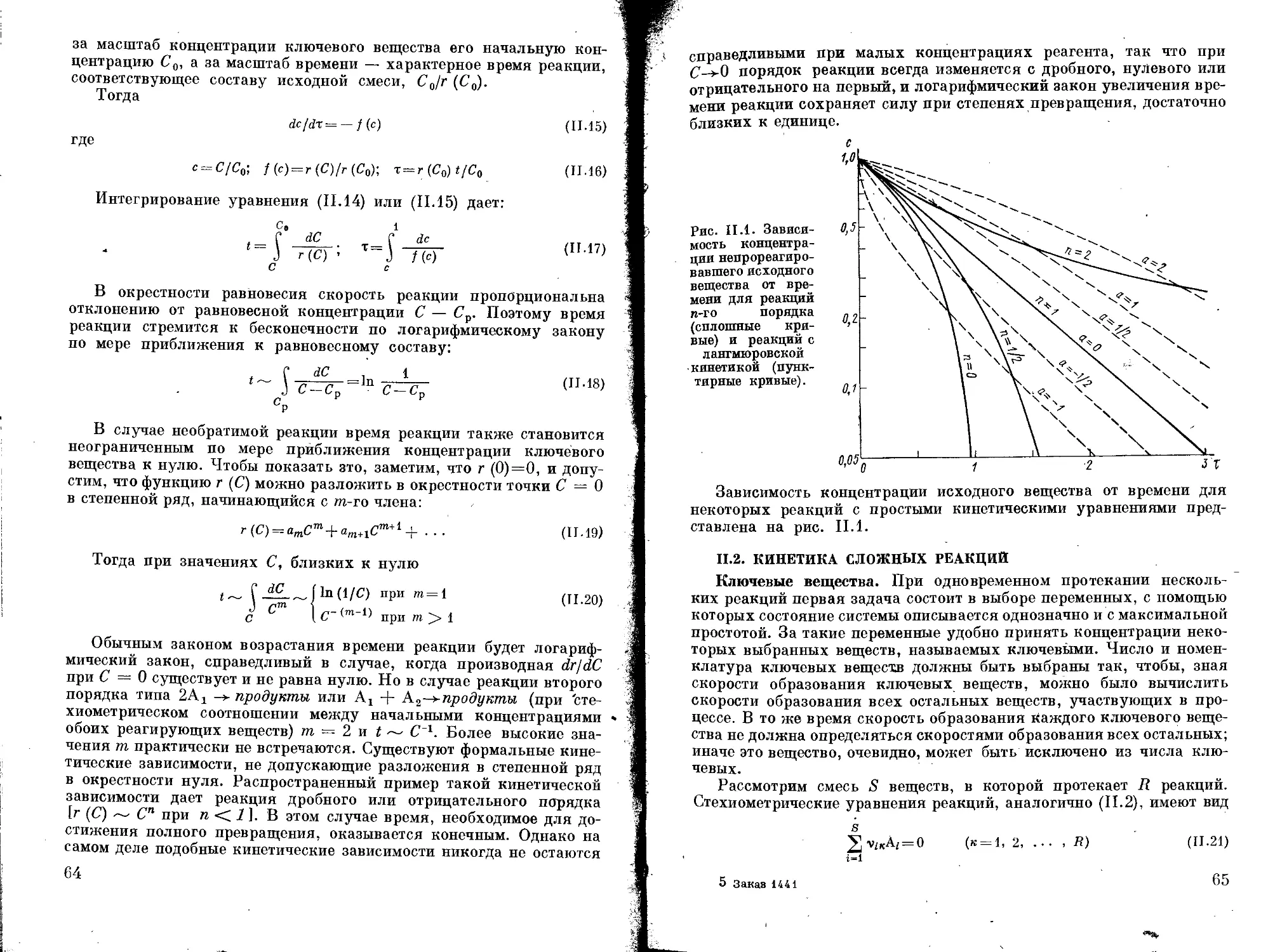
/







































































































































































































































Автор: Иоффе И.И. Письмен Л.М.
Теги: химическая технология химическая промышленность химия инженерная химия
Год: 1972




Текст
Сканировал: NeptUliyi
Магнитогорск
2008
И. И. ИОФФЕ
Л. М. ПИСЬМЕН
ИНЖЕНЕРНАЯ
ХИМИЯ
ГЕТЕРОГЕННОГО
КАТАЛИЗА
Издание 2-е,
переработанное и дополненное
ИЗДАТЕЛЬСТВО «ХИМИЯ»
Ленинградское отделение
1972
УДК 66.097
Сканировал: NeptUliyi
Магнитогорск
2008
Иоффе И. И., Письмен Л. М. Инженерная хи-
мия гетерогенного катализа. Изд-во «Химия»,
, Л., 1972, стр. 464, табл. 8, рис. 102.
В книге изложены теоретические основы гетеро-
генного катализа, кинетика каталитических реакций,
научный основы подбора катализаторов. Описаны
промышленные гетерогенно-каталитические процессы,
расчет и устройство каталитических реакторов, син-
тез катализаторов и методы исследования каталити-
ческих реакций.
Во втором издании учтены последние достижения
в области катализа, подробнее рассмотрены процессы
переноса в каталитических реакторах, методы рас-
шифровки кинетических уравнений по эксперимен-
тальным данным.
Книга рассчитана на широкий круг сотрудников
научно-исследовательских, проектных и промышлен-
ных организаций, связанных с синтезом и исследова-
нием катализаторов, проектированием и осуществле-
нием гетерогенно-каталитических процессов, а также
может служить пособием для студентов и аспирантов
вузов соответствующих специальностей.
3-14-2
25—72
ПРЕДИСЛОВИЕ К 2-му ИЗДАНИЮ
При подготовке нового издания, в связи со значительным
развитием исследований в области теоретического и приклад-
ного катализа и математического моделирования каталити-
ческих процессов и реакторов, авторам пришлось фундамен-
тально переработать книгу и внести в нее ряд важных допол-
нений. Заново написаны или полностью переработаны главы III,
IV, VI, VII, VIII и XI. В остальные главы внесены суще-
ственные изменения и дополнения. В данное издание не вклю-
чена глава о лабораторных исследованиях катализаторов и
глава о статистической обработке зкспериментальных данных
в связи с выходом ряда монографий и учебных пособий по
этим вопросам. По той же причине сокращен объем главы об
оптимизации каталитических реакторов. Таким образом, на-
стоящее издание в значительной мере надо рассматривать как
новую книгу.
Глава XI написана совместно с кандидатом технических
наук А. П. Дороховым, Б. Ю. Гуревичем, Т. Хуттером и
В. П. Пилявским, за что авторы приносят им благодарность.
Авторы выражают признательность своим коллегам по от-
делу инженерной кинетики Всесоюзного научно-исследователь-
ского института нефтехимических процессов и теоротделу Инсти-
тута электрохимии Академии Наук СССР за помощь в работе
и полезные обсуждения. Авторы благодарны редакторам на-
стоящего издания Ю. М. Левину и 3. И. Грива за большой
труд, который они вложили в его подготовку.
И. Иоффе, ВНИИНЕФТЕХИМ, Ленинград
Л. Письмен, ИЭлАН, Москва
1*
ИЗ ПРЕДИСЛОВИЯ К 1-му ИЗДАНИЮ
Предлагаемая книга представляет собой попытку сведения
воедино основных проблем, лежащих "в основе практического
применения гетерогенно-каталитических реакций в химической
промышленности. В связи .с этим материал, рассматриваемый
в книге, достаточно разнообразен и охватывает как вопросы
научных основ подбора и производства катализатора, Так
и кинетику гетерогенно-каталитических реакций, расчеты кон-
тактных аппаратов, лабораторные методы исследования ката-
лизаторов и каталитических реакций. Все эти вопросы авторы
старались рассматривать с точки зрения их практического
использования на разных стадиях разработки промышленных
каталитических процессов. На изложение материала не могли
не отразиться личный опыт и личные научные интересы авторов;
вследствие чего не все материалы и теоретические положения,
затрагиваемые в книге, освещены с одинаковой полнотой.
Естественно, что столь обширный материал, как основы тех-
нического катализа, не мог быть изложен без заметных
упущений. Поэтому авторы будут весьма благодарны всем,
кто поможет их устранить.
Относительно приведенной в книге литературы следует
оговорить, что авторы не ставили перед собой цели составления
подробной библиографии. Наоборот, авторы стремились огра-
ничиться минимумом необходимых ссылок на монографии,
обзорные статьи и узко специальные сообщения.
ВВЕДЕНИЕ
Химическая промышленность до недавнего времени в основном
базировалась на методах классической химии; каталитические про-
цессы были немногочисленны, а в органическом синтезе ограничи-
вались почти исключительно введением гомогенных катализаторов —
кислот или щелочей. В результате увеличения производства синте-
тических продуктов значительно возросло число каталитических,
и в частности гетерогенно-каталитических (контактных) процессов.
В ряде отраслей промышленности органического синтеза гетерогенно-
каталитические процессы, как технологически наиболее прогрессив-
ные, стали преобладающими. В настоящее время свыше 90% вводи-
мых в действие многотоннажных химических процессов являются
каталитическими, большей частью гетерогенно-каталитическими.
Поэтому разработка и обобщение теоретических основ технологии
промышленных гетерогенно-каталитических процессов — актуаль-
ная задача.
Под термином «катализ» обычно понимают явление ускорения
химической реакции в результате действия какого-либо вещества
(или веществ) — катализатора, количество которого практически
не изменяется в ходе реакции. Следует подчеркнуть, что основным
свойством катализаторов является способность вызывать реакции,
не протекающие с заметной скоростью без их участия.
В зависимости от наличия поверхности раздела между реаген-
тами и катализатором различают гомогенный и гетерогенный ката-
лизы. В первом случае реагенты и катализатор составляют одну фазу,
во втором — они находятся в разных фазах. В гетерогенном ката-
лизе возможны различные комбинации агрегатных состояний ката-
лизатора и реагентов, однако практический интерес представляет
почти исключительно катализ газообразных или жидких реагентов
твердыми катализаторами.
Каталитическими свойствами обладает значительное количество
твердых неорганических и органических соединений. Катализаторы
специфичны, т. е. каждый катализатор может ускорять только опре-
деленный круг химических превращений и, наоборот, для каждой
реакции катализаторами являются только определенные вещества.
Эффективность применения того или иного вещества в качестве
5
катализатора определяется всей совокупностью его химических и
физических свойств.
Сложность явлений в техническом гетерогенном катализе делает
необходимым его разностороннее изучение. Наука о реальном тех-
ническом процессе всегда будет относиться к области пограничных
наук, так как на реальные промышленные процессы влияют самые
различные факторы, изучение которых затрагивает различные области
знаний. В отношении химических и, в частности, гетерогенно-ката-
литических процессов это особенно существенно, поскольку они
определяются взаимодействием разнообразных химических и физи-
ческих явлений, а их описание требует специальных математических
методов. Кроме того, при разработке промышленных процессов и
управлении ими следует руководствоваться и экономическими кри-
териями. Поэтому нам кажется целесообразным для определения
науки по исследованию, разработке и управлению промышленным
химическим процессом ввести специальный термин — инженерная
химия.
Как и всякая прикладная наука, инженерная химия гетероген-
ного катализа должна строиться в соответствии с теми практическими
задачами, которые она призвана решать. В данном случае это раз-
работка гетерогенно-каталитических процессов для химической про-
мышленности. При этом могут разрабатываться как новые, ранее
не освоенные химико-технологические процессы, так и различные
модернизируемые варианты существующих промышленных произ-
водств. В общем случае разработка каждого каталитического про-
цесса * состоит из трех этапов: 1) выбор катализатора, 2) выбор
режима процесса и 3) выбор реактора. В отдельных случаях задача
может быть ограничена одним или двумя этапами.
Для выполнения первого этапа, кроме чисто эмпирических
сведений, необходимы представления о механизме гетерогенного
катализа и в первую очередь о связи его с химическими и физико-
химическими свойствами твердого вещества — катализатора — и
участников катализируемой реакции. Активный и селективный
катализаторы, кроме химических, должны, как правило, обладать
еще определенными структурными свойствами.
Химическая кинетика каталитической реакции определяет не
только оптимальный режим ее протекания, но и структуру катали-
затора, позволяющую реализовать его потенциальные химические
возможности. Следует также учитывать, что химические процессы
на гетерогенном катализаторе тесно связаны с рядом физических
процессов переноса вещества и тепла. Для совокупности всех этих
процессов в химической литературе пользуются термином «макро-
кинетика». Очевидно, что знание кинетических и макрокинетических
закономерностей необходимо как для выполнения упомянутого вто-
* В дальнейшем под термином «каталитический» будет пониматься «гетеро-
генно-каталитический», если это не требует специальных оговорок.
6
рого этапа разработки промышленного процесса, так и для заверше-
ния первого.
Третий, завершающий этап разработки промышленного катали-
тического процесса — выбор реактора — тесно связан со вторым
этапом, поскольку не только режим процесса определяет конструк-
цию реактора, но, в свою очередь, конструкция реактора наклады-
вает определенные требования и ограничения на условия проведения
реакции. Однако для выбора конструктивной схемы реактора тре-
буются дополнительные знания, связанные с физической кинетикой,
гидродинамикой и теплофизикой процессов в каталитических реак-
торах. Кроме того, создание работающего реактора требует оценки
его устойчивости в ходе эксплуатации. Наконец, среди многообразия
возможных конструктивных схем реакторов необходимо суметь
достаточно обоснованно выбрать наилучший, т. е. оптимальный'
вариант. Для решения двух последних вопросов следует ознако-
миться со специальным математическим аппаратом теории устой-
чивости и теории оптимального управления.
В соответствии с изложенными требованиями, инженерная химия
/ гетерогенного катализа должна включать в себя физико-химические
[ основы гетерогенного катализа, кинетику и макрокинетику гетеро-
генно-каталитических процессов, теорию каталитических реакторов.
I Все зти достаточно далеко отстоящие друг от друга отрасли знания
Хрбъединены общим объектом дрилажениц. —
Всякая прикладная' йЯ^йавключает в себя и экспериментальные
методы исследований. В инженерной химии они не очень специфичны
и в значительной своей части идентичны таковым, применяемым
в химической кинетике, прикладной гидродинамике и теплофизике.
Более специфическими являются скорее методы обработки экспери-
ментальных данных, которые разрабатываются с учетом возможности
использования информации, полученной с модельных или крупных
полупромышленных и промышленных установок. Эти эксперимен-
тальные и специфические математические методы обработки данных
экспериментов составляют неотъемлемую часть знаний, необходи-
мых для разработки промышленных каталитических процессов.
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ
С — совокупность концентраций (вектор);
С — концентрация, ML"3;
с — безразмерная концентрация;
D — коэффициент диффузии, L2?"1;
Е — энергия активации, 1ЛТ~ 2;
F — площадь, поверхность, L2;
h — -тепловой эффект реакции,
к — термохимический коэффициент;
Кр — константа равновесия;
к — константа скорости реакции;
L — длина реактора, L;
I ~ размер частицы, L',
Р — давление,
q — плотность теплоотвода, МТ'3-,
г — объемная скорость реакции;
'S — полное время контакта в реакторе, Г;
s — селективность, среднее время пребывания;
Т — температура;
t — астрономическде и текущее время, Г;
и — линейная скорость потока, LT’1;
V — объем, L3;
v— объемная скорость потока, L3?-1;
w — фильтрационная скорость, LT~i;
X — продольная координата, L;
х — безразмерная продольная координата;
Y — поперечная координата, L',
у — безразмерная поперечная координата;
Z — предэкспонента;
а — коэффициент теплопередачи,
Р — коэффициент массопередачи,
У — теплоемкость единицы объема, Л2д/-27,-20-1;
е — доля свободного объема;
т) — фактор эффективности;
0 — тепловой параметр;
9 — безразмерная температура;
и — динамическая вязкость жидкости, L~1MT~1',
х — константа скорости поверхностной реакции;
к — теплота адсорбции, L2MT'2;
р — кинематическая вязкость жидкости, L^T'1;
v — стехиометрический коэффициент;
р — плотность, ML~3, скорость поверхностной реакции;
<т — поверхность в единице объема, L'1-,
X — коэффициент теплопроводности, LMT'^Q'1, параметрическая чувстви-
тельность;
Y — модуль Тиле.
8
Часть
первая
ОСНОВЫ ТЕОРИИ
ГЕТЕРОГЕННОГО КАТАЛИЗА
Глава I.___________________________________________________
ТЕОРИЯ КАТАЛИТИЧЕСКОГО ДЕЙСТВИЯ
1.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О КАТАЛИЗЕ
Химические превращения в каталитических процессах отличаются
от обычных химических превращений по меньшей мере тем, что в них
всегда участвует один дополнительный компонент —' катализатор.
Именно этот компонент, не входящий в стехиометрические урав-
нения реакции, обусловливает специфику каталитических превра-
щений. Гетерогенные катализаторй, как правило, — твердые веще-
ства, поэтому необходимость учитывать физические и химические
свойства катализаторов для объяснения механизма их действия
ставит гетерогенный катализ в пограничную область между собственно
химией и физикой твердого тела.
Классическое выражение, связывающее скорость химической
реакции г с условиями ее протекания, имеет вид
Е
r = Ze~ RT f(C) ’ (1.1)
где Z — так называемый предэкспоненциальный множителя (пред-
экспонент); Е — энергия активации; R — газовая постоянная; Т —
абсолютная температура; / (0 — функция концентраций компо-
нентов реакционной смеси.
Понятие энергии активации определяется существованием потен-
циальных энергетических барьеров, с преодолением которых свя-
зано любое химическое превращение.
Каждой определенной конфигурации атомов реагирующих моле-
кул соответствует некоторое значение потенциальной энергии си-
стемы. Например, для реакции типа А + ВС АВ + С зависи-
мость потенциальной энергии системы от межъядерных расстояний
А — В и В — С может быть представлена поверхностью потенциаль-
ной энергии, топографическая карта и сечения которой приведены
на рис. 1.1. Устойчивым химическим соединениям соответствуют
минимумы на поверхности потенциальной энергии. Наиболее легкий
путь перехода от одного устойчивого состояния к другому, т. е. путь
9
реакции, ведет через «перевал» между двумя «долинами» поверхности.
Переходное состояние, в котором находятся реагирующие молекулы
в точке «перевала», называется активированным комплексом. Раз-
ность между энергией активированного комплекса и суммой энергий
реагирующих веществ называется энергией активации. Последняя
Рис. 1.1. Поверхность потенциальной энергии для реакции
А + ВС АВ + С
(Числа означают величину потенциальной энергии системы).
является, таким образом, той минимальной энергией, которую надо
сообщить реагирующим молекулам, чтобы химическое превращение
стало возможным. Обычным источником этой дополнительной энер-
гии является кинетическая энергия теплового движения молекул.
Доля «горячих» молекул, обладающих тепловой энергией, равной
или большей энергии активации, определяется статистикой Больц-
_ JL
мана и пропорциональна е RT, что и отражено в формуле k(I.l).
10
Предэкспонент в формуле (1.1) определяет вероятность взаимо-
действия молекул, обладающих энергией, достаточной для преодо-
ления потенциального барьера. Он равен произведению фактора
частоты А и энтропийного фактора еЛ8/й, зависящего от энтропии
активации (энтропия образования активированного комплекса) AS
Z=Ae й (1.2)
В гомогенных системах фактор частоты для мономолекулярных
реакций равен частоте колебаний вдоль пути реакции, а для реакций
взаимодействия между двумя молекулами (или большим их числом)—
частоте столкновений. Столкновение между молекулами реагиру-
ющих веществ, обладающими энергией, достаточной для образования
активированного комплекса, может, однако, и не привести к хими-
ческому превращению, если при столкновении не выполнены неко-
торые дополнительные условия, например, определенная взаимная
ориентация сталкивающихся молекул. Эти ограничения и учиты-
ваются в уравнении (1.2) энтропийным фактором.
Для гетерогенно-каталитических реакций предэкспоненциальный '
множитель определяется не только энтропией активации, но и числом
активных мест, приходящихся на единицу поверхности катализа-
тора. Последнее, в отличие от числа соударений в гомогенных систе-
мах, может колебаться в очень широких пределах и не поддается,
в общем случае, расчету. Поэтому в гетерогенно-каталитических
реакциях даже приближенная априорная оценка величины пред-
экспоненциального множителя невозможна.
Как это видно из формул (1.1) и (1.2), ускорение химической
реакции в принципе может быть достигнуто путем либо снижения
величины Е, либо увеличения AS. каталитическое ускорение шеакг-
ций идет, видимо, большей частью по пути "снижения- Е; Снижений
энергии активации под- дейивией" катализатора в общем случай
является следствием образования иных промежуточных соединений ।
и активированных комплексов и соответственно изменения формы
поверхности потенциальной энергии, благодаря чему открывается
новый путь-реакции, проходящий через «перевалы» меньшей высоты.
Значения энтропии активации для различных реакций могут
отличаться на много порядков. В настоящее время можно считать
установленным существование кинетического компенсационного эф-
фекта, выражающегося взаимозависимостью Е и Z
lnZ = a + pE« (1.3)
где а и Р — коэффициенты, постоянные для данного типа реакции;
п — показатель степени, обычно близкий к единице.
Компенсационный эффект проявляется обычно либо в определен-
ного типа реакциях сходных соединений (например, членов гомоло-
гического ряда) на одном и том же катализаторе, либо в данной
реакции при введении в катализатор модифицирующих добавок,
либо при проведении реакции на катализаторах сходного типа.
11
Во всех случаях может быть эмпирически найдена зависимости
типа (1.3).
Значение компенсационного эффекта нельзя недооценивать при
исследованиях по подбору катализаторов. Было показано, что изме-
нения предэкспоненциального множителя достигают 7—11 порядков,
так что скорость реакции может значительно увеличиваться, несмотря
на рост энергии активации [1 ]. Вопрос о существовании компен-
сационного эффекта в данном ряду катализаторов пока может быть
' решен только экспериментально.
Природа компенсационного эффекта недостаточно ясна. По одному
из объяснений, компенсационный эффект свойственен процессам
в конденсированных средах (к которым, конечно, принадлежат и
твердые катализаторы) и связан с локализацией элементарных актов
реакции в микроскопически малых областях и конечной скоростью
перераспределения энергии в системе [2 ]. По другому объяснению [3 ],
компенсационный эффект есть следствие неоднородности катали-
тической поверхности. Таким образом, возможно, что компенсацион-
ный эффект и не связан с энтропийными составляющими константы
скорости реакции.
Общность между техническим гетерогенным катализом и фер-
ментативными процессами указывает на возможность существования
энтропийных механизмов гетерогенного катализа, в которых скорость
или, что более существенно, направление реакции изменяются за
счет повышения вероятности образования промежуточных состояний
некоторых реакционных направлений. Иллюстрацией к этому могут
служить реакции стереоспецифического катализа и избирательные
синтезы на цеолитных катализаторах.
Одним из первых объяснений явления катализа была теория
промежуточных соединений. Положительной чертой этой теории
являлось то, что она подчеркивала химическую сторону катализа
и объясняла избирательность катализа образованием промежуточ-
ных соединений различной природы в зависимости от химического
сродства данных веществ. Однако зависимость активности катали-
заторов от способа их приготовления, а также явления отравления
и промотирования катализаторов заставили предположить, что ката-
литическую активность нельзя объяснить только химическим соста-
вом катализатора.
Несоответствие чисто химической теории промежуточных соеди-
нений всей совокупности экспериментального материала привело
к представлению о том, что катализ осуществляется не на всей .
поверхности, а только на так называемых активных центрах. Наибо-
лее четко эти представления были изложены еще Тэйлором.
Существенным подтверждением концепции активных центров
являются экспериментальные данные, полученные при изучении
промотирования и отравления катализаторов. Так, Облад с сотруд-
никами [4] установили, что в алюмосиликатном катализаторе кре-
кинга активны только около 4% всей поверхности. Однако Боресков
с сотрудниками [5, 6 ], а затем и другие исследователи [7 ] показали,
12
что представления о существовании активных центров не являются
универсальными. Так, различным образом приготовленная платина
оказалась практически одинаково активна (считая на единицу по-
верхности) в реакциях окисления S02 в S03 и этилена в окись эти-
лена. Силикагель разных способов приготовления и кристаллический
кремнезем обладают одинаковой удельной активностью в реакции
парофазного гидролиза хлорбензола. Из этих данных следует', что
в ряде случаев каталитическое действие в равной мере присуще всем
атомам поверхности катализатора.
В настоящее время можно считать общепринятым, что существует
несколько типов гетерогенно-каталитических превращений. Рогин-
ский показал целесообразность выделения в катализе вообще и в гете-
рогенном в частности двух больших классов: окислительно-восста-
новительного, или катализа с электронными переходами, и кислотно-
основного, или ионного катализа.
Катализ первого класса, сокращенно называемый «электронным
катализом», осуществляется на твердых телах — проводниках элек-
трического тока (металлах и полупроводниках). Эти тела обладают
рядом общих физико-химических свойств, связанных с наличием
в них подвижных электронов. Для тел-проводников характерна
электропроводность, окраска (т. е. заметное поглощение света в види-
мой области спектра), термоэлектронная эмиссия и внешний фото-
эффект. К этому классу относятся каталитические реакции окисле-
ния, восстановления, гидрирования, дегидрирования, объединяемые
в тип гомолитических. Все они сопровождаются разделением элек-/|
тронов в электронных парах молекул. Общий механизм действия I
катализатора сводится при этом к облегчению электронных перехо- I
дов в реагирующих молекулах за счет собственных электронов |
катализатора.
Катализ второго класса — ионный — протекает на твердых те-
лах, не имеющих свободных носителей тока в объеме, т. е. на изо-
ляторах. Электропроводность этих тел, заметная при высоких тем-
пературах, — ионная, аналогичная электропроводности электро-
литов. Катализаторы этого типа, как правило, не окрашены; реакции
происходят без разделения электронных пар и объединяются в тип
гетеролитических. Сюда относятся реакции изомеризации, присоеди-
нения (гидратации, аминирования), замещения (гидролиза), дез-
аминирования. Указанные два класса каталитических реакций не
1 включают в себя, однако, веех возможных механизмов катализа^
q Принципиально проблема гетерогенного катализа является тй;
’ кой же квантово-химической задачей, как и общая проблема реак- (
: ционной способности химических соединений. Однако в гетерогенном .
1катализе задача усложняется необходимостью учета квантовых 4
/состояний твердого тела. Как известно, в настоящее время кванто- *
вая химия еще не может преодолеть расчетные трудности, возника-
ющие при решении даже более простых задач. Поэтому современной
теории катализа в значительной мере приходится довольствоваться
выведением полуэмпирических закономерностей и обобщений.
13
Существенной причиной затруднений в создании теории гетеро-
генного катализа является то обстоятельство, что одни и те же
экспериментально наблюдаемые явления зачастую обусловливаются
процессами самой различной природы, что, естественно, затрудняет
их интерпретацию. Поэтому главное внимание исследователей
направлено пока не на попытки создания универсальной теории,
а на объяснение различных этапов каталитического процесса или
отдельных типов гетерогенно-каталитических реакций. Основные
современные теоретические, представления о явлениях гетерогенного
катализа и будут изложены в настоящей главе.
1.2. АДСОРБЦИЯ
Сам факт ускорения реакции твердыми катализаторами обуслов-
ливает предварительную адсорбцию реагентов на их поверхности.
Существует два вида адсорбции — физическая и химическая (или
хемосорбция). Последнюю иногда называют активированной адсорб-
цией.
Физическая адсорбция вызывается силами Ван-дер-Ваальса и
обычно не требует энергии активации. Хемосорбция, представляющая
собой химическую реакцию между молекулой вещества и атомами
поверхности твердого тела, как и всякая химическая реакция,
сопряжена с преодолением некоторого энергетического барьера.
Типичный график зависимости количества адсорбированного
вещества или степени заполнения поверхности 0 от температуры при
постоянном парциальном давлении сорбируемого вещества (изобара
адсорбции) и наличии обоих видов адсорбции представлен на рис. 1.2.
При низких температурах наибольшую роль играет быстрая физи-
ческая адсорбция. Ее равновесие с повышением температуры сме-
14
щается в сторону меньших заполнений, чему соответствует левая
нисходящая ветвь кривой. Одновременно растет скорость химической
адсорбции (для которой в типичном случае требуется энергия акти-
вации). Восходящая ветвь кривой соответствует той ситуации, когда
рост количества хемосорбированного вещества с температурой опе-
режает падение количества вещества, адсорбированного физически.
Хемосорбция в этих условиях уже играет преобладающую роль.
Весь участок изобары адсорбции до максимума кривой соответствует
ложному равновесию; истинное равновесие здесь не достигается
из-за малых скоростей хемосорбции. С дальнейшим ростом темпе-
ратуры равновесие обратимой хемосорбции — обратимой экзотерми-
ческой реакции — смещается в сторону десорбции вещества с по-
верхности, что изображается правой нисходящей ветвью кривой.
* Эта ветвь соответствует истинному равновесию. Хемосорбция может
быть как обратимой, так и необратимой (в том смысле, в каком
применяется понятие необратимой реакции). Процессы гетерогенного
катализа чаще связаны с обратимой хемосорбцией, а явления отрав-
ления катализаторов с необратимой адсорбцией. Как и всякая хими-
ческая реакция, хемосорбция специфична: она целиком определяется
природой адсорбента и адсорбата. Хемосорбция сопровождается
резким изменением электронной структуры сорбируемого вещества,
часто вплоть до ионизации и диссоциации последнего. Процесс хемо-
сорбции влияет и на состояние поверхности адсорбента.
Первые теоретические работы по хемосорбции на поверхности
твердого тела принадлежат Лангмюру [8]. В их основе лежат сле-
дующие постулаты:
1) хемосорбция происходит только в монослое молекул;
2) все адсорбционные центры идентичны (однородная поверх-
ность);
3) взаимодействие сорбированных молекул отсутствует, харак-
теристики связи молекул адсорбата с адсорбционными центрами
однозначны и постоянны;
4) число адсорбционных центров на единицу поверхности по-
стоянно на всех ее участках.
Можно привести много возражений против этих постулатов.
Тем не менее, лангмюровские представления во многих случаях
дают удовлетворительное приближение к действительности; в дру-
гих случаях они являются отправной точкой для построения более
Сложных физических моделей.
Рассмотрим сначала адсорбцию одного вещества. При принятых
допущениях, очевидно, скорость адсорбции пропорциональна доле
свободной поверхности (1 — 0) и концентрации вещества в объеме С,
а скорость десорбции пропорциональна доле занятой поверхности 0.
При наступлении адсорбционного равновесия эти скорости стано-
вятся равными
^адс^ (1 0) = ^дес0 (1'4}
где 7еадс и /сдес — константы скорости процессов адсорбции и десорб-
ции, соответственно.
15
Отсюда определяется доля занятой поверхности 9:
Q_____^адсС_______^£7_______)___
^дес Ч-^'адсб' 1-|-6(7 l-J-d/C
Здесь введены адсорбционный коэффициент
обратный адсорбционный коэффициент а = Ь~г.
коэффициент Ь представляет собой константу равновесия процесса
адсорбции; он связан с теплотой ад-
сорбции Л соотношением
х
b = bffi RT
где Ьо — константа.
С ростом температуры Ь умень-
шается, что соответствует сдвигу равно-
весия экзотермического процесса ад-
сорбции в сторону десорбции вещества
с поверхности. Обратный адсорбцион-
ный коэффициент равен
х
а — а^е RT
где а0 = Ьо1.
Согласно (1.5) коэффициент а равен концентрации в объеме,
соответствующей половинному заполнению поверхности. Уравне-
ние (1.5) описывает изотермическую адсорбцию на активной поверх-
ности и называется изотермой Лангмюра (рис. 1.3). При малых
концентрациях адсорбата в газовой фазе, пока ЬС <£ 1, оно дает
9 = ЬС, т. е. при малых степенях заполнения поверхности адсорбция
протекает по закону Генри (участок ОА на рис. 1.3). При больших
концентрациях, когда ЬС )§> 1, 0^1, что соответствует насыщению
поверхности адсорбента (участок BD на рис. 1.3). Чем больше Ь,
тем при меньших объемных концентрациях наступает насыщение
поверхности.
Если молекула диссоциирует при адсорбции на два осколка,
то уравнение (1.4) принимает вид
Рис. 1.3. Изотерма адсорбции
Лангмюра.
^адсГ (1 Ох — Оз) — ^дес01®2
Решение уравнения (1.8) дает:
I
<L5)
^адс/^дес И
Адсорбционный
(1.6)
(1.7)
(1-8)
Scrap-1]
где х = 02/91-
В области малых заполнений, когда ЬС <£ 1, формула (1.9) при-
водится к виду 0Х = (бС/Х)1/». В случае, когда достигается степень
заполнения, близкая к насыщению, формула (1.9) дает 0Х =
= (1 + х)-1 = const.
Рассматривая общий случай адсорбции смеси веществ, обозна-
чим через 0Z долю поверхности, занятую i-м веществом, и через
46
/садс о Адес i — константы скорости соответственно адсорбции и
десорбции /-го^ вещества. Аналогично (1.4) можно написать
^адс [Ci fl У 0/) = ^>дес/6/ (ЕЮ)
\ /=1 /
Разделив друг на друга любые два уравнения вида (1.10), найдем
*адс j Лдес i Cj _
— т—~ • -j.—г ‘ ~сг ”*
/£дес ] /садс i ''i
(Ы1)
Подставляя (1.11) в (1.10) и преобразуя, получаем окончательно
0/ = ед+ (1.12)
/ \ /=1 /
ГДе bi ^адс i/^дес /•
Существует ряд других выводов уравнения изотермы адсорбции,
в том числе термодинамический, однако все они приводят к тем же
результатам и для своего обоснования требуют тех же исходных
постулатов.
Многочисленные исследования показали, что в ряде случаев
наблюдаются значительные отклонения от изотермы адсорбции
Лангмюра. Причиной таких отклонений может быть невыполнение
любого из вышеприведенных постулатов. Одним из важнейших
явлений, не согласующихся с постулатами Лангмюра, является
уменьшение теплоты адсорбции при увеличении степени заполнения
поверхности.
Подобные отклонения можно объяснить двояко. Отказавшись от
постулата 3, приходим к представлению о хемосорбции на однород-
ной поверхности, сопровождающейся взаимодействием сорбирован-
ных частиц. Если это взаимодействие заключается во взаимном
отталкивании, теплота адсорбции должна уменьшаться с увеличе-
нием степени заполнения в согласии с опытными данными. Выбрав
некоторую зависимость коэффициента адсорбции Ъ [связанного
с теплотой адсорбции соотношением (1.6)] от степени заполнения
поверхности и подставив ее в уравнение (1.5),. можем аппроксими-
ровать таким образом любую экспериментальную изотерму адсорб-
ции. Отталкивание хемосорбированных молекул может являться
следствием квантово-механического обменного взаимодействия [9].
Силы кулоновского или диполь-дипольного взаимодействия играют
малую роль, так как они должны сказываться лишь при значитель-
ной плотности сорбированных молекул, между тем отклонения от
изотермы Лангмюра (или изотермы Генри) часто становятся -замет-
ными уже при очень малых степенях заполнения поверхности.
Весьма правдоподобно объяснение природы сил взаимодействия
сорбированных частиц через посредство электронного газа кристал-
лической решетки катализатора (см. постулат 3, а также работы
[9, 10]); сила такого взаимодействия незначительно уменьшается
2 Заказ 1441 17
с расстоянием и потому оно может быть заметным даже при малом
заполнении поверхности.
Другое объяснение опытных данных заключается в отказе от
постулата 2. Поверхность при этом считается неоднородной, т. е.
ее различные участки характеризуются разными теплотами адсорб-
ции и адсорбционными коэффициентами. Первые порции сорбата
заполняют участки с максимальными теплотами адсорбции, а после-
дующие — менее активные участки; в соответствии с этим теплота
адсорбции уменьшается с заполнением по закону, зависящему от
доли участков с различными теплотами адсорбции на поверхности
катализатора.
Кейер и Рогинский для доказательства неоднородности поверх-
ности провели опыты, известные под названием дифференциально-
изотопного метода [И ]. При адсорбции с первыми порциями сорби-
руемого газа впускаются меченые (радиоактивные) молекулы сорбата.
После достижения сорбционного равновесия сорбат откачивается,
причем меченые молекулы десорбируются в последнюю очередь,
что доказывает неоднородность поверхности. С другой стороны,
Хориути и Тойя [9] показали экспериментально, что вид функций
распределения по теплотам адсорбции водорода на никеле и воль-
фраме меняется с температурой в интервале 0—300° С. Это, по мне-
нию авторов, является доказательством против теории неоднородной
поверхности, поскольку энергия создания или перераспределения
дефектов на поверхности твердого тела значительно больше энергии
теплового движения атомов в рассматриваемом интервале темпера-
тур. Опыты Кейер и Рогинского авторы объясняют статистико-
вероятностными расчетами, которые дополнены представлением
о двух состояниях адсорбированного водорода и преимущественной
адсорбции последнего на одной из кристаллографических граней.
Математическая теория адсорбции на неоднородной поверхности
была развита Рогинским [12]. Неоднородная поверхность всегда
может быть представлена как совокупность микроскопических
участков, каждый из которых однороден, т. е. содержит адсорбцион-
ные центры, характеризуемые одной и той же теплотой адсорбции А
и, следовательно, одним и тем же адсорбционным коэффициентом Ь.
В пределе распределение по теплотам адсорбции А можно считать
непрерывным и следующим некоторой дифференциальной функции
распределения <р (А). Величина <р (A) dA равна доле поверхности,
приходящейся на участки с теплотой адсорбции, заключенной
в пределах от А до А + dA. Так как суммирование по всем возможным
значениям А дает полную величину поверхности, дифференциальная
функция распределения всегда должна быть нормирована к единице:
^макс
[ Ф(1)й=1 (1.13)
х
мнн
Для каждой совокупности участков с фиксированным А, как для
однородной поверхности, степень заполнения адсорбированным ве-
18
ществом определяется изотермой Лангмюра. Для поверхности в целом
суммарная степень заполнения 0С; выражается равенством
' Хмакс
ес(С)= f е (с, х) <р(м ак (i.i4)
МИН
Подставляя уравнения (1.5) и (1.7) в (1.14), получаем
9с <£) =
Хмакс
МИН
14
Ф.ормула (1-15) является
уравнением изотермы адсорб-
ции на неоднородной поверх-
ности. Форма этой изотермы
зависит от вида функции рас-
пределения <р (X). Наиболее
практически важными являются
следующие законы распределе-
ния:
экспоненциальный
ф(Х) = Яе““Х (1.16)
степенной
Ф
(1.15)
q> (X) ЛК
a^RT
' С
Рис. 1.4. Перемещение фронта адсорб-
ции на неоднородной поверхности.
Заштрихованная область относится к участ-
кам, заполненным адсорбатом.
| = ЯА» (1.17)
и равномерный (частный случай обоих законов при а - 0)
Ф(А) = Я (1-18)
где Н и а — константы.
Подставив (1.16), (1.17) или (1-18) в (1-15) и проинтегрировав,
найдем вид изотермы адсорбции, соответствующей этим функциям
распределения. Чтобы избавиться от непринципиальных аналити-
ческих трудностей и добиться максимально простых и наглядных
результатов, рассмотрим, следуя Рогинскому [12], более подробно
физическую картину адсорбции на неоднородной поверхности. Когда
степень заполнения даже на самых активных участках еще мала,
адсорбция повсюду следует закону Генри. Наклон соответствующей
прямой линии в координатах 0 — С тем круче, чем больше теплота
адсорбции на данном участке. Ширина области Генри для неодно-
родной поверхности, определяемая ее шириной для участков с макси-
мальными значениями теплоты адсорбции, очень мала. На активных
участках быстро наступает насыщение, на участках же с меньшими
значениями А, рост 0 остается линейным. Благодаря этому на графике
функции распределения (рис. 1.4) создается крутой фронт, отделя-
ющий участки с 0, близким к единице, от участков с 0, близким к нулю.
Этот фронт перемещается в сторону меньших значений А, почти не
2* ' 19
изменяя формы. Поэтому линию фронта адсорбции можно с доста-
точно хорошим приближением заменить прямой, параллельной оси
ординат, проходящей через точку, соответствующую тем участкам
Поверхности (с теплотой адсорбции, равной %*), для которых в дан-
ный момент степень заполнения равна 1/2. При этом мы пренебре-
гаем адсорбцией на участках поверхности, которые обладают X, мень-
шим %*, а степень заполнения участков с теплотой адсорбции больше
указанной величины принимаем за единицу, т. е.
0 = 0 при Х<Х*; 0=1 при Х>Х* (1-19)
Используя формулы (1.7) и (1.5), получаем выражение для теп-
лоты адсорбции %*, соответствующей половинному заполнению
поверхности:
№ = RT 1п(а0/С) (1.20)
Теперь можно написать вместо (1.14) и (1.15)
^макс
0с (0= f q>(X)dX. (1-21)
RT In (а„/С)
Подставляя в уравнение (1,21)- выражение (1.16) и интегрируя,
получаем известную изотерму Фрейндлиха
0с = ЛС“-Д * (1.22)
где п — aRT; А = (H/a)a0~aRT; В = (Н/а)е~аКмакс.
Подставив выражение степенной функции распределения (1.17)
в (1.21), при а —1 приходим к изотерме адсорбции вида
0 = —( ДГ 1П — )“+14- (1.23)
а + 1 \ а0 J 1 а + 1 _ ' '
При а = 0 (равномерное распределение) приходим к логариф-
мической изотерме Зельдовича—Рогинского
0С= A In С + В (1.24)
где А = HRT, В = ЯХмакс — Л In а0.
Все полученные результаты могут быть распространены и на
случай адсорбции нескольких веществ, хотя соответствующие мате-
матические выражения и будут весьма громоздкими. *
Изложенная математическая теория Рогинского формальна по
своему характеру: она позволяет объяснить любую эксперимен-
тальную изотерму адсорбции подбором надлежащих функций рас-
пределения ф (X), не касаясь природы последних. Как мы уже гово-
рили, любая экспериментальная изотерма может быть объяснена
и наличием взаимодействия сорбированных частиц и аппроксимиро-
вана уравнением (1.5) с адсорбционным коэффициентом, зависящим
от заполнения.
20
1.3. ЭЛЕКТРОННЫЕ ПРЕДСТАВЛЕНИЯ В КАТАЛИЗЕ
Для понимания каталитических процессов, протекающих на
поверхности твердых тел, необходимо иметь хотя бы качественное
представление о состоянии электронов поверхности катализатора
и сорбированных на ней атомов. Каталитические свойства поверх-
ности определяются ее способностью образовывать связи с молеку-
лами из газовой фазы. Эти вопросы должны являться предметом
исследования поверхности квантовой химией, однако современное
ее состояние еще не позволяет дать строгое решение такого рода
задач. Поэтому для объяснения и предсказания явлений, относя-
щихся к области окислительно-восстановительного гетерогенного
катализа, был применен ряд общих представлений о поведении
электронов в твердом теле.
По мнению ряда исследователей, хемосорбцию на металлах можно
объяснить, предположив, что образование связи между металлом
и молекулой сорбата определяется наличием у металла донорных
или акцепторных электронных уровней. Металлы с простой валент-
ной оболочкой, образующей s-зону, являются типичными донорами
электронов с малой плотностью уровней в зоне. Такие металлы
хорошо адсорбируют акцепторы электронов, т. е. молекулы окисли-
телей. Однако из-за большой прочности образующейся связи с пере-
ходом металла в другую фазу (окисел, сульфид и т. п.) такие металлы,
как правило, непригодны в качестве катализаторов.
Шваб исследовал каталитическую активность сплавов серебра
в реакции разложения муравьиной кислоты [13]. Им было показано,
что с заполнением свободных электронных уровней в сплаве актив-
ность катализатора падает, а энергия активации реакции увеличи-
вается.
Большой экспериментальный материал указывает на то, что.]
каталитическая активность металлов связана с наличием незапол- ।
ненных d-уровней, свойственных переходным элементам таблицы ’
Менделеева. В кристаллах металлов этого типа образуются сложные
валентные зоны, являющиеся результатом перекрытия s- и d-зон.
Часть связей в кристалле при этом осуществляется за счет неспа-
ренных d-электронов. Степень участия d-элёктронов в образовании ।
валентных связей называется, по Полингу, процентом d-состояния. |
Переходные металлы способны образовывать связи как с~электроио-|
донорными молекулами, так и с электроноакцепторными, причем
прочность этих связей варьирует в широких пределах. Биком [14]
и Кембеллом [15] была показана прямая зависимость активности
катализаторов в реакциях гидрирования и дейтерообмена от про-
цента d-состояния (рис. 1.5). Однако в других реакциях, например
при синтезе аммиака, такой прямой зависимости не наблюдается.
Кроме того, в работе Кокса, Лаули и Гуатми [16 ] установлено, что
у различных никельмедных сплавов связь активности с d-co-
стоянием для реакции разложения НСООН сохраняется только
для некоторых кристаллографических плоскостей. Здесь приведены
Л, 21
значения процентов d-состояния/ для некоторых металлов по По-
лингу [17]:
Сг..................39 Си ....................36
Мп .................40,1 Та ....................39
Fe..................39,7 W....................43
Со..................39,5 Pt ....................44
Ni..................40
Ввиду недостаточной разработанности теории состояния элек-
тронов в металлах нет основании считать, что механизм каталити-
ческих реакций на них исчерпывается простыми донорно-акцептор-
Рис. 1.5. Зависимость логарифма
скорости гидрирования этилена на
пленках от процента d-состояния
в металлах.
ными переходами. В связи с этим
затруднено и толкование результа-
тов электрических измерений,
проводимых на металлических ката-
лизаторах в различных условиях,
включая измерения в процессе ре-
акции.
Механизм каталитического дей-
ствия поверхности полупроводников
исследован Волькенштейном [18]*.
В основе его теории лежит пред-
ставление о хемосорбированных ча-
стицах как о поверхностных при-
месях, составляющих единую кван-
тово-механическую систему со всей
кристаллической решеткой катали-
затора. Согласно этим представле-
ниям, существуют две формы хемо-
сорбции одновалентного атома или радикала на поверхности кри-
сталла:
1) «слабая», при которой сорбированная частица (рассматриваемая
вместе со своим адсорбционным центром) остается электрически
нейтральной, а связь между частицей и решеткой осуществляется
без участия свободного электрона или свободной дырки кристал-
лической решетки;
2) «прочная», при которой сорбированная частица удерживает
около себя свободный электрон или свободную дырку кристалли-
ческой решетки и свободный электрон или свободная дырка прини-
мают непосредственное участие в хемосорбционной связи. '
В первом случае говорят о прочной и-связи, во втором — о проч-
ной p-связи. Эти формы связи представляют два предельных случая,
из которых первый соответствует чисто гомеополярной, а второй —
чисто ионной. В действительности, как правило, связь бывает про-
* Взгляды Волькенштейна являются более общими, чем электронные пред-
ставления, развиваемые, например, Хауффе и Вагнером [19].
22
межуточного типа. «Слабая» связь является однЙйсттронно’й (того же
типа, что в молекулярном ионе Н|). Связь между адсорбированным
атомом и кристаллом осуществляет валентный электрон адсорби-
рованного атома, в большей или меньшей степени «затянутый»
в кристаллическую решетку. Адсорбированный таким образом атом
обладает повышенной реакционной способностью и может реаги-
ровать как с молекулами, приходящими из объема, так и с молеку-
лами, сорбированными в приповерхностном слое.
Центром для адсорбции атома (молекулы) из газа может служить
не только ион металла Ме+, но также и анион R~. В последнем слу-
чае электронное облако адсорбированного атома затягивается
в решетку симметрично относительно иона R~, являющегося
адсорбционным центром.
ii В случае «прочной» связи свободный электрон или дырка лока-
|иизуется при ионе (адсорбционный центр), причем этот ион превра-
пцается как бы в нейтральный атом с неспаренным валентным элек-
|троном, т. е. в радикал. Если решетка кристалла состоит из много-
явалентных ионов, то могут образовываться не нейтральные радикалы,
[а ион-радикалы. Образующийся нейтральный атом (радикал), т. е.
^фактически свободная валентность на поверхности кристалла, может
[либо прочно адсорбировать на поверхности одновалентный атом
с образованием двухэлектронной связи, либо при взаимодействии
с молекулой перераспределить в ней связи по схемам, показанным
гна рис. 1.6.
К По схеме рис. 1.6, а поверхность обогащается радикалами, кото-
рые вступают в реакцию, как показано на рисунке, или могут за
ючет десорбции радикалов, слабо связанных с поверхностью, давать
•рачало гомогенным цепным реакциям. По схеме 1.6, б молекула,
^обладавшая в свободном состоянии двойной связью, в адсорбиро-
ванном виде приобретает свободную валентность и делается реак-
ционноспособным поверхностным радикалом. В обоих случаях сво-
бодная валентность остается ненасыщенной и после акта адсорбции,
рю перемещается от атома поверхности катализатора к молекуле,.
[участвующей в реакции. '
1 Наличие свободной валентности на поверхности является, с точки
шрения электронной теории, необходимым, хотя и не всегда доста-
точным условием протекания реакции в адсорбционном слое. Согласно ;
этой теории, в реакции участвуют не все хемосорбированные частицы, I
а только те, которые находятся в реакционноспособном состоянии, 1
определяемом для данной реакции ее механизмом. Поэтому скорость |
реакции определяется при прочих равных условиях относительным !
содержанием среди хемосорбированных молекул активной для
данной реакции формы. Волькенштейн, а затем Гарсиа де ля Банда
120] показали, что при установившемся электронном равновесии
относительные содержания различных форм хемосорбированных
частиц, а следовательно, и скорость каталитической реакции, свя-
занной с концентрацией реакционноспособных форм, зависят от по-
ложения уровня Ферми, энергетического интервала между валентной
23
зоной и зоной проводимости и положения локального уровня хемо-
сорбированных молекул.
Хемосорбированная частица, связанная с поверхностью прочной
связью, удерживает свободный электрон или свободную дырку
Рис. 1.6. Перераспределение валентностей на поверхности
полупроводникового катализатора.
электрического заряда на поверхности, а следовательно, к заряже-
нию поверхности и сдвигу положения уровня Ферми на поверхности
кристалла. Поэтому адсорбция молекул какого-либо газа может
затруднить дальнейшую адсорбцию молекул того же газа, что при-
водит к экспериментально наблюдаемому падению теплот адсорбции
с увеличением степени заполнения поверхности и к отклонению от
лангмюровской изотермы адсорбции.
Каталитические реакции в зависимости от характера влияния
на их скорость положения уровня Ферми могут быть (условно)
24
разделены на два класса — донорные и акцепторные. Донорные
реакции ускоряются при понижении уровня Ферми, а акцепторные
при его повышении.
Скорость реакции может принимать максимальное значение
при каком-то среднем положении уровня Ферми (например, если
реакционноспособной формой хемосорбции является слабая связь).
В этом случае при положении уровня Ферми ниже оптимального
данная реакция, как акцепторная, будет ускоряться донорной при-
месью и замедляться акцепторной, и после того как оптимальное
значение уровня Ферми будет превышено, влияние примесей на
скорость реакции станет обратным. Следовательно, одна и та же при-
месь может служить для данной реакции либо промотором, либр
ядом. Поэтому понятия промотирования и отравления можно объ-
единить общим термином — модифицирование.
В сложных параллельных, последовательных или параллельно-
последовательных реакциях донорные и акцепторные добавки могут
по-разному менять скорости составляющих реакций. Поэтому при
подборе полупроводниковых катализаторов нужно исследовать оба
класса добавок — донорные и акцепторные, а также проверять их
дозировку в достаточно широких интервалах, особенно в области
малых значений.
Электронные представления дают также некоторые направляющие
идеи в вопросе о влияний\«биографии» катализатора на его актив-
ность. Прежде всего, дефекты решетки, связанные с отклонениями
от стехиометрического состава, являются акцепторными или донор-
ными добавками в зависимости от их природы. Образование таких
дефектов тесно связано с условиями приготовления катализатора.
Так как во многих случаях дефектность решетки благоприятствует
каталитической активности, при приготовлении катализаторов жела-
тельно ее сохранить. Условия такого приготовления в общем соот-
ветствуют выдвинутому Рогинским в его «теории пересыщения» [21]
принципу, согласно которому активность катализатора повышается
с удалением условий приготовления катализатора от состояния
термодинамического равновесия. Это, впрочем, в настоящее время
может рассматриваться как тривиальный вывод из теории дефект-
ности твердого тела.
Несмотря на значение электронных представлений в катализе,
изложенная выше трактовка, основанная на «примесной» теории
проводимости, находится, в некотором противоречии с рядом экс-
периментальных данных. Вкратце укажем на основные из них.
1. Согласно представлениям о примесной проводимости, полу-
проводниковые катализаторы должны быть очень чувствительны
к различным добавкам и загрязнениям. На самом деле устойчивость
полупроводниковых катализаторов (окислов, сульфидов) к отравле-
нию намного превосходит устойчивость металлических катализа-
торов. В смешанных полупроводниковых катализаторах только
значительное содержание добавок изменяет свойства катали-
затора.
25
2. Каталитические реакции на полупроводниковых катализаторах
весьма часто проводятся в области столь высоких температур (400—
500° С), что участие примесных электронов в электропроводности
полупроводников невелико.
3. Наиболее активными, избирательными и технически при-
годными катализаторами являются окислы или сульфиды переход-
ных металлов, тогда как, исходя из представлений примесной теории,
каталитическими свойствами, должны обладать многие полупровод-
ники. Согласно представлениям этой теории, различного типа
модифицирование должно оказывать большее влияние на каталити-
ческие свойства полупроводника, чем его первоначальный химиче-
ский состав. На самом деле наблюдается обратное.
Кроме того, как указывает Волькенштейн [22], имеются значи-
тельные различия между идеальной и реальной поверхностью, что
может очень сильно исказить характер закономерностей, а следо-
t вательно, делает их толкование совершенно неоднозначным.
/ , Механизм окислительно-восстановительных реакций. В настоя-
Лущее время многие исследователи весьма скептически относятся
/ к идеям о связи каталитической активности с коллективными свой-
’Оствами электронов твердого тела (см., например, [23]) и вновь скло-
/ няются к чисто химическим концепциям, близким к теории проме-
/ жуточных соединений. Однако в рамках этих концепций, как ука-
зывалось в самом начале этой книги, нельзя объяснить многие
факты и наиболее фундаментальный из них — явление промотиро-
вания и модифицирования без образования новой фазы. Поэтому
более вероятным является широкая вариация механизмов катализа
от реакций, связанных, главным образом, с коллективными свой-
ствами электронов в твердом теле, до превращений, практически
идентичных с объемными гомогенными реакциями. Рассмотрим общий
подход к явлениям катализа на полупроводниках на примере наи-
более типичных для них окислительно-восстановительных превра-
щений. Для большей конкретности будет рассмотрен случай окис-
ления органических соединений.
Любая реакция контактного окисления должна включать с4$-
дующие стадии: V
1) образование электронной связи молекулы реагента с катали-
затором (хемосорбция реагента);
2) то же для молекулы кислорода (хемосорбция кислорода);
3) передача катализатором электронов от донора (реагента)
акцептору (кислороду); '
4) взаимодействие образовавшегося органического иона, ради-
кала или ион-радикала с ионом кислорода и возникновение новой
молекулы — продукта окисления; ' ,
5) десорбция продукта окисления.
Из сказанного вытекает, что катализатор окисления должен
быть способен -образовывать акцепторно-донорную связь с органи-
ческим реагентом и кислородом и каким-либо путем передавать
электроны от одной участвующей в реакции молекулы к другой.
26
Сам по себе механизм передачи электронов катализатором не имеет
значения и может быть различным для разных катализаторов
и реакций.
В отношении последовательных этапов реакций окисления при-
меним общий для случая сложных последовательных кинетических
процессов принцип лимитирующей стадии. Отсюда следует, что
в зависимости от величины скоростей составляющих стадий корре-
ляция между активностью катализатора и такими его свойствами,
как способность к комплексообразованию, электропроводность,
величина хемосорбции кислорода, может наблюдаться или отсутство-
вать. В силу этого возникает кажущаяся неоднозначность связи
каталитической активности твердого тела в реакциях окисления
с вышеперечисленными его свойствами.
Первые две стадии реакций контактного окисления, наряду с из-
ложенными выше механизмами, могут протекать по механизму
комплексообразования в тех случаях, когда катионы решезжи
сохраняют свою индивидуальность. Вервей [24] для обратных
шпинелей*, а затем Морин [251 — для окислов металлов с незапол-
ненными 3(?-уровнями электронов указали на такую возможность,
объяснив возникновение в таких соединениях электропроводности
присутствием в них ионов одного и того же металла в различных
валентных состояниях и в эквивалентных позициях кристаллической
решетки. Можно предполагать, что подобного рода механизм электро-
проводности возможен не только для окислов (в том числе и тройных
систем окислов [26]), но и для многих полупроводниковых соеди-
нений переходных металлов. Базируясь на этих представлениях,
Дауден [27 ] рассматривает хемосорбцию на поверхности и явления
замещения одного сорбента другим как реакции образования и пре-
вращения комплексов по механизму Sjvl- и SN2-замещения. Киселев
[28] также рассматривает адсорбцию как процесс поверхностного
комплексообразования, когда при возникновении донорно-акцеп-
торных связей неподеленная пара электронов лиганда оказывается
затянутой на внутренние орбитали атома решетки, являющегося
центром адсорбции. При таком механизме адсорбированные мо-
лекулы всегда будут в той или иной мере реакционноспособны.
Действительно, затягивание неподеленной пары лиганда на внутрен-
ние орбитали центрального атома приведет к деформации адсорби-
рованной молекулы и ослаблению внутримолекулярных связей.
Отметим попутно, что трактовка Киселева справедливо распростра-
няет электронные представления и на механизм кислотно-основного
гетерогенного катализа. Развивая представления теории поля ли-
гандов, Руней и Уэбб [29] показали, что механизм реакций дейтеро-
обмена, гидрирования и дегидрирования углеводородов на переходных
* Шпинелями называются такие окислы, в структуре которых атомы кисло-
рода образуют плотную кубическую решетку с октаэдрическими пустотами,
в которых расположены ионы двух- и трех^ледтных металлов. Обратная шпи-
нель имеет структуру Ме3+ [Ме2+ Ме3+] О4. ,
27
металлах также может быть хорошо объяснен, исходя из пред-
ставлений о поверхностных л-комплексах. Они считают, что поверх-
ностные атомы металлов обладают свойствами свободных атомов
или ионов,
Волькенштейн и Киселев подчеркивают, что при рассмотрении
системы адсорбент — адсорбат как единой квантовомеханической
системы электронный переход означает лишь переход носителя тока
(электрона, дырки) из одного энергетического состояния в другое
без фиксации геометрии перехода. Однако при сохранении ионами
решетки своих индивидуальных свойств и отсутствии зон проводи-
мости такая трактовка уже становится неприемлемой. В этом случае
переход электронов .от молекулы органического соединения к твер-
дому катализатору может привести к обычной реакции восстановле-
ния катиона переменной валентности, входящего в состав катализа-
тора, аналогично тому, как это происходит в гомогенном катализе
R3CH + Men+ —> R3C- +Me(n-1)+4-Н+
Соответственно, для сорбции кислорода процесс можно рассматри-
вать как окисление поверхностных ионов.
Мерой окислительной способности ионов в растворах является,
как известно, величина окислительно-восстановительного потен-
циала. По аналогии с уравнениями Бренстеда и Поляни — Семе-
нова [30, 31], можно предположить, что скорость стадии перехода
электрона от молекулы органического вещества к катализатору
выражается уравнением
lgA=24.-|-Blg£7 (1.25)
где U — окислительно-восстановительный потенциал катиона с об-
ратным знаком; к — константа скорости перехода; А и В — кон-
станты.
В таком случае при достаточной скорости окисления катиона
активность катализатора будет определяться значением U. Дей-
ствительно, мы знаем, что наиболее активными гомогенными ката-
лизаторами являются ионы марганца и кобальта, у которых наи-
более отрицательный окислительно-восстановительный потенциал.
Видимо, этот принцип можно распространить и на катализаторы,
у которых электроны решетки не обобщены. Как известно, кобальт-
содержащие катализаторы, в том числе и безванадиевые, обладают
высокой активностью в процессах с окислением связи С — Н.
Третья стадия процессов окисления — передача электронов от
донора к акцептору (от реагента к кислороду), в отличие от первых
двух, является специфичной для гетерогенного катализа и связывает
его с проблемами физики твердого тела. Принципиально проблема
подвижности электронов в адсорбционном комплексе не отличается
от проблемы подвижности электронов внутри молекулы, поскольку
- такая подвижность обусловливает реакционную способность системы.
Действительно, реакцию окисления какого-либо соединениц, напри-
мер SO2, на твердом катализаторе можно себе представить в виде
28
. . -ж ...
передачи электронов катализатора внутри адсорбционного комплекса,
аналогично передаче электронов внутри молекулы органического
соединения по системе двойных связей. При таком механизме, в кото-
ром в общем виде уже нельзя исключить пространственное передви-
жение электронов, лимитирующим фактором может оказаться по-
движность носителей тока, которая у полупроводниковых соеди-
нений невелика.
Если электропроводность объясняется перезарядкой ионов, зон-
ная теория полупроводников, по-видимому, в простейшем виде
неприменима; не происходит полного вырождения уровней валент-
ных электронов в отдельных ионах, а сохраняется периодичность
в энергетическом спектре валентных электронов кристалла. Катионы
решетки находятся в потенциальной яме, так что переход электрона
от катиона к катиону требует энергии активации, а длина свободного
пробега электрона соответствует междуатомным расстояниям в кри-
сталлической решетке. В таком случае энергия активации опреде-
ляется не только параметрами атома, образующего катион (т. е.
в конечном счете его положением в таблице Менделеева), но и меж-
атомными расстояниями в кристалле, что указывает на значение
геометрических параметров кристалла в отношении его каталити-
ческой активности.
Передача электронов катализатором может также осуществляться
прямым взаимодействием атома кислорода с электроном катиона
восстановленного катализатора, т. е. по механизму окисления-
восстановления катализатора.
Взаимодействие поверхностных радикалов или ион-радикалов
(их существование теперь доказано исследованиями с помощью
ИК спектров и методом ЭПР) с атомами кислорбда или между собою
могут протекать по разным детальным механизмам, но, очевидно,
с большой скоростью и поэтому не ~могут являться определяющими
стадиями.
Десорбция продуктов может быть связана с рекомбинацией
электронов и дырок, т. е. зависеть от подвижности носителей тока,
либо может протекать по механизмам, обратным механизмам адсорб-
ции, т. е. с энергетическими или пространственными переходами
электронов.
Как видно из изложенного, характер переходов электронов при
окислительно-восстановительных превращениях,' а следовательно,
и кинетические лимитирующие стадии процесса могут быть доста-
точно разнообразными. Поэтому мало вероятными представляются
однозначные корреляции каталитической активности с различными
электрофизическими параметрами катализатора при широкой вариа-
ции этих параметров и условий реакции. Соответственно, как пра-
вило, ни одно из указанных свойств твердого тела не является доста-
точным, а только необходимым для придания ему каталитической
активности.
29
1.4. ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ЭНЕРГЕТИЧЕСКИЕ
И ГЕОМЕТРИЧЕСКИЕ ФАКТОРЫ В КАТАЛИЗЕ
/ Промежуточные соединения. Как указывалось выше, ряд затруд-
I нений при объяснении явлений гетерогенного катализа с точки зре-
ния коллективных свойств электронов твердого тела, а также успехи
в идентификации поверхностных адсорбированных соединений при-
вели к возрождению чисто химических концепций в теории катализа,
; в общем аналогичных первоначальной теории промежуточных со-
} единений. Особое значение приобретают при этом индивидуальные
| свойства атомов и ионов в твердом теле, т. е. свойства, определяемые
/ положением элемента в периодической системе элементов. Соот-
I ветственно, как и в общей теории химических реакций велика роль
J энергетических параметров самого превращения.
1 Исследования инфракрасных спектров адсорбированных моле-
кул показали, что в процессе адсорбции, а следовательно, и в первой
стадии катализа, действительно происходит образование промежу-
точных соединений, которые во многих случаях идентичны или
близки к обычным химическим соединениям, постулировавшимся
в ряде механизмов каталитических превращений.
! Так, Сидоровым [32] было показано образование эфирных Свя-
’ зей при адсорбции метанола на силикагеле. Хироте, Фуэки и Сакаи
[33] установили наличие поверхностных координационных алю-
; миниевых комплексов при адсорбции метиламина на окиси алюми-
; ния, Захтлер с сотрудниками [34] показали, что первой стадией
I процесса окисления бензальдегида на окисных катализаторах яв-
/ ляется образование несимметричного бензоата металла. Эти данные
! свидетельствуют о том, что химические свойства веществ, участву-
? ющих в каталитических процессах, в значительной степени сохра-
] няются и в ходе поверхностных реакций, что может служить хорошей
' основой для раскрытия их механизма.
Принцип энергетического соответствия. Согласно принципу
энергетического соответствия Баландина [35] для разрыва связи
А—В в молекуле, адсорбированной на поверхности катализатора,
требуется меньшая энергия, чем в объеме, потому что части моле-
кулы А и В связываются с атомами катализатора с выделением
энергии. Энергетические барьеры при реакции
AB + CD = AD + BC
для адсорбции исходных молекул Еадс и десорбции продуктов
реакции Едес выражаются уравнениями
’ ч ъ
I + (1-26)
I di di
‘‘ s h
\ Елес= — (1-27)
где h — теплота реакции; s = (?АВ + <2cd + Cad + <2вс — сумма
энергий образующихся и разрываемых связей; q — Сак + Свк +
30
+ Qck + <2dk — адсорбционный потенциал, равный сумме энергии
связей катализатора с атомами и радикалами, образующимися в про-
цессе реакции.
Энергия активации реакции Е выражается по Баландину урав-
нением
Е=^-е, (1.28>~
4
где е — энергетический барьер наиболее эндотермической стадии;
3/4 — коэффициент, учитывающий, что при катализе происходит
деформация связей, а не их разрыв.
Рис. 1.7. Зависимость энергетиче-
ского барьера реакции от адсорб-
ционного потенциала катализатора
для случая, когда одна из стадий
эндотермична.
Рис. 1.8. Зависимость энергетического
барьера реакции от адсорбционного
потенциала катализатора для случая,
когда обе стадии экзотермичны.
Максимальная активность катализатора для данной реакции
достигается при
<7 = 4 (1-29)
что видно из рис. 1.7 и 1.8, на которых даны зависимости
^адс и Елес от q. При постоянных значениях q величина энер-
гетического барьера определяется наименьшей из двух величин h
или s, которые, согласно уравнениям (1.26),и (1.27), линейно зависят
от адсорбционного потенциала q, образуя на графике прямые, накло-
ненные к оси абсцисс под углом 45°, которые получили в специаль-
ной литературе наименование «вулканообразные кривые». Зависи-
мость энергетического барьера реакции от адсорбционного потен-
циала катализатора выражается на графиках сплошной ломаной
линией. Минимум значения Е лежит либо точно в точке s/2 (рис. 1.7),
когда одна из стадий реакции эндотермична, либо в окрестностях
точки s/2 в интервале ±h/2 (рис. 1.8), когда обе стадии экзотермичны
и реакция в области минимума не имеет энергетического барьера.
31
к
В соответствии с рис. 1.7 и 1.8 замена одного катализатора другим
ведет к ускорению реакции, когда
4г<0 <ьзщ
' Темкин [36] вывел уравнение (1.29) кинетическим, путем и пока-
зал, что оно не связана с геометрическими постулатами мультиплет-
ной теории Баландина (см. ниже), а является общим свойством реак-
ций типа АВ->А4-В. Стоит также отметить, что существование
оптимальной энергии адсорбции реагентов на катализаторе вытекает
из самых общих положений теории катализа: адсорбированная
молекула не может быть реакционноспособной, если ее связь с по-
верхностью слишком сильна или слишком слаба.
Большое развитие вопросы связи каталитических свойств твер-
дого тела с энергетическими характеристиками реакции и самого
твердого тела получили в работах Борескова [37], Ройтера [38]
и Захтлера [34]. Боресков при этом исходит из предпосылки, что
энергия связи кислорода с катализатором в поверхностном слое
окисла входит слагаемым в величину энергии активации реакций
окисления. Захтлер, изучая реакцию разложения муравьиной кис-
лоты на металлах, получил четко выраженную вулканообразную
кривую активности катализаторов по теплоте образования формиатов
металлов, промежуточное образование которых было доказано ИК
спектрами. Более подробно связь термодинамических параметров
с каталитической активностью рассмотрена в главе IV в связи с про-
блемой подбора катализаторов.
Принцип геометрического соответствия. Так как поверхностные
атомы катализатора принимают участие в образовании промежу-
точных активных комплексов, возможность образования последних
должна, очевидно, зависеть от взаимного расположения активных
атомов («центров»). Следовательно, существует определенное соот-
ветствие между геометрией расположения атомов на поверхности
катализатора и типом катализируемых им реакций. В наиболее за-
конченном виде принцип геометрического соответствия был выдви-
нут Баландиным в 1929 г. в связи с изучением реакции дегидриро-
вания циклогексана. В дальнейшем принцип геометрического соот-
ветствия являлся предметом многочисленных исследований, обзор
которых можно найти в литературе [35; 39].
Согласно теории Баландина, активными центрами являются
группы атомов — мультиплеты, представляющие собой не успев-'
шие разрастись кристаллизационные центры или небольшие участки
кристаллической решетки катализатора. Механизм реакции заклю-
чается в том, что в молекулах реагентов, адсорбированных на ката-
литической поверхности, происходит разрыв валентных связей
между атомами, притягиваемыми разными атомами поверхности ка-
тализатора, и одновременно образуются новые связи между атомами,
адсорбированными у одного атома катализатора. Это наглядно
иллюстрируется, например, схемами реакций дегидратации и де-
32 '
гидрирования этанола, протекающих на активных центрах, состоя-
щих из двух атомов (дублетах):
Н Н кат
I I •
Н— С-С=о + Н- н
I
Н кат
Н
Н Н
|. кат | ;
-С---С—г-н
Н . О—?-н
• кат :
кат
Н\ ’ /Н
\:=с/ +н-он
кат
Группы атомов, заключенные в рамки, находятся на поверхности
катализатора; части реагирующих молекул, не заключенные в рамки,
непосредственно не связаны с поверхностью. Согласно мульти-
плетной теории, должно наблюдаться соответствие между межатом-
ными расстояниями и расположением атомов катализатора в муль-
типлете, с одной стороны, и межатомными расстояниями и распо-
ложением атомов в молекулах реагентов, с другой.
Хотя в своем классическом виде мультиплетная теория сейчас
потеряла свое значение, однако сам факт влияния геометрии решетки
на каталитические свойства вряд ли может подвергаться сомнению
хотя бы из соображений структуры и соответственно энергетики
переходного комплекса и взаимосвязи состояния электронов в твер-
дом теле с геометрией решетки.
1.5. ИОННЫЙ КАТАЛИЗ НА ПОВЕРХНОСТИ
На поверхности гетерогенных катализаторов могут протекать
ионные реакции, аналогично тому, как они протекают в среде жид-
кого диэлектрика. Это указывает на существование особой, весьма
значительной группы гетерогенно-каталитических реакций, объеди-
няемых понятием «гетерогенного ионного катализа». Механизм
этой группы процессов в основном может быть объяснен на основе тех
в достаточной мере разработанных положений, которыми оперируют
в области гомогенного ионного катализа [40, 411. Наиболее хорошо
изучены гетерогенные каталитические реакции, катализируемые
твердыми протонными и апротонными кислотами или соответственно
основаниями. Гетерогенный ионный (но не кислотно-основной) ка-
тализ изучен гораздо меньше.
3 Заказ 1441
33
Кислотно-основпой катализ. Каталитическое действие кислот
на различные гетеролитические реакции в органической химии, та-
кие, как гидролиз, конденсация, изомеризация и другие, является
наиболее старым примером катализа. Ему посвящен ряд монографий
[42—441; здесь мы коснемся лишь основных положений теории, свя-
занных с переносом процессов на твердую поверхность. Обзоры по
этому вопросу изложены в работах [45—47 ].
Основная функция катализатора-кислоты состоит во введении
протона или положительного иона в реагирующую молекулу, а ка-
тализатора-основания — в удалении протона из молекулы или вве-
дении в нее аниона. Образующиеся таким путем заряженные комп-
лексы нестабильны и либо реагируют с другой молекулой, либо рас-
падаются; в обоих случаях происходит регенерация катализатора.
Процессы переноса протона называют прототропией, а анионов —
анионотропией.
Действие катализаторов в таких процессах может быть выражено,
например, такой протолитической схемой:
кислотный катализ
основной катализ
СН“ с=о+нв
;С=С—О+НВ \с = СОН+В-
СН—с=о+в~
Другим примером являются реакции с обратимым присоедине-
нием молекул, содержащих гидроксильную группу, к карбониль-
ной группе
ОН
НВ-|-\с=О+ВОН >С+ 4-ROH+B- \с<^ +НВ
ОН 0R
/°" \ /0Н
B"+V;=O+ROH —' 'ус +НВ +В“
z 7 \qr z \qr
Скорость каждой стадии в реакциях кислотно-основного ката-
лиза вследствие снижения энергии активации значительно выше,
чем у всего процесса в целом, когда тот протекает без ката-
лизатора.
Вышеприведенные механизмы относятся к так называемым брен-
стедовским кислотам или основаниям, содержащим, соответственно,
ион Н+ или ОН-. Как известно, в современной физической химии
понятие кислот и оснований распространяют на соединения, не
имеющие ионов Н+ и ОН-, но способные быть акцепторами (кис-
лоты) или донорами («снования) электронной пары. Такие кислоты
и основания называют льюисовскими. Ввиду общности действия
льюисовских кислот и оснований и дативных комплексов, в послед-
34
нее время намечается тенденция возврата к первоначальному опре-
делению кислот и оснований. Однако, поскольку химическое воз-
действие бренстедовских и льюисовских кислот в значительной мере
одинаково, мы будем рассматривать их совместно. В случае льюи-
совских кислот протолитические процессы заменяются промежуточ-
ным образованием комплекса из реагирующей молекулы и катали-
затора при участии неподеленной электронной пары реагента.
К гетерогенным катализаторам кислотно-основного типа отно-
сится ряд твердых кислот и оснований.
Твердые кислые катализаторы:
1) малолетучие минеральные кислоты Н3РО4, H2SO4 на твердых
пористых носителях;
2) твердые неорганические кислоты и продукты их полной и
неполной дегидратации (например, алюмосиликаты, алюмотитанаты,
гетерополикислоты, окислы кремния, алюминия, титана, вольфрама);
3) синтетические смолы — катиониты;
4) соли кислородсодержащих минеральных кислот, главным об-
разом с тяжелыми металлами (фосфаты, сульфаты, станнаты, вольф-
раматы);
5) галогениды трехвалентных металлов на пористых носителях.
Границу между первыми двумя группами провести затрудни-
тельно, так как адсорбированные кислоты часто реагируют с но-
сителем, образуя новые твердые кислоты, как это, вероятно, про-
исходит в случае «твердой фосфорной кислоты» (Н3РО4 на ки-
зельгуре).
Твердые основания:
1) твердые неорганические основания — гидроокиси и окиси
щелочных и щелочноземельных металлов как без носителей, так
и на носителях; амиды щелочных и щелочноземельных металлов;
2) смолы — аниониты;
3) щелочные и щелочноземельные соли слабых кислот (карбо-
наты, карбиды, сульфиды, нитриды, силикаты и т. п.).
Особо следует выделить молекулярные сита — цеолиты, пока
еще недостаточно изученные, но являющиеся по своему химическому
воздействию твердыми кислотами (см. раздел 1.6).
Важнейшей закономерностью, установленной для гомогенного
кислотного катализа, является уравнение Бренстеда. В наиболее
простом и общем виде оно имеет вид (для кислот)
lg fc = lg a-f-a 1g ки (1.31)
где к — константа скорости каталитической реакции; кя — кон-
станта ионизации кислоты; а — положительный показатель степени;
а — постоянная, сохраняющая в данном классе реакций одно и
то же значение для ряда кислот близкого строения.
Аналогичную форму имеет уравнение для катализа основаниями.
Существенным для подбора гетерогенных кислотно-основных
катализаторов является применимость к гетерогенному катализу урав-
нения Бренстеда. Ввиду отсутствия жидкой фазы, а следовательно,
3*
35
явления диссоциации, уравнение Бренстеда, аналогично случаю
с концентрированными кислотами, принимает вид
lgfc = lga+atf0 (1-32)
Здесь Но — функция кислотности Гаммета в логарифмической
форме, которая является мерой протонизации кислоты и выражается
формулой
T^-p/cB-lg-^gp- (1.33)
где р/св — обратный логарифм константы протонизации эталонного
основания (индикатора) в разбавленном растворе, а [ВН+]/[В°] экспе-
Рис. 1.9. Симбатность каталитичес-
кой активности и функции кислот-
ности для реакции крекинга изо-
пропилбензола на алюмосиликатном
катализаторе.
риментально определяемое (нап-
ример, по окраске) отношение кон-
центраций протонизированной и
депротонизированной формы ин-
дикатора.
Таким образом, сравнивая для
различных твердых катализаторов
точки перехода окраски индика-
торов, можно расположить твер-
дые кислые катализаторы в ряд по
силе протонизации и сравнить этот
ряд с каталитической активностью.
Имеется весьма большое коли-
чество работ, связывающих ката-
литическую активность твердых
катализаторов с их кислотностью,
так что рассматривать их здесь
не представляется возможным.
В качестве примера на рис. 1.9 показана связь крекирующей актива
ности алюмосиликатного катализатора с его общей кислотностью.
Более подробно это соответствие будет прослежено в главе IV в связи
с проблемой подбора катализаторов, здесь же мы остановимся лишь
на основных особенностях гетерогенного кислотно-основного
катализа.
В главных чертах механизм действия твердых кислот и основа-
ний должен быть аналогичен механизму действия кислот и основа-
ний в гомогенных жидкофазных системах. Для частного случая
минеральных кислот, адсорбированных на твердой поверхности,
это было показано Гольданским, Семеновым и Чирковым [48].
Для собственно твердых кислот, как показано рядом авторов [49—
51] на примере реакции крекинга на алюмосиликатных катализа-
торах, каталитическая активность находится в прямой зависимости
от количества, находящегося в катализаторах обменивающегося водо-
рода. Аналогия в строении и действии гомогенных и гетерогенных
кислых катализаторов указывает на возможность протекания реак-
ций по ионному механизму с ионом протона в качестве катализа-
36
&№ SSSerifeai л
тора. Реакции на алюмосиликатных и подобных катализаторах в этом
случае, видимо, проходят через стадию присоединения протона с об-
разованием иона карбония [52, 53]. Например, механизм реакции
гидратации этилена следующий:
С2Н4 4- Н+ —> СН3СН2+
СН3СН| + Н2О > СН3СН2ОН4-Н+
Еще Эванс [54] установил, что на поверхности активированного
флориндина, так же как и в растворе H2SO4, спектр 1,1-дифенил-
этилена имеет в ультрафиолетовой области максимум, весьма близ-
кий к максимуму поглощения иона трифенилкарбония, но очень
сильно отличается от спектра самого 1,1-дифенилэтилена. Позже
общность кислотно-основного взаимодействия в гомогенной средн
и на поверхности твердого катализатора показана во многих рабо-
тах методом инфракрасной спектроскопии как для углеводородов,,
так и для спиртов и органических оснований [33 ].
Для катализаторов щелочного типа также наблюдается корре-
ляция между основностью и каталитическими свойствами, например
селективностью в реакции Фишера—Тропша [55].
Следует подчеркнуть, что наряду с далеко идущей аналогией,
гомогенных и гетерогенных кислотно-основных катализаторов по-
следние, в общем случае, вследствие отсутствия растворителя и
объемной жидкой фазы имеют ряд особенностей.
Кислотно-основные свойства твердой поверхности, видимо, на-
ходятся в тесной связи с электроотрицательностью металлического’
иона, как это показано Танака и Озаки [56]. Последняя прибли-
женно может быть вычислена по формуле
X=(l + 2z)X0 (Д.34)’
где z — заряд иона; Хо — электроотрицательность нейтрального
атома по Полингу.
Для твердых кислот недостаточно выяснено различие и общность,
механизмов каталитического действия бренстедовских и льюисов-
ских кислот. Так, по данным одних авторов [57 ] реакция дегидра-
тации спиртов происходит только за счет апротонной кислотности.
Другие авторы [58] опровергают это утверждение. В ряде работг
например [59], указывается, что кислотные свойства гидратиро-
ванной окиси алюминия и силикагеля не связаны с водородом их
гидроксильных групп.
Весьма существенным (см. главу IV) является одновременное
присутствие центров разной кислотной силы.
В некоторых парофазных процессах каталитическим путем можно»
проводить реакции, для которых в Жидкой фазе кислота служит
реагентом, а не катализатором. Это является следствием самореге-
нерации твердой кислой поверхности в условиях высокотемпе-
ратурного гетерогенного процесса, что невозможно в жидкофазных
реакциях. Примерами такого рода переходов от жидкофазных:
37"
некаталитических процессов с кислотами в качестве реагентов к ге-
терогенным реакциям кислотного катализа являются, например,
конденсация бензола с фталевым ангидридом в антрахинон [60].
Катализ на ионообменных смолах. Ионообменные смолы (иониты),
которые катализируют разнообразные химические превращения,
протекающие по механизму кислотно-основного катализа, начинают
применять в промышленности. Применение ионитов в качестве ката-
лизаторов освещено в [61, 62]. Ниже кратко излагаются основные
'Сведения об этой несколько специфической области гетерогенного
катализа.
Иониты представляют собой сшитые полимеры, имеющие в своей
молекуле специфические функциональные ионогенные группы. Эти
группы могут генерировать как катионы, так и анионы, в зави-
•симости от чего иониты обладают свойствами полимерных твердых
кислот (катиониты) или оснований (аниониты). Общий тип структуры
ионитов виден из приводимых ниже формул полистирольных катио-
нита и анионита в активной форме
—СН—СН2-СН—СН2—СН-СН2—
SO3H SO3H SO3H
полистирольный катионит
-сн-сн2-----------------------сн-сн2-
I I
//\
г II + I II
^XCH2N(CH3)3OH- , ^XCH2N(CH3)SOH-
полистирольный анионит
Способные к обмену ионы, нейтрализующие заряд ионогенных
групп полимера, называют противоионами. Они подвижны и не за-
креплены в определенных местах полимерной молекулы. Противо-
ионы подобны соответствующим ионам в гомогенных средах и обла-
дают аналогичными каталитическими свойствами. При этом надо
учесть, что они находятся в ионитах в сольватированном состоянии.
Во многих случаях, когда нет обстоятельств, осложняющих про-
текание собственно химической реакции, наблюдаются совпадения
гомогенной и гетерогенной реакций, в отношении кинетических
порядков по реагентам.
Гескелл и Гаммет [63] ввели понятие коэффициента эффектив-
ности ионита, который определяется как отношение скорости гетеро-
генной реакции к скорости гомогенной при равной эквивалентной
.концентрации ионов в обоих случаях.
К факторам, определяющим каталитическую активность иони-
тов, аналогично неорганическим катализаторам, можно отнести
«следующие:
38
1) химическое строение;
2) абсорбционная и растворяющая способность по отношению1
к реагентам;
3) структура пор.
Химическое строение ионита определяет его силу как кислоты
или основания, т. е. термодинамическую активность соответствую-
щих противоионов, что в свою очередь определяет каталитическую
активность ионита.
Важное влияние на активность и избирательность ионитных
катализаторов оказывает их способность к абсорбции и в отличие от
неорганических катализаторов к объемному растворению реаген-
тов. Очевидно, что высокая растворимость реагентов в ионите спо-
собствует повышению его каталитической активности. Степень рас-
творимости реагента в ионите определяется как химической струк-
турой ионита, так и свойствами применяемого растворителя. Варьи-
руя то и другое, можно добиться высокой избирательности действия
ионитов как катализаторов, значительно превосходящей избиратель-
ность твердых неорганических кислот.
Пористая структура ионитов сказывается на их каталитической
активности в основном через явления внутренней диффузии. Однако
применительно к ионитам, видимо, надо различать диффузию в по-
рах ионита и диффузию в массе полимера. Диффузия в порах ионита
совершенно аналогична диффузии в порах прочих катализаторов
и подробно описана в главе III.
Диффузия в массе полимера связана с объемной растворимостью
компонентов реакционной смеси в ионите и определяется химиче-
ской структурой полимера, в частности расстояниями между поли-
мерными цепями и густотой поперечных сшивок. Эти расстояния
ограничивают возможность проникновения той или иной молекулы
внутрь полимера и тем оказывают важнейшее влияние на избиратель-
ность действия ионитов.
Иониты с высокой растворяющей и абсорбирующей способностью
по отношению к реагентам и хорошо развитой пористостью могут
обладать коэффициентом эффективности, превосходящим единицу.
Недостатком ионитных катализаторов является их относительно1
низкая термическая и механическая стойкость. Обычно они не вы-
держивают долго температуры выше 150—180° С, однако имеется
указание на возможность применения для катализа весьма термо-
стабильных ионитов [64].
1.6. ЦЕОЛИТНЫЕ КАТАЛИЗАТОРЫ*
По механизму своего действия, по-видимому, несколько особое-
место занимают цеолитные катализаторы. Применение их, начавшееся
в сравнительно недавнее время, произвело поистине революцию
* Структуре цеолитов посвящены монографии [65, 66]. Обзоры по этому
вопросу даны в работах [67—69].
39
в промышленных процессах каталитического крекинга. Имеются
указания на их использование в промышленном процессе гидрокре-
кинга, а также о перспективах их применения для таких технических
процессов, как изомеризация, алкилирование и др.
Цеолиты представляют собой кристаллические алюмосиликаты
общей формулы Ме2/пО • Al2O3-n SiO2-p Н2О, в которых Me обо-
значает либо одновалентные щелочные (п = 1), либо двухвалент-
ные щелочноземельные (п = 2), либо другие специально введенные
металлы. Кристаллическая решетка цеолитов образуется цепочками
тетраэдров [(Si, Al) О4], соединенных общими атомами кислорода.
Размеры и форма кристаллической решетки строго определены
для каждого вида цеолита. Металлические катионы находятся в пу-
стотах решетки цеолитов и способны обмениваться на другой металл
или водород.
Основные особенности цеолитов как катализаторов следующие.
1. Каталитической активностью обладают не только протони-
зированная
которые можно себе представить как бренстедовские или льюисов-
ские кислоты, но и замещенные металлами цеолиты. Последним та-
ких свойств, на первый взгляд, приписать нельзя.
2. Способность цеолитов, не имеющих в своей структуре метал-
лов переменной валентности и не обладающих полупроводниковыми
свойствами, катализировать не только реакции кислотно-основного
типа, но и окислительно-восстановительные. В качестве примера
приводим данные по гидрированию бензола на различных формах
морденита:
Катализатор . . LiMor NaMor КМог MgMor СаМог НМог
Выход циклогек-
сана, % ... . 87,7 98,3 11,0 69,4 98,6 19,7
3. Исключительно высокая активность и селективность цеоли-
тов в ряде реакций и высокая термическая стабильность их решетки.
К настоящему моменту нет единой точки зрения на механизм
действия цеолитов. Миначев и Рубинштейн с сотрудниками [70 ]
считают, что в цеолитах активные центры при катализе реакций с об-
разованием карбоний-иона расположены на шестичленных кольцах
алюмосиликатной решетки, не занятых катионами металлов. Такие
40
кольца несут избыток отрицательного заряда и могут присоединять
к себе протон.
Другие авторы склонны рассматривать каталитическую актив-
ность цеолитов как результат наличия в решетке бренстедовских
или льюисовских кислотных центров, причем последние сохраняются
и в замещенных цеолитах [71, 721. Влияние иона металла при этом
приписывается стабилизации им структуры цеолита [73 ].
Однако вышеуказанные механизмы пригодны только для гетеро-
литических реакций с участием карбоний-ионов, но не могут объяс-
нить окислительно-восстановительных превращений без участия
ионов переменной валентности. Можно высказать предположение,
что поскольку цеолитным структурам свойственна координационная
ненасыщенность, механизм их действия приближается к механизму
действия комплексообразующих катализаторов и потому достаточно
универсален.
Высокая селективность цеолитных катализаторов, кроме дру-
гих причин, связана еще с молекулярно-ситовым эффектом, вслед-
ствие которого во внутреннюю полость кристаллической решетки,
где осуществляется сам акт катализа, могут проникать, из-за гео-
метрических ограничений, только определенные молекулы или их
части. Так на цеолитах можно осуществлять селективное гидриро-
вание бутена в смеси его с изобутеном или дивинила в смеси с изо-
бутиленом. Вариация размеров окон в решетке при синтезе цеоли-
тов создает довольно широкие возможности для повышения селектив-
ности действия цеолитных катализаторов.
На цеолитных катализаторах можно осуществлять окислительно-
восстановительные реакции за счет ионов металлов переменной ва-
лентности. Для этого последние должны вводиться в решетку цео-
лита путем обмена с первоначальным ионом щелочного или щелочно-
земельного металла. В этом случае ион металла переменной валент-
ности сохраняет свою индивидуальность и механизм его действия
аналогичен таковому в гомогенном катализе. Восстановлением ионов
металла в решетке цеолитов удается получать металлы в атомнодис-
персном состоянии. Эти возможности представляются весьма ин-
тересными в отношении проведения на цеолитах высокоселективных
и высокопроизводительных процессов.
1.7. ОСОБЕННОСТИ ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ
РЕАКЦИЙ JB ЖИДКОЙ ФАЗЕ
Жидкофазные процессы имеют ряд особенностей по сравнению
с реакциями, протекающими в газовой фазе. Первое, что бросается
в глаза, — это значительное увеличение скорости превращений
при переходе от газо- к жидкофазным процессам. Скорость реакций
при этом увеличивается обычно на 1—2 порядка при перенесении
процесса из газовой фазы в фидкую при одновременном понижении
температуры на 100—200 град. Так, на наиболее производительных
катализаторах парофазного восстановления нитросоединений скорость
41
s
li
процесса (при практически полном восстановлении) составляет
величину порядка 10 молъ/ч на 1 кг катализатора при температуре
процесса 300—350° С. Тот же процесс в жидкой фазе протекает на близ-
ких по составу катализаторах со скоростями более чем 600 моль/ч ;
на 1 кг катализатора при 100—150° С. Аналогичную картину можно
наблюдать и для процессов окисления углеводородов и спиртов и
других процессов, протекающих как в газовой, так и жидкой
фазах.
Пока нет теоретического объяснения такого увеличения скорости
гетерогенных каталитических реакций в жидкой фазе по сравнению
лс газовой. Можно предполагать, что это явление в какой-то мере свя-
Iэано с тем, что жидкость является «полуупорядоченной системой»,
Г' к которой неприменимы принципы классической химической кине- >
тики, базирующейся на теории столкновений и кинетической теории
газов. Вследствие существования ближнего порядка в располо-
жении молекул, т. е. их определенной ориентации и взаимного влия-
ния, возможно, что понижение энтропии А5 при образовании
у активированного комплекса реагентов с катализатором составляет не-
! значительную величину; это резко сказывается на скорости процесса
(см. раздел 1.1). Точно так же объединение молекул в сольватацион-
1 ные комплексы может сопровождаться понижением энергии актива-
‘ ции реакции, подобно тому, как это имеет место при интермолекуляр-
ных превращениях.
В жидкой фазе молекулы растворителя, адсорбируясь на по-
верхности катализатора, непосредственно влияют на ее состояние,
заряд, свободную энергию и распределение электронов на поверх-
ностных уровнях. Наличие жидкой фазы определяет возможность
протекания реакций каталитического гидрирования и окисления
по электрохимическому механизму, т. е. путем прямого перехода
электронов реагирующих молекул к электроду-катализатору [74,
75]. Близость каталитических и электрохимических жидкофазных
процессов позволяет широко применять электрохимические методы
при исследованиях состояния катализатора в ходе реакции, меха-
низма реакции и кинетики процессов. Особенно много в этом на-
правлении сделано Сокольским [75, 76]. ?
На примере окисления углеводородов на гетерогенных окисных
катализаторах было установлено, что в жидкофазном процессе
в ряде случаев образуются иные продукты, чем в газофазном с той же
исходной системой [77, 78]. Продукты реакции при этом прибли-
жаются к продуктам реакции жидкофазного цепного окислений
•с гомогенными катализаторами из растворимых солей металлов пере-
менной валентности. Так, о-ксилол в газовой фазе окисляется на
i пятиокиси ванадия во фталевый ангидрид, а в жидкой — в о-толуи-
I ловую кислоту, которая получается при окислении о-ксилола в жид-
। кой фазе и с солями кобальта и марганца. В некоторых работах роль
I поверхности окисных катализаторов при жидкофазном окислении
I углеводородов сводят только к генерированию радикалов для цеп-
| ного процесса, протекающего в объеме [79, 80]. Однако исследования
1 42
к.
по окислению гептана, о-ксилола и циклогексана дают основание-1
считать, что это не так [78, 81]. Было установлено, что в реакциях
окисления с применением твердых катализаторов могут образовы-
ваться соединения, отсутствующие в составе продуктов реакции при
гомогенном цепном окислении. Так, при окислении гептана на сме-
шанном окисном ванадий-вольфрамовом катализаторе образуется
энантовый альдегид с выходом 8% и энантовая кислота с выходом
^7% от образовавшихся кислот и карбонильных соединений соот-
ветственно. Как известно, при цепном окислении атаке подвергаются
вторичные и третичные атомы углерода, а концевые метильные группы
в реакцию не вступают. Было показано, что в реакциях окисления
некоторых парафинов и олефинов на твердых полупроводниковых
и металлических катализаторах при добавке ингибиторов, например
гидрохинона, скорость реакции сначала падает пропорциональна
добавке ингибитора, но, начиная с некоторого момента, становится
постоянной и не равной нулю, что однозначно доказывает наличие
нецепного поверхностного процесса. Таким образом, в случае жидко-
фазного окисления углеводородов на твердых катализаторах мы
имеем дело с гетерогенно-гомогенным процессом, причем доля объем-
ного продолжения здесь весьма значительна. Действительно, ве-
роятность превращения промежуточного продукта на катализаторе
пропорциональна времени пребывания молекулы в адсорбированном
состоянии т. Время т определяется иэ уравнения Френкеля
т = тоех/Л1’ (1.35).
где т0 — период колебания адсорбированной молекулы в направле-
нии, перпендикулярном к поверхности; К — теплота адсорбции.
При адсорбции вещества < А.г, хотя бы за счет разности теплот
конденсации и растворения. Для сорбции из растворов неравенство
может соблюдаться только при эндотермичности процесса
сольватации. Таким образом, во многих случаях выполняется не-
равенство 2иж < 2vr, что благоприятствует десорбции промежуточных
продуктов реакции в условиях жидкофазных процессов. Эти сообра-
жения, вероятно, применимы также к выходу радикалов в объем.
Важно отметить, что поскольку гетерогенно-гомогенный меха-
низм для гетерогенно-каталитических реакций окисления в жидкой
фазе является общим случаем, а не частным, в отличие от газофаз-
ных процессов, этими реакциями можно управлять совместно при
помощи катализаторов и ингибиторов.
Для технического катализа указанные свойства жидкофазных
процессов могут оказаться выгодными в случаях, когда целевые
продукты являются промежуточными в цепи последовательных пре-
вращений, как то бывает, например, в большинстве процессов окис-
ления, неполного гидрирования и т. п.
Вместе с тем, надо иметь в виду, что условия диффузии в капил-
лярах внутри зерна катализатора в жидкой фазе менее благоприятны,
чем в газовой, за счет резкого уменьшения коэффициентов диффузии.
Поэтому при работе на тонкодисперсных катализаторах вследствие
43
перехода реакции в диффузионную область может наблюдаться об-
ратное явление: избирательность по промежуточным продуктам
реакции при переходе в жидкую фазу будет падать.
Кроме изложенных выше особенностей жидкофазных гетерогенно-
каталитических реакций, следует отметить, что они, как правило,
весьма чувствительны к природе растворителя. Растворитель может
вытеснять реагенты с поверхности катализатора, тормозя тем самым
каталитическую реакцию (см. раздел II.4). В других случаях кис-
лотно-основные свойства растворителя могут повлиять на характер
сорбции реагентов на поверхности катализатора, как это бывает
при сорбции водорода на металлах.
1.8. СЛОЖНЫЕ КАТАЛИЗАТОРЫ И КАТАЛИТИЧЕСКИЕ СИСТЕМЫ
Промышленные катализаторы большей частью представляют со-
бой многокомпонентные и многофазные системы. К такого рода со-
ставам пришли эмпирически, часто в результате длительного поиска
и последующего усовершенствования катализаторов. Одним из
оснований для создания сложных катализаторов были наблюдения,
что каталитическая активность двух или нескольких соединений
часто не аддитивна, а принимает экстремальные значения. Теоре-
тические основы механизма действия и подбора сложных катализа-
торов серьезно стали разрабатываться сравнительно недавно и пока
еще полностью не ясны. Здесь будут рассмотрены некоторые вопросы
теории сложных катализаторов, непосредственно связанные с об-
щей теорией катализа.
Сложность состава катализаторов может быть следствием трех
причин: промотирования или, точнее, модифицирования; приме-
нения в качестве катализаторов смеси нескольких веществ; приме-
нения катализаторов на носителях. На практике при приготовлении
катализаторов весьма часто применяют сразу все три приема.
Модифицированные катализаторы. Промотированием называют
добавку к катализатору небольшого количества другого вещества
(веществ) с целью повышения его активности. Хотя этот термин
укоренился в литературе по катализу, часто правильнее применять
более общий термин — модифицирование. Этот термин лучше пере-
дает механизм действия добавок, поскольку одна и та же добавка
в разных количествах часто может как повышать, так и понижать
активность катализатора. Влияние добавок на селективность до-
статочно сложно и обычно связано с уменьшением активности ката-
лизатора в одном направлении и увеличением в другом. Наконец
существуют добавки, влияющие на стабильность катализаторов.
Поэтому под модифицированием катализаторов мы будем понимать
введение в них небольших количеств добавок, изменяющих свойства
катализаторов в нужную сторону. Сами эти добавки мы будем на-
зывать модификаторами.
По механизму действия можно различать несколько видов мо-
дификаторов. Так действие электронных модификаторов сводится
44
к изменению электронных состояний в кристалле катализатора.
Примерами электронного модифицирования, вероятно, являются
повышение активности медного катализатора гидрирования неболь-
шими количествами никеля [82], окисновольфрамового катали-
затора (WO3) полного окисления углеводородов —- добавкой 1%
NaOH [83], окисномедного катализатора окисления пропилена в ак-
ролеин — добавкой селена [84]. Последний случай интересен тем,
что модификатор, ввиду его летучести в условиях реакции, следует
постоянно добавлять в реакционную газовую смесь. Этот прием,
называемый газовым промотированием, применяют и в других про-
цессах, например при окислении этилена в окись этилена, и он на-
чинает все более распространяться.
Фазовые модификаторы — это такие добавки, которые способст-
вуют образованию или сохранению в катализаторе фазы, обладаю-
щей наибольшей активностью. Согласно Жермену [17], так дейст-
вует СгО3 в медно-хромовых катализаторах Адкинса, где окись
хрома препятствует полному восстановлению окиси меди в неактив-
ную медь.
Структурные модификаторы стабилизируют желаемую, обычно
пористую, структуру катализатора, которая без модификаторов
может быть нарушена вследствие спекания. Наиболее хорошо та-
кой механизм действия изучен для добавки окиси алюминия к же-
лезному катализатору синтеза аммиака. В этом случае добавка
1% А12О3 приводит к увеличению поверхности восстановленного
железного катализатора от 0,5 до 10 кроме того, предотвра-
щает рост кристаллов a-Fe при отжиге. Адсорбционными измерениями
удалось показать, что при содержании 0,42% А12О3 в катализаторе
она покрывает 35% его свободной поверхности и образует на кри-
сталлах железа тончайший слой, препятствующйймих спеканию [17 ].
Смешанные катализаторы отличаются от модифицированных
тем, что добавки к основному компоненту вводят не в малых, а
в соизмеримых количествах, вплоть до случаев, когда нет оснований
считать один из компонентой основным. Естественно, что граница
между смешанными и модифицированными катализаторами весьма
условна, так же как и граница между смешанными катализаторами
и катализаторами на носителях. «
Теоретически возможен ряд причин, по котЗ^рм смешанные ка-
тализаторы могут обладать большей активностью' и лучшими свой-
ствами, чем однокомпонентные. Мы изложим две наиболее обосно-
ванные из них.
Во-первых, компоненты смешанного катализатора в процессе
его формирования могут реагировать между собой с образованием
нового, более активного соединения. В этом случае катализатор
фактически не является смешанным (например, висмут-молибдено-
вый катализатор в реакции синтеза нитрилакриловой кислоты
окислительным аммонолизом пропилена [85]).
Во-вторых, повышение активности смешанных катализаторов
может быть связано с механизмом или энергетикой перехода
45
электронов. В этом случае роль одного из компонентов аналогична
роли электронных промоторов. Обычно при этом смешанный катали-
затор представляет собой твердый раствор одного компонента в другом
или их сплав. Так, сложный окисный ванадий-молибденовый ката-
лизатор — это твердый раствор МоО3 в V2O8 со структурой пятиокиси
ванадия [86].
Катализаторы на носителях, вероятно, являются наиболее
распространенным типом сложных катализаторов. Они получили
широкое распространение благодаря удобству их практического-
применения (подробнее см. главу V). В этих катализаторах актив-
ную часть наносят на инертную подкладку — носитель. Таким об-
разом, эти катализаторы как минимум, двухфазны. Возможно троя-
кого рода влияние носителя на каталитический процесс: 1) увели-
чение поверхности катализатора; 2) влияние пористой структуры
носителя на активность и избирательность (эта проблема целиком
относится к вопросам, связанным с диффузионными явлениями)
и 3) специфическое действие носителя.
До сравнительно недавнего времени носитель рассматривали
как инертную составляющую катализатора. Обычно как доказатель-
ство инертности носителей приводится отсутствие у них каталити-
ческой активности. Однако, как указывалось несколько выше, и
у других типов сложных катализаторов один из компонентов может
не обладать каталитической активностью. Шваб [87 ] показал, что
при варьировании носителей для одного и того же активного ком-
понента изменяется не только удельная каталитическая активность
последнего, но и электрические свойства получаемого катализатора
(электропроводность). Следовательно, влияние носителя может иметь
электронную природу, что должно также вытекать из теории явле-
ний в пограничных слоях металлов и полупроводников.
Некоторые исследователи, например Кобозев и его сотрудники
[88], придавали большое значение вопросу концентрации активного
компонента на носителе. Во многих случаях установлено, что удельная
активность катализатора растет по мере понижения концентрации
активного компонента на носителе, а в некоторых случаях удалось
установить и точки максимума для этой зависимости [89].
При оценке результатов экспериментов по сравнительной актив-
ности катализаторов на носителях надо иметь в виду, что, как пока-
зали электронно-микроскопические исследования [90], активные
компоненты (металлы или окислы) часто образуют на носителе скоп-
ления кристаллов или сферолитов. '
Каталитические системы. В отличие от сложных катализаторов
под каталитическими системами мы будем понимать комбинацию
двух или нескольких самостоятельно действующих катализаторов
или катализаторов и ингибиторов, совместное действие которых может
привести к неаддитивному эффекту. Физически каталитические си-
стемы могут представлять собою как механические смеси, так и
совместно сформованные гранулы или совместно нанесенную на но-
ситель смесь каталитически активных веществ. Частным случаем
46
каталитических систем являются так называемые бифункциональные
катализаторы. Следует отметить, что трудно установить точную
границу между каталитическими системами и смешанными катали-
заторами. *
Еще в 1946 г. Наумовым [91 ], вероятно, впервые было строго
доказано на примере реакции изомеризации окиси этилена, что
механическая смесь двух компонентов (силикагеля и окиси алюми-
ния) в условиях, исключающих их взаимодействие (температура
200° С), обладает значительно более высокой активностью, чем
каждый из компонентов в отдельности. В 1958 г. метод механиче-
ского смешивания платинированного угля с алюмосиликагелем был
предложен для получения активных катализаторов гидродеалкили-
рования [92]. В 1964 г. Никс и Вейз показали эффективность такого
приема при проведении ароматизации парафинов на смеси алюмо-
силикатного и дегидрирующего платинового катализаторов [93].
В настоящее время полифункциональные катализаторы широко при-
меняют в основном в процессах превращения углеводородов [94,
95]. Чтобы провести сложное превращение веществ, приходится
иметь дело с многоступенчатым процессом, протекающим в виде
серии последовательных и параллельных реакций. В этом случае
часто недостаточно эффективно применять один катализатор, так
как при этом ускоряется лишь одна ступень процесса.
В природных ферментативных процессах для каждой стадии
сложного процесса существует свой весьма активный, но специфи-
ческий катализатор, так что весь процесс как бы передается по эста-
фете — «каталитической цепи» — от одного катализатора к другому,
за счет чего значительно выигрывает селективность превращения.
Аналогичное явление можно осуществить и в техническом катализе.
При этом создается возможность проведения в одном реакторе много-
стадийных процессов. Классическим примером простейшего
процесса каталитической системы может служить одностадийная
изомеризация парафинов на бифункциональном катализаторе из
окиси алюминия и платины, протекающая следующим образом
(применительно к бутану):
С4Н10 ^1* С4Н8+Н2
С4Н8 из0-с4н8
[Pt]
изо-С4Н8 ———> изо-С4Н10
На более сложной системе осуществлен достаточно’избирательный
синтез малеинового ангидрида из бутана по следующим стадиям
[96 ]:
c4h10+hci+i/2o2 1°^-» c4h9ci + h2o
С4Н9С1 с4Н8+НС1
C4Hs+3O2 [С0‘Оз'У.0Ч С4Н2О3+ЗН2О
47
Здесь, наряду с двумя твердыми катализаторами в виде меха-
нической смеси, в каталитическую систему входит й хлористый
водород, который играет в данном случае роль гомогенного катали-
затора.
Возможен синтез каталитических систем, состоящих из катали-
заторов и ингибиторов. При гетерогенно-каталитическом жидкофаз-
ном окислении олефинов среднего молекулярного веса в. соответ-
ствующие окиси было показано, что процесс в упрощенном виде
протекает согласно схеме:
Олефин—
----► перекисный радикал---► продукты полного разложения
[гетерогенный катализатор]
----------------------------► ОКИСЬ
При проведении процесса на каталитических системах из твер-
дого катализатора (СоаО3) и ингибитора — твердого (А1аО3) или
растворимого (гидрохинона) — за счет торможения реакции радика-
лообразования селективность процесса резко увеличивается [97,
98].
Очевидно, что факт неаддитивного влияния на сложные химиче-
ские процессы механической смеси катализаторов указывает на
обязательный переход в объем некоторого количества промежуточ-
ного соединения из данной цепи превращений. Следует иметь в виду,
что реакции, внешне протекающие по последовательным схемам,
например
Ai —>- Аа —>- Ад
в гетерогенно-каталитических системах фактически осуществляются
по схеме
Аг Аа А3
М —> А| —>
где s обозначает сорбированное состояние. Если время сорбцион-
ного состояния относительно велико, т. е. равновесие
А2 AJ
сдвинуто вправо, объемная концентрация Аа может быть исчезающе
мала. Однако и при этом может иметь место перенос вещества Аа
на поверхность другого компонента каталитической системы, что
приведет к изменению направления процесса, если на этом втором
компоненте протекает иная реакция, например Аа -> Bs. Бели
активные компоненты каталитической системы наносятся совместно
на одну инертную поверхность, то переход промежуточных соеди-
нений может происходить не только через объем, но и путем поверх-
ностной диффузии. Системы, состоящие из катализатора и ингиби-
тора, всегда могут дать выигрыш в селективности; для других
случаев вопрос о целесообразности и способе применения каталити-
ческих систем должен быть решен путем количественного расчета.
Сканировал: NeptUliyi
Магнитогорск
2008
48
1.9. ВЗАИМОДЕЙСТВИЕ КАТАЛИЗАТОРА И РЕАКЦИОННОЙ СРЕДЬН
При рассмотрении механизма действия катализаторов, оценке
их активности и особенно при составлении кинетических уравне-
ний процессов на данных катализаторах нужно иметь в виду тот
факт, что взаимодействие между катализатором и средой не ограни-
чивается влиянием катализатора на реакционную среду, а наблю-
дается и обратное влияние среды на катализатор. Четко вопрос о
влиянии среды на катализатор, вероятно, впервые рассматривался
Брунсом [99] и Боресковым [100].
Под воздействием среды в катализаторе могут изменяться: ,
1) только поверхностные свойства (соответственно и каталитиД
ческая активность);
2) химический состав и, следовательно, свойства всего объема
катализатора без образования новых фаз;
3) химический состав с образованием новых фаз.
Взаимодействие без образования новых фаз. Изменение ката-
литических свойств поверхности катализатора под влиянием среды
трудно рассматривать в отрыве от всего механизма гетерогенного
катализа. Адсорбция компонентов реакционной среды на поверх-
ности полупроводниковых катализаторов равнозначна внедрению
примесей в поверхность катализатора с появлением новых локальных
электронных уровней, сдвигом уровня Ферми и общим изменением
состояния электронно-дырочного газа. Следовательно, с точки зре-
ния электронной теории катализа данный тип влияния среды на
активность катализатора включается в общее рассмотрение меха-
низма гетерогенно-каталитических реакций.
Изменение химического состава катализатора под влиянием
среды весьма часто не приводит к образованию новой фазы, но за-
метно сказывается на каталитической активности. В окисных полу-
проводниковых катализаторах это связано с обогащением или обед-
нением окисла кислородом по сравнению со стехиометрическим со-
ставом. Такого типа явления наблюдались, например, в реакции
разложения метанола на окиси цинка [101 ] и в реакции окисления
водорода на окиси никеля [102]. Для кислотно-основных катализа-
торов такого типа влияние среды на,катализатор связано со степенью
гидратации, а следовательно, с величиной функции кислотности
катализатора. Действительно, при реакциях гидратации-дегидрата-
ции всегда существует равновесие гидратированных и дегидрати-
рованных форм катализатора:
MenOm + H2O Me„Om_i(OH)2 (1.36)
Если реакция, идущая по уравнению (1.36), в исследуемой об-
ласти достаточно чувствительна к парциальному давлению водя-
ного пара, а каталитическая активность гидратированной формы
катализатора заметно выше, чем негидратированной (наиболее часто
встречающийся случай), то активность катализатора является сама
по себе функцией состава реакционной среды и давления. Изменения
4 Заказ 1441 49
фазового состава катализатора при этом не происходит, так как для
большей части применяемых катализаторов, таких как силикагель,
алюмосиликат, кремнефосфорные кислоты и другие, превращения
по реакции (1.36) не сопровождаются перестройкой кристалличе-
ской решетки соединения;
Фазовые превращения. Наиболее заметно влияние реакционной
среды на катализатор в тех случаях, когда изменения в составе ката-
лизатора приводят к его фазовым превращениям. При этом, оче-
видно, каталитические активности разных фаз в общем случае могут
различаться сколь угодно сильно. Большей частью явление фазовых
превращений связано с окислением или восстановлением катали-
тических систем: высший окисел # низший окисел или окисел #
металл.
Превращения протекают по схемам:
МеО + -^- Ог МеО2
А
МеОг + R MeO-J-RO (1.37)
или
МеОг + Нг MeO-f-H2O
MeO + RO MeO2+R (1.38)
где R и RO — восстановленная и окисленная формы реагента.
Если вся система реакций (1.37), (1.38) обратима и близка к со-
стоянию равновесия, то состав катализатора, вне зависимости от
исходного, целиком определяется термодинамическими условиями
равновесия. В этом случае, если состав катализатора, например
окисла, является функцией давления одного из компонентов, на-
пример кислорода, то, в соответствии с условиями гетерогенного
равновесия, для всей области температур Т и парциальных давле-
ний Р, за исключением точки равновесия с определенными Т и Р,
катализатор будет представлять собой одну фазу. Если реакция
проводится в точке равновесия, то катализатор может быть двух-
фазным, однако практическое осуществление такого случая неве-
роятно. Иное дело, если протекающие в системе реакции, например
реакции контактного окисления органических соединений, практи-
чески необратимы, тогда фазовый состав работающего катализатора
целиком определяется кинетическими, а не термодинамическими
параметрами. При проведении обратимых реакций в условиях, да-
леких от равновесия (что большей частью бывает на практике),
фазовый состав катализатора также не определяется термодина-
микой.
Против возможности существования нескольких самостоятельных
фаз в катализаторе при взаимодействии его со средой выдвигался
ряд возражений [1031. Эти возражения базировались на попытке
Вагнера и Хауффе доказать однофазность катализатора в условиях
реакции на основе кинетических представлений [1041. В качестве
50 '
исходного постулата ими было принято уравнение скорости фазо-
вого превращения
r = kpl (1.39)
где г — скорость реакции фазового превращения катализатора;
к — константа; р — давление реагента; I — длина границы раздела
трех фаз — твердых и газообразной.
