторожно разлагают водой и 5%-ной соляной кислотой.
Бензольно-эфирный слой отделяют, высушивают и упаривают до 50 мл. После
непродолжительного времени выделяются кристаллы, которые отсасывают от маслянистых примесей,
промывают спиртом и перекристаллизовывают из горячего бензола; при этом нужно обратить
внимание на то, чтобы растворитель не слишком сильно испарялся, так как иначе могут
одновременно выкристаллизоваться и следы побочного продукта (очевидно, гекса-п-хлорфе-
нилдистаннана). Выделенный продукт отсасывают, промывают эсЬиром и еще раз
перекристаллизовывают из бензбла. Выход 38% [111], 51,3% [112]; т. пл. 199° С [111, 113].
Тетра-я-толилолово образуется с достаточно высоким выходом [87, 100,
114—118].
Получение тетра-#-толилолова [114]. В колбу емкостью 2 л, снабженную обратным
холодильником и капельной воронкой, вносят 30 г порошка магния, 250 мл абсолютного
эфира и медленно прибавляют 200 г л-бромтолуола в эфире, затем нагревают на водяной бане
еще 3 часа. На следующий день реакционную смесь охлаждают холодной водой и при
хорошем перемешивании вливают горциями 50 г хлорного олова в равном объеме петролейного
эфира. Смесь кипятят в течение 3 час. на водяной бане, эфир возможно полно отгоняют,
Синтез на основе производных четырехвалентного олова
217
полученный продукт разлагают раствором хлористого аммония. Осадок отфильтровывают,
промывают спиртом, затем эфиром, высушивают и экстрагируют кипящим хлороформом,
экстракт охлаждают снегом и фильтруют. Выход 63%; т. пл. 236—237° С [113,114].
Аналогично получают тетра-^-тол ил олово с выходом 29%, т. пл. 128,5° С
[100, 116, 119]; тетра-о-толилолово с выходом 35%, т. пл. 216—217° С
[100, 116, 120, 121]; тетра-^-трифторметилфенилолово с выходом 57%,
т. пл. 143—144° С [104, 122] и тетра-(7г-трифторметилфенил)олово с выходом
43%, т. пл. 150—150,7° С [104, 112].
Тетрабензилололово [100], тетра-(2-фенилэтил)олово [44] образуются
при действии бромистого бензила или 2-фенилэтилмагния на хлорное или
бромное олово. Аналогично могут быть получены и соединения этого типа
с заместителями в бензольном ядре [2]. Этим методом пользуются для
синтеза как четырехзамещенных продуктов, так и монохлоридов и продуктов
гидролиза. Следует иметь в виду, что монохлориды образуются в
значительных количествах или являются основными продуктами реакции даже
при избытке реактива Гриньяра.
Тетрабензиллолово можно получить, если постепенно прибавлять 4 моля
хлористого бензила к эфирному раствору хлорного олова (1 моль;
приготовление см. ниже) в присутствии порошкообразного магния (4 г-атома). Эфир
отгоняют, остаток нагревают до 100° С, после охлаждения обрабатывают
водой и подвергают отгонке с водяным паром. Твердый остаток отсасывают,
высушивают на глиняной тарелке и растворяют в петролейном эфире. Тет-
рабеизилолово выделяется при охлаждении льдом, т. пл. 42—43° С [113,
123].
Аналоги тетрабензлиолол'ова с заместителями в бензольном ядре
образуются с невысоким выходом. Представляет известные трудности их
отделение от монохлоридов.
Получение тетра-о-хлорбензилолова [2]. Бромистый г>-хлорбензилмагпий, полученный
из 20 г бромистого о-хлорбензила и 3 г магния, прибавляют к раствору 14,2 г бромного
олова в эфире в условиях, описанных ниже в синтезе окиси б/:/с-(три-о-бромбепзилолова).
Эфирный слой, отделенный после разложения реакционной смеси, встряхивают с 30%-
ным едким кали. Основная масса образовавшегося хлористого три-охлорбензилслова
превращается в окись бмс-(три-о-хлорбензилолова) и выпадает в осадок. Выход 31%,
считая на бромистый о-хлорбензил; т. пл. 133,5° С (из ацетона). Эфирный раствор
промывают 2 раза водой, сильно встряхивают с водным раствором фтористого калия. Остаток
хлористого три-о-хлорбензилолова выпздаетв виде фторида, который перекристаллизовы-
вают из смеси спирта с водой. Эфирный раствор отфильтровывают, высушивают,
концентрируют в вакууме на водяной бане до сиропообразной консистенции и оставляют
кристаллизоваться в течение нескольких недель. Кристаллы быстро промывают эфиром,
высушивают и для очистки растворяют в небольшом количестве теплого бензола; раствор
разбавляют пятикратным количеством теплого метанола. Получают чистое соединение
ст. пл. 96,5° С. Выход 20%, считая на бромстый о-хлорбензил [2].
С бромистым дифенилметилмагнием реакция протекает с
осложнениями, тетра(дифенилметил)олова не образуется [1241 (см. аномальную
реакцию с третичным хлористым бутилом и хлористым циклогексилом). Тетра-
(2-фенилэтил)олово [44] (выход 71%) при перегонке разлагается [125].
Тетра-я-ксилилолово (выход 50%, т. пл. 278° С) [116], тетра-ж-ксил ил олово
(выход незначительный, т. пл. 224° С) [116] и тетра-я-т/?£Я7-бутилфенилолово
[122] получены аналогично тетра-я-толилолову [118]. Тетрамезитилолово
[100], по-видимому, получить не^удается [126]. При синтезе оловооргани-
ческих соединений с заместителями в бензольном ядре, вполне
индифферентными к реактиву Гриньяра, во многих случаях реакция проходит с очень
плохим выходом. В ряде случаев вообще не удается получить как Ar4Sn,
так и продуктов частичного замещения, например Ar3SnX [127]. Тетра-
анизилолово получено с выходом 1,5% [128]. Из образовавшейся
смолистой массы обработкой спиртово-водным раствором фтористого калия
выделено весьма немного фтористого трианизилолова [111]. Тетра-я-фено-
218 Оловоорганические соединения. Лит. стр. 261—268
ксифенилолово (/?-C6H5OC6H4)4Sn (т. пл. 171° С) образуется с
незначительным выходом [111]. Предположение, что синтез протекает
неудовлетворительно из-за пространственных затруднений, едва ли обосновано, так как
те же соединения получены с хорошим выходом при помощи литийоргани-
ческих соединений (см. стр. 242).
Тетра-а-нафтилолово может быть получено действием хлорного олова
на бромистый а-нафтилмагний [100, 128] наряду со смолообразными
продуктами [127], из которых удается выделить лишь ничтожное количество
кристаллического хлористого три-а-нафтилолова [111]. Полученное
вещество по составу весьма близко к (a-C10H7)4Sn и после соответствующей очи-
стки может быть использовано в качестве исходного продукта для
получения индивидуальных кристаллических оловоорганических соединений
а-нафтильного ряда [129].
Оловоорганические галогениды ароматического ряда, как правило,
синтезируют деарилированием соединений Ar4Sn (гл. VIII). Методом Гри-
ньяра стремятся обычно получить продукты исчерпывающего арилирова-
иия. Однако при строгом соблюдении соотношения реагентов и
экспериментальных условий могут быть получены в качестве основных продуктов
моногалогениды или дигалогениды. Отделение их от продуктов иной
степени замещения, образование которых неизбежно, представляет известные
трудности. В общем для синтеза оловоорганических галогенидов метод имеет
ограниченное применение. Так, получены, например, соединения (C6F5)3SnCl
и (C6F5)2SnCl2. Это малоустойчивые соединения. Они гидролизуются даже
слабыми щелочами и вступают в реакции с потерей пентафторфенильной
группы [106, 106а]. Хлористое три-(о-толил)олово образуется с плохим
выходом; т. пл. 115,0—115,7° С (из метанола) [112].
Хлористое трибензилолово, в том числе и с заместителями в бензольном
ядре, может быть получено прямым действием магнийорганического
соединения на хлорное олово. В этом случае важно, чтобы хлорное олово в
течение всего синтеза было в избытке. Действительно, если хлорное олово
прибавляют к ледяному эфирному раствору хлористого бензилмагния,
хлористое трибензилолово образуется с выходом только 8% [32]. При об
ратном порядке смешения реактивов получают более удовлетворительные
результаты. Хлорное олово образует с эфиром твердый нерастворимый
эфират. В мелкораздробленном состоянии, что необходимо для успешного
проведения синтеза, его получают медленным прибавлением (разогревание!)
из капельной воронки (при сильном взбалтывании) одного объема
безводного хлорного олова в десять объемов абсолютного эфира при охлаждении
до 0° С [123]. Бромное олово растворимо в эфире, причем только со слабым
разогреванием. В последнем случае реакция протекает в гомогенной среде
[2].
Получение хлористого трибензилолова [123]. К 100 г суспензии безводного хлорного
олова в эфире прибавляют 28 г порошка магния и из капельной воронки вливают небольшое
количество хлористого бензила. Если реакция не начинается в течение нескольких минут,
то для ее ускорения добавляют небольшое количество отдельно приготовленного раствора
хлористого бензилмагния. Остальное количество хлористого бензила (всего 150 г, кратно
3 молям) добавляют медленно по каплям в течение 2 час, при этом колбу сильно
встряхивают и охлаждают льдом. После того как выделение тепла прекращается, реакционную
смесь кипятят с обратным холодильником 2 часа, эфир отгоняют, а остаток в течение 2 час.
нагревают до 100° С. Реакционную смесь после охлаждения медленно прибавляют к равному
объему воды. Твердый продукт отсасывают, промывают водой и кристаллизуют сначала из
ацетона, содержащего воду, а затем из ледяной уксусной кислоты до тех пор, пока точка
плавления не станет постоянной (143—145° С). Выход около 60% [113, 123].
Монохлорид [С6Н5С (CH3)2CH2]3SnCl (т. пл. 117,5—118,5° С) получен
с выходом 44%. В этом случае четырехзамещенного продукта образуется
Синтез на основе производных четырехвалентного олова
219
только 9% даже тогда, когда магнийорганическое соединение и хлорное
олово берут в соотношении 4 : 1 [129а].
Моногалогениды удобно выделять в виде окиси.
Получение окиси б"ис-(три-о-бромбензилолова) [2]. К раствору 21,8 г бромного олова
в 50 мл эфира при перемешивании и охлаждении ледяной водой прибавляют в течение 10
мин. магнийорганическое соединение, приготовленное из 50 г бромистого о-бромбензила и
5,5 г магния в 150—200 мл эфира. Эфир при перемешивании отгоняют, остаток нагревают
на кипящей водяной бане в течение часа. После охлаждения эфир прибавляют обратно,
реакционную массу разлагают водой и разбавленной в 2 раза соляной кислотой; при
появлении помутнения фильтруют. Водный слой отбрасывают. Эфирный слой смешивают,
осторожно встряхивая, со 100 мл 30%-ного едкого кали. Через некоторое время
начинает выкристаллизовываться окись бис-(три-о-бромбензилолова). Через день, когда
кристаллизация заканчивается, встряхивают энергичнее. Осадок отсасывают, промывают
ацетоном, высушивают и экстрагируют 2 раза 200—300 мл ацетона. Вещество
выкристаллизовывается практически количественно; т. пл. 158—159°С; выход 35% в пересчете
на бромистый о-бромбензил.
Аналогично получают окись бш>(три-о-хлорбензилолова) с выходом 31 %,
т. пл. 133,5° (из ацетона); окись бас-(три-о-фторбензилолова) с выходом 25%,
т. пл. 113° С (из ацетона). Последний продукт заметно растворим в эфире
[2].
Бромистое три-а-нафтилолово и его аналоги (RC10He)3SnX, где R —
2-метокси-(т. пл. 221° С), 2-этокси-(т. пл.217°С) [126], 2-я-пропокси-(т. пл.
169° С), 2-изопропокси-(т. пл. 236° С), 2-я-бутокси-(т. пл. 159° С), 2-изо-
бутокси-(т. пл. 170° С), 2-я-амилокси-(т. пл. 142° С), 2-я-гексилокси-(т.
пл. 110° С) [130], получают медленным прибавлением расплавленного
бромного олова к бромистому а-нафтилмагнию [125], выход 15—43%.
Бромистое три(бифенил-2)олово образуется наряду с двубромистымбас-(бифенил-2)-
оловом [112, 131]. Тетрабифенилолово может быть получено, если заменить
реактив Гриньяра на литийорганическое соединение (см. стр. 243).
Бромистое тримезитилолово (т. пл. 178° С) получено с выходом 60% [126].
Оловоорганические галогениды с объемистыми арильными группами
получить не удается. Применение даже большого избытка галоидного фе-
нантрилмагния приводит к смеси галоидсодержащих оловоорганических
соединений, разделить которую не представляется возможным [127].
Оловоорганические соединения ароматического ряда с винильной
группой в боковой цепи представляют интерес в качестве мономеров (см. стр.
231). Тетра-я-винилфенилолово получено из бромистого винилфенилмаг-
ния и хлорного олова в тетрагидрофуране. Вещество легко полимеризуется,
получить его в чистом виде трудно [132]. Тетра-(я-аллилфенил)олово
получено с выходом 45% [133]. Тетра(фенилэтинил)олово (C6H5C=C)4Sn
получают действием бромистого фенилацетиленидмагния на хлорное олово в
смеси тетрагидрофурана с бензолом (2 : 1). Это хорошо кристаллизующееся
чувствительное к влаге вещество с т. пл. 174° С [134].
В качестве примера получения оловоорганических соединений
гетероциклического ряда можно привести следующий.
Получение тетра-а-тиенилолова (a-C4H3S)4Sn. В реактив Гриньяра, полученный из 10 г
иодтиофена и 1,2 г магния в 50 мл абс. эфира, вносят порциями при взбалтывании взвесь эфи-
рата хлорного олова, полученную прибавлением по каплям 1,4г (45% вычисленного количества)
хлорного олова к 25 мл охлажденного сухого эфира. От каждой порции эфирата эфир
вскипает, и эфират, поскольку он не моментально растворяется, окрашивается в каштаново-ко-
ричневый цвет. Затем смесь кипятят 1 час на водяной бане. Уже при кипячении начинается
выделение белых кристаллических игл из слегка окрашенной жидкости. Избыток реактива
Гриньяра разлагают 15 мл ледяной воды и эфир отгоняют. Остаток отсасывают, промывают
водой, высушивают в вакууме над пятиокисью фосфора и многократно экстрагируют
кипящим эфиром. Для окончательной очистки вещество перекристаллизовывают сначала из
теплого эфира, затем из горячего спирта. Т. пл. 156° С [135].
220
Олсвоорганшеские соединения. Лит. стр. 261—268
Синтез олсвоорганических соединений типа R^SnR (je-j-3^4)
дзаимодействием реактива Гриньяра
с оловоорганическими галогенидами
Магнийорганические соединения применяются для доалкилирования
оловоорганических галогенидов с целью, как правило, получения
соединений со смешанными радикалами. В этом основное значение метода. Лучшие
результаты получают в случае действия избытком магнийорганического со-
динения:
RySnQ + R'MgCl -> R3SnR' + MgCio,
RaSnClo - 2 R'MgCl -> R2SnR2 + 2 MgCl,.
Тригалогениды RSnX3 менее доступны и в реакциях с магпийорганически-
ми соединениями редко применяются. С оловоорганическими гидроокисями
реакция протекает ступенчато [136]:
R3SnOH + R'MgX -> ReSnOMgX + R'll,
R3SnOMgX + R'Mg X -> R3Sn R'.
Станноксановая связь —Sn—О—Sn^- разрывается с образованием четы-
рехзамещенных соединений [136—1381.
Для получения оловоорганических галогенидов с различными
радикалами
RSnCfo + R'MgCl -, RR'SnClo -\- MgCb
R2SnCl2 -j- R'MgCl -> RoR'SnCl + MgCl2
метод имеет ограниченное применение. При частичном алкилировании
RSnCl3 и R2SnCl2 рассчитанным количеством R'MgX образуется смесь
продуктов. Индивидуальные соединения выделены с невысоким выходом
(см. стр. 241). Для синтеза оловоорганических соединений с тремя или
четырьмя различными радикалами применяют последовательное
исчерпывающее алкилирование и ступенчатое деалкилирование полученного
соединения галоидом. Галоид, как известно, отщепляет в первую очередь (см.
стр. 339) более легкие радикалы, связанные с оловом, что дает возможность
синтезировать соединения с асимметрическим атомом олова R'R"R'"SnX
[17, 139—141].
Смешанные соединения R3SnR/ описаны достаточно подробно. Их
получают, как правило, из моногалогенидов R3SnX.
Аналогично действием металлоорганических соединений, содержащих
галогенид магния в радикале R'3MR"MgX (М = Si, Sn и др.) на те же мо-
ногалогениды олова образуются соединения, содержащие в молекуле два
разных металла R3SnR"MR'3. С помощью димагнийорганических
соединений получены R3SnR'SnR3. Примесь дистаннана R3Sn—SnR3 может быть
значительной, особенно в ароматическом ряду.
Общий прием синтеза сводится к следующему [142]. К 2—3-кратному
избытку реактива Гриньяра прибавляют оловоорганический галогенид,
смесь кипятят в течение 1—2 час. с обратным холодильником, эфир
отгоняют, а остаток нагревают на кипящей водяной бане в течение 1—1,5 час.
К охлажденной массе добавляют отогнанный эфир, смесь разлагают водой.
После обычной обработки выходы равны 60—98%.
Синтезированы (CH3)3SnR, где R — (в скобках приведены т. кип. и
rc2D°) хлорметил (44—48°'С/15 мм\ 1,4860/25) [143]; бромметил (46—50е С/
11 мм) [143]; иодметил (53— 54° С/6,5 мм; 1,5510/25) [143]; оксиметил (58—
59° С/6,5 мм) [144]; тиоцианометил (104—105° С/4 мм; 1,5247/25]) [145];
Синтез на основе производных четырехвалентного олова
221
этил (106° С/764 мм; 1,4527) [23, 28, 1461; пропил (130,8° С/760 мм; 1,47410)
[147]; бутил (149—150° С/726 мм; 1,4560/21,5) [148]; изобутил (140—141° С/
726 мм; 1,4544/21,5) [149, 150]; амил (171—172° С/721 мм; 1,4559) [149];
изоамил (162—164° С/725 мм; 1,4470/21) [149]; октил (102—118° С/12 мм;
1,4587/25 [148, 151]; децил (67° С/0,5 мм; 1,4602/25) [151]; додецил (158-
160^ С/14 мм; 1,4610/25) [148, 151]; винил (99—100 С/760 мм; 1,4544/25)
[6, 144, 152]; аллил (125,5° С/642 мм; 1,4734) [6, 153]; циклопропил (129—
130° С/760 мм) [95]; фенил (203—208° С/760 мм; 88,5/16лш) [152, 154—1561;
перфторфенил [107, 108, ПО], я-хлорфенил (80 С/2—3 мм) [154, 157];
Ji-хлорфенил (96° С/3,5 мм) [154]; п-бромфенил (124° С/15 мм) [157, 1581;
бензил (90° С/9 мм) [152, 157]; n-хлорбензил, я-бромбензил [159], я-толил
[1571; а-нафтил, Р-нафтил [160]; диметилсилилметил (153—154° С/760 мм)
[1501; триметилсилилметил (165—166° С/760 мм; 1,4569/25) [20, 161].
В качестве исходных галогенидов применяют обычно хлориды или
бромиды. В случае моногалогенидов лучше пользоваться фторидами — их
можно получить в очень чистом виде [8]. Экспериментальные условия мало
отличаются от описанных ранее при синтезе R4Sn прямым действием
реактива Гриньяра на SnCl4.
При синтезе (CH3)3SnR для гидролиза следует применять насыщенный
раствор хлористого аммония. Смешанные оловоорганические соединения
более реакционноспособны, чем симметричные [148].
При действии реактива Гриньяра на оловоорганические галогениды,
содержащие, помимо, того, галоид в алкильной группе, последний в условиях
опыта не затрагивается.
Таким образом, четырехзамещенные оловоорганические соединения,
содержащие, например, хлорметильную или бромметильную группу, могут
быть получены обработкой реактивом Гриньяра оловоорганических
галогенидов, полученных с помощью диазометана (см. стр. 321).
Получение хлорметилтриметилолова [143]. Раствор 120 г (0,51 моля) хлористого хлор-
метилди метил олова в равном объеме эфира прибавляют к бромистому метилмагнию,
приготовленному из 36,6 г (1,5 г-атома) магния и 150 г (1,58 моля) бромистого метила в 400 мл
безводного эфира так, чтобы поддерживать умеренное кипение смеси. Реакционную смесь
кипятят с обратным холодильником еще 18,5 час, охлаждают до 0° С и гидролизуют 180 мл
насыщенного раствора хлористого аммония. После обычной обработки получают 80 г (73%)
хлорметилтриметилолова с т. кип. 44—48° С/15 мм. При атмосферном давлении хлорметил-
триметилолово при перегонке (т. кип. 146° С) несколько разлагается.
Также получены бромметилтриметилолово (выход 71%) [143], хлор-
метилтриэтилолово (выход 55,4%, т. кип. 84—85,5° С/10 мм) [162] и хлор-
метилтри-я-бутилолово (выход 80%; т. кип. 108—112° С/0,5 мм) [143].
Описаны соединения ряда (CoH5)3SnR, где R — (в скобках т. кип. и n2D°)
метил (162—163° С/760 мм; 1,4656) [28, 146], пропил (112° С/50 мм; 1,4726)
[44, 116, 146, 147, 163], изопропил (192—194° С/719 мм; 1,4772/19) [164],
бутил (73—75° С/4 мм; 1,4736) [137, 148, 149], изобутил (86° С/10 мм) [44],
амил (102° С/10 мм) [44], изоамил (111° С/18,5 мм) [44], неопентил (106—
108° С/17 мм) [66], гексил (102—104° С/0,75 мм) [148], октил (135—138° С/
3—А мм) [148], я-нонил (144—146° С/5 мм) [165], додецил (158—164° С/1,1
мм) [148], винил (174—175° С/760 мм; 1,4780; 60—62° С/12—13 мм; 1,4738)
[6, 42, 166], аллил (76—77° С/10 мм) [163], фенил (254° С/760 мм; 1,5653)
[38, 137, 167], я-бромфенил (150—151° С/бмм) [150, 168], я-иодфенил (174—
175° С/760 мм) [150, 168].
Описаны также (C3H7)3SnR, где R — метил (94—96° С/11 мм) [44], этил
(101° С/10 мм) [137], бутил (137—138° С/37 мм; 1 ,4741) [146], винил (90° С/
8,2 мм; 1,4776/25) [169], фенил (150° С/15 мм) [167], я-бромфенил (168° С/
А мм) [150, 168]; (n-C4H9)3SnR, где R — метил (121° С/10 мм) [441, этил
(129° С/10 мм) [44], изоамил (177—178° С/20 мм; 1,4715) [146], гексил
222
Оловоорганические соединения. Лит. стр. 261—268
(165° С/7 мм; 1,4762/17) [170], гептил (165—167° С/3 мм) [171], винил
(114°С/Злш) [169], аллил (155° С/17 мм) [163], 3-бутенил (91—93° С/
0,35 мм) [172], циклопропил (94—95° С/0,65 мм) [95], перфторфенил [107],.
диметилсилилметил (133° С/5 мм; 1,4764/25) [172], этоксидиметилсилилме-
тил (147° С/5 мм; 1,4682/25) [172], диметил-[2-(диметилхлорсилил)этил]-
сил ил метил [172].
Получение триэтил-я-октилолова[148]. В трехгорлой колбе на 750 мл, снабженной
обратным холодильником, мешалкой и капельной воронкой, приготовляют магнийорганиче-
ское соединение из 14,5 г (0,6 г-атома) стружек магния в 150 мл безводного эфира и 116 г
(0,6 моля) бромистого октила. К охлажденному раствору прибавляют по каплям при
перемешивании 72,5 г (0,3 моля) хлористого триэтилолова. Кипятят один час с обратным
холодильником, затем растворитель отгоняют на водяной бане в течение 1,5 час. Отогнанный
эфир возвращают в реакционную смесь и последнюю разлагают при охлаждении 20 мл воды
и 200 мл 5%-ной соляной кислоты. Эфирный слой отделяют, водный экстрагируют эфиром.
Эфирный раствор, высушенный сульфатом натрия, обрабатывают сухим аммиаком.
Образуется небольшое количество осадка, который отделяют фильтрованием. Получают 67,5 г
(70%) триэтил-я-октилолова с т. кип. 135—138° С/3—4 мм.
При введении радикала большего молекулярного веса выход
снижается. Например, (CH3)3SnC12H25 -ft и (C2H5)3SnC12H25-ft выделены с выходом
15,5 и 33%. Кроме того, в этом случае при получении реактива Гриньяра
образуются в значительных количествах углеводороды
2 R'Br + Mg -* R'-R' + MgBr2.
Удаление их дистилляцией становится затруднительным. Продукт
реакции употребляют в дальнейших синтезах без дополнительной очистки
[148].
Во многих случаях получают значительно лучшие выходы продуктов,
если эфир заменяют тетрагидрофураном. Обычно раствор оловоорганиче-
ской соли в гептане обрабатывают магнийорганическим соединением в тет-
рагидрофуране. Таким образом, из хлористого триметилолова и хлористого
я-децил-, я-додецил- и я-октилмагния получают (CH3)3SnR, где R
соответственно я-децил (т. кип. 67° С/0,05 мм, выход 73%), я-додецил (т. кип.
93—98° С/0,15 мм, выход 73%), я-октил (т. кип. 56—58° С/0,3 мм, выход
74%) [148, 151].
Четырехзамещенные соединения не образуются, если имеют место сте-
рические затруднения. Реакция дигалогенида ди-тр em-ал кил олова с
избытком реактива Гриньяра заканчивается на образовании галоидного три-
трет-ал кил олова [67]. Далее, например, хлористый ыеопентилмагний с
(C2H5)3SnCl, (C2H5)2RSnCl и C2H5R2SnCl дает с хорошим выходом смешанные
оловоорганические соединения алифатического ряда. Хлористое тринео-
пентилолово с бромистым этилмагнием дает этилтринеопентилолово, однако
с хлористым неопентилмагнием образуется R3SnSnR3 (т. пл. 275—277° С;
R — неопентил). Тетранеопентилолово (т. пл. 130—135° С) можно
получить с хорошим выходом действием на R3SnBr избытка
концентрированного реактива Гриньяра в бензоле (где возможно поднять температуру
до 100° С). Упомянутое образование дистаннана является, вероятно,
результатом реакции [66]
RsSnCl + 2 RMgCl> R3SnMgCl + MgCl2 +R2,
R3SnMgCl + R3SnCl -^ R3Sn—SnR3 + MgCl2.
Перфорированные группы при синтезе смешанных тетраалкильных
соединений вводят в тетрагидрофуране. Три-я-бутилпентафторэтилолово
получено с выходом 48% (т. кип. 48° С/0,035 мм) [173] аналогично R2Sn(CF2—
CF3)2 (см. стр. 234). Обычным методом получены (в скобках т. кип. и по)-
триизобутилэтил-(115° С/15 мм), триамилпропил-(163° С/10 мм) [44], три-
Синтез на основе производных четырехвалентного олова
223
зиоамилметил-(138—140° С/4 мм; 1,4700/15) [146], триизоамилгептил-
(158—160° С/3 мм; 1,4696) [164], триизоамил-я-нонилолово (218—220° С)
[171].
С галоидными тривинил- и триаллилоловом реакция протекает
аналогично с образованием (CH2=CH)3SnR, где R —бутил (77—78° С/8,6 мм;
1,4851/25) [6], гексил (57—61° С/0,03 мм; 1,4851/25) [1511, я-октил (90-
93° С/0,2 мм; 1,4819/25) [151, 166], я-децил (90—94° С/0,05 мм; 1,4820/25)
П51], фенил (73—75° С/0,45лш; 1,5478/25) [6, 166], 4-метил-1-нафтил [166],
и (СН2=СН—CH2)3SnC4H9 (116—119° С/10 мм; 1,5162/25) [151].
Соединения, содержащие алифатические и алициклические радикалы,
исследованы недостаточно. Реакция протекает сложно. Например, при
реакции бромистого трициклогексилолова с магнийорганическим
соединением наряду с основным продуктом.
(СеНЫз SnBr + RMgX -> (C6Hn)3 SnR + MgBr2
образуется до 18% тетрациклогексил олова. Соединения (C6Hn)3SnR, где
R —метил (т. кип. 221° С/15 мм), этил (т. кип. 227—228° С/15 мм) [74],
изопропил (т. кип. 191—192° С/3 мм), бутил (т. пл. 116—117° С), изобутил
(т. пл. 103—104° С), амил(т. кип. 201—202° С/3 мм), изоамил (т. пл.
57—58° С [196] я-гексил (т. кип. 233—234° С/8 мм), я-октил (т. кип.
233—235° С/3 мм), я-нонил (т. кип. 243—245° С/3 мм) я-децил (т. кип.
272—274° С/3 мм) [174], фенил (т. пл. 191—192° С), я-толил (т. пл. 111° С)
[741, я-метоксифенил (т. пл. 75° С), этоксифенил (т. пл. 70° С) [196],
получены с выходом от 45 до 65%. Соединения этого типа термически стойки,
при перегонке не наблюдается разложения и перераспределения радикалов
[174]. Описаны также три-1-циклопентадиенилвинилолово [166] и три-1-
циклопентадиенилфенилолово (т. пл. 64—65° С) [97].
Что касается оловоорганических соединений ароматического ряда, то,
например, соединения (C6H5)3SnR образуются с достаточно хорошими
выходами (рядом с названием радикала в скобках приведены точки
плавления в °С): метил (60—61, т. кип. 185° С/3 мм) [120, 175, 176], этил (56—58;
т. кип. 138° С/4 мм) [120, 167, 176], я-пропил (т. кип. 190° С/3 мм) [176],
изопропил (84) [176],я-бутил (61—62, т. кип. 222° С/3 мм) [177], изобутил
(т. кип. 183° С/3 мм) [176], я-гексил (54) [151], я-гептил (т. кип. 230—232° С/
2—Змм) [178], я-октил (54—55) [13, 179, 180], я-нонил (т. кип. 250—255° С/
2—Змм) [178], винил (45,2—45,4) [6, 90, 166, 169, 181], аллил (73,5—74,5)
[182], пентен-4-ил [182], циклопропил (67—68° С) [95], циклогексил (131 —
132) [74], 1-циклопентадиенил (130—131) [97], я-фторфенил (т. кип. 171° С)
[150, 168], я-хлорфенил (139—140), я-бромфенил (133—135) [111, 150,
168], я-иодфенил (143) [150, 168], ж-гидроксифенил (207, 208) [183], фе-
ноксифенил (161—162) [111], я-толил (124) [120], о-толил (165) [177], о-
метоксиметилфенил (94,5—95,5) [184], бензил (91—92) [185], 2-фенилэтил
(127—127,5) [13], 1-нафтил (125) [120, 129], 2-фурил (160—161) [97], 2-тие-
нил (206) [156], 1-инденил (129—130) [97], триметилсилилметил (т. кип.
162—163° С/2 мм, п23 1,5943), я-триметилсилилфенил (132,5—133,5) [ 20].
Получение метилтри-я-толилолова [175]. К реактиву Гриньяра, приготовленному из
0,17 г (7 ммоля) магния и бромистого метила, прибавляют 2,17 г (5,1 ммоля) хлористого три-
п -толи л олова. Реакционную смесь кипятят с обратным холодильником 3 часа, затем избыток
магнийорганического соединения разлагают водой, эфирный слой отделяют и высушивают.
Твердое вещество, оставшееся после отделения эфира, кристаллизуют из этанола. Получают
1,90 г (92%) метилтри-я-толилолова с т. пл. 69° С.
Примесью к основному продукту могут быть соединения со связью Sri—
Sn. Так, при реакции хлористого трифенилолова и бромистого этилмагния
образуется наряду с этилтрифекилоловом заметное количество гексафенил-
дистаннана [168]. Выход лучше в тетрагидрофуране. В этом случае я-гек-
224
Оловоорганические соединения. Лит. стр. 261—268
оилтрифенилолово (т. пл. 54° С), например, образуется с выходом 83% [148,
151].
Получение (#-октил) трифенилолова [13]. Реактив Гриньяра, полученный из 14,4 г
(0,60 моля) магния и 125 г (0,65 моля) бромистого октила в 150 мл эфира взаимодействует
с 15,6 г (0,30 моля) хлористого трифенилолова. Реакционную смесь разлагают обычным
путем. Получают 75,0 г (54%) (я-октил)трифгнилолова. После нескольких
перекристаллизации из этанола т. пл. 53,5—55° С.
Наличие и характер орто-заместителей в моногалогенидах
ароматического ряда существенно влияет на их реакцию с магнийорганическими
соединениями. В бромистом тримезитилолове сравнительно легко происходит
замещение брома первичными алкильными радикалами даже с относительно
длинными углеродными цепями; труднее происходит замещение вторичными
радикалами; замещения третичными радикалами не происходит.
Получены [2,4,6-(СН3)зС6Н2]з5пК, где R — (в скобках т. пл., °С) метил (155),
этил (143,5), я-пропил (130), я-бутил (118,5), я-гексил (115), я-октил (104,5),
изопропил (146,5), изобутил (100), изоамил (107), фенил (143) [126].
Магнийорганические соединения, содержащие в молекуле иной элемент,
например R3SiR'MgX [20, 144, 161, 186] или R2HSiR'MgX [187, 188],
реагируют с оловоорганическими галогенидами, образуя соединения,
содержащие олово наряду с другими элементами (см. стр. 239).
(CHsfoSiCHaMgCl Ь R3SnCl -» (CM3)3SiCH2SnRo -f MgCl2,
«-(CHsfoSiCeliiMgBr + R3SnCl -» ^-(CWafeSiCsI^SnRs \- MgBrCI.
Получены с выходом 58—72%. триметил(триметилсилилметил)олово
(т. кип.64—65° С/24 мм; п?и 1,4594) [20, 161 ], триэтил(диметилэтилсилил-
метил) олово (т. кип. 99—100° С/3,5 мм; dl'1,135, ^1,4800), триэтил(ди-
метилэтилсилил)олово (т. кип. 99—100°С/3,5жж) [186],триэтил-(/г-триэтил-
силилфенил)олово [189], трифенил-(/г-триметилсилилфенил)олово (т. пл.
132,5—133,5° С) [20, 161], триметил-(/г-триметилстаннилбензил)олово
(т. кип. 98—100° С/0,04 мм) [190].
Получение трифенил (триметилсилилметил)олова [20]. К хлористому триметплсилил-
метилмагнию, полученному из 12,2 г (0,1 моля) хлорметилтрнметилсилана и 2,4 г (0,1 моля)
магния в 50 мл эфира, прибавляют по каплям 9,3 г (0,05 моля) хлористого трифенилолова
в 50 мл эфира со скоростью, достаточной для поддержания кипения смеси. Смесь
перемешивают при комнатной температуре 30 мин, за это время коричневая окраска исчезает.
Затем нагревают с обратным холодильником 3 часа, гидролизуют 5%-ной соляной
кислотой, эфирный слой высушивают хлористым кальцием и перегоняют. Выход 62%; т. кип.
162—163°, С/2 мм; d2^ 1,2369; я^1, 5943. В тетрагидрофуранэ выход значительно выше
[161] (см. стр. 240).
Димагнийорганические соединения реагируют с моногалогенидами
R3SnX с образованием соединений, содержащих два атома олова в молекуле
[191—193]. Димагнийорганическое соединение вступает в реакцию
полностью (R = — (СН2)4—, — С6Н4—) [194, 195]:
2 (C6H5)8SnCl + BrMgRMgBr - (C8H5)3SnRSn(CeH5)3 + MgCl2 + MgBr.
Эта реакция находит применение для получения оловоорганических
ацетиленидов.
2R3SnX -г XMgC = CMgX -* R3SnC = CSnR3 -b 2MgX2.
Компоненты смешивают в эфире или, лучше, хлороформе. В хлороформе, в
противоположность эфиру, димагнийорганическое соединение хорошо
растворяется. Реакционную смесь оставляют на несколько дней при комнатной
температуре; при более высокой температуре магнийорганическое
соединение неустойчиво. Ацетилениды олова ароматического ряда, как и их рас-
Синтез на основе производных четырехвалентного олова
225
творы в органических растворителях, к воде на холоду устойчивы,
щелочами разлагаются. Алифатические соединения влагой воздуха медленно гид-
ролизуются [191].
Как указано выше, соединения (CH2=CH)3SnX реагируют с реактивом
Гриньяра подобно предельным соединениям (см. стр. 223).
До открытия метода получения галоидного винилмагния [88] было
известно только несколько отдельных представителей оловоорганических
соединений с непредельными группами [197, 198]. Теперь их получают
действием хлористого или бромистого винилмагния на оловоорганические га-
логениды или хлорное олово (см. стр. 213), например,
RaSnCl + CH3=CHMgCl -* R8SnCH=CH3 + MgCl2,
Выход 70—89% [6, 90, 199 — 201].
Получение винилтри-#-бутилолова [6]. К 14,6 г (0,61 г-атона) стружек магния в
500 мл тетрагидрофурана (последний для удаления воды и перекисей перегоняют над литий-
алюминийгидридом) прибавляют 61 г (0,61 моля) бромистого винила. Применяют
холодильник с сухим льдом. Для инициирования реакции вводят несколько капель йодистого метила.
Когда реакция образования магнийорганического соединения закончится, заменяют
холодильник на обычный (с водяным охлаждением) и вводят раствор 107,5 г (0,33 моля)
хлористого три-^-бутилолова в равном объеме тетрагидрофурана с такой скоростью, чтобы
поддерживать умеренное кипение. Затем смесь нагревают с обратным холодильником 19 час,
охлаждают до комнатной температуры и гидролизуют 100 мл насыщенного водного раствора
хлористого аммония. Органический слой декантируют, остаток экстрагируют несколькими
порциями серного эфира. Растворитель отгоняют при атмосферном давлении. Остаток
дистиллируют в вакууме. Получают 88,4 г (84,5%) винил-три-я-бутилолова с т. кип. 95°
С/1,5 мм.
Тем же методом получены CH2=CHSnR3, где R = СН3, С2Н5, С6Н5,
а также метилдиэтилвинилолово [6, 42, 91, 144, 152, 166].
Твердые продукты, естественно, выделяются из реакционной смеси
иначе [6]. Время, необходимое для проведения синтеза, значительно
сокращается, если его проводят при более высокой температуре. В качестве
растворителя применяют смесь тетрагидрофурана с углеводородами. Так
получены: три-я-пропилвинилолово, выход 87% [169]. три-я-бутилвинилолово,
выход 85% [90]; трифенилвинилолово, выход 79% [169]. Методика
приведена для синтеза ди-я-бутилдивинилолова (см. стр. 235).
Оловоорганические окиси и гидроокиси в реакциях с магнийорганиче-
скими соединениями, в том числе и реактивом Иоцича [202—204], описаны
в ряде примеров.
Получение три-#-бутилвинилолова [90].
СН2=СН—MgCl + [(tt-C4H9)3Sn]20 -> 2 (tt-C4H9)3Sn—CH=:CH2 + (MgCl)20.
К 592,1 г (1,0 моля) окиси бш>три-я-бутилолова в 2 л гептана в колбе емкостью Ъл
прибавляют при температуре 70—80° С 2,2 моля хлористого винилмагния в тетрагидрофура-
не. Смесь нагревают с обратным холодильником 3 часа. Получают 541 г (?5%) три-я-бутил-
винилолова с т. кип. 114° С/3 мм [90, 205].
Оловоорганические соединения, содержащие перфторвинильную
группу, получают действием бромистого или йодистого перфторвинилмагния на
галоидное триалкил- или триарилолово.
С йодистым перфторвинилом работают в диэтиловом эфире, с бромистым —
в тетрагидрофуране:
R3SnCl + CF3=CFBr .—_* R3SnCF=CF2 + MgBrCl.
(СН2)*о
15 Заказ № 207
226
Оловоорганические соединения. Лит. стр. 261—268
Триэтил-, гри-я-бугил~, трифенилперфторвинилолово, а также диметил-,
ди-я-бутил, дивинил-, дифенилдиперфторвинилолово образуются с
выходом 31—67% [92, 196, 206, 207].
При синтезе бромистого перфторвинилмагния в тетрагидрофуране
необходимы некоторые предосторожности в отношении температурных условий
и концентрации реагентов [208]. Перфторвинилирование протекает с
лучшими результатами по реакции Барбье. В этом случае смесь бромгрифтор-
этилена (в небольшом избытке) и галоидного оловоорганического соединения
в тетрагидрофуране прибавляют при интенсивном перемешивании к
стружкам магния в тетрагидрофуране [208] при температуре от —10 до —15° С.
Выход поднимается от 50 до 80%.
Получение триэтилперфторвинилолова [208]. В трехгорлую колбу, снабженную
мешалкой, капельной воронкой, холодильником с сухим льдом и трубкой для подвода азота,
помещают 18 г (0,74 г-атома) стружек магния и прибавляют столько сухого тетрагидрофурана,
чтобы покрыть их. Магний активируют 10 каплями 1,2-дибромэтана, затем вводят 200 мл
тетрагидрофурана и небольшое количество бромтрифторэтилена. После начала реакции
(окрашивание смеси в коричневый цвет) колбу охлаждают до —15° С и при хорошем
перемешивании медленно (в течение 4 час.) прибавляют раствор 109 г (0,68 моля)
бромтрифторэтилена и 121 г (0,5 моля) хлористого триэтилолова в 300 мл тетрагидрофурана. Затем
реакционную смесь в течение 3 час. перемешивают при —15° С и оставляют на короткое время при
комнатной температуре; охлаждают до 0° С и гидролизуют насыщенным раствором
хлористого аммония. Органический слой декантируют, неорганические соли промывают тетрагид-
рофураном. Комбинированный органический слой и тетрагидрофуран от промывания
быстро дистиллируют в вакууме в приемник, охлажденный до —78° С. Фракционная перегонка
этого дистиллята дает 115 г (80%, считая на хлористое триэтилолово)
триэтилперфторвинилолова с т. кип. 66—67° С/12 мм.
Метод дал хорошие результаты в синтезе трибутил- и трифенилперфтор-
винилолова, диэтилдиперфторвинилолова, н-бутл- и фенилгрифгорвинило-
лова (см. стр. 235).
Моно- и диаллильные производные олова описаны достаточно подробно.
Их получают обработкой алкил- или арилгалогенидов олова бромистым
аллилмагнием в эфире, тетрагидрофуране или смеси тетрагидрофурана с
углеводородами.
RsSnCl + CHa=CH—CH2MgBr -* R3SnCH2—CH=CH2 + MgClBr.
Соединения, где R — метил (г. кип. 128—130° С) [6, 91, 153, 209], этил
(т. кип. 60—61° С/7 мм) [163, 210], пропил (т. кип. 101—103° С/4—5 мм)
[211], бутил (т. кип. 89° С/0,3 мм) [163,210] и другие алифатические
радикалы [212, 213], а также фенил (г. пл. 72—74° С) [93, 182, 210, 214] полу-
чены с хорошими выходами.
Для получения, например аллилгри-/г-голилолова [210] бромистый
аллилмагний в этиловом эфире и хлористое три-/г-толилолово в бензоле
нагревают с обратным холодильником 18 час. Выход 72,5%, т. пл. 58—60° С
[210]. Соединения этого типа в некоторых случаях выгоднее получать
действием бромистого аллила на R3SnNa [215]. Металлильные соединения
СН2 - С (CH3)CH2SnR3, где R — этил (г. кип. 73—74° С/4,2 мм), бутил
(т. кип. 82—83° С/0,1 мм) [210], фенил (т. пл. 72—73 ° С) [210, 214], толил
(т. пл. 45,5—47° С) получены в тетрагидрофуране.
сх-Триметилоловостирол образуется при действии бромистого триметил-
олова на реактив Гриньяра, полученный из а-бромстирола [216]. Пропе-
нилтриметилолово (г. кип. 125—126° С) содержит 64% цис- и 36%-транс-
изомеров. В последнем случае выход значительно выше, если применять
литийорганические соединения [217].
Смешанные соединения Ar3SnR, где R — непредельный радикал,
описаны довольно полно (см. стр. 231). Трифенил-4-пентенилолово образуется
при действии бромистого 4-пентенилмагния на хлористое трифенилолово,
Синтез на основе производных четырехвалентного олова 227
но выделить его в чистом виде не удается. Полученное маслообразное
вещество разлагается при перегонке [182]. При действии бромистого пропар-
гилмагния на оловоорганические галогениды образуется смесь продуктов
R3SnCH2C==CH и R3SnCH=C=CH2. В случае бромистого триметилолова
выход 30—40 и 70% соответственно. В ароматическом ряду пропаргилтри-
арилолово является основным продуктом реакции. Выходы, например,
для (C6H5)3SnR, где R = — СН2С==еСН, 75%, для —СН(СН3)С= СН—
60% и для —СН2С = ССН3 — 60% [218—221].
Получение трифенилпропаргилолова [220]. В раствор бромистого пропаргилмагния,
полученный из 0,48 г (0,022 г-атома) магния и 1,6 мл бромистого пропаргила в 50 мл
эфира, вводят при 18—20° С 3 г (0,0063 моля) твердого йодистого трифенилолова. Через 2—3
часа реакционную массу гидролизуют насыщенным раствором хлористого аммония и
экстрагируют эфиром. После выпаривания растворителя продукт перекристаллизовывают из гекса-
на. Получают 1,8 г (75%) вещества с т. пл. 81—83°С. Второй продукт реакции (C6H5)3SnCH=
= С=СН2 (менее 10%) очень хорошо растворим в гексане.
Трибугилбугадиенилолово (г. кип. 80—90° С/0,4 мм) получено с выходом
76% действием бромистого бутадиенил-2-магния на гексабутилдистаннок-
сан [222].
Ацетиленовые производные олова получают действием галоидного триал-
кил(триарил)олова на ацетилениды магния, а также лития и натрия [7,
219, 223, 224] (см. стр. 253). Обычно последние берут в небольшом избытке.
Реакцию проводят в растворе эфира или тетрагидрофурана или в их смеси.
При температуре 20—50° С образование четырехзамещенного соединения
заканчивается в течение нескольких часов [219]. Выходы в алифатическом
ряду часто умеренные.
Получены соединения формулы (CH3)3SnC==CR, где R = Н (выход
15%; т. кип. 95—96° С) [218]; R - —СН = СН2 (г. кип. 46,5—47° С/10 мм)
[225]; R - —С (СН3) - СН2 (выход 30%; т. кип. 40—42° С/5 мм) [218,
2211; R = С6НИ [219]; R — циклопентенил (выход 20%; т. кип. 68—70° С/
Змм) [221]; R — циклогексенил (выход 20%; т. кип. 68—70е С) [218, 221].
Описано также гриэтил(бутек-3-ин-1-ил)олоЕО (т. кип. 89,5—90° С/10 мм)
[225]. Соединения этого типа в отличие от аналогичных соединений
ароматического ряда чувствительны к влаге Еоздуха [218, 221].
В ароматическом ряду выходы достигают 80—8596 [218]. Ряд
соединений получен по схеме
(C6H5)3SnBr + RC=CMgBr -> (C6H5)3SnC=CR + MgBr2,
где R = H (т. пл. 34°С);СН8 (85%; 74—75° С) [218, 219, 226];СН2ОН (80%;
66-68° С) [218]; СН-СН2 (58%; 58-59° С) [218, 221]; С(СН3)-СН2
(77%, 64° С); циклогексенил (66%, 105—106° С) [144, 218].
Получение трифенил-(1-пропинил-1)олова [218]. К бромистому этилмагнию, получен"
ному из 0,36 г (0,015 г-атома) магния и 1,6 г (0,0147 моля) бромистого этила в 20 мл тетра"
гидрофурана, прибавляют малыми порциями 30 мл 1 N раствора пропина (0,03 моля) в тет-
рагидрофуране и перемешивают 30 мин. при 20—25° С. Затем вводят 4,3 г бромистого
трифенилолова в 10 мл тетрагидрофурана, перемешивают в течение одного часа при 30—40° С,
гидролизуют насыщенным раствором хлористого аммония и экстрагируют эфиром.
Растворитель выпаривают досуха, остаток перекристаллизовывают из гексана. Получают 3 г
(85%) вещества с т. пл. 84—85° С.
Получен ряд ениновых оловоорганических соединений действием
галоидного гриалкилолова на бромистый винилацетиленидмагний в эфире [224,
225, 227], однако с небольшими выходами. Соединения этого типа удобнее
получать действием триалкилстаннилнатрия на алкенилбромацетилены
[228], или алкил- или арилстаннолов на реактив Иоцича. В последнем
случае выходы триэтилвинилэтинил-, трибутилвинилэтинил-, трифенилвинил-
этинилолова достигают 87% [229]. Во всех соединениях связь металла с
15*
228
Оловоорганические соединения. Лит. стр. 261—268
углеродом гораздо более чувствительна к основаниям, чем кислотам.
Наиболее нестойки соединения, в которых атом олова находится
непосредственно у тройной связи [219].
В тех же условиях образуются диацетилениды [(C6H5)3SnC = C]2R,
где R — тримегилен (выход 40%, т. пл. 105° С); тетраметилен (выход 50%,
г. пл. 148° С); 9, 10-антрацен (т. пл. 225°С).со,со-Диацетиленидтрифенилолово
получено с выходом 35%, т. пл. 245° С [218, 219].
Смешанные оловоорганические соединения, содержащие циклопента-
диенильные группы, получают действием циклопентадиенилмагния на
оловоорганические галогениды [97, 230].
Бромистый циклопентадиенилмагний получают с выходом 75—85% (по
титрованию кислотой) действием бромистого эгилмагния на циклопента-
диенил (см. получение гетрациклопентадиенилолова, стр. 215):
C2H5MgBr + С5Нб беНЗ°Л-эфи^ G5H5MgBr + С2Нб,
C5H5MgBr + (C6H5)3SnCl -> (G6H5)3SnG5H5 + MgClBr.
Все операции с реактивом Гриньяра выполняют в свободном от
кислорода азоте.
Получение трифенил-1-циклопентадиенилолова [97]. Раствор 20 г (0,05 моля) хлористого
трифенилолова в 200 мл эфира быстро прибавляют к раствору, содержащему 0,1 моля
бромистого циклопентадиенилмагния в 105 мл смеси бензола с эфиром (т. кип. 60° С).
Реакционную смесь нагревают с обратным холодильником 4 часа, затем концентрируют до пасты.
Пасту обрабатывают дважды порциями по 200 мл смеси (1:1) бензола с петролейным эфиром
(т. кип. 57—70° С). Раствор фильтруют горячим. Фильтрат концентрируют и охлаждают.
Получают 17,4 г (84%) трифенил-1-циклопентадиенилолова с т. пл. 124—129° С. После двух
перекристаллизации из петролейного эфира (т. кип. 57—70° С) получают 14,8 г (71,5%)
чистого продукта с т. пл. 130—131° С.
В спирте трифенил-1-циклопентадиенилолово гидролизуется уже при
комнатной температуре с образованием гидроокиси трифенилолова.
Поэтому при получении оловоорганических соединений, содержащих цикло-
пентадиенильные группы, реакционную смесь не гидролизуюг водой.
Оловоорганические соединения растворяются в смеси бензола и
петролейного эфира, в то время как соли магния и бромистый
циклопентадиенилмагний нерастворимы.
Взаимодействие магнийорганических соединений ароматического ряда
с галогенидами R3SnX имеет свои особенности. Если с моногалогенидами
алифатического ряда реакция протекает, как правило, легко и с
высокими выходами R3SnAr [155, 158, 231, 232] (см. стр. 221), го в алицикличе-
ском ряду выходы часто умеренные или даже низкие [74] (см. стр. 223).
Например (C6Hn)3SnC6H4X, где X = Н (т. пл. 191° С), р-СН3 (т. пл. 111 °С,
т-СН3 (г. пл. 104° С), р-С2Н5 (т. пл. 89° С) [154], ж-трифтормегил (г. пл.
108° С) [122], р-^-С4Н9 (г. пл. 139,5° С) [122, 154], р-С1 (т. пл. 116° С),
/п-С1(г. пл. 109,5° С), р-Вг (г. пл. 124° С), p-F (т. пл. 155° С), р-СН30 (г. пл.
151, 152° С), /я-СН30 (г. пл. 129, 130° С), р-С6Н5 (г. пл. 118° С) получены
с выходом от 35 до 90% [154, 231].
Несимметричные соединения ароматического ряда наиболее широко
исследованы на примере трифениларилолова (см. стр. 237).
Соединения формулы (C6H5)3SnAr, где Аг — перфторфенил (т. пл. 82—
84° С) [107, ПО], (86° С) [108]; бифенил (г. пл. 138° С) [2331; бензил (г.
пл. 108—109° С) [234]; 2-фенилэтил (г. пл. 127—127,5° С) [232]; трифе-
нилмегил (г. разл. 272—273° С) [235], а также три-(ж-оксифенил)фенилолово
{т. пл. 203—205° С) [183], гри-(ж-толил)-я-голилолово (т. пл. 103° С), три-
(ж-толил)-о-голилолово(т. пл. 168°); трибензилфенилолово (т. кип. 290°/5лш)
и трибензил-я-голилолово [177] образуются обычно с выходом до 50—60%.
Синтез на основе производных четырехвалентного олова
229
Соединения типа R3SnC6H4X (где X — бром) представляют особый
интерес. Они могут служить исходным материалом для получения
производных с иными заместителями в ядре, например кислот R3SnC6H4COOH [236].
Их получают действием галоидного триалкил- или триарилолова на
бромистый или йодистый д-бромфенилмагний [111, 168, 236, 237]
RsSnBr + BrMgC6H4Br -» R3SnC6H4Br + MgBr2.
Получение три фенил-/z-бромфенилолова [168]. К профильтрованному раствору магний-
органического соединения, полученного из 41,3 г (0,17 моля) п-дибромбензола, 5 г (0,206
г-атома) магния в 125 мл абсолютного эфира, при охлаждении снегом с солью и
перемешивании постепенно приливают раствор 38,5 г (0,1 моля) хлористого трифенилолова в 100 мл
эфирно-бензольной смеси. После 2 час. кипячения на водяной бане реакционную смесь
разлагают охлажденным раствором хлористого аммония и фильтруют от нерастворимого в
эфире осадка. Эфирный слой высушивают сульфатом натрия, растворитель отгоняют,
кристаллический остаток промывают холодным спиртом. Перекристаллизовывают из смеси
бутилового и этилового спиртов. Получают 34,7 г (68,1%) вещества в виде бесцветных
призм ст. пл. 130° С.
Из нерастворимого в эфире осадка изолировано тетрафенилолово и
(C6H5)3SnC6H4—C6H4Sn (C6H5)3. Образование этих соединений не отмечено
при синтезе соответствующих фтор- и хлорпроизводных, что, очевидно,
связано с различной подвижностью галоида в ядре. Тримегил-/г-бромфенилолово
[97], гриэгил-я-бромфенилолово (выход 48,2%) [168, 236], грипропил-я-
бромфенилолово (выход 36,3%) получены в сходных условиях. Последние
соединения очень чувствительны к кислотам [168].
Если применяют я-бромфениллигий вместо реактива Гриньяра, трифе-
нил-п-бромфенилолова не образуется, вероятно, из-за расщепления связи
С—Sn литийорганическим соединением. Перед реакцией с хлористым три-
фенилоловом я-бромфениллигий следует превратить в магнийорганическое
соединение.
Получение трифенил-я-бромфенилолова [237]. К 11,8 г (0,05 моля) п-дибромбензола
при 0° С прибавляют 0,05 моля я-бутиллития. Через 10 мин. к литийорганическому
соединению быстро прибавляют 0,05 моля йодистого магния (получен обработкой магния иодом в
безводном эфире) и перемешивают 10 мин. Тем временем реакционную смесь охлаждают
до —20° С и вводят 19,3 г (0,05 моля) хлористого трифенилолова в 75 мл сухого бензола.
К концу прибавления цветная проба Гилмана отрицательна. Через 3 мин. реакционную
смесь гидролизуют холодным водным раствором хлористого аммония. Эфирный слой
отделяют, высушивают сульфатом натрия, фильтруют и перегоняют. Получают 21,7 г (86%)
вещества с т. пл. 132—135° С. После перекристаллизации из 150 мл этанола в смеси с 50 мл
бензола получают 18,1 г (72%) трифенил-п-бромфенилолова с т. пл. 133—135° С [237].
Аналогично получены триметил-/2-хлорфенилолово [157], грифенил-/?-
фторфенилолово (выход 78,6%; т. пл. 171° С), грифенил-/г-хлорфенилолово
(выход 88,2%; г. пл. 141° С) [111, 168]. Соединения формулы C6F5SnR3,
где R — метил, бутил или фенил, могут быть получены как в эфире, гак и
тетрагидрофуране [107, 108, ПО] (см. стр. 237).
Толил-, бензил-, ксилил-, мезитилмагнийбромиды дают с Ar3SnX че-
тырехзамещенные соединения с достаточно хорошими выходами.
Получение три-о-толил(бензил)олова [234]. В колбу на 100 мл помещают 5 г (0,012 моля)
хлористого три-о-толилолова в 25 мл эфира. К нему прибавляют по каплям при хорошем
перемешивании 0,015 моля хлористого бензилмагния в 25 мл эфира. Реакционную смесь
кипятят с обратным холодильником 1 час. и гидролизуют. Из эфирного слоя получают после
двух кристаллизации из метанола 3,4 г (58,6%) три-о-толилбенз ил олова с т.пл. 108—109° С.
Реакция затруднена или не имеет места в тех случаях, когда возникают
пространственные затруднения. С атомом олова может быть связано не
более трех ди-орго-замещенных арильных групп. Соединения олова с
числом орто-заместителей больше чем шесть не образуется даже при
использовании самых активных реагентов (литий-, натрий- и калийарила) [130].
230 Оловоорганические соединения. Лит. стр. 261—268
Длина цепи орто-заместителя не имеет существенного значения. Более
заметное влияние оказывает разветвленность цепи: даже замена я-бутила на
изобугил вдвое снижает выход продукта [130]. Действием бромистого ме-
зигилмагния на соответствующие монобромиды получены грифенилмези-
тилолово (г. пл. 157° С), гри-о-толилмезитилолово(г. пл. 125° С), гри-/г-кси-
лилмезитилолово (т. пл. 186° С) и три-а-нафтилмезитилолово с выходом
40—50 ?6. С бромистым тримезитилоловом выделено фенил тримезигилолово
(т. пл. 143°), но о-голилгримезитилолово получить не удается [126, 130].
Получение трифенил-о-метоксиметилфенилолова [184]. К бромистому о-метоксиметил-
фенил магнию, приготовленному нагреванием с обратным холодильником в течение 6 час.
1,95 г (0,080 г-атома) магния и 15,1 г (0,075 моля) о-бромбензилового эфира в 150 мл эфира
и 60 мл бензола, прибавляют 20,0 г (0,0518 моля) хлористого трифенилолова. Смесь
перемешивают несколько часов и гидролизуют раствором хлористого аммония. Получают после
двух кристаллизации из смеси бензола с этанолом 8,5 г (35%) трифенил-о-метоксиметилфе-
нилолова с т. пл. 93—95° С. После перекристаллизации из того же растворителя т. пл.
94, 5—95,5° С.
Выход несколько выше (38%), если бромистый о-мегоксимегил фенил -
магний получают с помощью лигийорганического соединения 1184].
Вообще в синтезе оловоорганических соединений реактив Гриньяра
получают иногда из литийорганических соединений, так как последние не
всегда пригодны для алкилирования. В случае их применения возможен
обмен радикалами между олово- и литийорганическими соединениями (см,
стр. 453). Для магнийорганических соединений эта побочная реакция не
имеет места. Магнийорганические соединения образуются при простом
добавлении бромистого магния к литийорганическому соединению. Методом
пользуются в том случае, когда магнийорганическое соединение прямым
методом получить не удается.
В случае синтеза оловоорганических соединений с гидроксифенильными
группами реакция протекает по уравнениям
m-BrC6H4OLi + QH9Li -> m-LiC6H4OLi + C4H9Br,
m-LiOC6H4Li + MgBr3 -» m-LiOC6H4MgBr + LiBr,
m-LiOC6H4MgBr + (C6H5)3 SnCl -> (C6H5)3 SnC6H4OLi,
NH4C1
(C6H5)3SnC6H4OLi > (C6H5)3SnC6H4OH.
н2о
Получение трифенил-лг-оксифенилолова [183]. К холодному раствору 49,5 г (0;286
моля) лгбромфенола в 60 мл эфира при хорошем перемешивании прибавляют 0,572 моля
я-бутиллития в 830 мл эфира. Реакционную смесь перемешивают при комнатной
температуре 12 час, затем обрабатывают 0,572 моля безводного бромистого магния в 450 мл эфира.
Через 30 мин. прибавляют 57 г (0,148 моля) хлористого трифенилолова, суспензированного
в 250 мл. эфира. Смесь нагревают с обратным холодильником 3 часа, затем выливают в
охлажденный льдом раствор хлористого аммония. Органический слой отделяют и
высушивают сульфатом натрия. Эфир отгоняют, а остаток обрабатывают 100 мл 95%-ного этанола,
охлаждают и фильтруют для удаления веществ, не вступивших в реакцию. После двух
кристаллизации из хлороформа получают 34,3 г (49,5%) трифенил-л*-оксифенилолова с т.
пл. 207—208° С.
Как известно, кислоты, фенолы, меркаптаны и другие подобные
соединения (см. стр. 352) при высоких температурах разрывают связь олова с
углеродом. В описанном синтезе сырой продукт может содержать фенол,
потому следует избегать нагревания выше 40—50° С.
Особый интерес представляют оловоорганические соединения, способные
давать полимеры или сополимеры.
Стиролы с оловоорганическими заместителями в ядре получают
действием хлористого или бромистого /2-винилфенилмагния на галоидозамещен-
Синтез на основе производных четырехвалентного олова 231
ные оловоорганические соединения в тетрагидрофуране:
сн = сн2 сн = сн2
I
+ ClSn(C6H5)3^f J +MgCl2.
MgGl Sn (СбН5)3
Для инициирования реакции Гриньяра вводят бромистый или йодистый
алкил. В препаративном отношении этот метод является наиболее удобным.
Выход 60—80%. Полимер в реакционной среде отсутствует [238], тогда
как в эфире при доступе воздуха образуются только неплавкие твердые
продукты полимеризации [133], Трифенил-(я-винилфенил)олово [238—
242] получено с выходом 78,5% (т. пл. 112—113° С), дифенилди(-я-винил-
фенил)олово — с выходом 65%, т. пл. 108—109° С [243].
Для ft-винилфенилтрициклогексилолова (т. пл. 104—105° С) и /г-винил-
бифенилтрифенилолова (т. пл. 139—141° С) выход составляет 30 и 41 %. л-Ви-
нилфенилгрибензилолово выделено в виде полимера, легко
окисляющегося при стоянии [244].
При перегонке стирола с оловоорганическими заместителями в ядре
требуется осторожность. При недостаточно глубоком вакууме значительная
часть оловоорганического производного стирола полимеризуется в
перегонном кубе, что снижает выход.
Тримегил-(/г-винилфенил)олово получено с выходом 62%, т. кип. 68° С/
0,52 мм, п2£ 1,5643 [245] и с выходом 81%, т. кип. 53—54° С/0,0001 мм,
п2£ 1,5648 [246]. Для триэгил-(/г-винилфенил)олова выход 40%, г. кип.
ЮГ С/0,55 мм, п2£ 1,5530 [245], для гри-я-бугил-(/г-винилфенил)олова
выход 41,1%, т. кип. 130—131° С/3 лш [133], 126° С/0,04 мм [242] для^ триизо-
амил-(л-винилфенил)олова выход равен 37%, т. кип. 150—152° С/3 мм
[133].
Олозоорганические производные а-метилстирола образуются
аналогично. Тримегил-(/г-а-мегилвинилфенил)олово получено с выходом 61 % (т.
кип. 49—51° С/0,0003 мм), df 1,2743, п2£ 1,5574; гриэгил-(Аг-а-мегилвинил-
фенил)олово — с выходом 50% (г. кип. 136—136,5° С/3 мм; п2£ 1,5448)
[247, 2481; три-«-бутил-(/г-метилвинилфенил)олово — с выходом 39,1%
(т. кип. 170—172° С/3 мм) 1133]; триизоамил-(/г-а-метилвинилфенил)олово —
с выходом 49,9% (г. кип. 190—192° С/4 мм); гри-я-бугил-(Аг-бугенилфенил)-
олово —с выходом 35,7% (т. кип. 191—193° С/5 мм) [133].
Синтез оловоорганических стиролов взаимодействием бромистого
винила с бромфенилпроизводным олова [236] в присутствии безводных солей
кобальта по схеме
R3SnC6H4Br + CH2^CHBr > R3SnC6H4CH = СН2
СоС12
происходит с низким выходом; этот способ не позволяет получить стиролы
в достаточно чистом состоянии [199, 245]. Другим путем получения
стиролов с оловоорганическими заместителями в ядре является действие ацет-
альдегида на реактив Гриньяра, полученный из триэтил-(/г-бромфенил)оло-
ва. При этом с выходом 14,9% образуется соответствующий карбинол.
Однако выделить продукт дегидратации в чистом виде не удается [199].
Оловоорганические соединения с аллильными радикалами в бензольном
ядре образуются при действии бромистого аллилфенилмагния на
оловоорганические галогениды. Получен ряд соединений общей формулы R3SnC6Hr
СН2СН = СН2, где R — этил, выход 57%, т. кип. 150—152° С/5 мм [249],
я-пропил, т. кип. 172—175° С/4—5 мм [90]; я-бугил, выход 58%, т. кип.
О
232
Оловоорганические соединения. Лит. стр. 261—268
172—17573—4 мм; триизоамил, выход 60%, т. кип. 195—198° С/3—4 мм
[171]; фенил, т. пл. 218—220° С [250].
Соединения R3SnCH = CHAr [251] и R3SnC=CAr образуются
аналогично соответствующим соединениям алифатического ряда (см. стр. 227). Аце-
тилениды R3SnC = СС6Н5, где R — метил [219], этил, бутил [192, 229];
(C6H5)3SnC = САг, где Аг—фенил, /г-хлорфенил, я-толил [192, 219], антра-
нил-9-метил [219], д-бромфенил, антранил-9 [218], получены с выходом до
80%.
Симметричные ацетилениды R3SnC = CSnR3 получают
взаимодействием моногалогенидов R3SnX с б^с-магнийбромацетиленом в эфире или
хлороформе. Описаны соединения со следующими R: фенил [191], я-хлорфе-
нил, о- и я-голил, бензил [192,224] (см. стр. 227). [(C6H5)3SnC==C]2Ar,
где Аг —фенилен(т. пл. 205° С), 9,10-дизамещенный антрацен (т.пл. 225°С)
получены аналогично [218].
Инденильную группу вводят действием на оловоорганические галоге-
ниды тонкой суспензии бромистого инденилмагния [97]. Выход последнего
не определяют (смесь гетерогенная), берут его в избытке.
Получение тркфенил-1-инденилолова [97]. 38,5 г (0,1 моля) хлористого трифенилолова
б 300 мл эфира прибавляют к 0,2 моля бромистого 1-инденилмагния в 600 мл эфира. Смесь
нагревают с обратным холодильником 48 час, охлаждают до комнатной температуры и
медленно выливают в водный раствор хлористого аммония. Органический слой высушивают
сульфатом кальция в течение 24 час. и концентрируют. Не вступивший в реакцию инден
удаляют в вакууме, остаток кристаллизуют из смеси хлороформ — петролейный эфир
(т. кип. 57—70° С). Получают 23,9 г (54%) сырого продукта ст. пл. 111—123° С. После
двух кристаллизации из той же смеси растворителей получают 21,0 г (45,5%) трифенил-
1-инденилолова с т. пл. 129—130° С.
Аналогично дифенилди-1-инденилолово получают с выходом 29,9 %> т.
пл. 108—110° С. Трифенил-1-инденилолово и дифенилди-1-инденилолово к
гидролизу достаточно устойчивы [97].
В случае соединений, обладающих подвижными атомами водорода
(например фуран), наиболее доступны лигийорганическиесоединения,
получаемые путем металлирования. В тех случаях, когда синтез смешанных*олово-
органических соединений через литийорганическиое соединения протекает
с осложнениями (см. стр. 245), прибавляют MgJ2 и переводят литийорга-
ническое соединение в реактив Гриньяра.
[| J+ QH9Li -» || И ы + QH10,
О О
MJ- Li + MgJ2 -> [Mj- MgJ + LiJ,
О О
Г~|_- MgJ + (C6H5)3 SnCl - Г")]-- Sn (СвН6)з + MgJCl.
о о
Таким образом, трифенил-2-фурилолово получено из фурана и
хлористого трифенилолова с выходом 65% (т. пл. 158—159° С) 1252].
Галогениды оловоорганических соединений, содержащие два атома
олова в молекуле, мало исследованы. С реактивом Гриньяра они реагируют по
уравнению [252а]
(СбН5)2 Sn (CH2)„ Sn (C6H5)2 + 2RMgX -» (СбН5)2 Sn (CH2)„ Sn (CGH5)2 + 2 MgJ,
I
J J Я Я
'2
(R = C6H5; СвНц; я = 4~6).
Синтез на основе производных четырехвалентного олова
233
Дигалогениды R2SnX2 с избытком реактива Гриньяра реагируют с
образованием соединений R2SnR'2 [146, 253, 254]. Экспериментальные
условия не отличаются от описанных выше для аналогичных соединений R3SnX.
В качестве исходных возможно применение дигалогенидов любых типов,
однако выбор определяется их доступностью. Например, в синтезе
смешанных соединений с винильными, перфгорвинильными, аллильными,
стиральными и другими непредельными группами исходными, как правило,
являются дигалогениды предельных соединений.
Окиси R2SnO [50а, 90, 202], как и соединения (R2SnX)20 [138,255], могут
реагировать подобно дигалогенидам
(n-C4H9)2 SnO + 2СН2 = CHMgBr -» (я-С4Н9)2 Sn (СН = СН2)2 + (MgCl)2 О.
Последняя реакция имеет ограниченное применение. Моногалогениды
R2SnR'X в отдельных случаях могут быть выделены с достаточно
хорошим выходом. Однако удобнее получить четырехзамещенный продукт и
провести деалкилирование (см. гл. VIII). Многочисленные соединения
R2SnR'2 нормального или изосгроения, полученные взаимодействием маг-
нийорганических соединений с дигалогенидами R2SnX2, в условиях,
описанных для моногалогенидов (см. стр. 220), приведены ниже (в скобках г.
кип. и п% ): (CH3)2SnR2, где R — этил (131—132° С/760 мм; 1,4650/19)
[28, 147, 170, 253], перфторэгил (62—63/89 мм) [96], изопропил (68/29 мм;
1,4621/25), бутил (70° С/4,4 мм; 1,4640/25), втор-бутл (68° С/0,5 мм;
1,4738/25) [256], трет-бутл (84—85° С/40 мм; 1,4062/25) [257], я-амил
(68—70° С/1 мм) [258], трет-амил (119,5—120° С/29 мм; 1,4870/25 мм)
1257], окгил (121—122°С/0,2лш; 1,4659/25) [151], 2-эгилгексил (101—102° С
/0,2 мм; 1,4715/15) [151], винил (120—121° С/760 мм; 1,4720/25) [90], про-
паргил (62° С/5 мм) [258а], циклопенгил (76—77° С/0,3 мм; 1,5109/25)
[256], циклогексил (98°С/0,4жж) [256], гримегилсилилмегил (146,5—147,5°
С/65 мм; 1,4644/25) [20, 161].
(C2H5)2SnR2, где R—а-хлорэгил (114—115° С/5 мм; 1,5083) [259, 260],
пропил (84,5° С/10 мм) [253], изопропил (74—75° С/5 мм) [165], бутил
(112° С/10 жж; 1,4734) [28], амил (139—141° С/14 мм) [148], я-нонил (195—
200° С/5 мм) [165], аллил (99—100°С/17лш) [163, 165, 210], фенил (155—
157° С/4 мм) [19, 167, 253], бензил (223—224° С/20 мм) [123], аллилфенил
(г. пл. 183—186° С) [249].
(/z-C3H7)2SnR2, где R —аллил (191—193 °С) [211 ], фенил (160—16ГС/3 мм)
[167].
(n-C4H9)2SnR2, где R — пенгафгорэгил (42° С/1 мм) [96], изопропил
(102° С/2,9 мм; 1,4756/25) [256], трет-бутл (123—125° С/40 мм) [257];
трет-амил [257], винил (78—80° С/2 мм; 1,4824/25) [90, 166], аллил (93°С/
1,1 мм; 144—145° С/15 мм; 1,5025 [210]; 1,4986/25) [151, 163]; Р-мегаллил
(142—143° С/7 мм; 1,4986 [210], 4-пенгенил (117—118° С/0,5 мм; 1,4842
25) [172], циклопропил (79° С/0,4 мм, 1,4912/25) [95], циклопентил
(128° С/0,3 мм; 1,5067/25) [256], циклогексил (143° С/0,45 мм; 1,5126/25)
[256], фенил [255, 257], гримегилсилилмегил (98° С/0,45 мм; 1,4777/25)
[161], димегилсилилмегил (130/5 мм; 1,4810/25) [172], эгоксидимегилси-
лилмегил (186/15 мм; 1,4655/25) [172].
(СН2 = CH)2SnR2, где R — 2-мегил-З-бугенил [166].
Оловоорганический галогенид удобно вводить в раствор углеводорода,
что даег возможность повысить температуру реакционной смеси.
Получение диэтилди-#-бутилолова [28]. К 0,67 моля бромистого я-бутилмагния в 500
мл безводного эфира прибавляют по камплям 0,185 моля двухлористого диэтилолова в 250
мл смеси бензола с эфиром (1:1). Реакционную смесь нагревают 3 часа с обратным холодиль.
234 Оловоорганические соединения. Лит. стр. 261—26
ником, затем избыток магнийорганического соединения разлагают 1 л 3,5 М раствора
хлористого аммония. Эфирный слой отделяют и высушивают хлористым кальцием, растворитель
отгоняют при атмосферном давлении. Получают 18,36 г вещества с т. кип. 110—115° С/10 мм.
Перегоняют на колонке в 40 теорет. тарелок. Т. кип. 112° С/10 мм\ d22 1,1035; п2р 1,4734.
Перфгорированные группы вводят обычно в геграгидрофуране.
Получение диметил-б'#с-(пентафторэтил)олова [173]. Перфториодэтан (27,3 г; 0,11 моля)
прибавляют при перемешивании в течение 2 час. к смеси двухлористого диметилолова (12,3 г;
0,055 моля) и магния (2,7 г; 0,11 моля) в тетрагидрофуране (500 мл) при комнатной
температуре. Реакционную смесь перемешивают при 35—40° С в течение 15 час. и затем все
летучее при 10~~3 мм конденсируют в колбу для перегонки. Остаток после удаления тетрагид-
рофурана дает 7,3 г (34%) диметил-б^с-(пентафторэтил) олова (т. кип. 62—63° С/89 мм).
Ди-и-бугил-б«с-(пенгафгорэгил)олово (г. кип. 42° С/1 мм\ выход 16%)
получено аналогично [173]. Оловоорганические соединения R2SnR'2, где
R' — радикал изосгроения [164, 256, 261], образуются обычными
методами, выходы от 50 до 88% [151, 2561. С оловоорганическими дигалогенидами
нормального строения хлористый mpem-бугилмагний дает соединения того
же типа. Так получены димегилди-тре/я-бугилолово (т. кип. 84,5—87° С/
40 мм), димегилди-трет-амилолово (т. кип. 119,5—120° С/29 мм), ди-я-
бутилди-тре/я-бугилолово (т. кип. 123—125° С/400 мм). Это стабильные
бесцветные жидкости с раздражающим запахом или в случае высших
соединений неперегоняющиеся воскообразные вещества [256, 257].
Взаимодействие двугалоидногоди-/яр<?/я-ал кил олова даже с большим избытком
реактива Гриньяра заканчивается на образовании галоидного три-трет-
ал кил олова [66]. В последнем случае возможно образование также циклов
со связью олово — олово.
Получениеокта-/угр£АЯ-бутилтетрастанноциклобутана [86]. Магнийорганическое
соединение, полученное из 14 г магния и 47 г третичного хлористого бутила в 200 мл тетрагидрофу-
рана (для инициирования реакции прибавляют немного йодистого метила), фильтруют от
не вступившего в реакцию магния и при перемешивании в течение 20 мин. вводят 10 г
двухлористого ди-гарет-бутилолова. Смесь кипятят с обратным холодильником 4 часа,
охлаждают и фильтруют. Твердый продукт (4,3 г, 56%) промывают разбавленной соляной
кислотой, затем ацетоном и высушивают. Получают тетрамер в виде светло-желтых кристаллов
с температурой разложения 205° С. На солнечном свету окраска бледнеет. Вещество
несколько растворяется в горячем пиридине и горячем дибутиловом эфире, из которых малыми
количествами может быть перекристаллизовано.
Реакция образования циклов со связью олово — олово имеет
ограниченное значение. Уже с двухлористым дициклогексилоловом соединения,
аналогичного окга-тре/я-бугилгеграсганноциклобугану, не образуется [86].
Хлористый винилмагний легко вступает в реакцию с
оловоорганическими хлоридами, бромидами или окисями с образованием оловооргани-
ческих соединений [6, 90] (см. стр. 225).
R2SnCl2 + 2 СН2 = CHMgCl -> R2Sn (CH = СН2)2 + 2 MgCl2.
Это, за немногим исключением, бесцветные жидкости с приятным запахом.
Они отличаются хорошей термической устойчивостью, стабильны по
отношению к воздуху, воде и разбавленным кислотам. Указанные свойства
представляют особый интерес в связи с известной нестабильностью аллиль-
ных оловоорганических соединений [6, 91, 163]. С накоплением винильных
(как и аллильных) групп в молекуле оловоорганического соединения
термическая стабильность понижается [91, 93]. Выход винильных производных
составляет 70—89% [6, 90, 199, 200] для чегырехзамещенных соединений
и 50—56% при частичном замещении с образованием моногалогенида.
Результаты лучше, если в качестве растворителя применяют смесь геграгид-
рофурана с углеводородами.
Синтез на основе производных четырехвалентного олова
235
Получение ди-н-бутилдивинилолова [90]. В колбу на 5 л помещают 303,3 г (1,0 моля) дву-
хлористого ди-«-бутилолова в 2 л гептана. Туда же при нагревании с обратным
холодильником прибавляют в течение 2 час, 2,5 моля хлористого винилмагния в тетрагидрофуране.
Смесь нагревают с обратным холодильником еще 4 часа и обрабатывают, как указано при
синтезе тетравинилолова (см. стр. 213). Получают 254,0 г (89%) дибутил дивинил олова с т.
кип. 78—80° С/2 мм.
Аналогично получают диметилдивинилолово [6, 90, 169]. Если в
качестве исходного применяют окись дибутилолова, ди-я-дибутилдивинилолово
образуется с выходом 58% [90]. Диметил-, ди-я-бутил-, дивинил- и дифе-
нилдиперфторвинилолово получены по методике, приведенной для три-
зтилперфторвинилолова [92, 206, 207] (см. стр. 226). Однако результаты
хуже, чем при перфторвинилировании соединений R3SnX. Выход наиболее
низкий при исчерпывающем перфторвинилировании трехгалоидных оло-
воорганических соединений [208].
Оловоорганические соединения, содержащие перфторвинильные труп*
пы, представляют собой бесцветные жидкости или твердые вещества с
химическими свойствами, отличными отсоединений олова с обычными виниль-
ными или перфторалкильными группами. На воздухе они медленно поли-
меризуются с последующим частичным окислением и гидролизом
полимера. В случае диметилдиперфторвинилолова в качестве конечного продукта
разложения выделена окись диметилолова [92, 207]. Легкость, с которой
перфторвинильная группа отщепляется от атома олова, усложняет
очистку оловоорганических соединений с перфторвинильными группами. Как
известно (см. стр. 206), удобным методом очистки тетразамещенных
оловоорганических соединений, загрязненных небольшим количеством трех-
замещенного продукта, является обработка сырого продукта
водно-спиртовым раствором фтористого калия. Примесь R3SnX выделяется в виде
нерастворимого фторида [8]. Этот метод применим только для очистки
R3SnCF = CF2. Если в молекуле присутствуют две или больше
перфторвинильные группы, то метод может оказаться неприменимым вследствие
частичной или полной деструкции четырехзамещенного оловоорганического
соединения [208] (см. стр. 213). В этом случае примесь монохлорида может
быть отделена пропусканием сухого газообразного аммиака в раствор
вещества в эфире. Выпавший осадок отфильтровывают.
Жидкие оловоорганические соединения, содержащие перфторвинильные
группы, при стоянии медленно выделяют белое твердое вещество. Природа
этого разложения еще не установлена [208].
Моно- и диаллильные производные олова получают обработкой алкил-
и арилгалогенидов олова эфирным раствором бромистого аллилмагния
[163, 182, 210]. Их следует перегонять в вакууме в атмосфере инертного
газа [6, 163]. Перегнанный продукт при комнатной температуре может
храниться месяцами, не изменяя показателя преломления [6]. Диэтилдиал-
лилолово получено, по-видимому, в виде твердого тетрамера [165].
Смешанные оловоорганические соединения, содержащие аллильные
радикалы наряду с ароматическими радикалами, как правило, образуются с
умеренным выходом (25—60%) [90, 151, 163, 210]. Они, как и тетрааллил-
олово, в блоке не полимеризуются [90]. Термическая устойчивость
смешанных соединений, например диаллилдифенил- или аллилтрифенилолова,
по сравнению с тетрааллилоловом повышается [93, 94].
Получение диаллилдифенилолова [151]. К кипящему раствору 343,3 г (1,0 моля) дву-
хлористого дифенилолова в 2 л гептана прибавляют в течение 2 час. 2,2 моля хлористого
аллилмагния в тетрагидрофуране. Смесь нагревают с обратным холодильником еще 4 часа.
После охлаждения гидролизуют осторожным прибавлением 2%-ной водной соляной кислоты,
пока образуются два жидких слоя. Органический слой отделяют, высушивают углекислым
натрием, растворитель отгоняют при атмосферном давлении. Остаток фракционируют.
Получают 213 2 (60%) диаллилдифенилолова с т. кип. 120—124° С/0,005 мм [151], 170 — 173°
С1Ъ мм\ п$ 1,6008 [93, 182].
236
Оловоорганжеские соединения. Лит. стр. 261—268
Оловоорганические соединения, содержащие аллильные радикалы,
представляют собой бесцветные перегоняющиеся в вакууме жидкости,
устойчивые до 170° С, растворимые в органических растворителях;
обесцвечивают раствор брома и перманганата. С уменьшением числа аллильных
радикалов у олова температура кипения и поверхностное натяжение соединений
повышаются, удельный вес и показатель преломления понижаются [211].
Ди-я-бутилди-Р-металлилолово (выход 83,2%; т. кип. 142—3° С/7 мм\
п2£ 1,4986) получено в тетрагидрофуране [210].
Циклопропильные соединения олова получены с помощью бромистого
циклопропилмагния [95]. Циклоалкильные производные (C5H9)2SnR2,
(C6Hn)2SnR2, где R — метил, я-бутил или фенил образуются аналогично
[151, 254, 256].
Получение диметилдициклогексилолова [256]. К хлористому циклогексилмагнию,
приготовленному из 50 г (0,42 моля) хлористого циклогексила и 9,7 г (0,4 г-атома) стружек
магния в 200 мл тетрагидрофурана (для инициирования реакции вводят несколько капель
бромистого этила), прибавляют 35 г (0,16 моля) двухлористого диметилолова в 70 мл того же
растворителя с такой скоростью, чтобы поддерживать умеренное кипение. Реакционную
смесь перемешивают и нагревают еще 22 часа. После охлаждения гидролизуют насыщенным
раствором хлористого аммония. Органический слой декантируют, водный слой промывают
эфиром, растворитель отгоняют при атмосферном давлении. Получают 41,3 г (82%)
диметилдициклогексилолова с т. кип. 98° С/0,45 мм и 101° С/0,6 мм.
Применение в качестве реакционной среды смеси углеводорода с тет-
рагидрофураном дает возможность сократить время, необходимое для
завершения реакции [151].
При взаимодействии дигалогенида дициклогексилолова с реактивом
Гриньяра в эфире диалкилдициклогексилолово (алкил: пропил, изопро-
пил, я-бутил, изобутил, амил, изоамил) получено с выходом 71—91%
[261а].
Действием на двухлористое дифенилолово реактива Гриньяра получены
соединения (C6H5)2SnR2, где R — (в скобках т. кип., т. пл. и п2£) метил
(96—98° С/0,1 мм) [175], винил (121,5° С/0,4 мм; 1,5929/25; 167—170° С/
7 мм; 1,5947/25) [91, 181, 262, 263], аллил (120—124° С/0,005 мм) [93, 151,
182], я-гептил (251— 252° С/2 мм), я-октил (252° С/4 мм), я-нонил (241 —
242° С/2 мм), я-децил (270—275° С/4—Ъ мм) [178], пентен-4-ил (174—175° С/
1,5 мм; 1,5598) [182]; циклопентил (т. пл. 49—50,2° С), циклогексил (т.
пл. 119—120° С) [254, 256], циклогептил (253—4°С/3), циклооктил (255° С/3),
циклононил (262 °С/3), циклодецил (282—3° С/3) [264а], п-трет-бутилфепил
(т.пл. 156° С) [122],л*-оксифенил(т. пл. 189—190° С) [183, 256]; перфторфе-
нил (180—182° С/0,45 мм; т. пл. 84—86° С) [106а, 107, 108, 158]; 1-цикло-
пентадиенил (т. пл. 105—106° С) [97]; бифенил (т. пл. 156° С) [133]; 1-наф-
тил (т. пл. 209—210° С) [254]; 1-инденил (т. пл. 108—110° С) [97]; 2-тиенил
(т. пл.207—210 °С) [254]; я-метоксифенил (т. пл. 125—126° С) [254]; триме-
тилсилилметил (137° С/0,35 мм; 1,5499/25) [20, 161]; /г-триметилсилил-
фенил (т. пл. 95—96° С) [20].
Получение дифенилди-н-гептилолова. К неостывшему раствору реактива Гриньяра,
приготовленному из 1,02 г (0,0422 г-атома) магния и 7,52 г (0,042 моля) ^-бромистого гепти-
ла, при постоянном перемешивании добавляют небольшими порциями 5,20 г (0,012 моля)
сухого двуиодистого дифенилолова, причем наблюдается вскипание эфира. Смесь нагревают
2 часа. После отгонки эфира нагревание продолжают еще 1,5 часа при 100—110° С. К
реакционной массе приливают отогнанный эфир, после чего смесь разлагают сначала холодной
водой, затем 1%-ным раствором соляной кислоты. Эфирный слой отделяют, высушивают над
хлористым кальцием и после отгонки эфира остаток фракционируют в вакууме. Выделяют
4 г дифенилди-я-гептилолова [178].
Взаимодействием дизамещенных дихлоридов олова с бромистым пер-
фторфенилмагнием получен ряд производных диперфторфенилолова.
Бромистый перфторфенилмагний получают в этиловом эфире или тетрагидро-
Синтез на основе производных четырехвалентного олова
237
фуране. В дибутиловом эфире образование бромистого
перфторфенилмагния начинается только после прибавления равного объема этилового эфира.
При применении высококипящего растворителя (дибутилового эфира или
толуола) выход равен 60—85%.
Получение ди(пентафторфенил)дивинилолова [107]. К 1,50 г (62 мг-атома) стружек
магния в 100 мл сухого эфира прибавляют в течение 15—20 мин. 14,6 г (60 мг-молей) бром-
пентафторбензола. Реакция начинается без промедления. Через 2 часа большая часть магния
растворяется с образованием черного раствора бромистого перфторфенилмагния, но раствор
перемешивают еще в течение ночи. 6,1 г (25 мг-моля) двухлористого дивинилолова (т. кип.
62° С/25 мм) прибавляют по каплям к 50 ммолям бромистого пентафторфенилмагния в
100 мл эфира. Происходит разогревание, выделяется масло черного цвета. Реакционную
смесь нагревают с обратным холодильником в течение часа, затем этиловый эфир постепенно
заменяют дибутиловым эфиром. При температуре около 70° С черное масло исчезает, и около
130° С появляется серый осадок, раствор становится светло-коричневым. Кипятят с
обратным холодильником при 145° С°в течение 2 час. После охлаждения смесь гидролизуют
избытком насыщенного водного раствора хлористого аммония. Оловоорганическое соединение
экстрагируют гексаном. После удаления растворителя и фракционирования под вакуумом
лолучают 9,46 г (75%) бесцветной жидкости с т. кип. 107—109° С/0,3 мм; п2£ 1,5014.
Аналогично получены соединения (C6F5)2SnR2, где R — (в скобках т.
кип. и п™) метил (126° С/2,6 мм; 1,4912), бутил (139—14 1° С/2,0 мм;
1,4914); фенил (180—182° С/0,45 мм; т. пл. 84—86° С) [106а, 107, 108, ПО]
п-толил (т. пл. 73—75° С) [108, ПО], о-толил (т. пл. 174° С) [106а].
Вещества устойчивы на воздухе и не гидролизуются водой, но легко отщепляют
перфторфенильные группы [107, 108].
В случае синтеза соединений Ar2SnAr'2 выход зависит от строения
ароматических радикалов. Для оловоорганических соединений, содержащих
толильные, ксилильные, мезитильные, гидроксифенильные радикалы,
выход обычно не превышает 50—60%. Например, бас-(бифенил-2)-дибензил-
олово получено действием на двубромистое 6ш>(бифенил-2)-олово избытка
бромистого бензилмагния в эфирно-бензольной среде с выходом 59,7% (т.
пл. 142—143° С) [264].
Даже при действии на двубромистое диарилолово бромистого мезитил-
магния, в случае которого пространственные затруднения наиболее
вероятны, образуются четырехзамещенные соединения [1,3,5-СбН2(СНз)з]2
SnAr2, где Аг —фенил (158° С), /г-толил (211° С), /г-ксилил (~ 200° С),
а-нафтил (187° С), с выходом 25—40% [130]. Дифенилди-ж-оксифенилолово
получено с выходом 35%, т. пл. 189—190° С (если температура повышается
на ГС в минуту) [183].Ди-/г-метоксифенил-6ш>1-нафтилолово (т. кип. 98°
С/0,4 мм) [254,256], ди-/г-метоксифенил-6ис-тиенилолово [254], ди-1-нафтил-
б^с-тиенилолово [254] получены аналогично.
Четырехзамещенные соединения с общим числом орто-заместителей
больше шести получить не удается [130] (см. стр. 219).
Оловоорганические соединения, содержащие оксигруппы, не
растворяются в воде, но растворяются при добавлении едкого натра, причем с
увеличением числа оксифенильных групп растворимость увеличивается
[183].
Гетероциклические оловоорганические галогениды образуют
четырехзамещенные соединения с достаточно хорошим выходом
+ 2CH3MgBr-^| I Г [ + 2MgClBr.
Sn
/ \ / \
CI C1 СН3 СНз
Двойной избыток бромистого метилмагния в эфире перемешивают 2
часа при комнатной температуре, гидролизуют 1%-ной соляной кислотой.
238 Оловоорганические соединения. Лит. стр. 261—268
После обычной обработки получают бесцветное масло с т. кип. 130—135° С/
0,2 мм; п2% 1,6130; выход 73% [265].
Соединения R2SnAr2, содержащие двойные или тройные связи, описаны
достаточно подробно. Выход для диэтилди(аллилфенил)олова равен 43%
(т. пл. 210° С) [250], дифенилди-(1-инденил)олова — 29,9% (т. пл. 108—
110° С) [97], дифенилди-(1-циклопентадиенил)олова — 70% (т. пл. 105—
106 С) [97] (см. стр. 231).
Диацетилениды олова R'2Sn(C = CR)2 получают взаимодействием оло-
воорганических дигалогенидов с алкинилмагнийпроизводными в диэтило-
вом эфире
QHsMgBr + RC = СН -* RC = CMgBr + С2Н6,
2 RC = GMgBr + RgSnCb -> R2Sn (С = CR)2 + MgX2.
Получение диэтилдигексин-1-илолова [266]. К бромистому этилмагнию (из 21,82 г;
0,2 моля бромистого этила) в 40 мл сухого эфира медленно прибавляют при перемешивании
раствора 16,2 г (0,2 моля) гексина-1 в 30 мл сухого эфира. После того как выделение
этана прекратится, смесь кипятят 2 часа, затем после охлаждения реакционной смеси
прибавляют небольшими частями при перемешивании 24,8 г (0,1 моля) двухлористого диэтилоло-
ва в 100 мл сухого эфира, затем смесь кипятят 3 часа.
Эфирный слой декантируют, соли магния промывают легким петролейным эфиром (4
раза по 50 мл), растворитель удаляют в вакууме и остаток перегоняют. Получают 21,3 г-
(63%) диэтилдигексин-1-илолова с т. кип. 101° С/0,15 мм; п2р 1,4888.
Аналогично получены (C3H7)2Sn(C=CR)2, где R — С3Н7 (т. кип. 96° С/
(0,15 мм; п2£ 1,4881), С4Н9 (т. кип. 99° С/0,1 мм; п™ 1,4884); соединения
(C4H9)2Sn(C=CR)2, где R —С3Н7 г(т. кип. 109 —110°С/0,15 мм; п™ 1,4899),
С4Н9 (т. кип. 114° С/0,15лш; /z2D° 1,4827), С6Н5, а также (C6H*5)2Sn (C==CR)2,
где R -С2Н5, С3Н7, С4Н9, С6Н5 (т. пл. 71-72° С).
Диметилдифенилэтинилолово [221], по-видимому, лучше получать с
помощью литийорганического соединения [267] (см. стр. 242).
Оловоорганические соединения, содержащие фенилэтинильную группу, в некоторых
случаях мало стабильны, в связи с чем для их очистки нельзя применять
дистилляцию [266].
В галогенидах R2SnX2 действием рассчитанного количества реактива
Гриньяра можно заменить только один атом галоида на органический
радикал с образованием R2R'SnX. Недостатком метода также, как и при
аналогичной реакции с SnX4, является образование трудноразделимой смеси
веществ различной степени алкилирования (см. стр. 241) или даже четы-
рехзамещенного производного в качестве основного продукта реакции.
Так, хлористый я-октилмагний в тетрагидрофуране и двухлористое диме-
тилолово в гептане, взятое в молярных отношениях, дают диметилди-я-ок-
тилолово с выходом 72%. Часть исходного двухлористого диметилолова
остается неизменной.
С хлористым 2-этилгексилмагнием реакция протекает аналогично —
из реакционной смеси выделено диметилди-(2-этилгексил)олово с выходом
73% и исходное двухлористое диметилолово [151].
Оловоорганические галогениды со смешанными радикалами обычно
получают деалкилированием галоидами полнозамещенных оловоорганиче-
ских соединений со смешанными радикалами (см. гл. VIII)
R2SnX2 + 2 R'MgX -» R2SnR2 + 2 MgX2,
R2SnR.2 + X2-* R2RrSnX + R'X
(X ■— галоид).
Прямое алкилирование оловоорганических солей реактивом Гриньяра
с целью получения смешанных оловоорганических галогенидов требует тща-
Синтез на основе производных четырехвалентного олова
23$
тельной отработки экспериментальных условий и сравнительно редко
приводит к вполне удовлетворительным результатам. Однако в отдельных
случаях оловоорганические галогениды со смешанными радикалами получают
этим методом; например:
СН2 = CHMgCl + (л-QHe.fe SnCl2 -* (л-QHeb (СН2 = СН) SnCl + MgCl2.
Хлористое ди-я-бутил(винил)олово образуется с выходом 56% [90].
При действии эквимолекулярного количества бромистого аллилмагния
на двуиодистое ди-я-пропилолово в основном идет замещение одного атома
иода с образованием йодистого ди-я-пропилаллилолова с выходом 49%,
т. кип. 95—97° С/4—5 мм. Ди-я-пропилдиаллилолово получено действием
двойного избытка бромистого аллилмагния [211]. Бромистый аллилфенил-
магний (4 моля) в реакциях с двуиодистыми ди-я-бутилоловом или диизо-
амилоловом дает йодистое ди-я-бутил-(аллилфенил)олово (т. кип. 202—
205° С/3—4 мм) и йодистое диизоамил(аллилфенил)олово (т. кип. 225—
228° С/3—4 мм) с выходом 55%. Это жидкости, обесцвечивающие раствор
брома в хлороформе [171].
При синтезе оловоорганических бромидов не следует вводить ион хлора
в реакционную смесь, так как возможна реакция обмена с образованием
примеси хлорида.
Получение бромистого диэтил-л-бромфенилолова [19]. 213 г (0,63 моля) твердого дву-
бромистого диэтилолова прибавляют при охлаждении и перемешивании к магнийорганиче-
скому соединению, приготовленному из 15 г (0,62 г-атома) магния и 150 г (0,63 моля) /г-ди-
бромбензола в 150 мл безводного эфира. Реакционную смесь кипятят 2 часа и разлагают
раствором 90 г хлористого аммония в 300 мл воды. Из сухого эфирного раствора повторной
дистилляцией выделяют фракцию с т. кип. 150—160° С/1 мм; выход 30 г (12%).
Потенциометрическое титрование показывает, что образуется смесь бромистого и
хлористого диэтилди-л-бромфенилолова, С10Н14ВгЬ7С10,3 Sn.
Бромистое диэтил-/г-хлорфенилолово получено в виде бесцветного масла
с выходом 9%, т. кип. 122° С/0,2 мм (реакционную смесь в толуоле
кипятили в течение часа, избыток магнийорганического соединения разлагали
бромистым аммонием) [19]. Окись диалкилолова в реакциях с реактивом
Гриньяра исследована недостаточно. Соединения R2R'SnX (после
обработки реакционной смеси соляной кислотой) получены с выходом 18,5—29,5%.
При избытке реактива Гриньяра образуются четырехзамещенные
продукты R2SnR2 [50а].
С соединениями R3SiCH2MgX и R2HSiCH2MgX продукты
исчерпывающего замещения образуются с хорошим выходом (см. стр. 224).
Получение диметил-##с-(диметилсилилметил)олова [(CH3)2HSiCH2]2Sn(CH3)2 [172а].
Магнийорганическое соединение приготовляют обычным образом в колбе, снабженной
мешалкой и холодильником. 50 г (0,465 моля) диметилхлорметилсилана в 150 мл безводного
диэтилового эфира прибавляют по каплям к 12,4 г (0,465 моля) стружек магния с такой
скоростью, чтобы поддерживать кипение. К раствору Гриньяра прибавляют по каплям 40,8 г
(0,185 моля) двухлористого диметилолова в 300 мл диэтилового эфира. Затем смесь
нагревают с обратным холодильником 1 час, разбавляют 100 мл бензола, промывают раствором
5%-ной соляной кислоты, водой и высушивают сульфатом натрия. Растворитель удаляют
при атмосферном давлении, а остаток перегоняют в вакууме. Получают 48,2 г диметил-бш>
(диметилсилилметил) олова с т. кип. 101° С/40 мм\ dfl,\08; «^1,4743.
Аналогично получены дибутил-б^с-(диметилсилилметил)олово (выход
66,6%; т. кип. 130° С/5 мм; df 1,043; ri$ 1,4810) и диметил-бас-(диметил-
силилпропил) олово (выход 75,5%; т. кип. 146° С/15 мм\ df 1,052; п2£
1,4830 [172а]. Соединения этого типа описаны достаточно подробно [187,
1881, в тетрагидрофур^не выход значительно выше [161].
240 Оловоорганические соединения. Лит. стр. 261—268
Получение диметил-##£-(триметилсилилметил)олова [(CH3)3SiCH2]2Sn (СН3)з [161].
Хлористый триметилсилилмагний получают из 40 г (0,327 моля) хлор мети л три мети л си л а на
и 7,55 г (0,31 г-атома) магния в 200 мл тетрагидрофурана (тетрагидрофуран перед
употреблением перегоняют над литийалюминийгидридом). Для инициирования реакции вводят
несколько капель бромистого этила. К реактиву Гриньяра прибавляют раствор 27,5 г (0,125
моля) двухлористого диметилолова в 70 мл тетрагидрофурана с такой скоростью, чтобы
поддерживать умеренное кипение смеси. Реакционную смесь нагревают с обратным
холодильником 20 час, дают охладиться до комнатной температуры и гидролизуют 60 мл насыщенного
водного раствора хлористого аммония. Растворитель отгоняют при атмосферном давлении,
остаток фракционируют в вакууме. Получают 37,8 г (94%) вещества ст. кип. 49—52° С/
0,35 мм [161], т. кип. 146,5—147,3° С/65 мм\ df 1,073; 1,0559; ngl,4702; 1,4644 [20, 161].
Аналогично получены ди-я-бутил-бш>(триметилсилилметил)олово (т.
кип. 98° С/0,45 мм; /г^ 1,4777), дифенил-6ш>(триметилсилилметил)олово
(т. кип. 138—140° С/1,5 мм; d^l,1404; tif, 1,5425); дифенил-6ш>(м-триме-
тилсилилфенил)олово (выход 23,5%; т. пл. 95—96° С) [20, 161].
Дивинил-бас-(триметилсилилметил)олово получено из двубромистого
бш:-(триметилсилиметил)олова и бромистого винилмагния [161]. Реактивы
Гриньяра типа R3SnR'MgX находят значительное применение для синтеза
соединений, содержащих наряду с оловом кремний [268], свинец [189],
бор [144] и другие элементы (см. стр. 224).
Димагнийорганические соединения алифатического ряда в реакциях с
дигалогенидами приводят к образованию соединений с атомом олова в цикле
СН2 —СН2
/ \
(С2Н5)2 SnBr2 + Cl4g (СНо)5 MgCl -> (C2H5)2Sn CH2 + 2MgClBr,
\ /
СНо — СН2
(QH9)2 SnCl2 + [ClMgCH2Si (СН3Ы2 О -> (QH9)2 Sn [CH2Si (CH,)2]20 + 2 MgCi2.
Получение диэтилциклопентаметиленолова [269]. В реактив Гриньяра, полученный
из 112 г 1,5-дихлорпентана в 400 мл абсолютного эфира, вносят порциями при сильном
встряхивании 130 г двубромистого диэтилолова (половина вычисленного количества). После
внесения всего двубромистого диэтилолова смесь кипятят 30 мин. на водяной бане, затем
эфир отгоняют. Оставшуюся массу нагревают 1 час. на кипящей водяной бане. Отогнанный
эфир возвращают обратно, смесь осторожно разлагают водой (без подкисления). Эфирный
слой промывают водой, эфир отгоняют над хлористым кальцием. Оставшееся масло
фракционируют в вакууме в токе С02. Получают 23 г вещества с т. кип. 95° С/14 мм [269].
Аналогично получают диметилциклопентаметиленолово [269].
Получение циклоокси-б"#с-(диметилсилилметил)дибутилолова [172а]. 125 г (0,54 г
моля) окси-б&с-хлорметилдиметилсилана в 400 мл диэтилового эфира прибавляют по каплям
к 26,2 г (1,07 г-атома) магния с такой скоростью, чтобы поддерживать кипение. К
полученному реактиву Гриньяра прибавляют по каплям 153,3 г (0,505 моля) двухлористого дибутил-
олова в 200 мл эфира. Затем реакционную смесь нагревают 1 час с обратным холодильником,
промывают водой и 1%-ным раствором едкого натра до нейтральной реакции; прибавляют
1 г порошка едкого кали, растворитель удаляют при атмосферном давлении; остаток
нагревают при температуре бани 200° С при 5 мм. Фракционной дистилляцией получают с выходом
37% циклический продукт с т. кип. 170° С/25 мм; df 1,113; /г^1Д800 [172а].
С двухлористым диметил- и диэтилоловом в той же реакции получены
циклические олигомеры с мол. весом 800—850 [1726].
Избыток оловоорганического дигалогенида способствует образованию
соединений типа QSnR2 — R' —SnR2Cl. В случае димагнийорганическо-
го соединения, полученного из бромистого метилена [83] или я-дибромбен-
зола [270], продукт такого же типа может образоваться наряду с более
высокомолекулярными веществами, содержащими метиленовую или фени-
леновую группы.
Получение /г-фзнилгн-б'яс-^хлордиметилолова) [270]. Магнийорганическое соединение,
приготовленное из 86,6 г (0,36 7моля) /г-дибромбензола и 18 г (0,745 г-атома) магния в 400 мл
тетрагидрофурана, прибавляют по каплям при хорошем перемешивании к раствору 177 г
Синтез на основе производных четырехвалентного олова
241
(0,806 моля) двухлористого диметилолова в 100 мл тетра гидрофура на. Смесь кипятят с
обратным холодильником 2 часа, выпаривают досуха в вакууме и остаток экстрагируют
горячим бензолом. Растворитель из экстракта выпаривают, остаток кристаллизуют из лигроина.
Получают кристаллический продукт ст. пл. около 130° С. Раствор вещества в смеси эфира
с тетрагидрофураном обрабатывают насыщенным водным раствором хлористого аммония
(для удаления не вступившего в реакцию двухлористого диметилолова, которое растворимо
в последнем), выпаривают досуха и остаток кристаллизуют из лигроина. Получают с выходом
30% белое твердое вещество ст. пл. 160—162° С.
Тригалогениды RSnCl3 в реакциях с магнийорганическими
соединениями находят ограниченное применение. Их используют для
получения соединений RSnRg, RR^SnX и RSnRg (где X — галоид). Четырех-
замещенные соединения RS11R3 получают обычно из моногалогенидов
R^SnX и сравнительно редко из RSnX3. Выбор метода определяется в
основном доступностью исходного соединения. Экспериментальные условия
и выходы близки к описанным для хлорного олова или оловоорганиче-
ских галогенидов иных классов.
я-Бутилтри(трифторвинил)олово [208], я-бутилтри(циклопропил)олово
[95], я-бутилтри(фенилэтинил)олово [226], метил- и бутилтри(пентафторфе-
нил)олово [106, 108] получены действием магнийорганических соединений
на соответствующие тригалогениды алкилолова с выходами 50—70%. я-Бу-
тилтри(гексин-1-ил)олово представляет собой бесцветную жидкость,
которая при перегонке, по-видимому, полимеризуется [266].
Для синтеза четырехзамещенных соединений наибольшее применение
находит треххлористое фенилолово. В реакциях с соответствующими
магнийорганическими соединениями получены C6H5SnR3, где R — метил [175,
271], циклопропил (т. пл. 93° С/0,23 мм) [95], пентафторфенил (выход
85%, т. пл. 95—96° С) [106а, 1081, л^-трифторметилфенил, п-трет-бутил-
фенил (выход 31%, т. пл. 295° С) [122], /г-ал л ил фен ил (выход 73,2%,
т. пл. 148° С) [250]. /г-Толилтри(пентафторфенил)олово получено с
выходом 80%, т. пл. 107° С [108]. Фенилтри-(1-циклопентадиенил)олово
(т. пл. 43—60° С) образуется с выходом 59%, но очищенный продукт ст. пл.
64—65° С выделен с выходом 10%. На воздухе очищенное фенилтри-(1-цик-
лопентидиенил)олово через 4 часа вновь становится ржаво-коричневым
[97] (сравни тетра-1-циклопентадиенилолово, стр. 215).
Соединения, содержащие в бензольном ядре иод, получены действием
магнийорганического соединения на трехгалоидное /г-иодфенилолово:
JC6H4SnBr3 + 3 RMgX -* JC6H4SnR3 + 3 MgBrX.
Триэтил-п-иодфенилолово (т. кип. 174—175° С/760 мм) и трифенил-п-
иодфенилолово (т. пл. 143° С) [168] синтезированы с выходами 16 и 32%.
Частичное алкилирование оловоорганических тригалогенидов описано
на нескольких примерах:
СН2 = CHMgCl + n-C4H9SnClo -* (/г-QHe) (СН3=СН) SnCl2 + MgCl2
(выход 54%),
2 СН2 = CHMgCl + /z-C4H9SnCl3 -» (д-С4Н9) (СН2 = СН)2 SnCl + 2 MgCl2
(выхоа 50%).
Получение двухлористого я-бутилвинилолова [90]. К 282,3 а (1 моль) треххлористого
я-бутилолова в 2,5 л гептана (в колбе на 5 л) прибавляют в течение 2 час. 1 моль хлористого
винилмагния в тетрагидрофуране, поддерживая температуру реакционной смеси между
25—40° С. Смесь перемешивают 3 часа и фильтруют через воронку Бюхнера. Растворитель
после обычной обработки удаляют при атмосферном давлении, остаток тщательно
фракционируют в вакууме. Получают 146 г (53,5%) двухлористого я-бутилвинилолова с т. кип. 99—
101° С/3 мм.
16 Заказ № 207
242
Оловоорганические соединения. Лит. стр. 261—268
Для получения хлористого дивинил-я-бутилолова 2 моля хлористого
винилмагния вводят в раствор треххлористого я-бутилолова в гептане при
температуре кипения. Выход 50%, т. кип. 82—84° С/3 мм [90].
Оловоорганические соединения, содержащие винильные группы, в
отличие от аналогичных кремнийорганических соединений не присоединяют
галоида по двойной связи, а деалкилируются с образованием оловооргани-
ческих галогенидов (см. стр. 340).
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Остин [272] предложил использовать для синтеза оловоорганических
соединений реакцию органических соединений лития с хлорным оловом
4 RLi + SnCU -* R4Sn + 4 LiCl.
По сравнению с реактивом Гриньяра литийорганические соединения имеют
ограниченное применение. Их используют, например, в тех случаях, где
приготовление реактива Гриньяра связано с трудностями, или там, где,
необходим реагент с повышенной реакционной способностью. С RLi
удается получить малодоступные оловоорганические соединения. Это
относится прежде всего к R4Sn ароматического ряда с заместителями в бензольном
ядре [115, 237, 273—276]. Соединения алифатического ряда, такие как
тетра-т/?ет-бутилолово, не удается получить как с помощью магний-, так
и литийорганических соединений [63, 66]. Вообще для получения
алифатических оловоорганических производных литийорганическими
соединениями пользуются редко, например для синтеза стереоизомерных тетрапро-
пенильных и тетраизопропенильных соединений олова [35].
Реакцию проводят в растворителях, обычных для органических
соединений лития (эфир, углеводороды).
Получение тетра-цис-пропенилолова [217]. К 0,34 молям ^«г-пропениллития,
полученного из цис-бромпропена и лития, прибавляют в течение часа 33,0 г (0,075 моля) бромного
олова в 50 мл эфира и оставляют до утра. После гидролиза насыщенным раствором
бикарбоната натрия отделенный эфирный слой обрабатывают газообразным аммиаком и
фильтруют. Дистилляцией фильтрата получают 16,1 г (76%) вещества ст. кип. 57—59° С/0,2 мм;
п% 1,5178.
Тетра-т/га«опропенилолово (т. кип. 53—56° С/0,05 мм) получено с
выходом 83%. В этом случае реакционную смесь (после прибавления
бромного олова) кипятят с обратным холодильником в течение 12 час. [217].
Тетра(метоксиметил)олово образуется с небольшим выходом. Выход
выше, если в реакции с метоксиметиллитием применяют не хлорное олово,
а галоген иды метоксиметил олова [277а].
При взаимодействии хлорного олова и литийорганического соединения
в молярных отношениях 1 : 2 или 1 : 3 образуется смесь
оловоорганических галогенидов различной степени замещения, которые, как, например,
хлористое триизопропилолово и двухлористое диизопропилолово, разделяют
дистилляцией [63]. Хлористое три-трет-бутилолово получают
прибавлением трет-бутиллития к суспензии хлорного олова в эфире при 0° С
(обратный порядок прибавления ведет к образованию окрашенного
продукта со связями олово — олово). При соотношении реагентов 3,3 : 1
образуется продукт, содержащий 90% монохлорида, однако все же хлористое
три-т/?ет-бутилолово в чистом виде получить не удается (всегда имеется
примесь двухлористого ди-т/?ет-бутилолова) [63].
Тетрациклопропилолово получено с выходом 88% прибавлением
хлорного олова к избытку трициклопропиллития [277].Длятетрациклогексил-
Синтез на основе производных четырехвалентного олова
243
ацетиленида олова (C6HnC=C)4Sn (т. пл. 130° С) выход равен 44,3%
[278].
Получение тетрациклопентадиенилолова [279]. Взвесь 18,0 г (250 ммоля) пентадиенил -
лития в 50 мл бензола смешивают по каплям при наружном охлаждении с 16,0 г (61,4
ммоля) хлорного олова в 40 мл бензола. После окончания прибавления смесь еще 3 часа
перемешивают, отфильтровывают, остаток промывают 2 раза бензолом. Желтый бензольный
раствор концентрируют до появления обильной кристаллизации. Кристаллы отсасывают,
промывают небольшим количеством бензола и высушивают в высоком вакууме. Маточник не
используют. Выход 19,9 г (52,5%). Светло-желтый кристаллический осадок вполне стабилен,
если его сохранять под азотом. Т. пл. 81—82° С (вещество может быть нагрето до 200° С без
заметного разложения).
Оловоорганические соединения ароматического ряда с помощью литий-
органических соединений образуются легко и с хорошими выходами. Так,
например, тетра(пентафторфенил)олово получено действием пентафторфе-
ниллития на хлорное олово в эфире при —65° С с выходом 91,4% (т. пл.
220—222° С, вещество термически стабильно до 399° С) [109].
Получение тетра-я-толилолова (p-CH3C6H4)4Sn [272]. К избытку /г-толиллития при
энергичном перемешивании добавляют 1,3 г хлорного олова, растворенного в 25 мл бензола.
Реакция протекает быстро на холоду; после ее завершения смесь разлагают водой, эфирный
слой отделяют. Остаток после испарения эфира перекристаллизовывают из бензола. Выход
91%, т. пл. 234—235° С [272].
Таким же путем получено тетра-п-диметиламинофенилолово (выход
58,5%; т. пл. 198—199° С) [237, 272], тетра-ж-толилолово (выход 58%; т. пл.
128,4—129,6°С), тетра-о-толилолово (выход38%, т. пл. 217,5—219,5°С) [ 112],
тетра-(ж-трифторметилфенил)олово [104, 122]. Получены также орто- (т. пл.
300—301° С), мета-(т. пл. 145,5—145,8° С) и пара-(т. пл. 268,5° С) тетра-
бифенилолово с выходом до-77,5%. Гладкое образование орто-изомера
является несколько неожиданным [131].
Получение тетра-2-бифенилолова [131]. В трехгорлую колбу на 500 мл помещают 25 мл
сухого эфира 2 г (0,29 г-атома) кусочков лития, прибавляют несколько капель 2-бромби-
фенила и начинают перемешивание. Тотчас начинается энергичная реакция; в течение 1 часа
прибавляют по каплям 23,3 г (0,1 моля) 2-бромбифенила в смеси 50 мл эфира и 50 мл
бензола. Затем смесь нагревают в течение 1—1,5 час. на водяной бане. Бромистый литий
отделяют. Получают прозрачный раствор 2-бифениллития с выходом 89—94%. 175 мл эфирно-
бензольного раствора, содержащего 13,7 г (86 ммоля) 2-бифениллития, прибавляют по
каплям в течение 30 мин. при энергичном перемешивании к раствору 4 г (15 ммоля) хлорного
олова в 25 мл бензола. После отгонки 75—100 мл растворителя желтовато-серую суспензию
кипятят в течение 2 час, гидролизуют разбавленной бромистоводородной кислотой,
отсасывают, высушивают и перекристаллизовывают из кипящего о-дихлорбензола. Выход 71,2%;
т. пл. 299—300° С.
Взаимодействие 2-бифениллития с бромным оловом в молярном
соотношении 3,56 : 1 приводит в основном к бромистому три-(2-бифенил)олову;
примесь тетра-(2-бифенил)олова составляет 14,4% [131].
Тетразамещенные оловоорганические соединения ароматического ряда>
как правило, плохо растворимы; их очистка связана с большими
трудностями. Термически вещества устойчивы, но часто при нагревании
разлагаются, не плавясь.
Тетра-9-фенантр ил олово получено с выходом 55% в виде неочищенного
продукта. Кристаллизуют его из кипящего о-дихлорбензола.
Кристаллизация (лучше из о-дихлорбензола) тетра-1-нафтилолова связана с большими
потерями. Выход сырого продукта 20% [127]. По синтезу тетрамезитилоло-
ва получены противоречивые данные [126, 127]. Из реакционной среды
выделены хлористое тримезитилолово и окись димезитилолова [126].
Тетра-9-флуорилолово и тетра-1-инденилолово (выход 41 и 48%
соответственно) устойчивы к гидролизу. Этим они отличаются от несимметричных
16*
244 Оловоорганические соединения. Лит. стр. 261—268
оловоорганических соединений с теми же радикалами. Вообще
симметричные тетразамещенные оловоорганические соединения как в химическом* так
и термическом отношении устойчивее, чем соответствующие
несимметричные [280].
Тетра-(офеноксифенил)олово образуется с выходом 27% [281].
С помощью литийорганических соединений получены и
оловоорганические соединения, содержащие гидроксил в бензольном ядре:
т-ВгСбН4ОН + 2я-С4Н9и -» m-LiOC6H4Li + я-С4Н9Вг + я-С4Н10,
4 (/n-LiOC6H4Li) + SnCl4 -» (m-LiOC6H4)4 Sn + 4 LiCl,
NH4C1
(m-LiOC6H4)4 Sn —» (m-HOC6H4)4Sn.
н2о
Получение тетра-л*-оксифенилолова [183]. К охлажденному раствору 52 г (0,30
моля) .w-бромфенола в 50 мл эфира прибавляют при хорошем перемешивании 0,6 моля «-бу-
тиллития в 580 мл эфира. Реакционную смесь перемешивают при комнатной температуре
в течение 12 час, затем добавляют 15,5 г (0,058 моля) хлорного олова в 100 мл бензола,
кипятят с обратным холодильником 4 часа и выливают в охлажденный льдом раствор
хлористого аммония. Органический слой отделяют, водный слой экстрагируют 200 мл эфира,
полученные растворы соединяют и высушивают в течение 24 час. сульфатом кальция. Эфир
удаляют, бромистый бутил отгоняют в вакууме. Оставшееся темное масло кристаллизуют из
смеси хлороформа с петролейным эфиром (т. кип. 57—70° С). Получают 14,0 г (48,5%) тет-
ра-ти-оксифенилолова с т. пл. 170—182° С. После перекристаллизации из смеси тех же
растворителей (в отношении 1:1) получают 11,0 г вещества с т. пл. 180—184° С (с
разложением). Соединение не растворяется в воде, но растворяется в 5%-ной гидроокиси натрия.
Соединения, содержащие в молекуле наряду с оловом другие элементы,
образуются с хорошим выходом. Так, для тетра-(я-триметилсилилфенил)оло-
ва выход'равен 58% [273]. Оловоорганические; соединения, в которых атом
олова непосредственно связан с ядром барена, получены с помощью
литиевых производных баренов. Взаимодействием хлорного олова с фенилбаре-
ниллитием получена смесь ди- и тризамещенных соединений [282].
QHsC-^r-CH + >z-C4H9Li
BioHjo
CLi
Ф/
BwHfo
Сбн5С__а. + snci4 —*- (с6н5с—-c)2snci2 +
Взаимодействие хлорного олова с фенилбарениллитием [282]. Сухой эфир (30 мл) и затем
2,6 г (0,01 моля) хлорного олова прибавляют при 5—10° С при перемешивании к бензольному
раствору фенилбарениллития, приготовленному из 8,8 г (0,04 моля) фенилбарена.
Образуется осадок. Смесь нагревают с обратным холодильником 3 часа, охлаждают и
обрабатывают 50 мл разбавленной (1:4) хлористоводородной кислоты. Осадок, который не
растворяется в воде и в эфирно-бензольной смеси, отделяют и высушивают. Получают 2 г (25%)
хлористого три (фенилбаренил) олова с т. пл. 287—289° С. Эфирно-бензольный слой отделяют,
высушивают сульфатом магния и удаляют растворитель. После перекристаллизации из 1,2-
дихлорэтана получают 2,3 г (37%) двухлористого бг/с-(фенилбаренил)олова с т. пл. 216,5—
217,5° С.
Тетра(фенилбаренил)олово получить не удается, по-видимому, из-за
стерических препятствий [282].
Дилитийорганические соединения в реакциях с хлорным и бромным
оловом исследованы достаточно подробно. С дилитиевыми соединениями
Синтез на основе производных четырехвалентного олова 245
ароматического ряда образуются спиросоединения
(XX)
Выходы низки (12—23%). Реагенты следует смешивать при температуре —
—20^ 10° С и перемешивать некоторое время при комнатной температуре
[264]. При взаимодействии дилитий-бш>бензила с хлорным оловом
образуется четырехзамещенное соединение
которое применяют в реакциях без очистки [265]. Подобным же образом
спиросоединения образуются с 1,4-дил итий-1,2,3,4-тетрафенилбутадиеном
[283—285] и Ы-этил-2,2'-дилитийдифениламином [286]
с
к
С
к
с2н5
1
1
N
YY^i
K/V
Sn
СО
N
1
с2н5
Что касается реакций литийорганических соединений с оловоорганичес-
кими галогенидами, то по сравнению с реактивом Гриньяра они находят
меньшее применение. Их используют главным образом для введения ви-
нильных и ароматических групп [39, 193, 195, 237, 252, 254, 275, 276, 287—
289]. Следует отметить, что литийорганические соединения могут привести
к расщеплению оловоорганических соединений с разрывом связи олово —
углерод [183, 252, 289, 290, 291] (см. стр. 453). Однако эти побочные
реакции не имеют места, если литийорганическое соединение перед
взаимодействием с оловоорганическим галогенидом превращают в реактив Гриньяра
прибавлением галоидного магния. Этим приемом пользуются, кроме того,
в тех случаях, когда превращение в магнийорганические соединения
способствует получению более высоких выходов [183, 184, 252, 290, 292].
Литийорганические соединения реагируют с оловоорганическими моно-
галогенидами уже на холоду. Реакция может найти применение даже для
аналитических целей. Например, титр фениллития можно определить с
помощью хлористого трифенилолова. В этом случае образуется трудно
растворимое тетрафенилолово [293]. Однако при комнатной температуре реакция
во многих случаях не идет до конца. Так, при взаимодействии хлористого
трибутилолова с виниллитием трибутилвинилолово образуется с выходом
85%. Продукт содержит 8% хлористого трибутилолова, которое отделяют
СбН5 || ,| СбН5
CH.-l^JL-cHe
Sn
CeH5 •—rj |j— С6Н5
с6н5 " " QH5
246 Оловоорганические соединения. Лит. стр. 261—268
обычным образом, например обработкой раствором едкого натра или
газообразным аммиаком [193, 294]. Реакцию следует заканчивать нагреванием
реакционной смеси с обратным холодильником [295]. Взаимодействием три-
фторвиниллития с бромистым триметилоловом получено триметил(перфтор-
винил)олово (т. кип, 112—115° С) с выходом 64% [296]. Для трибутилаллил-
олова (т. кип. 94—95° С/0,55 мм) выход равен 69% [214].
Получение триметилпропенилолова [217]. В трехгорлую колбу, снабженную мешалкой,
обратным холодильником и капельной воронкой, помещают 200 мл сухого эфира, 3,36 г
(0,48 г-атома) нарезанного кусочками лития и прибавляют по каплям 30 г (0,249 моля)
1-бромпропена (смесь изомеров) в 45 мл эфира с такой скоростью, чтобы поддерживать
энергичное кипение. К полученному раствору литийорганического соединения прибавляют
в течение 30 мин. 49 г (0,2 моля) бромистого триметилолова в 50 мл эфира. Реакционную смесь
нагревают с обратным холодильником в течение 2 час, затем гидролизуют насыщенным
раствором хлористого аммония. Фракционной дистилляцией сухого органического слоя
получают 35,3 г (88%) триметилпропенилолова с т. кип. 124—126° С. Отношение цис-транс-изо-
меров 67:33 [217].
Аналогично, исходя из цис- 1-бромпропена, получено ^ш>пропенилтри-
метилолово с выходом 72%. Согласно хроматографическим данным, примесь
транс-изомера составляет около 1 % [217]. Примерно с тем же выходом
получены траяс-триметилпропенилолово (т. кип. 125—128° С; п2£ 1,4638) и
изопропенилтриметилолово (т. кип. 118—120° С; п2£ 1,4608) [217].
Перераспределения радикалов, часто неизбежного при избытке литий-
органического соединения, можно избежать, если вести реакцию осторожно
при 0° С, прибавляя литийорганическое соединение при хорошем
перемешивании к моногалогениду. В этих условиях при действии циклопропилли-
тия на хлористое триметилолово образуется циклопропилтриметилолово с
выходом 66% без примеси тетраметил олова и диметилдициклопроп ил олова
[277].
В синтезах с оловоорганическими галогенидами наиболее подробно
исследованы литийорганические соединения ароматического ряда. Для
завершения реакции необходимо нагревание. Выходы часто умеренные.
Соединения (CH3)3SnAr, где Аг — я-толил (т. кип. 80,5° С/3 мм), м-толил (т. кип.
65,7—67,° С/0,6 мм), 3,4,5-триметилфенил (т. кип. 80—81° С/высокий
вакуум; т. пл. 25° С), тримезитил (т. кип. 65—66° С/высокий вакуум; т. пл.
35—36°С),/г-анизил(т. кип. 125— 127°С/15лш),|3-нафтил(т. пл. 29—29,5° С);
(СвНц^пСвНД, где X — /-С3Н7 (т. пл. 74° С), p-(C6Hu)3Sn (т. пл. 336—
340° С), р -(CHa)2N (т. пл. 148° С) получены с выходом 35—80% [160, 297].
Получение триэтил-я-толилолова [297]. В эфирный раствор /г-толиллития (получен из
2,12 г лития и 22,0 г я-бромтолуола) при охлаждении и перемешивании добавляют 21,2 г
хлористого триэтилолова. Затем смесь кипятят 2 часа. Фракционированием выделяют
20,1 г (77%) триэтил-я-тол ил олова с т. кип. 91—93° С/1 мм, п2£ 1,5339.
Получение триметил-(о-фторфенил)олова [298]. К 92 мл 1,1 N бутиллития в пентане
при —78° С (под азотом) прибавляют в течение примерно 8 мин. 17,5 г (0,1 моля) о-бром-
фторбензола в 170 мл эфира. Температуру поддерживают ниже —50° С. Затем при —75° С
в течение 5 мин. вводят раствор 20 г (0,1 моля) хлористого триметилолова в 20 мл эфира так,
чтобы температура не превышала —45° С. Реакционную смесь перемешивают при —78° С
в течение 4 час, затем дают нагреться в течение ночи до комнатной температуры.
Гидролизуют насыщенным раствором хлористого аммония. Эфирный слой высушивают,
обрабатывают сухим газообразным аммиаком и фракционируют. Получают 15,6 г (60%) триметил-
(о-фторфенил)олова с т. кип. 65° С/4 мм\ п2£ 1,5180 [298].
Аналогично получены триметилмезитилолово (выход 68%, т. пл. 35—
36° С) [175], триметил-(4-бифенил)олово (выход 50%, т. кип. 118—119° С/
/0,25 мм) [298], а также другие соединения этого типа, например (C6H5)3SnR,
где R — 2,5-диметилфенил (выход 49,7%; т. пл. 97—99°С), 2,6-диметилфе-
нил (выход 44,5%; т. пл.118—119°С),2,4-диметилфенил (выход 71,2%; т. пл.
Синтез на основе производных четырехвалентного олова 247
113—114° С), мезитил (выход 74,2%; т. пл. 157—158° С) [231]. Три-(2-бифе-
нил)фенилолово (т. пл. 171,5—172° С) и три-(2-бифенил)-я-толилолово
(т. пл. 174,5° С) получены с выходом 74,5% [264].
Трифенил-п-аминофенилолово с помощью литийорганических
соединений, подобно оксисоединениям, получить не удается [183, 290], однако
реакции с диметиламинофениллитием находят значительное применение.
Получены триметил(диметиламинофенил)олово (т. пл. 40—41° С) [157],
трициклогексил-(п-диметиламинофенил)олово (т. пл. 148° С) [154], трифе-
нил-(.м-диметиламинофенил)олово (т. пл. 90—91° С) [299], трифенил-(2-ди-
метиламинофлуоренил)олово [194], трифенил-(/г-диметиламинофенил)оло-
во, трифенил-(о-диметиламинофенил)олово, дифенилди-(п-диметиламино-
фенил)олово. Это твердые вещества, растворимые в воде [39, 194,237, 290].
Оловоорганические соединения, содержащие третичную аминогруппу,
могут быть получены также действием диалкиламиногалогенидов на трифе-
нилстанниллитий (см. стр. 500).
Взаимодействие ArLi с оловоорганическими галогенидами в присутствии
фторбензола проходит, по-видимому, через стадию образования дигидро-
бензола. Действием фениллития на хлористое три-я-бутилолово получено
три-я-бутил-(о-хлорфенил)олово [300].
Для синтеза оловоорганических соединений типа R3SnC6H4X (где X —
активный заместитель в бензольном ядре) применяют обычно литийоргани-
ческие соединения, уже содержащие этот заместитель. Второй возможный
путь (исходя из R3SnC6H4Br) не приводит к цели. Например, при
взаимодействии трифенил-п-бромфенилолова с металлическим литием трифенил-п-ли-
тийфенил олова не образуется. С бутил литием происходит обмен металл
металл F959].
Получение трифенил-(о-оксифенил)олова [290]. В охлажденный раствор 0,246 моля
я-бутиллития в 490 мл эфира быстро вводят 21,3 г (0,123 моля) о-бромфенола в 25 мл эфира.
Затем прибавляют 47,7 г (0,123 моля) хлористого трифенилолова, перемешивают 2 часа,
реакционную смесь гидролизуют холодным раствором хлористого аммония. Эфирный слой
отделяют и высушивают сульфатом натрия. Эфир отгоняют, остаток для удаления непрореа-
гировавших веществ кипятят в течение 10 мин. с 100 мл этанола. Смесь охлаждают, осадок
отфильтровывают и кристаллизуют из хлороформа. Выход 31 г (57%). При нагревании
вещество разлагается (в зависимости от скорости нагревания) в пределах 176—182° С.
Аналогично получены триэтил-(о-оксифенил)олово, дифенилди-(о-окси-
фенил)олово, трифенил-(о-оксиметилфенил)олово, трифенил-(/г-оксиметил-
фенил)олово [290]. Если реакция осложняется перераспределением
радикалов, то литийорганическое соединение переводят в реактив Гриньяра с по,
мощью галоидного магния [183, 184, 290]. Безводный бромистый магний-
например, получают действием брома на большой избыток магниевых стру,
жек в эфире [290]. Таким опутем получены трифенил-(ж-оксифенил)олово-
дифенилди-(ж-оксифенил)олово, фенилтри-(ж-оксифенил)олово [ 183].
Стиролы с оловоорганическими заместителями в бензольном ядре
получают обычно с помощью магнийорганических соединений в тетрагидрофура-
не (см. стр. 231). Триэтил-(п-а, (З-дифтор-Р-хлорстирил)олово и трибутил-
(я-а, р-дифтор-Р-хлорстирил)олово получены в эфире действием я-литий-а-
(З-дифтор-Р-хлорстирола на хлористые триэтил- и трибутилолово при
температуре от — 60 до — 70° С. Выход не превышает 40% [301, 302].
Ацетилениды олова образуются при взаимодействии R3SnX с ацетилени-
дами лития в эфирно-бензольном растворе. Описаны, например, триметил
(фенилацетиленид)олово (т. пл. 67° С, т. кип. 60° С/0,2 мм) [267], три-я-бу-
тил(фенилацетиленид)олово (т. пл. 174° С) [288]. Выход около 58%. С трех-
хлористым бутилоловом, треххлористымфенилоловомили двухлористымди-
аллилоловом ифенилацетиленидомлития реакция протекает аналогично[303].
248 Оловоорганические соединения. Лит. стр. 261—2 68
Можно привести примеры получения оловоорганических соединений из
органических производных лития, полученных путем металлирования.
Получение (флуоренил-9)-трифенилолова [276]. В трехгорлую колбу, снабженную
мешалкой, обратным холодильником, капельной воронкой и газоподводной трубкой,
помещают раствор 4,15 г (0,025 моля) флуорена в 100 мл эфира и по каплям прибавляют
25 мл 1 М эфирного раствора фениллития. Происходит легкое разогревание с
образованием 9-флуорениллития, раствор окрашивается в яркий оранжево-красный цвет. К
раствору при перемешивании прибавляют по каплям в течение 30 мин. 9,6 г (0,025 моля)
хлористого трифенилолова в 200 мл эфира. Появляется мелкокристаллический осадок хлористого
лития. После непродолжительного стояния при энергичном перемешивании осторожно
прибавляют 10 мл воды. Эфирный слой отделяют, высушивают сульфатом натрия и сильно
упаривают. Полученную таким образом кристаллическую кашеобразную массу
кристаллизуют из небольшого количества петролейного эфира (т. кип. 60—100° С) в присутствии
активированного угля. Получают 8,7 г (67,7%) вещества с т. пл. 129—130° С.
Трифенил-(1-инденил)олово получено из индена, фениллития и
хлористого трифенилолова (т. пл. 128° С, выход 60%). С двухлористым дифеншюло-
вом и треххлористым фенилоловом также образуются четырехзамещен-
ные продукты (см. стр. 249) [97, 276].
Реакция не идет при наличии пространственных затруднений. Обработка
бромистого тримезитилолова мезитиллитием, мезитилнатрием или мезитил-
калием не приводит к образованию тетрамезитилолова [126].
Действием 2-фурил-, 3-пиридил- или 2-пиридиллития на хлористое три-
фенилолово получены соответственно 2-фурил-,3-пиридил- и 2-пиридилтри-
фенилолово с выходами 44, 58 и 12% [252]. В этих синтезах иногда получают
более удовлетворительные результаты, если литийорганическое соединение
переводят в магнийорганическое [252]. а-, у-Пиколины легко металлируются
в метильную группу, например фенил- или бутиллитием. При
взаимодействии полученного таким образом а-или у-пиколиллития с галогенидами триал-
килолова получены триэтил-(2-пиридилметил)олово (т. кип. 120—121° С/3—
4 мм\ п2£ 1,531), триэтил-(2-я-бутилпиридил-4-метил)олово (т. кип. 144—
145° С/3—4 мм\ /г^о 1,5364). Это бесцветные жидкости, перегоняющиеся в
вакууме, чувствительные к гидролизу. Особенно это относится к
производным а-пиколина. Триметил-(2-пиридилметил)олово полностью разлагается
уже влажным эфиром. Производные у-пиколина более устойчивы [39].
Литийорганические соединения, содержащие другой элемент в молекуле,
например германий, реагируют с оловоорганическими моногалогенидами
по схеме
(СН3)з SnBr + LiC6H4CH2Ge (CH3)3 -* (СН3)3 SnC6H4CH2Ge (CH3)3 + LiBr.
Триметил-(п-триметилгерманилбензил)олово получено с выходом 15% ,
т. кип. 114—115° С, п2£ 1,5278. Аналогично получены соединения,
содержащие наряду с оловом кремний или другие элементы [190]. Например, три-
я-пропилбаренилолово (т. кип. 148—150° С/1 мм), трифенил(фенилбаре-
нил)олово (т. кип. 186—187° С) образуются с выходом 35 и 84% при
действии соответствующего моногалогенида олова на фенилбарениллитий [282,
304].
При взаимодействии моногалогенидов R3SnX с /г-дилитий-, так же как
и с димагнийорганическими соединениями в реакцию вступают оба
атома лития с образованием веществ с двумя атомами олова в
молекуле (C6H5)3Sn — R — Sn (СбН5)з. Это бесцветные вещества с высокой
температурой плавления. R = р-С6Н4, т. пл. 289—292° С, выход 67%; р-С6Н4 —
С6Н4, т. пл. 235—236° С; — (СН2)4—, т. пл. 149—150,5° С [195].
С 2,2'-дилитийдифениленом реакция протекает более сложно [305]. Ба-
ренил-бас-(три-я-пропилолово) получено с выходом 30% [282].
Синтез на основе производных четырехвалентного олова
249
Действием литийорганических соединений на дигалогениды R2SnX2
получают обычно полнозамещенные соединения R2SnR2. Метод находит
применение в основном в ароматическом ряду. Общие правила работы и методики
те же, что и для моногалогенидов R3SnX. Продукты частичного замещения
R2SnR'X получают сравнительно редко; как правило, при действии
рассчитанного количества литийорганического соединения образуется
трудноразделимая смесь веществ различной степени замещения. Лишь в отдельных
случаях продукты реакции определяются соотношением реагентов.
Например, действием пентафторфениллития на двухлористое диэтилолово
получено бш>(пентафторфенил)диэтилолово. Хлористое диметил(пентафторфе-
нил)олово выделено при прибавлении пентафторфениллития к избытку дву-
хлористого диметилолова в эфире [306]. Соединения типа R2R'Sn (ОХ)иолу-
чены действием R'Li на R2SnCl (ОХ) (где R — метил или фенил, ОХ —
анион у-оксихинолина) [307].
С помощью литийорганических соединений получают обычно
полнозамещенные соединения; например, соединения формулы (CH3)2SnR9, где R —
/г-толил (т. кип. 110—112° С), мезитил (т. кип. 170—172° С; т. пл. 101° С),
3, 4, 5-триметилфенил (т. пл. 56° С) образуются с хорошим выходом [177].
Получение дифенил-^й^-(2-бифенил)олова [264]. Раствор 12,0 г (35 ммоля) двухло-
ристого дифенилолова в 100 мл эфира смешивают в течение часа с 130 мл (75 ммоля) раствора
бифенил-2-лития. Образовавшуюся бледно-желтую суспензию кипятят еще 1 час и гидроли-
зуют водой. После обработки смеси получают 13,9 г (68,9%) вещества с т. пл. 147—148° С,
которое из бутилового спирта кристаллизуется в виде призм ст. пл. 149,5° С.
Дифенилди-(9-флуоренил)олово (т.пл. 179° С) и дифенилди-(1-инденил)
олово (т. пл. (116—117° С) [276], дифенилди-(а-тиенил)олово (т. пл. 202—
210° С) [2541 образуются с выходами 60, 40 и 30% соответственно.
Оловоорганические ацетилениды исследованы достаточно подробно.
Получены соединения типов (C2H5)3Sn — С = С — Sn (C2H5)3 (т. кип. 123° С/
/0,05 мм) [308], С6Н5С= CSnR3 (R = С4Н9, т. кип. 174° С/4 мм) [228, 288] и
(С6Н5С = C)2SnR2, где R — метил (т. пл. 89°С, т. кип. 152° С/0,2 мм) [221,
267], я-бутил (т. пл. 14° С, n2Dl 1,588), фенил (т. пл. 82° С) [267], аллил
[303] с выходами 70—90%. Реакцию обычно проводят в жидком аммиаке и
эфире (см. стр. 253).
Число описанных соединений, содержащих атом олова в кольце, пока
остается незначительным. Они могут быть получены из соответствующих ди-
галогенидов [265]:
OTX)+2C-HiL^CX~X)+2ua
Sn Sn
/ \ / \
С1 С1 СвН5 СбН5
(выход 82,5%; полученный продукт — диморфен; т. пл. 136—137° С из
лигроина; 146—147° С — из спирта или после нагревания первой модификации
около точки плавления в течение нескольких минут) [265]. Те же продукты
могут быть получены действием соответствующих димагний- или дилитий-
органических соединений на дигалогениды R2SnX2 [265, 269, 283, 285] в
условиях, описанных для хлорного олова (см. стр. 209). Действием 1,5-дили-
тийпентана на двухлористое дифенилолово получено (C6H5)2Sn(CH2)5 (т. кип.
138—140°С/0,1 мм) [309]. Реакция находит большее применение в
ароматическом ряду.
R R
250
Оловоорганические ссединения. Лит, стр. 261—268
9,9-Дизамещенные дибензостаннолы образуются с хорошим выходом уже
при комнатной температуре, если раствор 2,2'-дилитийбифенила (его
удобнее получать из 2,2'-дибромбифенила и лития) прибавлять по каплям к
эфирным или бензольным растворам оловоорганических дигалогенидов R2SnX2.
В длительном кипячении нет необходимости* Длительное кипячение ведет
к перераспределению радикалов с образованием смеси продуктов, в которой
доказано присутствие R4Sn (выход 56—65%). Описаны, например, 9,9-ди-
замещенные дибензостаннолы, где R — этил (т. пл. 73° С), бутил (т. пл.
56° С), циклогексил (т. пл. 104° С), фенил (т. пл. 141,5° С), n-толил (т. пл.
108° С), бифенил-2 (т. пл. 196—196,5° С) [264, 305]. Заместители в
положении 9 могут быть и различными [264].
При взаимодействии дигалогенидов R2SnX2 с дилитийорганическими
соединениями могут образоваться не только циклические продукты с атомом
олова в цикле, но и высокомолекулярные соединения с атомом олова в цепи.
Поли-п-фенилендифенилолово получено взаимодействием двухлористого ди-
фенилолова с /г-дилитийбензолом [310]. С о, о'-дилитийдибензилом в
основном образуется также полимерный материал [265, 311]. При
взаимодействии двухлористого ди-я-бутилолова с баренилендилитием образуется димер-
ное ди-я-бутилбарениленолово с выходом 66% (т. пл. 186—186,5° С) [282]:
BiqHu,
Li~С-^т-CLi + (Az-C4H9)2SnCl2 *- (C4H9)2Sn Sn(C4H9)2 + 2L1C1.
В и С С
BioHlo
Фенилированные циклобутадиены с атомом олова в цикле получены
взаимодействием 1,4-дилитий-1,2,3,4-тетрафенилбутадиена с оловоорганически-
ми дигалогенидами [283—285]:
СбН5—С—С—СбНб
Li / \
СбНбСгггССбНб ""—^ СбНб—С С—СбНз —>
Эфир ч^ у
Li Li
СбН5—С—С—СбН5
RR'SnCl2 / \
> С6Н5-С C-CeH5 + 2LiCl.
XSn/
/ \
R R'
Соединения указанного типа, гдеИ = R' — метил, винил (т. пл. 158—159°С)
получены с выходом 69%, фенил (т. пл. 173—174° С) — 20% [283, 284].
Получение 1,1 -диметил-2,3,4,5-тетрафенилстаннола. Раствор 54 г (0,3 моля) дифенил-
ацетилена в 150 мл безводного эфира энергично перемешивают в атмосфере азота с 6 г мелко
нарезанных кусочков лития. Быстро образуется красный дилитийбутадиен, реакция
протекает со слабым разогреванием. Через 2 часа полученную смесь прибавляют при 0° С к
раствору 33 г (0,15 моля) двухлористого диметилолова в 250 мл безводного эфира.
Полученный желтый раствор фильтруют для удаления хлористого лития, фильтрат упаривают,
прибавляют 250 мл хлористого метилена и удаляют выпавший хлористый литий повторным
фильтрованием. Хлористый метилен заменяют безводным спиртом и оставляют в холодильнике.
Получают 57,6 г (77%) сырого продукта с т. пл. 185—195° С. Перекристаллизацией из смеси
хлористого метилена и спирта получают вещество с т. пл. 193—195° С [312].
Синтез на основе производных четырехвалентного олова
251
При взаимодействии двухлористого диметилолова с 2,2'-дилитийдифени-
ловым эфиром оловоорганическое соединение с атомом олова в цикле
О (CeH4)2Sn (CH3)2 образуется с выходом 2,4—5% (т. пл. 259—260° С) [313].
Для синтеза смешанных соединений R3SnR' тригалогениды RSnX3
применяют в тех случаях, когда моногалогенидыН'35пХ труднодоступны. Это
относится прежде всего к соединениям, где возможны пространственные
затруднения. Этим путем получены, например, метилтримезитилолово
(т. пл. 35—36° С), метилтри-(3,4,5-триметилфенил)олово (т. пл. 133° С)
[174], фенилтри-(9-флуоренил)олово. Следует иметь в виду, что соединения
этого типа отличаются сравнительно малой стабильностью. Например,
выделить фенилтри-(1-инденил)олово не удается. При обработке
реакционной смеси водой вещество гидролизуется. Из реакционной среды
выделены фенилстаннановая кислота и инден. При фракционировании
реакционной смеси вещество разлагается с выделением металлического олова [276].
Заметим, что тетра-(1-инденил)олово в тех же условиях стабильно [276].
При взаимодействии треххлористого я-бутилолова или треххлористого
фенилолова с фенилацетиленидом лития образуется я-бутилтри(фенилаце-
тиленид)олово (т. пл. 70°С) или,- соответственно, фенилтри (фенилацетиле-
нид)олово (т. пл. 105° С) с выходом 63 и 47% [303]. Другой метод синтеза
оловоорганических ацетиленидов см. стр. 253.
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ ЩЕЛОЧНЫХ МЕТАЛЛОВ
ИЛИ ПУТЕМ РЕАКЦИИ ТИПА ВЮРЦА
В синтезе оловоорганических соединений находят применение натрийор-
ганические соединения. Их получают в основном взаимодействием
углеводородов с металлическим натрием. Наиболее широко применяют ацетиле-
нид натрия, его производные [7, 10, 134, 218, 314] и другие соединения,
содержащие активный атом водорода [124, 276, 279, 315]
4RNa + SnCl4 -> R4Sn + 4NaCl.
Если в качестве исходных соединений применяют галоидные алкилы или
арилы, то реакция протекает по следующему уравнению:
4RX + 8Na + SnCl4 -» R4Sn + 4NaCl + 4NaX.
В этом случае имеет место реакция типа Вюрца; натрийорганические
соединения являются, по-видимому, промежуточными продуктами [19, 280,
316, 317].
Реакционной средой являются углеводороды, эфир или жидкий аммиак.
Металлический натрий применяют как в мелкораздробленном состоянии
{диаметр частиц < 1 мм) в легком петролейном эфире [316, 318—320], так и
в виде расплава в высококипящих углеводородах [267].
Четырехзамещенные соединения образуются в достаточно чистом виде,
примесь оловоорганических галогенидов незначительна (степень алкилиро-
вания около 3,95) [316]. По синтезу оловоорганических галогенидов этим
методом приводятся только патентные данные [3201.
В алифатическом ряду простейшие соединения R4Sn удобнее получать
с помощью реактива Гриньяра [316]. Для хлористых алкилов нормального
строения более высоких, чем бутил (но не с разветвленной цепью), в случае
применения реакции типа Вюрца выход повышается. В этом случае метод
имеет преимущество перед реактивом Гриньяра [19, 59].
Основными побочными продуктами являются соединения со связью
Sn — Sn, примесь которых в неочищенном продукте может быть
значительной [59, 316].
252
Оловоорганические соединения. Лит. стр. 261—268
Получение тетра-#-гексилолова [59]. В литровую круглодонную колбу помещают 23 г
натрия под слой ксилола (300 мл), нагревают до кипения и, энергично встряхивая,
превращают расплавленный натрий в мелкораздробленное состояние. Ксилол удаляют, натрий
промывают несколько раз петролейным эфиром (т. кип. 40—60° С) и переносят в трехгорлую
колбу, снабженную мешалкой, обратным холодильником и капельной воронкой. Натрий
заливают 300 мл петролейного эфира. В колбу, при нагревании до температуры кипения
петролейного эфира, прибавляют в течение часа смесь 32,6 г (14,6 мл\ 0,125 моля)
хлорного олова и 66,8 г (0,55 моля) хлористого я-гексила. Реакционную смесь нагревают с
обратным холодильником еще 6 час, хлористый натрий удаляют центрифугированием,
растворитель выпаривают и остаток фракционируют. Получают27,5 г (48%) тетра-я-гексилолова
с т. кип. 187—190° С/1,5 мм [59].
Побочным продуктом является дистаннан. Его примесь в неочищенных
продуктах, полученных по этому методу, может быть значительной. Так,
тетра-я-октилолово, из которого летучие примеси отгоняют в вакууме
(200° С/0,5 мм), содержит 25% гекса-я-октилдистаннана и около 4% гало-
генидов октилолова [59].
Тетрадецилолово [321] получают в высококипящих углеводородах
(расплавленный натрий и хлористый децил). Тетра(дифенилметил)олово с
помощью дифенилметилкалия синтезировать не удается [124].
Тетрациклопентадиенилолово получено из циклопентадиенилнатрия и
хлорного олова с выходом 67% [322].
Получение тетра(метилциклопентадиенил)олова [279]. К 5,40 г (52,5 ммоля) метилцик-
лопентадиенилнатрия, суспендированного в 20 мл бензола, прибавляют по каплям раствор
3, 10 г (11,9 ммоля) хлорного олова в 10 мл бензола. Реакционную смесь отфильтровывают,
растворитель удаляют. Остаток, представляющий собой желтое масло, не нуждается в
дополнительной очистке. Выход 5,20 г (82,5%).
Четырехзамещенные оловоорганические соединения ароматического
ряда обычно синтезируют взаимодействием арилгалогенида натрия и хлорного
олова в кипящем эфире [323], бензоле или толуоле [115, 280, 324, 324а, 325,
325а].
Получение тетрафенилолова [323]. 13 г хлорного олова медленно, при энергичном
перемешивании прибавляют к холодному абсолютному эфиру с целью получения твердого эфи-
рата в возможно мелкораздробленном состоянии. После добавления 23 мл бромбензола в
реакционную смесь малыми порциями вносят 10 г натриевой проволоки. Тотчас же
начинается реакция. После прибавления всего натрия реакционную смесь кипятят с обратным
холодильником 10 час. Затем эфир отгоняют, из остатка тщательно извлекают кипящим
бензолом тетрафенилолово. Выход вещества ст. пл. 223° С составляет 10 г.
Тетратол ил олово получено с выходом 45% взаимодействием хлортолуола
с хлорным оловом в присутствии натрия и ртути в петролейном эфире [325а].
Ацетилениды натрия с хлорным оловом образуют соединения типа
Sn(C=CR)4. Тетраэтинилолово при быстром нагревании взрывает [326].
Получение тетрапропин-1-илолова [323]. В суспензию 5 г натрия (2,1-Ю-1 г-атома) в
100 мл гексана барботируют пропин. При 25—30° С реакция протекает легко и
заканчивается в течение 2 час. К реакционной смеси прибавляют по каплям 13 г (0,5 • IQr1 моля)
хлорного олова в 20 мл гексана и кипятят 2 часа. Фильтруют через стеклянный фильтр и
растворитель удаляют. Перекристаллизовываютиз гексана. Получают 1 г (73%) тетрапропин-
1-илолова с т. пл. 150° С.
Взаимодействием хлорного олова и фенилацетиленида натрия (в смеси
тетрагидрофурана с бензолом 2 : 1) с выходом 65% получают тетрафенилэти-
нилолово. Это хорошо кристаллизующееся, чувствительное к влаге вещество
ст. пл. 139° С [134,314].
Тетра-(1-инденил)олово образуется с хорошим выходом лишь в том
случае, если инденил-1-натрий (полученный нагреванием индена с натрием при
150° С) свободен от металлического натрия [276].
Синтез на основе производных четырехвалентного олова
253
Оловоорганические галогениды при взаимодействии с галоидным алки-
лом и натрием алкилируются до четырехзамещенных соединений. Эта реакция
имеет значение для синтеза практически важного тетрабутилолова, которое
образуется с более высоким выходом, если вместо хлорного олова в
реакцию вводят двухлористое дибутилолово [316]:
(С4Н9)2 SnCl2 + 2QH9CI + 4Na -* (С4Н9)4 Sn + 4NaCl.
Получение тетрабутилолова [316]. К 4 г-атомам измельченного натрия (диаметр частиц
1 мм в бензине, т. кип. 40—60° С, в количестве 1 л на 0,74 моля двухлористого дибутилоло-
ва) прибавляют по каплям в течение 30 мин. смесь 1 моля двухлористого дибутилолова и
2 молей +10% избытка хлористого бутила. По окончании реакции через 0,5—1,5 часа смесь
кипятят еще 7 час. при 45—47° С. После обычной обработки выход тетрабутилолова равен
«7,9%.
Часть тетрабутилолова можно перевести в двухлористое дибутилолово по
реакции, обратной диспропорционированию, с тем, чтобы вновь ввести в
описанную выше реакцию. Применение высококипящих растворителей или
изменение степени измельчения натрия может привести к снижению выхода.
Побочным продуктом является гексабутилдистаннан (около 1,0%).
Аналогично тетра-я-октилолово получено действием металлического
натрия на смесь двухлористого ди-я-октилолова с хлористым я-октилом. В
качестве растворителя применяют также R4Sn (например, тетраметил-, тетра-
октилолово) [318].
Оловоорганические соединения, содержащие алициклические радикалы,
образуются с достаточно хорошим выходом.
Получение трициклогексил-я-бифенилолова [154]. Раствор 20 г бромистого трициклогек-
силолова и 16 г о-хлорбифенилолова в 150 мл петролейного эфира (т. кип. 60—80° С) кипятят
с 7 г натриевой проволоки в течение 7 час. Фильтруют через стеклянную вату, обрабатывают
водой, органический слой отделяют, промывают, высушивают сульфатом натрия и
концентрируют. Твердое вещество кристаллизуют из смеси этанола с бензолом (9:1). Получают три-
циклогексил-о-бифенилолово с выходом 60% (т. пл. ПО—111° С).
Трициклогексилфенилолово (выход 75%, т. пл. 191° С) [154] и триэтил-
фенилолово получены аналогично действием металлического натрия на
смесь бромистого трициклогексилолова в петролейном эфире или,
соответственно, йодистого триэтилолова и бромбензола в эфире [327, 328].
Алкилирование трехгалоидных производных ал кил олова представлено на
примере получения бутилтрифенилолова из фенилнатрия и треххлористого
бутилолова в толуоле [329].
Натрийорганические соединения RNa для синтеза оловоорганических
соединений находят значительное применение. Это относится прежде всего
к тем натрийорганическим соединениям, которые образуются путем замены
на натрий подвижного водорода. Их взаимодействие с оловоорганическими
галогенидами проводят в углеводородах, эфире или жидком аммиаке.
Например, к циклопентадиенилнатрию, суспендированному в ксилоле, прибавляют
хлористое триэтилолово при 40—60° С [322, 330]. Для получения дифенил-
ди(дифенилметил)олова аммиачный раствор дифенилметилкалия,
полученный из дифенилметана в жидком аммиаке, упаривают, заменяют аммиак на
эфир и кипятят с двухлористым дифенилоловом. Получают дифенилди(ди-
фенилметил)олово с выходом 58% [124].
В реакциях с оловоорганическими галогенидами наиболее подробно
исследованы ацетилениды натрия. Соединения типа R3SnC=CR или
R2Sn(C=CR)2 (R — радикал или Н) образуются с выходами порядка 50—
60%. Оловоорганические производные ацетиленового ряда обычно
представляют собой жидкости. Так, например, триметил(1-пропинил)олово имеет
т. кип. 25—26° С/0,2 мм [218].
254 Оловоорганические соединения. Лит. стр. 261—268
Получение диметилди(фенилэтинил)олова [218]. К суспензии 1 г (4 • 10-2 г-атома)
натрия в 25 мл гексана прибавляют 4,5 г (4 • 10~2 моля) фенилацетилена и через 2 часа —
6,2 г (2 • 10~2 моля) двубромистого диметилолова. Кипятят 2 часа. Перекристаллизовывают
из гексана. Получают 3,5 г (50%) вещества с т. пл. 66,7° С.
Трифенил(хлорацетиленид)олово получено с выходом 80% (т. пл. 80—
81° С)
(С6Н5)3 SnCl + NaCEECCl -» (С6Н5)3 SnteCCl + NaCl.
Получение трифенил(бромацетиленид)олова [218]. К раствору амида калия,
полученному из 0,9 г (2,5 • 10~2 моля) калия и 50 мл жидкого аммиака при —40° С, прикапывают
1,86 г (10~2 моля) 1,2-дибромэтилена в 4 мл тетрагидрофурана и перемешивают 20 мин. при
—40° С. Затем вводят 4,30 г (10~2 моля) бромистого трифенилолова в 10 мл растворителя,
перемешивают в течение часа, прибавляют 50 мл эфира и, чтобы уничтожить избыток
амида калия, 3,4 г хлористого аммония. Фильтруют через стеклянный фильтр, растворитель
удаляют в вакууме и остаток перекристаллизовывают из гексана или петролейного эфира.
Выход 75%, т. пл. 104° С.
Фенильные соединения этого типа устойчивы, в то время как
аналогичные им производные алифатического ряда в некоторых случаях
чувствительны к влаге воздуха [218]. Взаимодействие фторацетилена и амида натрия с
последующим прибавлением хлористого трифенилолова протекает более
сложно. Из реакционной смеси выделен (CeH5)3SnCH2CN [331].
Соединения общей формулы (CH3)3SnC~CR и (C2H5)3SnC=CR', где
R — (в скобках т. кип. и п%) — СН=СН2 (49° С/Юлим; 1,5058), —СН(СН3) —
—СН3 (50° С/10 мм; 1,4676); R' — СН-СН2 (90—91° С/10 мм; 1,5100),
СН(СНз) — СН3 (91,5° С/10 мм; 1,4778) получены с выходами до 80%
[228, 332]. Бутин-1-ол-4 в тех же условиях (амид натрия в жидком аммиаке)
реагирует с образованием смеси двух продуктов. Например, с хлористым
триэтилоловом получены следующие соединения: (C2H5)3SnOCH2CH2C=CH и
(C2H5)3SnOCH2CH2C=CSn (C2H5)3 с выходами 24 и 41 % соответственно [333].
Что касается соединений, содержащих двойные и тройные связи,
например R3SnC=C—СН=СН2, то они выделены в следующих условиях.
Получение ениновых оловоорганических соединений [224]. К винилацетилениду натрия
в жидком аммиаке добавляют абсолютный эфир, смесь нагревают до исчезновения запаха
аммиака. К полученной суспензии при охлаждении (лед + соль) прибавляют хлористое три-
этилолоео. После нагревания в течение 6 час. смесь разлагают насыщенным раствором
хлористого аммония и после обычной обработки получают триэтилвинилацетиленилолово.
Те же соединения могут быть получены с помошью бромистого винилаце-
тиленмагния (см. стр. 227), а также действием R3SnNa на алкилбромацети-
лены в жидком аммиаке (см. стр. 501).
Ениновые оловоорганические соединения представляют собой
бесцветные жидкости со специфическим запахом. В чистом виде без доступа воздуха
они достаточно устойчивы. Примесь галоидного триал кил олова вызывает
при хранении полимеризацию с образованием темных твердых продуктов.
На воздухе они довольно легко гидролизуются [224].
Описаны также диацетилениды олова R3SnC=C—C=CR. Они образуются
с выходом 40—70%.
Получение трифенил-(1-фенил-4-бутадиинил)олова [218]. В раствор амида натрия,
полученный из 0,23 г (10~2 г-атома) натрия и Г:0 мл аммиака, вводят при —50° С 0,7 г фенил-4-
бутадиина в 10 мл эфира. Через 3 мин. прибавляют 2г (0,42 • 102 моля) йодистого
трифенилолова и перемешивают 2 часа при —-'0° С. Продукт экстрагируют эфиром,
перекристаллизовывают из гексана. Получают 1,1 г (55%) вещества с т. пл. 88° С.
Трифенил-(1-этил-4-бутадиинил)олоьо (т. кип. 100° С/1 мм) и трифе-
нил-(1-метил-4-бутадиинил)олово получены аналогично [218, 314].
Синтез на основе производных четырехвалентного олова
255
В случае ацетилена и диацетилена возможны оловоорганические
соединения двух типов: R3SnC=CH и R3SnC = CSnR3. Этинильные
производные оловоорганических соединений получают взаимодействием оловоорга-
нических галогенидов с ацетиленидами щелочных металлов. В качестве
растворителя применяют эфир, гексан и другие углеводороды. Выход достигает
80%. Соединения представляют собой жидкости; триэтилэтинил олово имеет
т. кип. 57—58° С/11 мм, диэтилдиэтинилолово — т. кип. 32—35° С/0,1 мм,
трибутилэтинилолово — т. кип. 85—90° С/0,1 мм [326]. Триметилэтинил-
олово получено аналогично [334].
Получение трифенилэтинилолова требует большого избытка ацетиленида
натрия; кроме того, оловоорганический галогенид следует вводить в
реакционную смесь быстро. Медленное прибавление галогенида приводит к
образованию симметричного продукта
(С6Н5)3 SnJ + NaC=CH -* (C6H5)3 SiiCeeeCH + NaJ,
(С6Н5)3 SnC=CH + NaC=CH -* (C6H5)3 SnC=CNa + CHeeCH,
(G6H5)3 SnCEECNa + JSn (C6H5)3 -» (C6H5)3 SnC=CSn (C6H5)3 + NaJ.
Получение трифенилэтинилолова [218]. К раствору ацетиленида натрия, полученному
из ацетилена и 0,5 г (2 • 10~2 г-атома) натрия в 50 мл аммиака, прибавляют 20 мл эфира и
быстро вводят 7 г (1,5 • 10~2 моля) твердого йодистого трифенилолова. Добавляют
хлористый аммоний и оставляют на 3—4 мин. Прибавляют еще 50 мл эфира и аммиак удаляют.
Фильтруют через стеклянный фильтр, эфир отгоняют в вакууме. Остаток перекристалл и-
зовывают из минимального количества гексана. Получают 4 г (70%) вещества ст. пл. 37—
38° С. Вторым продуктом реакции является бис-(трифенилстаннил)ацетилен.
Для получения монозамещенного продукта пользоваться бромистым
трифенилоловом не следует, оно менее реакционноспособно.
Трифенилэтинилолово при обычных условиях неустойчиво и
разлагается с выделением ацетилена и образованием дизамещенного продукта. Три-
этил-(1-бутадиинил)олово получено с выходом 65% (т. кип. 67—68° С/2 мм)
[335].
Получение трифенил-(1-бутадиинил)олова (С6Н5)з5пС=С—teCH [218, 314]; 0,7 г
натрия (3 • 10"2 моля) растворяют в 80 мл жидкого аммиака, прибавляют 2,2 г 1,4-дихлор-
бутина (1,5 • 10~2 моля) в 20 мл эфира и через 15 мин. быстро вводят 4,76 г (10-2 моля)
йодистого трифенилолова при—50° С. Через 15—20 мин. выделяют трифенил(бутадиинил)
олово, перекристаллизовывают из гексана. Выход 27%, т. разл. 99° С. Одновременно
образуется дизамещенный продукт с т. пл. 245° С [218, 314].
Соединения R3SnC = CSnR3 получают действием моногалогенидов
R3SnX на ацетиленид натрия в жидком аммиаке [191, 192, 224, 308].
Получение б'ис-(триметилолово)ацетилена (CH3)3SnC=C Sn(CH3) [298]. К ацетилениду
натрия, приготовленному в жидком аммиаке из 0,63 г-атома натрия, прибавляют 0,49 моля
хлористого триметилолова в 70 мл эфира. Смесь перемешивают 30 мин., затем холодильник
с сухим льдом удаляют, аммиаку дают медленно испариться. Твердый остаток обрабатывают
700 мл эфира и фильтруют. Фильтрат выпаривают при —70° С/1 мм, остаток перегоняют
при 90—100° С/0,2 мм. Получают70 г (80%)быс-(триметилолово)ацетилена ст. пл. 59—60°С.
6ш>(Триарилолово)ацетилены, где арил — фенил [218], я-хлорфенил,
п-толил,о-толил, бензил [192], образуются с тем же выходом (см. стр. 238).
При синтезе 6ш>(триалкилолово)ацетиленидов из соответствующих станно-
Лов (RgSnOH) выход не превышает 62% [336]. С помощью динатрийдиацети-
лена в жидком аммиаке (см. выше) получены диацетилениды бш:-(триалкил-
олова) или диацетилениды бис-(трифенилолова) с выходом 70—80% [303,
337, 338]
R3SnCl + NaC=C—C=CNa -» R3SnC=C—C=CSnR3 + 2NaCl.
256
Оловоорганшеские соединения. Лит. стр. 261—268
Это твердые соединения или масла: R = СН3 (т. пл. 140° С), п-С4Н9
(п% 1,4946) [3031; я-С8Н17 (n2D° 1,4905) [338]; СбН5 (т. пл. 245° С). Три-
ацетиленовые производные олова R3SnC~C—С=С—C~CSnR3 могут быть
получены аналогично [218, 314].
В патенте [339] описаны соединения [R2SnCH2COO]n, которые
получают взаимодействием оловоорганических дигалогенидов с NaCH2COONa.
Натриевые производные малонового эфира, ацетил ацетона, фенилсульфонил-
ацетона (C6H5S02CHNaCOCH3) реагируют с двухлористым диэтилоловом
с образованием четырехзамещенных оловоорганических соединений,
например (C4H9)2Sn[CH (S02C6H5)COCH3]2 [340].
Калийорганические соединения для синтеза оловоорганических
соединений не нашли сколько-нибудь значительного применения, хотя, например,
триметилизопропилолово получено с помощью изопропилкалия с выходом
53% [340а].
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ ЦИНКООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Первые синтезы оловоорганических соединений были осуществлены с
помощью цинкалкилов. Так, тетраэтилолово и тетраметилолово [341]
были получены Франкландом [342, 343] и Буктоном [344] действием диал-
килцинка на хлорное олово еще в 1853—1859 гг.
2 (С2Н5)2 Zn + SnCl4-^ (C2H5)4 Sn + 2 ZnCl2.
Тетраэтилолово образуется и при действии диэтилцинка на двухлористое
олово [345] (видимо, в этом случае первоначально образуется диэтилолово,
которое затем диспропорционирует).
Несимметричные оловоорганические соединения образуются аналогично.
Метилтриэтилолово [346] и этилтриметилолово [139] получены действием
диметил- и диэтилцинка на йодистые триэтил- и триметилолово.
В ароматическом ряду оловоорганические соединения могут быть
получены этим методом с хорошим выходом.
Получение тетрафенилолова [85]. К бромистому фенилмагнию прибавляют постепенно
хлористый цинк в сухом эфире. Реакция происходит тотчас. Добавляют толуол, эфир
отгоняют на водяной бане. К охлажденной реакционной смеси прибавляют эквивалентное
количество хлорного олова в толуоле, смесь кипятят в течение часа. При охлаждении прибавляют
разбавленную соляную кислоту и фильтруют. Остаток на фильтре экстрагируют кипящим
бензолом. Толуольный слой соединяют с бензольным экстрактом. После концентрации
выделяют тетрафенилолово с выходом 91,2%; т. пл. 224—225° С.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ АЛЮМИНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Алюминийорганические соединения применяют для синтеза
четырехзамещенных оловоорганических соединений [292, 347—353] и отчасти
оловоорганических галогенидов [49, 348—351, 354].
3 SnCU + 4R3A1 -* 3R4Sn + 4AICI3,
SnCl4 + R3AI -> RsSnCl + AICI3.
В реакциях обычно используют R3A1, R2A1X, или R3A12X3 (X — галоид),
и сравнительно редко R4A1M (M — металл) [355, 356].
Хлорное и бромное олово в этом синтезе примерно равноценны, тетраал-
коксиолово [352], а также SnS2, SnS04, SnCl2 [357], SnO, Sn02, H2Sn03
[358] применяются редко. В реакциях находят применение также
оловоорганические галогениды [352, 359, 360], их ацилаты, окиси и сульфиды [357].
Синтез на основе производных четырехвалентного олова
257
Реакцию проводят или без растворителя, или в бензоле, гексане или
других инертных растворителях в атмосфере инертного газа.
Взаимодействие триэтил- и триизобутилалюминия с четыреххлористым
оловом протекает с выделением тепла [361], однако даже при избытке три-
алкилалюминия неполно [348, 351].
Получение тетраэтилолова [348]. 40,0 г хлорного олова добавляют в токе чистого азота
при перемешивании к раствору 34,0 г триэтилалюминия в 40 мл я-гексана (умеренное кипение
последнего за счет теплоты реакции). Кипячение продолжают еще 1,5 часа, реакционную
массу разлагают водой, водный раствор экстрагируют эфиром. После высушивания
хлористым кальцием и отгонки растворителя получают 25,8 г (71%) тетраэтилолова с т. кип. 51 —
51,5° С/8 мм] df 1,1990; п2£ 1,4738.
При перегонке остатка получают 7 г хлористого триэтилолова с т. кип.
86—88° С/12 мм; df 1,4085; n2D° 1,5060.
Аналогично из хлорного олова и триизобутилалюминия получены тет-
раизобутилолово (53,6%); т. кип. 128—129° С/8мм. Продукт содержит
примесь хлористого триизобутилолова [347, 348].
Оловоорганические галогениды остаются в полученном продукте даже
при избытке алюминийорганического соединения. Дело в том, что при ал-
килировании хлорного олова алюминийорганическими соединениями
образуется сложная смесь, в которой R4Sn, R3SnCl, R2SnCl2 [49, 352, 361, 362],
а также R2SnCl2-AlCl3 и R3SnCl-AlCl3 [352] находятся в равновесии с
галоидным алкилалюминием. Алкилирование хлорного олова протекает
полностью лишь в том случае, если образующийся в результате реакции
хлористый алюминий удаляют из реакционной смеси. Обычно его связывают в виде
комплекса МА1С14 прибавлением хлористых натрия, калия или лития
[349, 351, 352, 363], а также в виде C13A1-NR3 или C13A1-0R2 третичными
аминами или эфиром [352, 364, 365]:
3SnCl4 + 4R3A1 + 4NaCl -* 3R4Sn + 4NaAlCl4.
Введение в реакционную смесь галогенидов щелочных металлов дает еще
то преимущество, что сравнительно малоактивный двухлористыи алкилалю-
миний превращается в более реакционноспособный NaAl(C2H5)Cl3 [349].
Наиболее часто реакцию между алюминийорганическими соединениями
и хлорным оловом проводят в присутствии хлористого натрия. Комплексооб-
разующее действие последнего проявляется наиболее полно при
температурах выше 100° С. Перед началом реакции, например, атилалюминийсескви-
хлорид и хлористый натрий нагревают, а затем вводят хлорное олово.
Реакционная смесь превращается в гомогенную и хорошо перемешиваемую
массу.
120е С
4 (С2Н5)3А12С1з + 3 SnCl4 + 8 NaCl ► 3 (С2Нз)4 Sn + 8 NaAlCl*.
Получение тетраэтилолова целесообразнее осуществлять при более
высокой температуре, употребляя небольшой избыток этилалюминийсесквихло-
рида [349].
Получение тетраэтилолова [349].
а) 24 г хлористого диэтилалюминия и 18 г сухого хлористого калия нагревают в
атмосфере азота при перемешивании в течение 30 мин. до 100—120° С. К образовавшемуся
комплексу КА1(С2Н5)2С12 постепенно добавляют 23 г хлорного олова при температуре 80—110° С.
Смесь нагревают при 130—150° С в течение 10 мин., после чего тетраэтилолово отгоняют
непосредственно из реакционной массы в вакууме. После повторной перегонки получают
19,2 г (94%) тетраэтилолова с т. кип. 71—72° С/10 мм.
б) 80 г этилалюминийсесквихлорида [эквимолекулярная смесьС2Н5А1С12и (С2Н5)2А1С1]
нагревают с 58 г сухого хлористого калия при НО—120° С (30 мин.). К
образовавшемуся комплексу КгА12 (С2Н5)зС15 постепенно прибавляют в атмосфере азота и при
перемешивании 58 г хлорного олова, поддерживая температуру реакционной смеси на уровне 80—
17 Заказ № 207
258
Оловоорганические соединения. Лит. стр. 261—268
100 С. Затем смесь нагревают при 130—150° С в течение 20 мин. и в вакууме отгоняют тет-
раэтилолово; выход 46,5 г (94%); т. кип. 71—72° С/10 мм.
Следует иметь в виду, что триизобутилалюминий термически
относительно мало устойчив. Поэтому при получении изобутильных производных олова
избегают высоких температур во время добавления первых порций хлорного
олова вплоть до образования хлористого диизобутилалюминия
SnCl4 + (/-С4Н9)3А1 -» (/-QH9)2 A1C1 -f- /-QHoSnCls.
Дальнейшее добавление хлорного олова можно вести при повышенной
температуре [349].
Получение тетраизобутилолова [349]. К 34,8 г триизобутилалюминия при температуре
10—15° С постепенно прибавляют 13 г хлорного олова (при перемешивании в атмосфере
азота), затем сразу вводят 20 г сухого хлористого калия и нагревают при 100—120° С до
образования растворимого комплекса. К смеси при 80—100° С вновь добавляют 17 г хлорного
олова и нагревают,при 130—140° С в течение 20 мин., после чего отгоняют в вакууме
непосредственно из реакционной смеси 30 г тетраизобутилолова, т. кип. 127—128° С/8 мм [349].
Выход достигает 87% [364].
В качестве комплексообразователя удобно применять также дибути-
ловый эфир. В его присутствии тетраэтил-, тетра-я-бутил- и тетраоктил-
олово получены с почти количественными выходами. R4Sn в случае
соединений со сравнительно низкой температурой кипения отгоняют в вакууме
прямо из реакционной смеси (комплексы хлористого алюминия обычно
перегоняются при более высокой температуре) [352].
Получение тетраизобутилолова [364]. К 55,8 г (0,28 моля) триизобутилалюминия
(в пересчете на чистый продукт) при перемешивании (вначале медленно, затем быстрее)
прибавляют по каплям 52 г (0,20 моля) хлорного олова таким образом, чтобы температура
оставалась в пределах от—10 до 0° С. Затем, продолжая перемешивание, вводят 75 мл (0,31 моля)
дибутилового эфира, нагревают 1 час при 60—70° С и отгоняют тетраизобутилолово при 0,001
мм\ т. кип. вначале 75—77° С, затем поднимают до 92° С. Конденсат содержит некоторое
количество эфирата треххлористого алюминия (С4Н9)20 • А1С13, который разлагают 2 N
раствором едкого натра. Выход 58,7 г (84%); т. кип. 135—137° С, п2£ 1,4742.
Соединения олова с более длинными алкильными остатками не
смешиваются с эфиратами хлористого алюминия и могут быть отделены еще легче
[352]. Так, тетраоктилолово образуется с выходом 98%, если в хлорное
олово вводят триалкилалюминий, затем дибутиловый эфир и реакционную смесь
перемешивают при 60—70° С до расслоения [365]. Более тяжелый слой
представляет собой комплекс; его>отделяют, следы хлористого алюминия из оло-
воорганического соединения отмывают водой. Комплекс хлористого алюми-
миния можно удалить и гидролизом [352].
В случае применения алкоксисоединений олова [356] прибавление ком-
плексообразователей излишне
Д R3A1 + 3 Sn (OR)! -> 4 Al (ORV+ 3 R*Sn.
В бензоле реакция заканчивается образованием сильно ассоциированного
этилата алюминия, а тетраэтил олово с хорошим выходом отгоняется из
реакционной смеси. Алкоголяты олова легко получить из галогенидов,
например, с помощью этилата натрия [352].
При взаимодействии алюминийорганических соединений с атомом
алюминия в цикле с хлорным оловом происходит замена алюминия в цикле на
олово [366]. Так, 6ш:-(3,3-диметилпеитаметилен)олово (I) образуется с
выходом 68% (т. кип. 75—82° С/10~* мм; nf> 1,5182).
С оловоорганическими дигалогенидами реакция протекает аналогично.
Синтез на основе производных четырехвалентного олова
259
3,3-Диметилпентаметилендибутилолово (11) получено с выходом 65% (т. кип.
71— 77° С/10"4 мм\ п2£ 1,4948) [366, 367].
СН3 СН2—СНа '
\ / \
С
. / \ /
LCH3 СН2-СН2 -
СНз СНо—СН2 С4Ы9
\ / \ /
Sn G Sn
/ \ / \
2 сн3 сн2—сн2 с4н9
(I) (II)
Оловоорганические соединения ароматического ряда образуются с
хорошим выходом. В этом случае из реакционной смеси отгоняют эфират
треххлористого алюминия.
Получение тетрафенилолова [368]. Раствор 0,42 моля фенилалюминийсесквихлорида
в хлорбензоле медленно прибавляют к 0,3 моля хлорного олова. Температура
поднимается до 100° С. Затем полученную смесь очень осторожно и постепенно выливают в 65 г диэти-
лового эфира. Растворители отгоняют сначала при нормальном давлении, а затем в вакууме.
Получают 155 г эфирата треххлористого алюминия с т. кип. 72—80е С/10-4 мм. Остаток
промывают разбавленной соляной кислотой, водой и спиртом и экстрагируют бензолом.
Из бензольного раствора получают 120 г (92%) тетрафенилолова с т. пл. 220—222° С.
Сравнительная мягкость алкилирующего действия алюминийорганиче-
ских соединений позволяет получать с их помощью также продукты
неполного алкилирования [49, 361, 362], например двухлористое диэтилолово
[3491 или двубромистое диизобутилолово [350]. Однако при добавлении
этилалюминийсесквихлорида к хлорному олову даже в относительно мягких
условиях трудно получить, например, двухлористое диэтилолово без
примеси моно- и трихлорида. Кроме того, при пониженных температурах не
вполне проявляется комплексообразующее действие хлористого натрия
[349]. При синтезе оловоорганических галогенидов в одну стадию получают
удовлетворительные результаты только в отдельных случаях. Например,
хлористое триметилолово получено взаимодействием метилалюминийсескви-
хлорида и хлорного олова с выходом 69% [359].
Получение алюминийорганического соединения и алкилирование
галоидного олова может быть проведено в одну стадию. В этом случае алюминий
в виде стружек обрабатывают смесью галоидного алкила и галогенида олова
[349]
2 С2Н5Вг + 2 Al + SnBr4 -> (C2H5)2 SnBr2 + 2А1Вг8.
Получение двубромистего диэтилолсва. К 5,47 г алюминиевых опилок добавляют 2 мл
раствора бромного олова (65,7 г) в бромистом этиле (32,7 г). Воздух из прибора вытесняют
азотом, смесь нагревают до начала реакции и вводят бромное олово в бромистом этиле с
такой скоростью, чтобы температура экзотермической реакции удерживалась на уровне 90—
100° С. Реакционную смесь перемешивают 2 часа при 100° С, двубромистое диэтилолово
отгоняют. Выход 32,8 г (65%); т. кип. 233° С, т. пл. 62,5—63° С [349].
Оловоорганические галогениды с более высокими выходами могут быть
получены в ароматическом ряду. Продукт реакции определяется молярным
соотношением реагентов [367а, 368].
4 (С6Н5)8 А12С13 + 3 SnCL -» 3 (С6Н5)4 Sn + 8 А1С18,
(С6Н5)3 А1аС18 + SnCU ~> (СсН5)з SnCl + 2 А1С13,
2(С6Н5)3 А12С13 + 3 SriCl4 -* 3 (C6H,)2SnCl2 + 4А1С18,
(С6Н5)3 А12С13 + 3 SnCl4 -* 3 C6H5SnCl3 + 2 A1C1S.
Получение треххлористого фенилолсва [SG8]. К 1,2 молям хлорного олова медленно
прибавляют раствор 0,42 моля фенилалюминийсесквихлорида в хлорбензоле, смесь выливают
в 65 мл диэтилового эфира, растворитель отгоняют. В результате получают 307 г (84%)
треххлористого фенилолова с т. кип. 109—110° С/10 мм. Эфират треххлористого алюминия
остается в перегонной колбе.
17*
260 Оловоорганшеские соединения. Лит. стр. 261—268
Оловоорганические галогениды с помощью алюминийорганических
соединений могут быть переведены в четырехзамещенные соединения.
Экспериментальные условия не отличаются от описанных для хлорного олова
[359]. Из хлористых триэтил- или три-я-бутилолова действием триэтилалю-
миния или, соответственно, три-м-бутилалюминия получены тетраэтил- и
и тетра-я-бутилолово с выходом 89 и 87% [359]. Соединения RnSnR'4_n
получены аналогично [360], а также действием триал килалюминия на окись
или сульфид диалкилолова, гексаалкилдистанноксан или ацилаты алкил-
олова [357].
Продукты частичного алкилирования RnSnX4_n из ди-и тригалогенидов
алкилолова образуются с хорошим выходом. Так, хлористое триметилолово
при синтезе его из двухлористого диметилолова получено с выходом 69%.
В этих случаях лучше применять сесквихлориды алюминия; они менее
активны [360].
Получение хлористого триэтилолова [380]. К 75 г треххлористого этилолова в приборе,
заполненном азотом, прибавляют по каплям в течение 1,5 час. при температуре ^70° С 77 г
этилалюминийсесквихлорида. Массу перемешивают без нагревания и при ~~20° С добавляют
100 мл эфира, после чего осторожно гидролизуют 80 мл 5%-ной соляной кислоты.
Органический слой отделяют и перегонкой выделяют 52 г хлористого триэтилолова с п2£ 1,5042.
Подобным образом получены смеси хлористого триметилолова с тетра-
метилоловом, хлористого три-я-бутилолова с тетра-я-бутилоловом и
хлористого три-я-октилолова с тетра-я-октилоловом.
Синтез гидрида триалкилолова в одну стадию действием на хлорное олово
смеси триалкилалюминия и гидрида диалкилалюминия пока еще исследован
недостаточно [369].
Получение четырехзамещенных оловоорганических соединений
действием тетраалкилалюминита лития [355] или натрия [356] на хлорное олово
проводят в углеводородах, например бензоле. Выход достигает 85—93%.
При взаимодействии оловоорганических галогенидов с гидридами алкил-
алюминия образуются оловоорганические гидриды (см. стр. 455).
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПОСРЕДСТВОМ МЕТАЛЛООРГАНИ ВЕСКИХ СОЕДИНЕНИЙ
ДРУГИХ МЕТАЛЛОВ
Металлоорганические соединения серебра, ртути или свинца в реакциях
с хлорным оловом или оловоорганическими соединениями описаны на
частных примерах. Например, AgC^CCH(OC2H5)2 в ацетоне применяют для
синтеза (C6H5)3SnC=CCH(OC2H5)2 [370].
Ртутноорганические соединения с хлорным оловом реагируют по
уравнению
2 (С6Н5)2 Hg + SnCl4 -* (CeII5)2 SnCl2 + 2 C6H5HgCl.
Реакция практического значения не имеет (необходимо длительное
нагревание, относительно низкий выход — 33%) [371 ]. При избытке хлорного олова
возможно образование наряду с хлористой фенилртутью треххлористого
фенил олова [2, 10].
Взаимодействие оловоорганических соединений с хлорным оловом и
оловоорганическими галогенидами находит значительное препаративное и
производственное применение (см. стр. 199).
Реакция тетрафенилсвинца с хлорным оловом заканчивается на
образовании нерастворимого двухлористого дифенилсвинца [372, 373]
(С6Н5)4 Pb + SnCl4 -> (С6Н3)2 SnCl2 + (С6Н5)2 РЬС12.
Синтез на основе производных четырехвалентного олова
261
ЛИТЕРАТУРА
1. Р о 1 1 а г d F. Н., N i с к 1 е s s G., DolanD.N., Chem. a. Ind., 1965, 1027.
2. В a h r G., Z о с h e G., Вег., 88, 1450 (1955).
3. Maire J.- С, Ann., 6, 969 (1961).
4. Maire J.- С, Compt. rend., 251, 1292 (1960).
5. MaireJ. -С, Compt rend., 249, 1359 (1959).
6. S e у f e r t h D., Stone F.G., J. Am. Chem. Soc, 79, 515 (1957).
7. ИнгамР., РозенбергС, ГильманГ., РикенсФ., Оловоорганиче-
ские и германийорганические соединения. М., ИЛ, 1962.
8. Кг a use E., Вег., 51, 1447(1918).
9. KorschingH., Z. Naturforsch., 1, 219 (1946).
10. S t a v e 1 е у L. К., Р a g e t H. P., G о а 1 b у В. В., W а г г е п J. В., J. Chem.
Soc. 1950 2290.
11. We r n er A., PfeifferP., Z. anorg. allgem. Chem., 17, 82 (1898).
12. E d g e 1 1 W. F., W a r d С H., J. Am. Chem. Soc, 76, 1169 (1954).
13. V a n d e г К e r k G. J. M., NoltesJ. G., Z u i j t e n J. G. A., J. appl.
Chem., 7, 356 (1957).
14. С 1 a r k H., W i 11 i s С J., J. Am. Chem. Soc, 82, 1888 (1960).
15. К о ч к и н Д. А., Л у к ь я н о в а Л. В., Р е з н и к о в а Е. Б., ЖОХ, 33, 1945
(1963).
16. Е d g е 1 1 W. F., W a r d С. Н., J. Am. Chem. Soc, 77, 6486 (1955).
17. W a r i n g С. Е., HortonW.S, J. Am. Soc, 67, 540 (1945).
18. L i p p i n с о t t E. R., Tobi n M. C, J. Am. Chem. Soc, 75, 4141 (1953).
19. Van der KerkG. J.M., Z u i j t e n J. G. A., J. appl. Chem., 6, 49 (1956).
20. P a p e t t i S., PostH.W., J. Org. Chem., 22, 526 (1957).
21. P e d 1 e у J. В., S k i n n e r H. A., ChernickCL, Trans. Farad. Soc,
53, 1612 (1957).
22. M а н у л к и н 3. М., ЖОХ, 11, 386 (1941).
23. Н а у м о в С. Н., М а н у л к и н 3. М., ЖОХ, 5, 281 (1935).
24. S i e b e r t H., Z. anorg. allgem. Chem., 263, 82 (1950).
25. Б а л а н д и н А. А., К л а б у н о в с к и й Е. И., К о з и н а М. П., У л а н о-
в а О. Д., Изв. АН СССР, ОХН, 1957, 12.
26. D е 1 h а у е А., N a s i e 1 s k i J., P 1 a n с h о n M., Bull. Soc chim. Belg.,
69, 134 (1960).
27. P о 1 1 a r d F. H., NicklessG., С о о k e D. J., J. Chromatogr., 17, 472 (1965).
28. DillardC. R., McNeill E. H., Simmons D. E., V e 1 de 11 J. D., J.
Am. Chem. Soc, 80, 3607 (1958).
29. St a vel ey Z. A., Warren J. В., Paget H. P., DowrickD. J., J.
Chem. Soc, 1954, 1992.
30. В a m b у n e k W., F r e i s e V., Z. physik. Chem., 7, 317 (1956).
31. В а н-д е р К е р к Д ж., Л ю й т е н Д ж., Сб. «Синтезы органических
препаратов», т. 8. М., ИЛ, 1958, стр. 57.
32. Pfeiffer P., Schnurmann К., Вег., 37,319(1904).
33. Кочешков К. А., ЖОХ, 4, 1359 (1934).
34. К г a u s е Е., Grosse A., Die Chemie der metal lorganischen Verbindungen. Berlin
Gebr. Borutrager, 1937, S. 315.
35. Б о р и с о в А. Е., Н о в и к о в а Н. В., Изв. АН СССР, ОХН, 1959, 1370.
36. S a s i п R., S a s i n G. S., J. Org. Chem., 20, 770 (1955).
37. G e r s h b e i n L. L., I p a t i e f f V. N., J. Am. Chem. Soc, 74, 1540 (1952).
38. BowdenK, В r a u d e E. A., J. Chem. Soc, 1952, 1068.
39. ZimmerH, GoldH., Ber., 89, 712 (1956).
40. К r a u s С A., Sessions W.V., J. Am. Chem. Soc, 47, 2361 (1925).
41. S a s i n G. S., BorrorA.L, S a s i n R., J. Org. Chem., 23, 1366 (1958).
42. П е т р о в А. Д., МироновВ.Ф., Изв. АН СССР, ОХН, 1957, 1491.
43. В г i п с k m а п F. E., S t о п е F. G. A., J. Am. Chem. Soc, 82, 6218 (1960).
44. J о n e s W. J., E v a n s D. P., G u 1 w e 1 1 Т., G r i f f i t h s D. О., J. chem.
Soc, 1935, 39.
45. R a m s d e n H. E., DavidsonH., Англ. пат. 695610 (1953); С. А., 48, 10057
(1954).
46. R a m s d e n H. E., G 1 о s к е у С R., Пат. США 2675399 (1954); С. А., 49, 5512
(1955).
47. В о г b е 1 у-К uszmann A., N a g у J., Period, polytechn. chem. Engng., 6,
127 (1962).
48. R a m s d e n H., Пат. США 3067226 (1962); РЖХим., 1964, 16Н56П.
49. Van der KerkG. J. M., Z u i j t e n J. G. A., van Egmond J.C,
NoltesJ. G., Chimia, 16, 36 (1962).
50. С r a m e г С R., Tin and its uses, 46, 7 (1959).
262
Оловоорганические соединения
50а. Fol desi J., Acta. chim. Acad. Scient. hung., 45, 237 (1965).
51. 3 a x a p к и н Л. И., О х л о б ы с т и н О. Ю., Струнин Б. Н., ДАН СССР,
144, 1299 (1962).
52. 3 а х а р к и н Л. И., Охлобыстин О. Ю., Струнин Б. Н., Изв. АН
СССР, ОХН, 1962, 2002.
53. ШевердинаН. И., АбрамоваЛ. В., ПалееваИ. Е., Кочешков
К. А., Хим. пром., 1962, 707.
54. Беликин А. В., Сборник изобретений и рационализаторских предложений
НИИТЭХИМ, № 9, 21 (1962).
55. N е u m a n n W. Р., В u r k h a r d t G., Ann., 663, 11 (1963).
56. Ramsden H. E., Пат. США 2675397 (1954); С A., 49,8332(1955).
57. R a m s d e n H. E., Англ. пат. 701714 (1953); С. А., 49, 4011 (1955).
58. L e s b г е М., Dupont R., С. А., 49, 15729 (1955).
59. V a n der К е г к G. J. M., Z и i j t e n J. G. A., J appl. Chem., 7, 369 (1957).
60. V о i g t D., LesbreM., G a 1 1 a i s F., Compt. rend., 239, 1485 (1954).
61. К о р ш а к В.В., Рогожин С. В., Макарова Т. А., Высокомолек. соед.,
4,1297(1962).
62. М е а 1 s R. N., J. Org. Chem., 9, 211 (1944).
63. Р г i n с е R. H., J. Chem. Soc, 1959, 1759.
64. G г и t t n e r G., К г а и s е Е., Вег., 50, 1802 (1917).
65. Н е n g 1 е i n F. A., L a n g R., S с h m a ck L., Makromol. Chem., 22, 103 (1957).
66. Zimmer H., H e с h e n b 1 e i k n e r J., HombergO., DanzikM.,J.
Org. Chem., 29, 2632 (1964).
67. KrauseE., WeinbergK., Ber., 63, 381 (1930).
68. P f e i f f e r P., H e 1 1 e r J. , Ber., 37, 4618 (1904).
69. Eberiy K.C., S m i t h G. E. P., A I b e r t у Н. Е., Пат. США 2560042 (1951);
С. A., 4Gy 1581 (1952),
70. С a s t e 1 P., GrasG., Trav. soc. pharm. Montpellier, 17, 205 (1957); С. А , 52, 18990
(1958).
71. К г а и s С A., CallisCC, Пат. США 1639947 (1926); С. А., 21, 3180 (1927).
72. Li pp incott E.R., Мег с i e г Р., Т о b i n M. C, J. Phys. Chem., 57, 939 (1953).
73. Sasin G. S., J. Org. Chem., 18, 1142 (1953).
74. KrauseE., P о h 1 a n d R., Ber., 57, 532 (1924).
75. M и р о нов В. Ф., Е г о р о в Ю. П., Петров А. Д., Изв. АН СССР, ОХН, 1953,
1400.
76. Охлобыстин О. Ю., Захаркин Л. И., Струнин Б. Н., Авт. свид.
СССР 144484 (1963); РЖХим., 1964, 7Н55П; Бюлл. изобрет., № 15, 123 (1963).
77. A n d e r s о п Н. Н., V a s t a J. A., J. Org. Chem., 19, 1300 (1954).
78. S a s i n G. S., S a s i n R., J. Org. Chem., 20, 387 (1955).
79. S о I e r i о A., Gazz., 81, 664 (1951).
80. W a 1 1 а с e J. M., Пат. США 2888435 (1959); РЖХим., 1961, 8П173.
81. V a n der К e r k G. J. M., N о t 1 e s J. G., Z u i j t e n J. G. A., J. appl. Chem.,
7, 366 (1957) ,
82. Da vies H., К i p p i n g F. S„ J. Chem. Soc, 99, 296 (1911).
83. КотонМ.М., Киселева Т. М., ЖОХ, 35, 2036 (1965).
84. В 6 e s e k e n J., R u t g e r s J. J., Rec. trav. chim., 42, 1017 (1923).
85. С h a m b e r s R. F., SchererP.C, J. Am. Chem. Soc, 48, 1054 (1926).
86. F a r r a r W. V., S k i n n e r H. A., J. Organometal. Chem., 1, 434 (1964).
87. P f ei f f er P., Truski er P., Ber., 37, 1125 (1904).
88. И о ф ф е С. Т., Несмеянов А. Н., Методы элементоорганической химии.
Магний, бериллий, кальций, стронций, барий. М., Изд-во АН СССР, 1963, стр. 29.
89. Normant H., Bull. Soc. Chim. France, 1959, 1764.
90. RosenbergS. DM G i b b о n s A. J., Ramsden H. E., J. Am. Chem. Soc,
79, 2137 (1957).
91. К о т о н М. М., К и с е л е в а Т. М., 3 а п е в а л о в а Н. П., ЖОХ, 30, 186 (1960).
92. К а е s z H. D., S t a f f о г d S. L., S t о n e F. G., J. Am. Chem. Soc, 82, 6232
(1960).
93. К о т о н М. М., К и с е л е в а Т. М., ЖОХ, 27, 2553 (1957).
94. V i j а у а г a g h a v a n К. V., J. Ind. Chem. Soc, 22, 135 (1945).
95. S е у f e r t h D., С о h e n H., Inorg. chem., 1, 913 (1962).
96. G i e 1 e n M., N a s i e 1 s k i J., J. Organometal. Chem., 1, 173 (1963).
97. GilmanH., G i s t L. A., J. Org. Chem., 22, 250 (1957).
98. G i I m a n H., G i s t L. A., J. Org. Chem., 22, 689 (1957).
99. Иоффе С. Т., Несмеянов А. Н., Методы элементоорганической химии.
Магний, бериллий, кальций, стронций, барий. М., Изд-во АН СССР, 1963, стр. 46.
100. Lesb re M., Roques G., Compt. rend. 78-e Congr. socs. savantes Paris et depts.,
Sect., sci., 1953; 423; С. А., 49, 15768 (1955).
101. KrauseE., G г о s s e A., Die Chemie der metallorganischen Verbindungen.
Berlin. Gebr. Borutrager. 1937, S. 319.
Синтез на основе производных четырехвалентного олова
263
102. Кочешков К. А., Синтетические методы в области металлоорганических
соединений. М., Изд-во АН СССР, 1947, стр. 38.
103. 3 а х а р к и н Л. И., О х л о б ы с т и н О. Ю., С т р у н и н Б. И., Изв. АН СССР,
ОХН, 1961, 2254.
104. FuchsO., P о s t H. W., Rec. trav. chim., 78, 566 (1959).
105. Lorenz D. H., ShapiroP., S t e r n А., В е с k e г E. J., J. Org. Chem.,
28, 2332 (1963).
106. HolmesJ.M, P e а с о с k R. D., TatlowJ.C, Proc. chem. Soc, 1963, 108.
106a. H ol m es J. M., P e а с о с k R. D., TatlowJ.C, J. Chem. Soc, 1966,
A, 150.
107. Pohlmann J.L, В r i n с k m a n F. E., Tesi G., D о n a d i о R. E., Z.
Naturforsch,, 20b, 1 (1965).
108. С h a m b e r s R. D., С h i v e r s Т., J. Chem. Soc, 1964, 4782. •
109. TamborskiC, SoloskiE. J., Stanley M., J. Organometal. Chem., 4,
446 (1965).
110 Chambers R. D., Chi vers Т., Proc. chem. Soc, 1963, 208.
111. Kr a use E., Weinberg K., Ber., 62,2235(1929).
112. Stern A., Becker E., J. Org. Chem., 29, 3221 (1964).
113. G r i f f i t h s V. S., D e r w i s h G. A., J. molec spectr., 7, 233 (1961).
114. Кочешков К. А., НадьМ.М., ЖОХ, 5, 1158 (1935).
115. L e s b r e M., Roues J., Bull. Soc. chim. France, 18, 490 (1951).
116. К r a u s e E., Becker R., Ber., 53, 173 (1920).
117. Srivostova T. N., В h a 11 а с h a r у a S. N., J. Inorg. Nucl. Chem.,28, 1480(1966).
118. Pf ei f f er P., LehnardtR, Luf tenstei пег Н., PradeR.,
Schnurmann K., TruskierP., Z. anorg. Chem., 68, 102 (1910).
119. Кочешков К. А., Надь М. М. ЖОХ, 4, 1434 (1934).
120. KrauseE., SchmitzM., Ber., 52, 2150 (1919).
121. M о r r i s H., В у e r 1 у W., S e 1 w о о d P. W., J. Am. Chem. Soc, 64, 1727 (1942).
122. NevilleG., Canad. J. Chem., 41, 814 (1963).
123. S m i t h T. A., Kipping F. S., J. Chem. Soc, 101, 2553 (1912).
124. Sisido K., Takeda Y., Nazaki H., J. Org. Chem., 27, 2411(1962).
125. L ui j t en J. G. A., van der KerkG. J.M., J. appl. Chem., 11, 35 (1961).
126. ЛапкинИ. И., СедельниковаВ. А., ЖОХ, 30, 2771 (1960).
127. BahrG, G e 1 i u s R., Ber., 91, 818 (1958).
128. QuintinC, Ing. Chim., 14, 205 (1930); С. А., 26, 2182 (1932).
129. П и к и н а Е. И., Т а л а л а е в а Т. В., К о ч е ш к о в К- А., ЖОХ, 8, 1844 (1938).
129а. R e i с h 1 W., Inorgan. chem., 5, 87 (1966).
130. Л а п к и н И. И., Д у м л е р В. А., ЖОХ, 34, 3690 (1964).
131. BahrG., GeliusR., Ber., 91,812(1958).
132. D r e f a h 1 G., P 1 о t n e r G., LorenzD, Angew. Chem., 72, 454 (1960).
133. Я к у б о в а Ф. А., Р а ш к е с А. М., КучкаревА. Б., МанулкинЗ. М.,
ЖОХ, 35, 387 (1965).
134. HartmannH., R e i s s W., К a r b s t e i n В., Z. Naturwiss., 46, 321 (1959).
135. KrauseE., R e n w a n z G., Ber., 60, 1582 (1927).
136. С о n s i d i n e W. J., V e n t u r a J. J., Chem. a. Ind., 1962, 1683.
137. ШостаковскийМ. Ф., Комаров Н. В., МисюнасВ. К-. Изв.
АН СССР, ОХН, 1962, 368.
138. А1 1 est on D. L., D a v i e s A. G., H a n с о с k M., White R. F., J. Chem.
Soc, 1963, 5469.
139. P о p e W. J., P e а с h e у S. J., Proc. Chem. Sos., 16, 42 (1900).
140. Pope W. J., P each ey S. J., Proc, Chem. Soc, 16, 116 (1900).
141. R a m s d e n H. E., Пат. США 3010979 (1961); РЖХим., 1963, 15H89.
142. GruttnerG, KrauseE., Ber., 50, 1802 (1917).
143. SeyferthD., Rochow E.G., J. Am Chem. Soc, 77, 1302 (1955).
144. S e у f e r t h D., J. Am. Chem. Soc, 81, 1844 (1959).
145. S e у f e r t h D., J. Org. Chem., 22, 478 (1957).
146. МанулкинЗ.М., ЖОХ, 13, 46 (1943).
147. P о p e W. J., P e а с h e у S. J., Proc. Chem. Soc, 19, 290 (1903).
148. Van der KerkG. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 56 (1956).
149. M а н у л к и н 3. М., ЖОХ, 13, 42 (1943).
150. SchmidbaurH., WaldmannS, Ber., 97, 3381 (1964).
151. R о s e n b e r g S. D., Debreczeni E., Weinberg E. L, J. Am. Chem.
Soc, 81, 972 (1959).
152. P e d 1 e у J. В., S k i n n e r H. A., Trans. Farad. Soc, 55, 544 (1959).
153. П е т р о в А. Д., МироновВ.Ф, ДолгийИ.Е, Изв. АН СССР, ОХН,
1956, 1146.
154. Е a b о г п С, W a t е г s J. A., J. Chem. Soc, 1962, 1131.
155. В u 1 1 а г d R. H., R о b i n s о n W. R., J. Am. Chem. Soc, 49, 1368 (1927).
156. KrauseE., R e n w a n z G., Ber., 62, 1710(1929).
264 Оловоорганические соединения
157. Huang H. H., Hui К. М., J. Organometal. Chem., 2, 288 (1964).
158. Засосов В. А., Кочешков К. А., Сборник статей по общей химии, т. 1.
М., Изд-во АН СССР, 1953, стр. 290. '
159. В о t t R. W., E a b о г п С, Р а п d е К. С, Swaddle Т. W., J. Chem.
Soc, 1962, 1217.
160. BuchmannO., Grosjean M., N a s i e 1 s k i J., Bull. Soc. Chim. Belg.,
71,467(1962).
161. S e у f e r t h D., J. Am. Chem. Soc, 79, 5881 (1957).
162. Костяновский Р. Г., Прокофьев А. К., Изв. АН СССР, ОХН, 1965,
175.
163. J о п е s W. J., Davies W.C., BowdenS.T., EdwardsC, Davis
V. E., T h о m a s L. H., J. Chem. Soc, 1947, 1446.
164. МанулкинЗ.М, ЖОХ, 14, 1047 (1944).
165. T и л л я е в К. С, МанулкинЗ.М., ДАН УзССР, 2, 31 (1960).
166. RamsdenH.E., Пат. США 2873287 (1959); РЖХим, 1961, 14Л114.
167. L е s b г е М., Buisson R., LuijtenJ.G., van der KerkG. J., Rec.
trav. chim., 74, 1056(1955).
168. Засосов В. А., Кочешков К. А., Сборник статей по общей химии, т. 1.
М., Изд-во АН СССР, 1953, стр. 278.
169. Sai t ow A., RochowE. G., SeyferthD., J. Org. Chem., 23, 116 (1958).
170. МанулкинЗ.М., ЖОХ, 16, 235 (1946).
171. МанулкинЗ.М., Якубова Ф. А., КучкаровА. Б., РашкесА. М.,
Узб. хим. ж., 6, 52 (1962).
172. SeyferthD., WeinerM.A., J. Am. Chem. Soc, 84, 361 (1962).
172a. M e r k e r R. L., S с о t t M. J., J. Am. Chem. Soc, 81, 975 (1959).
1726. Г а л а ш и н а Л. М., Казина Г. В., Соболевский М. В.,
Пластмассы, № 1, 26 (1966).
173. S t о п е F. G., Т г е i с h е 1 P. M., Chem. a. Ind., 26, 837 (1960).
174. Р у б и н ч и к Г. Ф., МанулкинЗ.М, ЖОХ, 34, 949 (1964).
175. К и 1 а М. R., AmbergerE., Mayer К., Вег., 98, 634 (1965).
176. СнегурЛ.Н, МанулкинЗ.М., ЖОХ, 34, 4030 (1964).
177. К i p p i n g F. В., J. Chem. Soc, 1928, 2365.
178. СнегурЛ.Н., Манулкин 3. М. Узб. хим. ж., 5, 49 (1961).
179. К u i v i 1 а Н. G., Organometal. Chem., 1, 47 (1964).
180. F u с h s R., GilmanH, J. Org. Chem., 22, 1009 (1957).
181. R a m s d en H. E., Пат. США 2965661 (1960); РЖХим., 1962, 2Л105.
182. GilmanH., E i s с h J., J. Org. Chem., 20, 763 (1955).
183. GilmanH., G i s t L. A., J. Org. Chem. 22, 368 (1957).
184. GilmanH., ArntzenC, J. Am. Chem. Soc, 72, 3823 (1950).
185. T am b or ski G., FordF. E., LehnW.L, MooreG.J, Soloski
E. J., J. Org. Chem., 27, 619 (1962).
186. Ш о с т а к о в с к и й М. Ф., К о т р е л е в В. Н., К о ч к и н Д. А., Кузне-
цоваТ. И., Кал и нина С. П., Бор исенко В. В., ЖПХ, 31, 1434 (1958).
187. М е г k e r R. Z., Пат. США 3043858 (1962); РЖХим., 1964, 7Н60П.
188. М е г к е г R. L., Пат. США 2956045 (1960); РЖХим., 1962, 17П145.
189. GruttnerG., KrauseE., Ber., 50, 1559 (1917).
190. Bott R.W., EabornC, Walton D., J. Organometal. Chem., 2, 154 (1964).
191. BeermannC, HartmannH, Z. anorg. allgem. Chem., 276, 20 (1954).
192. H a r t m a n n H., HonigH., Angew. Chem., 69, 614 (1957).
193. SeyferthD., W e i n e r M. A., Chem. a. Ind. (London), 1959, 402.
194. ZimmerH, Angew. Chem., 65, 347 (1953).
195. ZimmerH., MosleH.G, Ber., 87, 1255 (1954).
196. РубинчикГ.Ф, МанулкинЗ.М., ЖОХ, 36, 748 (1966).
197. S о 1 e r i о A., Gazz., 85, 61 (1955).
198. Несмеянов А. Н., Борисов А. Е., Абрамова А. Н., Изд. АН СССР,
ОХН, 1946, 647.
199. К о л е с н и к о в Г. С, Д а в ы д о в а С. А., ЖОХ, 29, 2042 (1959).
200. ФрейдлинаР. X., Мартиросян Г. Т., Несмеянов А. Н., ДАН
СССР, 137, 1129 (1961).
201. М и р о н о в В. Ф., П е т р о в А. Д., М а к с и м о в а Н. Г., Изв. АН СССР,
ОХН, 1959, 1954.
202. ШостаковскийМ. Ф., Комаров Н. В., МисюнасВ. К., С к л я-
н о в а А. М., ДАН СССР, 161, 370 (1965).
203. ШостаковскийМ. Ф., Комаров Н. В., МисюнасВ. К., Заинч-
к о в с к а я М. К., Изв. АН СССР, ОХН, 1964, 1102.
204. G i b b о n s A. J., Rosenberg S. D., Пат. США 2962522 (1960); РЖХим, 1962,
11Л20.
205. S m о 1 i n E. M., Tetrahedron L., 1961, 143.
206. SeyferthD., Brandle K., R a a b G.f Angew. Chem., 72, 77 (1960).
Синтез на основе производных четырехвалентного олова
265
207. К а е s г Н. D., S t a f f о г d S. L., StoneF.G., J. Am. Chem. Soc, 81, 6336
(1959).
208. SeyferthD., RaabG., BrandleK., J. Org. Chem., 26, 2934 (1961).
209. PetrowA.D, MironowV.F., Angew. Chem , 73, 59 (1961).
210. SchwartzW., PostH.W., J. Organometal. Chem., 2, 357 (1964).
211. ТилляевК.С, МанулкинЗ.М, Узб. хим. ж., 5, 73 (1961).
212. Т и л л я е в К. С, МанулкинЗ.М, ДАН УзбССР, 1965, 45.
213. Т и л л я е в К- С, МанулкинЗ.М, Узб. хим. ж, 1963, 47.
214. SeyferthD., WeinerM., J. Org. Chem, 26, 4797 (1961).
215. Van d e г К e r k G. J. M, N о 1 t e s J. G, J. appl. Chem, 9, 106 (1959).
216. Бай Л. И, Якубович А. Я, М у л е р Л. И, ЖОХ, 34, 3696 (1964).
217. SeyferthD, VaughanL, J. Organometal. Chem, 1, 138 (1963).
218. LeQuanM, CadiotP, Bull. Soc. chim. France, 1965, 35.
219. LeQuanM, В i 1 1 i о t t e J. C, CadiotP, Compt. rend, 251, 730 (1960).
220. LeQuanM, CadiotP. N, Bull. Soc. chim. France, 1965, 45.
221. L e QuanM, CadiotP, Compt. rend, 254, 133 (1962).
222. AufdermarshC. A., J. Org. Chem, 29, 1994 (1964).
223. Ш о с т а к о в с к и й М. Ф, В л а с о в В. М, М и р с к о в Р. Г, ЖОХ, <33,
324 (1963).
224. ЗавгороднийВ. С, Петров А. А, ЖОХ, 32, 3527 (1962).
225. Петров А. А, ЗавгороднийВ. С, ЖОХ, 30, 1055 (1960).
226. HartmannH, HonigH., Angew. Chem, 70, 75 (1958).
227. HartmannH, EschenbachW., Naturwiss, 46, 321 (1961).
228. ЗавгороднийВ. С, Петров А. А, ДАН СССР, 143, 855 (1962).
229. ШостаковскийМ. Ф, Комаров Н. В, М и с ю н а с В. К-, Авт. свид.
СССР 163176 (1964); РЖХим, 1965, 7Н55Н; Бюлл. изобрет, № 12 (1984).
230. М а с k G. Р, Р а г к е г Е, Пат. США 2604483 (1952); С. А, 47, 4358 (1953).
231. GilmanH, Rosenberg S. D., J. Org. chem, 18, 680 (1953).
232. V a n d e г К e г к G. J. M, L u i j t e n J. G. A, Noltes J.G, Chem. a. Ind.
(London), 1956, 352.
233. П у ч и н я н Е. А, МанулкинЗ.М, ДАН УзбССР, 1961, 51.
234. GilmanH, Rosenberg S. D., J. Org. Chem, 18, 1554 (1953).
235. В a i 1 i e J. С, Iowa State Coll, J. Sci, 14, 8 (1939); С. А, 34, 6241 (1940).
236. С h a t t J., W i I 1 i a m s A. A, J. Chem. Soc, 1954, 4403.
237. GilmanH, W u T. C, J. Am. Chem. Soc, 77, 3228 (1955).
238. LeebrickJ.R., RamsdenH.E, J. Org. Chem, 23, 935 (1958).
239. КотонМ.М, К и с е л е в а Т. М, Ф л о р и н с к и й Ф. С, Изв. АН СССР,
ОХН, 1959, 948.
240. КотонМ.М, К и с е л е в а Т. М, Флоринский Ф.С, Высокомолекул.
соед, 2, 1639 (1960).
241. КотонМ.М, К и с е л е в а Т. М, Ф л о р и н с к и й Ф. С, Международный
симпозиум по макромолекулярной химии, секция 1, М, 1960, стр. 167.
242. GreberG., E g 1 е G, Makromolek. chem, 64, 207 (1963).
243. N ol t es J. G, Budding H. A., van der KerkG.J.M, Rec. trav.
chim, 79, 408 (1960).
244. КотонМ.М, ДокукинаЛ.Ф, ЖОХ, 34, 1757 (1964).
245. ПетровА.Д, ЧернышевЕ.А, Краснова Т. Л., ДАН СССР, 140,
837 (1961).
246. N о 1 t e s J. G, В u d d i n g H. A, van der Kerk G. J.M, Rec. trav. chim,
79, 1076 (1960).
247. КоршакВ.В, Полякова А. М, ТамбовцеваЕ. С, Изв. АН СССР,
ОХН, 1959, 742.
248. Полякова А. М, КоршакВ.В, ТамбовцеваЕ. С, Высокомолек.
соед, 3, 662 (1961).
249. Т и л л я е в К. С, МанулкинЗ.М, ДАН УзССР, 3, 45 (1961).
250. П у ч и н я н Е. А, МанулкинЗ.М, ДАН УзССР, 3, 49 (1962).
251. SeyferthD, VaughanL, Suzuki R., J. Organometal. Chem, 1, 437
(1964).
252. GilmanH, GoreanT.N, J. Org. Chem, 17, 1470 (1952).
252a. ZimmerH, M i 1 1 e r J, Naturwiss, 53, 38 (1966).
253. В u 1 1 a r d R. H, H о 1 d e n F. R, J. Am. Chem. Soc, 53, 3150 (1931).
254. БобашинскаяЧ.С, Кочешков К. А, ЖОХ, 8, 1850 (1938).
255. JohnsonO.H, FritzH.E., J. Org. Chem, 19, 74 (1954).
256. SeyferthD, J. Org. Chem, 22, 1599 (1957).
257. WestR, WebsterM.H, WilkinsonG, J. Am. Chem. Soc, 74, 5794
(1952).
258. Anderson H. H, Inorg. Chem, 1, 647 (1962).
266
Оловоорганические соединения
258а. С л а д к о в А. М., Л у н е в а Л. К., ЖОХ, 36, 553 (1966).
259. Я к у.б о в и ч А. Я., М а к а р о в С. П., Г а в р и л о в Г. И., ЖОХ, 22, 1788
(1952).
260. Я к у б о в и ч, А. Я., Макарове. П., ГинсбургВ. А.,
Таврило в Г. И., М е р к у л о в а Е. Н., ДАН СССР, 72, 69 (1950).
261. CaujolleF.,Lesbre М., В г u A., Meynier D., В г и J., Compt. rend.,
240, 1829 (1955).
261а. Р у б и н ч и к Г. Ф., Манулкин З.М., ЖОХ, 36, 261 (1966).
262. S е у f e r t h D., J. Am. Chem. Soc, 79, 2133 (1957).
263. HenryM. C, NoItesJ.G., J. Am. Chem. Soc, 82, 555 (1960).
264. GeliusR., Ber., 93, 1759 (1960).
264a. P у б и н ч и к Г. Ф., Манулкин З.М., ЖОХ, 36, 1301 (1966).
265. К u i v i 1 а Н., В е u m е 1 О. F., J. Am. Chem. Soc, 80, 3250 (1958).
266. IbekweS.D, N e w 1 a n d s M. J., J. Chem. Soc, 1965, 4608; РЖХим, 1966
5Ж373.
267. H a r t m a n n H., KarbsteinB., Schaper P., Reiss W., Natur
wiss., 50, 373 (1963).
268. SeyferthD., R о с h о w E. G., J. Org. Chem., 20, 250 (1955).
269. GriittnerG., KrauseE., WiernikM., Ber., 50, 1549 (1917).
270. LeusinkA.J., NoItesJ.G., Buddi ng H. A., van d e г KerkG.J.
M., Rec trav. chim., 83, 609 (1964).
271. SeyferthD., Alleston D. L, Inorg. Chem., 2, 417 (1963).
272. A u s t i n P. R., J. Am. Chem. Soc, 54, 3726 (1932).
273. MeenR.H, G i 1 m a n H., J. Org. Chem., 22, 564 (1957).
274. T а л а л а е в а Т. В., К о ч е ш к о в К- А., ЖОХ, 12, 493 (1942).
275. ZimmerH., Sparmann H. W., Naturwiss., 40, 220 (1953).
276. Zi mmer H., SparmannH.W, Ber., 87,645(1954).
277. SeyferthD., С о h e п Н., Inorg. Chem., 2, 625 (1963).
277a. SchollkonfU., Traenckner H.-J., J. Organometall. Chem., 5, 300 (1966).
278. H a r t m a n n H., Meyer K., Naturwiss., 52, 303 (1965).
279. F r i t z H. P., KreiterCG., J. Organometall. Chem., 4, 313 (1965).
280. S r i v a s t a v a T. N., TandonS. K, Indian J. Appl., Chem., 26, 171 (1963).
281. Poller R.C., J. Chem. Soc, 1963, 706.
282. Zakharkin L.J., BregadzeV. J., OkhlobystinO. Ju., J.
Organometall. Chem., 4, 211 (1965).
283. Leavitt F.C., ManuelT.A., JohnsonF., J. Am. Chem. Soc, 81, 3163
(1959).
284. Leavitt F.C., ManuelT.A., JohnsonF., Matternas L.U.,
L e h m a n n D. S., J. Am. Chem. Soc, 82, 5099 (1960).
285. Leavitt F. C, Johnson F., ГШ. США 3116307 (1963); РЖХим., 1965, 18H117.
286. GilmanH., Z u e с h E. A., J. Am. Chem. Soc, 82, 2522 (1960).
287. BachmanG. В., Carlson С. L., Robinson M., J. Am. Chem. Soc,
73, 1964 (1951).
288. H ar t m a n n H., NiemollerH., ReissW., KarbsteinB.,
Naturwiss., 46, 321 (1959).
289. GilmanH., MooreF.W., JonesR.G., J. Am. Chem. Soc, 63, 2482 (1941).
290. GilmanH., A r n t z e n С E., J. Org. Chem., 15, 994 (1950).
291. W о о d s L. A., GilmanH., Proc Jowa Acad. Sci., 48, 251 (1941); С. А., 36, 3492
(1942).
292. Несмеянов А. Н., Соколик Р. А., Методы элементоорганической химии.
Бор, алюминий, галлий, индий, таллий. М., «Наука», 1964, стр. 351.
293. GilmanH., Bull. Soc chim. France, 1963, 1356.
294. J u e n g e E. С, S e у f e r t h D., J. Org. Chem., 26, 563 (1961).
295. SeyferthD., WeinerM.A, J. Am. Chem. Soc, 83, 3583 (1961).
296. SeyferthD., WelchD, R a a b G., J. Am. Chem. Soc, 84, 4266 (1962).
297. ВязанкинН.С, РазуваевГ.А, К о р н е в а С. П., ЖОХ, 34, 2787
(1964).
298. SeyferthD., S a r a f i d i s С, E v n i n А. В., J. Organometal. Chem., 2, 417
(1964).
299. GilmanH., Rosenbergs., J. Am. Chem. Soc, 74, 5580 (1952).
300. Цзэн Чжао-лунь, ДунШи-хуа, ЧжанГ о-м инь. Kexue tongbao.,
2, 165 (1964); РЖХим., 1965, 7Ж322.
301. С о р о к и н а Р. С, Панов Е. М.Дочешков К. А., ЖОХ, 35, 1625 (1965).
302. К о ч е ш к о в К. А., Панов Е.М., Сорокина Р.С., Изв. АН СССР, ОХН,
1961, 532.
303. Hart mann H., Wagner H., Karbstein В., el A'ssarM. К.,
ReissW, Naturwiss., 51, 215 (1964).
Синтез на основе производных четырехвалентного олова 267
304. ЗахаркинЛ. И., БрегадзеВ. И., ОхлобыстинО. Ю., Изв. АН
СССР, ОХН, 1964, 1539.
305. G е 1 i u s R., Angew. Chem., 72, 322 (1960).
306. M a s s e у A. G., Randall E. W., ShawD, Chem. a. Ind., 30, 1244 (1963).
307. WestlakoA.H., MartinD. F., J. Inorg. Nucl. Chem., 27, 1579 (1965).
308. HartmannH., BeermannC, Пат. ФРГ 1062244 (1960); РЖХим., 1961,
24Л101.
309. BajerF., P о s t H., J. Org. Chem., 27, 1422 (1962).
310. Адрова Н. А., Котон М. М., Прохорова Л. К., Сб.
«Высокомолекулярные соединения. Гетероцепные высокомолекулярные соединения». М., «Наука»,
1963, стр. 9.
311. НепгуМ.С, N о t 1 es J. G., J. Am. Chem. Soc, 82, 561 (1960).
312. F г e e d m a n H. H., J. Org. Chem., 27, 2298 (1962).
313. KupchikE.J., UrsinoJ.A., Chem. a. Ind., 1965, 794.
314. MassonJ.C, Q u a n M. L., Chodkiewicz W., С a d i о t P., Compt.
rend., 257, 1111 (1963).
315. DepreeD., Пат. США 3161664 (1964); РЖХим., 1966, 11Н108П.
316. Van d e r KerkG. J. M.( Luijten J.G.A., J. appl. Chem., 4, 301 (1954).
317. P a n a i t e s с u M., PaltinE, Rev. с h i m. (RPR), 13, 724 (1962); РЖХим.,
1963, 24T361.
318. II e с h e n b 1 e i k n e г J., Molt К., Пат. США 3059012 (1962); РЖХим., 1964,
11H97.
319. T э н М и н-д а. РЖХим., 1960, 77427.
320. К u s с h k R., Пат. ГДР 20270 (1960); РЖХим., 1961, 24Л119.
321. G 1 о s к е у С. R. Пат. США 2805234 (1957); С. А., 52, 2050 (1958).
322. К а ц у м у р а Т., J. Chem. Soc. Japan, 83, 727 (1962); РЖХим., 1963, 16Ж254.
323. TabernD.L, О г n d о г f f W. R., Denni'sL.M., J. Am. Chem. Soc, 47,
2039 (1925).
324. HarrisJ.O., Пат. США 2431038(1947); С. А., 42, 1606(1948).
324a. Reindl E.f В о i d о 1 К., Пат. ФРГ 1027669 (1966); РЖХим., 1936, 2HI90.
325. Герм. пат. 508667 (1930); С. А., 25, 713 (1931).
325а. S r i v a s t a v а Т. N.. В h a t t а с h а г у a S. N., Z. anorg. allgem. Chem.,
344, 1021(1966).
326. J e п к ri e г Н., Пат. ФРГ 1152106 (1964); РЖХим., 1965, 8Н102П.
327. Ladenburg A., Ann., 159,251(1871).
328. Ladenburg A., Ber., 4, 17 (1871).
329. John so н E. W., Church J. M., Пат. США 2570686 (1951); С. А., 46, 5084,
(1952).
330. Кацумура Т., СудзукиТ., Японск. пат. 13374 (1963); РЖХим., 1965,
15Н83.
331. V i e h e H. G., Franchi mont E., Ber., 95, 319 (1962).
332. П е т р о в А. Д., М и н г а л е в а К. С, 3 а в г о р о д н и й В. С, ЖОХ, 34,
533 (1964).
333. ШостаковскийМ. Ф., Власов В. М., МирсковР. Г.,
Петрова В. Н., ЖОХ, 35, 47 (1965).
334. West R., Krai ha nz el С, Inorg. Chem., 1, 967 (1962).
335. ЗавгороднийВ. С, Петров А. А., ЖОХ, 35, 760 (1965).
336. Комаров Н. В., ШостаковскийМ. Ф., Гусева И. С, Авт. свид.
СССР, 148409 (1962); РЖХим., 1963, 9Н44; Бюлл. изобрет., № 16 (1962).
337. HartmannH., D i e t z E., KomorniczykK., ReissW., Naturwiss.,
48, 570 (1981).
338. HartmannH., К a r b s t e i n В., Reiss W., Naturwiss., 52, 59(1965).
339. J e n s e n К. А., С 1 a u s о n-K a s s N., Z. anorg. allgem. Chem., 250, 277 (1943).
340. M а с k G. P., ParkerE., Flat. ФРГ 939028 (1956); РЖХим., 1957, 67182.
340a. F б 1 d e s i I., G б m б г у P., Acta chim. Acad, scient. hung., 45, 231 (1965).
341. В u с k t о n G. В., Ann., 109, 218 (1859).
342. F r a n k 1 a n d E., Ann., 85, 329 (1853).
343. Frankland E., Ann., Ill, 54 (1859).
344. В u с k t о n G. В., Ann., 112, 220 (1859).
345. F r a n k 1 a n d E., L a w r e n с e A., J. Chem. Soc, 35, 130 (1879).
346. С a h о u r s A., Ann., 122, 48 (1862).
347. 3 a x a p к и н Л. И., О х л о б ы с т и н О. Ю., ДАН СССР, 116, 236 (1957).
348. 3 а х а р к и н Л. И., О х л о б ы с т и н О. Ю., Изв. АН СССР, ОХН, 1959, 1942.
349. 3 а х а р к и н Л. И., Охлобыстин О.Ю., С т р у н и н Б. Н., ЖПХ, 36,
2034 (1963).
350. 3 а х а р к и н Л. И., Охлобыстин О. Ю, ЖОХ, 31, 3662 (1961).
351. Англ. пат. 802796 (1958); С. А., 53, 9061 (1959).
352. Neumann W. P., Ann., 653, 157 (1962).
268
Оловоорганические соединения
353. М а ц уд а С, МацудаХ., Я м а н э Я., Polymer Applic, 13, 312(1964),
РЖХим., 1965, 11С330.
354. Англ. пат. 812787 (1959); С А., 53, 19879 (1959).
355. Dickson R.S., West B.O., Austral. J. Chem., 15, 710 (1962).
356. J e п к n e г Н., Пат. ФРГ 1153367 (1964); РЖХим., 1965, 4Н53П.
357. ManghamJ.R., Пат. США 3095433 (1963); РЖХим., 1965, 6Н87П.
358. Anderson R., Англ. пат. 871730 (1961); РЖХим., 1962, 6Л113.
359. Johnson W.( J. Org. Chem., 25, 2253 (1960).
360. Johnson W., Пат. США 3036103 (1962); РЖХим., 1964,8Н107П.
361. Takeda Y., Okuyama Т., Fueno Т., Furukawa J., Makromol.
Chem., 76, 209 (1964).
362. Van Egmond J.C., Janssen M. J., Lui j t en J. G. A., van d e г
KerkG.J.M., van d e r W a n t G. M., J. appl. Chem., 12, 17 (1962).
363. J e n к n e r H., S с h m i d t H. W., Пат. ФРГ 1048275 (1959); С. А., 1960, 19487a.
364. N e u m a n n W. P., NiermannH., S о m m e r R., Ann., 659, 27 (1962).
365. N e u m a n n W. P., Chem. Labor, und Betrieb., 15, 363 (1964).
366. P о 1 s t er R., Ann., 654, 20 (1962).
367. P о 1 s t e r R., Пат. ФРГ 1156807 (1964); РЖХим., 1966, 8H107.
367a. WittenbergD., Пат. ФРГ 1124947 (1965); РЖХим., 1966, 18Н166.
368. WittenbergD., Ann., 654, 23 (1962).
369. N e u m a n n W. P., NiermannH., Ann., 653, 164 (1962).
370. JohnsonO.H., H о 1 u m J. R., J. Org. Chem., 23, 738 (1958).
371. Ar onhei m В., Ann., 194,145(1878).
372. G od d ar d A. E., Ash 1 ey J. N., E v a n s R. В., J. Chem. Soc, 121,978(1922)
373. GoddardD, G о d d a r d A. E., J. Chem. Soc, 121, 256 (1922).
Г лава IV
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ВЗАИМОДЕЙСТВИЕМ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ОЛОВОМ, ЕГО СПЛАВАМИ ИЛИ ОЛОВООРГАНИЧЕСКИМИ
СОЕДИНЕНИЯМИ СО СВЯЗЬЮ ОЛОВО —ОЛОВО
Металлоорганические соединения лития, цинка или ртути реагируют с
оловом или его сплавами с образованием R4Sn или оловоорганических га-
логенидов. Реакцию обычно проводят в эфире или углеводородах. Так, при
нагревании эфирных растворов фениллития с тонкоизмельченным оловом
образуется тетрафенилолово (выход 19,6%). Значительно лучше выходы в
случае применения амальгамы олова [1, 21.
Получение тетрафенилолова из фениллития и амальгамы олова [1, 2]. Кипячением 4,76 2
фениллития в 50 мл эфирного раствора с амальгамой олова [6,7 г (0,056 моля) олова и 22 г
(0,11 моля) ртути] в течение 25 час. получают 4,1 г тетрафенилолова с т. пл. 225° С. Выход
67,7%. Увеличение количества олова в амальгаме снижает выход.
Соединения типа М [R4A1] (гдеМ — в основном щелочной металл)
действуют аналогично. Например, нагреванием до 50—60° С в течение 1,5 час.
Na[(C2H5)4All и мелкораздробленного олова в толуоле получают тетраэтил-
олово и триэтилалюминий [3].
Тетраэтилолово образуется также при нагревании до 150° С йодистого
этил цинка с порошком олова [41.
Взаимодействие ртутноорганических соединений с металлическим оловом
или сплавом олово—натрий приводит к оловоорганическим соединениям
типа R4Sn и R3SnX. Последние образуются, вероятно, согласно следующим
реакциям [51:
6C6H,HgCl + 3 Sn -> 3 (C6H5)2SnCl2 + 6 Hg,
3 (C6H5)2SnCl2 -* 2 (C6Hs)8SnCl + SnCl4,
SnCl4 + Sn -* 2 SnCl2.
Получение хлористого три-я-толилолова [5]. 3,3 г (0,01 моля) хлористой я-толил-
ртути и 8,5 г металлического олова в порошке нагревают до кипения в 20 мл ксилола
п течение 18 час. Ксилольный раствор выпаривают досуха и обрабатывают небольшим
количеством петролейного эфира. Выделяются кристаллы, которые после
перекристаллизации из спирта имеют т. пл. 97—98° С. Экстракцией металлического остатка
бензолом получают еще некоторое количество монохлорида. Общий выход 44%.
Аналогичным образом получают хлористое трифенилолово (47%),
хлористое трибензилолово (71%), хлористое три-п-карбэтоксифенилолово
(выделено в виде соответствующего сульфида) [5—7], бромистое три-о-то-
л ил олово, бромистое три-ж-трифторметилфенил олово [81.
Тетраарильные оловоорганические соединения получены при
взаимодействии сплава олово — натрий с RHgX:
6 C6H3HgCl + 3 Na2Sn -* 3 (С6Н ,)2Sn + 6 NaCl -f 6 Hg,
3 (C6H5)2Sn -» (C6H5)3Sn—Sn (C6H5)8 + Sn,
(C6H5)3Sn — Sn (C6H5)3 -* (CeH5)4Sn + (C6H5)2Sn.
270
Оловоорганические соединения.
Получение тетра-я-толилолова [б]. 2,5 г хлористой я-толилртути и 7,5 г сплава олово —
натрий (15% натрия) нагревают до кипения в 20 мл ксилола в течение 18 час. Из
сконцентрированного ксилольного раствора выделяются кристаллы в виде игл (вес 0,12 г). Из
бензольного раствора, полученного после экстракции остатка сплава, выделяют еще 0,25 г
этого же вещества. После перекристаллизации из хлороформа т. пл. 236—237° С. Выход
тетра-я-толилолова 41%, т. пл. 238° С.
Таким же образом получены тетрафенилолово (51%) и тетра-д-хлорфе-
нилолово [5]. Главным затруднением при этой методике является очистка
от примеси гексаарилдистаннана.
При переходе хлорвинильного радикала от атома ртути к атому олова
его геометрическая конфигурация не изменяется. Так, например, из
хлористой тр^шс-р-хлорвинилртути и сплава олово — натрий в бензольном
растворе образуется хлористое три-траж>Р-хлорвинилолово [9].
4 CHCl=CHHgCl + NaSn -» (C1CH=CH)3 SnCl + СН=СН + 4 NaCl + 4 Hg.
Взаимодействие хлористой транс-fi-хлорвинилртути со сплавом олово—натрий [9].
8 цилиндрическую трехгорлую колбу, снабженную газовводом, ртутным затвором и
холодильником, закрытым хлоркальциевой трубкой, вводят 19 г хлористой трш-/с-|3-хлорвинил-
ртути и 30 мл сухого бензола. В раствор добавляют 25 г сплава олово — натрий (с
содержанием 15% натрия). Реакционную смесь нагревают при 45—48° С в токе водорода в течение
3 час. при энергичном перемешивании. Реакция начинается быстро, образуется объемистый
серый осадок, и происходит легкое разогревание. По окончании реакции еще теплую массу
отфильтровывают от избытка сплава и выделившейся металлической ртути, шлам
промывают на фильтре сухим бензолом. Этот раствор соединяют с фильтратом и полностью
отгоняют растворитель в вакууме. Сухой остаток дважды перекристаллизовывают из петролей-
ного эфира (т. кип. 50—75° С). Получают 2,9 г (40%) хлористого три-/?7раж:-|3-хлорвинил-
олова с т. пл. 120—121° С.
В аналогичных условиях выход хлористого три-^ш>Р-хлорвинилолова
составляет 58%; т. кип. 119,5° С/1 мм. Образования гекса-Р-хлорвинилди-
станнана не наблюдается. Течение реакции, следовательно, иное, чем в
случае ароматических соединений [91.
Нагреванием ди(перфторфенил)ртути с оловом при 260° С в течение
9 дней получено тетра(перфторфенил)олово (C6F5)4Sn [10].
К реакциям получения несимметричных четырехзамещенных оловоорга-
нических соединений типа R3SnR' или R2SnR2' относятся следующие:
R2Hg + 2 (С2Н5)3 Sn-Sn (C2H5)8 -» 2 RSn (C2H5)3 + Hg,
R2Hg + [(С2Н5)2 Sn]л - R3Sn (C2H5)2 + Hg.
Этим путем могут быть получены оловоорганические соединения с
оксигруппой или замещенной аминогруппой.
Получение триэтил-я-диметиламинофенилолова (CH3)2NC6H4Sn(C2H5)3 [И, 12]. 8,24 г
(0,02 моля) гексаэтилдистаннана и 8,8 г (0,02 моля) ди-я-диметиламинофенилртути
нагревают сначала полчаса на кипящей водяной бане, затем 5 час. до 150—170° С на масляной
бане. Ртуть выделяется почти количественно. Слитую с металла жидкость подвергают
фракционированию в вакууме при 3 мм. Собраны следующие фракции: 1) 140—145° С, 2) 145—
160° С и 3) 160—175° С. При дальнейшей разгонке выделяют триэтил-я-диметиламинофенил-
олово с т. кип. 172—173° С/3 мм.
Таким же образом получены триэтилфенилолово и триэтил-о-оксифенил-
олово [11, 12].
Получение о-оксифенилтриэтилолова [И, 12]. 16,5 г (0,04 моля) гексаэтилдистаннана
нагревают на масляной бане до 130° С. в течение 2 час. с 15,4 г (0,04 моля) б^'с-о-оксифенил-
ртути. Выделение ртути протекает еще легче, чем в случае аминосоединения, и значительно
быстрее и легче, чем для дифенилртути. Оно проходит почти количественно. Жидкую
массу сливают с металлической ртути, разгоняют при 3 мм. Собирают фракции: 1) 140—190СС,
2) 190—195° С и 3) 195—200° С. При новом фракционировании выделяют фракцию 197—
200° С/3 мм, являющуюся практически чистым о-оксифенилтриэтилоловом и составляющую
Синтез взаимодействием металлоорганических соединений с оловом 271
главную часть продукта реакции. Вещество представляет собой жидкость (d^5 1,3239; п2^
1,5377) с характерным слабым запахом. Оно растворимо во всех органических
растворителях и в водной щелочи, но не в воде. Раствор щелочи скоро разлагается с выделением
твердых продуктов.
Получение диэтилдифенилолова [И, 12]. 8,5 г (0,05 моля) диэтилолова нагревают в
течение получаса на масляной бане с 17 г (0,05 моля) дифенилртути до 150° С. За это время
выделяется 7,5 г металлической ртути (75%). Реакционную массу по охлаждении обрабатывают
петролейным эфиром, paciBop отделяют от ртути, растворитель отгоняют и остаток
фракционируют; т. кип. 154—156° С/4 мм\ выход 63%.
Стереоизомеры ди-р-хлорвинилртути с гексаэтилдистаннаном реагируют
иначе. Выделены лишь хлористое триэтилолово, ацетилен и металлическая
ртуть [9].
Эфиры триалкилстаннилуксусной кислоты получены нагреванием
дистаннанов с эфнрами мер кур-бас-уксусной кислоты [13].
(С2Н5)3 Sn—Sn (C2H5)3 + Hg (CH2COOR)2 -> 2 (С2Н5)3 SnCH2COOR + Hg.
Получение метилового эфира триэтилстаннилуксусной кислоты [13]. 10,5 г гексаэтил-
дистаннана и 8,7 г метилового эфира меркур-б^с-уксусной кислоты нагревают 1 час при
160°С в атмосфере азота и декантируют с ртути (3,4 г\ 68%). При разгонке выделяют 5,6 г
(41%) метилового эфира триэтилстаннилуксусной кислоты с т. кип. 69—71° С/1,8 мм; п2£
1,4835.
ЛИТЕРАТУРА
1. Т а л а л а е в а Т. В., К о ч е ш к о в К. А., ЖОХ, 12, 493 (1942).
2. Т а л а л а е в а Т. В., К о ч е ш к о в К. А., ЖОХ, 8,Л831 (1938).
3. RobinsonG., Пат. США 3057894 (1962); РЖХим., 1964, 15Н68П.
4. L e t t s Е. А., С о 1 1 i e N., Proc. Chem. Soc, 2, 166 (1986).
5. Н а д ь М. М., К о ч е ш к о в К. А., ЖОХ, 8, 42 (1938).
6. К о ч е ш к о в К. А., Надь М. М., А л е к с а н д р о в А. П., ЖОХ, 6, 1672
(1936).
7. Kozeschkow K.A., NadjM.M., Al exandrow A. P., Ber., 67, 1348
(1934).
8. S t e r n А., В е с k e r E. I., J. Org. Chem., 29, 3221 (1964).
9. H e с м е я н о в А. Н., Борисов А. Е., А б р а м о в а А. Н., Изв. АН СССР,
ОХН, 1949, 570.
10. В u r d о n J., CoeP. L, FultonM., J. Chem. Soc, 1965, 2094.
11. Несмеянов А. Н., К о ч е ш к о в К. А., Пузырева В. П., ЖОХ, 7,118
(1937).
12. Kozeschkow К. А., N е s m e j a n о w A. N., P u s у г е w a W. Р., Вег.,
69, 1639 (1936).
13. Л у ц е н к о И. Ф., Б а у к о в Ю. И., X а с а н о в Б. Н., ЖОХ, 33, 2724 (1963).
Г лава V
ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПО КРАТНЫМ СВЯЗЯМ
Присоединение оловоорганических соединений по кратным связям
является простым и препаративно удобным методом синтеза различных
производных олова. В реакции присоединения вступают оловоорганические
гидриды, алкоксисоединения, станииламины, гексаалкилдистанноксаны, а
также соединения типа R3Sn — SnR3 и (R2Sn)n. Вторым компонентом могут
служить олефины, алкины или органические и неорганические вещества,
содержащие группы ^:С=0, ^C=S, ^C=N—, —N=-О, —N=N— и др.
Наиболее подробно эти реакции изучены на примере
оловоорганических гидридов. Последние легко присоединяются к олефинам и алкинам с
образованием новой связи олова с углеродом. В ряде случаев эти реакции
проходят самопроизвольно, но обычно требуется нагревание смеси до 60—
100° С в течение нескольких часов или применение катализаторов.
Преимущество этого метода перед синтезом через магнийорганические и другие
подобные соединения заключается в том, что он позволяет получать олово-
органические соединения с различными функциональными группами в
алифатическом радикале у атома олова.
Взаимодействием оловоорганических гидридов с альдегидами и кетонами
получены соответствующие алкоксисоединения, а в реакциях с азометином,
азо- и нитрозосоединениями, изоцианатами и изотиоцианатами— соединения
со связью Sn—N.
Соединения типа R3SnSnR3 и (R2Sn)n присоединяются по
фторированным двойным и тройным углерод-углеродным связям, но эти реакции
почти не изучены.
Алкоксисоединения олова [11 и станииламины [2] не реагируют с оле-
финами (например,. со стиролом) при длительном нагревании смеси, но они
легко присоединяются к ацетатам энолов и кетену, давая а-металлирован-
ные кетоны, сложные эфиры и NjN-диалкиламиды триалкилстаннилуксусной
кислоты.
Гексаалкилдистанноксаны, алкоксисоединения олова и станииламины
экзотермически реагируют с изоацианатами, изотиоцианатами,
альдегидами, нитрилами, двуокисью серы и карбодиимидами. Это направление
развивается в работах Дэзиса и сотр.
Согласно кратким сообщениям [2а, 26], оловоорганические соединения
могут вступать в реакции присоединения по связи С—Sn. Триэтилаллилоло-
во присоединяется к бензальдегиду по уравнению [2а]
СвНвСНСН.,СН=СН2
OSn (C2H5)3
о
/
С6Н5С + H2C=CHCH2Sn (СзЬУз -
н
Присоединение по кратным связям
273
При 150° С в отсутствие растворителя реакция заканчивается в течение
4 час, а при 200° С — за 2 часа. Сухой хлористый цинк действует как
катализатор, что указывает на полярный механизм реакции. Это
подтверждается увеличением ее скорости при введении в молекулу альдегида электроно-
акцепторных групп. Так, к n-трихлорметилбензальдегиду триэтилаллилоло-
во присоединяется уже при 20° С, а хлораль реагирует настолько
энергично, что требуется охлаждение смеси.
При нагревании до 150° С триэтилаллилолова с уксусным ангидридом
получены ацетат триэтилолова (выход 90%) и метилаллилкетон (выход 44%)
[2аJ. Из продуктов взаимодействия триэтил(фенилэтинил)олова с хлоралем
(10 час. при 150—200° С) с выходом 70% выделен 4,4,4-трихлор-1-фенил-3-
триэтилстанноксибутин-1 [26]
О
/
(С2Н5)3 SnC=CC6H5 + CC13C -*(СзН3)8 SnOCH (CC13) C=CC6H5.
Н
Триэтил(фенилэтинил)олово легко присоединяется также к циклогексанону
в присутствии хлористого цинка [2а]
у—ч /С^ССеНз
■=0 + (C2H5)3SnC=CC6H, - < X
N—/xOSn(C2H5)3
Соединения типа R3SnCH2X (X = CN,COCH3, COOC2H5, CONR2)
присоединяются к альдегидам и кетонам даже при обычной температуре [2в].
ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
К ОЛЕФИНАМ
В 1956 г. Ван-дер-Керк с сотр. [3] предложили новый метод синтеза оло-
воорганических соединений путем присоединения оловоорганических
гидридов к замещенным а-олефинам
R3SnH + CH2=CHR' -* R3SnCHaCHaR'
(R'=CN, OH, OR, OOCR и др.)
Аналогичным образом проходят реакции с оловоорганическими ди- и
тригидридами [4]:
R2SnH2 + 2 СНа=СН—R' -» R2Sn (CH2CH2R')2,
R3nH3 + 3 СН2=СН— R' -> RSn (CH3CH2R')3.
Существенное значение имеет степень чистоты применяющихся олефи-
нов. Например, присутствие стабилизатора в стироле уменьшает скорость
реакции [51.
Следует подчеркнуть, что гидриды фенилолова более активны, чем
соответствующие алкилзамещенные соединения [3, 6—11].
Термическая устойчивость оловоорганических гидридов уменьшается
с уменьшением числа органических радикалов у атома олова. Реакции с ди-
гидридом диизобутилолова можно проводить при 100—110° С [12, 13], но
большинство ди- и особенно тригидридов олова разлагается при этой
температуре. В некоторых случаях присоединение гидридов сопровождается
побочными процессами или не проходит вообще.
18 Заказ № 207
274
Оловоорганические соединения. Лит. стр. 317—318
Дальнейший успех реакции гидростаннирования связан с
использованием катализаторов. Как показано Нейманом [12—15], эффективными
катализаторами являются вещества, которые разлагаются при достаточно
низких температурах с образованием свободных радикалов (азодиизобутиро-
нитрил, гидразиновые соединения и др.). Каталитическое влияние на
присоединение оловоорганических гидридов к олефинам оказывает также в
некоторых случаях облучение смеси УФ-светом [8, 10, 14, 16—20].
Применение катализаторов позволяет существенно расширить область
применения реакции [12—14]. Скорость ее при прочих равных условиях
пропорциональна концентрации радикалов, образующихся при распаде
катализатора [5,14]. Катализатор целесообразно прибавлять к реакционной смеси
отдельными порциями или непрерывно в виде раствора. С быстро
реагирующими компонентами достаточно однократного прибавления.
Следует отметить, что тонкоизмельченные переходные металлы,
перекись бензоила или фенилазотрифенилметан не оказывают какого-либо
положительного влияния на присоединение оловоорганических гидридов к
олефинам [14, 21]. Это объясняется [5, 14] следующими причинами.
Индуцированное разложение перекиси бензоила, которое проходит очень легко,
подавляется конкурирующей реакцией обрыва цепи в результате
взаимодействия перекиси с оловоорганическими гидридами (см. стр. 490). При
комнатной температуре фенилазотрифенилметан не дает достаточного количества
радикалов. При более высоких температурах он способствует гидростанни-
рованию [5,12—14].
Вещества, активно реагирующие со свободными радикалами, заметно
замедляют реакцию гидростаннирования или прекращают ее вообще [5, 10].
Так, феноксил
х х
\_ _/
/_ \
X X
не реагирует с гидридом триэтилолова в заметной степени при нагревании
смеси до 70° С в течение 20 час. [5]. Однако это вещество в количестве,
эквимолекулярном концентрации азодиизобутиронитрила, полностью
прекращает гидростаннирование эфиров акриловой кислоты при 40° С [5]. Другим
примером может служить реакция между гидридом триэтилолова и акрило-
нитрилом. В присутствии катализатора радикалообразующего типа она
заканчивается при 60°С в течение часа, а после прибавления феноксила
проходит гораздо медленнее. Вероятно, в этом случае реакция не прекращается
полностью вследствие того, что радикалы образуются непосредственно из
акрилонитрила [5].
Влияние катализатора и феноксила на скорость присоединения гидрида
триэтилолова к стиролу
(С2Н3)з SnH + СН2=СН-СбН5 -^ (СзВДз SnCH2CH2CGH5
показано на рис. 2 [14]. Стабилизированный стирол гидростаннируется при
100° С в течение 7 час. (кривая 2). Нестабилизированный стирол вначале
реагирует значительно быстрее (кривая 3). Реакция заканчивается очень
быстро, если прибавить небольшое количество азодиизобутиронитрила
(кривая 4). Совершенно иначе действует прибавление небольшого
количества феноксила (кривая /). Таким образом, по крайней мере во многих случаях
гидростаннирование олефинов проходит по радикальному механизму. Этот
вывод сделан, помимо Неймана [5, 14], и другими авторами [10, И, 17, 20,
22].
Присоединение по кратным связям
275
Присоединение оловоорганических гидридов к олефинам начинается,
по-видимому, с атаки радикала R', образующегося при распаде
катализатора, на молекулу оловоорганического гидрида. В результате в реакционной
смеси появляется триал кил станнильный радикал. При некатализируемом
присоединении радикал R' образуется за счет термической активации оле-
фина [5, 9, 14]. Взаимодействие оловоорганического радикала с олефином
Рис. 2. Скорость реакции присоединения
гидрида триэтилолова к стиролу
О 120 " 240 360
Время, мин
приводит к свободному радикалу с нечетным электроном на углероде.
Последний с новой молекулой гидрида образует конечный продукт
присоединения и триалкилстаннильный радикал
R3SnH + R" -> R3Sn* + R'H,
R3SrT + C6H5CH=CH2 - R3SnCH2CHC6H5,
R3SnCH2CHCGH5 + R3SnH -» R3SnCH2CH2C6H5 + R3Sn\
Впоследствии было установлено, что в некоторых случаях гидростанни-
рование олефинов мЬжет проходить по ионному механизму [22а, 226J.
Детальное изучение продуктов гидростаннирования акрилонитрила методами
газо-жидкостной хроматографии, ИК-спектроскопии и спектров ЯМР
показало, что в результате реакции образуются два вещества:
R3SnH + H2C=CH-CN -» CH3-CH-CN + R3SnCH2CH2CN
R3Sn
(R = CH3; C2H5; ;-C3H7; C4H9; C6H5).
Из продуктов деалкилирования смеси бромом выделен а-бромпропионитрил
R3Sn Br
CH8-CH-CN + Br2 -> CH3-CH-CN + R3SnBr.
Дальнейшие исследования показали, что соотносительные количества
а- и р-аддуктов в смеси зависят от характера органических радикалов у
атома олова, полярности среды и катализаторов. Так, в присутствии азодиизо-
бутиронитрила а-аддукт вообще не образуется.
Изучение скорости реакции акрилонитрила с оловоорганическими
гидридами в бутиронитриле и декане показало, что природа растворителя
практически не влияет на скорость образования р-аддукта. Что касается а-ад-
дукта, то в полярном растворителе (бутиронитриле) он образуется гораздо
быстрее, чем в неполярном (декане). Таким образом, а-аддукт образуется по
ионной реакции, а |3-аддукт — по радикальной реакции.
Скорость ионной реакции гидростаннирования увеличивается в ряду:
(C6H5)3SnH < (CH3)3SnH ж (*-C3H7)3SnH < (QH9)3SnH < (QH^SnH <
< (CH3)3SnH. Если бы скорость реакции определялась только стерическими
факторами, то реакционная способность оловоорганических гидридов изме-
18*
276
Оловоорганические соединения. Лит. стр. 317—318
нялась бы следующим образом:
(С6Н5)3 SnH < (/-С3Н7)з SnH < (C4H9)3 SnH < (C2I15)3 Snll < (CH8)3 SnH.
Таким образом, активность оловоорганических гидридов зависит не только
от величины заместителя у атома олова, но и от его индукционного эффекта.
Электронодонорные заместители у атома олова способствуют реакции гид-
ростаннирования. С другой стороны, эта реакция проходит лишь в тех
случаях, когда у двойной углерод-углеродной связи находятся достаточно
сильные электроноакцепторные группы. Эти данные позволяют сделать
предположение о том, что скорость ионной реакции гидростаннирования
определяется стадией, на которой происходит атака атома водорода олово-
органического гидрида на атом углерода:
Н2С—СН—С ==N
/5
^SnR3 J
(-+-) ^~) быстро
*-R3Sn + Н3С—СН—CeeeN —+■ Н3С—СИ—C=N
SnR3
Следует отметить, что гидростаннирование олефинов проходит по ионному
механизму лишь с достаточно электрофильными реагентами. Вследствие
низкой электрофильности двойной углерод-углеродной связи и ее большой
реакционной способности в свободнорадикальных реакциях, в большинстве
случаев гидростаннирование олефинов проходит по свободнорадикальному
механизму. При гидростаннировании олефинов ионный механизм должен
рассматриваться как исключение [22а, 226].