Это уравнение, однако, не является достаточно общим.
Лангмюр еще в 1916 г. при кинетическом обосновании Правила
фаз показал сложность кинетических механизмов, приводящих к мно-
гофазной системе, удовлетворяющей условиям термодинамического
равновесия [105]. В соответствии с реальным механизмом топо-
химических реакций, скорость перехода фаз в катализаторе может
определяться как скоростью появления зародышей новой фазы,
так и скоростью диффузии атомов в кристаллической решетке и
скоростью перестройки последней. Для окисных ванадиевых ката-
лизаторов, например, переход У2О4 # У2О^ протекает по диффу-
зионному механизму [106].
Факт многофазности катализатора в условиях воздействия среды
на катализатор был экспериментально доказан в работе [107 ] на
примере окисления углеводородов на окисных ванадиевых катали-
заторах. Так же можно трактовать и данные исследований Тарама
и сотрудников [106 ]. Многофазность окисных медных катализаторов
в условиях протекания реакции окисления олефинов была показана
Поповой [108]. Соотношение между фазами окисных катализаторов
определяется стационарными условиями скорости окисления и вос-
становления катализатора, причем сами по себе зти процессы мо-
гут быть и не связаны непосредственно с катализируемой реакцией.
Поэтому вопрос о фазовом составе катализатора совсем не следует
ставить в зависимость от механизма основной катализируемой ре-
акции.
При значительных различиях в активности отдельных фаз ката-
лизатора зависимость его фазового состава от состава реакционной
смеси может существенно осложнить кинетику реакций.
Важно отметить, что поскольку окисление и восстановление ката-
лизатора не являются прямой и обратной реакциями, то добавки
к катализатору могут в различной степени ускорять эти реакции.
Результатом этого является возможность изменения стационарного
фазового состава катализатора за счет введения добавок, что при
разной активности фаз приводит и к изменению активности катали-
затора в реакции. Такой вид влияния на активность катализатора
можно назвать фазовым модифицированием, а сами добавки — фа-
зовыми модификаторами. Очевидно, фазовое модифицирование воз-
можно только в условиях воздействия среды на катализатор.
Если реакции превращения фаз катализатора не являются проме-
жуточными стадиями каталитической реакции, то их скорость может
быть значительно меньше скорости основного процесса. В зтом
случае время достижения катализатором стационарного состояния
достаточно велико и катализатор в течение значительного срока со-
храняет свойства, отличные от его свойств при стационарном составе.
1Это, по-видимому, является наиболее распространенной причиной
«разработки» катализаторов, а часто и кажущихся различий в удель-
'Цей—енггивности катализаторов, приготовленных различными мето-
дами. Этим же можно объяснить и часто наблюдаемый гистерезис
в изменении каталитической активности при изменении темпера-
| туры. Медленное установление стационарного состава приводит
| к тому, что в определенном интервале температур каталитическая
активность существенно зависит от того, приближаются ли к рабо-
чей температуре со стороны более высокой или со стороны более
низкой температуры.
1.10. ОТРАВЛЕНИЕ КАТАЛИЗАТОРОВ
Под отравлением катализаторов понимается понижение или пол-
ное исчезновение активности катализаторов под действием неболь-
ших количеств веществ, называемых «ядами». Вследствие важности
этого явления для практики катализа, а также довольно широкого
распространения неправильных представлений о нем, целесообразно
рассмотреть важнейшие стороны теории отравления катализаторов.
Детальные исследования показали необходимость дифференци-
ровать различные типы явлений, объединяемых общим понятием
«отравление». Прежде всего, целесообразно различать понятия отрав-
ления и блокировки. При отравлении наблюдается специфическое
действие яда в отношении данного катализатора и данной реакции.
Блокировка же представляет собой фактически механический про-
цесс экранирования поверхности катализатора в результате отло-
жения на ней примесей. Поэтому блокировка не специфична ни в от-
ношении реакции, ни в отношении катализатора. Однако, естест-
венно, блокировка резче сказывается на пористых катализаторах
вследствие забивки устьев пор. Наиболее часто встречающимся
видом блокировки катализаторов является отложение на их поверх-
ности высокомолекулярных углеродистых соединений при проведе-
нии различного рода органических реакций, в частности крекинга.
Такой процесс обычно называют зауглероживанием или закоксовы-
ванием катализатора. При блокировке в первом приближении не
меняются ни энергия активации катализатора, ни его избиратель-
ность (исключая процессы в диффузионной области), поскольку
действие блокирующего вещества сводится к механическому выклю-
чению отдельных участков поверхности. Блокировка, как правило,
является обратимым процессом, если при удалении блокиру-
ющего вещества не происходит разрушения или дезактивации ката-
лизатора. Так, углеродистые отложения удаляются простым выжи-
ганием (при условии должной термоустойчивости катализатора).
Далее, из общего понятия «отравление» целесообразно выделить
явление изменения структуры катализатора под действием компо-
52
Количество яда на 1 г катализатора t
ноль* 10^
Рис. 1.10. Зависимость степени
дезактивации катализатора от ко-
личества яда для реакции дезал-
килирования кумола на синтети-
ческом алюмосиликате:
1 — хинальдин; 2 — хинолин; 3 — пир-
рол; 4 — пиперидин; 5 — дециламин;
6 — анилин.
нентов реакции или примесей и связанное с этим падение активности
катализатора, например при отравлении алюмосиликатных катали-
заторов водяным паром [109, НО ]. В этом случае под влиянием водя-
ного пара происходит ускоренная рекристаллизация с перерожде-
нием структуры геля и уменьшением его внутренней поверхности.
, Яд как бы способствует спеканию катализатора.
Механизм собственно отравления, очевидно, связан с типом ката-
лиза и различается для процессов электронного катализа на полу-
проводниках и металлах и процессов
? ионного катализа. Рассмотрим сна-
: чала последний случай, как более
; простой.
» При ионном катализе действие
J яда сводится к связыванию располо-
| женных на поверхности каталити-
I чески активных ионов. В наиболее
jj распространенном случае кислотного
j ионного катализа отравление насту-
* пает при нейтрализации поверхност-
( ных центров кислотности. Поэтому
I основания, включая щелочи и ами-
I носоединения, являются каталити-
/ ческими ядами. Поскольку активные
I ионы таких кислотных катализато-
i ров, как алюмосиликаты, занимают
всего 2—3% общей поверхности ка-
тализатора, незначительного количе-
ства яда, например едкого натра
или пиридинового основания, доста-
* точно, чтобы отравить катализатор.
{ Зависимость между количеством яда
i и степенью дезактивации алюмоси-
' ликатного катализатора при отрав-
лении азотистыми соединениями пред-
ставлена на рис. 1.10. Из этой зависимости следует, что по степени
«ядовитости» азотистые соединения располагаются в следующий ряд
(с ее убыванием): хинальдин, хинолин, пиррол, пиперидин, деци-
ламин, анилин.
Если рассматривать эти вещества непосредственно с точки зре-
ния основности, то пиперидин должен быть наиболее сильным ядом
в этом .ряду. Однако некоторые вторичные причины, как-то: стери-
ческие факторы, силы Ван-дер-Ваальса для тяжелых молекул,
а возможно, частичное разложение некоторых соединений в условиях
реакции — приводят к несовпадению последовательности веществ
в рядах по дезактивирующему действию и силе основности.
Наиболее сложен механизм действия ядов на металлические
или полупроводниковые катализаторы. Металлы, особенно благо-
родные, значительно более чувствительны к ядам, чем окисные
53
полупроводниковые катализаторы. Мэкстед [111], рассматривая
отравление металлических катализаторов, делит яды на три группы:
1) молекулы, содержащие элементы основной подгруппы V и VI
групп периодической системы, а именно: N, Р, As, О, S, Se, Те,
включая элементы в свободном состоянии, кроме азота;
2) соединения металлов;
3) молекулы, содержащие кратные связи, например, окись уг-
лерода, цианистые соединения и т. п.
Свойства ядов первой группы Мэкстед связывает с наличием у них
неподеленных электронных пар, вследствие чего образуются проч-
ные хемосорбционные связи яда с металлом, обусловливающие
большую продолжительность жизни яда в адсорбированном состоя-
нии. Таким образом, яд, покрывая поверхность катализатора, де-
зактивирует его. Любарский [112] показал, что при покрытии моно-
слоем тиофена никелевого катализатора гидрирования наступает
полное отравление последнего. Если активная поверхность состав-
ляет лишь'часть общей поверхности катализатора, то количество
яда, вызывающее полное отравление, естественно, меньше, чем тре-
бующееся для образования монослоя. Роль неподеленных электрон-
ных пар при отравлении подтверждается тем, что соединения, в ко-
торых они отсутствуют, нетоксичны (см. табл. 1.1). Нужно только
иметь в виду, что нетоксичные соединения под влиянием реагентов
могут переходить в токсичные: например, арсенаты в условиях
гидрирования переходят в арсины.
Перевод яда в нетоксичное соединение является одним из мето-
дов детоксикации реагентов [111 ]. На этом же принципе основаны
также некоторые промышленные методы регенерации металличе-
ских катализаторов.
' Отравление ионами металлов свойственно платиновым, палла-
диевым и другим катализаторам из металлов VIII группы и благород-
ных металлов других групп. Было обнаружено, что каталитцческая
активность платиновых и палладиевых катализаторов гидрирова-
ния понижается в присутствии ионов ртути, свинца, висмута, олова,
кадмия, меди, железа и других. Сравнение токсичности ионов раз-
личных металлов по отношению к платиновым катализаторам гидри-
рования приводит к заключению, что токсичность свойственна, по-
видимому, тем металлам, у которых все пять орбит </:оболочки,
непосредственно следующих за s- и р-валентными орбитами, заняты
электронными парами или по крайней мере одиночными d-электро-
нами. По мнению Мэкстеда, отсюда вытекает, что отравление пла»
тины и подобных ей катализаторов ионами металлов включает, ве-
роятно, образование адсорбционных комплексов, которые можно
рассматривать как интерметаллические соединения с участием d-
электронов в образовании интерметаллических связей.
Каталитические яды третьей группы могут рассматриваться
как яды только при совместном присутствии их с другими реаген-
тами, поскольку сами по себе они претерпевают многочисленные
превращения на катализаторах. Очевидно, тормозящее действие
54
Таблица 1.1
Влияние электронной конфигурации на токсичность
Токсичны
Не токсичны
Сероводород Н : S : Н
Сульфат-ион
О
О : S : О
О
Сульфон RC : S : CR'
Меркаптаны R —С : S : Н
Сульфиды R—С : S : С—R'
Арсенат-ион
Арсин Н : As : Н
Н
Фосфин Н : Р Н
Фосфат-ион
О
Аммиак Н : N : Н
И
Ион аммония Н : N : Н
этих соединений на другие реакции объясняется их высокими ад-
сорбционными коэффициентами из-за наличия в их молекулах крат-
ных связей.
Систематических исследований по отравлению полупроводнико-
вых катализаторов ядами, добавляемыми к реагентам, пока нет.
По-видимому, это связано с отмечавшейся выше стабильностью
полупроводниковых катализаторов к такого типа отравлениям. Не
имеется также специальных теоретических обобщений по вопросу
об отравлений полупроводниковых катализаторов. Можно себе пред-
ставить, что устойчивость реальных полупроводниковых катализа-
торов к ядам частично связана с тем, что в катализаторах уже имеется
такое количество примесей, что небольшое их увеличение не при-
водит к модифицированию свойств катализатора. Известно значи-
тельное количество примеров дезактивации полупроводниковых
55
катализаторов введением в них относительно большого количества
(порядка 1%) примесей. Механизм такого отравления может трак-
товаться согласно общим принципам действия модификаторов.
Общепринято деление процессов отравления на обратимые и
необратимые. Однако такое деление является скорее вопросом прак-
тического удобства, так как, строго говоря, обратимо всякое адсорб-
ционное отравление. Практически адсорбционное отравление не-
обратимо только в том случае, когда адсорбированный яд образует
с катализатором химическое соединение. Хотя понятие обратимости
Адсорбиробаннеий тиофен, т • /0‘
Рис. 1.11. Кривая отравления тио-
феном платинового катализатора в
реакции гидрирования циклогексена:
k — константа скорости реакции на отрав-
ленном катализаторе; fe0 — то же на неот-
правленном катализаторе.
является относительным, принято
считать отравление обратимым
только тогда, когда при обработке
поверхности свежим, не содержа-
щим яда реагентом наступает от-
носительно быстрое восстановле-
ние активности катализатора.
Для исследования кинетики
отравления существенен источник
поступления каталитического яда.
Яд может либо поступать из реа-
гирующей смеси, где он содержится
в виде примеси, либо образовы-
ваться на самом катализаторе из
исходного вещества или продук-
тов реакции. В обоих случаях
кинетика отравления поверхности
будет совершенно различной, что
особенно сильно ощущается при
диффузионном торможении про-
цесса (см. раздел III.6).
Зависимость степени отравления катализатора от количества
поглощенного им яда для многих случаев в широких пределах имеет
линейный характер. Типичная кривая отравления катализатора
с широким интервалом линейной зависимости представлена на
рис. 1.11. Однако при неоднородности поверхности кривая отравле-
ния может иметь значительные отклонения от линейности. Величина
отклонения зависит от типа функции распределения поверхности по
теплотам адсорбции и от функции взаимосвязи теплоты адсорбции
яда и энергии активации реакции.
Следует отметить, что явления отравления используют на прак-
тике и для улучшения свойств катализаторов. Поскольку действие
яда неодинаково сильно сказывается на различных реакциях, про-
текающих на данном катализаторе в данной реакционной системе,
создается возможность применять так называемое селективное отрав-
ление для повышения избирательности катализатора. Широко
известно, например, селективное отравление серебряных катализа-
торов галогенами, когда реакция полного окисления этилена подав-
ляется сильнее, чем реакция образования окиси этилена, и изби-
56
рательность катализатора, таким образом, повышается. Метод селек-
тивного отравления, видимо, довольно широко применяют в про-
мышленности, однако конкретные случаи его применения мало ос-
вещены в литературе, если не считать недостаточно надежных па-
тентных указаний. Кроме того, вряд ли существует определенная
граница между селективным отравлением катализаторов и их моди-
фицированием.
ЛИТЕРАТУРА
1. В. Э. Вассербергер, Автореф. докт. дисс., ИОХ, 1965.
2. С. 3. Р б г и н с к и й, Ю. Л. X а и т, ДАН СССР, 130, № 2, 366
(1960).
3. Г. И.Лихтенштейн, Кинетика и катализ, 4, № 1, 35 (1963).
4. G. А. М i 11 s, Е. Н. В oedeker, A. G. О b 1 a d, J. Am. Chem.
Soc., 72, 1554, 2682 (1950).
5. В. С. Ч e с а л о в а, Г. К. Б о р е с к о в, ДАН СССР, 85, № 2,
377 (1952).
6. Г. К. Б о ресков, В. А. Дзисько, ЖФХ, 24, № 9, 1135 (1950).
7. А. А. Б е л а я, М. Я. Р у б а н и к, Кинетика и катализ, 3, № 2,
201 (1962).
8. G. Langmuir, J. Агд. Chem. Soc., 40, 1361 (1918); Trans. Faraday
Soc., 17, 617 (1922).
9. Ю. X о p и у т и, T. T О й я, Кинетика и катализ, 4, № 1, 3 (1963).
10. Т. Т о у a, J. Res. Inst. Catalysis Hokkaido Univ., 6, 308 (1958); 8,
209 (1960).
11. H. П. К e й e p, Усп. хим., 19, № 1, 59 (1950).
12. С. 3. Рогинский, Адсорбция и катализ на неоднородных поверх-
ностях, Изд. АН СССР, 1948.
13. G. М. Schwa b, Trans. Faraday Soc., 42, № 11, 689 (1946).
14. В е е k, Wheeler, L о 1 е, Discuss. Faraday Soc., № 8, 251 (1950).
15. C. Kemball, Proc. Roy. Soc., A 207, 539 (1951).
16. C. Cox, A. Loulis, J. G w a t h m e у, J. chim. phys., 61, 487
(1964).
17. Ж. Жермен, Гетерогенный катализ, ИЛ, 1961, стр. 89.
18. Ф. Ф. Волькенштейн, Электронная теория катализа на полу-
проводниках, Физматгиз, 1960.
19. К. Haufe, G. Wagner, Z. Electrochem., N 3, 651 (1958).
20. X. Гарсиа де ля Ванда, Сборник «Электронные явления
в адсорбции и катализе на полупроводниках», Изд. «Наука», 1969, стр. 135.
21. С. 3. Рогинский, ЖФХ, 15, 708 (1942). •
22. Ф. Ф. Волькенштейн, Сб. Электронные явления в адсорбции
и катализе на полупроводниках, Изд. «Наука», 1969, стр. 49.
23. Дж. М. Томас, В. Д ж. Т о м а с, Принципы гетерогенного ка-
тализа, Изд. «Мир», 1969.
24. Е. J.Verweya. oth., Phil. Res. rept, 3, 173 (1955).
25. F. J. M or in, Bell. Syst. Techn. J., 37, 1047 (1958).
26. И. T. Ш e ф т e л ь, А. И. 3 а с л а в с к и й, ' ФТТ, 3, № 9, 2712
\*«7U ly .
27. D. A. Dowden, D. W e 1 1 s, Proceedings of the 2 International
Congress of the Catalysis, Paris, 1963.
28. В. Ф. К и с e л e в, Электронные явления в адсорбции и катализе
на полупроводниках, Изд. «Наука», 1969, стр. 159.
29. A. R о о п е у, G. W е b b, J. of Catalysis, 3, № 6, 54 (1964).
N. Bronsted, К. Pedersen, Z. Phys. Chem., 108, №2, 185
31. Н. Н. Семенов, О некоторых проблемах химической кинетики
и реакционной способности. Свободные радикалы и цепные реакции, Изд. АН
СССР 1958.
32. А. Л. С и д о р о в, ЖФХ, 30, 995 (1956).
33. Л. Л и т т а, Инфракрасные спектры адсорбированных молекул, Физ-
матгиз, 1969.
34. В. М. 3 а х т л е р и др., IV Международный конгресс по катализу,
1968, препринт доклада № 34.
35. А. А. Баландин, Мультиплетная теория катализа, т. I и II, Изд.
МГУ, 1963-1964.
36. М. И. Темкин, ЖФХ, 31, № 3 (1957).
37. Г. К. Боресков, В. В. Поповский, В. А. Сазонов,
Основа предвидения каталитического действия. Труды IV Международного
конгресса по катализу, Изд. «Наука», вып. 1, 1970, стр. 343.
38- В. А. Ройте р, Г. И. Голодец, Ю. И. Пятницкий, Там же,
стр. 365.
39. А. А. Баландин, Усп. хим., 31, № 11, 1265 (1962).
40. И. И. Иоффе, ДАН СССР, 58, № 18, 1681 (1947).
41. С. 3. Р о г и н с к и й, сб. «Проблемы кинетики и катализа», вып. 6,
Изд. АН СССР, 1949.
42. А. И. Шатенштейн, Теория кислот и оснований, Госхимиз-
дат. 1949.
43. Д. Белл, Кислотно-основный катализ, ИЛ, 1955.
44. В. Ф. Л ю д е р, С. Ц у ф ф анти, Электронная теория кислот
и оснований, ИЛ, 1950, стр. 206.
45. А. П. Б а л л о д, Е. В. Т о и ч и в а, Усп. хим., 20, № 2, 161
(1951).
46. F. A. Long, М. А. Р a u 1, Chem. Bev., 57, N 5, 935 (1957).
47. И. И. И о ф ф е, С. 3. Р о г и н с к и й, ЖФХ, 31, № 3, 612 (1957).
48. В. И. Г о л ь д а н с к и й, Н. Н. Семенов, Н. М. Чирков,
ДАН СССР, 52, № 9, 783 (1946).
49. J. Turcevitsch, В. К. Smith, J. Chem. Phys., 16, 467 (1948).
50. Б. Л. М о л д а в с к иЛ, Я. С. Б е з д е л ь, ЖПХ, 16, 1633
(1946).
51. К. Г. Миесеров, ДАН СССР, 88, № 3, 503 (1Й53); 91, № 3, 553
(1953).
52. М. И. Р о з е н г а р т, Т. Ф. Буланова, ЖПХ, 37, 383 (1964).
53. В. W. В 1 п е, С. J. Engle, Ind. Eng. Chem., 43, № 2, 494 (1951).
54. A. E v a n s, Discuss. Faraday Soc., № 8, 302 (1950).
55. M. E. D г y, G. J. О о s t h n i z e n, J. of Catalysis, 3, № 1, 18
(1968).
56. К. J. T a n a k a, A. О z a k i, J. of Catalysis, 8, № 1, 1 (1967).
57. К. В. T о и ч и e в а, К. Ю. П и н, ЖФХ, 29, № 10, 1854 (1955).
58. R. A. R о s s, D. E. В e n n e t, J. of Catalysis, 8, № 3, 293 (1967).
59. T! В. Антипина, О. В. Б у л г а к о в, ДАН СССР, 179, № 4,
845 (1968).
60. A. N. S ас hahen, Р. D. Caesar, Ind. Eng. Chem., 38, № 1, 43
(1946).
61. А. Г. Полянский, Усп. хим., 35, № 9, 1046 (1962).
62. F. Н е 1 f е г i с h, Angew. Chem., № 9, 241 (1954).
63. V. С. Н a s k^e 11, G. Р. Н a m m е t, J. Am. Chem. Soc., 71, 1284
(1949).
64. J. Manassen, Sh. К h a 1 i f, J. of Catalysis., 7, № 2, 110 (1967).
65. O. G г u b n e r, P. Jiru, M. В a 1 e k, Molekularsiebe, Berlin,
1968.
66. Advances in Chemistry. Ser. 101. Molecular sieve Zeolites, Washing-
ton, 1971.
67. X. M. M и н а ч e в, Кинетика и катализ, 11, № 2, 413 (1970).
68. P. В. Venuto, P. S. Landis, Adv. in Catalysis, 18, 259 (1968).
58
69. J. Turkevich, Catalys. Rev., 1, 1 (1967).
70. Я. И. Исаков и др., Основы предвидения каталитического дей-
ствия. Труды IV Международного Конгресса по катализу, вып. II, Изд. «Наука»,
1970, стр. 135..
71. К. В. Топчиева и др., Там же, стр. 144.
72. Т. Мисидзава и др., IV Международный Конгресс по катализу,
1968, Препринт доклада № 55.
73. Дж. А. Рабой др., Основы предвидения каталитического действия.
Труды IV Международного Конгресса по катализу, Изд. «Наука», вып. II,
1970, стр. 115.
74. А. И. Ш л ы г и н, сб. «Каталитические реакции в жидкой фазе»,
Изд. АН Каз. ССР, Алма-Ата, 1963, стр. 185.
75. Д. В. Сокольский, Гидрирование органических соединений
в жидкой фазе, Изд. АН КазССР, Алма-Ата, 1962.
76. Д. В. Соколовский, сб. «Каталитические реакции в жидкой
фазе», Изд. АН КазССР, Алма-Ата, 1962.
77. И. И. Иоффе, Кинетика и катализ, 3, № 2, 178 (1962).
78. И. И. Иоффе, Н. В. Климова, Кинетика и катализ, 4, № 5,
779 (1963).
79. I.Burger, М. Golette, С. Genevieve, I. С. В a lace an и,
Compt. rend, № 15, 2235 (1961).
80. Э. А. Блюмберг, Л. Я. Марголис, Н. М. Эмануэль,
ДАН СССР, 150, № 5 (1961).
81. I. I. Ioffe, L. J а. М а г g о 1 i s, 3d International Congress on Cata-
lysis, Amsterdam, 1964, Preprints II-X.
82. V. N. I p a t i e f f, Bull. Soc. chim. France, 7, № 4, 281 (1940).
83. C. 3. Рогинский, сб. «Проблемы кинетики и катализа», вып. 6,
Изд. АН СССР, 1949, стр. 12.
84. Э. X. Е н и и к е е в, О. В. И с а е в, Л- Я. М а р г о л и с, Кине-
тика и катализ, 1, № 5, 431 (1960).
85. А. И. Ге льбштейн и др., Кинетика и катализ, 4, № 5, 764
(1963).
86. И. И. И о ф ф е, 3. И. Е ж к о в а, А. Г. Любарский, ЖФХ,
35, № 10, 248 (1961); В. Б. Казанскийи др., Кинетика и катализ, 2, № 6,
862 (1961).
87. G. М. Schwab, Angew. Chem., № 11, 399 (1961).
88. Н. И. Кобозев, Усп. хим.', 25, № 5, 545 (1956).
89. A. Gault, Compt. rend., 245, N 19, 1620 (1957).
90. А. Б. Ш e xt e p, ДАН СССР, 58, 684 (1947).
91. А. И. Наумов, Тезисы докладов Всесоюзной конференции по ката-
лизу, Госхимиздат, 1947, стр. 68.
92. Пат. США 2564727, 1958.
93. Р. S. N i х, Р. В. W е i s z, J. of Catalysis, 3, № 2, 179 (1964).
94. П. Вейс, сб. «Катализ, полифункциональцые катализаторы и слож-
ные реакции», Изд. «Мир», 1965, стр. 9.
95. J,. Н. Sin felt, Adv. Chem. Eng., № 5, 37 (1965).
96. P. А. А г а с и e в и др., Азерб. хим. журн., № 5, 128 (1969).
97. И. И. И о ф ф е, Н. В. Климова, И. Я. М а к р о у с о в а, ДАН'
СССР, 169, № 2, 389 (1966).
98. И. Я. М а к р о у с о в а, И. И. И о ф ф е, Каталитические реакции
в жидкой фазе. Труды 2 Всесоюзной конференции, Алма-Ата, 1967, стр. 526.
99. Б. П. Брунс, Автореф. докт. дисс., Физико-хим. ин-т им. Карпова,
1955.
100. Г. К. Б о р е с к о в, сб. «Гетерогенный катализ в химической про-
мышленности», Госхимиздат, 1955, стр. 710.
101. К. И. М а т в е е в, Г. К. Боресков, сб. «Проблемы кинетики
и катализа», вып. 8, Изд. АН СССР, 1955, стр. 165.
102. Г. К. Боресков, В. В. Поповский, Кинетика и катализ,
2, № 5, 657 (1961).
ЮЗ. Б. П. Брунс, ЖФХ, 21, № 9, 1011 (1947).
59
104. C. Wagner, К. Н а uf f е, Z. Elektrochem., 44, № 3, 172 (1938).
105. J. Langmuir, J. Am. Chem. Soc., 28, № 11, 2291 (1916).
106. Тарама К., Тэранеси Ш., Ю суи T., Когё кагаку дзасси,
60, 1222 (1957).
107. И. И. Иоффе, 3. И. Е ж к о в а, А. Г. Любарский, Кине-
тика и катализ, 3, № 2, 194 -(1962).
108. Н. И. Попевай др., Из. СО АН СССР, № 12, 78 (1960).
109. G. А. М i 11 s, Ind. Eng. Chem., 42, № 1, 182 (1950).
110. G. A. M i 11 s, H. A. S h a b e k e r, Petrol. Ref., 30, № 9, 97 (1951).
111. E. Мэкстед, сб. «Катализ, вопросы теории и методы исследова-
ния», ИЛ, 1955, стр. 100.
112. Г. Д. Любарский, Кинетика и катализ, 3, № 4 (1962).
Глава II
КИНЕТИКА ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
[1-6]
П.1. СКОРОСТЬ РЕАКЦИИ
Определение скорости реакции. Рассмотрим реакцию, протека-
ющую согласно стехиометрическому уравнению
VjAi-f-V2A2 + • • • -|-тпАп -—> тп+1Ап+1+ . . . -j-VgAg (И.1)
где vt — стехиометрические коэффициенты исходных веществ (1
- I - п) и продуктов реакции (п + 1 - i - S).
Перенеся все члены уравнения (П.1) в правую часть, можем за-
писать его в виде:
s
0=2vfA| (П.2)
i=i
В этой формуле, как и повсюду в дальнейшем, стехиометрические
коэффициенты исходных веществ принимаются отрицательными, а
продуктов реакции положительными.
Скорость реакции (II.2) по г-му веществу в гомогенной системе
определяется как количество i-го вещества, образующегося в еди-
нице реакционного объема- в единицу времени:
Здесь — количество г-го вещества; V — объем реакционной
зоны.
Если реакция протекает изохорически (например, в замкнутом
объеме), то, заменяя количество вещества Nt на его концентрацию
Ct — NJV приводим уравнение (П.З) к виду:
Величины для различных веществ, участвующих в реакции
(II.2), пропорциональны соответствующим стехиометрическим коэф-
фициентам и имеют тот же знак. Поэтому можно определить скорость
реакции (II.2) без ссылки на вещество, по которому она измерена,
с помощью соотношения:
г=г,/х{ (П-5)
Ясно, что г всегда больше нуля.
Скорость реакции — функция состава реагирующей смеси, дав-
ления и температуры в реакционной зоне. Поэтому величина ско-
рости реакции может служить характеристикой данного процесса
61
только в том е-лучае, когда оговорены условия, при которых эта
скорость наблюдалась.
Формула (П.З) может служить строгим определением понятия
скорости реакции только в том случае, если все условия реакции
постоянны во всем объеме реакционной зоны V. Если же темпера-
тура и концентрации реагентов различны в разных точках реакцион-
ного объема, формула (П-3) определяет лишь среднюю скорость
реакции в объеме V. Определение локальной скорости, т. е. истинной
скорости реакции в данной точке, получаем, заменяя V в уравнении
(П.З) малым объемом A V, внутри которого все условия реакции можно
считать постоянными. Формула (П.4) определяет локальную скорость
реакции, протекающей при постоянном объеме, если под Ct понимать
концентрацию i-го вещества в данной точке. В дальнейшем во всех
случаях, когда условия в реакционной зоне неоднородны, будем
пользоваться только понятием локальной скорости реакции.
Зависимость скорости реакции от концентраций реагентов и
температуры. Классическое выражение, связывающее скорость необ-
ратимой реакции с условиями ее протекания, имеет вид
r = k(T)F(Ci, С2, ...,Сп) (П-6)
где к — константа скорости реакции, зависимость которой от тем-
пературы Т выражается уравнением Аррениуса:
Е
k = Ze~RT (П-7)
Для гомогенных реакций входящий в формулу (П.6) концентра-
ционный множитель F, согласно основному постулату химической
кинетики, равен:
/ = П (11.8)
i=l
Это выражение базируется на теории столкновений. Величина
az (i = 1, 2, . . ., п) называется порядком реакции по i-му веществу.
Суммарный порядок реакции равен 2 а,- Если стехиометрическое
уравнение (II.1) отражает механизм реакции (т. е. процесс, описы-
ваемый этим уравнением, протекает в одну стадию), порядки az
должны совпадать по абсолютной величине со стехиометрическими
коэффициентами соответствующих исходных веществ vz. Если ре-
акция, описываемая уравнением (II.1), протекает с заметной ско-
ростью в обоих направлениях, ее скорость г определяется как раз-
ность скоростей прямой и обратной реакций (соответственно гг
и г.2):
Г=Г1 — Г2 (П-9)
62
Учитывая (П.6) и (П.8), а также принимая порядки реакций
равными абсолютным значениям соответствующих стехиометриче-
ских коэффициентов, записываем (II.9) в виде
п S
r = fciU cfi-h, П cv{i (11.10)
iM i~n+l
где кх и к2 — константы скорости прямой и обратной реакций.
По мере превращения исходных веществ скорость реакции падает,
обращаясь в нуль при достижении Химического равновесия. Соот-
ношение между равновесными концентрациями реагирующих ве-
ществ С/р можно получить, приравнивая скорость реакции нулю:
RT =КР (П.11)
г=»1 1 Р ^2 н
Здесь Кр — константа равновесия, зависящая только от темпе-
ратуры.
Интегрирование кинетических уравнений. Чтобы однозначно
характеризовать состояние смеси веществ, где протекает только
одна химическая реакция (которая может быть как обратимой, так
и необратимой), достаточно знать скорость образования или исчез-
новения только одного реагирующего вещества. Согласно уравне-
ниям (II.4) и (II.5), скорости образования всех веществ связаны
между собой простыми соотношениями:
I dCj __ 1 dCj __ di, _(11.12)
Vi dt Vj dt dt
Интегрируя уравнение (11.12), получаем следующие соотноше-
ния между концентрациями веществ, участвующих в реакции,-спра-
ведливые в любой момент времени:
(C/-C/0)/vy=(Cz-Ct0)/vi=.g (П.13)
Величина £ называется степеньюшолноты реакции и равна нулю
в начальный момент времени. С помощью соотношений (11.13) можно
выразить концентрации всех веществ через величину £ или через
концентрацию одного из веществ, которое принимается за ключевое.
При этом скорость реакции г может быть представлена как функция
только одной переменной — степени полноты реакции или концен-
трации ключевого вещества. За ключевое удобно принять исходное
вещество, имеющееся в начальный момент в относительном недо-
статке, т. е. вещество с наименьшей величиной С'го /vz. Стехиометри-
ческий коэффициент ключевого вещества можно без потери общности
принять равным —1. Тогда кинетическое уравнение (II.4) прини-
мает вид:
dC/dt = -r(C) (11.14)
Индекс при концентрации ключевого вещества здесь опущен.
Уравнение (11.14) можно привести к безразмерной форме, приняв
63
за масштаб концентрации ключевого вещества его начальную кон-
центрацию Со, а за масштаб времени — характерное время реакции,
соответствующее составу исходной смеси, С0/г (Со).
Тогда
dc/dx=~ / (с)
где
с-С/Сд', f (c) = r (С)/г (Сц); т — т(Со) t/Сд
Интегрирование уравнения (11.14) или (11.15) дает:
с. 1
[* dC [* de
t== j Т(су; T=j
С с
В окрестности равновесия скорость реакции пропорциональна
отклонению от равновесной концентрации С — Ср. Поэтому время
реакции стремится к бесконечности по логарифмическому закону
по мере приближения к равновесному составу:
(П.15)
(П.16)
(П.17)
В случае необратимой реакции время реакции также становится
неограниченным по мере приближения концентрации ключевого
вещества к нулю. Чтобы показать зто, заметим, что г (0)=0, и допу-
стим, что функцию г (С) можно разложить в окрестности точки С = О
в степенной ряд, начинающийся с m-го члена:
r(C) = amCm+am^Cm^l+ . . . (IT-19)
Тогда при значениях С, близких к нулю
1п(1/С) при m = l (П.20)
С" при m > 1
Обычным законом возрастания времени реакции будет логариф-
мический закон, справедливый в случае, когда производная drjdC
при С = 0 существует и не равна нулю. Но в случае реакции второго
порядка типа 2АТ -> продукты или Aj + А2—+ продукты (при 'сте-
хиометрическом соотношении между начальными концентрациями *
обоих реагирующих веществ) т = 2 и t ~ С'1. Более высокие зна-
чения т практически не встречаются. Существуют формальные кине-
тические зависимости, не допускающие разложения в степенной ряд
в окрестности нуля. Распространенный пример такой кинетической
зависимости дает реакция дробного или отрицательного порядка
[г (С) ~ Сп при п < 1 ]. В этом случае время, необходимое для до-
стижения полного превращения, оказывается конечным. Однако на
самом деле подобные кинетические зависимости никогда не остаются
64
справедливыми при малых концентрациях реагента, так что при
С->0 порядок реакции всегда изменяется с дробного, нулевого или
отрицательного на первый, и логарифмический закон увеличения вре-
мени реакции сохраняет силу при степенях превращения, достаточно
близких к единице.
Рис. II.1. Зависи-
мость концентра-
ции непрореагиро-
вавшего исходного
вещества от вре-
мени для реакций
к-го порядка
(сплошные кри-
вые) и реакций с
лангмюровской
кинетикой (пунк-
тирные кривые).
Зависимость концентрации исходного вещества от времени для
некоторых реакций с простыми кинетическими уравнениями пред-
ставлена на рис. II. 1.
П.2. КИНЕТИКА СЛОЖНЫХ РЕАКЦИЙ
Ключевые вещества. При одновременном протекании несколь-
ких реакций первая задача состоит в выборе переменных, с помощью
которых состояние системы описывается однозначно и с максимальной
простотой. За такие переменные удобно принять концентрации неко-
торых выбранных веществ, называемых ключевыми. Число и номен-
клатура ключевых веществ должны быть выбраны так, чтобы, зная
скорости образования ключевых веществ, можно было вычислить
скорости образования всех остальных веществ, участвующих в про-
цессе. В то же время скорость образования каждого ключевого веще-
ства не должна определяться скоростями образования всех остальных;
иначе это вещество, очевидно, может быть исключено из числа клю-
чевых.
Рассмотрим смесь 5 веществ, в которой протекает R реакций.
Стехиометрические уравнения реакций, аналогично (II.2), имеют вид
s
2*(,А(=0 (* = 1-2,...,/?) (11.21)
г=1
5 Заказ 1441 65
где v1K — стехиометрический коэффициент вещества Az в к-й реакции;
vlK < 0, если данное вещество является исходным в данной реакции,
и v1K > 0, если оно образуется в данной реакции как конечный
продукт.
Суммарная скорость образования любого i-ro вещества равна
R
(г= 1, 2, • • ., S) (11.22)
где гм — скорость к-й реакции.
Из всех величин г{ можно выбрать некоторое число независимых
величин — скоростей образования ключевых веществ, а скорости
образования всех остальных веществ выразить через скорости обра-
зования ключевых. Составим матрицу стехиометрических коэффи-
циентов процесса. Она представляет собой прямоугольную таблицу
с S рядами и R столбцами, в i-м ряду и к-м столбце которой стоит
стехиометрический коэффициент г-го вещества в к-й реакции v,K.
Переставляя строки и столбцы матрицы (т. е. меняя номера веществ
и реакций), можно добиться того, что в левом верхнем углу окажется
квадратная матрица порядка К (т. е. с К рядами и К столбцами),
определитель которой отличается от нуля.
Если все определители (7Г + 1)-го порядка, которые можно со-
ставить из матрицы стехиометрических коэффициентов, равны нулю,
то ненулевой определитель К го порядка А называется главным опре-
делителем, а число К — рангом матрицы. Ранг матрицы стехиоме-
трических коэффициентов и определяет число ключевых веществ Я,
достаточное для однозначного описания процесса. Число К не может
превышать меньшего из чисел R и S. Предположим сначала, что
S R, К = R, и покажем, как можно вычислить скорости образо-
вания S — К веществ, не входящих в число ключевых. Совместное
решение К уравнений из системы (11.22) дает
к
г(К> = 4“2А;л = С 2, . . ., /Г) (11.23)
i-i
где А(к — алгебраическое дополнение элемента viK определителя А„
т. е. умноженный на (—1)>+к определитель, получающийся из А
вычеркйванием i-й строки и к-го столбца.
Подстановка уравнений (П.23) в остальные S — К уравнений
системы (11.22) дает:
к к
(7=К+1.......S) (11.24)
к=11=1 ' —
Замечаем, что определитель Ai;-, получающийся из главного
определителя А заменой элементов i-й строки v<K на элементы у-й
строки VjK, равен:
= Ъ viKAiK (11.25)
К=1
Следовательно, можно представить скорость образования ве-
ществ, не входящих в число ключевых, в виде:
1=1 г=1
Полученная формула справедлива и при К < R. В этом случае
R — К реакций не независимы, и могут быть представлены как линей-
ные комбинации К реакций, входящих в состав главного определи-
теля матрицы стехиометрических коэффициентов. Скорости образо-
вания всех веществ могут быть выражены, аналогично (11.22), через
скорости этих основных реакций; тем самым задача сводится к опи-
санному выше случаю R = К. Отметим, что хотя общее число ключе-
вых веществ определено химизмом процесса, их конкретный выбор
в значительной мере произволен [7, 8]. Вместо концентраций ключе-
вых веществ в качестве определяющих переменных можно использо-
вать также степени полноты независимых реакций (i = 1, 2, . . .
. . ., К) *.
Скорость тепловыделения. Знание скоростей образования клю-
чевых веществ позволяет вычислить скорость изменения в ходе про-
цесса любой линейной функции состава реагирующей смеси. Важ-
нейшей функцией такого рода является энтальпия единицы объема
смеси Н, вычисляемая по формуле
8
H=~'^lqiCi (11.27)
i*=l
где qt — парциальная мольная энтальпия i-ro вещества.
Скорость убыли энтальпии смеси в ходе процесса равна скорости
выделения тепла реакции. Эту важнейшую для расчета химических
процессов величину нетрудно представить как линейную функцию
скоростей образования ключевых веществ.
Дифференцирование формулы (11.27)" по времени реакции t дает:
4г—<п2<»
1=1
* Вопросы описания сложных систем реакций впервые рассмотрены Пше-
жецким и Рубинштейном [8]. Их подробное изложение можно найти в книге
Ариса [7, глава II].
5*
67
Используя уравнения (11.22), можно переписать эту формулу
в виде
в
(IL29>
к-1
где hK — теплота к-й реакции:
s
Ьк~ (11.30)
С помощью формул (11.26) скорость тепловыделения можно
выразить через скорости образования ключевых веществ
где
s
= + 2 ?/Д»7
/=Н+1
Интегрирование кинетических уравнений. Совокупность К кон-
центраций ключевых веществ может быть иначе названа вектором со-
става. /С-мерный вектор состава С — основная переменная, харак-
теризующая состояние смеси, претерпевающей сложный химический
процесс. Скорости образования ключевых веществ г{ могут быть
представлены как функции только вектора С и температуры:
Т) (i = l, 2, ..., К) (11.31)
ul'
Действительно, если величины г(- зависят от концентраций веществ,
не входящих в число ключевых, последние всегда можно исключить
с помощью соотношений
к
(l = K+i, S) (11.32)
г-1
которые легко получить, интегрируя уравнения (11.26).
Интегрирование системы кинетических уравнений (11.31) может
быть выполнено аналитически лишь в простейших случаях *. Для
численного интегрирования системы (11.31) используют вычислитель-
ные машины.
Не производя интегрирования, можно качественно описать харак-
тер изменения во времени концентраций реагентов в процессах раз-
личных типов. К первому типу отнесем процессы, включающие ряд
* Интегралы большого числа кинетических уравнений приведены в ра-
боте [9].
68
параллельных необратимых реакций, например процесс, следующий
системе стехиометрических уравнений
Ai —> Vjkj (; = 2, 3, • • S) (11.33)
Концентрации конечных продуктов А/ монотонно возрастают со
временем t. Если все реакции (II.33) протекают по одинаковому по-
рядку а и константа скорости реакции образования вещества А;-
равна kj, то прирост концентрации любого конечного продукта за
время t равен:
t
Cj Cj Q = Vjkj J (£i) dti (11.34)
0
Разделив два любых равенства типа (11.34) друг на друга, полу-
чаем:
Су CjQ Vjkj
(П.35)
С[ Cio
Количества образующихся конечных продуктов, таким образом,
пропорциональны произведениям соответствующих стехиометриче-
ских коэффициентов и констант скоростей реакций. Концентрация
исходного вещества в любой момент времени определяется так же,
как для единственной необратимой реакции с константой скорости
к = 2 Л/, что дает возможность вычислить и концентрации всех
i
конечных продуктов.
Если порядки параллельных реакций (II.33) различны, то в ходе
процесса меняется его избирательность: первоначально образуются
в больших количествах вещества, реакции образования которых идут
по более высокому порядку, а затем, по мере убыли концентрации
исходного вещества, преимущество переходит к реакциям, протека-
ющим по меньшему порядку. Задача интегрирования кинетических
уравнений усложняется; аналитическое интегрирование осуществимо
лишь в простейших случаях.
К другому важному типу принадлежат процессы, включающие
цепь последовательных необратимых реакций, например:
viAi —> v2A2 —> . . . —> vsAs
(11.36)
Если первоначально в смеси присутствует только исходное веще-
ство Ах, то концентрации всех промежуточных веществ А,- [где i = 2,
3, . . ., (5 — 1) ] в ходе процесса сначала увеличиваются, а затем,
пройдя через максимум, начинают уменьшаться, асимптотически
приближаясь к нулю. Концентрация любого вещества Az достигает
максимума тем позже, чем выше его порядковый номер в схеме (11.36).
Процессы смешанного типа включают как параллельные, так и
последовательные реакции; все реакции или некоторые могут быть
обратимыми. В процессе, включающем только обратимые реакции,*
концентрации всех веществ при t -> оо приближаются к равновесным
69
(IL37)
(11.38)
(п:з9^
(11.40)
значениям С;р. Если какое-либо вещество или одно из веществ, на-
ходящихся с ним в равновесии, вступает в необратимую реакцию,">
то по прошествии достаточного промежутка времени его концентра-,
ция станет исчезающе малой. Наличие максимума у функции изме
нения концентрации во времени указывает, что данное вещество яв-
ляется промежуточным в цепи последовательных превращений.
Системы реакций первого порядка. Если процесс включает
только реакции первого порядка, система кинетических уравнений
(11.31) всегда может быть проинтегрирована аналитически *. Пусть
в смеси S веществ происходят все возможные реакции типа А£ А;-
(г, j = l, 2, . . ., S). Концентрации веществ в любой момент вре-
мени t определяются решением системы линейных дифференциальных
уравнений с постоянными коэффициентами
8
dCt
(4=1,2, , S)
где ki} j) — константа скорости реакции А;->А£, а
8
кц == кн
/-1
8
Так как, зная суммарную концентрацию У С, = const и концен- ’
i=l
трации S — 1 веществ, всегда можно вычислить концентрацию остав- :
шегося вещества, число ключевых веществ не превышает S — 1.
Исключать из системы уравнений (11.37) концентрацию какого-либо
вещества удобно, однако, только в том случае, если это вещество не
вступает ни в одну реакцию (например, конечное вещество в цепи
превращений). Поэтому рационально совместно решать систему урав- !
нений типа (11.37), описывающих изменение со временем концентра- =
ций S', веществ, вступающих хотя бы в одну реакцию дальнейшего
превращения. После определения функций изменения концентраций ,
этих веществ можно легко вычислить и концентрации остальных ве-
ществ.
Вводя матрицу констант скорости К, элемент которой, стоящий
в г-м ряду и j-м столбце, равен ki}, можно записать систему (II.37)
в компактной матричной форме
dC/dt = КС
где С'— ^'-мерный вектор состава с компонентами Ct.
Матричное уравнение (11.39) может быть проинтегрировано фор-
мально так же как если бы К и С были обычными скалярными вели
чинами. При этом получаем:
C(t) = eKtC0
* Общая система кинетических уравнений первого порядка была впервые ;
- проинтегрирована Зволинским и Эйрингом [10].
70
Задача решения системы уравнений (11.37) сводится, таким обра-
зом, к вычислению элементов кинетической матрицы Z, являющейся
экспоненциальной функцией матрицы констант скорости К:
Z(t)=eKl (11.41)
Вычисление функций от матриц производится на основании из-
вестной из' матричного исчисления теоремы Сильвестра. Кинетиче-
скую матрицу Z (0 вычисляют по формуле
S' . i
= м/ч (11.42)
<7-1
где
И,_П^ (11.43)
Здесь S' чисел — собственные числа матрицы К — являются
корнями векового уравнения
Д(Л)^|Х/-Я|=0 (П.44)
где А (X) — определитель порядка S', элемент которого, стоящий
в г-м ряду и )-м столбце равен k{j, а диагональные элементы
равны X — кц.
Символ I в формулах (11.43) и (11.44) обозначает единичную ма-
трицу, диагональные элементы которой равны единице, а все прочие—
нулю. Любой элемент кинетической матрицы Z (t) может, таким
образом, быть представлен в форме
s' . t
(11.45)
<7=1
где mltq — элемент матрицы Mq.
Раскрывая равенство (11.40), находим
S'
Сг(О = 2?г/(ОС/о (11.46)
/-1
или, учитывая (11.45),
s' S' , .
C((O = 2 2w’C/o (z = l, 2, • • ., S') (11.47)
/=i <?=i
Концентрации реагирующих веществ в любой момент времени
являются, таким образом, линейными функциями начальных концен-
траций Cj 0. Можно доказать, что все собственные числа kq действи-
тельны [11 ]. Это доказательство опирается на принцип микроскопи-
ческой обратимости (детального равновесия), согласно которому при
термодинамическом равновесии системы скорость каждой из реакций
должна обращаться в нуль. Иными словами, не может быть поло-
жения, когда, например, имеется цикл необратимых реакций типа
74
Ar—>-А2—>-А3—>-Аг, которые могли бы продолжать идти и по дости-.я
жении равновесия, не приводя к изменению концентраций веществ, Я
включенных в этот цикл. Согласно принципу детального равнове- я
сия, при любом г, j I
kilCj р= kj(Ci р (П-48) 1
Введем диагональную матрицу D с элементами dtl = Clp, dtj = 01
при i =£= }. Тогда равенства (11.48) можно записать в матричной форме ]
KD — DK* (II.49) |
где звездочка обозначает транспонированную матрицу, т. е. матрицу |
с элементами k*j = ksi. j
Умножая равенство (11.49) слева и справа на D~ /г, получаем: (
Р^Л’1/2ЯЛ,/2 = Л1/2Л,*1)-,/2 = Р* (11.50)
Таким образом, матрица Р = D~ симметрична, т. е. ее
элементы связаны равенствами = pti.
Таким образом, матрица констант скорости К подобна симметрич-
ной матрице Р. Известно, что собственные числа подобных матриц
одинаковы, а собственные числа симметричных матриц действительны.
Таким образом, все величины kq действительны и в рассматриваемой
системе реакций первого порядка не может возникнуть колебаний,
даже затухающих, и функции Ct (£) могут иметь только конечное
число экстремумов, не превышающее ранга матрицы констант ско-
рости К-
Другой подход к интегрированию кинетических уравнений [И]
состоит в преобразовании матрицы К в уравнении (11.39) к диагональ-
ной матрице Л с помощью преобразования подобия Л = U lKU.
Диагональными элементами матрицы Л являются собственные числа
матрицы Kkq. Вводя новую переменную — вектор Y = U~lC, преоб- )
разуем уравнение (11.39) к виду: ;
dY/dt — A.Y (11.51)
Это уравнение представляет собой просто совокупность независи- ’
мых скалярных уравнений, решение которых имеет вид у{ = yioe%lt
Основная расчетная работа при применении данного метода заклю-
чается в определении собственных чисел kq и матрицы С7, осуществля-
ющей необходимое преобразование подобия. Оба описанных метода
совершенно равнозначны и сводятся, в конечном счете, к одним и
тем же вычислениям. Так как по физическому смыслу задачи концен-
трация ни одного из веществ не может неограниченно возрастать
или убывать со временем, числа kq во всех случаях либо отрицательны,
либо равны нулю.
72
Параллельные и последовательные реакции. Как пример при-
ведем решение системы кинетических уравнений первого порядка
для простой параллельно-последовательной схемы реакций
. hi kt .
Ai ► Аа А3
I_______--------------1
Вещество А3 в реакции не вступает, так что S' = 2. Матрица К
принимает вид
I — (М+М) О II
Я= (II.52)
I М —^'2 II
Раскрывая вековое уравнение (11.44), получаем
откуда
(X-f-M) (Х-[-М-[-М)— 0 (11.53)
М——(м -ь м); М — м
(11.54)
Формулы (П.43) дают
Ж = -Г---1—Г-Г-
Л?1 — л?2 + ^3
— ^2 “Ь Л'3
-м
Mi
1
к% — к i — />'3
О
О
О
м—м—м
(11.55)
Подставляя элементы этих матриц и числа Х2 в (П.47) и пола-
гая С2о = С3о = О, С1о = 1, получаем:
Ci(i) = e'(ftl+ft3H (11.56)
Сг (i) = ^--ГТйГ (e-fe/-e-(ftl+ft3) 0 (11.57)
Ki — лг-г^з
Легко вычислить и концентрацию вещества А3 : С3 = 1 —
-(С1 + С2) .
С3(0 = 1+ . \ ’ll ----к1- е-к*‘ (11.58)
М— М-ЬМ М— М~РМ
Полученные выражения, очевидно, определяют безразмерные
концентрации реагентов ct = Cf/C10.
Важной характеристикой процесса, включающего параллельные
и последовательные химические превращения, является его селек-
тивность, определяемая как отношение количества образовавшегося
целевого продукта (пусть это будет вещество А2) к количеству про-
реагировавшего исходного вещества. Селективность з удобно пред-
ставлять в качестве функции безразмерной концентрации непро-
реагировавшего исходного вещества сг или степени его превращения
1 — сг Используя формулы (II.57) и (II.58), имеем
с2 су1/(1+?г) —£
5 = -77-^--i---X /Г V (11.59).
1 —(1— Y1 + Y2) (1—Q)
где Vi = к2]1С1, у2 = /Сд/Ар
73
Зависимость s (cj при различных значениях параметров
показана на рис. II.2. Данные по селективности параллельных и
последовательных реакций представлены также на рис. III.15 и
III.16 (стр. 144). На рис. II.2 ясно видно различие между процессами,
включающими параллельные и последовательные побочные реакции.
Чем больше отношение параметров Ti/Yg’ т- е- чем быстрее протекает
последовательная реакция! по
сравнению с параллельной, тем
сильней уменьшается селекти-
вность с увеличением степени
превращения 1 — сг.
Рис. II.2. Селективность последова-
тельно-параллельных реакций первого
порядка.
П.З. КИНЕТИКА РЕАКЦИЙ
В ПОТОКЕ
Основные уравнения. В от-
личие от реакций в замкнутом
объеме при осуществлении реак-
ций в потоке (с неизменным ре-
жимом течения) концентрации
реагентов повсюду остаются по-
стоянными во времени, но меня-
ются в пространстве. Для вы-
вода кинетического уравнения
реакции в потоке рассмотрим
поток реагирующей смеси через
бесконечно малый элемент объе-
ма реактора длиной dX (где X — координата, отсчитываемая по
ходу потока) *. Благодаря химическим превращениям, протека-
ющим в выделенном элементе объема, количество Nt i-ro вещества,
проходящее через единицу поперечного сечения реактора в единицу
времени, изменяется на величину dNt = rt dX, откуда
dNi/dX^ri (11.60)
Если реакция протекает без изменения объема, то, заменяя вели-
чину N[ концентрацией г-го вещества в реагирующем потоке Ct по
формуле Nt — иС;. (где и — линейная скорость потока), приводим
уравнение (11.60) к виду
udCildX = ri (II-61)
Это уравнение можно, в свою очередь, привести к виду (II.4),
если ввести вместо координаты X новую независимую переменную —
текущее время контакта t = Х]и. Как это ясно из определения,
величина t равна времени прохождения потока от входа реактора
до сечения с координатой X. Время, за которое поток проходит весь
* Более строгий анализ кинетики процессов в проточных реакторах про-
водится в главе VII.
74
реактор от входа до выхода, будем называть временем контакта.
Время контакта играет в проточных реакциях ту же роль, что время
реакции для процесса, идущего в замкнутом объеме, и, как мы видим,
кинетические уравнения процессов обоих типов полностью совпа-
дают, если химические превращения не сопровождаются изменением
объема реагирующей смеси.
Если реакция идет с изменением объема, условия ее протекания
в потоке и в замкнутом объеме существенно различны. В то время как
реакция в замкнутом сосуде идет при постоянном объеме, реакция
в потоке протекает при постоянном давлении. Вследствие этого видо-
изменяется форма кинетических уравнений процесса 112, 13]. В про-
цессе с изменением объема линейная и объемная скорости потока
меняются по длине реактора, и переход от уравнения (11.60) к (11.61)
уже невозможен.
В правой части уравнения (11.60) стоит выражение для скорости
реакции, зависящее не от количества вещества, а от его концентра-
ции. Связь между этими двумя переменными проста, если реагиру-
ющая смесь подчиняется законам идеального газа. Тогда
АГ, Ni Р NRT
С‘~ и ~ N ' RT ' U~ Р
(11.62)
где N — общее число молей смеси, проходящих через единйцу по-
перечного сечения реактора в единицу времени.
Единственная реакция. Рассмотрим необратимую реакцию со
стехиометрическим уравнением (II.2). В этом случае, аналогично
уравнению (11.13), изменение числа молей любого i-ro вещества всегда
может быть выражено через изменение числа молей любого другого
7-го вещества:
tf/-Ar/0=(v//vz) (JV€ — JV, 0) (П-63)
Суммируя выражение (П.63) по всем веществам, находим измене-
ние общего числа молей
у_уо=₽<(^-^-о) (П.64)
где
s
р<=—2v/ (IL65>
7=1
Подставляя уравнение (11.64) в (11.62), получаем:
Р N i
Cl = ^RT‘ ЛГ0+₽, (Ni-Nt о) (11 ‘66>
Подставив (II.66) в выражение для rh можно проинтегрировать
Уравнение (11.60). Например, в случае необратимой реакции первого
порядка Aj—>2А2 = —1 и кинетическое уравнение принимает
вид
75
где х = IcX/Uq — безразмерная координата;
непрореагировавшего исходного вещества; а
щества Ал в исходной смеси.
Интегрирование уравнения (11.67) дает:
п = NJNb 0 — доля I
= Ni, 0/No — Д°ля ве-1
ч
х= (1+ “) In (1/n) — а (1 — п)
(П-68) |
Зависимость п от х при различных значениях параметра а пред-
ставлена на рис. II.3. Как видно из рис. II.3, снижение скорости
реакции вследствие увеличения объема реагирующей смеси чувст-
Рис. II.3. Зависимость концентрации непро-
реагировавшего исходного вещества от коор-
динаты для реакции первого порядка в по-
токе.
Выразим из соотношения (II.66):
вуется особенно сильно
при больших степенях пре-
вращения. При разбавле-
нии реагента инертным
веществом величина а
уменьшается и влияние
увеличения объе'ма в ре-
зультате химических пре-
вращений на скорость ре-
акции становится все
слабей. В пределе а->0 ха-
рактеристики процесса бу-
дут те же, что и в случае
реакции в замкнутом
объеме.
Возможен и другой спо-
соб решения, при котором
расчетные уравнения для
реакций с изменением
объема приводят в фор-
мальное соответствие с
уравнением (11.61).
/V — (Ng fijNj о)
1 P/RT-faCt
Используем тождества
0 = oi Nq=UqCq', Cq—P]RT
'(11-69)
(П.70) §
где u0 — линейная скорость; С0 — ^Ci0 — плотность потока во
I *
входном сечении (в молях на единицу объема).
Соотношение (11.69) приводим к виду
откуда
V HpQ (l-PfQo/Co)
1 1-P,CZ/CO
uod-P/Qo/Co),^
dNi~ (1-ТГс;/с0)2 - dCi
(11.71)
(П.72)
76
Подставляя уравнения (11.71) и (11.72) в (11.60), получаем:
dCi
и°Чх
1-PiQo/Co r‘
(11.73)
Уравнение (11.73) формально не отличается от (11.61). Его правая
часть по-прежнему есть некоторая функция от концентрации и тем-
пературы. Более того, его можно привести к форме (II.4), если ис-
пользовать в качестве независимой переменной условное текущее
время контакта t = Х/и0. Таким образом, изменение объема в ходе
реакции может быть учтено при составлении функции скорости обра-
зования данного вещества, что в случае единственной реакции равно-
сильно введению в выражение для г( множителя (1 — Pf-C’l7C’z 0)2 :
: (1 - ₽,С(.0/С0).
Сложные реакции. При расчете сложного процесса с К ключе-
выми веществами связь между количествами молей У(- ключевых ве-
ществ, проходящими через единицу поперечного сечения реактора
в единицу времени, и их концентрациями Сг для идеальной газовой
смеси по-прежнему определяется формулой (11.62). Чтобы проинте-
грировать систему уравнений типа (11.60), остаётся только выразить
общее число молей N, проходящее через единицу поперечного сечения
реактора в единицу времени, через числа молей ключевых веществ.
Для веществ, не входящих в число ключевых, аналогично (11.32)
к
Ni-^о = 4“2Л^£“ЛГ/о) (/=« + !• •-.«) (П-74)
г=1
где Л — главный определитель матрицы стехиометрических коэф-
фициентов; Л£-;- — определитель, получающийся из А заменой стехио-
метрических коэффициентов i-ro вещества на стехиометрические коэф-
фициенты /-го вещества (см. раздел II.2). Суммируя прирост чисел
молей всех веществ, получаем
к
, (П.75)
i=l
где
8
Р/=1+4-2Л° (г=1>2' • • •’к) (п-7б>
з=к+1
Подставляя формулу (II.75) в (11.62), имеем:
Р N,
Cl = ~RT---------------- <П-77)
Уо + £₽< (N(-Ni0)
i=l
Уравнения (II.60) можно проинтегрировать, подставив в его пра-
вую часть соотношение (11.77). Другой путь состоит в решении
системы уравнений (11.77) относительно величины У,-; подстановка
77
полученных выражений в уравнение (11.60) приводит его в фор-
мальное соответствие с (II.61). В качестве примера рассмотрим две
последовательных реакции:
Ai —> V1A2 —> V2A3
Вещества Aj и А2 принимаем за ключевые. Вычислим коэффи-
циенты и Р2 по формуле (11.76):
д= -1 0 = vi; А13 — 0 V2 — — V1V2
Vi — V1 V1 — V1
(11.78)
Агз =
= —v2; Pi=—(v2 —1); 02 = — (v2/vi — 1)
V2 I
Если обе реакции идут по первому порядку, скорости образования
ключевых веществ равны соответственно:
Г1=—kiCi', г2 = Т1й1С1—к2С2 (П-79)
Подставляя уравнение (11.77) в (П.79) и используя (П.78), запи-
сываем расчетные уравнения (II.60) в виде
dN1 =_______________feiAj__________ ... 80>
0 dX 1 + 0i(Ai-Ai,o)/Ao+02A2/Ao 1
dA2___________vifciAi-feaAa______ . .
0 dX 1 +₽1 (Al ~^,o)/Ao+ 02^2^0 . 1 ’
где начальная концентрация вещества А2 принята равной нулю.
Разделив уравнение (II.81) на (II.80) и проинтегрировав получен-
ное уравнение, находим:
k\ — kz
(11.82)
Ai ki—k2 L\ Ai ] J
После подстановки уравнения (II.82) в (II.80) последнее легко
интегрируется
АдХ Г , Ai о 1 . Ад о । ^2 (v2 —1)—fci (vi —1) A* 0— Ai
— = p 4. (vi -1) —J In -I —I-
(V2 —vi)A? Alt 0 , Nj \кг/к'
* k2(k1—k2) ’ Ao \ AXi 0 J
В случае параллельных и последовательных реакций одинакового
порядка изменение объема в результате химических превращений^
влияет (как это, в частности, видно из рассмотренного примера)
только на скорость процесса, но не на его селективность. Чем выше
порядок реакции, тем сильнее влияет на ее скорцсть изменение объема
реагирующей смеси. Увеличение объема в ходе химических превраще-
ний приводит к повышению селективности процесса, если порядок
основной реакции меньше, чем побочной, и снижает селективность
в противоположном случае.
78
ИЛ. ОСОБЕННОСТИ КИНЕТИКИ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Каталитическое превращение на твердом катализаторе, в отличие
от гомогенных реакций, представляет собой сложный процесс, состоя-
щий из ряда последовательных стадий: 1) подведение реагентов
к внешней поверхности катализатора; 2) диффузия реагентов в порах
катализатора к внутренней его поверхности (для пористых'катализа-
торов); 3) адсорбция реагентов на поверхности катализатора; 4) соб-
ственно химической реакции; 5) десорбция продуктов реакции;
6) диффузия продуктов реакции с внутренней поверхности катали-
затора; 7) диффузия продуктов реакции с внешней поверхности ката-
лизатора.
В стационарных условиях для каждой стадии разности скоро-
стей процесса в прямом и обратном направлениях равны. Однако
сами скорости могут существенно различаться. Стадия процесса,
протекающая в данных условиях с наименьшей скоростью, назы-
вается лимитирующей. Скорость всего процесса в целом и его кинети-
ческие закономерности определяются скоростью лимитирующей
стадии. В предельных случаях различают кинетическую, адсорбцион-
ную и диффузионную области протекания гетерогенно-каталитиче-
ских реакций. Границы областей определяются условиями проведе-
ния процесса, и в каждой из них действуют особые закономерности,
свойственные лимитирующей стадии. Между названными предель-
ными областями лежат переходные,^ которые подчиняются законо-
мерностям смешанного характера.
Здесь мы рассмотрим закономерности кинетики гетерогенно-ката-
литических реакций в отсутствие диффузионного торможения, т. е.
три из перечисленных стадий — адсорбцию, собственно реакцию
и десорбцию. В обычных кинетических исследованиях эти стадии
неразличимы; тем не менее гетерогенно-каталитический процесс
остается по своей природе сложным. Этим и объясняются характер-
ные для гетерогенного катализа сложные и разнообразные кинетиче-
ские закономерности.
Определение скорости гетерогенной реакции. Скорость гете-
рогенных реакций теоретически правильно определять как количество
вещества, образующегося или реагирующего в единицу времени,
отнесенное не к единице объема, а к единице активной поверхности.
Эта величина, обозначенная здесь через р, связана со скоростью
реакции г, определенной ранее (см. раздел II.1), простым соотноше-
нием
r = po (11.83)
где ст — площадь активной поверхности в единице реакционного
объема.
Аналогично скорость реакции по i-му веществу, отнесенная к еди-
нице активной поверхности, определяется равенством:
гг = рго
(П.84)
79
Так как на твердом катализаторе превращение претерпевают адсор- |
бированные на поверхности молекулы, возможны три пути гетеро- ,
генно-каталитической реакции: 1) мономолекулярное. превращение,
обусловленное взаимодействием адсорбированных молекул с кристал-
лической решеткой катализатора; 2) взаимодействие адсорбирован-
ных молекул, «ползающих» по активной поверхности; 3) взаимодей-
ствие адсорбированных молекул с молекулами, «налетающими»
на поверхность катализатора из объема. Очевидно, в любом из назван-
ных случаев скорость химической реакции зависит от концентрации
реагентов (всех или, если происходит «налетание», — некоторых) нд
активной поверхности.
Количество вещества, хемосорбированного на поверхности ката-
лизатора, измеряется двумерной концентрацией т)г-, связанной с долей
поверхности 0,, занятой данным веществом, сорбированным в моно-
слое, соотношением
= (11.85)
где рг- — сорбционная емкость единицы активной поверхности, яв-
ляющаяся постоянной для данной системы реагент — катализатор.
Если хемосорбированные молекулы вступают в реакцию типа
(II.1), причем непременным условием превращения является пред-
варительная адсорбция всех реагентов, то, видоизменяя основной
постулат химической кинетики (П.8), можно написать следующее
выражение для скорости реакции на поверхности
р = *Пщ/ = *' Пб? (П-86)
2=1 2=1
где к' — ТсЙ (г“‘.
f=l
Соответствующее выражение для мономолекулярной реакции
имеет вид:
р= /ьт)== (11.87)
В том случае, когда некоторые реагенты Az (i = т + 1, . . ., п)
участвуют в реакции, не будучи сорбированы (т. е. при «налетании»),
для скорости реакции получается следующее выражение:
т п т п
Р=*ПП? П ср^к'Пвр П ср (П.88)
i=l i=m+i i=l i=m+i
Двумерные концентрации как и степени заполнения поверх-
ности ,0(., не принадлежат к числу непосредственно измеряемых вели-
чин, поэтому кинетические уравнения процесса должны быть выра-
жены через измеряемые концентрации реагентов в объеме. Для этого
в формулы (11.86)—(11.88) надо подставить выражения, связывающие
степень заполнения поверхности с концентрацией соответствующего
вещества в объеме. Для процесса с установившимся адсорбционным
равновесием это будут уравнения изотермы адсорбции, а для реакции, .
скорость которой лимитируется скоростью адсорбции или десорбции
80
реагентов двумерные концентрации веществ адсорбированных на по-
верхности, определяются более сложными соотношениями, завися-
щими от скорости самой химической реакции.
Результирующее кинетическое уравнение, таким образом, описы-
вает многостадийный процесс, поэтому неудивительно, что оно мо-
жет быть весьма сложным и качественно своеобразным. Различные
формы кинетических уравнений гетерогенно-каталитических реакций
будут рассмотрены ниже.
Лангмюровская кинетика. Рассмотрим реакцию на однородной -
активной поверхности. Пусть адсорбция реагентов следует изотерме
Лангмюра (см. раздел 1.2). Если адсорбированное вещество вступает
в мономолекулярную реакцию (достаточно медленную, так что адсорб-
ционное равновесие не нарушается), то скорость реакции, отнесенная
к единице активной поверхности, согласно формулам (11.87) и (1.2),
равна - '
Р“ I + 61C1 + 62C2 ( 89)
где blt Ь2 — адсорбционные коэффициенты; Сх, С2 — концентрации
исходного вещества и продукта реакции.
При малых заполнениях поверхности (bfij 1), когда адсорбция
следует закону Генри, уравнение (11.89) принимает вид:
p=/cpb1C1 (11.90)
Это уравнение описывает процесс так же, как и уравнение гомо-
генной кинетики: реакция протекает по первому порядку с констан-
той скорости, отнесенной к единице активной поверхности, и =
= В силу (II-7) и (1.3), зависимость константы скорости от
температуры выражается уравнением
— (В—X,)
, = Zpb0e^- (IL91>
где — теплота адсорбции реагента. ,
Температурная зависимость константы скорости реакции остается,
таким образом, аррениусовой, причем кажущаяся энергия активации
Е' равна Е — При больших теплотах адсорбции кажущаяся
энергия активации может даже стать отрицательной; мы встречаемся
при этом с весьма редким случаем замедления химической реакции
с ростом температуры, причиной которого является уменьшение рав-
новесных степеней заполнения поверхности, не компенсируемое уско-
рением самого химического взаимодействия.
В другом предельном случае 1 поверхность практически
полностью занята исходным веществом; тогда
р = 1ср (11.92)
Реакция идет по нулевому порядку и кажущаяся энергия актива-
ции равна истинной. При Ь2С2 ЬгС2 и Ь2С2 1 имеем:
P = fcpb1Ci/b2C2 (II.93)
6 Заказ 1441 81
В этом случае поверхность почти полностью покрыта продуктом
реакции, тормозящим каталитическую реакцию, и Е' = Е 4- Х2—
Кинетическая функция (11.89) при средних заполнениях поверх-
ности не может быть разбита, как (II.6), на два сомножителя, один
из которых зависел бы только от температуры, а другой — только
от концентрации. Если же все-таки представить скорость реакции
в каком-либо интервале температур и концентраций уравнением типа
р = хСя, то кажущаяся энергия активации реакции будет находиться
в пределах Е — <Z Е' <Z Е, а порядок реакции — в пределах
0<а < 1. Лангмюровская теория адсорбции объясняет, таким обра-
зом, распространенность дробных порядков в каталитических реак-
циях. Кажущаяся энергия активации, как и кажущийся порядок
реакции, физического смысла не имеют и пригодны лишь для аппрок-
симации кинетического уравнения в некоторой ограниченной об-
ласти; обе эти величины меняются с изменением температуры и кон-
центрации реагирующего вещества.
Уравнения лангмюровской кинетики могут быть написаны и для
произвольной реакции, которой соответствует стехиометрическое
уравнение (П.1). Подставляя выражение (1.12) в (11.86), получаем:
к п (pbiCifi
Р=--------------— (П.94)
/ S \ г
' 1 + 2 V4
\ i-1 /
В формулу (11.94) входят концентрации не только исходных ве-
ществ и продуктов реакции, но и всех веществ, присутствующих
у активной поверхности и сорбирующихся на ней. Согласно этому
уравнению, реакция замедляется при увеличении концентрации как
продуктов реакции, так и веществ, стехиометрически не участву-
ющих в реакции, но способных достаточно хорошо сорбироваться на
поверхности катализатора, закрывая доступ к ней молекулам реа-
гирующих веществ.
При малых степенях заполнения поверхности, когда адсорбция
всех веществ протекает по закону Генри, формула (11.94) принимает
вид:
р = к п (iHbif* П (II.95)
г-1 1-1 ‘
Это выражение эквивалентно уравнению (II.6). Если зависимость
всех коэффициентов bt от температуры выражается соотношениями,
аналогичными (1.3), и теплота адсорбции i-го вещества равна Xz, то
реакция идет с кажущейся энергией активации:
Е' = Е -2 «Ас 4 (11.96)
1=1
82
«' Если для некоторого вещества Ая величина Ъя Ся велика по
сравнению как с единицей, так и с величинами b[Ci для всех осталь-
ных веществ, данное вещество почти полностью покрывает всю по-
верхность, тормозя тем самым ход реакции. Кинетическое уравне-
ние (11.94) принимает при этом вид
' к п
Р=——------п----- (П.97)
S а-
(ЬяСя)*"1
Влияние блокирующего вещества на скорость реакции тем силь_
п
ней, чем выше ее суммарный порядок а = "S af. Кажущаяся энер-
i-i
гия активации процесса равна
£' = £-2а<(Х4-Хя) (11.98)
2=1
где %я — теплота адсорбции блокирующего вещества Ая.
Блокировать активную поверхность может продукт реакции или
одно из исходных веществ; блокирующее вещество может стехио-
метрически и не участвовать в реакции. Последний случай особенно
важен для каталитических реакций в жидкой фазе. Растворители,
в отличие от инертных газов в газофазных процессах, имеют, как пра-
вило, достаточно высокие адсорбционные^ коэффициенты, и замена
одного растворителя другим может привести к резкому изменению
кинетики каталитической реакции. Блокирование поверхности ис-
ходным веществом может вызвать специфическое для гетерогенного
катализа явление самоторможения процесса, когда одно из исходных
веществ, сильно адсорбируясь на поверхности катализатора, затруд-
няет доступ к ней остальных реагентов и тем самым замедляет ката-
литическую реакцию.
Такой эффект, в частности, может проявиться в реакции типа Ах +
+ А2—>- продукты. В условиях, когда вещество Ах адсорбируется
настолько сильно, что почти полностью покрывает активную поверх-
ность, скорость реакции будет обратно пропорциональна его кон-
центрации:
Азр.1р.2Ь2Ст2
Р Ь1С1
(11.99)
Отдельно следует рассмотреть важный случай адсорбции с дис-
социацией. Когда один из осколков вступает в мономолекулярную
реакцию, а адсорбцией остальных реагентов и продуктов реакции
можно пренебречь, выражение для скорости реакции легко получить,
используя формулу (1.9):
' (11400)
6*
83
При малых степенях заполнения (ЬС 1) эта формула дает по-
ловинный порядок по концентрации исходного вещества:
р=/ь7^с1/2 (п.ю1)
Однако эта формула не остается справедливой при концентрации
исходного вещества, как угодно близкой к нулю. Дело в том, что при
С-+0 скорость реакции, пропорциональная С 1г, убывает медлен-
нее, чем скорость адсорбции, пропорциональная С; поэтому при
достаточно малых концентрациях реакция ябязательно переходит
в адсорбционную область и ее порядок меняется с половинного на
первый.
Таким образом, лангмюровская кинетика объясняет самые разно-
образные формы кинетических закономерностей гетерогенно-катали-
тических реакций и достаточно хорошо приближается к действитель-
ности при описании многих процессов. Как будет показано ниже,
кинетика процесса часто остается лангмюровской даже при невыпол-
нении основных постулатов, принятых при выводе изотермы адсорб-
ции Лангмюра.
Кинетика реакций в отсутствие адсорбционного равновесия.
Если скорость химической реакции на поверхности катализатора
достаточно велика, то адсорбционное равновесие не достигается и
степени заполнения поверхности молекулами реагентов нельзя опре-
делить из уравнения изотермы адсорбции. В предельном случае,
когда адсорбция одного из реагентов является наиболее медленной
стадией, скорость процесса лимитируется скоростью адсорбции этого
реагента, и можно говорить о протекании реакции в адсорбционной
области. Скорость адсорбции определяется константой скорости
адсорбции и концентрацией сорбируемого вещества; следовательно,
кинетика процесса в адсорбционной области формально следует урав-
нению реакции первого порядка; Поэтому различить кинетическую
и адсорбционную области только по кинетическим измерениям нельзя
и при необходимости следует ставить специальные эксперименты по
измерению скорости адсорбции или применять другие прямые методы
исследования, например, спектроскопию адсорбированных молекул.
Если скорости стадий реакции и адсорбции сравнимы между
собой, то степени заполнения поверхности, а затем и скорость реак-
ции могут быть найдены из уравнений баланса потоков адсорбции
и десорбции реагентов и потока химической реакции. Для любого
вещества А-, вступающего в реакцию типа (П-1), уравнение баланса
имеет вид
/ 8 \
( 1 Of I —^дес/9/ V/р (П-102)
\ 4=1 /
где калс j и /сдес j — константы скорости соответственно адсорбции
и десорбции j-го вещества; р — скорость химической реакции на
поверхности, зависящая от стененей заполнения поверхности моле-
кулами реагентов О,.
84
F
Запишем уравнение баланса исходного вещества для частного
случая необратимой мономолекулярной реакции, считая что про-
дукты реакции быстро десорбируются с активной поверхности:
/садсс(1—е) = /сдесе+/ще ' (п.юз)
Здесь вместо р подставлена правая часть уравнения (11.87). Пре-
образуя выражение (11.103), находим:
0 = л , ЬС1. кп---- (!I • Ю4)
1 4~ ЬС ^|хЬ//садс
и
А /}-- (11.105)
14~ ьс 4~ к^ь / /садс
Выражение (11.105) отличается от соответствующего лангмюров-
ского уравнения (11.89) (при Ь2 = 0) лишь одним членом в знамена-
теле, содержащим отношение констант скоростей реакции и адсорб-
ции. Если это отношение мало, адсорбция перестанет быть лимити-
рующим фактором и вместо уравнения (11.105) становится справед-
ливой формула (11.89). Оба выражения почти эквивалентны, если не
считать того, что неравновесная кинетическая функция (11.105) дает
несколько более сложную зависимость скорости реакции от темпера-
туры. В области малых заполнений (ЬС 1) формула (11.105) дает
кинетическое уравнение реакции первого порядка с эффективной
константой скорости:
+ 1 (П.106)
\ k[lb А-адс )
Степени заполнения поверхности, близкие к единице, не дости-
гаются, если процесс лимитируется скоростью адсорбции исходного
вещества. Даже если равновесие адсорбции нацело смещено в сто-
рону адсорбции реагента (Ь = оо), но скорость адсорбции сравнима
со скоростью реакции, кинетика процесса определяется уравнением
лангмюровского типа:
Р= /сцДадо + С (П.107)
Кинетика реакций' на неоднородной поверхности. При невыпол-
нении одного из постулатов Лангмюра (см. раздел 1.2) вид изотермы
адсорбции меняется. Подставляя в формулы (11.88)—(11.90) уравне-
ние любой изотермы адсорбции, отличной от лангмюровской, по-
лучаем видоизмененные кинетические зависимости, характеризующие
процесс на неоднородной поверхности или при взаимодействии мо-
лекул в адсорбированном слое. Если адсорбционное равновесие не
достигается, соответствующие неравновесные зависимости получают,
заменяя уравнения изотерм адсорбции зависимостями степеней за-
полнения поверхности от концентраций реагентов в объеме, опре-
деленными из условия баланса потоков адсорбции, собственно реак-
ции и десорбции.
85
Особенности кинетики реакций на неоднородной поверхности не *
исчерпываются, однако, простым изменением формы изотермы адсорб-
ции. Поверхность, неоднородная по теплоте адсорбции, должна быть
неоднородна и кинетически. Будем считать, следуя Рогинскому fl4],
что в ходе процесса зависимость скорости реакции от концентраций
реагентов остается неизменной на всех участках и температурная
зависимость скорости реакции по-прежнему описывается уравнением
Аррениуса. При этом величина предзкспонента постоянна на всех
участках, а значение энергии активации распределено по некоторому
закону. Все эти допущения являются дискуссионными, но в первом
приближении они достаточны, так как главным эффектом действия
катализатора обычно бывает именно изменение энергии активации
реакции.
Скорость реакции сильно зависит от энергии активации; поэтому
при любой форме функции распределения энергии активации реак-
ция практически будет идти только на тех участках поверхности, где
значение Е минимально. Эти участки образуют так называемую кон-
тролирующую полосу. Если в ходе реакции не происходит никаких
изменений состояния поверхности, контролирующая полоса сохра-
няет свое положение и процесс идет фактически на однородной по-
верхности, точнее — на однородной части поверхности катализатора,
входящей в контролирующую полосу; остальная же поверхность
никакого участия в реакции не принимает. Соответственно и кинетика
процесса является чисто лангмюровской.
Чтобы выявились особенности кинетики на неоднородной поверх-
ности, контролирующая полоса должна прийти в движение. Это
происходит при отравлении активных участков. Оговоримся, что под
термином «яд» будем понимать реагент, продукт реакции или при-
месь, способные интенсивно сорбироваться на участках активной
поверхности, закрывая доступ к ним реагентов. Возможно сочетание
следующих условий: отравление обратимо или необратимо; энергия
активации реакции Е и теплота адсорбции яда %я меняются симбатно
или антибатно, или же корреляция между ними отсутствует. Случай
симбатности величин Е и %я малоинтересен. Яд сорбируется на наи-
менее активных участках, и отравления фактически не происходит,
пока концентрация яда не достигла критического (очевидно, весьма
высокого) значения. При отсутствии корреляции 1Я и £ яд сорби-
руется с одинаковой вероятностью на участках поверхности с раз-
личными значениями Е, контролирующая полоса остается неподвиж-
ной и только активность катализатора постепенно падает со временем <
при необратимом отравлении и приходит к пониженному стационар-
ному состоянию, зависящему от концентрации яда, при обратимом.
При том и другом характере взаимосвязи между Е и %я энергия акти-
вации сохраняет постоянное значение в течение всего процесса и ки-
нетика остается лангмюровской.
Если Е и %я меняются антибатно, яд в первую очередь оккупирует
наиболее активные участки поверхности. При необратимом отравле-
нии контролирующая полоса постепенно сдвигается в сторону боль-
86
щих значений E. Аналогичное положение создается при, «утомлении»
катализатора, когда первыми выходят из строя наиболее активные
участки.
Рассмотрим обратимое отравление при Е и Хя, меняющихся анти-
батно и связанных линейной зависимостью:
£ = £'О-РЛЯ (11.108)
В случае интенсивной адсорбции яда (когда произведение адсорб-
ционного коэффициента яда Ъя на его концентрацию в объеме Ся
велико по сравнению как с единицей, так и с величинами й£-С£- для
всех остальных веществ, участвующих в процессе) степень заполне-
ния ядом 0Я участков, входящих в контролирующую полосу, будет
близка к единице. Согласно уравнению (1.2)
6я=(Ц-«я/Ся)-1^1-«я/Ся (П.109)
Здесь ая — обратный адсорбционный коэффициент яда.
Скорость реакции р пропорциональна доле оставшейся поверх-
ности, равной Ля/Ся, и ее зависимость от Хя, согласно соотноше-
ниям (П.98) и (II.99), определяется выражением:
р = const-ехр[А,я(р — 1)/RT] (11.110)
При 0 > 1 скорость реакции растет с увеличением Хя и, следо-
вательно, даже при наличии отравления реакция в основном идет
на участках с минимальными значениями Е, и контролирующая
полоса сохраняет свое положение. По-прежнему действует лангмю-
ровская кинетика, и скорость реакции, как и в случае, когда спра-
ведлива формула (11.97), обратно пропорциональна Ся.
При 0 < 1 наиболее активные участки выключаются из процесса
по мере увеличения Ся. Контролирующая полоса перемещается
в сторону больших значений Е, примерно совпадая с фронтом адсорб-
ции яда: по одну сторону этой полосы ничтожна доля поверхности,
свободной от яда, а по другую — слишком велика энергия активации.
О порядке реакции по концентрации яда здесь нельзя говорить, так
как действие последнего проявляется в увеличении энергии актива-
ции реакции.
П.5. КИНЕТИКА СЛОЖНЫХ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Квазистационарность. Сложный гетерогенно-каталитический про-
цесс включает ряд стадий адсорбции и десорбции исходных веществ,
промежуточных и конечных продуктов и реакций взаимных превра-
щений веществ, адсорбированных на активной поверхности. Полное
число стадий может быть весьма велико, и, чтобы разобраться в ки-
нетике сложного процесса, необходимо учесть обычно наблюдаемые
резкие различия между скоростями отдельных стадий. Ключ к этому ‘
дает теория стационарных реакций Хориути—Темкина [16, 17], ко-
торая опирается на понятие квазистационарности реакций, впервые
. 87
введенное в теории цепных процессов Боденштейном и обоснованное
в работах Семенова [18] и Франк-Каменецкого [19].
Запишем общую стехиометрическую схему
s
У, viftAi = 0 (Л = 1, 2, . . ., R) (11.111)
г=1
и соответствующую ей систему кинетических уравнений
R
^L = \vikrw cs) (1=1, 2, • S) (П.112)
описывающую изменение во времени концентраций всех S веществ,
участвующих в сложном процессе (включая нестойкие промежуточ-
ные комплексы и адсорбированные формы) и учитывающую скорости
r(k) всех R элементарных стадий рассматриваемого процесса. При
этом стадии адсорбции и десорбции учитываются наравне со ста-
диями химических превращений. Например, однослойная адсорбция
без диссоциации описывается, как и обычная реакция, стехиометри-
ческим уравнением — Az— Z + А* = 0 (где Z — вакантное место на
поверхности; А* — адсорбированное вещество Az). Вакантные места
(которых может быть, в случае неоднородной поверхности, несколько
сортов) включаются, как и все другие вещества, в стехиометрическую
схему (11.111) и систему уравнений (11.112).
Детальная система кинетических уравнений (11.112) слишком
сложна для решения. Более того, в нашем распоряжении практически
никогда нет данных, необходимых для записи входящих в нее кине-
тических функций, поскольку значительная часть переменных —
концентраций адсорбированных комплексов и других нестойких про-
межуточных соединений — не может быть измерена обычными ме-
тодами. Для расчетных целей система (11.112) должна быть сведена
к более простой, в которую входили бы только непосредственно на-
блюдаемые переменные — концентрации веществ в объеме.
Введем основной временной масштаб — характерное время про-
цесса t* (т. е. время, за которое происходит существенное накопле-
ние конечных продуктов) — и перейдем к безразмерным переменным
т = t/t*, с{ = CJC*, где С* — масштаб концентрации i-ro вещества.
Тогда система уравнений (11.112) примет вид
R
= •••’ Cs) = (И-113Х
А=1 *
где = г(к> (С1; . . ., Cs)/r(ft’ ((?:, . . ., С*8).
Величина tik = С*/г^ (С*, . . ., C*s), имеющая размерность вре-
мени, представляет робой характерное время образования или исчез-
новения i-ro вещества за счет к-й реакции (стадии).
Выделим теперь новый временной масштаб — характерное время
быстрых реакций t*—и допустим, что все величины tik могут
88
быть разделены на две группы, в одну из которых входят величины
того же порядка, что и характерное время процесса t*, а в другую —
величины порядка Z* или еще меньшие. Тогда система уравне-
ний (11.113) распадается на две подсистемы — «медленную» и «бы-
струю»
в
dr- 'ЖП /*
’ ”’Cs) 2,...., 5‘) (II.H4)
*=i г
в
= "s) = + (11.115)
ft-i 1
где 8 =/*/<*< 1.
В «быструю» подсистему (11.115) входят «неустойчивые» вещества,
среди реакций исчезновения которых есть хотя бы одна с характер-
ным временем tlk, намного меньшим t* (т. е. принадлежащим ко
второй группе). Остальные вещества, не вступающие в быстрые
реакции исчезновения, входят в «медленную» подсистему (11.114).
Уравнения системы (11.115) содержат малый параметр 8 при про-
изводной по времени. Это значит, что характерное время изменения
соответствующих концентраций ct (i = S* + 1, . . ., 8) значительно
меньше характерного времени процесса t*. Член с производной по
времени в уравнениях (11.115) может быть значительным только в те-
чение короткого начального периода быстрого изменения концен-
траций «неустойчивых» веществ. После этого последние выходят на
квазистационарные значения, медленно изменяющиеся со временем
по мере изменения концентраций «устойчивых» веществ, которые
входят в «медленную» подсистему (11.114). Отбрасывая член с про-
изводной в уравнениях (11.115), получаем систему алгебраических
уравнений
к
(d, • • , cs) = 0 (i = 5*+l, .. ., S) (П-116)
ik
й=1
решение которой позволяет выразить концентрации всех «неустой-
чйвых» веществ с(- (j = 8* + 1, . . ., 8) через концентрации всех
остальных веществ в данный момент времени *. Подставляя найден-
ные величины сг (сг, . . ., cs*) в уравнения (11.114), получаем упро-
щенную систему кинетических уравнений
R
de • ’ЧП i*
••• М (1 = 1........^*) (П-117) (
й=1 г
* Хотя числе уравнений (11.116) в точности равно числу неизвестных,
эти уравнения не обязательно будут независимыми. В этом случае уравнения
из системы (II.116), представляющие собой линейную комбинацию остальных
Уравнений, заменяются инвариантными соотношениями, связывающими кон-
центрации различных «неустойчивых» веществ.
89
в которую входят только концентрации «устойчивых» , веществ.
Сложность исходной системы уравнений, однако, оставляет свой
след: в систему- (11.117) входят уже новые кинетические функции
ф**’ (Сх,. . ., Cs*) =/(ft’(C1, . . ., CS*CS* + 1) (clf . . .,cs*),. . . cs (clt.
которые могут быть гораздо более сложными, чем исходные кинети-
ческие функции /<4). Заметим, что концентрации «устойчивых» веществ,
входящие в упрощенную систему (11.117), обычно как раз и являются
непосредственно наблюдаемыми переменными, через которые экс-
периментаторы выражают наблюдаемые скорости химических пре-
вращений. Сложность получаемых при этом кинетических закономер-
ностей (особенно характерная для гетерогенного катализа) отражает
наличие множества стадий процесса, чьи скорости, стехиометрические
уравнения и состав образующихся промежуточных продуктов могут
оставаться совершенно неизвестными (и могут быть в действитель-
ности изучены только с помощью тонких физико-химических методов
исследования).
Упрощенной системе кинетических уравнений (11.117) соответ-
ствует и упрощенная стехиометрическая схема процесса, состоящая
из суммарных уравнений «маршрутов реакций» и включающая
только «устойчивые» вещества. Суммарные стехиометрические урав-
нения можно получить из общей схемы (11.111), умножая входящие
в нее уравнения на «стехиометрические числа» akr (их не следует
путать со стехиометрическими коэффициентами vik) и складывая их
таким образом, чтобы исключить промежуточные «неустойчивые»
вещества. Стехиометрические числа должны быть, следовательно,
выбраны так, чтобы удовлетворить равенствам:
в
2*Л = 0 (г = У* + 1, S) (11.118)
. й-1
Число линейно-независимых наборов Р стехиометрических чисел
равно рангу прямоугольной матрицы а, составленной из величин akr.
Согласно уравнению (11.118) (которое в матричной форме записы-
вается как va = 0), ранг матрицы а равен разности между полным
числом стадий и рангом матрицы v, т. е. числом линейно независимых
«неустойчивых» веществ. Каждому r-му набору стехиометрических
чисел соответствует некоторое суммарное стехиометрическое уравне-
ние, или маршрут реакции. Следует отметить, что, в то время как
число независимых маршрутов Р строго определено стехиометрией
процесса, их конкретный выбор произволен; при этом скорость
реакции по каждому маршруту будет зависеть от того, какой набор
независимых маршрутов выбран для описания процесса.
Стехиометрические коэффициенты «устойчивых» веществ в урав-
нениях маршрутов определяются равенствами:
R
. v;r=2vifea*r (/=1, (п.119)
ь=1
90
Ранг К новой матрицы стехиометрических коэффициентов v',
равный минимальному числу переменных, необходимых для одно-
значного описания кинетики процесса, согласно уравнению (11.119),
не может превышать ранга матрицы а, т. е. числа линейно-независи-
мых маршрутов. Если величина К меньше чем Р, то независимые
переменные, определяющие состояние системы — концентрации клю-
чевых веществ или степени полноты независимых реакций, выби-
раются так же, как в разделе II.2.
Реакции, протекающие с образованием промежуточного комплекса.
В качестве простейшего примера рассмотрим стадийную схему
A + Z А*; А* B + Z (11.120)
Нетрудно видеть, что эта схема описывает реакцию первого по-
рядка, протекающую через промежуточную стадию адсорбции на
активной поверхности (Z — вакантное место на поверхности; А* —
адсорбированное вещество А). Таким образом, в случае равновесной
адсорбции мы должны получить лангмюровскую кинетическую функ-
цию. (По такой же схеме могут протекать и гомогенные реакции;
в этом случае Z обозначает инициирующий реакцию радикал или мо-
лекулу гомогенного катализатора, а А* — промежуточный радикал
или активный комплекс). Обозначая концентрации веществ А, А*
и Z соответственно через С, ц, и ц', записываем систему кинетических
уравнений
4 ЯР
+ (П.121)
и и*
“!F==^r=~*1CT1' + (fc+^)T1 (П-122)
где а — площадь активной поверхности в единице объема (множи-
тель 1/а, естественно, появляется только в гетерогенных процессах,
в связи с разной размерностью объемной концентрации С и поверх-
ностных концентраций ц и ц').
Естественным масштабом концентрации исходного вещества слу-
жит его начальная концентрация Со, а масштабом концентраций ц
иц' — их постоянная сумма р. (сорбционная емкость единицы актив-
ной поверхности). Вводя безразмерные переменные
с = С/С0; 0 = т)/р = (р — т]')/р; T=fcipai
приводим систему уравнений (11.121) и (11.122) к виду
dc/dT= —с(1-0) + А0 (11.123)
ed0/dT = c(l-0)-A (1-}-6)0 (II-124)
где 8 = po/C0; К = ^/(к^), 6 = к/к[.
91
»
Параметр е равен доле имеющегося количества исходного веще-
ства, которая может быть одновременно сорбирована на активной
поверхности. Как мы видим, выполнение условия квазистационар-
ного протекания адсорбции 8 1 определяется только малой сорб-
ционной емкостью поверхности, независимо от соотношения скоро-
стей реакции и адсорбции, т. е. от величины параметра 6. Отбрасывая
член с производной в уравнении (11.124) и выражая 0 через с, легко
получить кинетическое выражение для реакции, лимитируемой ско-
ростью адсорбций исходного вещества или (при 6 1) лангмюров-
скую кинетическую функцию.
Так как в рассматриваемом случае имеется две стадии и одно
промежуточное «неустойчивое» вещество, здесь существует един-
ственный маршрут реакции А->В, которому соответствует набор
стехиометрических чисел (1; 1). Суммарное уравнение получают пу-
тем сложения двух стехиометрических уравнений (11.120).
Следующий пример несколько сложнее. Рассмотрим параллельно-
последовательную схему реакций, протекающих на поверхности ка-
тализатора. Полная стехиометрическая схема этого процесса имеет
вид:
1) 'Aj + Z А*; 2) A2+Z А?;
Й1
3) Аз + Z *4 А?; 4) А,* -> А?;
> зт «- 3. 1 (11.125)
5) А* —> AJ; 6) А* —► AJ
Не будем выписывать полной системы кинетических уравнений
и приводить ее к безразмерной форме; это нетрудно сделать, учиты-
вая, что масштабом объемных концентраций будет, как и в преды-
дущем примере, начальная концентрация исходного вещества Со,
а масштабом поверхностных концентраций — сорбционная емкость
поверхности по одному из. веществ (например, исходному) |х. Малая
величина относительной сорбционной емкости поверхности 8 =
= цо/С0 по-прежнему служит условием квазистационарного проте-
кания адсорбции. При 8 1 степени заполнения поверхности могут
быть найдены решением соответствующих стационарных уравнений.
Из четырех имеющихся «неустойчивых» веществ Z, А*, А2, А3
три независимы, так как сумма всех четырех двумерных концентра-
ций — величина постоянная. Поэтому здесь имеется Р = 6—3 =
= 3 независимых маршрута реакции: Ai—>А2, Ах->-А3, А2-> »
—>-А3, — которым соответствуют наборы стехиометрических чисел
(1; —1; 0; 1; 0; 0;), (1; 0; —1; 0; 0; 1), (0; 1; —1; 0; 1; 0). Однако число
ключевых веществ или независимых реакций К равно в данном
случае 2.
Если для всех веществ Ах, Аа, А3 существует адсорбционное рав-
новесие, то стационарные уравнения для двумерных концентраций
адсорбированных молекул и вакантных мест эквивалентны тем, ко-
торые использовались в разделе 1.2 при выводе лангмюровской изо-
92
термы адсорбции. Используя выражения (1.2), находим скорости
реакций:
п _ ,, „ а_______fciHibiCl____
pi fciMi 1+Ь1С1+Ь2С2+ЬзСз,
/С2Р-2^2^2
р2 А2И2е2 1 + ь1с1 + ь2с2+ь3с3 >
(П.126)
Л —Т, „ Й _____________________
Рз ЗР101 1 + &1С1 + Ь2С2+&зСз
Из этих выражений можно исключить концентрацию одного из
веществ (например, А3), получив тем самым кинетические функции,
зависящие от концентраций двух оставшихся ключевых веществ.
Формулы (11.126) перестают быть справедливыми, если имеется
хотя бы одна медленная адсорбционно-десорбционная стадия. Не
будем выписывать громоздких общих кинетических выражений для
случая, когда скорости всех стадий адсорбции, реакции и десорбции
сравнимы между собой (хотя получить их нетрудно), и рассмотрим
лишь один важный частный случай. Пусть исходное вещество Ах
находится в адсорбционном равновесии, а десорбция промежуточного
вещества А2 замедлена. Предположим также, что концентрация ве-
ществ А2 и А3 в объеме пренебрежимо малы, что может обеспечи-
ваться быстрым удалением десорбировавшихся продуктов из зоны
реакции. Тогда степени заполнения поверхности определяются урав-
нениями
^1^1(1—61 — 0г) == 01» !ЧМ1 = (Н2^2 + ^2дес) 02» 0з = О (11.127)
и скорость образования конечного продукта А3 равна:
РЗ = &2Ц202 + йзР101 =
PlblCi [/с3-|-Ц2^1^2/(^2дес 4~
l + bl^l [ 1 + р.1^1/(Л:2дес + Р-гЛг)I
(11.128)
При быстрой десорбции промежуточного продукта (й2дес ц2^2)
реакция протекает фактически по параллельной схеме Aj-^Ag,
А1-^-А3, так как вещество А2 десорбируется, не успев прореагиро-
вать. В обратном предельном случае медленной десорбции имеем:
Р1&1С1 (fci + fc3) ,
рз 1+biCi (I + M1/M2) ( ’
В этом случае промежуточное вещество практически не выходит
в объем и кинетические закономерности реакции таковы, как если бы
она протекала в одну стадию (А1-^-А3) с константой скорости
fe* = (^i + &3)/(1 + Hi^i/R2^2) при адсорбционном коэффициенте ис-
ходного вещества Ъ* =&!(! + 111кг/\12к2). Отсюда видно, как адсорб-
ционно-десорбционные стадии искажают кинетику реакции на актив-
ной поверхности. Наблюдателю, ничего не знающему о механизме
процесса, при разных соотношениях между скоростями химического
превращения и десорбции параллельно-последовательная схема
реакций в одном случае представляется параллельной, а в другом —
одностадийной.
93
Эволюционные реакции. Особую группу реакций, включенных
в сложный гетерогенно-каталитический процесс, составляют медлен-
ные побочные реакции, приводящие к изменению свойств катализа-
тора (модифицированию или отравлению) в результате его взаимодей-
ствия с реакционной средой. Как правило, характерное время таких
реакций £° значительно превышает основной временной масштаб t*,
т. е. характерное время основных реакций. В отличие от «медленных»
реакций, лимитирующих скорость основного процесса синтеза целе-
вых продуктов, будем называть зти наиболее медленные реакции эво-
люционными.
Естественно, что эволюционные реакции оказывают существенное
влияние на скорость основного процесса только в том случае, если
последний проводится в стационарном режиме (в проточном аппарате,
где поддерживается постоянная концентрация исходных веществ)
в течение достаточно долгого времени, сравнимого с временным мас-
штабом t°. Наличие эволюционных реакций приводит, разумеется,
к тому, что процесс в целом не является строго стационарным: ки-
нетические характеристики основных реакций медленно изменяются
со временем. На временах, значительно превышающих t°, может
установиться истинный стационарныйгрежим; если, однако, мы имеем
дело с необратимым отравлением, этот стационарный режим может
соответствовать полной потере активности катализатора.
В качестве простого примера рассмотрим схему реакций
ki кг
Аг ---> А2 --► А3 (11.130)
4
А4
г
где А 2 — не десорбирующийся промежуточный комплекс; А3 — це-
левой продукт; А4 — побочный продукт, блокирующий активную
поверхность.
Если образование побочного продукта происходит очень мед-
ленно (к3 к3), можно написать стационарные уравнения для сте-
пеней заполнения поверхности, соответствующих всем веществам,
кроме А4. Предполагая для простоты адсорбционное равновесие по
исходному веществу А4 и быструю десорбцию конечного продукта А3,
имеем:
biCj (1 — 01 —02 — 04) = 04; р1Л101 = р2А:202; р4 == 1X2*302 (11.131)
откуда
' л (*1Р1/*2р2) Ь1Р1 (1— 04)
2~ 1+61G (1+*ijxi/*2H2) ( }
04= 1 — exp-f — t .... . b1C1 I (11.133)
I *2[Х4 l + ^iCi (l-|-pi*i/[X2*2) J
94
Следовательно, скорость образования целевого продукта экспо-
ненциально затухает со временем
П ==„ ко ________Н1*1Ь1С1________f МзН1 ,v
Рз Р2 2 2 l + bjCHl + MlWa) ₽l fc2(x4 tX
Х 1 + Ь1С1(1Ь+НгЫМ2) }=р°ехр ( -(*зРо/^4) t} (П-134)
где Ро = k^CjJI.1 + b1C1 (1 + Цх^х/Иг^г) 1 — скорость реакции
в начальный момент (на незаблокированном катализаторе).
Скорость реакции убывает тем быстрей, чем больше отноше-
ние Zc3/Zc2, т. е. чем большая доля промежуточного вещества А2 рас-
ходуется на вредную побочную реакцию. Полное количество целе-
вого продукта, образующееся на единице активной поверхности за
в'ремя работы катализатора (до его прлной дезактивации), равно:
СО
f Рз(0[<» = -г-Р4 (II 135)
о
Следующий пример наиболее наглядно иллюстрирует модифици-
рование катализатора под действием реакционной среды. Пусть реак-
h
ция Aj + А2->А3 протекает на катализаторе, способном существо-
вать в двух модификациях (например, окисленной и восстановлен-
ной), которые обозначим символом Фх и Ф2. Предположим, что обе
модификации могут переходить друг в друга под действием исходных
веществ Ах и А2 по схеме:
Ъ , h
ПМОД1 МОД2
Ах+Фх > Ф2; А2+Ф2 >- Фх (11.136)
Если каталитически активна только модификация Фх, то ланг-
мюровское кинетическое выражение для скорости основной реакции
имеет вид
tpxfcpxP2blb2CxC2 /тт х<57\
Р (1 + Ь1Сх + ь2с2)2 ( • >
где <рх — доля поверхности, занятая модификацией Фх.
Согласно схеме превращений катализатора (11.136), стационар-
ное значение величины фх, равное ф1ст, определяется из уравнения
^МОД 1С1Ф1СТ — ^МОД 2^-2 (1-фхст) (П.138)
где &мод j, Агмод2 — константы скорости соответствующих реакций.
Отсюда находим стационарную скорость основной реакции:
^РхРгЬхЬгСхСа кмоя 4С4 ... .
Рст~ (1 + ЬхСх + Ь2С2)2 * fcMO«xCi+&2C2 ' • ’
Сравнивая рст с р, видим, что под влиянием реакций модификации
катализаторов кажущийся порядок реакции по веществу А2 повы-
шается, а по веществу Ах — снижается. В частности, при слабой
95
адсорбции обоих исходных веществ (fejCj, b2C2 1) кажущийся
порядок реакции по веществу Ах становится ниже, а по веществу А2 —
выше первого; наибольшее изменение порядка реакции (соответ-
ственно до нулевого и второго) наблюдается при &мод1 Сх Лмод2 С2,
чему соответствует малое равновесное содержание активной моди-
фикации катализатора.
В нестационарном режиме (в течение периода релаксации) доля
активной модификации определяется решением уравнения
d<Pl/dt = Л’мод 1С1Ф1 “Ь ^мод2С2Ф2 = (^мод 1^-1 + &мод 2С2) (ф1 — ф!ст) (П-140)
откуда
Ф1 = Ф1ст + (Ф1— ф1ст)ехР {-(*мод1С1 + *мод2С2)0 (П-141),
Р = Рст + (Ро —Рст) ехр {—(^мод^х + ^модгСг)/} (11.142)
Наблюдаемая скорость реакции изменяется со временем от на-
чальной величины р0 до стационарной рст; соответствует ли это из-
менение повышению или падению активности, зависит от того,
была ли исходная доля активной фазы меньше или больше стацио-
нарной величины <р1ст;
Если характерное время изменения свойств катализатора имеет
тот же порядок величины, что и время основной реакции, и процесс
проводится в нестационарном режиме, то приходится совместно ре-
шать дифференциальные уравнения, описывающие изменение со
временем концентраций реагентов и доли активной модификации
катализатора (или доли незаблокированной поверхности в преды-
дущем примере). В этом случае скорость процесса в каждый момент
времени будет зависеть от его предыдущей истории и, в частности,
от исходного состава реагирующей смеси.
Отметим, что исследование кинетики сложных каталитических
реакций чаще всего может дать основания лишь для неоднозначных
соображений о ее механизме, но, не будучи связано с более деталь-
ными физическими и физико-химическими исследованиями, не может
выявить характера элементарных стадий процесса. С другой стороны,
знание кинетики реакций, какой бы механизм ни лежал в их основе,
является необходимой предпосылкой всех расчетов промышленных
процессов. Для расчетных целей безразлично, соответствует ли форма
кинетических уравнений детальному механизму каталитического про-
цесса. Зависимость скорости реакций от концентраций реагентов и
температуры часто представляют (в некоторой ограниченной области)
выражениями типа (II.6)—(II.8) с эмпирическими коэффициентами;
при этом в формулу (П-8) должны также входить концентрации ве-
ществ, тормозящих реакцию, с отрицательными порядками az. Для
приближенного формального описания кинетики реакций в широком
интервале изменения значений переменных более пригодны уравне-
ния лангмюровского типа.
96
ЛИТЕРАТУРА
1. С. Л. Киперман, Введение в кинетику гетерогенных каталитиче-
ских реакций, Изд. «Наука», 1964.
2. Н. М. Эмануэль, Д. Г. Кнорре, Курс химической кинетики,
Изд. «Высшая школа», 1967.
3. С. Бенсон, Основы химической кинетики, Изд. «Мир», 1964.
4. К. J. L a i d 1 е г, Chemical Kinetics, New York, 1965.
5. J. C. J urgens, Cinetique chimique applique, Paris, 1958.
6. Дж. Томас, У. T о м а с, Гетерогенный катализ, Изд. «Мир», 1969.
7. Р. Арис, Анализ работы химических реакторов, Изд. «Химия», 1967.
8. С. Я. Пшежецкий, Р. Н. Рубинштейн, ЖФХ, 21, 659
(1947).
9. В. Загр адник, Усп. хим., 30, 1272 (1961).
10. В. Zwolinsky, Н. Е у г i n g; J. Am. Chem. Soc., 69, 2702 (1947).
11. Д ж. У э й, Ч. П р е т е р, сб. «Катализ. Полифункциональные ката-
лизаторы и сложные реакции», Изд. «Мир», 1965, стр. 68.
12. Г. М. П а н ч е н к о в, ЖФХ, 22, 209 (1948).
13. Г. М. П а н ч е н к о в, В. Р. Лебедев, Химическая кинетика
и катализ, Изд. МГУ, 1961.
14. С. 3. Рогинский, Адсорбция и катализ на неоднородной поверх-
ности, Изд. АН СССР, 1948.
15. Ф. Ф. Волькенштейн, Электронная теория катализа на полу-
проводниках, Физматгиз, 1960.
16. Д з. Хориути, сб. «Проблемы физической химии», вып. 2, 1959,
стр. 59.
17. М. И. Темки н, сб. «Научные основы подбора и производства ката-
лизаторов», Новосибирск, 1964, стр. 46.
18. Н. Н. Семенов, ЖФХ, 17, 187 (1943).
19. Д. А. Франк-Каменецкий, Усп. хим., 10, 373 (1941).
7 Заказ 1441
Глава III -------------------;------------------------------
МАКРОКИНЕТИКА КАТАЛИТИЧЕСКИХ РЕАКЦИЙ [1-3]
III.1. ЗАКОНЫ ТЕПЛО- И МАССОПЕРЕНОСА
Одной из обязательных стадий гетерогенно-каталитической реак-
ции является перенос вещества к активной поверхности. Типичный
гетерогенно-каталитический процесс идет на поверхности твердой
частицы, большей частью пористой, которая омывается потоком газа
или жидкости. Если химическая реакция протекает достаточно
быстро, скорость процесса может лимитироваться подводом реаген-
тов из ядра потока к внешней поверхности частицы, а также диффу-
зией реагентов в порах внутрь зерна катализатора. В этом случае
говорят соответственно о внешне- и внутридиффузионном торможе-
нии процесса.
Диффузионное торможение процесса обычно сопровождается
и затруднениями с отводом тепла реакции, ведущими к появлению
перепадов температуры внутри пористого зерна катализатора и между
поверхностью частицы и ядром потока. Реальные кинетические зако-
номерности каталитического процесса определяются как истинной
кинетикой реакции на активной поверхности, так и условиями
массо- и теплопереноса; их изучение составляет предмет макрокине-
тики химических процессов.
Массо- и теплопередача в порах. Наиболее важное значение
в процессах гетерогенного катализа имеет перенос вещества и тепла
внутри пористой частицы катализатора. Перенос вещества в порах
осуществляется исключительно путем молекулярной диффузии. Если
диаметр поры значительно превышает среднюю длину свободного
пробега, то молекулы диффундирующих веществ сталкиваются друг
с другом гораздо чаще, чем со стенками поры и последние не ока-
зывают существенного влияния на скорость диффузии в пористом
зерне. В этих условиях диффузия в порах протекает так же, как в объ-
еме неподвижной жидкости или газа и скорость переноса вещества
вдоль поры, отнесенная к единице ее поперечного сечения, опреде-
ляется законом Фика:
q~—DMdC/dX' (Ш-1)
Движущей силой диффузии является градиент концентрации диф-
фундирующего вещества С вдоль направления поры X', а поток ве-
щества направлен в сторону уменьшения концентрации. Величина
коэффициента молекулярной диффузии DM определяется как свой-
ствами самого диффундирующего вещества, так и составом среды,
в которой оно диффундирует. Подробный анализ процессов диффузии
можно найти в монографиях по кинетической теории газов [4]. По
98
Сканировал: Neptunyi
Магнитогорск
2008
порядку величины DM равен произведению средней скорости движе-
ния молекул v на среднюю длину свободного пробега X:
а<=4-;х (Ш.2)
О
Средняя скорость движения молекул зависит от температуры газа.
В равновесном газе, где распределение скоростей молекул опреде-
ляется законом Максвелла, величина v равна
где к — константа Больцмана; Т — абсолютная температура; га —
масса молекулы.
Выражение для длины свободного пробега, которое выводится
в элементарной кинетической теории газов, исходя из представления
о молекулах как об упругих сферах диаметра d, имеет вид
Х = (/2 лй)’1 (Ш-4)
где п — число молекул в единице объема.
Эта величина связана с давлением газа Р соотношением п = Р/кТ.
Таким образом, выражение (III.2) для коэффициента молекулярной
диффузии в газах может быть записано в виде:
D 2 рГ V/г for
°м 3 \ ят ) л№Р (1 1 ’
Величина DK обратно пропорциональна давлению и возрастает
с повышением температуры пропорционально Т 1г; чем больше масса
и диаметр молекулы, тем труднее она диффундирует. Зависимость
коэффициента молекулярной диффузии от свойств среды проявляется
в основном в изменении эффективного сечения столкновений. Опре-
деление коэффициентов молекулярной диффузии в многокомпонент-
ных смесях представляет собой чрезвычайно сложную задачу. При
расчете химических процессов зависимостью коэффициентов диффу-
зии от состава газовой смеси обычно можно пренебречь. Также не-
существенна в обычных условиях и зависимость коэффициента диф-
фузии от температуры: степенная зависимость DM (Г) не идет ни
в какое сравнение с экспоненциальной температурной зависимостью
константы скорости реакции, и при перепадах температуры, наблю-
даемых в каталитических процессах, коэффициент молекулярной диф-
фузии остается практически постоянным.
В жидкостях величины коэффициентов молекулярной диффузии
на несколько порядков ниже, чем в газах. Медленность молекулярной
диффузии в жидкостях сильно ограничивает возможность примене-
ния пористых катализаторов для проведения жидкофазных реакций.
По мере уменьшения диаметра пор столкновения молекул со стен-
ками поры начинают играть все большую роль в процессе молеку-
лярной диффузии. В предельном случае, когда диаметр поры намного
7* 99
меньше, чем средняя длина свободного пробега, и молекулы сталки-
ваются со стенками гораздо чаще, чем между собой, коэффициент
диффузии будет пропорционален уже не средней длине свободного
пробега, а диаметру поры dn:
1 - 2 , / 2кТ \ 1/2 /ггг
^кн- 3 dnv- 3 Ц—) (Ш.6)
Область А d„ называют) кнудсеновской областью, а вели-
чину £>кн — кнудсеновским коэффициентом диффузии.