Гидростаннирование диенов проходит, по-видимому, по следующей
схеме [14]:
R3SnH + R" - R3Sn* + RH,
R3Sn' + CH2=CH—СП=СН2 — R3SnCH2CHCH=CH2,
RgSnCHoCHCH =Ш2 ^ R8SnCH2CH==CHCH2,
R3SnCH2CHCrI=CH2 + R3SnII -> R8SnCH2CHaCH = CH2 + R3Sn\
R3SnCH2CH=CHCH2 + R3SnI I -» R8SnCHaCH=CHCH8 + R3Sn\
Как видно из приведенной схемы, в результате реакции образуются два
вещества [14, 23]:
R3SnH + H2C=CR'—CR"=CHRW -* R3SnCH2CR'=CR"CH2R'" +
+ R3SnCn2CHR/CR"=CHR"f.
Для анализа этой смеси важен тот факт, что первое соединение селективно
титруется иодом при 0—20° С с отщеплением непредельного радикала
[23].
Относительные количества этих веществ зависят от заместителей в
сопряженной системе и характера органического радикала у атома олова.
Если в приведенном выше уравнении R"' = Н, то с выходом 80%
образуется 1,4-аддукт, имеющий, по-видимому, гараяоконфигурацпю [14, 23].
В случае замещенных диенов станнильный радикал атакует
предпочтительно незамещенный конец сопряженной системы. Например, с 1,3-пен-
тадиеном количество продукта присоединения в положение 1,2
состарен-
Присоединение по кратным связям
277
вляет 35% [14, 23]. Аналогично реагирует дигидрид диизобутилолова [23].
Присоединение гидрида триметилолова к диенам (аллену, 3-метил-1,2-бу-
тадиену, 2,3-пентадиену, 2-метил-2,3-пентадиену и др.) подробно
рассмотрено в статье Куивилы с сотр. [24]. Если применять избыток оловооргани-
ческого гидрида, то можно осуществить также присоединение двух молекул
оловоорганического гидрида к диенам [4, 12, 13, 23].
2 R3SnH + CHo=rCH—R'—CII=CHa -* R3SnCH2Cn2R'Cn2CH2SnR3.
В литературе имеются указания о присоединении оловоорганических
гидридов к олефинам, у которых двойная связь находится в середине
молекулы [5,14]. Однако в этом случае реакция проходит лишь с достаточно
активными олефинами. Например, циклопентен и циклогексен не реагируют
с оловоорганическими гидридами даже в присутствии катализаторов или
при облучении смеси УФ-светом [5, 10, 14]. Этиловый эфир кротоновой
кислоты гладко присоединяет гидрид триэтилолова:
(С2Н,)Я Snll + СПаСП=СНСООС2Н5 -* (QJIfib SnCH (СН3) СН2СООС2Н5.
Для этой реакции точно установлен радикальный механизм: при 80е С она
проходит медленно и полностью прекращается в присутствии небольшого
количества феноксила. Напротив, при прибавлении 1 мол.% азодиизобутиро-
нитрила гидростаннировапие заканчивается в течение 30 мин. [5, 14].
Все сказанное верно для оловоорганических ди- и тригидридов.
Присоединение их к олефинам проходит ступенчато [14, 16, 17, 20, 25].
Как отмечено на стр. 462, при смешении оловоорганических дигидридов
и дигалогенидов происходит обратимая реакция. Нейман с сотр. [14, 26]
показали, что образующиеся при этом смеси веществ реагируют с
олефинами как индивидуальные (галоид)гидриды диалкилолова:
R2Sn (Н) X + СН2=СН—R' -» R2Sn (CH2CH2R') X.
Этот изящный способ гидростаннирования позволяет синтезировать
соответствующие оловоорганические галогениды в одну стадию. Реакция
проходит легко, в большинстве случаев экзотермически, при температуре 20—
40° С [261. Применение катализаторов радикалообразующего типа, если
это необходимо, дает положительный эффект.
Можно исходить и из (дигалоид)гидридов алкилолова. Так,
взаимодействием трехбромистого этилолова, гидрида триизобутилолова и фенилаце-
тилена, протекающим через стадию образования (дибром)гидрида этилолова,
получен [26] (З-(этилдибромстаннил)стирол:
C2H5SnBr3 + (/-C4H9)3SnH + С6Н5С ~ СН -» GSH5GH = CHSn (СзН5)Вг2 +
+ (r-QHob SnBr.
Методика проведения этих реакций описана очень кратко [26]. Гало-
генид алкил(или арил)олова смешивают с эквивалентным количеством
гидрида при 20° С в атмосфере аргона. К смеси прибавляют рассчитанное
количество или избыток ненасыщенного соединения. Можно прибавлять также
гидрид к смеси оловоорганического галогенида и ненасыщенного
соединения.
За ходом реакций присоединения удобно наблюдать с помощью ИКг
спектров (изменение интенсивности полос Sn—Н и С=С в области 1800—
1875 еж"1 и 1590—1670 см'1 соответственно) [10, 12, 13].
Скорость и направление реакций оловоорганических гидридов с
различными олефинами существенно зависят от природы последних, как это
можно видеть из следующих примеров.
278
Оловоорготические соединения. Лит. стр. 317—318
Незамещенные олефины. При нагревании до кипения
гидрида триметилолова сос-олефинами реакция проходит лишь в очень
незначительной степени (примерно на 2%) [22].
С я-октеном-1 гидриды триалкилолова реагируют только в присутствии
катализатора [4, 6, 12, 13].
Получение триэтил-я-октилолова [12]. 10,35 г (0,05 моля) гидрида триэтилолова и 11,22 г
(0,10 моля) я-октена-1 нагревают и перемешивают в течение 45 час. при 90—100° С. К
смеси прибавляют частями 0,92 г (0,0056 моля) азодиизобутиронитрила (при непрерывном при
бавлении катализатора продолжительность реакции значительно сокращается). Для
удаления побочного продукта — цианистого триэтилолова — смесь встряхивают с 2 мл 2 N
едкого натра и 30 мл воды, а затем перегоняют в вакууме. Получают 9,14 г (94%) триэтил-я-
октилолова с т. кип. 145—148° С/12 мм\ п2£ 1,4713.
Аналогичные результаты получены [12] при применении в качестве
катализатора фенилазотрифенилметана (температура 80—90° С), нитрила
фенилазоизомасляной кислоты (80—90°С) или перекиси т-трет-бутила
(100—110° С).
Присоединение гидрида триметилолова к 1,3-пентадиену без
катализатора проходит в незначительной степени. Анализ образующегося продукта
методом жидкостной хроматографии показывает, что в его состав входят
четыре соединения. Основными из них являются цис- и транс-изомеры
состава [22]
СН3 CH2CII2Sn(CH3)3 СНз Н
)>с==с<^ и Ус=°( '
Н 'Н Н CII2CHaSn (СНзЬ
В меньших количествах образуются цис- и транс-изомеры продукта
моноприсоединения, у которых группа триметилолова связана с Р-угле-
родным атомом
Sn(CH8)8
СНз СН—СНз СНз Н
V-G^ и УС:=С<^
П Н Н СН—СНз
I
Sn (СН3)з
Присоединение по у- или б-углеродному атому не имеет места [22].
Нагреванием до 70—80° С гидрида триэтилолова с двумя молями диал-
лила в присутствии катализатора получают [12] (C2H5)3Sn(CH2)4CH==CH2.
Вероятно, при этом образуется и 1,6-ди(триэтилстаннил)-я-гексан. С окта-
диеном реакция проходит за 25—30 час. при 70—80° С [12].
(С2Н3)3 SnH + СН2=СН—(СН2)4—СН=СПа -*
-> (C2H5)cSn (CII2)8Sn (С2П,-;)з + (C2H5)3Sn (CH2)eCH=CII2.
Гидрид триметилолова присоединяется при нагревании к пиперилену
[22, 27]. Анализ образующихся продуктов хроматографическими методами
показывает, что реакция проходит только по двойной связи, находящейся
у ос-углеродного атома, и приводит к смеси цис- и т/шяоизомера-д
CH3CH=---CHCH2CH2Sn (СП3)з (выход —15%) и в меньшем количестве— к
цис-и транс-СН3СН=СПСН (CH3)Sn(CH3)3. При температуре 100° С выход
этих веществ составляет 40%. Одновременно обнаружены водород, тетра-
метилолово и три нендептифицирозанных, чувствительных к кислороду
вещества типа R(CH3)2SnIi.
Гидриды триалкилолова присоединяются к стиролу как в присутствии
катализаторов [12, 13], так и без них [4]. С гидридом три-я-пропилолова
Присоединение по кратным связям
279
реакция проходит за 4 часа при 100° С [4]. Температуру можно снизить до
. 40—60° С, если гидростаннирование стирола проводить в присутствии азо-
диизобутиронитрила. Так получены [12] аддукты гидрида триэтилолова со
стиролом и р-метил стирол ом. Продукт взаимодействия эквимолекулярных
количеств гидрида триэтилолова с /г-дивинилбензолом полимеризуется
при перегонке.
Дигидрид диметилолова не реагирует с этиленом при нагревании смеси
реагентов до 50° С в защищенной от действия света ампуле в течение 180 час.
[25]. Реакция начинает протекать с заметной скоростью, если смесь
облучать УФ-светом. В этих условиях из эквимолекулярных количеств дигид-
рида диметилолова и этилена образуются неустойчивый гидрид диметил-
этилолова и диметилдиэтилолово с выходом 37 и 31% соответственно. Выход
диметилдиэтилолова повышается до 69%, если взять два моля олефина
на моль оловоорганического гидрида [25].
При нагревании до 50° С в течение 163 час. эквимолекулярных
количеств дигидрида диметилолова и бутадиена в защищенной от действия света
ампуле с выходом 34% получают неустойчивый гидрид диметил-(2-буте-
нил)олова [17]
(2(СН3 SnH2 + СН2=СН—СН=СН2 -> (СН3)з SnH (СН2СН=СНСН3).
Кроме того, образуется диметил-бш:-(2-бутенил)олово (выход 19%),
которое, согласно ИК-спектру, имеет m/шяс-конфигурацию. К этому
соединению могут приводить следующие реакции [17]:
(СН3)2 SnH (СН2СН=СНСН3) + СНа=СН—СН=СНа -> (СНз)а Sn (CH2CH=GHCH3j2,
2 (СН3)3 SnH (СНаСН=СНСН3) -» (СН3)2 SnH3 + (СН3)3 Sn (CH2CH=CHCH3)2.
Выход диметил-бнс-(2-бутенил)олова повышается до 71%, если смесь
дигидрида диметилолова и двух молей бутадиена нагревать до 50° С при
облучении в течение 23 час.
В аналогичных условиях дигидрид диметилолова не реагирует с цис-
и трапс'2-бутеном [17]. Другим примером инертности двойной связи,
находящейся в середине молекулы, может служить реакция гидрида
триэтилолова с 1-винилциклогексеном-З. При нагревании смеси до 80—90° С
в течение 50 час. с катализатором (азодиизобутиронитрил) присоединение
проходит только по винильной группе [12].
Значительно легче присоединяются дигидриды диалкилолова к стиролу.
В присутствии катализатора дигидрид диэтилолова реагирует со стиролом
за 12 час. при 60—70° С. С дигидридом диизобутилолова реакция проходит
за 2 часа при 100—110° С [12].
Гидриды фенилолова гидростаннируют олефины в более мягких
условиях. Трифенил-я-октилолово получено [6] при простом нагревании до
80° С гидрида трифенилолова с я-октеном-1 в течение 7 час. Выход 72%,
т. пл. 54—55°С [4, 6] (ср. [10]).
Получение (2-фенилэтил)трифенилолова [6]. 10,5 г (0,03 моля) гидрида трифенилолова
и 6,2 г (0,06 моля) стирола нагревают при 80° С в течение 30 мин. Образующуюся
кристаллическую массу промывают петролейным эфиром (т. кип. 60—80° С). Получают 11,2 г (82%)
вещества с т. пл. 122—125° С. После двух кристаллизации из петролейного эфира (т. кип.
60—80° С) температура плавления повышается до 127—127,5° С.
О реакции дигидридов диалкил- и диарилолова с диенами см. стр. 513.
Галоидзамещенные олефины. Оловоорганические
гидриды являются эффективными восстановителями галоидных алкилов и
арилов (см. стр.457). Поэтому получить продукты присоединения к галоид-
замещенным олефинам часто не удается.
Так, при нагревании до 80° С в течение 4 час. гидрида трифенилолова и
бромистого аллила с количественным выходом образуются бромистое три-
280 Оловоорганические соединения. Лит. стр. 317—318
фенилолово и пропен [6]
(CeH5)8SnH -f СНа=СЫСН2Вг -> (CeH5)8SnBr + СН2=СНСП3.
Кларк и сотр. [16] подробно изучили взаимодействие дигидрида диме-
тилолова с фторированными олефинами.
Дигидрид диметилолова образует с тетрафторэтиленом гидрид диметил-
(тетрафторэтил)олова (выход 52,3%) и диметил-б£/с-(тетрафторэтил)олово
(9,2%). Небольшое количество нелетучего остатка (10%) состоит из
металлического олова и тефлона. Скорость реакции значительно возрастает при
нагревании с одновременным облучением смеси УФ-светом. В зтих условиях
(50° С, 20 час.) получают 75% гидрида диметил(тетрафторэтил)олова и
1—2% диметил-бш:-(тетрафторэтил)олова. При изменении соотношения
реагентов с 1 : 1 до 2 : 1 в тех же условиях образуется 77% соединения
(СН3)2 Sri, (C2F4H)2 и 13% продукта моноприсоединения.
Гидрид диметил(тетрафторэтил)олова представляет собой бесцветную
жидкость с т. кип. 124° С. При хранении в запаянной эвакуированной
ампуле в темноте при 25° С он устойчив по меньшей мере в течение 3 месяцев.
При нагревании выше 100° С вещество диспропорциоипрует. Так,
нагревание гидрида до 140° С в течение 26 час. приводит к диметил-бш:-(тетра-
фторэтил)олову [16]. Второй продукт диспропорционировапня—дигидрид
диметилолова в этих условиях разлагается (см. стр. 463).
С несимметричными фторолефинами дшидрид диметилолова реагирует
более сложным образом. Так, с трифторэтиленом при температуре 25 или
50° С в темноте реакция не проходит вообще. Однако при облучении смеси
УФ-светом уже при 25° С за 20 час. в реакцию вступает —90% дигидрида.
Основными продуктами являются водород, трифторэтилен и шлш-дифтор-
этилен, а также значительное количество нелетучего остатка, содержащего
металлическое олово и двуфтористое диметилолово. Кроме того, получают
небольшое количество тетраметилолова и гидрида диметил(трифторэтил)оло-
ва (эти вещества идентифицированы по ИК-спектрам и спектрам ЯМР).
С 1,1-дифторэтиленом дигидрид диметилолова реагирует в
незначительной степени при нагревании смеси до 50° С в темноте [16]. Для этой
реакции предложена [16] следующая схема:
(СНз)2 SnH2 + CF2=CH2 -> (СН3)2 SnH (C2F2H3),
(СН3)2 SnH (C2F2H3) -> [(CH3)2 SnHF] + CII2=CHF,
2 [(CH3)2 SnHF] -> (CH3)2 SnF2 + (CH3)2 SnIIa.
При взаимодействии 1 моля дигидрида диметилолова с 2 молями 1,1-Ди-
фторэтилена образуются те же вещества и, кроме того, фтористое триметил-
олово и двуфтористое диметилолово.
При нагревании до 50° С эквимолекулярной смеси трифторбромэтилена
и дигидрида диметилолова в течение 21 часа в реакцию вступает 98% олово-
органического гидрида. Однако и в этом случае выделены лишь бромиды
метилолова [16]
(СНо)2 SnII2 + CF^CFBr -> (СНз)а SnH (C2F3BrH),
(СН3)2 SnH (C2F3BrH) -> [(CH3)a SnHBr] + C2F3H,
2 [(CH3)2 SnHBr] -> (CH3)2 SnH2 + (CH3)2 SnBr2.
Другие вещества — водород, металлическое олово и бромистое триметил-
олово — являются, вероятно, продуктами разложения дигидрида
диметилолова [16]
2 (СН3)2 SnH2 -* (СН3)4 Sn + Sn + 2Н2,
(СН3)4 Sn + (CH3)2 SnBr2 -* 2 (СН3)з SnBr.
Присоединение по кратным связям
281
Если ту же реакцию проводить с 2 молями трифторбромэтилена на моль ди-
гидрида диметилолова, то наряду с этими веществами образуется 13% ди-
метил-б^о(2-бромтрифторэтил)олова. Это — бесцветная жидкость, которая
разлагается при нагревании до 100° С [16].
(CI 1з)<> Sn (CFoCFBrI I)2 -> 2 CoF3H 4- (СНз)2 SnBr2.
При облучении УФ-светом эквимолекулярных количеств дигидрида ди-
я-бутилолова и тетрафторэтилена при температуре 25° С в течение 21 часа
получают гидрид ди-//-бутил(тетрафторэтил)олова с выходом 90% [20!.
Ди-я-бутил-6'^с-(тетрафторэтил)олово образуется с умеренным выходом при
нагревании рассчитанных количеств реагентов до 30° С в течение 4 час. [28]
или облучении смеси УФ-светом в течение 18 час. [20J.
Непредельные спирты. В присутствии катализаторов ал-
лиловый спирт легко присоединяет гидрид триэтилолова:
(С2135)3 SnH + CH2=CHCTIoOH -» (QjIIsb SnCn2CH2CH2OI I.
Получение триэтил(3-оксипропил)олова [12, 13]. 10,35 г (0,05 моля) гидрида т• иэтил-
олова, 5,81 г (0,10 моля) аллилового спирта и 0,4 г (0,0025 моля) азодиизобутироиитрила
нагревают до 35—40°С при перемешивании до исчезновения полосы связи Sn—Н в ИК-спектре
(продолжительность опыта около часа) и перегоняют в вакууме. Выход триэтил(3-оксипро-
пил) олова 89%, т. кип. 128—130°С/12 мм, п2£ 1,4960. В колбе остается небольшое
количество побочного продукта реакции — цианистого триэтилолова. Т. пл. 164—165° С (из петролей-
ного эфира с т. кип. 120° С).
Аналогичным образом получено [12] три-я-бутил(3-оксипропил)олово
(т. кип. 173—176° С/12 мм).
Гидрид трифенилолова присоединяется к непредельным спиртам и без
катализатора [4, 6].
Более сложно реагируют с непредельными спиртами дигидриды диал-
килолова. При нагревании до 40° С в течение 48 час. дигидрида ди-я-бутил-
олова с избытком аллилового спирта в присутствии азодиизобутироиитрила
получают 2,2-ди-я-бутил-1-окса-2-станнациклопентан (т. пл. 219—226° С)
[29]. По-видимому, реакция проходит следующим образом:
(С4Н9)2 SnH2 + НОСН2СН=СН2 -> (С4Н9)2 Sn (H) ОСН2СН=СН2 -f- Н2
1 _J {
(QH9)2 Sn (Н) СН2СН2СН2ОН -> (C4H9)2SnOCH2CH2CH2
С аллилмеркаптаном дигидрид ди-я-бутилолова образует бш>(аллил-
меркаптид) ди-я-бутилолова [29].
Простые ненасыщенные эфиры. Простые виниловые
эфиры присоединяют гидриды триалкилолова при нагревании смеси
реагентов до 60° С в течение 20—40 час. в присутствии
азодиизобутироиитрила [12].
RsSnll + CH2=CHOR'-+ R3SnCH2CH2OR\
Обычно в реакцию берут два моля олефина на моль оловоорганического
гидрида. Так получены [12] продукты присоединения гидрида триэтилолова
к этилвиниловому эфиру (продолжительность реакции 24 часа), я-бутил-
виниловому эфиру (43 часа), изобутилвиниловому эфиру (29 час), а также
гидрида триизобутилолова к я-бутилвиниловому эфиру (38—40 час).
Гидрид три-я-бутилолова присоединяется к изобутилвиниловому эфиру при
нагревании эквимолекулярной смеси реагентов до 70° С в течение 18 час
в присутствии катализатора. Выход 86%, т. кип. 105—107°С/0,25 мм.
282
Оловоорганические соединения. Лит. стр. 317—318
С гидридом трифенилолова реакция проходит при простом нагревании.
Получение (2-феноксиэти л) трифенилолова [6]. 9,96г (0,028 моля) гидрида трифенилолова
и 0,056 моля винилфенилового эфира нагревают до 80° С в течение 2 час. При охлаждении
выделяются кристаллы, которые отфильтровывают и промывают лигроином. Получают
10,91 г (89%) вещества ст. пл. 78—81° С. После двух перекристаллизации из лигроина т. пл.
82,5—83° С.
По патентным данным [30], оловоорганические гидриды присоединяются
к непредельным соединениям, содержащим эпоксигруппу, с образованием
соединений типа
i I
R3Sn (СНа)з ОСН2СНСН30.
Непредельные кетоны. Реакция оловоорганических
гидридов с непредельными кетонами может проходить по нескольким
направлениям. Гидриды трибутил- и трифенилолова присоединяются по
карбонильной группе винилфенилкетона и 1,3-дифенилпропенона [22а, 30а]
R3SnH + \:=С-С=0 -» ^СН—С-С—OSnR3.
7 I I 7 I I
При длительном нагревании этих веществ с большим избытком гидрида
трифенилолова получают насыщенные кетоны. Последние являются,
по-видимому, продуктами перегруппировки виниловых спиртов, образующихся
при гидрогенолизе 1,4-аддуктов гидридом трифенилолова
(СбН5)3 SnH + NcH—С=С-0—SnR3 — (СбН5)3 SnSnR3 + N>CH—C=C—OH -»
7 I I X I I
-> Nch—ch—c=o
x I
При нагревании метилвинилкетона с гидридом трифенилолова
образуется метилэтилкетон и соответствующий оловоорганический кетон [22а, 30а]
(C6H5)3SnH + H2C=CH—С—СН3
II
О
Гидрид трибутилолова гладко присоединяется по карбонильной группе
(СН3)2С^СНСОСН3, тогда как гидрид трифенилолова селективно
гидрирует двойную углерод-углеродную связь этого кетона с образованием
(СН3)2СНСН2СОСН3 и гексафенилдистаннана [31]. При реакции гидрида
трифенилолова с метилвинилкетоном или винилфенилкетоном
количественно образуется гексафенилдистаннан [4, 31]. Однако аллилацетон, в котором
карбонильная группа не сопряжена с двойной связью, гладко присоединяет
гидриды трибутил-[32] и трифенилолова [4, 32]
R3SnH + СЧ2=С iCH2CH2COCK8 -> RsSnC^CHoCfi.QIaCOOTs.
Облучение смеси УФ-светом ускоряет реакцию [32]. При нагревании гидрида
трибутилолова с Н(СН3)С=СН(СН2)3СОСН3 или (СН3)2С=СН(СИ2)2СОСН3
наблюдается только восстановление кетонов до соответствующих спиртов.
К аналогичному результату приводит и облучение смеси реагентов УФ-
светом. В случае же гидрида трифенилолова наряду со спиртами образуются
соответствующие алкоксисоединения трифенилолова [32].
Непредельные амины. Гидрид трифенилолова реагирует с
аллиламином уже при комнатной температуре [6], но продукта присоеди-
I > СНзСН2—С—СНз
&
> (C6H5)3SnCH2CH2—С—СН3
II
о
Присоединение по кратным связям
283
нения при этом не образуется; из реакционной смеси выделен только гек-
сафенилдистаннан.
Позже было показано, что в присутствии аминов происходит
каталитическое разложение оловоорганических гидридов. Эта реакция подробно
рассмотрена на стр. 477.
Оловоорганические соединения с аминогруппой в органическом
радикале у атома олова получают восстановлением соответствующих нитрилов
литийалюминийгидридом (см. стр. 447).
Непредельные кислоты. При комнатной температуре
гидриды триалкилолова образуют с непредельными кислотами ацилаты триал-
килолова [6, 12]
(СзН7)3 SnH + СН3=СН—СООН -> (СзИтЬ SnOOCCH =СН2 -!- Н2.
С оловоорганическими гидридами ароматического ряда реакция проходит
иначе. Гидрид трифенилолова реагирует с акриловой кислотой мгновенно
и количественно с образованием неустойчивой Р-трифенилстаннилпропио-
новой кислоты. Последняя сразу же отщепляет молекулу бензола [6]
(СвНз)з SnH + СЧа=СНСООН -> (С6Н5)3 SnCH2CH2COOH ->
-> (СвП3)2 Sn+GH2GH2GOO- + С6Н6.
Конечный продукт можно рассматривать как внутреннюю соль гидроокиси
(2-карбоксиэтил)дифенил олова.
Получение внутренней соли (2-карбоксиэтил)дифенилолова [6]. Смесь 14,0 г (0,04 моля)
гидрида трифенилолова и 4,3 г (0,06 моля) акриловой кислоты нагревают до 70° С. При этой
температуре происходит экзотермическая реакция, смесь закристаллизовывается. Избыток
акриловой кислоты отмывают петролейным эфиром (т, кип. 60—80° С). Получают 11,9 г
(93%) порошка светло-желтого цвета, нерастворимого в обычных органических
растворителях. Большая часть его растворяется в избытке разбавленного раствора едкого натра
(примерно 1 N). Вероятно, при этом образуется натриевая соль гидроокиси 2-карбоксиэтил-
дифенилолова. Раствор отфильтровывают и фильтрат подкисляют разбавленной соляной
кислотой по метилоранжу. Осадок отфильтровывают, промывают горячей водой и высушивают
в вакууме. Получают белое порошкообразное вещество с т. разл. 270° С.
Производные ненасыщенных кислот. Гидриды
триалкилолова легко присоединяются к эфирам акриловой кислоты [6, 331:
R3SnH + CH2-CHCOOR' -> R3SnCH2Cf~I2COOR\
Так, (2-карбоксиметилэтил)три-я-пропилолово получено [6]
нагреванием до 100° С гидрида три-я-пропилолова с двумя молями метилакрилата
в течение 4 час. с последующей перегонкой смеси в вакууме; т. кип. 145—
150° С/12 мм, выход 63%. С гидридом три-я-бутилолова реакция проходит
за 6 час. при 80° С; выход (2-карбоксиметилэтил)три-я-бутилолова
составляет 84%, т. кип. 140—141° С/0,4 мм.
Применение катализатора позволяет существенно снизить температуру
и сократить продолжительность реакции. Например, гидрид триэтилолова
присоединяется к метилакрилату лишь при нагревании смеси реагентов
до 100° С в течение 15 час, а в присутствии азодиизобутиронитрила — за
1 час при 40° С [3]. В аналогичных условиях метилметакрилат реагирует
с гидридами триэтил- и трибутилолова значительно медленнее.
В тех случаях, когда карбонильная группа значительно удалена от
непредельной связи, реакция проходит так же, как и с незамещенными оле-
финами. (со-Карбоксиэтил-я-ундецил)триэтилолово получают [12]
нагреванием до 60° С в течение 40—45 час. гидрида триэтилолова с двумя молями
этилового эфира я-ундециленовой кислоты. Катализатор (азодиизобутиро-
нитрил) прибавляют частями или непрерывно в виде раствора.
284
Оловоорганические соединения. Лит. стр. 317—318
Аналогично проходят реакции с оловоорганическими ди- и тригидрида-
ми:
RoSnH2 + 2СН2=СНСООСН5 -> R2Sn (СН2СН2СООСН8)2,
RSnH3 + ЗСН2=СНСООСН.ч -> RSn (CH2CH3COOCH8)3.
В качестве примеров можно указать на присоединение дигидрида диэтилоло-
ва к метилакрилату. С катализатором реакция заканчивается при 40—
50° С за 7 час. На 1 моль оловоорганического гидрида берут 3 моля метил-
акрилата, избыток олефина отгоняют в вакууме. В остатке получают ди-(2-
карбоксиметилэтил)диэтилолово с т. кип. 150—156° С/0,001 мм. Нагревание
до 40—50° С эквимолекулярных количеств дигидрида диэтплолова и метил-
метакрилата в присутствии азодиизобутиронитрила в течение 4 час.
приводит к гидриду (2-карбоксиметилпропил)диэтилолова. Выход 33%, т. кип.
63—65° С/0,5 мм [12].
Если же взять 3 моля метилметакрилата на моль дигидрида диэтилолова,
тс после 9 час. нагревания при 40—50° С в присутствии катализатора
получают ди-(2-капбоксиметилпропил)диэтилолово. Выход 95%, т. кип. 123—
127°С/0,03 мм [121.
Тригидриды я-бутил- и изобутилолова присоединяют 3 молекулы
метилметакрилата в присутствии азодиизобутиронитрила при 50—60° С в течение
5 час. Образующиеся соединения имеют т. кип. 150—155° С/0,001 мм и
165—167° С/0,01 мм соответственно.
Как уже отмечалось на стр. 273, гидриды фенилолова более реакционно-
способны, чем соответствующие алкилзамещенные соединения. (2-Карб-
оксиметилэтил)трифеиилолово получено [6] нагреванием до 80° С в течение
4 час. 10,5 г (0,03 моля) гидрида трифенилолова с 5,2 г (0,06 моля) метпл-
акрилата. Избыток метилакрилата удаляют в вакууме и остаток
кристаллизуют из этилового спирта. Получают 11 г (85%) кристаллов ст. пл. 46,5—
47,5° С. Из маточника выделяют еще 1,5 г менее чистого вещества с т. пл.
43—44° С.
С катализатором реакция между дигидридом дифенилолова и метилме-
такрилатом заканчивается при 40—50° С за 4 часа; соотношение компонентов
1 : 3. Выход ди-(2-карбоксиметилэтил)дифенилолова равен 95%, т. кип.
180—184° С/0,01 мм [12].
Азодиизобутиронитрил катализирует присоединение гидридов триал-
килолова к винилацетату [12]:
R3SnH + Н2С=СНООССН3 -> R8SnCHaCH3OOCCH8.
Как и в предыдущих случаях, здесь применяют избыток олефина (молярное
соотношение реагентов 1 : 2). С гидридом триэтилолова реакция
заканчивается при 30—40 ° С через 2 часа. При нагревании (2-ацетоксиэтил)три-
этилолова до 80°С вещество разлагается с выделением этилена и ацетата
триэтил олова. Поэтому синтезировать это соединение без катализатора не
удается. Аллилацетат реагирует с гидридом триэтилолова значительно
медленнее (20 час. при температуре 40—50° С). К гидриду триизобутилолова
аллилацетат присоединяется только при нагревании смеси до 70° С в
течение 22 час. [12].
С алкилиденмалоновыми эфирами оловоорганические гидриды
реагируют следующим образом [34]:
ОС2Н5
R'HC-C (X) СООСзНб + R3SnH -> R'CH2C (X)=c/
OSnR3
(X = NC; R' = CH3, CeH5, az-GH3OC6H4, /z-CIQIV.
X - C2H5COO; R' = CH8, C6H5).
Присоединение по кратным связям
285
Образующиеся О-станнилкетенацетали чрезвычайно реакционноспособ-
ны, что позволяет использовать их для синтеза различных веществ [35].
Присоединение оловоорганических гидридов к амидам или нитрилам
кислот проходит как в присутствии катализаторов, так и без них
R8SnH -I- CH2-=CHCONH2 -> R3SnCH,CH2CONH2,
R8SnH + CH2-=CHCN -» RsSnCHoCIIaCN.
Получение (2-карбамилэтил)три-#-пропилолова [6]. 7,50 г гидрида три-«-пропилолова
и 2,0 г акриламида нагревают при 80° С в течение 4 час. и смесь перегоняют в вакууме.
Получают 7,4 г (77%) вещества с т. кип. 155—16ГС/0,4 мм. При хранении оно закристал-
лизовывается, т. пл. 44—47° С.
Продукты присоединения гидрида триэтилолова к акрил- или мета-
криламиду получают [12] нагреванием до 40—50° С эквимолекулярной смр-
си реагентов в присутствии азодиизобутиронитрила.
При нагревании до 70° С в течение 2 час. эквимолекулярных количеств
гидрида трифенилолова и акриламида в метиловом спирте образуется
(2-карбамилэтил)трифенилолово [6]. Его выделяют следующим образом.
Метанол выпаривают, остаток кристаллизуют из петролейного эфира (т. кип.
60—80° С), а затем из смеси этилового спирта с водой (1 : 1). Выход 90%,
т. пл. 123—124° С.
При проведении реакции с нитрилами непредельных кислот применяют
2 моля олефина на моль оловоорганического гидрида.
Получение (2-цианэтил)три-#-бутилолова [6]. 45,0 г (0,15 моля) гидрида три-«-бутилоло-
ва и 15,9 г (0,30 моля) акрилонитрила нагревают до 80°С б час, смесь перегоняют в вакууме.
Собирают фракцию с т. кип. 125—145° С/0,2 мм. Повторная перегонка дает 35,9 г (70%)
вещества ст. кип. 132—138° С/0,2 мм,
(2-Цианэтил)триэтилолово и ди-(2-цианэтил)диизобутилолово
образуются при нагревании соответствующих оловоорганических гидридов с ак-
рилонитрилом в присутствии катализатора. Температура и
продолжительность реакции 50° С, 22 часа и 100—110° С, 1 час соответственно.
С гидридами трифенилолова реакция проходит достаточно легко и без
катализатора.
Получение (2-цианэтил)трифенилолова [6]. 10,5 г (0,03 моля) гидрида трифенилолова
и 3,2 г (0,06 моля) акрилонитрила нагревают при 80°С в течение 5 час. Избыток
акрилонитрила отгоняют в вакууме и остаток кристаллизуют из метилового спирта. Получают 10,3;
(85%) (2-цианэтил)трифенилолова с т. пл. 93—94° С. Из маточника выделяют еще 1,4 г
вещества ст. пл. 89—92° С.
Подобным же образом проводят реакцию гидрида трифенилолова с кро-
тононитрилом и цианистым аллилом.
Получение (1-цианпропил-2)-трифенилолова [6]. Смесь 10,5 г (0,03 моля) гидрида
трифенилолова и 4,0 г (0,06 моля) кротононитрила нагревают до 80° С. Примерно через 10 мин.
происходит экзотермическая реакция. Для завершения реакции смесь нагревают в течение
1,5 час. и избыток кротононитрила удаляют в вакууме. Твердый остаток последовательно
кристаллизуют из метилового спирта, циклогексана и вновь из метилового спирта.
Получают 11,1 г (89%) вещества ст. пл. 103—104° С.
Следует подчеркнуть, что в этом соединении группа трифенилолова
связана с асимметричным атомом углерода.
Получение (З-цианпропил)трифенилолова [6]. 10,5 г (0,03 моля) гидрида трифенилолова
и 4,0 г (0,06 моля) цианистого аллила нагревают при 90° С в течение 3 час. Избыток
цианистого аллила отгоняют в вакууме. Остаток обрабатывают 100 мл петролейного эфира (т. кип.
60—80° С). Раствор фильтруют от небольшого количества образовавшегося в качестве
побочного продукта тетрафенилолова. Маточник упаривают досуха, остаток дважды
кристаллизуют из метилового спирта. Получают 8,3 г (73%) белоснежных игл с т. пл. 80—81° С.
286
Оловоорганические соединения. Лит. стр. 317—318
Непредельные металлоорганические
соединения. Оловоорганические гидриды легко присоединяются к винильным
и /г-стирильным соединениям бора, кремния, германия и олова. Гидрид три-
этилолова и В-тривинил-Ы-трифенилборазин реагируют только в
присутствии азодиизобутиронитрила [36]
(CH2=CHBNC6H5)3 + 3 (С2Н5)3 SnH -» [(C2H5)3 SnCH2CH2BNCGH5]3.
Образующееся вещество кристаллизуется в виде бесцветных игл с т. пл.
87—88° С.
С гидридом трифенилолова та же реакция проходит без катализатора.
Нагреванием смеси реагентов в толуоле в течение 4 час. получают
кристаллическое вещество с т. ' пл. 219—221° С (из смеси толуола с метиловым
спиртом).
Присоединением гидридов олова к [(CH20)2B]2C==CR'B (OCH2)2 (R'=H
или СН3) получены [37] соединения состава R3SnCH(CH3)CH2B(OCH2)2.
/г-Фенилен-бао(диметилоловогидрид) легко присоединяется к моно-я-
стирильным производным германия, олова и свинца [38]
HSn (CH3)2CeH4Sn (СН3)2 Н + 2R3MIVCeH4CH=CH2->
СН3 СНз
^R3MIVC6H4CH2CH2SnC6H4SnCH2CH2C6H4MIV
СН3 СН3
I. R = CH3, MIV=Sn; II. R=CeH5, MIV=Sn;
III. R=C6H5, MIV=Ge; IV. R=C6H5, MIV=Pb.
Получение я-##с-[(2-/г-трифенилстаннилфенил) этилстаннил] бензола [38]. Смесь 2,25 г
(0,006 моля)/г-фенилен-б^с-(диметилоловогидрида) и 5,44 г (0,012 моля) трифенил-/г-стирил-
олова нагревают при 75° С в течение 18 час. и продукт кристаллизуют из ацетона.
Получают 5,85 г (76%) аморфного порошка с т. пл. 110—134° С. Кристаллизация из смеси
ацетона, этилацетата и воды не оказывает существенного влияния на температуру плавления.
Перекристаллизация из пропилового спирта дает 4,2 г (54%) чистого вещества с т. пл. 124—
136° С.
Аналогичным образом получены [38] соединения I, II и IV. Их синтез
проводят при температуре 85, 70 и 85° С; продолжительность реакций 26,
18 и 16 час; растворители при перекристаллизации — водный метиловый
спирт, смесь пропилового спирта с водой и водный ацетон соответственно.
Выходы продуктов составляют 40, 47 и 23%.
При реакции гидрида трифенилолова с моновинильными производными
кремния, германия и олова образуются соединения с двумя атомами
металла в молекуле [39]
(С0НБ)з SnH + (C6H5)S MCH=CH2 -> (CGH5)b SnCH2CH2M (C0H6)3
(М = Si, Ge, Sn).
Получение 1-трифенилстаннил-2-трифенилсилилэтана [39]. 5,0 г (0,017 моля) винил-
трифенилкремния и 7,0 г (0,02 моля) гидрида трифенилолова нагревают до 80° С в вакууме
в течение 4,5 час. Продукт растворяют в хлороформе и осаждают ацетоном. Получают
5,8 г (54%) кристаллов с т. пл. 203—205° С. Перекристаллизация из ацетсна повышает
точку плавления до 207—208° С.
Подобным же образом получают [39] 1-трифенилстаннил-2 трифенил-
гермилэтан (выход 32%) и бш>(1,2-трифенилстаннил)этак (50%).
Реакция между гидридом трифенилолова и винилтрифенилсвинцом
приводит к образованию гексафенилдистаннана и металлического свинца.
Аллилтрифенилкремний и аллилтрифенилгерманий не реагируют с
гидридом трифенилолова [39]. При нагревании аллилтрифенилолова с гидри-
Присоединение по кратным связям
287
дом трифенилолова в бензоле в течение 18 час. получают [4] только 18%
1,3-б^с-(трифенилстаннил)пропана, т. пл. 182—185° С (после нескольких
перекристаллизации из лигроина). Как и в случае винильных соединений,
аллилтрифенилсвинец восстанавливается до металлического свинца [39].
Взаимодействие 2 молей гидрида трифенилолова с дивинилдифенилкрем-
нием и дивинилдифенилгерманием приводит [39] к соответствующим
продуктам присоединения, содержащим три атома металла в молекуле:
СбН5
2 (С6Н5)3 SnH + (С6Н5)2 М (СН=СН2)2 -> (С6Н5)3 SnCH2CH2MCH2CH2Sn(CGH5)3
СбН5
(М= Si, Ge).
Получение дифенил-(?#с-[2'-(трифенилстаннил)этил] кремния[39]. Смесь 4,72г (0,02моля)
дивинилдифенилкремния и 14 г (0,04 моля) гидрида трифенилолова нагревают до 80 °С
в вакууме в течение 4,5 час. Твердый продукт реакции перекристаллизовывают из лигроина
и диметилформамида. Получают 16 г (86%) вещества с т. пл. 143—144°С.
Гидрид трифенилолова экзотермически реагирует с дивинилдифенил.
германием. После нагревания смеси при 70° С в течение часа и двух
кристаллизации из лигроина получают чистое вещество с т. пл. 139—142° С.
Выход 66%.
Реакция гидрида трифенилолова с дивинилдифенилоловом проходит
своеобразно с образованием 1,2-бш>(трифенилстаннил)этана [39].
Гидрид трифенилолова не реагирует с диаллильными соединениями
кремния и германия. С диаллилдифенилоловом он образует аллилтрифенилолово.
Проведены также реакции дигидрида дифенилолова с двумя
эквивалентами моновинильных соединений кремния, германия и свинца. Однако
в этом случае ожидаемые продукты присоединения не образуются, а
выделены только соединения состава (C6H5)3SnCH2CH2M(C6H5);> [39]. Вероятно,
это объясняется диспропорционированием оловоорганического дигидрида.
2 (СвН3)2 SnH2 -* (СвН5)3 SnH +.CeH6SnH3.
Для идентификации соединений с двумя и более атомами металла в
молекуле предложено применять метод ИК-спектроскопии [39].
О реакциях оловоорганических дигидридов с дивинильными и ди-я-
стирильными соединениями кремния, германия, олова и свинца см. стр. 513.
ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ К АЛКИНАМ
Оловоорганические гидриды алифатического ряда присоединяются к
алкинам легче, чем к алкенам [4, 14, 40]. В ряде случаев гидростаннирова-
ние алкинов проходит экзотермически.
Реакция эквимолекулярных количеств оловоорганического гидрида и
алкина является общим методом синтеза винильных соединений олова
R3SnHe+ CH==CR4 -> R3SnCH=CHR'.
Так, три-я-бутилвинилолово получено [41, 42] с выходом 75—80%
взаимодействием гидрида три-я-бутилолова с ацетиленом в бензоле при
избыточном давлении ацетилена 10,2—13,6 атм. В качестве катализатора
применяют 1% азодиизобутиронитрила.
Получение три-«-пропилгексенил-1-олова [4]. 9,3 г (0,035 моля) гидрида трипропилолова
и 4,9 г (0,06 моля) гексина-1 смешивают при комнатной температуре. Смесь нагревают при
90° С в течение б час и перегоняют в вакууме. Получают 9,5 г (82%) вещества с т. кип.
75—80° С/0,1 мм.
288
Оловоорганические соединения. Лит. стр. 317—318
Фенилацетилен экзотермически реагирует с гидридом трипропилолова.
Перегонкой смеси в вакууме получают [4] |3-трипропилстаннилстирол, т. кип.
119—120° С/0,0001 мм. Выход 75%^
Получение р-триэтилстаннилстирола [5]. 6,13 г (0,06 моля) дважды перегнанного фе-
нилацетилена и 6,21 г (0,03 моля) гидрида триэтилолова нагревают при 80 ± 0,2° С с 0,248 г
(0,0015 моля) азодиизобутиронлтрила. Происходит экзотермическая реакцчя, с
количественным выходом образуется вещество с т. кип. 87—97°С/0,15 мм, п2£ 1,5579. В его ИК-спе-
ктре наблюдается интенсивная полоса в области 988 см'1, характерная для траяс-изомера.
Реакция между гидридом триэтилолова и диэтиловым эфиром ацети-
лендикарбоновой кислоты проходит уже при 0° С [1]. Перегонка смеси в
вакууме даег 86% продукта присоединения, т. кип. 125—126° С/2 мм;
df 1,2660; по 1,4921.
Аналогично получены [1] продукты присоединения гидрида
триэтилолова к диметиловому эфиру ацетилендикарбоновой кислоты (выход 73%,
т. кип. 115—116° С/3 мм) и гидрида трипропилолова к диэтиловому эфиру
ацетилендикарбоновой кислоты (выход 66%, т. кип. 140° С/1 мм).
В том случае, когда тройная углерод-углеродная связь активирована
только одной сложноэфирной группой (эфир тетроловой кислоты), для
получения продукта с высоким выходом требуется более высокая температура.
Реакция гидрида триэтилолова с метиловым эфиром тетроловой кислоты [1]. К 13,85 г
гидрида триэтилолова прибавляют 6,57 г метилового эфира тетроловой кислоты. Смесь
нагревают до 120° С в течение 10 мин. и перегоняют в вакууме. Получают 13 г (65%) продукта
присоединения с т. кип. 80—82° С/2 мм\ df 1,2594; п2£ 1,4979.
Аналогично проходит реакция между гидридом трипропилолова и
метиловым эфиром тетроловой кислоты (выход 77,5%; т. кип. 116,5° С/2 мм).
Значительный интерес представляет разработанный Несмеяновым с
сотр. [43—45J метод синтеза биметаллических производных этилена путем
присоединения оловоорганических гидридов к металлорганическим моно-
ацетиленидам
R3SnH + R^IV^feCH -> R3SnCH=CHM"R„_r
Гидриды три-я-пропил- и три-я-бутилолова при нагревании легко
присоединяются к триэтилэтинилкремнию с образованием 1-триэтилсилил-2-три-
алкилстаннилэтилена.
Получение 1-триэтилсилил-2-три-я-пропилстаннилэтилена [44]. Слитые при комнатной
температуре 3,6 г тр и эти л эти нил к ремни я и 5 г гидрида три-«-пропилолова нагревают при
60—65° С в аргоне в течение 4 час. При перегонке в вакууме собирают фракцию с т. кип.
126° С/2 мм; df 1,0585; п™ 1,4860; выход 5 г (64%).
Получение 1-триэтилсилил-2-три-я-бутилстаннилэтилена [43]. Смесь 5,55 г
гидрида три-«-бутилолова и 2,90 г триэтилэтинилкремния нагревают при 90—95° С в течение
9 час. По охлаждении продукты реакции перегоняют в вакууме. Получают 3,36 г (41%)
вещества с т. кип. 144—146° С/2 мм; df 1,0282; п2£ 1,4851. Выделено 2,57 г исходного
гидрида три-«-бутилолова.
В аналогичных условиях с три-я-пропил- и три-я-бутилэтинилоловом
получены бш>1,2-(три-я-пропилстаннил)этилен (выход 42%; т. кип.
164° С/1,5 мм) и бш>1,2-(три-я-бутилстаннил)этилен (выход 73,6%; т. кип.
177—178° С/2 мм).
Исследованы реакции этих соединений с хлором, хлористым водородом
и хлорной ртутью. Интересно отметить, что в случае 1-триэтилсилил-2-
Присоединение по кратным связям.
289
триалкилстаннилэтилена образуется хлористое триалкилолово, тогда как
связь Si—С остается не затронутой. Молекула бш>(триалкилстаннил)-
этилена рвется в двух местах.
Гидрид три-я-бутилолова легко присоединяется и к три-я-бутилэтинил-
германию.
Получение 1-три-#-бутилстаннил-2-три-/^-бутилгермилэтилена [43]. 6,4 г гидрида
три-я-бутилолова и 5,91 г\ три-я-бутилэтинилгермания нагревают в токе аргона при
100° С в течение 9 час. и 4 часа при 125°С. При перегонке продуктов реакции в вакууме
собрана фракция с т. кип. 168—170° С/2 мм- df 1,0825; л$ 1,4897; выход 79,6%.
Изучение продуктов присоединения оловоорганических гидридов к эти-
нильным соединениям кремния, германия и олова методами ИК-спектров
и спектров ЯМР показывает, что реакция идет в ^с-положение с
образованием транс-изомеров этиленового ряда, возможно, через л-комплекс или
четырехчленное переходное состояние [43]
R3M H
R3MC==CH + R3SnH -* R3MC=CH -> R3MC=CCH \ /
1 i—-i _* c=C
HSnR3 H SnR3 / \
H SnR3
Исследована [45] реакция дигидрида ди-я-бутилолова с три-#-бутил-
этинилоловом. Найдено, что при нагревании реагентов до 125° С в токе ар-
гона в течение 2 час. процесс идет в двух направлениях. Перегонкой
реакционной массы выделен бш;-1,2-(три-я-бутилстаннил)этиленст. кип. 180—
182° С/2 мм\ по 1,5041. Спектр ЯМР указывает на присутствие 95% транс»
изомера и 5% ^ш>изомера.
Второй продукт реакции ди-я-бутил-б^с-ф-три-я-бутилстаннилвинил)-
олово имеетт. кип. 155—157° С/2-10~2лш; п™ 1,5176.
CH=CHSn (C4H9)3
(rc-C4HQ)2 SnH2 + 2 (/г-С4Н9)3 SnC=CH -* (я-С4Н9)2 Sn
\
CH=CHSn (QH9)3.
Реакция гидрида трифенилолова с дифенилэтинилстибином приводит к
1-трифенилстаннил-2-дифенилстибилэтилену [45а]:
(СбН5)2 SbC=CH + (QH5)3 SnH -► (C6H5)2SbCH=CHSn (СбН5)3.
Ди-я«бутилэтинилстибин реагирует с гидридом трифенилолова с
образованием 1-трифенилстаннил-2-ди-я-бутилстибилэтилена и 1,2-б«с-(три-
фенилстаннил)этилена. Последний возникает в результате обмена водорода,
связанного с атомом олова, на этинильную группу, соединенную с сурьмой
(QH9)2 SbC=CH + (СбН5)з SnH -> (QH9)2 SbH + (QH5)3 SnC=CH,
(C6H5)3 SnC-CH + (СбН5)з SnH -> (СбН5)3 SnCH=CHSn (СбН5)3.
Иначе реагирует ди-я-бутилэтинилстибин с гидридом триметилолова.
При этом выделены 1-(триметилстаннил)-2-(ди-я-бутилстибил)этилен и
триметилстаннилди-я-бутилстибин, который является продуктом
вторичного процесса, протекающего по радикальному механизму [45а]:
(QH9)2 SbC=CH + (СН3)з SnH -> (C4H9)2 Sb* + (СН3)3 Sn* + Н' + СН=С\
(С4Н9)2 Sb' + (СН8)з Sn* -* (С4Н9)2 SbSn (СН3)3,
СН=С + Н* -> СНееСН или СН=С—С=СН + Н2.
19 Заказ № 207
290
Оловоорганические соединения. Лит. стр. 317—318
Реакции оловоорганических гидридов с диинами проходят ступенчато.
Так, взаимодействием гидрида триэтилолова с эквимолекулярным
количеством диацетилена или этилдиацетилена получены 1-триэтилстаннилбутен-
1-ин-З (т. кип. 50—51° С/2 мм) и 1-триэтилстаннилгексен-1-ин-3 (т. кип.
88—89° С/2 мм) соответственно [46]. Гидрид триметилолова образует с
1,6-гептадиином только один продукт присоединения: (CH3)3SnCH^CH-
(CH2)3CH=CHSn(CH3)3 [27]. Из гидрида триметилолова и 1,8-нонадиина
получают [27] (СНз)з5пСН=СН(СН2)5СН=:СН5п(СНз)з наряду с примесью
(~5%) (CH3)3SnCH=CH (CH2)5C [Sn (СН3)3]=СН2 и очень небольшого
количества продукта моноприсоединения.
Реакции оловоорганических гидридов ароматического ряда с алкинами
описаны на небольшом числе примеров. При смешении гидрида трифенил-
олова с фенил ацетиленом происходит экзотермическая реакция. С выходом
94% образуется Р-трифенилстаннилстирол, т. пл. 119—120 ° С (из петро-
лейного эфира) [4].
Нагреванием до 60° С в течение 2 час. эквимолекулярных количеств
гидрида трифенилолова и пропаргилового спирта получено [4] (3-окси-
пропенил-1)-трифенилолово. Выход 51%, т. пл. 115—125° С.
Продукты присоединения алкина к эквимолекулярному количеству оло-
воорганического гидрида могут реагировать со второй молекулой гидрида
[4, 27]. Так, если ацетилен пропускать через избыток гидрида
трифенилолова, то он присоединяет две молекулы последнего. Аналогичным образом
проходят реакции фенилацетилена с двумя молекулами гидрида
трифенилолова, а также метилового эфира пропиоловой кислоты с двумя
эквивалентами гидридов трипропил- или трифенилолова. В результате этих реакций
образуются, вероятно, соединения следующего состава (R3Sn)2CHCH2C6H5
и (R3Sn)2CHCH2COOCH3 соответственно [4, 22а]. О реакции
оловоорганических дигидридов с соединениями ряда ацетилена см. стр. 514.
Механизм реакции гидростаннирования алкинов до последнего времени
оставался малоизученным. Как отмечено на стр. 287, азодиизобутиронитрил
ускоряет присоединение оловоорганических гидридов к ацетилену [41,
42] и другим алкинам [5, 15]. Ингибиторы радикалов и катализаторы ради-
калобразующего типа существенно влияют на скорость реакции фенил-
ацетилена с гидридом триэтилолова, что свидетельствует в пользу
радикального характера процесса [5, 14]. Впоследствии было установлено, что при
гидростаннировании алкинов имеют место как ионные, так и радикальные
реакции. Вывод сформулирован и обоснован Ван-дер-Керком и его школой
[47—49]. Исследованиям в этой области посвящена монография Лейсинка
[22аI. Изложение механизма реакции гидростаннирования алкинов ведется
здесь в основном по этой работе.
При установлении механизма химических реакций важную роль играет
изучение структуры образующихся соединений. Однако соответствующие
исследования начаты сравнительно недавно. В 1963 г. Сейферт и Вон [501
нашли, что продукт присоединения гидрида триметилолова к метилацети-
лену представляет собой смесь цис-$- и гар£шс-|3-аддукта с небольшой
примесью изопропенилтриметилолова. Согласно цитируемым Лейсинком данным
Фултона, присоединение оловоорганических моногидридов к
фенилацетилену приводит к цис-fi- и /пра/-/с-|3-аддуктам. Последний возникает в
результате перегруппировки первоначально образующегося цис-$-аддуктп:
Н II R3Sn H
\ / \ • /
RsSnH + 1 IC=CCeH5 -> С=С -> С-=С
/ \ / \
R3Sn С6Н5 Н С6Н5
цис-$- транс-$-
Присоединение по кратным связям 291
Исследована [51] также структура продуктов взаимодействия 2 молей
гидрида трифенилолова с 1 молем диацетилена или /г-диэтинилбензола. Их
ИК-спектры указывают на наличие группы
Н Н
\ /
/ \
SnR8
и исключают присутствие групп
R3Sn
\
С=СНо.
/
Следует отметить, что во всех рассмотренных выше примерах атом олова
присоединяется исключительно или почти исключительно к незамещенному
углеродному атому. Ван-дер-Керк с сотр. впервые установили, что при гид-
ростаннировании алкилацетиленмонокарбоксилатов образуются
значительные количества и других веществ. Впоследствии теми же авторами
детально исследовано строение продуктов взаимодействия гидридов триал-
кил- и трифенилолова с пропаргиловым спиртом, алкилпропиолатами, этил-
тетролатом, эфирами ацетилендикарбоновой кислоты и другими
замещенными ацетиленами [22а, 47—49]. Реакции проводились путем нагревания
смеси реагентов под азотом. За их ходом наблюдали по изменению
характеристической полосы Sn—Н в области 1800—1850 см"1 и по результатам
анализа смеси методом газо-жидкостной хроматографии. Полученные
соединения охарактеризованы элементарным анализом, ИК- и ЯМР-спектрами и
реакциями деалкилирования. Выходы различных продуктов рассчитывались
по данным газо-жидкостной хроматографии и спектров ЯМР. Найдено, что
нагревание до 100° С гидрида триэтилолова (1 моль) с пропаргиловым
спиртом (2 моля) приводит к смеси аддуктов состава
Н SnR3 H H R3Sn H
\ / \ / \ /
с=с
/ \
Н СН2ОН R3Sn
I
Их общий выход около 70%, относительные количества 1:11:111 =
= 25 : 45 : 30. Те же соединения получают и при реакции
эквимолекулярных количеств пропаргилового спирта с гидридом трифенилолова. В этом
случае отношение 1 : 11 : 111 = 5 : 45 : 50.
Гидриды триалкилолова очень активно реагируют с ацетилендикарбокси-
латами.В ИК-спектре, снятом сразу же после прибавления гидрида триметил-
олова к избытку (100%) диэтилового эфира ацетилендикарбоновой кислоты
при 0° С, отсутствует полоса связи Sn—Н. Спектр ЯМР указывает на
присутствие в смеси лвух аддуктов состава 1:1, образующихся с
количественным выходом в отношении IV : V = 9 : 1 [22а, 47, 49а]
СаНзООС SnR3
\ /
R3SnI I 4- C2H5OOCC=CCOOC2H5 -» С=С +
/ \
Н СООС2Н5
IV
И SnR3
+ с=с
/ \
ОаНбООС СООС2Н5
V
с=с
\
СНаОН
И
с=с
/ \
н сн2он
III
19*
292
Оловоорганические соединения. Лит. стр. 317—318
Более сложно проходит реакция между эквимолекулярными
количествами гидрида триметилолова и этилтетролата [22а, 47, 49а]
СНз SnR3
(СН3)з Snll -,'- CH3C=CCOOC2H5 -> С-С +
/ \
Н СООС2Н5
VI
Н SnR3 СНз Н
\ / \ /
+ с=с + с=с
/ \ / \
СНз СООСзНз R3Sn COOC2H5
VII VIII
С алкилпропиолатами гидриды триметил-, триэтил- и трипропилолова
образуют следующие соединения:
Н COOR' R3Sn COOR'
\ / \ /
RзSnCH2CH2COOR, С=С С=С
/ \ / \
Н SnR3 Н Н
IX X XI
Н COOR'
\ /
С=С и R3SnC=CCOOR'
/ \
R3Sn H
XII
При этом основными продуктами являются алкил-а-триалкилстаннилакри-
латы. Выход a:iкил-^«с-[3-триалкилстаннилакрилатов и алкил-транс-$-
триалкилстаннилакрилатов возрастает при переходе от гидрида
триметилолова к гидриду трипропилолова. В том же ряду увеличивается и скорость
реакции присоединения [22а, 49].
Гидрид триметилолова экзотермически реагирует с цианоацетиленом
уже при комнатной температуре. С выходом 90% получают а-триметил-
станнилакрилонитрил (т. кип. 78—80° С/18 мм) [22, 47]. Еще более
энергично проходят реакции между гидридами триметил- или триэтилолова и
дицианоацетиленом, в которых требуется применение растворителя [22а,
49а]. При этом, как и при гидростаннировании цианоацетилена, образуется
только одно соединение (выход более 80%):
NeeC SnR3
R3SnH + NC—СееС—CN -» C=C
/ \
H C=N
XIII
Детально исследовано также взаимодействие гидридов триалкилолова
с 1-гексином, фенилацетиленом, этоксиэтином и бутилтиоэтином [22а, 49а].
Нагревание до 50° С в течение 5 час. гидрида триэтилолова с 1-гексином
дает смесь трех соединений в отношении XIV : XV : XVI = 3 : 51 : 46
(общий выход 55%).
Н SnR3 H H R3Sn H
RsSnH + НСееСС4Н9 -> С=С + С=С + С=С
/ \ / \ / \
Н С4Н9 R3Sn QH9 Н С4Н9
XIV XV XVI
Присоединение пи кратным связям
293
Увеличение продолжительности реакции повышает выход продукта,
изменяя его состав. Так, если смесь нагревать 6 час. при 50° С, а затем оставить
ее на 16 час. при 20° С, то выход продукта составляет 75%, а отношение
компонентов XIV : XV : XVI = 3 : 33 : 64. Нагревание при 50° С той же
смеси в течение 12 час. с последующим 16-часовым выдерживанием при 20° С
дает 85% продукта с отношением 3 : 31 : 66. Аналогичным образом ведет
себя гидрид триметилолова. С гидридом трифенилолова образуются, по-
видимому, только соединения типа XIV и XV.
Интересно отметить, что с фенилацетиленом гидрид триметилолова
реагирует гораздо медленнее, чем с гексином (ср. с гидростаннированием
соответствующих алкенов, стр. 278). Даже после нагревания смеси до 50° С
в течение 66 час. в ней все еще присутствует около 25% исходного оловоор-
ганического гидрида. Как следует из данных газо-жидкостной
хроматографии, реакция приводит к трем аддуктам в отношении XVII : XVIII : XIX =
= 6 : 66 : 28
Н Sn(CH,)3 Н Н
\ / \ /
(CH8)8SnH + HC=CCeH6- С=С + С=С +
/ \ / \
Н СбН5 (CH3)3Sn С6Н5
XVII XVIII
(CH3)3Sn H
+ с=с
н сбн5
XIX
Гидрид триэтилолова с фенилацетиленом а-аддукта, по-видимому, не
образует. Что касается этоксиэтина, то при нагревании 80 мин. до 50° С он
образует с гидридом триэтилолова три продукта в отношении XX : XXI :
: XII*- 4 : 91 : 5
Н SnR3 H H R3Sn H
\ / \ / \ /
R8SnH+ НС=СОСН5-> С=С + С=С + С=С
/ \ / \ / \
Н ОС2Н5 R3Sn OC2H5 H ОСаН5
XX XXI XXII
Если гидрид триэтилолова нагревать с этоксиэтином в среде бутиронитрила,
то получают практически чистый г^с-Р-аддукт (XXI). Последний является
основным продуктом взаимодействия этоксиэтина с гидридом
трифенилолова.
Описанные выше результаты изучения структуры продуктов гидро-
станнирования алкинов позволяют заключить [22а], что обычно в
результате реакции образуется смесь различных соединений. При этом с моноза-
мещенными ацетиленами, содержащими сильные электроноакцепторные
заместители (COOR, CN), гидриды триалкилолова образуют в основном а-
аддукты. Если же в составе молекулы алкина имеются электронодонорные
(С4Н9,ОС2Н5) или слабые электроноакцепторные (СН2ОН,С6Н5) заместители,
то получают главным образом цис- и траяс-^-аддукты с примесью а-аддукта.
Что касается дизамещенных ацетиленов, то с гидридами триалкилолова они
дают продукты траяс-присоединения.
Дальнейшие опыты показали, что прибавление к смеси алкина и олово-
органического гидрида феноксила уменьшает количества р-аддуктов, но
почти не влияет на выход а-аддукта (в случае метилпропиолата). Напротив,
294
Оловоорганические соединения. Лит. стр. 317—318
азодиизобутиронитрил способствует образованию Р-аддукта за счет
уменьшения количества а-аддуктов. Таким образом, [3-аддукты образуются по
крайней мере частично по свободнорадикальному механизму. Образование
а-аддуктов, вероятно, проходит иным путем. С целью изучения этого
вопроса было исследовано влияние растворителя на гидростаннирование
ацетиленов, содержащих сильные электроноакцепторные заместители [22а,
48]. Исследование реакции гидрида триметилолова с метил пропиолатом
и цианоацетиленом в полярных и неполярных растворителях с различным
значением диэлектрической постоянной е показало, что скорость
образования а-аддукта гораздо больше в бутиронитриле (е = 20,3), чем в декане
(е = 1,99) или 1,2,4-триметилбензоле (е = 2,38). Скорость же
образования ^ис-Р-аддукта практически одна и та же в каждом из этих
растворителей. Более того, значительно отличается и форма кривых, выражающих
зависимость концентрации а- и Р-аддуктов от продолжительности реакции. В то
время как концентрация а-аддукта постепенно повышается со временем,
^с-Р-аддукт образуется практически сразу после смешения реагентов и
затем его количество не изменяется. Аналогичные результаты получены при
гидростаннировании метилпропиолата, цианоацетилена, диэтилацетилен-
дикарбоксилата и дицианоацетилена гидридом триэтилолова. Зависимость
скорости образования а-аддуктов в этих реакциях от полярности среды
служит веским доводом в пользу ионного механизма их образования. Этот
вывод подтверждается результатами, полученными при гидростаннировании
цианоацетилена (в бутиронитриле) и диэтилацетилендикарбоксилата (в
бутиронитриле или циклогексане) гидридом триэтилолова в присутствии
феноксила или азодиизобутиронитрила. Найдено, что феноксил практически
не влияет на скорость указанных реакций. Прибавление первых порций
азодиизобутиронитрила вне зависимости от применяемого растворителя
приводит к двойственному эффекту. Скорость образования а-аддукта не
изменяется, но наблюдается резкое и внезапное увеличение количества цис-$-
аддукта и одновременно в смеси появляется тра^с-Р-аддукт. Дальнейшее
прибавление азодиизобутиронитрила гораздо менее эффективно. Эти
данные свидетельствуют о том, что транс-$-аддукт образуется по свободноради-
кальной реакции. Как будет показано на стр. 299, транс-$-адду кты
возникают в результате вызываемой свободными радикалами перегруппировки
ifwc-p-аддуктов. Прибавление к смеси феноксила полностью прекращает
этот процесс.
Таким образом, при гидростаннировании ацетиленов, содержащих у
тройной связи сильные электроноакцепторные заместители, образование
а-аддуктов проходит по ионному механизму. цис-$-Аддукты образуются в
результате свободнорадикального присоединения оловоорганического
гидрида к алкинам. траяс-р-Аддукты являются продуктами свободнорадикаль-
ной изомеризации ^с-Р-аддукта.
В цитированной (на стр. 290) книге Лейсинка подробно рассмотрена
стереохимия ионной реакции присоединения. Автор подчеркивает тот факт, что
при гидростаннировании диэтилацетилендикарбоксилата в отсутствие
растворителя образуется почти исключительно продукт траяс-присоединения
(см. стр. 291). Аналогичные результаты получены [22а, 496] и при
проведении следующих реакций (также без растворителя):
Н Sn(C2H5)3 D Sn(C2H5)3
\ / \ /
(QH5)3snD + Hc=a:oocH3-> c=c + с=с
/ \ / \
D СООСНз И СООСНз
XXIII (93%) XXIV (7%)
Присоединение по кратным связям
295
Н Sn(C2H5)3 D Sn(C2H5)3
\ / \ /
(С2НзЬ SnD A- IICeeeCN -> С-С + С=С
/ \ / \
D CN H CNT
XXV (95%) XXVI (о%)
D SnR3 H SnR3 H SnR3
DC=GCN 86о/0 \ / \ / \ /
R3SnH + -> С=С + С=С + С=С
HCeeCCN 14% / \ / \ / \
Н CN D CM H CN
XXVII (82%) XXVIII (4%) XXIX (14%)
(R = СН3; С2Н5; С4Н9).
Изомеры XXIV, XXVI и XXVIII, присутствующие в реакционной смеси в
небольших количествах, образуются в результате перегруппировки
первичных продуктов реакции [22а, 496J. Эта изомеризация вызывается оловоор-
ганическими радикалами. Действительно, при нагревании до 60° С смеси
изомеров XXIII и XXIV (XXIII : XXIV = 78 : 22) в циклогексане в
течение 160 мин. изомеризации не наблюдается. Соотношение компонентов не
изменяется в заметной степени и в присутствии небольших количеств азоди-
изобутиронитрила или дейтерида триэтилолова. Однако при одновременном
прибавлении этих двух веществ образуется равновесная смесь состава
1 : 1.
Ряд экспериментов проводился в присутствии различных растворителей.
Изучена стереохимия следующих реакций: дейтерида триэтилолова с циано-
ацетиленом в бутиронитриле и метиловом спирте; гидрида триэтилолова с
диэтиловым эфиром ацетилендикарбоновой кислоты в бутиронитриле и
циклогексане; гидридов триметил- и триэтилолова с дицианоацетиленом в тех
же растворителях.
В каждом из приведенных выше случаев, как это следует из спектров
ЯМР, наблюдается транс-присоединение (—9596). Итак, в ионных
реакциях присоединения в растворителе или без него в основном проходит
шраяс-присоединение [22а, 496].
Существенное влияние на скорость ионной реакции присоединения
оказывает характер заместителя как в молекуле оловоорганического гидрида,
так и в молекуле алкина. Найдено [22а], что скорость ионной реакции гид-
ростаннирования цианоацетилена в бутиронитриле увеличивается в
следующем ряду (C6H5)3SnH << (CH3)3SnH < (C2H5)3SnH ж (C4H9)3SnH. Та
же последовательность наблюдается и при гидростаннировании диэтило-
вого эфира ацетилендикарбоновой кислоты в бутиронитриле или
циклогексане: (C6H5)3SnH < (CH3)3SnH <C (C2H5)3SnH << (C4H9)3SnH. Таким образом,
стерические факторы не играют здесь большой роли, так как иначе следовало
бы ожидать увеличения реакционной способности в ряду (C6H5)3SnH <^
< (C4H9)3SnH < (C2H5)3SnH < (CH3)3SnH.
Очевидно, что скорость ионных реакций присоединения существенно
зависит от индукционного эффекта. Прохождению реакции способствуют элект-
ронодонорные заместители у атома олова.
Заместители у тройной углерод-углеродной связи оказывают обратное
влияние. Как уже неоднократно отмечалось выше, ионная реакция гид-
ростаннирования проходит в заметной степени только в том случае, если
у тройной углерод-углеродной связи находятся электроноакцепторные
заместители.
Полученные результаты свидетельствуют в пользу нуклеофильной атаки
водорода оловоорганического гидрида на тройную углерод-углеродную связь
296
Оловоорганические соединения. Лит. стр. 317—318
алкина. Эта стадия, вероятно, определяет скорость ионной реакции
гидростаннирования.
Дальнейший свет на механизм ионных реакций гидростаннирования
проливает изучение кинетического изотопного эффекта [22а, 496].
Установлено, что ионные реакции гидростаннирования алкинов имеют
суммарный второй порядок и первый порядок по каждому из реагентов.
При измерении константы скорости гидростаннирования алкинов гидридом
и деитеридом триэтилолова оказалось, что во всех изученных случаях
Предполагая, что на стадии, определяющей скорость реакции, водород
переходит от атома олова к атому углерода, можно ожидать следующих
величин изотопного эффекта:
а) если в этом переходном состоянии водород все еще полностью
связан с оловом, то изотопный эффект будет несколько больше единицы;
б) если связь Sn—Н разорвана, но взаимодействие С—Н все еще слабое,
то в этом случае изотопный эффект должен быть больше единицы;
в) если водород полностью или почти полностью связан с углеродом
вследствие большого различия нулевой точки энергий связей Sn—Н и
Sn—D, изотопный эффект должен быть меньше единицы.
Из полученных экспериментально величин изотопного эффекта видно,
что перенос водорода от атома олова к атому углерода осуществляется на
стадии, определяющей скорость процесса.
На основании всех этих данных предлагается [22а, 496] следующая схема
ионной реакции гидростаннирования:
R3SnH + R'C=CR"
(-И R\ <"> быстро к\ / 6
-^R.iSn + h/C=C4r„ . HA=CxR..
Следует подчеркнуть, что приведенные на стр. 292—£96 результаты
относятся к гидростаннированию алкинов, содержащих электроноакцептор-
ные заместители.
Переходя к рассмотрению механизма гидростаннирования ацетиленов,
содержащих электронодонорные заместители, отметим, что с ними реакции
проходят так же, как и с алкинами, содержащими одновременно
электронодонорные и электроноакцепторные заместители. Механизм
гидростаннирования монозамещенных ацетиленов, содержащих слабые
электроноакцепторные заместители, не исследовался, но, по-видимому, здесь
характерны те же закономерности [22а].
Механизм гидростаннирования алкинов, содержащих
электронодонорные заместители, изучался на примере гексина, этилтетролата и этокси-
этина. Опыты, проведенные с гидридами триметил- и триэтилолова в
различных растворителях, показали, что скорость гидростаннирования этих
алкинов практически не зависит от полярности среды. Напротив,
прибавление к смеси небольших количеств азодиизобутиронитрила или фенок-
сила оказывает очень большое влияние на эти реакции. В присутствии
феноксила некоторые из них не проходят вообще. Прибавление первой
порции азодиизобутиронитрила приводит к резкому увеличению общего
количества образующего аддукта.
R'v
>с=с>
"•SnR.
Присоединение по кратным связям -
297
Дальнейшее добавление азодиизобутиронитрила гораздо менее
эффективно. Все эти результаты показывают, что рассматриваемые реакции
проходят по радикальному механизму.
Следует подчеркнуть, что при гидростаннировании нуклеофильных
(1-гексин) и слабо электрофильных (фенилацетилен)алкинов образуются
преимущественно Р-аддукты, а при гидростаннировании электрофильных
(цианоацетилен)алкинов — сс-аддукты. Таким образом, образование а-ад-
дуктов проходит в основном по ионной реакции, а Р-аддуктов — только по
радикальной реакции. (Учитывается возможность образования небольших
количеств а-аддукта при свободнорадикальном гидростаннировании
нуклеофильных алкинов.)
Как отмечено на стр. 293, Р-аддукты представляют собой обычно смесь
цис- и транс-изомеров. На примере гидростаннирования 1-гексина было
показано [22а], что во время реакции отношение между количествами цис-
и транс-$-аддуктов значительно уменьшается. Цис-транс-изомеризация
происходит также в процессе присоединения гидрида триметилолова к
этилтетролату. В этом случае уменьшается величина отношения цис-
и транс-а-изомеров, тогда как цис-^-ajxjxywi вообще не изомеризуется. Гид-
ростаннирование метилпропиолата гидридом триметилолова приводит
только к а- и tyttc-P-аддуктам. На этих и ряде других примеров установлено,
что первичными продуктами при гидростаннировании алкинов являются
^ис-аддукты, а транс-аддукты образуются в результате их
перегруппировки. Изучение этой перегруппировки показало, что она проходит под
влиянием триалкилстаннильных радикалов [22а]. Так, цис-^-аддукты,
полученные из гидрида триметилолова и метилпропиолата или цианоацетилена, изс-
меризуются только при одновременном прибавлении оловоорганического
гидрида и азодиизобутиронитрила. При гидростаннировании 1-гексина или
этилтетролата скорость изомеризации соответственно цис-§- и цис-а-аддук-
тов значительно увеличивается при прибавлении азодиизобутиронитрила.
Напротив, небольшие количества феноксила полностью прекращают этот
процесс. Например, при гидростаннировании этилтетролата отношение
цис- и транс-а-адду ктов равно 14. Эта величина не изменяется в
присутствии феноксила, а без него уменьшается в ходе реакции до значения 5,4.
После прибавления азодиизобутиронитрила отношение цис-а- к транс-а-
равно 3,8. Убедительное доказательство участия оловоорганического радикала
в этих процессах получено при изучении изомеризации ^шхх-аддукта
дейте р ид а триметилолова с метилпропиолатом. Этот аддукт не
перегруппировывается ни термически, ни под влиянием азодиизобутиронитрила.
В присутствии дейтерида триэтилолова образуется небольшее количество
транс-а- адд у кта.
Если же к цис-а-аддукту одновременно прибавить дейтерид
триэтилолова и азодиизобутиронитрил, то образуется равновесная смесь цис- и
транс-изомеров состава 1:1.
Таким образом, общая схема процессов, проходящих при радикальном
гидростаннировании алкинов, может быть представлена схемой [22а],
приведенной на стр. 298.
Аналогичная схема предложена и для свободнорадикального
гидростаннирования алкенов. В реакции инициирования R" — какой-либо
радикал, который отщепляет водород от оловоорганического гидрида.
Ненасыщенные реагенты не претерпевают теломеризации или полимеризации до
тех пор, пока в смеси имеется непрореагировавший оловоорганический
гидрид. Это означает, что реакции (1) и (4) протекают быстрее, чем процессы
полимеризации или теломеризации, соответственно.
Скорость гидростаннирования алкинов определяется одной из следую-
щих стадий (см. схему, стр. 298).
2d8
Оловоорганические соединения. Лит. стр. 317—318
R3Sn' -
-СееС— ^R3Sn—C=C
k-.
(3)
R3Sn—C=C + R8SnH -Л R3Sn—C=C—H + R3Sn. (4)
Если &_з<С k± [R3SnH], то скорость реакции определяется стадией (3).
Если же k-з 5^> kA [R3SnH], то она определяется стадией (4).
СХЕМА
R3SnH + R1' > R3Sn* + R"H
(i)
H SnR3
\ /
R3Sn* + C=C
t
RsSnH
H I SnR3
RgSn'+П—C=C—R'-
>C=C
(2)
II
(3)N
R'
►C=C
R3Sn
(4)
R'
R3SnII
H
\ /
R3Sn* + C=C
R3Sn R'
цис-$
!R3Sn
\ /
R3Sn*+ C=C
шранс-$
Выяснение этого вопроса проводили следующим образом. К алкину
одновременно прибавляли два различных оловоорганических гидрида,
образующиеся соединения анализировали методами газо-жидкостной
хроматографии и ИК-спектроскопии. Установлено, что скорость реакции
присоединения гидридов триалкилолова кметилпропиолату определяется,
вероятно, стадией (3). При свободнорадикальном гидростаннировании метилпро-
пиолата гидрид триметилолова менее реакционноспособен, чем гидрид
триэтилолова: (CH3)3SnH <C (C2H5)3SnH. По отношению к 1-гексину и этокси-
этину гидриды триметил- и триэтилолова обладают примерно одинаковой
реакционной способностью, тогда как гидрид трифенилолова более
активен: (C6H5)3SnH > (CH3)3SnH ж (C2H5)3SnH. Следует отметить, что при
одновременном гидростаннировании алкина двумя различными оловооргани-
ческими гидридами может иметь место равновесие:
R3Sn' + R3SnH ^± R3SnH + R3Sn'
k-ъ
Присоединение по кратным связям
299
Это означает, что отношение аддуктов (а именно, |3-(CH3)3Sn : P-(C2H5)3Sn)
определяется состоянием этого равновесия и константой скорости реакции
(3) (см. схему). Если R—алкил, a R'—фенил, то равновесие между
R3Sn' и R'3Sn* может быть значительно сдвинуто в правую сторону.
Следовательно, опыты со смесью гидридов триалкил- и трифенилолова не
позволяют сделать каких-либо выводов о реакции (3). Можно предполагать, что
при гидростаннировании 1-гексина смесью гидридов триметил- и триэтил-
олова константа равновесия \ будет близка к 1. При замене 1-гексина или
этоксиэтина метилпропиолатом отношение |3-(С2Н5)3 Sn/|3-(CH3)3Sn
увеличивается от 1 до 1,6. Вероятно, это обусловлено реакцией (3). Различие
реакционной способности триалкилстаннильных радикалов по отношению к
нуклеофильным (например 1-гексину, (CH3)3SrT > (C2H5)3Sn') и электро-
фильным ацетиленам (например, метилпропиолату, (CH3)3Sn' < (C2H5)3Sn*)
может быть объяснено одной из следующих схем:
\ . \ \ (-)
С =С <-* С=С <-» С=С
I. \ ' \ \ m (-> \ \ ' \
—Sn* R — Sn'+) R — Sn(^ R
/ / /
\ \ 4
c=c ~ c=c <+ c=c^+)
—Sn* R — Sn^ R — Sn(-} R
/ / /
Влияние полярности заместителя играет, по-видимому, важную роль при
присоединении оловоорганических гидридов к электрофильным и
нуклеофильным ацетиленам. Наблюдаемая зависимость между относительными
количествами продуктов гидростаннирования и природой алкинов приводит
к следующему выводу. При гидростаннировании метилпропиолата скорость
реакции определяется стадией (3), а в случае этоксиэтина или 1-гексина —
стадией (4).
Заканчивая рассмотрение механизма, по которому проходит свободно-
радикальное гидростаннирование алкинов, необходимо остановиться на
механизме реакции изомеризации. Для его выяснения оловоорганический
гидрид R3SnH смешивался с аддуктом состава R3SnCH=CH—R". За
проходящими при этом реакциями обмена, изомеризации и
гидростаннирования наблюдали с помощью газо-жидкостной хроматографии. Найдено, что
при прибавлении гидрида триметилолова к ф^-|^триэтилстаннилэтоксиэти-
лену, помимо изомеризации, наблюдается главным образом реакция
обмена
(С2Н5)3 SnCH=CHOC2H5 + (СНз)з SnH -> (CH3)3 SnCH=CHOC2H5 + (C2H5)3 SnH.
Аналогичным образом проходит взаимодействие гидрида триметилолова
с 1-триэтилстаннил-1-гексеном. С метил ^ш>р-триэтилстаннилакрилатом
гидрид триметилолова реагирует по следующей схеме:
(С2Н5)3 SnCH=CHCOOCH3 + (СНз)з SnH—
► (СН8)з SnC4=CHCOOCH8 + (C2H5)3SnH
(СНз)з Sn
СНСН2СООСН3
(C2H5)3Sn
Скорость этих реакций увеличивается в присутствии азодиизобутиронитри-
ла и замедляется феноксилом. Таким образом, они проходят, вероятно, по
300
Оловоорганические соединения. Лит. стр. 317—318
свободнорадикальному механизму. Возможные здесь процессы изображены
ниже [22а]:
нч /н
R,Sn'
С=С + R,Sn
V
I цис
+
R3sn н
С=С + R Sn'^=
II D' 3
И Н
R,Sn-C-C
R"
1. транс
I \ „ -Sn-H
R3Sn R" I
Нч
^С=С + R3Sn
R-Sn R"
И. л
1! И
i I I
R.Sn-C-C—Н + —Sn#
R,Sn R
r;sh i i
RoSn + C=C
П. транс
При R"=OC2H5 I^ac-аддукт присутствует в смеси в большем
количестве, чем П^ис-аддукт. Продукт состава 2 : 1 в этом случае вообще не
образуется, т. е. &_4 ^> k-з^* k* [SnH]. При R"=C4H9 гидростаннирования
не наблюдается и, следовательно, k2 [SnH] ^ /г_4> &_3. Эти результаты
соответствуют данным, полученным при гидростаннировании двойной
углерод-углеродной связи, находящейся в середине молекулы (см. стр. 277). В
приведенной выше схеме константа &_4 должна иметь тот же порядок, что и
к_ъ т.е. fcJSnH] <C&_i- Таким образом, при гидростаннировании
неактивированной двойной углерод-углеродной связи перенос водорода от атома
олова к атому углерода является стадией, определяющей скорость
процесса.
Тот факт, что в реакциях обмена между гидридом триметилолова и
^ш>Р-триэтилстаннилэтоксиэтиленом или метил ^ш>|3-триэтилстаннилакри-
латом цис-аддукт I превращается главным образом в ^оаддукт II,
указывает на предпочтительность некоторых конформационных положений олово-
органической группы в свободнорадикальном промежуточном аддукте [496].
Суммируя все данные по механизму гидростаннирования алкинов,
можно сделать следующие выводы [22а].
1. Гидростаннирование углерод-углеродной тройной связи может
проходить как по ионному, так и по свободнорадикальному механизму.
Гидростаннирование ацетиленов, содержащих сильные электроноакцепторные
группы, проходит в основном по ионному механизму. Гидростаннирование
алкинов, содержащих электронодонорные или слабые
электроноакцепторные группы, проходит преимущественно по свободнорадикальному
механизму.
2. При ионном гидростаннировании монозамещенных ацетиленов
образуются только а-аддукты. Радикальное гидростаннирование этих алкинов
приводит к преимущественному образованию Р-аддуктов.
3. Ионное гидростаннирование проходит по шранс-механизыу. Нуклео-
фильная атака атома водорода оловоорганического гидрида на атом
углерода является первой стадией, определяющей скорость процесса. Затем
оловоорганический катион, образующийся на этой стадии, вступает в транс-
присоединение к винильному карбаниону. В ионных реакциях присоедине-
Присоединение по кратным связям
301
ния важную роль играет стабилизация оловоорганического катиона. Эта
стабилизация может происходить вследствие внешней или внутренней
сольватации. В первом случае катион стабилизируется вследствие образования
пентакоординационного^ комплекса. Во втором случае основную роль
играет электростатическое взаимодействие, что видно из существенного
влияния полярности среды на скорость реакции.
4. Свободнорадикальное гидростаннирование проходит по транс-ме-
ханизму и приводит к цис-аддуктам. Последние могут
перегруппировываться в шранс-аддукты. В реакциях присоединения и изомеризации первая
стадия начинается с атаки оловоорганического радикала на атом углерода.
Затем в реакциях присоединения происходит перенос водорода
оловоорганического гидрида от атома олова к свободнорадикальному
промежуточному продукту. В реакции изомеризации вторая стадия заключается в
отщеплении оловоорганического радикала. Прохождение этой стадии
зависит от различных факторов. Один из них определяется предпочтительностью
некоторых конформационных положений оловоорганической группы в сво-
боднорадикальном промежуточном аддукте.
ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
К АЛЬДЕГИДАМ И КЕТОНАМ
Гидриды триалкилолова присоединяются к алифатическим альдегидам
и бензальдегиду при нагревании смеси реагентов до 70° С. Реакция не
проходит до конца и осложняется побочными процессами [14, 52]. Однако в
присутствии азодиизобутиронитрила легко и с хорошим выходом
образуются алкоксисоединения олова [9, 14, 15, 52]:
О
/
R'C + HSnR3 -> R'CH2OSnR3
(R'=C2H5, C3H7, C6H5, р-СН3ОСбН4 и др.).
Так, при нагревании до 70° С в течение 9 час. 16,3 мл (0,018 моля) изо-
масляного альдегида, 14,8 мл (0,09 моля) гидрида триэтилолова и 50 мг
азодиизобутиронитрила получают [52] 24,1 г изобутокситриэтилолова с т. кип.
94—96° С/11 мм\ df 1,165; по 1,4678. Так же ведут себя гидриды трибутил-
и триизобутилолова, а среди альдегидов — масляный альдегид, бензальде-
гид и анисовый альдегид.
Гидриды триалкилолова энергично реагируют с хлоралем даже при 0° С.
В зависимости от порядка прибавления реагентов и их молярного
отношения образующееся трихлорэтокситриалкилолово может или присоединять
еще одну молекулу хлораля, или восстанавливаться гидридом в дихлор-
этокситриалкилолово:
о°с
R3SnH -fO=CH—CC13 > R3SnOCH2CCl3
R3Sn—О—СН—О—СН2СС13 R3SnOCH2CHCl2 + R8SnCl
I
СС13
(R=CH3, QHs, C4H9).
302
Оловоорганжесше соединения. Лит. стр. 317—318
Аналогичным образом проходит гидростаннирование пентафторбензальде-
гида:
20° С
R3SnH + 0=CH-C6F5 > R3SnOCH2CeF5
+OCHCeF6 I -OCHCeF6
RsSnOCH—OCHaCeFg
I
QF5
На примере присоединения гидрида триэтилолова к бензальдегиду или
гидроксибензальдегиду показано, что каталитическое действие на
гидростаннирование альдегидов оказывает бирадикал 02 [52].
Скорость присоединения оловоорганических гидридов к альдегидам
заметно увеличивается и при введении в реакционную смесь небольшого
количества спиртов или фенолов [14, 52]. Например, бензилокситриэтилолово
получено [52] смешением эквимолекулярных количеств гидрида
триэтилолова, бензальдегида и метилового спирта. Реакция заканчивается примерно
через 15 мин. Бензилокситриэтилолово представляет собой бесцветную
жидкость с т. кип. 99—101° С/0,5 мм\ по 1,5272.
Изомеры оксибензальдегида реагируют с гидридами триалкилолова и без
катализатора [52]. При смешении эквимолекулярных количеств
салицилового альдегида и гидрида триэтилолова происходит экзотермическая
реакция и смесь полностью закристаллизовывается. Образующееся о-(окси-
метил)фенокситриэтилолово имеет т. пл. 51° С (из гексана). Нагреванием
до 80—90° С в течение 8 час. эквимолекулярных количеств гидрида триизо-
бутилолова и салицилового альдегида в небольшом количестве гексана
получены бесцветные кристаллы о-(оксиметил)фенокситриизобутилолова с
т. пл. 33—34° С (из гексана). Выход 63%. В этом случае реакция гидростан-
нирования сопровождается перегруппировкой образующегося вещества
с переходом триалкилстаннильной группы к более кислому кислородному
атому фенола
СН2—О—SnR3 CH2—ОН
п
ОН | О—SnR3
О'
Впоследствии было установлено, что эти два соединения находятся
между собой в состоянии подвижного равновесия [22а]. Положение
равновесия зависит от температуры, растворителя и характера органического
радикала у атома олова. Например, при R = С2Н5 в CDC13 при комнатной
температуре образуется равновесная смесь состава 1:1.
Л1-(Оксиметил)фенокситриэтил- и лг-(оксиметил)фенокситрибутилолово
получены [52] нагреванием до 70—80° С в течение 30 мин.
^-оксибензальдегида с эквимолекулярным количеством оловоорганического гидрида. Этиль-
ное соединение кристаллизуется в виде пластинок ст. пл. 63 С (из цикло-
гексана). ж-(Оксиметил)фенокситрибутилолово при 0° С представляет собой
маслянистую жидкость.
В отличие от других алкоксисоединений олова о-(оксиметил)фенокси-
триэтилолово, как и аналогичное мета-соединение, довольно устойчиво на
воздухе. Возможно, эта устойчивость обусловлена образованием
внутримолекулярного комплекса между атомом олова и кислородом гидроксиль-
ной группы [52].
Гидрид трифенилолова реагирует с бензальдегидом в присутствии
метилового спирта уже при комнатной температуре. Однако образующийся
Присоединение по кратным связям
303
продукт присоединения расщепляется второй молекулой гидрида, так что
в конечном счете получают бензиловый спирт, гексафенилдистаннан и не-
прореагировавший альдегид. При реакции гидрида трифенилолова с
салициловым альдегидом гексафенилдистаннан образуется лишь в качестве
примеси к о-(оксиметил)фенокситрифенилолову (после перекристаллизации
из толуола т. пл. 195—196° С).
Для реакций оловоорганических гидридов с альдегидами доказан
автокатализ [14, 52]. При использовании разбавленных растворов реагентов в
циклогексане реакция начинается лишь через длительный промежуток
времени. Скорость реакции заметно увеличивается при прибавлении к смеси
соответствующего алкоксисоединения.
Изомасляный, бензиловый и салициловый альдегиды реагируют с ди-
гидридом диизобутилолова уже при комнатной температуре и без
катализатора [12, 52]:
О
RC + (i-QHeb SnH2 -> RCH2OSn (H) (C4H9-/)2 -* (/-С4Н9)2 Sn (OCH2R)2.
\
н
Получение диизобутоксидиизобутилолова [52]. К изомасляному альдегиду при
энергичном перемешивании прибавляют по каплям дигидрид диизобутилолова (молярное от
ношение реагентов 4:1). Смесь оставляют на 12 час. при 20° С и перегоняют в вакууме.
Выход диизобутоксидиизобутилолова равен 47%; т. кип. 96° С/0,002 мм; т. пл. 20° С.
Взаимодействием стехиометрических количеств салицилового альдегида
и дигидрида диизобутилолова в небольшом количестве циклогексана
получено [52] ди-(о-оксибензилокси)диизобутилолово с выходом 94%\ т. пл.
146° С.
Эффективным катализатором гидростаннирования ароматических
альдегидов является сухой хлористый цинк [9, 14, 52] (алифатические альдегиды
в этих условиях полимеризуются). Действие хлористого цинка объясняется
[14, 52] дополнительной поляризацией карбонильной группы вследствие
комплексообразования. Так, фурфурол экзотермически реагирует с гидридом
триэтилолова в присутствии небольшого количества хлористого цинка
(молярное отношение реагентов 50 : 50 : 3). Реакция заканчивается в течение
5 мин. Фурфурилокситриэтилолово представляет собой бесцветную
жидкость с т. кип. 130° С/12лш, по 1,5072.
Аналогичным образом проходит присоединение гидрида триэтилолова
к бензальдегиду в присутствии хлористого цинка.
Нагревание оловоорганических гидридов с кетонами приводит к
образованию смеси вторичных спиртов и алкоксисоединений олова [8, 53].
Реакция присоединения оловоорганических гидридов к кетонам становится
основной при облучении смеси УФ-светом [8, 14, 18, 53]. Так, облучение
смеси двух молей циклогексанона и 1 моля гидрида трибутилолова в течение
20 час. приводит к циклогексилокситрибутилолову (выход 73%) [8].
Подобным же образом получены [8] продукты присоединения гидрида
трибутилолова к диэтилкетону и ацетофенону, но в этих случаях образуется
лишь около 30% алкоксисоединений олова. Во всех реакциях наряду с
продуктами присоединения образуется небольшое количество вторичных
спиртов. Влияние температуры на соотношение продуктов присоединения
и восстановления показано на рис. 3 [8].
Опыты по присоединению оловоорганических гидридов к кетонам в
присутствии катализаторов радикалообразующего типа не дали
удовлетворительных результатов [52]. В основном, а иногда исключительно при этом
304
Оловоорганические соединения. Лит. стр. 317—318
проходит восстановление кетонов в спирты или разложение оловооргани-
ческого гидрида с выделением водорода.
Эффективным катализатором присоединения оловоорганических
гидридов алифатического ряда к кетонам является сухой хлористый цинк [9, 14,
52]. Правда, реакция не проходит самопроизвольно, как это имело место с
100|
50h
.- "О-.
• '"Ч
"Ч. V
и ' /
I / /
Г У
[ 1о — '* /
у
S
\ **
L х ~- ~~ *"*
[ Их-—~*~~~~х
-20
50
100 150
Температура. Г
Рис. 3. Температурная
зависимость выхода цикл ore кс а нол а (/)
и
циклогексилокситрибутилолова (//) в реакции гидрида три-
бутилолова с цикл ore ксаноном
ароматическими альдегидами, однако при нагревании смеси до 60—80° С в
течение 5—10 час. алкоксисоединения олова образуются с достаточно
высокими выходами [521. Так получены, например, изопропокситриэтилолово
(выход 60%; т. кип. 83° С/(12лш; По 1,491), в/дар-бутокситриэтшюлово (96%;
т. кип. 102° С/0,35 мм\ п™ 1,476) и др. [52]. Присоединение
оловоорганических моно- и дигидридов к гексафторацетону проходит экзотермически и в
отсутствие катализатора [54]
R3SnH + (CF3)2 CO -> (СНз)з SnOCH (CF3)2.
Реакция описана на примере гидрида триметилолова и дигидридов диметил-
и ди-я-бутилолова. Также легко проходит присоединение гидридов три-
метил- и триэтилолова к трифторацетофенону [22а]:
CF3
R3SnH + CF3—С—С6Н5 -> R3Sn—О—СН
О
\
СвНв
Изучен механизм реакции гидростаннирования хлорал я, пентафторбенз-
альдегида, 2,2,2-трифторацетофенона и салицилового альдегида [22а].
Найдено, что скорость этих реакций значительно увеличивается с ростом
полярности среды, а также при переходе от гидрида трифенилолова к
гидриду триметилолова: (C6H5)3SnH<^: (C4H9)3SnH < (CH3)3SnH < (C2H5)3SnH.
Согласно Лейсинку [22а], гидростаннирование этих электрофильных
карбонильных соединений проходит по ионному механизму:
!
— Sn — Н + 0=С
у медленно
-Sn + Н—С-
-О
(-) быстро
I I
— Sn—О—С-
Присоединение по кратным связям
305
Аналогичная схема предложена для объяснения каталитического
действия хлористого цинка:
Предполагается, что и в этом случае решающую роль играет нуклеофиль-
ная атака водорода оловоорганического гидрида на атом углерода.
ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
К АЗОМЕТИНАМ, А30- И НИТРОЗОСОЕДИНЕНИЯМ,
ИЗОЦИАНАТАМ И ИЗОТИОЦИАНАТАМ
Оловоорганические гидриды не присоединяются по связи C=N без
катализатора [14, 55]. Однако в присутствии радикалообразователей или
сухого хлористого цинка реакция проходит легко [9, 14, 15, 55]
Шз~<СЗ~~М CH3-^^-N-SnR3
I + HSnR3^ |
/~^>-сн /^А_сн2
Полученные соединения представляют собой бесцветные масла, которые
перегоняются в вакууме без заметного разложения.
По-видимому, описанное Лоренцом и Бекером [19] восстановление бен-
зальанилина в N-бензиланилин избытком гидрида трифенилолова также
проходит через промежуточное образование продукта присоединения [501.
Провести этим путем гидростаннирование азометинов с алифатическими
радикалами, у атома азота (бензал метил амина и бензальбутиламина),
а также анисальдегиданила или бензальазина не удается 155]. Также не
проходит реакция с пиридином по крайней мере в присутствии катализаторов
радикалообразующего типа.
Гидрид триэтилолова присоединяется к дициклогексилкарбодиимиду
при нагревании до 70° С в присутствии азодиизобутиронитрила 155]
CeHi1N=C=NC0H1i + R8SnH - C6H1IN=CHN (SnR8) QHU.
Ы-Циклогексил-Ы-триэтилстаннил-КГ-циклогексиламидин получен с
выходом 73 %.
Оловоорганические гидриды не присоединяются к мононитрилам ни в
присутствии азодиизобутиронитрила, ни в присутствии хлористого цинка
или эфирата трехфтористого бора [55]. Однако гидриды триалкилолова
экзотермически реагируют с динитрилом алкилиденмалоновои кислоты, давая
с количественным выходом N-станнилкетенимины [34]
CN
R'HC=С + R8SnH - R'HC— C=C=N—SnR3
\ W
CN H CN
(R-C2H5, C4H9; R'=CH8, CaHsO, C6H5, n-(CH3)2 NC6H4,
n-CH3OC6H4, /z-02NC6H4, я-С1С6Н4, фурил).
Исследованы [35] некоторые реакции полученных соединений.
20 Заказ № 207
306
Оловоорганические соединения. Лит. стр. 317—318
Гидростаннирование по связи—N=N—производных азобензола и эфи-
ров азодикарбоновой кислоты проходит даже без катализаторов [56]
C6H5N-NC6H5.^ HSnR3 - C8H5N (SnR3)-NHC8H5.
Образующиеся соединения могут вступать в реакцию со второй молекулой
оловоорганического гидрида [56]
[C6H5N (SnR3)-NHC6H5 + HSnR3 -> G6H5NH-NHC6H5 + R3SnSnR3.
Так, нагреванием до 110° С азобензола с двумя молями гидрида триэтил-
олова получают гидразобензол и гексаэтилдистаннан. Соответствующий
продукт присоединения в индивидуальном состоянии не выделен, но его
образование доказано следующим образом [56].
Эквимолекулярные количества реагентов нагревают в бензоле в течение
5 час. Растворитель отгоняют и продукт реакции гидролизуют кипячением
с 90%-ным этиловым спиртом. Получают гидразобензол и гидроокись три-
этилолова (94%):
н2о
(QHsb SnH + C6H5N-NG6H5 - (С2Н5)3 SnN (C6H5) NHC6H5 ►
-» (СзНБ)зSnOH + CeH5NHNHCeH5.
При смешении эквимолекулярных количеств диэтилазодикарбоксилата
и гидрида триэтилолова в гексане происходит экзотермическая реакция [56].
Раствор оранжевого цвета постепенно обесцвечивается и из него
выделяются бесцветные кристаллы диэтилгидразиндикарбоксилата. Выпариванием
фильтрата получают Ы,Ы/-б^с-(триэтилстаннил)-Ы,Ы/-ди-(этоксикарбонил)-
гидразин. Таким образом, соответствующий продукт присоединения
неустойчив и легко диспропорционирует
-(СоН5)3 Sn Н
(С2Н5)3 SnH + C2H5OC—N=N—С—ОС2Н5
• II II
О О
/
N—N
/ \
С2Н50—С С—ОС2Н5
о о
(C2H5)3Sn Sn(C2H5)3
\ /
-* N—N ОС2Н5 + С2Н5ОС— NH—NH— С—ОС2Н5.
/ \ / II II
С2Н50—С С 0 0
II II
о о
N,N,-бwc-(Tpиэтилcтaннил)-N,N/-ди(этoкcикapбoнил)гидpaзин не
реагирует с гидридом триэтилолова даже при 4-часовом нагревании до 110° С
без растворителя. Однако гидридом трифенилолова вещество легко гидро-
генизуется с образованием диэтилгидразиндикарбоксилата (выход 98%)
и 1,1,1-триэтил-2,2,2-трифенилдистаннана (95%). Реакция завершается за
15 мин. при температуре 90° С.
При нагревании до 110° гидрида триэтилолова с я-аминоазобензолом
последний восстанавливается до я-фенилендиамина [55].
Гидрид трифенилолова легко присоединяется к азобензолу.
Получение ^трифенилстаннил-Ы,Ы'-дифенилгидразина [56]. К нагретому до кипения
раствору 2,40 г (0,013 моля) азобензола в 10 мл бензола прибавляют по каплям 4,60 г (0,013
моля) гидрида трифенилолова в 10 мл бензола. Через 2 часа из почти бесцветного раствора
отгоняют 10 мл бензола и остаток медленно охлаждают до 0° С, при этом на стенках колбы
выделяются бесцветные кристаллы. Маточный раствор удаляют, к остатку добавляют 10 мл
бензола и 5 мл пентана, после чего раствор быстро охлаждают до 0° С. Выпавший осадок
отфильтровывают, промывают пентаном и высушивают в вакууме 0,01 мм. Получают 5,80 г
(85%) вещества с т. чл. 130° С (нечетко, с предварительным смоканием).
Присоединение по кратным связям
307
При нагревании до 90° С азобензола с двумя молями гидрида трифенил-
олова в течение 1,5 час. получают 94% гексафенилдистаннана(т. пл. 231 —
233° С) и 82% гидразобензола (т. пл. 124—126° С). Эти вещества разделяют
фракционированной кристаллизацией из этилового спирта [56].
Гидриды триалкилолова легко реагируют с изоцианатами [55, 57—60]
и арилизоцианатами [59, 60]. Анализ образующихся продуктов методами
И К- и УФ-спектроскопии, а также спектров ЯМР показывает, что в
зависимости от природы ненасыщенного реагента и оловоорганического гидрида
наблюдается тот или иной тип присоединения [60].
Присоединение оловоорганических гидридов к изоцианатам проходит
по полярному механизму [57, 58]. Скорость реакции значительно
возрастает с увеличением полярности растворителя. Небольшие количества фе-
ноксила или катализаторов радикалообразующего типа на нее не влияют.
Реакционная способность оловоорганических гидридов по отношению к
изоцианатам увеличивается в следующем ряду: (C6H5)3SnH<^ (CH3)3SnH<
<< (C4H9)3SnH и (C2H5)3SnH. Гексилизоцианат менее активен, чем фенил-
изотиоцианат. Все эти данные ясно указывают на нуклеофильную атаку
водорода оловоорганического гидрида на углеродный атом изоциана-
та [57].
Для реакций гидростаннирования изоцианатов доказан автокатализ [22а];
так как образующиеся аддукты имеют большой дипольный момент (\i =
= 3,5—4D), то, по-видимому, автокатализ обусловлен сольватацией
ионного промежуточного продукта. Исследование реакций гидрида и дейте-
рида триэтилолова с фенилизоцианатом методом газовой хроматографии
показало, что скорость реакции является функцией концентрации обоих
реагентов. Однако вследствие автокаталитических эффектов и побочных
процессов порядок реакции точно установить не удается.
Изотопный эффект в реакциях гидростаннирования изоцианатов
незначительный : /Сн/ДЪ~1- Как показано на стр. 296, это свидетельствует в
пользу того, что нуклеофильная атака атома водорода оловоорганического
гидрида на атом углерода изоцианатной группы является первой стадией,
определяющей скорость процесса. В этом случае (в зависимости от степени
переноса водорода от олова к углероду в переходном состоянии) отношение
KhIKd может иметь все значения между 3,7 и 0,4.
При изучении гидростаннирования изотиоцианатов получена та же
качественная картина, что и при гидростаннировании изоцианатов.
По-видимому, эти реакции проходят по одному и тому же механизму,
общая схема которого может быть представлена следующим
образом [22а]:
R4SnH + R—N=C=X
R'_N-^-c^^X
Q
SnR,
R—N—CH=X
(+) 1
'R3Sn + I
R'—N=CH—X(_)
x=o
быстро
SnRo
*- R— N— CH=0
x = s
— R-N=CH— S-SnR,
Тоже различие в активности оловоорганических гидридов и изоцианатов
наблюдается и при проведении реакции без растворителя.
20*
308 Оловоорганические соединения. Лит. стр. 317—318
Нагреванием до 90° С гексилизоцианата с гидридом триэтилолова с
последующей перегонкой смеси в вакууме получают [60] вещество с т. кип.
105—106° С/0,1 мм; п2о 1,4910. По данным анализа и ИК-спектра, оно
представляет собой триэтил-(М-гексилкарбамоил)олово.
(С2Н5)3 SnH + C6H13N=C=0 - (C2H5)3 Sn—C-NHC6H13.
II
О
Кроме того, образуются N-гексилформамид и гексаэтилдистаннан.
Реакция между гидридами триалкилолова и арилизоцианатами проходит
при комнатной температуре и сопровождается значительным выделением
тепла. Исследование выделенных продуктов показывает, что и в этом
случае присоединение гидрида проходит по связи C=N [60]. При замене гидрида
триэтилолова соответствующим дейтеридом в ИК-спектре вещества
наблюдается отчетливая адсорбция связи С—D [14]. Однако в результате реакции
гидрида триэтилолова с арилизоцианатами образуются соединения со
связью Sn—N, a HeSn—С, как это имеет место в случае гексилизоцианата
[14, 56, 60]
О
У
R3SnH + R'—N=C=0 -* R3SnN (R') С
\
H
Эти вещества легко очищаются перекристаллизацией или перегонкой в
вакууме. Так, смешением при обычной температуре эквимолекулярных
количеств гидрида трибутилолова и фенилизоцианата получено с выходом 82%
трибутил-(Г\[-фенилформамидо)олово (т. кип. 170° С/1 мм\ т. пл. 64° С).
Аналогично получено триэтил-(]М-фенилформамидо)олово (т. кип. 171 —
172° С/13 мм\ т. пл. 50—53° С). В его ИК-спектре наблюдается слабая
адсорбция в области 1695 см-1. Предполагается [60], что она обусловлена
присутствием следов N-фенилформамида, образующегося при гидролизе аддук-
та. Нейман и Гейман [55] в результате той же реакции выделили
кристаллическое соединение с т. пл. 63° С, в ИК-спектре которого выше 1640 см~{
адсорбция карбонильной группы полностью отсутствует.
Сходным образом проводят реакцию и в том случае, когда в бензольном
кольце изоцианата находится заместитель, например хлор или нитрогруппа.
При этом никаких побочных процессов не наблюдается [60].
Получение триэтил-[^(/г-хлорфенил)формамидо]олова [60]. Раствор 6,21 г (0,03 моля)
гидрида триэтилолова и 4,55 г (0,03 моля) я-хлорфенилизоцианата кипятят в 25 мл сухого
бензола в течение часа. Перегонка продукта реакции дает 9,30 г (87%) бесцветной жидкости,
которая постепенно закристаллизовывается (т. пл. 77—79° С).
Нагреванием 7,45 г (0,036 моля) гидрида триэтилолова и 5,90 г (0,036
моля) я-нитрофенилизоцианата в 60 мл бензола в течение 2 час. получают
[60] триэтил-[М-(я-нитрофенил)формамидо]олово. Выход 10,9 г (82%);
т. пл. 116—119° С.
Взаимодействие я-фенилендиизоцианата с двумя молями оловооргани-
ческого гидрида приводит к п-бис- [М-(триэтилстаннил)формамидо]бензолу.
Получение /г-^#£-[Ы-(триэтилстаннил)формамидо]бензола [60]. К раствору 2,0 г (0,0125
моля) /х-фенилендиизоцианата в 150 мл сухого бензола прибавляют 5,20 г (0,0250 моля)
гидрида триэтилолова. При нагревании бесцветного раствора до температуры кипения начи-
Присоединение по кратным связям
309
нает выделяться осадок. Его отфильтровывают, промывают пентаном и высушивают.
Получают 6,40 г (90%) кристаллов с т. пл. 191—195° С.
При нагревании до 45° С в течение 2 час. дигидридадиэтилоловасдвумя
молями фенилизоцианата с количественным выходом получают 6uc-(N-
фенилформамидо)диэтилолово [60].
Взаимодействие гидрида триэтилолова с метилизотиоцианатом проходит
своеобразно. Основным продуктом экзотермической реакции является
сульфид бш>(триэтилолова) [60]. Однако при нагревании в бензоле
эквимолекулярных количеств гидрида триэтилолова и фенилизотиоцианата в
течение 2 час. получают 85% 5-триэтилстаннил-1М-фенилтиоформимидата
(т. кип. 115° С/0,2 мм)
(С2Н5)з SnH + C6H5N=C=S -> (С2Н5)з SnSCH=N—C6H,.
Связь олова с азотом легко гидрогенизуется второй молекулой олово-
органического гидрида. Гидриды триалкилолова менее эффективны в
реакциях гидрогенолиза по сравнению с гидридом трифенилолова[56]. Например,
гидрид триэтилолова не реагирует с триэтил-(1Ч-фенилформамидо)оловом,
если смесь нагревать при 130° С в течение 2 час, тогда как реакция триэтил-
(М-фенилформамидо)олова с эквимолекулярным количеством гидрида три-
фенилолова завершается через 20 мин. при температуре 90° С [60]
О О
(С6Н5)8 SnH + (С2Н5)3 SnN (С6Н5) С - (С2Н5)3 SnSn (C6H5)3 + CeH5NHC
\ \
н н
Показано [58, 61], что эта реакция проходит по полярному механизму.
Небольшие количества феноксила или азодиизобутиронитрила на нее не
влияют. Однако скорость реакции значительно возрастает с увеличением
полярности растворителя.
Скорость гидрогенолиза различных производных со связью Sn—N
замедляется электроноакцепторными и ускоряется электронодонорными
заместителями. Эти данные указывают на электрофильную атаку атома
водорода оловоорганического гидрида на атом азота (ср. с реакциями
присоединения гидридов к изоцианатам, стр. 307).
Вероятно, в присутствии электрофильных реагентов (е) связь Sn—Н
поляризуется в соответствии со схемой (I), а в присутствии нуклеофильных
реагентов (N) —со схемой (II) [61]:
R R
R—Sn+5—Н"5 е R—Sn"5—Н+5 N.
/ /
R R
(О (П)
Эти схемы позволяют объяснить различную активность оловоорганических
гидридов в реакциях присоединения к изоцианатам и при гидрогенолизе
связи Sn—N. В реакциях первого типа более активны гидриды
триалкилолова, так как электронодонорные алкильные радикалы увеличивают
плотность электронов на атоме олова и способствуют поляризации связи Sn—Н
по схеме (I). Напротив, в реакциях гидрогенолиза активнее гидрид трифе-
нилолова [61].
Восстановление изоцианатов в 'соответствующие арилформамиды можно
провести и в одну стадию. Так, нагреванием фенилизоцианата и а-нафтил-
изоцианата с двумя эквивалентами гидрида трифенилолова получают
соответствующие арилформамиды с выходом 40—50% [19]:
310
Оловоорганические соединения. Лит. стр. 317—318
О
/
Аг—N=C=0 + 2 (CeH5)3 SnH -> ArNHG + (С6Н5)3 SnSn (C6H5)3
\
Н
(Аг = СвН5, а-С10Н7).
Такой тип восстановления не имеет места при использовании
эквимолекулярного количества гидрида триэтилолова [60]. Однако гексилизоцианат и гек-
силизотиоцианат восстанавливаются в незначительной степени гидридом
триэтилолова с образованием гексаэтилдистаннана и N-гексилформамида
или N-фенилтиоформамида соответственно [59].
N-Арилформамиды образуются и при гидролизе продуктов
присоединения [60]
О О
/ /
R3Sn—N—С + Н20 -* R3SnOH + n-XC6H4NHC
\ \
н н
я-ХСбН4
Нагревание до 90° С в течение 3 час. фенилизотиоцианата с избытком
гидрида трифенилолова приводит к гексафенилдистаннану и сульфиду бис-
(трифенилолова) [19]. Таким образом, по отношению к гидрогенолизу
гидридом трифенилолова связь углерода с серой более подвижна, чем связь
углерода с кислородом |19].
Оловоорганические гидриды легко реагируют также с нитрозосоедине-
ниями. При постепенном прибавлении нитрозобензола к гидриду
триэтилолова происходит экзотермическая реакция и образуется триэтилстаннил-
фенилгидроксиламин [14, 55]:
C6H5NO + HSnR3 -> C6H5NHOSnR3.
Как и незамещенный фенилгидроксиламин, он легко реагирует со второй
молекулой оловоорганического гидрида с образованием азоксисоединения.
Последнее гладко получается в одну стадию, если исходить из стехиометри-
ческих количеств реагентов [14, 55].
/г-Нитрозодиметиланилин образует с гидридом триэтилолова бис-(п-
диметиламиноазоксибензол) и гексаэтилдистанноксан [55]. Диэтилнитрозо-
амин не реагирует с гидридом триэтилолова даже в присутствии хлористого
цинка или азодиизобутиронитрила.
М-Циклогексил-М-триэтилстаннил-КГ-циклогексиламидин (т. кип. 126° С/
/0,0001 мм; по 1,5233) получен нагреванием до 70° С в течение 24 час.
гидрида триэтилолова с эквимолекулярным количеством дициклогексилкарбо-
диимида в присутствии азодиизобутиронитрила. Сходным образом из бен-
зальанилина и гидрида триэтилолова получен [55] с выходом 72% бензил-
фенилтриэтилстанниламин (т. кип. 149—151° С/0,002 мм; п™ 1,5907).
ПРИСОЕДИНЕНИЕ АЛКОКСИСОЕДИНЕНИЙ ОЛОВА
ПО КРАТНЫМ СВЯЗЯМ
Как показано Несмеяновым и Луценко с сотр. [62—64],
оловоорганические алкоксисоединения легко присоединяются к ацегату энола и кетену
с образованием а-металлированных кетонов и сложных эфиров
соответственно.
Присоединение по кратным связям
311
При сливании эквивалентных количеств метокситриалкильных
соединений олова и ацетата энола происходит умеренное разогревание и из
реакционной смеси могут быть отогнаны алкилацетат и оловоорганическое
соединение. Подробное изучение полученных таким образом оловоорганических
соединений с помощью ИК-спектра и спектра комбинационного рассеяния
показало [62], что они представляют собой не эноляты триалкилолова, а
изомерные им а-металлированные кетоны.
Реакция проходит по следующему суммарному уравнению:
R3SnOCH3 + СН3С=СН2 -* CH3COCH2SnR3 + СН3СООСН3.
I
ооссн3
Впоследствии было установлено, что алкокситриалкилолово образует с
энолами эфиров не только станнилированные кетоны, но и изомерные
им энолы [64а].
Получение триэтилстаннилацетона [62]. К 11,8 г (10,05 моля) метокситриэтилолова
прибавляют 5 г (0,05 моля) изопропенилацетата. Реакционная смесь заметно разогревается.
После получасового нагревания при 40—50° С отгоняют метилацетат и остаток перегоняют в
вакууме. Получают 12,3 г (95%) вещества с т. кип. 100,5—101° С/6 мм\ df 1,2875; п2£ 1,4991.
Аналогично получены [62] трипропилацетонилолово (т. кип. 98 —
100° С/1 мм\ df 1,1983; rto 1,4865), трибутилацетонилолово (т. кип. 130—
132° С/2 мм\ df 1,1255; п2о 1,4842) и другие а-металлированные кетоны.
При добавлении триэтилстаннилацетона к эфирному раствору бензаль-
дегида происходит следующая реакция [63]:
(С2П5)3 SnCHoCOCH8 + С6Н5СНО — СбЧ,СНСН2СОСН8.
I
0>П(С2Н;,)3
Структура вещества надежно доказана данными элементарного анализа,
реакцией со спиртовым раствором хлористого водорода (образование
хлористого триэтилолова и бензальацетона) и ИК-спектром.
Получение фенилацетонилкарбинолята триэтилолова [63]. К 20,6 г
триэтилстаннилацетона в 25 мл сухого эфира прибавляют при охлаждении до 0° С 8,4 г бензальдегида. При
перегонке в вакууме получают 21,2 г (73%) фенилацетонилкарбинолята триэтилолова с т. кип.
124—126° С/2 мм] df 1,2582; п™ 1,5202; MRD: найдено 89,20; вычислено 89,95.
Алкоксисоединения олова легко реагируют с винилацетатом [62]. При
сливании эквивалентных количеств метокситриэтил- или метокситрибутил-
.олова и винилацетата смесь заметно разогревается, из нее может быть
количественно отогнан метилацетат. Однако ожидаемый R3SnCH2CHO
разлагается при перегонке в вакууме (остаточное давление 1—1,5 мм).
При пропускании избытка кетена в растворы алкокситриалкилолова
в эфире происходит энергичная реакция и с выходом 70—80% образуются
эфиры триалкилстаннилуксусной кислоты [64]
R3SnOR' + СН2=С=0 -» R3SnCH2COOR\
Строение этих соединений доказано с помощью ИК-спектров.
Получение метилового эфира триэтилстаннилуксусной кислоты [64]. В раствор 13,3 г
метокситриэтилолова в 25 мл сухого эфира пропускают в течение 15 мин. 0,1 г-моля кегена;
наблюдается разогревание реакционной смеси. Эфир отгоняют и остаток перегоняют в
вакууме. Получают 11 г (70%) вещества с т. кип. 71—72° С/2 мм\ df 1,2958; ri$ 1,4833.
312
Оловоорганические соединения. Лит. стр. 317—318
Так же получены метиловый эфир трипропилстаннилуксусной кислоты
(т. кип. 92—93°С/1 мм; cf 1,2064; n2D 1,4818), этиловый и пропиловый эфи-
ры трипропилстаннилуксусной кислоты (т. кип. 105—107° С/1 мм и ПО—
111°С/1лш; п2о 1,4784 и 1,4772 соответственно) и другие оловоорганические
сложные эфиры.
Для получения устойчивых выходов этих соединений (70—80%)
реакцию необходимо проводить с избытком кетена [33, 64, 65]. Если в реакцию
с кетеном вводить диалкоксисоединения диалкилолова, то образуются
соответствующие эфиры диалкилстаннил-бш>уксусной кислоты
R2Sn (OR')2 + 2CH2=C=0 -> R2Sn (CI I2COOR')2.
Полученные таким образом эфиры охарактеризованы элементарным
анализом и ИК-спектрами.
Получение метилового эфира диэтилстаннил-б'ас-уксусиой кислоты [66]. В раствор
12,3 г диметоксидиэтилолова в 30 мл сухого эфира при 0—5° С пропускают избыток
кетена. Наблюдается разогревание реакционной смеси. Посла отгонки растворителя
остаток перегоняют в вакууме. Получают 12 г (72%) вещества с т. кип. 109 —111° С/1 мм,
df 1,3771; /i^o 1/.932.
Аналогично получены этиловый эфир диэтилстаннил-б^с-уксусной
кислоты (выход 69%; т. кип. 106—108° С/1 мм; df 1,3012, tin 1,4850)";
«-пропиловый эфир диэтилстаннил-бис-уксусной кислоты (50%; 134—136° С/1 мм;
d\ 1,2370; п2о 1,4820) и этиловый эфир ди-я-пропилстаннил-бис-уксусной
кислоты (71 %; 135—137° С/1 мм; df 1,2407; njj 1,4823).
Исходя из того, что димер кетена имеет Р-лактонное строение при
действии на него алкоксисоединений триалкилолова, можно было ожидать
получения т-триалкилоловянного производного ацетоуксусного эфира
167]. По-видимому, это соединение действительно образуется па первой
стадии реакции. Однако далее оно перегруппировывается в более устойчивый
триалкилоловянный енолят ацетоуксусного эфира
R3SnOR' + СН2—С=-СП2 -> R8SnCHoCOCHoCOOR'^
I
НО—СМ—О
R3SnCH2C=CHCOOR'
I
ОН
сн
/ \
СНз—С С—OR'
I II
О О
\ у
R3Sn
Полученные оловоорганические производные Р-кетоэфиров и ацетил-
ацетона обладают лабильной связьюолово—кислород. Так, они легко гидро-
лизуются даже влагой воздуха с образованием ацетоуксусного эфира и
гидроокиси триалкилолова. Уксусная кислота превращает триалкилоловян-
ные еноляты ацетоуксусного эфира в ацетаты триалкилова и ацетоуксусный
эфир. С хлористым бензоилом эти соединения образуют хлористое триалкил-
олово и енолацетат.
Получение триэтилоловяиного енолята ацетоуксусного эфира [67]. К 12,1 г этокситри-
этилолова в 15 мл сухого эфира приливают по каплям 4,1 г димера кетена в 5 мл эфира.
Реакционную смесь нагревают при 40—50° С в течение 30 мин. После отгонки растворителя
остаток перегоняют в вакууме. Получают 10,8 г (67%) вещества с т. кип. 92—94° С/2 мм;
df 1,2632; п2£ 1,4920.
Присоединение по кратным связям
313-
О другом методе синтеза соединений этого типа см. стр. 353.
Алкоксисоединения олова присоединяются также к эфирам
ацетилендикарбоновой кислоты, в которых тройная связь активирована сложно-
эфирными группами [1].
Так, при нагревании до 100° С в течение часа метокситриэтилолова
с диметиловым эфиром ацетилендикарбоновой кислоты образуется продукт
присоединения с выходом 80—85%:
СН3ООСС=ССООСН3 + (C2H5)3SnOCH3 -> СН3ООСС=ССООСН3.
I I
(CH3)3Sn ОСН3
Реакция метокситриэтилолова с диметиловым эфиром ацетилендикарбоновой кислоты
[1]. К 14,2 г метокситриэтилолова прибавляют 0,7 г диметилового эфира
ацетилендикарбоновой кислоты. Смесь нагревают при 100° С в течение часа и перегоняют в вакууме.
Получают 19 г (83%) продукта присоединения с т. кип. 136,5° С/2 мм\ с^° 1,3265; п2£
1,4988*.
Сходным образом проходят реакции этокси- и пропокситриэтилолсва
с диметиловым эфиром ацетилендикарбоновой кислоты. Выходы продуктов
74 и 73% соответственно; т. кип. 149—150° С/1 мм и 145—146° С/2 мм.
В случае диэтилового эфира ацетилендикарбоновой кислоты требуется
более длительнее нагревание (8 час.) и еыходы несколько ниже (50—70%).
Реакция метокситриэтилолова с диэтиловым эфиром ацетилендикарбоновой кислоты
[1]. К 7,09 г метокситриэтилолова прибавляют 5 г диметилового эфира ацетилендикарбоновой
кислоты. Смесь нагревают при 100—110° С в течение 8 час. При перегонке в вакууме
получают 6,2 г (52%) продукта присоединения ст. кип. 145—147°С/3мм\ df 1,2690; л^ 1,4919.
Аналогично получены (C2H5)3SnC(COOC2H5) =С(ССОС2Н5)ОС2Н5
(выход 47%; т. кип. 156—158° С/3 м; rff 1,2424; п% 1,4901) и соединение
(C3H7)3SnC(COOC2H5)=C(COOC2H5)OCHs (выход 35%; т. кип. 159—160°
С/1 мм; df 1,2045; по 1,4881).
Алкоксисоединения олова реагируют [68—71] и с другими соединениями
с полярной кратной связью: изоцианатами, изотиоцианатами,
альдегидами, нитрилами, двуокисью углерода, сероуглеродом, двуокисью серы и
карбодиимидом. Эти реакции сопровождаются исчезновением адсорбции
двойной связи в ИК-спектре молекулы акцептора и характерным
изменением в спектре ПМР [68]. Присоединение к указанным выше соединениям
(кроме нитрилов) проходят очень быстро и экзотермически уже при обычной
температуре. Выходы аддуктов близки к количественным. Наиболее
подробно в литературе описано присоединение алкоксисоединений триалкило-
лова к изоцианатам [68]
R3SnOR' + R"N=C=0-* R3SnNR"COOR'.
Реакции проводят путем простого смешения эквимолекулярных количеств
реагентов при обычной температуре с последующей перегонкой смеси в
вакууме. Так получены, например, следующие соединения:
(C2H5)3SnN(QH9)COOC2H5, т. кип. 57,5° С/0,01 мм;
(QH9)3SnN(QH5)COOCH3, т. кип. 98— 100°С/0,01 мм;
(C4H9)3SnN(C2H5)COOCH3, т. кип. 89-С00 С/0,05 мм.
Полученные вещества охарактеризованы элементарным анализом, ИК-
и ЯМР-спектрами, а также химическими реакциями. При обработке этих
соединений изоцианатами образуются (C4H9)3Sn(NRCO)AZCCOR/ (где п =--
i ,
= 1,2), а затем циклические тримеры состава R'NCONRCONRCO [72].
314
Оловоорганические соединения. Лит. стр. 317—318
Интересно отметить, что при нагревании до 100° С в вакууме 0,01 мм
продукт присоединения фенокситрибутилолова к фенилизоцианату
диссоциирует на исходные соединения. Также не удается выделить в чистом виде
соединения типа R2Sn[N(R') COOR"]2 [68]. Метокситрибутилолово легко
реагирует с гексахлорацетоном. Образующееся вещество при нагревании
до 55° С в течение 6 час. разлагается по уравнению [73]
(С4Н9)3 SnOC (СС13)2 ОСН3 -» (С4Н„)з SnCCl3 -f 0=COCH3
I
СС13.
Реакции между алкоксисоединениями триалкилолова и азодикарбоно-
вым эфиром проходят своеобразно [1]. При добавлении азодикарбонового
эфира к этокситриэтилолову наблюдается значительное разогревание,
сопровождающееся выделением окиси углерода и азота. При разгонке
смеси получены с высокими выходами диэтиловыи эфир угольной кислоты и
этокситриэтилолово. Образование этих веществ объяснено [11 следующей
схемой:
^<^_ ОС2Н5 || /ОС2Н5
\ N -1 *>- С( 4 N2 + СО + (C2H5)3SnOC2H5
Ч / -Sn(C2H5)3 \ОС2н5
н f'
о
Таким образом, реакция сводится к разложению азодикарбонового
эфира в присутствии алкоголята оловоорганического соединения на эфир
угольной кислоты, окись углерода и азот.
ПРИСОЕДИНЕНИЕ СТАННИЛАМИНОВ ПО КРАТНЫМ СВЯЗЯМ
В реакциях присоединения станниламины ведут себя подобно алкокси-
соединениям олова. Джонс и Лапперт [74] действием кетена па диметила-
минотриметилолово получили окрашенное в желтый цвет вещество,
которому приписано строение М,Ы-диметиламида триметилстаннилуксусной
кислоты. Указанные авторы провели также реакцию изопропенилацетата с
диметиламинотриэтилоловом. Однако из образующихся продуктов ими
выделены только диметилацетамид, ацетат триэтилолова и высококипящий
неидентифицированный полимер.
Реакция диалкиламинотриалкилстаннанов с кетеном и изопропенилаце-
татом подробно исследована Луценко и Пономаревым с сотр. [651.
Полученные результаты не подтвердили данных Джонса и Лапперта. Найдено,
что при пропускании избытка кетена в эфирный раствор диалкиламинотри-
алкилстаннана происходит энергичная реакция и с хорошим выходом
образуются Ы,Ы-диалкиламиды триалкилстаннилуксусной кислоты
R3SnN(CH3)2 + СН2=С=0 -> RsSnCHaCON (CH3)2.
Полученный таким образом М,1М-диметиламид триметилстаннилуксусной
кислоты представляет собой прозрачную жидкость с т. кип. 68—69° С/мм,
а не порошок желтого цвета.
Реакция изопропенилацетата с диметиламинотриэтилоловом проходит
по следующей схеме [65]:
(СаН5)3 SnN (СНз)2 + СН2=С—СН3 -> R3SnCH2COGH3 +CH3CON (GH3)2.
I
ООССНз
Присоединение по кратным связям
315
Диметиламинотриметилолово образует с диазометаном
триметилстаннилдиазометан (т. кип. 64—67° С/1 мм) [74а]:
(CH3)3SnN (СНз)2 + CH2N2 -» [(GH3)3Sn—CH—N=N—N(CH3)2.
(CH3)3Sn—CH—N=N+
Триметилстаннилдиазометан обладает высокой реакционной способностью,
как это можно видеть из следующей схемы 174а]:
(CH3)3Sn—CH — N=N+
н2о(он )
f/2[(CH3)3Snl20 + CH2N
НС1
(CHOjSnCl
(CH,)3SnBr + N,+ С -г МВг
(CH3)3SnOCH3 + N2 (CH;3)4Sn + (CH3)3SnCN + N2
V2((CH3)3Sn]20- + N2+ CH4, C211Г), C3HS
В литературе описано также присоединение станниламинов к двуокиси
углерода, сероуглероду, фенилизо- и изотиоцианату, бензонитрилу, ди-я-
толилкарбодиимиду, двуокиси серы и N-тиоанилину 12, 75]. Реакции гладко
проходят при обычной температуре. Выходы продуктов практически
количественные.
Диэтиламинотрибутилолово не реагирует с окисью 1-бутена при
нагревании смеси до кипения [75а]. Однако в присутствии диэтиламида лития
гладко образуется 1-этил-2-диэтиламиноэтокситрибутилолово:
С2Н5-СН-СН2 + (C4H9)3SnN (C2H5)2-,C2H5CMCH2N(C2H5)2.
\/ I
О 05п (QH9)3
Аналогичным образом диэтиламинотрибутилолово ведет себя по
отношению к окисям стирола и циклогексена. Образующиеся продукты
представляют собой прозрачные жидкости, хорошо растворимые в обычных
органических растворителях. При вакуумной перегонке они выделяются из
смеси с выходом 65—75%.
Нагреванием азида трибутилолова с диэтиловым эфиром ацетилендикар-
боновой кислоты в бензоле в течение 2,5 час. получен [76] 1-трибутилстаннил-
4,5-диэтоксикарбонил-1,2,3-триазол:
N С—СООСзН5
(С4Н9)3 SnN3 + QH5OOCC=CCOOC2H5 -» || ||
N С—СООСгНз
\ /
N
Sn (C4H9)3
316
Оловоорганические соединения. Лит. стр. 317—318
С ацетилендикарбоновой кислотой реакция проходит иначе:
2 (QH9)3SnN3 + НООСС=ССООН -» (С4Н9)3 SnOOCC=CC003n (QH„)8 Лг 2NII3.
j По патентным данным [77] азиды фенилолова образуют с фосфинами
соединения состава (C6H5)3SnNP[N(CH3)2]3 и C6H5Sn(NPC6H5)3.
Присоединение гексаалкилдистаннанов, гексаалкилдистанноксанив
и соединений типа (R2Sn)^ по кратным связям
Реакции этого типа описаны лишь на отдельных примерах.
При облучении УФ-светом в течение 72 час. раствора дифенилолова в тет-
рафторэтилене получено [78] белое кристаллическое вещество ст. пл. 128° С.
По данным анализа, оно соответствует дифенилстаннилтетрафторэтану
(С6Н5)2 SnC2F4. В результате аналогичной реакции гексаметилдистаннана
с тетрафторэтиленом образуется гигроскопичное вещество, которое
разлагается на воздухе. По данным анализа и ИК-спектру, оно представляет
собой 1,2-бис-(триметилстаннил)тетрафторэтан [78]. Найдено [79], что при
70° С и облучении УФ-светом гексаметилдистаннан и тетрафторэтилен
реагируют между собой по ряду параллельных процессов. В двух основных
реакциях участвует радикал (CH3)3SnCF2CF2\ способный отрывать водород от
групп СН или присоединять гексаметилдистаннан. В результате
образуются (CH3)3SnCF2CF2H и (CH3)3Sn (CF2)2Sn(CH3)3 соответственно.
Аналогичным образом проходит реакция гексаметилдистаннана с гексафторпропеном
[79, 80].
Облучение УФ-светом смеси гексаметилдистаннана и гексафтор-2-бу-
тина приводит к (CH3)3SnC(CF3)=C (CF3)Sn (CH3)3, т. кип. 53° С/10-3 мм
[79—81]. Согласно спектру ЯМР и ИК-спектру, это вещество представляет
собой чистый транс-изомер. При облучении УФ-светом гексафтор-2-бутина
и гексабутилдистаннана образуется сложная смесь продуктов, из
которой выделено дибутил-бис-(1,1,1,4,4,4-гексафтор-2-бутенил-2)-олово —
(С4Н9)2 Sn[C(CF3)--^C(CF3)H]2, т. кип. 60—63° С/10_3лш. Вещество
охарактеризовано ИК-спектром и спектром ЯМР [81]. В реакции присоединения к
перфторалкенам вступает также (CH3)3SnMn (CO)5 [79, 80].
Гексаалкилдистанноксаны экзотермически реагируют с изоцианатами
[82]. Обработкой образующихся продуктов первичным амином, а затем
двуокисью углерода получают карбаматы триалкилолова:
R'NH2
R3SnOSnR3 + R'NCO-> R3SnNR'COOSnR3 > 2R'NHCOOSnR8.
C02
При нагревании оба соединения превращаются в R3SnNR'CONR'SnR3.
С хлоралем гексабутилдистанноксан образует продукт присоединения
состава 1:1, который разлагается при 150° С по следующей схеме [73]:
(QH9)3SnOCH (CCI3)OSn(C4H9)3 -» (С4Н9)3 SnCCl3 + (С4Н9)3 SnOCH=0.
Продукт присоединения гексабутилдистанноксана к бромалю менее
устойчив. В 1 М растворе в четыреххлористом углероде он разлагается уже при
комнатной температуре с выделением трибутил(трибромметил)олова и
формиата трибутилолова [73].
В реакции присоединения вступают и некоторые другие
оловоорганические соединения. Хлорфенилтрибутилолово получено [83] с выходом
47,9% присоединением хлористого трибутилолова по тройной связи дегид-
робензола.
Кратко сообщается (без деталей эксперимента ) [84] о присоединении
дифенил(трифенилстаннил)фосфина к хлористому аллилу, стиролу и
Присоединение по кратным связям
317
фенилацетилену. В результате этих реакций получены соединения состава
(C6H5)3SnCH2CH IP (С6Н5)2]СН2С1, (C6H5)3SnCH2CH IP (СбН5)2]С6Н5 и
(C6H5)3SnCH=C (C6H5) P (С6Н5)2 соответственно.
ЛИТЕРАТУРА
1. Л у цен ко И. Ф., П о н о м а р е в С. В., ПетрийО. П., ЖОХ, 32, 896
(1962).
2. G е о г g е Т. A., J о п е s К-, L а р р е г t M. F., J. Chem. Soc, 1965, 2157.
2а. К 6 n i g К., N е u m a n n W. P., Tetrahedron L., 1967, 495.
26. M и р с к о в Р. Г., В л а с о в В. М.,' ЖОХ, 36, 562 (1966).
2в. Noltes J. G., Creemers Н. М. J. С, van der К е г k G. J. М., J. Orga-
nometal. Chem., 11, Р21 (1968).
3. V a n der KerkG.J.M., L u i j t e n J. G. A., Noltes J.G., Chem. a. Ind.,
1956, 352.
4. V a n der KerkG.J.M., Nol t es J.G.,J. appl. Chem., 9, 106 (1959).
•5. N e u ra a n n W. P., SommerR, Ann., 675, 10 (1964).
6. V a n der KerkG.J.M., Noltes J.G., L u i j t e n J. G. A., J. appl. Chem.,
7, 356 (1957).
7. NoltesJ.G., vanderKerkG.J.M., Chimia, 16, 122 (1962).
8. CalasR., ValadeJ, Pommier J.— C, Compt. rend., 255, 1450 (1962).
9. N e u m a n n W. P., HeymannE, Angew. Chem., 75, 166 (1963).
10. D e 1 F r a n с о G. J., ResnickP., D i 1 1 a r d C.R, J. Organometal. Chem.,
4, 57 (1965).
11. S e v es t r e J., Peintures, pigments, vernis, 41, 361 (1965); РЖХим., 1966, 6C286.
12. N e u m a n n W. P., NiermannH., S о m m e r R., Ann., 659, 27 (1962).
13. Neumann W. P., Chem. Labor, und Betrieb., 15, 9, 363 (1964); РЖХим., 1965,
7Ж321.
14. Neumann W. P., Angew. Chem., 76, 849 (1964).
15. Neumann W. P., Angew. Chem., 73, 768 (1961).
16. С 1 a r к H. C, FurnivalS.G., KwonJ.T, Canad. J. Chem., 41, 2889 (1963).
17. ClarkH.C, К w о n J. Т., Canad. J. Chem., 42, 1288 (1964).
18. ValadeJ., Pommier J.— C, Bull. Soc. Chim. France., 1963, 199.
19. LorenzD.H., В е с к e r E. I., J. Org. Chem., 28, 1707 (1963).
20. BarnetsonC, ClarkH.C, KwonJ.T., Chem. a. Ind., 1964, 458.
21. Fuc'hsR., Gilman H., J. Org. Chem., 22, 1009 (1957).
22. NicklessG., Pollard F.H., Chem. a. Ind., 1963, 1493.
22a. L ё и s i n к A. J., Hydrostannation. A mechanistik study Utrecht (Nederland) Scho-
tanus and Jens Utrecht. N. Y., 1966.
226. L e u s i n к A. J., N о 1 t e s J. G., Tetrahedron L., 1966, 335.
23. N'e u m a n n W. P., SommerR., Angew. Chem., 76, 52 (1964).
24. К u i v i 1 a H. G., RahmanW, Fish R. H., J. Am. Chem. Soc, 87, 2835
(1965).
25. ClarkH.C, KwonJ.T., ReevesLW., W e 1 1 s E. J., Canad. J. Chem.,
41, 3005 (1963).
26. N e u m a n n W. P., Pedain J., Tetrahedron L., 1964, 2461.
27. Pollard F. H., NicklessG., С о о к е D. J., J. Chrornatogr., 17, 472 (1965).
28. KrespanC.G., E n g e 1 h a r d t V. A., J. Org. Chem., 23, 1565 (1958).
29. LaliberteB.R., Davidsohn W., HenryM.C, J. Organometal. Chem.,
5, 526 (1966).
30. Becker R., WendeA., Пат. ФРГ 1158974 (1964); РЖХим., 1965, 14H133.
30a. L e u s i n к A. J., NoltesJ.G., Tetrahedron L., 1966, 2221.
31. PereyreM, ValadeJ., Compt. rend., 260, 580 (1965).
32. PereyreM., ValadeJ., Compt. rend., 258, 4785 (1964).
33. R i j к e n s F., J a n s s e n M. J., D r e n t h W., van der Kerk G. J. M.,
J. Organometal Chem., 2, 347 (1964).
34. N e u m a n n W. P., SommerR., Muller E., Angew. Chem., 78, 545 (1966),
35. SommerR,, Neumann W. P., Angew. Chem., 78, 546 (1966).
36. S e у f e r t h D., TakamizawaM., Inorg. Chem., 2, 731 (1963).
37. В r a u n J., Compt. rend., 250, 218 (1965).
38. Leusink A.J., NoltesJ.G., В u d d i n g H. A., v a n d e г К е г к G.
J. M., Rec. trav. chim., 83, 609 (1964).
-39. H e n г у M. C, NoltesJ.G., J. Am. Chem. Soc, 82, 558 (1960).
40. N о 1 t e s J. G., vanderKerkG.J.M, Rec. trav. chim., 81, 41 (1962).
41. S m о 1 i n E. M., Tetrahedron L., 1961, 143.
42. Smolin E.M., O'Connor M. N., Пат. США 3074985 (1963); РЖХим., 1964,
20H69.
318
Оловоорганические соединения
43. Н е с м е я н о в А. Н., Б о р и с о в А. Е., ДАН СССР, 174, 96 (1967).
44. Несмеянов А.Н., Борисов А. Е., В а н Ш и - х у а, Изв. АН СССР,
серия хим., 1967, 1141.
45. Несмеянов А.Н., Борисов А. Е., Изв. АН СССР, серия хим., 1967, 226.
45а. Несмеянов А. Н., Борисов А. Е., Новикова Н. В., ДАН СССР*
172, 1241 (1967).
46. 3 а в г о р о д н и й В. С, П е т р о в А. А., ЖОХ, 35, 1313 (1965).
47. L е u s i n k A. J., Marsman J. W., В u d d i n g H. A., Rec. trav. chim.,
84, 295 (1965).
48. Leusink A. J., Marsman J, W., Rec. trav. chim., 84, 1123 (1965).
49. SeyferthD., Vaughan LG., J. Organometal. Chem., 1, 138 (1963).
49a. Leusink A. J., Budding H. A., MarsraanJ. W., J. Organometal. Chem-
9, 285 (1967).
496. Leusink A. J., Budding H. A., Drenth W., J. Organometal. Chem.,
9, 295 (1967); 11, 541 (1968).
50. L e и s i n к A. J., M a r s m a n J. W., Buddi ng H. A., Nol t es J.G., van
der KerkG. J.M., Rec. trav. chim., 84, 567 (1965).
51. L e a v i t t F. C, Mat t ernas L.U., J. Polymer Sci., 62, 68 (1962).
52. N e и m a n W. P., HeymannE, Ann., 683, 11 (1965).
53. P о m m i e r J. C, V a 1 a d e J., Bull. Soc. chim. France, 1965, 975.
54. С и 1 1 e n W. R., S t у a n G. E., Inorg. Chem., 4, 1437 (1965).
55. N e и m a n n W. P., HeymannE., Ann., 683, 24 (1965).
56. N о 1 t e s J. G., Rec. trav. chim., 83, 515 (1964).
57. L e и s i n к A. J., Noltes J.G., Rec. trav. chim., 84, 585 (1965).
58. L e и s i n к A. J., В и d d i n g H. A., Noltes J. G., Rec. trav. chim., 85, 151
(1966).
59. N о 1 t e s J. G., JanssenM.J, Rec. trav. chim., 82, 1955 (1963).
60. N о 1 t e s J. G., J a n s s e n M. J., J. Organometal. Chem., 1, 346 (1964).
61. CreemersH.M. J. C, Noltes J. G., Rec. trav. chim., 84, 590 (1965).
62. H e с м е я н о в А. Н., Луценко И. Ф., Пономарев СВ., ДАН СССР,
125, 1073 (1959).
63. П о н о м а р е в С. В., Л у ц е н к о И. Ф., ЖОХ, 34, 3450 (1964).
64. Л у ц е н к о И. Ф., П о н о м а р е в С. В., ЖОХ, 31, 2025 (1961).
64а. Ре г е у г е М., В е 1 1 eg а г d е В., Mendelsohn J., Valade J., J.
Organometal. Chem., 11, 97 (1968).
65. Пономарев С. В., Л и с и н а 3. М., Луценко И. Ф., ЖОХ, 36, 1966.
66. По ном а рев С. В., Б а у к о в Ю. И., Л у цен ко И. Ф., ЖОХ, 34, 1928-
(1964).
67. П о н о м а р е в С. В., М а ч и г и н Е. В., Л у ц е н к о И. Ф., ЖОХ, 36, 518
(1966).
68. В 1 о о d w о г t h A. J., D a v i e s A. G., J. Chem. Soc, 1965, 5238; 6858.
69. В 1 о о d wor t h A. J., Da vies A. G., Proc. Chem. Soc, 1S63, 264,315.
70. flavies A. G., Harrison P. G., J. Chem. Soc, 1967C, 1313.
71. D a v i e s A. G., S у m e s W. R., Chem. Communs., 1965, 25; J. Chem. Soc, 1967C,
1009.
72. В 1 о о d w о r t h A. I., Davies A.G., Chem. Communs., 1965, 24.
73. D a v i e s A. G., SymesW.R., J. Organometal. Chem., 5, 394 (1966).
74. Jones K., L a p p e r t M. F., J. Chem. Soc, 1965, 1944.
74a. LappertM. F., LorberthJ., Kurzreferate Abstracts 3. Internationales
Symposium uber metallorganische Chemie. Miinchen, 1967, S. 162.
75. Jones K-, LappertM. R, Proc Chem. Soc, 1962, 358.
75a. T z s с h а с h A., R e i s s E., J. Organometal. Chem., 8, 255 (1967).
76. L u i j t e n J. G. A., van der KerkG.J.M., Rec trav. chim., 83, 295 (1964).
77. Washburn R. N., В a 1 d w i n R. А., Пат. США 3112331 (1963); РЖХим., 1965,
16H61.
78. В e g M. А., С 1 a r k H. С, Chem. a. Ind., 1962, 140.
79. С 1 а г k H. С, Chem. Eng. News, 43, 53 (1965).
80. С 1 а г к Н. С, Т s a i J. H., Chem. Communs., 1965, 111.
81. С u 1 1 en W. R., Dawson D.S., Sty an G. E., J. Organometal. Chem., 3,
406 (1965)
82. В 1 о о d w о r t h A. J., D a v i e s A. G., J. Chem. Soc, 1965, 6245; 1966, 299.
83. T s e n g С h a o-I u n, T u n g S h i-h u а, С h a n g К u о М i n g, Scientia sinica,
13, 1170 (1964); РЖХим., 1965, 17Ж275.
84. S с h u m a n n H., J u t z i P., SchmidtM., Angew. Chem., 77, 912 (1965).
Глава VI
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ЧЕРЕЗ ДВОЙНЫЕ ДИАЗОНИЕВЫЕ, ГАЛОГЕНОНИЕВЫЕ СОЛИ
И АЛИФАТИЧЕСКИЕ ДИАЗОСОЕДИНЕНИЯ
Синтез оловоорганических соединений в ароматическом ряду может
быть осуществлен при разложении двойных диазониевых солей типа
(ArN2Cl)2-SnCl4 порошками металлов. Реакция получения металлооргани-
ческих соединений путем разложения двойных диазониевых солей была
найдена Несмеяновым [1], а в области оловоорганических соединений
исследована Несмеяновым и Кочешковым с сотр. [2, 3]. С помощью этого
метода можно получить в основном соединения типа Ar2SnX2 (X — галоид).
Ароматическое ядро может содержать активные заместители, однако в этом
направлении (которое дает преимущество по сравнению с приемами
получения через реактив Гриньяра или через литийорганические соединения)
метод пока менее развит.
Однако в более поздних работах Несмеянова, Реутова и сотрудников
для целей синтеза было успешно применено разложение C6H5N2-BF4 и
галогенониевых солей порошком олова.
Что касается алифатического ряда, то в развитие «реакции Геллермана»
[4] Якубовичем с сотр. [5, 6] было изучено действие диазометана (или
аналогичных соединений) на галоидное четырехвалентное олово (или
галоидные оловоорганические соединения). Метод позволяет получить в основном
соединения типа (ClCH2)nSnXw (n и т = 1—3). С диазоуксусным эфиром
четыреххлористое олово реагирует с образованием энолята состава
[NaC = C(OCaH5)0]2SnCla [7].
Действие оловоорганических гидридов на разнообразные
алифатические диазосоединения нашло применение в работах последнего времени [8, 9].
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ТИПА Ar2SnX2
ПУТЕМ РАЗЛОЖЕНИЯ ДВОЙНЫХ ДИАЗОНИЕВЫХ СОЛЕЙ
(ArNoCl)2-SnCl4
а) Получение двойной соли. Хлористый арилдиазоний получают диазотированием
твердым нитратом натрия 1 моля соответствующего амина в 300 мл крепкой соляной кислоты с
300 г льда и прибавлением 5%-ной соляной кислоты доводят объем смеси до 1 л.
Полученный 1%-ный раствор хлористого арилдиазония сливают с раствором хлорного олова в
концентрированной соляной кислоте. На 1 моль диазония берут V2 моля хлорного олова.
Осевшую соль отсасывают и промывают холодной 5%-ной соляной кислотой, спиртом и эфиром
и просушивают на воздухе. Это обычно мелкокристаллические вещества; температура
разложения, например, для (C6H5N2Cl)«SnCl4 78° С [2, 3, 10].
б) Разложение двойной соли. В взвесь порошка металла (медь, цинк или олово) в
доведенном до нужной температуры растворителе (например, этилацетат при температуре его
кипения), при энергично работающей мешалке, постепенно всыпают сухую диазониевую
соль с такой скоростью, чтобы поддерживать температуру кипения. Обычно это занимает
I—5 мин. Реакция протекает очень энергично. Раствор отсасывают от осадка
неорганических соединений олова и избытка металла — восстановителя. От фильтрата отгоняют
растворитель. Остаток экстрагируют по возможности полно петролейным эфиром, и из такого
раствора при его упаривании и охлаждении выкристаллизовывают оловоорганическое
соединение. Для отделения от примесей оловоорганическое соединение растворяют в спирте
320
Оловоорганжеские соединения. Лит. стр. 324
и высаживают аммиаком (окись диарилолова). Осадок отсасывают, промывают спиртом и
эфиром. Выходы при различных радикалах таковы: фенил 23%,0-толил 20%, /г-хлорфенил
5%, л-бромфенил и о-анизил 7,5%.
По Уотерсу [11] оловоорганическое соединение образуется при действии
порошка олова в ацетоновом растворе прямо на хлористый фенилдиазоний.
Лучшие выходы Ar2SnX2 (до 40%) дает взаимодействие арилдиазоний-
борфторидов с хлористым оловом и цинковой пылью в раствору ацетона,
В качестве примеси образуется некоторое количество хлоридов три-
арилолова и моноарилолова. Метод дает хорошие результаты в случае
фенила, несколько заниженные для его гомологов и галоидозамещенных.
Разложение арилдиазонийборфторидов [12]. В раствор 0,0375 моля SnCl2-2H20 в 50 мл
ацетона, помещенный в стакан диаметром 4,5 см, снабженный термометром и
соответствующей по размерам стакану весьма энергично работающей мешалкой Витта, при наружном
охлаждении и перемешивании всыпают попеременно небольшими порциями 0,0375 г-атома
цинковой пыли и 0,025 моля свежеприготовленного арилдиазонийборфторида. Тотчас же
начинается повышение температуры и выделение азота. Приблизительно через час, когда
выделение азота прекращается, баню со льдом отставляют и перемешивание продолжают
при комнатной температуре. На следующий день ацетон отгоняют на водяной бане, остаток
извлекают петролейным эфиром, который затем также отгоняют. Жидкий остаток
разбавляют этиловым спиртом и обрабатывают избытком 25%-ного аммиака. Образовавшийся
осадок оловоорганических оксидов отфильтровывают, промывают спиртом и эфиром. Не
растворившийся в спирте и эфире осадок диарилстанноксида идентифицируют переведением
в диарилртуть или в двухлористое диарилолово. Из массы, остающейся по испарении
растворителей из спиртово-эфирного раствора, выделяют окись триарилолова, отделяя ее от
арилстанноновой кислоты обработкой 10%-ным раствором едкого натра, в котором первая
нерастворима. Из щелочного раствора арилстанноновую кислоту высаживают действием
С02. Выход Ar2SnX2, где Аг —фенил 41%, я-толил 35,8%, о-толил 27,3%, л-бромфенил
13,4%, я-хлорфенил 13,1%, [3-нафтил 11%.
Выход достигает 76—82%, если оловоорганические соединения
получают через двойные иодониевые соли двухлористого олова:
(Ar2JCl)2.SnCl2 + Sn -* Ar2SnCl2 + 2 ArJ + 2 SnCl2.
Получение окиси дифенилолова [13]. 20 г (0,063 моля) (C6H5)2JCl вносят в 25 мл
ацетона, затем при интенсивном перемешивании присыпают 20 г (0,1 моля) безводного
двухлористого олова и 10 г (0,084 г-атома) порошка олова. Через некоторое время
реакционная смесь разогревается почти до кипения. После интенсивного
перемешивания в течение 4 час. неорганический слой отфильтровывают. Оставшееся после испарения
ацетона масло промывают соляной кислотой (1:1), растворяют в спирте и спиртовый раствор
выливают на холоду в 10%-ный раствор едкого натра. Выпавший осадок окиси дифенилолова
отфильтровывают, промывают водой, спиртом и эфиром. Выход 7 г (76%).
Аналогично получены окиси: ди-л-тол ил олова (67%), ди-я-хлорфенил-
олова (82%), ди-л-бромфенил олова (47%), ди-л-иодфенилолова (39%).
Хлориды диарилиодония (m-02NC6H4)2JCl или (/tz-C2H5OCOC6H4)2JC1
двойных солей с двухлористым оловом не образуют.
Оловоорганические соединения с электроноакцепторными группами в ароматическом
ядре получают разложением порошком олова в присутствии двухлористого
олова несимметричных солей диарилиодония, например таких, как
(р-СН3ОС6Н4) (/7-C2H5OCOC6H4)JCl [14].
Разложение хлористого #-карбэтоксифенил-/г-анизилиодония порошком олова [14].
4,2 г (0,01 моля) хлористого я-карбэтоксифенил-я-анизилиодония и 1,9 г (0,01 моля)
двухлористого олова разлагают 1,2 г порошка олова в 15 мл ацетона. После перемешивания в
течение 5 час. реакционную смесь оставляют на ночь. Непрореагировавшее олово
отфильтровывают, растворитель испаряют, остаток обрабатывают соляной кислотой (1:1) и
экстрагируют бензолом. Бензол испаряют, а оставшееся масло растворяют в спирте и гидролизуют
разбавленным раствором аммиака. Выпавший осадок (/?-C2H5OCOC6H4)2SnO
отфильтровывают, промывают спиртом и большим количеством эфира. Выход 0,8 г (36%).
Аналогично получены Ar2SnO, где Аг = С6Н5, С2Н5ОСОС6Н4 и о-СН3С6Н4
с выходом 34, 40 и 53% соответственно. При разложении несимметричных
Синтез через двойные диазониевые соли
321
солей иодония, содержащих нитрогруппы, происходит осмоление. Олово-
органические соединения не выделены [14].
Реакции разложения порошком олова двойных солей галогенидов ди-
фенилбромония (соответственно хлорония) и хлорного олова проводят в тех
же условиях. Двухлористое дифенилолово получено с выходом около 55%
[15].
ПОЛУЧЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛИФАТИЧЕСКИХ ДИА30С0ЕДИНЕНИЙ
Синтез оловоорганических соединений с помощью диазометана был
осуществлен и развит Якубовичем с сотр. [5, 6]. Авторы нашли, что хлорное
и бромное олово (но не SnF4) при температуре от 0° до минус 5° С реагируют
в бензоле с диазометаном, диазоэтаном или диазобутаном с образованием
всей гаммы возможных а-галоидалкильных производных олова:
CH2N2 CH2N2 CH2N2
SnCI4 > ClCH2SnCl3 > (CICH2)2 SnCI2 -» > (C1CH2)3 SnCl->
—N2 —N2 -N2
CH2N2
»(ClCH2)4Sn.
-N.
С увеличением числа радикалов, связанных с оловом, трудности
дальнейшего алкилирования возрастают. Образование тетразамещенного
продукта обычно протекает очень медленно и только в присутствии избытка
диазометана. Количества первичного, вторичного или третичного
производных в смеси зависят от молярного соотношения диазосоединения и га-
логенида олова [5, 6, 16]. Даже если берут на 1,5 моля хлорного олова 1 моль
диазометана, то наряду с треххлористым хлорметилоловом (т. кип. 72,5—
73° С/5 мм образуется двухлористое ди(хлорметил)олово (т. пл. 89,5—90° С)
и хлористоетри(хлорметил)олово (т. кип. 138—140° С/5 мм). Последние два
соединения имеют близкие температуры кипения и разделяются с трудом [5].
При действии на хлорное олово или ClCH2SnCl3 избытка диазометана
тетрахлорметилолово образуется с выходом 50—60% (т. пл. 49—49,5° С,
т. кип. 148,5° С/8 мм). В случае применения бромного олова выход
несколько повышается.
При молярном соотношении реагентов основным продуктом реакции
является двубромистое ди(бромметил)олово и трехбромистое бромметилолово
[5, 6].
Взаимодействие диазометана с бромным оловом [5]. К раствору 91 г (0,2 моля) бромного
олова в 60 мл бензола прибавляют бензольный раствор 17,5 г (0,4 моля) диазометана в
400 мл сухого бензола, высушенного перед опытом над кусочками едкого кали в течение 2
час. Прибавление диазометана ведут при 1—2° С. Реакция проходит энергично, при сильном
выделении азота. Смесь оставляют на ночь, затем отфильтровывают от небольшого осадка,
и бензол отгоняют под вакуумом. Смесь (около 78 г), полученную после отгонки бензола,
перегоняют в вакууме при 5 мм. При этом выделяются фракции: I) т. кип. 70—135° С —
21,5 г, II) т. кип. 155—160° С —44 г, III) т. кип. 165—180° С — 0,5 г, остаток — твердая
темная смола — 12,5 г. После вторичной разгонки первой фракции выделяют 5 г трехбро-
мистого бромметилолова, т. кип. 109° С/5 мм; ^3,251 (тяжелая бесцветная жидкость,
которая смешивается с водой и растворяется в обычных органических растворителях). При
155—160° С перегоняется бесцветное быстро закристаллизовывающееся двубромистое ди-
(бромметил)олово (44 г), т. пл. 87° С (из бензола). Тетра(бромметил)олово получено из дву-
бромистого ди(бромметил)олова и диазометана (т. пл. 57° С).
С диазоэтаном реакция протекает аналогично [5, 17], однако, если даже
берут 1 моль диазоэтана на 2 моля хлорного олова, основным продуктом
реакции является двухлористое ди-(а-хлорэтил)олово (т. кип. 112° С/4 мм)9
наряду с хлористым три-(а-хлорэтил)оловом (т. кип. 130° С/3 мм). Треххло-
р'истое а-хлорэтилолово (т. кип. 69—71° С) получить трудно.
21 Заказ jsfi 207
322
Оловоорганшеские соединения. Лит, стр. 324
При обработке хлористого три-(а-хлорэтил)олова большим избытком
диазометана образуется тетра-(а-хлорэтил)олово (т. кип. 142° С/2 мм).
При обработке 1 моля хлорного олова 2 молями диазобутана основным
продуктом реакции является двухлористое ди-(а-хлорбутил)олово [5].
Реакция протекает достаточно легко не только с SnX4 или продуктами
его взаимодействия с диазометаном, например (XQH2)3SnX, но и с RSnX^
и R2SnX2 (где X —CI, Br, J; R — незамещенная алкильная группа). Моно-
галогениды R3SnX того же типа в реакцию не вступают 118, 19].
Метод дает возможность синтезировать соединения олова с различными
алкильными группами [5, 6, 14, 20, 21J:
ClCH2SnCl3 + 2 CH3CHN2 -> ClCH2SnCl (СНС1СН3)2 + N2,
(СН3)2 SnCl2 + CH2Na -» (СН3)2 SnCl (CHaCl) + N2.
Получение хлористого хлорметилдиметилолова [18]. В трехгорлую колбу на 3 л,
снабженную механической мешалкой, обратным холодильником и литровой капельной воронкой,
помещают 50 г (0,228 моля) двухлористого диметилолова в 750 мл сухого эфира, охлаждают
до —50° С и медленно при хорошем перемешивании прибавляют раствор диазометана в
эфире (0,35 моля CH2N2 в 500 мл) при —5° С. Раствор диазометана при контакте с раствором
двухлористого диметилолова моментально обесцвечивается. Наблюдается выделение азота.
После завершения прибавления диазометана реакционная смесь окрашивается в
светло-желтый цвет. Перемешивание продолжают, реакционной смеси дают нагреться до комнатной
температуры. Эфир отгоняют при атмосферном давлении. Остаток переносят в круглодонную
колбу на 100 мл и перегоняют в колонке, снабженной головкой обратной конденсации.
Получают 38,7 г (72,8%) хлористого хлорметилдиметилолова с т. кип. 76—79° С/10,5 мм.
С двубромистым или дву йодистым диметил оловом, дву хлор истым ди-
этилоловом, двухлористым ди-я-бутилоловом или треххлористым я-бутил-
оловом реакция протекает с выходом 75—80% [21]. Жидкие продукты
обладают кожнонарывным действием, а также являются сильными лакрима-
торами [21].
Хлористое трифенилолово с диазометаном не реагирует. С двухлористым
дифенилоловом и треххлористым фенилоловом реакция протекает в
присутствии порошка меди [22].
Получение хлористого хлорметилдифенилолова [22]. 34 г (0,1 моля) двухлористого
дифенилолова смешивают в 300 мл (0,21 моля) эфирного раствора диазометана в
присутствии порошка меди. Остаток, полученный после обычной обработки, перегоняют.
Получают 16 г (45%) хлористого хлорметилдифенилолова в виде бесцветного масла; т. кип.
152—154° С/0,07 мм;п2£ 1,6168.
Треххлористое фенилолово реагирует с избытком диазометана с
образованием смеси продуктов: двухлористого хлорметилфенилолова,
хлористого ди(хлорметил)фенилолова и три(хлорметил)фенилолова [22]. Все
вещества представляют собой относительно стабильные на воздухе
жидкости.
Треххлористое фенилолово и диазометан. 30,2 г (0,1 моля) треххлористого фенилолова
смешивают на холоду с 300 мл (0,28 моля) раствора диазометана в эфире. Затем в окрашенный
в желтый цвет раствор прибавляют медный порошок, эфир отгоняют, а остаток
фракционируют. Получают 5>г^(16%) двухлористого хлорметилфенилолова с т. кип. 115° С/0,1 мм,
я^ 1,5900, 8 г (24%) хлористого ди(хлорметил)фенилолова, т. кип. 126—128° С/0,1 мм,
п2£ 1,6020 и 13г(38%)три(хлорметил)фенилолова ст. кип. 140—142°С/0,1 мм, п2£ 1,6031
[22].
Двухлористое олово реагирует с диазометаном с образованием
полиоловянных производных сложного строения [6].
Оловоорганические соединения с функциональными группами у сс-уг-
леродного атома получены взаимодействием оловоорганических гидридов*
с диазосоединениями.
Диазометан реагирует с гидридами триалкилолова в присутствии по-
Синтез через двойные диазониевые соли
323
рошка меди [8,9] или при облучении смеси УФ-светом [8]. Так, из
гибрида три-я бутил олова и диазометана получено [8] метилтри-я-бутил олово:
(С4Н9)3 SnH + CH2N2 -> (C4H9)3 SnCH3 + N2.
Следует отметить, что в тех же условиях хлористое три-я-бутилолово
не реагирует с диазометаном и может быть количественно выделено из
реакционной смеси.
Получение метилтрипропилолова [8]. К 6 г гидрида трипропилолова при
перемешивании прибавляют частями 5,5 г диазометана в 150 мл эфира (эфирный раствор диазометана
предварительно высушивают гранулированным едким кали и оттитровывают как обычно).
При прибавлении к смеси порошка меди начинается выделение азота. На следующий день
избыток диазометана разлагают 1 г сульфата меди в 20 мл бензола. Смесь фильтруют, эфир
отгоняют и остаток перегоняют в вакууме. Метилтрипропилолово имеет т. кип. 94—96° С/
/11 мм) ^з 1,125. Аналогично получено метилтрибутилолово.
В сходных условиях дигидрид диэтилолова дает с избытком диазометана
диметилдиэтилолово:
(С2Н5)2 SnH2 + 2CH2N2 -> (C2H5)2 Sn (CH3)2 + N2.
Таким образом можно получить всю гамму смешанных тетраалкильных
соединений олова в достаточно чистом виде.
В среде бензола диазоуксусный эфир реагирует с гидридами триалкил-
олова очень энергично (в эфире реакция не идет) [8]:
R3SnH + N2CHCOOC2H5 -» R3SnCH2COOC2H5 -f N2.
Полученные соединения представляют собой маслянистые жидкости,
слегка окрашенные в желтый цвег. Они достаточно устойчивы к
нагреванию. Так, (2-карбэтоксиметил)три-я-пропилолово перегоняется при
обычном давлении без разложения, т. кип. 217—219° С.
Получение (2-карбэтоксиметил)три-#-пропилолова [8]. 7 г диазоуксусного эфира и
10 г гидрида три-я-пропилолова смешивают в 40 мл бензола. Реакция начинается при
нагревании до 65° С и продолжается более часа. Бензол отгоняют и остаток перегоняют в вакууме.
Собирают фракцию с т. кип. 136—140° С/11 мм. Вещество представляет собой слегка
желтоватое масло, которое при обычном давлении имеет т. кип. 217—218° С.
Аналогично проводят реакцию с гидридом три-я-бутилолова (смесь нагревают до 100° С).
Диазокетоны реагируют с оловоорганическими гидридами, однако с
худшим выходом. В данном случае необходимо избегать соприкосновения
с порошком меди, который вызывает самопроизвольное разложение диазо-
кетонов.
Взаимодействием диазоацетона с гидридом три-я-бутилолова Лесбр [8]
получил трибутилстаннилацетон, впрочем, как позже было указано [23]f
в не вполне чистом состоянии:
(С4Н9)3 SnH + CH8COCHN2 -* (C4H9)3 SnCH2COCH3 + N2.
Сходным образом получен трипропилстаннилацетофеион.
Эти соединения дают хорошо кристаллизующиеся семикарбазоны. С ди-
азоацетонитрилом и гидридом три-я-бутилолова получено [8] три-я-бутил-
цианометилолово:
(С4Н9)3 SnH + NoCHCN -» (C4H9)3 SnCH2CN + N2.
21 *
324
Оловоорганические соединения
ЛИТЕРАТУРА
1. Н е с м е я н о в А. Н., ЖРФХО, 61, 1393 (1929); Вег., 62, 1010 (1929).
2. Коч еш ков К. А., Несмеянов А.Н., Климова В. А., ЖОХ, 6, 167
(1936).
3. Nesmejanow A.N., К о с h e s h k о w К. A., Klimova V. А., Вег., 68,
1877 (1935).
4. Н е 1 1 е г m а п п L., N е w n а п М. D., J. Am. Chem. Soc, 54, 2859 (1932).
5. Я кубов и ч А. Я., Макарове. П., Г а в р и л о в Г. И., ЖОХ, 22, 1788
(1952).
6. Якубович А. Я., Макарове. П., Гинсбург В. А., Гаврилов
Г. И., М е р к у л о в а Е. Н., ДАН СССР, 72, 69 (1950).
7. Н е с м е я н о в А. Н„ С е г а л е в и ч А. Е., Изв. АН СССР, ОХН, 1942, 8.
8. LesbreM., Buisson R., Bull. Soc. chim. France, 1957, 1204.
9. К г a m e г К. A., W г i g h t A. N., J. Chem. Soc, 1963, 3604.
10. P e у т о в О. А., П т и ц ы н а О. А., П а т р и н а Н. Д., ЖОХ, 28, 588 (1958).
11. W a t е г s W. A., J. Chem. Soc, 1939, 864.
12. Н е с м е я н о в А. Н., Макарова Л.Г., ДАН СССР, 87, 421 (1952).
13. П т и ц ы н а О. А., Р е у т о в О. А., Т у р ч и н с к и й М. Ф., ДАН СССР, 114,
ПО (1957).
14. П т и ц ы н а О. А., Р е у т о в О. А., Т у р ч и н с к и й М. Ф., Науч. докл.
высш. школы. Химия и хим. технол., 1, 138 (1959).
15. Несмеянов А. Нм Р е у т о в О. А., Т о л с т а я Т. П., ПтицынаО. А.,
Исаева Л. С, ТурчинскийМ. Ф., Б о ч к а р е в а Г. Л., ДАН СССР,
125, 1265 (1959).
16. V а п d е г К е г k G. J. M., L u i j t е п J. G. A., J. appl. Chem., 6, 93 (1956).
17. GilmanH., L e e p e г R. W., J. Org. Chem., 16, 466 (1951).
18. S e у f e г t h D., Rochow E. G., Inorganic Syntheses, 6, 37 (1960).
19. Vander KerkG. J.M., Noltes J.G., L и i j t e n J. G. A., J. appl. Chem.,
7, 356 (1957).
20. К о с т я н о в с к и й Р. Г., П р о к о ф ь е в А. К., Изв. АН СССР, ОХН, 1965,
175.
21. S е у f e r t h D., RochowE.G., J. Am. Chem. Soc, 77, 1302 (1955).
22. Kramer K., Wright N., Ber., 96, 1877(1963).
23. H e с м е я н о в А. Н., Л у ц е н к о И. Ф., П о н о м а р е в С. В., ДАН СССР,
125, 1073 (1959).
Глава VII
ВВЕДЕНИЕ ОЛОВА НА МЕСТО АТОМА ВОДОРОДА
Гексаалкилдистанноксаны, алкоксисоединения олова и станниламины
легко реагируют с производными ацетилена и другими соединениями,
содержащими подвижный атом водорода. Например:
R3SnOSnR3 + 2 HC=eCR' -» 2 R3SnC=CR' + Н20.
Впервые этот метод синтеза оловоорганических соединений описан Лу-
ценко с сотр. [1]. При нагревании до 140° С в течение 18 час. фенилацети-
лена с метокситриэтил оловом ими получено триэтилфенилэтинилолово с
выходом 75%.
Шостаковским с сотр. [2—13] подробно изучено взаимодействие гекса-
алкилдистанноксанов и алкоксисоединений олова с ацетиленом, алкил- и
арилацетиленами, ацетиленовыми спиртами, ацеталями и эфирами. Ими
найдено, что скорость реакции и выходы образующихся продуктов
возрастают по мере увеличения кислотности ацетиленового водорода. Если
взаимодействие ацетилена с гидроокисями триалкилолова и гексаалкилди-
станноксанами протекает под давлением и при температуре 100—120° С,
то с фенил- и винилацетиленами реакция проходит в обычных условиях при
комнатной температуре [4].
Получение три-#-пропилэтинилолова [4]. В пробирочный автоклав емкостью 0,25 л
помещают 31,8 г гидроокиси три-я-пропилолова, подают ацетилен под давлением 15—18 атм
и нагревают при 100—120° С до прекращения поглощения ацетилена. Перегонкой
образующегося продукта в вакууме получают 8,85 г (37%) вещества с т. кип. 91° С/10 мм; $£
1,1555; п2£ 1,4780; MRD найдено 67,1; вычислено 67,16.
Аналогично получены [4] триэтилэтинилолово и три-я-бутилэтинилолово.
Получение триэтилвинилэтинилолова [4]. В круглодонную колбу емкостью
50 мл, снабженную механической мешалкой и газоотводящей трубкой, присоединенной к
ловушке с сухим льдом, помещают 11,14 г гидроокиси триэтилолова и 7 г предварительно
охлажденного до —5 —0° С винил ацетилена. Смесь перемешивают при обычной
температуре в течение 6—8 час. и перегоняют в вакууме. Получают 8,07 г (62,8%)
триэтилвинилэтинилолова с т. кип. 78—79° С/3 мм.
Аналогично получены [3, 4] три-я-пропилвинилэтинилолово (т. кип.
100—101° С/3 мм) и три-я-бутилвинилэтинилолово (т. кип. 135—136° С/
/3,5 мм). Выходы продуктов составляют 50—60%.
Фенилацетилен реагирует с гидроокисью триэтилолова в более жестких
условиях (5 час. при 100—140° С). Триэтилфенилэтинилолово, выделенное
с выходом 52,7%, имеет т. кип. 193° С/0,35 мм [5].
326
Оловоорганические соединения. Лит. стр. 328
Реакции первичных, вторичных и третичных ацетиленовых спиртов с ме-
токситриэтилоловом могут проходить в двух направлениях [2]:
I > (С2Н5)3 SnOXC=CH + CH3OH
> (C2H5)3SnOXC=CSn (С2Н5)3 + СНзОН
II
(Х = СН2, —СН2СН2-, СН(СНз) и С (СНзЬ).
2 (QHsJs SnOCH3 + HOXCeeCH ■
При действии на метокситриэтилолово 2—4-кратного избытка
ацетиленового спирта при охлаждении до —10° С или при обычной температуре в
основном образуются ацетиленовые эфиры типа I, тогда как нагревание
смеси до 100—110° С или длительное перемешивание ее при обычной
температуре приводит к эфирам типа II (в случае третичных ацетиленовых
спиртов для этого требуется температура порядка 190—200° С).
Аналогичные результаты получены [7] при замене метокситриэтилолова гидроокисью
триэтилолова или гексаэтилдистанноксаном [2]. Выходы продуктов
заметно увеличиваются, если образующуюся в результате реакции
воду отгонять в виде азеотропной смеси с бензолом или толуолом.
Оловоорганические ацетиленовые эфиры при длительном хранении или
нагревании диспропорционируют по следующей схеме [2, 7]:
2 (QH5)3 SnOXC = СН -* (С2Н5)3 SnOXC = С5п (С2Н5)3 + HOXG ее СН.
На скорость этой реакции существенное влияние оказывает природа
исходного ацетиленового спирта [7].
Фенилацетилен гладко реагирует с оловоорганическими ацетиленовыми
эфирами с образованием триалкилфенилэтинилолова [7J:
(QHsb SnOXC ее СН + НС ее ССбН5 -> (С2Н5)3 SnC ее CCGH5 + НОХС ее СН.
При применении избытка фенил ацетилена и нагревании эта реакция
проходит почти количественно.
Оловоорганические а-ацетиленовые соединения типа R3SnOXC=CSnR3
реагируют с йодистым метилом при нагревании до 100° С в запаянной
ампуле:
R3SnOXC ее CSnRg + CH3J -> R3SnJ + R3SnC = СХОСН3.
Выход оловоорганических ацетиленовых простых эфиров составляет 30—
50%. В тех же условиях высшие галоидные ал килы в эту реакцию не
вступают [11].
Несимметричные ацетиленовые ацетали первичных, вторичных и
третичных спиртов реагируют с метокситриэтилоловом следующим образом
[4, 8, 91:
НСееСХО (C2H5)3SnCEECXO
(QH5)3 SnOCH3 + СНСН3 -> СНСН3 + СН3ОН.
с4н9о с4н9о
Реакции проводят путем нагревания смеси реагентов до 100—120° С в
течение 3 час. Выходы продуктов составляют около 70%. Те же соединения
образуются и при применении вместо метокситриэтилолова гексаэтилди-
станноксана. В этом случае для достижения высоких выходов необходимо
применять двойной избыток ацеталя или отгонять образующуюся в
результате этой реакции воду в виде азеотропной смеси с растворителем [8, 10].
Введение олова на место атома водорода
327
Получение 1-[4-(триэтилстаннил)бутин-3-окси]-1-бутоксиэтана [8]. 10,08 г (0,025 моля)
гексаэтилдистанноксана, 8,5 г (0,05 моля) 1-(бутин-3-окси)-1-бутоксиэтана и 8 г сухого
бензола нагревают при 80—120° С в течение б час. до полной отгонки бензола и выделившейся
в результате реакции воды. Получают 9,8 г (52,2%) вещества, имеющего при повторной
перегонке следующие константы: т. кип. 111—112° С/3 мм; d^° 1,1290; гг^ 1,4749.
Аналогично из гексаэтилдистанноксана или метокситриэтилолова
получены [9] оловоорганические ацетиленовые простые эфиры и оловоорга-
нические ацетиленовые виниловые эфиры. Например:
(С2Н3)з SnOSn (С2Н5)3 + 2 НСееССНоОСНз -» 2 (С2Н5)3 SnC=CCH2OCH3 -f П,0,
(С2Н5)3 SnOSn (С3Н5)8 + 2 НС=ССН2СН2ОСН==СН2 -»
-» (С2Н5)3 SnC=CCH2CH2OCH==CH2 + Н20.
Получение 3-метокси-1-(триэтилстаннил)пропина-1 [9]. Смесь 26,03 г (0,061 моля)
гексаэтилдистанноксана и 5,2 г (0,075 моля) З-метоксипропина-1 нагревают при 60—80° С
в течение 4 час. При перегонке в вакууме выделяют8,57г (25,6%) вещества,имеющего после
двух перегонок следующие константы: т. кип. 62—63° С/0,55 мм; df 1,2404; л^ 1,4878;
MRD найдено 63,84, вычислено 64,66.
Получение 4-винилокси-1-(триэтилстаннил)бутина-1 [9]. Смесь 10,68 г (0,025 моля)
гексаэтилдистанноксана и 4,8 г (0,05 моля) винилбутин-1-илового эфира нагревают при 90—
110° С в течение 8 час. и перегоняют в вакууме. Получают 8,46 г (56,3%) 4-винилокси-1-(три-
этилстаннил)бутина-1, имеющего после второй перегонки следующие константы: т. кип.
107,5—108°С/2 мм; df 1,1993; /г^ 1,4925; MRD найдено 72,87; вычислено 73,48.
Метилтриэтилстаннилпропиолат получен [14] взаимодействием
метокситриэтилолова с метиловым эфиром пропиоловой кислоты. Вещество
легко гидролизуется, превращаясь в пропиолат триэтилолова.
С гексаэтилдистанноксансм реакция проходит иначе [10]:
(С2Н5)3 SnOSn (СаН5)3 -!- НС=СС112ОСОСН3 -»
-» (С2Н5) SnOCH2C=CH 4- (C2M5)3SnOOCCIl3.
Кратко сообщается [15] (без деталей эксперимента) о реакциях гексаалкил-
дистанноксанов с флуореном, метилмалоновым эфиром и динитрилом
малоновой кислоты.
Ацетилен и его гомологи активно реагируют со станниламинами [16, 17].
Нагреванием диметиламинотриметилолова с пропил- и бутилацетиленом
получены [161 пентинилтриметилолово (т. кип. 172° С/760 мм) и гексинил-
триметилолово (т. кип. 82° С/12 мм) соответственно. Диметиламинотриме-
тилолово экзотермически реагирует с фенилацетиленом. Выход фенилэти-
нилтриметилолова 92%, т. кип. 68° С/0,3 мм. Так же получены фенилэти-
нилтриэтилолово (т. кип. 88° С/0,1 мм) и фенилэтипилтрифенилолово
(т. пл. 62° С).
При пропускании ацетилена через раствор 13,99 г диметиламинотри-
я-бутилолова в 50 мл петролейного эфира (т. кип. 30—40э С) в течение
5 час. образуется бш>(три-я-бутилстаннил)-ацетилен.В тех же условиях из
бш>(диметиламино)-ди-я-бутилолова получают полимерный продукт
состава (RySnC^Q^ (т. размягчения 128° С).
Применение станниламинов в качестве исходных веществ позволяет
распространить реакцию станнилирования и на другие соединения с
подвижным атомом водорода [16]:
R3SnN(CH3)2 + |^J -> RsSn-/^j + (CH3)2NH,
(C3H5)3SnN(CH3)2 + f Jj J -> (QH5)3Sn-<^J + (CH3)3NH.
328
Оловоорганические соединения
Получение циклопентадиенилтрифенилолова [16]. К раствору 17,3 г диметиламинотри-
фенилолова в 150 мл петролейного эфира (т. кип. 80—100° С) прибавляют 3,5 г циклопен-
тадиена. Смесь нагревают до кипения в течение часа и раствор фильтруют горячим. По
охлаждении выпадает 13,0 г (71, 3%) циклопентадиенилтрифенилолова с т. пл. 129° С.
Аналогично получено циклопентадиенилтриметилолово с т. кип. 56—
60° С/1 мм. Реакция между диметиламинотри-я-бутилоловом и циклопентадие-
ном проходит уже при обычной температуре. Выход циклопентадиенил-
три-я-бутилолова 98,5%, т. кип. 134° С/0,8 мм.
Получение (1-инденил)трифенилолова [16]. К 8,81 г диметиламинотрифенилолова в
50 мл петролейного эфира (т. кип. 60—80° С) прибавляют 2,60 г индена. Смесь нагревают в
течение 30 мин., растворитель удаляют при 50° С/15 мм и остаток перекристаллизовывают
из петролейного эфира. Получают 7,70 г (74%) (1-инденил)трифенилолова с т. кип. 129° С.
Диалкил(триалкилстаннил)амины реагируют с ацетонитрилом [16, 18]
фенил- и дифенилацетонитрилом [16], нитрометаном [16] и ацетоном [16].
Структура образующихся продуктов точно не установлена.
О реакции алкоксисоединений олова и станниламинов с кетеном и изо-
пропенилацетатом см. стр. 311.
С хлороформом и трихлорэтиленом диметиламинотриметилолово
образует (СНз)35пСС13 и (СН3)з5пСС1=--СС12 соответственно [19, 20].
ЛИТЕРАТУРА
1. Л у ц е н к о И. Ф., Пономарев СП., ПетрийО. П., ЖОХ, 32, 896
(1962).
2. Ш о с т а к о в с к и й М. Ф., Власов В. М., М и р с к о в Р. Г., ЖОХ, 34,
1354 (1964).
3. К о м а р о в Н. В., Ш о с т а к о в с к и й М. Ф., Г у с е в а И. С, Авт. свид.
СССР 158573 (1963); РЖХим., 1965, 16Н60; Бюлл. изобр., №22, 12 (1963).
4. Шостаковский М. Ф., Комаров Н. В.> Гусева И. С, М и с ю-
н а с В. К., ДАН СССР, 158, 918 (1964).
5. Шостаковский М. Ф., Власов В. М., МирсковР. Г., К о р о т а-
е в а И. М., ЖОХ, 35, 401 (1965).
6. Шостаковский М. Ф., Власов В. М., Мирсков Р. Г., ЖОХ, 35,
790 (1965).
7. Шостаковский М. Ф., Власов В. М., МирсковР. Г.,
Петрова В. Н., ЖОХ, 35, 47 (1965).
8. Шостаковский М. Ф., Власов В. М.,Мирсков Р. Г.,
Логинов а И. Е., ЖОХ, 34, 2843 (1964).
9. Ш о с т а к о в с к и й М. Ф., В л а с о в В. М., М и р с к о в Р. Г., ДАН СССР,
159, 869 (1964).
10. Шостаковский М.Ф., В л а с о в В. М., Мирсков Р. Г., ЖОХ, 34,
3178 (1964).
11. Шостаковский М. Ф., Власов В. М., Мирсков Р. Г., ЖОХ, 35, 1121 (1965).
12. Шостаковский М. Ф., К о м а р о в Н. В., Гусева И. С, М и с ю-
н а с В. К., С к л я н о в а А. М., Б у р н а ш о в а Т. Д., ДАН СССР, 163,
390 (1965).
13. Ш о с т а к о в с к и й М. Ф., В л а с о в В. М., М и р с к о в Р. Г., Авт. свид.
СССР 173757 (1965); Бюлл. изобр., № 9 (1965).
14. Neumann W.P., К 1 е i n e r F. G., Tetrahedron L., 1964, 3779.
15. L e u s i n k A. J., Marsman J. W., В u d d i n g H. A., Noltes J. G.,
van d e r KerkG.J.M., Rec. trav. chim., 84, 567 (1965).
16. J о n e s K., LappertM. R, J. Organometal. Chem., 3, 295 (1965).
17. J о n e s K., LappertM.R, Proc. Chem. Soc, 1964, 22.
18. G e о г g e K., Jones K., LappertM.R, J. Chem. Soc, 1965, 2157.
19. DaviesA. G., M i t с h e 1 1 T. N., J. Org. Chem., 6, 559 (1955).
20. С h i v e r s Т., D a v i d В., J. Organometal. Chem., 13, 177 (1958).
Глава VIII
ДЕАЛКИЛИРОВАНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Большинство описанных выше методов синтеза приводит к соединениям
типа R4Sn. Они обычно являются исходными веществами при получении
соединений с различными функциональными группами у атома олова. Для
этой цели осуществляют последовательное превращение тетраалкил- или
тетраарилолова вгалогениды моно-, ди- или триалкил-(соответственноарил-)-
олова, а последних — в другие производные.
Для синтеза оловоорганических галогенидов с одинаковыми
радикалами у атома олова наиболее важное препаративное значение имеет деалки-
лирование хлорным или бромным оловом. Оловоорганические соединения
с различными радикалами у атома олова деалкилируют свободными
галоидами или галоидоводородами. Гораздо реже в качестве деалкилирующих
агентов применяют кислоты, спирты, меркаптаны, соли различных
металлов, серу, селен, теллур и другие вещества.
ДЕАЛКИЛИРОВАНИЕ ГАЛОИДНЫМИ СОЛЯМИ ОЛОВА
Существенное значение для синтеза оловоорганических соединений типа
R3SnX и R2SnX2 в алифатическом ряду, а также галогенидов всех трех
возможных типов в ароматическом ряду имеют следующие разработанные Ко-
чешковым реакции [1—5]:
3R4Sn + SnX4-*4R3SnX,
R4Sn + R2SnX2-*2R3SnX,
R4Sn + SnX4 -* 2R2SnX2,
R3SnX + RSnX3-* 2R2SnX2?
R4Sn + 3SnX4 -* 4RSnX3,
R2SnX2 + SnX4 -> 2RSnX3.
Впоследствии этот метод был использован для получения трехгалоидных
соединений олова и в алифатическом ряду (см. ниже).
По сравнению с другими способами деалкилирования метод позволяет
полностью использовать все органические радикалы, связанные с атомом
олова. Выходы продуктов 80—90%. Кроме того, в тех случаях, когда при
синтезе оловоорганических соединений, например реакцией типа Вюрца
или действием галоидных алкилов на олово и его сплавы, образуется смесь
галогенидов олова различной степени алкилирования, ее превращают в
оловоорганическое соединение определенного состава нагреванием с
рассчитанным количеством R4Sn или SnX4 [6].
При синтезе оловоорганических галогенидов этим методом необходимо
обращать особое внимание на стехиометрическое соотношение реагентов
и температуру реакционной смеси, так как это оказывает решающее
влияние ка состав конечного продукта.
330
Оловоорганические соединения. Лит. стр. 370—375
Получение соединений типа R3SnX
Характерной чертой реакций с участием неорганических галоидных
солей олова в алифатическом ряду является самопроизвольное повышение
температуры до 70—100° С при смешении реагентов. Высказано
предположение [5], что это вызвано заменой первого галоида в SnX4 на алифатический
радикал. Действительно, исследование механизма реакции с помощью
ядерного магнитного резонанса [7] и химическими методами [8] показало, что ее
первая стадия может быть выражена уравнением
R4Sn + SnX4-> R3SnX + RSnX3.
При 0—20° С эта реакция заканчивается в течение 30—40 мин. [8—10].
Таким путем получены, в частности, галогениды этил-, я-бутил-и изобу-
тилолова [8], а также я-пропил-, я-пентил-, 3,3,5-триметилгексилолова
[10] и я-октилолова [9]. При нагревании на водяной бане
эквимолекулярной смеси йодного олова с тетраметилоловом образуется йодистое триметил-
олово и трехиодистое метилолово [11].
Препаративное значение этой реакции ограничено, так как в результате
образуется смесь двух продуктов. Выделение из нее чистых веществ связано
с известными трудностями. При нагревании они могут реагировать друг
с другом с образованием соединений типа R2SnX2. Для разделения олово-
органических моно- и тригалогенидов предложено использовать их
различную растворимость в воде [12] или разбавленной кислоте [13, 14].
Тригалогениды алкилолова реагируют с тетраалкильными
соединениями олова по уравнению
RSnX3 + 2R4Sn -* 3R3SnX.
При нагревании до 200° С треххлористого этилолова с тетраэтилоловом в
течение часа образуется 98,6% хлористого триэтилолова. Интересно
отметить, что при 150° С реакция заканчивается только через 24 часа [8].
Обычно соединения типа R3SnX получают взаимодействием хлорного
(или бромного) олова с тремя эквивалентами R4Sn [3, 4, 15—26]. Так,
нагреванием смеси бромного олова с тетраметилоловом при 120° С в течение
3 час. с последующим повышением температуры до 155° С получают
бромистое триметилолово. Выход 89%; т. кип. 160—165° С [15].
Получение хлористого триэтилолова [3, 4]. 21,1 г (0,09 моля) тетраэтилолова смешивают
с 7,8 г (0,03 моля) свежеперегнанного хлорного олова. При смешении в течение первых
секунд никаких изменений не наблюдается, смесь имеет слабокоричневую окраску, а затем
внезапно с некоторым просветлением окраски происходит разогревание, достигающее 70° С.
Смесь постепенно нагревают на водяной бане в течение 45 мин., а затем 3 часа на масляной
бане при 190—210° С. Перегонка продукта реакции дает с выходом 82—83% хлористое
триэтилолово с т. кип. 89—91° С/12 мм; т. пл. 12—14° С. В перегонной колбе остается дву-
хлористое диэтилолово с т. пл. 84° С (из петролейного эфира).
Аналогично получены бромистое триэтилолово [3, 4], хлористое три-я-
пропил-[17, 23] и триизопропилолово [20], бромистое триизопропилолово
[16].
В некоторых случаях синтез соединений типа R3SnX сопровождается
разложением вещества. Например, при получении хлористого три-я-бу-
тилолова происходит следующий побочный процесс [16, 18]:
2(C4H9)3SnCl -^ 3QHi0 + 3QH8 + Sn + SnCl2.
Потери составляют около 20%. Нагреванием при 230° С тетра-я-бутил-
олова с хлорным оловом в течение 3 час. получают 73,3% хлористого три-я-
бутилолова [19]. Выход увеличивается, если пользоваться следующим
приемом.
Деалкил ирован ие
331
Получение хлористого три-#-бутилолова [18]. Смесь 590г неочищенного тетрабутилолова
{1,38% хлора) и 129 г (58 мл) хлорного олова нагревают при температуре бани 220—230° С
в течение 1,5 час, после чего охлаждают сначала на воздухе до 100° С, а затем водой
до 30° С (на охлаждение требуется примерно 45 мин.)- После повторного 1,5-часового
нагревания при 220—230° С смесь фильтруют и перегоняют в вакууме. Получают 680,5 г (94,5%)
хлористого три-я-бутилолова с т. кип. 140—152° С/10 мл.
Синтез хлористого тривинилолова необходимо вести при более низкой
температуре. Нагревание при 185—200° С в течение 1,5 часа тетравинил-
олова (0,134 моля) и хлорного олова (0,045 моля) приводит к смеси
металлического олова с хлористым оловом [24]. При 90—100° С образуется смесь
хлористого тривинилолова (46,6%) и двухлористого дивинилолова (22%)
[24] и лишь в том случае, если реакцию проводить при 30° С, хлористое три-
винилолово образуется практически с количественным выходом [22, 27]*
Получение хлористого тривинилолова [22]. К 600 г (2,64 моля) тетравинилолова при
перемешивании в течение часа прибавляют 229,0 г (0,88 моля) хлорного олова (температура
бани 30° С). Смесь перемешивают 2 часа при той же температуре и по возможности быстро
перегоняют в вакууме (во время медленной перегонки возможно частичное диспропорциони-
рование хлористого тривинилолова). Получают 795 г (96%) хлористого тривинилолова
с т. кип. 59—60° С/6 мм.
Сравнительно редко галогениды триалкилолова получают нагреванием
соединений типа R2SnX2 с R4Sn, В отличие от предыдущих случаев здесь
никаких внешних или температурных изменений не происходит даже при
длительном стоянии смеси при комнатной температуре. Однако при
нагревании до 200—215° С в течение 2—3 час. образуются соединения типа
R3SnX с выходом 70—75%.
В качестве примеров можно указать на синтез хлористого и бромистого
триэтилолова из двухлористого (или двубромистого) диэтилолова 13, 4].
Получение хлористого триэтилолова из двухлористого диэтилолова[3, 4]. 24,8 г (0,1 моля)
двухлористого диэтилолова прибавляют к 23,5 г (0,1 моля) тетраэтилолова. Смесь
нагревают на масляной бане до 210—215° С в течение 2 час. и перегоняют в вакууме; т. кип. 100—
ЮГ С/16 яя; выход 75%.
Кратко сообщается [28] о синтезе хлористого диметил(пентафторфенил)-
олова по реакции
(CH3)2Sn(C6F5)2 + (CH3)3SnCl2 -» 2(СН3)2 (QF5) SnCl.
Соединения типа R3SnX (R — алкил) представляют собой, как правило,
жидкости с характерным резким и неприятным запахом. Они являются
слабыми лакриматорами.
Аналогично проводят реакцию в ароматическом ряду, но обычно она не
проходит до конца. Например, нагреванием при 220 + 10° С
тетрафенилолова с рассчитанным количеством хлорного олова в течение часа
получают [29] только 60—70/6 хлористого трифенилолова. Для очистки от не-
прореагировавшего тетрафенилолова предложено ряд методик [17, 18,
30—33].
Получение хлористого трифенилолова [17, 30, 32, 33]. 128,1 г (0,3 моля) тетрафенилолова
смешивают с 26,8 г (0,1 моля) свежеперегнанного хлорного олова. Реакция заметно
протекает уже при комнатной температуре; первоначально образовавшаяся густая масса
постепенно становится более жидкой. Смесь постепенно нагревают на масляной бане до 205° С,
3—4 часа при 205—210° С и 3 часа при 150—160° С. Содержимое колбы представляет собой
теперь коричневую жидкость, которая при охлаждении постепенно закристаллизовывается.
Для очистки от примесей сырой продукт измельчают и длительно экстрагируют эфиром.
Через 12 час. из эфирного раствора выпадает небольшое количество тетрафенилолова. Эфир
отгоняют досуха и остаток дважды кристаллизуют из спирта. Выход 85%; т. пл. 105—106° С.
332
Оловоорганические соединения. Лит. стр. 370—375
Синтез хлористого трифенилолова можно проводить также в
присутствии инертных органических растворителей [34].
Аналогично получают бромистое трифенилолово [30, 33, 35], хлористое
три-/г-толилолово (выход 80%) [33, 36, 37], хлористое три-ж-толилолово
[30, 33] (75%) и хлористое три-о-толилолово [30, 33] (72%).
Получение хлористого три-(/г-фторфенил)олова [38]. В круглодонную колбу,
снабженную магнитной мешалкой и обратным холодильником, помещают 8,1 г (10,032 моля) четырех-
хлористого олова и 42,8 г (0,097 моля) тетра-(/г-фторфенил)олова. Смесь нагревают при 190—
200° С в течение 3 час. Образовавшийся продукт закристаллизовывается при прибавлении
циклогексана. После двух перекристаллизации из гексана и циклогексана получают
33,6 г (60%) вещества с т. пл. 118, 2—120,5° С.
Галогениды триарилолова представляют собой белые кристаллические
вещества, практически без запаха, хорошо растворимые в ацетоне, этил-
ацетате и хлороформе; в петролейном эфире плохо растворимы на холоду,
умеренно при нагревании. Введение метильной группы в бензольное кольцо
несколько снижает растворимость этих соединений.
Получение соединений типа R2SnX2
Как уже было отмечено ранее, на первой стадии реакции тетраалкиль-
ных соединений олова с хлорным (или бромным) оловом образуется смесь
соединений типа R3SnX и RSnX3. При нагревании они реагируют между
собой, давая соответствующие ди галоген иды.
С помощью газовой хроматографии показано [8], что хлориды моно-,
ди- и трибутилолова присутствуют в смеси даже при 200° С. При этой
температуре соединения типа R2SnX2 частично диспропорционируют,
превращаясь в R3SnX. Высказано предположение [8], что реакция между моно-и
тригалогенидами алкилолова является обратимой. Впоследствии этот
вывод подтвержден кинетическими исследованиями, проведенными методом
ядерного магнитного резонанса [7]. Но соединения типа RSnX3 термически
неустойчиЕЫ и поэтому при достаточно высоких температурах равновесия
не наступает [7, 8].
Нагреванием (CH3)3SnBr с CH3SnBr3 при 195—206° С в течение 2 час.
получено двубромистоедиметилолово почти с количественным выходом [4,5].
Гораздо чаще соединения типа R2SnX2 получают нагреванием тетраал-
кильных соединений олова с хлорным (или бромным) оловом.
Получение двухлористогодиэтилолова [4, 5]. К 4,7 г (0,02 моля)
тетраэтилоловаприбавляют 5,2 г (0,02 моля) хлорного олова. Смесь окрашивается в слабокоричневый цвет и в
течение 1—2 сек. сохраняет комнатную температуру. Затем сразу происходит просветление
раствора, сопровождающееся повышением температуры до 110° С. Смесь нагревают в
течение 1,5 часа до 200—210° С. По охлаждении содержимое колбы застывает в сплошную
кристаллическую массу, имеющую слабый запах моногалоидного соединения. Вещество
кристаллизуется из лигроина в виде шелковистых игл ст. пл. 84°С. Выход близок к количественному»
Так же получают двубромистое диметил- и диэтилолово [4, 5]„
двухлористое [4, 5, 39] и двубромистое ди-я-пропилолово [4, 5]. Двухлори-
стое ди-я-бутилолово получают нагреванием до 240° С тетрабутилолова с
хлорным оловом в течение 3 час; выход 95%, т. пл. 43°С [18, 401. При более
продолжительном нагревании или при дальнейшем повышении температуры
вещество разлагается [41]. При понижении температуры или сокращении
времени реакции выход основного продукта снижается [41]. Так, при
нагревании тетрабутилолова с хлорным оловом до 200—215° С в течение 3 час.
образуется 74% (вместо 95%) двухлористого дибутилолова [42].
Нагреванием при 200° С тетрабутилолова с бромным оловом в течение 3 час.
получают 75% двубромистого ди-я-бутилолова с т. кип. 96—98° С/0,04 мм
Деалкил ирован ие
333
[43]. При получении двухлористого диизобутилолова образуется примесь
моногалоидного соединения. Разделить их перегонкой не удается.
Целесообразнее дополнительное прибавление хлорного олова.
Получение двухтористого диизобутилолова [8]. 139,0 г (0,40 моля) тетраизобутил-
олова и 104,2 г (0,40 моля) хлорного олова нагревают при 200° С в течение 20 час.
Затем добавляют еще 5 мл SnCl4, нагревают при той же температуре 30 мин. и смесь
лерегоняют в вакууме. Получают 180,8 г (74,4%) двухлористого диизобутилолова с
т. кип. 67—71° С/0,25 мм. Избыток хлорного олова собирают в приемнике, который
охлаждают жидким воздухом.
Двухлористое ди-я-гексилолово получают [44] нагреванием на водяной
бане эквимолекулярной смеси реагентов в течение 1,5 часа, а затем 3 часа
лри 220—225° С. Продукт очищают перегонкой в вакууме; т. кип. 134—-
148° С/1,5 мм; т. пл. 44—46° С; выход 87,5%.
Получение двухтористого ди-(2-этил-я-гексил)олова [44]. Смесь 75 г неочищенного
тетра-(2-этил-я-гексил)олова и 31,3 г хлорного олова нагревают при 225—230° С в
течение 3 час. После охлаждения прибавляют 250 мл петролейного эфира (т. кип. 60—80° С),
фильтруют, раствор выпаривают и остаток перегоняют в вакууме. Получают 69 г (65%)
масла желтого цвета с т. кип. 157—162° С/0,3 мм.
Аналогично получены двухлористое ди-я-октилолово [40, 44, 45]
(выход 83,5%; т. пл. 45,5—46,5° С) и двухлористое ди-3,5,5-триметил-я-гексил-
олово (выход 75%; т. кип. 154—157° С/0,1 мм) [44].
Дигалогениды диалкилолова получают также взаимодействием галоге-
нидов триалкилолова с рассчитанным количеством хлорного или бромного
олова. Например, при нагревании до 190—195° С (C2H5)3SnCl (0,2 моля) с
хлорным оловом (0,1 моля) образуется двухлористое диэтилолово
практически с количественным выходом [4, 5].
Вследствие термической неустойчивости двухлористого дивинилолова
синтез его проводят при 20—30° С [22, 27].
Получение двухлористого дивинилолова [22] (ср. [46]). В колбу, снабженную капельной
воронкой и обратным холодильником с хлоркальциевой трубкой, помещают 340,5 г (1,5 моля)
тетравинилолова. В течение 2Va часа прибавляют 390,8 с (1,5 моля) хлорного олова при
температуре бани 30° С. Смесь перемешивают 2 часа и перегоняют в вакууме. Получают
718,8г (98%) двухлористого дивинилолова с т. кип. 54—56° С/3 мм; т. пл. 74,5—75° С. Если
реакцию проводить при 100° С, то выход снижается до 70%.
Дигалогениды диалкилолова — белые кристаллические вещества со
своеобразным, довольно резким запахом, который, однако, легко отличить от
запаха моногалоидных соединений. Они хорошо растворимы в обычных
органических растворителях, кроме холодного гексана и петролейного эфира.
В алициклическом и ароматическом ряду реакцию проводят так же,
как и в алифатическом.
Получение двухлористого дициклопропилолова [47]. Синтез вещества проводят в
атмосфере азота. В колбу ня 25 мл% снабженную обратным холодильником и термометр >м,
помещают 4,85 г (0,0171 моля) тетрациклопропилолова, охлаждают льдом и из пипетки
прибавляют 2 мл (0,0172 моля) хлорного олова. Происходит экзотермическая реакция
(температура повышается до 110° С). Смесь нагревают до 220° С в течение часа и 4,5 часа при 220—
225° С. Летучие продукты удаляют в вакууме и остаток перегоняют также в вакууме.
Выход 7,9 г (86%); т. кип. 205—206° С/0,1 мм; т. пл. 59—60° С. После кристаллизации из
гексана т. пл. 60—60,5° С.
Аналогично получают двубромистое дициклопропилолово.
Нагреванием до 220° С эквимолекулярных количеств тетрафенилолова
и бромного олова в течение 4 час. получают [48] двубромистое дифенил-
олово. Смесь перегоняют в вакууме, основную фракцию охлаждают сухим
льдом, постепенно нагревают до 0° С, жидкую часть декантируют и остаток
кристаллизуют из петролейного эфира. Т. пл. 37° С.
334
Олоеоорганические соединения. Лит. стр. 370—375
Двухлористое дифенилолово образуется практически с количественным,
выходом при нагревании до 220° С в течение 1,5 часа эквимолекулярных
количеств хлорного олова и тетрафенилолова [1, 2]. Более длительное
нагревание снижает выход [48] (вместо нагревания предложено [49] облучать
смесь кварцевой лампой до образования гомогенной жидкости). Вещества
кристаллизуют из петролейного эфира; т. пл. 42—44° С.
Соединения толильного ряда получают нагреванием соответствующей
смеси на кипящей водяной бане в течение 2 час. и затем 2 часа при 200—
205° С [50—52].
В отличие от тетрафенилолова, которое реагирует с хлорным оловом
при комнатной температуре в очень малой степени, реакция между тетра-
м- или тетра-я-толилоловом и четыреххлористым оловом заметно идет с
выделением тепла при смешении реагирующих веществ. В случае орто-сое-
динения даже через 2 месяца смесь не становится жидкой и однородной,
что служит признаком медленно протекающего процесса [51, 52]. Однако
после соответствующего нагревания реакция проходит до конца и выход
близок к теоретическому.
Подобным же образом получают и другие соединения типа Ar2SnX2.
Получение двухлористого ди-#-анизилолова [53]. 3,1 г (1 моль) тетра-я-анизилолова и
1,5 г (1 моль) безводного хлорного олова запаивают в пробирке. Уже при смешении заметна
растворение тетра-я-анизилолова с легким разогреванием и постепенным окрашиванием в
темно-красный цвет. Пробирку помещают в глицериновую баню, нагретую до 80° С
(содержимое пробирки делается жидким и однородным). Нагревают при 80—145° С в течение
15 мин., при 145—160° С — 2 часа, при 160—185° С — 1 час. По охлаждении вещество за-
кристаллизовывается. Его кристаллизуют из петролейного эфира (при сильном охлаждении).
Получают 4 г (87%) двухлористого ди-/г-анизилолова ст. пл. 76° С. После второй
кристаллизации из того же растворителя т. пл. 84° С.
Аналогично получают [531 двубромистое ди-я-анизилолово (выход 74%),
двухлористое ди-я-фенетилолово (66%), двухлористое ди-я-бифенилолово
и двухлористое ди(циклогексилфенил)олово [54].^
Получение двубрсмистого ди-я-бифенилолова [55]. В пробирке запаивают 1,4 г (1 моль)
тетра-/г-бифенилолова и 0,88 г (1 моль) бромного олова. Нагревают при 90—165° С 30 мин.,
при 165—185° С— 1 час, а при 200—210° С — 2 часа. После охлаждения вещество
кристаллизуют из лигроина. Получают 1,6 г (70, 6%) двубромистого ди-я-бифенил олова с
т. пл. 144—145° С.
В некоторых случаях синтез соединений типа Ar2SnX2 проводят в
растворителе [46, 55, 56]. Например, двухлористое дибензилолово получают
нагреванием (C6H5CH2)3SnCl с хлорным оловом в бензоле или толуоле [56].
При получении двухлористого ди-а-нафтилолова из Ar4Sn и SnCl4 в
качестве растворителя применяют ксилол [55].
Получение двухлористого ди-о-феноксифенилолова [57]. 10,0 г тетра-о-феноксифенилоло-
ва и 3,62 г хлорного олова кипятят в 10 мл ксилола в течение часа и растворитель отгоняют.
Для очистки от побочно образовавшегося треххлористого о-феноксифенилолова смесь
нагревают при 280° С/0,5 мм. Остаток кристаллизуют из петролейного эфира (т. кип. 60—80° С).
Получают 10,44 г (79%) двухлористого ди-о-феноксифенилолова с т. пл. 98—99° С.
Соединения типа Ar2SnX2 представляют собой белые кристаллические
вещества, хорошо растворимые в обычных органических растворителях.
Умеренно, но все же заметно растворимы в холодном гексане и петролейном
эфире. Бромиды растворяются несколько хуже соответствующих хлоридов.
Деалкил ирован ие
335
Получение соединений типа RSnX3
Алифатические соединения типа RSnX3 получают с выходом 66%
нагреванием R4Sn с двумя эквивалентами хлорного олова. Побочным продуктом
является двухлористое диалкилолово [8]:
R4Sn + 2SnX4-* 2RSnX3 + R2SnX2.
Получение треххлористого этилолова и двухлористого диэтилолова [8]. 265 г (1,015 моля)
хлорного олова медленно сливают с 117,5 г (0,50 моля) тетраэтилолова при перемешивании
и температуре 0° С. Таким образом избегают нежелательного взаимодействия тетраэтилолова
с треххлористым этилоловом (см. стр. 330). Смесь перемешивают в течение 2 час; за это
время в осадок выпадает часть двухлористого диэтилолова. Перегонкой на колонке получают
246 г (65%) треххлористого этилолова с т. кип. 36—38° С/1 мм. В остатке — двухлористое
диэтилолово. Его перегоняют с воздушным холодильником. Выход 123 г, т. кип. 102—107°
С/12 мм. Избыток хлорного олова собирается в ловушке, охлажденной жидким воздухом.
Реакцию можно проводить также в бензоле. Из 1,5 моля тетраэтилолова
и 3 молей четыреххлористого олова после перемешивания в течение 24 час.
в 1 л бензола получают 64,6 мол.% C2H5SnCl3, 32,1% (C2H5)2SnCl2 и 3,3%
(C2H5)3SnCl.
С четырехбромистым оловом реакцию проводят при нагревании.
Получение трехбромистого этилолова и двубромистого диэтилолова [8]. К 177,0 г (0,404
моля) бромного олова при перемешивании в течение 3 час. прибавляют по каплям 47,5 г
(0,202 моля) тетраэтилолова (температура 100° С). Затем перемешивают при той же
температуре еще 12 час. Количества реагентов должны точно соблюдаться, так как избыток бромного
олова отделяется с трудом. Перегонкой смеси на колонке получают 171,6 г трехбромистого
этилолова с т. кип. 46° С/0,1 мм. С помощью газовой хроматографии установлено, что
продукт содержит 15,6 г двубромистого диэтилолова. Его отделяют перегонкой на эффективно
действующей колонке. Получают 156,0 г чистого трехбромистого этилолова (66,6% в расчете
на введенное в реакцию количество олова). Трехбромистое этилолово — жидкость. Данные
Дрюса [58, 59] о том, что это твердое вещество, видимо, были ошибочными.
Перегонкой остатка с воздушным холодильником получают двубромистое диэтилолово;
т. кип. 69—71° С/0,1 мм\ выход 33%.
Сходным образом получают треххлористое я-бутил- и изобутилолово.
Получение треххлористого я-бутилолова и двухлористого ди-я-бутилолова [8]. К 53,5 г
(0,203 моля) хлорного олова при 100° С и перемешивании прибавляют по каплям 34,7 г
(0,10 моля) тетра-я-бутилолова в течение 30 мин. Смесь перемешивают 2 часа при 100° С.
При более длительном нагревании вещество разлагается. Перегонкой в вакууме получают
53,4 г (63%) треххлористого я-бутилолова, т. кип. 44—45° С/0,1 мм (при перегонке
применяют колонку высотой 20 см); в охлажденной жидким воздухом ловушке собирается
небольшое количество четыреххлористого олова. Остаток представляет собой двухлористое ди-я-бу-
тилолово. Выход 29,9 г (33%); т. кип. 90—95° С/0,1 мм.
Синтез треххлористого изобутилолова проводят [8] путем прибавления
тетраизобутилолова (0,26 моля) к нагретому до кипения хлорному олову
(0,52 моля). Смесь перемешивают при температуре бани 200° С в течение
30 мин. и перегоняют в вакууме. Выход треххлористого изобутилолова 61 %;
т. кип. 35° С/0,25 мм. Наряду с ним получают 33,1 % двухлористого диизо-
бутилолова с т. кип. 69—71° С/0,25 мм.
Отщепление трех радикалов от тетраалкильных соединений олова в
обычных условиях не происходит. Например, нагревание рассчитанных
количеств тетра-я-бутилолова и хлорного олова в запаянной ампуле не
приводит к образованию треххлористого я-бутилолова 117]. Найдено [8], что
при нагревании до 120—130° С двухлористого диэтилолова с четырех-
хлористым оловом в течение 4 дней образуется только 7,7% треххлористого
этилолова. Применение избытка хлорного олова не влияет на выход веществ.
Однако при проведении реакции тетраалкилолова с хлорным оловом в
среде оксихлорида фосфора с присутствии фосфорного ангидрида
треххлористое этилолово получено с выходом 91,6%:
(QHs)4Sn + 3SnCl4-* 4C2H5SnCl3.
336 Оловоорганические соединения. Лит. стр. 370—375
Получение треххлористого этилолова [8]. К 156,5 г (0,6 моля) хлорного олова при 0° С
и перемешивании прибавляют по каплям 23,5 г (0,1 моля) тетраэтилолова. Через 2 часа
добавляют 184,0 г (1,2 моля) РОС13 и 21,3 г (0,15 моля) Р205. Смесь нагревают при 130° С в
течение 48 час, разбавляют 100 мл бензола и фильтруют. Отгоняют при атмосферном
давлении бензол, РОС13, SnCl4 и РОС13 • SnCl4. Остаток перегоняют в вакууме. Получают 86,5 г
треххлористого этилолова с т. кип. 76—78° С/12 мм (температура бани 180° С). Остаток
(66,3 г) объединяют с предыдущим отгоном и кипятят со 100 г SnCl4 в течение 6 час.
Аналогичная обработка дает 96,0 г C2H5SnCl3. Общий выход 94,3%.
Вероятно, каталитическое действие хлорокиси фосфора объясняется
двумя причинами [8]. Как полярный растворитель хлорокись фосфора
способствует реакции, проходящей, несомненно, по ионному механизму.
Кроме того, дальнейшее диспропорционирование двухлористого диэтил-
олова осуществляется благодаря нуклеофильной атаке на атом олова.
Интересно отметить, что в отличие от оксихлорида фосфора реакция с
хлористым сульфурилом идет совсем в ином направлении, а именно 18]:
у (QHsbSnCIa + S02C12 -* CaHsSnCis + НС1 + S02 + C2H4.
Введенное в реакцию хлорное олово остается без изменения.
Иногда соединения типа RSnX3 получают по следующей схеме [60—
62]:
CH3Sn(C6H5)3 + 3SnCl4 -» CH3SnCl3 + 3C6H5SnCl3.
Получение треххлористого метилолова [60, 61]. 36,5 г (0,1 моля) трифенилметилолова
осторожно смешивают с 78,3 г (0,3 моля) хлорного олова. Смесь нагревают в запаянной
ампуле в течение 1,5 часа до 185—190° С (необходимо обеспечить равномерность нагрева
трубки, в противном случае происходит частичное осмоление). По удалении не вошедшего в
реакцию хлорного олова собирают фракцию, застывающую в приемнике в кристаллическую
массу. После кристаллизации из тщательно высушенного петролейного эфира получают
продукт ст. пл.45—46° С. Выход 65%. Остаток в колбе представляет собой в основном
треххлористое фенилолово. После повторной перегонки в вакууме т. кип. 142—143°
С/25 мм.
При нагревании до кипения в течение 24 час. двухлористого диметил-
олова (0,05 моля) с эквимолекулярным количеством четыреххлористого
олова в 70 мл диметилсульфоксида с выходом свыше 90% получено [59а]
комплексное соединение треххлористого метилолова с растворителем, т. пл,
188—190° С (из этилового спирта и хлороформа или эфира).
Винильные и арильные радикалы отщепляются гораздо легче алкильных.
При нагревании до 70° С рассчитанных количеств тетравинилолова и
хлорного олова в течение 2 час. образуется 86% треххлористого винилолова
[22, 27].
Получение треххлористого винилолова [22]. К 50,0 г (0,22 моля) тетравинилолова при
перемешивании в течение 21/* часа прибавляют по каплям 187,6 г (0,66 моля + 10%)
хлорного олова (температура бани 30° С). Смесь перемешивают 2 часа при 70° С и избыток
хлорного олова отгоняют при обычном давлении (32 г; т. кип. 115—125° С). Перегонка остатка
в вакууме дает 190,0 г (86%) треххлористого винилолова с т. кип. 64—65° С/15 мм.
Вследствие некоторой термической нестойкости вещества повышение
температуры до 100° С несколько снижает выход конечного продукта [241.
Соединения типа ArSnX3 получают нагреванием тетраарильных
соединений олова с хлорным оловом при более высокой температуре [1,2, 28,
50—52, 63, 64].
Получение треххлористого фенилолова [1, 2]. 42,7 г (0,П моля) тетрафенилолова и
71,2 г (0,3 моля) хлорного олова нагревают до 210—220° С в запаянной ампуле в течение 1,5—
2 час. Содержимое трубки — коричневая, тяжелая, дымящая на воздухе жидкость (беречь
от влаги воздуха!). После повторной перегонки в вакууме получают треххлористое фенил-
олово с т. кип. 142—143° С/25 мм. Выход 80%.
Деалкил ирован ие
337
Также получены треххлористоео-толилолово (выход 65%) и треххлори-
сгое п-тол ил олово (40%) [51, 52].
Ароматические соединения типа ArSnX3 можно получать и реакцией ди-
галогенидов диарилолова с хлорным оловом. Таким образом, например,
были получены оловоорганические соединения с заместителями в бензольном
кольце.
Получение треххлористого я-хлорфенилолова [63, 64]. 8,24 г (0,02 моля) двухлористого
ди-я-хлорфенилолова и 5,2 г (0,02 моля) хлорного олова нагревают до 150° С в запаянной
ампуле в течение 2 час. Содержимое ампулы растворяют в абсолютном эфире и
отфильтровывают. Растворитель отгоняют и остаток обрабатывают петролейным эфиром.
Прозрачный раствор сливают с темноокрашенного масла, концентрируют до небольшого объема и
охлаждают снегом и солью. Выпавший кристаллический осадок быстро отсасывают и
высушивают в вакуум-эксикаторе; т. пл. 39° С, выход почти количественный. Вещество
чрезвычайно легко гидролизуется.
Аналогично получены треххлористое /г-бромфен ил олово и трехброми-
стое я-иодфенилолово.
Получение треххлористого а-нафтилолова [55]. 4,44 г (0,01 моля) двухлористого ди-
а-нафтилолова и 2,61 г (0,01 моля) хлорного олова нагревают до 150° С в запаянной ампуле
в течение 3 час. Продукт реакции представляет собой кристаллическое вещество ст. пл.
77—78° С (из петролейного эфира). Выход 91%.
1ДЕАЛКИЛИРОВАНИЕ ГАЛОИДАМИ
В зависимости от условий проведения реакции при действии галоидов на
тетраалкильные соединения олова образуются соединения типа R3SnX,
R2SnX2 или, наконец, SnX4. Отщепление первого и второго радикалов
проходит при различной температуре [65—67]. Третий и четвертый радикалы
отщепляются в близких условиях, поэтому получить таким путем
соединения типа RSnX3 обычно не удается [68] (ср. [69]).
В качестве деалкилирующего средства обычно применяют бром или
иод. Хлор и особенно фтор используются редко.
Насильским с сотр. [70—76] проведены подробные кинетические
исследования реакций оловоорганических соединений с бромом и иодом. В
частности, ими установлено [70], что реакция тетраалкильных соединений олова
с бромом и иодом в полярных и малополярных растворителях имеет
суммарный второй порядок и первый порядок по каждому из реагентов. В
полярных растворителях скорость реакции определяется главным образом
пространственным взаимодействием вступающего галогена и уходящей соль-
ватированной группы R3Si и пространственными затруднениями,
препятствующими сольватации. Скорость реакции уменьшается в ряду
СН3 > QHs > п-С3Н7 > i-СзНт
и возрастает с увеличением ионной силы раствора. Пространственный
эффект резко преобладает в реакциях тетраалкильных соединений олова с
иодом и сравним по величине с индуктивным эффектом при реакциях с бромом.
В малополярных растворителях влияние индуктивного эффекта
сказывается сильнее, скорость реакции изменяется в порядке
СНз < СаН5 > я-СзН7 < *-С3Н7. [70]
Отмечено [71], что скорости реакций (k) бромидов триалкилолова с
бромом в уксусной кислоте уменьшаются в следующем ряду:
^CH3 > ^С,Ы5 > /гС3Н7 .
22 Заказ № 207
338
Олово органические соединения. Лит. стр. 370—375
Это изменение скоростей реакций объяснено возрастающей ролью
индуктивного эффекта.
Теми же авторами изучено [72, 73] действие иода на соединения типа
ArSnR3 (где R = СН3, /-С3Н7 и п-С4Н9) в метиловом спирте:
ArSnR3 + J2-> R3SnJ + ArJ.
Найдено, что эта реакция подчиняется кинетическому уравнению второго
порядка (константы скорости измерены при 25° С). В случаях, когда
Аг — /?-СН3ОС6Н4, р- и /л-СН3СбН4, p-(CH3)3SnC6H4, C6H5, p-(CH3)3SiC6H4 и
/?-ВгС6Н4, эти реакции подчиняются уравнению Гаммета для электрофиль-
ного замещения. Поскольку это реакции второго порядка, то вероятность
нуклеофильного катализа под действием ионов иода отпадает. Когда же
Аг — 1- или 2-нафтил, 9-фенантрил, 3-пиренил, константы скорости резко
меняются в зависимости от природы радикала (они уменьшаются в ряду
СН3 > С4Н9 > /-С3Н7).
На примере реакций замещенных производных триметилфенилолова типа
XC6H4Sn (СН3)3 (где X = Н, /?-СН3, р-СН30, р-Br и m-CH3) исследовано
[74] влияние структурных факторов на скорость электрофильного
бимолекулярного процесса у ароматического атома углерода. Для всех
производных изучена температурная зависимость констант скоростей реакций,
вычислены параметры уравнения Аррениуса. Показано [75], что в реакциях
оловоорганических соединений с иодом важную роль играют стерические
факторы.
Изучая кинетику реакции иода с соединениями (CH3)3SnC6H4Sn(CH3)3 и
(CH3)3SnC6H6Si(CH3)3 Насильский и сотр. [76] пришли к выводу о том, что
электронодонорный индуктивный эффект группы (CH3)3Si и электроноакцеп-
торный эффект металла в ней взаимно погашают друг друга. В группе
(CHg)gSn преобладает донорный индуктивный эффект. Кинетика реакции иода
с соединениями типа R3SnC6H4X исследована и другими авторами [77].
Для препаративных целей действие галоидов на оловоорганические
соединения с одинаковыми радикалами у атома олова используется
сравнительно редко. Бромистоетриметил- [78—81],триэтил- [82], трициклогексил-
[83] и три-(2-фенилэтил)олово [84] получены прибавлением рассчитанного
количества брома к соответствующим тетраалкильным соединениям олова
при температуре от 0° до минус 30—40° С.
Получение бромистого триэтилолова [82]. 11,2 г (0,07 моля) брома постепенно
прибавляют к 16,4 г (0,07 моля) тетраэтилолова (смесь охлаждают снегом с солью). Продукт
очищают перегонкой в вакууме. Получают 17,6 г (88%) бромистого триэтилолова с т. кип.
105—107° С/15 мм.
Второй алкильный радикал отщепляется при более высокой температуре*
Двубромистое диметилолово получено бромированием тетраметилолова при
комнатной температуре [79] или при нагревании смеси до 50—80° С [80].
Получение двубромистого дициклогексилолова [85]. К 3 г тетрациклогексилолова
прибавляют 1,34 г брома в 50 мл сероуглерода. Происходит экзотермическая реакция. Через
48 час. растворитель отгоняют. Для удаления бромциклогексана остаток нагревают до 70° С
в вакууме и продукт кристаллизуют из спирта, а затем эфира. Т. пл. 58° С. То же вещество
образуется при длительном кипячении тетрациклогексилолова с бромом в хлороформе или
четыреххлористом углероде [83].
Иод реагирует с тетраалкильными соединениями олова только при
нагревании. Например, для тетраметилолова температура реакции 100° С,
выход йодистого триметил олова 86,4%. Для тетраэтилолова температура
реакции от 80° до 130—140° С, выход йодистого триэтилолова 84,7% [68].
Гораздо чаще иодирование проводят нагреванием соответствующих реаген-
Деалкил ирован ие
339
тов в среде инертных растворителей. Так, йодистое триметил олово получают
в нагретом до кипения эфирном [68] или бензольном [86] растворе.
Получение йодистого триметилолова [68]. В колбу на 250 мл помещают 25 г (0, И моля)
предварительно перегнанного над натрием тетраметилолова в 50 мл сухого эфира и
небольшими порциями прибавляют 35,5 г (0,14 моля) измельченного и тщательно высушенного
иода. Иод вводят в течение 10 час, после чего смесь кипятят еще 2 часа. Растворитель
отгоняют, остаток отфильтровывают и перегоняют в вакууме. Получают 38,9 г (96%) йодистого
триметилолова с т. кип. 68—69° С/15 мм.
Аналогично получено йодистое триэтилолово [68].
Для отщепления более тяжелых радикалов требуется более высокая
температура. Тетра-я-пропил-, тетра-я-бутил- и тетраизобутилолово не
реагируют с иодом в эфире [87]. В хлороформе реакция проходит медленно,
гораздо лучше в бензоле или толуоле при температуре их кипения. Легче
всего иодирование этих соединений проходит в кипящем ксилоле.
Получение йодистого три-я-пропилолова [87]. В круглодонную колбу на 100 мл,
соединенную с обратным холодильником, помещают свежеперегнанные над натрием 11 г (0,04
моля) тетра-я-пропилолова и 10 мл ксилола. Затем небольшими порциями прибавляют 9,6 г
(0,04 моля) ьода. Смесь кипятят в течение 8 час, фильтруют, ксилол отгоняют и остаток
перегоняют в вакууме. Получают 3,5 г (95,3%) йодистого три-я-пропилолова с т. кип. 140—
141е С/15 мм] л^1, 5435.
Аналогично получены [87] йодистое три-я-бутилолово, йодистое триме-
зитилолово и ряд других.
Получить в чистом виде соединения типа R2SnJ2 непосредственно из
R4Sn не удается. Например, нагревание до 110° С бензольного раствора
иода с тетрациклогексилоловом в запаянной ампуле приводит к смеси
продуктов [85]. Поэтому оловоорганические дииодиды получают иодированием
соединений типа R3SnJ.
Получение двуиодистого диметилолова [68]. В колбу Вюрца на 25 мл, соединенную
с коротким холодильником, помещают 7 г (0,024 моля) йодистого триметилолова и при
нагревании до 1С0° С прибавляют 6,1 г (0,024 моля) иола в течение 10 час. По окончании реакции
содержимое колбы закристаллизовывается. Пссле перекристаллизации из лигроина
получают кристаллы дииодида с т. пл. 30—31° С. Выход 65%.
Аналогично получены [88] двуиодистое диэтил- и ди-я-пропилолово..
Действие галоидов на соединения типа R4Sn с различными радикалами
у атома олова приводит к преимущественному отрыву одного из них.
Порядок отщепления различных радикалов галоидами исследован рядом авторов
[22, 68, 87, 89—108]. В порядке легкости отрыва от атома олова они
располагаются следующим образом: о-толил >> п-толил > фенил > бензил >
^>винил>метил> этил > пропил > изобутил > амил > гексил > гептил >
^>октил. Найдено [93], чтоаллильная группа отщепляется от олова бромом
при — 50° С, фенильная и винильная при 0° С, а метильная и бутильная
при 40° С. Перфторвинильная группа легко отрывается иодом при комнатной
температуре [109].
Указанная закономерность в отрыве радикалов позволяет
синтезировать оловоорганические бромиды или иодиды с различными радикалами
у атома олова. Отщепление радикалов проходит в тех же условиях, что и при
действии галоидов на симметричные тетраалкильные соединения олова. Так,
при нагревании соединений типа R2SnR2' (где R —СН3 или QH5; R' —
бутил, амил, гексил и т. д.) в эфире с избытком иода происходит
преимущественный отрыв одной метильной или этильной группы [110,111].
Получение йодистого диэтил-я-октилолова[110]. Раствор 51 г (0,2 моля) иода в 500 мл
эфира кипятят с 64 г (0,2 моля) триэтил-я-октилолова в течение 10 час. Непрореагировавший
иод удаляют встряхиванием с небольшим количеством водного раствора бисульфата натрия.
22*
340 Оловоорганические согдлнзния. Лит. стр. 3/0—375
Эфирный раствор высушивают и эфир отгоняют. Остаток перегоняют на небольшой колонке.
Бромистое диэтил-я-октилолово имеет т. кип. 132—135° С/0,9 мм (перегонку ведут при
возможно более низкой температуре, чтобы избежать диспропорционирования продукта).
Выход 69%.
Подобным образом получены [98] соединения (CH3)(G>H5)(C3H7)SnJ,
(CH3)2(C2H5)SnJ и др.
При действии рассчитанного количества брома на триметил-я-децил-,
триметил-я-додецил- и триметил-я-октилолово в четыреххлористом углероде
происходит отрыв метильной группы [93]. По методике, описанной для
синтеза йодистого три-я-пропилолова (см. стр. 339) получены [90] следующие
соединения: (C2H5)2(C3H7)SnJ (т. кип. 132—134° С/16 мм), (C3H7)2(C4H9)SnJ
(т. кип. 160° С/24 мм), (n-C4H9)2(i-C5H11)SnJ (т. кип. 195° С/12 мм).
Взаимодействием диэтил- и ди-я-бутилдинеопентилолова с одним
эквивалентом брома при —30° С получены бромиды алкилдинеопентилолова [112].
При реакции диалкилдинеопентильных соединений олова с двумя
эквивалентами брома в кипящем хлороформе образуется двубромистое динеопентил-
олово. Аналогично из триэтилнеопентилолова получают бромистое ди-
этилнеопеитилолово и двубромистое этилнеопентилолово. Интересно
отметить, что при действии хлора на триметилтрифторметилолово отщепляется
метальный, а не трифторметильный радикал [15, 1131:
(CH3)3SnCF3 + С12 -> [(CHabSnCFaCla] -> (СН3)2 (CF3)SnCl + СН3С1.
Таким образом, решающим фактором, определяющим легкость отрыва
того или иного радикала, является энергия связи Sn—С, а не
«электроотрицательность» соответствующего радикала, как это предполагалось
ранее.
Получение хлористого диметилтрифторметилолова [113]. В трубку Кариуса помещают
9,5 г (0,041 моля) три мети лтрифторметил олова и конденсируют 2,68 г (0,038 моля) хлора.
Ампулу эвакуируют и запаивают. Смесь охлаждают до —46° С и постепенно повышают
температуру до комнатной в течение 2 дней. За это время окраска хлора исчезает. Летучие
вещества (смесь хлороформа и хлористого метила с небольшой примесью трифторхлорметана)
удаляют в вакууме. Получают остаток в виде белого кристаллического вещества. Его
очищают возгонкой в вакууме при 60—70° С. Получают 8 г (77%) хлористого
диметилтрифторметилолова с т. пл. 46—47° С. При комнатной температуре вещество не реагирует с хлором
в течение 10 дней.
Винильный радикал отщепляется гораздо легче алкильных.
Получение бромистого дивинил-я-октилэлова [93]. К охлажденному до 0° С раствору
115,2 г (0,368 моля) тривинил-я-октилолова в 184 мл четыреххлористого углерода при
перемешивании прибавляют 58,6 г (0,368 моля) брома в 184 мл четыреххлористого углерода
в течение 2 час. Растворитель отгоняют в вакууме водоструйного насоса при 60° С. Выход
бромистого дивинил-я-октилолова 107,6 г (80%); т. кип. 82—86° С/0,01 мм.
Аналогично получены [93] бромистое дивинил-я-децил-, дивинил-я-гек-
силолово и др.
Бромирование дибутилдивинилолова проводят при комнатной
температуре без растворителя. Выход бромистого дибутилвинилолова 68,4%; т. кип.
72—73° С/0,03 мм [22]. При действии брома на тетрааллилолово при
температуре —15° С образуется смесь бромистого триаллилолова и двуброми-
стого диаллилолова [114]. Последовательное отщепление аллильных групп
достигается понижением температуры реакции до —50° С [93]. Из бутил-
триаллилолова, дибутилдиаллилолова и дифенилдиаллилолова в этих
условиях получены [93] бромистое я-бутилдиаллил-, дибутилаллил- (т. кип.
85—89° С/0,03 мм) и дифенилаллилолово. В качестве растворителя
применяют хлороформ.
Деалк ил ирован ие
341
Йодистое диметилвинил- (т.кип. 57,5—59° С5 мм), дибутилви-
нил- (т. кип. 108,5—110° С/1,75 мм) и тривинилолово (т. кип. 60° С/1,8—
1,9 мм) образуются при нагревании в течение 8 час. эфирных растворов
соответствующих диалкилдивинильных соединений олова или тетравинил-
олова с рассчитанным количеством иода [94].
Следует отметить, что для высших алкильных радикалов приведенный
на стр. 339 порядок отрыва различных групп становится менее заметным.
Например, гептильный и октальный радикалы отщепляются иодом почти
в тех же условиях, что и бутильный или изоамильный [89]. Иодирование три-
этилизопропилолова приводит к смеси двух продуктов:
■(QH5y>nJ + «-C3lI7J
(GJWsSnCsHW -h h —
(/-CsH7) (QH,)2SnJ + C2H,J
Также не наблюдается разницы в скорости отрыва изопропильной и втор-
бутильной групп [115]. Это объясняется, очевидно, более жесткими
условиями проведения реакции, а не структурой соответствующих оловоорга-
нических соединений. Действительно, при|нагревании диметилди-я-бутил-
олова с иодом в бензоле также образуется смесь продуктов [115], тогда как
в эфире получают йодистое метилди-я-бутилолово [111]. Алкилциклогек-
сильные соединения олова не реагируют с иодом в эфире [115]. При
нагревании смеси в бензоле отщепляются как алкильный, так и циклогексиль-
ный радикалы.
Реакция диметилдициклогексилолова с иодом [115]. К раствору 34,0 г (0,108 моля) ди-
метилдициклогексилолова в 300 мл бензола прибавляют 27,45 г (0,108 моля) иода. Смесь
кипятят в течение часа и бензол отгоняют. Перегонка остатка в вакууме дает 10,3 г йодистого
циклогексила (т. кип. 42° С/1 мм\ п2^ 1,5462); небольшую промежуточную фракцию;
йодистое диметилциклогексилолово (17,1 г; т. кип. 86° С/0,65 мм); небольшую промежуточную
фракцию и, наконец, йодистое метилдициклогексилолово; т. кип. 136—140° С/0,65 мм;
выход 20,8 г.
Галоидирование а-замешенных олсвсорганических зфирсв или
нитрилов приводит, как правило, к отрыьу замешенного радикала. Однако,
если заместитель находится по отношению к атому олова в |3- или в еще
более удаленном положении, то отщепляется алкильный радикал без
заместителя [18, 116—119]. Например:
(QH9)3SnCH2CN + J2 -> (C4H9)3 SnJ + JCH2CN,
(GHekSnCHiCHtCN -f Его -* (QH9)2 (CNCHlCHo) SnBr -f CH9Br.
Получение брсмистсго (2-кар£скскметил5ТИл),ркбути.ролсва [116]. 20,8 г (0,13 моля)
брома прибавляют по каплям к охлажденному до 0° С растЕору 49,0 г (0,13 моля) (2-карбо-
ксиметилэтил)трибутилолова в 250 мл хлороформа. Хлороформ отгоняют и остаток
перегоняют в вакууме. Получают 46,0 г (£С%) вещества с т. кип. 125—128° С/0,002 мм.
Отрыв незамещенного алкильного радикала проходит и в том случае,
если с атомом олова связаны два или три замещенных радикала,в которых
заместитель находится в Р- или у-положении по отношению к атому олова,
например [116]:
QHoSn(CH3CH2COOCH8)3 + Br -» BrSn(CH2CH2COOCH3)3 + QH8Br.
Получение брсмистсго три-(2-карбскси^етилзтил)олсва [116]. 6,55 г (0,015 моля) три-
(2-карбоксиметилэтил)бутилолова смешиЕакт при С0 С с 2,40 г (0,015 меля) брома в
хлороформе. Продукт реакции очищают перегонкой в высоком вакууме. Выход 5,9 г (84%); т.
кип. 154—158° С/0,005 мм.
Подобно этому при обработке двумя эквивалентами брома получено дву-
бромистое £шс-(2-карбоксиметилэтил)олсЕО. Выход 46%; т. пл. 138—140° С
{из спирта).
342 Оловоорганические соединения. Лит. стр. 370—375
Аналогичное влияние на прочность связи олова с углеродом оказывают
заместители, находящиеся в винильной группе [120]:
Нч /Sn(GH3)3 Нч .Br
>С=С< +Вг2-* >С=С< +(CH3)aSnBr,
W хСООСН3 Н/ хСООСН3
нч н нч м
>С=С< + Вг2-> )с=с\ +СН3Вг.
(CH3)3Sr/ xCOOGH3 (CH3),SnBr XGOOGH3
Деалкилирование соединений.типа [(CH3)3SiCH2]4Sn и R2Sn[CH2Si(CH3)3]2
(где R — СН3 или п-С4Н9) бромом и иодом проходит в чрезвычайно жестких
условиях, что обусловлено, по-видимому, стерическими факторами [121].
Диалкилди(триметилсилилметил)олово не реагирует с иодом в кипящем
эфире или бензоле, поэтому реакцию проводят в ксилоле. Таким путем
получены [(CH3)3SiCH2]2(CH3)SnJ и [(CH3)3SiCH2]2(C4H9)SnJ. Высказано
предположение [121], что реакция иодирования этих соединений представляет
собой свободнорадикальный процесс. Нагреванием при 175° С смеси иода и те-
тра(триметилсилилметил)олова получено йодистое три(триметилсилилме-
тил)олово. Действие брома на [(CH3)3SiCH2]2(CH3)2Sn при 0° С в четырех-
хлористом углероде приводит к смеси продуктов — [(CH3)3SiCH2]2(CH3)SnBr
и [(CH3)3SiCH2](CH3)2SnBr.
Фенильные радикалы отщепляются в более мягких условиях.
Получение двухиодистого ди(триметилсилилметил)олова [121]. Раствор^ЗО г (0,067
моля ) ди (три метил сил ил метил )дифенилолова и 34,2 г (0,134 моля) иода в 150 мл бензола
кипятят в течение дня. Бензол отгоняют при обычном давлении, а образовавшийся в
результате реакции иодбензол — в вакууме. Остаток разбавляют петролейным эфиром,
обрабатывают древесным углем и кристаллизуют при температуре —75° С. Выход 30,5 г (83%); т. пл.
34,6—35,4° С (из петролейного эфира).
Действие галоидов на соединения типа (C6H5)nSnR4_n (где R —
алифатический радикал, который может содержать заместители, или винильная
группа) приводит к отщеплению ароматического радикала.
Получение хлористого метилдифенилолова [122J. К нагретому до кипения раствору 72,8 г
(0,20 моля) метилтрифенилолова в 350 мл хлороформа прибавляют по каплям 50,5 г (0,20
моля) иода в 1 л хлороформа. Для завершения реакции смесь кипятят в течение часа,
растворитель удаляют и остаток растворяют в эфире. Эфирный раствор трижды экстрагируют
20%-ным водным раствором едкого натра, затем промывают дистиллированной водой и
трижды 10%-ной соляной кислотой. Эфирный раствор высушивают сульфатом натрия и эфир
отгоняют. Остаток дважды перегоняют в вакууме. Получают 29,4 м (44,2%) хлористого
метилдифенилолова' с т. кип. 129—130° С/0,15 мм; п2р 1,6140.
Обработка дифенилдивинилолова рассчитанным количеством брома в
четыреххлористом углероде при комнатной температуре приводит к отрыву
одного фенильного радикала [93]. Очистка йодистого дифенилвинилолова
связана с известными трудностями. Поэтому продукт выделяют в виде
соответствующего фторида [94].
При иодировании трифенилцианметилолова отщепляется фенильная, а
не цианметильная группа (ср. с уравнением на стр. 340).
(QHsbSnCHgCN + J2 -» (C6H5bSn (J) (CH2CN) + C6H5J.
Получение йодистого дифенил(цианметил)олова [18]. Растворы 3,90 г (0,01 моля)
трифенил(цианметил)олова в 60 мл бензола и 2,54 г (0,01 моля) иода в 40 мл бензола
охлаждают до 0° С и смешивают при этой температуре. Смесь оставляют при 0° С на 45
час. и затем нагревают при 35° С в течение 3 час. Смесь фильтруют, выпаривают и остаток
промывают петролейным эфиром (т. кип. 40—60° С). Получают 2,3 г (53%) вещества с
т. пл. 99—100° С.
Д °алкилированиг
343
Подобно этому получены [116] йодистое (2-карбоксиметилэтил)дифе-
нилолово и йодистое (2-фенилэтил)дифенилолово.
Йодистое (2-цианэтил)дифенилолово получено иодированием (2-дициан-
этил)трифенилолова в хлороформе при комнатной температуре. Раствор
оставляют до обесцвечивания, растворитель отгоняют и продукт
кристаллизуют из петролейного эфира; т. пл. 121 —122,5 ° С.
Получение йодистого [2-(2'-оксопирролидин)этил]дифенилолова [123]. К
охлажденному льдом раствору 2,31 г (0,005 моля) [2-(2/-оксопирролидин)этил]трифенилолова в
хлороформе при перемешивании медленно прибавляют 1,27 г (0,005 моля) иода в 60 мл
хлороформа. Иод сразу обесцвечивается. Раствор выпаривают, остаток растворяют в
бензоле, высаживают петролейным эфиром (т. кип. 60—80° С) и кристаллизуют из 80%-ного
водного ацетона. Получают белые иглообразные кристаллы ст. пл. 151—152° С.
Получение бромистого (З-оксипропил)дифенилолова [116]. 0,16 г (0,002 моля) брома в
25 мл четыреххлористого углерода прибавляют по каплям к раствору 0,82 г (0,002 моля) (3-
оксипропил) трифенилолова в 25 мл того же растворителя. После удаления растворителя
остаток дважды кристаллизуют из смеси бензола с петролейным эфиром (1 :3). Выход чистого
вещества 0,31 г (36%); т. пл. 127—128,5° С.
Получение двубромистого (2-карбоксиметилэтил)фенилолова [116]. К раствору 3,07 г
(0,007 моля) (2-карбоксиметилэтил)трифенилолова в 75 мл бензола прибавляют по
каплям 2,24 г (0,014 моля) брома. После удаления растворителя остается бесцветное масло,
кристаллизующееся при добавлении небольшого количества петролейного эфира (т. кип.
60—80° С). После перекристаллизации из лигроина и циклогексана получают 2,62 г (86%)
блестящих иглообразных кристаллов с т. пл. 111 —114° С.
Аналогично получено двубромистое (З-цианпропил)фенилолово; т. пл.
101—102°С (из петролейного эфира).
Температурные условия отщепления галоидами радикалов от тетраариль-
ных соединений олова весьма близки. Например, при кипячении в течение
часа тетра-/2-толилолова с хлором, бромом или иодом в хлороформе
отщепляются все толильные группы [124]. Для получения моногалоидных
соединений требуется соблюдение особых условий, как это видно из следующего
примера.
Получение бромистого трифенилолова [125—127]. 85,4 г (0,2 моля) тетрафенилолова
растворяют в 700 мл горячего пиридина и раствор при перемешивании возможно скорее
охлаждают погружением в лед. Таким образом получают тетрафенилолово, которое большей
частью опять выпадает из раствора в виде суспензии мелких кристаллов (в форме,
чрезвычайно удобной для реакции). Затем при постоянном перемешивании и при охлаждении до
—20° С вводят в течение 2,5 час. шестью порциями 32 г брома в 150 г пиридина (раствор
брома охлаждают до —48° С вбрасыванием внутрь твердой углекислоты). В процессе
реакции суспендированное бромистое трифенилолово постепенно исчезает и получается
прозрачный раствор. Пиридин отгоняют на масляной бане при 175° С и непродолжительное время
нагревают в вакууме для удаления бромбензола. Остаток (до его застывания) растворяют в
эфире и обрабатывают в делительной воронке разбавленной соляной кислотой. Эфирный
слой промывают 30%-ной щелочью и отфильтровывают. Эфирный раствор гидроокиси
трифенилолова обрабатывают разбавленной бромистоводородной кислотой. Выход чистого
вещества 90%; т. пл. 125,5° С.
Аналогично получены [128] бромистое три-ж-толил и три-я-толилолово.
При обработке бромистого трифенилолова бромом выход двубромистого
дифенилолова составляет 93% при 25° С, 47% при —25° С [129]. Бромистое
трифенилолово деарилируется бромом и при температуре —70° С [130].
Получение йодистого трифенилолова [82, 128]. В колбу емкостью 1,5 л помещают
150 г тетрафенилолова в 975 г хлороформа. Раствор нагревают до кипения и к нему
небольшими порциями прибавляют 90 г иода. Для завершения реакции смесь нагревают в течение
часа. Хлороформ и иодбензол отгоняют (под конец в вакууме) и из остатка сырое йодистое
трифенилолово экстрагируют эфиром в аппарате Сокслета. После кристаллизации из
петролейного эфира (т. кип. 77—117° С) получают йодистое трифенилолово с т. пл. 119—121° С.
Выход 70%.
При действии иода на тетра-офеноксифенилолово образуется оиодди-
фениловый эфир и двуйодистое ди-офеноксифенилолово [571. Последнее
существует в двух формах: а-модификация с т. пл. 121 —122° С, которая
344 Оловоорганические соединения. Лит. стр. 370—3/5
при нагревании до температуры несколько выше 122° С превращается в
более устойчивую Р-форму с т. пл. 139—140° С. Получить йодистое три-офе-
ноксифенилолово обработкой тетразамещенного соединения одним
эквивалентом иода при 75—145° С не удается. В результате реакции образуется
смесь двуиодистого ди-о-фечоксифенидолова и непрореагировавшего
исходного вещества.
Получение двуиодистого ди-о-феноксифенилолова [57]. 10,0 г тетра-о-феноксифенилолова
растирают в ступке с 6,38 г иода. Смесь нагревают при 65° С в течение 2 час, а затем 1 час
при 120° С и перегоняют в вакууме. Отгоняют 4,1 г (55%) о-иоддифенилового эфира, т. кип.
136—138° С/0,6 мм\ т. пл. 53,5—56° С. Остаток кристаллизуют из нитрометана. Получают
6,6 г (74%) двуиодистого ди-(о-феноксифенил)олова с т. пл. 119—121° С. При дальнейших
кристаллизациях образуется или чистая а-форма с т. пл. 121—122° С, или чистая Р-форма
с т. пл. 139—140° С. Анализы и ИК-спектры обеих форм обнаруживают большое сходство.
Если а-форму нагреть несколько выше 122° С, она вновь затвердевает и плавится при 139—
140° С.
Иногда для деарилирования оловоорганических соединений применяют
галоид в связанном состоянии. Так, при нагревании тетрафен ил олова и,
N-бромсукцинимида в четыреххлористом углероде в течение 16 час.
получены гексафенилдистанноксан, окись дифенилолова и сукцинимид [131].
По-видимому, реакция проходит по следующей схеме:
О О
(C6H5hSn + Br—N/J -* (CeHobSn—N< + C6H5Br,
О О
(I)
о
У-,
(I) + H20 -> (C6H5)3SnOH + H — N«
</
Гексафенилдистанноксан и окись дифенилолова образуются в результате
реакции дегидратации и диспропорционирования.
При действии бромсукцинимида на дибензилдифенилолово происходит
отщепление фенильных радикалов с образованием бромбензола [107].
Треххлористое а-стильбенилолово, а-стильбенилстанноновая кислота
и окись ди-а-стильбенилолова дают с диоксандибромидом ^ис-а-бромстиль-
бен [132].
Оловоорганические соединения легко и гладко арилируют треххлори-
стый иод в солянокислом растворе. С треххлористым фенилоловом реакция
проходит по стадиям [133]:
JC13 + C6H5SnCl3 -* C6H5JC12 + SnCl4,
JC13 + 2C6H5SnCl3 -> (C6H5)2JC1 -f 2SnCl4,
C6H5JC12 + C6H5SnCl3 -* (C6H5)2JC1 + SnCl4.
Вторая стадия может служить превосходным способом синтеза солей
колония. Дифенилиодоний образуется и при действии иодвинилиодидхлорида на
треххлористое фенилолово [134].
В хлороформе реакция протекает иначе. Пятичасовое нагревание трех-
хлористого иода с тетрафенилоловом в среде хлороформа приводит к
образованию двухлористого дифенилолова (84%), бензола и иодбензола [135].
В некоторых случаях галоидирование ароматических соединений
происходит с низкими выходами соответствующих продуктов или не идет вооб-
Деалкилирование
345
ще. Так, при бромировании тетра-я-фторфенилолова при —15° С выход
(p-FC6H4)3SnBr составляет лишь 4% [129]. Бромистое три(бифенилил-2)оло-
во и двухлористое ди(бифенилил)олово существенно не изменяются при
кипячении с растворами брома в четыреххлористом углероде или иода в
бензоле [136].
Отрицательные результаты получены при длительном кипячении
раствора тетра(фенантрил-9)олова в хлороформе с бензольным раствором иода
[137] и при действии иода на тетра-отолил, тетра-^-трифторметилфенил- и
тетра-п-трифторметилфенилолово [138] в четыреххлористом углероде.
При действии галоидов на гетероциклические оловоорганические
соединения происходит раскрытие цикла [139, 1401:
Вг,
СНзч /СН2-СН2ч /СН2—СН2ч /СН3
/К >Sn<v / х
СНз7 ХСН2-СН2/ хСНа—СН3/ ХСН3
2Вг2
СНз
СНзч /СН2-СН2ч /СН2-СН2ССН2СН2Вг
-> \с< >Sn/ |
СИ/ ХСН2-СНо/ ХВг СНз
СНз Вг СНз
I I I
-» BrCH2CH2CCH2CH2SnCH2CH2CCH2CH2Br
I I I
СНз Вг СНз
Получение бромистого диэтил-5-бромамилолова[141]. К 12,3 г (0,05 моля) диэтилцикло-
пентаметиленолова в 50 мл метилацетата при встряхивании прибавляют 8,0 г (ОД моля)
брома в 25 мл того же растворителя (смесьгохлаждают льдом). Растворитель отгоняют и
остаток перегоняют в вакууме. Выход практически количественный; т. кип. 189—192° С/16 лш
(основная часть переходит при 190,5° С/16 мм).
Аналогично получаютдиметил-5-бромамилолово ст. кип. 167,5° С/15лш.
Обработка циклопентаметилендифенилолова иодом в растворе четырех -
хлористого углерода дает йодистое циклопентаметиленфенилолово с т. кип.
219° С (баня)/0,45—0,85 мм. Структура вещества подтверждена [142] его
ИК-спектром.
Фридманом [143] подробно изучено действие галоидов на 1,1-диметил-2,3,
4,5-тетрафенилстаннол. Обработка растворов этого вещества в
четыреххлористом углероде одним эквивалентом брома или иода приводит к раскрытию
цикла:
СбНб СеНб СбН.-> С6Нб
\_/ \ /
H5Q-^ ^-С6Н5 ~Л НБСв-> Vc6H5
Sn SnX X
СНз СНз СНз СНо
Обработкой того же станнола двумя молями хлора или брома получают
1,4-дихлор-^ш:-^ш> 1,2,3,4-тетрафенилбутадиен и 1,4-дибром-^с-г^с-1,2,
3,4-тетрафенилбутадиен соответственно. Их синтез можно провести поста-
дийно. С иодом реакция заканчивается на стадии образования йодистого
(4-иод-1,2Д4-тетрафенил-^ш>^ш:-1,3-бутадиенил)диметилолова. Последнее
не реагирует с избытком иода при нагревании смеси реагентов до 100° С.
При более высоких температурах вещество разлагается. Обработкой стан но-
346
Оловоорганические соединения. Лит. стр. 370—375
ла эквимолекулярным количеством иода с последующим прибавлением
•брома получен [143] 1-бром-4-иод-1,2,3,4-тетрафенил-^с,^с-бутадиен:
сбн
ъ СбНб
\ /
H5C6-f \-С6Н5
+ 0,5Вг2-^
Sn—J J
/\
СНз
СНз
С6Н5 С6Н5
\ / •]
Н5С6-(^ Y-CCH5
Sn—Br J
L /\ J
СНз СНз
0,5J2
CSH5
C6H5
H5C6-/ \-QH5
Br
+ (СНзЬЭпВгг
При действии иода на соответствующий хлорид с хорошим выходом
образуется 1-хлор-4-иод-1,2,3,4-тетрафенил-^ш:,^ш;-бутадиен [143].