Выражение (III.6) было получено Кнудсеном для пор прямой ци-
линдрической формы; оно, однако, будет приближенно соблюдаться
и для узких пор произвольной формы, если модифицировать его, вос-
пользовавшись выражением для среднего гидравлического радиуса
поры, т. е. отношения ее объема к поверхности гг = е/о (где е — доля
свободного объема пористой частицы ио — площадь поверхности
пор, отнесенная к единице объема зерна).'Так как гидравлический
радиус цилиндрической поры равен 1/4 ее диаметра, формула (III.6)
принимает вид:
_ 8 е / 2кТ у/а
кн— 3 ’ о \ лп )
(Ш.7)
Отношение к/m в формуле (III.7), как и в предыдущих формулах,
может быть заменено на равное ему отношение R)M, где R — газо-
вая постоянная и М — молекулярный вес.
При фиксированном диаметре поры переход в кнудсеновскую
область диффузии происходит при уменьшении давления; переход
этот является плавным, поэтому в довольно широком интервале дав-
лений оба предельных выражения (III.5) и (III.6) неприменимы и
для определения коэффициента диффузии пользуются различными
приближенными переходными формулами, например, формулой гар-
монического среднего:
1 1 I 1
В* “ DM + D,
i(III.8)
При исследовании макрокинетики химических реакций в пори-
стом зерне нерационально рассматривать процесс в отдельной поре.
Поры реальной частицы катализатора неодинаковы по размеру и,
пересекаясь друг с другом, образуют запутанную сеть; более того,
форма свободного объема частицы может напоминать скорее совокуп-
ность каверн неправильной формы, чем сеть капилляров. Поэтому
пористое зерно рационально рассматривать как квазигомогенную
среду, характеризуя скорость диффузии реагентов эффективным коэф-
фициентом диффузии D, а скорость химической реакции — эффектив-
ной кинетической функцией г (С, Т). Последняя выражает зависи-
мость скорости реакции в единице объема пористого зерна от кон-
центраций реагентов и температуры в данной точке объема зерна и
связана со скоростью реакции на единице активной поверхности р
соотношением г = op (С, Т).
100
Если сравнить эффективный коэффициент диффузии D с истин-
ным коэффициентом молекулярной диффузии DK (или DKn, илиО^),то
становится ясным, что D < DM. Это объясняется, во-первых, тем,
что в пористом зерне доступно для диффузии не все сечение, а лишь
часть, занятая порами, равная доле свободного объема е, и, во-вто-
рых, вследствие удлинения пути диффузии в извилистых порах. При
случайном распределении направления пор линия, проведенная
вдоль поры, на разных участках пойдет под различными углами к пря-
мой, соединяющей точки начала и конца пути [5]. Доля пор, пере-
секающих плоскость, перпендикулярную направлению диффузии,
под углом 6 к нормали составляет в изотропной среде 2 cos 6 sin 6 d 6.
Путь по такой поре удлиняется в 1/cos 6 раз. Вычисляя среднее
удлинение пути, находим:
Л/2
D = eDM/^ -^y2cos0sin0</0=eZ)M/2 (Ш.9)
о
Это простое соотношение, конечно, далеко НВ всегда выполняется.
В общем случае эффективный коэффициент диффузии можно пред-
ставить в виде D — aDM (где а — множитель, зависящий от струк-
туры зерна). Величина а обычно определяется экспериментально
[2, 3, 6]. Ряд работ [7] посвящен теоретическому предсказанию
величины D, ее типичные значения представлены в табл. V.1 (стр. 175).
В кнудсеновской области величина а падает с уменьшением раз-
мера пор. При определенных условиях в мелких порах проявляется,
однако, дополнительный механизм переноса вещества за счет по-
верхностной диффузии молекул, адсорбированных на стенках пор.
Этот эффект, в принципе, может приводить к величинам я, превы-
шающим единицу.
Существенное влияние на величину D в катализаторах, содер-
жащих узкие поры, оказывает распределение пор по размерам. При
резко неоднородном распределении размеров пор само понятие эф-
фективного коэффициента диффузии теряет определенность [8].
Представим себе частицу, свободный объем которой состоит из сети
широких транспортных макропор и множества отходящих от них
узких капилляров, работающих в кнудсеновской области. Зерна та-
кой структуры, которые образуются при спрессовывании мелких
микропористых гранул катализатора, находят себе широкое приме-
нение, поскольку они сочетают хорошо развитую внутреннюю по-
верхность с относительно высокой скоростью диффузии, обеспечи-
ваемой системой транспортных макропор (см. главу V). Измерение
величины D в подобном составном зерне (путем измерения скорости
диффузии через зерно вещества, не вступающего в химические пре-
вращения) даст, очевидно, лишь величину D в макропорах. Между
тем, химическая реакция, протекающая в основном в капиллярах,
на которые приходится преобладающая часть внутренней поверх-
ности катализатора, может лимитироваться гораздо более медленной
диффузией в кнудсеновских микропорах.
101
При описании макрокинетики каталитической реакции на ссн|
ставных зернах применяют «двойную» диффузионную модель, вводя!
отдельные эффективные коэффициенты диффузии для системы транс-1
портных макропор и для микропор в мелких гранулах [9]. При этом!
сначала определяют зависимость скорости реакции в мелких грану-1
лах от локальных концентраций реагентов в транспортных макро-1
порах, а затем вычисляют макроскопическую скорость реакции!
в зерне в целом с учетом диффузионного торможения в макропорахл
Описывать составное зерно как квазигомогенную среду с зффектив-1
ным коэффициентом диффузии, найденным в отсутствие химической!
реакции, можно только в предельных случаях, когда реакция либо!
не тормозится диффузией в микропорах, либо протекает настолько!
быстро, что локализуется на внешней поверхности малых гранул.1
В пористом катализаторе перенос тепла осуществляется как с по- j
мощью молекулярного теплопереноса в порах, так и за счет тепло-1
проводности самого катализатора. В газах коэффициент молекуляр-’
ной теплопроводности %м примерно равен коэффициенту молекуляр- ’
ной диффузии Da, умноженному на теплоемкость единицы объема
газа у. Эффективный коэффициент теплопроводности пористой ча-
стицы можно представить формулой
X = Xo-W„ (Ш.10)
где Хо — теплопроводность данной частицы в вакууме, которая
определяется переносом тепла по твердой фазе.
При наличии газовой фазы теплоперенос по твердой частице пре-
обладает, так что отношение
Х/7^=1+Хо/«Хи ' (Ш.И)
может значительно превышать единицу. Типичные значения эффек-
тивных коэффициентов диффузии и теплопроводности в пористых
зернах приведены в табл. V.I. Экспериментальному определению эф-
фективного коэффициента теплопроводности пористого катализатора
посвящены работы [10].
Внешняя массо- и теплопередача. Помимо процессов диффузии
и теплопередачи внутри пористой частицы, существенное влияние на
макроскопическую скорость каталитической реакции может оказы-
вать массо- и теплообмен между внешней поверхностью частиЦЕг и
омывающим ее потоком. Гетерогенно7каталитический процесс всегда
проводится в условиях интенсивного движения реагирующей смеси;
при этом в основной части («ядре») потока молекулярная диффузий
играет пренебрежимо малую роль по сравнению с конвекцией, благо-
даря которой происходит выравнивание состава и температуры
смеси. У твердой поверхности скорость потока обращается, однако,
в нуль; поэтому вблизй поверхности перенос вещества будет опре-
деляться молекулярной диффузией реагентов. В первых работах по
диффузионной кинетике гетерогенных реакций, принадлежащих
Нернсту [11 ], принималось, что вблизи поверхности существует слой
неподвижной жидкости толщиной б и диффузия через этот слой ли-
102
митирует скорость переноса вещества к активной поверхности. При
этом, согласно закону Фика (III. 1)
9 = (£>м/й)(СОо—со) (ПГ.12)
где д — количество вещества, диффундирующего через единицу по-
верхности в единицу времени; Ом/б = р — множитель, называемый
коэффициентом массопередачи; Сх — С 0 — перепад концентраций
между ядром потока и поверхностью.
На самом деле скорость потока плавно спадает по мере прибли-
жения к твердой поверхности, так что представление о существовании
неподвижного диффузионного слоя не соответствует действитель-
ности. Чтобы найти поток вещества, диффундирующего на твердую
поверхность, необходимо решить уравнение конвективной диффузии
с граничными условиями, заданными на этой поверхности [12].
В случае ламинарного движения стационарное распределение кон-
центрации вещества определяется уравнением конвективной диф-
фузии:
ЭС I ЭС I дС П д2С /ТТТ 4<П
дХ ^~Uy dY +Uz dZ 3X2 (Ш.13)
Здесь ux, uy, uz — компоненты вектора скорости и, являющиеся
функциями координат X, Y, Z.
Величину и направление скорости в каждой точке определяют ре-
шением уравнений гидродинамики. В правой части уравнения (III.13)
оставлена вторая производная только по координате X, нормальной
к поверхности, так как по всем другим направлениям перенос веще-
ства молекулярной диффузией пренебрежимо мал. Граничные усло-
вия для уравнения (III.13) определяются тем, что диффузионный по-
ток на твердую поверхность катализатора равен скорости химической
реакции, а на достаточном удалении от поверхности концентрация
равна Ст.
При больших скоростях движения практически весь перепад ско-
рости сосредоточен в тонком гидродинамическом пограничном слое
толщиной б0, а перепад концентрации — в диффузионном погранич-
ном слое толщиной б. Величина б будет различной на разных участ-
ках поверхности, являющейся неравнодоступной в диффузионном
отношении. То же относится и к толщине гидродинамического погра-
ничного слоя б0. Отношение б0/б тем выше, чем больше отношение
кинематической вязкости вещества v к коэффициенту молекулярной
диффузии Du. В жидкостях, где v/DM '^> 1, диффузионный погранич-
ный слой гораздо тоньше гидродинамического. В этом случае при
решении уравнения (III.13) можно воспользоваться достаточно про-
стыми выражениями для скорости потока вблизи твердой поверх-
ности, что позволяет найти аналитическое решение уравнения (III.13)
при протекании быстрой гетерогенной реакции или реакции первого
порядка на поверхности частиц простой геометрической формы (пла-
стина или шар) [12, 13]. В газах толщины диффузионного и гидро-
динамического пограничных слоев — величины одного порядка и
103
уравнение (III.13) может быть решено только приближенными мето-
дами [14]. Аналогичным способом — путем решения уравнения кон-
вективной теплопроводности — определяют и тепловой поток на
твердую поверхность
Решение уравнения (III.13) позволяет представить поток веще-
ства на активную поверхность в виде (III.12) с эффективной толщи-
ной диффузионного слоя б, зависящей от скорости и физических
свойств вещества. Кроме того, величина б оказывается зависящей
и от скорости гетерогенной реакции [12]. Это связано с тем, что при
конечной скорости реакции концентрация реагирующего вещества
изменяется вдоль неравнодоступной активной поверхности, что,
в свою очередь, влияет на условия массопереноса. Только в том слу-
чае, когда гетерогенная реакция протекает практически мгновенно,
приповерхностная концентрация Со будет повсюду равна нулю, если
реакция необратима, или некоторой равновесной концентрации в слу-
чае обратимой реакции; при этом величина б является вполне опре-
деленной и не зависит от кинетики процесса.
При обтекании тел сложной геометрической формы уравне-
ния (III. 13) становятся практически неразрешимыми. Было бы бес-
смысленно, например, пытаться таким образом определить диффу-
зионный поток на поверхность частиц, уложенных в плотный зерни-
стый слой. В этом случае эффективная толщина диффузионного слоя
или коэффициент массопередачи р = Дм/б могут быть определены
только экспериментально. Опыты по измерению коэффициента массо-
передачи проводят в условиях, когда гетерогенная реакция про-
текает практически мгновенно и скорость исследуемого процесса
лимитируется исключительно переносом вещества к активной по-
верхности. Экспериментальные данные удобно представлять в виде
функциональной зависимости между безразмерными параметрами
Num=-^~ = 4-; Ргм = -тг-; Ие = -^- (III-14)
о ’ Р'м V
Здесь NuM — диффузионное число Нуссельта (или число Шер-
вуда Sh); Ргм — диффузионное число Прандтля (иногда его назы-
вают числом Шмидта Sc); Re — число Рейнольдса; I — характерный
линейный размер (обычно диаметр твердой частицы или ее гидравли-
ческий радиус).
При исследовании теплообмена между движущимся веществом
и активной поверхностью, тепловой поток дт, аналогично (III.12),
может быть выражен формулой
9т=а(Г00-7’о) (III.15)
где а — коэффициент теплопередачи; — То — перепад темпера-
тур между веществом и твердой поверхностью.
Зависимость коэффициента теплопередачи от скорости и физи-
ческих свойств вещества может быть выражена с помощью
104
соотношений между тепловыми числами Нуссельта и
Прандтля
NuT= —• Ргт = ^- (Ш.16)
Хм * Хм
и числом Рейнольдса Re. Здесь у — теплоемкость единицы объема
вещества и /м — его коэффициент молекулярной теплопроводности.
Вместо диффузионного и теплового чисел Нуссельта часто исполь-
зуют факторы массо- и теплопередачи:
NuM _ Р / у у/з. NuT НИ 17)
“ КеРг4/3 " \ Ри / ’ 7т Repri/3 \ Хм ) 1 7
Функциональные зависимости, описывающие процессы массо- и
теплопередачи, почти не отличаются друг от друга. Аналитически
эти зависимости для зернистого слоя сферических частиц выра-
жаются формулами [15]:
0,725 . _ 1,10 /пт 1яч
7м Reo,4i_ о,15’ 7т Reo.41 — 0,15 1 '
При больших числах Рейнольдса /м ~ /т — Re“°>4. Ряд крите-
риальных зависимостей, предложенных различными исследовате-
лями, приведен в монографии Аэрова и Тодеса [16].
Как уже указывалось, процессы массопередачи и химической ре-
акции не являются полностью независимыми. Поэтому в случае
неравнодоступной поверхности нельзя вычислять макроскопическую
скорость Гетерогенного процесса, просто приравнивая величину q
[см. уравнение (III.12) ] к скорости реакции на активной поверх-
ности. Так как локальная скорость массопередачи к различным участ-
кам поверхности неодинакова, при этом возникает тем большая
ошибка, чем больше перепад концентрации реагента вдоль активной
поверхности. К счастью, в наиболее важном для технологии случае —
в случае процессов, протекающих в зернистом слое катализатора,
поверхность твердых частиц можно, по-видимому, с достаточной
степенью точности считать равнодоступной в диффузионном отноше-
нии. Эксперименты по определению р в зернистом слое [16 ] показы-
вают, что локальные значения изменяются вдоль поверхности до-
вольно нерегулярно, оставаясь, однако, величинами одного порядка.
Исключение составляют небольшие участки близ точек соприкосно-
вения твердых частиц, практически не участвующие в процессе из-за
очень медленного подвода к ним реагентов. Приближение равно-
доступной поверхности [1 ] приводит к единственному практически
приемлемому методу расчета процессов в зернистом слое, и мы будем
в дальнейшем широко им пользоваться.
При предельно точном расчете скорости массо- и теплопередачи
следует учитывать такие явления, как термодиффузия и диффузи-
онная теплопроводность, возникающие при наложении и взаимном
влиянии процессов переноса вещества и тепла, а также изменение
физических свойств реагирующей смеси под влиянием химических
105
превращений. В условиях обычных каталитических процессов эти
явления играют, однако, малую роль. То же относится и к стефа-
новскому потоку, возникающему вследствие неравной скорости
диффузии исходных веществ и продуктов реакции или вследствие
изменения объема в результате химических превращений. Влияние
стефановского потока может быть существенным только в случае
реакций в неразбавленных газовых смесях, идущих со значительным
изменением объема.
Ш.2. ОБЛАСТИ ПРОТЕКАНИЯ РЕАКЦИИ
Основные уравнения. Чтобы понять основные закономерности
диффузионного торможения каталитических реакций, начнём с про-
стейшего случая — необратимой изотермической реакции первого
порядка [17, 18]. Пусть эта реакция протекает на частице катали-
затора, имеющей форму пластины толщиной 21, торцы которой от-
крыты для подачи реагента, а боковые грани «запечатаны». Если
такое зерно однородно, то концентрация реагирующего вещества С
будет изменяться только в одном направлении — вдоль оси X, пер-
пендикулярной к торцам пластины. В согласии со сказанным в раз-
деле III.1, будем рассматривать пористый катализатор как гомоген-
ную среду, а перенос вещества в порах характеризовать эффектив-
ным коэффициентом диффузии D. Тогда стационарное распределение
концентрации реагента по толщине пористой пластины будет описы-
ваться одномерным диффузионным уравнением
D^~kc=° (Ш.19)
где к — константа скорости реакции, отнесенная к единице объема
пористого катализатора.
Очевидно, величина к связана с константой скорости и, отнесен-
ной к единице активной поверхности, соотношением к = хо (где
<т — площадь активной поверхности в единице объема пористой ча-
стицы). Если открытая внешняя поверхность зерна равнодоступна
в диффузионном отношении, гранитные условия на торцах пластины
могут быть записаны в виде:
Р(СЮ— С) = ±DdCldX (приХ=±1) (Ш.20)
Здесь Р — коэффициент массопередачи и Ст— концентрация
реагента в потоке, омывающем частицу катализатора.
Граничные условия (III.20) получают из баланса количества ве-
щества, подводимого к торцам пластины масс опер ед ачей из ядра
потока и отводимого внутрь диффузией в порах катализатора. Так
как вся рассматриваемая система симметрична относительно цен-
тральной плоскости пластины, одно из граничных условий может
быть заменено на условие симметрии: dC/dX — 0 при X = 0.
Нетрудно написать решение задачи (III.19), (III.20). Мы, однако,
не будем делать этого, а обратим внимание на качественные законо-
106
мерности процесса при различных соотношениях между его харак-
терными параметрами. С этой целью запишем уравнение (III. 19) и
граничные условия (III.20) в безразмерной форме. Вводя безразмер-
ную координату х = Х/l и безразмерную концентрацию с = С/С^,
имеем:
d*c/dx* = Wc (III.21)
Bi (i—с (Д)) = (de/dx)x=1 (dc/dx)x=0 =0 ' (Ш.22)
В уравнение (III.21), (III.22) входят два безразмерных параметра:
модуль Тиле Т = I]/ k/D и число Био Bi = $l/D. Модуль Тиле
равен корню из отношения характерного времени диффузии l2/D
к характерному времени реакции к~г. Иначе его можно определить
как отношение характерного размера зерна I к эффективной глубине
проникновения реакции в толщу пористого катализатора ^D/k.
При Т 1 наиболее медленной стадией процесса является хими-
ческая реакция; при этом диффузия в порах протекает достаточно
быстро, глубина проникновения реагента в поры катализатора ве-
лика и его концентрация должна быть практически постоянной по
всему объему зерна. При Т 1 скорость процесса должна лимитиро-
ваться скоростью диффузии вещества в порах; проникнув в поры,
исходное вещество быстро вступает в химическую реакцию и его кон-
центрация спадает практически до нуля в тонком слое близ внешней
поверхности частицы.
Другой характерный параметр процесса — число Био опре-
деляет соотношение между внешнедиффузионным и внутридиффузион-
яым сопротивлением. При Bi > 1 основные транспортные затрудне-
ния должны быть связаны с диффузией в порах катализатора, а при
Bi 1 — с переносом вещества из ядра потока к внешней поверх-
ности частицы. Последний случай практически нереален. Действи-
тельно, учитывая, что Р = Ом/6 (см. раздел III.1), имеем:
Bi=-^- = -L.-^- = NuM/a»l (III.23)
Справедливость этого неравенства очевидна, так как в обычных
для каталитических процессов условиях интенсивного протока газа
толщина диффузионного пограничного слоя 6 много меньше харак-
терного размера зерна I (NuM= 1/6 1), a D всегда меньше Da
(а = D/Du < 1). Таким образом, перенос вещества к внешней по-
верхности зерна всегда протекает гораздо быстрее, чем диффузия
в порах на глубину порядка I, и, следовательно, при увеличении
скорости химической реакции внутридиффузионное сопротивление
всегда должно начать сказываться раньше, чем внешнедиффузионное.-
Если, однако, константа скорости реакции вырастет настолько, что
реакция локализуется в тонком приповерхностном слое, сравнимом
по своим размерам с толщиной диффузионного пограничного слоя, то
внешнее сопротивление переносу вещества может стать уже заметным
фактором, лимитирующим скорость всего процесса. Исходя из гра-
ничного условия (III.20), оценим относительную величину перепада
107
концентраций вовне и внутри пористой частицы; отношение этих?
перепадов служит мерой сравнения внутри- и внешнедиффузионного'
сопротивлений. Так как по порядку величины dC/dX ~ С]/ k/D (на-,
помним, что величина V D/к представляет собой характерную глу-
бину проникновения реакции в толщу катализатора), то
Ст-С ~VkD _ Y
С р Bi
(III.24)
и, так как Bi 1, внешний перепад концентраций может стать
сравнимым с внутренним только при Т 1.
По мере увеличения скорости химической реакции и, соответ-
ственно, величины Т может начать сказываться еще один фактор —
микроструктура зерна. Если глубина проникновения реакции в ка-
тализатор уменьшится настолько, что станет сравнимой с харак-
терным размером пор dn, то пористый материал уже нельзя будет
рассматривать как квазигомогенную среду. В этом предельном слу-
чае реакция пойдет только на внешней поверхности частицы, как
если бы она вовсе не была пористой. Если глубина шероховатостей
намного меньше толщины диффузионного пограничного слоя, то эту
поверхность можно считать равнодоступной в диффузионном отно-
шении.
Предельные режимы. Описанная физическая картина процесса
позволяет выделить четыре .предельных режима реакции на пористом
катализаторе [1, 17].
I. Внутрикинетический режим; ]/D/к I (Т <х 1); лимитиру-
ющая стадия — химическая реакция в порах. Концентрация реагента
постоянна по всей толще зерна.
II. Внутридиффузионный режим; D/k<^ I (Т ^>1), но D/k^>
а8 (T/Bi 1) и I^D/k '^> da. Лимитирующая стадия — диффузия
в порах. Концентрация реагента спадает в относительно тонком слое,
значительно превосходящем, однако, размеры диффузионного по-
граничного слоя или размеры пор.
III. Внешнекинетический режим; j/D/Zc^dn, х р. Так как
к = хо ~ х dn1, последнее неравенство можно заменить на усло-
вие p/Zc )§> dn. Лимитирующая стадия — реакция на внешней по-
верхности катализатора. Вся внутренняя часть зерна не работает.
IV. Внешнедиффузионный режим; ад |/D/k (/kD/$ = *F/Bi )§>
1). Лимитирующая стадия — перенос вещества из ядра потока
к внешней поверхности частицы. Концентрация реагента спадает
до весьма малых значений уже на внешней поверхности зерна.
Помимо указанных предельных режимов, имеются, конечно, со-
стояния, промежуточные между I и II, II и III, II и IV, III и IV.
Нерационально пытаться выводить общие формулы, описывающие
сразу все возможные режимы процесса, тем более, что к одному из
них — внешнекинетическому — неприменимо даже диффузионное
уравнение (III.19) или (III.21). Вполне достаточно вывести более
108 •
простые зависимости, описывающие лишь пары предельных режйМов
вместе с разделяющими их промежуточными областями. Начнем
с режимов, наиболее характерных для каталитических процессов —
внутрикинетического и внутридиффузионного.
Реакция в пористом зерне. Как было показано выше, при не
слишком больших значениях модуля Тиле Т внешнедиффузионное
сопротивление не оказывает заметного влияния на процесс в пори-
стой частице. Поэтому уравнение (III.21) можно решать в рассматри-
ваемом случае с более простыми граничными условиями:
с(1) = 1; (dc/dz)x=0 = 0 (III.25)
Решение задачи (III.21), (III.25) имеет вид:
c = ch(1F;r)/ch'F (II 1.26)
В предельных случаях Т 1 и Т 1 с помощью формулы
(III.26) можно подтвердить полученные ранее выводы о распределе-
нии концентраций внутри частицы при внутрикинетическом и вну-
тридиффузионном режимах. Действительно, при Т 1 функция ги-
перболического косинуса близка к единице и с 1. При Т 1
гиперболический косинус можно заменить на экспоненту и с =
= е ', т. е. концентрация реагента экспоненциально спадает
по мере удаления от внешней поверхности зерна.
Найдем теперь наблюдаемую скорость процесса, которая, оче-
видно, равна потоку реагирующего вещества на торцы пластины:
q=D(^-\ = (Ш.27)
\ dX /х~1 I \ dx J 00
Во внутрикинетическом режиме (Т 1) и q— кСт1-
Это идеальное значение скорости реакции соответствует полному
использованию всей активной поверхности катализатора.
Во внутридиффузионном режиме (Т ~^> 1) th Т 1 и q = kDCa:.
В этом случае полная скорость процесса пропорциональна уже не
объему частицы катализатора, как во внутрикинетическом режцме,
а ее внешней поверхности, так как катализатор, находящийся в глу-
бине зерна, фактически не работает.
Важнейшей характеристикой процесса на пористом катализаторе
является фактор эффективности ц, равный отношению наблюдаемой
скорости реакции к скорости реакции в отсутствие диффузионного
торможения, т. е. во внутрикинетическом режиме. Из формулы
(III.27) следует:
T] = g/fcC0OZ = th Ч/Ч (III.28)
Очевидно, 1 во внутрикинетическом режиме (’Р’ <1) и я»
>=« Т’1 во внутридиффузионном режиме (Т 1). С ростом модуля
Тиле величина фактора эффективности монотонно уменьшается.
Влияние формы зерна. Аналогичные зависимости нетрудно полу-
чить и для частиц другой формы. Если зерно катализатора имеет
109
форму шара или цилиндра с «запечатанными» торцами, то распре-'
деление концентрации внутри частицы можно найти, решая урав-
нение
(IIL29>
их \ их /
где т = 1 для цилиндра и т — 2 для шара. Безразмерная коорди-
ната х направлена вдоль радиуса цилиндра или шара. В качестве
масштаба длины рационально выбрать гидравлический радиус ча-
стицы, т. е. отношение ее объема V к площади внешней поверхности,
открытой для доступа реагента, F. Эта величина равна половине ра-
диуса цилиндра или трети радиуса шара. Таким образом, коорди-
ната х изменяется в пределах 0 sj х х0 (где х0 = 2 для цилиндри-
ческой частицы и х0 — 3 для сферической). В случае, когда сопро-
тивлением массопередаче на внешнюю поверхность частицы можно
пренебречь, уравнение (II 1.29) следует решать с граничными усло-
виями [ср. (III.25)]:
с(х0) = 1; (dcldx)x=Q = 0 (III.30)
В случае сферической частицы решение задачи
имеет вид
_ 3 sh Чх
с~~‘ sh3Y
а в случае цилиндрической частицы
.. /о(^)
Zo(2Y)
(III.29), (Ш.ЗО)
(Ш.31)
(Ш.32)
где 70 — модифицированная функция Бесселя нулевого порядка.
Соответствующие выражения для фактора эффективности
имеют вид
kCmV ¥2 k dx
3Ycth 3Y-1-
Цшар — extra
(Ш.ЗЗ)
ц,1Ил= y/o(2^- (Ш-34)
*
где Ij — модифицированная функция Бесселя первого порядка.
При Т < 1 и Т > 1 формулы (III.31)—(III.34), как и формулы
(II 1.26), (II 1.28), подтверждают основные качественные закономер-
ности протекания реакции в кинетическом и внутридиффузионном
режимах.
Несложный анализ этих выражений показывает, что форма зерна
оказывается несущественной ни при малых, ни при больших значе-
ниях модуля Тиле. В первом случае, независимо от геометрической
110
формы частицы, концентрация реагента постоянна по всему объему
пористого зерна, а величина фактора эффективности близка к еди-
нице. Во втором случае концентрация реагента экспоненциально
спадает по мере удаления от внешней поверхности и фактор эффек-
тивности изменяется обратно пропорционально величине модуля
Тиле. Независимость наблюдаемой скорости реакции от формы зерна
в обоих предельных режимах понятна из физических соображений.
В кинетическом режиме геометрические факторы пе^имеют никакого
значения, так как диффузия не является лимитирующим фактором.
Во внутридиффузионном режиме реакция проходит-практически до
Рис. III.1. Зависимость
фактора эффективности
от модуля Тиле ¥ для
изотермической реакции
первого порядка на зер-
нах различной формы:
1 — пластина; 2 — цилиндр;
3 — шар.
конца в тонком слое, много меньшем характерного размера зерна;
кривизна этого слоя несущественна, и поэтому процесс всегда про-
ходит так же, как если бы поверхность частицы была плоской. Форма
частицы катализатора влияет на скорость процесса наиболее сильно
в области, промежуточной между кинетической и внутридиффузион-
ной. Однако, как это видно из рис. III. 1, функции (II 1.28), (III.33),
(III.34) не слишком сильно отличаются друг от друга даже в об-
ласти Т ~ 1, где различие между ними наиболее заметно. Это от-
метил Арис [19].
Реакция на внешней поверхности. При дальнейшем увеличении
модуля Тиле процесс переходит во внешнекинетический или внешне-
диффузионный режим. Первый из них встречается в процессах на по-
ристых катализаторах довольно редко. Нетрудно видеть, что два
вышеупомянутых условия осуществления внешнекинетического ре-
жима (стр. 108) могут быть одновременно удовлетворены только
в том случае, если выполнено неравенство D/da Пм/6. Последнее
возможно либо при настолько быстром обтекании пористой частицы
потоком газа, что толщина диффузионного пограничного слоя. 6
будет много меньше характерного размера неоднородности зерна <4,
либо в случае, когда пористость частицы незначительна и величина
а ~ D/DK аномально мала. При D/dn Z)M/6 процесс переходит
непосредственно из внутридиффузионной области во внешнедиффу-
зионную, минуя внешнекинетическую; для частиц с развитой пори-
стостью этот случай является наиболее типичным.
111
Переход от внешнекинетической или внутридиффузионной области Я
к внешнедиффузионной легко проследить с помощью уравнения ба-Я
ланса вещества на внешней поверхности частицы: Я
zC = p(C'oo-C) (Ш.35) I
В случае, когда внешнекинетическая область протекания реакции Д
не осуществляется и прослеживается непосредственный переход 1
из внутридиффузионной области во внешнедиффузионную, константу J
скорости реакции к в уравнении (III.35) надо заменить на вели- 1
чину ]/ kD. Разумеется, соотношение (II 1.35) можно записать, лишь 1
предположив, что внешняя поверхность частицы равнодоступна [1 ]. з
Выражая из него концентрацию реагента у активной поверхности, |
находим: J
С=рСоо/(х + Р) (Ш-36) I
Наблюдаемая скорость реакции равна: 1
9 = х + Р Соэ (III.37) ।
Таким образом, реакция идет по первому порядку с эффективной 4
константой скорости, определяющейся соотношением: 1
4—4+f <>"•»» J
Из формулы (III.38) видно, что обратная величина эффективной 4
константы скорости реакции и* равна сумме кинетического [к-1 |
или (kD)~ /г] и диффузионного (Р"1) «сопротивлений». |
Пользуясь формулами (III.36) — (III.38), не следует забывать, |
что они получены исходя из предположения о равнодоступности |
внешней поверхности частицы. При обтекании одиночной частицы I
потоком вещества это предположение явно несправедливо, так ч
как условия массопередачи на участки передней и тыльной сторон J
частицы резко различны. Единственным строгим методом учета влия- h
ния внешней массопередачи на скорость гетерогенной реакции яв-
ляется, как отмечалось в разделе III. 1, решение уравнения (III. 13). |
Неравнодоступность поверхности будет сказываться особенно сильно |
в сложных процессах, включающих последовательные реакции, 1
приводя к уменьшению выхода промежуточного продукта. |
Завйсимость скорости реакции от температуры и скорости потока. 1
Выделенные четыре основных области протекания реакции на по- я
ристой частице позволяют проследить характер зависимости на- Я
блюдаемой скорости реакции от внешних параметров — температуры 1
частицы (которая здесь считается постоянной по всему ее объему) j
и скорости потока. Рассмотрим реакцию с аррениусовской темпера- 1
турной зависимостью константы скорости к: 1
ч k=Ze~EIRT (III.39) |
112
В кинетическом режиме наблюдаемая энергия активации равна
истинной энергии активации Е. При повышении температуры про-
цесс переходит во внутридиффузионный режим. Так как скорость
реакции в этом режиме равна
q = VkDCm =VZDCoae-E/2RT
(111.40)
то наблюдаемая энергия активации снижается до половины истин-
пой. Это понижение происходит постепенно в области, промежуточ-
ной между кинетической и внутридиффузионной. При дальнейшем
повышении температуры реакция может
перейти во внешнекинетический режим,
в котором наблюдаемая энергия актива-
ции вновь возрастает до ее истинного
значения Е, либо, минуя этот режим,
сразу перейти во внешнедиффузионную
область, где скорость реакции практи-
чески перестает зависеть от температуры.
Соответствующие аррениусовские диа-
граммы в координатах Inc? — 1/77 приве-
дены на рис. III.2.
Зависимость от скорости потока начи-
нает проявляться по мере возрастания
роли внешнедиффузионного торможения
и становится максимальной во внешне-
диффузионном режиме, где скорость реак-
ции пропорциональна коэффициенту мас-
сопередачи к внешней поверхности зерна.
Рис. III.2. Аррениусовская
диаграмма для реакции на
пористом зерне катализа-
тора:
I — внутрикинетический режим;
II — внутридиффузионный ре-
жим; III — внешнекинетиче-
ский режим; IV — внешнедиф-
фузионный режим.
Ш.З. ВНЕШНЕДИФФУЗИОННОЕ ТОРМОЖЕНИЕ
И РАЗОГРЕВ ВНЕШНЕЙ ПОВЕРХНОСТИ КАТАЛИЗАТОРА
Баланс вещества на равнодоступной поверхности. Рассмотрим
общий случай гетерогенной реакции, протекающей согласно стехио-
метрическому уравнению
£vzAz = 0 (III.41)
i
Сначала рассмотрим процесс, локализованный на равнодоступ-
ной внешней поверхности, и будем исследовать режимы процесса
в области, переходной между внешнекинетической и внешнедиффу-
зионной. Если скорость химической реакции на единицу внешней
поверхности частицы равна р (С, Т) (где С — совокупность при-
поверхностных концентраций реагирующих веществ и Т — темпера-
тура катализатора), то баланс вещества на внешней поверхности
зерна катализатора записывается в виде
8 Заказ 1441
Pz (Cz-CZoo) = vzp(C, Т)
(.Ш.42)
113
где рг = DiK/6 — коэффициент массопередачи для г-го вещества^
и Cioa — его концентрация в ядре потока.
Из уравнений (III.42) находим, что концентрации всех веществ,
участвующих в реакции, связаны между собой линейными соотно- «
шениями:
Wv£)(Cz-Cioo) = (p//v/)(C/-С/оо) (III.43)
Выразив с помощью этих соотношений концентрации всех ве- »
ществ через концентрацию какого-лйбо одного из них Сг, можно
из уравнений (III.42) и (III.43) найти приповерхностные концен-
трации реагентов и наблюдаемую скорость реакции. Учитывая со-
отношение (III.43), находим полную производную функции р (С)
по концентрации вещества Ct
Ф gp v/ gp
dCi Zl dCj ' dCi Vi Za ₽/ dCj
i
где суммирование распространяется на все вещества, участвующие
в реакции. В типичном для химических процессов случае реакция
ускоряется исходными веществами (др/дС; >-0 при v,- < 0); ко-
нечные продукты либо замедляют реакцию, либо не влияют на ее
скорость (др/дС; 0 при Vy >-0). Поэтому в указанном «нормаль-
ном» случае полная производная правой части уравнений (III.42)
по концентрации любого вещества Су всегда отрицательна. Так
как производная левых частей этих уравнений положительна, урав-
нения (III.42) в рассматриваемом случае всегда имеют единственное
решение относительно Ct. Уравнения (III.42) могут иметь несколько
решений только в «аномальных» случаях, когда процесс тормозится
одним из исходных веществ или ускоряется одним из продуктов
реакции.
По мере увеличения температуры поверхности или уменьшения
скорости потока (соответственно — увеличения скорости реакции
или уменьшения коэффициента массопередачи) приповерхностные
концентрации исходных веществ уменьшаются. В случае необрати-
мой реакции во внешнедиффузионном режиме приповерхностная -»
концентрация хотя бы одного из исходных веществ должна быть '
близкой к нулю. Однако концентрации всех исходных веществ мо-
гут, как это видно уже из соотношений (III.43), одновременно об- ,
ратиться в нуль только при соблюдении условия диффузионной
стехиометрии:
PiCfooM—PA-oo/v/ (III.45)
Если коэффициенты массопередачи всех исходных веществ оди-
наковы, это равенство выполняется в стехиометрической смеси
(т. е. в смеси, где концентрации всех исходных веществ пропорцио-
нальны их стехиометрическим коэффициентам). Если же коэффи-
циенты массопередачи реагентов отличаются друг от друга, то
условие (III.45) может быть выполнено лишь при определенном со-
ставе смеси, отличном от стехиометрического. Очевидно, что соблю-
дение условия диффузионной стехиометрии маловероятно. Поэтому
114
обычно при протекании реакции (III.41) во внешнедиффузионной
области близка к нулю приповерхностная концентрация только
одного из исходных веществ, — у которого величина PfC/co/v/ ми-
нимальна, т. е. наиболее трудно диффундирующего или присутству-
ющего в реакционной смеси в непропорционально малых количествах.
Наблюдаемая скорость реакции, равная скорости подвода к поверх-
ности этого вещества, будет пропорциональной его концентрации
в ядре потока и независимой от концентраций всех остальных ис-
ходных веществ.
Если рассматриваемая реакция — обратимая, то во внешнедиф-
фузионном режиме приповерхностные концентрации всех реагентов
близки к равновесным. Их можно определить, используя термодина-
мическое соотношение между равновесными концентрациями
Пср=Кр(Т) (Ш.46)
(где Кр — константа равновесия, зависящая от температуры поверх-
ности) и линейные соотношения между концентрациями реагиру-
ющих веществ (III.43). Очевидно, наблюдаемая скорость реакции во
внешнедиффузионном режиме не зависит от химической кинетики
процесса и определяется только условием равновесия (III.46) и
скоростью диффузии реагентов к активной поверхности.
Тепловой баланс. До сих пор мы не занимались определением
температуры активной поверхности. Между тем, как отмечалось
в разделе III. 1, в условиях, когда скорость процесса лимитируется
диффузией реагентов, следует также ожидать затруднений с тепло-
обменом между активной поверхностью и ядром потока. Темпера-
тура зерна катализатора, на внешней поверхности которого проте-
кает химическая реакция с тепловым эффектом h, определяется из
уравнения теплового баланса:
а(7’-г’оо) = МС1 П (Ш.47)
Уравнение (III.47) имеет в точности ту же форму, что и уравне-
ния (III.42), отличаясь от последних только коэффициентами. Можно
сказать, что тепловой эффект h играет роль «стехиометрического
коэффициента» температуры; в экзотермическом процессе h О
и тепло можно рассматривать как «продукт реакции», а в эндотер-
мическом процессе /г < 0 и тепло играет роль «исходного вещества».
Выше отмечали, что, если реакция ускоряется одним из ее продук-
тов, то можно ожидать, что решение системы уравнений (III.42)
будет неоднозначным. При этом одному и тому же набору значений
характерных параметров будет соответствовать несколько возможных
стационарных режимов процесса. Именно такая ситуация возникает
в экзотермических процессах.
Используя уравнения (III.42) и (III.47), можно выразить припо-
верхностные концентрации всех реагирующих веществ через темпе-
ратуру активной поверхности Т:
ci = Cioa + И/М (Т - Л») Д11 -48)
115
8*
Подставляя уравнение (III.48) в (III.47), получаем единственное*
уравнение, определяющее температуру активной поверхности ТА
Это уравнение удобно решать графически, определяя искомую тем-4;
пературу как абсциссу точки пересечения кривой тепловыделения)
Ф (Т) = hp [С (Т), Т] и прямой теплоотвода с тангенсом угла на- j
клона а (рис. Ш.З). Функция ф (Т) должна обращаться в нуль при)
исчерпании хотя бы одного из исходных веществ. Естественно, что)
первым будет исчерпано
Рис. Ш.З. Графическое реше-
ние уравнения (III.47):
1—5 — прямые скорости теплоот-
вода, соответствующие различным
значениям Т^; А, В и с — точки
стационарных тепловых режимов.
лимитирующее вещество, для которого ве- <
личина ^iCloolvi минимальна. Поэтому
функция ф (Т) обращается в нуль при ;
т т h min (
(HI-49) J
Формула (III.49) определяет макси- )
мальный возможный перепад темпера- •
тур между активной поверхностью и ;
ядром потока. В потоках газа, где диф- J
фузионное и тепловое числа Прандтля
близки к единице (см. раздел III.1), ;
коэффициенты массо- и теплопередачи
связаны между собой соотношением i
а = уР (где у — теплоемкость объема !
смеси). При этом максимальный пере- -
пад температур равен: 3
у-min (111.50) .
T — T
Это выражение представляет собой не что иное, как разогрев
реагирующей смеси, достигаемый при полном превращении лимити-
рующего реагента в адиабатических условиях.
Множественные режимы и гистерезис. Исследуем зависимость
температуры активной поверхности от температуры ядра потока. '
Наиболее интересным является случай, когда наклон прямой теп- '
лоотвода меньше максимального наклона кривой ф (Т) (см. рис. Ш.З). i
Если прямая находится в положении 1 или 5 (увеличение Тт при- ]
водит к сдвигу прямых вправо), уравнение теплового баланса (III.47) 1
имеет только одно решение, отвечающее в первом случае кинетиче- I
скому режиму с малым разогревом поверхности, а во втором — вне-
шнедиффузионному с разогревом, близким к максимальному. Пря-
мая 3 пересекается с кривой тепловыделения в трех точках А, В,
С, из которых А и С соответствуют кинетическому и внешнедиффу-
зионному режимам, а режим, соответствующий точке В, оказывается
неустойчивым и потому не может быть реализован.
Неустойчивость промежуточного режима можно доказать сле-
дующим образом. Допустим, что температура поверхности соответ^
ствует промежуточному режиму В. Если под действием какого-
либо случайного фактора температура несколько повысится, то,
116
как это видно из рис. Ш.З, скорость тепловыделения возрастет
сильнее, чем скорость теплоотвода, что вызовет, в свою очередь,
дальнейшее повышение температуры. Напротив, при малом сниже-
нии температуры скорость теплоотвода превысит скорость тепловыде-
ления и температура будет продолжать снижаться. Таким образом,
рассматриваемый режим является неустойчивым: любое малое от-
клонение от него имеет тенденцию разрастаться со временем. Если
температура поверхности соответствует стационарному режиму А
или С, то при ее случайном повышении скорость тепловыделения
становится меньше, а при снижении — больше скорости теплоотвода.
В обоих случаях процесс, будучи выведен малым случайным воз-
мущением из стационарного режима, возвращается к первоначаль-
ному состоянию. Таким образом, стационарный режим неустойчив,
если производная скорости тепловыделения по температуре поверх-
ности больше производной скорости теплоотвода:
dy/dT>a (III.51)
Заметим, что при выводе условия неустойчивости (III.51) мы
неявно предполагали, что концентрации реагирующих веществ свя-
заны с температурой соотношениями (III.48). Эти соотношения,
однако, были выведены для стационарного режима и остаются спра-
ведливыми только при возмущениях специального вида. Но для
устойчивости режима требуется, чтобы система возвращалась к нему
после любого малого возмущения температуры или концентраций
реагентов; поэтому выводы о том, что стационарный режим, не удов-
летворяющий условию (III.51), устойчив, сделать, строго говоря,
еще нельзя. Для строгого доказательства устойчивости стационар-
ных режимов требуется более тонкий анализ условий нестационар-
ного протекания процесса. Эти вопросы будут подробно рассмотрены
в главе VIII; забегая вперед, можно, однако, сказать, что в данном
случае реакции на внешней поверхности твердой частицы стационар-
ный режим действительно всегда устойчив, если производная ско-
рости тепловыделения меньше производной скорости теплоотвода,
т. е. если неравенство (III.51) нарушено.
Аналогичным образом можно доказать неустойчивость промежу-
точных стационарных режимов и в том случае, когда множествен-
ные решения появляются вследствие сильного ускорения реакции
ее продуктами или торможения исходными веществами.
Вернемся к рис. Ш.З. Если постепенно повышать температуру
ядра потока Тт, то реакция останется в кинетической области до
тех пор пока прямая теплоотвода, сдвигаясь вправо, не достигнет
положения 4, после чего произойдет скачкообразное повышение
температуры и процесс перейдет во внешнедиффузионный режим.
Это явление называется «зажиганием» реакции. Обратную картину
мы наблюдаем, начав с прямой 5 и снижая температуру потока.
Когда прямая теплоотвода сдвинется до положения 2, реакция скач-
ком перейдет из внешнедиффузионного режима в кинетический
с соответствующим резким снижением температуры поверхности
117
Рис. 111.4. Изменение темпера-
туры поверхности Т при повы-
шении и понижении темпера-
туры ядра потока Т .
(«затухание» реакции). В промежутке между прямыми 2 и 4, где <
существуют два устойчивых стационарных состояния, реакция идет
в диффузионном или кинетическом режиме, в зависимости от перво-
начальной температуры активной поверхности. Здесь мы наблюдаем
типичное явление гистерезиса: скорость процесса зависит не только
от условий, действующих в данный момент, но и от предыдущего
состояния системы. Характерная гистерезисная петля показана
на рис. III.4, изображающем зависимость температуры поверхности
от температуры ядра потока. Промежуточные температуры поверх-
ности, заключенные между абсциссами точек касания прямых 2
и 4 с кривой тепловыделения (рис.
III.3), никогда не реализуется,
так как они соответствуют неустойчи-
вым стационарным режимам.
В случае, когда максимальный нак-
лон кривой тепловыделения меньше
наклона прямой теплоотвода, всегда
существует только один стационарный
режим. При этом температура поверх-
ности плавно изменяется при повы-
шении температуры ядра потока и пе-
реход из кинетического режима во
внешнедиффузионный происходит по-
степенно, проходя весь спектр проме-
жуточных состояний.
Экзотермическая реакция первого
порядка. Рассмотрим простейший при-
мер возникновения множественных ре-
реакцию первого порядка [11. В этом
жимов — экзотермическую
случае уравнение (III.47) принимает вид:
« (r-roo) = ^ (П [£«, ~(«/Ьр) (Г-^со)] (Ш-52)
Здесь приповерхностная концентрация исходного вещества вы-
ражена соотношением (III.48). Пусть зависимость константы ско-
рости реакции от температуры выражается уравнением Аррениуса.
Тогда, вводя безразмерную температуру поверхности
Е (Т-Тт)
е= дуг 007 (III.53)
со
имеем
х (T)=Ze-E/RT = K (Гто) exp ГА АД- - А)1 =
L \ со /J
а
= х (Уоо) ехР 0 х (А) (III.54)
Последнее преобразование в формуле (III.54) выполнено с уче-
том того, что в обычных химических процессах RT/Е 1. Это
118
приближение ценно тем, что оно позволяет получить Простое и фи-
зически понятное решение, не нарушая специфических. качествен-
ных свойств рассматриваемого процесса и не внося в расчет заметной
ошибки. В дальнейшем будем часто пользоваться приближенной
формулой (III.54).
Учитывая (III.53), (III.54), приводим уравнение (III.52) к без-
размерному виду
H = (Ш.55)
где
В ^ЕСт
(IIL56)
Безразмерный параметр ц характеризует относительную ско-
рость процессов массопередачи и химической реакции. Параметр 0
равен безразмерному максимальному разогреву активной поверх-
ности; в этом легко убедиться, подставив в формулу (III.49) разность
Т—Тт из выражения (III.13). Величина <р (0) в уравнении (III.55)
неограниченно возрастает при 0->О и обращается в нуль при 0 = 0.
Поэтому уравнение (III.55) всегда имеет решение, лежащее в интер-
вале 0 < 0 < 0. Очевидно, что, если функция <р (0) монотонно убы-
вает в этом интервале, то существует только одно стационарное
состояние. Число решений уравнения (III. 55) может превышать еди-
ницу только если функция <р (0) имеет экстремумы. При этом, оче-
видно, точки экстремумов отвечают критическим условиям перехода
от одного режима к другому. Дифференцируя правую часть уравне-
ния (III.55) по 0 и приравнивая производную к нулю, получаем сле-
дующее соотношение, которое должно быть выполнено в критической
точке:
02/(0-1) = 0 (Ш.57)
Отсюда легко найти безразмерную температуру поверхности
в критической точке
0 = А-0 (1±/1—4/0) (III.58)
Знак плюс в формуле (III.58) дает безразмерную температуру
поверхности во внешнедиффузионном, а знак минус — в кинети-
ческом режиме в критических точках, соответственно, затухания
и зажигания реакции. При малых значениях параметра максималь-
ного разогрева (0 4) функция <р (0) монотонна и существует
только один стационарный режим процесса. При этом безразмерная
температура поверхности 0 плавно возрастает с уменьшением пара-
метра (т. е. увеличением температуры ядра потока) от 0 = О
при ц = со до 0 = 0 при ц = 0. При 0 >> 4 температура поверх-
ности непрерывно возрастает только до достижения критической тем-
пературы поверхности, определяемой меньшим значением 0 в фор-
муле (III.58), после чего скачком повышается до температуры, соот-
ветствующей верхнему температурному режиму. Если активная
119
поверхность первоначально была горячей, то с увеличением пара/
метра р. (уменьшением происходит пбстепенное снижение f
до точки затухания реакции, после чего процесс скачком переходи!
на низкотемпературный режим. Безразмерные температуры поверх-
ности, лежащие в интервале
у 0(1 +/1-4/0) >0> у0 (1 —/1 — 4/0) (Ш.59)
никогда не реализуются. Чем больше максимальный разогрев 0,
тем шире этот интервал: меньше разогрев поверхности в низкотем-
пературном режиме и больше — в высокотемпературном. Если
величина 0 велика (0 4), что наблю-
дается в реакциях с большим тепловым
эффектом и высокой энергией актива-
ции, формула (III.58) дает 9 0 на
верхнем температурном режиме и
0 «=! О — на нижнем. Таким образом,
при больших значениях 0 реакция мо-
жет протекать либо в чисто кинетичес-
ком, либо в чисто внешнедиффузион-
ном режиме; переходной же области
соответствуют неустойчивые режимы,
Рис. Ш.5. Критические усло-
вия зажигания (кривая /) и
затухания (кривая 2) для экзо-
термической реакции первого
порядка:
I — кинетическая область; II —
внешнедиффузионная область; III—
область множественных режимов.
и она не может быть реализована.
Уравнения (III.55), (III.57) дают в
параметрической форме соотношение
между параметрами ц и 0 в кинетичес-
ких точках скачкообразного перехода
между режимами:
р=е’/(0—1); 0 = 02/(0 — 1) (Ш.60)
График зависимости In ц от 0 представлен на рис. II 1.5. В об-
ласти, заключенной между кривыми / и 2, возможны два устойчи-
вых режима реакции, а вне ее — только один: или кинетический
(ниже кривой 2), или внешнедиффузионный (выше кривой 1).
Локальные разогревы. При протекании реакции на неравно-
доступной поверхности возможно, что на одних участках поверхности
реакция идет во внешнедиффузионном, а на других — в кинети-
ческом режиме. Соответственно могут резко различаться темпера-
туры участков поверхности. Как указывалось в разделе III.1, в зер-
нистом слое поверхность твердых частиц почти равнодоступна, за
исключением застойных участков близ точек соприкосновения со-
седних зерен, диффузия к которым сильно затруднена. В работе [20 ]
вычислен локальный разогрев в точках соприкосновения, возника-
ющий в случае, когда процесс на основной открытой поверхности
протекает в кинетическом режиме, а в застойных участках — во
внешнедиффузионном. Разогрев в точках контакта зерен ответстве-
нен за такие явления, как спекание частиц в зернистом слое при из-
120
меряемых температурах, меньших температуры плавления катали-
затора.
Подробный анализ режимов экзотермической реакции показывает,
что реакция на различных участках поверхности может протекать
в разных режимах даже в том случае, когда поверхность равнодо-
ступна. В работе [21 ] была показана возможность существования
асимметричных режимов гетерогенной реакции, при которых одна
сторона пластины катализатора работает в кинетическом, а противо-
положная во внешнедиффузионном режиме. Интересно, что при
определенных условиях режим процесса на одной из граней пластины,
являясь неустойчивым, стабилизируется под влиянием теплового
потока через катализатор.
Ш.4. ВНУТРИДИФФУЗИОННОЕ ТОРМОЖЕНИЕ
И ВНУТРЕННИЙ РАЗОГРЕВ КАТАЛИЗАТОРА
Изотермическая реакция. Исследуем теперь условия протекания
реакции (III.41) в пористом зерне катализатора. Если скорость хими-
ческой реакции в единице объема пористой частицы равна г (С, Т),
то распределение концентрации Ct любого вещества Az внутри ча-
стицы произвольной формы определяется решением системы диф-
ференциальных уравнений
^Ci + iVi/D^riC, Т)=0 (Ш.61)
где V2 — оператор Лапласа; Dt — эффективный коэффициент диф-
фузии i-ro компонента.
Сопротивление массопередаче из ядра потока к внешней поверх-
ности, как было показано в разделе III.2, всегда мало по сравнению
с внутридиффузионным сопротивлением и начинает играть заметную
роль только в глубокой внутридиффузионной области протекания
реакции. Поэтому граничные условия для уравнений (III.61), за-
данные на внешней поверхности частицы Г, можно записать в виде
Ci(D = Cioo (III.62)
где С{оо — концентрация i-ro вещества в потоке, обтекающем частицу
катализатора.
Умножая каждое i-e уравнение (III.61) на D^Vi и попарно вы-
читая одно из другого, находим, что величины
wij=(Dilvi)Cl-(Dilvj)Cj (Ш.63)
определяются решением уравнения
V2»i/ = 0 (Ш.64)
с граничным условием
^/^n = pi/vjc(.oo-(Z>;-/V/)C/OOS^~ (Ш.65)
Единственным решением уравнения (III.64) при условии (III.65)
является wtj = const = wff и, следовательно, концентрации всех
121
веществ, участвующих в реакции, связаны друг с другом линейнымиЗИ
соотношениями: ,'Я
(Z>z/vJ (Cz-CZoo) = (^/v/) (Q-C/oo) (III-66) Я
Эти соотношения аналогичны соотношениям (III.43), выведенным 'Я
для процесса на внешней поверхности катализатора. Используя Я
равенства (III.66), можно, как и в разделе III.3, выразить концен- Я
трации всех веществ через концентрацию одного ключевого реагента. Я
В качестве последнего рационально выбрать то исходное вещество, Я
которое в данном процессе будет лимитирующим, а именно то, у ко- Я
торого величина DC^Jv является наименьшей. Таким образом, полу- Я
чаем единственное уравнение для концентрации ключевого вещества С: Я
V2C + (v/Z>)r(C, Т) = 0 (III.67) Я
Индексы при стехиометрическом коэффициенте ключевого ве- Я
щества (который будет отрицательным) и его коэффициенте диф- Я
фузии здесь опущены; предполагается также, что концентрации Я
всех веществ, от которых зависит скорость реакции г, выражены Я
через концентрацию ключевого вещества с помощью соотношений Я
>(111.66). I
Приведем уравнение (III.67) к безразмерному виду. Для этого II
за масштаб концентрации примем концентрацию ключевого вещества Я
во внешнем потоке Ст, за масштаб длины — характерный размер Я
частицы I (например, ее гидравлический радиус), а за масштаб ско- Я
рости реакции — скорость, соответствующую составу обтекающего 1
частицу потокаг!Г. = г (С^, Т). Тогда уравнение (III.67) приводится -Я
к виду я
V2c_4f2/(c) = 0 (III.68) 1
где с = C/Cm — безразмерная концентрация ключевого вещества; 1
/ (с) = т/Гоо — безразмерная скорость реакции (оператор Лапласа V2 1
включает теперь дифференцирование по безразмерной координате).
В уравнении (III.68) выделен основной безразмерный параметр — Я
модуль Тиле я
/\v\r(Coo, Т) Я
---(Ш.69) |
Другие параметры, зависящие от концентраций различных ве- 'а
ществ в потоке и от их коэффициентов диффузии, будут входить я
в выражение для безразмерной скорости реакции / (с, Т). Границ- Я
ное условие для уравнения (III.68): с (Г) = 1. *1
Будем решать уравнение (III.68) для зерна плоской формы. ’1
Напомним, что, как было показано в разделе III.2, изменение формы
зерна (при сохранении гидравлического радиуса) лишь в слабой |
степени влияет на наблюдаемую скорость реакции. Температуру 3
будем пока считать постоянной по всему объему частипы. В случае *
плоского зерна уравнение (III.68) принимает вид I
d2C/dx2=4f2/(c) (III. 70) j
122 1
где х — безразмерная координата, направленная перпендикулярно
торцам пластины и равная 0 в ее середине.
В качестве масштаба длины I принимается, как и в разделе III.2,
полутолщина пластины. Граничные условия:
с(1) = 1; (dc/dx)x=0=o
(III.71)
Для интегрирования уравнения (III.70) введем новую переменную
р = dc/dx. Так как d2c/dx2 = dp/dx = pdp/dc = у d (p2)/dc, урав-
нение (III.70) можно переписать в виде:
d(p2)/de = 2Y2/ (с)
(III.72)
Интегрируя это уравнение с учетом граничного условия в центре
пластины, имеем
V dx
de
/2 ¥
f (cl)dci
(Ш.73)
где Ci — концентрация Ключевого вещества в центре пластины,
которая пока неизвестна.
Если бы значение cz было известно, исходя из формулы (III.73)
можно было бы сразу определить величину фактора эффективности:
(III.74)
Таким образом, теперь задача сведена к определению величины cz.
Повторно интегрируя уравнение (III. 73) имеем:
V~2 ¥х= J J/(ci)*i
<7 и,
-у»
dc2
(Ш.75)
Соотношение (III. 75) дает в неявном виде зависимость концен-
трации ключевого вещества от координаты х; оно, однако, содержит
неизвестную величину cz, которую надо определить, используя гра-
ничное условие при х = 1. Подставляя х ~ \ и с = 1 в формулу
(III.75), получаем трансцендентное уравнение для определения ве-
личины ср.
/2¥=; p(C1)*i /г
(III.76)
dc = (f (ci)
Уравнение (III.76) удобно решать графически (рис. III.6). При
этом величина cz определяется как абсцисса точки пересечения кри-
вой <р (cz) и горизонтальной прямой линии с ординатой У 2 W. Можно
показать, что это уравнение всегда имеет по крайней мере одно
решение. Если функция ф (cz) монотонно убывает во всем интервале
123
0<cz<l, то решение уравнения (III.76) всегда единственное; npj
этом концентрация ключевого вещества в центре пластины монотонно!
убывает с увеличением модуля Тиле Чг. Теоретически возможны,!
однако, случаи, когда функция <p (cz) не является монотонной, что’
влечет за собой множественность стационарных режимов процесса^
и связанных с этим скачкообразных переходов из внутрикинетиче-^
Рис. III.6. График функ-
ции ф (cz) [см. уравне-
ние (III.76)], в случае,
когда имеется одно (кри-
вая 1) или три (кри-
вая 2) стационарных ре-
жима. Стрелки указы-
вают направление скач-
кообразных переходов
ской области во внутридиффузионную и об-.
ратно с возникновением гистерезисных пе-
тель.
Естественно, возникает вопрос о том,
при каких условиях возможно появление
экстремумов функции <р (cz). Найдем произ-
водную функции <р (с;):
Йф
dci
f (ci) dci
If (c)-f(ci)]
Из формулы (III.77) видно, что при мо-
нотонной возрастающей функции / (с) всегда
d^fdcl <Z 0. Таким образом, монотонное воз-
растание скорости реакции с концентрацией
ключевого вещества является достаточным
между режимами. условием отсутствия множественных режимов
замечаем,
(III.44) ]
процесса. Учитывая соотношения (III.66),
что производная функции / (с) равна [аналогично
df ___ D vi дг
de ~ vrTC дс(
i
l
(Ш.78)
где суммирование распространяется на все вещества, участвующие
в реакции. Если, как это обычно и бывает, dr/dCi >0 при vz < О
и drldCi^G при vz >0, производная (III.78) всегда положительна;
Согласно сказанному выше, при этом всегда dtp/dct < 0 и процесс
на пористой частице имеет только один стационарный режим. Мно-
жественные режимы, в принципе, возможны только в «аномальных^
случаях, когда процесс тормозится одним из исходных веществ или
ускоряется одним из продуктов реакции. Так, численные расчеты
показали возможность появления множественных режимов реакции
типа А + В—^-продукты, с лангмюровской кинетикой в условиях,
когда процесс сильно тормозится одним из исходных веществ [22 J.
Неизотермическая реакция. Рассмотрим еще одну переменную —
температуру катализатора, которая до сих пор считалась постоянной.
В действительности скорость отвода (подвода) тепла реакции, выде-
124
ляющегося (поглощающегося) внутри пористой частицы, — вели-
чина конечная, так что проведение реакции со значительным тепло-
вым эффектом может вызвать заметный перепад температуры по
толщине частицы, который, в свою очередь, влияет на скорость про-
цесса на пористом катализаторе. Изменение температуры катализа-
тора определяется уравнением теплового баланса
У2Г + (/г/Х)г(С, 7’) = 0 (Ш.79)
которое должно решаться совместно с уравнениями (III.61), опреде-
ляющими концентрации реагирующих веществ в объеме пористого
зерна. Здесь % — эффективный коэффициент теплопроводности по-
ристого зерна и h — тепловой эффект реакции.
Сопротивление теплопередаче из ядра потока к внешней поверх-
ности катализатора, как будет показано в разделе III.5, оказывает
на скорость процесса гораздо большее влияние, чем внешнее со-
противление массопередаче. Мы пока не будем обсуждать этоТо
вопроса и зададим граничное условие для температуры на внешней
поверхности зерна в форме, не учитывающей внешнего теплового
сопротивления:
Т(Г) = ТОО (Ш.80)
Уравнение (III.79) и граничное условие (III.80) имеют ту же
форму, что и уравнение (III.61) и граничные условия (III.62), отли-
чаясь от последних только коэффициентами. Умножив уравнение
(III.79) на yjh и сложив его с уравнением (III.67), получим линейную
связь между концентрацией ключевого вещества и температурой:
(Z>/v)(C-Coo) = (x//0(2’-2’oo) (III-81)
Подставляя температуру, найденную из этого соотношения, в вы-
ражение для скорости реакции г (С, Т) и переходя к безразмерным
переменным, мы получаем, как и раньше, кинетическую функцию,
зависящую только от одной переменной с. Таким образом, и в неизо-
термическом случае задача расчета реакции на пористой частице
сводится к решению только уравнения (III.68). Однако учет изме-
нения температуры по толщине катализатора'может привести к ка-
чественному изменению характера решения. Напомним, что появле-
ние множественных режимов возможно, если в некотором интервале
концентраций кинетическая функция / (с) убывает с увеличением
концентрации ключевого вещества и соответственным изменением
всех других переменных, связанных с нею линейными соотношениями
(III.66), (III.81). Выражение (III.78) надо теперь дополнить слагае-
,, т hD dr '•
мым учитывающим зависимость скорости реакции от
температуры. Так как дг/дТ >0 и v < 0, указанное дополнительное
слагаемое отрицательно в экзотермическом и положительно — в эндо-
термическом процессе. Таким образам,' в случае экзотермической
реакции производная df/dc может при определенных условиях стать
отрицательной, что, в свою очередь, может вызвать появление
125
экстремумов у функции <р (cz) и, следовательно, возникновение мно-
жественных режимов процесса, переход между которыми будет про-
исходить скачкообразно при плавном изменении внешних условий.
Уравнение (III.68) или (III.70) не решается аналитически ни
в случае изотермической реакции с порядком, отличным от первого,
ни в случае неизотермической реакции (даже первого порядка).
Решение этого уравнения для зерен плоской и сферической формы
при различной кинетике процесса проводили численными методами
с помощью ЭЦВМ [23 ] *. Основной целью расчета является вычисле-
ние фактора эффективности как функции определяющих параметров
процесса.
В качестве примера приведем результаты расчета неизотермиче-
ской реакции первого порядка с аррениусовской температурной
зависимостью константы скорости реакции к. В этом случае уравне-
ние теплового баланса (III.79) удобно преобразовать к безразмерному
виду, введя безразмерную температуру 0 = Е (Т—T^/RT2^. Вы-
ражение для безразмерной скорости реакции принимает вид:
.. г (С, Т) к (Г) С Г Е ( 1 1 \]
/( )~'-(Соо’2’оо) Ч^ооИоо СеХР[/г(2’оо dj~
= С 6ХР 1+ (RT^/E) 9- СеЧ (И 1.82)
Здесь, как и в разделе Ш.З, использовано приближенное пре-
образование выражения для константы скорости химической реак-
ции, учитывающее тот факт, что в обычных химических процессах
RTт/Е <§( 1. Расчетные уравнения записываются теперь в виде
V2C_ 4rW' = 0; v20-|-0YW = O (1П. 83)
Граничные условия:
с(Г) = 1; 0(Г) = О (Ш.84)
Уравнения (III.83) содержат два безразмерных параметра: мо-
дуль Тиле Т и параметр максимального разогрева 0:
чг=г/Ч2’оо)/Ч e=hECmD/KRT>a3 (Ш.85)
Физический смысл параметра 0 ясен из линейного соотношения
между безразмерными концентрацией и температурой, которое легко
получить, умножив первое из уравнений (III.83) на 0 и сложив его
со вторым: '
0 = 0(1-с) (Ш.86)
Величина 0 очевидно, равна максимальной безразмерной темпе-
ратуре, достижимой внутри пористого зерна при полном выгора-
* Приближенное решение при малых отклонениях от кинетического режима
получено в работах [24]. В работе [25] учтено изменение объема реагирующей
смеси в результате реакции.
126
нии реагента. С учетом соотношения (III.86) задача решения урав-
нений (Ш.83) при условии (III.84) сводится к виду
у20-|-¥2(0-0)^ = 0; 0(Г) = О (Ш.87)
Различие между уравнениями (III.87) и (III.68) заключается в том,
что вместо концентрации ключевого вещества теперь в качестве ос-
новной переменной выступает безразмерная температура. Для зерна
плоской формы уравнение (III.87) можно, как и раньше, решить
Рис. II 1.7. График функции <р (0/)
[см. уравнение (111.87)].
Рис. III.8. Зависимость фактора эффектив-
ности т] от модуля Тиле ¥ для неизотерми-
ческой реакции первого порядка на пори-
стой пластине.
в квадратурах. При этом, аналогично (III.74) и (III.76), величина
фактора эффективности определяется соотношениями
1 I d0 I
П ~ | dx |^=±1
в
/2¥ = J
О
' 6z I"1/2
J ««дв-едйб! d& = <р (0Z)
.в
(Ш.88)
(III.89)
где 9г = 0(1 — сг) — безразмерная температура в центре пластины.
Вид функции <р (0J при различных значениях параметра макси-
мального разогрева 0 представлен на рис. III.7. Эта функция имеет
экстремумы при 0 >4,5. На рис. III.8 показана зависимость ц
от Чг при различных значениях 0. Аналогичный вид имеет зависи-
мость т] от V и 0 для зерен сферической формы. Зависимость между
значениями параметров Y и 0 в критических точках перехода между
режимами (для зерна плоской формы) показана’ на рис. III.9. Как
и на аналогичной диаграмме, характеризующей экзотермическую
реакцию на внешней поверхности зерна (см. рис. III.5), два устой-
чивых стационарных режима существуют в области, заключенной
между двумя ветвями критической кривой.
127
Характерно, что в экзотермических процессах наблюдаются»
значения фактора эффективности, превышающие единицу, причем!
при больших значениях параметра 0, когда возникают множествен-!
ные режимы, увеличение фактора эффективности за счет внутреннего!
разогрева зерна может быть весьма значительным. Такое увеличение!
фактора эффективности характерно только для необратимых реак-|
пий, так как в случае обратимой экзотермической реакции внутрен-|
Рис. Ш.9. Критические условия
скачкообразнрго перехода между
режимами для экзотермической
реакции первого порядка на по-
ристой пластине:
I — внутрикинетическая область; II —
внутридиффузионная область; III —
область множественных режимов.
Lo
нии разогрев приводит к смещению 1
равновесия в нежелательную сто- 1
рону. I
Скорость реакции во внутридиф- 1
фузионном режиме. Во внутридиф- |
фузионной области протекания реак-
ции задача вычисления фактора эф-
фективности значительно упроща-
ется, и зависимость его от основ-
ных параметров процесса может быть
получена в аналитической форме.
Действительно, если реакция необ-
ратима, то во внутридиффузионном
режиме концентрация ключевого (ли-
митирующего) вещества в центре
пластины близка к нулю; поэтому
формулу (III.74) можно переписать
в виде:
/2 Г С I 7'
. Пдиф = -йг- \f(c)dc (Ш-90)
Заметим, что это выражение пригодно не только для плоского
зерна, но и для зерна произвольной формы, поскольку во внутри-
диффузионном режиме реакция проходит практически до конца
в тонком слое, кривизна' которого несущественна. Если реакция
обратима, то состав смеси в центре пластины близок к равновесному.
'В этом случае величину cz можно определить с помощью равновесного
соотношения (III.46) между концентрациями реагентов и темпера-
турой и линейных соотношений (III.66), (III.81) между этими пере-
менными.
В качестве примера рассмотрим изотермическую реакцию и-го
порядка. В этом случае / (с) = сп и формула (III.90) дает
Соответственно, наблюдаемая скорость реакции равна
q = = K2W/(n-|-l) С(”+1)/2 (III.92)
Таким образом, наблюдаемый порядок реакции во внутридиф-
фузионном режиме равен среднему арифметическому между истин-
128
ным и первым. Если константа скорости реакции к зависит от тем-
пературы по уравнению Аррениуса, то наблюдаемая энергия акти-
вации, как и для реакции первого порядка (см, раздел III.2), равна
половине истинной.
Другой пример — изотермическая реакция с лангмюровской
кинетикой. В этом случае (см. раздел II.4)
kb±C± kb^Ci
Г l + + 1 [С2оо +(Z>2/-01) Cloo] + f*l— ^2 (Di/Dj)} Ci
(III.93)
где индекс 1 относится к исходному веществу и индекс 2 — к про-
дукту реакции.
Последнее равенство написано с учетом соотношения (III.66).
Переходя к безразмерным переменным, имеем
г(С1; С2)_________с_____
Т() '•(С'юо- С2оо) (1 + г)62 ’
ш =; 1 / г _ 7 Г________kbl_______Т7’.
V ^!ОО L^^ + Vloo+^oo) J ’
. ri + M^oo + W^)^] ГА
1 + Ъ1С1со + Ь2С2оо
и из формулы (III.90) получаем
14? 1/
[у—In (1 + V)] /2 (III.95)
Исследуем эту формулу в случае С2оо = 0. Вводя безразмерные
параметры
“1 = \61оо, “2 = i2c1oo(D2/7J1); ^=lVkh/D'i (HI-96)
имеем: ^*=4^*^= + 1 <р(у); (1+«г) /2 ф(^)=т^[у-1п(1+т)]1/2; y=?~“2 <ш-97) 1 Y । 1 -На2
В различных предельных случаях эту формулу можно упростить,
учитывая, что <р (у) = 1 при у = 0, ф (у) — 2/у при у > 1 и
9 Заказ 1441 129
q* 1
q* Y 2«i >> 1
q* « a, Y2 In а2/а2 > 1
q* (14-аЛ /21п а2/а2 <‘1
(III.98)
Ф (у) 1^2 In [1/(1 + у)] при у, близких к —1 (величины у < —1г
очевидно, невозможны):
I • • • 1» «1, 1» а2
II ... «1» а2, аг > 1
III . . . а2> а, > Va2 > 1
IV . . . а2 > а2 1
Первый случай соответствует слабой адсорбции как исходного, I
так и конечного продуктов, когда порядок реакции близок к пер- 1
вому. Как видно из соотношений (III.98), повышенные значения ч
фактора эффективности наблюдаются при сильной адсорбции исход- ]
ного вещества, а пониженные — при сильной адсорбции продукта
реакции. |
В рассмотренном выше случае неизотермической реакции пер- |
вого порядка выражение для фактора эффективности во внутридиф- *
фузионном режиме можно получить, учитывая, что безразмерная
температура в центре зерна 0г должна быть близка к величине без-
размерного максимального разогрева 0. После интегрирования фор-
мулы (III.88) получаем: "
Пдиф = -р- (ев-®-1)'/2 (Ш-99)
Согласно этой формуле, величина фактора эффективности во
внутридиффузионном режиме монотонно возрастает с увеличением
параметра 0. Зависимость фактора эффективности от модуля Тиле,
соответствующая формуле (III.99), изображается пунктирными пря-
мыми на рис. III.8. Эта зависимость практически совпадает с точ-
ным решением даже при Т ~ 1, причем оба решения — приближен-
ное и точное — тем ближе друг к другу, чем больше величина пара-
метра 0.
Ш.5. РАСЧЕТ ЭКЗОТЕРМИЧЕСКОЙ РЕАКЦИИ
С УЧЕТОМ ВНЕШНЕГО И ВНУТРЕННЕГО
ТЕПЛО- И МАССОПЕРЕНОСА
Исследуем теперь решение системы уравнений материального
и теплового балансов экзотермической реакции на пористом зерне
катализатора »
Dt v2Cz + vzr(C, Т) = 0 (III.100)
%y*T + hr(C, Т) = 0 (III.101)
при наиболее общих граничных условиях, учитывающих сопротив-
ление переносу вещества и тепла на внешней поверхности зерна Г
[26]:
-Dt (acj3n)r = ₽l-(Cz0-cZoo)
-7.(бГ/аге)г = а(Г0-Гоо)
130
(III.102)
(Ш.103)
Здесь С[ — концентрации веществ, участвующих в реакции; Т —
температура; г — скорость реакции в единице объема пористого
катализатора; % — эффективные коэффициенты диффузии и теп-
лопроводности в пористом зерне; — стехиометрический коэффи-
циент Z-ro вещества (vz <; 0 для исходных веществ и тг > 0 для про-
дуктов реакции); h — теплота реакции; V2 — оператор Лапласа;
С1о = С{ (Г), Та = Т (Г) — концентрации реагентов и температура
на внешней поверхности зерна; Сгоо, — значения соответству-
ющих переменных в ядре потока, омывающего частицу катализатора;
р(-, а — коэффициенты массо- и теплопередачи из ядра потока к внеш-
ней поверхности зерна; п — направление внешней нормали к по-
верхности Г.
Заметим, что 0, — DiJb и Dt = а£>1м(где Dia — коэффициент
молекулярной диффузии j-ro вещества, 6 — толщина диффузионного
пограничного слоя и а — коэффициент, зависящий от структуры
пористого катализатора). Величины 6 и а можно с достаточной сте-
пенью точности считать одинаковыми для всех веществ, участву-
ющих в реакции. Аналогично вышеизложенному (см. раздел III.4),
система уравнений (III.100) может быть сведена к ,единственному
уравнению для концентрации одного из реагирующих веществ,
которое принимают за ключевое. Введем с этой целью вспомогатель-
ную величину
«7/=(Я/М) Ci-iDjlvj) Cj (III.104)
Из уравнений (III.100) и граничных условий (III.102) следует,
что величина определяется решением задачи
у2и?;/=0; (dwtjldn)r = (aS}-^(w^—wf?) (III.105)
Очевидно, решение этой задачи имеет вид w{j = const = w^.
Из формулы (III.104) теперь следует, что концентрации всех ве-
ществ, участвующих в реакции, связаны между собой линейными
соотношениями
(DilVi) (Ci-Cia3) = (Di/vf) (C,-CIoo) (III.106)
и, таким образом, концентрации всех реагентов можно выразить
через концентрацию одного ключевого вещества. Уравнения и гра-
ничные условия,- определяющие концентрацию ключевого вещества
и температуру в пористой частице катализатора, удобно записать
в следующей безразмерной форме:
V2C_YJ/(c, 0)=О (III.107)
V20+ бо’Л!/ (с,0) = О (III.108)
сп (дс/дх)т = BiM (1 — сп) (III.109)
(Т*/Т*) (50/5z)r= BiT0n (III.НО)
где х — безразмерная координата (с масштабом Z), направленная
вдоль внешней нормали к поверхности, и BiM = fil/D, BiT = aZ/% —
диффузионное и тепловое числа Био.
9* 131
Выбор безразмерных переменных в уравнениях (III.107), (Ш.10&)Я
несколько необычен. За масштаб концентрации ключевого вещества?*®
внутри пористого зерна мы приняли концентрацию ключевого ве-®
щества на внешней поверхности зерна, так что с = С/Сй. Аналогично,®
безразмерная температура внутри зерна определяется как 0 = ®
= (Т — Т 0)/Т* (где Tq — масштаб внутренней температуры, за- Я
висящий от температуры внешней поверхности зерна). В соответ- Я
ствии с этим параметрыТо = I [г (Со, Т0)/DC0]1/г, &0= hCJDj%T^ Я
(параметр максимального разогрева), а также безразмерная скорость Я
реакции / (с, 0) = г (С, T)/r (С0, Т 0) зависят от значений переменных |
на внешней поверхности зерна. Поскольку эти значения неизвестны 1
заранее, величины Т0 и 0 0, как и другие параметры, которые могут 1
входить в выражение для / (с, 0), не являются характерными пара- ч
метрами рассматриваемого процесса. Преимущество такого выбора Я
безразмерных переменных заключается, однако, в том, что в даль- 1
нейшем можно воспользоваться результатами исследования хорошо Я
изученной задачи расчета экзотермической реакции на пористом и
зерне катализатора при фиксированных значениях концентрации
и температуры на внешней поверхности зерна. Нетрудно видеть,
что Т о и 0 0 являются характерными параметрами этой задачи. В гр а- I
ничных условиях (III.109), (III.НО) участвуют безразмерная кон-
центр ация ключевого вещества на внешней поверхности частицы I
сп = о/^оо и безразмерная температура внешней поверхности |
0П=(ТО — Тт)/Т*, масштабы которых связаны уже только со
значениями переменных в ядре потока. j
Пользуясь уравнениями (III.107), (III.108) и граничными уело- 1
виями (III.109), (III.НО), можно получить оценку условий, при ко-
торых существует заметный перепад концентрации и температуры
между поверхностью катализатора и внешней средой. Как было по-
казано в разделе III.2, внешнее сопротивление массопередаче на-
чинает сказываться только, когда реакция локализуется в тонком
слое, толщина которого сравнима с толщиной диффузионного по-
граничного слоя 6. Действительно, поскольку величина 4% является ?
мерой проникновения реакции в глубь пористого катализатора,
так что dc/dz ~ 4%, из граничного условия (III.109) следует: j
Так как всегда BiM 1 [см. уравнение (III.23)], внешний пере- *1
пад концентрации сравним с внутренним только при Чг0^> 1. |
Для оценки перепада температуры воспользуемся линейной |
связью между концентрацией и температурой внутри пористой ча- |
стицы, которая следует из уравнений (III.106), (III.107) i
e=0o(i—с) (ш.иг) \
откуда ;
—d0/dr = 6odc/dr—0ОЧГО (III.113) :
132
Принимая во внимание, что для газовой среды а = %м/6 (где %м —
' коэффициент молекулярной теплопроводности газа) и используя
граничное условие (III.110), находим:
= (III.114)
Так как перенос тепла в пористом катализаторе идет в основном
по твердой фазе, то %/%м 3> 1. Для сильно экзотермических реакций
величина 0О может принимать значения порядка нескольких единиц.
Поэтому величина 0П, характеризующая влияние внешнего сопро-
тивления теплоотводу, может быть существенной (0П й 1) при сравни-
тельно небольших значениях Т 0, когда внешнедиффузионное со-
противление еще не оказывает заметного влияния на процесс.
Учитывая, что тепловой поток на внешнюю поверхность катали-
затора —у^дТ/дп равен скорости тепловыделения в результате ре-
акции, граничное условие (III.103) или (III. 110) можно преобразо-
вать, введя фактор эффективности т), вычисленный для процесса
с фиксированными значениями концентрации и температуры на
внешней поверхности катализатора:
BiT9n=(7o*/7’*)0o4fgi1 = <p(en) (III.115)
Используя значения фактора эффективности т), найденные [(см.
раздел (III.4)] путем решения задачи с фиксированными С0, Т 0,
можно вычислить правую часть уравнения (III.115) <р (0П); после
этого значения внешнего разогрева 0П определяются как точки
пересечения графика функции <р (0П) с прямыми с наклоном BiT,
проведенными через начало координат.
Поскольку величины 4% и 0О зависят от поверхностных значе-
ний концентрации и температуры и потому не являются характер-
ными параметрами процесса, они должны быть выражены через
параметры Чг = I [г (С^, T^fDC^]1^, ^ — hC^D/yT*, отнесенные
к условиям в ядре потока. Установлено, что при не слишком боль-
ших внешних разогревах 0П Со «=; и, следовательно, (Т*/Т*) 0О «=#
0, 4% = Wa (0П) (где о (0П) — сильно возрастающая функция 0П).
В случае, когда зависимость г] (Ч^) неоднозначна (см. рис. III.8),
график зависимости правой части <р (0П) уравнения (III.115) от 0П
имеет форму, показанную на рис. III.10. При увеличении 0П функ-
ция <р (0П) резко возрастает за счет сильной зависимости Чг0 от 0П.
При дальнейшем увеличении 0П, когда начинает сказываться внешне-
диффузионное торможение процесса, функция ф (0П) проходит через
максимум и резко спадает до нуля. Решения уравнения (III.115)
определяются как точки пересечения кривой ф (0П) и прямой с на-
клоном BiT. Из рис. III.10 видно, что уравнение (III.115) может
иметь до пяти решений, причем при значениях числа BiT меньше
некоторых критических, решения, соответствующие малым внешним
разогревай, исчезают. При увеличении температуры потока, а сле-
довательно, параметра Т кривая ф (0П) сдвигается вверх, и при
значениях Чг, больших некоторых критических, также исчезают
133
Рис. II 1.10. Графическое решение
уравнения (III.115).
низкотемпературные решения. Участку кривой ср (0П) слева от точки 1
соответствует внутрикинетический, а между точками 2 и 3 — вну-
тридиффузионный режим протекания реакции. В задаче с фикси-
рованными значениями поверхностных концентраций и температуры
внутрикинетический режим существует левее точки 4, а внутридиф-
фузионный — правее точки 5. Наличие сопротивления теплоотводу
на внешней поверхности зерна приводит к расширению области
промежуточных неустойчивых состоя-
ний. Кроме того, при наличии внеш-
него сопротивления теплоотводу
множественные режимы возникают
и в том случае, когда зависимость
Я о) однозначна.
Реакция первого порядка. Рас-
смотрим более подробно необрати-
мую экзотермическую реакцию пер-
вого порядка на пористом катализа-
торе, имеющем форму пластины
толщиной 21 с «запечатанными» боко-
выми гранями. Это простейший при-
мер, на котором видны все харак-
терные особенности рассматриваемой
задачи. Пусть г (С, Т) = Ze~E,RTC.
Если принять за масштаб температуры Т величину = ИТЦЕ, то
/(с, 0)=Сехр^0 +—(RT0!E<d) (III.116)
4%= I Vk (To)/D, e0=hEC0D/xRTl
Величины Tq и 0O связаны с характерными параметрами задачи
Т = Z ]/ A (Toaj/D, & = hECcoD/xRT2co следующими соотношениями,
вытекающими из формул (III.109), (III.110)
Ч^Ч'Л/2 (III.117)
0о = (Гоо/То)2®-Оп/т (III.118)
где m = yfi/Da > 1.
При записи параметра 0 за масштаб внешней температуры при-
нята величина Т* = RT2OJE. При не слишком больших 0П (0П 1)
величины Tq и Т* практически совпадают и, так как m 1, 0О 0.
Заметим, что из формулы (III. 118) следует существование макси-
мально возможного разогрева 0П, макс = ®п —
Используя соотношение (III.112) между концентрацией и темпе-
ратурой внутри катализатора, сведем систему (III.107), (III.108)
к единственному уравнению относительно безразмерной темпера-
туры 0
d20/dz2-|-,Fo (0о-0)ее = 6о (III.119)
где х — безразмерная координата, принимающая значения х — 0
в центре пластины и х = ±1 — на ее поверхности.
134
Переходя к интегрированию уравнения (III.119), будем считать
величину 0П, а следовательно, и значения параметров IF 0 и <90 из-
вестными. Далее можно найти величину 0Л, подставляя полученное
решение в граничное условие (III.НО). Таким образом, будем решать
уравнение (III.119) с граничными условиями:
(йо/^)Хс0 = о, е(±1)=о (ш.120)
Метод решения задачи (Ш.119), (III.120) изложен в . разделе
III.4, ее решение выражается формулами (III.88) и (III.89):
г0/ Ч*/«
/2 f й
(е°-0М
Lo
ez Гб/
/2 'Fo = j M j (0o-0i) e91 dOi
о Le
(III.121)
(III.122)
Используя формулы (III.121) и (III.117), записываем граничное
условие (III.115) в виде
М = У2
Л/2
0п
-б/ -I1/»
J (00-0) е9</0
-о
(III.123)
где
jf=BiT/T = (a/X) VDfk(T^) (III.124)
безразмерный параметр теплопередачи.
Исследуем сначала область 0Л 0П — 1, в которой можно пре-
небречь внешнедиффузионным торможением. Тогда 0О 0 и задача
сводится к совместному решению уравнений (III.123) и (III.122)
относительно неизвестных величин 0Z и 0Л.
В двух предельных случаях из этой системы уравнений можно
исключить величину 0Z. В кинетической области, считая 0Z @в,
получим из формулы (III.122):
К2 ¥А/2=2К0//0О . (Ш.125)
Подставляя 0Z, найденное из уравнения (III.125), в (III.123)
получим в том же приближении:
Л! = Чг0оееп/0п (III.126)
Во внутридиффузионной области можно положить в уравнении
(III.123) 0Z «з @о- При этом получаем:
1И = —к—V2 (ге° —0О-1) (III-127)
vn
Правая часть формулы (III.126) имеет минимум, равный еТво,
при 0П = 1, а минимум выражения (III.127) достигается при 0Л = 2.
Из выражения (III. 127) следует, что во внутридиффузионном режиме
135
правая часть уравнения (III. 123) не зависит от параметра Т и, сле-1
довательно, значение 0П в этой области определяется только пара- ?*
метрами М и 0О. • |
Зависимость 0П (М) при известных значениях ¥ и 60 может быть
построена с помощью графика функции ф (0Z) (см. рис. III.7). Дей-
ствительно, задавая значение 0Z, можно по графику определить
Чг0 = Чгееп/2 и, подставляя полученные значения 0Н и 0Z в формулу
(III.123), построить зависимость 0П (М).
Рис. III.11. Зависимость внешнего ра-
зогрева 0П от параметра М-.
0 = 5; ту = 0,37(a); 0,30(6); 0.20(e).
Рис. III.12. Зависимость внешнего
разогрева 0П от параметра М:
0 = 4(a—в); iF= 0.47(a), 0,45(6), 0.40(e);
0 = 3(г,6); 4f= 0,50(г), 0,45(6); $
0 = 2; У = 0.53(e).
На рис. III.11 и III.12 приведен ход кривых 0П (7И) в области
0П ~ 1 Для нескольких значений 0 и Т. Пересекая полученные кри-
вые прямыми М = const, можно найти соответствующие этим зна-
чениям параметра теплопередачи безразмерные перепады темпера-
туры между поверхностью катализатора и внешним потоком. Обра- •
тимся к изучению зависимости 0П (М) при различных комбинациях ;
параметров 0 иТ. Представленное на рис. III.11 семейство кривых !
соответствует 0 = 5 и нескольким значениям Т. В этом случае, 7
как уже указывалось, зависимость 0Z’ от Vq = Те6^2 может быть »
неоднозначной, поэтому кривая М (0П) тоже может быть неодно-
значной в некоторой области изменения 0П. Границы области неодно-
значности кривой М (0П), отмеченные точками 4 и 5 на рис. III.11,
в которых ) dM]dQn | = оо, соответствуют точкам максимума и ми-
нимума функции ф (0Z). Указанная неоднозначность М (0П) при
0 > 4,5 проявляется в том случае, когда Т < Чгмакс, где 1/2Чгмакс
значение ф (0Z) в максимуме (см. рис. III.7). При этом точкам 4 на
рис. III.11 соответствуют 0П = 2 In (Чгмакс/Чг), а точкам 5 0П =
136
= 2 In (Ч'мии/Т) (где у 2ТНИИ — значение <p (9Z) в минимуме).
Если Т < Ч'мин, т0 на графике М (0П) присутствуют точки типа 5
и 4; при Тмнн < V < Чумаке точки типа 5 отсутствуют, и при
щ ^>Ч^макс зависимость М (0П) является однозначной.
Кривые М (0П), соответствующие разным Т, могут иметь в об-
ласти 0П— 1 минимумы (точки 1 и 3) и максимумы (точки 2) и вы-
ходят во внутридиффузионной области на единую асимптотическую
кривую, определяемую формулой (III.127). Наличие на кривой М (0П)
экстремумов означает существование в рассматриваемой задаче
множественных режимов, причем значения 0П, при которых dM)dQr, —
— О (точки 1—3 на рис. III.11 и III.12), разделяют чередующиеся
области устойчивых и неустойчивых режимов. Заметим, что точки 4
и 5 (соответствующие значениям Т 0, при которых в задаче с задан-
ными значениями поверхностных концентраций С 0 и температуры Т 0
происходил перескок между режимами) попадают в область неустой-
чивых режимов, разделяющих внутрикинетическую и внутри-
диффузионную области. Таким образом, наличие сопротивления
теплоотводу на внешней поверхности катализатора приводит к рас-
ширению области неустойчивых режимов. Это проявляется также в
возникновении неустойчивой области при 0 < 4,5, когда задача без
внешнего сопротивления вообще не имеет неустойчивых режимов.
Скачкообразные переходы во внутренних режимах могут наблю-
даться, как это видно из рис. III.12, до 0 = 2.
При значениях параметра М, меньших абсолютного минимума
функции М (0П) (этот минимум может соответствовать точкам 1
и 3, см. рис. III.11, III.12), в рассматриваемой области 0 < 0П 2
режимов с небольшими внешними разогревами не существует и про-
цесс переходит во внешнедиффузионный режим (0П — 0П). Во внешне-
диффузионном режиме существенным становится перепад концен-
трации у поверхности катализатора, и поэтому для получения зави-
симости М (0П) в этой области значений 0П необходимо учитывать
связь 0О с 0П, определяемую формулой (III.118). Поскольку при
этом реакция локализована в узком слое бдиз поверхности катали-
затора из-за большого внутридиффузионного торможения, ход кри-
вой М (0П) во внешнедиффузионной области можно получить из
формулы (III.127) с учетом зависимости (III.118). При больших 0П
можно также заменить приближенную кинетическую функцию ее^2
на exp 0п/2 (1 + 60п) (где Ъ — RT^IE) и учесть различие между
величинами T*t и Т*.
При увеличении 0П от значений порядка единицы до 0П ~ 0П М(0П)
проходит через максимум, высота которого тем больше, чем больше 0П
или т, а при 0П-*0П функция М (0„) спадает до нуля. При умень-
шении т максимум М (0П) снижается, и, начиная с некоторых зна-
чений т, максимум исчезает и переход во внешнедиффузионную об-
ласть при изменении параметра М происходит плавно.
Таким образом, исследование зависимости 0П (7И) показывает,
что при заданнрм значении параметра М в рассматриваемой системе
137
возможны одно, три или пять соответствующих ему значений 0П
и соответственно одно, три или пять стационарных решений. Этим
решениям отвечают внутрикинетический режим I, внутридиффузион-
ный II и внешнедиффузионный III режимы и чередующиеся с ними
два промежуточных неустойчивых режима. Выпишем возможные
режимы процесса при 0 >4,5 и m > 1, а также переходы между
ними при изменении гидродинамических условий:
Уменьшение М Увеличение М
Случай 1
» 2
» 3
¥>¥
макс
макс
II —> III
¥МЙН>¥
а) I----> II----> III
б) I----> III
а) I----> II----> III
б) I----> III
III----> II *
III----► II
III----> II
III----> I
III----> I**
¥
Характер переходов между режимами зависит от того, попа-
дает ли величина Т в интервал Тмин < Т < Ч\,акс, где задача с фик-
сированными значениями Со, То имеет множественные решения.
Возможные режимы при 4,5 > 0 >2 таковы же, как и в случае 3.
Для исследования переходов между режимами, связанных с из-
менением температуры потока и, следовательно, изменением пара-
метра Т, удобно пользоваться кривыми, изображающими зависимость
параметра Т от значения температуры в центре катализатора 0Z
при фиксированных прочих параметрах задачи BiT, 0, m.
Рассмотрим область небольших внешних разогревов 0п S 2,
где несущественно внешнедиффузионное сопротивление и 0О^ 0.
Из уравнения (III. 122) получаем:
КГчгееп/2;--ф(0/) (III.128)
Подставляя это соотношение в формулу (III.123), определим 0П
через температуру в центре катализатора 0Z и параметры BiT, 0О, Т:
-6/ i'/<
J (0О - 0) е0 dO
-0
n Iffy)
0П~ BiT
(III.129)
Выразив в уравнении (III.122) Чг0 через Т и воспользовавшись
соотношением (III.129), находим искомую связь между 0Z и Т:
К2 ¥ = <р (0/) ехр
ф (0г)
2BiT
-0/
J (00-6)
-о
(III.130)
Из формулы (III.130) следует, что при отсутствии сопротивления
теплопередаче на поверхности катализатора (BiT—> оо) зависимость Т
от 0г совпадает с (III. 122) и имеет вид, изображенный на рис. III.7.
При наличии внешнего теплового сопротивления начинает сказы-
ваться экспоненциальный фактор в формуле (III.130) и кривая T (0Z)
* В случае 1 кинетический режим I отсутствует.
** В случае 36 на внутридиффузионный режим II невозможно попасть
при изменении одного параметра М.
138
проходит ниже соответствующей кривой прй BiT = оо. Во цнутри-
диффузионной области, где Т (0/) принимает большие значения,
функция T (0Z) проходит через максимум, после чего спадает до
малых значений (см. рис. III.13). Значение функции в максимуме
нетрудно определить, если учесть, что этот максимум находится
в области значений 0Z, весьма близких к 0О. При этом с высокой точ-
ностью можно считать 0Z = 0О под знаком радикала в уравнении'
(П1Л30): 1(0) I
}Л2 Т = <р (0Z) exp] - [А-00-И1/2 ) (Ш.131)
Тогда для значения Т (0Z) в максимуме
получаем соотношение:
<р (0z) = 2BiT (ее° -0О- 1)“‘/г (III.132)
Величина Т в максимуме может быть
получена подстановкой соотношения
(III.132) в (III.131):
/2 Ч\,акс=-^~- (ее°-0о-1)-‘/2 (III.133)
Чтобы выяснить дальнейший ход кри-
вой Т (0Z) в области, соответствующей
значительным внешним перепадам темпе-
ратуры со, необходимо учесть, что макси-
мально возможный внутренний разогрев
0о с ростом 0П падает, и согласно фор-
муле (III.118), 0о -> 0 при 0П-> 0П- При
зтих значениях 0П 0Z^0O и, пользуясь
соотношениями (III.131) и (III.118),
можно найти следующую зависимость
V (0z):
У2 4r = mBiT(0-0z) (eВ 9Z-0z-1)“‘/2
Рис. III.13. Зависимость
разогрева в центре пласти-
ны от модуля Тиле:
- 0=5, BiT = со(а); 20(6),
10(e). Стрелки указывают на-
правление скачкообразных пе-
реходов между режимами.
т
exp~2-(0z-0) (III.134)
В случае, когда величиной Ь0П нельзя пренебречь по сравнению
с единицей, показатель экспоненты в формуле (III.134) следует
заменить на т (0Z — 0)/2 [1 + Ът (0 — 0Z)]. Из формулы (III.134)
следует, что при т 1 функция T (0Z) резко спадает до весьма ма-
лых значений при уменьшении 0Z и, переходя через минимум, под-
нимается вдоль оси' ординат при 0Z 1.
Таким образом, задавая значения параметра Т, можно определить
стационарные температуры в центре катализатора, пересекая гра-
фик Т (0Z) прямыми Т = const. На рис. III.13 изображена зависи-
мость Т от 0Z при 0 = 5 и BiT = oo, 20 и 10. При различных зна-
чениях параметра Y может существовать одно, три или пять соот-
ветствующих ему значений 0Z и соответственно одно, три или пять
стационарных решений, которым отвечают внутрикинетический
139
режим I, внутридиффузионный II и внешнедиффузионный III pi
жимы и два промежуточных неустойчивых решения.
* Переходы между режимами при изменении параметра Т (измене-
нии внешней температуры) зависят от относительной высоты двух
максимумов на кривой (0Z). Для каждого 0 > 4,5 существует
такое значение параметра BiT = Bi?, что при BiT < Bi* значение
функции в правом максимуме, которое линейно зависит от BiT,
станет меньше значения функции в левом максимуме (см. рис. III.13).
Оценку этого значения BiT можно получить, считая, что вследствие
слабого влияния экспоненциального фактора в формуле (III. 130)
в области левого максимума, значение Т в нем не зависит от BiT
и равно Тогда, используя (III.133), получаем:
1 Г~Г---------
Bi? е |/ у (ев-0- 1) (III-135)
Возможны следующие схемы переходов между стационарными
режимами процесса при изменении температуры среды и, следова-
тельно, параметра
Увеличение W Уменьшение W
Случай 1 . . . BiT>Bi? I--> II--> III III--> I
» 2 . . . BiT<Bi? II-----> III III----> I
Интересной особенностью переходов между режимами в рассмо-
тренной системе, является то, что при некоторых условиях внутри-
диффузионный режим, хотя и существует, но не проявляется ни при
увеличении, ни при уменьшении температуры и скорости потока.
III.6. СЕЛЕКТИВНОСТЬ СЛОЖНЫХ РЕАКЦИЙ
И ОТРАВЛЕНИЕ КАТАЛИЗАТОРА
ПРИ ДИФФУЗИОННОМ ТОРМОЖЕНИИ ПРОЦЕССА
Последовательные реакции. При обсуждении особенностей диф-
фузионной кинетики сложных процессов следует прежде всего под-
черкнуть, что понятие области протекания реакции имеет смысл
применительно к каждой отдельной реакции, но не к процессу в це-
лом. Действительно, один и тот же процесс может включать как мед-
ленные, так и быстрые реакции, которые при одних и тех же условиях»
могут протекать в различных областях — диффузионных или кине-
тических. Одной из главных характеристик процесса, состоящего
из нескольких одновременно протекающих реакций является его
селективность (избирательность), т. е. отношение скорости обра-
зования целевого продукта к скорости расходования исходного
вещества. На избирательность процессов, включающих последо-
вательные реакции, определяющее влияние оказывает соотношение
скоростей диффузии и дальнейшего превращения промежуточных
140
продуктов. Это может быть проиллюстрировано на примере двух
последовательных мономолекулярных реакций
Ai —Аг — * А3
где А 2 — целевой продукт.'
Пусть процесс протекает на внешней поверхности частицы и
константы скоростей обеих реакций равны соответственно и х2.
Тогда скорости образования веществ А2 и А3 (соответственно q2
и </з) определяются формулами:
52 = 02 (Сг— С2оо); 5з='СгСг (III.136)
Отношение дг/Чз определяет избирательность процесса, которая
равна gzK^z + <?з)- Очень простую характеристику — локальную
селективность — получаем, принимая концентрацию вещества А2
в ядре потока С2оо равной нулю. Используя выражение (III.136),
находим:
5г/53 = 02/Х2 (III.137)
Таким образом, локальная селективность процесса определяется
только отношением константы скорости диффузии целевого продукта
и константы скорости его дальнейшего превращения и тем выше,
чем больше отношение 02/х2. При переходе реакции А2->А3 во
внешнедиффузионную область (х 2 ~2> Р г) целевой продукт вовсе
не образуется.,
Рассмотрим теперь те же две последовательные реакции первого
порядка, протекающие внутри пористой частицы. Распределение
безразмерной концентрации вещества (ci= С JC юо) в зерне плоской
формы описывается формулой (III.26); оно зависит только от вели-
чины модуля Тиле для первой реакции Т = I kJD Уравнение,
определяющее концентрацию вещества А2, можно записать в без-
размерном виде
Аг/^2—у2^2С2 + хрг ch (’P.z)/ch’P = О
(Ш.138)
Т>2
где сг =
Решение
Cloo’ 7 V -кГ’-D^-
этого уравнения дает:
ch (уЧ^) . 1 /ch (уЧ^г) ch (Ч'я) \
С2 - Th (у¥) + 1-у2 \ chyT chT )
(III.139)
где£= (£>2/£>!) (С 2 со /С1оо).
Отсюда легко найти поток вещества А 2 на внешнюю поверхность
пористой частицы:
g2 = jgi^i°° =С2ооГйО2 th (у¥)-С1ООГ^ОГ (YY)
(III.140)
141
Положив в уравнении (III. 140) С2со = 0 и разделив величину —qg
на поток исходного вещества
<71=Cjoo th Y
(HI.141)
получаем следующую формулу для локальной селективности про-
цесса:
s =____ / dc-2 \ ,/ dcr \ 1 — у th (yTj/th ¥ .
qi \ dz /х=1'\ dz )x=l 1 —у2
(III. 142)
Рис. 111,14. Зависимость локальной се-
лективности s от параметра у для после-
довательных реакций первого порядка.
Зависимость s (у) при различных значениях Чг показана на
рис. III. 14. Исследуем эту зависимость в различных предельных
случаях. Если обе реакции протекают во внутрикинетическом ре-
жиме (Чг 1, уЧ’’ С 1), то
th 4f 4f, th (уЧ^) уЧг и s ^1.
Если основная реакция про-
текает во внутридиффузионном
режиме, а побочная — во
внутрикинетическом (Чг )§> 1, но
у¥ < 1), то s~(l- у2Чг):
: (1 — у2), и так как в этих
условиях у 1, селективность
процесса по-прежнему близка
к единице. Если обе реакции
протекают во внутридиффузион-
ном режиме (Ч*1 1, уЧ*1 )>> 1,
th Ч*1 th (уЧ*1) 1), то s =
= (1+?)'1 и селективность про-
цесса зависит от величины у.
Следует особо отметить, что в отличие от рассмотренного выше
процесса на внешней поверхности частицы, селективность процесса
в порах катализатора может оставаться значительной и после пере-
хода обеих реакций в диффузионный режим. Параметр у, определяю-
щий локальную селективность процесса, имеет смыЬл отношения глу-
бины проникновения первой реакции в толщу пористого катализа-
тора, VD i/ki к глубине проникновения второй реакции V £> 2/к 2-
Селективность процесса падает с увеличением глубины проникно-
вения в пористую частицу реакции Aj -► А2, так как при этом удли-
няется путь, который необходимо пройти образовавшимся молеку-
лам целевого продукта, чтобы вырваться из пор катализатора, не
вступив в реакцию дальнейшего превращения. Чем больше глубина
проникновения в пористую частицу реакции А2 -> А3, т. е. чем меньше
ее скорость и быстрее диффундирует вещество А2, тем меньшее коли-
чество целевого продукта вступает в последовательную реакцию
и тем, следовательно, выше локальная селективность процесса.
При переходе обеих реакций во внутридиффузионный режим ло-
кальная селективность процесса остается при малых у близкой
к единице. При этом замедление диффузии исходного вещества при
прочих равных условиях приводит к повышению селективности.
142
Если рассматриваемый процесс проводится в изотермическом
реакторе идеального вытеснения, концентрации реагентов в потоке
Cica изменяются по длине реактора согласно уравнениям
^£^ = гГ(С1Оо, С2оо) (j = l, 2) (Ш.143)
где г* — скорость образования i-ro вещества в единице объема ре-
актора. Эта величина связана с потоком вещества на внешнюю по-
верхность пористого зерна соотношением г* = (где о — пло-
щадь внешней поверхности зерен в единице объема слоя). Исполь-
зуя выражения для потоков (III.140) и (III.141), находим, что связь
между безразмерными концентрациями веществ А( и А2 в потоке
cin=Cloo/C*, с2п = С2т/С* (где С* — концентрация исходного
вещества на входе в реактор) определяется уравнением
^С2п _£2П . бу th (уТ) _ 1—У th (уТ)/th V
^1П с1п' th Т 1 у2
(III.144)
где б = D z/D р
Решение уравнения (III. 144), соответствующее начальному усло-
вию с2п = 0 при с1п — 1, имеет вид:
1-у th (y¥)/th Y eft ‘MvW)/th У_С1п
1-у2 ' l-6yth(y4f)/th4f
(III.145)
Исходя из этой формулы, можно найти и полную селективность
процесса s* = с2п/(1 — с1п) как функцию от доли непрореагиро-
вавшего исходного вещества Cj . Выпишем выражение для селектив-
ности в предельных случаях, когда обе реакции протекают во вну-
трикинетическом (Чг 1, уТ 1) или внутридиффузионном (Т Э> 1,
уф' 1) режиме
Л*'1—1
* _ С1П С1П 1
кин 1-С1П ‘ 1—у*
(III.146)
6v-l A А
* С1П С1П —1 . . С1П _______С1П____—1________
ДИф 1-С1П ' (1 + у) (1-бу) 1-С1П ’ (1 + К776) (1-/^6)
(Ш-147)
где у* = бу2 = A2/Ai.
В кинетическом режиме селективность, естественно, не зависит
от эффективных коэффициентов диффузии обоих веществ и опреде-
ляется только отношением констант скорости обеих реакций у*.
Селективность процесса во внутридиффузионном режиме при 6 = 1
равна:.
*дИф= 1П /Чин (III.148)
cfn — 1
Исследование формулы (III.148) показывает, что селективность
процесса в диффузионном режиме всегда меньше, чем в кинетическом,
причем отношение .УдифДкин уменьшается с понижением доли
143
непрореагировавшего исходного вещества с1п при у* < 1 и увеличи-
вается — при у* > 1 (рис. III.15, а). С уменьшением 6 селектив-
ность процесса во внутридиффузионном режиме падает при малых
степенях превращения и возрастает — при больших (рис. III.15, б).
Селективность при параллельных реакциях. Совсем иной характер
носит влияние диффузионного торможения на процесс, включа-
ющий параллельные реакции. В этом случае и в диффузионных об-
ластях истинная химическая кинетика реакций на поверхности мо-
жет остаться единственным фактором, определяющим селективность
я , 6
Рис. III.15. Зависимость полной селективности s * от
степени превращения 1—с± для последовательных реакций
первого порядка:
а — при в = 1 и различных значениях V*; б — при V* = 1 и различ-
ных значениях 6. I — внутрикинетический режим; II — внутридиф-
фузионный режим.
процесса. Если истинный порядок параллельных реакций один и
тот же, относительные количества образующихся продуктов никак
не зависят от диффузионных факторов и определяются только от-
ношением констант скорости параллельных реакций при данной тем-
пературе. В случае параллельных реакций разного порядка при пе-
реходе в одну из диффузионных областей, вследствие уменьшения
концентрации исходного вещества у активной поверхности, относи-
тельные скорости реакций меняются в пользу реакции, протекающей
по меньшему порядку. Этот эффект, очевидно, выражен более резко
во внешнедиффузионной области, чем во внутридиффузионной.
Рассмотрим в качестве примера две параллельные реакции
A t А 2, 2А t А3, протекающие во внутридиффузионном режиме.
Распределение концентрации С вещества At внутри зерна опреде-
ляется из уравнения
Dd^C/dX^ = k1C + 2kiC^ (III. 149)
где kit к2 — константы скорости реакций первого и второго порядка.
144
Введем безразмерные переменные с = С'/С00, | = X^fk^D я
параметр у = 4^k2Cca/ki, определяющий селективность процесса.
Тогда уравнение (III.149) примет вид:
, , 3
l + yyc
(Ш.150)
Будем считать, что ось £ направлена от внешней поверхности
в глубь катализатора. На внешней поверхности зерна задано гра-
ничное условие с (0)= 1. Второе граничное условие при протека-
нии реакции во внутридиффузионном режиме можно задать в форме:
с->0 при оо. Вводя вспомогательную переменную р = dc/dt,,
имеем:
4й
(p2)/dc = c (1 -ру ус^
(III.151)
Интегрирование этого уравнения с учетом граничного условия:
р — 0 при с = 0 (т. е. при >оо), — даеТ:
dc/dg= -с /Г+^Г (III.152)
После повторного интегрирования находим распределение кон-
центрации исходного вещества с (|):
с = 4{(2 + у+2/ГиГ)еЕ-2у+ (2+у-2 /1 + у) е’Ч’1 (Ш.153)
Долю исходного вещества, превратившуюся в продукт реакции А 2,
или селективность процесса, определяют теперь по формуле:
Селективность процесса во внутридиффузионном режиме, опре-
деленная по формуле (III.154), всегда выше, чем селективность того
же процесса в кинетическом режиме:
«кнн = *1Соо/(*1Ссо +2*2^) = (1 + ~ у)’1 (III.155)
Обе функции показаны на рис. III.16. Еще большая селективность
достигается во внешнедиффузионном режиме, где, вследствие ма-
лости концентрации исходного вещества у активной поверхности,
практически все исходное вещество будет вступать в реакцию пер-
вого порядка А1-^-А2.
Аналогично, в случае параллельных реакций типа А4->-А2,
А, + А3->А4 выход вещества А2 возрастает по мере перехода
в диффузионный режим, причем тем сильнее, чем медленней диф-
фундирует вещество А3.
Ю Заказ 144 1
145
(111.156}
Рис. III.16. Селективность парал-
лельных реакций разного порядка
в кинетическом |И внутридиффу-
зионном режиме.
Неизотермичность пористого зерна может приводить к сильно
изменению селективности процесса, благодаря различию энерг
активаций основной и побочной реакций [27 ]. Этот эффект окажете!
значительным для процессов, включающих как последовательные:
так и параллельные реакции. Часто побочные реакции идут при оп
тимальной температуре процесса с малой скоростью, обладая боль4
шой энергией активации. Разогрев катализатора в диффузионно^
режиме ускоряет такие реакции сильнее, чем основную реакцию
ведущую к образованию целевого продукта, вследствие чего селек
тивность процесса резко падает.
Отравление активной поверхности. Реакция адсорбции яда-
протекающая при отравлении поверхности катализатора (см. главу I),
представляет собой фактически до-
полнительную реакцию, конкури-
рующую с основной реакцией син-
теза целевого продукта. Адсорбция
яда на поверхности пористого ката- ?
лизатора, как и всякая реакция,
тормозится диффузией реагента в по-
рах, и характер отравления поверх-
ности будет зависеть от того, проте-
кает ли реакция отравления в кине-
тической или диффузионной области.
При анализе кинетики процесса
на отравляющемся катализаторе воз- -
никают особые трудности, связанные
с тем, что показатели такого процесса ,
изменяются со временем по мере на- •
копления адсорбированного катали-
тического яда на активной поверх-
ности. В случае, когда яд поступает
в зерно катализатора из потока реагентов, его концентрация в
порах катализатора Ся и количество адсорбированного яда, отнесен-
ное к единице активной поверхности, <о определяются уравнениями:'
Af1
со, с, ту,
~ = ря(Ся,<г,С, Т)
Здесь е — доля свободного объема пористой частицы; £>я — эффек-
тивный коэффициент диффузии яда; о — площадь активной поверх-';
ности в единице объема зерна; ря — скорость адсорбции яда,
отнесенная к единице активной поверхности.
Величина ря в общем случае зависит от концентрации свобод-
ного и адсорбированного яда, состава реагирующей смеси в дан-
ной точке С и температуры Т.
146
И?. Скорость основной реакции будет зависеть от количества адсор-»
ВТ бированного яда; поэтому при расчете процесса необходимо решать
В- уравнения (III.156) совместно с уравнениями материального баланса
В’ реагентов и баланса тепла. Вследствие постепенного накопления
В яда наблюдаемая скорость процесса будет уменьшаться со време-
F' нем вплоть до достижения равновесной степени отравления или пол-
ной потери активности катализатора.
F Когда адсорбция яда протекает медленно — его диффузия в по-
ь pax не является лимитирующим фактором — концентрация яда Ся
- станет постоянной по всему объему зерна. В этом случае отравление
1 поверхности будет практически равномерным (различия локальных
значений <о, появляющиеся вследствие изменения концентраций
реагирующих веществ по глубине зерна, как правило, не играют
заметной роли). Отравление такого типа равносильно уменьшению
эффективной константы скорости к основной реакции. Так как ско-
рость реакции во внутридиффузионном режиме пропорциональна к !г
(и, соответственно, фактор эффективности т)—к~ /г), равномерное
отравление снижает скорость реакции во внутридиффузионном
режиме слабее, чем в кинетическом. Аналогичным образом воздей-
ствует на скорость основной реакции и обратимое отравление актив-
ной поверхности. В обоих случаях основная часть яда сорбируется
во внутренних областях зерна, которые при переходе во внутридиф-
фузионный режим становятся практически недоступными для ре-
агентов.