О получении 1,2-дигалоидтетрафенилбензолов действием галоидов на оло
воорганические соединения см. статью Сейферта с сотр. [144].
ДЕАЛКИЛИРОВАНИЕ ГАЛОИДОВОДОРОДАМИ
В алифатическом ряду этот способ деалкилирования имеет ограниченное
препаративное значение. Более широко он применяется в ароматическом
ряду, особенно для соединений, содержащих сложные ароматические
радикалы (см. стр. 348, 349).
В зависимости от условий хлористый водород отщепляет от тетраалкиль-
ных соединений олова один или два алифатических радикала. Так,
нагреванием тетраметилолова с хлористым водородом в хлороформе получено
[91] хлористое триметилолово с выходом 70%; избыток хлористого водорода
не влияет на реакцию. В более жестких условиях возможно провести
отщепление от атома олова двух алкильных радикалов. Двухлористое метил-
этилолово [92, 145] и двухлористое этилпропилолово [92] получены при
пропускании хлористого водорода в нагретое до 140—150° С диметилдиэтил-
или диэтилдипропилолово соответственно.
Из продуктов реакции хлористого водорода с тетраэтилоловом
индивидуальные соединения не были выделены [146].
Хлористое тридодецил-, тритетрадецил-, тригексадецил- и триоктадецил-
олово получены [147] взаимодействием хлористого водорода с R4Sn в эфире.
Получение хлористого триоктадецилолова [147]. Эфирный раствор 2,2 г (0,00194 моля)
тетраоктадецилолова насыщают сухим хлористым водородом и раствор оставляют на ночь.
Эфир отгоняют и остаток кристаллизуют из этилацетата, а затем из эфира. Получают 1,1 г
(62%) вещества с т. пл. 61—62° С.
Порядок отщепления от атома олова различных радикалов примерно тот
же, что и при деалкилировании галоидами, а именно [91, 94, 97, 148—152]:
ot-тиенил >я-анизил>а-нафтил> д-толил > перфторвинил ^ фенил>пен-
тафторфенил > метил > этил > я-пропил > я-гексил :> перфторэтил.
Например, хлористое диметилэтилолово получают [91] пропусканием
хлористого водорода в кипящий раствор триметилэтилолова в хлороформе;
выход 80%.
Винильные группы отщепляются легче алкильных, но с большим трудом,
чем фенильные радикалы. Например, хлористое ди-я-бутилвинилолово и
Деалкилирование
347
хлористое диметилвинилолово, а также соответствующие бромиды
получены [94] действием галоидоводородов на диалкилдивинильные соединения
олова.
При нагревании триметилтрифторметилолова с сухим хлористым
водородом выделяется 80% метана [153].
Непредельные радикалы отщепляются гораздо легче алкильных.
При взаимодействии тетрааллилолова со спиртовым раствором
хлористого водорода при комнатной температуре происходит энергичная реакция с
образованием пропилена и хлорного олова [154]:
5 (C3H5)4Sn + 4HC1 -> 4С3Нб + SnCl4,
Еще легче происходит отщепление перфторвинильных радикалов. Так,
при действии бромистого водорода на дибутилдиперфторвинилолово при
0° С получено [109] двубромистое ди-я-бутилолово с выходом 85%.
С целью сравнения реакционной способности винильной и перфторви-
нильной групп изучено [149] действие хлористого водорода на
соответствующие соединения типа R4Sn, а также надивинилдиперфторвинилолово.
Применяли следующую методику.
Навеску вещества помещают в стеклянный шарик емкостью 170 мл,
соединенный с высоковакуумной системой. В шарик конденсируют
хлористый водород в количестве, примерно эквивалентном навеске оловоорга-
нического соединения. Через определенный промежуток времени летучие
продукты удаляют, непрореагировавший хлористый водород отделяют
пропусканием газа над сухим едким натром и после высушивания остаток
анализируют методами газовой хроматографии и ИК-спектроскопии. Оловоорга-
ническое соединение вновь обрабатывают хлористым водородом и
продукты анализируют тем же способом.
Найдено [149], что при 100° С от тетраперфторвинилолова отщепляется
в течение 60 час. 96% органических радикалов, а от тетравинилолова в тех
же условиях — 82%.
При действии галоидоводородов на арилалкильные соединения олова
преимущественно отщепляется арильный радикал.
Получение двухлористого бгас-(дифенилметил)олова [155]. К 5 N раствору хлористого
водорода в этиловом спирте прибавляют частями при непрерывном встряхивании 15,2 г
(0,025 моля) дифенил-бш>(дифенилметил)олова. Смесь нагревают при 50° С в течение 3 час.
На следующий день осадок отфильтровывают, промывают этиловым спиртом, петролейным
эфиром (т. кип. 30—50° С) и перекристаллизовывают из смеси циклогексана с бензолом
(3:2). Получают 10,5 г(80%) двухлористого бш>(дифенилметил)олова ст. разл. 115—-117° С.
Исследована [72, 73, 156] кинетика реакции триалкиларильных
соединений олова с хлористым водородом в метиловом спирте. При этом
наблюдаются те же закономерности, что и придеалкилировании галоидами (см. стр. 337).
Газообразный бромистый водород при реакции с циклопентаметиленди-
фенилоловом образует двубромистое циклопентаметиленолово с т. кип.
166° С (температура бани) 0,40—0,45 мм [142]. Двубромистое ди(триме-
тилсилилметил)олово получено [121] реакцией бромистого водорода с ди(три-
метилсилилметил)дифенилоловом в хлористом метилене при температуре
—78° С. При действии бромистого водорода на диметил- и ди-я-бутилди(три-
метилсилилметил)олово отщепляется по одному радикалу разного вида
[121].
Спиртовый раствор хлористого водорода превращает дибензилдифенил-
олово в двухлористое дибензилолово с т. пл. 163° С [107].
Приведенный выше порядок отрыва различных радикалов от атома олова
относится к деалкилированию соединений типа R4Sn. Однако при отрыве
второй и последующих групп сказывается влияние атома галоида, уже свя-
348 Оловоорганшеские соединения. Лит. стр. 370—375
занного с оловом. Отмечено [149], что при взаимодействии дифенилдипер-
фторвинилолова с эквивалентным количеством хлористого водорода
образуются примерно равные количества бензола и трифторэтилена. При
добавлении второго эквивалента хлористого водорода бензола образуется меньше.
Дальнейшее прибавление хлористого водорода приводит к
преимущественному отрыву перфторвинильных групп.
В зависимости от условий проведения реакции и характера
органического радикала, связанного с атомом олова, при действии галоидоводородов на
тетраарильные соединения олова образуются галогениды типа R3SnX,
R2SnX2 или SnX4.
Получение хлористого трифенилолова [157]. В суспензию 20 г тонко измельченного тет-
рафенилолова в 100 г хлороформа пропускают энергичный ток хлористого водорода. Смесь
слегка разогревается и через 30—40 мин. образуется почти прозрачный раствор. Небольшое
количество непрореагировавшего тетрафенилолова растворяют при встряхивании и
растворитель отгоняют. Остаток почти полностью закристаллизовывается при охлаждении до
0° С. Его отфильтровывают, дважды промывают холодным петролейным эфиром и
кристаллизуют из лигроина. Выход чистого вещества 13,5 г (75%); т. пл. 106° С.
В тех же условиях от тетра(бифенилил-2)-олова отщепляются два ариль~
ных радикала [1361:
(Ci2H9)4Sn + 2HC1 -» (Ci2Hc,)2SnCl2 + 2С12Ню.
Получение двухлористогоб#с-(бифенилил-2)-олова [136]. В суспензию 11,5 г (0,0157
моля) тонко измельченного тетра-(бифенилил)-2-олова в 200 мл сухого хлороформа
пропускают в течение часа энергичный ток хлористого водорода. В процессе реакции
суспендированное тетра(бифенилил-2)-олово постепенно растворяется и образуется почти прозрачный
раствор, из которого избыток хлористого водорода удаляют пропусканием азота.
Небольшое количество непрореагировавшего вещества отфильтровывают и растворитель отгоняют
(под конец в вакууме). В остатке бесцветное масло, которое быстро
закристаллизовывается. Вещество очищают переосаждением из небольшого количества горячего
бензола 6—8-кратным избытком горячего этилового спирта. Выход 83,4%. Аналитически чистый
препарат получают переосаждением продукта с последующим длительным высушиванием
в вакууме при 100° С.
Аналогично получено! 158] двубромистоебш>(бифенилил-2)-олово ст. шк
172,5—173° С (из я-бутилового спирта).
Кипячением в течение 4 час. тетра(фенантрил-9)-олова в хлороформе
с сухим хлористым водородом приводит к полному отрыву всех арильных
групп (из продуктов реакции выделен фенантрен с выходом 72,6%) [137]:
(C14H9)4Sn + 4HC1 -> SnCl4 + 2С14Ню.
Однако при соответствующей дозировке хлористого водорода основным
продуктом является двухлористое ди(фенантрил-9)-олово (в смеси сфенан-
треном и хлористым три(фенантрил-9)-оловом:
(CnHobSn + 2HC1 -» 2(Ci4He)2SnCIa + 2С14Ню.
Получение двухлористого ди(фенантрил-9)-олсва [137]. В суспензию 1 г (0,0012 моля)
тетра(фенантрил-9)-олова в 50 мл хлороформа при энергичном перемешивании пропускают
сухой| хлористый водород в течение 45 мин. Уже через 20 мин. образуется прозрачный
раствор. На следующий день растЕоритель отгоняют (остаток в вакууме). Фенантрен
отделяют нагреванием вешестЕа с теплым эфиром. Остаток растворяют в небольшом
количестве уксусноэтилового эфира, фильтрат упаривают дссуха и вещество высушивают при
100° С. Выход чистого продукта 381 мг.
При обычной температуре 9,9-бмс-(бифенилил-2)-дибензостаннол (I)
реагирует с хлористым водородом в эфире с образованием смеси хлор исто-
Дтлкилирование
349
го три(бифенилил-2)-олова и двухлористого £шс-(бифенилил-2)-олова [1581:
(D
Ох/0 м сх 9 ™ oi-o
(ГО (^л) 0 0
(II) . (Ш) (IV)
(С12Н9 = бифенил-2)
Нагреванием смеси реагентов в этиловом спирте получают в качестве
основного продукта двухлористое бис-(бифенил-2)-олово.
Действие на 9,9-*>#с-(бифенилил-2)-дибензостаннол хлористого водорода [158]. а)
Суспензию 1,000 г (0,00173 моля) 9,9-б«с-(бифенилил-2)-дибензостаннола в 20 мл сухого эфи-
та встряхивают в течение 2 час. с 2 мл эфира, насыщенного хлористым водородом (380 мг
НС1; 0,0104 моля). Смесь разбавляют 10 мл бензола и избыток хлористого водорода удаляют
встряхиванием с водой. Органический слой отделяют, высушивают хлористым кальцием и
растворитель отгоняют в вакууме. Остаток представляет собой масло, которое через 2 дня
полностью закристаллизовывается. Вакуум-возгонкой отделяют 94 мг (0,00061 моля) би-
фенила с т. пл. 68—69,5° С (смешанная проба без депрессии). Остаток после возгонки
кристаллизуют из сухого этилового спирта, а затем из смеси бензола с этиловым спиртом.
Получают 302 мг (28,4%) хлористого три(бифенилил-2)-олова с т. пл. 155—156,5° С и 217 мг
{25,3%) двухлористого бш>(бифенилил-2)-олова с т. пл. 164—168° С. Из этих двух веществ
легче растворяется второе.
б) В суспензию 1,000 г (0,00173 моля) 9,9-б//с-(бифенилил- 2)-дибензостаннола в 75 мл
сухого этилового спирта при частом встряхивании в течение 2 час. пропускают ток сухого
хлористого водорода (синтез проводят в отсутствие влаги воздуха). Для завершения
реакции смесь нагревают, отфильтровывают 20 мг непрореагировавшего исходного вещества и
прозрачный раствор оставляют на ночь в холодильнике. Выпадает 502 мг двухлористого
9,9-б«с-(бифенилил-2) -олова в виде блестящих квадратиков с т. пл. 169,5—170,5° С.
Упариванием фильтрата примерно наполовину получают еще 200 мг вещества с т. пл. 169—170° С.
Общий выход ~83,4%.
Спиро-9,9'-бш?-(дибензостаннол) (II) в мягких условиях реагирует с
одним эквивалентом хлористого водорода с образованием, вероятно, 9-хлор-
9-(бифенилил-2)-дибензостаннола (II) (выход 84%). В более жестких
условиях (горячий спиртовый раствор хлористого водорода) наряду с (III)
образуется только двухлористое бш;-(бифенилил-2)-олово и небольшое
количество бифенила.
Действие хлористого водорода на спиро-9,9'-*>#с-(дибензостаннол) [158]. а) 1,000 г
(0,00236 моля) вещества растворяют в 30 мл сухого бензола и прибавляют 0,50 мл сухого
эфира, насыщенного хлористым водородом (94 мг НС1; 0,0026 моля). После двухдневного
стояния при комнатной температуре растворители отгоняют в вакууме. Остаток желтовато-
белого цвета промывают метиловым спиртом. Получают 910 мг (83,8%) 9-хлор-9-(бифени-
лил-2)-дибензостаннола (III) в виде мелкокристаллического порошка с т. пл. 226—230° С.
Для очистки его растворяют в уксусноэтиловом эфире и высаживают этиловым спиртом;
температура плавления повышается до 230—232° С.
б) В суспензию 0,95 г (0,00225 моля) спиро-9,9'-5^е-(дибензостаннола) в 50 мл сухого
этилового спирта пропускают ток сухого хлористого водорода в течение 4 час. В отличие от
предыдущего случая твердая фаза не исчезает, но внешний вид осадка изменяется (тонко
измельченный осадок становится более крупнодисперсным, увеличивается и его
количество). Смесь нагревают до кипения и фильтруют горячей. На фильтре остается 0,85 г порошка
грязно-белого цвета с т. пл. 192—215° С. Его обрабатывают эфиром. Из нерастворившегося
в эфире остатка получают 180 мг (17,4 %) III с т. пл. 230—232° С.
350 Оловоорганические соединения. Лит. стр. 370—375
Существенно отличаются по отношению к хлористому Еодороду также
соединения инденового и флуоренового ряда. (Инденил-1)-трифенилолово
реагирует с отщеплением индена, в то время как от (флуоренил-1)-трифе-
нилолова отщепляется фенильный радикал [48]:
(QH.-OsSnCl 4 ЯН ^ИНДеПИЛ: RSn(CeH5)3 + HC1
I R—флуоренил
R(CeH5)2SnCl + СсНб
Получение хлористого (флусренил-9)-дифенилолова [48]. В суспензию (флуоренил-9)-
трифенилолова в 50 мл эфира пропускают в течение 3—4 мин. ток хлористого водорода,
причем образуется прозрачный растьор. Примерно через 30 мин. начинается кристаллизация
продукта реакции; т. пл. 140—141° С (из петролейного эфира).
Аналогично из ди(флуоренил-9)дг4енило^ова получено хлористое ди(флу-
оренил-9)-фенилолово с т. пл. 143° С (из бензола) [48].
Спиртовый раствор хлористого Еодорода уже при сбычкой температуре
превращает аллилтрифенилолово в хлорное олоео [159]. Издиалилдкфенил-
олова в тех же условиях образуются пропилен, бензол, двухлористое ди-
фенилолово и хлорное олово [154].
ДЕАЛКИЛИРОВАНИЕ КИСЛОТАМИ, СПИРТАМИ И МЕРКАПТАНАМИ
Взаимодействие оловоорганических соединений с минеральными
кислотами описано очень кратко. Кипячением тетра-п-толилолова с азотной
кислотой получен n-нитротолуол с выходом 55%. В аналогичных условиях, а
также при обычной температуре серная кислота отщепляет практически все
тол ильные группы [124).
(2-Диметиламинофлуоренил-9)-трифенилолово при примерно 12-часовом
нагревании с 0,4 N H2S04 в спиртовом растворе остается без изменения.
При обработке его эфирного раствора хлористым водородом образуется
хлористое трифенилолово и 2-диметиламинофлуорен [153].
Изучено [151] действие соляной кислоты на дициклогексилдифенил-,
дифенилди-п-анизил-, ди-а-нафтилди-я-анизил- и
дифенилди-а-нафтилолово. По легкости отщепления радикалы располагаются следующим образом:
n-анизил, а-нафтил, фенил, циклогексил [151].
Двухлористое бензилфенилолово получено [96] нагреванием трибензил-
фенилолова с концентрированной соляной кислотой в течение нескольких
минут; более длительное нагревание приводит к полному отрыву всех ариль-
ных радикалов.
О кинетике реакции фенилтрициклогексилолова с соляной или хлорной
кислотами см. статьи Иборна с сотр. [160—162].
Чрезвычайно легко отщепляются кислотами аллильные группы [163] и
радикалы с ацетиленовой связью, находящейся в а-положении по
отношению к атому олова [164, 165]. Например, при действии на 1,1-диэтокси-З-
(трифенилстаннил)-2-пропин даже 1%-ной соляной кислоты образуется
хлористое трифенилолово [164].
Нагревание гексаизопропил- или гекса-я-бутилдистанноксапа с метан-
сульфокислотой приводит к б^с-(метансульфонатам) диалкилолова [166].
Карбоновые кислоты отщепляют от тетраалкильных соединений олова,
как правило, только один радикал. Относительная реакционная
способность оловоорганических соединений уменьшается в ряду СН3, С2Н5, С3Н7,
С4Н9. Существенное влияние имеет константа ионизации кислоты [167] и
природа растворителя [168]. При температуре кипения ксилола в присутст-
Деалкилирование
351
вии силикагеля кислоты с константой ионизации, меньшей чем у адипиновой
кислоты, не реагируют с тетраалкильными соединениями олова [167].
Изучена [169] реакция тетраэтил-, тетра-я-пропил- и тетраизопропил-
олова с кислотами с константами ионизации 10"1—10~10. При кипячении
с сильными органическими кислотами выход ацилатов триалкилолова
составляет 55—60%.
Получение трифторацетата тризтилолова [169]. К 4,7 г (0,02 моля) тетраэтилолова
в течение 15 мин. прибавляют тремя равными порциями 0,02 моля трифторуксусной кислоты.
Смесь кипятят 15 мин. и непрореагировавшие вещества отгоняют в вакууме. Получают 3,7 г
трифторацетата тризтилолова с т. кип. 218° С/760 мм; т. пл. 122—123° С (из петролейного
эфира).
Аналогично получены моно- и дихлорацетат триэтил- и три-я-пропилоло-
ва и др. Получить таким путем производные слабых органических кислот
не удается вследствие чрезвычайно низкого выхода [169].
При действии трифторуксусной или перфторпропионовой кислот на
диметилди-я-амилолово или диметилди-я-бутилолово образуется трудно
разделимая смесь продуктов [111].
Отщепление второго радикала органическими кислотами описано лишь
для некоторых частных случаев. При нагревании тетраизопропилолова с
моно- или дихлоруксусной кислотой образуются соответствующие диацилаты
диизопропилолова [170].
Получение димонохлорацетата диизопропилолова [170]. К 5,8 г (0,02 меля)
тетраизопропилолова прибавляют равными порциями 0,02 моля монохлоруксусной кислоты в течение
15 мин. при температуре кипения смеси. Смесь кипятят 30 мин. После охлаждения получают
кристаллы ст. пл. 54—55° С. Выход 47%.
Циклические продукты типа
(C2H5)2SnOOC(CH2)nCOOSn(C2H5)2COO(CH2)AZCOO
получены нагреванием до 165—170° С тетраэтилолова с двухосновными
кислотами в течение 6—10 час. [171, 172].
Получение адипината диэтилолова [171]. 4,2 г тетраэтилолова и 2,6 г адипиновой
кислоты нагревают при температуре бани 165—170° С в течение 8,5 часа. За это время
выделяется два эквивалента этана. После охлаждения кристаллическую массу промывают 10 мл
сухого спирта. Выход 5,2 г (90%); т. пл. 143—144° С. Вешество хорошо растворяется в
хлороформе на холоду, при нагревании — в бензоле, толуоле, ксилоле, дихлорэтане и че-
тыреххлористом углероде. В метиловом и этиловом спиртах, ацетоне и петролейном
эфире не растворяется даже при нагревании.
Аналогично получены азелаинат и себацинат диэтилолова. Во всех
случаях применения избытка двухосновной кислоты время реакции
значительно сокращается. Однако образовавшийся в этом случае продукт трудно
очистить от избытка кислоты, и выходы более низкие 1171].
Винильный [23, 94] и аллильный [93, 154] радикалы отщепляются
гораздо легче алкильных. Реакцию тетравинилолова с органическими
кислотами предложено исгользовать ^ля синтеза тетрааиилатов олова [172а].
Получение ди(монобромацетата)диметилолова [23]. В кслбу Зрленмсйера, снабженную
воздушным холодильником, помещают 1 г (0,00494 моля) диметилдивинилолова и 1,37 г
(0,0088 моля) монобромуксусной кислоты. Смесь нагревают на еодяной бане в течение 5 мин.
и вещество перекристаллизоЕЫЕггст из смеси дихлорэтана с петрслейным эфиром. Получают
2,05 г (97,9%) вещества с т. пл. 159—160° С.
Следует отметить, что при действии кислот на диалкилдивинильные
соединения олова отщепляются сразу две винильные группы. Получить чистые
ацилаты диметилвинилолова удается только в случае монохлоруксусной
352 Оловэорганические соединения. Лит. стр. 370—375
и сс-бромпропионовой кислот. В остальных случаях вещество содержит
примесь диацилата диметилолова [94]. Еще легче происходит отрыв перфтор-
винильных радикалов [109, 149, 173]. Напротив, соединения R2Sn(C2F5)2
устойчивы к действию кислот. Например, при нагревании до 80° С
ди-я-бутилди(пентафторэтил)олова с трифторуксусной кислотой в течение
15 час. выделяется только 6,7% бутана. В тех же условиях от тетра-я-
бутилолова отщепляется 46,5% бутильных групп. Таким образом, по
отношению к кислотам диалкил-бш:-(пентафторэтильные) соединения олова
более похожи на дигалогениды диалкилолова, чем на тетраалкильные
соединения [150].
Вероятно, устойчивость перфторалкильных радикалов к действию
кислот объясняется индуктивным влиянием атомов фтора, приводящем к
повышению электронной плотности на нефторированном атоме углерода по
сравнению с фторированным [153]. Напротив, в случае перфторвинильных
радикалов имеет место следующий тип сопряжения [174]:
cf =cf2
СН2=СН— Sn —*- CF=CF2
СН =СН2
Фенильные радикалы отщепляются кислотами гораздо легче алкильных.
Например, при нагревании до 60° С пентафторэтилтрифенилолова с
трифторуксусной кислотой в течение 30 мин. выделяется 99,7% бензола [153].
Тетрафенилолово до 100° С устойчиво к действию органических кислот.
Однако при 100—120° С происходит отрыв всех фенильных радикалов с
образованием основных солей олова [175, 176]. Хлорангидриды реагируют с
тетрафенилоловом более энергично. Так, при 50°С из хлористого ацетила
и тетрафенилолова образуется двухлористое дифенилолово. При 100—
130° С происходит полное разложение тетрафенилолова с образованием аце-
тофенона и хлорного олова [175].
При действии сильных органических кислот на тетра-/г-фторфенилолово
получены соответствующие ацилаты триарилолова.
Получение трифторацетата три-л-фторфенилолова [129]. Смесь 1,14 г (0,01 моля)
трифторуксусной кислоты и 5 г (0,01 моля) тетра-я-фторфенилолова нагревают на водяной бане
в течение 30 мин. После охлаждения экстрагируют 100 мл эфира. Из эфирного раствора
получают 2,0 г кристаллов с т. пл. 312—322° С.
С гептафтормасляной кислотой реакция проходит в более мягких
условиях.
Получение гептафторбутирата три-л-фторфенилолова [129]. Раствор 5 г (0,01 моля)
тетра-я-фторфенилолова и 2,14 г (0,01 моля) гептафтормасляной кислоты в 15 мл бензола
нагревают в течение 15 мин., после чего бензол отгоняют. Для удаления непрореагировав-
ших веществ остаток экстрагируют 50 мл бензола. Получают 2,0 г (33%) вещества с т. пл.
277—278° С.
Кратковременное нагревание гетероциклического оловоорганического
соединения — 1,1-диметил-2,3,4,5-тетрафенилстаннола — с разбавленным
раствором ледяной уксусной кислоты в спирте приводит к отщеплению
ненасыщенного углеродного цикла, связанного с атомом олова. В результате
реакции количественно образуется 1,2,3,4-тетрафенилбутадиен [ 143].
Описана реакция алкильных [167, 169] и арильных[129, 177, 178]
соединений олова сфенолами и меркаптанами. Меркаптидытриэтилолова получены
[169] нагреванием эквимолекулярных количеств тетраэтилолова с
меркаптанами в течение 30 мин. Арилмеркаптаны являются более сильными деалкили-
рующими агентами по сравнению с алкилмеркаптанами, но во всех случаях
выход продукта реакции не превышает 40%.
Деалкилирование
353
Получение Р-нафтилмеркаптида триэтилолова [169]. К 4,7 г (0,02 моля) тетраэтилолова
прибавляют 0,02 моля Р-нафтилмеркаптана при умеренном кипении смеси. Смесь кипятят еще
30 мин. и непрореагировавшие вещества отгоняют. Получают 2,93 г Р-нафтилмеркапти-
да триэтилолова с т. кип. 189—190° С/1 мм: d204l,3231; /1^1,5308.
Аналогично получены фенилмеркаптид триэтилолова (выход 38%),
бензилмеркаптид триэтилолова (39%) и др. Выход алкилмеркаптидов
триэтилолова не превышает 20%.
При кипячении спиртового раствора ди-я-бутилдиперфторвинилолова
в течение 20 час. образуется [109] диэтоксидибутилолово; выход
практически количественный. В тех же условиях триэтилперфторвинил- и перфтор-
винилтрифенилолово со спиртом не реагируют. Однако при реакции три-я-
бутилперфторвинилолова со спиртовым раствором этилата натрия с
хорошим выходом получено этокситри-я-бутилолово [109].
При взаимодействии тетраал лило лова с этиловым спиртом при 130СС
происходит полный распад оловоорганического соединения с образованием
пропилена и гидрата окиси олова [154]:
(C3H5)4Sn + 4С2Н5ОН -» 4С3Н6 + Sn(OC2H5)4,
Sn(OC2H5)4 + 4H20 -> Sn(OH)4 + 4C2H5OH.
Фенолы [178] итиофенолы [177] полностью деарилируют тетрафенилоло-
во при нагревании смеси реагентов до 150°С в течение 6 час.
Кетоны и сложные эфиры, содержащие атом олова в а-положении по
отношению к карбонильной группе, обладают лабильной связью олово —
углерод, которая легко расщепляется при действии соединений,
содержащих подвижный атом водорода [179—183]:
(C3H7)3SnCH2COCH3 + СНзСООН -* (C3H7)3SnOOCCH3 + СНзСОСНз,
(C2H5)3SnCH2COCH3 + СН3ОН -> (C2H5)3SnOCH3 + СН3СОСН3,
(C2H5)3SnCH2COCH3 + С6Н5С = СН -» (C2H5)3SnC = ССвН5 + СН3СОСН3,
(C3H7)3SnCH2COOC2H3 4- СН3СОСН3СООС2Н5 -» СНзС=СНСООС2Н5 + CHsCOOC2H5.
03п(С3Н7)з
Например, кипячение метилового эфира трипропилстаннилуксусной
кислоты с метиловым спиртом дает 80% метокситрипропилолова
с т. кип. 84—85°С/3 мм, по 1,4750 [180]. Обработкой метилового эфира ди-
этилстаннил-бис-уксусной кислоты метиловым спиртом получают диметок-
сидиэтилолово и метилацетат. Взаимодействие этилового эфира диэтилстан-
нил-бш>уксусной кислоты с муравьиной кислотой приводит к диформиату
диэтилолова и этил ацетату [182].
Реакция этилового эфира трипропилстаннилуксусной кислоты с ацетоуксусным эфиром
[183]. К 17,9? .тилового эфира трипропилстаннилуксусной кислоты приливают 7,2 г аце-
тоуксусного эфира, нагревают 2 часа при 140—160° С с отгонкой образующегося этилацетата
(4 г, 85%). При перегонке остатка в вакууме получают 10,1 г (50%) трипропилоловянного
енолята ацетоуксусного эфира с т. кип. 114—116° С/2 мм; ^1,1766; я^1,4849.
ДЕАЛКИЛИРОВАНИЕ СОЛЯМИ
Описано действие на оловоорганические соединения солей серебра,
золота, ртути, таллия, железа и некоторых других элементов (о действии солей
олова см. ранее стр. 329). Наиболее подробно изучено взаимодействие ал-
кильных и арильных соединений олова с сулемой, которая отщепляет
органические радикалы от олова более интенсивно, чем иод или хлористый водо-
23 Заказ № 207
354 Оловоорганическые соединения. Лит. стр. 370—375
род. Так, при прибавлении тетраметилолова к спиртовому раствору
хлорной ртути при комнатной температуре происходит экзотермическая
реакция. После кипячения смеси в течение 3 час. получают [91] хлористое три-
метилолово и двухлористое диметилолово с выходом 70 и 12,5%
соответственно. Избыток сулемы отщепляет два метальных радикала уже при
комнатной температуре; выход двухлористого диметилолова составляет 86%.
С увеличением алкильного радикала, связанного с атомом олова, скорость
реакции заметно уменьшается. Тетраэтилолово не реагирует с сулемой при
комнатной температуре. После кипячения смеси в течение 3 час. получают
хлористое триэтилолово (35%) [91]. Если же продолжительность
нагревания повысить до 16 час, то выход хлористого триэтилолова составляет 78%
[111]. С бромной или йодной ртутью реакция проходит медленно (19 и 23
часа соответственно). Выход бромистого триэтилолова 73%, йодистого 68%
[111]. При нагревании тетраоктилолова с хлорной ртутью образуются
хлористая октилртуть (44%) и оловоорганическое соединение состава (С8Н17)Х
SnCly [91].
Как и другие деалкилирующие агенты, сулема отщепляет от
несимметричных оловоорганических соединений наиболее легкий алкильный
радикал. Например, нагреванием спиртового раствора сулемы с (СН3)з5пС2Н5
или (CH3)2Sn(C2H5)2 в течение 2 час. получены соответственно соединения
(CH3)2(C2H5)SnCl (76%) и (C2H5)2SnCl2 (85%) [91].
Получение хлористого трициклопропилолова [47]. Синтез проводят в атмосфере азота.
В трехгорлую колбу, соединенную с аппаратом Сокслета, помещают 14,14 г (0,05 моля)
тетрациклопропилолова в 200 мл эфира, а в экстрактор — 13,37 г (0,05 моля) хлорной
ртути. Реакцию проводят в течение 3 дней. Выделившиеся блестящие кристаллы хлористой цик-
лопропилртути отфильтровывают. Фракционированной перегонкой фильтрата с
последующим фильтрованием и повторной перегонкой получают 9,2 г (66%) хлористого
трициклопропилолова с т. кип. 84—85° С/0,4 мм; я^1,5415.
Аналогично из тетрациклопропилолова и бромной ртути получено
бромистое трициклопропилолово, а из циклопропилтрифенилолова — трех-
хлористое и трехбромистое циклопропилолово [47]. В тех же условиях при
нагревании бутилтривинилолова с рассчитанным количеством галоидных
солей ртути в течение 20 час. отщепляется одна винильная группа [184].
Подобно этому из трибутилперфторвинилолова и HgCl2 получена
хлористая перфторвинилртуть и хлористое трибутилолово [109].
Хлориды фенилолова реагируют с кипящим спиртовым раствором
сулемы практически моментально и количественно. Вследствие плохой
растворимости тетрафенилолово реагирует несколько медленнее j 185, 186]. В
зависимости от относительных количеств реагентов образуются моно-, ди-
или треххлористое фенилолово. Основная реакция всегда сопровождается
побочными, поэтому продукт содержит также примесь других галогенидов
олова. Следует отметить, что единственным ртутноорганическим
соединением является хлористая арилртуть; диарилртуть не образуется.
Следовательно, хлористая фенилртуть по отношению к оловоорганическим
соединениям в данных условиях уже не является деарилирующим агентом.
Аналогично проходят реакции между триэтиларилоловом и диацетатом
ртути в тетрагидрофуране [186а]:
(C2H0)GSnC6H4X + Hg (ООССНзЬ-^ rC2H,)3SnOOCCH3+ XC6H4HgOOCCH3
(Х = р-СНп, p-Cl, р-СНзО, Н, ш-СНз, /я-СН30, т-С1)
Исследование влияния заместителей на скорость этой реакции показало,
что она является реакцией электрофильного замещения. На стадии,
определяющей ее скорость, промежуточный продукт ионного типа не образуется
1186а].
Дсалкилирование
355
Картина меняется, если реакцию вести в щелочной среде, где
действующим неорганическим агентом будет уже окись ртути. В этом случае
единственным ртутноорганическим продуктом является всегда дифенилртуть.
В щелочной среде уравнения реакций приобретают следующий вид:
(C6H5)2SnO + HgO + 2NaOH -* (C6H5,4Hg + Na2Sn03+ Н20,
IQHoSnOOH + HgO + 4NeOH -» (C6H6)£Hg + 2Na2Sn08 + 2H3O.J
Таким образом, гидроокись фенилртути по отношению к олоЕСОрганиче-
ским соединениям в щелочкой среде должна яеляться деарилирукщим
агентом. Действительно, в щелочной среде хлористая фенилртуть
реагирует с фенильными соединениями слова с образованием дкфенилртути [185,
1861:
2C6H5HgCl + (CeH5)2Sr.Clr + 6NaOH -» 2(CcH5)2Hg + Na2Sn08 + 4NaCl + 3H20,
2C6H,HgCl + 2C6H5SnOOH + 6NaOH -> 2(CeH6)2Hg + 2Na2Sn03 + 2NaCl + 3H20.
Подобно этому двубромистое диизопропенилолово образует с бромной
ртутью в среде сухого спирта бромистую изопропенилртуть, а в водном-
щелочном растворе — диизопропенилртуть [187]. Реакция галоидных солей
ртути с оловоорганическими соединениями широко применяется для
идентификации последних [55, 136, 158, 188].
При переходе органического радикала от атома олова к ртути его
геометрическая конфигурация не изменяется. Реакции ртутных солей с изопро-
пенильными соединениями олова использованы Несмеяновым при
обосновании сформулированного им правила о сохранении стереохимической
конфигурации органического радикала при гетеролитическом или
гемолитическом переходе его от одного металла к другому [189—192].
В литературе описаны также реакции оловсорганических соединении с
солями и других металлов. Бромистое серебро, галоидные соли
двухвалентного олова, меди, тантала и палладия, а также трифторацетат
двухвалентного олова отщепляют от тетраэтилолова один органический радикал 1111J.
При нагревании тетраэтилолова с хлористым серебром или трифторацетатом
серебра получают двухлористое диэтилолово и бмс-(трифторацетат)диэтил-
олова соответственно. С фтористым олоеом, бензоатом олова (II), йодистой
ртутью, муравьинокислым таллием, йодистым серебром и окисью серебра
тетраэтилолово не реагирует [III]. В зависимости от молярного
соотношения реагентов при действии на тетраметил- и тетраэтилолово азотистокис-
лой ртути образуется или нитрат триалкилолова, илидинитратдиалкилолова
1193].
Нагревание тетрафенилолова с хлоркой медью, хлористыми солями
кобальта, никеля или железа в этилцеллозольве ведет к полному деарилиро-
ванию' олоЕоорганического соединения и образованию хлорного олова
[1941'
(C6H,)4Sn + 8CuCl2 -» SnCl4 + 4CeH5Cl + fCuCl.
С хлорным железом реакция проходит следующим образом:
(C6H5)4Sn + 4FeCl3 -» SnCl4 + 4C6H5FeCl2,
QHeFeClo-^QHs'+FeCla.
В хлороформе хлористое железо отщепляет от тетрафенилолова только один
органический радикал [195]. Тетргфенилолово инертно по отношению к
хлористому марганцу [107].
В некоторых случаях олоЕоорганические соединения применяют в
качестве арилирующего (реже алкилирующего) средства.
23*
356 Оловооршнические соединения. Лит. стр. 370—375
AgNo3 не реагирует с тетраалкильными соединениями олова [196],
но легко отщепляет ацетилен от соединений типа R3Sn — С не С— SnR3
[197] и фенильные радикалы [198—200]. Изучено [199,200] действие
азотнокислого серебра в среде сухого этилового спирта на тетраэтил-,
триэтилфенил-, диэтилдифенил- и тетрафенилолово. При 25° С тетраэтил-
и тетрафенилолово совершенно устойчивы, в то время как три остальных
соединения расщепляются, но различным образом. Триэтилфенил- и ди-
этилдифенилолово реагируют с азотнокислым серебром следующим
образом:
2(C2H5)3SnQIl5 + 3AgN03^ 2(C2H5)3SnN03 + (CeH.Agb-AgNOa,
(QsHibSnfCeHJa + 3AgN03-* (C2H5)2Sn(N03)2 + (C6II5Ag)3.AgN08.
Двойное соединение фенилсеребра с азотнокислым серебром при
нагревании или под действием света постепенно разлагается с образованием ди-
фенила, металлического олова и азотнокислого серебра.
В тех же условиях с этилтрифенилоловом сразу образуется тонко
измельченное серебро и дифенил:
2C2H5Sn(C6H5)3 -f- 2AgN03 -» (С2Н.) (C6H5)aSn(N08) + 2Ag + CGH,-CGTT5.
Различный ход этих реакций объяснен [199, 200] положительным
индукционным эффектом алкильных групп.
Трехфтористый бор реагирует с тетраметил- и тетрафенилоловом по
схеме [201, 202^:
R4Sn + 2BF3-> R3Sn3F4+ R3F2.
Описано отщепление галогенидами бора алкильных [203], винильных
[203, 204], перфторвинильных [173, 205] и перфторфенильных [206, 207]
радикалов от атома олова. В порядке легкости отрыва органические радикалы
располагаются следующим образом [152]: я-толил > фенил >> перфторфе-
нил > алкил >> перфторалкил.
Так, при действии треххлористого бора на триметилперфторэтилолово
при обычной температуре наблюдается отрыв только метальных групп.
Перфторэтильный радикал остается незатронутым [153].
Треххлористый и трехбромистый бор отщепляют от тетраалкилолова не
более двух органических радикалов [208]. В случае тетрафенилолова можно
осуществить ступенчатое отщепление всех ароматических групп, причем
наиболее трудно отрывается последний радикал [209].
Данные по действию солей алюминия на оловоорганические соединения
противоречивы. В статье Джонсона [210] указывается, что после
трехчасового нагревания при 130СС треххлористого алюминия с тетрабутилоловом
последнее остается практически без изменения. По другим данным [211]
в хлороформе треххлористый алюминий отщепляет от тетраалкильных
соединений олова один или два радикала уже при комнатной температуре.
В тех же условиях из тетрафенилолова образуется неорганическая соль
[195, 211]. Возможно, в этом случае деалкилирующим агентом является
хлороформ (см. стр. 366).
Треххлористый галлий реагирует с тетраметилоловом уже при 0'С
.2121:
(CH3)jSn +GaCl3-* (CH3)3SnCl.C48GaCl2.
То же соединение образуется и при замене тетраметилолова на хлористое
триметилолово [212].
Подробно изучено [191, 195, 213, 214] взаимодействие треххлористого
таллия с оловоорганическими соединениями, например тетраэтил-, тетрабу-
Деалкилирование
357
тил-, тетрафенил-, тетра-о-толил-, тетра-я-толил-, тетра-о-анизилоловом
и стереоизомерными тетрапропенильными, тетраизопропенильными и тет-
равинильными соединениями олова. Несмеяновым с сотр. [191] установлено,
что геометрическая конфигурация изопропенильного радикала при переходе
от атома олова к таллию не изменяется:
Т1Х3+
СН3ч /Н
SnX2->
2
ГСН3ч /И,]
„Ж
TlX + SnX4
Интересно отметить, что m/шяс-тетрапропенилолово реагирует гораздо
быстрее, чем его стереоизомер [213].
Тетраметилолово образует с четыреххлористым кремнием хлористое
триметилолово и хлористый метил [7]. При нагревании до 186—190°С в
течение 24 час. эквимолекулярных количеств тетраэтилолова и треххлористого
фенилкремния получают хлористое триэтилолово и двухлористое диэтилолово
с выходом 41,4 и 42,4% соответственно [215]. С треххлористым этилкрем-
нием тетраэтилолово не реагирует [111].
При нагревании в хлороформе эквимолекулярных количеств тетрафенил-
олова и четыреххлористого кремния или четыреххлористого титана
образуется хлористое трифенилолово [216].
Тетраэтилолово и хлориды этилолова энергично реагируют с
четыреххлористым титаном с выделением непредельных углеводородов, хлористого
Еодорода и треххлористого титана [217]. Кратковременное нагревание
тетраэтилолова с четыреххлористым титаном приводит к хлористому триэтил-
олову (выход 42%) [111].
С четыреххлористыми солями циркония и тория тетраэтилолово
практически не реагирует [217].
О реакции тетраалкильных соединений олова с галогенидами германия
см. стр. 56.
Деалкилирование оловоорганических соединений галогенидами фосфора
изучено рядом авторов [111, 202, 2i8, 219]. Тетраэтилолово не реагирует с
треххлористым фосфором при нагревании смеси до температуры кипения
[111, 219]. Однако при нагревании с трехбромистым или трехиодистым
фосфором получены [219] бромистое и йодистое триэтилолово соответственно.
Взаимодействием 2,0 г тетрафенилолова с 4,0 г пятифтористого фосфора
при 135°С получают [202] четырехфтористый фенилфосфор и твердый
осадок, в котором по данным ИК- и ЯМР-спектров содержится гексафторфосфат.
При обычной температуре тетраметил- и тетрафенилолово с пятифтористым
фосфором не реагируют [218].
Нагреванием до 95°С дибутилдивинилолова с трехбромистым мышьяком
получен [220] бромистый дивинилмышьяк:
(C4H9)2Sn(CH^:CH2)2-f[AsEr3^ (C4H9)2SnBr2 + (CH2=CH)2AsEr.
Пятифтористый мышьяк полностью восстанавливается тетрафенил
оловом до трехфтористого мышьяка. Производные фенилмышьяка в продуктах
реакции не обнаружены [202].
При нагревании тетрафенилолова с треххлористой сурьмой в хлороформе
получено [195] хлористое трифенилолово с выходом 52%.
Взаимодействием тетраэтилолова с треххлористым висмутом в
хлороформе получена [221] смесь хлористого триэтилолова (30%) и двухлористого
диэтилолова (6%). При нагревании до 120°С тетрабутилолова или трипро-
пилбутилолова с треххлористым висмутом без растворителя образуются
двухлористое дибутилолово и двухлористое пропилбутилолово
соответственно.
В зависимости от молярного соотношения реагентов тетрафенилолово
образует с треххлористым висмутом в хлороформе или хлористое трифенил-
353
Оловоорганические соединения. Лит. стр. 370—375
олово, или двухлористое дифенилолово [222]. В этой реакции хлороформ
играет роль не только растворителя, но и галоидирующего агента, тогда
как треххлористый висмут является только катализатором.
ДЕЙСТВИЕ ОКИСЛИТЕЛЕЙ
НА ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Действие кислорода на оловоорганические соединения описано очень
кратко. В патентной литературе [223] описан способ получения спиртов
окислением оловоорганических соединений с последующим гидролизом
образовавшихся тетраалкоксильных производных олова. Окисление
проводят при 40—60°С и давлении до 136 атм в течение 10—60 мин. В качестве
катализатора применяют соли кобальта, магния или железа.
Спектральным методом изучено [224] взаимодействие тетраалкильных
производных олова с хромовым ангидридом. Установлено, что на одну
молекулу оловоорганического соединения максимально расходуется 12
атомов кислорода, т. е. алкильные группы окисляются до кетонов или кислот.
Исследование кинетики реакции тетра-я-бутилолова с хромовым
ангидридом при различных соотношениях компонентов показало, что это реакция
второго порядка (первого по каждому компоненту). Определены константы
скорости реакций тетраалкильных производных олова с хромовым
ангидридом и показано, что они существенно зависят от природы оловоорганического
соединения.
Отщепление органических радикалов наблюдается также при
получении некоторых перекисных соединений триалкил- [225] или триарилолова
[225, 226]. Неустойчивость последних объяснена [226] склонностью к
превращению по следующей схеме:
R OR
I /
R2Sn—0-> R2Sn
I \
OOGR' OOCR'
Гидроокись трибензилолова разлагается на воздухе с образованием окиси
дибензилолова и бензальдегида [56]. Описано также отщепление кислородом
некоторых замещенных ароматических радикалов. Так, растворы (2-диме-
тиламинофлуоренил-9)трифенилолова при доступе воздуха окрашиваются
в интенсивный красный цвет. Выпариванием смеси получают гидроокись
трифенилолова с т. пл. 119—120°С [48]. Хлористое три- и ди-(д-метоксибен-
зил)олово разлагаются на воздухе с образованием массы
красно-коричневого цвета [56]. На отрыв одной из ароматических групп при действии перман-
ганата калия на трифенил-о-оксиметилфенилолово указывается в статье
Гилмана [82]. Легко окисляются циклопентадиенильные соединения олова
[227].
ДЕАЛКИЛИРОЗАЧИЕ ФОСЮРОМ, СЕРОЙ, СЕЛЕНОМ
И ДРУГИМИ ЭЛЕМЕНТАМИ
Шуман и Шмидт с сотр. [228—235] исследовали взаимодействие
оловоорганических соединений с бором, алюминием, галлием, таллием,
индием, фосфором, серой, селеном и теллуром.
С бором, алюминием, галлием, индием и таллием реакция начинается
лишь при температуре выше 370° С и проходит в соответствии с
уравнением [228]:
(QH5)4Sn + М -> 2С6Н5—С6Н5 + SnM.
Выход дифенила уменьшается при переходе от бора к таллию.
Двалкилированиг
359
Деалкилирование серой и селеном подробно изучено на примере бу-
тильных и фенильных соединений олова.
Тримерный сульфид ди-я-бутилолова получен нагреванием при 150—
190° С тетрабутилолова с двумя эквивалентами серы в течение 24 час.
1230]. Можно применить также реакцию сульфида бш>(трибутилолова)
с эквимолекулярным количеством серы. Вещество очищают перегонкой
в вакууме. Иногда продукт окрашен в оранжевый цвет. В этом случае
его оставляют стоять на длительное время, фильтруют и снова
перегоняют в вакууме.
Взаимодействием серы с хлористым трибутилоловом получен продукт
состава ClSn(C4H9)2SSn(C4H9)2Cl [231].
Реакция "хлористого трибутилолова с серой [231]. Синтез проводят в колбе с обратным
холодильником, верхний шлиф которого соединен с двумя ловушками. Первую охлаждают
смесью метилового спирта с сухим льдом, вторую — жидким воздухом. В колбу помещают
65,1 г (0,2 моля) хлористого трибутилолова и 12,8 г (0,4 моля) серы. Смесь нагревают при
180—190° С, перемешивая магнитной мешалкой, в течение 6 час. За это время сера полностью
растворяется. Нагревают при 190° С еще 8 час. В первой ловушке конденсируется дибу-
тилсульфид (образования сероводорода не наблюдается). Содержимое колбы дважды
перегоняют на колонке в вакууме 1 мм. Получают 5,0 г дибутилсульфида (т. кип. 76—78° С/1 мм),
непрореагировавшее хлористое трибутилолово, 3,0 г (4%) г хлористого бутил мерка пто-
дибутилолова и 28,0 г (49%) ClSn^H^SSn^H^Cl (т. кип. 110—115° С/1 мм; т. пл. 35° С).
Подобно сере, селен отщепляет от тетрабутилолова два органических
радикала, но реакция проходит при более высокой температуре.
Получение селенида дибутилолова [232]. Смесь 70 г (0,2 моля) тетрабутилолова и 32 г
{0,4 моля) черного селена нагревают при 190—210° С в течение 6 час. Образовавшиеся в
результате реакции ди-я-бутилселен, дибутилдиселен и непрореагировавшее тетрабутилолово
отгоняют в вакууме 1 мм (т. кип. 38—40, 88—90 и 136—139° С соответственно). При
повышении температуры до 215° С отгоняется дибутилдиселен. После охлаждения до
комнатной температуры к остатку прибавляют бензол. Селен и селенид олова отфильтровывают.
Прозрачный фильтрат, обладающий резким неприятным запахом, нагревают до 150° С в
высоком вакууме в течение 20 час. В остатке получают тримерный селенид дибутилолова.
Выход 42 г.
Триэтилстаннилацетон активно реагирует с серой уже при обычной
температуре с образованием сульфида бш>(триэтилолова) и смолообраз-
ного продукта [183].
В зависимости от условий проведения реакции при действии серы на
тетрафенилолово отщепляется 1, 2 или более ароматических радикалов.
Нагреванием тетрафенилолова с двумя эквивалентами серы получен
[230] тримерный сульфид дифенилолова
3(C6H5)4Sn + 63 -* [(CdH,)aSnS]3 + 3(C6H5)2S.
Предложен [230] следующий механизм этой реакции, имеющий место и
в других описанных выше случаях [231, 232]. Расплавленная сера
поляризует тетрафенилолово. Поляризованные молекулы взаимодействуют
между собой по схеме
R3Sn+5 — IT5 +-S—Sx—S+-> RgSnSS^SR
(R = C6H5).
Образовавшееся сернистое соединение под действием избытка
тетрафенилолова превращается в фенилмеркаптид трифенилолова.
360
Оловоорганические соединения. Лит. стр. 370—375
Например, при х = 2 происходит следующая реакция:
■ 4R3SnSR.
R—SnR3 ! R—SnR3
i
R3Sn—S S S —— SR -
: R—SnR3;
Фенилмеркаптид трифенилолова присоединяет новую цепь серы
R R
RS—Sn+5 —R~5 + -S—Sx—S+ -» RS—Sn—S—S —SR.
I I
R R
В свою очередь это соединение реагирует с избытком фенилмеркаптида
трифенилолова с образованием дифенилмеркаптида дифенилолова.
Последний в условиях реакции (190—210° С) неустойчив и разлагается с
выделением дифенилсульфида и сульфида дифенилолова
R—R2SnSR
RS—Sn—S
I
R
R—R2SnSR
-S—
R—R2SnSR
R
4R2Sn(SR)2,
3RS—Sn—SR -» 3R2S + (R2SnS)3.
I
R
Предложенный механизм реакции подтверждается тем, что синтезированный
независимым способом дифенилмеркаптид дифенилолова количественно
разлагается при нагревании до 110° С в соответствии с приведенным выше
уравнением. Те же продукты образуются и при нагревании до 190—210° С
фенилмеркаптида трифенилолова с серой.
Получение сульфида дифенилолова [230]. 21,4 г (0,5 моля) перекристаллизованного из
бензола тетрафенилолова и 3,2 г (0,1 моля) серы растирают в ступке в отсутствие влаги
воздуха. Смесь запаивают в толстостенную стеклянную ампулу и нагревают при 190—210° С
в течение 2 час. После охлаждения содержимое ампулы растворяют в бензоле. Бензол и
образовавшийся в результате реакции дифенилсульфид отгоняют. Остаток растворяют в
трихлорэтилене и высаживают петролейным эфиром. После двух переосаждений получают
кристаллы с т. пл. 183—184° С. Вещество хорошо растворяется в бензоле, трихлорэтилене,
пиридине и диметилформамиде.
Тримерный сульфид дифенилолова получен также нагреванием
сульфида бис-(трифенилолова) с двумя эквивалентами серы. Выход 98,3%
[230].
При длительном нагревании до 200° С тетрафенилолова с тремя экви-
Деалкилирование
361
валентами серы образуются бесцветные кристаллы полимерного сульфида
дифенилолова [233]
1
3(C6H5)4Sn + 8S-
[(C6H5)4Sn3S4]„-^4(C6H5)2S.
Высказано предположение [233], что вещество имеет следующее строение:
R R
Я R
\/
Sn
s/Xs
R4 I I /R
>Sn Sn<
s
• R R
\/
Sn
s/4s
R4 I A
)Sn Sn<
s
Наличие групп
(C6H5)2Sn
/
i
\
Sn
s/4s
R4 I I A
>Sn Sn<
/ \/ X^-
к S
S—
и C6H5Sn—S—
\
s—
подтверждается тем, что при кипячении продукта с соляной кислотой
(1 : 1) образуется двухлористое дифенилолово и треххлористое фенил-
олово.
Получение сульфида фенилолова [(CeHs^SnsSJx [233]. 42,7 г (0,1 моля) тетрафенилолова
и 9,6 г (0,3 моля) серы сплавляют в вакууме в толстостенной стеклянной ампуле. Смесь
нагревают при 190—210° С в течение 9 час. После охлаждения до комнатной температуры
к содержимому ампулы прибавляют бензол, фильтруют, бензол и образовавшийся в
результате реакции дифенилсульфид отгоняют. Остаток закристаллизовывается при прибавлении
небольшого количества эфира. Вешество растворяют в небольшом количестве пиридина и
высаживают петролейным эфиром. Получают бесцветные кристаллы с т. разл. 250° С.
Аналогично получен продукт состава [(С6Н5)2 Sn2S3]x. Нагревание
до 200° С тетрафенилолова с большим избытком серы (примерно
9 эквивалентов) приводит к дальнейшему отрыву фенильных групп.
Анализ продукта показывает, что в среднем каждый атом олова еще
содержит одну фенильную группу и связан с тремя другими атомами олова
через серу. При нагревании вещества с соляной кислотой образуется
треххлористое фенилолово и сероводород [233].
Нагревание до температуры выше 220° С тетрафенилолова [233, 236],
а также описанных выше продуктов [233] с серой приводит к отщеплению
всех фенильных групп. Например,
[(C6H5)2SnS]3 + 3S -> 3SnS + 3(C6H5)2S.
О реакции тетра-я-толилолова с серой с целью получения п-дитолил-
дисульфида см. статью Боста и Бейкера [124].
С хлоридами фенилолова сера реагирует более сложно, чем с
соответствующими соединениями алифатического ряда. При нагревании до
190—200° С хлористого трифенилолова с двумя эквивалентами серы в
течение 10 час. получены дифенилсульфид, двухлористое дифенилолово
и оловоорганическое соединение состава (C6H5)5Sri3S5Cl. Реакция
проходит по следующей общей схеме [231]:
15(C6H5)3SnCl + 24S-> 3(C6H5)5SnrS;Cl + 8(C6H;)2SnCl2 + 9(CGH3)2S.
Двухлористое дифенилолово и треххлористое фенилолово реагируют с
серой лишь при температуре выше 220° С. По-видимому, при этом проис-
362
Оловоорганические соединения. Лит. стр. 370—375
ходит разрыв углерод-углеродных связей. Из реакционной смеси
наряду с сульфидом олова и сероводородом выделены только продукты
крекинга [231]. Взаимодействие двухлористого дифенилолова с серой
сопровождается реакцией диспропорционирования
3(C6H5)2SnCl2 + 3S -> 2С6Н6 + 2C6H5SnCl3 + SnS +
Ч^чя/Ч
Треххлористое фенилолово не реагирует с серой до 240° С. Из реакционной
смеси выделены только продукты диспропорционирования — хлорное
олово и двухлористое дифенилолово [231].
При нагревании тетрафенилолова с селеном образуется фенилселе-
нид трифенилолова [232]:
(C6H5)4Sn + Se -» (C6H5)3SnSeC6H5.
Реакция тетрафенилолова с селеном проходит примерно так же, как
и с серой (см. стр. 359) [232]. Вначале происходит нуклеофильное
расщепление селеновой цепи тетрафенилоловом и далее реакция протекает по
схеме:
(C6M5)4Sn + -Se—Sex—Se+ -* (QHsbSnSeSe^SeCeHs.
Образовавшееся несимметричное селеноловоорганическое соединение
вступает в дальнейшую реакцию с фенильным анионом поляризованного
тетрафенилолова. Например, при х = 2:
R—SnR3
R3Sn—Se!
Se!
i
i
R—SnR3
R—SnR3
•Se!
SeR->4R3SnSeR
(R=CeH6).
В противоположность сернистому аналогу, фенилселенид трифенилолова
является устойчивым соединением и не вступает в дальнейшую реакцию
с селеном.
Получение фенилселенида трифенилолова [232]. 43 г (0,1 моля) тетрафенилолова и 16 г
(0,2 моля) красного селена помещают в толстостенную стеклянную ампулу и сплавляют
в вакууме. Смесь нагревают при 190—210° С в течение 60 час. Содержимое ампулы
представляет собой после этого маслянистую жидкость желтого цвета, в которой суспендировано
небольшое количество непрореагирозавшего тетрафенилолова и черного селена. К смеси
прибавляют бензол, фильтруют и бензол выпаривают. Образовавшийся в результате
реакции дифенилселен отгоняют в вакууме 1 мм, остаток растирают с эфиром. При охлаждении
до —4° С выделяются бесцветные иглообразные кристаллы фенилселенида трифенилолова.
После нескольких перекристаллизации из этилового спирта получают химически чистое
вещество ст. пл. 87—88° С. Выход 24 г (47,1%). Вещество устойчиво к действию влаги
и воздуха, хорошо растворяется в ароматических углеводородах, дихлорметане и диметил-
формамиде. Плохо растворимо в эфире и тетрагидрофуране.
При нагревании до 240° С тетрафенилолова с избытком селена
получить оловоорганическое соединение не удается. Наряду с металлическим
оловом с хорошим выходом образуются только дифенилселен и дифенил-
диселен [232].
При нагревании тетрафенилолова с фосфором образуются
оловоорганические фосфины [228, 234, 235]. Их структура зависит от молярного
соотношения реагентов, температуры и продолжительности опыта.
Деалкилирование
363
Нагревая до 235—250° С тетрафенилолово с двумя эквивалентами
фосфора в запаянной под вакуумом ампуле, получают [235] фенилфосфин
дифенилолова [(C6H5)2SnPC6H5]3 и тетра-(трифенилстаннил)дифосфин
l(C6H5)3Sn]2P-P[Sn(C6H5)3]2.
При других соотношениях фосфора и тетрафенилолова и температуре
порядка 260—280° С образуются более сложные вещества:
(C6H5SnP)„,
(C6H5)2P-Sn-P-
I I
с6н5
(C6H5-P-Sn-)2PC6H5
или
СбН5
, [(C6H5)2PSn(C6H5)2PC6H5]n,
[(C6H5P_Sn-)3P]n.
Выделить эти соединения в чистом виде не удается. Их присутствие
доказано [235] окислением реакционной смеси кислородом воздуха с
последующим разделением образующихся веществ.
При нагревании тетрафенилолова с фосфором выше 300° С образуются
трифенилфосфин и фосфиды олова состава Sn3Pn [228]. Аналогичные
реакции с мышьяком или сурьмой начинаются выше 320° С или,
соответственно, 360° С. Висмут реагирует с тетрафенилоловом при температуре
выше 475° С с образованием дифенила и SnBin [228].
ДЕАЛКИЛИРОВАНИЕ ВОДОЙ И ЩЕЛОЧАМИ
Незамещенные алкильные или арильные радикалы, связанные с
атомом олова, устойчивы к действию воды и щелочей. Например, при
кипячении тетрафенилолова со спиртовым раствором едкого кали (— 15%)
в течение 3 час. вещество остается практически без изменения [82].
Введение электрофильных групп к а-углеродному атому поляризует
связь олова с углеродом, снижая тем самым ее устойчивость к действию
воды и Щелочей.
Интересная разница наблюдается при гидролизе четырех
гомологических цианистых соединений: цианистого трифенилолова, (цианметил)-,
(2-цианэтил)- и (З-цианпропил)трифенилолова [237]. Первый член этого
ряда гидролизуется даже влагой воздуха с образованием гидроокиси
трифенилолова и синильной кислоты [237, 238]. Цианметильная группа от-
ноносительно устойчива, но легко отщепляется при действии щелочи
[237]. При гидролизе цианогруппы, находящейся у |3- или у-углеродного
атома, образуются соли соответствующих оловоорганических кислот
{см. стр. 448). Та же картина наблюдается и в случае (цианметил)три-я-
бутилолова [18, 145], а также (карбоксиалкил)трифенильных соединений
олова.
Триэтилацетонилолово легко и быстро (с заметным разогреванием)
гидролизуется водой с образованием гидроокиси триэтилолова и ацетона [179].
Также легко реагируют с водой и другие соединения, содержащие атом олова
ва-положении по отношению к карбонильной группе [180—182]. Оловоор-
ганические соединения, имеющие карбонильные группы в |3-положении, с
водой не реагируют [239].
Хлористое диметилтрифтормегилолово легко растворяется в воде с
образованием устойчивых кислых растворов, которые могут быть оттитрова-
364
Оловоорганшеские соединения. Лит. стр. 370—375
ны водным раствором щелочи. Однако образующаяся гидроокись
чрезвычайно неустойчива и самопроизвольно разлагается на окись диметилолова и
фтороформ [15]
(CH8)2CF8SnCl + Н20 -> (CH3)2Sn (CF8)(OH) + НС1,
(CH3)2Sn(CF3)(OH) -> (CH3)2SnO + CF3H.
Отщепление перфторалкильных радикалов щелочью описано и другими
авторами [150, 153].
Еще легче отщепляются перфторвинильные группы, которые
неустойчивы даже по отношению к влаге воздуха [240]. В случае диметилдипер-
фторвинилолова наряду с гидролизом происходит полимеризация вещества
с последующим гидролизом или окислением полимера. В конечном счете
образуется окись диметилолова [149].
Сравнительная устойчивость винильной и перфторвинильной групп к
действию воды и щелочи видна из табл. 8 [149].
Таблица 8
Действие воды и щелочи на тетравинил-
и тетраперфгорвинилолово
Реагент
20%-ный NaOH
Вода
Вода
Условия
время,
часы
60
50
50
ратура, °с
100
25
100
Степень разложения, %,
определенный по количеству
выделившегося
CF2=CFH из
(CF2=CF)4Sn
96,5
3
66
СН2=СН2 из
(CH2=CH)4Sn
6,5
0
7
Действие водно-спиртового раствора фтористого калия на неочищенное ди-
этил- или ди-я-бутилдиперфторвинилолово, содержащее примесь хлористого
ди-я-бутилперфторвинил олова, приводит к полному отрыву перфторвиниль-
ных групп [109]:
2R2Sn (CF=CF3)C1 + 2F- + Н20 -» RaSn—О—SnRa + 2С1" + 2CFS=CFH.
Как отмечено выше, винильные соединения олова относительно
устойчивы к действию воды и щелочи. Однако введение атома хлора в виниль-
ную группу резко снижает ее устойчивость. Так, при действии 15%-ного
раствора щелочи на стереоизомеры хлористого три-|3-хлорвинилолова
выделяется ацетилен. Отмечено, что с транс-транс-транс-изомером реакция
проходит значительно легче [190].
При действии воды и щелочей от оловоорганических соединений легко
отщепляются также ацетиленовые [164, 165, 197, 241—245], циклопента-
диенильные [246, 247] и некоторые другие радикалы [48, 82].
В отличие от перфторалкильных групп, перфторфенильные радикалы,
связанные с атомом олова, довольно устойчивы к действию воды и щелочей
[152, 206, 248]. Например, тетрапентафторфенилолово остается практически
без изменения, если его оставить в смеси ацетона с водой на 26 дней при
обычной температуре. При кипячении вещества c6iV соляной кислотой на
протяжении 5 час. оно также не изменяется. Лишь при 5-часовом
нагревании с 10%-ным едким натром наблюдается образование пентафторбензола
[2483.
Деалкилирование
365
Интересно отметить, что ионы галоида или циана являются
катализаторами в реакциях отщепления перфторфенильных радикалов водой [152,
206]. Так, триметилпентафторфенилолово практически не изменяется в
водно-спиртовом растворе. Однако при прибавлении к этому раствору
кристаллика фтористого калия сразу же выпадает осадок гидроокиси три-
метилолова и образуется пентафторбензол. Предложена [152] следующая
схема этой реакции:
(CH3)3SnC6F5 + Х- -» [(CH3)3(C6F5)SnX]-
H2Oj|
(CH3)sSnOH + C6F5H + Х- 4- [(CH3)3C6F5Sn(OH)2X]-
Аналогично ведут себя и другие пентафторфенильные производные олова.
ТЕРМО- И ФОТОРАСПАД.
РЕАКЦИИ С ГАЛОИДНЫМИ АЛКИЛАМИ
Термическое разложение оловоорганических соединений изучено
недостаточно. Уоринг и Гортон [249] исследовали кинетику термического
разложения тетраметилолова при 440—493° С. Показано, что при начальном
давлении 80 мм реакция описывается кинетическим уравнением первого
порядка. Понижение давления повышает порядок реакции (ср. [250]). О
термическом распаде тетраметилолова и двухлористого диметилолова см.
статьи Лонга [251] и Прайса с сотр. [252]. При облучении
ультрафиолетовым светом чистого тетраэтилолова в начальной стадии наблюдается отрыв
этил-р адикалов [253—255 ] :
(C2H5)4Sn -* С2Н5* + (C2H5)3Sn* -> (C2H5)3$n3n(C3H5)3.
Димеризация (а не распад) радикала (C2H5)3Sn* объясняется тем, что связь
Sn—Sn несколько прочнее связи Sn—С [253, 256]. При дальнейшем
фотораспаде тетраэтилолова без доступа воздуха образуются этан, этилен,
бутан и окрашенные оловоорганические соединения сложного строения [257].
Совершенно иначе проходит фотолиз оловоорганических соединений при
контакте с кислородом воздуха, а также без доступа воздуха, но в
растворах галоидных алкилов [258]. В первом случае фотораспад подавляется
реакциями фотохимического окисления, во втором наблюдается
взаимодействие оловоорганических соединений с галоидопроизводными.
Например, в ходе реакции тетраэтилолова с четыреххлористым углеродом
образуются хлористое триэтилолово, гексахлорэтан, хлороформ, хлористый
этил и смесь газообразных углеводородов, содержащая 81,1% этана, 18,3%
этилена и 0,6% бутана [253, 258]. Для первой стадии реакции предложена
следующая схема:
(C2H5)4Sn _»(C2H5)3Sn* + С2Н5',
ссц-1 cci3-+ cr,
(C2H5)3Sn- + СС14 -» (C2H5)3SnCl + GCU',
CCI3' + (C2H5)4Sn -* (C2H5)3SnC2H4 + CHC13,
(C2H5)33nC2H4-* (C2H5)3Sn* + C2H4,
C2H4 + ССЦ - ClCH3CH3CCl8t
2CC13'-*C2C!6.
Образующееся хлористое триэтилолово подвергается дальнейшему
фотолизу, в результате чего основным продуктом взаимодействия оказывается
не (C2H5)3SnCl (выход по олову 12,1%), a (C2H5)2SnCl2 (23,3%).
366 Оловоорганические соединения. Лит. стр. 370—375
Подтверждением этому служит тот факт, что при облучении чистого
хлористого триэтилолова лампой ПРК-7 в течение 90 час. 29,7% его
превращается в двухлористое диэтилолово. Одновременно в реакционной смеси
обнаружен^ неизменившиеся хлористое триэтилолово, металлическое
олово и смесь газообразных углеводородов.
Фотохимическая реакция тетраэтилолова с бромистым пропилом также
протекает по свободнорадикальному механизму [259]. Наряду с бромистым
триэтилоловом образуются этан, этилен, пропан и пропилен. Соотношение
между этими продуктами существенно зависит от соотношения исходных
веществ.
Фотолиз тетра-я-пропилолова с избытком четыреххлористого углерода
приводит к (C3H7)2SnCl2 (выход 15%), пропану, пропилену и 1,1,1,3-тетра-
хлорбутану [258].
Аналогично проходит фотолиз тетраметил- и тетрапропилолова в
хлороформе [237].
Реакцию тетраалкильных соединений олова с галоидными алкилами
можно инициировать также органическими перекисями [260] или трех-
хлористым алюминием [261—263]. Так, прибавление А1С13 к растворам
тетраэтилолова в галоидных алкилах с добавкой бензола приводит к
экзотермической реакции с образованием галогенида триэтилолова [261, 262].
Изучено влияние добавок перекиси бензоила на реакцию тетраэтил-»
диметилдиэтил- и хлористого триэтилолова с четыреххлористым углеродом,
а также тетраэтилолова с бромистым я-пропилом [259, 260]. Во всех
случаях при 75—80° С начинается свободнорадикальная цепная реакция.
Наиболее подробно исследовано взаимодействие перекиси бензоила со смесью
тетраэтилолова и четыреххлористого углерода. Основными продуктами этой
реакции являются хлористое триэтилолово, этилен и хлороформ. Наряду с
(QHrjJsSnCl изолировано двухлористое диэтилолово. Следовательно, и в
этом случае хлористое триэтилолово может подвергаться тем же
превращениям, что и исходное тетраэтилолово. Это подтверждается реакцией
перекиси бензоила со смесью хлористого триэтилолова и четыреххлористого
углерода. При этом цепной характер взаимодействия сохраняется, хотя
выход основных продуктов снижается [260].
При нагревании или облучении смеси тетраалкильных соединений олова
с трифториодметаном или пентафториодэтаном большинство перфторметиль-
ных или перфторэтильных групп отрывают водород от алкильных радикалов
с образованием фтороформа или пентафторэтана [153, 264].
Несколько менее подробно изучена реакция оловоорганических
соединений ароматического ряда с галоидными алкилами [54, 107, 124, 265—267].
В присутствии перекиси бензоила тетрафенилолово реагирует с
четыреххлористым углеродом с образованием хлорного олова, хлорбензола, гекса-
хлорэтана, бензойной и фталевой кислот [266]. Перекись ацетила не
инициирует реакцию между четыреххлористым углеродом и тетрафенилоловом
[267].
Тетрафенилолово в хлороформе при действии света распадается с
образованием двухлористого дифенилолова, бензола и гексахлорэтана по
уравнению [268]:
(C6H5)4Sn + 4СНС13 -> (C6H5)2SnCl2 + 2С6Н5' + С13ССС13 + 2(СНС12)2.
При фотохимических реакциях диэтилдифенилолова с четыреххлористым
углеродом, хлороформом и метиловым спиртом происходит отрыв фениль-
ного радикала, который далее реагирует с растворителем [107].
Аналогичная картина наблюдается и при фотореакциях дибензилдифе-
нилолова с четыреххлористым углеродом и хлороформом [107]. Тетра-я-то--
Деалкилирование
367
лилолово не реагирует с бромистым этилом или бромистым изопропилом при
кипячении смеси в течение 36 час. При 80-часовом кипячении тетра-я-толил-
олова с избытком третичного бромистого бутила наряду с непрореагиро-
вавшими исходными веществами выделены бромистый водород, изобутан
и незначительное количество толуола [124]. О реакции галоидных арилов
с оловоорганическими ссединениями см. стр. 452.
РЕАКЦИИ С ОРГАНИЧЕСКИМИ ПЕРЕКИСЯМИ
Тетраалкильные соединения олова реагируют с ацильными и алкиль-
ными перекисями лишь при температуре их распада. Нагревание до 95° С
тетраэтилолова с 10-кратным молярным избытком перекиси бензоила дает
в качестве основных продуктов бензоат триэтилолова, этилен и бензойную
кислоту. Кроме того, образуются двуокись углерода и этан [253, 269—271],
Аналогично проходят реакции с тетра-я-пропил- и тетраизопропил-
оловом [272]. Замена одного алкильного радикала на другой в основном
отражается на соотношении продуктов реакции; характер же продуктов
остается неизменным [272].
Если вместо перекиси бензоила взять перекись ацетилбензоила, то
образуется эквимолекулярная смесь ацетата и бензоата триэтилолова:
(C2H5)4Sn + С6Н,СООООССН3 -> (C2H5)3SrOCCC6H5 +
+ (CaHsbSnOOCCHs + С2Н6-+ С6НьССОН -f CH4.
Вероятно, ацетат образуется за счет разложения промежуточного
комплекса, сопровождающегося выделением С2Н5' и С6Н5ССО* радикалов [253,
2541:
(C2H5)3Sn-C2H5
\ -» (С2Н5)з5пООССНз + С6Н5СОО* + С2Н5'
I l .
Q Q С6Н5' + С02
I! II
СНз—С—00—С—С6Н5
Равновероятен аналогичный комплекс, образующий бензоат
триэтилолова. Свободные радикалы (СН3", СвН5СОО', С6Н5') могут атаковать
молекулы исходного соединения с отрывом атомов водорода:
(C2H5)4Sn + R* -> (C2H5)3SnCHCH3 + RH.
Сложный оловоорганический радикал распадается на этилен и радикал
триэтилолова. Это подтверждается большим содержанием в реакционной
смеси этилена по сравнению с этаном [253].
С перекисью трет-бутила реакция проходит в более жестких условиях
(16 час. при 145° С). Основными продуктами реакции являются трет-буто-
кситриэтилолово, m/?£m-бутиловый спирт и этилен [269, 271]. По-видимому,
эти три вещества возникают при гомолитическом распаде промежуточно
образующегося активного пятичленного комплекса:
СН—СН3
(C2H5)3Sn H
(C2H,)4Sn + (СН3)< СООС (СН3)з -> (СН8)3СО—6С(СН3)3 -»
-> (C2H5)8SnOC (СН3)з + (СНз)8СОН + С2Н4.
Это уравнение описывает лишь основное направление реакции.
Параллельно с ним, вероятно, проходит свободнорадикальнсе взаимодействие пере-
368
Оловоорганические соединения. Лит. стр. 370—375
киси с тетраэтилоловом, в результате которого в реакционной смеси
обнаруживаются этан, следы метана, бутана, металлического олова и гексаэтил-
дистаннана. га/?е/п-Бутилпербензоат образует с тетраэтилоловом бензоат
триэтилолова и /гг/?£га-бутокситриэтилолово [273]. При замене одной этильной
группы на хлор, бром или остаток бензойной кислоты реакция несколько
меняется [253, 254, 270, 274]. В случае хлористого триэтилолова
наблюдается образование смеси двухлористого диэтилолова и дибензоата диэтил-
олова. Кроме того, образуются этан и этилен за счет диспропорционирования
вытесненных этил-радикалов [253, 2701:
(C2H5)3SnCl + (СвН5СОЭ)3 -* (CaH5)2Sn (CI) (OOCG6H,) + С2Н4 + С2Н6
I
(QHsbSnCU + (C3H5)2Sn (OOCC6H5)2.
Такая же реакция наблюдается при взаимодействии перекиси с бензоа-
том триэтилолова. Однако одновременно с этаном и этиленом образуется
0,3 моля эфира бензойной кислоты [253, 270, 274]:
(C2H5)2Sn (ООСС6Н5)2 + С2Н4 + С2Н6
/
(C2H5)3SnOOCC6H5 + (С3Н5СОО)2
(G2H5)2Sn (ООССбН5)2 + GGH5GOOG2H5.
Совершенно иначе перекись бензоила реагирует салкоксисоединениями
триэтилолова [253, 254, 274, 275]. В этом случае уже при растворении
перекиси в оловоорганическом соединении наблюдается бурная
экзотермическая реакция, которая в более мягких условиях (в среде сухого бензола в
атмосфере азота при 20° С) проходит по уравнению:
(C2H5)3SnOR + (СбН5СОО)2 -* (C2H5)2Sn (OR) (OOCC6H5) + CeH5COOR
(R=CH3, C2H5, C6H3CH2, циклогексил, СбН3).
Состав образующихся веществ доказан превращением их в дибензоаты тет-
раэтилдистанноксана и определением спирта в продуктах гидролиза.
Кроме того, обработкой (алкокси)бензоатов диэтилолова бензойной кислотой
или аммиаком получены соответственно дибензоат и окись диэтилолова
[253].
Отщепление третьего органического радикала ацильными перекисями
не происходит. При многодневном хранении дибензоата диэтилолова с
перекисью бензоила трибензоат этилолова не образуется [275].
ДЕАЛКИЛИРОВАНИЕ ГАЛОИДНЫМИ АЦИЛАИИ
Образование кетонов при действии галоидных ацилов на оловооргани-
ческие соединения проходит в очень жестких условиях и с выходами
продуктов, не превышающими 5—10%. Однако в присутствии хлористого
алюминия тетраарильные соединения олова в очень мягких условиях и с
выходами до 70% реагируют с галоидными ацилами [265, 276] (ср. [277]) по
схеме:
(C6H5)4Sn + 4 С6Н5СОС1 -> SnCU + 4 С3Н,СОСзН5.
Исходным материалом могут служить и тетраалкильные соединения
олова. Так, из тетраэтилолова и бензоилхлорида получают этилфенилкетон с
выходом 47,2% [277].
При ацилирозании триэтилстаннилацетона хлористым ацетилом
получены [183] хлористое триэтилолово и изопропенилацетат. Ацетилаце-
тон в продуктах реакции не обнаружен. Реакция идет в положение 1,4
Деалкилирование
369
с переносом реакционного центра:
(C2H5)3SnCH2COCH3 + СН3СОС1 -* (C2H5)3SnCl + СН2=С (ООССН3) СН8.
При нагревании метилового эфира трипропилстаннилуксусной кислоты с
хлористым ацетилом из реакционной смеси выделены [183] метилацетат,
1-метоксивиниловый эфир уксусной кислоты, метиловый эфир ацетоуксусной
кислоты и хлористое трипропилолово. Состав продуктов показывает, что
реакция протекает по двум направлениям: в положение 1,2 и 1,4.
В случае ацилирования этилового и пропилового эфиров
трипропилстаннилуксусной кислоты хлористым ацетилом выделены только
соответствующие алкилацетаты, эфиры ацетоуксусной кислоты и хлористое
трипропилолово.
Выше показано (см. стр. 353), что этиловый эфир
трипропилстаннилуксусной кислоты реагирует с ацетоуксусным эфиром с образованием три-
пропилоловянного енолята ацетоуксусного эфира. Исходя из этих данных,
а также на основании анализа состава продуктов, выделенных при
взаимодействии эфиров трипропилстаннилуксусной кислоты с хлористым
ацетилом, авторы [183] предлагают следующую схему этих реакций:
г 2 3^°4 ,-М-> СН2=С (OR) ОСОСНз
(C3H7)3SnCH3C +сн3сос1
\ -(CaH7)3SnCl
—-CH3COCH2COOR
in u \ с гн гс\с\ъ +CH,COCH2COOR rurr^r, +CH3COCI
(C3H7)3SnCH2COOR —-си goor > CH3C=CHCOOR >
OSn (C3H7)3
-* (C3H7)3SnCl + (CH3CO)2CHCOOR.
Эфир ацетилацетоуксусной кислоты, вероятно, осмоляется в процессе
реакции и при фракционировании 1183].
ГИДРОГЕНИЗАЦИЯ
Гидрогенизация тетраалкильных соединений олова описана очень
кратко. Нагревание до 300—310° С тетраэтил- и тетраизобутилолова в
течение 20 час. при начальном давлении водорода 100 атм приводит к
выделению металлического олова и соответствующих углеводородов
1278]. В случае тетраэтилолова образуется 67% этана, небольшое
количество этилена и лишь следы пропана и бутана. Тетраизобутилолово
дает 79% изобутана, 9% изобутилена и около 2% С8-углеводородов
[278].
Тетрафенилолово под давлением водорода начинает разлагаться
около 200° С [278], а без водорода устойчиво до 375° С [279—281].
Изучено [278] влияние растворителя, температуры и продолжительности
нагревания на разложение тетрафенилолова при начальном давлении
водорода 60 атм. Во всех случаях основными продуктами являются
металлическое олово и бензол, содержащий лишь примесь дифенила. Ни в одном
из опытов не наблюдается полной гидрогенизации (выделяется 38,6—
76,3% металлического олова).
Применение катализаторов — порошков металлов — позволяет
значительно снизить температуру разложения тетрафенилолова [279—281].
В отсутствие водорода по силе действия они располагаются в следующий
ряд: Pd >> Ag > Au > Ni. Под давлением водорода этот ряд несколько
изменяется: Pd >> Ni > Ag > Au > Си.
24 Заказ № 207
370
Оловоорганшеские соединения
Катализаторы оказывают существенное влияние и на направление
реакции разложения. В присутствии никеля количественно образуется
бензол [281]:
(C6H5)4Sn + 2 Н2 -> Sn + 4 СбНб.
Прибавка палладия приводит к иному типу разложения
(C6H5)4Sn -* 2 С6Н5 - С6Н5 + Sn.
ДЕАЛКИЛИРОВАНИЕ ОКИСЛАМИ АЗОТА
Тетраметилолово активно реагирует с двуокисью азота [282].
Реакция обычно сопровождается самовоспламенением и взрывом даже при
температуре — 78° С. Однако, если реагенты разбавить равным объемом
этилацетата, реакция гладко проходит при — 78° С с образованием дини-
трата диметилолова [282].
Азотистый ангидрид (в присутствии окиси азота) реагирует с тетра-
фенилоловом и хлоридами фенилолова по схеме [283]:
C6H5SnCl3 + 2 N203 -* C6H5N2N03 + N03SnCl3.
Выход солей диазония составляет 50% и выше.
ДЕАЛКИЛИРОВАНИЕ ПЕНТАКАРБОНИЛОМ ЖЕЛЕЗА
Нагреванием пентакарбонила железа с диалкилдивинил- или диал-
килдифенилоловом в среде этилциклогексана получены [284] циклические
соединения состава [R2SnFe(CO)4]2-
ЛИТЕРАТУРА
1. Кочешков К. А., ЖРФХО, 61, 1385 (1929).
2. Kozeschkow К. А., Вег., 62, 996 (1929).
3. Кочешков К. А., ЖОХ, 4, 1359 (1934).
4. Kozeschkow К. А., Вег., 66, 1661 (1933).
5. Кочешков К- А., ЖОХ, 5, 211 (1935).
6. Е b е г 1 у К. С, SmithG. Е., А 1 b e r t H. Е., Пат. США 2560042 (1951);
С. А., 46, 1581 (1951).
7. G г a n t D., van Wazer J.R..J. Organometal. Chem., 4, 229 (1965).
8. N e u m a n n W. P., BurkhardtG, Ann., 663, 11 (1963).
9. Luijten J.G. A., Rec trav. chim., 85, 873 (1966).
10. Англ. пат. 739883 (1955); РЖХим., 1958, 54879.
11. Pope W.J., P each у S. J., Chem. News, 87, 253 (1903).
12. DorfeltC, Пат. ФРГ 1152693 (1964); РЖХим., 1965, 6H130.
13. G 1 о s k e у С. R., Англ. пат. 734371 (1955); С. А., 50, 8709 (1956).
14. G 1 о s k e у С. R., Пат. США 2718522 (1955); РЖХим., 1957, 42361.
15. С 1 а г к Н. С, W i 1 1 i s С. J., J. Am. Chem. Soc, 82, 1888 (1960).
16. Van der KerkG. J.M., L u i j t e n J. G. A., J. appl. Chem., 6,49(1956).
17. V a n der KerkG.J.M., N о 1 t e s J. G., L u i j t e n J. G. A., J. appl. Chem.,
7, 366 (1957).
18. V a n der К e г к G. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 93 (1956).
19. Jones W.J., DaviesW.C, BowdenS. Т., EdwardsC, Da vies
V. E., Thomas L. H., J. Chem. Soc, 1947, 1446.
20. P r i n с e R. H., J. Chem. Soc, 1959, 1783.
21. В 1 а к e D., Coates С E.Jate J.M., J. Chem. Soc, 1961, 618.
22. R о s e n b e r g S. D., Gi bbons A. J, J. Am. Chem. Soc, 79, 2138 (1957).
23. S a i t о w A., R о с h о w E. G., S e у f e r t h D., J. Org. Chem., 23, 116 (1958).
24. Seyferth D., StoneF.G.A, J. Am. Chem. Soc, 79, 515 (1957).
25. NitzscheS., RiedleR., Пат. США 2868820 (1959); С. А., 53, 13057 (1959).
26. Пат. ФРГ 957483 (1957); С. А., 53, 18865 (1959).
27. R о s e n b e r g S. D., G i b b o n s A. J., Пат. США 2873288 (1959); РЖХим.,
1960, 58120.
Дсалкилирование
371
28. М a s s е у A. G., R a n d а 1 1 Е. W., ShawD., Chem. a. Ind. 1963, 1244.
29. G i 1 m a n H., Rosenberg S. D., J. Am. Chem. Soc, 74, 5580 (1952).
30. К о z e s с h к о w K. A., NadjM.M, Alexandrow A.P., Ber., 67, 1348
(1934).
31. GilmanH, MelvinH.W, J. Am. Chem. Soc, 71, 4050 (1949).
32. P a p e t t i S., PostH.W., J. Org. Chem., 22, 526 (1957).
33. К оч еш к о в К. А., НадьМ.М., А л е к с а н д р о в А. П., ЖОХ, 6, 1672
(1936).
34. Reindl Е., Gel ber t Н., Пат. ФРГ 1100630 (1961); РЖХим., 1962, 20Л51.
35. Р е d 1 е у J. В., Skinner H.A., Trans. Faraday Soc, 55, 544 (1959).
36. В r own M. P., FowlesG.W. A., J. Chem. Soc, 1958, 2811.
37. Л а п к и н И. И., Д у м л е р В. А., ЖОХ, 34, 3690 (1964).
38. Lorenz D. Н, S h а р i г о P., S t е г п А., В е с к е г Е. I., J. Org. Chem., 28,
2332 (1963).
39. Johnson О. Н., F г i t z Н. Е., Н а 1 v о г s о n D. О., Evans R. L, J.
Am. Chem. Soc, 77, 5857 (1955).
40. H e с h e n b 1 e i к n e r I., Molt K.R., Пат. США 3059012 (1962); РЖХим.,
1964, 11H97.
41. V a n d e r KerkG.J.M., L u i j t e n J. G. A., J. appl. Chem., 4, 301 (1954).
42. J о h n s о п О. Н., FritzH.E.,J. Org. Chem., 19, 74 (1954).
43. A 1 1 e s t о n D. L., DaviesA.G., J. Chem. Soc, 1962, 2050.
44. V a n der KerkG.J.M, L u i j t e n J. G. A., J. appl. Chem., 7, 369 (1957).
45. К о р ш а к В. В., Р о г о ж и н С. В., Макарова Т. А, Высокомолек. со-
ед., 4, 1297 (1962).
46. LangerH. G., J. Inorg. Nucl. Chem., 21, 297 (1961).
47. Sey f er t h D., Cohen H. M., Inorg. Chem., 2, 652 (1963).
48. Z i m m e r H., S p a r m a n n H. W., Ber., 87, 645 (1954).
49. P а з у в а е в Г. А., Сб. «Синтезы органических соединений», т. 1. М.— Л., Изд-во
АН СССР, 1950, стр. 41.
50. К о ч е ш к о в К. А., НадьМ.М., ЖОХ, 4, 1434 (1934).
51. К о ч е ш к о в К- А., НадьМ.М, ЖОХ, 5, 1158 (1935).
52. К о z e s с h k о w К. А., N a d j М. М, Вег., 67, 717 (1934).
53. Т а л а л а е в а Т. В, ЗайцеваН.А, Кочешков R.A., ЖОХ, 16, 901
(1946).
54. П у ч и н я н Е. А., М а н v л к и н 3. М., ДАН УзбССР, 12, 51 (1961).
55. П и к и н а Е. И., Т а л а л а е в а Т. В., Кочешков К- А., ЖОХ, 8, 1844 (1938).
56. S i s i d о К., TakedaG, KinugawaZ, J. Am. Chem. Soc, 83, 538 (1961).
57. P о 1 1 e r R. C, J. Chem. Soc, 1963, 706.
58. D r u с e J. G. F., J. Chem. Soc, 119, 758 (1921).
59. D r u с e J. G. F., J. Chem. Soc, 121, 1859 (1922).
59a. L a n g e r H. G., Tetrahedron L., 1967, 43.
60. П а в л о в с к а я М. Е., К о ч е ш к о в К. А., ДАН СССР, 49, 263 (1945).
61. В е a t t i e I. R, D u i 1 t a n G. P. M. J. Chem. Soc, 1963, 1519.
62. KulaM.R, KreiterC.G, I о r b e r t h J., Ber., 97, 1294 (1964).
63. НесмеяновА.Н, К о ч е ш к о в К. А., ЖОХ, 1, 219 (1931).
64. Kozeschkow К. А, N esmejanow A. N, Вег., 64, 628 (1931).
65. Gru t tn er G., KrauseE, Ber., "50, 202 (1917).
66. GriittnerG, KrauseE., Ber., 50,1802(1917).
67. Harada Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 35, 290 (1939); С. А., 33,
5357 (1939).
68. МанулкинЗ. М., ЖОХ, 11, 386 (1941).
69. Abel F. W., Brady D. В., J. Chem. Soc, 1965, 1192.
70. GielenM., N a s i e 1 s k i J., J. Organometal. Chem., 1, 173 (1963).
71. G i e 1 e n M., N a s i e 1 s k i J., Bull. Soc chim. Belg., 71, 601 (1962).
72. BuchmanO, Grosjean M, N a s i e 1 s k i J., Bull. Soc. chim. Belg., 72,
286 (1963).
73. Delhaye A, Nasielski J, PlanchonM-, Bull. Soc chim. Belg., 69r
134 (1960).
74. BuchmanO., GrosjeanM, Nasielski J., Helv. Chim. Acta, 47, 1679
(1964).
75. BuchmanO, GrosjeanM., N a s i e 1 s k i J., Helv. Chim. Acta, 47, 1688
(1964).
76. BuchmanO., GrosjeanM., Nasielski J., Helv. Chim. Acta, 47, 2037
(1964).
77. Bott R.W, EabornC., WatersJ.A, J. Chem. Soc, 1963, 681.
78. EabornC, WatersJ.N, J. Chem. Soc, 1962, 1131.
79. К о ч к и н Д. А., К о т р е л с в В. Н., Кал ин-ина СП, Кузнецова
24*
372
Оловоорганшеские соединения
Г. И., Л айне Л. В., Ч е р в о в а Л. В., Б о р и с о в а А. И., Б о р и с е н-
к о В. В., Высокомолек. соед., 1, 1507(1959).
80. К г a u s С. A., S e s s i о n s W. V., J. Am. Chem. Soc, 47, 2361 (1925).
81. Pollard F. H.,. NicklessG., С о о k e D. J., J. Chromatogr., 17, 472 (1965).
82. G i 1 m a n H., ArntzenCE., J. Org. Chem., 15, 994 (1950).
83. KrauseE, Pohland R, Ber., 57, 532 (1924).
84. Lui j t en J. G. A., van d er Kerk G. J. M., J. appl. Chem., 11, 35 (1961).
85. G r u t t n e r G., Ber., 47, 3257 (1914).
86. S e у f e r t h D., К a h 1 e m N., J. Org. Chem., 25, 809 (I960).
87. M а н у л к и н З. М., ЖОХ, 13, 42 (1943).
88. T и л я е в К- С, М а н у л к и н 3. М., ДАН УзбССР, 3, 45 (1961).
89. М а н у л к и н 3. М., ЖОХ, 14, 1047 (1944).
90. М а н у л к и н 3. М, ЖОХ, 13, 46 (1943).
91. М а н у л к и н 3. М., ЖОХ, 16, 235 (1946).
92. В и 1 1 а г d R. Н., Н о 1 d e n F. R., J. Am. Chem. Soc, 53, 3150 (1931).
93. R о s e n b e r g S. D., D e b г е с z e n i E., Wei nberg E. L, J. Am. Chem.
'Soc, 81, 972 (1959).
94. S e у f e r t h D., J. Am. Chem. Soc, 79, 2133 (1957).
95. S e у f e r t h D., Naturwiss., 44, 34 (1957).
96. KippingF. B.( J. Chem. Soc, 1928, 2365.
97. F r a n k 1 a n d E., Ann., 109, 44 (1859).
98. H а у м о в С. Н., МанулкинЗ.М, ЖОХ, 5, 281 (1935).
99. Якубова Ф. А., Рашкес A.M., КучкаревА. Б., Манулкин З.М.,
ЖОХ, 35, 387 (1965).
100. S m i t h Т. А., К i p p i n g F. S., J. Chem. Soc, 101, 2553 (1912).
101. ТиляевК.С, МанулкинЗ.М., Узб. хим. ж., 3, 47 (1963).
102. Morgu nof f N.. Ann., 144,157,(1868).
103. Cahours A., Ann., 114, 378 (I860).
104. С a h о u r s A., Ann., 122, 60 (1862).
105. LadenburgA., Ber., 4, 17 (1871).
106. P о p e W. J., P e а с h v S. J., Proc Chem. Soc, 16, 42 (1900).
107. РазуваевГ. А., ФетюковаВ., ЖОХ, 21, 1010 (1951).
108. СнегурЛ.Н., МанулкинЗ.М., ЖОХ, 34, 4030 (1964).
109. S е у f e r t h D., R a a b G., В r a n d 1 e К. A., J. Org. Chem., 26, 2934 (1961).
110. Vander KerkG.J.M., L u i j t e n J. G. A., J. appl. Chem., 6, 56 (1956).
111. An d erson H. H., Inorg. Chem., 1, 647(1962).
112. Z\ m m er H., H e с h e n b 1 e i k n e r I., HombergO.A., D a n z i k M.,
J. Org. Chem., 29, 2632 (1964).
113. С h a m b e r s R. D., ClarkH.C, WillisC.J., Canad. J. Chem., 39, 131
(1961).
114. Vi j ar a j a r a gh a v a n K., J. Indian Chem. Soc, 22, 135 (1945).
115. S e у f er t h D.f J. Org. Chem., 22, 1599 (1957).
110. V a n d e г К e r k G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 179 (1959).
! 17. V a n d e r Kerk G.J.M., Noltes J. G., L u i j t e n J. G. A., J. appl
Chem., 7, 356 (1957).
118. Neumann W. P., S о m m e r R., Ann., 675, 10 (1964).
119 К а а б а к Л. В., Т о м и л о в А. П.; ЖОХ, 33, 2808 (1963).
120. Leusink A. J., M a r s m a n J. W., В u d d i n g H. A., Rec trav. chim., 84,
689 (1965).
121. Seyferth D., J. Am Chem. Soc,. 79, 5881 (1957).
122. GilmanH, Ca r t ledge F. K., S i m S. Y., J. Organometal. Chem., 4, 332
(1965).
123. Noltes J. G., L и i j t e n J. G. A., J. appl. Chem., 11, 38 (1961).
124. В os t R. W., В a ker H. R., J. Am. Chem. Soc, 55, 1112 (1933).
125. KrauseE., Ber., 51, 912 (1918).
126. KrauseE., Ber., 53, 185 (1920).
127. KrauseE., WeinbergK, Ber., 62, 2235 (1929).
128. Chambers R. F., Scherer P. C, J.Am. Chem. Soc, 48, 1054 (1926).
129. F и с h s О., Р о s t H., Rec. trav. chim., 78, 566 (1959).
130. N e и m a n n W. P., К б n i g K., Ann., 677, 1 (1964).
131. К и р с h i k E. J., L a n i g a n Т., J. Org. Chem., 27, 3661 (1962).
132. Несмеянов А. Н., Борисов А. Е., Волькенау Н. А., Изв. АН СССР,
OXH, 1956, 162.
133. ФрейдлинаР.Х, Несмеянов А. Н., ДАН СССР, 29, 566 (1940).
134. БрайнинаЭ.М, ФрейдлинаРЛ, Изв. АН СССР, ОХН, 1947, 627.
135. МанулкинЗ.М., Узб. хим. ж., 2, 66 (1960).
136. В a h r G., G е 1 i u s R., Ber., 91, 812 (1958).
137. BahrG., G e 1 i u s R., Ber., 91, 818 (1958).
Деалкилирование
373
138. S t e r n А., В е с к e r E. I., J. Org. Chem., 29, 3221 (1964).
139. P о 1 s t e r R., Ann., 654, 20 (1962).
140. Polster R., Пат. США 1150388 (1964); РЖХим., 1966, 5H100.
141. GruttnerG., К г a u s e E., Ber., 50, 1549 (1917).
142. BajerF.J., PostH.W., J. Org. Chem., 27, 1422 (1962).
143. F r e e d m a n H. H., J. Org. Chem., 27, 2298 (1962).
144. Seyferth D., Saraf i dis C, Evnin А. В., J. Organometal. Chem., 2, 417
(1964).
145. L e s b г е М., В u i s s о n R., Bull. Soc. chim. France, 1957, 1204.
146. Buckton G. В., Ann., 109, 22 (1859).
147. N ea 1 s R. N., J. Org. Chem., 9, 211 (1944).
148. Bullard R. H, Rob'inson W.R', J. Am. Chem., Soc, 49, 1368 (1927).
149. KaeszH.D., Staff ord S.L., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6232
(1960).
150. StoneF.aA., T r e i с h e 1 P. M., Chem. a. Ind., 1960, 837.
151. Бобашинская Ч. С, Кочешков К. А., ЖОХ, 8, 1850 (1938).
152. ChambersR.D., С h i v e r s Т., J. Chem. Soc, 1964, 4782.
153. KaeszH.D., P h i 1 i p p s J. P., S t о n e F. G. A., J. Am. Chem. Soc, 82,
6228 (1960).
154. КотонМ.М, КиселеваТ.М, ЖОХ, 27, 2553 (1957).
155. S i s i d о К., T a k e d a G., NozakiH., J. Org. Chem., 27, 2411 (1962).
156. BuchmanO., Grosjean M., N a s i e 1 s k i J., Helv. Chim. Acta., 47,
1695 (1964).
157. В a h r G. Z. anorgan. Chem., 256, 107 (1948).
158. Gel i us R., Ber., 93, 1759 (1960).
159. КотонМ.М., К и с е л е в а Т. М., 3 а п е в а л о в а Н. П., ЖОХ, 30, 186 (1960).
160. EabornC, WatersJ.A., J. Chem. Soc, 1961, 542.
161. BottR.W., EabornC, Walton D. R.M., J. Organometal. Chem., 2, 154
(1964).
162. BottR.W., EabornC, Greasley P.M., J. Chem.; Soc, 1964, 4804.
163. SisidoK., Takeda Y., J. Org. Chem., 26, 2301 (1961).
164. JohnsonO.H,HoluniJ.R,J. Org. Chem., 23, 738 (1958).
165. IbekweS.D., N e w 1 a n d s M. J., J. Chem. Soc, 1965, 4608.
166. Anderson H. H., Inorg. Chem., 3, 108 (1964).
167. LesbreM., Dupont R., Compt. rend. 78е Congr. Socs. Savantes Paris et depts.
Sect. Sci, 1953, 429; С А., 49, 15730 (1955).
168. LesbreM., Dupont R., Compt. rend., 237, 1700 (1953).
169. SasinR., SasinG.S., J. Org. Chem., 20, 770 (1955).
170. SasinG.S., BorrorA.L, SasinR., J. Org. Chem., 23, 1366 (1958).
171. Земляне к-и й Н. Н., ПановЕ.М., Кочешков К- А., ЖОХ, 32, 291
(1962).
172. ФайзиН. А., СловохотоваН. А., Землянский Н Н., Панов
Е. М., Кочешков К- А., ЖОХ, 33, 2610 (1963).
172а. Henderson А., Н о 1 1 i d а у А. К., J. Organometal. Chem., 4, 377 (1965).
173. Kaesz H. D., S t a f f or d S. L., S t one F. G. A., J. Am. Chem. Soc, 81, 6336(1959).
174. Стерлин Р. Н., Л и В э й - г а н, К н у н я н ц И. Л., ДАН СССР, 140, 137 (1961).
175. К от он М. М., ЖОХ, 26, 3212 (1956).
176. Котон М. М., ЖОХ, 22, 643 (1952).
177. К ото н М. М., Мо с к в и н а Е. П., Ф л о р и н с к и й Ф. С, ЖОХ, 20, 2093
(1950).
178. КотонМ.М., Москвина Е.П., Ф л о р и н с к и й Ф. С, ЖОХ, 19, 1675
(1949).
179. Н е с м е я н о в А. Н., Л v ц е н к о И. Ф., Пономареве В., ДАН СССР,
125, 1073 (1959).
180. Л у ц е н к о И. Ф., Пономарев СВ., ЖОХ, 31, 2025 (1961).
181. ЛуценкоИ. Ф., Б а у к о в Ю. И., ХасаповБ. Н., ЖОХ, 33, 2724 (1963).
182. П о н о м а р е в С. В., Б а у к о в Ю. И., ЛуценкоИ. Ф., ЖОХ, 34. 1938
(1964).
183. П о н о м а р е в С В., ЛуценкоИ. Ф., ЖОХ, 34, 3450 (1964).
184. Seyferth D., J. Org. Chem., 22, 478 (1957).
185. Кочешков К. А., Н е с м е я н о в А. Н., ЖОХ, 4, 1102 (1934'..
186. NesmejanowA.N., Kozeschkow К. А., Вег., 67, 317 (1934).
186а. Hashimoto H., MorimotoY., J. Organometal. Chem., 8, 271 (1Э67).
187. H e с м е я н о в А. Н., Б о р и с о в А. Е., Новикова Н.В., Изв. ЛИ СССР,
ОХН, 1959, 259.
188. KrauseE., RenwanzG., Ber., 65, 777 (1932).
374
Оловоорганшеские соединения
189. Несмеянов А. Н., Борисов А. Е., Абрамова А. Н., Изв. АН СССР,
ОХН, 1946, 647.
190. Несмеянов А. Н., Борисов А. Е., Абрамова А. Н., Изв. АН СССР,
ОХН, 1949, 570.
191. Н е с м е я н о в А. Н., Борисов А. Е., Н о в и к о в а Н. В., Изв. АН СССР,
ОХН, 1959, 1216.
192. Н е с м е я н о в А. Н., Б о р и с о в А. Е., Новикова Н. В., ДАН СССР, 119,
504 (1958).
193. TagliaviniG., Cattalini L., Belluco U., Rec. Sci. Rend., Sez A2,
286 (1962).
194. РазуваевГ. А., Федотов M. С, ЗайченкоТ. Н.Дульвинская
H. А., Сборник статей по общей химии, т. 2, М., Изд-во АН СССР, 1953, стр. 1514.
195. Манулкин 3. М., ТатаренкоА. Н., Юсупов Ф. Ю., Труды
Ташкентом, фармацевт, ин-та, 1, 291 (1957).
196. KrauseE, SchmitzM., Ber., 52, 2150 (1919).
197. BermannC, Hartmann H., Z. anorgan. allgem. Chem., 276, 20 (1954).
198. G i 1 m a n H., W о о d s L. A., J. Am. Chem. Soc, 65, 435 (1943).
199. LesbreM., В u i s s о n R., L u i j t e n J. G. A., van d e г К e r k G. J. M.,
Rec. trav. chem., 74, 1056 (1955).
200. Van d e г Ker k G. J.M., Lu i j ten J. G. A., N ol tes J. G., Angew. Chem.,
70, 298 (1958).
201. В u г g А. В., S p i e 1 m a n J. R., J. Am. Chem. Soc, 83, 2667 (1961).
202 S h а г p D. W. A., W i n f i e 1 d J. M., J. Chem. Soc, 1965, 2157.
203. В г i n с k m a n F. E., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6218 (1960).
204. BrinckmanF.E., S t о n e F. G. A., Chem. a. Ind., 1959, 184.
205. S t a f f о г d S. L., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6238 (1960).
206. ChambersR.D., ChiversT., Proc Chem. Soc, 1963, 208.
207. ChambersR.D., ChiversT., J. Chem. Soc, 1965, 3933.
208. GerrardW., MooneyE.F, R e e s R. G., J. Chem. Soc, 1964, 740.
209. Burch J. E., GerrardW., Howarth M., Mooney E.F., J. Am. Chem.
Soc, 79, 5884 (1957).
210. JohnsonW.K, J. Org. Chem., 25, 2253 (1960).
211. M а н у л к и н 3. М., ЖОХ, 18, 299 (1948).
212. Schmidbaur H. Findeiss W.. Angew. Chem., 76, 752 (1964).
213. Б о р и с о в А. Е., Новикова Н. В., Изв. АН СССР, ОХН, 1959, 1670.
214. К о ч е т к о в А. К., Ф р е й д л и н а Р. X., Изв. АН СССР, ОХН, 1950, 203.
215. Шостаковский М. Ф., Соколов Б. А., М а н ц и в о д а Г. П., ЖОХ,
33, 3779 (1963).
216. Пучинян Е. А., Труды Ташкентск. фармацевт, ин-та, 2, 311 (1960).
217. О с и п о в О. А., К а ш и р е н и н о в О. Е., ЖОХ, 32, 1717 (1962).
218. Т г е i с h e I P. M., Goodrich R. A., Inorg. Chem., 4, 1424 (1965).
219. S а с с о A., Atti accad. nazl Lincei, Rend., Classe Sci. fis. mat. e nat., 11, 101 (1951);
С. А,, 49, 158 (1955).
220. MaierL, Seyferth D., S t о n e F. G. A., R о с h о w E. G., J. Am. Chem.
Soc, 79, 5884 (1957).
221. M а н у л к и н 3. М., ЖОХ, 20, 2004 (1950).
222. Пучинян Е. А., Труды Ташкентск. фармацевт, ин-та, 1, 310 (1957).
223. W а 1 d e R. А., Пат. США 3016397 (1962); РЖХим., 1963, 12Н23.
224. Deb land ге С, G i е 1 е п М., N a s i e 1 s k i J., Bull. Soc chim. Belg., 73,
214 (1964).
225. AllestonD.L, DaviesA.G., J. Chem. Soc, 1962, 2465.
226. ШушуновВ.А, Б р и л к и н а Т. Г., ДАН СССР, 141, 1391 (1961).
227. Gilman Н., LewisA. G., J. Org. Chem., 22, 250 (1957).
228. Schumann H., Kopf H., Schmidt M., Z. anorgan. allgem. Chem.,
331, 200 (1964).
229. SchmidtM., Schumann H., Z. Naturforsch., 19b, 74 (1964).
230. SchmidtM., D e r s i n H. J., Schumann H., Ber., 95, 1428 (1962).
231. Schumann H., SchmidtM. Ber., 96, 3017 (1963).
232. SchmidtM., Schumann H. Ber., 96, 780 (1963).
233. SchmidtM., Schumann H., Ber., 96, 462 (1963).
234. Schumann H., KopfM., SchmidtM., Angew. Chem., 75, 672 (1963).
235. S с h u m a n n H., К б p f M., S с h m i d t M., Ber., 97, 1458 (1964).
236. P а з у в а е в Г. А., КотонМ.М, ЖОХ, 5, 361 (1935).
237. Van d e г К e r k G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 113 (1959).
238. Zimmer H., LiibkeK., Ber., 85, 1119 (1952).
239. V a n d e г К e r k G. J. M., N о 1 t e s J. G., Tin and its uses, 37, 10 (1956).
240. Seyferth D., Brandle K-, R a a b G., Angew. Chem., 72, 77 (1960).
Деалкилирование
375
241. HartmannH., HonigH., Angew. Chem., 70, 75 (1958).
242. HartmannH., KarbsteinB., Schaper P., Reiss W., Naturwiss.,
50, 373 (1963).
243. HartmannH., Dietz E., Komorniczyk K-, Reiss W.,
Naturwiss., 48, 570 (1961).
244. L e QuanM., BillioteJ. -С, С a d i о t P., Compt. rend., 251, 730 (I960).
245. Петров А. А., Завгородний В. С, ЖОХ, 30, 1055 (1960).
246. Т а ц у о К., J. Chem. Soc. Japan, Pure Chem. Sec, 83, 727 (1962); РЖХим., 1963,
16Ж254.
247. GilmanH., GistLA., J. Org. Chem., 22, 250 (1957).
248. TamborskiC, SoloskiE. J., DecS. M., J. Organometal. Chem., 4, 446
(1965).
249. WaringCE., Horton W.S., J. Am. Chem. Soc, 67, 540 (1945).
250. Sathyamurthy T. W., SwaminathanS., Yeddanapalli L. M.,
J. Indian. Chem. Soc, 27, 509 (1950).
251. Long L. H., J. Chem. Soc, 1956, 3410.
252. PriceS. J. W., Trotma n-D ickenson A. F., Trans. Faraday Soc, 54, 1630
(1957).
253. P а з у в а е в Г. А., Ж- Всесоюзн. хим. общ. им. Д. И. Менделеева, 7, 325 (1962).
254. Razuvaev G. A., Vyasankin N. S., Shshepetkova О. A.,
Tetrahedron., 18, 667 (1962).
255. ВязанкинН. С, РазуваевГ. А., Дергунов Ю. И., Щепетко-
ва О. А., Труды по химии и хим. технологии (Горький), 1961, 150.
256. Рабиновичи. Б., Тельной В. И., Николаев П. Н., Разуваев
Г. А., ДАН СССР, 138, 852 (1961).
257. ВязанкинН. С, РазуваевГ. А., Дергунов Ю. И., Щепетко-
в а О. А., Труды по химии и хим. технологии (Горький), 1961, 58.
258. Разуваев Г. А., ВязанкинН. С, ГладышевЕ. Н., Бородавко
И. А., ЖОХ, 32, 2154 (1962).
259. РазуваевГ. А., ВязанкинН. С, Дергунов Ю. И., Гладышев
Е. Н., Изв. АН СССР, ОХН, 1964, 848.
260. Разуваев Г. А., Дергунов Ю. И., В я з а н к и н Н. С, ДАН СССР, 145,
347 (1962).
261. РазуваевГ. А., ВязанкинН. С, Дергунов Ю. И.,
Дьячковская О. С, ДАН СССР, 132, 364 (1960).
262. РазуваевГ. А., Вязанкин Н. С, Дьячковская О. С, Киселева
И. Г., Д е р г у н о в Ю. И., ЖОХ, 31, 4056 (1961).
263. Da о-Н uy-G ia о, Compt. rend., 260, 6937 (1965).
264. KaeszH.D., P h i 1 i p p s J. P., StoneF.G.A., Chem. a. Ind., 1959, 1409.
265. LesbreM., Roques G., Compt. rend 78е Congr. Socs. savantes Paris et depts.,
Sect. Sci., 1953, 423; С. А., 49, 15768 (1955).
266. Ольдекоп Ю. А., Соколова Р.Ф., ЖОХ, 23, 1159 (1953).
267. О л ь д е к о п Ю. А., М о р ы г а н о в Б. Н., ЖОХ, 23, 2020 (1953).
268. Разуваев Г. А., Фетюкова В. В., Шубенко М. А., Уч. зап. Горьк.
ун-та, 15, 91 (1949).
269. РазуваевГ. А., ВязанкинН. С, Дьячковская О. С, ЖОХ, 32,
2161 (1962).
270. РазуваевГ. А., Дьячковская О. С, ВязанкинН. С,
Щепетко в а О. А., ДАН СССР, 137, 618 (1961).
271. РазуваевГ. А., ВязанкинН. С., Сб. «Химия перекисных соединений».
М., Изд-во АН СССР, 1963, стр. 283.
272. ВязанкинН. С, РазуваевГ. А., БревноваТ. Н., ЖОХ, . 34, 1005
(1964).
273. ВязанкинН. С, РазуваевГ. А., БревноваТ. Н., ЖОХ, 35, 2033
(1965).
274. ВязанкинН. С, РазуваевГ. А., Кругла я-Щ епеткова О. А.,
Дьячковская О. С, Сб. «Химия перекисных соединений». М., Изд-во АН СССР,
1963, стр. 298.
275. ВязанкинН. С, РазуваевГ. А., Дьячковская О. С, Щепет-
кова О. А., ДАН СССР, 143, 1348 (1962).
276. С к о л д и н о в А. П., К о ч е ш к о в К-А., ЖОХ, 12, 398 (1942).
277. Малиновский М. С, Труды Горьк. пед. ин-та, 1940, 39.
278. GerschbeinL L, I p a t i e f f V. N.. J. Am. Chem. Soc, 74, 1450 (1952).
279. КотонМ.М, ЖОХ, 2, 345 (1932).
280. KotonM. M., Ber., 66, 1213 (1933).
281. КотонМ.М., ЖОХ, 4, 653 (1934).
282. A d d i s о n С. С, SimpsonW.B, W a 1 k er A., J. Chem. Soc, 1964. 2360
283. Макарова Л. Г., Несмеянов А. Н., ЖОХ, 9, 771 (1939).
284. К i n g R. В., S t о n e F. G. A., J. Am. Chem. Soc, 82, 3833 (1960).
Г лава IX
ДИСПРОПОРЦИОНИРОВАНИЕ.
ПЕРЕРАСПРЕДЕЛЕНИЕ РАДИКАЛОВ
Под диспропорционированием подразумеваются здесь процессы,
происходящие под влиянием нагревания часто в присутствии восстановите-
телей (реактив Гриньяра, амальгама натрия, сплавы олова с натрием,
металлическое олово, гидразингидрат и др.) и приводящие к тому, что
часть атомов олова обогащается радикалами, часть же атомов теряет их.
Так, диэтилолово распадается во время перегонки на металлическое
олово и тетраэтилолово [1]. При 100° С в присутствии 2—3 вес. % А1Вг3
диэтилолово диспропорционирует за 5 час. При этом возникают
тетраэтилолово и металлическое олово в соотношениях, точно отвечающих
уравнению [2]
2 (C2H5)2Sn -* (C2H5)4Sn + Sn.
По данным Неймана с сотр. [3] при диспропорционировании диэтил-
олова в мягких условиях образуются окрашенные соединения типа
(C2H5)3Sn [Sn(C2H5)2]nSn (C2H5)3 или [(C2H5)3Sn]2Sn (C2H5) [Sn(C2HB)2lw
Sn (С2Н5)3- Чем более разветвлена цепь из атомов олова в молекуле, тем
глубже окраска соответствующих соединений. Соли металлов, например
хлорное железо, олово или алюминий, а также бромистый этилмагний и
трибутоксибор действуют как катализаторы [3]. Окрашенные масла того
же типа образуются и при диспропорционировании
гексаэтилдистаннана. Проходящие при этом процессы подробно изучены Разуваевым с
сотр. [4—8]. Чистый гексаэтилдистаннан не изменяется при нагревании
до 235° С в течение 10 час. Разложение вещества наблюдается лишь при
260 С. На основании изучения электронных спектров показано, что
распад гексаэтилдистаннана может быть изображен следующим образом:
(QHsbSnSn (С2Ы5)3 -* (C2H5)4Sn + (C2H5)2Sn,
(C2H5)2Sn ->JSn + 2 C2H,'
Более энергично диспропорционирование проходит в присутствии
бромистого триэтилолова — слабого катализатора реакции
перераспределения радикалов [7]. При термостатировании смеси гексаэтилдистаннана
и хлористого триэтилолоза при 200—210° С в течение 10 час. наблюдается
выделение металлического олова и тетраэтилолова. Еще энергичнее
(1 час при 200° С) диспропорционирование проходит в присутствии 3
молей двухлористого диэтилолова и 2 молей гексаэтилдистаннана.
Возникающее тетраэтилолово реагирует с двухлористым диэтилоловом, в
результате чего суммарное уравнение реакции приобретает следующий вид:
2 (C2H5)3SnSn;(C2H,)3 + 3 (QHobSnCb-> 6 (QsHsfoSnClJK- Sn.
Диспропорционирование
377
Следует подчеркнуть, что это уравнение описывает лишь конечный
результат. Механизм реакции, по-видимому, сложен [7].
Гораздо быстрее диспропорционирование проходит в присутствии
солей алюминия [4, 5]. Проведение процесса в запаянных эвакуированных
ампулах, соединенных с ртутным манометром, показывает, что реакция не
сопровождается газообразованием и может быть описана уравнением
2 (C2Hr))3SnSn(C2H5)3 —я-> 3 (C2H5)4Sn + Sn.
При проведении реакции в мягких температурных условиях (70—75° С)
образование промежуточных веществ можно констатировать по
появлению интенсивной вишневой окраски и последующему расслоению
реакционной смеси. Анализ верхнего бесцветного слоя показывает, что он
состоит в основном из тетраэтилолова. Анализ отслоившейся тяжелой
вязкой окрашенной в темно-красный цвет жидкости свидетельствует о
том, что она по составу соответствует диэтилолову, содержащему
некоторое количество катализатора. Анализом на хлор показано равномерное
распределение катализатора в верхнем и нижнем слоях.
В конечном результате единственными продуктами диспропорциони-
рования гексаэтилдистаннана являются олово и тетраэтилолово [4,5].
Аналогично проходит диспропорционирование органостаннанов типа
(C2H5)3Sn [Sn(C2H5)2]nSn (С2Н5)3 и других оловоорганических
соединений, содержащих разветвленную цепь из атомов олова. Образование в
результате этих реакций тетраэтилолова может служить качественной
реакцией на связь Sn—Sn [9].
В отсутствие катализатора гексаметилдистаннан достаточно
устойчив к диспропорционированию [10]. При нагревании до 100° С в течение
38 час. вещество остается практически без изменения. Однако в
присутствии трехфтористого бора оно диспропорционирует уже при комнатной
температуре
(CHsfoSnSnfCHsb — - (CH3)4Sn;+-[(CH3)2Sn]/2.
В свою очередь образовавшееся диметилолово диспропорционирует в
гораздо менее метилированное соединение состава [(CH3)2Sn5lx- Более
слабыми катализаторами диспропорционирования гексаметилдистаннана
являются диборан и фтористый диметилбор [10].
При действии хлористого водорода на раствор гексаметилдистаннана
в метиловом спирте при 25° С в присутствии треххлористого золота
образуется тетраметилолово и [(CH3)2Sn]/7 [ 11]. В отсутствие хлористого
водорода реакция проходит иначе и приводит к смеси хлористого триметил-
олова и двухлористого диметилолова.
Дифенилолово в присутствии реактива Гриньяра при температуре
около 100° С претерпевает следующее диспропорционирование [12]:
3 (C645)2Sn -V(QH5)3SnSn (C6H5)3 + Sn.
Следует заметить, что с более сложными радикалами этот процесс не
идет. В результате диспропорционирования дифенилолова может
образоваться также тетрафенилолово и додекафенилпентастаннан [13].
В тетрагидрофуране дифенилолово диспропорционирует уже при
комнатной температуре [14]. Нагретый до кипения эфирный раствор
бромистого фенилмагния не реагирует в заметной степени с додекафенилцик-
логексастаннаном. При обработке последнего мелкодисперсным литием в
тетрагидрофуране выделяется металлическое олово и образуется
тетрафенилолово [14]. Действие лития на двухлористое дифенилолово в нагре-
378 Оловоорганические соединения. Лит. стр. 379—380
том до кипения тетрагидрофуране приводит к тетрафенилолову,
элементарному олову и октафенилтристаннану [15!.
При нагревании до 160°С порошка олова (цинка,, алюминия или
железа) и двухлористого диэтил-, дипропил- или дибутилолова с водой с
хорошим выходом образуются хлориды триалкилолова [15а]. В среде
метилового спирта или тетрагидрофурана выход значительно ниже. В
отсутствие олова или при понижении температуры до 100° С этот процесс
вообще не имеет места. Дихлориды диалкилолова в присутствии
металлического олова превращаются с хорошим выходом в Alk3SnCl и в том
случае, если диспропорционирование проводить в смеси хлористого алкила
с mpera-амином. Замена указанных металлов цинком, алюминием,
магнием или натрием приводит к смеси AlkoSnCl и A!k4Sn. Нагревание до
160° С хлористого трибутилолова с цинком и триэтиламином дает тетра-
бутилолово с примесью гексабутилдистаннана. В тех же условиях из
хлористого триэтилолова и хлористого бутила получают тетраэтилолово
(38 %), триэтилбутилолово (20 %), диэтилдибутилолово (4 %), этилтри-
бутилолово (3 %) и тетрабутилолово (2 %). Рассмотрена возможная
схема этих проиессов [15а].
Диспропорционирование типа
3 (C6H5)2SnCl2 -> 2 (C6H5)3SnCl + SnCl4
в присутствии амальгамы натрия [16] или азотистой кислоты (выход
незначительный) наблюдал Аронгейм [17]. Невелики и выходы
гидроокиси трифенилолова при действии гидразин-сульфата в присутствии
соды в спиртовой среде на двухиодистое дифенилолово [18]. Вследствие
лучших выходов препаративный интерес представляет нагревание ди-
фенилолова со сплавом олово — натрий [19].
Гидроокиси триалкил- [20—25] и триарилолова [26—29] легко дис-
пропорционируют до (RaSnO)^ при нагревании.
Получение окиси дифенилолова из гидроокиси трифенилолова [28]. 40 г (0,0558 моля)
гексафенилдистанноксана кипятят при перемешивании в 300 мл ксилола в течение 2 дней.
Образовавшийся белый осадок отфильтровывают, промывают 5 раз и высушивают. Получают
15,0 г (98%) окиси дифенилолова ст. пл. 370—2° С. Вместо ксилола можно применять спирт
или ацетонитрил [29].
К реакции диспропорционирования можно отнести и превращение ал-
килстанноновых кислот при нагревании со щелочью в окиси диалкилолова,
гидроокиси триалкилолова и оловянную кислоту [30—35].
Превращение этилстанноновой кислоты в окись диэтилолова [32]. 5 г этилстанноновой
кислоты постепенно прибавляют к 100 мл кипящего 10%-ного водного раствора едкого
натра. Осадок отсасывают, промывают водой до отсутствия щелочной реакции и высушивают.
Гидроокись триэтилолова получена при перегонке в вакууме окиси
диэтилолова [22]. При термическом разложении (145° С/15 мм, 1 час) ди-
гидроокиси ди-(о-феноксифенилолова) выделен гекса-о-феноксифенил-
дистанноксан и небольшое количество вещества, анализ которого
соответствует о-феноксифенилстанноновой кислоте. Более подробно это
соединение не исследовалось [36].
Нагревание с гидразинсульфатом в присутствии соды в спиртовой
среде превращает йодистое трифенилолово (57 час. кипячения, выход 2,9%)
и хлористое трифенилолово (48 час. кипячения, выход 6%) в тетрафе-
нилолово [18]. Со значительно лучшим выходом (37%) протекает такого
рода диспропорционирование со сплавом олово — натрий [19].
Нагревание смесей тетраалкильных производных олова в
присутствии небольшого количества (2—3%) катализатора, подобного хлори-
Диспропор ц ион ирован ие
379
стому алюминию, в растворителе (например, пентане) или без него до
невысокой температуры (например, 60° С) в течение нескольких часов ведет
к перераспределению радикалов, так что образуются все возможные
комбинации тетраалкильных производных олова. Так, например,
эквимолекулярная смесь тетраэтилолова и тетраметилолова в результате такого
перераспределения образует равновесную смесь, содержащую 8,4 мол.%
тетраэтилолова, 28,6% метилтриэтилолова, 38,4% диэтилдиметилилова
и 24,6% смеси тетраметилолова и триметилэтилолова [34, 35].
Облучение у-лучами диметилдиэтилолова приводит к смеси
метилтриэтилолова и триметилэтилолова [37].
Реакция перераспределения радикалов описана и в патентной
литературе [38, 39]. Так, при нагревании до 190° С в присутствии треххло-
ристого алюминия смеси тетрабутил- и тетрафенилолова получены [38]
трибутилфенилолово, дибутилдифенилолово и бутилтрифенилолово.
Аналогичная картина наблюдается и при нагревании в тех же условиях
смеси тетраэтил- и тетраизобутилолова, а также тетрабутил- и тет-
раамилолова.
При перегонке трифенил-я-гептилолова и трифенил-я-нонилолова
происходит частичное перераспределение радикалов [40]. Вероятно,
при этом образуется тетраалкилолово и тетрафенилолово. Подобный
процесс наблюдается и при перегонке йодистого дивинилфенилолова
141]. При этом образовавшееся на первой стадии реакции вещество дис-
пропорционирует, превращаясь в смесь йодистого тривинилолова и
йодистого трифенилолова:
2 (СвН5) (CH2=CH)2SnJ -> 2 (CH2=CII)3SnJ + (C6H5bSnJ:J
Получение йодистого тривинилолова [41]. К раствору 50 г (0,153 моля) дивинилдифенил-
олова в 400 мл эфира прибавляют 38,8 г (0,153 моля) иода. Смесь кипятят в течение 8 час.
Эфир отгоняют и остаток перегоняют в вакууме. Получают 27,5 г иодбензола и 28,2 г
йодистого тривинилолова, т. кип. 54—60° С/1,3—0,7 мм. Твердый остаток представляет собой
йодистое трифенилолово. Повторная перегонка второй фракции дает 25 г чистого йодистого
тривинилолова с т. кип. 60—62° С/1,8 мм.
Аналогично ведет себя при нагревании бромистое дивинилфенилолово.
Хлористое ди-я-бутилвинилолово перегоняется в вакууме без всяких
изменений [41].
При нагревании винилтрифенилолова в бромбензоле вещество
медленно диспропорционирует с образованием тетрафенилолова [42].
Аллильные производные олова устойчивы до 170° С и лишь при
длительном нагревании при 170° С часть диаллилдифенилолова
превращается в тетрафенил- и тетрааллилолово. Последнее разлагается до моно-
аллильного производного олова, двуокиси олова и диаллила.
По своей термической устойчивости аллильные производные олова
образуют ряд: аллилтрифенилолово ^> диаллилдифенилолово ^>
тетрааллилолово [43].
ЛИТЕРАТУРА
1. Krause E., von Grosse A., Die Chemie der metallorganischen Verbindungen.
Berlin, Gebr. Borutrager, 1937, S. 354.
2. В я з а н к и н Н. С, Р а з у в а е в Г. А., К о р н е в а С. П., ЖОХ, 33, 1041 (1963).
3. NeumannW.P, Pedain J., Ann., 672, 34 (1964).
4. P а з у в а е в Г. А., ВязанкинН. С, Дергунов Ю. И.,
Дьячковская О. С, ДАН СССР, 132, 364 (1960).
5. Разуваев Г. А., Вязанкин Н. С, Дергунов Ю. И., Вышинский
Н. Н., ЖОХ, 31, 1712 (1961).
6. Р а з у в а е в Г. А., В я з а н к и н Н. С, Щ е п е т к о в а О. А., ЖОХ, 31, 3762
(1961).
380
Оловоорганические соединения
7. Р а з v в а е в Г. А., Д е р г у н о в Ю. И., В я з а н к и н Н. С, ЖОХ, 32, 2515
(1962)/
8. Вязанкин Н. С, Круглая О. ГА., Дергунов Ю. И., Разуваев
Г. А. Сб. «Каталитические реакции в жидкой фазе». Алма-Ата, Изд-во АН КазССР,
1963, стр. 416.
9. В я з а н к и н Н. С, Р а з у в а е в Г. А., Круглая О.А., Изв. АН_ СССР,
ОХН, 1962, 2008.
10. В u r g А. В., S p i е 1 m a n J. R., J. Am. Chem. Soc, 83, 2667 (1961).
11. TagliaviniG., BellucoU., PilloniG., Ricerca scient., 3, Parte 2,
Sez. A., 889 (1962).
12. KrauseE., BeckerR., Ber., 53, 173 (1920).
13. В о e s e k e, n J., Rutgers J.J., Rec. trav. chim., 42, 1017 (1923).
14. N e u m a n n W. P., К б n i g K., Ann., 677, 1 (1964).
15. N e u m a n n W. P., К 6 n i g K., Ann., 677, 12 (1964).
15a. Sisido K., Kozima S., J. Organometal. Chem., 11, 503(1968).
16. Aronheim В., Ann., 194, 171 (1878).
17. Ar onheim В., Ber., 12, 509 (1879).
18. G i 1 m a n H., Barnet t M.M., Rec trav. chim., 55, 563 (1936).
19. НадьМ.М., Кочешков К. А., ЖОХ, 8, 42 (1938).
20. К r a u s С. A., Bu-llard R. H., J. Am. Chem. Soc, 51, 3605 (1929).
21. Schmitz-DuMontO., Z. anorgan. Chem., 248, 289 (1941).
22. H a r a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 36, 497 (1939).
23. С a h о u r s A., Ann., 114, 355 (1860).
24. H a r a d а Т., Sci. Papers Inst. Phys., Chem. Res. (Tokyo), 36, 501 (1939).
25. К о ч к и н Д. А., Котрелев В. Н., Калинина С. П., Кузнецова
Г. И., ЛайнеЛ. В., ЧервоваЛ. В., Борисова А. И., Борисенко
В. В., Высокомолек. соед., 1, 1507 (1959).
26. Chambers R. F., Scherer Р.С., J. Am. Chem. Soc, 48, 1054 (1926).
27. R e i с h 1 e W. Т., J. Org. Chem., 26, 4634 (1961).
28. R e i с h 1 e W. Т., J. Polymer Sci., 49, 521 (1961).
29. KupchikE.J., L a n i g a n Т., J. Org. Chem., 27, 3661 (1962).
30. P f e i f f e r P., Ber., 35, 3305 (1902).
31. Pfeiffer P., Lehnhardt R., Ber., 36, 1054 (1903).
32. DruceJ.G. F., J. Chem. Soc, 119, 758 (1926).
33. DruceJ.G. F., J. Chem. Soc, 121, 1859 (1922).
34. С a 1 1 i n g a e r t G., В e a t t у H. A., N e a 1 H. R., J. Am. Chem. Soc, 61, 2755
(1939).
35. Ca 1 1 i nga er t G., S or о os H.,. H n i z d a V., J. Am. Chem. Soc, 62, 1107
(1940).
36. Poller R. C, J. Chem. Soc, 1963, 706.
37. Prosch U., Z о p f 1 H. J., Z. Chem., 3, 97 (1963).
38. JohnsonE.W., ChurchJ. M., Пат. США 2599557 (1952); С. А., 47, 1728
(1953).
39. Johnson E. W., С h u г с h J. M., Пат. США 2608567 (1952); С. А., 47, 8089 (1953).
40. С н е г у р Л. П., Манулкин 3. М., Узб. хим. ж., 1961, 49.
41. S е у f e r t h D., J. Am. Chem. Soc, 79, 2133 (1957).
42. R о t h m a n L. А., В е с k e r E. I., J. Org. Chem., 25, 2203 (1960).
43. К о т о н М. М., К и с е л е в а Т. М., ЖОХ, 27, 2553 (1957).
Глава X
ГИДРОЛИЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Гидролитическая устойчивость связи олова с различными
функциональными группами существенно зависит от их природы. По отношению к
гидролизу водой наиболее устойчивы оловоорганические галогениды. Аци-
латы алкил- и арилолова гидролизуются в более мягких условиях.
Например, при гидролизе (хлор)ацетата диметил- [1] или (бром)ацетата ди-я-бу-
тилолова [2] остаток уксусной кислоты количественно заменяется на гид-
роксил, в то время как атом галоида остается незатронутым. Еще легче
проходит гидролиз станниламинов, алкокси-, перекисных и некоторых
других типов оловоорганических соединений, при работе с которыми должна
быть исключена влага воздуха.
Большое влияние на легкость гидролиза оказывают число и характер
органических радикалов, связанных с атомом олова. Галогениды триал-
килолова с водой практически не реагируют. Так, хлористое триметилоло-
во легко растворяется в воде. Образования гидроокиси триметилолова не
происходит, даже если раствор хранить в течение недели при комнатной
температуре [31. При нагревании водных или водно-спиртовых растворов
дигалогенидов диалкилолова образуются основные соли R2Sn (OH)X [4—6].
Еще легче гидролизуются соединения типа RSnCl3 [7—10].
Для препаративных целей важное значение имеют продукты
исчерпывающего гидролиза, которые обычно получают действием водных
растворов щелочи или аммиака на оловоорганические галогениды. Образующиеся
при этом гидроокиси являются неустойчивыми соединениями и легко
дегидратируются. По-видимому, скорость дегидратации увеличивается при
переходе от соединений типа R3SnX к RSnX3. Известны некоторые
простейшие гидроокиси триалкил- и триарилолова (см. стр. 382). Дигидроокиси
диалкилолова образуются лишь в отдельных случаях (третичный
алифатический радикал у атома олова). Тригидроокиси алкил- или арилолова не
получены. Как правило, гидролиз соединений типа R2SnX2 и RSnX3
сопровождается гидролитической поликонденсацией с образованием, в
конечном счете, смеси полимерных продуктов:
п R2SnX2 + 2/i NaOH -* (R2SnO)„ + 2/i NaX + n H20,
n RSnX3 + 3/z NaOH -»(RSnOOH)„ + 3/i NaX + n H20.
Гидроокиси триалкилолова или окиси R3SnOSnR3 представляют собой
низкоплавкие кристаллические вещества или жидкости, хорошо
растворимые в обычных органических растворителях. Окиси диалкилолова —
белые аморфные вещества, не растворимые в органических растворителях и
щелочах. Алкилстанноновые кислоты растворяются в спирте и
концентрированных щелочах. Из щелочного раствора они легко выделяются при
382 Оловоорганические соединения. Лит. стр. 394—395
прибавлении кислоты или пропускании углекислого газа. Различную
растворимость продуктов гидролиза предложено [11, 12] использовать для раз-
деления смеси соединений олова различной степени алкилирования.
Следует отметить, что в некоторых случаях реакция гидролиза
сопровождается отщеплением органических радикалов, связанных с атомом
олова (см. стр. 363).
ГИДРОЛИЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ КЛАССА R3SnX
Как уже было отмечено ранее, галогениды триалкилолова устойчивы
к действию воды, но легко гидролизуются водными растворами щелочи или
аммиака [13—24] (в редких случаях применяют влажную окись серебра).
Образующиеся гидроокиси триалкилолова, кроме гидроокиси триметилоло-
ва, являются неустойчивыми соединениями и легко дегидратируются,
превращаясь в гексаалкилдистанноксаны (окиси гексаалкилдистаннанов):
2 RgSnOH -> R3SnOSnR3 + Н20.
Скорость этой реакции зависит от характера органического радикала,
связанного с атомом олова. Гидроокись триметил олова отщепляет воду
только при действии металлического натрия [20, 21]. Гексаэтил- и гекса-
пропилдистанноксаны образуются при высушивании соответствующих
гидроокисей над концентрированной серной кислотой в вакууме.
Дегидратация гидроокисей трибутил-, триизобутилолова и высших алкильных
соединений олова происходит, по-видимому, в процессе гидролиза. Возможна
и обратная реакция. Так, при прибавлении к гексаметил-, гексаэтил- и
гексапропилдистанноксанам воды образуются соответствующие гидроокиси
[22, 23]. Высшие члены ряда с водой не реагируют.
Гидроокись триметилолова хорошо растворяется в воде и выделить ее из
водных растворов экстрагированием органическими растворителями не
удается. При ее получении используют плохую растворимость вещества в
концентрированном растворе хлористого натрия.
Получение гидроокиси триметилолова [25]. К раствору 15,95 г (0,08 моля) хлористого
триметилолова в 10 мл воды при перемешивании прибавляют по каплям 3,2 г (0,08 моля)
едкого натра в 10 мл воды. Смесь перемешивают в течение 2 час, осадок отделяют и
высушивают. Вещество экстрагируют петролейным эфиром (т. кип. 40—60° С) в течение 6 час.
Экстракт отфильтровывают и выпаривают. Остаток представляет собой смесь гидроокиси
триметилолова и гексаметилдистанноксана. К нему прибавляют несколько капель воды,
образующуюся массу измельчают и высушивают в вакуум-эксикаторе над хлористым кальцием.
Выход 11,95 г (82,75%); т. пл. 118—119° С (в запаянном капилляре).
Получение гексаметилдистанноксана [26]. Синтез и очистку вещества проводят при
полном отсутствии влаги воздуха. Применяемую посуду и бензол тщательно высушивают. К
раствору 30 г гидроокиси триметилолова в 100 мл горячего бензола прибавляют небольшими
кусочками 2,5 г натрия. После того как прекратится выделение водорода, смесь охлаждают.
Раствор сливают с небольшого количества вязкой жидкости, бензол отгоняют и остаток
перегоняют в вакууме. Получают 25 г (78%) гексаметилдистанноксана с т. кип. 84—80° С/22—
18 мм.
Аналоги хлористого триметилолова плохо растворяются в воде. Поэтому
гидролиз проводят, встряхивая при комнатной температуре эфирный
раствор оловоорганического галогенида с рассчитанными или несколько
большим количеством 30—50%-ного раствора щелочи. Чем далее находится
оловоорганическое соединение в гомологическом ряду, тем хуже его
растворимость в воде, тем более длительное время требуется для завершения
реакции [23]. Часто операцию повторяют с половинным объемом щелочи.
Получение гидроокиси триэтилолова [23]. Раствор 407 г хлористого триэтилолова в
2,5 г эфира встряхивают в делительной воронке со 110 г (25% избытка) едкого кали в 260 мл
воды. Органический слой отделяют и встряхивают с раствором 50 г щелочи в 140 мл воды.
Гидролиз
38а
Эфирный слой фильтруют и эфир отгоняют. Из остатка, представляющего собой смесь
гидроокиси триэтилолова и гексаэтилдистанноксана, удаляют остатки эфира, помещая его ь
вакуум-эксикатор над концентрированной серной кислотой. Одновременно вся имеющаяся
гидроокись триэтилолова превращается в гексаэтилдистанноксан.- При прибавлении
рассчитанного количества воды (15 мл) происходит экзотермическая реакция с регенерацией
исходной гидроокиси. Ее выдерживают над 50%-ным раствором едкого кали. Выход 363 г
(96%); т. пл. 49—50° С.
Аналогично проводят гидролиз хлористого три-^-пропилолова [23].
Продукт реакции представляет собой бесцветное масло, частично
кристаллизующееся при хранении. Кристаллы гидроокиси отфильтровывают и
отжимают между листами фильтровальной бумаги. Т. пл. 34—35° С.
Фильтрат представляет собой гекса-^-пропилдистанноксан.
Подобно этому получены гексаизопропил-, гекса-^-бутил-, гекса-^-гек-
сил- и гекса-я-октилдистанноксаны [23]. Для получения значительных
количеств гекса-я-бутилдистанноксана удобно пользоваться следующей
методикой.
Получение гекса-#-бутилдистанноксана [23]. В трехгорлую колбу (2 л), снабженную
мешалкой, обратным холодильником и капельной воронкой, помещают 119 г (17% избытка)
едкого кали в 509 мл воды. Раствор нагревают до 90—95° С и при этой температуре
прибавляют в течение 15 мин. при энергичном перемешивании 823 г хлористого три-я-бутилолова.
Смесь перемешивают 30 мин. при той же температуре, охлаждают и центрифугируют.
Верхний слой фильтруют. Выход чистого вещества 662 г (88%); т. кип. 186,5—187° С/3 мм;
п^01,4862 [24].)
Для получения больших количеств гексаалкилдистанноксанов
предложено [27] обрабатывать галогениды триалкилолова твердой щелочью при
70—110° С (желательно под давлением). Выходы продуктов составляют
75—85%.
Гексаалкилдистанноксаны с различными радикалами у атома олова,
например R2R'SnOSnR'R2, изучены недостаточно. В литературе имеются
сведения о синтезе этих соединений [23, 28—33], но физические константы
приведены [34] лишь для отдельных представителей этого типа веществ:
ошж-диметилтетра-я-амилдистанноксан (т. кип. 170—172° С/1 мм,
d|°l ,193; п2® 1,4855); шжж-тетраметилди-я-амилдистанноксан (т. кип.
107— 109° С/1 мм; d%"1,317; лЬ 1,4867); шлш-диметилтетра-н-бутилдистан-
ноксан (т. кип. 135—137° С/1 мм; df 1,251; я# 1,4868) и некоторых других.
В большинстве случаев образование этих соединений доказывалось
косвенным путем. Например, шлш-тетраэтилди-я-бутил-, шлш-тетраэтилди-я-
октил-, шлш-тетраметилди-я-бутилдистанноксан и другие получены [28}
с выходом порядка 70% встряхиванием эфирных растворов соответствующих
иодидовс20%-ным раствором едкого кали (теоретическое количество, а
затем половина). Эфирный раствор высушивают сульфатом натрия и эфир
отгоняют. Остатки растворителя удаляют, помещая вещество в вакуум-
эксикатор над концентрированной серной кислотой. В остатке получают
оловоорганические окиси типа (R2R'Sn)20 в виде прозрачного масла,
которое обработкой уксусной кислотой превращают в соответствующие ацетаты
[31].
Получить гидроокись ди-я-бутилвинилолова не удается, так как
вещество легко диспропорционируется даже при комнатной температуре [35].
Общая схема реакции описывается следующим уравнением:
он ~
(С4Н9)2(СН2=СН) SnCl > [2 (С4Н9)2(СН2=СН) SnOH] ->
-> (C4H9)2SnO -j- (QH9)2Sn (CH=CH2)2 + H20.
Гидрокоокись тривинилолова является относительно устойчивым
соединением.
Получение гидроокиси тривинилолова [36]. К раствору 25,0 г (0,625 моля) едкого натра
в 300 мл воды при перемешивании в течение 30 мин. прибавляют 100,0 г (0,425 моля) хло-
384
Оловоорганически эсоединения. Лит. стр. 394—395
ристого три винил олова. Сразу же выпадает белый осадок. Смесь перемешивают в течение
часа, осадок отфильтровывают в атмосфере азота и высушивают в вакуум-эксикаторе над
едким кали. Получают 68 г (74%) гидроокиси тривинилолова.
По патентным данным [37] при гидролизе хлористого тривинилолова в
тех же условиях образуется тетрагидрат гидроокиси тривинилолова.
Безводное вещество получают высушиванием гидрата в течение 90 час. над
едким кали в вакууме.
Гидролиз бромистого (2-карбоксиэтил)дибутилолова двумя
эквивалентами едкого кали сопровождается реакцией конденсации с
образованием внутренней соли (2-карбоксиэтил)-ди-я-бутилолова [38]:
(C4H9)3Sn (Br) (СН2СН2СООСН3) + 2 КОН ->
-* (C4H9)3Sn (ОН) (СН2СН2СООК) + КВг + СН3ОН,
(QHe)aSn (ОН) (СН3СН2СООК) -> [(QH9)2Sn (ОН) (СН3СН2СООН)] -»
-* (С4Н9)2£пСН2СН2СОО- + Н20.
Мономерная структура конечного продукта (C^g^SnCHUCHUCOO
маловероятна вследствие его плохой растворимости в обычных
органических растворителях и довольно высокой температуры плавления (ПО—
113° С). Действительно, определение молекулярного веса по методу Раста
дает величину порядка 1600 [38]. Другой метод синтеза соединений типа
(RsSnCHsCHsCOO),, описан на стр. 448.
Гидролиз галогенидов трифенилолова приводит, по-видимому, к смеси
гидроокиси трифенилолова и гексафенилдистанноксана [39—44]. Для
получения чистых соединений применяют следующие методики.
Получение гидроокиси трифенилолова [45]. 1,16 кг (3,0 моля) хлористого
трифенилолова растворяют в 4 л ацетона, и, если это необходимо, раствор фильтруют. В течение 10 мин.
к нему прибавляют 180 г (4,5 моля) едкого натра в 600 мл воды. Температура смеси
несколько повышается, выпадает объемистый белый осадок. Смесь перемешивают в течение 3 час,
разбавляют 3 л воды и фильтруют. Осадок промывают кипящей водой до отрицательной
реакции на ион хлора и высушивают на воздухе. Получают 996,5 г (90,6%) вещества с
т. пл. 115—122° С (с разложением). Перекристаллизация из 95%-ного этилового спирта дает
белые кристаллы с т. пл. 119° С, разлагается при 121° С.
Получение гексафенилдистанноксана [45]. 73,4 г (0,2 моля) гидроокиси трифенилолова
кипятят в 200 мл толуола до отгонки теоретического количества воды (прибор Дина —
Старка). Раствор фильтруют горячим, охлаждают до комнатной температуры и снова
фильтруют. Через 24 часа выделяется 36,5 г гексафенилдистанноксана. Его высушивают в
вакуум-эксикаторе над фосфорным ангидридом. Т. пл. 122—123,5° С.
Для того чтобы различить гидроокись от соответствующей окиси,
обычные методы идентификации не применимы. Однако эти вещества ведут себя
различно по отношению к реактиву Фишера [45] (раствор иода и сернистого
ангидрида в смеси пиридина и метилового спирта):
(CeH5)3SnOH + J2 + S02 + СН3ОН -» (C6H5)3Sn J + H J + HSO4CH3,
(C6H5)3SnOSn(C6H5)3 + J2 + S02 + СЫ3ОН -* 2 (C6H5)3Sn J + HSO4CH3.
Обе реакции проходят количественно. В первом случае на каждый атом
олова приходится 1 моль иода, во втором — половина моля. Эта реакция
успешно применена для анализа смеси гексафенилдистанноксана и
гидроокиси трифенилолова [46]. Существенно отличаются также ИК-спектры
этих соединений [45].
Следует подчеркнуть, что во многих случаях не проводилось столь
подробного исследования продуктов гидролиза галогенидов триарилолова.
Гидролиз
385
Получение гидроокиси три-ж-толилолова [47]. Раствор 13,5 г бромистого три-ж-тол ил
олова в 200 мл эфира встряхивают в течение часа с раствором 4 г едкого кали в 100 мл воды.
Эфирный слой промывают водой, высушивают сульфатом натрия и эфир отгоняют. В остатке
получают маслянистую жидкость, представляющую собой чистую гидроокись три-л*-толил-
олова. Выход 11,1 г (95%).
Аналогично или при обработке соответствующих галоидонроизводных
концентрированным аммиаком получены гидроокиси трициклогексил-
[47], трибензил- [18, 47, 48], три-(2-фенилэтил)-, три-о-толил- [39, 49],
три-д-толил- [39], три-ос-нафтил-, фенилбензил-п-толил- и я-бутилбензил-
фенилолова [5].
Гидроокись трибензилолова превращается в соответствующую окись
при нагревании. Этот переход удобно наблюдать под микроскопом [50].
Получение гекса-д-фторфенилдистанноксана [51]. Бромистое три-я-фторфенилолово
получают, прибавляя по каплям 1,76 г (0,01 моля) брома в 75 мл четыреххлористого
углерода к охлажденному до 0° С раствору 5,52 г (0,011 моля) тетра-(/г-фторфенил)олова в
25 мл того же растворителя. Растворитель отгоняют и остаток кипятят в течение 30 мин.
с 50 мл 5%-ного раствора едкого кали. Смесь охлаждают до комнатной температуры и
фильтруют. Осадок промывают водой, эфиром и высушивают при 100° С. Выход гекса-я-
фторфенилдистанноксана составляет 28%.
Согидролиз смеси хлористого триметилкремния (3—5 моля) и 1 моля
хлористого три-я-пропил- или три-я-бутилолова в бензоле небольшим
избытком водного аммиака с последующей нейтрализацией смеси приводит к
соединениям типа R3SnOSi(CH3)3 [52].
Гидроокиси триалкил- и триарилолова получают также гидролизом
смеси продуктов реакции четыреххлористого олова с рассчитанным
количеством реактива Гриньяра (см. стр. 211).
ГИДРОЛИЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
КЛАССА R2SnX2
Гидролиз оловоорганических дигалогенидов и диацилатов водой
проходит ступенчато [53]. По-видимому, на первой стадии реакции образуются
соединения типа R2Sn (OH)X [4—6]. Последние чрезвычайно неустойчивы
и легко дегидратируются:
R2SnX2 + Н20 -* R2Sn (ОН) X,
2 R2Sn (ОН) X -^ (XSnR2OSnR2X)2 + Н20.
Так, двубромистые тетраалкилдистанноксаны получают [54]
кипячением водно-спиртовых растворов оловоорганических бромидов] в течение
30 мин. Структура соединений типа (XR2SnOSnR2X)2 рассмотрена на стр.
434.
Более удобный способ синтеза соединений типа (XSnR2OSnR2X)2
заключается в гидролизе оловоорганических галогенидов в присутствии
оснований: триэтиламина [6, 55—57], пиридина [54] или метилата натрия
[58]. В некоторых случаях образующуюся при гидролизе кислоту связывают
рассчитанным количеством щелочи [56, 59].
Двухлористый тетраметилдистанноксан получен [54] прибавлением
3,9 г пиридина в 30 мл этилового спирта к раствору 11 г двухлористого диме-
тилолова в 80 мл воды. Осадок отфильтровывают, промывают спиртом и
высушивают при 105° С. Получают 5,7 г вещества с т. пл. ^> 200° С.
Получение двухлористого тетраэтилдистанноксана154]. К раствору 2,0 г двухлористого
диэтилолова в 15 мл этилового спирта прибавляют 0,64 г пиридина в 6 мл спирта.
Прибавление к раствору нескольких капель воды приводит к образованию белого осадка (вероятно,
комплекс с пиридином), который растворяется при нагревании смеси на водяной бане в
течение нескольких минут. Из прозрачного раствора получают кристаллы двухлористого
тетраэтилдистанноксана с т. пл. 175,5—176,5° С.
25 Заказ № 207
386
Оловоорганические соединения. Лит. стр. 394—395
Аналогично получены соответствующие я-пропильное и я-бутильное
производные олова.
В тех же условиях с оловоорганическими дибромидами гидролиз идет
дальше с образованием, в конечном счете, (гидрокси)бромидов тетраалкил-
дистанноксана, например [BrSn(C2H5)2 OSn(C2H5)20H]2 [54]. Замена
пиридина триэтиламином позволяет получить соответствующие двубромистые
тетраалкилдистанноксаны.
Получение двубромистого тетра-я-бутилдистанноксана [56]. К раствору 2,02 г (1 моль)
триэтиламина в 20 мл этилового спирта при перемешивании медленно прибавляют 7,86 г
двубромистого ди-я-бутилолова в 50 мл эфира. Почти сразу выпадает осадок (C2H5)3N •
• НВг (3,51 г, 98%), его отфильтровывают. Фильтрат концентрируют при пониженном
давлении. Осадок отфильтровывают и перекристаллизовывают из этилового спирта. Получают
3,22 г (50%) вещества с т. пл. 101,5—104° С. Двубромистый тетра-я-бутилдистанноксан
хорошо растворяется в ароматических и алифатических углеводородах.
Аналогично получен диацетат тетра-я-бутилдистанноксана [56].
Вследствие хорошей растворимости вещества в обычных органических
растворителях его выделяют следующим образом. Из реакционной смеси
растворитель отгоняют в вакууме, остаток встряхивают со смесью воды с эфиром и
вещество выделяют из эфирного слоя. Дибензоаты тетраметил- и тетра-
этилдистанноксана образуются уже при кристаллизации соответствующих
диацилатов диалкилолова из 96%-ного этилового спирта [60].
Дифенокситетра-я-бутилдистанноксан получен [61] перемешиванием
дифеноксиди-я-бутилолова с избытком воды в течение 2 час. Продукт
высушивают и кристаллизуют из гексана.
Соединения типа (XSnR2OSnR2X)2 получают также сплавлением олово-
органических галогенидов, ацилатов или феноксисоединений с окисями
диалкилолова, а также обработкой последних рассчитанным количеством
фенолов или органических кислот (см. стр. 400, 403, 419).
При гидролизе димеров (XSnR2OSnR2X)2 в эфире рассчитанным
количеством разбавленного раствора едкого натра или, лучше, водой в
присутствии, органических оснований происходит замена одного галоида на гидрок-
сил [54, 62]. Те же соединения получены [53] непосредственным гидролизом
оловоорганических дихлоридов водой в присутствии триэтиламина.
Образующиеся соединения даже после нескольких перекристаллизации
плавятся в широком интервале температур, вероятно, разлагаясь при плавлении.
При перекристаллизации (гидрокси)галогенидов тетраалкилдистаннокса-
на из спирта происходит следующая обратимая реакция [53, 54, 62]:
(XSnR2OSnR2OII)2 *=S (XSnR2OSnR2OR')2.
Н2О
Гидролитическая устойчивость алкоксипроизводных уменьшается с
увеличением алкильной группы, связанной с атомом олова или с кислородом
[54]. Как и соответствующие гидроксисоединения, эти вещества, вероятно,
разлагаются при плавлении.
Получение ClSn(CH3)20Sn(CH3)20R (R = Н, СН3) [54]. 3,8 г двухлористого тетра-
метилдистанноксана и 2,5 мл пиридина кипятят в А№мл метилового спирта в течение 2 час.
Спирт отгоняют в вакууме и продукт кристаллизуют из того же растворителя. Получают
2,4 г кристаллов (хлор)метокситетраметилдистанноксана. На воздухе вещество гидроли -
зуется в течение нескольких дней, превращаясь в соответствующее гидроксисоединение.
Получение (хлор)гидрокситетраэтилдистанноксана [54]. К раствору 5 г двухлористого
тетраэтилдистанноксана в 60 мл влажного метилового спирта прибавляют 3,6 мл пиридина
и смесь нагревают на водяной бане в течение нескольких минут. Через несколько дней из
раствора начинают выделяться кристаллы, которые после промывания метиловым спиртом
имеют т. пл. 214—217° С. Продукт представляет собой смесь гидрокси- и алкоксисоединений.
Перекристаллизацией из смеси воды с ацетоном получают чистое гидроксипроизводное.
Гидролиз
387
Также синтезированы [54] и другие соединения этого типа.
Гидролиз дииодидов диалкилолова позволяет получать соединения типа
(JSnR2OSnR20H)2, минуя стадию выделения двуиодистых тетраалкилди-
станноксанов. Так, 2 г двуиодистого диэтилолова и 0,6 г пиридина в 15 мл
влажного метилового (или этилового) спирта нагревают до температуры
несколько ниже температуры кипения спирта в течение нескольких минут.
Из раствора через несколько дней получают вещество, которое после
кристаллизации из спирта представляет собой (алкокси)иодид тетраэтилди-
станноксана. На воздухе он гидролизуется за несколько дней, превращаясь
B[HOSn(C2H5)2OSn(C2H5)2J]2 [54] (ср. [63]).
Гидролиз диацилатов диалкилолова водой приводит к смесям соединений
с различной структурой и молекулярным весом. По патентным данным
[64] при перегонке с паром диацилатов диалкилолова образуется смесь
органостанноксаноЁ со средней степенью конденсации я, равной трем:
CH3COO(SnR20)„OCCH3. При длительной обработке (18—20 час.) горячим
влажным воздухом в вакууме средняя степень поликонденсации достигает
10 [64].
Гидролизом диацетатов дизтил- и ди-я-пропилолова водой при 0, 20 и
50° С также получены смеси низкомолекулярных соединений станноксаново-
го ряда [65]. Обработка полученных смесей горячим влажным воздухом в
вакууме не приводит к значительному увеличению молекулярного веса.
Высказано предположение [65], что наряду с образованием линейных поли-
органостанноксанов отдельные цепи молекул связываются друг с другом
кислородными атомами с образованием «сшитых» полимеров.
Индивидуальные станноксановые соединения с достаточной длиной
цепи и активными концевыми группами могут быть получены гидролизом
диацилатов диалкилолова рассчитанным количеством воды в присутствии
диазоалканов [66, 67]:
2 (C4H9)2Sn (ООССНзЬ + Н20 + 2 CI I2N2 ->
-* CHbCOOSn(C4He)2OSn (QH9)oOOCCH3 + 2СН3СООСН3 + 2 Na.
Сам диазоалкан в построении конечных продуктов реакции не участвует,
а лишь связывает образующуюся в результате гидролиза кислоту и тем
предотвращает обратную реакцию. Этот прием позволяет:
а) осуществить синтез индивидуальных соединений по стадиям;
б) получить линейные соединения с активными концевыми группами,
создающими возможность проведения дальнейших ступеней.
Изменяя количество воды и диазоалкана, можно по желанию закончить
реакцию на образовании индивидуальных соединений с определенным
молекулярным весом. Так, гидролизом диацетата ди-я-бутилолова получен
димер—диацетаттетра-я-бутилдистанноксан и из того же диацетата
ди-я-бутилолова непосредственно получен тетрамер.
Получение to,со-диацетата октабутилтетрастанноксана [67]. К раствору 16,7 г (0,048
моля) диацетата дибутилолова в 50 мл сухого ацетона прибавляют 1,5 мл (0,и83 моля) воды
и по каплям в течение 15 мин. приливают раствор 3,028 г (0,072 моля) диазометана в 130 мл
эфира. Через полчаса бесцветный раствор упаривают в вакууме примерно наполовину;
выпавшие кристаллы отфильтровывают и высушивают в вакууме. Получают 8,5 г (65,1%)
со, со-диацетата октабутилтетрастанноксана ст. пл. 137—138° С (с разложением). Ди-
ацетат октабутилтетрастанноксана хорошо растворим на холоду в хлороформе, при
нагревании в бензоле, спирте и четырех хлор истом углероде. Умеренно растворим при
нагревании в гексане, ацетоне, метилэтилкетоне.
Дальнейший гидролиз проходит значительно медленнее или требует
некоторого повышения температуры.
25*
388 Оловоорганические соединения. Лит. стр. 394—395
Получение ю,ю-диацетатапексадекабутилоктастанноксана [67]. К раствору 5,0 г. (0,0046
моля) со,со-диацетата октабутилгетрастанноксана в 400 мл сухого метилэтилкетона
прибавляют 0,3 мл (0,0166 моля) воды и 0,2022 г (0,0048 моля) диазометана и 7,5 мл эфира. Через
6 час. бесцветный раствор упаривают в вакууме примерно наполовину, выпавший осадок
отфильтровывают и высушивают в вакууме. Получают 3,0 г (63%) со, со-диацетата гекса-
декабутилоктастанноксана с т. разл. около 210° С. Вещество хорошо растворимо при
нагревании в хлороформе и бензоле, умеренно в ацетоне, метилэтилкетоне и гексане.
Получение ю,ю-диацетата дотриаконтабутилгексадекастанноксана [671. К раствору 5,2 г
(0,0047 моля) со,со-диацетата октабутилтетрастанноксана в 250 мл сухого метилэтилкетона
прибавляют 0,1 мл (0,0055 моля) воды и раствор 0,3058 г (0,0073 моля) диазометана в 14 мл
эфира. Диазометан при 37—40° С обесцвечивается в течение 2 час. По охлаждении
выделяется осадок (0,93 г), который отфильтровывают. Раствор упаривают в вакууме примерно на
х/з- Получают 2,3 г вещества с т. разл. выше 330° С. Вещество умеренно растворимо в
горячем ксилоле.
При повышении температуры до 40—45° С возможно получение
индивидуальных станноксанов и с более высоким молекулярным весом.
Исчерпывающий гидролиз оловоорганических соединений типа R2SnX2 щелочью
приводит к полимерным окисям диалкил- и диарилолова (R2SnO)^.
Для препаративных целей в качестве исходных соединений применяют
легко доступные оловоорганические галогениды.
Исследован [68—70] щелочной гидролиз двухлористого диметилолова.
Найдено, что в водных растворах вещество диссоциирует; при любых
концентрациях раствор имеет кислую реакцию вследствие незначительного
гидролиза хлорида (двухлористое диметилолово в 0,064 молярных водных
растворах при 25° С гидролизуется на 10,5% [68]):
(CHafeSnCla + п Н20 -> (CH3)2Sn (Н30)^+ + 2С1~,
(CH3)2Sn (Н20)2+ + Н20 -> (CH3)2Sn (НзО^ОН* + Н30+.
При прибавлении 0,5 N едкого натра к 4,4 г двухлористого
диметилолова в 150 мл воды окись диметилолова не осаждается из раствора до тех пор,
пока рН среды не достигнет значения 5,7. В этой точке окись диметилолова
образуется медленно и рН уменьшается до 5,32. При дальнейшем
прибавлении щелочи в осадок выделяется вся окись диметилолова, которая остается
в виде суспензии, пока рН не станет равным 11,5. При дальнейшем подщела-
чивании (рН 11,52—11,55) окись переходит в (CH3)2Sn(OK)2- Окись
диметилолова лучше всего получать прибавлением рассчитанного количества
едкого натра к раствору двухлористого диметилолова или прибавлением
оловоорганического галогенида к щелочному буфферу при рН8—9 [68].
В этих условиях окись диметилолова выпадает в виде хорошо
образованного белого порошка.
Аналоги двухлористого диметилолова плохо растворяются в воде.
Соответствующие окиси обычно получают гидролизом спиртовых растворов
оловоорганических хлоридов или бромидов типа R2SnX2 15—20%-ным
раствором щелочи [17, 18, 21, 22]. Выделившийся осадок
отфильтровывают и промывают водой до отрицательной реакции на ион галоида. Дигало-
гениды дигексил-, диоктилолова и высших алкильных соединений олова
плохо смачиваются водой. Поэтому их гидролиз проводят в присутствии
эмульгаторов, применяя особую методику очистки.
Получение окиси ди-#-гексилолова [71]. К нагретому до кипения раствору 31,5 г (0,79
моля) едкого натра и 0,5 мл ализаринового масла в 160 мл воды при энергичном
перемешивании прибавляют в течение часа 124 г (0,34 моля) расплавленного двухлористого ди-я-гек-
силолова. Смесь нагревают при перемешивании 45 мин. После охлаждения осадок окиси
отфильтровывают промывают водой и высушивают в вакууме при 50° С. Выход
количественный.
Гидролиз
389
Иногда наряду с эмульгаторами применяют растворители — спирт,
ацетон и др. В этом случае окись диалкилолова очищают последовательным
выщелачиванием минеральных примесей водой и растворимых оловоор-
ганических соединений спиртом.
Получение окиси ди-я-октилолова [71]. К нагретому до кипения раствору 140 г едкого
кали и 1 мл ализаринового масла в смеси 350.мл этилового спирта и 350 мл воды при
энергичном перемешивании прибавляют в течение 45 мин. 350 мл расплавленного двухлористого
ди-я-октилолова. Смесь кипятят при перемешивании 30 мин., охлаждают и фильтруют. К
осадку прибавляют 500 мл воды и оставляют на 3 часа, фильтруют, приливают 50 мл
этилового спирта и смесь оставляют на ночь, а затем еще на 3 часа с новой порцией этилового
спирта. Вещество отфильтровывают, высушивают в вакуум-эксикаторе, измельчают и
оставляют на ночь под слоем воды, а затем на 3 часа со спиртом. Окись ди-я-октилолова
отфильтровывают и высушивают при 40—50° С в течение 13 час. Выход 291 г (96%).
Аналогично получена окись ди-(2-этил-я-гексил)олова [71]. Окись ди-
(3,5, 5-триметил-я-гексил)олова получена [71] с выходом 96,5% постепенным
прибавлением раствора 100 г (0,225 моля) двухлористого ди-(3,5,5-три-
метил-я-гексил)олова в 600 мл ацетона к нагретому до кипения раствору
29,5 г (0,525 моля) едкого кали в смеси 840 мл воды и 1 л ацетона. Смесь
кипятят в течение 1,5 часа, осадок отфильтровывают, промывают смесью
ацетона с водой (2 : 1) и высушивают в вакууме при 70° С.
При разветвленном алкильном радикале, по всей вероятности, имеют
место пространственные затруднения, в результате чего образуется не окись,
а дигидроокись диалкилолова, достаточно стойкая для возможности ее
выделения в индивидуальном состоянии.
Получение дигидрсокиси ди-тр^т-бутилолова [72]. К двубромистому ди-трет-бутжо-
лову (полученному действием брома на ди-ягрет-бутилолово в эфире) прибавляют 15%-ный
водный раствор едкого кали. Плохо растворимая в эфире дигидроокись выпадает в виде
мелких кристаллов; ее отфильтровывают, промывают водой и эфиром; перекристаллизовывают
из теплого эфира.
Аналогично получена дигидроокись дд-треш-амилолова [72]. Вероятно,
окись дилаурилолова также существует в виде соответствующей
дигидроокиси.
Получение окиси дилаурилолова [73]. Раствор двухлористого дилаурилолова
обрабатывают при взбалтывании избытком \N раствора едкого натра. Сразу же наблюдается
помутнение, которое быстро переходит в желатинообразный осадок, содержащий некоторое
количество воды. Продукт промывают водой до полного исчезновения адсорбированных ионов,
потом взбалтывают несколько раз со спиртом и высушивают в вакууме над хлористым
кальцием. Получаемая окись дилаурилолова умеренно растворима в эфире, четыреххлористом
углероде и сероуглероде. Определение молекулярного веса показывает, что вещество моно-
мерно.
Интересно отметить, что продукт щелочного гидролиза двухлористого
дибензилолова растворим в избытке щелочи. Это объяснено [74]
следующими реакциями:
(CGH*CH?)2Sr;Cl2 + 2 КОН -» (C6H5CHo)£Sn (ОН)2 + 2 КС1,
(C6RCH2)2Sn (ОН), + 2 КОН -» (CcH6CH2)2Sn (ОК)2 + Н20.
Индивидуальные соединения из раствора выделить не удается. Более того,
если осадок предполагаемой дигидроокиси дибензилолова
отфильтровывать, то он перестает растворяться в избытке щелочи [74].
Гидролиз дигалогенидов диарилолова можно иллюстрировать
следующими примерами.
Получение окиси дифенилолова [75]. Раствор 25 г (0,0733 моля) двухлористого
дифенилолова в 50 мл бензола прибавляют к раствору 6,15 г (0,154 моля) едкого натра в 200 мл
воды. Эмульсию отфильтровывают, осадок промывают водой и высушивают. Полученная
таким путем окись дифенилолова имеет т. пл. 323—4° С, разлагается при 360° С. Выход 19,8 г
(94%).
390 Оловоорганические соединения. Лит. стр. 394—395
Условия проведения гидролиза оловоорганических соединений с
заместителями в бзнзольном кольце зависят от природы заместителей. Так, при
получении окисей типа (XQjH^SnO (где X— хлор, бром или иод)
применяют 5%-ный раствор щелочи [76, 77].
Получение окиси ди-лг-толилолова [48]. 1 г двухлористого ди-^-толилолова растворяют
в 6—7 мл спирта и к раствору при взбалтывании постепенно прибавляют избыток аммиака.
Через 30 мин. раствор нагревают до кипения, осадок отсасывают, промывают горячей водой,
небольшим количеством спирта, затем эфира и высушивают при 60—70° С. Окись ди-ж-то-
лилолова представляет собой белый аморфный порошок, неплавкий и лишь темнеющий при
нагревании.
Двуиодистое ди-(о-феноксифенил)олово гидролизуется лишь в очень
жестких условиях, при которых вещество разлагается [78].
Свойства продуктов гидролиза двухлористого ди-о-феноксифенилолова
изменяются в зависимости от условий гидролиза, но ни в одном случае не
получено нерастворимое или неплавкое вещество. Так, при гидролизе
небольшим избытком водного раствора кислого углекислого натрия при 0° С
образуется кристаллическое вещество, элементарный анализ и число гид-
роксильных групп которого соответствуют дигидроокиси тетра-(о-фенокси-
фенил)дистанноксана HO[Sn(C6H40C6H5-o)20)]2H.
Получение дигидроокиси тетра-(о-фгнэксифгнил)дистанноксана [78]. Раствор 6,0 г
двухлористого ди-о-феноксифенилолова в 100 мл эфира охлаждают смесью льда с солью и
при перемешивании к нему прибавляют 2,1 г кислого углекислого натрия в 100 мл воды.
Через 2 мин. осадок отфильтровывают, промывают водой и эфиром и высушивают. Получают
5,0 г вещества с т. пл. 164° С. Для очистки его растворяют в хлороформе и осаждают
избытком петролейного эфира (т. кип. 40—60° С); температура плавления повышается до 165—166°
С. Криоскопическое определение молекулярного веса дает невоспроизводимые результаты,
что, вероятно, обусловлено неустойчивостью соединения в органических растворителях.
Действительно, из бензольного раствора вещества через 3 дня выпадают бесцветные
кристаллы с т. пл. 162—180° С, которые по данным элементарного анализа, ИК-спектру и
содержанию гидроксильных групп сходны с исходным тетра-(о-феноксифенил)дистанноксаном. По-
видимому, они являются ассоциированной формой последнего.
При гидролизе двухлористого ди-(о-феноксифенил)олова избытком
едкого натра образуются устойчивые на воздухе бесцветные кристаллы
дигидроокиси ди-(о-феноксифенил)олова. При растворении или нагревании
вещество разлагается и очистить его перекристаллизацией не удается.
Получение дигидроокиси ди-(о-фгноксифенил)олова [78]. К раствору 2,0(Уг
двухлористого ди-(о-фзноксифзнил)олова в 30 мл эфира прибавляют 40 мл 2N водного раствора едкого
натра. Смесь встряхивают в течение нескольких минут и оставляют на час при комнатной
температуре. Осадок отфильтровывают, промывают водой и эфиром и высушивают на
воздухе. Получают 1,35 г дигидроокиси ди-(о-фэноксифэнил)олова с т. пл. 141—142° С (с
разложением).
При помощи гидролиза двубромистого ди(триметилсилилметил)олова —
[(СНзЬ SiCH2]2SnBr2 [79] и гетероциклического оловоорганического
соединения [80]
получают соответствующие полимерные окиси.
Окиси диалкил- и диарилолова представляют собой обычно
нерастворимые неплавкие порошки белого цвета. В 1960 г. Райхл [75] доказал, что
они представляют собой линейные полимеры типа (I^SnO)^ Отдельные
цепи этих молекул, очевидно, связаны координационными связями [53] так,
Гидролиз
391
что образуется сетчатый полимер:
RoSn
) 0-
\ /
R2Sn
i
t
0
-R2Sn SnRc
\ /
0
1
R2Sn
/ \ / \
1 SnR2—0 0—
0
R2Sn
/ \
> 0-
R2Sn
R2Sn
i
1
0
-R2Sn SnR2-
\ /
0
R2Sn
/ \
j—0 0-
\ /
R2Sn
I
1
0
R2Sn Sr
0
1
R2Sn
/ \
-0 0-
\ /
R2Sn
О наличии в окисях диалкил- и диарилолова пятикоординационных атомов
олова свидетельствуют спектры Мэссбауэра этих соединений [81, 82].
Райхл исследовал методами рентгенографии и ИК-спектров строение
окисей диметил-, ди-я-бутил-, ди-я-октил- и дифенилолова. Вследствие
плохой растворимости этих полимерных веществ определить молекулярный
вес удалось лишь для окиси ди-я-октилолова, умеренно растворимой в
горячем ксилоле. Средний молекулярный вес этой окиси оказался равным
18 000. Высказано предположение [75], что средний молекулярный вес и
других окисей имеет примерно тот же порядок, хотя и зависит от
органического радикала, связанного с атомом олова, условий проведения гидролиза
и других факторов. Действительно, средняя степень полимеризации
растворимых в тетрагидрофуране и хлороформе окисей ди-#то/?-бутилолова и
ди-т/?ет-бутилолова, по осмометрическим данным, соответственно равны 2,3
и 1,4 [83]. Отмечено [75], что положение и интенсивность линий на
рентгенограмме окиси дифенилолова зависит от метода синтеза вещества. Это
обусловлено, по-видимому, различными концевыми группами и
молекулярным весом образующихся продуктов. Во всех случаях выделить из
окисей диалкил- или диарилолова индивидуальные соединения со станноксано-
вой связью (тример, тетрамер и т. д.) не удается. О термостабильности оло-
воорганических окисей см. стр. 515.
При согидролизе хлористого триметил кремни я с двухлористым
диметил-, диэтил-, ди-я-пропил- или ди-я-бутилоловом образуются соединения
состава (CH3)3SiO(SnR20)2Si(CH3)3 [84]. Если реакцию проводить с
избытком хлористого триметил кремния, то выход продукта практически
количественный. С эквимолекулярными количествами реагентов он
составляет только 30%.
Получение <?#с-(триметилсилокси)тетраметилдистанноксана [84]. Реакцию проводят
в трехгорлой колбе, снабженной мешалкой, обратным холодильником и капельной
воронкой. Примерно двойное количество водного аммиака смешивают с 200 мл петролейного
эфира, охлаждают льдом и при энергичном перемешивании постепенно прибавляют раствор
6,6 г двухлористого диметилолова и 19,5 г хлористого триметилкремния в 150 мл бензола.
Затем добавляют 200 мл воды и смесь перемешивают 10 мин. Органический слой отделяют,
дважды промывают водой, высушивают сульфатом натрия и растворитель отгоняют при
пониженном давлении. Для удаления гексаметилдисилоксана остаток нагревают в вакууме
и вещество кристаллизуют из гексана или петролейного эфира; т. пл. 167—168° С,
Аналогично получены и другие бш;-(триметилсилокси)тетраалкилди-
станноксаны. Эти соединения хорошо растворяются в алифатических и
392
Оловоорганические соединения. Лит. стр. 394—395
ароматических углеводородах. В кислородсодержащих растворителях,
таких как спирт, эфир, диоксан или ацетон, происходит медленное
разложение с образованием окиси диалкилолова. Реакция разложения ускоряется
в присутствии воды.
Убедительным доказательством дистанноксановой структуры этих
соединений является образование тех же веществ при согидролизе хлористого
триметилкремния с дихлортетраалкилдистанноксанами [84].
При использовании эквивалентного количества реагентов образуются
соединения типа (CH3)3SiOSnR2OSi(CH3)3- Последние легко разлагаются по
следующей схеме:
2 (CH3)3SiOSnR203i (СНз)з - (CH3)3Si03i (QH3)3 + (CH8)*SiO [SnR20]2Si (СН3)з.
ГИДРОЛИЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ КЛАССА RSnX3
Гидролиз соединений класса RSnX3 проходит ступенчато. Так, треххло-
ристое этил-, бутил- и октилолово при продолжительном контакте с влажным
воздухом превращаются в гидратированные гидроксидихлориды алкилоло-
ва состава RSn(OH)Cl2-H20 [85]. Этильное соединение имеет т. пл.
94—96° С, бутильное и октильное — 80—87 и 45—46° С
соответственно.
С одним или двумя эквивалентами водной щелочи трихлориды алкил-
олова образуют дигидроксихлориды алкилолова RSn(OH)2Cl. При
высушивании их в вакууме они теряют молекулу воды с образованием
полимерных продуктов состава [RSn(0)Cl],i. Полимеры того же типа, содержащие
вместо галоида остаток органической кислоты, получены [86] при
перекристаллизации триацилатов я-бутил- или фенилолова из 95%-ного спирта.
В алифатическом ряду реакция обратима: при нагревании оксимоноацила-
тов я-алкилолова с соответствующей кислотой образуется исходный оло-
воорганический триацилат. Оксимоноацилаты фенилолова с кислотами
дают тетраацилат олова и бензол [86].
Обработка соединений класса RSnX3 тремя эквивалентами или
избытком щелочи или аммиака приводит к полимерным станноновым кислотам
[76, 77, 87—92]:
RSnCls + 3 NH4OH -» (RSnOOH)rt 4- 3 NH4CI + Н20.
Так получены, например, метил- [87], этил- [85, 88] и бутилстанноновая
кислоты [85]. При обработке тремя эквивалентами щелочи треххлористого
октилолова выпадает осадок, молекулярный вес и анализ которого
соответствуют формуле
/z-C8H17Sn (OH)2OSn (ОН) С1 (С8Н17-п).
Конечный продукт гидролиза — октилстанноновая кислота — образуется
только в том случае, если треххлористое октилолово кипятить в течение
нескольких часов с тремя эквивалентами щелочи [85].
Алкилстанноновые кислоты не растворяются в обычных органических
растворителях, кроме низших алифатических спиртов. При растворении в
спиртах, вероятно, образуются ортоэфиры RSn(OR')3 или полимерные
продукты состава (RSnOOR')/2[93]. Этилстанноновая кислота хорошо
растворяется также в воде [84].
Алкилстанновые кислоты легко превращаются при действии избытка
щелочи в соли щелочных металлов, а последние могут быть переведены и в
некоторые другие соли.
Гидролиз
393-
Получение калиевой соли этилстанноновой кислоты [88]. Часто встряхиваемую смесь
1 г едкого кали в 25 мл воды и 1 г этилстанноновой кислоты отфильтровывают от избытка
последней и фильтрат выпаривают в эксикаторе до появления кристаллов. Эти кристаллы
калиевой соли по данным анализа соответствуют формуле C2H5SnOOK. Растворяясь в воде,
они дают щелочной, вследствие гидролиза, мутный раствор.
Аналогично получают и натриевую соль. Обменом 3 г калиевой соли
этилстанноновой кислоты с 1 а хлорной меди готовится медная соль, обменом
с сульфатом магния — магниевая соль и с хлористым барием — бариевая
соль. Однако, с Ag+, Zr2+, Hg2+, Co2+, Ni2+ получены [88] только основные
соли. Например, при прибавлении азотнокислого серебра к раствору
калиевой соли этилстанновой кислоты выпадает осадок коричневого цвета
состава Ag20-C2H5SnOOAg. Соли кальция, стронция и бария получены при
прибавлении избытка раствора натриевой или калиевой соли я-пропилстан-
ноновой кислоты к растворам соответствующих хлоридов [89].
Изучение алкилстанноновых кислот рентгеноструктурными методами
указывает на отсутствие в них кристаллической структуры [94].
По-видимому, они имеют следующее строение [93]:
R R R
I I I
_0—Sn О—Sn—О—Sn—О—
i i i
—О—Sn—R R—Sn—О—Sn—О—
1 ' i
Возможные структурные формулы алкилстанновых кислот рассмотрены и
другими авторами [95—97].
Трихлориды арилолова растворяются в воде, давая растворы, имеющие
сильно кислую реакцию [90]. При кипячении с водой выпадает белый
аморфный осадок — продукт частичного гидролиза. Водный аммиак
высаживает белую аморфную арилстанноновую кислоту, нерастворимую в избытке
осадителя.
Получение я-толилстанноновой кислоты [91, 92]. 1,7 г треххлористого н-толилолова
растворяют на холоду в небольшом количестве воды (2—3 мл). Раствор обрабатывают
избытком 20%-ного едкого кали (выпадающий вначале осадок растворяется). Прозрачный
раствор почти нейтрализуют уксусной кислотой, а затем пропускают углекислый газ.
Выпавший белый осадок отсасывают, промывают несколько раз водой и во влажном состоянии
обрабатывают спиртом при 60—70° С. Раствор фильтруют и концентрируют при 40—45° С
под небольшим вакуумом, кислоту по выпаривании отсасывают.
Аналогично получены фенилстанноновая кислота [7, 8],г о-толилстан-
нововая кислота [91, 92], ж-толилстанноновая кислота [90],п-хлорфенилстан-
ноновая кислота [76, 77], я-бромфенилстанноновая кислота [76, 77], п-
иодфенилстанноновая кислота [76, 77] и а-нафтилстанноновая кислота
[98].
Арилстанноновые кислоты являются очень слабыми кислотами и
высаживаются из щелочных растворов при прибавлении уксусной кислоты или
при пропускании в раствор углекислого газа. В отличие от станноновых
кислот жирного ряда они хорошо растворяются в-большинстве
органических растворителей (в этиловом и метиловом спиртах, хлороформе и этил-
ацетате). Умеренно растворяются в бензоле, не растворяются в Еоде и петро-
лейном эфире.
394
Оловоорганические соединения
ЛИТЕРАТУРА
1. О к a w a r a R., RochowE., J. Am. Chem. Soc, 82, 3285 (1960).
2. A 1 1 e s t о n D. L., D a v i e s A. G., J. Chem. Soc, 1962, 2050.
3. Z i m m e r H., G о 1 d H., Ber., 89, 712 (1956).
4. Aronheim В., Ann., 194, 145 (1878).
5. К i p p i n g F. В., J. Chem. Soc, 1928, 2365.
6. G i b b ons A. J., Sawyer A. K-, Ross A., J. Org. Chem., 26, 2304 (1961).
7. Кочешков К-А., ЖРФХО, 61, 1385 (1929).
8. Kozesch kow К. A., Ber., 62, 996 (1929).
9. H e с м е я н о в А. Н., К о ч е ш к о в К. А., ЖРФХО, 62, 1795 (1930).
10. N е s m e j a n о w A. N., К о z e s с h k о w К- А., Вег., 63, 2496 (1930).
11. Англ. пат. 797976 (1958); С. А., 53, 3061 (1959).
12. G 1 о s к е у С. R., Пат. США 2862944 (1958); РЖХим., 1960, 53764.
13. N i t z s с h e S., R i e d 1 e R., Пат. США 2868820 (1959); С. А., 53, 13057 (1959).
14. N i tzsche S., R i edle R., Пат. США 2770611 (1956); С. А., 51, 7761 (1957).
15. D б г f e 1 t С, G i 1 b e г t H.f Пат. ФРГ 1084722 (1960); РЖХим., 1962, 5Л110.
16. Пат. ФРГ 957483 (1957); С. А., 53, 18865 (1959).
17. Dane к О., Collect. Czechosl. Chem. Communs., 26, 2035 (1961).
18. S i s i d о К., Т а к e d a Y., К i n u g a w a Z., J. Am. Chem. Soc, 83, 538 (1961).
19. КочкинД.А., ВашковВ. И., ДремоваВ. П., ЖОХ, 34, 325 (1964).
20. Н а г a d а Т., Sci Papers Inst. Phys. Chem. Res. (Токуо), 38, 115 (1940).
21. В г о w n M. P., W e b s t e r D. E., J. Phys. Chem., 64, 698 (1960).
22. H a r a d а Т., Repts. Sci. Res. Inst. (Japan), 24, 177 (1948); С. А., 45, 2356 (1951).
23. Vander KerkG.J.M., L u i j t e n J. G. A., J. appl. Chem., 6, 49 (1956).
24. КочкинД.А., КотрелевВ. Н., Калинин а С. П., Кузнецова
Г. И., Лайне Л. В., Ч е р в о в а Л. В., Б о р и с о в а А. И., Б о р и с е н к о
В. В., Высокомолек. соед., 1, 1507 (1959).
25. L u i j t e n J. G. A., Rec. trav. chim., 82, 1179 (1963).
26. KriegsmannH., Hoffmann H., PischtschanS., Z. anorgan.
allgem. Chem., 315, 283 (1962).
27. Gel ber t H., Пат. ФРГ 1089757 (1961); РЖХим., 1962, 11Л91.
28. V a n d e г К e r k G. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 56 (1956).
29. V a n d e г К e r k G. J. M., Nol tes J.G., J. appl. Chem., 9, 179 (1959).
30. N о 1 t e s J. G., L u i j t e n J. G. A.,, vander К e r k G. J. M., J. appl. Chem.,
11, 38 (1960).
31. Rosenbergs. D., DebreczeniE., Weinberg E. L., J. Am. Chem.
Soc, 81, 972 (1959).
32. Я к у б о в а Ф. А., Р а ш к е с А. М., К у ч к а р е в А. Б., МанулкинЗ. М.,
ЖОХ, 35, 387 (1965).
33. С н е г у р Л. М., М а н у л к и н 3. М., ЖОХ, 34, 4030 (1964).
34. Anderson H. IT., Inorg. Chem., 1, 647 (1962).
35. С о n s i d i п е W. J., V e n t u r a J. J., Kushlefsky B. G., Ross A.,
J. Organometal. Chem., 1, 299 (1964).
36. RosenbergS.D., G i b b о n s A. J., J. Am. Chem. Soc, 79, 2138 (1957).
37. Rams den H. E., RosenbergS.D., Пат. США 2892856 (1959); С. А., 53,
19881 (1959).
38. V a n d e r KerkG.J.M, Nol tes J. G., J. appl. Chem., 9, 113 (1959).
39. К оч еш к об К. А., НадьМ. М., Александров А. П., ЖОХ, 6, 1672
(1936).
40. С h a m b e r s R. F., S с h e r e г Р. С, J. Am. Chem. Soc, 48, 1054 (1926).
41. WestR., BaneyR.H., PowellD.L, J. Am. Chem. Soc, 82, 6269 (1960).
42. GilmanE, MillerL.S., J. Am. Chem. Soc, 73, 2367 (1951).
43. S с h m i t z-D u m о n t O., Z. anorgan. allgem. Chem., 248, 289 (1941).
44. Friebe E., Kelker H., Z. analyt. Chem., 192, 267 (1963).
45. Kushlefsky В., Simmons I , R о s s A., Inorg. Chem., 2, 187 (1963).
46. К u s h 1 e f s k у В., Ross A., Analyt. Chem., 84, 1666 (1962).
47. L u i j t e n J. G. A., v a n d e г К e r k G. J. M., J. appl. Chem., 11, 35 (1961).
48. Надь М. М., К о ч е ш к о в К. А., ЖОХ, 8, 42 (1938).
49. S t e r n А., В е с k e r E. I., J. Org. Chem., 29, 3221 (1964).
50. Bahr G., Zoche G. Ber., 88, 1450 (1955).
51. F u с h s О., Р о s t H. W., Rec. Trav. Chim., 78, 566 (1959).
52. О k a w a r a R., S u g i t a K., J. Am. Chem., Soc, 83, 4480 (1961).
53. Al les ton D. L., DaviesA. G., Hancock M., J. Chem. Soc, 1964,
5744.
54. Ok a war a R., W a d a M., J. Organometal. Chem., 1, 81 (1963).
55. J о h n s о n O. H., Fritz H.E., Halvorson D.O., EvansR.L, J. Am.
Chem. Soc, 77, 5857 (1955).
Гидролиз
395
56. А 1 1 е s t о n D. L., D a v i e s A. G., HancockM., White R. F. M., J. Chem.
Soc., 1963, 5469.
57. Alleston D. L, DaviesA.G., Chem. a. Ind.. 1961, 949.
58. J о h n s о n O. H., Fritz H. E., J. Org. Chem., 19, 74 (1954).
59. Кацумура Т., Японск. пат. 3777 (I960); РЖХим., 1962, 13Л94.
60. Р а з у в а е в Г. А., Щ е п е т к о в а О. А., В я з а н к и н Н. С, ЖОХ, 32,
2152 (1962).
61. С о п s i d i n e W. J., V e n t u r a J. J., J. Org. Chem., 28, 221 (1963).
62. A 1 1 es t on D. L., DaviesA.G., FiggisB. N., Proc. Chem. Soc, 1961,
457.
63. H a r a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 36, 497 (1939).
64. M а с k G. P., P a r k e г Е., Пат. США 2628211 (1953); С. А., 47, 5165 (1953).
65. Ж и в у х и н С. М., Д у д и к о в а Э. Д., К и р е е в В. В., ЖОХ, 31, 3106 (1961).
66. К о ч е ш к о в К. А., П а н о в Е. М., 3 е м л я н с к и й Н. Н., Изв. АН СССР,
ОХН, 1961, 2255.
67. 3 е м л я н с к и й Н. Н., П а н о в Е. М., СловохотоваН. А., Шамаги-
наО. П., Кочешков К. А., ДАН СССР, 149, 312 (1963).
68. RochowE.G., S е у f e r t h D., J. Am. Chem. Soc, 75, 2877 (1953).
69. T о b i a s R. S., О g r i n s I., N e v e t t B. A., Inorg. Chem., 1, 638 (1962).
70. Gi ngol d W., RochowE.G., S e у f e r t h D., SmithA.C, WestR.,
J. Am. Chem. Soc, 74, 6306 (1952).
7b V a n d e г К e r k G. J. M. L u i j t e n J. G. A., J. appl. Chem., 7, 369 (1957).
72. KrauseE., W e i n b e r g K-, Ber., 63, 381 (1930).
73. S о 1 e r i о A., Cazz. chim. itai., 81, 664 (1951).
74. S m i t h F. А., К i p p i n g T. S., J. Chem. Soc, 101, 2553 (1913).
75. R e i с h 1 e W. Т., Je Polymer Sci., 49, 521 (1961).
76. H e с м е я н о в А. Н., Кочешков К. А., ЖОХ, 1, 219 (1931).
77. К о z e s с h k о w К. А., N е s m e j a n о w A. N., Вег., 64, 628 (1931).
78. Poller R. G., J. Chem. Soc, 1963, 706.
79. S e у f e r t h D., J. Am. Chem. Soc, 79, 5881 (1957).
80. К u i v i 1 a H., В e u m e 1 O., J. Am. Chem. Soc, §0, 3250 (1958).
81. Гольданский В. И., Макаров Е. Ф., СтуканР. А., Трухтано
В. А., X р а п о в В. В., ДАН СССР, 151, 351 (1963).
82. Александров А. Ю., Митрофанов К. П., О х л о б ы с т и н О. Ю.,
П о л а к Л. С, Ш п и н е л ь В. С, ДАН СССР, 153, 370 (1963).
83. В a u m G., С о n s i d i n e W. J., J. Polymer Sci., 1963, Bl, 517.
84. О k a w a r a R., WhiteD.G., Fujitani K., S a t о H., J. Am. Chem. Soc,
83, 1342 (1961).
85. L u i j t e n J. G. A., Rec trav. chim., 85, 873 (1966).
86. Anderson H. H., Inorg., Chem., 2, 912 (1964).
87. P f e i f f e r P., L e h n a r d t R., Ber., 36, 1054 (1903).
88. D r u с e J. G. F., J. Chem. Soc, 119, 758 (1921).
89. D r u с e J. G. F., J. Chem. Soc, 121, 1859 (1922).
90. Кочешков К. А., НадьМ.М, ЖОХ, 4, 1434 (1934).
91. К о ч е ш к о в К. А., НадьМ.М., ЖОХ, 5, 1158 (1935).
92. К о z e s с h k о w К. А., N a d j М. М., Вег., 67, 717 (1934).
93. A t 1 a s S. М., М а г к Н. F., Angew. Chem., 72, 249 (1960).
94. S о 1 е г i о A., Gazz. chim. ital., 85, 61 (1955).
95. LambourneH., J. Chem. Soc, 121, 2533 (1922).
96. Lambourne H., J. Chem. Soc, 125, 2013 (1924).
97. Ж и в у х и н С. М., Д у д и к о в а Э. Д., П ш и я л к о в с к а я Н. Бм ЖОХ,
33, 2598 (1963).
98. Пики на Е. И., Т а л а л а е в а Т. В., Кочешков К. А., ЖОХ, 8, 1844
(1938).
Глава XI
РЕАКЦИИ ПРЕОБРАЗОВАНИЯ ФУНКЦИОНАЛЬНЫХ ГРУПП
У АТОМА ОЛОВА
Связь олова с остатками минеральных и органических кислот, алкокси-,
меркапто-, амино- и другими функциональными группами имеет типично
ковалентный характер. Для синтеза этих соединений применяются два
основных метода. Один из них заключается в действии кислот и других
соединений с достаточно подвижным атомом водорода на соответствующие
кислородсодержащие соединения олова. Например:
R2SnO + 2 НХ -> R2SnX2 + Н20,
4 R2SnO + 4НХ -» (XSnR2OSnR2X)3 + 2 Н20.
Однако в ряде других случаев оловоорганические соединения проще
получать реакцией обмена между оловоорганическими галогенидами и солями
(алкоголятами, меркаптидами и др.) калия, натрия или свинца.
Наряду с этими методами для синтеза оловоорганических соединений с
различными функциональными группами у атома олова применяют
действие соединений с активным атомом водорода на алкоксисоединения
олова, станниламины или оловоорганические галогениды. В последнем случае
реакции проводят в присутствии акцепторов галоидоводорода.
Алкоксисоединения олова, гексаалкилдистанноксаны и станниламины
легко присоединяются по кратным связям. Эти реакции, которые также
приводят к преобразованию функциональных группу атома
олова,рассмотрены на стр. 313—316.
ДЕЙСТВИЕ КИСЛОТ И ДРУГИХ СОЕДИНЕНИЙ
С ПОДВИЖНЫМ АТОМОМ ВОДОРОДА НА ОЛОВООРГАНИЧЕСКИЕ ГИДРООКИСИ,
ОКИСИ ИЛИ СТАННОНОВЫЕ КИСЛОТЫ
Оловоорганические галогениды, сульфаты, алкилсульфонаты, нитраты,
карбонаты, сульфиды, селениды и ацилаты получают действием
минеральных или органических кислот на оловоорганические гидроокиси, окиси или
станноновые кислоты.
Наиболее подробно эти реакции изучены в ряду соединений типа R3SnX.
Так получены, например, галогениды триалкил- [1—4], трициклогексил- [5],
трибензил- [6—8] и триарилолова [9—14], сульфаты триметил- и триэти-
лолова [15], алкил- и арилсульфонаты триалкилолова [16, 17], нитрат три-
метилолова [18], цианаты триэтил- [1] и трифенилолова [19], изотиоционат
триэтилолова [17], а также соединения со связью Sn—О—В [20—22],
Sn—О—Р [23—25] и Sn—О—As [26, 27]. В последних трех случаях вместо
свободных кислот можно применять их ангидриды или эфиры. Эти реакции
часто проходят количественно, что позволяет использовать их для
аналитических целей [28—30].
Реакции преобразования функциональных групп
397
Гексаалкилдистанноксаны (кроме метильного соединения)
адсорбируют углекислый газ воздуха, превращаясь в карбонаты бас-(триалкилоло-
ва) [3, 31,32]. Карбонат бш>(триэтилолова) образуется [33] и при
пропускании углекислого газа в триметилсилокситриэтилолово. Карбонаты триал-
килолова при нагревании до 140—150° С в вакууме количественно
превращаются в исходные гексаалкилдистанноксаны [3].
Гексаэтилдистанноксан образует с сероводородом сульфид бис-(триэтил-
олова) с т. кип. 133—137° С/1 мм 11, 34, 35]. Реакция проходит,
по-видимому, в две стадии с образованием в качестве промежуточного продукта
тиола триэтилолова [35]. В индивидуальном состоянии это соединение не
выделено.
Изоциановая кислота быстро и почти количественно реагирует с гек-
саалкилдистанноксанами при 70—80° С [36]:
R3SnOSnR3 + 2 HNCO -> 2 R3SnNCO + И20.
Сплавление гексабутил- или гексафенилдистанноксана с циануровой
кислотой при 130° С приводит к оловоорганическим триазонам,
обладающим высокой термической и хорошей гидролитической устойчивостью [36]:
(NCOH)s + 2 R3SnOSnR3 — (NCOSnR3)3 + 3 Н20.
Оловоорганические гидроокиси и гексаалкил(арил)дистанноксаны легко
и гладко реагируют с предельными кислотами жирного ряда [2, 20, 35,
37—45], ненасыщенными кислотами [41, 46—57], кислотами с
ацетиленовой связью [54, 58], галоидозамещенными кислотами [2, 35, 39, 41, 59, 60],
ароматическими кислотами [39—41, 55, 61—63], аминокислотами [64, 65],
N-ациламинокислотами [64] и другими карбоновыми кислотами [66—71].
Исследовано [35] взаимодействие гексаэтил- и гекса-я-пропилдистаннок-
сана с органическими кислотами с константой диссоциации Ю-"1—10~10. Во
всех случаях ацилаты триалкилолова образуются с хорошим выходом при
нагревании гексаалкилдистанноксана с избытком кислоты.
Так же получены ацетаты триметил- (т. Щ1. 197° С) [2, 40], триэтил-
(119°С)[35], трибутил-(85°С) [40], триоктилолова (47—48°С) [40], моно-
хлорацетаты триметил- (148°С) [40] и триэтилолова (111°С) [39].
Получение метакрилата трибутилолова [47—49]. К раствору 207,6 г гексабутилдистан-
ноксана в 600 мл бензола медленно прибавляют 60 г метакриловой кислоты при
температуре не выше 25° С. Смесь постепенно нагревают и выдерживают при 30° С. Образующуюся в
результате реакции воду удаляют в вакууме. Потерю бензола возмещают добавлением его
через капельную воронку. После того как раствор станет прозрачным, реакцию продолжают
в течение еще 1 часа (общая продолжительность реакции 5 час.) и бензол полностью
отгоняют. К остатку приливают 200 мл петролейного эфира и раствор охлаждают до —20° С.
Вещество выделяется в виде длинных крупных прозрачных кристаллов с т. пл. 18° С.
Выход практически количественный.
При прибавлении 25,1 г акриловой кислоты к раствору 103,8 г гекса-
бутилдистанноксана в 300 мл бензола образуется акрилат трибутилолова
[47]. Для завершения реакции смесь нагревают при 50° С в течение часа,
а затем кипятят 3,5 часа. Бензол выпаривают, продукт выдерживают при
112° С на протяжении 2 час. К остатку, представляющему собой вязкую
жидкость, прибавляют петролейный эфир. Выход акрилата трибутилолова
•близок к количественному, т. пл. 74,5—75°С.
Нагреванием гидроокисей триалкилолова или гексаалкил(фенил)дистан-
ноксанов с ацетилендикарбоновой, пропиоловой или фенилпропиоловой
кислотами в бензоле или смеси эфира с бензолом получены [54]
оловоорганические ацетиленкарбоксилаты. Триэтил-, трипропил- и трибутилстанни-
.льные производные ацетилендикарбоновой кислоты представляют собой
.кристаллические вещества, которые при нагревании до 160—180° С раз-
398 Оловоорганические соединения. Лит. стр. 424—431
лагаются, давая с хорошим выходом дизамещенные ацетилены [54]:
R3SnOOCC=CCOOSnR8-> 2С02 + R8SnC=CSnR8.
Интересно отметить, что триалкилстаннильные эфиры аминокислот
существенно отличаются от производных карбоновых кислот и N-ацилами-
нокислот по своей химической активности. При контакте с водой, этиловым
и бензиловым спиртами или с я-крезолом они легко разлагаются с
выделением свободных аминокислот [64].
Ацилаты триалкилолова с различными радикалами у атома олова
хорошо растворяются в обычных органических растворителях и очистить их
перекристаллизацией часто не удается. Чистые вещества получают
растворением гексаалкилдистанноксана в небольшом избытке кислоты с
последующим высушиванием в вакууме над твердым едким кали до постоянного
веса [6, 44, 72—74]. Перфторацилаты диметил-я-бутил- и диметил-л-ами-
лолова могут быть очищены перегонкой в вакууме [75].
Замена одного из алифатических радикалов на фенилыюе кольцо снижает
растворимость соответствующих ацилатов и тем самым облегчает их
очистку, как это можно видеть из следующего примера.
Получение ацетата фенилдизтилолсва [40]. К окиси б^с-(фенилдиэтилолова),
полученной обычным образом из 6,7 г сырого бромистого фенилди этил олова, прибавляют 1 г (0,017
моля) ледяной уксусной кислоты. Образовавшуюся твердую массу кристаллизуют из 3 мл
этилового спирта. Осадок промывают петролейным эфиром и высушивают в вакууме над
хлористым кальцием. Получают 2,7 г (43%) вещества с т. пл. 93,5—94,5° С.
В ароматическом ряду реакцию проводят так же, как и в
алифатическом. Так, ацетат трифенилолова получен [40] нагреванием до 130° С 3 г
гидроокиси трифенилолова и 0,5 г ледяной уксусной кислоты. После
охлаждения твердую массу экстрагируют этиловым спиртом в течение 3 час.
Спиртовый раствор (40 мл) выпаривают наполовину, охлаждают,
выделившиеся кристаллы отфильтровывают и высушивают в вакууме. Получают
2,1 г (62%) ацетата трифенилолова с т. пл. 122° С.
Получение ацетата три-л^-тслилолсва [76]. 11,1 г гидрсскиси три-^-тслилолсва и 1,7 г
ледяной уксусной кислоты растворяют в 10 мл бензола. Бензол и выделившуюся в
результате реакции воду отгоняют. В остатке получают масло, которое постепенно закристаллизо-
вывается. Получают 11,4 г (93%) ацетата три-^-толилолова с т. пл. 91—96° С (из петролей-
ного эфира).
Аналогично получены [40, 76] ацетаты три-я-тслил- (т, пл. 114—116°С)>
трибензил- (т. пл. 118,5—119,5° С), три-2-фенилэтил- и три-а-нафтило-
лова.
Синтез полимеризующихся оловоорганических производных п-винил-
бензойной кислоты, N-виниламида фталевой кислоты и N-(o-, я-карбокси-
фенил)-акрил(метакрил)амидов проводят в более мягких условиях [55, 77].
Получение /г-винилбензсата трифенилслсва [55]. К раствору 1,48 г (0,01 моля) /г-ви-
нилбензойной кислоты в 30 мл эфира прибавляют по каплям 3,67 г (0,01 моля) гидроокиси
трифенилолова в 30 мл эфира. Смесь перемешивают в течение часа и эфир отгоняют.
Получают бесцветные кристаллы с т. пл. 81—83° С; выход 4,7 г (95%).
Помимо минеральных и органических кислот оловоорганические
гидроокиси и гексаалкил(арил)дистанноксаны реагируют также с перекисями,
амидами [77а] и имидами кислот. Так, гидроокиси триуетил-и трифенилолова
образуют с перекисью водорода соединения состава RSr.OOH [78]. При
действии на гексаэтилдистанноксан рассчитанного количества перекиси
водорода в сухом этиловом спирте в присутствии сульфата натрия
получена [78, 79] перекись бмс-(триэтилолова) ^Hs^SnOOSn^Hsb.
Реакции преобразования функциональных групп
399
Другой метод синтеза перекисей указанного типа описан на стр. 416.
При наличии достаточного избытка перекиси водорода образуется пер-
гидрат гидроперекиси триэтилолова — [(C2H5)3SnCOH]2-H202.3TO
вещество медленно разлагается уже при комнатной температуре (продукты
разложения взрывчаты при 150—180° С) [81]. Гексаэтилдистанноксан,
перекись водорода и спирт в гексане в присутствии сульфата натрия образуют
перекись этил триэтилолова — (С2Н5)з5пООС2Н5 [82]. Из
эквимолекулярных количеств надуксусной или надпропионовой кислот и гидроокиси
трифенилолова в тщательно высушенном CHsOH получены [80, 83]
продукты состава (C6H5)2Sn(OH)(OCCR) (или (C6H5)2SnO-(C6H5)2Sn(OOCR)2).
Как уже отмечено на стр. 397, гексаалкилдистакноксаны реагируют с
изоциановой кислотой с образованием изоцианатов триалкилолова. Для
синтеза этих соединений вместо изоциановой кислоты можно применять
. мочевину или биурет [36]. Реакции проводят при 130—140 и 160—170° С
соответственно. Выходы продуктов близки к количественным: [
R3SnOSnR3 + 2 (H2N)2CO -> 2 R3SnNCO + 2 NH3 + H20,
R3SnOH -1- H2NCONHCONH2 ^ R3SnNHCONHCONH2 ■£ R3SnNCO.
При нагревании гидроокиси триэтилолова с я-тол>олсульфонамидом или
имидом фталевой кислоты образуются [40] кристаллы триэтильных
соединений олова со связью Sn—N; их т. пл. 69—70 и 71—73° С соответственно.
Обладая щелочными свойствами [84, 85], гидроокиси триалкилолова и
гексаалкилдистанноксаны реагируют и с другими соединениями,
содержащими подвижный атом водорода: спиртами [56, 86], фенолами [35, 39, 40,
87, 88], силанолами [56, 89], меркаптанами [1, 35, 39, 90—94] и эфирами
меркаптокислот [95—104]. Так, фенокситриэтилолово (т. кип. 147, 5—148°С/
/12 мм) и д-нитрофенокситриэтилолово получены [40] растворением
гидроокиси триэтилолова и фенола в эфире. Фенокситри-я-пропил-, фенокси-
и д-крезолокситрифенилолово образуются практически с количественным
выходом при нагревании гексапропил- или гексафенилдистанноксана с
избытком фенола в течение 10 мин. [39].
С ацетилацетоном гексаэтилдистанноксан образует ацетилацетонат
триэтилолова; т. кип. 92° С/0,3 мм; т. пл. 40° С [105].
Гексаэтилдистанноксан реагирует с этилмеркаптаксм уже при обычной
температуре. После нагревания смеси в течение 10 мин. получают этилмер-
каптид триэтилолова с т. кип. 125—126° С/12 мм. Аналогично получены
[35, 92] изопропил-, я-, изо- и гарет-бутилизоамил-, фенил- и я-толил-
меркапти/ы триэтил- и три-я-пропилолова.
К реакциям рассматриваемого типа можно отнести также
взаимодействие оловоорганических гидроокисей или гексаалкил(арил)дистаннокса-
нов с азотистыми гетероциклическими соединениями [38, 106, 107]:
R3SnOSnR3 + 2 R'H -* 2 R3SnR' + Н20,
где R'— остаток пиррола, пиразола, имидазсла или бензтриазола. Так,
Ы-триметилстаннил-1, 2, 4-триазол получен [106] смешиЕанием водных
растворов хлористого триметилолова, едкого натра и триазсла. Смесь
перемешивают в течение 45 мин., осадок отфильтровывают, высушивают и
обрабатывают горячим ацетоном. Выход чистого вещества 84%; т. пл. 277—
278° С (с разложением).
При смешивании растворов гексабутилдистанноксана с рассчитанным
количеством имидазола в ацетоне образуется N-трибутилстаннилбензимида-
зол с т. пл. 137,5—139° С (из этилацетата).
Аналогичным образом или при нагревании смеси реагентов в бензоле
или спирте получены [106] N-трипропилстаннилимидазол (т. пл. 149—
400 Оловоорганические соединения. Лит. стр. 424—431
150° С), и N-трифенилстаннилимидазол (т. пл. 310—311° С), N-трифенил-
станнилбензимидазол (т. разл. 298° С), Ы(1)-трифенилстаннил-1,2,3-
бензтриазол (т. пл. 270,5—272° С) и другие соединения этого типа.
Переходя к приложению рассматриваемого метода в ряду соединений
типа R2SnX2 и RSnX3, необходимо прежде всего отметить, что щелочные
свойства оловоорганических соединений уменьшаются с уменьшением числа
органических радикалов у атома олова [84]. В том же ряду снижается и
гидролитическая устойчивость связи олова с различными функциональны-
мы группами (см. стр. 381). Поэтому спирты, фенолы и некоторые другие
указанные выше соединения реагируют с окисями диалкил- и диарилолова
более сложно, чем с гексаалкил(арил)дистанноксанами.
При растворении окисей диалкил- или диарилолова в галоидоводород-
ных кислотах образуются оловоорганические дигалогениды. В отдельных
случаях эти реакции проходят уже на холоду. Так, при встряхивании
эфирного раствора дигидроокиси ди-т/?ет-бутилолова с бромистоводородной
или соляной кислотой получены [108] двубромистое и двухлористое ди-
mpem-бутилолово соответственно.
Получение двуиодистого дибутилолова[109]. 15,0 г окиси ди-я-бутилолова при
перемешивании растворяют в смеси 36 мл 54%-ной иодистоводородной кислоты и 60 мл бензола в
течение 30 мин. Смесь перемешивают еще час и оставляют на ночь. Нижний слой отделяют,
промывают водой, водным раствором тиосульфата натрия и высушивают сульфатом натрия.
Получают 14,9 г двуиодистого дибутилолова с т. кип. 124—134° С/0,2 мм.
Гораздо чаще синтез галогенидов типа R2SnX2 проводят при
нагревании [39, ПО—115]. Так получены, например, двухлористое диметил- [2],
диэтил- [15, 116], ди-«-бутил- и дибензилолово [117]. В ароматическом ряду
вместо соляной кислоты целесообразно применять хлористый тионил
[118]:
Ar2SnO + SOCl2 -* Ar2SnCl2 + S03.
При выпаривании в эксикаторе над едким кали раствор окиси диметил-
или диэтилолова в небольшом избытке разбавленной серной кислоты
выделяются прекрасно образованные кристаллы соответствующих сульфатов
[119].
Взаимодействием окисей диалкилолова с алкил- [118] или арилсульфо-
кислотами [120] получены бш>(этилсульфонаты) диэтил- и ди-я-пропилоло-
ва, бис-(метилсульфонат) ди-^-пропилолова и бш>(п-толуолсульфонат)
ди-я-бутил олова.
С рассчитанным количеством азотной кислоты окиси алкилолова
образуют динитраты или (гидрокси)нитраты диалкилолова. Интересно отметить,
что последние с трудом конденсируются в дистанноксаны [18], тогда как
с (гидрокси)хлоридами и (гидрокси)ацилатами диалкилолова эта реакция
проходит очень легко (см. стр. 385).
В литературе, преимущественно патентной, описаны также реакции
окисей диалкил- и диарилолова с сероводородом [121, 122], эфирами борной
[20—22], фосфорной [23] и арсоновой [27] кислот. К оловоорганическим
соединениям, содержащим фосфор, приводит также действие фосфорной
кислоты фосфорного ангидрида или хлорокиси фосфора на окиси
диалкилолова [24, 123].
Результат реакции органических кислот с окисями диалкилолова
определяется молярным соотношением реагирующих веществ. Нагревание
эквимолекулярных количеств оловоорганической окиси и кислоты приводит
к со, (о-дианилатам тетраалкилдистанноксана [61, 124—126]:
2 R2SnO + 2 R'COOH -> R'COO (SnR20)2OCR' + H20.
По-видимому, в качестве промежуточных продуктов при этом образу-
Реакции преобразования функциональных групп
401
ются (гидрокси)ацилаты диалкилолова [127], которые обработкой вторым
молем кислоты могут быть превращены в соединения с двумя различными
ацильными остатками^ у атома олова [128].
Харада [126] взаимодействием органических кислот с избытком олово-
органической окиси в спирте получил соединения следующего состава:
R'0(R2SnO)3R' и R2Sn(OOCR")2.
При растворении окисей диалкилолова в рассчитанном количестве или
избытке органической кислоты образуются диацилаты диалкилолова
[20, 49, 52, 61, 67, 119, 125, 126, 128—137].
Впервые Кауром [119] получены этим методом диацетаты диметил-
и диэтилолова.Он описал их в виде правильно образованных призм (без
констант). Позже из продуктов реакции окиси диметилолова с уксусной
кислотой был выделен [129] только о),со -диацетат тетраметилдистанноксана.
Диацилаты диметил- (т. пл. 67°С) и диэтилолова (т. пл. 44°С) получены
[136] нагреванием соответствующих окисей диалкилолова с избытком
уксусного ангидрида с последующим удалением его в вакууме. Синтез
и очистку этих веществ проводят в атмосфере азота.
Диацилаты диметилолова и сильных органических кислот, например
диформиат (т. пл. 184—185°С) или диметакрилат (т. пл. 140—144°С)
диметилолова, являются устойчивыми соединениями и легко получаются при
растворении окиси диметилолова в кислоте [129, 130].
Дипропил-, дибутил- и высшие алкильные соединения олова образуют
устойчивые соединения и со слабыми органическими кислотами. Низшие
члены ряда легко очищаются перегонкой в вакууме. Например, диацетат
ди-я-бутилолова имеет т. кип. 146—147° С/10 мм [138].
Малеаты, лауринаты и другие производные высших карбоновых
кислот представляют собой маслянистые жидкости; при нагревании в
вакууме они разлагаются, не перегоняясь. Эти соединения удобно получать
следующим образом [132].
По первому методу в колбу емкостью 1 л, снабженную мешалкой и
обратным холодильником, помещают 2,5 моля органической кислоты. К
нагретой до 60—70° С кислоте при перемешивании прибавляют 1,25 моля
окиси ди-я-бутилолова и смесь нагревают при той же температуре в течение
2—2,5 час. Образовавшуюся в результате реакции воду отгоняют под
вакуумом. Отгонку воды можно заменить сушкой реакционной смеси.
Предварительно растворенную в хлороформе или другом инертном растворителе
смесь пропускают через колонку высотой 350 и диаметром 25 мм,
заполненную хроматографической окисью алюминия. При этом, помимо
поглощения воды, осуществляется очистка конечного продукта от смолы.
Растворитель отгоняют от полученного вещества под вакуумом. Выход продукта
95—98%. Метод применим для получения дикаприлата, дилаурината, ди-
стеарата и диолеата ди-я*бутилолова.
Второй метод состоит в следующем [132]. В круглодонную колбу с
мешалкой, обратным холодильником и термометром помещают 1 моль
ангидрида кислоты и 150 мл толуола. Смесь нагревают до 60—80° С, добавляют
1 моль оловоорганической окиси и температуру в колбе поддерживают на
уровне 60—80° С до образования прозрачного раствора. Под вакуумом
отгоняют толуол. Выход продукта 95% и выше. Метод применим, например,
для получения малеата ди-я-бутилолова.
Нагревание окисей диалкилолова с двухосновными кислотами приводит
к полимерным продуктам [139, 140]. Двухосновные кислоты с короткой
цепью, например янтарная или адипиновая, образуют циклические
продукты с небольшим молекулярным весом (от 2 до 4 звеньев). Так, сукцинат
ди-я-бутилолова представляет собой циклический тетрамер ст. пл. 187—
187,5° С (из бензола) [140].
26 Заказ М 207
402 Оловоорганжеские соединения. Лит. стр. 424—431
При проведении реакции с себациновой, итаконовой или терефталевой
кислотой возможность циклизации уменьшается. Образующиеся
продукты имеют молекулярный вес от 3000 и выше [140].
По патентным данным [141—144] при нагревании окисей диалкилолова
с эквимолекулярным количеством жирной кислоты и 0,5 молями
малеинового ангидрида образуются соединения типа (=CHCOOSnR2OOCR')2.
Замена малеинового ангидрида на моноэфир малеиновой кислоты приводит к
(алкилмалеат)ацилатам диалкилолова [145], тогда как из окиси дибутило-
лова, малеинового ангидрида и малеиновой кислоты получены [146]
полимерные продукты.
Что касается диацилатов диарилолова, то они известны пока лишь на
отдельных примерах. Диацетат дифенилолова (т. пл. 116—117° С) получен
с выходом 57% растворением окиси дифенилолова в уксусной кислоте-при
комнатной температуре с последующим удалением избытка кислоты в
вакууме (температура не выше 35° С). Аналогично получен дипропионат
дифенилолова — белое кристаллическое вещество ст. пл. 75—76° С. В
бензоле оба этих соединения мономерны [146а]. Взаимодействие окиси
дифенилолова с винной кислотой [1466], вероятно, осложняется наличием окси-
групп (см. стр. 405).
Диацилаты дифенилолова получены также взаимодействием окиси
дифенилолова с диацилатами кремния (см. стр. 420).
К соединениям, у которых атом олова одновременно связан с
галоидом и остатком органической кислоты, приводит следующая реакция
[129, 147]:
ClSnRoOSnRoCl + R'COOH -» R2Sn(Cl)(OOCR') + Н20.
Получение (хлор)ацетата диэтилолсва [147]. 1 г дихлорида тетраэтилдистанноксана и
3 мл уксусной кислоты нагревают на водяной бане в течение 10 мин. Образуется прозрачный
раствор. Избыток кислоты отгоняют в вакууме и остаток кристаллизуют из уксусной кислоты
или возгоняют; т. пл. 94° С. Выход практически количественный.
Аналогично получены другие (хлор)ацетаты [147] и (хлор)формиаты
[129, 147] диалкилолова. Последние кристаллизуются с молекулой воды.
Другой метод синтеза соединений этого типа состоит в
нагревании.окисей диалкилолова с галоидангидридами кислот [124]. Так, нагревание 2,3 г
окиси диметилолова с 1,1 г хлористого ацетила в 15 мл сухого бензола в
течение 10 час. дает (хлор)ацетат диметилолова с выходом 67,8%. О синтезе
(галоид)ацилатов диалкилолова см. также стр. 422.
Что касается других производных кислот, то при нагревании окисей
диалкилолова с алкилгидроперекисями получены [148] хорошо
кристаллизующиеся соединения типа R4Sn2(OOR,)2 О.
При сплавлении окиси дибутилолова с мочевиной выделяются аммиак
и вода, после чего при 170—180° С образуется воскообразный продукт
(т. пл. 195 — 215° С), состав которого соответствует формуле
[(C4He)2SnOSn(C4H9)2(NCO)2]„ [36].
Согласно патентам данным [149—151], при нагревании окисей
диалкилолова со спиртами в толуоле или циклогексане образуются диалкокси-
диалкильные соединения олова. Однако позже было показано, что реакция
проходит более сложно и приводит к смеси олигомеров состава
R'0(SnR20)rtR' [152, 153] (ср. [154]). Молекулярный вес этих продуктов
зависит от условий проведения реакции. Окись ди-я-бутилолова не
реагирует в заметной степени с я-бутиловым спиртом при нагревании
эквивалентных количеств реагентов в эфире, циклогексане или толуоле. При
применении большого избытка спирта в отсутствие растворителя получают
продукт со средней степенью конденсации п =2,5 -ч- 5,7. Аналогичные
результаты получены [152] с я-гексиловым и я-амиловым спиртами.
Реакции преобразования функциональных групп
403
При нагревании окиси дибутилолова с этилен гликолем образуется
циклический продукт состава [(С^^пОСНгСНгО^ [155, 156], а не
(С4Н9)25пОСН2СН20, как это предполагалось ранее [150].
Систематические исследования реакции окисей диалкилолова с диолами,
проведенные Воронковым и Ромоданом [156а], показали, что она
преимущественно протекает по общей схеме:
О
R2SnO + HOR'—ОН •£. R2Sn R' + Н20
v
или
О— R'—О
/ \
R2SnO + НО— R'—ОН •£ R2Sn SnR2 + Н20.
\ /
О—R'—О
В случае 1,2- и 1,3-гликолей получают циклические простые эфиры
соответствующих диалкилстаннандиолов, обладающих пяти-, шести- или деся-
тичленным циклом. Например, продуктом взаимодействия окиси
диэтилолова с пропандиолом-1,3 является 2,2-диэтил-2-станна-1,3-диоксан:
ОСН2
/ \
(C2H5)2Sn СН2.
осн2
Расщепление окиси диэтилолова пропандиолом-1,2 и бутандиолом-2,3 дает
циклические продукты, которые имеют, по-видимому, следующую
структуру:
OCHR'CHR"0
/ \
R2Sn SnR2
X /
OCHR'CHR"0
В то же время нагревание окисей диалкилолова с диэтиленгликолем
приводит к легко перегоняющимся в вакууме мономерным 2,2-диалкил-2-станна-
1,3,6-триоксациклооктанам [156а]:
ОСНоСНо
/ \
R2Sn О
ОСН2СН2
Гетероциклические оловоорганические соединения получены [156] и при
нагревании окиси ди-я-бутилолова с транс- и ^ис-1,2-циклогександиолом
или Л4&зо2,3-бутандиолом.
По патентным данным [157] нагревание оловоорганических окисей с
моноэфирами глицерина приводит к циклическим продуктам типа
R2SnOCH2CHOCH2OOCR' или [OCH2CH2CH2OOCR'OSnR2]n.
Фенолы образуют с окисями диалкилолова диарилокситетраалкил-
дистанноксаны [158]:
2 R3SnO + 2 АгОН -* [АЮ (SnR20)2Ar]2 + Н20.
Получение дифенокситетрабутилдистанноксана [158]. В трехгорлую колбу, снабженную
механической мешалкой, обратным холодильником и прибором Дина — Старка, помещают
47,1 г (0,5 моля) фенола, 124,5 г (0,5 моля) окиси дибутилолова и 500 мл толуола. Смесь ки-
26*
404 Оловоорганические соединения. Лит. стр. 424—431
пятят при перемешивании в течение 2 час. Отгоняется 4,5 мл воды. Мутный раствор
фильтруют и толуол отгоняют в вакууме. В остатке 159,0 г (95%) масла желтого цвета, которое
постепенно закристаллизовывается; т. пл. 137—139,5° С (из гексана).
Аналогично получены [158] ди-трет-бутилфенокси* и ди-Р-нафтокси-
тетрабутилдистанноксаны. Вещества, образующиеся в результате
реакции окиси ди-я-бутилолова с я-нитрофенолом и гидрохиноном, не
растворяются в толуоле и выделяются из смеси практически в чистом виде. Дистан-
ноксаны, полученные из гидрохинона или 2,2-б^с-4-(оксифенил)пропана и
окиси ди-я-бутилолова имеют, вероятно, полимерную природу.
Во всех случаях применение 2 молей фенола на моль оловоорганической
окиси не приводит к соединениям типа R2Sn(OAr)2. Из реакционной смеси
выделены непрореагировавший фенол и соответствующие диетанноксаны
[158].
По патентным данным [159] при нагревании до 80—120° С оловооргани-
ческих окисей с моно- или дигидрофенолами в среде углеводородов
образуются соединения состава [R2Sn(OH)]mX,^, где X — остаток исходного фе-
нола,
С ацетил ацетоном и 1,3-бутандионом окись диметилолова образует
соответствующие соединения типа R2SnX2 [160].
В отличие от спиртов, меркаптаны гладко реагируют с окисями диал-
килолова с образованием димеркаптидов диалкилолова [152]. Эта реакция
широко освещена в патентной литературе Г92—94, 161, 162]. Помимо
меркаптанов, применялись меркаптоспирты [163, 164], меркаптокислоты [165],
эфиры меркаптокислот [95—104, 166—173] и другие замещенные
меркаптаны [174—178]. Реакции проводят путем нагревания смеси реагентов в
толуоле или без растворителя.
Нагревание эквимолекулярных количеств меркаптана, органической
кислоты и окиси дибутилолова приводит к (меркапто)ацилатам дибутил-
олова [179, 180].
Реакции алкил- и арилстанноновых кислот с кислотами, спиртами и
меркаптанами описаны очень кратко.
Галоидоводородные кислоты превращают алкилстанноновые кислоты в
тригалогениды алкилолова [181—183]. Интересно отметить, что винил-
иг аллилстанноновые кислоты образуют с бромистоводородной кислотой
тримеры состава (CH2=CHSnBr3)3 (т. пл. 119° С) и (CH2=CHCH2SnBr3)3
(т. пл. 109° С) соответственно [184] (ср. [185]).
а-Стильбенилстанноновая кислота при обработке хлористым тионилом
Дает треххлористое а-стильбенилолово.
Получение треххлористого а-стильбенилолова [118]. К суспензии Зг (0,009 моля)
а-стильбенилстанноновой кислоты в 10 мл петролейного эфира прибавляют 4,5 г (0,036 моля)
хлористого тионила в 25 мл петролейного эфира. Смесь нагревают при 60—70° С в течение
1,5 часа, после чего растворитель и избыток хлористого тионила отгоняют в вакууме.
Получают 3,2 г (88%) треххлористого а-стильбенилолова с т. пл. 109—110° С (из гептана).
С серо- и селеноводородом алкилстанноновые кислоты реагируют по
схеме [182, 186]:
2 RSnOOH -f 3H2Se -> (RSn3e)3Se -f 4H20
Обработка метил- или винилстанноловой кислоты органическими кислотами
с последующей очисткой продукта реакции перегонкой или
перекристаллизацией приводит к соединениям состава
CH3Sn(OOCR)203n (СН3) (03CR) ОЗп (СН3) (OOCR)2 [187]
И
[CH2=C4Sn (О) OOCR]3 [184]
Реакции преобразования функциональных групп
405
соответственно. Однако при кипячении 7 г ледяной уксусной кислоты с 5 г
ангидрида бутилстанконоЕой кислоты в течение часа с выходом 89%
выделен [188] триацетат я-бутилолова с т. кип. 125—129° С/1 мм. Аналогично
получен [188] и трипропионат я-бутилолоЕа. Триацилаты, алкил- и фенил-
олова целесообразнее получать реакцией обмена (см. стр. 407).
В патентной литературе описан ряд соединений типа RSn (SR')3,
которые получают нагреванием до 125-—150° С алкил- и арилстанноновых
кислот с меркаптанами [92, 94, 189—192], меркаптсспиртами [95] или зфирами
меркаптокислот [98, 1С0, 101,103, 104, 167].
РЕАКЦИЯ ОБМЕНА
Реакции оловоорганических окисей и станноновых кислот с
соединениями, содержащими подвижный атом Еодорода, не всегда дают надежные
результаты. При синтезе оловоорганических ацилатов из окисей и кислот
иногда наблюдаются побочные процессы, например [72]:|
[(C4H9)r(CHt=CHCH2)Sri]bO + 2 СНс[ССОИ -»
-> СН3СОО [Sn (QH9)20]2OCCH3 -f 2 С3Нб.
Метод не приводит к цели при синтезе ацилатов замещенных кислот [59], а
также алкоксипроизводных олова (см. стр. 402) и других нестойких к
гидролизу оловоорганических соединений.
Во многих случаях олоЕООрганические соединения проще получать
реакцией обмена между оловоорганическими галогенидами и солями калия,
натрия, серебра и др. Например, для этильных соединений олова описан
[1] следующий ряд превращений: R3SnSCH3 и R3SnSSnR3 -» R3SnJ ->
— R3SnBr ~> R3SnCN ~> R3SnNCS -> R3SnCl -> R3SnOSnR3 -> R3SnNCO ->
-> R3SnOOCCH3 -> R3SnF.
Каждое последующее соединение может быть получено из предыдущего
нагреванием его с соответствующей солью серебра. Подобный же рял
превращений осуществлен [193] и в ароматическом ряду, исходя из йодистого
трифенилолова.
Реакцию обмена можно вести без растворителя. Например, цианистое
триэтилолово получено [194] из цианистого серебра и йодистого триэтил-
олова. Обмен чаще ведут в растЕорителе.При этом желательно, чтобы один
из продуктов выпадал в осадок.
Примером обмена в водно-спиртоЕОЙ среде могут служить
многочисленные случаи получения оловоорганических фторидов [5—7, 42, 108, 109,
195—198] типа R3SnF и R2SnF2 B алифатическом и ароматическом рядах.
Получение двуфтористсго дкбутилолсва [109]. К раствору 6,08 г дву хлор истого дибу-
тилолова в 25 мл этилового спирта приливают раствор 2,32 г фтористого калия в 10 мл воды.
Сразу же выпадает объемистый белый осадок. Смесь разбавляют 75 мл воды и перемешивают
в течение 5 мин. Осадок (5,12 г; 95%) отфильтровывают, промывают водой, высушивают при
120° С и перекристаллизовывают из 150 мл этилового спирта. Температура плавления
чистого вещества 157—160° С.
Оловоорганические фториды представляют собой кристаллические
вещества, не растворимые в обычных органических растворителях. Их
применяют для выделения различных оловоорганических соединений, когда
последние не удается очистить обычными методами [6, 197].
Реакцию между хлористым трифенилоловом и ионом фтора
предложено [199—201] использовать для ксличесгЕенгого определения последнего в
растворе.
Если, как это имеет место в сл^чге йодистых и роданистых солей,
исходная соль натрия или калия хорошо растворяется в спирте и ацетоне, то
406 Оловоорганические соединения. Лит. стр. 424—431
обмен удобно вести в этих растворителях, наблюдая ход реакции по
выпадению хлористого натрия.
Получение йодистого трициклопропилолова [42]. К 6,52 г (0,0203 моля) хлористого три-
циклопропилолова в 10 мл ацетона прибавляют раствор 0,0203 моля йодистого натрия в
15 мл ацетона. Осадок хлористого натрия отфильтровывают, ацетон отгоняют и остаток
перегоняют в вакууме. Получают 6,3 г (84%) вещества с т. кип. 97,5—98°С/0,4 мм; п2£ 1,5908.
Аналогично получены двуиодистое диметил- [115] и дициклопропилоло-
во [42], йодистое тримезитилолово [202], двуиодистое ди-я-хлорфенил-, ди-
я-бромфенил- и ди-я-иодфенилолово [203, 204].
Обменом оловоорганических хлоридов с соответствующими солями
натрия или серебра в спирте или ацетоне получены арилсульфонаты
триэтилолова [43], бш>(метилсульфонаты) диалкилолова [17], нитрат и перхлорат
триметилолова [205], нитраты триэтил- [206] и трифенилолова [207, 208],
диизотиоцианаты диметил-[115, 209], диэтил-[209], дибутил-[115, 209,
210], дициклопропил- [42] и дивинилолова [120] и другие [193, 211 —
213], а также диизотиоцианаты и (гидрокси)изотиоцианаты тетраалкил-
дистанноксанов (из ClSnR2OSnR2Cl и ClSnR2OSnR2OH соответственно)
1209]. ИК-спектры оловоорганических изотиоцианатов указывают на
наличие в них связи Sri—N [209, 211, 212].
Получение диизотиоцианата дипропилолова [209]. К раствору 2,8 г дву хлористого
дипропилолова в 6 мл этилового спирта прибавляют 1,6 г роданистого натрия в 13 мл
спирта. Сразу же выпадает осадок хлористого натрия, его отфильтровывают. Выпариванием
фильтрата в вакууме получают 2,9 г вещества с т. пл. 135—136° С (из бензола).
Действием цианистого калия или цианистого серебра на
оловоорганические галогениды получены изоцианиды триметил- [214], триэтил-[1, 34, 40,
213] и дипропилолова [215]. В качестве растворителя применяют бензол.
Синтез изоцианида триэтилолова лучше всего проводить в смеси эфира с
водой, тогда как в случае диизоцианида диметил олова присутствие воды
в системе приводит к побочным реакциям [216, 217].
Фульминат трифенилолова получен [218] из хлористого трифенилолова
и фульмината калия в жидком аммиаке.
Вследствие слабой диссоциации связи Sn—S образование
оловоорганических сульфидов происходит даже в том случае, когда образуются хорошо
растворимые продукты реакции. Например, сульфиды бш>(триалкил)-,
диалкил-, трикумил- и дикумилолова получены [20, 122, 219—222] обменом
сульфидов натрия или аммония с раствором (или суспензией)
оловоорганических галогенидов в воде. Подобным жэ образом получены сульфиды
состава (RSn)2S3 [223]. С дву хлористым дифенил оловом реакцию проводят
в смеси ацетона с водой [224].
Получение сульфида ди-ж-толилолова [225]. К раствору 1 г двухлористого ди-лс-толил-
олова в 2 мл сухого спирта прибавляют спиртовый раствор щелочи, насыщенный
сероводородом (0,4 г едкого кали растворяют в 7,6 мл спирта и раствор насыщают сероводородом).
Выпавший осадок отфильтровывают и высушивают при 60° С. Вещество очищают
следующим образом: растворяют в хлороформе, фильтруют и высаживают спиртом. После
двух кристаллизации т. пл. 121,6—122° С. Выход 84%.
Аналогично получены [203, 204, 226] и другие сульфиды диарилолова,
а также сульфид ди-(триметилсилилметиленЪлова [227].
Селениды диметил- и бис-(трифенилолова) получены [228, 229]
взаимодействием селенида натрия с оловоорганическими хлоридами в сухом
бензоле. Их синтез и очистку проводят в атмосфере сухого азота. Чистые
вещества устойчивы на воздухе и не гидролизуются в заметной степени водой.
По патентным данным [122] синтез сульфидов и селенидов бис-(три ал кил-
и б^с-(триарилолова) можно проводить в водном или спиртовом растворе.
Реакции преобразования функциональных групп
407
Обменом оловоорганических галогенидов с солями калия, натрия или
свинца получены ацилаты три- [64, 202, 230—238] и диалкилолова [129,
138, 217, 238—242]. При получении ацилатов триалкилолова удобно
пользоваться солями свинца [230]. Последние могут быть получены растворением
свинцового глета в кислоте и не нуждаются в дополнительной очистке.
Примером синтеза диацилатов диалкилолова может служить получение
диацетата диизобутилолова обменом ацетата натрия или калия с двухло-
ристым диизобутилоловом [138, 239]. Выход чистого вещества 76%; т. кип.
123—124° С/3 мм.
Обменом водных растворов двухлористого диметилолова с кислым
фталатом калия или салицилатом натрия получены [217] соответственно
фталат (т. разл. 360° С) и салицилат диметилолова (т. пл. 205—206° С).
В тех же условиях из двухлористого диметилолова и бензоата аммония
образуется (CH3)2SnO.(CH3)2Sn(OOCC6H5)2 (т. пл. 236—237° С). В случае
легко гидролизующихся оловоорганических ацилатов в качестве
растворителя применяют соответствующую кислоту. Так получен [129], например,
диформиат диметилолова (т. пл. 185° С).
Реакция обмена применяется и для синтеза оловоорганических
производных замещенных органических кислот, например окси-[59] или цианук-
сусной кислоты [243].
Изучено [50, 243] декарбоксилирование некоторых из этих соединений.
В алифатическом ряду реакция часто осложняется побочными процессами.
Так, основными продуктами разложения этилмалонатов триэтил- и
трибутилолова, а также цианацетата триэтилолова являются тетраэтил- и тетра-
бутилолово [243]. Декарбоксилирование цианацетата трибутилолова дает
около 20% (цианметил)трибутилолова (жидкость ст. кип. 140—144° С/
/1,3 мм).
Несколько лучше проходит реакция в ароматическом ряду. При
нагревании до 140° С в вакууме а-цианпропионата трифенилолова в течение 45
мин. получают [50] (1-цианэтил)трифенилолово с выходом 27%.
Аналогично проходит декарбоксилирование цианацетата трифенилолова [243].
Синтез триацилатов алкил- и арилолова можно иллюстрировать
следующими примерами.
Получение триизо5утирата бутилолова [188]. 9,0 г треххлористого бутилолова и 26 г
(33% избытка) изобутирата серебра нагревают в 50 г четыреххлористого углерода в течение
90 мин. Смесь фильтруют, осадок промывают четыреххлористым углеродом и из фильтрата
растворитель отгоняют при 60 мм. Перегонка остатка в вакууме дает 2,9 г вещества с т.
кип. 137—139° С/1 Af/if затем основную фракцию (6,322:4. кип.'Ш—141° С/1 мм\ df 1,213;
n2Q 1,4627) и 2,4 г вещества с несколько более высокой температурой кипения.
Аналогично получены триацетат (т. пл. 46° С), трипропионат (п2£
1,470), трибутират (п2о 1,463) и тривалериат (п2о 1,4627) бутилолова.
Получение трипропионата фенилолова [188]. 22 г треххлористого фенилолова и 52 г
(30% избытка) пропионата серебра нагревают в 100 г четыреххлористого углерода в течение
часа. Осадок отфильтровывают, промывают и растворитель удаляют. Перегонка остатка
в вакууме 1 мм при температуре примерно 195 °С дает 25 г (82%) трипропионата фенилолова
в виде переохлажденной жидкости, которую закристаллизовывают охлаждением до —78° С.
Перекристаллизацией из бензола, а затем из хлористого бутила получают чистый
трипропионат фенилолова ст. пл. 66—67° С.
Аналогично получены [188] триацетат фенилолова (т. пл. 76° С после
пяти кристаллизации из" хлористого бутила), триизобутират фенилолова
(т. пл.50,5°С) и три-я-бутират фенилолова (вязкая жидкость с df 1,355;
/22D° 1,5105).
408 Оловоорганические соединения. Лит. стр. 424—431
В отдельных случаях оловоорганические ацилаты получают действием
кислот на алкоголят натрия с последующим обменом образующейся
натриевой соли с оловоорганическими галогенидами [109]. Тот же прием
используют и при синтезе алкилпероксипроизводных три- и диалкилолова [244—
249]. С увеличением числа перекисных групп у атома олова
гидролитическая и термическая устойчиеость оловоорганических перекисей
уменьшается [244].
Реакция оловоорганических галогенидов с алкоголятами щелочных
металлов является основным методом синтеза алкоксисоединений олова
[62, 109, 232, 250—274]. Последние легко гидролизуются, поэтому их
синтез и очистку необходимо проводить при полном отсутствии влаги,
применяя тщательно высушенные спирты. Следует отметить, что при действии ме-
тилата натрия на хлористое триметилолово получены противоречивые
результаты [253, 258]. По-видимому, это объясняется недостаточной
термической устойчивостью метокситриметилолова (см. стр. 417).
Получение диметоксидиметилолова [253]. Синтез и очистку вещества проводят в
атмосфере сухого азота. К алкоголяту натрия (из 6,08-г (0,262 г-атома) натрия в 80 мл сухого
метилового спирта) при 0° С и перемешивании прибавляют по каплям 28,6 г (0,131 моля) дву-
хлористого диметилоловл. Температуру постепенно повышают до комнатной и смесь
перемешивают до нейтральной реакции. Осадок хлористого натрия отфильтровывают и
метиловый спирт отгоняют (под конец в вакууме). Остаток возгоняют в высоком вакууме при 60—
70° С. Получают 6,0 г (20%) диметоксидиметилолова с т. пл. 86—87° С (в запаянном капил-
(ляре).
Метокситриэтил- (т. кип. 73—74° С/13 мм), метокситрипропил- (т. кип.
87—88° С/3 мм) и метокситрибутилолово (т. кип. 101—102° С/2 мм)
получены [250] с выходом 70—80% из соответствующих хлоридов и метилата
натрия в сухом метиловом спирте.
Выделяющийся в результате этих реакций хлористый натрий отделяется
с трудом, что усложняет выделение алкоксисоединений олова. В ряде
статей предлагается проводить реакцию в смеси спирта с углеводородами или
в среде чистых углеводородов. Так получены, например, этокситриэтил-
1258], диэтокси- [257] и дифеноксидибутилолово.
Получение дифеноксидибутилолова [275]. Реакцию проводят в атмосфере сухого азота
в трехгорлой колбе емкостью 1 лу снабженной мешалкой, обратным холодильником,
соединенным с прибором Дина — Старка, капельной воронкой и газоотводной трубкой. К
свежеприготовленному раствору метилата натрия (46 г натрия в 3 л сухого метилового спирта)
прибавляют 188,2 г (2 моля) фенола и смесь кипятят в течение 2 час. Затем приливают 1 л
сухого гептана, метиловый спирт отгоняют и отделяют от гептана в приборе Дина — Старка
(для полного удаления метилового спирта требуется примерно 18 час.) Потерю гептана
возмещают периодически добавлением его к раствору. После удаления всего метилового спирта
выпадает твердый белый осадок (фенолят натрия). При непрерывном перемешивании
прибавляют раствор 303,8 г (1 моль) двухлористого дибутилолова. Смесь кипятят в течение 4 час.
По охлаждении осадок хлористого натрия отфильтровывают, промывают 250 мл гептана, и
из объединенных гептановых вытяжек растворитель удаляют в вакууме. В остатке —
маслянистая жидкость, кристаллизующаяся при охлаждении. Выход 90%; т. пл. 45—48° С
(из гептана); т. кип. 161° С/0,35 мм.
Получить феноляты некоторых замещенных фенолов (например, галои-
дированных) с помощью натрия, амида натрия или литийорганических
соединений не удается. Для этой цели применяют [261] гидрид натрия.
Синтез галоид- и нитрофенокситрифенилолова проводят в среде тетрагид-
рофурана. Выходы продуктов близки к количественным.
Получение 2-хлорфенокситрифенилолова[261]. 1, 268 г (0,01 моля) 2-хлорфенола в 10 мл
сухого тетрагидрофурана постепенно смешивают с 0,3 г (0,012 моля) гидрида натрия, а
затем к смеси сразу прибавляют 3,855 г (0,01 моля) хлористого трифенилолова в небольшом
количестве тетрагидрофурана. После начала реакции (что видно по выделению бесцветного
Реакции преобразования функциональных групп
409
осадка) смесь кипятят в течение 30 мин. и оставляют на ночь. Осадок отфильтровывают и
фильтрат упаривают. Кристаллизацией остатка из смеси бензола с петролейным эфиром (1:30)
получают бесцветные иголочки 2-хлорфенокситрифенилолова. Выход 4,32 г (90,5%); т. пл.
59—60° С. Вещество хорошо растворяется в тетрагидрофуране, диоксане, бензоле и сухом
этиловом спирте, умеренно — в петролейном эфире.
^Аналогично получены [261] 4-бромфенокси-, 2,4,6-трихлорфенокси-
2-бромфенокси-, 2,4,6-трибромфенокси- и другие (галоидфенокси)трифе
нильные соединения олова, а также о, м- и п-нитрофенокситрифенилолово.
По патентным данным [276, 277], оловоорганические галогениды при
действии на них смеси алкоголята натрия и солей органических кислот
превращаются в соединения типа R2Sn (OR')(OOCR").
Ацетилацетонат таллия (I) [278] или натрия [160] образует с двухлори-
стым дифенилоловом ацетилацетонат дифенилолова (т. пл. 123—125° С).
Аналогично получен [278] дибензоилацетонат дифенилолова (т. пл. 239—
24Г С).
Взаимодействием оловоорганических галогенидов с меркаптидами
натрия [91, 279, 280], свинца [281] или ксантогенатами натрия [280, 282, 283]
получены соединения типа R3SnSR', R2Sn (SR')2 и RnSn(SCSOR')4-/2
соответственно. В патентной литературе [284—288] описан ряд производных
три- и диалкилолова, в которых атом олова связан с одной из следующих
групп: SC(S)SR, SC(S)NR2 или SC(NR)SR.
Реакция обмена успешно применена и для синтеза станниламинов,
которые играют важную роль в химии оловоорганических соединений
(см. стр. 314,327, 417,481). Синтез станниламинов типа RnSn(NR/2)4-rt из
оловоорганических хлоридов и диалкиламидов лития исследован рядом
авторов [289—295]. Константы этих веществ приведены в обзорной статье
Джонса и Лапперта [296].
Получение диэтиламинотриметилолова [3]. Эфирный раствор диэтиламида лития
(приготовлен из 0,76 г лития, 6,9 г бромистого я-бутила и 4,0 г диэтиламина в 30 мл эфира)
прибавляют по каплям к раствору 7,3 г бромистого триметилолова в 30 мл эфира. Для
завершения реакции смесь кипятят в течение 3 час, растворитель отгоняют и остаток
перегоняют при обычном давлении. Собирают фракцию с т. кип. 156—162° С. Выход 61%.
Другой способ синтеза соединений этого типа состоит в реакции
оловоорганических галогенидов с избытком бромистого диэтиламиномагния в
тетрагидрофуране [31]: |
?R3SnX + (C2H3)2NMgBr -» R3SnN (С2Н5)2 + MgBrX
(R = C2H5, /г-С3Н7, /г-С4И9).
Следует отметить, что с солями первичных аминов реакция иногда
осложняется побочными процессами, приводящими к JST-алкилдистанназанам
[291]:
R3SnNHR' + R3SnX -> (R3Sn)2NR' + НХ,
2R3SnNHR'-* (R3Sn)2NR'+ R'NH2.
При действии амида лития или натрия на галогениды триалкилолова
образуются три(триалкилстаннил)амины [3]. С амидом лития реакцию
можно проводить в аммиаке, диэтиловом эфире или тетрагидрофуране. Амид
натрия реагирует с галогенидами триалкилолова только в жидком аммиаке.
В диэтиловом эфире, тетрагидрофуране или пиридине реакция не проходит
даже при температуре кипения соответствующего растворителя. Три(триал-
килстаннил)амины легко окисляются и гидролизуются на воздухе, поэтому
их синтез проводят в атмосфере сухого инертного газа.
410 Оловоорганические соединения. Лит. стр. 424—431
Получение т<ри(триметилстаннил)амина [3]. Амид лития получают из 0,56 г (0,08
г-атома) лития и 50 мл жидкого аммиака в присутствии 0,01 г азотнокислого железа. К
нему добавляют 50 мл сухого эфира и для удаления аммиака смесь выдерживают при
комнатной температуре, а затем кипятят в течение часа с обратным холодильником. К
образовавшейся суспензии прибавляют 12,1 г (0,05 моля) бромистого триметилолова в 30 мл сухого
эфира. Смесь кипятят в течение 3 час. и перегоняют в вакууме. Получают 7,0 г (83%)
вещества с т. кип. 130° С /14 мм\ т. пл. 22—24° С.
Аналогично получены три(триэтилстаннил)- и три(три-я-пропилстан-
нил)амины.
Три(триметилстаннил)амин образуется при действии нитрида лития на
хлористое триметилолово в тетрагидрофуране [297]:
Li-3N + 3 (CH3)3SnCl -* [(CH3)3Sn]3N -f 3 LiCl.
К бш:-(триметилсилил)аминотриметилолову (т. кип. 58—59° С/1 мм;
т. пл. 20—22° С) [298, 299] и бш;-(триметилсилил)аминотрибутилолову
[299] (т. кип. 140—145° С/1 мм) приводит следующая реакция:
l(CH3)3Si]2NNa + R3SnCl -> R3SnN [Si (СН3)3]2 + NaCl.
Реакцией обмена получен ряд других соединений со связью Sn—N. Так,
, хлористое триметил-[106, 107, 300], трибутил-[106, 107], трифенил- [106,
107, 300—302], трибензил- [303] и тритолилолово 1*303] образуют с азидом
натрия соответствующие оловоорганические азиды.
Получение азида триметилолова [302]. Раствор 1,55 г хлористого триметилолова в 50 мл
дистиллированной воды смешивают с 1,0 г азида натрия в 50 мл 0,2 N соляной кислоты.
Смесь оставляют на час при обычной температуре, а затем трижды экстрагируют эфиром
(порциями по 100 мл). После удаления растворителя получают 0,93 г (58%) вещества с т. пл.
120—121° С.
Взаимодействием хлористого трифенилолова с цианамидом натрия
получено [107] соединение состава (C6H5)3SnN=C=NSn (С6Н5)3-
Натриевые соли сульфонамидов образуют с оловоорганическими гало-
генидами сульфонамиды три- [40, 304, 305] и диалкилолова [304]. Также
легко проходит реакция между натриевой солью сукцинимида и хлористым
трифенилоловом, но выделить образующееся вещество в чистом виде не
удается [306].
Обмен хлоридов триалкил- и трифенилолова с N-натриевыми или
N-калиевыми солями имидазола, триазола, бензтриазола и других
гетероциклических соединений азота приводит к соответствующим соединениям
со связью Sn—N [106, 107, 291, 307]. Их синтез проводят в среде жидкого
аммиака. Так получены [307], например, N-триметилстаннилимидазол,
N-триметилстаннил- 1, 2, 3-бензтриазол, N-триметилстаннил-!, 2, 4-триазол,
N-триметилстаннилбензимидазол, N-трибутилстаннил пиррол, N-трибу--
тилстаннилбензпиразол, З-амино-М-триэтилстаннил-1,2,4-триазол и
М(1)-трифенилстаннил-1,2,3-триазол.
Вместо калиевых солей можно применять магниевое производное:
R3SnCl + R'NMgBr -> R3SnNR' + MgClBr.
Так получены N-трифенилстаннилпиррол и N-триметилстанилбензимида-
зол. Выходы продуктов не превышают 10%.
Получение диэтилди-1,2,4-триазолилолова [307]. К нагретому раствору 6,9 г (0,1
моля) 1, 2, 4-триазола в 175 мл петролейного эфира (т.кип. ПО—135° С) прибавляют
небольшими кусочками 3,91 г (0,1 г-атома) натрия. Реакция проходит медленно, образуется
суспензия коричневого цвета. К ней прибавляют нагретый раствор 12,38 г (0,05 моля) двухло-
ристого диэтилолова в 100 мл петролейного эфира, смесь кипятят в течение 7,5 часа и осадок
отфильтровывают. Получают 27,25 г порошка темно-коричневого цвета. 10 г вещества экст-
Реакции преобразования функциональных групп
411
рагируют горячим пропиловым спиртом в течение б час. К экстракту прибавляют
активированный уголь, фильтруют и фильтрат выпаривают. Получают 5,7 г диэтилди-1, 2, 4-триа-
золилолова в виде порошка светло-коричневого цвета.
Аналогично получено дибутилди-(1,2,4-триазолил)олово. Оба
соединения легко гидролизуются.
Оловоорганические производные 1,3-дифенилтриазина состава
(CH3)2SnX2, C6H5Sn(X)2Cl и (C6H5)3SnX (X—остаток 1,3-дифенилтри-
азина)получены [308] с выходом 90—100% встряхиванием галогенидов
олова с небольшим избытком соответствующей соли серебра в сухом
эфире.
Помимо соединений со связью Sn—N в литературе описан ряд олово-
органических соединений со связью Sn—В, Sn—Р, Sn—Sb, Sn—As и др.
Как уже отмечено на стр. 356, при действии трехфтористого бора на тетр-
аметилолово образуется тетрафторборат триметилолова. Более удобный
способ синтеза этого соединения состоит в обмене бромистого триметилолова
с тетрафторборатом серебра [309, 310]. Аналогично получены [309, 311]
(CH3)3SnAsF6 и (CH3)3SnSbF6.
Об оловоорганических соединениях со связью Sn—As или Sn—Sb
впервые кратко упоминается в патенте 1936 г. [312]. Позже эти соединения
получены взаимодействием галогенидов триалкилолова с натрийдифенилар-
сином (соответственно, натрийдифенилстибином) [26]:
R3SnX + (С6Н5)2ЭМа -> R3Sn3 (C6H5)2 + NaX
(3 = As, Sb).
Тем же путем осуществлен синтез соединений со связью Sn—Р[25, 32, 313,
314]:
R3SnBr + (C6H5)2PNa -> R3SnP(C6H5)2 + NaBr.
Обработка фенилфосфинонатрия бромистым триэтилоловом дает фе-
нил-бш>(триэтилстаннил)фосфин [25]. Также реагирует с бромистым три-
метилоловом метилфосфинонатрий и фосфинонатрий [315, 316]:
3 (CH3)3SnBr + 3 NaPH2 -» [(CH3)3Sn]3 P + 3 NaBr + 2 РН3.
В ряде случаев оловоорганические арсины и фосфины целесообразнее
получать другими методами (см. стр. 417).
Кратко сообщается [317] о синтезе соединения со связью Ge—Si—Sn из
[(C6H5)3Ge]3SiLi и хлористого трифенилолова.
Взаимодействием хлоридов алкил- или фенилолова с натрийкарбониль-
ными соединениями кобальта, железа или марганца получены [318—323]
оловоорганические кобальт, железо- и марганецкарбонилы.