Совсем другой характер носит отравление катализатора при бы-
строй необратимой адсорбции яда. В этом случае реакция отравле-
ния протекает в диффузионном режиме, и яд сначала оседает в устьях
пор. По мере отравления участков активной поверхности, близких
к внешней поверхности частицы, яд начинает сорбироваться на
более труднодоступных участках, и, таким образом, область отрав-
ленного катализатора расширяется в глубь пористой частицы.
Рассмотрим процесс отравления плоского зерна катализатора,
считая, что максимальное количество яда, которое может адсорби-
роваться на единице поверхности катализатора, равно соо и что уча-
сток поверхности, адсорбировавший такое количество яда, стано-
вится полностью неактивным. Если адсорбция яда протекает необ-
ратимо и мгновенно, скорость отравления катализатора определяется
скоростью диффузии яда в порах. При этом скорость адсорбции яда
в момент, когда уже отравлен слой катализатора толщиной 6, равна
dQldt = DnC^b | (III.157)
где Ся — концентрация яда в потоке.
Здесь не учитывается сопротивление массопередаче из ядра по-
тока к внешней поверхности катализатора, играющее заметную роль
только на начальной стадии процесса.
Количество адсорбированного яда на единицу внешней по-
верхности зерен Q, соответствующее полному отравлению слоя
10* — 147
толщиной 6, равно бсгсо 0- Учитывая это соотношение, получаем из
(III.157) уравнение, определяющее скорость перемещения границы
отравленной зоны
где ц = £>яСя/о<о0.
Интегрирование этого уравнения с начальным условием 6 (0) = О
дает:
(Ш.159)
Скорость основной реакции отлична от нуля только в неотравлен-
ной зоне, т. е. на глубине, превышающей 6.'Легко видеть, что отрав-
ление катализатора в рассматриваемом случае снижает ско-
Рис. III. 17. Зависимость скорости реак-
ции на отравляющемся катализаторе
от безразмерного времени т.
рость реакции не только за
счет уменьшения площади ак-
тивной поверхности, но и
вследствие того, что молекулам
реагентов, прежде чем достиг-
нуть активной поверхности, не-
обходимо пройти отравленную
зону, создающую дополнитель-
ное диффузионное сопротив-
ление.
Скорость реакции можно
определить, решая обычные
уравнения баланса реагентов
в порах неотравленной зоны;
для реакции первого порядка
зто будет уравнение (III.19)
или (III.21) с граничным .ус-
ловием
Р/6) (Сео - С) = D (dC/dX)^^
(III.160)
заданным на границе неотравленной зоны. В простейшем случае изо-
термической реакции первого порядка безразмерная концентрация
исходного вещества в неотравленной зоне с = С/Сао равна
/т Ysh [Т(1-/т)14-сЬ[’Р(1-/т)1
где х = ХЦ — безразмерная координата; У — I ]/k/D — модуль
Тиле; т = 2£>яСяг/<тсо 0Z2 — безразмерное время.
При записи формулы (III.161) учтена зависимость толщины отрав-
ленной зоны от времени (Ш.159). Отношение наблюдаемой скорости
реакции к скорости реакции на неотравленном катализаторе равно:
= /(’Pth4f) = {/T Tth Ч'+th’F/th [Т(1-/т)])-1
?0 \ ' х-1- Vx
(Ш.162)
Сканировал: Neptunyi
Магнитогорск
2008
148
При ¥ 1 (в кинетическом режиме) ц* «=* 1 — Т/т и, таким
образом, скорость реакции снижается пропорционально доле отрав-
ленной поверхности. При V 1 (внутридиффузионный режим)
справедливы приближенные формулы:
при Y (1 - У7) > 1
Пднф=1/(ЧГ /т +1) (III.163)
при Ч1, (1 — ]/r) <£ 1
* ¥(1-/т)
^“Ф 1 + ^2 /7(1-/7)
Зависимость ц* (Ут) при различных значениях модуля Тиле
представлена на рис. III.17.
В ряде процессов отравление катализатора происходит не за счет
адсорбции яда, поступающего в зерно извне, а вследствие образова-
ния веществ, блокирующих активную поверхность в результате
одной из побочных реакций. Именно такая ситуация возникает в про-
цессах каталитического крекинга, где происходит закоксовывание
катализатора, т. е. его отравление образующимися высокомолеку-
лярными продуктами. В процессах такого типа скорость отравления
обычно пропорциональна локальной скорости основной реакции.
Задача расчета макроскопической скорости такого процесса
на пористом зерне катализатора математически очень сложна и,
по-видимому, не может быть решена аналитически, даже в простей-
ших случаях. Численные результаты были получены в работах [28J.
ЛИТЕРАТУРА
1. Д. А. Франк-Каменецкий, Диффузия и теплопередача в хи-
мической кинетике, Изд. «Наука», 1967.
2. Е. Е. Petersen, Chemical Reaction Analysis, 1965.
3. C. N. Satterfield, T. К. Sherwood, The Role of Diffusion
in Catalysis, Palo Alto — London, 1963; C. N. Satterfield, Mass Transfer
in Heterogeneous Catalysis, Cambridge, 1969.
4. С. Ч e п м e h, T. К а у л и и г, Математическая теория неоднород-
ных газов, ИЛ, 1960; Дж. Кларк, М. Макчесни, Динамика реальных
газов, Изд. «Мир», 1967; Дж. ГиршфеЛьдер, Ч. Кертисс, Р. Берд,
Молекулярная теория газов и жидкостей^ ИЛ, 1961.
5. А. Уилер, сб. «Катализ, вопросы теории и методы исследования»,
ИЛ, 1955, стр. 479.
6. D. S. S с о 11, F. A. L. D u 11 i е n, Am. Inst. Chem. Eng. J., 8,
113 (1962); J. Hoogschlagen, Ind. Eng. Chem., 47, 906 (1955);
P. F. D e i s 1 e r, R. H. Wilhelm, Ind. Eng. Chem., 45, 1219 (1953);
E. W i c k e, W. BrStz, Chem. Ing. Techn., 21, 219 (1949); E. L. P i r e t et
al., Chem. Eng. Progr., 47, 628 (1951); P. L. Walker, F. Runisko, E. Ha-
ats, J. Phys. Chem., 59, 245 (1955); R.M. Barret, T. Gal or, Proc.
Roy. Soc., 251A, 353 (1959); M. F. Johnson, W. E. К r eg e r, H. E r i k-
s о n, Ind. Eng. Chem., 49, 283 (1957); R. H. V i 11 e t, R. H. Wilhelm,
Ind. Eng. Chem., 53, 837 (1961); P. B. W e i s z, C. D. Prater, Adv. in Cata-
lysis, 6, 143 (1954); P. B. Weisz, A. B. S c h w a r t z, J. of Catalysis,
1, 309 (1962); N. W a k а о, P. W. Seiwood, J. M. S m i t h, Am. Inst.
Chem. Eng. J., 8, 478 (1962); В. А. Рейтер и др., ЖФХ, 24, 459 (1950);
28, 1638, 1812 (1954); 29, 1073 (1955); J. М. S m i t h et al., Am. Inst. Chem.
149
Eng. J., 7, 10 (1961); 8, 217 (19'62); M. F. L. J о h n s о n, W. F. S t u a r t, J.
oi Catalysis, 4, 248 (1965); C. N. Satterfield, S. K. S a r a f, Ind. Eng.
Chem. Fund., 4, 451 (1965).
7. Б. В. Дерягин, ДАН СССР, 50, 623 (1946); E. A. Mason,
R. B. Evans, G.M. Watson, J. Chem. Phys., 35, 2076 (1961); 36, 1894
(1962); 38, 1808 (1963).
8. N. W а к а о, Препринты IV Международного конгресса по катализу,
1968.
9. I. О. М i n g 1 е, J. М. S m i t h, Am. Inst. Chem. Eng. J., 7, 243
(1961).
10. S. Masamune, J. M. Smith, J. Chem. Eng. Data, 8, 54 (1963);
R. A. M i s c h k e, J. M. S m i t h, Ind. Eng. Chem. Fund., 1, 288 (1962);
R. A. S e h r, Chem. Eng. Sci., 9, 145 (1958).
11. W. Nernst," E. Brunner, Z. Phys. Chem., 60, 46 (1907).
12. В. Г. Л e Bin, Физико-химическая гидродинамика, Физматгиз, 1959.
13. P. Chambre, А. А с г i v о s, J. Appl. Phys., 27, 1322 (1956).
14. D. Rosner, Am. Inst. Chem. Eng. J., 9, 321 (1963); Chem. Eng.
Sci., 19, 1 (1964).
15. J. d e A c e t i s, G. T h о d о s, Ind. Eng. Chem., 52, 1003 (1960).
16. M. Э. А э p о в, О. M. T о д e с, Гидравлические и тепловые основы
работы аппаратов со стационарным и кипящим зернистым слоем, Изд. «Химия»,
1968.
17. Я. Б. Зельдович, ЖФХ, 13, 163 (1939).
18. Е. W. Thiele, Ind. Eng. Chem., 31, 916 (1939).
19. R. Aris, Chem. Eng. Sci., 6, 265 (1957).
20. С. И. К у ч а н о в, Л. M. Письмен, ДАН СССР, 167,1335 (1966).
21. Л. М. Письмен, Ю. И. X а р к а ц, ДАН СССР, 178, 901 (1968).
22. G. W. R о b е г t s, С. N. Satterfield, Ind. Eng. Chem. Fund.,
5, 317 (1966).
23. P. B. Weisz, J. S. Hicks, Chem. Eng. Sci., 17, 265 (1962);
E. E. Petersen, Chem. Eng. Sci., 17, 987 (1962); J. D. Tinkler,
А. В. M e t z n e r, Ind. Eng. Chem., 53, 663 (1961); J. J. Carberry, Am.
Inst. Chem. Eng. J., 7, 350 (1961); G. W. Roberts, C. N. Satterfield,
Ind. Eng. Chem. Fund., 4, 288 (1965); V. Hlavacek, M. Marek, Chem.
Eng. Sci., 23 , 506, 865, 1083 (1968); D. W. D г о 11, R. A r i s, Chem. Eng.
Sci., 24, 541 (1969).
24. R. E. S c h i 1 s о n, N. R. A m u n d s о n, Chem. Eng. Sci., 13,
226, 237 (1961); J. В e e k, Am. Inst. Chem. Eng. J., 7, 237 (1961); J. D. Tin-
kler, R. L. Pigford, Chem. Eng. Sci., 15, 326 (1961); D. J. Gunn,
Chem. Eng, Sci., 21, 383 (1966).
25. V. W. Weekman, J. of Catalysis, 4, 260 (1965); 5, 44 (1966).
26. В. Г. Л e в и ч, Ю. И. X а р к а ц, Л. М. Письмен, ДАН СССР,
171, 406 (1966); Л. М. П и с ь м е н, Ю. И. X а р к а ц, В. Г. Л е в и ч,
Препринты IV Международного конгресса по катализу, 1968.
27. К. Ostergaard, Proceedings of the III International Congress
on Catalysis, Amsterdam, 1964.
28. S. Masamune, J. M. Smith, Am. Inst. Chem. Eng. J., 12,
384 (1966); Y. Murakami et al„ Ind. Eng. Chem. Fund., 7 , 599 (1968).
Часть
втора я
ОСНОВЫ СИНТЕЗА
ПРОМЫШЛЕННЫХ КАТАЛИЗАТОРОВ
Глава IV ________________________________________________
МЕТОДЫ ПОДБОРА
ХИМИЧЕСКИ АКТИВНЫХ КАТАЛИЗАТОРОВ
В отношении задачи подбора гетерогенных катализаторов оче-
видно нет оснований выдвигать слишком жесткие требования о рас-
четном изыскании новых катализаторов. Пути решения следует
искать в сочетании теоретического подхода с наиболее эффективными
методами использования накопленного экспериментального мате-
риала.
Следует подчеркнуть, что сегодня задачу подбора катализаторов,
в силу накопленного большого опыта, понимают большей частью
не просто как нахождение катализаторов, но как нахождение ката-
лизаторов, обладающих лучшими показателями по сравнению с уже
известными или удовлетворяющих каким-либо специальным химиче-
ским или технологическим требованиям.
Наличие химической каталитической активности у твердого соеди-
нения является необходимым, но не достаточным условием создания
активного и селективного катализатора. Следует отличать подбор
катализатора, т. е. нахождение для данной реакции каталитически
активного вещества, от умения приготовить из него приемлемый для
практических целей катализатор. В главе III освещено влияние по-
ристой структуры зерна катализатора на скорость и направление
текущих в нем реакций. Неудачная структура зерна может свести на
нет потенциально высокие свойства самого катализатора. Важней-
шее значение для технического катализа также имеет стабильность
катализатора по отношению к термическому режиму, отравлению и
механическому воздействию. Однако эти требования вторичны и
в значительной степени решаются вариацией условий приготовления
катализаторов. Поэтому задачей подбора, в первую очередь, является
прогнозирование химической активности катализатора.
В проблеме подбора катализаторов следует различать две сто-
роны: 1) качественные принципы подбора и 2) количественные методы
оценки активности или селективности.
Рассмотрим эти задачи раздельно.
151
IV. 1. КАЧЕСТВЕННЫЕ ПРИНЦИПЫ |
ПОДБОРА КАТАЛИЗАТОРОВ |
Первым качественным принципом, очевидно, является рацио4
нальная классификация катализаторов, которая дает возможность»
связывать определенные типы катализаторов с соответствующими
типами реакций и тем самым ограничивать круг веществ, обследуемых]
на каталитическую активность для каждой конкретной реакции.]
В основу такой классификации можно положить изложенные в главе I]
соображения Рогинского. , ]
Второе исходное положение в вопросе подбора гетерогенных ката-;
лизаторов — принцип тесной аналогии или точнее единства гомоген-]
ных и гетерогенно-каталитических реакций. Было показано, что,
реакции, протекающие с гомогенными катализаторами в жидкой фазе,]
могут быть проведены в газовой иди жидкой фазе с применением]
тех же или аналогично действующих веществ в качестве катализато-
ров, но уже гетерогенного типа [1 ]. Поскольку гомогенный катализ,
базирующийся на образовании промежуточных соединений, изучен
гораздо полнее и систематичнее, указанная аналогия может оказы-
вать существенную помощь в изыскании новых гетерогенных ката-
лизаторов.
Наконец, третьим принципом подбора следует признать принцип,
выдвинутый Паулингом [2], об ответственности d-состояний за ката-
литическую активность элементов во многих каталитических реак-
циях. Есть основания считать, что этот принцип можно распростра-
нить и на /-состояния [3].
Рассмотренные принципы не столько открывают новые явления,
сколько систематизируют многочисленные наблюдения исследовате-
лей в области катализа и более четко формулируют их интуитивный,
подход.
Попробуем обобщить принципы качественного подхода к подбору
активных и селективных катализаторов, поскольку они не потеряли
актуальности.
В настоящее время очевидно, что первоначальную классифика-
цию Рогинского следует несколько дополнить.
По свойствам и механизмам протекающих на них превращений
гетерогенные катализаторы целесообразно разделить на четыре типа:
1) кислотно-основные; 2) комплексообразующие; 3) электронные полу-
проводники; 4) металлы.
Для всех групп катализаторов в соответствии с их химическими
и дополнительными физическими свойствами существуют определен-
ные области катализируемых ими реакций, хотя эти области и пере-
крываются.
Кислотно-основные катализаторы. В соответствии с теорети-
ческими представлениями, изложенными в главе I, твердые кислоты
и основания катализируют гетеролитические превращения. В част-*
ности, гетерогенные катализаторы кислотного типа применимы
для ускорения реакций изомеризации, крекинга, полимеризации
152
Mr алканов, галогенирования, присоединения воды и других полярных
Иу молекул по двойной связи, алкилирования, арилирования, различ-
Ю яых конденсаций с отщеплением воды, аммиака, хлористого водо-
Вг рода и других полярных молекул, гидролиза, бекмановских перегруп-
В= пиров ок и перегруппировок других типов (например, Фриса, пина-
Р"’ колинов ой, аминофенольной), конденсаций альдегидов и кетонов
>с фенолами и ариламинами и ряда подобных реакций.
I Гетерогенные катализаторы основного типа применимы для реак-
? ций конденсации карбонильных соединений, ацетиленовых конден-
( саций, а также реакций Перкина, Фаворского и др.
; Аналогично, конечно, катализируются гетеролитические превра-
* щения неорганических веществ.
Таким образом, в отношении твердых кислот и оснований пол-
: ностью реализуется принцип аналогии гетерогенного и гомогенного
катализа.
Комплексообразующие катализаторы. В настоящее время еще
• ведутся споры о самом определении «комплексные соединения».
Мы примем следующую терминологию: акцепторно-донорными ком-
плексами будем называть комплексы, образуемые донорной связью
с участием s- и р-орбиталей, а координационными — комплексы, об-
разованные дативными координационными связями с участием d- и
/-орбиталей.
Очевидно, что комплексообразователи акцепторно-донорного
типа являются льюисовскими кислотами либо основаниями и, наобо-
рот, соответствующие кислоты и основания будут комплексообразо-
вателями. Таким образом, здесь скорее только терминологическое
различие и специальное рассмотрение этих комплексообразователей
не требуется. _
Общий механизм каталитического действия координационных
комплексов сводится, как указывалось, к облегчению электронных
переходов в общей системе электронов и ядер внутри комплекса,
по сравнению с переходами между отдельными молекулами. В этом
плане следует считать, что стадия образования координационных
комплексов может ускорять как реакции окисления -восстановления,
так и реакции перераспределения валентных связей (интра- и интер-
молекулярные), поскольку между различными молекулами, входя-
щими в координационную сферу комплекса в качестве лигандов,
взаимодействие облегчается. Этим правилом с большой вероятностью
можно руководствоваться при подборе катализаторов.
В последнее время особое внимание уделяют л-комплексам в ка-
тализе, роль которых в гомогенных каталитических превращениях
ненасыщенных соединений очень велика (см., например, статью
Моисеева [4]). Хотя данные о гетерогенном катализе газофазного
гидроформилирования на сульфиде рутения [5] недостаточно одно-
значны из-за возможности протекания параллельной гомогенной
реакции с летучими карбонилами металла, однако, из активности ме-
таллического палладия в реакциях газофазного окисления этилена
в ацетальдегид и бензола в ацетилфенол [6, 7 ], можно сделать вывод,
153
что и для этого случая приложим принцип аналогии гомогенного и
гетерогенного катализов. Координационные комплексообразова-
тели — соли металлов переходной валентности — способны выпол-
нять функции гетерогенных катализаторов для многочисленных
реакций присоединения возможных лигандов к ненасыщенным угле-
водородам или реакций, протекающих через стадию такого при-
соединения.
Электронные полупроводники. Хотя в настоящее время по вопро-
сам полупроводниковой теории катализа имеется обширнейшя я
литература, однако работ, посвященных проблеме прогнозирования
Каталитических свойств полупроводников, немного.
Рассмотрим работы, характеризующие современное состояние дан-
ного вопроса. Так, Дауден с сотрудниками [8, 9] связывает актив-
ность полупроводниковых катализаторов, в противоположность теоре-
тическим взглядам Волькенштейна, не с коллективными свойствами
электронов в полупроводнике, а с электронным строением атома
металла в окисле. Рассматривая хемосорбцию как процесс образова-
ния поверхностных комплексов и применяя расчеты на основе теории
кристаллического поля, Дауден показал, что для реакций, в которых
лимитирующей стадией является адсорбция реагента или десорбция
продукта, за счет энергии стабилизации кристаллическим полем сле-
дует ожидать минимальных скоростей у ионов с d0-, d5- и й10-оболоч-
кой (в слабом поле) и с d°- и й10-оболочкой (в сильном поле). Соответ-
ственно этому максимальной активностью должны обладать ионы
с de и cP-оболочками (в слабом поле). Этот эффект связан с наиболь-
шими энергетическими выгодами при адсорбции (экзотермическая
стадия) или десорбции (эндотермическая стадия). Наличие такой
двухпиковой активности было установлено в работе [10 ] для изотоп-
ного обмена Н2 D2, Диксоном и др.—для диспропорционирования
циклогексена [11 ] и Марголис — для полного окисления пропи-
лена [12]. Крылов в работе [13] солидаризируется, в общем, со
взглядами Даудена и подтверждает двухпиковую зависимость на
примере окисления СО в СО2. Таким образом, эта закономерность,
по-видимому, может быть использована для качественных прогнозов
каталитической активности окислов переходных металлов йримени-
тельно ко многим реакциям окислительно-восстановительного типа,
простым по своему механизму или включающим простую лимитирую-
щую стадию. Однако двухпиковая закономерность является отнюдь
не универсальной. Она неприменима практически для всех реакций
неполного окисления углеводородов, и нет указаний на ее примени-
мость для других сложных реакций. Одна из причин этого — непри-
годность схемы Даудена при хемосорбции через стадию л-комплекса.
Марголис [12] указала на существенную связь избирательности
окисных катализаторов с такими факторами, как наличие недо-
строенной d-оболочки у иона металла, работа выхода электрона
окисла, геометрия кристаллической решетки. Гельбштейн с сотруд-
никами [14] предлагает в качестве определяющих критериев при
подборе катализаторов окисления олефинов энергию связи кислород—
154
металл в решетке окисла и работу выхода электрона. Зависимости,
приведенные в работах [12, 14], достаточно обоснованы. Однако мно-
гочисленность комбинаций указанных факторов, а также ряд исклю-
чений из них не дают возможности вывести, основываясь на указанных
принципах, общие качественные рекомендации в отношении подбора
катализаторов окисления.
В терминах подвижности кислорода решетки качественная оценка
селективности окисных катализаторов неполного окисления дана
в работах Захтлера с сотрудниками [15 ] и Трифиро, Иру и сотрудни-
ков [16]. В работе [15] установлена четкая симбатность между селек-
тивностью реакции окисления бензальдегида в бензойную кислоту
и параметром д АН (х)/дх (где АН — энтальпия окисла; х — степень
восстановления окисла в долях). В работе [16] для реакций окисли-
тельного аммонолиза пропилена и его окисления показана обратная
зависимость селективности от скорости диффузии кислорода в ре-
шетке окисла. Обе эти работы свидетельствуют о том, что селектив-
ность реакций неполного окисления связана с вероятностью дальней-
шего взаимодействия первичного продукта реакции за время его
сорбционного состояния с поверхностным кислородом окисла. Такое
взаимодействие тем менее вероятно, чем меньше подвижность кисло-
рода решетки. Соответственно окислы с очень высокой подвижностью
кислорода мало селективны в реакциях неполного окислеция.
Другой подход к качественному подбору полупроводниковых
(точнее, окисных) катализаторов окисления предложен в работе [17 ].
На основе имеющихся экспериментальных данных катализаторы
делятся на две группы: катализаторы, активирующие разрыв С—Н
связи (о-активирующие катализаторы), и катализаторы, активирую-
щие разрыв С = С связи (л-активирующие катализаторы).
К первой группе относятся окислы металлов переходной валент-
ности, у которых катионы решетки сохраняют свои индивидуальные
свойства. В этом случае можно ожидать аналогию в механизме реак-
ций гетерогенно-каталитического окисления на окислах и гомоген-
ного химического окисления в растворах. Для данной группы ката-
лизаторов обоснован механизм с первичным взаимодействием за счет
водорода органической молекулы и кислорода окисла металла [18],
например:
В2СН2 + СгО3 -* В2СН—СгО2
I
ОН
или
В2СН2 + СгО3 В2СН. + СгО2ОН
При достаточно быстром реокислении твердого окисла механизм
этих уравнений приводит к каталитическому окислению органиче-
ской молекулы через стадии попеременного окисления-восстановле-
ния окисла, а активность последнего связывается с окислительно-
восстановительным потенциалом его катиона.
Действительно, для правых 3d окислов Со, Мп, Ni, катализирую-
щих разрыв С—Н связи в олефинах [19 ] или окисление парафинов
155
[20], характерны как отсутствие взаимного перекрывания электрон-
ных уровней в кристаллической решетке [21], так и высокий отри-
цательный окислительно-восстановительный потенциал.
Наличие зонной энергетической структуры электронов решетки
окисла может существенно изменить механизм взаимодействия по- ;
следнего с органической молекулой. Наличие низкорасположенных ;
основных или примесных уровней и ловушек электронов в виде дефек- :
Сопротивление V2 О5
Рис. IV. 1. Зависимость активности и селек-
тивности промотированных ванадиевых
катализаторов в реакциях окисления от
электрического сопротивления:
□ — конверсия о-ксилола во фталевый ангидрид;
V — конверсия бутенов в малеиновый ангидрид;
0 — селективность малеинового ангидрида по
бензолу.
тов и дырок приводит, как
известно, к большей вероят-
ности затягивания электро-
нов в решетку, что особенно
легко протекает для л-элек-
тронов [22]. В этом случае
первым актом катализа будет
разрыв С = С связи, и ката-
литический процесс, как по-
казано Рейтером [23 ] для
окисления нафталина на
V2O5, полностью протекает
в сорбционном слое, не зат-
рагивая решетку.
Синтез избирательных ।
о-активирующих катализа-
торов можно вести одним из
двух способов. В первом спо-
собе синтеза исходят из оки-
слов с изолированными ка-
тионами решетки, высоким
окислительным потенциалом
и малой подвижностью
кислорода в решетке. Для ,
увеличения скорости пере-
дачи электронов, чтобы она i
не стала лимитирующей, к
катализатору надо добавить
ионы переходных элементов,
способствующих образованию зон проводимости (V, Mo, Ti и др.).
Второй способ синтеза о-активирующих катализаторов базируется
на окислах элементов с высоким окислительным потенциалом, обла-
дающих высокой проводимостью, добавляя к ним катионы Р, Bi, *
Mg и других непереходных элементов для разрыва зон проводи-
мости.
Для улучшения избирательности л-активирующих катализаторов
следует повышать их электропроводность, расширяя зоны проводи-
мости, и увеличивать добавками их акцепторные свойства.
На рис. IV. 1 представлены зависимости избирательности и актив-
ности промотированных ванадиевых катализаторов от электриче-
ского сопротивления активной составляющей катализатора для реак^
156
ций окисления бензола и бутиленов в малеиновый ангидрид и о-кси-
лола во фталевый ангидрид.
Как видно из рис. IV. 1, несмотря на некоторый разброс точек,
совершенно четко можно проследить противоположный Характер за-
висимости для реакций с разрывом о- и л-связей. Сами по себе эти за-
висимости соответствуют рассмотренным выше рекомендациям.
Металлические катализаторы. Что касается металлических ката-
лизаторов, то несмотря на большой объем экспериментальных мате-
риалов, изложенных, например, в книге Бонда [24], теоретических
оснований для качественных прогнозов активности и избирательности
катализаторов здесь, видимо, меньше, чем для других типов.
Состояние вопроса можно вкратце свести к следующим положе-
ниям. Благодаря достаточной подвижности электронов в металличе-
ских структурах металлы хорошо катализируют гомолитические
реакции: гидрирование, восстановление, окисление. При этом, в про-
тивоположность распространенному мнению, в определенных усло-
виях, например при жидкофазных реакциях, металлы являются
нейлохими катализаторами неполного окисления органических соеди-
нений [25].
Твердые металлические поверхности сохраняют способность ката-
лизировать реакции присоединения, связанные со стадией образова-
ния комплексов, например реакции с участием СО.
В работах Бика [26], Кембэла [27] и других была показана
связь гидрирующей способности металлов и сплавов с процентом
d-состояния, что свидетельствует о комплексообразовании как пер-
вичном акте в этих реакциях. Таким образом, наличие вакантных
d-электронных орбит является условием каталитической активности
металлов в реакциях окисления, восстановления и ряде реакций при-
соединения.
Свободные щелочные металлы благодаря своим высоким электро-
нодонорным свойствам способны катализировать различные гетеро-
литические реакции в закритических условиях, исключающих гомо-
генный механизм катализа [28]. Так, литий катализирует присоеди-
нение этилена к циклогексану при температурах до 450° С и этилена
к аммиаку при температурах до 175—200° С.
Представления о мере эффективности изложенных выше каче-
ственных соображений по подбору катализаторов может проиллю-
стрировать следующий «эксперимент» над массивом существующей
информации.
Из справочника Ройтера [28], в котором сведены данные о ката-
лизаторах для 1017 реакций, по таблице случайных чисел выбрали
29 примеров. Для каждого примера были сравнены соображения о
типе гетерогенных катализаторов на основе изложенных принципов
и по данным справочника. При этом не оказалось значительных
расхождений между результатами испытания и изложенными прин-
ципами. Количество расхождений составило менее 27%.
Таким образом, имеющиеся теоретические соображения и на-
копленный экспериментальный материал позволяют для каждого
157
данного типа реакций довольно надежно намечать набор веществ,
обладающих нужным каталитическим действием. Очевидно, сегодняш-
нее состояние проблемы подбора в целом заключается в более точном
определении состава приближенно оптимальных катализаторов для
данной частной реакции и уточнении их параметров (активности,
селективности) в реальных условиях, а также в выработке надежных
правил для способа приготовления этих катализаторов.
IV.2. КОЛИЧЕСТВЕННЫЕ МЕТОДЫ ПРОГНОЗИРОВАНИЯ
АКТИВНОСТИ И СЕЛЕКТИВНОСТИ КАТАЛИЗАТОРОВ
Как^указывалось выше, количественные методы подбора и прогно-
зирования катализаторов носят полуэмпирический характер и по-
этому не универсальны, а ограничены более или менее узкими клас-
сами превращений и катализаторов. Фактически к настоящему мо-
менту определялось три основных направления в разработке этих
методов.
1. Использование линейных корреляций между термодинамиче-
скими или другими физико-химическими величинами и активностью
и селективностью катализаторов.
2. Приложение приближенных квантово-химических расчетов
к оценке относительной активности (селективности) катализаторов.
3. Применение математической теории распознавания для про-
гноза каталитических свойств твердых веществ на основе существую-
щих экспериментальных данных и информации о физико-химических
параметрах этих веществ и реагирующих молекул.
Линейные корреляции формулируются как принцип линейных
соотношений свободной энергии (ЛССЭ), который применяется для
создания количественной теории органических реакций [29, 30].
Эта теория базируется на трех известных уравнениях: уравнении
Бренстеда, связывающем скорость Каталитической реакции с кон-
стантой диссоциации катализирующей кислоты (основания); уравне-
нии Гаммета — Тафта, связывающем скорости однотипных реакций
с индуктивными, стерическими и другими эффектами заместителей
в гомологическом ряду соединений; уравнении Поляни—Воевод-
ского—Семенова, связывающем энергию активации взаимодействия
радикала и молекулы с тепловым эффектом этой реакции в ряду одно-
типных превращений.
Хотя упомянутые уравнения включают в себя некоторые эмпири-'
ческие коэффициенты, однако они находятся в соответствии с фунда-
ментальными представлениями современной химической кинетики
и теории строения органических соединений.
Фактически идея применения. ЛССЭ к гетерогенному катализу
была впервые развит^ Баландиным в 1946 г. в форме его принципа
энергетического соответствия в катализе. Рассмотрим трактовку
этого принципа с точки зрения ЛССЭ. В этом случае его можно свести
к следующим основным положениям.
158
1. Скорость гетерогенной химической реакции определяется ско-
ростью образования или разрушения адсорбционного комплекса.
2. К любому из двух названных лимитирующих этапов прило-
жимы уравнения ЛССЭ.
3. Для оптимального катализатора, т. е. для максимальных
скоростей реакции, должно иметь место равенство констант образова-
ния и разрушения адсорбционного комплекса.
Из указанных соображений следует, что для оптимального катали-
затора
^адс аадс 1g ^адс = дес ^Сдес 1g ^дес (IV. 1)
где Лиа — эмпирические коэффициенты; — константы рав-
новесия образования адсорбционного (десорбционного) комплекса.
Если принять, что Лиа приближенно равны для обеих стадий,
то
^с = *Йс (IV.2)
откуда соответственно вытекает
Лад? ~ 4г + Т ^адс) (IV.3)
где Ладо — теплота адсорбции для оптимального катализатора; Q и
АА — тепловой и энтропийный эффекты реакции; ААадс — энтро-
пийный эффект адсорбции.
Для многих реакций разность — ААадс) мала, тогда
(IV.4)
Баландин, исходя иэ представлений о компенсационном эффекте,
принимал, что
1 Я = -|-Ладс (IV-5)
где Е — энергия активации каталитической реакции.
Отсюда следует, что по мере удаления от точки оптимума энергия
активации реакции будет увеличиваться либо за счет адсорбционной,
либо за счет десорбционной стадии. Это приводит к наличию макси-
мума активности по отношению к данной реакции в ряду однотипных
катализаторов или в серии однотипных реакций на данном катализа-
торе. В монографии [31 ] даны примеры принципа применения энер-
гетического соответствия к подбору оптимальных катализаторов и
оценке направления реакций. Оказалось, что метод эффективен в от-
ношении дегидратации и дегидрировании спиртов, дегидрирования
циклогексановых углеводородов и фурановых соединений и ряда
Других реакций.
В обзоре Крауса [32 ] указывается на принципиальные ограниче-
ния при применении ЛССЭ к гетерогенным каталитическим реак-
циям. Они проистекают, если отвлечься от явлений переноса, от двух
159
причцн. Во-первых, как указывалось в главе I, имеет место взаимна
влияние как катализатора на реакцию, так и реакции на катализатор»
Вследствие этого при протекании реакции с новыми компонентами
линейные соотношения не сохраняются. Во-вторых, в большинстве
случаев гетерогенно-каталитические реакции описываются извест -
ным кинетическим уравнением Лангмюра (см. раздел II.4) -
М>арА
i + ^biPi
i
где к — константа скорости поверхностной реакции, фактически пред-
ставляющая собой произведение константы скорости поверхностной^
реакции на единичном активном центре (кр) на число активных цен- j
тров (А). По своему физическому смыслу ЛССЭ приложимо только ►
к кр, поэтому экспериментально линейные соотношения можно i
наблюдать только когда 1 3> 2 ^iPi L — const. Все же ЛССЭ
можно использовать достаточно широко и в области лангмюровской ;
кинетики, поскольку последнее равенство сохраняется достаточно
часто, а выполнение первого неравенства возможно при относительно
низких парциальных давлениях реагента. Кроме того, используя от-
ношение скоростей реакций в одинаковых условиях, можно рассчи-
тывать на исключение знаменателя из уравнения (IV. 6).
Представлялось заманчивым сохранить для гетерогенных катали-
тических реакций значения констант в уравнениях Гаммета и Тафта,
определенных по данным гомогенных реакций. В обзоре [32] обра-
ботано значительнее число опубликованных экспериментальных дан-
ных и показано, что для ряда алкильных заместителей (СН3, С2Н6,
С8Н7, С4Н9, С8Н6С8Н4) уравнение Тафта применимо с сохранением
литературного значения константы индукционного эффекта о*.
В работе [33 ] показано, что реакция дегидратации ряда насыщен-
ных алифатических спиртов С5—С7 на окислах Al, Zr и Si хорошо
описывается простейшим уравнением Тафта с сохранением литера-
турных значений а*. В то же время коэффициент чувствительности р*
для различных окислов меняется симбатно с теплотой адсорбции
органических кислородсодержащих соединений, таких как дизтило- ?
вый эфир, а также линейно связан с чувствительностью катализатора |
по отношению к отравлению пиридином. Это указывает на связь р*/?
с сорбционной характеристикой катализатора. Авторы работы подчер-
кивают, что при подборе катализаторов необходимо раздельно оцени*
вать интенсивные (химические) и экстенсивные (число активных цен-
тров) свойства катализаторов.
Применить литературные значения а* удается, однако, не всегда,
что можно объяснить как различием механизмов гетерогенных и гомо-
генных реакций, так и температурой их проведения. Для ряда арома-
тических соединений применение уравнения Гаммета не дает удовлет-
ворительных результатов, хотя здесь, возможно, сказывается спе-
цифика переноса реакции в жидкую фазу.
160
Г Весьма четко прослеживается применимость принципа ЛССЭ
для реакций с кислыми катализаторами. Обстоятельные исследова-
ния данного вопроса провели Ионеда с сотрудниками [34—36 ].
В их' работах приведены данные по кинетике реакций деалкилиро-
вания и изомеризации на алюмосиликатных, алюмоборатных, суль-
фаталюминиевыХ и магнийборатных катализаторах. Было показано,
что указанные реакции на перечисленных катализаторах хорошо
подчиняются уравнениям ЛССЭ. Так, например, для реакции деалки-
лирования бензольных производных имеет место четкая линейная
связь между константой о* и теплотой образования карбоний-иона
(ДЯ), в соответствии с этим константа скорости реакции деалкилиро-
вания выражается уравнением'
18^деалк =-2 ----1-'® (IV.7)
I а для реакции изомеризации
v \Нт
(IV.8)
L где е и у — эмпирические константы; А и В — константы; к™ —
[ константа скорости миграции группы с АНт — 0; АНт — энтальпия
। образования карбоний-иона для мигрирующей группы.
Константы АН илиАНт, в свою очередь, могут быть выражены
в уравнениях (IV.7) и (VI.8) через константу скорости деалкилиро-
вания изопропилбензола как эталона и эмпирические постоянные.
Совместное решение уравнений (IV.7) и (IV.8) дает возможность рас-
. считывать не только активность, но и селективность кислых катали-
заторов в реакциях изомеризации различных алкилбензолов.
Существенный вклад в теорию применения ЛССЭ к гетерогенному
f катализу вносит учет функции распределения каталитических центров
по коррелирующему параметру, например по функции кислотности
Гаммета. В этом случае, в соответствии с основным уравнением Ро-
гинского для неоднородных поверхностей, скорость реакции для
кислых катализаторов, если предположить,-что активность катализа-
г тора зависит только от силы кислоты и вид кинетической зависимости
одинаков для всех центров, выразится уравнением
; N
ri = fp(P)Z kijVll (IV.9)
i-i.
где / (Р) — функция парциального давления реагентов; N — число
вариантов центров по активности (1 j N); ц>£/ — доля J-x цен-
тров в общем количестве активных центров i-ro катализатора.
Константа скорости реакции кц для каждого /-го центра опреде-
ляется из уравнений
11 Заказ 1441
кц — кд/ 4- kij
(IV.10)
(IV.ll)
161
где (Нц) — функция кислотности Гаммета для каждого /-го центра;
а — эмпирический параметр.
Из уравнений (IV.9) — (IV. 11) следует, что:
N
(IV.12)
7-1 ' .
Уравнение (IV. 12) объясняет многие кажущиеся отклонения
от линейного закона связи активности катализатора с его кислот-
ностью и, очевидно, может быть распространено на другие, не кислот-
ные, катализаторы. При этом функция кислотности заменяется соот-
ветствующей характеристикой, например окислительно-восстанови-
тельным потенциалом, как это сделано в работе [361 для окисного
никелевого катализатора.
Экспериментальные результаты по реакции крекинга углеводо-
родов на алюмосиликатном катализаторе, подтверждающие уравнение
(IV.12), приведены в работе [36]. Аналогичный метод был применен
Бакарджиевым, Шнобелем и Венке к реакции дегидратации изопро-
панола на том же катализаторе [37 ].
В упомянутой серии работ Ионеда развивает обобщение метода
ЛССЭ для случая совместного изменения как катализатора, так и тем-
пературы реакции. Комбинируя уравнение (IV.8) для любого i-ro
катализатора при 273° К и это же уравнение для любой температуры
на J-м катализаторе, можно определить значения к- (7) и представить
их в форме аррениусовской зависимости. Тогда для любых катализа-
торов и температур получим уравнение
lg*z(T) = IgZ,—-(IV-13)
из которого можно определить Zz, Et и эмпирическую константу
Таким образом, из зависимостей, выражаемых уравнениями
(IV.7)—(IV.13) появляется возможность для расчета активности и се-
лективности катализаторов.
В работах Ройтера, а также Голодца с сотрудниками [38—41]
рассмотрены результаты по применению ЛССЭ к реакциям гетероген-
но-каталитического окисления. Авторы установили наличие хорошей
линейной взаимосвязи между теплотой хемосорбции кислорода на
катализаторе и активностью последнего в реакциях полного окисле-
ния углеводородов, а также наличие восходящей и нисходящей
ветвей в такой зависимости. Аналогичные результаты получены
Боресковым и сотрудниками для реакции окисления СН4 и Н2 в от-
ношении теплоты десорбции кислорода для ряда окисных катализа-
торов [42].
Установлено [411, что трудно определяемую величину теплоты
сорбции кислорода на окисле можно в первом приближении заменить
на более доступную теплоту окисления низших окислов в высшие,
хотя четкость корреляции при этом несколько снижается. Поскольку
теплоты сгорания углеводородов, отнесенные к одному атому угле-
162
рода, находятся в интервале 48—60 ккал/г-ат, то оптимальным ката-
лизаторам полного окисления должна соответствовать теплота сорб-
ции, равная 25—30 ккал/г-ат, что и имеет место на самом деле. Ско-
рости окисления различных углеводородов на одном катализаторе
хорошо коррелируются со значением энергии углеродной связи, раз-
рываемой в лимитирующей стадии. При этом показано, что последняя
не может являться С—Н-связью, а только одной из л-связей или наи-
менее прочной С—С-связью.
Наряду с термодинамическими характеристиками, мерой проч-
ности связи кислорода с решеткой могут служить и такие кинетиче-
ские характеристики, как начальная скорость восстановления окис-
лов водородом или скорость гомомолекулярного или гетерогенного
изотопного обмена кислорода на окислах. Первый метод был приме-
нен Захтлером и Дебуром [43], а второй широко развит Боресковым
и его школой [42, 44, 45]. Слабой стороной использования кинети-
ческих параметров является, то, что по ним имеется мало данных, они
не поддаются приближенным расчетам и для своего определения тре-
буют эксперимента, вполне сравнимого по сложности с прямым опре-
делением активности и селективности катализатора.
Квантово-химические расчеты. Приложение квантово-химических
расчетов к количественной оценке каталитических свойств веществ
находится в начальной стадии. Ионеда и другие применили разрабо-
танный Фукуи [46 ] индекс реакционности о-электронных систем —
степень делокализации электрона — в качестве коррелирующего
параметра в уравнениях типа ЛССЭ [47, 48 ]. В основу были положены
представления, что в этих сложных реакциях имеет место одна лими-
тирующая стадия, в которой активные центры катализатора отры-
вают радикал от молекулы RX. При этом была установлена хорошая
сходимость экспериментальных и расчетных величин для реакции
дегидрогенизации различных спиртов и для дегидрирования цикло-
гексану и его гомологов на окисных катализаторах. Нет сомнений, что
аналогичные корреляции удастся осуществить и для л-электронных
систем по таким общепринятым индексам, как л-электронная плот-
ность, индекс свободной валентности и т. п.
В последнее время началось развитие квантово-химических ме-
тодов расчета энергии сорбции. Так Дункен и Опитц [49 ] и Шопов,
Андреев и Петков [50 ] провели квантово-химические расчеты энер-
гии адсорбции водорода, этилена и циклогексана на никелевом ката-
лизаторе и получили результаты, не сильно отличающиеся от экспе-
риментальных. Такого рода квантово-химические расчеты совместно
с изложенными выше положениями Баландина, Темкина и Ройтера
создают возможность подбора с помощью вычислительных машин
оптимальных катализаторов из серии аналогичных соединений.
Это, конечно, может значительно уменьшить объем эксперименталь-
ной работы при подборе катализаторов.
Близко к квантово-химическим методам стоит метод прогнози-
рования каталитической активности металлов или их ионов на базе
теории промежуточных ионов, предложенной Гарднером [51—53],
11* , 163
хотя в настоящее время эта теория оперирует только эмпирическими Я
величинами. Я
Статистические методы. Как было показано выше, до сих пор Ц
все еще нет достаточно универсального метода прогнозирования я
каталитических свойств твердых веществ и металлов. В то же время я
имеется значительный, но недостаточно точный экспериментальный 1
материал о каталитических свойствах многочисленных катализа- ;
торов для различных химических превращений. По нашему мнению, J
это можно объяснить двумя причинами: ;
1. Существует столь много различных факторов й их комбинаций,
влияющих на каталитические свойства и механизм реакций, что ;
обоснованные предсказания, базирующиеся на подробном анализе
физической сущности явления, становятся невозможными.
2. Одно и то же явление ускорения химической реакции вызы-
вается различными процессами, протекающими на атомном и моле- *
кулярном уровнях. J
В таких случаях следует проводить анализ явлений на основе
вероятностно-статистических представлений, как в различных раз-
делах физики и биологии. Сравнительно простые статистические ме- ;
тоды, однако, не дают эффективных результатов из-за слишком боль- ;
шого количества переменных и, главным образом, неточности имею- ;
щихся данных. Как ясно из вышеизложенного, корреляционные
методы, базирующиеся на гипотезе Гаммета—Тафта, обладают очень
ограниченной способностью к экстраполяции их прогнозов за пре-
делы узкого круга катализаторов, соединений и реакций. Можно
ожидать, что современные более мощные методы статистического
анализа, базирующиеся на математической теории распознавания,
окажутся более эффективными. Аналогичные математические методы
прогнозирования успешно применяются в медицине, геологии и
метеорологии.
Как известно, существуют различные методы и алгоритмы в тео-
рии распознавания [54—56]. Некоторые из них от начала до конца 4
оперируют статистическими представлениями, другие также исхо-
дят из этих представлений, но в ходе решений используют геометри-
ческую интерпретацию, носящую формально детерминистский ха-
рактер. Нам представляется, что изложение проблемы будет более
понятно в терминах второй интерпретации методов теории распозна-
вания. Однако при решении конкретных задач следует применять ,
и другие наиболее эффективные для данного случая алгоритмы рас-
познавания, в частности базирующиеся на теории прецептрона.»
Рассмотрим математическую трактовку задачи. Пусть имеется
множество объектов произвольной природы, называемых реализа- .
циями. В нашем случае — исследуемые катализаторы. Каждой реа-
лизации соответствует набор чисел, называемых признаками, харак-
теризующими свойства катализатора. Геометрически объект изобра-
жается точкой в гиперпространстве признаков. Вводится понятие
класса, которое образуется множеством реализаций, обладающих
общим свойством. В нашем случае классами могут быть катализа-
164
торы, обладающие высокой или низкой активностью или селектив-
ностью. В пространстве признаков классы ограничены гиперповерх-
ностями. Решение задачи сводится к определению по значениям
данной реализации того, к какому классу она принадлежит.
Объекты — реализации, по которым определяются гиперповерх-
ности, ограничивающие классы, называются обучающей последо-
вательностью. При отсутствии строгого качественного различия
в поведении объектов границы классов выбираются произвольно.
При делении объектов на классы теряют некоторую информацию
о количественных значениях их каталитической активности, но зато
сводят практически трудно решаемую задачу многомерной корре-
ляции для большого множества признаков к менее сложной задаче
распознавания образов. Кроме того, при использовании метода
распознавания можно учитывать информацию о качественных при-
знаках объектов, что невозможно осуществить методами корреляции.
В работах [57, 58 ] рассматривается применение математической
теории распознавания к( проблеме прогнозирования каталитических
свойств на примере двух задач: •
1. Окисление окиси углерода на ряде различных окислов ме-
таллов.
2. Неполное окисление углеводородов на пятиокиси ванадия.
Для первой задачи были использованы взятые из литературы [59 J
данные для 26 окислов о значении их удельной каталитической
активности при 150° С. Были выбраны 22 параметра, характери-
зующие наиболее существенные термодинамические кристаллогра-
фические и электронные свойства твердого тела. Обследуемые ката-
лизаторы по активности были разбиты на три класса.
В основу алгоритма решения задачи подбора катализаторов окис-
ления СО была положена геометрическая интерпретация теории
распознавания в виде метода потенциалов. С этой целью в простран-
стве признаков было введено понятие потенциала, создаваемого
точкой ж,- в точке х (потенциал точки). Потенциал точки вычисляли
по формуле
UXXi = -^~ (IV.14>
где Rxx. — эвклидрво расстояние между точками х и xt.
Предварительно, чтобы исключить влияния выбора единиц изме-
рения, численные значения признаков были нормализованы по сле-
дующей формуле:
Здесь xtj — безразмерное значение /-го признака для i-ro катали-
затора; Xfj размерное значение /-го признака для i-ro катали-
затора; Xj — среднее размерное значение /-го признака для всех
катализаторов; п;- — среднеквадратичное отклонение размерного зна-
чения /-го признака по всем катализаторам.
165
Само решение состояло из двух стадий: 1) выделение признаков, 1
влияющих на активность и их ранжирование и 2) собственно опреде-1
ление принадлежности катализатора к классу («распознавание»'!
катализатора). Для первой стадии был разработан специальный J
алгоритм. На последующей стадии в соответствующих точках опре-1
делился потенциал класса как среднее арифметическое потенциалов I
точек данного класса в выбранной точке. Данный объект относился!
к тому классу, по отношению к которому он имел наибольший по- |
тенциал. Наивысший достигнутый процент узнавания по этой схеме I
для реакции окисления СО для 26 окислов составил 84%. С прак-4;
тической точки зрения, такой результат представляется достаточно
высоким. Для контроля эта же задача была решена при помощи |
другого алгоритма («Кора») [56 ] по двухклассной схеме и было ?
достигнуто узнавание свыше 90%. В табл. IV. 1 приведены резуль-
таты последних расчетов.
Таблица IV.1 Результаты машинного распознавания каталитической активности оиислов в реакции окисления СО (окислы ; расположены в порядке убывания их активности) '
Kf Класс А Правиль- Кз Класс В+С (мало- Правиль-
по пор. (активные катализаторы) ность ответа по пор. и неактивные катализ аторы) НОСТЬ ответа
1 Мо02 + 12 ZnO +
2 СоО + 13 ТЮ2 4-
3 CO3O4 + 14 Fc2O3 +
4 МпО 15 MgO +
5 CdO + 16 СеОз +
6 Ag2O + 17 v205 +
.7 NiO + 18 Сг2О3 4-
8 CuO + 19 ZrO2 +
9 SnO — 20 HgO 4-
10 Cu2O +• 21 wo3 4-
11 CO2O3 22 23 24 25 26 ThO2 BeO GeO A12O3 SiO2 4—1—1—H4~
Из описанного можно получить некоторые следствия для теории
катализа, вытекающие из значимости различных свойств окислов
в отношении влияния на каталитическую активность последний.
Оказалось, что только 8 параметров из первоначальных 22 влияют
на каталитическую активность. Они представлены в табл. IV.2,
из которой видно, что каталитическая активность окислов в реак-
циях окисления вызывается наличием подвижных электронов. Суще-
ственно, что их подвижность может быть вызвана различными при-
чинами. Наличие подвижных (обменивающихся) электронов является
необходимым, но не достаточным свойством, так как стерический
•фактор доступности может свести к нулю потенциальную каталити-
166
Таблица IV.2
Свойства окислов и реагентов, в наибольшей степени»
влияющих на каталитическую' активность'
Объекты Физико-химические свойства
Окислы — ка- тализаторы окисления СО в СО2 1. Плотность 2. Цвет 3. Температура плавления 4. Отношение свободного объема кристаллической ре- шетки к объему атомов металла 5. Параметр зоны проводимости 6. Магнитная восприимчивость 7. Количество d-, /-электронов, не участвующих в ва- лентной связи металл—кислород 8. Потенциал ионизации, усредненный по числу валент- ных связей металла в решетке окисла
Окисляемый углеводород 1. Молярный дипольный момент 2. Сумма угловых напряжений 3. Молярная магнитная восприимчивость 4. Структурный фактор j гс-с а0 где Z—длина связи С—С d0—диаметр атома кислорода в решетке катализа- тора 5. Число узлов на молекулярных орбиталях 6. Высший л-электронный уровень 7. Низший л-электронный уровень 8. Полное число валентных р-состояний 9. Полное число операций симметрии молекул 10. Максимальный индекс свободной валентности 11. Минимальный индекс свободной валентности
Продукты окисления < углеводородов 1—9. То же, что и для углеводородов, кроме 4 10. Молярный потенциал ионизации 11. Структурный фактор m 4 1С~° Т-1 . «0 где /со — длина С—О связи 12. Минимальная л-электронная. плотность
ческую активность соединения. Значимость температуры плавления
характеризует зависимость каталитической активности от стабиль-
ности связей в кристалле и этот же параметр независимо был пред-
ложен Ройтером для его корреляционных уравнений [40].
Для решения второй задачи был применен алгоритм «Кора».
50 реакций окисления различных углеводородов на катализаторе
V2O6 были разделены на три класса: А — высокоселективный (выход
более 50%), В — среднеселективный (выход 10—50%) и С — мало-
селективный (выход менее 10%). 46 энергетических, стереохимиче-
ских, квантово-химических и других свойств реагентов и продук-
тов были избраны в качестве параметров, например, такие как
167
Таблица IV.3 Результаты машинного распознавания селективностиЯ| реакций неполного окисления углеводородов на VaOgSB дд
№ Но пор. Реагент Продукт ' Правиль- Я кость Я ответа Я
1 2 3 4 5 -В 7 8 9 10 И 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 168 о-Ксилол Нафталин Антрацен Толуол Бензол м- Ксилол Пентен-1 Пентен-2 Бутадиен Нафталин Антрацен п-Ксилол л-Ксилол п- Ксилол 2-Метилбутен-2 м- Ксилол Толуол 2-Метил бутен-2 2-Метилбутен-1 2-Метилбутен-2 2-Метилбутен-1 Бутен-1 Бутен-2 З-Метилбутен-1 Пентен-1 Пентен-2 Пентен-2 2-Метилбутен-2 Пропилен Пентен-1 2-Метилбутен-1 Пентен-2 2-Метилбутен-1 З-Метилбутен-1 Пентен-1 Пентен-1 Пентен-1 Пентен-1 Пентен-2 З-МетиЛбутен-1 Пропилен Пентен-2 Пентен-2 З-Метилбутен-1 Класс А Фталевый ангидрид Фталевый ангидрид Антрахинон Бензойная кислота Малеиновый ангидрид Класс В Малеиновый ангидрид Ацетальдегид Ацетальдегид Малеиновый ангидрид Нафтохинон-1-4 Фталевый ангидрид Малеиновый ангидрид n-Толуиловый альдегид Терефталевый альдегид Ацетон л-Толуиловый альдегид Бензальдегид Ацетальдегид Ацетон 2,3-Эпокси-2-метилбутан Ацетальдегид Малеиновый ангидрид Малеиновый ангидрид Ацетон Ацетон Пропионовый альдегид Метанол З-Метилбутан-2-он Класс С Формальдегид Бутанон 2,3-эпокси-2-метилбутан 2,3-эпоксипентан Бутанон Ацетальдегид Пропионовый альдегид Масляный альдегид 2,3-Эпоксипентан Метанол Кротоновый альдегид Бутанон Ацетальдегид Бутанон Ацетон 2,3-Эпокси-2-метилб утан + 1 +++++++++++++ .1 III +++++ 1 +++++++++++ 1 1 + +++++
Продолжение-
- ПО пор. Реагент Продукт Правиль- ность ответа
’ 45 46 47 ' 48 49 50 п-К си лол Бутадиен З-Метилбутен-1 Пентен-2 2-Метилбутен-2 Пентен-2 n-Толуиловая кислота Формальдегид- З-Метилбутан-2-он Бутанол-1 Бутаной Этанол + -Н-+ i 1'
«угловое напряжение», минимум и максимум л-электронной плотно-
сти, число операций симметрии молекулы и т. п. Результаты про-
гноза представлены в табл. IV.3. Как видно из табл. IV.3, правильное
распознавание в классах А и С превышало 95%, а в В 70%. Досто-
верность предсказания в каждой операции дихотомии *, с помощью
которых проводился весь расчет, составляла 82—88%. Перечень
наиболее влиятельных признаков молекул реагентов для второй
задачи также представлен в табл. IV.2. Из этих данных видно, что
их число составляет только 11—12 и что основная часть этих пара-
метров относится к квантово-химическим свойствам. '
Из данных работ [57 , 581 можно сделать вывод, что путем при-
менения статистических методов распознавания к подбору катали-
заторов можно осуществлять достоверные полуколичественные про-
гнозы каталитической активности твердых веществ и направлений
реакций, ими катализируемых. Основой для таких прогнозов может
быть либо существующие экспериментальные данные, имеющиеся
в литературе, либо результаты новых экспериментов, проведенных
по специально разработанному плану. При использовании методоа
распознавания могут быть установлены новые и широкие корреля-
ционные зависимости, что, конечно, будет способствовать повыше-
нию надежности прогнозов и решению ряда теоретических проблем
катализа.
IV.3. ПОДБОР КАТАЛИТИЧЕСКИХ СИСТЕМ
Из материала, изложенного в разделе 1.8, вытекает, что подбор-
каталитической системы базируется на том, что каталитический
процесс происходит по стадиям. Стадии эти могут и не совпадать
с механизмом процесса, протекающего на одном катализаторе.
Существенно, чтобы они были совместимы и приводили к селектив-
ному выходу целевого продукта. Например, деалкилирование пири-
дина водяным паром на никелевом катализаторе протекает согласно
схеме:
ACHs+H2O -X Л+со + 2н2
\ /
* Дихотомией называют операцию разделения множества-па два класса.
16»
Селективность процесса может быть повышена при проведении
процесса на каталитической системе V2O5—МоО3—TiO2 + Ni с уча-
стием воздуха [60].
Процесс при этом протекает по стадиям:
/О
f\CH3 +0* (V»O»)- f \С"' +Н2О
N N
(Ni)
II 1с\ —Ч 1+СО
N N
Таким образом, подбор компонентов каталитической системы
•сводится к подбору селективных катализаторов для отдельных
стадий.
В некоторых случаях проведение процесса в одну стадию на ката-
литической системе выгодно даже, если оно и не приводит к измене-
нию его механизма. Например, при протекании процесса по схеме
1 2
А В —> С
1'
когда целевым является продукт С, введение катализатора, уско-
ряющего реакцию 2, сдвигает равновесие А # В вправо и процесс
превращается как бы в систему двух необратимых реакций без тер-
модинамических ограничений по выходу. При протекании процесса
по схеме
когда целевым является продукт D и порядок реакции 3 ниже,
чем 2, введение катализатора, ускоряющего превращение В, сни-
жает его концентрацию и повышает выход D по сравнению с двух-
стадийным процессом, для которого во второй стадии неизбежны
повышенные концентрации вещества В.
Возможны также случаи со сложной лангмюровской кинетикой,
когда проведение процесса в одну стадию с каталитической систе-
мой заведомо выгоднее, чем проведение его в несколько стадий
с отдельными катализаторами, даже когда в целом механизм процесса
не меняется. Подробнее эти вопросы рассмотрены в монографии [61].
О целесообразности применения систем из катализатора и инги-
битора уже говорилось в разделе 1.8. Подбор ингибитора базируется»
на общей теории ингибиторов, рассмотрение которой выходит за
рамки этой книги. Твердые ингибиторы, т. е. поверхности, обры-
вающие цепи, в общем, удобнее применять, чем растворимые, ко-
нечно, при сравнимой эффективности. К сожалению, теория твердых
ингибиторов разработана гораздо хуже, чем растворимых.
Подбор каталитических систем не ограничивается качественным
определением состава компонентов; требуется также определить и
их соотношение. При знании кинетики" составляющих процесс реак-
170
ций это соотношение можно определить из условий максимизации
выхода целевого продукта, аналогично тому, как это делается для
определения оптимальной температуры процесса. Однако, как легко
понять, при этом состав катализатора должен меняться по длине
реактора, что не всегда удобно. Кроме того, взаимная адсорбция
компонентов может изменить кинетику отдельных составляющих
реакций при совместном протекании процесса. Поэтому практически
при подборе соотношений компонентов каталитической системы при-
ходится большей частью исходить из некоторых средних концентра-
ций компонентов реакции и решать задачу приближенно, уточняя
состав системы экспериментально, методами направленного поиска.
ЛИТЕРАТУРА
1. И. И. Иоффе, ДАН СССР, 58, № 8, 1681 (1947).
2. L. Pauling, Proc. Roy. Soc., A196, 343 (1949).
3. В. N. Figgis, Introduction to Ligand fields, New York, 1966.
4. И. И. Моисеев, сб. «Проблемы кинетики и катализа». Вып. XIII,
Изд. «Наука», 1968, стр. 36.
5. Пат. США 3487112. Offical Gazett, 1969, 869 (5) 1606.
6. А. Ф. М и л л и д ж, Англ. пат. 1012011; РЖХим, 1966, 23Н81.
7. Франц, пат. 1518312, 1968; Chem. Absr., 70, № 21, 96402 (1969).
8. D. A. Dowden, D. Wells, Aetas du 2 eme Congress Interna-
tionale de Catalyse, Paris, 1961, p. 1489.
9. D. A. D о w d e n, N. McKenzi, В. M. T r a p n e 1 1, Proc. Roy.
Soc., A237, 245 (1956).
10. Д. А. Дауден, Основы предвидения каталитического действия. Труды
IV Международного конгресса по катализу, том. I, Изд. «Наука», 1970, стр. 39.
11. G. М. D‘i х о n, D. N i с h о 11 s, Н. S t е i п е г, Proceedings of the
III International Congress on Catalysis, vol. 2, Amsterdam, 1956, p. 185.
12. Л. Я. Марголис, Основы предвидения каталитического действия,
Труды IV Международного конгресса по катализу, т. 1, стр. 225, Изд. «Наука»,
1970.
13. О. В. Крыло в, Катализ неметаллами, Изд. «Химия», 1967.
14. А. И. Г е л ь б ш т е й н и др., Основы предвидения каталитического
действия. Труды IV Международного Конгресса по катализу, вып. I, изд. «Нау-
ка», 1970, стр. 251.
15. В. М. 3 а х т л е р и др., Там же, стр. 355.
16. Ф. Т р и ф и р о и др., Там же, стр. 218.
17. И. И. И о ф ф е, 3. И. Е ж к о в а, А. Г. Л ю б а р с к и й, ДАН
СССР, 154, № 4, 903 (1964).
18. Т. И..Т емникова, Курс теоретических основ органической хи-
мии, Госхимиздат, 1959.
19. Б. Л. М о л дав с кий, Ю. Д. К е р н о с, Кинетика и катализ, 1,
№ 3, 267 (1960).
20. L. I. Н о f е г, Р. G u s s е у, R. В. A n d е г s о n, J. of Catalysis,
3, 451 (1964).
21. F. J. Morin, Semiconductors, Ed. by N. Hannay, N. Y., 1959.
22. Ф. ф. В о л ь кенште йн, Электронная теория катализа, Физмат-
гиз, 1959.
23. В. А. Р о й т е р, Каталитическое окисление нафталина, Изд. «Нау-
кова Думка», Киев, 1963.
24. J. S. Bond, Catalysis by Metals, London, 1962.
25. И. И. И о ф ф е, Н. В. К л и м о в а, И. Я. М о к р у с о в а, ДАН
СССР, 169, № 2 (1966).
26. Веек, Wheller, Lole, Disc. Faraday Soc., № 8, 251 (1950).
27. C. Kemhall, Proc. Roy. Soc., A207, 539 (1951).
171
I
28. Каталитические свойства веществ. Справочник, под ред. В. А. Р
те ра, Изд. «Наукова Думка», Киев, 1968.
29. В. А. Пальм, Основы количественной теории органических реак|
ций, Изд. «Химия», 1967.
30. R. W.T aft, Steric Effects in Organic Chemistry, Ed. by M. S. New-’
man, New
31. A.
ка», 1964.
32. M.
33. K.
York, 1957. Я
А. Баландин, Мультиплетная теория катализа, Изд. «НауЯ
Kraus, Advances in Catalysis, XVII, New York, 1967, p. 75Я
К о x л e ф л, M. К p а у с, В. Б а ж а н т, Основы предвидений
каталического действия. Труды IV Международного Конгресса по катализуЯ
Изд. «Наука», 1970, стр. 418. Я
34. J-. Yoneda, J. of Catalysis, 9, № 1, 51 (1967). Я
35. Н. М a t s u m о t о, I. I. T a k e, I. Yoneda, ibid., 10, № 3,-Я
211 (1968). Я
36. T. U s h i j i m a, M. T a k a n a s h i, J. Y о n e d a, ibid., 10Л
№ 4, 403 (1968). Я
37. I. В a k a r d j i e v, К. H. S c h n о b e 1, K. Wencke, Z. Phys3
Chem., 239, № 112,_ 65 (1968). Я
38. В. A. P о й т e p, co. «Катализ и катализаторы», т. 4, Изд. «Наукова!!
Думка», 1968, стр. 17. Я
39. Г. И. Голодец, Ю. И. Пятницкий, Там же, стр. 25. я
40. Г. И. Голодец, Ю. И. Пятницкий, В. В. Гончару к,Я
Теор. экспер. химия, № 3, .602 (1967). 1
41. В. А. Р о й т е р, Г. И. Г о л о д е ц, Ю. И. Пятницкий, Ос-1
новы предвидения каталитического действия. Труды IV Международного кон-1
гресса по катализу, т. II, Изд. «Наука», 1970, стр. 365. |
42. Г. К. Боресков, В. В. П о и о в с к и й, В. А., С а з о н о в, |
Там же, стр. 343. . • . ’
43. W. М. Н. S а с h 11 е г, J. Н. de Boer, Proceedings of the III Inter-J
national Congress on Catalysis, vol. 1, Amsterdam, 1965, p. 252. 1
44. G. K. Boreskov, Disc. Faraday Soc., 41, 263 (1966). Э
45. Г. К. Б о p e с к о в, В. А. С а з о н о в, В. В. Поповский.
ДАН СССР, 176, № 6, 1331 (1967). i
46. Сб. «Современная квантовая химия», т. I, Изд. «Мир», 1968, стр. 59.-1
47. I. М о s h i d a, Y. V о n е d a, J. of Catalysis, 9, № 1, 57 (1967). =
48. T. Hishida, T. Ushijima, Y. Voneda, ibid'., 11, Ml, 71 (1968). J
49. X. X. Д у н к e н, X. О и и ц, Основы предвидения каталитического!
действия. Труды IV Международного Конгресса по катализу, вып. I, Изд.|
«Наука», 1970, стр. 73. |
50. D. S h о р о v, A. A n d г е е v, D. Р е t к о v, J. of Catalysis, 13, |
№ 2, 123 (1969). Я
51. Р. А. Гарднер,
Труды IV Международного
1970t стр. 399.
A. G а г d п е г,
A. G а г d п е г,
S. Sebestyen, Decision-making Process in Pattern Recognition, |
1962. J
И. Васильев, Распознающие системы, Изд. «Наукова Думйа», |
I
1
Основы предвидения каталитического действия.!
Конгресса по катализу, вып. II, Изд. «Наука», |
J. of. Catalysis, 3, № 1, 7 (1964). 3
ibid., J., 10, № 1, 290 (1968). I
52. R.
53. R.
54. G. I
New York,
55. B.
Киев, 1969.
56. M. M. Б о н г a p д, Проблемы узнавания, Изд. «Наука», 1967. 1
57. И. И. Иоффе и др., Основы предвидения каталитического действия.
Труды IV Международного Конгресса по катализу, вып. II, Изд. «Наука», ;
1970, стр. 210.
58. И. И. И о ф ф е и др., ДАН СССР, 189, № 6, 1290 (1969).
59. О. В. Крылов, Кинетика и катализ, 3, № 4, 502 (1962).
60. Л. Я. Л е й т и с и др., Изв. АН ЛатвССР, № 5, 566 (1969).
61. Дж. М. Томас, В. Дж. Томас, Принципы гетерогенного ката-:
лиза, Изд. «Мир», 1969.
172
I
ОСНОВЫ ТЕХНОЛОГИИ
ПРОМЫШЛЕННЫХ КАТАЛИЗАТОРОВ
V.I. ХАРАКТЕРИСТИКА И СТРУКТУРА
ПРОМЫШЛЕННЫХ КАТАЛИЗАТОРОВ
Требования к промышленным катализаторам. К промышленным
катализаторам, кроме требования достаточной активности и селек-
тивности, предъявляют специальные требования:
1. Желательно, чтобы катализатор был не слишком чувствитель-
ным к ядам и примесям, содержащимся в реагентах. Особенно важно,
чтобы-' не наблюдалось быстрой необратимой потери активности
катализатора.
2. Катализатор как в отношении химической активности, так
и механической прочности не должен быть очень чувствительным
к перегревам. В промышленных условиях желательно, например,
чтобы перегрев на 50—100 град выше регламентируемой темпера-
туры процесса не приводил бы к необратимой дезактивации ката-
лизатора.
3. Зерна катализатора не должны раздавливаться под тяжестью
слоя катализатора и разрушаться при свободном падении с высоты,
несколько превышающей высоту контактного аппарата.
4. Катализатор не должен заметно истираться потоком газа,
обтекающим его зерна. Для катализаторов, работающих в кипящем
слое, требования к прочности на истирание значительно повышены.
5. Катализатор не должен разрушаться или терять механиче-
скую прочность под действием процессов, протекающих на его по-
верхности.
6. Общий срок службы катализатора, независимо от причин его
определяющих, не должен быть слишком малым. Вряд ли можно
считать пригодным для нормальной промышленной эксплуатации
катализатор, который надо выгружать из аппарата чаще, чем один
раз в месяц. »
Если катализатор предназначен для работы в суспендированном
состоянии, в кипящем или движущемся слое с непрерывной цирку-
ляцией между реактором и регенератором, то для каждого конкрет-
ного процесса определяется допустимая потеря катализатора в еди-
ницу времени. При этом, естественно, нецелесообразно добиваться
высокой устойчивости катализатора к истиранию, если его дезакти-
вация по другим причинам наступает раньше его механического
разрушения. Эти качественные соображения могут быть подкреп-
лены более строгим экономическим обоснованием.
Структура катализатора. После химического состава важней-
шими факторами, определяющими активность и избирательность
катализатора, являются величина его удельной поверхности и струк-
тура пор гранул.
В настоящее время известно, что по первичной структуре пор
катализаторы и носители делятся на две группы.
Гранулы (зерна) первой группы являются вторичными образова-
ниями («вторичными» структурами), представляющими собой си-
стемы корпускулярного строения, в которых поры образованы про-
межутками между первичными мелкими частицами различной формы
(«первичными» структурами). К этой группе относятся ксерогели
(стекловидные силикагель, алюмогель и другие гели), керамика,
в том числе корундовая и др. Следует отметить, что ксерогели имеют
глобулярное строение, т. е. получаются в результате агломерации
Рис. V.I. Модели глобулярных систем:
а — мелкопористой, малые частицы упакованы с числом касаний 6; б— крупнопористой,
большие частицы упакованы с числом касаний 3.
первоначальных сферических частиц геля, образующихся в момент
осаждения [1 ]. Корпускулярное, в том числе глобулярное строение
гранул катализатора дает возможность связать величину пор с ти-
пом упаковки частиц (координационным числом) и размером перво-
начальных частиц [2] (рис. V.1).
У второй, сравнительно небольшой группы катализаторов и но-
сителей поры представляют собой каналы, образовавшиеся за счет
выделения газов при сушке, выгорании или растворении компонен-
тов твердого тела. К этой группе относятся активные угли, скелет-
ные катализаторы и возможно некоторые другие системы.
В табл. V.1 представлены величины удельной поверхности и
диаметра пор некоторых широко применяемых катализаторов и но-
сителей.
Особую группу катализаторов в отношении структуры состав-
ляют цеолиты — молекулярные сита. В них нет пор в обычном
смысле этого слова, а диффузия реагентов в глубь катализатора
происходит по внутрикристаллическим пустотам.
174
Таблица V.1
Характеристика пористой структуры некоторых
катализаторов и носителей
Катализатор или носитель Удельная поверх- ность, м2 / г Средний радиус пор, А Эффективный коэффициент внутренней диффузии по Аг, см2 /сек Эффективный коэффициент теплопро- водности, ккал/(м»чх Хград)
Активный уголь БАУ 5501050 10-15 0,0036 0,0018
Алюмосиликатный катализатор
крекинга 180-200 30-40 — 0,0024
Гель окиси хрома 20 — —
Двуокись титана 7-9 — -— —
Катализатор риформинга .... 160 80 0,027 0,0015
Корундовая керамика 1 1000 — —
Никель-хромовый катализатор 180 90 0,006 —
Окись алюминия, прокаленная
при:
450° С 244 15 —. —
900° С 121 17-34 0,027 —
1100° С 80 40-80 — —
Прессованная серебряная чернь — 150 -— 0,61
Силикагель:
крупнопористый 320 100 -— —
тонкопористый 320-400 50 0,003 —
Вторичные частицы (гранулы), являющиеся бидисперсными, по-
лучаются агломерацией тем или иным технологическим приемом
(см. раздел V.2) частиц с первичной структурой.
Для каждого процесса может быть подобрана некоторая опти-
мальная пористая структура зерен катализатора, определяемая соот-
ношением скоростей химических превращений и диффузии веществ,
участвующих в данном процессе. Для реакций с участием веществ
малого молекулярного веса, ведущих в условиях реакции к стабиль-
ным целевым продуктам, целесообразно применять катализаторы
с большой удельной поверхностью и, следовательно, с сильно раз-
витой системой пор малого диаметра. Повышение молекулярного
веса реагентов или продуктов реакции приводит к понижению ско-
рости их диффузии, вследствие чего для проведения быстрых реак-
ций требуются более широкопористые катализаторы, поверхность
которых обычно невелика.
Особо чувствительны к характеру пористости частиц реакции
контактного окисления. В этом случае скорости реакций, как пра-
вило, велики, а целевые продукты неустойчивы в условиях реакции.
Это приводит к тому, что при появлении внутридиффузионного тор-
можения усиливается дальнейшее окисление целевого продукта и
селективность процесса падает. С количественной стороны этот во-
прос будет рассмотрен в разделе V.3.
175
V.2. КРАТКИЕ СВЕДЕНИЯ
О ТЕХНОЛОГИИ ПРОИЗВОДСТВА КАТАЛИЗАТОРОВ
Классификация катализаторов. Основными технологическими опе-
рациями в производстве гетерогенных катализаторов различных ти-
пов являются осаждение, пропитка, фильтрация, промывка осадка,
сушка, прокалка, формовка. Наиболее распространены из них две:
1) осаждение активной части катализатора в виде кристаллического
осадка или геля при взаимодействии водных растворов двух или
нескольких химических соединений; 2) пропитка каталитически
неактивного твердого вещества — носителя — раствором (обычно
водным) активных соединений. Для получения катализаторов при-
меняют также и другие, специальные способы, например, термиче-
ское разложение соединений, выщелачивание растворимых частей
сплавов или природных материалов и др.
По методам приготовления и химической природе промышлен-
ные катализаторы можно разделить на следующие группы:
1. Осажденные (солевые, окисные, кислотно-основные). Катали-
заторы этой группы представляют собою либо монолитные гели,
либо вторичные формованные структуры.
2. Катализаторы на носителях, или импрегнированные катали-
заторы (солевые, окисные, кислотно-основные, металлические). Ката-
лизаторы этого типа получают либо на готовых зернах носителя,
либо также методом формования.
3. Плавленые * (металлические и окисные).
4. Скелетные (металлические).
5. Цеолитные (молекулярные сита).
6. Органические (ионообменные смолы).
Конечно, приведенная классификация неполна; например, она
не включает коллоидные катализаторы, природные катализаторы и
некоторые другие.
Надо также иметь в виду, что граница между носителями и ката-
лизаторами весьма условна.
Осужденные гелевые катализаторы. Отличительной особенностью
осажденных катализаторов является, во-первых, то, что в основу
технологии их приготовления положен метод соосаждения активных
составляющих катализатора, а, во-вторых, то, что в составе катали-
затора отсутствует носитель, т. е. инертное твердое вещество, обра-
зующее самостоятельную фазу, на поверхность которого наносят
активные составляющие катализатора. Соосаждение составных ком-
понентов катализатора приводит к образованию либо монолитной
гелеобразной структуры, которой присуща механическая прочность,
либо кристаллических осадков или дробленых частиц аморфной
структуры, требующих дальнейшей обработки для превращения
их в прочные гранулы катализатора.
* Все монолитные металлические катализаторы заданной формы (сетки,
цилиндры, проволочные спирали) относятся нами к плавленым.
176
Наиболее распространенными монолитными гелеобразными ката-
лизаторами являются алюмосиликат и силикагель; последний ча!це
используют как носитель. Технология производства этих двух ката-
лизаторов наиболее подробно описана. Ознакомимся на их примере
с общей технологией производства монолитных гелей.
Горячий
Рис. V.2. Схема производства шарикового алюмосиликатного
катализатора:
1 — распределительный конус; 2 — формовочная колонна; з — фильтр; 4 —
емкость для закалки; 5 — емкость для активации и промывки; 6 — сушилка;
7 — печь для прокаливания.
В настоящее время распространенной технологией получения
силикагеля и алюмосиликагеля является коагуляция в капле.
При этом частицы катализатора получаются в удобной для большин-
ства процессов сферической форме. Кроме того, метод коагуляции
в капле дает возможность легко организовать производство катали-
затора по непрерывной схеме, а также избежать применения формо-
вочных машин. Применительно к алюмосиликатному катализатору
технология производства по методу коагуляции в капле (рис. V.2)
сводится к следующему [3, 4].
Раздельно готовят растворы сульфата алюминия и жидкого
стекла (силиката натрия). Чтобы образовался гель высокопрочной
структуры, требуется медленное протекание коагуляции (5—15 сек).
Мгновенная коагуляция приводит к образованию рыхлых меловидных
12 Заказ 1441
177
гелей, не сохраняющих сферической формы. Скорость коагуляции
определяется температурой, величиной pH раствора, в котором
происходит осаждение. Она уменьшается с повышением кислот-
ности раствора, а кроме того, зависит от концентрации соосаж-
даемых солей.
Для процессов коагуляции в капле особое значение приобретает
точная дозировка растворов, так как от этого зависит не только
качество получаемого продукта, но и возможность образования
частиц определенной формы и размера.
Смешение реагентов осуществляется либо с помощью механиче-
ских мешалок, либо в струе: в кислый раствор сульфата алюминия
подается с высокой скоростью раствор жидкого стекла, что обеспе-
чивает хорошее их смешение. Образовавшийся в результате смеше-
ния золь поступает на распределительный конус 1, имеющий ряд
продольных желобков, по которым раствор стекает в виде отдельных
струек в основной аппарат — формовочную колонну 2. В верхней
части колонна заполнена циркулирующим минеральным маслом.
Струйки золя с распределительного конуса попадают в масло, где
и разбиваются на отдельные капли. Величина капель, определяю-
щая величину готовых гранул катализатора, зависит от диаметра
желобков, скорости струек, поверхностного натяжения и вязкости
масла. Коагуляция должна протекать за время падения капли через
слой масла.
Пройдя масло, шарики скоагулировавшегося геля попадают
в водный раствор солей, который получается от промывки предыду-
щих партий геля. Раствор солей циркулирует под слоем масла,
проходя нижнюю часть колонны снизу вверх и унося с собой шарики
геля в следующие аппараты. При промывке в емкосте 5 гель претер-
певает синерезис, при котором происходит выделение интермицел-
лярной воды. Теряя воду, шарики геля сжимаются в радиальном
направлении, что ведет к появлению в них тангенциальных напря-
жений, способных разрушить шарик. Для предотвращения этого
шарики геля предварительно подвергаются термической обработке
(закалке) горячим раствором солей, что упрочняет структуру геля.
Термическая обработка не должна сопровождаться вымыванием
солей, а потому горячий раствор содержит соли в концентрации,
соответствующей концентрации солей в интермицеллярной жидкости.
Следующая технологическая стадия — активация — носит менее
общий характер и связана с механизмом действия алюмосиликат-
ного катализатора. Алюмосиликатные катализаторы представляют
собой твердые кислоты. В процессе осаждения происходит замеще-
ние активных атомов водорода натрием, что полностью снимает
каталитическую активность геля. Поэтому в дальнейшем гель под-
вергается активации в емкосте 5 либо слабой кислотой, либо раство-
ром сульфата алюминия, причем последний предпочтителен. Следы
натрия окончательно удаляются промывкой раствором сульфата
аммония. После активации шарики подвергаются тщательной про-
мывке очищенной водой до практически полного удаления солей.
178
Ответственной операцией в производстве монолитных гелей
является сушка. Как и скорость диффузии солей при промывке,
скорость диффузии паров воды при сушке должна быть ограничен-
ной, поэтому во избежание нарушения прочности шариков катали-
затора сушку проводят в токе водяного пара при постепенном повы-
шении температуры. Растрескивание шариков уменьшается при
введении в воду для промывки геля поверхностно-активных веществ,
снижающих поверхностное натяжение жидкости [5].
Во время прокаливания катализатора удаляется вся остаточная
влага и шарики приобретают значительную механическую проч-
ность. С повышением температуры прокаливания механическая
прочность катализатора возрастает, однако одновременно умень-
шается его удельная поверхность, а следовательно, и каталитиче-
ская активность. Без ущерба для активности алюмосиликатного
катализатора температуру его прокаливания можно довести до
750° С [6].
Описанная технология является типичной для всей группы моно-
литных гелеобразных катализаторов. Производство сферического
силикагеля состоит из тех же стадий (за исключением активации),
только в качестве реагирующих растворов применяют не жидкое
стекло и сульфат алюминия, а жидкое стекло и серную кислоту.
Осажденные формованные катализаторы. Если по своим физико-
химическим свойствам осаждаемый катализатор не образует моно-
литного геля или имеет кристаллическую структуру, или, наконец,
если структура монолитного геля нежелательна, ввиду значительного
внутридиффузионного торможения проводимой реакции, осаждение
катализатора ведут обычными методами. Полученные осадки отфиль-
тровывают от маточного раствора и затем промывают. При использо-
вании в качестве реагентов соединений, образующих в виде побочных
продуктов термически нестойкие соли, например нитрат аммония,
стадия промывки может быть или совсем исключена, или проведена
не полностью. Дальнейшая технология зависит от природы осадка
и требований к прочности катализатора. В редких случаях (при
проведении контактных реакций в жидкой фазе) осадок размалы-
вают и катализатор применяют в виде порошка.
Наиболее универсальный метод формования гранул катализа-
тора — таблетирование порошков. В этом случае осадок после от-
мывки солей (в случае, если эти соли термически стойки) сушат,
прокаливают (обычно при температуре не ниже температуры реак-
ции) и тщательно размалывают. Размолотый порошок таблетируют
на специальных машинах, где давление достигает 300 ат и более.
Если порошок катализатора плохо таблетируется, к нему добавляют
тальк, графит или другие связующие материалы.
В качестве примера технологии таблетированных катализаторов
можно привести схему производства железо-хромового катализа-
тора (рис. V.3). Согласно описанию [71, сульфат железа (II) и хро-
мовый ангидрид растворяют в воде и готовый раствор насосом пере-
качивают в осадитель 2, снабженный воздушным барботером для
12*
179
перемешивания массы. В осадитель добавляют раствор едкого натра
и дают острый пар для подогрева. Полученный шлам гидроокисей
железа и хрома перекачивают насосом в стальной чан 3. Массу
в чане подогревают паром и промывают декантацией до отрицатель-
ной реакции промывных вод на примеси. Отмытый шлам насосом
прокачивают через фильтр-пресс 4. Осадок из пресса вновь возвра-
щают в чан, перемешивают с водой и насосом подают в буферный
Смесь окислов
(Ре2О3 +СГ2О3)
Рис. V.3. Схема производства железо-хромового таблетированного катализа-
тора:
1 — емкость для раствора; 2 — чан для осаждения; 3 — чан для промывки; 4 — фильтр-
пресс; 5 — чан-хранилище; 6 — распылительная сушилка; 7 — бункер-хранилище; 8 —
смеситель; 9 — таблеточная машина.
чан-хранилище 5. В зависимости от назначения катализатора даль-
нейшую обработку полученных гидроокисей ведут по одной из двух
технологических схем.
По первому варианту (получение катализатора А) суспензия
гидроокисей из чана поступает в распылительную сушилку 6, обо-
греваемую топочными газами. Полученный в сушилке порошок
окисей железа и хрома подают в бункер-хранилище 7, а затем —
в смеситель S, где происходит смешение с добавками, облегчающими
таблетирование. Полученную в смесителе массу таблетируют на
машинах 9 в таблетки диаметром 9—12 мм и направляют на упа-
ковку.
По второму, более сложному, варианту (получение катализа-
тора В) шлам из чана-хранилища поступает на фильтр-пресс. Отжа-
тый осадок сушат, прокаливают, обрабатывают на бегунах со свя-
зующими добавками, гранулируют и затем таблетируют на таблеточ-
ной машине. Необходимость предварительного гранулирования свя-
180
зана с тем, что в ряде случаев при таблетировании мелких гранул
диаметром — 1 мм можно получить более прочные таблетки, чем при
таблетировании непосредственно из порошка окислов.
Таблетированные катализаторы, как правило, уступают боль-
шинству других по прочности; они сильнее подвержены эрозии пото-
ком газа и термическому растрескиванию. Поэтому таблетирование
катализаторов в значительной степени вытеснено другими видами
формования, среди которых наиболее распространены экструзия и
вмазывание.
При экструзии влажный осадок, часто со связующим веществам,
выдавливают в виде шнура из непрерывно действующего шнеко-
вого или гидравлического пресса. Форму и поперечный размер шнура
определяют по отверстию в формующей головке пресса. На выходе
из головки шнур катализаторной массы разрезают, а полученные
червеобразные гранули катализатора сушат и прокаливают.
Методом вмазывания производят, например, алюмосиликатный
катализатор, применяемый для различных процессов в виде непо-
движного слоя [3, 4]. Первоначальные стадии технологической
схемы аналогичны стадиям описанного выше производства гелевого
алюмосиликата, с той разницей, что коагуляцию производят в аппа-
рате с мешалкой (пропеллерной или турбинной), куда одновременно
подают растворы жидкого стекла и сульфата алюминия. При таком
осаждении гель образует с водой однородную массу-шлам. Шлам
геля после синерезиса направляют на отмывку и активацию ката-
лизатора и затем отфильтровывают на непрерывно действующих
фильтрах или фильтр-прессах. Полученную мелкозернистую влаж-
ную массу, состоящую из мельчайших частиц алюмосиликагеля,
формуют на специальных машинах для получения гранул катали-
затора в виде цилиндриков. При различной конструкции машин
процесс в них принципиально один и тот же: пасту осадка вмазывают
в отверстия перфорированного стального листа или стенок полого
барабана. На рис. V.4 показан общий вид формовочной машины
вмазывания барабанного типа. Вмазанный катализатор подвергается
сушке. Высушенные цилиндрики катализатора либо выбивают спе-
циальным штампом, либо выдавливают сжатым воздухом, а затем
досушивают и прокаливают. В отличие от монолитных гелевых
катализаторов, сушка формованных катализаторов не требует боль-
ших предосторожностей. При быстром синерезисе частиц геля,
склеенных в гранулу, не могут возникнуть опасные напряжения
из-за слишком малой величины частиц.
Кроме экструзии на прессах и вмазывания удобным методом фор-
мования катализаторов является гранулирование на тарельчатом
грануляторе. Частицы катализатора получаются при этом сфериче-
ской формы. Технологическая схема производства катализатора
с применением тарельчатого гранулятора для целей формовки опи-
сана в работе [8].
Для микросферических катализаторов, применяемых в процессах
с кипящим слоем, формование гранул можно вести путем распыления
181
шлама осадка на распылительной сушилке. Технология производ-
ства такого катализатора сводится, например к следующему [9].
Из раствора жидкого стекла серной кислотой осаждают гель крем-
невой кислоты, его отфильтровывают на барабанных фильтрах,
отмывают от солей, разбалтывают в растворе сульфата алюминия и
затем на взвеси силикагеля производят осаждение алюмогеля аммиа-
ком. Суспензию смешанного геля подают на распылительную су-
Рис. V.4. Формовочная машина вмазывания
барабанного типа.
шилку, частицы катализа-
тора после сушки имеют
вид мелких сферических
гранул. Полученные на
распылительной сушилке
гранулы прокаливают.
Производство микросфери-
ческого алюмосиликатного
катализатора с формова-
нием на распылительной
сушилке описано в рабо-
тах [10, 11].
Кроме приведенных
выше окисных и кислот-
ных катализаторов (алюмо-
кремневая кислота), в про-
изводстве находят приме-
нение и осажденные соле-
вые катализаторы. Так,
в производстве фенола
парофазным гидролизом
хлорбензола применяют
катализатор, получаемый
осаждением трикальций-
фосфата аммиаком из ра-
створов хлорида кальция
и фосфата натрия [12].
Катализаторы на носи-
телях. Наиболее распрост-
раненным технологическим приемом производства катализаторов
на носителях является пропитка. Для пропитки, как правило,
применяют водные растворы солей активного компонента катализа-
тора. В общем случае пропитка гранулированного носителя со-»
стоит из следующих стадий: эвакуации газа из носителя, обработки
носителя раствором, удаления избытка раствора, сушки и про-
калки.
Эвакуация газа из носителя перед его погружением в пропиты-
вающий раствор приводит к более равномерному распределению
активного компонента в катализаторе, поскольку находящийся
в порах воздух препятствует проникновению раствора. Однако из-за
усложнения технологии при использовании стадии эвакуации в про-
182
мышленности ее избегают. Равномерность пропитки обеспечивают
длительным пребыванием носителя в подогретом пропитывающем рас-
творе или добавкой к нему поверхностно-активных веществ.
Пропитку гранулированного носителя осуществляют различными
способами. Часто применяют метод пропитки в избытке раствора.
В этом случае предварительно определяют адсорбционное равнове-
сие между раствором разных концентраций и носителем. Особо сле-
дует обратить внимание на возможность избирательной адсорбции
компонентов из раствора носителем. Пропитывающий раствор гото-
вят такой концентрации, чтобы поглощенное по расчету количество
солей создавало в готовом катализаторе нужную концентрацию актив-
ного компонента. Пропитку гранул (в том числе и таблеток) носи-
теля можно осуществлять достаточно примитивно в чанах или чашах
с последующим отделением избытка раствора на нутч-фильтрах или
центрифугах. Более рациональным, однако, является применение
специальных пропиточных машин [13], представляющих собою
движущуюся бесконечную ленту, на которой подвешены сетчатые
корзины из нержавеющей стали или другого материала. Носитель
загружают из бункера в корзины. При движении ленты корзины
опускаются на некоторое время в короб с пропитывающим раство-
ром, а затем приподнимаются и перемещаются в обратном направ-
лении над коробом, давая раствору стечь в него. Далее лента машины
с подвешенными корзинами может, например, последовательно про-
ходить тоннельные сушилку и прокалочную печь.
Для производства небольших количеств катализатора при ис-
пользовании малых избытков пропитывающего раствора применяют
метод пропитки с последующим испарением.
Можно осуществлять пропитку носителя расплавом окислов.
В работах [14, 15] описывается применение катализаторов из ко-
рунда, пропитанного расплавом пятиокиси ванадия или смеси пяти-
окиси ванадия с трехокисью молибдена, соответственно для окисле-
ния нафталина и о-ксилола во фталевый ангидрид и бензола в мале-
иновый ангидрид.
В ряде случаев раствором активных компонентов пропитывают
не гранулированный носитель, а порошкообразный, а затем фор-
муют гранулы. Так, катализатор дегидрирования и гидроформинга
получают пропиткой порошка окиси алюминия раствором молибдата
аммония [16 ]. Пропитку порошка проводят либо в смесительном ба-
рабане, если процесс периодический, либо на ленточном транспор-
тере — при непрерывном производстве. Пропитанный порошок но-
сителя сушат, активируют, а затем таблетируют.
Своеобразным видом пропитки носителя активной составляющей
является введение последней в виде пара, например нанесение ионов
хлора или трехфтористого бора на подкладку из окиси алюминия [17 ]
или алюмосиликата [18].
Как уже отмечалось, пропитывающий раствор большей частью
содержит не активные компоненты катализатора как таковые, а со-
единения, переходящие в эти компоненты, после тех или иных
183
химических превращений. Когда на носитель желательно нанести
окислы, пропитку ведут раствором термически нестойких солей, таких
как нитраты, формиаты, оксалаты, аммониевые соли, иногда при-
меняют также легко разлагающиеся кислоты, например хромовую
при производстве хромсодержащих катализаторов. В ряде случаев
для нанесения активного компонента приходится комбинировать
методы пропитки и осаждения, например при получении катализа-
торов из нерастворимых солей (фосфатов, карбонатов, силикатов
и др.) на инертных носителях. При этом обычно пропитывают носи-
тель растворимой солью осаждаемого металла, сушат его и затем
вносят пропитанный и просушенный носитель, содержащий осажда-
емую соль, в раствор осадителя, например фосфата натрия.
Иногда, например при относительно низкой растворимости солей,
внести нужное количество активных компонентов в носитель удается
только путем многократных пропиток. При этом после каждой про-
питки лучше перевести пропитывающее соединение в нерастворимое
состояние — термически нестойкую растворимую соль путем про-
калки превратить в нерастворимый окисел. В противном случае при
каждой последующей пропитке произойдет вымывание содержимого
из носителя и невозможно будет достичь желаемого эффекта. Коли-
чество пропиток определяется составом катализатора, однако много-
кратное их повторение сильно усложняет технологический процесс
и удорожает катализатор.
Удобным методом производства импрегнированных катализаторов
является пропитка носителя рассчитанным количеством раствора
без его избытка. Пропитку производят разбрызгиванием раствора на
носитель, перемешиваемый во вращающемся барабане или бетоно-
мешалке. В случае необходимости барабан или бетономешалку снаб-
жают обогревом.
Для получения металлических катализаторов на носителях тре-
буется восстановление окислов или солей газом (водородом, парами
спирта) либо восстанавливающим раствором. В первом случае через
катализатор, предварительно прокаленный для перевода солей
в окислы, пропускают газ-восстановитель при повышенной темпера-;
туре. Очень часто процесс восстановления ведут непосредственно
в реакторе. Примером металлических катализаторов на носителе,
восстанавливаемых из солей растворами, являются платиновые ката-
лизаторы на окиси алюминия и на силикагеле. Для восстановления
соединений платины используют аммиачный раствор формальдегида
[19]. При приготовлении платино-силикагелевого и аналогичных
катализаторов надо иметь в виду, что непосредственная пропитка
геля раствором часто приводит к растрескиванию геля. Причина
этого, вероятно, кроется в возникновении при быстрой гидратации
внутренних напряжений в геле, аналогичных возникающим во время
ускоренной дегидратации, или в более простом эффекте эа счет дав-
ления сжимаемого в капиллярах зерна воздуха. Для устранения рас-
трескивания гель перед пропиткой насыщают водой, пропуская через
него сильно увлажненный воздух [16 ].,
184
К катализаторам, получаемым методом импрегнирования, следует
отнести и так называемую «твердую фосфорную кислоту», хотя дан-
ный катализатор имеет существенные особенности. В этом случае
пропитывающее соединение, т. е. ортофосфорная кислота, взаимо-
действует с носителем (силикагелем, кизельгуром), образуя новое
соединение — кремнефосфорную кислоту, которая и является актив-
ным компонентом катализатора. В отличие от ранее описанных при-
меров, в ряде вариантов приготовления этого катализатора коли-
чество пропитывающего соединения может превосходить количество
носителя. Имеется несколько методов промышленного производства
«твердой фосфорной кислоты» [20, 21]. По одному из них кизельгур,
или лучше синтетический кремнезем, в количестве—30% от веса
катализатора замешивают в обогреваемом до 170—180° С смесителе
с упаренной ортофосфорной кислотой (с содержанием 78—79% Р2О6),
взятой в количестве —70% от веса катализатора. Далее массу выдер-
живают при 320° С, растирают, увлажняют и формуют в виде «чер-
вяков».
Плавленые, скелетные и прочие катализаторы. К числу плав-
леных катализаторов прежде всего относятся металлические ката-
лизаторы ненанесенного типа. Отдельные представители группы
плавленых катализаторов, как, например, катализаторы синтеза и
окисления аммиака, получили широкое распространение. Другие
катализаторы — металлокерамические — только начинают находить
применение. В целом, однако, этот класс катализаторов в настоящее
время менее распространен, чем осажденные катализаторы и катали-
заторы на носителях.
Плавленые катализаторы делятся на два типа: окисные и метал-
лические. Технологию производства плавленых окисных катализа-
торов лучше всего рассмотреть на примере производства катализато-
ров синтеза аммиака, получаемых путем сжигания железа в пламени
кислорода с образованием расплава магнитной окиси железа.
По патентам Баденской анилиновой и содовой фабрики [221 катали-
затор готовят сжиганием в кислородном пламени железа ^высокой
степени чистоты с добавками специальных промоторов. Получаемый
сплав размельчают до частиц нужных размеров.
Обычно плавленые металлические катализаторы применяют в виде
стружек, сеток или проволочных спиралей. Платиновые катализа-
торы такого типа, как известно, используют для окисления аммиака
в азотную кислоту. Что касается производства таких катализаторов,
то, очевидно, специальной операцией может быть только составление
сплава нужного состава, если, конечно, это необходимо. Однако,
кроме того, плавленые катализаторы могут подвергаться и обработке
для разрыхления поверхности с целью увеличения их активности.
Так, плавленый никелевый катализатор гидрирования активируют
либо анодным окислением, либо -окислением гипохлоритом [23].
Большое значение, в основном для малотоннажных производств,
приобрели скелетные катализаторы, так называемые катализаторы
Ренея. Готовят их выщелачиванием сплава каталитически активного
185
металла с металлом, растворимым в щелочах или кислотах. В ка-
честве активных металлов нашли применения никель, кобальт, медь
и железо. Растворимыми компонентами являются алюминий, магний,
цинк. Особенно широкое применение, в ряде промышленных процес-
сов, приобрел никель Ренея, получаемый выщелачиванием Ni—Al
сплава 20—30% раствором едкого натра. Для практических целей
применяют исходный Ni—Al сплав с содержанием 35—50% никеля.
Способы приготовления никеля Ренея для промышленных целей из-
ложены, например, в монографии [24]. Готовый катализатор пред-
ставляет собой тонкий порошок, который во избежании самовозгора-
ния хранят и транспортируют в виде масляной суспензии.
Большое распространение наряду с катализатором Ренея получил
катализатор Бага — кусочки сплава Ni—Al, выщелоченные лишь
частично [25]. Баг с сотрудниками нашли, что, например, для гидро-
генизации жиров наибольшая активность катализатора достигается
при удалении из сплава всего 8% алюминия. Катализатор Бага,
в отличие от катализатора Ренея, обладает тем преимуществом, что
может применяться в проточных аппаратах в виде неподвижного слоя.
Катализаторы типа Ренея не поддаются регенерации. Несколько
лучше в этом отношении катализаторы Бага, которые можно неко-
торое время активировать, дополнительно обрабатывая раствором
щелочи, однако и они недолговечны.
Скелетным катализаторам посвящен ряд монографий и обзорных
статей [23, 26, 27].
Одной из групп катализаторов, находившей до недавнего времени
довольно широкое применение, являются природные катализаторы —
глины. Природные глины: гумбрин, флоридин, каолин и другие —
естественные алюмосиликаты калия, натрия и магния. Их применяют
в качестве катализаторов процессов крекинга нефти, а также для
других процессов, катализируемых твердыми кислотами. Кислые
природные алюмосиликаты, например гумбрин, требуют только раз-
мола или размола и формования. Нейтральные глины, такие как
каолин, необходимо предварительно активировать. Активирование
глин заключается в обработке их горячей кислотой, в результате
чего происходит растворение щелочных компонентов глины, а не-
растворимая часть превращается в гидратированный кислый алюмо-
силикат, содержащий способные к обмену атомы водорода.
В последнее время в качестве катализаторов начинают исполь-
зовать ионообменные смолы. Пока нет сведений о специальной ре-
цептуре для производства ионообменных смол-катализаторов. Об-4
щие же методы приготовления ионообменных смол приведены в не-
которых обзорах [28]. Наряду с синтетическими ионообменными смо-
лами в качестве катализатора можно применять и сульфоуголь [29].
Особое место среди катализаторов занимают молекулярные сита —
цеолиты. Их свойства, как указывалось в главе I, обусловливают
большую перспективность этого типа катализаторов. Принцип по-
лучения цеолитов заключается в рекристаллизации аморфных гелей
щелочных или щелочноземельных алюмосиликатов путем гидротер-
186
мальвой обработки. Полученные таким образом осадки кристалли-
ческих алюмосиликатов фильтруют. В ряде случаев первоначальный
осадок до сушки подвергается обработке раствором солей с целью
обмена катиона. Необходимую кристаллическую структуру цеоли-
тов получают при их приготовлении вариацией многочисленных
параметров — температура и время гидротермальной обработки, со-
став исходного геля, щелочность среды и т. п. Более подробно прин-
ципы синтеза цеолитов и технология их производства описаны в мо-
нографиях [30, 31].
Носители для катализаторов. Значительное количество ката-
лизаторов готовят на гелевых, носителях, которые сами по себе яв-
ляются катализаторами для ряда процессов. Это, прежде всего, си-
ликагель, окись алюминия, алюмосиликагель. Методы получения
этих носителей были рассмотрены выше.
В качестве носителей для катализаторов применяют широкий круг
веществ, которые обычно специально не получают для целей катализа,
а только подвергают некоторой очистке и активации. Сюда относятся,
например, активный уголь, пемза, кизельгур, асбест и др.
Относительно недавно в качестве носителей стали использовать
специальным образом приготовленную керамику. Применяют кера-
мику на основе а-окиси алюминия (корунда), окиси циркония, си-
ликата циркония (циркона), карборунда, динаса, муллита. Кера-
мические носители инертны, температуростойки и могут изготов-
ляться с диаметром пор 2000—3000 А. Возможность получения ши-
роко- и малопористых носителей особенно важна при синтезе ката-
лизаторов для получения целевых продуктов, являющихся промежу-
точными в системе последовательных необратимых реакций, напри-
мер в реакциях окисления. Характеристики основных керамических
носителей даны в работе [32].
Метод приготовления корундового носителя сводится к следую-
щему: мелко измельченный и предварительно прокаленный глинозем
(температура прокаливания порядка 1400° С) формуют в гранулы
нужного размера и прокаливают при более высокой температуре
(1500° С и выше). Механизм спекания первоначальных частичек свя-
зан с диффузией вещества в точках касания зерен. Рекомендуемые
добавки (TiO2, MgO), очевидно, способствуют образованию дефектов
в решетке окиси алюминия и тем самым понижают температуру спе-
кания. Поверхность корундового носителя колеблется от десятых
до нескольких квадратных метров на 1 г, а пористость — примерно
от 5 до 40% в зависимости от условий приготовления.
V.3. ОЦЕНКА ЭФФЕКТИВНОСТИ
И ОПТИМИЗАЦИЯ ПРОМЫШЛЕННЫХ КАТАЛИЗАТОРОВ
Эффективность катализаторов определяется технико-экономиче-
скими показателями производств, в которых они применяются.
В общем виде эффективность катализатора можно оценить крите-
рием, который имеет экономическую природу и выражает прибыль
187
каталитического производства, отнесенную к единице веса расхо- i
дуемого катализатора в единицу времени. Такой критерий описы- ;
вается формулой ;
—L (V.i)
где П — годовая прибыль производства; Q — годовой объем произ-
водства по целевому продукту, т; Ц — цена 1 т целевого продукта;
Ф — себестоимость 1 т целевого продукта; q — годовой расход
катализатора в производстве, т.
В таком общем виде критерий не пригоден для решения конкрет-
ных задач выбора оптимального катализатора. Раскроем значения
величин, входящих в уравнение (V.1). Объем производства нагляднее
всего выражается через весовую производительность единицы объема
катализатора
<? = S - 8760 (———WKaT (V.2)
\ 1рабТ грег /
где G — часовая производительность катализатора по целевому про-
дукту, т/м3; £ — коэффициент, определяющий долю времени экс-
плуатации производства в течение года; ipa6 — длительность рабо-
чего цикла катализатора; iper — длительность цикла регенерации
катализатора; Укат — объем загрузки катализатора, м3.
Расход катализатора бцределяется как величиной производитель-
ности катализатора,'так и общей длительностью его работы (сроком
службы) и отношением полезного и бесполезного времени при экс-
плуатации реактора.
Себестоимость продукта включает в себя все расходные статьи
калькуляции, такие как расходы на сырье, энергию, капитальные
затраты на сооружение установки, а также прочие статьи расхода.
Расходы по сырью Фс удобно выразить через расходные коэффи-
циенты, отражающие селективность катализатора
Фс=6Укат{2 (V.3)
где v}- — цена J-го вида сырья; — расходный коэффициент по
/-му виду сырья.
В общем случае составляющие критерия оптимальности зависят
от всех факторов, управляющих свойствами катализатора: химиче-
ского состава, структуры пор и размера и формы частиц. Строгое
решение задачи оптимального подбора катализатора с отысканием
глобального максимума критерия по всем факторам должно произ-
водиться по уравнениям математической модели процесса с учетом
режима эксплуатации катализатора, поскольку последний суще-
ственно влияет не только на кинетику химической реакции, но и на
общую длительность работы катализатора, частоту и длительность
его регенераций. При этом еще надо учитывать концентрации целе-
вого и побочного продуктов реакции в катализате, поскольку это отра-
жается на расходах по стадиям выделения и очистки целевого про-
188
дукта. Это, однако, требует очень большого объема эксперименталь-
ных исследований, математически недостаточно разработано и пока
не практикуется. Для решения конкретных вопросов задачу при-
ходится расчленять, подбирая раздельно оптимум по каждому из
названных факторов.
Все элементы критерия оптимальности зависят от химического
состава катализатора *. Методами, изложенными в главе IV, или
чисто эмпирическим поиском удается наметить один или несколько
вариантов состава химически активного катализатора. Однако для
экономически обоснованного выбора катализатора следует уточнить
зависимость критерия оптимизации от состава катализатора для вы-
бранных вариантов. Такую зависимость можно выявить дополни-
тельной постановкой специально спланированных направленных экс-
периментов и выразить величины G, ф, £раб, £рег и другие как функ-
ции состава катализатора, например в виде полиномов. Либо, что
менее строго, но требует меньше времени, произвести расчет кри-
терия для ряда вариантов состава катализатора. В первом случае
оптимизацию по критерию можно провести методами математического
программирования, а во втором просчетом и сравнением значения
критерия оптимизации при различных вариантах. При этом, конечно,
исследования должны проводиться с максимальным исключением
влияния диффузионных факторов на результаты. Тогда оптимизацию
структуры и формы катализатора можно проводить для данного
состава как второй этап решения общей задачи оптимизации ката-
лизатора.
Пористая структура и размеры зерна катализатора через диффу-
зионные явления, прежде всего длияют на активность и избиратель-
ность катализатора. Эти вопросы рассматривались в главе III. Од-
нако структура катализатора влияет не только на эти свойства.
Она определяет в значительной мере механическую прочность ката-
лизатора и тем влияет на егодолговечность. Скорость зауглероживания
катализатора и скорость регенерации, также зависят от структуры
пор катализатора. Форма и размер зерен определяют и гидрав-
лическое сопротивление слоя катализатора и следовательно энерге-
тические затраты на транспорт потока. В отношении активности и
селективности катализатора и сопротивления слоя можно в более
или менее строгой форме применять теоретически обоснованные ме-
тоды оптимизации структуры и формы, в отношении же остальных
свойств, на которые влияют структура и форма, приходится при-
менять названные выше методы эмпирической оптимизации или рас-
четного сравнения отдельных вариантов.
В отношении активности, вероятно, впервые вопрос оптимальной
пористой структуры катализатора был рассмотрен Боресковым [33].
Анализируя размерности, Боресков пришел к выводу, что при невы-
соких давлениях оптимальной является неоднородная структура,
* Приближенно, без учета стадии выделения, объем капитальных затрат
обратно пропорционален производительности катализатора.
189
т. е. система пор катализатора должна состоять из длинных и широ-
ких транспортных пор, задачей которых является подвод реагирую-
щих веществ в глубь зерна, и коротких и тонких капилляров, где
происходит превращение реагирующих веществ. Такая структура
обеспечивает полное использование внутренней поверхности катали-
затора и в несколько раз увеличивает скорость реакции. При высо-
ких давлениях, когда длина свободного пробега молекул газа зна-
чительно уменьшается, оптимальной окажется однородная тонко-
пористая структура катализатора. Такой подход сводит проблему
оптимизации структуры к достижению коэффициента использования
объема зерна катализатора, равного 1.
Однако при более строгой постановке вопроса оптимизация пори-
стой структуры и размеров зерна катализатора не может быть отор-
вана от задачи оптимизации реактора в целом, так как только в по-
следнем и могут быть реализованы те или иные качества самого зерна
[34]. За основу расчета реактора можно принять следующий крите-
рий оптимальности (см. главу IX)
Р--- рвыхм вых) ГВХИ (Свх) X
(V.4)
где vBX и г’вых — объемные скорости потока на входе и выходе реак-
тора; и (Свх) и и (Свых) — ценности единицы объема входного и вы-
ходного потоков, как функции вектора состава потока газа; X — экс-
плуатационные расходы, включающие расходы на катализатор, ам-
мортизационные отчисления на контактный аппарат и транспортные
расходы.
Приближенно можно считать что:
X АР
(V-5)
где Ур — объем реактора; v — объемная скорость потока газа;
L — длина реактора; АР — перепад давления на единицу длины
реактора, являющийся функцией формы и размера зерен катализа-
тора, линейной скорости и физических свойств потока, газа; и
Х2 — коэффициенты стоимости.
Считая, что объемная скорость потока вдоль реактора постоянна
и равна v и ценность единицы объема потока является линейной функ-
цией состава, можно записать функцию критерия (V.4) в виде
f==maxP=max
т, Т, R,l х, T,R,l
v
' h
У, щ (Cr —Cjo) —^1Т— АР
.1-1
(V-6)
Таким образом, при заданных реакторе, размерах и форме зерна
максимум критерия (V.6) будет функцией начальной концентрации
реагентов, времени контакта, последовательности температур вдоль
реактора, размера (радиуса) зерна R и параметра характеризую-
190
щего пористую структуру зерна (средний радиус пор, мода функции
распределения пор и т. п.).
Как показано в главе IX, конечной целью определения оптималь-
ной температурной последовательности (ОТП) в реакторе является
оптимальная селективность процесса в каждом сечении аппарата.
Но на селективность сложной химической реакции, протекающей на
пористом катализаторе, а также на производительность единицы
объема катализатора можно оказать влияние, варьируя пористую
структуру катализатора. В случае изменения пористой структуры
катализатора при фиксированной температуре кинетика химической
реакции будет переходить из одной кинетической области в другую,
например, из внутрикинетической во внутридиффузионную или на-
оборот. Соответственно изменится и селективность сложной реакции.
В общем случае для определения оптимальной области протекания
реакции, с точки зрения селективности, необходимо решить внутри-
диффузионную задачу в виде системы уравнений
V(^*VCz) = r/(C, Г) («=1, 2, . . К) (V.7)
с соответствующими граничными условиями на поверхности и в центре
зерна.
Здесь важно отметить, что при учете параметров R и £ скорости
реакции для ключевых компонентов следует выражать в обобщенном
виде, который охватывал бы внутрикинетическую, переходную, и
внутридиффузионную области протекания реакций. Такие выраже-
ния, если их вообще удается получить, оказываются чрезвычайно
громоздкими й мало пригодными для анализа и решения. Поэтому
в случае сложных реакций такой подход оказывается практически
неприемлемым. Другой метод решения поставленной задачи, при-
годный при отсутствии внешнедиффузионного торможения, поясним
на примере последовательной реакции Ах—>А2—>А3и модели
структуры зерна в виде прямолинейных цилиндрических пор.
Как показано в главе III для реакции А1 -> А2 -> А3 внутрики-
нетический режим обеспечивает наибольшую селективность в отно-
шении А2. Соответственно гг и г2 — скорости изменения концентра-
ций компонентов — будут функциями только температуры и концен-
трации в ядре потока, а от радиуса зерна катализатора не будут
зависеть. Поэтому задачу определения ОТП и времени контакта
можно в данном случае рассматривать независимо от задачи опре-
деления оптимального значения радиуса зерна и решать ее так, как
изложено в главе IX.
После выбора температуры в реакторе или на отдельных его участ-
ках можно приступить к определению оптимального значения пара-
метра пористой структуры катализатора (в нашем случае радиуса
пор). Задача заключается в том, чтобы при заданной температуре Топт
определить такое значение радиуса пор (гп), которое обеспечивало бы
максимально развитую внутреннюю поверхность катализатора при
условии, что реакции протекают во внутрикинетическом режиме.
191
В рассматриваемом случае для реакций 1-го порядка система
уравнений, описывающая динамику химического превращения в од-
ном капилляре имеет вид
= (v-8)
(V.9)
алл Гд
с граничными условиями:
снаружи зерна
Х = 0; С. = (7? (/=1,2)
в центре зерна
у_ n. dCi п
Х R’ dX “°
Решение этой системы уравнений приводит к следующему выра-
жению для скорости превращения компонента Аа, отнесенной к еди-
нице объема катализатора в случае внутрикинетического режима
протекания реакции
г2кин=---- ~ (к^С^з к^С^з) (V. 10)
т п
здесь С1з — концентрация реагентов у поверхности зерна; е — по-
розность слоя.
Соответственно для диффузионного режима имеем
г2диф=—Ц -^2С13^ — + l-S/s)7'2] (v-n)
где
S — удельная внутренняя поверхность катализатора.
Из уравнений (V.10) и (V.11) и условия
г2кИИ==Г2диф (V-12)
можно либо определить R при заданном гп, либо оптимальное гп
при выбранном R. При выбранном R для решения уравнений (V.10)
— (V.12) относительно гп необходимо знать функциональную зави-
симость коэффициента диффузии от радиуса пор. Если переход вну-
трикинетического режима во внутридиффузионный происходит при
кнудсеновской диффузии в порах катализатора, то г2Диф не зависит
от гп и решение уравнения (V.12) может быть проведено графически,
как это представлено на рис. V.5. В противном случае решение урав-
нений (V.10) — (V.12) проводится численными методами.
Точка пересечения функций г2Кин и г2диф определяет оптималь-
ное значение радиуса капилляра. В действительности не существует
резкой границы между внутрикинетическим и внутридиффузионным
192
г2диф
rl кин
режимами, и определенное таким образом значение радиуса капил-
ляра будет несколько завышенным. Выбор диаметра зерна произ-
водят из следующих соображений.
При известных времени контакта и диаметре аппарата линейная
скорость потока газа является определенной величиной. Решая ме-
тодами, изложенными в главе IX, уравнения (V.4)—(V.6) в предпо-
ложении кинетической области совместно с уравнением сопротивле-
ния зерненного слоя, например уравнением Эргана (см. главу VII),
находим оптимальное значение R для выбранного диаметра кон-
тактного аппарата. Если из уравнений (V.10)
— (V.12) получаются при этом неприемлемые
значения гп опт, следует задаться новым диа-
метром аппарата и повторить расчет. На
практике вариации значений гП невелики и,
поэтому, объем вычислительной работы не-
значителен.
Если окажется, что оптимальным, с точки
зрения селективности, является внутридиф-
фузионный режим, то ход решения в общем
не изменяется. При другой, не капиллярной
модели пористости зерна следует заменить
уравнения (V.8) и (V.9) соответствующими,
что, впрочем, пока не разработано. Слинько
[35] для бидисперсной глобулярной модели
фактически заменяет оптимизацию структуры
зерна катализатора определением верхней границы диаметра гло-
бул, при которой практически все промышленные реакции проте-
кают в кинетической области. Эта величина примерно равна 100 мк
и обеспечивает полное использование поверхности глобул и соз-
дание макропор для молекулярного течения реагентов от наружной
поверхности зерна к поверхности глобул.
л. опт гп —►
Рис. V.5. График для
определения оптималь-
ного значения гп в обла-
сти кнудсеновской диф-
фузии по уравнениям
(V.10) — (V.12).
V.4. МЕТОДЫ УПРАВЛЕНИЯ СТРУКТУРОЙ
И ФОРМОЙ КАТАЛИЗАТОРОВ
Определение оптимального химического Состава катализатора,
как указывалось, относится к проблеме его подбора. Теория же при-
готовления катализаторов рассматривает способы наиболее полной
реализации потенциальных возможностей в отношении активности
и селективности для катализатора данного состава.
Практически параметрами, которыми можно управлять в про-
цессах приготовления катализаторов, являются:
1) фазовая структура (фазовый состав) катализатора;
2) собственно структура поверхности;
3) структура пор и вторичных образований *;
4) форма и размер зерен катализатора.
* Для этих свойств более правильно было бы применять наименование
«текстура», однако этот термин плохо прививается.
13 Заказ 1441
Фазовый состав катализатора. В результате подбора задаются
химический состав и соотношения исходных компонентов катализа-
тора. Достижение же нужного фазового состава реализуется в ходе
приготовления катализатора, а иногда даже в процессе его работы.
Если в ходе приготовления катализатора меняется его фазовый
состав, то имеем дело с топохимическими процессами, которые под-
чиняются законам топохимической кинетики. Соответственно с этим,
научными основами получения катализаторов цвляются закономер-
ности топохимических превращений неорганических веществ, сравни-
тельно хорошо разработанные в области технологии силикатов. К со-
жалению, вследствие широкой номенклатуры катализаторов, ее
изменчивости и относительно малой мощности производств катали-
заторов топохимические основы этих производств разработаны пока
недостаточно.
Фазовые превращения могут определять время и температуру
прокалки катализаторов. Например, термодинамическая граница
превращения у-А12О3 в а-А12О3 лежит около 1200° С. Это усло-
вие определяет нижнюю границу температуры прокалки при при-
готовлении корундовых носителей. Для увеличения скорости реак-
ции температуру приходится поднимать, так как при 1200° С реак-
ция фазового перехода протекает слишком медленно. Однако
скорость превращения возрастает и при увеличении дефектности
решетки исходной формы окисла, как это известно из топохимии.
Поэтому добавка к у-А12О3 небольших количеств ТЮ2 позволяет
ускорить реакцию превращения у-А12О3 в а-А12О3 и тем сни-
зить температуру прокалки. Ограничения на температуру прокалки
катализаторов из окисей металлов (CuO, Ni2O3) на окиси алюминия
определяют температурой разложения исходных солей (нижний пре-
дел), например нитратов, а также температурой фазового перехода
механической смеси окислов в шпинели (верхний предел). Время и
температура восстановления цйнк-хромового катализатора опреде-
ляется скоростью превращения хромата цинка в хромит и необхо-
димостью отвода тепла при зтой сильно экзотермической реакции.
Важно то, что определение режимных параметров прокалки или
другой топохимической переработки исходных компонентов катали-
затора должно базироваться на кинетических соображениях и опи-
раться на экспериментальные данные по фазовому составу катализа-
тора в ходе его приготовления.
Структура поверхности. Вопрос о получении оптимальной струк-
туры поверхности катализатора гораздо яиенее четок, чем другие
стороны производства катализаторов. В общем, проблема, видимо,
сводится к созданию достаточно дефектной поверхности, обладающей
поэтому относительно большим количеством энергетически богатых
участков (активных центров). Это касается, очевидно, катализаторов,
работающих по сорбционному, а не химическому механизму, в пер-
вую очередь, металлов. Методы достижения зтой цели, если не ка-
саться химического промотирования, базируются, в общем, на теории
пересыщения Рогинского (см. главу I). К этим же методам, вероятно,
194
следует отнести и протравливание поверхности плавленых металли-
ческих катализаторов (см. раздел V.4).
Структура пор катализатора, а следовательно, и величина его
удельной поверхности формируется на разных этапах приготовления
катализатора и различным путем, в зависимости от типа катализатора.
Для распространенных гелевых катализаторов и носителей регулиро-
вание структуры пор осуществляется либо на стадиях осаждения,
промывки и прокалки, либо путем специальной гидротермальной
обработки. Установлено [36, 37], что увеличению пористости сили-
кагеля способствует pH 7 при осаждении и щелочная реакция про-
мывных вод. Этими же работами установлено, что повышение темпера-
туры прокаливания свыше 600° С приводит к уменьшению удельной
поверхности силикагеля и сокращению диаметра пор. Ряд аналогич-
ных исследований проведен для алюмосиликагеля [38, 391 и алюмо-
хромогеля [40]. Варьирование условий осаждения в отношении pH
растворов, их концентрации, скорости осаждения.и времени вызрева-
ния осадков, а также температуры осаждения дает возможность пу-
тем изменения величины кристаллитов изменять пористость катали-
заторов, получаемых из кристаллических осадков. Исследованиями
Кагановой с соавторами [41] показано для силикагелей и алюмоси-
ликагелей, что наблюдаемые зависимости пористости готового ката-
лизатора от pH среды, температуры сушки, а также содержания
окиси алюминия сводятся к взаимосвязи пористой структуры геля
со степенью его синерезиса. Гели с равной степенью синерезиса, т. е.
с равными отношениями количества воды к количеству окислов, имеют
одинаковую пористую структуру.
Существенное влияние на пористую структуру ксерогелей может
оказать величина поверхностного натяжения межмицеллярной жид-
кости в период синерезиса геля. Так, Топчиевой с сотрудниками [42]
было показано, что вытеснение воды в алюмосиликагелях бутанолом
или пентанолом, имеющими меньшую величину поверхностного натя-
жения, дает возможность получить широкопористые гели (диаметр
пор примерно 100 А) при сохранении большой удельной поверх-
ности (400 м2/г).
В ряде случаев пористая структура катализаторов зависит от фа-
зового состава первоначально получаемого геля. Так, удельная по-
верхность и пористость активной окиси алюминия зависят от фазо-
вого состава исходной гидроокиси. Окись, полученная из байерита,
имеет большую удельную поверхность и гораздо меньший диаметр
пор, по сравнению с окисью, полученной из бемита.
Весьма эффективным способом влияет на структуру монолитных
гелей гидротермальная обработка, которая заключается в обработке
геля водяным паром при повышенных температурах и давлениях.
При этом происходит рекристаллизация геля, исчезновение узких
пор, образование широких пор и общее уменьшение удельной поверх-
ности. Так, в силикагеле путем гидротермальной обработки можно
увеличить средний размер пор до 200—300 А, а удельную поверх-
ность снизить до 50—60 №/2.
.13*
195
Структура немонолитных гелей и кристаллических осажденных 1
катализаторов и носителей регулируется как на стадии осаждения, •
так и на стадии формования. При этом на первой стадии формируются :
первичные глобулы, а на второй вторичная структура зерна, т. е.
происходит упаковка глобул и образуются каналы между ними.
При осаждении гелевых катализаторов и носителей в виде шлама
структура образующихся при этом первичных глобул подчиняется
приведенным выше закономерностям для монолитных гелей. Струк-
тура кристаллических осадков подчиняется общим закономерностям
процессов кристаллизации.
Получаемые при осаждении немонолитных катализаторов осадки
должны быть каким-либо способом скреплены. Простейшим методом
скрепления с одновременным формованием является таблетирование,
при котором порошок катализатора под давлением в несколько сот
атмосфер сжимается в форме (матрице) в таблетку.
Другой основной вид скрепления осадков — формование во влаж-
ном состоянии, при котором используются разные методы придания
прочности сформованным частицам. Некоторые осадки полуколло-
идного характера, например, фосфаты и сульфаты металлов, сфор-
мованные во влажном состоянии, после сушки «схватывают», в ре-
зультате чего готовые гранулы обладают достаточной прочностью.
Этот процесс «схватывания» обусловливается аналогично таблетиро-
ванию силами молекулярного сцепления высокодисперсных частиц
осадка или же связыванием частиц осадка растворенными в маточном
растворе солями. В других случаях (применительно к окислам),
когда простого влажного формования недостаточно, используют так
называемый «метод пептизации», который заключается в том, что
к отмытому осадку добавляют небольшие количества растворяющего
его реагента, разрушающегося при прокаливании. В процессе пепти-
зации образуются соли, которые остаются в массе после формования,
а при сушке и прокаливании разлагаются с регенерацией исход-
ного окисла, связывающего частицы осадка в прочный единый моно-
лит. Такой способ применяется, например, для получения высоко-
прочной окиси алюминия, когда к отмытому и отфильтрованному
осадку гидроокиси алюминия добавляют 1—3% концентрированной
азотной кислоты. Полученный нитрат алюминия прокаливают,
а окись алюминия, образующаяся при этом, склеивает первоначально
образовавшиеся из гидроокиси глобулы окиси алюминия. Для ката- -<i
лизаторов, получаемых методом влажного формования, также харак- 5
терна обратная взаимосвязь между прочностью гранул и их пори» ч
стостью. Величина вторичных пор этих катализаторов определяется •
как дисперсностью первичных глобул или кристаллитов, так и кон-
центрацией солей в маточном растворе осадка или при пептизации.
Регулирование структуры, синтетических носителей типа кера-
мики и алюмокерамики производят варьированием степени помола
составляющих шихты и температуры прокалки (спекания) массы и
добавками специальных веществ. Повышение дисперсности исходных
частиц и увеличение температуры и длительности прокаливания спо-
196
собствует получению более плотного, малопористого носителя с ма-
лой удельной поверхностью.
В общем, пористость катализаторов или носителей, получаемых
в результате «склеивания», спекания или таблетирования первона-
чальных частиц, находится в прямой зависимости от диаметра по-
следних. Чем меньше размеры исходных частиц, тем меньше пори-
стость готовых гранул и диаметр пор.
Пористость катализаторов неглобулярной структуры, таких как
скелетные металлические катализаторы Бага, активированные глины
и некоторые другие, получаемых методом выщелачивания, регули-
руют путем вариации концентрации выщелачивающего раствора,
температуры и длительности выщелачивания.
В катализаторах, получаемых пропиткой носителя, пористая
структура последнего в основном определяет структуру готового
катализатора и величину удельной поверхности, хотя по абсолют-
ным величинам они могут сильно отличаться друг от друга. Так, для
катализатора серебро на <х-А12О3 имеют место следующие соотно-
шения:
Поверхность носителя, м2/г .... 170 121 80 10
» катализатора, м^/г . . 100 73 39 6
Абсолютные значения удельной поверхности катализатора в це-
лом, поверхность активного компонента и функция распределения
размера пор определяются концентрацией активного компонента
в катализаторе, а следовательно, и количеством пропиток. При
определении числа пропиток надо учитывать, что носители с раз-
витой пористостью быстро насыщаются вносимым реагентом и зна-
чительное количество пропиток здесь неэффективно. При обра-
ботке же малопористых носителей каждая пропитка приводит к не-
которому увеличению содержания солей (окислов) в катализаторе
и полного насыщения долго не наступает. В этом случае применение
многократных пропиток целесообразно. "Сказанное выше подтвер-
ждается данными о результатах пропиток при приготовлении нике-
левых, хромовых и кобальтовых катализаторов, на различных но-
сителях (табл. V.2) [16].
В катализаторах на носителях необходимо следить за структурой
слоя активного компонента, покрывающего носитель. Так, Шехтер,
Рогинский и Исаев [43] показали съемкой в электронном микроскопе,
что в платино-асбестовом катализаторе платина находится на ас-
бесте в виде сферолитов различной величины. Адлер и Кивней [44]
нашли для платино-глиноземного катализатора, что в зависимости
от метода нанесения платина различным образом располагается на
окиси алюминия, образуя монослой при пропитке и сферические дис-
кретные частицы при соосаждении. В общем, дисперсность актив-
ного компонента в нанесенных катализаторах может варьироваться
в достаточно широких пределах и тем самым определять свойства
катализатора. Поэтому для таких катализаторов нужно иметь
197
Таблица V.2
Влияние числа пропиток на содержание активных
компонентов в катализаторе (в пересчете на металл) i
Носитель Активный компонент Содержание активного компонента, % Коэффициент обогащения
число пропиток число пропиток
1 2 3 4 2 3 4
Алунд Co(NO3)3 6,7 9,6 12,2 1,43 1,27
Алюмогель (NH4)2CrO4 13 23 32 — 1,77 1,39 —
Кизельгур табле- Ni(NO3)3 10 18 24 28 1,8 1,33 1,17
тированный Муллит Ni(NO3)3 2,4 4,8 6,8 8,5 2,0 1,42 1,47
характеристику как общей поверхности, так и поверхности актив-
ного компонента.
Для катализаторов, работающих в кипящем и движущемся слоях,
особую роль играет прочность к абразивному воздействию соседних
частиц. В связи с этим структура, а также форма таких катализаторов
в значительной степени определяются требованиями прочности.
Широко распространен метод приготовления прочных к истиранию
катализаторов путем коагуляции в капле, описанный подробно выше.
В этом случае гранулы катализатора приобретают сферическую :
форму, гладкую поверхность и мало поддаются истиранию. Имеются
сведения о производстве катализаторов для кипящего слоя сушкой
гелевых суспензий или специальных масс в распылительных сушил-
ках с получением микросферических частиц [45]. Наконец, при
производстве катализаторов для кипящего слоя применяют высоко-
прочные носители типа корунда, алюмосиликагеля. Заполняя поры
носителя активными компонентами путем пропитки раствором, рас-
плавом или высокодисперсной суспензией, получают «армирован-
ные катализаторы», роль носителя в которых сводится только
к роли скелета, препятствующего разрушению собственно контакт-
ной массы.
Размер и форма частиц. Получение частиц катализатора задан-
ных размеров и формы для использования в неподвижном слое —
задача чисто технологическая и конструктивная; решается она в зна-
чительной степени подбором соответствующего оборудования — таб- j
леточных машин, экструзионных прессов и т. п. Наименьшее сопро-
тивление потоку оказывает слой частиц, обладающих наименьшей >
площадью сечения в плоскости, перпендикулярной направлению
потока, и оставляющих большую долю поперечного сечения реак-
тора свободной для прохода реагирующего потока. Примером таких
частиц являются правильно упакованные кольца Рашига. Описание
методов формования фасонных зерен катализатора можно найти в ра- '
'боте [461.
198
Сканировал: Neptunyi
Магнитогорск
2008
г
Размер частиц, применяемых в кипящем слое, обычно примерно
на порядок ниже, чем в неподвижном слое, он почти не влияет на
гидравлическое сопротивление потоку; применение слишком мелких
частиц ограничивается, однако, опасностью уноса катализатора из
слоя. Обычно используют частицы сферической формы, как наиболее
устойчивые к истиранию. Регулировку размера частиц производят
в ходе получения гранул при коагуляции (см. раздел V.2) или ско-
ростью распыления при получении гранул на распылительной су-
шилке. Сферическая форма гранул, очевидно, определяется самой
технологией получения катализатора.
Наиболее мелкие частицы применяют при работе с суспендиро-
ванным катализатором в жидкой фазе, поскольку здесь желательна
получить максимально однородную систему. Величину частиц опре-
деляют либо степенью выщелачивания для скелетных металлических
катализаторов, либо степенью помола, либо условиями осаждения.
Стабильность структуры. Особым вопросом при разработке
научных основ технологии производства катализаторов является
создание структур, повышающих стабильность катализатора. Если
стабильность по отношению к ядам является в основном функцией
химического состава активных компонентов катализатора, то ста-
бильность поверхности и пористой структуры определяется комплек-
сом физико-химических свойств всех составных частей катализатора.
Эти элементы структуры меняются под влиянием температуры, спе-
цифических реагентов (например, водяного пара) или вследствие
самого каталитического процесса (каталитическая коррозия).
Чувствительность катализаторов к воздействию высоких темпера-
тур связана с рядом различных явлений. Прежде всего повышение
температуры «размораживает» дефекты решетки катализаторов (как
полупроводниковых, так и металлических), приближая систему
к равновесию. Такое изменение дефектного состояния решетки не-
избежно приводит к изменению активности катализатора: в боль-
шинстве случаев к ее понижению [47]. Далее, повышение темпера-
туры и приближение ее к температуре плавления материала вызывает
значительное ускорение самодиффузии в твердом веществе и, как
следствие этого, — явление спекания, приводящее к уменьшению-
поверхности катализатора. Как указывалось ранее, это во многих
случаях приводит к понижению активности катализатора. Примеров
такого рода явлений описано очень много; можно указать на работу
Борескова с сотрудниками по катализатору парофазного гидролиза
хлорбензола [48] и работу Битепаж по алюмосиликатным катализа-
торам [49]. Еще одним следствием повышения температуры может
быть превращение каталитически активных соединений в неактивные.
Например, при температуре выше 500° С в смешанном катализаторе-
окисления нафталина во фталевый ангидрид происходит взаимодей-
ствие сульфата калия с сульфатом ванадия и образуется каталити-
чески неактивный ванадат калия. Кроме указанных явлений, при
высоких температурах может происходить растрескивание или рас-
плавление всей массы катализатора, или носителя.
199»
Вследствие разнообразия причин, вызывающих термическую дез-
активацию катализаторов, часть из которых связана с самой сущ-
ностью каталитических явлений, нет возможности наметить общие
пути повышения термостабильности катализаторов. Как правило,
катализаторы на носителях более термостойки. Это объясняется тем,
что в качестве носителей применяют высокоплавкие соединения, а на-
несенные на них пленки металлов или низкоплавких окислов при
повышении температуры близко к температуре плавления все же
не дают явлений спекания. Однако если активные компоненты не
образуют пленку на носителе, а осаждаются друзами кристаллов или
сферолитами, то применение носителя может и не привести к повыше-
нию термостабильности.
Боресков и сотрудники обратили внимание на то, что твердые ве-
щества с ковалентными связями лучше противостоят релаксационным
явлениям, спеканию, выравниванию дефектов, чем вещества с ион-
ным типом связей [50 ]. Это, вероятно, сможет стать в некоторой сте-
пени принципом подбора носителей и структур для стабилизации
катализаторов, однако пока вопрос еще недостаточно разработан.
Таким образом, в большинстве случаев не остается ничего другого,
как следовать очевидному принципу, что при прочих равных усло-
виях для приготовления катализатора следует применять возможно
более высокоплавкие и термостойкие соединения.
В отношении таких явлений, как гидротермальная рекристалли-
зация и каталитическая коррозия, общие методы стабилизации струк-
тур еще не выработаны.
Придание механической прочности катализаторам. Придание
частицам катализатора должной механической прочности — доста-
точно сложная и многосторонняя проблема. Прочность гранул на
раздавливание особо важна для катализаторов, применяемых в не-
подвижном слое. Хотя при 3-метровой высоте контактного слоя дав-
ление на опорную решетку не превышает обычно 0,3 кгс)смг, однако
благодаря точечному характеру нагрузки последняя может дости-
гать значительно больших величин и носить скорее раскалывающий,
чем раздавливающий характер. Кроме того, во время загрузки ап-
парата зерна катализатора подвергаются ударной нагрузке при па-
дении на опорную решетку или уже загруженную часть катализатора.
Все же требования к этому типу прочности для неподвижных ката-
лизаторов относительно невелики. Такой малопрочный материал,
как пемза, имеет допустимое напряжение на раздавливание
12 кгс/см2, что примерно на порядок превышает раздавливающую'
нагрузку на катализатор в слое.
Для катализаторов на носителях важным является вопрос о проч-
ности сцепления активной составляющей с носителем. Активная со-
ставляющая катализатора не должна удаляться с носителя под
эродирующим влиянием потока газа, что иногда имеет место при
применении непористых носителей.
Как упоминалось выше, простейшим методом скрепления порош-
ков катализатора с одновременным формованием является таблети-
200
рование, при котором порошок катализатора сжимается в форме
(матрице) в таблетку при давлениях в несколько сот и более атмосфер.
Оказалось, однако, что прочные таблетки образуют далеко не все
порошки. Стекло, силикагель, например, совершенно не таблети-
руются, а порошки с пластичными частицами таблетируются хорошо.
Очевидно, что скрепление таблетки происходит за счет межмолеку-
лярных сил сцепления, которые действуют на весьма малом расстоя-
нии. Пластичные частицы при больших давлениях деформируются,
образуя между собой достаточно большие поверхности полного со-
прикосновения, обеспечивающие прочное сцепление частиц. С жест-
кими частицами этого не происходит и они либо разрушаются, либо
сохраняют между собою точечный контакт, недостаточный для при-
дания прочности таблетке. Поэтому при таблетировании часто при-
меняют инертные и стабильные пластичные добавки, такие как тальк,
графит, глины. Щукин и Ребиндер [51 ] с сотрудниками показали,
что значительное упрочнение таблеток может быть достигнуто путем
предварительного, до таблетирования, виброуплотнения порошков.
Конструкции такого типа машин разрабатываются. Нужно только
иметь в виду, что излишнее уплотнение таблеток может привести
к падению активности и селективности катализатора из-за умень-
шения диаметра макропор.
ЛИТЕРАТУРА
1. А. В. Киселев, Усп. хим., 25, № 6, 705 (1956).
2. А. П. Карнаухов, Кинетика и катализ, 3, № 4, 583 (1962).
3. В. И. Оборин, Синтетический алюмосиликатный катализатор,
Облгиз, Грозный, 1949.
4. А. В. Агафонов, Алюмосиликатные катализаторы, Гостоптех-
издат, 1952.
5. Д. М. Р аппопор т, сб. «Научные основы подбора и производства
катализаторов», Изд. СО АН СССР, Новосибирск, 1964, стр. 432.
6. Р. М. Масагутов и др., ХТТ и М, № 1, 69 (1959).
7. Ch. Placek, Ch. Z. Pedigo, Ind, Eng. Chem., 51, № 4, 94 (1959).
8. П. И. К а нове ц и др., сб.«Научпые основы подбора и производства
катализаторов», Изд. СО АН СССР, Новосибирск, 1964, стр. 480.
9. РЖХим, 1954, № 16, 38593.
10. В. С. А л и е в, С. А. Е ф и м о в а, сб. «Научные основы подбора
и производства катализаторов», Изд. СО АН СССР, Новосибирск, стр. 438.
И. Е. И. К и с л я к о в а, С. В. Капацинс кий, сб. «Научные
основы подбора и производства катализаторов», Изд. СО АН СССР, Новоси-
бирск, стр. 444.
12. Т1. Н. Ворожцов (мл.), Хим. пром., № 6, 176 (1947).
13. W. D. Stilwell, Ind. Eng. Chem., 49, № 2, 245 (1957).
14. S. Iwinsky, J. Zarnot a, Przem. Chem., 38, № 10, 621 (1959).
15. А. Г. Любарский, Автореф. канд. дисс. НИОПиК, 1962.
16. F. G. С i а р е t t е, С. D. А е 1 m, L. L. В а г а 1, Commercial Pre-
paration of Industrial Catalysis, in «Catalysis in Practice», New York, 1957, p. 49.
17. E. U. В a 1 1 о n a. oth., J. Phys. Chem., 65, № 9, 1639 (1961).
18. T. В. Антипина, E. H. Авдонина, ЖФХ, 33, вып. 1,
192 (1959).
19. R. W. M a a t m a n, Ind. Eng. Chem., 65, № 9, 1639 (1961).
20. A. D u n s t о n, F. H о w e s, J. Inst. Petrol. Eng., 347 (1936).
201
21. И. Э. Г е л ь м с и др., сб. «Научные основы подбора и производства
катализаторов», Изд. СО АН СССР, Новосибирск, 1964, стр. 295.
22. Герм. пат. 247852 Ch. Z. 1912 II. 157, 254437 Ch. Z. 1913. I. 195, 263612
' Ch. Z. 1913. II. 1437.
23. Б. M. Богословский, 3. С. Казакова, Скелетные катали-
заторы, их свойства и применение в органической химии, Госхимиздат, 1957,
стр. 144.
24. Р. Н. Emmet, Catalysis, vol. 3, New York, 1955, p. 417.
25. А. Б а г, T. E г у п о в а, Д. В о л о к и т и н, Пром, орган, хим.,
2, 141 (1936).
26. Е. Л и б е р, Ф. М о риц, Скелетные катализаторы и их применение,
сб. «Катализаторы органических реакций», ИЛ, 1955.
27. Р. Ш р е т е р, Методы препаративной органической химии, ИЛ,
1950.
28. А. Г. Полянский, Усп. хим., 31, № 9, 1046 (1952).
29. И. И. И о ф ф е и др., сб. «Проблемы кинетики и катализа», вып. X,
Изд. АН СССР, 1960, стр. 294.
30. Advances in Chemistry. Ser. 101. Molecular Siev — Zeolites, Washin-
gton, 1971. ,o
31. O. Griibner, P. J e r u, M. К a 1 e k, Molecularsiebe, Berlin,
1968.
32. M. P. Грэй, Ф. Паулсон, Инженер-нефтяник, 36, № 6, 71
(1964).
33. Г. К. Боресков, Хим. пром., № 1,1 (1960).
34. W. A. Rescheto w, I. I. Ioffe, Chem. Techn., 23, № 3, 132
(1971).
35. M. Г. С л и н ь к о, О. А. Малиновская, В. С. Б е с к о в,
Хим. пром., № 9, 641 (1967).
36. Г. К. Боресков и др., ЖПХ, 22, № 5, 603 (1948).
37. И. Е. Н е й м а р к, Усп. хим., 25, № 6, 744 (1956).
38. 3. 3. В ы с о ц к и й, И. Е. Н е й м а р к, Укр. хим. ж., 20, № 5,
513 (1954).
39. Ю. С. Никитин, ХТТиМ, № 3, 35 (1960).
40. А. М.’ Рубинштейн, А. Л. Клячко-Гурвич, В. М. Аки-
мов, Изв. АН СССР, ОХН, № 5, 780 (1961).
41. Э. М. К а г а н о в а, Т. Е. Ш а х о в а, А. Е. П а н и т к о в а,
Коллоидн. ж., 23, № 5, 568 (1961).
42. К. В. Топчиева и др., Кинетика и катализ, 1, № 6, 987 (1961).
43. А. Б. Ш е х т е р, С. 3. Р о г и н с к и й, Б. М. И с а е в, Изв. АН
СССР, ОХН, № 4, 322 (1945).
44. > S. F. A d 1 е г, I. I. К е a v п е у, I. Phys. Chem., 64, № 2, 208 (1960).
45. S. Gabriel, Chem. a. Ind., 1960, № 46, 1401.
46. А. В. Марковский, сб. «Научные основы подбора и производства
катализаторов», Изд. СО АН СССР, Новосибирск, 1964, стр. 195.
47. S. Z. Roginsky, Acta physicochim. URSS, 4, № 5, 729 (1936);
ДАН СССР, 27, № 5, 454 (1940).
48. Г. К. Б о р е с к о в и др., ДАН СССР, 62, № 5, 642 (1948).
49. ГО. А. Бите паж, ЖОХ, 17, № 2, 199 (1947).
50. Г. К. Б о р е с к о в и др., ДАН СССР, 189, № 5, 1031 (1969).
51. М. Р. Спасский и др., ДАН СССР, 189, № 4, 767 (1969).
Часть
третья
ОСНОВЫ РАЗРАБОТКИ
И ОПТИМИЗАЦИИ
КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
Глава VI-----------------------------------------------
ЗЕРНИСТЫЙ СЛОЙ КАТАЛИЗАТОРА
VI.1. РАСПРЕДЕЛЕНИЕ ВРЕМЕНИ ПРЕБЫВАНИЯ В РЕАКТОРЕ
Проточные реакторы. Большинство современных промышленных
процессов проводится в непрерывно действующих проточных реак-
торах. Такой реактор представляет собой открытую систему, взаимо-
действующую с внешней средой: в аппарат непрерывно подаются
исходные вещества и отводятся продукты реакции и выделяющееся
тепло. На показатели работы реактора влияют, наряду с химической
кинетикой и макрокинетикой процесса, новые, специфические фак-
торы: конвективный поток реагентов и теплообмен с внешней средой.
Расчет и теоретический анализ работы реактора с учетом взаимодей-
ствия и взаимного влияния всех этих факторов — далеко не простое
дело. Число параметров и переменных, необходимых для «точного»
расчета, в практически важных случаях может быть чрезвычайно
большим и превосходить возможности даже самых быстродейству-
ющих вычислительных машин. Дополнительную сложность вносят
типичные для крупномасштабных систем явления статистической не-
упорядоченности и случайного разброса характеристик процесса.
Эти явления нельзя рассматривать как внешнюю, досадную помеху;
они связаны с самой природой процесса и должны обязательно при-
ниматься во внимание при анализе его работы. Непременным зало-
гом успеха при расчете промышленных химических реакторов яв-
ляется предварительный анализ основных факторов, влияющих на
процесс в данных условиях. Только таким путем можно выделить
основные связи из сложной и запутанной картины взаимодействия
различных процессов переноса и химической реакции, не отягощая
расчет излишними и зачастую обманчивыми уточнениями и в то же
время не упуская из виду существенных, хотя, может быть, и труд-
ных для анализа, действующих факторов.
Влияние случайного разброса времени пребывания в реакторе
на степень превращения. Уже при осуществлении первых непре-
рывных химических процессов, когда теория химических реакторов
была еще в зародыше, замечали, что эффективность процесса резко
203
снижалась при переходе на непрерывный режим. Рассмотрим в каче-
стве простейшего примера гомогенную необратимую изотермическую
реакцию первого порядка с константой скорости к. Если такая реак-
ция протекает в замкнутом объеме (в периодически действующем
реакторе) в течение времени s, то доля непрореагировавшего исход-
ного вещества равна (см. раздел II. 1):
^wx = C(S)/CBX=e-^ (VI.1)
Если ту же реакцию проводить в непрерывно действующем реак-
торе объемом V, выбрав объемную скорость подачи реагентов v таким
образом, чтобы среднее время контакта
s = Vjv
(VI.2)
было таким же, как время периодического процесса, то долю непро-
реагировавшего исходного вещества уже невозможно определять по
формуле (VI. 1). Как сейчас убедимся, величина сВь1Х в этом случае
всегда окажется больше, а значит, эффективность процесса — ниже,
чем в периодическом процессе, причем это ухудшение показателей
может быть весьма значительным. Дело в том, что при сохранении
среднего времени реакции неизменным истинное время пребыва-
ния в реакторе различных элементов потока неодинаково и может
подвергаться значительному статистическому разбросу. Диффузия
реагентов, турбулентные вихри, принудительное перемешивание, не-
одинаковость скоростей потока в различных частях реактора (в част-
ности, образование застойных зон) приводят к тому, что время пре-
бывания в проточном аппарате не определено однозначно и может
рассматриваться как случайная величина. Введем дифференциальную,
функцию распределения <р (т) случайной величины — времени пре-
бывания в реакторе. Вероятность того, что молекула, попавшая в ре-
актор, останется там в течение промежутка времени длительностью от
т до т + йт, равна <р (т) dr; поскольку вероятность «когда-нибудь»
выйти из реактора равна единице (достоверности), функция <р (т)
должна удовлетворять следующему условию нормировки:
J ф(т)Л=1 (VI.3)
О
Если вещество может вступать в реакцию первого порядка, то, 4
согласно формуле (VI. 1), вероятность того, что молекула, пребывав-
шая в зоне реакции время т, осталась непрореагировавшей, равна e~h^. J
Безразмерную концентрацию исходного вещества на выходе реак- ;
тора, равную средней вероятности проскока, можно, очевидно, найти,
усредняя эту вероятность по распределению времени пребывания
в реакторе * *:
СО
свых= J е"^ф(т)йт (VI.4)
. о
* Формула (VI.4) впервые указана Данквертсом [1].
204
z. В предельном случае, когда все молекулы проводят в реакторе
одинаковое время т = s, функция распределения времени пребыва-
ния в реакторе описывается так называемой fl-функцией Дирака
б (т — $). Эта функция равна нулю при т =£ s, а при т = s обращается
в бесконечность; основное свойство ее выражается формулой:
J Ф(т)6(т-s)dr= ф(s) (VI.5)
— со
Учитывая уравнение (VI.5) замечаем, что при <р (т) = 6 (т — s)
концентрации, вычисленные по формулам (VI.1) и (VI.4), совпадают;
таким образом, в этом — и только в этом — случае совпадают по-
казатели непрерывного и периодического реакторов. Если функция
распределения <р (т) отличается от 6-функции, то при одном и том же
среднем времени контакта доля непрореагировавшего исходного ве-
щества в непрерывном процессе всегда больше, чем в периодическом.
Характеристическая функция и статистические моменты. Интег-
рал в формуле (VI.4) представляет собой, не что иное, как преобразо-
вание Лапласа от функции распределения. В теории вероятности
интеграл
£(р) = J *“РТФ (т) dx (V1.6)
о
рассматриваемый как функция комплексной переменной р, назы-
вается характеристической функцией* (см., например, [2]). Харак-
теристическая функция содержит полную информацию о распреде-
лении времени пребывания в реакторе, так как, зная ее, всегда можно
найти функцию распределения <р (т) с помощью обратного преобра-
зования Лапласа. Хотя характеристическая функция лишена на-
глядности, свойственной функции распределения, пользоваться ею,
как правило, гораздо удобнее. Если имеется теоретическая модель
течения в реакторе, то с ее помощью всегда проще вычислить харак-
теристическую функцию, чем функцию распределения, и во многих
случаях аналитическое выражение может быть найдено только для
первой из них. Характеристическая функция оказывается более по-
лезной и при практических расчетах. Как это следует из формулы
(VI.4), степень превращения вещества в реакции первого порядка
непосредственно определяется характеристической функцией, взя-
той от действительного положительного аргумента р = к.
С помощью характеристической функции можно найти и основные
статистические характеристики распределения — статистические мо-
менты
«/= /°т'ф(т)йт (/ = 0,1,2,...) (VI.7)
о
* Указанное определение характеристической функции применимо, когда
случайная величина т принимает только положительные значения.
205
Нетрудно убедиться, что моменты ссу- получаются как коэффи-
циенты разложения характеристической функции (VI.6) в ряд Тей-
лора в окрестности точки р = 0:
СО
= тг^ (Vi-8)
/-о
В силу условия (VI.3) нулевой момент а0 равен единице. Первый
момент ах равен среднему времени пребывания в реакторе:
(VI.9)
Второй момент а2 связан с важнейшей статистической характери-
стикой распределения — дисперсией х2, которая служит основной
мерой «размытости» распределения:
СО
ха= J (т —s)2(p(T) йт = а2—s2= — S2 (VI.10)
о
Вместо формулы (VI.8) бывает удобно пользоваться разложением
в ряд логарифма характеристической функции:
lng(p) = 2<—В'jy-P7 (VI. 11)
/-1
Коэффициенты этого разложения х;- называются семиинвариан-
тами. Первые два семиинварианта совпадают соответственно со сред-
ним временем пребывания в реакторе и дисперсией, а остальные
могут быть выражены через линейные комбинации статистических
моментов. Удобство использования семиинвариантов связано с тем,
что у наиболее часто встречающихся в теории вероятности распре-
деления — нормального, или гауссова, — все семиинварианты, на-
чиная с третьего, равны нулю. Вследствие этого высшие семиинва-
рианты могут служить мерой отклонения распределения от нормаль-
ного закона распределения.
Используя разложение в ряд (VI.И), можно переписать фор-
мулу (VI.4) в виде: .
со
1 - 2 (-1)'
/=2
S
СВЫХ—®ХР \ ks
(VI. 12)
Если распределение близко к нормальному, то можно ограни-
читься только первым членом суммы, т. е.
свых = ехр[—ь(1— x2k/2s)l (VI.13)
206
' Иэ этой формулы видно, что концентрация непрорёагировавшего
исходного вещества увеличивается с ростом дисперсии х2. Предельно
низкая концентрация с = e~ks достигается при %2 = т- е- когда
время пребывания в реакторе строго фиксировано. Если дисперсия
невелика, так что x2fc/s 1, отклонение времени пребывания в реак-
торе от среднего значения приводит лишь к небольшому уменьшению
степени превращения.
Диффузионная модель. Рассмотрим теперь причины, приводящие
к появлению случайного разброса времени пребывания в реакторе.
Все эти причины можно свести к одной — разбросу мгновенных зна-
чений продольной компоненты скорости «элемента потока» на его
траектории, связывающей вход и выход реактора. Этот разброс ско-
ростей может быть вызван попаданием в различные области реактора,
где скорость движения неодинакова. Например, в случае ламинар-
ного потока в трубе скорость сильно изменяется по сечению аппарата,
будучи малой около его стенок и значительно превышая среднюю
скорость движения у центра трубы. В реакторе с насадкой локальная
скорость мала близ твердой поверхности; кроме того, в этом случае
могут возникнуть значительные вариации скорости, связанные с об-
разованием каналов и застойных зон вследствие неоднородности упа-
ковки твердых частиц. При попадании в застойные зоны с малой ско-
ростью движения потока значительную роль начинает играть и мо-
лекулярная диффузия. В турбулентном потоке локальные скорости
изменяются не только в пространстве, но и во времени, и турбулент-
ные пульсации и вихри становятся основной причиной случайного
разброса времени пребывания в реакторе.
Вычисление функций распределения времени пребывания в реак-
торе с учетом всех перечисленных факторов, как правило, невоз-
можно; часто функцию распределения получают экспериментально,
подавая на вход реактора импульс трассирующего вещества или дру-
гой переменный сигнал и измеряя концентрацию трассирующего ве-
щества на выходе реактора как функцию времени. Однако и в зтом
случае получаемую функцию распределения стремятся выразить
через небольшое число параметров (по возможности имеющих не-
посредственный физический смысл), зависимость которых от гидро-
динамики и физических свойств потока можно было бы найти в по-
ставленной серии экспериментов.
Таким образом, для успешного решения задачи определения функ-
ции распределения времени пребывания в реакторе необходимо
«огрубление» истинной гидродинамики процесса, позволяющее оце-
нить суммарное влияние всех многообразных действующих факто-
ров на перемешивание потока. Здесь приходит на помощь основное
свойство распределений случайных величин, выражаемое централь-
ной предельной теоремой теории вероятности. Согласно этой теореме,
распределение случайной величины, подверженной влиянию много-
численных слабых факторов, должно быть близко к нормальному
закону. Установления распределения, близкого к нормальному, сле-
дует ожидать в достаточно протяженных системах, где «элемент
207
потока» за время прохождения реактора успеет побывать в зонах
с различной скоростью движения и многократно испытать действие
различных случайных факторов.
Характеризовать распределение времени пребывания с помощью
нормального закона очень удобно, так как этот закон содержит только
два параметра: среднее время пребывания «и дисперсию х2. Согласно
формуле (VI.13), х2 определяет степень ухудшения характеристик
процесса, к которому приводит наличие случайного разброса. Широ-
кая распространенность нормальных распределений и удобство при-
менения их в практических расчетах являются (хоть это зачастую
и не осознается) основной причиной, вызвавшей к жизни так называ-
емую диффузионную модель химических реакторов *, которая, как
будет показано ниже, дает функцию распределения времени пребы-
вания в аппарате, близкую к нормальному закону. '
При выводе уравнений диффузионной модели предполагается,'
что перенос вещества осуществляется двумя путями: конвекцией
с постоянной скоростью и и диффузией с эффективным коэффициентом
диффузии D, величина которого также не зависит от координаты.
При этом уравнение материального баланса, описывающее измене-
ние концентрации реагента по длине реактора при стационарном про-
текании химической реакции первого порядка, имеет вид
Dd^C/dX* — udC/dX — кС=А (VI. 14)
где X — продольная координата.
Чтобы найти функцию распределения времени пребывания в реак-
торе, необходимо решить нестационарное уравнение, описывающее
процесс распространения химически инертного трассирующего ве-
щества:
Мгновенному импульсу трассирующего вещества на входе реак-
тора (при X = 0) соответствует начальное условие:
С(Х,0) = 46(Х) (VI.16)
Согласно этому условию, в начальный момент t = 0 концентра-
ция равна нулю повсюду, кроме входного сечения реактора, где
сосредоточено все введенное трассирующее вещество, полное коли-
чество которого на единицу поперечного сечения реактора равно А.
Будем решать уравнение (VI.15), пренебрегая возмущающим дей-
ствием границ, т. е. считая, что трассирующее вещество вводится
в сечении X = 0 бесконечно длинной трубы (рис. VI. 1). Тогда реше-
нием задачи (VI. 15) при условии (VI. 16) является:
C(X,t) =—iDt (VI.17)
2VaDt
* Эта модель впервые введена Халбертом [3].
208
Этой формуле можно придать двоякий смысл. Прежде всего, она
описывает пространственное распределение трассирующего веще-
ства по длине аппарата в любой момент времени t 0. Согласно фор-
муле (VI.17), расстояние X, которое проходит мблекула введенной
примеси за фиксированный промежуток времени t, является случай-
ной величиной, распределенной по нормальному закону со средним
значением ut и дисперсией 2Dt. Иначе говоря, «центр тяжести» про-
странственного распределения примеси смещается со средней ско-
ростью потока и, причем само распределение постепенно «размы-
вается», так что его дисперсия
линейно возрастает со временем
(рис. VI.1).
Рис. VI.2. Функция распределения
времени пребывания в реакторе, опи-
сываемом диффузионной моделью.
Цифры на кривых указывают значения
числа Пекле.
Рис. VI.1. Распределение концентрации
трассирующего вещества (метки) по
длине реактора в различные моменты
времени (t2 = 2tx).
Если фиксировать координату X = L — «выход реактора», — то
концентрация С (L, f), определенная по формуле (VI. 17), совпадает,
с точностью до множителя, с функцией распределения времени пре-
бывания в реакторе <р (т). Функцию распределения, удовлетворяю-
щую условию нормировки (VI.3), можно получить, положив А = и.
Вводя безразмерное время пребывания 0 = uxfL и число Пекле
Ре = uL/D, имеем:
<р(^)=ИОТе (VI.18>
Графически функция распределения (VI. 18) представлена на
рис. VI.2. Эта функция тем более «размазана», чем меньше число
Пекле, т. е. чем большую роль в переносе вещества играет диффузия
.хО сравнению с конвекцией. По мере увеличения числа Пекле функ-
ция распределения (VI. 18) сужается, обращаясь в пределе (Ре -> оо)
в 6-функцию.
Статистические моменты распределения времени пребывания в ре-
акторе легче всего определить, исходя не из формул (VI.17) или
14 Заказ 1441 209
(VI.18), а как отмечалось выше, из соответствующего выражения для
характеристической функции. Последнее можно получить, выпол-
няя преобразование Лапласа в уравнении (VI. 15). Учитывая началь-
ное условие (VI. 16), находим, что лапласовский образ концентрации
трассирующего вещества
С (р, Х) = f е~Р‘С (X, t) dt
о
определяется решением уравнения:
л d*C dC AS.,VX
D dX* u dX pC~ Лб(Х)
(IV.19)
(VI-20)
Решение этого уравнения в области X ^>0, удовлетворяющее
условию ограниченности при X ->оо, имеет вид:
l(X- [-£ У*71?-1)] ,и'г”
Если теперь положить X = L и А — и, то формула (VI.21) даст
характеристическую функцию
гИ=т=^«р[-Й-(/^¥-‘)] 'v,-22)
удовлетворяющую условию нормировки g (0) = 1. По аналогии
с формулами (VI.4) и (VI.6) то же выражение, с заменой р на к опре-
деляет безразмерную концентрацию непрореагировавшего исход-
ного вещества на выходе реактора сВых (в случае протекания реакции
первого порядка). Вводя безразмерные параметры Ре = uL/D и
к = kL/u, записываем формулу для свых:
свых= (1 +4X/Pe)-1/s exp [-у Ре (/l-HW ~)] (VI.23)
Функцию распределения (VI; 18) можно получить из (VI.22) с по-
мощью обратного преобразования Лапласа. Отметим, что такой спо-
соб вычисления функции распределения является обычно наиболее
простым. Разлагая логарифм характеристической функции (VI.22)
в ряд Тейлора по р, находим:
(VI.24)
(VI.25)
210
Мерой отклонения распределения от нормального закона служат
величины:
х//х£/2 ~ Ре ( 2 ' 1) (VI.26)
При Ре 1 все высшие семиинварианты малы и распределение
близко к нормальному.
При небольших значениях числа Пекле (порядка единицы и
меньше) формулы (VI.21)—(VI.26) становятся неточными, так как
в этих условиях оказывается необходимым учитывать возмущающее
действие границ реактора, вводя соответствующие граничные усло-
вия на входе и выходе аппарата. Вопрос о граничных условиях для
уравнения (VI.14) или (VI.15) в свое время широко обсуждался.
Данкверте [1 ] предложил граничные условия следующего вида:
DdC/dX = u(C — C0) (при Х = 0) (VI.27)
, dC/dX = Q (при X — L)
Наиболее строгое обоснование зтих условий дано в работе [4].
Точность, вносимая граничными условиями (VI.27), является, од-
нако, обманчивой. Дело в том, что при их выводе предполагается,
что диффузионная модель справедлива повсюду, в том числе и для
процессов переноса на малых расстояниях. На самом деле, однако,
не существует систем, в точности описывающихся уравнением кон-
вективной диффузии (VI.14) или (VI.15) с постоянными значениями
линейной скорости потока и коэффициента диффузии. В случае тур-
булентного потока в реакторе без насадки скорость потока почти
постоянна по всему сечению аппарата (кроме тонкого слоя близ его
стенки), однако коэффициент турбулентной диффузии является пере-
менной величиной, увеличиваясь пропорционально расстоянию от
стенки реактора. В ламинарном потоке перенос вещества осуществ-
ляется молекулярной диффузией, так что коэффициент диффузии
постоянен. Однако основная причина случайного разброса времени
пребывания в реакторе — сильное различие локальных скоростей
потока на различных расстояниях от стенки аппарата. Наконец,
в реакторах с насадкой, отклонение времени пребывания в реакторе
от среднего значения вызывается образованием турбулентных вихрей
в промежутках между твердыми частицами, разбросом локальных
скоростей потока за счет неоднородности упаковки слоя и задержкой
вещества в застойных зонах. Во всех этих случаях распределение '
времени пребывания в реакторе делается близким к нормальному,
если длина аппарата достаточно велика, и только в зтих условиях
диффузионная модель становится пригодной для приближенного
описания процесса.
Однако применимость диффузионной модели для приближенного
расчета процесса в целом вовсе не означает ее пригодности для опи-
сания локальных характеристик процесса, проявляющихся на ма-
лых расстояниях. Так, в рамках диффузионной модели возмо-
жен перенос вещества против течения потока; такая возможность
14»
211
учитывается при выводе граничных условий (VI.27). На самом деле в
ламинарном потоке, а также в реакторах с зернистым слоем такого
обратного движения нет. В таком случае возможно лишь отставание
«элементов потока», попавших в участки с малой скоростью движе-
ния или в застойные зоны, от центра тяжести пространственного рас-
пределения, который перемещается со средней скоростью потока и.
В этих условиях гранич-
ные условия (VI.27) уже
не являются строгими.
Значение эффективного
коэффициента диффузии и
пределы применимости
диффузионной модели в
каждом случае должны
определяться на основе
анализа физических осо-
бенностей процессов пере-
носа вещества в рассмат-
риваемой системе.
Как правило, если диф-
фузионная модель справед-
лива, т. е. в достаточно
протяженных реакторах,
число Пекле оказывается
Рис. VI.3. Зависимость концентрации непро- велико. Более того, для
реагировавшего исходного вещества от па- применимости диффузион-
раметра Л при различных значениях числа * „ "
г 1 г г Пекпе. нои модели необходимо,
чтобы существенное изме-
нение концентраций происходило на достаточно больших масшта-
бах; можно показать' (см. раздел VI.2), что это последнее условие
выполняется при Х/Ре = kD/u 1. При соблюдении указанных
условий можно вести расчет, используя вместо уравнения (VI.27)
упрощенные граничные условия:
С(0) = Со; С (оо) < оо (VI.28)
Решение уравнения (VI. 14) с граничными условиями (VI.28) дает
(рис. VI.3):
«вых=4^- = ехр [-^(/1 + 4Х/Ре-1)] (VI.29)
Гидродинамические режимы. С формой функции распределения
времени пребывания в реакторе связано понятие о гидродинамиче-
ском режиме аппарата. Принято выделять два предельных гидро-
динамических режима: идеального вытеснения и идеального смеше-
ния. В режиме идеального вытеснения время пребывания в реакторе
одинаково для всех элементов потока; соответственно, функция
распределения времени пребывания имеет вид б-функции б (т—s).
В этом режиме продольное перемешивание потока отсутствует и
212
стационарное распределение концентрации по длине реактора опре-
деляется решением уравнения:
и dC/dX = —кС
В режиме идеального смешения концентрации реагентов по-
стоянны по всему объему аппарата. Непрерывный переход от режима
идеального вытеснения к режиму идеального смешения можно про-
следить в рамках диффузионной модели, решая уравнение (VI. 14)
или (VI. 15) с граничными условиями (VI.27) и оценивая изменение
степени превращения и статистических характеристик распределе-
ния при уменьшении числа Пекле. Режиму идеального вытеснения
соответствует предельный случай Ре—>оо, а режиму идеального
смешения — Ре —> 0. Все промежуточные режимы иногда опреде-
ляют как режимы неполного смешения. Согласно сказанному выше,
диффузионная модель далеко не всегда пригодна для описания
работы реакторов в режиме неполного смешения. При расчете труб-
чатых реакторов она оказывается справедливой только при больших
числах Пекле, когда гидродинамический режим реактора прибли-
жается к режиму идеального вытеснения; при этом расчет реактора
в приближении идеального вытеснения обеспечивает обычно доста-
точную для технологических целей точность результатов, и влияние
продольного перемешивания потока может быть учтено как малая
поправка. При расчете реакторов малой протяженности, где про-
дольное перемешивание особенно заметно и могут наблюдаться
сильно «размазанные» функции распределения, необходимо уже
учитывать реальную физическую картину процессов переноса веще-
ства, так как диффузионная модель в этих условиях не применима.
VI.2. ГИДРОДИНАМИКА И ПРОЦЕССЫ ПЕРЕНОСА
В ЗЕРНИСТОМ СЛОЕ
Реакторы с неподвижным зернистым слоем катализатора — наи-
более распространенный тип аппаратов для проведения каталитиче-
ских процессов. Зернистый слой представляет собой совокупность
беспорядочно уложенных частиц катализатора через промежутки,
между которыми протекает поток жидкости или газа, т. е. своеобраз-
ную крупнозернистую среду, которая должна быть в сильной сте-
пени подвержена воздействию различных случайных факторов, свя-
занных с неоднородностью упаковки частиц и распределения потока
реагирующей смеси.
Вследствие относительно большого размера частиц катализатора,
значительное влияние на скорость химических превращений в зер-
нистом слое оказывают процессы переноса вещества и тепла внутри
твердых частиц. Процессы на изолированном зерне катализатора
изучались в главе III; знание макроскопической скорости реакции
на отдельном зерне в зависимости от концентраций реагентов и
температуры потока в данной точке слоя — необходимый элемент
математического описания процессов в зернистом слое. Другим
213
важнейшим элементом является описание процессов переноса веще-
ства и тепла в зернистом слое в целом.. Зернистый слой катализато-
ра — статистическая система, макроскопическое поведение которой
определяется взаимодействием множества элементарных процессов.
Структура упаковки. Пространство между частицами в зернистом
слое со случайной упаковкой имеет весьма сложную форму, которую
трудно представить наглядно. Некоторое представление о форме по-
рового пространства можно составить, рассматривая простейшие
способы правильной упаковки шаров одинакового размера. Воз-
можны различные правильные структуры — от кубической упаковки
с пористостью е = 0,48 и координационным числом (т. е. числом
соседей каждого шара), равным 6, до плотнейшей упаковки с долей
свободного объема е = 0,26 и координационным числом 12. В неупо-
рядоченном слое сферических частиц ’могут встречаться отдельные
области, приближающиеся к различным способам правильной упа-
ковки, а также локальные дефекты, вызванные отсутствием какой-
либо частицы на положенном месте и образованием «сводов», которые
оберегают участки с повышенной локальной пористостью от давле-
ния лежащих выше частиц. Еще более сложным может быть харак-
тер упаковки слоя, состоящего из частиц неправильной формы или
зерен различного размера. Вибрация зернистого слоя способствует
переходу от менее плотных к более плотным структурам.
Для характера течения в зернистом слое чрезвычайно важно,
что при любом способе упаковки зернистого слоя его поровое про-
странство состоит из перемежающихся областей с различным «живым
сечением»: узкие каналы, соединяясь между собой, переходят в от-
носительно широкие каверны в промежутках между твердыми части-
цами. Извилистость каналов и ширина каверн изменяются при пере-
ходе от одного типа упаковки к другому, однако указанное общее
свойство порового пространства слоя остается характерным как для
упорядоченных способов упаковки, так и для случайных.
Некоторое влияние на характер упаковки слоя, естественно,
оказывают стенки реактора. Особенно сильно оно чувствуется в слу-
чае правильных структур. Локальная пористость слоя правильно
уложенных шаров в непосредственной близости к стенке, должна
быть равной единице. Далее, на расстоянии от стенки, равном поло-
вине диаметра частицы, пористость минимальна. При дальнейшем
удалении от стенки наблюдается затухающие колебания пористости,
исчезающие на расстоянии нескольких диаметров частицы от стенки
реактора. В неупорядоченном слое или в слое частиц неправильной
формы влияние стенок приводит только к некоторому увеличению
пористости в их ближайшей окрестности и перестает ощущаться
уже на расстоянии одного диаметра зерна от стенки [5].
Гидродинамика потока. Характерные черты гидродинамики по-
тока в зернистом слое непосредственно связаны с его геометрией.
В этой книге будем рассматривать только особенности течения
жидкости или газа через зернистый слой, которые непосредственно
влияют на процессы переноса вещества и тепла. При умеренных
214
н высоких скоростях потока, обычных для каталитических процес-
сов, конвекция является основным фактором, определяющим ско-
рость всевозможных процессов переноса, причем решающее значение
имеет «внутренняя гидродинамика» зернистого слоя, т. е. локаль-
ное распределение скоростей потока в различных точках порового
пространства. Поле скоростей в зернистом слое нельзя найти рас-
четным путем. Эту задачу решать было бы столь же бессмысленно,
как пытаться определять индивидуальные скорости газовых молекул
вместо статистического описания свойств газа в целом. Однако для
исследования зернистого слоя, представляющего собой случайную
среду с крупномасштабными неоднородностями, недостаточны отно-
сительно простые методы, применяемые в теории обычных пористых
сред. Единственным пока способом анализа особенностей «внутрен-
ней гидродинамики» зернистого слоя остается качественный физиче-
ский анализ основных свойств данной системы, соединенный с экспе-
риментальными исследованиями.
Говоря о «скорости потока в зернистом слое», часто имеют в виду
совершенно различные величины; зта неопределенность связана
с тем, что имеется несколько уровней и способов усреднения скорости
потока. Самое детализированное описание гидродинамики потока
дает задание истинных локальных скоростей в каждой точке свобод-
ного объема зернистого слоя. Истинная локальная скорость потока
обращается в нуль у поверхности твердых частиц. При скоростях
потока, обычных для промышленных каталитических процессов,
близ твердой поверхности наблюдается резкий перепад скорости,
сосредоточенный в тонком гидродинамическом пограничном слое,
толщина которого мала по сравнению с характерным размером твер-
дых частиц или промежутков между ними. Поле истинных локальных
скоростей близ твердой поверхности определяет скорость массо-
и теплообмена межДу потоком и поверхностью твердых частиц (см.
главу III). Влияние распределения истинных локальных скоростей
потока близ твердой прверхности на процессы переноса в слое в целом
сказывается лишь в том, что участки близ твердой поверхности, где
скорость потока близка к нулю, могут играть роль «застойных зон»,
в которых происходит задержка и накопление вещества, распро-
страняющегося по слою с движущимся потоком. Особенно сильные
застойные эффекты должны наблюдаться в областях близ точек со-
прикосновения твердых частиц (рис. VI.4). Эти области эквивалентны
узким и глубоким каналам; турбулентные пульсации в них не про-
никают, истинная локальная скорость потока близка к нулю, и
перенос вещества осуществляется только с помощью медленного
процесса молекулярной диффузии.
Область за пределами гидродинамического пограничного слоя и
застойных зон близ точек соприкосновения частиц принято назы-
вать ядром потока. Истинные локальные скорости в ядре потока
также изменяются в пространстве (в зависимости от локальной
геометрии слоя), а при турбулентном режиме течения подвержены и
нерегулярным колебаниям во времени. Гидродинамическая картина
215
образуются турбулентные
Рис. VI.4. Застойная зона у
точки соприкосновения частиц.
течения в ядре потока определяется указанной выше характерной
структурой порового пространства зернистого слоя, состоящего
из относительно широких каверн, соединенных узкими каналами.
Известно, что при обтекании одиночной сферы уже при числах
Рейнольдса порядка нескольких десятков у тыльной стороны сферы
вихри. В зернистом слое, состоящем
из плотно уложенных сфер или частиц
другой формы, эти вихри «запираются»
в кавернах. Образование вихря в про-
межутке между частицами хорошо видно
на рис. VI.5 [6]. В указанных усло-
виях нельзя еще говорить о развитой
турбулентности, а только о вихревом
режиме с преобладанием инерциальных
сил. Полный спектр турбулентных
пульсаций в зернистом слое не может
наблюдаться, так как максимальный воз-
можный размер вихрей определяется
характерным размером каверн. Кроме вихрей возможного наиболь-
шего размера, в слое образуются и меньшие вихри, на существование
которых указывают как измерение локальных коэффициентов массо-
передачи между потоком и твердыми частицами в зернистом слое [7 ],'
Рис. VI.5. Распространение метки в зернистом слое при ламинарном (6) и вих-
ревом (а) режимах течения.
так и непосредственное исследование спектра турбулентных пульса-
ций [8]. Определяющую роль в переносе вещества в слое играют
вихри максимального размера. Такой вихрь не может выйти из ка-
верны, в которой он возник, из-за относительно малого диаметра*
каналов, соединяющих отдельные каверны. В самих узких каналах
возможно только возникновение маломощных мелкомасштабных
вихрей. Поэтому в зернистом слое практически отсутствует перенос
вещества из одной каверны в другую против хода потока. Такой
характер течения подтверждается рис. VI.6 [6], где ясно показано
различие между реальной картиной распространения метки в зер-
нистом слое (затемненная область) и той, которая наблюдалась бы
при переносе вещества против течения, согласующемся с диффузион-
216
ной моделью (см. раздел VI. 1). (В последнем случае нижняя граница
распространения метки совпала бы с указанной на рис. VI.6 ли-
нией а).
Таким образом, можно нарисовать следующую картину «вну-
тренней гидродинамики» зернистого слоя. Поровое пространство
слоя состоит из каверн-ячеек, соединенных узкими каналами. Круп-
номасштабные вихри, «запертые» в кавернах, осуществляют интен-
сивное перемешивание во всем их объеме, кроме застойных зон.
В узких каналах размеры вихрей малы, поэтому вещество не пере-
носится из одной каверны в другую против хода потока.
Очень важно отметить дискретный
характер течения в слое, согласующийся
с геометрией порового пространства.
Вихрь, «запертый» в каверне, образует
основной элемент этой дискретной струк-
туры — ячейку идеального смешения. По-
следний термин указывает на интенсив-
ность перемешивания в основном объеме
ячейки; смешение потока в ячейке может,
однако, и не быть полным вследствие за-
держки вещества в застойных зонах, об -
разования мелких вихрей и пр. Тем не
менее, и в этих более сложных случаях
сохраняется дискретность ячеек, степень
же перемешивания потока внутри ячеек
можно учесть, введя функцию распреде-
ления времени пребывания в ячейке, вид
которой будет определяться процессами
конвективного и диффузионного перено-
Рис. VI.6. Распространение
метки в зернистом слое.
Линия а указывает нижнюю
границу области распростране-
ния метки для диффузионной
модели.
сов, протекающими в различных частях каверны-ячейки.
Размер ячеек в неупорядоченном зернистом слое может быть
различным, случаен и способ их соединения между собой; следова-
тельно, и скорости потока в разных ячейках будут различными.
Усредняя скорость потока на масштабе отдельной ячейки, мы можем
ввести понятие средней локальной скорости (или локальной скорости
потока), равной отношению характерного размера ячейки к сред-
нему времени пребывания потока в данной ячейке. Локальная ско-
рость потока является случайной величиной, принимающей различ-
ные значения в разных областях слоя. Если, однако, зернистый
слой статистически однороден, то вероятность обнаружить то или
иное значение локальной скорости не зависит от пространственного
положения ячейки. Помимо того, в статистически однородном слое
локальные скорости потока в соседних ячейках являются (с хоро-
шей степенью точности) статистически независимыми.
Статистическая однородность зернистого слоя нарушается при
каналообразовании, когда основная часть потока проходит или ско-
рее «пробивает» слой по нескольким резко выделенным направ-
лениям — каналам, а весь остальной объем реактора остается
217
практически застойным. Каналообразование совершенно недопустимо
в технологическом процессе, так как оно приводит, с одной стороны,
к проскоку непрореагировавших исходных веществ и, с другой, —
к резкому нарушению условий процесса в застойных областях, в ре-
зультате чего появляются сильные разогревы и т. п. Как правило,
каналообразование — это не просто ухудшение показателей, но пол-
ный срыв процесса. Вероятность образования каналов особенно
сильна в неглубоких слоях (в реакторах малой длины) или при мелко-
дисперсном зернистом материале. Опасность «пробоя» — каналооб-
разования — один из основных факторов, влияющих на допустимые
параметры зернистого слоя. В хорошо спроектированном процессе
не должен проявляться и другой фактор, способный нарушить ста-
тистическую однородность слоя, — неравномерность распределения
реагирующей смеси по сечению аппарата. Следует заметить, однако,
что при достаточно большом отношении длины слоя к его диаметру,
сам зернистый слой сглаживает эту неоднородность, которая объяс-
няется обычно неудачной конструкцией распределительного устрой-
ства. Естественным фактором, нарушающим статистическую одно-
родность зернистого слоя, является влияние стенок аппарата, од-
нако, как отмечалось выше, уже на расстоянии порядка диаметра
зерна от стенки это влияние перестает ощущаться и практически
им всегда можно пренебречь. Таким образом, статистически одно-
родный ячеистый характер течения свойственен любому правильно
спроектированному аппарату с зернистым слоем.
Усреднение локальных скоростей потока по всему сечению реак-
тора дает среднюю скорость потока, цоторая является основным
наблюдаемым параметром, определяющим закономерности течения
в зернистом слое. Вместо средней скорости потока и часто исполь-
зуют фильтрационную скорость w — ей (где е — доля свободного
объема слоя). Все эмпирические зависимости коэффициентов пере-
носа от гидродинамики потока выражают через среднюю или чаще
фильтрационную скорость. Обе эти величины, конечно, не полностью
определяют характер течения. Неудивительна поэтому невысокая
точность большинства эмпирических закономерностей, что объяс-
няется наличием случайных факторов в неупорядоченных крупно--
зернистых средах.
Гидродинамическое перемешивание. Разброс значений истинных
локальных скоростей потока приводит к тому, что время пребыва-
ния в реакторе с зернистым слоем является случайной величиной.
Если на вход аппарата подать импульс трассирующего вещества,»
то нг( выходе получим более или менее размытую кривую изменения
концентрации во времени, совпадающую с дифференциальной функ-
цией распределения времени пребывания в слое. Аналогично, струя
трассирующего вещества, введенная в какую-либо точку зернистого
слоя, постепенно размывается по всему его сечению. Оба эти,
явления определяются гидродинамическим перемешиванием потока,
или переносом вещества в продольном и поперечном направ-
лениях.
218
Рис. VI.7. Экспериментальные зави-
симости эффективного продольного
числа Пекле от числа Рейнольдса
для жидкостей (2) и газов (2).
В рамках диффузионной модели (см. раздел VI. 1) процессы
гидродинамического перемешивания характеризуются эффективными
коэффициентами продольной и поперечной диффузии D± или
числами Пекле Рец = ul/Dn, Ре± = ul/DY (I — диаметр зерна).
Имеющиеся экспериментальные данные по продольному перемеши-
ванию свидетельствуют о различии в характере зависимости числа
Пекле от числа Рейнольдса для потоков жидкости и газов (рис. VI.7).
В газах при числах Рейнольдса от 30 и выше значение Рец прак-
тически постоянно и равно 2 [9, 10]. Иная картина наблюдается
в случае жидких потоков. Экспериментальные данные, полученные
различными исследователями [9—12], показывают, что в жидко-
стях при Re = 304-200 наблюдаются значительно меньшие вели-
чины Рец, чем в газах. С ростом
обнаруживает тенденцию к увели-
чению, и при Re > 10® достигается
предельное значение Ре ц 2. При
малых скоростях потока, когда
перенос вещества в слое осущест-
вляется, в основном, путем моле-
кулярной диффузии, число Пекле
линейно возрастает с увеличением
числа Рейнольдса. Интересно от-
метить, что разброс эксперимен-
тальных значений Ре ц для потоков
жидкости особенно велик и, напри-
мер, при Re = 30 достигает 100%.
С ростом числа Рейнольдса эти
расхождения постепенно уменьшаются. Многие авторы отме-
чают [10, 11 ], что в случае потоков жидкости функции распределения
времени пребывания в слое обнаруживают длинные устойчивые
«хвосты». Такое же поведение характерно и для потоков газа в слое
пористых частиц или частиц, хорошо адсорбирующих трассирующее
вещество [13]. С ростом числа Рейнольдса «хвосты» проявляют тен-
денцию к уменьшению и при достаточно больших Re могут совер-
шенно исчезнуть. Появление «хвостов» вызывает увеличение дис-
персии функции распределения и, соответственно, увеличение эф-
фективного коэффициента продольной диффузии и уменьшение
числа Re ||.
Экспериментальные данные различных авторов по поперечному
перемешиванию потока [14, 15] показывают, что при числах Рей-
нольдса от нескольких десятков и выше число Ре± практически
не зависит от Re как для потоков газа, так и жидкости и в обоих
случаях равно 10—12. При малых числах Re начинает проявляться
различие между потоками газа и жидкости, связанное с различной
скоростью молекулярной диффузии в жидкостях и газах. В области,
где основную роль играет молекулярная диффузия, зависимость Pe_L
от Re становится линейной. При увеличении скорости потока воз-
растание эффективного коэффициента поперечной диффузии и выход
219
его на предельное постоянное значение происходит после отрыва
пограничного слоя от твердой поверхности и образования вихрей
в промежутках между частицами, т. е. при переходе от случая (б)
к случаю (а) на рис. VI.5.
Для объяснения экспериментальных Данных по гидродинамиче-
скому перемешиванию был выдвинут ряд моделей зернистого слоя.
Наиболее удачной оказалась дискретная ячеистая модель, которая
согласуется с описанной выше гидродинамической картиной течения
в слое. Первоначальным вариантом дискретной модели была модель
ячеек идеального смешения [12, 16], хорошо объяснившая данные
по продольному перемешиванию в потоках газа. Для описания про-
дольного перемешивания в потоках жидкости, где наблюдаются более
сложные зависимости эффективного коэффициента продольной диф-
фузи от скорости потока, были выдвинуты различные варианты
моделей с застойными зонами. Первой моделью этого типа была
модель Тернера—Ариса [17 ]. Согласно этой модели зернистый слой
рассматривали как канал постоянного поперечного сечения, харак-
теризующийся определенными значениями линейной скорости по-
тока и коэффициента продольной диффузии, от стенок которого
отходят тупиковые каналы-ответвления, где по предположению,
конвекция отсутствует и перенос вещества осуществляется только
путем молекулярной диффузии. В последующих работах [18] за-
стойные явления рассматривали в рамках ячеистой модели. Метод
анализа таких систем, использующий аппарат характеристических
функций, был указан в работе Каца [19]. Расчеты по различным
вариантам моделей с застойными зонами позволили объяснить на-
блюдаемые в потоках жидкости пониженные значения числа Ре ц и
наличие «хвостов» у функций распределения времени пребывания
в слое. Недостатком этих работ является, однако, то, что физический
смысл застойных зон в них не конкретизируется; вследствие этого
оказалось невозможным выявить непосредственную связь характе- .
ристик продольного перемешивания с параметрами зернистого слоя
и провести количественное сравнение теории с экспериментом.
Готтшлих [20], пытаясь придать модели Тернера—Ариса физиче-
ское содержание, предположил, что роль тупиковых каналов или
застойных зон играет диффузионный пограничный слой у поверх-
ности твердых частиц. Оценка толщины диффузионного слоя, необ-
ходимой для объяснения экспериментальных данных по продоль-
ному перемешиванию, не совпала, однако, с толщиной диффузион-
ного пограничного слоя, оцениваемой на основе измерения коэффи-»
циента массопередачи (см. раздел VI.3). Это несоответствие быйо
отнесено автором на счет влияния распределения толщины диффу-
зионного слоя на неравнодоступной поверхности твердых частиц.
Экспериментальное исследование локальных коэффициентов массо-
передачи в зернистом слое показывает [7 ], что в нем имеются области,
массопередача к которым резко затруднена — зоны близ точек со-
прикосновения твердых частиц. Расчет по модели ячеек с застойными
зонами близ точек соприкосновения твердых частиц [21 ] позволил
220
получить хорошее соответствие с экспериментальными 'данными
по продольному перемешиванию в зернистом слое (см. раздел VL3).
В работах [22] для объяснения данных по гидродинамическому
перемешиванию в зернистом слое была выдвинута модель капилля-
ров случайного размера и ориентации. Эти модели, однако, не согла-
суются с реальной структурой насыпного зернистого слоя.
Простейшие геометрические соображения, основанные на дискрет-
ной модели зернистого слоя, позволили также объяснить наблюдае-
мые значения эффективного коэффициента поперечной диффузии [15 ].
Впоследствии на основе ячеистой модели были рассчитаны харак-
теристики нестационарного процесса распространения примеси в по-
перечном направлении [23].
На работу реального зернистого слоя существенное влияние
оказывают случайные факторы. По-видимому, именно эти факторы
в значительной мере ответственны за имеющийся значительный
разброс экспериментальных данных, опубликованных различными
исследователями *. Гидродинамическое перемешивание в слое со слу-
чайной структурой исследовано в работах [24], этот вопрос подробно
рассматривается в разделе VI.4.
Основной метод теоретического определения эффективных коэф-
фициентов переноса в зернистом слое, которым мы будем пользо-
ваться в последующих разделах этой главы, состоит в следующем.
На основе выбранной модели слоя рассчитывают статистические
характеристики процесса переноса трассирующего вещества в зер-
нистом слое. В наиболее интересных случаях нельзя найти функцию
распределения времени пребывания слоя или пространственного
положения трассирующего вещества в явном виде. Этого, однако,
и не требуется для решения поставленной задачи, так как наиболее
удобной характеристикой процессов гидродинамического перемеши-
вания являются статистические моменты, определяемые с помощью
метода характеристических функций. Эффективные коэффициенты
переноса определяются из сравнения вычисленной дисперсии рас-
пределения с дисперсией, соответствующей диффузионной модели
слоя. Вычисление высших статистических моментов, характеризую-
щих отклонение формы распределения от нормального закона, дает
возможность установить пределы применимости диффузионной мо-
дели.
Анализ дискретной ячеистой модели позволяет объяснить и те
свойства функций распределения времени пребывания в слое, кото-
рые не описываются диффузионной моделью. Оставаясь в рамках
диффузионной модели, нельзя объяснить функций распределения
с длинными «хвостами», наблюдаемых в потоках жидкости; такие
распределения, существенно отличающиеся от нормального закона
распределения, вообще нельзя характеризовать единственным
* Другой причиной является то, что в условиях, когда диффузионная мо-
дель неточна, различные способы обработки экспериментальных данных приво-
дят к разным значениям эффективных коэффициентов переноса.
221
параметром — эффективным коэффициентом диффузии. Анализ ди-^
скретной ячеистой модели позволяет найти основные характеристики]
продольного и поперечного переноса вещества в зернистом слое
с учетом перемешивания потока в ячейках, застойных эффектов и
неоднородности упаковки слоя и определить условия, в которых эти
процессы переноса адзкватно описываются диффузионной моделью
123-25].
Перенос тепла в зернистом слое во многом аналогичен переносу
вещества. Различие между обоими процессами состоит в том, что
тепло может переноситься не только по движущейся фазе (жидкость
или газ), но и по неподвижной (твердые частицы). При анализе про-
цессов переноса тепла в потоках газа при достаточно больших тем-
пературах необходимо учитывать также лучистый теплообмен между
частицами. \
Экспериментальным исследованием поперечной теплопроводности
зернистого слоя занимались многие авторы [26—28]. Перенос тепла
в зернистом слое осуществляется тремя путями [27 , 28]:. движу-
щейся жидкостью или газом, через твердые частицы и точки их со-
прикосновения и смешанный перенос через твердые частицы и обте-
кающий их поток. Пренебрегая последним способом переносом тепла
и считая два первых аддитивными, Аэров [27 ] предложил следующую
формулу для определения аффективного коэффициента поперечной
теплопроводности
%j.7%=Xox/x + *j. Рг (VI.30)
где % — молекулярная теплопроводность потока; %0±, — экспе-
риментальные параметры; Рг = v/% — тепловое число Прандтля.
Обработка опытных данных показывает, что коэффициент /с±, кото-
рый при Re —> оо совпадает с обратной величиной эффективного
числа Пекле для поперечной теплопроводности, не зависит от числа
Рейнольдса во всей исследованной области, вплоть до Re = 3500.
Численные значения найденные в работах [27, 28], лишь незна-
чительно отличаются для частиц различной формы и составляют
примерно 0,1, что согласуется с значениями обратного .числа Пекле
для поперечного переноса вещества. При Re < 100 существенную
роль играет перенос тепла через твердую фазу. Влияние материала
частиц слабо проявляется даже при малых числах Рейнольдса, так
как основное сопротивление переносу тепла через твердые частицы
локализуется в точках их соприкосновения, где тепло передается,
главным образом, через тонкий слой неподвижного газа, примы-*
кающий к точке контакта зерен. Этот факт был установлен экспери-
ментально в работе [29], где изучалась теплопроводность слоя сфе-
рических частиц в отсутствие движения газа и была обнаружена
сильная зависимость теплопроводности слоя от давления газа.
В работах [30] приведены результаты исследования стационар-
ного распределения температуры в зернистом слое в направлении,
противоположном движению потока, от точки впуска тепловой метки.
Результаты опытов, проведенных при Re < 50, обнаруживают зкспо-
222
ненциальный спад температуры по мере удаления от точки впуска
метки. Эффективный коэффициент продольной теплопроводности
определяется из значения коэффициента зкспоненциального зату-
хания. Полученная зависимость %ц от Re описывалась формулой,
аналогичной (VI.30). Как и следовало ожидать %0± — %оц, так как
при отсутствии потока зернистый слой становится изотропным.
Величина /сц отличается от к± и равняется приблизительно 0,7—0,8.
Следует, однако, отметить, что точность полученных результатов
невысока. Это особенно сильно проявляется с увеличением числа
Рейнольдса, когда основной перепад температур происходит на рас-
стояниях порядка размера отдельного зерна. При Re >> 50 опыты
вообще не удалось провести, так как перенос тепла против потока
становится в этих условиях незначительным. Так как, однако, при
средних и больших значениях числа Рейнольдса основное значение
имеет перенос тепла движущимся газом или жидкостью, для оценки
эффективных коэффициентов теплопроводности можно использовать
данные по исследованию эффективных коэффициентов диффузии,
считая соответствующие числа Пекле одинаковыми. Роль твердых
частиц в зтих условиях сводится только к созданию емкостных аф-
фектов и должна проявляться лишь в нестационарном режиме. Ана-
лиз процессов продольной теплопроводности с учетом различных
механизмов задержки тепла был проведен на основе квазигомоген-
ной модели в работах [31 ]. Продольную теплопроводность зерни-
стого слоя исследовали с помощью ячеистой модели [321: были
учтены различные механизмы переноса тепла — через движущуюся
и твердую фазы и смешанный механизм, — и задержка тепла твер-
дыми частицами. Расчет коэффициентов продольной и поперечной
теплопроводности в стационарном режиме с учетом трех названных
механизмов переноса тепла был проведен в работе [33]. Эксперимен-
тальные данные по структуре, гидродинамике и коэффициентам
переноса вещества и тепла в зернистом слое собраны и систематизи-
рованы в монографии Аэрова и Тодеса [34].
X.
VI.3. ПРОДОЛЬНОЕ ПЕРЕМЕШИВАНИЕ
‘В ЗЕРНИСТОМ СЛОЕ
Основные соотношения. В этом разделе рассматривается про-
дольное перемешивание в зернистом слое на основе дискретной
ячеистой модели.
Время пребывания в отдельной ячейке является случайной вели-
чиной с дифференциальной функцией распределения <р (т), которая
определяется процессамй перемешивания в отдельной ячейке; в даль-
нейшем будем ее называть микрораспределением. Будем сначала счи-
тать все ячейки идентичными и, следовательно, имеющими одина-
ковую функцию микрораспределения <р (т) *. При исследовании
продольного перемешивания^ очевидно, достаточно ограничиться
* Случайный разброс параметров ячеек учитывается в разделе VI.4.
223
анализом одномерной модели слоя. При этом слой рассматривается
как одномерная цепочка идентичных ячеек, каждая из которых имеет
индекс п, указывающий ее положение в цепочке. Так как перенос
вещества против течения практически отсутствует, перемещение
частиц трассирующего вещества по цепочке будет происходить
только в одном направлении, т. е. из п-й ячейки в (п + 1)-ю.
Время пребывания в слое равно сумме времен пребывания в ячей-
ках, последовательно проходимых частицей примеси. Так как вре-
мена пребывания в отдельных ячейках взаимонезависимы, характе-
ристическая функция времени пребывания в цепи N ячеек (G) равна
произведению характеристических функций распределения времени
пребывания в отдельных ячейках (g)
G^P)=gN(p)
СО со
где Gn (р) = J е"ОтФЛ’ (т) dr; g (р) = J ф (т) e~p~dr; Фу (т) —
о о
функция распределения времени пребывания в цепи из N ячеек.
Так как при сложении независимых случайных величин их се-
миинварианты * складываются, семиинварианты функции рас-
пределения времени пребывания в слое ФДт (т) равны семиинвариан-
там микрораспределения, умноженным на число ячеек N по длине
слоя. Первый семиинвариант равен среднему времени пребывания
в слое S = Ns (где s — среднее время пребывания в отдельной
ячейке). Второй семиинвариант х2 равен дисперсии времени пребы-
вания в слое и служит основной характеристикой процесса продоль-
ного перемешивания потока. Зная третий семиинвариант z3, можно
вычислить коэффициент асимметрии Sk = x3z-3/2, характеризую-
щий отклонение функции распределения от нормального закона.
Для нормального распределения х3 = Sk = 0 и все высшие семиин-
варианты также обращаются в нуль. Распределения с Sk =^= 0 яв-
ляются несимметричными, причем распределения с положительным
коэффициентом асимметрии должны обладать длинными «хвостами»
при больших t. Так как х3 и х2 пропорциональны N, то Sk(N)—
— N~l/‘ Sk(1) (где SkU) — коэффициент асимметрии микрораспре-
деления). Очевидно, Sk(2V) —> 0 при N ->оо. Величины (/ =
= 3, 4, . . .) также стремятся к нулю с увеличением N, так что при
N -> оо распределение Ф]у (т) всегда приближается к нормальному
закону, независимо от вида микрораспределения.
Этот факт непосредственно следует из центральной предельно?
теоремы теории вероятностей. Однако дисперсия предельного за-
кона, а также число ячеек, на котором происходит приближение
к нормальному распределению, существенно зависят от свойств
микрораспределения. Из определения коэффициента асимметрии
следует, что Sk(2V) 1 при N Ns (где ]/лГ. = Sk(1)). Очевидно,
* Семиинварианты случайной величины определяют по формуле (VI. 11).
224
Ns может существенно превышать единицу только в том случае,
когда Sk(1) > 1, т. е. когда микрораспределение обладает «аномаль-
ной» асимметрией.
Для вычисления статистических характеристик распределения
времени пребывания в слое необходимо, таким образом, задаться
моделью ячейки и определить функцию микрораспределения. Наи-
более приемлемой моделью для описания перемешивания в потоках
жидкости и газа при средних и больших числах Рейнольдса пред-
ставляется модель ячеек идеального смешения с застойными зонами.
Наблюдаемое на опыте различие в поведении потоков жидкости и
газа указывает на то, что перенос вещества в застойных зонах дол-
жен быть диффузионным.
Ячейки с застойными зонами. Пусть С — концентрация ней-
тральной примеси в свободном объеме ячейки и Сх — ее концентра-
ция в застойной зоне. Если в момент t = 0 в ячейку подан б-импульс
f примеси, то изменение концентраций С и Сг со временем опреде-
ляется решением системы уравнений
' 4г==------— (VI .31)
dt s0 \ dv JV ' '
= (VL32)
с граничными и начальными условиями
С1(Г) = С; (dCt/dv^^O (VI.33)
С(О) = Со; Сх(0)=0 (VI.34)
Здесь s0 — среднее время пребывания в свободном объеме ячейки;
D — коэффициент молекулярной диффузии; о — площадь поверх-
| ности раздела Г между ячейкой и застойной зоной, отнесенная к еди-
нице свободного объема ячейки; Гх — поверхность частиц; v и vx —
направления внешних нормалей к поверхностям Г и Гх.
Решение задачи (VI.31)—(VI.34) определяет (с точностью до мно-
жителя) функцию микрораспределения <р (т).
Рассмотрим сначала застойную зону, совпадающую с диффузион-
ным пограничным слоем у поверхности твердых частиц. Такую
застойную зону можно считать плоской, с толщиной б, много мень-
шей диаметра частицы I. Тогда уравнение (VI. 32) и граничные усло-
вия (VI.33) принимают вид:
dx* D dt ’ С1(б, I)— с,
=0 (VI.35)
Переходя к трансформантам Лапласа, С (/?), Сх (р, х), имеем
Й2С1 _ р
dx* D >
15 Заказ 1441
С1(б) = С; =0
\ dx /к=о
(VI.36)
225
откуда
(VI.37)
Уравнение для трансформанты Лапласа С (р) принимает вид
Со = (р 4-SJ1) С + Со V^D th V~p[D 6 (VI .38)
откуда
C(p) = Co(p+sf1 + o/p5th/p7o6)-i (VI.39)
Трансформанта С с точностью до нормировочного множителя
совпадает с характеристической функцией микрораспределения.
Вводя множитель i/Coso (соответствующий нормировке функции
распределения на единицу), имеем
S(p) = [l + pMl + aoth iG^/TT^) I"1
(VI.40)
где а0 = об — отношение объема застойной зоны к свободному объему
ячейки; tD = 82/D — характерное время проникновения в застой-
ную зону.
Семиинварианты функции распределения времени пребывания
в цепи N ячеек с характеристической функцией микрораспределе-
ния g (р) определяют по формуле:
х^)=лг(-1)/ (dHng/api)p^
(VI.41)
Используя уравнение (VI.40), имеем
XW) = Vs0 (1 + а0) = Vs; = Vs® (1 +-^-atD/S }
“(Ws)2]
^N~> = 2Vs3 Г1 + at D js + -|-
(VI.42)
где a = a0/(i + a0) и s = s0 (1 + a0) — среднее время пребывания
в ячейке.
В случае цепи ячеек идеального смешения дисперсия функции
распределения времени в слое равна Ns2. Оценим величину
относительной добавки
ваемая застойная зона
ности твердых частиц:
к дисперсии atjj/s, считая, что рассматри-
совпадает с диффузионным слоем у поверх-
atD б и 62 _ Re-Рг 1
s I ’ I ' D Nu3 Res^-i
(VI.43)
Здесь I — характерный размер ячейки (диаметр частицы); и — ли-
нейная скорость потока; Re = ul[v\ Рг = v/D; Nu = Z/б. В послед-
нем из соотношений (VI.43) использована зависимость числа Нус-
сельта от чисел Рейнольдса и Прандтля Nu = RemPr z*. Так как
показатель т лежит в пределах от 0,5 до 1, относительная добавка
к дисперсии за счет рассматриваемых застойных зон уже при сред-
226
них числах Рейнольдса ничтожно мала для потоков как газа, так
и жидкости.
Рассмотрим теперь застойную зону близ точки соприкосновения
сферических частиц (см. рис. VI.4). Если принять ширину устья
застойной зоны равной 260 (где 60 — толщина вязкого подслоя),
то глубина ее у о будет равна V I б0. Так как I > б о, то уо/&о =
= РЖо » I-
В такой узкий и глубокий канал турбулентные пульсации могут
проникнуть лишь на незначительную глубину, сравнимую с шири-
ной его устья 80. Диффузионное уравнение для застойной зоны опи-
санной формы решается в вырожденных биполярных координатах.
Опуская подробности решения [21 ], выпишем окончательные вы-
ражения для семиинвариантов микрораспределения:
4®’ = №,(l+-jy“o«p/*) (VI.44)
4W) = 2Ns3 j^l -g- a<ito/s-f- 12g яо (гв/3)2 J
Формулы (VI.44) отличаются от формул (VI.42) только числен-
ными коэффициентами, хотя различие в значениях коэффициентов,
вызванное различие»! формы застойных зон, достигает иногда почти
двух порядков. Более важным обстоятельством является, однако,
то, что величины а0 и tD в рассматриваемом случае зависят от ха-
рактеристик потока иначе, чем в случае плоских застойных зон.
Для качественной оценки порядка величин а6 и tD воспользуемся
соотношениями, полученными в обычной теории пристеночной тур-
булентности [35 ]:
ао _ -М. ₽ ( А-У = ( . JLV Re-2K1 (VI.45)
Ds D v D и*
Здесь u* — динамическая скорость; к = (и*/и)2 — безразмерный
коэффициент сопротивления.
Величина к при турбулентном режиме течения слабо зависит
от числа Рейнольдса и при Re = 102 н-103 равна примерно 0,1—0,03.
Из формул (VI.15), (VI.46) видно, что в потоках жидкости (Рг =
= v/D — 103) относительная добавка к дисперсии за счет задержки
в застойных зонах aotD/s значительна при Re — Ю2; при Re ~ 103
эта добавка уже не должна играть заметной роли. В газах вели-
чина atDjs всегда мала.
Сравнение ячеистой и диффузионной моделей. Сравним характери-
стики функций распределения времени пребывания для исследован-
ной дискретной модели слоя с соответствующими характеристиками
15*
227
квазигомогенной модели, описываемой диффузионным уравнением
дС _ дС'
dt _jLll dX2 и дХ
(VI.47)
где D [| — эффективный коэффициент продольной диффузии; X —
продольная координата.
Эквивалентная форма уравнения конвективной диффузии
s
дС
dt
D* д2С
"ll "dX2
дС
dX
(VI.48)
где е — доля свободного объема; w = ей — фильтрационная ско-
рость.
Между коэффициентами и D*\ имеет место соотношение
Д*1 = eZ> л; числа Пекле в обоих случаях будут, очевидно, одина-
ковыми.
Функция распределения времени пребывания в слое, полученная
решением уравнения (VI.47) в приближении бесконечной среды, и
выражения для семиинвариантов приведены в разделе VI. 1. Учиты-
вая, что длина слоя L связана с числом ячеек N соотношением L =
= Nl = Nus и опуская малые добавочные члены, обратно пропор-
циональные числу Пекле Ре = УРец, перепишем формулы для
семиинвариантов (VI.24), (VI.25):
xPT)=Vs; ^N) = 22Vs2/Pe(|; *(3N) — W/Pe^ (VI.49)
Величину эффективного коэффициента продольной диффузии D ц
или число Пекле Рец определяют из сравнения дисперсий обеих
моделей Очевидно, значения xW из формул (VI.49) и (VI.44)
совпадают, если принять:
Реп=2(1 + 1г49 nn=T-(1+i-'T-) (VL50)
Из приведенных выше оценок величины atjjjs следует, что в потоках
жидкости при Re — 102 должны наблюдаться повышенные значения
эффективного коэффициента продольной диффузии. В потоках га-
зов, а также в потоках жидкости при Re 103, Ре = 2, как и
в простой модели ячеек идеального смешения^ и застойные зоны не
вносят существенного вклада в продольное перемешивание потока.
Эти оценки согласуются с экспериментальными данными (см.
рис. VI.7).
Сравним теперь коэффициенты асимметрии функций распределе-
ния, полученных из ячеистой и диффузионной моделей. Для диффу-
зионной модели Sk <iV) — 1/ ]/VPe II и распределение близко к нор-
мальному при N > Ns — Pe"j* — 1, т. е. нормальное распределение
устанавливается в слое относительно небольшой длины. Заметим,
что при меньших длинах слоя (V — 1) вообще бессмысленно вво-
дить квазигомогенную модель.
228
Для ячеистой модели из формул (VI.44) следует:
qfcW- 2 [j! “• 1 “ а tD\3'2 М7Т Rn
Sk -^[1 + ~Г~+128 Ж ]/ (1+l2—) (VL51)
В потоках газов tD/s ~1 4-10 и SkCV) = 2/ VN как и в мо-
дели ячеек идеального смешения. В жидкостях при Re ~ Ю2,
когда относительная добавка к дисперсии равна по порядку вели-
чины единице (a tD/s 10), tD/s^> 1 и Sk(;v) = tD/(s j/TV). В этом
случае Ns~ (tD/s)2^> 1 и нормальный закон распределения мог бы
установиться только в слоях очень большой длины (N ~ 107).
Поэтому практически в этих условиях всегда должны наблюдаться
асимметричные распределения с длинными «хвостами». Интересно,
что даже при Re ~ 103, когда застойные зоны уже не вносят суще-
ственного вклада в дисперсию, асимметрия распределения может
быть еще значительной, так что при умеренном числе ячеек «хвосты»
функций распределения исчезнут лишь при Re 104. Сильное раз-
личие коэффициентов асимметрии для ячеистой и диффузионной
моделей в потоках жидкости при Re = 102 4-103 говорит о том, что
в этих условиях диффузионная модель является неадекватной.
Следует заметить, что условие малости коэффициента асимметрии
является, вообще говоря, только необходимым, но не достаточным
условием установления нормального распределения. Чтобы опре-
делить достаточные условия приближения распределения к нор-
мальному закону, необходимо рассмотреть высшие семиинварианты
всех порядков. Как и при определении коэффициента асимметрии,
для приведения семиинвариантов к безразмерному виду естественно
использовать дисперсию распределения, взятую в соответствующей
степени. Исходя из вида характеристической функции (VI.40),
нетрудно показать, что при выполнении условия Sk С 1 все вели-
чины также будут близки к нулю и, следовательно, выпол-
нение условия Sk 1 в рассматриваемом случае не только необхо-
димо, но и достаточно для установления нормального закона рас-
пределения времени пребывания в слое.
Аналогичным образом может быть описан процесс продольной
теплопроводности в зернистом слое. В потоках газов перенос
тепла идет в основном по движущейся фазе при Re > 102; в жидко-
стях тепло переносится исключительно движущейся фазой уже при
весьма малых числах Рейнольдса. Твердые частицы в этих условиях
выступают в роли застойных зон, и при оценке характеристик функ-
ции распределения можно воспользоваться формулами (VI.42) (см.
раздел VI.5).
Расчет стационарных режимов. Большое практическое значение
имеет вопрос о применимости различных моделей к расчету химиче-
ских реакций, протекающих в стационарном режиме. В частности,
интересно выяснить, в каких условиях оправдано и рационально
применение диффузионной модели слоя и каково значение эффектив-
ного коэффициента продольной диффузии, при котором расчет
229
процесса с помощью диффузионной модели дает наиболее точный;
результат. Этот вопрос отнюдь не тривиален, поскольку в условиях, ;
когда диффузионная модель не точна, нельзя без дополнительных;
доказательств утверждать, что одно и то же значение эффективного'
коэффициента диффузии будет давать наилучший результат при
расчете различных — стационарных и нестационарных — характе-
ристик процесса, например, дисперсии функции распределения вре-
мени пребывания в слое и степени превращения реагента в стацио-
нарном процессе.
Для того чтобы провести сравнение различных моделей в стацио;
парном режиме, удобно рассмотреть необратимую гомогенную реак-
цию первого порядка с константой скорости к [25 ]. Будем считать,
что концентрация реагирующего вещества С изменяется только
по длине реактора и постоянна по его сечению. Тогда концентрации
реагента на выходе и входе реактора СВыХ и Свх связаны соотноше-
нием (см. раздел VI. 1)
JZ5J“-=C ф(т)е-*Мт = б(к) (VI .52)
^ВХ J
О
где Ф (т) — функция распределения времени пребывания в слое;
G (к) — соответствующая характеристическая функция, взятая от
действительного положительного аргумента к.
Из формулы (VI.52) видно, что для совпадения результатов,
полученных с помощью различных моделей, достаточно, чтобы соот-
ветствующие характеристические функции были близки друг к другу
на действительной положительной полуоси.
В простейшем случае, когда реактор описывается моделью идеаль-
ного вытеснения, функция распределения времени пребывания в слое
и ее характеристическая функция определяются формулами:
<D(t) = 8(t-NZ/u); G(k) = exp(—Nkl/u) (VI.53)
Характеристическую функцию для диффузионной модели (VI.47)
легче всего найти, решая уравнение
<VI-54>
с приближенными граничными условиями (см. раздел VI. 1)
С(О) = СВХ; С(оо)<оо (VI-55X
Учитывая соотношение (VI.52), находим:
G(к) = exp [-gL (1 -/1+4WH/^)] =
= exp pV (1 - Vl + 4X/Pe|| )J VI.56)
где X = kljw, Рец = uljD ц.
230
Будем считать, что X 1; это соответствует предположению
о том, что на масштабах, сравнимых с размером отдельной ячейки,
не происходит существенного изменения концентрации исходного
вещества. Очевидно, что, если это предположение не выполняется,
то описывать процесс с помощью квазигомогенной'модели вообще
бессмысленно. Так как число Рец — величина порядка единицы,
показатель экспоненты в формуле (VI.56) можно разложить в ряд
Тейлора по малому параметру Х/Рец. Ограничиваясь членами не
выше второго порядка малости по X, имеем:
G(fc) = exp [—IV (X —Х2/Рец)] (VI.57)
Сравнение формул (VI.53) и (VI.57) показывает, что расчет сте-
пени превращения по диффузионной модели дает лишь относительно
малую поправку к расчету, выполненному на основе модели идеаль-
ного вытеснения.
Получим теперь соответствующие результаты для модели ячеек
идеального смешения с застойными зонами. В этом случае выраже-
ние для характеристической функции G (к) можно представить
в виде
G(A) = [1-M (fc)]’N = exp { — ЛГ1п [1 +А (Л)]) (VI.58)
где А (к) = g~l — 1; согласно формуле (VI.40):
। ____________________________________ ________!_
A(fc)=fcs —а (1 — th]/ktD / ]/fctj>)] (VI.59)
Здесь a = a0/(i + a0) — доля объема застойных зон.
На основании формулы (VI.59), нетрудно заметить, что всегда
А (к) < ks, а так как, согласно предположению, ks 1, то A <<^ Ф.
Разлагая показатель экспоненты в формуле (VI.58) в ряд Тейлора
по А и ограничиваясь членами не выше второго порядка малости,
имеем:
G (к) =ехр £ — N (д-(VI.60)
Сравнение формул (VI.57) и (VI.60) позволяет выразить пара-
метры диффузионной модели через параметры дискретной ячеистой
модели. Как мы убедимся, результаты существенно зависят от соот-
ношения между константой скорости реакции и двумя временными
масштабами, имеющимися в рассматриваемой системе — средним
временем пребывания в ячейке s и характерным временем проникно-
вения в застойную зону
Предположим сначала, что выполняется соотношение
s<^k-l<^tD (VI.61)
В этом случае, согласно формуле (VI.59)
A (k)^ks (1— a) = fcs0 (VI.62)
где s0 — среднее время пребывания в свободном объеме ячейки.
231
Параметры диффузионной модели, определяемые из сравнения
формул (VI.57) и (VI.60) оказываются следующими:
u = Z/s0; Рец =2; Z)||=Z2/2s0 (VI.63)
В обратном предельном случае
(VI.64)
выражение для Д (&) следует разложить в ряд Тейлора по малому
параметру ktD. Ограничиваясь членами второго порядка малости,
найдем:
X—Х2/Ре„ =ks---l-kW — 4-astD№ (VI.65)
" о
Формула (VI.65) приводит к следующим значениям параметров
диффузионной модели:
/ 1 1 V1
« = Z/s; Рен =(^—+ —;
= (Z2/2s) (1+4 aiD/s} (VI.66)
Легко заметить, что в обоих рассмотренных случаях выполняется
условие малости параметра Х/Рец, использованное при выводе фор-
мулы (VI. 57).
Различие между формулами (VI.63) и (VI.66) физически легко
объяснимо. В случае, когда выполнено условие (VI.61), реакция
практически завершается за время, много меньшее того, которое
необходимо частицам реагента для проникновения в застойные
зоны. Поэтому в таком процессе влияние застойных зон на превра-
щение реагента не чувствуется и параметры диффузионной модели
должны быть такими же, как в случае, если бы застойные зоны были
отгорожены от проточной части ячеек непроницаемыми перегород-
ками. Другими словами, поровое пространство зернистого слоя
в этом случае может рассматриваться как совокупность ячеек идеаль-
ного смешения без застойных зон, объем которых равен объему
проточной части зернистого слоя. Если же реакция идет настолько
медленно, что выполняется условие (VI.64), то за время, необходи-
мое для достижения в реакторе заметной степени превращения,
успевает установиться динамическое равновесие между частицами
реагента, находящимися внутри и вне застойных зон. При этом
застойные зоны, естественно, влияют на величины параметров и
aD ц, определяемые формулами (VI.66). Неравенства (VI.61) и (VI.64)
можно переписать в более удобной форме, введя в них вместо кон-
станты скорости реакции к число ячеек по длине реактора N. Эти ве-
личины тесно связаны между собой, так как заметная степень пре-
вращения исходных веществ может быть достигнута на временах
порядка к"1 и длинах L = NI — и/к. Положив в формуле (VI.53)
Свых/^вх = In G (к) = 1, находим, что, зта степень превращения
232
достигается при N = (les) *. Подставляя это соотношение в нера-
венства (VI.61), (VI.64), получаем эквивалентные им. соотношения
ЛГ»<р/« (VI.67)
Следует заметить, что условие N tjy/s слабее, чем полученное
выше общее условие N Ns установления нормального распреде-
ления с коэффициентом диффузии Лц — см. формулу (VI.66).
При тех значениях параметров, когда неравенство N tD/s выпол-
нено, но общие условия установления нормального распределения
не соблюдены, различие между описанием процесса с помощью ячеи-
стой и диффузионной моделей может проявиться при расчете неста-
ционарных процессов.
Результаты, полученные для простых гомогенных реакций, не-
трудно распространить на гетерогенные реакции, а также процессы,
включающие произвольное число реакций первого порядка. В слу-
чае гетерогенной реакции эффективной константой скорости к будет
произведение константы скорости гетерогенной реакции, отнесенной
к единице активной поверхности, на площадь активной поверхности
в единице объема ячейки. Для этой величины останутся справедли-
выми все выделенные выше соотношения. Реакция на пористой ча-
стице катализатора может рассматриваться как гомогенная реакция,
протекающая только в застойных зонах, но не в проточной части
ячейки. Степень превращения, достигаемая в результате такой реак-
ции, по-прежнему определяется формулой (VI.58), но функция
Д (к) в этом случае должна быть связана с функцией распределения
времени пребывания только в застойных зонах, а не во всем объеме
ячейки. Эта видоизмененная функция Д (к), в отличие от (VI.59),
будет иметь вид:
А (к) = as th (VI.68).
Заметим, что величина YktD совпадает в данном процессе с мо-
дулем Тиле, а отношение Д (k)[aks с выражением для фактора эф-
фективности изотермической реакции первого порядка на плоской
частице катализатора (см. главу III). При определении параметров
диффузионной модели в рассматриваемом случае предположим, что
и = l/s, считая тем самым величину и равной фильтрационной ско-
рости потока. Из сравнения формул (VI.57) и (VI.60) можно опре-
делить эффективную константу скорости й* и эффективный коэффи-
циент продольной диффузии D ||. В предельных случаях ktD 1
и ktD < 1, соответствующих внутридиффузионному и внутрикине-
тическому режимам протекания реакции, получаем:
при ktD >1
‘• = -7-/-^-. <v,m>
при ktD 1
к* = ак-, D.. 2 (VI.70)
И 2s. \ 3 as /
233
В обоих случаях параметр %/Рец мал.
В случае, когда кинетические зависимости нелинейны, фор-
мула (VI.52) неприменима, поскольку вероятность химического
превращения зависит при этом не только от времени пребывания,
но и от траекторий частиц реагентов в зоне реакции. Если условия
реакции в проточной части слоя и в застойных зонах одинаковы и
описываются одной и той же кинетической функцией г (С), то ха-
рактерным временем реакции служит величина С/r (С) и можно
ожидать, что параметры квазигомогенной модели будут определяться
формулами (VI.63) или (VI.66), в зависимости от соотношения между
временами C/r (С) и tD. В случае, когда реакции с нелинейными
кинетическими зависимостями протекают в системе с локально не-
однородными условиями протекания реакции, нельзя вывести эф-
фективное квазигомогенное уравнение только из анализа гидроди-
намических процессов переноса. В зтом случае необходимо отдельно
решать уравнения для различных частей слоя (например, свобод-
ного объема и застойных зон), отличающихся друг от друга усло-
виями протекания химической реакции.
Следует подчеркнуть, что во всех рассмотренных случаях пара-
метр %/Рец мал и, следовательно, модель идеального вытеснения
является хорошим первым приближением при расчете химических
процессов в зернистом слое. Если параметр А, = кзне является малой
величиной, то описывать зернистый слой с помощью квазигомоген-
ной модели невозможно. Что же касается диффузионной модели
с параметрами, определенными соотношениями (VI.63) или (VI.66),
то она дает правильное второе приближение при расчете процесса
в зернистом слое только в том случае, если выполнены неравен-
ства (VI.61) или (VI.64). В промежуточных случаях (при к — t~^)
эффективный коэффициент продольной диффузии зависит от кон-
станты скорости реакции к, вследствие чего диффузионную модель
применять бесполезно.
VI.4. ПРОДОЛЬНОЕ И ПОПЕРЕЧНОЕ ПЕРЕМЕШИВАНИЯ
В ЗЕРНИСТОМ СЛОЕ С НЕПРАВИЛЬНОЙ УПАКОВКОЙ
При исследовании продольного перемешивания потока в зерни-
стом слое, описываемом ячеистой моделью, предполагалось, что
все ячейки идентичны, а параметры функции распределения^ вре-
мени пребывания в отдельной ячейке строго фиксированы. В реаль-
ном зернистом слое с неправильной упаковкой зерен условие иден-
тичности ячеек, очевидно, нарушается, и параметры микрораспре-
деления могут рассматриваться как случайные величины. В этом
разделе мы рассмотрим процесс продольного и поперечного переноса
нейтральной примеси в зернистом слое без предположения об иден-
тичности ячеек. Будем считать, что слой однороден, а параметры
ячеек, последовательно проходимых частицей примеси, статисти-
чески независимы. Это допущение (которое должно выполняться
в хорошо спроектированном процессе (см. раздел VI.2), где отсут-
234
ствуют такие резко нежелательные явления, как каналообразование)
означает, что ячейки с различными значениями параметров «хорошо
перемешаны», так что внутри слоя нет флуктуаций пористости
с протяженностью, значительно превышающей размер ячейки.
Пусть функция распределения времени пребывания в ячейке
(микрораспределение) <р (т[а) и соответствующая характеристиче-
ская функция
00
?(pr») = J е-Р-'ф (т I s) dT
о
зависят от случайного вектора s. Компонентами этого вектора
являются различные параметры микрораспределения, меняющиеся
случайным образом от ячейки к ячейке. В простейшем случае ячеек
идеального смешения есть только один такой параметр s=Vfv.
Распределение случайного вектора s будем характеризовать функ-
цией распределения / (s), определяющей плотность^ вероятности
попадания частицы примеси (метки) в ячейку с данным значением
вектора s. Функцию / (s) будем далее называть параметрическим
распределением.
Длина и направление шага при переходе между соседними ячей-
ками также будут рассматриваться как случайные величины.
Функция макрораспределения. Введем функцию макрораспреде-
ления Ф (£, х), имеющую смысл плотности вероятности обнаруже-
ния частицы гидродинамически нейтральной примеси (трассирую-
щего вещества) в момент времени t в ячейке с координатой х. Эта
функция полностью определяет процесс перемешивания в рассматри-
ваемой системе, и ее значение позволяет найти все интересующие нас
характеристики продольного и поперечного перемешивания при-
меси. Если Wn (t) dt — вероятность совершить ровно п переходов
между ячейками за интервал времени t, a Vn (х) d3x— вероятность
попадания из начала координат в точку х в результате п перехо-
дов, то
Ф(х, t)=<S *М‘)ГП(«)>=£ <Wn(t)>Vn(x} (VI.71)
n=0 n=0
Здесь < > — усреднение по параметрическому распределению.
При записи последнего выражения в формуле (VI.71) предполагается
статистическая независимость параметров ячеек, с одной стороны,
и длины и направления шага при переходе между ними, с другой.
Для дальнейшего исследования удобно провести в формуле
(VI.71) преобразование Лапласа по t и преобразование Фурье по х
G (р, <о) = J e-ptdt $ eitt,x<D (х, t) d3x =
О -со
= 5 <Wn(p)>4n(«>) (VI.72)
n-0
235
где
Wn(p) = $ e-PtWn(t)df,
О
¥„(w) = J eiaixVn(x)d»x
—oo
(VI,
(VI.74)
(VI.75)
С помощью функции G (p, <в) — трансформанты Лапласа от хара
теристической функции макрораспределения — легче всего опреде
лить интересующие нас статистические характеристики перемеши
ния примеси в зернистом слое.
Чтобы вычислить функцию < Wn (р) } , рассмотрим некоторую»
фиксированную траекторию частицы примеси, проходящую чере
ячейки со значениями Sj (j = 1, 2, . . ., п) случайного вектора s»
Очевидно, вероятность к моменту времени t совершить точно п пере-
ходов (т. е. функция Wn (t) для данной траектории) равна
t
Wn (t) = j Фп («11*0’ «Ь ’-’«л-1) 1 —
о
— J <р(т|«п)Л dt!
о
Здесь Ф„ (Zt) — плотность вероятности прохождения цепи п ячеек
за время Zlt т. е. функция распределения времени пребывания в рас-
сматриваемой цепи ячеек; множитель в квадратных скобках опредет
ляет вероятность остаться в n-й ячейке в течение остального проме-
жутка времени t—Так как последовательные переходы взаимо-
независимы, лапласовский образ функции Wn (i) определяется 4И
формулой
wn (p) = 1 g (pkn) П g (Phi)
г
3=0
Чтобы найти функцию < Wn (р) > , достаточно теперь’усреднитьвыра-
жение (VI.74) по всем траекториям:
<IV„(p)> =
- I . . . \ [1 — gn. (p|s)l П gj (p|s) П ф/ (.S') dsj =
’ 1=0 з=о
= р"1 (l—<g (P)»<g(p)>n
Здесь интегрирование распространяется на всю область изменения
набора параметров s.
Вычислим теперь функцию (со). Пусть v (г, 9) — плотность,
вероятности перехода между ячейками с шагом г под углом 0 к на-?
правлению движения потока.
.236
Соответствующая характеристическая функция равна
4(ю|| , ю_|_) = 2л J J v(r, Q) егю II '’СО80/О (}z2 ®xr sin &) sin Or2 dr dO (VI.76)
о 0
где Jo — функция Бесселя; и2 = и2! + 2®^.
Согласно условию нормировки функции у (г, 9), Т = 1 при
<о || = <»j = 0. Так как последовательные переходы предполагаются
статистически независимыми, то
Yn(®) = r (®ц, ®x)=<ei“ll,J0(l/T®J_Zi)>n (VI.77)
где I = г cos 9 и 11 = г sin 9 — шаг при переходе между ячейками
соответственно в продольном и поперечном направлениях, а угловые
скобки обозначают усреднение по распределению v {г, 9).
Подставляя функции (VI.75) и (VI.77) в формулу (VI.72), находим:
ОО
п=0
= A/p(l + A-<eiM|l'jo (l/2w±Z1)>) (VI.78)
где Д (р) = < g > ~1 — 1.
Статистические моменты. Лапласовские образы статистических
моментов продольного и поперечного распределений примеси опре-
деляются дифференцированием функции (VI.78) по соц и (Ojj
= (diG/dai)^0 (VI.79)
Используя формулу (VI.76), находим:
й" (р) = <1>/рЛ
. (vi.so)
Ра1- (р)= <Л!>/рД
Воспользуемся формулой обратного преобразования Лапласа
И (0=^- J eP^(p)dp (VI.81)
a-ioo
и исследуем асимптотическое поведение статистических моментов
Ц (t) при t ->оо, не конкретизируя пока функции g (р). Вид функ-
ции р, (/) определяется, как известно, особыми точками подинте-
грального выражения в формуле (VI.81). Мы будем считать, что все
эти особые точки — полюсы, и нумеровать их в порядке убывания
действительных частей Ро, Pi, • • •, Р/- В теории функций ком-
плексного переменного полюсом функции называется точка, где
функция обращается в бесконечность. Из формул (VI.89) следует,
что полюсы функций р, (р) совпадают с нулями функции Д (р),
237
точка р = 0 также всегда является полюсом. Если в окрестности
полюса р = р/ функция изменяется как (р—Р/)~", то данный полюс
называется полюсом n-го порядка. Вычетом функций z (р) в полюсе
n-го порядка называется величина
Res z (р) = (/>-/>/)« {dn-^ldpn-i)p^p. (VI.82)
Выражение (VI.81) можно представить теперь в виде ряда
Р (t) = 2 Res (р) dP] (VI.83)
>0
Асимптотическое поведение функций р (£), как нетрудно видеть,
определяется положением точки р0. Можно показать, что, незави7
симо от вида микрораспределения, всегда ро=0. Действительно,
полагая р = х + iy и учитывая, что функция микрораспределе-
ния ф (т) положительна и нормирована на единицу, получаем сле-
дующее неравенство для действительной части функции g (р) в об-
ласти х = Re р Ss 0:
СО оо
Re g (р) = j е"*т cos ртф (т) dr<Z J <р (т) dx — 1
о о
Таким образом, функция р (I) не имеет особых точек в правой
полуплоскости и на мнимой оси, и асимптотическое выражение
для р (f) определяется, согласно формуле (VI.83), вычетом в точке
Ро=0.