Несмеяновым, Анисимовым с сотр. [324, 325] описан ряд смешанных
биметаллических производных типа С5Н5 (CO)3MSn (C6H5)3 "и
C5H5(CO)3M2Sn (С6Н5)2 (M=Cr, Mo, W). Соединения этого типа получены
взаимодействием натриевых солей трикарбонил-я-циклопентадиенил-
молибдена (вольфрама, хрома) с хлористым трифенилоловом или двухло-
ристым дифенилоловом в тетрагидрофуране. Аналогично получены [325,
326] (C6H5)3SnRe (CO)5, (C6H5)2Sn[Re (CO)5]2, C6H5Sn[Fe(CO)2(K-C5H5)]3
и R2Sn [Fe (СО)2(я-С5Н5)]2 (R =С2Н5, С5Н5 и С6Н5). Последовательным
замещением атомов галоида в оловоорганических дигалогенидах получены
[327—329] соединения, в которых атом олова одновременно связан с
карбонилами двух различных металлов.Вследствие своеобразной природы
этих веществ их синтез и реакции рассмотрены в главе XVI (см. стр. 505).
Среди соединений со связью Sn—О—Э наиболее подробно изучены
алкил- и арилстанносилоксаны. Их получают согидролизом олово- и крем-
нийорганических галогенидов (см. стр. 385), реакцией гетерофункциональ-
412
Оловоорганические соединения. Лит. стр. 424—431
ной конденсации (стр. 516), действием силанолов на оловоорганические
гидроокиси (стр. 399) или хлориды (стр. 414). Один из наиболее удобных
методов синтеза этих соединений основан на обмене триалкилсиланолятов
щелочных металлов или триалкилсиланолята тетраалкилсурьмы с олово-
органическими галогенидами [330—336]:
(CH3)nSnCl4_„ + (CH3)3SiOLi -> (CH3)nSn [OSi (СН8)8]4_Л + NaCl.
Реакции проводят в сухом эфире при полном отсутствии влаги воздуха.
Естественно, что при получении алкилстанносилоксанов можно
исходить из триалкилстаннолятов лития и алкилхлорсиланов [332, 336]:
(CH3)3SnOLi + (CH3)3SiCl -* (CH3)3SnOSi (CH3)3 -f LiCl,
2 (CH3)3SnOLi -ф- (CH3)2SiCl2 -* (CH3)2Si [OSn (CH3)3]3 + 2 LiCl.
Андриановым с сотр. [337] исследовано взаимодействие двухлористого
диметилолова сдинатриевыми солямидиметилсилоксанов№0[5ЦСН3)20]пЫа,
где п =2, 3,10, 20. Найдено, что независимо от условий проведения
реакции и степени полимеризации п, образуются полимерные продукты
с молекулярным весом порядка 3000.
Помимо органостанносилокеанов в литературе описан ряд соединений
со связью Sn—О—Al, Sn—О—Р, Sn—О—As и др.
Исследовано [338, 339] взаимодействие хлоридов фенилолова с
NaHAl(OC2H5)3 в эфире. Отмечено, что термическая устойчивость
образующихся соединений уменьшается с уменьшением числа органических
радикалов у атома олова.
При реакции солей арсоновых кислот с хлористым трифенилоловом
образуются соединения со связью Sn—О—As [340]:
2 (C6H5)3SnCl + C6H5AsO (ONa)2 -> [(C6H5)3SnO]2As (О) С6Н5 + 2 NaCl,
2 (C6H5)3SnCl + KH2As04 -* [(C6H5)3SnO]3AsO + 2 KCl + 2 H2As04,
(CH3)2SnCl2 + 2 (CH3)3AsOOAg -> (CH3)2Sn [OAs (CH3)20]2 + 2 AgCl.
При сливании водных растворов двухлористого диметилолова и арсоно-
вой кислоты выделяется осадок бис-(фениларсоната) диметилолова.
Аналогично получены [216, 217] арсенат, молибдат, вольфрамат
диметилолова и некоторые производные фосфорных кислот.
Получение фенилфосфоната дивинилолова [120]. К 2,0 г (0,0082 моля) двухлористого
дивинилолова в 30 мл воды приливают водный раствор фенилфосфоновой кислоты. Выпавший
осадок отфильтровывают, промывают водой и высушивают. Выход 2,13 г (79%). При
нагревании до 355° С вещество разлагается не плавясь.
Аналогично получены [120, 284—288] фенилфосфат, фенилдифосфат,
фосфат и диметафосфат диметилолова, а также ряд соединений типа
R3SnOP(0)(OR'), (R3SnO)3P(0)OR\ R3SnSP(S)(OR)2, R2Sn [SP(S)(OR)2]2,
R3SnSP(S)(SR)2 и R2Sn [SP(S)(SR)2]2.
О реакции эфиров кислот трехвалентного фосфора с оловоорганически-
ми галогенидами см. статьи Арбузова [341—343], Малатесты [344, 345] и
Пудовика с сотр. [346].
Взаимодействием двухлористого диметилолова с хроматом аммония
получен [217] продукт состава (CH3)2SnO-(CH3)2SnCr04.
Реакции преобразования функциональных групп
413
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГАЛОГЕНИДОВ С КИСЛОТАМИ,
ПЕРЕКИСЯМИ, СПИРТАМИ, МЕРКАПТАНАМИ И ДРУГИМИ СОЕДИНЕНИЯМИ,
СОДЕРЖАЩИМИ ПОДВИЖНЫЙ АТОМ ВОДОРОДА
Реакции оловоорганических галогенидов с соединениями, содержащими
подвижный атом водорода, проходят самопроизвольно лишь в отдельных
случаях. Так, действием бромисто- или иодистоводородной кислоты на олово-
органические хлориды получены трехиодистое метилолово [182], трехбро-
мистое [347—350] и трехиодистое [347—350] фенилолово.
При пропускании сероводорода в водный раствор двухлористого
[351, 352] или двубромистого [219] диметилолова выпадает осадок тримерно-
го сульфида диметилолова с т. пл. 148° С (из петролейного эфира).
Аналогично реагирует с сероводородом хлористое триметилолово [353]. С трига-
логенидами метил- [182] и арилолова [203, 204] сероводород образует
сульфиды типа (RSnS)2S.
Аналогично получены [219] селениды бш:-(триметилолова),диметил- и
метил олова.
Интересно отметить, что при действии сероводорода на органостан-
ноксаны состава (XSnR2OSnR2X)2 (R = C4H9; X = C1,NCS, OOCCH3; или
R =CeH5; X =С1) образуется смесь соединений типа R2SnS и R2SnX2
[353а].
Реакции оловоорганических галогенидов с органическими кислотами,
гидроперекисями, спиртами и меркаптанами проходят, как правило, только
в присутствии акцепторов галоидоводородов. Примером может служить
образование диацилатов тетраметилдистанноксана при действии на водный
раствор двухлористого диметилолова пропионовой, масляной, циан-,
хлор- или бромуксусной кислот и аммиака [354]. Со щавелевой, меркапто-
уксусной и бензойной кислотами реакция проходит иначе и приводит к диа-
цилатам диметилолова.
Интересно отметить, что галогениды триалкилолова не реагируют с
органическими гидроперекисями в присутствии аммиака или органических
оснований, тогда как галогениды триарилолова легко вступают в эту
реакцию [245]:
Ar3SnCl + ROOH + NH3 - Ar3SnOOR + NH4C1.
Получение /тгр£/тг-бутилперокситрифенилолова [245]. К раствору 3,8 г хлористого
трифенилолова в 90 мл эфира при перемешивании прибавляют по каплям 1 г гидроперекиси
трет-бутила в 10 мл эфира. Одновременно через реакционную смесь пропускают сухой
аммиак. Реакция заканчивается примерно через 20 мин., что видно по прекращению
выделения осадка хлористого аммония. После удаления эфира в вакууме получают 3,6 а (83%)
вещества ст. пл. 63—65° С (из пентана).
Аналогично получены и другие перекисные соединения трифенилолова.
При замене органических гидроперекисей на перекись водорода
получают другой интересный тип оловоорганических перекисей, в которых два
атома олова связаны через перекисную группу [245]:
2 (C6H5)3SnCl + 2 Н202 + 2 NH3 - (CsH5)3Sn-00-Sn (CSH5)3 + 2 NH4C1.
Соединения состава R4Sn2 (OOR')20 получены [148] взаимодействием
диацетатов тетраалкилдистанноксанов с органическими гидроперекисями
в присутствии триэтиламина. В тех же условиях дистанноксаны,
содержащие в качестве концевых групп атомы хлора, реагируют с
гидроперекисями следующим образом:
C13nR203nR2Cl -f R'OOH + (C2H5)3N -> C13nR203nR2OOR' + (CiHsfoN-HCl.
Аналогично проходит реакция дихлортетраалкилдистанноксанов со
спиртами [355]. Взаимодействие спиртов с оловоорганическими галогени-
414
Оловоорганические соединения. Лит. стр. 424—431
дами описано лишь на отдельных примерах [356—358]. Более подробно оно
изучено в ряду фенолов и гетероциклических спиртов.
В реакциях оловоорганических дихлоридов с фенолами такие
акцепторы галоидоводородов, как металлический натрий или карбонат натрия
неэффективны [359]. Однако в присутствии амида натрия эти реакции
проходят легко, например:
О
Г LoH + (С4Н9Ь$пС12 + 2 NaNH2 -* [' j[ Sn (C4H9)2 + 2 NaCl + 2 NH3-
О
Получение (о-фенилендиокси)дибутилолова [359]. Синтез вещества проводят в атмосфере
азота. К 0,125 моля двухлористого дибутилолова и 0,25 моля амида натрия в нагретом до
кипения эфире при энергичном перемешивании прибавляют по каплям раствор 0,125 моля
пирокатехина в эфире. Смесь перемешивают до тех пор, пока не прекратится выделение
аммиака (32 часа). Осадок серого цвета отфильтровывают и возгоняют в вакууме при
260° С. Получают белое кристаллическое вещество с т. пл. 272°С. (о-Фенилендиокси)ди-
бутилолово не растворяется в обычных органических растворителях, кроме горячего
пиридина, тетрагидрофурана и диметилформамида.
То же вещество получено при замене амида натрия пиридином.
Аналогично получены [359] (о-фенилендиокси)диметилолово и (2,2'-бифенилен-
диокси)диметил олово.
Двухлористое диметил- и диэтилолово образуют с а-нитрозо-р-нафто-
лом бас-(а-нитрозо-(3-нафтокси)тетраалкилдистанноксаны [360].
Аналогично проходит реакция двухлористого диметилолова с катехолом [354], тогда
как действие на двухлористое дипропил- или дибутилолово |3-нафтола
приводит к (хлор)(|3-нафтокси)тетраалкилдистанноксанам [360].
При смешивании стехиометрических количеств оловоорганических
дихлоридов с 8-оксихинолином в спирте образуются бм>(8-оксихиноляты)
диалкил- и дифенилолова [278, 361, 362]:
RaSiiCb + 2 C9H7NO -> R2Sn (C9H6NO)2 + 2 HCi.
а-Гидрокси-со-триметилсилоксидиметилсилоксан ведет себя подобно
другим гидроксилсодержащим соединениям [363]:
(CHsbSnCla + 2 НО [Si (CH3)20]nSi (CH3)3 + 2 (C2H5)8N -»
-* (CH3)2Sn (О [Si (CH3)20]nSi (CH3)3}2 + 2 (C2H5)3N -HCI.
Реакция между ацетил ацетон ом и двухлористым дифенилоловом проходит
более сложно [278, 364].
Действие на оловоорганические галогениды меркаптанов в присутствии
акцепторов галоидоводородов является одним из основных методов
синтеза оловоорганических меркаптидов [98, 99, 169, 170, 281, 365—369]. Так
получены, например, моно-, ди- и тримеркаптиды метил- и этилолова
[368].
В тех же условиях дитиолы образуют с оловоорганическими дигалоге-
нидами гетероциклические соединения с аюмом олова в кольце [281,
370—374]. Например:
R S—СИ*
CH2SH \ /
R2SnBr2 + | -°*L_* Sn
CH2SII / \
R S-CI
Реакции преобразования функциональных групп
415
Реакция между двухлористым дибутилоловом и тиолпропионовой кислотой
проходит следующим образом [242]:
S-CH2
(QH9)2SnCl3 + HSCH2CH2COOH NaHC°3-> (C4H9)2Sn
0-CH2
К реакциям рассматриваемого типа следует отнести также взаимодействие
оловоорганических хлоридов с дифенилфосфином в присутствии триэтил-
амина [375, 376]:
R3SnCl + (С6Н5)2РН + (C2H5)cN -* R3SnP (C6H5)2 + (QH5),N -HCl,
R2SnCl2 + 2 (CeH3)3PH + 2 (C2H5)SN -» R2Sn [P (C6H5)2]2 + 2 (QHr>)8N-HCl,
RSnCl3 + 3 (CeH3)2PH + 3 (C2H6)sN -» RSn [P (C6H5)2]3 + 3 (C2H5)SN • HC1.
Станнилфосфины получены также взаимодействием оловоорганических
галогенидов с дифеиилфосфинонатрием (см. стр. 411). Следует отметить,
что вещества, полученные этими двумя методами, часто неидентичны.
Так, три-я-бутилстаннилдифенилфосфин описан в виде жидкости с т. кип.
192° С/0,6 мм [25] и кристаллического вещества (т. пл. 90—96° С), которое
разлагается при нагревании до 150° С [376]. Также противоречивы данные
о дифенилстаннил-бш>(дифенилфосфике) (т. пл. 78—80° С [376] и 144—
147° С [25]).
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ АЦИЛАТОВ,
АЛКОКСИСОЕДИНЕКИЙ И СТАННИЛАМИКОВ
Среди различных оловоорганических соединений особое место занимают
алкоксисоединения олова и станниламины. Благодаря своей высокой
реакционной способности они широко применяются для синтеза различных
производных олова. Преимущество этого метода синтеза заключается в
том, что образующиеся в качестве побочных продуктов спирт или амин
легко удаляются из реакционной смеси, что значительно облегчает очистку
основного вещества. Кроме того, применение алкоксисоединений или стан-
ниламинов как исходных соединений позволяет проводить реакции при
низких температурах.
Алкоксисоединения олова [377, 378] и станниламины [31, 32] активно
реагируют с минеральными и органическими кислотами или их
ангидридами. Обработка алкоксисоединений олова галоидангидридами кислот
[377, 379], треххлористым бором [380] или алкилхлорсиланами [381]
приводит к оловоорганическим галогенидам. Интересно отметить, что олово-
органические соединения обменивают алкоксигруппы на атомы галоида
при нагревании их с бромистыми или йодистыми алкилами [382].
При получении оловоорганических перекисей в качестве исходных
веществ удобно применять алкоксисоединения олова [244—248].
Преимущество этого способа заключается еще и в том, что при получении солей
гидроперекисей как исходных соединений, последние в ряде случаев
подвергаются каталитическому разложению [383, 384].
Получение декагидро-9-нафтилперокситрибутилолсва [244]. Смешивают растворы 1,79 г
гидроперекиси декагидро-9-нафтила и 3,28 г метокситрибутилолова в 40 мл эфира.
Растворитель удаляют в вакууме. Остаток заметно густеет, но не кристаллизуется, при охлаждении
до —80° С. Продукт, который не удается перегнать при 70° С (баня) /0,01 мм,
отфильтровывают от небольшого количества твердого вещества. Получают декагидро-9-пафтилперокси-
трибутилолово в виде маслянистой жидкости с п2£ 1,4952.
416
Оловоорганические соединения. Лит. стр. 424—431
Тем же путем получены 1-метил-1-фенилэтилперокситрибутилолово
[244], декагидро-9-нафтилперокситрифенилолово [244], тетралинперокси-
трипропилолово [245] и др.
С перекисью водорода алкоксисоединения триалкилолова образуют
перекиси бш;-(триалкилолова):
R3SnOR' + H202-> R3Sn—00—SnR3 + R'OH.
Получение перекиси б"ас-(трипропилолова) [245]. 5,6 г метокситрипропилолова в 20 мл
эфира смешивают с 0,35 г перекиси водорода в 10 мл эфира при 0° С. Смесь оставляют на
несколько часов при обычной температуре и эфир отгоняют. Остаток представляет собой
чистую перекись б«с-(трипропил олова).
Перекиси бас-(триал кил олова) чрезвычайно неустойчивы [79, 245,
384]. Нагревание до 60° С перекиси бш:-(триэтилолова) уже через 2—3 мин.
заканчивается взрывом. Изучение разложения этого вещества в растворе
нонана показало, что реакция может быть описана кинетическим
уравнением первого порядка до значительной глубины превращения [79, 384].
Алкоксисоединения олова активно реагируют и с другими
соединениями, содержащими подвижный атом водорода. Так, нагревание метокситри-
этилолова с ациламидами или карбаматами приводит к соединениям со
связью Sn—N[256]:
у° /°
(C2H5)3SnOCH3 + RNHC— X -» (C2H5)3SnN(R) С—X + СН3ОН
(R = C6H5, п-СбН13; Х = Н, СНз, ОСН3)
Соединения того же состава образуются и при присоединении оловооргани-
ческих гидридов к арилизоцианатам (см. стр. 307).
Алкоголяты триалкилолова реагируют с уретаном при нагревании до
60—70° С по следующему уравнению [384а]:
R3SnOCH3 + H0NCOOC2H5 -* R3SnNCO + CH3OH + С2Н5ОН.
Таким образом получены изоцианаты триэтилолова (выход 90%, т. кип.
113—114° С/18 мм) и трипропилолова (выход 87%, т. кип. 78—78,5° С/
/3- 10~2лш).
Тиоациламиды и тиокарбаматы реагируют с метокситриэтилоловом
следующим образом [256]:
S X
f I
(C2H5)3SnOCH3 + C6H5NHC—X -* (C2H5)3SnSC=NC6H5 + CH3OH
(X == H, СНз, ОСН8).
С бензилселенидом изобутокситриэтилолово образует бензилселенид
триэтилолова [378].
В литературе имеются указания на синтез соединений типа R2Sn(OR')X
(X =F, OOCR, N(R)OOSR) действием фтористого бензоила или
соединений с подвижным атомом водорода на диалкоксипроизводные диалкило-
лова [267, 273, 379]. Следует отметить, что с органическими кислотами ди-
метоксидибутилолово реагирует по следующей схеме [385]:
2 (C4H8)2Sn(OCH3)2 + 2 RCOOH -> (C4H9)2Sn (OCH3)2.(C4H9)2Sn (OOCR)2 + 2 CH3OH.
В ряде случаев синтез оловоорганических соединений различных типов
осуществляют исходя из станниламинов. Так, реакция станниламинов с
синильной кислотой является простым и препаративно удобным методом
синтеза оловоорганических цианатов [289]:
R„Sn [N (C2H5)2]4_„ + 2/х HCN -> R„Sn (CN)^ Чг п [H2N (C2H5)2] CN-.
Реакции преобразования функциональных групп 417
Как уже отмечено на стр. 408, при действии метилата натрия на
хлористое триметилолово получены противоречивые результаты. Метокситриме-
тилолово получено с выходом 95% осторожным метанолизом диэтиламино-
триметилолова в эфире при —50° С [386]:
(CH3)3SnN (С2Н5)2 + СН3ОН -* (CH3)3SnOCH3 + (C2H5)2NH.
Вещество очищают возгонкой в вакууме. При 45° С оно разлагается [386].
Аналогично получены и другие алкоксисоединения олова [32, 253, 290,
387], в том числе соединения типа RSn (OR')3 [290]: триметоксиметил- (т.пл.
195°С), триметоксиэтил-(т. пл. 165°С), трцметоксибутил- (т. разл. 250°С),
триэтоксибутил- (т. кип. 130° С/0,1 мм), триметоксифенил- (т.пл. 212° С)
и триэтоксифенилолово (т. пл. 119°С).
Найдено [32, 313], что станниламины легко реагируют с дифенилфосфи-
ном и дифениларсином с образованием соединений со связью Sn—Р и Sn—As.
Получение триметилстаннил(дифенилфосфина) [32]. К 2,16 г ?диметиламинотриметил-
олОва прибавляют при обычной температуре 1,94 г дифенилфосфина и смесь перегоняют в
вакууме. Получают 2,90 г (80%) вещества с т. кип. 150° С/0,8 мм.
Аналогично из 1,41 г диметиламинотриметилолова и 1,02 г дифенилар-
сина получено [32] 1,20 г (77%) триметилстаннил(дифениларсина) с т. кип.
136° С/0, 05 мм.
Частным случаем реакций рассматриваемого типа являются реакции
переацидирования, переалкоксилирования и переаминирования.
Для получения ацилатов три- и диалкилолова реакция переацидирования
применяется сравнительно редко [39, 139, 140, 388]. Изучение этой
реакции на примере триэтильных и трипропильных соединений олова показало
[39], что процесс является равновесным:
[RsSnOOCR + R'COOH ;± R3SnOOCR' + RCOOH.
Действием двуосновных кислот на диацетаты диэтил- [139] и дибутилолова
[140] получены полимерные продукты. Отмечено [140], что замена диацетата
дибутилолова окисью дибутилолова не приводит к изменению их состава
(см. стр. 401).
При нагревании до 90° С метокситри- или диметоксидиал кил олова с
высшими спиртами происходит реакция переалкоксилирования [273, 279,
293]:
IRsSnOCHg + R'0№-* R3SnOR' + СН3ОН.§
Этот метод применен, в частности, для синтеза гликолятов [257] и ацетил-
ацетоната [389] дибутилолова:
О
|'. J4H9)2Sn(OC2H5)3 + НО (CH2)rtOH -* (C4H9)2Sn (СН2)Л + 2 С2Н5ОН.
о
Реакцию проводят нагреванием раствора диэтоксидибутилолова в
бензоле с рассчитанным количеством гликоля. Образующийся в результате
реакции этиловый спирт отгоняют в виде азеотропной смеси с растворителем.
После завершения реакции раствор выпаривают в вакууме. Остаток
очищают перегонкой или возгонкой в вакууме. Так получены, например, 1,3-
пропандиоксидибутилолово (т. возгонки 184—185° С/0,3 мм), 1,4-бутан-
диоксидибутилолово (т. кип. 175—178°С/0,4 мм) и гексилендиоксидибутило-
лово (т. кип. 163—165° С/0,7—0,8 мм).
27 Заказ № 207
418
Оловоорганшеские соединения. Лит. стр. 424—431
При действии меркаптанов на меркаптиды или ацилаты триалкилолова
имеют место следующие равновесные процессы [39]:
R3SnSR + R'SH -^ R3SnSR' + RSH,
R3SnOOCR + R'SH ^ R3SnSR' + RCOOH.
С алкоксипроизводными олова эта реакция проходит более гладко [389а].
При нагревании диалкокситетраалкилдистанноксанов с меркаптанами ал-
коксильные группы количественно заменяются на остаток меркаптана [390].
Станниламины легко вступают в реакцию переаминирования [292]:
R3SnNR; + RgNH -» R3SnNR2' + R;NH.
Высококипящий амин вытесняет амин с более низкой температурой кипения,
так что относительный ряд замещения следующий:
(C4H9)2N- > (C2H5)2N- > (CH8)3N.
Другой тип переаминирования включает в себя как первичные, так и
вторичные амины. Вероятно, здесь основную роль играют стерические факторы,
а не летучесть соответствующих веществ [292]:
(CH3)3SnNR2 + R'NH2-> (CH8)3SnNIIR'+R2NH,
2 (CH3)3SnNR2 + R'NH2 -> [(CH3)3Sn]2NR' -|- 2 R2NH.
Вторая реакция особенно легко проходит в случае низших алкильных
производных. Взаимодействие метил- и этил амина с диметиламинотриметилоло-
вом приводит только к соответствующим дистанназанам. Превратить их в
N-алкиламинотриметилолово обработкой избытком амина не удается.
Однако диметиламинотриметилолово образует с анилином N-фенилтри-
метилстанниламин, а не N-фенилгексаметилдистанназан. При действии на
диметиламинотрибутилолово т/?£т-бутиламина выделено только трет-бу-
тиламинотрибутилолово.
Обработка бифункциональных оловоорганических соединений
первичными аминами приводит к циклостанназанам [292]:
п (CH3)2Sn [N (СН3)2]2 + п RNH2 -> [(CH3)2SnNR]„ + In (CH3)2NH.
К третьему типу переаминирования можно отнести реакции с участием
аммиака [292]:
3 (CH3)3SnN(CH3)2 + NH3 -> [(CH3)3Sn]3N + 3 (CH3)2NH.
Диэтиламинотриметилолово реагирует с гексаметилдисилазаном обычно
[392]:
(CH3)3SnN(C2H5)2 + HN [Si (CH3)3]2 -^ (CH3)3SnN [Si (CH8)8]a + (C2H5)2NH.
Заменить все аминогруппы в бм;-(диметиламино)диметилолове и три-
(диметиламино)метилолове на остатки гексаметилдисилазана не удается.
В результате этих реакций получают соединения следующего состава
(CH3)2NSn(CH3)2N [Si (СН3)з]2 и [(CH3)2N]2Sn(CH3)N[Si(CH3)3]2 [391].
Реакции преобразования функциональных групп
419
Нагревание бис-(диэтиламино)диметилолова с соответствующими
диаминами приводит к циклическим соединениям [392]:
(CH3)2Sn [N (QHsbb + (CH8)8SiNH (CH2)3NHSi (CH3)3
I
Si(CH3)3
(CH3)2Sn (CH2)3
Si (CH3)3
Аналогично проходит реакция с o-(CH3)3SiNHC6H4NHSi(CH3)3'
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ТИПА RgSnOSnR3, R3SnSSnR3 и (R2SnO)„
Как показано Андерсеном [84] на примере гексаэтилдистанноксана и
сульфида бггс-(триэтилолова), соединения этих типов легко реагируют с
галоидными солями бора, кремния, германия и другими веществами (табл. 9).
При действии бромистого магния на гидроокись триэтилолова образуется
бромистое триэтилолово [393]. Гексаэтилдистанноксан более активен, чем
сульфид бас-(триэтилолова) [84]. Так, гексаэтилдистанноксан реагирует с
ацетатом и роданидом триметилкремния и цианистым триэтилкремнием,
тогда как сульфид не вступает ни в одну из этих реакций (ход реакций не
зависит от отгонки низкокипящего вещества).
Интересно отметить, что тетрабутоксититан образует с гидроокисью
трифенилолова тетра(трифенилстаннокси)титан (выход 20%) [394].
Нагревание окисей диалкилолова с соединениями типа R2SnX2 приводит к дистан-
ноксанам состава (XSnR2OSnR2X)2 (X — галоид, остаток органической
кислоты или другая функциональная группа) [141, 209, 212, 275, 395 — 401].
Реакции можно проводить в среде инертных органических растворителей,
например в бензоле, толуоле или ксилоле. Применение избытка оловоорга-
нического галогенида не влияет на структуру образующихся веществ.
Напротив, 3 моля оловоорганической окиси с 1 молем дигалогенида образуют
соединения типа (XSnR2OSnR2OH)2[398, 402]. О синтезе соединений этого
типа см. также стр. 386.
Окиси диалкилолова легко реагируют также с тригалогенидами или три-
ацилатами алкилолова [402а]:
R2SnO + X3SnR'-> XR2SnOSnR'X2.
При нагревании в бензоле окисей диалкилолова с галогенидами триалкил-
олова проходит следующая реакция:
R2SnO + ClSnR3' -* ClR2SnOSnR3'.
При замене оловоорганических галогенидов на четыреххлористое или че-
тырехбромистое олово наблюдается только реакция обмена: .
R2SnO + X4Sn -* R2SnX2 + OSnX2.
Соединения типа RJV\OSnR2X получены нагреванием окисей
диалкилолова с металлоорганичестими галогенидами ртути, таллия, германия или
свинца [403].
27*
420
Оловоорганшеские соединения. Лит. стр. 424—431
Таблица 9
Реакции обмена окиси и сульфида б'йС-триэтилолова
Исходные
C2H5OBCI2
С2Н5ОСОСН3
СзНзОСОСбНз
(CH3)2Si(OOCCF3)2
(CH8)3SiOOCCH8
CH3OSi(NCO)3
(CH3)3SiNCS
(C2H5)3SiCN
CH3SiCl3
SiBr4
C12H25SU.3
(C6H5)2SiF2
(/-C3H7)3GeOH
(/x-C3H7)3GeF
GeCl4
SnCU
SnBr4
#-C4H9SnCl3
SnCl2
(C2H3)2SnCl2
PC13
AsCl3
SbCl3
SiBr4
C12H25S1J3
GeCl4
SnCli
SnCI2
PCI3
AsCl3
вещества
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H3)3Sn)20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(CaH3)8Sn]aO
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20 1
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(C2H5)3Sn]20
[(CaHOaSnJaO
[(C2H3)3Sn]20
[(C2H5)3Sn]2S
[(C2H5)3Sn]2S
[(C2H5)3Sn]2S
[(C2H5)GSn]2S
[(C2H5)3Sn]2S
[(C2n5)3Sn]2S
[(C2H3)3Sn]2S
Продукты реакции
(C2H5OBO)n
СаН5ОСаНб
C2H5OC2H5
[(CH3)2SiO]n
[(CH3)3Si]20
(CH3OSiO)20
[(CH3)3Si]20
[(C2H5)3Si]20
(CH3SiO)20
Si02
(Ci2H25SiO)20
[(C6H5)2SiO]n
(/-C3H7Ge)20
[(AZ-C3H7)3Ge]20
Ge02
Sn02
Sn02
[/z-dHeSnOJaO ,
SnO
[(C2H5)2SnO]n
p4o6
As406
Sb203
SiS2
(C12H23SiS)2S
GeS2
SnS2
Sn$
P4S10+P
ASaS3
(CaH5)8SnCl
(C2H5)3SnOOCCH3
(C2H5)3SnOCOC6H5
(C2H5)3SnOOCCF3
(C2H5)3SnOOCCH3
(CaH5)8SnNCO
(C2H5)3SnNCS
(CaH3)3SnCN
(C2H5)3SnCl
(C2H5)3SnBr
(C2H5)3SnJ
(C2H5)3SnF
(CaH3)3SnOH
(CaH-JsSnF
(CaH5)aSnCl
(C2H3)3SnCl
(C2H3)3SnBr
(CaH.i)3SnCl 1
(CaH-JeSnCl
(C2H3)3SnCl
(C2H,)3SnCl
(CaH3)3SnCl
(C2H5)3SnCl
(C2H5)3SnBr
(C2H5)3SnJ
(CaH5)8SnCl
(CaH3)3SnCl
(CaH5)3SnCl
(C2H5)3SnCl
(C2H3)3SnCl
Выход,
%
37
92
36
63
98
90
86
__
77
91
88
—
—
—
93
99
84
92
95
86
74
88
87
88
83
91
88
74
92
89
При нагревании сульфидов диалкилолова с соединениями типа R2SnX2
нолучены [4026] дистантианы типа XR2SnSSnR2X (X ==F, CI, Br, J, CNS,
OCH3,OOCR). В отличие от кислородных аналогов эти соединения в растворе
мономерны.
С треххлористым или трехбромистым алкилоловом сульфиды
диалкилолова экзотермически реагируют по следующей схеме:
R2SnS + R'SnX3-> XR2SnSSnR'X2.
С четыреххлористым или четырехбромистым оловом реакция проходит
иначе [4026]:
R2SnS + SnX4 -> R2SnX2 + (SSnX2).
Окись дифенилолова реагирует с диацетатом диэтилкремния согласно
уравнению [146а]:
(C6H5)2SnO + (GsHsbSi (OOCCH3)2-* (C6H3)2Sn (OOCCH3)2 + [(G2H5)2SiO]ll#
Реакции преобразования функциональных групп
421
Диацетат дифенилолова выкристаллизовывается из смеси с выходом 90%
в виде белоснежных игл с т. пл. 115—117° С. Аналогично получен дипропио-
нат дифенилолова (выход 52%, т. пл. 75—76° С) [146а].
Важное препаративное значение имеют реакции оловоорганических
окисей или гексаалкил(арил)дистанноксанов с сульфидом натрия [224, 404].
Получение сульфида ^яс-Стрифенилолова) [403]. К раствору 5 г (0,00698 моля) гекса-
фенилдистанноксана в 75 мл этилового спирта прибавляют 6,71 г (0,02795 моля) Na2S •
• 9Н20 в смеси 50 мл воды и 100 мл этилового спирта. Прозрачный раствор оставляют на
час при комнатной температуре, после чего к нему прибавляют 3,36 г (0,0459 моля)
уксусной кислоты. Выделяется осадок желтого цвета. Смесь выпаривают в вакууме, остаток
промывают и высушивают. Получают 5,0 г порошка серого цвета. После двух кристаллизации
из ацетонитрила получают белое кристаллическое вещество с т. пл. 145,5—147° С.
Аналогично реагируют с сульфидом натрия окиси диалкил- и
[дифенилолова [224, 404]:
2 S2~ + R2SnO + 4 Н+ —2 [R2SnS] + НаО + H2S.
Получение сульфида диметилолова [404]. Смесь 8,25 г (0,05 моля) окиси диметилолова
и 24,0 г (0,1 моля) Na2S • 9Н20 в 200 мл воды со следами (примерно 5 мл) этилового спирта
перемешивают в течение часа при обычной температуре. Образуется прозрачный раствор.
К нему постепенно прибавляют 12 г (0,2 моля) уксусной кислоты. Выпадает осадок белого
цвета, появляется запах сероводорода (при дальнейшем прибавлении кислоты никаких
изменений не происходит). Осадок отфильтровывают и дважды промывают водой. Получают
8,92 г (98,7%) тримерного сульфида диметилолова. Для очистки вещество растворяют в
горячем циклогексане, прибавляют активированный уголь, фильтруют и смесь охлаждают.
Получают 6,8 г (75%) вещества с т. пл. 149,5—151° С.
Аналогично получены [404] тримерные сульфиды дибутил- и диоктил-
олова. Сульфид дибутилолова выделяется в виде порошка или маслянистой
жидкости. При кристаллизации из холодного ацетона или при нагревании
в органических растворителях кристаллическая форма вновь превращается
в жидкую. Последняя при хранении в течение нескольких месяцев за-
кристаллизовывается; т. пл. 63—69° С.
С окисью дифенилолова реакция проходит значительно медленнее
(примерно 48 час.) [224].
Райхле [404] предложил своеобразный способ синтеза
оловоорганических сульфидов, который состоит в нагревании окисей диалкилолова с
сероуглеродом. Так получены, например, сульфиды диметил-, дибутил- и диок-
тилолова и др. Тот же путь применим и для синтеза сульфидов бис- (триал -
килолова) [404] и бис-(трифенилолова) [404, 405].
Аналогично проходит реакция бензиламина с гексафенилдистаннокса-
ном в сероуглероде [405]:]
(C6H5)8SnOSn(C6H5)3; + 2 C6H5CH2NH2 + 2 CS2 -* 2 C6H5CH2NHC (S) SSn (C6H3)3 + H20.
Взаимодействием гексаметилдистанноксана с амидом натрия получено
[389] аминотриметилолово, которое при нагревании превращается в три-
(триметилстаннил)амин. Выход последнего составляет 70—80%, т. пл. 26—
28° С.
Гексаалкилдистанноксаны образуют с литийорганическими
соединениями тетраалкил(соответственко, арил)олово. При прибавлении
эфирного раствора метиллития к гексаметилдистанноксану при обычной
температуре образуется тетраметилолово и триметилстаннолят лития [336]:
(CH3)3SnOSn(CH3)3 + CI I3Li -> (CH3)3SnOLi + (CH3)4Sn.
422 Оловоорганические соединения. Лит. стр. 424—431
Триметилстаннолят лития — легко гидролизующееся вещество с т. разл.
200° С. Оно хорошо растворяется в эфире, бензоле, циклогексане и четырех-
хлористом углероде. В разбавленном бензольном растворе это соединение
существует в виде гексамера.
О ПЕРЕРАСПРЕДЕЛЕНИИ ФУНКЦИОНАЛЬНЫХ ГРУПП У АТОМА ОЛОВА
Как показано Аллестоном и Дэвисом [109], при смешении двубромистого
дибутилолова с двухлористым или диацетатом дибутилолова происходит
следующая реакция:
(C4H9)2SnX2 + (C4H9)2SnBr2-> 2 (C4H9)2Sn(X)(Br).
Аналогично реагируют бис-(8-окс^Йколяты) и дигалогениды диалкил-
и дифенилолова [406, 407], а также оловоорганические гидриды и
галогениды (см. стр. 462).
При смешивании дигалогенидов и димеркаптидов диалкилолова имеет
место следующий равновесный процесс [91]:
(CjHgbSnCls + (C4H9)2Sn(SR)2 7t 2 (C4H9)2Sn(SR)Cl.
Напротив, соединения типа f(C2H5)2Sn(X)(OOCC6H5) (X=C1 или Вг),
полученные действием перекиси бензоила на галогениды триэтилолова
[408, 409] или нагреванием окиси диалкилолова с хлористым бензоилом [61,
124], выделить в чистом виде не удается, так как они разлагаются с
выделением соответствующего диацилата и дигалогенида:
2 (C2H3)2Sn(X)(OOCC6H5) -> (C2H3)2SnX2 + (QH5)2Sn(OOCC6H3)2.
Алкоксисоединения олова реагируют с оловоорганическими галогенида-
ми и ацилатами с образованием комплексов состава R2Sn(OR)2 • R2SnX2
(X — галоид или остаток органической кислоты) [385, 410]. Имеющиеся в
литературе данные о синтезе (галоид) метоксидиалкильных соединений олова
нуждаются,по-видимому, в дополнительной проверке. Так, (бром)метокси-
дибутилолово имеет те же константы, что и комплекс (С4Н9)25п(ОСН3)2 *
• (C4H9)2SnBr2 [382, 410, 411].
ДРУГИЕ РЕАКЦИИ, ПРИВОДЯЩИЕ К ПРЕОБРАЗОВАНИЮ
ФУНКЦИОНАЛЬНЫХ ГРУПП У АТОМА ОЛОВА
Высокая подвижность связи олова с различными функциональными
группами обусловливает возможность синтеза оловоорганических соединений и
другими, отличными от описанных выше методов, способами. Эти реакции
имеют менее общий характер и описаны лишь на отдельных примерах.
Так, сульфид бис- (трифен ил олова) получен [405] при обработке
аммиаком раствора бромистого трифенилолова в сероуглероде:
(C6H5)3SnBr + 4 NH3 + CS2 -> (C6H5)3SnSSn (C6H5)3 + NH4SCN + 2 NH4Br.
Бромистое триметилолово образует с N-этиламинотриметилкремнием
комплекс состава 1:1, который при нагревании превращается в этиламинотриме-
тилолово (выход 72%, т. кип. 153° С) [294]. Аналогичные комплексы
третичных кремнийорганических аминов с оловоорганическими галогенидами не
разлагаются при нагревании и могут быть очищены возгонкой в вакууме
или перекристаллизацией из сухого хлороформа. Различная устойчивость
этих координационных соединений объясняется стерическими факторами
[294].
Реакции преобразования функциональных групп
423
Галоидацетаты и галоидпропионаты триалкилолова при нагревании с
эквимолекулярным количеством бш>(я-диметиламинофенил)ртути образуют
четвертичные аммониевые основания состава
[p4CH3)2NC6H4HgC6H4N(CH3)2CH2COOSnR3]+X-
(Х = Вг или J).
Аналогично проходят реакции и в том случае, когда в орто-положении фе-
нильного кольца ртутноорганического соединения стоит метильная группа
или хлор. Соединения того же типа получены и на основе бш>(3-пиридил)-
ртути и фенил-3-пиридилртути [60].
Обработка дихлоридов тетраалкилдистанноксана бромом приводит
к (хлор)бромидам диалкилолова [412, 413]. Так получен, например, (хлор)-
бромид дибутилолова.
Исследованы [368, 369] некоторые реакции оловоорганических меркап-
тидов. При действии на меркаптиды триалкилолова треххлористого бора,
мышьяка или фосфора, брома или иода образуются галогениды
триалкилолова.
Йодистый метил гладко реагирует с метил мер капти дом триметилолова по
следующей схеме:
(CH3)3SnSCH3 + CH3J -» [(CH3)3SnS (СН3)а] J.
Нагревание до 270° С в течение 24 час. метилмеркаптида триметилолова дает
тетраметилолово и сульфид диметилолова; кроме того, выделено [368]
небольшое количество сернистого олова и низкокипящих углеводородов.
Нагревание метилмеркаптида триметилолова с серой не приводит к оло-
воорганическому дисульфиду. Вместо него с хорошим выходом получают
сульфид бис-(триметилолова). Изоцианид триметилолова присоединяет серу
при 150—160° С с образованием изотиоцианата триметилолова (выход 51%)
[214].
Чрезвычайно активными соединениями являются оловоорганические фос-
фины, арсины и стибины. Кислородом воздуха (быстрее спиртовым
раствором перекиси водорода) они легко окисляются в алкил- или арилстаннил-
дифенилфосфонаты [25, 376, 414, 415]:
R„Sn [Р (С6Н5)2]4_П ~ 02 -> R„Sn [OP (О) (С6Н,)2]4_/Г
Аналогично проходит окисление дифенил(триалкилстаннил)арсинов [26]:
R3SnAs (С6Н3)2 + 02 -> R3SnOAs (О) (С6НГ))2.
Окисление дифенил(триалкилстаннил)стибинов приводит к твердому
клейкому продукту [26]. Выделить из него индивидуальные соединения не
удается.
При гидролизе станнилфосфинов образуются гексаалкилдистанноксаны
или гидроокись трифенилолова [25, 376].
Йодистый метил превращает дифенил(триалкилстаннил)фосфины в
соответствующие фосфониевые соли [25, 376]:
, R3SnP(C6H5)2 + CH3J -> [R3SnP(C6H5)2 (CH3)] J.
При нагревании дифенил-(триалкилстаннил)арсинов или -стибинов с
йодистым метилом в эфире получают йодистое триалкилолово [26].
424
Оловоорганические соединения
ЛИТЕРАТУРА
1. AndersonEE, V a s t a J. A., J. Org. Chem., 19, 1300 (1954).
2. Oka war a R., WebsterD. E., RochowE. G., J. Am. Chem. Soc, 82,
3287 (1960).
3. SisidoK., К о z i m a S., J. Org. Chem., 29, 907 (1964).
4. Пат. ФРГ 957483 (1957); С. А., 53, 18865 (1959).
5. К г a u s e E., P о h 1 a n d R., Ber., 57, 532 (1924).
6. К i p p i n g F. В., J. Chem. Soc, 1928, 2365.
7. KrauseE., SchlottigO., Ber., 63, 1381 (1930).
8. В a h r G., Z о с h e G., Ber., 88, 1450 (1955).
9. KrauseE., Ber., 61, 912 (1928).
10. KrauseE., BeckerR., Ber., 53, 173 (1920).
11. KrauseE., WienbergK., Ber., 62, 2235 (1929).
12. G e 1 i u s R., Ber., 93, 1759 (1960).
13. S t e r n А., В е с к e r E. I., J. Org. Chem., 29, 3221 (1964).
14. С h a m b e r s B. F., SchererP.C, J. Am. Chem. Soc, 48, 1054 (1926).
15. С a h о u r s A., Ann., 114, 354 (1860).
16. СэйдзокоМ., Японск. пат. 6765 (1963); РЖХим., 1965, 15Н130.
17. Anderson H. H., Inorg. Chem., 3, 108 (1964).
18. Yasuda К., Okawara R., J. Organometal. Chem., 3, 76(1965); 6, 528 (I960).
19. ZimmerE, LiibkeK., Ber., 85, 1119 (1952).
20. D a n e k O., Collect. Czechosl. chem. Communs., 26, 2035 (1961).
21. Англ. пат. 772646 (1957); С. А., 51, 1551 (1957).
22. R a m s d e n H. E., Пат. США 2867641 (1959); РЖХим., 1961, 11Л123.
23. К u b о Н., Agr. Biol. Chem. (Tokyo), 29, 43 (1965); С. А., 63, 7032 (1965).
24. С h u г с h J. M., Пат. США 2630436 (1953); С. А. 48, 1420 (1954).
25. С a m p b e 1 1 I. G. M., F о w 1 e s G. W. A., N i x о n L. A., J. Chem. Soc, 1964,
1389.
^.Campbell I.G.M., Fowles G. W.A., NixonLA., J. Chem., Soc, 1964,
3026.
27. W a 1 d e A. W., V a n E s s e n H. E., Z b о r n i k T. W., Пат. США 2762821(1956);
РЖХим. 1958, 78652.
28. S с h w e i t z e r G. К», McCartyS.W., J. Inorg. Nucl. Chem., 27, 191 (1965).
29. В о с k R., В u r k h a r d t P., Angew. Chem., 73, 114 (1961).
30. Bock R., N i e der a u er H. Т., В eh rends К., Z. Analyt. Chun., 190, 33
(1962).
31. S i s i d о К., К о z i m a S., J. Org. Chem., 27, 4051 (1962).
32. JonesK., LappertM.R, J. Organometal. Chem., 3, 295 (1965).
33. Okawara R., Sugita K., J. Am. Chem. Soc, 83, 4480 (1961).
34. К u 1 m i z P., J. prakt. Chem., 80, 60 (1860).
35. S a s i n G. S., J. Org. Chem., 18, 1142 (1953).
36. S t a m m W., J. Org. Chem., 30, 693 (1965).
37. Okawara R., О h a r a M., Bull. Chem. Soc, Japan, 36, 623 (1964).
38. Okawara R., OharaM., J. Organometal. Chem., 1, 360 (1964).
39. S a s i n G. S., SasinR., J. Org. Chem., 20, 387 (1955).
40. V a n d e г К e r k G. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 49 (1956).
41. Cummins R. A., Dunn P., Austral. J. Chem., 17, 185 (1964).
42. S e у f e r t h DM Cohen H.M., Inorg. Chem., 2, 652 (1963).
43. P а з у в а е в Г. А., Дергунов Ю.И., В я з а н к и н Н. С, ЖОХ, 32, 2515
(1962).
44. V а п der К е г k G. J. M., L u i j t e n J. С. A., J. appl. Chem., 6, 56 (1956).
45. N о s е к J., Collect. Czechosl. chem. Communs., 29, 597 (1964).
46. К о т о н М. М., К и с е л е в а Т. М., П а р и б о к В. А., ДАН СССР, 125, 1263
(1959).
47. Montermoso J. С, Andrews Т. М., М а г i n е 1 1 i L. P., J. Polymer Sci.,
32, 523 (1958).
48. МонтермозоД. Ч., ЭндрьюзТ. М., Маринелли Л. П., Л а л а й-
б е р т е Б. Р., Каучук и резина, 1960, 61.
49. Rubber J. and Internat. Plast., 139, 11 (1960).
50. V a n der К e r k G. J. M., N о 1 t e s J. G., L u i j t e n J. G. A., J. appl. Chem.,
7, 356 (1957).
51. Ш о с т а к о в с к и й М. В., Котрел ев В. Н., К а л и н и н а С. П.,
Кузнецова Г. И., ЛайнеЛ. В., Борисова А. И., Высокомолек. соед., 3, 1128
(1961).
52. Ш о с т а к о в с к и й М. Ф., Калинина СП., КотрелевВ. Н., К о ч-
к и н Д. А., К у з н е ц о в а Г. И., Л а й н е Л. В., Б о р и с о в а А. И., Сб.
«Международный симпозиум по макромолекулярной химии», Москва, 1960. Секция I.
М.. Изд-во АН СССР, 1960, стр. 160.
Реакции преобразования функциональных групп
425
53. М о n t е г m о s о J. С, М а г i n е 1 1 i L P., Andrews T.M., Пат. США 3016369-
(1962); РЖХим., 1963, 8Т65.
54. L u i j t e n J. G. A., v a n d e r KerkG.J.M., Rec. trav. Chim., 83, 295 (1964).
55. К о т о н М. М., К и с е л е в Т. М., Изв. АН СССР, ОХН, 1961, 1783.
56. ШостаковскийМ. Ф., Котрелев В. Н., К о ч к и н Д. А.,
Кузнецова Г.И., К а л и н и н а С. П., Б о р и с е н к о В. В., ЖПХ, 31, 1434 (1958).
57. К о т о н М. М., Киселева Т. М., Ф л о р и н с к и й Ф. С., Сб.
«Международный симпозиум по макромолекулярной химии», Москва, 1960, 'Секция I. M.,
1960, стр. 167.
58. Leusink A. J., Marsmann J. W., Budding H. A., N о 1 t e s J. G., van
d e r KerkG.J.M., Rec. trav. Chim., 84, 567 (1965).
59. A n d e r s о n H. H., J. Org. Chem., 22, 147 (1957).
60Miller V.L, Chan J. K., J. Org. Chem., 28, 1938 (1963).
61. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б р е в н о в а Т. Н., ЖОХ, 34, 1005
(1964).
62. Р а з у в а е в Г. А., В я з а н к и н Н. С, Д ь я ч к о в с к а я О. С, ЖОХ, 32,
2161 (1962).
63. К о о р m a n s M. J., Пат. США 3097999 (1963); РЖХим, 1965, 21Н402.
64. F r a n k е 1 М., GertnerD., Wagner D., Z i 1 к h a A., J. Org. Chem., 30,
1596 (1965).
65. К о ч к и н Д. А., В е р е н и к и н а С. Г., Труды Всес. научно-исслед.
витаминного ин-та, 8, 39 (1961).
66. К о ч к и н Д. А.. В е р е н и к и н а С. Г., Ч е к м а р е в а И. Б., ДАН СССР,
139, 1375 (1962).
67. К о ч к и н Д. А., Ч е к м а р е в а И. Б., Труды Всес. научно-исслед. витаминного
ин-та, 7, 37 (1961).
68. К о ч к и н Д. А., Ч е к м а р е в а И. Б., ЖОХ, 31, ЗОЮ (1961).
69. KoopmansM. J., Голл. пат. 96805 (1961); РЖХим, 1962, 7Л451.
70. К е 1 s о R. G., Пат. США 3095427 (1963); РЖХим., 1965, 9Н131.
71. О s b о г n S. W., J u n-S h e n Y u А., Англ. пат. 973318 (1964); РЖХим., 1966,
6C195.
72. Rosenbergs. D., Debreczeni E., Wein berg'E. L, J. Am. Chem.
Soc, 81, 972 (1959).
73. N о 1 t e s J. G., L u i j t e n J. G. A., v a n d e r Kerk G.J.M., J. appl. Chem.,
11, 38 (1961).
74. Якубова Ф. А., Р а ш к е с А. М., К у ч к а р е в А. Б., М а н у л к и н 3. М.,
ЖОХ, 35, 387 (1965).
75. An derson H. H., Inorg. Chem., 1, 647 (1962).
76. L u i j t е n J. G. A., v a n d e г К e r k G. J. M., J. appl. Chem., 11, 35 (1961).
77. К и с е л е в а Т. М., К о т о н М. М., Ч е т ы р к и н а Г. М., Изв. АН СССР, ОХН,
1962, 1798.
77а. Davis A. G., Mitchell Т. N., Symes W. R., J. Chem. Soc, 1966C, 1311.
78. Da nn ley R. L., Aue W., J. Org. Chem., 30, 3845 (1965).
79. Александров Ю. А., Труды по химии и хим. технологии (г. Горький), 1960,
642; Александров Ю. А., Шушунов В. А., ДАН СССР, 140, 595 (1961).
80. Александров Ю. А., Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Сб. «Химия .
перекисных соединений». М., Изд-во АН СССР, 1963, стр. 291.
81. Александров Ю. А., Шушунов В. А., ЖОХ, 35, 115 (1965).
82. А л е к с а н д р ов Ю. А., Труды по химии и хим. технологии (Горький), 1962,
485
83. Ш*у ш у н о в В. А., Б р и л к и н а Т. Г., ДАН СССР, 141, 1391 (1961).
84. A n d е г s о n H. H., J. Org. Chem., 19, 1766 (1954).
85. Р г i n с е R. H., J. Chem. Soc, 1959, 1783,
86. КочкинД. А., Котрелев В. Нм Калинина С. П., Кузнецова
Г. И. и др. Высокомолек. соед., 1, 1507 (1959).
87. G г i f f i t h s V. S., Derwish G. A. W., J. Molec Spectr., 7, 233 (1961).
88- К о ч к и н Д. А., К отр ел ев В. Н., К а л и н и на С. П., Кузнецова Г. И.,
Борисенко В.В., Высокомолек. соед., 1, 482 (1959).
89. К о ч к и н Д. А., Ч и р г а д з е Ю. Н., ЖОХ, 32, 4007 (1962).
90. Sey f er th D., J. Am. Chem. Soc, 79, 2133 (1957).
91. Гольденштейн И. П., Землянский Н. Н., Гурьянова Е. Н., С ю-
т к и н а О. П., П а н о в Е. М., Кочешков К. А., Изв. АН СССР, серия хим.,
1967, 2201.
92. J о h n s о n E. W., Пат. США 2891922 (1959); РЖХим., 1960, 70500.
93. J о h n s о n E. W., Пат. США 2801258 (1957); С. А., 51, 18007 (1957).
94. Ramsden Е Е., В и с h a n a n Н. W., С h u г с h J. M., J о h n s о n E. W.,
Англ. пат. 719421 (1954); С. А., 50, 397 (1956).
426
Оловоорганические соединения
95. L e i s t n e r W. Е., Н е с к е г А. С, Пат. США 2883363 (1959); С. А., 53, 14586
(1959).
96. L e i s t n e r W. Е., К п о е р к е О. Н., Пат. США 2641588 (1953); С. А., 48, 5207
(1954).
97. Англ. пат. 743304 (1956); С. А., 50, 16828 (1956).
98. L e i s t n е г W. Е., Н е с к е г А. С, Пат. США 2870119 (1959); С. А., 53, 10844
(1959).
99. Wei n berg E. L., JohnsonE. W., Пат. США 2648650 (1953); С. А., 48,
10056 (1954).
100. WienbergE.L, JohnsonE.W., Пат. США 2832750 (1958); РЖХим., 1960,
70501.
101. WeinbergE.L, Johnson E. W., Пат. США 2832751 (1958); РЖХим., 1960,
37046П.
102. М а с к G. Р., Пат. США 3115509 (1963); РЖХим., 1965, 12Н86.
103. Leis tner W. Е., НескегА. С, Пат. США 2872468 (1959); РЖХим., 1960,
70499.
104. L e i s t n e r W. Е., Н е с к е г А. С, Пат. США 2704756 (1955); С. А., 50, 2687
(1956).
105. N е u m a n n W. Р., К 1 е i n e r F. G., Tetrahedron L., 1964, 3779.
106. L u i j t e n J. G. A., J a n s s e n M. J., van d e г К е г к G. J. M., Rec trav.
chim., 81, 202 (1962).
107. Van d e г К е г к G. J. M., L u i i t en J. G. A., J a n s s e n M. J., Chimia,
16, 10 (1962).
108. KrauseE., WeinbergK., Ber., 63, 381 (1930).
109. A 1 les ton D. L.f DaviesA.G., J. Cnem. Soc, 1962, 2050.
110. MatsudaS., MatsudaH., J.Chem. Soc. Japan, Ind. Chem. Sec, 63, 114(1960).
РЖХим., 1961, 8Ж229.
111. D r u с e J. G. F., Chem. News., 120, 229 (1920).
112. С a hours A., Compt. rend., 77, 1403 (1873).
113. P f e i f f e r P., L e h n a r d t R., Ber., 36, 1054 (1903).
114. P о p e W. J., P e а с h e у S. J., Proc. Roy. Soc. (London), 72, 7 (1903).
115. S e у f e r t h D., RochowE.C, J. Am. Chem. Soc, 77, 1302 (1955).
116. LowigC, Ann., 84, 308 (1852).
117. S m i t h Т. А., К i p p i n g F. S., J. Chem. Soc, 101, 2553 (1912).
118. H e с м е я н о в А. Н., Макарова Л. Г., ДАН СССР, 87, 421 (1952).
119. CahoursA., Demarcay E., Compt. rend., 88, 112 (1879).
120. S е у f e r t h D., S t о n e F. G. A., J. Am. Chem. Soc, 79, 515 (1957).
121. Poller R.C., J. Chem. Soc, 1963, 706.
122. Weinberg E. L.,Tomka L. А., Пат. США 2858325 (1958); РЖХим., 1960,
37184.
123. И т о Т., К а ц у м у р а Т., Японск. пат. 22484 (1963); РЖХим., 1966, 8С290.
124. Р а з у в а е в Г. А., ЩепетковаО. А., ВязанкинН. С, ЖОХ, 32, 2152
(1962).
125. К р и ж а н с к и й Л. М., О х л о б ы с т и н О. Ю., Попов А. В., Рогозев
Б. И., ДАН СССР, 160, 1121 (1965).
126. Н а г a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 57, 25 (1963).
127. ИтоТ, Японск. пат. 7686 (1960); РЖХим., 1961, 9Л108.
128. G 1 о s k e у С. R., Пат США 2838554 (1958); РЖХим., 1960, 14530.
129. О k a w a r a R., R о с h о w E. G., J. Am. Chem. Soc, 82, 3285 (1960).
130. S a i t о w A., R о с h о w E. G., S e у f e r t h D., J. Org. Chem., 23, 116 (1958).
131. Van d e г К е г к G. J.M., Lui j ten J. G. A., J. appl. Chem., 7, 369 (1957).
132. ШевердинаН. И., Абрамова Л. В., ПалееваИ. Е., Кочешков
К- А., Хим. промышленность, 1962, 707.
133. ТэнМи н-д a, Hua xue shi jie, 13, 533 (1958); РЖХим., 1960, 77427.
134. Мацуда С, М а ц у д а X., Я м а н э Я-, J. Chem. Soc Japan, Ind. Chem. Sec,
67, 467 (1964); РЖХим., 1964, 23C46.
135. Англ. пат. 787930 (1957); С. А., 52, 7774 (1958).
136. Мае da Y., Oka w а га R., J. Organometal. Chem., 10, 247 (1967).
137. Франц. пат. 1105652 (1955); С. А., 53, 6082 (1959).
138. К о т о н М. М., К и с е л е в а Т. М., ДАН СССР, 130, 86 (1960).
139. К о р ш а к В. В., Р о г о ж и н С. В., Макарова Т. А., Высокомолек. соед.,
4, 1297 (1962).
140. A n dr ews Т. М., Bower F.A., L a L i b e r t е В. R., М о n t e г m о s о J. С,
J. Am. Chem. Soc, 80, 4102 (1958).
141. Barker R.L, Англ. пат. 761568 (1956); РЖХим., 1959, 55524.
142. Gloskey С. R., Пат. США 2826597 (1958); РЖХим., 1960, 33053.
143. G 1 о s k e у С. R., Англ. пат. 751499 (1956); С. А., 51, 1659 (1957).
.144. WeinbergE.L, Пат. США 2857413 (1958); С. А., 53, 4135 (1959).
Реакции преобразования функциональных групп
427
145. Кубота X., Они си X., Японск. пат. 2365 (1960); РЖХим., 1961, 15Л145.
146. О н и с и X., Т а с и р о Т., Японск. пат. 5983 (1960); РЖХим., 1961, 5Л91.
146а. Кочеш'ков К. А., Г о л ь д е р В. Г., П а н о в Е. М., Землянский
Н. Н., С ю т к и н а О. П., Изв. АН СССР, Серия хим., 1967, 1171.
1466. Q u i n t i n С, Ing. Chem., 14, 205 (1930); С. А., 26, 2182 (1932).
147. WadaM., Shindo M., Okawara R.f J. Organometal. Chem., 1, 95(1963).
148. D a v i e s A. G., Graham I. F., Chem. a. Ind., 1963, 1622.
149. M а с k G. P., P а г k e г Е., Пат. США 2938013 (1960); РЖХим., 1962, 8П126.
150. В u г t S. L., Пат. США 2583084 (1952); С. А., 47, 146 (1953).
151. Англ. пат. 664133 (1952); С. А., 46, 11230 (1952).
152. Рыбакова Н. А., ТайковаН. К-, ЗильберманЕ. Н., Труды по
химии и хим. технологии (Горький), 1959, 183.
153. Б е р л и н А. А., П о п о в а 3. В., Я н о в с к и й Д. М., ЖПХ, 33, 871 (1960).
154. М а с k G. Р., Р а г k e r E., Пат. США 2626953 (1953); С. А., 47, 11224 (1953).
155. В о г n s t e i n J., LaLiberte В. R., Andrews Т., Montermoso
J., J. Org. Chem., 24, 886 (1959).
156. С о n s i d i n e W. J., J. Organometal. Chem., 5, 263 (1966).
156a. Воронков М. Г., РомоданЮ. П., Химия гетероцикл. соед., 1966, 892.
157. Ramsden H. E., Пат. США 2744876 (1956); РЖХим., 1958, 26101.
158. ConsidineW. J., Ventura J.J., Gibbons A. J., Ross A., Canad.
J. Chem., 41, 4239 (1963).
159. HulseR., Twitchett H. J., Англ. пат. 957841 (1964); РЖХим., 1965,
2 4C2 77.
160. M с G г a d у M. M.f T о b i a s R. S.f J. Am. Chem. Soc, 87, 1909 (1965).
161. BestCE., Пат. США 2731484 (1956); РЖХим., 1958, 58348.
162. Англ пат. 728953 (1955); С. А., 49, 13693 (1955).
163. R a m s d e n H. Е., Пат. США 2885415 (1959); С. А., 53, 18972 (1959).
164. Ramsden Н. Е., Англ. пат. 743313 (1956); С. А., 50, 16829 (1956).
165. Gregory W. А., Пат. США 2636891 (1953); С. А., 48, 3397 (1954).
166. Weinberg E. L., Johnson E. W., Пат. США 2832752 (1958); РЖХим., 1960,
50104.
167. Leistner W.E., Н е с k e r А. С, Пат. США 2641596 (1953); С. А., 48, 5208 (1954).
168. Leistner W. Е., Нескег А. С, Пат. США 2870182 (1958); С. А., 53, 13057
(1959).
169. Leistner W. Е., SetzlerW.E., Пат. США 2752325 (1956); РЖХим., 1958,
26102.
170. Ramsden Н. Е., Англ. пат. 759382 (1956); С. А., 51, 13918 (1957).
171. Mack G. Р., Пат. США 3069447 (1962); РЖХим., 1964, 20Н60.
172. Leistner W. Е., Н е с к е г А. С, Пат. США 2680107 (1954); С. А., 49, 4713
(1955).
173. Англ. пат. 797113 (1958); С. А., 53, 12185 (1959).
174. RamsdenH.E., Англ. пат. 742975 (1956); С. А., 51, 2033 (1957).
175. Ramsden Н. Е., Англ. пат, 750106 (1956); С. А., 51, 1247 (1957).
176. Ramsden H. Е., Пат. США 2904569 (1959); РЖХим., 1961, 8Л118.
177. W e i n b e r g E. L., Johnson E.W., RamsdenH.E., Пат. ФРГ 1178853
(1965); РЖХим., 1966, 11Н98.
178. Пат. ФРГ 1020338 (1957); С. А., 53, 19881 (1959).
179. Ма с к G. P., Pa r ker E., Пат. США 2998441 (1961); РЖХим., 1962, 16Л58.
180. М а с к G. Р., Р а г к е г Е., Пат. США 2914506 (1959); РЖХим., 1962, 1П151.
181. Druce J. G. F.f J. Chem. Soc, 121, 1859 (1922).
182. P f e i f f e r P., LehnardtR, Ber., 36, 3027 (1903).
183. Smith A.C, R о с h о w E. G., J. Am. Chem. Soc, 75, 4105 (1953).
184. Sol er i о A., Gazz chim ital., 85, 61 (1955); РЖХим., 1955, 55176.
185. J ones W. J., DaviesW.C, BowdenS.T., EdwardsC, Davies V. E.,
Thomas L. H., J. Chem. Soc, 1947, 1446.
186. TchakarianA., BevillardP., Bull. Soc. chim. France, 1950, 1300.
187. Lam bourne H., J. Chem. Soc, 121, 2533 (1921).
188. A n d e r s о n H. H., Inorg. Chem., 3, 912 (1964).
189. S t e f 1 E. P., В e s t С. Е., Пат. США 2713580 (1955); РЖХим., 1958, 44261.
190. S t e f 1 E. P., BestCE., Пат. США 2731482 (1956); С. А., 51, 13918 (1957).
191. BestCE., Пат. США 2713585 (1955); С. А., 50, 5725 (1956).
192. Англ. пат. 728954 (1955); С. А., 49, 13693 (1955).
193. S r i v a s t a v а Т. N., TandonS. К., Indian J. Appl. Chem., 26, 171 (1963).
194. С a h о u r s A., Ann., 114, 50 (1862).
195. С a s s о 1 A., MagonL, В a r b i e r i R., J. Chromatogr., 19, 57 (1965).
196. Krause E., Ber., 51, 1447 (1918).
197. V a n d e г К е г к G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 179 (1959).
198. Johnson W. K-, Пат. США 3036103 (1962); РЖХим., 1964, 8H107.
428
Оловоорганические соединения
199. А 1 1 е п N.. FurmanN.E, J. Am. Chem. Soc, 54, 4625 (1932).
200. BallczoE, SchiffnerH., Mikrochim. acta, 1956, 1829.
201. В a 1 1 с z о H., S с h i f f n e r H., Z. analyt. Chem., 152, 3 (1956).
202. ЛапкинИ.И, СедельниковаВ. А., ЖОХ, 30, 2771 (1960).
203. Несмеянове., К о ч е ш к о в К. А., ЖОХ, 1, 219 (1931).
204. Kozechkow К. A., Nesmeianov А. N.. Вег., 64, 628 (1931).
205. С 1 а г к Н. С, О'В г i e n R. J., Inorg. Chem., 2, 740 (1963).
206. С 1 а г к Н. С, О'В г i e n R. J., P i с к а г d A. L.,J. Organometal.Chem.,4,43(1965).
207. S h а р i г о P. J., В е с к е г Е. I., J. Org. Chem., 27, 4668 (1962).
208. T s a i Т. Т., С u t 1 e г A., LehnW.L, J. Org. Chem., 30, 3049 (1965).
209. WadaM., NishinoM., О к a w а г a R., J. Organometal. Chem., 3, 70 (1965).
210. AllestonD.L, DaviesA.G., H a n с ос к М., W h i t e R. F. M., J. Chem.
Soc, 1963, 5469.
211. Cummi ns R.f Dunn P.. Austral. J. Chem., 17, 411 (1964).
212. GreenB.S., S о w e r b у D. В., W i n к s n e K- J-, Chem. a. Ind., 1963, 1306.
213. С a hours A., Ann., 122, 48 (1862).
214. S e у f e r t h D., К a h 1 e m N., J. Org. Chem., 25, 809 (1960).
215. D r u с e J. G. F., Rec. trav. chim., 44, 340 (1925).
216. Gingold K., R о с h о w E. G., S e у f e r t h D., S m i t h A. C, W e s t R., J.
Am. Chem. Soc, 74, 6306 (1952).
217. R о chow E. G., Seyferth D., Smith A. C, J. Am. Chem. Soc, 75, 3099 (1953).
218. Beck W., SchwiererE., Ber., 97, 3517 (1964).
219. К r i e g s m a n n H., Hoffmann H., Z. Chem., 3, 268 (1963).
220. H a r a d а Т., Repts. Sci. Res. Inst. (Japan), 24, 177 (1948); С. А., 45, 2356 (1951).
221. Weinberg E.L.Ramsden H. E, Flat. США 2746946 (1956); С. А., 50, 12541
(1956).
222. Weinberg E. L., Tomka L. А., Пат. США 2832753 (1958); С. А., 52, 13307 (1958).
223. Forstner J. A., Muetterties E. L., Inorg. Chem. 5, 552 (1966).
224. ReichleW. Т., J. Polymer Sci., 49, 521 (1961).
225. К о ч е ш к о в К. А., НадьМ.М., ЖОХ, 4, 1434 (1934).
226. Т а л а л а ев а Т. В., Зайцева Н.А., К о ч е ш к о в К. А., ЖОХ, 16, 901
(1946).
227. S е у f e r t h D., J. Am. Chem. Soc, 79, 5881 (1957).
228. S с h m i d t M., R u f H., Angew. Chem., 73, 64 (1961).
229. Schmidt M, R u f H., Ber., 96, 784 (1963).
230. ЗемлянскийН.Н, ПановЕ.М., К о ч е ш к о в К. А., ЖОХ, 32, 2195
(1962).
231. Юй-чжень Лин, Scientia, 1959, 89; РЖХим., 1960, 20760.
232. ShishidoM., Kinukawa J., Японск. пат. 6428 (1954); РЖХим., 1958, 8942.
233. Кацумура Т., Японск. пат. 3777 (1960); РЖХим., 1962, 13Л94.
234. Zimmer H., Miller J.J., Naturwiss., 53, 38 (1966).
235. D 6 г f e 1 t С, R e i n d 1 E., HartelK., Пат. ФРГ 1072814 (I960);? РЖХим.,
1962, 9Л107.
236. Франц. пат. 1107726 (1956); РЖХим., 1958, 34427.
237. В a v 1 е у А., К n u t h С. J., Пат. США 2972595 (1961); РЖХим., 1962, 16П205.
238. Ь Ъ е г 1 у К. С, Пат. США 2560034 (1951); С. А., 46, 7583 (1952).
239. Н е n g 1 е i n F. A., LangR., SchmackL, Makromol. Chem., 22, 103 (1957).
240. В a v 1 e у A., KnuthC. J., Пат. США 2936299 (1960); РЖХим., 1962, 1П150.
241. W e i n b e r g E. L. Англ. пат. 753998 (1956); С. А., 51, 6683 (1957).
242. Fry e A. H., Horst R. W., Pa 1 iobagis M. A., J. Polymer Sci., A2,
1765 (1964).
243. Van d e г К e r k G. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 93 (1956).
244. Al lest on D. L., DaviesA.G., J. Chem. Soc, 1962, 2465.
245. R i e с h e A., D a h 1 m a n n J., Ann., 675, 19 (1964).
246. R i e с h e А., В е г t z Т., Angew. Chem., 70, 507 (1958).
247. R i e с h e А., В е г t z Т., Пат. ФРГ 1081891 (1960); РЖХим., 1962, 5Л111.
248. R i е с h е А., В е г t z Т., Пат. ГДР 20284 (1960); РЖХим., 1962, 4Л116.
249. Magel i О. L., Наг г ison J. В., Пат. США 3152156 (1964); РЖХим., 1966,
7Н135.
250. Несмеянов А. Н., ЛуценкоИ. Ф,, Пономареве. В., ДАН СССР,
124, 1073 (1959).
251 ЛуценкоИ.Ф., Пономарев СВ., ЖОХ, 31, 2025 (1961).
252. П о н о м а р е в С. В., Б а у к о в Ю. И., Л у ц е н к о И. Ф., ЖОХ, 34, 1938 (1964).
253. A m b е г g е г Е., К и 1 а М. R., Вег., 96, 2562 (1963).
254. РазуваевГ. А., ВязанкинН. С, ЩепетковаО. А., ЖОХ, 30, 249а
(1960).
255. Gerrard W., MooneyE.R, ReesR. G., J. Chem. Soc, 1964, 740.
256. NoltesJ.G., Rec trav. Chim., 84, 799 (1965).
Реакции преобразования функциональных групп
429
257. Mehrotra R. С, Gupta V. D., J. Organometal. Chem., 4, 145 (1965).
258. Mehrotra R. С, Gupta V, D., J. Organometal. Chem., 4, 237 (1965).
259. К r a u s С A., N e a 1 A. M., J. Am. Chem. Soc, 51, 2403 (1929).
260. Ж и в ухи н СМ., Дуд и ков а Э. Д., Тер-Сар кисян Э.М., ЖОХ, 32,
3059 (1962).
261. D' A n s J., G о 1 d H., Вег., 92, 3076 (1959).
262. LadenburgA., Ann. (Spl.), 8, 79 (1872).
263. La den burg A., Ber., 3, 353 (1870).
264. Aronheim В., Ann., 194, 145 (1878).
265. К а ц у м у р а Т., J. Chem. Soc, Japan, Pure Chem. Sec, 89, 729 (1962); РЖХим.,
1963, 16Ж252.
266. Mack G., Parker E., Пат. США 2700675 (1955); РЖХим., 1956, 7790.
267. М а с k G., P a r k e r E., Пат. ФРГ 953079 (1956); РЖХим., 1958, 26100.
268. М а с к G. Р., Р а г к е г Е., Пат. США 2727917 (1955); РЖХим, 1956, 76050.
269. Mack G. Р., Пат. США 2745820 (1956); РЖХим., 1958, 59335.
270. Б р и л к и н а Т. Г., Сафонова М. К., С о к о л о в Н. А., ЖОХ, 36, 2202
(1966).
271. Н а г г i п g t о n R. С, Smith J. L., Пат. США 3038877 (1962); РЖХим., 1964,
8С355.
272. F a u 1 k n e r D., M i 1 n e J. N., Пат. США 2583419 (1952); G. А., 47, 146 (1953).
273. АтавинА. С, Дубов а Р. И., Васи лье в А. П., ЖОХ, 36, 1506 (1966).
274. Cleverdon D., S t a u d i n g e r J. J. P., F а и 1 к n e r D., M i 1 n e J. N.. Пат.
США 2623892 (1952); С. А., 47, 3036 (1953).
275. С о n s i d i n e W. J., V e n t и r a J. J., J. Org. Chem., 28, 221 (1963).
276. M а с к G. P., S e v a r e s e F. В., Пат. США 2684973 (1954); С. А., 49, 10359 (1955).
277. М а с к G. P., S e v a r e s e F. В., Пат. США 2631990 (1953); С. А., 47, 5718 (1953).
278. Nelson W.H., MartinD.F., J. Inorg. Nucl. Chem., 27, 89 (1965).
279. В 1 а к e E., С о a t e s G. E., TateJ.M., J. Chem. Soc, 1961, 618.
280. Англ. пат. 781905 (1957); С. А., 52, 2049 (1958).
281. Davidson W. E., Hills К., H enr у М. С, J. Organometal. Chem., 3, 285 (1965).
282. Англ. пат. 740397 (1955); С. А., 59, 13987 (1956>.
283. L e i s t n e r W. E., H e с к е г А. С, Пат. США 2759906 (1956); РЖХим., 1959,
29605.
284. Англ. пат. 737392 (1955); С. А., 50, 9010 (1956).
285. С h u г с h J. M., Ramsden H. E., H i r s с h 1 a n d H., Buchanan H.W.,
Пат. США 2630442 (1953); С. А., 48, 1420 (1954).
286. Church J.M., Ramsden H. E., HirschlandH., Buchanan H. W.
Пат. США 2743257 (1956); С.А., 50, 11056 (1956).
287. М с D e r m о t t J. Р., Пат. США 2786812—14 (1957); С. А., 51, 10892 (1957).
288. Англ. пат. 800168 (1958); С. А., 53, 7023 (1953).
289. Lor b er t h J., Ber., 98, 1201 (1965).
290. L о r b e r t h J., К u 1 a M. R., Ber., 97, 3444 (1964).
291. JonesK., L a p p e r t M. F., Proc. Chem. Soc, 1962, 358.
292. Jones K., L a p p e r t M. F., J. Chem. Soc, 1965, 1944.
293. К u 1 a M. R., К r e i t e г С G., L о r b e r t h J., Ber., 97, 1294 (1964).
294. Abel E. W., В r a d у D., L e r w i 1 1 B. R., Chem. a. Ind., 1962, 1333.
295. Wi berg E., R ieger R., Пат. ФРГ 1121050 (1962); РЖХим., 1964, 16H57.
296. Jones R., Lappert M. F., Organometal. Chem. Rev., 1, 67 (1966).
297. LehnW.L, J. Am. Chem. Soc, 86, 305 (1964).
298. S с h e r e r O. J., SchmidtM., J. Organometal. Chem., 1, 490 (1964).
299. SchererO., SchmidtM., Angew. Chem., 75, 642 (1963).
300. ThayerJ.S., WestR., Inorg. Chem., 4, 114 (1965).
301. R e i с h 1 e W. Т., Inorg. Chem., 3, 402 (1964).
302. ThayerJ.S., WestR., Inorg. Chem., 3, 406, 889 (1964).
303. Srivastava T. N., Bhattacharya S. N., J. Inorg. Nucl. Chem., 28, 1480
(1966).
304. M а с к G. P., P a r к е г Е., Пат. США 2634281 (1953); С. А., 48, 1420 (1954).
305. M а с к G. P., P a r к е г Е., Пат. США 2618625 (1952); С. А., 47, 1977 (1953).
306. К и р с h i к Е. J., L a n i g а п Т., J. Org. Chem., 27, 3661 (1962).
307. L u i j t е п J. G. A., v a n d e r KerkG.J.M., Rec trav. Chim., 82, 1181 (1963).
308. В r i n с к m a n F. E., H a i s s H. S., Chem. a. Ind., 1963, 1124.
309. ClarkH.C.,0' BrienR.J., Inorg. Chem., 2, 1020 (1963).
310. H a t h a w а у B. J., WebsterD.E., Proc Chem. Soc, 1963, 14.
311. ClarkH.C, O'BrienR.J., Proc. Chem. Soc, 1963, 113.
312. Англ. пат. 445813 (1935); Ch. Zbl., 11, 1287 (1936).
313. JonesK., Lappert M. F., Proc. Chem. Soc, 1964, 22.
314. К u с h e n W., Buchwald H., Ber., 92, 227 (1959).
.315. Брукер А. Б., Балашова Л. Д., СоборовскийЛ. 3., ДАН СССР, 135,
843 (1960); ЖОХ, 5, 2207 (1965).
430
Оловоорганические соединения
316. Б р у к е р А. Б., Б а л а ш о в а Л. Д., Соборовский Л. 3., Авт. свид.
170976 (1965); РДХим., 1966, 7Н112; Бюлл. изобр., № 10, 29 (1965).
317. WittenbergD., GilmanH., Quart. Rev., 13, 116 (1959).
318. К a h n О., В i g о r g n e M., Compt. rend., 261, 2483 (1965).
319. G о r s i с h R. D., J. Am. Chem. Soc, 84, 2486 (1962).
320. В о n a t i F., WilkinsonG., J. Chem. Soc, 1964, 1789.
321. HeinF, JehnW, Ann., 684, 4 (1965).
322. HieberW, BreuR., Ber., 90, 1270 (1957).
323. H i e b e r W., В г е u R., Angew. Chem., 68, 679 (1956).
324. Несмеянов А. Н., АнисимовК. Н., Колобова Н. Е., Захарова
M. Я», ДАН СССР, 156, 612 (1964).
325. Несмеянов А. Н., Анисимов К- Н., Колобова Н. Е., X а н д о ж-
к о В. Н., ДАН СССР, 156, 383 (1964).
326. Несмеянов А. Н., Анисимов К- Н., Колобова Н. Е., Скрипкин
B. В., Изв. АН СССР, ОХН, 1966, 1292.
327. Несмеянов А. Н., Анисимов К. Н., Колобова Н. Е.,
Захарова М. Я., Изв. АН СССР, ОХН, 1965, 1122.
328. Bonati F., С е n i n i S., М о г е 1 1 i D., U g о R., J. Chem. Soc, 1966A, 1052.
329. P a t i 1 H. R. H., G r a h a m W. A. G., J. Am. Chem. Soc, 87, 673 (1965).
330. PapettiS., PostH. W., J. Org. Chem., 22, 526 (1957).
331. Schmidbaur H., SchmidtM., J. Am. Chem. Soc, 83, 2963 (1961).
332. Schmidbaur H., Hussek H., J. Organometal. Chem., 1, 244 (1964).
333. ThiesC, К i n s i n g e r J., Inorg Chem., 3, 551 (1964).
ob4. Tatlock W.S., RochowE.G., J. Org. Chem., 17, 1555 (1952).
335. Seyferth D., AllestonD.L, Inorg. Chem., 2, 418 (1963).
336. SchmidbaurH., HussekH., Angew. Chem., 75, 575 (1963).
337. Андрианов К. А., Васильева Т. В., Нудельман 3. Н., X а н а н а-
швили Л.М., К о ч е т к о в а А. С, Ч е р е д н и к о в а А. Г., ЖОХ, 32, 2307
(1962).
338. Schmitz-Dumont О., Bungard G., Ber., 92, 2399 (1959).
339. S с h m i t z-D u m о n t О., В u n g a r d G., Angew. Chem., 67, 208 (1955).
340. ChamberlandB. L., McDiarmid A.G., J. Chem. Soc, 1961, 445.
341. A p б у з о в Б. А., П у д о в и к А. Н., ЖОХ, 17, 2158 (1947).
342. А р б у з о в Б. А., Г р е ч к и н Н. П., ЖОХ, 17, 2166 (1947).
343. Арбузов Б. А., Г р е ч к и н Н. П., Изв. АН СССР, ОХН, 1956, 440.
344. М а 1 a t e s t a L., Gazz. chim. ital., 80, 527 (1950).
345. MalatestaL., Sacco A.,Ormezzano L, Gazz. chim. ital., 80, 658 (1950);
C. А. 46, 4473 (1952).
346. ПудовкинА.Н, Муратов А. А., ДАН СССР, 158, 419 (1964).
347. Кочешков К. А., Ж РФХО, 61, 1385 (1929).
348. Kozesckov К. А., Вег., 62, 996 (1929).
349. Несмеянов А. Н., Кочешков К. А., ЖРФХО, 62, 1795 (1930).
350. NesmejanovA. N.. К о z e s с h k о v К. А., Вег., 63, 2496 (1930).
351. Brown М. Р., О к a w a r a R., R о с h о w E. G., Spectorchim. Acta, 16, 595
(1960).
352. К о z e s с h к о v К. А., Вег., 66, 1661 (1933).
353. ZimmerH., GoldH., Ber., 89, 712 (1956).
353а. Р о 11 е г R. С, S p i 1 1 m a n G. E., J. Organometal. Chem., 7, 259 (1967).
354. О h a r a M., О к a w a r a R., N а к a m u r a Y., Bull. Chem. Soc Japan, 38, 1379
(1965).
355. О к a w a r a R., WadaM, J. Organometal. Chem., 1, 81 (1963).
356. Шостаковский M. Ф., А т а в и н А. С, В я л ы х Е. П., Трофимов
Б. А., ЖОХ, 35, 751 (1965).
357. R a m s d e n Н. Е., В a n k s С. К., Пат. США 2789994 (1957); РЖХим., 1959, 39597.
358. Weinberg E. L., Пат. США 2796412 (1957); С. А., 51, 16537 (1957).
359. EmeleusH.J., Zuckermann J. J., J. Organometal. Chem., 1, 328 (1964).
360. T a n а к а Т., U e e d a R., W a d a M., О к a w a r a R., Bull. Chem. Soc,
Japan, 37, 1554 (1964).
361. TanakaT., Komura M., Kawasaki Y., О к a w a r a R., J.
Organometal. Chem., 1, 484 (1964); Ka wa к a m i K., Okawara R., J. Organometal. Chem.,
6, 249 (1966)
362. В 1 а к e D., С о a t e s G. E., T a t e J. M., J. Chem. Soc, 1961, 756.
363. Андрианов К- А., ЯкушкинаС. Я-, ЖОХ, 35, 330 (1965).
364. Nelson W. H., Martin D. F., J. Organometal. Chem., 4, 67 (1965).
365. SchmidtM., D e r s i n H. J., Schumann H., Ber., 95, 1428 (1962).
366. LeistnerW.E., К n о e p k e О. Н., Пат. США 2726254 (1955); С. А., 50, 6095
(1956).
367. Abel E.W., A r i m t a g e D. А., В r a d у D. В., J. Organometal. Chem., 5,
130 (1966).
Реакции преобразования функциональных групп
431
368. A b е 1 Е. W., В г a d у D. В., J. Chern. Soc, 1965, 1192.
369. Abel E. W., В г a d у D. В., С г о s s е В. С, J. Organornetal. Chern., 5, 260 (1966).
370. Голл. пат. 109491 (1964); РЖХим., 1966, 5Н99.
371. WieberM., S с h rn i d t M., J. Organornetal. Chern., 1, 336 (1964).
372. WieberM., SchmidtM., Z. Naturforsch., 18b, 846 (1963).
373. Poller R. C, Proc. Chem. Soc, 1963, 312.
374. WieberM., SchmidtM., J. Organornetal. Chem., 2, 129 (1964).
375. SchumannH, К 6 p f H., SchmidtM., Angew. Chem., 75, 672 (1963).
376. S с h u m a n n H., К о p f H., S с h m i d t M., J. Organornetal. Chem., 2, 159 (1964).
377. V a 1 a d e J., PereyreM., Compt. rend., 254, 8693 (1965).
378. N e u m a n n W. P., HeymannE, Ann., 683, 11 (1965).
379. Я к у б о в и ч А. А., М а к а р о в С. П., Г и н з б у р г В. А., ЖОХ, 28, 1036
(1958).
380. Gerrard W., Rees R. G., J. Chem. Soc, 1964, 3510.
381. P о m m i e r J. -С, Р е г е у r e M., V a 1 a d e J., Compt. rend., 260, 6397 (1965).
382. PommierJ.-C, ValadeJ., Compt. rend., 260, 4549 (1965).
383. К a rn о j i t z k у V., Chimie et Industrie, 85, 92 (1961).
384. Александров Ю. А., БрилкинаТ. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1961, 3; 19G5, 228.
384а. Ч а у з о в В. А., Л и т в и н о в а О. В., Б а у к о в Ю. И., ЖОХ, 36, 952 (1966).
385. Землянский Н. Н., Гольдштейн И. П., Гурьянова Е. Н., Ш а -
м а г и н а О. П., П а н о в Е. М., С л о в о х о т о в а Н. А., Кочешков К. А.,
Изв. АН СССР, ОХН, 1967, 728.
386. Amberger Е., К и 1 а М. R., L о г b e r t h J., Angew. Chem., 76, 145 (1964).
387. A m b e r g e r E., К и 1 a M. R., Ber., 96, 2560 (1963).
388. Землянский Н. Н., Шамагина О. П., Панов Е. М., Кочешков
К. А., ЖОХ, 35, 1029 (1965).
389. Scherer O.J., S с h m i d t J. F., SchmidtM., Z. Naturforsch., 19b, 447
(1964).
389a. Sukhani D., Gupta V. D., Mehrotra R. C, J. Organornetal. Chem., 7,
85 (1967).
390. M а с k G. P., P a r k e г Е., Пат. США 2809956 (1957); С. А., 52, 3863 (1958).
391. L о r b e r t h J., К u 1 a M. R., Ber., 98, 520 (1965).
392. Scherer O. J., Sc hmi d t J., Wo k u 1 a t J., SchmidtM., Z. Naturforsch.,
20b, 183 (1965).
393. ШостаковскийМ. Ф., Комаров Н. В., Мисюнас В. К., 3 а и н-
ч к о в с к а я М. К., Изв. АН СССР, ОХН, 1964, 1102.
394. Cohen H. J., J. Org. Chem., 25, 154 (1960).
395. Strecker A., Ann., 105, 306 (1858).
396. S t г е с k e r A., Ann., 123, 365 (1862).
397. A 1 1 e s t о n D. L., D a v i e s A. G., Chem. a Ind., 1961, 949.
398. P f e i f f e г Р., В г а с k O., Z. anorgan. allgem. Chem., 87, 229 (1914).
399. G i b b о n s A. J., SawyerA.K, RossA, J. Org. Chem., 26, 2304 (1961).
400. H a r a d а Т., Sci. Papers Inst. Phys. Chem., Res. (Tokyo), 35, 290 (1939); С. А., 33,
5357 (1939).
401. Англ. пат. 711564 (1954); С. А. 49, 14797 (1955).
402. Alleston D.L., D a v i e s A. G., HancockM, J. Chem., Soc, 1964, 5744.
402a. D a v i e s A. G., Harrison P. G., J. Organornetal. Chem., 7, P13 (1967).
4026. D a v i e s A. G., Harrison P. G., J. Organornetal. Chem., 8, P19 (1967).
403. Da vies A. G., Harrison P. G., J. Organornetal. Chem., 10, P33 (1967).
404. R e i с h 1 e W. Т., Inorg. Chem., 1, 650 (1962); J. Org. Chem., 26, 4634 (1961)
405. К u p с h i k E. J., С a 1 a b r e t t a P. J., Inorg. Chem., 3, 905 (1964).
406. W a d a M., Kawasaki K., Oka wa r a R., J. Organornetal. Chem., 4, 159(1965).
407. W e s t 1 a k о A. H., M a r t i n D. F., J. Inorg. Nucl. Chem., 27, 1579 (1965).
408. РазуваевГ. А., Дьячковская О. С, ВязанкинН. С, Щепет-
кова О. А., ДАН СССР, 137, 618 (1961).
409. Razuvaev G. A., Vyasankin N. S., Shshepetkova О. A.,
Tetrahedron, 18, 667 (1962).
410. Гольдштейн И. П., Землянский Н. Н., Шамагина О. П.,
Гурьянова Е. Н., Панов Е.М., СловохотоваН. А., Кочешков К. А.,
ДАН СССР, 163, 880 (1965).
411. Davies A. G.. Harrison P. G.., J. Chem. Soc, 1967—С, 298.
412. JohnsonO.H, FritzH.E., J. Org. Chem., 19, 74 (1954).
413. JohnsonO.H., FritzH.E., Halvorson D.O., Evans R.L., J. Am.
Chem., Soc, 77, 5857 (1955).
414. Schumann H., К 6 p f H., SchmidtM., Ber., 97, 1458 (1964).
415. S с h u m a n n H., К 6 p f H., S с h m i d t M., Ber., 97, 2395 (1964).
Г лава XII
КООРДИНАЦИОННАЯ СПОСОБНОСТЬ
ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Наличие вакантных d-орбит на внешней оболочке атома олова
определяет его способность к координации и оказывает существенное влияние на
направление химических реакций [1,2], каталитическую активность [3],
устойчивость [4, 5], структуру и свойства оловоорганических соединений.
Оловоорганические соединения легко образуют комплексы с различного
рода лигандами, имеющими в своем составе гетероатомы с неподеленной
парой электронов (азот, кислород, сера и др.) [6—27]. В ряде случаев эти
комплексы представляют собой высокоплавкие кристаллические вещества. Их
применяют для выделения и очистки оловоорганических соединений
различной степени алкилирования.
Образование соединениями ди- и триэтилолова окрашенных комплексов
с дитизоном использовано [28] для их определения колориметрическим
методом. Строение этих комплексов рассмотрено в статье Ирвинга и Кокса
[29].
При комплексообразовании валентное состояние атома олова меняется.
Молекулы оловоорганических галогенидов имеют форму тетраэдра, в
центре которого расположен атом олова [30]. Такой структуре соответствует
5/73-гибридизация олова.
Методами ИК-спектров и рентгеноструктурного анализа установлено,
что комплекс хлористого триметилолова с пиридином имеет конфигурацию
григональной бипирамиды [31—33]. Этой конфигурации для соединений с-
координационным числом 5 соответствует 5/?3й-гибридизация [34].
На примере комплекса двухлористого диметилолова с бипиридилом
показано [17, 31], что шестикоординационные соединения олова имеют ок-
таэдрическую конфигурацию, т. е. здесь осуществляется 5/?3^2-гибридиза-
ция [34]. Для рассматриваемого комплекса возможны следующие стерео-
и оптические изомеры [17]:
Акцепторные свойства атома олова существенно зависят от характера
окружающих его групп. Так, склонность к комплексообразованию
оловоорганических хлоридов изменяется в следующем ряду [13, 35, 36]:
C6H5SnCl3 > (QH9)2SnCl2 > (CsH0)2SnCl2 > (C6H5)3SnCl > (C4H9)3SnCl.
В том же ряду уменьшается электронная плотность на атоме олова [37—
41]. Помимо галогенидов, комплексы образуют и другие оловоорганические
Координационная способность
433
соединения, например, диизотиоцианаты диалкилолова [37, 42, 43], дифуль-
минат дифенилолова [42], нитрат и перхлорат триметилолова [23], трибутил-
станнилбензимидазол [44], диэтиламинотриметилолово [45].
Соединения, содержащие связи Sn—S, не образуют комплексов с
обычными донорами. Вероятно, в этих соединениях вакантные d-орбиты олова
участвуют во взаимодействии с неподеленной парой электронов атома серы
[46, 47]. Сопряжение типа рп — d„ имеет место и в (триалкилстаннил)фос-
финах, (триалкилстаннил)арсинах, гексафенилдистаннане и некоторых
других соединениях олова [48—50].
О влиянии растворителей на устойчивость оловоорганических
комплексов см. статьи Окавары с сотр. [17, 51].
Наряду с образованием комплексов с различными органическими лиган-
дами, оловоорганические соединения способны к внутримолекулярной
координации и межмолекулярной ассоциации. Такие типы координации
возможны в тех случаях, когда молекула имеет в своем составе электронодонор-
ную группу (OR, OOCR, NR2 и др.). Например, гидроокись триметилолова
в твердом состоянии ассоциирована [52—54]. Ассоциация обусловлена не
водородными связями, а образованием мостиков типа Sn—O...Sn.
Следует отметить, что природа связи олова с кислородом длительное
время оставалась неясной. В литературе высказываются две точки зрения
относительно характера этой связи в ацилатах три- и диалкилолова. На
основании анализа ИК-спектров в некоторых работах [55—57] делается вывод
о том, что эти соединения имеют ионную структуру. Другие авторы [4, 58—
60] рассматривают ацилаты алкилолова как ковалентные соединения,
построенные аналогично органическим эфирам. Сравнительно малые величины
дипольных моментов (2,0D для ацилатов триалкилолова и 1,4D для диаци-
латов диалкилолова) однозначно свидетельствуют в пользу ковалентного
характера связи Sn—О в этих соединениях [61].
В твердом состоянии ацилаты триалкилолова являются
координационными полимерами, где ацетатные группы играют роль мостиков между
атомами олова [4, 58, 62, 63]. В растворах ацетаты триалкилолова мономер-
ны [4, 58, 61]. В случае же формиатов триалкилолова полимерная структура
частично сохраняется и в растворе [62]. Следует отметить, что измерение
ядерного магнитного резонанса растворов ацилатов триалкилолова не
подтверждает вывод об ассоциации этих веществ [37].
Что касается диацилатов диалкилолова, то они имеют, по-видимому,
следующую структуру [61] (ср. [64]):
Как видно из приведенной схемы, координационные возможности атома
олова здесь полностью использованы (две донорноакцепторные связи).
Поэтому следует ожидать, что диацилаты диалкилолова будут обладать меньшей'
способностью к межмолекулярному взаимодействию. Действительно, ИК-
спектры диацетата дибутилолова не изменяются при переходе от его
разбавленных растворов к жидкому состоянию [61].
28 Заказ № 207
434
Оловоорганические соединения. Лит. стр. 435—437
Подобную же структуру имеют ацетилацетонаты диалкил- [65, 66] и
дифенилолова [67]. Подробно исследованы [68] донорноакцепторные
свойства фенокситриэтил- и фенокситриметилолова.
Внутримолекулярная координация осуществляется и в бш>(оксихино-
лятах) диалкилолова. По данным ИК-спектров они могут иметь одну из
представленных ниже структур или представлять собой смесь этих
изомеров [17]:
СН3 СН3 СН3
?$*у W*
р^.—U->г- ч^-—L-Ъ*
N
IN-
Сопоставляя имеющиееся в литературе данные по исследованию
соединений со связью Sn—О—Sn следует прежде всего отметить, что гексаалкил-
дистанноксаны имеют молекулярные веса, соответствующие мономерным
формам молекул [61], в то время как соединения типа XSnR2OSnR2X (X —
электроотрицательная группа, например, галоид или остаток органической
кислоты) существуют в димерной форме [61, 69—72]. Это различие в
поведении дистанноксанов объясняется, по-видимому, тем, что наличие электро-
ноакцепторных групп при атоме олова повышает его акцепторную
способность и способствует образованию донорноакцепторных связей.
В литературе дискутируются две структуры димеров (XSnR2OSnR2X)2
R2SnX2
т
О
R2Sn SnR2
\ /
О
1
R2SnX2
1
R2SnX
1
О
XR2Sn SnR2X
\ /
О
1
R2SnX
II
Нетрудно убедиться, что структура I с четырехчленным циклом кова-
лентных связей Sn—О мало вероятна из-за стерических препятствий.
Более вероятна с этой точки зрения вторая из рассматриваемых структур [61].
Сравнительно небольшие дипольные моменты этих соединений
свидетельствуют о высокой степени симметрии, что вполне согласуется со структурой
II. Той же точки зрения на строение дистанноксанов указанного типа
придерживаются и другие авторы [70—72].
В димерах [XSnR2OSnR2OH]2 может быть дополнительная слабая
межмолекулярная ассоциация между кислородом гидроксильной группы и
атомом олова [70].
Рентгеноструктурный анализ показывает, что соединения состава
(CH3)3SiO(SnR20)2Si(CH3)3 построены следующим образом [71]:
Sn =
==Sn—О
t I
О—Sn=
/
=Sn
Интересно отметить, что соединения состава ClSn(C4H9)2 SSn(C4H9)2 CI [73]
и R«Sn[OSi(CH3)3]4-n [74] в растворе мономерны.
Координационная способность
435
Как показано Ван-дер-Керком с сотр. [4, 5], соединения типа R3SnR'
(R — алифатический или ароматический радикал, a R' — группа,
связанная с атомом олова через азот) имеют следующую структуру:
н сн3 й сн.
нс-^-сн сн3 сн3 нс—-сн сн3 сн3
Эти соединения исключительно устойчивы, еслиИ'—ненасыщенная группа,
содержащая два атома азота в стерически благоприятном положении, а
именно, когда второй атом азота находится в р-положении к первоначально
присутствующей NH-rpynne.
Известен ряд случаев, когда два различных оловоорганических
соединения могут образовывать между собой комплексы за счет донорных групп
одного из компонентов и акцепторных способностей атома олова в другом.
Гексаметилдистанноксан и гидроокись триметилолова дают с галогени-
дами триметилолова комплексы состава [(СН3)з5п]20 • (СН3)з$пВг [75,
76], [(CH3)3SnOH]2 . (CH3)3SnX [76—81] и (CH3)3SnOH - (CH3)3SnX - Н20
[77, 79, 82]. Комплексы последнего типа известны [82] и для гидроокиси
триэтил олова.
Методами криоскопии, дипольных моментов и инфракрасной
спектроскопии изучено [2, 83] взаимодействие в системах R2SnX2—R2Sn(OR)2 и R2Sn-
(OR)2—SnCl4 (X — хлор, бром или остаток органической кислоты; R = СН3
или С4Н9). Показано, что во всех этих системах происходит образование
комплексов состава 1:1. Их образование обусловлено донорными свойствами
алкоксигрупп и акцепторной способностью оловоорганических галогенидов
или ацилатов.
Оловоорганические соединения склонны также к образованию
координационных связей через атом галоида [4, 84—91]. Рентгеноструктурные
исследования показали [84], что фтористое триметилолово имеет структуру
координационного полимера с мостиковыми связями между молекулами.
Аналогичный тип координации имеет место в случае хлористого и бромистого
триметилолова [92] и некоторых других соединений типа R3SnX [93—96],
но он отсутствует у хлористого трифенилолова [97].
По патентным данным [98] при нагревании до 100—120° С замещенных
аммониевых солей типа R4NX с оловоорганическими галогенидами
образуются четвертичные аммониевые оловоорганические соли.
ЛИТЕРАТУРА
1. A b е 1 Е. W., Brady D., L e r w i 1 1 В. R., Chem. a. Ind., 1962, 1333.
2. Гольдштейн И. П., Землянский Н. Н., Шамагина О. П.,
Гурьянова Е. Н., Панов Е. М., Кочешков К. А., ДАН СССР, 163, 880 (1965).
3. D у е г Е., Р i n k e r t о n R. В., J. appl. Polymer Sci., 9, 1713 (1965).
4. Van derKerk G.J. M.,Luij ten J. G. A., Janssen M. J.,Chimia, 16, 10(1962).
5. L u i j t e n J. G. A., J a n s s e n M. J., van d e г К е г к G. J. M., Rec. trav.
chim., 81, 202 (1962).
6. К о ч е ш к о в К- А., Уч. зап. МГУ, 3, 297 (1934).
7. К о z e s с h к о w К. А., N a d j М. М., Вег., 67, 717 (1934).
8 Кочешков К. А., Надь М. М., ЖОХ, 5, 1158 (1935).
9. Р f е i f f е г P., F г i e d m a n n В., В е k a t e Н., Ann., 376, 310 (1910).
10. DruceJ.G. R, Chem., News, 119, 229 (1920).
11. DruceJ.G.R, J. Chem. Soc, 120, 1758 (1921).
12. D r u с e J. G. R, J. Chem. Soc, 121, 1859 (1922).
13. Гольдштейн И. П., Гурьянова Е. Н., Дел инская Е. Д.,
Кочешков К. А., ДАН СССР, 136, 1079 (1960).
28*
436
Оловоорганические соединения
14. А 1 1 е s t о n D. L., D a v i e s A. G., J. Chem. Soc, 1962, 2050.
15. Р f e i f f е г P., Z. anorgan. Chem., 133, 91 (1924).
16. AllestonD.L, DaviesA.G., Chem. a. Ind., 1961, 551.
17. T a n а к а Т., К о m u r a M., Kawasaki Y., О к a w a r a R., J.
Organometal. Chem., 1, 484 (1964).
18. Blake D., CoatesG. E., TateJ.M., J. Chem. Soc, 1963, 756.
19. П у д о в и к А. Н., Муратова А. А., С е м к и н а Э. П., ЖОХ, 33, 3350
(1963).
20. Y a s u d а М., Т о b i a s R. S., Inorg. Chem., 2, 207 (1963).
21. RoncucciL., Faraglia G., Barb ieri R., J. Organometal. Chem., 1,
427 (1964).
22. К r a u s С A., G r e e r W. N.. J. Am. Chem. Soc, 45, 3078 (1923).
23. Clark H.C., O'Brien R. J., Pickar d A. L, J. Organometal. Chem., 4,
43 (1965).
24. Joshi K.K., WyattP.A. H., J. Chem. Soc, 1959, 3825.
25. С a h о u r s A., Ann., 122, 48 (1862).
26. Werner A., Pfeiffer P., Z. anorgan. Chem., 17, 82 (1898).
27. Matwyoff N. A., D r a g о R. S., Inorg. Chem., 3, 337 (1964).
28. AldrideW. N., CremerJ.E., Analyst., 82, 37 (1957).
29. Irving H., Cox J. J., J. Chem. Soc, 1961, 1470.
30. Landolt H., В о r n s t e i n R., Physikalisch-chemische Tabellen, 2, 19 (1951).
31. В e a t t i e I. R., McQuillan G. P., J. Chem. Soc, 1963, 1519.
32. Beattie I. R., McQuillan G. P., HulmeR., Chem. a. Ind., 1962, 1429.
33. Hulme R., J. Chem. Soc, 1963, 1524.
34. К i m p b a 1 1 G. E., J. Chem. Phys., 8, 188 (1940).
35. Г о л ь д ш т е й н И. П., Ф а й з и Н. А., С л о в о х о т о в а Н. А.,
Гурьянова Е. Н., Викторова И. М., Кочешков К. А., ДАН СССР, 138, 839
(1961).
36. Г о л ь д ш т е й н И. П., Гурьянова Е. Ы., Кочешков К. А., Сб.
«Синтез и свойства мономеров». М., «Наука», 1964, стр. 109.
37. Poller R. С, J. Organometal. Chem., 3, 321 (1965).
38. Poller R. С, Proc Chem. Soc, 1963, 312.
39. M a i r e J.-C, С a s s a n J., Compt. rend., 260, 3931 (1965).
40. G r i f f i t h s V. S., Dervish G. A. W., J. Molec. Spectr., 3, 165 (1959).
A\. Gr i f f i th s V. S., Dervish G. A. W., J. Molec. Spectr., 5, 148 (1960).
42. M u f f i A. S., P о 1 1 e r R. C, J. Organometal. Chem., 3, 99 (1965).
43. W a d a M., NishinoM., Okawara R., J. Organometal. Chem., 3, 70 (1965).
44. JanssenM. J., LuijtenJ. G. A., van der KerkG. J.M., J.
Organometal. Chem., 1, 286 (1964).
45. JonesK., L a p p e r t M. F., J. Organometal. Chem., 3, 295 (1965).
46. С u m p e г С W. N., M e 1 n i k о f f A., V о g e 1 A. I., J. Chem. Soc, Inorg. Phys.
Theor., 1966, 242.
47. С u m p e г С W. N., M e 1 n i k о f f A., V о g e 1 A. I., J. Chem. Soc, Inorg. Phys.
Theor., 1966, 246.
48. Campbell I.G.M., F о w I e s G. W. A., N i x о n L. A., J. Chem. Soc, 1964,
1389.
49. Campbell I. G.M., F о w 1 e s G. W. A., N i x о n L. A., J. Chem. Soc, 1964,
3026.
50. H a g u e D. N., P r i n с e R. H., Proc. Chem. Soc, 1962, 300.
51. К о m u r a M., Kawasaki Y., TanakaT., Okawara R., J. Organometal.
Chem., 4, 308 (1965).
52. К r i e g s m a n n H., Hoffmann H., P i s с h t s с h a n S., Z. anorgan. allgem.
Chem., 315, 283 (1962).
53. Okawara R., Y a s u d a K., J. Organometal. Chem., 1, 356 (1964).
54. К a s a i N., Y a s u d a K., Okawara R., J. Organometal. Chem., 3, 172 (1965).
55. F r e e m a n n J. P., J. Am. Chem. Soc, 80, 5954 (1960).
56. Okawara R., Webster D.E., RochowE.G., J. Am. Chem. Soc, 82,
3287 (1960).
57. Okawara R., RochowE.G., J. Am. Chem. Soc, 82, 3285 (1960).
58. JanssenM. J., LuijtenJ. G. A., van der Kerk G. J.M., Rec trav.
chim., 82, 90 (1963).
59. В e a t t i e I. R., G i 1 s о n Т., J. Chem. Soc, 1961, 2585.
60. Cum m i ns R. A., Dunn P., Austral J. Chem., 17, 185 (1964).
61. ЗемлянскийН. Н., Гольдштейн И.П., Гурьянова Е. К.,
Панов E. M., С л о в о х о т о в а Н. А., Кочешков К. А., ДАН СССР, 156, 131
(1964).
62. Okawara R., О h а г а М., J. Organometal. Chem., 1, 360 (1964).
Координационная способность
437
63. О к a w a r a R., О h a r a M., Bull. Chem. Soc. Japan, 36, 623 (1963).
64. W a d a M., ShindoM., О к a w a r a R., J. Organometal. Chem., 1, 95 (1963).
65. MehrotraR.C, Gupta V.D., J. Organometal. Chem., 4, 237 (1965).
66. S m i t h J. A. S., W i 1 к i n s E. J., Chem. Communs, 1965, 381.
67. Nelson W.H., MartinD. F., J. Inorg. Nucl. Chem., 27, 89 (1965).
68. Matwiyoff N.A., Drago R.S., J. Organometal. Chem., 3, 393 (1965).
69. AllestonD.L, D a v i e s A. G., H a n с о с к М., W h i t e R. F. M., J. Chem.
Soc, 1963, 5469.
70. О к a w a r a R., W a d a M., J. Organometal. Chem., 1, 81 (1963).
71. О к a w a r a R., Proc. Chem. Soc, 1961, 383.
72. ConsidineW. J., Ventura J. J., Gibbons A. J., Ross A., Canad.
J. Chem., 41, 1239 (1963).
73. S с h u m a n n H., SchmidtM., Ber., 96, 3017 (1963).
74. S с h m i d b a u r H., H u s s e к Н., J. Organometal. Chem., 1, 244 (1964).
75. Harada Т., Bull. Chem. Soc. Japan, 15, 455 (1940).
76. К r i e g s m a n n H., Hoffman H., Z. anorgan. allgem. Chem., 321, 224 (1964).
77. H a r a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 36, 504 (1939); С. А., 34,
3675 (1940).
78. Harada Т., Bull. Chem. Soc. Japan, 6, 240 (1931).
79. K'rausC.A, Harada Т., J. Am. Chem. Soc, 47, 2416 (1925).
80. К r a u s С A., CallisCC, J. Am. Chem. Soc, 45, 2624 (1923).
81. К r a u s С. А., В u 1 1 a r d R. H., J. Am. Chem. Soc, 52, 4056 (1930).
82. H a r a d а Т., Sci Papers Inst. Phys. Chem. Res. (Tokyo), 36, 497 (1939); С. А., 33,
5357 (1939).
83. 3 e м л я н с к и й Н. Н., Гольд штейн И. П., Ш а м а г и н а О. П.,
Гурьянова Е. Нм Панов Е.М., Словохотова Н. А., Кочешков К-А.,
Изв. АН СССР, серия хим., 1967, 728.
84. Clark Н. С, О' BrienR.J., Т г о t t e r J., Proc. Chem. Soc, 1963, 85.
85. Neumann W. P., Schick R., К 6 s t e r R., Angew. Chem., 76, 380 (1964).
86. О с и п о в О. А., К а ш и р е н к о в О. Е., ЖОХ, 32, 1717 (1962).
87. К а ш и р е н и н о в О. Е., Сб. «Материалы 4-й научной конференции аспирантов».
Ростовский ун-т, Ростов-на-Дону, 1962, стр. 144; РЖХим., 1963, 14В60.
88. С a s s о 1 А., В а г b i e r i R., Ann. chimica, 55, 606 (1965); РЖХим., 1966, 6В61.
89. Т a g 1 i a v i n i G., Z a n e 1 1 a P., J. Organometal. Chem., 5, 299 (1966).
90. GielenM., N a s i e 1 s k i J., G e r n a u x R., Bull. Soc, chim. Belg., 72, 594
(1963).
91. Y a s u d a K., Kawasaki Y., К a s a i N., T a n a k а Т., Bull. Chem. Soc.
Japan, 38, 1216 (1965).
92. Kriegsmann II, Pisch tschan S., Z. anorgan. allgem. Chem., 308, 212
(1961).
93. С 1 a r k H. С, О' В г i e n R. J., Inorg. Chem., 2, 1020 (1963).
94. ClarkH.C, O'Brien R. J., Proc Chem. Soc, 1963, 113.
95. О k a w a r a R., H a t h a w а у B. J., W e b s t e r D. J., Proc, Chem: Soc, 1963,
13.
96. Y a s u d а К., О k a w a r a R., J. Organometal. Chem., 3, 76 (1965).
97. Kriegsmann H., Geissler H., Z. anorgan. allgem. Chem., 323, 170 (1963).
98. Seyferth D., Пат. США 3070615 (1962); РЖХим., 1965, 1H325.
Глава XIII
РЕАКЦИИ СОЕДИНЕНИЙ ТИПА
R3SnSnR3 И (RaSn)n
В соединениях типа (R2Sn)„ и R3SnSnR3 связь Sn—Sn легко
расщепляется при действии различных окислителей, перекисей ацилов, галоидных
алкилов и некоторых других реагентов. Для этих соединений характерны
также реакции диспропорционирования (см. стр. 376). Менее подробно
изучено их деалкилирование и способность к присоединению по кратным
связям (см. стр. 316).
Кислород воздуха превращает диалкильные и диарильные соединения
олова в соответствующие полимерные окиси [1—12].
2К25п + 02-*2(К25пО)л.
Скорость окисления зависит от характера органических радикалов,
связанных с атомом олова. Чем меньше молекулярный вес радикала, тем легче
проходит реакция (свет действует ускоряющим образом). Например, чистое
дифенантрилолово на воздухе практически не изменяется [1]. Дифенилолово
окисляется довольно медленно [2], а диметилолово иногда со взрывом [2,
3]. В растворах реакция проходит значительно быстрее.
Окисление дифенилолова [11]. К раствору 5,44 г (0,02 моля) дифенилолова в 15 мл
бензола при взбалтывании в течение 15 мин. приливают 2,5 г (0,022 моля) перекиси
водорода. Реакция проходит с некоторым разогреванием. Через 1,5 часа осадок
[отфильтровывают, промывают бензолом и высушивают в вакууме. Выход окиси дифенилолова 97%.
Дифенилолово воспламеняется при обработке дымящей азотной кислотой
[9]. Окрашенное в темно-красный цвет диэтилолово обесцвечивается при
растворении в хлороформе. Из раствора получают кристаллы с т. пл. 216° С,
состав которых отвечает формуле HO[(C2H5)2SnO]3H -(C^HsbSnCla [12].
В растворе бромистого этила диэтилолово дает бромистое соединение того
же типа с т. пл. 210° С [12]. Гексаалкилдистаннаны окисляются значительно
медленнее [13—16]. Изучено [17, 18] окисление гексаэтилдистаннана
кислородом в растворе я-нонана при начальной концентрации раствора от 10 до
100 мол. %. Реакция проходите заметной скоростью при 50° С. С повышением
температуры ее скорость значительно возрастает. Скорость окисления
линейно возрастает с увеличением концентрации, что указывает на первый
порядок реакции. В качестве основных продуктов выделены окись диэтилолова,
гексаэтилдистанноксан, ацетальдегид и вода [17].
Высказано предположение [18], что промежуточным продуктом этой
реакции является перекись триэтилолова. Обнаружить ее в реакционной
смеси не удается вследствие большой скорости взаимодействия перекиси с гек-
саэтилдистаннаном. Если же в качестве окислителя применять чистую
перекись триэтилолова, то она уже при комнатной температуре энергично
реагирует с гексаэтилдистаннаном. Единственным продуктом является гекса-
Реакции соединения типа RgSnSnR8 и (RtSn)n 439
этилдистанноксан. Предложен [18] следующий механизм реакции:
(C2H5)3SnO- + (C2H5)3SnSn (С2Н5)3-> (C2H5)8SnOSn (ОДЬ + (C2H5)3Sn-,
(C2H5)3Sn- + (C2H5)3Sn-00—Sn (C2H5)3 -* (C2H5)3SnOSn (QH^ + (Q^bSnCT,
(C2H5)3SnO* + (C2H5)3Sn--^ (QHsbSnOSn (C2H5)3,
2 (C2H5)3Sn- -> (C2H5)3SnSn (C2H5)8.
Стабильные кислородные радикалы (А) моментально взаимодействуют с
гексаэтилдистанном, о чем можно судить по исчезновению сигнала при
контроле за реакцией методом ЭПР [19].
-О'
(А)
Гексаарилдистаннаны с кислородом воздуха не реагируют [10, 15, 20].
Гексафенилдистаннан легко окисляется перманганатом калия в ацетоне при
комнатной температуре. С количественным выходом выделяется гидроокись
трифенил олова:
3 (C6H5)2SnSn (СбН5)3 + 2 КМпО* + ЗН20 -* 6 (C6H5)3SnOH.
Предполагается, что промежуточными продуктами этой реакции являются
перекисные соединения олова, но выделить их не удается [21]. Рассмотрен
механизм окисления гексафенилдистаннана кислородом воздуха в тетраги-
дрофуране в присутствии метилата натрия [21а]. В бензольном растворе гек-
сафенантрил-8-дистаннан сразу восстанавливает раствор перманганата
калия в ацетоне на холоду [1].
При действии серы на диарильные соединения олова образуются
соответствующие сульфиды. Например, сульфид дифенилолова получен
кипячением соответствующей смеси в толуоле в течение часа. При комнатной
температуре реакция проходит значительно медленнее.
Реакция дифенилолова с серой [11]. Раствор 0,80г (0,25 моля) серы в 10 мл толуола
прибавляют к 5,45 г (0,020 моля) дифенилолова в 15 мл толуола. Смесь кипятят в течение 3,5
час, охлаждают до комнатной температуры и отфильтровывают от незначительного
количества осадка (0,025 г). Фильтрат концентрируют до 15 мл и фильтруют. Получают 4,37 г
сульфида дифенилолова с т. пл. 179—180,5° С. Дальнейшее концентрирование раствора до
объема 3 мл дает еще 0,81 г вещества с т. пл. 177—178,5° С. Общий выход 85%.
Сульфид состава
получен [22] из соответствующего оловоорганического соединения и серы в
бензоле. Реакция проходит при комнатной температуре в течение
нескольких дней. Выход 69%; т. пл. 274—275° С (из хлористого метилена).
Аналогично получен сульфид б^с-(триметилолова) [16].
При действии галоидов на соединения типа R3SnSnR3 [4, 9, 10, 13, 16,
23—30] и (R2Sn)^ [2, 3, 6, 7, 9, 10, 31—33] образуются оловоорганические
моно- или дигалогениды. Специальными опытами с хлоридами этилолова и
гексаэтилдистаннаном показано, что при—60н—70° С хлор и бром
действуют только на связи Sn—Sn. Связь олова с углеродом остается
незатронутой. Действие хлора на диалкильные соединения олова удобно
применять для выяснения структуры последних [33]. В случае арильных
соединений олова для той же цели используют реакцию с иодом в бензоле [24].
440 Оловоорганические соединения. Лит. стр. 445—446
Обработкой гекса- или нонамерного диэтилолова хлором получено [33]
двухлористое диэтилолово, не содержащее примесей хлористого триэтил-
олова или треххлористого этилолова.
Реакция октадекаэтилциклононастаннана с хлором [33]. Реакцию проводят в приборе
Шленка,[предварительно заполненном сухим азотом или аргоном. К охлажденному до —70° С
раствору 0,5 г октадекаэтилциклононастаннана в дихлорметане прибавляют по каплям
предварительно охлажденный раствор хлора в том же растворителе. Хлора прибавляют
небольшой избыток, так чтобы образовался раствор темно-желтого цвета. Смесь перемешивают в
течение 2 мин. и избыток хлора удаляют, пропуская в охлажденную до —70° С реакционную
смесь ток пропилена. Затем растворитель удаляют в вакууме и остаток анализируют методом
газовой хроматографии.
Окта-т/?£/п-бутилциклотетрастаннан легко растворяется в бензольном
растворе иода с образованием кристаллического продукта, который по
данным анализа соответствует формуле J[Sn(C4H9)2]4J [34].
Аналогично реагируют с галоидами и диарильные соединения олова.
Реакция дифенилолова с хлором [11]. В раствор 2,72 г (0,01 моля) дифенилолова в
четыреххлористом углероде пропускают ток хлора (из перманганата калия и хлористого
водорода. Теоретический выход хлора 0,015 моля). Раствор выпаривают примерно наполовину,
отфильтровывают от небольшого количества осадка и выпаривают досуха. В остатке —
двухлористое дифенилолово с т. пл. 40—41° С. Выход 98%.
Бромированием дифенилолова в бензоле получено двубромистое
дифенилолово с выходом 48% [11].
При иодировании дифенилолова рассчитанным количеством иода
получены противоречивые результаты. По данным Крафта и Бородиной [35]
реакция проходит следующим образом:
(CGH5)2Sn + 2 J -* (CeH5)aSnJo,
п (CeH5)aSn + (C6H5)2SnJ2 _> J [Sn (CeH5)a]nJ.
Нейман и Кениг [36] обработкой додекафенилциклогексастаннана
эквимолекулярным количеством иода получили небольшое количество двуиодис-
того дифенилолова, тогда как большая часть исходного вещества осталась
без изменения.
Взаимодействием додекафенилциклогексастаннана с рассчитанным
количеством иода в толуоле получено [36] двуиодистое дифенилолово. Оно
образуется также с выходом 86% при прибавлении иода к суспензии
дифенилолова в метиловом спирте. Для завершения реакции смесь нагревают до
50—55° С [И].
Избыток иода деарилирует дифенилолово. Из реакционной смеси
выделены трехиодистое фенилолово и четырехиодистое олово. Одновременно
образуется иодбензол [35]. Иодирование дифенантрилолова также
сопровождается отрывом арильных радикалов. Образуется смесь продуктов, которая
плавится в широком интервале температур [1].
Буэ с сотр. [37] исследовали кинетику реакции гексаметил- и гексабутил-
дистаннана с иодом. В метиловом спирте эти реакции проходят очень быстро
и с измеримой скоростью в ацетоне, содержащем йодистый натрий.
Установлено, что каталитическое влияние оказывает окись диалкилолова, обычно
присутствующая в качестве примеси в исходном гексаалкилдистаннане.
Кинетика и механизм реакций гексаалкил- и гексаарилдистаннанов с иодом
подробно исследованы Тальявини с сотр. [38].
Растворы гексафенилдистаннана и гекса-я-хлорфенилдистаынана
обесцвечивают иод при комнатной температуре, превращаясь виодиды триарил-
олова [23].
Реакции соединения типа R3SnSnR3 и (RaSn)n
441
Бромирование гексафенилдистаннана [9]. К охлажденному до —30° С раствору 2 г
гексафенилдистаннана в 100 г хлороформа прибавляют по каплям охлажденный до той же
температуры титрованный раствор брома в хлороформе. Каждая капля тотчас
обесцвечивается, пока не добавлено 0,46 г брома. Хлороформ отгоняют. В остатке — бесцветные
кристаллы бромистого трифенилолова с т. пл. 120,5 С (из спирта).
Соединения типа R3SnSnR3 и (R2Sn)n восстанавливают азотнокислое
серебро [1, 4, 7—10, 14, 39] и сулему [16, 40—42] до свободного металла:
R3SnSnR3 + HgCl2 -> 2 R3SnCl + Hg,
R2Sn + HgQ3 - RaSnCla -f Hg.
Реакция диэтилолова с сулемой [40, 41]. 5,3 г (0,03 моля) диэтилолова смешивают с
3,1 г (0,03 моля) сулемы. Тотчас начинается экзотермическая реакция. Смесь нагревают 30
мин. на водяной бане. Выделение ртути количественное. Ее отделяют и вещество
кристаллизуют из петролейного эфира. Получают 5,8 г (80%) двухлористого диэтилолова с т. пл.
84° С.
Реакция дифенилолова с сулемой [11]. К 1,98 г (0,0072 моля) сулемы в 70 мл эфира
прибавляют 2,0 г (0,0073 моля) дифенилолова. Сразу же начинается реакция, что видно по
образованию серого осадка, содержащего капельки ртути. Смесь фильтруют, фильтрат
концентрируют до 3 мл, снова фильтруют и охлаждают в холодильнике. Получают 1,24 г (58%)
двухлористого дифенилолова с т. пл. 35—38° С. Перекристаллизация из минимального
количества петролейного эфира дает 0,81 г кристаллов с т. пл. 41—42° С.
Дифенантрилолово с сулемой не реагирует, тогда как гексафенантрил-
дистаннан восстанавливает ее до каломели [43].
О реакции диалкильных соединений олова с ртутноорганическими
соединениями см. стр. 270.
Реакция между дибутилоловом и двухлористым дибутилоловом
приводит к хлористому трибугилолову и металлическому олову [43 а]. Дву-
иодистое олово восстанавливается дибутилолоЕом до металла [43 6].
Дифенилолово реагирует с хлорным оловом и двухлористым дифенил-
оловом следующим образом:
(CeH5)3Sn + SnCl4 — (C6H,)2SnCl2 + SnCl2.
(C6H5)2Sn + 2 (C6H5)2SnCl2 -> 2 (CeH5)8SnCl + SnCl2.
Реакция дифенилолова с хлорным оловом [11]. К охлажденному льдом раствору 1,67 г
(0,006 моля) дифенилолова в смеси 10 мл бензола и 5 мл петролейного эфира (т. кип. 60—
90° С) в течение 15 мин. прибавляют раствор 1,16 г (0,006 моля) хлорного олова в 17 мл
бензола. Смесь кипятят 30 мин., охлаждают и фильтруют. Осадок промывают бензолом и
высушивают в течение часа при 110° С. Получают 1,14 г хлористого олова (теоретический
выход 1,16 г). Фильтрат концентрируют до образования маслянистой жидкости, приливают
7 мл петролейного эфира (т. кип. 40—60° С), фильтруют, концентрируют до 3 мл, и
охлаждают. Выход двухлористого дифенилолова 1,29 г (76%); т. пл. 39,5—41° С.
Реакция дифенилолова с двухлористым дифенилоловсм [11]. 1,72 г (0,006 моля)
дифенилолова и 4,34 г (0,012 моля) двухлористого дифенилолова растирают в ступке и смесь
переносят в колбочку на 50 мл. Нагревают на водяной бане до образования гомогенного раствора
(10 мин.), а затем 5 час. при 130—140° С. По охлаждении приливают 25 мл лигроина (т. кип.
60—90° С). Смесь нагревают до кипения и фильтруют горячей. По охлаждении раствора
выделяется 3,53 г (77%) хлористого трифенилолова с т. пл. 103—105° С.
Интересно отметить, что по данным Крауса и Сэшона [16] при
высушивании растворов гексаметилдистаннана хлористым кальцием образуется
хлористое триметилолово.
Соединения типа R3SnSnR3 и (R2Sn)n активно реагируют с органическими
перекисями с образованием оловоорганических ацилатов. Эту реакцию
предложено [44] использовать для оценки строения органостаннанов. Так,
взаимодействием диэтилолова с перекисью бензоила получен дибензоат
диэтилолова [45].
При реакции дифенилолова с рассчитанным количеством перекиси
бензоила образуется [11] со,со-дибензоат тетрафенилдистаннана:
2 (C6H5)2Sn + (С6Н5СОО)2-* CGIi5COOSn (C6H5)3Sn (CeIl5)2OOCCeH5.
442
Оловоорганические соединения. Лит. стр. 445—446
Его состав доказан элементарным анализом, отсутствием депрессии
температуры плавления с веществом, полученным независимым путем (см. стр.
466), и реакцией с бромом:
C6H5COOSn (C6H5)2Sn (СбН5)200ССбН5 + Вг2 -> 2 (C6H5)2Sn (Br) (ООССбН5).
Получение о),о)-дибензоата тетрафенилдистаннана [11]. К раствору 0,77 г перекиси
бензоила в 10 мл бензола прибавляют 1,70 г дифенилолова. Реакция сопровождается
незначительным выделением тепла. Через 15 мин. из раствора начинают выделяться первые
кристаллы. Смесь оставляют на ночь, осадок отфильтровывают и высушивают. Получают 2,0 г
(81%) вещества с т. пл. 172—177° С. После перекристаллизации из бензола т. пл. 184—185° С
и при дальнейших кристаллизациях не изменяется.
Как показано Разуваевым с сотр. [19,46—49], гексаэтилдистаннан в
бензоле реагирует с перекисью бензоила, ацетилбензоила, циклогексильным
эфиром надугольной кислоты и mpem-бутилпербензоатом:
(CaHsbSnSn (СзН5)з + (СвНйСОО)2 -* 2 (C2H5)3Sn (ООССвН5),
(C2H5)8SnSn (С2Н5)3 + С6Н5СООООССН3 -» (QH5)3SnOOCCeH5 + (QH5)3SnOOCCH3,
(C2H5)3SnSn (С2Н5)3 + (СвНцООО)з -> 2 (C2H5)3SnOC6H11 + С02.
Реакции проходят при комнатной температуре. Бензол в реакциях с
перекисями не участвует. При замене бензола четыреххлористым углеродом
растворитель уже не является инертным. Так, при взаимодействии гексаэтилди-
станнана с перекисью бензоила, кроме бензоата триэтилолова, образуется
заметное количество (17,5%) хлористого триэтилолова.
Перекись трет-бутнлэ. реагирует с гексаэтилдистаннаном отлично от
ацильных перекисей [47, 48, 50]. Реакция проходит при температуре
разложения перекиси (130—140° С). При этом на 1 моль распавшейся перекиси
образуется 0,54 моля этана, 1,56 моля этилена, 0,02 моля метана, 0,03 моля
бутана, 0,87 моля пгрет-бутловото спирта, 0,94 моля /прет-бутокситриэтил-
олова и полимерные продукты, содержащие олово.
Кроме того, Разуваевым и Федотовой [51] изучен распад бензолазотри-
фенилметана и нитрозоацетанилида в растворе четыреххлористого углерода
в присутствии дифенилолова. Установлено, что фенильные радикалы,
образующиеся из бензолазотрифенилметана, присоединяются к добавленному
в реакционную смесь дифенилолову, переводя его в гексафенилдистаннан:
(C6H5)SCN2C6H5 -> (СбН5)3С + N2 + СбН'5,
(C6H5)3Sn + СбН-5 -> (C6H5)3Sn\
При нагревании нитрозоацетанилида и дифенилолова в четыреххлорис-
том углероде образуется тетрафенилолово. С гексафенилдистаннаном нитро-
зоацетанилид практически не реагирует [51].
Соединение типа (R2Sn)n и R3SnSnR3 вступают также в реакции с
галоидными алкилами. Взаимодействие диалкильных соединений олова с ал кил-
или арилгалогенидами может проходить в двух направлениях [10, 43а, 52, 53]:
^Sn + R'X^RoR'SnX,^
R33n -f 2 R'X -* R3SnX2 —S-> R3SnX.
Так, при нагревании до 140° С диэтилолова с йодистым этилом образуется
[52] йодистое триэтилолово. Аналогично проходят реакции дибутилолова с
бромистым «-бутилом и бромбензолом (вне зависимости от молярного
отношения реагентов) [53]. Нагреванием дибутилолова с эквимолекулярным
количеством бромистого аллила или бромистого бензила получено бромистое
дибутилаллил- и дибутилбензилолово соответственно. При применении
избытка этих реагентов образуется двубромистое дибутилолово. С трифенил-
хлорметаном дибутилолово образует двухлористое дибутилолово.
Реакции соединения типа ReSnSnR5 и (R2Sn)n
443
Аналогично ведут себя дигалоидные алкилы. Так, дибутилолово
экзотермически реагирует с 1, 2-дибромэтаном с образованием двубромистого
дибутилолова и этилена (выход 70 и 96,3% соответственно). При 5—10° С
реакция заканчивается за 30 мин. В отличие от этого 1,4-дибромбутан
реагирует с дибутилоловом лишь в очень жестких условиях (24 часа при 140—
150° С) [53]:
(C4H9)2Sn + Br (CH2)4Br -> (QH9)2Sn(Br) (CH2)4Br.
Более сложно проходят реакции между дифенилолозом и йодистыми
(или бромистыми) алкилами [53 а].
Скорость и направление реакций гексаалкилдистаннанов с галоидными
алкилами также зависят от природы последних. Так, нагревание до 180° С
гексаэтилдистаннана с йодистым метилом приводит к метил триэти л олову
и йодистому триэтилолову [42]. При фотолизе гексаэтилдистаннана с
бромистым этилом [54], бромистым амилом [54] или бромистым бензилом [54, 55]
основными оловоорганическими продуктами оказываются бромистое триэтил-
олово и двубромистое диэтилолово. Соединения типа R3SnR' не образуются.
Гексаэтилдистаннан реагирует также с 1,4-дибромбутаном, 1,5-дибромпен-
таном, р-бромэтилбензолом; хлористым бензилом и трифенилхлорметаном
[19, 56]. Наиболее легко проходит реакция с трифенилхлорметаном (4,5
часа при 100° С):
(QHsJgSnSn (С2Н5)3 + 2 (СбН5)3СС1 -> 2 (C2H5)3SnCl + 2 (СбН5)3С.
Трифенилметильные радикалы образуются даже при избытке оловооргани-
ческого соединения, что указывает на отсутствие взаимодействия между
ним и возникающими радикалами даже при нагревании. С хлористым
бензилом реакция проходит в жестких условиях (4 часа при 190—200° С).
Основными продуктами являются хлористое триэтилолово и дибензил. В
наиболее жестких условиях проходят реакции гексаэтилдистаннана с (5-бром-
этилбензолом, 1,4-дибромбутаном и 1,5-дибромпентаном (7—14 час. при
190—200° С). В этом случае параллельно с основным процессом образования
бромистого триэтилолова (выходы 70,5, 72,5 и 82,4% соответственно)
наблюдается диспропорционирование гексаэтилдистаннана.
Нагревание до 80° С гексаэтилдистаннана в четыреххлористом углероде
в присутствии кислорода воздуха приводит к смеси хлористого триэтилолова
и двухлористого диэтилолова [47, 57]. Инициирующее действие кислорода
может быть заменено добавками перекиси бензоила. Предложена [47, 57]
следующая схема этой реакции:
(C2H5)3SnSn(C2Ho)3 + CClg -» (C3H5)3SnSn (С2Н5)2СНСН3 + СНС13,
(C3H5)3SnSn(C2H5)2CHCH3--* (QHsbSn' + (C2H5)2Sn + С2Н4,
(C2H5)3Sn' + CCI4 -> (QjHsbSnCl + CCf3,
(C2H5)2Sn + ССЦ -* (C2H5)2SnCl2,
При нагревании или облучении УФ-светом гексаалкилдистаннанов с
йодистыми перфторалкилами образуются триалкилперфторалкильные соединения
олова [58—63]:
R3SnSnR3 + R'X -> R3SnR' + R3SnX.
Получение триметилтрифторметилолова [60]. В трубку Кариуса емкостью 100 мл
помещают 15,7 г (0,048 моля) гексаметилдистаннана. Охлаждают, эвакуируют и конденсируют
28,9 г (0,153 моля) трифториодметана. Смесь нагревают при 80° С в течение 2 час. и затем
фракционируют. Получают 19,2 г (0,098 моля) непрореагировавшего трифториодметана,
фракцию с т. кип. 100—104° С и 10,5 г (0,036 моля) йодистого триметилолова (т. кип. 165—
169° С). Повторная перегонка второй фракции дает 9,8 г (0,039 моля)
триметилтрифторметилолова с т. кип. 100—101° С.
444 Оловоорганические соединения. Лит. стр. 445—446
По другому методу [58, 59] в толстостенную трубку из стекла «пирекс»
(толщина стенок 3,5 мм) помещают раствор гексаметилдистаннана в
избытке трифториодметана. Смесь облучают ультрафиолетовой лампой мощностью
200 вт, помещенной на расстоянии 20 см от ампулы. Время реакции 6 час,
выход чистого вещества 85%.
Триметилтрифтометилолово получено также взаимодействием
гексаметилдистаннана с йодистым трифторацетилом [63а]:
CF3COJ + (CH3)3SnSn (СНз)з -> (CH3)3SnCF3 + (CH3)3SnJ + CO.
С хлористым трифторацетилом реакция проходит иначе и приводит к смеси
хлористого триэтилолова и двухлористого диэтилолова.
Триметилтрифторметилолово представляет собой бесцветную жидкость
со слабым запахом, сходным с запахом тетраметил олова. При нагревании до
150° С в течение 20 час. в атмосфере инертного газа вещество разлагается
с выделением гексафторциклопропана [60]:
CF2—GF2
2 (CH8)8SnCF8 -> (CH8)3SnF + \ /
CF2
Те же продукты образуются и при термическом разложении триметилтри-
фторметилолова в присутствии тетрафторэтилена [60].
Получение пентафторэтилтрифенилолова [62]. 20 г (0,0286 моля) гексафенилдистаннана'
и 7,5 г (0,0305 моля) пентафториодэтана нагревают при 220° С в течение 15 час. в стальной
бомбе емкостью \00 мл. После охлаждения до комнатной температуры летучие вещества
удаляют. Твердый продукт реакции переносят в аппарат Сокслета и экстрагируют 500 мл
петролейного эфира (т. кип. 30—60° С) в течение 5 час. Образовавшийся раствор титруют
иодом в петролейном эфире до тех пор, пока не прекратится обесцвечивание иода. При этом
непрореагировавшая часть исходного гексафенилдистаннана количественно превращается
в йодистое трифенилолово. Его отделяют пропусканием через раствор сухого аммиака.
Осадок отфильтровывают и фильтрат концентрируют. Получают 3,1 г (22%)
пентафторэтилтрифенилолова с т. пл. 127—128° С.
Как показано на примере бром- и иодбензола, галоидные арилы не
реагируют в заметной степени с гексаэтилдистаннаном при нагревании смеси
до 180—190° С в течение 20—30 час. [19, 56].
Связь Sn—Sn расщепляется также рядом других реагентов. Например,
нагреванием дифенилолова с дибензилдисульфидом в бензоле получен дитио-
бензилат дифенилолова:
2 (C6H5)2Sn + CeH5CH3SSCHaCeH5 -> 2 (C6H5)2Sn (SCH2C6H5)2.
Получение дитиобензилата дифенилолова [11]. Смесь 5,46 г (0,02 моля) дифенилолова»
4,93 г (0,02 моля) дибензилдисульфида нагревают в 20 мл бензола в течение 7,5 часа. После
охлаждения фильтруют и бензол отгоняют в вакууме. Остаток представляет собой светло-
желтую жидкость. Выход 9,6 г (92%); п2£ 1,6610.
С бензохиноном дифенилолово образует полимерный продукт состава
[(C6H5)2SnOC6H40]„.
Реакция дифенилолова с бензохиноном [11]. 2,71 г (0,01 моля) дифенилолова в 10 мл
бензола прибавляют к раствору 1,07 г (0,01 моля) бензохинона в 10 мл того же растворителя.
Через несколько минут раствор окрашивается в темно-зеленый цвет, появляется осадок того
же цвета, и 5 мин. спустя жидкость окрашивается в зеленовато-желтые тона. Через 30 мин.
выпадает осадок белого цвета. Смесь оставляют при комнатной температуре на 8 час, затем
нагревают на водяной бане 15 мин. и отфильтровывают. Осадок промывают бензолом и
высушивают. Выход продукта 3,24 г (86%).
Описаны реакции гексаэтилдистаннана с тетраацетатом свинца [46, 47,
55], азодиизобутиронитрилом [46, 47, 55], нитрозоацетанилидом [46, 47, 55],
хлористым бензоилом [19], бензолсульфохлоридом [19], я-толуолсульфохло-
Реакции соединения типа R3SnSnR3 и (R2Sn),
445
ридом [19]. Наиболее легко и практически мгновенно проходит
взаимодействие гексаэтилдистаннана с тетраацетатом свинца:
(QsHgfoSnSn (С2Н5)3 + РЬ(ООССН3)4 -> 2 (QsHgfoSnOOCCHg + РЬ(ООССП3)2.
Реакция гексаэтилдистаннана с тетраацетатом свинца [46]. Раствор 1,11 г (0,0025 моля)
тетраацетата свинца в 25 мл сухого бензола приливают к 1,03 г (0,0025 моля)
гексаэтилдистаннана. Немедленно выпадает творожистый осадок,содержащий ацетат свинца и ацетат
триэтилолова. Реакционную смесь нагревают до кипения и фильтруют. Из фильтрата при
охлаждении кристаллизуется 0,84 г (63,6%) ацетата триэтилолова; т. пл. 133—134° С
(из бензола).
Гораздо сложнее протекает реакция гексаэтилдистаннана с нитрозоацет-
анилидом в бензоле. Несмотря на мягкие условия проведения реакции
(комнатная температура) наблюдается обильное смолообразование.
Единственным идентифицированным продуктом является ацетат триэтилолова [46].
В растворах четыреххлористого углерода образуется ацетат триэтилолова,
хлористое триэтилолово и хлористый фенилдиазоний [46].
Реакции соединений типа R3SnSnR3, проходящие без разрыва связи
,Sn—Sn, остаются мало изученными. При кипячении в течение 15 час. гекса-
•фенилдистаннана со смесью ледяной уксусной кислоты и уксусного
ангидрида получено [64] соединение состава (CH3COO)3SnSn(OOCCH3h с
т.разл.^>300° С. Описаны реакции ди-я-бутилолова с кислотами и
гидроперекисями [65].
ЛИТЕРАТУРА
1. Bahr G., GeliusR., Вег., 91, 829 (1958).
2. С h a m b e г s R., S с h e г е г P., J. Am. Chem. Soc, 48, 1054 (1926).
3. К г a u s C, G г e e r W., J. Am. Chem. Soc, 47, 2568 (1925).
4. В 6 e s e k e n J., Rutgers J.J., Rec. trav. chim., 42, 1017 (1923).
5. S a w у e г А. К-, J. Am. Chem. Soc, 87, 537 (1 э>65).
6. F г a n k 1 a n d E., Ann., 85, 329 (1853).
7. P f e i f f e г P., Ber., 44, 1269 (1911).
8. LowigC, Ann., P4, 308 (1852).
9. KrauseE., BeckerR., Ber., 53, 173 (1920).
10. KrauseE., PohlandR., Ber., 57, 532 (1924).
И. К u i v i 1 a H. G., J a k u s i k E. R., J. Org. Chem., 26, 1430 (1961).
12. Ha г a da Т., J. Sci. Res. Inst. (Tokyo), 43, 31 (1948).
13. В г о w n M. P., Fo w 1 es G. W. A., J. Chem. Soc, 1958, 2811.
14. Harada Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 35, 290 (1939); С. А.,
33, 5357 (1939).
15. KrausCA, Bullard R. H., J. Am. Chem. Soc, 48, 2131 (1926).
16. KrausCA., SessionsW.V., J. Am. Chem. Soc, 47, 2361 (1925).
17. А л e к с a н д p о в Ю. А., Р а д б и л ь Б. А., Ш у ш у н о в В. А., Труды по
химии и хим. технологии (Горький), 1960, 388.
18. А л е к с а н д р о в Ю. А., Ш у ш у н о в В. А., ДАН СССР, 140, 595 (1961).
19. Р а з у в а е в Г. А., Дер гу нов Ю. И., В я з а н к и н Н. С, ЖОХ, 32, 2515
(1961).
20. Oilman H., R о s e n b е г g S. D., J. Org. Chem., 18, 1554 (1953).
21. Bahr G., Z. anorgan. allgem. Chem., 256, 107 (1948).
21a. T s a i Т. Т., LehnW.L, J. Org. Chem., 31, 2981 (1966).
22. К u i v i 1 a H., В e u m e 1 O., J. Am. Chem. Soc, 80, 3250 (1958).
23. НадьМ.М, К о ч е ш к о в К. А., ЖОХ, 8, 42 (1938).
24. Law К. К-, J. Chem. Soc, 1926, 3243.
25. L a d e n b u г g A., Ber., 3, 353 (1870).
26. Krause E., Weinberg K-, Ber., 62, 2235 (1929).
27. С a hours A., Ann., 114, 227 (1880).
28. Gilman H., Gerow С W., J. Org. Chem., 22, 334 (1957).
29. KrausCA., E a t о u g h H., J. Am. Chem. Soc, 55, 5008 (1933).
30. KrausCA., FosterLS., J. Am. Chem. Soc, 49, 457 (1927).
31. 3 e м л я н с к и й Н. Н., ПановЕ.М., К о ч е ш к о в К- А., ДАН СССР, 146,
1335 (1962).
32. N е u m a n n W. Р., К б п i g K-, Angew. Chem., 76, 892 (1964).
446
Оловоорганические соединения
33. NeumannW.R, P e d a i n J., Ann., 672, 34 (1964).
34. F a r г a r W. V., S к i n n e г H. A., J. Organometal. Chem., 1, 434 (1964).
35. К р а ф т М. Я-, БородинаГ.М, ЖОХ, 32, 1665 (1962).
36. NeumannW.R, К б n i g K., Ann., 677, 1 (1964).
37. BoueS., GielenM., Nasielski J., Bull. Soc. chim. Belg., 73, 864 (1964).
38. T a g I i a v i n i G., F a I e s с h i n i S., P i I I о n i G., P I a z z о g n a G., J.
Organometal. Chem., 5, 136 (1966).
39. T s a i Т. Т., С u t 1 e r A., LehnW.L, J. Org. Chem., 30, 3049 (1965).
40. Несмеянов А.Н., Кочешков К. А., П у з ы р е в а В. П., ЖОХ, 7, 118
(1937).
41. Kozeschkow К. A., Nesmejanow A. N., Pusyrewa W. Р., Вег.,.
69, 1639 (1936).
42. G r u t t n e r G., Вег., 50, 1808 (1917).
43. В a h r G., G e I i u s R., Вег., 91, 825 (1958).
43 а. Si si do К., Kozi ma S., I si b asi Т., J. Organometal. Chem., 10, 439 (1967).
436. Si si do K., Kozi ma S., H an a d а Т., J. Organometal. Chem., 9, 99, (1967).
44. P а з у в а е в Г. А., Щепеткова О.А., В я з а н к и н Н. С, ЖОХ, 31, 1401
(1961).
45. В я з а н к и н Н. С, Р а з у в а е в Г. А., К о р н е в а С. П., ЖОХ, 33, 1041 (1963)
46. Р а з у в а е в Г. А., В я з а н к и н Н. С, Щ е п е т к о в а О. А., ЖОХ, 30, 2498
(1960).
47. Р а з у в а е в Г. А., Ж- Всесоюзн. хим. общ. им. Д. И. Менделеева, 7, 325 (1962).
48. Разуваев Г. А., Вязанкин Н. С, Сб. «Химия перекисных соединений».
М., Изд-во АН СССР, 1963, стр. 283.
49. В яз а н к и н Н. С, Р а з у в а е в Г. А., Б р е в н о в а Т. Н., ЖОХ, 35, 2033
(1965).
50. В я з а н к и н Н. С, Р а з у в а е в Г. А., К р у г л а я О. А., Изв. АН СССР,
ОХН, 1962, 2008.
51. Разуваев Г. А., Федотова Е. И., ЖОХ, 21, 1118 (1951).
52. LadenburgA., Вег., 4, 19 (1871).
53. Б ы ч к о в В. Т., В я з а н к и н Н. С, ЖОХ, 35, 687 (1965).
53а. Sisido К., Miyanisi Т., Nabika K.,Kozima S., J. Organometal. Chem.,
11, 281 (1967).
54. P а з у в а е в Г. А., Вязанкин H.C., Гладышев Е.Н., Б о р о д а в к о
И. А., ЖОХ, 32, 2154 (1962).
55. Вязанкин Н. С, Разуваев Г. А., Дергунов Ю. И., Щепеткова
О. А., Труды по химии и хим. технологии (Горький); 1961, 58.
56. Разуваев Г. А., Вязанкин Н. С, Дергунов Ю. И., П у н ч у к Н. М.,
Ж- Всесоюзн. общ. им. Д. И. Менделеева, 5, 707 (1960).
57. Р а з у в а е в Г. А., Д е р г у н о в Ю. И., В я з а н к и н Н. С, ДАН СССР, 145,
347 (1962).
58. Chambers R. D., Clark Н. С, W i 1 1 i s С. J., Chem. a. Ind., 1960, 76.
59. ChambersR.D., С 1 a r k H. C, Willis С J., Canad. J. Chem., 39, 131
(1961).
60. С 1 a r k H. C, W i 1 1 i s С J., J Am. Chem. Soc, 82, 1888 (1960).
61. KaeszH.D., P h i 1 i p p s J. P., StoneF.G.A., Chem. a. Ind., 1959, 1409.
62. К a e s z H. D., P h i 1 i p p s J. P., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6228
(1960).
63. L a g о w s k i J. J., Quart Rev., 13, 233 (1959).
63a. С u 1 1 e n W. R., S t у a n G. E., Canad. J. Chem., 44, 1225 (1966).
64. WibergE., BehringerE, Z. anorgan. allgern. Chem. 329, 290 (1964).
65. В я з а н к и н Н. С, Б ы ч к о в В. Т., ЖОХ, 36, 1684 (1966).
Глава XIV
ИЗМЕНЕНИЕ В РАДИКАЛЕ
Существуют два основных типа реакций оловоорганических соединений,,
приводящих к изменению в органическом радикале у атома олова. В
реакциях первого типа исходными служат оловоорганические соединения,
содержащие атомы галоида, нитрильную или сложноэфирную группу в
органическом радикале. Менее характерны реакции присоединения к винильным
или аллильным производным олова.
Примером реакций первого типа может служить взаимодействие хлор-
метильных соединений олова с йодистым натрием, этиленимином калия или
калиевой солью пиррола. Так получены, например, иодметилтрифенилолово
(т. кип. 180° С/0,05 мм) [1], тетра(иодметил)олово (т. пл. 76° С) [2], этиленими-
нометилтриэтилолово (т. кип. 43,5—44,5° С/0,2 мм) [3] и пиррилметилтри-
этилолово (т. кип. 86,2° С/0,2 мм) [3]. (Диметиламинометил)триэтилолово и
морфолинометилтриэтилолово образуются при нагревании (хлорметил)три-
этилслова и соответствующих аминов в метиловом спирте. Выходы
продуктов составляют 80—90% [3]. О кинетике реакции между (иодметил)триметил-
оловом и йодистым натрием, меченным J181, см. статью Хепнера и Вальки-
витца [4].
Триметил(трифторметил)олово реагирует с йодистым натрием иначе.
При нагревании смеси реагентов до 80° С образуется йодистое триметил олово
с выходом 90% [5].
Действие электрофильных или нуклеофильных реагентов на
оловоорганические соединения с заместителями у а-углеродного атома приводит к
отщеплению замещенных радикалов. Однако если функциональные группы
находятся у (3-углеродного атома или дальше, то они легко вступают в
характерные для них реакции, в то время как остальная часть молекулы
остается незатронутой.
Так, при действии литийалюминийгидрида на цианметилтрибутилолово
или метиловый эфир трибутилстаннилуксусной кислоты образуется гидрид
трибутилолова [6]. (2-Цианэтил)трипропилолово и (2-карбоксиметилбутил)-
трибутилолово гладко восстанавливаются до (З-аминопропил)трипропило-
олова и (З-оксипропил)трибутилолова соответственно [7, 8]:
LiAIH,
(C3I-l7)3SnCH2CH2CN > (G3H7)3SnCH2CH2NH2,
LiAlH4
(C4H9)3SnCH2CH2COOCH3 > (QH9)3SnCH2CH2CH2OH.
Аналогично получены [7] (4-оксибутил)трифенилолово] и (5-оксиамил)-
трибутилолово.
Примером реакций рассматриваемого типа может служить также
взаимодействие реактива Гриньяра с оловоорганическими соединениями,
содержащими в радикале нитрильную или сложноэфирную группу [7]:
CH3MgJ
(C3H7)3SnCH2CH2CN > (C3H7)3SnCH2CH2COCH3,
CH3MgJ
(C4H9)3SnCH2CH2COOCH3 > (C4H9)3SnCH2CH2C(CH3)2OH.
44S Оловоорганшеские соединения. Лит. стр. 455—456
Образующиеся соединения вступают в обычные реакции, характерные
для гидроксильной или карбонильной группы [7, 9]. Выделены и
охарактеризованы [7] оксим и семикарбазон (З-кетобутил)трипропилолова, 2,4-ди-
нитрофенилгидразоны (З-кетобутил)трипропил олова и (2-бензоилэтил)-
трипропилолова.
Эфиры триалкилстаннилуксусной кислоты и триалкилцианметильные
соединения олова превращаются щелочами в гидроокиси триалкилолова
(см. стр. 363). Если же нитрильная или сложноэфирная группы находятся
у [5-углеродного атома, то при их омылении образуются соли
соответствующих кислот. Например, (2-карбоксиметилэтил)трипропил олово гидроли-
зуется щелочью уже при обычной температуре с образованием аморфного
гигроскопического вещества, хорошо растворимого в воде, бензоле,
хлороформе, метиловом и этиловом спиртах [8]:
(С3г17)35пС 12СЧ2СООС 13 + NaOH-> (С3Ч-)з^пСЧ2СМ2СОЭХа + С 13ОЧ.
Аналогично получены [8] динатриевая соль бш>(2-карбоксиэтил)дипро-
пилолова и тринатриевая соль три-(2-карбоксиэтил)пропилолова. Их
растворимость в воде повышается с увеличением числа COONa-групп в
молекуле.
При действии минеральных кислот на водные растворы этих солей
выделяются свободные кислоты.
Получение (2-карбоксиэтил)три-я-бутилолова [8]. К 15,1 г (0,04 моля) (2-карбоксиме-
тилэтил)три-я-бутилолова прибавляют 1,6 г (0,04 моля) едкого натра в 400 ^л 75%-ного
этилового спирта. Смесь оставляют на 2 дня при комнатной температуре и упаривают досуха
в вакууме. Остаток растворяют в 200 мл воды и для удаления возможной примеси непрореа-
гировавшего исходного вещества раствор дважды экстрагируют эфиром (порциями по 50 мл).
Маслянистую жидкость, образующуюся при подкислении водного раствора 1 N соляной
кислотой, дважды экстрагируют 100 мл эфира; эфирный раствор высушивают и растворитель
отгоняют. В остатке получают 9,8 г (68%) бесцветного масла, которое по данным анализа
соответствует (2-карбоксиэтил)три-я-бутилолову.
Перегнать вещество в вакууме не удается, так как при нагревании до
120° С оно разлагается с выделением бутана и образованием внутренней соли
гидроокиси (2-карбоксиэтил)ди-я-бутил олова:
(С4Ч9)з5пСЧ2СЧ2СООт1 -» (С4Чо)2зп^СН2СЧ2СОО- + С4Н10.
Другой способ синтеза этого соединения приведен на стр. 283, 384.
О скорости гидролиза (2-этоксикарбонилэтил)- и (3-этоксикарбонилпро-
пил)триэтилолова см. статью Ван-дер-Керка с сотр. [10]. Устойчивость
(со-карбоксиалкил)трифенильных соединений олова существенно зависит
от положения карбоксильной группы по отношению к атому олова.
При кипячении (2-цианэтил)трифенилолова с водно-спиртовым раствором
щелочи в течение нескольких часов выделяется аммиак и образуется
натриевая соль (2-карбоксиэтил)трифенилолова [8]. Подкислением суспензии этой
соли в воде получают продукт, который по данным анализа соответствует
внутренней соли (со-карбоксиэтил)дифенилолова:
(C6H;.)3SnCFI2CirI2COONa + HC1 -» [(С6ЧД3'5пСН2СН2СООН1 ->
-> (C6H5)2Sn+CH2CH2COO- + СбЧб.
Вещество не плавится при нагревании до 300° С т не растворяется в обычных
органических растворителях. Растворение в разбавленном растворе едкого
натра приводит, по-видимому, к натриевой соли гидроокиси (2-карбокси-
этил)дифепил олова.
(З-Цианпропил)трифенилолово гидролизуется щелочью с образованием
растворимой в воде натриевой соли (З-карбоксипропил)трифенилолова, при
Изменение в радикале
449
подкислении которой выделяется свободная кислота — (3-карбоксипропил)-
трифенилолово. Ее можно проэтерифицировать диазометаном в (3-карбо-
метоксипропил)трифенил олово. (4-Карбоксибутил)трифенилолово
совершенно устойчиво. Наличие в этом соединении свободной карбоксильной
группы доказывается реакцией с диазометаном, приводящей к (4-карбомето-
ксибутил)трифенил олову.
Более сложно проходит омыление (2-ацетоксиэтил)трифенилолова [8].
Основным продуктом этой реакции является гидроокись трифенилолова
наряду с небольшим количеством (2-оксиэтил)трифенилолова. Последнее
образуется также при окислении перекисью водорода в щелочной среде бо-
разина состава [(C6H5)3SnCH2CH2BNC6H5]3 ШЬ Из продуктов окисления
1(СН3)35пСН2]3В выделена [12] лишь окись диметилолова.
Станносилоксаны типа [H(CH3)2Si(CH2)mLSnR4-,2 превращаются этила-
том натрия в [C2H50(CH3)2Si(CH2)]„SnR4-,i. При гидролизе
водно-спиртовым раствором щелочи они образуют циклические шестичленные силоксаны
[13]. Например:
NaOH
[Н (CH3)2SiCH2]2SnR2 -\- Н20 > (CH3)2SiCH2SnR2CH2Si (СН3)20.
С2Н5ОН | I
Аналогично получены соответствующие дисилоксаны:
2 H(CH3)2SiCH2Sn(C4H9)3 + Н20-* [(C4H9)3SnCH2Si (CH3)2]20.
Переходя к рассмотрению реакций присоединения к винильным и ал-
лильным соединениям олова, необходимо подчеркнуть, что галоиды и галои-
доводороды действуют как деалкилирующие агенты (см. стр. 340, 347).
Полисульфид водорода среднего состава H2S5,2 при нагревании его
с аллильными или |3-метилаллильными соединениями триалкил- и триарил-
олова также действует как деалкилирующий агент, например, [14]:
(/>-CH3C6H4)3SnCH2CH=CH2 —Л [(p-CH3C6H4)3SnS]3.
180°С
При обычной температуре реакция проходит иначе:
R3SnCH2CH=CH2 + H2S5,2 -> [R3SnCH2CH (СН3)]25Х.
Оловоорганические алкилтиолы получены [15] присоединением
сероводорода к винил- или аллилтрибутилолову при —70° С и облучении смеси
реагентов УФ-светом:
(C4H9)3SnCH2CH-=CH2 + H2S -» (C4H,)3SnCH2CH2CH2SH.
Те же соединения образуются при облучении УФ-светом смеси оловоорга-
нических соединений с тиоуксусной кислотой с последующим омылением
образующихся продуктов [15]:
о°с
(С4Н9)35пСН2СН=СН2 -f CHsCOSH —> (C4H9)3SnCH2CH2CH2SCOCH3,
он-
(C4H9)3SnCH2CH2CH2SCOCH3 > (C4H9)3SnCH2CH2CH2SH.
Сейферт [16] сообщив о присоединении полигалоидметанов к триэтил-
винилолову. Реакции проходят при 90—95° С в присутствии перекиси бен-
зоила:
(G2H5)3SnCH=CH2 + CC18Z -» (C2H5)3SnCHZGH2CCl3.
29 Заказ № 207
450
Оловоорганические соединения. Лит. стр. 455—456
Продукты присоединения образуются с умеренным выходом (Z = Н, С1
или Вг). Трихлорбромметан легко присоединяется к винилтрифенил олову
в присутствии перекиси бензоила [17]:
(C6H5)3SnCH=CH2 + CCl3Br -> (C6H5)3SnCHBrCH2CCl3.
При действии щелочи на 7,Т,Т-тРихлор-а-бромпропилтрифенилолово
происходит отщепление бромистого водорода [17]:
(CGH5)3SnCHBrCH2CCl3 -> (C6H5)3SnCH=CHCCl3.
Триэтилстаннилвинилацетилен присоединяет трифенилметильные
радикалы почти исключительно по двойной связи [18]:
(C2H5)3SnCEEC—СН=СН2 + 2 (СвН5)3С -> (C2H5)3SnC=C-CH--CH2-C (СбН5)3.
С (СбН5)3
Значительно труднее проходит эта реакция с триэтилизопропенилоловом.
Исследование образующегося продукта с помощью ИК-спектра показало*
что в данном случае трифенилметильные радикалы присоединяются в
положение 1,4 с образованием соединения состава
(C2H5)3SnC=C=C-CH2-C (C6H3)o.
1
С (СвН3)з
Варьирование методики проведения реакции, изменение температуры»
применение в качестве источника свободных радикалов вместо гексафенил-
этана, трифенилхлорметана и ртути не влияет на направление
присоединения. Однако в последнем случае продукт реакции содержит примесь енино-
вого производного ртути, образующегося в результате реакции деалкили-
рования [18].
При нагревании триэтилстаннилвинилацетилена с диазометацом в эфире
образуется соответствующий пиразолин [19]:
(C2H5)3SnC=C—СН=СН2 + CH2N2
■(C2H5)3SnC=C—СН—СН2 -i
I I
N CH2
\/
N
-* (C2H5)3SnC=C—С—CH2
II I
N CH2
\/
NH
Такой ход реакции подтвержден с помощью ИК-спектров. При нагревании
пиразолина до 100—120° С он превращается в триэтилстаннилциклопропил-
ацетилен:
(C2H5)3SnC=C— С—СН2-* (C2H5)3SnC=CCH—CH2 + N2.
II I X /
N СН2 СН2
\ /
NH
К триэтилстаннилизопропенилацетилену диазометан присоединяется
Изменение в радикале
451
лишь при нагревании смеси реагентов в эфире в течение 20 час.
Образующийся пиразолин не претерпевает изомеризации:
сн3
(C2H5)3Sn—С=С С=СНа
N СН3
\/
N
(C2H5)3SnC=C—C=CH2 + CH2N2 —
I
СНз
СНз
I
(C2H,)3Sn—С=С—С—СН2
N СН2
термическое I \./
разложение i ^
СНз
(C2H5)3Sn—С=С—С CI12
сн2
Строение полученных соединений подтверждено [19] ИК- и ЯМР-спек-
трами.
На примере триметил-, триэтил- и трифенилвинилолова показано [20],
что оловоорганические соединения способны присоединяться к циклонам
при нагревании смеси в запаянных ампулах в атмосфере инертного газа.
В качестве растворителя применяют бензол. Температурный режим и
продолжительность реакции зависят от структуры циклона и винилоловянного
соединения. О завершении реакции судят по исчезновению характерной
окраски раствора циклона в бензоле.
Наиболее легко в реакцию типа Дильса — Альдера с винильными
соединениями олова вступает фенциклон. При 120—140° С в течение 5—6 час.
полностью заканчивается реакция с триметил- и триэтилвинидоловом.
При применении в качестве исходного вещества винилтрифенилолова
требуется гораздо более длительное нагревание смеси. Во всех случаях
получены циклические оловоорганические соединения с эндокарбонильным
мостиком. Они представляют собой белые кристаллические вещества с т. пл.
~ 200° С:
сн9
СН —SnRo
Реакции диенового синтеза тетрациклона и ацециклона с винилоловянными
соединениями проходят более сложно и выделить соответствующие аддукты
в чистом виде не удается [20]. К тому же выводу пришли Ротман и Беккер
[21, 22] при изучении взаимодействия тетрациклона с трифенилвинилоло-
вом. Напротив, трифенил-1-циклопентадиенилолово легко присоединяется
к малеиновому ангидриду, диэтилмалеату и диэтилацетиленкарбоксилату
[23].
Сейферт с сотр. [24] изучил реакцию тетрациклона с оловоорганическими
производными ацетилена. Нагреванием 26,5 г тетрациклона с 2.5 г бис-
(триметилстаннил)ацетилена в 100 мл ксилола в течение 138 час. ими полу-
29*
452
Оловоорганические соединения. Лит. стр. 455—456
чен 1,2-бш>(триметилстаннил)тетрафенилбензол (т. пл. 232—233° С). Выход
практически количественный:
СбН5 Sn(CH3)3 СбП5
с6н5
сбн5
^\ с
>=о + ill
I
C6H--|^Y-Sn(CH3)3 , со
СбН5-Ц^-5п(СНз)з '
СвН3
Sn(CH3)3
I
сбн5
Аналогично из тетрациклона и триметил(фенилэтинил)олова или триметил-
пропинилолова получены [24] (триметилстаннил)пентафенилбензол (т. пл.
320—326° С) и 1-(триметилстаннил)-2-метилтетрафенилбензол (т. пл. 193—
196° С) соответственно.
1-Иод-2-(триметилстаннил)тетрафенилбензол (получен взаимодействием
1,2-бш>(триметилстаннил)тетрафенилбензола с одним эквивалентом иода)
образует с тетрациклоном йодистое триметилолово и 2-иод-2-(триметилстан-
нил)октафенилбифенил (выход 80%). К тому же соединению приводят
следующие реакции:
с6н5 сбн5 сбн5 сбн5
свн6
I
СвНз-^-Бп (СН3)з
CeHe-^-J
CgH5
* СбН5
~\.
СбН5 Sn(CH3)3 J
_)-QH3 + (CH3)3SnJ
QH6
t
сбн5
C6H5 с
CoH,
I
I
CeH5
CeHj
I
—Sn (СН3)з J—^\-С0Н-,
Sn(CH3)3 J-L J-C,H6
I
C.HS
Аналогично реагируют с 1,2-быс-(триметилстаннил)тетрафенилбензолом и
другие галоидные арилы [24]. Так, при действии на это вещество п-иодто-
луола или ле-иодтолуола образуются два различных (1-триметилстаннил)-
2-толилтетрафенилбензола:
QH5-
С«Н5-
СбН5
I
^V-ceHs
с,н.
. U_Sn(CH8)3
с
I
сн3
свн5-
сбн5-
-с6н5
-Sn(CH3)3
-сн3
Однако эта реакция сочетания проходит лишь с достаточно
разветвленными оловоорганическими соединениями [24].
Триэтилвинилолово при нагревании до 95° С в течение 17 дней
присоединяет трихлорсилан [161. Хотя аддукт в чистом виде выделить и не удается,
при обработке его метилатом натрия в эфире получают чистое р-триметокси-
силилтриметилолово [16].
В ряде случаев провести реакции присоединения не удается. Так,
попытки каталитического гидрирования винильных, аллильных и фепильных
Изменение в радикале 453
соединений олова или оловоорганических соединений с ацетиленовыми
радикалами оказались неудачными [16, 25—29] (ср. [30]).
Алкил- и ариллитиевые соединения отщепляют непредельные радикалы
от атома олова. Например, при действии на трибутилаллил- или трифенил-
аллилолово фениллития образуется [31, 32] аллиллитий:
R3SnCH2CH=CHa + C6H5Li -* R3SnCGH5 -f CH2=CHCH2Li.
Этот метод широко применяется для получения литийорганических
соединений. Так получены, например, виниллитий [32—34], аллиллитий [32,
35—37], металлиллитий [35,36], перфторвиниллитий [38—40], изопропил-
литий и другие литийорганические соединения [41—44]. В ряде случаев
эти реакции являются обратимыми [23]. Они подробно рассмотрены в
соответствующем томе настоящего издания.
Аналогично, при нагревании (C6H5)3SnCH2C=CH с бромистым этилмаг-
нием в эфире, получено [30] этилтрифенилолово.
К категории реакций, вносящих изменение в алифатический радикал
у атома олова, следует отнести и наблюдающуюся в присутствии перекисей
145] или под действием УФ-облучения [46] перегруппировку хлористого
транс-транс-транс-три-^-хлорвкпклолова (т. пл. 120—121° С) в жидкий
цис-цис-цис-изомер. Такое же явление наблюдается [46] при облучении
УФ-светом двухлористого ди-|3-хлорвинилолова. Известны многочисленные
случаи превращения под влиянием химических воздействий обычно более
богатых энергией ^^с-изомеров в более устойчивые транс-изомеры. Все они
отличаются от данного тем, что здесь направления перегруппировки под
влиянием облучения УФ-светом и химического воздействия одинаковы.
Обычно же эти направления противоположны [45]. Показано, что в случае
оловоорганических соединений это превращение имеет цепной характер.
Пропенилтриметилолово (71% цис- и 29% транс- изомер а) при нагревании
в присутствии перекиси бензоила или азодиизобутиронитрила изомери-
зуется в незначительной степени [47]. Более удачными оказались опыты
с УФ-облучением [47].
Известен ряд реакций, приводящих к изменению в ароматическом
радикале у атома олова. И здесь обычно исходят из соединений, уже содержащих
заместители в фенильном кольце.
Так, нагреванием 2 г тетра-я-бензальдегидэтиленацетальолова в смеси
90 мл тетрагидрофурана и 30 мл воды в присутствии каталитических
количеств я-толуолсульфокислоты получают тетра-я-бензальдегидолово [48].
Последнее легко реагирует при обычной температуре с трифенилфосфинме-
тиленом с образованием тетра-я-винилфенилолова. /2-Триметилстаннил-
и ft-триэтилстаннилбензойная кислота получены [49, 50] карбонизацией
соответствующих магнийорганических соединений, приготовленных из
n-бромфенилтриметил- или и-бромфенилтриэтилолова. О константах
диссоциации этих кислот см. статью Чатта и Вильямса [50].
Другой способ синтеза бензойной кислоты с оловоорганическими
заместителями в ядре заключается в окислении соответствующих спиртов:
(CGH5)3SnC6H4CH2OH-Ai -\- KMn04 -» (C6H5)3SnC6H4COOK-n -> (C6H3)3SnCeH4COOH-/z.
Получение я-трифенилстаннилбензойной кислоты [51]. К 16,0 г (0,035 меля) трифенил-
я-оксиметилфенилолова в 150 мл очищенного от спирта ацетона в течение 4 час. прибавляют
небольшими частями 7,37 г (0,0467 моля) перманганата калия. Смесь перемешивают до
исчезновения окраски марганцевокислого калия (час или больше), осадок отделяют и
промывают 20 мл ацетона. Затем осадок, состоящий из двуокиси марганца и калиевой соли /г-три-
фенилстаннилбензойной кислоты, высушивают над хлористым кальцием в вакуум-эксикаторе
и трижды экстрагируют 95%-ным этиловым спиртом (порциями по 75 мл). Спиртовым
экстракт охлаждают и осторожно подкисляют соляной кислотой по метилоранжу. Затем раствор
подщелачивают(метиловыйоранжевый),хлористый калий отделяют и раствор концентрируют
454
Оловоорганические соединения. Лит. стр. 455—456
до объема порядка 100 мл.Слегка нагретый раствор вновь подкисляют и медленно разбавляют
водой до образования значительного количества осадка. По охлаждении смеси его отделяют
и высушивают. Получают 9,1 г сырой я-трифенилстаннилбензойной кислоты с т. пл. 164—
166° С. Вещество очищают перекристаллизацией из смеси этилового спирта с водой. Выход
7,2 г (44%); т. пл. 166—168° С.
Гилманом с сотр. [51—53] изучены реакции сочетания оловоорганических
оединений с солями диазония. (д-Диметиламинофенил)трифенилолово дает
оответствующие азосоединения с я-хлор-, я-бром- и л-нитрофенилдиазоний-
лоридом [51, 53]:
(C6H5)3Sn-^^>-N (CH3)2 + X-<^^>-N(+) _>
-+ (G6H5)3Sn-<^^>-N(CH3)24+ H<+)
Выход повышается, если исходить из (ж-диметиламинофенил)трифенил-
олова и фторборатов диазония.
Получение [3-диметиламино-6-(4/-нитрофенилазо)фенил]трифенилолова [53]. К 8,0 г
(0,017 моля) (ж-диметиламинофенил)трифенилолова е 100 мл диоксана прибавляют 4,0 г
(0,017 моля) фторбората я-нитрофенилдиазония в 500 мл смеси диоксана с водой (1:1).
Реакционную смесь разбавляют водой, выпавший осадок отфильтровывают и высушивают. Затем
его экстрагируют 400 мл петролейного эфира (т. кип. 11—120° С) и фильтруют горячим.
После охлаждения фильтрата получают 5,7 г [3-диметиламино-6-(4'-нитрофенилазо)фенил]-
трифенилолова с т. пл. 205—206° С. В кристаллическом состоянии вещество окрашено
в зеленый цвет, а в аморфном — в красный. Выпаривание маточника дает еще 1,3 г вещества
с т. пл. 205—205,5° С. Общий выход 7,0 г (66%).
Оловоорганические соединения, содержащие одну или несколько групп
четвертичного аммониевого основания, получены [54, 55] обработкой
соединений типа RAZSn[C6H4N(CH3)2]4-A2 йодистым метилом или диметилсульфа-
том. В последнем случае образуются хорошо растворимые в воде соединения
олова.
Непосредственное введение заместителей в фенильное кольцо у атома
олова обычно осуществить не удается.
Результаты, полученные при нитровании арильных соединений олова,
противоречивы. Форлендер [56] сообщил о нитровании тетрафенилолова
дымящей азотной кислотой при —5—0° С. Как показывает последующая
обработка бромом, нитрогруппа находится преимущественно в
пара-положении. Нитрование окиси дифенилолова при —15° С происходит в основном
в мета-положении [57]. По данным других авторов под действием азотной
кислоты или окислов азота происходит только расщепление тетраарильных
соединений олова (см. стр. 370). Окисление метальной группы в толильных
соединениях олова перманганатом калия или K3Fe(CN)6 приводит к
глубокому изменению молекулы [58, 59].
Гладко идет хлорирование оловоорганических соединений, содержащих
иод в бензольном ядре с образованием иодидхлоридов, например
(Cl2 JC6H4)2SnCl2 [60, 61].
Кратко сообщается (без деталей эксперимента) [62] об окислении цикло-
пентадиенилтрифенилолова. Проведение реакции в условиях переноса
кислорода при —20° С дает эндоперекись (выход 50%). Последняя разлагается
уже при 0° С. Восстановлением вещества натрийборгидридом или водородом
на никеле Ренея получают устойчивый цис-лпол:
_ -Sn(C6H5)3
(С6Н5)35п-Л^ J> ^~^>l~Sn(C6H5)3 HO-J^ уУ~ОН
Изменение в радикале
455
Трифенил-2-фурилолово не вступает в реакции, характерные для фу-
рана (конденсация с малеиновым ангидридом, кротоновым альдегидом и др.).
Также понижена активность пиридинового кольца в трифенил-3-пиридил-
олове [63]. Метиллитий не присоединяется по азомети новой связи, если
реакцию проводить в течение 4 час. при —35° С. Вероятно, при комнатной
температуре проходят следующие реакции:
С J + CHsLi "* % J ' + (СвНОзЭпСНз, Ц ) + CM8Li - (^ jLCH8
NN NN
1,1-Диметил-1-станнациклогексан легко реагирует с фенил(бромдихлор-
метил)ртутью с образованием 3-дихлорметил-1,1-диметил-1-станнациклогек-
сана (выход 50%, п% 1,5330) [64]:
СНз СН3
CeH5HgCCl2Br+ Sn< \->CeH3HgBr + Sn< >
/ ^-^ / ^-f
CH3 CH3 '
GHC12
О соединениях типа R3Sn(CH2)^MgX см. стр. 224.
ЛИТЕРАТУРА
1. KramerK., WrightN., Ber., 96, 1877 (1963).
2. Н 6 р р п е г К., W а 1 k i e w i t z D., Z. Chem., 2, 23 (1962).
3. К о с т я н о в с к и й Р. Г., П р о к о ф ь е в А. К., Изв. АН СССР, ОХН, 1965, 175.
4. НбррпегК., W а 1 k i e w i t z D., Z. Chem., 3, 227 (1963).
5. Seyferth D., Y ick-Pui M. J., GordonM.E., В u r 1 i t h J. M., J. Am.
Chem., Soc, 87, 681 (1965).
6. LesbreM., Buisson R., Bull., Soc. Chim. France, 1957, 1204.
7. V a n d e г К е г к G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 176 (1959).
8. Van der Kerk G. J. M., Noltes J. G., J. appl. Chem., 9, 113 (1959).
9. Becker R., Wen d e А., Пат. ГДР 26569 (1963); РДХим., 1964, 22H70.
10. R i i j к e n s F., J a n s s e n M. J., van der Kerk G.J. M., J. Organometal.
Chem., 2, 347 (1964.)
11. Seyferth D., T а к a m i z a w a M., Inorg. Chem., 2, 731 (1963).
12. Seyferth D., J.Am. Chem. Soc, 81, 1844 (1959).
13. M e r r e r R. L., S с о t t M. J., J. Am. Chem. Soc, 81, 975 (1959).
14. S с h w a r t z W. Т., PostH.W., J. Organometal. Chem., 2, 425 (1964).
15. Stamm W., J. Org. Chem., 28, 3264 (1963); Пат. CJJA 3206488 (1965); РЖХим.,
1966,23H86.
16. Seyferth D., J. Org. Chem., 22, 1252 (1957).
17. Фрейдлина P. X.,Мартиросян Г. Т., Несмеянов А. Н., ДАН СССР,
137, 1129 (1961).
18. 3 а в г о р о д н и й В. С, П е т р о в А. А., ДАН СССР, 149, 846 (1963).
19. 3 а в г о р о д н и й В. С, П е т р о в А. А., ЖОХ, 34, 1931 (1964).
20. А р б у з о в Б. А., Ш а п ш и н с к а я Л. А., К у д р я в ц е в а М. И., Изв. АН
СССР, ОХН, 1961, 2160.
21. RothmanLA., BeckerE. I., J. Org. Chem., 24, 294 (1959).
22. Rothman L. А., В е с к е г Е. I., J. Org. Chem., 25, 2203 (1960).
23. GilmanH., GistLA., J. Org. Chem., 22, 250 (1957).
24. Seyferth D., S a r a f i d i s C, E v n i n А. В., J. Organometal. Chem., 2, 417
(1964).
25. 3 а в г о р о д н и й В. С, П е т р о в А. А., ДАН СССР, 143, 855 (1962).
26. S i s i d о К., Т а к е d a Y., J. Org. Chem., 26, 2301 (1961).
27. Фрейдлина Р. X., Жукова И. Ф., Миронов В. Ф, Изв. АН СССР,
ОХН, 1960, 2258.
28. S p i а 1 t e r L., В и е 1 1 G. R., H a r r i s С. W., J. Org. Chem., 30, 375 (1965).
29. J о h n s о п О. Н., Н о 1 и m J. R-, J. Org. Chem., 23, 738 (1958).
30. L e Q и a n M., С a d i о t P., Bull. Soc. chim. France, 1965, 45.
31- Seyferth D., W e i n e r M. A., J. Org. Chem., 24, 1395 (1959).
32. J ohnson С So Weiner M. A., Waugh J. S., Seyferth D., J. Am. Chem.
Soc, 83, 1306 (1961).
456
Оловоорганические соединения
33. S е у f e r t h D.f W e i n e r M. A., J. Am. Chem. Soc, 83, 3583 (1961).
34. Seyferth D., Пат. США 3085119 (1963); РЖХим., 1965, 6H68.
35. Seyferth D., W e i n e r M. A., Org. Chem., 26, 4797 (1961).
36. Seyferth D., WeinerM.A., Пат. США 3085120 (1963); РЖХим. 1965, 12H84.
37. Seyferth D., V a u g h a n L. G., J. Am. Chem. Soc, 86, 883 (1964).
38. Seyferth D., R a a b G., В г a n d 1 e K. A., J. Org. Chem., 26, 2934 /(1961).
39. S e у f e r t h D., WelchD.E., R a a b G., J. Am. Chem. Soc, 84, 4266 (1962).
40. Seyferth D., W a d а Т., R a a b G., Tetrahedron L, 22, 20 (1960).
41. G i 1 m a n H., Rosenberg S. D., J. Org. Chem., 24, 2063 (1959).
42. Seyferth D., Suzuki R., M u r p h у С. J., SabetC. R., J. Organometal.
Chem., 2, 431 (1964).
43. Seyferth D., CohenH.M, Inorg. Chem., 2, 625 (1963).
44. Seyferth D., V a u g h a n L. G., Suzuki R., J. Organometal. Chem., 1, 437
(1964).
45. Несмеянов А. Н., Борисов А. Е., ВильчевскаяВ. Д., Изв. АН
СССР, ОХН, 1949, 578.
46. Н е с м е я н о в А. Н., Б о р и с о в А. Е., Абрамов А. Н., Изв. АН СССР,
ОХН, 1949, 570.
47. Seyferth D., V a u g h а п L. G., J. Organometal. Chem., 1, 138 (1964).
48. D r e f a h 1 G., LorenzD, J. prakt. Chem., 24, 106 (1964).
49. Засосов В. А., Кочешков К. А., Сборник статей по общей химии, т. 1,
М., Изд-во АН СССР, 1953, стр. 290.
50. С h a t t J., W i 1 1 i a m s A. A., J. Chem. Soc, 1954, 4403.
51. GilmanH., Arntzen С. Е., J. Org. Chem., 15, 994 (1950).
52. G i 1 m a n H., G i s t L. A., J. Org. Chem., 22, 368 (1957).
53. Gilman H., Rosenberg S. D., J. Am. Chem. Soc, 74, 5580 (1952).
54. GilmanH., Wu Т. С, J. Am. Chem., 77, 3228 (1955).
55. EabornC, Waters J.A., J. Chem. Soc, 1962, 1131.
56. V о r 1 a n d e r D., Ber., 58, 1893 (1925).
57. ChaJlengerF., RothsteinE., J. Chem., Soc, 1934, 1258.
58. К о ч е ш к о в К. А., Н а дь М. М., ЖОХ, 5, 1158 (1935).
59. Kozesch kow K. A., NadjM.M., Ber., 67, 717 (1934).
60. К о ч е ш к о в К- А., Несмеянов А.Н, ЖОХ, 1, 219 (1931).
61. NesmejanowA.N, К о z e s с h k о w K. A., Ber., 64, 628 (1931).
62. S с h e n с k G. O., Koerner E., Gustorf К. Н., Angew. Chem., 73, 707 (1961).
63. GilmanH., G о r e a u T. N., J. Org. Chem., 17, 1470 (1952).
64. Seyferth D., W a s h b u r n e S. S., J. Organometal. Chem., 5, 389 (1966).
Глава XV
СИНТЕЗ И РЕАКЦИИ
ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
Первый метод синтеза оловоорганических гидридов предложен Краусом
и Гриром [1] еще в 1922 г., но длительное время эти соединения не
привлекали особого внимания. Интенсивное исследование оловоорганических
гидридов начинается примерно с 1956 г., когда Ван-дер-Керк с сотр. [2]
применили их для синтеза оловоорганических соединений, содержащих
различные функциональные группы в органическом радикале.
Впоследствии оловоорганические гидриды приобрели большое значение
для получения различных типов оловоорганических соединений. Кроме
того, эти гидриды могут применяться и в качестве восстановителей.
СИНТЕЗ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
Метод синтеза оловоорганических гидридов, предложенный Краусом
и Гриром [1], заключался во взаимодействии натрийоловоорганических
соединений с хлористым или бромистым аммонием в жидком аммиаке.
Впоследствии этот метод был использован и другими авторами [3—5]:
R3SnNa + NH4X -* R3SnH + NaX + NH3,
R2SnNa2 -1- 2 NH4X -> R2SnH2 + 2 NaX + 2 NH3.
Вместо соединений типа R3SnNa можно применять и R3SnLi, как это
показано на примере синтеза гидридов трибутил-[6, 7] и трифенилолова [5].
В 1947 г. Финхолт с сотрудниками предложили более удобный метод,
который состоит в восстановлении оловоорганических галогенидов литий-
ал юминийгидридом [8, 9]:
4 R3SnX + LiAlH4 -» 4 R3SnH + LiX + A1X3,
2 R2SnX2 + LiAlH4 -» 2 R2SnH2 + LiX + A1X3,
4 RSnX3 + 3 LiAlH4 -> 4 RSnH3 + 3 LiX + 3 A1X3.
Этот метод в настоящее время широко применяется для получения
оловоорганических гидридов [10—29]. Синтез проводят, прибавляя оловооргани-
ческое соединение к избытку литийалюминийгидрида в эфире, диоксане или
тетрагидрофуране. Смесь перемешивают в течение нескольких часов, не-
прореагировавший литийалюминийгидрид разлагают водой и вещество
очищают перегонкой в вакууме.
Выбор растворителя оказывает существенное влияние на выход продукта
реакции. Так, при получении дигидрида диметилолова в диоксане
максимальный выход составляет только 55% [16]. Кроме тогодигидрид
диметилолова не удается полностью очистить от диоксана. Выход повышается и
вещество получается в чистом виде при замене диоксана эфиром.
Восстановлением 66,1 г (0,30 моля) двухлористого диметилолова 21,1 г (0,056 моля)
458 Оловоорганшеские соединения. Лит.. стр. 49 2—494
литийалюминийгидрида в 150 мл ди-я-бутилового эфира получают дигидрид
диметилолова с выходом 81% [16].
Оловоорганические гидриды легко окисляются кислородом воздуха,
поэтому их синтез проводят в атмосфере сухого азота или аргона.
Получение гидрида триэтилолова [12]. В трехгорлую колбу, снабженную механической
мешалкой, обратным холодильником и капельной воронкой, помещают 6,0 г (0,15 моля)
литийалюминийгидрида в 250 мл сухого, очищенного от перекисей эфира. К образовавшейся
суспензии при энергичном перемешивании постепенно прибавляют 38,5 г (0,15 моля)
хлористого триэтилолова в 100 мл эфира. Для завершения реакции смесь кипятят в течение
2 1/2 час. По охлаждении прибавляют 500 мг хинола, осторожно разлагают 12 мл воды и
добавляют 300 мл 20%-ного раствора виннокислого натрия. Органический слой отделяют,
водный — экстрагируют 200 мл эфира; объединенные экстракты высушивают сульфатом
натрия. Эфир отгоняют и остаток перегоняют в вакууме. Собирают фракцию с т. кип. 79—
— 81° С/92 мм. Выход 66%.
Аналогично получены гидриды три-я-пропил-[28] (выход 75%), три-я-
бутил- [12] (74%), ди-я-пропил- (72%) и ди-я-бутилолова (66%). Выход
продуктов увеличивается до 80—90%, если пользоваться следующим
приемом. После разложения реакционной смеси водой органический слой
отделяют, фильтруют через прокаленный сульфат натрия, эфир отделяют на
колонке, продукт перегоняют в вакууме.
Получение гидрида триизобутилолова [30]. К суспензии 5,7 г (0,125 моля)
литийалюминийгидрида в 100 мл сухого эфира при энергичном перемешивании и температуре —30 -г-
~ —40° С прибавляют по каплям 81,4 г (0,25 моля) хлористого триизобутилолова в 250 мл
сухого эфира. Смесь кипятят 2 часа, избыток исходного гидрида разлагают дистиллированной
водой и образовавшуюся гидроокись алюминия растворяют прибавлением 20%-ного раствора
виннокислого натрия. Эфирный слой отделяют, водный — экстрагируют эфиром (2 раза по
100 мл), объединенные эфирные вытяжки высушивают сульфатом натрия. Эфир отгоняют на
колонке и продукт перегоняют в вакууме (без колонки). Получают 64 г (88%) гидрида
триизобутилолова с т. кип. 101—104°С/12лш; df 1,09; п2£ 1,4697. Полученное вещество не
содержит хлора или эфира.
Так же получены ди- и тригидрид изобутилолова.
п-Фенилен-бис-(диметил оловогидрид) получен [31] прибавлением раствора ClSn(CH3)2C6H4SnX
X (СН3)2С1 в тетрагидрофуране к суспензии LiAlH4 в эфире. Дальнейшую
обработку проводят, как это описано для гидрида триэтилолова. Вещество
представляет собой бесцветную жидкость с т. кип. 93—94° С/0,2 мм (112—
113° С/1 мм); df 1,599; п% 1,5732; выход 53%.
Для получения гидридов наряду с хлоридами можно применять и другие
оловоорганические соединения. Так, гидрид три-я-бутилолова образуется
[32] с выходом 88% при восстановлении гексабутилдистанноксана литий-
алюминийгидридом. Гидриды трибутил- и трифенилолова получены [6]
действием литийалюминийгидрида на соединения типа R3SnSnR3 в среде
тетрагидрофурана.
В ароматическом ряду реакцию проводят так же, как и в алифатическом.
Гидрид трифенилолова получен [12] по методике, аналогичной синтезу
гидрида триэтилолова Вещество представляет собой вязкую жидкость
с т. кип. 168—172° С/0,5 мм. При 0° С она постепенно закристаллизо-
вывается. Из метилового спирта получают белые иглы ст. пл. 26—28° С [12].
Получение гидрида три-л*-толилолова[33]. В трехгорлую колбу, снабженную мешалкой,
обратным холодильником и капельной воронкой, помещают 2,0 г (0,053 моля)
литийалюминийгидрида и 40 мл эфира. При перемешивании в течение 30 мин. прибавляют по
каплям 3,4 г (0,0079 моля) хлористого три-ж-толилолова в 60 мл эфира. Смесь перемешивают
при комнатной температуре в течение 3,5 час, осторожно разлагают водой и выливают в
водный раствор виннокислого натрия. Органический слой отделяют, промывают водой и
высушивают сульфатом натрия. Растворитель отгоняют в вакууме и остаток перегоняют
также в вакууме. Получают 2,0 г (65%) вещества с т. кип. 194—195° С/1,3 мм.
Синтез и реакции оловоорганических гидридов
459
Гидрид три-о-толил олова получен [33] восстановлением 1,4 г бромистого
три-о-толилолова в 25 мл эфира 2,0 г литийалюминийгидрида в 40 мл эфира.
Реакцию проводят так же, как и в предыдущем случае, но после удаления
эфира остаток возгоняют при 90° С в вакууме (0,2 мм) и затем
кристаллизуют из метилового спирта. Выход чистого гидрида 77%, т. пл. 98,6—99,4° С.
При действии на хлористое три-п-толилолово (21,4 г в 250 мл эфира)
•литийалюминийгидрида (2,0 г в 150 мл эфира) образуется гидрид три-
л-толилолова [34]. Для завершения реакции смесь кипятят в течение
2,5 час. и затем перемешивают 30 мин. без нагревания. Дальнейшую
обработку проводят как обычно. После удаления эфира в вакууме получают
88% вещества с т. пл. 81,8—84,4° С. Перекристаллизация из смеси
метилового спирта с водой повышает температуру плавления до 82,3—84,9° С.
Аналогично получены гидриды тримезитил-[34] и три-2-бифенилилоло-
ва [33]. Гидриды три-п-фторфенил-, три-п-хлорфенил- и три-ж-трифторметил-
фенилолова неустойчивы. При получении гидрида три-п-фторфенилолова
образуется 33% гидрида и 50% гекса-п-фторфенилдистаннана [34]. Получить
гидриды три-ж-трифторметилфенил- и три-п-хлорфенилолова не удается.
Восстановление соответствующих хлоридов приводит к гексаарилдистан-
нанам [35].
Получение дигидрида дифенилолова [35]. К суспензии 1,90 г (0,050 моля)
литийалюминийгидрида в 35 мл сухого эфира при 0° С прибавляют по капля?л 17,2 г (0,05 моля) двухло-
ристого дифенилолова в 50 мл эфира в течение 30 мин. Смесь перемешивают при охлаждении
30 мин. и избыток литийалюминийгидрида осторожно разлагают 100 мл воды. Органический
слой отделяют, трижды промывают ледяной водой (порциями по 100 мл) и высушивают
хлористым кальцием. Раствор концентрируют при пониженном давлении и следы эфира
удаляют, выдерживая остаток в вакууме, меньшем 1 мм, в течение нескольких часов.
Получают 12,2 г (89%) бесцветной или светло-желтой жидкости. Перекристаллизация из
смеси петролейного эфира (т. кип. 30—60° С) с хлористым метиленом (1:1) при охлаждении до
— 70° С дает гроздь белых игл с т. пл. —21 -*- —20° С; п2£ 1,6128.
В некоторых случаях получить этим методом оловоорганические
гидриды в достаточно чистом виде и с хорошим выходом не удается. Кроме
того, метод требует применения значительных количеств легко кипящих
растворителей.
При получении оловоорганических гидридов в качестве восстановителя
удобно применить гидрид ди ал кил алюминия [36—38]. Побочным продуктом
в этом случае является галогенид диалкилалюминия, который можно
использовать для синтеза исходных оловоорганических соединений (см. гла-
ву I):
R3SnCl + R^AIH -> R3SnH + R^AICI,
R2SnCl2 + 2 RgAlH -> R2SnH2 + 2 R^AICl,
RSnCl3 + 3 R^AIH -> RSnHg + 3 R^AICI.
В этих реакциях можно применять и другие галогениды или алкокси-
соединения олова. Синтез гидридов проводят в эфире, инертном
растворителе (например, пентане или декалине) или без растворителя.
Порядок прибавления реагентов практически не влияет на выход
продуктов реакции. Обычно к охлажденному до —20 -ч 30° С гидриду
диалкилалюминия прибавляют частями оловоорганическое соединение [37].
В ряде случаев оловоорганический гидрид является самой легколетучей
частью реакционной смеси и может быть отогнан из нее в вакууме. Иногда
полученные продукты содержат примесь исходных галогенидов, которые
способствуют разложению гидридов при хранении. Для очистки вещество
встряхивают с небольшим количеством насыщенного раствора карбоната
натрия или смесью диоксана с водой (1 : 1), фильтруют, высушивают
сульфатом натрия и перегоняют [37].
460 Оловоорганические соединения. Лит. стр. 492—494
Особенно чистые гидриды алкилолова образуются при восстановлении
оловоорганических фторидов. Вероятно, это объясняется более высокими
температурами кипения фторидов диалкилалюминия, являющихся
побочными продуктами. Можно исходить также из алкоксисоединений олова [37]
или станниламинов [39]. Например, гидрид триэтилолова получен с выходом
95,5% восстановлением этокситриэтилолова гидридом диэтилалюминия.
Получение гидрида триэтилолова из хлористого триэтилолова. [37]. В трехгорлую»
колбу, снабженную обратным холодильником, магнитной мешалкой (оболочка из тефлона)
и капельной воронкой, помещают 8,6 г (0,1 моля) гидрида диэтилалюминия. Охлаждают до
—20° С и при перемешивании медленно прибавляют 24,1 г (0,1 моля) хлористого
триэтилолова, а затем 10,5 мл диэтилового эфира или 16,6 мл ди-я-бутилового эфира. Для
завершения реакции смесь перемешивают 2 часа при 0° С. Продукт отгоняют в вакууме (0,2 мм)
при температуре от0° до —10° С в охлажденную ловушку (остаток представляет собой
комплекс хлористого диэтилалюминия с эфиром). Выход 18,4 г (89%); п2р 1,4709. При
повторной перегонке т. кип. 148—150° С/760 мм.
Получение гидрида триэтилолова из фтористого триэтилолова [37]. К 4,3 г (0,05
моля) гидрида диэтилалюминия при перемешивании и температуре —30° С постепенно
прибавляют 11,2 г (0,05 моля) тонко измельченного фтористого триэтилолова, которое
растворяется по мере прохождения реакции. Смесь перемешивают 2 часа при 20° С и гидрид
отгоняют при —10° С/0,2 мм в охлажденную ловушку. Выход 10,0 г (97%). Как показывают
контрольная перегонка и ИК-спектр,препарат химически чистый. Разложение его соляной
кислотой дает теоретическое количество водорода.
Взаимодействием хлористого триэтил-, триизобутил- или трифенил-
олова с дейтеридом диэтилалюминия получены [40] оловоорганические дей-
териды (т. кип. 35—37° С/11 мм, 104—106° С/11 мм, 152—156° С/0,002 мм
соответственно).
При получении оловоорганических ди- и тригидридов температуру
кипения алюминийорганического соединения повышают прибавлением к
реакционной смеси соответствующего комплексообразователя, например эфира
или амина, в количестве примерно эквивалентном количеству гидрида
диалкилалюминия. Естественно, можно с самого начала проводить реакцию
в высококипящем эфире (например, в ди-я-бутиловом эфире) или применять
в качестве восстановителя гидрид диизобутилалюминия, который дает высо-
кокипящий галогенид. Последний способ удобен при получении ди- или
тригидридов алкилолова.
Получение дигидрида диэтилолова [37]. К раствору 57,0 г (0,4 моля) гидрида
диизобутилалюминия в 100 мл ди-я-бутилового эфира прибавляют по каплям 49,6 г (0,2 моля)>
двухлористого диэтилолова в 150 мл того же растворителя. Продукт отгоняют
непосредственно из реакционной смеси при —20°С/0,2—0,5 мм. Выход 34,8 г (97%); п2£ 1,4679. ИК-
спектр вещества показывает, что оно не содержит эфира. При обработке дигидрида
диэтилолова соляной кислотой выделяется теоретическое количество водорода.
Дигидрид ди-я-бутилолова получен [37] постепенным прибавлением
51,6 г двухлористого ди-я-бутилолова в 150 мл эфира к охлажденному до
0° С раствору 29,3 г (0,34 моля) гидрида диэтилалюминия в 100 мл того же
растворителя. Для завершения реакции раствор нагревают в течение 2 час.,,
после чего осторожно разлагают при 0° С смесью воды с диоксаном (1:1).
Гидроокись алюминия отфильтровывают, фильтрат высушивают
прокаленным сульфатом натрия, растворитель отгоняют и остаток перегоняют в
вакууме. Дигидрид ди-я-бутилолова имеет т. кип. 85° С/19 мм. Выход 85%.
Получение тригидрида этилолова [37]. К охлажденному до 0°С раствору 85,-Гг (0,6
моля) гидрида диизобутилалюминия в 50 мл ди-я-бутилового эфира прибавляют по каплям
50,8 г (0,2 моля) треххлористого этилолова в 50 мл того же растворителя. Смесь слегка
разогревается и окрашивается в темно-зеленый цвет. Ее перемешивают 2 часа при —1И°С
и продукт перегоняют при —20е С/0,5 мм в охлажденную жидким воздухом ловушку.
Дистиллят затвердевает в белый порошок, который при комнатной температуре плавится в
легкоподвижную бесцветную жидкость. Выход 29,3 г (97%); т. кип. 22—23°С/745 мм; nf} 1,4991.
Синтез и реакции оловоорганических гидридов
461
Аналогично из14,1 г треххлористого я-бутилолова в 250 мл эфира и 12,9 г
гидрида диэтилалюминия в 50 мл эфира получен тригидрид я-бутилолова.
Выход 62%; т. кип. 98—100° С/7 мм.
Применение этого метода синтеза в ароматическом ряду ^ожно
иллюстрировать следующими примерами.
Получение дигидрида дифенилолова [37]. 10,3 г (0,03 моля) двухлористого дифенилолова
растворяют в 100 мл диэтилового эфира и при —5° С прибавляют по каплям к раствору
5,2 г (0,06 моля) гидрида диэтилалюминия в 100 мл эфира. Смесь перемешивают 2 часа при
—20°С и разлагают смесью воды с диоксаном (1:1). Осадок отфильтровывают, фильтрат
высушивают прокаленным сульфатом натрия, эфир отгоняют и остаток перегоняют в вакууме.
Выход 6,7 г (81%); т. кип. 89—93° С/0,3 мм; п2® 1,5950. При охлаждении ниже 0° С продукт
закристаллизовывается. ИК-спектр указывает на присутствие в веществе следов эфира.
Однако для многих синтезов эта примесь не имеет значения. Очистить вещество
кристаллизацией не удается.
Получение дигидрида ди-я-толилолова [41]. К 26,3(0,31 моля) гидрида диэтилалюминия
в 150 мл эфира при —40° С в течение часа прибавляют по каплям 36,2 г (0,097 моля)
двухлористого ди-л-толилолова в 100 мл сухого эфира. Смесь перемешивают в течение часа при
комнатной температуре, охлаждают до —20° С и при этой температуре осторожно разлагают
водой. Органический слой отделяют, водный слой экстрагируют 50 мл эфира и объединенные
эфирные вытяжки высушивают хлористым кальцием. Фильтруют эфир отгоняют в вакууме,
к остатку прибавляют 100 мл петролейного эфира (т. кип. 30—50° С) и размешивают с
небольшим количеством активированного угля. После выпаривания раствора получают 27,5 г
(94%) дигидрида ди-я-тол ил олова ст. пл. 24—25° С. Вещество хорошо растворяется в
обычных органических растворителях.
Аналогично получены [41] дигидриды ди-п-этоксифенил- (т. пл. 53—
54° С), ди-п-бифенилил- (т. пл. 140—141° С), ди-а-нафтил- (т. пл. 83°С)
и ди-[3-нафтилолова (т. пл. 100—102° С).
Тригидрид фенилолова получен [37] восстановлением 30,4 г (0,1 моля)
треххлористого фенилолова 53 г (0,03 моля) гидрида диизобутилалюминия.
Реакцию проводят в ди-я-бутиловом эфире при —30° С. Вещество
содержит примесь растворителя, отделить которую перегонкой не удается.
Известны и другие методы синтеза оловоорганических гидридов, но они
имеют, по-видимому, менее общее значение. Так, гидриды триалкилолова
получены [42] восстановлением оловоорганических галогенидов
амальгамированным алюминием. В случае дигидрида ди-я-пропилолова этот путь не
приводит к цели [42].
Гидриды триэтил-, три-я-бутил- и трифенилолова, а также дигидрид
диэтилолова синтезированы [43] взаимодействием диборана с алкоксисоеди-
нениями олова. Вместо алкоксисоединений в этой реакции можно применять
и станниламины [39].
Кратко сообщается [44] о получении гидридов три-я-пропил- и три-я-бутил-
олова разложением соответствующих формиатов в вакууме при 160—180° С:
RaSnOOCH -> RgSnH + С02.
Обработка натрийтригидрида олова в жидком аммиаке йодистыми
алкилами приводит к тригидридам алкилолова [45]:
2 ЗпНЦ + 2 Na -» 2 H3SnNa -f H2,
HaSnNa + RJ -» RSnH3 + NaJ.
Неустойчивые тригидриды метил-[24] и фенилолова [46] рекомендуется
получать восстановлением оловоорганических хлоридов натрийборгидридом.
Оловоорганические гидриды и галогениды олова легко вступают с друг
с другом в реакции обмена [47, 48]:
RoSnHo -}- RgSnXa ^ R2SnX3 + R^SnHs,
2 RaSnH + RgSnXa ^ 2 R3SnX + RgSnlb,
3 R3SnII + R'SnXa U 3 R3SnX + R'SnII3
(X = F, CI, Br или J).
462
Оловоорганические соединения. Лит. стр. 492—494
Эти реакции используют [47] для синтеза низкокипящих
оловоорганических гидридов из более высококипящих. Стехиометрические количества
реагентов смешивают при комнатной температуре и продукт отгоняют в
вакууме. При другом способе синтеза оловоорганических гидридов исходят
из органостанноксанов [47, 49]:
R3SnOSnR3 + R2'SnH2 -» 2 R3SnH + -^- (R2SnO)n.
Если R и R' — алифатические радикалы, то при 0—25° С реакция проходит
очень быстро (примерно за минуту). В большинстве случаев образующаяся
окись диалкилолова выпадает в мелкокристаллическом состоянии и легко
отделяется от гидрида фильтрованием. С ароматическими станноксанами
реакция проходит медленнее [47].
В литературе описано несколько своеобразных оловоорганических
гидридов состава R2Sn(H)X (X — галоид или остаток органической
кислоты) и H[SnR2LH.
Гидриды типа R2Sn(H)X получают взаимодействием оловоорганических
галогенидов [48, 50—53] или диацилатов диалкилолова [54, 55] с оловоор-
ганическими гидридами:
R3SnH + R2SnX2-> R3SnX + R2Sn(II)X,
R2SnII2 -f R2SnX2 ^ 2 R2Sn(H)X.
Реакция является обратимой и чаще всего отделить образующиеся галоид-
ил и (ацилокси)гидриды от исходных веществ не удается. При перегонке
(хлор)гидрида ди-я-бутилолова в вакууме количественно отгоняется
соответствующий дигидрид [501. При низких температурах равновесие практически
полностью сдвинуто в сторону R2Sn(H)X [48, 53, 54].
При смешении дигидрида ди-я-бутилолова с диацетатом ди-я-бутил-
олова реакция проходит за несколько минут и после нескольких
перекристаллизации из эфира при —70° С получают бесцветное вещество, которое по
данным анализа соответствует (гидрид)ацетату ди-я-бутилолова [54].
При достаточно низких температурах (гидрид)ацетат представляет
собой индивидуальное соединение без примеси исходных веществ. В пользу
этого говорит [54] постоянство состава вещества после нескольких
перекристаллизации. Мало вероятно, чтобы смесь продуктов сохраняла постоянный
состав в этих условиях. Кроме того, соединение имеет т. пл. 17—20° С, тогда
как диацетат дибутилолова плавится при 8,5—9,0° С, а дигидрид дибутил-
олова не кристаллизуется при длительном хранении при 0° С. В ИК-спектре
(гидрид)ацетата ди-я-бутилолова наблюдается интенсивная полоса при
1880 смГ1, отнесенная к связи Sn — Н. При хранении вещество разлагается
с выделением со,со-диацетата тетрабутилдистаннана (о синтезе этого
соединения см. раздел «Реакция оловоорганических гидридов с кислотами»).
(о,со-Дигидрид тетра-я-бутилдистаннана приготовлен [56]
восстановлением соответствующего дихлорида литийалюминийгидридом в эфире.
Вещество представляет собой бесцветную жидкость с п2о 1,5205. При обработке
этого соединения уксусной кислотой выделяется рассчитанное количество
водорода и образуется со,оз-диацетат тетра-я-бутилдистаннана. Подобным
образом получены [48] и другие гидриды этого типа.
Чаще гидриды типа Н [SnR2]„H получают разложением дигидридов
в присутствии тонко измельченного олова [36]. При п = 8 полоса Sn—Н
в области 1780 см'1 исчезает и образуется циклический продукт (R2Sn)8
[36]. Более подробно эти реакции рассмотрены на стр. 477.
Большинство оловоорганических гидридов представляет собой
бесцветные прозрачные жидкости. Их устойчивость уменьшается при переходе от
моно- к тригидридам [57] и от алкильных соединений к арильным [58].
Синтез и реакции оловоорганических гидридов
463
В атмосфере инертного газа моногидриды могут храниться как угодно долго,
дигидриды — многие месяцы, а оловоорганические тригидриды уже через
2—3 дня превращаются в полимерные продукты. Особенно легко
разлагаются тригидриды винил-[25] и фенил олова [37]. Устойчивость гидридов
R2Sn(H)X (X — галоид или остаток органической кислоты) меньше, чем
у оловоорганических дигидридов [48, 50, 53—55].
Каталитическое влияние на процесс разложения оловоорганических
гидридов оказывают применяющиеся в качестве смазок силиконы и
шероховатые поверхности металлов. Аналогично действуют примеси галоидных
солей алюминия, в присутствии которых оловоорганические гидриды
разлагаются следующим образом [37]:
4 R3SnH -> R4Sn + SnH4 -* 3 R4Sn + Sn + 2 H2,
2 R2SnH3 -» R4Sn + SnH4 -> R4Sn + Sn + 2 H2,
4 RSnH3 -> R4Sn + 3 SnH4 -> R4Sn + 3 Sn + 6 H2.
Оловоорганические ди- и тригидриды легко разлагаются также при
нагревании. При перегонке этих соединений температура бани должна быть не
выше 50° С, так как иначе может произойти внезапное разложение гидрида
с образованием окрашенного масла, а чаще даже с выделением
металлического олова и водорода [37]. Гидриды изобутилолова устойчивее
соответствующих я-бутильных соединений [30].
Систематически изучено [16] разложение дигидрида диметилолова при
нагревании и облучении УФ-светом. При 25° С в темноте в запаянной под
вакуумом ампуле дигидрид диметилолова совершенно устойчив. При
нагревании до 120° С в темноте в течение 6 час. наблюдается выделение
металлического олова. Через 45 час. вещество разлагается полностью с
образованием гидрида триметилолова (50,7%), гексаметилдистаннана (6,1%),
металлического олова (39%) и водорода (—-60%), содержащего незначительную
примесь метана. Таким образом, разложение вещества проходит по
следующей схеме:
3 (CH3)2SnH2 -> 2 (CH3)3SnH + Sn + 2 Н2.
В свою очередь, гидрид триметилолова, хотя и в меньшей степени, разла-
гается с образованием гексаметилдистаннана и водорода:
2 (CPIskSnH -> (CII3)LSnSn(CH3)3 + Н2.
Облучение УФ-светом значительно ускоряет этот процесс. При 130° С
реакция практически заканчивается через 6 час. Основными продуктами
являются тетраметилолово (20%), гидрид триметилолова (40%), водород (53%)
и металлическое олово. Если вещество нагревать при 110° С с
одновременным облучением УФ-светом в течение 40 час, то разложение проходит
глубже. В этих условиях гидрид триметилолова из продуктов разложения
не выделен вообще. Выход тетраметилолова (21,9%), водорода (55% с
примесью незначительного количества метана) и металлического олова (49,1%)
изменяется незначительно. По-видимому, под влиянием УФ-света наряду
с приведенной выше схемой, имеет место и иной тип разложения [16]:
2 (CH3)2SnH2 -> (CH3)4Sn + Sn + 2 Н2.
В рассмотренных условиях (40 час.) одновременно проходят все три
процесса. Механизм их сложен и для его выяснения необходимы подробные
кинетические исследования [16].
Фотораспад дигидрида дибутилолова в отсутствие кислорода воздуха
приводит к окрашенным оловоорганическим соединениям сложного состава.
В их основе лежат разветвленные цепи из атомов олова [59].
464
Оловоорганшсские соединения. Лит. стр. 492—494
Исследование механизма разложения гидрида трифенилолова в октане,
бензоле или этиловом спирте при 80—120° С показало [27], что реакция
разложения проходит по свободнорадикальному механизму с образованием
в качестве основных продуктов гексафенилдистаннана и тетрафенилолова.
Каталитическое влияние на процесс разложения оказывают кислород и
катализаторы радикалообразующего типа [27, 60].
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С НЕОРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
Оловоорганические гидриды легко окисляются кислородом воздуха.
Как показано Борисовым и Абрамовой [60], в случае гидрида
трифенилолова реакция проходит следующим образом:
4 (C6H5)3SnH + 02 - 2 (QH5)3SnSn (С6Н5)3 + 2 Н20.
Следы перекиси бензоила несколько ускоряют, а гидрохинона — замедляют
эту реакцию. Описаны реакции гидрида триэтилолова с серой, селеном
и теллуром [63а]..
При действии галоидов на оловоорганические гидриды проходит замена
атома водорода на галоид, например [16]:
(CH3)2Sn (H) (C2F4H) + С1а -+ (CH3)2(C2F4H) SnCl + IIC1.
При нагревании эквимолекулярных количеств бромсукцинимида и
гидрида трифенилолова в бензоле в течение 8 час. образуется [61] сукцинимид
и бромистое трифенилолово. Тот же результат получен при проведении
реакции в присутствии пиридина.
Соляная кислота превращает оловоорганические гидриды в
соответствующие хлориды [5, 35, 37, 53, 62]. С оловоорганическими дигидридами
эту реакцию можно провести ступенчато [53]:
(C4H9)2SnH2 -|- НС1 - (C4H9)2Sn (H) С1 + Н2,
(CiH9)2Sn(H)Cl + HC1 -* (C4H9)2SnCl2 + Н2.
По данным Фритца и Шера [63, 636], тригидрид этилолова при —78° С
образует с одним эквивалентом бромистого водорода бром(дигидрид) этил-
олова — C2H5SnH2Br. Это белое кристаллическое вещество, которое
разлагается уже при —65° С с выделением водорода и окрашенного в желтый
цвет полимерного продукта состава (C2H5SnBr)n.
При взаимодействии тригидрида этилолова с двумя эквивалентами
бромистого водорода при —78° С реакция заканчивается на стадии образования
(бром)дигидрида этилолова. Затем при —70 --. 60° С имеют место
побочные реакции, в результате которых получается полимерное вещество со
связями Sn — Sn. Обработка тригидрида этилолова тремя эквивалентами
бромистого водорода приводит к неоднородному продукту сложного состава.
Совершенно иная картина наблюдается в реакциях бромистого водорода
с гидридом трифенилолова при —78° С. С одним молем бромистого водорода
гидрид трифенилолова реагирует по уравнению:
(CeH5)8SnH + HBr-* (C6H5)2SnHBr + C6HG.
Выделить (бром)гидрид дифенилолова не удается, так как при нагревании
до комнатной температуры наблюдаются вторичные процессы по группам
SnHBr. В конечном счете получают нерастворимый в обычных органических
растворителях продукт со связями Sn — Sn (Sn : Br : C6H5 =1:1: 1,6).
Двумя или тремя эквивалентами бромистого водорода гидрид
трифенилолова превращается при —78° С в (дибром)гидрид фенилолова:
(C0H5)2SnHBr + НВг -> C6H5SnHBr2 + С6Н(
Синтез и реакции оловоорганических гидридов
465
Повышение температуры до комнатной приводит к соединениям со связями
Sn — Sn [63, 636].
Обработка гидрида трифенилолова серной кислотой дает сульфат бис-
(трифенилолова) [62]. При действии на гидрид трифенилолова фенилсуль-
фоновой или фенилсульфиновой кислот образуются соответствующие
производные трифенилолова [64].
Более сложно проходит взаимодействие гидрида трифенилолова с
азотной кислотой. По-видимому, здесь вначале образуется неустойчивый
нитрат трифенилолова, который затем перегруппировывается в окись дифенил-
олова и нитробензол [65] (ср. [66]).
(QH5)3SnH + HN03 -> [(C6H5)3SnON02] -> QH5N02+ (C6H5)2SnO.
О кинетике реакции гидрида трибутилолова с кислотными и щелочными
агентами см. статьи Кунвилы и Левинса [67] и Десси с сотр. [68].
Андерсоном [14] изучено взаимодействие гидрида триэтилолова с гало-
генидами и окисями тринадцати переходных элементов и семи элементов
главных групп Периодической системы. Во всех случаях проходят реакции
восстановления с образованием соединений меньшей степени окисления,
а в некоторых случаях свободных элементов. Средний выход продуктов
составляет 88%. Некоторые реакции, например с двухлористым палладием,
треххлористым мышьяком, бромной ртутью и четыреххлористым оловом,
легко проходят в бензоле при 25° С. В других случаях требуется
непродолжительное нагревание смеси до температуры кипения.
Гидрид трифенилолова реагирует с треххлористым фосфором, треххло-
ристой сурьмой и сулемой с выделением соответствующего свободного
элемента, галоидоводорода и гексафенилдистаннана [60]. Кондуктометрическое
титрование раствора натрия в жидком аммиаке дигидридом диметилолова
указывает на образование диметилстаннилендинатрия, (CH3)2SnNa2, диме-
тилстаннилнатрия, (CH3)2SnHNa, и шлш-тетраметилстаннилдинатрия
NaSn (CH3)2Sn'CH3 2Na. При соответствующих условиях образуется также
аминодиметилстаннилнатрий (CH3)2Sn(NH2)Na [19].
k РЕАКЦИИ^ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С ОРГАНИЧЕСКИМИ КИСЛОТАМИ И ГИДРОПЕРЕКИСЯМИ
При действии органических кислот на гидриды триалкилолова
выделяется водород и образуются ацилаты триалкилолова. С дигидридами диалкил-
олова реакцию можно провести ступенчато. При этом основными
продуктами являются или со,со-диацилаты тетраалкилдистаннана или диацилаты
диалкилолова. С оловоорганическими гидридами ароматического ряда
реакция обычно проходит более сложно и приводит к со,со-диацилатам тетрафенил-
дистаннана. Получить таким путем диацилаты дифенилолова не удается.
Нагревая до кипения растворы гидрида трифенилолова в гептане
с уксусной или пропионовой кислотами, получают [69] соответствующие
ацилаты трифенилолова, вне зависимости от молярного соотношения
реагентов:
(CeH5)8SnH + RCOOH _► (CeH3)3SnOOCR + Н2.
Однако при взаимодействии эквимолекулярных количеств гидрида
трифенилолова и сс-тиофенкарбоновой кислоты, наряду с соответствующим
ацилатом трифенилолова (выход 32%), образуется со,со-дитиофенкарбонат
тетрафенилдистаннана (21%). Последний является единственным
продуктом реакции двух молей гидрида с одним молем кислоты [69].
Аналогично реагирует гидрид трифенилолова с а-фуранкарбоновой
кислотой [69]. И здесь, наряду с ацилатом трифенилолова, образуется соот-
30 Заказ № 207
466
Оловоорганические соединения. Лит. стр. 492—494
ветствующий дистаннан. Количество последнего растет при увеличении
молярного отношения между гидридом трифенилолова и а-фуранкарбоновой
кислотой. Взаимодействие гидрида трифенилолова с бензойной кислотой
ведет только к со,со-дибензоату тетрафенилдистаннана.
Характерной чертой диацилатов тетрафенилдистаннана является
отсутствие адсорбции карбонильной группы в ИК-спектре твердого вещества
[И, 69].
При действии двух молей уксусной кислоты на дигидрид дибутилолова
образуется диацетат дибутилолова [20, 54, 55].
Применяя эквимолекулярные количества реагентов, получают [20, 54,
55] со,со-диацетат тетрабутилдистаннана. Продукт идентифицирован
анализом и превращением в (бром)ацетат дибутилолова [20]:
2 (QH9)2SnH2 + 2 СНзСООН -> CH3COOSn (C4H9)2Sn (C4H9)2OOCCH3,
CH3COOSn (C4H9)2Sn (QH9)2OOCCH3 + Br2 -► 2 (QH^Sn (OOCCH3) (Br).
Получение о),О)-диацетата тетра-я-бутилдистаннана [20]. К 4,70 г (0,0176 моля) ди-
гидрида ди-я-бутилолова прибавляют 1,20 г (0,02 моля) ледяной уксусной кислоты.
Спустя 5 мин. начинается выделение водорода, которое завершается через 10 час. Обработка
продукта бромом указывает на присутствие 68% дистаннана (в расчете на дигидрид ди-я-бу-
тилолова). Продукт смешивают с 12 мл сухого эфира и оставляют на ночь при —70° С.
Получают 3,35 г (65%) бесцветных кристаллов с т. пл. —7 -. 4° С; п2р 1,5060.
Аналогично проходит реакция дигидрида ди-я-бутилолова и с другими
органическими кислотами [10, 54, 55, 70].
Исследовано [20] действие кислот различной силы (от бензойной до три-
фторуксусной) на дигидрид дифенилолова. Хотя в большинстве случаев
кислоты брались в избытке, единственным выделенным продуктом является
соответствующий дистаннан. Его выход увеличивается, если применять
избыток дигидрида дифенилолова.
Реакцию дигидрида дифенилолова с органическими кислотами можно
иллюстрировать следующими примерами (гидрид получался
восстановлением двухлористого дифенилолова литийалюминийгидридом и после
гидролиза смеси к эфирному слою прибавляли кислоту).
Получение о),со-дикапроната тетрафенилдистаннана [20]. К 13,74 г (0,05 ммоля)
дигидрида дифенилолова в 120 мл эфира прибавляют 0,1 моля капроновой кислоты. Спустя
несколько минутначинается выделение водорода и через 8 час. реакция заканчивается.
Концентрированием раствора получают 14,8 г (77%) кристаллов с т. пл. 58—72° С. Вещество
очищают перекристаллизацией из петролейного эфира (т. кип. 40—60° С). Выход чистого
со, со-дикапроната тетрафенилдистаннана 9,55 г (49%); т. пл. 85—87° С.
Так же проводят реакцию дигидрида дифенилолова с трихлоруксусной,
трифторуксусной, каприловой и ооксибензойной кислотами [20]. В
отдельных случаях применяют более концентрированные растворы дигидрида
дифенилолова.
Получение О),0)-ди(дихлорацетата) дифенилолова [20]. К 0,05 моля дигидрида
дифенилолова в 25 мл эфира прибавляют 0,04 моля дихлоруксусной кислоты. Реакция
заканчивается за 3 часа при комнатной температуре. Выделившийся осадок отфильтровывают и
кристаллизуют из хлороформа. Получают 2,81г (75%) вещества с т.пл. 169° С (с разложением).
Аналогично ведет себя дигидрид дифенилолова по отношению к уксусной
и муравьиной кислотам.
Изучение взаимодействия оловоорганических дигидридов с
органическими кислотами с помощью ИК-спектров и измерением количества водорода,
выделившегося по мере прохождения реакции, показывает, что она может
Синтез и реакции оловоорганических гидридов
467
быть представлена следующей схемой [54, 55]:
2 R2SnH2 + 2 R'COOH
2 Н2 + [2 R2SnHOOCR' ^ R2SnH2 + R2Sn(OOCR')2]
2R/COOH
2 R2Sn(OOCR/)2 + 2 H2 R'COOSnR2SnR2OOCR' + H2
Образование в качестве промежуточного продукта диацетата дибутил-
олова подтверждается следующей реакцией [20, 54, 70]:
(C4H9)2SnH2 + (C4H9)2Sn(OOCCH3)2 -> CH3COOSn (C4H9)2Sn (QI I9)2OOCCH3 + Ha.
Смешение гидроперекиси трет-бутилг с эквимолекулярным количеством
гидрида триэтилолова ведет к образованию mpem-бутилоЕОГо спирта и гек-
саэтилдистанноксана [59]. Несколько иначе проходит реакция гидрида
трибутилолова с избытком той же гидроперекиси [21]. Если исходить из
двух молей гидроперекиси и одного моля гидрида, то получают mpem-бу-
тилперокситри-я-бутилолово:
(C4H9)sSnH + ROGH -> (QH9)£SnOR + Н20,
(C4H9)3SnOR + ROOH -> (QH9)cSnCOR + ROM
(R= f-CjUg).
Тригидрид я-бутилолова очень энергично реагирует с избытком
гидроперекиси mpem-бутила, но образующийся продукт вьтделцть в чистом виде
не удается из-за его неустойчивости к гидролизу:
C4H9SnH3 + 3 г-С4Н9ООН -» C4H9Sn (OQHe-0s + 3 Н20.
О реакции оловоорганических гидридов с перекисями ацилов см. стр. 490.
[ПРИСОЕДИНЕНИЕ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
ПО КРАТНЫМ СВЯЗЯМ
Оловоорганические гидриды вступают в реакции присоединения к оле-
финам, алкинам, альдегидам и кетонам, изоткоцианатам и др.:
R3SnH + СН2=СН—R' -> R3SnCH2CH2R',
R3SnH + CH=C—R' -* R3SnCH2=CHR',
R3SnH + R^O -* RsSnOCHR^,
RsSnH + CeH5N=NC6H5-» R3SnN (C0H5) NIIQH-,,
R3SnH + R'N=C=0 -> RoSnCONHR',
R3SnH + R'N=C=S -> R3SnSCH=NR',
где R — органический радикал, в состав которого могут входить различные
функциональные группы. Эти реакции рассмотрены в гл. V.
зо*
4G8
Оловоорганические соединения. Лит. стр. 492—494
ВОССТАНОВЛЕНИЕ ОЛОВООРГАНИЧЕСКИМИ ГИДРИДАМИ
ГАЛОИДНЫХ АЛКИЛОВ И АРИЛОВ
Как уже отмечено на стр. 279, гидрид трифенилолова восстанавливает
бромистый аллил в пропилен. Аналогично хлористый изобутилен и гидрид
трифенилолова дают изобутен и хлористое трифенилолово [71].
Взаимодействием бромистого я-бутила и бромбензола с гидридом трифенилолова
получены [71] бромистое трифенилолово и бутан или бензол соответственно.
Восстановление оловоорганическими гидридами алкил- и арилгалогенидов
исследовано рядом авторов [29, 33, 59, 60, 72—82]. По сравнению с другими
восстановителями гидриды олова имеют некоторые преимущества, что
позволяет использовать эти соединения в препаративной органической химии.
Реакции восстановления представляют значительный интерес и для
изучения химических свойств оловоорганических гидридов.
Реакционная способность алкил- и арилгалогенидов уменьшается в
следующем ряду [29, 34, 72—74]
RJ>RBr> RC1>RF.
Например, хлористые алкилы восстанавливаются оловоорганическими
гидридами гораздо труднее бромистых [72]. Йодистый я-гептил
восстанавливается легче бромистого я-октила. Бромистый бензил активнее хлористого
бензила. При смешении бензотрихлорида с гидридом три-я-бутилолова
происходит экзотермическая реакция, тогда как бензотрифторид не
реагирует с этим гидридом при нагревании смеси до 80° С в течение нескольких
часов [72]. Скорость реакции оловоорганических гидридов с галоидирован-
ными бензолами уменьшается при переходе от иод- к хлорбензолу [73, 74].
Галоидные арилы инертнее галоидных алкилов в реакциях с
оловоорганическими гидридами. На восстановление арилгалогенидов существенное
влияние оказывают заместители в бензольном кольце. Так, хлор- и фтор-
бензол не восстанавливаются гидридами трифенил- и три-я-толилолова
в обычных условиях [34]. При нагревании до 100° С хлорбензола и гидрида
трифенилолова в течение 10,5 час. никакой реакции не наблюдается, а при
более высокой температуре (135°С) образуется тетрафенилолово [60]. В то же
время обработка о- или я-дихлорбензола гидридом трифенилолова дает
хлорбензол и хлористое трифенилолово. Взаимодействием гексафторбензола
с гидридом трифенилолова получают фтористое трифенилолово и пентафтор-
бензол с выходом 19%.
Изучены реакции замещенных бромбензолов и галоиднафталинов с
гидридами трифенил- и три-я-толилолова. При 154° С восстановление
проходит практически количественно. Выходы продуктов составляют 85—100%.
Понижение температуры до 124° С повышает селективность действия
гидрида.
Наличие в орто- или пара-положении по отношению к атому брома
электроно до норных групп уменьшает скорость реакции соответствующих
бромбензолов с гидридами трифенил- или три-я-толилолова. Напротив,
присутствие электроноакцепторных групп способствует восстановлению.
Интересно отметить, что если в качестве восстановителя применять гидрид
три-я-фторфенилолова, то выходы продуктов не зависят от природы
заместителя в молекуле бромбензола.
При восстановлении галоиднафталинов существенную роль играют,
по-видимому, стерические факторы. Их влиянием объяснена [341 большая
легкость восстановления 1-иоднафтал-ина по сравнению с 2-иоднафталином,
а также повышенная реакционная способность 4-бром-1-метилнафталина и
2-бром-2-метил нафталина.
Характерной чертой реакций оловоорганических гидридов с алкил- и
арилгалогенидами является возможность ступенчатого восстановления
геминальных полигалогенидов [72, 75]. Гидрид трифенилолова
экзотермически реагирует с четыреххлористым углеродом и с количественным выходом
образуются хлороформ и хлористое трифенилолово [60, 76]. Менее энергично
проходит реакция с хлороформом [60, 76], который восстанавливается тем же
Синтез и реакции оловоорганических гидридов
469
гидридом за 24 часа лишь на 20% [76], а хлористый метилен — на 2% [76].
Однако последние две реакции проходят до конца при 18-часовом нагревании
смеси оловоорганического гидрида с соответствующим галоидметаном.
По-видимому, применение вместо гидрида дейтерида трифенилолова
позволило бы ступенчато вводить атомы дейтерия в молекулу полигалогенида [76].
Другим примером реакций этого типа может служить взаимодействие
гидрида трифенилолова с бензотрихлоридом, хлористым бензилиденом и
хлористым бензилом [60]. С бензотрихлоридом гидрид энергично реагирует
уже при обычной температуре, а с хлористым бензилом — лишь при
нагревании до 80° С. Таким образом, по скорости течения реакции гидрида
трифенилолова с органическими полигалоидными соединениями последние
можно расположить в ряд [60]:
ССЦ > RCC13 > RCHC12 > RCH2C1 > CHCI3.
Для восстановления полигалоидметанов можно применять и гидриды
триалкилолова [75, 77, 83]. Например, взаимодействием гидрида три-я-
бутилолова с четыреххлористым углеродом получен хлороформ с выходом
85% [75]. Тем же путем осуществлено превращение хлороформа в
хлористый метилен. Бромоформ восстановлен в бромистый метилен (выход 62%)
и трибромфторметан — в дибромфторметан (69%). В последних двух
случаях выход бромистого трибутилолова составляет около 95%. Это различие
обусловлено частичным восстановлением в бромистый метил и бромфторме-
тан соответственно [75].
Гидрид три-я-бутилолова экзотермически реагирует с гглг-дибромцикло-
пропаном и с хорошим выходом (70—85%) образуются замещенные цик-
лопропилбромиды [75]:
У Вг У н
| \,с( Ц. (CiH,)«SnH -* | V/ + (QHi,)sSnBr.
Так же легко провести полное восстановление замещенных дибромцикло-
пропанов. Так, 1,1-дибром-2,2,3,3-тетраметилциклопропан превращен
двумя эквивалентами гидрида три-я-бутилолова в 1,1,2,2-тетраметилцикло-
пропан с выходом 69%. Реакция проходит ступенчато [75].
Восстановление хлорциклопропанов проходит в более жестких условиях.
Замена одного атома галоида на водород в 7,7-дихлорбицикло-[4,1, 01-геп-
тане требует нагревания до 140° С [75].
В тех случаях, когда имеется возможность образования стереоизомеров,
получается их смесь. По данным спектра ЯМР восстановление 7,7-дибром-
бицикло-[4,1,0]-гептана гидридом три-я-бутилолова приводит к смеси
цис- (I) и т/?аяс-7-бромбицикло-[4,1,0]-гептана (II) в отношении 2,5 : 1.
Интересно отметить, что с увеличением кольца стереоспецифичность
реакций повышается. Так, монобромид, полученный из 9,9-дибромбицикло-
[6,1,0]-нонана представляет собой цис-изомер (III). Восстановление 1,1-ди-
бром-2,2,3-триметилциклопропана гидридом три-я-бутилолова дает смесь
изомеров (IV) и (V). Их количества относятся между собой как 4 : 1 [75]:
вг Г >.^н ' - 'Вг
н КУ ^вг \ / \н
(II) ("И
470 Оловоорганические соединения. Лит. стр. 492—494
(IV)
Заканчивая рассмотрение вопроса о значении характера галоидных
алкилов и арилов на реакции восстановления, необходимо отметить, что
оловоорганические гидриды дегалоидируют вицинальные галогениды [72].
Так, двубромистый мезо-сшлъбен образует с гидридом три-я-бутилолова
транс-стшьбен. Аналогично проходит реакция с 1,2-дибромпропаном [72]:
СН3СНВгСН2Вг + 2 (QH9)3SnHj-> СНа=СНСН8 + 2 (QH9)3SnBr + Н3.
Влияние природы оловоорганического гидрида на скорость
восстановления изучено недостаточно. По предварительным данным [72] исследованные
гидриды по своей реакционной способности располагаются следующим
образом:
(C6H5)2SnH2 » n-C4H9SnH3 > (C6H5)3SnH да (n-QH9)2SnH2 > (n-QH9)3SnH.
Найдено [77], что при переходе от гидрида триметилолова к гидриду
три-я-бутилолова скорость реакции с четыреххлористым углеродом и
хлороформом заметно уменьшается. Однако в вопросе об относительной
реакционной способности оловоорганических гидридов остается много неясного [72].
Восстановление тригидридом я-бутилолова проводилось в условиях,
близких к температуре его разложения. Неизвестна также роль смешанных
(галоид)гидридов олова [72].
Существенную роль при восстановлении оловоорганическими гидридами
арилгалогенидов играют катализаторы. Радикальный'катализ значительно
расширяет область применения реакции и позволяет провести ее в более
мягких условиях. Например, в присутствии 1,5 мол. % азодиизобутирони-
трила хлористый циклогексил восстанавливается гидридом три-я-бутилолова
более чем в 70 раз быстрее, чем без катализатора [72]. Хлорбензол,
который инертен по отношению к гидриду трифенилолова при 150° С,
может быть восстановлен с хорошим выходом, хотя для этого требуются
значительные количества азодиизобутиронитрила [72]. Прибавление того же
катализатора или перекиси бензоила к смеси гидрида трифенил- или три-
(я-фторфенил)олова и 4-бромбифенила существенно увеличивает выход
бифенила [34].
На основании этих данных сделано заключение [58, 84], что
восстановление галоидных алкилов и арилов оловоорганическими гидридами проходит
по радикальному механизму и схематически его можно выразить
следующим образом:
\ \
—SnH+ Q ->— Sn' + QH,
/ /
2Sn'->Sn—Sn,
\ \
—Sn* + RX _► -SnX + R\
/ /
2R'-*R-R или R(+H) + R(-H),
/
R + HSn— -+ Sn' + RH,
Синтез и реакции оловоорганических гидридов
471
где Q — свободный радикал или молекула, которые способны отщеплять
водород от оловоорганического гидрида.
В пользу этой схемы говорят структурные изменения, которые
наблюдаются при восстановлении некоторых типов галоидных алкилов [58, 78,
84]. Например:
СН3-СН=СН—CHoCl + (CeH5)8SnH -* ГСН8СН=СНСН3 Л + (C6H5)3SnCl
CH2=:CH-CH(CI)CH3 + (C6H5)3SnH -» 1сНа=СНСНаСН3/ + (CeH5)8SnCl
То, что обе эти реакции приводят к смеси бутенов, можно объяснить
промежуточным образованием аллильного радикала:
СЪзсн=снсн; ^=^ сн3снсн=сн,
Аналогичным образом при восстановлении бромистого пропаргила,
вероятно, образуется пропаргиловый радикал:
HC=CCH2Br + (C4H9)3SnH -» [НСееССН^ z± Н*С=С=--СН2] -> Н2С=С=СН2 +
+ НС=ССН3 -f (C4H9)3SnBr.
Примером подобных перегруппировок может служить также следующая
реакция [58, 78, 84]:
Р г
+ (C,I-Iy)3SnH *-/ / / +- / JL /7 + (C4H9)3SnBr
Перегруппировкой сопровождается и восстановление эпибромгидрина.
Основным продуктом реакции является аллиловый спирт, что можно
объяснить раскрытием окисного кольца 2-эпоксипропильного радикала [58]:
\
— SnH ч
СН2—СН—СН1 -* 'ОСН3СН=СНо - , — Sn* + СН3=СНСНаОН.
Таким образом, по меньшей мере, в ряде случаев восстановление галоидных
алкилов и арилов проходит по радикальному механизму [58, 75, 78, 79].
В то же время ряд фактов не согласуется с гомолитической схемой
реакции [34, 62]. Так, прибавление гидрохинона не оказывает существенного
влияния на восстановление 4-бромбифенила гидридами триарилолова,
а при длительном нагревании смеси даже увеличивает выход бромистого
триарилолова.
Небольшие добавки трифенилбора способствуют восстановлению,
причем этот катализатор в диметиловом эфире диэтиленгликоля эффективнее,
чем в ксилоле. Более того, на примере двух гидридов и шести растворителей
показано [34], что выход увеличивается по мере увеличения диэлектрической
постоянной растворителя. Следовательно, последний играет важную роль
в реакциях восстановления.
Лоренц с сотр. [34] считает, что некатализируемые реакции
восстановления являются скорее гетеролитическими, чем гомолитическими. К тому же
выводу пришли и Борисов и сотр. [62] при исследовании взаимодействия
гидрида трифенилолова с бромистым тропилием.
472
Оловоорганические соедиенения. Лит. стр. 492—494
Более простой способ восстановления алкил- и арилгалогенидов
предложил Куивила с сотр. [72]. Этот способ основан на том, что в ряде случаев
оловоорганические гидриды являются более эффективными
восстановителями, чем литийалюминийгидрид. Поэтому оловоорганический гидрид можно
применять как «переносчик водорода», например, следующим образом:
LiAlH4 + RaSnX -* LiAlX4 + R3SnH
T
I
R3SnH + R'X -* R'H + R3SnX
Примером реакций этого типа является восстановление бромциклогек-
сана. К нагретому до кипения раствору бромциклогексана в эфире
прибавляют избыток литийалюминийгидрида и оловоорганический галогенид. Этот
способ удобен для восстановления галогенидов, не содержащих
функциональных групп, которые легко восстанавливаются литийалюминийгидридом.
Другой способ, который ближе не исследовался, состоит в
восстановлении оловоорганического галогенида точно одним эквивалентом
литийалюминийгидрида с последующим прибавлением к смеси алкил- или арилгало-
генида. Обе методики позволяют проводить восстановление, минуя стадию
выделения оловоорганических гидридов [72].
При проведении реакций восстановления необходимо учитывать, что
оловоорганические гидриды могут реагировать с различными
функциональными заместителями, а также присоединяться к алкенам и алкинам. Так,
при восстановлении Р-бромстирола гидридом трибутилолова в качестве
побочного продукта образуется 2-фенилэтилтрибутилолово [72]. Однако
восстановление 1,1-дибром-2-винилциклопропана показывает, что в
присутствии неактивированной олефиновой связи гидрид преимущественно
реагирует по связи С—Вг [75].
Оловоорганические гидриды легко реагируют по карбонильной группе,
но при восстановлении ос-галоидкетонов она остается незатронутой [58].
ВОССТАНОВЛЕНИЕ ОЛОВООРГАНИЧЕСКИМИ ГИДРИДАМИ
АЛЬДЕГИДОВ И КЕТОНОВ
Реакции оловоорганических гидридов с альдегидами и кетонами могут
проходить по двум направлениям. В присутствии катализаторов
оловоорганические гидриды присоединяются по карбонильной группе с образованием
алкоксисоединений олова. Эти реакции рассмотрены на стр. 301.
Нагревание стехиометрических количеств реагентов в течение
нескольких часов позволяет провести восстановление карбонильной группы в
спиртовую. Оловоорганические моно-, ди- и тригидриды при этом превращаются
в соединения типа R3SnSnR3, (R2Sn)„ и (RSn)„ соответственно [56, 69, 71,
74, 85—88]. Например:
2 R3SnH + \с=0 -+ R3SnSnR3 + \CH0H,
R2SnH2+\c=0-> (R2Sn)„ + \CH0H.
По способности восстанавливать альдегиды и кетоны оловоорганические
гидриды располагаются в ряд [58, 85]:
(C6H5)2SnH2 > (C4H9)2SnH2 > n-QHeSnHa > (C6H5)3SnH > (/z-C4H9)3SnH.
Так, гидрид три-я-бутилолова реагирует с бензальдегидом лишь при
нагревании смеси до 140° С, тогда как с гидридом трифенилолова та же реакция
проходит при 100° С [58, 85].
Синтез и реакции оловоорганических гидридов
473
Изучено [86] восстановление циклогексанона, о, м- и я-метилциклогекса-
нона избытком гидрида трифенилолова. При нагревании смеси реагентов
в течение 5 час. получают соответствующий спирт и гексафенилдистаннан.
Количество продукта восстановления существенно зависит от температуры.
Так, если реакцию проводить при 100°С в течение 5 час, то образуется 27%
спирта, а при 150° С — 56%. Дальнейшее повышение температуры приводит
к разложению гидрида, в результате чего выход продукта восстановления
снижается. Например, нагревание смеси реагентов до 200° С в течение
5 час. дает 46% спирта.
Восстановление метилциклогексанонов приводит к смеси стереоизомер-
ных спиртов. С ометилциклогексаноном получают 39% ^^с-метилцикло-
гексанола и 61% транс-изомера. В случае м- и я-метилциклогексанонов
выход цис- и транс-изомеров составляет 83 и 17, 19 и 81% соответственно
[86].
При нагревании до 146° С эквимолекулярных количеств гидрида
трифенилолова и метил-2-тиенилкетона в течение 2 час. получают [69] 7% метил-
2-тиенилкарбинола. Если же взять 5 молей гидрида на моль кетона, то
выход карбинола увеличивается до 20%.
Целесообразнее применять в качестве восстановителей оловоорганиче-
ские дигидриды. Дигидрид ди-я-бутилолова экзотермически реагирует
с альдегидами, а при восстановлении кетонов требуется незначительное
нагревание [58, 85]. Образующееся в качестве побочного продукта ди-я-
бутилолово представляет собой маслянистую жидкость, от которой спирт
легко отделить отгонкой. Ди-я-бутил олово можно также окислить на
воздухе и затем смесь проэкстрагировать эфиром. Интересно отметить, что при
обработке воздухом полученного таким образом препарата ди-я-бутилолова
образуется бесцветное кристаллическое вещество, анализ которого
соответствует дигидроокиси ди-я-бутилолова. Окисление проводят в метиловом
спирте при 40—50° С [85].
При восстановлении метилвинилкетона дигидридом дифенилолова
происходит следующая реакция [85, 87]:
(CGH5)2SnH2 + СМ2=СНСОСН3 -» [(C6H5)2Sn]rt + CH2=CIICH (ОН) СН3.
Продукты легко разделяются, так как образующаяся модификация
дифенилолова не растворяется в эфире. Чистый метилвинилкарбинол выделен
с выходом 59%.
Восстановление дигидридом дифенилолова в ряде случаев легко
проходит при комнатной температуре. Реакции проводят с избытком олово-
органического соединения, который затем разлагают прибавлением к смеси
диэтиламина. Последний способствует также превращению дифенилолова
в менее растворимую модификацию. Вследствие термической нестойкости
дигидрида дифенилолова возможности его применения ограничены [85].
Иногда дигидрид дифенилолова разлагается в присутствии простых
кетонов, таких как ацетон, антрахинон и 2-ацетилгексанон [58]. Это
обусловлено, по крайней мере частично, присутствием случайных примесей
в исходных веществах, так как бензохинон, циклогексанон и бензофенон
восстанавливаются этим гидридом с хорошим выходом [58].
Дигидриды удобнее всего использовать для восстановления простейших
альдегидов и кетонов, с которыми реакции проходят гладко и с хорошим
выходом.
а, ^-Ненасыщенные карбонильные соединения селективно
восстанавливаются в ненасыщенные спирты [85, 87]. Однако выходы продуктов
с дигидридом ди-я-бутилолова гораздо ниже, чем с дигидридом
дифенилолова. Вероятно, это объясняется присоединением гидрида к образующимся
ненасыщенным спиртам.
474
Оловоорганические соединения. Лит. стр. 492—494
Тригидрид я-бутилолова разлагается при комнатной температуре. Если
применять большой избыток этого гидрида, то он действует как
восстановитель, например, циклогексанона [88], 4-метилциклогексанона [85] или
4-т/?ет-бутилциклогексанона [85].. Во время реакции образуется также
соединение тёмнокрасного цвета. После удаления летучих продуктов оно
представляет собой стеклообразное вещество, которое в тонко измельченном
состоянии самовоспламеняется на воздухе. Элементарный анализ этого
соединения отвечает формуле C4H9Sn. К его образованию приводят,
по-видимому, следующие реакции:
2 C4H9SnH3 + 3 R2CO -^ 2 / п (C4HPSn)rt + 3 R2CHOH,
2 C4H9SnH3 -» 2 / п (C4H§Sn)rt + 3 Н2.
Еще быстрее разлагается тригидрид фенилолова, поэтому полученные с ним
результаты не очень отчетливы. Этим гидридом восстановлены [85]
следующие кетоны: циклогексанон, 4-т/?£/я-бутилциклогексанон и (/)-метон.
Существенное влияние на реакции восстановления оказывает и природа
альдегида или кетона. Например, дигидрид дифенилолова восстанавливает
камфору очень медленно, тогда как с бензальдегидом происходит
экзотермическая реакция [85, 87]. Гидрид трифенилолова восстанавливает бен-
зальдегид при нагревании смеси на водяной бане в течение 15 час, а
циклогексанон не полностью восстанавливается в тех же условиях даже за 2 дня.
Большую роль в реакции играют стерические факторы, что, в частности,
отражается на низкой реакционной способности камфоры [85, 87]. Бензил,
а-дикетон, восстанавливается очень легко двумя молями дигидрида
дифенилолова в гидробензоин. Диметилгидрорезорцин и 2-ацетилциклогекса-
нон, Р-дикетоны просто ускоряют разложение дигидрида дифенилолова.
Бензохинон восстанавливается с хорошим выходом в гидрохинон, но антра-
хинон не восстанавливается [85, 87]. Стерические факторы проявляются
и при восстановлении ди гидр и дом дифенилолова кетостероидов [58].
Следует отметить, что в реакциях дигидрида дифенилолова с л*-нитро-
бензальдегидом, д-нитробензальдегидом и ж-нитроацетофеноном [85]
вначале происходит восстановление нитрогруппы (при реакции нитробензола
с большим избытком гидрида трифенилолова образуется анилин с выходом
38% [71]).
Механизм реакций восстановления альдегидов и кетонов оловоорганиче-
скими гидридами не ясен. Возможно, гидрид присоединяется по
карбонильной группе, а затем проходит конденсация образующегося алкоксисоеди-
нения с избытком оловоорганического гидрида. Однако вопрос этот требует
дополнительного исследования [58, 89].
ВОССТАНОВЛЕНИЕ ОЛОВООРГАНИЧЕСКИМИ ГИДРИДАМИ
АЦИЛГАЛОГЕНИДОВ
Активность хлорангидридов органических кислот по отношению к олово-
органическим гидридам примерно такая же, как у бромистых алкилов [58].
Поскольку взаимодействие альдегидов с оловоорганическими
моногидридами проходит довольно медленно, этот путь иногда можно использовать
для синтеза альдегидов из хлористых ацилов [58, 60, 90]. Однако
синтетические возможности метода ограничены, так как в результате реакции
наряду с альдегидом образуется эфир [58]. Найдено, что последний
является основным продуктом восстановления хлорангидридов карбоновых
кислот гидридом трифенилолова. Выход альдегида при этом не превышает
16% [90а]. Без растворителя основным продуктом является бензил-
бензоат. Механизм этих реакций подробно рассмотрен Куивилой и Уолшем
Синтез и реакции оловоорганических гидридов
475
[91, 92]. Предложена [58] следующая схема процесса:
\ \
—Sn' + RCOC1 -* — SnCl + RG=0,
/ /
О
\ II \
RC=0 + — SnH -> RC—H + —Sn',
/ /
О
II
:-с-
RC
-0-
0
II
=0 + RC—H -
\
-CR + —SnH -
0
II
-> RG—О
0
II
» R-C-
-CR,
0—GH2R +
—Sn*.
/
В пользу этого механизма свидетельствует, в частности, тот факт [58], что
при обработке смеси бензальдегида и хлористого изобутирила оловооргани-
ческим гидридом образуется бензилизобутират, а не изобутилбензоат.
Напротив, при действии гидрида трибутилолова на смесь хлористого бензоила
и изомасляного альдегида получают [58] изобутилбензоат, а не
бензилизобутират.
При восстановлении хлористого бензоила гидридом трифенилолова
получены противоречивые результаты [60, 93, 94]. Установлено [94], что
в качестве основных продуктов при этом образуются бензилбензоат и
хлористое трифенилолово, а также небольшое количество бензола. Реакция
проводилась как при обычной температуре, так и при нагревании в среде
инертных растворителей или без них. На скорость реакции существенное
влияние оказывают катализаторы радикалообразующего типа, а также
ингибиторы свободных радикалов. Ни в одном случае образования
бензальдегида не наблюдалось.
По данным других авторов [60, 93] хлористый бензоил восстанавливается
гидридом трифенилолова до бензальдегида.
Этиловый эфир хлормуравьиной кислоты очень медленно
реагирует с гидридом три-я-бутилолова при 80° С [58]. Прибавление 2 мол.%
азодиизобутиронитрила заметно ускоряет реакцию. Если тем же гидридом
восстанавливать трифенилацетилхлорид при 100° С, то образуется трифенил-
ацетальдегид, трифенилметан и окись углерода.
Р,|5,|3-Трифенилэтилтрифенилацетат, если и образуется, то в
незначительной степени. Образование окиси углерода и трифенилметана в этой
реакции вызвано, по-видимому, декарбонилированием промежуточно
образующегося ацильного радикала в более устойчивый трифенилметиль-
ный радикал. Последний затем отщепляет водород от молекулы оловоорга-
нического гидрида [58]:
(С6Н5)зС-С=0 -* СО + (CeHsbC
При смешении эквимолекулярных количеств гидрида три-я-бутилолова и
хлорангидрида янтарной кислоты происходит экзотермическая реакция [90].
После двухчасового нагревания до 40° С и перегонки продукта в вакууме
получают вещество, эквивалент нейтрализации и анализ на хлор которого
соответствует хлорангидриду формилуксусной кислоты. Однако по данным
ИК-спектра продукт восстановления представляет собой у-хлор-у-бутиро-
476
Оловоорганические соединения. Лит. стр. 492—494
лактон. Исходя из этого, общая схема реакции может быть выражена
следующим образом [90]:
СН2СОС1
CHoCOCl
+ (C4H9)3SnH -
СН2СНО 1
I
СН2СОС1J
сн2—с
н
/
СН2—С—С1
сн2—с
\
с
у
О + (C4H9)3SnCi.
о
\
о
/
Сн2—С
|\
о- ci
Циклизация проходит достаточно быстро, так как тот же продукт получают
и при восстановлении хлорангидрида янтарной кислоты двумя молями
гидрида три-я-бутилолова. Образования янтарного альдегида не наблюдается
[90].
Восстановлением хлорангидрида фталевой кислоты двумя молями
гидрида три-я-бутилолова получен фталид с выходом 55%. Легкость замены
второго атома галоида на водород объяснена [90] тем, что хлор теперь
находится в бензильном радикале, и, следовательно, восстанавливается легче,
чем в у-хл°р-У"бутиролактоне.
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С СОЕДИНЕНИЯМИ, СОДЕРЖАЩИМИ СЕРУ
Реакции этого типа описаны очень кратко [29, 64]. Панг и Бекер [64]
исследовали восстановление различных функциональных групп,
содержащих серу, гидридом трифенилолова. Бензилсульфид образует с гидридом
трифенилолова сульфид быс-(трифенилолова), толуол, сероводород и бен-
зилмеркаптан:
(Cgh5ch2s)2 + 2 (C6H5)3snH -»(C6H5)3snssn(CcH5)3 -НСбН5сн3:+ c6h5ch2sh + h2s.
Взаимодействием гидрида трифенилолова с бензилмеркаптаном получены
сульфид бш>(трифенилолова), толуол и сероводород. Тиобензофенон
восстанавливается до дифенилметана:
С6Н5СС6Н5 + 2 (C6H5)3SnH -> (С6Н5)2СН? + (C6H5bSnSSn(C6H5)3.
II
S
При действии на гидрид трифегилолова сероуглерода с незначительным
выходом (13%) образуется сульфид бш>(трифенилолова). Другие продукты
этой реакции не идентифицированы.
Связь S—Н в метилмеркаптане инертна к действию (C6H5)3SnH даже при
нагревании смеси реагентов до 160° С. Аналогично ведет себя связь С—S в
дифенилсульфиде и тиантрене. С фенилдисульфидом реакция проходит
легко, но при этом образуется лишь фенилмеркаптид трифенилолова:
CGH5SSCGH5 + 2 (CeHsfeSnH -> 2 (CGH5)3SnSC645 + Н2.
Синтез и реакции оловоорганических гидридов
£11
При действии на гидрид трифенилолова тиофенола, 2-тионафтола или фенил-
сульфенилхлорида образуются арилмеркаптид трифенилолова и хлористое
трифенилолово соответственно [64]:
ArSH + (C6H5)3SnH -» (CGH5)3SnSAr + Н2,
CGH5SCl -f- 2 (CGH5)3SnH -* (CGH5)3SnCl + (GGT I0)35n5C6H5 + H3.
Дифенилсульфон, дифенилсульфоксид и метил-я-толуолсульфонат с
гидридом трифенилолова не реагируют [64].
РЕАКЦИИ, ПРИВОДЯЩИЕ К ОБРАЗОВАНИЮ СОЕДИНЕНИЙ
LCO СВЯЗЬЮ Snj— Sn
Оловоорганические гидриды широко применяются для синтеза
соединений со связью Sn — Sn. Этот синтез можно осуществить двумя путями.