Непосредственно из определения характеристической функции
g (р) следует, что g (0) = 1. Воспользовавшись разложением харак-
теристической функции микрораспределения g (р | s) в ряд по мо-
ментам
g(pi«)=i+2<-i)Z -v-p* (vi-84>
находим:
Д=<«1>Р+ ( <ai>2-4-<«2>) Ра+- • • (VI.85)
Легко заметить, что величина < a i ) представляет собой не что
иное, как усредненное по всем ячейкам значение среднего времени
пребывания в ячейке. Очевидно, функции pjl (р) и р^ (р) имеют
в точке р = 0 полюс второго порядка, а функция pH (р) — полюс'
третьего порядка. Вычисляя соответствующие вычеты и отбрасывая
члены, не зависящие от времени, получаем асимптотические фор-
мулы:
’ Ц* (<)=</> Res («Pz/M)p-o= < 1> Ч < Я1 >
(«)= <i!)4<ai>
238
Эффективные коэффициенты диффузии. При описании зернистого
слоя квазигомогенной моделью функция макрораспределения опре-
деляется решением уравнения конвективной диффузии
^-L У 'бУ V 6У ) + jD| № “ дХ ~~ dt (VI-87)
с начальным условием Ф (X, Y, 0) = 6 (Л") 6 (У). Параметры, вхо-
дящие в уравнение (VI.87), можно найти путем сравнения первого
и второго моментов макрораспределений, вычисленных на основе
непрерывной и дискретной моделей. Выполняя в уравнении (VI.87)
преобразование Фурье по пространственным координатам, получаем:
G (<о,, (0^,/) = ехр [(ги©! —£>,<0^ —2Z>±(i>5.) i] (VI.88)
Дифференцируя выражение (VI.88) по Иц, <о_ц находим:
ц} (t)=ut, (xs> (<) = u2<2 + 2Z>i<, =4Drt (VI.89)
Сравнение формул (VI.86) и (VI.89) дает:
»=<! >/<«!> (VI.90)
=4-< «I >-! [ < z >2 ( < «2 > / < «I >2—2)4- <Z2>] (VI.91)
А*
Z*±=T<Z?> /<“1> ' (VI.92)
Формулы (VI.91) и (VI.92) можно модифицировать, приняв
во внимание статистическую независимость длины и направления
шага. Вводя эффективные продольные и поперечные числа Пекле,
основанные на средней длине шага в продольном направлении
< I > = < г cos 9 > , получим:
рс “<Z> <^2> \ , <Г*> <СО826>-1-1
1 D, L\<ai>2 <r>2 <cose>2j (VI.93)
Формулы (VI.90)—(VI.94) были выведены при самых общих
предположениях о зернистом слое как дискретной случайной среде,
без каких-либо специальных предположений о геометрической струк-
туре слоя и характера перемешивания внутри ячеек. Для опреде-
ления численных значений коэффициентов переноса необходимо
конкретизировать рассматриваемую модель. Рассмотрим сначала
формулу для эффективного коэффициента продольной диффузии D ц.
В системе идентичных ячеек идеального смешения < > =s и < а2 > =
= 2s2. Поэтому первый член в квадратных скобках в формуле (VI.91)
обращается в нуль. Если шаг в продольном направлении I строго
фиксирован, формула (VI.93) дает Рец = 2. Увеличение эффектив-
ного коэффициента продольной диффузии и уменьшение числа
Пекле Рец может быть вызвано, вообще говоря, тремя причинами.
239
Во-первых, при сохранении упорядоченности геометрической струк- д
туры и идентичности ячеек его величина возрастает благодаря силь- Я
ной задержке примеси в застойных зонах внутри ячеек. Этот случай Я
был рассмотрен в разделе VI.3, было показано, что соответствующая Я
добавка к эффективному коэффициенту продольной диффузии суще- 11
ственна для жидкостей при Re = 102 4-103 и несущественна для Я
газов. Второй причиной, которая может вызвать отклонение вели- я
чины < а 2 > —2 < at > 2 от нуля даже в случае системы ячеек идеаль- - Я
ного смещения, является разброс средних времен пребывания в от- 1
дельных ячейках. Действительно, считая шаг в продольном направ- |
лении фиксированным, а перемешивание потока внутри ячеек — |
идеальным, можно переписать формулы (VI.91) и (VI.93) в виде ;1
Ре,=2/(1 + 2у); £>,=(Z2/2<s>)(1 + 2y) (VI.95) 1
где введен параметр j
Y=-hT~1=^-u(s)('-<so>)2^ (vi.96) :
\ 6о / 6о J «Я
0
характеризующий степень размытости параметрического распреде-
ления. Чем больше этот разброс, .тем выше величина эффективного
коэффициента продольной 'диффузии. Наконец, третьей причиной
увеличения дисперсии макрораспределения является случайный раз-
брос расстояния между ячейками в продольном направлении: при
этом увеличение тем сильней, чем больше разброс. Все три назван-
ных эффекта вызывают отклонение эффективного продольного числа
Пекле от «идеального» значения Рец = 2, причем их вклады в эф-
фективный коэффициент продольной диффузии аддитивны.
Эффективный коэффициент поперечной диффузии D± не зависит
ни от характеристик перемешивания в отдельных ячейках, ни от
разброса параметров микрораспределения и определяется только
средним по слою значением среднего времени пребывания в ячейке.
Когда шаг в поперечном направлении 1^ строго фиксирован, попе-
речное число Пекле Ред равно 4(Z/Z1)2. Увеличение эффективного
коэффициента поперечной диффузии и, соответственно, уменьше-
ние Рех может быть вызвано только случайным разбросом расстоя-
ния между ячейками в поперечном направлении.
В системе идентичных ячеек идеального смешения ( < а 2 > =
=2 < a j ) 2) отношение продольного и поперечного чисел Пекле равно
Pej_ _ < cos2 0 )
Ре, ~ (sin20>
(VI.97)*
и определяется только распределением угла смещения 0 при пере-
ходе между ячейками. Наблюдаемое на опыте большее значение Ред
по сравнению с Рец (см. раздел VL2) связано с асимметрией распре-
деления v (0). Благодаря наличию направленного движения в слое,
наиболее вероятным будет смещение в направлении потока, т. е.
с углом 0 = 0. С увеличением 0 плотность вероятности v (0) должна
240
монотонно уменьшаться и, так как смещение метки в сторону, про-
тивоположную движению потока, практически невозможно, v (0) =
= 0 при л/2 0 л. Из сказанного следует, что в случайной
среде среднее квадратичное смещение в направлении потока всегда
будет больше, чем в поперечном направлении и, соответственно,
Ре£>Рец. Для установления количественного соотношения ме-
жду Рец и Ре± необходимо задать распределение v (0). Так, напри-
мер, если считать, v (0) — cos'" 0 (0 sS 0 sg л/2), то
р _2(т+1) И+З) р 2 (m+lp(m-|-3) .
II (m + 2)2 > -L (m+2)2 ’
Ре
—i = m+l (VI.98)
lell
При m = 0 Pe± = Ре у, как это и должно быть в случае изотроп-
. ной системы. Наблюдаемые на опыте значения продольного и попе-
речного чисел Пекле соответствуют значению параметра т — 4.
Исходя из общей формулы для функции макрораспределения
(VI.78), можно вычислить также высшие моменты продольного и
поперечного распределений примеси и найти значения высших се-
миинвариантов, характеризующих отклонение формы макрораспре-
деления от нормального закона, соответствующего решению урав-
нения конвективной диффузии (VI.87). Такое исследование показы-
вает, что установление нормального распределения концентрации
примеси вещества замедляется под действием тех же причин, кото-
рые приводят к повышенным значениям эффективного коэффициента
продольной диффузии.
VI.5. ТЕПЛОПРОВОДНОСТЬ ЗЕРНИСТОГО СЛОЯ
В СТАЦИОНАРНОМ РЕЖИМЕ
Механизм теплопередачи в зернистом слое. В потоках газов
с понижением числа Re твердые частицы начинают играть активную
роль в теплопроводности зернистого слоя; при этом нарушается
подобие процессов тепло- и массопереноса, имеющее место при
больших числах Re. Для анализа процесса переноса тепла в зерни-
стом слое необходимо учесть три механизма теплообмена: 1) перенос
тепла движущимся газом; 2) теплопроводность по твердой фазе через
точки контакта частиц и 3) смешанный механизм теплопередачи
по газовой и твердой фазам через поверхность их раздела. При вы-
соких температурах необходимо учесть также лучистый теплообмен;
мы, однако, ограничимся диапазоном температур, характерным для
каталитических процессов, в котором лучеиспусканием можно пре-
небречь пр сравнению с остальными механизмами переноса тепла.
Вклад трех видов теплообмена в теплопроводность зернистого
слоя можно . учесть путем анализа схемы, представленной на
рис. VI.8. В дальнейшем будем называть частицы «твердыми ячей-
ками». Описание зернистого слоя с помощью показанной на рисунке
к
16 Заказ 1441 241
двумерной схемы оправдано тем, что все направления в плоскости,
перпендикулярной движению потока, равноправны.
Стационарное распределение температур в рассматриваемой мо-
дели под действием стационарного источника тепла мощностью Q,
Рис. VI.8. Схема тепло-
переноса между газо-
выми ячейками (Q) и
твердыми частицами (•):
тип — номер ячейки или
частицы в поперечном и
продольном направлениях.
П-2 П П+2
Рис. VI.9. Схема тепловых потоков между
газовыми и твердыми ячейками.
включенного в газовой ячейке с индексами т — п = 0, определяется
решением линейной системы конечноразностных уравнений
аг [^'2' (^n-1, т-1 + ^п-1, т+1) — ^п<п^| +
+ Р (T’n-I, tn + П+i, т + т+1Ч~^п, m-i) = о (VE99)
ат (^п, zn-x + П, zn+l + ^n+2, т-1~\~1'п+2, т+1) —^n+1,
+ ₽ (^nm-j-^n+1, nt-l+^n-f-l, zn+l + ^n+2, т) — ^п+1> nil = О (VI.100)
При составлении этих
уравнений каждой стрелке
, на рис. VI.8 сопоставля-
ется тепловой поток, про-
порциональный разности
средних температур Тг и
Тт соответствующих газо-
вых и твердых ячеек. Ко-
эффициент аг характери-
зует конвективный тепло-
перенос между газовыми
ячейками, коэффициент
ат — теплоперенос между
твердыми частицами через точки их контакта и коэффициент |3—тепло-
перенос между твердой и газовой фазами через поверхность их раз-
дела. Если температура внутри каждой частицы или ячейки практи-
чески постоянна и основной перепад температуры происходит на'
их границе, коэффициенты аг и (3 можно оценить по формулам
aT=uyT/l; p = fcTa (VI.101)
где уг — теплоемкость единицы объема газа; кт — коэффициент
теплопередачи от потока к твердой частице; о — площадь поверх-
ности твердых частиц, отнесенная к единице свободного объема
слоя.
242
Коэффициент ат определяет теплопроводность слоя’ в отсутствие
конвекции. Чтобы учесть конечную теплопроводность твердых ча-
стиц, выражение для ₽ надо заменить на
₽ = (1/М+12/Хт)-! (VI.102)
где хт — теплопроводность твердых частиц.
Аналогичным образом можно учесть конечную теплопроводность
газовых ячеек.
Поперечная теплопроводность. Рассмотрим сначала процесс по-
перечной теплопроводности. Будем решать систему конечнораз-
ностных уравнений (VI.99), (VI.100) в бесконечной среде. Введем
характеристические функции
7Ъ(®)= 2 W“>n;
п=-оо
^(«а)= 2 ТтП^шП (VI. 103)
Умножая уравнения (VI.71), (VI.72), на eiran и суммируя по п,
находим:
тг [у +1)-7* ] -}- [1- (ViT^+ХП +1'Лг-1+^т+1)-
-?rm] = Q6OT0 (VI.104)
VT (^_х+Vn+1+Х2^_х + h-VTn+J - \тЪ ] 4-
4-[4’ (^+^-1+ ^+1+^)-^ ]=0 (VI.105)
где X = e~ta>, vr = аг/£, vT — ат/0.
Будем искать частные решения системы (VI.104), (VI.105) в виде:
Г^,= г>0« (VI. 106)
Величина О определяется решением алгебраического уравнения
Ай + 1 = 0 (VI.107)
где
__ 2Х[Х2 (2угУт + 2ут + 1) + (6УгУт+4Уг + 2утЧ-1)1
X2 (2vrvT—1) —2vrvT
(VI. 108)
-Х*4-2Х2(10угуг4-8ут4-8ут+6)+4угут-1
X2 (2vrvT —1)4- 2УгУ-г
Уравнение (VI.107) имеет две пары взаимно обратных корней.
Вычислив зти корни и определив соответствующие значения коэф-
фициентов а и Ъ, можно найти распределение температур в рас-
сматриваемой системе. Однако, как ясно из дальнейшего, для вычис-
243
пения эффективного коэффициента поперечной теплопроводности
нет надобности искать точное решение системы (VI.104), (VI.105).
Величину определяют, сравнивая распределение температуры
в рассматриваемой дискретной модели с распределением темпера-
туры, получающимся иэ решения уравнения конвективной диффузии:
Х±тЙ'-Т“-^ = Сб(Х)в(У) (VI.109)
Сравнение удобно провести не для самих распределений, а для
их Фурье-образов. Пусть
Т(У, (0) = -^- J Т(Х, Y)eia>x/ldX
“СО
Уравнение для Фурье-образа Т имеет вид:
х±^ + -^т=еб(У)
Откуда _______
“Yr 1 у /
(VI.НО)
(VI.111)
(VI.112)
В формуле (VI. 112) выбирается значение корня с отрицательной
действительной частью, соответствующее ограниченности темпера-
туры при |У| ->ро.
Распределение температуры в дискретной модели слоя, очевидно,
имеет смысл сравнивать с распределением (VI.112) только при до-
статочно больших значениях индексов т, п. Характер распределения
при больших п определяется поведением соответствующего Фурье-
образа при малых со. Нетрудно видеть, что при а> = 0 (и соответ-
ственно X = 1) уравнение (VI.107) имеет корень 0=1. Поэтому
при малых со корень уравнения (VI. 107) естественно искать в виде
О = 1 ± 8 (где 8 < 1). Кроме двух корней, близких к единице,
уравнение (VI. 107) имеет еще пару корней. Частные решения, соот-
ветствующие этим корням, однако, быстро затухают с ростом т и,
следовательно, при т <£ 1 их вкладом в искомое распределение
можно пренебречь.
Подставляя в уравнение (VI. 107) X = 1 — гео и О' = 1 ± 8,
находим ___ _____________
е= ± К—i<oKvr/(vr-hvT+l) (VI.113),
откуда
Г5,«о) = аехр(/
^(W) = 6exp(j/—
(VI.114)
Сравнение показателей экспонент в формулах (VI.112) и (VI.114)
(с учетом соотношения Y = mli) дает следующие выражения для
244
эффективного коэффициента поперечной теплопроводности Хх и
числа Пекле Рех:
Ре - и1уг г-( 1 Y 2Уг
± Х± \ 11 J l + vr+vT
Используя соотношение (VI.101), можно переписать формулы
(VI.115) в виде:
Xj. = -у- + М + “т )
(VI.116)
Ре =2(4-^ /(.1 + — +—)
J-, \ 11 7 / \ иуг “Уг 7
Из формул (VI.116) видно, что все три рассмотренных механизма
теплопередачи вносят аддитивный вклад в теплопроводность зер-
нистого слоя. При и =. 0 коэффициент теплопередачи кт ничтожно
1
мал и Хх — Ххо = -% Пиело Пекле Рех равно нулю при и = 0
и монотонно возрастает с ростом скорости потока. Так как ~ иа
и а < 1, то с увеличением и число Ре± приближается к предельному
постоянному значению Pe_L = 2 (Z/Zi)2. Эта величина совпадает
со значением числа Ре^ для процесса поперечной диффузии, по-
скольку при больших скоростях потока преобладают процессы кон-
вективного переноса, а остальные виды переноса тепла становятся
несущественными. Эти результаты согласуются с имеющимися экс-
периментальными данными по поперечной теплопроводности зерни-
стого слоя (см. раздел VI.2).
Продольная теплопроводность. При рассмотрении процесса про-
дольной теплопроводности наибольший интерес вызывает распреде-
ление температур в бесконечном слое, установившееся под действием
стационарных источников тепла, которые равномерно распределены
в плоскости п = 0. В этом случае можно рассмотреть лишь одно-
мерную модель слоя. Система конечноразностных уравнений, соот-
ветствующая этой модели, имеет вид (см. рис. VI.9):
4 (ТЙ-1+^+i) -Тгп + vr (Т£_2-Trn) = Q8n о <VI. 117)
у (П-П-2)-П-1+ vT [4 (Гп+1-^-з) - П-х] = 0 (VI. 118)
Частные решения системы (VI.117), (VI.118) будем искать в форме:
Трп = а$п-, TTn = btin (VI.119)
Величина О' определяется решением уравнения:
(l + 2vr+2vTvr)O6 — 2(3vrvT+2v}.+2vT4-l) О44-
+ (6vrvT+4vr + 2vT+D fla — 2vrvT=0 (VI.120)
245
Можно показать, что при любых значениях параметров vr, vT
все корни уравнения (VI. 120) действительны, причем три из них
всегда положительны, а три — отрицательны. Физический смысл
имеют только положительные корни:
•6'1=1
'6'2,3 =
_ Г-(4угут + 4уг + 2ут-|-1) ± РЛ(4угТт + 4уг + 2ут+1)2 —8угУт(2угУт + 2ут-Р1) 11 >2
2(2vrvT-j-2yT+l)
(VI.123)
(VI.124)
(VI.121)
Анализ формулы (VI.121) показывает, что при любых значениях
параметров vr, vT корни й2, й3 удовлетворяют неравенствам
0< 68< 1 <6’2< оо (VI.122)
Принимая во внимание неравенства (VI.122), записываем реше-
ние системы уравнений (VI.117), (VI.118), ограниченное при п -> оо
и обращающееся в нуль при п—>~— оо, в виде:
^ = «АП+Мз" 1 Тт = а2Щ
= ) при ’ T^bt&n
Величины и аг (г = 1,2,3) связаны между собой соотношением
bi _ 2 [6'?(14-УГ) —уг]
fy(l-H?)
Для определения трех неизвестных постоянных а2, а3 служат
два условия непрерывности функций Тгп и Тп при п= 0 и требова-
ние, чтобы решение, определяемое формулами (VI.123), удовлетво-
ряло неоднородному уравнению (VI.117) при п — 0. Окончательная
формула, описывающая стационарное распределение температур
вдоль зернистого слоя, имеет вид:
тп=Qo [(“2 — »з) + (“I — «2) о з ]
Т*п = Qo [“1 («2 — аз) + “з (“1 + “2) б?]
T^Qo^i-^W )и
П=<?о“2(а1-аз)й2п J ПРИ
(VI.125)
где <2о = <2 [«1 (а2 — а3) + а2 (а3 — + а3 (он — а2) йзр1.
Исследование формулы (VI.124) с учетом (VI.121) показывает:
«1=1; — оо < а3 <1 <а2 <оо (VI.126)
Из формул (VI. 125), (VI. 126) видно, что температура газовой
фазы Тгп монотонно возрастает по ходу потока. Температура твердой
фазы 7ft также монотонно возрастает с увеличением п при а3 >0;
при а3 < 0 величина Тт максимальна при п = 0 и убывает с ростом
| п | (рис. VI. 10). При п —оо температуры обеих фаз асимптотически
приближаются к постоянному значению Trm = Т*т = Qo ( а2 — а3).
246
Чтобы найти значение эффективного коэффициента продольной
теплопроводности, сравним полученное распределение температур
с решением
квазигомогенного уравнения:
d2T dT
Х' = (VI.127)
удовлетворяющее граничным условиям Т (— оо) = 0,
имеет вид:
Т(Х) = ! QlUyr (приХ^О)
I (CA'Yr) exp (иугХ/х#) (при X -sC 0)
, для описания теплопереноса в зернистом слое уравне-
Решение,
(VI.128)
О х
Рис. VI.10. Распределение температуры
газовой и твердой фаз по длине слоя:
I и II — твердая фаза; III —газовая фаза.
Очевидно,
нием (VI. 127) необходимо, чтобы температуры обеих фаз мало отли-
чались друг от друга и замет-
ное изменение температур
происходило бы на расстоя-
ниях, больших по сравнению
с размером отдельной ячейки.
Нетрудно заметить, исходя
из формул (VI. 125), что для
выполнения указанных тре-
бований необходимо, чтобы
величины а2 и й2 были до-
статочно близки к единице.
Можно показать, что это
условие выполняется только
при vr <£ 1, ч1о соответствует
достаточно малым скоростям
потока. В этом случае корни
ft 2 и $3 оказываются близкими
соответственно к единице и нулю. Вследствие малой величины й3 уже
на расстоянии в несколько ячеек вниз по потоку от источник а уста-
новится постоянная и одинаковая для обеих фаз температура. Вверх
по потоку от нулевой ячейки будет наблюдаться постепенное экспо-
ненциальное понижение температуры. Именно такое распределение
температур наблюдалось в опытах К унии и др.. [30 ]. Значение эффек-
тивного коэффициента продольной теплопроводности определяется
из сравнения показателей экспонент в формулах (VI.125) и (VI.128).
Так как при vr 1
6’2 l+2vr/(l + 2vT)
(VI.129)
то
±Yr£ = nln fl2=*
Xii I 1 + 2v-
и, следовательно:
Уги1 I 1
(VI.130)
/ста
~2~
Равенство (VI.130) написано с учетом соотношений (VI.101). Сле-
дует отметить, что формула (VI. 130) не содержит члена, описываю-
щего перенос тепла газовым потоком. В условиях, когда, возможно
247
сравнение дискретной модели с квазигомогенной, этот член должея-
быть мал. Однако его можно добавить к выражению (VI. 130) бла-
годаря тому, что вклады различных механизмов в теплопроводность,
согласно этой формуле аддитивны. Окончательные формулы для Хц
и Рец = и/тг/Х|| будут иметь вид:
Хц=^(«т+-^- + -^-) (VI.131)
Ре.. =2/(1 + -^-+-^-')
II I \ иуг 1 иуг J
Формулы (VI.131) следует рассматривать как интерполяционные,
поскольку они дают правильное значение Х|| и Р®|| как ПРИ малых,
так и при больших числах Re. Необходимо отметить сходство между
формулами1 (VI. 131) и (VI.116). Обе показывают, в согласии с имею-
щимися экспериментальными данными (см. раздел (VI.2), монотонное
увеличение Хц и Рец с ростом скорости потока и дают в пределе линей-
ную зависимость Хц от н и асимптотическое приближение числа Ре
к постоянному значению, которое совпадает с соответствующей вели-
чиной для процесса продольного или поперечного перемешивания
вещества. Отклонение от линейной зависимости х (м) при малых чис-
лах Re, вызванное учетом смешанного механизма теплопередачи, не
может быть пока проверено на опыте из-за неточности имеющихся
экспериментальных данных. Следует ожидать (см. разделы VI.3 и
VI.4), что в случае нестационарного процесса величина Хх останется
без изменения, а в выражении для хц появится добавочное слагаемое,
связанное с задержкой тепла в частицах.
VI.6. СТАЦИОНАРНЫЕ РЕЖИМЫ ПРОЦЕССОВ
В ЗЕРНИСТОМ СЛОЕ
Влияние ячеистой структуры слоя на режимы экзотермической
реакции. Исследование экзотермических процессов на изолирован-
ных частицах катализатора (см. главу III) показывает, что при опре-
деленных условиях могут наблюдаться скачкообразные переходы
между различными стационарными режимами процесса при плавном
изменении состава и температуры потока, омывающего частицу.
Если описывать зернистый слой катализатора в приближении идеаль-
ного вытеснения, то локальные условия перескока между режимами
будут такими же, какд в случае изолированной частицы. Например,'
если концентрации реагентов и температура в данной точке слоя
таковы, что в этих условиях кинетического режима процесса на изо-
лированной частице не существует, то частица, катализатора, поме-
щенная в данную точку слоя, будет работать в диффузионном режиме.
Причиной появления перескоков между режимами частицы, помещен-
ной в слой, в условиях, когда на изолированной частице эти пере-
скоки не наблюдаются, может быть только перенос тепла против тече-
ния потока, не учитываемый в приближении идеального вытеснения.
248 г
Сканировал: Neptunyl
Магнитогорск
2008 , ,
Поскольку в зернистом слое при Re = ul/v^i 102 йеренос веще-
ства и тепла против течения происходит только на расстояниях,
сравнимых с размером отдельной ячейки, при исследовании влияния
гидродинамики слоя на положение критических точек перескока
между различными режимами рационально пользоваться ячеистой
моделью слоя. При этом, благодаря отсутствию переноса вещества
и тепла между ячейками в направлении, противоположном движению
потока, для вывода локальных условий перехода между режимами
процесса достаточно исследовать режимы работы отдельной ячейки
при заданных значениях концентраций и температуры на ее
входе [36 ].
Рассмотрим каталитическую реакцию, протекающую на внешней
поверхности зерен. Если считать поверхность зерен равнодоступной,
а жидкость или газ в ячейке идеально перемешанными, то уравнения
материального и теплового балансов ячейки записываются в виде
СВХ-СЯ=№(СЯ-СП) ' (VI. 132)
Тя—Твх~(а/у)ая (Тп — Тя) (VI.133)
Р(С«-Сп) = Р(Сп. Тп) (VI.134)
а(2’п-2’я)=Ар(Сп, Тп) ДVI. 135)
где С — концентрация исходного вещества; Т — температура;
р — скорость реакции, отнесенная к единице внешней поверхности
частиц; h — тепловой эффект реакции; а и 0 — коэффициент тепло-
и массопередачи от Дадра потока к твердой поверхности; у — тепло-
емкость единицы объема потока; ст — площадь внешней поверхности
частиц в единице объема слоя. Индексы «вх» отмечает значения
переменных на входе ячейки, «я» — в ее объеме и «п» — на поверх-
ности зерен.
Отметим, что, поскольку движение потока внутри ячейки весьма
нерегулярно, следует ожидать, что различные участки поверхности
в среднем не имеют преимущества друг перед другом и гипотеза
о равнодоступности оправдывается с той же степенью точности, как и
гипотеза об идеальном перемешивании в ячейке. Исключение пред-
ставляют застойные зоны близ точек соприкосновения зерен, где
локальные коэффициенты массопередачи падают практически до
нуля (см. раздел VI.2). Эти застойные зоны, однако, не могут внести
существенного вклада в суммарное превращение и тепловыделение
в ячейке, так что при исследовании интегральных характеристик
процесса их можно не учитывать.
Проведем тождественное преобразование системы (VI.132) —
(VI.135). Исключая из нее Ся, Тя, имеем:
^*(Свх-Сп) = р(Ся, 7П) (VI.136)
«*(2’п-2’вХ) = Лр(Сп, Тп) (VI.137)
где
l/₽*=l/₽ + os; l/a*=l/a + (is7y (VI.138)
/ 249
Тогда уравнения (VI.132) и (VI.133) приводятся к виду:
С'вх-С'я = р*о«(Свх-<?п) (VI. 139)
Тя Т’вх = (а*/у)^(Гп Гвх) (VI-140)
Число возможных режимов работы ячейки при заданных опреде-
ляющих параметрах и условиях Свх, Твх равно числу решений нели-
нейной системы алгебраических уравнений (VI.136), (VI.137). Эти
уравнения формально эквивалентны хорошо исследованным уравне-
ниям процесса на равнодоступной поверхности изолированного
зерна (см. раздел Ш.З), отличаясь от них только заменой истинных
коэффициентов тепло- и массопередачи а и 0 на эффективные (мень-
шие) величины а* и 0*. Скорость переноса вещества к поверхности
ячейки меньше, чем скорость подачи вещества к равнодоступной по-
верхности изолированного зерна, так что переход ячейки от кинети-
ческого к диффузионному режиму должен происходить при больших
числах Re или меньших температурах потока.
Оценим относительную величину поправки к коэффициенту мас-
сопередачи: '
Bas = — as —al —^>al Re-0 »рг~2/3 (VI.141)
' и RePr •
При оценке величины Nu здесь использована эмпирическая зави-
симость фактора массопередачи числа Re (см. раздел III.1). Примерно
такая же оценка получается для поправки к коэффициенту теплопере-
дачи, если заменить в уравнении (VI.141) 0 на a/у и диффузионные
числа Nu и Рг на тепловые. Безразмерный фактор формы al — вели-
чина порядка нескольких единиц (al = л для простой кубической
упаковки шаров и al 4 для объемно-центрированной упаковки).
Из формулы (VI.141) видно, что при обычных скоростях потока
(Re 102) поправки к коэффициентам переноса незначительна для
жидкостей (Рг 1). Для газов (Рг — 1) относительная поправка
0os может составлять при Re — Ю2 30—40% ; с увеличением числа Re
эта величина уменьшается, хотя и довольно медленно. Легко заме-
тить, что величина 0os характеризует максимальную степень превра-
щения исходного вещества в одной ячейке, достижимую, когда реак-
ция протекает в диффузионном режиме. Так как 0as < 1, в кинети-
ческом режиме (к 0) степень превращения в одной ячейке всегда
мала.
При исследовании режимов работы ячейки можно, ввиду отме^
ченной эквивалентности уравнений, использовать все результаты
исследования режимов работы изолированного зерна. Поскольку
0* < 0 и а* < а, под влиянием перемешивания в ячейке переход
в диффузионный режим наступает при меньших температурах, чем
на изолированном зерне. Однако, в силу уравнения (VI.141),
максимальный возможный сдвиг критической температуры (в газах
при Re ~ 102) в реакциях с обычными значениями энергии актива-
ции может составить лишь несколько градусов.
250
Переходу в диффузионный режим способствует уменьшение ско-
рости потока. Поскольку на практике зернистый слой не имеет пра-
вильной структуры, в различных ячейках локальные скорости
потока неодинаковы и не совпадают со средней скоростью и. При
этом может создаться ситуация, когда в некоторых («надкритиче-
ских») ячейках реакция идет в диффузионном режиме, а в других
(«подкритических») — в кинетическом. Переход (надкритических
ячеек в диффузионный режим способствует «зажиганию» соседних
ячеек. Поэтому в слое со случайной структурой переход в диффузион-
ный режим происходит быстрее (при меньших температурах), чем
в упорядоченном слое. Это подтверждается численным расчетом мо-
дели зернистого слоя, со случайным распределением потока между
Рис. VI.11. Режимы проте-
кания реакции в зернистом
слое с упорядоченной (в) и
неупорядоченной (а и б)
структурой при одинаковых
значениях характерных па-
раметров процесса:
ф — ячейки, работающие в
диффузионном режиме; О —
ячейки, работающие в кинети-
ческом режиме.
ячейками [37] (рис. VI.И). Из рис. VI.11 видно, что под влиянием
неоднородности зернистого слоя происходит ускорение перехода
в диффузионный режим.
Аналогичное исследование работы отдельной ячейки может быть
выполнено в том случае, когда реакция идет внутри пористых зерен.
При этом уравнения материального баланса ячейки по-прежнему
записывают в форме уравнений (VI.132)—(VI.135) или (VI.136),
(VI.137), (VI.139), (VI. 140), где под р (Сп, Т„) надо понимать эффек-
тивную скорость реакции в пористом зерне, отнесенную к единице
его внешней поверхности.
Примыкание зерна к разным ячейкам несущественно вследствие
слабого влияния внешних градиентов на эффективность работы по-
ристой частицы [38]. Задача исследования режимов ячейки, как и.
в случае реакции на внешней поверхности зерен, сводится к тем же
уравнениям, что и уравнения процесса на изолированном зерне, с той
лишь разницей, что истинные коэффициенты массо- и теплопередачи
на внешнюю поверхность р и а заменяются на эффективные величины
Р* и а*. Влияние внешних коэффициентов переноса на режимы пори-
стого зерна было рассмотрено в разделе III.5. Полученные резуль-
таты применимы, после указанной замены, и к частице, помещенной
в зернистый слой. В условиях, когда'внешнедиффузионное торможе-
ние не влияет на процесс внутри пористой частицы, влияние ячеис-
той структуры не сказывается и подавно из-за малости дополнитель-
ного сопротивления os.
251
Стационарные режимы слоя в целом. Выведенные выше условия
перехода между различными режимами отдельной ячейки носят ло-
кальный характер и зависят от концентраций реагентов и темпера-
туры потока в данной точке зернистого слоя. Чтобы найти условия,
при которых в тех или иных частях реактора достигаются концентра-
ции реагентов и температуры, соответствующие названным точкам
перехода, необходимо решить систему уравнений, описывающих рас-
сматриваемый процесс. Если на входе реактора реакция идет в кине-
тическом режиме и требуется выяснить, перейдет ли она в диффу-
зионный под действием выделяющегося тепла, то вплоть до самой
точки перехода можно пользоваться приближением идеального вытес-
нения. Это оправдано тем, что в кинетическом режиме степень превра-
щения, достигаемая в одной ячейке, всегда мала, а в этих условиях
приближение идеального вытеснения является достаточно точным.
Аналитическое решение уравнений, описывающих неизотермиче-
ский процесс в реакторе, обычно недоступно. Однако иногда удается
получить качественные оценки условий, в которых должны наблю-
даться скачкообразные переходы между различными режимами про-
цесса. Рассмотрим сначала адиабатическую реакцию первого по-
рядка. В этом случае поперечные градиенты отсутствуют, а концен-
трация исходного вещества и температура в любом сечении реактора
вязаны между собой линейным соотношением
(Л/у)(Свх-С) = Т-Твх (VI. 142)
Если реакция идет на внешней поверхности зерна, точки пере-
скока между режимами определяются соотношением (III.60) между
параметрами ц = Р*//г.(7’) и 0 = ^>*hEC/a*RT2. Это соотношение
представлено на рис. III.5. Полагая к (71) = к (Твх) е® и используя
соотношение (VI.142), находим связь между параметрами ц и 0 на
адиабатическом пути реакции
1пц = (1пцвх —а8вХ) + а0 (VI. 143)
где рвх = $*/к (Твх), 0ВХ = $*hECBX/a*RT*x, а = а*/0*у.
Таким образом, адиабатический путь процесса приближенно
изображается в координатах ц и 0 прямой с наклоном а (а *=» 1 в газах
и а >1 в жидкостях). В точке пересечения адиабатической прямой
с кривой «зажигания» происходит переход в диффузионный режим.
Естественно, что обратный переход в экзотермическом адиабатиче-
ском процессе невозможен. Проведя прямую с наклоном а через
критическую точку 0 = 4, In ц = 2 (пунктирная линия на рис. Ш.5),'
легко убедиться, что при начальных условиях, соответствующих об-
ласти левее этой прямой, скачкообразные переходы между режимами
не будут наблюдаться.
На рис. III.9 представлены критические условия перехода из
кинетического режима во внутридиффузионный (нижняя кривая)
и обратно (верхняя кривая) для реакции первого порядка на пори-
стой пластине катализатора. Этот процесс определяется параметрами
0= hEDC/yRT2, |i = D/k(T) I2 = V2. Адиабатический путь про-
252
цесса в координатах In р — 0 изображается прямой (VI.143) с накло-
ном а = yJDy > 1. Переход во внутридиффузионный режим проис-
ходит в точке пересечения адиабатического пути с нижней кривой
рис. III.9, наклон которой всегда меньше единицы. Каки в случае
реакции на внешней поверхности зерна, при начальных условиях,
лежащих в области слева от адиабатического пути, который прохо-
дит через точку возникновения множественных режимов, скачкооб-
разные переходы невозможцы.
При расчете сложных процессов также можно выявить критиче-
ские поверхности в пространстве С, Т, на которых происходят пере-
ходы между различными режимами, и найти, пересекается ли с ними
кривая, изображающая адиабатический путь процесса. При этом
существенно, что в случае сильно экзотермических реакций при
расчете процесса вплоть до самой критической точки можно пользо-
ваться кинетическими зависимостями скоростей реакции от С и Т,
не учитывающими диффузионного торможения процесса.
Рассмотрим теперь более сложный и со всех точек зрения самый
интересный случай процесса с теплоотводом на стенку реактора.
Здесь поперечные градиенты уже существенны. Реакция в цилин-
дрическом аппарате описывается (в приближении идеального вы-
теснения) уравнениями
(™44’
+ (VI.14S)
В условиях, когда тепло передается в основном движущейся жид-
костью или газом, Хх/Т = — wl/m, (где m «а, 10). Граничные
условия на стенке реактора (при у — Ьг)
дС/ду-О-, Li(dT/dy)=—ВЦТ-ТС) (VI.146)
где Тс — температура стенки; Bi = aZj/xj. — число Био; a — коэф-
фициент теплопередачи от потока к стенке реактора.
В центре реактора (при у = 0) должны быть выполнены очевид-
ные условия
dC/dy = Q-, дТ/ду — 0 (при д = 0) (VI-147)
Если Bi 1, при расчете процесса можно считать температуру
постоянной во всем сечении и меняющейся только у стенки. Как
следствие этого концентрация по всему сечению реактора также по-
стоянна. Таким образом, приходим к приближению «одномерного»"
реактора идеального вытеснения:
<vb148’
г(С, Г) (V1.149)
253
В большинстве случаев, однако, сопротивление теплопередаче
внутри зернистого слоя значительно превышает сопротивление
теплопередаче от потока к стенке реактора, так что Bi 1*. При
этом второе граничное условие (VI.146) принимает вид Т (Lj) = Тс.
В случае Bi 1 возникают существенные поперечные перепады тем-
пературы и приближение «одномерного» реактора неприменимо..
Для оценки стационарных режимов зернистого слоя в целом необ-
ходимо, таким образом, хотя бы качественно исследовать характер
решений уравнений (VI.144) и (VI.145). Заметим, что первые два
члена этих уравнений описывают перенос вещества и тепла, соответ-
ственно в поперечном и продольном направлениях. Возможны два
предельных режима теплопереноса [36]. Первый — почти адиабати-
ческий, когда отвод тепла на стенку незначителен и практически все
тепло реакции уходит на нагревание реагирующего потока. В этом
режиме первый член уравнения (VI.145) пренебрежимо мал повсюду,
кроме ближайшей окрестности стенки реактора. Переход трубчатого
реактора в почти адиабатический режим является крайне нежелатель-
ным, поскольку при этом не решается главная задача аппарата этого
типа — обеспечение отвода тепла реакции на стенку — и температура
в центре реактора быстро возрастает, вызывая угрозу перехода про-
цесса в диффузионный режим. Желательным обычно является другой
предельный режим работы реактора, который можно назвать почти
изотермическим. В этом режиме тепло реакции отводится в основном
на стенку, а изменение температуры по длине реактора мало. Соот-
ветственно второй член уравнения (VI.145) мал по сравнению с пер-
вым и в первом приближении может быть отброшен. Из сравнитель-
ной оценки обоих членов ясно, что условие работы реактора в почти
изотермическом режиме имеет вид:
yw /X। Li 1 Li
1 (Vl.laO)
Здесь под L надо понимать характерную длину, на которой про-
исходит существенное изменение температуры в продольном направ-
лении. Из условия (VI.150) следует, что эта величина должна быть
велика по сравнению с диаметром трубки.
Перенос вещества в трубчатом реакторе не подобен переносу тепла,
вследствие различия граничных условий на стенке реактора (VI.146).
Поскольку сток вещества на стенке отсутствует, перепад концентра-
ций по сечению реактора может быть вызван только различием скоро-
стей реакции, связанным с перепадом температур. В первом прибли-^
жении можно считать концентрацию постоянной по всему сечению
реактора. Тогда изменение концентрации по длине реактора опреде-
ляется уравнением
wd<C>/dx=<r(C, Г)> (VI.151)
где < > — усреднение по сечению реактора.
* Обзор экспериментальных данных по теплопередаче на стенку реактора
с зернистым слоем дан в монографии Аэрова и Тодеса [34]. Непосредственное из-
мерение профиля температур поперек слоя проведено Ханратти [39].
254
Уравнение (VI.151) легко получить из уравнения (VI.144), интег-
рируя его по поперечной координате и учитывая граничное усло-
вие (VI. 146).
Следует отметить, что обратное предположение о наличии значи-
тельного изменения концентрации по сечению и, соответственно,
малости второго члена в уравнении (VI.144) по сравнению с первым
приводит к абсурду. Отбрасывая член с производной по длине в урав-
нении (VI.144) и замечая, что г {С, Т) = 0 при С = 0, легко видеть,
что это уравнение не имеет отличных от тождественного нуля реше-
ний с производной дС/ду, обращающейся в нуль на стенках трубы и
на ее оси.
Исследуем условия существования решений упрощенного темпе-
ратурного уравнения:
JSr+7-<+iT(<C>':r,=0 <VL,52)
В уравнении (VI.152) единственной переменной остается темпера-
тура, и само зто уравнение, независимо от кинетики процесса, имеет
ту же форму, что и уравнение, описывающее стационарную реакцию
нулевого порядка в цилиндрическом пористом зерне или сосуде без
конвекции [40]. Полагая г = к(Т^) ев / (С), приводим уравнение
(VI. 152) и граничное условие Т = Тс к виду
4S-+-U# + He(< = 0, 0(1)=О (VI.153)
где Н = Ehf (С) к(Тс) Ll/^RTf.
Решение задачи (VI.153) имеет вид [40, 41].
О
0 = 1пЯ(е^+?2е-^)2 (VI. 154)
где параметр X определяется соотношением:
ch2X —2/Я (VI.155)
Очевидно, рассматриваемая задача имеет решение только при
Н < ЕГ^ = 2. Максимальный возможный разогрев, достигаемый
в критической точке Н = 2 на оси реактора, равен 9макс = In 4
1,38. Если решать уравнение (VI.153) с более общим граничным
условием (VI.146), учитывающим сопротивление теплопередаче на
стенке реактора, решение также может быть представлено в виде
уравнения (VI. 154), но зависимость между параметрами X и Н и фор-
мулы для критического значения параметра Н = принимают более
сложный вид, зависящий от величины числа Био [36, 41 ].
При Н почти изотермический режим работы реактора с ма-
лым изменением температуры по длине слоя невозможен. Эффектив-
ность теплопередачи на стенку уже недостаточна для отвода тепла ре-
акции, и процесс становится почти адиабатическим. Дальнейшее раз-
витие процесса может иметь двоякий характер. Во-пёрвых, вследствие
255
резкого повышения температуры процесс в некотором сечении ре-
актора может перейти в диффузионную область. Точку этого пере-
хода можно приближенно оценить, считая процесс адиабатическим,
методами, описанными выше. Точность адиабатического приближе-
ния тем выше, чем больше величина Н. Другая возможность состоит
в том, что раньше, чем произойдет переход в диффузионную область,
вследствие выгорания исходного вещества начнет выполняться не-
равенство Н < ЕГ^. Тогда дальнейшее возрастание температуры
может прекратиться, и переход в диффузионную область так и не
произойдет. На рис. III.5 и III.9 равенство Н = ЕГ^ изображалось бы
прямой, параллельной оси ординат. Переход в диффузионную об-
ласть не произойдет, если адиабатический путь пересечет линию
Н = ЕГ^ раньше, чем кривую «зажигания» реакции.
Из-определения параметра Н легко видеть, что наиболее опасным
местом, где следует проверят^ неравенство Н < ЕГ^, является вход
реактора. В дальнейшем, по мере уменьшения концентрации исход-
ного вещества,, величина Н монотонно уменьшается (если только не
считать случаев автокатализа и торможения реакции исходным веще-
ством). Согласно сказанному выше, при Н > ЕГ^ (на входе реактора)
должны наблюдаться температурные пики в лобовом слое (Явление
практически весьма распространенное). Вопрос О том, приведет ли
образование пика к переходу процесса в диффузионный режим, зави-
сит от того, будет ли точка «зажигания» реакции достигнута раньше,
чем начнет выполняться неравенство Н < ЕГ^.
При осуществлении сложного процесса, включающего несколько
реакций, по-прежнему возможны оба описанных предельных режима
реактора — почти адиабатический и почти изотермический. В по-
следнем случае производная температуры в продольном направлении
должна быть мала, а концентрации реагентов — почти постоянны по
сечению аппарата. Условия существования почти изотермического
режима определяются исследованием уравнения типа (VI.152).
В этом уравнении скорость тепловыделения уже не будет зависеть от
температуры по экспоненциальному закону и будет иметь вид суммы
нескольких экспонент с различными показателями. В такой форме
это уравнение не решается аналитически, но приближенные оценки
можно получить ла основе результатов, полученных для единствен-
ной реакции, если аппроксимировать скорость тепловыделения в не-
которой ограниченной области законом Аррениуса. Если в сложном
процессе наибольшим тепловым эффектом обладает реакция с уча-
стием промежуточного продукта, то наибольшая опасность перехода
в почти адиабатический режим может наблюдаться не во входном
сечении, а там, где превращение промежуточного продукта будет
идти с достаточной скоростью.
Наличие резкого перехода от почти изотермического к почти адиа-
батическому режиму, сопровождающееся возникновением ярко вы-
раженных температурных пиков, подтверждается численными рас-
четами, выполненными как в теории теплового взрыва (42 ] (где воз-
256 '
никает математически эквивалентная задача), так и непосредственно
для ,реакторов с зернистым слоем 143]*. Влияние случайного
разброса локальных скоростей потока, приводящее при определен-
ных условиях к «маскировке» закономерного поперечного профиля
температур сильными флуктуациями, возникающими вследствие
локального перехода в диффузионный режим, исследовано в ра-
боте [37].
ЛИТЕРАТУРА
1. Р. у. Dankwert s, Chem. Eng. Sci., 2, 1 (1953).
2. Б. В. Гнеденко, Курс теории вероятностей, Физматгиз, 1961.
3. Н. М. Hulburt, Ind. Efig. Chem., 36, 1012 (1944).
4. J. F. Wehner, R. H. Wilhelm, Chem. Eng. Sci., 6, 89 (1956).
5. L. H. R о b 1 e e, R. M. Baird, J. W. Tierney, Am. Inst. Chem.
Eng. J., 4, 460 (1958).
6. J. M. H i b y, Proc. Symp. on the Interaction between Fluids and Parti-
cles, The Inst, of Chem. Eng., p. 312 (1962). *
7. M. Э. А э p о в и др., ЖТФ, 29, 924 (1959); J. Rhodes, F. Р е -
е b 1 е s, Am. Inst. Chem. Eng. J., 11, 481 (1965).
8. К. K. J о 11 s, T. J. Hanratty, Chem. Eng. Sci., 21, 1185 (1966).
9. О. Л e в e н ш п и л ь, Инженерное оформление химических реакций,
Изд. «Химия», 1969; О. Levenspiel, К. В. Bischoff, Advances in Che-
mical Engineering, vol. 4, New York — London, 1963, p. 95.
10. K. W. Me Herny, R. H. Wilhelm, Am. Inst. Chem. Eng. J., 3,
83 (1957); R. H. Wilhelm, Pure a. Appl. Chem., 5, 403 (1962).
11. J. Ji Carberry, R. H. B. retton, Am. Inst. Chem. Eng. J., 4,
367 (1958); E. E. Ebach, R. R. White, Am. Inst. Chem. Eng. J., 4,
161 (1958); D. A. Strang, C. J. G e a n k о p 1 i s, Ind. Eng. Chem., 50,
1305 (1958); A. W. Liles, C. J. Geankojilis, Am. Inst. Chem. Eng.
J., 6, 591 (1960).
12. H. Kramers, G. A1 b e r d a, Chem. Eng. Sci., 2, 173 (1953).
13. J. J.’Van D e e m p t e r, Chem. Eng. Sci., 13, 143 (1961); M. B. G 1 a-
ser, L. Mitchell, Am. Inst. Chem. Eng. J.t 9, 103 (1963).
14. R. А. В e r n a r d, R. H. W> 1 h e 1 m, Chem. Eng. . Progr., 46,
233 (1950); E. Singer, R. H. Wilhelm, Chem. Eng. Progr., 46, 343
(1950); В. А. Баум, Изв. АН СССР. ОХН, № 2, № 9 (1952); R. Н. W i I -
helm, Chem. Eng. Progr., 49, 150 (1953); A.KliAkenberg, H.J.Kra-
i e n b r i n k, Ind. Eng. Chem., 45, 1202 (1953); W. L. Towle, T. K. S h e r -
wood, Ind. Eng. Chem., 31, 457 (1939); R. W. F a h i о n, J. M. S m i t h,
Am. Inst. Chem. Eng. J., 1, 28 (1955); D. S. Plants, H. E. J ohnst one,
Am. Inst. Chem. Eng. J., 1, 193 (1955); M. E. Hartmann, С. J. H. W e -
vers, H. Kramers, Chem. Eng. Sci., 9, 80 (1958); F. E. G r a n e,
G. H. F. G a r d n e r, J. Chem. Eng. Data, 6, 283 (1961); M. Э. А э p о в,
H. H. Умник, ЖПХ, 27, 265 (1954).
15. Т. В а г о n, Chem. Eng. Progr., 48, 118 (1952); W. E. R a n z, Chem.
Eng. Progr., 48, 247 (1952).
16. N. R. A m u n d s о n, R. A r i s, Am. Inst. Chem. Eng. J., 3, 280
(1957).
17. G. A. T u r n e r, Chem. Eng. Sci., 7, 156 (1958); 10, 14 (1959); R. A r i s,
Chem. Eng. Sci., 10, 80 (1959); 11, 194 (1959).
* О численных расчетах реакторов с зернистым слоем см. также работы
[44, 45] и общие руководства по теории химических реакторов, указанные
в литературе к главе УП. Расчеты с помощью ячеистой модели провёдены в ра-
ботах [37, 46].
17 Заказ 1^41 257
18. H. A. Deans, Soc. Petrol Eng. J., 3, 49 (1963); В. Г. Л e в и ч,
В. С. Маркин, Ю. А. Ч и з м а д ж е в, ДАН СССР, 166, 1401 (1966);
168, 1364 (1966); Chem. Eng. Sci., 22, 1357 (1967).
19. S. Katz, Chem. Eng. Sci., 9, 61 (1958).
20. C. G о t t s c h 1 i c h, Am. Inst. Chem. Eng. J., 9, 88 (1963).
21. В. Г. Левин, Л. M. Письмен, С. И. Кучаиов, ДАН
СССР, 168, 392 (1966). .
22. В. Н. Николаевский, IIMM, 23, 1042 (1959); Изв. АН СССР.
Сер. мех. и маш., № 4, 146 (1959); Р. G. Sat f man, J. Fluid Meeh., 6,
321 (1959); 7, 194 (1960).
23. Л. M. П и с ь м e н, С. И. К у ч а н о в, В. Г. Левин, ДАН
СССР, 174, 650 (1967); С. И. К у ч а н о в, В. Г. Л е в и ч, Л. М. Письмен,
ПМТФ, № 3, 45 (1967).
24. С. И. К у ч а н о в, Л. М. П и с ь м е н, ПМТФ, № 4, 45 (1967);
С. И. К у ч а н о в, Л. М. П и с ь м е и, В. Г. Л е в и ч, Труды III Всесоюз-
ного совещания по тепло- и массообмену, т. 5, Минск, 1968, стр. 57.
25. С. И. К у ч а н о в, Л. М. П и с ь м е н, ТОХТ, 1, 116 (1967).
26. В. Г. Б а р у х ов, Г. К. В о р е с к о в, ЖПХ, 20, 721 (1947);
D. Y. В u n n е 11 a. oth.. Ind. Eng. Chem., 41, 1977 (1949); M. Э. А э р о в,
Н. Н. Умник, ЖТФ, 11, 1345 (1951); С. А. С о b е г 1 у, W. R. Mar-
sh а 11, Chem. Eng. Progr., 47, 141 (1951); J. О. Н о и g h е n, Е. L. Р i -
ret, Chem. Eng. Progr., 47, 287 (1951); J. M. Campbell, R. Hunting-
ton, Petrol. Ref., 31 (2), 123 (1951); D. F. M о 1 i n o, J. О. H о u g h e n,
Chem. Eng. Progr., 48, 147 (1952); R. W. S h u 1 e г, V. P. S t a 11 i n g s,
J. M. Smith, Chem. Eng. Progr. Symp. Ser, 48, № 4, 19 (1952); W. В. A r g o,
J. M. Smith, Chem. Eng. Progr., 49, 443 (1953); D. A. P 1 u t z,
H. F. Johnstone, Am. Inst. Chem. Eng. J., 1, 193 (1955); J. H. Quin-
ton, J. A. Storrow, Chem. Eng. Sci., 5, 245 (1956); P. H. Cal de r-
b a n k, L. A. P о g о r s k i, Trans. Inst. Chem. Eng., 35, 195 (1957);
R. F. В a d d о u г, С. I. Y о о n, Chem. Eng. Progr. Symp. Ser., 57, № 32
(1961).
27. M. Э. Аэров, H. H. Умник, ЖТФ, 11, 1351 (1951).
28. S. Y a g i, D. Kunii, Am. Inst. Chem. Eng. J., 3, 373 (1957); 6,
97 (1960); S. Yagi, N. Wakao, Am. Inst. Chem. Eng. J., 5, 79 (1959);
S. Y a g i, D. К u n i i, K. Endo, J. Heat a. Mass Transfer, 7, 333
(1964).
29. S. M a s a m u n e, J. M. S m i t h, Ind. Eng. Chem. Fund., 2, 136
(1963).
30. S. Y a g i, D. К u n i i, N. W a k a o, Am. Inst. Chem. .Eng. J., 6,
543 (1960); D. Kunii, Y. M. Smith, Am. Inst. Chem. Eng. J., 7, 31 (1961).
31. R. Aris, Proc. Roy. Soc., A245, 268 (1958); D. W. Green,
R. H. P e r r y, R. E. В a b с о c k, Am. Inst. Chem. Eng. J., 10, 645 (1964).
32. С. И. К у ч а н о в, Л. M. П и с ь м e и, ТОХТ, 1, 374 (1967).
33. С. И. Кучанов, Л. М. Письмен, В. Г. Левин, ТОХТ, 2, 576
(1968).
34. М. Э. А э р о в, О. М. Т о д е с, Гидравлические и тепловые основы
работы аппаратов со стационарным и кипящим зернистым слоем, Изд. «Химия»,
1968.
35. В. Г. Левин, Физико-химическая гидродинамика, Физматгиз,
1959.
36. В. Г. Л е в и ч, Л. М. Письмен, ДАН СССР, 176, 150 (1967).
37. В. Г. Л е в и ч, Л. М. Письмен, Э. Д. Г е р м а и, ТОХТ, 1,
366 (1967).
38. Л. М. Письмен, ДАН СССР, 167, 151 (1966); К. В. Bischoff,
Chem. Eng. Sci., 23, 451 (1968).
39. T. J. Hanratty, Chem. Eng. Sci., 3, 209 (1954).
40. Д. А. Франк-Каменецкий, Диффузия и теплопередача в хи-
мической кинетике, Изд. «Наука», 1967.
41. В. В. Б а р з ы к и н, А. Г. Мержанов, ДАН СССР, 120, 1271
(1958).
258
42. А. Г. Me ржа и ов, В. Г. А б р а м о в, В. Т. Г о и т к о в с к а я,
ДАН СССР, 148, 156 (1963); В. В. Барзыкин и др., ПМТФ, № 3, 118
(1964); В. Г. Абрамов, В. Т. Гонтковская, А. Г. Мержанов,
Изв. АН СССР. ОХН, № 4, 5 (1966).
' 43. Н. S. М i с k 1 е у, R. W. L е 11 s, Canad. J. Chem. Eng., 41, 273
(1963); 42, 21 (1964); В. С. Б e с к о в, В. А. К у з и я, М. Г. С л и я ь к о,
Хим. пром., № 7, 508 (1964); V. Hlavacek, М. Marek, Coll. Czech.
Chem. Comm., 32, 3291, 3309 (1967).
44. J. В e e k, Adv. in Chem. Eng., 3, 204 (1962).
45. J. К j a e r, Mesurement and Calculation of Temperature and Conver-
sion in Fixed-ned Catalytic Reactors, Copengahen, 1958.
46. M. L. M c gn ire, L. L ap i d us, Am. Inst. Chem. Eng. J., 11,
85 (1965).
17*
Глава VII______, _____________________________' _______
РАСЧЕТ РЕАКТОРОВ
ПО МАТЕМАТИЧЕСКОЙ МОДЕЛИ *
VII.1. ПРИНЦИПЫ МАТЕМАТИЧЕСКОГО МОДЕЛИРОВАНИЯ
КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
Математическое моделирование реакторов для гетерогенно-ката-
литических процессов — частный случай математического модели-
рования химических реакторов. Рассмотрим основные принципы ме-
тода математического моделирования и приложения этого метода
к расчету каталитических реакторов.
Метод математического моделирования заключается в том, что
явления, протекающие в заданном объекте, и их взаимосвязь коли-
чественно описываются системой математических уравнений, которая
и представляет собою математическую модель объекта. Для катали-
тических реакторов математическая модель в общем случае должна
включать в себя всю систему уравнений кинетики, макрокйнетики,
гидродинамики и теплообмена, которым посвящены главы I —III
и VI. Численные значения коэффициентов модели могут меняться при
изменении масштаба реактора, но структура модели остается неиз-
менной. Значения коэффициентов модели, таких, как кинетические
константы, коэффициенты диффузии и тепло- и массопереноса могут
определяться как экспериментальным путем при лабораторных или
стендовых исследованиях, так и расчетно-теоретическим путем» При
наличии модели и известных значениях коэффициентов с примене-
нием ЭВМ могут быть исследованы различные варианты реактора для
заданного процесса и проведена его оптимизация.
В инженерном и экономическом отношении существенно знать
количество стадий, необходимых для надежного перехода от лабо-
раторных исследований к промышленным установкам. При отсутст-
вии надежного и достаточно полного математического описания про-
цесса число таких переходных стадий может быть довольно велико и
к тому же не избавляет от существенных ошибок при промышленной
реализации новых процессов. Метод математического моделирования
может значительно улучшить это положение. Однако не следует
думать, что при этом во всех случаях будут исключены стадии про-
межуточной проверки процесса между лабораторной разработкой
и промышленным внедрением.
Всякая математическая модель обладает двумя принципиаль-
ными недостатками: 1) возможно, что в нее не включены все явления,
протекающие в реальном объекте и 2) уравнения модели не доста-
точно точно будут аппроксимировать процессы, протекающие
* По вопросам математического моделирования и расчета см. монографии
П-8].
260 . z '
в объекте, при значительном изменении граничных условий, связан-
ном с переходом от лабораторного исследования к промышленной
реализации. Эти недостатки тем более должны учитываться, по-
скольку каждая математическая модель неизбежно является в боль-
шей или меньшей степени упрощенной. Поэтому создание ее — итера-
тивный процесс, котором следует различать синтетическую и ана-
литическую стадии. На первой стадии проводится синтез модели на
основе лабораторных исследований и теоретических расчетов; на
второй — проверка адекватности модели либо на специальной уста-
новке, либо на действующем производстве, когда ведутся работы по
его коренному усовершенствованию. В случае необходимости модель
со стадии проверки должна возвращаться на доработку. Таким обра-
зом, хотя метод математического моделирования не исключает в об-
щем случае промежуточной стадии исследования между лабораторией
и промышленной реализацией, однако Он, во-первых, резко умень-
шает число этих переходных стадий, а во-вторых, что весьма суще-
ственно, повышает надежность проектных решений для промышлен-
ного объекта. .
Следует отметить, что математическая модель может быть создана
только для заданной принципиальной схемы реактора. Выбрр такой
схемы обычно производится с учетом уже имеющегося промышлен-
ного опыта и возможностей химического машиностроения. Поэтому
приступая к разработке каталитических реакторов, обязательно
следует иметь сведения об опыте уже действующих производств.
VII.2. ПРОМЫШЛЕННОЕ ОСУЩЕСТВЛЕНИЕ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Основные требования к промышленным реакторам. Высокая
экономическая эффективность многотоннажного промышленного про-
цесса обычно может быть достигнута только в том случае, когда
процесс проводится непрерывно. Особенно зто относится к газофаз-
ные процессам, так как вследствие низкой плотности газов такой про-
цесс бывает рентабелен лишь при малой продолжительности реакции,
не превышающей нескольких секунд. Жидкофазные гетерогенно-
каталитические процессы иногда осуществляются в промышленном
масштабе и периодическим способом, однако наиболее многотоннаж-
ные и рентабельные из них являются непрерывными.
Конструкция реайтора должна обеспечивать поддержание опре-
деленных значений следующих основных параметров процесса: 1) вре-
мени реакции; 2) температуры в различных точках реакционной
зоны; 3) давления в реакторе; 4) скорости массопередачи к актив-
ной поверхности катализатора и между фазами (для многофазных
процессов); 5) активности катализатора.
Если какое-то время контакта является оптимальным, то,
очевидно, всякое отклонение от него снижает эффективность про-
цесса. С этой точки зрения, чаще всего наиболее благоприятен
261
гидродинамический режим идеального вытеснения. Приближение
к нему обычно достигается при проведении процесса в узких длин-
ных аппаратах. Гидродинамический режим, однако, тесно связан
с тепловым режимом реактора и его устойчивостью, поэтому один
только характер оптимальной функции распределения времени кон-
такта не является достаточным основанием для выбора типа реактора.
Обеспечение заданной температуры в реакционной зоне — по-
жалуй, наиболее важная и трудная задача при конструировании
промышленного реактора. Большинство практически осуществляе-
мых процессов сопровождается выделением или поглощением теплд, и
для поддержания температуры в реакторе на заданном уровне необ-
ходимо, соответственно, отводить или подводить тепло извне.
По тепловому режиму реакторы можно разделить на адиабатиче-
ские аппараты и реакторы с теплообменом в реакционной зоне (вну-
тренним теплообменом). В адиабатическом режиме тепло отводится
либо самим реагирующим потоком, либо движущимся катализа-
тором. В газофазных процессах, где теплоемкость реагирующего
потока мала, проведение реакции в адиабатическом режиме приводит
к появлению значительного перепада температуры по длине слоя ка-
тализатора. Чтобы этот перепад не превышал допустимых значений,
реактор приходится разделять на ряд зон — адиабатических слоев,
в промежутках между которыми поток охлаждается или нагревается
до требуемой температуры. Изменение температуры реагирующей
смеси может достигаться либо с помощью промежуточных теплооб-
менников, либо путем добавления холодного (горячего) сырья или
инертного вещества.
Удобнее проводить адиабатические процессы в жидкой фазе,
где поток обладает большей теплоемкостью. Теплоносителем с весьма
высокой теплоемкостью является сам катализатор в процессах с дви-
жущимся или кипящим циркулирующим, слоем, однако конструк-
тивные и эксплуатационные трудности в подобных процессах
велики.
При проведении реакций со значительным тепловым эффектом
обычно приходится применять аппараты с отводом тепла непосред-
ственно из зоны реакции. В этом случае реактор охлаждается или
нагревается с помощью специального теплоносителя. В качестве
последнего можно применять также и реагирующую смесь.
Проблемы поддержания необходимого давления в реакционном
аппарате и создания в нем этого давления носят чисто конструктив-
ный характер. Если эффективность процесса возрастает с увелйче- '
нием давления, предел повышению рабочего давления в реакторе
ставит лишь одновременное удорожание аппарата. Удачное конструк-
тивное решение позволяет поднять допустимый предел давления и тем
самым интенсифицировать промышленный процесс. Перепад давле-
ний внутри реакционной зоны может быть вызван гидравлическим
сопротивлением слоя катализатора. Отрицательный эффект послед-
него, однако, связан в основном не с созданием градиента давлений,
а с увеличением энергетических затрат на движение потока.
262
Интенсивность массопередачи к внешней поверхности зерен ката-
лизатора зависит от конструкции контактного аппарата. Ее можно
повысить, увеличив линейную скорость потока. Однако одновременно
возрастает гидравлическое сопротивление слоя. Скорость внутренней
диффузии зависит только от структуры пористого катализатора и
свойств реагирующей среды. Уменьшение размера зерен снижает
отрицательные последствия внутридиффузионного торможения,
позволяя полнее использовать реакционный объем. Однако при этом
также повышается гидравлическое сопротивление слоя частиц.
При переводе процесса в кипящий слой, где можно использовать
мелкие частицы, не повышая гидравлического сопротивления слоя,
возникают специфические затруднения с диффузией реагентов между
различными частями потока газов.
Задача поддержания постоянной активности катализатора
возникает при проведении промышленных процессов на катализа-
торах, быстро меняющих свои свойства в ходе реакции. Чаще всего
причиной падения активности катализатора является отложение вы-
сокоуглеродис^тых соединений на его поверхности. Катализатор,
потерявший активность таким образом, может быть регенерирован
путем выжигания углеродистых соединений. Газофазный процесс на
неподвижном катализаторе, активность которого быстро падает, по
необходимости становится циклическим, т. е. состоящим из переме-
жающихся стадий реакции и регенерации, разделенных периодами
продувки системы инертным компонентом. Конструкция реактора
должна быть приспособлена к условиям проведения процесса на
обеих его стадиях, зачастую резко различных. Экономически и кон-
структивно оказалось целесообразным во многих случаях перейти от
временных циклов к пространственным. В последнем случае реакция
и регенерация катализатора проводятся одновременно, но в разных
аппаратах или их частях. Естественно, что осуществить простран-
ственные циклы можно только с подвижным катализатором, т. е.
проводя процесс в кипящем или движущемся слое. В таких процессах
свежий катализатор непрерывно вводят в слой взамен потерявшего
активность, благодаря чему в реакторе устанавливается стационар-
ная активность катализатора. Преимущества непрерывной регенера-
ции сказываются тем сильней, чем выше скорость зауглероживания.
Однако, в установках с кипящим и движущимся катализатором воз-
никают неблагоприятные гидродинамические режимы потока, исти-
рается катализатор, встречаются и конструктивные трудности.
Ни один из возможных промышленных реакторов не свободен от
недостатков. Поэтому какой-либо универсальной конструкции не
существует, различные типы реакторов в разной мере пригодны
для промышленного осуществления того или иного процесса. Выбор
наилучшей схемы реакторного узла зачастую может быть осу-
ществлен лишь путем тщательного технико-экономического анализа.
В реакторах всех типов может также осуществляться рецирку-
ляция непрореагировавшего сырья. Этот технологический прием при-
меняют в тех случаях, когда невыгодно или невозможно добиться
263
высокой степени превращения хотя бы по одному из исходных ве-
ществ; его целесообразность определяется либо экономией сырья,
либо (в процессах, идущих под высоким давлением) уменьшением
энергетических затрат. Обязательным условием практического осу-
ществления рециркуляции сырья является легкость выделения про-
дуктов реакции.
Реакторы для газофазных процессов с неподвижным катализа-
тором. При проведении газофазных реакций наибольшее распро-
странение получили реакторы с неподвижным зернистым слоем ка-
тализатора. Реакторы с неподвижным слоем могут работать как
в адиабатическом режиме, так и с теплоотводом через стенку аппарата.
Из адиабатических аппаратов наиболее простойконструкцией обла-
дают так называемые емкостные контактные аппараты, обычно пред-
ставляющие собой металлические цилиндры; в их нижней части на-
ходится решетка, на которую насыпают катализатор в виде различ-
ного типа гранул. Газы предпочтительно направлять в аппарат сверху
вниз. Возможность полезно использовать значительную часть объема
таких аппаратов и предельная простота конструкции делают их очень
дешевыми в изготовлении. Отсутствие приспособлений для теплооб-
мена ограничивает применение емкостных контактных аппаратов.
Они пригодны для проведения: 1) процессов с небольшим тепловым
эффектом; 2) процессов, не очень чувствительных к изменению тем-
пературы; 3) процессов с малой степенью превращения за проход.
Как правило, эндотермические процессы легко осуществимы в ем-
костных реакторах. В нефтехимической промышленности емкостные
адиабатические реакторы применяют, например, для таких много-
тоннаЖных процессов, как платформинг.
К процессам, избирательность которых не зависит или слабо за-
висит от температуры в широких пределах ее изменения, относятся
некоторые экзотермические реакции, протекающие во внешнедиф-
фузионной области, например, реакции окисления аммиака в азот-
ную кислоту, метанола в формальдегид и др. В процессах этого рода
на поверхности зерен катализатора автоматически устанавливается
температура адиабатического разогрева (см. раздел III.3); адиабати-
ческий режим становится при этом не только рациональным, но и
единственно возможным.
На практике емкостные аппараты применяют иногда и для про-
ведения экзотермических реакций в кинетической области, если они
дают термически устойчивые продукты и, следовательно, не теряют
избирательности в широких температурных пределах. Такой процесс
заведомо не оптимален; применение емкосГного реактора оправды-
вается лишь простотой его конструкции.
Возможности проведения в емкостных аппаратах процессов на ка-
тализаторах с быстро падающей активностью ограничены, так как
в отсутствие теплообменных устройств тепло, выделяющееся в сильно
экзотермическом процессе регенерации, может привести к спеканию
катализатора и потере им активности. Если температура разогрева
при регенерации превосходит предел термостойкости катализатора,
ее снижения можно добиться, разбавляя регенерирующий агент
(воздух) азотом или водяным паром.
Высота слоя катализатора в емкостном контактном аппарате опре-
деляется кинетическими параметрами процесса с учетом гидродина-
мики потока. Наиболее тонкий слой становится «двумерным» и может
заменяться сеткой из каталитического материала. Это имеет место
, при проведении весьма быстрых реакций во внешнедиффузионной
области, например при окислении аммиака до окислов азота.
Емкостные аппараты в виде толстостенных цилиндров являются
- наиболее удобными для проведения реакций при высоких давлениях,.
так как всйкое усложнение конструкции в этом
случае сильно удорожает ее.
Если для данного процесса высота слоя
катализатора в обычном емкостном аппарате
велика и создает слишком большое гидравличе-
ское сопротивление, целесообразно применять
емкостные аппараты с радиальным вводом газа.
В таких аппаратах гидравлическое сопротивле-
ние мало и не зависит от объема катализа-
тора. Это свойство делает их особо пригодными
для агрегатов очень большой мощности. «Прин-
цип устройства такого аппарата ясен из
рис. VII. 1. Аппараты с радиальным вводом газа
рассчитывают таким образом, чтобы создать рав-
номерный поток газа по всему слою катализа-
Рис. VII.1. Схема
реактора с ра-
диальным распре-
делением газа.
тора.
Более сложной является конструкция по-
лочных контактных аппаратов (рис. VII.2 и
VII.3), пригодных для проведения реакций,
обладающих заметным тепловым эффектом. В полочных реакторах
катализатор находится на нескольких расположенных друг над дру-
гом перфорированных полках. Тепло' реакции отводится или под-
водится в теплообменниках, через которые проходят реакционные
газы, переходя с полки на полку. Такие теплообменники устанавли-
вают либо внутри аппарата (рис. VII.2), либо вне его (рис. VII.3).
В полочных реакторах по высоте каждого слоя неизбежно возникает
перепад температуры. Последний можно свести к минимуму, умень-
шая высоту слоев, однако это неизбежно приводит к увеличению
числа полок и соответственно к усложнению и удорожанию аппарата.
Кроме того, слишком низкие слои зернистого катализатора обычна
непригодны, так как, если высоту слоя можно сравнить с размером
частиц катализатора, могут возникать нежелательные явления из-за
поперечной неоднородности слоя (местные перегревы и проскоки
газа в местах с наименьшим гидравлическим сопротивлением), веду-
щие к ухудшению показателей или к срыву процесса. При проведении
процессов в полочных реакторах вместо устройства промежуточных
теплообменников* иногда применяют промежуточный ввод холодного
(горячего) сырья или инертного компонента.
265
Полочные адиабатические аппараты обладают большим числом
степеней свободы проектирования и управления: в каждом проме-
жуточном теплообменнике реагирующая смесь может нагреваться
или охлаждаться до любой выбранной температуры, что позволяет
проводить процесс в разных слоях на различных температурных
Рис. VII.2. Схема полочного реактора
с внутренними теплообменниками:
1 — катализатор; 2 — теплообменники.
уровнях, добиваясь максималь-
ной его эффективности. Преи-
муществами полочных аппара-
тов являются относительная
простота конструкции (что осо-
бенно существенно для аппа-
ратов большой мощности),
Продукт
Рис. VII .3. Схема полочного реактора
с выносными теплообменниками:
1 — катализатор; 2 — теплообменники.
возможность применять для охлаждения и нагревания газов
между полками высокоэффективные теплообменные устройства,
а также небольшое гидравлическое сопротивление слоя катализатора.
К недостаткам полочных аппаратов относятся: затруднения с вы-
грузкой и загрузкой аппарата, трудность достижения равномерного
распределения потока газов по сечению аппарата цри малой высоте
слоя катализатора на полке, что часто препятствует проведению в та-
ких аппаратах быстрых необратимых экзотермических процессов.
Большое распространение для работы с зернистым катализатором
266
получили контактные ап-
Чаще всего для этой цели
Паро-газовая
смесь
Продукт
17///'
при проведении экзотермических гетерогенно-каталитических реак-
ций со значительным тепловым эффектом
параты с теплоотводом из зоны реакции,
применяют трубчатые реакторы.
Они представляют собой различ-
ного вида теплообменники, в труб-
ках (реже — в межтрубном про-
странстве) которых находится ка-
тализатор (рис. VII.4). В качестве
теплоносителя применяют газы,
высококипящие органические теп-
лоносители, расплавленные ме-
таллы (натрий, ртуть, сплавы),
расплавленные соли. Температу-
ру в кипящих банях регули-
руют, изменяя давление инертного
газа (азота) над уровнем тепло-
носителя в бане. Если тепло-
носитель не является кипящей
жидкостью, применяют искусствен-
ную циркуляцию (либо прокачи-
вают теплоноситель через систему
реактор — теплообменник, либо
устанавливают мешалку в самом
реакторе). Из-за малой теплоем-
кости и низких коэффициентов теп-
лоотдачи газы в качестве теплоно-
сителей применяют только для
проведения реакций с относительно
малым тепловым эффектом.
При работе с высокими давле-
ниями конструкции трубчатых ре-
акторов сильно усложняются. Пока
применяют только реакторы с ох-
лаждением катализатора газами,
поступающими на реакцию.
В трубчатых реакторах имеются
хорошие условия для отвода
тепла от катализатора. Это объяс-
няется тем, что отношение поверх-
ности теплоотдачи к объему ката-
лизатора в них весьма велико. Кроме того, в трубчатых реакторах
применяют большей частью высокие слои катализатора и соот-
ветствующие им большие линейные скорости потока газа, что обеспе-
чивает приемлемые значения констант тепло- и массопередачи. Ука-
занные преимущества позволяют осуществлять в трубчатых реак-
торах сильно экзотермические процессы) например, различные
реакции каталитического окисления).
Рис. VII.4. Трубчатый реактор с
отводом тепла реакции расплавом
солей, прокачиваемым через меж-
трубное пространство:
1 — трубки, заполненные катализатором;
2 — температурные компенсаторы; 3 —
штуцер для ввода водяного пара или азота;
4 — перфорированная тарелка; 5 — гильзы
для термопар; в — штуцер для ввода рас-
плава солей; 7 — трубные решетки; « —
штуцер для слива расплава солей (при чи-
стке и ремонте аппарата); 9 — штуцер для
вывода расплава солей.
267
В большинстве современных трубчатых реакторов температура;
бани почти постоянная по всей длине реакционной зоны. Это, однако,
приводит к «установлению весьма невыгодного в некоторых случаях
распределения температур по длине трубки: реагирующий поток
сперва разогревается за счет тепла реакции, а затем, когда скорость
тепловыделения становится меньше скорости теплоотвода, темпера-
тура в реакторе начинает падать, асимптотически приближаясь
к температуре бани. При этом в точке температурного максимума
появляется опасность оплавления и дезактивации катализатора
или скачкообразного перехода 'процесса во внешнедиффузион-
ную область; у выхода из реактора процесс замедляется из-за
слишком малой температуры. Если при проведении сложного про-
цесса температура распределяется так, как описано выше, то зто
может привести к сильному, или даже полному, падению избира-
тельности.
Принципиально проблема поддержания в трубчатом реакторе
заданного (оптимального) температурного профиля по длине трубки
не может быть решена для любого виДа реакции. -
Основным недостатком трубчатых реакторов является сложность
их изготовления. Для достижения постоянства температуры по сече-
нию трубок последние приходится делать весьма узкими. Увеличение
диаметра трубок ведет к резкому возрастанию температурных пере-
падов по их сечению. Поскольку производительность промышлен-
ного реактора должна быть весьма велика, приходится применять
аппараты с очень большим числом таких трубок (до нескольких тысяч).
При проведении экзотермических процессов, как адиабатических,
так и с внутренним теплообменом, иногда применяют автотермиче-
ские реакционные узлы, конструкция которых позволяет осуществ-
лять охлаждение реагирующей смеси в промежуточных теплообмен-
никах или в. зоне реакции с помощью теплообмена с холодной исход-
ной смесью, одновременно нагревающейся до температуры реакции.
Теплообмен между входящим и выходящим из реактора потоками
может быть осуществлен и в емкостных (одностадийных) адиабати-
ческих реакторах. В отдельных случаях, когда допустим значитель-
ный перегрев хотя бы одного из реагентов (например, водяного пара),
подобный принцип применим и при проведении эндотермических
процессов. Преимуществом автотермических реакционных узлов яв-
ляется уменьшение затрат на теплообмен, а также определенные кон-
структивные удобства, особенно важные при проведении реакций
под давлением. Основным недостатком этих схем является возник*
новение явлений неустойчивости и скачкообразного перехода между'
различными режимами процесса.
Кроме трубчатых, возможны и другие конструкции аппаратов
с неподвижным слоем катализатора и теплообменом в зоне реакции.
В процессах, связанных с периодической регенерацией катализатора,
конструкции реакторов особенно, разнообразны. Это вызвано необ-
ходимостью приспособить конструкцию к зачастую резко различным
условиям реакции и регенерации.
Реакторы для газофазных процессов с кипящим слоем катали-
затора*. Явление псевдоожижения (флюидизации) заключается в том,
что при продувании газа снизу через слой мелкодисперсных твердых
частиц все они приходят в беспорядочное движение, в результате
чего слой расширяется, принимает вид кипящей жидкости и приобре-
тает свойство текучести. Переход слоя в псевдоожиженное состояние
происходит скачком при некоторой линейной скорбсти потока, назы-
ваемой критической скоростью.
Основные преимущества кипящего слоя определяются интенсив-
ным движением составляющих его частиц. Он текуч, как жидкость,
благодаря чему становится возможным непрерывное удаление из
слоя на регенерацию частиц катализатора, потерявшего активность,
и ввод в реактор свежего катализатора. Циркулирующие частицы
могут также служить теплоносителем. Перемешивание твердых ча-
стиц вызывает уравнивание средней активности катализатора во
всем объёме реактора.
Интенсивное перемешивание твердых частиц обусловливает высо-
кую эффективную теплопроводность кипящего слоя. Перенос тепла
в нем осуществляется, главным образом, самими частицами, нагре-
вающимися или охлаждающимися у теплообменной поверхности и
перемещающимися внутрь слоя. Благодаря этому в кипящем слое
создается почти полная изотермичность как по длине, так и по се-
чению.
Другое важное преимущество кипящего слоя связано с.возмож-
ностью использовать мелкие частицы катализатора, не увеличивая
гидравлического сопротивления слоя. Благодаря применению мел-
ких частиц устраняется внутридиффузионное торможение.
Однако кипящий слой обладает и рядом недостатков. Наибо-
лее важным из них является неоднородность слоя. Значительная
часть потока газа проходит сквозь него в виде газовых пузырей и
струй, составляющих как бы особую фазу, в которой отсутствуют
химические превращения. Диффузия реагентов из пузырей в проме-
жутки между твердыми частицами затруднена, вследствие чего воз-
никает дополнительное — межфазное — сопротивление массообмену
между потоком газа и поверхностью катализатора.
Кроме того, недостатком кипящего слоя является значительное
перемешивание потока газа при движении твердых Частиц, что создает
неблагоприятный гидродинамический режим, иногда приближа-
ющийся к идеальному смешению по газу. Именно наличие меж-
фазного сопротивления и продольного перемешивания потока объ-
ясняет часто наблюдаемое на практике снижение скорости и избира-
тельности каталитических процессов при переводе их в кипящий
слой.
Кипящему слою свойственны и недостатки технологического
характера. Вследствие постоянного трения частиц друг о друга
* По процессам в кипящем слое катализатора см. монографии [9—12].
269
катализатор разрушается и выносится из реактора. Поэтому прихо-
дится создавать специальные, особо прочные катализаторы. Эрозии
в кипящем слое подвержены не только катализатор, но и все распо-
ложенные в слое части реакционного аппарата, что вызывает необ-
ходимость специальной их защиты и частых ремонтов реактора.
Из-за выноса катализатора в уста-
новках для работы с кипящим слоем
приходится предусматривать системы
улавливания (циклоны, фильтры, элек-
трофильтры) или увеличивать объем
реактора для создания сепарационной
зоны. И то, и другое ведет к удорожа-
нию оборудования.
Кипящий слой катализатора наибо-
лее целесообразно применять в процес-
сах, требующих частой регенерации ка-
тализатора. Для проведения реакций
с большим тепловым эффектом кипя-
щий слой особенно рационален в аппа-
ратах большой, мощности, поскольку
в этом случае другие конструкции ста-
новятся практически неприемлемы.
Реакторы для работы с кипящим
слоем катализатора обычно представ-
ляют собой цилиндрические аппараты
с распределительной решеткой внизу.
В случае экзо- или эндотермических
реакций аппарат снабжается внутрен-
ним Теплообменником. Вследствие прак-
тического отсутствия перепада темпера-
туры по сечению аппарата и благопри-
ятных условий тепловой устойчивости,
в процессах с кицящим слоем можно
допускать весьма значительную раз-
ность между температурами катализа-
тора и теплоносителя.
Различают несколько типов контактных аппаратов и узлов для
работы с кипящим слоем катализатора.
Аппараты без циркуляции катализатора применяют в том случае,
когда катализатор не требует непрерывной регенерации. Такогр
типа аппараты могут быть просто емкостными, например, как на
рис. VII.5, либо разделенными на секции. Секционирование осуще-
ствляется либо перфорированными перегородками, либо многослой-
ным рядом колосников. Секционированные аппараты значительно
более эффективны, так как в них улучшается массообмен и устра-
няется осевое перемешивание.Однако конструкция секционных ап-
паратов сложнее, а сами секционирующие элементы подвергаются
повышенному истиранию.
Продет Воздух на оздуд-
^3
4
Нагретое
сырье
• На по-
догрей
-^—Badt
$ ^—Boda
— Сырье
Рис. VII.5. Аппарат с кипя-
щим нециркулирующим слоем
катализатора:
1 — водяной теплообменник; 2 —
газовый теплообменник; з —
фильтры; 4 — слой катализатора.
270
Имеется достаточно много различного рода конструктивных схем
установок с циркулирующим кипящим слоем и непрерывной регене-
рацией катализатора [13, 14]. По одной из наиболее распространен-
ных схем (рис. VII.6) работают следующим образом: реакционные
Рис. VII.6. Установка с кипящим циркулирующим слоем
катализатора:
1 — циклоны; 2 — реактор; 3 — отпарник (десорбционная камера);
4 — перегородка; 5 — регенератор; 6 — стояк для спуска регенериро-
ванного катализатора.
газы по трубе попадают в катализаторопровод, соединяющий регене-
ратор с реактором, и за счет своей энергии эжектируют регенериро-
ванный катализатор в реактор 2. Контактные газы выходят из реак-
тора через систему циклонов 1 и направляются в узел улавливания
продуктов реакции. Катализатор в псевдоожиженном состоянии по-
стоянно вытекает из реактора через отпарник 5, в котором псевдо-
ожижение производится водяным паром, одновременно десорбиру-
ющим продукты реакции с катализатора. Из отпарника катализатор
271
поступает в катализаторопровод, по которому он вспомогательным по- |
током воздуха эжектируется в регенератор 5. Одновременно в ниж-
нюю часть регенератора с помощью воздуходувки подают основную [
массу воздуха. В регенераторе происходит выжигание углеродистых
ч
Продукты
реакции
Воздух.
воздух
Рис. VII.7. Схема установки
с циркулирующим гранулиро-
ванным катализатором:
1 — регенератор; 2 — теплообмен-
„ ники; з — питатель для регенери-
рованного катализатора; 4 — пита-
тель для отработанного катализа-
тора; 5 — распределительное уст-
ройство для подачи катализатора;
6 — реактор; 7 — эрлифт отрабо-
танного катализатора; 8 — эрлифт
регенерированного катализатора.
отложений на катализаторе в условиях, 1
обеспечивающих изотермичность слоя. 1
Температуру в регенераторе можно 1
регулировать количеством подаваемого 1
воздуха. Отработанный воздух через !
циклон 1 выводится в атмосферу. Отно- |
шение объемов реактора и регенератора ]
определяется отношением длительности I
рабочего цикла катализатора к длитель- |
ности регенерации. I
Один дз вариантов установки с дви- 1
жущимся зерненым катализатором I
представлен на рис. VI 1.7. Реакция (гид- I
реформинг нефтяных фракций) проте- 1
кает в колонном реакторе 6, в верхнюю 1
часть которого по распределительному I
устройству 5 поступает катализатор в |
виде непрерывного потока зерен. Ката- 1
лизатор под действием силы тяжести 1
сплошной массой движется по аппарату 1
сверху вниз и выводится в нижней части 1
реактора. Реагенты выводятся в несколь- I
ких точках аппарата, расположенных I
на различной высоте. Вывод продуктов I
реакции осуществляется также в ряде |
точек, расположенных несколько выше I
мест ввода реагентов. Катализатор из 1
реактора через эрлифт 7 поступает в I
подъемную трубу и далее в питатель 4 |
регенератора 1. Регенератор также I
представляет собой аппарат колонного 1
типа, снабженный теплообменниками 2. ]
Регенерация осуществляется воздухом 1
паром, поступающим в нижнюю часть |
выходит сверху и направ- I
или смесью его с водяным
регенератора. Отработанный воздух
ляется на очистку. Катализатор в регенераторе движется сверху*
вниз сплошной массой. Температура в регенераторе поддержи-
вается за счет теплоты сгорания углеродистых отложений на ката-
лизаторе. Регенерированный катализатор поступает в эрлифт 8,
откуда возвращается в реактор через питатель 3. Следует под-
черкнуть, что конструкции как различных узлов установок по-
добного типа (распределительных устройств, питателей для катали-
затора и других), так и самих аппаратов далеко не просты. Более
подробное описание их можно найти в работе [15].
272
J
Преимущество систем с движущимся гранулированным катали-
затором — непрерывность процесса и независимость температурных
режимов реакции и регенерации при сохранении благоприятного
гидродинамического режима потока, отсутствие межфазного тормо-
жения процесса и некоторых технологических неудобств, связанных
с применением кипящего слоя катализатора. Преимуществом дви-
жущегося слоя перед кипящим является также то, что степень по-
тери активности катализатора, выходящего из реактора, вполне опре-
делена и регулируется скоростью движения слоя, в то время как из
кипящего слоя, полностью перемешанного по катализатору, наряду
с отработанным, удаляется и определенная доля свежего.
Основные недостатки систем с движущимся слоем связаны с кон-
структивной сложностью аппаратуры и сложностью управления про-
цессом. При осуществлении процессов в движущемся слое трудно
поддерживать пространственную однородность слоя, всякое наруше-
ние которой ведет к перераспределению потока газа внутри аппа-
рата, проскоку непрореагировавшего реагента через места наимень-
шей плотности слоя и усилению коксообразования на катализаторе
в местах его застоя. Для придания однородности слою катализатора
в аппаратах устанавливают специальные направляющие, которые
одновременно могут служить для теплоотвода. Тем не менее, и при
этом плотность слоя катализатора остается неконтролируемой и раз-
личной в разных частях аппарата;
Жидкофазные промышленные процессы обладают следующими
преимуществами перед газофазными.
1. Большая, как правило, производительность катализатора и еди-
ницы реакционного объема, обусловливаемая высокой, по сравнению
с газами, концентрацией реагентов при сравнимом времени реакции.
2. Протекание реакций при более низких, чем в газовой фазе,
температурах, что дает возможность применять каталитические про-
цессы для термически недостаточно стабильных соединений.
3. Улучшение условий теплопередачи в слое катализатора и,
следовательно, простота и малые размеры теплообменников; возмож-
ность отвода тепла реакции путем испарения одного из компонентов
реагирующей смеси.
К недостаткам жидкофазных методов следует отнести возника-
ющие в ряде случаев затруднения с отделением катализатора от про-
дуктов реакции, сложность подбора прочных в условиях жидкофаз-
ных реакций формованных катализаторов и носителей. Возможность
применения жидкофазных процессов ограничивается и тем, что во
многих случаях для сохранения жидкого состояния реагентов при
температуре реакции необходимо применять высокие давления, а это
приводит к усложнению и удорожанию аппаратуры.
Наиболее частым вариантом жидкофазных процессов являются
процессы, в которых реакционный поток состоит из двух или несколь-
ких фаз. К ним относятся газо-жидкостные процессы и процессы
с несмешивающимися жидкостями. В этих случаях возникает не-
обходимость обеспечить достаточно высокие скорости межфазного
18 Заказ 1441 273
массообмена, что достигается энергичным перемешиванием потока ре- Я
агентов или всей реакционной массы, включающей суспендирован- 'Я
ный катализатор. Практически для различных вариантов жидко- 3
фазных процессов применяют одну и ту же реакционную аппаратуру. 3
Периодические методы осуществления жидкофазных гетерогенно-
каталитических реакций используют в промышленности достаточно •]
широко при производстве относительно малотоннажных продуктов: 5
фармацевтических препаратов, душистых веществ и т. п. Аппараты 1
для периодического проведения гетерогенно-каталитических реак-
ций не отличаются от реакторов периодического действия для про-
ведения некаталитических реакций. Реакторы должны оснащаться "
устройствами, обеспечивающими хорошее перемешивание реакцион-
ной смеси, — мешалками или выносными циркуляционными кон-
турами. Это особенно важно при проведении газо-жидкостных реак-
ций. Если реакция проводится при кипении жидкости, как, напри-
мер, этерификация с твердыми катализаторами, то перемешивание
осуществляется за счет кипения и специальной мешалки не требуется.
Естественно, что реакционные аппараты должны быть снабжены
устройствами для подвода или отвода тепла к реакционной массе
в виде теплообменников или рубашки. Если процесс проводится под
давлением, аппараты представляют собой автоклавы, конструкция
которых зависит от величины давления. Для высоких давлений осо-
бенно удачны бессальниковые автоклавы с экранированным двига-
телем и принудительной внутренней циркуляцией, обеспечиваемой
винтовым насосом, помещенным внутри аппарата.
Для проведения непрерывных жидкофазных процессов обычно
применяют либо емкостные аппараты с мешалкой, либо аппараты
колонного типа, снабженные (и те и другие), если надо, теплообмен-
ными устройствами. В первом варианте пока используют только сус-
пендированный катализатор, во втором можно применять и гранули-
рованный неподвижный катализатор.
VII.3. РЕАКТОРЫ ИДЕАЛЬНОГО СМЕШЕНИЯ
С ОДНОФАЗНЫМ ПОТОКОМ РЕАГЕНТОВ *
Режим идеального смешения в реакторе может быть достигнут
только путем интенсивного принудительного перемешивания реаги-
рующей смеси с помощью мешалки или внешнего циркуляционного
контура.
Если поток реагентов однофазный, то уравнения математической
модели реактора могут быть написаны в квазигомогенном прибли-
жении как для мелкодисперсного суспендированного, так и для зер-
нистого неподвижного катализатора. Если поток реагентов много-
фазный, то естественно, квазигомогенная модель не приложима и
расчет ведется по методам, изложенным в разделе VII.7. Однако,
* Первые работы по расчету реакторов идеального смешения принадлежат
Кириллову [16] и Макмаллину и Веберу [17].
274 '
если, например, проводится газо-жидкостная реакция ,с большим
избытком газофазного реагента, то процесс можно рассчитывать как
для однофазного потока и описывать квазигомогенной моделью.
Объем реакционного пространства Vp при этом приравнивают объему
жидкой фазы
' Гр=Гп(1-е)
где Vn — полезный объем реактора; е — газосодержание (см. раз-
дел VII.7).
Менее строго, но с приемлемой для практических расчетов степенью
точности, моделью идеального смешения могут быть описаны про-
цессы, протекающие в тонких слоях катализатора, например, на
каждой полке полочного реактора.
Материальный и тепловой балансы. В режиме идеального сме-
шения температуры и концентрации реагентов одинаковы по всему
объему реактора; такой же состав и температуру имеет й поток, вы-
ходящий из реактора. Состав исходной смеси, которая подается
в реактор, будет, конечно, другим, так что у входа в реактор идеаль-
ного смешения все переменные изменяются скачкообразно. Уравне-
ния материального баланса реактора идеального смешения, рабо-
тающего в стационарном режиме, легко получить, приравнивая раз-
ность между количеством i-ro вещества, входящим и выходящим
из реактора в единицу времени, скорости изменения количества дан-
ного вещества в результате химических реакций
(Ci-Ci0)V=Vri(C,!r) (VII.1)
где Ci0, С[ — концентрации i-ro вещества на входе и выходе реак-
тора; v — объемная скорость потока; V — объем аппарата; rt — ско-
рость образования i-ro вещества в единице объема реактора.
В случае единственной реакции, скорость которой равна f (С, 71),
ri = vir (С, Т), где vt- — стехиометрический коэффициент i-ro ве-
щества (положительный — для продуктов реакции и отрицательный —
для исходных веществ). Учитывая это, приводим уравнения (VII.1)
к виду:
P(Q-C/0) = vzr(C, Т) (VI 1.2)
Здесь Р — v/V. Обратная величина S — V/v называется време-
нем контакта; как будет видно из дальнейшего, она равна среднему
времени пребывания в реакторе. Заметим, что выражение для ско-
рости реакции г (С, Т) может определяться не только кинетическими,
но и диффузионными факторами. Например, в случае, если реакция
идет на взвешенном в потоке твердом катализаторе, это выражение
должно быть составлено с учетом торможения массо- и теплопере-
дачи к активной поверхности (см. главу III).
Аналогичным образом составляется уравнение теплового баланса
реактора:
V(T-T0)v=hr(C Т) V-q (VIL3)
Левая часть этого уравнения представляет собой разницу между
количеством теплоты, уходящим из реактора с реагирующей смесью
18* 275
и входящим в него со смесью исходных веществ. Здесь у — тепло-
емкость единицы объема потока; предполагается, что она не меняется
в результате химических превращений. Первый член в правой части
уравнения (VI 1.3) равен скорости выделения тепла в результате
химической реакции с тепловым эффектом h. Величина q равна ско-
рости отвода тепла от зоны реакции. Если реактор охлаждается внеш-
ним теплоносителем с постоянной температурой Тс, то
q = krF(T — Tc) (VI1-4)
где кт — коэффициент теплопередачи от реагирующей смеси к тепло-
носителю; F — площадь поверхности теплообмена.
Учитывая формулу (VII.4), можно привести уравнение (VII.3)
к виду
а(Т-Г») = Лг(С, Г) (VII.5)
где
(vn-6)
V \ уР J ’ l-|-fcTP/YP х ’
В частном случае адиабатического реактора, когда все тепло
отводится с движущимся потоком, кт = 0 и, соответственно, а =
= ур и Г* = То.
Систему уравнений (VII.2), (VII.4) можно свести к единственному
уравнению для концентрации одного из веществ, которое прини-
мают за ключевое, или для температуры реакции. Разделив каждое
уравнение (VII.2) на v{, а уравнение (VII.2) — на h и приравнивая
их левые части, получаем линейные соотношения между концентра-
циями реагирующих веществ и температурой:
(₽/vZ) (Ct-Ci0) = (а/Л) (Т-Т*) (VII.7)
С помощью этих соотношений можно выразить скорость реакции
через какую-либо одну из переменных С{ или Т, а затем решить соот-
ветствующее уравнение баланса.
В случае нескольких одновременно протекающих реакций урав-
нения материального и теплового балансов имеют вид:
₽(G-Ci0) = 2vi/r/(C. Т)
3
a(T-T*)=^h1rl(C, Т)
з
(VII.8)
Здесь vtj — стехиометрический коэффициент i-ro вещества в ;-й реак-
ции; Г] — скорость /-й реакции; hj — ее тепловой эффект. ‘
Систему уравнений (VII.8) можно упростить, сведя ее к меньшему
числу уравнений для концентраций ключевых веществ, число ко-
торых равно рангу матрицы стехиометрических коэффициентов
(см. раздел II. 2).
Основные свойства решений. При технологическом расчете
реакторов идеального смешения можно, задав значения температуры
реакции и времени контакта S (или, что то же самое, величины
276
Р = S"1), вычислить концентрации реагентов е помощью уравнений
материального баланса (VII.2) или (VII.8).
Найденный состав реагирующей смеси определяет и скорость
тепловыделения в результате реакции. После этого из уравнения
теплового баланса можно найти скорость теплоотвода q, необходимую
для поддержания заданной температуры. Параметры, характеризу-
ющие интенсивность теплоотвода — а и Т*,—определяются из урав-
нений (VII.5) или (VII.8). Уравнение (VII.5), очевидно, 1дает следу-
ющее соотношение между этими параметрами:
Г*=7’-(й/а)г (С, Т) (VI 1.9)
Если температура исходной смеси задана, то величины Т* и а
можно менять, варьируя температуру теплоносителя Тс и площадь
поверхности теплообмена F. Здесь остается дополнительная степень
свободы: каждая из величин Тс я F или Т* и а может принимать
различные значения, достаточно лишь, чтобы было выполнено соот-
ношение (VII.9). При некоторых значениях параметров рассчитыва-
емый режим может, однако, оказаться неустойчивым к малым случай-
ным возмущениям и, следовательно, практически трудноосуществи-
мым. Поэтому необходимым элементом расчета реактора является
проверка устойчивости выбранного режима.
При фиксированных значениях параметров процесса концентра-
ции реагентов и температура в реакторе определяются совместным
решением уравнений (VII.2), (VII.5) или (VII.7), (VII.8). Легко за-
метить, что эти уравнения полностью эквивалентны уравнениям ма-
териального и теплового балансов на внешней равнодоступной по-
верхности катализатора (см. раздел ПГ.З). Согласно полученным там
результатам, при определенных условиях система уравнений ма-
териального и теплового балансов может иметь несколько решений,
соответствующих однозначно заданному набору характерных пара-
метров процесса. Появление множественных режимов возможно
в случае, когда реакция ускоряется одним из ее продуктов или тор-
мозится одним из исходных веществ, а-также в случае экзотермиче-
ской реакции со значительным тепловым эффектом. В этих условиях
при плавном изменении температуры исходной смеси или теплоноси-
теля температура реактора изменяется скачком в критических точ-
ках перехода между режимами; поэтому на графике зависимости Т
от Т* появляется характерная гистерезисная петля (как на рис. III.4).
Заметим, что, в отличие от процессов на внешней поверхности зерна,
при проведении процесса в реакторах идеального смешения возможна
ситуация, когда не только промежуточный, но й один из крайних
режимов становится неустойчивым. Рассуждения, основанные на
анализе стационарных уравнений, которые привели к условию не-
устойчивости (III.51), доказывают только неустойчивость промежу-
точного режима, но еще не свидетельствуют об устойчивости тех ре-
жимов, для которых неравенство (III.51) не удовлетворяется. Более
того, существует область значений параметров процесса, в кото-
рой имеющийся единственный стационарный режим реактора
277
оказывается неустойчивым и процесс переходит в резко нестационар-
ный и технологически недопустимый режим нелинейных колебаний.
Возможность потери устойчивости — один из существенных не-
достатков реакторов идеального смешения. Еще более очевидный их
недостаток заключается в необходимости сильного увеличения сред-
него времени контакта для достижения заданной степени превраще-
с временем периодического процесса или
ния сырья, по сравнению
Рис. VII.8. Зависимость обратной
скорости реакции от концентра-
ции реагента.
временем пребывания в реакторе
идеального вытеснения. Сравним две
формулы:
„ с0-с
s~~^cT
. (VIL10)
t—
(VII.11)
с
Формула (VI 1.10) получена из
уравнения материального баланса
реактора идеального смешения
(VII.2), а (VII.11) — интегрирова-
нием кинетического уравнения (11.14)
при постоянной температуре. В обоих
случаях предполагается, что за клю-
чевое вещество принято то исходное
вещество, которое имеется в относительном недостатке, и кон-
центрации всех остальных веществ, влияющих на скорость реак-
ции г (С), выражены через концентрацию ключевого вещества.
Стехиометрический коэффициент последнего принят равным —1,
а индекс при его концентрации опущен. В «нормальном» случае,
когда отсутствуют явления автокатализа и торможения исходными
веществами, dr/dC < 0. При этом кривая, выражающая зависимость
1/гот С, при постоянной температуре будет такой, как на
рис. VIII.8.
Из формулы (VII.11) следует, что время периодического процесса
или, что равнозначно, процесса в реакторе идеального вытеснения,
равно площади под этой кривой, заключенной между абсциссами С
и С0. Величина же времени контакта S равна площади прямоуголь-
ника ABKD и, таким образом, всегда S > t. По мере приближения
к равновесной концентрации Ср (71) (или по мере исчерпания лими-
тирующего исходного вещества в необратимой реакции) величина 1/г,
а следовательно, и S, возрастает как (С — Ср)-1, а величина t, вы-
ражающаяся интегральной формулой (VII.11), — как — In (С — Ср).
Поэтому отношение S/t всегда становится неограниченно большим
при приближении к равновесной степени превращения. Если функ-
ция г (С) имеет максимум (такой функции соответствует пунктирная
кривая на рис. VII.8), то неравенство S > t нарушается; однако и
в этом случае оно начнет выполняться при степени превращения,
достаточно близкой к равновесной.
278
С помощью рис. VII.8 нетрудно убедиться, что показатели про-
цесса можно существенно улучшить, используя вместо одного реак-
тора идеального смешения цепочку последовательно соединенных
реакторов. Суммарное время контакта, необходимое для достижения
заданной степени превращения в последовательности, состоящей из
двух реакторов, равно площади двух прямоугольников AEFG и
GHKD (где GFH — ордината, соответствующая концентрации клю-
чевого вещества в первом реакторе С i). Увеличивая число последо-
вательно соединенных реакторов N, можно и далее уменьшать сум-
марное время контакта, приближаясь в пределе (N -> оо) к времени
периодического процесса t.
Распределение времени пребывания в реакторе. Относительно
низкая степень превращения, достигаемая в реакторах идеального
смешения, объясняется сильной размытостью функции распределения
времени пребывания в аппарате. Очевидно, доля молекул, выходя-
щая из реактора в единицу времени, равна v/V = р. Следовательно,
за некоторый малый промежуток времени Дт молекула может выйти
из реактора с вероятностью (3 Дт или остаться в нем с вероятностью
1 — рДг. Вероятность остаться в реакторе до момента т = тЛт и
выйти из него к моменту т = (т + 1) Дт равна рДт (1 — рДт)т.
Переходя к пределу т -> оо Дт -> 0, получаем:
<p(T) = Pe"P't (VI 1.12)
При всех расчетах (см. главу VI) удобней пользоваться не функ-
цией распределения (VI 1.12), а характеристической функцией:
j (т) dt
о
Р 1
РЧ-P 1 + ps
(VII.13)
Исходя из этого выражения, легко найти статистические моменты
распределения. В частности, среднее время пребывания в реакторе
и дисперсия х2 определяются формулами:
СО
(4?) = С T<p(r)dT=p-i = s
\ &Р /р=о J
о
со
('t-s)2<₽c')^=s2
о
(VII.14)
Сама характеристическая функция, как было показано в раз-
деле VI.1, непосредственно.дает безразмерную концентрацию непро-
реагировавшего исходного вещества на выходе реактора
СО
с=^=( e-*t<p(T)rfT=g(fc) = —1— (VII.15)
Со J 1 + ks
о
где к — константа скорости реакции первого порядка.
279
Функция распределения времени пребывания в цепочке последо-Я
вательно, соединенных реакторов оказывается гораздо менее «раз- Я
мазанной», чем функция распределения времени пребывания в'оди- Ц
ночном реакторе идеального смешения. Полное время пребывания Я
в цепочке реакторов т равно сумме N случайных величин — вре- Я
мени пребывания в каждом из реакторов т„ (ге=1, 2, . . .,N). Ц
Очевидно, все величины т„ статистически независимы. В теории ве- 1
роятностей доказывается, что характеристическая функция распре- J
деления суммы независимых случайных величин GN (р) равна произ- Я
ведению характеристических функций распределения всех слагаемых, я
Следовательно, Ц
' NN |
^(р)=П&(р)=П1^ I
/ м
где s„ — среднее время пребывания в n-м реакторе. • I
В частном случае, когда все величины s„ равны, т. е. sn— S, 3
имеем:
Gn(p) = (A+pS)~n (VII.17) |
Статистические моменты распределения легче всего найти, диф- 1
ференцируя по р логарифм характеристической функции. Нетрудно |
видеть, что все семиинварианты распределения равны сумме coot- I
ветствующих семиинвариантов для каждого реактора цепочки. <
В частности, среднее время пребывания в [цепочке реакторов [s.y J
и дисперсия н2 равны:
«2=5*» (VII.18) j
n~l n~l ;
В одиночном реакторе отношение дисперсии к квадрату среднего ]
времени пребывания в реакторе, которое может служить основной |
мерой размытости распределения, согласно формуле (VII. 14), равно ]
единице. В то же время для цепочки реакторов получаем: |
N I / N \1 I
<1 (VII.19) I
n~l I \n«l / J
Если какой-либо реактор в цепочке заменить на два с тем же сум- |
марным средним временем контакта, то числитель в уравнении |
(VII.19) уменьшится, в то время как знаменатели не изменится. I
Таким образом, распределение всегда сужается с увеличением числа 1
последовательно включенных реакторов. Распределение окажется 1
наиболее узким, если все величины s„ одинаковы. Чтобы убедиться 1
в этом, найдем минимум дисперсии при постоянном суммарном вре- j
280 1
мени контакта 5. Эта вариационная задача на условный экстремум
сводится к задаче определения минимума величины
- XS = 2 <s»- (VII -20)
n=l
где A — неопределенный множитель Лагранжа.
Дифференцируя формулу (VI 1.20) по каждой из переменных sn,
находим условие минимума s„ = Х/2. Множитель А определяют ис-
ходя из условия 2 s„ = -VA/2 = S. Таким образом, прификсирован-
П=1
ном S дисперсия будет минимальной в случае sn = s = S/N.
Если температура, а значит, и константа скорости реакции одина-
ковы во всех реакторах цепочки, то безразмерная концентрация не-
прореагировавшего исходного вещества на ее выходе равна GN (к).
Эта величина, как легко убедиться варьированием lnGjv(fc)no sn
при фиксированном S, также минимальна, если все величины s„
одинаковы. С увеличением числа реакторов в цепочке N (и постоян-
ном S) концентрация с,у монотонно уменьшается, асимптотически
приближаясь при N -> оо к величине ст = e~KS, характерной для
периодического реактора или реактора идеального вытеснения.
В общем случае сложной системы реакций первого порядка всегда
имеется некоторое оптимальное значение времени пребывания в реак-
торе , при котором вероятность превращения молекулы исходного ве-
щества в молекулу целевого продукта будет максимальной. Среднее
время контакта S может быть выбрано таким образом, чтобы оно сов-
падало с оптимальным. Тогда любое отклонение времени пребывания
в цепочке реакторов от средней величины вызовет ухудшение показа-
телей процесса. Последние будут наилучшими при минимальной дис-
персии распределения, т. е. при одинаковых s„ в случае, когда S и N
фиксированы и при максимальном N, когда фиксировано только сум-
марное время контакта S. Этот результат впервые получен (более
сложным способом) в работе [18].
Функцию распределения времени пребывания в цепочке N реак-
торов Ф.у (т) можно получить из характеристической функции с по-
мощью обратного преобразования Лапласа. При sn = S имеем
Ф)у(т) =-------Ке~х (VII.21)
или
NN~N-1
ФН*) = -(лПГ1)Те^Т (VII.22)
где т = x/NS — безразмерное время пребывания в реакторе.
График функции (VI 1.21) при различных значениях N представ-
лен на рис. Vll.9. При N >1 эта функция имеет максимум в точке
*^макс 1 — N *. С увеличением N наиболее вероятное время
281
пребывания в реакторе тмакс = NsrMaKC приближается к среднему
времени контакта S: максимум функции распределения ста-
новится все острей, и при N со она асимптотически приближается
к 6-функции, 6(т — NS) или 6 (т — 1), характерной для режима иде-
ального вытеснения. Последовательное соединение реакторов иде-
ального смешения позволяет, таким образом, избавиться от основ-
ного недостатка реакторов этого типа — размытости функции рас-
пределения времени пребывания в аппарате, сохранив их главное
технологическое преимущество — хорошие условия для теплообмена.
VII.4. РЕАКТОРЫ ИДЕАЛЬНОГО ВЫТЕСНЕНИЯ
С ОДНОФАЗНЫМ ПОТОКОМ
Основные уравнения. Режим идеального вытеснения характери-
зуется пренебрежимо малой ролью диффузии и теплопроводности
в продольном (т. е. параллельном движению реагирующей смеси)
направлении. Соответственно, каждый «элемент потока», проходя
реактор, не взаимодействует со своими соседями, вошедшими в реак-
тор раньше и позже него, и остается в аппарате, перемещаясь вдоль
него со скоростью и, строго фиксированное время т = L/u, необхо-
димое для прохождения длины реактора L. Если, кроме того, зна-
чение концентраций реагентов С,- и температуры Т постоянны по
сечению аппарата, независимо от расстояния До его стенок, то ста-
ционарный режим реактора описывается при и = const в квазигомо-
282
генном приближении системой обыкновенных дифференциальных
уравнений:
udCildX’=ri{C, Т) (VII.23)
yudT/dX = rT(C, Т)~д(Т, Тс) (VH.24)
Здесь X — продольная координата; г{ — скорость образования г-го
вещества; гт — скорость тепловыделения в единице объема реактора;
у — теплоемкость единицы объема реагирующей смеси; q — скорость
теплоотвода от единицы объема реакционной зоны, зависящая от тем-
пературы в зоне реакции Т та температуры теплоносителя Тс. Вы-
ражение для скорости г должно учитывать диффузионное торможение
реакций на отдельном зерне [см. формулу (VII.49)].
Уравнения (VII.23) достаточно записать только для ключевых
веществ, так как концентрации всех остальных реагентов можно
выразить через ключевые с помощью линейных соотношений (см.
раздел II.2). При расчете процесса с неподвижным катализатором под
и надо понимать фильтрационную скорость потока w, т. е. скорость,
рассчитанную на полное сечение аппарата, равную истинной средней
скорости, умноженной на долю свободного объема е. Выражения для
функций г; и гт в случае гетерогенного процесса должны быть состав-
лены с учетом как кинетических, так и диффузионных факторов;
поэтому для квазигомогенной модели расчет реактора всегда должен
быть предварен анализом процессов на отдельном зерне катализатора,
позволяющим установить макроскопическую скорость процесса в еди-
нице объема слоя.
Как указывалось в главе VI, для зернистого слоя характерно прак-
тическое отсутствие смешения вещества в направлении, обратном
движению потока. Поэтому реакторы с неподвижным зернистым слоем
катализатора хорошо описываются моделью идеального вытеснения.
Проточные аппараты с однофазным потоком могут рассчитываться
по модели идеального вытеснения, когда число Пекле ^Ре = ^-^
составляет порядка 102 и более. К аппаратам последнего типа могут,
например, относиться колонны, через которые проходит поток жид-
кости со взвешенным катализатором.
При наличии теплоотвода к стенке аппарата слабым местом мо-
дели, выраженной уравнениями (VII.23), (VII.24), может стать пред-
положение о независимости переменных от поперечной координаты.
В случае гомогенных процессов это предположение выполняется при
турбулентном режиме течения; но в гетерогенных реакторах с зер-
нистым слоем поперечные градиенты могут быть весьма существен-
ными (см. раздел VII.5).
Уравнения (VII.23), (VII.24) полностью эквивалентны уравнениям
периодического процесса и Зтановятся идентичными последним, если
ввести вместо координаты X текущее время контакта t — Х]и. Для
расчетов часто более рационально использовать в качестве не-
зависимой переменной текущее время контакта I, чем координату X.
Так, если процесс идет в кинетической области, концентрации
283
реагентов в сечениях реактора идеального вытеснения с равными t
остается неизменными при увеличении или уменьшении длины реак-
тора L и линейной скорости потока и. (Процессы, идущие в диффу-
зионной или переходной области, не обладают этим свойством, по-
скольку их скорость зависит от и). В дальнейшем будем использовать
обе формы выражения текущей координаты.
Чисто изотермические процессы почти не встречаются на прак-
тике. В тех немногих случаях, когда изотермический процесс яв-
ляется хорошим приближением к реальному, применимы формы
уравнений, рассмотренных в главе II разделах II.1 и II.2.
Там же (в разделе П.З) был рассмотрен и случай процессов с из-
менением объема реагирующей смеси в ходе реакции. Для точных
расчетов в неизотермических процессах следует учитывать измене-
ние объема с температурой и величину и заменяют на и (Т), которую
вводят под знак дифференцирования. Уравнения, приведенные
в главе II, оперируют с постоянными давлениями вдоль слоя катали-
затора. В действительности, однако, слой катализатора оказывает
гидравлическое сопротивление потоку и изменяет его объем при газо-
фазных процессах.
Адиабатические реакторы. В адиабатическом процессе отсут-
ствует теплообмен с внешней средой (q == 0). В случае, когда имеется
только одна реакция (обратимая или необратимая), расчетные уравне-
ния принимают вид:
dC/dt = wdC/dX=—г (С, Т) (VII.25)
dT/dt = wdT/dX = (h/y)r(C. Т) (VII.26
Здесь С — концентрация ключевого вещества, за которое обычно
принимают исходное вещество, имеющееся в относительном недо-
статке; г — скорость реакции в единице объема слоя.
Разделив уравнения (VII.25), (VII.26) друг на друга, имеем
dT/dC = —A/у (VII.27)
откуда
(Л/у) (Со -С) = Т-То (VII.28)
где индекс нуль отмечает значение переменных на входе реактора.