Первый из них заключается в каталитическом дегидрировании
оловоорганических гидридов:
2 R3SnH -^ R3SnSnR3 + Н2,
п R2SnH2 -> (R2Sn)n + п Н2.
Второй метод состоит во взаимодействии оловоорганических моно- или ди-
гидридов с соединениями типа R3SnX и R2SnX2 (например, оловоорганиче-
скими галогенидами, алкоксисоединениями, станниламинами и др.). Этот
путь позволяет синтезировать соединения, содержащие разветвленную
цепь из атомов олова.
Примером реакций первого типа может служить образование гексаметил-
дистаннана при нагревании до 70° С соответствующего гидрида в присутствии
диборана [95]. Разложением гидрида триэтилолова тонко измельченным
порошком олова получен [36] гексаэтилдистаннан. В случае гидрида
трифенилолова эффективными катализаторами являются .алифатические или
ароматические амины [96, 971 (ср. [71, 811).
Дегидрирование оловоорганических дигидридов подробно изучено
Нейманом с сотр. [41, 98—101] и другими авторами [35, 102, 103].
Броун и Морган [103] получили полидиметилолово без предварительного
выделения соответствующего гидрида в чистом виде.
Получение полидиметилолова [103]. К раствору 10,4 г (0,047 моля) двухлористого ди-
метилолова в 50 мл эфира прибавляют 10 мл сухого триэтиламина. Смесь охлаждают льдом
и при перемешивании прибавляют в течение 45 мин. 2,8 г литийалюминийгидрида в 225 мл
эфира. Для завершения реакции смесь перемешивают в течение 2 час. Выделившийся осадок
отфильтровывают и из фильтрата эфир отгоняют в вакууме. Остаток представляет собой
масло желтого цвета, из которого постепенно выделяется твердое вещество ярко-желтого
цвета. Его отделяют, промывают холодным бензолом и затем растворяют в горячем бензоле.
Спектр ЯМР этого раствора почти идентичен со спектром, определенным для циклического
шестичленного диметилолова.
При нагревании до 90° С дигидрида диэтилолова в течение 37 час. полного
отщепления водорода не наблюдается [101]. Молекулярный вес проб,
взятых из реакционной смеси, постепенно повышается. Одновременно в ИК-
спектре исчезает адсорбция связи Sn—Н при 1822 еж-1 и появляется новая
полоса 1780 см'1. На промежуточных стадиях разложения дигидрида
диэтилолова образуются олигомеры состава H[Sn(C2H5)2]rcH. Степень
конденсации п постепенно повышается до значения 8—9. Последние остатки групп
Sn—Н удаляются с трудом. Только после месячного хранения дигидрида
диэтилолова в запаянной ампуле на солнечном свету исчезает адсорбция
связи Sn—Н при 1780 еж"1. Образующаяся модификация диэтилолова
представляет собой вязкую жидкость лимонно-желтого цвета. Вещество
чрезвычайно легко окисляется на воздухе [101].
478
Оловоорганические соединения. Лит. стр. 492—494
Оловоорганические дигидриды конденсируются гораздо быстрее и при.
более низкой температуре, если в качестве катализатора применять амины.
Каталитическое действие последних возрастает в следующем ряду [101]:
диметилформамид, диэтиламин, триэтиламин, пиперидин, пиридин и анилин.
Каталитическая активность аминов заметно увеличивается в присутствии
оловоорганических галогенидов [101]. Дегидрирование тщательно
очищенного дигидрида диэтилолова в присутствии пиридина даже при нагревании
проходит очень медленно и не полностью. Скорость реакции увеличивается
и она идет с выделением тепла, если к раствору дигидрида диэтилолова
прибавить небольшое количество комплекса (CsHs^SnClg^CsHsN. Правда,
при этом наблюдаются побочные процессы.
В соответствии со всем сказанным общую схему каталитического
дегидрирования дигидрида диэтилолова можно представить следующим образом
[1011:
| 9 (C2H5)2SnH2 -> Н [Sn(C2H5)2]9H + 8 Н2,
н [Sn(c2H5)2]9H:l> [Sn(c2H5)2]9 + н2.
Скорость реакции, а также молекулярный вес полученных таким образом
препаратов диалкилолова определяется природой алкильных групп у атома
олова и характером катализатора. Так, конденсацией дигидрида
диэтилолова в присутствии пиридина и двухлористого диэтилолова получен [101]
октадекаэтилциклононастаннан [(C2H5)2Sn]9. Если же реакцию проводить
с диэтиламином в присутствии небольшого количества безводного
хлористого цинка, то образуется додекаэтилциклогексастаннан [(C2H5)2Sn]6 [99].
Применение в качестве катализатора метилата натрия в тетрагидрофуране
приводит к смеси полимергомологов.
Получение октадекаэтилциклононастаннана [101]. Синтез и очистку вещества проводят
в атмосфере сухого инертного газа. Воздух из применяемых растворителей должен быть
предварительно удален. В двухгорлую колбу, снабженную магнитной мешалкой (оболочка
из тефлона) и соединенным с газометром обратным холодильником., помещают 38,0 г (0,21
моля) дигидрида диэтилолова, 100 мл пиридина и 100 мл толуола (без толуола из смеси
выделяется во время реакции гидрид состава Н [Sn(C2H5)2]9H). Затем прибавляют 0,6 г (0,024
моля) двухлористого диэтилолова. Сразу же начинается выделение газа и через 24 часа
реакция заканчивается (последние 2 часа смесь нагревают до 80—100° С). Прозрачный раствор
оставляют на 5 дней в холодильнике. Смесь разбавляют 200 мл метилового спирта и
охлаждают до —20° С. Осадок отфильтровывают, промывают 7 раз метиловым спиртом (порциями
по 15 мл) и высушивают при 20° С/0,2 мм до постоянного веса. Получают 35,5 г (94%)
кристаллов золотисто-желтого цвета ст. разл. 150°С (в предварительно нагретом блоке вещество
разлагается при 220° С с выделением олова). Вещество очень хорошо растворяется в цикло-
гексане, очень плохо в метиловом и этиловом спиртах; не растворяется в воде.
Гекса- и нонамерное диэтилолово и его растворы окрашены в желтый
цвет. В УФ-спектре этих соединений отсутствуют какие-либо специфические
полосы. Причиной окраски является, очевидно, взаимодействие между
атомами олова в молекуле [101].
Следует отметить, что устойчивость оловоорганических дигидридов
увеличивается при переходе к высшим членам гомологического ряда [99].
Отщепление водорода от дигидридов ди-я-бутил- и диизобутилолова
проходит медленно, требуется нагревание смеси до 80—100° С. Ди-н-бутилолово
кристаллизуется в виде циклического гексамера, тогда как диизобутилоло-
во — в нонамерной форме.
Получение додекабутилциклогексастаннана [99]. Раствор 39,9 г дигидрида ди-я-бутил-
олова в смеси 45 мл пиридина и 50 мл толуола нагревают при 60° С в течение 7 час, затем
6 час. при 80° С и 10 час. при 90—100° С. За это время выделяется 90% теоретического
количества водорода. Для завершения реакции смесь оставляют на несколько месяцев при
обычной температуре. Отщепление водорода проходит быстрее, если к смеси прибавить 1—3 моль,
% двухлористого ди-я-бутилолова (в расчете на дигидрид). Однако выделяющееся при этом
вещество менее однородно. После удаления растворителя остается масло золотисто-желтого
цвета, которое закристаллизовывается в течение нескольких недель. Выход 35 г (88%).
Синтез и реакции оловоорганических гидридов
479
Если в качестве катализатора применять 2 мол. % метилата натрия
в тетрагидрофуране, то реакция заканчивается уже при 20° С за 14 час.
Растворитель удаляют, остаток промывают метиловым спиртом и
высушивают в вакууме. Получают додекабутилциклогексастаннан в виде масла
золотисто-желтого цвета, которое закристаллизовывается в течение 2 час.
Выход 94%.
Конденсацией дигидрида дибензилолова в диметилформамиде в
присутствии следов двухлористого дибензилолова получен [22] октабензилтетра-
станнан, т. пл. 226—228° С (из смеси толуола с лигроином; при плавлении
вещество частично разлагается). В пиридине из того же гидрида образуется
смесь высших гомологов.
Конденсация оловоорганических дигидридов в присутствии
каталитических количеств комплекса R2SnCl2-2C5H5N проходит и в ароматическом
ряду.
Так, циклический додекафенилциклогексастаннан получен [98—100]
конденсацией дигидрида дифенилолова в присутствии пиридина:
6 (C6H5)2SnH2 _»[(C6H5)2Sn]G + 6 Но.
Как и в случае дигидрида диэтилолова, с чистым, тщательно перегнанным
дигидридом дифенилолова реакция проходит очень медленно. Скорость ее
заметно увеличивается при прибавлении небольшого количества
двухлористого дифенилолова. Вероятно, способ действия катализатора основан на
обмене водорода с галоидом [100]. Образующийся (хлор)гидрид
дифенилолова в присутствии амина быстро конденсируется с выделением водорода.
Вещество кристаллизуется из толуола в виде прямоугольных
пластинок, содержащих 11—13% растворителя, что соответствует примерно
2,6 моля толуола на моль гексамерного дифенилолова. Последнее, в отличие
от диэтилолова, устойчиво на воздухе в течение месяца. Структура вещества
доказана реакцией с иодом (количественное образование двуиодистого
дифенилолова) и определением молекулярного веса осмометрическим методом.
В качестве побочного продукта в результате реакции образуется более
высокомолекулярная модификация дифенилолова, представляющая собой
порошок светло-желтого цвета. Количество последнего заметно
увеличивается, если реакцию проводить выше 50° С или с более сильными основаниями,
например, триэтиламином [100].
Получение додекафенилциклогексастаннана [100]. Синтез и очистку вещества проводят
в атмосфере сухого инертного газа. В двухгорлую колбу емкостью 100 мл помещают 6,0 г
дигидрида дифенилолова (перегнанного в вакууме 10~4 мм) и к нему прибавляют 50 мл
свежеперегнанного пиридина, из которого предварительно удаляют воздух. При
перемешивании сразу же начинается выделение водорода, который собирают в газовой бюретке. Смесь
перемешивают при комнатной температуре в течение 5 час, а затем нагревают 10 мин.
до 70° С. Выделившийся осадок отсасывают и высушивают в вакууме. Выход сырого продукта
5,8 а (98%). Для очистки его дважды перекристаллизовывают из толуола, применяя каждый
раз по 200 мл растворителя и небольшое количество активированного угля. Получают
бесцветные прямоугольные кристаллы, которые высушивают при 110° С/0,2 мм в течение 2
час. (во время высушивания выделяется 340 .мг толуола). Получают 2,6 г (44%) вещества
с т. разл. 270° С. Обработка маточника дает еще одну порцию кристаллов.
Додекафенилциклогексастаннан с трудом растворяется в пиридине и
ароматических углеводородах, в других обычных органических
растворителях не растворяется или растворяется плохо.
После шестимесячного хранения молекулярный вес вещества не
изменяется.
При замене пиридина диметилформамидом реакция проходит только
в присутствии двухлористого дифенилолова [100]. С неперегнанным
дигидридом дифенилолова в диметилформамиде получен циклический пентамер —
480 Оловоорганшеские соединения. Лит. стр. 492—494
декафенилциклопентастаннан с т. пл. 285° С (из толуола) [98—100].
Однако воспроизвести этот результат не удается. Вероятно, ход реакции
зависит от присутствия следов сокатализатора [100].
По-видимому, еще одна модификация дифенилолова со средней степенью
конденсации 8,6 ±1,1 [35] получена прибавлением диэтиламина к эфирному
раствору неочищенного дигидрида дифенилолова. Менее растворимый
продукт образуется при замене триэтиламина три-я-бутиламином [35].
Нейман и Кениг [100] отмечают, что при дегидрировании дигидрида
дифенилолова диэтиламином наблюдаются также побочные реакции.
Помимо аминов, катализаторами конденсации дигидрида дифенилолова
являются и другие вещества. Никель Ренея отщепляет водород от
дигидрида дифенилолова, но при этом выделяется также металлическое олово
[100]. Аналогично действуют тонко измельченные переходные металлы.
Метиловый спирт не реагирует с чистым дигидридом дифенилолова,
но с сырым продуктом проходит следующая реакция [98—100]:
6 (CeH6)a5nH2 -* Н [5п (С6Н5)2]бН.
Та же реакция наблюдалась ранее Куивилой [35], но он считал, что при
этом образуется еще одна модификация дифенилолова.
Получение гидрида Н [Sn(C6H5)>]eH [100]. Синтез проводился соответственно
описанной в литературе [35] методике. К 5,6 г неперегнанного дигидрида дифенилолова прибавляют
100 мл сухого эфира и 10 мл сухого метилового cnnpia. Смесь выдерживают в защищенной
от действия света колбе в течение 24 час. Выпадает 4,6 г (82%) осадка светло-желтого цвета.
В ИК-спектре вещества имеется полоса 1790 см'1, характерная для группы —Sn—Sn—Н.
Определение молекулярного веса показывает, что продукт состоит в основном из
соответствующего гексамера.
Обработка полученного гидрида пиридином в присутствии двухлористого
дифенилолова дает додекафенилциклогексастаннан [98—100] (ср. [35]).
Таким образом, синтез последнего проходит через стадию полимерного
продукта типа Н [Sn (С6Нб)2]бН, а не мономерного дифенилолова.
Подобным образом проходит конденсация и других ароматических ди-
гидридов. Так, при нагревании до 70° С 6,4 г дигидрида ди-я-толилолова
в 80 мл диметилформамида образуются бесцветные ромбические кристаллы
ди-/г-толилолова [41]. Вещество плохо растворяется в обычных органических
растворителях и определить его молекулярный вес не удается. Вероятно,
его строение сходно с додекафенилциклогексастаннаном. В отличие от
последнего ди-А1-толилолово и другие описанные ниже диарильные соединения
олова не дают кристаллогидратов с толуолом.
Получение ди-(/г-этоксифенил)олова [41]. 3,45 г дигидрида ди-(я-этоксифенил)олова
и 150 мл диметилформамида оставляют на 24 часа при 25° С. Для завершения реакции смесь
нагревают до 70° С. Выпавший осадок кристаллизуют из 250 мл ди-я-бутилового эфира.
Получают 2,3 г (67%) кристаллов светло-желтого цвета с т. разл. 235—240° С. Вещество
плохо растворяется в обычных органических растворителях.
Ди-я-бифенилилолово получено [411 дегидрированием соответствующего
дигидрида в пиридине. Реакция заканчивается за 3 часа при 20—30° С.
Из 2,3 а дигидрида ди-Р-нафтилолова и 40 мл диметилформамида в
присутствии следов двухлористого ди-р-нафтилолова получают игольчатые
кристаллы ди-Р-нафтилолова. Реакция проходит при 40—50° С за 7 час.
Отщепление водорода от дигидрида ди-а-нафтилолова приводит к
образованию ди-а-нафтилолова с количественным выходом. Правда,
образующийся сырой продукт растворяется очень плохо и очистить его не удается [41].
К образованию органостаннанов приводит и гетерофункциональная
конденсация оловоорганических гидридов с соединениями типа R3SnX или
R2SnX2 [47,. 49, 89, 99, 101 — ПР. Скорость этих реакций существенно
зависит от характера функциональной группы X у атома олова [1041. На-
Синтез и реакции оловоорганических гидридов
481
пример, в случае трибутильных соединений олова она увеличивается в
следующем ряду:
—SeSn (С4Н9)3» — SSn (С4Н9)3 < —OAlk ж —Р (С4Н9)2 <
< -OSn (С6Н5)3 < - OSnAlk3 < —N (С2Н5)2.
Так, гидрид трибутилолова не реагирует в заметной степени с сульфидом
и селенидом бш>(трибутилолова) даже при нагревании смеси до 160° С.
С метокситрибутилоловом реакция заканчивается при 110° С за 180 час,
а с диэтил(триэтилстаннил)фосфином при той же температуре за 130 час.
Диэтиламинотрибутилолово экзотермически реагирует с гидридом
трибутилолова уже при обычной температуре. Важную роль играет также
характер органического радикала у атома олова. По своей реакционной
способности оловоорганические гидриды располагаются следующим образом:
(s^-QH9)3SnH < (/-CiH9)3SnH < (/i-CiHgfoSnH < (C6H5)3SnH.
Примером реакций рассматриваемого типа может служить образование
симметричных и несимметричных гексаалкилдистаннанов, октаалкилтри-
станнанов или полистаннанов при действии оловоорганических гидридов на
гексаалкилдистаннаны или окиси диалкилолова [49, 89, 104, 105]:
2 R3SnH + RgSnOSnRg-* 2R3SnSnRg + H20,
2 RgSnli + R2SnO-> R3SnSnR2SnR8 + H20,
R2SnH2 + R2SnO -* 2 / n (R2Sn)rt + H20.
При нагревании до 100°С стехиометрических количеств гидрида три-я-
бутилолова и гекса-я-бутилдистанноксана в течение 114 час. получают [89]
гекса-я-бутилдистаннан с выходом 90%, т. кип. 166—167° С/0,4 мм.
Вещество охарактеризовано элементарным анализом, определением
молекулярного веса и титрованием бромом.,
Дигидрид ди-я-бутилолова реагирует с окисью ди-я-бутилолова примерно
за 12 час. при 100° С. При этом с количественным выходом образуется
полимерное ди-я-бутилолово, представляющее собой вязкую жидкость желто-
зеленого цвета [89].
Нагреванием до 120° С гексаэтилдистанноксана с дигидридом диэтилоло-
ва получен [47] октаэтилтристаннан с выходом 50%. Кроме того, в качестве
побочного продукта образуется гексаэтилдистаннан.
Получение окта-я-бутилтристаннана [89]. 7,26 г (0,025 моля) гидрида три-я-бутилолова
нагревают при 100° С с 3,11 г (0,0125 моля) окиси ди-я-бутилолова в течение 26 час. Во
время реакции отгоняется 0,0129 моля годы. Остаток представляет собой маслянистую
бесцветную жидкость, элементарный анализ и определение молекулярного веса которой
соответствует окта-я-бутилтристаннану. При нагревании в вакууме вещество разлагается не
перегоняясь.
Если исходить из стехиометрических количеств дигидрида
ди-я-бутилолова и гекса-я-бутилдистанноксана, то на первой стадии проходит реакция
обмена с образованием гидрида три-я-бутилолова и окиси ди-я-бутилолова
[89]. Последние реагируют между собой при нагревании до 100° С как
описано выше.
С ароматическими соединениями реакция проходит легче, чем с
алифатическими [49].
Аналогично реагируют гидриды со станниламинами [49, 99, 104—106]:
R3SnN(C2H5)2 + HSnRg -» RaSnSnRg + HN(C2H5)2,j
2 R3SnN(C2H5)2 + HaSnRg-* RsSnSnR^SnRg + 2 (C2H5)2NH,
R2Sn [N(C2H5)o]2 + 2 HSnRg -» R3SnSnR2SnR3 + 2 (QHr^NH.
31 Заказ Х> 207
482
Оловоорганычсские соединения. Лит. стр. 492—494
В случае, когда у атома азота находится алифатический радикал,
реакция проходит при температуре от 0 до 50° С. Конденсацией станнилендиами-
нов с оловоорганическими дигидридами получают шестичленные
циклические продукты [99, 106]:
3 R2SnH2 + 3 R2Sn [N(C2H5)2]2-* (R2Sn)G + 6 (C2H5)2NH.
Синтез проводят, прибавляя по каплям дигидрид к станнилендиамину.
Осадок отфильтровывают, промывают сухим метиловым спиртом и
кристаллизуют из бензола. Другой способ очистки состоит в высаживании
продукта реакции из диэтиламина метиловым спиртом. В случае
необходимости вещество кипятят с изопропилозым спиртом и осаждение
повторяют. Об окончании реакции судят по исчезновению полосы связи
Sn — Н в ИК-спектре [106].
Органосганнаны получают также взаимодействием оловоорганических
гидридов с производными (Ы-фенилформамидо)олова [107, 108].
Дигидриды диалкил- или диарилолова реагируют с триалкил-(М-фе-
нилформамидо)оловом по схеме [107]:
RsSnN (СвН6) СНО + RgSnH2-> R^nSnR^H + CeH5NHCHO.
Изменяя соотношение реагентов, можно получить октаалкилтристаннаны
[107—108а]:
2 R3SnN(CeH3)C IO -I- R2SnH3-* R35nSnR2SnR3-f 2CeH5NHCHO.j
К тристаннанам приводит и следующая реакция [108а]:
R2Sn [N (СбПс) СНО]2 4- 2 (CeH5)3SnH -*
(CeH5)3SnSnR3SrnCeH5)3 + 2 C6l!5NHCnO.
Получение 1,!, 1,3,3,3-гексабутил-2,2-дифенилтристаннана [108л]. К 8,92 г (0,02
моля; трибутил-(\т-фенилформамидо)олова вводят шприцем 2,75 г (0,02 моля) дигидрида
дифенилолова. Смесь нагревают при 50—60° С в течение 30 мин., а затем 20 мин. при 90° С.
Продукт реакции переносят в 100 мл гексана и охлаждают до 0° С. Выделившийся осадок
отфильтровывают и фильтрат упаривают в вакууме. Получают 5,5 г маслянистой жидкости,
которую хроматографируют на коленке с окисью алюминия. Пос.е удаления растворителя
в вакууме получают 4,1 г слегка желтоватой жидкости "с п^ 1,5886. Мол. вес. найдено 807,
вычислено 850.
Линейные тетрастаннаны синтезированы конденсацией соответствующих
гидридов в присутствии диэтиламина или пиридина [107]:
2 RsSnSnR^H -> RaSnRgSnSnR^SnRa + Н2.
Тетрастаннаны с разветвленной цепью получены [108] взаимодействием
тригидрида фенилолова с триалкил-(Ы-фекилформамидо)оловом:
\2 R3SnN(C6H5)CHO + C6H5SnH3 -> C6H5Sn(SnR3)3 + 3 C6H5NHCHO.
Если взять 2 моля трибутил-(1М-фенилформамидо)олова на 1 моль
тригидрида фенилолова, то образуется вещество состава [(C6H5)3Sn]2Sn(C6H5)H
[107].
Присоединением гидридов типа R3SnSnR'2H к фенилизоцианату с
последующей обработкой образующегося продукта оловоорганическими дигид-
ридами получены [107] пентастаннаны:
2R3SnSnR2N(C6H5)CHO+R2SnTl2 -» RsSnSnRaSnRgSnRaSnRsH- 2CeH6NHCHO.
К линейным гексастаннанам приводят следующие реакции [107]:
R3SnSnR2N(C6H5)CHO + R2SnH2-> R3SnSnR2SnR2H + C6H6NHCHO,
2 R3SnSnR2SnR2H -» R3Sn (SnR2)4SnR3 + H3.
Синтез и реакции оловоорганических гидридов
483
Полимерные органостаннаны, содержащие чередующиеся диалкил- и
дифенилстаннильные группы, синтезированы [108, 108а] следующим обра-
зом:
п R2Sn [N(C6H5)CHO]2 + (C6H5)2SnH2 -*
[—Sn(C6H5)2SnR2—]-+ n CeH5NHCHO.
Получение полимерного диэтилдифенилдистаннана [108а]. При прибавлении к 11,8 г
(0.028 моля) 6«с-(1Ч-фенилформамидо) диэтилолова 7,78 г (0,028 моля) дигидрида дифенилоло-
ва происходит экзотермическая реакция. Смесь нагревают при 50° С в течение 2 1/а час. и
затем N-фенилформамид удаляют, дважды обрабатывая смесь сухим этиловым спиртом (20
мл). Образующийся маслянистый продукт растворяют в 30 мл сухого бензола и
отфильтровывают (на фильтре остается около 0,8 г осадка). Из фильтрата после удаления
растворителей получают 11,1 г (90%) вязкого продукта светло-желтого цвета. Его растворяют в 25 мл
сухого бензола и этот раствор медленно прибавляют к 500 мл петролейного эфира.
Получают 5,3 г (45%) вещества с т. пл. 230—233° С (с разложением).
Оловоорганические гидриды вступают также в реакцию конденсации
с алкоксисоединениями [49, 89] или галогенидами [ 49, 104] ал кил- и арил-
олова:
(С4Н9)23п(ОСН3)2 + (QUgfeSnHa-> 2/л [(QH^bSn],, + 2СН3ОН,
(CeH5)3SnH + (C2H5)3SnCl + (C2H5)3N-> (C2rb)3SnSn (C6H5)3 + (C2H5)3N.HC1.
При нагревании в ксилоле гидрид триэтилолова реагирует с двухлористым
диэтилоловом и без акцепторов галоидоводородов [109—111]:
(Q>H5)2SnCl2 + 2 (QH^SnH -» 2 (C2H5)3SnCl + (C2H5)2Sn + H2.
Однако эта реакция не проходит до конца и осложняется побочными
процессами [ПО, 111].
Подробно изучен [108а, 111а, 1116] механизм гидросташюлиза связей
Sn—N, Sn — Р, Sn — As, Sn—О и Sn — S. В частности, было установлено,
что азодиизобутиронитрил и феноксил практически не влияют на реакцию
гидрида трифенилолова с триэтил-(М-фенилформамидо)оловом, но ее
скорость существенно зависит от полярности среды. При замене циклогексана
(е = 2,0) бутиронитрилом (е = 23,3) она возрастает более чем в 100 раз.
Скорость гидростаннолиза связи Sn—N увеличивается, если у атома
азота находятся электронодонорные группы и уменьшается, если эти группы
обладают электроноакцепторными свойствами. Важное значение имеет и
характер органического радикала у атома олова. Замена в триэтил-(Ы-фе-
нилформамидо)олове этильной группы на фенильную уменьшает
относительную скорость реакции в бутиронитриле от величины > 100
до 1,5.
Что касается заместителей у атома олова в оловоорганическом гидриде,
то здесь наблюдается обратное явление. Это можно видеть из следующего
примера. Гидрид триэтилолова практически не реагирует с триэтил-
(Ы-гексилформамидо)оловом при 40°С в циклогексане. С гидридом
трифенилолова через 10 мин. степень превращения достигает 50%. По сравнению
с гидридом трифенилолова гидрид три-(п-анизил)олова в несколько раз
менее активен.
Исследование кинетики реакции (диэтиламино)триэтилолова с гидридом
триэтилолова показало, что она имеет суммарный второй порядок и первый
порядок по каждому из реагентов. Наконец, изучение изотопного эффекта
этой реакции дало величину kHlkD = — 1,2.
7i*
/■U
Олсвоорганические соединения. Лит. стр. 492—494
Совокупность полученных экспериментальных данных находится в
хорошем соответствии со следующей схемой реакции [108а]:
R„SnNR9 + R3SnH
медленно
R3Sn*
.N*
Н
Sn«
5+
RlSn+N/+ R8Sn-
R3SnN-
H
+ R3Sn- 6_™ RaSnSnR; + R^Fi.
Аналогично проходит гидростаннолиз связи Sn—О [108а, 111а, 1116].
В тех случаях, когда атом олова связан с серой, фосфором или мышьяком,
преимущественно происходит реакция обмена [111а, 1116]:
R3SnX + R^SnH ^ R3SnH + RgSnX.
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С МЕТАЛЛООРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
При действии литийорганических соединений на оловоорганические
гидриды получены противоречивые результаты. По данным Виттига с сотр.
[5], при реакции гидрида трифенилолова с метиллитием гладко образуется
трифенилстанниллитий (выход не указан):
(C6H5)3SnH + CH3Li -» (C6H5)GSnLi + СН4.
Гилман и Мелвин [112] взаимодействием гидрида трифенилолова с фенил-
литием получили тетрафенилолово с выходом 90%.
При нагревании до 100° С в течение 9 час. диэтилцинка с гидридом три-
этилолова образуются металлический цинк, бензол, тетраэтилолово и гек-
саэтилдистаннан с выходом 83,5, 54,2, 70,4 и 19,2% соответственно. Реакция
протекает, по-видимому, по двум направлениям, из которых второе является
преимущественным [113]:
(C2H5)3SnH + (С2Н 02Zn -» C6H6 -f Zn + (C3H5)4Sn,
2 (C2H5)3SnR + (C2H5)2Zn -> 2C3II6 + Zn + (C2H5)3SnSn (С2Н5)3.
Гидрид триэтилолова экзотермически реагирует с хлористой этилртутью
и ацетатом метилртути [111]. В первом случае возникает ртуть, хлористое
триэтилолово и этан. Выходы составляют 77,7, 40,9 и 62,5% соответственно.
Во втором случае выделены ацетат триэтилолова, метан и ртуть с выходами
64,6, 76,9 и 70,0%.
При действии гидрида трифенилолова на диэтилцинк или диметилкадмий
в пентане или бензоле также выделяется свободный металл [108а, 113а].
Однако если реакцию между димети л кадмием и гидридом трифенилолова
проводить в диметоксиэтане при низкой температуре, то с выходом 30%
получают комплекс бш>(трифенилстаннил)кадмия с диметоксиэтаном:
Н2С — СНа
I I
Н3С—О О—СНз
2 (C6H5)3SnH + Cd
СНз
СН8
П2С—СН2
I I
Н3С—О О—СН3
\ /
Cd
/ \
(C6H5)3Sn Sn (C6H5)3
+ 2СН4.
Синтез и реакции оловоорганических гидридов
485
Прибавление 2,2'-бипиридила к раствору этого комплекса в тетрагидрофу-
ране дает вещество оранжевого цвета, представляющее собой комплекс
бш>(трифенилстаннил)кадмия с тетрагидрофураном. То же соединение
получено с выходом 87% при прибавлении гидрида трифенилолова к раствору
комплекса диметилкадмия с 2,2'-бипиридилом в диметоксиэтане:
2 (C6H5)3SnH +
—60° С
+ 2СН4.
(C645)3Sn Sn (С6Н5)з
Тот же метод использован для синтеза соединений со связью Sn—Zn [108a].
Получение комплекса #йС-(трифенилстаннил)кадмия с 2,2'-бипиридилом [108а].
В двухгорлую колбу емкостью 25 мл, снабженную обратным холодильником и соединенную
с газовой бюреткой> помещают 0,75 г(0,005 моля) диметилкадмия в 2 мл диметоксиэтана.
При прибавлении 0,82 г бипиридила в 3 мл диметоксиэтана смесь сразу же окрашивается в
желтый цвет. Раствор охлаждают до —70° С и к нему прибавляют 3,6 г (0,01 моля) гидрида
трифенилолова. При постепенном повышении температуры до —28° С начинает выделяться
газ и смесь окрашивается в оранжевый цвет. Через 165 мин. температура повышается до
комнатной, а в бюретке собирается 231 мл газа (теоретическое количество 224 мл).
Реакционную смесь разбавляют 10 мл петролейного эфира, выделившийся осадок
оранжево-красного цвета отфильтровывают и высушивают в вакууме. Получают 4,36 г (86,8%) вещества с
т. пл. 154° С (с разложением).
Аналогично проходит реакция между комплексами хлористого этил-
цинка или хлористого метилкадмия и гидридом трифенилолова:
D D
i I
RMC1 + (CHJsSnH -* (C64,)3Sn-M-Cl + RH.
Г T
D D
Реакция между дигидридом дифенилолова и комплексами диметилкадмия
проходит более сложно. Например, в диметоксиэтане выделяется лишь
небольшое количество газа и образуется металлический кадмий.
Интересные результаты получены [108а] при действии гидрида
трифенилолова на магнийорганические соединения. Коричневая окраска комплекса
диметилмагния с 2,2'-бипиридилом изменяется при этом на пурпурную,
причем продукт очень быстро обесцвечивается на воздухе. Аналогично
реагирует комплекс диметилмагния и Ы,Ы,Ы',Ы'-тетраметилендиамина с
гидридом трифенилолова. Гидролиз образующегося продукта дает метан,
гидрид трифенилолова и окись магния. Таким образом, одна из метильных
групп остается у атома магния:
D D
<CH3)2Mg ' + (C6H5)3SnH *• (C6H5)3SnMgCH3 + СН.
Ч /' t
D D
Более четкий результат получен при использовании в этой реакции комплекса
C2H5MgBr» (C2H5)3N. В этом случае выделено теоретическое количество этана.
Остаток представляет собой маслянистую жидкость, которая при нагревании
в вакууме закристаллизовывается. Данные анализа этого кристаллического
вещества соответствуют бромистому трифенилстаннилмагнию:
UH5MgBr.(C2H5)3 N+ (С6Н5)3 SnH ——--> [(С6Н5)3 SnMgBr]2 + С2Н6.
—(C2H5)3N
486
Оловоорганшеские соединения. Лит. стр. 492—494
То же соединение получено при реакции бромистого этилмагния с гидридом
трифенилолова в диэтиловом эфире.
Получение бромистого трифенилстаннилмагния [108а]. К охлажденному до —65° С
раствору бромистого этилмагния (из 1,2 г бромистого этила и 0,24 г магния) в эфире
прибавляют 3,51 г гидрида трифенилолова. Температуру смеси повышают в течение 6 час. до
+ 10° С. При—12° С начинается выделение газа. После того как. реакция закончится
(температура не выше 10° С), смесь разбавляют 10 мл бензола, раствор фильтруют и
концентрируют до 1/б первоначального объема. Затем прибавляют 20 мл петролейного эфира.
Получают маслянистую жидкость, которая закристаллизовывается при высушивании в
вакууме. Выход 3,13 г; т. пл. 230° С (с разложением). Мол. вес. найдено 949, вычислено 908.
Реакция гидрида триэтилолова с симметричными ртутноорганическими
соединениями проходит в более жестких условиях (100—140° С) [111].
Она описывается одним из следующих уравнений:
R:Hg + 2(C2H5)3SnH -
2 R'H + Ug + (С2Н5)з SnSn (QjM.,)3 (1)
H2 + Hg + 2(C2H5)3SnR' (2)
Так, диэтил- и дибензилртуть реагируют по первому пути при любых
соотношениях реагентов [109, 111, 113]. Дифенил- и ди-п-толилртуть реагируют по
уравнению (2). По тому же направлению реагирует с гидридом
триэтилолова и трифенил висмут. Реакция проходит при нагревании смеси до 140° С
в течение 75 час. [111].
С фенил(бромдихлорметил)ртутью гидрид трибутилолова реагирует
следующим образом [114]:
(С4Н9)з SnH + QHsHgCClaBr -» (QH9)3 SnBr + CGH5HgCHCl2.
Луценко и сотр. [115—117] взаимодействием оловоорганических гидридов
с эфирами меркур-бш>уксусной кислоты получили ряд эфиров ди- и
триал килстаннилуксусной кислоты:
RsSnH + Hg (CH2COOR')2 -> R3SnCH2COOR' + Hg + CH3COOR',
R2SnH2 + Hg (CH2COOR')2 -> R2Sn (CH2COOR')2 + Hg + 2CH3COOR'.
Полученные соединения Охарактеризованы элементарным анализом и ИК-
спектрами.
Получение пропилового эфира триэтилстаннилуксусной кислоты [115]. 24,3 г пропило-
вого эфира меркур-б^с-уксусной кислоты и 12,5 г гидрида триэтилолова нагревают при 75° С
в атмоссЬере азота в течение 1,5 час. Жидкую часть декантируют с осадка металлической
ргути (11,5 г, 96%) и перегоняют в вакууме. Получают 13,2 г (73%) пропилового эфира
триэтилстаннилуксусной кислоты с т. кип. 102—105° С/3,5 мм; df 1,2226: n2Q 1,4779.
Аналогично получен метиловый эфир триэтилстаннилуксусной кислоты.
Выход 68%, т. кип. 71—73° С/2 мм; df 1,2962; п% 1,4831.
Получение метилового эфира диэтилстаннил-(?и£-уксусной кислоты [116]. 28 г метилового
эфира меркур-бис-уксусной кислоты и 8,4 г дигидрида диэтилолова нагревают в атмосфере
азота до начала реакции, затем кипятят еще 30 мин. и декантируют с осадка металлической
ртути (15,6 г, 97%). При разгонке выделяют 10,7 г (82%) метилового эфира диэтилстаннил-
ш/с-уксусной кислоты с т. кип. 117— \\9° С/2.5 мм; df 1.-V813; п% 1,4940.
Аналогично получены диэтиловые эфиры диэтилстаннил-бш>уксусной
кислоты (выход 60%; т. кип. 112—114° С/1,5 мм; df 1,3007; п$ 1,4858) и
ди-«-пропилстаннил-бш>уксусной кислоты (выход 71%; т. кип. 121 —
12?° С/1,5 мм; df 1,3088, /iS 1,4909).
Синтез и реакции оловоорганических гидридов
487
Так же энергично реагируют с эфирами меркур-бис-уксусной кислоты
(хлор)гидриды диалкилолова [117]:
С1
-fHg(CH2COOR')2 /
Г R2SnH2 + R2SnCl2 * R2Sn(H)Cl _Hg; _СНзСООс1> R2Sn
CHoCOOR
Образующиеся эфиры диалкилхлорстаннилуксусной кислоты легко гидро-
лизуются влагой воздуха, превращаясь в димеры состава ClSnR2OSnR2Cl.
Получение метилового эфира дипропилхлорстаннилуксусной кислоты [117]. 4,8 г ди-
гидрида дипропилолова смешивают в атмосфере азота с 5,6 г двухлористого дипропилолова
и к полученному (хлор)гидриду дипропилолова через 15 мин. прибавляет 13,2 г метилового
эфира меркур-б^с-уксусной кислоты. Смесь перемешивают в течение 30 мин. и декантируют
с выделившейся ртути (7,8 г, 97%). При разгонке органической част выделяют 9,8 г (77%)
вещества с т. кип. 110—114° С/1,5 мм; df 1,4343; п^ 1,5165.
Аналогично получен метиловый эфир диэтилхлорстаннилуксусной
кислоты (т. кип. 104—107° С/3 мм\ df 1,5498; n2S 1,5264). При действии алюми-
нийорганических соединений на оловоорганические гидриды атомы
водорода замещаются на органический радикал [40, 47, 118, 119].
RsSnH+RgAl-* R3SnR'+ R2A1H,
R2SnH2 + 2 R3AI _> R2SnR2 + 2 R2A1H.
Например, стехиометрические количества этильных соединений реагируют
при 80° С уже за 20 мин. [119]. Также быстро обмениваются я-бутильный
и я-октильный радикалы, изобутильный — медленнее. По-видимому,
реакция проходит через стадию активированного четырехчленного комплекса,
как это установлено [40, 47], с оловоорганическим дейтеридом:
г.
D
/
R3SnD -f R2A1H ^ I R2Sn A1R2
R3SnH + R2A1D.
При прибавлении к смеси оловоорганического гидрида и алюминийорга-
нического соединения а-олефина вновь образуется триалкилалюминий
[119]. В результате фактически проходит гидростаннирование сх-олефиноз,
а алюминийорганическое соединение действует как катализатор [119].
Небольшие количества переходных металлов, например тонко
измельченного никеля, ускоряют этот процесс [47].
Вместо триалкилалюминия можно применять также гидрид диалкил-
алюминия, LiAlR4 и даже литийалюминийгидрид [85]. В этих веществах
радикалы и олефин должны соответствовать друг другу, иначе вначале
вытесняется нежелательный радикал.
Преимущество этого способа гидростаннирования заключается [47,
119] в особо гладком присоединении алифатических оловоорганических
гидридов к незамещенным а-олефинам, винилиденовым соединениям и
диенам. Установлено также [47] легкое гидростаннирование аллильных
соединений олова, которое без катализатора проходит с умеренным выходом
(см. стр. 287); последний не улучшается и в присутствии катализаторов
радикалообразующего типа.
Реакции в большинстве случаев проходят в течение 3 час. [119]. Первый
водородный атом тригидрида алкилолова реагирует быстрее, чем дигидриды
диалкилолова, а последние быстрее, чем гидриды триалкилолова. Правда,
этот тип реакций ограничен выбором олефинов, так как алюминийалкилы
ре^глэуют почти со всеми функциональными группами. Однако можно
488
Оловоорганические соединения. Лит. стр. 492—494
исходить из соединений, содержащих вторую двойную связь, находящуюся
в середине молекулы, которую затем, например эпоксидированием или
другими обычными реакциями, превращают в ту или иную функциональную
группу.
При взаимодействии гидридов триалкил- или трифенилолова с диэтил-
амидом или N-фенилпропионамидом диэтилалюминия соединения со связью
Sn—А1 не образуются [109а].
На стр.481, 482 описаны реакции оловоорганических гидридов с олово-
органическими N-фенилформамидами и станниламинами. При этом с хорошим
выходом образуются соединения со связью Sn—Sn. Тот же метод с успехом
применен и для синтеза соединений со связью Sn—Ge. Следует отметить,
что реакция между германийорганическими аминосоединениями и олово-
органическими гидридами проходит в гораздо более жестких условиях. Так,
диэтиламинотриалкилгерманий реагирует с гидридом трифенилолова лишь
при длительном нагревании смеси до 120° С в бутиронитриле [108а, 119а]:
Alk3GeN (C2H5)2 + (С6Н5)3 SnH -* Alk3GeSn (CeH5)3 -f (С2Н5)2 NH.
бш>(Диэтиламино)диалкилгерманий не может быть использован в этих
реакциях, так как оловоорганические дигидриды легко разлагаются диэтил-
амином. Однако вследствие летучести диметиламина с бмс-(диметиламино)-
диалкилгерманием реакции проходят легко. Так получен, например, гидрид
трибутилгермилдибутилолова:
(С4Н9)3 GeN (СН3)2 + (С4Н9)2 SnH2 -> (QH9)3 GeSn (C4H0)2 H + (CH3)2NH.
При каталитическом разложении этого гидрида диэтиламином образуется
соответствующее тетраметаллическое производное:
(С2Н5)2 NH
2 (С4Н9)3 GeSn (С4Н9)2 Н > (С4Н9)3 GeSn (С4Н9)2 Sn (QH9)2 Ge (C4119)3 + 112.
Аналогично получены и другие соединения со связью Sn—Ge [108а, 119а]:
(С6Н5)3 GeSn (С2Н5)2 N (С2Н5)2 + (QH5)3 SnH -> (QT l5)3 GeSn (C2H5)2 Sn (СвН5)з + (QI b)2 NH.
Нежелательного действия диэтиламина избегают, проводя реакцию при
90° С в вакууме. При обработке (C6H5)3QeSn(C2H5)2N(C2H5)2 N-фенил-
формамидом, а затем дигидридом диэтил- или дифенилолова образуются
соединения состава (C6H5)3GeSn(C2H5)2SnR2Sn(C2H5)2Qe(C6H5)3.
Соединения этого типа могут быть выделены из реакционной смеси при обработке
ее петролейным эфиром, в котором N-фенилформамид не растворим.
Линейное гексаметаллическое производное получено в результате следующих
реакций:
(СвН5)зGeSn (C2II5)2 N(C6H5) СНО + (CeH5)2 SnII2 ->
-> (C6H5)3GeSn (CaH5)2Sn (C6H5)2H+HN(C6H5) CHO,
(C2H5)2NH
2 (C6H5)3 GeSn(C2H5)2 Sn (C6H5)2 H >
-* (C6H5)3GeSn(CaH5)2Sn(C6H5)2Sn(C6H5)2Sn(C2H5)2Ge(CeH5)3 + H2.
Метод позволяет синтезировать разнообразные германийоловоорганиче-
ские соединения с линейной или разветвленной цепью из атомов металла.
Его можно иллюстрировать уравнениями следующих реакций.
N(C6H5)C(0)H Sn (QH5)3
I I
(C6H5)3 GeSn-C>H5 + 2 (C6H5)3 SnH -* C2H5-Sn-Ge (C6H5)3+2HN (C6H,)CHO,
I I
N(CeH6)C(0)H Sn(C6H5)3
Синтез и реакции оловоорганических гидридов
48,)
Ge(C6H5)3 (C6H5)3Ge Ge(C6H5)3
I I I
2 C2H5S11N (C6H5) CHO + R2SnH2 -> QH6Sn—SnR2—SnQHe + 2 HN (С0Н5)СНО,
I I I
Ge (C6H3)3 (CeH5)3 Ge Ge (C6H5)3
Ge (С6Н3)з
I
C2H5SnN(C6H3)CHO-
Ge (CeH3)s
(C6H3)2SnH2-
- Ge (С6Н5)Я
C2H5Sn-Sn (C6H5)2 H
I
Ge (C6H3)3
Ge (C6H5)3
Ge (СбН5)з
I I
■ C2H5Sn—Sn (C6H5)2—Sn (C6H5)2—Sn—C2H5
Ge(CeH6)s
Ge (C6H5)3
При реакции имидазолов или ацетатов триалкилсвинца вместо гидростанно-
лиза происходит обмен группами между атомами олова и свинца. Эта
реакция рассмотрена на стр. 571.
Постепенное прибавление тетра(диметиламино)титана (4 моля) к гидриду
трифенилолова (1 моль) приводит к смолообразному продукту, из которого
выделен тетра(трифенилстаннил)титан. Гидрид трибутилолова в этих
условиях превращается в гексабутилдистаннан.
При обработке 1 моля тетра(диэтиламино)титана 2 молями гидрида
трифенилолова в вакууме при 80° С образуется [(C6H5)3Sn]2TiN(C2H5)2.
Аналогично получен ряд соединений со связью Sn—Zr. Гидрид
трифенилолова прибавляют к тетра(диэтиламино)цирконию„ при 60° С в вакууме.
В зависимости от молярного соотношения реагентов образуются соединения,
в которых атом циркония связан с одним, двумя, тремя или четырьмя три-
фенилстаннильными группами [108а]; например:
Zr[N(C2H5)2]4 + 4(CG4,)3SnH
Zr[N (С21 Ь);]4 + 2 (С6Т Ь)3 SnI I
[(C6H;))3Sn]4Zr + 4(C,H-J)2N;I,
[(QI I5)3 Sn]2 Zr[N(C2H5)2]2 + 2 (C2H,)2 NH.
Обработка этих веществ N-фенилформамидом, а затем дигидридом дифенил-
олова приводит к соединениям более сложного состава [108а]:
2[(С6Ч3)3 SnZ]3ZrN (С6Н5) СНО + (С6НГ))2 SnH2 -»
- [(С6Н3)з Sn]3 ZrSn (C6H,)2 Zr [Sn (С0Н5)3]з + 2 HN (C6' I,) CHO,
п [(C8H5)3Sn]3 Zr [N (C6H3) CHOJa -|- n (C6H5)2 SnII2-*
Sn(C6H5)3
I
Zr—Sn (C6H3)2-
L Sn (C6H3)3
2^IIN(C3H,)CHO.
Если исходить из оловоорганических гидридов алифатического ряда,
то выделить индивидуальные соединения со связью Sn—Zr не удается.
Гидриды триалкил- и трифенилолова обладают различной активностью
и по отношению к сурьма- и висмуторганическим соединениям. С гидридами
триалкилолова реакция проходит только при длительном нагревании смеси
(10 час. и более) до 100—170° С [120]:
3 (C2H,)3SnH -}- (С2Н.,)3М _» 3 С2Н6 + [(C2H3)3Sn]3 М
(M=Bi, Sb).
Гидрид трифенилолова реагирует в более мягких условиях [108а]. Так,
при нагревании до 65° С 1 моля триэтилвисмута с 3 молями гидрида трифенил-
490
Оловоорганические соединения. Лит. стр. 492—494
олова наблюдается энергичное выделение газа и через 1V2 часа реакция
заканчивается:
(С2Н5)зВ1 + 3 (СвН5)3 SnH -^ [(С6Н5)8 Sn]3Bi + 3 С2Нб.
Триэтилсурьма образует с гидридом трифенилолова (молярное отношение
реагентов 1 : 3) три(трифенилстаннил)сурьму. Это белое кристаллическое
вещество с т. пл. 153° С (с разложением).
Соединения со связью Sn—Fe получены обработкой комплекса бис-
(диэтиламино)железа с 2,2'-бипиридилом гидридом трифенилолова [108а].
Соединения, содержащие в молекуле два или более атомов различных
металлов, получены и другими методами (см. стр. 502).
Дифенилхлорфосфин гладко восстанавливается гидридом трифенилолова
[120а].
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С СОЕДИНЕНИЯМИ РАДИКАЛООБРАЗУЮЩЕГО ТИПА
Как уже неоднократно отмечалось ранее, на реакции гидростаннирова-
ния и восстановления существенное влияние оказывают катализаторы
радикалообразующего типа. С целью выяснения механизма реакций изучено
взаимодействие оловоорганических гидридов с соединениями этого типа.
Разложение перекиси бензоила гидридом триэтилолова при 40° С
проходит очень быстро и зависит только от скорости смешения реагентов.
Бензоилоксирадикал, лишь только образовавшись, сразу же реагирует с
гидридом триэтилолова [47,121] .Образующаяся кислота дает с другой молекулой
гидрида водород и бензоат триэтилолова. Лишь небольшая часть бензо-
ильных радикалов разлагается обычным образом до двуокиси углерода
и фенильных радикалов. Сходным образом ведет себя перекись диацетила
[47, 121]:
H3C003nR3
+ И/СОСУ
\
Н3С—COO—SnR3
+ H3COO'j
\
0-0
соз + сп
R3SnH
■СНа-С
\
V г/
О О
С-СНз
RjSnH
R3Sn+Cn3COOH
RaSn'+CH*
RaSnH+HaCCOOH
R3SnOOCCH3 + H2
Примерно 1,5 г перекиси диацетила, будучи смешаны с двойным
молярным количеством гидрида триэтилолова, взрывает при 25° С (сама перекись
устойчива до 80° С) [47]. Больше 70% образующихся короткоживущих
радикалов поглощается оловоорганическим гидридом. Остальная часть
разлагается до двуокиси углерода и метильных радикалов, которые затем
превращаются в метан. Взаимодействие гидрида трифенилолова с перекисью
бензоила приводит к со,со-дибензоату тетрафенилдистаннана [11, 69]. По
другим данным [27] эта реакция проходит более сложно с образованием
бензоата трифенилолова, оз,(о-дибензоата тетрафенилдистаннана и
небольшого количества полимерного продукта.
(о,оз-Дибензоат тетрафенилдистаннана образуется и при реакции
перекиси бензоила с дигидридом дифенилолова [55]. В зависимости от молярного
Синтез и реакции оловоорганшеских гидридов
491
соотношения реагентов реакция дигидрида дибутилолова с перекисью бен-
зоила может проходить в двух направлениях [59]:
(С4Н9)2 SnH2 -Ь (СвН5СОО)2 -> Н2 + (QH9)2 Sn (ООСС6Ч,)2,
2 (QH9)2 SnH3 + (СбН5СОО)2 -> 2 Н2 + СвН5СООЗп (С4Н9)2 Sn (C4H9)2 ООССвЧ5.
В обоих случаях выходы продуктов близки к количественным.
Таким образом, ацильные перекиси не могут применяться в качестве
катализаторов гидростаннирования [74] (см. стр. 274).
Азодиизобутиронитрил и динитрил азоциклогексилкарбоновой кислоты
разлагаются сами по себе с такой же скоростью, как и в присутствии
гидридов триалкилолова [47, 121]. Общая схема реакции выражается
следующим образом:
R3SnCN -f смола ~]
Н
+ Na +
CN
R3Sn—N=
CN
Сходным образом ведет себя азодиизобутиронитрил, который чаще всего
применяется как катализатор различных реакций оловоорганических
гидридов.
Разложение нитрила фенилазоизомасляной кислоты проходит медленно
даже при 100°С. Поэтому с гидридами триал килолова в этом случае
проходит только гидростаннирование азогруппы. Образующийся продукт очень
быстро превращается в фенилгидразон ацетона [47, 121]:
C6H5N=NC (СН3)а С\ + R3SnH -» C3H5NHN(3nR3)C(CH3)2 CN -*
-» R3SnCN + C0H5NHN=C (CH3)2.
Ион тропилия легко отрывает водород от гидрида триэтилолова,
превращаясь в циклогептатриен [122]. Трифенилметильные радикалы реагируют с
гидридом триэтилолова с кратковременным ярко-красным окрашиванием
реакционной смеси и постепенным образованием гексаэтилдистаннана и
трифенилметана [59].
РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ ГИДРИДОВ
С ДИАЗОСОЕДИНЕНИЯМИ
Как показано на стр, 486, оловоорганические соединения с
функциональными группами у а-углеродного атома могут быть получены взаимодействием
оловоорганических гидридов с эфирами меркур-бм>уксусной кислоты.
Другой способ синтеза этих соединений заключается во взаимодействии олово-
органических гидридов с диазосоединениями:
RgSnI-I + N2CHCOOC2H3 -> R,SnCH2COOC2H5 -f N2,
RoSnII + CH3COCHN2 -> R3$nCH2COCH3 + N2,
R3SnH + N2CHCN -* R3SnCH2CN2+N2.
Эти реакции рассмотрены на стр. 323.
492
Оловоорганические соединения
ЛИТЕРАТУРА
1. KrausC.A., Greer W.N., J. Am. Chem. Soc, 44, 2629 (1922).
2. V a n d e г К e г к G. J. M., L u i j t e n J. G. A., N о 1 t e s J. G., Chem. a. Ind.,
1956, 352.
3. С h a m b e r s R. F., SchererP.C, J. Am Chem. Soc, 48, 1054 (1926).
4. Bullard R. H., V i n g e e R. A., J. Am. Chem. Soc, 51, 892 (1929).
5. W i t t i g G., M e у e e F. J., L a n g e G., Ann., 571, 167 (1951).
6. В a u m G. А., С о n s i d i n e W. J., J. Org. Chem., 29, 1267 (1964).
7. T a m b о r s к i C, F о r d F., S о 1 о s к i E., J. Org. Chem., 28, 237 (1963).
8. Finholt A. E., BondA.C, W i 1 z b а с h K. E., Schlesinger H. J.,
J. Am. Chem. Soc, 69, 2692 (1947).
9. Finholt A. E., Bond A.C., Schlesi nger H. I., J. Am. Chem. Soc, 69,
1199 (1947).
10. Lide D., J. Chem. Phys., 19, 1605 (1951).
11. GilmanH., Eisch J., J. Org. Chem., 20, 763 (1955).
12. V a n d e г К e г к G. J. M., No 1 t e s J. G., L u i j t e n J. G. A., J. appl. Chem.,
7, 366 (1957).
13. LesbreM., Buisson R., Bull. Soc. chim. France, 1957, 1204.
14. A n d e r s о n H. H., J. Am. Chem. Soc, 79, 4913 (1957).
15. D i 1 1 a r d С R., McNeill E.H., SimraonsD.E, V e 1 d e 1 1 J. D., J.
Am. Chem. Soc, 80, 3607 (1958).
16. Clark И. C, Furnival S. G., К w о n J. Т., Canad. J. Chem., 41, 2889 (1963).
17. А д р о в а Н. А., К о т о н М. М., К л а г е с В. А., Высокомолек. соед. 3, 1041
(1961).
18. НепгуМ.С, N о 1 t e s J. G., J. Am. Chem., Soc, 82, 558 (I960).
19. К е t t 1 е S. F. A., J. Chem. Soc, 1959, 2936.
20. SawyerA.K, Kuivila H. G., J. Org. Chem., 27, 610 (1962).
21. A 1 1 es t on D. L., D a v i e s A. G. , J. Chem. Soc, 1962, 2465.
22. N e u m a n n W. P., К б n i g K-, Angew. Chem., 76, 892 (1964).
23. M a t h i s-N о ё 1 R., L e s b r e M., d e R о с h I. S., Compt. rend., 243, 257 (1956).
24. F 1 i t с г о f t N., К a e s z H. D., J. Am. Chem. Soc, 85, 1377 (1963).
25. В r i n с k m a n F. E., S t о n e F. G. A., J. Inorg. Nucl., Chem., 11, 24 (1959).
26. M a d d о x M. L., Flitcroft M., К a e s z H. D., J. Organometal. Chem., 4,
50 (1965).
27. D e 1 Franco G. J., Re s n i с k P., D i 1 1 a r d C. R., J. Organometal. Chem.,
4, 57 (1965).
28. К r a m e r K. A. W., W r i g h t A. N., J. Chem., Soc, 1963, 3604.
29. P о 1 1 a r d F. H., N i с k 1 e s s G., С о о k e D. J., J. Chromatogr., 17, 472 (1965).
30. N e u m a n n W. P., N i e r m a n n H., S о m m e r R., Ann., 659, 27 (1962).
31. Leusink A. J., Noltes J. G., Budding H. A., van der Kerk G. J.M.,
Rec trav. chim., 83, 609 (1964).
32. С о n s i d i n e W. J., V e n t u r a J. J., Chem. a. Ind., 1962, 1683.
33. S t e r n А., В е с k e r E. I., J. Org. Chem., 29, 3221 (1964).
34. Lorenz D. H., S h a p i г о P., S t e r n А., В е с k e r E. I., J. Org. Chem.,
28, 2332 (1963).
35. К u i v i 1 a H. G., SawyerA.K, А г m о u г A. G.f J. Org. Chem., 26, 1426
(1961).
36. Neumann W. P., Angew. Chem., 73, 542 (1961).
37. N e u m a n n W. P., NiermannH, Ann., 653, 164 (1962).
38. J enkner H., Пат. ФРГ 1117577 (1962); РДХим., 1963, 13H89.
39. К u 1 a M. R., L о r b e r t h J., A m b e r g e r E., Ber., 97, 2087 (1964).
40. N e u m a n n W. P., SommerR, Angew. Chem., 75, 788 (1963).
41. N e u m a n n W. P., К 6 n i g K-, Ann., 677, 12 (1964).
42. V a n der К e r k G. J. M., N о 1 t e s J. G., L u i j t e n J. G. A., Chem. a. Ind.
1958, 1290.
43. Am b er ger E., К u 1 a M. R., Ber., 96, 2560 (1963).
44. Ohara M., О k a w a r a R., J. Organometal. Chem., 3, 484 (1965).
45. E m e 1 e u s H. I., К e t t 1 e S. F., J. Chem. Soc, 1958, 2444.
46. A m b e r g e r E., F r i t z H. P., KreiterCG., KulaM.R, Ber., 96, 3270
(1963).
47. N e u m a n n W. P., Angew. Chem., 76, 849 (1964).
48. N e u m a n n W. P., P e d a i n J., Tetrahedron L., 1964, 2461.
49. N e u m a n n W. P., S с h n e i d e r B. Angew. Chem., 76, 891 (1964)
50. Sawyer A. K., Kuivila H.G., Chem. a. Ind., 1961, 260.
51. S a w у e г А. К., В г о w n J. E. J. Organometal. Chem., 5, 438 (1966).
52. G i b b о n s A. J., S a w у e r A. K-, R о s s . A., J. Org. Chem., 26, 2304 (1961).
53. Sawyer A. K., Brown J.E., Hanson E.L, J. Organometal. Chem., 3, 464
(1965).
Синтез и реакции олсвоорганических гидридов
493
54. S a w у е г А. К., К u i v i 1 а Н. G., J. Org. Chem., 27, 837 (1962).
55. S a w у е г А. К., К и i v i 1 a H. G., J. Am. Chem. Soc, 82, 5958 (I960).
56. S a w у е г А. К., К и i v i 1 a H. G., J. Am. Chem., Soc, 85, 1010 (1963).
57. V a n d e r KerkG.J.M., NoltesJ.G., J. appl. Chem., 9, 106 (1959).
58. К и i v i 1 a H. G., Advances in Organometallic Chemistry, v. 1. Academie Press,
New York — London, 1964, p. 47.
59. В я з а н к и н Н. С, БычковВ. Т., ЖОХ, 35, 684 (1965).
60. Борисов А. Е., Абрамова А. Н., Изв. АН СССР, ОХН, 1964, 844.
61. Kupchik E.J., LaniganT., J. Org. Chem , 27, 3661 (1962).
62. Б о р и с о в А. Е., А б р а м о в а А. Н., П а р н е с 3. Н., Изв. АН СССР, ОХН,
1964, 941.
63. Fritz G., Scheer H., Z. Naturforsch., 19b, 537 (1964).
бза. Вязанкин Н. С, Бочка рев М. Н., Санина Л. П., ЖОХ, 36, 166(1966).
бзб. F r i t z G., S с h e e r H. , Z. anorgan. allgem. Chem., 338, 1 (1965).
64. P a n g M., В е с k e r E. I., J. Org. Chem., 29, 1948 (1964).
65. S h a p i г о P. J., В е с k e r E. I., J. Org. Chem., 27, 4668 (1962).
66. T s a i Т. Т., С u t 1 e r A., LehnW.L, J. Org. Chem., 30, 3049 (1965).
67. Kuivila H. G., Levins P. L, J. Am. Chem. Soc, 86, 23 (1964).
68. D e s s у R. E., H i e b e г Т., PaulicF., J. Am. Chem. Soc, 86, 28 (1964).
69. W e b e r S., В е с k e r E. I., J. Org. Chem., 27, 1258 (1962).
70. Sawyer A. K-, К u i v i 1 a H. G., Пат. США 3083217 (1963); РЖХим., 1965
3H102.
71. NoltesJ.G., van d e г К a r k G. J. M., Chem. a. Ind., 1959, 294.
72. Kuivila H. G., M e n a p а с e L. W., J. Org. Chem., 28, 2165 (1963).
73. R о t h m a n L. А., В е с k e r E. I., J. Org. Chem., 24, 294 (1959).
74. R о t h m a n L. А., В е с k e r E. I., J. Org. Chem., 25, 2203 (1960).
75. Seyferth D., YamazakiH., Alleston D. L, J. Org. Chem., 28, 703
(1963).
76. LorenzD., BeckerE.L, J. Org. Chem., 27, 3370 (1962).
77. Kriegmann H., U 1 b r i с h t K., Z. Chem., 3, 67 (1963).
78. К u i v i 1 a H. G., M e n a p а с e L. W., WarnerCR., J. Am. Chem. Soc,
84, 3584 (1962).
79. KupchikE.J., KieselR.J., J. Org. Chem., 29, 764 (1964).
80. Kupchik E.J., Kiesel R. J., Chem. a. Ind., 1962, 1654.
81. KupchikE.J., С о n n о 1 у R. E., J. Org. Chem., 26, 4747 (1961).
82. G r i e s b a u m K., J. Am. Chem. Soc, 86, 2301 (1964).
83. К r i e g s m a n n H., U 1 b r i с h 1 K-, Z. anorgan. allgem. Chem., 328, 90 (1964).
84. M e n a p а с e L. W., Kuivila H. G., J. Am. Chem. Soc, 86, 3047 (1964).
85. К u i v i 1 a H.G., Beumel O.F., J. Am. Chem. Soc, 83, 1246 (1961).
86. V a 1 a d e J., PereyreM, С a 1 a s R., Compt. rend., 253, 1216 (1961).
87. Kuivila H.G., BeumelO. F., J. Am. Chem. Soc, 80, 3798 (1958).
88. Smolin E. M., Tetrahedron L., 1961, 143.
89. Sawyer A. K-, J.Am. Chem. Soc, 87, 537 (1965).
90. Kuivila H. G., J.Org. Chem., 25, 284 (1960).
90a. Kupchik E. J., Kiesel R. J., J. Org. Chem., 31,456(1966).
91. К u i v i 1 a H. G., W a 1 s с h E. J., J. Am. Chem. Soc, 88, 571 (1966).
92. W a 1 s с h E. J., Kuivila H.G., J. Am. Chem. Soc, 88, 576 (1966).
93. Van d e г К e r k G. J. M., N о 1 t e s J. G., L u i j t e n J. G. A., J. appl. Chem.,
7, 356 (1957).
94. KupchikE. I., Kiesel R., J. Org. Chem., 29, 3690 (1964).
95. В u r g А. В., S p i e 1 m a n J. R., J. Am. Chem. Soc, 82, 2667 (1961).
96. S t e r n А., В е с k e r E. I., J. Org. Chem., 27, 4052 (1962).
97. V a n d e r KerkG.J.M., NoltesJ.G., L u i j t e n J. G. A., Rec trav.
chim., 81, 853 (1962).
98. N e u m a n n W. P., К о n i g K., Angew. Chem., 74, 215 (1962).
99. Neumann W. P., P e d a i n J., S о m m e r R., Ann., 694, 9 (1966).
100. N e u m a n n W. P., KonigK, Ann., 677, 1 (1964).
101. N e u m a n n W. P., P e d a i n J., Ann., 672, 34 (1964).
102. Kuivila H.G., BeumelO., J. Am. Chem. Soc, 80, 3250 (1958).
103. В г о w n T. L., M о r g a n G. L., Inorg. Chem., 2, 736 (1963).
104. N e u m a n n W. P., S с h n e i d e г В., S о m m e r R., Ann., 692, 1 (1966).
105. S о m m e r R., S с h n e i d e г В., N e u m a n n W. P., Ann., 692, 12 (1966).
106. SommerR., Neumann W. P., Schneider В., Tetrahedron L., 1964, 3875.
107. Creemers H. M. J. C, NoltesJ.G., Rec. trav. chim., 84, 382 (1965).
108. CreemersH.M. J.C., NoltesJ.G., van d e г К e r k G. J. M., Rec.
trav. chim., 83, 1284 (1964).
108a. Creemers H. M. J.C., Hydrostannolysis. A general method for establishing tin-
metal bonds. Utrecht (Nederland). Schotanus and Jens Utrecht. N. Y., 1967.
109. Вязанкин Н. С, Р а з у в а е в Г. А., К о р н е в а СП., ЖОХ, 33, 1041 (1963).
ПО. Вязанкин Н. С, Р а з у в а е в Г. А., К о р н е в а С. П., ЖОХ, 33, 1046 (1963).
494
Олсвоорганические соединения
111. Вязанкин Н. С, Разуваев Г. А., Корнева С. П., ЖОХ, 34, 2787 (1964).
Ilia. Creemers H. M. J. С, N о 1 t es J. G., Rec. trav. chim., 84, 1589 (1965).
1116. Creemers H. M. J. C, V e r b с e k F., N о 1 t e s J. G., J. Organometal. Chem.,
8, 469 (1967).
112. G i 1 m a n H., M e 1 v i n H. W., J. Am. Chem. Soc, .71, 4050 (1949).
113. Вязанкин H.C., Разуваев Г. А., К о р н е в а С. П., Круглая О. А.,
Г а л и у л и н а Р. Ф., ДАН СССР, 158, 884 (1964).
113а. Tombe F. J. A. des, van der KerkG. J. M., Creemers H. M. J. C,
Noltes J.G., Chem. Commun., 1966, 914.
114. S e у f e r t D., SimmonsH.D, T о d d L. J., J. Organometal. Chem., 2, 282
(1964).
115. ЛуценкоИ.Ф, Б а у к о в Ю. И., X а с а н о в Б. Н., ЖОХ, 33, 2724 (1963).
116. П о н о м а р е в С. В., Б а у к о в Ю. И., ЛуценкоИ.Ф., ЖОХ, 34, 1938
(1964).
117. Б а у ков Ю. И., Б е л а в и н И. Ю., Л у ц е н к о И. Ф., ЖОХ, 35, 1121 (1965).
118. Neumann W.P., Tin and its uses, 61, 5 (1964).
119. Neumann W. P., NiermannH., Schneider В., Angew. Chem., 75,
790 (1963).
119a. CreemersH.M.J.C, NoltesJ.G., J. Organometal. Chem., 7, 237 (1967).
120. Вязанкин Н. С, Круглая О. А., РазуваевГ. А., Семчикова
Г. С, ДАН СССР, 166, 99 (1966).
120а. Seyferth D., Sato Y., Takamizawa M., J. Organometal. Chem., 2, 367
(1964).
121. Neumann W. P., SommerR., Lind H., Angcw. Chem., 76, 5S7 (1964).
122. ParnersZ.N, VolpinM. E., Kursanov D.N., Tetrahedron L., 196k
20.
Г[лава XVI
СИНТЕЗ И РЕАКЦИИ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
СОДЕРЖАЩИХ ДВА И БОЛЕЕ АТОМОВ МЕТАЛЛА В МОЛЕКУЛЕ
Среди различных классов оловоорганических соединений важное место
занимают вещества, содержащие в молекуле два или более атомов
различных металлов. Последние могут быть или непосредственно связаны между
собой, или разделены углеводородными радикалами.
В настоящей главе рассматриваются только соединения со связью Sn —
М, где М — металл, иной чем олово. Наиболее подробно изучены
соответствующие производные лития и натрия типа R3SnM и R2SnM2. Они легко
вступают в реакции типа реакции Вюрца с галоидными алкилами, а также
галогенидами бора, кремния, германия, олова и свинца. Этим методом
получены, например, станнилсиланы RgSnSiRg и R3SnSiR2SnR3, станнилгерма-
ны RgSnGeRJ, станнилплюмбаны Pb(SnR3)4, а также оловоорганические арси-
ны, стибины и фосфины. Последние могут быть получены и другими
методами.
Известны соединения, в которых атом олова связан с металлами второй
группы периодической системы (см. стр. 484).
Значительный интерес представляют оловоорганические соединения, в
которых атом олова связан с карбонилами металлов VI—VIII групп
периодической системы. Систематические исследования в этой области проведены
Несмеяновым с сотрудниками. Обращает на себя внимание устойчивость
связи Sn — Mb этих веществах по отношению к электрофильным реагентам.
Например, при действии хлора или хлористого водорода на трифенилоло-
вопентакарбонилрений отщепляется фенильная группа, тогда как связь
Sn—Re остается не затронутой.
Что касается оловоорганических соединений, в которых два атома
металла разделены между собой углеводородными радикалами, то их получают
или реакцией Гриньяра (см. стр. 224), или присоединением
оловоорганических гидридов к винильным соединениям металлов (см. стр. 286).
Несмеяновым с сотрудниками получен ряд биметаллических
производных этилена по реакции:
R3SnHj+|RnMC = СН -» R3SnCI I=CtIMR„
(M=JSi, Ge, Sn, Sb)-
Синтез и реакции этого нового класса веществ рассмотрены на стр. 288.
СИНТЕЗ СОЕДИНЕНИЙ ТИПА R3SnM и R2SnM2
(где М — щелочной металл)^
Соединения указанного типа обычно получают действием щелочных
металлов на оловоорганические галогениды или реакцией двухлористого олова с
тремя эквивалентами литийорганического соединения.
496 Оловоорганические соединения. Лит. стр. 510—511
В большинстве случаев образующиеся вещества не выделяют в
индивидуальном состоянии, а используют в виде растворов. Для анализа
растворов триалкилстанниллитиевых соединений предложен [1] метод двойного
титрования с использованием бромистого аллила.
При действии лития на растворы оловоорганических хлоридов
образуются триалкил- или триарилстанниллитиевые соединения. Реакция проходит
ступенчато с образованием в качестве промежуточных продуктов гексаал-
кил- или гексаарилдистаннанов соответственно [2—8]:
RsSnCl + 2 Li -> R3SnLi + LiCl,
R3SnLi + R3SnCl -> R3SnSnR3 + LiCI,
R3SnSnR3 + 2 Li -» 2R3SnLi.
Растворитель играет существенную роль в этих реакциях. Например,
получить триалкилстанниллитиевые соединения в эфире не удается [4],
но в тетрагидрофуране синтез проходит легко, как это можно видеть из
следующего примера.
Получение триметилстанниллития [4]. К суспензии 13,9 г (2г-тпома) лития в 150 мл
тетрагидрофурана прибавляют по каплям 39,8 г (0,2 1уоля) хлористого триметилолова в
150 мл того же растворителя при температуре около 5° С. Реакция проходит с выделением
тепла, раствор окрашивается в темно-зеленый цвет. Для завершения реакции смесь
перемешивают в течение 3 час. и отфильтровывают.
При перемешивании 0,2 моля хлористого трибутилолова и 2,0 г-атома
лития в течение часа трибутилстанниллитий не образуется. Однако при
прибавлении 100 мл тетрагидрофурана смесь разогревается и окрашивается в
темно-зеленый цвет. Реакция заканчивается после перемешивания смеси в
течение 2 час.
Аналогично проводят реакцию и в ароматическом ряду.
Получение трифенилстанниллития [5]. 6,15 г (0,016 моля) хлористого трифенилолова
в 17 мл сухого тетрагидрофурана при перемешивании прибавляют по каплям к суспензии
0,62 г (0,08 г-апюма) лития в 16 мл тетрагидрофурана. Реакционная смесь слегка
разогревается, выпадает осадок белого цвета. Проба Гилмана отрицательна. Через 30 мин. осадок
исчезает, и после 3-часового перемешивания образуется раствор темно-зеленого цвета,
который дает положительную пробу Гилмана. Раствор отфильтровывают через стеклянную
вату.
Так же получен метилдифенилстанниллитий [9].
Как уже отмечено выше, промежуточными продуктами этих реакций
являются гексаалкил- или гексаарилдистаннаны. Естественно, можно
исходить непосредственно из этих веществ и действием на них лития получать
литийоловоорганические соединения. Так получены, например, три-я-
бутил-[4] итрифенилстанниллитий [3, 8]. В качестве растворителя
применяют тетрагидрофуран.
Кратко сообщается [10] о синтезе бис- (трифенилстаннил)магния из
хлористого трифенилолова и магниевой стружки в тетрагидрофуране. Реакция
проходит при обычной температуре в присутствии каталитических количеств
бромистого этила. Образующийся раствор серовато-зеленого цвета дает
положительную пробу Гилмана:
2 (С6Н5)з SnCl + Mg — ^ (С6Н5)з SnSn (CGH5)3 + MgCl2,
C2H5Br
(C6II5)3SnSn (CeH3)8 + Mg > [(C6H5)3Sn]2Mg.
Двухлористое дифенилолово образует с четырьмя эквивалентами лития в
тетрагидрофуране дифенилстаннилендилитий [11]:
(С6Н5)2 SnCi2 + 4 Li -* (C6H5J2 SnLi2 + 2 LiCl.
Оловоорганические соединения с двумя и более атомами металла 497
Синтез натрийоловоорганических соединений проводят в среде жидкого
аммиака [12—22].
Краусом с сотрудниками, в частности Булларом, а также Шерером
установлено, что галоидные триалкилы и триарилы олова при действии на их
раствор в жидком аммиаке металлического натрия дают окрашенные в
желтые или красные тона растворы триалкил-(соответственно, триарил)стан-
нилнатрия RgSnNa, электролитически диссоциирующие на Na"1" и анион
R3Srf. (Об измерении электропроводности растворов
триметилстаннилнатрия в жидком аммиаке см. работы Крауса с сотр. [23, 24].) Те же вещества
образуются и при действии натрия на гексаалкил- или гексаарилдистаннаны,
которые в предыдущей реакции являются промежуточными веществами.
Оловоорганические дигалогениды в тех же условиях вначале
превращаются в соединения типа (R2Sn)n. При действии на них натрия
образуются динатрийтетраалкил(или арил)дистаннаны, а затем динатрий-
диалкильные (соответственно арильные) соединения олова:
R2SnCl2 + 2 Na -> (R2Sn)n + 2 NaCl,
2 (Я25п)п + 2 Na -> NaSnR2SnR2Na,
NaSnR2SnR2Na + 2 Na -* 2 R2SnNa2.
Оба этих типа натрийоловоорганических соединений также ионизированы,
окрашены в желто-красные тона и растворимы в аммиаке.
Примером реакций этого типа может служить образование динатрийтет-
раметилдистаннана и диметилстаннилендинатрия при постепенном
прибавлении натрия к раствору двубромистого диметилолова в жидком аммиаке.
При малом количестве введенного натрия образуется желтый осадок,
количество которого возрастает до достижения соотношения Ша: 1Вг. Осадок
представляет собой поли(диметилолово) — [(CH3)2Sn]rt. При дальнейшем
введении натрия проходит обратимая реакция образования
диметилстаннилендинатрия. Это соединение легко растворяется в жидком аммиаке с
образованием интенсивно красного раствора, мутного при высоких
концентрациях. По виду определить конец реакции образования этого вещества
нельзя, поэтому, по прибавлении всего необходимого количества натрия,
приходится продолжать перемешивание в течение часа или больше.
Позже динатрийтетраметилдистаннан и диметилстаннилендинатрий
получены [25] при кондуктометрическом титровании раствора натрия в жидком
аммиаке дигидридом диметилолова. Однако, по данным Кэтла, их растворы
в жидком аммиаке окрашены в зеленовато-лимонные тона. По-видимому,
наблюдаемый прежде красный цвет этих растворов обусловлен образованием
аминодиметилстаннилнатрия [25]. Его получают также быстрым
прибавлением дигидрида диметилолова к натрию в жидком аммиаке при — 45°С
[25]:
2 (CH3)2SnH2 + 2 Na + 2 NH3 -> 2 (CH3)2Sn (NH2)Na + 3H2.
Вещество может быть выделено в чистом виде при нагревании растворов
диметилстаннилендинатрия до комнатной температуры.
Другой метод синтеза триалкил- и триарилстанниллитиевых соединений
заключается во взаимодействии тонко измельченного хлористого олова с
тремя молями литийорганических соединений в эфире при — 10° С [22,
26—31]:
SnCl2 + 3 RLi -> R3SnLi + 2 LiCl.
Реакцию можно иллюстрировать следующими примерами:
Получение триэтилстанниллития [27]. К охлажденной до —10° С суспензии 8,54 г (0,045
моля) тонко измельченного хлористого олова в 100 мл эфира прибавляют по каплям 0,135
32 Заказ X» 207
498 Оловоорганические соединения. Лит. стр. 510—511
моля этиллития в \62 мл эфира. После прибавления первого эквивалента этиллития смесь
окрашивается в темно-оранжевый цвет, двух эквивалентов — в темно-красный и трех
эквивалентов— в коричневый. Проба Гилмана I отрицательна, пока не добавлено два
эквивалента этиллития. Проба Гилмана II остается отрицательной до добавления трех
эквивалентов этиллития.
Аналогично получены трибутил-[27], трифенил-[26, 28—30] и изомеры
тритолилстанниллития [29].
Промежуточными продуктами этих реакций являются соединения типа
(R2Sn)n. Взаимодействием дифенилолова с эквимолекулярным количеством
фениллития Виттиг с сотр. [32, 33] получили и выделили в кристаллическом
состоянии трифенилстанниллитий:
(CeII-,)2Sn -f Cc/I,Li -> (C6M5)3SnLi.
Получение трифенилстанниллития [33]. К раствору 2,7 г (0,01 моля) дифенилолова в
5 мл бензола приливают 12,2 мл 0,82 N раствора фениллития, не содержащего бромистого
лития. После смешения реагентов проба Гилмана положительна. Реакция проходит с
незначительным разогреванием, цвет раствора изменяется от темно-красного до коричневого.
Раствор оставляют на 4 дня при комнатной температуре и декантируют с выделившихся
кристаллов трифенилстанниллития. Вещество промывают смесью эфира с бензолом
и почти бесцветные теперь кристаллы высушивают при 60° С в течение 2 час. Образуется
порошок светло-желтого цвета, представляющий собой комплекс трифенилстанниллития с
1 молем эфира.
Кратко сообщается (без деталей эксперимента) о присоединении натрий-
ацетиленидов [34] и диацетиленна'грия [35] к диэтилолову в жидком
аммиаке:
(QsH5)3Sn + NaC=CR-- (ColI-).:Sn(C=CR)Na.
Строение образующихся продуктов доказано обработкой их бромистым
этилом. Выделены и идентифицированы соединения типа (C2H5)3SnC^CR и
(C2H5)3SnC =C-Czl CSn(C2H5)3.
К образованию натрий-и литииоловоорганическнх соединений приводят
и другие реакции. Тетраметкл- [14, 15] и тетраэтилологю [ 181 реагируют с
растворами натрия в жидком аммиаке с образованием соединений типа R3SnNa:
R4Sn -f 2 Na ~ NHo -> R3SnNa -\- NaNII2 + RII.
Триметилстаниллитий образуется и при действии двух эквивалентов
лития на раствор /г-хлорбензилтриметилолова в тетрагидрофуране [36]:
n-ClC6H4CHoSn (СН3)3 + 2 Li — »-CICGH4CH2Li -.- (C:I3),/;nLi.
Что касается тетрафенилолова, то при действии на его раствор в жидком
аммиаке натрия можно провести ступенчатое отщепление одного или двух
ароматических радикалов [14,15,37]:
(CeHri)4Sn + 2Na-> (C6II5)3SnNa + C6'I.,\a,
(QI I5)3 SnNa + 2 Na -> (CGT I5)2 SnNa-, -f G0? l.-,Xa,
C6H5Na -!- NIIP -» CGITG + NaNI io.
Трифенилстаннилнатрий получен также взаимодействием нафталиппат-
рия с гексафенилдистаннаном [20], бромистым [20] и хлористым [38] три-
фенилоловом или тетрафенилоловом:
(C6II5)3SnSn (CeH5)3-f 2C10H8Na^ 2(Cerb):jSnNa -;- 2Q,! Is,
(C6H5)3 SnX -f- 2Ci0H8Na ^ (CGH5)3 SnNa -{ Na X 2C10H«,
(C6H5)4 Sn -r Ci3H8Na -> (CeII5)3 SnNa + C64G -f C10' h-
Следует подчеркнуть, что полученные различными методами растворы
литииоловоорганическнх соединений различаются своей устойчивостью
Оловоорганические соединения с двумя и более атомами металла 499
[3, 8, 9, 201 и свойствами. Так, трифенилстанниллитий, полученный из
хлористого трифенилолова и лития в тетрагидрофуране, дает положительную
пробу Гилмана I [4]. Если же трифенилстанниллитий получен из хлористого
олова и фениллития, то проба Гилмана отрицательна [26, 28].
Другим примером различного поведения этих соединений может служить
образование тетрафенилолова при гидролизе или карбонизации трифенил-
станниллития, полученного из хлористого олова и фениллития [28, 29].
С трифенилстанниллитием, полученным из хлористого трифенилолова и
металлического лития, эти реакции проходят иначе [8,20]. Приготовленные
различными методами растворы R3SnLi ведут себя неодинаково и по
отношению к галоидным ал килам и арилам (см. стр. 502). Это различие свойств
литийоловоорганических соединений объяснено [8] тем, что при синтезе
трифенилстанниллития из хлористого олова и фениллития одновременно
проходят следующие реакции:
SnCl2 + 2 QHgLi -» (С6Н,)2 Sn,
(С6Н5).2 Sn -> (СвН3)з SnSn (CeH5)3 -f Sn,
(CeH,h SnSn (С6Н,-,)з -h C6TI5Li ~> (CeH5)8 SnLi + (CGH5)4 Sn.
Строение литий- и натрийоловоорганических соединений почти не
изучено. Высказано предположение [39], что триалкил- и триарилстаннилли-
тиевые соединения представляют собой комплексы, которые в растворах
диссоциируют на составляющие их компоненты:
RaSnLi z± R2Sn+ RLi.
Это предположение основано на следующих данных. При реакции этих
соединений с галоидными алкилами и арилами или галогенидами триалкил-
(или арил)олова наряду с ожидаемыми соединениями типа R3SnR' и
R3SnSnR3 образуются незначительные количества окисей (R2SnO)„ и тетра-
алкильных (соответственно арильных) соединении олова [26, 27, 29, 33].
Бромированием трифенилстанниллития при — 25°С с количественным
выходом выделено двубромистое дифенилолово [29]. Указания на
комплексную природу этих соединений содержатся в работах и других авторов [20,
32, 33]. Однако против предположения о диссоциации
трифенилстанниллития по приведенной выше схеме имеются серьезные возражения [3,28].
Так, карбонизация этого вещества не приводит к образованию бензойной
кислоты [3,28]. Трифенилстанниллитий не вступает и в другие реакции,
характерные для фениллития.
Аналогичным образом при реакциях трифенилстаннилнатрия с
углекислым газом, двуокисью серы, бензофеноном и хлористым бензоилом
образуется лишь гексафенилдистаннан [201.
РЕАКЦИИ ЛИТИЙ- И НАТРИЙОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С КИСЛОРОДОМ, СЕРОЙ, СЕЛЕНОМ И ТЕЛЛУРОМ
Окисление литий-и найтрийоловоорганических соединений почти не
изучено. Высказано предположение, что при окислении трифенилстаннилнатрия
образуется трифенилстаннолят натрия, который затем гидролизуется до
гидроокиси трифенилолова [15, 40]. По данным других авторов [20], три-
фенилстаннилнатрий превращается кислородом в гексафенилдистаннан.
При прибавлении к раствору трифенилстанниллития в
тетрагидрофуране эквимолекулярного количества серы сразу же происходит
экзотермическая реакция и смесь окрашивается в красно-коричневый цвет [5,6]. На
дне колбы выделяются бесцветные кристаллы. После 1,5-часового
перемешивания проба Гилмана становится отрицательной. Выделившийся осадок
32*
500
Оловоорганические соединения. Лит. стр. 510—511
почти полностью растворяется при прибавлении к смеси бензола. Раствор
отфильтровывают от небольшого количества осадка и из фильтрата
растворитель удаляют в вакууме. Остаток коричневого цвета промывают сухим
этиловым спиртом. Получают бесцветные кристаллы литийтрифенилстаннил-
сульфида. Вещество не плавится при нагревании до 250°С, не смачивается
холодной водой. Лишь при нагревании с водой до кипения оно разлагается
с образованием сульфида лития и сульфида бис-(трифенилолова). В
бензольном растворе вещество димеризовано.
Реакция образования литийтрифенилстаннилсульфида проходит
ступенчато [5,6]:
(С6Н5)3 SnLi -|- S8 -> (С6Н-,Ь Sn38Li,
7 (CeH5)8 SnLi + (С6Н5)з SnSLi -* 8 (СбН5)8 SnSLi.
Селен и теллур превращают трифенилстанниллитий в (C6H5)3SnSeLi
и (C6H5)3SnTeLi соответственно.
Дифенилстаннил-бис-(литийсульфид) получен [11] действием на дилитий-
дифенилолово двух эквивалентов серы.
Соединения типа (C6H5)3SnXLi (X = S, Se, Те) и (C6H5)2Sn(SLi)2 легко
реагируют с хлористым бензоилом и галогенидами олова, германия и
свинца:
(СбН,з)з SnSLi + CGH5CC)C1 -» (СбН5)з SnSCOCGH5 + LiCl,
(СбН5)з SnXLi + BrGe (CGH5)8 -> (C6H5)8 SnXGe (C6H5)3 + LiBr,
(C6H5)3SnXLi + CISn (CeH5)8-» (CeH5)8SnXSn (C6H5)8 + UCV
(C6H5)3 SnXLi + QPb (СбНГ))з-> (CGH5)3SnXPb (CGH5)3 + LiCl,
(C6H5)2'Sn (SLi)2+ 2CGH5C0C1 -> (C6II5)2 Sn (SCOC6H5)2 + 2 LiCl.
При действии на трифенилстаннилнатрий дифенилсульфида образуются
гексафенилдистаннан, фенилмеркаптид трифенилолова и фенилмеркаптид
натрия [20].
РЕАКЦИИ ЛИТИЙ- И НАТРИЙОЛОВООРГАНИ 1ЕСКИХ СОЕДИНЕНИЙ
С АЛКИЛ- И АРИЛГАЛОГЕНИДАМИ
Соединения типа R3SnM и R.2SnM2, полученные из оловоорганических
галогенидов, легко реагируют с галоидными алкилами и арилами обычным
образом [2—4, 8, 15—17, 19, 20, 27, 41—43]:
R3SnLi + R'X-> R8SnR'-!-LiX.
Так получены, например, триметилэтилолово [4], этилтрифенилолово [3,
20], аллилтрифенилолово [8,42], бензилтрифенилолово [2, 3, 8, 20] и другие
четырехзамещенные оловоорганические соединения. Галоидный алкил или
арил постепенно прибавляют к растворам литий- или натрийоловооргани-
ческих соединений в тетрагидрофуране или жидком аммиаке
соответственно. Выходы продуктов составляют 60—98%.
Отмечено [8], что при реакции трифенилстанниллития с хлористыми
алкилами образуется небольшое количество тетрафенилолова. Выделить
последнее не удается, если хлориды заменить соответствующими бромидами. При
реакции трифенилстанниллития с бромистым пропаргилом образуется
алленовое соединение (выход 30%) [44, 45]:
(CsH5)3SnLi + ВгСН2С=СН -* (СвН5)з SnCH==C=CH2.
Триэтилстаннилнатрий образует с 1-бромбутадиеном-1,3 триэтилстаннил-
винилацетилен [46]. К тому же соединению приводит взаимодействие
Оловоорганические соединения с двумя и более атомами металла 501
триэтилстаннилнатрия с винилацетиленом:
(СгНзЬ SnNa + НС=С—СН=СЬЬ —,
Ц (C2H3)3SnC=C—СН=СН2.
(C2H5)3SnNa + BrCH=CH—CH=CH2 -1
Интересно отметить, что в молекуле монобромацетилена атом водорода
более подвижен, чем атом брома [47]:
R3SnNa R3SnNa
НСееСВг > R3SnC=CBr > R8SnC=CSnR8.
Так получены, например, 1-триметилстаннил-2-бромацетилен (т. кип.
67—68°С/14 мм), 1-триэтилстаннил-2-бромацетилен (т. кип. 63°С/1 мм) и
соответствующие бш>(триалкилстаннил)ацетилены. Полученные
соединения охарактеризованы ИК-спектрами и спектрами ЯМР [47]. Трифенилстан-
нилнатрий гладко реагирует с дииодацетиленом в жидком аммиаке с
образованием бш>(трифенилстаннил)ацетилена [48].
Оловоорганические соединения типа R3SnC^CCR'=CHR'' (Rr=CH3
или С2Н5; R' = Н или СН3) получены [21, 49, 50] с хорошим выходом при
действии триалкилстаннилнатрия на алкенилбромацетилены в жидком
аммиаке. Этот путь особенно удобен для синтеза ениновых оловоуглеводоро-
дов из гомологов винилацетилена.
Реакцию проводят следующим образом. В трехгорлую колбу,
снабженную холодильником капельной роронкой и мешалкой, наливают аммиак
и к нему по каплям прибавляют хлористое триалкилолово. В полученную
молочно-белую суспензию в течение часа вносят небольшими кусочками
металлический натрий (25% избытка). После прибавления около половины от
рассчитанного количества натрия раствор становится прозрачным и
окрашивается в салатный цвет, что указывает на образование гексаалкилдистан-
нана. При введении оставшегося количества натрия раствор приобретает
темно-зеленую окраску и под конец избытком натрия окрашивается в синий
цвет. К полученному раствору триалкилстаннилнатрия прибавляют по
каплям соответствующий алкенилбромацетилен (25% избытка). Реакция
вначале идет очень бурно, причем окраска смеси изменяется до серой. После
перемешивания в течение часа к смеси прибавляют эфир. На другой день
кашицеобразную массу нагревают с обратным холодильником на водяной
бане до удаления аммиака, центрифугируют и жидкую часть разгоняют:
эфир отгоняют при обычном давлении, а остаток перегоняют в вакууме.
Перед разгонкой его еще раз отделяют от осадка. Все операции проводят в
атмосфере азота.
1,3-Ениновые оловоуглеводороды представляют собой бесцветные сильно
преломляющие свет жидкости со специфическим запахом. В чистом виде они
могут храниться без доступа воздуха продолжительное время. На воздухе
они довольно легко гидролизуются [21, 49].
Триалкилстаннилдиацетилены получены [35] действием бромдиацетилена
на триалкилстаннилнатриевые соединения.
Трифенилстаннилнатрий легко реагирует с монохлорацетатом натрия в
жидком аммиаке [15, 51]. Однако выделить в чистом виде натриевую соль
трифенилстаннилуксусной кислоты не удается вследствие расщепления
связи олова с углеродом в процессе очистки вещества [51—53].
Взаимодействием триалкил- или триарилстаннилнатриевых соединений
с хлористым метиленом получены [54, 55] соединения состава (R3Sn)2CH2.
Более сложно проходит реакция триметилстаннилнатрия с хлороформом
[56—58]. При смешении триметилстаннилнатрия с 1,2-дихлорэтаном
происходит лишь отщепление этилена и образование гексаметилдистаннана. Четы-
реххлористый углерод превращает натрийтриметил- и трифенилолово в сое-
$02 Олсвоорганические соединения. Лит. стр. 510—511
динения типа R3SnSnR3 [55, 56]. С n-дихлорбензолом получен [14] п-бис-
(триметилстаннил)бензол. Реакция с галоидными ал килами и арилами
проходит более сложно, если исходить из литийоловоорганических
соединений, полученных действием 3 молей RLi на хлористое олово. Например,
при применении растворов трифенилстанниллития, наряду с обычными
продуктами реакции, образуются значительные количества тетрафенилолова
[26, 28]. Кроме того, с орто-замещенными йодистыми арилами наблюдается
следующий побочный процесс [28]:
(CeH5)8SnLi + o-JCGII4R -> (CGH5)3SnJ -[- o-LiC6H4R.
Взаимодействием три-ж-толил- и три-п-толилстанниллития с
соответствующими арилгалогенидами получены [29] тетра-ж-толилолово (выход
74%) и тетра-п-толилолово (80%). Однако при реакции три-о-толилстан-
ниллития с о-иодтолуолом вместо тетра-о-толилолова образуется гекса-о-
толилдистаннан с выходом 45%.
Аналогично реагирует три-о-толилстанниллитий с хлористым бензилом.
В результате образуется смесь три-о-толилбензилолова (2%), гекса-о-
толилдистаннана (38%) и тетра-о-толилолова (7%). Очевидно, в этих
реакциях существенную роль играют стерические факторы.
Помимо реакций с алкил- и арилгалогенидами, изучено взаимодействие
трифенилстанниллития с окисью этилена [19,29], эпихлоргидрином [29],
диэтилкарбонатом [30], этилхлорформиатом [30] и флуореном [8, 39, 59].
Окись этилена и эпихлоргидрин образуют с трифенилстанниллитием (2-
оксиэтил)трифенилолово (выход 44,8%; т. пл. 66—67°С) и трифенил-2-
окси-3-хлорпропилолово (выход 27%; т.пл. 97—99°С) соответственно. Из
продуктов реакции трифенилстанниллития с диэтилкарбонатом или
этилхлорформиатом выделен только гексафенилдистаннан и небольшое количество
тетрафенилолова.
Трифенилстанниллитий металлирует флуорен с образованием 9-флуо-
рениллития. После карбонизации реакционной смеси получают флуорен-9-
карбоновую кислоту с выходом 19,5% и 31% гексафенилдистаннана [59].
РЕАКЦИИ ЛИТИЙ- И НАТРИЙОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ГАЛОИДНЫМИ СОЛЯМИ И МЕТАЛЛООРГАНИЧЕСКИМИ СОЕДИНЕНИЯМИ
Триалкил- и триарилстанниллитиевые соединения легко реагируют с
галоидными солями бора, кремния, германия, олова, свинца, фосфора,
мышьяка, сурьмы и висмута с образованием оловоорганических соединений
со связью Sn—Э.
Взаимодействием триэтилстанниллития с диметиламиноборхлоридами в
эфире получены [60] [(CH3)2N]2BSn(C2H5)3 (т. кип. 32—35°С/0,001 мм),
(CH3)2NB(Cl)Sn(C2H5)3 (т. кип. 26—28°С/0,001 мм) и (CH3)2NB[Sn(C2H5)3]2
(т. кип. 91—93°С/0,001 мм). Трибутил- и трифенилстанниллитий реагируют с
хлористым триметилкремнием в эфире при — 20°С с образованием
соединений типа R3SnSi(CH3)3. Выходы продуктов составляют 60—70% [61].
При повышении температуры выход станнилсиланов снижается вследствие
побочных реакций. Синтез соединений типа R3SnSiR3 описан рядом
авторов [3, 4, 20, 26, 28, 62].
Получение триметилсилилтрибутилолова [4]. К раствору трибутилстанниллития
(приготовлен из 0,1 моля гексабутилдистаннана) в тетрагидрофуране при перемешивании
прибавляют 23,9 г (0,22 моля) хлористого триметилкремния в 100 мл того же растворителя
при температуре около 0° С. Растворитель и хлористый литий удаляют и остаток
перегоняют в вакууме. Получают 56,6 г (77,8%) вещества с т. кип. 88° С/0,2 мм.
Оловоорганические соединения с двумя и более атомами металла 503
С трифенилстанниллитием при 24 °С реакция проходит иначе.
Взаимодействием хлористого триметилкремния с трифенилстанниллитием получен [3]
гексафенилдистаннан с выходом 68,7%.
Станнилсиланы состава R3SnSiR2SnR3 получены [7] взаимодействием
триэтил-, трибутил- или трифенилстанниллития с двухлористым диметил-
и дифенилкремнием.
Трифенилстанниллитий образуете четыреххлористым германием в эфире
или тетрагидрофуране тетра(трифенилстаннил)германий. Аналогично
получено [7] соответствующее соединение свинца:
РЬС12 -}, 2 (С6Н,)з SnLi -» {[(CeH5)3 Sn]2 Pb} -> [(С6Н5)3 SnJ4 Pb + Pb.
Побочным продуктом этих реакций является гексафенилдистаннан.
Реакция между трифенилстанниллитием и хлористым трифенилгерма-
нием приводит к сложной смеси продуктов [63]. Однако трифенилгермил-
трифенилолово легко получается при прибавлении хлористого трифенило-
лова к трифенилгермилкалию.
Диксон и Вест [31] исследовали взаимодействие триэтилстанниллития с
двухлористым дициклопентадиенилтитаном.
Как уже отмечено на стр. 496, при синтезе литий-и натрийоловооргани-
ческих соединений из оловоорганических галогенидов в качестве
промежуточных продуктов образуются соединения типа R3SnSnR3 и (R2Sn),2. Если
реакции проводить с рассчитанном количеством щелочного металла, то они
могут быть выделены в индивидуальном состоянии. Этот путь является
одним из основных методов синтеза гексаалкил- и гексаарилдистаннанов.
Получение гексаметилдистаннана [14, 64]. К раствору 40 г бромистого триметилолова
в 200 мл жидкого аммиака прибавляют 4,1 г натрия в 100 мл того же растворителя. Аммиак
выпаривают и остаток экстрагируют эфиром. Эфирный раствор промывают теплой водой,
высушивают сульфатом натрия и остаток перегоняют в вакууме. Получают 16г (71%)
вещества с т. кип. 181 — 182° С/760 мм и 85—88° С/45 мм.
Вместо бромистого триметилолова в этой реакции можно применять
хлористое триметилолово [65]. Аналогично получен [18] гексаэтилдистаннан.
Другой способ синтеза того же вещества заключается в нагревании до 130°С
40 г хлористого триэтилолова и 5 г порошка натрия в 50 мл изоамилового
эфира в течение 5 час. Растворитель отгоняют и остаток перегоняют в
вакууме. Выход гексаэтилдистаннана 70%, т. кип. 160°С/23 мм [66,67]. Гекса-
этил-, гекса-я-пропил-, гексаизобутил-, шлш-тетраэтилди-л-пропил- и симм-
тетраэтилдиизобутилдистаннаны синтезированы [68] нагреванием
соответствующих оловоорганических бромидов с натрием в запаянной ампуле в
среде эфира.
Примером проведения этих реакций в ароматическом ряду может служить
образование гексафенилдистаннана при действии трифенилстанниллития на
фтористое [3], хлористое [8, 26, 28] или бромистое [20] трифенилолово.
Более удобный способ синтеза этого соединения заключается в действии
натрия на хлористое трифенилолово в смеси бензола со спиртом.
Получение гексафенилдистаннана [69]. К раствору 3 г хлористого трифенилолова в
смеси 35 мл бензола и 35 мл сухого спирта постепенно прибавляют кусочки натрия (всего
4—5 г). Реакционная смесь закипает, прибавлением натрия поддерживают кипение (если
под конец реакция замедляется, добавляют новую порцию спирта). По растворении натрия
смесь выливают в 1 л воды. Взвешенное вещество отсасывают или извлекают бензолом.
Т. пл. 237° С.
Тем же способом получены гекса-я-толил-, гекса-я-ксилил-, гекса-я-
хлорфенил- [70], гексабензил- [71] и гексациклогексилдистаннан [72].
504 Оловоорганические соединения. Лит. стр. 510—511
Получение гекса-о-толилдистаннана [29]. В колбу емкостью 125 мл помещают 4,0 г
(0,0094 моля) хлористого три-о-толилолова, 3,0 г натрия, 10 мл бензола и 30 мл ксилола.
Смесь кипятят в течение часа и фильтруют горячей. Растворитель удаляют пропусканием
тока воздуха над поверхностью раствора. Твердый остаток дважды кристаллизуют из
минимального количества бензола. Получают 2,3 г (62,1%) гекса-отолилдистаннана с т. пл.
298—300° С.
Синтез гекса(бифенилил-2)-дистаннана проводят в ксилоле [73].
Взаимодействием в жидком аммиаке AlkgSnNa с Ar3SnCl получены триал-
килтриарилдистаннаны. В этом случае большое значение имеет порядок
прибавления реагентов. Необходимо исходить из алифатического натрийоло-
воорганического соединения и галоидного триарилолова. При обратном
порядке прибавления образуется смесь гексаалкил- и гексаарилдистаннана
с триалкилтриарилдистаннаном [42]. Реакцию можно проводить также в
среде органических растворителей.
Получение 1,1,1-триметил-2,2,2-трифенилдистаннана [41. К раствору • триметил-
станниллития (приготовлен из 0,1 моля хлористого триметилолова) прибавляют раствор
38,5 г (0,1 моля) хлористого трифенилолова в 75 мл тетрагидрофурана при температуре
около 0° С. Образовавшуюся смесь темно-коричневого цвета гидролизуют насыщенным
раствором хлористого аммония. Органический слой объединяют с эфирным экстрактом водного
слоя и высушивают сульфатом магния. Выпаривание растворителя дает твердое белое
вещество, которое обрабатывают горячим этиловым спиртом. Получают 11,3 г (32,3%) гекса-
фенилдистаннана с т. пл. 230—234° С. Из фильтрата выделяют 26,2 г (50,9%) 1,1,1-триме-
тил-2,2,2-трифенплдистаннана с т. пл. 103—106J С. Перекристаллизация из этанола дает
белые кристаллы с т. пл. 107—108,5° С.
Получение 1,1,1,-триметил-2,2,2-три-я-толилдистаннана описано
Брауном и Фоулесом [74].
При действии натрия на растворы оловоорганических дигалогенидов
в жидком аммиаке образуются соединения типа (R2Sn)„. Таким путем
получены диметил- [13, 65], диэтил-[75] и дифенилолово [151. Однако позже
показано, что образующиеся при этом соединения имеют более сложную
структуру [76, 77]. По-видимому, при получении этих веществ частично проходит
их изомеризация. Очевидно, эти процессы имеют место и при получении
диэтилолова из двухлористого диэтилолова и амальгамы натрия в эфире
[76, 78]. При действии на двухлористое дифенилолово в тетрагидрофуране
мелкодисперсного лития, сплава натрия с калием, пылью цинка или
магния происходит частичное расщепление связей олова с углеродом [77].
Отщепление галоида от двухлористого дифенилолова проходит легко,
если раствор двухлористого дифенилолова в тетрагидрофуране медленно
смешивать с нафталиннатрием в том же растворителе. При этом с хорошим
выходом образуется бесцветный додекафенилциклогексастаннан [77]:
6 (С6П5)2 SnCl2 + C10H8Na -» [(C6H,)2Sn]6 + 12LNaCl.
Тем же путем получено [38] ди-я-бифенилилолово.
О получении соединений типа Cl(SnR2)nCl см. статью Живу хина с сотр.
[79].
Взаимодействием натрий- или литийоловоорганических соединений с
галогенидами олова получены также соединения типа R3Sn(SnR2).4SnR3 и
органостаннаны с разветвленной цепью из атомов олова, например окта-
метил-[65] и октафенилтристаннан [38]:
2 (СН3)з SnCl + (CH3)2 SnNa2 -> (CH3)3 SnSn (CH8)2 Sn (CH3)8 + 2 NaCl,
2 (С6Ч,)3 SnNa + (QH5)2 SnCl2 -> (ССН5)3 SnSn (CeH5)2 Sn (CeH5)8 + 2 NaCl.
При взаимодействии диметилстаннилендинатрия с двубромистым диме-
тилоловом образуется динатрийгексаметилтристаннаи [13]:
2 (CH3)2SnNa2 + (СН3)2 SnBr2 -» Na [Sn (C4S)2]8 Na + 2 NaBr.
Оловоорганические соединения с двумя и более атомами металла 505
Это вещество может быть, в свою очередь, проалкилировано действием
галоидного алкила, что дает октаалкилтристаннан. Очевидно, также можно
получить этот тип соединений с различными радикалами у атома олова.
С галоидным триметилоловом получен додекаметилпентастаннан [13]
2 (CH3)3SnX + Na [Sn (СН3)2]з Na -» (СН3)3 Sn [Sn (CH3)2]3 Sn(CH3)3 + NaX.
Декаметилтристаннан синтезирован сливанием аммиачных растворов динат-
рийтетраметилдистаннана и галоидного триалкилолова [40]:
2 (СН3)3 SnX + Na [Sn (СН3)2]2 Na -* (CHS)3 Sn [Sn (CK3)2]2 Sn (CH3)3 + 2 Na X.
Взаимодействием трифенилстанниллития с хлористым оловом в тетрагидро-
фуране получено [80] соединение состава [(C6H5)3Sn]3SnLi, которое с хлорным
оловом дает тетра(трифенилстаннил)олово — Sn[Sn(C6H5)3]4. Последнее
образуется также в результате следующих реакций [7,80,81]:
lSnCl4 + 4 (СбН5)3 SnLi -» Sn [Sn (C6H5)3]4 + 4 LiCl,
4 (С6Н5)3 SnCl + 8 Li + SnCl4 -> Sn [Sn (C6H5)3]4 -f (C6II5)3 SnSn (CGH,)3.
Соединения со связью Sn—Sn могут быть получены и другими методами
SnCl2 + 2 RLi -» (R2Sn)n + 2 LiCl,
R2SnH2 + R2Sn (NRg)2 -» (R2Sn)„ + 2 R^NH.
Второй путь позволяет синтезировать вещества с разветвленной цепью
из атомов олова. Эти реакции рассмотрены на стр. 193 и 477 соответственно.
Реакциям соединений типа R3SnSnR3 и (R2Sn)„ посвящена глава XIII.
В литературе приведены противоречивые данные о продуктах реакции
трифенилстанниллития с фенилхлорфосфинами и треххлористым фосфором
[22, 82]. Описаны [83, 83а] реакции трифенилстанниллития с хлористыми
солями мышьяка, сурьмы и висмута;
3 (С6Н5)3 SnLi + МС13 -> [(0,115):» Sn]3 M -f 3 LiCl.
Выходы продуктов составляют около 80%.
Триметил- и трифенилстанниллитий образуют с три-я-бутилфосфатом
в тетрагидрофуране я-бутилтриметилолово [4] и я-бутилтрифенилолово [3]
соответственно.
Целесообразнее оловоорганические фосфины, арсины и стибины получать
по реакции:
R3SnX + (C6H-)23Na -» RsSn3 (OH5)2 + NaX
(Э = Р, As, Sb)
ИЛИ
RsSnNRg + (C6H5)2 ЭМ -* R3Sn3 (C6H,)2 + Я2Ш\.
Эти реакции рассмотрены на стр. 411 и 417.
СИНТЕЗ СОЕДИНЕНИЙ ТИПА R3SnM(CO)^ и R2Sn[M(CO)J2
(где М — металл VI—VIII групп)
Би- и полиметаллические соединения олова с карбонилами переходных
металлов систематически изучены в работах Несмеянова, Анисимова с сотр.
[84—87]. Взаимодействием натриевых солей трикарбонил-я-циклопента-
диенилмолибдена, а также соответствующих производных вольфрама и хрома
с хлористым трифенилоловом или двухлористым дифенилоловом в
тетрагидрофуране получены [84] биметаллические производные следук щего состава:
506 Оловоорганические соединения. Лит. стр. 510—511
(C6H5)3SnM(CO)3C5H5 и (C6H5)2Sn [M(CO)3C5H5]2 (M=Cr, Mo, W). Это белые
или зеленовато-желтые кристаллические вещества, хорошо растворимые в
обычных органических растворителях.
Получение трикарбонил-л-циклопентадиенилмолибденоловотрифенила [84]. В круг-
лодонную колбу на 250 мл, снабженную мешалкой, обратным холодильником и вводом для
азота помещают 0,25 г дисперсного натрия в тетрагидрофуране и при постоянном
перемешивании прибавляют небольшими порциями 0,7 г свежеперегнанного циклопентадиена.
К полученному раствору циклопентадиенилнатрия прибавляют 2,7 г Мо(СО)6. Реакционную
смесь нагревают при 60—70° С до прекращения выделения СО. К охлажденной до комнатной
температуры смеси прибавляют при перемешивании 3,9г хлористого трифенилолова и \Ъмл
тетрагидрофурана. Выделившийся хлористый натрий отфильтровывают, растворитель
удаляют и оставшуюся массу подвергают сублимации (75° С/1 мм), в результате чего выделяют
0,1 г Mo (CO)g- Остаток после отделения карбонила молибдена растворяют в смеси петролей-
ного эфира и хроматографируют на А1203. После перекристаллизации из смеси петролейного
эфира и толуола получают 3,5 г трикарбонил-л-циклопентадиенилмолибденоловотрифенила
ст. пл. 212,5—215° С.
Аналогично из 10 а двухлористого дифенилолова в 30 мл
тетрагидрофурана, циклопентадиенилнатрия (из 1,7 г натрия и 5,2 г циклопентадиена в
тетрагидрофуране) и 19,5 а Мо(СО)6 получено 8,5 г (C6H5)2Sn[Mo(CO)5C5H5]9
с т. пл. 190—191,5°С.
Синтез оловоорганических производных карбонилов хрома и
вольфрама ведут в среде диглима.
Получение трикарбонил-л-циклопентадиенилвольфрамоловотрифенила [84]. 17,6 г
W(CO)g прибавляют к циклопентадиенилнатрию, приготовленному из 1,15 г натрия и 3,5|г
циклопентадиена в диглиме. После выделения СО при комнатной температуре прибавляют
8 г хлористого трифенилолова в 30 мл диглима. После сублимации, хроматографирования и
двукратной перекристаллизации из смеси гептана с толуолом выделяют 6 г вещества ст. пл.
229—230,5° С.
Тем же методом получены (QHs^SnfW^CO^CsHsL (т. пл. 207—209°С)
и (С6Н5)з5пСг(СО)3С5Н5 (т. пл. 220—222°С).
Ренийпентакарбонилнатрий с хлористым трифенилоловом и двухлори-
стым дифенилоловом образует трифенилоловоренийпентакарбонил и
дифенилоловоди(ренийпентакарбонил) соответственно [85]. Это
кристаллические вещества, хорошо растворимые в полярных органических
растворителях, в воде нерастворимы.
Получение трифенилоловоренийпентакарбонила [85]. При взаимодействии 2,9 г (0,0075
моля) хлористого трифенилолова с NaRe (CO)5 (в 30 мл тетрагидрофурана), полученным
из 2,45 г (0,0038 моля) Re2(CO)10 и 50 мл 0,8 % амальгамы натрия при комнатной
температуре выделено 3,87 г (77 %) белого кристаллического вещества с т. пл. 142—143°С.
Аналогично при прибавлении 2,8 г (0,08 моля) двухлористого
дифенилолова (40 мл тетрагидрофурана) к NaRe(CO)5 (из 5,6 г Re2(CO)10 и 100 мл
0,8% амальгамы натрия с последующим перемешиванием смеси при 20—
25°С в течение 25 час. получен [85] дифенилоловоди(ренийпентакарбо-
нил). Выход 3,8 г (51%); т. пл. 139°С.
Несмеяновым, Анисимовым и сотрудниками получен также ряд
полиметаллических соединений, в которых атом олова одновременно связан с кар-
бонилами двух различных металлов VI—VIII групп периодической системы
[86]. Например:
С6Н-} С0Н;,
I I
(СО).-, Мп—Sn—CI + NaAl (CO)n -» (СО), Мп—Sn—M (СО)п
СбтЬ С3Н5
(Vl = Co, Re)
Оловоорганические соединения с двумя и более атомами металла 507
ИЛИ
0,1 Ь <«"*
/ /
(СО), Mn-Sn-Cl + NaM' (СО)3 (л-С5Н5) -» (СО)3 Mn-SnM' (СО), (л-С,Н,)
\ \
СбН3 0бН5
(M'=Mo,W).
Известны и соответствующие производные диметилолова [88].
В результате реакции треххлористого фенилолова или двухлористого
дифенилолова с NaFe(CO)2C5H5 в тетрагидрофуране получены соединения
[jt-C6H6Fe(CO)a]8SnCeH6 (т. пл. 243-245°С) и l^-C5H5Fe(CO)2]2Sn(C6H5)2
(т. пл. 148— 150°С), а исходя из [n-C5H5Fe(CO)2]2SnCl2 получены
[tt-C5H5Fe(CO)2]2Sn(C2H5)2 (т. пл. 87—89°С) и [tt-C5H5Fe(CO)2],Sn(C5H5)2
(т. пл. 149— 150°С) [87].
Оловоорганические марганец-, железо- и кобальткарбонилы описаны и
другими авторами [88—94]. Так, при взаимодействии хлористого триметил-,
триэтил- или три-я-бутилолова в инертной атмосфере с водным раствором
Fe(CO)42 получены [89] соединения состава Fe(CO)4(SnR3)2. В тех же
условиях из дигалогенидов диалкилолова образуются димеры состава
[Fe(CO)4(SnR2)2l2. Синтез соответствующих соединений ароматического
ряда осуществлен обменом гидроокиси трифенилолова или двухлористого
дифенилолова с Ca[HFe(CO)J2 [90].
Натрийдикарбонил-я-циклопентадиенилжелезо легко реагирует с оло-
воорганическими галогенидами алифатического и ароматического рядов.
Так получены, например, соединения * (CH3)3SnFe(CO)2(C5H5-tt) [88],
(C2H5)2Sn[Fe(CO)2C5H5-n]2 [91] и (C6H5)3SnFe(CO)2(C5H5-n) [92].
К биметаллическим соединениям марганца и олова приводит
взаимодействие оловоорганических галогенидов с эквивалентным количеством нат-
риймарганецпентакарбонила в тетрагидрофуране [92]:
R4-n SnC1n + п NaMn (С°)а ~* Vn Sn tMn (с°)з]я + п NaCl.
(«=1,2).
Так получены (C6H5)3SnMn(CO)5 (т. пл. 148— 150°С), (CH3)2Sn[Mn(CO)5b
(т. пл. 102—104°С) и (C6H5)2Sn[Mn(CO)5]2 (т. пл. 137—139°С). Эти
соединения легко растворяются в полярных растворителях, например хлористом
метилене, эфире, ацетоне и тетрагидрофуране; они хорошо растворяются
также в этиловом спирте, бензоле и других ароматических углеводородах
и умеренно или плохо — в алифатических углеводородах, например,
гексане.
Оловоорганические галогениды гладко реагируют и с натрийкобальттет-
ракарбонилом [90, 93, 94]:
R4_„ SnCln + п NaCo (CO)4 -> (C0I I5)4-n Sn ICo (CO)4]„ -|- n NaCl.
Так, из продуктов реакции двухлористого диметилолова с NaCo(CO)4 в
нейтральной водной среде выделены кристаллы (CH3)2Sn[Co(CO)4]2.
Соответствующие производные три- и дибутилолова представляют собой маслянистые
жидкости желтого цвета [94]. Трифенилоловокобальттетракарбонил имеет
т. пл. 123°С, а дифенилоловоди(кобальттетракарбонил) разлагается, не
плавясь, при 139— 141°С [90].
Другой метод синтеза оловоорганических кобальткарбонилов основан
на взаимодействии соединений типа [Co(CO)3L]2SnX2 (L = СО, (С6Н5)3Р,
(СбН50)3Р, (я-С4Н9)3Р; X =С1, Вг или J) с реактивом Гриньяра [91, 95].
Исходные вещества получают в полярных растворителях по схеме:
SnX2 - [Со (СО)з L]2 -^ [L(CO)3Co]2 SnX2.
508
Оловоорганические соединения. Лит. стр. 510—511
Выходы продуктов, как правило, достаточно высокие. Реакцию можно
иллюстрировать следующим примером.
Получение дифенилоловоди(кобальттетракарбонила) [95]. К окрашенному в оранжевый
цвет раствору двухлористого ди(кобальттетракарбонил)олова (1 г) в 60 мл эфира
прибавляют 8 мл эфирного раствора бромистого фенилмагния (15,6 г бромистого фенилмаг-
ния в 100 мл эфгра). Раствор окрашивается в темно-коричневый цвет. На следующий
день растворитель удаляют и остаток кристаллизуют из этилового спирта. Получают 0,31 г
вещества с т. разл. 140° С.
Аналогично получено диэтилди(кобальттрикарбонилтри-я-бутил-
фосфин)олово (т. пл. 77°С). При действии на дигалогениды ди(трикар-
бонилтри-я-бутилфосфинкобальт)олова бромистого фенилмагния на
органический радикал замещается только один атом галоида, что, по-видимому,
объясняется стерическими факторами [95].
РЕАКЦИИ СОЕДИНЕНИЙ
ТИПА R3SnM(CO)„ и R2Sn[M(CO)J2
Характерной особенностью оловоорганических производных карбонилов
хрома, молибдена, вольфрама и других металлов VI—VIII групп
периодической системы является устойчивость связи Sn—М к действию электро-
фильных реагентов. Реакции оловоорганических соединений
рассматриваемого типа с галоидами и галоидоводородами подробно изучены
Несмеяновым, Аписимовым и сотр. [84—87]. Так, при действии хлористого водорода на
С5Н5(СО)зМо5п(С6Н5)з и (C6H5)3SnW(CO)3C5H5 в растворе четыреххлористого
углерода при — 15°С в течение 30 мин. были выделены [84] почти с
количественными выходами С5Н5(СО)зМо5п(С6Н5)С12 и CsHsCCO^WSnf^CeHs)^.
Аналогичное действие хлористого водорода на (CeHsbSnlMo^O^CsHsU и
(C6H5)2Sn[W(CO)3C5H5]2 привело к получению [C5H5(CO)3Mo]2SnCl2 и
[C5H5(CO)3W]2SnCl2. Все эти галоидопроизводные соединения представляют
собой кристаллические вещества светло-желтого цвета, устойчивые на
воздухе. Они хорошо растворяются в обычных органических растворителях.
Получение фенилдихлороловотрикарбонил-зт-циклопентадиенилмолибдена С12(СсН5)-
SnMo(CO)3C3H5 [84]. В охлажденный до —15° С раствор 3 г (С6Н5)з5пМо(СО)зСзН5 в
20 мл четыреххлористого углерода пропускают в течение 15 мш. ток сухого хлористого
водорода. Выпавший небольшой осадок отделяют, фильтрат упаривают в вакууме и
остаток пзрекристаллизовывагат из смеси я-гептана с бензолом. Получают 2,1 г (95 %)
вещества с т. пл. 109—110° С.
Получение фенилдихлороловотрикарбонил-л-циклопентадиенилвольфрама Cl2(CGH5)
SnW(CO)3C5H5 [84]. При действии сухого хлористого водорода на охлажденный до-
—15° С раствор 1,5 г С5Н5(СЭ)з\Узп(СзНз)з в 30 мл четыреххлористого углзрода в
течение 15 мин. выделено 0.65 г (52%) кристалл 1ческого вэщкггва желтого цвета с т.
пл. 112—114° С (из гептана).
Аналогично ведут себя по отношению к хлору, брому и хлористому
водороду ренийоловоорганические соединения [85] .И в этом случае связь металл—
металл сохраняется, но происходит полное или частичное замещение фе-
нильных радикалов на атомы галоида. Так, хлор и хлористый водород
действуют на трифенилоловопентакарбонилрений с образованием трихлор-
оловопентакарбонилрения. Действием избытка брома на трифенилолово-
пентакарбонилрений и на дифенилоловоди(пентакарбонилрений) в смеси
гептана с бензолом были выделены трибромоловопентакарбонилрений и
дибромоловоди(пентакарбонилрений). При применении рассчитанного
количества брома оказалось возможным провести и ступенчатое отщепление
фенильных групп от атома олова.
Оловоорганические соединения с двумя и более атомами металла 509
Получение фенилдибромоловопентакарбонилрения Br2(C6H5)SnRe(CO)5 [85]. При
взаимодействии 0,5 г (0,00075 моля) трифенилоловопентакарбонилрения в 30 мл смеси
гептана с бензолом (1:1) с 0,24 г (0,0015) моля брома при охлаждении до —10° С в
течение 30 мин. получают 0,32 г (64%) вещества с т. пл. 129—130° С.
Обработка избытком брома монофенилоловотри(пентакарбонил)рения
дает монобромидоловотри(пентакарбонил)рения. Последний с натрийпента-
карбонилом рения в тетрагидрофуране образует кристаллическое вещество
светло-желтого цвета, аналитические данные которого согласуются с
формулой [(CO)5Re]3SnSn[Re(CO)5]3 [85].
Метод позволяет синтезировать соединения с тремя различными
заместителями у атома олова. При гидрохлорировании (C6H5)2Sn[Co(CO)4][Mn(CO)5]
выделены [86] кристаллы состава С1(СбН5)5п[Со(СО)4][Мп(СО)б] с т. пл.
87—88°С.
Интересно отметить, что при деарилировании оловоорганических
соединений со связью Fe—Sn галоидоводородами отщепляется фенильная
группа [87,92], тогда как хлором легко разрывается связь металл — металл
192].
Реакции биметаллических соединений, содержащих связь Sn — Мп с
галоидами и галоидоводородами, также могут проходить или с разрывом
связи металл—металл, или с отщеплением органического радикала от атома
олова [92]. То или иное направление реакции определяется природой
радикала и числом групп Мп(СО)5 в молекуле. Например, избытком хлора или
брома в четыреххлористом углероде при 25°С трифенилоловомарганецпен-
такарбонил деарилируетея до X3SnMn(CO)5 (X = С1, Вг). При
хлорировании (C6H5)2Sn[Mn(CO)5]2 образуются Cl3SnMn(CO)5, хлорбензол и С1Мп(СО)4
выделенный в виде С12Мп2(СО)8. Хлористый водород действует более
селективно. В хлористом метилене при 25°С он отщепляет от (C6H5)2Sn[Mn(CO)5]2
только фенильные группы, давая Cl2Sn[Mn(CO)5]2 (выход 69%) и бензол.
Галоидопроизводные трифенилоловомарганецпентакарбонила, которые
не удается получить прямым галоидированием, могут быть получены
реакцией, обратной диспропорционированию. Так, например, соединение состава
(C0H5)2ClSnMn(CO)5 получено нагреванием до 148°С в течение 45 мин. сте-
хиометрических количеств (C6H5)3SnMn(CO)5 и Cl3SnMn(CO)5 [92]:
2 (С6Н5)3 SnAIn (СО); + Cl8SnMn (СО)3 -> 3 (С6Н5)2 CISnMn (CO)5.
Этот метод особенно удобен при синтезе галоидопроизводных
биметаллических соединений алифатического ряда.
В литературе описаны также некоторые реакции замещения
карбонильной группы в соединениях типа R3SnM(CO)„ (M—■ Re, Mn, Со). Так, при
действии трифенилфосфина, трифениларсина и трифенилстибина на три-
фенилоловопентакарбонил рения происходит замена группы СО на
соответствующий лиганд [96]. Скорость и полнота замещения зависит от природы
лиганда (и количественного соотношения компонентов) в
последовательности
(С6Н5)3 Р > (С6Н3)з As > (СвН5)3 Sb.
Получение (СвН5)з5пКе(СО)4Р(СбН5)з [96]. При нагревании до 220° С в течение 3 час.
0,68 г (C6H5)3SnRe (СО)р и 1,56 г трифенилфосфина выделен белый проект с т. пл.
218—220° С.
Аналогично получены [92,94,96]'(C6H5)3SnRe(CO)4As(C6H5)3,(C6H5)3SnRe-
(CO)4Sb(C6H5)3, (C4H9)3SnCo(CO)3P(C6H5)3, (C4H9)2Sn[Co(CO)3P(C6H5)3]2,
(C6H5)3SnMn(CO)4P(C6H5)3(C6H5)3SnMn(CO)4As(C6H5)3 и другие соединения
этого типа.
510
Оловоорганические соединения
При нагревании до 180—194°С трифенилоловомарганецпентакарбонила
с эквимолекулярным количеством тетрафенилциклопентадиенона получено
соединение состава:
(C6H5)3Sn ЧСО)3
Структура вещества подтверждена его ИК-спектром.
ЛИТЕРАТУРА
1. GilmanH., Bull. Soc. chim. France, 1963, 1356.
2. T a m bo r sk i С, FordF.E., LehnW.L, MooreG.J., SoloskiE.J.,
J. Org. Chem., 27, 619 (1962).
3. TamborskiC, FordF.E., Soloski E. J., J. Org. Chem., 28, 181 (1963).
4. TamborskiC, FordF.E., S о 1 о s k i E. J., J. Org Chem., 28, 237 (1963).
5. S с h u m a n n H., T h о m K. F., S с h m i d t M., J. Organometal. Chem., 1, 167 (1963).
6. S с h u m a n n H., ThomK.F., SchraidtM., Angew. Chem., 75, 138 (1963).
7. WillemsensL. C, v a n der KerkG. J.M., J. Organometal. Chem., 2, 260
(1964).
8. G i 1 m a n H." MarrsO. L, SimS. Y., J. Org. Chem., 27, 4232 (1962).
9. GilmanH., Ca r t 1 edge F. K., Si mS. Y., J. Organometal. Chem., 4, 332 (1965).
10. T a m b о r s k i C, S о 1 о s k i E. J., J. Am. Chem. Soc, 83, 3734 (1961).
11. Sch uma nnH.,Tho m K. F.,Sch m i d tM., J. Organometal. Chem., 2, 97 (1964).
12. К г a u s С A., G г е е г W. N., J. Am. Chem. Soc, 44, 2629 (1922).
13. К r a u s С A., G r e e r W. N., J. Am. Chem. Soc, 47, 2568 (1925).
14. К r a u s С A., S e s s i о n s W. V., J. Am. Chem. Soc, 47, 2361 (1925).
15. С h a m b e r s R. F., S с h e r e r P. C, J. Am. Chem. Soc, 48, 1054 (1926).
16. BullardR.H., RobinsonW.R, J.Am. Chem. Soc, 49, 1368 (1927).
17. В u 1 1 a r d R. H., H о 1 d e n F. R., J. Am. Chem. Soc, 51, 3150 (1931).
18. H a r a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 35, 290 (1939); С. А., 33, 5357
(1939).
19. G i 1 m a n И., А г n t z e n С. Е., J. Org. Chem., 15, 994 (1950).
20. В 1 a k e D., С о a t e s G. E., T a t e J. M., J. Chem. Soc, 1961, 618.
21. 3 а в г о р о д н и й В. С, П е т р о в А. А., ЖОХ, 32, 3527 (1962).
22. С a m p b е 1 1 I. G. M., F о w 1 е s G. М. А., N i х о n L. A., J. Chem. Soc, 1964, 1389.
23. К г a u s С. A., J о h n s о n E. G., J. Am. Chem. Soc, 55, 3542 (1933).
24. К r a u s С. А., К a h 1 e r W. H., J. Am. Chem. Soc, 55, 3537 (1933).
25. К e t t 1 e S. F. A., J. Chem. Soc, 1959, 2936.
26. GilmanH., RosenbergS. D, J. Am. Chem. Soc, 74, 531 (1952).
27. G i 1 m a n H., R о s e n b e r g S. D., J. Am. Chem. Soc, 75, 2507 (1953).
28. G i 1 m a n H., R о s e n b e r g S. D., J. Org. Chem., 18, 680 (1953).
29. GilmanH., RosenbergS. D., J. Org. Chem., 18, 1554 (1953).
30. GilmanH., GistL. A., J. Org. Chem., 22, 689 (1957).
31. DicksonR.S.,WestB.O, Austral. J. Chem., 14, 555 (1961).
32. W i t t i g G., Angew. Chem. 62, 231 (1950).
33. W i t t i g G., M e у e r F., L a n g e G., Ann., 571, 167 (1951).
34. П е т р о в A. A., 3 а в г о р о д н и й В. С, ЖОХ, 34, 2806 (1964).
35. 3 а в г о р с д н и й В. С, Петр о в А. А., ЖОХ, 35, 760 (1965).
36. В о t t R. W., E a b о r n C.Wal tonD. R.M., J. Organometal. Chem., 2, 154 (1964).
37. T а л а л а е в а Т. В., К о ч е ш к о в К. А., ЖОХ, 8, 1831 (1938).
38. N е u m a n n W. Р., К 6 n i g K., Ann., 677, 12 (1964).
39. D'AnsJ., ZimmerH., E n d r u 1 a t E., L u b k e K., Naturwiss., 39, 450 (1952).
40. К r a u s С A., N e a 1 A. M., J. Am. Chem. Soc, 51, 2406 (1929).
41. В u 1 1 a r d R. H., V i n g e e R. A., J. Am. Chem. Soc, 51, 892 (1929).
42. К r a u s С A., BullardR.H., J. Am. Chem. Soc, 48, 2131 (1926).
43. V a n der К e r k G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 106 (1959).
44. L e Q u a n M., С a d i о t P., Compt. rend., 254, Ш (1962).
45. L e Q u a n M., С a di о t P., Bull. Soc. chim. France, 1965, 45.
Оловоорганические соединения с двумя и более атомами металла 511
46. 3 а в г о р о д н и й В. С, П е т р о в А. А., ЖОХ, 33, 2791 (1963).
47. 3 а в г о р о д н и й В. С, П е т р о в А. А., ЖОХ, 35, 931 (1965); 36, 1480(1966).
48. BeermannC, HartmannH., Z. anorgon. allgem. Chem., 276, 20 (1954).
49. 3 а в г о р о д н и й В. С, П е т р о в А. А., ДАН СССР, 143, 855 (1962).
50. П е т р о в А. А., М и н г а л е в а К. С, 3 а в г о р о д н и й В. С, ЖОХ, 34, 533
(1964).
51. V а п d е г К е г к G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 113, (1959).
52. К о ч к и н Д. А., Н о в и ч е н к о Ю. П., Авт. свид. СССР 139833 (1961); РЖХим.,
1961, 6П248; Бюлл. изобр., № 14, 48 (1961).
53. К о ч к и н Д. А., Н о в и ч е н к о Ю. П., Авт. свид. СССР 139834 (1961); РЖХим.;
1961, 6П266; Бюлл. изобр., № 14 (1961).
54. К г a u s С. А., N е а 1 А. М., J. Am. Chem. Soc, 52, 695 (1930).
55. К г a u s С. А., Е a t о u g h H., J. Am. Chem. Soc, 55, 5014 (1933).
56. К г a u s С A., N e a 1 A. M., J. Am. Chem. Soc, 52, 4426 (1930).
57. PapettiS., PostH. W., J. Org. Chem., 22, 526 (1957).
58. К a e s z H. D., J. Am. Chem. Soc, 83, 1514 (1961).
59. G i 1 m a n H., MarrsO.L, T г е р к a W. J., D i e h 1 J. W., J. Org. Chem., 27,
1260 (1962).
60. N 6 t h H., H e r m a n n s d 6 г f e г К. Н., Angew. Chem., 76, 377 (1964).
61. Wiberg E., Stecher O., AndrascheckH. J., Kreuzbichler L,
StandeE., Angew. Chem., 75, 516 (1963).
62. К r a u s С A., E a t о u g h H., J. Am. Chem. Soc, 55, 5008 (1933).
63. GilmanH., GerowC. W., J. Org. Chem., 22, 334 (1957).
64. С 1 a r к H. C, W i 1 1 i s С J., J. Am. Chem. Soc, 82, 1888 (I960).
65. В г о w n T. L., Morgan G. L., Inorg. Chem., 2, 736 (1963).
66. H e с м е я н о в А. Н., К о ч е ш к о в К. А., П у з ы р е в а В. П., ЖОХ, 7, 118
(1937).
67. К о z e s с h k о w К. A., NesmejanowA. N., PusyrewaW. P., Ber., 69,
1639 (1936).
68. GriittnerG., Ber., 50, 1808 (1917).
69. К г a u s e E., vanGrosseA., Die Chemie der metal lorganischen Verbindungen.
Berlin, Gebr. Borutrager, 1937, S. 357.
70. KrauseE., WeinbergK., Ber., 62, 2240 (1929).
71. Law K. K., J. Chem. Soc, 1926, 3243.
72. К r a u s e E., P о h 1 a n d R., Ber., 57, 532 (1924).
73. В a h r G., G e 1 i u s R., Ber., 91, 825 (1958).
74. В г о w n M. P., FowlesG.W. A., J. Chem. Soc, 1958, 2811.
75. H a r a d а Т., Sci. Papers Inst. Phys. Chem. Res. (Tokyo), 38, 146 (1940); C. A., 35, 2470
(1941).
76. Neumann W. P., Pedain J., Ann.. 672, 34 (1964).
77. Neumann W. P., К 6 n i g K., Ann., 677, 1 (1964).
78. P f e i f f er P., Ber., 44, 1269 (1911).
79. Ж и в у х и н С. М., Д у д и к о в а Э. Д., К о т о в А. М., ЖОХ, 33, 3274 (1963^
80. G i 1 m a n Н., С а г t 1 е d g e F. К., Chem. a. Ind., 1964, 1231.
81. G i 1 m a n Н., С а г t 1 е d g e F. К., J. Organometal. Chem., 5, 48 (1966).
82. Schumann Н., К 6 р f H., S с h m i d t M., Ber., 97, 2395 (1964).
83. S с h u m a n n H., SchmidtM., Angew. Chem., 76, 344 (1964).
83a. Schumann H., Osterman Т., Schmidt M., Ber., 99, 2057(1966); J.
Organometal. Chem., 8, 105 (1967).
84. Несмеянов А.Н., АнисимовК. И., Колобова Н. Е., Захарова М. Я.,
ДАН СССР, 156, 612 (1964).
85. Несмеянов А. Н., АнисимовК. Н., Колобова Н. Е., X а и д о ж -
к о В. Н., ДАН СССР, 156, 383 (1964).
86. Несмеянов А. Н., А н и с и м о в К. Н., К о л о б о в а Н. Е., 3 а х а р о-
в а М. Я., Изв. АН СССР, ОХН, 1965, 1122.
87. Н е с м е я п о в А. Н., А н и с и м о в К. Н., Колобова Н. Е., Скрип-
к и н В. В., Изв. АН СССР, ОХН, 1966, 1292.
88. Р a t i 1 Н. R. H., G r a h a m W. A. G., J. Am. Chem. Soc, 87, 673 (1965).
89. KahnO., BigorgneM, Compt. rend., 261, 2483 (1965).
90. H e i n F., J e h n W., Ann., 684, 4 (1965).
91. BonatiF., WilkinsonG., J. Chem. Soc, 1964, 179.
92. Gorsich R. D., J. Am. Chem. Soc, 84, 2486, (1962).
93. H i e b e r W., В г е u R., Angew. Chem., 68, 179 (1956).
94. H i с b e r W., Breu R., Ber., 90, 1270 (1957).
95. В о n a t i F., S e n i n i S., M о r e 1 1 i D., U g о R., J. Chem. Soc. 1966A, 1052.
96 H e с м e я ii о в A. H., Колобова H. Ем А н и с и м о в К. Н., X а н л о ж-
к о В. Н., Изв. АН СССР, ОХН, 1966, 163.
Глава XVП
ОЛОВСОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
В литературе описаны полимерные соединения, содержащие атом олова
как в основной, так и в боковой цепи полимерной молекулы. Их получают,
^соответственно, полимеризацией оловоорганических соединений,
содержащих в своем составе винильные группы, поликонденсацией или с помощью
реакций присоединения.
ПОЛИМЕРЫ С ОЛОВООРГАНИЧЕСКИМИ ЗАМЕСТИТЕЛЯМИ
[В БОКОВОЙ ЦЕПИ
По патентным данным [1], винильные и аллильные соединения олова
полимеризуются в присутствии катализаторов типа Циглер—Натта. При
анионной полимеризации триэтилвинилолова под действием я-бутиллития
получены твердые гомополимеры среднего молекулярного веса. Показано, что
триэтилвинилолово в присутствии н-бутиллития способно вступать в
реакцию сополимеризации со стиролом и триметилвинилкремнием [2]. В
условиях же радикальной полимеризации эти соединения практически не
полимеризуются [3—6] (стр. [7,8)1. Также неудачными оказались попытки запо-
лимеризовать триэтил- и трифенилвинилолово под воздействием ^-облучения
[9]. Сополимеризация диаллилдиэтилолова со стиролом или метилметакри-
латом в растворе при 60°С в присутствии перекиси бензоила проходит с
небольшим выходом! 10]. Молекулярный вес сополимеров не превышает 16 000.
Низкомолекулярные продукты образуются и при сополимеризации триал-
килаллильных соединений олова с метилметакрилатом [11].
Введение между винильной группой и атомом олова бензольного кольца
или карбоксильной группы создает благоприятные условия для
полимеризации соответствующих оловоорганических соединений. Так, я-трифенилстан-
нилстирол или метакрилат трифенилолова полимеризуются с большей
скоростью, чем незамещенный стирол и метилметакрилат [5]. Наличие в
полимере оловоорганического остатка повышает его термостойкость [12].
/г-Триметилстаннилстирол легко полимеризуется в присутствии
инициаторов свободно-радикального типа [13]:
C(R)=CHe
-C(R)-CH2-
Sn (СНз)з Sn (СН3)з
Полимеризация л-триметилстаннилстирола[13].В /г-триметилстаннилстироле растворяют
0,10 мол. % азодиизобутиронитрила. 1—2 г раствора запаивают в заполненный азотом
ампуле и тер моста тир уют при 70° С в течение нескольких часов. Образующийся продукт
растворяют в 20 мл бензола, и раствор прибавляют по каплям к 400 мл метилового спирта.
Осадок отсасывают, промывают метиловым спиртом и высушивают в вакууме до постоянного
веса.
Оловоорганшеские полимеры
513
Проведение полимеризации я-триметил- и я-триэтилстаннилстирола под
давлением 6000 атм в присутствии инициатора позволяет увеличить выход
полимеров и повысить вязкость их растворов [14, 15]. Большое значение
имеет также природа органического радикала у атома олова [16, 17].
Изучена полимеризация д-винилфенилтрифенилолова. Полученные
результаты позволяют сделать вывод о том, что природа заместителя у атома
металла практически не влияет на скорость полимеризации. Удлинение
боковой цепи в случае я-винилбифенилтрифенилолова привело к
уменьшению реакционной способности в условиях радикальной полимеризации [18].
В литературе описан [16, 19—21] ряд сополимеров, полученных на основе
стиролов, содержащих в ядре группу R3Sn. Исследована сополимеризация
и рассчитаны константы сополимеризации следующих пар мономеров:
я-винилфенилтрифенилолово — стирол; я-винилфенилтрициклогексилоло-
во — стирол; д-винилбифенилилтрифенилолово—стирол [19]. Показано, что
по активности в сополимеризации со стиролом оловоорганические мономеры
образуют ряд: я-винилфенилтрифенилолово > я-винилбифенилилтрифени-
лолово > я-винилфенилтрициклогексилолово.
Значительно легче проходит полимеризация [5, 22—31] и
сополимеризация [5, 23—29, 32—35] оловоорганических акрилатов и метакрилатов:
R3SnOOC(R')=CH2 -* -[-^Sx\OOC(K)-CH2-\n-.
Существенное влияние на свойства образующихся полимеров оказывают
природа радикала, связанного с атомом олова, и условия проведения
полимеризации [30].
Также легко проходит полимеризация и сополимеризация я-винилбензо-
атов три- [36, 37] и диалкилолова [37]. Полимеризацию га-винилбензоата три-
фенилолова можно провести в блоке или в растворе в присутствии
инициаторов радикальной полимеризации или без них при 80—150°С [38]. Полимеры,
полученные в блоке, представляют собой прозрачные бесцветные или слегка
окрашенные в желтый цвет термопластичные массы, не растворимые в
обычных органических растворителях.
Интересно отметить, что мономеры (I) и (II)
\—CONHCH=CH2 f\—NHCOCH=CH
Ji-COOSn (СвН5)з ЦJ-COOSn (С6Н5)3
(I) (И)
образуют полимеры с различной термостойкостью, а именно более стабилен
полимер, полученный из соединения (II) [39].
Оловоорганические заместители можно вводить также в полимерный
продукт. Так, взаимодействием я-литийполистирола (или орто-изомера)
с R3SnCl получены [40] соответствующие замещенные в ядре полистролы.
ПОЛИМЕРЫ С АТОМОМ ОЛОВА В ОСНОВНОЙ ЦЕПИ
В главе V описана реакция присоединения оловоорганических
гидридов к ненасыщенным соединениям. Взаимодействием дигидридов диалкил-
или диарилолова с диенами получают линейные полимеры с атомом олова
в основной цепи [41—47]:
R2SnH2 + СН2=СН—R'-CH=CH2
г R
I
-Sn-CH2CH2R'CH2CH2-
, I
L R
33 Заказ № 207
514 Оловоорганические соединения. Лит. стр. 516—518
Алифатические диены, не имеющие заместителей, поляризующих
двойную связь, реагируют с оловоорганическими дигидридами только в
присутствии катализатора [41, 43, 44]. С замещенными диенами, например 1,4-ди-
винилбензолом или металлоорганическими производными стирола, реакция
проходит без катализатора [45,48].
Дигидрид дифенилолова [46] и п-фенилен-бис-(диметилоловогидрид)
[49] образуют с 1,4-дивинилбензолом или 3,9-дивинилспиро-бис-(л*-диокса-
ном) твердые стеклообразные продукты присоединения. Взаимодействием
тех же гидридов с дифенилди-я-стирильными соединениями германия, олова
или свинца получают полимерные стеклообразные продукты [46, 49]:
(C6H5)2SnH2 + (С6Н5)2 М (СбН4СН=:СН2)2 -*
Г СбНб СбНб ~~
I I
—Sn—СН2СН2СвН4МС6Н4СН2СН3-
I I
СбНб СбНб
(M = Ge, Sri, Pb).
Реакция дигидрида дифенилолова с дифенилди-/г-стирилгерманием [13]. 4,33 г (0,010
моля) дифенилди-я-стирилгермания и 2,79 г'(0,010 моля) дигидрида дифенилолова в 15 мл
сухого бензола нагревают при 70° С до образования однородной смеси. Бензол отгоняют
и смесь нагревают при 75°С в вакууме в течение 15 час, а затем 24 часа при 115° С. Остаток
представляет собой прозрачный стеклообразный полимер с температурой размягчения
примерно 100° С. Для очистки его растворяют в 45 мл бензола и при энергичном перемешивании
прибавляют по каплям к 400 мл метилового спирта. Осадок отфильтровывают, промывают
метиловым спиртом и высушивают в вакууме до постоянного веса. Выход практически
количественный, температура размягчения 105—110° С. Мол. вес около 27 000 (степень
полимеризации равна примерно 45).
Большое влияние на ход реакций присоединения оказывают стерические
факторы [50]. При реакции дигидрида дифенилолова с дивинилдифениль-
ными соединениями кремния и германия полимерные продукты образуются
лишь в качестве примесей [51].
Для получения полимерных продуктов используют также присоединение
оловоорганических гидридов к производным ацетилена. Оловоорганические
гидриды присоединяются по двойной и тройной связям с различной
скоростью. Поэтому синтез полимерных продуктов этим методом можно
проводить двумя различными способами [47].
Первый из них заключается во взаимодействии эквимолекулярных
количеств дигидрида диалкил-или диарилолова и диина, содержащего обе
тройные связи на концах молекулы. Реакция проходит в мягких условиях и
заканчивается после гидрирования тройной связи до двойной.
По второму способу исходят из эквимолекулярных количеств
моноацетиленового соединения и оловоорганического дигидрида. Здесь различаются
две стадии реакции.
Вначале дигидрид присоединяется по тройной связи с образованием
R2(R'CH=CH)SnH. При нагревании этот гидрид присоединяется по
двойной связи другой молекулы [47].
Присоединение п-фенилен-бш>(диметилоловогидрида) к 1,5-гексадиину
или 1,8-нонадиину приводит к хорошо растворимым в бензоле каучукооб-
разным продуктам со средним молекулярным весом порядка 100 000 [49].
При реакции оловоорганических дигидридов с диэтинилбензолами наря-
Оловоорганические полимеры
515
ду с полимерными продуктами [52] получены [50] циклические соединения
состава:
v
Sn
. • Sh
, /\
R R
Дигидрид дифенилолова образует с диацетиленом относительно
низкомолекулярный продукт [53].
Примером присоединения оловоорганических дигидридов к
моноацетиленовым соединениям может служить реакция дигидрида дипропилолова с фе-
нилацетиленом.
Присоединение дигидрида дипропилолова к фенилацетилену [54]. Раствор 14,48 г
(0,070 моля) дигидрида дипропилолова и 7,15 г (0,070 моля) фенил ацетилена нагревают в
50 мл пентана в течение 5 час. После удаления растворителя получают 21,6 г бесцветной
подвижной жидкости. Ее нагревают при 100° С в течение 8 час, а затем 24 часа при 130° С.
Образующийся продукт растворяют в бензоле, фильтруют и растворитель выпаривают
(последние следы растворителя удаляют продолжительным нагреванием при 130° С в вакууме
10~3 мм). Получают прозрачную слегка желтоватую вязкую жидкость с молекулярным
весом порядка 3400.
Реакция дигидрида дифенилолова с фенилацетиленом проходит
своеобразно. Основным продуктом является циклический димер [51].
Для получения полимеров были проведены опыты по присоединению
дигидрида дифенилолова к ненасыщенным эфирам [49].
В патентной литературе описано [55, 56] присоединение
оловоорганических дигидридов и к другим соединениям, содержащим в своем составе
ненасыщенные группы.
При реакции n-дилитийбензола с двухлористым дифенилоловом
образуется неплавкий низкомолекулярный продукт состава—[-С6Н4 Sn(C6H6)2—\п—
(п = 2—3). Вещество не растворяется в обычных органических
растворителях [57].
Кроме углерода и олова в основной цепи полимера могут находиться
также кислород, кремний, сера и другие элементы. К таким полимерам
приводят, например, следующие реакции [58, 59]:
(CH3)2SnCl2+ NaCH2COONa -» — [—(CH3)2SnCH2COO—]n— + 2 NaCl,
(C6H5)2SnCl2 + KOR'OK-> -[-(C6H6)2SnOR/0-]„- +^2 KC1.
Нагреванием дигидридов ди-я-бутил- или дифенилолова с дивинилсуль-
фоном получены низкомолекулярные продукты, имеющие, по-видимому,
следующее строение [60]:
-[-(C6H5)2SnCH2CH2S02CH2CH2—]л—.
Полиорганостанноксаны — станноксановые аналоги силоксанов —
получают гидролизом оловоорганических галогенидов (см. стр. 388). Баум и
Консидаин [61] изучили термостабильность некоторых из этих полимерных
продуктов. Найдено, что наибольшей термостабильностью обладают окиси
диметил- и дифенилолова. У окисей дибутилолова она уменьшается в
следующем ряду:
(n-C4H9)2SnO > (s-C4H9)2SnO > (/-C4H0)2SnO.
33е
516
Оловоорганические соединения
Интересно отметить, что кристалличность окиси диизобутилолова после
расплавления и охлаждения не изменяется, тогда как в случае окиси ди-
ятор-бутилолова она убывает вследствие дальнейшей полимеризации [61].
К образованию органостанноксанов приводит также конденсация диаци-
латов диалкилолова с алкоксисоединениями олова [62—64]. Тем же путем
получены [65—67] соединения со связью Sn — О— Si. Другой способ их
синтеза заключается в согидролизе соединений типа R2SnCl2 с алкил- и арилхлор-
силанами [68,69] или в нагревании органосилоксанов с оловоорганическими
окисями [70—73] и ацильными производными олова [74]. Описаны и
другие станноорганосилоксаны [75].
Полимеры, содержащие в основной цепи фосфор и олово, получают
[76] конденсацией первичных фосфинов RPH2 с оловоорганическими
соединениями типа CH2-C(R')(CH2)„ SnR2(CHa)nC(R') = CH2.
К полимерам со связью Sn — О — As приводят следующие реакции [77]:
х (CH3)2SnCl2 + х RAsO (ONa)2 -> 2х NaCl + —[—Sn (CH3)2 OAs (R) 00— ]x—,
3x (CH3)2SnCl2 + 2x Ag3As04 -> 6x AgCl + -[—OAs <[OSn (CH3)20]2 > AsOOSn (GH8)alX-
По патентным данным [78] нагревание оловоорганических гидроокисей
или окисей с этилен-бас-тиогликолем, органической кислотой и меркапто
соединениями (исключая меркаптокарбоновую кислоту) приводит к полиди-
бутилстаинилмеркаптосоединениям.
В литературе описаны и другие оловоорганические соединения, имеющие
полимерную структуру, например соединения типа (R2Sn)„ (см. стр. 480) или
(R2SnS)„ (см. стр. 413). Отмечено [61], что термостабильность сульфида ди-я-
бутилолова выше, чем у окиси ди-я-бутилолова.
О продуктах реакции окисей или диацилатов диалкилолова с
двухосновными кислотами см. стр. 401, 417.
ЛИТЕРАТУРА
1. N a t t a G., Mazzan t iG., LongiP., Bernardini R, Австрал. пат. 236677
(1961); РЖХим., 1963, 11T34; Пат. ГДР 24665 (1963), РЖХим., 1964, 6С201.
2. Платэ Н. А., Мальцев В. В., Давыдова С. Л., Каргин В. А. Высокомо-
лек. соед., 8, 1890(1965); Платэ Н. А., Мальцев В. В., Колесников Г. С,
Давыдова С. Л., Авт. свид. СССР 176408(1965); Бголл. изобр., № 12 (1965).
3. КотонМ.М., К и с е л е в а Т. М., ЖОХ, 27, 2553 (1957).
4. К о т о н М. М., К и с е л е в а Т. М., 3 а п е в а л о в а Н. П., ЖОХ, 30, 186 (1960).
5. Котон М. М., К и с е л е в а Т. М., Ф л о р и н с к и й Ф. С., Сб. «Международный
симпозиум по макромолекулярной химии». Москва, 1960. Секция I. M., Изд-во АН
СССР, 1960, 147; J. Polvmer Sci., 52, 237 (1961).
6. Колесников Г. С, Давыдова С. Л., ЖОХ, 29, 2042 (1959).
7* А с а и X., Vinyl Japan, 2, 53 (1962); РЖХим., 1963, 22Т72.
8. A u f d e r m a r s с h С. А., Р а г i s е г R.f J. Polvmer Sci., A2, 4727 (1964).
9. Н а у a k a wa К., К a wa se К., М a t s u d at., Nature, 206, 1038 (1965).
10. К о л е с н и к о в Г. С, Д а в ы д о в а С. Л., Ермолаева Т. И., Шилова
Н. Д., Б ы х о в с к а я М. Б., Высокомолек. соед. 2, 567 (1960).
11. КоршакВ. В., Полякова А. М., Миронов В. Ф., П е т р о в А. Д., Изв.
АН СССР, ОХН, 1959, 178.
12. Котон М. М., Киселева Т. М., ФлоринскийФ. С, Высокомолек. соед.,
2, 1639 (1960).
13. N о 1 t е s J. G., В u d d i n g H. A., van d e г К е г k G. J. M., Rec. trav. chim.,
79, 1076 (1960).
14. Коршак В. В., Полякова А. М.,ТамбовцеваЕ. С, Изв. АН СССР, ОХН,
1959, 742.
15. Полякова А. М., Сахарова А. А., Чернышев Е. А.,
Краснова Т. Л., К о р ш а к В. В., П е т р о в А. Д., Высокомолек. соед., 5, 353 (1963).
16. ЧернышевЕ. А., ПетровА. Д., Краснова Т. Л., Синтез и свойства
мономеров. М., «Наука», 1964, стр. 103.
17. П о л я к о в а А. М., К о р ш а к В. В., Т а м б о в ц е в а Е. С, Высокомолек. соед.,
3, 662 (1961).
18. К о т о н М. М., Д о к у к и н а Л. Ф., ДАН СССР, 152, 1357 (1963).
19. К о т о н М. М., Д о к у к и н а Л. Ф., Высокомолек. соед., 6, 1791 (1964).
Оловоорган ические полимеры
517
20. G r e b e r G., E g 1 е G., Makromol. Chem., 67, 207 (1963).
21. S a n d 1 е г S. R., D a n n i n J., T s о и К. С, J. Polymer Sci., A3, 3199 (1965).
22. M о н т е р м о з о Дж. Ч., ЭндрьюзТ. М., Маринелли Л. П., Л а л а й-
бертеБ. Р., Каучук и резина, 9, 61 (1960).
23. Ш о с т а к о в с к и й М. Ф., Котрелев В. Н., Калинина С. П.,
Кузнецов а Г. И., Л а й н е Л. В., Б о р и с о в а А. И., Высокомолек. соед., 3, 1128 (1961).
24. К о ч к и и Д. А., КотрелевВ. Н., Калинина С. П., Кузнецова Г. И.
ЛайнеЛ. В., Ч е р в о в а Л. В., Б о р к с о в а А. И., Б о р и с е н к о В. В.,
Высокомолек. соед., 1, 1507 (1959).
25. Ш о с т а к о в с к и й М. Ф., Калинина С. П., КотрелевВ. Н., К о ч-
к и н Д. А., Кузнецова Г. И., Л а й н е Л. В., Б о р и с о в а А. И., Б о р и-
сенко В. В., Сб. «Международный симпозиум по макромолекулярной химии»,
Москва, 1960. Секция I. M., Йзд-во АН СССР, 1960, стр. 160.
26. К о т о п М. М., К и с е л е в а Т. М., П а р и б о к В. А., ДАН СССР, 125, 1263 (1959)
27. Юй-чжень, Лин. Scientia, 3, 89 (1959); РЖХим., 1960 20760.
28. Ш о с т а к о в с к и й М. Ф., Котрелев В. Н., Калинина С. П.,
Кузнецов а Г. И., Л а й н е Л. В., Б о р и с о в а А. И., Высокомолек. соед., 3, 1131 (1961).
29. Sh о s t a k о v s к i i M. R, Kalinina S. P., Kotrelev V. N.. Koc-
hkinD. A., KuznetsowaG. I., Laine L. V., BorisovaA. I.,J. Polymer
Sci., 52, [157], 223 (1961).
30. Montermoso J. C, Andrews T. M., M a r i n e 1 1 i L. P., J. Polymer Sci., 32,
[125], 523 (1958).
31. M о n t e r m о so J. С, М a r i n e 1 1 i L. P., Andrews Т. М., Пат. США
3016369 (1962); РЖХим., 1963, 8T65.
32. КочкинД. А., КотрелевВ. Н., ШостаковскийМ. Ф., К а л и н и-
н а С. П., Кузнецова Г. И., БорисенкоВ. В., Высокомолек. соед., 1, 482
(1959).
33. К о л е с н и к о в Г. С, Г у р г е н и д з е Г. Т., Изв. АН СССР, ОХН, 1962, 1275.
34. Ma r i п е 1 1 i L. P., AndrewsT. M., Montermoso J.C., Пат. США 3012018
(1961); РЖХим., 1963, 23Т130.
35. М а с k G. Р., Пат. США 3069394 (1962); РЖХим., 1965, 2С206.
36. L е е Ъ г i с к J. R., W a s у 1 с i w J., К е е d i s h N., J. Polymer Sci., 54, 835 (1961).
37. LeebrickJ. R., Пат. США 3167532 (1965); РЖХим., 1966, 8C273.
38. К qT о н М. М., К и с е л е в а Т. М., Изв. АН СССР, ОХН, 1961, 1783.
39. К и с е л е в а Т. М., К о т о н М. М., Ч е т ы р к и н а Г. М., Изв. АН СССР, ОХН,
1962, 1798.
40. L e a v i t t F. С, M a t t e r n a s L. U., J. Polymer Sci., 45, 249 (1960).
41. N e u m a n n W. P., Angew. Chem., 75, 225 (1963).
42. N e u m a n n W. P., Angew. Chem., 76, 849 (1964).
43. N e u m a n n W. P., N i e r m a n n H., Angew. Chem., 75, 790 (1963).
44. N e urn a n n W. P., N i e r m a n n H., Som m er R., Ann., 659, 27 (1962).
45. А д р о в а Н. А., К о т о н М. М., К л а г е с В. А., Высокомолек. соед., 3, 1041 (1961).
46. N о 1 t e s J. G., v a n d e г К e r k G. J. M., Rec. trav. chim., 80, 623 (1961).
47. N о It e s J. G., v a n d e г К e r k G. J. M., Chimia, 16, 122 (1962).
48. А д р о в а Н. А., К о т о н М. М., К л а г е с В. А., Высокомолек. соед., 5, 1817 (1963).
49. L e u s i n k A. J., N о It e s J. G.. В u d d i n g H. A., van d e г К e r k G. J. M.,
Rec. trav. chim., 83, 609 (1964).
50. L e u s i n k A. J., NoltesJ. G., Budding H.A., van d e r KerkG. J. M.,
Rec. trav. chim., 83, 1036 (1964).
51. H e n г у M. C, N о 1 t e s J. G., J. Am. Chem. Soc, 81, 561 (1960).
52. Лунева Л. К., С л а д к о в А. М., К о р ш а к В. В., Высокомолек. соед., 7, 427
(1965).
53. L e a v i t t F. С, M a t t e r n a s L. U., J. Polymer Sci., 62, [174], 68 (1962).
54. NoltesJ.G., van d e r KerkG.J.M., Rec. trav. Chim., 81, 41 (1962).
55. N i e b e r g a 1 1 H. R., Англ. пат. 923584 (1963); РЖХим., 1964, 22C155.
56. R ei h s L. J., Англ. пат. 935740 (1963); РЖХим., 1965, 7C220.
57. Адрова Н. А., КотонМ. И., Прохорова Л. К., Сб. «Высокомолекулярные
соединения. Гетероцепные высокомолекулярные соединения». М., «Наука», 1963, стр. 9.
58. W i 1 s о п G. R., Пат. США 3184430 (1965); РЖХим., 1966, 11С299.
59. D ер гее D. О., Пат. США 3161664 (1964), РЖХим., 1966, 11Н108.
60. Р е а г с е Е.М., J. Polymer Sci., 40, 273 (1959).
61. Baum G. A., Considine W. J., J. Polymer Sci., Bl, [9] 517 (1963).
62. К о т о н М. М., К и с е л е в а Т. М., ДАН СССР, 130, 86 (1960).
63. Живу хин С. М., Дуди ков а Э. Д., Т е р-С а р к и ся н Э. М., ЖОХ, 32, 3059
(1962).
64. Землянский Н. Н., Шамагина О. П., Панов Е.М., Кочешков К. А.,
ЖОХ, 35, 1029 (1965).
65. Henglein F. A., Lang R., SchmackL, Makromol. Chem., 22, 103 (1957).
518
Оловоорганические соединения
66. И с и ц у к а Т., М о м о н о н М., Ф у д з и т а Р., Японск. пат. 12945 (1960); РЖХим.,
1962, 14Л122.
67. Т h i e s С, К i n s i n g e r J., Inorg. Chem., 3, 551 (1964).
68. А н д р и а н о в К. А., Г а н и н а Т. Н., X р у с т а л е в а Е. Н., Изв. АН СССР,
ОХН, 1956, 798.
69. А н д р и а н о в К. А., Я к у ш к и н а С. Я., ЖОХ, 35, 330 (1965).
70. Пат. ФРГ 1104705 (1931); РЖХим., 1963, 7Т92.
71. И с и ц у к а Т., М о м о н о н М., Ф у д з и т а Р., Японск. пат. 12948 (1960); РЖХим.,
1962, 14Л122.
72. F о s t e r W. Е., К о е n i g P. E., Пат. США 2998440 (1961); РЖХим., 1963, 5Т39.
73. F о s t e r W. Е., К о е n i g P. E., Пат. США 2998407 (1961); РЖХим., 1962, 23П408.
74. И с и ц у к а Т., МомононМ., Ф у д з и т а Р., Йосида К., Японск. пат. 12946
(1960); РЖХим., 1962, 17Л71.
75. N i e b e r g а 1 1 Н., Пат. ФРГ 1087810 (1961); РЖХим., 1963, 15Т128.
76. N i e b e r g а 1 1 Н., Пат. ФРГ 1086896 (1961); РЖХим., 1962, 8П148.
77. С h a m b е г 1 a n d В. Е., М с D i a r m i d A. G., J. Chem. Soc, 1961, 445.
78. О н и с и X., T а с и р о Т., Японск. пат. 18387 (1960); РЖХим., 1962, 24П435.
Г лава XVIII
ПРИМЕНЕНИЕ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Оловоорганические соединения широко применяются в синтезе и
переработке полимерных материалов, отчасти в качестве физиологически
активных веществ. По данным Люитена [1] их мировое производство в 1948 г.
составляло несколько сотен килограммов, в 1955 г. — 300 т, а в 1962 г.—
3000 т и, судя по многочисленным патентам, за пять истекших лет еще более
выросло. Применению оловоорганических соединений в различных областях
промышленности посвящены ряд обзорных статей [2—17] и отдельные главы
монографий [18, 19]. Подробное рассмотрение этого вопроса не входит в
задачи настоящего издания. В данном разделе мы лишь кратко
останавливаемся на основных областях применения оловоорганических соединений.
Самым значительным промышленным применением оловоорганических
соединений является стабилизация пластиков на основе поливинилхлорида
[2—18, 20, 21]. Как отмечено рядом авторов, оловоорганические соединения
имеют ряд преимуществ по сравнению с другими стабилизаторами. В
частности, они повышают термическую устойчивость и светостойкость полимера и
являются антиоксидантами. Стабилизаторы, не содержащие олова, после
выполнения своей функции часто ускоряют деструкцию поливинилхлорида.
Продукты разложения оловоорганических соединений играют роль
инертной добавки [22]. Следует отметить, что оловоорганические соединения
являются не только термо- и светостабилизаторами. Они применяются и при
защите поливинилхлорида от ионизирующей радиации [23—26], а также для
придания ему погодоустойчивости [27, 28].
Стабилизаторами могут служить многие оловоорганические соединения,
например тетраалкильные и тетраарильные производные олова [29], гексаал-
килдистанноксаны [30], алкокси- и арияоксисоединения олова [31—33] и
■продукты их конденсации [34, 35], сульфонамиды три- и диалкилолова
[36, 37], оловоорганические ацилаты [38—47]. В последнем случае обычно
применяют производные высших карбоновых кислот, например лауриновой.
Применение ацилатов низших карбоновых кислот ограничено вследствие
их летучести и гидролитической неустойчивости [48]. Более эффективны ор-
ганостанноксаны R'COO[SnR20]„OCR' [48, 49] или (:CHCOOSnR2OOCR')2
(50—52].
В качестве стабилизаторов поливинилхлорида широкое распространение
получили соединения со связью Sn—S [53—61], такие, как меркаптиды [62—
67], сульфиды [68, 69], а также соединения типа R„Sn(SR'COOR")4~,t
170—72], R'S(SnR20),,SnR2SR' [73] или R'S(SnR2S)„R' [71]. Для придания
поливинилхлориду большей термической устойчивости оловоорганические
соединения используют в композиции с другими веществами [74—83].
Иногда стабилизатор вводят в полимер путем сополимеризации исходного
мономера с винильными соединениями олова [84] или метакрилатами триал-
килолова [85].
520
Оловоорганические соединения. Лит. стр. 521—525
Приведенные в литературе данные по сравнительной эффективности
действия различных оловоорганических стабилизаторов скудны и не
позволяют сделать каких-либо общих выводов по этому вопросу. Отмечено [86],
что по термостабилизируюшему действию ацилаты ди-я-бутилолова мало
отличаются от алкоксисоединений. Значительно выше оно у меркаптссоеди-
нений. Светостабилизирующее действие лучше у алкоксисоединений и
особенно у меркаптидов слова [86, 87].
При применении оловоорганических соединений в качестве
стабилизаторов поливинилхлорида в некоторых случаях необходимо учитывать их
токсичность [88, 89]. С этой точки зрения наибольшее внимание привлекают
малотоксичные производные ди-я-октилолова [90, 91], например при
использовании поливинилхлорида в качестве тары для пищевых продуктов.
Механизм действия оловоорганических стабилизаторов окончательно
не выяснен [92—95]. По-видимому, он включает в себя ряд параллельно
идущих процессов [3,18].
Оловоорганические соединения являются стабилизаторами не только
поливинилхлорида, но и других полимерных материалов, например
поливинил ацетал ей и поливинилкеталей [69], полиолефинов [96—100],
облученного полиэтилена [101], смесей из НК, бутадиенстирольного или бутадиен-
нитрильного каучуков [102], полифениленов [103], жидких
хлорированных диэлектриков [104, 105].
Другой важной областью применения оловоорганических соединений
является использование их в качестве катализаторов ряда химических
процессов.
Тетраалкильные соединения олова применяются в качестве компонентов
комплексов (совместно с четыреххлористым титаном или другими
галоидными солями переходных металлов) в полимеризации олефинов [106—138].
На активность катализатора и свойства образующихся полимеров
существенное влияние оказывает природа алкильного радикала у атома олова [139].
В ряде работ [140—148] исследована полимеризация этилена в
гомогенной каталитической системе, которая образуется при смешении в
определенных соотношениях тетрафенилолова, треххлористого (или трехбромистого)
алюминия и небольшого количества галогенида ванадия в циклогексане или
другом углеводороде.
Оловоорганические соединения являются катализаторами и других
процессов, например полимеризации эфиров а-хлоракриловой кислоты [149],
диаллилфталата [150] и других винильных мономеров [151—156], а также
при полимеризации формальдегида [157], в реакциях этерификации [3,158],
образования полиэфиров [159] и полиэфироамидов [160]. В работе японских
авторов [161] показано, что в присутствии катализатора, содержащего (/)-
ментокситриэтилолово в сочетании с треххлористым алюминием и
хлористым триэтилоловом, из бензофурана образуется оптически активный по
лимер.
Оловоорганические соединения являются катализаторами синтеза
полиуретанов [162—173]. Они также весьма активны при получении пенопластов
[174—180]. В этом случае их каталитическая активность изменяется
в следующем ряду [181]:
R2SnX2 > R2SnO > R3SnX > RSnOOH > R4Sn
(R=CH3, C4H9 или C6H5).
Различные соединения олова нашли применение в качестве антиоксидан-
тов [182—187], присадок к смазочным маслам [188, 189], вулканизаторов
[190—194], противостарителей резин [195, 196] и для других целей [197—205].
Физиологическая активность оловоорганических соединений изучена
Применение оловоорганических соединений
521
довольно подробно (см., например, литературу, цитированную в
обзорной статье [206]). Найдено [207, 208], что оловоорганические
соединения в большинстве случаев токсичны и влияют на нервную систему.
Наиболее активны соединения типа R3SnX. Их используют, в частности, как
фунгициды [208—223], инсектициды [224, 225], средства для сохранения
древесины [226—229] или дезинфицирующие средства [230].
ЛИТЕРАТУРА
1. L u i j t e n J. G. A., Metall, 18, 814 (1964); РЖХим., 1965, 5A38.
2. В о n f i g 1 i о G., Materie plast, 24 365 (1958).
3. RossA., Ann. N. Y. Acad. Sci., 125, 107 (1965).
4. RiethmayerS.A., Kunstoff-Rundschau, 10, 277 (1963).
5. W a j n г у b M., Przem. Chem., 42, 16 (1963).
6. BelpaireR, Rev. beige matieres plast., 6, 201 (1965).
7. S m i t h H. V., Brit. Plast., 27, 215 (1954).
8. RiethmayerS.A., Kunststoff-Rundschau, 10, 345 (1963).
9. T i с h у V., Chem. listy, 55, 154 (1961).
10. PiecosR., Przem. Chem., 44, 423 (1965).
11. J oh n son W. A., Bedrijten techn., 17,676 (1962).
12. Sm i t h H. V., Brit. Plast., 37, 445 (1964).
13. F e r n 1 e у A. M., Plastica, 29, N 320, 66 (1964); РЖХим., 1965, 5C329.
14. Sm i t h H. V., Brit. Plast., 29, 337 (1956).
15. FreyH., Kunststoffe, 52, 667 (1962).
16. ThiniusK., SchlimperR., Plaste and Kautschuk, 9, 165 (1962).
17. Smith H. V., Rubber J. and Internat. Plastics, 138, 966 (1960).
18. Старение и стабилизация полимеров. М., «Наука», 1964.
19. Н а г w о о d J. H., Industrial Applications of the Organometallic Compounds. London,
Chapman and Hall, 1963.
20. HedgesE.S., Angew. Chem., 70, 135 (1958).
21. Von ReussH. H., Angew. Chem., 70, 135 (1958).
22. Fibres and Plastics, 21, 47 (1960).
23. Ш те д и н гМ. Н., КарповВ.Л., Высокомолек. соед., 4, 1806 (1962).
24. G a u t г о n R., W i р р 1 е г С, Ind. plast. mod., 12, 37 (1960); РЖХим., 1961, 20П20.
25. Франц. пат. 1280061 (1961); РЖХим., 1963, 9Т282.
26. W i р р 1 е г С, Bull. Soc. Industr. Mulhouse, 1959, 114; РЖХим., 1960, 86757.
27. CooverH.W., McConnell R. L, NewlandG. C, TamblynJ. W.,
Plast. Technol., 6, 45 (1960); РЖХим., 1961, 6П99.
28. F о r s s e n E., Plast.-Varlden, 9, 542 (1959); РЖХим., 1960, 86759.
29. M а с k G. P., P a r k e r E., Пат. США 2745819 (1956); РЖХим., 1958, 66086.
30. N i t z s с h e S., R i e d 1 e R., Пат. США 2868820 (1959); С. А., 53, 13057 (1959).
31. С 1 e v e r d о n D., S t a u d i n g e r J. J. P., F a u 1 k n e r D., M i 1 n e J. N., Пат.
США 2623892 (1952); С. А., 47, 3036 (1953).
32. M а с k G. P.,Пат. США 2745820 (1956); РЖХим., 1958, 59335.
33. H a r r i n g t о n R. С, SmithJ.L, Пат. США 3038877 (1962); РЖХим., 1964,
8C355
34. M а с k G. P., P а г k e г Е., Пат. США 2592926 (1952); С. А., 46, 11767 (1952).
35. M а с k G. P., P a r k e г Е., Пат. ФРГ 915939 (1954); РЖХим., 1957, 52736.
36. М а с k G. P., P a r k e r E., Пат. США 2618625 (1952); С. А., 47, 1977 (1953).
37. М а с к G. Р., Р а г к е г Е., Пат. США 2634281 (1953); С. А., 48, 1420 (1954).
38. AndersonR., К е 1 s о R., Пат. США 3037040 (1962); РЖХим., 1963, 21Н64.
39. D у s о n G. М., Н о г г о с к s J. A., F e r n b у А. М., Plasttechnik 1960; РЖХим.,
1962, 16П26.
40. Chem. Process (USA) 27, 33 (1964); РЖХим., 1965, IC246.
41. Sc h 1 egel H., Kunstoffe Plastics, 11, 1 (1964); РЖХим., 1964, 19C322.
42. Японск. пат. 13368 (1963); РЖХим., 1965, 14С318.
43. S q u i r e s S., G о о d i е г К., Англ. пат. 881578 (1961); РЖХим., 1963, 5Т23.
44. К у б о т а X., О н и с и X., Японск. пат. 2365 (1960); РЖХим., 1961, 15Л145.
45. К а ц у м у р а Т., Японск. пат. 9147 (1959); РЖХим., 1963, 11Т83.
46. И то Т., Японск.пат. 284 (1961); РЖХим., 1963, 17Т58.
47. МацудаС, МацудаХ., Я м а н э Я., J. Chem. Soc. Japan, Ind. Chem. Sec, 67,
467 (1964); РЖХим., 1964, 23C243.
48. M а с k G. P., P a r k e г Е., Пат. США 2628211 (1953); С. А., 47, 5165 (1953).
49. И т о Т., Японск. пат. 7686 (1960); РЖХим., 1962, 0Л1О8.
50. G 1 о s k e у С. R., Пат. США 2826561 (1958); РЖХим., 1960, 55015.
51. G 1 о s к е у С. R., Пат. США 2826597 (1958); РЖХим., 1960, 33053.
522
Оловоорганические соединения
52. О н и с и X., Т а с и р о Т., Японск. пат. 5983 (1960); РЖХим., 1962, 9Л51.
53. М а с к G. P., P a r k е г Е., Пат. США 2914506 (1959); РЖХим., 1962, 1П1Л.
54. R a m s d e n Н. Е., Англ. пат. 743313 (1956); С. А., 50, 16829 (1956).
55. Hartmann А., Пат. ФРГ 1073496 (1960); РЖХим., 1961, 14Л115.
5Р. В а г к е г R. L., Англ. пат. 855214 (1960); РЖХим., 1961, 6П396.
57. S t e f 1 Е. Р., С h г i s С. Е., Пат. США 2731441 (1956); РЖХим., 1958, 16291.
58. Франц. пат. 1107726 (1956); РЖХим., 1958, 34427.
59. М а с к G. Р., Р а г к е г Е., Пат. США 2998441 (1961); РЖХим., 1962, 16Л58.
60. Т h i n i u s К., S с h г б d е г Е., Пат. ГДР 21727 (1961); РЖХим., 1962, 8П235.
61. В о b о 1 i| Е., К a m i о n s к a J., К г е m к у Е., Р a z g a n А., Польск. пат. 47960
(1964); РЖХим., 1965, 6Н137.
62. JohnsonE.W., Пат. США 2891922 (1959); РЖХим., 1960, 70500.
63. S t e f 1 Е. Р., В е s t С. Е., Пат. США 2713580 (1955); РЖХим., 1958, 44261.
64. В е s t С. Е., Пат. США 2731484 (1956); РЖХим., 1958, 58348.
65. S t e f 1 Е. Р., В е s t С. Е., Пат. США 2731440 (1956); РЖХим., 1958, 3127.
66. Horun S., Rev. Chim. (RPR), 12, 575 (1961); РЖХим., 1962, 16П27.
67. W a 1 1 а с e J. M., Пат. США 2888435 (1959); РЖХим., 1961, 8П173.
68. W e i n b e r g E. L., R a m s d e n H. E., Пат. США 2746946 (1956); РЖХим., 1958,
55851.
69. HeckmaierJ., ZoebeleinH., Пат. ФРГ 1028776 (1962); РЖХим., 1963,
23П337.
70. W e i n b е г g E. L., J о h n s о n E. M., Пат. США 2832750 (1958); РЖХим., 1960,
70501.
71. W e i n b e r g E. L., JohnsonE.W., Пат США 2832751 (1958); РЖХим., 1960,
37046.
72. М а с k G. Р., Пат. США 3069447 (1962); РЖХим., 1964, 20Н60.
73. М а с k G. Р., Н е i g h t s J., P a r к е г Е., Пат. США 2809956 (1957); РЖХим., 1959,
51802.
74. К и мура Т., Sci. and ind,, 39, 725 (1965); РЖХим., 1967, 12С398.
75. Mezz A., Kunststoffe-Plast, 6, 169 (1959); РЖХим., 1960, 40871.
76. С h u г с h J. M., R a m s d e n H. E., H irschlandH., В u с h a n a n H. W.,
Пат. США 2743257 (1956); РЖХим., 1958, 26103.
77. Гельфман Я. А., Земля некий Н. Н., Лаурис И. В., Сютки-
н а О. П., Ку с к о в а В. П., П а н о в Е. М., Пласт, массы, 9, 10 (1966).
78. Т о у A. D. F., R a t t e n b u г г у К. Н., Н i n k е п s Т. М., Пат. США 2946764
(1960); РЖХим., 1962, 8П237.
79. Франц. пат. 1269236 (1961); РЖХим., 1962, 24П589.
80. S m i t h H. V., Англ. пат. 799775—6 (1956); РЖХим., 1960, 15696.
81. Е с к е 1 m a n n А., К u s с к R., Пат. ГДР 14024 (1957); РЖХим., 1959, 59171.
82. Woo ten W. С, Grossman J.M., Пат. США 3063963 (1962); РЖХим., 1964
17С509.
83. С а э к и Я., Japan Plast, 13, N 10, 8 (1962); РЖХим., 1963, 22Т71.
84. RamsdenH. E., Пат. США 3143531 (1964); РЖХим., 1966, 4С171.
85. О к a g а Т., N a g a n о Т., К a t о Е., Y a m о m о t о Т., Японск. пат. 696 (1962),
С. А., 58, 11538 (1963).
86. Б е р л и н А. А., П о п о в а 3. В., Я н о в с к и й Д. М., ЖПХ, 33, 871 (1960).
87. П о п о в а 3. В., Я н о в с к и й Д. М., Сб. «Международный симпозиум по макромо-
лекулярной химии». Москва, 1960, секция 3. М., Изд-во АН СССР, 1960, стр. 372.
88. FernleyA., H о г г о с k s J. A., Plastteknik, 1960; РЖХим., 1962, 16П26.
89. Н о u s к а М., Z. Lebensmittel — Untersuch und Forsch., 114, 373 (1961).
90. L u i j t e n J. G. A., P e z а г г о S., Brit. Plast., 30, 183 (1957); РЖХим., 1959, 13734.
91. Sm i th H. V., Plastic, 12, 134, 137, 140 (1962); РЖХим., 1963, 3T174.
92. W i n к 1 e г D. E., J. Polymer Sci., 35, N 128 (1959).
93. F г у e A. H., H о r s t R. W., P a 1 i о b a g i s M. A., J. Polymer Sci., A2, 1765
(1964).
94. Fry eA. H.,H orst R. W.,P a 1 i ob agi sM. A., J. Polymer Sci., A2, 1785 (1964).
95. M а с к G. P., Modern Plastics Encyclopedia, 37, 333 (1959).
96. LeistnerW.E., К n о е р к е О. Н., Н е с к е г А. С, Пат. США 3015644 (1962);
РЖХим., 1963, 3T196.
97. S a 1 у е г I. О., К e n у о n A. S., Пат. США 2985617 (1961); РЖХим., 1963, 8T139.
98. И т о Т., К а ц у м у р а Т., Японск. пат. 22484 (1963); РЖХим., 1966, 8С390.
99. К о б а я с и Ю., Ф у к у м а Н., К и т а о к а А., Японск. пат. 8479 (1962); РЖХим.,
1964, 15С298.
100. КобаясиЮ.,ФукумаН., КитаокаА., Японск. пат. 8178 (1962); РЖХим.,
1964, 8С358.
101. К а р п о в В. Л., Л е щ е н к о С. С, Ш е в е р д и н а Н. И., Ф и н к е л ь Э. Э.,
Абрамов а Л. В., Авт. свид. СССР 138043 (1961); РЖХим., 1962, 11П43; Бюлл. изобр.,
№ 9 (1961).
Применение оловоорганических соединений
523
102. R a m s d e n H. E., W e i n b e r g E. L., TomkaA.L, Пат. США 2790785 (1957);
РЖХим., 1959, 62996.
103. Hechenbleikner I., BresserR. Е., Н от b e г g О., Пат США 3196129
(1965); РЖХим., 1966, 17С387.
104. Н е d g e s E. S., Tin and its uses, 38, 8 (1957).
105. R a a b E. L., G о r s 1 i n e F. С.,Пат. США 2734926 (1956); С. А., 50, 8945 (1956).
106. А к и е с и С, А с о Т., О м у р a D., J. Chem., Soc. Japan, Ind. Chem. Sec, 64, 497
(1961); РЖХим., 1963, 1C51.
107. AshikpriN., H о n d a M., Bull Chem. Soc. Japan, 34, 767 (1961).
108. T а к а м и Я-, J. Chem. Soc. Japan, Ind. Chem. Sec, 64, 2049, (1961); РЖХим., 1963,
10C74.
109. T а к а м и Я., J- Chem. Soc. Japan, Ind. Chem. Sec, 64, 1841 (1961); РЖХим., 1962,
10P53.
НО. Белы. пат. 557505 (1960); РЖХим., 1961, 16П109.
111. Белы. пат. 556683 (I960); РЖХим., 1961, 15П109.
112. WibergE., StecherO., Andrascheck H.-J., Kr uizb ichler L,
StaudeE., Angew. Chem., 75, 516 (1963).
113. N i en b b ur H. J., BohmH., Her beekR., Пат. ФРГ 1105167 (1961); РЖХим.,
1962, 20П171.
114. T а к а м и Я-, Repts. Govt. Chem. Industr. Res. Inst. Tokyo, 57, 213 (1962); РЖХим.,
1963, 4C41.
115. GreschamW.F, M e г с k 1 i n g N. G., Пат. США 3036269 (1962); РЖХим.,
1963, 9T45.
116. T а к а м и Я., J. Chem. Soc, Japan, Ind. Chem. Sec, 65, 234 (1962); РЖХим., 1963,
22C77.
117. J о у n e r F. В., Пат. США 3072631 (1963); РЖХим., 1964, 19С153.
118. A s h i k a r i N., H i r a t a H., Rev. Electr. Commun. Lab., 11, 164 (1963); РЖХим.,
1964, 9C65.
119. T а к а м и Я., Японск. пат. 13532 (1963); РЖХим., 1965, 9С143.
1?0. Белы. пат. 623911 (1963); РЖХим., 1965, 24С169.
121.Белы. пат. 620081 (1963); РЖХим., 1965, 18С142.
122. К а ш и р е н и н о в О. Е., Сб. «Материалы 4-й научной конференции аспирантов».
Ростовский ун-т. Ростов-на-Дону, 1962, стр. 144; РЖХим., 1963, 14В60.
123. An dersonA. W., Br uce J. M., M e с k 1 i n g N. G., T г ue 11 W. L., Пат. США
3050471 (1962); РЖХим., 1964, 2C171.
124. И т о и К., Японск. пат. 1837 (1964); РЖХим., 1966, 9С228.
125. L о е b W. Е., Пат. США 3166547 (1965); РЖХим., 1966, 11С208.
126. J о у п е г F. В., S h е а г е г N. Н. ,Пат. США 3086964 (1963); С. А., 59, 777 (1963).
127. Я н а г а т а М., Я м а д а А., Ямамуро Й., Коба яс и Э., Японск. пат. 24599
(1963); РЖХим., 1966, 8С225.
128. М i 1 1 t о w п Т. Т. L., Пат. США 3163629 (1964); РЖХим., 1966, 11С205.
129. NewbergR. G., Miller F. D., WagensommerJ., Австрал. пат. 251755
(1964); РЖХим., 1966, 11С210.
130. К е n t о n J. R., A d a m s L. М., Пат. США 3168590 (1965); РЖХим., 1966, 10С194.
131. I s b e n j i a n H., Пат. США 2898330 (1959); РЖХим., 1961, 13П211.
132. А г i e s R. S., Пат. США 2900374 (1959); РЖХим., 1961, 13П212.
133. Франц. пат. 1388997 (1965); РЖХим., 1966, 6С159.
134. Белы. пат. 547618(1959); РЖХим., 1961, 16П123.
135. Р о w 1 а п d S. P., P r i t с h e t t E. G., Пат. США 2916480 (1959); РЖХим., 1961,
23П149.
136. Франц. пат. 1150061 (1958); РЖХим., 1960, 90610.
137. В е г g e r M. N., Chem. a. Ind., 1959, 677.
138. В a um A. L. J., А 1 е х а п d e r D. А., Англ. пат. 858581 (1961); РЖХим., 1962,
15П297
139. A s h i k a r i N., Н i r a t a H., Electr. Commun. Lab. Techn. J., 11, 1029 (1962);
РЖХим., 1964, 9C65.
140. С т о ц к а я Л. Л., Т о п ч и е в А. В., К р е н ц е л ь Б. А., ДАН СССР, 146, 372
(1962).
141. Т о п ч и е в А. В., С т о ц к а я Л. Л., К р е н ц е л ь Б. А., Пласт, массы, 12, 3
(1962).
142. С т о ц к а я Л. Л., Кренцел ь Б. А., ДАН СССР, 151, 595 (1963).
143. С т о ц к а я Л. Л., ЛещеваИ. Ф., Кренцель Б. А., Нефтехимия, 4, 43
(1964).
144. С а г г i с k W. L., J. Am. Chem. Soc, 80, 6455 (1958).
145. Carrie к W. L., Kluiber R. W., Bonner E. R, Wartman L. H.,
BuggF.M., S m i t h J. J., J. Am. Chem., Soc, 82, 3883 (I960).
146. CarrickW. L.,ChasarA.G.,SmithJ.JMJ.Am. Chem. Soc, 82, 5319 (1960).
147. P h i 1 1 i p s G. W., С a r r i с к W. L., J. Polymer Sci., 59, № 168, 401 (1962).
524
Оловоорганшеские соединения
148. GreshamW.F., M е с к 1 i n g N. G., Пат. США 2900372 (1959); РЖХим., 1961,
13П133
149. AnsponH. D., P schorr F. Е., Пат. США 2683705 (1954); РЖХим., 1956, 11180.
150. А и м и М., Н а к а д а Т., Японск. пат. 10344 (1963); РЖХим., 1965, 13С217.
151. VandenbergE. L, Пат. США 3159613 (1964); РЖХим., 1965, 7С246.
152. D и с к Е. W„ W a t t е г m a n J. А., Голл. пат. 106385 (1963); РЖХим., 1965, 8С204.
153. J е п к i n s L. Т., Пат. США 3068212 (1962); РЖХим., 1964, 17С172.
154. R iec he А., В er t z Т., Пат. ФРГ 1081891 (1960); РЖХим., 1962, 5Л111.
155. R i е с h е А., В е г t z Т., Пат. ГДР 20284 (1960); РЖХим., 1962, 4Л116.
156. С a n t e r i n о P. J., Franzus В., Пат. США 3050510 (1962); РЖХим., 1964,
4С161.
157. L о t z R., Пат. ФРГ 1110863 (1962); РЖХим., 1963, 7Т45.
158. А 1 1 е s t о n D. L., D a v i e s A. G., J. Chem. Soc, 1962, 2050.
159. Caldwell J. R., Пат. США 2720507 (1955); РЖХим., 1958, 9834.
160. С о n g i u n d i N. R., J e n к i n s L. Т., Пат. США 3160609 (1964); РЖХим., 1966,
8С258.
161. TakedaY., HayakawaY., FuenoT., FurukawaJ., Makromol. Chem.,
83, 234 (1965).
162. H о s t e t t 1 e r F., С о x E. F., Пат. США 3084177 (1963); РЖХим., 1965, 6H61.
163. E 1 e h a s о n P. Т., L i с a r i J. J., Insulation, 8, № 7, 33 (1962); РЖХим., 1963,
8T243.
164. BritainJ. W.( Ind. Eng. Chem., Prod. Res. and Developm, 1, 261 (1962); РЖХим.,
1964, 3C236.
165. HostettlerR, CoxE.R, Ind. Eng. Chem., 52, 609 (1960).
166. Fridman L., Пат. США 3159605(1964); РЖХим., 1986, 15C518.
167. G u d g e о n H., T r a p p e G., T w i t с h e t t H. J., Англ. пат. 887288 (1961);
РЖХим., 4963, 9T176.
168. HostettlerR, Австрал. пат. 236924 (1962); РЖХим., 1963, 24Т224.
169. К г a u s e К. Н., Kunstoff-Rundschau, 7, 473 (1960); РЖХим., 1962, 1П43.
170. W a t s о п J. W., Н а г t 1 е С, Англ. пат. 976902 (1964); РЖХим., 1966, 10С278.
171. Мег ten R., LoewG., Пат. ФРГ 1110858 (1962); РЖХим., 1963, 11Т226.
172. Н u I s e R., T w i t с h e t t H. J., Англ. пат. 957841 (1964); РЖХим., 1965, 24С277.
173. Н и 1 s e R., T w i t с h e t t H. J., Англ. пат. 899948 (1962); РЖХим., 1964, 7С352.
174. MertenR., LoewG., Пат. ФРГ 1111406 (1962); РЖХим., 1963, 8Т167.
175. В u i s t J. M., M a g s о n M. S., Англ. пат. 980968 (1965); РЖХим., 1966, 8С486.
176. HostettlerR, В а г n e s R., М с L a u g h l i n R., Пат. США 3073788 (1963);
РЖХим., 1965, 10С453.
177. G m i t t e r G. Т., В r a i d r i с h E. V., Пат. США 3050477 (1962); РЖХим., 1964,
5C272.
178. Франц. пат. 138897; (1965); РЖХим., 1966, 9С513.
179. MertenR., В а у е г О., SimmlerW., LoewG., Пат. ФРГ 1111378 (1962);
РЖХим., 1963, 4Т267.
180. MertenR., LoewG., В а у е г О., Пат. ФРГ 1111377 (1962); РЖХим., 1963,
8Т165.
181. М а с к G. P., Chem. Process (USA), 23, 30, 34 (1960); РЖХим., 1961, 6П62.
182. F a t h J., Пат. США 2943548 (1960); РЖХим., 1962, 7П320.
183. W e i n b e r g E. L., Т о m к a L. А., Пат. США 2858325 (1958); РЖХим., 1960, ЗТ184.
184. W e i n Ъ е г g E. L., Т о m к a L. А., Пат. США 2832753 (1958); РЖХим., 1959, 73407
185. В е г е п b a u m М. В., В е г t о z z i E. R., Пат. США 3029267 (1962); РЖХим., 1963,
15Н57.
186. R a m s d e n H. Е., Пат. США 2912448 (1959); РЖХим., 1962, 1Л96.
187. R a m s d e n Н. Е., В а п к s С. К., Пат. США 278994 (1957); РЖХим., 1959, 39597.
188. С о h e n G., M u r p h у С. М., O'R e a r J. G., R a v n e r H., Z i s m a n W. A., Ind.
Eng. Chem., 45, 1766 (1953).
189. AntlerM., Ind. Eng. Chem., 51, 753 (1959).
190. НовиковА. С, К а л у ж н и н а К. Ф., НудельманЗ. Н., Каучук и
резина, 5, 16 (1959).
191. Н о в и к о в А. С, Н у д е л ь м а н 3. Н., Авт. свид. СССР 128461 (1960); РЖХим.,
1962, 4П444; Бюлл. изобр., № 10, 19 (1960).
192. М i п с к 1 е г L. S., С о t t 1 е D. L., L e m i s z к а Т., Пат. США 2967167 (1961);
РЖХим., 1962, 7П360.
193. М i п с к 1 е г L. С, С о t t 1 е D. L., Zemiszkal, Пат. США 2918456 (1959);
РЖХим., 1961, 18П261.
194. Ф у р у к а в а Д., Я м а с и т а С, М а ц у с и т а X., О к а С, J. Soc Rubber Ind.
Japan, 36, 701 (1963); РЖХим., 1964, 15C519.
195. T о m k a L. A., W e i n b e r g E. L., Пат. США 2789106 (1957); РЖХим., 1958, 8942.
196. R a m s d e n H. E., Пат. США 2965661 (1960); РЖХим., 1961, 2Л105.
Применение оловоорганических соединений
525
197. М е г к е г R. L., Пат. США 2956045 (1960); РЖХим., 1962, 17П145.
198. ИсицукаТ., МомононМ., ФудзитаР., Японск. пат. 12945, 12948 (1960);
РЖХим., 1962, 14Л122.
199. F о s t е г W. Е., К о е n i g P. E., Пат. США 2998440 (1961); РЖХим., 1963, 5Т39.
200. PrestonJ., Пат. США 3019204 (1962); РЖХим., 1963, 17Т134.
201. А и 1 t L. H., S а 1 i s b u r у L., Пат. США 3076726 (1963); РЖХим., 1964, 23С463.
202. С и с и д о К., Японск. пат. 6428 (1954); РЖХим., 1958, 8942.
203. LuzW. D., Пат. ФРГ 1103570 (1961); РЖХим., 1963, 9Т491.
204. М е г k e r R. L., Пат. США 2920060 (1960); РЖХим., 1962, 7Л117.
205. D 6 г f е 1 t С, W о 1 f f W., Пат. ФРГ 1084101 (1960); РЖХим., 1962, 22П558.
206. Barnes J. M., Mados L., Organometal. Chem. Rev., 3, 137 (1968).
207. S a 1 q u a i n J., Teintex, 26, 615, 617, 621, 623, 627 (1961); РЖХим., 1962, 10П621.
208. Van d e г К er к G. J. M., Chem. Weekbl., 56, 339 (1960).
209. NoltesJ.G., L u i j t e n J. G. A., van der KerkG. J. M., J. appl. Chem.,
11, 38 (1961).
210. Van der KerkG. J. M., L u i j t e n J. G. A., van EgmondJ.C, Nol-
t e s J. G., Chimia, 16, 36 (1962).
211. Van der К e г к G. J. M., L u i j t e n J. G. A., J. appl. Chem., 6, 56 (1956).
212. Van der К e г к G. J. M., L u i j t e n J. G. A., J. appl. Chem., 11,35(1961).
213. S t e с к e r H. C, Tin and its uses, 41, 13 (1957).
214. К о ч к и н Д. А., В а ш к о в В. И., Д р е м о в а В. П., ЖОХ, 34, 325 (1964).
215. В а г n e t t R. F., Rubber and Plast. Age., 46, 260 (1965); РЖХим., 1965, 20C209.
216. Dor f e 1 tC, Gi 1 ber t H., Пат. ФРГ 1084722(1960); РЖХим., 1962, 5Л110.
217. К о о p m a n s M. J., Пат. США 3097999 (1963); РЖХим., 1965, 21Н402.
218. Голл. пат. 99430 (1961); РЖХим., 1962, 16Л339.
219. Н u e с kv H. J., L u i j t e n J. G. A., Tin and its uses, 45, 8 (1958).
220. Sr i vas t a va T. N.. T a ndon S. K., Indian J. appl. Chem., 27, 116 (1964);
РЖХим., 1966, 5H531.
221. С a m p b e 1 1 I. G. M., F о w 1 e s G. W. A., N i x о n L. A., J. Chem. Soc, 1964, 1389.
222. Hartel K., Angew. Chem., 70, 135 (1958).
223. Lew i s W. R., Chem. Prod., 21, 431 (1958).
224. В 1 и m M. S., В о w e r F. A., J. Econ. Entomol., 50, 84 (1957).
225. G i r a i t i s A. P., К о b e t z P., S h a p i г о Н., Пат. США 3147293 (1964); РЖХим.,
1966, 12H108.
226. Ф у сэ Г., J. Japan Wood. Res. Soc, 7, 151 (1961); РЖХим., 1962, 9Л406.
227. RoosenP., Rev. agric (Belg.), 13, 82 (1960); РЖХим., 1961, 7Л482.
228. D 6 r f e 1 t С, R e i n d 1 e E., Hartel К., Пат. ФРГ 1072814 (1960); РЖХим.,
1962, 9Л107.
229. L e e b r i с к J. R., Пат. США 3167473 (1965); РЖХим., 1966, 11C300.
230. G г и п L.f F г i с к е г Н. H.f Tin and its uses, 61, 8 (1963).
Г лава XIX
АНАЛИЗ ОЛОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Простейшей качественной реакцией на олово является появление
серовато-белого налета двуокиси олова при сжигании вещества на шпателе [1J
или платиновой проволоке [2].
Соединения типа RSnX3 и R2SnX2, а также ионы двух-и
четырехвалентного олова образуют с «пирокатехиновым фиолетовым» окрашенные
комплексы [3]. При прибавлении этого реагента к раствору испытываемого
вещества в небольшом количестве метилового спирта или ацетона он сразу же
окрашивается в интенсивный синий цвет. Если это необходимо, окрашенный
раствор титруют 0,1 N раствором «комплексона III» (динатриевая соль
этилендиаминтетрауксусной кислоты). При этом комплексные оловоорга-
нические соединения распадаются на исходные вещества и раствор
окрашивается в характерный для «пирокатехинового фиолетового» желтый цвет.
Комплексы неорганических солей олова устойчивы по отношению к «комп-
лексону III» [3].
Другой способ применим в основном для определения соединений
ароматического ряда. Исследуемое вещество (около 40 мг) помещают в
небольшую пробирку, прибавляют 1 мл четыреххлористого углерода и три капли
брома. Смесь упаривают при нагревании почти досуха и избыток брома
удаляют током воздуха. К остатку прибавляют несколько капель воды,
нагревают примерно 10 сек., после чего прибавляют три капли разбавленной
соляной кислоты (1:1) и небольшой кусочек магния. Через 30 сек. кашпо
вещества переносят стеклянной палочкой на пропитанную фосфорномолиб-
деновокислым аммонием полоску бумаги; голубое пятно является
положительной пробой [4, 5].
Количественное определение олова чаще всего проводят разложением
вещества до двуокиси олова. Для этой цели предложено применять
дымящую азотную кислоту [6], концентрированную серную кислоту [7, 8], смесь
концентрированной серной кислоты и перекиси водорода [9, 10] или
концентрированной серной и азотной кислот [5,7,11,12].
Для определения олова в нелетучих оловоорганических соединениях
наиболее прост прием Краузе и Беккера [И], в том видоизменении, в
котором его применили в своих работах Кочешков и Несмеянов [12, 13].
Навеску оловоорганического соединения (примерно 0,5 г) помещают в кварцевую
пробирку длиной 12—15 см, внутренним диаметром 2,5 см и весом около 30 г, взвешиваемую
с помощью специального колечка из алюминиевой проволоки. В наклонно поставленной
пробирке навеску осторожно заливают, в случае надобности при наружном охлаждении
снегом, 20 каплями заранее приготовленной смеси из 1 объема дымящей серной кислоты
(например, с 3—8% S03) и 1 объема азотной кислоты (уд. в. 1,50—1,52). Обе кислоты
должны быть проверены на отсутствие зольных примесей. Обычно наступает энергичная
реакция. (В случае появления пламени опыт следует считать испорченным; требуется более
интенсивное охлаждение.) По затихании реакции пробирку, укрепленную наклонно (угол
Анализ
527
приблизительно 20—30° к горизонтальной плоскости), нагревают сначала очень
осторожно, прямо на голом пламени горелки, давая закончиться реакции окисления, что узнается
по полному просветлению потемневшей сначала реакционной массы. В случае необходимости,
каждый раз охлаждая, добавляют новую порцию (по 5—10 капель) той же смеси кислот
и опять греют. После просветления нагревают сначала до полного удаления азотной кислоты
(прекращение выделения окислов азота), а затем и до полного удаления серной кислоты
(прекращение выделения дымящихся паров). Наклонное положение и большая длина
пробирки предохраняют при аккуратной работе от выбрызгивания и позволяют удаление
кислот вести в довольно энергичном темпе, что значительно сокращает время по сравнению с
работой в тигле. Операция удаления кислот занимает около получаса. Под конец пробирку
прокаливают на паяльной горелке до постоянного веса, дают остыть в эксикаторе и
взвешивают Sn02.
Одним из вариантов этого метода является разложение вещества смесью
серной кислоты и нитрата аммония [14].
Летучие оловоорганические соединения вначале обрабатывают
небольшим количеством брома, а затем анализируют как обычно [15—17]. По
другому способу их пропускают в токе инертного газа в смесь серной и азотной
кислот [18] или вещество переносят током кислорода в силикагелевую
трубку, запаивают и сжигают [19].
Содержание олова в оловоорганических соединениях может быть
определено и другими методами. В их основе лежит разложение вещества с
последующим превращением его в растворимое соединение двух- или
четырехвалентного олова. Ион Sn2l~ или Sn4" определяют обычными способами
[20—28]. Примером может служить следующая методика [20].
К навеске вещества прибавляют 6 мл концентрированной серной кислоты и к смеси
при нагревании прибавляют 20 объемов перекиси водорода, пока раствор не станет
совершенно прозрачным. Затем его нагревают до выделения паров серного ангидрида. Если при
этом наблюдается появление коричневой окраски, то добавляют еще несколько капель
перекиси водорода. Образующийся прозрачный раствор разбавляют 150 мл воды, нагревают до
кипения и олово восстанавливают до двухвалентного состояния прибавлением 0,6 г
металлической сурьмы, после чего раствор титруют стандартным раствором KJ—KJ03,
применяя в качестве индикатора крахмал. Для предотвращения окисления воздухом титрование
проводят в атмосфере инертного газа.
В тех случаях, когда оловоорганическое соединение содержит кремний,
определение олова проводят обычным образом, а затем двуокись кремния
удаляют с помощью фтористоводородной кислоты [29—31].
По другому способу [30] смесь окисей кремния и олова обрабатывают
едким натром и серой. При этом образуется растворимый тиостаннат
натрия. Раствор фильтруют и из нагретого до кипения фильтрата сульфид
олова осаждают уксусной кислотой. Последовательной обработкой едким
натром, перекисью водорода и азотной кислотой его превращают в станноновую
кислоту, которую прокаливают до двуокиси олова [30].
Для очистки и идентификации оловоорганических соединений в
различных смесях использованы методы полярографии [32—40], фотометрии [41],
бумажной [42—47] и газо-жидкостной [47—58] хроматографии. Анализ
оловоорганических соединений упоминается в ряде обзорных статей [59—62]
и монографии Губен-Вейля [63].
Разработаны способы определения олова в стабилизированном оловоорга-
ническими соединениями поливинилхлориде [64—68], полистироле [64],
а также в биологических материалах [3, 69—71], в воздухе [72] и в растворах
[73].
Углерод, водород и азот в оловоорганических соединениях определяют
обычными методами. Однако микроопределение углерода и водорода не
всегда дает надежные результаты. Предложено [74] при определении этих
элементов сожжением заменять часть наполнителя (окись меди — хромат
свинца) цилиндром из платиновой сетки, в который помещают
гранулированный свинцовый сурик. При полумикрометоде определения углерода и водо-
528
Оловоорганические соединения
рода в качестве катализатора сожжения применяют ванадат серебра [75].
Анализ на азот в оловоорганических соединениях проводят по
модифицированному Циммерманом [76] методу Прегля — Дюма.
Непредельные связи в оловоорганических соединениях определяют по
родановому числу [77, 78], а подвижный водород в соединениях типа
R„Sn(CH2—С^СН)4~,.г — бензоатом серебра [79].
Общее содержание галоида в оловоорганических соединениях можно
определять по Кариусу [80] или сожжением вещества над платиновым
катализатором с поглощением галогена серебряной сеткой [81].
Что касается галоида, непосредственно связанного с атомом олова, то
он может быть определен потенциометрическим титрованием раствора
вещества в этиловом спирте или ацетоне 0,1 N раствором азотнокислого серебра с
использованием каломельного и платинового электродов [82, 83]. Анализ
можно проводить и прибавляя избыток азотнокислого серебра к навеске
вещества в смеси воды с эфиром с последующим потенциометрическим
титрованием соли серебра 0,1 N соляной кислотой [84]. В ряде случаев
целесообразнее поступать следующим образом [85]
Навеску вещества (0,1—0,5 г) помещают в колбу Эрленмейера на 100 мл и к ней
приливают 5 мл этилового спирта и 10 мл 10%-ного спиртового раствора едкого кали. Колбу
нагревают в течение 10 мин. на водяной бане при периодическом встряхивании. Затем смесь
переносят в делительную воронку, экстрагируют эфиром, водный слой отделяют,
нейтрализуют концентрированной азотной кислотой и дальнейшее определение проводят как
обычно.
Другой способ заключается в обработке вещества избытком 0,1 N
раствора щелочи; последний оттитровывают 0,1 N соляной кислотой по
фенолфталеину [86].
Определение в оловоорганических соединениях остатков органической
кислоты [87, 88] или алкоксигрупп [89] проводят простым титрованием
раствора вещества кислотой или щелочью. Серу в оловоорганических меркап-
тидах определяют титрованием подкисленного соляной кислотой спиртового
раствора оловоорганического соединения титрованным раствором иода
[90].
ЛИТЕРАТУРА
1. И н г а м Р., Р о з е н б е р г С, Г и л ь м а н Г., Р и к е н с Ф., Оловоорганические и
германийорганические соединения. М., ИЛ, 1962.
2. S p i а 1 t er L., В а 1 1 es t er M., Analyt. Chem., 34, 1183 (1962).
3. BiirgerK.,Z. Lebensmittel — Untersuch. und Forsch., 114, 1 (1961).
4. G i 1 m a n H., G о r e a u T. N.. J. Org. Chem., 17, 1470 (1952).
5. S i s i d о K., T a k e d a Y., К i n u g a w a Z., J. Am. Chem. Soc, 83, 538 (1961).
6. PfeifferP., LehnardtR., LuftensteinerH., PradeR., Schnur-
mannK, T r u s k i e r P., Z. anorg. Chem., 68, 102 (1910).
7. G i 1 m a n H., R о s e n b e r g S. D., J. Am. Chem. Soc, 75, 3592 (1953).
8. О k a w a r a R., R о с h о w E. G., J. Am. Chem. Soc, 82, 3285 (1960).
9. S t r a f f о г d N.. Mikrochim. Acta, 2, 306 (1937).
10. D i 1 1 a r d C. R., McNeillE.H., SimmonsD.E, G e 1 d e 1 1 J. В., J. Am.
Chem. Soc, 80, 3607 (1958).
11. KrauseE, BeckerR., Ber., 53, 173 (1920).
12. К ozesc h k о w K- A., Ber., 61, 1659 (1928).
13. К о ч е ш к о в К. А., Н е с м е я н о в А. Н., ЖРФХО, 62, 1795 (1930).
14. К о h a m a J. W., Bull. Chem. Soc Japan, 36, 830 (1963).
15. Gilman H., KingW.B., J. Am. Chem. Soc, 51, 1213 (1929).
16. V a n d er KerkG.J.M., NoltesJ.G., LuijtenJ. G. A., J. appl. Chem,
7, 366 (1957).
17. Neumann W. P., Ann. 653, 157 (1962).
18. P r i с e J. W., Tin nad its uses, 41, 5 (1957).
19. Brown M. P., FowlesG. W. A., Analyt. Chem., 30, 1689 (1958).
20. С a m p b e 1 1 I. G. M., F о w 1 e s G. W. A., N i x о n L. A., J. Chem. Soc, 1964, 1389.
Анализ
529
21. G е у е г R., S e i d 1 i t z H. J., Z. Chem., 4, 468 (1964).
22. F a r n s w о r t h M., P e к о 1 a J., Analyt. Chem., 31, 410 (1959).
23. С 1 a r к R. E. D., Analyst, 61, 242 (1936).
24. С 1 a r к R. E. D., Analyst, 82, 182 (1957).
25. F a r n s w о r t h M., P e к о 1 a J., Analyt. Chem., 26, 735 (1954).
26. ReverchonR., Chim. analyt., 47, 70 (1965).
27. R u s s e 1 1 А. В., Analyst, 84, 712 (1959).
28. FritzG., ScheerH., Z. anorgan. allgem. Chem., 331, 151 (1964).
29. H e n g 1 e i n F. A., L a n g R., SchmackL, Makromol. Chem., 22, 103 (1957).
30. P a p e t t i S., P о s t H. W., J. Org. Chem., 22, 526 (1957).
31. О к a w a r a R., WhiteD.G., F u j i t a n i K-, SatoH., J. Am. Chem. Soc, 83,
1342 (1961).
32. J e h r i n g H., M e h n e r H., Z. Chem., 3, 34 (1963).
33. J e h r i n g H., M e h n e r H., Z. Chem., 4, 273 (1964).
34. M e h n e r H., J e h r i n g H., Z. Chem., 3, 472 (1963).
35. В о с к R., G о r b а с h S., О е s e r H., Angew. Chem., 70, 272 (1958).
36. N a n g n i о t P., M a r t e n s P. H., Anal. Chim. Acta, 24, 276 (1961).
37. D о e r i n g W. v. E., Hoffmann A. K„ J. Am. Chem. Soc, 76, 6162 (1954).
38. Costa G., Ann. Chim. (Rome), 41, 207 (1951); С. А., 45, 10094 (1951).
39. Costa G., Gazz. chim. ital., 80, 42 (1950); С А., 44, 9826 (1950).
40. T о р о п о в а В. Ф., СайкинаМ. К-, Сборник статей по общей химии, т. 1. М.
Изд-во АН СССР, 1953, стр. 210.
41. S k е е 1 R. Т., В г i с к е г С. Е., Analyt. Chem., 33, 428 (1961).
42. W i 11 i a m s D. J., P r i с e J. W., Analyst, 85, 579 (1960).
43. W i 1 1 i a m s D. J., P r i с e J. W., Analyst, 89, 220 (1964).
44. В a r b i e r i R., В e 1 1 u с о U., T a g 1 i a v i n i G., Ann. chim. (Rome), 48, 940 (1958).
45. P e у т о в О. А., ПтицынаО.А, Т у р ч и н с к и й М. Ф., ДАН СССР, 139, 146
(1961).
46. Т а н а к а Й. М о р и к а в а Т., Japan Analyst, 13, 753(1964); РЖХим., 1965, 5Г217.
47. F r a n с J., W u r s t М., М о и d r у V. С, Collect. Czechosl. Chem. Communs, 26, 1313
(1961).
48. PollardF.H., NicklessG., CookeD.L, J. Chromatogr., 13, 48 (1964).
49. M а ц у д a X., M а ц у д а С, J. Chem. Soc. Japan, Industr. Chem. Sec, 63, 1960 (1960);
РЖХим., 1961, 13Д223.
50. A b e 1 E. W., N i с k 1 e s s G., P о 1.1 a r d F. H., Proc. Chem. Soc, 1960, 288.
51. ProschU., Z б p f 1 H.-J., Z. Chem., 3, 97 (1963).
52. SteinmeyerR.D, F e n t i m a n A. F., К a h 1 e r E. J., Analyt. Chem., 37, 520
(1965).
53. T о n g e B. L., J. Chromatogr., 19, 182 (1965).
54. К a e s z H. D., P h i 1 i p p s J. P., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6228
(1960).
55. К a e s z H. D., P h i 1 i p p s J. R., S t о n e F. G. A., Chem. a. Ind., 1959, 409.
56. G e r r a r d W., M о о n e у E. F., R e e s R. G., J. Chem. Soc, 1964, 740.
57. N e u m a n n W. P., Bur khar d tG., Ann., 663, 11 (1963).
58. GeisslerH., KriegsmannH., Z. Chem., 4, 354 (1964).
59. В e 1 с h e r R., GibbonsD., S у k e s A., Mikrochemie ver Mikrochim. Acta, 40,
76 (1952).
60. S у k es A., Mikrochim. Acta, 1956, 1155.
61. Pri ceJ. W., Chem. Trade. J. and Chem. Engr., 141, 1503 (1957).
62. NeumannW.P, Angew. Chem., 75, 225 (1963).
63. Г у б е н - В е й л ь, Методы органической химии, т. 2. Методы анализа. М., Госхим-
издат, 1963.
64. Б е з у г л ы й В. Д., П р е о б р а ж е н с к а я Е. А., Д м и т р и е в а В. Н., ЖАХ,
19, 1033 (1964).
65. ChapmanA.H, Duckworth M. W., P r i с е J. W., Brit. Plastics, 32, 78
(1959).
66. A d a m s о n J. H., Analyst, 87, 597 (1962).
67. N e u b e r t G., Z. analyt. Chem., 203, 265 (1964).
68. S с h r 6 d e r E., M a 1 z S., Plaste a. Kautschuk, 5, 416 (1958).
69. H a r d о n H. J., В r u n i n k H., v a n d e г Р о 1 E. W., Analyst, 85, 847 (1960).
70. G о r b а с h S., В о с k R., Z. analyt. Chem., 163, 429 (1958).
71. G о d a r E. M., A 1 e x a n d e r O. R., Ind. Eng. Chem., Analyt. Ed., 18, 681 (1946).
72. С е л и в о х и н П. И., Вестн. техн. и экон. информ. Н.-и. ин-та техн.-экон. исслед.
Гос. ком-та хим. пром-ти при Госплане СССР, 8, 27 (1964).
73. А ш б е л ь Ф. Б., П а р ш и н а А. М., Г о й з м а н М. С, Ж и ж и н а Л. И., Куп*
цоваК.М, Зав. лаб., 31, 1062 (1963).
74. Si 1 b er t F. С, К i r n er W. R., Ind. Eng. Chem., Analyt. Ed., 8, 353 (1930).
34 Заказ Ко 207
530
Оловоорганические соединения
75. С о 1 a i t i s D., L e s b r e M., Bull. Soc. chim. France, 1952, 1069.
76. Zimmerman W., Mikrorhemie ver. Mikrochim. Acta, 31, 42 (1943).
77. Б у г о р к о в а А. А., М и р о н о в В. Ф., П е т р о в А. Д., Изв. АН СССР, ОХН,
1960, 474.
78. Р е t г о w A. D., M i г о п о w W. F., BugorkowaA. A., Fette, Seifen, Anstrich»
mittel, 62, 1107, (1960); РЖХим., 1961, 19Ж15.
79. L e Q u a n M., В i 1 1 i о t t e J.-C, С a d i о t P., Compt. rend., 251, 730 (1960).
80. G r u t t n e r G., К г a u s e E., W i e r n i k M., Ber., 50, 1549 (1917).
81. SaffordH. W., StragandG. L, Analyt. Chem., 23, 520 (1951).
82. V a n d e г К e r k G. J. M., N о 1 t e s J. G., L u i j t e n J. G. A., J. appl. Chem.,
7, 356 (1957).
83. V a n d e г К e r k G. J. M., N о 1 t e s J. G., J. appl. Chem., 9, 179 (1959).
84. Borbely-KusczmannA., NadyJ., Periodica Politech., 6, 127 (1962).
85. V a n d e г К e r k G. J. M., L u i j t e n J. G. A., J. appl. Chem., 7, 369 (1957).
86. SisidoK., TakedaY.,J. Org. Chem., 26, 2301 (1961).
87. В я з а н к и н Н. С, Р а з у в а е в Г. А., Б р е в н о в а Т. Н., ЖОХ, 34, 1005 (1964).
88. F г а п к е 1 М., G е г t п е г D., W a g п е г D., Z i 1 к h a A., J. Org. Chem., 30, 159t>
(1965).
89. С о n s i d i n e W. J., VenturaJ.J, J. Org. Chem., 28, 224 (1963).
90. S a s i n G. S., S a s i n R., J. Org. Chem., 20, 387 (1955).
СВИНЦОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ВВЕДЕНИЕ
Первое свинцовоорганическое соединение— гексаэтилдиплюмбан—-
было получено Левигом в 1853 г. при реакции сплава свинец—натрий с
йодистым этилом. В дальнейшем химия свинцовоорганических соединений
получила свое развитие в классических работах Грютнера и Краузе, которые
описали соединения типа R4Pb, R3PbX, R2PbX2 и R6Pb2.
Открытие в 1923 г. Мидгли антидетонационного действия тетраэтилсвин-
ца послужило стимулом для промышленного синтеза этого соединения.
По сравнению с другими металлоорганическими соединениями IV
группы периодической системы область свинцовоорганических соединений
развивается значительно медленнее. За последние 20—30 лет были открыты новые
типы свинцовоорганических соединений, например R3PbLi (Гилман с сотр.),
R3PbRe(CO)5 (Несмеянов с сотр.), АгРЬХ3, где X — остаток органической
кислоты (Кочешков с сотр.).
Как правило, свинцовоорганические соединения устойчивы на воздухе.
Соединения типа Alk4Pb — жидкости, Аг4РЬ — твердые вещества, имеющие
определенные температуры кипения или плавления, нерастворимые в воде,
но хорошо растворимые в органических растворителях; Alk3PbX и Alk2PbX2
(X — галоид, остаток органической кислоты или гидроксильная группа)
хорошо растворяются в воде и обычных органических растворителях.
Галогениды три- и диарилсвинца не растворяются в органических
растворителях и не плавятся.
Характерной особенностью свинцовоорганических соединений является
то, что галогениды, ацилаты и другие производные три- и диалкилсвинца
значительно менее устойчивы, чем соответствующие соединения
ароматического ряда. Что касается соединений типа А1кРЬХ3, то о синтезе их имеется
ряд противоречивых данных. Очевидно, они легко разлагаются в момент
образования.
В ароматическом ряду соединения типа АгРЬХ3 (X — галоид) не
получены, так как они разлагаются в момент образования с выделением АгХ
и РЬХ2. Триацилаты арилсвинца по сеоим свойствам приближаются к тет-
раацетату свинца.
Соединения типа (Alk2Pb)„ не выделены. Старые данные о диарилсвинце
(Краузе) в последнее время были подвергнуты критическому рассмотрению
(Ван-дер-Керк).
Для синтеза соединений типа R4Pb, сссбенно получаемых в
промышленных масштабах (тетраэтилсвиыец), главным методом является действие
галоидных алкилов на сплавы Pb—Na в присутствии катализаторов
(например, амш:ов, гльдегилсв, кетоков, сложных зфиров и др.).
34*
532
Свинцовоорганические соединения. Лит. стр. 537—540
Перспективным методом, но пока применяемым в ограниченных
масштабах, является электролиз комплексных металлоорганических соединений на
свинцовом аноде. Электролизу могут быть подвергнуты NaAlR4, NaBR4,
RMgX и т. п. (R — везде алкил).
Из препаративных методов наибольшее применение имеет использование
магнийорганических соединений. Так как четыреххлористый свинец
неустойчив и является сильным окислителем, в магнииорганическом синтезе
его не применяют. Действуют обычно магнийорганическими соединениями
на соли двухвалентного свинца. Реакция, вероятно, отвечает схеме:
2 RMgX + PbX2 -> (R2Pb)„ + 2 MgX2,
(R2Pb)„-*R3PbPbR3,
2R3PbPbR3->3R4Pb+Pb.
В алифатическом ряду в случае тетрапропилсвинца и его высших гомологов
в результате реакции образуется смесь гексаалкилдиплюмбанов и тетраал-
килсвинца. Ее обрабатывают галоидом, а затем вновь реактивом Гриньяра.
В алициклическом и ароматическом рядах соединения типа R3PbPbR3
могут быть выделены из смеси в индивидуальном состоянии.
Методы синтеза свинцовоорганических соединений с помощью литий-,
цинк- или алюминийорганических соединений пока разработаны очень
мало.
Метод разложения двойных солей диазония, широко применяемый для
синтеза ряда металлоорганических соединений, для свинцовоорганических
соединений дает плохие выходы и приложим лишь к простейшим
ароматическим соединениям.
Диазометан и диазоэтан реагируют с хлористым триэтилсвинцом и дву-
хлористым диэтилсвинцом лишь в присутствии катализаторов (медная
бронза) с образованием свинцовоорганических соединений, имеющих галоид в
радикале:
Си С1РЬ(С2Нв)3
CH2N2 > [СН2] > (С2Н5)3РЬСН2С1,
[СН2] [СН2]
(С2Н5)2РЬС12 > [(С2Н5)2РЬС1 (СН2С1)] > (С2Н5)2РЬ (СН2С1)2.
Использование ртутноорганических соединений дает возможность
получить свинцовоорганические соединения классов ArPb(OOCR)3, а также
Ar2Pb(OOCR)2:
2 Ar2Hg + Pb (OOCR)4 -> Ar2Pb (OOCR)2 + 2 ArHgOOCR,
Ar2Hg + Pb (OOCR)4 -» ArPb (OOCR)3 + ArHgOOCR.
Разделение продуктов легко достигается прибавлением к смеси
спиртового раствора хлористого водорода, причем в первую очередь осаждается
ArHgCl. Метод дает возможность получать свинцовоорганические
соединения этих типов с различными заместителями в ароматическом радикале.
Соединения с заместителями в алифатическом радикале легко получаются
при действии на трифенилплюмбиллитий циклических соединений типа
(СН2)лХ (X — кислород, сера, аминогруппа; п = 2, 3 или 4):
(C6H5)3PbLi + (СН2)„Х -U (C6H5)3 Pb (CH2)„XH.
Введение
533
С Р-пропиолактоном образуются свинцовоорганические соединения,
имеющие в радикале карбоксильную группу:
СН2
(QHsfoPbLi + СН2 С=0 -> (С6Н5)зРЬСН2СН2СООЫ.
ч</
В последние годы была открыта интересная реакция присоединения к
кетену гидроокиси и ацетата трифенилсвинца, приводящая к получению
свинцовоорганических соединений с карбоксильной группой в
алифатическом радикале:
(С6Н5)3РЬОН + 4 СН2=С=0 -> [(С6Н5)зРЬСН2СО]20 + (СН3СО)20,
с2н6он
(С6Н5)зРЬООССН3 + СН2=С=0 > (С6Н5)3РЬСН2СООС2Н5 + CHgCOOH
По методу синтеза свинцовоорганических соединений присоединением
свинцовоорганических гидридов к алкенам и алкинам в литературе имеются
пока лишь краткие сообщения. По-видимому, эта реакция для
свинцовоорганических соединений протекает значительно легче, чем для соединений
кремния, германия и олова.
Для свинцовоорганических соединений, так же как и для других метал-
лоорганических соединений IV группы периодической системы, характерны
реакции деалкилирования, гидролиза, реакции, связанные с изменением
функциональных групп у атома свинца, и реакции, связанные с изменением
в радикале — последние развиты очень мало.
Деалкилирование свинцовоорганических соединений с помощью
галоидов и галоидоводородных кислот проходит легко уже при низких
температурах с отрывом одного или двух радикалов. В ароматических соединениях
при действии галоидов происходит отрыв сразу двух радикалов и только
при применении комплекса брома с пиридином удается получить Аг3РЬХ.
Органические кислоты дают в алифатическом ряду Alk3PbOOCR (в
присутствии силикагеля) или сразу Ar2Pb(OOCR)2 в.случае соединений
ароматического ряда.
Из солей металлов наиболее важное препаративное значение в синтезе
свинцовоорганических соединений путем деалкилирования имеет диацетат
ртути, который дает возможность ступенчато отщепить все ароматические
радикалы от Аг4РЬ. Деалкилирование с помощью тетраацетата свинца
описано на отдельных примерах и недостаточно четко.
Гидролиз свинцовоорганических соединений раствором щелочи или
влажной окисью серебра приводит к соответствующим гидроокисям, окисям или
арилплюмбоновым кислотам.
Реакции, связанные с изменением функциональных групп у атома
свинца, например реакции нейтрализации и реакции обмена, описаны на ряде
примеров. Этими методами получены свинцовоорганические производные
минеральных и органических кислот типа R3PbX и R2PbX2 в
алифатическом и ароматическом рядах, алкоксисоединения триалкилсвинца, меркап-
тиды триалкил- и триарилсвинца, а также некоторые соединения со связью
РЬ—N.
Взаимодействием бромидов триалкилсвинца с триалкилсиланолятами
натрия получены соединения типа триалкилсилилокситриалкилсвинца.
Следует, однако, отметить, что различные типы свинцовоорганических
соединений представлены значительно беднее, чем для германия и олова.
Химия свинцовоорганических гидридов начала развиваться лишь в
последние годы. В настоящее время известны только соединения типа
534 Свинцовоорганические соединения. Лит. стр. 537—540
А1к3РЬН, получаемые восстановлением галогенидов триалкилсвинца литий-
алюминийгидридом или метанолизом боранатов триалкилсвинца:
R3PbBH4 + 3 CH3OH-*R3PbH + В (ОСН3)3 + 3 Н2.
Свойства этих соединений изучены очень мало, но уже из имеющихся
данных можно сказать, что они гораздо более активны в реакциях
присоединения, чем соответствующие гидриды германия и олова.
Соединения типа R3PbLi обычно получают действием металлического
лития на галогениды триалкил- и триарилсвинца:
R3PbCl + 2 Li -> R3PbLi + LiCl,
а также действием ароматических литийорганических соединений на двухло
ристый свинец:
3 ArLi + РЬС12 -> A 3PbLi + 3 LiCl.
RgPbLi, так же как и в случае других металлоорганических соединений,
получены только в растворах (окрашенные растворы). Они легко реагируют
с галоидными алкилами и с галогенидами алкил- и арилсвинца, образуя
соединения типа R3PbR' и R6Pb2, однако только в том случае, когда
используются соединения с одинаковыми радикалами. Соединения типа R3PbPbR'3
или R2R'PbPbR'R2 получить не удается.
Номенклатура свинцовоорганических соединений
Формула евин-
цовооргани-
ческого
соединения
Название, принятое
в настоящей книге
Другое,
встречающееся в русской
литературе название
Английское название
Немецкое
на вание
R3PbH
R2PbX2
R3PbX
R4Pb
RPbOOH
R2PbO
R3PbOPbR3
R3PbOH
RsPbLi
RaPbP ^R3
RsPbOR
R3PbSR
Гидрид
триалкилсвинца
Двугалоидпый диал-
килсвинец
Галоидный триалкил-
свинец
Тетраалкилсвинец
А ри л пл юм бо нова я
кислота
Окись диалкилсвипца
Гексаалкилдиплюм-
боксан
Гидроокись
триалкилсвинца
Три алкил плюм 'ил-
литий
Гексаалкилдиплюм-
бан
Алкокситриалкил-
свинец
Алкилмеркалтид
триалкилсвинца
Диалкилдигалоген-
плюмбан
Триалкилгалоген-
илюл'ан
Теграаткнл 1Л'Омбан
То же
То же
Окись триалкилсвин
ца
Трналкилплюмбанол
Литий триалкилсви-
нец
Trialkyllead 4ydride
Trialkylplumbane
Dialkyllead dihalide
Lead dialkyl dihalide
Dialkyldihaloplumba-
ne
Trialkyllead halide
Lead trialkylhalide
Tnalkylhaloplumbane
Tetraalkylleid
Lead tetraalkyl
Arylplumbonic acid
Dfalkyllead oxide
Load dialkyl oxide
Trialkyllead oxide
Trialkyllead hydroxide
Lead trialkyl
hydroxide
Trialkylplumbyllithi-
um
Trialkylleadlithium
Lithium trialkylplum-
bide
Hexaalkyldiplumbane
Hexaalkyldilead
Trialkyllead
Lead trialkyl
Trialkyllead alkoxidc
(Alkylthio) trialkyllead
Alkvl trialkyllead
Sulphide
Trialkylbleihydrid
Dialkylbloidihalo-
gen id
Trialkylbleihaloge-
nid Trialkylhalo-
genplumban
Tetraalkyl blei
BleitetraalkyI
Tetranlkylplu.nban
Dialkylbieioxid
Trialkylbleioxid
Trialkylbleihydro-
xid
Lithiumtriphenyl-
nlumban
Lithiumtriphenyl-
blei
Hexaalkyldiplum-
bart
Trialkylblef
Trialkylblefalkoxi-
de
Введение
53GT
Свинцовоорганические соединения изучались с применением различных
физико-химических методов. Исследовались ультрафиолетовые спектры
П—6], инфракрасные спектры поглощения [4, 7—26], спектры
комбинационного рассеяния [8, 23, 27, 28], спектры ядерного магнитного резонанса
[4, 29—39], спектры электронного парамагнитного резонанса [40].
Проведены рентгеноструктурные исследования [41—43] некоторых ароматических
свинцовоорганических соединений. Измерялись дипольные моменты [4, 44—
501, молекулярный вес [30, 51], растворимость [52, 53], рефракция [53, 54].
Проведены исследования термодинамических свойств
свинцовоорганических соединений: определены теплоты сгорания [8], теплоты образования
[55—60] и некоторые другие константы [8, 23]. Исследовались магнитные
свойства [61—68]. Проведен расчет частоты связиС=Свацетиленидах
свинца [69]. Методом полярографии изучено поведение галогенидов и
гидроокисей триалкилсвинца при их восстановлении на ртутном капельном
электроде [70—75]. Измерена электропроводность свинцовоорганических
соединений в жидком безводном хлористом водороде [76]. Методом дифракции
электронов исследовалась структура тетраметилсвинца [77]. Изучались масс-
спектры [78, 79] и некоторые другие физико-химические свойства
свинцовоорганических соединений [80, 81].
Комплексообразующая способность свинцовоорганических соединений
изучена очень мало. Динитрат, дихлорид и дибромид дифенилсвинца
образуют с пиридином нестабильные комплексы состава (C6H5)2PbX2.4C5H5N
(X — N03, C1 и Вг). Более устойчивы координационные соединения дигало-
генидов дифенилсвинца с двумя молекулами донора [82]. Описано
образование в растворах комплексов 1:1 хлористого триметил- и триэтилсвинца с
основаниями Люиса [83]. Хилл и Генри [84] получили комплексы
хлористого трифенилсвинца, двухлористого и двубромистого дифенилсвинца с
пиридином, диметилсульфоксидом, М,]М-диметилформамидом, 2,2'-дипириди-
лом, 1,10-фенантролином и хлористым тетраметиламмонием.
Комплексы состава [PbAlk4(NO)2]2+(N03)r получены с хорошим выходом
при нитровании тетраэтил- и тетрапропилсвинца с помощью N204 [85].
Своеобразный тип свинцовоорганических комплексов описан в работах
итальянских авторов [86—90].
Что касается применения свинцовоорганических соединений, то
наиболее широкое использование находит тетраэтилсвинец, применяемый в
качестве антидетонатора. По данным 1965 г., ежегодная выработка
тетраэтилсвинца составляет около 200 000 т. Как указывалось, антидетонационные
свойства тетраэтилсвинца были открыты в 1923 г. [91—93] и с этого времени
проведены многочисленные исследования и имеется обширная патентная
литература, посвященная этому вопросу. Механизм антидетонационного
действия тетраэтилсвинца до сих пор не совсем ясен, хотя по этому вопросу
предложено несколько теорий [94, 95].
Сделано много попыток использовать свинцовоорганические соединения
в качестве катализаторов и инициаторов полимеризации непредельных
соединений [96]. В литературе описано использование свинцовоорганических
соединений в следующих реакциях: тетраэтилсвинец использовался в
качестве инициатора полимеризации этилена [97—100], пропилена [100, 101],
винилхлорида [102, 103], винилацетата, метилметакрилата [103],
полимеризации дегидрированных кислых смол [104], теломеризации этилена с
толуолом [105]; тетравинилсвинец — в полимеризации акрилонитрила [106];
тетрациклогексилсвинец — в полимеризации винилацетата [107]. Сделаны
попытки полимеризации олефинов на ряде свинцовоорганических
соединений [108]. Соединения типа R4Pb использовались в качестве катализаторов
получения полиэфиров [109]. Пропилен полимеризовался на комплексах
A(MR'R"R'"),rAeA — Rb, K;M —Pb [1101. Полимеризация 1-нитропропилена
536
Свинцовоорганжеские соединения. Лит. стр. 537—540
была проведена на тетраэтилсвинце [111]. Имеется большая литература,
главным образом патентная, касающаяся вопроса применения свинцово-
органических соединений в качестве компонентов (совместно с галогени-
дами металлов, главным образом переходных) смешанных катализаторов
полимеризации этилена [112—121], пропилена [122—124], акрилонитрила
[125], стирола и винилхлорида [126]. Смесь тетраэтилсвинца и алюминийор-
ганических соединений использовалась в качестве катализатора
полимеризации этилена [127] и пропилена [128]. Тетраэтилсвинец совместно с акри-
лонитрилом [129] или бутиллитием [130] является активным катализатором
полимеризации этилена. Имеются указания об использовании свинцовоор-
ганических соединений в качестве стабилизаторов, например полиэтилена и
полипропилена при термообработке [131], стабилизатора галоидсодержа-
щих смол [132].
Тетрафенилсвинец вводился в стирол с целью получения материалов
для сцинтилляционных детекторов [133]. Сделаны попытки применения
свинцовоорганических соединений и в качестве присадок к минеральным
маслам [134]. Запатентован и ряд других способов применения
свинцовоорганических соединений [135—137].
Литература, приведенная по вопросу применения свинцовоорганических
соединений, не является исчерпывающей.
Полимерные материалы, имеющие в молекуле свинец, пока не нашли
своего практического применения [138]. Тетравинилсвинец в литературе описан,
однако он термически мало стоек и неспособен к полимеризации [139].
Триэтилвинилсвинец и диэтилдивинилсвинец разлагаются с выделением
металлического свинца под действием перекисных катализаторов при 120—
130°С [140]. Попытка получить продукты сополимеризации триэтилвинил-
свинца со стиролом и а-метилстиролом привела к получению материала,
содержащего от 4,5 до 5% свинца [140,141]. Сделана попытка
полимеризации аллилтрифенилсвинца в присутствии перекисей или азотсодержащих
соединений, но уже при 100 — 120° С вещество разлагается с выделением
металлического свинца. Брло установлено, что аллилтрифенилсвинец инги-
бирует полимеризацию ванильных мономеров (стирол, метилметакрилат)
даже в присутствии инициаторов полимеризации [142]. Попытка полимери-
зовать виниловые мономе-р-ы типа (СН3)зРЬ(СН2)зСН=СН2 на алюминий-
органических соединениях в смеси с четыреххлористым титаном показала,
что полимеризация проходит с очень низким выходом и до низкого
молекулярного веса [143].
Что касается полимеризации свинцовоорганических производных
стирола, то эти соединения легко полимеризуются под влиянием катализаторов
типа азодиизобутиронитрила. Проведена полимеризация триметил-д-
стирилсвинца и триметил-а-метил-д-стирилсвинца 1144], триэтшых-метил-
стирилсвинца [145, 146] и триэтил-я-стирилсвинца [147]. Ряд работ
посвящен изучению полимеризации трифенил-д-стирилсвинца [147—153].
Проведена также сополимеризация трифенил-д-стирилсвинца со стиролом
[154, 155] и винилтолуолом [155]. Присоединение дигидрида дифенилолова
к дифенилдистирилсвинцу привело к образованию полимерного материала
[156].
Помимо проведенных опытов по полимеризации непредельных
свинцовоорганических соединений Котоном с сотрудниками была проведена
полимеризация различных свинцовоорганических ацилатов: метакрилата трифе-
нилсвинца [150, 151], акрилата трифенилсвинца [150], диметакрилата ди-
фенилсвинца [157, 158] и д-винилбензоата трифенилсвинца [157—159].
Получены свинцовоорганические производные ряда азотсодержащих
непредельных кислот, изучена их способность к полимеризации и некоторые
свойства полимеров [160].
Введение
537
Свинцовоорганическим соединениям посвящены специальные
обзорные статьи Гилмана с сотр. [161] и Виллемсенса [162]. Кроме этого, в ряде
обзорных статей отдельные свойства свинцовоорганических соединений
описаны вместе с германий-и оловоорганическими соединениями [163—
[170]. В литературе имеется несколько обзоров, посвященных специально
методам синтеза и свойствам тетраэтилсвинца [171 —174].
ЛИТЕРА ТУ РА
1. La P a g 1 i a S. R., J. Moles. Spectrosc, 7, 427 (1961).
2. BowdenK, В r a n d e E. A., J. Chem. Soc, 1952, 1068.
3. В а о С. N. R., Ramachandran J., BalasubramanianA., Canad.
J. Chem., 39, 171 (1961).
4. D a v e L. D., E v a n s D. F., W i 1 k i n s о n G., J. Chem. Soc, 1959, 3684.
5. D r e n t h W., J a n s s e n M., J., v a n d e r Kerk G.J. M., V 1 i egent-
h a r t J. A., J. Organometal. Chem., 2, 265 (1964).
6. H a g u e D. N., P r i n с e R. H., J. Chem. Soc, 1965, 4690.
7. H e n г у M. C, N о 1 t e s J. C, J. Am. Chem. Soc, 82, 555 (1960).
8. Lippincott E. R., Tobin M. C, J. Am. Chem. Soc, 75, 4141 (1953).
9. T a i m s a 1 u P., W о о d J. L., Trans. Faraday Soc, 59, 1754 (1963).
10. ВышинскийН.Н., РудневскийН. К-, Сб. «Спектроскопия». М., «Наука»,
1964, стр. 115.
11. N ol t es J. G., HenryM.C, JanssenM.J, Chem. Ind (London), 1959, 298.
12. W e s t R., В a n e у R. H., J. Phys. Chem., 64, 822 (1960).
13. J anssenM. J., L u i j t e n J. G. A., van d e r Kerk G. J. M., Rec. Trav.
Chim., 82, 90 (1963).
14. О k a w a r a R., S a k i у a m a M., Kagaku No Ryoiki Zokan, 45, 127 (1958); C. A., 55,
17211a (1961).
15. F r i t z H. P., FischerE.O., J. Chem. Soc, 1961, 547.
16. F r i t z H. P., S с h n e i d e r R., Ber., 93, 1171 (1960).
17. О k a w a r a R., S a t о H., J. Inorg. Nucl. Chem., 16, 204 (1961).
18. О и к а в a X., J. Chem. Soc Japan, Pure Chem. Sec, 84, 1 (1963); РЖХим., 1964,
9Б138.
19. Вышинский Н. Н., РудневскийН. К-, Оптика и спектроскопия, 10, 797
(1961).
20. Богомолове. Г., ВеселковаИ. А., ЛодочниковаВ. И., Труды
Комиссии по спектроскопии АН СССР. М., Изд-во АН СССР, 1964, вып. 1, стр. 475.
21. О б р е и м о в И. В., Ч у м а е в с к и й И. А., Ж- структурн. химии, 5, 59 (1964).
22. ВышинскийН. Н., Рудневский Н. К-, Труды по химии и хим. технол.
(Горький), 1960, 538.
23. CrowderG.A., GorinG., К г u s e F. H., S с о t t D. W., J. Molec Spectrosc,
16, 115 (1965).
24. S h e 1 i n a R. K., P i t z e r K. S., J. Chem. Phys., 18, 595 (1950).
25. ВышинскийН. Н., Александров Ю. А., РудневскийН. К., Изв.
АН СССР, серия физ., 26, 1285 (1962).
26. К a w a s a k i Y., T a n а к а Т., О к a w a r a R., Bull. Chem. Soc. Jap&iv 37, 903
(1964); РЖХим., 1965, 8Ж19.
27. S i e b e r t H., Z. anorgan. allgem. Chem., 263, 82 (1950).
28. J а с к s о n J. A., N i e 1 s e n J. R., J. Molek. Spectrosc, 14, 320 (1964).
29. F 1 i t e г о f t N., К a e s z H. D., J. Am. Chem. Soc, 85, 1377 (1963).
30. M ii 1 1 e r E., G u n t e r F., S с h e f f 1 e г К-, F e t t e 1 H. Ber., 91, 2888 (1958).
31. KloseG., Ann. Phys. (DDP), 10, 391 (1963).
32. S m i t h G. W., J. Chem. Phys., 42, 4229 (1965).
33. В r u g e 1 W., A n к е 1 Th., К г u с к e b e r g F., Z. Elektrochem., 64, 1121 1960).
34. N a r a s i m h a n P. Т., RogersM.T, J. Chem. Phys., 34, 1049 (1961).
35. П е т р о в А. А., Л е б е д е в В. Б., ЖОХ, 32, 657 (1962).
36. F r i t z H. P., S с h w a r z h a n s К. Е., J. Organometal. Chem., 1, 297 (1964).
37. D r a g о R. S., Matwiyof f N. A., J. Organometal. Chem., 3, 62 (1965).
38. S с h n e i d e r W. G., В u с к i n g h a m A. D., Disc. Faraday Soc, 1962, 147.
39. Singh G., J. Organometal. Chem., 11, 133 (1968).
40. M u 1 1 e r E., G u n t e r F., S с h e f f 1 e r K., FettelH., Ber., 91, 2888 (1958).
41. ЖдановГ.С, И с м а и л з а д е И. Г., ДАН СССР, 68, 95 (1949).
42. Ж Д а н о в Г. С, И с м а и л з а д е И. Г., ЖФХ, 21, 1495 (1950).
43. Т е р 1 у J., М а 1 у J., Chem. Zvesti., 7, 463 (1953); С. А., 48, 10427b (1954).
44. S m у t h С. P., J. Am. Chem. Soc, 63, 57 (1941).
538
Свинцовоорганические соединения
45. L e w i s G. L., О e s p e r P. F., S m у t h С P., J. Am. Chem. Soc, 62, 3243 (1940).
46. Weiss E., Z. anorgan. allgem. Chem., 286, 236 (1956).
47. StrohmeierW, M i 1 t e n b e r g e г К-, Z. Physik. Chem. (Frankfurt), 17, 274
(1958).
48. FischerE.O., SchreinerS., Ber., 92, 938 (1959).
49. КаширениновО. Е., Сб. «Материалы 4-й научной конференции аспирантов.
Ростовский ун-т». Ростов-на-Дону, 1962, стр. 144; РЖХим., 1963, 14В60.
50. М а 1 a t es t a L., Gazz. chim. ital., 73, 349 (1943); С. А., 41, 1137h (1947).
51. StrohmeierW., H u m p e n e г К-, M i 1 t e n b e г g K., S e i f e r t F., Z. Ele-
ctrochem., 63, 537 (1959).
52. StrohmeierW., Miltenberger K., Ber., 91, 1357 (1958).
53. Fel d h a k eC. J., St evens CD., J. Chem. and Engng. Data, 8, 196 (1963);
РЖХим., 1964, 8Б622.
54. V о g e 1 A. J., С r e s s w e 1 1 W. Т., L e i с e s t e r J., J. Phys. Chem., 58, 174 (1954).
55. С r e s s w e 1 1 W. Т., L e i с e s t e r J., V о g e 1 A. I., Chem. a. Ind., 1953, 19.
56. LautschW.R, Chem. Tech. (Berlin), 10, 419 (1958).
57. S с о t t D. W., G о о d W. D., W a d d i n g t о n G., J. Phys. Chem., 60, 1090 (1956).
58. GoodW. D., ScottD.W., LacinaJ.L., McCul lough J. P., J. Phys.
Chem., 63, 1139 (1959).
59. G о о d W. D., S с о t t D. W., Pure Appl. Chem., 2, 77 (1961).
60. ТельнойВ.И., Р а б и н о в и ч И. Б., ЖФХ, 39, 2076 (1965).
61. В о t h о г е 1 P., Ann. Chim. (Paris), [13], 4, 669 (1959).
62. F i s с h е г Е. О., Р i e s b e r g e n U., Z. Naturforsch., lib, 758 (1956).
63. F i s с h e r E. O., G r u b e r t H., Z. anorgan. allgem. Chem., 286, 237 (1956).
64. P г е с k e 1 R., S e 1 w о о d P. W., J. Am., Chem. Soc, 62, 2765 (1940).
65. Д о р ф м а н Я. Г., ДАН СССР, 125, 765 (1959).
66. M о r r i s H., S e 1 w о о d P. W., J. Am. Chem. Soc, 63, 2509 (1941).
67. P г е с k e 1 R., S e 1 w о о d P. W., J. Am. Chem. Soc, 62, 2765 (1940).
68. К a d о m t z e f f I., Compt. rend., 226, 661 (1948).
69. Г а с т и л о в и ч Е. А., Ш и г о р и н Д. Н., К о м а р о в Н. В., Оптика и спектро-
копия, 16, 46 (1964).
70. Costa G., Ann. chim., 40, 541 (1950); С. А., 45, 6507 (1951).
71. Торопова, Сайкина М. К-, Сборник статей по общей химии, т. 1. М., Изд-во
АН СССР, 1953, стр. 210.
72. Costa G.,"Ann. chim., 41, 207 (1951); С. А., 45, 10094 (1951).
73. R i с с о b о n i L., Gazz. chim. ital., 72, 47 (1942); С. А., 37, 574 (1943).
74. С а й к и н а М. К., Уч. зап. Каз. ун-та, 116 (2), 129 (1956).
75. К о р ш у н о в И. А., М а л ю г и н а Н. И., ЖОХ, 31, 1062 (1961).
76. R е а с h М. Е., W a d d i п g t о п Т. С, J. Chem. Soc, 1961, 1238.
77. В г о с k w а у U. О., J e n k i n s H. О., J. Am. Chem. Soc, 58, 2036 (1936).
78. DibelerV.H., J. Res. Natl. Bur. Standards, 49, 235 (1952).
79. H о b г о с k B. G., К i s e r R. W., J. Phys. Chem., 65, 2186 (1961).
80. F r e n с h F. A., R a s m u s s n R. S., J. Chem. Phys., 14, 389 (1946).
81. T e г а о К-, Japan. J. appl. Phys., 2, 364 (1963); РЖХим., 1964, 16Б595.
82. P f e i f f e r P., T r u s k i e r P., D i s s e 1 k a m p P., Ber., 49, 2445 (1916).
83. M at wi у of f N. A., Dr ago R.S. Inorg. Chem., 3, 337 (1964).
84. H i 1 1 s K., H e n г у М., J. Organometal. Chem., 3, 159 (1965).
85. H e t n a s k i В., U r b a n s k i Т., Tetrahedron, 19, 1319 (1963).
86. С г о a t t о U., В a r b i e r i R., Ricerca Scient, 8, Sez A, Parte 2 (1965); РЖХим.,
1966, 12B189. ^
87. BarbieriR., Ricerca Scient., 3, Sez A, Parte 2, 635 (1963); РЖХим., 1964, 8Б742.
88. G i n s t i n i a n i M., F a r a g 1 i a G., BarbieriR., J. Chromatogr., 15, 207
(1964).
89. В a r b i e r i R., Far agli aG., Gi usti ni an i M., Ricerca Scient., 4, Sez. A.,
Parte2, 109 (1964); РЖХим., 1966, 1B61.
90. BarbieriR., F a r a g 1 i a G., G i u s t i n i a n i M., R о n с u с с i L., J. Inorg.
and Nucl. Chem., 26, 203 (1964).
91. M i d g 1 e у Т., Н о с h w a 1 t С. А., С а 1 i g a e r t G., J. Am. Chem. Soc, 45, 1821
(1923).
92. G a r r e t t А. В., J. Chem. Educ, 39, 414 (1962).
93. N i с k e r s о n S. P., J. Chem. Educ, 31, 560 (1954).
94. R о s s A., R i f k i n E. В., Ind. Eng. Chem., 48, 1528 (1956).
95. R i f k i n E. B. R., Walcutt C, Ind. Eng. Chem., 48, 1532 (1956).
96. HarwoodJ.H., Industr. Chemist., 36, 437 (1960).
97. С h r i s t 1 R. J., R о e d e 1 M. J., Пат. США 2897183 (1959); РЖХим., 1961, 13П129.
98. Г о л ь д ш т е й н А. Л., Л а п и с о в а Н. П., 3 о р и н а Н. П., Авт. свид. СССР
138040 (1961); РЖХим., 1963, 4П147; Бюлл. изобр., № 9, 45 (1961).
99. Англ. пат. 811865 (1959); С. А., 53, 17576е (1959).
Введение
539
100. J о у n e r F. В., Пат. США 3072631 (1963); РЖХим., 1964, 19С153.
101. К о о j m а п P. L., G h i j s e n W. L., Rec. Trav. Chim., 66, 247 (1947).
102. H а к ам у р а X., ФудзитаЮ., НагатаХ., Японск. пат. 17995 (1961);
РЖХим., 1964, 20С144П.
.103. FurukawaJ., TzurutaT., Bull. Inst. Chem. Res. Kyoto Unv., 38, 319 (1960);
РЖХим., 1961, 21P69.
104. BreslowD.S., Пат. США 2554810 (1951); С. А., 45, 8809 (1954).
105. ChenicekJ. A., BlochH. S. (Universal Oil Products Co.), Пат. США 2867673
(1959); С. А., 53, 14053h (1959).
106. A r n о 1 d L. E. (Rohm and Haas Co.), Пат. США 2991276 (1961); С. А., 55, 26535g
(1961).
107. К о т о н М. М., ДАН СССР, 88, 991 (1953).
108. С en t eri i n о P. J., F r a n z u s В., Пат. США 3050510 (1962); РЖХим., 1964,
4С161.
109. С а 1 d w e 1 1J. R., W е 1 1 rn a n J. W., Пат. США 2720505 (1955); РЖХим., 1958,
9838.
ПО. Англ. пат. 914144 (1962); РЖХим., 1964, 4Н7П.
111. Козлов Л. М., Драбкина Л. С, БурмистроваВ.И., Труды Казанск.
хим.-технол. ин-та, вып. 30, 115 (1962).
112. I s b е п j i a n H., Пат. США 2898330 (1959); РЖХим., 1961, 13П211.
113. R а у R. I., S i s t r u n k Т. О., Пат. США 2868772 (1959); РЖХим., 1961, 8П139.
114. ГольдштейнА. Л., Л а п и с о в а Н. П., 3 о р и н а Н. П., Пластмассы, 11,
3 (1960).
115. Англ. пат. 838227 (1961); С. А., 55, 6039h (1961).
116. В и а Е., М а 1 a t e s t а А., N е g г о т a n t i A. (Montecatini), Итал. пат. 531219
(1960); С. А., 54, 4048d (1960).
117. В е s s a n t К. Н. С, L а с h о w i с z S. К. (Distillers Co.), Англ. пат. 795882 (1958);
С. А., 52, 21242е (1958).
118. Пат. США 3332927 (1967); Organomet. Compd., 1, 178 (1967).
119. Union Carbide Corp., Англ. пат. 873498 (1962); С. А., 56, 3654i (1962).
120. Phillips Petroleum Co., Англ. пат. 796530 (1959); С. А., 53, 1836 (1959).
121. BruceJ.M. (du Pont), Пат. США 2992190 (1961); С. А., 55, 27978 (1961).
122. N i e n b u r g H. J., В 6 h m H., HerbeckR. (BASF), Пат. ФРГ 1105167 (1961)
С. А., 55, 26541b (1961).
123. S о b u e H., S a i t о Y., J. Chem. Soc. Japan, 62, 1110 (1959); С. А., 55, 13901e (1961).
124. J о у n e r F. B. (Eastman Kodak), Пат. США 3072631 (1963); С. А., 58, 7006e (1963).
125. MarvelCS, Wool for d R. GM J. Am. Chem. Soc, 80, 830 (1958).
126. Solvay and Cie, Бельг. пат. 545968 (1960); С. А., 54, 16924 (1960).
127. Rohm and Haas Co, Англ. пат. 771746—7 (1957); С. А., 51, 12542 (1957).
128. FreimillerLR., McKeeverC. H. (Rohm and Haas), Пат. США 2786035
(1957); С. А., 51, 9210f (1957).
129. H e i 1 i g m a n n R. G., S t i с k n e у Р. В. (Bordon Co), Пат. США 2765297; С. А., 51,
405li (1957).
130. В u e n i n g R. (Dimanig — Nobel), Пат. ФРГ 1144483 (1963); С. А., 58, 11481h (1963).
131. S a 1 у e r I. О., К e n у о n A. S., Пат. США 2985617 (1961); РЖХим., 1963, 8T319.
132. RichardW.R., Пат. США 2477349 (1949); С. А., 44, 4718 (1949).
133. Б а р о н и Е. Е., К и л и н С. Ф., Л ебеадз.еТ. Н., РозманИ.М., Ш о-
ния В. М., Атомная энергия, 17, 497 (1964).
134. А 1 t a m i r a M. S., Пат. США 2493213 (1950); С. А., 44, 2229^4250).
135. С h е п i с е k J., В 1 о с h H. S., Пат. США 2867673 (1959); РЖХим., 1961, 4Л103.
136. R i d g w a y J. A., Industr. and Engng. Chem., 50, 1531 (1958).
137. AkermanK., S z u с h n i к A., Internat. J. appl. Radiat. and Isotopes, 15, 319
(1964); РЖХим., 1966, 11H2.
138. Б о н н о И., Усп. хим., 34, 850 (1965).
139. J u e n g е Е. С, С о о к S. Е., J. Am. Chem. Soc, 81, 3578 (1959).
140. К о р ш а к В. В., П о л я к о в а А. М., С у ч к о в а М. Д., Высокомолек. соед., 2,
13 (1960).
141. Montecatini, Итал. пат. 589299 (1961); С. А., 55, 5034с (1961).
142. К о т о н М. М., К и с е л е в а Т.М., Запевал оваН. П., ЖОХ, 30, 186 (1960).
143. LongiP., BernardiniF., Colombo L., Rent. 1st. Lombardo Sci. e lettere
CI. mat., fis., chim. e geol., 93, 134 (1959); РЖХим., 1962, 10P60.
144. N о 1 t e s J. G., В u d d i n g H. A., v a n d e r KerkG.J.M., Rec. Trav. Chim.,
79, 1076 (1960).
145. КоршакВ. В., Полякова А. М., ТабовцеваЕ. С, Высокомолек.
соед., 1, 1021 (1959).
146. Полякова A.M., К о р ш а к В.В., ТамбовцеваЕ. С, Высокомолек. соед.,
3, 662 (1961).
540
С винцовоорганические соединения
147. Р а г s H. G., G r a h a m W. A. G., A t к i п s о п E.R., MorganC.R., Chem. Ind.
(London), 1960, 693.
148. Котон М. М., Киселева Т. М., Флор и некий Ф. С, Изв. АН СССР, ОХН,
1959,948.
149. Котон М. М., Киселева Т. М., ФлоринскийФ. С, Сб.
«Международный симпозиум по макромолекулярной химии», секция I. M., Изд-во АН СССР, 1960,
стр. 167.
150. К о т о н М. М., Киселева Т. М., Ф л о р и н с к и й Ф. С, Высокомолек. соед.,
2, 1639 (1960).
151. К о т о н М. М., Киселева Т. М., А р х и п о в а И. Л., Высокомолек. соед., 6,
1496 (1964).
152. К о t о п М. М., К i s е 1 е v а Т. М., F Ьо г i п s k i j F. S., J. Polymer Sci., 52, 237
(1961).
153. КотонМ.М, ФлоринскийФ. С, ЖОХ, 32, 3057 (1962).
154. К о т о н М. М., Д о к у к и н а Л. Ф., Высокомолек. соед., 6, 1791 (1964).
155. S t а п 1 е у R. S., D a n п i п J., T s о п К. С, J. Polymer Sci., A3, 3199 (1965).
156. N о 1 t e s J. G., v a n d e r KerkG.J.M., Rec. Trav. Chim., 80, 623 (1961).
157. К о ч к и н Д. А., Н о в и ч е н к о Ю. П., К у з н е ц о в а Г. И., Л а й н е Л. В.,
Авт. свид. СССР 133224 (1960); РЖХим., 1962, 8П150; Бюлл. изобр., № 21, 47 (1960).
158. К о ч к и н Д. А., ДАН СССР, 135, 857 (1960).
159. КотонМ.М. К и с е л е в а Т. М., Изв. АН СССР, ОХН, 1961, 1783.
160. К и с е л е в а Т. М., К о т о н М. М., Четыркина Г. М., Изв. АН СССР, ОХН,
1962, 1798.
161. LeeperR. W., Summers L.,GilmanH., Chem. Rev.* 54, 101 (1954).
162. W i 1 1 e m s e n L. С, Organolead Chemistry. New York, International Lead Zinc
Research Organization, 1964.
163. В я з а н к и н Н. С, К р У г л а я О. А., Усп. хим., 35, 1388 (1966).
164. Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Усп. хим., 35, 1430 (1966).
165. A i t k е п I. D., S h е 1 d о п R., S t а р 1 е t о n G. В., Brit. Plast., 34, 662 (1961).
166. Б р а у н Д. В., Усп. хим., 31, 769 (1962).
167. М а г 1 е t t E. M., Ann. N. Y. Acad. Sci., 125, 12 (1965).
168. Н е п г у М. С, D a v i d s о n W. E., Ann. N.* Y. Acad. Sci., 125, 172 (1965).
169. SchmidbauerH, Angew. Chem., 77, 206 (1965).
170. Luijten J. G. A., Ricjkens F., van der Ker к G. J. M., Advances Orga-
nometal. Chem., Vol. 3, New York — London, 1965, S. 397.
171. ShapiroH., Advan. Chem. Ser., 23, 290 (1959).
172. КоршакВ.В., Колесников Г. С, Тетраэтил свинец. М., Госхимиздат, 1946.
173. КоршакВ.В., К о л е с н и к о в Г. С, Усп. хим., 15, 325 (1946).
174. ХиратаКачакуКогё, Chem. Ind., (Japan), 7, 226 (1956); РЖХим., 1958,
74841.
Глава I
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ ГАЛОИДНЫХ АЛКИЛОВ НА СВИНЕЦ,
ЕГО СПЛАВЫ И СОЛИ
В литературе имеются только отдельные указания на то, что
металлический свинец может реагировать с галоидоуглеводородами с образованием
свинцовоорганических соединений [1, 2]. Синтетического значения эти
реакции не имеют.
Неизмеримо более важную роль в химии свинцовоорганических
соединений играет взаимодействие сплавов свинца и щелочных металлов (чаще всего
натрия) с галоидными алкилами. Открытая еще в 1853 г. [3,4], эта реакция
в дальнейшем приобрела значение в качестве промышленного метода
получения тетраалкильных соединений свинца, именно тетраэтилсвинца. В
простейшей форме она может быть выражена уравнением:
4 RX + 4 PbNa -* R4Pb -f 4 NaX + 3 Pb.
На самом деле процесс протекает, несомненно, гораздо сложнее. Его
рассмотрению посвящены специальные обзоры [5,6], поэтому здесь этот метод
изложен очень кратко. Патентная литература в данной главе приведена не
исчерпывающе.
Синтез тетраэтилсвинца проводят нагреванием до 80—90° С хлористого
зтила и сплава Pb—Na. В патентах указаны иногда и другие
температурные условия проведения реакции [7—10]. Имеется ряд патентов [11—19],
в которых предложены различные способы приготовления сплава, причем
содержание в них натрия колеблется в самых широких пределах. Описано
применение и тройных сплавов Na — Pb — К [20 — 22], Na — Pb —Mg
[23—25], а также сплавов свинца с литием [26, 27], магнием [28] или
кальцием [29, 30]. Проведены реакции с бромистым и йодистым этилом [30а].
Найдено, что реакции сплавов Pb — Na, чаще всего с хлористыми
алкилами, значительно ускоряют каталитические количества различных
органических веществ, например альдегидов [31],кетонов [32, 33], триалкил- и
триарилфосфитов [34], органических соединений кремния, титана, алюминия
или циркония [35—36] и т.п. [37—40]. Рекомендуется также прибавлять
к смеси сплява и галоидного алкила небольшие количества поверхностно-
активных веществ [41—43].
В отдельных случаях тетраэтилсвинец получают нагреванием сплава
Pb — Na с диэтилсульфатом [44] или с этиленом под давлением водорода
[45]. С изопропиловым спиртом или ацетоном в сернокислой среде сплав
Pb — Na образует окрашенные соединения, окисление которых приводит,
по-видимому, к смеси гидроокисей три- и диизопропилсвинца [46, 47].
Предложены различные технологические схемы ведения процесса [48—
54] и выделения продуктов реакции (отгонка с паром, экстракция избытком
галоидного алкила или другим органическим растворителем и др.) [55—68].
Метод был применен для синтеза тетраметил-[69], тетраэтил-[70], тетра-я-
пропил-[70] и тетраизобутилсвинца 169] в лабораторных масштабах.
542 Свинцовоорганические соединения. Лит. стр. 543—544
Получение тетраэтилсвинца [79]. Сплав свинца с натрием приготовляют следующим
образом. 390 г металлического свинца расплавляют в железном тигле и к нему прибавляют
110 г натрия порциями примерно по 5 г. После каждого прибавления порции натрия тигель
закрывают крышкой. После добавления всего количества натрия смесь перемешивают,
очищают с поверхности и расплавленном виде выливают в цилиндрический сосуд
соответствующих размеров. Синтез тетраэтилсвинца проводят в атмосфере сухого инертного газа.
400 г сплава измельчают и по возможности быстро переносят в трехгорлую колбу емкостью
1 л, снабженную мешалкой, обратным холодильником и капельной воронкой. Сплав
заливают 300 мл бромистого этила, прибавляют 32 мл пиридина и смесь нагревают на водяной
бане, добавляя с интервалами в 1 час 2—3 капли воды. После 8-часового нагревания
остаток сплава разлагают водой и смесь перегоняют с водяным паром. Нижний слой
дистиллята отделяют, промывают разбавленным едким натром и затем разбавленной серной
кислотой, высушивают хлористым кальцием и перегоняют. Получают 95 г тетраэтилсвинца е
т. кип. 83* С/13 мм.
В ароматическом ряду также можно применить подобную реакцию
[71, 72]. Однако она мало разработана (выходы продуктов незначительны).
Метод синтеза свинцовоорганических соединений взаимодействием дига-
логенидов свинца с галоидными алкилами остается малоизученным.
Например, йодистый цетил образует с двухлористым свинцом в присутствии
активированного серебром цинка тетрацетилсвинец с выходом 30% [73].
Получение тетрацетилсвинца [73]. Смесь 194 г двухлористого свинца, 105 г йодистого
цетила и 65 г активированного цинка быстро прибавляют к 200мл нагретой до 60СС воды,
причем температура повышается до 80—90° С. Смесь перемешивают в течение 2—3 час.
при 90° С и отфильтровывают. Фильтрат и осадок трижды экстрагируют 200 мл бензола, а
затем также трижды — 200 мл гексана. После удаления растворителя сырой продукт
растворяют в 500—700 мл ацетона и оставляют кристаллизоваться. Получают 24,7 г (30%)
тетрацетилсвинца с т. пл. 40,5—42° С.
Применяющийся в этой реакции цинк активируют следующим образом: 65 г цинка
суспендируют в 800 мл воды и при перемешивании и температуре 80—90° С к нему прибавляют
6,5 г азотнокислого серебра в 50 мл воды. Затем цинк отсасывают и трижды промывают
декантацией 800 мл воды.
Что касается реакции плюмбита калия (или натрия) с галоидными
алкилами, то в этом вопросе остается еще много неясного. На стр. 190 описан
предложенный Дрюсом метод синтеза оловоорганических соединений
класса AlkSnX3, взаимодействием йодистых алкилов со станнитом калия.
Распространить этот метод синтеза на соединения класса А1кРЬХ3 Дрюсу не
удалось [74].
Однако Лесбр [75—77] утверждает, что метилплюмбоновая кислота
может быть получена по схеме:
CH3J + PbOOHNa -> CHgPbOOH + NaJ.
Лесбр описал также синтез иодидов согласно уравнению [78]:
RJ + PbCl2.CsCl2 — RPbCl3 + CsJ,
RPbCl8 + 3 CsJ -> RPbJ3 + 3CsCl.
Следует отметить, что в обоих случаях методика неясна, выходы и анализы
продуктов реакции не приведены. Воспроизвести эти опыты не удалось [79].
Синтез действием галоидных алкилов на свинец
543
ЛИТЕРАТУРА
1. С a h о и г s A., R i с h e E., Compt. rend., 36, 1001 (1863).
2. G г о h n H., P a u d е г t R., Z. Chem., 3, 89 (1963).
3. L б w i g С., J. prakt. Chem., 60, 304 (1953).
4. В u с к t о n G. В., Ann., 109, 278 (1959).
5. КоршакВ.В., Колесников Г. С, Тетраэтилсвинец. М., Госхимиздат, 1946.
6. S h a p i г о Н., Advan. Chem. Ser., 23, 290 (1959).
7. Англ. пат. 719913 (1954); РЖХим., 1957, 52378.
8. D a u d t H. W., Пат. США 1749567 (1929); Ch. Ztb., 1, 3354 (1930).
9. D a u d t H. M., Пат. США 2091114 (1937); С. А., 31, 7446 (1937).
10. Англ. пат. 453271 (1936); С. А., 31, 1043 (1936).
11. С а 1 i n g a e r t G., Пат. США 1622233 (1927); Ch. Ztb., 1, 1709 (1928).
12. Y о t z M. А., Пат. США 1658544 (1928); Ch. Ztb., 1, 1913 (1928).
13. MonroeK. P., Пат. США 1661809 (1928); Ch. Ztb., 1, 2304 (1928).
14. К r a u s С. А., Пат. США 1694268 (1928); Ch. Ztb., 1, 2355 (1929).
15. CalcottW.S., Пат. США 1944167 (1934); Ch. Ztb., 1, 2654 (1934).
16. CalcottW.S., Пат. США 1962173 (1934); Ch. Ztb., 11, 3842 (1934).
17. Англ. пат. 290444 (1928); Ch. Ztb., 11, 1214 (1929).
18. T a n n e r H. M., Англ. пат. 707075 (1954); РЖХим., 1955, 4849.
19. T a n n e г Н. М., Пат. США 2635105 (1953); РЖХим., 1955, 8319.
г0. С а 1 с о t t W. S., P a r m e 1 e e A. E., L о г r i m a n F. R., Англ. пат.' 280169 (1927);
Ch. Ztb., 1, 1459 (1928).
21. С a 1 с о t t W. S., P а г m e 1 e e A. E., L о г г i m a n F. R., Пат. США 1664021 (1928);
Ch. Ztb., 1, 2989 (1928).
22. П о р и н И. Г., СыскаевИ. M., Авт. свид. СССР 131350 (1960); РЖХим., 1961,
8Л116; Бюлл. изобр., № 17, 20 (1960).
23. Down i ngF. В., В a k e L. S., P arm el e e A. E., Пат. США 2061267 (1937); С, А.,
31, 652 (1937).
24. С а 1 i n g а е г t G., S h a p i г о Н., Пат. США 2535190 (1950); С. А., 45, 3864 (1951).
25. Р i г 1 о t M. G., Пат. ФРГ 945450 (1956); РЖХим., 1957, 38873.
26. W i с z e г S. В., Пат. США 2960515 (1960); С. А., 55, 9282 (1961).
27. Англ. пат. 854776 (1960); С. А., 55, 11362 (1961).
28. Shapiro H., Пат. США 2535235—7 (1950); С. А., 45, 3865 (1951).
29. К г о h п I. Т., S h a p i г о Н., Пат. США 2594183 (1952); С. А., 47, 145 (1953).
30. S h a p i г о Н., К г о h n I. Т., Пат. США 2594225 (1952); С. А., 47, 146 (1953).
30а. Ми сима С, РЖХим., 1963, 1SB29.
31. С 1 е m W. J., Р 1 и п к е t t R. J., Пат. США 2515821 (1950); С. А., 44, 9978 (1950).
32. Н о 1 b г о о к G. Е., Пат. США 2464397—9 (1949); С. А., 43, 4287 (1949).
33. М е 1 v i n W. S., N i с h о 1 s A. F., Пат. США 2907780 (1959); РЖХим., 1961, ЗЛ68.
34. В i г i t z L. F., Пат. США 3108127 (1961); РЖХим., 1965, 21Н76.
35. В i r i t z L. F., Пат. США 3048611 (1962); С. А., 57, 16655 (1962).
35а. Beaird F. M., Kobetz P., Пат. США 322(5408—9 (1965); РЖХим., 1967,
5Н110, 112.
36. Bivitz L., Пат. США 3057892 (1962); С. А., 58, 5723 (1963).
37. S h a p i г о Н., D e W i t t E. G., Пат. США 2575323 (1951); С. А., 46, 5073 (1952).
38. Р 1 и п к е t t R. J., Пат. США 2477465 (1949); С. А., 43, 8398 (1949).
39. С 1 е m W. J., Р о d о 1 s к у Н., Пат. США 2426598 (1947); С. А., 42, 203 (1948).
40. Р е а г s а 1 1 Н. М., Пат. США 2414058 (1947); С. А., 41, 2430 (1947).
41. Англ. пат. 899697 (1962); РЖХим., 1963, 11Н76.
42. Итал. пат. 621656 (1961); С. А., 57, 15152 (1962).
43. Н о b b sC. L., Пат. США 2686799 (1954); РЖХим., 1955, 47197.
44. М i 1 1 е г L. А., Пат. США 2982778 (1961); РЖХим., 1963, ЗН94.
45. Pines H., Пат. США 2850513 (1958); РЖХим., 1959, 88036.
46. Fich ter F., Stei n J., Helv. chim. Acta, 14, 1205 (1931).
47. G о 1 b г а с h A., Helv. chim. Acta, 14, 1436 (1931).
48. StantonR., Канад. пат. 499635 (1954); РЖХим., 1955, 56355.
49. N e h e г С. M., W e i m e г Р. Е., Пат. ФРГ 940297 (1956); РЖХим., 1957, 42363.
50. М a t t i s о n E. L., Пат. США 2744126 (1956); РЖХим., 1959, 5669.
51. К о м а ц у С, О г а в а Д., Н а к а м о р и X., Японск. пат. 2317 (1956); РЖХим.,
1959 43313.
52. С о о'k S. Е.", S i s t г и п к Т. О., Пат. США 3049558 (1951); С. А., 57, 16656 (1962).
53. J arvi eJ.M. S., Schul erM. J., S t e г t i n g J. D., Пат. США 3048610(1962);
С. А., 58, 550 (1963).
54. T и 1 1 i о V., Пат. США 3072694 (1963); С. А., 58, 13992 (1963).
55. R и d у D. D., Пат. США 2723227 (1955); РЖХим., 1958, 61903.
56. Англ. пат. 945118 (1963); РЖХим., 1965, ЗН71.
544
Свинцовоорганшеские соединения
57. В 1 i t z е г S. М., В г о w п О. М., G г а п d j e a n W. В., Пат. США 2777866 (1957);
РЖХим., 1959, 2207.
58. G i г a i t i s A. P., G г a n d j e a n W. В., NeherC.M., Пат. США 2777867 (1957);
РЖХим., 1960, 66375.
59. L u g g R. R., Пат. ФРГ 940298 (1956); РЖХим., 1957, 38874.
60. Англ. пат. 724155 (1955); РЖХим., 1957, 55506.
61. Белы. пат. 538376 (1959); РЖХим., I960, 54437.
62. Krohnl. Т., Канад. пат. 498258 (1953); РЖХим., 1955, 44253.
63. G г а п d j e a n W. В., Пат. США 2994712 (1961); РЖХим., 1962, 16Л59.
64. Р a d g i t t F. L., Пат. США 2765328 (1956); РЖХим., 1959, 5668.
65. С а 1 i n g a e г t G., S h a p i г о Н., Пат. США 2535193 (1950); С. А., 45, 3865 (1951).
66. CalingaertG., Shapiro H., Пат. США 2562856 (1951); С. А., 46, 1581 (1952).
67. С а 1 i п g а е г t G., S h a p i г о Н., Пат. США 2558207 (1951); С. А., 46, 131 (1952).
68. HeddenG. D., Пат. США 3110719 (1963); РЖХим., 1965, 15Н80.
69. Н е а р R., S a u n d е г s В. С, J. Chem. Soc, 1949, 2983.
70. S a u n d e r s В. С, S t а с е у G., J. Chem. Soc, 1949, 919.
71. Poli sA., Ber., 20, 716 (1887).
72. Poli s A., Ber., 21, 3424 (1888).
73. N о s e k J., Collect. Czechosl. Chem. Communs, 29, 537 (1964).
74. D r u с e J.., Chem. News, 124, 215 (1922).
75. LesbreM., Compt. rend., 200, 559 (1935).
76. LesbreM., Compt. rend., 206, 1481 (1938).
77. LesbreM., Compt. rend., 210, 535 (1940).
78. LesbreM., Compt. rend., 204, 1822 (1937).
79. CalingaertG., Shapiro H., D у k s t r a F. J., HessL., J. Am. Chem. Soc,
70, 3902 (1948).
Г лава II
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ СИНТЕЗА
СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В литературе описано несколько электрохимических методов синтеза
свинцовоорганическпх соединений. В лабораторной практике они не
получили широкого распространения, но, судя по многочисленным патентным
заявкам, представляют значительный интерес для техники.
Электрохимические методы применяются главным образом для синтеза
тетраалкилсвинца. Кроме того, они используются и для получения гексаал-
килдипл юмбанов.
Синтез тетраалкилсвинца можно осуществить двумя путями. Первый
из них заключается в электролитическом восстановлении кетонов или
галоидных алкилов на свинцовом катоде. Так, при электролизе ацетона в
сернокислом растворе на свинцовом катоде выделяется тетраизопропилсвинец и
окрашенные соединения, в состав которых входит, по-видимому, диизопро-
пилсвинец [1,2]. Электролизом метилэтилкетона и диэтилкетона получены
[3] тетра-#то/?-бутилсвинец и тетра-(3-ауил)свинец соответственно. Ксвин-
цовоорганическим соединениям приводит также электролиз а, (З-ненасыщен-
ных кетонов [4—7], метилизоамилкетона [8] и пропионового альдегида [9].
Состав образующихся при этом продуктов не исследован.
Вероятно, общую схему реакций, проходящих на катоде, можно
выразить следующим образом [10, 11]:
R2CO <- :v> 3H+ r2gh + II20,
4R2CII + Pb->Pb(CHR2)4.
При электролитическом восстановлении галоидных алкилов (метод
применялся к йодистому и бромистому метилу, этилу и изоамилу) на
свинцовом катоде в растворе водной или спиртовой щелочи образуются четырех-
замещенные свинцовоорганические соединения. Процесс ведется с
диафрагмой с небольшими плотностями тока при температуре несколько ниже
температуры кипения галоидного алкила. Анодом служит графит. Механизм
реакции не изучался, но по бурой окраске реакционной смеси можно
заключить, что вначале образуются соединения типа (R2Pb)«, которые при
температуре опыта переходят в R4Pb с выделением металлического свинца.
Тетраалкилсвинец отделяют перегонкой с водяным паром [12—14].
Дальнейшего развития отмеченные выше методы не получили. Гораздо
более важное значение имеет электролиз комплексных металлоорганических
соединений на свинцовом аноде. Первые работы в этом направлении
принадлежат Хейну с сотр. [15—17]. Впоследствии метод подробно исследован
Циглером с сотр. [18—27], применившим для синтеза тетраалкилсвинца
легко доступные комплексные соединения алюминия. Характеристика
некоторых свойств этих веществ приведена в табл. 10.
35 Заказ № 207
543
Свинцовоорганшеские соединения. Лит. стр. 547—548
Таблица 10
Свойства комплексных металлоорганических
электролитов [22]
Электролит
NaF-Al(C2H5)3
NaF.2Al(C2H3)3
KF.A1(C2H5)3
KF-2A1(C2H3)8
Na-A1(C2H5)4
K-A1(C2H5)4
Т. пл., °C
72—73
35
56—58
129
124
73
Электропроводность
при 100 °C, &-'-см-1
0,002
0,04
0,02
0,07
0,033 (экстраполяция)
0,08
Для сравнения отметим, что электропроводность расплава хлористого
натрия (850°С) 3,66 Ог^см'1.
Электролиз этих комплексов рассмотрен в обзорных статьях Циглера
[28] и Марлетта [10]. Если исходить из NaF*2Al(C2H5)3, то на катоде
выделяется алюминий, а на аноде — этильные радикалы [10]:
анод: [(C2H5)3A1.F.A1 (С2Н5)зГ-» С2Н5 -j- (C2H5)3A1 + (C2FI5)2A1F + ё,
катод: [(С2Н5)зА1 • F• А1 (С2Н5)зГ + 3 е-» Al + (CaH5)8AlF- + 3С2Н".
Для того чтобы избежать вторичных процессов, электролиз ведут в
вакууме или катодное и анодное пространство разделяют диафрагмой [10, 25].
Следует также отметить, что большая глубина превращения электролита
значительно затрудняет выделение чистого тетраалкилсвинца [27].
При электролизе натрий -или калийтетраалкилалюминия на катоде
выделяется щелочной металл, а на аноде — смесь тетраалкилсвинца и триал-
килалюминия [19]:
4 NaAlRsQHs + Pb -» 4 Na + Pb (C2H5)4 + 4 A1R3.
Все операции проводят в атмосфере сухого инертного газа.
Выделяющийся на катоде щелочной металл целесообразно отделять в виде
амальгамы ртути. В этом случае реакцию проводят в проточной системе,
так как иначе тетраалкилсвинец оседает на поверхности анода и
препятствует прохождению электрического тока [25].
Температурный режим процесса определяется двумя факторами:
устойчивостью образующегося свинцовоорганического соединения и
электропроводностью электролита (последняя возрастает с повышением температуры
[24]). Обычно электролиз ведут при температуре порядка 100°С [24—27].
Вследствие высокой температуры плавления тетраметилалюмината натрия
тетраметилсвинец получают в среде тетрагидрофурана [29].
Иногда выделяющиеся на аноде тетраалкилсвинец и триалкилалюминий
Г>азделяются с трудом. В этом случае к смеси прибавляют фтористый натрий
30], цианистые соли или гидриды калия и натрия [31—35] или третичный
амин [36,] с которыми триалкилалюминий образует устойчивые комплексы
[37]. Другой способ заключается в прибавлении к электролиту алкокситри-
алкилалюмината натрия или калия [25, 26, 38]. Выделяющиеся на аноде
тетраалкилсвинец и алкоксидиалкилалюминий легко разделяются
перегонкой. К аналогичному результату приводит обработка смеси
тетраалкилсвинца и триалкилалюминия алкокситриалкилалюминатом натрия [25, 39,40].
Электрохимические методы синтеза
547
Свинцовоорганические соединения получают также электролизом водных
растворов тетраалкилборнатрия [41]. И в этом случае необходимо работать
в атмосфере инертного газа, так как образующийся триэтилбор
самовоспламеняется на воздухе. Реакция подробно исследована на примере синтеза
тетраэтил свинца. Найдено, что лучше всего исходить из 50%-ного раствора
тетраэтил бор натрия и вести электролиз до тех пор, пока концентрация не
уменьшится до 10%-ной (в этом интервале концентраций электропроводность
раствора максимальна и имеет постоянное значение 5—6-10"2 ом'1-см'1).
Оптимальная плотность тока 3,5 а/дюйм2 при напряжении 1,8 в
(расстояние между электродами 6 мм). При более высокой плотности тока анод
покрывается слоем маслянистого продукта, препятствующим прохождению
тока. При прибавлении к раствору изооктана плотность тока может быть
повышена до 21 а/дюйм2, но полностью избавиться от маслянистого продукта
не удается.
Электролиз тетраэтилборнатрия приводит к нерастворимой в воде смеси
тетраэтилсвинца и триэтилбора. При достаточно низкой плотности тока эта
смесь самопроизвольно отделяется от поверхности свинца в виде капель или
отдельного слоя, который поднимается на поверхность раствора (плотность
смеси РЬ(С2Н5)4 + 4(С2Н5)3В меньше 1).
Сравнивая синтез тетраэтилсвинца из комплексных алюминийоргани-
ческих соединений и тетраэтил бор натрия, Циглер [41] отмечает, что
преимуществом последнего является его большая устойчивость по отношению к
кислороду воздуха. Кроме того, температуры кипения тетраэтилсвинца и
триэтилбора значительно отличаются между собой, так что они легко
разделяются перегонкой. К недостаткам метода относится меньшая доступность
тетраэтилбор натрия, а также его странное поведение при работе с высокими
плотностями тока.
По патентным данным [42], электролиз тетраалкилборнатрия можно
проводить также в расплаве или в среде органических растворителей.
Помимо комплексных металлоорганических соединений алюминия или
бора в качестве электролита применяют также растворы алюминий-[43] или
магнийорганических соединений [44—50]. Для предотвращения отложения
магния на катоде предложено к раствору RMgX прибавлять галоидный ал-
кил [45—48]. Обычно электролиз проводят в среде низших алкиловых эфи-
ров моно- или диэтиленгликоля при 45—60°С.
Описаны [50—53] промышленные установки по синтезу тетраалкилсвин-
ца этим методом.
В патентной литературе имеется указание, что при пропускании дуги
высокого напряжения (25 000 в) между свинцовыми электродами в среде
жидких углеводородов образуются свинцовоорганические соединения, которые
можно выделить перегонкой в вакууме [54—56].
Электролиз свинцовоорганических соединений с целью синтеза гексаал-
килдиплюмбанов описан очень кратко. При электрохимическом
восстановлении хлористого триэтилсвинца в щелочном растЕОре получен [57] гексаэтил-
диплюмбан. Выход 80—90% [58].
ЛИТЕРАТУРА
1. Та f elJ., Ber., 44, 323 (1911).
2. TafelJ., Ber., 39, 3626 (1906).
3. RengerG., Ber., 44, 337 (1911).
4. LawH., J Chem. Soc, 101, 1016 (1912).
5. LawH., J. Chem. Soc, 101, 1544 (1912).
6. LawH., Proc. Chem. Soc, 28, 98 (1912).
7. L a w H., Proc Chem. Soc, 28, 162 (1912).
8. TafelJ., Ber., 42, 3146 (1909).
35*
548
Свинцовоорганшеские соединения
9. S с h e p p s W., Вег., 46, 2564 (1913).
10. М а г 1 е t t Е. М., Ann. N. Y. Acad. Sci., 125, 12 (1965).
11. Т ом и л о в А. П., ФиошинМ.Я., Усп. хим., 32, 60 (1963).
12. Р 1 u m p R. Е., Н a m m e t L. В., Trans. Electrochem. Soc, 73, 523 (1938).
13. С a 1 i n g a e r t G. H. F., Пат. США 1539297 (1925); С. А., 19, 2210 (1925).
14. Mea d В., Пат. США 1567159 (1925); Ch. Ztb., 1, 2052 (1926).
15. Н е i n F., Z. Elektrochem., 28, 469 (1922).
16. Hei nF., Petzschner W. K-, S e g i t z F. A., Z. anorgan. allgem. Chem., 141,
161 (1924).
17. H e i n F., S e g i t z F. A., Z. anorgan. allgem. Chem., 158, 153 (1926).
18. ZieglerK., LehmkuhlH., Angew. Chem., 67, 424 (1955).
19. Z i e g 1 e r K., Angew. Chem., 71, 628 (1959).
20. Z i e g 1 e r K., Angew. Chem., 72, 565 (I960).
21. Gri m me W., LehmkuhlH., ZieglerK., Zosel К, К о b sH.-D., Scha-
e f f e r R., Bull. Soc. Chim. France, 1963, 1456.
22. ZieglerK., Chem. Ingr.-Techn., 35, 325 (1963).
23. ZieglerK., Англ. пат. 81460Э (1959); С. А., 53, 17733 (1959).
24. ZieglerK., Англ. пат. 848364 (1960); С. А., 55, 5199 (1961).
25. Ц и г л е р К., Л е м к у л ь Г. Пат. СССР 132136 (1960); РЖХим., 1962, 9Л98; Бюлл.
изобр., № 18, 83 (1960).
26. Z i e g 1 е г К., L e h m k u h 1 Н., Пат. ФРГ 1127900 (1962); РЖХим., 1964, ЗН103.
27. Z i e g 1 е г К., L e h m к u h 1 Н., Пат. ФРГ 1114816 (1962); РЖХим., 1963, 20Н48.
28. ZieglerK., Organo-alutninium compounds. В кн.: H.Zeiss. Organometallic
Chemistry. Reinhold Publ., New York, 1960.
29. ZieglerK-, Бельг. пат. 617628 (1962); С. А., 60, 3008 (1964).
30. Z i e g 1 e г К., L e h m k u h 1 H., Пат. ФРГ 1153372 (1964); РЖХим., 1965, 8H96.
31. Zi egl er К., Lehm kuh 1 H., Пат. ФРГ 1153369 (1964); РЖХим., 1935, 12Н83.
32. Z i e g 1 е г К., Бгльг. пат. 622150 (1963); С. А., 59, 10117 (1963).
33. М с К а у Т. W., Пат. CdR 3038885 (1963); РЖХим., 1965, 11Н99.
34. М с К а у Т. W., Пат. США. 3033957 (1953); РЖХим., 1965, 7Н56.
35. М с К а у Т. W., Пат. ФРГ 1120443 (1961); С. А., 57, 11234 (1962).
36. Z i e g 1 е г К., Пат. ФРГ 1134572 (1962); С. А., 58, 550 (1963).
37. Несмеян овА. Н., Сокол и к Р. А., Методы элемектоорганической химии. Бор,
алюминий, пллий, индий, таллий. М., «Наука», 1964, стр. 342.
38. Gi га i ti sA. Р., Пат. COU 3177130 (1965); РЖХим., 1966, 11H110.
39. Z i e g I e г К. L e h m k u h 1 H., Пат. ФРГ 1153754 (1964); РЖХим., 1965, 17Н108.
40. ZieglerK., Англ. пат. 864394 (1961); С. А., 55, 20962 (1961).
41. Z i e g 1 е г К., S t e u d е 1 O.-W., Ann., 652, 1 (1962).
42. PinkertonR.C, Пат. США. 3028325 (1962); С. А., 57, 4471 (1962).
43. Англ. пат. 8420ЭЭ (I960); С. А., 55, 5199 (1951).
44. В г a i t h w a i t e D. G., Пат. CUR 3007857 (1961); РЖХим., 1963, 5H67.
45. В r a i t h w a i t e D. G., Пат. США. 3007853 (1961); РЖХим., 1962, 3H95.
45. LinskJ., MayerleE.A., Пат. СОИ 3155502 (1964); РЖХим., 1966, 9Н103.
47. L i n s k J., Пат. СШ\ 3116303 (1953); РЖХим., 1985, 14H92.
48. Li nsk J.,Ca r 1 R. W.,Fi el d E., Пат. США. 3164537 (1965); РЖХим., 1966, 12H109.
49. В о t t L. L., Hydrocarbon Process and Petrol. Refiner, 44, 115 (1965); РЖХим., 1966,
9H102.
50. G i r a i t i s A. P., Пат. ФРГ 1046517 (1958); С. А., 55, 383 (1961).
51. Chem. Eng. News, 39 (38), 36 (1961).
52. Chem. Eng. News, 42, (49), 52 (1964).
53. Chem. Eng. News, 41 (6), 26 (1963).
54. S u 1 1 i v a n F. W., Пат. США. 1933547 (1933); Ch. Ztb., 11, 384 (1934).
55. S u 1 1 i v a n F. W., Пат. США. 2037650 (1937); С. А., 31, 6454 (1937).
56. R i с e F. О., Англ. пат. 407036 (1934); С. А., 28, 4847 (1934).
57. M i d g 1 e у Т., Н о с h w a 1 t С. А., С а 1 i n g a e r t G., J. Am. Chem. Soc, 45, 1821
(1923).
58. H e i n F., К I e i n A., Ber., 71, 2381 (1938).
Глава III
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МЕТАЛЛСОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА СОЛИ ДВУХВАЛЕНТНОГО СВИНЦА
ИЛИ МЕТАЛЛИЧЕСКИЙ СВИНЕЦ
Наиболее распространенный в лабораторных условиях метод синтеза
соединений типа R4Pb заключается в действии реактива Гриньяра на дву-
хлористый свинец.
В качестве алкилирующих (или арилирующих) агентов применяют
также натрий-, литий-, цинк- или алюминииорганические соединения, а
вместо хлористого свинца иногда используют окись или сульфид
двухвалентного свинца [1—3].
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ МАГНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Действие магнийорганических соединений на соли двухвалентного
свинца позволяет получать соединения типа R4Pb в алифатическом, алицик-
лическом, ароматическом и гетероциклическом рядах, а также соединения
типа R3PbPbR3 в алициклическом и ароматическом рядах.
При прибавлении хлористого свинца к реактиву Гриньяра образуются
окрашенные в красный цвет соединения (см. стр. 554), которые диспропорцио-
нируют при нагревании, последовательно превращаясь в R3PbPbR3, а затем
в R4Pb [4, 5]:
PbCl2 + 2RA'gX^(R2Pb)rt
3(R2Pb)rt-*R3PbPbR3 + Pb
3R3PbPbR3->3R4Pb + Pb
2 РЬС12 + 4 RMgX -> R4Pb+ Pb + 4 MgX2.
Следует отметить, что эта схема отражает только общее направление
процесса, который проходит, по-видимому, более сложно.
При получении тетраалкильных соединений свинца лучше всего
исходить из хлористых алкилов. С бромистыми алкилами выходы продуктов
несколько ниже; при иодидах еозможны побочные реакции и загрязнение
образующихся веществ высококипящими йодистыми алкилами [6].
Получение тетраметилсвинца [7]. К зфирнсму раствору хлористого метилмагния (2
моля) при периодическом встряхивании и внешнем охлаждении холодной водой прибавляют
небольшими частями 1 г-моль сухого мелко растертого двухлористого свинца. Смесь
нагревают в течение нескольких часов и по охлаждении к ней осторожно прибавляют воду до
разделения слоев. Органический слой отделяют, Еысушивают хлористым кальцием и эфир
отгоняют. Остаток перегоняют с масляной бани при обычном давлении. Если, как это
часто бывает, сырой продукт содержит еще гексаметилдиплюмбан, то при медленной
перегонке последний разлагается, отлагая темное зеркало свинца и образуя тетрамети л свинец.
Повторная перегонка фракции с т. кип. ПО—115° С дает чистое вещество с т. кип.
110° С/760 лш.
550 Свинцовоорганические соединения. Лит. стр. 556—557
Впоследствии эта методика была воспроизведена и другими авторами
[8, 9]. Найдено [10], что при проведении этой реакции в присутствии
йодистого метила выход тетраметилсвинца улучшается, металлический свинец
выделяется в весьма незначительном количестве.
Начиная с синтеза тетраэтилсвинца, удаление гексаалкилдиплюмбанов
требует многократной перегонки, сопровождающейся выделением
металлического свинца, или применения во время синтеза высококипящих
растворителей. Так, тетраэтилсвинец получен с хорошим выходом взаимодействием
бромистого этилмагния с хлористым свинцом в смеси эфира с диметиловым
эфиром этиленгликоля [11].
Устойчивость гексаалкилдиплюмбанов к нагреванию возрастает с
увеличением радикала у атома свинца. При синтезе высших гомологов
алифатического ряда образуется смесь гексаалкилдиплюмбанов и тетраалкилсвин-
ца. Эту смесь последовательно обрабатывают галоидом, а затем избытком
реактива Гриньяра. Таким образом, метод синтеза соединений типа R4Pb
часто сводится к следующему [7]:
Br2 RMgX
R3PbPbR3 > 2 R3PbBr > R4Pb.
Этот прием используется, например, при синтезе тетра-я-пропилсвинца.
Получение тетра-и-пропилсвинца [7]. К магнийорганическому, соединению,
приготовленному из 100 г хлористого w-пропила в 500 мл эфира, при охлаждении и периодическом
встряхивании прибавляют 200 г двухлористого свинца. Довольно энергичную реакцию,
сопровождающуюся выделением металлического свинца, заканчивают многочасовым
нагреванием на водяной бане. Избыток реактива Гриньяра разлагают водой, органический слой
отделяют и эфир отгоняют. Остаток растворяют примерно в'пятикратном количестве этил-
ацетата и обрабатывают эфирным раствором брома при —75° С до тех пор, пока окраска
не перестанет исчезать. Продукт реакции вновь обрабатывают хлористым я-пропилмагнием.
После обычной обработки остающееся масло перегоняют в вакууме под азотом. Получают
50 г тетра-я-пропилсвинца с т. кип. 126° С/13 мм, 75—80° С/0,01 мм [8].
В качестве примера синтеза тетраалкильных соединений свинца со
вторичными радикалами можно привести следующий.
Получение тетраизопропилсвинца [12]. В реактив Гриньяра, полученный из 40 г
хлористого изопропила в 400 мл эфира, постепенно вводят 100 г двухлористого свинца.
Происходит энергичная реакция, сопровождающаяся выделением свинца и окрашиванием
смеси в темно-красный, а затем зеленый цвет. После 10 часового кипячения смесь разлагают
водой, органический слой отделяют, высушивают хлористым кальцием и эфир отгоняют.
Уже при первой перегонке остатка вся жидкость кипит около 120° С/14 мм. Однако для
получения чистого вещества необходима вторичная перегонка. Выход тетраизопропилсвинца
20 г; т. кип. 120° С/14 мм. При хранении вещество медленно разлагается.
Кроме описанных выше соединений, этим методом в алифатическом ряду
получены тетра-я-бутил- (т. кип. 140°С/1 мм\ 108— 110°С/0,01 мм) [8,
ISIS], тетраизобутил-(т. пл. — 23°С) [16], тетра-я-амил- (т. кип. 170° С/1 мм)
и тетраизоамилсвинец [13, 16].
При синтезе тетравинилсвинца, из-за его неустойчивости, избегают
стадии разложения избытка реактива Гриньяра.
Получение тетравинилсвинца [17]. К бромистому винилмагнию (из 24,3 г магния и
107 г бромистого винила в 1 л тетра гидрофура на) прибавляют небольшими частями 139 г
двухлористого свинца, поддерживая равномерное кипение эфира, а затем смесь кипятят
еще 1 час. Осадок металлического свинца отфильтровывают, промывают несколько раз
тетрагидрофураном и из фильтрата растворитель отгоняют на колонке в небольшом вакууме
(температура бани не выше 90° С). Затем колбу соединяют через насадку Кляйзена с
охлажденной сухим льдом ловушкой, дно колбы погружают в нагретую до 70—85° С баню и тет-
равинилсвинец постепенно отгоняют в вакууме 0,5—1 мм в течение 24 час, а затем
перегоняют на колонке при 30—40° С/1—2 мм. Получают 32,3 г (41%) вещества с n2J'5 1,5408. При
Синтезы на основе солей двухвалентного свинца
551
повторной перегонке температуру бани поддерживают дай 85—95° С (но не выше 95° С),
Хранить вещество следует в ампулах из темного стекла при температуре не выше —26° С,
При комнатной температуре оно постепенно разлагается с выделением осадка желтого цвета,
Тетравинилсвинец получен также с несколько меньшим выходом из
бромистого винилмагния и диацетата свинца или хлористого свинца и
хлористого винилмагния [17] (ср. [18]).
Двухлористый свинец активно реагирует с хлористым триметилсилил-
метилмагнием в тетрагидрофуране при — 10°С. Выход тетра(триметилси-
лилметил)свинца 37%, т. кип. 104—105° С/0,01 мм [19].
Получение тетрадиклопропилсвинца [20]. К реактиву Гриньяра (из 5,08 г магния и 25 г
бромциклопропана в 175 мл тетрагидрофурана) при перемешивании прибавляют в течение
25 мин. 42? ^двухлористого свинца. Смесь нагревают при 70—80° С в течение 15 час. и
растворитель отгоняют в вакууме при температуре бани 40—45° С. Затем колбу соединяют с
охлажденной до —80° С ловушкой, давление в системе понижают до 0,1 мм, а температуру
бани повышают до 90° С. Через 30 час. в ловушке собирается 10,4 г вещества. Перегонка его
в вакууме дает 7,54 г тетрациклопропилсвинца с т. кип. 57—58° С/0,1 мм; п2^ 1,5505.
Интересна исключительная устойчивость к нагреванию гексациклогек-
силдиплюмбана. При кипячении в ксилоле в течение 6 час. вещество не
изменяется. Лишь при нагревании до 190°С наблюдается выделение
металлического свинца, но тетрациклогексилсвинец при этом не образуется [21].
Последний можно получить только из бромистого трициклогексилсвинца и
бромистого циклогексилмагния [22].
Тетрабензилсвинец резко отличается от других металлоорганических
соединений IV группы своей неустойчивостью по отношению к окислителям.
Неочищенный продукт самовоспламеняется на воздухе. Синтез и очистку тет-
рабензилсвинца проводят в атмосфере инертного газа, защищая от действия
света [23—25].
Получение тетрабензилсвинца [24]. Суспензию 32 г двухлористого свинца в 100 мл
эфира охлаждают льдом и к ней при энергичном перемешивании прибавляют эфирный
раствор хлористого бензилмагния (из 64 с хлористого бензила и 14 г магния в 300—400 мл
эфира). Смесь перемешивают при обычной температуре в течение часа, нагревают до кипения,
быстро охлаждают и разлагают при перемешивании 200 мл воды. Эфирный раствор темно-
желтого цвета декантируют в заполненную азотом колбу, емкостью 2 л, водный слой дважды
экстрагируют эфиром. К объединенным эфирным вытяжкам (~1 л) приливают 500 мл воды,
из которой удален кислород воздуха, и эфир отгоняют при пониженном давлении. После
удаления основной части эфира над слоем воды остается масло желтого цвета. Температуру бани
постепенно повышают до тех пор, пока при давлении не больше 20 мм отгонится примерно
200 мл воды. При этом одновременно с водой отгоняются побочные продукты реакции (бенз-
альдегид, толуол, дибензил и др.) тогда как тетрабензилсвинец оседает на дне колбы в виде
тяжелого масла желтого цвета. Сырой продукт при хранении под слоем воды постепенно
закристаллизовывается (охлаждение ускоряет кристаллизацию). Затвердевшую массу
промывают метиловым спиртом, затем растворяют при нагревании в метиловом спирте и раствор
отфильтровывают. Из маточника тетрабензилсвинец выделяется в виде крупных кристаллов
или призм золотистого цвета (концентрация раствора не должна быть слишком велика,
иначе тетрабензилсвинец выделяется в виде масла). Кристаллы отфильтровывают, промывают
метиловым спиртом и высушивают в вакууме. Выход 30 г; т. пл. 65,5—66,5° С. Вещество
хорошо растворяется в обычных органических растворителях, кроме холодного спирта.
Хранить его следует в атмосфере азота, применяя ампулы из темного стекла.
Аналогично получены тетра-о-фторбензилсвинец (т. пл. 74—75°С), тетра-
о-хлорбензилсвинец (т. пл. 99,5°С) и тетра-о-бромбензилсвинец (т. пл. 122°С).
Гексабензил- и гекса-о-фторбензилдиплюмбаны выделить в чистом виде не
удается, тогда как соответствующие хлор- и бромзамещенные соединения
относительно устойчивы [24].
В ароматическом ряду реакция проходит до конца только при получении
тетрафенил-[4,26—32] и тетра(аллилфенил)свинца [33]. Синтезтетрафенил-
свинца целесообразно проводить в смеси эфира с бензолом, так как в эфире
его выход несколько ниже [4, 26—30].
552
Свинцовоорганичвские соединения. Лит. стр. 556—557
Получение тетрафенилсвинца [v31, 32] (ср. [34]). К энергично перемешиваемому
раствору бромистого фенилмагния (из 1,5[моля бромбензола в 500 мл эфира) прибавляют 500 мл
чопуъш и 181 г (0,65 моля) двухлористого свинца. Последний добавляют тремя порциями
с 5-минутными интервалами. Смесь перемешивают при умеренном кипении в течение 5 час.
и затем выливают в толченый лед с хлористым аммонием (сильное перемешивание). Эфирно-
толуольный слой декантируют, водный слой со взвешенным в нем осадком
отфильтровывают и сухой осадок извлекают 400 мл хлороформа. Выход тетрафенилсвинца 82—83%; т.
пл. 225—226° С.
В маточных растворах может быть констатирована примесь гексафенил
диплюмбана [35], наряду с некоторым количеством хлористого трифенил-
свинца [27].
Если же реакцию проводить при комнатной температуре, то в качестве
основного продукта получают гексафенилдиплюмбан [36]. Двухлористый
свинец (40 г) вносят небольшими порциями в реактив Гриньяра (из 50 г
бромбензола). После перемешивания смеси в течение 3 час. ее разлагают
кусочками льда, эфир отгоняют в вакууме, а оставшуются в водном слое
смесь металлического свинца, двухлористого свинца и
гексафенилдиплюмбана отсасывают, промывают водой и спиртом и высушивают в вакууме над
фосфорным ангидридом. Многократной экстракцией полученной массы
серого цвета горячим бензолом и концентрированием экстрактов в вакууме
получают кристаллы гексафенилдиплюмбана, содержащие одну молекулу
кристаллизационного бензола. На воздухе кристаллы тотчас выветриваются,
окисления при этом не происходит.
Аналогично получены [21, 35, 37] гекса-я-толил- (т. пл. 244—245° С),
гекса-отолил- (т. пл. 248—250° С), гекса-ж-толил- (т. пл. 109° С), гекса-я-
ксилил- (т. разл. 225° С), гексамезитил- (т. разл. 235° С), гекса-я-анизил-
(т. разл. 198—200° С), гекса-о-анизил- (т. разл. 198—201° С), гекса-я-
фенетил- (т. разл. 170—171° С) и гекса-а-нафтилдиплюмбаны. Тетратолил-
и тетраксилилсвинец могут быть получены с удовлетворительными
выходами лишь через соответствующие бромиды триарилсвинца [35]. Однако
в гетероциклическом ряду можно получить соединения типа R4Pb
непосредственно из двухлористого свинца [38, 39].
Получение свинцовоорганических галогенидов действием реактива
Гриньяра на соли двувалентного свинца описано очень кратко [7, 27, 31].
Из продуктов реакции бромистого додецил- или фенилмагния с небольшим
избытком хлористого свинца в эфире выделены хлористый тридодецил-
[40] и хлористый трифенилсвинец [27] соответственно. Общую схему этих
реакций изображают следующим образом [5]:
2 РЬХ2 + 3 RMgX -> R3PbX + Pb + 3 MgX2.
Более подробно этот вопрос не исследован.
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ НАТРИЙ- И ЛИТИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Применение литий- или натрийорганических соединений не дает каких-
либо преимуществ перед реактивом Гриньяра, кроме тех случаев, когда
последний мало доступен [41]. Поэтому для синтеза свинцовоорганических
соединений они используются сравнительно релко [3, 10, 42—44].
Двуиодистый свинец реагирует с метиллитием в присутствии йодистого
метила следующим образом [10]:
4 CHsLi + 2 PbJ2 -» (CH3)4Pb+Pb-f 4 LiJ,
2 CH3J + Pb - (CH3)2PbJ2,
2 CH3Li + (CH3)2FbJ2 -> (CH3)4Pb + 2 LiJ.
Синтезы на основе солей двухвалентного свинца
553
Предложенная схема подтверждается тем, что выделяющийся вначале
металлический свинец в дальнейшем вступает в реакцию. Аналогично
проходят реакции двухлористого свинца с ариллитием и йодистыми арилами
[41, 45].
Получение тетрафенилсвинца [41]. Раствор фениллития (из 126 г бромбензола и 12 г
лития в 350 мл эфира) при перемешивании постепенно прибавляют к смеси 69,5 г
двухлористого свинца и 56 г иодбензола в 200 мл эфира, поддерживая равномерное кипение эфира.
Для завершения реакции смесь нагревают в течение 2 час. Затем ее охлаждают снегом с
солью, осторожно разлагают водой, осадок отфильтровывают и высушивают на воздухе.
Тетрафенилсвинец экстрагируют из продукта реакции 350 мл хлороформа в аппарате Сок-
слета в течение 24 час. Получают 103 г вещества ст. пл. 227—229° С.
Аналогично из 0,120 моля д-диметиламинофениллития, 0,040 моля
двухлористого свинца и д-иоддиметиланилина (0,040 моля + 10% избытка) в
эфире получен [41, 45] тетра-п-диметиламинофенилсвинец. Выход 63%,
т. пл. 187—189° С (из петролейного эфира).
О реакции двухлористого свинца с тремя молями фениллития см.
стр. 624.
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ЦИНКОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Одним из наиболее ранних и сейчас уже оставленных в лабораторной
практике методов синтеза свинцовоорганических соединений является
взаимодействие двухлористого свинца с цинкорганическими соединениями.
Описанный Бэктоном еще в 1859 г., он использован для получения тетра-
метил- [46] и тетраэтилсвинца [46, 47]. Получить по этому методу тетраизо
пропилсвинец не удалось [48].
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛЮМИНИЙ- И БОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Пр^и получении свинцовоорганических соединений с помощью алюми-
нийтриалкилов или его галогенидов необходимо учитывать, что как алкил-
алюминийгалогениды, так и образующийся во время реакции треххло
ристый алюминий обладают по отношению к тетраалкилсвинцу деалкилиру-
ющим действием [49]. Поэтому реакцию с двухлористым свинцом
целесообразно проводить в присутствии комплексообразователей, например
галогенидов щелочных металлов, или применять в качестве исходного
вещества фтористый свинец [50,51]. Исследовано [52] взаимодействие метил-
алюминийсесквихлорида с двухлористым свинцом в присутствии галогени-
да щелочного металла. Выход тетраметилсвинца возрастает в зависимости
от применяемых солей в следующем порядке: NaCl, NaF, КО. Самый
высокий выход достигнут при 130° С и продолжительности реакции 8 час.
По другому способу двухлористый свинец вводят в реакцию с тетраэтил-
алюминатом лития [53, 54] или с эквивалентным количеством алюминий-
органического соединения и йодистого алкила [55].
Тетраэтилсвинец образуется с хорошим выходом при алкилировании
сухого диацетата свинца триэтилалюминием [49, 56—58]:
6 РЬ (ООССН3)2 + 4 R3A1 -> 3 R4Pb + 3 Pb + 4 А1 (OOCCH3)a.
Получение тетраэтилсвинца [49]. К 35 г безводного диацетата свинца в 60 мл бензола
прибавляют 12,0 г триэтилалюминия (температура 65—70° С за счет теплоты реакции).
Смесь кипятят при перемешивании в течение 2,5 час, разлагают водой и тетраэтилсвинец
отгоняют с водяным паром. Получают 11,9 г (68,8%) вещества с т. кип. 84—85° С/15 мм;
п$ 1,5200.
554 Свинцовоорганические соединения. Лит. стр. 556—557
Аналогично получен [49] тетраизобутилсвинец. Выход 25%, т. кип,
101—102° С/1,5 мм.
Триэтилбор [59, 60] образует с окисью двувалентного свинца в водно-
спиртовой среде или с нафтенатом свинца в тетра гидрофура не тетраэтил-
свинец с выходом 42 и 27% соответственно. Действие тригексилбора на
нафтенат свинца в диметиловом эфире этиленгликоля дает лишь 17%
тетрагексилсвинца. Для получения свинцовоорганических соединений
предложено применять также тетраалкилборнатрий [2, 61, 62].
К СИНТЕЗУ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
КЛАССА (R2Pb)„
Тот факт, что при прибавлении реактива Гриньяра к хлористому
свинцу наблюдается появление переходящей красной окраски раствора,
позволяет предполагать [7, 63], что последняя обусловлена образованием на
промежуточной стадии реакции соединений типа (R2Pb)„.
Попытки выделения этих веществ из растворов предпринимались
неоднократно. В алифатическом ряду они оказались безуспешными [64].
Впрочем, дициклопентадиени л свинец получен взаимодействием цикло-
пентадиенилнатрия с азотнокислым свинцом в диметилформамиде, в
котором эта соль хорошо растворяется [65—70].
Получение дициклопентадиенилсвинца [65]. Синтез и очистку вещества проводят в
атмосфере сухого инертного газа. В трехгорлую колбу, емкостью 500 мл, снабженную
мешалкой и обратным холодильником, помещают 250 мл сухого гареш-бутилового спирта и к
нему прибавляют 3,45 г (0,15 г-атома) мелко нарезанного металлического натрия и 6 мл
свежеперегнанного циклопентадиена. Смесь нагревают на водяной бане в течение полутора
часов. Затем ее охлаждают до 55° С, прибавляют еще 6,4 г циклопентадиена (всего в
реакцию вводят 12,4 г (0,15 моля) циклопентадиена) и смесь нагревают до тех пор, пока весь
натрий не вступит в реакцию (на это требуется примерно еще полтора часа). Во время реакции
в осадок выпадает небольшое количество трет-бутилата натрия. Для полного его
превращения в циклопентадиенилнатрий добавляют еще 2 мл циклопентадиена и смесь нагревают на
водяной бане в течение 15 мин. Если при этом не образуется прозрачный раствор, то
приливают 2 мл бензола. Затем обратный холодильник заменяют на нисходящий и mpem-бутло-
вый спирт отгоняют с водяной бани. Получают окрашенный в красный цвет порошок цикло-
пентадиенилнатрия. К нему прибавляют 25,0 г (0,075 моля) высушенного при 100° С
азотнокислого свинца в 130 мл диметилформамида (растворитель высушивают 3-часовым
кипячением с карбидом кальция с последующей перегонкой). Смесь нагревают в течение часа на
водяной бане и растворитель отгоняют в вакууме при 60—70°С до тех пор, пока выделяющаяся
твердая масса не станет крошиться. Полное удаление растворителя не рекомендуется, так
как в этом случае вещество с трудом извлекается из колбы. Осадок отделяют и возгоняют в.
высоком вакууме при температуре бани 100—130° С в течение 3—4 час. Полученный продукт
может быть очищен повторной возгонкой при 80—120° С. Чистое вещество (выход 25%)
окрашено в желтый цвет. Оно хорошо растворяется в бензоле, ацетоне, эфире и петролейном
эфире. Не растворяется в воде и не реагирует с ней на холоду. При нагревании до 138—139° С
оно смокает, не плавясь. Дальнейшее нагревание приводит к разложению,
сопровождающемуся углублением первоначальной окраски.
Дициклопентадиенилсвинец получен с выходом 85% из двухлорис-
того свинца и дициклопентадиенилмагния [70а].
Что касается соединений типа (AnjPb)^, то несмотря на ряд работ [36,
71—74], в вопросе об их синтезе остается еще много неясного. С одной
стороны, Краузе и Рейсо [36] из продуктов реакции двухлористого свинца с
бромистым арилмагнием (2 моля) выделили дифенил- и ди-я-толилсвинец.
Предложенная этими авторами методика синтеза заключается в следующем.
Рассчитанное количество двухлористого свинца прибавляют к реактиву
Гриньяра при + 2° С. Реакционную смесь перемешивают в течение 2 час.
при 0° С и разлагают льдом. Всю массу экстрагируют бензолом,
экстракты объединяют и упаривают в вакууме до 50 мл. Выпавший гексафенил-
диплюмбан отфильтровывают и из фильтрата, приливанием его в спирт, оса-
Синтезы на основе солей двухвалентного свинца
555
ждают диарилсвинец в виде порошка красного цвета. Полученные
соединения охарактеризованы элементарным анализом и химическими
реакциями.
С другой стороны, было найдено [73, 74], что первоначальные продукты
реакции двухлористого свинца с 2 молями галоидного арилмагния легко
разлагаются водой и спиртом. Состав образующихся веществ зависит от
условий проведения гидролиза. Так, при прибавлении воды, из которой
удален кислород воздуха, к окрашенному в темно-красный цвет раствору
(полученному взаимодействием двухлористого свинца с 2 молями
бромистого фенилмагния в эфире при — 15-1 20° С) наблюдается
обесцвечивание смеси и образование окиси свинца [74]. (Если же гидролиз проводить
водой, содержащей небольшое количество перекиси водорода, то
образуется тетра(трифенилплюмбил)свинец; методика синтеза этого вещества
приведена на стр. 625.)
Аналогичные результаты получены и другими авторами [73]. При
прибавлении сухого спирта или воды к окрашенному в темно-красный цвет
раствору (из 3 молей бромистого фенилмагния 1 моля двубромистого свинца
в тетрагидрофуране) смесь обесцвечивается и из нее выделяется основной
бромид свинца с выходом 90% (в расчете на взятый в реакцию свинец). В
другом опыте двубромистый свинец вводился в реакцию с 2 молями
бромистого фенилмагния в тетрагидрофуране. После удаления растворителя и
экстрагирования остатка хлористым метиленом получен твердый продукт
оранжево-красного цвета, который легко разлагается при обычной
температуре с выделением металлического свинца. Он очень чувствителен также
к действию воздуха и воды. Высказано предположение, что этот продукт
представляет собой смесь дифенилсвинца, бромистого трифенилплюмбил-
магния, бромистого магния и тетр а гидрофура на. Аналогично двубромистый
свинец реагирует с бромистым мезитилмагнием, но образующийся при этом
продукт более устойчив к нагреванию. Например, при нагревании в
течение 4 час. двубромистого свинца с избытком реактива Гриньяра в
тетрагидрофуране заметного выделения металлического свинца не наблюдается.
Однако при обработке смеси водой удается выделить лишь следы свинцово-
органического соединения, что еще раз свидетельствует о
гидролитической неустойчивости соединений типа (Аг2РЬ)„.
По-видимому, при реакции бромистого мезитилмагния с бромистым
свинцом образуется следующая равновесная смесь [73]:
R3PbMgBr ;± R2Pb + RMgBr.
При пропускании через нее углекислого газа равновесие сдвигается в
правую сторону. Действительно, обработка смеси бромом при низких
температурах приводит к бромистому тримезитилсвинцу. Если же в исходный
окрашенный раствор пропустить углекислый газ и затем его пробромировать,
то получают двубромистый димезитилсвинец [73].
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА МЕТАЛЛИЧЕСКИЙ СВИНЕЦ
Реакции металлоорганических соединений с металлическим свинцом
имеют в основном принципиальный интерес.
Как показано Талалаевой и Кочешковым [75, 76], фениллитий образует
с металлическим свинцом при комнатной температуре тетрафенилсвинец.
Его выход увеличивается при замене фениллития бромбензолом и
металлическим литием [75, 77]. Реактив Гриньяра со свинцом в заметной степени
556
Свинцовоорганические соединения
не.реагирует [76]. По патентным данным [78], реакция между кадмийоргани-
ческими соединениями и металлическим свинцом приводит к соединениям
типа R4Pb.,
Тетраэтилсвинец получен [79] при алкилировании металлического
свинца тетраэтилалюминатом натрия, тетраэтилборнатрием, триэтилцинкна-
трием, триэтилфторалюминийнатрием, триэтилметоксиалюминийнатрием
или этилтриэтоксиалюминийнатрием в присутствии хлористого этила. В
качестве побочного продукта выделен гексаэтилдистаннан. Реакции
проводят с мелкодисперсным свинцом (взятым из реакции галоидного алкила
со сплавом свинец — натрий) в среде алифатических эфиров, тетрагидро-
фурана или тетрагидропирана при 70—90° С:
2 NaAl (С2Н5)4 + РЬ + 2С2Н5С1 -> (С2Н5)4РЬ + 2 NaCl + 2 (С2Н5)3А1,
2 Na (C2H5)3A1F + Pb + 2 С2Н5С1 -> (C2H5)4Pb + 2 NaCl + 2 (C2H5)2A1F.
Понижение температуры снижает скорость реакции, тогда как при более
высоких температурах тетраэтилсвинец частично разлагается.
Существенное влияние на выход тетраэтилсвинца оказывает также растворитель. В
среде диизопропилового эфира или ароматических эфиров выход
тетраэтилсвинца уменьшается. В пиридине преобладают, по-видимому,
побочные процессы [79].
ЛИТЕРАТУРА
l.Blitzer S.M., Pearson Т.Н., Пат. США 2859225—6,2859228—32(1958);
РЖХим., 1960, 70498.
2. К о b е t z Р., Р i п к е г t о n R. С, Пат. США 3068261 (1962); РЖХим., 1964, 14Н104.
3. В 1 i t z е г S. М., Р е а г s о п Т. Н. Пат. США 2989558 (1961); РЖХим., 1962, 16Л60.
4. KrauseE., vonGrosseA., Die Chemie der metallorganischen Verbindungen.
Berlin. Gebr. Borutrager, 1937.
5. WillemsensL.C, Organolead Chemistry. New York, 1964, p. 51.
6. KrauseE., Ber., 62, 1877 (1929).
7. GruttnerG., KrauseE., Ber., 49, 1415 (1916).
8. AmbergerE., Honigschmid-GrossichR., Ber., 98, 3795 (1965).
9. SiebertH., Z. anorgan. allgem. Chem., 263, 82 (1950).
10. G i 1 m a n H., J о n e s R. G., J. Am. Chem. Sec, 72, 1760 (1950).
11. F г е у F. W., С о о к S. E., J. Am. Chem. Soc, 82, 530 (1960).
12. G r u t t n e r G., К r a u s e E., Ber., 50, 574 (1917).
13. JonesW.J., E v a n s D. P., G u 1 w e 1 1 Т., G r i f f i t h s D. С, J. Chem. Sec,
1935 39
14. D a n z e'r R., Monatsh., 46, 241 (1925).
15. S a u n d e r s B. C, S t а с е у G. J., J. Chem. Soc, 1949, 919.
16. G r u t t n e r G., К г a u s e E., Ber., 50, 278 (1917).
17. J u e n g e E. С, С о о к S. E., J. Am. Chem. Soc, 81, 3578 (1959).
18. Ma i er L., Tetrahedron L., 6, 1 (1959).
19. S e у f e r t h D., F г е у e r W., J. Org. Chem., 26, 2604 (1961).
20. J u e n g e E. С, Н о u s e к R. D., J. Org. Chem., 29, 2040 (1964).
21. Gi 1 m a nH., В a i 1 i e J.C., J. Am. Chem. Soc, 61,731 (1939).
22. KrauseE., Ber., 54, 2060 (1921).
23. К r a u s e E., S с h 1 6 t t i g O., Ber., 63 1381 (1930).
24. В a h r G., Z о с h e G., Ber., 88, 542 (1955).
25. В a h r G., Z о с h e G., Ber., 90, 454 (1957).
26. P f e i f f e r P., T r u s к i e r P., Ber., 37, 1125 (1904).
27. К r a u s e E., S с h 1 6 t t i g O., Ber., 58, 427 (1925).
28. Gi 1 m a nH., R о b i nso n J., J. Am. Chem. Soc, 49, 2315 (1927).
29. К о ч е ш к о в К. А., Б о р о д и н а Г. М., Изв. АН СССР, Отд. ест. наук, 1937, 569.
30. М с D. В 1 a i e r J., В г у с е - S m i t h D., P e n d i 1 1 у В. W., J. Chem. Sec, 1959,.
3174.
31. Set ze r W. C, L e e p e r R. W., G i 1 m a n H., J. Am. Chem. Soc, 61, 1609 (1939).
32. G i 1 m a n H., M e 1 s t г о m D. S., J. Am. Chem. Soc, 72, 2953 (1950).
33. П у ч и н я н Е. А., МанулкинЗ. М., Труды Ташкентск. фармацевт, ин-та, 3,
427 (1962).
34. F о s t e r L. S., D i x W. M., G r u n f e s t I. J., J. Am. Chem. Soc, 61, 1685 (1939).
Синтезы на основе солей двухвалентного свинца
557
35. К г a u s е Е., S с h m i t z N., Ber., 52, 2165 (1919).
36. KrauseE., ReissausG.G., Ber., 55, 888 (1922).
37. Lesb reM., Sa tge J., Voi g t D., Compt. rend., 246, 594 (1958).
38. KrauseE., RenwanzG., Ber., 60, 1582 (1927).
39. G i 1 m a n H., T о w n e E. В., Rec. trav. chim., 51, 1054 (1932).
40. MealsR. N.. J. Org. Chem., 9, 211 (1944).
41. Gi 1 ma n H., Summer sL, Lee per R. W., J. Org. Chem., 17, 630(1952).
42. A u s t i n P. R., J. Am. Chem. Soc, 54, 3726 (1932).
43. A u s t i n P. R., J. Am. Chem. Soc, 55, 2948 (1933).
44. В i n d s с h a d 1 e r E., G i 1 m a n H., Proc. Iowa Acad. Sci., 48, 273 (1941); C. A., 36,
1593 (1942).
45. SummersL, Iowa State Coll. J. Sci., 26, 292 (1952); С. А., 47, 8673 (1953).
46. В u с к t о n G. В., Ann., 109, 218 (1859).
47. M e у e r M., Chem. News, 131, 1 (1925).
48. T a f e 1 J., Ber., 44, 323 (1911).
49. 3 a x a p к и н Л. И., О х л о б ы с т и н О. Ю., Изв. АН СССР, ОХН, 1959, 1942.
50. В 1 i t z е г S. М., Р е а г s о п Т. Н., Пат. США 2985675 (1961); РЖХим., 1962, 15Л81.
51. J e n k e r H., Z. Naturforsch., 12b, 909 (1957).
52. PasynkiewiczS., Malinowski S., BitterJ., Przem. Chem., 44, 500
(1965).
53. D i с к s о n R. S., W e s t В. О., Austral. J. Chem., 15, 710 (1962).
54. В 1 i t z e r S. M., P e a r s о n T. H., Пат. США 2859227 (1958); РЖХим., 1961, 10Л129.
55. DobratzE.H., Пат. США 2816123 (1957); РЖХим., 1959, 54365.
56. D a h 1 i g W., P a s у п к i e w i с z S., W a z у n s к i К., Przem. chem., 39, 436 (1960)
57. Р е а г s о п Т. Н., В 1 i t z e r S. М., С а г 1 е у D. R., М с К а у Т. W., R а у R. L.,
S i m s L. L., Z i e t z J. R., Abstracts of Papers 131st Meeting of the American
Chemical Society, Miami, 1957, p. 36L.
58. SimsL. L., В li tzerS. M., С a r 1 e у D. R., С о о к S. E., J u e n g e E. C, L о w-
ranceB.R., Pearson Т. Н., Abstracts of Papers 133rd Meeting of the American
Chemical Society, San Francisco, 1958, p. 46L.
59. H о n e у с u t t J. В., R i d d 1 e J. M., J. Am. Chem. Soc, 82, 3051 (1960).
60. R i d d 1 e J. M., Пат. США 2950301 (1950); РЖХим., 1961, 23Л98.
61. H oney cu t t J. В., R i d d 1 eJ.M., J. Am. Chem. Soc, 83, 369 (1961).
62. R i d d 1 e J. M., Пат. США 2950302 (1960); РЖХим., 1961, 22Л71.
63. M 6 1 1 e r S., P f e i f f e г Р., Ber., 49, 2441 (1916).
64. С a 1 i n g a e r t G., Chem. Revs., 2, 43 (1926).
65. F i s с h e r E. O., G r u b e r t H., Z. anorgan. allgem. Chem., 286, 237 (1956).
66. D a v e L. D., E v a n s D. F., W i 1 к i n s о n G., J. Chem. Soc, 1959, 3684.
67. Weiss E., Z. anorgan. allgem. Chem., 287, 236 (1956).
68. F i s с h e г Е. О., Р i e s b e r g e n U., Z. Naturforsch., lib, 758 (1956).
69. F i s с h e r E. O., S с h r e i n e r S., Ber., 92, 938 (1959).
70. FritzH.P., Fi scher E.O., J. Chem. Soc, 1961, 547.
70a. Reid A. F., Wailes P. C, Austral. J. Chem., 19, ЗЭ9 (1986).
71. AppersonL.D., Iowa State Coll. J. Sci., 16, 7 (1941).
72. J e n s e n К. А., С 1 a u s о n- К a a s N., Z. anorgan. allgem. Chem., 250, 277 (1943).
73. Gl ock li ngF., H oo ton K., Ki n g s t о n D., J. Chem. Soc, 1961, 4405.
74. W i 1 1 e m s e n s L. C, v a n d e r KerkG.J.M., J. Organometal. Chem., 2, 271
(1964).
75. T а л а л а е в а Т. В., К о ч е ш к о в К. А., ЖОХ, 12, 403 (1942).
76. Т а л а л а е в а Т. В., К о ч е ш к о в К. А., ЖОХ, 8, 1891 (1938).
77. LewisF. В., Англ. пат. 854776 (1960); РЖХим., 1961, 18Л91.
78. С a li ngaer tG.,.Sha pi r о Н., Пат. США 2591509 (1952); С. А., 46, 11229 (1952).
79. Frey F. W., К о b e t z P., RobinsonG.C, S i s t r u n k Т. О., J. Org. Chem.,
26, 2950 (1961).
Глава IV
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ДЕЙСТВИЕМ МЕТАЛЛООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
НА СОЛИ ЧЕТЫРЕХВАЛЕНТНОГО СВИНЦА
Соединения класса R4Pb обычно получают прямым методом (см. стр.
541), электролизом (стр. 545) или действием металлоорганических
соединений на соли двухвалентного свинца (стр. 549). Применение реактива
Гриньяра и четыреххлористого свинца (или его комплексов) для этой цели
нетипично и описано лишь на отдельных примерах [1—7]. Так, тетраэтил-
свинец получен [8] при действии на гексахлорплюмбат калия или аммония
этиллития, бромистого этилмагния, диэтилцинка или сесквибромида этил-
алюминия. Выход тетраэтилсвинца максимален (65%) при использовании
в качестве алкилирующего агента этиллития или бромистого этилмагния
в среде диметилового эфира этиленгликоля. На основании полученных
экспериментальных данных предложена следующая схема реакции:
К2РЬС16 + 2 С2Н5М -» 2 С2Н5РЬС13 + 2 МС1 + 2 КС1,
С2Н5РЬС13 -> С2Н5С1 + РЬС12.
2 РЬСЬ + 4 С2Н5М -» (C2H5)4Pb + Pb -f 4 MCI
(M = Li, MgBr, ZnC2H5).
Таким образом, процесс сводится, по существу, к алкилированию двухло-
ристого свинца.
Большую роль в синтезе свинцовоорганических соединений играют
реакции алкилирования или арилирования тетраацилатов свинца и
свинцовоорганических галогенидов.
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ МАГНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
При смешении магнийорганических соединений с тетраацилатами
свинца или триацилатами арилсвинца прежде всего имеет место реакция
обмена с галоидным магнием с образованием РЬХ4 или АгРЬХ3 (X — галоид),
которые разлагаются с выделением дигалогенидов свинца [9].
Следовательно, и в этих случаях в реакции с магнийорганическими соединениями
вступают галогениды двувалентного свинца. С бромистым фенилмагнием или
бромистым толилмагнием выделены тетрафенилсвинец и гекса-о-толил-
диплюмбан соответственно. Здесь, как и обычно в реакциях с друхлористым
свинцом, побочным продуктом является металлический свинец [9].
Для синтеза соединений с различными радикалами у атома свинца,
например R3PbR', широко применяется действие реактива Гриньяра на свин-
цовоорганические галогениды. Необходимым условием для получения
этих веществ в достаточно чистом виде является небольшой избыток
реактива Гриньяра и отсутствие в исходном свинцовоорганическом соединении
примеси неорганической соли свинца. Чаще всего исходят из свинцовоор-
Синтезы на основе солей четырехвалентного свинца
559
ганических хлоридов или бромидов. Иодиды применяются сравнительно
редко [10], так как они обычно содержат небольшое количество двуиодис-
того свинца [И].
Прибавлением хлористого триметил- или триэтилсвинца к реактиву
Гриньяра в эфире с последующим нагреванием смеси получены [11—13]
соединения типа (CH3)3PbR и (C2H6)3PbR, где R—СН3, С2Н5, я-С3Н7,
i -C3H7, я-С4Нб, s-C4H9, ^-C4H8, я-С5Ни и t-C5Hn. Синтез триэтилвинил-
свинца проводят нагреванием при 70—75° С в течение 4—5 час. бромистого
винилмагния (0,1 моля) с хлористым триэтилсвинцом (0,05 моля) в 65 мл
тетрагидрофурана. Выход 70%, т. кип. 57—57,5° С/5 мм [14].
Взаимодействие галогенидов триалкилсвинца с димагниевыми
производными 1,5-дихлорпентана приводит к веществам, содержащим 2 атома свинца
в молекуле, например (СН3)3РЬ(СН2)5РЬ(СН3)3 (т. кип. 166,5° С/14 мм) [15],
С соединением (CH3)2SiHCH2MgCl хлористый триметилсвинец образует в
эфире (CH3)3PbCH2SiH(CH3)2. Выход 86%; т. кип. 26—27°С/1 мм [16].
Синтез триэтилбензилсвинца, как и тетрабензилсвинца (см. стр. 551)»
проводят в атмосфере азота [17]. Из 83 г хлористого триэтилсвинца и
хлористого бензилмагния (из 50,6 г хлористого бензила в 200 мл эфира)
получают 65 г вещества с т. кип. 80° С/0,05 мм.
Соединения состава (CH3)3PbR и (C2H5)3PbR(R = C6H5, р- и о-СН3С6Н4)
получают [18, 19] действием реактива Гриньяра (из 0,3 моля галоидного
арила) на бромистый триалкилсвинец (0,2 моля). Смесь кипятят в течение
часа, разлагают водой, эфирный слой промывают 10%-ной щелочью и
серной кислотой, эфир отгоняют и остаток перегоняют в вакууме.
При прибавлении хлористого триэтилсвинца к отфильтрованному от
непрореагировавшего магния раствору бромистого п-бромфенилмагния
(1,5 моля) в эфире образуется триэтил-п-бромфенилсвинец [20]. Смесь
перемешивают при обычной температуре в течение часа и выливают в
охлажденный до 0°С раствор хлористого аммония. Из органического слоя выделяют
вещество с т. кип. 143°С/0,002 мм. Выход 30%.
Свинцовоорганические производные стирола и а-метилстирола легко
полимеризуются (см. стр. 536), что необходимо учитывать при синтезе этих
соединений.
Получение триметил-^-винилфенил)свинца [21]. К 0,1 моля хлористого гг-винилфенил-
магния в 50 мл тетрагидрофурана прибавляют по каплям 24,9 г (0,075 моля) бромистого
триметилсвинца в 75 мл тетрагидрофурана. Происходит экзотермическая реакция. Внешним
охлаждением поддерживают температуру не выше 30° С. Затем смесь нагревают до 35° С в
течение 2 час, прибавляют 100 мг п-трет-бутшкатехола и избыток реактива Гриньяра
разлагают охлажденным до 0° С насыщенным раствором хлористого аммония. Органический
слой отделяют, водный экстрагируют эфиром и объединенные экстракты высушивают
сульфатом натрия. Растворители отгоняют в вакууме и остаток по возможности быстро
перегоняют в вакууме. (При медленной перегонке вещество полимеризуется. Если его достаточно
много, например 100 г, то может произойти взрыв.) Получают 22 г сырого продукта с т. кип.
77,5° С/3 • Ю-4 мм. Повторная перегонка на колонке дает 17,9 г (67%) слегка окрашенного
в желтый цвет масла с т. кип. 60—61° С/0,0015 мм\ п2^ 1,6070.
Отмечено [22], что триэтил-(д-винилфенил)свинец лучше Есего выделять
перегонкой в высоком вакууме при обычной температуре. Триметил-(я-
а-метилвинилфенил)свинец (т. кип. 69—70° С/0,001 мм) получен [21] с
выходом 56% взаимодействием 0,1 моля хлористого д-а-метилвинилфенилмаг-
ния в 60 мл тетрагидрофурана с 0,06 моля бромистого триметилсвинца.
Смесь нагревают при 50°С в течение 2 час. и обрабатывают как обычно.
Получение триэтил-(#-а-метилвинилфенил,свиниа [23, 24]. Синтез Естества проводят
в атмосфере сухого азота. К реактиву Гриньяра (из 1,22 г магния и 9,85 г л-бром-а-мечил-
стирола в 15 мл тетрагидрофурана) при 50° С постепенно прибавляют 10,5 г хлористого
триэтилсвинца в 25 мл тетрагидрсф>ураыя Зятем смесь нагрегают до кипения в течение 4 час.
560
Свинцовоорганические соединения. Лит. стр. 566—567
и при охлаждении до 0° С гидролизуют раствором хлористого аммония. Органический слой
отделяют,водный слой экстрагируют эфиром и объединенные вытяжки высушивают сульфатом
натрия. Эфир и тетрагидрофуран отгоняют, оставшееся масло разбавляют небольшим
количеством сухого эфира и охлаждают сухим льдом. Выпавшие в осадок примеси к основному
продукту отфильтровывают и вымораживание повторяют дважды. После трехкратной
перегонки остатка в вакууме получают вещество с т. кип. 148—149° С/3 мм. Выход 30%.
Дигалогениды диалкилсвинца при внесении в реактив Гриньяра
сбиваются в комок, что значительно снижает скорость реакции. Для
преодоления этого затруднения их следует прибавлять к реактиву Гриньяра в виде
кашицы в эфире. Так получены [25, 26] соединения ряда (CH3)2PbR2 и
(C2H5)2PbR2 (где R-CH3, n-C3H7, t-C3H7, n-C4H9, /-QHe и C5Hn), a
из 0,55 моля бромистого винилмагния и 0,23 моля двухлористого диэтил-
свинца в 800 мл тетрагидрофурана получен диэтилдивинилсвинец (т. кип.
30° С/1 мм [4], 74°С/13 мм [14]).
Синтез соединений типа R2PbR'R" и RR'PbR"R'" осуществлен
постепенной заменой радикалов на галоид с последующей обработкой реактивом
Гриньяра [27, 28].
Свинцовоорганические галогениды ароматического ряда реагируют
подобно алифатическим соединениям. При действии на них бромистого ал-
килмагния в эфире получены метилтрифенилсвинец (т. пл. 60°С) [29],
этилтрифенилсвинец (т. пл. 42°С) [30], пропилтрифенилсвинец (т. пл. 69—
70°С) [31], амилтрифенилсвинец (т. пл. 16—17°С) [31] и пропилфенилди-о-
толилсвинец (т. пл. 49—50°С) [31]. т/?ет-Бутилтрифенилсвинец(т. пл. 47°С)
[32] и ди-т/?ет-бутилдифенилсвинец (т. пл. 177°С) [33] образуются только
при обратном порядке введения реагентов: бромистый т/?ет-бутилмагний
прибавляют к избытку свинцовоорганического бромида.
Синтез винилтрифэнилсвинца проводят в тетрагидрофуране [34, 35].
Смесь нагревают в течение 24 час, гидролизуют насыщенным раствором
хлористого аммония, растворитель отгоняют и остаток дважды
экстрагируют 100 мл горячего гексана. Экстракт отфильтровывают от непро-
реагировавшего хлористого трифгнилсвинца, гексан отгоняют и остаток
очищают молекулярной перегонкой при давлении 0,05—0,06 мм и
температуре бани 160—170°С. Отгоняющаяся бесцветная жидкость
постепенно закристаллизовывается. Выход 23,6%; т. пл. 33—34°С.
Аллилтрифенилсвинец (т. пл. 76—77°С) получен [36, 37] нагреванием в
течение 4 час. 57 г хлористого трифенилсвинца с бромистым аллилмагнием
(из 92 г бромистого аллила и 40 г магния в 750 мл эфира). Смесь оставляют
стоять на 12 час, разлагают ледяным раствором хлористого аммония, эфир
отгоняют и остаток перекристаллизовывают из эфира.
Гилман с сотр. [38] описал синтез и свойства бутенил-3-трифенилсвинца
(т. пл. 84—86°С) и Р-трифенилстирилсвинца. Реакция между бромистым
пропаргилмагнием и (С6Н5)3РЬХ приводит к смеси (С6Н5)3РЬСН=С=СН2
(90%) и (С6Н5)3РЬСН2С=СН [39].
Реактив Гриньяра, полученный из триметил-5-бромамилолова, образует
с хлористым трифенилсвинцом соединение, содержащее в молекуле
одновременно атомы свинца и олова [15].
Галогениды триарилсвинца гладко реагируют с галогенидами цикло-
гексил-[30], бензил-[40] и арилмагния [20, 29, 30, 41—46]. Хлористый три-
фенилсвинец в условиях реакции Либрикаи Рамсдена образует с хлористым
д-винилфенилмагнием трифенил-(д-винилфенил)свинец [22, 47—49]. Из
20,0 г хлористого трифенилсвинца получают 17,5 г (77%) кристаллов
с т. пл. ПО—111°С (из этилового спирта) [48].
Синтез соединений типа (C6H5)2PbR2 можно иллюстрировать
следующими примерами.
Синтезы на основе солей четырехвалентного свинца
561
Получение дивинилдифенилсвинца [50]. 25 г (0,058 моля) двухлористого дифенилсвин-
ца прибавляют частями к 0,П7моля бромистого винилмагния в 150 мл тетрагидрофурана.
Осадок постепенно растворяется, раствор окрашивается в зеленый цвет. После обработки
смеси получают маслянистую жидкость желтого цвета, которая перегоняется со
значительным разложением. Дивинилдифенилсвинец представляет собой бесцветную жидкость с т. кип.
111 —114° С/0,002 мм. Выход 9,8 г (41%). Чистое вещество может храниться длительное
время без заметного разложения.
Выделить в чистом виде диаллилдифенилсвинец не удается [51].
При действии на двухлористый дифенилсвинец хлористого я-винил-
фенилмагния в тетрагидрофуране образуется с выходом 65% дифенилди-
(д-винилфенил)свинец, т. пл. 96—97°С (из этилового спирта) [48]. Дицик-
логексилдифенилсвинец (т. пл. 178—180°С) [52], дифенилди(аллилфенил)сви-
нец (т. пл. 194—196°С) [43], дифенилди(циклогексилфенил)свинец (т. пл.
170°С) [44], дифенилди-(9-фенантрил)свинец (т. пл. 208—210°С) [41], ди-
фенилди-о-толилсвинец (т. пл. 134—135° С) [53], дифенилди-2,5-ксилилсви-
нец (т. пл. 94°С) [29] и дифенилди-а-нафтилсвинец (т. пл. 197°С) [38]
получены взаимодействием двухлористого или двубромистого дифенилсвинца с
соответствующими магнийорганическими соединениями в эфире.
Из смешанных соединений гетероциклического ряда известны следующие:
трифенил-2-тиенилсвинец (т. пл. 206—207°С) [54,55], трифенил-2-фурилсви-
нец (т. пл. 166—167°С) [55], три-я-анизил-2-фурилсвинец (т. пл. 83°) [56],
трифенил-2-пиридилсвинец (т. разл. 220°С) [57], дифенилди-2-фурилсвинец
(т. пл. 118°С) [55], дифенилди-2-тиенилсвинец (т. пл. 185°С) [55], ди-2-фу-
рилди-я-анизилсвинец (т. пл. 77—79°С) [56], ди-2-фурилди-2-тиенилсвинец
(т. пл. 117—119°С) [55] и ди-2-тиенилди-2-селениенилсвинец (т. пл. 147°С)
[581.
Диэтилциклопентаметиленсвинец (т. кип. 11 ГС/13 мм) получен [59]
по следующей схеме:
СН2
/ \
(C2H5)2PbGl2'4- GIMg (CH2)5MgCl-^(C2H5)2Pb (CH2)3 + MgCl2.
X /
СН2
Реакция между димагниевым производным бромистого метилена и диацета*
том дифенилсвинца проходит совершенно иным путем [60]:
(СбН5)2РЬ (ООССНз)2 -J- BrMgGH2MgBr -> НОРЬ(С6Н5)2 [СН2РЬ (С6Н5)2]лСН2РЬ(СбН5)20Н.
Конечный продукт представляет собой нерастворимый и неплавкий
порошок белого цвета [60].
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ ЛИТИЙ- И НАТРИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Алкиллитий- и алкилнатрий, за редким исключением [61], применяют
только для синтеза свинцовоорганических ацетиленидов. При нагревании
хлористого триэтилсвинца с ацетиленидами щелочных металлов в пентане
получены [39] следующие неустойчивые на воздухе соединения: ацетиленид
триэтилсвинца (т. кип. 110°С/2,5 мм), метил ацетиленид (т. кип. 81°С/1,5 мм),
циклопентенил-1-ацетиленид (т. кип. 95°С/1,5л1м) и фенилацетиленид
триэтилсвинца (т. кип. 66°С/1лш). Хлористый триметил- итриэтилсвинецсцик-
лопентадиенилнатрием в сухом бензоле образуют неустойчивые соединения,
которые быстро разлагаются уже при обычной температуре [62]. Также
36 3 аказ № 1С 7
562 Свинцовоорганические соединения. Лит. стр. 566—567
не удается выделить в чистом виде бис-(триалкилплюмбил)ацетилены
[63], тогда как в циклогексильном и ароматическом рядах эти соединения
легко образуются при действии ацетиленида натрия на
свинцовоорганические галогениды в жидком аммиаке [63—66]. Аналогично получены [39, 66)
диацетиленид трифенилсвинца, метил- и фенилацетилениды трифенилсвин-
ца и бис-(трифенилплюмбил)диацетилен. Взаимодействием хлористого
трифенилсвинца с ацетиленидами натрия или лития в сухом пентане получены
[39] алкил-, алкенил- и фенилзамещенные ацетилениды трифенилсвинца.
Гораздо чаще литийорганические соединения применяются в качестве
арилирующих агентов. Так получены, например, трициклогексилфенилсви-
нец (т. пл. 146—147°С), трициклогексил-п-анизилсвинец (т. пл. 78°С), три-
циклогексил-п-толилсвинец (т. пл. 97—98°С), трициклогексил-п-хлорфенил-
(т. пл. 102—103°С), трициклогексил-^-хлорфенилсвинец (т. пл. 73—74°С)
и трициклогексил-п-диметиламинофенилсвинец (т. пл. 125°С) [67].
Как уже отмечено на стр. 558, реакция магнийорганических соединений
с тетраацилатами свинца сопровождается выделением металлического
свинца. С литийорганическими соединениями она проходит иначе [9]:
Pb (OOCR)4 + 4 ArLi -> Ar4Pb + 4 RCOOLi,
Таким образом были получены тетрафенил- [9] и тетрапентафторфенил-
свинец [68].
Взаимодействием свинцовоорганических галогенидов с
литийорганическими соединениями получены триэтилфенилсвинец [69], трифенил-д-
толилсвинец (т. пл. 124—125°С) [70], трифенил-9-флуоренилсвинец (т.
пл. 118—120°С) [71], трифенил(бенз-а-актрацен-7-ил)свинец (т. пл.
204 —205°С) [41], трифенил-3-инденилсвинец (т. пл. 122° С) [71],
дифенилди-п-толилсвинец (т. пл. 121—122°С) [70], дифенилди-9-флуоренил-
свинец (т. пл. 138—140°С) [71], дифенилди(бенз-а-антрацен-7-ил)сви-
нец (т. пл. 295—296°С) [41], дифенилди-3-инденилсвинец (т. пл. 107— 110°С)
[71], а из оптически активного /?-LiCGH4OC8H17-s оптически активный
(П'С3Н7)(0'СН3С6Н^) P'(s-C8HvOCGH^)Pb 131]. Реакции проводят путем
кратковременного перемешивания свинцовоорганического галогенида с литий-
органическим соединением с последующим гидролизом смеси и
перекристаллизацией выделившегося осадка. Ариллитий необходимо брать в точно
вычисленном количестве, иначе замещается не только галоид, но и радикал у
атома свинца.
Применение литийорганических соединений позволяет ввести в
свинцовоорганические соединения ароматические радикалы, содержащие гидро-
ксил-, амино- или сульфамидную группу [20, 41, 42, 70, 72, 73]. Для
достижения хороших выходов продуктов все операции с литийорганическими
соединениями должны проводиться достаточно быстро. Так, при действии
о-литийфенолята лития на эквимолекулярное количество хлористого
трифенилсвинца в эфире образуется 70% трифенил-(о-оксифенил)свинца, если
гидролиз проводить спустя 3 мин. после смещения реагентов [72]. Если же
до гидролиза к смеси прибавить бензол и нагреть ее в течение часа, то выход
снижается до 12%[41].
Получение трифенил-/г-аминофенилсвинца [72]. К суспензии 14,2 г (0,03 моля)
хлористого трифенилсвинца в эфире при перемешивании приливают за один раз растЕор /г-литий-
анилина (из 0,031 моля /?-броманилина в 30 мл эфира и 0,093 моля я-бутиллптия в 200 мл
эфира). Через 3 мин. смесь выливают в ледяную воду. Органический слой отделяют и непро-
реагировавший n-броманилин экстрагируют разбавленной соляной кислотой, в которой
трифенил-я-аминофенилсвинец не растворяется. Эфирный раствор высушивают и затем в него
пропускают ток сухого хлористого водорода. Осадок солянокислого трифенил-п-аминофенил-
свинца отфильтровывают, суспендируют в воде и превращают в свободный амин обработкой
разбавленным раствором аммиака. Получают 9,5 г (60%) вещества с т. пл. 172° С (из смеси*
бензола с петролейным эфиром).
Синтезы на основе солей четырехвалентного свинца
563
Аналогично получены [20, 41, 42, 72] трифенил-о-аминофенилсвинец
(т. пл. 164—165°С), трифенил-я-Ы-аминофенилсвинец (т. пл. 97—98°С),
трифенил-0-диметиламинофенилсвинец (т. пл. 101—102°С) и орто-, мета-
и пара-изомеры трифенилоксиметилфенилсвинца (т. пл. 134—136, 113—114
и 98—100°С соответственно). Выходы продуктов составляют 40—60%.
Попытки ввести в свинцовоорганические соединения карбоксильную
группу взаимодействием литиевой соли n-карбоксифениллития с хлористым
трифенилсвинцом оказались неудачными [20, 42].
n-Фенилендилитий превращает хлористый трифенилсвинец в п-бис-
(трифенилплюмбил)бензол (т. пл. 285—288°С):
2 (СбН5)зРЬС1 + р-ЫСеНДЛ -> p-(C6H5)3PbQH4Pb (С6Н5)8 + ЫС1.
То же вещество образуется с небольшим выходом (9%) и при работе с
эквимолекулярными количествами реагентов [20, 42].
Дигалогениды дифенилсвинца с дилитиевыми соединениями образуют
гетероциклические соединения, содержащие атомы свинца в цикле [74,
75]:
СН2
(C6H5)2PbCl2 + Li (CH2)5Li -> (CeH5)2Pb (CH2)3+2 LiCl,
\„{
<^\ C2H5
C2H5-N + (СвН6)2РЬВг!!.-» f J f J + 2LiBr.
СвНб CeHs
Следует отметить, что применение литийорганических соединений в
качестве арилирующих реагентов не всегда дает надежные результаты
вследствие возможной здесь побочной реакции [20]:
(C6H5)sPbR + RLi -» (CeH5)2PbR2 + C6H5Li.
Аналогично ведет себя диэтилбарий [76].
При действии на хлористый трифенилсвинец 2,2'-дилитийдифенила
получают тетрафенилсвинец (выход 49%) и 30% бесцветного кристаллического
вещества с т. пл. 136—137,5°С[77]. Его ИК- иУФ-спектры, а также
продукты реакции с хлористым водородом соответствуют 9,9-дифенилдибензоплюм-
болу:
%/\ /СвН5
I рь<
О7 • •
Для избежания этих вторичных процессов в отдельных случаях
пользуются следующим приемом. К раствору ариллития прибавляют избыток
бромистого магния и образующееся магнийорганическое соединение вводят
в дальнейшие реакции. Так получены, например, трифенил-о-оксифенил-
свинец [72], трифенил-я-аминофенилсвинец [78] и йодистый дифенил-2-
пиридилсвинец (из двуиодистого дифенилсвинца, бромистого магния и 2-
пиридиллития в эфире) [57].
36*
"564 Свинцовоорганжеские соединения. Лит. стр. 566—567
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ РТУТНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Несмеяновым с сотр. [79, 80] при взаимодействии на холоду в среде
сухого хлороформа ди-(траяс-|3-хлорвинил)ртути и тетраацетата свинца с
хорошим выходом получен диацетат ди-|3-хлорвинилсвинца:
2 (ClCH=CH)2Hg + Pb (OOCCH3)4 ->
(С1СН=СН)2РЬ (ООССН3)2 + 2ClCH=CHHgOOCCH8.
Реакция проходит с небольшим разогреванием и полностью заканчивается
в течение 2 час. Следует подчеркнуть, что в результате реакции образуется
только один стереоизомер нового хлорвинильного соединения, причем
конфигурация его хлорвинильной группы тождественна исходной.
Интересно отметить, что хлористая траяс-|3-хлорвинилртуть или ацетат
|3-хлорвинилртути, как было показано специальными опытами, с тетрааце-
татом свинца не реагируют.
Получение диацетата ди-|3-хлорвинилсвинца [79]. Раствор 15 г (0,046 моля) дя-(транс-
р-хлорвинил)ртути в 17 мл сухого хлороформа смешивают с раствором 10,3 г (0,023 моля)
тетраацетата свинца, содержащего небольшое количество уксусной кислоты, в 35 мл сухого
хлороформа. Раствор желто-оранжевого цвета. Через 30—40 сек. с небольшим разогреванием
начинается выделение кристаллического осадка и одновременное обесцвечивание раствора.
Через 2,5—3 часа наступает полное обесцвечивание раствора, реакционную массу охлаждают
льдом, осадок отфильтровывают, промывают 10 мл сухого холодного хлороформа. Это почти
чистый ацетат р-хлорвинилртути. При 110° С вещество сереет и при 118—120° С чернеет, не
плавясь. Выход 11,8 г (78%).
Фильтрат упаривают в вакууме без доступа влаги воздуха досуха (температура бани
30° С). Сухой остаток высушивают в эксикаторе над едким натром до исчезновения запаха
уксусной кислоты, после чего перекристаллизовывают из смеси сухих хлороформа и эфира
(хорошее охлаждение). Выход перекристаллизованного продукта 7,4 г (72%). После
повторной перекристаллизации продукт выделяет пузырьки ацетилена при 115—130° С, желтеет
при 140° С, при 185° С чернеет, не плавясь.
Диацетат ди-Р-хлорвинилсвинца очень хорошо растворим в хлороформе, умеренно в
сухом спирте и этилацетате, плохо в бензоле, нерастворим в эфире и четыреххлористом
углероде.
Кочешковым и Надь [81] подробно изучена реакция тетраацилатов
свинца с диарилртутью:
2 Ar2Hg + Pb (OOCR)4 -> Ar2Pb (OOGR)2 -> 2 ArHgOOCR.
Предложенный ими метод позволяет получать свинцовоорганические
соединения, содержащие различные заместители в бензольном ядре. Реакции
проводят в хлороформе при комнатной температуре.
Получение диацетата ди-о-анизилсвинца [81]. 4,14 г (0,01 моля) ди-о-анизилртути в
15 мл сухого хлороформа смешивают с 2,21 г (0,005 моля) тетраацетата свинца в 20 мл того
же растворителя. Полученный раствор оранжевого цвета оставляют стоять при комнатной
температуре в течение 4 дней, после чего хлороформ отгоняют досуха на водяной бане.
Остаток экстрагируют петролейным эфиром, в котором диацетат ди-о-анизилсвинца почти
нерастворим. Его очищают перекристаллизацией из спирта. Выход 1,7 г (63%); т. пл. 191 —
193°С.
Аналогично получены диацетаты дифенил- (т. пл. 200—20ГС), ди-ос-
нафтил- (т. пл. 236°С) и ди-я-карбэтоксифенилсвинца (т. пл. 207—208°С).
Впоследствии Кочешковым, Пановым и Лодочниковой [82—87]
разработан простой и удобный метод синтеза соединений класса АгРЬХ3
основанный на реакции:
Ar2Hg + Pb (OOCR)4 -> ArPb(OOCR)3 + ArHgOOCR.
Удаление побочного продукта легко достигается путем прибавления
рассчитанного количества спиртового раствора хлористого водорода (хлорид
арилртути выпадает при этом в осадок).
Синтезы на основе солей четырехвалентного свинца 565
Получение триацетата фенилсвинца [82]. К раствору 1,7 г тетраацетата свинца в 12 мл
сухого хлороформа приливают при перемешивании 1,17 г дифенилртути в 10 мл хлороформа.
Через 30 мин. реакция заканчивается, что видно по прекращению выделения бурого осадка
двуокиси свинца при прибавлении воды к капле реакционной смеси. Раствор охлаждают
смесью снега с солью и прибавляют по каплям 1,25 мл спиртового раствора хлористого
водорода (один эквивалент). Тотчас выпадает осадок хлористой фенилртути (т. пл. 258° С). Из
фильтрата, помещая его в вакуум-эксикатор, удаляют растворитель и остаток
кристаллизуют из этилацетата с добавлением одной капли уксусной кислоты. Т. пл. 101 —102° С; выход
близок к теоретическому.
Аналогично получены [82—88] триизобутират фенилсвинца (т. пл. 77—
78°С), триацетат я-толилсвинца (т. пл. 87°С), триацетат а-нафтилсвинца
(т. пл. 168—169°С), триизобутират Р-нафтилсвинца (т. пл. 99,5—100°С),
триацетат Р-нафтилсвинца (т. пл. 108—109°С), трипропионат Р-нафтил-
свинца (т. пл. 84—85°С), триацетат n-анизилсвинца (т. пл. 139—14ГС),
трипропионат n-анизилсвинца (т. пл. 75—76°С), триацетат п-иодфенилсвинца
(т. пл. НО—112°С), трипропионат п-иодфенилсвинца (т. пл. 96°С) и
триизобутират п-иодфенилсвинца (т. пл. 78°С).
Триацилаты фенилсвинца при действии на них диарилртути могут быть
превращены в диацилаты дифенилсвинца [85, 86].
Действие дифенилртути на двухлористый дифенилсвинец с целью
получения хлористого трифенилсвинца удалось Остину [89] только на одном
простейшем примере. Уже в толильном ряду реакция оказалась неприменимой.
Хлористый трифенилсвинец с дифенилртутью ке реагирует.
Триэтилалюминий гладко реагирует с тетраацилатами свинца в толуоле.
Выход тетраэти л свинца составляет 60—70% [8]. Аналогично получен тетра-
фенилсвинец. Выход 95,8%.
Мы считаем целесообразным привести здесь методику синтеза
тетраацетата свинца.
Получение тетраацетата свинца [90]. В трехгорлую колбу, емкостью 2 л, снабженную
термометром и механической мешалкой с ртутным затвором, помещают смесь 1080 г ледяной
уксусной кислоты и 360 г уксусного ангидрида и затем постепенно добавляют небольшими
порциями 600 г сухого сурика, быстро перемешивая смесь. Нужно следить, чтобы
температура не поднялась выше 65° С. К концу реакции осторожно подогревают колбу для
окончательного превращения окисла в ацетат. После охлаждении смеси осадок тетраацетата
свинца отсасывают через большую воронку (раствор фильтруется медленно) и промывают ледяной
уксусной кислотой. Выход около 300 г.
Тетраацетат свинца кристаллизуется в виде бесцветных призм с т. пл. 175—180° С.
Вещество неустойчиво на воздухе, быстро гидролизуется с выделением двуокиси свинца
коричневого цвета. Тетраацетат свинца растворим в хлороформе, четыреххлористом углероде
и бензоле, и если растворитель достаточно сухой, то вещество можно извлечь неизмененным.
Умеренно растворим в холодной уксусной кислоте, хорошо — в горячей.
Тетрапропионат свинца и тетраизобутират свинца могут быть получены
аналогично, но отделить их от соли двувалентного свинца сложнее.
Например, тетраизобутират свинца отделяют от диизобутирата свинца обработкой
реакционной смеси хлором при 0°С. Образовавшийся двухлористый свинец
отфильтровывают, из фильтрата выделяются кристаллы тетраизобутирата
свинца с т. пл. 109°С. Тетрапропионат свинца, полученный аналогично,
имеет т. пл. 132°С [91].
Тетрапропионат, тетраизобутират и соли высших карбоновых кислот
целесообразнее получать реакцией переацидирования [85, 92]:
РЬ (ООССН3)4 + 4 RCCOH ~> Pb (OOCR)4 + 4 СН3СООН.
Уксусную кислоту отгоняют в вакууме при 50—60°С. Из остатка, после
охлаждения, выкристаллизовывается соответствующая соль свинца.
566 Свинцовоорготические соединения
ЛИТЕРАТУРА
1. Gru tt nerG., Krau seE., Ber., 50, 1559(1917).
2. M a i e r L., Angew. Chem., 71, 161 (1959).
3. D г e f a h 1 G., L о r e n z D., J. prakt. Chem., 24, 106 (1964).
4. В a r t о с h а В., G г а у М. Y., Z. Naturforsch., 14b, 350 (1959).
5. D r e f a h 1 G., P 1 6 t n e r G., Angew. Chem., 72, 454 (1960).
6. В 1 i t z e r S. M., P e a r s о n Т. Н., Пат. США 3007955 (19ol); РЖХим., 1963, 7H74.
7. R a m s d e n H. E., ShannonH.F., Пат. США 3156716 (1964); РЖХим., 1966,
8Н110.
8.- F г е у F. W., С о о к S. E., J. Am. Chem. Soc, 82, 530 (1960).
9. Л о д о ч н и к о в а В. И., П а н ов Е. М., К о ч е ш к о в К. А., ЖОХ, 37, 547 (1967).
10. С а 1 i n g a e r t G., S о г о о s H., J. Org. Chem., 2, 535 (1938).
11. GriittnerG., К г a u s е Е., Вег., 49, 1125 (1916).
12. G r u t t n e r G., К г a u s е Е., Вег., 50, 574 (1917).
13. GilmanH., SweeneyO.R., R о b i n s о n J., Rec. trav. chim., 49, 205 (1930).
14. КоршакВ.В., ПоляковаА. М., СучковаМ. Д., Высокомолек. соед.,
2, 13 (1960).
15. G r u t t n e r G., К г a u s e E., W i e r n i k M., Вег., 50, 1549 (1917).
16. S с h m i d b а и r H., W a 1 d m a n n S., Ber., 97, 3381 (1964).
17. GlocklingF., KingstonD., J. Chem. Soc, 1959, 3001.
18. G г и t t n e r G., G г и t t n e r G„ Ber., 51, 1293 (1918).
19. BowdenK, В г а и d e E. A., J. Chem. Soc, 1952, 1068.
20. GilmanH., M e 1 s t г о m D. S., J. Am. Chem. Soc, 72, 2953 (1950).
21. NoltesJ.G., BuddingH.A., van d e r KerkG.J.M., Rec. trav. chim.,
79, 1076 (1960).
22. ParsH.G., G r a h a m W. A. G., A t к i n s о n E. R., MorganC.R., Chem. a.
Ind., 1960, 693.
23. КоршакВ. В., ПоляковаА. М., Т а м б о в ц е в а Е. С, Высокомолек. соед.,
1, 1021 (1959).
24. П о л я к о в а А. М., КоршакВ.В., Т а м б о в ц е в а Е. С, Высокомолек. соед.,
3, 662, (1961).
25. GriittnerG., К г а и s e E., Вег., 49, 1546 (1916).
26. Н е а р R., S а и n d e r s В. С, S t а с е у G. J., J. Chem. Soc, 1951, 658.
27. G г и t t n e r G., К г а и s e E., Ann., 415, 338 (1918).
28. G г и t t n e r G., К г а и s e E., Вег., 50, 202 (1917).
29. К г а и s e Е., S с h 1 6 t t i g О., Вег., 58, 427 (1925).
30. К г а и s e E., S с h m i t z N., Ber., 52, 2150 (1919).
31. Austi nP. R., J. Am. Chem. Soc, 55, 2948 (1933).
32. G i 1 m a n H., S w e e n e у O. R., К i r b у J. E., Iowa State Coll. J.Sci., 3, 1 (1928).
33. GilmanH., BalassaL., Iowa State Coll. J. Sci., 3, 105 (1929).
34. J и e n g e E. С, С о о k S. E., J. Am. Chem. Soc, 81, 3578 (1959).
35. S e у f e r t h D., WeinerM.A., J. Am. Chem. Soc, 84, 361 (1962).
36. А и s t i n P. R., J. Am. Chem. Soc, 53, 3514 (1931).
37. КотонМ. М., Киселева Т. М., ЗапеваловаН.П., ЖОХ, 30, 186 (1960).
38. G i 1 m a n H., T о w n e E. В., J о n e s H. L., J. Am. Chem. Soc, 55, 4689 (1933).
39. MassonJ .-C., LeQuan M., Chodkiewicz W., С a d i о t P., Compt. rend.,
257, 1111 (1961).
40. TamborskiC, Ford F.E,Lehn W.L, M о о r e G. J., S о 1 о s к i E. J.,
J. Org. Chem., 27, 619 (1962).
41. G i 1 rn a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
42. Melstrom D. S., Iowa State Coll. J. Sci., 18, 65 (1943); С. А., 38, 727 (1944).
43. П у ч и н я н Е. А., Манул кинЗ.М., ДАН УзбССР, 3, 47 (1962).
44. П у ч и н я н Е. А., М а н у л к и н 3. М., ДАН УзбССР, 12, 51 (1961).
45. 3 а с о с о в В. А., К о ч е ш к о в К. А., Сборник статей по общей химии, т.1.М.,Изд-во
АН СССР, 1953, стр. 278.
46. A u s t i n P. R., J. Am. Chem. Soc, 53, 1548 (1931).
47. КотонМ.М, КиселеваТ.М, ФлоринскийФ. С, Изв. АН СССР, ОХН,
1959, 948.
48. N о 1 t e s J. G., В u d d i n g H. A., v a n d e r KerkG.J.M., Rec. trav. chim.,
79, 408 (1960).
49. К о т о н М. М., Киселева Т. М., ФлоринскийФ. С, Высокомолек. соед.,
2, 1639 (1960).
50. Н е п г у М. С, N о 1 t e s J. G., J. Am. Chem. Soc, 82, 555 (1960).
51. Gi 1 m a nH, E i sch J., J. Org. Chem., 20, 763 (1955).
52. GruttnerG, Ber., 47, 3257 (1914).
53. LedererK, Ber., 49, 349 (1916).
54. К r a u s e E., R e n w a n z G., Ber., 62, 1710 (1929).
Синтезы на основе солей четырехвалентного свинца 567
55. GilmanH., TowneE. В., Rec. trav. chim., 51, 1054 (1932).
56. G i 1 m a n H., T о w n e E. В., J. Am. Chem. Soc, 61, 739 (1939).
57. GilmanH., Gregory W. A., SpatzS.M., J. Org. Chem., 16, 1788 (1951).
58. ЮрьевЮ. К., ГальберштамМ.А, К а н д р о р И. И., ЖОХ, 34, 4116
(1964).
59. G r u t t n e r G., К г a u s e E., Вег., 49, 2666 (1916).
60. К о т о н М. М., К и с е л е в а Т. М., ЖОХ, 35, 2036 (1965).
61. GlocklingF., J. Chem. Soc, 1955, 716.
62. F r i t z H. P., SchwarzhansK.E, J. Organometal. Chem., 1, 297 (1S64).
63. В е е г m a n n C, HartraannH., Z. anorgan allgem. Chem., 276, 20 (1954).
64. H а г t m a n n H., KomorniczykK., Naturwiss., 51, 214 (1964).
65. H a r t m a n n H., E s с h e n b а с h W., Naturwiss., 46, 321 (1959).
66. Hartmann H., DietzE., KomorniczykK., Reiss W., Naturwiss., 48,
570 (1961).
67. E a b or nC, PandeK.C, J. Chem. Soc, 1961, 3715.
68. Tamborski C, S о 1 о s к i E. J., DecS.M., J. Organometal. Chem., 4, 446
(1965).
69. EabornC, PandeK.C, J. Chem. Soc, 1960, 1566.
70. А и s t i n P. R., J. Am. Chem. Soc, 54, 3726 (1932).
71. D'AnsJ., Zimmer H., v. В г а и с h i t s с h M., Ber., 88, 1507 (1955).
72. G i 1 m a n H., S t и с к w i s с h С G., J. Am. Chem. Soc, 72, 4553 (1950).
73. Fel dha к eC. J., StevensC.D., J. Chem. and Engng. Data, 9, 241 (1964).
74. В a j e r F. J., P о s t H. W., J. Org. Chem., 27, 1422 (1962).
75. G i 1 m a n H., Z и е с h E. A., J. Am. Chem. Soc, 82, 2522 (1960).
76. G i 1 m a n H., H а и b e i n A. H., O'D о n n e 1 1 G., W о о d s L. A., J. Am. Chem.
Soc, 67, 922 (1945).
77. GeliusR., Angew. Chem., 72, 322 (1960).
78. GilmanH., S t и с к w i s с h C. G., J. Am. Chem. Soc, 64, 1007 (1942).
79. H e с м е я н о в А. Н., Ф р е й д л и н а Р. X., К о ч е т к о в А. К., Изв. АН СССР,
ОХН, 1948, 127.
80. Н е с м е я н о в А. Н., Б о р и с о в А. Е., ДАН СССР, 60, 67 (1948).
81. Н а д ь М. М., К о ч е ш к о в К. А., ЖОХ, 12, 409 (1942).
82. Панов Е.М., Лодочникова В. И., К о ч е ш к о в К. А., ДАН СССР, 111,
1042 (1956).
83. Л о д о ч н и к о в а В. И., ПановЕ.М., К о ч е ш к о в К. А., Изв. АН СССР,
ОХН, 1957, 1484.
84. Л о д о ч н и к о в а В. И., Труды XX Годичн. научн. сессии (Свердл. мед.) ин-та,
22, 90 (1957).
85. Л о дочн и нова В. И., ПановЕ.М., К о ч е ш к о в К. А., ЖОХ, 29, 225
(1959).
86. Лодочникова В. И., ПановЕ.М., К о ч е ш к о в К. А., ЖОХ, 33, 1199
(1963).
87. Л одочн и ко в а В. И., ПановЕ.М., К о ч е ш к о в К. А., ЖОХ, 34, 4022
(1964).
88. С г i eg ее R., D.i m ro t h P., Sc hem pf R., Ber., 90, 1137 (1957).
89. A usti nP. R., J. Am. Chem. Soc, 54, 3287 (1932).
90. Dimroth0.fSchweizerR.t Ber., 56, 1375 (1923); Сб. «Неорганические
синтезы», т. 1. М., ИЛ, 1951, стр. 50.
91. A b e g g R., Handbuch der anorganischen Chemie, Bd. 3. Leipzig, 1909, S. 741, 758.
92. Y u k a w a Y., S a k a i M., Bull. Chem. Soc Japan, 36, 761 (1963).
Глава V
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛИФАТИЧЕСКИХ ДИАЗОСОЕДИНЕНИЙ
И СОЛЕЙ ДИАЗОНИЯ
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ АЛИФАТИЧЕСКИХ ДИАЗОСОЕДИНЕНИЙ
Хлористый триэтилсвинец и двухлористый диэтилсвинец не реагируют
с эквимолекулярным количеством диазометана и диазоэтана при комнатной
температуре (среда — бензол) [1,2] или даже при нагревании (среда —эфир)
[2, 3]. Если же в смесь добавить медную бронзу, то при 10—15°С
происходит реакция и образуются свинцовоорганические соединения [2, 3]:
Си fClPb(C2H5b
CH2N2 —> [СН2] > (С2115)зРЬСН2С1,
ГС HI [ С Н 1
(С2Н5)2РЬС12 — U [(СаН5)2РЬС1 (СН,С1)] '-, (С2Н,)2РЬ (СП2С1)2.
В качестве побочного продукта образуется некоторое количество тетраэтил-
свинца. Неустойчивый триэтил-а-хлорэтилсвинец выделить из
реакционной смеси не удается. Реакция диазометана с хлористым
свинцом, четыреххлористым свинцом, (NH4)2PbCl6 и тетраацетатом свинца не
привела к образованию свинцовоорганических соединений [2,3].
Получение диэтилди(хлорметил)свинца [2]. Смесь из 24 г двухлористого диэтилсвинца
с небольшим количеством (0,1—0,2 г) медной бронзы в 300 мл сухого эфира охлаждают до
0° С. При этой температуре и хорошем перемешивании прибавляют половину эфирного
раствора диазометана, взятого из расчета 2 моля диазометана на 1 моль двухлористого
диэтилсвинца с 10%-ным избытком. Температуру поднимают до 10—15° С. При этом наблюдается
появление пузырьков газа, количество двухлористого диэтилсвинца уменьшается и раствор
постепенно теряет свою начальную желтую окраску. Затем раствор снова охлаждают до 0° С
и вносят вторую порцию диазометана. Когда раствор становится чуть желтым и прекращается
выделение газа, перемешивание заканчивают, реакционную смесь оставляют на ночь.
Затем отфильтровывают от осадка (4,3 г) и фильтрат разгоняют. Оставшуюся после отгонки
эфира жидкость с небольшим количеством осадка и бурой смолки разгоняют в вакууме с
дефлегматором. После многократной вакуумной перегонки получают диэтилди(хлорметил)-
свинец (т. кип. 96° С/2 мм\ d 1,9890); бесцветная, легко подвижная жидкость со
своеобразным запахом.
Аналогично получают и триэтилхлорметилсвинец (т. кип. 65,5—66,5°С/
Ъ мм\ df 1,7917; n2D° l, 5434).
Гидриды триалкилсвинца Alk3PbH (Alk = СН3, С2Н5) реагируют с ди-
азоэтаном при —80°С в эфире. Но выходы продуктов незначительны
вследствие идущего разложения гидридов триалкилсвинца [4].
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ПОМОЩЬЮ СОЛЕЙ ДИАЗОНИЛ
Метод разложения двойных солей арилдиазония порошками металлов
[5], примененный Несмеяновым, Кочешковым, Реутовым и другими для
синтеза органических производных ряда металлов, в случае свинца, вслед-
Синтез с помощью диазосоедин-ений
569
ствие малых выходов, не имеет практического значения [6]. Так, при
разложении в эфирной среде соли формулы C6H5N2Cl«PbCl2 цинковой пылью был
получен хлористый трифенилсвинец (идентифицирован по температуре
плавления). Из соли состава C6H5N2CbPbCl2 (с примесью РЬС12) при
действии порошка меди в среде ацетона было получено немного двухлористого
дифенилсвинца [71.
При разложении порошком металлического свинца фенилдиазонийбор-
фторида удалось получить выход тетрафенилсвинца, несколько превышающий
15% [8]. Если для разложения арилдиазонийборофторидов в ацетоне
применять вместо порошка свинца сплав свинец — натрий [9] (содержание
натрия 10%), то тетрафенилсвинец получается с выходом 30%. Этим путем был
получен также и тетра-п-толилсвинец [15%].
Получение тетрафенилсвинца [9]. В охлаждаемую ледяной водой взвесь Юг фенил-
диазонийборофторида в 60 мл сухого ацетона при энергичном перемешивании вносят в
течение полутора часов 7,5 г сплава свинец — натрий (содержание 10% Na). Происходит
выделение азота. Начальная температура +4° С, максимальная +13° С. По окончании
внесения сплава смесь перемешивают при охлаждении ледяной водой в течение 4 час. На
следующий день ацетон отгоняют, остаток промывают эфиром и экстрагируют хлороформом.
По испарении хлороформа получают тетрафенилсвинец. После кристаллизации из
хлороформа т. пл. 225° С. Выход 2,07 г (30% от теорет.).
Аналогично получают тетра-д-толилсвинец; выход 15,6%; т. пл. 240°С
[9].
ЛИТЕРАТУРА
l.GilmanH., L ее р е г R. W., J. Org. Chem., 16, 466 (1951).
2. Якубов и ч А. Я., Меркулова Е. Н., М а к а р о в С. П., Г а в р и л о в Г. И.,
ЖОХ, 22, 2060 (1952).
3. Якубович А. Я., Макарове. П., ГинсбургВ. А., Гавр и лов Г. И.,
М е р к у л о в а Е. Н., ДАН СССР, 72, 69 (1950).
4. В е с k e r W. Е., С о о k S. Е., J. Am. Chem. Soc, 82, 6264 (I960).
5. Н е с м е я н о в А. Н., Вег., 62, 1010 (1929); ЖРФХО, 61, 1393 (1929).
6. Мака р о в а Л. Г., Реакции и методы исследования органических соединений, т. III.
М., ГНТИХЛ, 1954, стр. 73.
7. К о ч е ш к о в К. А., НесмеяновА.Н., Г и п п Н. К., ЖОХ, 6, 172 (1936).
S. НесмеяновА.Н, К о ч е ш к о в К. А., НадьМ.М., Изв. АН СССР, ОХН,
1945, 522.
9. Н е с м е я н о в А. Н., М а к а р о в а Л. Г., Изв. АН СССР, ОХН, 1954, 380.
Глава VI
ПРИСОЕДИНЕНИЕ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ПО КРАТНЫМ СВЯЗЯМ
Присоединение свинцовоорганических гидридов, гидроокисей или аци-
латов к олефинам, алкинам или кетену приводит к соединениям с
различными заместителями в алифатическом радикале у атома свинца. Другими
методами эти вещества могут быть получены лишь в единичных случаях.
Свинцовоорганические гидриды активно реагируют с олефинами при
низких температурах и в отсутствие катализаторов. Триметилэтилсвинец
получен с выходом 92% взаимодействием гидрида триметилсвинца с
этиленом при 0°С и давлении 17 атм [1]. Повышение давления до 100 атм позволяет
понизить температуру до —50°С [2], тогда как при 1 атм реакция не
проходит вообще [3]. Как уже отмечено на стр. 278, оловоорганические гидриды
присоединяются к незамещенным олефинам лишь при длительном
нагревании смеси в присутствии азодиизобутиронитрила.
Нейман и Кюлейн [4] осуществили присоединение гидрида трибутилсвин-
ца к акрилонитрилу, метилакрилату, стиролу, фенилацетилену и фенилизо-
цианату (при 0°С):
(С4Н9)3РЬН + CH2=C4CN -* (C4II9hPbCH2CfI2CN,
(С4Н0)яРЬН + СН3=СНСОЭСЧз -> (С4Н9)3РЬСЧ2СНоСОЭСЧ3,
(С4Н9)8РЬН + СвИлСН=СЧ2 -» (С4Нэ)зРЬСН2СН3СвНз,
(Ci49)8PbH -r С3Н5С=СЧ - (С4Ч9)3РЬСЧ = СЧС0Н5,
(С4н9;3рьн + ce45N=c=o ->
[(C4H9)3PbN (С6Н->) СНО] -> втор 1чный поодукт рзакции.
Выходы продуктов составляют 90—100%. С акрилонитрилом процесс
заканчивается за 8 час. В тех же условиях гидрид трибутилолова инертен по
отношению к акрилонитрилу [4].
Реакция свинцовоорганических гидридов с алкинами исследована Лей-
синком и Ван-дер-Керком [51. Найдено, что гидрид триметилсвинца
присоединяется к циаиоэтину при —70°С с образованием а-цис-$- и транс-$-
триметилплюмбилакрилонитрила:
Н РЬ(СЧ3)з
\ /
(С1з)зРЬН + НС==С—CN -> С=С -{-
/ \
II C = N
Н Н (С-18)зРЬ Н
\ / \ /
+ С^С - С-С
/ \ / \
(СКз)зРЬ C = N Н C=N
Присоединение по кратным связям
571
С диэтилацетилендикарбоксилатом гидрид триметилсвинца образует в
основном продукт траяс-присоединения:
с2н,оос н
\ /
(СН3>РЬИ + С2Н5ООСС=ССООС2Н5 -* С=С +
(СНз)зРЪ СООС2Н5
(СНз)зРЬ Н
\ /
+ с^с
с2н5оос чсоос2н5
Гидроплюмбирование метилпропиолата приводит почти исключительно
к £{ш>|5-триметилплюмбилметилакрилату. Наряду с ним получают
небольшие количества транс-$- и ос-триметилплюмбилметилакрилата:
Н РЬ(СН3)з
\ /
(СН3)зРЫ I + НС=С—СООСН^ С=С +
/ \
Н СООСНз
Н Н (СНз)зРЬ Н
\ / \ /
+ с=с + с=с
(СНз)зРЬ СООСНз Н СООСНз
Рассмотрен механизм этих реакций.
Установлено, что ос-аддукты образуются по полярному, а р-аддукты по
радикальному механизму [5].
Присоединение свинцовоорганических гидридов к алкинам может быть
осуществлено также следующим образом. Ацетат трибутилсвинца
смешивают с гидридом трифенилолова в тетрагидрофуране при — 80°С и затем
прибавляют алкин. Вариантом того же метода является прибавление оловоор-
ганического гидрида к охлажденной до — 20°С смеси свинцовоорганическо-
го соединения и алкина [6, 7]:
н н
\ /
{QH9)3PbOOCCH3 + (С6Н5)3 SnH + НС=С—CN -> С=С +
/ \
(QH9)3Pb C=N
цис-§-
H РЬ (С4НЭ)3
\ /
+ С^=С + (C6H5)3SnOOCCH3,
/ \
Н a C=N
(С4Н9)3РЬООССНз + НС=ССООСН3 + (C2H,)aSnH -»
Н Н
\ /
-^ С=С + (C2H5)3SnOOCCH3
(QH9)3Pb СООСНз
Вместо ацетата можно использовать и свинцовоорганические имидазолы.
Образующиеся в качестве побочных продуктов оловоорганические ацетаты
или имидазолы удаляют из реакционной смеси, прибавляя к ней петролей-
ный эфир с последующим выпариванием растворителей. Выход
свинцовоорганических аддуктов составляет 60—90%.
572 Свинцовоорганические соединения
Реакция N-трибутилплюмбилимидазола с гидридом трифенилолова в присутствии циа-
ноэтина [7]. К охлажденному до—80° С раствору 2,3 г (0,005 моля)
N-трибутилплюмбилимидазола в 3,5 мл тетрагидрофурана при —80° С прибавляют 1,81 г (0,005 моля) гидрида
трифенилолова. Смесь перемешивают при той же температуре в течение часа и затем к ней
прибавляют 0,39 г (0,007 моля) цианоэтина. Смесь перемешивают еще час и оставляют
нагреваться до комнатной температуры. N-Трифенилстаннилимидазол осаждают 15 мл петролей-
ного эфира, фильтрат упаривают и перегоняют в вакууме. Получают смесь а- и ^/с-|3-трибу-
тилцианоэтинилсвинца с т. кип. ПО—111° С/0,2—0,3 мм\ п2£ 1,5174. Выход 75%.
Ацетат триэтилсвинца, этокситриметилсвинец, хлористый трифенилсви-
нец, диацетат и окись дифенилсвинца, а также гексафенилдиплюубан не
реагируют в заметной степени с кетеном при 10°С. В тех же условиях с
гидроокисью трифенилсвинца он образует (трифеиилплюмбил)уксусный
ангидрид:
2 (С6Н5)8РЬОН -f- 4 CHL=C=0 -» [(C6H?)8PLCH2CO]iO + (СНьСО)20.
Получение (трифенилплюмбил)уксусного ангидрида [8]. В охлажденную до 5° С
суспензию 45,5 г (0,1 моля) гидроокиси трифенилсвинца в 300 мл сухого эфира пропускают в
течение 2 час. ток кетена. Осадок отфильтровывают и промывают эфиром. Получают 41,0 г
(84%) вещества с т. разл. 100—110° С. Вещество обычно выделяется в чистом виде и не
нуждается в дополнительной очистке. Если это необходимо, его можно очистить растворением
1,0 г вещества в 5 мл хлороформа с последующим осаждением из раствора 25 мл петролей-
ного эфира (т. кип. 40—60° С).
При действии кетена (0,375 моля) на суспензию ацетата
трифенилсвинца (0,5 г) в этаноле (25 мл) при когнатной температуре с последу-
к щим управлением образукщегося раствора досуха получают 2,0 г
этилового эфира трифенилплюмбилуксусиой кислоты с т. пл. 61—62°С [8].
Согласно краткому сообщению [9], гексафенилдиплюмбоксан, меток-
ситрибутил- и метокситрифенилсвинец присоединяется к арилизоциана-
там, сероуглероду, хлоралю, трихлорацетонитрилу и динафтилкарбоди-
имиду. С гексахлорацетоном реакция проходит следующим образом:
[(С6Н5)зРЬОСН3 + (СС1з)2СО^(С6НГ))3РЬОС(СС1з)20СН8-> (СвНб)8РЬСС1а+СС1яСООС11з.
Описывается получение Р-оксиалкилтриалкилсвинца взаимодействием
галогенидов триалкилсвинца с окисями олефинов в жидком аммиаке с
последующим разложением образовавшегося алкоголята водой [10].
ЛИТЕРАТУРА
1. В е с k e r W. Е., С о о k S. Е., J. Am. Chem. Soc, 82, 5264 (I960).
2. В li tzerS. M., FarrarM. W., PearsonT.H., ZietzJ.R., Пат. США
3136795 (1964); РЖХим., 1966, 3H109.
3. D u I i у R., F e e n e у J., H о 1 1 i d а у А. К., J. Chem. Soc, 1962, 1144.
4. N e u m a n n W. P., KuhleinK., Angew. Chem., 77, 808 (1965).
5. Le'usinkA.J, van der KerkG. J.M., Rec. trav. chim., 84, 1617 (1965).
6. С r e e m e r s H. M. J. C, LeusinkA.J., NoltesJ.G., van der Kerk
G. J. M., Tetrahedron L., 1966, 3167.
7. CreemersH.M. J.C., Hydrostannolysis. A general method for establishing
tin-metal bonds. Utrecht (Nederland). Schotanus and Jens Utrecht. N. Y., 1967.
8. W i 1 1 e m s e n s L. C, v a n der KerkG. J.M., J. Organometal. Chem., 4, 241
(1965).
9. Da vies A. G., Pudderhatt R. J. Organometal. Chem., 5, 590(1966).
10. П о л е е с Б. М., Б р у к е р А. Б., С о б о р о в с к и й Л. 3., Авт. свид. СССР 148404
(1962); РЖХим., 1963, 7Н73; Бюлл. изобр., № 13, 16 (1962)
Глава VII
ВВЕДЕНИЕ СВИНЦА НА МЕСТО ВОДОРОДНОГО АТОМА
(плюмбирование)
Реакция плюмбирования позволяет синтезировать соединения типа
RPb(OOCR)3 в одну стадию:
R—II ;- Pb(OOCR')4-> RPb(OOCR')3 -r R'COOH.
Возможность такого типа реакций впервые показана Пановым и Кочешковым
Ш.Методпока не получил широкого распространения, что частично
объясняется неустойчивостью соединений типа RPb(OOCR)3. В ряде статей [2—7]
высказывается предположение о том, что они являются промежуточными
продуктами при окислении различных органических веществ тетраацетатом
свинца, но в индивидуальном состоянии их удалось выделить лишь в
отдельных случаях.
При нагревании тетраацетата свинца с анизолом [8] получен триацетат
?г-анизилсвинца.
Получение триацетата /г-анизилсвинца [8]. 22 г тетраацетата свинца и 7,5 г анизола
нагревают при 80° С в 200 мл уксусной кислоты в течение 4 дней. Растворитель удаляют в
вакууме, непрореагировавший тетраацетат свинца разлагают 100 мл воды и раствор
экстрагируют бензолом. При прибавлении к экстракту петролейного эфира (т. кип. 40—60° С)
выпадает осадок светло-коричневого цвета. Его перекристаллизовывают из бензола,
содержащего несколько капель уксусной кислоты. Выход триацетата /г-анизилсвинца 24%.
Триацетат 2,5-диметоксифенилсвинца получают [2] нагреванием при 80°С
50 г диметилового эфира резорцина и 177 г тетраацетата свинца в 850 мл
бензола в течение 65 час. Выход 39 г; т. пл. 146—149°С (с разложением).
Плюмбирование проходит по месту наибольшей электронной плотности
в молекуле органического соединения и представляет собой, по-видимому,
реакцию электрофильного замещения [7].
Более сложные процессы имеют место при металлировании тиофена тет-
раизобутиратом свинца [1]. Первоначально образуется нестойкий триизо-
бутират а-тиенилсвинца:
ПН- рь[ооссн(сНзЬ]4 - П_РЬ1ооосн(сн,ы. + ^ьснсоон,
S
который далее подвергается диспропорционированию:
2{^^!—РЫОООСН (СН3)2]з
S
РЬ [ООССН(СН3)2]2 + РЬ[ООССН (СН,)2]2.
574
Свинцовоорганическые соединения
Другой возможный механизм образования диизобутирата ди-а-тиенилсвин-
ца может быть связан с металлирующим действием триизобутирата а-
тиенилсвинца на тиофен.
Плюмбирование тиофена [1]. 6 г тетраизобутирата свинца (предварительно промытого
петролейным эфиром) растворяют в 10 мл тиофена и оставляют на 10 дней при комнатной
температуре. Для разложения непрореагировавшего тетраизобутирата свинца приливают 4 мл
сухого спирта и оставляют на 2 дня. Тиофен и спирт удаляют почти досуха в вакууме при
комнатной температуре. Остаток обрабатывают 5 мл бензола, отфильтровывают от
диизобутирата свинца и фильтрат разбавляют 7,5 мл гексана. Выпадает белый кристаллический
осадок, т. пл. 192° С (с разложением). Его анализ отвечает диизобутирату ди-а-тиенилсвинца.
Кроме того, состав продукта реакции подтвержден независимым синтезом (из тетра-а-тиенил-
свинца и изомасляной кислоты) и реакцией переацидирования.
Как показано Шостаковским с сотр. [9], реакция гидроокисей
триалкилсвинца с ацетиленовыми соединениями протекает так же, как и с
гидроокисями триалкилолова (см. стр. 327):
RsPbOH + HC=CR' -* R3PbC=ECR' + М20
(R = CH8> C2H5; R' = H, CH2=CII, C6H5, CH2C1).
Следует отметить, что активность гидроокисей триалкилосвинца по
отношению к ацетиленовым соединениям выше, чем у соответствующих оловоорга-
нических соединений. Очевидно, это обусловлено более высокой
полярностью связи металл — кислород при переходе от олова к свинцу,
достигающей в случае R3PbOH ионного состояния. Действительно, потенциометри-
ческие измерения показали, что рН 0,01 N раствора гидроокиси триэтил-
свинца в воде составляет 11,64, в то время как для 0,01 N водного раствора
едкого натра рН 12[9].
Приведенные факты позволяют сделать следующий выеод.
Взаимодействие гидроокиси триалкилсвинца с ацетиленом представляет собой реакцию
нейтрализации сильного основания слабой кислотой, что, по-видимому,
является следствием нуклеофильной атаки ацетилена ионом гидроксила:
R3Pb+ + ОН-+ H+[CeeCRT-* RsPdCeeCR' + Н20.
Необходимо отметить, что при проведении этой реакции с ацетиленом в-
жестких условиях образуется только симметричный био(тркалкилсвинец)-
ацетилен.
Получение триметилвинилэтинилсвинца [9]. В колбу емкостью 150 ло
помещают 4 г гидроокиси триметилсвинца и 95 мл сухого бензола. Образовавшуюся суспензию
охлаждают до 7° С и прибавляют 10 мл винилацетилена, охлажденного сухим льдом.
Наблюдается исчезновение осадка. Смесь перемешивают в течение часа и перегоняют в вакууме.
Получают 3 г триметилвинилэтинилсвинца с т. кип. 75—76е С/13 мм; d^° 1,8747; п2£ 1,5618;
MRD найдено 52,46; вычислено 52,86.j
Аналогично получены тризуетилфенилэтинилсЕинец (т. кип. 123"С/
1,5 мм) и триэти^-З-хлсрпрсгикихсгиьец (т. кип. 1ССсС/1 мм).
ЛИТЕРАТУРА
1. П а н о в Е. М., К о ч е ш к о в К. А., ДАН СССР, 123, 295 (1958).
2. PreussF.R.,JanshenI., Arch. Pharmazie, 293/65, 933 (1960).
3. Р г е u s s F. R., J a n s h e n I., Arch. Pharmazie, 295/67, 284 (1962).
4. OuletteR.J., S h a w D. L., J.Am. Chem. Sec, 86, 1651 (1964).
5. О u 1 e t t e R. J., S о u t h A., S h a w D. L., J. Am. Chem. Sec, 87, 1602 (1965).
6. MoonS., J. Org. Chem., 29, 3456 (1964).
7. D a v i e s D. I., J. Chem. Soc., 1963, 2351.
8. H a r v e у D. R., N о г m a n R. O. C, J. Chem. Soc, 1964, 4860.
9. ШостаковскийМ. Ф., К о м а р о в Н. В., Е р м о л о в а Т. И., ДАН СССР,
175, 1079 (1967).
Глава VIII
ДЕАЛКИЛИРОВАНИЕ
СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Сравнивая химические свойства германий-, олово- и
свинцовоорганических соединений класса R4Me, Ван-дер-Керк с сотр. [1] отметили, что в
названном ряду стерическое влияние органических радикалов и их
экранирующее влияние на атом металла от германия к свинцу уменьшается, в
результате чего связь металл — углерод становится более доступной действию
различных реагентов. Этому способствует также увеличение
поляризуемости связи металл ■—углерод при переходе от германия к свинцу. Кроме того,
увеличение межатомных расстояний и соответственно этому
ослабление внутримолекулярных связей приводит к постепенному уменьшению тер-
мо- и фотоустойчивости этих металлоорганических соединений.
Характерной чертой свинцовоорганических соединений является
неустойчивость соединений типа R3PbX и R2PbX2, например в алифатическом ряду.
Деалкилирование тетраалкилсвинца часто сопровождается реакцией дис-
пропорционирования, в результате чего образуется смесь
свинцовоорганических соединений различной степени алкилирования.
Для препаративных целей в качестве деалкилирующих агентов
применяют галоиды или галоидоводороды. Этим путем получены свинцовооргани-
ческие галогениды типа R3PbX и R2PbX2. Органические кислоты
отщепляют от свинцовоорганических соединений один алифатический или сразу два
ароматических радикала.
Что касается синтеза соединений типа RPbX3, то они образуются при
действии диацилатов ртути на диацилаты диарилсвинца. (Однако
целесообразнее получать их взаимодействием диарилртути с тетраацилатами свинца,
см. стр. 564.)
ДЕАЛКИЛИРОВАНИЕ ГАЛОИДАМИ
Деалкилирование полнозамещенных свинцовоорганических соединений
галоидами позволяет получать соединения типа R3PbX и R2PbX2
алифатического и ароматического рядов. При галоидировании тетраалкилсвинца
при температурах — 75 ~\ 65СС образуется галоидный триалкилсвинец;
для отщепления второй алкильной группы необходимо поднять
температуру до —20 ч 10°С. Так, хлористый триметилсвинец получен [2]
пропусканием медленного тока хлора в охлажденный до — 70°С 10—20%-ный
раствор тетраметилсвинца в этилацетате. Реакцию ведут до тех пор, пока не
перестанет исчезать окраска хлора. Температура при этом не должна
подниматься выше — 50СС. Раствор фильтруют и концентрируют в вакууме.
Аналогично получены хлористый триэтил- [2,3],трипропил-[21 и триизобутилсви-
нец [4].
576 Свннцовоорганические соединения. Лит. стр. 590—594
Получение двухлористого диметилсвинца [2]. Вначале получают, как описано выше,
раствор хлористого триметилсвинца и продолжают насыщение хлором без дальнейшего
охлаждения. Когда температура достигнет —10° С, ток хлора прекращают и жидкость после
кратковременного стояния декантируют с осадка.
Также получены [2,4] двухлористый диэтил-, дипропил-, диизобутил- и
диизоамилсвинец.
В аналогичных условиях проходит деалкилирование свинцовоорганиче-
ских соединений бромом. Подробно исследовано взаимодействие брома с
тетраметил- и тетраэтилсвинцом. Методом кулонометрии определены
константы скорости этих реакций в метиловом спирте при 25°С в присутствии
бромистого натрия [5].
Бромистый триметилсвинец получают [2] приливая к эфирному
раствору тетраметилсвинца эфирный раствор брома до тех пор, пока цвет брома не
будет удерживаться в течение короткого времени. После нагревания до
комнатной температуры эфир отгоняют и остаток перекристаллизовывают из
этилацетата, т. пл. 133° С. Аналогично получают бромистый триэтилсвинец,
т. пл. 103—104°С. Его не следует перекристаллизовывать из теплых
растворителей. Лучше всего, растворив его в эфире, добавить бензин и дать
медленно испариться эфиру.
Получение двубромистого диметилсвинца [2]. 50 г тетраметилсвинца бромируют в 500
мл этилацетата описанным выше для бромистого триметилсвинца способом. Затем при
постоянном перемешивании приливают без дальнейшего охлаждения еще столько же брома в
эгилацетате и дают смеси принять комнатную температуру. Весь двубромистый диметилсви-
нец выпадает в виде белого кристаллического порошка. Небольшой избыток брома не
вреди г. Осадок отсасывают. Перекристаллизация излишня (даже в растворителях при 50° С
происходит разложение с выделением этана). Хранить вещество следует в ампулах из темного
стекла без доступа воздуха.
Аналогично получают двубромистый диэтилсвинец. Его нельзя
перекристаллизовывать вследствие неустойчивости, еще большей, чем у низшего
гомолога.
При действии иода на тетраметил- [6] или тетраэтилсвинец [7] (эфирная
среда, температура — 60 ч 65°С) образуются иодиды триалкилсвивда.
Выход 60 и 73% соответственно.
От соединений с различными алифатическими радикалами у атома
свинца галоиды отщепляют в первую очередь более легкую группу. Например,
бромирование триметилэтилсвинца при — 70°С приводит к бромистому
диметилэтилсвинцу [4]. Если же реакцию вести при — 10°С, то образуется
двубромистый метилэтилсвинец. Из диметил-я-пропилизоамилсвинца в
соответствующих условиях получают бромистый метил-я-пропилизоамилсви-
нец или двубромистый я-пропилизоамилсвинец. Подобным же образом
получены [4,6] (С2Нъ){1-СъНп)РЪСи,{п-С^Щ(1-СъНп)РЪ02, (CH5)(i-C4H9)PbCl2,
(C2HB)(/i-C4Ho)PbBra и (/z-C3H7)(t-C4H9)PbBr2.
Хлорированием тетравинилсвинца в уксусной кислоте при обычной
температуре получен двухлооистый дивинилсвинец. Выход 72%, т. пл.> 300°
[8].
Галогениды циклогексилсвинца получают из легко доступного
гексациклогексилдиплюмбана, а не тетрапроизводного, как обычно. Бромирование
проводят при — 10°С в хлороформе, применяя рассчитанное количество
брома [9].
Получение йодистого три циклогексилсвинца и двуиодистого дициклогексилсвинца [9].
10 г гексацилогексилдиплюмбана нагревают в 300 мл бензола до кипения, охлаждают до
комнатной температуры и при перемешивании прибавляют бензольный раствор иода,
который тотчас обесцвечивается. По добавлении одной трети вычисленного количества иода
снова нагревают до кипения (для растворения остатков гексациклогексилдиплюмбана), опять
Деалкилирование
577
охлаждают и добавляют одну треть йодного раствора, после чего повторяют нагревание и
снова прибавляют по охлаждении оставшийся иод. По добавлении 2,8 г иода остается неис-
чезающий его цвет. В вакууме отгоняют бензол до остатка 30 мл и разбавляют 130 мл
горячего 94%-ного спирта. При охлаждении выкристаллизовывается иодид в виде золотисто-
желтых призм с т. пл. 91,7° С. Вещество прекрасно растворимо в бензоле и хлороформе,
легко в эфире и горячем спирте, трудно в холодном спирте. Если к бензольному раствору иоди-
да добавить бензольного же раствора иода, то взаимодействия сначала незаметно.
При нагревании внезапно происходит обесцвечивание иода. Чтобы избежать разложения
продукта, через несколько секунд после начала реакции смесь надо охладить льдом и
разбавить двойным объемом холодного спирта. Двуиодистый дициклогексилсвинец
кристаллизуется в виде тончайших игл светло-желтого цвета, чернеющих при 98° С. Вещество трудно
растворимо в бензоле и эфире, почти нерастворимо в спирте. Мало устойчиво и при
хранении разлагается.
От свинцовоорганических соединений ароматического ряда, даже при
температуре —75°С, бром отщепляет сразу два радикала. Бромистый три-
фенилсвинец образуется в небольших количествах [10]. Моногалоидные
соединения удается получить только бромированием тетраарилсвинца
раствором брома в пиридине при— 15°С [11] (ср. [12]).
Получение бромистого трифенилсвинца [11]. К охлажденной до —15° С суспензии 51,4 г
(0,1 моля) тонко измельченного тетрафенилсвинца в 1 л пиридина приливают при
размешивании раствор 0,2 моля брома в 200 мл пиридина (также охлажденный до —15° С). Смесь
перемешивают без дальнейшего охлаждения до тех пор, пока не исчезнет оранжевая
окраска и почти весь осадок не перейдет в раствор. Профильтрованный раствор упаривают при
16 мм почти досуха, остаток обрабатывают метиловым спиртом и 10% -ной
бромистоводородной кислотой и затем промывают водой и метиловым спиртом. Выход около 90%; т. пл.
166° С.
Аналогично получен [13] бромистый три-л*-толилсвинец (выход 78%;
т. пл. 146—147°С).
Грютнер [11] объясняет гладкое получение бромистого триарилсвинца в
пиридине тем, что бром образует с последним молекулярное соединение,
которое обладает более мягкими деарилирующими свойствами, чем сам бром.
Получение йодистого трифенилсвинца [13]. К взвеси 23 г (0,045 моля) тетрафенилсвин-
ца в 200 мл хлороформа при энергичном перемешивании прибавляют 11,43 г (0,045 моля)
иода в 150 мл хлороформа. Выход йодистого трифенилсвинца 88%; т. пл. 138—139° С.
Приливанием иода в сероуглероде к раствору тетра-/г-толилсвинца в
том же растворителе получен двуиодистый ди-я-толилсвинец [ 14,15].
При действии галоидов на соединения свинца, содержащие
одновременно алифатические и ароматические группы, первыми, как правило,
отщепляются ароматические радикалы. Например, метилтрифенилсвинец и этил-
трифенилсвинец дают соответственно бромистый метилдифенилсвинец и
бромистый этилдифенилсвинец, а метилциклогексилдифенилсвинец —
бромистый метилциклогексилфенилсвинец [16, 17]. Интересно отметить, что при
действии хлора на (3-хлор-2-оксипропил)трифенилсвинец отщепляется фе-
нильная группа, тогда как хлористый водород отщепляет замещенный ал-
кильный радикал [18]. Однако бромирование (трифенилплюмбил)уксусно-
го ангидрида [(C6H5)3PbCH2CO]20 приводит к бромистому трифенилсвинцу
[19].
Изучена [20] кинетика реакции триметилфенилсвинца с иодом в
метиловом спирте в присутствии избытка йодистого калия. Высказано
предположение, что она проходит с образованием промежуточного соединения, для
которого предложена структура сг-комплекса:
/Г— $
РЬ(СН3)3
37 Заказ № 207
578
Свинцовоорганические соединения. Лит. стр. 590—594
В противоположность тому, что имеет место в алифатическом ряду, ь
случае смешанных ароматических соединений первой отщепляется более
тяжелая группа. Например, трифенил-я-толилсвинец, трифенил-я-ксилил-
свинец, (я-оксиметилфенил)трифенилсвинец и трифенил-а-нафтилсвинец
образуют с бромом бромистый трифенилсвинец [16, 21].
По патентным данным [221 свинцовоорганические соединения,
содержащие атомы свинца в цикле, реагируют с галоидами следующим образом:
С(С6Н5)=С(С6Н5) С6Н5
(СН3)2РЬ
С(СбН5)=С(СбН5) Вг
+ Вг2 -* С=С(С6Нб)~С(С6Н5)=С (СбН5)РЬ (Вг) (СНз)2,
Для деалкилирования свинцовоорганических соединений можно
применять также треххлористый иод. При нагревании его с тетрафенилсвин-
цом в хлороформе получен хлористый трифенилсвинец, наряду с двухлори-
стым дифенилсвинцом [23].
ДЕАЛКИЛИРОЗАНИЕ ГАЛОИДОВОДОРОДАМИ
Газообразные галоидоводороды являются более мягкими деалкилиру-
ющими агентами по сравнению с галоидами. В зависимости от условий
эксперимента и молярного соотношения реагентов они могут отщепить один или
два радикала от атома свинца. В качестве деалкилирующего агента обычно
применяют хлористый водород [3,12, 24—26] и только в отдельных случаях —
бромистый водород [13].
Галогениды триалкилсвинца, полученные этим методом, часто содержат
примесь дигалогенидов диалкилсвинца [27, 28]. Последние могут
образоваться, например, в результате деалкилирования галогенидов триалкилсвинца
избытком хлористого водорода или реакции диспропорционирования (см.
стр. 597).
Для получения чистых веществ реакцию ведут на холоду, подбирая
растворитель таким образом, чтобы образующийся хлорид выпадал в осадок.
Так, при пропускании хлористого водорода через 5—10%-ный раствор тет-
раметилсвинца в гексане выделяются кристаллы хлористого триметилсвин-
ца. Выход 60% [7].
Получение хлористого триэтилсвинца [28]. 64,8 г тетраэтилсвинца постепенно
прибавляют к охлажденному льдом насыщенному раствору хлористого водорода в 250 мл сухого
эфира. Смесь выдерживают при той же температуре в течение 20 мин., а затем полчаса при
обычной температуре. Осадок отфильтровывают, промывают сухим эфиром и высушивают
Хлористый трипропилсвинец хорошо растворяется в обычных
органических растворителях. Его получают, пропуская в течение 2—3 час. ток
хлористого водорода через охлажденный льдом 10%-ный раствор тетрапропил-
свинца в петролейном эфире, в котором хлористый водород практически не
растворим. По окончании реакции растворитель быстро отгоняют и остаток
кристаллизуют из смеси спирта с водой. Т. пл. 133—134°С [271. Аналогично
получены [8, 27, 281 хлористый три-я-бутил- (т. пл. 106,5—108,5°С) триизо-
бутил- (т. разл. 122°С) и тривинилсвинец (т. пл. 119—12ГС). Позже эти
методики синтеза хлоридов триэтил-, трипропил- и трибутилсвинца
использованы и другими авторами [29]. Хлориды триэтил- и трипропилсвинца
очищались возгонкой в вакууме.
Синтез дигалогенидов диалкилсвинца можно иллюстрировать
следующими примерами.
Деалкил ирован ие
579
Получение двухлористого диэтилсвинца [30]. В нагретый до 90° С раствор 12 мл тетра-
этилсвинца в 100 мл сухого толуола пропускают ток сухого хлористого водорода. Примерно
через 20 мин. начинается выделение осадка. Хлористый водород пропускают еще в течение
часа, раствор фильтруют горячим, осадок промывают горячим толуолом и высушивают в
вакууме. Получают 16 г (80%) двухлористого диэтилсвинца с т. пл. 220° С.
Двухлористый ди-я-пропилсвинец получен [27] с выходом 82%
пропусканием тока сухого хлористого водорода через охлажденный эфирный
раствор тетра-я-пропилсвинца. Вещество перекристаллизовывают из
небольшого количества этилового спирта.
Порядок отщепления различных радикалов от смешанных свинцовоор-
ганических соединений исследован рядом автором [7, 30—37]. Найдено, что
легче всего происходит отрыв аллильной, Р-стирильной и некоторых
замещенных алкильных групп. Более устойчивы по отношению к хлористому
водороду арильные, алкильные и 3-бутенильный радикалы. Из алкильных
радикалов, по-видимому, в первую очередь отщепляются наиболее легкие
группы [7, 30, 31]. Следует подчеркнуть, что обычно реакция деалкилирова-
ния свинцовоорганических соединений с различными алифатическими
радикалами у атома свинца сопровождается диспропорционированием
образующихся хлоридов, так что в конечном счете получают смесь продуктов [7, 30,
31].
Более устойчивы свинцовоорганические хлориды, содержащие
одновременно алифатические и ароматические радикалы у атома свинца. Например,
из я-пропилфенилди-о-толилсвинца и я-пропилтрифенилсвинца получены
[33] хлористый я-пропилдифенил- и я-пропилфенил-о-толилсвинец
соответственно. Дальнейшая обработка хлористого я-пропилдифенилсвинца
хлористым водородом приводит кдвухлористомуя-пропилфенилсвинцу [33].
Установлено, что реакция триметилфенилсвинца с хлористым водородом в
метиловом спирте при 289—307СК является реакцией второго порядка [20].
Гладко идет образование свинцовоорганических галогенидов
ароматического ряда [21, 38—41].
Получение хлористого трифенилсвинца [40]. В нагретый до50—v60bC раствор
тетрафенилсвинца в хлороформе (15 мл растворителя на 1 г тетрафенилсвинца) при перемешивании
пропускают ток хлористого водорода до появления белого осадка двухлористого дифенилсвин-
ца. Смесь нагревают до кицения и отфильтровывают. Из фильтрата растворитель отгоняют
на водяной бане и остаток экстрагируют сухим горячим этиловым спиртом. Спиртовый
экстракт охлаждают до 0° С и осадок отфильтровывают. Выход хлористого трифенилсвинца 75—
80%, т. пл. 207° С (из этилового спирта).
Краузе и Шлоттиг [17] получали хлористый трифенилсвинец нагреванием
при 170—180°С в течение 12 час. сухой смеси 5 г тетрафенилсвинца и 20 г
хлористого аммония. Избыток хлористого амуония отмывают водой и
хлористый трифенилсвинец извлекают горячим спиртом. Выход около 75%.
Метод, видимо, удобен лишь в приложении к небольшим количествам.
Хлористый трифенилсвинец образуется также при нагревании до 130°С
тетрафенилсвинца с солянокислым триметиламином [42].
Хлориды три-/г-толил-(т. пл. 140—14ГС) [43], три-о-толил- (т. пл. 141—
142°С) [43] и три-я-анизилсвинца (т. пл. 152— 153°С с разложением) [44]
получены взаимодействием хлористого водорода с соответствующими тетра-
замещенными соединениями свинца в хлороформе.
Получение двухлористого дифенилсвинца [8]. 89,9 г тетрафенилсвинца растворяют в
хлороформе и в раствор при перемешивании пропускают ток сухого хлористого водорода.
Спустя 5—15 мин. выделяется обильный осадок. Хлористый водород пропускают еще в
течение 5—10 мин., осадок отфильтровывают, промывают хлороформом и высушивают на
воздухе. Выход 73 г (97%).
37*
580 Свинцовооргаяшеские соединения. Лит. стр. 590—594
При пропускании хлористого водорода через нагретый почти до кипения
бензольный раствор тетра-д-анизилсвинца получен [44] двухлористый ди-д-
анизилсвинец с выходом 98,6%.
От смешанных ароматических соединений свинца первой отщепляется
более тяжелая группа [33, 45—47]. Например, обработкой дифенилди-9-
фенантрилсвинца хлористым водородом в нагретом до кипения хлороформе
получают двухлористый дифенилсвинец [47].
а-Нафтил- отрывается ранее фенильного радикала, нос большим трудом,
чем а-тиенил- или а-фурил- [45].
При действии хлористого водорода на ди-(2-тиенил)ди(2-селениенил)сви-
нец образуется как тиофен, так и селенофен [48].
ДЕАЛКИЛИРОВАНИЕ КИСЛОТАМИ,
ФЕНОЛАМИ И МЕРКАПТАНАМИ
Минеральные и органические кислоты обычно отщепляют от свинцово-
органических соединений один алифатический или сразу два ароматических
радикала. В алифатическом ряду реакция иногда осложняется побочными
процессами, например диспропорционированием образующихся вначале
веществ.
Умеренное нагревание тетраалкилсвинца с галоидоводородными
кислотами приводит к галогенидам триалкилсвинца. Так, с концентрированной
соляной кислотой тетраэти л свинец при 35°С дает хлористый триэтилсвинец
с выходом 85% [49—51]. При продолжительном кипячении смеси
происходит полное разложение свинцовоорганического соединения с выделением
хлористого свинца. Так же реагируют с галоидоводородными кислотами тет-
раметил- и тетра-я-пропилсвинец [52,53].
К дигалогенидам диалкилсвинца приводит и обработка динитрата тет-
раалкилдинитрозилсвинца галоидоводородными кислотами [54, 55]:
[R4Pb (NO)2]2+ (NO,)*- + НХ -» R2PbX2.
Реакцию проводят путем прибавления кислоты к раствору свинцовоорга-
ндаеского соединения в воде или ацетоне. При этом сразу же выпадает
кристаллический осадок дигалогенида диалкилсвинца.
Получение динитрата тетраэтилдинитрозилсвинца [54]. 32,2 г (ОЛ моля) тетраэтилсвин-
ца растворяют в 100 мл сухого эфира и полученный раствор охлаждают твердой
углекислотой. При энергичном перемешивании по каплям прибавляют 22,2 г (14,2 моля) N204 в 60 мл
сухого эфира. При 0° С выпадает белый кристаллический осадок. Перемешивание
продолжают в течение часа при 0° С и затем без охлаждения до тех пор, пока температура
реакционной смеси не достигает комнатной. Получают 38 г продукта, который очищают растворением
в холодном ацетоне и отгонкой ацетона в вакууме при комнатной температуре. После
нескольких кристаллизации т. пл. 104—105° С. Вещество растворимо в воде, спирте, ацетоне, мало
растворимо в эфире и неполярных растворителях. Авторы доказывают наличие связи С—РЬ
реакцией полученного соединения с диацетатом ртути в нейтральной среде, при этом
происходит образование хлорида алкилртути. Также получают и динитрат тетрапропилдинитро-
зилсвинца с т. пл. ПО—111° С.
Получение двубромистого диэтилсвинца [54]. К раствору 2 г динитрата
тетраэтилдинитрозилсвинца в 2—4 мл воды прибавляют 2 мл 46—48%-ной бромистоводородной кислоты.
Получают 1,7 г двубромистого диэтилсвинца, который очищают перекристаллизацией из
ацетона.
Аналогично получены [54, 55] двухлористый диэтил- и ди-я-пропилсви-
нец, двубромистый ди-я-пропилсвинец, сульфаты диэтил- и ди-я-пропил-
свинца.
С влажным сернистым ангидридом тетраэтилсвинец образует сульфит
диэтилсвинца — (С2Н5)2РЬ50з[27, 30, 56, 57], а в случае азотной кислоты —
Деалкил ирован ие
581
динитрат диэтилсвинца [58, 59]. Мононитрат триэтилсвинца удается
выделить только частично.
Джонс, Эванс с сотр. [60] получали динитраты диалкилсвинца с
радикалами тяжелее этила действием концентрированной азотной кислоты на
тетраалкилсвинец. Найдено, что каталитическое влияние на эту реакцию
оказывают прибавляемые к смеси силикагель и небольшие количества эфира.
Аналогично при нагревании на водяной бане в течение 20—30 мин. 24 г
тетраэтилсвинца с 12 г /г-толуолсульфокислоты и несколькими
кусочками силикагеля получают 23 г n-толуолсульфоната триэтилсвинца с т. пл.
167—168°С [61]. Так же получены и другие арилсульфонаты триэтил-,
три-я-пропил- и триизобутилсвинца [28].
Реакция органических кислот с тетраалкилсвинцом в присутствии
силикагеля широко применяется для синтеза ацилатов триалкил свинца. Метод,
подробно разработан на большом числе примеров с разнообразными карбо-
новыми кислотами алифатического, ароматического и гетероциклического
рядов [7, 28,30, 49,56,62]. Так получены, например, следующие ацилаты
триэтилсвинца: ацетат (т. пл. 160°С), пропионат (т. пл. 141—142°С), капро-
нат (т. пл. 94,7—95,8°С), каприлат (т. пл. 85—87°С), изобутират
(т. пл. 120—122°С), фторацетат (т. пл. 180,5°С), хлорацетат (т. пл.
146°С), дихлорацетат (т. пл. 114°С), трихлорацетат (т. пл. 135—
138°С), бромацетат (т. пл. 12ГС) и дибромацетат (т. пл. 99—102°С). С
сильными кислотами, например хлор- или фторуксусной, реакция идет настолько
энергично уже на холоду, что приходится применять охлаждение или
разбавлять смесь эфиром. Слабые кислоты, например стеариновая, реагируют
медленно даже при температуре кипения смеси.
Скорость реакции уменьшается и с увеличением алкильного радикала у
атома свинца (см. табл. 11) [27, 63].
Таблица 11
Скорость реакций тетраалкильных соединений
свинца с уксусной кислотой
Соединение
(СН3)4РЬ
(С2Н,)4РЬ
(С3Н7)4РЬ
(С4Н9)4РЬ
(/-C5Hn)4Pb
/Ci-10\ сек-1
24,9 °С
1,16 ±0,03
0,92 ±0,06
0,67 ±0,08
0,225±0,013
0,311+0,005
0,300±0,015
49,8 °С
16,4±0,2
11,3±0,6
10,5±0,7
3,1±0,2
4,3±0,1
4,2±0,3
60,0 °С
41,2±1,3
28,7±1,8
27,74-1,1
8,2±0,4
10,6±0,4
12,2±0,5
Ацетат триэтилсвинца получают нагреванием до 90°С тетраэтилсвинца,
уксусной кислоты и силикагеля в течение часа [49]. В случае тетрапропил-
и тетраизопропилсвинца требуется нагревание смеси до той же температуры
в течение 2 час. 128]. Следует отметить, что в более жестких условиях
проходит отрыв всех алкильных групп от атома свинца. Так, тетраэтилсвинец
при нагревании с уксусной кислотой выше 90°С дает вместо ацетата
триэтилсвинца диацетат свинца [49, 64].
В некоторых случаях при обработке тетраалкильных соединений свинца
органическими кислотами образуются диацилаты диалкилсвинца или смесь
ацилатов три- и диалкилсвинца [30, 57, 62]. Например, нагреванием на
водяной бане в течение 30 мин. тетраэтилсвинца с бензойной кислотой получен
[30] дибензоат диэтилсвинца, т. пл. 168°С (из бензола).
582 Свинцовоорганические соединения. Лит. стр. 590—594
Тиоуксусная кислота реагирует с тетраэтилсвинцом на холоду с
образованием тиоацетата триэтилсвинца (С2Н5)зРЬ5СОСН3, т. пл. 45°С. При
нагревании смеси до 100°С без силикагеля получают соответствующие ацилаты
три- и диэтилсвинца, а также бш>(тиоацетат)свинца. Последний является
единственным продуктом, если реакцию проводить в присутствии
силикагеля [301.
Та же картина наблюдается и при обработке тетраэтилсвиица л*-нитро-
бензойной кислотой [62]. При проведении реакции в этиловом спирте
образуется ж-нитробензоат триэтилсвинца, а в бензоле — ди-ж-нитробензоат
диэтилсвинца.
Винильная и циклопропильная группы отщепляются от атома свинца
легче, чем алкильные. Смесь эквимолекулярных количеств уксусной
кислоты и тетравинилсвинца в присутствии силикагеля через 20 мин. дает 81%
ацетата тривинилсвинца (т. пл. 168—170°С). Тетраэтилсвинец в этих
условиях остается практически без изменения даже в течение 24 час. [8]. Реакция
между тетравинилсвинцом и трихлоруксусной кислотой проходит
экзотермически. При использовании эквивалентных количеств реагентов получают
трихлорацетат тривинилсвинца. Выход 74%, т. пл. > 300°С.
Обработка тетрациклопропилсвинца рассчитанным количеством
концентрированной соляной кислоты приводит к хлористому трициклопропилсвин-
цу (т. пл. 173—174°С) [651. Так же легко тетрациклопропилсвинец реагирует
с органическими кислотами. Интересно отметить, что с уксусной, монохлор-
уксусной или дихлоруксусной кислотами образуются диацилаты
дициклопропилсвинца, тогда как в случае трихлоруксусной кислоты получают
смесь ацилатов три- и дициклопропилсвинца.
Получение б'ис-(дихлорацетата) дициклопропилсвинца [65]. 0,2 г (0,54 моля)
тетрациклопропилсвинца и 0,07 г (0,54 моля) дихлоруксусной кислоты смешивают в 5 мл эфира.
Примерно через 3 часа начинается выделение осадка. Через 20 час. его отфильтровывают,
промывают несколько раз эфиром (порциями по 5 мл) и высушивают в вакууме при комнатной
температуре. Получают 0,13 г вещества с т. пл. 179—180° С (с разложением).
При нагревании ароматических четырехзамещенных соединений свинца
с минеральными или органическими кислотами отщепляются сразу два
ароматических радикала и образуются соединения типа Аг2РЬХ2. Например,
при внесении тетрафенилсвинца в нагретую до кипения азотную кислоту
получен динитрат дифенилсвинца с выходом 65% [12, 66, 67].
Исследовано[68]взаимодействие тетрафенилсвинца и хлористоготрифенил-
свинца с производными арсиновых кислот. Выделено и охарактеризовано ряд
соединений состава (C6H5)2Pb[OAs(0)(OH)R2] и (C6H5)2Pb(Cl)OAs(0)(OH)R.
Согласно данным Несмеянова и Фрейдлиной [69, 70], фениларсин деари-
лирует дигалогениды диарилсвинца до дигалогенидов свинца:
2 (С6Н,)2РЬХ2 + 2 CeH5AsH2 -» 2 РЬХ2 + 4 С6Н6 + C6H5AsAsCeH3.
Тетраэтил- и тетрафенилсвинец восстанавливаются этим реагентом до метал
лического свинца.
Деарилирование свинцовоорганических соединений органическими
кислотами впервые описано еще в 1887 г. Полисом [12] и впоследствии подробно
исследовано Котоном [71, 72]. Низшие одноосновные карбоновые кислоты
(муравьиная, уксусная, а также пропионовая, масляная, а-оксипропионовая
а-оксимасляная) легко отщепляют две ароматические группы от атома
свинца. При действии избытка кислоты при 100°С реакция заканчивается в
течение 1—2 час. [71—75].
Деалкилирование
583
Получение диизобутирата дифенилсвинца [74]. В трехгорлую колбу с термометром
и механической мешалкой помещают 20,6 г (1 моль) тетрафенилсвинца и 35 г (10 молей) изо-
масляной кислоты. Смесь нагревают при 115—120° С. Через 25 мин. тетрафенил свинец
растворяется, после чего перемешивают еще полтора часа при той же температуре. Горячую
реакционную массу разбавляют 60 мл бензина (фракция 75—100° С). Выпадает обильный
кристаллический осадок чистого диизобутирата дифенилсвинца. Выход 15,3—17,6 г (72—
84%); т. пл. 204—205° С (из бензола). Диизобутират дифенилсвинца хорошо растворяется
в дихлорэтане и уксусной кислоте на холоду; умеренно — на холоду и хорошо при
нагревании в бензоле, ксилоле и хлороформе; в эфире и петролейном эфире очень плохо
растворяется даже при нагревании.
Нагреванием в течение 5 мин. 1,58 г тетрафенилсвинца с 20 мл тиоуксус-
ной кислоты получают 1,55 г (60%) бм>(тиоацетата) дифенилсвинца с т. пл.
94—95°С (из этилового спирта) [76]. Тиосалициловой кислотой тетрафенил-
свинец полностью деарилируется при 130°С в течение 3 час. [77].
С увеличением молекулярного веса кислоты выход диацилатов
дифенилсвинца уменьшается. С кислотами от муравьиной до масляной реакция идет
как в растворе, так и без растворителя; с изовалериановой и капроновой
кислотами — только в отсутствие растворителя, а стеариновая кислота
вообще не реагирует с тетрафенилсвинцом [71,73]. Диацилаты диарилсвин-
ца и высших карбоновых кислот обычно получают реакцией переацидирова-
ния (см. стр. 609).
Диацилаты диарилсвинца образуются также и при действии
органических кислот на хлористый трифенилсвинец [68] или гексаарилдиплюмбаны
[78]. Правда, в последнем случае выход невелик.
При получении соединений типа Аг2РЬХ2 в n-толильном ряду наиболее
целесообразно исходить из легко доступного гекса-/г-толилдиплюмбана
[78]:
3 (СН3С6Н4)зРЬРЬ (СбН4СН3)3+8 HN03 -> 6 (СН3С6Н4)зРЬШз + 2 NO + 4 Н20,
(СН3С6Н4)зРЬШ3 + HN03 -> (CH3C6H4)2Pb (N03)2 + C6H5CH3.
Динитрат ди-/г-толилсвинца, в отличие от соответствующего дигалоге-
нида, растворим в горячем спирте и легко превращается спиртовым
раствором едкого кали в окись ди-/г-толилсвинца, а последняя — в диацилаты ди-
/г-толилсвинца.
Получение диацетата ди-/г-толилсвинца [78]. В 75 мл концентрированной азотной
кислоты, нагретой до кипения, маленькими порциями при постоянном встряхивании вносят
7,5 г гекса-/г-толилдиплюмбана, на что требуется примерно 20—25 мин. Реакция протекает
очень энергично, каждая новая порция гекса-/г-толилдиплюмбана вызывает вспенивание
смеси и выделение бурых паров окислов азота. Азотная кислота желтеет, на дне колбы
появляется осадок динитрата ди-/г-толилсвинца. После введения всего количества гекса-я-толил-
диплюмбана нагревание продолжают еще несколько минут, затем реакционную смесь
охлаждают, динитрат ди-/г-толилсвинца отсасывают и промывают водой.
Полученный таким образом сырой динитрат ди-/г-толилсвинца растворяют при
нагревании в 80 мл спирта и к кипящему раствору прибавляют 2,2 г едкого кали в 25 мл спирта.
Выпавший желтоватый осадок отсасывают и промывают несколько раз спиртом и эфиром.
Получают 5,25 г окиси ди-/г-толилсвинца.
3,2 г неочищенной окиси ди-п-толилсвинца смешивают с 3 мл ледяной уксусной кислоты.
Для более полного растворения осадка смесь осторожно нагревают, причем часть осадка все-
же остается нерастворенной. К смеси прибавляют 15 мл этилацетата, нагревают до кипения
и отфильтровывают. Фильтрат охлаждают в смеси снега с солью. Получают 3,08 г (64% в
расчете на исходный гекса-п-толилдиплюмбан) диацетата ди-п-толилсвинца с т. пл. 209—210°
С. Вещество очень хорошо растворяется на холоду в хлороформе и уксусной кислоте,
хорошо — в метиловом и этиловом спиртах. В бензоле, ацетоне и этилацетате на холоду
растворяется умеренно, при нагревании — хорошо. В гексане и эфире растворяется очень
плохо даже при нагревании.
Аналогично получен [78] диизобутират ди-/г-толилсвинца, т. пл. 202—
203°С.
При действии минеральных кислот на свинцовоорганические
соединения с ароматическими и алифатическими остатками у атома свинца прежде
5S4
Свинцовоорганические соединения. Лит. стр. 590—594
всего отщепляется арильная группа [17, 32, 79]. Так, обработкой у-диэтил-
'аминопропилтрифенилсвинца соляной кислотой получен солянокислый
хлористый у-диэтиламинопропилдифенилсвинец, в котором атом свинца связан
с тремя различными группами [80]:
(C6H5)3PbCH2CH2CH2N(C2H5)2 + 2 НС1 -» (СбН5)2РЬ (CI) CH2CH2CH2N (C2H5)2HC1 + СвНб.
Методом спектрофотометрии измерена [81] скорость расщепления
некоторых соединений типа (С6НП)3РЬС6Н4Х (X = Н, /?-СН3, /?-СН30, /?-С1 и
m-Cl) хлорной кислотой в смеси спирта с водой. Установлено, что степень
влияния заместителей на скорость расщепления свинцовоорганических
соединений меньше, чем при расщеплении аналогичных соединений олова, и
много меньше, чем при расщеплении триалкиларилгермания и триалкил-
арилкремния. Найдено, что влияние заместителей в арильной группе
аналогично влиянию заместителей при обычном электрофильном замещении в
ароматическом ядре.
Как показано на примере трифенил-(/г-диметиламинофенил)свинца,
уксусная кислота отщепляет от атома свинца в первую очередь замещенный
фенильный радикал [25].
Реакции кислот с гетероциклическими свинцовоорганическими
соединениями остаются пока мало изученными. Нагреванием тетра-а-тиенилсвинца
с изомасляной кислотой получен диизобутират ди-а-тиенилсвинца. При
прибавлении к концентрированной серной кислоте вещество разлагается с
выделением тиофена [82].
Реакции свинцовоорганических соединений с фенолами описаны очень
кратко. /г-Нитрофенол отщепляет от тетраэтилсвинца два этильных
радикала (нагревание в ксилоле в течение часа) [27]. Однако выделить
образующееся вещество в достаточно чистом виде не удается. С пирогаллолом и а-наф-
толом тетрафенилсвинец при 130°С практически не реагирует [83].
Тетраэтилсвинец при нагревании до 100°С деалкилируется фенилмеркап-
таном [84]. Состав образующихся при этом продуктов не выяснен.
При нагревании тетрафенилсвинца с фенилмеркаптаном в хлороформе
образуется небольшое количество (около 2%) фенилмеркаптида трифенил-
свинца, тогда как большая часть вещества остается без изменения [76].
В более жестких условиях (130—150°С) происходит полное разложение
молекулы свинцовоорганического соединения [85].
ДЕАЛКИЛИРОВАНИЕ СОЛЯМИ
Действием солей металлов на свинцовоорганические соединения
получены соединения класса R3PbX и R2PbX2 алифатического и
ароматического рядов и RPbX3 в ароматическом ряду. Впрочем, целесообразнее эти
вещества получать другими методами (см. стр. 564, 581, 605). Однако деалки-
лирующие свойства солей металлов изучены довольно подробно.
Для препаративных синтезов свинцовоорганических соединений иногда
применяют реакции тетраалкил- или тетраарилсвинца с солями ртути или
свинца. Так, при прибавлении сулемы к раствору тетраэтилсвинца в спирте
происходит экзотермическая реакция и образуется хлористый триэтилсви-
нец [86—88]. Нагревание окиси ртути и тетраэтилсвинца в спирте с
рассчитанным количеством фосфорной кислоты приводит к фосфатам три- или
диэтилсвинца [89].
По патентным данным [90], от тетраэтилсвинца можно отщепить сулемой
и три «этильных радикала, но образующийся треххлористый этилсвинец
неустойчив и сразу же разлагается с выделением двухлористого свинца.
Следует отметить, что побочным продуктом этих реакций является соль
Деалкилирование
585
этилртути. С Hg2(N03)2 реакция проходит иначе. В зависимости от
молярного отношения реагентов здесь образуются нитраты три- или диалкилсвин-
ца, диалкилртуть и металлическая ртуть [91].
От тетрафенилсвинца сулема отщепляет один или два радикала [92, 93].
Реакция проходит медленно из-за плохой растворимости исходного
тетрафенилсвинца. Деарилирование тетрафенилсвинца хлорной (или бромной)
ртутью можно провести ступенчато [92—94]. Так, двухлористый дифенил-
свинец получен действием сулемы на хлористый трифенилсвинец. Отщепить
третий ароматический радикал не удается. Двухлористый дифенилсвинец
при трехчасовом кипячении с избытком спиртового раствора сулемы остается
неизменным и только после 12-часового кипячения дает некоторое
количество хлористой фенилртути и двухлористого свинца [92, 93].
Как показано Кочешковым и Пановым [74, 75, 95].t: при добавлении
раствора соли ртути в органической кислоте к свинцовоорганическому
соединению, также растворенному в органической кислоте, гладко происходит
отщепление одного, двух, трех или четырех радикалов:
(C6H5)4Pb + Hg (OOCR)2 -> (C6H5)3PbOOCR + C6H5HgOOCR,
(CeH3)3PbOOCR + Hg (OOCRb -> (C6H5)2Pb (OOCR)2 + C6H5HgOOCR,
(C8H5)2Pb (OOCR)2 + Hg (OOCR)2 A C6H5Pb (OOCR)3 + CeH5HgOOCR,
C0H5Pb (OOCR)3 + Hg (OOCR)2 -» Pb (OOCR)4 + CeH5HgOOCR.
Интересно отметить, что в алифатическом ряду реакция заканчивается
на стадии образования диацилатов диалкилсвинца при любом избытке
уксуснокислой ртути [75].
Получение триацетата фенилсвинца [95]. 1,92 г (1 моль) уксуснокислой ртути
растворяют при слабом нагревании в 40 мл ледяной уксусной кислоты и сюда после охлаждения
до комнатной температуры прибавляют 2,88 г (1 моль) диацетата дифенилсвинца. Через
сутки, когда реакция практически закончится, к реакционной массе прибавляют по каплям
1,28 мл 4,7 N раствора хлористого водорода в спирте (что отвечает 1 молю) для превращения
образовавшегося ацетата фенилртути в хлористую фенилртуть. Осадок отфильтровывают
и фильтрат выпаривают в вакуум-эксикаторе над едким кали. Твердый остаток в количестве
3,63^г растворяют в 15.ил горячего сухого свежеперегнанного этилацетата и отфильтровывают.
Фильтрат охлаждают смесью соли и льда. Выделившиеся кристаллы отсасывают и
промывают гексаном. Получают 2,19 г (79%) триацетата фенилсвинца с т. пл. 101—102° С. Триацетат
фенилсвинца хорошо растворяется на холоду в метиловом и этиловом спиртах, хлороформе
и уксусной кислоте; умеренно растворяется на холоду и хорошо при нагревании в этилаце-
тате и бензоле; в эфире растворяется плохо, а в гексане не растворяется даже при
нагревании.
Аналогично получены [75,78,95] триизобутират фенилсвинца (т. пл.
77—78°С) и триацетат /г-толилсвинца (86—88°С).
Реакция, обратная диспропорционированию, очень хорошо
разработанная для синтеза оловоорганических соединений (см. стр. 329), в ряду свин-
цовоорганических соединений длительное время оставалась мало
исследованной. Впоследствие Ван-дер:Керк и Уиллемсенс получили триацетат
фенилсвинца и диацетат дифенилсвинца нагреванием до 70°С в уксусной
кислоте тетрафенилсвинца с тетраацетатом свинца в присутствии
каталитических количеств диацетата ртути [96]. Проведение реакции в
алифатическом ряду дало противоречивые результаты [96, 97]. Двухлористый
дифенилсвинец образует с тетрафенилсвинцом хлористый
трифенилсвинец согласно следующему уравнению [98]:
(СбН5)4РЬ-Ь(С6Н5)2РЬС12->2(С6Н5)3РЬС1.
В случае этильного производного выход продукта значительно снижается,
а с тетратолилсвинцом реакция не проходит вообще [98]. При действии на
диизобутират дифенилсвинца тетраизобутирата свинца последний
разлагается с выделением диизобутирата свинца [95].
586
Свинцовоорганические соединения. Лит. стр. 590—594
Образование соединений низшей степени валентности или даже
свободного металла имеет место и при деалкилировании свинцовоорганических
соединений азотнокислым серебром [99—111], треххлористым золотом
[112—115], азотнокислой [99—103] и хлорной медью [116], четыреххло-
ристым титаном [49, 62, 117], хлорным железом [116, 118] и платинохло-
ристоводородной кислотой [118—120]. От свинцовоорганических
соединений при этом отщепляется один (Ag*, Cu#, Au34", Ti4+) или два органических
радикала (Cu2+, Fe3+). В более жестких условиях соли золота, меди и
железа деалкилируют свинцовоорганические соединения до двухлористого
свинца. Интересно отметить, что от тетрафенилсвинца при 12-часовом
кипячении в этилцеллозольве с хлористым марганцем отщепляется одна фе-
нильная группа, тогда как тетрафенилолово в аналогичных условиях
практически не изменяется [116].
Примером реакций рассматриваемого типа может служить действие
азотнокислого серебра в среде сухого спирта на тетраметил-, тетраэтил- или
тетрафенилсвинец [99—103]:
R4Pb + AgN03 -* R3PbN03 + R" + Ag"
Реакция проходит при обычной температуре или при кипячении смеси с
обратным холодильником. Наиболее легко в нее вступают соединения типа
R3PbR' [104—108, 111], а также ацетилениды бш> (триал кил)- или трифе-
нилсвинца [108]. Промежуточными продуктами этих реакций являются
органические соединения серебра [105, 106, 109].
Утверждается [121], что взаимодействием четырехх лор истого (или че-
тырехбромистого) титана с тетраалкильными соединениями свинца при
—80° С могут быть получены титанорганические соединения типа RTiX3-
Свинцовоорганические соединения применяют в качестве алкилирующих
(или арилирующих) агентов и при получении других элементоорганических
соединений. Так, взаимодействием тетраметил- или тетраэтилсвинца с
треххлористым бором получен триалкилбор [122]:
3 R4Pb -f 2 ВС13 ~* 3 R2PbCl2 + R3B.
Избытком треххлористого бора тетраалкилсвинец деалкилируется до
двухлористого свинца [122].
Тетраметилсвинец легко деалкилируется боргидридом алюминия до
металлического свинца. Предполагается, что в качестве промежуточных
продуктов образуются соединения со связью РЬ—В, например соединение
(СН3)3РЬВН4 [123].
Реакция тетрафенилсвинца с треххлористым таллием протекает по
уравнению [73]:
(С6Н5)4РЬ + Т1С13 -> (С6Н5)2Т1С1 + (С6Н5)2РЬС12.
Аналогично тетрафенилсвинец реагирует с AsCl3, SbCl3, SbCl5, давая
(C6H5)2AsCl, (C6H5)2SbCl и (C6H5)2SbCl3 соответственно, причем во всех
случаях деарилирование тетрафенилсвинца заканчивается на стадии
образования двухлористого дифенилсвинца [73].
Деалкилирование свинцовоорганических соединений галогенидами
фосфора [124—128], мышьяка [124, 126] и сурьмы [126] описано и другими
авторами. Более подробно эти реакции рассмотрены в соответствующих
томах настоящего издания. О синтезе этим методом германий- и оловоорга-
нических соединений см. стр. 57 и 260 соответственно.
Деалкилирование свинцовоорганических соединений треххлористым
висмутом проходит своеобразно. При действии треххлористого висмута на
тетраэтилсвинец в отсутствие растворителя получают хлористый триэтил-
Деалкил ирован ие
587
свинец с выходом 15% [129]. Если же тетрафенилсвинец нагревать с
треххлористым висмутом в хлороформе, то деарилирующим агентом
является растворитель. В этом случае треххлористый висмут играет роль
катализатора [1201.
Аналогично ведет себя четыреххлористый кремний [49, 130].
Треххлористый алюминий деалкилирует тетраэтилсвинец в хлороформе' до дву-
хлористого свинца, а в гексане или петролейном эфире образуется хлористый
триэтилсвинец [118—120].
ДЕАЛКИЛИРОВАНИЕ СЕРОЙ И МЫШЬЯКОМ
В отличие от оловоорганических соединений (см. стр. 359), тетраалкил-
или тетраарилсвинец при нагревании с элементарной серой не образуют
свинцовоорганических сульфидов. Тетрабутил свинец не реагирует с серой
ниже 125° С [131]. Постепенное повышение температуры приводит к
энергичной реакции с выделением дибутилсульфида и сульфида свинца.
Аналогично ведет себя тетрафенилсвинец при 150° С [131]:
(С6Н5)4РЬ + 2 S -V2 (C6H-)2S Ь PbS.
Тетрафенилсвинец полностью деарилируется мышьяком при 300°С [132].
ДЕЙСТВИЕ ОКИСЛИТЕЛЕЙ
НА СВИНЦОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Окисление свинцовоорганических соединений подробно исследовано на
примере тетраэтилсвинца.* В отсутствие освещения тетраэтилсвинец в
растворе окисляется с заметной скоростью выше 80°С [133—135]. Окисление
тетраэтилсвинца протекает с самоускорением. Сначала процесс
развивается очень медленно (период индукции), и поглощения кислорода в течение
некоторого времени не происходит. Затем скорость этого процесса
увеличивается, достигает максимального значения и под конец уменьшается.
Окисление тетраэтилсвинца является сложной гетерогенной реакцией,
включающей, кроме химических стадий, ряд физических процессов
(растворение кислорода, выделение из реакционной смеси осадка и газообразных
продуктов и др.). В продуктах окисления тетраэтилсвинца
идентифицированы этан, этилен, бутан, ацетальдегид, гидроокись триэтилсвинца, дигид-
роокись диэтилсвинца, окись и перекись свинца. Предложена возможная
схема этой реакции [135].
Кислород воздуха вносит резкие изменения и в процесс термического
разложения тетраэтилсвинца [136, 137]. В тех случаях, когда реакции
окисления и термораспада могут конкурировать между собой, первый
процесс полностью вытесняет второй [137].
Изучено [138, 139] жидкофазное окисление углеводородов в присутствии
тетраэтилсвинца. Найдено, что свинцовоорганическое соединение ускоряет
этот процесс, что связано с диссоциацией тетраэтилсвинца на этильный и
этилсвинцовые радикалы. Из продуктов реакции выделены твердые
соединения свинца, которые ближе не исследовались. Анализ кинетических
кривых показывает, что они образуются в результате взаимодействия
тетраэтилсвинца с образующимися в процессе окисления гидроперекисями
углеводородов.
Озонолиз тетраэтилсвинца легко проходит уже при — 68°С [140]. В
результате образуется неидентифицированный продукт, изменяющий свой
цвет от белого до кремового-бурого при нагревании реакционной смеси от
— 68 до + 20°С. Конечные продукты в этом случае—гидроокись триэтил-
588
Свинцовоорганические соединения. Лит. стр. 590—594
свинца, окись диэтилсвинца, этокситриэтилсвинец, ацетальдегид и спирт.
Выход продуктов реакции мало зависит от температуры опыта.
В литературе отмечается неустойчивость по отношению к кислороду
воздуха некоторых типов свинцовоорганических соединений, например
растворов тетрамети л свинца в четыреххлористом углероде [141], фенил этил тетра-
производных свинца [142], свинцовоорганических соединений с вторичными
алкильными [10] или бензильными радикалами [142, 143]. Состав
образующихся при этом продуктов не исследован.
ДЕАЛКИЛИРОВАНИЕ ВОДОЙ И ЩЕЛОЧАМИ
Подобно соединениям кремния, германия и олова, тетраалкильные и
тетраарильные соединения свинца устойчивы к действию воды и щелочей.
Однако свинцовоорганические ацетилениды легко разлагаются водой.
Алкоголиз ацетиленида бис-(триэтил свинца) сухим метиловым спиртом
заканчивается за 12 час. [108].
О деалкилировании соединений типа (R2Pb)„ водой см. стр. 555.
ТЕРМО- И ФОТОРАСПАД. РЕАКЦИИ С ГАЛОИДНЫМИ АЛКИЛАМИ
Термическое разложение тетраалкильных соединений свинца изучено
довольно подробно. Панет с сотр. [144—151] доказал образование этильных
радикалов при термическом разложении тетраэтилсвинца в газовой фазе:
(С2Н5)4РЬ ^ РЬ* + С2Н,\
Впоследствии работы в этом направлении проводились и другими авторами
[136, 152—157]. С помощью газо-жидкостной хроматографии в статической
системе изучена [158] кинетика пиролиза тетраэтилсвинца при 233—267°С и
давлении 5—15 мм рт. ст. Обнаружено 17 продуктов реакции, содержащих
от 1 до 6 атомов углерода, причем на начальных стадиях образуются только
я-бутан, этилен, этан и водород. Взаимодействие этилена с различными
радикалами приводит к сложной смеси продуктов, обнаруживаемой на
поздних стадиях процесса.
Разуваевым и его школой [137, 159—161] подробно исследована кинетика
термического распада тетраэтилсвинца и его растворов в изооктане в
области температур 105—170°С. Установлено, что жидкофазное термическое
разложение тетраэтилсвинца является сложным цепным процессом,
протекающим с промежуточным образованием менее алкилированных соединений —
гексаэтилдиплюмбана и диэтилсвинца:
(С2Н5)4РЬ -> (С2НГ))зРЬРЬ (С2Н5)3 -* (С2Н5)2 РЬ -> РЬ.
Выделяющийся металлический свинец оказывает каталитическое влияние
на разложение гексаэтилдиплюмбана и диэтилсвинца. Это позволяет считать
термический распад тетраэтилсвинца автокаталитическим процессом [137,
160].
Напротив, следы кислорода воздуха и продуктов неполного окисления
тетраэтилсвинца ингибируют процесс его термораспада [137, 162—164].
Более сильными ингибиторами являются галоидные алкилы или продукты
их взаимодействия с тетраэти л свинцом [137, 160]. По патентным данным,
ингибиторами термического разложения тетраэтилсвинца являются также
смеси этилендибромида, например, со спиртами [165], высококипящими
углеводородами [166], талловым маслом [167], терпинеолом [168] или
пиперидином [169] и другие вещества [170—177].
Свинцовоорганические соединения легко разлагаются и при действии
на них УФ-света. Фотолиз тетраметилсвинца в газовой фазе дает свинец и
Деалкил ирование
589
этан [178]. Разуваемым и сотрудниками показано, что тетрафенилсвинец в
растворе бензола при нагревании или действии УФ-света распадается с
выделением металла и дифенила. Кроме того, специальными опытами в
меченном С14 бензоле или с радиоактивным тетрафенилсвинцом в обычном
растворителе ими же отмечено, что в образовании дифенила активное участие
принимает растворитель. Высказано предположение, что реакция проходит
по следующей схеме [179]:
(С6Н:))4РЬ + 2С*4Н6- [(С6Н5)4РЬ.2С£4Н6] -> 2С6Н5 -С^НЪ + 2С6Н6 + РЬ.
Обмен радикалов между свинцовоорганическим соединением и
растворителем в процессе термо- и фотораспада тетрафенилсвинца в бензоле
практически не имеет места [179—182].
Аналогично проходит фотолитическое разложение тетрафенилсвинца в
изопропилбензоле [183]. Из продуктов реакции выделены 2- и 3-изопропил-
дифенил.
Выше отмечено, что небольшие количества галоидоуглеводородов инги-
бируют процесс термического разложения тетраэтилсвинца. При
нагревании тетраэтилсвинца с эквимолекулярными количествами
галоидоуглеводородов без растворителя имеет место взаимодействие реагентов,
приводящее к сложной смеси продуктов [184, 185]. Так, при нагревании до 135°С в
течение 5 час. эквимолекулярных количеств тетраэтилсвинца и 1,2-дибром-
этана получают бромистый свинец, этан, этилен и бутан. Выход бромистого
триэтилсвинца составляет 24,6% от теорет., или 61,4% от вступившего в
реакцию тетраэтилсвинца. Двубромистого свинца образуется лишь 1,6%.
Реакционная способность взятого в реакцию бромистого алкила зависит
от его структуры и падает в ряду [184]:
С6Н5СНВгСНВгС6Н5 > ВгСН2СН2Вг > СН;}СНВгСНВгСН3 > С2Н,Вг >
> Вг (СН2)4Вг ж Вг (СН2)5Вг.
Реакция между тетраалкильными соединениями свинца и галоидными
алкилами может проходить и в ином направлении. При нагревании до 260°С
в вакууме в течение 2 час. тетраметилсвинца с избытком C14H3J получают
тетраметилсвинец, меченный С14. Ниже 200°С обменная реакция не имеет
места [186]. При нагревании до 150°С в течение 20 час. тетраэтилсвинца с
С142Н5Вг обмен этильными радикалами составил около 20% [187, 188].
Высказано предположение [189], что в этом случае реакция протекает через
стадию образования шестичленного комплекса, в котором чередуются
рвущиеся и образующиеся связи:
(С2Н5)4РЬ + С;4Н5Вг^==Г (C2H5)3Pbf <*а1Вг
-c^Rr + 'n2H,),PbcI4H5 + AlBr,
По патентным данным [190], нагревание тетраметил- и тетраэтилсвинца
с дибромметиленом или 1,2-дибромэтаном в присутствии А1С13, BF3 или
сплавов алюминия приводит к соединениям состава R3Pb(CH2)„ PbR3 (n= 1,2).
С йодистыми перфтор ал килами обмен радикалами проходит и в
отсутствие катализаторов. Триметилтрифторметил- и
триметилпентафторэтилсвинец получены [191, 192] нагреванием или облучением УФ-светом смеси тет
раметилсвинца с йодистым перфторметилом или этилом соответственно.
Реакции свинцовоорганических соединений ароматического ряда с
галоидными алкилами описаны очень кратко [193—195]. Найдено, что при нагре-
590
Свинцовоорганические соединения
вании тетрафенилсвинца с четыреххлористым углеродом и небольшим
количеством перекиси ацетила реакция проходит только между перекисью и
растворителем, а тетрафенилсвинец остается без изменения. В присутствии же
значительно больших количеств перекиси бензоила образуются двухлори-
стый дифенилсвинец и небольшое количество дибензоата дифенилсвинца.
Кроме того, из смеси выделены хлорбензол, гексахлорэтан, бензойная и
фталевая кислоты. Предложена возможная схема этой реакции [195].
ДЕАЛКИЛИРОВАНИЕ ПЕРЕКИСЯМИ АЦИЛОВ
Деалкилирование свинцовоорганических соединений перекисями аци-
лов описано очень кратко. Найдено [196, 197], что реакция перекиси
бензоила с тетраэти л свинцом принципиально не отличается от реакции этой
перекиси с этильными соединениями олова, но она проходит в более мягких
условиях. В результате образуются бензоат триэтилсвинца, этан, этилен,
бутан и следы двуокиси углерода.
ДЕАЛКИЛИРОВАНИЕ ОКИСЛАМИ АЗОТА
Тетрафенилсвинец реагирует с трехокисью азота (в присутствии N0),
давая динитрат дифенилсвинца и нитрат фенилдиазония с количественным
выходом по уравнению 1198]:
(С6Н5)4РЬ + 4 N203 - (C6l I,)2Pb (N03)2 + 2 CeH5N2N08.
О реакции тетраалкильных соединений свинца с двуокисью азота см. стр.
580.
ГИДРОГЕНИЗАЦИЯ
Тетраметил- и тетраэтилсвинец под давлением водорода начинают
выделять металл при 100—125°С [199—202]. В отсутствие водорода при этой
температуре разложение тетраал кил свинца не имеет места.
Тетрафенилсвинец количественно выделяет свинец при температуре 175—225°С и давлении
водорода 60 атм [203, 204]. В присутствии палладия, никеля, золота, сереб-
оа или меди разложение проходит при более низких температурах.
ЛИТЕРАТУРА
1. Van der KerkG. J.M., LuijtenJ.G. A., JanssenM. J., Chimia, 16, 10
(1962).
2. G г u t t n e г G., К г a u s e E., Ber., 49, 1415 (1916).
3. P f e i f f e r P., Truskier P., Ber., 37, 1125 (1904).
4. G r u t t n e г G., К г a u s e E., Ber., 50, 278 (1917).
5. GielenM., Nasi el ski J., DuboisJ.G., FresnetP., Bull. Soc. biol.
belg., 73, 293 (1964).
6. С a 1 i ngaertG., SoroosH., J. Org. Chem., 2, 535 (1938).
7 С a 1 i n g a e r t G., D у k s t г a F. J., S h a p i г о Н., J. Am. Chem. Soc, 67, 190
(1945).
8. J u e n g e E. С, С о о k S. E., J. Am. Chem. Soc, 81, 3578 (1959).
9. KrauseE., Ber., 54, 2060 (1921).
10. GruttnerG., KrauseE., Ber., 50, 574 (1917).
11. Gru ttnerG., Ber., 51, 1298 (1918).
12. P о is A., Ber., 20, 3331 (1887).
13. GilmanH., BailieJ.C, J. Am. Chem. Soc, 61, 731 (1939).
14. P о I i s A., Ber., 21, 3424 (1888).
15. F 1 о о d E. A., H о v i t z Z., J. Am. Chem. Soc, 55, 2534 (1933).
16. KrauseE., G г о s s e A., Die Chemie der metalloorganischen Verbindungen.
Berlin, Verlag Gebr. Borutrager, 1937, S. 416, 419.
Деалкилироеание
591
17. К г a u s e E., S с h I 6 t t i g О., Ber., 58, 427 (1925).
18. WillemsensL.C, van d e г К е г к G. J., M., J. Organometal. Chem., 4, 34
(1965).
19. WillemsensL.C, van d e r KerkG. J.M., J. Organometal. Chem., 4, 241
(1965).
20. D e 1 h а у e A., N a s i e 1 s к i J., P I a n с h о n M., Bull. Soc. chim. belg., 69, 134
(1960).
21. G i 1 m a n H., M e 1 s t г о m D. S., J. Am. Chem. Soc, 72, 2953 (1950).
22. FreedmanH, Пат. США 3090797 (1963); РЖХим., 1965, 8H103.
23. M а н у л к и н 3. М, Узб. хим. ж., 2, 66 (1960).
24. В и с к t о п G. В., Ann., 112, 220 (1859).
25. G i I m a n H., R о b i n s о n J., J. Am. Chem. Soc, 52, 1975 (1930).
26. Y а к u b о v i с h A. I., P e t г о v I., J. praKt. Chem., 144, 67 (1935).
27. S a u n d e r s B. C, S t а с е у G. J., J. Chem. Soc, 1949, 919.
28. H e a p R., S a u n d e r s B. C, J. Chem. Soc, 1949, 2983.
29. AmbergerE., H oni gschmi d-Grossi chR., Ber., 98, 3795 (1965).
30. H e a p R., S a u n d e r s B. C, S t а с е у G. J., J. Chem. Soc, 1951, 658.
31. С a 1 i n ga er t G., Sor о os H., S h a pi г о H., J. Am. Chem. Soc, 62, 1104 (1940).
32. H u r d C. D., A u s t i n P. R., J. Am. Chem. Soc, 53, 1543 (1931).
33. A u s t i n P. R., J. Am. Chem. Soc, 55, 2948 (1933).
34. К о т о н М. М., К и с е л е в а Т. М., 3 а п е в а л о в а Н. П., ЖОХ, 30, 186 (1960).
35. GilmanH., TowneE.B., JonesH.L, J. Am. Chem. Soc, 55, 4689 (1933).
36. L e e p e r R. W., Iowa State Coll. J. Sci., 18, 57 (1943); С. А., 38, 726 (1944).
37. Stuckwisch C.G., Iowa State Coll. J. Sci., 18, 92 (1943); С. А., 38, 728 (1944).
38. GilmanH., R о b i n s о n J., J. Am. Chem. Soc, 51, 3112 (1929).
39. Bahr G., Z. anorgan. Chem., 253, 330 (1947).
40. L i e b e r E., R а о С. N. R., К e a n e F. M., J. Inorg. Nucl. Chem., 25, 631 (1963).
41. LieberE., К e a n e F. M., Chem. a. Ind., 1961, 747.
42. КотонМ. М., ЖОХ, 18, 936 (1948).
43. AustinP. R., J. Am. Chem. Soc, 53, 3514 (1931).
44. G i 1 m a n H., T о w n e E. В., J. Am. Chem. Soc, 61, 739 (1939).
45. G i 1 m a n H., T о w n e E. В., Rec trav. chim., 51, 1054 (1932).
46. К h a r a s с h M. S., R e i n m u t h О., М а у о F. R., J. Chem. Educ, 11, 82 (1934).
47. G i 1 m a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
48. Юрьев Ю. Км Га л ьберштамМ. А., К а н д р о р И. И., ЖОХ, 34, 4116
(1964).
49. В г о w n е О. Н., R e i d E. E., J. Am. Chem. Soc, 49, 830 (1927).
50. Polis A., Ber., 20, 716 (1887).
51. Cat 1 i n W. E., Iowa State Coll. J. Sci., 10, 65 (1935); С. А., 30, 935 (1936).
52. В u с к t о n G. В., Ann., 109, 218 (1859).
53. С a h о u r s A., Ann., 122, 48 (1862).
54. H e t n a r s к i В., UrbanskiT., Tetrahedron, 19, 1319 (1963).
55. H e t n a r s к i В., UrbanskiT., Roczn. chem., 37, 1073 (1963).
56. SaundersB.C, S t а с е у G. J., J. Chem. Soc, 1948, 1773.
57. McCombieH.,SaundersB.C,Nature, 159,491 (1947).
58. Austin P. R., J. Am. Chem. Soc, 53, 1548 (1931).
59. M с С 1 e a r у R. F., DegeringE.F, Ind. Eng. Chem., 30, 64 (1938).
60. J о n e s W. J., E v a n s D. P., G u 1 w e 1 1 Т., G r i f f i t h s D. С, J. Chem. Soc,
1935 39.
61. GiimanH.,RobinsonJ., Rec trav. chim., 49, 766 (1930).
62. GilmanH., SpatzS.M., К о 1 b e z e n M. J., J. Org. Chem., 18, 1341 (1953).
63. R о b i n s о n G. C, J. Org. Chem., 28, 843 (1963).
64. J о n e s L. W., W e r n e r L. P., J. Am. Chem. Soc, 40, 1257 (1918).
65. J u e n g e E. С, Н о u s e к R. D., J. Org. Chem., 29, 2040 (1964).
66. КочешковК.А, Б о р о д и н а Г. М., Изв. АН СССР, Отд. ест. наук, 1937,
569.
67. S е t z е г W. С, L е е р е г R. W., G i I m а п Н., J. Am. Chem. Soc, 61, 1609 (1939).
68. НепгуМ. С, Inorg. Chem., 1, 917 (1962).
69. Н е с м е я н о в А. Н., Ф р е й д л и н а Р. X., ЖОХ, 5, 53 (1935).
70. N е s m e j a n о w A. N., F r e i d 1 i n a R. C, Ber., 67, 735 (1934).
71. Котон М. М., ЖОХ, 11, 376 (1941).
72. К о т о н М. М., ЖОХ, 9, 2283 (1939).
73. G о d d a r d A. E., A s h 1 e у J. N., E v a n s R. В., J. Chem. Soc, 121, 978 (1922).
74. ПановЕ.М, КочешковК.А, ЖОХ, 25, 489 (1955).
75. ПановЕ.М, К о ч е ш к о в К. А., ДАН, СССР, 85, 1037 (1952).
76. Н е п г у М. С, К г е b s A. W., J. Org. Chem., 28, 225 (1963).
77. КотонМ. М., ЖОХ, 22, 643 (1952).
78. К о ч е ш к о в К. А., П а н о в Е. М., Изв. АН СССР, ОХН, 1955, 718.
r}g9 Свшщовоорганические соединения
79. М б 1 1 е г S., Р г е i f f е г P., Ber., 49, 2441 (1916).
80. G i 1 m a n H., S u m m e r s L., J. Am. Chem. Soc 74 5924 (19o2).
81. E a b о r n C, P a n d 1 e К. С J. Chem. Soc 1961, 3715
82 П а н о в Е. М., К о ч е ш к о в К- А., ДАН СССР, 123, 2У5 (1Уо«).
83'. К о т о н М. М.„ ЖОХ, 17, 1307 (1947).
84. Oilman Н., N е 1 s о n J. F., J. Am. Chem. Soc., 59 935 (1937). 2(Ш(195())
85 КотонМ.М., Москвина Е. П., Ф л о р и н с к и и Ф. С, ЖОХ, 20, ДШ \\ЧЫ).
86. Англ. пат. 331494 (1929); Ch. Ztb., 1, 2688 (1930).
87. М а н у л к и н 3. М., ЖОХ, 16, 237 (1946)
88. К h a r a s с h M. S., Пат. США 1987685 1929); Ch. Ztb И, 437 (1935).
89. A i n 1 е у A. D., Е 1 s о n L. A., S extonW- A. J. Chem. Soc, 1946, 776.
90. D i е t z H.-J., Пат. ГДР 32275 (1964); РЖХим., 1966 3H524_
91. Tagli avini G., Belluco U., Ricerca scient-, 2A, 76 (1962), РЖХим., \мьб,
92. Кш к о в К. А., Н е с м е я н о в А. Н. ЖОХ 4, 1102 (1934).
93. N е s m е j а п о w А. N.. К о z е s с h к о w К. А., Вег., 67,37(ВД
94. Н е i п R S с h n e i t е г Н., Z. anorgan. а11д^^™ЙХН 955 Til"
95. К о ч е ш к о в К. А., П а н о в Е. М., Изв. АН СССР, ОХН, 1У^, /п.
96. WillemsensL.С., van der Kerk G. J. M.f J. Organometal. Chem., 13,
357 (1988).
97. К о ч е т к о в А. К., Ф Р е й д л и н а Р. X., Уч. зап. МГУ им. М. В. Ломоносова,
орг. хим., 1 2, 144 (1952); Изв. АН СССР, ОХН, 1950, 203.
98. A u s t i n P. R., J. Am. Chem. Soc, 54, 3287 (1932).
99. G i 1 m a n H., W о о d s L. A., J. Am. Chem. Soc, 65, 43* П943).
100. SemeranoG, R i с с о b о n i L., Ber., 74, 1089 П94П.
101. SemeranoG., RiccoboniL, Ricerca scient., 11 269 (1940)- Ch. Ztb., 1, 350
(1941).
102. SemeranoG., R i ccob oni L, Cal I i gari R, Rer.. 74. 1W (1941).
103. LichtenwalterM., Iowa State Coll. J. Sci., С. А. 34 6241 (19*0)
104. KrauseE, SchmitzN., Ber., 52, 2150 (1919).
105. G 1 о с к 1 i n g F., J. Chem. Soc, 1955, 716.
106. GlocklingF., J. Chem. Soc, 1956, 3640.
107. GlocklingF., К i n g s t о n D., J. Chem. Soc, 1959, 3001.
108. В e e r m a n C, H a r t m a n n H., Z. anorgan. allgem. Chem., 276, 20 (1954)
109. BawnC. E.H., WhitbyF. J., Disc Faraday Soc, 2, 228 (1947).
110. Spi ceJ. E., T wi st W., J. Chem. Soc, 1956, 3519.
111. R ei с h R., Compt. rend., 177, 322 (1923).
112. R i с с о b о n i L., BellucoU., T a g 1 i a v i n i G., Ricerca scient., 2A, 323
(1962); РЖХчм., 1933, 24Ж310.
113. TagliaviniG., BellucoU., Cattalini L., Ricerca scient. 2A, 350 (1962)
РЖХим., 1964, ЗЖ284.
114. T a g 1 i a v i n i G., S с h i a v о n G., В e 1 1 u с о U., Gazz. chim. ital., 88, 746(1958)
РЖХим., 1959, 53489.
115. В e 1 I u с о U., R i ceo b о n i L., T a g 1 i a v i n iG., Ricerca scient., 30, 1255 (1960)
РЖХим., 1961, 13Ж265.
116. Разув а ев Г. А., Федотов М. С, Зайченко Т.Н., Кульвин-
с кая Н. А., Сборник статей по общей химии, т. 2. М., Изд-во АН СССР, 1953, стр. 1514.
117. М а 1 a t e s t a A., Canad. J. Chem., 37, 1176 (1959).
118. М а н у л к и н 3. М., ЖОХ, 18, 299 (1948).
119. G i 1 m a n Н., А р р ег son L. D., J. Org. Chem., 4, 162 (1939).
120. С а 1 i n g a e r t G., В e a t t у H. A., SoroosH, J. Am., Chem. Soc, 62, 1099
(1940).
121. В a w n С E. H., G 1 a d s t о n e J., Proc Chem. Soc, 1959, 227.
122. StoneLS., Пат. США 2923740 (1960); РЖХим., 1961, 21Л72.
123. H о 1 1 i d а у А. К-, J e f f e r s W., J. Inorg. Nucl. Chem., 6, 134 (1958).
124. Kh arasch M. S., J en sen E. V., WeinhouseS., J. Org. Chem., 14,429 (1949)
125. Зиновьев Ю. М., СоборовскийЛ. З., ЖОХ, 34, 929 (1964).
126. MaierL, Tetrahedron L., 6, 1 (1959).
127. S e у f e r t h D., F г е у e r W., J. Org. Chem., 26, 2604 (1961).
128. В a r t о с h а В., G г а у М. Y., Z. Naturforsch., 14b, 350 (195*1).
129. МанулкинЗ.М, ЖОХ, 20, 2004 (1950).
130. МанулкинЗ.М, Узб. хим. ж., 2, 41 (1958).
131. S с h m i d t M., S с h u m a n n H., Z. anorgan. allgem. Chem., 325, 130 (1963).
132. SchurowA. E., R a s u w a j e w G. A., Ber., 65, 1507 (1932).
133. Ш у ш у н о в В. А., Б р и л к и н а Т. Г., А л е к с а н д р о в Ю. А., Труды по
химии и хим. технологии (Горький), 1959, 329.
134. Александров Ю. А., Б р и л к и н а Т. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1961, 3.
Деалкилирование
593
135. Брил кинаТ. Г.,ШушуновВ. А., Реакции металлоорганических соединений
с кислородом и перекисями. М. , «Наука», 1963, стр. 215.
136. Б е р л и н Л. Е., Труды Научно-исследовательского ин-та. М., Госхимиздат, 1958,
стр. 35.
137. Р а з у в а е в Г. А., В я з а н к и н Н. С, В ы ш и н с к и й Н. Н., ЖОХ, 30, 4099
(1960).
138. Ш и м о н а е в Г. С, Р о ж к о в И. В., ЖФХ, 29, 791 (1955).
139. Ш и м о н а е в Г. С, Р о ж к о в И. В., ЖФХ, 31, 387 (1957).
140. Александров Ю. А., ШеяновН. Г., ЖОХ, 36, 953 (1966).
141. Н е i n e F., N е b е Е., R e i m a n n W., Z. anorgan. allgem. Chem., 251, 125 (1943).
142. К г a u s e E., S с h 1 6 t t i g О., Ber., 63, 1381 (1930).
143. В a h г G., Z о с h e G., Ber., 88, 542 (1955).
144. PanethF, HofeditzW., Ber., 62, 1335 (1929).
145. PanethF., LautschW., Nature, 125, 564 (1930).
146. PanethR.LautschW., Naturwiss., 18, 307 (1930).
147. PanethR.LautschW., Ber., 64, 2702 (1931).
148. PanethR.LautschW., Ber., 64, 2708 (1931).
149. P a n e t h R, H e r z f e 1 d K., Z. Elektrochem., 37, 577 (1931).
150. PanethRA., L о 1 e i b i t H., J. Chem'. Soc, 1935, 366.
151. P a n e t h F. A., Hof edi tzW., Wunsch A., J. Chem. Soc, 1935, 372.
152. TaylorH.S., JonesW.H.J. Am. Chem. Soc, 52, 1111 (1930).
153. M e i n e г t R. N., J. Am. Chem. Soc, 55, 979 (1933).
154. Cramer P. L., J. Am. Chem. Soc, 56, 1234 (1934).
155. L e e r m а к е г s J. A., J. Am. Chem. Soc, 55, 4508 (1933).
156. С r a m e г P. L., J. Am. Chem. Soc, 60, 1406 (1938).
157. W i d m a i e r O., Brennstoff-Chem., 34, 83 (1953).
158. P r a t t G. L., P u г n e 1 1 J. H., Trans. Faraday Soc, 60, 519 (1964).
159. P а з у в а е в Г. А., ВязанкинН.С, В ы ш и н с к и й Н. Н., ЖОХ, 29, 3662
(1959).
160. ВязанкинН. С, РазуваевГ. А., Дергунов Ю. И., Труды по химии и
хим. технологии (Горький), 1961, 652.
161. Р а з у в а е в Г. А., В я з а н к и н Н. С, В ы ш и н с к и й Н. Н., ЖОХ, 30, 967
(1960).
162. Т h о m a s W. Н., С о о k S. Е., Пат. США 3133095 (1964); РЖХим., 1966, 9Н104.
163. Т h о m a s W. Н., С о о к S. Е., Пат. США 3133101 (1964); РЖХим., 1966, 4Н69.
164. С о о к S. Е., Т h о m a s W., Пат. США 3133097 (1964); РЖХим., 1966, 9Н105.
165. Т h о m a s W. Н., С о о к S. Е., Пат. США 3133102 (1964); РЖХим., 1966, 4Н70.
166. С о о к S. Е., Пат. США 3147294 (1964); РЖХим., 1966, 7Н118.
167. С о о к S. E.,Thom as W. Н., Пат. США 3133098 (1964); РЖХим., 1966,9Н106.
168. С о о к S. Е., Т h о m a s W. Н., Пат. США 3133103 (1964); РЖХим., 1966, 4Н71.
169. Т h о m a s W. Н., С о о к S. Е., Пат. США 3097223 (1963); РЖХим., 1965, 12Н82.
170. С о о к S. Е., S h a p i г о Н., Пат. США 3038918 (1962); РЖХим., 1964, 1Н50.
171. С о о к S. Е., ThomasW.H., Пат. США 3098089 (1963); РЖХим., 1965, 8Н104,
172. С о о к S. Е., S h a p i г о Н., Пат. США 3038919 (1962); РЖХим., 1963, 22Н57.
173. С о о к S. Е., Т h о m a s W. Н., Пат. США 3133099 (1964); РЖХим., 1966, 4Н72.
174. Т h о m a s W. Н., С о о к S. Е., Пат. США 3098090 (1963); РЖХим., 1965, 8Н105.
175. G i r a i t i s A. P., ShapiroH., J ohnstonJ.D., Пат. США 3133105 (1964V.
РЖХим., 1966, 9H107.
176. Франц. пат. 1389553 (1965); РЖХим., 1966, 10Н115.
177. С а 1 i n g a e r t G., Пат. США 2660592 (1953); РЖХим., 1955, 30032.
178. L e i g h t о n P. A., M о r t e n s e n R. A., J. Am. Chem. Soc, 58, 448 (1936).
179. P а з у в а е в Г. А., Петухов Г. Г., К а п л и н Ю. А., ЖОХ, 33, 2394 (1963).
180. Р а з у в а е в Г. А., П е т у х о в Г. Г., К а п л и н Ю. А., Д р у ж к о в О. Н.,
ДАН СССР, 152, 1122 (1963).
181. Коршунов И. А., О р л о в а А. А., ЖОХ, 28, 45 (1958).
182. Назарова Л. М., Харламова Е. Н., Александрова Г. Е., ЖОХ,
31, 3309 (1961).
183. BlairMcDonaldJ., Bryce-Smith D., PendillyB. W., J. Спэт.
Soc, 1959, 2174.
184. РазуваевГ. А., Дергунов Ю. И., ВязанкинН. С, ЖОХ, 31, 998 (1961).
185. Ст а с и н е в и ч Д. С, ГольдштейнА. Л., Труды по химии и хим.
технологии (Горький), 1960, 209.
186. TagliaviniG., BellucoU., SchiavonG., RiccoboniL., Ricerca
scient., 2A, 2349 (1958); РЖХим., 1959, 53515.
187. К о р ш у н о в И. А., Баталов А. П., О р л о в а А. А., Радиохимия, 1, 079
(1959).
188. К о р ш у н о в И. А., А м е н и ц к а я Р. В., О р л о в а А. А., Б а т а л о в А. П.
ЖОХ, 29, 1992 (1959).
38 Заказ № 207
594
Свинцовоорганические соединения
189. Б а т а л о в А. П., К о р ш у н о в И. А., ЖОХ, 31, 1649 (1961).
190. W i с z e r S. В., Пат. США 2447926 (1948); С. А., 42, 7975 (1948).
191. KaeszH.D., P h i l i p p s J. R., S t о n e F. G. A., Chem. a Ind., 1959, 1409.
192. KaeszH.D., P h i 1 i p p s J. R., S t о n e F. G. A., J. Am. Chem. Soc, 82, 6228
(1960).
193. P а з у в а е в Г. А., Ф етю ко в а В. В., Ш у б е н к о М. А., Уч. зап. Горьк. ун-та,
15, 91 (1949).
194. О л ь д е к о п Ю. А., М о р ы г а н о в .Б. Н., ЖОХ, 23, 2020 (1953).
195. ОльдекопЮ. А.,Соколова Р. Ф., ЖОХ, 23, 1159 (1953).
196. Р а з у в а е в Г. А., Дьячковская О. С, Вязанкин Н. С, Щепет-
к о в а О. А., ДАН СССР, 137, 618 (1961).
197. Р а з у в а е в Г. А., Вязанкин Н. С, Дьячковская О. С, ЖОХ, 32,
2161 (1962).
198. Несмеянов А. Н., Макарова Л. Г., ЖОХ, 9, 771 (1939).
199. Ипатьев В. Н., Р а з у в а е в Г. А., Б о г д а н о в И. Ф., ЖРХО, 61, 1791 (1929)
200. Ип ат ь ев В. Н., Р а з у в а ев Г. А., Б о гд а н о в И. Ф., ДАН СССР, 1929, 155.
201. R a z u v a e v G. А., К о t о n M. М., Вег., 66, 854 (1933).
202. KotonM. M., Вег., 66, 1213 (1933).
203. ZartmanW.H., A d k i n s H.,' J. Am. Chem. Soc, 54, 3398 (1932).
204. GerschbeinL. L, I p a t i e f f V. N., J. Am. Chem. Soc, 74, 1540 (1952).
Г лава IX
РЕАКЦИИ ДИСПРОПОРЦИОНИРОВАНИЯ,
ТЕРМИЧЕСКОГО РАСПАДА И ПЕРЕРАСПРЕДЕЛЕНИЯ
РАДИКАЛОВ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Процессы диспропорционирования играют весьма важную роль в химии
свинцовоорганических соединений. Достаточно напомнить, что они позволя-
к т синтезировать тетраалкил- и тетраарилсвинец исходя из дигалогенидов
свинца и реактива Гриньяра (см. стр. 549). Существенное влияние реакции
диспропорционирования оказывают и на состав продуктов деалкилирования
свинцовоорганических соединений (см. стр. 579).
Однако, несмотря на большой фактический материал по диспропорциони-
рованию различных производных свинца, систематические исследования
в этой области начаты лишь в последние годы.
Разуваевым и его школой [1—5] подробно изучено диспропорционирова-
ние чистого гексаэтилдиплюмбана и его растворов в я-нонане. Установлено
[1], что в отсутствие кислорода воздуха вещество устойчиво в течение
нескольких суток. Однако при прибавлении к нему 1—1,5 вес. % треххлори-
стого алюминия оно полностью диспропорционирует за 2—3 часа при
обычной температуре с образованием тетраэтил свинца и металлического свинца
[2]. Аналогичное влияние на гексаэтилдиплюмбан оказывают бромистый
триэтилсвинец, двубромистый диэтилсвинец, галоидные соли ртути и
алюминия, хлористая и бромистая этилртуть и хлористое триэтилолово [ 1—3, 5].
Проведение этих реакций в эвакуированных ампулах, соединенных с
ртутным манометром, показало практически полное отсутствие газообразования.
Соотношение между количествами свинца и тетраэтилсвинца согласуется с
уравнением:
2 (C2H5)3PbPb (С2Н5)з -> 3 (С2Н5)4РЬ + РЬ.
Интересно отметить, что диспропорционирование
гексаэтилдиплюмбана вызывают и галоидные алкилы, например 1,2-дибромэтан [1, 4].
Дальнейшие исследования, проведенные теми же авторами, показали, что
в этом случае катализаторами диспропорционирования являются,
по-видимому, не сами галоидные алкилы, а первичные продукты их
взаимодействия с гексаэтилдиплюмбаном — бромистый триэтилсвинец и двубромистый
диэтилсвинец. Совокупность полученных данных позволила допустить, что
диспропорционирование гексаэтилдиплюмбана является частным случаем
реакции перераспределения радикалов [1] (см. стр. 598). Действительно,
гексаэтилдиплюмбан можно считать несимметричным соединением типа
(C2H5)3PbR [R = (С2Н5)3РЬ]. Если допустить, что и этильные радикалы и
группа (С2Н5)3РЬ будут одинаково легко участвовать в реакции
перераспределения, то в присутствии указанных выше катализаторов будет
образовываться равновесная смесь соединений: (С2Н5)4РЬ (А), (С2Н5)зРЬРЬ(С2Н5)а
(Б), (С2Н5)2РЬ[РЬ(С2Н5)3]2 (В), С2Н5РЬ[РЬ(С2Н5)зЬ (П и РЬ[РЬ(С2Н5)4]
(Д). Поскольку соединения Б—Д также имеют несимметричные атомы
за*
596 Свинцовоорганшеские соединения. Лит. стр. 599—600
свинца, процесс усложнения будет продолжаться. Вместе с тем он не может
быть бесконечным, так как соединения, содержащие в своем составе цепь
из атомов свинца, должны,, быть крайне неустойчивы. Таким образом,
наряду с процессом усложнения будет протекать распад усложненных
молекул/например
СаН3
(QHsbPb-Pb-Pb (СвН5)з -> 2 (СаН5)4РЬ + РЬ,
I
с2н,
вследствие чего единственными продуктами реакции перераспределения
радикалов гексаэтилдиплюмбана являются тетраэти л свинец и
металлический свинец [1].
В работах Разуваева с сотр. [1,3] неоднократно подчеркивается, что дис-
пропорционирование гексаэтилдиплюмбана проходит только в
присутствии каталитических количеств указанных выше веществ (в случае
дибромэтана от 0,11 до 5 вес.%). Повышение концентрации дибромэтана уменьшает
скорость диспропорционирования, а при эквимолекулярных количествах
реагентов смесь не изменяется при обычной температуре в течение 40—
45 час. При повышении температуры наблюдается уменьшение
индукционного периода и появление конкурирующей реакции взаимодействия
гексаэтилдиплюмбана с галоидным ал килом (см. стр. 615).
Аналогичная картина наблюдается при смешении эквимолярных
количеств гексаэтилдиплюмбана и бромистого триэтилсвинца. При этом
происходит распад обоих компонентов, приводящий к тетраэтилсвинцу, двуброми-
стому свинцу и металлическому свинцу [3]. Реакция протекает без выделения
газообразных веществ. При комнатной температуре она заканчивается за
5—8 мин. и, по-видимому, может быть описана следующими уравнениями:
(С2Н5ЬРЬРЬ(С2Н5Ь + 2 (С2Н5)3РЬЗг -> 3 (С2Н5)4РЬ + РЬЗг,,
2 (C2H5)3PbPb (GHsfeHH (С2Н5)4РЬ + РЬ.
С эквимолекулярным количеством двубромистого диэтилсвинца гексаэтил-
диплюмбан образует тетраэтилсвинец, бромистый триэтилсвинец, двубро-
мистый свинец и металлический свинец [3].
По патентным данным [6, 7] превращению гексаалкилдиплюмбана в
тетраалкилсвинец способствуют и каталитические количества силикагеля
или активированного угля. Особый вид диспропорционирования,
сопровождаемый перераспределением радикалов, имеет место при нагревании до
100 + 5°С смеси гексаметил- и гексаэтилдиплюмбана [8]. В конечном итоге
получают смесь следующих соединений (%): ди мети л диэтилсвинца (23),
тетрамети л свинца (18), метилтриэтилсвинца (31), триметилэти л свинца (15)
и тетраэтилсвинца (13).
Скорость диспропорционирования гексаарилдиплюмбанов
определяется природой ароматического радикала у атома свинца и условиями
проведения эксперимента. Так, при нагревании гексафенилдиплюмбана выделяются
металлический свинец и тетрафени л свинец [9]. При фотооблучении этот
процесс идет дальше. В продуктах реакции, кроме указанных выше веществ,
обнаружен дифенил, который образуется за счет дальнейшего разложения
тетрафенилсвинца. Проведением реакции в полностью дейтерированном
бензоле доказано отсутствие взаимодействия между гексафенилдиплюмбаном
и растворителем [9, 10].
Склонность к диспропорционированию уменьшается с увеличением
радикала у^атома свинца. Она, кроме того, зависит и от их строения (в толильном
ряду орто-изомеры более устойчивы). Так, диспропорционирование гекса-
-отолилдиплюмбана проходит с трудом [11—14], а гекса-/г-толилдиплюмба-
Диспропорционирование
597
на гораздо легче [11, 12]. При ксилильных или циклогексильных
радикалах этот переход не осуществляется.
Радикалы по степени их уменьшающейся стабильности (кипящий ксилол)
при диспропорционировании могут быть расположены следующим образом:
мезитил, циклогексил, а-нафтил > о-этоксифенил, о-метоксифенил, о-
толил ^> я-этоксифенил, п-метоксифенил, n-толил ^> ж-толил, фенил ^>
р> этил, метил [13, 15].
При действии восстановителей на свинцовоорганические галогениды
имеет место диспропорционирование другого рода. Например, по Гилману
и Барнетту [16] 30-часовое кипячение хлористого трифенилсвинца с 2 г
гидразинсульфата (в присутствии соды) дает тетрафенилсвинец с выходом
70%. Двухлористый дифенилсвинец переходит в аналогичных условиях в
тетрафенилсвинец (выход 53%). Наблюдались превращения двухлористого
дифенилсвинца в хлористый трифенилсвинец, двухлористого ди-о-анизил-
свинца — в хлористый три-о-анизилсвинец (под влиянием порошка
металлического свинца или сплава Pb — Na) [17]. Диспропорционирование свин-
цовоорганических галогенидов может проходить и в отсутствие
восстановителя. В частности, пятичасовое кипячение хлористого диметилэтилсвинца в
ацетоне привело к смеси всех возможных в этом случае тетраалкильных
соединений свинца, хлористого триметилэтилсвинца и хлористого
диметилэтилсвинца, как главных продуктов, и, наконец, небольших количеств
хлористого триэтилсвинца и хлористого метилдиэтилсвинца [18]. При
нагревании хлористого триэтилсвинца образуется смесь тетраэтилсвинца и
двухлористого диэтил свинца [191.
Аналогичный процесс наблюдается и в ароматическом ряду. Например,
шестичасовое нагревание хлористого трифенилсвинца в бутиловом спирте
дает 10% тетрафенил свинца и двухлористый дифенилсвинец [20].
Следует подчеркнуть, что подробного исследования всех продуктов
реакции обычно не проводилось. Вместе с тем, как подчеркивается в работах
Разуваева с сотрудниками, необходимо строго различать реакцию диспро-
порционирования, которая вызывается действием катализатора, от реакции
термического распада. Авторами [5, 21, 22] установлено, что, например, для
гексаэтилдиплюмбана, помимо рассмотренной на стр. 595 реакции диспро-
порционирования, характерна следующая реакция разложения:
Ш (с2н5)3рьрь (С2Н5)3-> (С2Н5)4рь +рь -г 2;с2н5*.
Изучением состава образующихся продуктов и кинетических
закономерностей протекания этой реакции доказано, что она проходит сложным путем с
промежуточным образованием диэтилсвинца [5, 21—23].
Такой тип разложения присущ и другим классам свинцовоорганических
соединений, особенно алифатического ряда. Проведены подробные
кинетические исследования термического разложения гидроокиси триэтилсвинца
в я-нонане [24]. В продуктах реакции (150° С) обнаружены дигидроокись
диэтилсвинца, тетраэтилсвинец, вода, этилен, этан, бутан. Кроме того, при
этом выделяется твердый осадок, в котором качественно доказано
присутствие перекиси свинца. Авторы [24] предполагают, что вначале
образуются тетраэтилсвинец и дигидроокись диэтилсвинца, термический распад
которой и приводит к конечной сложной смеси продуктов.
Гексаэтилдиплюмбоксан разлагается в более мягких условиях (70—90°С)
в соответствии с уравнением [25, 26]:[
(СаН5)3РЬОРЬ (С2Н5)з -> (С2Н5)4РЬ + (С2Н5)2РЬО + С2Н6 VC2H4.
Нагревание до 100° С в ксилоле гексаэтилдиплюмбоксана приводит к тетра-
фенилсвинцу и окиси дифенилсвинца [27].
598 Свинцовоорганические соединения. Лит. стр. 599—600
В литературе приведены также данные по термическому разложению
хлоридов, бромидов и гидроокисей три- и диэтилсвинца [28, 29], двухлори-
стого дибутилсвинца [30] и свинцовоорганических ацилатов [31—33]. В
последнем случае реакция может проходить, по-видимому, в двух
направлениях. Так, ацетат триэтилсвинца разлагается по следующей схеме [31]:
2 (С2Н5)зРЬООССН3 -» (С2Н5)2РЬ (ООССН3)а + (С2Н5)4РЬ.
В свою очередь, д и ацетат диэтилсвинца диспропорционирует до ацетата
триэтилсвинца и триацетата этилсвинца. Последний неустойчив и сразу же
распадается с выделением диацетата свинца и этилацетата [31].
Аналогичным образом проходит разложение димонохлорацетата диэтилсвинца
[32, 33]:
2 (С2Н5)аРЬ (ООССН2С1)2 -*
-» (С2Н5)3РЬООССН2С1 -|- РЬ(ООССПаС1)2 + CH2CICOOC2H5.
Совершенно иным путем проходит разложение свинцовоорганических
ацилатов, легко выделяющих углекислый газ. Кочешков и Александров [34]
подвергли термическому распаду этилмалонат трифенилсвинца и его бен-
зильного производного. Этот распад протекает при 130—165°С по
уравнениям:
(С6И5)зРЬООССН2СООС2Н5 -* (СвН5)зРЬСНаСООСаН5 +С02,
(СвН5)3РЬООССИ (СНаСвН6)СООС2Н6 - (СбН5)зРЬСН (СН2СбН5) COOQH6 + С02.
Выходы продуктов составляют 40 и 60%.
Получение этилового эфира трифенилплюмбилуксусной кислоты [34]. 9,5 г (0,0095 моля)
этилмалоната трифенилсвинца нагревают при 160—165° С в вакууме (15 мм). Вещество
плавится, наблюдается энергичное выделение углекислого газа. Спустя 20—25 мин. после
начала нагревания выделение углекислого газа прекращается. По охлаждении образуется
кашицеобразная масса. Ее многократно промывают горячим петролейным эфиром (65 мл),
фильтруют горячей от непрореагировавшего исходного вещества и фильтрат упаривают.
Получают белое кристаллическое вещество ст. пл. 59—60° С. Этиловый эфир
трифенилплюмбилуксусной кислоты хорошо растворяется в ацетоне, хлороформе, бензоле и этилацетате,
умеренно в петролейном эфире. В метиловом и этиловом спиртах с трудом растворяется на
холоду, несколько лучше при нагревании.
Впоследствии эта методика была воспроизведена Уиллемсенсом и Ван-
дер-Керком [35]. Состав конечного продукта доказан ими независимым
синтезом.
О термо- и фотораспаде тетраалкильных и тетраарильных соединений
свинца см. стр. 588.
Реакции перераспределения радикалов в ряду свинцовоорганических
соединений легко проходят при нагревании до сравнительно низкой
температуры (80° С) смеси двух тетраалкилсвинцов в присутствии катализаторов
(особенно эффективны BF3 и его эфират, А1С13, СН3А1С12, (СН3)2А1С1,
SnCl4, (С2Н5)3РЬС1 и (С2Н5)3РЬВг, менее активны ZnF2, HgCl2, AlBr3,
ZrCl4) [18, 36—40]. При этом устанавливается равновесие и образуется
смесь всех возможных комбинаций тетраалкильных производных свинца
(изомеризации радикалов не наблюдается). Так, смесь эквимолекулярных
количеств тетраметил- и тетраэтилсвинца дает 6,25 мол.% тетраметил-
и столько же тетраэтилсвинца, по 25% триметилэтил- и метилтриэтилсвин-
ца и 37,5 мол.% диметилдиэтилсвинца. Ароматические радикалы
подвергаются перераспределению столь же легко. Этот прием использован в синтезе
практически важных смесей тетраалкильных соединений свинца с
метальными и этильными радикалами [41—43].
В аналогичных условиях взаимодействия перераспределяют свои
радикалы смеси диэтилртути и тетраметилсвинца (содержащие эквивалентные
Диспропорционирование
599
количества метильных и этильных радикалов ртути и свинца). Из
рассмотрения состава этих равновесных систем следует, что ртуть обнаруживает
относительно большее сродство к метильным радикалам, чем к этильным по
•сравнению со свинцом [44, 45].
Своеобразное перераспределение радикалов имеет место в совершенно
иных условиях, чем описано выше (эфирно-бензольная среда, отсутствие
катализатора) [46, 47]:
(С6Н5)4РЬ + 4 /i-C4H9Li ■£ (ft-C4H9)4Pb + 4 C6H5Li.
После нагревания смеси до 60° С в течение 1 часа и карбонизации получено
75% бензойной кислоты наряду с 75% тетра-я-бутилсвинца. Бромистый
я-бутилмагний и ароматические натрийорганические соединения не вступают
в подобного рода реакцию с тетрафенилсвинцом.
Что касается относительной легкости обмена различных радикалов, то
она характеризуется, например, следующим направлением реакции [46]:
(C6H5)2Pb (CeH4CHs-/?)2 -f /1-C4H9L1 -> p-CH3C6H4Li.
Ряд реакций рассматриваемого типа между тетраэтил- и тетрафенилсвинцом
и ртутно-, магний- или алюминийорганическими соединениями был
исследован с применением меченых атомов С14 [48, 49]. Высказано
предположение [50] о связи между способностью металлоорганических соединений
обменивать радикалы и их каталитической активностью при
полимеризации непредельных соединений.
ЛИТЕРАТУРА
1. Р а з у в а е в Г. А., В я з а н к и н Н. С, Д е р г у н о в Ю. И., ЖОХ, 30, 1310 (1960).
2. Р а з у в а е в Г. А., ВязанкинН. С, Дергунов Ю. И., Дьячков-
с к а я О. С, ДАН СССР, 132, 364 (1960).
3. Р а з у в а е в Г. А., Д е р г у н о в Ю. И., В я з а н к и н Н. С, ЖОХ, 31, 998 (1961).
4. В я з а н к и н Н. С, Р а з у в а е в Г. А., Д е р г у н о в Ю. И., Труды по химии и
хим. технологии (Горький), 1961, 652.
5. Р а з у в а е в Г. А., В я з а н к и н Н. С, Дергунов Ю. И., Вышин-
скийН.Н., ЖОХ, 31, 1712 (1961).
6. М с D у е г W., С 1 о s s о п R. D., Пат. США 2571987 (1951); С. А., 46, 3556 (1952).
7. G i t t i n s Т. W., M a t t i s о n E. J., Пат. США 2763673 (1956); РЖХим., 1959, 2206.
8. CalingaertG., SoroosH, ShapiroH., J. Am. Chem. Soc, 64, 462 (1942).
9. P a 3 у в а е в Г. А., П е т у х о в Г. Г., К а п л и н Ю. А., ЖОХ, 33, 2394 (1963).
10. Р а з у в а е в Г. А., П е т у х о в Г. Г., К а п л и н Ю. А., Д р у ж к о в О. Н., ДАН
СССР, 152, 1122 (1963).
11. Кг a u seE., R ei ss a u s G. G., Вег., 55, 888 (1922).
12. К г a u s е Е., Вег., 54, 2060 (1921).
13. G i 1 m а п Н., В a i 1 i e J. С, J. Am. Chem. Soc, 61, 731 (1939).
14. A u s t i n P. R., J. Am. Chem. Soc, 53, 1548 (1931).
15. С a 1 i n g a e r t G., S о г о о s H., J. Org. Chem., 2, 535 (1938).
16. G i 1 m a n H., В a r n e t t M. M., Rec trav. chim., 55, 563 (1936).
17. H а д ь М. М., К о ч е ш к о в К. А., ЖОХ, 12, 409 (1942).
18. С а 1 i nga er t G., Sor 00s H., Sh a p i г о Н., J. Am. Chem. Soc, 62,1104 (1940).
19. С a 1 i n g a e r t G., Chem. Revs., 2, 43 (1925).
20. A u s t i n P. R., J. Am. Chem. Soc, 54, 3287 (1932).
21. P a 3 у в а е в Г. А., В я з а н к и н Н. С, Вышинский Н. Н., ЖОХ, 29, 3662
(1959).
22. Р а з у в а ев Г. А., В я з а н к и н Н. С, В ы ш и н с к и й Н. Н., ЖОХ, 30, 967
(1960).
23. Разуваев Г. А., ВязанкинН. С., Вышинский Н. Н., ЖОХ, 30, 4099
(1960).
24. А л е к с а н д р о в Ю. А., Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Труды по
химии и хим. технологии (Горький), 1959, 623.
25. А л е к с а н д р о в Ю. А., Б р и л к и н а Т. Г., Ш у ш у н о в В. А., ДАН СССР,
136, 89 (1961).
600
Свинцовоорганшеские соединения
26. А л е к с а н д р о в Ю. А., Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Труды по
химии и хим. технологии (Горький), 1960, 381.
27. Б р и л к и н а Т. Г., С а ф о н о в а М. К., Ш у ш у н о в В. А., ЖОХ, 32, 2684 (1962).
28. GilmanH., AppersonLD, J. Org. Chem., 4, 162 (1939).
29. С a 1 i n g a e r t G., S h a p i г о H., D у k s t r a F. J., H e s s L., J. Am. Chem. Soc,
70, 3902 (1948).
30. Evans D. P., J. Chem. Soc, 1938, 1466.
31. А л е к с а н д р о в Ю. А., М о к е е в а Т. И., Труды по химии и хим. технологии
(Горький), 1961, 365.
32. М с С о m b i e H., S a u n d e r s В. С, Nature, 159, 491 (1947).
33. Н е а р R., S a u n d е г s В. С, S t а с е у G. J., J. Chem. Soc. 1951, 658. .
34. К о z e s с h k о w К. A., A 1 e x a n d г о w A. P., Ber., 67, 527 (1934).
35. WillemsensL. C, van der KerkG. J. M., J. Organometal. Chem., 4, 241
(1965).
36. С a 1 i n g a e r t G., В e a t t у H. A., J. Am. Chem. Soc, 61, 2748 (1939).
37. С a 1 i n g a e r t G., В e a t t у H. A., N e a 1 H. R., J. Am. Chem. Soc, 61, 2755 (1939),
38. С a 1 i n g a e r t C, Bea tty H. A., Soroos H., J, Am. Chem. Soc, 62, 1099
(1940).
39. С a 1 i n g a e r t G., В e a t t у H. A., HessL, J. Am. Chem. Soc, 61, 3300
(1939).
40. С a 1 inga er t G., SoroosH., J.Am. Chem. Soc, 61, 2758 (1939).
41. Ari mot oF. S., Пат. США 3151142 (1964); РЖХим., 1966, 11H111.
42. ArimotoF. S., Пат. США 3151141 (1964); РЖХим., 1966, 4H110.
43. W a 1 1 H. J., Пат. США 3158636 (1964); РЖХим., 1966, 8Н111.
44. С а 1 i n g a e r t G., S о г о о s H., T h о m s о п W., J. Am. Chem. Soc, 62, 1542 (1940).
45. С a 1 i n g a e r t G., S о г о о s H., S h a p i г о H., J. Am. Chem. Soc, 63, 947 (1941).
46. G i 1 m a n H., M о о r e F. W., J. Am. Chem. Soc, 62, 3206 (1940).
47. G i 1 m a n H., M о о r e F. W., J о n e s R. G., J. Am. Chem. Soc, 63, 2482 (1941).
48. НазароваЛ.М, ЖОХ, 29, 2671 (1959).
49. НазароваЛ.М, ЖОХ, 31, 1119 (1961).
50. Н а з а р о в а Л. М., Александрова Г. Е., Высокомолек. соед., 3, 1823 (1961).
Глава X
ГИДРОЛИЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
По отношению к гидролизу свинцовоорганические соединения
напоминают соединения олова. Подобно последним они легко гидролизуются
растворами щелочи и аммиака, причем устойчивость к гидролизу
уменьшается с уменьшением числа органических радикалов у атома свинца, а также
при переходе от галогенидов к алкоксисоединениям или ацилатам свинца.
В присутствии диазометана диацилаты диарилсвинца гидролизуются водой
до диплюмбоксанов состава RCOO(Ar2PbO)2OCR.
Характерной особенностью свинцвооорганических соединений является
относительная устойчивость гидроокисей три- и диалкилсвинца.
ГИДРОЛИЗ СОЕДИНЕНИЙ КЛАССА R3PbX
Как уже отмечено на стр. 382, гидролиз оловоорганических соединений
класса R3SnX приводит или к гидроокисям триалкилолова, или к гекса-
алкилдистанноксанам. Состав продукта реакции определяется природой
алкильного радикала у атома олова. Гидролиз галогенидов триалкилсвин-
ца в этом плане остается мало изученным.
Для получения гидроокисей триалкилсвинца обычно применяют 30% -ный
раствор щелочи или влажную окись серебра. Так, обработка эфирного
раствора бромистого триметилсвинца влажной окисью серебра дает
иглообразные кристаллы гидроокиси триметилсвинца [1].
Получение гидроокиси триэтилсвинца [2]. 10,0 г (0,03 моля) хлористого триэтилсвинца
в 150 мл бензола встряхивают в делительной воронке с 56,0 г (1,4 моля) едкого натра в 84 мл
воды в течение 20 мин. Органический слой с суспендированной в нем гидроокисью
триэтилсвинца отделяют и к нему прибавляют 150 мл бензола, нагретого до 70° С. Смесь встряхивают
(примерно 30 сек.) и быстро фильтруют. По охлаждении льдом выделяются белоснежные
кристаллы гидроокиси триэтилсвинца. Бензол декантируют и используют для дальнейшей
экстракции. Операцию повторяют до полного растворения гидроокиси триэтилсвинца.
Осадок промывают гексаном и растворившуюся в бензоле и гексане часть вещества выделяют
выпариванием раствора в вакууме при 50—60° С до 100 мл и охлаждением его ледяной
водой. Выход 78%.
Гидролиз галогенидов триэтилсвинца можно проводить также в эфире
[2—7], но в этом случае гидроокись триэтилсвинца содержит примесь
исходного свинцовоорганического галогенида [2].
С металлическим натрием гидроокись триэтилсвинца образует гекса-
этилдиплюмбоксан [8,9]. Интересно отметить, что дегидратация
гидроокиси триэтилолова проходит уже при ее высушивании над
концентрированной серной кислотой (см. стр. 383).
Гидроокиси три-я-пропил-, три-я-бутил-, триизобутил-, три-я-амил-
и триизоамилсвинца получены [1, 10] обработкой хлористого, бромистого
602 Свинцовоорганические соединения. Лит. стр. 604
или йодистого триалкилсвинца в эфире 30%-ной щелочью. В отличие от
этильного соединения, образующиеся гидроокиси не выпадают в осадок, а
остаются в виде раствора в эфирном слое. Отмечено [10—12], что в ряде
случаев вместо щелочи целесообразнее применять влажную окись серебра.
Полученные таким образом водные растворы гидроокисей триалкилсвинца
использованы в дальнейших реакциях без дополнительной очистки [10].
При встряхивании бензольного раствора йодистого трициклогексил-
свинца с 30%-ным едким кали выпадает белый аморфный порошок
гидроокиси трициклогексилсвинца [13]. Аналогично получают гидроокись
трифенилсвинца [14—18]. К раствору 1,89 г хлористого трифенилсвинца в 40 мл
этилового спирта прибавляют 0,4 г едкого кали в 4 мл воды. Смесь кипятят
в течение 3 час. и фильтруют горячей. По охлаждении выделяется
гидроокись трифенилсвинца с т. разл. 300—310° С. Выход количественный [18].
Известна и гидроокись три-п-толилсвинца [1].
Обработка гидроокиси трифенилсвинца в бензоле натрием приводит к
гексафенилдиплюмбоксану.
Получение гексафенилдиплюмбоксана [19]. Синтез вещества проводят при полном
отсутствии влаги воздуха. В круглодонную колбу емкостью 0,5 л помещают 20—25 мл
очищенного от окисной пленки натрия и 100—150 мл сухого нона на. Содержимое колбы нагревают
на масляной бане до температуры плавления натрия. Путем энергичного встряхивания
диспергируют расплавленный металл, после чего декантируют основную часть жидкости, натрий
промывают сухим гексаном, последний сливают и остатки жидкости отгоняют при
30—40° С и пониженном давлении (0,2—0,3 мм). Оставшийся в колбе натрий заливают сухим
бензолом и сюда же вводят 5—10 г гидроокиси трифенилсвинца. Колбу с реакционной
смесью энергично встряхивают при обычной температуре в течение 20—30 мин. При этом
малорастворимая в бензоле при комнатной температуре гидроокись трифенилсвинца
превращается в хорошо растворимый гексафенилдиплюмбоксан. Смесь фильтруют через
плотный стеклянный фильтр и из фильтрата при пониженном давлении и обычной температуре
отгоняют бензол. Получают хорошо образованные пластинчатые кристаллы
гексафенилдиплюмбоксана.
Гексафенилдиплюмбоксан — белое кристаллическое вещество, хорошо
растворимое в толуоле и ксилоле. Он легко гидролизуется водой до
гидроокиси трифенилсвинца. Гидролиз лучше всего проводить в водном спирте
или водном диоксане, так как в этих растворителях хорошо растворяется
как исходное, так и образующееся соединение.
ГИДРОЛИЗ СОЕДИНЕНИЙ КЛАССА R2PbX2
Согласно литературным данным [2, 10, 20—22], обработка дигалогенидов
диалкилсвинца щелочью или влажной окисью серебра приводит к дигидро-
окисям диалкилсвинца. Так получена, например, дигидроокись диэтилсвин-
ца. Вещество хорошо растворяется в щелочи [2], поэтому гидролиз проводят
влажной окисью серебра:
(СзНэЬРЬСЬ + 2 AgOH -* (C2H5)2Pb (OH)2 + 2 AgCl.
Дигидроокись диэтилсвинца выделена и в виде гексагидрата (С2Н5)2РЬ(ОН)2 •
• 6Н20. Последний дегидратируется уже при обычной температуре,
превращаясь в полимерную окись диэтилсвинца [2]. Дигидроокись
диэтилсвинца, подобно другим диалкильным соединениям свинца, легко
разлагается при нагревании. В отсутствие растворителей и воздуха разложение
протекает со взрывом [22].
Обработка щелочью соединений типа Аг2РЬХ2 приводит к полимерным
окисям (Аг2РЬО)„ [23—25]. Последние представляют собой нерастворимые
неплавкие порошки белого цвета. Их строение не изучалось.
Гидролиз
603
Свинцовоорганические ацилаты гидролизуются гораздо легче
соответствующих галогенидов. При хранении диацилатов диарилсвинца на воздухе
их температура плавления снижается, появляются примеси, не
растворимые в органических растворителях [26]. Водой гидролизуется уже
значительная часть свинцовоорганического соединения. Например, из смеси
ацетона с водой 15—20% вещества постепенно выделяется в виде (гидрокси)-
ацетата дифенилсвинца — (СбН5)2РЬ(ОН)ООССН3 [27]. При гидролизе
Ar2Pb(OOOCR)2 рассчитанным количеством воды в присутствии диазометана
это вещество образуется только в качестве промежуточного продукта.
Реакция идет дальше с отщеплением воды и образованием соединений с плюмбо-
ксановой связью [27—29]:
Ar2Pb(OOCR)2 + Н20 + 2 CH2N2 -» RCOOPbAr2OPbAr2OCCR -J- 2RCOOCH3 r 2 N2.
Реакция проходит так же, как и в случае оловоорганических соединений
(см. стр. 387). Она начинается с гидролиза диацилата диарилсвинца.
Диазоалкан не участвует в'построении конечного продукта, а лишь
связывает выделяющуюся в результате гидролиза кислоту. Гидролиз диацетата
или диизобутирата дифенилсвинца в присутствии диазометана, диазоэтана
или диазобутана приводит к соединениям одного и того же состава.
Получение со, ю-диацетата тетрафенилдиплюмбоксана [27]. К раствору 2,88г (0,06 моля)
диацетата дифенилсвинца в 150 мл ацетона при перемешивании приливают несколько
капель воды (около 0,1 мл у 0,006 моля) и частями в течение 5 мин. 0,504 г (0,021 моля)
диазометана в 14 мл эфира. Диазометан быстро обесцвечивается, выделяется газ. Если в
растворителях имеется вода в количестве, достаточном для гидролиза, то дополнительное ее
введение излишне. После прибавления 2 молей диазометана на 1 моль диацетата дифенилсвинца
желтый цвет раствора сохраняется в течение продолжительного времени. Через несколько
минут после начала реакции на стенках колбы появляются первые кристаллы осадка. Через
несколько часов его отфильтровывают, промывают ацетоном и высушивают в вакууме.
Выход 2,06 г (96%); т. пл. 218—219° С (с разложением).
Аналогично получены [27—29] диизобутират, дипропионат, димонохлор-
ацетат и дибензоат тетрафенилдиплюмбоксана, а также диацетаты тетра-Р-
нафтил- итетра-/г-анизилдиплюмбоксана. со, со-Диацилаты тетраарилдиплюм-
боксана представляют собой белые кристаллические вещества с
температурой разложения выше 200° С. Они не растворимы в обычных органических
растворителях, за исключением хлороформа.
ГИДРОЛИЗ СОЕДИНЕНИЙ КЛАССА RPbX3
Свинцовоорганические соединения типа RPbX3 в алифатическом ряду
неустойчивы и легко разлагаются. Имеющиеся в литературе [30] данные об
алкил-, аллил- и бензилплюмбоновой кислоте нуждаются в дополнительной
проверке.