Из соотношения (VI 1.28) следует, что адиабатический путь про-
цесса представляет собой прямую линию в координатах С, Т. Под-
ставляя (VII.28) в (VII.26) и интегрируя, получаем для единичной
реакции расчетную формулу
т
IX _ С dT
w J Рад (Г)
То
где гад (Г) = (/i/y)’rJ[C0F— (у/Л) (Т.— То)] — адиабатическая
скорость реакции.
284
(VII.29)
В случае необратимой реакции первого порядка интеграл
в формуле (VI 1.29) может быть выражен через известную интеграль-
ную экспоненциальную функцию:
2
— СО
((VI 1.30)
Чтобы получить этот результат, введем безразмерную темпера-
туру 9 = Е (Т — Тo)/(RTo) (где Е — энергия активации реакции и
R — газовая постоянная) и воспользуемся приближенным выра-
Рис. VII.10. Зависи-
мость безразмерной кон-
центрации от безразмер-
ной продольной коорди-
наты.
жением для константы скорости реакции к (Т) = к (Т0)е9 [анало-
гично соотношению (III.54)]. Тогда
г = Л(Г0)евС; С = Со(1—0/6) (VII.31)
где 0 = hEC0](yRTl) — безразмерный максимальный адиабати-
ческий разогрев, достигаемый при полном превращении лимити-
рующего исходного вещества.
Уравнение (VII.29) приводится теперь к виду
е
ж= (-£-^- = в-ЧЁ1(6)-Ш (6-0)1 (VII.32)
J О— и
0
где х = Хк (Т0)/w.
Зависимость безразмерной концентрации с = 1 — 0/0 от коор-
динаты х при различных значениях параметра 0 показана на
рис. VII.10. Аналогичные, но только более громоздкие формулы
получаются и в том случае, если использовать точное выражение для
аррениусовской температурной зависимости константы скорости
реакции. Формула (VII.29) выражается через интегральную экспо-
ненциальную функцию и в случае необратимых реакций с целочис-
ленным порядком, отличным от первого. В более сложных случаях
возможно только численное интегрирование, которое проводят,
обычно, по уравнениям (VII.25) и (VII.26).
285
При значительных тепловых эффектах зону реакции в адиабати-
ческих реакторах приходится чередовать с зоной теплообмена, в ко-
торой тепло снимается или через стенку или добавкой в реакционную
смесь холодного реагента или инертного вещества.
В качестве примера рассмотрим расчет адиабатического реактора
гидрокрекинга тяжелых бензиновых фракций в газовой фазе на
твердом катализаторе. Реактор должен удовлетворять следующим
условиям.
Содержание парафиновых углеводородов на выходе из реактора —
не менее 95%. Объемная скорость подачи сырья (в жидком виде) —
не менее 1,5 ч -1. Температура реакции — минимальная.
Учитывая, что исходное сырье состоит из смеси тяжелых пара-
финовых, нафтеновых и ароматических углеводородов, была принята
следующая схема процесса:
1 2
С1 -->• (?2 —> Сз
Здесь Ci—суммарная концентрация тяжелых углеводородов;
С2 — то же для парафинов; С3 — то же для легких парафинов (С6 и
ниже).
По работе [19] течению процесса гидрокрекинга соответствует
математическая модель
dCi/dt = —Zi exp (—Ei/RT) Cf
dC2/dt= - Z^^(-E^/RT') +Z1 eXp (~Ei/RT) С-1 (VII.33)
,,.Z2ew(-E2lRT) C2
dC*ldt ----------------
dT!dt=-\T^-^- '
где t — условное время контакта, рассчитанное по объемной ско-
рости подачи жидкого сырья; а = 0,954 — коэффициент торможе-
ния реакции; А^ад = 44,5 град — температура адиабатического ра-
зогрева смеси, приближенно рассчитываемая по превращению тяже-
лых парафинов.
Кинетические константы модели, рассчитанные по данным четы-
рех серий изотермических-опытов, проведенных на лабораторном ин-
тегральном реакторе, равны: In Zj = 185; Ei — 22 500 кал/молъ',
In Z2 = 141; E-i = 17 900 кал/моль- n = 1.
Поскольку адиабатический разогрев смеси при полном превра-'
Щении не превышает заданных ограничений по температуре, то был
принят реактор без промежуточного теплообмена. Интегрирование
системы уравнений (VII.33) производили с начальными условиями:
Ci = 0,316; С2 = 0,679; Т = 290, 300 и 310° С.
На рис. VII.11 представлены результаты машинного интегриро-
вания уравнений (VII.33), из которых видно, что заданные требова-
ния к выходным параметрам процесса могут быть выполнены при
начальной температуре процесса 300° С в реакторе с объемом ката-
286
Рис. VII.11. Расчетные кривые изменения
безразмерной концентрации парафиновых
углеводородов в зависимости от условного
/ 1 А
времени контакта I £усл=тН~ / •
\ "об /
лизатора 0,50 .и3 на 1 .м3/ч подаваемого сырья, что соответствует
объемной скорости подачи сырья 2,00 ч-1.
Реакторы с внутренним теплообменом. Перепады температуры,
возникающие при адиабатическом протекании реакции, часто бы-
вают слишком велики; поэтому множество промышленных процес-
сов проводится с отводом тепла из зоны реакции, если последняя идет
с выделением тепла или подводом его — в случае эндотермической
реакции. Подвод или отвод тепла, как правило, осуществляют путем
теплообмена реагирующей смеси с теплоносителем. Направление
движения реагирующей смеси и теплоносителя может совпадать (при
прямотоке) или быть проти-
воположным (при противо-
токе), а в качестве теплоно-
сителя можно использовать
либо некоторое постороннее
вещество, либо смесь исход-
ных веществ, одновременно
нагревающуюся до темпера-
туры, при которой реакция
идет с заметной скоростью.
В общем случае для расчета
реакторов с внутренним теп-
лообменом необходимо ре-
шать уравнения материаль-
ного и теплового балансов
реагирующих смесей (VI Г. 23),
(VII.24) совместно с уравне-
нием теплового баланса теп-
лоносителя. Если, однако,
теплоносителем служит ки-
пящая жидкость или происходит интенсивное перемешивание
теплоносителя, температура последнего Тс будет постоянной. Если
градиенты температуры по сечению аппарата малы, то за основу
можно принять одномерную модель. Этот простейший случай рас-
смотрим в первую очередь.
Скорость теплообмена между реагирующей смесью и теплоноси-
телем q может быть представлена в виде
q--=(k^Rr)(T-Tc) (VII.34)
где кт — коэффициент теплопередачи; 7?г—гидравлический радиус
реактора, т. е. отношение площади его поперечного сечения к пе-
риметру поверхности теплообмена.
Используя это выражение, перепишем уравнения (VIL23),
(VII.24) для случая, когда имеется только одна химическая реакция
iedC/dX= —r{C, Т) (VII.35)
wdTjdX = (Л/у)г (С, Т)-а(Т-Тс) (УП.36)
где а = kT!Rr.
287
Система уравнений (VII.35), (VII.36) не решается аналитически
даже для процессов с простейшей кинетикой. Тем не менее, ее ана-
лиз позволяет установить некоторые особенности решения. При
расчете экзотермического процесса наиболее интересной величиной
является максимальный разогрев, достигаемый в «горячей точке»
реактора. Если в реактор поступает исходная смесь с температурой,
близкой к температуре теплоносителя Тс, то в сечениях, близких
к входному, теплоотвод окажется незначительным и процесс будет
проходить в почти адиабатических условиях. В дальнейшем, по мере
повышения температуры реагирующей смеси скорость теплообмена
возрастает и в некотором сечении сравняется со скоростью тепловы-
деления. После этого температура реакции, пройдя через максимум,
начнет убывать. Верхнюю оценку для достигаемой максимальной
температуры можно найти, считая, что процесс протекает адиабати-
чески вплоть до самой «горячей точки». Тогда верхняя оценка тем-
пературы, при которой скорости тепловыделения и теплоотвода срав-
няются, может быть найдена по точке пересечения прямой теплоот-
вода q = а (Т — Тс) и кривой тепловыделения ср (71) = hr (Т).
Последнюю строят с учетом соотношения между концентрацией и
температурой (VII.28), которое выполняется в адиабатическом про-
цессе. Кривая тепловыделения и прямая теплоотвода изображены
на рис. III.3; они пересекаются в несколькцх точках, и верхнюю
оценку максимальной температуры дает точка пересечения, соответ-
ствующая наименьшей температуре. По мере увеличения температуры
теплоносителя Тс прямая теплоотвода сдвигается вправо, и при не-
котором критическом значении Tz низкотемпературная точка пере-
сечения исчезает. При зтом верхняя оценка температуры в «горячей
точке» резко повышается. Формально значение максимальной тем-
пературы, конечно, не может измениться скачком. Из теории обык-
новенных дифференциальных уравнений следует, что решение си-
стемы уравнений (VII.35), (VII.36) непрерывно изменяется с измене-
нием всех параметров, в том числе и Тс (см. также раздел VII.2).
Однако в области значений параметров, близкой к той, где кривая
тепловыделения касается прямой теплоотвода (рис. II 1.3, прямая 4),
следует ожидать сильной чувствительности температуры в «горячей
точке» к изменению параметров процесса.
VII.5. УСЛОЖНЕННЫЕ МОДЕЛИ ДЛЯ РЕАКТОРОВ
С ОДНОФАЗНЫМ ПОТОКОМ
В приведенных выше расчетах реакторов не были учтены неко-
торые факторы, существенно усложняющие расчеты. Например, к ним
относятся такие факторы, как изменение объема потока в связи с из-
менением температуры реакции и гидравлическим сопротивлением
- слоя катализатора или вследствие протекания Химической реакции,
возникновение радиальных градиентов температуры в слое катали-
затора и т. п. Далее, выражение скорости реакции формальными
уравнениями с эффективными коэффициентами хорошо оправды-
288 '
вается в кинетической области, но в переходных и диффузионных
областях для сложных реакций это может дать сильные искажения.
Поэтому* с развитием электронно-вычислительной техники все
больше используют усложненные модели реакторов, по которым
можно проводить более детальные расчеты.
.Изменение объема в ходе реакции. Изменение объема потока,
а следовательно, и его скорости в ходе реакции, т. е. по длине слоя
катализатора, может произойти из-за изменения температуры, дав-
ления, вследствие гидравлического сопротивления слоя и от измене-
ния общего числа молей в ходе реакции.
Общий вид системы уравнений для реакции с переменным объемом
следующий:
Г) = 0 (VII.37)
V(X*vn — V(ywT) + 2ЙЛ =° (VI 1.38)
Здесь —
д . д . д
V — -;—Fir + зГ- (VII.38а).
ох ‘ ду ’ dz ' 1
Уравнение для w получают сложением уравнений (VI 1.37) для
всех (-компонентов
у + (VII.39)
где Р — полное давление в системе.
В систему уравнений (VI 1.37)—(VI 1.39) входят концентрации
всех веществ, а не только ключевых, и поэтому она отражает общее
изменение числа молей. Эффективные коэффициенты диффузии D*
и теплопроводности %* не выносят из-под знака дифференцирования,
так как они зависят от скорости потока. От вида вещества коэффи-
циенты D* не зависят, поскольку они являются эффективными коэф-
фициентами для турбулентного режима.
Для окончательного расчета процесса, идущего только в одну
реакцию, достаточно использовать уравнение (VII.37) для ключевого
вещества и уравнение (VI 1.39) для скорости потока. В этом случае
ri = vir и S vir — ₽г [гДе Р — коэффициент расширения объема
потока {см. раздел П.З)].
Если в результате реакции не происходит изменения общего числа
молей (0 = 0), скорость потока определяют из уравнения:
<™">
Как указывалось в главе VI, для зернистого слоя диффузионным
членом массопереноса можно пренебречь и тогда для одномерной
задачи
(VII-41)
ад \ л 1 J
19 заказ 1441 . 289
Изменение давления по слою катализатора определяют уравне-
нием
4v= (VII.42) '
где t — коэффициент сопротивления.
Из уравнений (VII.41), (VII.42) следует, что изменение объема
потока под влиянием изменения температуры и роста гидравличе-
ского сопротивления выражается уравнением:
dw w j , w dT
(VII.43)
Коэффициент / в уравнениях (VII.42) и (VII.43) определяют по
формуле Эргана, которая имеет вид:
/-V(1-75 + 15»-!Rr)
где е — доля свободного объема зернистого слоя (порозность); Re =
= ------число Рейнольдса, в котором G — весовой расход газа (жид-
кости) на единицу площади поперечного сечения реактора; d — диа-
метр частицы катализатора (или диаметр сферы, имеющей то же от-
ношение поверхности к объему, что и частица катализатора).
Двумерная модель цилиндрических реакторов. Поскольку в прак-
тике применяют почти исключительно цилиндрические, трубчатые
или колонные реакторы, то распределение температур и концентра-
ций в поперечном сечении реактора в квазигомоГенном приближении
описывается уравнениями двумерной модели
„ (d*Ct . 1 дС(\ д .
У' дУ ) дХ Г‘
(VII.44)
со следующими краевыми условиями:
при X =0
С = С0; Т ~Т0
при Y — О
^=0; ^- = 0
ЗУ ’ дУ
при Y=R
где X — координата вдоль потока; Y координата по радиусу ре-
актора; D ± и — эффективные коэффициенты диффузии и тепло-
проводности в поперечном направлении.
Численные методы решения уравнений (VII.44), (VII.45) даны
в работе [20 ]. Для расчета эффективных коэффициентов поперечной
290
диффузии и теплопроводности по газовой фазе можно рекомендовать
следующие приближенные формулы:
D 7 (VII-46)
х И ’ х 11
Ввиду различия в механизмах массо- и теплопереноса в зернистом
слое возможны случаи, когда реактор с неподвижным слоем следует
рассматривать как аппарат идеального вытеснения по веществу и
неполного смешения по теплу. В этом случае уравнение (VII.45)
в системе (VII.44), (VII.45) заменяется на уравнение
(V.1.47)
Приближенное значение %ц определяют из формулы, аналогич-
ной (VII. 46):
Хц=у-^ (VI 1.48)
Более детально вопрос о значениях коэффициентов переноса
в зернистой среде рассматривался в главе VI. К этому можно добавить,
что в процессах низкого давления отношение теплопроводно-
стей < 50, так что в уравнении (VII.47) и %ц должны рассчи-
АТв
тываться с учетом теплопроводности по твердой фазе, и при малых
значениях критерия Рейнольдса (Re < 50) можно принять, что
= XII • Для давлений Р > 10 ат или для Re >• 50, долей переноса
тепла через твердую фазу можно пренебречь и ¥= %ц.
Двухфазная модель реакторов с зернистым слоем. До сих пор
часто в математической модели реакторов члены уравнений мате-
риального и теплового балансов, выражающие скорость химических
реакций, аппроксимируются уравнениями формальной химической
кинетики с некоторыми эффективными значениями кинетических кон-
стант. Недостатками такого приближения, во-первых, является то,
что эффективные константы должны определяться для каждого раз-
мера зерна и каждой структуры катализатора, а, во-вторых, в этом
случае модель обладает слабой экстраполирующей способностью,
особенно для быстрых и сильно экзотермических реакций, где велика
роль процессов переноса.
Модель проточного реактора с зернистым слоем катализатора,
в которой учитываются процессы внутри зерна и на его границе,
фактически представляет собою двухфазную модель, хотя и усред-
няющую условия в каждой фазе. Эта модель включает в себя уравне-
ние, описывающее перенос вещества внутри зерна катализатора,
перенос вещества и тепла между катализатором и потоком, а также
уравнения материального и теплового балансов для потока. Ввиду
достаточно большой теплопроводности материала зерен, последние
можно считать изотермическими и составлять баланс тепла для зерна
в целом.
19*
291
Двухфазная модель реактора с зернистым слоем в одномерном’
приближении без учета изменения объема потока описывается для
сферического зерна системой уравнений
дС,
w----
дХ
Wf 1
962 б
^~)=FBHPi(C*,T)
“П? aFr(T Тс)—a**FH (Т Ткат)
а**(7’-Ткат)=-^
с граничными
при х = 0
условиями
Ct = C°t; Т=Т<>
при 6=0
^L = 0
96
при 6 = 7?
дС1
D** =
(VII.49)
(VII.50)
(VII.51)
(VII.52)
РЛ, (С<-Cf п)
R
о
где w — линейная скорость потока; С{ — концентрация i-ro компо- ?
нента в потоке; С* и С1п— концентрации i-ro компонента внутри и
на поверхности зерна; 0 — коэффициент массопередачи к поверх-
ности зерна; FB — наружная поверхность катализатора, отне-
сенная к единице длины слоя; D** — эффективный коэффициент
диффузии в порах катализатора; 6 — координата по радиусу зерна j
катализатора; FBB — внутренняя поверхность катализатора на еди- j
ницу объема зерна; pz — скорость образования i-ro вещества, отне-
сенная к единице внутренней поверхности зерна; у — теплоемкость
потока; Т — температура потока; а — коэффициент теплоотдачи ’
к поверхности теплоотвода; FT — удельная поверхность теплоотвода;
Тс — температура стенки (поверхности) теплоотвода; а** — зффек-
тивный коэффициент теплоотдачи к поверхности зерна; Ткат — тем- * 1
пература зерна катализатора; R — радиус зерна; р} — скорость ‘
j-й реакции. k
Систему уравнений (VI 1.49)—(VI 1.52) решают численными мето- J
дами на ЭВМ. \ 4
В качестве примера применения расчета реактора с зернистым • ]
слоем по двухфазной модели рассмотрим расчет реактора для силь- '
но экзотермической реакции окисления о-ксилола во фталевый ангид-
рид, приведенный в работе [20]. <
292 ‘ ‘
Кинетическая схема процесса имеет вид:
А -±~^в
где А — о-ксилол; В — фталевый ангидрид; С — СО2 + Н2О.
Система кинетических уравнений
имеет вид:
-'д=(*14-М*’а/’о1 (VIL53)
гв = ^1Ла^>о2 ^з^в^Оа (VII.54)
ГС = ^'РАРОа + ^вРО. (VII.55)
Здесь Р( — парциальное давление реа-
гента; rt — скорость реакции.
В присутствии большого избытка
кислорода каждая из реакций описы-
вается псевдопервым порядком.
Поскольку в системе уравнений
(VI 1.53)—(VI 1.55) два ключевых веще-
ства и скорости реакции отнесены к
единице веса катализатора, то система
уравнений (VI 1.49)—(VI 1.52) принимает
вид
Фа=РМра-'Ра) (VII.56)
^в=Р^н(Рв-рв) <VIL57)
’ Фа=^а; Фв=^в (vii.58)
uY - aFT (Г - Tc) = a**FH (Т - Ткат)
, (VII.59)
(R
-hi j rB62rf6+ ^2 J [ (VII.60)
0 0 J
где ф, — левые части уравнения (VII.49); <pz — левые части уравне-
ния (VII.50); £—плотность катализатора.
Коэффициенты а, а** и (J в уравнениях (VII.56) и (VII.60) рассчи-
тывают по значениям критериев Re и Рг для зерен катализатора диа-
метром 3 • 10~3 м, а эффективный коэффициент диффузии определяется
по формуле
D**=^DM
где 8 — порозность частиц катализатора; к' — коэффициент извили-
стости пор; Dn — 0,795----молекулярный коэффициент диффу-
зии о-ксилола в воздухе.
Длина реактора
Рис. VII. 12. Зависимость кон-
версии о-ксилола, выхода
фталевого ангидрида и селек-
тивности процесса от длины
реактора.
-----расчет по двухфазной модели
(температура бани 375° С);----
— расчет по квазигомогенной мо-
дели (температура бани 350° С).
R }
293
На рис. VII.12 приведены кривые значений конверсии о-ксилола
во фталевый ангидрид по длине реактора для трубчатого реактора
диаметром трубки 25 мм, рассчитанные по однофазной и двухфазной
модели. Для обоих случаев принято, что D** = Q,Q^D№. В обоих
случаях расчет проведен для температуры на 15 град ниже темпера-
туры теплового взрыва реактора, определенной для соответствующей
модели. Как видно из рис. VII.12, расчет по двухфазной модели
показывает возможность увеличения выхода ангидрида с 0,57 до
0,66 за счет повышения рабочей температуры процесса.
VII.6. КАТАЛИЗАТОРЫ С ПЕРЕМЕННОЙ АКТИВНОСТЬЮ
И РЕГЕНЕРАЦИЯ КАТАЛИЗАТОРОВ
Нестационарная активность катализатора. Свойства большей
части (если не всех) реальных катализаторов меняются со временем,
и при этом изменяются показатели процесса.
Обычно работу катализатора делят на три периода: разработку,
стационарный период и падение активности. Исследователь и техно-
лог заинтересованы в максимальной длительности стационарного
периода, для которого обычно и ведется расчет реактора и выбор
оптимального режима. В ряде производств, однако, приходится
применять катализаторы, непрерывно меняющие свою активность и
все расчеты проводить для нестационарного режима.
Возможны две формы падения активности катализатора в непо-
движном слое. Во-первых, изменение активности может происходить
равномерно во всем объеме реактора, что, очевидно, означает незави-
симость скорости падения активности от нагрузки на катализатор *.
Такое изменение катализатора, определяемое только временем его
работы, называют старением. Если ход изменения во времени всех
кинетических характеристик катализатора известен, расчет процесса
на стареющем катализаторе для каждого момента времени может
быть проведен по тем же формулам, что и расчет обычного процесса.
При неизменном составе исходной смеси и режиме процесса состав
реагирующего потока на выходе аппарата меняется; в некоторый мо-
мент, когда активность катализатора или избирательность процесса
падают настолько, что дальнейшая работа становится невыгодной,
процесс останавливают и катализатор заменяют или регенерируют.
Скорость падения активности может зависеть от локальной
скорости процесса в данной точке; такое изменение называют утомле-
нием. Это явление наиболее ярко выражено при необратимом отрав-»
лении или блокировании активной поверхности продуктами реакции.
При этом скорость падения активности обычно весьма велика и наи-
более сильно ощущается нестационарный характер процесса. По-
скольку механизмы отравления и характер изменения кинетических
характеристик катализатора разнообразны и большей частью недо-
статочно известны, нерационально стремиться к максимальной общ-
* Этот случай впервые исследован Тодесом [21].
294
ности описания процесса на утомляющемся катализаторе (недости-
жимой и в силу математической сложности проблемы). Рассмотрим
простой и практически важный случай. Будем считать, что в аппарате
протекает единственная необратимая реакция и превращение одного-
моля исходного вещества ведет к выводу из строя (например, в ре-
зультате осаждения твердых продуктов реакции) активной поверх-
ности площадью ц', что равносильно выводу из строя объема слоят
равного ц = |л'/ао (гДе ао — активная поверхность единицы объема
неотравленного катализатора). Величину ц будем называть эффек-
тивным коэффициентом падения активности. Если каждый участок
поверхности работает до полного исчезновения активности, то,
очевидно, в реакторе объемом V можно превратить в конечные про-
дукты Е/ц молей исходного вещества. Время работы реактора t* со-
ставит
t»=V/nCop (VII.61)
где v — объемная скорость потока.
Из уравнения (VII.61) следует, что для продления периода работы
катализатора необходимо увеличивать объем слоя; оптимальное ре-
шение достигается с учетом экономических соображений.
Приближенная теория процесса на утомляющемся катализатора
исходит из допущения, что в каждый момент времени реакция про-
текает только в тонком слое близ границы, разделяющей уже отрав-
ленную и еще работоспособную части реактора. Это предположении
оправдывается с тем большей точностью, чем интенсивнее идет реак-
ция на неотравленном катализаторе и чем быстрее происходит era
отравление. Локализации процесса в тонком слое способствует и
разогрев зоны реакции при адиабатическом протекании реакции
с положительным тепловым эффектом. Нетрудно убедиться, что с мо-
мента начала работы реактора через время t координата работающего-
слоя равна
%макс=Р,^'0^и’ (VII.62^
где w — фильтрационная скорость потока.
Это выражение аналогично формуле для скорости адсорбционной
волны. Процесс дезактивации заканчивается, когда «волна утомле-
ния» доходит до конца реактора.
Реакция первого порядка. Для реакции первого порядка, про-
текающей на утомляющемся катализаторе в изотермических усло-
виях (или для реакции, скорость которой не зависит от температуры)г
можно дать точное математическое описание, не использующее пред-
положения о локализации процесса в тонком слое. Найдем распре-
деление концентрации исходного вещества и активности катализа-
тора в реакторе в любой момент времени, используя методы, развитые
в теории хроматографического процесса [22].
Процесс описывается двумя уравнениями: кинетическим и урав-
нением изменения активности катализатора. Так как концентрация
исходного вещества С и активность катализатора, характеризуемая
295
константой скорости реакции к, отнесенной к единице объема реак-
тора, зависят от двух переменных — времени t, прошедшего с момента
начала процесса, и расстояния от входа в реактор X, — оба эти
уравнения будут уравнениями в частных производных:
wdC/dX = — кС
k^dkjdt^ — \kkC
(VI 1.63)
(VI 1.64)
Вывод уравнения (VI 1.64) требует некоторых пояснений. Если
в единице объема реагирует kCdt молей исходного продукта, то при
этом выходит из строя поверхность do = y,'kCdt. Соответствующее
изменение к равно dk = —х0 do. Если незаблокированные участки
поверхности продолжают оставаться активными, то величина хс не
меняется со временем и сохраняет свое начальное значение х0 —
— &0/о0. Заменяя pi.'/cr0 на эффективный коэффициент падения актив-
ности ц, приходим к уравнению (VII.64). В уравнение (VII.63) не
входит производная концентрации по времени. Это связано с тем,
что существенное изменение активности катализатора происходит,
как правило,- на временах, значительно превышающих время кон-
такта. Поэтому основная реакция протекает квазистационарно, так
что член с производной концентрации по времени пренебрежимо мал
и может быть отброшен.
Уравнения (VII.63) и (VII.64) должны быть проинтегрированы
с граничными условиями
при X = 0, t О
при t = О, X О
С = С0
к=кд
(VI 1.65)
Приведем систему уравнений (VII.63), (VII.64) к безразмерной
форме, введя новые переменные с = С/Со, у — к/кй, х — k^X/w,
т =|xC'oAot Тогда задача (VI 1.63)—(VI 1.65) сведется к удобному виду,
не содержащему параметров:
дс!дх=—ус (VII.66)
ду1дх——ус (VI 1.67)
при х = 0, т Э1 0 .
(VII.68)
при т = 0, х 0 i/=l
Вычитая уравнение (VII.67) из (VII.66), получаем:
дс/дх — ду]дх = О (VI 1.69)
Из уравнения (VII.69) следует, что существует некоторая функция
Ф (х, т), обладающая тем свойством, что
с — дФ/дХ', у = дф/дх
(VII.70)
296
Действительно, подстановка уравнения (VII.70) в (VII.69) обра-
щает последнее в' тождество. Подставляя уравнения (VII.70) в любое
из уравнений (VII.66), (VII.67), приводим его к форме:
Э2Ф . ЭФ ЭФ
дхдт ' дх дт
(VII.71)
Это уравнение решаем, вводя новую вспомогательную функцию
Т (х, т), определяемую соотношением:
O = lnV (VII.72)
С помощью подстановки уравнения (VII.72) в (VII.71) последнее
обращается в уравнение типа волнового
d2V/5x5T = 0 (VI 1.73)
решение которого имеет вид суммы двух функций / (х) и <р (т), каждая
из которых зависит только от одной переменной. Функция Ф (х, т),
следовательно,- имеет вид:
Ф (х, т) = 1и [/ (х) + <р (т)] (VII.74)
Функции / (х) и ф (т), входящие в формулу (VIL74), должны быть
определены из граничных условий. Последние, согласно выражениям
(VII.74), (VII.70) и (VII.68), записывают в виде
/о+ф ’ dx
1___tf_=1
/4-фо dx
(VII.75) .
(VII.76)
где /о и ф0 — значения функций / (х) и ф (т) соответственно при х = О
и т = 0.
Условия (VII.75) и (VII.76) — обыкновенные дифференциальные
уравнения, интегрирование которых дает:
ф--/о + (/о+фо)ех (VII.77)
/ = -фо + (/о + фо)е* (VII.78)
Подставляя (VII.77) и (VII.78) в (VII.74), получаем:
Ф(я, т)=1п(/о + фо)+1и (е*4-ех — 1) (VII.79)'
Функция Ф (х, т), как видим, определена с точностью до произ-
вольного постоянного слагаемого, стоящего как^первый член в пра-
вой части (VII.79); для решения нашей задачи этого, очевидно, доста-
точно. Дифференцируя (VII.79) по т и х, находим с помощью формул
(VII.70) поле концентрации исходного вещества и активности катали-
затора в реакторе в любой момент времени:
r (VII.80)
(VII.81).
297
Из формул (VII.80) и (VII.81) видно, что, как это и требуется по
физическому смыслу задачи, концентрация исходного вещества яв-
ляется в каждый момент времени монотонно убывающей функцией
координаты х и в каждой точке реактора (в том числе и па выходе) —
монотонно возрастающей функцией времени т. Безразмерная кон-
станта скорости реакции у монотонно убывает с увеличением т при
фиксированном х и монотонно возрастает с увеличением х при фикси-
рованном т. Отношение скорости реакции кС в сечении с координа-
той х в момент т к максимальной скорости реакции к0С0 (скорости
реакции во входном сечении при т = 0), согласно (VII.80) и (VII.81),
равно:
кС е* + т gk'X/w+lbk'Cvt
-ус~ (ех+ ег _ i)2 — (efe„x/w+ep.feoCot _ (VII.82)
Чтобы найти координату жмакс сечения, в котором скорость
реакции максимальна в данный момент т, надо приравнять к нулю
частную производную по т правой части уравнения (VII.82). Диффе-
ренцируя, приходим к уравнению
ет_/макс_1 = 0
откуда:
^макс=1п (е'1 —1); Хмакс = ^-1п(^»Сог-1) (VI 1.83)
Из уравнений (VII.83) следует, что максимум интенсивности про-
цесса остается в лобовом сечении вплоть до момента т0 = 1п 2. [жмакс,
определенное из (VII.83) при г < тй, меньше нуля, т. е. до этого мо-
мента времени скорость реакции монотонно убывает с ростом х].
При г > т0 скорость перемещения максимума уменьшается со време-
нем; при больших г, когда ё1 1, формула (VI 1.83) становится
(после перехода к размерным переменным) неотличимой от формулы
(VII.62), а скорость перемещения максимума постоянной и равной 1.
Чем больше Соц и к0, тем ближе значения Хмакс, определенные из
уравнений (VII.62) и (VII.83), что подтверждает сказанное ранее об
условиях, способствующих локализации процесса в тонком слое.
Скорость реакции в точке ее максимальной интенсивности получаем,
подставляя (VII.83) в (VII.82):
1
(?/с)макс “ (1-е-г)-1 (VI 1.84)
Максимальная скорость реакции, таким образом, убывает со вре-
менем, асимптотически приближаясь к величине х/4 к0С0. Зависи-
мость концентрации непрореагировавшего исходного вещества от
продольной координаты в различные моменты времени представлена
на рис. VII. 13 *.
* Численные расчеты режимов неизотермической реакции на отравля-
ющемся катализаторе проведены в работе [23].
298
Сканировал: ЧерЙШУ
Магнитогорск
2008
Рис. VII.13. Зависимость концент-
рации исходного вещества от коор-
динаты реактора для различного'
времени работы катализатора.
Регенерация катализатора, проводимая обычно продувкой ката-
лизатора кислородсодержащими газами, является нестационарным
процессом, формально аналогичным процессам на катализаторах с па-
дающей активностью *. Она может протекать в кинетическом или
диффузионном режиме. Первый случай характерен для относительна
низких температур (порядка 400—500° С) и высокополимерных, аро-
матического характера углеродсодержащих отложений. В кинетиче-
ском режиме скорость реакции
равна
гр = ^“2С|г (VI 1.85)
где СОе ж Суг — концентрация ки-
слорода в газовой фазе и углерода
в катализаторе.
Константа скорости реакции в
уравнении (VII.85) имеет обычный
аррениусовский вид, а значения
энергии активации составляют
обычно 50 000—70 000 кал/молъ.
Часто для регенерации приме-
няют значительные избытки воз-
духа или воздуха в смеси с водя-
ным паром, так что концентрацию
кислорода можно считать постоян-
ной по всей длине реактора. Тогда
процесс регенерации в кинетичес-
кой области может быть описан
квазигомогенной моделью как пе-
риодический для всего реактора в целом системой из двух уравне-
ний — материального и теплового балансов. Решение этой системы
вполне аналогично системе (VII.25), (VII.26) или (VI 1.49), (VII.50)
для реактора идеального вытеснения. Условия устойчивости и пара-
метрической чувствительности здесь также аналогичны периодичес-
кому реактору или реактору идеального вытеснения и рассматри-
ваются в главе VIII.
В случае незначительного избытка кислорода и значительного
изменения его концентрации по длине слоя катализатора для расчета
регенерации при первом порядке по кислороду (наиболее частный
случай) следует использовать систему уравнений (VII.63), (VII.64)
и вытекающие из нее последующие выражения. При этом вместо
величины к и к0 следует подставить величины кС^ и &0£уг
Если процесс регенерации проводят при более высоких темпера-
турах, порядка 550—700° С или углеродистые отложения весьма
реакционны, то регенерация протекает в диффузионной области.
В зависимости от структуры зерна и скорости потока при этом может
* Обзор по кинетике окислительной регенерации дан в работе [24].
299
преобладать внутреннее или внешнее диффузионное торможение. 1
В таком случае процесс сосредотачивается в более или менее'коротком |
участке слоя катализатора, перемещающемся со временем вдоль 1
потока. Для расчета режима регенерации при этом можно использо- 1
вать методику, предложенную Тодесом [25] для расчета движущейся |
адсорбционной волны.
В этом случае переменные t — астрономическое время ъ х — длина -i
слоя катализатора заменяют новыми переменными t и у = х — wt j
(где w — скорость движения фронта выгорания). Такая замена зкви- ]
валентна переходу к новой системе координат, которая движется
относительно слоя катализатора со скоростью w в направлении дви-
жения потока. В этой системе возможно стационарное решение урав- '
нений материального и теплового балансов, которое, опуская выводы,
имеет вид:
иеС®,
“’ = usC»i + (li8) С»г (VII.86)
„ «Ъ (*~т)
(VII.87)*
Г и
«Р=4 , <vi1-88)
где и — линейная скорость потока; е — порозность слоя катализа-
тора; у — теплоемкость газа; у — теплоемкость единицы объема слоя
катализатора; X — теплопроводность слоя катализатора; h — теп-
лота выгорания углеродистых отложений; С®', — концентрация
кислорода на входе в реактор; С®г — начальная концентрация углеро-
дистых отложений в катализаторе; F — внешняя поверхность тепло-
обмена; АГ = Т — Тс — перепад температуры между слоем
и стенкой реактора; tp — время регенерации; L — длина слоя
катализатора.
В формулу (VII.87) входит величина и7макс, которая представляет
собою скорость движения фронта тепловой волны, и приближенно
определяется выражением:
а’макс = 11 (VII.89)
V
Величина АТ1 дает возможность оценить степень перегрева зерна 4
катализатора. Если последняя превышает допустимые пределы, то
следует изменить либо концентрацию О2, либо предельную степень
зауглероживания катализатора С®г. Обе зти величины можно опре-
делить из уравнений (VI 1.86) и (VI 1.87), задавшись величиной АГ.
* Формула (VII.87) выведена для случая, когда температура входящего
газа равна Тс. Вывод уравнений (VII.86) — (VII.89) проведен совместно о Гур-
фейном и Михлиной.
300
Если углеродистые отложения практически полностью забивают
поры, то процесс выгорания может идти либо во внешнекинетическом,
либо в диффузионном режиме, но в обоих случаях описывается урав-
нением топохимической реакции газ — твердое тело. При избытке
кислорода для сферического зерна это уравнение имеет вид
ГО Га
Л*
где t — время выгорания; R — радиус зерна; к* — эффективная кон-
станта скорости выгорания углеродистых отложений.
VII.7. РЕАКТОРЫ С ДВУХФАЗНЫМ ПОТОКОМ
(ДЛЯ ЖИДКОФАЗНЫХ РЕАКЦИЙ)
Принципиальные отличия жидкофазных реакций от газофазных
проявляются тогда, когда поток реагентов становится двух- или
многофазным, т. е. когда реакции на катализаторе протекают между
жидкостью и газом или двумя несмеши-
вающимися жидкостями. Здесь будем рас-
сматривать только эти случаи, поскольку
к однофазному потоку жидкости прило-
жимы закономерности и уравнения, выведен-
ные для газофазных гетерогенно-каталйти-
ческих процессов. Для качественного рас-
смотрения таких процессов можно принять
за основу сумму явлений, происходящих при
газо-жидкостной реакции типа А + В С
на твердом катализаторе, когда вещество А
находится в газовой фазе, В — в жидкости,
соответствующей таким реакциям, как гид-
рирование, алкилирование ароматики низ-
шими олефинами и т. п. На рис. VII. 14 зти
процессы представлены в виде схемы. Газ А
для того, чтобы достигнуть твердой поверх-
ности, на которой протекает реакция, дол-
жен перейти из дисперсной (газовой) фазы
в сплошную (жидкую), т. е. раствориться в
ней. После этого газ А должен продиффундировать
капилляров в зерне катализатора и наконец сорбироваться на по-
верхности катализатора. Вещество В из жидкой фазы должно только
продиффундировать через капилляры и затем сорбироваться на
поверхности. Образовавшееся на последней вещество С должно десор-
бироваться и, продиффундировав через капилляры, выйти в объем
жидкости или газа.
Таким образом, в многофазных жидкостных системах возни-
кает принципиально новая стадия — стадия межфазной диффузии,
способная частично или полностью определять общую скорость
301
Газовая
дюза
5 g
I е
Й g
Жидкая |
(раза сЭ g
Рис. VII. 14. Схема пе-
реноса
торе
с
веществ в реак-
двухфазным по-
током.
через систему
каталитического процесса. Скорость этой стадри зависит как от по-
верхности контакта между фазами, так и от величины эффективных
коэффициентов диффузии компонентов реакции из одной фазы
в другую.
Из сказанного следует, что в случае многофазной газо-жидкостной
реакции происходит значительное число физических процессов,
осложняющих собственно химическую реакцию. Ход же реального
процесса, как обычно, определяют соотношения между скоростями
отдельных его стадий. Для рассматриваемого случая ограничимся
в начале стационарным изотермическим процессом, протекающим
в реакторе непрерывного действия, и напишем для него систему урав-
нений материального баланса веществ нахединицу сечения реактора:
[(1 _ 61 _ е2) «ж^Ая: — (1 — ex — s2)Z>; =
= Р1о (W^-^-Wa^Ah) (VII.90)
+ G’-A, В,С) (VII.91)
k (J 1
--^(ещгС( Г) = ^№С, ж-С, г) (i = A, в, C) (VII.92)
CIA
[\1 ~ 81 — s2) ИЖС,- ж - (1 - 81 - e2)Z); =
= /Жж-4н) О = в, С) (VII.93)
Здесь X — координата вдоль реактора; ех и е2 — доли сечения реак-
тора, занимаемые газом и катализатором; иж и иг — линейные ско-
рости потока в жидкости и газе; D* — эффективный коэффициент
продольной диффузии в жидкой фазе; — коэффициент массопере-
дачи между фазами; о — поверхность раздела фаз в единице объема
реактора; ф — обратное значение коэффициента Генри; FH — удель-
ная внешняя поверхность катализатора в пересчете на единицу
длины реактора и единицу сечения; D** — эффективный коэффициент
диффузии в капиллярах катализатора; б — координата по радиусу
зерна; Ръ.л — внутренняя поверхность зерна катализатора; р — ско-
рость реакции по компоненту А в пересчете на единицу внутренней
поверхности катализатора; vB, vc — стехиометрические коэффи-
циенты (уА —1).
Граничные условия системы уравнений (VII.90) — (VII.94) отли-
чаются для случаев прямотока и противотока.
Для прямотока:
А=0; сАг=с°Аг; СВж=С°Вж; сСж=о
« = 0; -^2-=0
ab
302
б — /?; C[ BH — C{ H
X—H, сл, сAr ВЬ1Х, СВж = СВж выхСсж = сж_ вых
Для противотока:
Х — 0, сАг с»Аг, сВж=сВж_ вых сСж= сСж_ вых
Х—Н', САг= САг. вых‘> ^Вж= <'Вж<-’сж = 0
Остальные условия те же, что для прямотока.
Система уравнений (VII.90)—(VII.94) является общей для непре-
рывных процессов, т. е. «открытых систем». Однако значения входя-
щих в нее параметров различны для разного конструктивного типа
реакторов. Это связано с особенностями протекающих в них физи-
ческих процессов. Рассмотрим три типа реакторов: прямоточные
трубчатые или колонные реакторы с неподвижным катализатором,
те же реакторы с суспендированным катализатором, непрерывно
действующие мешалки с суспендированным катализатором. Переход
от расчета непрерывных реакторов к реакторам периодического дей-
ствия с суспендированным катализатором не сложен.
Анализ уравнений (VII.90)—(VII.94) в дополнение к уже изло-
женному указывает еще на ряд особенностей макрокинетики реакций
в двухфазной системе.
Первая связана с различием в скоростях движения сплошной
(жидкой) и дисперсной (газовой) фаз.
Для двухфазных газо-жидкостных и жидкость-жидкостных си-
стем величина иг для дисперсной фазы определяется не объемной
скоростью потока, а зависит от гидродинамических режимов потоков.
Области существования последних определяются отношением объем-
ных скоростей дисперсной и сплошной фаз. Для реакций под повышен-
ным давлением, которое обычно применяется в случаях газо-жидко-
стных каталитических реакций, наиболее часто встречается режим
пузырькового течения. В этом случае скорость всплывания пузырей
определяется разностью плотностей сплошной и дисперсной фаз,
диаметром пузыря, зависящим от типа и размера распределительного
устройства и от величины поверхностного натяжения на границе
раздела фаз. В качестве примера формулы, видимо, приемлемой для
расчета колонных аппаратов с суспендированным катализатором,
можно привести приближенную формулу для скорости всплывания
пузырьков в объеме жидкости при ламинарном движении [26 ]
где dn — диаметр газового пузыря, ж; g — ускорение силы тяжести,
м/с^к2-, рж — плотность жидкости, кг/л(3; рж — коэффициент 'динами-
ческой вязкости жидкости, кг/(ж-ч).
303
В свою очередь, диаметр пузыря, согласно [26], можно опреде-
лить по приближенной формуле
s/6a*rfc
=^= 1/ '
г pxg
(VI 1.96)
где dc — диаметр отверстия сопла (распределительной решетки), м;
о* — поверхностное натяжение суспензии катализатора (в н/м), ко-
торую в первом приближении ввиду малой концентрации катализа-
тора, можно приравнять поверхностному натяжению жидкости.
Если иг найдена, то ех можно определить из выражения
Ё1 = ^- (VII.97)
где Wr — объемный расход газа; Рр — полное сечение реактора.
Для случая суспендированного катализатора можно принять,
что е2 = 0, и тогда определяется по формуле
(VII.98)
/'рЦ—ех)
где Wx — объемный расход жидкости.
Поскольку объем газа меняется по высоте слоя жидкости за счет
поглощения реагента и изменения гидростатического давления, то
система уравнений (VI 1.90)—(VII.95), (VII.97) должна быть допол-
нена для случая большой степени поглощения газа или значительных
изменений гидростатического давления уравнением
Wr=
х I Р
WOr-Fpm^A (С) dX ^2-
о J р
(VII.99)
где Wor — объем газовой фазы на входе в реактор; т — коэффициент
пересчета внутренней поверхности катализатора на единицу объема
реактора; рд (С) — скорость реакции по компоненту А на единицу
внутренней поверхности катализатора; С — вектор концентрации
компонентов реакции; Ро, Рр — начальное и текущее гидростатиче-
ское давление в реакторе, а также уравнением
3 / IV
= у -jj— (VII.100)
где d°n — диаметр пузыря на выходе из сопла (решетки), рассчитай-
ный по уравнению (VI 1.96).
Для неподвижного зернистого слоя катализатора, утопленного
в жидкости, по данным [27 ], газосодержание
<Р=-Ат (VII.101)
1 — ь2
не зависит в первом приближении от скорости и физических свойств
жидкости. Это объясняется, видимо, тем, что газовые пузыри, подни-
304
маясь по насадке, принимают средний размер каналов между части-
цами катализатора. Другими словами
dn S ^экв
где tZ3KB — эквивалентный диаметр частиц катализатора.
В соответствии с этим с точностью до ±15% наблюдается корре-
ляция
<р __ 1
1-ф “1 + 1,3 KFrz
(VI 1.102)
где Fr2 = ----критерий Фруда для насадки; и-г — фильтра-
£“экв
ционная скорость газа, т. е. скорость газа, рассчитанная на полное
сечение реактора.
Из уравнений (VII.101) и (VII.102) рассчитывают ех при известном
е2 (обычно е2 = 0,35—0,45), а затем определяют и иг. При сильно
меняющемся объеме газа надо учитывать уравнение (VII.99).
При проведении реакций в непрерывно действующих аппаратах
с твердым катализатором необходимо учитывать зависимость коэф-
фициента продольной диффузии от скорости движения дисперсной
фазы.
По Железняку и Ландау коэффициент продольной диффузии
в барботажных колоннах удовлетворительно описывается выраже-
нием
1g^-=0,816 + 0,987 1gВеэкв+0,814 lgei+3,891g р (VII.103)
'’ж
где D* — эффективный коэффициент диффузии в сплошной фазе
(жидкости), см^/сек', vx — коэффициент кинематической вязкости
жидкости, см2/сек', ц = | +3р/ (гдер/ и ц " — коэффициенты дина-
мической вязкости соответственно для сплошной и дисперсной фаз).
Критерий Re3KB вычисляют для приведенного диаметра, учиты-
вающего линейные размеры аппарата и газовых пузырей и вычисляе-
мого по формуле
Пп = -PpCl-ei) (VI 1.104)
где Пр — диаметр реактора.
При этом надо иметь в виду, что, как указывалось, объемный рас-
ход дисперсной фазы Wr определяют по уравнению (VII.99).
Формулами (VII.103) и (VII.104), видимо, можно пользоваться
для случая колонных аппаратов с суспендированным катализатором,
где доля катализатора в общем реакционном объеме незначительна.
Для случая неподвижного слоя катализатора в работе 128] предло-
жены формулы
/ D v-0,31
Реж = 0,11 (Вег)°’4Б (Кеж)0’47 -у2- (VII.105)
\ ®экв /
20 Заказ 1441
305
где <7экВ — эквивалентный диаметр зерна катализатора. ’
Рр _шжбгэкв. О _ “ж^эквРж . р «г^эквРг _ /VTT <АС\ ’
1Сж ~ R1D* ’ Н ж~ ’ г“ еП?’ (VII.106)
Существенные параметры, в значительной мере определяющие 1
макрокинетику каталитических реакций в жидкой фазе, входят в 1
члены уравнений (VII.90)—(VII.94), описывающие массообмен между
газовой и жидкой фазами. В них входят, кроме концентраций, три я
величины а, 0 и ф, определение которых в достаточной степени J
сложно. |
Если принять за основу формулу (VII.96) или (VII.100) для |
приближенной оценки dn, то удельная поверхность контакта фаз *
определится выражением [261
6Wrff
0 =---5--
Urwn
(VII. 107)
где Н — высота слоя жидкости.
Недостатком уравнений (VII.95)—(VII.107) является неучет коа-
лесценции и дробления пузырей. Хотя имеются предложения в отно-
шении правил учета этих явлений на основе статистико-вероятно-
стных представлений [29], однако эта методика пока не проверена
и достаточно сложна.
Значения коэффициента ф в настоящее время определяют экспе-
риментально. Сложность, вносимая этим коэффициентом в решение
уравнений макрокинетики газо-жидкостных реакций, заключается
в том, что в общем случае он не остается неизменным в течение реак-
ции, как это принималось и принимается сейчас во многих работах,
а зависит от физических свойств реагентов и продуктов реакции и,
следовательно, от глубины протекания реакции.
Зависимость ф от состава реакционной смеси должна определяться *
экспериментально. Удобнее всего это делать в зависимости от глу-
бины превращения реагентов.
Поскольку в аппаратах с твердым катализатором реакция идет
на поверхности последнего, то перенос массы на границе фаз протекает
в отсутствие химической реакции. Поэтому для определения значе-
ний коэффициентов межфазного переноса в аппаратах с суспендиро-
ванным катализатором, где велика доля жидкой фазы, инертной в
отношении химической реакции, можно пользоваться формулами,
принятыми для расчета массообменных аппаратов, например прй- «
веденными в монографии [30]. Для неподвижного слоя .катализатора i
за неимением более точных и обоснованных выражений в первом при-
ближении, видимо, можно к значениям коэффициентов переноса,рас-
считанным без учета химической реакции, вводить поправочные коэф-
фициенты, учитывающие этот последний фактор ацалогично тому, как
это делается, например, в работах [31] и [32].
Правая часть уравнения (VII.93) описывает скорость массопере-
носа к поверхности катализатора. Она содержит кроме легко опреде-
ляемой величины удельной внешней поверхности катализатора F„
306
еще значение коэффициента массообмена р2 между твердой поверх-
ностью и жидкостью при наличии газовых пузырей в потоке.
Для суспендированного катализатора вследствие того, что
Л,₽2»оф (VI 1.108)
4
вопрос о транспорте вещества к, поверхности частиц катализатора
может не рассматриваться. Для неподвижного зернистого слоя ката-
лизатора обоснованным, видимо, является использование формул мас-
сопереноса к поверхности, применяемых в расчетах процессов рас-
творения.
Чтобы представить в явном виде уравнения (VII.90)—(VII.94)
нужно определить коэффициенты D** и раскрыть структуру зависи-
мости скорости поверхностной реакции от вектора концентраций.
Следует отметить, что приведенные выше выражения для расче-
тов dn, Vr, 0 и а дают недостаточно точные результаты. Поэтому
в практических целях желательно зти величины определять экспе-
риментально.
Для случая неизотермических реакторов система уравнений
(VII.90)—(VII.94) должна быть дополнена уравнениями теплового
баланса для каждой фазы. Они аналогичны уравнениям матери-
ального баланса,но в уравнении для сплошной фазы присутствует
член, учитывающий теплообмен с внешней средой.
Система уравнений (VII.90)—(VII.94), тем более дополненная
уравнениями теплового баланса, слишком сложна даже для числен-
ных решений на современных ЭВМ. Поэтому систему уравнений
(VII.90)—(VII.94) неизбежно приходится упрощать. (Ошибки в опре-
делении коэффициентов модели обычно значительно превосходят
неточности от упрощения модели). В первую очередь, сплошную
фазу с катализатором рассматривают как квазигомогенную, анало-
гично тому, как это делается для однофазных реакторов с зернистым
слоем катализатора. Принимают, что скорость теплообмена между
фазами бесконечно велика. Далее, по возможности, принимается
наличие предельных гидродинамических режимов (идеальное вытес-
нение или смешение) и постоянство объема Потоков и, наконец, если
это допустимо, пренебрегают уносом газом компонентов жидкой фазы.
Тогда для таких простейших случаев в приближении идеального
вытеснения по обеим фазам система уравнений принимает вид (для
реакции А -(- В -> С)
(1-81) «ж = ₽Аа 0|>САж-САг)-г (С) (VII.109)
dCAr „л
®1Ur ~'dX =Р1а (’^Лж-САг)
^Аж ^Вж
dX dx
Vu~^hr (С)-aFT (Г-7C)
21*
307
где h — тепловой эффект реакции; у — усредненная теплоемкость
обеих фаз; и — усредненная линейная скорость потока в целом.
Из рассмотрения системы (VII.109) видно, что помимо кинетики
реакций в активной фазе, на ход и результаты процесса в двухфаз-
ном потоке влияет диффузия реагентов между фазами и внутри актив-
ной фазы. Как и в процессах с однофазным потоком и твердым катали-
затором для процессов в двухфазном потоке возможны следующие
предельные области.
1. Диффузионная, или точнее межфазнодиффузионная: реакция
в активной фазе протекает очень быстро и процесс лимитируется диф-
фузией реагентов из пассивной фазы в активную.
2. Кинетическая или точнее внутрифазнокинетическая скорость
процесса всецело определяется кинетикой реакций в активной фазе.
Однако в отличие от однофазных потоков значительное превыше-
ние объемного коэффициента массопередачи (0о) над константой ско-
рости реакции является только необходимым, но не достаточным
условием протекания процесса в кинетической области. Действи-
тельно для предельного случая к —> 0, соответствующего абсорберу,
из уравнения материального баланса следует, что в кинетической
области, т. е. при Саж = Сдж. РавН будет иметь место следующее
равенство:
(VI 1.110)
Из уравнения (VII.110) можно вывести, что кинетическая область
в прямоточных реакторах может наступить лишь при условии:
Wx 1
Wr ' ip
(VII.111)
Отсюда для колонных прямоточных аппаратов чисто кинетическая
область достигается только на высоте одной теоретической тарелки,
а в противоточных аппаратах она вообще практически не дости-
гается.
Рассмотрим теперь системы уравнений для непрерывных и полу-
периодических мешалок. При этом примем перемешивание очень
энергичным, в результате чего происходит идеальное смешение по
обеим фазам. Тогда для непрерывной мешалки и реакции А -|- В -> G
в квазигомогенном приближении для реакционной фазы будем иметь
систему алгебраических уравнений
И7» (Саож-^аж) = Г (С)-0КАж~САж)] Й
^г(САог-Саг) = -₽^ОКАж-САж) Й (VII.112)
W'«(CBo!k-M=-V(C)9
(T—T0) = hr (С) Q-aTFT (Т-Тс)
где £2 — объем активной фазы.
Для полупериодической мешалки с непрерывной подачей газа
система уравнений будет состоять из алгебраических и дифферен-
308
циальных уравнений. Поскольку непрерывные и полупериодические
мешалки работают практически только с суспендированным катали-
затором, к ним приложимы соображения, изложенные на стр. 305.
Значения ((За) для некоторых систем мешалок имеются в литературе,
в частности для аппаратов с внутренним циркуляционным контуром
они даны в работе [33 ].
В качестве примера рассмотрим расчет лабораторного изотерми-
ческого колонного реактора гидрирования додецена в растворе тя-
желого бензина на суспендированном никелевом катализаторе [34].
Реактор представляет собою трубу из стекла длиной 2000 мм и
диаметром 76 мм, обогреваемую для компенсации теплопотерь элек-
трообмоткой. За основу была принята модель изотермического ап-
парата идеального перемешивания по жидкости. Степень перемеши-
вания была определена специальными опытами с трассером. Реакция
проводилась при 30—70° С с большим избытком водорода. Давление
водорода на выходе из реактора равнялось атмосферному.
Кинетическое уравнение реакции определялось из опытов при
низких температурах и больших скоростях перемешивания и имело
вид
г = kfCKmCHt 0 + (VI1.113)
где г—скорость реакции, моль)(л-мин)', к — константа скорости
реакции в пересчете скорости реакции на 1 а катализатора,
л/(мин-моль-г катализатора)', / — поправочный коэффициент актив-
ности катализатора для различных его партий; Скат — концентра-
ция катализатора, г/л; Сн2) С — концентрация водорода и до-
децена, моль! л-, КА — адсорбционный коэффициент додецена,
л/моль.
Величины к и КА определялись из опытов при трех разных тем-
пературах и выражались формулами:
к = 4,05- ЮН ехр ( — 15900/Я7) (VII.114)
КА = 4,26- 105 exp (7300/-RT) (VII.115)
С учетом практического постоянства концентраций водорода
в дисперсной и следовательно в сплошной фазах реактора, система
уравнений (VII.112) преобразовывалась к следующему конкретному
виду
---^Нг =Рп(кРН1-СН8) (VII.116)
Wx (C0~C)=VpkfCKaTC^ ——- (VII.117)
\ А /
(VII.118)
где — часовая
объем водорода на
подача жидкости в
17» _ Ц7ВЫХ
22,4 =И7ж(£о-С)
объемная подача водорода; И7™* — часовой
выходе из реактора; И7Ж — часовая объемная
единицу времени; Vp — объем реактора.
309
В системе уравнений (VII.116)—(VII.118) значение коэффици-
ента К находят и из таблицы экспериментальных данных по зависи-
мости растворимости водорода в бензине от температуры и давления.
Давление водорода Рнг принимали равным гидростатическому дав-
лению в середине реактора. Поверхность фазового раздела опреде-
ляли из следующей формулы для сферического пузыря:
Пф.Р = 6^]е (VII.119)
Здесь е — газосодержание (определяли экспериментально по раз-
ности высот реакционной жидкости при подаче водорода и без нее).
Диаметр пузыря dn определяли из экспериментальной кривой
зависимости dn от скорости подачи газа, приведенной в работе [22 ].
Значение 0 определяли по формуле Кэлдербенка и Моо—Юнга [35]
Р Sc*''* = 0,42 [ <Рж —Л (VII.120)
L Рж J
где Sc = -тг---число Шмидта, в котором Z?ab — эффективный коэф-
Дав
фициент диффузии газа в жидкости; рж, рг — плотность жидкости
и газа.
Некоторые результаты сравнения расчетных и эксперименталь-
ных данных приведены в табл. VII. 1.
Таблица VII.1
Сравнение экспериментальных и расчетных данных
производительности лабораторного реактора
гидрирования додецена (по водороду)
№ по пор. Т, °C С, молъ/л 1 ^кат’ г/л Подача додецена, мл/мин Производительность реактора, нл Нг/лин
эксперимен- тальная расчетная
1 40 1,22 17,6 88 1,61 1,44
2 50 0,407 2,6 178 0,083 0,098
3 60 0,407 4,4 87 0,71 0,70
4 70 0,407 4,4 86 0,76 0,69
Как видно из табл. VII.1, расхождения между эксперименталь-
ными и расчетными данными не столь велики и кроме общих неточ-
ностей в определении коэффициентов, видимо, связаны с недооцен-
кой влияния химической реакции на коэффициент межфазного
массообмена р.
VII.8. РЕАКТОРЫ С КИПЯЩИМ СЛОЕМ
В этой книге мы не будем заниматься анализом физических про-
цессов в кипящем слое, связанным с проблемой определения различ-
ных коэффициентов переноса, ограничившись описанием методов
310
кинетического расчета на основе известных характеристик процес-
сов переноса и химической реакции. Неоднородность кипящего слоя
вызывает резкие различия гидродинамических условий и условий
протекания реакций в разных частях потока газа; поэтому можно
говорить о газе, проходящем в пузырях, и газе, просачивающемся
сквозь плотный слой твердых частиц как о двух разных фазах по-
тока газа. В дальнейшем эти две фазы будем называть, соответственно,
пассивной и активной. Пассивная фаза является прерывной, а актив-
ная — сплошной, что иногда используется в качестве их наименова-
ний.
Гидродинамика потока в активной фазе подобна гидродинамике
всего потока в критической точке перехода от неподвижного слоя
к кипящему. В первом приближении можно предполагать, что ско-
рость потока в активной фазе иакт равна критической скорости икр,
а весь избыток газа сверх необходимого для начала псевдоожижения
проходит сквозь слой в пузырях (в пассивной фазе). При этом доля
газа, проходящего в активной фазе, равна (если и — скорость всего
потока газа):
(P = “kp/u (VII.121)
Гидродинамический режим пассивной фазы принято считать близ-
ким к идеальному вытеснению; отклонения от идеальности являются,
главным образом, следствием различия скоростей подъема пузырей
разного размера. Более сложен вопрос о перемешивании потока в ак-
тивной фазе. В плотном слое твердых частиц, при относительно малых
линейных скоростях потока, турбулентные пульсации не играют за-
метной роли и перемешивание потока может быть следствием только
взаимодействия потока с подвижными твердыми частицами. Механизм
перемешивания газа в активной фазе кипящего слоя состоит в увле-
чении твердыми частицами молекул реагентов, находящихся у по-
верхности частиц и внутри пор и адсорбированных на поверхности.
Если основная часть переносимого вещества адсорбирована на по-
верхности частиц, константа равновесия между ядром потока и при-
поверхностным слоем связана с удельной поверхностью частиц о
и сорбционными свойствами реагентов соотношением
= (VII.122)
где Г — сорбционный коэффициент.
При малом Г или о (слабая адсорбция или применение непори-
стых частиц с малой поверхностью) Кр -> <х> и перемешивания
реагентов нрактически не происходит. Различие адсорбционных
коэффициентов различных веществ определяет избирательность
перемешивания потока твердыми частицами. При определенных усло-
виях в кипящем слое может достигаться почти идеальное смешение
по одним реагентам и почти идеальное вытеснение — по другим.
Модели реакторов с кипящим слоем. Как указывалось в раз-
деле VII.2, аппараты с кипящим слоем могут быть секционированные
и емкостные (несекционированные), со стационарным и движущимся
311
слоем. Математические модели будут разными для различных типов
аппаратов, однако в основу их в настоящее время, как правило,
кладутся общие представления о квазидвухфазной физической мо-
дели кипящего слоя, изложенные выше. Рассмотрим сперва модель
емкостного аппарата со стационарным слоем.
Вследствие высокой температуропроводности реакторы с кипящим
слоем являются аппаратами идеального смешения по теплу и тепло-
вой баланс для них составляется в целом по реактору. Поэтому ос-
нову математической модели реакторов составляет система уравне-
ний материального баланса веществ в изотермических условиях.
Общая система уравнений, отнесенная к единице площади по-
перечного сечения реактора, для процесса в кипящем слое, тормо-
зимого как межфазной, так и внешней диффузией к частицам катали-
затора, включает дифференциальные уравнения переноса вещества
из газовых пузырей через поток газа в плотном слое к приповерх-
ностному слою:
(u икр) dCr_ n/dX-f- Роб (Сг_ п Сп_ ф) = 0 (VII.123)
икр^Сп ф/йХ—мКр (Сг п Сп. ф) + РнОн (Сп. ф — Скат/Г) =0 (VI 1.124)
2>кат-^Г + ₽н-^(сп.ф—£^Л+р(Скат)=О (VII.125)
Здесь Сп ф — концентрация вещества в плотной фазе; Сг п — кон-
центрация вещества в газовом пузыре; пкр — объемный коэффициент
массопередачи между фазами; — коэффициент массопередачи
к поверхности катализатора; о и он — соответственно полная и
внешняя удельная поверхность твердых частиц, отнесенная к еди-
нице объема всего слоя; р — скорость образования данного вещества,
отнесенная к единице поверхности катализатора; Скат — двухмер-
ная концентрация вещества на катализаторе; DKaT — коэффициент
перемешивания твердых частиц.
В уравнении (VII.125) не учитывается внутридиффузионное тор- с
можение процесса. Обычно это оправдано, так как размеры частиц
в кипящем слое всегда малы. Принято также, что адсорбция на по-
верхности частиц подчиняется закону Генри с коэффициентом Г.
В случае слабой адсорбции при движении частиц может, тем не
менее, увлекаться вещество, находящееся в порах. Тогда, вместо
уравнения (VII. 125) следует использовать при расчете уравнение
2?кате'd2CKaT/dX24- Рн (Сп. ф — Скат/Г) ан-|-ор (Скат) =0 (VII. 126)
где е' — отношение свободного объема внутри пористых частиц
к полному объему слоя.
Величина Скат обозначает в уравнении (VII.126) уже не двухмер-
ную, а обычную концентрацию вещества в порах. Величина Г в этом
случае носит эффективный характер. Уравнения (VII.125) и (VII.126)
не совсем точны, так как приповерхностные концентрации могут быть
неодинаковы в частицах различного размера, проведших в реакторе
неодинаковое время или двигавшихся по различным траекториям.
312
Естественно, что в предельных ситуациях, соответствующих различ-
ным областям протекания реакции, система уравнений (VII.123)—
(VII.125) упрощается. Наиболее частым является случай, когда
(5 -> сю и соответственно Сп ф = С^/Г.
Уравнения (VII.123)—(VII.126) напоминают систему уравнений
(VII.90) — (VII.94) для двухфазного .потока, однако они имеют ряд
особенностей, которые вскрываются при практическом расчете реак-
торов с кипящим слоем. Дело в том, что кипящий слой представляет
собою динамическую систему, в которой отношение высоты работаю-
щего кипящего слоя (Я) к высоте слоя в спокойном состоянии (Н0)
определяется диаметром частиц катализатора (dKaT) их плотностью
(ркат) и линейной скоростью потока газа и. Далее критическая ско-
рость инр не является произвольной величиной, а также является
функцией </кат и ркат.
Объемный коэффициент массопередачи Роб определяется как ве-
личиной удельного, на единицу поверхности, коэффициента массо-
передачи между фазами, так и поверхностью межфазного контакта,
т. е. числом и размером пузырей. Он выражается формулой
₽об=Уш$Руд (VII.127)
где N — число .пузырей; dn — их диаметр; Руд — удельный коэф-
фициент межфазной массопередачи. Поскольку
Л's 3 (VII.128)
4n;a*
то
Роб^А.-^pL руд (VII.129)
При этом надо учесть, что dn зависит от конструктивных размеров
распределительной решетки.
Значения и не могут выбираться вполне произвольно, так как
лежат в пределах между критической скоростью псевдоожижения (нкр)
и скоростью витания частиц (ПвнТ). Последняя превосходит первую
на 1,5—2 порядка. Для устойчивой работы аппаратов необходимо,
чтобы отношение и/иК9 — число псевдоожижения, — было не
меньше 3—5. Это связано в первую очередь с полидисперсностью
состава реальных катализаторов.
В связи с указанными особенностями кипящего слоя расчеты
емкостных реакторов целесообразно проводить следующим образом.
Определяются кинетические зависимости для катализатора вы-
бранной дисперсности в неподвижном слое и строится кинетическая
модель реакции. Интегрированием уравнений- кинетической модели
для изотермических условий и аппарата идеального вытеснения при-
ближенно определяют объем катализатора. Рассчитывается крити-
ческая скорость псевдоожижения для данного катализатора, выби-
рается число псевдоожижения и по нему определяется значение и.
Из заданной производительности установки, режимных параметров
процесса и скорости и определяют диаметр аппарата. После этого
313
определяют приближенно рабочую высоту слоя катализатора, ко-
торая обычно составляет 1,3—1,5 таковой для слоя в спокойном
состоянии. Если конструктивные размеры аппарата получаются не-
приемлемыми, то следует менять в допустимых пределах число псевдо-
ожижения или переходить к катализатору другой дисперсности. При
ориентировочно приемлемых конструктивных размерах аппарата пов-
торяют расчет на основе системы уравнений (VII.123)—(VII.125)
или (VII.123), (VII.124), (VII.126) с добавкой уравнений для опре-
деления рабочей высоты слоя Н, коэффициента 0об и уравнения теп-
лового баланса. В зависимости от результатов уточненного расчета
либо принимают его как окончательный, либо вносят изменения
в исходные и конструктивные параметры и расчет повторяют вновь.
Имеется большое количество работ, посвященных расчету и экс-
периментальному определению коэффициентов межфазного пере-
носа, диффузии, частиц, диаметру пузырей, расширению слоя
и других параметров кипящего слоя. Обзор этих результатов дан,
например, в монографии Аэрова и Тодеса [3]. Приведем для иллю-
страции некоторые расчетные формулы из этой монографии.
Наиболее простой вид для оценки критической линейной скорости
псевдоожижения пкр имеет формула
где
_ _______Аг_____
вкР~ 1400 + 5,22 У Аг
(VII.130)
= “кр^кат _ Ar = gdKaT ' Рт—рг
КР V ’ V2 Рг
V
Здесь dKaT — средний диаметр частиц катализатора; рг и v — плот-
ность и динамическая вязкость потока; рт — плотность твердых
частиц.
Формула (VII.130) дает оценку с точностью до ±30%. Для даль-
нейшего уточнения лучше проводить экспериментальные исследова-
ния. Для скорости витания предлагается формула
г> Аг
Нбвт»т==
18+0,6 УАг
(VII.131)
Эффективный коэффициент диффузии твердых частиц приходится
определять экспериментально по методикам, описанным в [3]. По-
скольку коэффициент Г, особенно для пористых веществ, также экс-
периментальная величина, удобнее экспериментально определять
О
кат эффективный коэффициент продольной диффузии газа, свя-
занный не с С^, а Сп. ф. Некоторые значения Z)£aT приведены в вы-
шеуказанной монографии.
Расширение кипящего слоя можно определить, например, по
упрощенной формуле
(VI 1.132)
где Но — высота слоя к началу псевдоожижения.
И [ «кр
Н0 \ мрит------мкр
314
По данным монографии Дэвидсона и Харрисона .[10], диаметр
пузырей можно оценить из уравнения:
0.711 W1'1
“ —“кр
Яр
Я-Яо
(VII.133)
В монографии [10] выведено уравнение для определения удель-
ного коэффициента межфазной массопередачи расчетным путем. Оно
имеет вид
Роб =0,975 (VII.134)
где Dr — коэффициент молекулярной диффузии газа.
Процессы на отравляющемся катализаторе. Интенсивность дви-
жения твердых частиц в кипящем слое позволяет непрерывно вводить
в реактор свежий катализатор взамен равного количества отработан-
ного. Благодаря этому в кипящем слое можно проводить в стацио-
нарном режиме процессы, идущие на катализаторах с быстро падаю-
щей активностью *. При не слишком больших скоростях подачи све-
жего катализатора смешение частиц в кипящем слое можно считать
идеальным, и функция распределения времени пребывания частицы
в слое определяется формулой (см. раздел VII.3)
<р(Ткат)=т’|е'Ткат/ТсР. (VII.135)
где тср — среднее время пребывания частицы катализатора в реак-
торе, равное объему слоя, деленному на объемную скорость подачи
свежего катализатора (и вывода отработанного).
Той же формулой определяется и распределение «возраста» Ча-
стицы ф (1).
Активность каждой частицы катализатора, описываемая констан-
той скорости реакции к, отнесенной к единице объема зерна, спадает
со временем пребывания либо линейно
к = к0—аткат (VI 1.136)
либо экспоненциально
' к=кве'а^ат (VII.137)
где к0 — константа скорости реакции на свежем катализаторе;
а — некоторая постоянная, характеризующая скорость падения ак-
тивности катализатора.
Случай экспоненциального падения активности наиболее важен.
Он наблюдается при «утомлении» катализатора, например при
необратимом отравлении или блокировании активной поверхности
* Средняя активность яастнц катализатора в кипящем слое рассчитана
в работах [36]. Расчет средней активности при различных функциях распреде-
ления времени пребывания частиц в слое (включая и режим идеального выте-
снения, осуществляющийся в движущемся слое, — см. следующий раздел) про-
веден Петерсеном [37]. «
315
продуктами реакции (см. раздел VII.6). Экспоненциальный закон
(VII.137) является решением уравнения
Л/Лгкат= — ак (VII.138)
В отличие от аналогичного уравнения (VI 1.64) коэффициент при к
в уравнении (VI 1.138) постоянен, так как вследствие интенсивного
движения частиц в кипящем слое каждая частица за время своего
пребывания в реакторе успевает побывать в различных его точках,
и даже в отсутствие идеального смешения потока газа скорость паде-
ния активности одинакова для всех частиц и определяется средними
значениями концентраций реагентов в слое. Так, в процессе, вклю-
чающем единственную необратимую реакцию первого порядка,
а = рА0С (VI 1.139)
где С — средняя концентрация исходного вещества в реакторе;
п — эффективный коэффициент падения активности (см. раздел VII.6).
Средняя активность катализатора в кипящем слое вычисляется
по формуле:
J (ткат) ф (Гкат) ^Гкат (VII. 140)
О
Подставляя в эту формулу соотношение (VII.135) и (VII.136) и
интегрируя, получаем:
к = W(l + <»*Cp) (VI 1,141)
Так как величина параметра а, входящего в формулу (VII.141),
зависит, согласно (VI 1.139), от средней концентрации реагента в слое,
для определения средней активности катализатора необходимо вос-
пользоваться решением уравнений материального баланса. В случае
реакции первого порядка, протекающей в кинетической области в ре-
жиме идеального вытеснения, имеем
с = е~ук (VII.142)
где с—С1Сй — безразмерная концентрация , исходного вещества;
х = кйХ/и — безразмерная координата и у'— к/к0.
Интегрируя соотношение (VII.142), находим среднюю безразмер-
ную концентрацию
%
ё=~£~ J e'yxdx= -L- (1-е-^) (VII.143)
0 У
где к — k0L]u", L — высота кипящего слоя.
G учетом соотношения (VII.139) формула (VII.141) приводится
к безразмерному виду
гГ=(1+М^)-1 (VII.144)
где М = иСйи/иср; иср = L/xcp,
316
Безразмерная средняя активность катализатора у, а затем и кон-
центрация непрореагировавшего исходного вещества на выходе реак-
тора с = crib , определяются совместным решением уравнений
(VIL143), (VII.144). Подставляя величину с, выраженную форму-
лой (VII. 143), в соотношение (VII. 144) и выполняя простые преобра-
зования, получаем трансцендентное уравнение
у М — 1+у
(VI 1.145)
Это уравнение удобно решать графически, определяя величину у
как абсциссу точки пересечения графика функции / (у) с горизонталь-
ной прямой X = const (см. рис. VII.15). При М 1 функция / (у)
Рис. VII.15. Графическое решение
уравнения (VII.145.)
РиЬ. VII.16. Зависимость безразмерной
концентрации исходного вещества от X.
стремится к бесконечности при у 0 и обращается в нуль при у = 1.
При М < 1 функция / (у) убывает от бесконечно больших значений
до нуля в интервале 1 — М у 1, а вне этого интервала либо
отрицательна, либо лишена смысла. Производная
*L&L = -(у)-2 1ц---—=- — =---------=; (VII.146)
dy М — 1+у у (М — 1+р)
отрицательна в интервале 0 у =51 при М > 1 и в интервале
1 — М у <Z 1 — при М < 1. Поэтому в указанном интервале
всегда существует единственное решение уравнения (VII.145). Оче-
видно, при М < 1 активность катализатора не может падать ниже
определенной величины, соответствующей средней величине безраз-
мерной константы скорости рёакции у=1—М, асимптотически
приближаясь к ней при увеличении длины слоя.
Зависимость безразмерной концентрации непрореагировавшего ис-
ходного вещества на выходе реактора от параметра X при различных
317
значениях М представлена на рис. VII.16. При Л/>1иХ^>1
решение уравнения (VII.145) дает:
1 ’ С = е'^ 1--^- (VII.147)
Таким образом, при М 1 максимальная степень превращения
в реакторе 1 — с не может превысить величины М~1, каким бы боль-
шим ни было время пребывания реагирующей смеси в зоне реакции.
При М < 1 и Л Э> 1 имеем:
/ j^l-М; c = e-K(l~M) (VII.148)
В этих условиях, увеличивая X, можно добиться степени превра-
щения, как угодно близкой к единице. Значение параметра М = 1
является, таким образом, критическим, и в правильно спроектиро-
ванном процессе скорость подачи свежего катализатора обязательно
должна быть такой, чтобы величина М не превышала единицы.
Идеальное смешение частиц в кипящем слое приводит к тому, что
частицы, только недавно вошедшие в реактор, и частицы, уже почти
потерявшие свою активность, имеют равную вероятность покинуть
слой. Этот общий недостаток систем с идеальным смешением потока
приводит к снижению среднего значения константы скорости к по
сравнению со значением, соответствующим идеальному вытеснению
в потоке катализатора,. Этот последний режим, как увидим, осуще-
ствляется в процессе с движущимся слоем.
VII.9. РЕАКТОРЫ С ДВИЖУЩИМСЯ СЛОЕМ
Основные уравнения. К описанию движущегося слоя полностью
применима схема двухфазного потока, рассмотренная в разделе VII.7.
Пассивной фазой является поток газа, а активной — газ, находя-
щийся в порах твердых частиц и сорбированный на активной поверх-
ности. Соответственно, эффективная константа скорости межфазной
диффузии равна коэффициенту массопередачи 0, умноженному на
внешнюю поверхность единицы объема твердых частиц он. Гидроди-
намический режим обеих фаз близок к идеальному вытеснению. Если
адсорбция на поверхности твердых частиц следует закону Генри,
уравнения баланса вещества в пассивной и активной фазах движу-
щегося слоя записываются в виде
uTdCT]dX + 0аи (Сг-Скгп1Г)=0 (VII. 149)
-|-0<7И Г — Сг)—р (Скат) 0 — 0 (VI 1.150)
где о — удельная поверхность частиц (полная) на единицу объема
слця; Г — постоянная закона Генри; р — скорость образования дан-
ного вещества, отнесенная к единице поверхности катализатора;
икат и ит — линейная скорость движения твердых частиц (при про-
тивотоке частиц и газа величину следует считать отрицательной)
и потока газов; Сг — концентрация реагента в потоке газа.
318
Роль концентрации в активной фазе здесь играет двухмерная
концентрация молекул, сорбированных на активной поверхности Ска,..
Внутридиффузионное торможение реакции здесь не учитывается.
Как и в кипящем слое, перенос вещества движущимися твердыми
частицами не играет роли, если реакция протекает во внешнедиффу-
зионной области или если частицы катализатора обладают малой
удельной поверхностью, плохо сорбирующей реагент. В кинети-
ческой области устанавливается сорбционное равновесие между фа-
зами Сг = Сках/Г; складывая (VII. 149) и (VII. 150), приходим в этом
случае к уравнению
(игЦ-Гаикат) dCr/dX — ра = 0 . (VII. 151)
Это уравнение формально подобно уравнению баланса однофаз-
ного потока с фиктивной линейной скоростью
и* = иг 4- Лшкат (VII. 152)
Поскольку скорость движения частиц обычно на несколько по-
рядков ниже скорости потока газа, величины и* и иг отличаются
значительно только в том случае, если частицы катализатора обла-
дают развитой поверхностью и хорошо сорбируют реагент. Анало-
гично (VII.149)—(VII.151) составляются уравнения теплового ба-
ланса слоя. В кинетической области протеканий реакции, когда
температура потока газа и твердых частиц равны, суммарное уравне-
ние баланса тепла принимает вид
(Тг^г+Ткат^кат) dT/dX —rr-|-g=O (VI 1.153)
где уг и укат — теплоемкость единицы объема соответственно потока
газа и твердых частиц; гг — скорость выделения тепла в единице
реакционного объема; q — плотность теплоотвода.
Более точное уравнение теплового баланса должно учитывать
перенос тепла вдоль слоя за счет теплопроводности, а также перепад
температуры по сечению, отнюдь не исключенный при проведении
процесса в движущемся слое.
Относительная роль процессов переноса тепла потоком газа и
твердыми частицами определяется величиной слагаемых угиг и
Ткат^кат- Так как теплоемкость твердых частиц весьма велика, то
при не слишком малой скорости движения они способны переносить
вдоль слоя значительные количества теплоты, сглаживая перепад
температуры по длине реактора. Это позволяет проводить процессы
в движущемся слое адиабатически, используя в качестве теплоноси-
теля сам катализатор, перегреваемый перед вводом в реактор в эндо-
термических процессах и охлаждаемый — в экзотермических.
Процессы на отравляющемся катализаторе. Особый интерес
представляет процесс в движущемся слое катализатора с падающей
активностью. В наиболее практически важном случае снижение
активности катализатора является следствием необратимого отрав-
ления или блокирования активной поверхности одним из продуктов
процесса (см. раздел VII.6). Если под о в уравнениях (VII.150),
319
(VII.151) понимать действующую (неотравленную) поверхность ка-
тализатора, расчет процесса на утомляющемся катализаторе может
вестись по обычным уравнениям баланса, дополненным уравнением
баланса активной поверхности
uKaTda/dX + ra = 0 (VII.154)
где га — скорость отравления катализатора в единице объема слоя.
Рассмотрим единственную необратимую реакцию, протекающую
в изотермических условиях на катализаторе переменной активности.
Будем для простоты пренебрегать переносом вещества движущимися
твердыми частицами; мы уже говорили, при каких условиях это
полностью оправдано. Мерой активности катализатора является эф-
фективная константа скорости реакции к, равная произведению кон-
станты скорости х0, отнесенной к единице активной поверхности, на
площадь неотравленной поверхности а; соответственно скорость обра-
зования исходного вещества в единице реакционного объема ра
равна —кС. Уравнение материального баланса исходного вещества
запишем в безразмерной форме, введя безразмерные переменные
с — CICQ, у = к/к0 и х = к0Х]и (где Со — исходная концентрация
реагента и к0 — константа скорости реакции на неотравленном ката-
лизаторе)
dc/dx — —ус (VII.155)
Безразмерное уравнение баланса активности записывается в виде
dy!dx=±Myc (VII.156)
Это уравнение можно получить из уравнения (VI 1.154), полагая,
к = XqO - коа/с!о, ра = р/сС/Оо-
Безразмерный параметр М определяется при этом соотношением
М=МС„и/иш. Знак плюс в уравнении (VII.156) соответствует
противотоку реагирующей смеси и катализатора, а знак минус —
прямотоку.
В случае прямотока газовой и твердой фаз уравнения (VII.155),
(VII.156) решаются с граничными условиями
с(0) = 1; у(0) = 1 (VII.157)
Разделив уравнение (VII.156) на (VII.155), получаем:
dy/dc = M (VI 1.158)
откуда, с учетом граничных условий (VII.157), следует
у = 1 — м (1 - с) (VII.159)
Подставив формулу (VII.159) в (VII.155)', получим уравнение
dc/dx= — с [1 — М (1— с)] ‘ (VII.160)
интегрируя которое, находим безразмерную концентрацию непрореа-
гировавшего исходного вещества, а затем, с помощью соот-
320
ношения
реакции
(VII.159) — и безразмерную константу скорости
1 М , _1 М fVTT
Выходу реактора соответствует значение безразмерной коорди-
наты х=каЬ1и='к. Зависимость безразмерной концентрации на
выходе от параметра А представлена на рис. VII.17а. При X 1 фор-
мула (VII. 161) дает:
при М > 1
с (%) 1-1/М
при Л/<1
с(Х)^(1-М)е-(1-м,Х (VII.162)
Рис. VII.17. Зависимость безразмерной концентрации исходного
вещества от безразмерной координаты X:
а — при прямоточном движении газа и катализатора; б — при противоточ-
ном движении газа и катализатора.
Таким образом, при М > 1 в реакторе, каким бы он ни был длин-
ным, не может быть достигнута степень превращения 1 — с, превы-
шающая М~ *.
При противотоке фаз граничное условие у = 1 задано при х = X.
Временно вводя граничное условие у (0)=у0, записываем решение
системы уравнений (VI 1.155), (VI 1.156) в виде
уЛм '
удр(у,+ М)х +м’
У о (уо +
у0 + Ме-{у°+м)х
(VII.163)
Неопределенная постоянная у0 должна быть найдена теперь ре-
шением уравнения
__ (Уо + М) у о
у0 + Ме~(у° + м)}-
Это уравнение удобно решать графически, переписав его пред-
варительно в форме
1 м
Х =—;—гчт 7Г = /(г/о) (VII.164)
Уо+М Уо(.Уо + М — 1) '"и/ ч '
21 Закав 1441
321
Поведение функции / (г/0) качественно то же, что и функции / (у)
в уравнении (VII.145). Уравнение (VII.164) всегда имеет единствен-
ное решение, лежащее в интервале 0 < у0 1 при М 1 и в интер-
вале 1 — М < у0 1 при М < 1. При М > 1 и X '^> 1 величина у0
и концентрация непрореагировавшего исходного вещества на выходе
реактора с (X) приближенно равны
а при М < 1 и X 1:
с(Х)
(VII.165)
(VII.166)
Зависимость безразмерной концентрации на выходе реактора
от параметра % при ^различных значениях М представлена на
рис. VII.17, б.
Интересно сравнить степень превращения, достигаемую при оди-
наковых значениях параметров М, X, в трех случаях, рассмотренных
в этом и предыдущем разделах: при идеальном смешении по катали-
затору (т. е. в кипящем слое), при прямотоке и противотоке реагирую-
щей смеси и катализатора. Во всех трех случаях существует мини-
мальное значение скорости подачи свежего катализатора, соответ-
ствующее критическому значению параметра М = 1, при котором
возможно достижение степени превращения, как угодно близкой
к единице в достаточно протяженном реакторе. При М > 1 во всех
трех рассмотренных случаях максимальная достижимая степень пре-
вращения 1 — с равна М~1. Сравнение кривых на рис. VII.16 и
VII.17, а также асимптотических формул (VII.148), (VII.162) и
(VII.166) показывает, что при одинаковых значениях параметра М
наименьшая эффективность процесса наблюдается в кипящем слое,
а наибольшая — в движущемся слое при противотоке реагирующей
смеси и катализатора.
ЛИТЕРАТУРА
1. Р. Арис, Анализ процессов в химических реакторах, Изд. «Химия»,
1967.
2. О. Левеншпиль, Инженерное оформление химических реакций,
Изд. «Химия», 1969.
3. М. Э. А э р о в, О. М. Т о д е с, Гидравлические и тепловые основы
работы аппаратов со стационарным и кипящим зернистым слоем, Изд. «Химия»,
1968.
4. В. В. К а ф а р о в, Методы кибернетики в химии и химической
технологии, Изд. «Химия», 1968.
5. С. В е й л а с, Химическая кинетика и расчет промышленных реак-
торов, Изд. «Химия», 1967.
6. X. Крамере, К. Вестертерп, Химические реакторы. Рас-
чет и управления ими, Изд. «Химия», 1967.
7. К. Г. Д е н б и г, Теория химических реакторов, Изд. «Наука, 1968.
8. Г. К. Боресков, Катализ в производстве серной кислоты, Госхим-
издат, 1956.
9. И. П. Мухленов и др., Катализ в кипящем слое, Изд. «Химия»,
1971.
322
10. И. Ф. Д э в и д с о н, Д. X а р р п с о н, Псевдоожижение твердых
частиц, Изд. «Химия», 1965.
11. М. Лева, Псевдоожижение, Гостоптехиздат, 1961.
12. Н. И. Сыромятников, Л. К. В а с а н о в а, ГО. Н. Ш и м а н-
с к и й, Тепло и массообмен в кипящем слое, Изд. «Химия», 1967.
13. J. S е е Ъ о 1 d a. oth., Oil and Gas J., 51, № 2, 111 (1952).
14. R. S. Gannes, Chem. Eng. Progr., 49, № 3, 113 (1953).
15. Технологические схемы процессов переработки нефти в США» Гостоп-
техиздат, 1956.
16. М. И. Кириллов, ЖПХ, 13, 987 (1940).
17. R. В. McMallin, М. Weber, Trans. Am. Inst. Chem. Eng.,
31, 409 (1935).
18. W. F. G r ii 11 e г, В. H. M e s s i k о m e r, Helv. chim. acta, 44,
285 (1961).
19. А. В. Ф p о с т, В e с т н. МГУ, № 3—4, III (1946).
20. D. L. С r e s s w e 11, W. R. P a te rson, Chem. Eng. Sci., 25,
1415 (1970).
21. О. M. Тодес, Изв. АН СССР. ОХН, 5, 483 (1946).
22. Г. К. Томас, сб. «Хроматография», ИЛ, 1949.
23. J. В. В u 11, D. М. R о h a n, Chem. Eng. Sci., 23, 4 (1968).
24. P. А. Б у я н о в, Закоксовывание и регенерация катализаторов де-
гидрирования при получении мономеров, Изд. «Наука», 1968.
25. О. М. Т о д е с, Я. М. Б и к с о н, ДАН СССР, 75, № 5, 727 (1950).
26. Т. X о б л е р, Массопередача и абсорбция, Изд. «Химия», 1964.
27. Г. К. 3 и г а н ш и.н, А. Ермакова, ТОХТ, 4, № 4, 594 (1970).
28. А. А. Б е з д е н е ж н ы х, В. И. Т а р а н о в, А. П. Орлов,
ТОХТ, 5, № 1, 163 (1971).
29. М. Р е г г u t, R. L о u t a t у, Chim. et ind. Gen. chim., 103, № 19,
2545 (1970).
30. В. В. К а ф a p о в, Основы массопередачи, Изд. «Высшая школа»,
31. В. В. Д и л ь м а н, Б. Б. Б р а н д т, ТОХТ, 5, № 2, 326 (1971).
32. Б. И. Б р а у н ш т е й н, А. С. Ж е л е з н я к, сб. «Процессы жид-
костной экстракции и хемосорбции», Изд. «Химия», 1966, стр. 81.
33. А. В. О с и п о в, Автореф. кандид. дисс. ВНИИНЕФТЕХИМ, 1970.
34. K.-Sh. Т. Matsuura, Zur Reaktionstechnik von Blasen-saulenreak-
toren, Diss. Technische Universitat, Berlin, 1965.
35. P. H. С a 1 d e r b a n k, M, В. Moo-Yung, Chem. Eng. Sci., 16,
39 (1961).
36. S. L. Andersen, R. J. M i k о v s k y, Chem. Eng. Sci., 15,
161 (196 ).
37. E. E. Peterson, Am. Inst. Chem. Eng., J., 6, 488 (1960).
21*
Глава VIII______________________________________________
УСТОЙЧИВОСТЬ КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
VIII.1. УСТОЙЧИВОСТЬ РЕАКТОРОВ
ИДЕАЛЬНОГО СМЕШЕНИЯ
Понятие устойчивости. Ни один реальный реактор не работает
в строго стационарном режиме. Флуктуации состава исходной смеси,
колебания внешних условий и другие малые случайные возмущения
непрерывно выводят процесс из стационарного состояния. Очевидно,
что процесс может протекать нормально только в том случае, если
малые внешние воздействия ведут и к малым отклонениям режима
процесса от стационарного; в противном случае любое слабое некон-
тролируемое возмущение приведет к нарастающему удалению от за-
данного стационарного состояния, т. е. к немедленному срыву про-
цесса.
Если система, выведенная каким-либо малым внешним воздей-
ствием из стационарного состояния, по . прекращении действия
возмущающего фактора возвращается в первоначальное состояние,
данное состояние называется устойчивым.
Чтобы ответить на вопрос об устойчивости стационарного режима
химического процесса, необходимо, таким образом исследовать пере-
ходные процессы в реакторе, которые описываются системой неста-
ционарных уравнений материального и теплового баланса. Уравне-
ния эти нелинейны и даже в простейших случаях не могут быть
решены аналитически. Задачу, однако, можно существенно упростить,
учитывая то, что для анализа устойчивости достаточно исследовать
лишь малые отклонения от стационарного состояния. Поэтому
нелинейные кинетические функции, входящие в уравнения материаль-
ного и теплового балансов, можно разложить в ряд Тейлора в окрест-
ности стационарного режима и, пренебрегая высшими членами раз-
ложения, представить их в виде линейных функций отклонения пере-
менных от их стационарных значений. В результате получаем гораздо
более простую систему линейных уравнений, правильно описыва-
ющую переходные процессы в области, достаточно близкой к стацио-
нарному состоянию. Эту линейную систему в ряде случаев удается
решить или исследовать аналитически, определив тем самым общие
условия устойчивости процесса.
В этом разделе рассмотрим вопрос об устойчивости стационарных
режимов реакторов идеального смешения — простейшей из систем,
исследуемых в теории химических реакторов. В режиме идеального
смешения (см. раздел VII.3) значения всех переменных одинаковы
по всему объему реактора. В соответствии с этим стационарный ре-
жим реакторов данного типа описывается алгебраическими, а неста-
ционарный — обыкновенными дифференциальными уравнениями.
Такие системы принято называть системами с сосредоточенными пара-
324
метрами. Исследование их устойчивости наиболее просто, так как
здесь используются хорошо изученные методы, впервые/развитые
Ляпуновым [1 ] и описанные во многих учебниках по теории обык-
новенных дифференциальных уравнений (см. например [2]), а также
в специальных монографиях [3,4] *. Гораздо более сложную задачу
представляет собой исследование устойчивости таких процессов, где
значения переменных изменяются не только во времени, но и в про-
странстве. К этому классу принадлежат процессы на пористом зерне
катализатора и в системах реактор — теплообменник. Стационарный
режим таких процессов описывается дифференциальными уравнени-
ями (в простейших случаях — обыкновенными), а нестационарный —
уравнениями в частных производных; их принято называть (хотя
это название нельзя признать удачным) системами с распределен-
ными параметрами. Анализ устойчивости распределенных систем
редко удается довести до конца, используя традиционные анали-
тические методы; ряд результатов, имеющих общий характер, здесь
может быть получен с помощью наиболее мощных методов совре-
менного функционального анализа.
Единственная реакция. Рассмотрим подробно простейшую си-
стему— реактор идеального смешения, в котором протекает един-
ственная химическая реакция с общим стехиометрическим уравне-
нием: 4
2vA = 0 (VIII.1)
I
Здесь vz — стехиометрический коэффициент z-го вещества; обычно
считают, что vz > 0 для продуктов реакции и vz < 0 для исходных
веществ.
Стационарный режим реакции в аппарате идеального смешения
описывается уравнениями (VII.2) и (VII.5). Перепишем эти уравне-
ния, обозначив стационарные значения переменных индексом «ст»:
₽ (С/ ст - Ci о) = vzr (Сст, Гст) (VIII.2)
а(7’ст-?’*) = Лг(Сст,7’стП (VII 1.3)
Соответственно, нестационарные уравнения материального и теп-
лового баланса будут иметь вид:
zZCz/dt=-₽(Cz-Czo) + vir(C, Т) (VIII.4)
dT)dt = -(a/y)(T~T*) + (h/y)r(C, Т) (VIII.5)
* Задача исследования устойчивости стационарных режимов химических
реакторов впервые поставлена Ван Хирденом [5]. Общие условия устойчивости
реакторов идеального смешения выведены Билоусом и Амундсоном [6]. В рабо-
тах Ариса и Амундсона [7] было проведено подробное исследование поведения
нестационарных решений в окрестности стационарного режима. Ряд последу-
ющих публикаций [8] касался, в основном, конкретизации основных резуль-
татов. Траектории решений в фазовой плоскости исследованы Вольтером и Саль-
никовым [9]. Ряд более поздних работ [10], опирающихся на новейшие матема-
тические методы [4], был посвящен анализу устойчивости реакторов идеального
смешения к сильным возмущениям.
325
Здесь Сi и С{й — концентрация i-го вещества соответственно
в реагирующей и исходной смеси; 0 — отношение объемной скорости
потока к объему реактора (т. е. величина, обратная времени кон-
такта); г — скорость реакции в единице объема аппарата, зависящая
от совокупности концентраций реагентов С и температуры Т; у —
теплоемкость единицы объема . реагирующей смеси; h — теплота
реакции; t — время; а и Т* — параметры, определенные формулами
Линеаризуем уравнения (VIII.4), (VIII.5) в окрестности стацио-
нарного режима, разложив кинетическую функцию г {С, Т) в ряд
Тейлора:
г<С, П -г «•„, Г„)+2 (^,)„<C<-<;<CT)+(»c,<r-r«)+ • • •
(VIII.6)
Здесь все производные вычисляются при значениях концентра-
ций реагентов С(- ст и температуры Тст, соответствующих исследуе-
мому стационарному режиму процесса. Другими членами ряда
(VIII.6) в достаточно близкой окрестности стационарного режима
можно пренебречь.
Подставляя (VIII.6) в (VIII.4), (VIII.5) и учитывая стационар-
ные уравнения (VIII.2), (VIII.3), находим, что отклонения концент-
раций и температуры от их стационарных значений = С( — CiCT,
т = Т — Т„ определяются решением уравнений:
>--«,+*2 (-^)„W”<(£V (vni-,)
(vin.s)
Деля каждое из уравнений (VIII.7) на v( и попарно вычитая их
друг из друга, находим, что отклонения концентраций различных
веществ от их стационарных значений связаны между собой соот-
ношениями
<vin.»
интегрирование который дает
S/=E/oe-₽J+(vz/v/)(^“5/oe'₽<) (VIII.10)
где — отклонения от стационарного режима в начальный мо-
мент времени t = 0.
Используя соотношения (VIII. 10), можно выразить все величины
Zj через отклонение от стационарного значения концентрации ка-
кого-либо одного вещества, которое принимается за ключевое.
В качестве ключевого вещества выберем, как обычно, один из
исходных реагентов; его стехиометрический коэффициент v( можно
326
при этом принять равным —1. Тогда уравнения (VIII.7), (VIII.8)
записываются в виде
dydt = —(^+rc) гтт4-ае-₽< (VIII.11)
dx/dt = (h/^)r(£>+(hrT/y—a/f)x + Ье'^ (VIII.12)
где
Гг=(^)ст’ Г^-2^(^)сТ (VIII.13)
Величина гс представляет собой полную производную скорости
реакции по концентрации ключевого вещества, вычисленную в усло-
виях стационарного режима с учетом линейных соотношений между
стационарными концентрациями:
£/ст= Cj0 -|- (Vj/vi) (Ci ст Сю) (VIII.14)
В «нормальном» случае, когда нет явлений автокатализа или
торможения исходными веществами, величина г'с положительна.
Параметры а и Ъ в уравнениях (VIII.11), (VIII.12) выражаются
через линейные комбинации исходных отклонений концентраций
реагирующих веществ от их стационарных значений. В случае на-
чального возмущения специального вида, когда сохраняются со-
отношения между концентрациями (VIII.14), величины а и b равны
нулю. Однако и в случае произвольно малого исходного возмущения
имеющиеся в начале отклонения от соотношений (VIII.14) быстро
ослабевают, «вымываясь» из реактора движущимся потоком. Мате-
матически зто выражается тем, что последние члены в уравнениях
(VIII.11) и (VIII.12), содержащие параметры а и Ь, экспоненциально
уменьшаются со временем. Поэтому отклонения от соотношений
между концентрациями реагентов (VIII.14) не влияют на характер
устойчивости процесса, и при анализе устойчивости соответствующие
члены в уравнениях (VIII.И), (VIII.12) можно опустить.
Таким образом, мы свели задачу исследования устойчивости
стационарных режимов к решению системы двух линейных обыкно-
венных дифференциальных уравнений с постоянными коэффициен-
тами (VIII.И), (VIII.12). Решение этой системы выражается линей-
ной комбинацией двух экспоненциальных функций ем и eKit (где
^-2 — корни уравнения):
+ Р + гс ГТ
— hrc/y I —hrT/x + а/у ~ ~
= + (гс — hrт/т + а/у 4- Р) л+ (а₽/у — Л₽гг/у-|-агс/у)=0 (VIII. 15)
Уравнение (VIIL15), в зависимости от соотношений между вхо-
дящими в него параметрами, может иметь либо два действительных
корня, либо пару комплексно-сопряженных корней. Если оба корня
действительны и отрицательны, то отклонения от стационарного
режима экспоненциально затухают со временем (рис. VIII.1,
327
кривые I). Если же хотя бы один из корней положителен, то отклоне-
ния будут, напротив, экспоненциально возрастать со временем, пока
процесс не выйдет из окрестности стационарного режима (рис. VII 1.1,
кривые II). В этом случае рассматриваемое стационарное состояние,
очевидно, неустойчиво к малым отклонениям. При комплексных
значениях будут наблюдаться колебания процесса вокруг стацио-
нарного режима, причем эти колебания будут затухающими, если
действительная часть X отрицательна (рис. VIII, кривая III) и на-
растающими — в обратном случае (рис. VIII.1, кривая IV). Таким
образом, для устойчивости исследуемого стационарного режима
необходимо и достаточно,
чтобы оба корня уравне-
ния (VIII. 15) имели отри-
цательные действительные
части.
Известно, что коэффи-
циент при X в квадратном
уравнении (VIII. 15) равен
сумме обоих корней, взя-
той с обратным знаком, а
^свободный член этого урав-
нения равен произведению
его корней. Оба корня
будут иметь отрицатель-
ные действительные части
Рис. VIII.1. Изменение отклонений от ста-
ционарного режима со временем при раз-
личных значениях X:
>. действительное отрицательное {кривая 1) и положи-
тельное (кривая II); X комплексное с отрицательной
(кривая III) и положительной (кривая IV’) действи-
тельной частью.
в том и только в том
случае, если оба эти коэффициента положительны. Таким обра-
зом, условия устойчивости стационарного режима имеют вид:
hrT —агс/$< а
hrT — yrc < а + ур
(VIII.16)
(VIII.17)
Первое из условий устойчивости имеет непосредственный физи-
ческий смысл. Его левая часть представляет собой полную про-
изводную скорости тепловыделения hr (С, Т) по температуре с уче-
том соотношения между стационарными значениями концентрации
ключевого вещества и температуры Сст = Со — (а//ф) (Гст — Т*)
[см. формулу (VII.7)]. Правая часть неравенства (VIII.16) равна
производной скорости теплоотвода по температуре (с учетом отвода
тепла как движущимся потоком, так и с помощью внешнего тепло-
носителя). Неравенство, обратное (VIII.16), таким образом, совпа-
дает с условием неустойчивости, выведенным в разделе III.3 (как
было показано в разделе VII.3, оно применимо и к реакторам идеаль-
ного смешения). При выводе этого условия отмечалось, что на его
основании можно делать заключение только о неустойчивости про-
цесса, но нельзя заключать, что процесс, в котором условие неустой-
чивости не выполнено, обязательно будет устойчивым. Действительно,
строгий анализ, основанный на исследовании нестационарных урав-
328
нений, показал, что для устойчивости стационарного режима не-
обходимо выполнение не только неравенства (VIII.16), но и второго
условия устойчивости (VIII.17). Это последнее условие учитывает
возможность возникновения в исследуемой системе нарастающих
колебаний. Действительно, если корни уравнения (VIIL15) — ком-
плексно-сопряженные, то условие (VIII.16) будет выполнено (так
как при этом = (х -ф- iy) (х — iy) = я2 + у2 >0). В то же время
условие (VIII.17) не выполняется в случае комплексно-сопряженных
корней с положительной действительной частью.
Легко заметить, что при гс>0 условия устойчивости всегда
выполняются для эндотермических реакций {h <0) и реакций, ско-
рость которых слабо зависит от температуры (гт «=< 0; например, для
реакций, идущих на взвешенном в потоке катализаторе во внешне-
диффузионном режиме). В случае обратимой экзотермической реак-
ции, идущей при оптимальной температуре, гт = 0 и стационарный
режим также будет устойчивым.
Возникновение неустойчивости возможно в экзотермических
процессах, а также в процессах,- где имеют место явления автока-
тализа или торможения исходными веществами и, вследствие этого,
гс <0. В тех же случаях возможно возникновение множественных
режимов процесса. Оба явления — неустойчивости и неоднознач-
ности решений — тесно связаны между собой. На рис. III.3 видно,
что условие (VIII.16) перестает вьйголняться в точке касания кривой
тепловыделения и прямой теплоотвода; в этой же точке изменяется
число стационарных решений. Когда прямая теплоотвода на рис.
III.3, сдвигаясь вправо, переходит через положение 2, появляются
два новых решения, одно из которых оказывается неустойчивым.
Эта связь между нарушением условий единственности и устойчи-
вости решений сохраняется и в пространственно распределенных
системах.
Экзотермическая реакция первого порядка. Рассмотрим более
подробно условия устойчивости стационарных режимов экзотерми-
ческой реакции первого порядка с аррениусовской температурной
зависимостью константы скорости к. На этом примере удобно про-
следить связь условий устойчивости со значениями характерных
параметров процесса. Введем, как в разделах III.3 и VII.3, без-
размерные стационарные температуру и концентрацию 6 = Е (Гст —
— T*)/RT\ с = Сст/Сй и безразмерные параметры
п=___Р__• о —= =/1 1 (VIII 18)
Тогда:
гс = А-(7)^ре0/ц
(VIII.19)
329,
Здесь использовано приближенное преобразование закона Ар-
рениуса к(Т) А: (7’*)ев[см. уравнение (III.54)]. Условия устой-
чивости (VIII.16), (VIII.17) принимают теперь вид
е0 (вс—1) < р.
е9 (вс — S) < р(14-£)
'(VIII.20)
(VIII.21)
Стационарные значения переменных определяются уравнениями:
Рис. VIII.2. Области устойчивости
экзотермической реакции первого
порядка в координатах 0 — в.
лотами 0 = 0 — <§ и 0=1
с=1—(VIII.22)
S Учитывая их, можно исключить
из условий устойчивости стационар-
ную концентрацию с и параметр р,
сведя их к виду
e2-ee+e>o (viii.23)
02-(в+1)е-|-в(1 + <£)>О (VIII.24)
На рис. VIII.2 возможные стацио-
нарные температуры процесса лежат
ниже прямой 0 = 0, а область, в
которой не выполняется неравенство
(VIII.23), лежит справа от кривой 1,
представляющей собой гиперболу
с асимптотами 0 = 1 и 0 = 0—1.
Расположение области, где нару-
шено второе условие устойчивости
(VIII.24), зависит от величины пара-
метра S. Граница этой области пред-
ставляет собой гиперболу с асимп-
+ <§. Согласно формуле /(VIII.18),
величина <§ изменяется в пределах от 1 в адиабатическом
реакторе до 0 при предельно интенсивном теплообмене с внешним
теплоносителем. При <§ — 1 область, где нарушено неравенство
(VIJI.24), лежит справа от кривой 2. Очевидно, во всей этой области
не выполняется и условие устойчивости (VIII.23). Таким образом,
в ^адиабатическом процессе условие (VIII. 23) является не только
необходимым, но и достаточным для устойчивости исследуемого
стационарного режима. Это справедливо и в общем случае реакции
с произвольной кинетикой. Действительно, подставляя а =
в неравенство (VIII.17), видим, что, если условие (VIII.16) выпол-
нено, то условие (VIII. 17) выполняется и подавно. Физически это
связано с тем, что в адиабатическом режиме невозможно возникно-
вение колебательных режимов.
По мере увеличения интенсивности теплообмена с внешним тепло-
носителем и, соответственно, уменьшения параметра <S кривая 2
330
сдвигается влево; при <§ <1 она пересекается с кривой 1 в
точке
0 = 1/(1 — S); e=l/S(l—S) (VIII.25)
В области между кривыми 1 и 2 слева от кривой 1 процесс не-
устойчив из-за нарушения условия (VIII.24); при этом отклонения
от стационарных значений переменных будут увеличиваться со вре-
менем путем нарастающих колебаний. В пределе при <§ -> 0 область,
где нарушается условие (VIII.24), ограничена прямыми 0 = 0
и 0 = 1; таким образом, только при 0 < 1 устойчивость процесса
гарантирована при любом значении параметра <§.
Заметим, что при 0 >4 процесс может иметь три стационарных
решения (см. раздел II 1.3). Область множественных режимов огра-
ничена кривой 3. Точкам, лежащим между верхними ветвями кри-
вых 1 и 3, соответствует высокотемпературный режим, точкам,
лежащим между нижними ветвями этих кривых — низкотемпера-
турный, а точкам, заключенным между двумя ветвями кривой 1 —
промежуточный режим, неустойчивый в силу условия (VIII.23)
или (III.51). При уменьшении параметра <§ вначале теряет устойчи-
вость высокотемпературный режим в области больших значений 0,
затем, по мере движения точки пересечения кривых 1 и 2 влево,
область неустойчивости высокотемпературного режима сдвигается
в сторону меньших значений 0. При <§ <9/16 кривая 2 заходит
в область слева от кривой 3, где существует только один стационар-
ный режим. В этой области значений параметров процесс, таким
образом, не будет иметь ни одного устойчивого стационарного ре-
жима. Наконец, при <§ <1/2 кривые 1 и 2 начинают пересекаться
ниже точки 0 = 4, 0 = 2, разделяющей высокотемпературную
и низкотемпературную ветви кривой 1. В этих условиях появляются
неустойчивые низкотемпературные режимы процесса, причем по
мере уменьшения <§ такие режимы становятся возможными при все
больших значениях параметра 0.
Исключив безразмерную температуру 0 из неравенств (VIII.23),
(VIИ.24) с помощью первого из соотношений (VIИ.22), можно вы-
делить области устойчивости в плоскости ц, 0. Пример разбиения
плоскости р, 0 (при £ — 0,4) показан на рис. VIII.3. В области I
процесс имеет один устойчивый стационарный режим; в области
II — один неустойчивый, в области III — два устойчивых (высоко-
и низкотемпературный) и один неустойчивый (промежуточный), в об-
ласти IV — один устойчивый (низкотемпературный) и два неустой-
чивых (высокотемпературный и промежуточный); наконец, в очень
узкой области V (в масштабе рисунка она неразличима) все три име-
ющихся стационарных режима неустойчивы.
Траектории в фазовой плоскости. Проведенный анализ, основан-
ный на линеаризации кинетических функций в окрестности стацио-
нарного режима, еще не позволяет полностью понять особенностей
поведения реактора в нестационарных условиях. В частности, оста-
ются невыясненными два важных вопроса: 1) каков максимальный
331
размер возмущений, после которых система возвращается в исход-
ное устойчивое стационарное состояние, 2) как протекает процесс
в том случае, если не существует ни одного устойчивого стационар-
ного режима. Ответ на эти вопросы может дать решение системы
нелинейных уравнений, описывающих данный процесс. Результаты
решения удобно изображать траекториями в фазовой плоскости С,
Т или в безразмерных координатах 1 — с, 0 (рис. VIII.4). В случае,
когда имеется два устойчивых стационарных режима (точки А и С),
в фазовой плоскости можно
выделить две области исходных
состояний, начавшись с кото-
рых, процесс выходит на каж-
дый из имеющихся устойчивых
Рис. VIII.3. Области устойчивости Рис. VIII.4. Траектории в фазовой
экзотермической реакции первого плоскости в случае трех стационар-
порядка в координатах р — 0 пых режимов (Л, В, С).
(S = 0,4).
режимов. Линия DE, разделяющая эти области, называется
сепаратрисой} эта линия всегда проходит через точку В, соот-
ветствующую неустойчивому стационарному режиму. Очевидно,
для того, чтобы процесс перешел из одного устойчивого состояния
в другое, необходимо такое возмущение, при котором точка, изо-
бражающая состояние системы, пересекла бы сепаратрису, войдя
в «область притяжения» другого устойчивого стационарного режима.
Очевидно, что все траектории процесса в фазовой плоскости
не могут выйти за пределы некоторой ограниченной области. Дей-
ствительно, концентрация исходного вещества заключена в преде-
лах от нуля до концентрации на входе в реактор Со, а температура
от То или Тс (температура теплоносителя) до температуры макси-
мального разогрева, достижимого при выгорании всего исходного
вещества в адиабатических условиях. В частности, в рассмотрен-
ном выше случае безразмерные концентрация и температура заклю-
чены в пределах 0 <с <1, 0 <9 <0. Если траектории не вы-
332
F
Рис. VIII.5. Траектории в
фазовой плоскости в случае,
когда имеется неустойчивый
стационарный режим (5) и
предельный цикл (показан
~ пунктиром).
ходят за пределы ограниченной области и в то же время не существует
ни одного устойчивого стационарного состояния, в котором эти тра-
ектории могли бы оканчиваться, то единственная возможность за-
ключается в том, что с течением времени траектории будут прибли-
жаться к некоторому устойчивому предельному циклу (рис. VIII.5).
При этом единственном возможным режимом процесса является
колебательный режим, при котором концентрации реагентов и тем-
пература реактора периодически изменяются со временем, зачастую
в весьма широком диапазоне*. Такой режим, очевидно, с техцоло-
гической точки зрения является совер-
шенно недопустимым.
Регулирование. В случае, когда ре-
жим процесса оказывается неустойчивым,
он может быть стабилизирован с помощью
надлежащим образом выбранной системы
автоматического регулирования. В общем
случае регулятор воздействует на пара-
метры процесса, изменяя их в зависи-
мости от измеряемых отклонений от ста-
ционарного режима. Чаще всего конт-
ролируется температура реакции, а регу-
лирование осуществляется путем измене-
ния температуры теплоносителя Тс. Если
последняя изменяется пропорционально
(с коэффициентом пропорциональности Л)
отклонению температуры от стационар-
ного значения, то:
Т’* = Т’5т + Л (7-Гст) (VIII.26)
Подставляя это выражение в уравнение (VIII.5), видим, что
после линеаризации в уравнениях (VIII.8) и (VIII.12) появляется
дополнительный член — (а/у)Лт. При этом условия устойчивости
(VIII.16), (VIII.17) принимают вид:
hrT — (а/Р) гс<ц(Ц-Л)
(VIII.27)
hrT — yrc<a (1+А) + Т0
(VIII. 28)
Очевидно, при достаточно большом А неравенства (VIII.27),
(VIII.28) всегда будут выполнены. Такое идеальное пропорциональ-
ное регулирование с достаточно большой константой обратной
связи А не всегда, однако, практически осуществимо. Поэтому иногда
применяют и более сложные схемы регулирования, при которых
температура теплоносителя изменяется пропорционально линей-
ной комбинации отклонения температуры, производной отклоне-
ния по времени и интегралу отклонения или даже некоторой
* Существование режима нелинейных колебаний установлено Вольтером
и Сальниковым [9].
333
нелинейной функции этих величин. Точный расчет таких регуля-
торов должен учитывать запаздывание регулирующего воздействия,
связанное с переходными процессами в теплообменнике и самой регу-
лирующей схеме.
Сложные реакции. В случае сложного процесса, включающего
несколько одновременно протекающих реакций, исследование устой-
чивости ведется в принципе теми же методами, что были описаны
выше. Получаемые критерии устойчивости будут иметь при этом,
однако, более сложную форму. Уравнения материального и тепло-
вого балансов сложного процесса имеют вид:
-^-=Р(С,0-Сг) + 2^/г/
(VIII.29)
— = a(T*-T) + ^hlri (VIII.30)
/
Здесь г, — скорость /-й реакции; vtj — стехиометрический коэф-
фициент i-ro вещества в у-й реакции; hj — ее тепловой эффект.