Что касается соединений типа ArPb(OOCR)3, то в отношении гидролиза
они больше напоминают диацилаты диарилсвинца, чем соли
четырехвалентного свинца. Триацилаты арилсвинца гидролизуются влагой воздуха, точка
плавления снижается и становится нечеткой, что говорит об изменении
химического состава. При прибавлении воды к раствору, например,
триацетата фенилсвинца в ацетоне происходит частичный гидролиз. Он идет полнее
в присутствии диазометана, который связывает образующуюся в результате
гидролиза кислоту. Из раствора выделяется не растворимое в обычных
органических растворителях соединение, анализ которого отвечает формуле
lArPb(0)OOCCH3U31].
Водным аммиаком соединения типа ArPb(OOCR)3 гидролизуются
полностью с образованием арилплюмбоновых кислот [32—36]. Реакция
протекает очень легко: спиртовый раствор свинцовоорганического соединения
604
Свинцовоорганические соединения
выливают в 5%-ный раствор аммиака. Выпавший осадок отделяют,
промывают водой, спиртом, эфиром и высушивают.
{ArPb(OOCR)8 + 3JNH4OH -» (АгРЬООН)л + 3]RCOONH4.
Арилплюмбоновые кислоты представляют собой неплавкие
нерастворимые порошки слегка желтоватого цвета. Они имеют, вообще говоря, амфо-
терный характер. Как основания, эти соединения легко растворяются в
минеральных и органических кислотах. Более слабо выражен кислотный
характер арилплюмбоновых кислот, которые не растворяются в соде,аммиаке и
относительно трудно растворяются в щелочах. Например,
свежеприготовленная фенилплюмбоновая кислота растворяется в 15—20%-ной щелочи, а
после высушивания — лишь в 40—50%-ной щелочи [33]. При длительном
высушивании арилплюмбоновых кислот возможна их ангидризация
[33, 35,36].
ЛИТЕРАТУРА
1. Кг a use E., Pohland E., Вег., 55, 1282 (1938).
2. С а 1 i п g a e r t G., D у k s t r a F. J., Shapiro H., J. Am. Chem. Soc, 67,
190 (1945).
3. BucktonG. В., Ann., 112, 220 (1859).
4. К 1 i p p e 1 J., Jahresber., 1860, 380.
5. LowigC, Ann., 88, 318 (1853).
6. CahoursA, Ann., 122, 48 (1862).
7. M i d g 1 e у Т., Н о с h w a It С. А., С а 1 i n g a e r t G., J. Am. Chem. Soc, 45, 1821
(1923).
8. Александров Ю. А., БрилкинаТ. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1960, 381.
9. Александров Ю. А., БрилкинаТ. Г., Шушунов В. А., ДАН СССР*
136, 89 (1961).
10. S a u п d е г s В. С, S t а с е у G. J., J. Chem. Soc, 1949, 919.
11. К 1 i р р е U. J., prakt. Chem., 81, 287 (1860).
12. Р f е i f f е г P., T r u s k i e r P., Dissel kapmP., Ber., 49, 2445 (1916).
13. KrauseE, Ber., 54, 2060 (1921).
14. Bahr G., Z. anorgan. allgem. Chem., 253, 330 (1947).
15. LieberE., К e a n e F. M., Chem. a. Ind., 1961, 747.
16. GriittnerG., Ber., 51, 1298 (1918).
17. L i e b e r K., R а о С N. R., К e a n e F. M., J. Inorg. Nucl. Chem., 25, 631 (1963).
18. West R.,B aney R. H.,Powel 1 D. L.,J. Am.Chem.Soc,82,6269(1960).
19. Б р и л к и н а Т. Г., С а ф о н о в а М. К., Ш у ш у н о в В. А., ЖОХ, 32, 2684 (1962).
20. Н е а р R., S a u n d e r s В. С, S t а с е у G. J., J. Chem. Soc, 1951, 658.
21. С а 1 i n g a er t G., S h a p i г о Н., D у k s t r a F. J., H es s L., J. Am. Chem. Soc,
70, 3902 (1948).
22. Александров Ю. А., Брилкина Т. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1959, 623.
23. Р о 1 i s A., Ber., 20, 332 (1887).
24. Z е с h m e i s t e r L., С s a b а у J., Ber., 60, 1617 (1927).
25. Коч к и н Д. А., Л у кь я н о в а А. В., Р е з н и к о в а Е. Б., ЖОХ, 33, 1945,
(1963).
26. К о ч е ш к о в К- А., ПановЕ.М, Изв. АН СССР, ОХН, 1955, 718.
27. П а н о в Е. М., 3 е м л я н с к и й Н. Н., К о ч е ш к о в К. А., ДАН СССР, 143, 603
(1963).
28. К о ч е ш к о в К. А., П а н о в Е. М., 3 е м л я н с к и й Н. Н., Изв. АН СССР, ОХН,
1961, 2255.
29. 3 е м л я н с к и й Н. Н., ПановЕ. М., Л о д о ч н и к о в а В. И., К о ч е ш-
к о в К. А., ЖОХ, 35, 843 (1965).
30. LesbreM, Cornpt. rend., 200, 559 (1935).
31. Л о д о ч н и к о в а В. И., Диссертация. М., МГУ, 1964.
32. П а н о в Е. М., К о ч е ш к о в К. А., ДАН СССР, 85, 1037 (1952).
33. П а н о в Е. М., К о ч е ш к о в К. А., ДАН СССР, 85, 1293 (1952).
34. К о ч е ш к о в К. А., П а н о в Е. М., Изв. АН СССР, ОХН, 1955, 711.
35. Л о д о ч н и к о в а В. И., ПановЕ.М., К о ч е ш к о в К. А., Изв. АН СССР,
ОХН, 1957, 1484.
36. Л-о д о ч н и к о в а В. И., ПановЕ.М., К о ч е ш к о в К. А.» ЖОХ, 33. 1199
(1963).
Г лава XI
РЕАКЦИИ ПРЕОБРАЗОВАНИЯ ФУНКЦИОНАЛЬНЫХ ГРУПП
У АТОМА СВИНЦА
К преобразованию функциональных групп у атома свинца приводят те
же реакции, которые используются в ряду оловоорганических соединений.
Так, при прибавлении минеральных или органических кислот к растворам
свинцовоорганических гидроокисей в воде или спирте образуются
соответствующие соединения типа R3PbX или R2PbX2. Этим методом получены,
например, галогениды три-[1—5] и диалкилсвинца [6], сульфаты трипропил-
[4] и ди-я-бутилсвинца [6], арилсульфонаты триалкилсвинца [5], нитрат
трифенилсвинца [71, азиды три- и дифенилсвинца [8—10], ацилаты три-
[2,11—14] и диалкилсвинца [5]. Гидроокись трифенилсвинца использована
для аналитических целей при разделении различных анионов [15]. Следует
отметить, что иногда реакция между органическими кислотами и
гидроокисями триалкилсвинца сопровождается деалкилированием свинцовоорганиче-
ского соединения. Например, никотиновая, /г-хлорбензойная и ж-нитро-
бензойная кислота превращает гидроокись триэтилсвинца в
соответствующие диацилаты диэтилсвинца [11]. В ряде случаев галогениды и ацилаты типа
R3PbX в алифатическом ряду целесообразнее получать реакцией деалкили-
рования (см. стр. 578, 581).
Что касается реакции гидроокисей триарилсвинца с органическими
кислотами, то она имеет важное препаративное значение. В качестве типичного
примера можно указать на получение изобутирата трифенилсвинца
смешением эквимолекулярных количеств гидроокиси трифенилсвинца и изомасля-
ной кислоты в спирте. Полученное вещество кристаллизуется в виде
бесцветных иголочек ст. пл. 188—190° С [16]. Нагревание в спирте в течение часа
5,5 2 гидроокиси трифенилсвинца с 1,84 г я-винилбензойной кислоты дает
д-винилбензоат трифенилсвинца. Выход 51,7%, т. пл. 136—138°С(из гекса-
на) [17]. Аналогично получены [17—26] и другие ацилаты трифенилсвинца.
Окиси диарилсвинца [16, 23] и арилплюмбоновые кислоты [27—31]
при нагревании с органическими кислотами превращаются с хорошим
выходом в соответствующие ацильные производные.
Помимо кислот, свинцовоорганические гидроокиси легко реагируют с
меркаптанами [12, 32], фтальимидом [12] и арилсульфамидами [12, 13, 33],
но они инертны по отношению к фенилнитрометану, оксимам и спиртам [12].
Напротив, гексаэтилдиплюмбоксан активно реагирует со спиртами [34,35]:
(С2:-15)3РЬОРЬ (С2Н5)з + ROH -> (С2Н5),РЬОН + (C2H5)3PbOR
(R = CH3, QH5, C0H5CH2f (СНзЫСбТЦС).
Аналогично ведет себя гексаэтилдиплюмбоксан по отношению к перекисям
трет-бутла и а-кумила [34, 36]:
(С2Н5)зРЬОРЬ(С2Н5)з + ROOH -> (C2H6)3PbOOR + (QH^PbOH.
606
Свинцовоорганические соединения. Лит. стр. 610—611
Обе реакции проходят с большой скоростью уже при — 10° С [34—36].
Гидроокись трифенилсвинца образует с перекисями неустойчивые соединения,
которые сразу же перегруппировываются с выделением фенола и окиси ди-
фенилсвинца [36, 37].
Второй метод, приводящий к преобразованию функциональных групп у
атома свинца, заключается в обмене свинцовоорганических галогенидов с
солями (алкоголятами, меркаптидами и т. д.) калия, натрия, серебра или
свинца. Этот метод используют при получении свинцовоорганических
фторидов [3], иодидов [38, 39], цианидов [12, 40], цианатов, тио- и селеноциа-
натов [12, 40] и тиогликольамидов [41].
Получение сульфида б'ас-(триэтилсвинца) [38]. К охлажденному до 0° С раствору 13,9 г
(0,04 моля) хлористого триэтилсвинца в 700 мл воды прибавляют по каплям 10,6 г (0,04
моля) Na2S • 9Н20 в 350 мл воды. Выделившееся масло растворяют в эфире, фильтруют, эфир
выпаривают и операцию повторяют. В остатке получают маслянистую жидкость
желто-зеленого цвета, с ^э2,05; п ^ 1,6249. Вещество легко окисляется на воздухе, превращаясь
в соответствующий сульфат.
Реакция между хлористым трифенилсвинцом и сульфидом натрия
проходит своеобразно и приводит к натрийтрифенилплюмбилсульфиду
(C6H5)3PbSNa [32]. Это неустойчивое вещество легко разлагается даже
при температуре порядка 30 °С:
2 (C6H5)3PbSNfa -> (C6H)>PbSPb(G8H5)3 + Na2S.
С йодистым метилом натрийтрифенилплюмбилсульфид образует меркаптид
трифенилсвинца. Более удобный способ синтеза свинцовоорганических мер-
каптидов основан на реакции:
2 (С6Н5)3РЬС1 + Pb(SR)2 -* 2 (C6H5)3PbSR + РЬС12.
Выходы продуктов близки к количественным. Полученные соединения
охарактеризованы элементарным анализом и химическими реакциями.
Получение я-пропилмеркаптида трифенилсвинца [32]. 4,86 г хлористого
трифенилсвинца и 1,79 г ди-«-пропилмеркаптида свинца нагревают при перемешивании в 100 мл бензола
в течение 3 час. Выделившийся осадок двухлористого свинца отфильтровывают, фильтрат
выпаривают и остаток кристаллизуют из спирта. Получают 4,84 г (95%) вещества с т. пл.
57—58° С.
Для синтеза свинцовоорганических ацилатов алифатического ряда
реакции обмена галоидопроизводных свинцовоорганических соединений
применяются сравнительно редко [5,12,14], так как образующиеся ацильные
производные свинца с трудом отделяются от исходных галогенидов [5].
Более гладко эти реакции проходят в ароматическом ряду. Например,
ацетат три-/г-толилсвинца получают [20] смешением спиртового раствора
хлористого три-/г-толилсвинца с избытком водного раствора диацетата свинца.
Выпавший осадок двухлористого свинца отделяют, из разбавленного
фильтрата кристаллизуется ацетат три-/г-толилсвинца (т. пл. 161—162°С):
2 (я-СН3С6Н4)зРЬС1 4- РЬ(ООССЧ3)2 -* 2 (л-СН3СвЧ4)зРЬОЭССНз + РЬС12.
Соединения типа R3PbOOR' получают взаимодействием
свинцовоорганических галогенидов с натриевыми солями алкильных гидроперекисей [42,
43]. Последние лучше готовить из гидроперекисей и амида натрия:
R'OOH + NaNH2 -* R'OONa -f- NH3,
R3PbX + R'OONa-* R3Pb03R' + NaX.
В ароматическом ряду приводит к цели и смешение бромистого
трифенилсвинца, гидроперекиси и амида натрия [42]:
(С6Н6)3РЬВг + ROOH + NaNH2 -* (C6H3)3PbOOR + NaBr + NH3.
Реакции преобразования функциональных групп
607
Энергичная реакция наблюдается при взаимодействии хлористого три-
этилсвинца с натриевыми производными нитрометана, фенилнитрометана
или метилатом натрия в среде метилового спирта или бензола. Однако
выделить из смеси индивидуальные соединения не удается [12]. Что касается
свинцовоорганических алкоксисоединений, то они были получены
взаимодействием соответствующих галогенидов с алкоголятами натрия в эфире, тетра-
гидрофуране или бензоле [44, 44а, 45]:
R3PbBr + R'ONa -* R3PbOR' + NaBr.
Так получены [44] метокситриметил-, метокситриэтил-, метокситри-я-
пропил-, метокситри-я-бутил и метокситрифенилсвинец. Метокситриал-
кильные производные свинца, кроме метокситриметилсвинца, хорошо
растворяются в обычных органических растворителях. Определение
молекулярного веса в бензоле показывает, что в растворе они мономерны. В то же
время исследование их ИК-спектров указывает на ассоциацию этих веществ в
твердом состоянии. Следует отметить, что метокситриметилсвинец по своим
свойствам существенно отличается от других метокситриалкильных
производных свинца. Так, его температура разложения (250—260° С) гораздо
выше, чем у других членов гомологического ряда (50—60° С).
Метокситриметилсвинец растворим только в полярных средах (хлороформ, метиловый
спирт, несколько труднее в тетрагидрофуране). В хлороформе он
ассоциирован.
Получение метокситриэтил-, метокситри-я-пропил- и метокситри-я-бутилсвинца [44].
Синтез метокситриалкильных соединений свинца проводят при полном отсутствии влаги
воздуха. К избытку метилата натрия в эфире или тетрагидрофуране прибавляют за один раз
хлористый триалкилсвинец. Смесь кипятят при перемешивании в течение 10—24 час. и
фильтруют. Осадок промывают эфиром, из фильтрата растворители отгоняют и остаток перегоняют
или возгоняют в вакууме (10~2 мм) при 50—80° С (для метокситриэтилсвинца), 45—65° С
(для метокситри-я-пропилсвинца), или 70—100° С (для метокситри-я-бутилсвинца). При
проведении реакции в тетрагидрофуране выход метокситриэтилсвинца 93%, метокситри-я-
пропилсвинца — 81%. При замене тетрагидрофурана эфиром выход этильного соединения
составляет 59%, я-нропильного — 70% и метокситри-я-бутилсвинца—91%.
Свинцовоорганические соединения со связью РЬ — N остаются пока
мало изученными. Взаимодействием хлористого триметил- [46] или трифе-
нилсвинца [9, 47] с азидом натрия получены свинцовоорганические азиды.
Отмечено [9], что азид трифенилсвинца лучше получать из гидроокиси
трифенилсвинца и азотистоводородной кислоты.
При нагревании в бензоле хлористого триметилсвинца с бис-(триметил)-
силил амидом натрия образуется триметилплюмбил-бш>(триметилсилил)-
амин [48]:
(СНз)зРЬС! }- NaN[Si(CH3)8]2 -» (CH3)3PbN[Si (CH3)3]2 + NaCl.
Вещество представляет собой бесцветную жидкость с т. кип. 85—87° С/3 мм.
К соединениям со связью РЬ—N приводит также реакция
свинцовоорганических галогенидов с натриевыми солями арилсульфамидов
[13, 14, 33]:
ArS02NHNa + R3PbCl -> ArS02NHPbR3 + NaCl.
Арилсульфамидотриалкильные соединения свинца хорошо растворяются
в обычных органических растворителях.
Получение я-толуолсульфамидотриэтилсвинца [33]. 3,4 г я-толуолсульфамида
растворяют в спирте и к раствору прибавляют 0,5 г натрия. Выделившийся осадок
отфильтровывают, промывают спиртом и высушивают. К суспензии этой соли (1,93 г) в спирте
прибавляют 3,3 г хлористого триэтилсвинца и смесь кипятят в течение 20 мин. По охлаждении смесь
фильтруют и из фильтрата продукт осаждают водой. Осадок отделяют, промывают водой и
кристаллизуют из бензола. Чистое вещество имеет т. пл. 127° С.
1э08
Свинцовоорганические соединения. Лит. стр. 610—611
Аналогично получены [33] фенилсульфамидотриэтилсвинец (т. пл. 132° С),
я-толуолсульфамидотри-я-пропилсвинец (т. пл. 100—101° С) и бифенилсуль-
фамидотри-я-пропилсвинец (т. пл. 96° С).
Значительный интерес представляют биэлементоорганические
соединения, в которых атом свинца связан с атомами рения [49], вольфрама [50],
молибдена [50], кобальта [51, 52], марганца [53] или железа [54—57]. Их
получают взаимодействием свинцовоорганических галогенидов с
натриевыми производными карбонилов-соответствующих металлов:
R3Pb XJ+ NaM (CO)„ ->eR3PbM (CO)^+NaX.
Синтез и реакции этих веществ рассмотрены в главе XV (см. стр. 629).
Известны и соединения со связями РЬ—О—Si [58—60], Pb—О—Р
[61, 62] или РЬ—О—As [63]. Так, смешением эквимолекулярных количеств
бромистого тримети л свинца и триметилсиланолята натрия в эфире при
обычной температуре получен триметилсилокситриметилсвинец (т. кип.
73° С/10 мм) [58]:
(СИз)зРЬВг +KCH3)3SiONa -* (СН3)зРЬ051(СН3)з + NaBr.
Аналогично получены триэтилсилокситриэтилсвинец (т. кип. 100° С/1 мм),
триметилсилокситриэтилсвинец (т. кип. 111,5°С/10 мм) и триэтилсилокси-
триметилсвинец(т. кип. 65С71 мм). Алкилплюмбосилоксаны — чрезвычайно
реакционноспособные бесцветные подвижные жидкости с резким
неприятным запахом. При работе с ними необходимо учитывать высокую
токсичность этих соединений [58]. С фторфосфонатом серебра хлористый триэтил-
свинец образует бш>(триэтилплюмбил)фторфосфонат— [(C2H5)3PbO]2P(0)F
(т. пл. 260°С) [61]. С триэтилфосфитом бромистый триэтил- и трифенилсвинец
не* реагируют. При нагревании смеси до температуры кипения наблюдает-
ся'только диспропорционирование исходного свинцовоорганического
соединения [64].
Взаимодействием натриевой соли диметиларсиновой кислоты с хористам
трифенилсвинцом получен (С6Н5)зРЬОАя(0)(СН3)2. Нагревание той же
соли с двухлористым дифенилсвинцом ведет к (C6H5)2Pb[OAs(0)(CH3)2]2
или (C6H5)2Pb(Cl)[OAs(0)(CH3)2] [63].
Вариантом рассматриваемого метода является получение нерастворимых
в воде свинцовоорганических соединений из растворимых гидроокисей,
ацетатов или нитратов обменом с соответствующей солью калия или натрия.
Так, цианид три-я-пропилсвинца [5], фульминаты трипропил- и трифенил-
свинца [65] получены прибавлением натриевых солей к водному раствору
свинцовоорганической гидроокиси:
R3PbOH + NaCNO -^ R3PbCNOi-f NaOH.
Из динитратов диарилсвинца и галогенидов натрия получены двубромистый
дифенил- [66, 67] и двуиодистый ди-л*-нитрофенилсвинец [7]. С цианистым
калием динитрат дифенилсвинца образует (C6H5)2Pb(OH)(CN) [68, 69],
с фосфорнокислым натрием—[(С6Н5)2РЬ]з(Р04)2, с содой образуется
[(СбНб)2РЬОН]2С03, с хроматом калия — (С6Н5)2РЬСг04 [70]. Обменом
диацетата дифенилсвинца с роданистым калием получен дитиоцианат
дифенилсвинца, с сероводородом — сульфид дифенилсвинца [68, 71].
Тот же способ используется и для синтеза свинцовоорганических ацила-
тов. Например [11]:
(СзН3),РЬОЭССН3 + RCOONa -> (C2H;))3PbOOCR -f CH3C03Na.
Водный раствор натриевой соли органической кислоты смешивают с
водным же раствором ацетата триэтилсвинца. Почти сразу же выпадает осадок
соответствующего ацилата триэтилсвинца.
Реакции преобразования функциональных групп
609
Для синтеза свинцовоорганических ацилатов, трудно доступных иными
путями, применяют реакцию переацидирования. Эта реакция подробно
разработана в ряду соединений типа R2PbX2 [27, 72, 73] и АгРЬХ3 [28, 29,
74—76]. Например, при прибавлении органических кислот к спиртовому
раствору диацетата дифенилсвинца получены [27, 72] дистеарат (т. пл.
100—ЮГ С), диметакрилат (т. разл. 205° С), димонохлорацетат (т. разл.
185° С) и дибензоат дифенилсвинца (т. пл. 240° С). Прибавлением спиртового
раствора хлористого водорода к спиртовому же раствору диацетата ди-|3-
хлорвинилсвинца получен [77] двухлористый ди-Р-хлорвинилсвинец (т. разл.
163—167°С). В тех же условиях серная кислота превращает диацетат ди-|3-
хлорвинилсвинца в сульфат (т. разл. 171—172°С), а бензойная — в
дибензоат ди-Р-хлорвинилсвинца (т. разл. 204—204,5°С). Спиртовый раствор
хлористого водорода легко вытесняет остатки органической кислоты и из
других свинцовоорганических ацилатов [14, 78, 79].
Что касается соединений типа ArPb(OOCR)3, то по своим реакциям,
например с йодистым калием, они сходны с тетраацилатами свинца. Однако
если тетраацетат свинца реагирует с йодистым калием с одновременным
выделением двух эквивалентов иода, то с ArPb(OOCR)3 реакция протекает
более сложно и свободный иод выделяется в весьма незначительном
количестве. Основное направление этой реакции описывается следующими
уравнениями
ArPb(OOCR)3 + 3KJ-* ArPbJ3 + 3RCOOK,
ArPbJ3 -» ArJ + PbJ2.
Как уже отмечено выше, триацилаты арилсвинца легко вступают также в
реакцию переацидирования.
Получение трибензоата фенил свинца [28]. 0,55 г (1 моль) триизобутирата фенилсвинца
вносят в горячий раствор 0,44 г (1 моль + 20% избытка) бензойной кислоты в 8 мл гексана.
Раствор нагревают до кипения и оставляют для кристаллизации до следующего дня.
Выпавший осадок отсасывают, промывают последовательно горячим гексаном, спиртом, эфиром.
Осадок (0,4 г) перекристаллизовывают из 1,5 мл сухого бензола, промывают и высушивают
в вакууме при 61° С. Выход 0,27 г (41,5%); т. пл. 149,5—151° С. Трибензоат фенилсвинца —
белое кристаллическое вещество, на холоду очень хорошо растворяется в хлороформе,
хорошо в этилацетате и бензоле, в спирте умеренно растворяется на холоду и хорошо — при
нагревании. В лигроине и гексане не растворяется.
Аналогично получены трибензоат а-нафтилсвинца (т. пл. 173—174° С)
[29], триметакрилат я-толилсвинца [74], трибензоат /г-анизилсвинца (т. пл.
178—179°С) [30] и трипропионат иодфенилсвинца (т. пл. 84—85° С) [76]
К преобразованию функциональных групп у атома свинца приводят и
некоторые другие реакции. Например, фосфат, формиат и ацетат
триэтилсвинца получены при действии соответствующей кислоты на карбонат
триэтилсвинца. Тот же способ применен и для синтеза диацилатов
свинца [13]:
(C2H5)2PbS03 + 2 СН2С1СООН -* (QHs^Pb (ООССН2С1)з + S02 + Н20.
При получении свинцовоорганических перекисей в качестве
исходных веществ применяют алкоксисоединения свинца [42]:
R3PbOR' + R"OOH-* R3PbOOR" + R'OH,
(CeH5)8PbOR + H202-> (C6H5)3Pb—OO—Pb(C6H5)8 + ROH.
Двухлористый дифенилсвинец с 8-оксихинолином в метиловом спирте
образует бш>(8-оксихинолят) дифенилсвинца [81]. Аналогично получен
бис-(8-оксихинолят) дибутилсвинца и соединения с двумя различными груп-
39 Заказ № 207
610
Сви нцовоорганические соединения
нами у атома свинца: (C4H9)2PbBr(C9H6NO), (C4H9)2Pb(OOCCH3) (C9HeNO),
(C6H6)2Pb(Cl)(C9H6NO) или (C6H5)2Pb(OOCCH3)(C9H6NO).
В присутствии акцепторов галоидоводородов свинцовоорганические
галогениды вступают в реакции с меркаптанами [82] и фосфинами [83].
Получение этилмеркаптида триэтилсвинца [82]. 4,4 г (0,07 моля) этилмеркаптана и 17,8 г
(0,06 моля) хлористого триметилсвинца встряхивают в водном растворе едкого кали в
течение часа. Выделившееся тяжелое масло высушивают сульфатом магния и перегоняют в
вакууме. Получают 10,2 г (53%) вещества с т. кип. 36° С/0,05 мм; п2£ 1,5918.
При прибавлении триэтиламина к смеси хлористого трифенилсвинца и
дифенилфосфина в бензоле сразу же выделяется осадок солянокислого
триэтиламина [83]:
(С6Н5)3РЬС1 + (С6Н5)2РН + (C2H5)3N -* (С6Н5)3РЬР (СбН5)2 + (CaH5)3N.HCl.
Образующиеся соединения легко окисляются на воздухе и работать с ними
необходимо в атмосфере сухого инертного газа. Водно-спиртовым
раствором перекиси водорода они количественно окисляются до соответствующих
фосфонатов:
(С6Н5)3РЬР (С6Н5)2 + 02 -* (СбН5)8РЬОР (О) (С6Н5)2.
£ис-(дифенилфосфинат) дифенилсвинца, полученный аналогично, очень
неустойчив и выделить его в индивидуальном состоянии не удается [83].
ЛИТЕРАТУРА
1. LowigC, Ann., 88, 318 (1853).
2. К 1 i p p e 1 J., J. prakt. Chem. 81, 287 (1860).
3. KrauseE., PholandE., Ber., 55, 1282 (1922).
4. P f e i f f e r P., T r u s k i e r P., D i s s e 1 k a m p. P., Ber., 49, 2445 (1916).
5. S a u n d e r s В. С, S t а с е у G. J., J. Chem. Soc, 1949, 919.
6. JonesW.J., E v a n s D. P., G u 1 w e 1 1 Т., G r i f f i t h s D. С, J. Chem. Soc,
1935 39.
7. Gil'manH., LeeperR. W., J. Org. Chem., 16, 466 (1951).
8. LieberE., К e a n e F. M., Chem. a. Ind., 1961, 747.
9. ThayerJ.S., WestR, Inorg. Chem., 3, 406 (1964).
10. LieberE., RaoCN.R., К e a n e F. M., J. Inorg. Nucl. Chem., 25, 631 (1963).
11. Gi 1 m an H., S p at zS. M., К о 1 b e z e n M. J. J., Org. Chem., 18, 1341 (1953).
12. H e a p R., S a u n d e r s В. С, J. Chem. Soc, 1949, 2983.
13. McCombieH., Saunders В. С, Nature, 159, 491 (1947).
14. H e a p R., SaundersB.C, S t а с е у G. J., J. Chem. Soc, 1951, 658.
15. SchweitzerG.K., M с С a r t у S. W., J. Inorg. Nucl. Chem., 27, 191 (1965).
16. КотонМ.М., ФлоринскийФ.С, ЖОХ, 32, 3057 (1962).
17. К о т о н М. М., К и с е л е в а Т. М., Изв. АН СССР, ОХН, 1961, 1783.
18. К и с е л е в а Т. М., КотонМ.М., Ч е т ы р к и н а Г. М., Изв. АН СССР, ОХН,
1962, 1798.
19. A u s t i n P. R., J. Am. Chem. Soc, 53, 3514 (1931).
20. A u s t i n P. R., J. Am. Chem. Soc, 53, 1548 (1931).
21. К о ч к и н Д. А., Ч е к м а р е в а И. Б., ЖОХ, 31, ЗОЮ (1961).
22. Кочки н Д. А., Л у кь я нов а Л. В., Р е з н и к о в а Е. Б., ЖОХ, 33, 1945
(1963).
23. К о ч к и н Д. А., ДАН, СССР, 135, 857 (1960).
24. К о ч к и н Д. А., ЧекмареваИ. Б., Труды Всесоюзн. н.-и. витамин, ин-та, 7,
37 (1960).
25. КочкинД. А., ВереникинаС. Г., Труды Всесоюзн. н.-и. витамин, ин-та, 8,
39 (1961).
26. К о ч к и н Д. А., В е р е н и к и н а С. Г., Ч е к м а р е в а И. Б., ДАН СССР, 139,
1375 (1961).
27. П а н о в Е. М., К о ч е ш к о в К. А., ДАН СССР, 85, 1293 (1952).
28. К о ч е ш к о в К. А., П ановЕ.М., Изв. АН СССР, ОХН, 1955, 711.
29. Л од оч н и ко в а В. И., ПановЕ.М., К о ч е ш к о в К. А., Изв. АН СССР,
ОХН, 1957, 1484.
Реакции преобразования функциональных групп
611
80. Л о д о ч н и к о в а В. И., П а н о в Е. М., К о ч е ш к о в К. А., ЖОХ, 33, 1199 (1963).
31. Л о д о ч н и к о в а В. И., П а н о в Е. М., К о ч е ш к о в К. А., ЖОХ, 34, 4022
(1964).
32. Н е п г у М. С, К г е b s A. W., J. Org. Chem., 28, 225 (1963).
33. SaundersB.C, J. Chem. Soc, 1950, 684.
34. Александров Ю. А., БрилкинаТ. Г., Шушунов В. А., ДАН СССР,
136, 89 (1961).
35. Александров Ю. А., БрилкинаТ. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1960, 381.
36. А л е к с а н д р о в Ю. А., Б р и л к и н а Т. Г., Ш у ш у н о в В. А., Сб. «Химия
перекисных соединений». М., Изд-во АН СССР, 1963, стр. 291.
37. Ш у ш у н о в В. А., Б р и л к и н а Т. Г., ДАН, СССР, 141, 1391 (1961).
38. С а 1 i п g a e r t G., D у k s t r a F. J., S h a p i г о Н., J. Am. Chem. Soc, 67, 190
(1945).
39. G r u t t n e r G., К г a u s e E., Ber. 49, 1415 (1916).
40. Е m e 1 е u s H. J., E v a n s P. R., J. Chem. Soc, 1964, 510.
41. В a 1 1 i n g e г Р., Пат. США 3081325 (1963); РЖХим., 1964, 24H89.
42. R i e с h e A., D a h 1 m a n n J., Ann., 675, 19 (1964).
43. R i e с h e A., D a h 1 m a n n J., Monatsber. Dtsch. Akad. Wiss. Berlin, 1, 491 (1959).
44. AmbergerE., Honigschmid-GrossichR., Ber., 98, 3795 (1965).
44a. DaviesA.G., Pudderhatt R. J., J.Organometal. Chem., 5, 590 (1966).
45. G i r a i t i s А., К о b e t z P., ShapiroH., Пат. США 3147293 (1964); РЖХим.,
1966, 12H108.
46. T h а у e r J. S., W e s t R., Inorg. Chem., 3, 889 (1964).
47. R e i с h 1 e W. Т., Inorg. Chem., 3, 402 (1964).
48. Sch er er O. J.,Schm i d M., J. Organometal. Chem., 1,490(1964).
49. Несмеянов А. Н., АнисимовК. Н., Колобова H. E., X андожкоВ. Н.*
ДАН СССР, 156, 383 (1964).
50. Несмеянов А. Н., Анисимов К. Н., Колобова Н. Е.,
Захарова М. Я., ДАН СССР, 156, 612 (1964).
51. HeinR, Jehn W., Ann., 684, 4 (1965).
52. H e i n R, К 1 e i n e r t P., J e h n W., Naturwiss., 44, 34 (1957).
53. GorsichR.D., J. Am. Chem. Soc, 84, 2486 (1962).
54. К a h n О., В i g о r g n e M., Compt. rend., 261, 2483 (1965).
55. HeinF., PoblothH., Z. anorgan. allgem. Chem., 248, 84 (1941).
56. H e i n F., H e u s e r E., Z. anorgan. allgem. Chem., 254, 138 (1947).
57. HeinF., Schnei terH.,Z. anorgan. allgem. Chem., 259, 183 (1949).
58. Schrnidbaur H., Hussek H., J. Organometal. Chem., 1, 257 (1964).
59. S с h m i d b a u r H., S с h m i d t M., J. Am. Chem. Soc, 83, 2963 (1961).
60. SchmidbaurH., Angew. Chem., 77, 206 (1965).
61. SaundersB.C, S t а с е у G. J., W i 1 d F., W i 1 d i n g I. G. E., J. Chem. Soc,
1948, 699.
62. H a r t 1 e R. J., Пат. США 3055925 (1962); РЖХим., 1964, 11H90.
63. H e n г у М. С, Inorg. Chem., 1, 917 (1962).
64. А р б у з о в' Б. А., П у д о в и к А. Н., ЖОХ, 17, 2158 (1957).
65. В е с k W., S с h w i е г е г Е., Ber., 97, 3517 (1964).
66. Р о 1 i s A., Ber., 20, 716 (1887).
67. S е t z е г W. С, L е е р е г R. W., G i 1 m a n H., J. Am. Chem. Soc, 61, 1609 (1939).
68. Polis A., Ber., 20, 3331 (1887).
69. Z e с h m e i s t e r L., С s a b а у J., Ber., 60, 1617 (1927).
70. Polis A., Ber., 21, 3424 (1888)
71. К о ч е ш к о в К. А., Б о р о д и н а Г. М., Изв. АН СССР, Отд. ест. наук, 1937, 569.
72. П а н о в Е. М., К о ч е ш к о в К. А., ЖОХ, 25, 489 (1955).
73. П а н о в Е. М., К о ч еш к о в К. А., ДАН, СССР, 123, 295 (1958).
74. К о ч е ш к о в К. А., П а н о в Е. М., Изв. АН СССР, ОХН, 1955, 718.
75. Панов Е. М., Л о д о ч н и к о в а В. И., К о ч е ш к о в К. А., ДАН СССР,
111, 1042 (1956).
76. Л о Д о ч н и к о в а В. И., П а н о в Е. М., К о ч е ш к о в К. А., ЖОХ, 29, 2253
(1959).
77. Несмеянов А. Н.,Фрейдлина Р. X.,Кочетков А., Изв. АН СССР, ОХН,
1948, 127.
78. Н а д ь М. М., К о ч е ш к о в К. А., ЖОХ, 12, 409 (1942).
79. 3 е м л я н с к и й Н. Н., ЛодочниковаВ. И., Панов Е. М.,
Кочешков К. А., ЖОХ, 35, 843 (1965).
80. Л о д о ч н и к о в а В. И., П а н о в Е. М., К о ч е ш к о"в К. А., ЖОХ, 34, 946 (1964).
81. Н u b e r F., E n d e r s M., Z. Naturforsch., 20b, 601 (1965).
82. A b e 1 Е. W., В г a d у D. В., J. Chem. Soc, 1965/ 1192.
83. Schumann H., SchwabeP., Schmidt M., J.Organometal. Chem., 1, 366
(1964).
39*
Глава XII
РЕАКЦИИ СОЕДИНЕНИЙ ТИПА R3PbPbR3
Гексаалкил- и гексаарилдиплюмбаны, подобно оловоорганическим
аналогам, легко реагируют с окислителями, кислотами, солями, галоидными
алкилами и другими веществами. Однако с свинцовоорганическими
соединениями реакции обычно проходят более сложно и приводят к смеси
продуктов. Эта особенность объяснена [1—4] наличием равновесия:
R3PbPbR3^R4Pb + R2Pb.
Присутствие диэтилсвинца в препаратах гексаэтилдиплюмбана доказано и
Разуваевым с сотр. [5, 6]. Соединения типа R3PbPbR3 по связи РЬ — РЬ
вероятно, не диссоциируют [7,8], хотя в литературе приведены
противоречивые данные по определению их молекулярного веса [9—12].
При работе с гексаалкилдиплюмбанами необходимо учитывать, что они
легко окисляются на воздухе. Поэтому все операции с ними необходимо
проводить в атмосфере сухого инертного газа.
Подробно изучено [13—16] окисление кислородом
гексаэтилдиплюмбана как в чистом состоянии, так и в растворах. Найдено, что этот процесс
проходит сложно, о чем свидетельствуют своеобразные особенности
кинетических закономерностей реакции и многообразие образующихся при этом
веществ. Рассмотрен возможный механизм реакции [16].
Гексациклогексил- [9, 17] и гексаарилдиплюмбаны [10, 11, 17]
устойчивы по отношению к кислороду воздуха. Из продуктов фотохимического
окисления растворов гексациклогексилдиплюмбана кислородом выделены
окись дициклогексилсвинца, окись и перекись свинца [17]. Показано, что в
качестве промежуточного продукта при этом образуется гексациклогексил-
диплюмбоксан. Окисление возможно осуществить и с помощью пермангана-
та калия. В ацетоне гексафенил- [18], гекса-я-толил- и гекса-о-толилдиплюм-
баны [19] превращаются этим окислителем в гидроокиси триарилсвинца.
С серой гексафенилдиплюмбан образует в аналогичных условиях сульфид
бис-(трифенилсвинца) [2]. Кроме того, из реакционной смеси выделены тетра-
фенилсвинец и сульфид дифенилсвинца.
Разуваевым с сотр. [20] изучено окисление гексаэтилдиплюмбана
гидроперекисью изопропилбензола и дигидроперекисью дг-диизопропилбензола.
Найдено, что реакция легко начинается в растворе уже при 15—20° С. При
невысокой (до 0,3 моль/л) начальной концентрации реагирующих веществ
и в отсутствие избытка гидроперекиси эта реакция в растворе я-гексана,
и-нонана и трихлорбензола сопровождается выделением гидроокиси триэтил-
свинца. Например:
(С2Н5)зРЬРЬ(С2Н5)з+С6Н5(СНз)2СООН-^(С2Н5)зРЬОН+(С2Н5)зРЬОС(СНз)2С6Н5.
С повышением концентрации гидроперекиси относительно
гексаэтилдиплюмбана выход гидроокиси триэтилсвинца понижается. Высказано предполо-
Реакции соединений типа R3PbPbR3
613
жение, что в реакционной смеси, наряду с приведенной выше реакцией,
протекают следующие процессы:
(С2Н5)3РЬОН + С6Н5(СН3)2СООН ■£. (С2Н5)3РЬООС (СНз)2СбНб + Н20,
(С2Н5)3РЬОС (СН3)2С6Н5 + СбН5(СНз)2СООН -» (С2Н5)3РЬООС (СНз)2С6Н5 + С6Н5 (СН3)2СОН.
Кроме того, в реакции с гексаэтилдиплюмбаном могут вступать и перекис-
ные свинцовоорганические соединения:
(С2Н5)3РЬРЬ (С2Н5)3 + (С2Н5)3РЬООС (СН3)2С6Н5 -> (С2Н5)3РЬОРЬ (С2Н5)3 +
+ (СаНБ)8РЬОС(СН8)2СвНБ.
Теми же авторами [20, 21] показано ^что гексаэтилдиплюмбан легко
реагирует и с перекисями ацилов:
(С2Н5)3РЬРЬ(С2Н5)з + (С6Н5СОО)2-*2 (С2Н5)3РЬООСС6Н5.
Гладко проходит галоидирование гексаалкил(арил)диплюмбанов [10, 22—
24]. Реакции проводят в условиях, аналогичных описанным на стр. 439.
Например, хлорированием гексаэтилдиплюмбана при — 75°С получают
хлористый триэтилсвинец; из брома и гексаметилдиплюмбана —
бромистый триметилсвинец [23]. Синтез свинцовоорганических галогенидов
ароматического ряда этим методом можно иллюстрировать следующими
примерами.
Получение бромистого три-я-ксилилсвинца [10]. К охлажденному до —50е С раствору
10 г гекса-я-ксилилдиплюмбана в 400 мл пиридина при перемешивании прибавляют 1,6 г
брома в 50 мл пиридина. Продукт перекристаллизовывают из спирта. Т. пл. 177° С. При
—10° С реакция идет далее и происходит отщепление одной арильной группы.
Ряд соединений типа Аг3РЬХ получают при более высокой температуре.
Это относится, например, к йодистому три-о-анизил- и три-я-фенетилсвин-
цу. В случае гекса-я-анизилдиплюмбана основными продуктами реакции
являются тетра-я-анизилсвинец (50,6%) и двуиодистый ди-п-анизилсви-
нец (7,1%) [23]. О галоидировании гексациклогексилдиплюмбана см.
стр. 551.
Гексаалкил(арил)диплюмбаны превращают в свинцовоорганические
галогениды и другими методами. Так, при прибавлении к гексаэтилдиплюм-
бану эквимолекулярного количества S02C12, SOCl2, SC12 или S2C12 в толуоле
при обычной температуре получают 65—97% хлористого триэтилсвинца
[25]. Его выход снижается в ряду S02C12 — SOCl2 — SC12 — S2C12 симбатно
нуклеофильности атома серы. Кроме того, в реакционной смеси обнаружены
небольшие количества (C2H5)2Pb2h и РЬ2+ [25].
При действии на гексаэтилдиплюмбан Cu2Cl2, CuCl2, AuCl3, Hg2Cl2
или FeCl3 в метиловом спирте образуется хлористый триэтилсвинец,
тетраэтилсвинец и двухлористый свинец [1]. Азотнокислое серебро и сулема
восстанавливаются гексаэтилдиплюмбаном до металла или Hg2Cl2. Свинцо-
воорганическое соединение при этом превращается в нитрат и хлористый
триэтилсвинец соответственно [1]. Смешением бензольных растворов тетра-
ацетата свинца и гексаэтилдиплюмбана получен [21, 26] ацетат
триэтилсвинца. Сложный окислительно-восстановительный процесс имеет место при
реакции гексаэтилдиплюмбана с хлористым триэтилоловом или двухлори-
стым диэтилоловом [27]. Например, при непрерывном встряхивании смеси
0,0091 моля гексаэтилдиплюмбана и 0,0208 моля хлористого триэтилолова
в течение 24 час. в продуктах реакции обнаружено 0,0006 моля хлористого
триэтилсвинца, 0,0073 моля двухлористого свинца, 0,0036 моля тетра-
этил свинца, 0,0055 моля хлористого триэтилолова и 0,0073 моля тетраэтил-
олова.
614
Свинцовоорганические соединения. Лит. стр. 615—616
Гексаарилдиплюмбаны легко восстанавливают азотнокислое серебро
до свободного металла [111. С треххлористым алюминием они реагируют
согласно уравнению [28, 291:
АгзРЬРЬАгд +:А1С1з -* [Аг3РЬ2С]2 -* Аг4РЬ + РЬС12.
Нагревание гексафенилдиплюмбана с безводными СиС12, СоС12 и
порошкообразными Си, Ni в этилцеллозольве приводит к хлоридам три- и дифенил-
свинца и двухлористому свинцу. Кроме того, выделяется бензол [30].
Относительно подробно исследовано взаимодействие гексаалкил-[29, 31,
32] и гексаарилдиплюмбанов [19, 24, 29, 33] с галоидоводородами. При
действии хлористого водорода на, гексаметил-, гексациклогексил- или
гексаарилдиплюмбаны в этиловом спирте образуется хлористый триалкил(арил)-
свинец и двухлористый свинец [29]:
R3PbPbR3 + 3 НС1 -* RgPbCl + PbCl2 + 3 RH
(R = CH3, с-СвНц, т-,р- и о-СН3СбН4)
В хлороформе реакция проходит иным путем [29]:
R3PbPbR3 + 2 HCl -* R4Pb2Cl2 -* R4Pb + РЬС12
ИЛИ
R3PbPbR3 + 3 НС1 -* R3Pb2Q3 -> R3PbCl + PbCl2.
При взаимодействии гексаэтилдиплюмбана с хлористым ацетилом или
хлористым бензоилом в метиловом спирте получают тетраэтилсвинец,
хлористый триэтилсвинец и двухлористый свинец [4].
Гексафенилдиплюмбан не реагирует с уксусной кислотой (2 моля) в
среде бензола или я-гептана при обычной температуре [2]. При нагревании
раствора до кипения образуются бензол (выход 90% в расчете на уксусную
кислоту), диацетат свинца (41 %), тетрафенилсвинец (25%) и ацетат трифенил-
Свинца (18%). Небольшая часть (около 3%) гексафенилдиплюмбана
остается в неизменившемся состоянии. Нагревание гексафенилдиплюмбана с
уксусной кислотой без растворителя приводит к диацетату свинца и диаце-
тату дифенилсвинца. Аналогично реагирует гексафенилдиплюмбан с тиоук-
сусной кислотой. В соответствии с полученными продуктами авторы [2]
предлагают следующую схему реакции:
(СбН5)3РЬРЬ(СбН5)3^ [2(С6Н5)3РЬ] -* (С6Н5)4РЬ + [(СбН5)2РЬ],
(C6H5)4Pb + CHgCOOH -> (С6Н5)3РЬООССН3 + С6Н6,
;с6н5)3рьооссн3 + сндсоон -> (сбн5)2рь (ооссн3)2 + сбн6,
[(С6Н5)2РЬ] + 2 СН3СООН -» Pb (00ССН3)2 + 2 СбНб.
О действии азотной кислоты на гекса-я-толилдиплюмбан см. стр. 583.
Согласно данным Гилмана и Липера [34], гексафенилдиплюмбан
присоединяется при нагревании к малеиновому ангидриду:
о о
СН—С (С6Н5)3РЬСН—С
(С6Н5)3РЬРЬ(С6Н5)з + О -* О
СН-С (СбН5)3РЬСН—С
о о
Строение продукта реакции подтверждается тем, что он образует со щелочью
натриевую соль. Кроме того, его температура разложения (265—270°С)
Реакции соединений типа R8PbPbR;
615
существенно отличается от температур плавления двух других возможных
здесь веществ: кислого малеата трифенилсвинца (С6Н5)3РЬООССНСНСООН
(т. пл. 207° С) и малеата бис-(трифенилсвинца) (т. пл. 198—199° С).
Как показано Разуваевым с сотр. [35], реакции гексаэтилдиплюмбана
с галоидными алкилами (бромистым этилом, 1,2-дибромэтаном и 1,2-ди-
бромпропаном) могут проходить в двух направлениях. В случае
эквимолекулярных количеств галоидного алкила и гексаэтилдиплюмбана имеет
место сложный процесс, приводящий к большому числу разнообразных
продуктов. Если же реакцию проводить с каталитическими количествами
галоидного алкила, .то наблюдается резкое уменьшение термической
стабильности гексаэтилдиплюмбана и, как результат этого, диспропорцио-
нирование по уравнению:
2 (С2Н5)3РЬРЬ (С2Н5)3-* 3 (С2Н5)4РЬ + РЬ.
Образование тетраэтилсвинца при нагреваний гексаэтилдиплюмбана с
йодистым этилом [36] или 1,2-дибромэтаном [2] отмечено и другими
авторами.
Гексациклогексилдиплюмбан реагирует с четыреххлористым и четырех-
бромистым углеродом уже при обычной температуре [17]. Из продуктов
реакции выделены галогениды три- и дициклогексилсвинца. Найдено, что
скорость этих реакций заметно увеличивается в присутствии кислорода
воздуха.
Реакции диспропорционирования и термического распада гексаалкил-
(арил)диплюмбанов рассмотрены на стр. 595.
ЛИТЕРАТУРА
1. В е 1 1 и с о U., T a g 1 i a v i n i G., Ricerca scient., 2A, 2, 102 (1962); РЖХим., 1963,
12Ж290.
2. К г е b s A. W., H e n г у М. С, J. Org. Chem., 28, 1911 (1963).
3. В е 1 1 и с о U., С a 11 a 1 i n i L., T a g 1 i a v i n i G., Ricerca scient., 2A, 2, 110
(1962); РЖХим., 1963, 12Ж47.
4. BellucoU., P e 1 о s о А., С a t t a 1 i n i L., T a g 1 i a v i n i G., Ricerca scient.,
2A, 2, 269 (1962); РЖХим., 1963, 20Ж251.
5. P a з у в a e в Г. А., ВязанкинН.С, В ы ш и н с к и й Н. Н., ЖОХ, 29, 3662
(1959).
6. Р а з у в а е в Г. А., В я з а н к и н Н. С, Вышинский Н. Н., ЖОХ, 30,
967 (1960).
7. BellucoU., T a g 1 i a v i n i G., F a v e г о P., Ricerca scient., 2A, 2, 98 (1962);
РЖХим., 1963, 12Ж46.
8. M u 1 1 e r E., G u n t e r E., S с h e f f 1 e г К., Ber., 91, 2888 (1958).
9. KrauseE., Ber., 54, 2060 (1921).
10. К r a u s e E., S с h m i t z M., Ber., 52, 2165 (1919).
11. KrauseE., R e i s s a u s G. G., Ber., 55, 888 (1922).
12. Foster L. S., Dix W. M., Gr un t f es t I. J., J. Am. Chem. Soc, 61, 1685 (1939).
13. Александров Ю. А., Брилкина Т. Г., Шушунов В. А., Труды по
химии и хим. технологии (Горький), 1961, 3.
14. Ш у ш у н о в В. А., Б р и л к и н а Т. Г., А л е к с а н д р о в Ю. А., Труды по
химии и хим. технологии (Горький), 1959, 329.
15. Александров Ю. А., Вышинский Н. Н., Труды по химии и хим.
технологии (Горький), 1961, 656.
16. Брилкина Т. Г., ШушуновВ. А., Реакции металлоорганических соединений
с кислородом и перекисями. М., «Наука», 1966.
17. HeineR, N е b e E., ReimannW., Z. anorgan. allgem. Chem., 251, 125 (1943).
18. В a h r G., Z. anorgan. allgem. Chem., 253, 330 (1947).
19. A u s t i n P. R., J. Am. Chem. Soc, 53, 1548 (1931).
20. Александров Ю. А., Брилкина Т. Г., Квасов А. А., Р а з у в а е в Г. А.,
Ш у шу н о в В. А., ДАН СССР, 129, 321 (1959).
21. Вязанкин Н. С, РазуваевГ. А., Дергунов Ю. И., Щепетко-
в а О. А., Труды по химии и хим. технологии (Горький), 1961, 58.
616
Свинцоеоорганические соединения
22. ВязанкинН. С, РазуваевГ. А., Дергунов Ю. И., Труды по химии и
хим. технологии (Горький), 1961, 652,
23. KrauseE., von GrosseA., DieChemie der metal lor ganischen Verbindungen.
Berlin, Gebr. Borutrager, 1937, S. 180.
24. G i 1 m a n H., В a i 1 i e J. C, J. Am. Chem. Soc, 61, 731 (1939).
25. BellucoU., Cattalini L, Peloso A., Tagl i a viniG., Ricerca scient.,
2A, 3, 1107 (1963); РЖХим., 1964,23Ж232.
26. P а з у в а е в Г. А., ВязанкинН.С, Щ е п е т к о в а О. А., ЖОХ, 30, 2498
(1960).
27. РазуваевГ. А., Дергунов Ю. И..ВязанкинН. С, ЖОХ, 32, 2515 (1962).
28. G i 1 m а п Н., А р р е г s о n L. D., J. Org. Chem., 4, 162 (1939).
29. EmeleusH.J., EvansP.R., J. Chem. Soc, 1964, 511.
30. РазуваевГ. А., Федотов М. С, Заварова Т. Б., Баженова Н. Н.,
Труды по химии и хим. технологии (Горький), 1961, 622.
31. Т a g 1 i a v i n i G., В е 1 1 и с о U., P i 1 1 о n i G., Ricerca scient., 2A, 3, 889 (1963);
РЖХим., 1965, 4Ж345.
32. М i d g 1 е у Т., Н о с h w a 1 t С. А., С a 1 i n g a e r t G., J. Am. Chem. Soc, 45, 1821
(1923).
33. G i 1 m a n H., T о w n e E. В., J. Am. Chem. Soc, 61, 739 (1939).
34. G i 1 m a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
35. P a з у в a e в Г. А., ВязанкинН.С, Д е р г у н о в Ю. И., ЖОХ, 30, 1310
(1960).
36. К г о h n I. Т., S h a p i г о Н„ Пат. США 2555891 (1951); С. А., 46, 523 (1952).
Глава XIII
РЕАКЦИИ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
СВЯЗАННЫЕ С ИЗМЕНЕНИЯМИ В РАДИКАЛЕ
Реакции свинцовоорганических соединений, связанные с изменениями
в радикале, разработаны очень мало. Присоединение галоидов и галоидово-
дородных кислот к алкенильным производным свинца обычно приводит к
разрыву связи С—РЬ. Например, попытка присоединить бром к триэтил-2-
метилпропенил-2-свинцу оказалась неудачной, при бромировании
выделился лишь 1,2,3-трибром-2-метилпропан [1]. При осторожном бромировании
бром удалось присоединить лишь к дифенилди-(я-аллилфенил)свинцу,
при этом был получен дифенил-(п-аллилфенил) (я-2,3-дибромпропилфенил)-
свинец [2].
Гидрид трифенилолова не присоединяется к трифенилвинилсвиниу,
при этом был выделен лишь металлический свинец и гексафенилдистанан [3].
К трифенил-п-стирилсвинцу присоединение гидрида трифенилолова
происходило нормально:
(С6Н5)зРЬ-<^^>-СН^СН2 + (C6H5)3SnH -» (C6H5)3Pb^<^^>-CH2CH2Sn(C6H=
Исходя из дифенилдистирилсвинца и дигидрида дифенилолова были
получены полимерные вещества, имеющие в молекуле свинец и олово [4]:
(С6Н5)4РЬ (С6Н5СН=СН2)2 + (CeH5)2SnH2 -
Г СбН5 СбН5
—Sn—СН2СН2С6Н4РЬСбН4СН2СН2-
I I
СбН5 СбН5
В литературе имеется указание, что при действии фениллития на тетра-
винилсвинец [5,6] и трифенилвинилсвинец [7] образуется виниллитий и
тетрафенилсвинец. Однако выделение виниллития из раствора
сопровождается рядом экспериментальных трудностей.
Остин безуспешно пытался окислить толильные производные свинца
перманганатом калия в ацетоне [8]. Более успешно протекало окисление
трифенилаллилсвинца (С6Н5)зРЬСзН5, который образовывал трифенил-2,3-
пропандиолсвинец (С6Н5)3РЬСН2СН (ОН)СН2ОН — белый кристаллический
порошок с т. пл. 124—125° С [3].
В дальнейшем Гилман с сотр. [9] провели окисление перманганатом калия
в ацетоне (Аг-оксиметилфенил)трифенилсвинца и получили с выходом 25%
(я-карбоксифенил)трифенилсвинец. При окислении аналогичного орто-про-
изводного помимо окисления гидроксильнои группы происходил отрыв
одного фенильного радикала с образованием гидроокиси (о-карбоксифенил)-
дифенилсвинца. Обработкой полученного соединения хлористым водородом
был выделен соответствующий хлорид и затем хорошо кристаллизующийся
618
Свинцовоорганические соединения. Лит. стр. 619
метиловый эфир после обработки диазометаном:
(СвН5)зРЬ-/~^^°1(С6Н5)2РЬ- ^ НС1
СН2ОН 0Н
// \ ™L tCMA^Ph-// \
(C6H5)2Pb-<f > Г12_1. (CHsJsPb-
J
соон ^ соосн
3
Из продуктов окисления мета-производного чистого продукта выделить не
удалось.
Попытки ввести в молекулу свинцовоорганического соединения
альдегидную группу окислением некоторых соединений (гидроксиалкил)-три-
фенилсвинца также оказались неудачными. По предположению авторов
одновременно с окислением происходило и деалкилирование [9].
Реакции свинцовоорганических соединений, имеющих галоид в
радикале, с магнием и литием для получения соответствующих металлоорганиче-
ских соединений в литературе почти не разработаны. Имеется только
указание, что бром, находящийся в алифатическом радикале, связанном со
свинцом, реагирует с магнием нормально с образованием соответствующего
магнийорганического соединения. Эта реакция была проведена на примере
получения (С2Н5)зРЬ(СН2)5 MgBr [10]. Что касается галоида ароматического
радикала, то бром в (я-бромфенил)трифенилсвинце не реагирует с магнием
в эфире, а реагирует в эфире со сплавом магний—медь, однако попытки
провести карбонизацию и выйти к соответствующим карбоксильным
соединениям оказались неудачными [9]. Это удалось лишь с выходом 1,5% при
действии диэтилбария на (я-бромфенил)трифенилсвинец [11]:
(C6H5)3Pb (C6H4Br-p) (^BQ- (С6Н5)3РЬСбН4СООН-р
Попытки нитрования тетрафенилсвинца смесью дымящейся азотной и
серной кислот при — 50° С привели к получению лишь сульфата свинца и
нитробензола. Кроме того, был выделен непрореагировавший тетрафенил-
свинец [12]. Нитрование тетраэтил- и тетрапропилсвинца с помощью N204
при 0°С приводит не к реакции с изменением в радикале, а к образованию
динитрата тетраалкилдинитрозилсвинца [PbAlk4(NO)2]2+(N03)2 с хорошим
выходом [13].
Что касается нитрования динитрата дифенилсвинца, то при нагревании
его с азотной кислотой (уд. в. 1,51) в запаянной трубке при 100°С
происходит нитрование обеих фенильных групп в мета-положение [14]. Нитрование
смесью азотной (уд. в. 1,52) и концентрированной серной кислот при
—15-; 20° С приводит к образованию смеси из 93—96% мета-изомера
3 — 4% орто-изомера [15—17]. Описано также и нитрование диацетата
дифенилсвинца с помощью азотной кислоты (уд. в. 1,5).
Восстановление нитрогруппы в двубромистом ди-ж-нитрофенилсвинце
в кислой, нейтральной и щелочной средах приводит к превращению
нитрогруппы в аминогруппу с одновременным отщеплением ароматического
радикала от свинца.
Если в ароматических свинцовоорганических соединениях имеется
аминогруппа, то она, аналогично простым аминам, способна к реакциям диазо-
тирования и сочетания. Так, я-аминофенилтрифенилсвинец был продиазо-
тирован и полученное соединение сочеталось с р-нафтолом, при этом было
Реакции, связанные с изменениями в радикале
619
получено азосвинцовое соединение, которое в кислых растворах окрашено в
красный цвет, а в щелочных в зеленый [18]. Однако выход полученного
красителя был очень мал. Попытка увеличить выход применением в качестве
диазотирующего агента изоамилнитрита при 0°С в растворе спирта,
насыщенного хлористым водородом, не привела к увеличению выхода красителя
после сочетания с Р-нафтолом [18]. Несколько лучшего выхода(35%) удалось
достигнуть при диазотировании трифенил-л-аминофенилсвинца в ледяной
уксусной кислоте нитритом натрия.
Свинцовоорганические красители получены реакцией сочетания диазо-
ниевой соли с оксиарильными соединениями свинца [19].
Получение трифенил[2-окси-3,5-ди-(/г-нитрофенилазо)фенил]свинца [19]. 50 г я-нит-
роанилина диазотируют обычным образом. Полученный раствор диазониевой соли
прибавляют к смеси 5,3 г (0,1 моля) трифенил-я-оксифенилсвинца, 20 мл этилацетата, 20 мл
95%- ного спирта и 40 мл 10%-ного раствора едкого натра. Смесь быстро перемешивают до
исчезновения всего этилацетата. На это требуется около 2 час. Твердый остаток собирают на
фильтре, промывают водой и 50%-ным спиртом. Полученный краситель темно-красного цвета
перекристаллизовывают из спирта, содержащего небольшое количество уксусной кислоты.
Получают 7,1 г (86%) вещества с т. разл. 180° С.
ЛИТЕРАТУРА
1. G 1 о с к 1 i n g F., К i n g s t о n D., J. Chem. Soc, 1959, 3001.
2. Л у ч и н я н Е. А., М а н у л к и н 3. М., ДАН УзбССР, 19, 47 (1962).
3. Henry M. С, N о 1 t e s J. G., J. Am. Chem. Soc, 82, 558 (I960).
4. N о 1 t e s J. G., v a n d e r KerkG.J.M., Rec. Trav. chim., 80, 623 (1961).
5. J u e n g e E. C, S e у f e r t h D., J. Org. Chem., 26, 563 (1961).
6. Англ. пат. 876008 (1962); С. А., 56, 10188a (1962).
7. S e у f e r t h D., W e i n e r M. A., J. Am. Chem. Soc, 84, 361 (1962).
8. A u s t i n P. R., J. Am. Chem. Soc, 53, 3514 (1931).
9. GilmanH., MelstromD. S., J. Am. Chem. Soc, 72, 2953 (1950).
10. G r i\ t t n e r G., KrauseE., Ber., 49, 2666 (1916).
11. GilmanH., HaubeinA.H., O'D о n n e 1 1 G., WoodsLA., J. Am. Chem.
Soc, 67, 922 (1945).
12. G i 1 m a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
13. H e t n a r s k i В., U r b a n s k i Т., Tetrahedron, 19, 1319 (1963).
14. VorlanderD., Ber., 58, 1893 (1925).
15. Challenger F.f RothsteinE., J. Chem. Soc, 1934, 1258.
16. К о ч е ш к о в К. А., Б о р о д и н а Г. М., Изв. АН СССР, Отд. ест. наук, 1937, 570.
17. Sc h m i d t H., Med. und Chem. Abhandl med.-chem. Forschungsstatten I. G. Farben-
ind, 3, 418 (1936); C. A., 31, 5866 (1937).
18. G i 1 m a n H., S t u с k w i s с h С G., J. Am. Chem. Soc, 64, 1007 (1942).
19. G i 1 m a n H., S t u с k w i s с h C.,G., J. Am. Chem. Soc, 72, 4553 (1950).
Г лава XIV
СИНТЕЗ И РЕАКЦИИ
СВИНЦОВООРГАНИЧЕСКИХ ГИДРИДОВ
Из трех возможных типов свинцовоорганических гидридов соединения
RPbH8 пока не известны, a R8PbH и R2PbH2 получены только в
алифатическом ряду. Это неустойчивые вещества, которые разлагаются даже при
низких температурах. Их получают восстановлением галогенидов свинца
литийалюминийгидридом или натрийборгидридом при температуре от — 60°
до — 100° С. Другой метод заключается в действии диборана на метокситри-
алкилсвинец с последующей обработкой смеси метиловым спиртом.
Свойства свинцовоорганических гидридов описаны очень кратко.
Следует отметить, что устойчивость металлоорганических гидридов IV группы
уменьшается при переходе от кремния к свинцу. В том же ряду
увеличивается их химическая активность.
СИНТЕЗ СВИНЦОВООРГАНИЧЕСКИХ ГИДРИДОВ
Свинцовоорганические галогениды активно реагируют с
литийалюминийгидридом с образованием соответствующих гидридов. Реакцию проводят
в среде эфира при температуре от — 90 до — 110° С [ 1 ], в диметиловом эфире
при—78°С [2] или диметиловом эфире диэтиленгликоля при—60°С [3]. Так
получены гидриды триметил- (т. пл. —106°С) [1,2], триэтил- (т. пл. —145°С)
[2], три-я-пропил-^рь-н 1675 см"1) [2], три-я-бутил- (vPb_H 1680 см"1),
TpHH3o6yran-(vpb_-H 1675 см"1) [3] и трициклогексилсвинца (vPb_H 1650 см"1),
а также дигидриды диметил- (vPb_H 1709 см"1) [1, 2], диэтил- [2] и ди-я-
бутилсвинца (vPb_H 1640 см"1) [3].
Более сложно проходит реакция между галогенидами триалкилсвинца и
калийборгидридом в жидком аммиаке [4—6]. По-видимому, в качестве
промежуточного продукта образуется боранат триалкилсвинца. После удаления
аммиака из эквимолекулярной смеси хлористого триметилсвинца и калий-
боргидрида при — 78°С остается хлористый калий и осадок белого цвета
состава (СН3)зРЬВН4-хЫН3 (х > 2). При — 55°С он разлагается в
соответствии с уравнением:
(СН3)зРЬВН4-хЫНз^ (СНз)зРЬН + (*-1) NH3 + NH3.BH3.
Перегонкой смеси при —100° С выделен [4—6] чистый гидрид
триметилсвинца. Выход 75%, т. пл.— 104° С. Аналогично получен [6] гидрид триэтилсвин-
ца. Выход 25—30%.
Целесообразнее получать гидриды триалкилсвинца взаимодействием
метокситриалкилсвинца с дибораном [7]. При смешении реагентов при
— 78° С в эфире практически с количественным выходом образуются
нерастворимые в эфире боранаты триалкилсвинца:
4 R3PbOCH3 f 3 (ВН3)2 -» 4 R3PbBH4 -\- 2 НЕ(ОСН3)2.
Синтез и реакции свинцовоорганических гидридов
621
Первый член ряда — боранат триметилсвинца — медленно разлагается
уже при — 78° С с выделением гидрида триметилсвинца и диборана:
(СН3)зРЬВН4 -» (СНз)зРЬН + 0,5 (ВН3)2.
Боранаты триэтил-, трипропил- и трибутилсвинца значительно устойчивее.
В связи с этим интересно отметить, что из соединений типа А1к3РЬОСН3
наиболее устойчив метокситриметилсвинец.
Свинцовоорганические боранаты превращают в соответствующие
гидриды обработкой метиловым спиртом при — 78° С:
R3PbBH4 + 3 СН3ОН -» R3PbH + В(ОСН3)з + 3 Н2.
Так получены гидриды триэтил-, трипропил- и трибутилсвинца.
Получение боранатов триалкилсвинца. [7]. Синтез и очистку веществ проводят в цель-
нопаянной вакуумной системе. Воздух и другие газы из применяемых растворителей
предварительно удаляют в вакууме. В сосуд Шленка помещают свежеприготовленный метокси-
триалкилсвинец и конденсируют эфир. При нагревании смеси до комнатной температуры
образуется прозрачный раствор. Его замораживают и конденсируют избыток диборана.
Температуру постепенно повышают до —78° С и смесь перемешивают при этой температуре
длительное время. При этом количество осадка значительно увеличивается (выпавший при
вымораживании смеси метокситриалкилсвинец растворяется в эфире при —78° С не полностью).
После завершения реакции удаляют в вакууме диборан при —100-ь —110° С, эфир, остатки
растворенного *в нем диборана и диметоксиборан при —78ч 65° С. Остаток представляет
собой боранат триалкилсвинца. Даже при длительном выдерживании в вакууме при —78 -г-
—65° С боранат триалкилсвинца содержит остатки метоксиборанов.
Получение гидридов триалкилсвинца [7]. Метиловый спирт, из которого
предварительно удаляют в вакууме растворенные в нем газы, конденсируют в сосуд Шленка, содержащий
боранат триалкилсвинца. Смесь постепенно нагревают до —78° С. В этих условиях гидрид
триалкилсвинца выделяется в виде тяжелой маслянистой жидкости. В некоторых опытах
наблюдается незначительное выделение металлического свинца, что указывает на частичное
разложение свинцовоорганического гидрида. Вещество идентифицируют, измеряя
количество водорода, выделившегося при метанол изе соответствующего бораната. Чистые гидриды
триалкилсвинца медленно разлагаются при —30 -. 20° С с выделением свинца и
окрашенной в желтый цвет маслянистой жидкости, представляющей собой, по-видимому, смесь тет-
раалкилсвинца и гексаалкилдиплюмбана.
Устойчивость свинцовоорганических гидридов возрастает с
увеличением алкильного радикала у атома свинца [3, 7]. Гидриды триметил- и три-
этилсвинца начинают разлагаться даже ниже — 78° С [4, 7] и при — 20-4-
— 30° С этот процесс проходит с заметной скоростью [1,2,5—7]. Как
показывает анализ образующихся веществ, ббщую схему разложения гидрида
триметилсвинца можно представить следующим образом [6]:
(СН3)зРЬН-> (СНз)зРЬ* + Н\
Н' + Н*->Н2,
(СН3)зРЬН + И" -> (СНз)зРЬ* + Hi,
^РЬСНз + Н* ->^РЬ*+ СН4,
2 (СН3)8РЬ* -> (СНз)зРЬРЬ (СН3)з,
2 (СНз)зРЬ' -» (СН3)4РЬ + (СН3)2РЬ,
3 (СН3)2РЬ -> РЬ + (СНз)зРЬРЬ (СН3)з,
2(СН3)зРЬРЬ (СНз)з -* ЗРЬ (СН3)4 + РЬ.
Гидриды трипропил-, трибутил- и трициклогексилсвинца могут храниться
при 0°С в темноте по меньшей мере в течение нескольких дней [3].
В заключение отметим, что попытки синтеза гидридов триарилсвинца
взаимодействием триарилплюмбилнатрия с бромистым аммонием [8] или
каталитическим гидрированием гексафенилдиплюмбана [9] не увенчались
успехом.
622
Свинцовоорганические соединения
РЕАКЦИИ СВИНЦОВООРГАНИЧЕСКИХ ГИДРИДОВ
Свинцовоорганические гидриды значительно активнее гидридов германия
и олова. Так, гидрид триметилсвинца реагирует с кислородом воздуха со
взрывом [2]. Тот же гидрид образует с жидким аммиаком окрашенные в
зеленые, а затем в красные тона растворы [5]. Одновременно наблюдается
выделение метана, тетраметилсвинца и металлического свинца. Проходящие
реакции можно выразить следующей схемой:
(СН3)зРЬН + ЫНз-*Ш4[РЬ(СН3)з] (вещество зеленого цвета),
NH4 [РЬ(СН3)з] ->:(СН3)2РЬ + СН4 + NH3,
NH4 [Pb (СН3)з] + Pb (CH3)2 -* NH4 [Pb2 (CH3)5] (вещество красного цвета),
NH4 [Pb2(CH3)5] -> Pb + (CH3)4Pb + СН4 + NH3,
2 NH4[Pb2 (CH3)5] -> Pb + (CH3)4Pb + (СНз)зРЬРЬ (СН3)3 + 2 NH3 + Н2.
При прибавлении к раствору NH4[Pb(CH3)3] хлористого триметилсвинца
образуются практически с количественным выходом гексаметилдиплюмбан
и хлористый аммоний [5].
Подобно гидридам олова, свинцовоорганические гидриды легко
реагируют с хлористым водородом [4, 6]:
(СН3)зРЬИ + НС1 -» (СН3)3РЬС1 + Н2.
При— 112° С выход хлористого триметилсвинца составляет 96%, а при
—78°С — только 78%. В качестве побочного продукта в этом случае
выделяется гексаметилдиплюмбан [6].
Алкилгалогениды быстро и экзотермически восстанавливаются свинцово-
органическими гидридами [3]:
R3PbH + R'J -» RsPbJ + R'H
С низшими галоидными алкилами реакция проходит количественно, что
может быть использовано и для аналитических целей.
Взаимодействием гидридов триалкилсвинца с 1,2-дибромэтаном, четырех-
хлористым углеродом и хлористым триэтилоловом получены, соответственно,
этилен, хлороформ и гидрид триэтилолова. Бензальдегид и нитробензол
восстанавливаются гидридами триалкилсвинца при 0°С [3]. Как уже
отмечено на стр. 472, с оловоорганическими гидридами эти реакции проходят в
более жестких условиях.
Присоединение свинцовоорганических гидридов по кратным связям и
их реакции с диазометаном рассмотрены на стр. 570 и 568.
ЛИТЕРАТУРА
1. AmbergerE., Angew. Chem., 72, 494 (1960).
2. BeckerW.E., GookS.E., J. Am. Chem. Soc, 82, 6264 (1960).
3. Neumann W.P., KuhleinK., Angew. Chem., 77, 808 (1965).
4. D u f f у R., H о 1 1 i d а у A. K., Proc. Chem. Soc, 1959, 124.
5. D u f f у R., H 6 1 1 i d а у А. К., J. Chem. Soc, 1961, 1679.
6. D u f f у R., F e e n e у J., H б 1 1 i d а у A. K., J. Chem. Soc, 1962, 1144.
7. AmbergerE., Honigschmid-GrossichR., Ber., 99, 1673 (1966).
8. G i 1 m a n H., В a 1 i e J. C, J. Am. Chem. Soc, 61, 731 (1939).
9. P о d a 1 1 H. E., P e t r e e H. E., Z i e t z J. R., J. Org. Chem., 24, 1222 (1959).
Глава XV
СИНТЕЗ И СВОЙСТВА СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ,
СОДЕРЖАЩИХ ДВА И БОЛЕЕ АТОМОВ МЕТАЛЛА В МОЛЕКУЛЕ
В литературе описан ряд соединений типа R3PbM (где М — щелочной
металл), R3PbM(CO)„ (М = Re,Mo, W,Mn, Co), [(R3Pb)2Fe(CO)4]2, а также
[(R2Pb)2Fe(CO)4]2 и R2Pb[M(CO)J2 (M=Co, Мп). Подробно изучены иоловоор-
ганические аналоги этих веществ (см. стр. 495). По своим методам синтеза и
свойствам би-элементоорганические производные олова и свинца имеют
много общего. Так, литийтриалкил- и литийтриарилсвинец получают теми
же методами, что и R3SnLi, а именно взаимодействием свинцовоорганических
галогенидов с литием или прибавлением 3 молей фениллития к двухлористо-
му свинцу. Как и в случае трифенилстанниллития (см. стр. 498), растворы
трифенилплюмбиллития, полученные этими двумя способами, имеют
различные свойства.
Триалкил- и триарилплюмбиллитий (соответственно, -натрий)
применяются для синтеза свинцовоорганических соединений с различными
радикалами у атома свинца (в том числе замещенных в алкильном или арильном
радикале), а также гексаалкил- и гексаарилдиплюмбанов.
Что касается соединений, в которых атом свинца связан с рением,
кобальтом, молибденом, вольфрамом или железом, то главный метод их синтеза
основан на реакции обмена карбонилов металлов со свинцовоорганически-
ми галогенидами:
R3PbX + NaM (CO),, -* R3PbM (CO)„ + NaX.
Аналогично получают и соединения типа R3SnM(CO)n. Однако свойства
этих соединений существенно различаются между собой. Так, бромирование
трифенилсвинецренийпентакарбонила приводит к выделению бромпентакар-
бонила рения и двубромистого свинца, тогда как в случае трифенилоло-
ворений пентакарбонила наблюдается отщепление только фенильного
радикала. Связь олова с рением остается при этом не затронутой [1].
Известны и соединения, в которых два атома металла разделены между
собой углеводородными радикалами. Однако методы синтеза и свойства
этих веществ остаются пока мало изученными (см. стр. 560, 617).
СИНТЕЗ СОЕДИНЕНИЙ ТИПА R3PbM
(М — щелочной металл)
Действие щелочных металлов на хлористый триметил- [2] и тривинилсви-
нец [3] описано очень кратко. Более подробно исследована реакция лития с
хлористым трифенилсвинцом [4—6]. Во время синтеза вещество частично
диспропорционирует, что видно по различной окраске образующихся
растворов (от темно-зеленой до черной).
624 Свинцовоорганические соединения. Лит. стр. 630—631
Получение трифенилплюмбиллития из хлористого трифенилсвинца и лития [4]. К 21,16 г
(0,04 моля) хлористого трифенилсвинца и 0,7 г (0,1 г-атома) лития приливают 150 мл тет-
рагидрофурана. После небольшого индукционного периода происходит экзотермическая
реакция, сопровождающаяся изменением окраски. Смесь перемешивают в течение 4 час. при
обычной температуре и отфильтровывают. Обработка аликвотной части раствора хлористым
бензилом дает 98,2% бензилтрифенилсвинца.
Реакция хлористого трифенилсвинца с литием проходит в несколько
стадий [4, 6]:
(СаН5)зРЬС1~Ь 2 Li -> (C6H5)3PbLi -f LiCl,
(C6H5)3PbLi + (С6Н,)зРЬС1 -> (С8Н5)3РЬРЬ (С8Ч,)3 |- LiCl,
(С6Н5)3РЬРЬ (С6Н5)3 + 2 Li -* (С6Ы5)з PbLi.
Применение в качестве исходного вещества гексафенилдиплюмбана
позволяет повысить выход и степень чистоты трифенилплюмбиллития [5, 7].
При действии металлического натрия в жидком аммиаке на галогениды
триалкил- и трифенилсвинца вначале образуются соединения типа
R3PbPbR3, а затем окрашенные в темно-красный цвет растворы R3PbNa
[8—10]. Гексафенилдиплюмбан реагирует и с другими щелочными и
щелочноземельными металлами: литием, калием, рубидием, кальцием, стронцием и
барием [11]. Выходы продуктов несколько уменьшаются при переходе от лития
к рубидию и от кальция к барию. Интересно отметить, что
щелочноземельные металлы ведут себя в этих реакциях как одновалентные элементы [11].
Со смесью магния и бромистого магния гексафенилдиплюмбан реагирует
более сложным образом [8].
Другой метод синтеза соединений типа R3PbLi заключается в действии
3 молей фениллития в эфире при — 10° С на хлористый свинец [4, 12—16].
РЬС12 + 2 C6H5Li -* (C6H5)2Pb + 2 LiCl,
(C6H5)2Pb + C6H5Li ^ (CeH5)3 PbLi.
Диссоциация приготовленного таким образом трифенилплюмбиллития
доказана выделением бензойной кислоты из продуктов, образующихся при
карбонизации реакционной смеси [12, 17]. При карбонизации
трифенилплюмбиллития, полученного из хлористого трифенилсвинца и лития в тетрагидро-
фуране, бензойная кислота не образуется [4, 13]. Следовательно, в этом
случае равновесие трифенилплюмбиллития с фениллитием, если оно имеет
место, полностью сдвинуто в сторону трифенилплюмбиллития.
Получение трифенилплюмбиллития из хлористого свинца и фениллития [12]. К
охлажденной до —10° С суспензии 11,1 г (0,04 моля) хлористого свинца в 30 мл эфира при
перемешивании прибавляют по каплям 0,120 моля фениллития в 110 мл эфира. После
прибавления первых 5—10 мл раствора фениллития смесь окрашивается в светло-желтые тона. После
добавления всего количества фениллития образуется суспензия, состоящая из окрашенного
в "светло-серый цвет осадка в слегка желтоватом растворе.
Позже этот метод использован и другими авторами [17, 18]. Найдено [19],
что при замене эфира тетрагидрофураном реакция быстро проходит даже
при —40° С
В ряде случаев соединения типа R3PbNa получают по следующей схеме
[10, 14]:
R4Pb + 2 Na + NH3 -* R3PbNa + RH + NaNH2.
В порядке уменьшения легкости отрыва от атома свинца оргднические
радикалы располагаются следующим образом [10]: СН2СН = СН2, *-С4Н9,
я-С4Н9, С2Н5, СН3, С6Н5, p-(CH,)2NC6H4. Таку при действии, ратрия на три-
этилфенилсвинец образуется диэтилфенилплюмбил натрий Щ1. Тетравинил-
свинец деалкилируется литием до металлического свинца [$01.
Свинцовоорганияеские соединения с двумя и более атомами металла 625
В статье Гилмана и Биндшадлера [14] приведена сравнительная
характеристика различных методов синтеза соединений со связью Pb—Na*
Отмечено, что при получении триэтилплюмбилнатрия наиболее целесообразно
исходить из тетраэтилсвинца и натрия в аммиаке. Образующийся в качестве
побочного продукта амид натрия можно удалить фильтрованием смеси. Во
многих случаях, например при проведении реакций триэтилплюмбилнатрия
с галоидными алкилами, в этом нет необходимости. Алкилирование амида
натрия проходит гораздо медленнее, чем триэтилплюмбилнатрия. Трифенил-
плюмбилнатрий лучше всего получать из гексафенилдиплюмбана, так как
при этом не образуется побочных продуктов [14].
РЕАКЦИИ СОЕДИНЕНИЙ ТИПА R3PbMj
Соединения типа R3PbM чрезвычайно чувствительны к действию
кислорода и влаги воздуха. Поэтому все операции с ними необходимо проводить
в атмосфере сухого инертного газа. Продукты окисления и гидролиза три-
фенилплюмбиллития подробно исследованы Уиллемсенсом и Ван-дер-Кер-
ком [18]. Трифенилплюмбиллитий получался взаимодействием хлористого
свинца с фениллитием в эфире. Выделившийся осадок, представляющий
собой смесь трифенилплюмбиллития и хлористого лития, отфильтровывался
и высушивался в вакууме. Под влиянием кислорода и влаги воздуха он
быстро изменяется, окрашиваясь в красный цвет. Значительно быстрее этот
процесс проходит при обработке осадка смесью снега и соли, содержащей
небольшое количество перекиси водорода. Образующееся вещество
представляет собой тетра(трифенилплюмбил)свинец — Pb[Pb(C6H5)3J4. Из
продуктов его иодирования выделены йодистый трифенилсвинец и йодистый свинец
с выходом 92 и 93% соответственно. Отсутствие в продуктах реакции
двуйодистого дифенилсвинца 'доказано методом тонкослойной хроматографии.
Получение тетра(трифенилплюмбил)свинца [18]. К суспензии 5,6 г (0,02 моля)
хлористого свинца в 500 мл эфира при охлаждении смесью сухого льда с ацетоном и
перемешивании прибавляют 0,06 моля фениллития в 75 мл эфира. Температуру постепенно повышают
до —10° С и смесь перемешивают при этой температуре в течение часа. Выделившийся
осадок отфильтровывают и высушивают в вакууме. Затем его охлаждают сухим льдом с
ацетоном и при энергичном перемешивании прибавляют 150 мл смеси снега с солью (температура
этой смеси —10 -. 15° С), содержащей 5 мл 30% -ной перекиси водорода. Внешнее
охлаждение удаляют и смесь перемешивают в течение часа. Образуется продукт красного цвета.
Интенсивность окраски углубляется по мере перемешивания. Экстрагирование смеси
хлороформом дает окрашенный в красно-коричневый цвет раствор, который быстро
становится мутным. Выпаривание его в вакууме сопровождается разложением вещества. Как только
появятся первые кристаллы, раствор фильтруют и фильтрат вновь выпаривают в вакууме.
Получают 2,5 г тетра(трифенилплюмбил)свинца. Его очищают перекристаллизацией из
хлороформа описанным выше способом. Вещество хорошо растворяется в хлороформе и
бензоле, не растворяется в спирте и петролейном эфире,
В присутствии кислорода и влаги воздуха тетра(трифенилплюмбил)-
свинец разлагается на протяжении нескольких дней с выделением
гексафенилдиплюмбана и окиси свинца. Значительно быстрее разложение проходит
в растворе. Поэтому удаление эфира из трифенилплюмбиллития до его
гидролиза играет весьма существенную роль [18]. Действитетельно, при
гидролизе трифенилплюмбиллития (из 12,2 г бромистого свинца) в эфире смесью
воды и тетрагидрофурана в отсутствие кислорода образуются гексафенил-
диплюмбан (0,2 г), бифенил (0,1 г) и основной бромид свинца ЗРЬО*РЬВг2
(1—3 Н20) [19]. О продуктах гидролиза бромистого тримезитилплюмбил-
магния см. стр. 555.
Как и соответствующие соединения германия и олова (см. стр. 150),
трифенилплюмбиллитий присоединяет серу, селен и теллур [6]. Реакции прово-
40 Заказ № 207
626
Свинцовоорганические соединения. Лит. стр. 630
дят путем простого смешения эквимолекулярных количеств трифенилплюм-
биллития с этими элементами в тетрагидрофуране. При этом в случае серы
цвет раствора изменяется до оранжево-красного, с селеном сохраняется
первоначальная темно-зеленая окраска, а растворы (C6H5)3PbTeLi окрашены
в черный цвет. Хлористый бензоил, хлористое трифенилолово и хлористый
трифенилсвинец превращают (C6H5)3PbSLi в (C6H5)3PbSC(0)C6H5 (выход
60%; т. пл. 92°С), (C6H5)3PbSSn(C6H5)3 (выход 47%; т. пл. 137° С) и
(C6H5)3PbSPb(C6H5)3 (58%; т. пл. 139— 140°С) соответственно.
Аналогично получены (С6Н5)зРЬ8е8п(С6Н5)3 (выход 35%; т. пл. 138°С),
(C6H5)3PbSePb(C6H5)3 (выход 40%; т. пл. 10ГС) и (С6Н5)3РЬТеРЬ(С6Н5)3
(выход 60%; т. пл. 128—129°С).
О синтезе (C6H5)3PbSNa из хлористого трифенилсвинца и сульфида
натрия см. стр. 606.
Триэтил- и трифенилплюмбилнатрий легко реагируют с алкил- и арил-
галогенидами. Например, обработкой триэтилплюмбилнатрия бромистым
я-бутилом в смеси жидкого аммиака с эфиром получают практически с
количественным выходом триэтил-я-бутилсвинец (т. кип. 81—83°С/5 мм) [13].
С бромистым втор-бучтом реакция проходит медленнее, и выход триэтил-
#/ж?/?-бутилсвинца несколько ниже (82%). При действии бромистого трет-
бутилолова на триэтилплюмбилнатрий выделено лишь небольшое
количество (около 30%) тетраэтилсвинца.
Хлористый аллил образует с триэтилплюмби л натрием триэтилаллил-
свинец с выходом 72%; т. кип. 85—86°С/8 мм [14].
Получение триэтил-2-метилпропенил-2-свинца [21]. 9,5 г (0,413 г-атома) натрия в
600 мл жидкого аммиака прибавляют в течение 2 мин. к 44 г (0,226 моля) тетраэтилсвинца в
250 мл сухого эфира при —80 -; 100° С. К образующемуся раствору
триэтилплюмбилнатрия быстро прибавляют 30 мл (0,2 моля) З-хлор-2-метилпропена в 100 мл эфира. Аммиак
выпаривают, остаток промывают водой и водный раствор экстрагируют эфиром.
Объединенные эфирные вытяжки высушивают, растворитель отгоняют и остаток перегоняют в вакууме.
Получают 30 г (43%) триэтил-2-метилпропенил-2-свинца с т. кип. 45—50° С/0,1 мм.
Вещество быстро разлагается на воздухе с выделением металлического свинца.
Завгородний и Петров [22] взаимодействием триэтилплюмбилнатрия с
алкенилбромацетиленами получили с хорошим выходом 1,3-ениновые
свинецуглеводороды:
(C2H5)3PbNa + BrC=C—CR=CH2^ (C2H5)3PbC=CCR=CH2 + NaBr
(R = H, СН3).
Эти соединения, как и соответствующие производные олова, представляют
собой бесцветные сильно преломляющие свет жидкости с неприятным
запахом. В чистом виде в отсутствие влаги воздуха они могут храниться
длительное время без заметного разложения.
С помощью триэтилплюмбилнатрия получены также соединения,
содержащие аминогруппу в алифатическом радикале у атома свинца:
(2-диэтиламиноэтил)- и (З-диэтиламинопропил)триэтилсвинец [23].
Реакция триэтилплюмбилнатрия с хлористым бензилом происходит
своеобразно и приводит в основном к стильбену. Выход триэтилбензил-
свинца не превышает 10% [14]. В связи с этим интересно отметить, что
при действии на триэтилплюмбилнатрий 2-бромстироЛа получают 40%
триэтилплюмбилстирола (т. кип. 94°С/10~2 мм) [21].
Аналогично реагирует триэтилплюмбилнатрий с галоидными арилами.
Так, при прибавлении к нему иодбензола получают 15% триэтилфенил-
свинца (т. кип. 127—137° С/7 мм). Замена иодбензола на бромбензол
позволяет повысить выход этого вещества до 77% [14]. Триэтил-и-три-
фторметилфенилсвйнец получен [21] из 22,5 г я-бромтрифторметилбензо-
ла, 39,0 г тетраэтилсвинца и 5,5 г натрия в жидком аммиаке. /г-Хлор-
Свинцовоорганические соединения с двумя и более атомами металла 627
трифторметилбензол в этих условиях с триэтилплюмбилнатрием не
реагирует [21].
Трифенилплюмбилнатрий менее активен, чем триэтилплюмбилнатрий.
При действии на него бромистых алкилов или галоидбензилов реакция
проходит достаточно легко и с хорошим выходом. Так получены, например,
этилтрифенилсвинец [14] и бензилтрифенилсвинец [14], а также бензилтри-
м-толил- (т. пл. 81—82°С), бензилтри-я-фенетил-(т. пл. 76—77°С) и бензил-
три-я-анизилсвинец (т. пл. 80—8ГС) [8, 24].
Трифенилплюмбилнатрий в отличие от этильного соединения не
реагирует с галоидными арилами [14].
Трифенилплюмбиллитий вступает в реакции как с галоидными алкилами,
так и с галоидными арилами. Так, с йодистым этилом или бромистым
я-пропилом он образует этилтрифенил- и я-пропилтрифенилсвинец с
выходом 45 и 58% соответственно [12].
Уиллемсенс и Ван-дек-Керк [25] взаимодействием трифенилплюмбилли-
тия с хлористым метиленом, хлороформом и четыреххлористым углеродом
получили с хорошим выходом ди-, три- и тетра(трифенилплюмбил)-замещен-
ные метаны. Эти соединения образуются при смешении рассчитанных
количеств реагентов в тетрагидрофуране при — 60°С. Для завершения реакции
смесь перемешивают 2 часа при обычной температуре и затем нагревают до
кипения в течение 30 мин. Тетра(трифенилплюмбил)метан выделяют
экстрагированием горячим хлороформом, из которого он кристаллизуется с 4
молекулами растворителя. Из ацетона получают чистое вещество с т. разл.
292—294° С. Три- и ди(трифенилплюмбил)метаны имеют т. пл. 166 и 97—
97,5° С соответственно [25].
При действии на трифенилплюмбиллитий хлорбромметиленов
образуется смесь продуктов, выделить из которой чистые трифенил-со-галоид-
алкильные соединения не удается. Даже при применении избытка три-
фенилплюмбиллития продукт состоит в основном из соединений типа
(С6Н5)3РЬ(СН2).РЬ(С6Н5)з [15, 26].
С 3-диэтиламинопропилхлоридом трифенилплюмбиллитий дает 91%
продукта алкилирования. Вещество представляет собой некристаллизую-
щееся масло, которое при нагревании в вакууме разлагается, не
перегоняясь. Для очистки его обрабатывают йодистым метилом или диметилсульфа-
том. В последнем случае образуются кристаллы следующего состава
[(C6H5)3PbCH2CH2CH2N(C2H5)2CH3]+[OS03CH3]", хорошо растворимые в воде,
метиловом спирте и этиленгликоле [15, 26].
Уиллемсенс и Ван-дер-Керк [7] исследовали ряд реакций типа:
(C6H5)3PbLi + (СН2)ПХ -* (CeHe)8Pb(CH2)nXLi —*(СвН5)зРЬ(СНа)яХН
(Х = 0, S или NR; /г = 2, 3 или 4).
Реакции проводят путем прибавления раствора трифенилплюмбиллития в
тетрагидрофуране к охлажденному до — 60° С раствору циклического
соединения. Затем смесь постепенно нагревают до обычной температуры,
нейтрализуют ледяной уксусной кислотой и добавляют смесь серного эфира
со льдом. Органический слой отделяют, высушивают, выпаривают досуха
и остаток очищают перекристаллизацией. С трехчленными циклами,
содержащими кислород или серу, реакции проходят гладко и с хорошим
выходом. Так, из окиси этилена или этиленсульфида получены 2-оксиэтил-
и 2-меркаптоэтилтрифенилсвинец (т. пл. 72 и 104—106°С соответственно).
Выходы продуктов составляют 90—91%.
Этиленимин и N-бутиленимин не реагируют с трифенилплюмбиллитием
даже при нагревании смеси до 115°Св запаянной ампуле. С ацилированными
этилениминами реакции проходят обычным образом, давая 2-(ациламино-
40*
628 Свинцовоорганические соединения. Лит. стр. 630
Э1ил)трифенильные производные свинца. Так получен, например, 2-(аце-
тиламино)этилтрифенилсвинец (выход 90%; т. пл. 113°С).
Трифенилплюмбиллитий активно реагирует также с замещенными окиси
этилена. Так, из эпихлоргидрина и трифенилплюмбиллития получают с
количественным выходом (3-хлор-2-оксипропил)трифенилсвинец. Его
структура подтверждена спектром ЯМР и химическими реакциями.
Интересно отметить, что окись триметилена легко превращает
трифенилплюмбиллитий в 3-оксипропилтрифенилсвинец (т. пл. 99°С), тогда как с
триметиленсульфидом при обычной температуре реакция не проходит
вообще. При нагревании образуется маслянистая жидкость, выделить из
которой индивидуальные соединения не удается.
Взаимодействие трифенилплюмбиллития с |3-пропиолактоном протекает
своеобразно: сн
/ \
Н2С С=0 + (C6H5)3PbLi -> (CGH5)3PbC[i2CH2COOLi.
\ /
О
Подкисление продукта приводит к отщеплению бензола и образованию
полимерного продукта состава [(С6Н5)2РЬ4СН2СНСОО"']Аг.
С пятичленными гетероциклами, например тетрагидрофураном,
трифенилплюмбиллитий не реагирует даже при нагревании смеси до 115° С в
течение 6 час [7].
Проведено металлирование флуорена трифенилплюмбиллитием с
последующей карбонизацией реакционной смеси. С трифенилплюмбиллитием,
полученным из хлористого трифенилсвинца и лития, образуется только
0,5% флуоренил-9-карбоновой кислоты [13]. Ее выход составляет 52%,
если для металлирования флуорена применять (C6H5)3PbLi, полученный из
двухлористого свинца и фениллития в эфире [17].
Реакции соединений типа R3PbM с металлоорганическими галогенидами
почти не изучены. Свинцовоорганические галогениды, как и
соответствующие оловоорганические соединения, образуют с рассчитанным количеством
натрия в жидком аммиаке гексаал кил (соответственно, арил)диплюмбаны
[8, 9, 24, 27]. Интересно отметить, что в алифатическом ряду вместо
щелочных металлов можно применять алюминий. Так, гексаэтилдиплюмбан
получен [28, 29] взаимодействием щелочного раствора хлористого триэтил-
свинца с алюминием.
Получение гексаэтилдиплюмбана [29]. К раствору 7 г хлористого триэтилсвинцг в 50 мл
2,5 N едкого кали прибавляют 1,5 г алюминиевого порошка. Смесь выдерживают при 15—
18° С в течение 20 час, после чего гексаэтилдиплюмбан отделяют от водного слоя, быстро
фильтруют, освобождают от кислорода воздуха в вакууме и высушивают сульфатом натрия.
Приготовленный таким образом гексаэтилдиплюмбан в отсутствие кислорода и влаги
воздуха не изменяется в течение 5—6 дней. Выход количественный; dj® 1,9473.
Подробное исследование реакции показало, что она приводит к
образованию смесей гексаэтилдиплюмбана и диэтилсвинца [29, 30]. Состав смесей
можно варьировать изменением продолжительности эксперимента или
кратковременным нагреванием реакционной смеси.
Получение гексафенилдиплюмбана [8]. К взвеси 2,24 г (0,004 моля) йодистого
трифенилсвинца в 100 мл жидкого аммиака прибавляют небольшими порциями 0,093 г (0,04 г-атома)
натрия. После удаления аммиака остаток извлекают хлороформом и гексафепилдиплюмбан
осаждают спиртом. Выход 94%; т. пл. 155° С.
Аналогично получен гекса-^-толилдиплю^мбан. Гексаарилдиплюмбаны
целесообразнее получать из хлористого свинца и реактива Гриньяра (см.
стр. 552).
Опыты по получению соединений типа R2R'PbPbR'R2 и (R2Pb)„ не
увенчались успехом [31]. Также не удается выделить чистые вещества из
. Свинцовоорганшеские соединения с двумя и более атомами металла 629
продуктов реакции триэтилплюмбилнатрия с двухлористым дициклопен-
тадиенилтитаном [32].
СИНТЕЗ И РЕАКЦИИ СОЕДИНЕНИЙ ТИПА
R3PbM(CO)„ и R3Pb[M(CO)„]2
В литературе описан ряд свинцовоорганических соединений с карбони
лами металлов VI—VIII групп периодической системы. Несмеяновым,
Анисимовым с сотр. [1, 33] разработан метод синтеза и исследованы реакции
соединений, в которых атом свинца связан с молибденом, вольфрамом или
рением. Взаимодействием 1,45 г хлористого трифенилсвинца с натрийпен-
такарбонилом рения (из 1 г Re2(CO)i0 и 20 мл 0,8%-ной амальгамы натрия в
50 мл тетрагидрофурана) при 50°С в течение 1,5 часа получены белоснежные
кристаллы трифенилсвинецпентакарбонила рения с т. пл. 131—133°С.
Выход 78% [1].
Получение трифенилсвинец-л-циклопентадиенилтрикарбонилвольфрама
С5Н;з(СО)зДМРЬ(СбН5)з [33]. К циклопентадиенилнатрию, полученному из 1,15г натрия и 3,5 г
циклопентадиена,'в 30 мл диглима прибавляют 17,6 г гексакарбонила вольфрама,
растворенного в 40 мл диглима. Реакционную смесь нагревают при 120—130° С до прекращения
выделения СО. Затем при комнатной температуре прибавляют 10,5 г хлористого
трифенилсвинца в \0 мл диглима. После сублимации, хроматографии и многократной
перекристаллизации (гептан — бензол) выделяют 4 г кристаллов желтого цвета с т. пл. 210—211,5° С .
Из продуктов реакции хлористого трифенилсвинца с NaMo(CO)3C5H5
в тетрагидрофуране выделены желтые кристаллы С5Н5(СО)3МюРЬ(С8Н5)з с
т. пл. 205°С [33]. Найдено [33], что при действии на эти соединения электро-
фильных реагентов происходит разрыв связи металл—металл. Как показано
теми же авторами, в случае соответствующих оловоорганических
соединений эта связь сохраняется, а происходит отщепление фенильного радикала
(см. стр. 508).
Галогениды триалкил- и диалкилсвинца легко реагируют с натриймарга-
нецпентакарбонилом в среде тетрагидрофурана [34]:
R4_nPbCln + n NaMn (CO)5 -> R4_„Pb [Mn[(CO)5y+ n NaCl.
Так получены (С2Н5)2РЬ[Мп(СО)5]2 (т. пл. 77—79°С), (С2Н5)3РЬМп(СО)5
(т. кип. 70—73° С/0,1 мм), (СН3)3РЬМп(СО)5 (т. пл. 30—31°С), а также
(СН3)2РЬ[Мп(СО)5]2 (т. пл. 108—110°С). Хлористый водород атакует связь
РЬ — Мп в трифенилсвинецпентакарбонилмарганцепри 25° С с образованием
ClMn(CO)r), бензола или хлорбензола и неидентифицированных
содержащих свинец продуктов. Аналогично хлорирование (С6Н5)2РЬ[Мп(СО)5]2
в четыреххлористом углероде при 5°С приводит к С1Мп(СО)5 и твердому
продукту, содержащему остатки три- и диалкилсвинца [34].
Свинцовоорганические кобальткарбонилы получены [35,36] обменом
гидроокиси трициклогексилсвинца, галогенидов три- и дифенилсвинца с
NaCo(CO)4 в петрюлейном эфире. Это кристаллические вещества, хорошо
растворимые в петролейном эфире. Трифенилсвинецкобальттетракарбонил
(т. пл. 123°С) окрашен в желтый цвет, а (С6Н5)2РЬ[Со(СО)4]2 (т. разл. 103—
106°С) — в темно-красный.
Взаимодействием гидроокисей триалкилсвинца с Ca[HFe(CO)4l2 в
петролейном эфире получен ряд железокарбонилов диалкилсвинца [37—40].
Высказано предположение [39], что железотетракарбонилы триалкилсвинца
неустойчивы и легко диспропорционируют. Авторы считают возможной
последовательность следующих реакций:
2 R3PbOH -j- H2Fe (СО)4 -» (R3Pb)2Fe (СО)4 + Н20,
(R3Pb),>Fe(CQ)4-* R4Pb + R2PbFe(CO)4.
630
Свинцовоорганические соединения
В алициклическом и ароматическом рядах были выделены и соединения
типа (R3Pb)2Fe(CO)4, которые в предыдущем случае являются
промежуточными продуктами. Впоследствии было установлено [41], что ион Fe(CO)4~
с галогенидами три- и диалкилсвинца образует [(R3Pb)2Fe(CO)4]2 и
[Fe(CO)4(PbR2)2]2 соответственно. Реакции проводили в водном растворе при
обычной температуре.
Найдено [40], что при действии на бис-(трифенилплюмбил)тетракарбо-
нилжелезо двухлористой (или двубромистой) ртути выделяется галогенид
трифенилсвинца и HgFe(CO)4. Если же к раствору [(C6H5)3Pb]2Fe(CO)4
прибавить CdJ2, CuCl2-2H20, СоС12»6Н20 или BiBr3, то соединения типа
MnFe(CO)4 не образуются. В этих случаях из смеси выделен тетрафенилсви-
нец, что указывает на диспропорционирование исходного вещества.
ЛИТЕРАТУРА
1. Несмеянов А. Н., Анисимов К. Н., Колобова Н. Е., Хандож-
к о В. Н., ДАН СССР, 156, 383 (1964).
2. Н о 1 1 i d а у А. К., Р a s s G., J. Chem. Soc, 1958, 3485.
3. Н о 1 1 i d а у А. К., Р е n d 1 е b u г у R. E., J. Chem. Soc, 1965, 6659.
4. G i 1 m а п Н., М а г г s О. L., S i m S. Y., J. Org. Chem., 27, 4232 (1962).
5. T amborskiC, FordF. E., LehnW.L, Moore G. J., SoloskiE. J.,
J. Org. Chem., 27, 619 (1962).
6. Sc h urn a n n H., T horn K. F., Sc h m i d t M., J. Organometal. Chem., 4, 28(1965).
7. WillemsensL.C, van der KerkG. J. M., J. Organometal. Chem., 4, 34
(1965).
8. G i 1 m a n H., В a i 1 i e J. C, J. Am. Chem. Soc, 61, 731 (1939).
9. FosterL.S., Dix W.M.,Grunt festl. J., J. Am. Chem. Soc, 61, 1685 (1939).
10. В i n d s с h a d 1 e r E., Iowa State Coll. J. Sci., 16, 33 (1941); С. А., 36, 4476 (1942).
11. G i 1 m a n H., L e e p e r R. W., J. Org. Chem., 16, 466 (1951).
12. GilmanH., SummersL., LeeperR. W., J. Org. Chem., 17, 630 (1952).
13. GilmanH., MarrsO.L, T r e p k a W. J., D i e h U. W., J. Org. Chem., 27,
1260 (1962).
14. G i 1 m a n H., В i n d s с h a d 1 e r E., J. Org. Chem., 18, 1675 (1953).
15. SummersL., Iowa State Coll. J. Sci., 26, 292(1952).
16. В i ndsch ad 1 er E., GilmanH., Proc. Iowa Acad. Sci., 48, 273 (1941); С. А.,
36, 1595 (1942).
17. D'AnsJ., ZimmerH, EndrulatE., LiibkeK., Naturwiss., 39, 450 (1952).
18. WillemsensL.C, van der KerkG. J.M., J. Organometal. Chem., 2, 271
(1964).
19. GlocklingR, H о о t о n К., К i n g s t о n D., J. Chem. Soc, 1961, 4405.
20. J и e n g e E. C, S e у f e r t h D., J. Org. Chem., 26, 563 (1961).
21. GlocklingR, KingstonD., J. Chem. Soc, 1959, 3001.
22. 3 а в г о р о д н и й В. С, П е т р о в А. А., ДАН СССР, 143, 855 (1962).
23. G i 1 m a n Н., М е 1 s t г о m D. S., J. Am. Chem. Soc, 72, 2953 (1950).
24. В a i 1 i e J. C, Iowa State Coll. J. Sci., 14, 8 (1939); С. А., 34, 6242 (1940).
25. W i 1 1 e m s e n s L. C, v a n der Kerk G. J. M., Rec trav. chim., 84, 43 (1965).
26. G i 1 m a n H., SummersL, J. Am. Chem. Soc, 74, 5924 (1952).
27. С a 1 i n g a e r t G., S о г о о s H., J. Org.Chem., 2, 535 (1938).
28. HeinR, KleinA., Ber., 71, 2381 (1938).
29. P а з у в а е в Г. А., ВязанкинН.С, В ы ш и н с к и й Н. Н., ЖОХ, 29, 3662
(1959).
30. Р а з у в а е в Г. А., В я з а н к и н Н. С, В ы ш и н с к и й Н. Н., ЖОХ, 30, 967
(1960).
31. А р р е г s о п L. D., Iowa State Coll. J. Sci., 16, 7 (1941); С. А., 36, 4476 (1942).
32. D i с k s о n R. S., W e s t B. O., Austral. J. Chem., 14, 555 (19dl).
33. Несмеянов A. H., Анисимов К. Н., Колобова Н. Е., Захар о-
ва М. Я., ДАН СССР, 156, 612 (1964).
34. G о г s i с h R. D., J. Am. Chem. Soc, 84, 2486 (1962).
35. HeinF., Kleinert P., J e h n W., Naturwiss., 44, 34 (1957).
36. Hein F., Jehn W., Ann., 684, 4 (1965).
37. HeinF., PoblothH., Z. anorgan. allgem. Chem., 248, 84 (1941).
38. H e i n F., H e u s e r E., Z. anorgan. allgem. Chem., 254, 138 (1947).
39. H e i n F., H e u s e r E., Z. anorgan. allgem. Chem., 255, 125 (1947).
40. H e i n F., Sc h n e i t e r H., Z. anorgan allgem. Chem., 259, 183 (1949).
41. К a h n О., В i g о r g n e M., Compt. rend., 261, 2483 (1965).
Глава XVI
АНАЛИЗ СВИНЦОВООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для качественного определения свинца в свинцовоорганических
соединениях применяют сожжение образца на металлической или лучше
платиновой пластинке в пламени горелки. При наличии свинца на пластинке
образуется серый налет [1].
Способность дитизона образовывать окрашенные комплексы с свинцово-
органическими соединениями (комплекс с R3PbX окрашен в желтый цвет,
R2PbX2 — в оранжевый, РЬХ2 — в красный) может быть использована для
качественного определения этих соединений [2—4].
Количественное определение свинца в свинцовоорганических
соединениях сводится к различным методам минерализации органического
соединения и определения неорганических соединений свинца весовым, объемным
методом или рядом физико-химических методов [5].
Для летучих свинцовоорганических соединений рекомендуют
разложение 68%-ной азотной кислотой в запаянных трубках по Кариусу. Для
предупреждения взрыва трубки следует вскрыть после нагревания в течение
12 час. до 150° С, выпустить газы и, снова запаяв, нагревать еще 12 час. до
200° С [6].
Для более высококипящих веществ применим метод, состоящий в
разложении свинцовоорганического соединения крепкой серной кислотой и
окислении перманганатом калия [7,8]. К навеске исследуемого вещества
приливают 10 мл охлажденной льдом дымящейся серной кислоты (10% олеума),
причем раствор может оставаться прозрачным или будет выделяться
небольшой бесцветный осадок. Затем прибавляют по каплям концентрированный
водный раствор перманганата калия, сначала при сильном охлаждении до
тех пор, пока окраска перманганата не будет сохраняться в течение
нескольких минут даже при нагревании, после чего раствор выпаривают
досуха. Сульфат свинца определяют весовым путем.
Удобно применять и разложение с помощью галоидов, особенно для
летучих свинцовоорганических соединений [6]. Определенное количество
вещества помещают в колбу, растворяют примерно в 10-кратном количестве
чистого четыреххлористого углерода и смешивают при охлаждении с
большим избытком 10%-ного раствора брома в четыреххлористом углероде.
Смесь выпаривают на водяной бане почти досуха, приливают немного
абсолютного спирта, кипятят некоторое время и хорошо охлаждают.
Фильтруют через тигель с фильтрующим дном и промывают выпавший в осадок
чистый двубромистый свинец небольшим количеством охлажденного льдом
спирта.
Можно применять следующий метод [9]. Свинцовоорганическое
соединение (0,2 г) осторожно смешивается с несколькими миллилитрами брома. После
энергичной реакции прибавляют 20 мл воды и избыток брома выпаривают.
632
Свинцовоорганические соединения Лит. стр. 633—635
Прибавляют 4 N раствор аммиака, пока весь свинец не выделится в виде
гидроокиси. Полученную гидроокись растворяют в 10%-ной уксусной
кислоте и из горячего раствора осаждают хромат свинца. Свинец далее определяют
объемным методом как обычно.
Некоторые свинцовоорганические соединения, например тетрафенил-
свинец, рекомендуют разлагать и определять объемным методом, действуя
на бензольный раствор избытком раствора иода при нагревании с
одновременным облучением лампой мощностью 300 вт. Реакция протекает по
уравнению:
РЬ(СвН3)4 + 3 J2 -* PbJ2 + 4C,5H3J.
Избыток иода титруют раствором тиосульфата натрия без добавления
крахмала не до полного обесцвечивания, а до слабо-розовой окраски [10].
Наиболее удобным и широко применяемым методом анализа нелетучих
свинцовоорганических соединений является метод Гилмана и Робинзона
[11]. Навеску около 0,5 г помещают в стакан из тугоплавкого стекла емкостью
400 мл и добавляют 7,5 мл концентрированной серной кислоты. Стакан,
покрытый часовым стеклом, очень осторожно греют на небольшом пламени.
Слишком бурное течение реакции предупреждают, отставляя пламя и
приводя жидкость в круговое движение, как только появятся первые признаки
реакции (50—90°С). Осторожно греют до тех пор, пока в серной кислоте
совсем не исчезнут взвешенные крупинки вещества и не соберется на дне
осадок сернокислого свинца. Тогда содержимое греют до появления белого дыма.
По охлаждении примерно до 80° С добавляют 1—1,5 мл азотной кислоты
(уд. в. 1,42). По прекращении экзотермической реакции удаляют часовое
стекло и осторожно греют до появления белых паров. Операция повторяется
с 1 мл азотной кислоты и так до просветления серной кислоты и полного
растворения сернокиолого свинца в горячей серной кислоте. По
охлаждении до 30°С, 5—10 мл воды смывают частицы осадка со стенок,
смешивают воду с кислотой, снова греют до появления белых паров и так
дважды. Выпаривание лучше всего вести на слабо нагретой
электрической плитке. В охлажденный раствор приливают 150жлводы я 100 мл 95%-
ного спирта. Через час осадок фильтруют через тигель с пористым дном,
промывают 10%-ной серной кислотой и 95%-ным спиртом до удаления
серной кислоты. Тигель сушат при 110° и взвешивают сернокислый
свинец.
Отдельные авторы [12] применяют также следующий метод
определения свинца в свинцовоорганических соединениях: 0,15—0,2 г вещества
сжигают на газовой горелке с концентрированной азотной кислотой до
прекращения выделения паров N02 (полное удаление N02 имеет
существенное значение). К осадку азотнокислого свинца прибавляют 100 мл
воды и затем AN раствор аммиака до полного осаждения гидроокиси
свинца. Затем прибавляют 10% раствор уксусной кислоты до получения
прозрачного раствора, который нагревают до 90°С. Из бюретки
прибавляют 1 % раствор двухромовокислого калия. После каждого
прибавления раствор встряхивают и повторно нагревают. Когда образование осадка
и коагулирование прекратится, прибавляют 25%-ный избыток
раствора двухромовокислого калия и смесь нагревают 15 мин. при
постоянном встряхивании. Если жидкость над осадком не будет совсем
прозрачна, то добавляют еще 2 мл двухромовокислого калия и нагревание
повторяют. Осадок хромата свинца собирают на фильтре, промывают
горячим 5%-ным раствором ацетата аммония. Фильтровальную бумагу
прокалывают и осадок смывают горячей разбавленной соляной кислотой
в коническую колбу. Затем раствор охлаждают и титруют 0,1 N раствором
сульфата железа, используя в качестве индикатора фенантролинжелезо.
Анализ
633
Определение углерода, водорода и галоида в свинцовоорганических
соединениях проводится обычным методом [5]. Иногда рекомендуют при
анализе свинцовоорганических соединений разбавлять пробу мелкоизмельчен-
ной окисью меди и кварцевым песком [13].
Для определения углерода и водорода в металлоорганических, в том
числе и свинцовоорганических, соединениях иногда рекомендуют метод
сожжения на Со304 и серебряной сетке [14], а также и другие методы [15].
Так как тетраэтилсвинец широко применяют в качестве антидетонатора
моторного топлива, то методы его определения в бензине очень
разнообразны. Здесь можно применять как химические методы [5,16—20], так и
различные физико-химические методы [21—49].
Определение примеси гексаалкилдиплюмбана в тетраэтилсвинце для
промышленности имеет важное значение, поэтому в литературе разработан
ряд методов его определения [50—57].
В связи с особой токсичностью тетраэтилсвинца разработан ряд
аналитических определений его малых количеств в воде и в воздухе [55—63].
Разработано несколько методов обнаружения и удаления
свинцовоорганических соединений из воздуха, воды и бензина [64—76].
Наряду с широко разработанными методами определения
тетраэтилсвинца, анализ других свинцовоорганических соединений разработан
относительно мало. Имеются отдельные работы по газо-жидкостной
хроматографии [77, 78], бумажной хроматографии [79, 80], по спектрофотометриче-
скому методу [81, 82]. Проведен масс-спектрометрический анализ тетра-
метил- и тетраэтилсвинца [83], изотопный анализ тетраметилсвинца [84],
определение свинца в органических материалах методами колориметрии [85],
а также амперокулонометрический метод анализа тетраметил- и
тетраэтилсвинца [86].
ЛИТЕРАТУРА
1. Sp i al ter L., В a 1 lesterM., Analyt. Chem., 34, 1183 (1962).
2. BarbieriR.TagliaviniG., BellucoU., Ric. Sci., 30, 1963^(1960); C. A.,
55, 14159f (1961).
3. Hen der sonS. R., SnyderL.Y., Anal. Chem., 33, 1172 (1961).
4. I r v i n g H., С о x J. J., J. Chem. Soc, 1961, 1470.
*5. Губен-Вейль. Методы органической химии, т. 2. Методы анализа. М., ГНТИХЛ,
1963, стр. 76.
6. G г u t t n е г G., К г a u s е Е., Вег., 49, 1129 (1916).
7. Р о 1 i s А., Вег., 19, 1025 (1886); 20, 718 (1887).
8. К о т о н М. М., ЖОХ, 9, 2283 (1939).
9. Е m е 1 е u s H. I., E v a n s P. R., J. Chem. Soc, 1964, 510.
10. Н ei n Fr., К 1 ei n A., MeseeH. J., Z. analyt. Chem., 115, 182 (1938).
11. G i 1 m a n H., R о b i nso n J., J. Am. Chem. Soc, 50, 1714 (1929).
12. S a u n d e r s B. C, S t а с е у G. J., J. Chem. Soc, 1949, 919.
13. H e i n Fr., HeuserE., Z. anorgan. allgem. Chem., 254, 143 (1947).
14. Gawargions Y.,A., M а с d о n a 1 d A. M. G., Analyt. chim. Acta, 27, 119 (1962);
РЖХим., 1963, 72152.
15. С о 1 a i t i s D., L e s b r e M., Bull. Soc. chim. France, 1952, 1069.
16. M i d g 1 e у Th., H о с h w a 1 t С. А., С a 1 i n g a e r t G., J. Am. Chem. Soc, 45, 1822
(1923).
17. Lunge-Berl, Chemisch-technische Untersuchungsmethoder, Erganzungswerk zur
8 AuflL, Teil 2, Springen. Berlin, 1939, S. 32.
18. J a h r K. F., К г о 1 1 e r E., P e r n о 1 1 I., Chem., 2, 88 (1950).
19. J a h r K. F., P e r n о 1 1 I., Z. analyt. Chem., 129, 339 (1949).
20. KrolerE., Z. analyt. Chem., 129, 6 (1949).
21. GarciaEscolar N., Vino Martinez E., Anales Real Soc, Espan, Fis. Quim.
(Madrid), Ser. B, 53, 161 (1957); С. А., 53, 6986 12647 (1959); 54, 441 (1958).
22. AnzaldiO., Bol. Inform. Petrol. (Buenos Airos), 312, 170 (1959); С. А., 53, 19361
(1959).
23. Пташинский И. А., Фролова М. К., Труды Всесоюзн. н.-и. ин-та по пере-
$i работке нефти, газа и получения искусств, жидк. топлива, 1957, 181; С. А., 53 15538d
(1959).
634
Свинцовоорганические соединения
24. С h г i s t о f f e r s о n G. D., В е а с h J. Y., Collog. Spectros. Intern. 9th (Lyons, .1961).
3, 491 (1962); С. А., 58, 13670h (1963).
25. LoroiieD., P a u 1 G. Rev. Inst. Franc. Petrole Ann. Combust. Liquides, 17, 830
(1962); С A., 57, 11452c (1962).
26. LovelockJ.E., Z 1 a t к i s A., Anal. Chem., 33, 1958 (1961).
27. ParkerW.W., SmithG.Z., HudsonR.L, Anal. Chem., 33, 1170 (1961); 35,
1334 (1963).
28. Ho war d H. E., Fer guso n W. C, Sny der L. R., Anal. Chem., 32, 1814 (1960).
29. VanRysselbergeJ., LeysenR., Nature, 189, 478 (1961).
30. В uel 1 B. E., Am. Soc. Testing Mater., Spec. Tech. Publ., 269, 157 (1960); С. А., 55,
4936a (1961).
31. О к a d о Т., U e d а Т., К о h z u m а Т., Bunko Kenkyu, 4, 30 (1956); С. А., 53, 6586a
(1959).
32. F a r к a s M., F о d о r n i C. P., Magyar kern, folyoirat, 69, 407 (1963); РЖХим., 1964,
152187
33. W о г к P. L., J u 1 i a r d A. L., Anal. Chem., 28, 1261 (1956).
34. КоямаК., ТагутиЮ., Э г у т и С, Бунсэки кагаку, Japan Analyst., 12, 435
(1963); РЖХим, 1964, 12239.
35. A n z a 1 d i О., Bol. inform, petrol., 1959, 170; РЖХим., 1960, 28268.
36. G a r с i a E. L., P a z С. М., Inform, quim. analit., 18, 66 (1964); РЖХим., 1965, 5Г155.
37. В uel 1 В. Е., Sympos. Spectroscopy, Philadelphia, Pa, Am. Soc. Test. Mater., 1960, 157;
РЖХим., 1964, 6Г223.
38. L о v e 1 о с k J. E., Z 1 a t k i s A., Anal. Chem., 33, 1958 (1961).
39. В e 1 1 о m о A., D'AmoreG., Chem. Tech. (Berlin), 14, 341 (1962).
40. LovelockJ.E., GregoryN.L, Gas. Chromatog. Intern. Simp. (1961), 3, 219
(1962); С. А., 58, 3865h (1963).
41. DawsonH. J., Anal. Chem., 35, 542 (1963).
42. Дмитриевский В. С, Химия и технология топлив и масел, 3, 59 (1958); С. А.,
53, 3671h (1959).
43. BraunK. H., Chem. Tech. (Berlin), 10, 159 (1958).
44. В r a n d t M., v a n der В e r g R. H., Anal. Chem., 31, 1921 (1959).
45. S а о r i H., Shoseki Giho, 2, 182 (1958); С. А., 53, 15538e (1959).
46. У в а р о в а Е. И., В а н я а р к и н а Н. М., Зав. лаб., 26, 1097 (1960).
47. G е 1 i u s R., Prenssner К. R., Chem. Tech. (Berlin), 15, 290 (1963).
48. В 1 u m e n t h a 1 A., Mitt. GebieteLebensm. Hyg., 51, 159 (1960); С. А., 55, 2071 (1961).
49. T a g 1 i a v i n i G., Chim. Ind. (Milan), 49, 902 (1957).
50. Bahr С Z. anorg. Chem., 253, 335 (1947).
51. Гольдштейн А. Л., Л а п и с о в а Н. П., Штифман Л. Мм ЖАХ, 17, 143
(1962).
52. Л а п и с о в а Н. П., Г о л ь д ш т е й н А. Л., ЖАХ, 16, 508 (1961).
53. В е 1 1 и с о U., T a g 1 i a v i n i G., В a r b i e r i R., Ric. Sci., 30, 1675 (1960); С. А.,
55, 14175d (1951). A
54. T a gl i a v i n i G., В e 1 1 uco U., R iccoboni L., Ric. Sci., Rend. Sez., A, [2] if
338 (1961); С. А., 57, 5306g (1962).
55. D e V r i e s J. E., L a u w - Z e с h a A., P e 1 1 e с е г A., Anal. Chem., 31, 1995 (1959).
56. ВертиулинаЛ. Н., Коршунов И. А., Хим. наука и промышленность, 4,
136 (1959).
57. Р у д н е в с к и й Н. К., В ы ш и н с к и й Н. Н., Изв. АН СССР, серия физ., 23, 1228
(1959).
58. ХрусталеваВ. А., Информ. бюлл. Моск. н.-и. ин-та санитарии и гигиены, 1961,
11; С. А., 54, 5328в (1960).
59. ДулинскийВ., КозлингЗ., Газ, вода и техническая санитария, 34, 258 (1960);
С. А., 55, 6747е (1961).
60. X р у с т а л е в а В. А., Гигиена и санитария, 25, 57 (1960).
61. ХрусталеваВ. А., Сб. «Методы определения вредных веществ в воздухе». М., 1961,
стр. 105; РЖХим., 1962, 18Д77.
62. Snyder L. J., H endersonS. R., Anal. Chem., 33, 1175(1961); Пат. США 3071446
(1963); С. А., 58, 6200 (1963).
63. Г е р н е т Е. В., Труды Горьк. н.-и. ин-та гигиены труда и проф. болезней, 6, 5 (1957);
С. А., 54, 6391 i (1960).
64. Cho udh ur i В. К., V i s w a n a t h a n С. R., VatsS.S., A i у а г A. R.,
Defence Sci., J. (New Delbi), 2, 34, (1961); С. А., 56, 6286b (1962).
65. В г е у n a n Т., Angew. Chem., 62, 430 (1950).
66. С о о k E. В., S m i t h R. W., В r i n k 1 e у S. R., U. S. Bur. Mines, Rept. Invest.,
N 4947 15pp (1953); С. А., 47, 6127f (1953).
67. S h a p i г о А., О 1 s о n D. A. H. (Socony Mobie Oil Co); Пат. США 2969320 (1960);
С. А., 55, 14898b (1961).
68. К о з ю р а А. С, М и р н а я А. П., Л а д М. Д., Труды объединенного семинара по
гидротехнике и водохоз. стр-ву, вып. 2. Харьков, 1959 (1960), 64; РЖХим., 1962, 9И317.
Анализ
635
69. Е м е л ь я н о в Б. В., Ш е м я к и н а 3. Н., X а л я в и н М. Н., Пат. СССР 128466
(1960); С. А., 54, 15430в (1960).
70. КозюраА. С, Смирнова А. Н., Мирназ А. П., Труды объединенного
семинара по гидротехнике и водохоз. стр-ву, вып. 3. Харьков, 1961, 55; РЖХим., 1962,
12И358.
71. Е м е л ь я нов Б. В., Шемякина 3. Н., X а л я в и н М. Н., Пат. СССР 127070
(1960); С. А., 54, 18842в (1960).
72. 3 е м с к о в И. Ф., X а л я в и н М. Н., Хим. пром., 1958, 500.
73. 3 е м с к о в И. Ф., Хим. пром. 1961, 290.
74. Земсков И. Ф., СидельниковаГ. И., Труды по химии и хим. технологии
(Горький), 1, 16 (1960).
75. ЗемсковИ. Ф., СидельниковаГ. И., Труды по химии и хим. технологии
(Горький), 3, 20 (1960).
76. 3 е м с к о в И. Ф., С и д е л ь н и к о в а Г. И., ЖПХ, 35, 469 (1962).
77. A b е 1 Е. W., N i с к 1 е s s G., Р о 1 1 а г d F. H., Proc. Chem. Soc, 1960, 288.
78. В о n e 1 1 i E. J., H а г t m a n n H., Anal. Chem., 35, 1980 (1963).
79. SchaferH., Z. analyt. Chem., 180, 15 (1961).
80. В a r b i e r i R., В e 1 1 u с о U., T a g 1 i a v i n i G., Ann. Chim. (Rome), 48, 940 (1958);
С. А., 53, 9901h (1959); Ric. Sci., 30, 1671 (1960); С. А., 55, 14175c (1961).
81. HendersonS.R., S n у d e r L. J., Anal. Chem., 33, 1172 (1961).
82. С u г г у R. P., Anal. Chem., 31, 960 (1959).
83. G h a t e M. R., В h i d e K. N., Indian J. Chem., 2, 243 (1964); РЖХим., 1965, ЗГ224.
84. В a t e G. L., M i 1 1 e r D. S., К u 1 p J. L., Anal. Chem., 29, 84 (1957).
85. G a g e J. C, Analyst., 80, 789 (1955); С. А., 50, 1518 (1956).
86. P i 1 1 о n i G., P 1 a z z о g n a G., Ricerca scient., 4, 27, Parte 2, Ser. A (1965); РЖХим.,
1965, 5Г218.
АВТОРСКИЙ УКАЗАТЕЛЬ *
Абдуллаев Н. Д. 43 (121, 124), 66 (35, 37а)
Абрамова А. И. 200 (44), 225 (198), 270, 271
(9), 355 (189, 190), 364 (190), 453 (46),
464, 465 (60, 62), 468, 469 (60), 471 (62),
474, 475 (60)
Абрамова Л. В. 182 (21—26), 186 (21), 209,
211 (53), 401 (132), 520 (101)
Авдеева В. И. 56 (62), 71 (48)
Авербух Б. С. 182 (26)
Адрова Н. А. 250 (310), 457 (17), 513 (45),
514 (45, 48), 515 (57)
АзербаевИ. Н. 175 (200)
АимиМ. 520(150)
Акиеси С. 520 (106)
Александров А. П. 269(6, 7), 331, 332 (30,
33), 384, 385 (39), 598 (34)
Александров А. Ю. 165 (133, 136, 137), 391
(82)
Александров Ю. А. 15 (20), 165 (54, 71, 130,
134 135), 398 (79), 399 (80—82), 415 (384),
416 (79, 384), 438 (17, 18), 439 (18), 535
(25), 587 (133, 134, 140), 597 (24—26),
598 (31), 601 (8, 9), 602 (22), 605, 606 (34—
36), 612 (13—15), 613 (20)
Александрова Г. Е. 589 (182), 598 (50)
Алексеева В. П. 160 (25)
Алиев М. И. 66 (35)
Али-Заде И. Г. 66 (37а)
АменицкаяР. В. 589(188)
Андрианов К. А. 189 (82), 412 (337), 414 (363),
516 (68, 69)'
Анисимов К. Н. 109 (101—103), 155, 156 (55,
56), 157(55), 165(124, 125), 411(324—
327), 505 (84—87), 506 (84—86), 507 (87),
508 (84—87), 509 (96), 608 (49, 50), 623,
628 (1), 629(1, 33)
Антонова А. Б. 109(101, 102), 155, 156(56)
Арбузов Б. А. 412 (341—343), 451 (20), 608
(64)
Армейская Л. В. 168 (194)
АрхиповаИ. Л. 536 (151)
Асаи X. 512 (7)
Асо Т. 520(106)
Асланов И. А.'43 (ИЗ—120, 122, 123), 66
(36, 37)
Атавин А. С. 408 (273), 414 (356), 416, 417
(273)
Ашбель Ф. Б. 527 (73)
Баженова Н. Н. 614 (30)
Бай Л. И. 226 (216)
Баландин А. А. 165 (171), 207, 208 (25)
Балашова Л. Д. 411 (315, 316)
Барони Е. Е. 536(133)
Барчуков А. И. 15 (10), 16 (89)
Баталов А. П. 589 (187—189)
БатуевМ. И. 15 (12)
Бауков Ю. И. 15 (65), 55 (56—59), 56 (58,
62, 63), 57 (59), 71 (48, 49), 119 (38—40),
138 (53—56), 165(120), 271 (13), 312(66),
353, 363 (181, 182), 408(252), 416 (384а),
486 (115—117), 487 (117)
Безуглый В. Д. 527 (64)
Белавин И. Ю. 55, 57 (59), 138 (55), 486,
487 (117)
Беликин А. В. 209 (54)
Берлин А. А. 402 (153), 520 (86)
Берлин Л. Е. 587, 588 (136)
Бирюков И. П. 15 (61)
Бобашинская Ч. С. 233, 236, 237, 249 (254),
346, 350 (151)
Богданов И. Ф. 590 (199, 200)
Богомолов С. Г. 535 (20)
Бонно И. 159 (1), 175 (222), 536 (138)
Борисенко В. В. 224 (186), 338 (79), 378 (25),
382, 383 (24), 397 (56), 399(56, 86, 88),
513 (24, 25, 32)
Борисов А. Е. 41(107), 47(6, 7), 48 (6),
69 (44), 120 (42), 165 (36), 199 (40—42),
200(40, 44—46, 48), 201 (40, 41), 202 (41,
43, 46, 53), 208, 213, 216 (35), 225 (198), 242
(35), 270, 271 (9), 288 (43—45), 289 (43, 45а),
344 (132), 355 (187, 189—192), 356, 357
(191, 213), 364 (190), 453 (45, 46), 464,
465 (60, 62), 468, 469 (60), 471 (62), 474,
475 (60), 564 (80)
Борисов С. Н. 160 (29)
Борисова А. И. 188 (76), 338 (79), 378 (25),
382, 383 (24), 397 (51, 52), 399 (86), 401 (52),
513 (23—25, 28, 29)
Бородавко И. А. 365, 366 (258), 443 (54)
Бородина Г. М. 197(35), 440(35), 551 (29),
582 (66), 608 (71), 618 (16)
Боршаговский Б. В. 165 (125)
Бочкарев М. Н. 131 (36, 37), 165 (96), 464
(62а)
Бочкарева Г. Л. 321 (15)
Брайнина Э. М. 344 (134)
Брауер Г. 195 (23)
Браун Д. В. 16 (106), 175 (220), 537 (166)
Бревнова Т. Н. 367 (272), 368 (273), 397,
400, 401, 422(61), 442(49), 528(87)
Брегадзе В. И. 165 (130), 248 (304)
Брегер А. X. 182(26)
Брилкина Т. Г. 8 (12), 16 (104), 175 (208),
358 (226), 399 (80, 83), 408 (270), 415, 416
* Цифры без скобок обозначают страницы, где даются ссылки на работы авторов;
цифры в скобках — порядковые номера ссылок, приводимые в конце соответствующих
глав.
Авторский указатель
637
(384), 537 (164), 587 (133—135), 597 (24—
27), 601 (8, 9), 602 (19, 22), 605, 606 (34—
36), 606 (37), 612 (13, 14, 16, 20), 613 (20)
Бродская Э. И. 165 (39)
Брукер А. Б. 411(315, 316), 571, 572
(7), 572 (10)
Брюханов В. А. 165 (128)
БуачидзеМ. А. 66 (31,32,34)
Бубнов Ю. Н. 68 (39)
Бугоркова А. А. 116(15, 16), 117(15), 162
(11, 12), 528 (77, 78)
Бурлаченко Г. С. 15 (65), 55 (58, 59), 56 (58,
59, 62, 63), 71 (48, 49), 119 (38), 165 (120)
Бурмистрова В. И. 536(111)
Бурнашова Т. Д. 325, 326 (12)
Быховская М. Б. 159 (7), 512 (10)
Бычков В. Т. 140 (63, 64, 64а, 65), 154, 155
(51), 155 (57, 58), 442, 443 (53), 445 (65),
463, 467, 468, 491 (59)
Вайнштейн Б. И. 182 (26)
Ван Ши-хуа 288 (44)
Ваняаркина Н. М. 634 (46)
Васильева Т. В. 189 (82), 412 (337)
Варшавский С. Л. 168 (193)
Варшев Н. А. 165 (175)
Вашков В. И. 382 (19), 521 (214)
Вахомчик Л. П. 28 (36), 83 (48)
Вдовин В. М. 160 (26)
Вердиева С. Ш. 66 (36)
Вереникина С. Г. 605 (25, 26)
Верещинский И. В. 182 (25, 26а)
Вертиулина Л. Н. 633 (56)
Веселкова И. А. 535 (20)
Взенкова Г. Я- 15(12), 19(10, И), 39(81,
95), 60(3), 116(17), 117 (17, 23), 122(6),
125 (6, 7), 126 (6), 162 (3)
Викторова И. М. 432 (35)
Вилков Л. В. 16 (80), 71 (56)
Вильчевская В. Д. 453 (45)
Виноградова В. Н. 54 (55), 92 (47)
Виталина М. Д. 162 (5, 6)
Власов В. М. 227 (223), 254 (333), 272, 273
(26), 325(2, 6—11, 13), 326 (2, 7, 8—11),
327 (8—10)
Волков В. Ф. 165 (51)
Волькенау В. А. 200 (48), 344 (132)
Вольпин М. Е. 15 (22), 71 (50—56), 491 (122)
Воронков М. Г. 8 (10, 11), 15 (81), 403 (156а)
Вышинский Н. Н. 15 (20, 32), 165 (25, 48,
51, 54, 71, 76), 376, 377(5), 535 (10, 19,
22, 25), 587 (135), 588 (137, 159, 161),
595 (5), 597 (5, 21—23), 612 (5, 6, 15), 628
(29, 30), 633 (57)
Вязанкин Н. С. 14(5), 16(103), 87(61),
131 (36, 37), 139 (57, 58, 62), 140 (63—65,
67, 68), 147 (14, 14д), 151 (14д, 33а), 152,
153 (14), 154, 155 (14, 45, 46, 51—53, 57,
58), 165 (96), 175 /207), 246 (297), 365 (254,
255, 257, 258), 366 (258—262), 367 (269—
272), 368 (270, 273—275), 376 (2, 4—8),
377 (4, 5, 7, 9), 386 (60), 397 (43, 61, 62,
65, 66), 400(61, 124), 401 (61), 402 (124),
408 (62, 254), 422 (124, 408 (409), 439
(19), 441 (44, 45), 442 (19, 46, 48—50, 53),
443 (19, 53—57), 444 (19, 46, 55, 56),
445 (19, 46, 65), 463 (59), 464 (62а), 467,
468 (59), 480, 483 (109—111), 484 (111, ИЗ),
486 (109, 111, ИЗ), 490 (120), 528 (87), 537
(163), 587 (137), 588 (137, 159—161), 589
(184), 590 (196, 197), 595 (1—5), 596 (1, 3),
597 (5, 21—23), 612 (5, 6), 613 (21, 22, 26,
27), 615 (35), 628 (29, 30)
Вялых Е. П. 414 (356)
Гаврилов Г. И. 233 (259), 319, 321, 322 (5, 6),
568 (2, 3).
Гайворонская Г. К. 116(22)
Галашина Л. М. 240 (1726)
Галиулина Р. Ф. 139 (62), 154, 155 (50), 484,
486 (113)
Гальберштам М. А. 561 (58), 581 (48)
Гальперин Е. Г. 16 (78)
Ганина Т. Н. 516 (68)
Гар Т. К. 15(40), 16 (ПО), 20 (15), 38(84),
60(8, 10, 11, 13—15), 61 (10, 11, 18), 63
(8, 14), 64 (15, 21), 67, 69 (14), 134 (41а),
136 (45—49)
Гастилович Е. А. 15 (34), 165 (38, 42), 535 (69)
Гвердцители И. М. 65 (30), 66 (31—34), 114 (7)
Гельфман Я. А. 519 (77)
Гер нет Е. В. 633 (63)
Гинсбург В. А. 233(260), 319, 321, 322(6),
415, 416 (379), 568 (3)
Гипп Н. К. 569 (7)
Гладштейн Б. М.'26(10), 82(36), 97 (40)
Гладышев Е. Н. 139 (57, 58), 147 (14, 14а),
151 (14а, 33а), 152, 153(14), 154, 155(14,
45, 46), 365 (258), 366 (258, 259), 443 (54)
Гойзман М. С. 527 (73)
Голованова Н. И. 165 (39)
Гольданский В. И. 165 (125—130), 194 (20),
391 (81)
Гольдер В. Г. 402, 420, 421 (146а)
Гольдин В. А. 182 (26)
Гольдштейн А. Л. 535(98), 536(114, 118),
589(185), 633(51, 52)
Гольдштейн И. П, 165 (161), 399, 409 (91),
416(385), 422(91, 385, 410), 432(2, 13,
35, 36), 433, 434 (61), 435 (2, 83)
Голубева Е. И. 202 (53)
Городинский Г, М. 165 (126)
Горохов Л. Н. 16 (82)
Горшкова Г. Н. 165 (19)
Гречкин Н. П. 412 (342, 343)
Губен-Вейль 162 (4), 527 (63), 631 (5)
Гунцадзе Т. П. 65 (30), 66 (33), 114 (7)
Гургенидзе Г. Т. 159 (15), 513 (33)
Гурикова Т. Г. 139 (58), 154, 155 (46)
Гурьянова Е. Н. 165 (161), 399, 409 (91),
416(385), 422(91, 385, 410), 432(2, 13,
35, 36), 433, 434 (61), 435(2, 83)
Гусева И. С. 255 (336), 325 (3, 4, 12), 326 (4,
12)
Гусейнзаде Б. М. 43 (119, 121)
Давыдова Л. С. 159 (16), 225, 231, 234 (199),
512 (2, 6, 10)
Давыдова С. Л. 27, 31 (20), 39 (93), 81 (8),
101 (52—55), 117(30), 159 (6, 7, 9, 13, 14)
Джуринская Н. Г. 38 (85), 52 (41), 60 (4—9),
61 (4), 62 (5, 7), 63 (5—9), 65 (7), 115 (12,
14), 117(27), 136(43—45, 47)
Делинская Е. Д. 432 (13)
Делягин Н. Н. 165 (128, 133, 134)
Дергее Д. О. 515 (59)
Дергунов Ю. И. 87 (61), 365 (255, 257), 366
(259—262), 376 (4, 5, 7, 8), 377 (4, 5, 7),
638
Авторский указатель
397, 406 (43), 439, 442 (19), 443 (19, 55—
57), 444 (19, 55, 56), 445 (19), 589 (160,
184), 595 (1—5), 596 (1, 3), 597 (5),
613 (21, 22, 27), 615 (35)
Дмитриева В. Н. 527 (64)
Дмитриевский В. С. 633 (42)
Докукина Л. Ф. 231 (244), 513 (18, 19), 536
(154)
Долгий И. Е. 19 (9, И), 21 (9), 39 (90, 95), 116
(17, 22), 117(17, 26), 122, 125(7), 162(13),
221, 226 (153)
Дорфман Я. Г. 165 (135, 155), 535 (65)
Драбкина Л. С. 536 (111)
Дремова В. П. 382 (19), 521 (214)
Дружков О. Н. 589 (180), 596 (10)
Дубов С. С. 15 (16), 28 (36), 83 (48), 165(10,41)
Дубова Р. И. 408, 416, 417 (273)
Дудикова Э. Д. 387 (65), 393 (97), 408 (260),
504 (79), 516 (63)
Думлер В. А. 165(5, И), 219, 229, 230, 237
(130), 332 (37)
Дургарьян С. Г. 39 (95а), 40 (Ю2д), 48, 50
(14а, 146)
Дулова В. Г. 15 (22), 71 (52, 54—56)
Думенский В. 633 (59)
Дун Ши-хуа 247 (300)
Дьячковская О. С. 87(61), 366(261, 262),
367 (270), 368 (270, 274, 275), 376, 377 (4),
397, 408 (6), 422 (408), 590 (196, 197), 595
(2)
Дядюша Г. Г. 15 (68), 165 (87)
Егоров Ю. П. 15 (19, 33, 41, 51, 52, 68), 60 (3),
101(53), 117(24), 122, 125, 126(6), 165
(32, 50, 83, 84, 87, 88), 210 (75)
Егорочкин А. Н. 15 (56, 62, 63), 165 (95, 96)
Емельянов Б. В. 633 (69, 71)
Емельянова Л. И. 54 (54, 55), 76 (8). 92 (46,
47), 93(55)
Ермолаева Т. И. 159 (6, 7), 512 (10), 574 (9)
Жданов Г. С. 165 (149, 150), 535 (41, 42)
Живухин С. М. 387 (65), 393 (97), 408 (260),
504 (79), 516 (63)
Жижина Л. И. 527 (73)
Жукова И. Ф. 114 (5), 453 (27)
Забродина К. С. 162 (9)
Заварова Т. Б. 614 (30)
Завгороднин В. В. 165 (31, 37, 159), 195 (25),
227 (224, 225, 228), 232 (224), 249 (228),
254 (224, 228, 332), 255 (224, 335), 290 (46),
364 (245), 450 (18, 19), 451 (19), 453 (25),
497 (21), 498 (34, 35), 500 (46), 501 (21, 35,
47, 49, 50), 626 (22)
Заинчковская М. К. 225 (203), 419 (393)
Зайцева Н. А. 334 (53), 406 (226)
Зайченко Т. Н. 355 (194), 586 (116)
Запевалова Н. П. 213, 226, 234, 236 (91), 350
(159), 512(4), 536(142), 560(37), 579(34)
Засосов В. А. 221 (158, 168), 223(168), 228
(158), 229 (168), 236 (158), 241 (168), 453
(49), 560 (45)
Захаркин Л. И. 30 (54, 55), 51 (61), 165 (130),
187 (70), 208 (51, 52), 211 (76) 212 (51, 52),
216 (51, 52, 103), 248 (304), 256 (347—350),
257 (347—349), 258 (349), 259 (349, 350),
553, 554 (49)
Захарова М. Я. 411 (324, 327), 505, 506, 508,
(84, 86), 509 (86), 608 (50), 629 (33)
Звездин В. Л. 140 (65), 155 (58)
Землянский Н. Н. 165 (161), 193, 194 (1), 351
(171. 172), 387(66, 67), 388 (67), 399(91),
402 (146а), 407(230), 409(91), 416(385),
417(388), 421 (146а), 422(91, 385, 410),
432 (2), 433, 434 (61), 435 (2, 83), 439 (31),
516 (64), 519 (77), 603 (27—29), 609 (79)
Земсков И. Ф. 633 (71—76)
Зильберман Е. Н. 402, 404 (152)
Зиновьев Ю. М. 586 (125)
Зорина Н. П. 535 (98), 536 (114, 118)
Зуева Г. Я. 15(56, 62), 20 (На—Ив),
160 (29)
Иванов А. Ю. 182 (26а)
Изэн Чхао-лунь 247 (300)
Имото Хидэдзи 175 (206)
Иосида К. 516 (74)
Иоффе С. Т. 213 (88), 215 (99), 225 (88)
Ипатьев В. Н. 590 (199, 200)
Исаева Л. С. 321 (15)
Исицука Т. 516 (66, 71, 74>, 520 (198)
Исмаилзаде И. Г. 165 (147—150), 535 (41, 42)
Ито Т. 519 (46, 49), 520 (98)
Итон К. 520 (124)
Каабак Л. В. 168 (191, 192), 341 (119)
Казина Г. В. 240 (1726)
Калинина С. П. 188 (76), 224 (186), 338 (79),
378(25), 382, 383(24), 397 (51, 52, 56),
399 (56, 86, 88), 401 (52), 513 (23—25, 28.
29, 32)
Калужнина К. Ф. 520 (190)
Кандрор И. И. 561 (58), 580 (48)
Каплин Ю. А. 589 (179, 180), 596 (9, 10)
Карасев А. Н. 165 (124)
Каргин В. А. 512 (2)
Карпов В. Л. 519 (23), 520 (101)
Карцев Г. Н. 16 (87, 88, 90)
Карягин С. В. 165 (126)
Карякин Н. В. 15 (76)
Касаточкин В. И. 165 (19)
Кацумура Г. 252, 253 (322), 519 (45)
Кацумура Т. 187 (72), 252 (322), 253 (322,
330) ,"385 (59), 400 (123), 407 (233), 408 (265),
519 (45), 520 (98)
Каширенинов О. Е. 357(217), 435(86, 87),
520 (122), 535 (49)
Квасов А. А. 612, 613 (20)
Кечина А. Г. 20(116)
Киккава С. 183 (32)
Килин С. Ф. 536 (133)
Кимура Т. 519 (74)
Кирев Г. Г. 15 (19, 33), 165 (32, 83)
Киреев В. В. 387 (65)
Киселев В. Г. 68 (39)
Киселева И. Г. 87 (61)
Киселева Т. М. 212 (83), 213 (91), 214 (93),
226(91, 93), 231(239—241), 234(91, 93),
235 (93), 236 (91, 93), 347, 350, 351, 353 (154)
350 (159), 366 (262), 379 (43), 397 (46, 55,
57), 398 (55, 77), 401, 407 (138), 512 (3—5,
12), 513 (5, 26, 38, 39), 516 (62), 536 (142,
148—152, 160), 560 (37, 47, 49), 561 (60), 579
(35), 605 (17, 18)
КитаокаА. 520(99, 100)
Клабуновский Е. И. 165 (171), 207, 208 (25)
Авторский указатель
639^
Клагес В. А. 457 (17), 513 (45), 514 (45, 48)
Климентова Н. В. 27, 31 (20), 81 (8), 101 (52,
54, 55), 152 (13, 14, 16)
Климова В. А. 162 (5, 6, 9), 319 (2, 3)
Кнунянц И. Л. 28 (36), 83 (48), 352 (174)
Кобаяси Э. 520 (127)
Кобаяси Ю. 520 (99, 100)
Козина М. П. 165 (171), 207, 208 (25)
Козминг 3. 633 (59)
Козлов Л. М. 536 (111)
Козюра А. С. 633 (68,70)
Колесников Г. С. 27, 31 (20), 39 (93), 62 (196),
81 (8), 101 (52—55), 117(30), 159(6, 7, 9,
13—16), 225, 231, 234 (199), 512 (2, 6, 10),
513 (33), 537 (172, 173), 541 (5)
Колесников С. П. 15 (67), 61 (16, 17), 62 (19—
20), 134 (416), 160 (22—26)
Колобов Н. Е. 109 (101—103), 155, 156 (55,
56). 157 (55), 165 (124. 125), 411 (324—327),
505 (84—87), 506 (84—86), 507 (87), 508
(84—87), 509 (85—87, 96), 608 (49, 50),
623, 628(1), 629 (1, 33)
Комаров Н. В. 15 (34), 43 (ИЗ, 114), 165 (38,
39, 42), 220, 221 (137), 225 (202, 203), 227,
232 (229), 233 (202), 255 (336), 325 (3, 4, 12),
326 (4, 12), 419 (393), 535 (69), 574 (9)
Комарова С. Д. 50 (26)
Комацу С. 541 (51)
Корешков Ю. Д. 71 (52)
КорневаС. П. 139 (62), 147 (14, 14а), 151 (14а,
33а), 152, 153 (14), 154 (14, 50), 155 (14),
246 (297), 376 (2), 441 (45) 480, 483 (109—
111), 484(111, 113), 486(109, 111, 113)
Коротаева И. М. 325 (5)
Коротаевский К. Н. 168 (194)
Коршак В. В. 40 (102), 41 (104), 159 (4, 5, 11,
12), 160 (18, 25, 26), 209 (61), 231 (247, 248),
333(45), 401, 417(139), 512(11), 513(14,
15, 17), 514 (52), 536 (140, 145, 146), 537
(172, 173), 541 (5), 559 (14, 23, 24), 560 (14)
Коршунов И. А. 535(75), 589 (181, 187—
189), 633(56)
Корытко Л. А. 165(126, 128)
Костяновский Р. Г. 221 (162), 322 (20), 447 (3)
Котов А. М. 504 (79)
Котон М. М. 212 (83), 213 (91), 214 (93),
226 (91, 93), 231 (239—241, 244), 234 (91,
93), 235 (93), 236 (91, 93), 240 (83), 2501(310),
347 (154), 350 (154, 159), 351 (154), 352
(175—178), 353 (154, 177, 178), 361 (236),
369 (279—281), 379 (43), 397 (46, 55, 57),
398(55, 57), 401, 407(138), 457(17),
512 (3—5, 12), 513 (19, 26, 38, 39, 45), 514
(45, 48), 515 (57), 516 (62), 518 (19, 26,
38, 39, 45), 535 (107), 536 (142, 148, 154, 159,
160), 560 (37, 47, 49), 561 (60), 579 (34, 42),
582 (71, 72), 583 (77), 584 (83, 85), 590 (201,
202), 605 (16—18), 631 (8)
Котрелев В. Н. 188 (76). 224 (186), 338 (79),
378(25), 382, 383(24). 397 (51, 52, 56),
399 (56, 86, 88), 401 (52). 513 (23—25, 28,
29, 32)
Кочетков А. К. 202 (54), 302 (55, 56), 356 (214),
564 (79), 609 (77)
Кочеткова А. С. 189 (82), 412 (337)
Кочешков К. А. 8 (1), 165 (161), 182 (21—26),
183 (29, 30), 186 (21), 193, 194 (1), 199 (37—
39), 200 (37, 38), 201 (37—39, 50—52),
202(37), 208—211 (33), 216(102, 114), 217
(119), 218(129), 221(158, 168), 223 П29,
168, 254), 228 (158), 229 (168), 236 (158, 254),
237(254), 241(168), 242(274), 247(301,
302), 249 (254), 269 (1, 2, 5—7), 270 (5, 11),
271 (11, 12), 319 (2, 3), 329 (1—5), 330 (3—5),
331 (3, 4, 30, 33), 332 (3—5, 30, 33), 333
(1, 2, 4,5), 334 (1, 2, 50—53, 55), 336 (1, 2,
50—52, 60, 63, 64), 337 (51, 52, 55, 63, 64),
346, 350 (151), 351 (171, 172), 354 (185,
186), 355 (55, 185, 186), 368 (276), 378 (19),
381 (7—10), 384 (39), 385 (39, 48), 387 (66,
67), 388 (67), 390 (48, 76, 77), 392 (76, 77,
90—92), 393 (7, 8, 76, 77. S0—92, 98),
399 (91), 401 (132), 402 (146а), 406 (203,
204, 225, 226), 407 (230), 409 (91), 413 (203,
204, 347—350, 352), 416 (385), 417 (388),
420, 421 (146а), 422(91, 385, 410), 432
(2, 6—8, 13, 35, 36), 433, 434 (61), 435 (2, 83),
439 (23, 31), 440 (23), 441 (40, 41), 453 (49),
454 (58—61), 498 (37), 503 (66, 67), 516 (64),
526 (12, 13), 551 (29), 555 (75, 76), 556 (76),
558 (9), 560 (45), 562 (9), 564 (81—83, 85—
87), 565 (82, 83, 85—87), 569 (7, 8), 573 (1),
582 (66, 74, 75), 583 (74, 78), 584 (82), 585
(74, 75, 78, 92, 93, 95—97), 597 (17), 598
(34), 603 (26—29, 32—36), 604 (33, 35, 36),
605 (27—31), 606, 607 (27—30), 608(27—
30, 71), 609 (27—30, 72—76, 78—80), 618
(16)
Кочкин Д. А. 175 (200), 207 (15), 224 (186),
338(79), 378(25), 382 (19, 24), 383(24),
397 (52, 56, 66—68), 399 (56, 86, 88, 89),
401 (52, 67), 501 (52, 53), 513 (24, 25, 29, 32),
521 (214), 536 (157, 158), 602 (25), 605 (21—
26)
Кояма К. 633 (34)
Кравцова И. Д. 15 (51)
Кравченко А. Л. 15 (45, G3), 16 (88, 90),
26, 38, 40, 42 (6), 50 (28), 52 (28, 40), 57
(67), 60, 64(12)', 80,81(1), 87(56), 118,
119 (34, 35), 129 (35)
Краснова Т. Л. 40(99, 101), 159(11), 231
(245), 513 (15, 16)
Крафт М. Я. 197(35), 440(35)
Кренцель Б. А. 520 (140—143)
Крижанский Л. М. 165 (126), 400, 401 (125)
Круглая О. А. 16 (103), 139 (62), 140 (67. 68),
154,155(50, 52, 53). 165 (96), 175 (207),
200 (47), 376 (8). 377 (9), 442 (50), 484, 486
(113), 490(120).. 537(163)
Круглая-Щепеткова О. А. 368 (274)
Кубота X. 402 (145), 519 (44)
Кудрявцева М. И. 451 (20)
Кузнецова Г. И. 188 (76), 224 (186), 338 (79),
378(25), 382, 383(24), 397(51, 52, 56),
399 (56, 86, 88), 401 (52), 513 (23—25, 28,
29, 32), 536 (157)
Кульвинская Н. А. 355 (194), 586 (116)
Кулюлин И. П. 97 (40)
Куприевич В. А. 15 (68), 165 (87)
Купоцва К. М. 527(73)
Курсанов Д. Н. 71 (50—54, 56), 491 (122)
Кускова В. П. 519 (77)
Кучкарев А. Б. 36 (76. 76а). 37 (80), 40 (76),
219, 231, 236(133), 339 (99), 383 (32),
398 (74)
Кучкаров А. Б. 222, 223, 232, 239 (171)
640
Авторский указатель
Лад М. Д. 633 (68)
Лайкин И. И. 165 (5, И), 217 (126), 219 (126,
130), 224(126), 229(130), 230 (126, 130),
237 (130), 213, 248 (126), 332 (37)', 406, 407
(202)
Лайне Л. В. 188(76), 338 .'79), 339 (86),
378 (25), 382, 383 (24), 397 (51, 52), 399 (86),
401 (52), 513 (23—25, 28, 29), 536 (157)
Лалайберте Б. Р. 397 (48), 513 (22)
Лаписова Н. П. 535 (98), 536 (114, 118), 633
(51, 52)
Лаурис И. В. 519 (77)
Лебеадзе Т. Н. 536 (133)
Лебедев В. Б. 165 (121), 535 (35)
Лейтес Л. А. 165 (50, 83, 88)
Лепендина О. Л. 165 (135)
Лещева И. Ф. 520 (143)
Лещенко С. С. 520 (101)
Ли Вей-Чан 28 (36), 83 (48), 352 (174)
Лившиц Б. Л. 15 (39), 165 (49)
Лисина 3. М. 312, 314, 315 (65)
Литвинова О. В. 416 (384а)
Логинова И. Е. 325—327 (8)
Лодочникова В. И. 535 (20), 558, 562 (9),
564, 565(82—87), 603(29, 31, 35, 36),
604 (35, 36), 605(29—31), 609 (29, 30, 75,
76, 79, 80)
Лопатин Б. В. 15 (17)
Лукевиц Э. Я. 8 (10, 11)
Лукьяькина Н. В. 20(116, Ив)
Лукьянова Л. В. 207(15), 602 (25), 605(22)
Лунева Л. К. 41(104), 42(111), 160(18),
165 (19), 233 (258а), 514 (52)
Луценко И. Ф. 15 (65), 55 (56—59), 56 (58,
62—64), 57(59), 71(48, 49), 119(38—40),
138 (53—56), 165 (120), 271 (13), 272, 288
(1), 310, 311 (62—64), 312(64—67), 314,
315 (65), 323 (23), 325 (1), 353 (179—183),
359 (183), 363 (179—182), 368, 369 (183),
408(250—252), 486(115—117) 487(117)
Лысенко Е. Н. 168 (194)
Макаров Е. Ф. 165 (125, 126, 128), 391 (81)
Макаров С. П. 233(259, 260), 319, 321, 322
(5, 6), 415, 416 (379), 568 (2, 3) .
Макарова Л. Г. 54 (54, 55), 76 (8), 92 (46, 47),
93(55), 320(12), 370(283), 400, 404(118),
569 (6, 9), 590(198)
Макарова Т. А. 209(61), 333(45), 401, 417
(139)
Максимова Н. Г. 39 (91), 225 (201)
Малиновский М. С. 368 (277)
Малюгина Н. И. 535 (75)
Мальцев В. В. 512 (2)
Манаков М. Н. 20(12), 50(29), 114(9), 160
(19—21)
Манулкин 3. М. 36 (76, 76а), 37, 39, (956),
40(79, 80), 42(112), 82(30), 115(13),
207, 208 (22, 23), 209, 210 (22), 219, 221
(23, 146, 149, 164, 165), 222 (170, 171), 223
(146,164,165,171,174,176,178,196),226(196,
211—213), 228 (233), 231 (133, 249), 232 (171,
250), 233 (146, 170, 211, 249), 234 (164, 165),
236(133, 178, 211, 261а, 264а), 238 (250),
239 (171, 211), 241 (250), 251 (174), 334, 336
(54), 337, 338 (68), 339 (68, 87—91, 99, 101,
108), 340(90, 98), 341(89), 344(135),
346, 354 (91), 355 (195), 356 (195, 211), 357
(195, 221), 379 (40), 383 (32, 33), 398 (74),
551 (33), 560, 561 (43, 44), 578 (23), 584 (87),
586(118), 587(118, 129, 130), 617(2)
Манцивода Г. П. 357 (215)
Марков Л. К. 163 (15)
Маркова С. В. 15 (30, 36)
Мартиросян Г. Т. 225, 234 (200), 450 (17)
Мастрюков В. С. 16 (80), 71 (56)
Матвеева А. Д. 15 (12)
Мацусита X. 520 (194)
Мачигин Е. В. 312 (67)
Маянц Л. С. 16 (78)
Медведев Ф. А. 16 (81)
Меркулова Е. Н. 233 (260), 319, 321, 322 (6),
568 (2, 3)
Мехмандарова Н. Т. 43(119, 120, 123), 66
(36, 37)
Мещеряков А. П. 116 (22)
Мингалева К. С. 165 (159), 254 (332), 501 (50)
Мирецкий В. Ю. 182 (25, 26а)
Мирная А. П. 633 (68, 70)
Миронов В. Ф. 15 (19 40, 45, 51, 63, 73), 16
(87, 90, ПО), 19 (9, И), 20 (15), 21 (9),
24 (7, 8), 26 (6), 38 (6, 84, 85, 87), 39 (90, 92,
95), 40, 42 (6), 50 (28), 52 (28, 40, 41), 57
(67), 60 (4—7, 10, 13—15), 61 (4, 7, 10,
12, 18), 62 (5, 7), 63 (4—9, 14), 64 (15, 21),
65(7), 67 69(14), 80(1), 87(1, 56), 114
(5), 115(12, 14), 116(15—17), 117(15, 24,
27), 118, 119 (34, 35), 122, 125 (7), 129 (35),
134 (41а), 136 (43, 47), 137 (48, 49), 159 (4,
5), 160(26), 162(11—13), 165(8, 83, 88),
175 (197), 208 (42), 210 (75), 221 (42, 153),
225 (42, 201). 226 (153, 209), 453 (27),
512(11), 528(77, 78)
Мирсков Р. Г. 227 (223), 254 (333), 272, 273
(26), 325(2,5—11, 13), 326 (2, 7), 327(8—
Мисима С. 541 (30а)
Мисюнас В. К. 165 (39), 220, 221 (137), 225
(202, 203), 227, 232 (229), 233 (202), 325,
326 (4, 12), 419 (393)
Митрофанов К. П. 165 (133—135), 391 (82)
Митрофанова Е. В. 140 (68), 154, 155 (53)
Михайлов Б. М. 68 (39)
Мокеева Т. И. 598 (31)
Момонон М. 516 (66, 71, 74), 520 (198)
Монастырский Л. М. 168 (194)
Морикава Т. 527 (46)
Морыганов Б, Н. 366 (267), 589 (194)
Москвина Е. П. 352, 353 (177, 178), 584 (85)
Мулер Л. И. 226 (216)
Муратова А. А. 412 (346), 432 (19)
Нагата X. 535 (102)
Надь М. М. 216(114), 217(119), 269(5—7),
331, 332 (30, 33), 334, 336 (50, 51), 337 (51,
52), 378 (19), 384 (39), 385 (39, 48), 390 (48),
392, 393 (90—92), 406 (225), 432 (8), 439,
440 (23), 454 (58, 59), 564 (81), 569 (8),
597 (17), 609 (78)
Назарова Л. М. 165 (183), 589 (182), 598
(48—50)
Накада Т. 520 (150)
Накамори X. 541 (51)
НакамураМ. 181, 182, 184(11)
Накамура X. 535 (102)
Наметкин. Н. С. 39 (95а), 40 (102а), 48, 50
(14а, 146), 62 (20)
Авторский указатель
641
Наумов С. Н. 207, 208, 221 (23), 339, 340 (98)
Нейман М. Б. 186 (62)
Несмеянов А. Н. 41 (107), 47(6, 7), 48(6),
54 (55), 69 (44), 76 (8), 92 (47), 109 (101—
103), 120 (42), 155, 156 (55, 56), 157 (55),
199 (37—43), 200 (37, 38, 40, 44—48),
201 (37—41, 49—52),v 202 (37, 41, 43, 46,
53, 54), 203 (55), 213 (88), 215 (99), 225 (88,
198, 200), 234 (200), 245, 256 (292), 270,
271(9, 11, 12), 281(9, 10), 288(43—45),
289(43, 45а), 310, 311 (62), 319(1—3, 7),
320 (12), 321 (15), 323 (23), 336, 337 (63, 64),
344 (132, 133), 353 (179), 354 (185, 186), 355
(185—187, 189—192), 356,357 (191),363(179),
364 (190), 370 (283), 390, 392, 393 (76, 77),
400, 404(118), 406(203), 408(250), 411
(324—327), 413 (203, 349, 350), 441 (40, 41),
450 (17), 453 (45, 46), 454 (60, 61), 503 (66,
67), 505 (84—87), 506 (84, 86), 507 (87),
508 (84—87), 509 (85—87, 96), 526 (13),
546 (37), 564 (79, 80), 568 (5), 569 (7—9),
582 (69, 70), 585 (92, 93), 590 (198), 608
(49, 50), 609 (77), 623, 628 (1), 629 (1, 33)
Нефедов В. Д. 165 (175)
Нефедов О. М. 15(67), 20(12), 50(29), 61
(16, 17), 62(19, 19а, 196, 20), 114(9), 134
(416), 160 (19—24)
Николаев П. Н. 365 (256)
Новиков А. С. 520 (190, 191)
Новикова Н. В. 47 (6, 7), 48 (6), 199 (40—43),
201(40, 41), 202(41, 43), 208, 213, 216,
242 (35), 289 (45а), 355 (187, 191, 192), 356,
357 (191, 213)
Новиченко Ю. П. 501 (52, 53), 536 (157)
Нудельман 3. Н. 189 (82), 412 (337), 520
"(190, 191)
Обреимов И. В. 15 (37, 38), 165 (35), 535 (21)
Огава Д. 541 (51)
Оикава X. 15 (28, 29), 49, (22), 81 (21), 535
(18)
Ока С. 520 (194)
Ольдекоп Ю. А. 366 (266, 267), 589 (194), 590
(195)
Омура Д. 520 (106)
Ониси X. 402 (145, 146), 516 (78), 519 (44, 52)
Орлова А. А. 589 (181, 187, 188)
Осипов О. А. 357 (217), 435 (86)
Осипова М. А. 199, 200, 201 (40)
Охлобыстин О. Ю. 30 (54, 55), 55 (61), 165
(129, 130, 136, 137), 187(70), 208(51, 52),
211(76), 212(51, 52), 216(51, 52, 103),
244 (282), 248 (282, 304), 250 (282), 256
(347—350), 257(347—349), 258 (349),
259 (349, 350), 391 (82), 400, 401 (125), 553,
554 (49)
Павлова И. Д. 165 (50)
Павловская М. Е. 336 (60)
Палеева И. Е. 209, 211 (53), 401 (132)
Панов Е. М. 165 (161), 193, 194 (1), 247 (301,
302), 351 (171, 172), 387 (66, 67), 388(67),
399 (91), 402 (146а), 407 (230), 409 (91), 416
(385), 417 (388), 420, 421 (146а), 422 (91, 385,
410), 432 (2), 433, 434 (61), 435 (2, 83),
439 (31), 516 (64), 519 (77), 558, 561 (9),
564, 565 (82, 83, 85—87), 573 (1), 582 (74,
75), 583 (74, 78), 584 (82), 585 (74, 75, 78,
95), 603 (26—29, 32—36), 604 (33, 35, 36),
605 (27—31), 609 (27—30, 72—76, 79,
80)
Парнес 3. Н. 464, 465, 471 (62)
Паршина А. М. 527 (73)
Патрина Н. Д. 319 (10)
Петрий О. П. 272, 288, 313, 314 (1), 325 (1)
Петров А. А. 16(81), 42(109), 115(11), 165
(31, 37, 121, 159), 195 (25), 227 (224, 225,
228), 232 (224), 249 (228), 254 (224, 228),
255 (224, 335), 290 (46), 364 (245), 450 (18,
19). 451 (19), 453 (25), 497 (21), 498 (34, 35),
500 (46), 501 (21, 35, 47, 49, 50), 535 (35),
626 (22)
Петров А. Д. 1 (9), 15(19), 19(11), 20(12),
21 (9), 39 (87, 90—92, 95), 40 (99, 101), 50
(26, 29), 52 (40), 60 (4—8, 12), 61 (4, 5, 7, 17)
62 (5, 7), 63 (4—8), 64 (12), 65 (7, 30), 66
(33), 115 (12, 14), 116 (15, 17), 117, (15,
17, 24, 26), 118, 119(35), 122, 125(7),
136(43—45), 159(4, 5, 11), 160(19, 20,
26, 27), 162 (11—13), 165 (83), 175 (197), 208,
(42), 210 (75), 221 (42, 153), 225 (42, 201),
226 (153, 209), 231 (245), 254 (332), 512 (11),
513(15, 16), 528 (77, 78)
Петров И. 578 (26)
Петров Ю. Н. 16 (89)
Петрова В. Н. 254 (333), 325, 326 (7)
Петровская Л. И. 15 (65), 165 (120)
Петухов В. А. 15 (45, 73), 165 (8)
Пикина Е. И. 218, 223 (129), 334, 337, 355
(55), 393 (98)
Пинчук Н. М. 443, 444 (56)
Писаренко В. И. 20 (Па)
Платэ Н. А. 512 (2)
Плотникова М. В. 165 (135)
Погорелов А. Г. 20 (На)
Поздеев В. В. 182 (25, 26а)
Полак Л. С. 165 (124, 133—136), 391 (82)
Полеес Б. М. 571, 572(7, 10)
Полякова А. А. 16 (81)
Полякова А. М. 15 (15), 40 (102), 159 (4, 5,
11, 12), 160(26, 28), 231 (247, 248), 512
(11), 513(14, 15, 17), 536(140, 145, 146),
559 (14, 23, 24), 560 (14)
Пономарев С. В. 56 (64), 272, 288 (1), 310, 311
(62—64), 312 (64—67), 313 (1), 314 (1, 65),
315(65), 323(23), 325(1), 353(179—183),
359 (183), 363 (180, 182), 368, 369 (183),
408 (250—252), 486 (116)
Пономаренко В. А. 15 (12, 56, 62), 19 (10, 11,
Па), 20(116), 39(81, 95), 60(3), 116(17),
117(17, 23, 25), 122, 125(6,7), 126(6),
160 (29), 162 (13), 163 (15)
ПоповА. В. 400, 401 (125)
Попова 3. В. 14 (5), 520 (86» 87), 402 (153)
Порин И. Г. 541 (22)
Преображенская Е. А. 527 (64)
Прокофьев А. К. 221 (162), 322 (20), 447 (3)
Прохоров А. М. 15 (10)
Прохорова Л. К. 250 (310), 515 (57)
Пташинский И. А. 633 (23)
Птицина О. А. 319 (10), 320 (13, 14), 321 (14,
15), 322 (14), 527 (45)
Пудовик А. И. 412 (341, 346), 432 (19), 608
(64)
Пуяырева В. П. 270, 271 (11), 441 (40, 41), 503
(66, 67)
41 Заказ № 207
642
Авторский указатель
Пучинян Е. А. 228 (233), 232, 238, 241 (250),
334, 336 (54), 357 (216, 358, 222), 551 (33),
560, 561 (43, 44)
Пшиялковская Н. Б. 393 (97)
Рабинович И. Б. 165 (170), 365 (256), 535 (60)
Радбиль Б. А. 438 (17)
Разуваев Г. А. 15 (56, 62, 63, 76), 87 (61), 139
(57, 58, 62), 140 (63—65, 67, 68), 147 (14, 14а)
151 (14а, 33а), 152, 153 (14), 154 (45, 46,
50—53), 155(14,45,46, 50—53, 57, 58), 165
(95,96), 246 (297),334 (49), 339,344,347 (107),
355 (107, 194), 361 (236), 365 (254—258) 366
(107, 259—262, 268), 367 (269—272), 368
(273—275), 376 (2, 4—8), 377 (9), 386 (60),
397(43, 61, 62), 400 (61, 124). 401 (61), 402
(124), 408 (254), 422(61, 124, 408, 409),
439 (19), 441 (44, 45), 442 (19, 46—51),
443 (19, 54—57), 444 (46, 47, 109), 445 (19,
46), 480, 483 (109—111), 484(111, 113),
486(109, 111, 113), 490(120), 528(87),
586 (116), 587 (132, 137), 588 (137,159—161),
589 (179, 180, 184, 193), 590 (196, 197, 199—
201). 595 (1—5), 596 (1—5, 9, 10), 597
(5, 21—23), 612(5, 6,20), 613(20—22,26,
27), 614 (30), 615 (35), 628 (29, 30)
Рашкес А. М. 219 (133), 222, 223 (171), 231
(133), 232 (171), 236 (133), 239 (171), 339
(99), 383 (32), 398 (74)
Резникова Е. Б. 207 (15), 602, 605 (22)
Реутов О. А. 319 (10), 320 (13, 14), 321 (14,
15), 322 (14), 527 (45)
Рогов В. Я. 194 (20)
Рогожин С. В. 209 (61), 333 (45), 400 (125),
401 (125, 139), 417 (139)
Родэ В. В. 26 (10), 82 (36)
Рожков И. В. 587 (138, 139)
Розман И. М. 536 (133)
Ромодан Ю. П. 403 (156а)
РочевВ.Я. 165(127, 129)
Рубинчик Г. Ф. 223 (174, 196), 226 (196),
236 (261а, 264а), 251 (174)
Рудневский Н. К. 15 (20), 165 (25, 48, 51, 54),
535 (10, 19, 22, 25), 633 (57)
Рыбакова Н. А. 402, 404 (152)
Рябухин Ю. С. 182 (25)
Рясный Г. К. 165 (137)
Савельева И. С. 202 (53)
Сайкина М. К. 527 (40), 535 (71, 74)
Салимое М. А. 66 (37а)
Санина Л. П 131 (36, 37), 464 (62а)
Саранкина С. А. 36 (76, 76а, 80), 37 (79),
40(76), 42 (112), 82 (30), 115 (13)
Сафин И. А. 15 (61)
Сафонова М. К. 408 (270), 597 (27), 602 (19)
Сахарова А. А. 159 (И), 513 (15)
СаэкиЯ. 519 (83)
Свирежева С. С. 15 (56)
Свиридова Н. Г. 160 (29)
Седельникова В. А. 217, 219, 224, 230, 243,
248 (126), 406, 407 (202)
Сегалевич А. Е. 319 (7)
Селивохин П. И. 527 (72)
Семкина Э. П. 432(19)
Семчикова Г. С. 490 (120)
Сидельникова Г. И. 633 (74—76)
Снсидо К. 520 (202)
Склянова А. М. 225, 233 (202), 325, 326 (12)
Сколдинов А. П. 368 (276)
Скрипкин В. В. 165(125), 411(326), 505,
507—509 (87)
Сладков А. М. 41(104), 42(111), 160(18),
165 (19), 233 (258а), 514 (52)
Словохотова Ы. А. 351 (172), 387, 388 (67),
416 (385), 422 (385, 410), 432 (35), 433,
434 (61), 435 (83)
Смирнов Ю. Д. 168 (193)
Смирнова А. Н. 633 (70)
Смолян 3. С. 168 (194)
Снегова А. Д. 20 (11а), 163 (15)
Снегур Л. Н. 223 (176, 178), 236 (178), 339
(108), 37? (40), 383 (33)
Соболевский М. В. 240 (1726)
Соборовский Л. 3. 26(10), 82(36), 97(40),
411 (315, 316), 571, 572 (7, 10), 586 (125)
Соколик Р. А. 245, 256 (292), 546 (37)
Соколов Б. А. 357 (215)
Соколов Н. А. 408 (270)
Соколова Р. Ф. 366 (266), 590 (195)
Солодовников С. П. 15 (42), 165 (92)
Сосин С. Л. 160 (25)
Сорокина Р. С. 247 (301, 302)
Ставицкий И. К. 160 (29)
Стадничук М. Д. 16 (81), 42 (109), 115(11)
Стасиневич Д. С. 589 (185)
СтерлинР. Н. 15 (16), 28 (36), 83 (48), 165 (41),
352 (174)
Стопка я Л. Л. 520 (140—143)
Струков О. Г. 165 (10)
Струнин Б. Н. 30(54, 55), 208(51, 52), 211
(76), 212(51, 52), 216(51, 52, 103), 256—
259 (349)
Стручков Ю. Т. 71 (56)
Стукан Р. А. 165 (125), 391 (81)
Судзуки Т. 253 (330)
Суздалев И. П. 165 (126, 128)
Сучкова М. Д. 536 (140), 559, 560 (14)
Сыркин Я. К. 16 (87, 88, 90)
Сыркус Н. П. 182 (26)
Сыскаев И. М. 541 (22)
Сэйдзоко М. 396 (16)
Сюткина О. П. 165 (161), 399 (91), 402 (146а),
409 (91). 420, 421 (146а), 422 (91), 519 (77)
Тагути Ю. 633 (34)
Тайкова Н. К. 402, 404 П52)
Таками Я. 520(108, 109,' 114, 116, 119)
Талалаева Т. В. 218, 223, (129), 242 (274),
269 (1, 2), 334 (53, 55), 337, 355 (55), 393
(98), 406 (226), 498 (37), 555 (75, 76)
Тамбовцева Е. С. 40 (102), 159 П2), 231 (247,
248), 513 (14, 17), 536 (145, 146), 559 (23,
24)
Танака Й. 527 (46)
Тасиро Т. 516 (78), 519 (52)
Татаренко А. Н. 355—357 (195)
Тацуо К. 364 (246)
Тельной В. И 15 (76), 165 (170) 365 (256),
535 (60)
Темкин А. Я. 165 (135)
Тер-Саркиеяп Э. М. 408 (260). 516 (63)
Тиляев К. С. 221(165), 226(211—213), 231
(249), 233(165, 211, 249), 235(165), 236,
239 (211), 339 (88, 101)
Тихова Н. В. 14 (5)
Авторский указатель
643
Тихонова Л. И. 39 (95а), 40 (102а), 48, 50
(14а, 146)
Толстая Т. П. 321 (15)
Томилов А. П. 168(191—193), 341(119),
545(11)
Топчиев А. В. 520 (140, 141)
Торопова В. Ф. 527 (40), 535 (71)
Трофимов Б. А. 414 (356)
Трухтанов В. А. 391 (81)
Турсунбаев Г. Л. 39 (956)
Турчинский М. Ф. 320 (13, 14), 321 (14, 15),
"322 (14), 527 (45)
Тэн Мин-да 251 (319), 401 (133)
Уварова Е. И. 633 (46)
Уланова О. Д. 207, 208 (25)
Ульянова С. Д. 165 (171)
Файзи Н. А. 351 (172), 432 (35)
Федин Э. И. 15 (65), 165 (120)
Федотов М. С. 355 (194), 586 (116), 614 (30)
Федотов Н. С. 24 (7, 8),
Федотова Е. И. 442 (51)
Фетюкова В. 339, 344, 347, 355 (107), 366
(107, 268), 589 (193)
Финкель Э. Э. 520 (101)
Фиошин М. Я. 545 (11)
Флоринский Ф. С. 101 (51), 231 (239—241),
352, 353 (177, 178), 397 (57), 512 (5, 12),
513 (5), 536 (148—150, 152, 153), 560
(47, 49), 584 (85), 605 (16)
Фрейдлин Л. X. 114 (5)
Фрейдлина Р. X. 202 (54), 203 (55, 56), 225,
234 (200), 344 (133, 134), 356 (214), 450 (17),
453 (27), 564 (79), 582 (69, 70), 585 (96, 97),
609 (77)
Фролова М. К. 633 (23)
Фудзита Р. 516 (66, 71, 74), 520 (198)
Фудзита Ю. 535 (102)
Фукума Н. 520 (99, 100)
Фурукава Д. 520 (194)
Фусэ Г. 521 (226)
Халявин М. Н. 633 (69. 71, 72)
Хананашвили Л. М. 189 (82), 412 (337)
Хандожко В. Н. 109(103), 155—157(55),
411 (325), 505, 506, 508(85), 509(85, 96),
608 (49), 623, 628, 629 (1)
Хасапов Б. И. 55(57), 119(39), 138(53),
271 (13), 353, 363 (8, 18), 486 (115)
Харламова Е. Н. 589 (182)
Хачатуров А. С. 61 (17)
Хаяси Д. 182, 184 (18)
Хидекель М. Л. 15 (56, 62, 63), 165 (95)
Хирата 537 (174)
Хмельницкий Р. А. 16 (81)
Хосака X. 181, 182, 184 (14)
Храпов В. В. 165(126, 127, 129, 130), 194
(20), 391 (81)
Хрусталева В. А. 633 (58, 60, 61)
Хрусталева Е. Н. 516 (68)
Чаузов В. А. 416 (384а)
Чекмарева И. Б. 397 (66—68), 401 (67), 605
(21,24,26)
Чельцова М. А. 50 (26)
Червова Л. В. 338 (179), 378 (25), 382, 383
(24), 399 (86), 513 (24)
Чередникова А. Г. 189 (82), 412 (337)
Чернышов Е. А. 15 (42), 40 (99, 101), 159 П1),
165 (92), 231 (245), 513 (15, 16)
Четыркина Г. М. 398 (77), 513 (39), 536 (160),
605 (18)
Чжан Го-минь 247 (300)
Чиргадзе Ю. Н. 399 (89)
Чубарова М. А. 165 (19)
Чумаевский Н. А. 15 (15, 18, 37—39), 160 (28),
165 (33, 35, 36, 49), 535 (21)
Шаляпин Н. К- 182 (26)
Шамагина О. П. 387, 388 (67), 416 (385),
417 (388), 422 (385, 410), 432 (2), 435 (2, 83),
516 (64)
Шапшинская Л. А. 451 (20)
Шевердина Н. И. 182 (21—26), 186 (21), 209,
211 (53), 401 (132), 520(101)
Шейнкер Ю. Н. 15 (67)
Шейченко В. И. 15 (67), 62 (19а)
Шемякина 3. Н. 633 (69, 71)
Шепелева Р. И. 200, 202 (46)
Шергина ft. И. 165 (39)
Шеянов Н. Г. 587 П40)
Шигорин Д. Н. 15 (34), 165 (38, 42), 535 (69)
Шилова Н. Д. 159 (7), 512 (10)
Шимоиаев Г. С. 587(138, 139)
Ширяев В. И. 160 (21)
Шитикова Н. А. 162 (9)
Шихеева М. И. 66 (37а)
Шихиев И. А. 43(113—124), 66 (35—37а)
Шония В. М. 536 (133)
Шорыгин П. П. 15(73), 165(8)
Шостаковский М. Ф. 43(113, 114), 165(39),
188 (76), 220, 221 (137), 224 (186), 225 (202,
203), 227 (223, 229), 232 (229), 233 (202),
254 (333), 255 (336), 325 (2—13), 326 (8—12),
327(8—10), 357(215), 397(51, 52, 56),
399 (56), 401 (52), 414 (356), 419 (393), 513
(23, 25, 28, 29, 32), 574 (9)
Шпинель В. С. 165 (124, 128, 133—136), 391
(82)
Штединг М. Н. 519 (23)
Штифман Л. М. 633 (51)
Шубенко М. А. 366 (268), 589 (193)
Шуров А. Е. 587 (132)
Шушунов В. А. 8(12), 16(104), 175(208),
186 (62), 358 (226), 398 (79), 399 (80, 81, 83),
415(384), 416(79, 384), 438 (17, 18), 439
(18), 537 (164), 587 (133—135), 597 (24—27),
601 (8, 9), 602 (19, 22), 605, 606 (34—37),
612 (13, 14, 16, 20), 613 (20)
Щепеткова О. А. 365 (254, 255, 257), 367 (6,
254, 270), 368 (254, 270, 275), 386 (60),
400,402 (124), 408 (254), 422 (124, 408, 409),
441(44), 442(46), 443(55), 444(46, 55),
590(196), 613 (21, 26)
Эгути С. 633 (34)
Эскин И. Т. 201 (52)
Юй Чжень-лин 407(231), 513(27)
Юсупов Ф. Ю. 355—357 (195)
Юсуфов Б. Г. 43 (115—117)
Юрьев Ю. К. 561 (58), 580 (48)
Яковлев И. П. 15(17)
Яковлева Т. В. 165 (31)
4t*
644
Авторский указатель
Якубова Ф. А. 219 (133), 222, 223 (171), 231
(133), 232(171), 236(133) 239(171), 339
(99), 383 (32), 398 (74)
Якушкина С. Я. 414 (363), 516 (69)
Ямада А. 520 (127)
Ямамуро Й. 520 (127)
Abegg R. 565 (91)
Abel E. W. 16 (83), 26 (11), 163 (14), 165 (154),
337 (69), 409 (294), 414 (367—369), 422
(294), 423 (368, 369), 432 (1), 527 (50), 609
(82),' 633(77)
Adams L. M. 520 (130)
Adamson J. H. 527(66)
Addison С. С. 165 (21), 370 (282)
Adkins H. 590 (203)
Ahrens J. U. 40 (103)
Ainley A. D. 584 (89)
Aitken I. D. 16 (105), 175 (219), 537 (165)
Aiyar A. R. 633 (64)
Akerman K. 20 (16, 17), 35 (69), 536 (137)
Alberty H. E. 210 (69), 329 (6)
Aldride W. N. 432 (28)
Alexander D. A. 520 (138), 527 (71)
Allen N. 405 (199)
Alleston D. L. 118(33), 220, 233(138), 241
(271), 333 (43), 358 (225), 381 (2), 385 (53,
56, 57), 386(53, 56, 62). 400, 405 (109),
406 (210), 408 (109, 244), 412 (335), 415,
416 (244), 419 (397, 402), 422 (109), 432 (14,
16), 434 (69), 457, 467 (21), 468, 469, 471, 472
(75), 520 (158)
AllredA.L. 89(1), 89,90(5), 105(74,77), 150(290)
Alssar M. E. 48 (13)
Altamira M. S. 536 (134)
Amberger E. 128, 134 (32), 165 (106, 111, 113),
223, 236. 241, 246(175), 408(253), 417
(253, 386, 387), 460 (39), 461 (39, 43, 46), 550
(8), 578 (29). 607 (44), 620, 621 (1, 7)
Anderson A. 165 (70)
Anderson A. W. 520 (123)
Anderson H. H. 27 (21, 26, 27), 30 (21), 31 (26.
27), 81 (2, 5, 7), 82 (7), 89 (7), 90 (21—23).
91(7, 38—40, 43), 93(51), 95(1, 3, 4),
96 (4, 5, 16, 18, 19, 22, 25, 31, 33, 35, 38),
97(5, 16, 18, 35—37, 41), 99 (1, 3—5,33,
38), 100(3—5, 16, 31, 33, 37, 38, 41, 49),
107 (88), 110 (5, 16, 18, 31, 36, 37, 105), 124
(12), 125 (12, 23), 127 (30), 128 (31), 130
(12), 133 (12, 23, 31), 134 (12, 30), 135 (12),
211 (77), 233 (258), 339, 341 (111), 350 (166),
351, 354, 355, 357 (111), 383 (34), 392 (86),
396 (1, 17), 397 (1, 59), 398 (75), 399 (1, 84),
400(84), 405 (1, 59, 188), 406 (1, 17), 407
(59, 188), 419 (84), 457. 465 (14)
Anderson R. 256 (358), 519 (38)
Andrascheck H. J. 502 (61), 520 (112)
Andrews Т. М. 397(47, 48, 53), 401, 402,
(140), 403 (155), 417 (140), 513 (22, 30, 31, 34)
Ankel Th. 535 (33)
Anspon H. D. 520 (149)
Antler M. 520 (189)
Anzaldi O. 633 (22, 35)
Apperson L. D. 554(71), 586, 587(119), 598
(28), 614 (28), 629 (31)
Ятани Т. 181, 182, 184 (8)
Яманэ Я. 226 (353), 401 (134), 519 (47)
Ямасита С. 520 (194)
Янагата М. 520 (127)
Яновский Д. М. 402 (153), 520 (86, 87)
Aranyine-Halmos Т. 162 (8)
Aries R. S. 520 (132)
Arimoto F. S. 598 (41, 42)
Armitage D. A. 414 (367)
Armour A. G. 194 (6), 459, 464, 477, 480 (35)
Arnold L. E. 535 (106)
Arntzen С. Е. 186 (66, 67), 223, 230 (184), 245,
247 (184, 290), 338, 343, 358, 363, 364 (82),
453, 454 (51), 497, 500, 502 (19)
Aronheim B. 260 (371), 378 (16, 17), 381, 385
(4), 408 (264)
Arons J. R. 15(44)
Ashikari N. 520 (107, 118, 139)
Ashley J. N. 203'(57), 260 (372), 582, 583, 586
(73)
A'ssar M. K. 247, 249, 251, 255, 256 (303)
Atkinson E. R. 536 (147), 559, 560 (22)
Atlas S. M. 392, 393 (93)
Ault L. H. 520 (201)
Austin P. R. 242, 243\272), 552 (42, 43), 560
(31, 36, 41), 562 (70)', 565 (89), 579 (32, 33,
43), 580(33), 581 (58), 584(32), 585, (98),
596 (14), 597 (20), 605 (19, 20), 606 (20),
612, 614 (19), 617 (8)
Aufdermarsh С. А. 227 (222), 512 (8)
Bachman G. B. 245 (287)
Bahr G. 194(5, 9), 195(9), 197(5, 9, 36),
198 (5, 9), 206, 208(2), 217—219 (2, 127, 131),
220 (2), 243(127, 131), 345(136. 137),
348 (136, 137, 157), 355 (136), 385 (50),
396 (8), 438 (1), 439 (21), 440 (1), 441 (1, 43)
504 (73), 551 (24, 25), 579 (39), 588 (143),
602 (14), 612 (18), 633 (50)
Bailie J. C. 228 (235), 551, 552 (21), 577, 578
(13), 596, 597 (13), 613, 614 (24), 621
624 (8), 627 (24), 628 (8, 24)
Bajer F. J. 49(23), 249(309), 345, 347(142),
563 (74)
Bake L. S. 541 (23)
Baker A. G. 165 (181)
Baker H. R. 343, 350, 361, 366, 367(124)
Balassa L. 560 (33)
Balasubramanian A. 535 (3)
Baldwin R. A. 109 (98), 316 (77)
Ballester M. 162 (1), 526 (2), 631 (1)
Ballczo H. 405 (200, 201)
Ballinger P. 606 (41)
Bambynek W. 165(176, 180), 207(30)
Bandet M. 107 (91)
Baney К. Н. 15 (14), 81 (18), 89 (9), 165 (179),
384 (41), 535 (12), 602 (18)
Banks C. F. 414 (357), 520 (187)
Bao С N. R. 535 (3)
Barbieri R 175 (216), 405 (195), 432 (21).
435 (88), 527 (44), 535 (86—90), 63Г (2),
633 (53, 80)
Barker R. L. 402, 419 (141), 519 (56)
Авторский указатель
645
Barnes J. M. 521 (206)
Barnes R. 520 (176)
Barnetson С. 274, 277, 281 (20)
Barnett M. M. 378 (18), 521 (215), 597 (16)
Bartocha B. 165 (82), 558 (4), 586 (128)
Basileiados K. 181, 182, 184 (4, 5)
Bate G. L. 633 (84)
Bauer H. 27, 36 (29), 31 (59), 38 (82), 54 (50),
77 (1), 90 (27), 93 (53), 95, 97, 99 (7), 95,
99 (8), 98 (24), 145 (6)
Baum A. L. J. 520(138)
Baum G. A. 108 (92), 391 (83), 457, 458 (6),
515, 516 (61)
Bavley A. 407 (237, 240)
Bawn С. Е. H. 586 (109, 121)
Bayer O. 520 (179, 180)
Beach J. Y. 633 (24)
Beaird F. M. 541 (35a)
Beattie I. R. 336 (61), 378, 379 (34), 432 (31, 32),
433 (59), 586, 587 (120), 598 (36—39)
Beck W. 99 (48), 406 (218), 608 (65)
Becker E. I. 216(105, 112), 217—219, 243
(112), 269(8), 274, 305, 309, 310(19),
332 (38), 345 (138), 379 (42), 385 (49), 396
(13), 406(207), 451(21, 22), 458(33), 459
(33, 34), 465 (64, 65, 69), 466 (69), 468 (33,
34, 73, 74, 76), 469 (76), 472 (69, 74), 473
(69), 476 (64), 477 (34, 64, 96), 480 (34).
490 (69), 491 (74)
Becker R. 194—198 (10), 216, 217, 221
(116), 282(30), 377(12), 396(10), 438—
441, 448(9), 526(11)
Becker W. E. 568 (4), 570 (1), 620 —622 (2)
Beears W. L. 64 (24)
Beermann С 51 (33),224, 225, 232 (19П, 249
(38), 255(38, 191), 356,364 (197), 501 (48),
562 (63), 586, 588 (108)
Beg M. A. 316 (78)
Behrends K. 396(30)
Behringer H. 445 (64)
Bekate H. 432 (9)
Belcher R. 527 (59)
BellegardeB. 311 (64a)
Bellomo A. 633 (39)
Belluco U. 355(193), 377(11), 527 (44), 585
(91), 586 (112—115), 589 (186), 612 (1, 3, 4,
7), 613 (1, 25), 614 (4, 31), 631 (2), 633 (53,
54, 80)
Belpaire F. 519 (6)
Benkeser R. A. 49 {21), 117(32)
Berenbaum M. B. 520 (185)
Berg van der R. H. 633 (44)
Berger M. N. 520 (137)
Bernardini F. 512 (1), 536 (143)
Bertozzi E. R. 520 (185)
Bertz T. 408, 415 (246—248), 520 (154, 155)
Bessant К. Н. С 536(117)
Best С. Е. 190 (90), 404 (161), 405 (189—191),
519 (64—65)
Better J. 553 (52)
Beumel O. F. 196 (34), 238, 245, 249, 250 (265),
390 (80), 439 (22), 472—474 (85, 87),
477 (102), 487 (85)
Bevillard F. 404 (186)
Bhattacharya S. N. 216(117), 252 (325a),
410 (303)
Bhide K. N. 633 (83)
Bigorgne M. 411 (318), 507 (89), 608 (54), 530
(41)
Billiotte J. С 227, 228, 232 (219), 364 (244),
528(79)
Bills J. L. 15 (77)
Bindschadler E. 552 (44), 624 (10, 14: 16),
625-627(14)
Birr К. Н. 48, 50 (8)
Blair McD. J. 551 (30), 589 (183)
Blake D. 330 (21), 414 (362), 432 (18), 497-500,
502, 503 (20)
Blake E. 409, 417(279)
Blitzer S. M. 186 (61, 64), 541 (57), 549 (1, 3),
552 (3), 553 (50, 54, 57, 58), 558 (6), 570
(2)
Bloch H. S. 535 (105), 536 (135)
Blofeld R. E. 26 (13). 163 (16)
Bloodworth A. J. 313 (68, 69, 72), 314 (68),
316(82)
Blum M. S. 521 (224)
Blumenthal A. 633 (48)
Boboli E. 519 (61)
Bock R. 396 (29, 30), 527 (35, 70)
Boeseken J. 196(32), 212(84), 377(13), 438,
439, 441 (4)
Boeters H. 128 (32)
Bohm H. 520 (113), 536 (122)
Boidol K. 252 (324a)
Bonati F. 411 (320, 328), 507 (91, 95), 508 (95)
Bond A. C. 124(10), 457(8, 9)
Bonelli E. J. 633 (78)
Bonfiglio G. 519 (2)
Bonner E. F. 520 (145)
Bonnot J. 175 (221)
Booth R. B. 152, 154, 155 (39)
Borbely-Kuszmann A. 208, 211, 216 (47),
528 (84)
Bornmann P. 96 (28)
Bornstein J. 403 (155)
Bornstein R. 432 (30)
Borror A. L. 208, 210 (41), 351 (170)
Bost R. W. 343, 350, 361, 366, 367'(124)
Bothorel P. 535 (61)
Bott L. L. 547 (49)
Bott R. W. 34 (64), 52 (39), 83 (42—46, 50),
124 (19), 221 (159), 224, 248 (190), 338 (77),
350(161, 162), 498 (36)
Bottel R. S. 16 (97)
Boue S. 440 (37)
Bowden K. 165 (14), 208, 221 (38), 535 (2),
559(19)
Bowden S. T. 221, 222, 226, 233—235 (163),
330 (19), 404 (185)
Bower F. A. 401, 402, 417 (140), 521 (224)
Brack O. 419 (398.)
Bradv D. 409, 422 (294), 432 (1)
Brady D. B. 337 (69), 414 (367—369), 423 (368,
369), 609 (82)
Braidrich E. V. 520 (177)
Braithwaite D. G. 547 (44, 45)
Brandle K. 40 (96), 226, 235 (206, 208), 241
(208), 364 (240)
Brandle K. A. 339, 347, 352—354 (109), 453 (38)
Brandt M. 633 (44)
Brauchitsch M. 562 (71)
Braude E. A. 165 (14), 208, 221 (38), 535
(2), 539 (19)
BraunD.39 (88)
Braun J. 68 (40), 286 (37)
Braun К. Н. 633 (43)
646
Авторский указатель
Bregadze V. J. 244, 248, 250 (282)
Breslow D. S. 535 (104)
Bresser R. E. 520 (103)
Breu R. 411 (322, 323), 507 (93, 94), 509 (94)
Brevnan T. 633 (65)
Bricker С. Е. 527 (41)
Brinckman F. E. 54(52), 127(28), 159(8),
165(82), 214(43), 216, 221, 222, 228, 229,
236, 237(107), 356(203, 204), 411 (308),
457, 463 (25)
Brinkley S. R. 634 (66)
Britain J. W. 520(164)
Brockway U. O. 535 (77)
Brook A. G. 36 (73), 51 (34), 81 (19), 89 (8),
97(39), 100(50), 103(64), 124, 134(18),
146, 147(12), 148(20), 150(12)
Brown С L. 78 (1), 81( 11), 91 (44), 106 (86),
162 (2)
Brown D. H. 165 (69)
Brown J. 35 (72)
Brown J. E, 462, 463 (51, 53), 464 (53)
Brown M. P. 15 (50, 53), 90, 91 (19, 33), 105
(76), 145(8), 165(68, 98), 332(36), 382,
388(21), 413(351), 438, 439(13), 504 (74),
527 (19)
Brown О. М. 541 (57)
Brown T. L. 165 (79), 477 (103), 503, 504 (65)
Browne О. Н. 580, 581, 586, 587 (49)
Bru A. 234 (261)
Bruce J. M. 520 (123), 536 (121)
Brugel W. 535 (33)
Brunink H# 527 (69)
Bryce-Smith D. 551 (30), 589 (183)
BuaE. 536(116)
Buchanan H. W. 409, 412 (285, 286), 519 (76)
Buchmann O. 221, 246 (160), 337, 338 (72,
74—76), 347 (72, 156), 399, 404, 405 (94)
Buchwald H. 411 (314)
Buckingham A. D. 535 (38)
Budding H. A. 40 (98, 100), 48, 52 (18a),
159 (10), 231 (243, 246), 240 (270), 286 (38),
290 (47, 50), 291, 292 (47, 49a), 294—296,
300 (496), 305 (50), 307, 309 (58), 327 (15),
342 (120), 397 (58), 458 (31), 512 (13),
514 (13, 49, 50), 515 (49), 536 (144), 559 (21),
560, 561 (48)
Buell В. Е. 633 (30, 37)
Buell G. R. 453 (28)
Buening R. 536 (130)
Bugg F. M. 520 (145)
BuisW. J. 162(7)
Buisson R. 221, 223, 237(167), 319, 323(8),
346 (145), 356 (199), 363 (145), 447 (6),
457 (13)
Buist J.M. 520(175)
BuIIard R. H. 221, 228 (155), 233 (253), 339,
346 (92), 346 (148), 378 (20), 435 (81), 438,
439 (15), 457 (4), 497, 500 (16, 17), 500 (41),
500, 504 (42)
Bungard G. 412 (338, 339)
Burch J. E. 356 (209)
Burdon J. 270(10)
Burg A. B. 356 (201), 377 (10), 477 (95)
Burgel K. 526, 527(3)
Burger H. 15 (26)
Burke J.J. 165 (94)
Burkhardt G. 209 (55), 330, 332, 333, 335 (8),
527 (57)
Burkhardt P. 396 (29)
Burlith J. M. 447(5)
Burschkies K. 27 (29), 31 (59), 36 (29), 38 (82),
54 (50), 77 (1), 90 (27), 93 (53, 54, 56),
95 (7, 8), 96 (24), 97 (7), 99 (7, 8), 145 (6)
Burt S.L. 402, 403(150)
Burton R. 51 (36)
Bush R. P. 16 (83)
Bush R. T. 165 (154)
Butcher F. K. 165 (55, 70)
Butcher S. 165 (89)
Byerly W. 217 (121)
Cadiot P. 227 (218—221), 228, 232 (218, 219),
238, 249 (221), 251 (218, 314), 252 (314),
253 (218), 253, 254 (218, 314), 255, 256 (218),
258 (314), 364 (244), 453 (30), 500 (44, 45),
528 (79), 560—562 (39)
Cahill P. 165 (89)
Cahours A. 181, 182, 184(2,3), 186(48—50,
54), 256 (346), 339 (103, 104), 378 (23),
396(15), 400(15, 112, 119), 401 (119), 405
(194), 406 (213), 432 (25). 439 (27), 541 (1),
580 (53), 601 (6)
Calabretta T. J. 421, 422 (405)
Calas R. 273, 274, 303 (8), 472, 473 (86)
Calcott W. S. 541 (15, 16, 20, 21)
Caldwell J. R. 520 (159), 535 (109)
Callingaert G. 378, 379 (34, 35), 535 (91), 541
(11, 24, 65—67), 542 (79), 545 (13), 547 (57),
554 (64), 556 (78), 559 (10), 576 (6, 7), 578(7),
579 (7, 31), 586, 587 (120), 588 (177), 596 (8),
597 (15, 18, 19), 598 (14, 18, 29, 36—40, 45),
601 (2, 7), 602 (2, 21), 606 (38), 614 (32), 628
(27) 633 (16)
Callis С. С. 186 (58, 59), 210 (71), 435 (80)
Campbell I. G. M. 396, 411 (25, 26), 415 (25),
423 (25, 26), 433 (48, 49), 521 (221), 527 (20)
CanterinoP. J. 520 (156)
Carl R. W. 547(48)
CarleyD. R. 553 (57, 58)
Carlson C. L. 245 (287)
Carrick W. L. 520 (144—147)
Cartledge F. K. 149 (26), 151 (32), 196 (33),
342 (122), 496, 499 (9), 505 (80, 81)
Cartmell E. 15 (50)
Cassan J. 15 (69), 165 (7, 63), 432 (39)
Cassol A. 405 (195), 435 (88)
Castel R. 186 (60), 210 (70)
Catlin W. E. 580 (51)
Cattalini L. 355(193), 586(113), 612(3,4),
613 (25), 614 (4)
Caujolle F. 234 (261)
Cawley S. 15 (57, 59)
Cenini S. 411 (328)
CenterlinoP. J. 535 (108)
Challenger F. 454 (57), 618 (15)
Chamberland B. L. 412 (340), 516 (77)
Chambers R. D. 54(53), 216, 221, 228, 229
(108,110), 236(108), 237(108, 110), 241
(108), 340(113), 346(152), 356(152, 206,
207), 364, 365 (206), 438, 439 (2), 443, 444
(58, 59)
Chambers R. F, 188 (79), 196 (30), 212, 256 (85),
343(128), 378(26), 384(40), 396 (14)/457
(3), 497—501, 504 (15)
Chan J. K. 397, 423 (60)
Chandra G. 103 (65), 111 (106)
Chandra S. P. 16 (79)
Chang Kuo Ming 316 (83)
А в шорский указа/пел ь
647
Chapman A. H. 527(65)
Chasar A. G. 520 (146)
Chatt J. 34 (63), 117 (31), 229, 231 (236), 453
(50)
Chenicek J. R. 535 (105), 536 (135)
Chernick С L. 165 (168), 207 (21)
Chipperfield J. R. 90 (15)
Chivers T. 216, 221, 228, 229(108, 110), 236
(108), 237(108, 110), 241 (108), 346 (152),
356 (152, 206, 207), 364, 365 (152, 206)
Chodkiewicz W. 251, 252, 254, 258 (314), 560—
562 (39)
Choudhuro В. К. 633 (64)
Chris С. Е. 519 (57)
Christl R. J. 535 (97)
Christofferson G. D. 634 (24)
Christopher P. M. 16 (94)
Church J. A. 409, 412 (286)
Church J. M. 253 (329), 379 (38, 39), 396 (24),
399 (94), 400 (24), 404, 405 (94), 409, 412
(285), 519 (76)
Claridge R. F. С 165 (182)
Clark H. C. 23 (4, 5), 165 (73, 103, 145), 207
(14), 274 (16, 17, 20), 277 (16, 17, 20, 25), 279
(17, 25), 280 (16), 281 (16, 20), 316 (78—80),
330(15), 340(15, 113), 364(15), 406(205,
206), 411 (309, 311), 432, 433 (23), 535 (84,
93, 94), 443, 444 (58—60), 457, 458, 463,
464 (16), 503 (64)
Clark R. E. D. 527 (23, 24)
Clauson-KaasN. 554 (72)
Clem W. J. 541 (31, 39)
Cleverdon D. 408 (274), 519 (31)
Closson R. D. 596 (6)
Coates G. E. 8 (8, 9), 195 (27), 330 (21), 409
(279), 414 (362), 417 (279), 432 (18), 497—
500, 502, 503 (20)
Coc P. L. 270 (10)
Cohen G. 520 (188)
Cohen H. 116(21), 214, 221—223, 233, 236,
241 (95), 242, 246 (277)
Cohen H.J. 419 (394)
Cohen H. M. 333, 354 (47), 397, 405, 406 (42),
453 (43)
Colambo L. 536 (143)
Colaitis D. 528 (75), 633 (15)
Collie N. 186 (53), 269 (4)
Congiundi N. R. 520 (160)
Considine W. J. 220 (136), 383 (35), 386 (61),
391 (83), 403 (156, 158), 404 (158), 408, 419
(275), 434 (72), 457 (6), 458 (6, 32), 515,
516 (61), 528 (89)
Cook E. B. 633 (66)
Connoly R. E. 468, 477 (81)
Cook S. E. 536(139), 541(52), 550(11,17),
553 (58), 558 (8), 560 (34), 565 (8), 568 (4),
570 (1), 576—579, 582 (8), 588 (162—174),
620—622 (2)
Cooke D. J. 207, 278, 290 (27), 338 (81),
457, 468, 476 (29), 527 (48)
Coover H. W. 14 (4), 519 (27)
Costa G. 527 (38, 39), 535 (70, 72)
Cottle D. L. 520 (192, 193)
Cotton F. A. 15 (77)
Cox E. F. 520 (162, 165)
Cox J. J. 432 (29), 632 (4)
CoyleT. D. 15 (55), 165 (119)
Cramer С R. 208, 216 (50)
Cramer P. L. 588 (154, 156)
Creemers H. M. J. С 140 (70, 71), 273 (2в),
309(61), 480(107), 482 (108a), 483 (108a,
Ilia, 1116), 484 (108a, 113a), 485, 486 П08а),
488 (108a, 119a), 489, 490 (108a)
Cremer J. E. 432 (28)
Cresswell W. T. 165 (2, 6), 535 (54, 55)
Criegee R. 565 (88)
Croatto U. 175 (216), 535 (86)
Cross R. J. 27, 32/35 (31), 81 (9), 124, 125, 136
(14), 139 (58a)
Crosse В. С. 414, 423 (369)
Crowder G. A. 535 (23)
Csabav J. 602(24), 608 (69)
Culleri W. R. 304 (54), 316 (81), 444 (63a)
Cummins R. A. 165 (3, 27, 28, 53, 80), 397 (41),
406 (41, 211), 433 (60)
Cumper С W. N. 433 (46, 47)
Cunningham J. 54 (53)
Curry R. P. 633 (82)
Cutler A. 406 (208), 441 (39), 465 (66)
Dahlig W. 553 (56)
Dahlmann J. 101 (58, 61), 408, 413, 415, 416
(245), 606 (42, 43)
D'Amore G. 633 (39)
Danek O. 185 (44), 382 , 388 (17), 396, 397,
400,401,406(20)
Danley R. L. 398 (78)
Dannin J. 513 (21), 536 (155)
D'Ans J. 408, 409 (261), 499, 502 (39), 562 (71),
624, 628 (17)
Danyluk S. S. 15 (57, 59)
Danzer R. 550 (14)
Danzik M. 195(28), 210, 221, 222, 234, 242
(66), 340(112)
Dao-Huy Giao 87 (62), 114 (1), 366 (263)
Daudt H. W. 541 (8, 9)
Dave L. D. 165 (18), 203 (58), 535 (4), 554 (66)
David B. 328 (20)
Davidsohn W. 281 (29)
Davidson W. E. 16 (108), 104 (72), 105 (73, 82),
106 (72, 82), 159 (3), 175 (223), 409, 414 (281),
537 (168)
Davies A. G. 101 (56, 57), 220, 233 (138), 311
(68), 313 (69—72), 314 (68, 73), 316 (73, 82),
328 (19), 333 (43), 358 (225), 381 (2), 385 (53,
56, 57), 386 (53, 56, 62), 390 (53), 398 (77a),
400 (109), 402 (148), 405 (109), 406 (210),408
(109, 244), 413 (148), 415 , 416 (244), 419
(397, 402, 402a, 403), 420 (4026), 422 (109,
110a), 432 (14, 16), 434 (69), 457, 476 (21),
520 (158), 572 (9), 607 (45a) 434 (69), 457,
467 (21), 520 (158), 572 (9), 607 (45a)
Davies D. I. 573 (7)
Davies H. 212 (82)
Davies W. С 206, 216 (45), 330 (19), 404 (185)
Davis V. E. 221, 222, 226, 233—235 (163),
330 (19), 404 (185)
Dawson D. S. 316 (81)
Dawson H. J. 633 (41)
Deblandre С 358 (224)
DeBoerC. E. 49 (21), 117 (32)
Debrezeni E. 221—224, 233—236, 238 (151),
339, 340, 342, 351 (93), 383 (31). 398, 405 (72)
Dec S. M. 48 (11), 364 (248), 562 (68)
Degering E. F. 581 (59)
Del Franco D. J. 273, 274, 277, 279 (10), 457,
464, 490 (27)
DelhayeA. 207 (26), 337, 338, 347 (73), 577 (20)
648
Авторский указатель
Demarsay E. 186 (49, 50), 400, 401 (119)
Dennis L. M. 26(1,2), 27(16, 18), 29(16),
36 (18), 50 (31), 53 (43), 54 (5П, 81 (10),
82 (32, 33), 83 (33, 38), 90 (16), 93 (52), 95
(10, 13), 96 (23), 252 (323)
Depree D. 251 (315)
Dersin H. J. 358—360 (230), 414 (365)
Derwish G. A. W. 165 (16, 44, 52), 216, 217,
218 (113), 399 (87), 432 (40, 41)
Dessy R. E. 465 (68)
DeVries J. E. 633 (55)
De Witt E. G. 541 '(37)
Dibeler V. H. 53 (44), 165 (140), 535 (78)
Dickson R. S. 256, 260 (355), 497, 503 (31),
553 (53), 629 (32)
Didtschenko R. 19 (3), 31 (58)
Diehl J. W. 151 (31), 502 (59), 624, 626, 628 (13)
Dietz E. 51 (32), 255 (337), 364 (243), 562 (66),
584 (90)
Dillard C. R. 165 (24, 65, 167), 207—209, 221,
233 (28), 273, 274, 277, 279 (10), 457 (15,
27), 464, 490 (27), 526 (10)
Dimroth O. 565 (90)
Dimroth P. 565 (88)
Disselkamp P. 535 (82), 602 (12), 605 (4)
Dix W. M. 551, 552 (34), 612 (12), 624,628 (9)
Dobratz E. H. 553 (55)
Doering W. E. 527 (37)
Dolan D. N. 206 (1)
Domange L. 165 (67)
Donadio R. E. 216, 221, 222, 228, 229, 236,
237 (107)
Dorfelt С 330(12), 382(15), 407(235), 520
(205), 521 (216, 228)
Downey M. F. 64, 66—68 (25)
Downing F. B. 541 (23)
Dowrick D. J. 207, 208 (29)
Drago R. S. 432 (27), 434 (68), 535 (37. 83)
Drefahl G. 219 (132), 453 (48), 558 (3, 5)
Drenan J. W. 29 (48)
Drenth W. 15 (71), 68 (38), 75 (7), 165 (12),
283 (33), 294—296. 300 (496), 312 (33), 535 (5)
Drew H. D. K. 29, 32 (39), 90» 91, 93 (28), 99
(44), 145 (3)
Druce J. G. F. 190 (88, 91, 92), 335 (58, 59),
378 (32, 33), 392, 393 (88, 89), 400 (111),
404 (181), 406 (215), 432 (10—12), 542 (74)
Dubac J. 36, 40 (78), 51 (37, 38), 81 (23, 24),
82(23), 114 (3), 124 (13)
Dubois J. G. 576 (5)
Duck E. W. 520 (152)
Duckworth M. W. 527 (65)
Duffy R. 570 (3), 620—622 (4—6)
Duiltan G. P. M. 336 (61)
Dunn P. 165 (3), 397 (41), 406 (211), 433 (60)
Dupont R. 209(58), 350(167, 168), 351, 352
(167)
Durig J. R. 15 (44)
Dyer E. 432 (3)
Dykstra F. J. 542 (79), 576—579 (7), 598 (29),
601 (2), 602 (2, 21), 606 (38)
Dyson G. M. 519 (39)
Eaborn C. 34 (64—66), 41 (108), 48 (16—18),
50 (17), 52 (16, 17, 18, 39), 83 (40—47, 49,
50), 124 (19), 221 (154, 159), 224 (190), 228,
247 (154), 248 (190), 253 (154), 338 (77, 78),
350 (160—162), 454 (55), 498 (36), 562 (67,
69), 584(81)
Eataugh H. 439 (29), 501 (55), 502(55, 62)
Eberly К. С. 190 (90), 210 (69), 329 (6), 407(238)
Eckelmann A. 519 (81)
Edgell W. F. 165 (60), 207 (12, 16)
Edwards C. 221, 222, 226, 233—235 (163),
330 (19), 404 (185)
EgleG. 231 (242), 513 (20)
Egmond J. С van 175 (198), 208, 216, 256 (49),
257, 259 (49, 362), 521 (210)
Eisch J. 68 (42), 223, 226, 227, 235, 236 (182),
457, 466, 490(11), 561 (51)
Elehuson P. T. 520 (163)
Eller W. 182, 184 (15)
Elson L. A. 584 (89)
Emeleus H. I. 414 (359), 461 (45), 606 (40),
614 (29), 631 (9)
EmmertB. 182, 184 (15^
Enders M. 609 (81)
Endrulat E. 499, 502 (39), 624, 628 (17)
Engelhardt V. A. 281 (28)
English W. D. 16 (95)
Epstein L. M. 165 (132)
Erdeyne-Schneer A. 162 (8)
Eschenbach W. 237 (227), 562 (65)
Essen H. E. van 396, 400 (27)
Evans D. F. 165 (18), 203 (58), 535 (4), 554 (66)
Evans D. P. 208, 209, 217, 221, 222 (44), 550
(13), 581 (60), 598 (30), 605 (6)
Evans J. V. 165 (53)
Evans P. R. 606 (40), 614 (29), 632 (9)
Evans R. B. 203 (57), 260 (372), 582, 583, 586
(73)
Evans R. L. 332 (39), 387 (55), 423 (413)
Evnin A. B. 246, 255 (298), 346(144), 45U
452 (24)
Faleschini S. 440 (38)
Faraglia G. 432 (21), 535 (88—90)
Farkas M. 633 (32)
Farnsworth M. 527 (22, 25)
Farrar M. W. 570 (2)
Farrar W. V. 194 (16), 212, 213, 234 (86), 440
(34)
Fassel V. A. 15 (8), 29 (43)
Fath J. 520 (182)
Faulkner С J. 188 (74)
Faulkner D. 408 (272, 274),r519 (31)
Favero P. 612 (7)
Feeney J. 570 (3), 620—622 (6)
Feldhake C. J. 535 (53), 562 (73)
Fentiman A. F. 182, 183 (27), 527 (52)
Fenton D. E. 48 (10), 87 (55)
Ferguson W. C. 633 (28)
Fernby A. M. 519 (39)
Fernlev A. M. 519 (13), 520 (88)
Fettel "H. 535 (30, 40)
Fichter F. 541 (46)
Field E. 547 (48)
Figgis B. N. 386 (62)
Findeiss W. 87 (58), 356 (212)
Finholt A. E. 124, 125 (10), 457 (8, 9)
Fischer E. O. 165 (78), 194 (2), 535 (15, 48, 62,
63), 554 (65, 68—70)
Fish R. H. 277(24)
Fisher L. 60 (1)
Flitcroft N. 165 (64, 105), 457 (24, 26), 461 (24),
535 (29)
Flood E. A. 22 (1, 2), 27 (15), 36 (77), 81, 82,
84 (3), 90 (20), 91, 92 (37), 93 (50), 95 (2),
Авторский указатель
649
96 (17), 106 (17, 87)t 122, 125 (2), 145, 146,
151, 153 (7), 577(15)
Fodorne С. P. 633 (32)
Foldesi J. 208, 233, 239 (50a), 256 (340a)
Ford F. E. 145, 146 (10), 223 (185), 457 (7),
496 (2, 34), 499 (34), 500 (2, 34), 502, 503,
505 (34), 560 (40), 623, 624 (5)
Forssen E. 519 (28)
Forstner J. A. 406 (223)
Foster L. S. 22 (2), 29' (48), 53 (46), 81 (12),
89, 90 (14), 95, 106 (9), 122 (1), 145—147, 150,
154, 155 (4), 439 (30), 551, 552 (34), 612 (12),
624 , 628 (9)
Foster W. E. 516 (72, 73), 520 (199)
FowlesG. W. A. 15 (50), 145 (8), 332 (36),
396, 411 (25, 26), 415 (25), 423 (25, 265), 433
(48, 49), 438, 439 (13), 497 (22), 504 (74),
505 (22), 521 (22), 527 (19, 20)
Franc J. 527 (47)
Franchimont E. 254 (331)
Frankel M. 397, 398, 407 (64), 528 (88)
Frankland E. 181, 182, 184 (1), 256 (342, 343,
345), 339, 346 (97), 438, 439 (6)
Franzus B. 520 (156), 535 (108)
Freedman H. H. 82 (29), 250 (312), 345, 346,
352 (143), 578 (22)
FreimillerL. R. 536(128)
Freise V. 165 (176, 180), 207 (30)
Freeman J. P. 433 (55)
French F. A. 535 (80)
Fresnet P. 576 (5)
Frey F. W. 550(11), 556(79), 558, 565 (8)
Frey H. 519(15)
Freyer W. 551 (19), 586 (127)
Fricker H. H. 521 (230)
Fridman L. 520 (166)
Friebe E. 165 (62), 384 (44)
Friedmann B. 432 (9)
Fritz G. 464, 465 (63, 63a), 527 (28)
Fritz H. E. 233 (255), 332 (39, 42), 385 (55, 58),
423 (412, 413)
Fritz H. P. 165 (78,99, 106), 243, 251, 252
(279), 461 (46), 535 (15, 16, 36), 554 (70),
561 (62)
Frohning C. D. 20 (13b), 47 (2a), 89 (6a)
Frve A. H. 407, 415 (242), 520 (93, 94)
Fuchs O. 216, 217, 243 (104), 343, 345, 352
(129), 385 (51)
Fuchs R. 27 (22), 30 (56), 36 (74), 47 (5), 49 (19),
64(23), 90(26), 95(6), 124, 135 (11), 223
(180), 274 (21)
Fueno T. 257, 259 (361), 520 (161)
Fujitani K. 392 (84), 527 (31)
Fulton M. 270 (10)
FurmanN. H. 405 (199)
Furnival S. G. 274, 277, 280, 281 (16), 457,
458, 463, 464 (16)
Furukawa J. 257, 259 (361), 520 (161), 535 (103)
Fusari S. A. 35 (72)
Gage J. C. 633(85)
Gallais F. 165 (151), 209 (60)
Garcia E. L. 633(21, 36)
Garrett A. B. 535 (92)
Gautron R. 519 (24)
Gawargions Y. A. 633 (14)
Gebbic H. A. 165 (70)
Geissler H. 435 (97)
Gelbert H.332 (34),382 (15),383 (27), 521 (216)
GeldellJ. B. 526 П0)
Gelius R. 194 (5,'9), 195(9), 197, 198(5,9),
217, 218(127), 219(127, 131), 237(264),
243 (127, 131), 245, 247 (264), 248 (305), 249
(264),250 (264, 305), 345 (136, 137), 348 (136,
137, 158), 349 (158), 355 (136, 158), 396 (12),
438—440 (1), 441 (1, 43), 504 (73), 563 (77),
633 (42)
George K. 328 (18)
George M. V. 146, 151, 152 (9), 154 (43)
George T. A. 272, 315 (2)
Gernaux R. 435 (90)
Gerow C. W. 27, 32, 34 (14), 36, 38, 40 (75),
47 (3), 51 (35), 52 (3), 64 (22), 78 (4), 137 (51),
146,(11), 147(13, 15, 16), 148, 149(19),
150(15, 29), 151 (16,30,33), 153(11, 13,41),
154(19, 41,43), 155 (13, 41), 439.(28), 503 (63)
Gerrard W. 165 (55, 70), 356 (208, 209), 408
(255), 415 (380), 527 (56) •
Gershbein L. L. 208, 210, 216 (37), 369 (278),
590 (204)
Gertner D. 397, 398, 407 (64), 528 (88)
Geyer R. 527 (21)
Ghate M. R. 633(83)-
Gibb Т. С 165 (123)
Gibbons A. J. 213, 223 (90), 225 (90, 204), 231,
233—235, 239, 241, 242 (90), 330 (22), 331,
333, 336 (22, 27), 339, 340 (22), 381 (6),383
(36), 385(6), 403, 404(158), 419(399), 434
(72), 462, 463 (52), 527 (59)
Gielen M. 175 (224), 214, 233 (96), 337 (70, 71),
358 (224), 435 (90), 440 (37), 576 (5)
Gilman H. 8 (6), 9 (2), 16 (99, 100), 26 (14),
27 (19, 22), 30 (56), 32, 34 (14), 36 (73—75),
38, 40 (75), 47 (3, 5), 48 (9, 14), 49 (19, 25),
51 (34,35), 52 (3), 64 (22, 23), 68 (42), 69
(43), 78 (4), 81, 82 (6), 89 (8), 90 (26), 95 (6),
100(50), 124, 135(11), 137(51, 52), 144(1,
2), 146(9,11,12), 147(12, 13, 15,16), 148
(19), 149 (19, 23, 26), 150 (12, 15, 29), 151 (9,
16, 30—33), 152(9), 153(11, 13, 41), 154
(19, 23, 41, 43), 155 (11, 13, 41), 164(1),
175(1, 196, 209, 210), 183 (31). 186 (66),
190 (31), 194 (14), 196 (33), 206 (7), 214 (97,
98), 215 (98), 223 (97, 180, 182—184), 226
(182), 227 (7, 182), 228 (97, 183, 231, 234),
229 (234, 237), 230 (183, 184), 232 (97, 252),
235 (182), 236 (97, 182, 183), 237 (183), 238
(97), 241 (97), 242 (237, 273), 243 (237), 244
(183, 273), 245 (183, 184, 237, 252, 286, 289—
291, 293, 299), 246 (231), 247 (183, 184, 237,
252, 290, 299), 248 (97, 252), 251 (7), 274 (21),
321 (17), 331 (29, 31), 338 (82), 342 (122),
343 (82), 356 (198), 358 (82, 227), 363 (82),
364(82,247), 378 (18), 384(42), 411 (317),
439 (20, 28), 451 (23), 453 (23, 41, 51), 454
(51—54), 455(63), 457, 466(11), 484(112),
490 (11), 496 (1, 8, 9), 497 (19, 26—30), 498
(26—30), 499 (8, 9, 26—29), 500 (8, 19, 27),
502 (8, 19, 26, 28—30, 59), 503 (8, 26, 28,
63), 504 (29), 505 (80, 81), 526 (1, 4, 7), 527
(15), 537(161), 550(10), 551(21,31,32),
552 (10, 21, 31, 32, 39, 41, 44), 559 (13, 20),
560 (20, 32, 33, 38, 41), 561 (38, 41, 51, 55—
57), 562 (20, 41, 72), 563 (20, 41, 57, 72, 75,
76, 78), 568 (1), 578 (13, 21, 25), 579 (21, 35,
38, 44), 580 (44, 45, 47), 581 (61,62), 582 (62,
67), 584(25, 80, 84), 586 (62, 99, 119), 587
'650
Авторский указатель
(119), 596 (13), 597 (13, 16), 598 (28, 46, 47),
605(7, 11), 608(7,11,67), 613(24), 614
(24, 28, 33, 34), 617 (9), 618 (9, 11, 12), 619
(\8, 19), 621 (8), 623 (4), 624 (4, 8, 11—14,
16), 625 (14), 626 (13, 14), 627 (12, 14, 26),
628 (8, 13), 632 (И)
Gilson Т. 433 (59)
Gingold W. 388 (70), 406, 412 (216)
Giraitis A. P. 168 (190), 521 (225), 541 (58),
546 (38), 547 (50), 588 (175), 607 (45)
Gist L. А. 214 (97, 98), 215 (98), 223/228 (97,
183), 230 (183), 232 (97), 236 (97, 183), 237
(183), 241 (97), 244, 245, 247 (183), 248 (97),
364(247), 451, 453 (23), 454 (52), 497,
, 498, 502 (30)
Gittins Т. W. 596 (7)
Giustiniani M. 535 (88—90)
Gladstone J. 586 (121)
Glarum S. N. 122 (4), 147 (17)
Glockling F. 15(27), 16(102), 27(30, 31), 29
(30), 32, 35 (31), 78 (7), 81 (9), 109 (100),
124, 125, 136(14), 148(21, 22), 149(25),
150, 151 (21), 152 (36), 153 (21), 154 (21, 36),
155 (21), 554, 555 (73), 559 (17), 561 (61)
586 (105—107), 617, 618 (1), 624, 625 (19),
626, 627(21)
Gloskey C. R. 208, 212 (46), 252 (321), 330 (13,
14), 382 (12), 401 (128), 402 (142, 143), 519
(50, 51)
Glujsen W. L. 535 (101)
Goalby В. В. 206, 207, 251, 260 (10)
Godar E. M. 527 (71)
Goddard A. E. 203 (57), 260 (372, 373), 582,
583, 586 (73)
Goddard D. 260 (373)
Godfrey K. L. 22 (2)
Gohlke R. S. 71 (57, 58)
Golbrach A. 541 (47)
Gold H. 208, 245, 247, 248 (39), 381 (3), 408,
409 (261), 413 (353)
Goldfarb f. D. 15 (21)
Gomory P. 256 (340a)
Good W. D. 535 (57, 59)
Goodier K. 519 (43)
Goodrich R. A. 357 (218)
Gorbach S. 527 (35, 70)
Gordon M. E. 447 (5)
Goreau T. N. 232, 245, 247, 248 (252), 455 (63),
526 (4)
Gorgv W. 165 (93)
Gorin G. 535 (23)
Gorsich R. D. 411 (319), 507, 509 (92), 608 (53),
629 (34)
Gorsline F. С 520 (105)
Graham I. F. 402, 413 (148)
Graham W. A. G. 165 (115), 411 (329), 507 (88),
536 (147), 559, 560 (22)
Grandjean W. B. 541 (57, 58, 63)
Grant D. 57 (66), 330, 332, 357 (7)
Gras G. 186 (60), 210 (70)
Gray M. Y. 558 (4), 586 (128)
Graybeal J. D. 165(139)
Greaslev P. M. 83 (44), 350 (162)
Greber G. 231 (242), 513 (20)
Green В. S. 406, 419(212)
Greenlee K. W. 35 (72)
Greenwood N. N. 165 (123)
Greer W. N. 432 (22), 438, 439 (3), 457 (1),
497 (12, 13), 504, 505 (13)
Gregory N. L. 633 (40)
Gregory W. A. 404 (165), 561, 563 (57)
Gresham W. F. 520 (115, 148)
Griesbaum K. 468 (82)
Griffiths D. С 208, 209, 217, 221, 222(44),
550 (13), 581 (60), 605 (16)
Griffiths J E. 15 (25), 90 (18), 99 (46), 123,
125 (9)
Griffiths V. S. 165 (16, 44, 52), 216—218 (113),
399 (87), 432 (40, 41)
Grim S. O. 109 (99), 153, 154, 155 (40)
Grimm A. 186 (55)
GrimmeW.545(21)
Grohn H. 20 (14), 184 (37), 185 (37—39), 186
(37, 39), 541 (2)
Grosjean M. 175(224), 221, 246(160), 337,
338 (72, 74—76), 347 (72)
Gross R. J. 15 (27), 148, 150—155 (21)
Grosse A. 8 (3), 195, 196, 198 (26), 208 (34),
215 (101), 376 (1), 503 (69), 549, 551 (4),
577, 578 (16), 613 (23)
Grossman J. M. 519 (82)
Grubert H. 194 (2), 535 (63), 554 (65)
Grun L. 521 (230)
Gruntfest I.J. 551,552,(34),612 (12),624, 628 (9)
Gruttner G. 182, 184, (16), 210 (64), 220 (142),
224 (189), 240 (189, 269), 249 (269), 337 (65,
66), 338, 339(85), 345(141), 441, .443 (42),
503 (68), 520 (177), 528 (80), 549 (7), 550
(7, 16), 552, 554 (7), 558 (1), 559 (11, 12, 15,
18), 560 (15, 25, 27, 28), 561 (52, 59), 575,
576 (2,4), 577(10, 11), 578(10), 602(16),
606 (39), 618 (10), 631 (6)
Gudgeon H. 520 (167)
Gulwell T. 208, 209, 217, 221, 222 (44), 550 (13),
581 (60), 605 (6)
Gunter F. 535 (30, 40). 612 (8)
Gupta V. D. 98 (43b), 408 (257, 258), 417 (257),
418 (389a), 434 (65)
Gussler H. 527 (58)
Gustorf К. Н. 454 (62)
Guy J. 165 (67)
Hague D. N. 15 (72), 165 (9), 433 (50), 535 (6)
Haiss H. S. 411 (308)
Hall C. D. 101 (56, 57)
Halvorson D. O. 332 (39), 385 (55), 423 (413)
Hammet L. B. 545 (12)
Hanada T. 185 (39a), 441 (436)
Hance F. E. 26 (1), 53 (43), 82, 83 (33)
Hancock M. 220, 233 (138), 385, 386 (53, 56),
390 (53), 406 (210), 419 (402), 434 (69)
Hanson E. L. 462—464 (53)
Happe J. A 165(100)
Harada T. 186 (47. 65, 68), 337 (67), 378 (22,
24), 382, (20, 22), 386, 387 (63) 388 (22), 400,
401 (126), 406 (220), 419 (400), 435 (75, 77—
79, 82), 438 (12, 14), 441 (14), 497, 498, 503
(18)
HardonH.J.527(69)
Harrington R. С 408 (271), 519 (33)
Harris C. W. 453 (28)
Harris D. M. 29 (42, 44), 32 (42, 60), 81 (14, 15),
96 (21), 124, 125 (16). 126 (25. 26), 134 (25),
137 (50), 147 (18)
Harris J. 0.252 (324)
Harrison J. B. 408 (249)
Harrison P. G. 313 (70),J419 (402a, 403), 420
(4026), 422 (410a)
Авторский указатель
651
Hartel К. 407 (235), 521 (222, 228)
Hartle С. 520 (170)
Наг tie R. J. 608 (62)
HartmannA. 519 (55)
Hartmann H. 40 (103), 48 (12, 13), 51 (32, 33),
219(134), 224(191, 192), 225(191), 227
(226, 227), 232 (191, 192), 238 (267), 241
(226), 243 (278), 245 (288), 247 (267, 288,
303), 249 (267, 288, 303, 308), 251 (134, 267,
303), 252 (134), 255 (192, 303, 308, 337, 338),
256 (303, 338), 356 (197), 364 (197, 241—243),
501 (48), 562 (63—66), 586—588 (108), 633
(78)
Harvey D. R. 573 (8)
Harwood J. H. 519 (19), 535 (96)
Harzdorf C. 135 (42)
Hashimoto T. 83 (50), 352 (186a)
Hatkaway B. J. 165(75), 411 (310), 435(95)
Haubein A. H. 563 (76), 618 (11)
Hayakawa K. 512 (9)
Hayakawa Y. 520 (161)
Hayes M. C. 165 (138)
Heap R. 541 (69), 560 (26), 578 (28), 579, 580
(30), 581 (28, 30), 582 (30), 598 (33), 602
(20), 605—607 (12, 14), 609 (14)
Hechenbleikner I. 195 (28), 210, 221, 222, 234,
242 (66), 251, 253 (318), 332, 333 (40), 340
(112), 520 (103)
Hecker A. C. 399 (95, 103, 104), 404 (95, 98,
103, 104, 172), 405 (95, 98, 103, 104), 409
(283), 414 (98), 520 (96)
Heckmaier J. 519 (69)
Hedden G. D. 541 (68)
Hedges E. S. 519 (20), 520 (104)
Heights J. 519 (73)
Heiligmann R. G. 536 (129)
Hein F. 411 (321), 507 (90)', 545 (15—17), 547
(58), 585(94), 588(141), 608(51, 52, 55—
57), 612, 615 (17), 628 (28), 629 (35, 36),
629 (37—40), 630(40), 632 (10), 633 (13)
Heller I. 189, 190(84), 210(68)'
HellermannL. 319 (4)
Helling J. F. 51 (36)
Hemmert F. 165 (104)
Henderson A. 351 (172a)
Henderson S. R. 631 (3), 633 (62, 81)
Henglein F. A. 210 (65), 407 (239), 516 (65),
527 (29)
Henry M. C. 15 (11, 23), 16 (108), 29 (46, 47),
39(47), 64, 66—68 (25), 104(72), 105(73,
82), 106(72, 82), 116(19, 20), 159 (3), 165
(40), 175 (223), 236 (263), 250 (311), 281 (29),
286, 287(39), 409, 414 (281), 457(18), 514,
515 (51), 535 (7, 11, 84), 537 (168), 561 (50),
582 (68), 583 (68, 76), 584 (76), 605, 606 (32),
608 (63), 612, 614, 615 (2),( 617 (3)
Herbeck R. 520 (113), 536 (122)
Herber R. H. 165 (122, 131, 132)
Hermannsdorfer К. Н. 502 (60)
Hess L. 542 (79), 598 (29, 39), 602 (21)
Hetnarski B. 535 (85), 580 (54, 55), 618 (13)
Heuser E. 608 (56), 629 (38, 39), 633 (13)
Heuther С. Н. 81 (4)
Herzfeld K. 588 (149)
Hevmann E. 273, 275, 301, 303—305 (9, 55),
306—308, 310 (55), 415, 416 (378)
Hieber T. 465 (68)
Hieber W. 411(322, 323), 507 (93, 94), 509 (94)
Hills K. 105 (73), 409, 414 (281), 535 (84)
Hinkens Т. М. 519 (78)
Hirata H. 520 (118, 139)
Hirschland H. 409, 412 (285, 286), 519 (76)
Hobbs С L. 541 (43)
Hobe D. von 165 (158)
Hobrock B. G. 535 (79)
Hochwalt С. А. 535 (91), 447 (57), 601 (7), 614
(32), 633 (16)
Hofeditz W. 588 (144, 151)
Hoffmann A. K. 527 (37)
Hoffmann H. 382 (26), 406, 413 (219), 433 (52),
435 (76)
Hofmann H. P. 51 (36)
HolbrookG. E. 541 (32)
Hoi den F. R. 233 (253), 339, 346 (92), 497,
500 (17)
Holliday A. K. 351 (172a), 570 (3), 586 (123),
620—622 (4—6), 623 (2, 3), 623 (3)
Holmes J. M. 216 (106), 236, 237 (106a), 241
(106, 106a)
Holum J. R. 260 (370), 350, 364 (164), 453 (29)
Homberg O. A. 195 (28), 210, 221, 222, 234,
242 (66), 340 (112), 520 (103)
Honda M. 520 (107)
Honig H 224 (192), 227 (226), 232 (192), 241
(226), 255 (192), 364 (241)
Honigschmid-Grossich R. 550 (8), 578 (29),
607 (44), 620, 621 (7)
Honeycutt J. B. 87 (60), 554 (59, 61)
Hooton K. 554, 555 (73), 624, 625 (19)
Hooton K. A. 27, 29 (30). 78 (7), 105 (74). 109
(100), 149 (25), 152; 154 (36)
Hoppner K. 447 (2, 4)
Horrocks J. A. 519 (39), 520 (88)
Horst R. W. 407, 415 (242), 520 (93, 94)
Horton W. S. 207, 220 (17), 365 (249)
Horun S. 519 (66)
Hostettler F. 520 (162, 165, 168, 176)
Houben J. 8 (2)
Housek R. D. 551 (20), 582 (65)
Houska M. Z. 520 (89)
Hovitz Z. 577 (15)
Howard H. E. 633 (28)
Howarth M. 356 (209)
Huang H. H. 165 (156), 221, 229, 247 (157)
Huber F. 609 (81)
Hudson R. L. 633 (27)
Hueck H. J. 521 (219)
Hugel G. 165 (174)
Huggins M. L. 15 (75)
Hughes M. B. 27, 32, 34 (14), 47, 52 (3), 147,
151 (16)
Hui К. М. 165 (156), 221, 229. 243 (157)
Huizda V. 378, 379 (35)
Hulme K. 432 (32, 33)
Hulse R. 404 (159), 520 (172, 173)
Humpener K. 16 (92), 27 (17), 535 (51)
Hunt H. R. 103 (62)
Hurd C. D. 579 584 (32)
Hurd D. T. 8 (7)
Hussek H. 412 (332, 336), 421 (336\ 434 (74),
608 (58)
Hutton R! E. 181, 182, 184 (13)
Ibekwe S. D. 42 (110), 238, 241 (266), 350, 364
(165)
Ingham R. K. 9-(2), 164 (1), 175 (1, 196), 194
(14), 206, 227, 251 (7), 526 (1).
Ingold K. 89 (2)
Ipatieff V. N. 208, 210, 216 (37), 369 (278), 590
(204)
652
Авторский указатель
Ireland J. 187 (73)
Irving H. 432 (29), 631 (4)
Isbenjian H. 520 (131), 536 (112)
Isibasi Т. 443 (43a)
Jackson J. A. 535 (28)
Jacobs G. 53 (49), 78 (3)
Jahnson F. 49 (24), 72 (57, 58)
Jahr K. F. 634 (18, 19)
Jakusik E. R. 438—442, 444 (11)
Janshen I. 573 (23)
JanssenM. J. 15 (11, 71), 29 (46), 68 (38), 75 (7),
84, 85, (52), 90 (24), 98 (43д), 165 (12, 178),
257, 259 (362), 283, 307 (59, 60), 308, 309 (60),
310 (59, 60), 312 (33), 399, 410 (106, 107),
432 (4, 5), 433 (4, 44, 58), 435 (4, 5), 448 (10),
535 (5, 11, 13), 575 (1)
Jarvie J. M. S. 541 (53)
Jeffers W. 586 (123)
Jeffrey D. A. 182, 183 (27)
Jehn W. 411 (321), 507 (90), 608 (51, 52), 629
(35, 36)
Jehnng H. 527 (32—34)
Jenkins H. O. 535 (77)
Jenkins C. R. 16 (83), 165 (154)
Jenkins L. T. 510 (160), 520 (153)
Jenkner H. 252, 255 (326), 256 (356), 257 (363),
258, 260 (356), 459 (38), 553 (51)
Jensen E. V. 586 (124)
Jensen K. A. 196, 197 (31), 256 (339), 554 (72)
Johnson С S. 453 (32)
Johnson E. G. 497 (23)
Johnson E. W. 253 (329), 379 (38, 39), 399
(92—94, 99—101), 404 (92—94, 99—101,
177), 405, (92, 94, 100, 101), 114 (99), 519
(62, 70, 71)
Johnson F. 71 (57, 58), 245 (283—285), 249
(283, 285), 250 (283—285)
Johnson О. Н. 9 (1), 29 (42, 44), 32 (42, 60),
47 (1, 4), 81 (14—16), 89 (11, 12), 90 (12),
91, 92 (41), 95 (12), 96 (21, 34), 99 (34), 124,
125 (15, 16), 126 (25), 127 (27), 130 (15), 134
(15, 25, 27), 137 (50), 147 (18), 233 (255), 260
(370), 332 (39, 42), 350, 364 (164), 385 (55,
58),'423 (412, 414), 453(29)
Johnson W. 256 (359, 360), 259 (359), 260 (359,
360)
Johnson W. A. 519 (11)
Johnson W. K. 356 (210), 405 (198)
Johnston J. D. 588 (175)
Jones E. R. H. 41 (106)
Jones H. L. 560, 561 (38), 579 (35)
Jones K. 175 (211), 272 (2), 314 (74), 315 (2,
75), 327 (16, 17), 328 (16, 18), 397 (32). 409
(291, 292, 296), 410 (291), 411 (32, 313), 415
(32), 417 (32, 313), 418 (292), 433 (45)
Jones L. V. 47 (4), 91, 92 (41),' 93 (49), 127, 134
(27), 581 (64)
Jones R. G. 245 (289), 550, 552 (10), 598 (47)
Jones W. H. 588 (152)
Jones W. J. 208, 209, 217 (44), 221 (44, 163),
222 (44, 163), 226, 233—235 (163), 330 (19),
404 (185), 550 (13), 581 (60), 605 (6)
Joshi К. К. 432 (24)
Joyner F. B. 520 (117, 126), 535(100), 536 (124)
Juenge E. С 246 (294), 536 (139), 550 (17),
551 (20), 553 (58), 560 (34), 576—579, 582 (8),
582 (65), 617 (5), 624 (20)
Juliard A. L. 633 (33)
Jun-Shen Yu A. 397'(71)
Jutzi P. 316 (84)
Kadomtzeff I. 165 (152, 153), 535 (68)
Kaesz H. D. 165 (45, 64, 82, 105), 214 (92), 226
(92, 207), 235 (92, 207), 346 (149), 347 (149,
153), 348 (149), 350 (153), 352 (149, 153, 173),
356 (153, 173), 364 (149, 153), 366 (153, 264),
443 (61, 62), 444 (62), 457 (24, 26), 461 (24),
501 (58), 527 (54), 535 (29), 589 (191, 192}
Kahlen N. 82 (35), 99 (47), 109 (104), 339 (86),
406,423(214)
Kahler E. J. 527 (52)
Kahler W. H. 497 (24)
KahnO. 411 (318), 507 (89), 608 (54), 630 (41)
Kamionska J. 519 (61)
Karantassis T. 181, 182, 184 (4, 5)
Karbstein B. 219 (134), 238 (267), 245 (288),
247, 249 (267, 288, 303), 251 (134, 267, 303),
252 (134), 255, 256 (303, 338), 364 (242)
Karnatak R. 16 (79)
Karnojitzky V. 101 (60), 175 (213), 414 (383)
Kasai N. 433 (54), 435 (91)
Kato E. 519 (85)
Kawakami K. 165, (17, 61), 414 (361), 429 (406)
Kawasaki Y. 165 (29, 57, 61), 432 (17), 433 (17,
51), 434 (17), 435 (91), 535 (26)
Kawase K. 512 (9)
Keane F. M. 579 (40,41), 602 (15, 17), 605 (8, 10)
Keedish N. 513 (36)
KelenG. P. van 15 (64), 165 (110, 114, 118)
Kelker H. 165 (62), 384 (44)
Kelso R. G. 397 (70)
Kelso R. 0.519(38)
Kenton J, R. 520(130)
Kenyon A. S. 520 (97), 536 (131)
Kettle S. F. A. 461 (45), 457, 465 (19), 497 (25)
Kharasch M. S. 580 (46), 584 (88), 586 (124)
Kiesel R. J. 468 (79, 80), 471 (79), 474 (90a),
475 (94)
Kilbourn В. Т. 165 (144)
Kimpball G. E. 432 (34)
King R. B. 370 (284)
King W. B. 527 (15)
Kingston D. 554, 555 (73), 559 (17), 586 (107),
617, 618 (1), 624, 625 (19), 626, 627 (21)
Kinsinger J. 412 (333), 516 (67)
Kinugawa Z. 185 (41), 334, 358 (56), 382, 385,
388 (18), 407, 408 (232), 526 (5)
Kipping F. S. 212 (82), 217. 218 (123), 223 (123,
177), 228, 249 (177), 339 (96, 100), 350 (96),
381, 385 (5), 389 (74), 396, 398 (6), 400 (117),
405 (6)
Kirby J. E. 560 (32)
Kircher J. F. 182, 183 (27)
Kirner W. R. 527 (74)
Kiser R. W. 535 (79)
Klang P. 60 (2)
Klein A. 547 (58), 628 (28), 632 (10)
Kleiner F. G. 327 (14), 399 (105)
Kleinert P. 608 (52), 629 (35)
Klippel J. 601 (4), 602(11), 605(2)
KloseG. 165(116). 535(31)
Kluiber R. W. 520 (145)
Knaub E. W. 162 (10)
Knoepke O.H.399, 404 (96), 414 (366), 520 (96)
Kobetz P. 168 (183), 203 (61), 521 (225), 541
(35a), 549, 554 (2), 556 (79), 607 (45)
Авторский указатель
653
KobsH. D. 545 (21)
Kochler I. S. 15 (6), 26 (3), 165 (22)
Koenig P. E. 516 (72, 73), 520 (199)
Koerner E. 454 (62)
Kohama J. W. 527(14)
Kohzuma T. 633 (31)
Kolbezen M. J. 581. 582, 586 (62), 605, 608 (11)
Koll M. 175 (218)
Komorniezyk K. 51 (32), 255 (337), 364 (243),
562 (64, 66)
Komura M. 414 (361), 432 (17), 433 (17, 51),
434—439 (17)
Konig K. 165 (13), 194 (4, 7, 8, 21), 195—197
(4, 21) 199 (4), 272, 273 (2a), 343 (130),
377 (14), 378 (15), 439 (32), 440 (36), 457
(22), 461 (41), 477 (41, 98, 100), 479 (22, 98,
100), 480 (41, 98, 100), 498, 504 (38, 77)
KooijmanP. L. 535 (101)
Koopmans M. J. 397 (63, 69), 521 (217)
Kopf H. 359, 362, 363 (228), 415 (375), 415,
423 (376), 423 (414, 415), 505 (82)
Kopf M. 358 (234, 235), 362 (234, 235), 363 (235)
Korsching H. 206, 207 (9)
Koster R. 435(85)
Koster-Pelugmacher A. 108 (93)
Kozuma S. 182, 184 (20a), 185 (39a), 378 (15a),
396 (3), 397 (3, 31), 409 (3, 31), 410 (3),
415 (31), 442 (43a, 436), 443 (53a)
Kraft D. 48, 50 (8) •
Kraihanzel C. 255 (334)
Kramer K- 74 (2), 75 (3, 4), 322 (22), 447 (1)
Kramer K. A. W. 319, 323 (9), 457, 458 (28)
Kraus С. А. 26 (15), 30 (52), 53 (46), 78 (1),
81 (3, 11, 12), 82, 84 (3), 89 (14), 90 (14, 20,
29), 91 (44), 95(2,9, 11), 101 (11), 106 (9,
11, 86), 122 (1—4), 125 (2), 134 (1), 145 (4, 5,
7), 146 (7), 147 (4, 17), 150 (4), 151 (5, 7, 34),
152 (5, 35, 38. 39), 153 (7, 34), 154 (4, 35, 38,
39), 155 (4), 162 (2, 3), 186 (58, 59), 208 (40),
210 (71), 338 (80), 378 (20), 408 (279), 432
(22), 435 (79—81), 438 (3, 6, 15), 439 (3, 15,
16, 29, 30), 441 (16), 457 (1), 497 (12—14, 23,
24), 498 (14), 499 (40), 500 (42), 501 (54, 55,
56), 502 (14,55,56, 62), 503 (14). 504 (13, 42),
505 (13, 40), 541 (14)
Krause E. 8 (3), 30 (52), 182, 184 (16), 194 (10,
11), 195 (10,24, 26), 196 (10, 24, 26), 197 (10),
198 (10, 26), 206 (8), 208 (34), 210 (8, 64, 67,
74), 212, 214 (67), 215 (101), 216 (111, 116),
217 (111, 116, 120), 218 (111), 219 (135),
220 (142), 221 (8, 116, 156), 222 (67), 223
(74, 111, 120, 156), 224 (189), 228 (74), 229
(111), 235 (8), 240 (189, 269), 249 (269), 337
(65,66), 338 (83), 343 (125—127), 345 (141),
355 (188), 356 (196), 376 (1), 377 (12), 389
(72), 396 (5, 7, 9—11), 400 (108), 405 (5, 7,
108, 196), 416 (7), 438 (9, 10), 439 (9, 10, 26),
440 (9), 441 (9, 10), 442 (10), 503 (69, 70, 72),
526 (11), 528 (80), 549 (4, 6, 7), 550 (7, 12,
16), 551 (4, 22, 23, 27), 552 (7, 27, 35, 36, 38),
554 (7, 36), 558 (1), 559 (11, 12, 15), 560 (15,
25, 27—30, 54, 59), 561 (29, 59), 575 (2, 4),
576 (2, 4, 9), 577 П0, 16, 17), 578 (10, 16),
579, 584 (17), 586 (104), 588 (142), 596, 597
(11,12),601 (1),602 (1,13),605 (3),606(3,39),
612 (9—11), 613 (10, 11,23), 618 (10), 631 (6)
Krayse К. Н. 520 (169)
Krebs A. W. 583, 584 (76), 605, 606 (32), 612,
614, 615(2)
Kreiter C. G. 165 (99, 106, 108), 243, 251, 252
(279), 336 (62), 409, 417 (293), 461 (46)
Kremky E. 519 (61)
Krespan C. G. 281 (28)
Kreuzbichler L. 502 (61), 520 (112)
Kriegsmann H. 165 (59),382 (26), 406, 413 (219),
433 (52), 435 (76, 92, 97), 468—470 (77),
527 (58)
Krohn I. T. 541 (29, 30, 62), 615 (36)
Kroller E. 633 (18, 20)
Kross R. D. 15 (8), 29 (43)
Kruckeberg F. 535 (33)
Kruse F. H. 535 (23)
Kubo H. 396, 400 (23)
Kuchen W. 411 (314)
Kuhlein K. 139 (60, 61), 154 (48, 49), 570 (4),
620—622 (3)
Kuivila H. G. 175 (203), 194 (6), 196 (34), 223
(179), 238, 245, 249. 250 (265), 277 (24), 390
(80), 423 (411), 438 (11), 439 (11, 22), 440—
442, 444 (11), 457 (20), 459 (35), 462 (50, 54—
56, 58), 463 (50, 54, 55), 464 (35), 465 (67),
466, 467 (20, 54, 55, 70), 468 (72, 78), 470
(58, 72, 84), 471 (58, 72, 78, 84), 472 (56, 58,
72, 85, 87), 473 (58, 85), 474 (58, 85, 87,
90—92), 475 (58, 90), 476 (90), 477 (35, 102),
480 (35), 487 (85), 490 (54*, 55)
Kula M. R. 165 (106, 108, 109, 111—113), 223,
236, 241, 246 (175), 336 (62), 408 (253), 409
(290, 293), 417 (253, 290, 293, 368, 387), 418
(391), 460 (39), 461 (39, 43, 46)
Kulmiz P. 397, 406 (34)
Kulp J. L. 633(84)
Kupchik E. J. 251 (313), 344 (131), 378 (29),
410, 411 (306), 421, 422 (405), 464 (61), 468
(79, 80), 471 (79), 474 (90), 475 (94)
Kusck R. 251 (320), 519 (81)
Kushlefsky B. G. 383 (35), 384 (45, 46)
Kuuth С J. 407 (237, 240)
Kwon J. T. 165 (103), 274 (16, 17, 20), 277 (16,
17, 20, 25), 279 (17, 25), 280 (16), 281 (16, 20),
457, 458, 463, 464 (16)
Labarre J. F. 16 (84), 28 (34), 39, 40 (94)
LachowiczS. K. 536(117)
Lacina J. Z. 535 (58)
Ladenburg A. 186 (51, 56), 253 (327, 328), 339
(105), 408 (262, 263), 439 (25)', 442 (52)
Lagowski J. J. 443 (63)
Laliberte B. R. 281 (29), 401, 402 (140), 403
(155), 417 (140)
Lambourne H. 393 (95, 96), 404 (187)
Landolt H. 432 (30)
Lang R. 407 (239), 516 (69), 527 (29)
Lange G. 194, 195 (3), 210 (65), 457, 484 (5),
498, 499 (33)
Langer H. G. 333, 334 (46), 336 (59a)
Lanigan T. 344 (131), 378 (29), 410,411 (306),
464 (61)
La Paglice S. R. 15 (70)
Lappert M. F. 175 (211), 272 (2), 314 (74), 315
(2, 74a, 75), 327 (16, 17), 328 (16, 18), 397
(32), 409 (291, 292, 296), 410 (291), 411 (32,
313), 415 (32), 417 (32, 313), 418 (292), 433
(45)
Laurie V. W. 15 (13)
Lauterbur P. С 165 (94)
Lautsch W. 588 (145—148)
Lautsch W. F. 535 (56)
654
Авторский указатель
Lauw-Zecha A. 633 (55)
Lavigne A. A. 104 (67)
Law H. 545 (4—7)
Law К. К. 439 (24), 503 (71)
Lawrence A. 256 (345)
Lawson J. R. 165 (24)
Leavitt F. С, 49 (24), 245 (283, 284, 285), 249
(283, 285), 250 (283,284, 285), 291 (51), 513
(40), 515(53)
Lederer К. 561 (53)
Leebrick J. R. 40 (97), 231 (238), 513 (36, 37),
521 (229)
Leeper R. W. 27 (19), 48 (9), 81, 82 (6), 183,
190 (31), 321 (17), 537 (161), 551, 552 (31),
552, 553 (41), 560—563 (41), 568 (1), 579 (36),
580 (47), 580 (67), 605, 608 (7), 608 (67), 614
(34), 618 (12), 624 (11), 624, 627 (12)
Leermakers J. A. 588 (155)
Lehman D. S. 245, 250 (284)
Lehmkuhl H. 168 (184, 186), 545 (18, 21), 545,
546 (25—27), 546 (30, 31, 39)
Lehn W. L. 108 (92), 145, 146 (10), 223 (185),
406 (208), 410 (297), 439 (21a). 441 (39), 465
(66), 496, 500 (2), 560 (40). 623, 624 (5)
LehnardtR.190(89),216,2l7(118),378(31),392
(87), 400 (113), 404, 413 (182), 526 (6)
Leicester J. 165 (2, 6), 535 (54, 55)
Leighton P. A. 589 (178)
Leistner W. E. 399 (95, 96, 98, 103, 104), 404
(95, 96, 98, 103, 104, 167, 168, 169, 172),
405 (95, 98, 103, 104, 167), 409 (283), 414 (98,
169), 520 (96)
Lemiszka T. 520 (192)
Lemmon D. H. 15 (31)
Lengel J. H. 53 (44)
Lepretre B. 165 (63)
Le Quan M. 227 (218—221), 228 (218, 219),
232 (218, 219), 238 (221), 249 (221), 251—256
(218). 364 (244), 453 (30), 500 (44, 45), 528
(79), 560—562 (39)
Lerwill B. R. 409, 422 (294), 432 (1)
Lesbre M. 16 (85, 101), 23 (3), 27 (23, 25), 28
(33), 30 (57), 35 (67, 68), 38 (68, 83, 86), 39
(33), 51 (316, 38), 64 (26), 65, 67 (29), 68 (29,
41), 69 (41, 45), 70 (41, 46, 47), 75 (5), 81
(23), 82 (23, 25, 26, 27, 34), 83 (25), 84 (26),
96, 97,99,105, 110 (32), 107 (91), 114(1, 4, 6),
115 (10, 10a), 116 (10), 118 (36), 120 (10a),
125 (20—22), 126 (21, 22), 128 (33), 131 (38,
39, 41), 132(39, 41), 133 (38), 134 (33),
135 (33, 38), 136 (33), 165 (58, 151), 189 (86),
190 (94), 209 (58, 60), 215 (100), 216 (100,
115), 217, 218 (100), 221, 223 (167), 234
(261), 237 (167), 242, 252 (115), 319, 323 (8),
346 (145), 350 (167, 168), 351, 352 (167), 356
(199). 363 (145), 366, 368 (265), 447 (6), 457
(13, 23), 528 (75), 542 (75—78), 552 (37),
603 (30), 633 (15)
Letts E. A. 186 (53), 269 (4)
Leusink A. J. 48, 52 (18a), 240 (270), 275,
276 (22a, 226), 282 (22a, 30a), 286 (38), 290
(22a, 47, 48, 50), 291 (22a, 47,48, 49a, 50),
292 (22a, 47, 49a), 293 (22a), 294 (22a, 48,
496), 295, 296 (22a, 496), 297, 299 (22a). 300
(22a, 496), 304 (22a), 305 (50), 307 (57, 58),
309 (58), 327 (15), 342 (120), 397 (58), 458 (31),
514 (49, 50), 515 (49), 570, 571 (5)
Levins P. L. 465 (67)
Lewinsohn M. 24 (6), 29 (50), 81, 84 (20), 92 (48),
189 (86)
Lewis A. G. 358 (227)
Lewis F. B. 188 (80), 555 (77)
Lewis G. L. 535 (45)
Lewis R. N. 8 (7)
Lewis W. R. 521 (223)
Leysen R. 633 (29)
Leyshon K. 48, 52 (18), 83 (49)
Licari J. J. 520 (163)
Lichtenwalter M. 586 (103)
Lide D. R. 165 (90), 457, 466 (10)
Lieber E. 579 (40, 41), 602 (15, 17), 605 (8, 10)
Lind H. 490, 491 (121)
Linsk J. 547 (46—48)
Lippincott E. R. 15 (7, 48), 26 (4), 165 (23, 66),
207 (18), 210 (72), 535 (8)
Loeb W. E. 520 (125)
LoewG. 520 (171, 174, 179, 180)
Lohmann D. H. 165 (46)
Loleibit H. 588 (150)
LongL. H. 365 (251)
LongiP. 512(1), 536 (143)
Loranc D. 633 (25)
Lorberth J. 165 (108, 109, 112, 160), 315 (74a),
336 (62), 409 (289, 290, 293), 416 (289), 417
(290, 293, 368), 418, 460, 461 (39)
Lorenz D. H. 216 (105), 219 (132), 274, 305, 309,
310 (19), 332, (38), 453 (48), 459 (34), 468
(34, 76), 469 (76), 477, 480 (34), 558 (3)
Lorriman F. R. 541 (20, 21)
Lotz R. 520 (157)
Lovelock J. E. 633 (26, 38, 40)
Lowig С 186 (46), 400 (116), 438, 441 (8), 541
(3), 601 (5), 605 (1)
LowranceB. R. 553 (58)
Lubke K. 363 (238), 396 (19), 499, 502 (39),
624, 628 (17)
Luftensteiner H. 216, 217 (118), 526 (6)
LuggR.R.541 (59)
Luijten J. G. A. 57 (65), 165 (178), 175 (195, 198,
201, 211a), 182, 184 (17), 185 (42), 187, 188
(17), 194 (15), 207 (13, 19, 31), 208, (19, 31,
49), 209, (19, 31, 59), 210 (19, 59), 212 (81),
216 (49), 217, 219 (125), 221 (148, 167), 222
(148), 223 (13, 167), 224 (13, 148), 228 (232),
233 (19, 148), 237 (167), 239 (19), 251 (19, 59,
316), 252 (59), 253 (316), 256 (49), 257, 259
(49, 362), 273 (3, 6), 278—282 (6), 283 (3, 6),
284, 285 (6), 315 (76), 321 (16), 322 (19),
330 (9, 16—18), 331 (17, 18), 332 (18, 41).
333 (44), 335 (17), 338 (84), 339 (110), 341
(18, 117), 342 (18), 343 (123), 356 (199,200),
363 (18), 382 (23, 25), 383 (23, 28, 30), 385
(47), 388, 389 (71), 392 (85), 397 (40, 44, 50,
54), 398 (40, 44, 54, 73, 76), 399 (40, 106,
107), 401 (131), 406 (40), 407 (50, 243), 410
(40, 106, 107, 307), 414 (366), 432 (4, 5),433
(4, 44, 58), 435 (4, 5), 457 (2, 12), 458 (12),
461 (42), 475 (93), 477 (97), 519 (1), 521 (209—
212, 219), 527 (16), 528 (82, 85), 535 (13), 537
(170), 575 (1)
Luz W. D. 520 (203)
Macdonald A. M. G. 633 (14)
Mack G. P. 228 (230), 256 (340), 387 (64), 399
(102), 402 (149, 154), 404 (102, 171, 179,
180), 408 (266—269), 409 (276, 277), 410
Авторский указатель
655-
(304, 305), 418 (390), 513 (35), 519 (29, 32,
34—37, 48, 53, 59, 72, 73), 520 (95, 181)
МаскауК. М. 15 (58)
Maddox M. L. 165 (64), 457 (26)
Maeda Y. 401 (136)
Mageli О. L. 408 (249)
Magnuson J. A. 162 (10)
Magon L. 405 (195)
Magos L. 521 (206)
MagsonM. S. 520 (175)
Maier L. 357 (220), 551 (18), 558 (2), 586 (126)
Maine J. С 15 (69), 165 (7, 63, 104), 206 (3—5),
207 (3), 208 (3—5), 210 (3, 5), 432 (39)
Malatesta L. 165 (162), 412 (344, 345), 535 (50),
536 (116\ 586 (117)
Malinowski E. R. 165 (117), 553 (52)
Malkolm С. Н. 51 (31a)
Maly J. 165 (141, 142), 535 (43)
Malz S. 527 (68)
Mangham J. R. 203 (63), 256, 260 (357)
Manuel G. 23 (3), 38 (83), 72 (59)
Manuel T. A. 49 (24), 245 (283, 284), 249 (283),
250 (283, 284) '
Marchand A. 165 (30. 56, 72)
Marinelli L. P. 397 (47, 48, 53), 513 (22, 30,'31,
34)
Mark H. F. 175 (217), 392, 393 (93)
Marlett E. M. 16 (107), 537 (167), 545, 546 (10)
MarreS. 51 (316), 115, 120 (10a)
Marrot J. 15 (69), 165 (7, 63)
Marrs O. L. 151 (31), 496, 499, 500 (8), 502 (8,
59), 503 (8), 623 (4), 624 (4, 13), 626, 628 (13)
Marsman J. W. 290'(47, 48, 50), 291 (47, 48,
49a), 292 (47, 49a), 294 (48), 305 (50), 327
(15), 342 (120), 397 (58)
Martens P. H. 527 (36)
Martin D. F. 249 (307), 409, 414 (278, 364),
422, (407), 434 (67)
Marvel C. S. 536 (125)
Massey A. G. 48 (Щ\ 87 (55), 165 (102), 249
(306), 331, 336 (28)
Massol M. 38 (86), 68—70 (41), 70 (46, 47), 114,
115 (8), 118 (36)
Masson J. C. 251, 252, 254, 258 (314), 560—
562 (39)
Mathis-Noel R. 125, 126 (22), 165 (58), 457 (23)
Matsuda H. 181 (6, 7, 9—12), 182, 184 (6, 7,
9—12, 18—20), 186 (6, 7, 36), 226 (353), 400
(110), 401 (134), 519 (47), 527 (49)
Matsuda S. 181 (6, 7, 8—12), 182 (6, 7, 8—12,
18—20), 183 (32), 184 (6, 7, 8—12, 18—20,
36), 186(6, 7). 226(353), 400(110), 401
(134), 519 (47), 527 (49)
Matsuda T. 512 (9)
Mathur S. 98 (43a, 436), 103 (436, 65), 111
(436, 106—108)
Matternas L. U. 245, 250 (284), 291 (51), 513
(40), 515 (53)
Mattison E. L. 541 (50), 596 (7)
Matwiyoff N. A. 432 (27), 434 (68), 535 (37,
83)
Mav L. 165 (65)
Maver К. К. 165 (113), 223, 236, 241, 246 (175)
Mayerle E. A. 547 (46)
Mayo F. R. 580(46)
Mazerolles P. 16 (84—86), 23 (3), 27 (23—25,
28), 28 (33—35), 30 (35), 35 (67, 68, 70, 71),
36 (35, 78), 38 (68, 83), 39 (33, 94), 40
(78, 94), 41 (105), 51 (316. 37, 38), 72 (59),
81 (22—24), 82 (23, 25, 26, 28, 34), 83 (25),
84 (26, 53), 91 (42), 96, 99 (20), 114 (1—3, 6),
115(2, 10, 10a), 116(10), 117(28, 29), 118
(37), 119 (29, 37), 120 (2, 10a, 41), 124 (13),
125, 126, 130. 134, 136 (24), 160(31)
Mazzanti G. 512 (1)
McCarty S. W, 396 (28), 605, 606 (15)
McCleafy R. F. 581 (59)
McCombie H. 580, 581(57), 598(32), 605,.
607, 609 (13)
McConnell R. L. 519 (27)
McCormic C. G. 165 (93)
McCullough J. P. 535 (58)
McDermott J, P. 409, 412 (287)
McDiarmid A. G. 412 (340), 516 (77)
McDyer W. 596 (6)
McGrady M. M. 165 (74), 404, 409 (160)
McKav T. W. 546 (33—35), 553 (57)
McKefver С. Н. 536 (128)
McKinnev D. S. 15 (6),'26 (3), 165(22)
McLaughlin R. 520 (176)
McNeill E. H. 165(167), 207—209, 221, 233"
(28), 457 (15), 526 (10)
McQuillan С. Т. 432 (31, 32)
Mead B. 545 (14)
Meals R. N. 209 (62), 552 (40)
MecklingN. G. 520 (123, 148)
Meen R. H. 48 (14), 69 (43), 242, 244 (273)
Mehner H. 527 (32—34)
Mehrata R. C. 98 (43a, 436, 43b), 103 (436,
65), 111(436, 106—108), 408(257, 258),
411 (257), 418 (389a), 434 (65)
Meinert R. N. 588 (153)
Meluikoff A. 433 (46, 47)
Melstrom D. S. 551, 552(32), 559(20), 560^
(20, 42), 562 (20, 42), 563 (20, 42)
Melvin H. W. 331 (31), 484 (112), 520 (123)
Melvin W. S. 541 (33)
Menapace L. W. 468 (72, 78), 470 (72, 84),
471 (72, 78, 84), 472 (72)
Mendel H. 165 (146)
Mendelsohn J. C. 149(27), 165(30, 56, 72),
311 (64a)
Menge K. 60 (2)
Mercier P. 15 (48), 165, (66), 210 (72)
Merckling H. G. 520(115)
Merker R. L. 224, 239 (172a, 187, 188), 240'
(172a), 449 (13), 520 (197, 204)
Merten R. 520 (171, 174, 179, 180)
Merz E. 165 (182)
Mesee H. J. 632 (10)
Metlesics W. 78 (6), 81 (17), 91, 92 (45)
Metras F. 149 (27)
Meyer F. J. 194, 195 (3) 457, 484 (5), 498,.
499 (33)
Meyer G. 190 (87)
Meyer K. 48 (12), 243 (278)
Meyer M. 553 (47)
Meynier D. 234 (261)
Mezz A. 519 (75)
Midgley T. 535 (91), 547 (57), 601 (7), 614
(32), 633 (16)
Miles D. 27 (22), 47 (5)
Miller D. S. 634 (84)
Miller F. A. 15 (31)
Miller F. D. 520 (129)
Miller J. J. 232 (252a), 407 (234)
Miller L. A. 541 (44)
656
Авторский указатель
Miller L. S. 384 (42)
Miller V. L. 397, 423 (60)
Milligan J. G. 152, 154(38)
Milltown Т. Т. L. 520 (128)
Miltenberger K. 16 (92, 93), 27 (17), 29 (45),
535(47, 51, 52)
Milne J. N. 408 (272, 274), 519 (31)
Minckler L. S. 520 (192, 193)
Moedritzer K. 20 (13a), 160 (30), 165 (101)
Mitchell T. N. 328 (19), 398 (77д)
Miyamsi T. 443 (53a)
Mohammed A. 165 (69)
Moller S. 554 (63), 584 (79)
Molt K. 251, 253(318)'
Molt K. R. 332, 333 (40)
Mondry V. С 527 (47)
Monrol K. P. 541 (13)
Montecatini 536 (141)
Montermoso J. C. 397(47, 48, 53), 401, 402
(140), 403 (155), 417 (140), 513 (22, 30, 31,
34)
Moon S. 573 (6)
Mooney E. F. 165 (55, 70), 356 (208, 209), 408
(255), 527(56)
Moore D. W. 165 (100)
Moore F. W. 245 (289), 598 (46, 47)
Moore G. J. 145, 146 (10), 223 (185), 496, 500
(2), 560 (40), 623, 624 (5)
Moore L. 27(22), 47 (5)
Morgan C. R. 536 (147), 559, 560 (22)
Morgan G. L. 165 (79), 477 (103), 503, 504 (65)
Morgan G. T. 29, 32 (39), 90, 91, 93 (28), 99
(44), 145(3)
Morel li D. 411 (328), 507, 508 (95)
Morgunoff N. 339 (102)
Morimoto Y. 354 (186a)
Morris H. 217 (121), 535 (66)
Morris W. С 165 (181)
Mortensen R. A. 589 (178)
Mortimer С. Т. 165 (166)
Mosle H. G. 224, 245, 248 (195)
Muetterties E. L. 406 (223)
Muffi A. S. 433 (42)
Miiller E. 284, 305'(34), 535 (30, 40), 612
(18)
Miiller J. H. 29 (40), 53 (45), 90 (30), 95, 99
(14)
Murphy C. J. 453 (42)
Murphy С. М. 520 (188)
Nabika K. 443 (53a)
Nagano T. 519(85)'
Nadj M. M. 432 (7)
Nagv J. 208, 211, 216 (47), 528 (84)
Nakamura Y. 413, 414 (354)
Nangniot P. 527 (36)
Narasimhan P. T. 15 (54), 535 (34)
Nash G. A. 165 (173)
Nasielski J. 175 (224), 207 (26), 214 (96), 221
(160), 233(96), 246(160), 337(70—76),
338 (72—76), 347 (72, 73, 156), 358 (224),
435 (90), 440 (37), 576 (5), 577 (20)
Nasutavicus W. A. 71 (58)
Natta G. 512 (1)
Neal A. M. 408 (259), 499 (40), 501 (54, 56), 505
(40)
Neal H. R. 378. 379 (34), 598 (37)
Neals R. N. 346 (147)
Nebe E. 588 (141), 612, 615 (17)
Nebergall W. H. 29 (44), 32 (60), 47 (1), 81
(15), 89, 90(12), 99(34), 124, 125(15,16),
130, 134 (15)
Negromanti A. 536 (116)
Neher С. М. 541 (49, 58)
Nelson J. F. 584 (84)
Nelson W. H. 409 (278), 414 (278, 364), 434(67)
Neubert G. 527(67)
Neumann W, P. 139 (59—61), 140 (66, 69),
154 (47—49, 54), 165 (13, 91), 175 (199, 202),
194 (4, 7, 8, 12, 17, 21, 22), 195 (4, 12, 21,
22). 196, 197(4, 21), 199(4), 209(55), 256
(352), 257, 258 (352, 364, 365), 260 (369),
272 (2a), 273 (2a, 5, 9, 12, 13), 274 (5, 12—
15), 275 (5, 9, 14), 276 (14, 23), 277 (5, 12—
14, 23, 26), 278 (12, 13), 279 (12), 281 (12, 13),
283 (12), 284 (12, 34), 285 (12, 35), 287 (14),
288 (5), 290 (5, 14, 15), 301 (9, 14, 15, 52),
302 (14), 303 (9, 12, 14, 52), 304 (9, 14, 52),
305 (9, 14, 15, 34, 35, 55), 306, 307 (55),
308, 310, (14, 55), 327 (14), 330, 332, 333,
335(8), 341(118), 343(130), 376(3), 377
(14), 378 (15), 399 (105), 415, 416 (378),
435 (85), 439 (32, 33), 440 (33, 36), 457 (22),
458 (30), 459 (36, 40), 460 (37, 40), 461 (37,
41, 47, 48), 462 (36, 47—49), 463 (30, 37, 48),
464(37), 477(37,41,98—101), 478(101),
479 (22, 98—100), 480 (41, 47, 49, 98—100,
104—106), 481 (47, 49, 99, 104—106), 482
(99. 106), 483 (49, 104), 487 (40, 47, 118,
119), 490, 491 (47, 121), 498 (38), 504 (38,
76, 77), 513 (41—44), 514 (41, 43, 444 527
(17, 57, 62), 570 (4). 620—622 (3)
Nevett B. A. 388 (69)
Neville R. G. 217, 228, 241, 243 (122)
Newberg R. G. 520 (129)
Newland G. С 519 (27)
Newlands M. J. 42 (110), 238, 241 (266), 350,
364 (165)
Newnan M. D. 319 (4)
Neymann E. 301—304 (52)
Nichols A. F. 541 (33)
Nicholson D. A. 150 (29a)
Nickerson S. P. 535 (93)
Nickless G. 26(11), 163(14, 19), 206(1), 207
(27), 274 (22), 278 (22, 27), 290 (27), 292 (22),
338 (81), 457, 468, 476 (29), 527 (48, 50)
633 (77)
Niebergall H. R. 515 (55), 516 (75, 76)
Niederauer H. P. 396 (30)
Nielsen J. R. 535 (28)
Niemoller H. 245, 247, 249 (288)
Nienburg H. J. 520(113), 536(122)
Niermann H. 273, 274, 277—279, 281, 283—
285, 300 (12), 458 (30), 459—461 (37), 463
(30, 37), 464, 477(37), 487(119), 513,
514 (43, 44)
Nino M. E. 633 (21)
Nishino M. 406, 419 (209), 433 (43)
Nitzsche S. 330 (25), 382 (13, 14), 519 (30)
Nixon L. A. 396, 411 (25, 26), 415 (25), 423 (25,
26), 433 (48, 49), 497, 505 (22), 521 (221),
527 (20)
Noltes J. G. 15 (11, 23), 29 (46), 29, 39 (47),
40 (98, 100), 48, 52 (18a), 65 (28), 116 (18—
20), 140(70), 159 (10, 17), 165(40)/ 175
(195, 198, 205, 214), 194 (15)
Notles J. G. 15 (11, 23), 29 (46, 47), 39 (47),
40 (98, 100), 48, 52 (18a), 65 (28), 116 (18—
Авторский указатель
657
20), 140 (70), 159 (10, 17), 165 (40), 175
(195, 198, 205, 214), 194 (15), 207 (13), 208
(49), 212 (81), 216 (49), 223, 224 (13), 226
(215), 228(232), 231 (243, 246), 236 (263),
240(270), 250(311), 256, 257, 259 (49),
273 (2в, 3, 4, 6. 7), 274 (4), 275, 276 (226),
277 (4), 278, 279, 281 (4, 6), 282 (4, 6, 30а),
283 (3, 6), 284, 285 (6), 286 (38, 39), 287 (4,
39, 40), 288 (4), 290 (4, 50), 305 (50), 306
(56), 307 (56—60), 308 (56), 309 (56, 58,
60, 61), 310 (59, 60), 322 (19), 327 (15),
330, 331, 335 (17). 341 (116, 117), 343 (116,
123), 356 (200), 363 (237, 239), 366 (237),
383 (29, 30), 384 (38), 397 (50, 58), 398 (73),
405 (197), 407 (50), 408, 416 (256), 447, 448
(7, 8), 449 (8), 457 (2, 12, 18), 458 (3, 12),
461 (42), 462 (57), 468, 472, 474 (71), 475
(93), 477 (71, 97), 480, 482(107, 108), 483
(108, 111а, 1116), 484 (111а, 1116,113а),
488 (119а), 500 (43), 501 (51), 512 (13),
513 (46, 47), 514 (13, 46, 47, 49—51), 515
(19, 51, 54), 521 (209, 210), 527(16), 528
(82, 83), 535 (7, И), 536 (144, 156), 559 (21),
560 (48), 561 (48, 50), 617 (3, 4)
Norman R. О. С. 573 (8)
Normani H. 39 (89), 213 (89)
Nosek J. 185 (43), 189 (43, 85), 397 (45), 542
(73)
Noth H. 165 (160), 502 (60)
Nozaki H. 186 (45), 217, 252, 253 (124), 347
(155)
Nutting H. S. 145, 151, 152 (5)
Oakes V. 181, 182, 184(13)
O'Brien R. J. 165 (73, 145), 406 (205, 206),
411 (309, 317), 432, 433(23), 435 (84, 93,
94)
O'Connor M. N. 287, 290 (42)
O'Donnell G. 563(76), 618(11)
Oeser H. 527 (35)
Oesper P. F. 535 (45)
Ogrins I. 388 (69)
Ohara M. 397 (37, 38), 399 (38), 413, 414 (354),
433 (62, 63), 461 (44)
Okada T. 519 (85), 633 (31)
Okawara R. 90, 91 (19), 165 (17, 20, 29, 57, 68),
381 (1), 385 (52, 54), 386, 387 (54), 392 (84),
396 (2, 18), 397 (2, 33, 37, 38), 399 (38),
400 (2, 18), 401 (129, 136), 402 (129, 147),
406 (209), 407 (129), 413 (351, 354, 355), 414
(354, 360, 361), 419 (209), 422 (406), 432 (17),
433(17, 43, 51, 53, 54, 56, 57, 62—64),
434 (17, 70, 71), 435 (95, 96), 461 (44),
526 (8), 527 (3), 535 (14, 17, 26)
Okkaba X. 165 (43)
Okuyama T. 257, 259 (361)
Olson D. A. H. 633 (67)
Olson D. H. 194 (18, 19)
Onyszchuk M. 15 (24), 90 (18), 99 (46)
O'Rear J. G. 520 (188)
Ormezzano L. 412 (345)
Orndorff W. K. 27 (16, 18), 29 (16), 36 (18),
50 (31), 54 (51), 81 (10), 82 (32), 93 (52), 95
(10, 13), 96 (23), 252 (323)
Osborn S. W. 397 (71)
Osterman T. 633 (83a)
Osthoff R. С 16 (91), 19 (5)
Oulette R. J. 573 (4, 5)
Padgitt F. L. 541 (64)
Paget H. P. 206(10), 207(10, 29), 208(29),
251, 260(10)
Paglia la S. R 165 (15), 535 (1)
Paliobagis M. A. 407, 415 (242), 520 (93, 94)
Paltin E. 251 (317)
Panaitescu M. 251 (317)
Pande К. С. 34 (64—66), 48 (17, 18), 50 (17),
52 (17, 18, 39), 83 (40, 42, 49), 221 (159),
562 (67, 69)
Paneth F. 588 (144—151)
Pang M. 465, 476, 477 (64)
Papetti S. 207, 221, 223, 224(20), 231 (32),
233, 236, 240 (20), 412 (330), 501 (57), 527
(30)
Pariser R. 512 (8)
Parisi G. L. 165 (132)
Parker E. 228 (230), 256 (340), 402 (149, 154),
404 (179, 180), 408 (266—268), 410 (304,
305), 416 (267), 418 (390), 519 (29, 34—37,
48, 53, 59, 73)
Parker W. W. 633 (27)
ParmeleeA. E. 541 (20, 21, 23)
Pars H. G. 536 (147), 559, 560 (22)
Pass G. 623, 628 (2)
Pasynkiewicz S. 553 (52, 56)
Patil H. R. H. 165 (115), 411 (329), 507(88)
Patnode W. I. 26 (2), 83 (38), 90 (16)
Paudert R. 20 (14), 184 (37), 185 (37—39), 186
(37, 39), 541 (2)
Paul G. 633 (25)
Paulic F. 465 (68)
Paz С. М. 534 (36)
Pazgan A. 519 (61)
Peach M. E. 16 (96), 165 (163)
Peachey S. J. 220(139, 140), 221, 233(137),
256(139), 330(11), 339(106), 400 (114)
Peacock R. D. 216 (106), 236, 237 (106a), 241
(106, 106a)
Pearce E. M. 515 (60)
Pearsall H. W. 541 (40)
Pearson Т. Н. 549 (1, 3), 552 (3), 553 (50, 54,
57, 58), 558 (6), 570 (2)
Pedain J. 194, 195(22), 277 (26), 376(3),
439, 440 (33), 461—463 (48), 477, 478 (99,
101), 479—482 (99), 504 (76)
Peddle D. J. 81 (19), 103 (64), 124, 134 (18),
148, 150 (20)
Pedley J. B. 165 (168, 172), 207 (21), 221, 225
(152), 332 (35)
Pekola J. 527 (22, 25)
Pellecer A. 633 (55)
Peloso A. 612 (4), 613 (25), 614 (4)
Pendiliv B. W. 551 (30), 589 (183)
Pendlebury R. E. 623 (3)
Pereyre M. 282(31, 32), 311 (64a), 415(377,
381), 472, 473 (86)
Pernoll J. 633 (18, 19)
Petersen E. 165 (91)
Peterson D. J. 146, 151, 152 (9)
Petree H. E. 621 (9)
Petzschner E. 545 (16)
Pezarro S. 520 (90)
Pfeiffer P. 186 (57), 189 (83, 84), 190 (89),
194, 195(13), 206 (11), 208(32), 210(32,
68), 212(87), 216(32, 87, 118), 217 (118),
218(32), 378(30, 31), 392 (87), 400(113),
404, 413 (182), 419 (389), 432 (9, 15, 26), 438,
439, 441(7), 504 (78), 526 (6), 535(82),
42 Заказ № 207
658
Авторский указатель
551 (26), 554 (63), 575, 578 (3), 584 (79),
602 (12), 605 (4)
Phillips С. S. G. 26 (13), 33 (61), 50 (30), 82 (31),
163 (16—18)
Phillips G. M. 520(147)
Phillips J. P. 347, 350, 352, 356, 364 (153),
366 (153, 264), 443 (61, 62), 527 (54, 55),
589(191, 192)
Pickard A. L. 406 (206), 432, 433 (23)
Piecos R. 519 (10)
Piesbergen U. 535 (62), 554 (68)
Pieters H. 162 (7)
Pietrusza E. W. 128 (34)
PikeR. M. 89 (10), 104(67)
Pilloni G. 377(11), 440(38), 614(31), 633
(86)
Pines H. 541 (45)
Pinkerton R. 168 (188), 432 (3), 547 (42), 549,
554 (2)
Pirlot M. G. 541 (25)
Pischtschan S. 382(26). 433(52), 435(92)
Pitzer K. S. 535 (24)
Pizzotti R. 165 (162)
Planchon M. 207(26), 337, 338, 347(73), 577
(20)
Plazzogna G. 440 (38), 634 (86)
Plotner G. 219 (132), 558 (5)
Plump R. E. 545 (12)
Plunkett R. J. 541 (38)
Pobloth H. 608 (55), 630 (37)
Podall H. 81 (4)
Podall H. E. 621 (9)
Podolsky H. 541 (39)
Pohland E. 601, 602 (1), 605, 606 (3)
Pohland R. 195, 196 (24), 210, 223, 228 (74),
338(83), 396, 405(5), 438, 439, 441, 442
(10), 503 (72)
Pohlmann J. L. 216, 221, 222, 228, 229, 236,
237 (107)
Pol E. W. van der 527 (69)
Polis A. 188(78), 542(71, 72), 577(12, 14),
578 (12), 580 (50), 582 (12),' 602 (23), 608
(66, 68, 70), 631 (7)
Polkinhorne H. 186 (69, 77)
Pollard F. H. 26(11), 163(14, 19), 206(1),
207 (27), 274 (22), 278 (22), 290 (27), 292
(22), 457, 468, 476 (29), 527 (48, 50), 633
(77)
Poller R. С 165(77), 175(215), 244(281),
334, 343, 344 (57), 378 (36), 390 (78), 400
(121), 413 (353a), 414 (373), 432 (37, 38),
433 (37, 42)
Polster R. 258 (366), 259 (366, 367), 348 (139,
140)
Polter P. E. 165 (81)
Pommier J. С 273 (8), 274 (8, 18), 303 (8, 18,
53), 415 (381, 382), 422 (382)
Pope E. A. 165 (169)
Pope W. K. 220(139, 140), 221, 233(147),
256(139), 330(11), 339(106), 400(114)
Post H. W. 49 (23), 165 (34), 207 (20), 216,
217 (104), 221—224 (20), 226 (210), 233 (20,
210), 235 (210), 236 (20, 210). 243 (104),
249 (309), 331 (32), 343 (129), 345 (129, 142),
347 (142), 352 (129), 385 (51), 412 (330),
449 (14), 501 (57), 527 (30), 563 (74)
Potter P. E. 15 (43)
Powell D. L. 81 (18), 89 (9), 165 (179), 384
(41), 602 (18)
Powell H. M. 165 (144)
Powland S. P. 520 (135)
Prade R. 216, 217 (118), 526 (6)
Pratt G. L. 588 (158)
Pratt L. 15 (43), 165'(81)
Preckel R. 535 (64, 67)
Prenssner K. R. 633 (47)
Preston J. 520 (200)
Preuss F. R. 573 (2, 3)
Price J. W. 527 (18, 42, 43, 61, 65)
Prince R. H. 15(72), 90(15), 165(9, 177),
210, 242 (63). 330(20), 399(85), 433(50),
535 (6)
PrinceS. J. W. 365 (252)
Pritchett E. G. 520 (135)
Prokai B. 139 (58a)
Prosch U. 379 (37), 527 (51)
Pschorr F. E. 520 (149)
Pudderhatt R. J. 572(9), 607 (45a)
Purnell J. H. 588 (158)
Pusyrewa W. P. 270, 271 (12)
Quan M. L. 251, 252, 254, 258 (314)
Quane D. 16 (97)
Quintin C. 217, 218 (128), 402 (1466)
Raab E. L. 520 (105)
Raab G. 40 (96), 153—155 (40), 226, 235 (206,
208), 241 (208), 246 (296), 339, 347, 352—
354 (109), 364 (240), 453 (38—40)
Rahman W. 277 (24)
Rai A. K. 103(65), 111 (106)
Rarnachandran J. 535 (3)
Ramsden H E. 40 (97), 53, 54, (48), 208 (45,
46, 48), 209 (56, 57), 212 (46, 48), 213 (90),
220 (141), 221 (166), 223 (90, 166, 181),
225 (90, 166), 231 (90, 238), 233 (90, 166),
234, 235 (90), 236 (181), 239, 241, 242 (90),
384 (37), 396 (22), 399 (94), 400 (22),
403(157), 404 (94. 163, 164, 170, 174—
177), 405(94), 406(221), 409, 412 (285,
286), 414 (170, 357), 519 (54, 68, 76, 84,
102, 186, 187, 196), 520 (68, 102, 186, 187,
196), 558 (7)
Randall E. W. 165 (102), 249 (306), 331, 336
(28)
Rao C. N. R. 579 (40), 602 (17), 605 (10)
Rasmussen R. S. 535 (80)
Rattenbnrrv К. Н. 519 (78)
Ravner H."520 (188)
Ray R. J. 14 (3), 536 (113)
Ray R. L. 553(57)
Reach M. E. 535 (76)
Redman H. E. 186 (64)
Rees R. G. 165 (55, 70), 356 (208), 408 (255),
415 (380), 527 (56)
Reeves L. W. 165 (103), 277, 279 (25)
Reich R. 586 (111)
Reichle W. T. 96 (26), 109 (97), 165 (131),
219 (129a), 378 (27, 28) 389—391 (75),
406 (224), 410 (301), 421 (224, 404), 607 (47)
ReidA. F. 196 (29), 554 (70a)
Reid E. E. 580 (49)
Reihs L. J. 515 (56)
Reimann W. 588 (141), 612, 615 (17)
Reindl E.252(324a),332(34),407(235),521 (228)
Reinhardt W/28, 31 (37)
Reinmuth O. 580 (46)
Авторский указатель
659
Reiss E. 315 (75а)
Reiss W. 51 (32), 219 (134), 238 (267), 245
(288), 247, 249 (267, 288, 303), 251 (267, 303),
255 (303, 337, 338), 256 (303, 338), 364
(242, 243), 562 (66)
Reissans G. G. 552, 554(36), 596, 597(11),
612, 613 (11)
Renger G. 545 (3)
Renwanz G. 30(52), 194(11), 219(135), 221,
223(156), 355(188), 552(38), 561(54)
Resnick P. 273, 274, 277, 279 (10), 457, 464,
490 (27) '
Reuss H. H. von 519 (21)
Reverchon R. 527 (26)
Riccoboni L. 535 (73), 586(100—102, 112,
115), 589 (186), 633 (54)
Rice F. O. 547 (56)
Richard W. R. 536 (132)
Riche E. 541 (1)
Riddle J. M. 203 (59 60), 554 (59—62)
Ridgway J. A. 536 (136)
Rieche A. 101 (58, 59, 61), 102 (61), 408 (245—
248), 413 (245), 415 (245—248), 416 (245),
520(154, 155), 606 (42, 43)
Riedel H. J. 165(182)
Riedle R. 330(25), 382(13, 14), 519(30)
Rieger R. 409 (295)
Reimschneider R. 60 (2)
Riethmayer S. A. 519 (4, 8)
Rifkin E. B. R. 535 (94, 95)
Rijkens F. 9 (2), 57 (65), 68 (38), 75 (7), 84
(52), 85 (52, 54, 54a), 86 (54,54a), 90 (24, 25),
98 (43г, 43д), 108, 110 (43r), 164(1), 175
(I, 24a), 194 (14), 206, 227, 251 (7), 283, 312
(33), 448 (10), 526 (1), 537 (170)
Rindle R. E. 194 (18, 19)
Riviere P. 75 (3a), 129 (33a)
Robinson G. С 186 (64), 269 (3), 556 (79), 581
(63)
Robinson J 551 (28), 559 (13), 578 (25), 579
Г38), 581 (61), 584 (25), 633 (11)
Robinson M. 245 (287)
Robinson R. E. 49 (21), 117(32)
Robinson W. R. 221, 228 (155), 346 (148), 497,
500 (16)
Roch J. S. de 165 (58), 457 (23)
Rochow E. G. 8 (7), 16 (91), 19 (1—5, 7, 8),
20(2), 26(8), 30 (58), 38 (84a), 61, 74(1),
81 (13), 89 (1—6), 90 (5, 19, 31—35), 91
(19), 105 (75—77), 165 (68, 164, 165), 184
(34, 35), 188—190 (81), 220 (143), 221 (143,
169), 222, 223, 225, 229, 235 (169), 240 (268),
322 (18, 21), 330, 351 (23), 357 (220), 381
(1), 388 (68, 70), 396, 397 (2), 400 (2, 115),
401 (129, 130), 402 (129), 404 (183), 406
(115, 216, 217), 407(129, 217), 412 (216,
217, 334), 413(351), 433(56, 57), 526(8)
Rochow T. G. 19 (4), 90 (32)
Roedel M. J. 535(97)
Roetheli В. Е. 168 (189)
Rogers M. T. 15 (54), 535 (34)
Rogues G. 215—218 (100)
Roha M. E. 64 (24)
Roncucci L. 432 (21), 535 (90)
Rones J. 216, 242, 252(115)
Roosen P. 521 (227)
Roques G. 366, 368 (265)
Rosenberg S. D. 9(2), 164(1), 175(1, 196),
194 (14), 206 (7), 221, 222 (51), 223 (51, 90),
224(51), 225(90, 204), 227 (7), 228(231,
234), 229 (234), 231 (90), 233—235 (51, 90),
236, 238 (51), 239, 241, 242 (90), 246 (231),
247 (299), 251 (7), 330 (22), 331 (22, 27, 29),
333, 336 (22, 27), 339, 340 (22, 93), 342, 351
(93), 383 (31, 36), 384(37), 398, 405(72),
439 (20), 453 (41), 454 (53), 497—499 (26—
29), 500 (27), 502 (26, 28, 29), 503 (26),
504 (28, 29), 526(1, 7)
Ross A. 381 (6), 383 (35), 384 (45, 46), 385 (6),
403, 404 (158), 419 (399), 434 (72), 462,
463 (52), 519, 520 (3), 535 (94)
Rothermundt M. 93 (54)
Rothman L. A. 379(42), 451 (21, 22), 468
(73, 74), 472, 491 (74)
Rothstein E. 454 (57), 618 (15)
Rubber J. 397, 401 (49), 519(17)
Rudy D. D. 541 (55)
Ruf H. 106 (83, 84), 406 (228, 229)
Ruidisch J. 30, 38 (53), 47(2), 50(27), 90
(17), 96 (27—30), 97 (42, 43), 99 (27, 30,
45), 104 (71), 106 (85), 107 (89), 108 (94)
Runge F. 8 (4)
Rupprecht H. 165 (111)
Russell A. B. 527 (27)
Rutgers J. J. 196 (32), 212 (84), 377 (13), 438,
439, 441 (4)
Russelberge J. van 633 (29)
Sabet С R. 453 (42)
SaccoA. 357(219), 412(345)
Sacher R. E. 15 (31)
Safford H, W. 528 (81)
Saito Y. 536 (123)
Saitow A. 221—223, 225, 229, 235 (169), 330,
331 (23), 401 (130)
Sakai M. 565 (92)
Sakiyama M. 535 (14)
Salisbury L. 520 (201)
Salquain J. 521 (207)
Salyer I. O. 520 (97), 536 (131)
Sandler S. R. 513(21)
Saori H. 633 (45)
Sarafidis С 246, 255(298), 346(144), 451,
452 (24)
Sasin G. S. 208(36, 41), 210(41, 73), 211
(78), 351 (169, 170), 352, 353 (169), 397,
399 (35, 39), 400, 417, 418 (39), 528 (90)
Sasin R. 208(36, 41), 210(41), 211(78),
351 (169, 170), 352, 353 (169), 397, 399, 40,0,
417, 418 (39), 528 (90)
Satge J. 16 (98), 30 (57), 38 (86), 64 (26, 27),
65 (26, 27, 29), 66 (27), 67 (27, 29), 68
(27, 29, 41), 69(41, 45), 70(41, 46, 47),
75 (3a, 5, 6), 76 (6), 82 (27), 96, 97, 99 (32),
103(63), 105(32), 107(91), 114(4,8),
115 (8), 118 (36), 123, 124 (8), 125 (8, 20—
22), 126(8, 21, 23), 127(8), 128(33), 129
(33a), 130 (8), 131 (38, 39, 41), 132 (39—
41), 133 (8, 38), 134 (8, 33), 135 (8, 33, 38),
136 (8, 33), 138 (8), 154 (44), 552 (37)
Sathyamurthv T. W. 365 (250)
Sato H. 392 (84), 527 (31), 535 (17)
Sato Y. 489 (120a)
Saunders В. С 541 (69, 70), 550 (15), 560 Г26),
578 (27, 28), 579 (27, 30), 580 (27, 30, 56, 57),
581 (27, 28, 30, 56, 57), 582 (30), 584 (27),
598 (32, 33), 601 (10), 602 (10, 20), 605 (5,
12—14, 33), 606 (5, 12, 14), 607 (12—14, 33),
42*
660
А ьторский указатель
608 (5, 33, 61), 609 (13, 14), 632 (12)
SauveD. M. 49 (21), 117(32)
Savidan L. 165 (86)
Sawodny W. 15 (26)'
Sawyer A. K. 194 (6). 381, 385 (6), 419 (399),
423(411), 438(5), 457(20), 459 (35), 462
(50—56), 463 (50—55), 464 (35,53), 466,
467 (20, 54, 55, 70), 472 (56), 477 (35),
480 (35, 89), 481, 483 (89), 490(55)
Sayre R. 165 (4)
Scattebol L. 41 (106)
Schaeffer R. 545 (21)
Schafer H. 633 (79)
Schaper P. 238, 247, 249, 251 (267), 364 (242)
Scheer H. 464, 465 (63a), 527 (28)
Scheffler K. 535 (30, 40), 612 (8)
Schempf R. 565 (88)
Schenck G. O. 454 (62)
Schepps W. 545 (9)
Scherer O. J. 15 (35), 107 (90), 410 (298, 299),
417, 421 (389), 418, 419 (392), 607 (48)
Scherer P. C. 188 (79), 196 (30), 212, 256 (85),
343 (128), 378 (26), 384 (40), 396 (14), 438,
439(2), 457 (3), 497—501, 504(15)
Schick R. 435 (85)
Schiavon G. 586 (114), 589 (186)
Schiffner H. 405 (200, 201)
Schlegel H. 519 (41)
Schlesinger H. I. 457 (8,9)
Schlesinger H. T. 124 (10)
Schlimper R. 519 (16)
Schlottig O. 396, 405, 416 (7), 551 (23, 27),
552 (27), 560, 561 (29), 577, 579, 584 (17),
588 (142)
Schmack L. 210 (65). 407 (239), 516 (65),
527 (29)
Schmall E. A. 81 (16), 89 (11), 95(12)
Schmeisser E. 81 (51)
Schmeisser M. 78 (5)
Schmertzing H. 81 (4)
Schmidbaur H. 15 (60), 16 (109), 34 (62). 87
(58), 96 (27, 28), 99 (27), 104 (68—71),
175(212), 221, 223(150), 356(212), 412
(331, 332, 336), 421(336), 434(74), 537
(169), 559 (16), 608 (58—60)
Schmidt H. 618 (17)
Schmidt H. W. 257 (363)
Schmidt J. 8 (5), 15 (35), 20 (136), 30, 38
(53), 47 (2), 50 (27), 87 (59), 90 (17), 96
<27—30), 97 (42, 43), 99 (27, 30, 45), 103
(66), 104 (68, 71), 105 (78—81), 106 (83—
85), 107(90), 108(94), 150(28, 28a), 418,
419 (392)
Schmidt J. F. 417, 421 (389)
Schmidt M. 316 (84), 358 (228—235), 359 (230—
232), 360(230), 361(231, 233), 362 (228,
23 Г, 232, 234, 235), 363 (228, 235), 406
(228, 229), 410 (298, 299), 412 (331), 414
(365, 371, 372, 374), 415 (375, 376), 417
(389), 418, 419 (392), 421 (389), 423 (376,
414, 415), 434 (73), 496(5, 6, 11), 499 (5,
6), 500(5, 6, 11), 505 (82—83a), 587(131),
607 (48), 608 (59), 609 (83), 623, 625 (6)
Schmitz M. 217, 223 (120), 356 (196), 552
(35), 560 (30), 612, 613 (10)
Schmitz-DuMont O. 378 (21), 384 (43), 412
(338, 339)
Scheider B. 140 (69), 154 (54), 462 (49), 480,
481 (49, 104—106), 482 (106), 483 (49, 104),
487(119)
Schneider R. 535 (16)
Schneider W. G. 535 (38)
Schneiter H. 585 (94), 608 (57), 630 (40)
Schnurmann K. 208, 210(32), 216(32, 118),
217 (118), 218 (32), 526 (6)
Schollkopf U. 242 (277a)
Schott G. 135 (42)
Schreiner S. 535 (48), 554 (69)
Schroder E. 519 (60), 527 (68)
Schuierer E. 99 (48)
Schuler M. J. 541 (53)
Schumann H. 87 (59), 150 (28, 28a), 316 (84),
358 (228—235), 359 (230—232), 360 (230),
361 (231, 233), 362 (228, 231, 232, 234, 235),
363 (228, 235), 414 (365), 415 (375, 376),
423 (376, 414, 415), 434 (73), 496 (5, 6, 11),
499 (5,6), 500(5, 6, 11), 505 (82—83a),
587(131), 609 (83), 623 , 625 (6)
Schwabe P. 609 (83)
Schwartz W. T. 165 (34), 226, 233, 235, 236
(210), 449 (14)
Schwarz R. R. 28 (37), 29 (50), 31 (37), 78 (5),
81 (20), 84 (20, 51)
Schwarzhans К. Е. 535 (36), 561 (62)
Schweitzer G. K. 396 (28), 605, 606 (15)
Schweizer R. 565 (90)
Schwierer E. 406 (218), 608 (65)
Scott D. W. 535 (23, 57—59)
Scott M. J. 239, 240 (172a), 449 (13)
Segitz F. A. 545 (16, 17)
Seidlitz H. J. 527(21)
Seifert F. 16 (92), 27 (17), 535 (51)
Selwood P. W. 217 (121), 535 (64, 66, 67)
Semerano G. 586 (100—102)
Semlyen J. A. 26 (13), 33 (61), 50 (30), 82
(31), 163(16—18)
Senini S. 507, 508 (95)
Sessions W. V. 208 (40), 338 (80), 438, 439, 441
(16), 497, 498, 502, 503 (14)
Setzer W. С 551, 552 (31), 582 (67), 608
(67)
Setzler W. E. 404, 414 (169)
Sevarese F. B. 409 (276, 277)
Sevestre J. 175 (204), 273, 274 (11)
Seyferth D. 28 (32), 38 (84a), 39 (32), 40 (96),
48 (15), 49 (20), 51 (36), 74 (1), 82 (35), 87
(57), 89(2), 99(47), 109(99, 104), 116
(21), 118(33), 139 (58a), 149(24), 153, 154
(40, 42), 155 (40), 206, 213 (6), 214 (95),
220 (143—145), 221 (6, 95, 143, 144, 161,
169), 222 (95, 169. 172), 223, (6, 95, 169),
224 (144, 161, 193), 225(6, 144, 169), 226
(6, 206, 208, 214, 217), 227 (144), 229 (169),
232(251), 233(95, 161, 172, 256), 234(6,
256), 235(6, 169, 206, 208), 236 (95, 161,
256, 262), 237 (256), 239 (161), 240 (144,
161, 268), 241 (95, 208, 271), 242 (217, 277),
245 (193), 246 (193, 214, 217, 277,
294—296, 298), 255 (298), 286 (36),
290—292 (49), 322 (18, 21), 330 (23, 24),
331 (24), 333 (47), 336 (24), 339 (86, 94, 95,
109), 341(94, 115), 342(94, 121), 346(94,
144), 347 (94, 109, 121), 351 (23, 94), 352
(94,' 109), 353 (109), 354 (47, 109, 184), 357
(220), 364 (240), 379 (41), 388 (68, 70), 390
(79), 397(42), 399(90), 400 (115, 120), 401
(130), 405(42), 406 (42, 115, 120, 214, 216,
217, 227), 407 (217), 412 (120, 216, 217, 335),
423 (214), 435 (98), 447 (5), 449 (11, 12, 16),
Авторский указатель
661
451 (24), 452 (16, 24), 453 (16, 31—40, 42—
44, 47), 455 (64), 468, 469, 471, 472(75),
486(114), 489 (120а), 551(19), 560 (35),
586 (127), 617 (5, 7), 624 (20)
Sexton W. А. 584 (89)
Shackelford J. M. 81 (4)
Shannon H. F. 558 (7)
Shapiro A. 633 (67)
Shapiro H. 521 (225), 537 (171), 541 (6, 24,
28—30, 37, 65—67), 542 (79), 556 (78),
576—578 (7), 579 (7, 31), 588 (170 172,
175), 596 (8), 597 (18), 598 (18, 29, 45),
601 (2\ 602'(2, 21), 606 (38), 607 (45), 615
(36)
Shapiro P. 216 (105), 332 (38) 459, 468, 477,
480 (34)
Shapiro P. J 406 (207), 465 (65)
Sharp D. W. A. 165 (69), 356, 357 (202)
Shaw D. 165 (102), 249 (306), 331, 336 (28)
Shaw D. L. 573 (4, 5)
Shearer N. H. 14 (4), 520(126)
Sheldon R. 16 (105), 175(219), 537(165)
Shelina R. K. 535 (24)
Sherman С S. 151, 153 (34)
Shindo M. 402 (147), 433 (64)
Shishido K. 185 (41), 407, 408 (232)
Siebert H. 15 (46, 47), 26 (19), 165 (85), 186
(52), 207 (24\ 535 (27), 550 (9)
Silbert F. С 527 (74)
Sim S. Y. 149 (26), 342 (122), 496, 499 (8,9),
500, 502, 503 (8), 623, 624 (4)
Sirnmler W. 520 (179)
Simrnonin M. P. 165 (107)
Simmons D. E. 165 (167), 207—209, 221, 223
(28), 457 (15), 526 (10)
Simmons H. D. 486 (114)
Simmons J. 384 (45)
Simons J. K. 29 (40, 49), 53 (45, 47), 83 (39),
90 (30), 95 (14, 15), 99 (14)
Simpson J. B. 168 (189)
Simpson W. B. 165 (21), 370 (282)
Sims L. L. 553 (57, '58)
Sindler В. М. 90 (31)
Singh G. 535 (39)
Sisido K. 182 (20a), 183 (28), 184 (20a), 185
(39a, 40), 186 (28, 40, 45), 217, 252, 253
(124), 334 (56), 347 (155), 350 (163), 358
(56), 378 (15a)', 382, 385, 388 (18), 396 (3),
397, 409 (3, 3.1), 410 (3). 415 (31), 441 (436),
442 (43a), 443 (53a), 453 (26), 526 (5),
528 (86)
Sistrunk Т. О. 14 (3), 536 (113), 541 (52), 556
(79)
Skeel R. T. 527 (41)
Skinner H. A. 165 (168, 169, 172, 173), 194
(16), 207 (21), 212, 213 (86), 221, 225 (152),
234 (86), 332 (35), 440 (34)
Smith A. C. 89 (2), 184 (34), 188—190 (81),
388 (70), 404 (183), 406 (216,217), 407
(217), 412 (216, 217)
Smith F. A. 184 (33), 389 (74)
Smith F. B. 152, 154 (35), 162 (3)
Smith G. E. P. 210 (69), 329 (6)
Smith G. W. 15 (66), 165 (97), 535 (32)
Smith G. Z. 634 (27)
Smith H. V. 519 (7, 12, 14, 17, 80), 520 (91)
Smith J. A. S. 434 (66)
Smith J. J. 520 (145, 146)
Smith J. L. 408 (271), 519(33)
Smith R. W. 633 (66)
Smith T. A. 217, 218, 233 (123), 339 (100),
400(117)
Smith T. D. 203 (62)
Smitz N. 586 (104)
Smolin E. M. 225 (205), 287 (41, 42), 290 (42),
472, 473 (88)
Smvth C. P. 535 (44, 45)
Snyder L. J. 631 (3), 634 (62, 81)
Snyder L. R. 633 (28)
Sobue H. 536 (123)
Solerio A. 190 (93), 212 (79), 225 (197), 389
(73), 393(94), 404(184)
Soloski E. J. 48 (11), 145, 146 (10), 216 (109),
223 (185), 243 (109), 364 (248), 457 (7), 496
(2—4, 10), 499 (3, 4, 10), 500 (2—4), 502
(3, 4), 503 (3), 504 (4), 505 (3, 4), 560 (40),
562 (68), 623, 624 (5)
Sornmer L. H. 128 (34)
Sommer R. 140(66, 69), 154(54), 165(91),
257, 258 (364), 273, 274 (5, 12), 275 (5),
276 (23), 277 (5, 12, 23), 278, 279, 281, 283
(12), 284 (12, 34), 285 (12, 35), 288, 290.(5),
303(12), 305 (34, 35), 341(118), 458(30),
460 (40), 463 (30), 477—479 (99), 480, 481
(99, 104—106), 482 (99, 106), 483 (104),
487 (40), 490, 491 (121), 513, '514 (44)
Soroos H. 378, 379 (35), 559 (10), 576 (6), 579
(31), 586, 587 (120), 596(8), 597(15, 18),
598 (18, 38, 40, 44, 45), 628 (27)
South A. 573 (5)
SowerbyD. B. 406, 419 (212)
Sparmann H. W. 242, 245 (275, 276), 248,
249, 251, 252 (276), 333, 334, 350, 358,
364 (48)
Spatz S. M. 561, 563 (57), 581, 582, 586 (62),
605, 608(11)
Spengler G. 175 (218)
Spialter L. 162 (1), 453 (28), 526 (2), 631 (1)
Spice J. E. 586 (110)
Spielman J. R. 356 (201), 377 (10), 477 (95)
Spill man G. E. 413 (353a)
Squires S. 519 (43)
Srivastava T. N. 15 (24), 216 (117), 244, 251
(280), 252 (280, 325a), 405, 406 (193), 410
(303), 521 (220)
Stacey G. J. 541 (70), 550 (15), 560 (26), 578
(27), 579 (27, 30), 580, 581 (27, 30, 56), 582
(30), 584 (27), 598 (33), 601 (10), 602 (10,
20), 605, 606 (5, 14), 607 (14), 608 (5, 61),
609 (14), 632 (12)
Stack W. F. 165 (173)
Stafford S. L. 15(55), 165(119), 214, 226,
235 (92), 226, 235 (207), 346—348, 352,
364 (149), 352, 356 (173), 356 (205)
Strafford N. 526 (9)
Stafford S. L. 15(55), 165(119), 214, 226,
235 (92, 207), 346—348 (149), 352 (149, 173),
356 (173, 205), 364 (149)
StandeE. 502(61), 520(112)
Stanley M. D. 216, 243 (109)
Stanley R. S. 536 (155)
Stanton R. 541 (48)
Stanun W. 399, 402 (36), 449 (15)
Stapleton G. B. 16 (105), 175(219), 537 (165)
Staudinger J. J. P. 408 (274), 519 (31)
Staveley L. A. K. 206 (10), 207 (10, 29) 208
(29), 251, 260(10)
StecherO. 502 (61), 520(112)
662
Авторский указатель
Sleeker H. С. 521 (213)
Stefl E. Р. 405 (189, 190), 519 (57, 63, 65)
Stein J. 541 (46)
Steingross W. 15 (9)
Steinmeyer R. D. 527 (52)
Stendel O. W. 547 (41)
Stern A. 216(105, 112), 217—219, 243 (U2),
269 (8), 332 (38), 345 (138), 385 (49),' 396
(13), 458 (33), 459, 468 (33, 34), 477 (34, 96),
480 (34)
Sterting J. D. 541 (53)
Stevens C. D. 535 (53), 562 (73)
Stickney P. B. 536 (129)
Stockier H. A. 165 (122, 131)
Stone F. G. 15 (55), 54 (52), 127 (28), 159 (8),
165 (45, 82, 119), 206 (6)', 208 (43), 213 (6),
214 (43, 92), 222 (173), 223, 225 (6), 226
(92, 207), 234 (173), 235 (6, 92, 207), 330
(24), 331, 336(24), 346(149, 150), 347
(149, 153), 348 (149), 350 (153), 352 (149,
150, 153, 173), 356(153, 173, 203—205),
257 (220), 364 (149. 150, 153), 366 (153,
264), 370 (284), 400, 406, 412 (120), 443
(61, 62), 444 (62), 457, 463 (25), 527 (54,
55), 589 (191, 192)
Stone L. S. 586 (122)
Stragand G. L. 528 (81)
Straub D. 165 (132)
Strecker A. 419 (395, 396)
Strohmeier W, 16 (92, 93), 27 (17), 29 (45),
165(158), 535 (47, 51, 52)
Stuckwisch С G. 562 (72), 563 (72, 78), 579
(37), 619 (18, 19)
Styan G. E. 304 (54), 316 (81), 444 (63a)
Sugita K. 385 (52), 397 (33)
Syjishi S. 15 (21)
Sukkani D. 98 (43b), 418 (389a)
Sullivan F. W. 547 (54, 55)
Summers L. 78 (2), 537 (161), 552 (41), 553
(45), 584 (80), 624 (12, 15), 627 (12, 15, 26)
Suzuki R. 232 (251), 453 (42, 44)
Swaddle T. W. 34 (64), 52 (39), 83 (42, 46), 221
(159)
Swaminathan S. 365 (250)
Sweeney O. R. 559 (13), 560 (32)
Swiger E. D. 165 (139)
Sykes A. 527 (59, 60)
Svmes W. R. 313 (71), 314, 316, (73), 398 (77a)
Szuchnik A. 20 (16, 17), 35 (69), 536 (137)
Tabern D. L. 27 (16, 18), 29 (16), 36 (18), 50
(31), 54 (51), 81 (10), 82 (32), 93 (52), 95
. (10, 13), 96 (23), 252 (323)
Tacupo T. 402 (146)
Tafe! J. 545 (1, 2, 8), 553 (48)
Taglievini G. 355(193), 377(11), 435(89),
440(38), 527 (44), 585(91), 586(112—115),
589(186), 612(1, 3, 4), 613(1, 25), 614
(4, 31), 631 (2), 633 (49. 53, 54, 80)
Taimsahi P. 165 (26, 47), 535 (9)
Takamizawa M. 286(36), 449(11), 489 (120a)
Takeda Y. 183 (28), 185 (40), 186 (28, 40, 45),
217, 252, 253 (124), 257, 259 (361), 334
(56), 347 (155), 350 (163), 358 (56), 382,
385, 388 (18), 453 (26), 520 (161), 526 (5),
528 (86)
Talukdar P. T. 154 (43)
Tamblyn J. W. 519(27)
Tamborski С 48(11), 108(92), 145, 146(10),
216 (109), 223 (185), 243 (109), 364 (248),
457 (7), 496 (2—4, 10), 499 (3, 4, 10), 500
(2—4), 502, 503, 505 (3, 4), 560 (40), 562
(68), 623, 624 (5)
Tanaka T. 159 (2), 165 (29, 57, 61), 414 (360,
361), 432 (17), 433 (17, 51), 434 (17), 435
(91), 535 (26)
TandonS. K. 244, 251, 252 (280), 405, 406 (193),
521 (220)
Tanner H. M. 541 (18, 19)
Tapley C. G. 186 (69), 188 (77)
Tate "J. M. 330(21), 409(279), 414(362),
417 (279), 432 (18), 497—500, 502, 503 (20)
Tatlock W. S 412 (334)
Tatlow J. C. 216 (106), 236, 237 (106a), 241
(106, 106a)
Taylor H. S. 588 (152)
Tchakirian A. 24 (6), 92 (48), 189 (86), 409
(186)
Teal G. K. 122 (3)
Teplv J. 165 (141, 142), 535 (43)
Terao K. 535 (81)
Termin E. 108 (93)
Tesi G. 216, 221, 222, 228, 229, 236, 237 (107)
Thaver J. S. 103 (95), 108 (96), 410 (300, 302),
605, 607 (9), 607 (46)
ThiesC. 412 (333), 516 (67)
Thinius K. 519 (16, 60)
Thorn K. F. 150 (28), 496 (5, 6, 11), 499 (5, 6),
500 (5, 6, 11). 623, 625(6)
Thomas A. B. 81 (13), 89 (3, 4), 165 (164, 165)
Thomas L. H. 221, 222, 226, 233, 234, 235 (163),
330 (19), 404 (185)
Thomas W. H. 588 (162—165, 167—169, 171,
173, 174)
Thomson W. 598 (44)
Tichy V. 519 (9)
Tobias R. S. 165 (74), 388 (69), 404, 409 (160),
432 (20)
Tobin M. С 15 (7, 48), 26 (4), 165 (23, 66),
207 (18). 210 (72), 535 (8)
Todd L. J. 486 (114)
Tombe F. J. A. 484 (113a)
Tomka L. A. 400 (122), 406 (122, 222), 520
(102, 183, 184, 195)
Tonge B. L. 527 (53)
Towne E. B. 522 (39), 560 (38), 561 (38, 55,
56), 579 (35, 44), 580 (44, 45), 614 (33)
Toy A. D. F. 519 (78)
Treichnel P. M. 222, 234 (173), 346, 352 (150),
357(218), 364(150)
Trappe G. 520 (167)
Treichel P. M. 222, 234 (173), 346, 352, 364
(150), 357 (218)
Trepka W. J. 151 (31), 502 (59), 624, 626,
628 (13)
Trotman - Dickenson A. F. 365(252)
Trotter J. 165(145), 435 (84)
Truett W. L. 520 (123)
Truskier P. 212 (87), 216 (87, 118), 217 (118),
526 (6), 535 (82), 551 (26), 575, 578 (3),
602 (12), 605 (4)
Tsai J. H. 316 (80)
Tsai Т. Т. 406 (208), 439 (21a), 441 (39),
465 (66)
Tseng Chao-Jun 316 (83)
Tson К. С 513 (21), 536 (155)
Tsuruta T. 535 (103)
Авторский указатель
663
TullioV. 541 (54)
Tung Shi-hua 316 (83)
Tuzi Т. 182, 184 (20a)
Twist W. 586 (110)
Twitchett H. J. 404 (159), 520 (167, 172, 173)
Tzschach A. 315 (75a)
Uden P. C. 163 (19)
Uedd T. 633 (31)
Ue-eda R. 414 (360)
Ugo R. 411 (328), 507, 508(95)
Ulbricht K. 165 (59), 468—470 (77), 469 (83)
Umo T. 400 (123), 401 (127)
Urbanski T. 535 (86), 580 (54, 55), 618 (13)
UrsinoJ.A. 251 (313)
Valade J. 149 (27), 165 (30, 56, 72), 273 (8),
274 (8, 18), 282 (31, 32), 303 (8, 18, 53),
311 (64a), 415 (377, 381, 382), 422 (382),
472, 473 (86)
Vandenberg E. I. 520 (151)
Van der Kerk G. J. M. 15 (71), 40 (98, 100),
48, 52 (18a), 65 (28), 68, (38), 75 (7), 84
(52, 54), 85 (52, 54, 54a), 86 (54a), 90 (24,
25), 98 (43г, 43д), 108, 110 (43r), 116
(18), 152, 154 (37), 159 (10, 17), 165 (12),
175(195, 198, 205, 211a, 214), 182, 184
(17), 185 (42), 187 (17), 188 (17, 75), 194
(15), 207 (13, 19, 31), 208 (19, 31, 49), 209
(19, 31, 59), 210 (19, 59), 212 (81), 216 (49),
217, 219 (125), 221 (148, 167), 222 (148) 223
(13, 167), 224 (13, 148), 226 (215), 228 (232),
231 (243, 246), 233 (19, 148), 237 (167),
239 (19), 240 (270), 251 (19, 59, 316), 252
(59), 253 (316), 256 (49), 257, 259 (49, 362),
273 (3, 4, 6, 7, 26), 274, 277 (4), 278, 279,
281, 282 (4, 6), 283 (3, 6, 33), 284, 285 (6),
286 (38), 287 (4, 40), 288 (4), 290 (4, 50),
305(50), 312(33), 315(76)/321 (16), 322
(19), 327 (15), 330 (16—18), 331 (17, 18),
332 (18, 41), 333 (44), 335 (17), 338 (84),
339(110), 341(18, 116, 117), 342(18),
343 (116), 356 (199, 200), 363 (18, 237, 239),
366 (237), 382 (23), 383 (23, 28—30), 384
(38), 385 (47), 388, 389 (71), 397 (40, 44, 50,
54, 58), 398 (40, 44, 54. 73, 76), 399 (40,
106, 107), 401 (131), 405 (197), 406 (40),
407(50, 243), 410(40, 106, 107, 307),
432 (4, 5), 433 (4, 44, 58), 435 (4, 5), 447 (7,
в), 448(7, 8, 10), 449(8). 457(2, 12), 458
(12, 31), 461 (42), 462 (57), 468, 472, 474
(71), 477(71, 97), 480, 482, 483(108),
484 (113a), 496 (7), 500 (43), 503, 505 (7), 512
(13), 513 (46, 47), 514 (13, 46, 47, 49, 50),
515 (49, 54), 521 (208—212), 527 (16), 528
(82, 83, 85), 535 (5, 13), 536 (144, 156), 537
(170), 554, 555 (74), 559 (21), 560, 561 (48),
570 (5), 571 (5, 6), 572 (8), 575 (1) 577 (18,
19), 598 (35), 617 (4), 624 (7, 18), 625 (18),
627 (7, 25), 628 (7)
Varma I. D. 124 (19)
Vasta J. A. 211 (77), 396, 397, 399, 405, 406 (1)
Vats S. S. 633 (64)
Vaughan L. G. 48 (15), 226 (217), 232 (251),
242, 246 (217), 290—292 (49), 453 (37, 44,
47)
Veldell J. D. 207—209, 221. 233 (28), 457 (15)
Ventura J. J. 220 (136). 383 (35), 386 (61),
403, 404 (158) 408, 419 (275), 434 (72),
458 (32), 528 (89)
VerbeekF. 483, 484 (111)
Verdonck L. 15(64), 165(110, 114, 118)
Viehe H. G.'254 (331)
Vijayaraghavan K. V. 214, 235 (94), 340 (114)
VingeeR. A. 457(4), 500(41)
Viswanathan С R. 633 (64)
Vladimiroff T. 165 (117)
Vliegenthart J. A. 15 (71), 535 (5)
Vogel A. I. 165 (2, 6), 433 (46, 47), 535 (54, 55)
Voigt D. 16 (84, 86), 27 (23, 24), 165 (151),
209 (60), 552 (37)
Vondel D. van de 15 (64), 20 (13), 26 (7), 83
(37), 127(29), 165(110)
VorlanderD. 454 (56), 618 (14)
Wada M. 165 (17), 385—387 (54), 402 (147),
406 (209), 413 (355), 414 (360), 419 (209),
422 (406), 433 (43, 64), 434 (70)
Wada T. 153, 154 (42), 453 (40)
Waddington G. 535 (57)
Waddington Т. С 16 (96), 165 (163), 535 (76)
Wagensommer J. 520 (129)
Wagler K. 545 (16)
Wagner D. 397, 398, *407 (64), 528 (88)
Wagner E. С 29 (40), 53 (45), 90 (30), 95, 99
(14)
Wagner H. 247, 249, 251, 255, 256 (303)
Wailes P. С 196 (29), 554 (72a)
Wainrub M. 519 (5)
Walcutt С 535 (95)
WaldeA. W. 396, 400(27)
Walde R. A. 358 (223)
Waldmann S. 34 (62), 221, 223 (150), 559 (16)
Walker A. 165(21), 370(282)
Walker G. R 26 (13), 33 (61), 50 (30), 82 (31),
163 (16, 18)
Walkiewitz D. 447 (2, 4)
Wall H. J. 598 (43)
Wallace J. M. 212 (80), 519 (67)
Walsch E. J. 474 (91,92)
Walton D. R. M. 41 (108), 48, 52 (16), 83
(43, 45, 47), 224, 248 (190), 350 (161), 498
(36)
Want G. M. van der 257, 259 (362)
Ward С. Н. 165 (60), 207 (12, 16)
Waring С. Е. 207, 220 (17), 365 (249)
Warner С R. 468, 471 (78) '
Warren J. B. 206(10), 207(10, 29), 208
(29). 251, 260(10)
Wartman L. H. 520 (145)
Washburn R. M. 109 (98)
Washburn R. N. 316 (77)
Washburne S. S. 455 (64)
Wasylciw J. 513 (36)
Waters D. N. 15 (49), 26 (5)
Waters J. A. 221, 228, 247, 253(154), 338,
(77), 350 (160), 454 (55)
Waters J. M. 338 (78)
Waters W. A. 320 (11)
Watson J. W. 520(170)
Watt R. 15 (58)
Watterman J. A. 520 (152)
Waugh J. S. 453 (32)
Wazer J. R. 57 (66), 160 (30), 165 (101), 330,
332, 357 (7)
Wazynski K. 553 (56)
Weber S. 465, 466, 472, 473, 490 (69)
Webster D. E. 15 (53), 165 (75, 98), 382, 388
(21), 396, 397, 400(2), 411 (310), 433 (56)
664
Авторский указатель
Webster D. J. 435(95)
Webster M. H. 233, 234 (257)
WeimerP. E.541 (49)
Weinberg E. L. 187 (71), 221—224, 233—236,
238 (151), 339, 340, 342, 351, (93), 383 (31),
398 (72), 399 (99—101), 400 (122), 402 (144),
404 (99—101, 166, 177), 405 (72, 100, 101),
406 (122, 221, 222), 407 (241), 414 (99, 358),
519 (68, 70, 71), 520 (68, 102, 183, 184, 195)
Weinberg K. 210, 212, 214 (67), 216—218 (111),
222 (67), 223, 229 (111), 343 (127), 389 (72),
396 (11), 400, 405, (108), 439 (26), 503 (70)
Weiner M. A. 49 (20), 222 П72), 224 (193),
226 (214), 233 (172), 245 (193), 246 (193,
214, 295), 453 (31—33, 35, 36), 560 (35)s
617 (7)
Weinhouse S. 586 (124)
Weiss E. 165 (157), 535 (46), 554 (67)
Welch D. E. 246 (296), 453 (39)
Wellman J. W. 535 (109)
Wells E. J. 165 (103), 277, 279 (25)
Welzel D. 51 (31a)
WendeA. 282(30), 448 ф)
Werner A. 186 (57), 206 (11), 432 (26)
Werner L. P. 581 (64)
West В. О. 256, 260 (355), 497, 503 (31), 553
(53), 629 (32)
West R. 165 (179), 233, 234 (257), 255 (334),
384 (41), 388 (70), 406 (216), 410 (300, 302),
412 (216), 535 (12), 602 (18), 605 (9), 607
(9, 46)
West R. C. 15 (14), 19 (3), 28 (38), 30 (51),
31 (58), 32 (51), 60 (1), 81 (18), 89 (2, 9, 13),
91 (13), 103 (62), 108 (95, 96), 122 (5), 124,
125 (17), 134 (5)
Westlako A. H. 249 (307), 422 (407)
WheatleyP. J. 165 (143)
Whipple R. O. 103 (62)
Whitby F. J. 586 (109)
White D. G. 392(84), 527(31)
White R. F. M. 220, 233 (138), 385, 386 (56),
406 (210), 434 (69)
Whiting M. С 41 (106)
WhitmoreF. C. 128 (34)
Wiberg E. 409 (295), 445 (64), 502 (61), 520
fe (112)
WidmaierO. 588 (157)
Wieber M. 20 (136), 47 (2a), 89 (6a), 103 (66),
105 (78—81), 414 (371, 372, 374)'
Wienbreczeni E. 398, 405 (72)
Wiernik M. 240, 259 (269), 528 (80), 559,
560 (15)
Wiezer S. B. 541 (26), 589 (190)
Wild F. 608 (61)
Wilding J. G. E. 608 (61)
Wilkins E. J. 434 (66)
Wilkinson G. 15 (43), 165 (18, 81), 203 (58),
233, 234(257), 411 (320), 507 (91), *535
(4), 554 (66)
Willemsen L. C. 152, 154 (37), 496, 503, 504
(7), 549 (5), 554, 555 (74), 571 (6), 572 (8),
577 (18, 19), 598 (35), 624 (7, 18), 625 (18),
627 (7, 25), 628 (7)
Williams A. A. 34 (63), 117 (31), 229, 231 (236),
453 (50)
Williams D. J. 527(42, 43)
Willis С J. 23 (4, 5), 207 (14), 330 (15), 340,
(15, 113), 364 (15) 443, 444 (58—60), 503
(64)
Willis H. A. 165 (55, 70)
Williston A. F. 29 (48)
Wilson G. R. 515 (58)
Wilzbach К. Е. 124 (10), 457 (8)
Winfield J. M. 356, 357 (202)
Winkler С 53 (42)
Winkler D. E. 520 (92)
Winksne K. J. 406, 419 (212)
Wippler C. 519 (24, 26)
Wiss Z. 185, 186 (39)
Wittenberg D. 16 (99), 144 (2), 175 (209), 259
(367a), 368), 411 (317)
WittigG. 194, 195 (3), 457, 484 (5), 498 (32. 33),
499 (33)
Wokulat J. 418, 419 (392)
Wolff W. 520 (205)
Wood J. L. 165(26, 47), 535 (9)
Woods L. A. 245(291), 356(198), 563(76),
586(99), 618 (11)
Woodward L. A. 15 (49), 26 (5)
Woolford R. G. 536 (125)
Wooster С. В. 90 (29), 95, 101, 106 (11)
Wooten W. C. 519 (82)
Work P. L. 633 (33)
Worral D. E. 29 (41)
Wright A. N. 74 (2), 75 (3, 4), 319, 323 (9),
457, 458 (28)
Wright N. 322 (22). 447 (1)
Wu Т. С. 229, 242'. 243, 245, 247 (237), 454 (K4
Wunsch A. 588 (151)
Wurst M. 527 (47)
Wyant R. E. 182, 183 (27)
Wyatt P. A. H. 432 (24)
Yamamoto T. 519 (85)
Yamazaki H. 468, 469, 471, 472 (75)
Yasuda K. 396, 400 (18), 433 (53, 54), 435 (91,
96)
Yasuda M. 432 (20)
Yeddanapalli L. M. 365 (250)
Yeldell J. B. 165 (167)
Yick-Pui M. J. 447(5)
Young C. W. 15 (6), 26 (3), 165 (22)
Youtz M. A. 541 (12)
Yukawa Y. 565 (92)
Zablothna R. 20 (16, 17), 26 (12), 35 (69)
Zanella P. 435 (89)
Zartman W. H. 590 (203)
Zbornik T. W. 396 (27)
Zechmeister L. 602 (24), 608 (69)
Zeiss H. 78 (6), 81 (6), 91 (45), 92 (45)
Zemiszka T. 520 (193)
Ziegler K. 168 (184—187), 545 (18—27),
546 (28—32, 36, 39, 40), 547 (41)
Zietz J. R. 165 (64), 553 (57), 570 (2)
Zilkha A. 397 (64)
Zimmer H. 207 (28), 208 (39), 210 (66), 224
(194, 195), 232 (252a), 242 (275, 276), 334
(48), 340 (112), 363 (238), 376 (3), 396 (19),
407 (234), 413 (353)
Zisman W. A. 520 (188)
Zlattis A. 633 (26, 38)
Zoche G. 206 (2), 396 (8)
Zuckermann J. J. 414 (359)
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ *
Ал кил герма ний
галогениды, реакции с
органическими гидроперекисями 101
серебряными v солями органических
кислот 100
сероводородом 104
тригидриды 127
N-Алкилдистанназаны 409
Алкилолово
(дигалоид)гидрид, присоединение к алке-
нам 277
дигидроксихлорид 392
селенид 404
сульфид 404, 406, 413
трехгалоидное 189, 404
реакции с
алифатическими диазосоединениями
322
литийорганическими соединениями
251
тримеркаптиды 405
б«с-(Алкилперокси)диалкилолово 408
Алкилперокситриалкилолово 408
Алкилперокситриалкил(или арил)свипец 606
Алкилстаннилдифенилфосфонаты 423
Алкилстанноновые кислоты 393
диспропорционирование 378
Алкил-а-триалкилстаннилакрилаты 292
Алкил-траяс-Р-триалкилстаннилакрилаты
292
Алкил-^ш:-р-триалкилстаннилакрилаты 292
Ал ко кси герма ний треххлористый,
присоединение к кетену 71
Алкоксисоединения олова, реакции с
алюминийорганическими соединениями 258
реактивом Гриньяра 206
Алкокситриалкилгерманий, реакция с алкил-
меркаптанами НО
Алкокситриалкилолово 399
присоединение к
двуокиси серы 313
двуокиси углерода 313
изотиоцианатам 313
изоцианатам 313
карбодиимиду 313
кетену 311
сероуглероду 313
эфирам ацетилендикарбоновой кислоты
313
реакции с
димером кетена 312
перекисью водорода 416
энолами эфиров 311
Алкокситриарилолово 408
Алкокситриэтилсвинец 605
Алленилтрифенилсвинец 560
Аллилбензолы с оловоорганическими
заместителями в ядре 231
Аллилгерманий треххлористый 21
Аллилдифенилолово бромистое 340
Аллилолово трехбромистое, тример 404
Аллилстанноновая кислота 190
реакция с бромистоводородной кислотой
404
Аллилтриалкилгерманий, присоединение
брома 115
меркаптанов 115
Аллилтриарилолово, присоединение
полисульфида водорода 449
Аллилтриметилгерманий 39
Аллилтри-/г-толилолово 226
Аллилтрифенилолово 235, 500
реакции с
фениллитием 453
хлористым водородом 350
термическая устойчивость 379
Аллилтрифенилсвинец, полимеризация 536
Аллилтриэтилгерманий 39
окисление 116
присоединение тиоуксусной кислоты 115
Аллилфенилциклотетраметиленгерманий 40
реакция с дихлоруксусной кислотой 83
Аллильные соединения олова,
каталитическое гидрирование 453
Амилгерманий треххлористый 30
я-Амилтрифенилгерманий 151
я-Амилтрифенилсвинец 560
Аминодиметилстаннилнатрий 465, 497
(З-Аминопропил)трипропилолово 447
Аминотриметилолово 421
3-Амино-1\[-триэтилстаннил-1,2,4-триазол 410
п- Аминофенилтрифенилсвинец, диазотирова -
ние 618
Ангидриды кислот
арилгерманиевых 93
реакция с сероводородом 97
бензилгерманиевой 54
я-бутилгерманиевой 93
л-диметил (или этил) аминофенилгермание-
вой 77
/г-монометиламинофенилгерманиевой 77
/2-тол ил германиевой 54
фенилгерманиевой 54, 76
я-Анизилсвинец
триацетат 565, 573
трипропионат 565
Арилгерманий, галогениды, реакции с
карбонилами марганца 109, 155
* Жирным шрифтом набрана цифра,
пропись получения данного соединения.
указывающая страницу, на которой приведена
666
Предметный указатель
карбонилами рения 109, 155
органическими гидроперекисями 101
Арилолово
сульфид 406, 413
тримеркаптиды 405
Арилстаннилдифенилфосфонаты 423
Арилтриэгилгерманий 34, 51
(2-Ацетиламино)этилтрифенилсвинец 628
Ацетилениды олова, каталитическое
гидрирование 453
(2-Ацетоксиэтил)триэтилолово 284
Баренил-бис-(три-я-пропилолово) 248
Бензилокситриэтилолово 302
реакция с перекисью бензоила 368
Бензилокситриэтилсвинец 605
Бензилтрибутилгерманий 35
Бензилтри-/г-толилсвинец 627
Бензилтри-/г-фенетилсвинец 627
Бензилтрифенилолово 500
Бензилтрифенилсвинец 627
Бензилфенилолово двухлористое 350
Бензилфенилтриэтилстанниламин 310
б«с-(2-Бифенил)дибензилолово 237
Бифенилди-1-инденилолово 232
2,2-Бифенилендиоксидиметилолово 414
9-(Бифенилил-2)-дибензостаннол хлористый
349
б^с-(Бифенилил)олово двухлористое 348, 349
реакция с галоидами 345
б«с-(2-Бифенил)олово 198
Бифенилсульфамидотри-я-пропилсвинец 608
Бромметилолово трехбромистое 321
/z-Бромфенилолово треххлористое 337
/z-Бромфенилтрифенилсвинец, реакция с ди-
этилбарием 618
Бромфенокситрифенилолово 409
1,4-Бутандиоксидибутилолово 417
7-Бутенилтриметилгерманий 119
Бутенил-3-трифенилсвинец 560
mpem-Бутиламинотрибутилолово 418
я-Бутилбензилфенилолово, гидроокись 385
я-Бутилвинилолово двухлористое 241
Бутилгерманий
бромгидрид 127
иоддигидрид 127
трехбромистый 96
трехиодистый 23
треххлористый 30, 84, 86
я-Бутилдиаллилолово бромистое 340
я-Бутилдинеопентилолово бромистое 340
Бутилди(триметилсилилметил)олово
йодистое 342
Бутилолово
гидроксидихлорид 392
оксимоноацилат 392
полимер 474
трехиодистое 189
треххлористое 184, 212, 335
триацетат 405, 407
трибутират 407
тривалериат 407
тригидрид 461
присоединение к метилметакрилату 284
реакции с
гидроперекисью трет-бутила 467
кетонами 473, 474
триизобутират 407
трипропионат 405, 407
mpem-Бутилперокситрибутилолово 467
mpem-Бутилперокситрифенилолово 413
Бутилстанноновая кислота 392
Бутилтриаллилово 223
реакция с бромом 340
Бутилтривинилолово, реакция с галоидными
солями ртути 354
я-Бутилтри(гексин-1-ил)олово 241
трет-Бутилтриметилгерманий 38
я-Бутил(триметилсилилметил)олово двубро-
мистое 347
Бутилтри(пентафторфенил)олово 241
я-Бугилтри(трифторвинил)олово 241
я-Бутилтрифенилгерманий 151
Бутилтрифенилолово 253
mpem-Бутилтрифенилсвинец 560
я-Бутилтри(фенилэтинил)олово 241, 251
я-Бутилтрифторфенилолово 226
я-Бутилтри(циклопропил)олово 241
я-Бутилтриэтилгерманий 35, 38
emop-Бутокситриэтилолово 304
/z-Винилбифенилтрифенилолово,
полимеризация 513
Винилгерманий треххлористый 54, 61
4-Винилокси-1 -(триэтилстаннил)бутин-1 327
Винилолово
трехбромистое, тример 404
треххлористое 336
Винилстанноновая кислота 190
реакции с
бромистым водородом 404
органическими кислотами 404
Винилтриалкилгерманий, присоединение
брома 115
бромистоводородной кислоты 115
меркаптанов 115
хлороформа 116
четыреххлористого углерода 116
этилового эфира бромуксусной кислоты
116
Винилтрибутилгерманий, реакция с бромом
82
Винилтрифениловово 225
диспропорционирование 379
присоединение
трихлорбромметана 450
циклонов 451
Винилтрифенилсвинец 560
Винильные соединения олова,
каталитическое гидрирование 452
Гексаалкилдигерман 50
Гексаалкилдигерманооксан, реакции с
изотиоцианистой кислотой 96
органическими кислотами 96
синильной кислотой 96
Гексаалкилдиплюмбаны 628
диспропорционирование 615
диссоциация 612
реакции с
галоидами 613
серой двухлористой 613
сульфурилом хлористым 613
тионилом хлористым 613
хлористым водородом 614
Гексаалкилдистаннаны 503
окисление 438
реакции с
Предметный указатель
667
галоидами 439, 440
натрием 497
органическими перекисями 441
перфторалкилами йодистыми 443
серебром азотнокислым 441
сулемой 441
эфирами меркур-бш>уксусной кислоты
271
Гексаалкилдистанноксаны 382, 383, 434
присоединение к изоцианатам 316
реакции с
алкинами 325 и ел.
амидами 398, 399
динитрилом малоновой кислоты 327
кислотами 396
метилмалоновым эфиром 327
мочевиной 399
силанолами 399
спиртами 399
углекислым газом 397
фенолами 399
флоуореном 327
Гексаамилдигерманооксан 86, 90
Гекса-о(или я)-анизилдиплюмбан 552
Гексаарилдиплюмбаны 628
диспропорционирование 596, 615
реакции с
алюминием треххлористым 614
галоидами 613
золотом хлорным 614
органическими кислотами 583
серебром азотнокислым 613
серой двухлористой 613
сульфурилом хлористым 613
тионилом хлористым 613
хлористым водородом 614
Гексаарилдистаннаны 194, 503
реакции с
иодом, кинетика 440
натрием 497
органическими перекисями 441
серебром азотнокислым <*41
сулемой 441
Гексабензилдигерман 82, 145
Гексабензилдигерманооксан 90
Гексабензилдистаннан 503
Гекса(бифенилил-2)-дистаннан 504
Гекса-«-бутилдигерманооксан 90
Гекса-«-бутилдистаннан 481
реакции с
гексафтор-2-бутином 316
иодом, кинетика 440
Гексан-«-бутилдистанноксан 383
присоединение к
бромалю 316
хлоралю 316
реакции с
метансульфокислотой 350
органическими кислотами 397
циануровой кислотой 397
1,1,1,3,3,3-Гексабутил-2,2-дифенилтристан-
нан 482
Гексавинилдигерман 32
Гекса-«-гексилдигерманооксан 90
Гекса-«-гексилдистанноксан 383
Гексадекабутилоктастанноксан, диацетат 388
Гексаизобутилдистаннан 503
Гексаизопропилдистанноксан 383
реакция с метансульфокислотой 350
Гексаксилилдиплюмбан 552
Гекса-/г-ксилилдистаннан 503
Гексамезитилдиплюмбан 552
Гексаметилдигерманооксан 90
реакции с
дифторангидридом метилфосфиновой
кислоты 97
окислами металлов 96
фениллитием 97
Гексаметилдистаннан 477
диспропорционирование 377
присоединение к
гексафтор-2-бутину 316
гексафторпропену 36
тетрафторэтилену 316
реакции с
иодом, кинетика 440
серой 439
трифторацетилом йодистым 444
трифторацетилом хлористым 444
трифториодметаном 443, 444
Гексаметилдистанноксан 382
комплексы 435
реакция с метиллитием 421
Гекса-а-нафтилдиплюмбан 552
Гексанеопентилдистаннан 222
Гекса-«-октилдистанноксан 383
Гекса-«-пропилдигерманооксан 90
Гексапропилдистаннан 503
Гекса-«-пропилдистанноксан 383
реакций с
органическими кислотами 397
фенолами 399
Гекса-я-толилдигерман 29, 145
Гекса-я-толилдигерманооксан 90
Гекса-ж-толилдиплюмбан 552, 628
Гекса-отолилдиплюмбан 552, 558
диспропорционирование 596
окисление 612
Гекса-я-толилдиплюмбан 552
окисление 612
реакция с кислотами 583
Гекса-отолилдистаннан 504
Гекса-я-толилдистаннан 503
Гексафенантрилдистаннан
окисление 439
реакция с сулемой 441
Гекса-я-фенетилдиплюмбан 552
Гексафенилдигерман 32, 33, 145
Гексафенилдиплюмбан 552, 628
диспропорционирование 596
окисление 612
присоединение к малеиновому ангидриду
614
реакции с
кобальтом хлористым 614
магнием 624
медью двухлористой 614
органическими кислотами 614
щелочными и щелочноземельными
металлами 624
Гексафенилдиплюмбоксан 572, 602
присоединение к
гексахлорацетону 572
динафтилкарбодиимиду 572
нафтилизоцианату 572
сероуглероду 572
трихлорацетонитрилу 572
хлоралю 572
668
Предметный указатель
термическое разложение 597
Гексафенилдистаннан 195, 197, 198, 282, 283,
286, 303, 307, 473, 493, 499, 503
окисление 439
реакции с
галоидами 440, 441
литием 496
нафталиннатрием 498
пентафториодэтаном 444
уксусной кислотой 445
Гексафенилдистанноксан 384
реакции с
реактивом Фишера 384
фенолами 399
циануровой кислотой 397
Гексафторизопропокситриметилолово 304
Гекса-/г-фторфенилдистанноксан 385
Гекса-/г-хлорфенилдистаннан 503
реакция с иодом 440
Гексациклогексилдиплюмбан 551
реакции с
галоидами 576, 577
четыреххлористым углеродом 615
фотохимическое окисление 612
Гексациклогексилдиплюмбоксан 612
Гексациклогексилдистаннан 214, 503
Гексаэтилдигерманооксан 90
реакции с
меркаптоуксусной кислотой 96
фенилмеркаптаном 96
Гексаэтилдиплюмбан 547, 556, 628
диспропорционирование 595, 596
диссоциация 612
окисление 612
реакции с
ал килами галоидными 615
ацетилом хлористым 614
бензоилом хлористым 614
бромом 613
гидроперекисью /г-диизопропилбензола
612
гидроперекисью изопропилбензола 612
диэтилоловом двухлористым 613
диэтилсвинцом двубромистым 596
железом треххлористым 613
золотом треххлористым 613
медью хлористой 613
медью хлорной 613
перекисями ацилов 613
ртутью хлористой 613
серебром азотнокислым 613
серой двухлористой 613
сулемой 613
сульфурилом хлористым 613
тетраацетатом свинца 613
тионилом хлористым 613
триэтилоловом хлористым 613
хлором 613
термическое разложение 597
Гексаэтилдиплюмбоксан 601
реакции с
органическими гидроперекисями 605
спиртами 605
Гексаэтилдистаннан 186, 306, 308, 477
диспропорционирование 376, 377
реакции с
азодиизобутиронитрилом 444
арилами галоидными 444
бензилом хлористым 443
бензоилом хлористым 444
бензолсульфохлоридом 444
Р-бромэтилбензолом 443
mpem-бутилпербензоатом 442
1,4-дибромбутаном 443
1,5-дибромпентаном 443
кислородом, механизм реакции 438, 439
метилом йодистым 443
нитрозоацетанилидом 445
перекисями ацилов 442
тетраацетатом свинца 445
/г-толуолсульфохлоридом 444
трифенилхлорметаном 443
циклогексильным эфиром надугольной
кислоты 442
четыреххлористым углеродом 443
фотохимические реакции 443
Гексаэтилдистанноксан 383
реакции с
ацетилацетоном 399
ацетиленовыми ацеталями спиртов 326
ацетиленовыми спиртами 326
«-бутилоловом треххлористым 420
Ь(бутин-З-окси)- 1-бутоксиэтаном 327
винилбутин-1-иловым эфиром 327
диметилкремнием, бш>(трифтор
ацетатом) 420
дифенилкремнием двуфтористым 420
диэтилоловом двухлористым 420
додецилкремнием трехиодистым 420
кислотами 396, 397
кремнием, (метокси)триизоцианатом 420
кремнием четырехбромистым 420
меркаптанами 399
метилкремнием треххлористым 420
метилпропиолатом 327
З-метоксипропином-1 327
оловом бромным 420
оловом двухлористым 420
оловом хлорным 420
перекисью водорода 398
сероводородом 397
сурьмой треххлористой 420
триизопропилгерманием, гидроокисью
420
триметилкремнием, ацетатом 420
триметилкремнием, изотиоцианатом 420
три-я-пропилкремнием фтористым 420
триэтилкремнием цианистым 420
фенилацетатом 420
фосфором треххлористым 420
этилацетатом 420
Гексилгерманий треххлористый 30, 60
«-Гексилдивинилолово бромистое 340
Гексилендио ксидибутилолово 417
«-Гексилтривинилолово, реакция с бромом
340
«-Гексилтрифенилолово 224
N-Гексилформамидотриэтилолово 416
Гептил герма ний
треххлористый 30
тригидрид, присоединение к
винилбутиловому эфиру 66
гептину-1 65
«-Гептилтрифенилолово, перераспределение
радикалов 379
«-Гептилтриэтилгерманий 35
Германий двуиодистый
присоединение к
Предметный указатель
669
ацетилену 71
толану 71
реакции с
2,3-диметилбутадиеном 72
изопреном 72
Германий четыреххлористый, реакция с
метиловым эфиром триалкилстаннилуксус-
ной кислоты 56
Германий-оловоорганические соединения 488,
489
Германийорганические алкоксисоединения
103
Германийорганические гидроокиси, реакция
с органическими кислотами 96
Германийорганические перекиси 102 и ел.
Германо-5-сшг/:о-(4,4)-нонан 28
Германо-6-с/шро-(5,5)-ундекан 28
Гермилтетрауксусная кислота, метиловый
эфир 57
Гидрид германия треххлористого
присоединение к
акрилнитрилу 63
алкилзамещенным циклопропана 62
аллилацетату 63
аллилгерманию треххлористому 64
аллилкремнию треххлористому 64
аллилу бромистому 63
аллилу хлористому 63
я-амилциклопропану 62
анизолу 62
арилзамещенным циклопропана 62
ацетилениду триметилгермания 64
ацетилену 61
бутену-1 61
винилацетату 63
винилу хлористому 62
галоидным алкилам 62
гексену-1 60
диеновым соединениям 61
трд«с-дихлорэтилену 63
изопрену 61
металлилу хлористому 63
1-метил-2-«-бутилциклопропану 62
метилвинилгерманию двухлористому 64
1-метилнафталину 62
нафталину 62
октену-1 61
пипирилену 62
производным нор корана 62
пропаргилу бромистому 63
пропаргилу хлористому 63
пропилену 61
стиролу 61
тетрахлорэтилену 63
фенетолу 62
фенилциклопропану 62
циклопентадиену 62
этилену 60
реакции с
аллилом йодистым 63
металл ил ом бромистым 63
тетраметилоловом 129
тетраэтилоловом 129
тетраэтилсвинцом 129
эфират, присоединение к ацетилену 61
Гидриды ал кил германия
присоединение к
аллиламину 67
ацетилену 65
гексину-1 65
фенилацетилену 65
реакции с
водой 131
галоидами 134
галоидными ал килами 136
галоидоводородными кислотами 134
гидрохиноном 132
гликолями 132
диазоацетоном 75
диазоацетофеноном 75
диазоуксусным эфиром 75
кислородом 129
меркаптанами 132
меркур-бис-ацетальдегидом 138
металлоорганическими соединениями 137
минеральными кислотами 135
ненасыщенными альдегидами 133
ненасыщенными кетонами 133
органическими кислотами 132
спиртами 131
фенолами 132
хлорангидридами кислот 134
щелочами 135
щелочными металлами 147
эфирами меркур-бис-уксусной кислоты
138
Гидростаннирование
азобензола 306
азометинов 305
акриламида 285
акриловой кислоты 283
акрилонитрила 274., 275, 285
алкенов, механизм реакции 275, 276
алкилиденмалоновых эфиров 284
алкилпропиолатов 292, 294
алкинов, механизм реакции 290—301
аллилацетата 284
аллилацетона 282
аллилового спирта 281
альдегидов 301
альдегидов и кетонов, механизм реакции
304
анисового альдегида 301
арилизоцианатов 308
ацетилена 287, 290
ацетона 304
ацетофенона 303
бензальанилина 310
бензальдегида 301, 303
бутадиена 279
2-бутена 279
«-бутилвинилового эфира 281
винилацетата 284
виниловых эфиров 281
винилтрифенилгермания 286
дициклогексилкарбодиимида 305, 310
диэтилацетилендикарбоксилата 288
диэтилкетона 303
диэтинилбензола 291, 514
изобутилвинилового эфира 281
изомасляного альдегида 301, 303
изотиоцианатов, механизм реакции 307
изоцианатов, механизм реакции 307
кротононитрила 285
«-масляного альдегида 301
метакриламида 285
метила кр и л ата 283, 284
метилацетилена 290
670
Предметный указатель
метилвинилкетона 282
метилметакрилата 283, 284
метилпропиолата 290, 297
Р-метилстирола 279
метилтетролата 288
метилэтилкетона 304
ненасыщенных спиртов 281
ненасыщенных эфиров 515
нитрозобензола 310
1,8-нонадиина 290
ж-оксибензальдегида 302
октадиена 278
я-октена-1 278, 279
пропаргилового спирта 290, 291
салицилового альдегида 302, 303
стирола 274, 275, 278, 279
тетрафторэчилена 280, 281
три-я-бутилэтинилгермания 289
три-«-бутилэтинилкремния 288
три-«-бутилэтинилолова 288, 289
В-тривинил-]М-трифенилборазина 286
триметил-/г-винилфенилолова 286
три-«-пропилэтинилолова 288
трифенил-/г-винилфенилгермания 286
трифенил-/г-винилфенилолова 286
трифенил-я-винилфенилсвинца 286
трифторацетофенона 304
трифторбромэтилена 280, 281
триэтилэтинилкремния 288
фенялацетилена 288, 290, 293, 515
фенилизотиоцианата 309
фенилизоцианата 308
фурфурола 303
хлораля 301
цианистого аллила 285
цианоацетилена 292, 294, 297
циклогексанона 303
циклогексена 277
циклопентена 277
Элилвинилового эфира 281
этилдиацетилена 290
этилена 279
этилкротоната 277
этилового эфира «-ундециленовой кислоты
283
этилтетролата 292, 296, 297
этоксиэтина 293, 296
эфиров акриловой кислоты 283
Декагидро-9-нафтилперокситрибутилолово
415
Декагидро-9-нафтилперокситрифенилолово
416
Декаметилтетрастаннан 505
«-Децилдивинилолово бромистое 340
«-Децилтривинилолово, реакция с бромом
340
«-Децилтрифенилгерманий 151
/г-Диазофенилтрифенилсвинец, азосочетание
с ^-нафтолом 618, 619
бис-(Диалкиламино)диалкил(или арил)-
олово 4С9
Диалкиламинотриалкилолово 409
присоединение к
бензонитрилу 315
двуокиси серы 315
двуокиси углерода 315
ди-/г-толилкарбодиимиду 315
кетену 314
сероуглероду 315
N-тиоанилину 315
фенилизотиоцианату 315
фенилизоцианату 315
реакции с
аминами 418
ацетонитрилом 328
ацетоном 328
нитрометаном 328
Диалкиламинотриарилолово 409
Диалкиламинотриметилолово, реакция с
аминами 418
Диалкилгерманий
диалкилсульфонаты 110
диацилаты ПО
реакция с эфирами меркур-б&с-уксус-
ной кислоты 138
иодгидрид, реакция с эфирами меркур-бис-
уксусной кислоты 138
окись, реакции с
ангидридами органических кислот 97
органическими кислотами 97
сульфаты ПО
хлоргидрид, реакция с эфирами меркур-
бис-уксусной кислоты 138
Диалкилдиарилолово 233
Диалкилдиацетилениды олова 238
бис-(0,0-Диалкилдитиофосфат)диалкилолова
412
0,0-Диалкилдитиофосфат триалкилолова 412
Диалкилолово
(алкилмалеат)ацилаты 402
(алкокси)ацилаты 409
(галоид)ацилаты 423
(галоид) гидр иды 462
присоединение к олефинам 277
(гидрокси)нитрата 400
двубромистое 182, 332
двугалоидное
алкилирование до моногалогенидов
реактивом Гриньяра 234, 238
гидролиз 381 и ел.
с заместителями в радикале 183, 184, 341,
342
реакции с
алкоголятами щелочных металлов 408
дилитийорганическими соединениями
249—251
дитиолами 414
ксантогенатом натрия 409
литийорганическими соединениями 249
меркаптидами металлов 409
натрийорганическими соединениями
253, 256
солями органических кислот 407
сульфонамидом натрия 410
двуиодистое 181, 406
двуфтористое 405
двухлористое 332, 400
реакции с
диалкиламидом лития 409
калием фтористым 405
натрием 497
натрием йодистым 405, 406
натрием роданистым 406
8-оксихинолином 414
согидролиз с хлористым триметилкрем-
нием 391
Предметный указатель
671
диацилаты 401, 407, 433
реакции с
алкоксисоединениями кремния 516
алкоксисоединениями олова 422, 516
органическими кислотами 417
ди(/г-винилбензоат), полимеризация 513
дигидриды 458—460
реакции с
диазометаном 323
триалкилалюминием 487
триал кил-(Ы-фенилформамидо)оловом
482
диизотиоцианаты 406
комплексы 433
кса нто ген а ты 409
меркаптиды 404, 409
бис-(метилсульфонаты) 406
нитраты 400
окись 388—391
реакции с
алкилгидроперекисями 402
алюминийорганическими
соединениями 260
галогенидами металлов 419
галоидангидридами органических
кислот 402
диацилатами диалкилолова 419
дигалогенидами диалкилолова 419
дигидрофенолами 404
диолами 403
кислотами 400—402
меркаптанами 404
моноэфирами глицерина 403
реактивом Гриньяра 239
сероводородом 400
спиртами 402
сульфидом натрия 420
триацилатами алкилолова 419
тригалогенидами алкилолова 419
фенолами 403, 404
фосфорной кислотой 400
фосфорным ангидридом 400
хлорокисью фосфора 400
эфирами арсоновои, фосфорной или
борной кислот 400
сульфид 400, 406, 421
реакции с
алкилоловом трехгалоидным 420
алюминийорганическими
соединениями 260
диацилатами диалкилолова 420
дигалогенидами диалкилолова 420
диизоцианатами диалкилолова 420
диметоксидиалкилоловом 420
оловом хлорным 420
б^с-(сульфонамиды) 410
Диалкилолово, гексамер 482
Диалкилолово, полимер 481
диспропорционирование 195, 376
окисление 438
реакции с
алкилгалогенидами 442
галоидами 439
органическими перекисями 441
серебром азотнокислым 441
сулемой 441
хлором 439
Ди-тре/я-алкилолово двугалоидное,
реакции с
mpem-бутилмагнием хлористым 234
реактивом Гриньяра 222
Диалкилперокситетраалкилдистанноксаны
402, 413
Диалкилсвинец
железокарбонилы 630
нитрат 585
Диалкилстаннандиолы 403
2,2-Диалкил-2-станна-1,3,6-триоксацикло-
октан 403
Диалкилстаннилендинатрий 497
Диалкилциклотриметиленгерманий, реакция
с галоидоводородными кислотами 82
Диалкоксидиалкилгерманий, реакции с
гликолями 111
органическими кислотами 111
Диалкоксидиалкилолово 408, 417
присоединение к кетену 312
реакции с
N-алкиламидами алкилсульфокислот 416-
органическими кислотами 416
Диалкокситетраалкилдистанноксаны,
реакция с меркаптанами 418
Диаллилдиметилгерманий 39
Диаллилдифенилолово 235
реакции с
бромом 340
хлористым водородом 350
термическая устойчивость 379
Диаллилолово двубромистое 183, 340
Ди-трет-амилолово
двубромистое 213
дву хлор истое 213
дигидроокись 213, 389
Ди-/г-анизилолово
двубромистое 334
дву хлор истое 334
Ди-о-анизилсвинец
двухлористый, диспропорционирование 597
диацетат 564
Ди-я-анизилсвинец
двуиодистый 613
двухлористый 580
Диарилокситетраалкилдистанноксаны 403
Диарилолово
окисление 438
реакции с
галоидами 439, 440
галоидными арилами 442
органическими перекисями 441
серебром азотнокислым 441
серой 439
сулемой 441
Диарилолово
двугалоидное, получение через диазосое-
динения 319
реакции с
дитиолами 414
меркаптидами металлов 409
двуиодистое 406
двуфтористое 405
двухлористое, реакции с
диалкиламидом лития ^09
натрием 497
натрием йодистым 405, 406
натрием роданистым 406
8-оксихинолином 414
сульфидом натрия 406
димеркаптиды 409
672
Предметный указатель
окись, получение через двойное иодоние-
вые соли двухлористого олова 320
реакции с
сероводородом 400
тионилом хлористым 400
эфирами арсоновой, борной или
фосфорной кислот 400
сульфид 406
Диарилсвинец двугалоидный, реакция с фе-
ниларсином 582
Диарилстаннилендинатрий 497
Диацетиленид трифенилсвинца 562
Дибензилдибутилгерманий 35
Дибензилдифенилолово, реакции с
N-бромсукцинимидом 344
хлористым' водородом 347
Дибензилолово
двубромистое 200
двухлористое 185, 200, 334, 347, 400
реакция с едким кали 389
дигидрид 479
Дибензостаннолы, 9,9-дизамещенные 250
Ди-/г-бифенилолово
двубромистое 334
двуиодистое 197
двухлористое 334
дигидрид 461
Ди-л-бифенилолово, полимер 197, 480
Ди(бифенил-2)-олово двубромистое 348
Ди(бромметил)олово двубромистое 321
Ди-л-бромфенилолово
двубромистое 201
двуиодистое 406
двухлористое 201
окись 390
Дибутилаллилово бромистое 340
Ди-я-бутил(аллилфенил)олово йодистое 239
Ди-я-бутилбарениленолово, димер 250
Ди-я-бутилвинилолово
бромистое 340, 347
гидроокись 383
йодистое 341
хлористое 239, 346
Ди-я-бутил германий
двуиодистый 53
двухлористый 84, 86, 96
дигидрид 123
присоединение к
винилметилкетону 67
фенилацетилену 65
диизоцианат 99
иодгидрид, присоединение к октену-1 69
хлоргидрид 128
присоединение к
акрилнитрилу 70
ал лил ацетату 70
аллиловому спирту 70
аллилу хлористому 70
винилбутиловому эфиру 70
диметил-2,3-бутадиену-1,3 69
диэтилацетальакролеину 70
метакролеину 70
метилакрилату 70
метилметакрилату 70
октену-1 69
Ди-я-бутилди ал кил олово 233
Ди-я-бутилдиаллилолово 233
реакция с бромом 340
Дибутйлдиацетилениды олова 238
Ди-я-бутилди-гарет-бутилолово 234
Ди-я-бутилдивинилолово 233, 235
реакции с
галоидами 340, 341
галоидоводородами 347
мышьяком трехбромистым 357
Ди-я-бутилди(диметилсилилметил)олово 233
Дибутил-бис-(диметилсил ил метил) олово 239
Ди-я-бутилдинеопентилолово, реакция с
бромом 340
Ди-я-бутилди(пентафтор этил) олово 234
реакция с трифторуксусной кислотой 352
Ди-я-бутилди-4-пентенилолово 233
Ди-я-бутилдиперфторвинилолово 226
реакции с
бромистым водородом 347
этанолом 353
Дибутилди(триметилсилилметил)олово 233
реакции с
бромистым водородом 347
галоидами 342
Ди-я-бутилдифенилолово 233
Ди-трет-бутилдифенилсвинец 563
Ди-я-бутилдициклоамилолово 233
Ди-я-бутилдициклогексилолово 233
Ди-я-бутилдициклопропилолово 233
Ди-я-бутилдиэтинилгерманий 51
Ди-я-бутилизоамилолово йодистое 340
Дибутил(карбэтоксиметил)германий, гидрид
75
Ди-я-бутилолово
ацетилацетонат 417
ацетиленид, полимер 327
(бром-)ацетат 466
гидролиз 381
(гидрид)ацетат 462
гликоляты 417
двубромистое 182, 332, 347
двуиодистое 180, 189, 400
двуфтористое 405
двухлористое 184, 187, 211, 212, 332, 335,
400
диспропорционирование 378
согидролиз с триметилкремнием
хлористым 391
диацилаты 401, 402, 466, 491
дигидрид 458, 460
присоединение к
гексафторацетону 304
тетрафторэтилену 281
три-я-бутилэтинилолову 289
реакции с
аллилмеркантаном 281
аллиловым спиртом 281
гексабутилдистанноксаном 481
диацетатом дибутилолова 467
бш>(диметиламино)диалкилгерманием
488
диметиламинотрибутилгерманием 488
диметоксидибутилоловом 483
а,Р-ненасыщенными карбонильными
соединениями 473
окисью ди-я-бутилолова 481
органическими кислотами 466
соляной кислотой 464
перекисью бензоила 491
фотораспад 463
дигидроокись 473
Предметный указатель
673
диизотиоцианат 406
дилауринат 401
(меркапто)ацилаты 404
бш;-(метансульфонат) 350
окись 391
реакция с хлористым винилмагнием 235
термостабильность 513
селенид 359
сульфид 359, 421
бш>(я-толуолсульфонат) 400
(хлор)бромид 422
(хлор)гидрид 462
(хлор)меркаптид 422
Ди- н-бутил олово
гексамер 478, 479
полимер 187, 193, 194, 473
реакции с
аллилом бромистым 442
бензилом бромистым 442
бромбензолом 442
я-бутилом бромистым 442
гидроперекисями 445
1,4-дибромбутаном 443
1,2-дибромэтаном 443
дибутилоловом двухлористым 441
кислотами 445
оловом йодистым 441
трифенилхлорметаном 442
Ди-втор-бутилолово, окись,
термостабильность 515
Ди-трет-бутилолово
двубромистое 212, 400
дву хлор истое 213, 400
дигидроокись 213, 389
окись
степень полимеризации 391
термосгабильность 515
Ди-трет-бутилолово, тетрамер, реакция
с иодом 440
Дибутилоловоди(кобальттетракарбонил) 507
Ди-«-бутилсвинец
дигидрид 620
сульфат 605
Ди-я-бутил-бис-(тетрафторэтил)олово 281
Ди-я-бутил(тетрафторэтил)олово, гидрид 281
Ди-я-бутил-бис-ф-три-я-бутилстаннилвинил)-
олово 289
Ди-я-бутил-б«с(триметилсилилметил)олово
240
Ди-трет-бутилфенокситетрабутилдистаннок-
сан 404
Дибутилциклотриметиленгерманий 51, 53
Ди-я-бутилэтинилгер маний 51
Дивинил-я-бутилолово хлористое 242
Дивинилди-(2-метил-3-бутенил)олово 233
Дивинилдиперфторвинилолово 226
реакция с хлористым водородом 347
Дивинилдифенилсвинец 561
Дивинилолово
двубромистое 202
двухлористое 202, 333
диизотиоцианат 406
фенилфосфонат 412
Дивинилсвинец двухлористый 576
Дивинил-бис-(триметилсилилметил)олово
240
Ди-о-галоидбензилолово 197
Ди-я-гексадецилолово двуиодистое 189
Ди-я-гексил олово
двубромистое 182
двухлористое 333
окись 388
Ди-я-гептилолово двубромистое 182
Ди-я-додецил олово двуиодистое 181
Диизоамилаллилфенилолово йодистое 239
Диизоамилсвинец двухлористый 576
Диизобутилолово
двухлористое 333, 335
диацетат 407
дигидрид 458
присоединение к
алкенам 285
альдегидам 303
диеновым соединениям 276
Диизобутилсвинец двухлористый 576
Диизобутоксидиизобутилолово 303
Диизопропенилолово двубромистое 199, 202
Диизопропилгерманий
двубромистый 95
двуиодистый 95
двухлористый 95
дигидрид 126
диизоцианат 99
Диизопропилолово
бш>(метансульфонат) 350
ди мо но хлор ацетат 351
Диизопропилсвинец 545
Ди-/г-иодфенилолово
двубромистое 201
двуиодистое 406
двухлористое 201
реакция с хлором 454
окись 390
Ди-(2-карбоксиметилпропил)диэтилолово
284
Ди-(2-карбоксиметилэтил)диэтилолово 284
Ди-(2-карбоксиметилэтил)дифенилолово284
Ди-(-2-карбоксиметилэтил)диэтилолово 284
Ди-(2-карбоксиметил)олово
двубромистое 183
двуиодистое 182
Ди-я-карбэтоксифенилолово
двухлористое 201
окись 320
Ди-/г-карбэтоксифенилсвинец, диацетат 564
Дикротилолово двубромистое 183
Ди-/г-ксилилолово 197
Дилаурилолово
двухлористое 212
окись 389
Дилитийдифенилолово 496
реакция с серой 500
Димезитилдиарилолово 237
Димезитилсвинец двубромистый 555
Димеркаптотетраалкилдистанноксаны 418
Диметил-я-амилолово, перфторацилаты 398
бш> (Диметиламино)ди-я-бутилолово,
реакция с ацетиленом 327
(Диметиламинометил)триэтилолово 447
[3-Диметиламино-6-(4'-нитрофенилазо)фе-
нил]трифенилолово 454
Диметиламинотри-я-бутилолово, реакции с
ацетиленом 327
циклопентадиеном 328
Диметиламинотриметилолово
присоединение к кетену 314
реакции с
алкинами 327
43 Заказ № 207
674
Предметный указатель
аминами 418
диазометаном 315
трихлорэтиленом 328
хлороформом 328
циклопентадиеном 328
Диметиламинотрифенилолово, реакция с
инденом 328
фенилацетиленом 327
циклопентадиеном 328
Диметиламинотриэтилолово
присоединение к изопропенилацетату 314
реакция с фенилацетиленом 327
я-Диметиламинофенилтрифенилгерманий 49
(Диметиламинофенил)трифенилолово,
реакция азосочетания 454
Диметил-5-бромамилолово бромистое 345
Диметил-бш>(2-бромтрифторэтил)олово 281
Диметил-бш>(2-бутенил)олово 279
Диметил-(2-бутенил)олово, гидрид 279
Диметил-я-бутилолово, перфторацилаты 398
Диметилвинилолово
бромистое 347
йодистое 341
хлористое 347
Диметилгерманий
двубромистый 80
двухлористый 20
гидролиз 89, 90
реакции с
азидом натрия 108
метиламином 107
хлорирование 117
окись 91
селенид 106
сульфид 105
Диметил-я-децил олово бромистое 340
Диметилдиалкилолово 233
Диметилдиаллил германий, полимеризация
159
Диметилди-трет-амилолово 234
Диметилдиамилсвинец 560
Диметилди-трет-бутилолово 234
Диметилди-я-бутилсвинец 560
Диметилдиви нилолово 233, 235
Диметилдиизопропилсвинец 560
Диметилдимезитилолово 249
Диметил-бис-(диметилсилилметил)олово 239
Диметил-бмс-(диметилсилилпропил)олово
239
Диметилди-я-октилолово 238
Диметилдиперфторвинилолово 226
реакция с водой 364
Диметилди-я-пропилгерманий 35
Диметилди-я-пропилсвинец 560
Диметилди-я-тол ил олово 249
Диметилди(триметилсилилметил)олово 233
Диметилди-(3,4,5-триметилфенил)олово 249
Диметилди(фенилэти нил) олово 238, 254
Диметилдициклогексилолово 233, 236
Диметилди-(2-этилгексил)олово 238
Диметилдиэтилгерманий 36
Диметилдиэтилолово 279, 323
Диметилдиэтилсвинец 560
Диметил-я-додецилолово бромистое 340
Диметил-я-октилолово бромистое 340
Диметилолово
арсенат 412
бис-(ацетилацетонат) 404
вольфрамат 412
двубромистое 332, 338
двуиодисюе 181, 339, 406
двухлористое 184, 187—189, 203, 354, 400
комплексы 432
реакции с
диметил-бис-(пентафторфенил)оловом
331
оловом хлорным 336
термическое разложение 365
диацетат 401
дибензоат 413
дигидрид 457, 465
присоединение к
бутадиену 279
гексафторацегону 304
тетрафторэтилену 280
трифторбромэтилену 280, 281
этилену 279
реакции с
2-бутеном 279
1,1-дифторэтиленом 280
натрием 407
-трифторэтиленом 280
диизотиоцианат 406
диизоцианид 406
димеркаптиды 414
ди(меркаптоацетат) 413
диметакрилат 401
диметафосфат 412
ди(монобромацетат) 351
диформиат 401, 407
молидбат 412
окись 388
салицилат 407
селенид 406, 413
сульфат 400
сульфид 413, 421
бис-(фениларсонат) 412
фенилдифосфат 412
фенилфосфат 412
фосфаг 412
фталат 407
(хлор)ацетат 402
гидролиз 381
Диметилолово
пентамер 505
полимер 477
диспропорционирование 377
окисление 438
Диметилоловоди(кобальттетракарбонил) 507
Диметилоловоди(марганецпентакарбонил) 507
3,3-Диметилпентаметилендибутилолово 259
био(3,3-Диметилпентаметилен)олово 258
Диме гил (пентафторфенил)олово хлористое
249, 331
Диметил-бш>(пенгафторэтил)олово 234
Диметил-я-пропилизоамилсвинец, реакция
с бромом 576
Диметилсвинец
двубромистый 576
двухлористый 576
дигидрид 620
Диметилсвинец-бис-марганецпентакарбонил
629
1 ,1-Диметил-1-станнациклогексан, реакция
с бромдихлорметилртутью 455
Диметилстаннилендинатрий 465, 497
силш-Диметилтетра-я-амилдистанноксан 383
Предметный указатель
675
силш-Диметилтетра-я-бутилдистанноксан 383
1,1-Диметил-2,3,4,5-тетрафенилстаннол 250
реакции с
галоидами 345, 346
уксусной кислотой 352
Диметил-бис-(тетрафторэтил)олово 280
Диметил-(тетрафторэтил)олово, гидрид 280
диспропорционирование 280
реакция с хлором 464
Диметил-бис-(триметилсилилметил)олово 240
Диметил(триметилсилилметил)олово
бромистое 342
Диметилтрифтор метил олово хлористое 340
реакции с
водой 364
хлором 340
Диметил(трифторэтил)олово, гидрид 280
Диметил хлор мети л германий хлористый 47
Диметилциклогексилолово йодистое 341
Диметилциклопентилолово 233
Диметилэтилолово
гидрид 279
йодистое 340
хлористое 346, 354
Диметилэтилсвинец
бромистый 576
хлористый 597
Диметоксидиал кил олово, реакции с
бензоилом фтористым 416
спиртами 417
Диметоксидибутил герма ний 103
Диметоксидиметилолово 408
присоединение к кетену 312
Ди-я-метоксифенил-бис-1-нафтилолово 237
2,5-Диметоксифенилсвинец, триацетат 573
Ди-я-метоксифенил-бис-тиенилолово 237
Динатрийгексаметилтристаннан 504
Динатрийтетраметилдистаннан 497
Ди-а-нафтил олово
двубромистое 201, 202
двухлористое 200, 201, 334
реакция с хлорным оловом 337
дигидрид 461
Ди-а-нафтил олово, полимер 197, 480
Ди-Р-нафтилолово двухлористое 201
Ди-(3-нафтилолово, полимер 480
Ди-а-нафтилсвинец, диацетат 564
Ди-1 - нафтил-бис-тиенилолово 237
Ди-(3-нафтокситетрабутилдистанноксан 404
Динеопентилолово двубромистое 340
Ди-;и-нитрофенилсвинец двуиодистый 608
Ди-я-нонилолово двубромистое 182
Ди-(о-оксибензилокси)диизобутилолово 303
Ди-я-октилолово
двубромистое 182
двуиодистое 189
двухлористое 333
окись 389
сульфид 421
Ди(пентафторфенил)диалки(или арил)олово
237
Ди(пентафторфенил)дивинилолово 237
Диперфтор вини л дифенил олово 226, 235
реакция с хлористым водородом 348
Диперфторфенилолово двухлористое 218
Дипропенилолово двугалоидное 199, 202
Ди-я-пропилаллилолово йодистое 239
Дипропилбутилолово йодистое 340
Ди-я-пропилгерманий
двубромистый 96
двуиодистый 96
двуфтористый 96
двухлористый 96
диацетат 97
Ди-я-пропилдиаллилолово 233, 239
Дипропилдиацетилениды олова 238
Ди-я-пропилдифенилолово 233
Ди-я-пропил олово
двубромистое 182, 332
двуиодистое 339
двухлористое 187, 332
диспропорционирование 378
согидролиз с хлористым тримети л
кремнием 391
дигидрид 458
диизотиоцианат 406
диизоцианид 406
бис-(метилсульфонат) 400
бис-(этилсульфонат) 400
Ди-я-пропилсвинец
двубромистый 580
двухлористый 576, 579, 580
сульфат 580
Ди-я-пропилстаннил-бис-уксусная кислота,
диэтиловый эфир 312, 486
Дипропилхлорстаннилуксусная кислота,
метиловый эфир 487
Дипропоксидиэтилолово 312
Ди-а-стильбенил олово
двухлористое 200
окись, реакция с диоксандибромидом 344
Ди-Р-стирилолово двухлористое 200
Ди-а-тиенилсвинец, диизобутират 584
Ди-2-тиенил-2-селенилсвинец 561
реакция с хлористым водородом 580
Ди-/2-толилгерманий, окись 54
Ди-jw -тол ил олово
двухлористое 334
окись 390
сульфид 406
Ди-о-толилолово двухлористое 200, 334
Ди-/2-толилолово
двубромистое 201
двухлористое 200, 201, 202, 334
дигидрид 461
Ди-/2-толилолово, полимер 197, 480
Ди-/г-толилсвинец 554
двуиодистый 577
диацилаты 583
динитрат 583
окись 583
Ди-(1,2,4-триазолил)дибутилолово 411
Ди-(1,2,4-триазолил)диэтилолово 410
Ди-3,5,5-триметил-я-гексилолово
двухлористое 333
окись 389
Ди-(гриметилсилилметил)олово
двубромистое 347
двуиодистое 342
окись 390
сульфид 406
Ди(трифенилплюмбил)метан 627
Ди-(9-фенантрил) олово 197, 198
окисление 438
реакции с
иодом 440
сулемой 441
Ди-(9-фенантрил)олово двухлористое 348
43*
676
Предметный указатель
Ди-(я-фенетил)олово двухлористое 334
Дифенил-(я-аллилфенил) (я-2,3-дибромпро-
пилфенил)свинец 617
Дифениларсинотриалкилолово 411
Дифенил-бис-(2-бифенил)олово 249
Дифенилгерманий 78
двубромистый 24, 81
гидролиз 91
реакция с сульфидом натрия 106
двуиодистый 96
двухлористый 86
Дигидрид 126
реакции с
диэтил ртутью 139, 154
фениллитием 148
окись 92
сульфид 96
хлоргидрид 129
Дифенилдиалкилолово 236
Дифенилдиаллилгерманий 40
Дифенилдиаллилолово 236
Дифенилдиаллилсвинец 561
Дифенилди-(я-аллилфенил)свинец 617
Дифенилдиарилолово 236
Дифенилдиацетилениды олова 238
Дифенилди(бенз-а-антрацен-7-ил)свинец 562
9,9-Дифенилдибензоплюмбол 563
Дифенилди-(я-бромфенил)германий 38
Дифенилдивинилгерманий 40
Дифенилдивинилолово 236
Дифенилди-(я-винилфенил)олово 231
Дифенилди-(я-винилфенил)свинец 561
Дифенилди-«-гексилгерманий 38
Дифенилди-«-гептилгерманий 38
Дифенилди-«-гептилолово 236
Дифенилди-«-децилгерманий 38
Дифенилди-(я-диметиламинофенил) олово 247
Дифенилди(дифенилметил)олово 253
Дифенилдиизоамилгерманий 38
Дифенилдиизобутилгерманий 38
Дифенилди(изопропилфенил)германий 37
Дифенилдиинденилгерманий 36
Дифенилди-(1-инденил)олово 232, 236, 238
Дифенилди-3-инденилсвинец 562
Дифенилди-2,5-ксилилсвинец 561
Дифенилди(я-метоксифенил) гер маний 37
Дифенилди-а-нафтилсвинец 561
Дифенилди-«-нонилгерманий 38
Дифенилди-(л*-оксифенил)олово 237, 247
Дифенилди-(о-оксифенил)олово 247
Дифенилди-«-октилгерманий 38
Дифенилди(пентен-4-ил)олово 236
Дифенилди-«-пропилгерманий 38
Дифенилди-(а-тиенил)олово 236, 249
Дифенилди-2-тиенилсвинец 561
Дифенилди-о-толилгерманий 37
Дифенилди-о-(или л)-толилсвинец 562
Дифенилди(триметилсилилметил)олово 236
Дифенилди-(#-триметилсилилфенил)олово
236
Дифенилди-(9-фенантрил)свинец 561
реакция с хлористым водородом 580
Дифенилди-(я-феноксифенил)германий 38
Дифенилди-9-флуоренилсвинец 562
Дифенилди-2-фурилсвинец 561
Дифенилдициклоалкилолово 236
Дифенилди(циклогексилфенил)свинец 561
Дифенилди-(1-циклопентадиеиил)олово 236,
238
Дифенилдиэтил гер маний 36
Дифенилди-(я-этилфенил)германий 37
Дифенилди-(л-этоксифенил)германий 38
бис- (Дифени л метил) олово двухлористое 347
Дифенилолово, гексамер 479, 480
реакция с иодом 440, 479
Дифенилолово
ацетилацетонат 409, 434
(бром)гидрид 464
двубромистое 333, 343
двуиодистое 197
диспропорционирование 378
двухлористое 200, 202, 203, 334, 344, 354
диспропорционирование 377, 378
реакции с
ацетилацетоном 414
я-дилитийбензолом 515
литием 496
нафталин натрием 504
серой 361, 362
диацетат 402, 421
дибензоилацетонат 409
дигидрид 456, 461, 466
присоединение к алкенам 284
реакции с
винилтрифенилкремнием 287
винилтрифенилсвинцом 287
диметилкадмием 485
дифенилдистирилсвинцом 617
метанолом 480
метилвинилкетоном 473
а,р-ненасыщенными карбонильными
соединениями 473
органическими кислотами 466
перекисью бензоила 490
дипропионат 402, 421
дифульминат, комплексы 433
окись 320, 389
сульфид 360, 406, 421
реакция с серой 361
Дифенилолово, пентамер 480
Дифенилолово, полимер 194, 196, 197, 473,
480
диспропорционирование 197, 377
окисление 438
присоединение к тетрафторэтилену 316
реакции с
азотной кислотой 438
ал килами бромистыми 443
ал килами йодистыми 443
бензолазотрифенилметаном 442
бензохиноном 444
бромом 440
дибензилсульфидом 444
дифенилоловом двухлористым 441
иодом 440
нитрозоацетанилидом 442
оловом хлорным 441
перекисью бензоила 441
серой 439
сулемой 441
фениллитием 498
хлором 440
Дифенилоловоди(кобальттетракарбонил) 507,
508
Дифенилоловоди(марганецпентакарбонил) 507
реакции с
хлористым водородом 509
хлором 509
Предметный указатель
677
Дифенилоловоди(ренийпентакарбонил) 411,
506
Дифенил-2-пиридилсвинец йодистый 563
Дифенилсвинец 554
азид 605
ал килдиар сенат 582
(гидрокси)ацетат 603
(гидрокси)цианид 608
двубромистый 608
двуйодистый 563, 608
двухлористый 579, 580, 585, 614
диспропорционирование 597
диацетат 564, 585, 609, 614
нитрование 618
реакция с димагниевым производным
бромистого метилена 561
дибензоат 609
дигидроксикарбонат 608
диизобутират 583
реакция с тетраизобу тир атом свинца
585
диметакрилат 609
димонохлорацетат 609
динитрат 582, 590, 618
дистеарат 609
дитиоцианат 608
быс-(дифенилфосфинат) 610
сульфид 608, 612
бш>(тиоацетат) 583
фосфат 608
хромат 608
Дифенилсвинецди(кобальттетракарбонил)
629
Дифенилсвинецди(марганецпентакарбонил)
629
реакция с хлором 629
Дифенилстаннил-бш>(литийсульфид) 500
реакция с хлористым бензоилом 500
Дифенилстаннилтеграфгорэтан 316
бш>(1,3-Дифенилтриазин)диметилолово 411
1,3-Дифенилтриазинтрифенилолово 411
Дифенил(триалкилстаннил)арсины 411, 417
окисление 423
реакция с йодистым метилом 423
Дифенил(триалкилстаннил)стибины 411
окисление 423
реакция с йодистым метилом 423
Дифенил(триалкилстаннил)фосфины 415, 417
реакция с йодистым метилом 423
Дифенил(трифенилстаннил)фосфин,
присоединение к
стиролу 316
фенилацетилену 317
хлористому аллилу 316
Дифенил-бш;-[2 -(трифенилстаннил)эгил]-
кремний 287
Дифенил-(9-флуоренил)олово 249
Дифенилхлоралкиларсенаг свинца 582
Дифенилциклопенгаметиленгерманий 37
Дифенилциклотетраметиленгерманий 37
реакция с бромом 81
Дифенилэтинилгерманий 51
Дифеноксиди-я-бутилолово 408
Дифенокситетра-я-бутилдистанноксан 386,
403
Ди-о-феноксифенилолово
дву хлор истое 334
двуиодистое 343, 344
дигидроокись 378, 390
Ди(флуоренил-9)-фенилолово хлористое 350
Ди-2-фурилди-/2-анизилсвинец 561
Ди-2-фурилди-2-тиенилсвинец 561
Ди-(а-хлорбутил)олово дву хлор истое 322
Ди-Р-хлорвинилолово двухлористое 199, 200,
202, 203
Ди-(3-хлорвинилсвинец
двухлористый 609
диацетат 564, 609
дибензоат 609
сульфат 609
Дихлоргермил-бис-уксусная кислота, мети*
ловый эфир 56
Ди-/2-хлорфенилолово, полимер 201
Ди-/2-хлорфенилолово
двубромистое 201
двуиодистое 406
окись 390
а,(3(илир,(3-)-Дихлорэтилгерманий треххло-
ристый 63
Ди-(а-хлорэтил)олово двухлористое 321
Ди-(2-цианэтил)диизобутилолово 285
Дициклогексилдиалкилолово 236
Дициклогексилдифенилолово 236
Дициклогексилдифенилсвинец 561
Дициклогексилолово двубромистое 338
Дициклогексилолово, полимер 196
Дициклогексилсвинец
двуиодистый 576
окись 612
Ди(циклогексилфенил)олово двухлористое
334
Дициклопентадиенил олово 194, 196, 203
Дициклопентадиенилсвинец 554
Дициклопентилдиалкилолово 236
Дициклопентилдифенилолово 236
Дициклопропилолово
двубромистое 333
двуиодистое 406
двухлористое 333
диизотиоцианат 406
Дициклопропилсвинец
диациалаты 582
бш>(дихлорацетат) 582
Y-Диэтиламинопропилдифенилсвинец
хлористый 584
(З-Диэтиламинопропил)триэтилсвинец 626
Диэтиламинотри-я-бутил олово 409
реакции с
окисью 1-бутена 315
окисью стирола 315
окисью циклогексена 315
Диэтиламинотриметилолово 409
комплексы 433
Диэтиламинотри-я-пропилолово 409
Диэтиламинотриэтилолово 409
(2-Диэтиламиноэтил)триэтилсвинец 626
Диэтил-5-бромамилолсво бромистое 345
Диэтил-я-бромфенилолово бромистое 239
Ди-(2-этил-я-гексил)олово
двухлористое 333
окись 389
Диэтилгерманий
двубромистый 96
двухлористый 20
бромгидрид, присоединение к
аллилу бромистому 70
бутадиену 69
октену-1 69
678
Предметный указатель
диизоцианат 99
окись, реакция с фенилмеркаптаном 97
хлоргидрид 128
присоединение к
аллилнитрилу 70
аллилу хлористому 70
ацетилену 69
бутадиену 69
винилацетату 70
винилметилкетону 70
гексину-1 70
диметил-2,3-бутадиену-1,3 69
изопропенил ацетату 70
фенилацетилену 70
этилакрилату 70
Диэтилдиал кил олово 233
Диэтилдиаллилолово 233
сополимеризация со стиролом 512
Диэтилди(аллилфенил)олово 233, 238
Диэтилдиамилсвинец 560
Диэтилдибензилолово 233
Диэтилди-«-бутилгерманий 36
Диэтилди-«-бутилолово 233
Диэтилди-«-бутилсвинец 560
Диэтилдивинилгерманий 39
Диэтилдивинилсвинец 560
полимеризация 536
Диэтилдигексин-1-илолово 238
Диэтилдиизопропилсвинец 560
Диэтилдиперфгорвинилолово 226
Диэтилди-«-пропилсвинец 560
Диэтилдифенилдистаннан, полимер 483
Диэтилдифенилолово 233, 271
реакция с азотнокислым серебром 356
Диэтилди(хлорметил)свинец 568
Диэтилдиэтинилолово 255
Диэтилизоамилгерманий, гидрид 122
Диэтилнеопентилолово бромистое 340
Диэ1ил-«-октилолово йодистое 339
Диэтилолово
адипинат 351
азелаинат 351
(алкокси)бензоаты 368
двубромистое 182, 187, 259, 332, 335
двуиодистое 180, 339
двухлористое 188, 199,332,333,335,354, 400
диспропорционирование 378
реакции с
оловом хлорных 335, 336
сульфурилом хлористым 336
тетраэтилоловом 331
согидролиз с триметилкремнием
хлористым 391
диацетат 401
дигидрид 460, 461
присоединение к алкенам 279, 284
реакции с
гексаэтилдистанноксаном 481
эфирами меркур-бш>уксусной
кислоты 486
диизотиоцианат 406
димеркаптиды 414
окись, диспропорционирование 378
себацинат 351
сульфат 400
(хлор) ацетат 402
(хлор)гидрид, реакция с эфирами меркур-
бип-уксусной кислоты 4$7
бис-этилсульфонат 400
Диэтилолово
гексамер 478
реакция с хлором 440
нонамер 478
реакция с хлором 440
полимер 186, 194, 195, 477
диспропорционирование 376
окисление 438
реакции с
диацетиленнатрием 498
натрийацетиленидом 498
перекисью бензоила 441
сулемой 441
эгилом йодистым 442
Диэтилоловоди(кобальттрикарбонилтри-я-
бутилфосфин) 508
Диэтил-«-пропилолово йодистое 340
Диэтилсвинец 628
двубромистый 580, 595
двухлористый 576, 579, 580
реакция с диазоалканами 568
диацилаты 581, 582, 605
дигидрид 620
дигидроокись 597, 602
окись 602
сульфат 580
сульфит 580
фосфат 584
Диэтилсвинец бис-марганецпентакарбонил
629
5,5-Диэтил-10, \0'-спиро-бис-(5,10-дигидро-
феназогермин) 49
2,2-Диэтил-2-станна-1,3-диоксан 403
Диэтилстаннандиолы 403
Диэтилстаннил-бис-уксусная кислота, эфиры
диметиловый 312, 486
реакция с метанолом 353
ди-я-пропиловый 312
диэтиловый 312, 486
реакция с муравьиной кислотой 353
Диэтилфенилолово, ацетат 398
Диэтилхлорстаннилуксусная кислота,
метиловый эфир 487
Диэтил-л-хлорфенилолово бромистое 239
Диэтилциклопентаметиленгерманий 37
Диэтилцилопентаметиленолово 240
Диэтилциклопентаметиленсвинец 561
Диэтилциклотетраметиленгерманий 37
Диэтилциклотриметиленгерманий, реакция
с бромом 81
Диэтоксидибутилолово 408
Диэтоксидиэтилолово 408
присоединение к кетену 312
Ди-(2-этоксикарбонилэтил)олово
двубромистое 183
Ди-я-этоксифенилолово дигидрид 461
Ди-я-этоксифенилолово, полимер 480
Додекабутилгексастаннан дигидрид 480
Ениновые
254, 501
оловоорганические соединения
Изоамилгерманий,тригидрид 122
Изобутилолово
треххлористое 335
тригидрид 458
присоединение к метилметакрилату 284
Изобутокситриэтилолово 301
Предметный указатель
679
(2-Изобутоксиэтил)три-я-бутилолово 281
Изопропенилтриметилолово 246
Изопропилстанноновая кислота 190
Изопропокситриэтилолово 304
(1-Инденил)трифенилолово 328
реакция с хлористым водородом 350
Иодметилтрифенилолово 446
2-Иод-2-(триметилстаннил)октафенилбифе-
нил 452
1-Иод-2-(триметилстаннил)тетрафенилбензол,
реакция с тетрациклоном 452
jw-Иодфенилгерманий трехбромистый 24
я-Иодфенилолово трехбромистое 337
я-Иодфенилсвинец
триацетат 565
триизобутират 565
трипропионат 565, 609
(2-Карбамилэтил)три-я-пропилолово 285
(2-Карбамилэтил)трифенилолово 285
(о-Карбметоксифенил)дифенилсвинец
хлористый 617, 618
[4-Карбоксибутил]трифенилолово, реакция с
диазометаном 449
(2-Карбоксиметилбутил)трибутилолово,
реакция с литийалюминийгидридом 447
Карбоксиметилвинилтриметилолово,реакция с
бромом 342
(2-Карбоксиметилпропил)диэтилолово,
гидрид 284
(2-Карбоксиметилэтил)дибутилолово
бромистое 341
(2-Карбоксиметилэтил)дифенилолово
йодистое 343
бис-(2-Карбоксиметилэтил)олово двуброми-
стое 341
(2-Карбоксиметилэтил)три-я-бутилолово 284
реакции с
бромом 341
едким натром 448
метилмагнием йодистым 447
(2-Карбоксиметилэтил)три-я-пропилолово 283
реакция с едким натром 448
(2-карбоксиметилэтил)трифенилолово 284
реакция с бромом 343
{2-карбоксиметилэтил)фенилолово двубро-
мистое 343
(З-Карбоксипропил)трифенилолово 449
реакция с диазометаном 449
(З-Карбоксипропил)трифенилолово,
натриевая соль 448
(о-Карбоксифенил)дифенил свинец,
гидроокись 617
(/?-Карбоксифетшл)трифенилсвйнец 617
{2-Карбоксиэ;ил)ди-я-бутилолово,
внутренняя соль 384, 448
бш>(2-Карбоксиэтил)дипропилолово, динат-
риевая соль 448
(2-Карбоксиэтил)дифенилолово, внутренняя
соль 283, 448
(2-Карбоксиэтил)три-я-бутилолово 448
(2-Карбоксиэтил)трипропилолово, натриевая
соль 448
(2-Карбоксиэтил)трифенилолово, натриевая
соль 448
(со- Карбоксиэтил-я-у ндецил)триэтилолово 283
(4-Карбометоксибутил)трифенилолово 449
(З-Карбометоксипропил)трифенилолово 449
(2-Карбэтоксиметил)три-я-пропилолово323
(З-Кетобутил)трипропилолово 448
я-Крезолокситрифенилолово 399
Лаурилстанноновая кислота 190
Литийтриметилгерманий селенид 106
Литийтрифенилплюмбил селенид 626
реакции с
трифенилоловом хлористым 626
трифенилсвинцом хлористым 626
Литийтрифенилплюмбилсульфид 626
реакции с
бензоилом хлористым 626
трифенилоловом хлористым 626
трифенилсвинцом хлористым 626
Литийтрифенилплюмбилтеллурид 626
реакция с хлористым трифенилсвинцом 626
Литийтрифенилстаннилселенид 500
Литийтрифенилстаннилсульфид 500
реакция с хлористым бензоилом 500
Литийтрифенилстаннилтеллурид 500
З-Меркаптопропилтри-я-бутилолово 449
2-Меркаптоэтилтри-я-бутилолово 449
2-Меркаптоэтилтрифенилсвинец 627
Метакрилметилтриметилгерманий 118
Металлилгерманий треххлористый 21
присоединение брома 115
Металлилтриметилгерманий 39
Метилацетиленид триэтилсвинца 561
(2-Метилбутин-3-ол-2)-4-триметилгерман 43
Метилгерманий
трехбромистый 24
трехиодистый 22
треххлористый, хлорирование 117
тригидрид 122
Метилди-я-бутилолово йодистое 341
Метилди(триметилсилилметил)олово
бромистое 342
Метилдифенилолово хлористое 342
Метилдифенилсвинец бромистый 577
Метилдифенилстанниллитий 496
Метилдициклогексилолово йодистое 341
Метилдиэтилвинилолово 225
Метилолово
селенид 413
трехбромистое 183
реакция с бромистымтриметилоловом 332
трехиодистое 181, 189, 330, 413
треххлористое 189, 336
реакция с
иодистоводородной кислотой 413
селеноводородом 413
тригидрид 461
тримеркаптиды 414
Метилплюмбоновая кислота 542
Метил-я-пропил изоамилсви нец бромистый
576
Метилстанноновая кислота 190, 392
реакция с органическими кислотами 404
Метилтри-я-бутилолово 323
Метилтримезитилолово 251
Метил-(триметилсилилметил)олово
двуброми стое 347
Метил-(3,4,5-тримегилфенил)олово 251
Мегилтри(пентафгорфенил)олово 241
Мегилтрипропилолово 323
Метилтри-я-толилолово 223
Мегилтрифенилгерманий 151
Метилтрифенилолово, реакции с
иодом 342
хлорным оловом 336
680
Предметный указатель
Метилтрифенилсвинец 560
реакция с бромом 577
Метилтриэтилгерманий 35
Метилтриэтилолово 256
Метилтриэтилстаннилпропиолат 327
Метилфенил-а-нафтилгерманий 124
1 -Метил-1 -фенилэтилперокситрибутилолово
416
Метилциклогексилдифенилсвинец 577
Метилэтилолово двухлористое 346
Метилэтилпропилолово йодистое 340
Метокситрибутил олово 408
присоединение к
винил ацетату 311
гексахлорацетону 314
изопропенилацетату 311
фенилизоцианату 313
этилизоцианату 313
Мегокситрибутилсвинец 572, 607
присоединение к
гексахлорацетону 572
динафтилкарбодиимиду 572
нафтилизоцианату 572
сероуглероду 572
трихлорацетонитрилу 572
фенилизоцианату 572
хлоралю 572
Метокситриметилолово 408, 417
Метокситриметилсвинец 607
Метокситрипропилолово 408
присоединение к
диэтилацетилендикарбоксилату 313
изопропенилацетату 311
кетену 312
Метокситри-«-пропилсвинец 607
Метокситрифенилсвинец 572, 607
присоединение к
гексахлорацетону 572
динафтилкарбодиимиду 572
нафтилизоцианату 572
сероуглероду 572
трихлорацетонитрилу 572
фенилизоцианату 572
хлоралю 572
Метокситриэтилгерманий, присоединение к
кетену 71
Метокситриэтилолово 408
присоединение к
винилацетату 311
димегил(или этил)ацетилендикарбокси-
лату 313
изопропенилацетату 311
кетену 311
реакции с
алкинами 325, 326
ациламидами 416
карбаматами 416
метилпропиолатом 327
перекисью бензоила 368
Меюкситриэтилсвинец 605, 607
3-Метокси-1-(триэтилстаннил)пропин-1 327
Морфолинометилтриэтилолово 447
Натрийтрифенилплюмбилсульфид 606
реакция с йодистым метилом 606
термическое разложение 606
а-Нафтилгерманий треххлористый 54
а-Нафтилолово треххлористое 337
а-Нафтилсвинец
триацетат 565
трибензоат 609
(З-Нафтилсвинец
триацетат 565
триизобутират 565
трипропионат 565
а-Нафтилстанноновая кислота 393
бис-(а-Нитрозо-Р-нафтокси)тетраметил (или
этил)дистанноксан 414
1Ч-(я-Нитрофенил)формамидотриэтилолово
308
я-Нитрофенокситрифенилолово 409
я-Нитрофенокситриэтилолово 399
н-Нонилтрифенилолово, перераспределение
радикалов 379
(4-Сьсибутил)трифенилолово 447
(/2-Оксиметилфенил)трифенил свинец
окисление 617
реакция с бромом 578
(ж-Оксиметил)фенокситрибутилолово 302
(о-Оксиметил)фенокситриизобутилолово 302
(ж-Оксиметил)фенокситрифенилолово 303
(о-Оксиметил)фенокситриэтилолово 302
(З-Оксипропенил-1 )-трифенилолово 290
(З-Оксипропил)дифенилолово бромистое 343
(З-Оксипропил)трибутилолово 447
(З-Оксипропил)трифенилсвинец 628
бис-(8-Оксихинолят) диалкилолова 414, 434
бис-(8-Оксихинолят) дибутилсвинца 609
био(8-Оксихинолят) дифенилолова 414
бш>(8-Оксихинолят) дифенилсвинца 609
(8-Оксихинолят)бромид дибутилсвинца 610
(2-Оксиэтил)трифенилолово 502
2-Оксиэтилтрифенилсвинец 627
[2-(2/-Оксопирролидин)этил]дифенилолово
йодистое 343
Октаалкилтристаннаны 482, 505
Октабензилтристаннан 479
Октабутилтетрастанноксан, диацетат 387
Окта-«-бутилтристаннан 481
Октадекаизобутилциклононастаннан 478
я-Октадецилдифенил-2-фенилэтил германий
151
Октаметил(или фенил)тристаннан 504
Октаэтилтристаннан 481
Октилгерманий треххлористый 30
я-Октилдивинилолово бромистое 340
Октилолово
гидроксидихлорид 392
трехгалоидное 330
Октилстанноновая кислота 392
Октилтривинилолово, реакция с бромом 340
я-Октилтрифенилолово 224
Олово бромное 218, 219, 243, 244, 256
реакции с
диазометаном 321
реактивом Гриньяра 206, 216
Олово двубромистое, реакция с ртутноорга-
ническими соединениями 199, 201
Олово двухлористое 194,; 195
реакции с
алюминийорганическими соединениями
203
борорганическими соединениями 203
диазометаном 322
литийорганическими соединениями 193
реактивом Гриньяра 194—197
Предметный указатель
681
ртутноорганическими соединениями
199—201
таллийорганическими соединениями 202,
203
Олово хлорное
реакции с
алюминийорганическими соединениями
256
ацетиленидами натрия 252
mpem-бутилмагнием галоидным 210, 212
диазоалканами 321
дилитийорганическими соединениями 244
дифенилметилмагнием бромистым 217
литийорганическими соединениями 242
натрийорганическими соединениями 251
неопентилмагнием хлористым 210
реактивом Гриньяра в безэфирной среде
208, 211, 212, 216
ртутноорганическими соединениями 260
свинцовоорганическими соединениями
260
смесями магнийорганических соединений
206
тетраалкилалюминатом лития 260
фенилбарениллитием 244
цинкорганическими соединениями 256
реакции типа реакции Вюрца 251, 252
Оловоорганические алкоксисоединения 301—
304, 408, 417
реакции с
алкилами галоидными 415
алкилхлорсиланами 415
бором треххлористым 415
меркаптанами 418
органическими гидроперекисями 415
Оловоорганические ацетилениды 249, 253,
255, 325—328, 398, 500, 501
Оловоорганические галогениды, синтез с
помощью металлоорганических соединений
241, 242, 259, 260
реакции с
аллилмагнием бромистым 235
аллилфенилмагнием бромистым 231
алюминийорганическими соединениями
260
ариллитием 246
ацетиленидами натрия 253
дилитийорганическими соединениями
248
дифенилфосфином 415
литийорганическими соединениями 245
меркаптанами 414
неопентилмагнием хлористым 222
перфторвинилмагнием 225
спиртами 414
тиофосфорными кислотами и их солями
412
фосфорными кислотами и их солями 412
эфирами кислот трехвалентного фосфора
412
частичное алкилирование реактивом
Гриньяра 220
Оловоорганические гидриды
реакции с
гексаалкилдистанноксанами 481
диазокетонами 323
окисями диалкилолова 481
станниламинами 481
устойчивость 462
Оловоорганические гидроокиси, реакция с
магнийорганическими соединениями 220,
225
Оловоорганические дигалогениды, реакции с
mpem-бутилмагнием хлористым 234
винилмагнием хлористым 234
2,2 -дилитийбифенилом 250
дилитийорганическими соединениями
250
димагнийорганическими соединениями
240
перфторфенилмагнием бромистым 236
реактивом Гриньяра 233
циклопропилмагнием бромистым 236
Оловоорганические железокарбонилы 507
Оловоорганические кобальткарбонилы 507
Оловоорганические марганецкарбонилы 507
Оловоорганическиемоногалогениды, реакции с
арилмагнийгалогенидами 228
ацетиленидами лития 247
димагнийорганическими соединениями
224
/2-литийполистиролом 513
бис-магнийбромацетиленом 232
циклопентадиенилмагнием бромистым
228
Оловоорганические окиси, реакции с
винилмагнием хлористым 234
реактивом Гриньяра 225, 233
Оловоорганические соединения
анализ 526 и ел.
бумажная хроматография 527
газо-жидкостная хроматография 527
комплексы 432 и ел.
физиологическая активность 520, 521
Оловоорганические спиросоединения 245
Оловоорганические тригалогениды, реакция
с магнийорганическими соединениями 241
Оловоорганическими гидридами
восстановление
азобензола 306, 307
аллила бромистого 280
я-аминоазобензола 306
антрахинона 473
2-ацетилгексанона 473
ацетона 473
бензальдегида 472
бензила хлористого 469
бензилидена хлористого 469
бензилсульфида 476
бензоила хлористого 475
бензотрифторида 468
бензотрихлорида 468, 469
бензофенона 473
бензохинона 473
бромбензола 468
бромоформа 469
(3-бромстирола 472
бутила бромистого 468
галогенидов металлов 465
гексафторбензола 468
7,7-дибромбицикло[4, 1, 0]гептана 469
1,1 -дибром-2-винилциклопропана 472
1,2-дибромпропана 470
1,1-дибром-2,2,3-триметилциклопропана
469
геж-дибромциклопропана 469
дифенилдисульфида 476
682
Предметный указатель
дифенилсульфоксида 477
дифенилсульфона 477
дихлорбензола 468
камфоры 474
метилвинилкетона 473
метил-2-тиенилкетона 473
метил-я-толуолсульфоната 477
а,Р-ненасыщенных карбонильных
соединений 473
я-нитрозодиметиланилина 310
окислов металлов 465
пропаргила бромистого 471
мезо-стильбена двубромистого 470
тиобензофенона 476
трибромфторметана 469
тропилия бромистого 471
фенилсульфенилхлорида 477
фторбензола 468
хлорангидрида янтарной кислоты 475, 476
хлорбензола 468, 470
хлороформа 468—470
хлорциклогексана 470
четыреххлористого углерода 468—470
этилового эфира хлормуравьиной
кислоты 475
£шс-(Пентакарбонил-я-циклопентадиенилмо-
либден)оловодифенил 506
Пентаметиленгерманий двухлористый 28
бис- (Пентафторфенил)диэтилолово 249
Пентафторэтилтривинилолово, реакция с три-
фторуксусной кислотой 352
Перфторфенилгерманий трехбромистый 54
Пиридилтрифенилолово 248
Пиррилметилтриэтилолово 447
1,3-Пропандиоксиди-я-бутилолово 417
Пропаргилтрифенилолово, реакция с этил-
магнием бромистым 453
Пропаргилтрифенилсвинец 560, 627
Пропенилтриметилгерманий 48
Пропенилтриметилолово 226
^ис-Пропенилтриметилолово 246
я-Пропилгерманий треххлористый 31, 61
я-Пропилдифенилсвинец хлористый, реакция
с хлористым водородом 579
я-Пропилизоамил(или бутил)свинец двубро-
мистый 576
я-Пропилолово трехиодистое 189
я-Пропилстанноновая кислота 190
я-Пропилтрифенилгерманий 151
я-Пропилтрифенилсвинец 560
реакция с хлористым водородом 579
я-Пропилтриэтилгерманий 35
Пропилфенилди-о-толилсвинец 560
реакция с хлористым водородом 579
я-Пропилфенилсвинец двухлористый 579
я-Пропилфенил-о-толилсвинец хлористый,
реакция с хлористым водородом 579
я-Пропилфенилциклотетраметиленгерманий,
реакция с бромом 81
Пропокситриэтилолово, присоединение к ди-
метилацетилендикарбоксилату 313
Свинцовоорганические полимеры 536
Свинцовоорганические соединения
анализ 633 и ел.
бумажная хроматография 631
газо-жидкостная хррматография 633
комплексы 535
применение 535, 536
реакции с
бором треххлористым 586
висмутом треххлористым 586
галогенидами мышьяка 586
галогенидами сурьмы 586
галогенидами фосфора 586
железом хлорным 586
золотом треххлористым 586
медью азотнокислой 586
медью хлорной 586
органическими кислотами 582
перекисями ацилов 590
платинохлористоводородной кислотой
586
серебром азотнокислым 586
титаном четыреххлористым 586
Спиро-9,9 -бш>(дибензостаннол), реакция с
хлористым водородом 349
Станниламины 409
реакции с
дифениларсином 417
дифенилфосфином 417
кислотами 415, 416
спиртами 417
О-Станнилкетенацетали 285
N-Станнилкетенимины 305
Станнилфосфины 415
гидролиз 423
окисление 423
а-Стильбенилолово треххлористое 200, 404
реакция с диоксандибромидом 344
а-Стильбенилстанноновая кислота 200
реакции с
диоксандибромидом 344
тионилом хлористым 404
а-Стирилтриметилгерманий 50
Р-Стирилтриметилгерманий 40
Стиролы с оловоорганическими
заместителями в ядре 230
Тетраалкилдинитрозилсвинец, динитрат 618
Тетраалкилдистанноксаны
(гидрокси)галогениды 419, 434
(гидрокси)изотиоцианаты 406
диацилаты 400, 419, 434
реакция с органическими
гидроперекисями 413
дигалогениды 419, 434
реакции с
галоидами 423
гидроперекисями 413
органическими кислотами 402
роданистым натрием 406
спиртами 413
Тетраалкилолово
очистка от галоидного триалкилолова 206—
209
реакции с
бором треххлористым 356
бромом, кинетика 338, 340
иодом, кинетика 338
кислородом 358
серебром азотнокислым 356
хлористым водородом 347
хромовым ангидридом 358
Тетраалкилсвинец
перераспределение радикалов 598
реакции с
Предметный указатель
683
диацилатами ртути 585
уксусной кислотой, скорость реакции 581
Тетрааллилгерманий 28
Тетрааллилово 214
термическая устойчивость 379
Тетра-(/г-аллилфенил)олово 219
Тетра(аллилфенил)свинец 551
Тетра-я-амилолово 209
перераспределение радикалов 379
Тетра-«-амилсвинец 550, 552
Тетра-я-анизилдиплюмбоксан, диацетат 603
Тетраанизилолово 217
Тетра-о-анизилолово, реакция с треххлори-
стым таллием 357
Тетра-я^анизилолово, реакция с
тетрагалогенидами олова 334
Тетра-я-анизилсвинец 613
реакция с хлористым водородом 579, 580
Тетраацетат свинца 565, 573
реакции с
анизолом 573
диарилртутью 564, 565
ди-(тряяс-Р-хлорвинил)ртутью 564
Тетра-я-бензальдегидолово 453
реакция с трифенилфосфинметиленом 453
Тетрабензилгерманий 27
Тетрабензилолово 217
Тетрабензилсвинец 551, 559
Тетра-я-бифенилгерманий 29
Тетрабифенилолово 219
Тетра-ж-бифенилолово 243
Тетра-о-бифенилолово 243
реакция с галоидоводородами 348
Тетра-/г-бифенилолово 243
реакция с тетрагалогенидами олова 334
Тетра-о-бромбензилсвинец 551
Тетра(бромметил)олово 321
Тетра-я-бутилгерманий 27
реакции с
германием четыреххлористым 84, 85
изоамилом бромистым 87
изоамилом хлористым 87
оловом хлорным 57
серой 87
смлш-Тетрабутилдиаллилдистанноксан,
реакция с уксусной кислотой 405
Тетра-я-бутилдистаннан, диацетат 462, 466
реакция с бромом 466
Тетра-я-бутилдистаннан
дибензоат 491
дигидрид 462
Тетра-я-бутилдистанноксан
диацетат 386
реакция с сероводородом 413
двубромистый 386
двухлористый 386, 434
реакция с сероводородом 413
Тетрабутилдистаннтиан двухлористый 359,434
Тетра-я-бутилолово 187, 208, 209, 211, 253,
258, 260
реакции с
алюминием треххлористым 356
висмутом треххлористым 357
водородом 369
иодом 339
оловом бромным 332
оловом хлорным 330—332, 335
селеном 359
серой 359
таллием треххлористым 356
трифторуксусной кислотой 352
хромовым ангидридом 358
Тетр а-я-бути л свинец 550
реакция с серой 587
Тетра-втор-бутилсвинец 545
Тетра-я-трет-бутилфенилолово 217
Тетравинилгерманий 28, 32
реакция с сулемой 87
Тетравинилолово 213
реакции с
водой и щелочами 364
иодом 341
оловом хлорным 331, 333, 336
органическими кислотами 351
таллием треххлористым 357
хлористым водородом 347
Тетравинилсвинец 550, 551
реакции с
литием 624
органическими кислотами 582
фениллитием 617
хлором 576
Тетра-я-винилфенилолово 219, 453
Тетра-я-гексадецилгерманий 27
Тетра-я-гексадецилолово 209
реакция с хлористым водородом 346
Тетра-я-гексилолово 209, 252
реакция с хлорным оловом 333
Тетрагексилсвинец 554
Тетра-я-гептилолово 208, 209
Тетра-ж-гидроксифенил олово 244
Тетра-я-децилгерманий 27, 47
Тетра-я-децилолово 252
Тетра-/г-диметиламинофенилолово 243
Тетра-/г-диметиламинофенилсвинец 553
Тетра-я-додецилгерманий 27
Тегра-я-додецилолово 209
реакция с хлористым водородом 346
Тетраизоалкилолово 210
Тетраизоамилгерманий 27, 50
Тетраизоамилолово 210
Тетраизоамилсвинец 550
Тетраизобугилгерманий 55
Тетраизобухилолово 210, 257, 258
перераспределение радикалои 379
реакции с
иодом 339
оловом хлорным 333, 335
Тетраизобугилсвинец 541, 550, 554, 565
Тетраизобутират свинца 565
реакция с тиофеном 574
Тетраизопропенилолово, реакция с трех
хлористым таллием 357
Тетраизопропилгерманий 27
Тетраизопропилолово 210
реакции с
оловом бромным 330
оловом хлорным 330
органическими кислотами 351
перекисью бензоила 367
Тетраизопропилсвинец 545, 550
Тетра-1-инденилолово 243, 252
Тетра-(иодметил)олово 446
Тетр а-ж-(или я)-ксилилолово 217
Тетраксилилсвинец 552
Тетрамезитилолово 217
Тетраметилгерманий 26, 50, 53
реакции с
684
Предметный указатель
ал килами галоидными 87
галлием треххлористым 87
изопропилом бромистым 87
индием треххлористым 87
иодом 82
сшш-Тетраметилди-я-амилдистанноксан 383
сшш-Тетраметилди-я-бутилдистанноксан 383
Тетраметилдистанноксан
дву хлор истый 385
д и ацетат 401
дибензоат 386
дибутират 413
ди(монобромацетат) 413
ди(монохлорацетат) 413
дипропионат 413
ди(цианацетат) 413
Тетраметилолово 186, 207
перераспределение радикалов 379
реакции с
бором трехфтористым 356
бором треххлористым 356
бромом 338
галлием треххлористым 356
двуокисью азот а 370
иодом 338, 339
кремнием четыреххлористым 357
натрием 339, 498
оловом бромным 330, 332
оловом йодным 330
ртутью азотистокислой 335
сулемой 354
фосфором пятифтористым 357
хлористым водородом 346
термическое разложение 365
фотохимические реакции 366
Тетраметилсвинец 541, 546, 549, 550, 553
реакции с
боргидридом алюминия 586
бором треххлористым 586
бромом 576
водородом 590
дибромметиленом 589
иодом 576
метилом йодистым 589
перфторметилом йодистым 589
перфторэтилом йодистым 589
серебром азотнокислым 586
хлористым водородом 578, 580
хлором 575
силш-Тетраметилстаннилдинатрий 465
Те1ра(метилциклопентадиенил)олово 252
Тетра-(метоксиметил)олово 242
Тетра-а-нафтилгерманий 31
Тетра-3-нафтилдиплюмбоксан, диацетат 603
Те^ра-а-нафтилолово 218
реакция с хлорным оловом 334
Тетра-1-нафтилолово 243
Тетранеопентилолово 222
Тетра-я-окгадецилгерманий 27
Тетра-я-октадецилолово, реакция с
хлористым водородом 346
Тетра-я-октилгерманий 47
Тетра-я-октилолово 209, 252, 253, 258
реакции с
оловом хлорным 330, 333
сулемой 354
Тетра(пентафторфенил)олово 216, 243, 270
реакция с водой и щелочами 364
Тетрапентафторфенилсвинец 562
Тетраперфторвинилгерманий 28
Тетраперфторвинилолово 213
реакции с
водой и щелочами 364
хлористым водородом 347
Тетраперфторфенилгерманий 48
Тетра-тряяс-пропенилгерманий 47
Тетра-цис-пропенилгерманий 47
Тетра-тряяс-пропенилолово 242
Тетра-^ыс-пропенилолово 242
Тетра-я-пропилгерманий 27, 50
реакция с иодом 82
Тетра-я-пропилдистанноксан двухлористыйг
386
Тетра-я-пропилолово 186, 208
реакции с
иодом 339
оловом бромным 332
оловом хлорным 330, 332
органическими кислотами 351
перекисью бензоила 367
фотохимические реакции 366
Тетра-я-пропилсвинец 541, 550
нитрование 618
реакция с хлористым водородом 578—580
Тетрапропин-1-илолово 252
Тетрапропионат свинца 565
Тетра-р-стирилгерманий 50
Тетра-я-тетрадецилгерманий 27
Тетра-я-тетрадецилолово 209
реакция с хлористым водородом 346
Тетра-а-тиенилгерманий 30
Тетра-а-гиенилолово 219
Тетра-а-тиенилсвинец, реакция с изомаслян-
ной кислотой 584
Тетра-ж-толилгерманий 28, 53
Тетра-о-толилгерманий 29, 53
Тетра-л-толилгерманий 29, 50, 53
Тетратол ил олово 252
Тетра-ж-толилово 217, 243
реакции с
бромом 343
оловом хлорным 332, 334
Тетра-о-толилолово 217, 243
реакции с
иодом 345
оловом хлорным 332, 334, 337
таллием треххлористым 357
Тетра-л-тол ил олово 216, 243, 270
реакции с
азотной кислотой 350
галоидами 343
оловом хлорным 332, 334, 337
серой 361
таллием треххлористым 357
Тетра-я-толилсвинец 569, 577
реакция с хлористым водородом 59
Тетра-(3,5,5-триметил-я-гексйл)олово 210
реакция с хлорным оловом 333
Тетра(триметилсилилметил)олово, реакция с
галоидами 342
Тетра(триметилсилилметил)свинец 551
Тетра-(я-триметилсилилфенил)олово 244
Тетра-(трифенилплюмбил)метан 627
Тетра-(трифенилплюмбил)свинец 555, 625
окисление 625
Тетра(трифенилстаннил)германий 503
Тетра-(грифенилстаннил)олово 505
Тетра(трифенилстаннил)свинец 503
Тетра(трифенилстаннил)титан 489
Тетра-ж-трифторметилфенилолово 217, 243
Предметный указатель
685
реакция с иодом 345
Тетра-(л-трифторметилфенил)олово 217
реакция с иодом 345
Тетра-(9-фениантрил)олово 243
реакции с
иодом 345
хлористым водородом 348
Тетра-(я-фенетил)олово, реакция с хлорным
оловом 334
Тетрафенилгерманий 29, 48, 50, 53
реакции с
германием четыреххлористым 84
натрием 147
Тетрафенилдиплюмбоксан
диацетат 603
дибензоат 603
диизобутират 603
димонохлорацетат 603
дипропионат 603
Тетрафенилдистаннан
дибензоат 490
ди(дихлор ацетат) 466
дикапронат 466
Тетрафенилдистанноксан двухлористыи,
реакция с сероводородом 413
Тетрафеннлолово 188, 215, 216, 252, 256, 259,
269, 270
перераспределение радикалов 379
нитрование 454
реакции с
азотистым ангидридом 370
алюминием треххлористым 356, 358
ацетилом хлористым 352, 368
бором, тригалогенидами 356, 358
бромом 343
N-бромсукцинимидом 344
висмутом 363
висмутом треххлористым 357
водородом 369, 370
галлием треххлористым 358
едким кали 363
железом хлористым 355
железом хлорным 355
индием треххлористым 358
иодом 343
иодом треххлористым 344
кобальтом хлористым 355
кремнием четыреххлористым 357
марганцем хлористым 355
медью хлорной 355
мышьяком 363
мышьяком пятифтористым 357
натрием 498
нафггалиннатрием 498
никелем хлористым 355
оловом бромным 332
оловом хлорным 331, 334, 336
органическими кислотами 352
селеном 361
серебром азотнокислым 356
серой 359, 360
сулемой 354
сурьмой 363
сурьмой треххлористой 357
таллием треххлористым 357, 358
тиофенолами 353
титаном четыреххлористым 357
фенолами 353
фосфором 362, 363
фосфором пятихлористым 357
хлористым водородом 348
Тетрафенилсвинец 551, 552, 553, 555, 558,
563, 564, 569, 577, 597, 612, 614
нитрование 618
реакции с
азотной кислотой 582
аммонием хлористым 579
бромом 577
водородом 590
иодом 577
иодом треххлористым 578
кислотами 582, 583
марганцем хлористым 586
мышьяком 587
мышьяком треххлористым 586
серебром азотнокислым 586
серой 587
сулемой 585
сурьмой пятихлористой 586
сурьмой треххлористой 586
таллием треххлористым 586
тетраацетатом свинца 585
тиофенолом 584
трехокисью азота 590
триметиламином солянокислым 579
хлористым водородом 579
четыреххлористым углеродом 590
фотолиз 589
Тетра-(2-фенилэтил)олово 217
реакция с бромом 338
Тетрафенилэтинилгерманий 48
Тетрафенилэтинилолово 219, 252
Тетра-о-феноксифенилдистанноксан, дигидро-
окись 390
Тетра-о-феноксифенилолово 244
реакции с
иодом 343, 344
оловом хлорным 334
Тетра-я-феноксифенилолово 218
Тетра-9-флуорилолово 243
Тетра-о-фторбензилсвинец 551
Тетра-/г-фторфенилолово, реакции с
бромом 345
гептафтормасляной кислотой 352
оловом хлорным 332
трифторуксусной кислотой 352
Тетра-2-фурилгерманий 48
Тетра-о-хлорбензилолово 217
Тетра-о-хлорбензилсвинец 551
Тетра-(хлорметил)олово 321
реакция с йодистым натрием 447
Тетра-я-хлорфенилолово 216, 270
Тетрахлорэтилгерманий треххлористый 63
Тетра-(а-хлорэтил)олово 322
Тетрацетилсвинец 542
Тетрациклогексилацетиленид олова 243
Тетрациклогексилгерманий 47
Тетрациклогексилолово 214
реакция с бромом 338
Тетрациклогексилсвинец 551
Тетра(циклогексилфенил)олово, реакция
с хлорным оловом 334
Тетра-2-циклогексилэтилгерманий 47
Тетрациклопентадиенилолово 243, 252
Тетра-1-циклопентадиенилолово 214, 215
Тетрациклопентилолово 214
Тетрациклопропилолово 214, 242
реакции с
оловом бромным 333
оловом хлорным 333
686
Предметный указатель
ртутью бромной 354
сулемой 354
Тетрациклопропилсвинец 551
реакция с кислотами 582
Тетра-2-этилгексилгерманий 47
Тетра-/шс-(2-этил-«-гексил)олово 210
реакция с хлорным оловом 333
Тетраэтилгерманий 27, 47, 50, 53, 55, 151
реакции с
германием четыреххлористым 84
изопропилом хлористым 87
иодом 82
сшш-Тетраэтилди-«-бутилдистанноксан 383
сшш-Тетраэтилдиизобутилдистаннан 503
Тетраэтилдинитрозилсвинец, динитрат 580
реакция с бромистоводородной кислотой
580
силш-Тетраэтилди-я-октилдистанноксан 383
силш-Тетраэтилди-я-пропилдистаннан 503
Тетраэтилдистанноксан
(алкокси)иодид 387
(гидрокси)бромид 386
двухлористый 385
дибензоат 386
Тетраэтилолово 186—188, 207, 256, 257, 258,
260, 269
перераспределение радикалов 379
реакции с
бензоилом хлористым 368
бромом 332
mpem-бутилбензоатом 368
висмутом треххлористым 357
водородом 369
иодом 338
медью двугалоидной 355
меркаптанами 352, 353
натрием 339, 498
оловом бромным 330, 332, 335
оловом двугалоидным 355
оловом, диацилатами 355
оловом хлорным 330, 332, 335
органическими кислотами 351
палладием двугалоидным 355
перекисью ацетилбензоила 367
перекисью бензоила 367
перекисью mpem-бутила 367, 368
ртутью азотистокислой 355
ртутью бромной 354
ртутью йодистой 355
ртутью йодной 354
серебром азотнокислым 355
серебром бромистым 355
серебром йодистым 355
серебром, окисью 355
сулемой 364
таллием треххлористым 356
таллием, формиатом 355
танталом двугалоидным 355
тиофенолами 352, 353
титаном четыреххлористым 357
фенилкремнием треххлористым 357
фосфором, тригалогенидами 357
хлористым водородом 346
цирконием, тригалогенидами 357
этилкремнием треххлористым 357
фотохимические реакции 365, 366
Тетраэтилсвинец 541, 542, 547, 550, 553,
554, 556, 558, 568, 581, 614
окисление 587
пиролиз 587
реакции с
бором треххлористым 586
бромом 576
висмутом треххлористым 586, 587
водородом 590
германием четыреххлористым 57
двуокисью азота 580
дибромметиленом 589
1,2-дибромэтаном 589
иодом 576
кремнием четыреххлористым 587
натрием 625
нитратом закиси ртути 585
л-нитрофенолом 584
органическими кислотами 581, 582
перекисью бензоила 590
серебром азотнокислым 586
сернистым ангидридом 580
сулемой 584
тетраацетатом свинца 585
тиофенолом 584
хлористым водородом 578—580
этилом бромистым 589
фотолиз 588, 589
Тетраэтинилгерманий 51
Тетраэтинилолово 252
а-Тиенилгерманий трехбромистый 24
а-Тиенилсвинец, триизобутират 573, 574
ж-Толилгерманий трехбромистый 24
о(или л-)-Толилоловотреххлористое 337
я-Толилсвинец
триацетат 565, 585
триметакрилат 609
ж-Толилстанноновая кислота 393
о-Толилстанноновая кислота 393
я-Толилстанноновая кислота 393
л-Толилтри(пентафторфенил)олово 241
л-Толилтриэтилгерманий 38
л-Толуолсульфамидотри-я-пропилсвинец 608
л-Толуолсульфамидотриэтилсвинец 607
Триалкилаллилово, реакция с
полисульфидом водорода 449
Триалкиларилолово, кинетика реакций с
иодом 338
хлористым водородом 347
Триалкил-(/г-бромфенил)олово, реакция с
бромистым винилом 231
Триал килгерманий
алкилмеркаптиды ПО
йодистый, реакция с сернистым серебром 99
хлористый, реакция с
аминолитиевыми соединениями 107
аминомагниевыми соединениями 107
Триалкилолово
акрилаты, полимеризация 513
алкилсульфонаты 396
арилсульфонаты 396
ацетилениды 232
ацилаты 397, 407, 433
реакции с
меркаптанами 418
органическими кислотами 417
бромистое, кинетика реакции с бромом 337
/2-винилбензоат, полимеризация 513
галоидацилаты, реакция с бис-(диметил-
аминофенил)ртутью 423
галоидное 210, 211, 396
гидрид 260, 458, 460
присоединение к
алкилиденмалоновым эфирам 284
Предметный указатель
687
алкинам 287—297
альдегидам и кетонам 301—304
диенам 276
олефинам 275, 278, 281, 283—285
реакции с
азодиизобутиронитрилом 491
диазометаном 322
диазоуксусным эфиром 323
диалкиламинотриалкилоловом 484
динитрилом азоциклогексилкарбоно-
вой кислоты 491
диэтиламидом диэтилалюминия 488
кислотами 283, 465
нитрилом фенилазоизомасляной
кислоты 491
триалкилалюминием 487
N-фенилпропионамидом
диэтилалюминия 488
гидроокиси
диспропорционирование 378
реакции с
гетероциклическими соединениями
азота 399
магнием бромистым 419
меркаптанами 399
органическими кислотами 397
силанолами 399
спиртами 399
тетрабутоксититаном 419
фенолами 399
изотиоцианаты 406
изоцианаты 397, 399
изоцианиды 406
йодистое 405, 406
ксантогенаты 409
меркаптиды 399, 409
реакции с
бором треххлористым 423
бромом 423
иодом 423
меркаптанами 418
мышьяком треххлористым 423
фосфором треххлористым 423
метакрилаты, полимеризация 513
сульфонамиды 410
фтористое 405
хлористое, реакции с
азидом натрия 410
алкоголятами щелочных металлов 408
амидом лития 409
винилацетиленидмагнием бромистым 227
гидроперекисями 413
диалкиламидом лития 409
диэтиламиномагнием бромистым 409
литийалюминийгидридом 457 и ел.
меркаптанами 399
органическими кислотами 397
реактивом Гриньяра 220
роданистым натрием 406
солями гетероциклических соединений
азота 410
солями органических кислот 407
селенидом натрия 406
спиртами 399
сульфидом натрия 406
фтористым калием 405
цианистым натрием 406
щелочами 382, 383
щелочными металлами 496 и ел.
бис- (Триал кил олово)
диацетилениды 255
карбонаты 397
перекиси 416
селенид 406
сульфид 406
Триалкилсвинец
арилсульфамиды 605
боронаты 621
реакция с метанолом 621
галогениды, реакции с
калий бор гидридом 620
натрием 624
гидриды 621
реакции с
алкилами галоидными 622
бензальдегидом 622
нитробензолом 622
триэти л оловом хлористым 622
четыреххлористым углеродом 622
йодистый 606
меркаптиды 605
селеноцианат 606
тиогликольамид 606
тиоцианат 606
фтальимид 605
фтористый 606
цианистый 606
Триалкилсганнилацегон, присоединение к
альдегидам и кеюнам 273
Триалкилстаннилдиацетилен 501
Триалкилстанниллитий 193, 496, 499
анализ 496
бш>(Триалкилстаннил)метан 501
Триалкилстаннилнатрий 497
реакции с
алкенилбромацетиленами 501
бромдиацетиленом 501
метиленом хлористым 501
монобромацетиленом 501
Триалкилстаннилуксусная кислота
М,М-диалкиламид 314
присоединение к альдегидам и кетонам
273
эфиры, присоединение к альдегидам и
кетонам 273
б^с-(Триалкилстаннил)этилен, реакции с
сулемой 289
хлористым водородом 289
хлором 289
Триалкил-/г-стирилсвинец, полимеризация
536
Триалкилтриарилдистаннаны 504
Триалкил(цианметил)олово, присоединение
к альдегидам и кетонам 273
Триаллилметилгерманий 39
Триаллилолово бромистое 340
Триамилгерманий хлористый 86
Триамил(карбэтоксиметил)германий 75
Три-я-амилсвинец, гидроокись 401
Три-я-амилфенилгерманий 36
Три-о-анизилсвинец йодистый 613
Три-/г-анизилсвинец хлористый 579
Три-/г-анизил-2-фурилсвинец 561
Триарилолово
азид 410
ацилаты 398
галоидное 396
реакции с
б^с-магнийбромацетиленом 232
мезитилмагнием бромистым 230
688
Предметный указатель
гидроокиси, диспропорционирование 378
изотиоцианат 406
йодистое 405, 406
меркаптиды 409
фтористое 405
бш>(Триарилолово)ацетилен 255
бш>(Триарилолово)
селенид 406
сульфид 406
Триарилсвинец
гидроокись 612
йодистый 606
селеноцианат 606
тиогликольамид 606
тиоцианат 606
фтористый 606
цианистый 606
Триарилстанниллитий 193, 496, 499
бш>(Триарилстаннил)метан 501
Триарилстаннилнатрий 497
реакция с хлористым метиленом 501
Трибензилбутилгерманий 35
Трибензил-(я-винилфенил)олово 231
Трибензилгерманий бромистый 81
Трибензилолово
азид 410
ацетат 398
гидроокись 358, 385
хлористое 185, 218, 269
Трибензил-л-толилолово 228
Трибензилфенилолово 228
Трибензилэтилгерманий 151
Три-(2-бифенил)олово
бромистое 219, 243
гидрид 459
хлористое 349
Три-(2-бифенил)-я-толилолово 247
Три-(2-бифенил)фенилолово 247
бос-(Три-о-бромбензилолово), окись 219
Три-я-бутилалкилолово 221
Три-я-бутилаллилгерманий 39
Три-я-бутилаллилолово 222, 226, 246
реакции с
сероводородом 449
тиоуксусной кислотой 449
фениллитием 453
Три-«-бутил(аллилфенил)олово 231
Трибутилацетонилолово 311
Трибутилбутадиенилолово 227
Три-«-бутил(3-бутенил)олово 222
Три-«-бутил(л-бутенилфенил)олово 231
Три-н-бутилвинилгерманий 39
Три-я-бутилвинилолово 222, 225, 245, 287
реакция с сероводородом 449
Три-я-бутил-(я-винилфенил)олово 231
Трибутилвинилэтинилолово 227, 325
Трибугилгексенил-1 -германий 411
Трибутилгерманий
акрилат 101
бромистый 81
гидрид 123
присоединение к
акрилонитрилу 68
акролеину 67
аллиловому спирту 65
аллилмеркаптану 63
винил бутиловому эфиру 66
пропаргиловому спирту 65
триэтилэтинилкремнию 69
фенилацетилену 65
изоцианат 99
метакрилат 101
мегилсульфонат ПО
трифтор ацетат 97
хлористый 30, 84, 85
Трибутилгермилдибутилолово, гидрид 488
Три я-бутил(диметилсилилметил)олово 222
Трибутил-(я-а,(3-дифтор-(3-хлорстирил) олово
247
Трибутил(карбэтоксиметил) германий 75
Трибутилметаллилолово 226
Три-«-бутил-(я-а-метилвинилфенил)олово
231
Три-//-бутил-(3-оксипропил)олово 281
Трибутилолово
азид 410
присоединение к диэтилацетилендикарб-
оксилату 316
реакция с ацетилендикарбоновой
кислотой 315
акрилат 397
ацегат 397
гидрид 457, 458, 461
присоединение к
алкинам 287—289
альдегидам и кетонам 301—303
динитрилу алкилиденмалоновой
кислоты 305
олефинам 275, 281—283, 285
фенилизоцианату 308
реакции с
гекса-я-бутилдистанноксаном 481
гидроперекисью mpem-бутила 467
диэтиламинотрибугилоловом 481
диэ'1 ил(триэтилстаннил)фосфином 481
кислотными агентами 465
метокситрибутилоловом 481
окисью дибутилолова 481
тетра(диметиламино)титаном 489
щелочными агентами 465
йодистое 189, 339
метакрилат 397
хлористое 184, 187, 211, 260, 330, 331
присоединение к дегидробензолу 316
реакция с серой 359
термическое разложение 330
цианацетат, декарбоксилирование 407
этилмалонат, декарбоксилирование 407
Три-трет-бутилоловохлористое 242
Трибутилоловокобальттетракарбонил 507
Три-н-бутилпентафторэтилол олово 222
Три-н-бутилпентенил-1-германий 41
Трибутилперфторвинилолово 226
реакции с
сулемой 354
этилатом натрия 353
Три-я-бутилперфторфенилолово 222, 229
Три-я-бутилсвинец
ацетат 571
боронат 621
гидрид 620, 621
присоединение к
акрилонитрилу 570
метилакрилату 570
стиролу 570
фенилацетилену 570
фенилизоцианату 570
гидроокись 601
хлористый 578
б«с-(Три-я-бутилстаннил)ацетилен 327, 398
Предметный указатель
689
Трибутилстаннилацетон 323
N-Трибутилстаннилбензимидазол 399, 410
комплексы 433
1-Трибутилстаннил -4,5-диэтокси -1,2,3-триа-
зол 316
Трибутилстанниллитий 496, 498
реакции с
диметилкремнием двухлористым 503
дифенилкремнием двухлористым 503
триметилкремнием хлористым 502
N-Трибутилстаннилпиррол ^410
1-Три-я-бутилстаннил -2-три-я-бутилгермил-
этилен 120, 289
Трибутилстаннилуксусная кислота,
метиловый эфир, реакция с литийалюминий-
гидридом 447
бш:-1,2-(Три-я-бутилстаннил)этилен 288, 289
Трибутил-р-стирилгерманий, реакция с
бромом 82
Трибутил(трибромметил)олово 316
Трибутил(трихлорметил)олово 314, 316
Три-«-бутил(фенилацетиленид)олово 247
Трибутилфенилгерманий 36
Три-«-бутил(хлорметил)олово 221
Три-«-бутил-(о-хлорфенил)олово 247
Три-я-бутилцианометилолово 323
цыс-Р-Трибутилцианоэтинилсвинец 572
Три-я-бутилциклопентадиенилолово 328
Три-я-бутилциклопропилолово 222
Три-я-бутилэтинилгерманий 41
Три-я-бутилэтинилолово 255, 325
Три-«-бутил(этоксидиметилсилилметил)олово
222
Трибутоксибутилгерманий 98
реакция с бутиловым спиртом 98
Тривинилалкил(арил)олово 223
Тривинилолово
гидроокись 383
йодистое 341
хлористое 331
Тривиннлсвинец
ацетат 582
трихлорацетат 582
хлористый 578
реакция со щелочными металлами 623
Три-я-гексадецил олово
йодистое 189
хлористое 346
Три-я-гексилгерманий бромистый 30
Тригептилгерманий хлористый 86
Три-я-гептилфенилгерманий 36
Три-я-децилфенилгерманий 36
Три(диалкиламино)алкил(арил)олово 409
Тридодецилолово хлористое 346
Тридодецилсвинец хлористый 552
Триизоамилалкилолово 223
Триизоамил(аллилфенил)олово 232
Триизоамил-(л-винилфенил)олово 231
Триизоамил-(я-а-метилвинилфенил)олово
231
Триизоамилсвинец, гидроокись 601
Триизоамилфенилгерманий 36
Триизобутилолово, гидрид 458
присоединение к
альдегидам 301, 302
олефинам 275, 281, 284
Триизобутилолово, дейтерид 460
Триизобутилсвинец
гидрид 620
л-толилсульфонат 581
44 Заказ № 207
хлористый 575, 578
Триизобутилфенилгерманий 36
Триизобутилэтилолово 222
Триизопропилгерманий
гидрид 27
присоединение к
акрилнитрилу 68
метилакрилату 67
изоцианат 99
хлористый 31
реакция с серебряными солями
органических кислот 100
Триизопропилолово
бромистое 330
хлористое 330
Трииодгермилуксусная кислота, этиловый
эфир 23
Три-(2-карбоксиметилпропил)-«-бутилолово
284
Три-(2-карбоксиметилпропил)изобутилолово
284
Три-(2-карбоксиметилэтил)олово бромистое
341
Три-(2-карбоксиэтил)пропилолово, трина-
триевая соль 448
Трикарбонил-я-циклопентадиенилвольфрам-
оловотрифенил 411, 506
реакция с хлористым водородом 508
Трикарбонил-я-циклопентадиенилмолибден-
оловотрифенил 411, 506
реакция с хлористым водородом 508
Трикарбонил-я-циклопентадиенилхромолово-
трифенил 411, 506
Три-я-карбэтоксифенилолово хлористое 269
Три-я-ксилилмезитилолово 230
Три-л-ксилилсвинец бромистый 613
Тримезитилалкилолово 221, 222, 224
Тримезитил олово
бромистое 219
гидрид 459
йодистое 339, 406
хлористое 243
Тримезитилплюмбилмагний, гидролиз 625
Тримезитилсвинец бромистый 555
Тримезитилфенилолово 224, 230
Триметилаллилгерманий, полимеризация 159
Триметилаллилолово 221, 226
Триметил-я-амилсвинец 559
•Триметиларилолово 221, 246
Триметилбензилолово 221
Триметил(4-бифенил)олово 246
Триметил-(/г-бромбензил)олово 221
Триметил-л-бромфенилгерманий 34
Триметил-я-бромфенилолово 229
Триметил-я-бутилсвинец 559
Триметил-ятор-бутилсвинец 559
Триметил-трет-бутилсвинец 559
Триметилвинилгерманий 39
Триметилвинилолово 221, 225
реакции присоединения с циклонами 451
Триметил-(я-винилфенил)олово 231
Триметил-(я-винилфенил)свинец 559
Триметилвинилэтинилгерманий 42
Триметилвинилэтинилсвинец 574
Триметил(галоидметил)олово 221
Триметилгексинилолово 327
Триметилгерманий
азид 108
бромистый 80
изоцианат 99
690
Предметный указатель
йодистый 30
фтористый 82
хлористый 20, 38, 47, 83
реакции с
азидом натрия 108
серебром азотнокислым 99
серебряной солью ортомышьяковой
кислоты 99
серебряной солью ортофосфорной
кислоты 99
(Триметилгермилметил)циклопропан 116
/г-Триметилгермилтриметилсилилбензол 38
Триметилгермил-1-триметилсилилэтилен 53
Триметилгермил-2-триметилсилилэтилен 52
1,1-бш:-(Триметилгермил)этилен 52
Триметил(диметиламинофенил)олово 247
Триметил(диметилсилилметил)олово 221
Триметилизоамилсвинец 559
Триметилизобутилгерманий 34
Триметилизопропилолово 256
Триметилизопропилсвинец 559
Триметил(иодметил)олово 220
Триметилмезитилолово 246
Триметилг (/г-сс-метилвинилфенил)олово 231
Триметил-(а-метилвинилфенил)свинец 559
Триметил-а-метилстирилгерманий 40
Триметил(оксиметил)олово 220
Триметилолово
азид 410
ацетат 397
ацетилениды 227, 254
бромистое 188, 330, 338, 435
гексафторарсенат 411
гидрид
присоединение к
акрилннтрилу 275
алкинам 289—297
диенам 277, 278
кетонам 304
хлоралю 301
реакции с
метил-^ис-В-триэтилстаннилакрилатом
299
1-триэтилстаннил-1-гексеном 299
^ш>|3-триэтилстаннилэтоксиэтиленом
299
гидроокись 382, 433
комплексы 435
гидроперекись 398
изотиоцианат 423
изоцианид 406
присоединение серы 432
йодистое 181, 188, 330, 338, 339
меркаптиды 414
пиролиз 423
реакции с
метилом йодистым 423
серой 423
монохлорацетат 397
нитрат 396, 406, 433
перхлорат 406
комплексы 433
сульфат 396
тетрафторборат 411
хлористое 188, 259, 260, 346, 354, 435
гидролиз 381
комплексы 432
реакция с треххлористым галлием 356
бас-(Три мети л олово)
селенид 413
сульфид 413
бмс-(Триметилолово)ацетилен 255
Триметилоловомарганецпентакарбонил,
присоединение к перфторалкенам 316
а-Триметилоловостирол 226
Триметилпентинилолово 327
Триметилперфторвинилолово 246
Триметилперфторфенил олово 229
реакция с водой 365
Триметилперфторэтилолово, реакция с
треххлористым бором 356
Триметил-(2-пиридилметил)олово 248
а-^мс-Р-Триметилплюмбилакрилонитрил 570
тряяс-Р-Триметилплюмбилакрилонитрил 570
г^с-Р-Триметилплюмбилметилакрилат 571
/пряяс-а-Триметилплюмбилметакрилат 571
Триметилплюмбил-бш>(триметилсилил)амин
607
Триметилпропенилолово 246
Триметил-я-пропилгерманий 35
Триметил-я-пропилсвинец 559
Триметил-(1-пропинил) олово 253
реакция с тетрациклоном 452
Триметилсвинец
азид 607
боронат 620, 621
бромистый 559, 576
гидрид 568, 620, 621
присоединение к
диэтилацетилендикарбоксилату 571
цианоэтину 570
этилену 570
реакции с
аммиаком 622
диазоэтаном 568
кислородом 622
хлористым водородом 622
гидроокись 601
реакция с винилацетиленом 574
хлористый 559, 575, 578
реакции с
хлором 576
щелочными металлами 623
бш>(Триметилсилиламино)трибутилолово 410
б^с(Триметилсилиламино) триметилолово 410
Триметилсилилтрибутилолово 502
Триметилсилилтриметилгерманийацетилен 42
бмс-(Триметилсилокси)тетрабутилдисганнок-
сан 391
бш:-(Триметилсилокси)тетраметилдистаннок-
сан 391
бш>(Триметилсилокси)тетрапропилдистаннок-
сан 391
б^с-(Триметилсилокси)тетраэтилдистаннок-
сан 391
Триметилсилокситри-я-бутилолово 385
Триметилсилокситриметилолово 412
Триметилсилокситриметилсвинец 608
Триметилсилокситрипропилолово 385
а-Триметилстаннилакрилонитрил 292
бш:-(Триметилстаннил)ацетилен 501
присоединение к тетрациклону 451
N-Триметилстаннилбензимидазол 410
я-Триметилстаннилбензойная кислота 453
/г-б^с-(Триметилстаннил)бензол 502
М-Триметилстаннил-1,2,3-бензтриазол 410
1 -Триметилстаннил-2-бромацетилен 501
Триметилстаннилдиазометан 315
Триметилстаннил-ди-я-бутилстибил 289
1-(Триметилстаннил)-2-(ди-«-бутилстибил)эти-
лен 289
Предметный указатель
691
Триметилстаннилдифениларсин 417
Триметилстаннил(дифенилфосфин) 417
N-Триметилстаннилимидазол 410
Триметилстанниллитий 496
реакция с три-я-бутилфосфатом 505
1-(Триметилстаннил)-2- метилтетрафенилбен-
зол 452
Триметилстаннилнатрий
реакции с
/2-дихлорбензолом 502
1,2-дихлорэтаном 501
четыреххлористым углеродом 501
электропроводность 497
(Триметилстаннил)пентафенилбензол 452
я-Триметилстаннилстирол, полимеризация
512, 513
1,2-бис- (Триметилстаннил)тетрафенилбензол
452
реакция с галоидными арилами 452
1,2-бш:-(Триметилстаннил)тетрафторэтан 316
1Ч-Триметилстаннил-1,2,2-триазол 399, 410
Триметилстаннилуксусная кислота, N,N-
диметиламид 314
Триметилстаннолят лития 421, 422
реакция с кремнийорганическими галоге-
нидами 412
Триметил-я-стирилгерманий 40
Триметил(тиоцианометил)олово 220
Триметил-л-(триметилгерманилбензил)олово
248
Триметил-4-триметилсилилбензилгерманий 52
Триметил(триметилсилилметил)олово 221, 224
Триметил-(я-триметилстаннилбензил)олово
224
1,1,1-Триметил-2,2,2-три-/г-толилдистаннан
504
1,1,1 -Триметил-2,2,2-трифенилдистаннан
504
Триметилтрифторметилолово 443, 444
реакции с
йодистым натрием 447
хлористым водородом 347
хлором 340
Триметилтрихлорметилолово 328
Триметилфенилсвинец, реакции с
иодом, кинетика 577
хлористым водородом 579
Триметилфенилэтинилолово 247, 32 7
реакция с тетрациклоном 452
Триметилфенилэтинилсвинец 574
Триметил-(о-фторфенил)олово 246
Триметил-я-хлорбензилгерманий 34
Триметил-я-хлорбензилолово 221
реакция с литием 498
Триметилхлорметилгерманий 47
Триметил(хлорметил)олово 220, 221
Триметил-я-хлорфенилолово 229
Триметилциклопентадиенилолово 328
Триметилпиклопропилолово 221
Три мети лэтил олово 500
Триметилэтилсвинец 559, 570
реакция с бромом 576
Триметилэтинилгерманий 41
Триметилэтинилолово 255
Триметоксибутилолово 417
Триметоксигерманий хлористый, реакция с
метиловым эфиром трибутилстаннилук-
сусной кислоты 57
Триметоксиметил германий 103
Триметилксиметилолово 417
Р-Триметоксисилилтриметилолово 452
Триметоксифенилолово 417
Триметоксиэтилолово 417
Три-а-нафтилгерманий бромистый 32
Три-а-нафтилмезитилолово 230
Три-а- нафтилолово
ацетат 398
бромистое 219
гидроокись 385
Тринеопентилолово галоидное 195
Три-«-нонилфенилгерманий 36
Три-(л*-оксифенил)фенилолово 228
Триоктадецилолово хлористое 346
Три-я-октилолово
ацетат 397
галоидное 189, 330
Три-«-октилфенилгерманий 36
Три-я-октилфенилолово 279
Триперфторфенилолово хлористое 218
Трипропилалкилолово 221
Трипропилаллилолово 226
Три-«-пропил(аллилфенил)олово 231
Трипропилацетонилолово 311
реакции с
метанолом 353
уксусной кислотой 353
фенил ацетиленом 353
Три-«-пропилбаренилолово 248
Трипропил-(л-бромфенил) олово 221, 229
Трипропилбутилолово, реакция с треххло-
ристым висмутом 357
Три-я-пропилвинилолово 221, 225
Три-«-пропилвинилэтинилолово 325
Три-«-пропилгексенил-1-олово 287
Три-«-пропилгерманий
бромистый 81, 95
изоцианат 99
йодистый 95
реакции с
иодмеркурацетоном 55
метиловым эфиром мер
кур-бш>уксус ной кислоты 55
формиат 97
фтористый 95
хлористый 95
Трипропилгермилуксусная кислота,
метиловый эфир 55
Три-«-пропилолово
гидрид 458, 461
присоединение к
алкенам 278, 283, 285
алкинам 287, 288, 290, 292
гидроокись 383
реакция с ацетиленом 325
изоцианат 416
йодистое 339
меркаптиды 399
хлористое 330
бш>(Три-я-пропилолово)
окись 211
перекись 416
Трипропилоловянный енолят ацетоуксус-
ного эфира 353
Три-«-пропилсвинец
боронат 621
гидрид 620, 621
гидроокись 601
44*
692
Предметный указатель
сульфат 605
/г-толуолсульфонат 581
фульминат 608
хлористый 575, 578
цианистый 608
бис-(Трипропилстаннил)ацетилен 398
Трипропилстаннилацетофенон 323
N-Трипропилстаннилимидазол 399
Р-Трипропилстаннилстирол 288
Трипропилстаннилуксусная кислота, эфиры
метиловый 312
реакции с
метанолом 353
хлористым ацетилом 369
пропиловый 312
этиловый 312
реакции с
ацетоуксусным эфиром 353
хлористым ацетилом 369
бис-l,2-(Три-я-пропилстаннил)этилен 288
Три-«-пропилфенилгерманий 36
Три-я-пропилфенилолово 221
Три-я-пропилэтинилолово 325
Тритетрадецилелово хлористое 346
Три-о-толил(бензил)олово 229
Три-о-толилмезитилолов о 230
Тритолилметаллилолово 226
Тритолилолово, азид 410
Три-ж-тол ил олово
ацетат 398
бромистое 343
гидрид 458
гидроокись 385
хлористое 332
Три-о-толиловоло
бромистое 269
гидрид 456
гидроокись 385
хлористое 218, 332
Три-/г-толил олово
ацетат 398
бромистое 343
гидрид 456
гидроокись 385
хлористое 269, 332
Три-ж-толи л свинец бромистый 557
Три-о-толилсвинец хлористый 579
Три-/г-тол ил свинец
ацетат 606
хлористый 579
Тритолилстанниллитий 498
Три-о-толилстанниллитий, реакции с
о-иодтолуолом 502
хлористым бензилом 502
Три-льтолил-л-юлилгерманий 53
Три-л-толил-о-толилгерманий 53
Три-ж-толил-я-толилолово 228
Три(триалкилстаннил)амины 409, 410
Три(триметилсилокси)метилолово 412
Три(триметилстаннил)амин 410
Три(три-«-пропилстаннил)амин 410
Три(трифенилааннил)висмут 489, 505
Три(трифенилстаннил)мышьяк 505
Три(трифенилстаннил)станниллитий 505
Три(трифенилстаннил)сурьма 490, 505
Три(трифенилстаннил)сурьма 490, 505
Три-ж-трифторметилфенилолово
бромистое 269
хлористое, реакция с литийалюминий-
гидридом 456
Три(триэтилстаннил)амин 410
Три(триэгилстаннил)висмут 489
Три(триэтилстаннил)сурьма 489
Трифенилалкенилолово 223
Трифенилалкилолово 223
Трифенилаллилгерманий 40
Трифенилаллилово 226
Трифенилаллилсвинец, окисление 617
Трифенил(аллилфенил)олово 232
Трифенил-о-аминофенилсвинец 563
Трифенил-я-аминофенилсвинец 562, 563
Трифенил(арил)олово 223, 228, 246
Трифенил(бенз-а-антрацен-7-ил)свинец^ 562
Трифенил(бромацетиленид)олово 254
Трифенил-/г-бромфенилолово 229
Трифенил-(1-бутадиинил)олово 255
Трифенилвинилгерманий 39
Трифенилвинилолово, полимеризация 512
Трифенил(я-винилфенил)олово 231
Трифенил(я-винилфенил)свинец 560
Трифенилвинилэтинилолово 227
Трифенилгерманий
акрилат 101
алкилмеркаптиды 104
бромистый 81
реакции с
азидом натрия 108
амидом лития 106
аммиаком 106
натриевыми солями гидроперекисей 102
серебром азотнокислым 99
серебряной солью трифенилгермил-
уксусной кислоты 100
гидрид 122, 124, 125
присоединение к
акриламиду 68
акрилонитрилу 68
ацетилену 64
1,1-дифенилэтилену 64
метил акр и л ату 68
метилвинилкетону 67
октадецену-1 64
октену-1 64
стиролу 64
трифенилаллилкремнию 69
трифенилаллилолову 68
циклогексану 64
реакции с
аллилмагнием хлористым 149
станниламинами 140, 154
фениллитием 148
гидроокись 89
реакция с трифенилгермилмуравьиной
кислотой 97
изоцианат 99
метакрилат 101
тиол 104
хлористый 86
реакция с N-натрийдифениламином 108
бш>(Трифенилгерманий)
перекись 102
сульфид 104
Трифенилгер мил калий 146
присоединение к 1,1-дифенилэтилену 151
Трифенилгермилллитий 146
присоединение к 1,1-дифенилэтилену 151
реакции с
азобензолом 154
азоксибензолом 154
альдегидами 150
Предметный указатель
693
бензофеноном 150
германием двуиодистым 152
свинцом двухлористым 152
селеном 150
серой 150
теллуром 150
2-фенилэтилом бромистым 151
флуореном 151
Трифенилгермилнатрий
окисление 150
реакции с
ал килами галоидными 151
аммонием бромистым 154
арилами галоидными 152
бором треххлористым 152
метиленом хлористым 151
пентаметиленбромидом 152
триметиленбромидом 152
трихлорсиланом 152
N-Трифенилгермилпиррол 108
Трифенилгермилтрифенилолово 503
/г-Трифенилгермилфенилдифенилметан 36
Трифенилгермилферроцен 51
1,Г-бш>(Трифенилгермил)ферроцен 51
Трифенил-.и-гидроксифенилолово 230
Трифенил-.и-(или о-, я-)-диметиламинофе-
нилолово 247
Трифенил-о-диметиламинофенилсвинец 563
Трифенил-я-диметиламинофенилсвинец,
реакция с уксусной кислотой 584
Трифенил-(2-диметиламинофлуоренил)олово
247
Трифенил-(1-инденил)олово 223. 232, 248
Трифенил-3-инденилсвинец 562
Трифенил-/г-иодфенилолово 241
Трифенил-/г-ксилилсвинец, реакция с
мом 578
Трифенилмезитилолово 230
Трифенилметаллилолово 226
Трифенил-(1-метил-4-бутадиинил)олово
Трифенил-(о-метоксиметилфенил)олово
230
Трифенил-а-нафтилсвинец 578
Трифенил-1-[2-окси-3,5-ди-(я-нитрофенилазо]
фенилсвинец 619
Трифенил-о-оксиметилфенилолово 247
реакция с перманганатом калия 358
Трифенил-(/г-оксиметилфенил)олово 247
Трифенил-л*-(или о-, /г-)-оксиметилфенил-
свинец 563
Трифенил-ж-оксифенилолово 247
Трифенил-о-оксифенилолово 247
Трифенил-о-оксифенилсвинец 563
Трифенил-2-окси-З-хлорпропилолово 502
Трифенил-я-октадецилгерманий 36
Трифенилолово
азид 410
реакция с фосфинами 316
акрилат, полимеризация 513
арсонаты 412
ацетат 398
ацетилениды 227, 232
бромистое 332, 343
реакции с
бромом 343
нафталиннатрием 498
/г-винилбензоат 398
полимеризация 513
гидрид 457, 458, 461, 572
окисление 464
бро-
254
223,
присоединение к
азобензолу 306
алкенам 275, 279, 281—287
алкинам 289—291, 293
салициловому альдегиду 303
разложение 464
реакции с
аллиламином 282
аллитрифенилгерманием 286
аллилтрифенилкремнием 286
аллилтрифенилоловом 286
аллилтрифенилсвинцом 287
арилизоцианатами 310
бромистым водородом 464
бромсукцинимидом 464
винилтрифенилгерманием 287
винилтрифенилсвинцом 286
винилфенилкетоном 282
диаллилдифенилгерманием 287
диаллилдифенилкремнием 287
диаллилдифенилоловом 287
дивинилдифенилоловом 287
диметилвинилметилкетоном 282
диметил кадмием 484
дифенилхлорфосфином 490
диэтиламидом диэтилалюминия 488
бш>(диэтиламино)железом 490
диэтиламинотриалкилгерманием 488
диэтилцинком 484
кислотами 465, 466
магнийорганическими соединениями
485
метилкадмием хлористым 485
метиллитием 484
перекисью бензоила 490
тетра(диметиламино)титаном 489
тетра(диэтиламино)цирконием 489
N-трибутилплюмбилимидазолом 572
триэти л висмутом 489
триэтилоловом хлористым 483
триэтилсурьмой 490
триэтил-(Ы-фенилформамидо)оловом
483
фенилизотиоцианатом 310
фениллитием 484
N-фенилпропионамидом диэтиалюми-
ния 488
этилцинком хлористым 485
гидроокись 384
диспропорционирование 378
реакции с
надкислотами 399
перекисью водорода 398
реактивом Фишера 384
гидроперекись 398
дейтерид 460
йодистое 197, 343
диспропорционирование 378
метакрилат, полимеризация 512, 513
нитрат 406
фенилселенид 362
фенилсульфинат 465
фенилсульфонат 465
фтористое 405
фульминат 406
хлористое 200, 269, 331, 348, 354
диспропорционирование 378
реакции с
магнием 496
нафталиннатрием 498
694
Предметный указатель
перекисью водорода 413
серой 361
три(трифенилгермил)силиллитием 411
цианистое 396
гидролиз 363
цианацетат, декарбоксилирование 407
а-цианпропионат, декарбоксилирование 407
бис- (Трифенилолово)
перекись 413
селенид 406
сульфат 465
сульфид 421, 430
бш>(Трифенилолово), диацетилениды 255
Трифенилоловокобальттетракарбонил 507
Трифенилоловомарганецпентакарбонил 507
реакции с
галоидами 509
тетрафенилциклопентадиеноном 510
трихлороловомарганецпентакарбонилом
509
Трифенилоловоренийпентакарбонил 411, 506
реакции с
трифениларсином 509
трифенилстибином 509
трифенилфосфином 509
хрористым водородом 495, 508
хлором 495, 508
Трифенил-4-пентенилолово 226
Трифенилперфторвинилгерманий 40
Трифенилперфторвинилолово 226
Трифенилперфторфенилолово 229
Трифенил-3-пиридилолово, реакция с
метиллитием 455
Трифенил-2-пиридилсвинец 561
я-бш>(Трифенилплюмбил)бензол 563
Трифенилплюмбиллитий 623, 624, 625
гидролиз 625
реакции с
алкилами галоидными 627
арилами галоидными 627
гетероциклическими соединениями 627
3-диэтиламинопропилхлоридом 627
р-пропиолактоном 628
селеном 625
серой 625
теллуром 625
флуореном 628
эпихлоргидрином 628
Трифенилплюмбилмагний бромистый 555
бш>(Трифенилплюмбил)тетракарбонилжелезо
630
реакция с дигалогенидами ртути 630
Трифенилплюмбилуксусная кислота,
этиловый эфир 572, 598
(Трифенилплюмбил)уксусный ангидрид 572
реакция с бромом 577
Трифенилплюмбилуксусная кислота,
этиловый эфир 572, 598
Трифенил-2,3-пропандиолсвинец 617
Трифенилпропаргилолово 227
Трифенил-(1-пропинил-1)-олово 227
Трифенилсвинец
азид 605, 607
акрилат, полимеризация 536
ацетат 614
реакция с кетеном 572
бромистый 577
я-винилбензоат 605
гидрид 621
гидроокись 572, 602, 605
реакции с
кетеном 572
натрием 602
перекисями 606
дифенилфосфинат 610
изобутират 605
йодистый 577, 613
малеат 614
меркаптиды 606
матакрилат, полимеризация 536
нитрат 605
я-пропилмеркаптид 606
фульминат 608
этилмалонат, термическое разложение 598
хлористый 552, 560, 569, 579, 614
диспропорционирование 597
реакции с
ацетиленидами лития, натрия 562
литием 624
органическими кислотами 583
производными арсиновых кислот 582
сулемой 585
сульфидом натрия 606
/г-фенилендилитием 563
Трифенилсвинецкобальттетракарбонил 629
Трифенилсвинецмарганецпентакарбонил,
реакция с хлористым водородом 629
Трифенилсвинецренийпентакарбонил 629
бш>(Трифенилсвинец)сульфид 612
Трифенилсвинец-я-циклопент адиенилтри-
карбонилвольфрам 629
Трифенилсвинец-л- циклопентадиенилтри-
карбонил молибден 629
бис- (Тр ифени л ста нни л) аце1 и лен 501
N-Трифенилстаннилбензимидазол 400
я-Трифенилстаннилбензойная кислота 453
М-(1)-Трифенилстаннил-1,2,3-бензтриазол 400
1-Трифенилстаннил-2-ди-«-бутилс1ибил-
этилен 289
К-Трифенилстаннил-М,М-дифенилгидра-
зин 306
1-Трифенилстаннил-2-дифенилстибилэтилен
289
N-Трифенилстаннилимидазол 400
бш>(Трифенилстаннил)кадмий, комплекс с
2,2'-бипиридилом 485
Трифенилстанниллитий 496, 498, 499
гидролиз 499
карбонизация 499
реакции с
алкилами бромистыми 500
бромом 499
висмутом треххлористым 505
германием четыреххлористым 503
диметилкремнием двухлористым 503
дифенилкремнием двухлористым 503
диэтил карбонатом 502
мышьяком треххлористым 505
окисью этилена 502
пропаргилом бромистым 500
свинцом хлористым 503
селеном 500
серой 499, 500
сурьмой треххлористой 505
теллуром 500
три-я-бутилфосфатом 505
триметилкремнием хлористым 502, 503
трифенилгерманием хлористым 503
трифенилоловом галоидным 503
фенилхлорфосфинами 505
Предметный указатель 695
флуореном 502
фосфором треххлористым 505
эпихлоргидрином 502
этилхлорформиатом 502
Трифенилстаннилмагний бромистый 485, 486
бш>(Трифенилстаннил)магний 496
Трифенилстаннилнатрий 498
окисление 499
реакции с ■
бензоилом хлористым 499
бензофеноном 499
двуокисью серы 499
дииодацетиленом 501
дифенилсульфидом 500
монохлорацетатом натрия 501
углекислым газом 499
четыреххлористым углеродом 501
N-Трифенилстаннилпиррол 410
1,3-бис-(Трифенилстаннил)пропан 287
Р-Трифенилстаннилпропионовая кислота 283
/г-Трифенилстаннилстирол
полимеризация 512, 513
сополимеризация со стиролом 513
Р-Трифенилстаннилстирол 290
N (1)-Трифенилстаннил-1,2,3-триазол 410
1-Трифенилстаннил-2-трифенилгермилэтан286
1 -Трифенилстаннил-2-трифенилсилил- этан
286
я-[бш>(2-/г-Трифенилстаннилфенил)этил-
станнил]бензол 286
1,2-бис-(Трифенилстаннил)этан 287
1,2-бис-(Трифенилстаннил)этилен 289
Трифенил-/г-стирилгерманий 40
Р-Трифенилстирилсвинец 560
Трифенил-/г-стирилсвинец
полимеризация 536
реакция с гидридом трифенилолова 617
Трифенил-(2-тиенил)олово 223
Трифенил-(2-тиенил)свинец 561
Трифенил-/г-толилгерманий 36
реакция с бромом 81
Трифенил-я-толилсвинец 562
реакция с бромом 578
Трифенил(триметилсилилметил)олово
223, 224
Трифенил-я-(триметилсилилфенил)олово
223, 224
Трифенилтриэтилдигерман 153
Трифенил(фенилбаренил)олово 248
Трифенил-(1-фенил-4-бутадиинил) олово 254
Трифенил-9-флуоренилсвинец 562
Трифенил-/г-фторфенилолово 229
Трифенил-(2-фурил)олово 223, 232
Трифенил-(2-фурил)свинец 561
Трифенил(хлорацетиленид)олово 254
Трифенил-/г-хлорфенилолово 229
Трифенилциклогексилгерманий 36
Трифенилциклогексилолово 223
Трифенил-1-циклопентадиенилолово 223, 228
Трифенилциклопропилолово 223
Трифенил-(1-этил-4-бу1адинил)олово 254
Трифенилэтилгерманий 36
Три-(2-фенилэтил)олово
ацетат 398
бромистое 338
гидроокись 385
Трифенилэтинилгерманий 51
Трифенилэтинилолово 255
бис-(Три-о-фторбензилолово), окись 219
Трифтор метил германий трехиодистый 23
Три-/г-фторфенилолово
гептафторбутират 352
гидрид 456
трифтор ацетат 352
хлористое 332
бш>(Три-о-хлорбензилолово), окись 219
7,7,7 -Трихлор-а-бромпропилтрифенилолово,
реакция со щелочами 450
Три-траяс-у-хлорвинилолово хлористое 270
перегруппировка в ^ш>изомер 453
Три-^нс-р-хлорвинилолово хлористое 270
Трихлоргермилуксусная кислота, эфиры
метиловый 56, 71
этиловый 71
1,2-бш:-(Трихлоргермил)этилен 61
Трихлорметилфенилолово 322
Три-я-хлорфенилолово хлористое, реакция
с литийалюминийгидридом 456
2,4,6-Трихлорфенокситрифенилолово 409
Три-(а-хлорэтил)олово хлористое 321
Трициклогексилалкилолово 223
Трициклогексиларилолово 228, 246
Трициклогексил-о-бифенилолово 253
Трициклогексил-(/г-винилфенил)олово 231
Трициклогексил германий
гидрид 124
метакрилат 101
хлористый 47
Трициклогексилгерманол 31
Трициклогексил-(/г-диметиламинофенил)
олово 247
Трициклогексил олово
бромистое 214, 338
реакция с реактивом Гриньяра 223
галоидное 338, 396
гидроокись 385
Трициклогексилсвинец
гидрид 620, 621
гидроокись 602
йодистый 576
Трициклогексил-/г-толилсвинец 562
Трициклогексилфенилолово 253
реакция с кислотами, кинетика 350
Трициклогексилфенилсвинец 562
Трицилогексил-л*-(или
/г-)-хлорфенилсвинец 562
Трициклопентадиенилвинилолово 223
Три-1-циклопентадиенилфенилолово 223
Трициклопропилолово
бромистое 354
йодистое 406
хлористое 354
Трициклопропилсвинец хлористый 582
Триэтилалкилолово 221
Триэтилаллилолово 221, 226
присоединение к
бензальдегиду 272
/г-трихлорбензальдегиду 273
уксусному ангидриду 273
хлоралю 273
Тоиэтилаллилсвинец 626
Триэтил(аллилфенил)олово 231
Триэтил-/г-анизилолово, реакция с диаце-
татом ртути 354
Триэтиларилолово 221
Триэтилбензилсвинец 559
Триэтил я-бромфенилгерманий 117
Триэтил-л-бромфенилолово 229
Триэтил-п-бромфенилсвинец 559
Триэтил-(1-бутадиинил)олово 255
696
Предметный указатель
Триэтилбутанолгерманий 36
Триэтил-(бутен-3-ин-1 -ил)олово 227
Триэтил-(2-я-бутилпиридил-4-метил)олово
248
Триэтил-я-бутилсвинец 559, 626
Триэтил-#-бутилсвинец 559, 626
Триэтил-втор-бутилсвинец 559, 626
Триэтил-трет-бутилсвинец 559
Триэтилвинилгерманий 39
Триэтилвинилолово 221, 225
полимеризация 512
присоединение
полигалоидметанов 449
трихлорсилана 452
присоединение к циклонам 451
сополимеризация с олефинами 512
Триэтилвинилсвинец 559
полимеризация 536
Триэтил-(/г-винилфенил)олово 231
Триэтил-(Аг-винилфенил)свинец 559
Триэтилвинилэгинилолово 227, 325
Триэтил-(Ы-гексилкарбамоил)олово 308
Триэтилгептанолгерманий 36
Триэтилгерманий
акрилат 101
алкилмеркаптиды ПО
ацетиленид, присоединение брома 115
бромистый 81
реакция с углекислым серебром 99
я-валерат 100
гидрид 122, 124
присоединение к
акриловой кислоте 67
аллилацетату 68
аллиловому спирту 65
ацетилену 65
винилэтилкарбинолам 65
гексину-1 65
диметиловому эфиру аллилборной
кислоты 68
пропаргиловому спирту 65
триэтилаллилгерманию 68
эфиру винилуксусной кислоты 68
реакции с
диэтилкадмием 140, 154
диэтил ртутью 139, 154
диэтилцинком 139
селеном 131
серой 131
теллуром 131
триэтилвисмутом 140, 154
триэтилталлием 140, 154
хлористым сульфурилом 135
изоцианат 99
йодистый, реакции с
трибутилстаннилацетоном 57
трибутилстаннилкусусной кислоты
метиловым эфиром 57
метакрилат 101
монохлорацетат 97
фтористый 82
хлористый, реакция с натриевыми солями
гидроперекисей 102
а-хлорпропионат 100
1-Триэтилгерманилбутен-1 42
1-Триэтилгерманил-З-метилбутен -З-ин-1 42
1-Триэтилгерманил-3-метилбутен-3-ил-1 42
1 -Триэтилгерманилпентен-3-ил-1 42
Триэтилгермилацетон 76
N-Триэтилгермилимидазол 98
бш>(Триэтилгер мил) кадмий 140
Триэтилгермиллитий,реакция с аммиаком 153
Триэтилгермилметилфенилацетон 76
бш>(Триэтилгер мил) ртуть 139
трш>(Триэтилгермил)таллий 140
Триэтил-я-диметиламинофенилолово 270
Триэтил (диметилэтилсилилметил)олово 224
Триэтил (диметилэтилсилил)олово 224
Триэтил-(Аг-а-,|3-дифтор-р-хлорстирил)оло-
во 247
Триэтилизоамилсвинец 559
Триэтилизопропенилолово, присоединение
трифенилхлорметана 450
Триэтилизопропилсвинец 559
Триэтил-я-иодфенилолово 241
Триэтил (карбэтоксиметил)германий 75
Триэтилметаллилолово 226
Триэтил-(я-а-метилвинилфенил)олово 231
Триэтил-2-метилпропенил-2-свинец 626
реакция с бромом 617
Триэтилметилсвинец 559
Триэтил-а-метилстирилгерманий 40
Триэтил-я-метоксифенилэтинилгерманий 41
Триэтил-1-нафтилгерманий 51
Триэтил-2-нафтилгерманий 51
Триэтил-(3-оксипропил)олово 281
Триэтил-о-оксифенилолово 247, 270
Триэтил-«-октилолово 222, 278
Триэтилолово, колориметрическое
определение 432
Триэтилолово
арилсульфонаты 406
ацетат 397
ацетилен иды 254
ацетилацетонат 399
бензилселенид 416
бромистое 132, 187, 330, 331, 338, 354
гидрид 458, 460, 461
присоединение к
алкенам 274, 275, 277—279, 281,
283—286
алкинам 288, 290—296
альдегидам 301—303
арилизоцианатам 308, 309
бензальанилину 310
гексилизоцианату 308
диенам 278
динитрилам алкилиденмалоновой
кислоты 305
дициклогексилкарбодиимиду 305, 310
диэтилазодикарбоксилатам 306
кетонам 304
нитрозобензолу 310
реакции с
азобензолом 306
я-аминоазобеизолом 306
галогенидами металлов 465
гидроперекисью трет-бутила 467
дибензилртутью 486
дифенилртутью 486
диэтиламинотриэтилоловом 483
диэтилоловом двухлористым 483
диэтилртутью 486
диэтилцинком 484
ионом тропилия 491
метилизотиоцианатом 309
метилртутью ацетатом 484
/г-нитрозодиметиланилином 310
окислами элементов 465
перекисями ацилов 490
Предметный указатель
697
трифенилвисмутом 486
трифенилметильными радикалами 491
триэтилвисмутом 489
триэтил-(1М-гексилформамидо)оловом
483
триэтилсурьмой 489
этилртутью хлористой 484
эфирами меркур-бш>уксусной
кислоты 486
гидроокись 306, 382
комплексы 435
реакции с
ацетиленовыми спиртами ?26
винилацетиленом 325
фенилацетиленом 325
гидроперекись 398, 399
дейтерид .460
присоединение к
метилпропиолату 294
цианоацетилену* 295
изотиоцианат 396, 416
изоцианид 396, 405, 406
йодистое 181, 338, 339, 354
меркаптиды 353, 399, 414
монохлорацетат 397
нитрат 406
сульфат 396
тиол 397
/г-толуолсульфонамид 399
трифторацетат 351
фенилацетонилкарбинолят 311
фтальимид 399
фтористое 405
хлористое 188, 199, 211,260,330,331,354
диспропорционирование 378
реакции с
оловом хлорным 333
перекисью бензоила 368
фотолиз 365, 366
цианацетат, декарбоксилирование 407
этилмалонат, декарбоксилирование 407
этилмеркаптид 399
бш>(Триэтилолово)
карбонат 397
перекись 398, 416
сульфид 309, 397
реакции с
германием четыреххлористым 420
додецилкремнием трехиодистым 420
кремнием четырехбромистым 420
мышьяком треххлористым 420
оловом хлористым 420
оловом хлорным 420
фосфором треххлористым 420
Триэтилоловянный енолят ацетоуксусного
эфира 312
Триэтилперфторвинилолово 226
реакция с этанолом 353
Триэтил-(2-пиридилметил)олово 248
Триэтилплюмбиламилмагний бромистый 618
Триэтилплюмбилнатрий, реакции с
арилами галоидными 626
бензилом хлористым 626
2-бромстиролом 626
я-бутилом бромистым 626
вишглбромацетиленом 626
дипиклопентадиенилтитаном
двухлористым 629
иодбензолом 626
метил-2-винилбромацетиленом 626
Триэтилплюмбилстирол 626
бис-(Триэтилплюмбил)фосфонат 608
Триэтилпропаргилгерманий 41
Триэтилпропенилгерманий 48
Триэтил-я-пропилсвинец 559
Триэтилсвинец
ацетат 581, 609, 613
ацилаты 581, 582
боронат 621
бромацетат 581
бромистый 576, 595, 613
гидрид 568, 620
реакция с диазоэтаном 568
гидроокись 574, 601, 612
реакция с ацетиленом 574
термическое разложение 597
нитрат 585, 613
/г-толуолсульфонат 581
формиат 609
фосфат 584, 609
хлористый 547, 559,575,578,580,613,614
реакции с
ацетиленидами щелочных металлов 561
диазоалканами 568
натриевыми производными нитроме-
тана 607
сульфидом натрия 606
фторфосфонатом серебра 608
циклопентадиенилнатрием 561
этилмеркаптид 610
бис-(Триэтилсвинец), сульфид 606
Триэтилсвинецмарганецпентакарбонил 629
1-Триэтилсилил-2-триалкилстаннилэтилен,
реакции с
сулемой 288
хлористым водородом 288
хлором 288
1-Триэтилсилил-2-три-я-бутилстаннилэтилен
288
1-Триэтилсилил-2-три-я-пропилстаннилэтилен
288
Триэтилсилокситриэтилсвинец 608
бмс-(Триэтилстаннил)ацетилен 398, 501
Триэтилстаннилацетон 311, 315
присоединение к бензальдегиду 311
реакции с
ацетилом хлористым 368
водой 363
серой 359
/г-Триэтилстаннилбензойная кислота 453
1 -Триэтилстаннил-2-бромацетилен 501
1-Триэтилстаннилбутен-1-ин-3 290
1-[4-(Триэтилстаннил)бутин-3-окси]-1-бут-
оксиэтан 327
Триэтилстаннилвинилацетилен 500
присоединение
диазометана 450
трифенилметильных радикалов 450
1 -Триэтилстаннилгексен-1 -ин-3 290
М,1Ч-бш>(Триэтилстаннил)-]\[,]\Г-ди (эт-
оксикарбонил)гидразин 306
Триэтилстаннилизопропенилацетилен, при
соединение диазометана 451
Триэтилстанниллитий 497
реакции с
диметиламиноборхлоридами 502
диметилкремнием двухлористым 503
дициклопентадиенилтитаном
двухлористым 503
Триэтилстаннилнатрий, реакции с
€98
Предметный указатель
1-бромбутадиеном-1,3 500
винил ацетиленом 501
р-Триэтилстаннилстирол 288
л-Триэтилстаннилстирол, полимеризация
513
N-Триэтилстаннилтиоформимидат 309
Триэтилстаннилуксусная кислота, эфиры
метиловый 271, 311
пропиловый 486
Триэтилстаннилфенилгидроксиламин 310
8-Триэтилстаннил-]М-фенилтиоформимидат
309
П'бис- [1М-(Триэтилстаннил)формамидо] -
бензол 308
Триэтилстаннилциклопропилацетилен 450
Триэтил-я-стирилгерманий 40
Триэтил-л*-(или я-)-толилолово, реакция с
диацетатом ртути 354
1,1,1 -Триэтил-2,2-2-трифенилдистаннан
306, 309
Триэтил-/г-трифторметилсвинец 626
Триэтил-(я-триэтилсилилфенил)олово 224
Триэтилфенилолово 253, 270
реакции с
диацетатом ртути 354
серебром азотнокислым 356
Триэтилфенилсвинец 562, 626
реакция с натрием 624
Триэтилфенилэтинилолово 325, 327
присоединение к
хлоралю 273
циклогексанону 273
Триэтилхлорметилсвинец 568
Триэтил-3-хлорпропионилсвинец 574
Триэтил-л*-(или /г-)-хлорфенилолово,
реакция с диацетатом ртути 354
Триэтил-а-хлорэтилгерманий 117
Триэтил-а-хлорэтилсвинец 568
Триэтилциклопентадиенилгерманий,
реакция с бромом 82
Триэтилэтинилгерманий 41
Триэтилэтинилолово 255, 325
Триэтоксибутил германий 103
Триэтоксибутилолово 417
Триэтоксифенилолово 417
Фенилбензил-я-толилолово, гидроокись 385
Фенилгерманий
трехбромистый 24
трехиодистый 23
трех хлор истый 23, 24, 86
реакция с диазометаном 74
Фенилдибромидоловопентакарбонилрений
509
Фенилдихлороловотрикарбонил - я- циклопен-
тадиенилвольфрам 508
Фенилдихлороловотрикарбонил-я-циклопен-
тадиенилмолибден 508
я-Фенилен-бш>(диметилоловогидрид) 458
присоединение к
триметил-(я-винилфенил)олову 286
трифенил-(/г-винилфенил)германию 286
трифенил-(/г-винилфенил)свинцу 286
о-Фенилендиоксиди-л-бутилолово 414
о-Фениленидиоксидиметилолово 414
/г-Фенилен-бш>(хлордиметилолово) 240
Фенилолово
азид, реакция с фосфинами 316
(дибром)гидрид 464
оксимоноацилат 392
трехбромистое 413
трехиодистое 413
треххлористое 256, 336, 354
реакции с
азотистым ангидридом 370
галоидоводородными кислотами 413
диазометаном 322
иодом треххлористым 344
серой 361, 362
триацетат 407
три-«-бутират 407
тригидрид 461
реакции с
кетонами 474
триал кил-(Ы-фенилформамидо)оловом
482
триизобутират 407
трипропионат 407
Фенилоловотри(ренийпентакарбонил),
реакция с бромом 509
Фенилплюмбоновая кислота 604
Фенилсвинец
триацетат 565, 585
трибензоат 609
триизобутират 565, 585
Фенилстанноновая кислота 393
реакция с окисью ртути 355
Фенилсульфамидотриэтилсвинец 608
Фенилтриалкилолово 241
Фенилтриарилолово 241
N-Фенилтриметилстанниламин 418
Фенилтри-(ж-оксифенил)олово 247
Фенилтриперфторвинилолово 226
Фенилтри-((фенилэтинил)олово 251, 327
Фенилтри-(9-флуоренил)олово 251
Фенилтри-(1-циклопентадиенил)олово 241
Фенилтрициклопропилолово 241
Фенилтриэтилгерманий 38
Фенил-бш>(триэтилстаннил)фосфин 411
бш>(Ы-Фенилформамидо) диэтилолово 309
N-Фенилформамидотри-я-бутилолово 308
N-Фенилформамидотриэтилолово 308,416
(2-Фенилэтил)дифенилолово йодистое 343
(2-Фенилэтил)трифенилолово 279
Фенокситрибутилолово, присоединение к
фенилизоцианату 314
Фенокситриметилолово, донор
но-акцепторные свойства 434
Фенокситри-я-пропилолово 399
Фенокситрифенилолово 399
Фенокситриэтилолово 399
донорно-акцепторные свойства 434
реакция с перекисью бензоила 368
(2-Феноксиэтил)трифенилолово 282
(9-Флуоренил)дифенилолово хлористое 350
(9-Флуоренил)трифенилолово 248
ж-Фторфенилгерманий трехбромистый 24
2-Фурилтрифенилолово 248
Фурфурилокситриэтилолово 303
(Хлор)алкилперокситетраалкилдистаннок-
саны 413
Хлоргермилтриуксусная кислота,
метиловый эфир 56
(Хлор)гидрокситетраметилдистанноксан 386
(Хлор)гидрокситетраэтилдистанноксан 386
(Хлор)ди-1,3-дифенилтриазинфенилолово 411
Хлорметилгерманий треххлористый 74
Хлор метилдиметил (или фенил)олово
хлористое 322
Предметный указатель
699
Хлорметилтриметилгерманий 38
Хлорметилтрифенилолово, реакция с
йодистым натрием 447
Хлорметилтриэтилолово, реакции с
диметиламином 447
калиевой солью пиррола 447
морфолином 447
этиленимином калия 447
(Хлор)метокситетраметилдистанноксан 386
{Хлор)(р-нафтокси)тетрабутил (или пропил)-
дистанноксан 414
{3-Хлор-2-оксипропил)трифенилсвинец 628
реакции с
хлористым водородом 577
хлором 577
n-Хлорфенилолово треххлористое 337
Хлорфенилтрибутилолово 317
N-/z -Хлорфенилформамидотриэтилолово 308
2-Хлорфенокситрифенилолово 408
а-Хлорэтилгерманий треххлористый 117
Р-Хлорэтилгерманий треххлористый 117
дегидрохлорирование 117
а-Хлорэтилолово треххлористое 321
(Цианметил)дифенилолово йодистое 342
(Цианметил)трибутилолово, реакция с литий-
алюминийгидридом 446
(Цианметил)трифенилолово, реакция с
иодом 342
щелочами 363
(1-Цианпропил-2)-трифенилолово 285
(З-Цианпропил)трифенилолово 285
реакции с
бромом 343
щелочами 363, 448
(З-Цианпропил)фенилолово двубромистое 343
(2-Цианэтил)дифенилолово йодистое 343
(2-Цианэтил)три-#-бутилолово 285
(2-Цианэтил)трипропилолово, реакции с
литийалюминийгидридом 447
метилмагнием йодистым 447
(2-Цианэтил)трифенилолово 285
реакции с
иодом 363
щелочами 363, 448
(2-Цианэтил)триэтилолово 285
Циклогексилокситри-#-бутилолово 303
реакция с перекисью бензоила 368
К-Циклогексил-М-триэтилстаннил-]Ч-цикло-
гексиламидин 305, 310
Циклоокси-бш>(диметилсилилметил)дибутил-
олово 240
Циклопентадиенилтрифенилолово 328
окисление 454
Циклопентаметиленгерманий
гидрид, окисление 130
дигалогенид 36
Циклопентаметилендифенилгерманий 49
Циклопентаметилендифенилолово,
реакции с
иодом 345
хлористым водородом 347
Циклопентаметиленолово двубромистое 347
Циклопентаметиленфенилолово йодистое 345
Ци клопентенил-1 - ацетиленидтриэтил свинец
561
Циклоггропилолово
трехбромистое 354
треххлористое 354
Циклопропилтриметилолово 246
Циклотетраметиленгерманий
двухлористый 31, 96
дигалогениды 36
Этиламинотриметилолово 422
Этил-я-бутилсвинец двубромистый 576
Этилгерманий
трехиодистый 22
■ треххлористый 20, 23, 57, 60
реакции с
диметиламином 107
диэтиламином 107
хлорирование 117
тригидрид 122
триизоцианат 99
5-Этил-5,10-дигидро-10,Ю-дифенилфеназагер-
мин 49
Этилдинеопентшюлово бромистое 340
Эгилдифенилсвинец бромистый 577
Этилениминотриэтилолово 444
Этилизоамилсвинец двухлористый 576
Этилизобутилсвинец двухлористый 567
Этилнеопентилолово двубромистое 340
Этилолово
(бром)дигидрид 464
гидроксихлорид 392
(дибром)гидрид,присоединение к стиролу 277
трехбромистое 335
трехиодистое 181, 189
треххлористое 335, 336
тригидрид 460
тримеркаптиды 414
Этилперокситриэтилолово 399
Этил-emop-/г-тол и л германий 38
Этил-втор-пропилдифенилгерманий38
Этилпропилолово двухлористое 346
Этилстанноновая кислота 190, 392
диспропорционирование 378
соли 393
Этилтри-я-бутил германий 38
Этилтривинилгерманий 39
Этилтриметилгерманий 38
Этилтриметилолово 256
Эгилтрифенилгерманий 151
Этилтрифенилолово 500
реакция с азотнокислым серебром 356
Этилтрифенилсвинец 560, 627
реакция с бромом 577
Этилфенил-а-нафтилгерманий, гидрид 124
Этилфенил-/г-толилгерманий 38
(З-Этоксикарбонилпропил)триэтилолово,
скорость гидролиза 448
(2-Этоксикарбонилэтил)триэтилолово,
скорость гидролиза 448
Этокситри-я-бутилгерманий, реакции с
бензойной кислотой 111
этиленгл и колем 111
Этокситриэтилолово 408
присоединение к
бутилизоцианату 313
диметилацетилендикарбоксилату 313
диэтилацетилендикарбоксилату 313
реакции с
азодикарбоновым эфиром 314
димером кетена 312
перекисью бензоила 368
Этокситриэтилсвинец 605
Эфиры гермилуксусной кислоты 56
ОГЛАВЛЕНИЕ
Предисловие 5'
ГЕРМАНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Введение' g
Г л а в а I. Прямой метод синтеза германииорганических соединений из галоидных
алкилов и металлического германия 19
Г л а в а II. Синтез германииорганических соединений действием галоидных
алкилов на соли германия или их комплексы 22
Глава III. Синтез германииорганических соединений с помощью магнийорганиче-
ских соединений 26
Синтез германииорганических соединений типа R4Ge 26
Синтез германииорганических соединений типа R3GeX, R2GeX2 и RGeX3 . . 30
Синтез германииорганических соединений типа R3Ge — GeR3 32
Синтез несимметричных германииорганических соединений действием магний-
органических соединений на германийорганические соединения 34
Глава IV. Синтез германииорганических соединений с помощью металлооргани-
ческих соединений лития, натрия, цинка, ртути, алюминия, олова и свинца . . 47
Синтез германииорганических соединений с помощью литийорганических
соединений 47
Синтез германииорганических соединений с помощью натрийор ганических
соединений 50
Синтез германииорганических соединений с помощью цинкорганических
соединений 53
Синтез германииорганических соединений с помощью ртутноор ганических
соединений 54
Синтез германииорганических соединений с помощью алюминийорганических
соединений 55
Синтез германииорганических соединений с помощью олово- и свинцовоорга-
нических соединений 56
Г л а в а V. Синтез германииорганических соединений присоединением различных
германииорганических соединений и солей германия к кратным связям .... 60
Присоединение HGeX3 к непредельным соединениям 60
Присоединение гидридов алкил- и арилгермания к кратным связям .... 64
Присоединение R2GeHX к кратным связям 69
Присоединение алкоксисоединений германия к кетену 71
Присоединение двуиодистого германия к производным ацетилена и бутадиена 71
Глава VI. Диазометод синтеза германииорганических соединений 74
Синтез германииорганических соединений с помощью алифатических диазосое-
динений 74
Синтез германииорганических соединений с помощью солей диазония ... 76
Глав а VII. Синтез германииорганических соединений заменой водородного атсма
на атом германия (германирование) 77
Глав а VIII. Органические соединения германия типа (R3Ce)n, (R2Ce)^ и (RGe)^ 78
Г л а в а IX. Деалкилирование германийсрганических соединений 80
ГлаваХ. Гидрслиз германииорганических соединений 89
Главка XI. Реакции германииорганических соединений, связанные с
преобразованием функциональных групп у атома германия . . 95
Реакция германииорганических гидроокисей, окисей и ангидридов германий-
органических кислот 95
Оглавление 701
Реакции обмена германийорганических галоидопроизводных 98
Реакции обмена германийорганических ацилатов, алкокси- и аминосоединений 109
Г л а в а XII. Реакции германийорганических соединений, связанные с
изменениями в радикале 114
Г л а в а XIII. Синтез и реакции германийорганических гидридов 122
Синтез органических гидридов германия 122
Реакции органических гидридов германия 129
Реакции с кислородом, серой, селеном, теллуром 129
Реакции с соединениями, содержащими подвижный водород 131
Реакции с галоидами, галоидводородными и другими минеральными
кислотами, солями металлов и металлоидов 134
Реакции с галоидными алкилами 136
Реакции с металл оорганически ми соединениями 137
Реакции с литийорганическими соединениями 137
Реакции с магнийорганическими соединениями 137
Реакции с ртутноорганическими соединениями 138
Реакции с цинк- и кадмийорганическими соединениями 139
Реакции с алюминий-, висмут- и таллийорганическими соединениями 140
Реакции с оловоорганическими соединениями 140
Реакции с диазопроизводными 141
Реакции с непредельными соединениями 141
Г л а в а XIV. Синтез и реакции германийорганических соединений, содержащих
два различных металла 144
Синтез и реакции соединений типа R«GeM4 —«, где М — Li, Na, К . . . . 144
Реакции соединений типа R3GeM 149
Реакции с кислородом, серой, селеном, теллуром 150
Реакции с углекислым газом и карбонильными соединениями 150
Реакции с углеводородами 151
Реакции с галоидными алкилами и арилами 151
Реакции с галоидными металлами, металлоидами и металлоорганически-
ми соединениями 152
Реакции с аммиаком и азотсодержащими соединениями 153
Синтез и реакции соединений типов R3GeMnR«-i и R3GeM(CO)5 (M = Re, Мп) 154
Глава XV. Полимеризация германийорганических соединений 159
Глава XVI. Анализ германийорганических соединений 162
ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Введение 164
Г л а в а I. Синтез оловоорганических соединений действием галоидных алкилов на
олово, его сплавы или соли двухвалентного олова 181
Синтез оловоорганических соединений при действии галоидных алкилов на
металл 181
Синтез оловоорганических соединений при действии галоидных алкилов и ари-
лов на сплавы олова 186
Синтез оловоорганических соединений при действии галоидных алкилов на
соединения двухвалентного олова 188
Г л а в а II. Синтез оловоорганических соединений действием металлоорганических
соединений на двугалоидное олово 193
Получение оловоорганических соединений взаимодействием литийорганиче-
ских соединений с двухлористым оловом 193
Пол-учение оловоорганических соединений взаимодействием реактива Гринья-
ра с двухлористым оловом 194
Синтез оловоорганических соединений взаимодействием ртутноорганических
соединений с двухлористым оловом 199
Синтез оловоорганических соединений взаимодействием таллий,- свинцово- и
других металлоорганических соединений с двухлористым (двубромистым)
оловом 202
Глав а III. Синтез оловоорганических соединений действием
металлоорганических соединений на производные четырехвалентного олова 205
Синтез оловоорганических соединений посредством реактива Гриньяра . . 205
Взаимодействие реактива Гриньяра с галогенидами олова 206
Синтез оловоорганических соединений типа R*SnR'# (x + у =4)
взаимодействием реактива Гриньяра с оловоорганическими галогенидами . . . 220
702
Оглавление
Синтез оловоорганических соединений посредством литийорганических
соединений 242
Получение оловоорганических соединений посредством органических
соединений щелочных металлов или путем реакции типа Вюрца 251
Получение оловоорганических соединений посредством цинкорганических
соединений 256
Синтез оловоорганических соединений посредством алюминийорганических
соединений 256
Получение оловоорганических соединений посредством металлоорганических
соединений других металлов 260
Г л а в а IV. Синтез оловоорганических соединений взаимодействием
металлоорганических соединений с оловом, его сплавами или оловоорганическими
соединениями со связью олово — олово 269
Г л а в а V. Присоединение оловоорганических соединений по кратным связям . 272
Присоединение оловоорганических гидридов к олефинам 273
Присоединение оловоорганических гидридов к алкинам 287
Присоединение оловоорганических гидридов к альдегидам и кетонам .... 301
Присоединение оловоорганических гидридов к азометинам, азо- и нитрозо-
соединениям, изоцианатам и изотиоцианатам 305
Присоединение алкоксисоединений олова по кратным связям 310
Присоединение станниламинов по кратным связям 314
Присоединение гексаалкилдистаннанов, гексаалкилдистанноксанов и
соединений типа (R2Sn)/2 по кратным связям 316
Глава VI. Синтез оловоорганических соединений через двойные диазониевые,
галогенониевые соли и алифатические диазосоединения 319
Получение соединений типа Ar2SnX2 путем разложения двойных диазоние-
вых солей (ArN2Cl)2-SnCl2 319
Получение оловоорганических соединений с помощью алифатических диазо-
соединений 321
Глав а VII. Введение олова на место атома водорода 325-
Г л а в а VIII. Деалкилирование оловоорганических соединений 329
Деалкилирование галоидными солями олова 329
Получение соединений типа R3SnX 330
Получение соединений типа R2SnX2 332
Получение соединений типа RSnX3 335
Деалкилирование галоидами 337
Деалкилирование галоидоводородами 346
Деалкилирование кислотами, спиртами и меркаптанами 350
Деалкилирование солями 353
Действие окислителей на оловоорганические соединения 358
Деалкилирование фосфором, серой, селеном и другими элементами 358
Деалкилирование водой и щелочами 363
Термо- и фотораспад. Реакции с галоидными алкилами 365
Реакции с органическими перекисями 367
Деалкилирование галоидными ацилами 368
Гидрогенизация 369
Деалкилирование окислами азота 370
Деалкилирование пентакарбонилом железа 370
Глава IX. Диспропорционирование. Перераспределение радикалов 376
ГлаваХ. Гидролиз оловоорганических соединений 381
Гидролиз оловоорганических соединений класса R3SnX 382
Гидролиз оловоорганических соединений класса R2SnX2 385
Гидролиз оловоорганических соединений класса RSnX3 392
Г л а в а XI. Реакции преобразования функциональных групп у атома олова . . . 396
Действие кислот и других соединений с подвижным атомом водорода на
оловоорганические гидроокиси, окиси или станноновые кислоты 396
Реакция обмена 405
Реакции оловоорганических галогенидов с кислотами, перекисями, спиртами,
меркаптанами и другими соединениями, содержащими подвижный атом
водорода 413
Реакции оловоорганических ацилатов, алкоксисоединений и станниламинов 415
Реакции оловоорганических соединений типа R3SnOSnR3, R3SnSSnR3 и
(R2SnO)„ 419
О перераспределении функциональных групп у атома олова 422
Оглавление
703
Другие реакции, приводящие к преобразованию функциональных групп у
атома олова 422
Глав а XII. Координационная способность оловоорганических соединений . . 432
Глав а XIII. Реакции соединений типа R3SnSnR3 и (R2Sn)rc 438
Глава XIV. Изменение в радикале 447
Глава XV. Синтез и реакции оловоорганических гидридов 457
Синтез оловоорганических гидридов 457
Реакции оловоорганических гибридов с неорганическими соединениями . . . 464
Реакции оловоорганических гидридов с органическими кислотами и
гидроперекисями 465
Присоединение оловоорганических гидридов по кратным связям 467
Восстановление оловоорганическими гидридами галоидных алкилов и арилов 468
Восстановление оловоорганическими гидридами альдегидов и кетонов . . . 472
Восстановление оловоорганическими гидридами ацилгалогенидов 474
Реакции оловоорганических гидридов с соединениями, содержащими серу 476
Реакции, приводящие к образованию соединений со связью Sn—Sn 477
Реакции оловоорганических гидридов с металлоорганическими соединениями 484
Реакции оловоорганических гидридов с соединениями ради кал ообразующего
типа 490
Реакции оловоорганических гидридов с диазосоеди нения ми 491
Глава XVI. Синтез и реакции оловоорганических соединений, содержащих два и
более атомов металла в молекуле • • 495
Синтез соединений типа R3SnM и R2SnM2 (где М — щелочной металл) . . . 495
Реакции литий- и натрийоловоорганических соединений с кислородом, серой,
селеном и теллуром 499
Реакции литий- и натрийоловоорганических соединений с алкил- и арилгалоге-
нидами 500
Реакции литий- и натрийоловоорганических соединений с галоидными солями
и металлоорганическими соединениями 502
Синтез соединений типа R3SnM (СО)^ и R2Sn [М (СО)/г]2 (где М — металл
VI—VIII групп) 505
Реакции соединений типа R3SnM (СО)« и R2Sn [M (CO)rt]2 508
Глав а XVII. Оловоорганические полимеры • . • 512
Полимеры с оловоорганическими заместителями в боковой цепи 512
Полимеры с атомом олова в основной цепи 513
Глав а XVIII. Применение оловоорганических соединений 519
Глава XIX. Анализ оловоорганических соединений 526
СВИНЦОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Введение 531
Гл а в а I. Синтез свинцоврорганических соединений действием галоидных алкилов
на свинец, его сплавы и соли 541
Гл ав а П. Электрохимические методы синтеза свинцовоорганических соединений 545
Глава III. Синтез свинцовоорганических соединений действием металлооргани-
ческих соединений на соли двухвалентного свинца или металлический свинец 549
Синтез свинцовоорганических соединений с помощью магнийорганических
соединений ........ 549
Синтез свинцовоорганических соединений с помощью натрий- и литийорга-
нических соединений 552
Синтез свинцовоорганических соединений с помощью цинкорганических
соединений 553
Синтез свинцовгорганических соединений с помощью алюминий- и
борорганических соединений 553
К синтезу свинцовоорганических соединений класса (R2Pb)n 554
Синтез свинцовоорганических соединений действием металлоорганических
соединений на металлический свинец 555
Г л а в а IV. Синтез свинцовоорганических соединений действием металл
оорганических соединений на соли четырехвалентного свинца 558
Синтез свинцовоорганических соединений с помощью магнийорганических
соединений „* * * *^**
Синтез свинцовоорганических соединений с помощью литий- и натрийорга-
нических соединений 561
Синтез свинцовоорганических соединений с помощью ртутноорганических
соединений 564
704
Оглавление
Г л а в а V. Синтез свинцовоорганических соединений с помощью алифатических
диазосоединений и солей диазония 568
Синтез свинцовоорганических соединений с помощью алифатических
диазосоединений 568
Синтез свинцовоорганических соединений с помощью солей диазония . . . 568
Г л аваУ1. Присоединение свинцовоорганических соединений по кратным связям 570
Глав а VII. Введение свинца на место водородного атома (плюмбирование) . . . 573
Глав а VIII. Деалкилирование свинцовоорганических соединений 575
Деалкилирование галоидами 575
Деалкилирование галоидоводородами 578
Деалкилирование кислотами, фенолами и меркаптанами 580
Деалкилирование солями 584
Деалкилирование серой и мышьяком 587
Действие окислителей на свинцовоорганические соединения 587
Деалкилирование водой и щелочами 588
Термо-и фотораспад. Реакции с галоидными алкилами 588
Деалкилирование перекисями ацилов 590
Деалкилирование окислами азота 590
Гидрогенизация 590
Глав а IX. Реакции диспропорционирования, термического распада и
перераспределения радикалов свинцовоорганических соединений 595
ГлаваХ. Гидролиз свинцовоорганических соединений 601
Гидролиз соединений класса R3PbX 601
Гидролиз соединений класса R2PbX2 602
Гидролиз соединений класса RPbX3 603
Г л а в а XI. Реакции преобразования функциональных групп у атома свинца . . 605
Глава XII. Реакции соединений типа R3PbPbR3 612
Глава XIII. Реакции свинцовоорганических соединений, связанные с
изменениями в радикале 617
Г л а в а XIV. Синтез и реакции свинцовоорганических гидридов 620
Синтез свинцовоорганических гидридов 620
Реакции свинцовоорганических гидридов 622
Глава XV. Синтез и свойства свинцовоорганических соединений, содержащих два и
более атомов металла в молекуле 623
Синтез соединений типа R3PbM (M — щелочной металл) 623
Реакции соединений типа R3PbM 625
Синтез и реакции соединений типа R3PbM(CO)n и R2Pb[M(CO)n]2 629
Г л а в а XVI. Анализ свинцовоорганических соединений 631
Авторский указатель 636
Предметный указатель 665
Ксенофонт Александрович Кочешков, Николай Николаевич Землянский,
Наталья Ивановна Шевердина, Евгений Максимович Панов
Методы элементоорганической химии. Германий, олово, свинец
Утверждено к печати Ордена Ленина Институтом элементоорганических соединений
Академии наук СССР
Редактор Л. С. Поваров. Технический редактор Я. Ф. Егорова
Сдано в набор 15/П 1968 г. Подписано к печати 15/Х 1968 г. Формат 70xl08Vie- Бумага № 1
Усл. печ. л. 61,6. Уч.-изд. л. 64,2. Т-13263. Тираж 2400 экз. Тип. зак. 207. Цена 4 р. 20 к.
Издательство «Наука». Москва К-62, Подсосенский пер., 21
2-я типография издательства «Наука». Москва Г-99, Шубинский пер., 10
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Страница
28
29
76
212
296
314
322
343
382
401
484
562
572
572
597
626
Строка
2 СВ.
20 св.
9 св.
10 сн. -
20 св.
5 сн.
3 св.
4 св.
2 сн.
7 св.
13 сн.
26 сн.
26 сн.
27 сн.
2 сн.
20 св.
Напечатано
Т. КИП.
час.
бутилгермания
[67]
Sn —D
мм
диазометана
(2-дициан-
2,5 г
R' и R2
СвН6
РЬ
2,0
0,5
гексаэтилдиплюм-
боксана
I бутилолова
Должно быть
т. пл.
2 час.
этилгермания
[74]
С—Н
1 мм
диазоэтана
(2-циан-
2,5 л
R'-R2
СгН0
РЬ (С6Н5)
4,0
5,0
гексафенилдиплюм-
боксана
бутила
Copyleft ® San'ky incorporation
derevyaha + Q