При записи этой системы уравнений достаточно ограничиться
уравнениями для ключевых веществ, выразив через последние
концентрации всех остальных веществ, входящих в выражения для
кинетических функций гу. Отклонения от стационарных линейных
соотношений между концентрациями реагентов, как и в рассмотрен-
ном выше случае единственной реакции, не будут влиять на устой-
чивость процесса. Полное число расчетных уравнений будет, таким
образом, равно К + 1, где К — число ключевых веществ (которое
не превышает числа протекающих реакций).
Введя переменные T]z= Ct — ClCT(i = 1, 2, . . ., К). т]0= Т — Гст
и разлагая правые части уравнений (VIII.29), (VIII.30) в ряд Тей-
лора, получаем систему линеаризованных уравнений
d К
= (УШ- 31>
К=0
где
«--№•+2 ”«(££•)„;
(VIII.32),
(1 при i = к
F (i, к=1,2, . . . , К)
0 при г к
2/ дгЛ / дг{\
vz'W/CT; “zo=2A'W/CT (z=1’2> • • -
i i
“oo=a+2A/(irL
334
Общее решение системы уравнений (VIII.31) выражается через
линейную комбинацию экспоненциальных функций:
к
ц; = 2 (г=0,1, • . . , К) (VIII.33)
?=0
где liq, \ — постоянные коэффициенты.
Числа Х? определяются решением уравнения
Д(Х)=О • (VIII.34)
где Д (X)— определитель (К + 1)-го порядка с элементами X6zfc—alk.
Для устойчивости исследуемого стационарного режима необхо-
димо и достаточно, чтобы все корни уравнения? (VI 11.34) имели отри-
цательные действительные части. Раскрывая определитель Д (X) и
группируя члены с одинаковыми степенями X, всегда можно пред-
ставить это уравнение в виде:
^К+1 + 2рп Ак+1-п =0 (VIII.35)
п=1
Условия, при которых все корни
отрицательные действительные части,
критерия Рауса—Гурвица.
Составим
уравнения (VIII.35) имеют
определяются с помощью
определителей:
I Pi Ь
Pl 1
Рз Pi
Р1
Рз
Рз
1
Р2
Р4
о
1
Рз
ряд
Pi
Рз
Рв
Р?
1
Рг
Р4
Рб Рб
о
1
Рз
о
о
1
Р4
(VIII.36)
с п ~^>К + 1 должны быть
все члены рп
последний из них совпадает со свободным
В этих определителях
заменены нулями, так что
членом рк+1.Всего для уравнения (VIII.35) можно составить К + 1
таких определителей. Если все определители (VIII.36) положительны,
то все корни уравнения (VIII.35) имеют отрицательные действитель-
ные части и, следовательно, исследуемый процесс протекает в устой-
чивом режиме. Например, при К = 3 условия устойчивости прини-
мают вид:
п Pi 1 п
Р1>0, =Р1Р2 — Рз >0
I Рз Рг I
Р1 1 о (VIII.37)
Рз Рз 1 =Р1РгРз —Р1Р4—Рз>о< Р4>0
0 Pt Рз
Таким образом, мы получили К + 1 условие устойчивости для
процесса с К ключ&ыми веществами. К сожалению, даже при не-
большом К эти условия весьма громоздки. Сложный процесс может
иметь большое число стационарных режимов, устойчивых и неустой-
чивых, соответствующих однозначно заданному набору характерных
335
параметров процесса. К этим режимам надо добавить предель-
ные циклы, существование которых может быть обнаружено путем
численного решения системы нелинейных уравнений (VIII.29),
(VIII.30). Чрезвычайно сложная картина стационарных, а тем более
нестационарных режимов, возникающая при одновременном проте-
кании нескольких реакций, способных ускоряться под действием
выделяющегося в ходе процесса тепла, до сих пор совершенно не
изучена.
VIII.2. ПАРАМЕТРИЧЕСКАЯ ЧУВСТВИТЕЛЬНОСТЬ
ТРУБЧАТЫХ РЕАКТОРОВ
Основные уравнения.; Стационарный режим трубчатого реактора
в приближении «одномерного» реактора идеального вытеснения
(см. раздел VII.4) описывается системой уравнений
uaCildX=ri(C, T)(i= 1,2, .... X) (VIII.38)
У yudT/dX--rr(C, T)—q(T, Тс) (VIII.39)
Где и — линейная скорость потока; — концентрация; X — про-
дольная координата; ri — скорость образования i-ro ключевого
вещества в единице объема реактора; у — теплоемкость единицы
объема реагирующей смеси; гт — скорость тепловыделения; q —
скорость теплоотвода (зависящая от температуры реагирующей
смеси Т и температуры теплоносителя Тс).
При постоянной температуре теплоносителя Тс распределение
концентраций реагентов и температуры по длине реактора опреде-
ляется решением системы уравнений (VIII.38), (VIII.39) с граничными
условиями Сг(0) = С(0, Т(0) = То, заданными на входе аппарата,
т. е. решением задачи Коши для системы обыкновенных дифферен-
циальных уравнений. Известно, что в случае, когда правые части
уравнений зависят от переменных непрерывным образом, задача
Коши для системы обыкновенных дифференциальных уравнений
всегда имеет единственное решение (см., например, [2]). Более
того, это решение всегда устойчиво, так как в реакторе идеального
вытеснения возмущение стационарного режима в некотором сече-
нии реактора не влияет на реагирующую смесь в соседних сечениях
и любое бесконечно малое возмущение «вымывается» из реактора
за конечное время, не успевая разрастись до макроскопических
размеров. Таким образом, всегда имеется единственный устойчивый
стационарный режим реактора идеального вытеснения.
Сказанное остается справедливым и в случае, когда имеются
поперечные градиенты концентрации и температуры. И в этом слу-
чае задача Коши для системы параболических уравнений (VII.44)
и (VII.45) всегда имеет единственное устойчивое решение. Явления
неустойчивости могут в принципе возникнуть п<?д влиянием продоль-
ного перемешивания потока, однако в достаточно протяженных реак-
торах этот эффект незначителен. При исследовании процессов в зер-
нистом слое учет продольного перемешивания сводится к учету
336
дискретной структуры слоя, влияние которой на режимы процесса
было оценено в разделе VI.5.
При регулировании реального технологического процесса чрез-
вычайно существенным является, однако, вопрос о том, в какой
мере влияют случайные возмущения состояния исходной смеси
и различных параметров процесса на основные показатели последнего.
Выше утверждалось, что бесконечно малое возмущение стационар-
ного режима не может разрастись до макроскопических размеров,
т. е. «коэффициент усиления» возмущений всегда остается конеч-
ным. Этот факт, являющийся следствием непрерывной зависимости
решений систем обыкновенных дифференциальных уравнений от
параметров и начальных условий (см., например, [2]), обеспечивает
устойчивость стационарных режимов процесса. Однако «коэффици-
ент усиления», оставаясь конечным, может быть значительным по
абсолютной величине; при этом реально существующие малые (не
бесконечно малые) возмущения могут усилиться настолько, что нач-
нут существенно влиять на наблюдаемые показатели процесса.
Мерой влияния изменения какого-либо параметра П на пока-
затели процесса является совокупность производных температуры
и концентраций реагирующих веществ на выходе аппарата по дан-
ному параметру /0 = дТ{Ь)/дП, Х> — ЭС^Ь^/дП. Каждая из вели-
чин Xi(i = 0, 1, . . ., К) называется параметрической чувствитель-,
ностью соответствующей переменной по данному параметру. Это
понятие впервые было введено Амундсоном и Билоусом [11]*.
Чтобы получить уравнения для величин %г, продифференцируем
уравнения (VIII.38), (VIII.39) по параметру П. При этом надо
помнить, что значения переменных в каждой точке в силу системы
уравнений (VIII.38), (VIII.39) зависят от варьируемого параметра.
Вводя переменные — dCt[X)/dH, (X) = дТ(Х)/дП и вос-
пользовавшись правилом дифференцирования сложных функций,
получаем:
"4r-2^7E''+-&-s"+-&(‘=1-2...........К) (vin-®>
/-1 -
/-1
Здесь все производные вычисляются в условиях исследуемого ста-
ционарного решения и, если последнее известна, являются заданны- •
ми функциями координаты X. Величины параметрической чувстви-
тельности Хг = ?/ (А) вычисляются решением системы линейных
уравнений (VIII.40), (VIII.41) с граничными условиями
^(0) = ^BX/37Z; £о(О) = 0Твх/аЯ (VIII.42)
* Численный расчет параметрической чувствительности трубчатых реакто-
ров проводился в работах [12].
22 Заказ 1441 , 337
где ClBX, Твх—концентрации реагентов в исходной смеси и ее тем-
пература.
В частности, если варьируемым параметром П является темпе-
ратура исходной смеси 7^, то £г(0) = 0 (* = 1, 2, . . К), Ц (0) = 1,
а производные по параметру в правой части уравнений (VIII.40),
(VIII.41) пропадают, так что последние становятся однородными.
Если варьируемым параметром является температура теплоноси-
теля Тс, то все величины £z (0) (I = 0, 1, . . ., К) равны нулю, обра-
щаются в .нуль и производные дг^дП, дт\/дП, и только в правой ча-
сти уравнения (VIII.41) остается член дц]дТс, делающий задачу
неоднородной. Задача (VIII.40)—(VIII.42) всегда имеет единствен-
ное решение и, если это решение найдено, то изменение значений
переменных на выходе аппарата при малом отклонении параметра П
от его исходного значения Пст, определяющего исследуемое стацио-
нарное решение (Сгст, УСт), может быть вычислено по формулам:
С/ = С[ ст Xi (П П„)
(VIII.43)
Т = Тст + 7.0 (П Пст)
Естественно, что эти формулы остаются справедливыми только
до тех пор, пока отклонения от первоначального стационарного
режима настолько малы, что можно пренебречь членами, пропор-
циональными квадрату и более высоким степеням отклонения.
Система уравнений (VIII.40), (VIII.41) может быть записана
в компактной матричной форме
d£/dX = r' (X)t,+rn (VIII.44)
где t, — (К + 1)-мерный вектор с компонентами (i = 0, 1, . . .,
К); г'(Х)— матрица с элементами г'ц = drJdCj, r'oj = drJdCj,
r’i0 = drJdT (i, / = 1, 2, . . ., K), r00 = dr^dT — dq/dT и rn —
вектор с компонентами Тщ^дг^дП (i = 1,2,.. .,К), гоп=дгх/дП —
— dq/dll.
Введем матрицу Y (X), каждый элемент которой уц (X) равен чувст-
вительности i-й переменной в сечении X к изменению значения /’-й
переменной на входе реактора, т. е. Ун = dCt(X)/dClBx, у10 =дС{(Х):
:dTm, yOj=dT(X)/dCjBX (i,7==l, 2, . . ., К), уОо = дТ(Х)/дТвх.
Очевидно, матрица У(Х) определяется решением задачи
dY/dX = r'Y; Г(0) = 7 (VIII.45)
где I — единичная матрица (К + 1)-го порядка.
Если матрица У(Х) известна, то решение уравнения (VIII.44)
с произвольными граничными условиями t, (0) = можно записать
в виде:
х
Z(X)=Y(X)&± ^Y(X)Y-i(X’)rn(X')dX' (VIII.46)
о
В справедливости этой формулы легко убедиться непосредствен-
ной подстановкой. Таким образом, матрица У(Х) фактически содер-
338
жит полную информацию о чувствительности значений переменных
в каждом сечении реактора ко всем характерным параметрам про-
цесса. В частности, можно сразу отметить, что высокой чувстви-
тельности можно ожидать в случае, когда элементы матрицы У(Х)
будут возрастающими функциями продольной координаты. При этом,
как правило, области повышенной чувствительности к изменению
различных параметров или граничных условий должны совпадать.
Адиабатические реакторы. В качестве простейшего примера
исследования параметрической чувствительности рассмотрим адиа-
батический реактор, в котором протекает единственная химическая
реакция с произвольной кинетической функцией r(C, Т). В данном
процессе имеется только одно ключевое вещество, а скорость тепло-
отвода q равна нулю, так что стационарные значения температуры Т
и концентрации исходного ключевого вещества С определяются
совместным решением двух уравнений (VII.25) и (VII.26). Пере-
пишем эти уравнения, введя вместо координаты X текущее время
контакта Xjw.
dC/dt = r(C, Т) (VIII.47)
dT/dt=fir(C, Т) (VIII.48)
Здесь h = fe/y —Максимальный адиабатический разогрев; h—тепло-
вой эффект реакции.
Как было показано в разделе VII.4, обе переменных связаны
между собой соотношением
й(Свх-О--г_гвх (VIII-49)
так что система (VIII.47), (VI 11.48) ожет быть сведена к единствен-
ному уравнению
dT/dt^^(T) (VIII.50)
где гад — адиабатическая скорость реакции:
Гад (Г)=ЙГ [Свх—Я-1 (Г—Т’вх), Г] (VIII.51)
Дифференцируя уравнение (VIII.50) по температуре исходной
смеси Твх и учитывая формулу (VIII.51), получаем уравнение для
чувствительности температуры в сечении t к исходной температуре,
т = dT(t)/dTax
dt/dt = r'ant+rc (VIII.52)
где
гс=дг[дС', raR = dra№fdT =Ыг/дТ--dr/dC (VIII.53)
Используем уравнение (VIII.50) для замены независимой пере-
менной t в уравнении (VIII.52) на переменную Т:
dr _ г^(Г)
dT гад (Т) 1“ гад (Г)
(VIII.54)
Проведенная замена вполне оправдана, так как темпера-
тура адиабатического процесса является монотонной функцией про-
дольной координаты или текущего времени контакта t. Решение
22*
339
уравнения (VI11.54) дает связь между параметрической чувстви-
тельностью % = т(£) и температурой на выходе реактора ТВь1Х.
Уравнение (VIII.54) легко интегрируется в квадратурах:
Твых
Х= Гад((ТвЫх)"+Гал(Гвых,1 (VW-55)
Твх
Формула (VIII.55) позволяет непосредственно исследовать зави-
симость % от длины реактора или времени контакта S — L/u [13].
Рассмотрим сначала случай необратимой реакции. При S = О,
когда Твх= ТВЪ1Х, величина %, очевидно, равна единице. При очень
больших временах контакта, когда лимитирующее исходное веще-
ство практически исчерпывается, СвыХ = 0 и, согласно соотношению
(VIII.49), достигается температура максимального адиабатического
разогрева ТВых = Твх -|- Ж. При этом скорость реакции ^ад(^вых)
близка к нулю и можно положить г (С, Т) = к (Т)Ст, где т — номер
первой из производных функции г (Z7, Т) по С при С = 0, не равных
нулю. Соответственно, 1
Гад = ^(П(<?вх-—; г/ = ^(Г)(Свх- /вх)
/
и формула (VIII.55) дает: /
X = (Свх+ 7'в^г7'вых-)ТОу (Свх+ Гв" Г ) ( dT=i (VIII.56)
ТГХ
Заметим теперь, что, согласно уравнению (VIII.52),
= (VIII.57)
\ dS /хА \ dt /т=1 дТ
В случае необратимой реакции всегда дг/дТ > О, причем значе-
ние этой производной на выходе реактора стремится к нулю, по мере
исчерпания исходного вещества при S -* оо. Отсюда следует, что
параметрическая чувствительность в случае экзотермической реак-
ции (Й > 0) всегда превышаетХ единицу, проходя е изменением
параметра S через максимум. В\случае эндотермической реакции
(К 0) параметрическая чувствительность, напротив, проходит
при увеличении S через минимум\ Так как, согласно уравнению
(VIII.52) \ х Х
=(4г') (VIII.58)
\ аЬ /х-о \ dt /т-о G
и, за исключением «аномальных» случаев автокатализа и торможе-
ния исходным веществом, гс > 0, % не может дважды пройти через
нуль и, следовательно, остается повсюду положительной. Аналогич-
ным образом изменяется % с увеличением времени контакта S
и в случае обратимой эндотермической реакции.
340
В случае обратимой экзотермической реакции также X (0) = 1.
Производная дг/дТ положительна при малых и отрицательна при
больших температурах. Реальный процесс всегда проектируется
таким образом, что на входе реактора дг/дТ > 0, так как иначе
условия протекания процесса были бы очень далеки от оптимальных.
Поэтому, согласно формуле (VIII.55), при малых S параметрическая
чувствительность превышает единицу. Далее с увеличением S она
проходит через максимум и, уменьшаясь, пересекает горизонталь-
ную прямую х = 1, причем в точке пересечения дг/дТ <0. Так как
с дальнейшим ростом Т величина дг/дТ остается отрицательной, %
при дальнейшем увеличении времени контакта S остается меньшей
единицы, выходя при 5 -» оо на асимптотическое значение х =
= — Гс1г'л^ где производные гс и гал .вычисляются при равновесных
значениях С и Т, соответствующих заданному значению Твх. Непо-
средственно из формулы (VIII.55) видно, что, поскольку гс > 0,
параметрическая чувствительность всегда положительна. Отрица-
тельные значения х могут наблюдаться только в случае эндотермиче-
ской или обратимой экзотермической реакции при «аномальной»
зависимости скорости реакции от концентрации исходного веще-
ства (гс <0).
Точка пересечения кривой х (&) Для обратимой экзотермической
реакции с горизонталью X — 1 имеет особый смысл [13]. Нетрудно
убедиться, что она соответствует такому режиму процесса, при
котором концентрация ключевого вещества на выходе реактора
минимальна и, следовательно, достигается максимальный выход
конечного продукта. Действительно, из соотношения (VIII.49) сле-
дует, что при £ (Z) = дС (t)/dTBX = 0 X (£) = т (0 = 1- Если при
выборе оптимальной температуры на входе реактора учитывается
стоимость аппаратуры и катализатора, то в оптимальном режиме
должна достигать минимального значения функция
P=€+XS (VIII.59)
где % — положительный коэффициент (см. раздел IX.1).
Так как с увеличением ТБХ необходимое время контакта умень-
шается (dS]dTBX <0), в оптимальной точке, где дР[дТъх = 0, имеем
£<°; x=i—nt>i (VIII.60)
и, таким образом, чувствительность оптимального адиабатического
реактора к изменению исходной температуры превышает единицу.
В случае необратимой реакции целочисленного порядка с по-
мощью формулы (VIII.55) можно получить аналитические выраже-
ния для х- Если реакция идет по нулевому порядку, гс = 0 и х
равна отношению скорости реакции на выходе из реактора к ско-
рости реакции на его входе. Здесь, в кажущемся противоречии
с полученными выше общими результатами, х монотонно возрастает
с увеличением времени контакта, если процесс идет с выделением
тепла и убывает — в случае эндотермической реакции. Следует, однако,
помнить, что независимость скорости реакции от концентрации
341
и сходного вещества никогда не сохраняется в области малых кон-
центраций; поэтому по исчерпании лимитируйнцего реагента %
резко падает или возрастает до предельного значения, равного 1.
В случае необратимой реакции первого порядка г = к (Т) С,
где к — константа скорости реакции, зависимость которой от тем-
пературы определяется уравнением Аррениуса. Формула (VIII.55)
принимает при этом вид:
{Твых
,* I Г к (Тъх) _________dT___________
ЙСВХ Л(Г)[1-(Г-Гвх)/йСвх]2
Гвх
(VIII.61)
Рис. VIII.6. Параметрическая
чувствительность адиабатиче-
ского реактора (для экзотер-
мической реакции первого по-
рядка).
Эту формулу удобно привести к
безразмерному' виду, введя, как в раз-
деле VI 1.4, безразмерную температуру
0=2? (Т — T^IRTl*. и параметр мак-
симального разогрева 0 = hECBX/RTBX,
а также воспользовавшись приближен-
ной формулой для температурной зави-
симости константы скорости реакции
к{Т)^к (7ВХ)Л
0вых „
У = е0вых f _§вых \ 14-— f . е — .
Z V 0 / +0 J (1-0/6)2
L о -I
(VIII.62)
Интеграл в формуле (VIII.62) выра-
жается через интегральную экспонен-
циальную функцию, определение кото-
рой было дано в разделе VI 1.4:
Х=1 + (в-0вых)е-Св-0вых)[ЁГ (©)-
— Е1(0—0ВЫХ)] " (VIII.63)
Зависимость х (0Вых) при различных значениях параметра 0 пред-
ставлена на рис. VIII.6.
Реакторы с внутренним теплообменом. Если тепло отводится
из зоны реакции и скорость теплоотвода q(T, Тс), согласно формуле
(VII.34), пропорциональна разности температур реагирующей смеси
Т и теплоносителя Тс с множителем пропорциональности а, то урав-
нения (VIII.40), (VIII.41), определяющие чувствительность темпера-
туры и концентрации исходного вещества в каждом сечении — т (/)',
£ (t) — к температуре исходной смеси Твх, принимают вид
dx!dt = (hrT — сОт+йг^ (VIII.64)
= —гтТ —гс£
(VIII.65)
Здесь, как и при анализе адиабатического процесса, принято, что
в аппарате протекает только одна химическая реакция с произволь-
342
ной кинетической функцией г {С, Т), и введены текущее время кон-
такта t = Х/и и обозначения гс = дг/дС, гт = дг/дТ. Уравнения
(VIII.64), (VIII.65) должны решаться с граничными условиями:
т(0) =1; £(0) = 0 (VIII.66)
Аналитическое решение уравнений (VIII.64), (VIII.65) невоз-
можно даже в простейших случаях. Однако сам вид этих уравнений
позволяет сделать некоторые выводы о характере изменения %
с увеличением длины реактора или времени контакта t. Близ входа
реактора, в силу граничных условий (VIII.66), т>0и£<0, при-
чем значения величины т, превышающие единицу, будут наблюдаться
в том случае, если производная скорости тепловыделения по тем-
пературе hrT будет превышать производную скорости теплоотвода а.
Последнее возможно только в экзотермических реакциях с достаточ-
но большим тепловым эффектом. При очень больших значениях
времени контакта, когда достигается практически полное превраще-
ние лимитирующего исходного вещества, Гт 0 и, поскольку
в этих условиях всегда гс > 0, из уравнений (VIII.64), (VIII.65)
следует, что обе величины £, т экспоненциально спадают к нулю.
В случае необратимой или обратимой эндотермической реакции,
когда гт > 0, величина £ может, согласно уравнению (VIII.65),
стать в некотором сечении реактора положительной только при
т < 0. С другой стороны, в силу уравнения (VIII.64), величина т
может, пройдя нуль, стать отрицательной только при hrc < 0 и,
так как величина £ при т = 0 должна быть меньше нуля, изменение
знака обеих величин £, т возможно только прийти £>0, т. е. либо
в экзотермической реакции с «нормальной» зависимостью скорости
реакции от концентрации ключевого исходного вещества (Й > 0,
гс >0), либо в случае эндотермической реакции, тормозящейся
исходным веществом или ускоряющейся одним из своих продуктов
(Й < 0, гс < 0). При Й > 0, гс <0 или Й < 0, гс > 0 величина т
всегда остается положительной, а £ — отрицательной. В случае
обратимой экзотермической реакции таких выводов сделать уже
нельзя, поскольку производная гт может менять знак и, следова-
тельно, знак может измениться и при т > 0. Реакции нулевого
порядка всегда соответствуют положительные значения т.
Аналогичным образом чувствительность температуры и концен-
трации реагирующей смеси к изменению температуры теплоносителя
Тс определяется решением системы уравнений
dx/dt=(hrT~а) т + йгс? + а (VIII.67)
dUdt = -rTx-rcl (VIII.68)
с граничными условиями:
Т(0) = 0; £ (0) = 0 (VIII.69)
Согласно уравнениям ‘(VIII.67), (VIII.68) £ -> 0 и т 1 при
t оо; при^малых t т>0 и'С<0; изменение знака х в принципе
343
возможно в тех же условиях, что и в предыдущем примере, но яв- «
ляется менее вероятным, так как в точке перемены знака должно
быть теперь выполнено условие—hrct,<^a. s
Помимо значений параметрической чувствительности на выходе 5
реактора, которая при достаточно большой степени превращения j
не может быть высокой, существенную роль играет в данном про- 4
цессе чувствительность температуры в «горячей точке» к изменению 4
параметров процесса. Оценку величины Тс, при которой параметри- >
ческая чувствительность температуры в «горячей точке» к измене- •?
нию Тс должна быть аномально высокой, можйо провести, как было 4
показано в ра!зделе VII.4, исходя из положения, точки касания пря- <
мой теплоотвода на рис. III.3 с кривой тепловыделения в адиабати- Н
ческом режиме. Эта точка соответствует переходу от плавного
продольного температурного профиля к профилю с резким температур- "
ным пиком. В тех же условиях следует ожидать и высокой чувстви-
тельности температуры в «горячей точке» к изменению веек других
определяющих ^параметров процесса.
VIII.3. СТАЦИОНАРНЫЕ РЕЖИМЫ
И УСТОЙЧИВОСТЬ АДИАБАТИЧЕСКИХ РЕАКТОРОВ
С ВНЕШНИМ ТЕПЛООБМЕНОМ
Стационарные режимы. Адиабатический процесс, идущий с выде-
лением чепла, можно проводить в автотермических условиях, ис- '
пользуя горячую смесь продуктов для подогрева исходной смеси -
во внешнем теплообменнике (рис. VIII.7). В такой технологической
схеме, очевидно, появляется обратная связь между температурой •
смеси продуктов реакции и исходной смеси, которая может приво-
дить к неустойчивости некоторых стационарных режимов процесса ,
и появлению скачкообразных переходов между различными режи- ’
мами при плавном изменении характерных параметров процесса. 4
Выпишем уравнения, определяющие стационарный режим тепло-
обменника, предполагая, что горячий и холодный потоки движутся
противотоком в режиме идеального вытеснения. Обозначая через Т\
и Т2 температуру соответственно холодного и горячего потоков,
имеем
edT1/dX = (kTP/^v)(Tz-T1) (VIII.70)
ЛТг/« = (ЛтР7тО(Г2-Т1) (VIII-71)
где кт — коэффициент теплопередачи между горячим и холодным
потоками; Р — периметр поверхности теплообмена; v — объемная j
скорость горячего потока (равная объемной скорости реагирующей
смеси); у — теплоемкость холодного и горячего потоков (принята j
не зависящей от температуры и состава смеси); X — координата, 4
направленная по течению холодного потока; е — доля исходной J
смеси, пропускаемая через теплообменник (при отсутствии байпаса 4
е = 1).
344
Перепишем уравнения (VIII.70), (VIII.71), введя безразмерную
координату х = Х/L (где L — длина теплообменника) и безразмер-
ный параметр а = k^PL/yv.
dTi/dx = (a/s)(T2-T1) (VIII.72)
dTzldx = a (Г2-Г1) (VIII.73)
Очевидно, температура горячего потока на входе в теплообмен-
ник (при х = 1) равна температуре на выходе адиабатического реак-
тора Твых! а температура на входе адиабатического слоя Твх опреде-
ляется условием смешения потока исходной смеси, пропускаемого
через теплообменник, с байпасным потоком, остающимся при тем-
пературе исходной смеси Т*. Таким образом,
для уравнений (VIII.72) и (VIII.73) имеют вид;
Л(0)=т*; т2(1) = тВыХ;
е7’1(1)+(1-е)Г* = Твх (VIII.74)
Связь между величинами ТВых и Твх опре-
деляется характеристиками процесса в адиа-
батическом реакторе. Если начальная темпе-
ратура Твх фиксирована, то температура на
выходе из адиабатического слоя ТВых может
быть найдена решением задачи Коши для
уравнений типа (VIII.47) и (VIII.48). Согласно
данных разделов VII.4, VIII.2, каковы бы ни
были схема реакций и кинетика процесса, ука-
занная задача имеет единственное решение,
однозначно определяющее функцию Твых (Твх)
или АТ (Твх) (где А Г = ТВых — Твх — перепад
температуры в адиабатическом слое). Очевидно,
граничные условия
Рис. VIII.7. Адиаба-
тический реактор (7)
с внешним теплооб-
менником (2).
производная % = dTBMjdTBX представляет собой параметрическую
чувствительность температуры на выходе реактора к температуре
на входе (см. раздел VIII.2).
Связь между температурами ТВых и Твх, согласно (VIII.74)
определяется также процессами, идущими в теплообменнике. Тем-
пературу Твх можно выразить через ТВыХ, решив систему уравнений
(VIII.72) и (VIII.73) с граничными условиями (VIII.74). Умножая
уравнение (VIII.72) на —8 и складывая его с уравнением (VIII.73),
находим, что данная система имеет инвариант:
Z2—еТ1 = const = Т2 (1)— sTi (1) =ДТ —(1 —е) Т*
(VIII.75)
Используя соотношения (VIII.74), находим
Ь-Г—*)]
(VIII.76)
345
откуда
где
Твх-Т* = ДТ/(Хо-1)
(VIII.77)
1/е—ехр [ —а (1 —е)/е] Лых- Г*
%0 1—ехр [—а(1—е)/е] Ё(ТВХ — Т*)
(VIII.78)
Величина 1/(е%0) имеет смысл чувствительности температуры
холодного потока' на выходе
горячего потока на его входе.
из теплообменника к температуре
Легко заметить, что всегда %0 > 1.
В частном случае е — 1 формула
(VIII.78) обращается в неопределен-
ность, раскрытие которой дает:
Xo=l + a-i (VIII.79)
При уменьшении параметра е ве-
личина Хо монотонно возрастает,
причем Хо 00 ПРИ е -> 0. G умень-
шением параметра а величина Хп
также увеличивается от Хо ~
при а -> оо до х0 = оо при а = 0.
Температура на входе реактора
То определяется теперь решением
уравнения:
Рис. VIII.8. Графическое рейхе- дг(гвх)-(%о D (Твх Т*) (VIIL80)
ние уравнения (VIII.80). Это уравнение всегда имеет по
крайней мере одно решение. Дей-
ствительно, левая часть уравнения (VIII.80) представляет собой
ограниченную функцию, поскольку перепад температуры в адиаба-
тическом слое не может превосходить некоторой максимальной
величины, которая достигается в том случае, если все возможные
в данной системе реакции проходят до конца. В то же время правая
часть уравнения (VIII.80) может изменяться в любых, пределах;
поэтому всегда должно найтись хотя бы одно значение Твх, при кото-
ром равенство (VIII.80) будет удовлетворено. Это утверждение спра-
ведливо для процесса с произвольной стехиометрией и кинетикой.
Если производная левой части уравнения (VIII.80) по Твх пре-
вышает величину Хо — 1 в некоторой области значений Твх, т. е.
если параметрическая чувствительность больше критического зна-
чения х = Хо» уравнение (VIII.80) может иметь несколько решений,
причем число их обязательно нечетно.
Характер эволюции решений уравнения (VIII.80) при измене-
нии параметров процесса можно исследовать с помощью рис. VIII.8.
Кривая линия на нем представляет собой график левой части урав-
нения (VIII.80), а прямые 1—5 — график его правой части при раз-
личных значениях температуры исходной смеси Т *. Увеличению
Т * соответствует параллельный сдвиг прямых вправо. При малых
346
Т * (прямые 1—4) существует только одно низкотемпературное
решение. При дальнейшем росте Т * происходит скачкообразное
увеличение Тах и процесс переходит в высокотемпературный режим,
являющийся в этих условиях единственным. При понижении исход-
ной температуры Т * процесс остается в высокотемпературном
режиме вплоть до значения Т *, соответствующего прямой 2, после
чего процесс скачком переходит в низкотемпературный режим.
Здесь, очевидно, встречаемся с тем же явлением гистерезиса, ко-
торое наблюдается в экзотермических процессах на внешней по-
верхности частицы катализатора, в пористом зерне и в реакторе
идеального смещения (см. разделы III.3 — III.5, VII.3). Очевидно,
скачкообразные переходы невозможны, если наклон левой части
уравнения (VIII.80), равный % — 1, нигде не превышает величины
%0 — 1. В точках касания кривой АТ (Х'вх) и прямой линии вели-
чина % равна %0; это условие определяет, таким образом, крити-
ческие точки скачкообразного перехода между режимами. Анало-
гичным образом исследуется эволюция решений при изменении
величины %0 [которому соответствует изменение угла наклона
прямой, изображающей правую часть уравнения (VIII.80)] или при
изменении параметров процесса в адиабатическом слое, влияющих
на вид левой части уравнения (VIII.80). При сложной кинетике
химического процесса кривая и прямая линии могут пересекаться
не только в одной или трех, но и в большем числе точек, однако
во всех случаях скачкообразный переход между режимами
будет происходить при соблюдении условия касания обеих линий
X = Хо-
Если имеется три стационарных решения, то среднему из них
соответствует величина параметрической чувствительности % > у0.
Такой стационарный режим должен быть неустойчивым, поскольку
в этих условиях малые возмущения стационарного режима усилива-
ются, проходя реактор и теплообменник (так как %0 > 1). В общем
случае, когда имеется 2п И-1 точек пересечения кривой и прямой
линий на рисунке типа рис. VIII.8, п промежуточных решений обяза-
тельно должны быть неустойчивыми. Соблюдение неравенства
X < Хо является необходимым условием устойчивости процесса .*,
однако, чтобы доказать достаточность этого условия, нельзя ограни-
чиваться анализом одних только стационарных уравнений и необхо-
димо исследовать поведение процесса в нестационарных условиях
(см. ниже).
Единственная реакция. Рассмотрим более подробно случай,
когда в адиабатическом слое протекает только одна экзотермическая
реакция с произвольной кинетической функцией г (С, Т). В этом
случае уравнения процесса в адиабатическом слое интегрируются
* Необходимое условие устойчивости 7 Уо было получено в работах
[5, 14].
- 347
в квадратурах (см. раздел
мулу (VII.29) в виде
VII.4). Перепишем интегральную фор-
5 =
Твых
dT
(VIII.81)
где S — время контакта в адиабатическом слое; гад (Т) — адиабати-
ческая скорость реакции, определенная формулой (VIII.51).
Если учесть соотношение (VIII.77), то правая часть уравнения
(VIII.81) будет функцией только температуры на входе^адиабатиче-
ского слоя Твх. Само соотношение (VIII.81) при этом может рас-
сматриваться как уравнение для определения неизвестной величины
Твх, это уравнение, очевидно, полностью эквивалентно общему
уравнению (VIII.80). Функция гад (Т) обращается в нуль при при-
ближении к равновесию или полном выгорании лимитирующего
исходного вещества, которому, согласно соотношению (VIII.81),
соответствует достижение максимального перепада температур в
адиабатическом слое AT = hCBx. Если зависимость скорости необра-
тимой реакции от концентрации ключевого вещества С при С = О
аналитическая (а это на самом деле всегда так — см. раздел II.1),
то интеграл в формуле (VIII.81) всегда стремится к бесконечности
при АГ -> fi,CBx. Скорость обратимой реакции стремится к нулю
при достижении равновесной концентрации С = Ср (Т). Так как
в окрестности равновесия г — С — Ср интеграл в (VIII.81) обра-
щается в бесконечность при температуре Т^х, удовлетворяющей
уравнению
[Свх Ср (Т’вых (^вх))1 — (Хо 1) (^шс ^*)
(VIII.82)
При выводе этой формулы использованы соотношения (VIII.49)
и (VIII.77). При Твх = Г*, согласно формуле (VIII.77), АГ = О,
Гвых = Т* и правая часть уравнения (VIII.81) обращается в нуль.
Таким образом, это уравнение всегда имеет решение, лежащее
в интервале Т* < Твх < КСВХ для необратимой или Т* < Твх <^ТВХ
для обратимой реакции. Если зависимость правой части уравнения
(VIII.81) от Твх имеет экстремумы, то это уравнение может иметь
несколько решений.
В качестве примера рассмотрим реакцию n-го порядка. Тогда:
г (С, Т)^к(Т) Cn = Ze~E/RT Сп'>
(VIII.83)
гад(П = ^(7’)1Свх-Й-1(7’-7’вх)]Л '
Вводя безразмерную температуру 0 = Е (Т — T*)!RT* и пара-
метры
н = k{T*]Rs^hE (vin.84)
348
Сканировал: NeptUliyi
Магнитогорск
и принимая, как обычно, приближенную формулу для зависимости -
константы скорости реакции от температуры к (Т) = к (Т*) е9,
преобразуем уравнение (VIII.81) к безразмерной форме:
Х«0ВХ _д
Н= J (VIII-85)
Овх
При п = 0 зто уравнение принимает вид:
[х =е-9вх—е-х°9вх =<р (0ВХ) (VIII.86)
Дифференцируя уравнение (VIIL86), находим, что в критической
точке перехода между режимами, которой соответствует экстремум
функции <р (9ВХ), должно' быть выполнено соотношение:
евх = -^ТГ ’ (VIII.87)
В соответствии с этим существует два стационарных решения
при
= (Хо — 1) Xo"Z°/("Z“ “ 4) (VIII.88)
а при р, > ц* стационарных решений нет. Этот результат находится
в кажущемся противоречии с общим утверждением о числе решений.
Надо, однако, помнить, что реакция может идти по нулевому по-
рядку только до исчерпания исходного вещества; при С, близкой
к нулю, порядок реакции изменяется с нулевого на первый, поэтому
всегда существует еще один стационарный режим, при котором на
части реактора концентрация исходного вещества близка к нулю
и реакция практически не идет. То же относится ко всем реакциям
с порядком п 1.
При п = 1 уравнение (VIII.85) принимает вид
(х = е-(в+0в1с){ЁТ(0)-Ш[0-0вх(хо-1)]Ы<р(0вх) (VIII.89)
где Ei (х) — интегральная экспоненциальная функция, определен-
ная формулой (VI 1.30).
Для графического решения уравнение (VIII.81) удобно предста-
вить в виде
0+1пр,-|---= 1п [ЁТ(0)-ЁТ(0-Д0)1 = ф(Д0) (VIII.90)
Хо — 1
Правая часть этого уравнения зависит только от параметра 0,
а левая изображается прямой линией с наклоном 1/(%0 — 1) и
ординатой 0 + In (1 при Д0 = 0. Функция ф (ДО) в интервале
0 ДО < 1 монотонно возрастает, причем йф/йДО обращается в бес-
конечность при ДО -► 0 и ДО -> 0. Отсюда следует, что при любом
0 > 0 найдется такое значение Хо > 1, чт0 при 1 <С Хо <С Хо урав-
нение (VIII.89) будет иметь три решения в некотором интервале
И* <С Н < Р*. Очевидно, величина (хо—I)”1 равна производной
функции ф (ДО) в точке перегиба и возрастает с увеличением 0.
349
Анализ устойчивости. Для строгого обоснования условий устой-
чивости системы реактор — теплообменник необходимо исследовать,
как изменяются со временем малые возмущения стационарного
режима. Решим эту задачу для частного случая е = 1 (система без
байпаса) [15]. Очевидно, малое возмущение температуры холодного
потока на выходе теплообменника тх (1), возникшее в некоторый
момент времени t, после прохождения реактора усилится в % раз (где
— параметрическая чувствительность температуры на выходе
адиабатического слоя к температуре на его входе) и спустя время
5Х (равное суммарному времени прохождения потоком реактора и
трубопроводов, связывающих реактор с теплообменником) вызывает
возмущение температуры горячего потока на входе в теплообменник
т2 (1) = %тх (1). Связь между возмущениями т2 и тх определяется
уравнениями, описывающими нестационарный режим теплообмен-
ника. Если линейные скорости горячего и холодного потоков одина-
ковы, то нестационарные уравнения имеют вид:
Этх/91-|-атх/аг = а (т2—тх) ' (VIII.91)
dti/dt — dxi/dx = ~а (т2 — Ti) (VIII-92)
Эти уравнения записаны в безразмерной форме, аналогичной
(VIII.72) и (VIII.73), причем за масштаб времени t принято время
прохождения через теплообменник L/u. Граничные условия для
уравнений (VIII.91), (VIII.92) имеют вид
П(О, t) = 0; т2(1, t) = XTi(l, I-®) (VIII.93)
где со = Siii/L.
Будем искать решение задачи (VIII.91)—(VIII.93) в виде:
xk(x, t) = eKir\k(x) (к = 1,2) (VIII.94)
Тогда уравнения и граничные условия для функций т]1 (х),
•ц2 (х) принимают вид
dr]i/dx = a (Т]2 — 41) — Хгц (VIII.95)
dx\i/dx = a (р2 — 4i)-|“X'42 (VIII.96)
Ш(0)=0; р2(1) = хщ(1)е-Ял) (VIII.97}
Исследуемый стационарный режим устойчив, если задача
(VIII.95)—(VIII.97) имеет нетривиальные решения только при
ReX 0, т. е. если числа % имеют отрицательную действительную
часть.
Решение системы уравнений (VIII.95), (VIII.96), удовлетворя-
ющее граничному условию цх (0) = 0, имеет вид
iH = ash (/%2 + 2Ха г) (VIII.98)
42 = а { [(Л+ а)/а] sh (/V + 2Xa х) + [ />.2-|-2Ха/а] ch (1ЛА.2+2Ха х) j
где а — константа.
350
Используя теперь второе граничное условие (VIII.97), получаем
трансцендентное уравнение для определения числа X:
<р(Х) = (а+1)-1 (% + <*+ /Л.2+2Ш cth /А.2 +2Ха) = (х/%о)е-шХ (VIII-99)
где Хо = (а + 1)/а — критическое значение параметрической чув-
ствительности в точке скачкообразного перехода между стационар-
ными режимами процесса.
Поставленная выше задача сводится теперь к исследованию
условий, при которых все корни уравнения (VIII.99) находятся
в левой полуплоскости комплексной плоскости %.
Рассмотрим положение действительных корней уравнения
(VIII.99). Графиком левой части этого уравнения <р (А.) при действи-
тельных значениях А является кривая, которая может иметь в обла-
сти — 2а < А < 0 несколько ветвей, выходящих на вертикальные
асимптоты. (Число таких ветвей определяется величиной а.) При
положительном А функция <р (X) монотонно возрастает, причем <р (0) =
1. Кривая <р (А) пересекается экспоненциальной кривой, изобра-
жающей правую часть уравнения (VIII.99), в нескольких точках
(при малых а возможно пересечение только в одной точке), которые
дают положение действительных корней уравнения (VIII.99). Поло-
жение самого правого корня зависит от величины параметра %!
при % > Хо этот корень находится в правой полуплоскости, а при
X Хо все действительные корни уравнения (VIII.99) отрицательны.
Таким образом, анализ нестационарных уравнений подтверждает,
что условие X < Хо — (1 + а)/а является необходимым для устойчи-
вости стационарного режима, так как выполнение этого условия
обеспечивает отрицательность действительных корней уравнения
(VIII.99). Для полного решения поставленной задачи необходимо
определить положение комплексных корней и найти условия, при
которых все комплексные корни находятся в левой полуплоскости
комплексной плоскости ц. Довольно сложные выкладки, которые
здесь не будут приведены, доказывают, что при | X I < Хо правая
полуплоскость комплексной плоскости р. отображается правой
и левой частями уравнения (VIII.99) на неперекрывающиеся области
[15]. Таким образом, при X > 0 соблюдение неравенства х < Хо
является не только необходимым, но и достаточным условием устой-
чивости процесса.
Это утверждение перестает быть справедливым в случае х <С О-
Можно показать, что при больших отрицательных значениях х»
превосходящих по абсолютной величине Хо> области, на которые
отображают правую полуплоскость правая и левая части уравнения
(VIII.99), перекрываются. В этом случае всегда можно найти такое
значение со > 0, что уравнение (VIH-99) будет иметь комплексный
корень с. положительной действительной частью. Такой стационар-
ный режим является неустойчивым, причем нестационарные реше-
ния, соответствующие малым начальным возмущениям, удаляются
от него путем колебаний с нарастающей амплитудой. Однако в случае
единственной реакции с произвольной кинетикой параметрическая
351
чувствительность % всегда положительна. В случае сложного
процесса в принципе возможны отрицательные значения однако
появление таких значений, тем более превышающих по абсолютной
величине единицу, представляется весьма маловероятным.
Доказательство достаточности условия х < Хо для устойчивости
стационарного режима с положительной % может быть проведено
и в самом общем случае путем следующих рассуждений. Заметим,
что, если корень уравнения (VIII.99) (или другого, более сложного
уравнения, которое получится при анализе нестационарных урав-
нений теплообменника при е ¥= 1) с наибольшей действительной
частью Л.о является комплексным, то температурные возмущения
Рис. УЦ1.9. Номограмма
* для определения скорости
Хо затухания возмущений.
должны быть знакопеременными. Между тем, при % > 0 положи-
тельное возмущение температуры на входе в адиабатический слой
приводит И положительному возмущению температуры на выходе
из реактора или, что то же самое, температуры горячего потока на
входе в теплообменник. Из баланса теплообменника следует, что
возмущение температуры холодного потока на выходе из теплообмен-
ника, а следовательно, и температуры на входе в реактор также
будет положительным. Следовательно, возмущение остается знако-
постоянным в любой момент времени, а это может быть только в том
случае, если число Хо действительно. Действительное же число А.
может стать положительным только при х > Хо-
Скорость затухания возмущении. С помощью уравнения (VIII.99)
легко определить величину собственного числа с наибольшей дейст-
вительной частью Л.о, которая является мерой скорости затухания
возмущений в устойчивом процессе. Возмущения затухают со вре-
менем пропорционально еЛо< и при А.о, хотя и отрицательных, но ма-
лых по абсолютной величине, затухание может быть слишком мед-
ленным. Очевидно, этого можно ожидать при больших значениях
параметра а, когда критическое значение Хо близко к единице.
Величину Хо удобно определять графически с помощью рис. VIII.9
[13]. Кривые, изображенные на рис. VIII.9, представляют собой
графики функции q> (Л) — левой части уравнения (VIII.99) при раз-
352 >
личных значениях а. Приведены только ветви этих кривых, соответ-
ствующие наибольшему числу Хо, в области X < 0. Масштаб по оси
ординат — логарифмический, поэтому правая часть уравнения
(VIII.99) изображается прямой линией (пунктирной) с наклоном
— со и ординатой х/%0 при X = 0. Искомое значение Хо определяется
как абсцисса точки пересечения зтой прямой с кривой, соответству-
ющей данному значению а. Очевидно, величина | Хо | возрастает
с уменьшением параметров а, <о, При а > л и <о 1 достаточно
точной верхней оценкой для | Хо | служит величина
%» = а(1 —/1 —л2/а2) (VIII. 100)
поскольку при X -> X* <р (X) — оо. При х/%0 ~ 1 величину | Хо |
можно найти по следующей приближенной формуле, которая полу-
чается апроксймацией функции ф (X) при X, близких к нулю, прямой
с единичным наклоном:
А. I 1 ~Х/Хо 1 —« (X — 1)
0 1 + со = (1-|- а) (1 + со)
(VIII.101)
VIII.4. АВТОТЕРМИЧЕСКИЕ РЕАКТОРЫ
С ВНУТРЕННИМ ТЕПЛООБМЕНОМ
Экзотермические процессы часто проводят в трубчатых реакторах
с внутренним теплообменом, используя в качестве теплоносителя
исходную смесь (рис. VIII.10). При этом одновременно осуществляет-
ся отвод тепла из зоны реакции и
подогрев исходной смеси до темпе-
ратуры реакции, и процесс можно
проводить автотермически. Ис-
следуем стационарные режимы
противоточного реактора с внут-
ренним теплообменом, предпола-
гая, что как реагирующая смесь,
Исходная смесь |
ГТ ^^Теплоноситель Продукт
Реактор WW
< *-Теплоноситель Г
Рис. VIII.10. Охлаждение трубча-
того реактора.
так и исходная смесь — теплоно-
ситель движутся в режиме идеального вытеснения- и что в зоне
реакции протекает одна необратимая реакция с кинетической
функцией *
г (С, T)=Ze~ElRTF (С)
(VIII. 102)
где Z — предэкспоненциальный множитель; Е — энергия актива-
ции; R — газовая постоянная; F (С) — некоторая функция концен-
трации ключевого исходного вещества.
Уравнения, описывающие стационарный режим процесса в зоне
реакции, совпадают с (VII.35), (VI 1.36).
Приведем расчетные уравнения к безразмерному виду, введя
безразмерные переменные: концентрацию с = С/Свх, температуру
* Излагаемая здесь задача решена в работе [16х].
23 Заказ 1441
353
реагирующей смеси 0 = Е (Т — TBX)/RTlx и теплоносителя 0Х =
= Е (Тг — TBX)/RTlx и продольную координату х — Х/L. Исполь-
зуя, как обычно, приближенную экспоненциальную зависимость
константы скорости реакции от безразмерной температуры, приво-
дим уравнения, определяющие стационарный режим процесса, к виду
dc]dx = — (р./0) е0 / (с)
dQfdx — v.efij (с) — а (0 — 01)
dQ1/dx = — а (0 — 01)
(VIII.103)
(VIII. 104)
(VIII.105)
где
hELk (Твх) F(CBX) JiCBXE .
yuRT*x > yRT*x ’
k^LP
a — —---
yv
/(0 =
F(C)
F (CBX)
(VIII.106)
CEX и TBX — концентрация исходного вещества и температура исход-
ной смеси; L — длина реактора; v — объемная скорость потока;
Р — периметр поверхности теплообмена.
Граничные условия для безразмерных переменных имеют вид:
с(0)=1; O(O) = 0i(O); 0i(l) = O (VIII.107)
Умножая уравнение (VIII.103) на 0 и складывая уравнения
(VIII.103)—(VIII.105), находим, что эта система уравнений имеет
инвариант:
0с J-0 — 0i = const= 0 (VIII.108)'
Используя соотношение (VIII.108), можно выразить с через
0 — 0j. Вводя новую переменную — перепад температур между
реагирующей смесью и теплоносителем, у = 0 — 0Х, и вычитая
уравнение (VIII.105) из (VIII.104), получаем
^- = ue'‘f (е~У) (VIII.109)
dx • 0
0 = In[/p/(^)J (VIII.110)
Дифференцируя соотношение (VIII.НО) по х, имеем:
+ (VIII.111)
az у
Лодставляя соотношения (VIII.108) и (VIII.111) в уравнение
(VIII.104), сводим систему уравнений (VIII.103)—(VIII.105) к одному
уравнению относительно неизвестной функции у (ж)
(±Z£.)/0/(±Zl)J (у')2 + ауу' = 0 JVIII.112)
Граничные условия для уравнения (VIII.112):
г/(0) = 0; П1) = И/(--~^(1)-)^(» (VIII.113)
354
Анализ исходной системы уравнений показывает, что величина
у = 0(1— с)—монотонно возрастающая функция координаты х, по-
скольку с монотонно убывает. Для интегрирования уравнения
(VIII.112) понизим его порядок, введя новую переменную z (у) =
= У' {х):
г' = ° (VIII.114)
При переходе от уравнения (VIII.112) к (VIII.114) мы пользуемся
тем, что функция у (х) монотонно возрастает и, следовательно,
z = у' > 0. Интегрируя уравнение (VIII.114) с учетом второго
граничного условия (VIII.ИЗ), находим
2
^0 -1!
Р У\е yi dy-i
I /(V1
(VIII.115)
у
где у0 = у (1).
Разделяя переменные в уравнении (VIII.115) и
получаем в неявном виде зависимость у (х):
у
интегрируя,
е yidy1
0— У1
0
у,
' § У2е~Уг
У1
(VIII.116)
в —г/2
0
о
Здесь постоянная интегрирования выбрана так, что у (0) = 0. При-
нимая во внимание, что при х = 1 у должно быть равно у0, полу-
чаем из (VIII.116) трансцендентное уравнение для определения
неизвестной величины у0:
(VIII.117)
Исследуем вид функции <р (у0). При у0 = 0 <р (0) = 0. Для реак-
ции нулевого порядка / = 1 и у0 формально может принимать любые
положительные значения. В этом случае при у0 -> оо функция <р
стремится к асимптотическому значению:
I оо
Ф*= С dy . и (VIIL118)
J р + а (1 + у) е~У
о
Нетрудно убедиться, что при достаточно больших значениях
У о ф'О/о) <0 и, следовательно, функция <р (у0) имеет максимум.
График функции <р (у0) при а = 2 и р, = 0,25 представлен на
рис. VIII.11. Из рис. VII.11 видно, что при рассматриваемых значе-
ниях параметров задача имеет два стационарных решения. Если
23*
355
увеличить параметр р,, то максимальное значение функции ф (у0)
уменьшится и при некотором ц = Щ станет равным единице. При
Ц > система не имеет стационарных решений. Если уменьшить
параметр р,, то величина <р*(р) возрастает и при некотором р — ра
станет равной единице; при р, < ра система будет иметь только
одно стационарное решение. Результаты численного расчета крити-
ческих значений рц и р2 при различных значениях а представлены
на рис. VIII.12.
Рис. VIII.11. График функ-
ции <р (уо):
1 — нулевой порядок реакции;
а = 2, ц =s 0,25; 2 — первый
порядок, 6=5, а = 0,8, ц =
= 0,Г, з — первый порядок,
6 = 5, а = 0,2, ц = 0,2.
Рис. VIII.12. Критические значения параметров,
соответствующие точкам скачкообразного пере-
хода между режимами работы реактора с внут-
ренним теплообменом:
а — реакция нулевого порядка; б — реакция первого
порядка, 6=5.
Отметим, что, так как в действительности реакция прекращается
при выгорании лимитирующего исходного вещества, т. е. при у0 =
= 0, функция ф (у0) для реакции нулевого порядка (например,
изображенная на рис., VIII.11) должна быть дополнена вертикаль-
ной прямой, соответствующей у0 = 0. При этом оказывается, что
система имеет три решения при ц2 < [i < [ij и одно — при значе-
ниях параметра р,, лежащих вне этого интервала.
Для реакции n-го порядка (п > 0) / (с) = сп, и уй могут изме-
няться в пределах О уй < 0. В этом случае при i/0 -> 0 функция
<р (Уо) логарифмически расходится и при любых значениях парамет- 1
ров 0, р,, а существует по крайней мере одно стационарное решение.
При монотонном возрастании функции ф (у0) стационарное решение
всегда единственно (рис., VIII.И). Если же функция ф (у0) имеет
максимум и минимум, то существует область значений параметров, j
при которых число стационарных решений равно трем. При фикси-
рованных значениях параметров 0 и а можно найти значения пара- ’
метра (j,, при которых появляются множественные режимы, из I
условия: 1
фмакс (Pl) — I! Фмин(р2)=1 (VIII.119)
356
Результаты численного расчета зависимости щ и ц2 от а при
0 = 5 приведены на рис. VIII.12. Из рис. VIII.12л б видно, что обе
критические кривые сходятся в точке (акр, РкР), которая опреде-
ляется из условий:
ф(Уо) = 1; ф' (#о) = ф"(Уо) = 0 (VIII.120)
Иными словами, в этом случае значение функции <р (у0) в точке
перегиба должно равняться единице, а ее производная — нулю.
В области, расположенной между критическими кривыми, система
имеет три стационарных решения, а вне этой области — одно.
Не все найденные таким образом стационарные режимы явля-
ются устойчивыми относительно малых возмущений. Если считать,
как обычно, что в случае, когда существует единственное решение,
оно всегда устойчиво, а в случае, когда стационарных решений три,
устойчивы только два крайних — высоко- и низкотемпературное
решения, то выявляется следующая картина перехода между режи-
мами процесса при постепенном увеличении параметра ц. При
р, < ц2 существует единственное низкотемпературное решение. При
р,2 < р < щ существуют два устойчивых режима — высоко- и
низкотемпературный. Так как, однако, для перехода между этими
режимами необходимо возмущение большой амплитуды, реактор
будет оставаться в низкотемпературном режиме вплоть до значения
параметра р, = рп выше которого существует только высокотемпе-
ратурный режим. При обратном ходе (уменьшение параметра р.)
реактор скачком перейдет из высокотемпературного режима в низко-
температурный при р, = р,2.
Возникновение множественных режимов, переход между кото-
рыми происходит скачкообразно при плавном изменении параметров
процесса, и связанные с этим явления неустойчивости стационарных
состояний представляют собой органический недостаток автотерми-
ческих схем. Недостаток этот, очевидно, вызван характерным для
автотермических реакционных узлов переносом тепла теплоносите-
лем против течения реагирующей смеси, приводящим к задержке
и возможному разрастанию случайных возмущений температурного
режима процесса. Те же явления наблюдаются и в другой автотер-
мической/схеме, рассмотренной в разделе VIII.3, — адиабатическом
реакторе с внешним теплообменником. Неустойчивость режимов
возможна, хотя и значительно менее вероятна, и в тех технологиче-
ских схемах, где тепло реакции отводится с помощью независимого
теплоносителя.
Во всех случаях появление неустойчивых режимов тесно связано
с нарушением единственности решения уравнений, описывающих
стационарный режим процесса. Для любой технологической схемы
реактор — теплообменник можно доказать следующие утверждения
[17]: 1) цо крайней мере, один стационарный режим существует
всегда; 2) при изменении условий процесса могут попарно возникать
дополнительные стационарные режимы, причем один режим из
357
каждой пары обязательно будет неустойчивым. Строгое доказатель-
ство устойчивости стационарных режимов пространственно распре-
деленных систем наталкивается на значительные затруднения.
Главная трудность здесь состоит в решении вопроса о возможности
возникновения нарастающих колебаний. Однако в случае гетеро-
генных процессов этот вид неустойчивости маловероятен и обычно
можно считать, что потеря устойчивости может происходить только
при тех значениях параметров процесса, при которых изменяется
число стационарных решений («в точках ветвления»).
VIII.5. УСТОЙЧИВОСТЬ РЕАКЦИИ
НА ПОРИСТОМ ЗЕРНЕ КАТАЛИЗАТОРА
Точки ветвления. Рассмотрим реакцию на пористом зерне катали-
затора. Если сопротивлением массо- и теплопередаче на внешнюю
поверхность частицы можно пренебречь, то система стационарных
уравнений всегда может быть сведена (см. раздел III.4) к единствен-
ному уравнению для безразмерной температуры 0
у20_|_(Х/(0)=о (VIII.121)
с граничным условием 0 (Г) = 0. (Здесь р — квадрат модуля Тиле.)
Пусть при некотором значении параметра р = р0 решение уравне-
ния (VIII.121) имеет вид 0 = 0О (х). Если представить решение,
соответствующее какому-либо соседнему значению параметра р
(мало отличающемуся от р0) в виде 0 = 0О (х) + 0j (х) (где 0j (х) —
малая добавка), то получим линеаризованное уравнение для функ-
ции 0Х (ж):
V20i + fx/i(90(z)) 0i = 0 (VIII.122)
где (0О (ж)) = (<///</©) 9 = 9О(Ж).
Можно показать [181, что, если при р = р0 уравнение (VIII.122)
с граничным условием 0Х (Г) =0 имеет ненулевые решения, то это
значение параметра является точкой ветвления решений уравнения
(VIII.121), т. е. что в этой точке появляется пара новых решений.
Оба решения можно в окрестности точки ветвления представить
в виде
0 (z)=0o (z)-J-a г'е фо (z) (VIII.123)
где е = р — р0; Фо (х)— собственная функция уравнения (VIII.122)
при р = р0; а — параметр, который определяется по формуле
a=±J/-A (VIII.124)
где
n = J<₽oW/(0o(^))^ (VIII.125)
^з = У <гг> (ж)/2 (0o(z))dz
358
Рис. VIII.13. Зависи-
мость нормы решения
от параметра и.
I
/2 = — (d2//d92)e=9i) ;(ж) и оба интеграла берутся по всему объему
зерна. Если 11/13 < 0, то величина а — действительная и при е > О,
т. е. р, р,0 существуют два решения, а при р, <; ц0 решений нет
(точка ветвления I типа). Если 1Х113 >0, два решения имеются,
наоборот при р, < р,0, а при р, > р,0 решений нет (точка ветвления
II типа — см. рис. VIII. 13). В простейшем случае (соответствующем
тому, что разбирался в разделе III.4) между точками ветвления I
и II типа лежит ветвь неустойчивых решений.
Выведем необходимое условие появления точек ветвления (а сле-
довательно, и множественных режимов процесса на пористом зерне).
Рассмотрим с этой целью уравнение:
у2ф-!-ц/1 (ж) ф= Хф (VIII.126)
При р, = 0 все его собственные числа %
отрицательны. По мере увеличения р, спектр
уравнения (VIII.168) сдвигается вдоль дейст-
вительной оси. Для того чтобы собствен-
ные значения возрастали с увеличением р,
[так чтобы при некотором значении этого па-
раметра уравнение (VIII. 126) имело бы не-
нулевые решения при % = 0, когда оно сов-
падает с (VIII.122)1 необходимо, чтобы по
крайней мере в некоторой области внутри
зерна функция (х) была положительной.
Собственная функция <р0, соответствующая наибольшему собствен-
ному числу %0, всегда положительна и, так как в экзотерми-
ческом процессе / (0) > 0, интеграл 1Х положителен. При этом для
возникновения в рассматриваемой системе точки ветвления II типа
необходимо, чтобы интеграл 13 был также положителен. Так как
<р0 )> 0, для положительности интеграла 13 необходимо, чтобы функ-
ция /2 (ж) была положительной по крайней мере в некоторой области
внутри пористого зерна. Сказанное позволяет связать условия
существования точек ветвления с параметрами функции / (0) [18].
В случае реакции га-го порядка имеем:
/(0) = еО(0_9)п (VIII.127)
/1 = е® (0 — 0)п—1 (0 — 0 — га) - (VIII.128)
/2 = е9 (0 _ 9)п-2 [(0 _9)2 _2п (0 -0)+п (п- !)] (VIII.129)
Здесь 0 — безразмерный максимальный разогреваем, раздел III.4).
Из формулы (VIII.128) следует, что /1>0 при
О<0<0—га
а из формулы (VIII.129), что /2 >0 при
О<О<0— п—Vn
0 — n-\-Yn <0<0
(VIII.130)
(VIII.131)
359
Из формулы (VIII.130) видно, что точка ветвления может по-
явиться только при 0 п, так как в противном случае /х всегда
отрицательна. Формула (VIII. 131) позволяет усилить это условие.
Учитывая, что "точка ветвления II типа, в которой кончается непре-
рывная ветвь решений, соответствующих кинетическому режиму
протекания реакций, должна находиться в низкотемпературной
области, видим, что область температур, при которых /2 >> 0, может
появиться только при
e>n+Vn (viir. 132)
В частности, для реакции первого порядка это условие имеет
вид © ^> 2. Очевидно, выведенные условия не зависят от формы
зерна катализатора. При © sg п + решение единственно при
любых значениях параметра ц (или модуля Тиле = р^ц).
Условия устойчивости. При строгом анализе условий устой-
чивости процесса на пористом зерне катализатора мы должны,
как обычно, записать систему нестационарных уравнений и линеари-
зовать ее в окрестности исследуемого стационарного режима. Затем,
разыскивая решения в виде комбинации экспонент типа elt, прихо-
дим к некоторой задаче на собственные значения. Если эта задача
имеет ненулевые решения только при % с отрицательными действи-
тельными частями (или, иначе говоря, если ее спектр лежит в левой
полуплоскости комплексной плоскости %), то исследуемый стацио-
нарный режим устойчив.
Наиболее просто решается задача об устойчивости в случае,
если эффективные коэффициенты диффузии всех веществ и коэффи-
циент температуропроводности пористого зерна равны между собой
(разными методами эта задача решалась в работах [19—21]). В этом
случае все собственные значения действительны и потеря устойчи-
вости может наблюдаться только в точках ветвления. При очень
больших и очень малых значениях модуля Тиле процесс всегда
устойчив, и он должен оставаться устойчивым вплоть до самой точки
скачкообразного перехода в другой режим (т. е. до точки ветвления
решений). Промежуточные режимы; лежащие на ветви решений между
точками ветвления I и II (см. рис. VIII.13), всегда неустойчивы.
Случай равенства коэффициентов диффузии и температуропро-
водности, однако, нетипичен. Нарушение же этого условия сильно
усложняет задачу исследования устойчивости, так как при этом
собственные значения уже комплексные и, становится возможным
возникновение нарастающих колебаний. Здесь были аналитически
исследованы различные предельные случаи [22, 23], а также про-
ведены численные расчеты [22, 24]. Ниже приведен приближенный
расчет [2U, демонстрирующий, что колебательная неустойчивость
не должна возникать и при нарушении равенства коэффициентов
диффузии и температуропроводности (D и %/у).
Рассмотрим простейший случай реакции с участием одного исход-
ного вещества. Принимая во внимание, что в кинетической области
360
концентрация и температура внутри зернаТ катализатора почти
постоянны, будем считать производные безразмерной кинетической
функции (см. раздел II 1.4), вычисленные в условиях стационарного
режима, а = (дЦдс)„, Ъ = (5//50)ст, не зависящими от координаты.
Тогда задача на собственные значения записывается в виде
Vari—pavi—рЬг0 = Ьх; У1(Г) = О (VlII 133)
у2уо-|-вр,ау]1-|-0}1буо = рЛУо? го(Г) = О
где Р = yDly;, v-je^, voext — отклонения безразмерных концентра-
ции и температуры (соответственно) от их стационарных значений.
Собственные значения X задачи (VIII. 133) связаны с собствен-
ными значениями оператора Лапласа для данной формы зерна фл,
а собственные функции уравнений v — с собственными функциями
уравнения
у2фл+флфп=0 v (VIII.134)
Будем искать решение, соответствующее собственному значению
в виде:
v^=Bn^n (VIII.135)
Подставляя уравнения (VIII.135) в (VIII.133) и учитывая
(VIII.134), получаем:
А (^п+Ра+Фп)+^Ра=О
-Лвца+5(Хлр-ец& + %) = 0 1 >
Из условия разрешимости системы уравнений (VIII.136) находим
уравнение для определения Х„:
pxj+M’MP+i)—в(ье—P«)J-WS—w(b8—«)=о (viii.137)
Уравнение (VIII.137) имеет мнимые корни при
в+г
Н=^-ь#^р7 (VIII.138)
и
Р<а/56<1/Р (VIII.139)
Наименьшее значение р, при котором могут появляться мнимые
собственные значения, соответствует Р = 0, n = 1, и равно фх/Ь0.
Из условий (VIII.139) видно, что появление мнимых собственных
значений в кинетическом режиме практически не может наблюдаться.
Д1режде всего, обычные значения Р для пористых катализаторов
превосходят единицу. Кроме того, поскольку фх >1 (в частности,
для плоской пластинки фх = л3/4, а для сферической частицы
фх = л2), даже при Р 1 мнимым корням соответствуют значения
параметра ц, при которых нарушаются условия протекания реакции
в кинетическом режиме. Таким образом, на непрерывной ветви реше-
ний, начинающейся с р. = 0 и соответствующей кинетическому
режиму протекания реакции, не возникает явлений колебательной
неустойчивости и решения из этой ветви устойчивы вплоть до точки
ветвления решений стационарных уравнений. Хотя мы пользовались
361
при исследовании простыми граничными условиями v (Г) = О,
указанный результат является общим, поскольку в кинетическом
режиме внешняя тепло- и массопередача не играет существенной
роли (см. раздел III.2).
При анализе устойчивости процесса в диффузионном режиме
следует учесть, что в этом случае реакция локализуется в тонком слое
близ внешней поверхности пористой частицы. Благодаря большой
скорости химической реакции флуктуации концентрации должны
чрезвычайно быстро затухать вне этого слоя, и только флуктуации
температуры могут свободно распространяться по всему объему
зерна Путем теплопроводности. Переходные процессы в тонком
реакционном слое должны протекать весьма быстро; поэтому при
анализе устойчивости можно считать, что этот слой всегда работает
в стационарном режиме и учитывать только наиболее медленный
нестационарный процесс распространения тепловых флуктуаций
в объеме пористого зерна. Исследуя процесс, протекающий в диффу-
зионном режиме, следует уже учесть сопротивление тепло- и массо-
переносу на внешней поверхности зерна. Учитывая упомянутые
выше допущения, записываем уравнения, описывающие нестацио-
нарный процесс, протекающий в диффузионном режиме, в виде
дТ/dt' = (х/у) у2Т (VII1.140)
-Р/(С<00-^о) = т/гдиф(С,о. Го) (i = l, 2,- ..,S) (VIII.141)
-«(Гсо-То) = ^диф(С10, T0) + x(dT/dn)v (VIII.142)
Здесь индекс ,со отмечает значения переменных в потоке, омывающем
частицу катализатора, а индекс 0 — значения переменных на внеш-
ней поверхности частицы Г; рг и а — коэффициенты массо- и
теплопередачи от ядра потока к внешней поверхности частицы;
гДиф(Сго, У о) —скорость реакции, отнесенная к единице внешней
поверхности частицы.
Последний член в правой части уравнения (VIII.142) учитывает
теплообмен между тонким' реакционным слоем и внутренностью
частицы катализатора; п обозначает направление внешней нормали
к активной поверхности. Таким образом, при данной постановке
задачи уравнения процесса в тонком реакционном слое (VIII. 140),
(VIII.142) служат граничными условиями для уравнения тепло-
проводности (VIII.140). Вводя безразмерные переменные’и линеари-
зуя граничные условия (VIII.141), (VIII.142) в окрестности стацио-
нарного режима, имеем:
5t/5«=v2t (VIII. 143)
S-
Bi^o^vzjg-^^S/o + pvz-^ro 0 = 1,2............5) (VIII.144)
j-i
BiTT0 = ©p о + T° + (9V^)r (VIII. 145)'
/-1 1
362
Здесь Zb T — отклонения безразмерных концентраций и температуры
от их стационарных значений t = t'%/yZ2 — безразмерное время;
/Диф (сго, во) = гдиф/^г (Соо, Т’со) — безразмерная скорость реакции
p = Z2r (Coo, T^/D*^, 0 = hCoaD*/T*%, Bi, = ₽,//£»„ BiT = aZ/X -
безразмерные параметры, причем за масштаб скорости реакции при-
нята скорость реакции в кинетическом режиме.
Все производные вычисляются при стационарных значениях
безразмерных концентраций с, = С,/Соо и безразмерной температуры
9 = (Т - Тт)1Т*.
Решая систему S линейных алгебраических уравнений (VIII.144),
можно выразить все величины Zi о через т0; подстановка полученных
выражений в (VIII.145) приводит это граничное условие к виду
Лт0= (ат/аж)г (VIII.146)
где А — постоянная.
Полагая т — v (х)еи, приходим к краевой задаче
V2y_Ь=0; Av(T) = (dv/dx)r (VIII.147)
Задача (VIII.147) всегда имеет действительный спектр. Таким
образом, на непрерывных ветвях решений, соответствующих диффу-
зионному режиму, также не возникает явлений колебательной неустой-
чивости. При A < 0 все собственные значения отрицательны, и
соответствующий стационарный режим устойчив. При А )> 0 в спектре
задачи (VIII.147) есть положительное собственное значение, и ста-
ционарный режим неустойчив. При изменении параметра А смена
устойчивости происходит в результате перехода собственного значе-
ния через нуль в точке ^ветвления А = 0. Области А 0 соответ-
ствует область неустойчивых режимов, разделяющих внутри- и
внешнедиффузионную области протекания реакции.
ЛИТЕРАТУРА
1. А. М. Ляпунов, Общая теория устойчивости движения, ОНТИ,
1935.
2. Л. С. Понтрягин, Обыкновенные дифференциальные уравне-
ния, Физматгиз, 1961.
3. Р. Веллман, Теория устойчивости решений дифференциальных
уравнений, ИЛ, 1954.
4. А. М. Летов, Устойчивость нелинейных регулируемых систем,
Физматгиз, 1962.; Ж. Ла-Салл ь, С. Лефшец, Исследование устойчи-
вости прямым методом Ляпунова, Изд. «Мир», 1964.
5. С. Van Н ее г de n, Ind. Eng. Chem., 45, 1242 (1953); Chem. Eng.
Sci., 14, 1 (1958).
6. О. В i 1 о u s, N. R. Amundson, Am. Inst. Chem. Eng. J., 1,
513 (1955).
7. R. Aris, N. R. Amundson, Chem. Eng. Sci., 7, 121, 132, 148
(1958); 11, 199 (1958).
8. W. О ppelt, E. W i c k e, Grundlagen der chemischen Prozessrege-
lung, Munchen, 1964; E. D. Gilles, H. Hofmann, Chem. Eng. Sci., 15,
328 (1961); A. S. Foss, Chem. Eng. Progr. Symp. Ser., 55, № 25, 47 (1959);
L. L a p i d u s, Chem. Eng. Progr. Symp. Ser., 57, № 36 (1961); D. B. Spal-
ding, Chem. Eng. Sci., 11, 53 (1959); E. W i c k e, Z. Flektrochem., 65, 267
363
(1961); М. Т. С л и н ь к о, Кинетика и катализ, 1, 153 (1960); К. R. Wes-
ter te гр, Cherii. Eng. Sci., 17, 423 (1962); В. В. Кафа р ов, В. В. Е ре-
мейк о, ЖПХ, 35, 2251 (1962); Хим. пром., № 2, 45 (1963).
9. И. Е. С а л ь и и к о в, Б. В. В о л ь т е р, ДАН СССР., 152, 171
(1963); Б. В. В о л ь т е р, И. Е. С а л ь н и к о в, сб. «Моделирование и оп-
тимизация каталитических процессов, Изд. «Наука», 1965.
10. J. Е. L е a t h г u m, Е. F. J ohnson, L. L а р i d u s, Am. Inst.
Chem. Eng. J., 10, 16 (1964); J. S. Berger, D. D. Perlmutter, ibid.,
10, 233 (1964); R. B. W a r d e n, R. A r i s, N. R. A m u n d s о n, Chem.
Eng. Sci., 19, 149, 173 (1964).
11. N. R. A m u n d s о n, О. В i 1 о u s, Am. Inst. Chem. Eng. J., 2,
116 (1956).
12. J. Coste, N. R. A m и n d s о n, R. A r i s, Am- Inst. Chem.
Eng. J., 7, 124 (1961); S. L. Liu, N. R. Amundson, Ind. Eng. Chem.
Fund., 1, 200 (1962); S. L. L i u, R. A r i s, N. R. Amundson, Ind.
Eng. Chem. Fund., 2, 12 (1963); E. A. Grens, R. A. M с К e a n, Chem.
Eng. Sci., 18, 291 (1963).
. 13. Л. M. П ис ьме н, TOXT, 2, 63 (1968).
14. M. Г. С л и н ь к о, А. Л. M у л e р, Кинетика и катализ, 2, 467 (1961);
Дж. К. О р к а т т, Д. Е. Л е м б, Труды I конгресса ИФАК, Изд. АН
СССР, 1961.
15. Ю. И. X а р к а ц, Л. М. Письмен, В. И. Мукосей, ДАН
СССР, 174, 1385 (1967).
16. В. И. М у к о с е й, Л. М. П и с ь м е н, Ю. И. X а р к а ц, ИФЖ,
12, 703 (1967).
17. Л. М. Письмен, Физика гонения и взрыва, 2, 239 (1969).
18. Л. М. П и с ь м е н, Ю. И. X аркац, ДАН СССР, 168, 632 (1966)!
Л. М. Письмен, ' Ю. И. X а р к а ц, В. Г. Левин, Препринты
IV Международного конгресса по катализу, 1968.
19. J. Wei, Chem. Eng. Sci., 20, 729 (1965).
20. T. И. 3 е л е н я к, ДАН СССР, 171, 266 (1966); Дифф, уравнения,
3, 19 (1966).
21. Л. М. Письмен, Ю. И. Харкац, ДАН СССР, 179, 397 (1968).
22. N. R. A m u n d s о n, L.P. Raymond, Am. Inst. Chem. Eng.
J., 11, 339 (1965).
23. J. C. W. К u o, N. R. A m u n d s о n, Chem. Eng. Sci., 22, 49,
443, 1185 (1967).
24. M. L. M c G u i r e, L. Lapidns, Am. Inst. Chem. Eng. J., 11,
85 (1965).
Глава IX ________________________:_____________________
ОПТИМАЛЬНОЕ ПРОЕКТИРОВАНИЕ РЕАКТОРОВ [1-5]
IX. 1. ОПТИМАЛЬНЫЙ ТЕМПЕРАТУРНЫЙ ПРОФИЛЬ
Основные положения. В любое из расчетных уравнений хими-
ческого процесса входит ряд переменных: время контакта, темпера-
тура потока на входе в реактор и температура теплоносителя, ско-
рость потока, диаметр зерна катализатора и т. д., значения которых
можно изменять в более или менее широких пределах. Приступая
к проектированию химического реактора, необходимо выбрать
значения этих переменных так, чтобы добиться наилучшего резуль-
тата процесса. Число и номенклатура варьируемых переменных
определяются принятым типом реактора и его схемой. Последняя
также должна быть выбрана оптимальной, а этого в большинстве
случаев можно добиться только путем сравнения наилучших резуль-
татов процесса, достижимых в реакторах различных типов.
Понятие «наилучшего» должно обладать количественной мерой,
называемой критерием оптимальности. Любой применяемый кри-
терий оптимальности имеет экономическую природу и определяется,
во-первых, изменением состава, а следовательно, и стоимости реаги-
рующего потока в результате процесса и, во-вторых, затратами на
ведение процесса. Не все составляющие критерия оптимальности
имеют одинаковое значение. Некоторые из них могут быть настолько
малы, что их разумно не принимать во внимание, и в каждом кон-
кретном случае надо решать вопрос о том, каким' упрощенным ва-
риантом критерия оптимальности надо пользоваться.
Во многих случаях упрощение критерия оптимальности компен-
сируется введением дополнительных условий и ограничений, кото-
рым должно удовлетворять оптимальное решение. Простейшим,
чисто химическим критерием оптимальности может быть выход
целевого продукта.
Задача оптимального проектирования реактора определенного
типа сводится, таким образом, к разысканию максимального зна-
чения критерия оптимальности путем варьирования ряда независи-
мых переменных, допустимые значения которых обычно ограничены
технологическими пределами. Проведение процесса в рассчитанном
режиме даст наилучший результат, достижимый (в реакторе данного
типа) на данном катализаторе при принятых условиях и ограниче-
ниях. Сравнивая максимальные значения критерия оптимальности
для реакторов различных типов, можно ’ определить; какой тип
реактора предпочтителен для осуществления данного процесса.
Рассмотрим одну из простейших и в то же время теоретически
наиболее важную задачу оптимизации — определение оптимальной
температуры в каждом сечении реактора идеального вытеснения.
Состав смеси продуктов реакции на выходе трубчатого реактора
365
зависит от профиля температуры по длине аппарата. Очевидно,
должен существовать такой продольный температурный профиль,
при котором выход целевого продукта или, в общем случае, зна-
чение принятого критерия оптимальности будет максимальным.
Этот температурный профиль будет для данного процесса оптималь-
ным. Оптимальный температурный профиль (ОТП), как правило,
практически не может быть реализован,' однако к нему можно прибли-
зиться, применяя различного типа секционированные реакторы.
Теоретическая роль ОТП в реакторе идеального вытеснения состоит
в том, Что он дает наилучшие возможные показатели, достижимые
в процессе с данной кинетикой* — тот идеал, к которому следует
стремиться при проектировании промышленного процесса. Из ана-
логии между процессом в реакторе идеального вытеснения и пери-
одическим процессом следует, что в точности тот же результат будет
достигнут в периодическом реакторе с температурой, оптимальным
образом изменяющейся со временем. Подчеркивая эту аналогию,
будем использовать вместо продольной координаты X текущее время
контакта t = Х/и (где и — линейная скорость потока в реакторе
идеального вытеснения).
Верхний предел температуры. В некоторых случаях можно
сразу указать характер ОТП. Если рост температуры, ускоряя
процесс, увеличивает также, независимо от состава реагирующей
смеси, его избирательность (или, по крайней мере, не влияет на
последнюю), оптимальная температура должна быть как можно
более высокой.
Так, в случае единственной необратимой реакции повышение
температуры только увеличивает ее скорость, а в случае обратимой
эндотермической реакции — к тому же и смещает равновесие в сто-
рону образования целевого продукта. Если, помимо основной реак-
ции образования целевого продукта, имеется параллельная или (и)
последовательная побочная реакция с энергией активации, меньшей,
чем у основной, то повышение температуры увеличивает и скорость,
и избирательность процесса. Во всех зтих случаях температуру
процесса следует поддерживать на верхнем допустимом пределе Т*.
Эта предельная температура может определяться, например, услови-
ями скачкообразного перехода процесса в диффузионный режим, при
котором, вследствие сильного разогрева активной поверхности пла-
вится или дезактивируется катализатор или начинают идти незамет-
ные при низкой температуре побочные реакции. Другим фактором,
ограничивающим допустимую температуру процесса, может быть
возникновение при повышенных температурах нежелательных реак-
ций, идущих в объеме (вне поверхности катализатора) по цепному
механизму. Предельная температура Т* зависит от состава реаги-
* Исключение составляют «аномальные» реакции, тормозящиеся исход-
ными веществами или ускоряющиеся продуктами реакции, которые при опре-
деленных условиях выгоднее проводить в реакторе идеального смешения —
см. раздел VII.3.
366
рующей смеси и поэтому может изменяться по длине реактора. Так,
если необратимая сильно экзотермическая реакция первого порядка,
идущая на внешней поверхности непористых частиц, должна по
технологическим условиям проводиться в кинетическом режиме,
то верхний предел температуры определяется критическим соотно-
шением между параметрами р, и 0 в точке «зажигания» реакции,
выведенным в разделе III.3. Эта зависимость изображается кри-
вой 2 на рис. III.5; сами параметры р, и 0 определены формулами
(III.56), где под Сад й То, следует теперь понимать текущие значения
концентрации исходного вещества и температуры потока в данном
сечении реактора. Из зтих формул и вида кривой зажигания следует,
что максимально допустимая температура процесса повышается
с уменьшением концентрации исходного вещества. Если переход
в диффузионную область допустим, то после этого перехода скорость
реакции практически не зависит от температуры, так что дальней-
шее повышение температуры становится ненужным.
Обратимая экзотермическая реакция. В случае обратимой экзо-
термической реакции повышение температуры ускоряет обратную
реакцию сильнее, чем прямую, так что равновесие процесса сме-
щается в нежелательную сторбну. При некоторой температуре
скорость образования целевого продукта г (С, Т) проходит через
максимум; зта оптимальная температура зависит от состава реаги-
рующей смеси, уменьшаясь по мере уменьшения концентрации
исходного вещества. Таким образом, вначале, пока еще не накопи-
лось значительное количество конечного продукта, процесс выгодно
вести при высокой температуре, чтобы увеличить скорость прямой
реакции, а затем температуру следует снижать, чтобы, сместив равно-
весие в нужную сторону, добиться максимального выхода целевого
продукта. Температура в каждом сечении реактора должна быть
выбрана так, чтобы скорость образования целевого вещества в этом
сечении была максимальной, т. е., чтобы выполнялось равенство:
дг/дТ =0 ’ (IX.1)
Если температурная зависимость константы скорости реакции
определяется уравнением Аррениуса, то
Ei Es
r = Ze RT f(C)-Ze (IX.2
где Et, E2 — энергия активации; Zx, Z2 — предэкспоненциальные
множители соответственно прямой и обратной реакции; R — газо-
вая постоянная; / (С) и g (С) — кинетические функции прямой
и обратной реакций; в случае экзотермической реакции Е2 > Et.
Концентрации исходных веществ и продуктов реакции связаны
между собой линейными соотношениями (11.13); поэтому функции,
выражающие зависимость скорости прямой и обратной реакций от
концентраций реагентов, можно выразить через концентрацию С
одного из исходных веществ, которое принимается за ключевое.
Дифференцируя функцию (IX.2) по температуре и приравнивая
367
производную нулю, находим оптимальную температуру процесса
как функцию концентрации ключевого вещества
(,х-3)
где h = Е2 — Ег — теплота реакции.
Условие для оптимальной температуры можно привести к форме,
не зависящей от кинетических функций / (С), g (С) [6].' Обозначим
через Тр температуру, при которой смесь того же состава, что и в рас-
сматриваемом сечении, находилась бы в равновесии. Так как
Г(ГР) = О
’-“'/«"(й-Ягт)
Сравнение формул (IX.3) и .(IX.4) приводит к соотношению:
1 J_____R , е2
Т Тр hl Ei , (IX’5)
Конечно, переход от формулы (IX.3) к (IX.5) возможен только
в том случае, если равновесная температура, соответствующая дан-
ному составу, существует и кинетические функции / (С), g (С) пра-
вильно описывают кинетику процесса в окрестности равновесия.
Формулы (IX.3), (IX.5) показывают, что оптимальная температура,
так же, как и температура равновесия, снижается с увеличением
степени превращения. Так как при малых степенях превращения
обе величины неограниченно возрастают, в сечениях, близких
к входному, оптимальная температура не может быть достигнута
и температуру реакции следует фиксировать на верхнем пределе Т*.
В частности, в случае реакции первого порядка равновесная темпе-
ратура обращается в бесконечность при степени превращения т] =
= 1 — С/С^, равной
Т)? = (1+ВД)-1 . (IX.6)
и оптимальная — при степени превращения:
T)* = (l+E2Z2/£1Z1)-i<r1; (IX.7)
. Поэтому при степенях превращения ц < ц* повышение темпера-
туры^ всегда приводит к ускорению реакции.
Произвольная система реакций. Внимательный читатель заметит
нестрогость рассуждений, использованных выше при вычислении
оптимальной температуры. Выбор температуры в каком-либо сече-
нии реактора влияет на скорость реакции не только в данном сече-
нии, но и во всех сечениях реактора, лежащих ниже по течению
или (в случае периодического процесса) во все последующие*моменты
времени. Полученный результат для оптимальной температуры
обратимой экзотермической реакции оказывается, как это будет
ясно из нижеследующего, правильным. Однако, если пытаться,
например, искать оптимальное распределение температуры для про-
цесса, включающего две последовательных реакции Ах -> А2 -> А3,
368
также исходя из условия максимума скорости образования целе-
вого продукта А2 в каждом сечении, то придем к неверным выводам.
ОТП следует определять с учетом влияния температуры Т (t) в каж-
дом сечении реактора или в каждый момент времени t на характе-
ристики процесса при всех t' >> t. Задача поиска оптимума при этом
сильно усложняется, так как различные сечения реактора ие явля-
ются независимыми.
Рассмотрим процесс, включающий произвольное число химиче-
ских реакций и описываемый системой кинетических уравнений
dCijdt — ri (С, Т) (t=l, 2, . . ., К) (IX.8)
где Ct — концентрация i-ro ключевого вещества; г{ — скорость его
образования в единице объема реактора, определенным образом
зависящая от совокупности концентраций С К ключевых веществ
и температуры Т, подлежащей оптимальному выбору.
Зададим время контакта (или время реакции) 5^и будем искать
ОТП исходя из условия максимума линейной функции состава реаги-
рующей смеси на выходе аппарата:
к
U=2juz[Cf(5)-Cz(0)] (IX.9)
i-1
Величина U — критерий оптимальности — выражает прирост
ценности реагирующей смеси в результате химических превращений,
причем относительная ценность каждого из реагентов выражается
соответствующим коэффициентом щ. Если некоторые из продуктов
реакции не входят в число ключевых веществ, их концентрации
могут быть выражены через концентрации ключевых веществ с по-
мощью известных линейных соотношений (см. раздел II.2), что снова
приведет нас к линейной функции состава (IX.9). В частности, если
имеется только один целевой продукт (А,), его всегда можно принять
за ключевое вещество; тогда все коэффициенты щ, кроме одного,
будут равны нулю, а величину U], характеризующую целевой про-
дукт, можно без потери общности принять равной единице.
Величина критерия оптимальности U зависит от всего темпера-
турного профиля Т (t), т. е. является функционалом от функции
Т (t). Отыскание функции, приводящей заданный функционал
к максимуму, является основной задачей вариационного исчисления,
на основе которого и должна решаться поставленная в этом разделе
задача.
Задача отыскания максимума функционала U при фиксированном
времени контакта S эквивалентна задаче отыскания безусловного
максимума функционала
к
P=V-XS = ^ щ [Ct (S)-Ct (0)]-M (IX.10)
i-l
где X — множитель Лагранжа.
24 Заказ 1441 369
К задаче на максимум функционала Р приходим и в том случае,
если, наряду с ОТП определяется и оптимальное время контакта S,
причем учитываются дополнительные расходы на оборудование
и катализатор, связанные с увеличением времени контакта. Величина
S пропорциональна объему реактора и, если считать, что названные
затраты являются лцнейной функцией объема, придем к критерию
оптимальности (IX.10) с коэффициентом %, имеющим смысл «стои-
мости единицы времени контакта». Следует отметить, что учет затрат
на катализатор и амортизацию оборудования, даже при их относи-
тельно малой величине, почти всегда существенно влияет на опти-
мальное решение: если не принимать их во внимание, то для широ-
кого класса процессов придем, пытаясь одновременно выбрать ОТП
и оптимальное время контакта S, к нелепому выводу — оптимальный
процесс должен продолжаться бесконечное время при нулевой тем-
пературе. Всякое ограничение времени контакта верхним пределом
или введение с той же целью минимально допустимой температуры
было бы произвольным решением, совершаемым, в конечном счете,
исходя из тех же самых экономических соображений, но только не
выраженных количественно.
Учитывая уравнения (IX.8), можно записать критерий оптималь-
ности (IX. 10) в виде
S / к \
Р= J У, И/Г; —X dt
0 \г=1 /
Наивысшее значение критерия оптимальности
от исходного состава потока С0‘.
(IX.И)
зависит только
S
<р(С0)= max Р= max f
т (t), s т (0, s 0J
(IX.12)
Вывод уравнений, определяющих ОТП, может быть проделан
наиболее наглядно на основе метода динамического программирова-
ния Веллмана [7] *. Разделим интервал (0, S) на две части: короткую
первую секцию (0, $) и остаток (s, S). Воспользовавшись элементар-
ным свойством интеграла, можно переписать уравнение (IX. 12)
в виде
<р(С0)= max
т (О, s
' s / к \ s / к \
J I У uiri — X I <#+ j I у utri — X j dt
.0 \i=l / s \ i=l /
(IX.13)
Очевидно, сумма (IX.13) может быть максимальной только в том
случае, если второй интеграл принимает наивысшее значение, зави-
сящее только от состава потока С (я) в точке t = s. Следовательно,
, s 7 К \
<p(C0)=max f У utr[~ % <# + ф(С (s))
г (О. s[0J /
(IX.14)
* Задача определения ОТП впервые решена этим методом в работах Ариса
II, 8]. Та же задача решена классическим методом в работах [9, 10].
370
Максимум в (IX.14) достигается выбором оптимальной функции
в интервале (0, з) более узком, чем первоначальный интервал (О, S).
При малом з, разлагая интеграл и функцию <р (С (з)) в ряд Тейлора
по s, получаем
' ( к \ к
(p(C0)= max I У % *+<р (Со) + У -Jr- о$ + 0 ($2)
т (Г), s \ / ** dc,«o
L \i-»i / 1=1
(IX.15)
где индексом 0 отмечены значения переменных при t = 0; 0 (з2) —
сумма членов, зависящих от квадрата и более высоких степеней
малой величины з.
Функцию (р (Со) в правой части уравнения (IX.15) можно, оче-
видно, вынести из-под знака максимума, как не зависящую от Т (2)
и S. После деления на з и перехода к пределу з -> 0 получаем из
(IX.15):
max
т (о, s
f-и1) Г1 о~Л. =0
(IX.16)
Подобное рассуждение справедливо не только для точки 2 = 0,
но и для любой точки в интервале 0 2 ==: 5. Поэтому индекс 0
в (IX. 16) может быть опущен. Вводя вспомогательные, так называе-
мые сопряженные переменные
^^Ui^-d^/dCi (2 = 1,2,... К) (IX.17)
переписываем уравнение (IX.16) в виде:
/ к \
max S’l’zn-X =0 (IX.18)
tw,s ]
Таким образом, при оптимальном выборе температуры в каждом
сечении должна достигать наивысшего значения функция
к
Н = (IX.19)
1=1
причем величина Н повсюду постоянна и равна нулю.
Функция Н впервые введена в классическом вариационном
исчислении (см., например, [И]) и называется функцией Гамильтона
или гамильтонианом. Условие максимума гамильтониана может быть
получено и классическими вариационными методами, однако, в отли-
чие от них, метод Веллмана позволяет сделать важный вывод: опти-
мальному решению соответствует наивысшее значение гамильто-
ниана, достижимое в заданной ограниченной области допустимых
температур, причем это значение не обязательно должно соответство-
вать аналитическому максимуму. Другой метод, позволяющий дать
более строгий вывод условий оптимальности в ограниченной области,
предложен Понтрягиным [121. Принцип максимума Понтрягина
24*
371
эквивалентен выведенному условию максимума гамильтониана
(IX. 19).
Если величина Н неограниченно возрастает с повышением темпе-
ратуры, оптимальная температура реакции должна быть, очевидно,
фиксирована на верхнем пределе Т*. Выше уже упоминались при-
знаки и примеры процессов, для которых оптимальная температура
повсюду равна максимально допустимой. Теперь можно назвать
и математическое свойство этих процессов: гамильтониан Н должен
быть монотонно возрастающей функцией температуры или неогра-
ниченно возрастать с ее увеличением при любых значениях перемен-
ных С{, фг (i — 1, 2, ..., К). Если существует аналитический макси-
мум гамильтониана, то оптимальное значение температуры должно
удовлетворять обычному необходимому условию максимума функции,
которое получаем, дифференцируя уравнение (IX.19) по Т:
к
2М = ° (IX.20)
i=l
Здесь штрих обозначает частную производную по температуре.
Отметим, что в случае единственной реакции (К = 1) уравне-
ние (IX.20) эквивалентно условию максимума скорости реакции
в каждом сечении (IX. 1), так что полученный выше результат для
оптимальной температуры обратимой экзотермической реакции
является справедливым. Если же число реакций превышает единицу,
условие максимума локальной скорости образования целевого про-
дукта, очевидно, не приводит к оптимальному решению. Заметим
для дальнейшего, что дифференцирование по температуре в (IX.20)
может быть заменено дифференцированием по любой величине, явля-
ющейся однозначной монотонной функцией температуры и исполь-
зуемой как мера последней. В качестве такой меры часто удобно
использовать константу скорости одной из реакций.
Уравнение (IX.20) вместе с уравнением
л.
Я=24>гг,—Л=о (IX.21)
г=1
составляет систему уравнений первого порядка в частных производ-
ных, решением которой определяется ОТП в области, не стесненной
температурными ограничениями. От системы (IX.20), (IX.21) можно
перейти к более удобной для расчета системе обыкновенных диффе-
ренциальных уравнений, называемых характеристическими и опи-
сывающих изменение переменных Ct, ф, (i = 1, 2, ..., К), а также
оптимальной температуры по ходу потока. Из теории уравнений
в частных производных известно, что если уравнение выражено
в форме
Н (C’i, С%, . . . , Cg, фц фа, • • . , Ф^) = 0
372
его характеристические уравнения имеют вид
dCt ~ ЭН
dt — d^i
дН
dt ~ дС{
(1 = 1, 2.....К) (IX.22)
(i = l, 2, • • , К) (IX.23)
Дифференцируя гамильтониан по фг, видим, что характеристи-
ческими уравнениями типа (IX.22) служат кинетические уравнения
процесса (IX.8). Дифференцирование гамильтониана по Ct дает
динамические уравнения для сопряженных переменных
(IX.24)
1=1
где гу = drJdCj.
Для определения ОТП необходимо проинтегрировать систему
2К уравнений (IX.8), (IX.24). При этом, если наивысшее значение
гамильтониана Н достигается в точке, где он проходит через макси-
мум, то оптимальная температура в каждом сечении определяется из
уравнения (IX.20). Если же аналитический максимум гамильтониана
при данных значениях ф,-, С, не существует или же значение Н при
Т = Т* превышает его значение в точке максимума, то температура
фиксируется на верхнем пределе Т*.
Чтобы получить однозначное решение системы (IX.8), (IX.20),
(IX.24), нужно иметь 2К + 1 граничных условий (одно дополни-
тельное условие необходимо потому, что интервал интегрирования —
оптимальное время контакта — в общем случае не определен).
К граничных условий — начальные концентрации ключевых ве-
ществ — заданы на входе в реактор (при t = 0). На выходе из реак-
тора (при t = S) должны быть выполнены граничные условия
= (IX.25)
Эти условия следуют из определения сопряженных переменных
(IX.17), если учесть, что на выходе реактора производная в этом
выражении равна нулю. Граничное условие, которое должно быть
выполнено при оптимальном выборе времени контакта S — t, полу-
чаем, подставляя (IX.25) в (IX.21):
ч к
У, ищ—Х=о (IX.26)
i-i
Условие (IX.26) равносильно требованию равенства нулю ско-
рости прироста критерия оптимальности в выходном сечении реак-
тора, в чем можно убедиться, взяв производную от интеграла в выра-
жении (IX. 12) по верхнему пределу. Если бы левая часть равенства
(IX.26) была больше нуля, процесс было бы рационально продол-
жить, а если она отрицательна, это означало бы, что койец реак-
тора работает во вред йроцессу и должен быть отсечен.
373
Уравнение (IX.20) можно заменить при расчете на дифферен-
циальное уравнение для оптимальной температуры Т. Дифференци-
руя (IX. 20) по t, имеем
(IX-27)
получаем:
(IX. 28)
где. штрих обозначает производную по температуре.
Используя динамические уравнения (IX.8), (IX.24),
4г = 2 2 I 2
i=l i=l i=l
Граничное условие для оптимальной температуры, заданное на
конце реактора, с учетом условий (IX.20) и (IX.25) имеет вид
К :
2«^' = 0 (IX.29)
г-1
Следует помнить, что уравнение (IX.28), как и уравнение (IX.20), ;
из которого оно выведено, справедливо лишь в том случае, когда
оптимальная температура определяется из условия аналитического
максимума гамильтониана Н; в противном случае оно исключается
из расчета и температура фиксируется на предельном допустимом s
значении Т = Т*.
Даже в простейших случаях система уравнений (IX.8), (IX.24), -j
(IX.28) не поддается аналитическому интегрированию. Машинное 1
время, необходимое для численного решения, резко возрастает |
с увеличением числа ключевых веществ, поскольку граничные уело- |
вия для концентраций и сопряженных переменных заданы на разных 1
концах реактора и при решении приходится использовать метод «
последовательных приближений. Тем не менее, исследование расчет- i
ных уравнений часто позволяет, не решая их, сделать важные каче- 1
ственные выводы о форме ОТП. I
Последовательные и параллельные реакции. Исследуем форму s
ОТП на примере процесса, включающего две последовательных j
реакции Ат -> А2 -> А3 с аррениусовской температурной зависимостью
констант скорости обеих реакций *. Если А2 — целевой продукт
и энергия активации побочной реакции больше, чем основной,
гамильтониан (IX.19) с повышением температуры проходит через
максимум, и для решения можно воспользоваться уравнением
(IX.28). В качестве меры температуры выберем константу скорости
основной реакции к±. Так как, согласно уравнению Аррениуса
Ei Е,
ki^Zie RT ; k2 — Z2e RT (IX.30)
* Эта задача впервые решена Амундсоном и Билоусом [13].
374
то константа скорости побочной реакции может быть выражена
формулой:
k2 = pkf, е, = Ег)Е1, p = Z2Z;° (IX.31)
Таким образом, кинетические уравнения процесса принимают вид
dC1/dt=~kf(C1)=r1 (IX.32)
dC2/dt = /c/(Ci)-pA:Bg(C2) = r2 (IX.33)
где / (Ci), g (С 2)— кинетические функции, выражающие зависи-
мость скоростей обеих реакций от концентраций реагентов.
Индекс при константе скорости основной реакции здесь опущен.
Так как, согласно (IX.32), (IX.33)
г{=—/, r'2 = f—pek^g, ri=0,
г 2 = — pg(s — 1) k*~*g, r11=—kf, ri2=0,
Г21 = kf, r2i = — pk^g'
уравнение для оптимальной температуры (IX.28) принимает вид
(lx.34)
dt г g
где штрих в уравнении (IX.34) означает производную по С2-
Сопряженные переменные в это уравнение не входят; поэтому
ОТП может быть найден совместным интегрированием трех уравне-
ний (IX.32)—(IX.34). В случае реакций первого порядка (/= С1,
g — С2) число расчетных уравнений можно уменьшить, введя отно-
шение концентраций целевого и исходного веществ y—C^Ci.
Тогда вместо (IX.32)—(IX.34) имеем:
dyldt = k(l+y)-pk*y (IX.35)
dk/dt^—к^/гу (IX.36)
Из уравнений (IX.34), (IX.36) следует, что оптимальное значе-
ние константы скорости основной реакции (а значит, и температуры
Т) всегда уменьшается по ходу потока, причем скорость уменьше-
ния температуры максимальна в сечениях, близких к входному,
где концентрация целевого продукта мала*. При С2->0 к -> оо,
поэтому близ входа температура всегда должна быть фиксирована
на верхнем пределе Т*. Интересно отметить, что уравнения (IX.34),
(IX.36) формально не теряют смысла и при е < 1 (т. е. когда Е л > Е2),
однако в этом случае они определяют температурный профиль,
приводящий не к максимальному, а к минимальному значению
критерия оптимальности.
♦ Исключение представляет «аномальный» случай, когда побочная реакция
тормозится целевым продуктом, так что g' < 0.
375
В случае двух параллельных реакций кинетические уравнения
принимают вид:
dCi/df=-fc/(C1)-pfceg(Ci) = r1 , (IX.37)
dC2/di = fc/(Ci) = r2 (IX.38)
Здесь, как и раньше, константа скорости побочной реакции
с помощью соотношений (IX.31) выражена через константу скорости
основной реакции, принятую за меру температуры. Уравнение
(IX.28) в рассматриваемом случае приводится к виду
здесь штрих — производная по С t.
В это уравнение уже входит сопряженная переменная фьи потому
необходимо учесть определяющее ее уравнение:
<Й|>1/<Я = -fc/' + W'+pA’Hipi (IX.40)
Расчет ОТП ведется совместным решением уравнений (IX.37),
(IX.39), (IX.40). Как и в рассмотренном выше случае, это решение
имеет смысл только при е > 1. Концентрация целевого продукта
в эти уравнения не входит, так что вводить в расчет уравнение
(IX.38) нет необходимости. В случае, когда обе реакции идут по
первому порядку (/ = g = Ct, /' = g' = 1), система расчетных
уравнений упрощается, сводясь к виду:
dk/dt = fca/si|>i (IX.41)
*!>!/<#=—fcd-iM+pfcti (IX-42)
От концентрации исходного вещества С± расчетные уравнения
(IX.41), (IX.42) уже не зависят. В случае, когда непрореагировавшее
исходное вещество не регенерируется, п1 = 0, и на выходе реактора
Ipt = 0. Тогда, если исключить аномальные случаи f <; 0, g' <Z 0,
из уравнения (IX.40) следует, что величина ф.1 заключена в пределах
0 =5 < 1 и уменьшается по ходу потока. Следовательно, правая
часть уравнения (IX.39) или (IX.41) положительна и оптимальная
температура возрастает по ходу потока [10, 14], причем скорость
ее увеличения нарастает с уменьшением ф1, т. е. с приближением
к выходу реактора. На выходе ф1 0 и А: °о, так что в сечениях
реактора, близких к выходу, температуру следует фиксировать на
верхнем пределе Т*.
В процессе, включающем как последовательную, так и парал-
лельную побочные реакции, оптимальная температура может как
повышаться, так и понижаться по ходу потока [14], в зависимости
от соотношения между энергиями активации реакций. При этом,
если энергия активации основной реакции меньше двух других,
оптимальная температура повсюду остается конечной и верхний
предел допустимой температуры, как правило, не достигается.
376
Разрывные ОТП. В более сложных процессах возможна ситуация,
когда оптимальная функция Т (t) имеет разрыв. В качестве примера
рассмотрим две последовательных обратимых реакции первого
порядка
1 з
Ai Аг А3
2 4
с аррениусовской температурной зависимостью констант скорости
kt (i = 1, 4). Выражая, как выше, все kt (i = 2, 3, 4) через кон-
станту скорости основной реакции kit записываем кинетические
уравнения процесса в виде
dCi/dt^ -kCt+p^Ci (IX.43)
dCi/dt=kCi - (p2fce* + psfce«) C2 + p4fcs‘ (C - Ci - C2) (IX.44)
где C — постоянная суммарная концентрация веществ At, А2, А3 и
Pi=ZiZ~*‘ (IX.45)
Индексы при к, Е, р, е соответствуют номерам реакций на схеме;
индекс при ki в уравнениях (IX.43), (IX.44) опущен. Согласно
(IX.43), (IX.44) гамильтониан (IX.19) принимает вид:
Н (к) = (фг - ф4) kCj. - (ф2 - ф!) р2Аг'С2 -
-фгрз^Са+фгр^'ЧС-С!—С2) -% (IX.46)
Исследование функции Н (к) показывает, что при е2 е4> ₽3 > 1
или es е4^> е2 1 она может иметь два максимума, относительная
величина которых будет изменяться по длине реактора по мере
изменения величин С4, С2, фь ф2. Оптимальная температура опре-
деляется положением самого высокого максимума; поэтому в сечении,
где величины обоих максимумов сравняются, должен возникнуть
скачок оптимальной температуры. Разрывные ОТП могут наблю-
даться также при е4>е2, е3 >1. В этом случае функция Н (к)
неограниченно возрастает при А и, кроме того, может иметь
один аналитический максимум. Близ входа реактора, пока реагиру-
ющая смесь состоит почти исключительно из исходного вещества А 4,
оптимальная температура будет (как и при всех других соотношениях
между энергиями активации реакций) фиксирована на верхнем пре-
деле Т*. Затем, по мере накопления целевого продукта А2, оптималь-
ная температура, соответствующая аналитическому максимуму га-
мильтониана, будет опускаться. Далее, когда накопится достаточно
большое количество вещества А3, величина Н (к (Т*)) превысит
значение гамильтониана в максимуме и оптимальная температура
скачком повысится до предельного значения Т = Т*. Другие схемы
реакций, приводящие к разрывным ОТП, рассмотрены в работе [15].
Очевидно, наличие скачка оптимальной температуры указывает на
то, что данный процесс рационально проводить в последовательно
377
соединенных реакторах с промежуточным подогревом или охлажде- 1
нием реагирующей смеси. 1
Учет диффузионного торможения процесса. Выше мы отмечали, 1
что верхнее ограничение допустимой температуры часто оказывается |
связанным с опасностью перехода процесса в диффузионный режим. 1
Представляет интерес исследовать оптимальный температурный
профиль, учитывая диффузионное торможение процесса в явном J
виде. При этом оказывается возможным решить задачу, не вводя j
искусственного верхнего температурного предела. 1
Рассмотрим процесс, включающий две последовательные необра- i
тимые реакции первого порядка Aj -> А2 А3, протекающие на ]
внешней поверхности катализатора с константами скорости (Тп) (
И х2 (Т’п), зависящими от температуры активной поверхности Тп *.
Очевидно, именно в процессах, в которых возможно дальнейшее |
превращение целевого продукта, диффузионное торможение может i
играть наибольшую роль, оказывая сильное влияние на их избира- J
тельность. Если и С2 — концентрации веществ А! и А2 в потоке, I
С1П, С2п — концентрации тех же веществ у поверхности катализа- j
тора ир — коэффициент массопередачи, который будем считать I
одинаковым для обоих веществ, то уравнения баланса вещества J
у активной поверхности имеют вид: j
₽(С1-С1п) = Х1С1п, ₽(С2п-С2)=Х1С1п-Х2С2п (IX.47) >
Определив из этих уравнений скорости образования веществ Ai
и А 2, отнесенные к единице активной поверхности, записываем
кинетические уравнения процесса в виде
(1Х-48> 1
да Xi + р
= (IX.49)
Р \ Xi + Р / Х2“(-Р
где а — площадь активной поверхности в единице объема реактора.
Уравнения (IX.48), (IX.49) удобно привести к безразмерному
виду:
dC\/dx =—aCi = ri (IX.50)
dCi/dx = a (1 — Ь) Сг~bCz = r2 (IX.51) »
Здесь т — 0о7 — безразмерное текущее время контакта; а =
= p-i/(l + Hi), ъ = Иг /(1 + Нг), Hi = *i/₽, Н2 = *г/₽ — безраз-
мерные параметры.
Если температурная зависимость константы скорости реакции ki
и к2 определяется уравнением Аррениуса (IX.30), то:
Ц2 = РР-Це=-^
P=z2zr), (1Х,52)
* Эта задача решена совместно с В. И. Мукосеем.
378
Величины а и b являются монотонными функциями температуры
поверхности Тп (а следовательно, и температуры потока Т) и ме-
няются с ростом температуры от 0 до 1; при этом значения а и Ь,
близкие к нулю, отвечают кинетическому режиму протекания соот-
ветствующей реакции, а значения, близкие к единице, — диффузион-
ному режиму.
Величину а, являющуюся монотонной функцией Тп и Т, можно
использовать как меру температуры. При этом задача выбора ОТП
при заданном времени контакта S сводится к вычислению оптималь-
ной функции а (т), определенной в интервале 0 =5 т =5 «(где s= 0oS—
безразмерное время контакта). Если оптимальная функция а (т)
найдена, то оптимальная температура реагирующей смеси в каж-
дом сечении реактора может быть определена по формуле
г=гп-ЖШГп=М1п(у--^-) (ix.53)
где q (Тп) — скорость тепловыделения на единицу активной поверх-
ности при температуре катализатора Т„, а — коэффициент тепло-
передачи от поверхности катализатора к потоку.
В рассматриваемом случае гамильтониан (IX.19) равен:
Н = 4’1г1+’|’2Г2 = ('ф2— Ф1) aCi — грг (abCi+ЬСг) — (IX.54)
Из формулы (IX.54) следует, что в отличие от исследованного
ранее случая, когда процесс протекает в кинетической области,
гамильтониан Н не возрастает неограниченно с увеличением темпе-
ратуры даже при е < 1. Действительно, при Т -> °° величины а и Ъ
стремятся к единице и гамильтониан (IX.54) становится отрицатель-
ным. Поэтому в рассматриваемом случае всегда существуют огра-
ниченные решения, вариационной задачи. Используя кинетические
уравнения (IX.50), (IX.51), приводим уравнение для оптимальной
температуры (IX.26) к виду
da__ 8 — 1 а2(1 — а)2
~dx Г~ ’ а [1 + е (1 -26)] +у [8 (1-26) - (1 -2а)] (1Х’55)
где у = Cz/Ci.
При а 0 уравнение (IX.55) сводится к уравнению для опти-
мальной температуры процесса, идущего в кинетической области,
(IX.36). Уравнение для переменной у (т) легко выводится из (IX.50),
(IX.51):
dy/dx = (a —6) 1/4-а (1-6) (IX.56)
Оптимальная функция а (т) определяется совместным решением
уравнений (IX.55), (IX.56) с граничными условиями:
при т = 0
у = 9
при X — S
y = a(l—a)/eb — a (IX.57)
Второе условие выводится из (IX.29) с учетом зависимости b
от a (IX.52).
379
Качественное исследование уравнения (IX.55) показывает, что
при е > 1 оптимальная температура, как и в случае процесса в отсут-
ствие диффузионных ограничений, убывает по ходу потока. При в <
< 1 оптимальная температура, напротив, возрастает по ходу потока,
что является специфической чертой рассматриваемой системы.
Указанные выводы подтверждаются численным расчетом опти-
мальных режимов, результаты которого представлены на рис. IX.1,
IX.2. Оптимальным обычно является режим протекания реакции,
близкий к кинетическому (а < 1/г), однако оптимальный режим
Рис. IX.1. Оптимальная функция
а (т) (р = 1, s — 10).
Цифры у кривых указывают значения па-
раметра г.
Рис. IX.2. Оптимальная функция а (т)
(s = 10).
Цифры у кривых указывают значения пара-
метра р.
сдвигается в сторону более высоких температур и больших значений
параметра а при уменьшении относительной скорости побочной
реакции р и времени контакта S.
В случае сильно экзотермических реакций, когда одновременно
существует несколько стационарных режимов процесса на активной
поверхности, может оказаться, что выбранное оптимальное значение
соответствует неустойчивому режиму гетерогенной реакции. В этом
случае надо учесть верхнее ограничение температуры, определяемое
условием «зажигания» реакции (см. раздел Ш.З).
IX.2. ОПТИМИЗАЦИЯ СТАДИЙНЫХ СХЕМ
Методы оптимизации стадийных схем. При проектировании
реальных технологических процессов оптимальному выбору подле-
жит лишь ограниченное число параметров. В этом состоит существен-
ное отличие практических задач оптимизации от задачи определения
ОТП, рассмотренной в разделе IX.1. В гибкой и эффективной реак-
380
торной схеме число варьируемых параметров Ф может, однако,
быть весьма большим. Примером такой схемы является цепочка
последовательно соединенных реакторов, размеры и режим работы
которых должны быть выбраны оптимальным способом.
Классический метод поиска максимума функции Ф переменных
состоит, как известно, в следующем. Определяются и приравни-
ваются нулю частные производные функции по всем независимым
переменным; в результате получается Ф уравнений, совместное
решение которых дает искомое положение максимума. Этот метод
чрезвычайно громоздок при большом Ф, а, кроме того, часто неосу-
ществим но той причине, что аналитический вывод уравнений, опре-
деляющих точку оптимума, невозможен. Другой причиной непри-
годности классического метода является наличие технологических
пределов варьирования независимых переменных. Может оказаться,
что критерий оптимальности вовсе не имеет максимума в аналити-
ческом смысле, а его наивысшее значение достигается на одной из
границ разрешенной области, т. е. когда одна или несколько неза-
висимых переменных фиксированы на предельных значениях.
Наиболее общим, но и самым трудоемким методом расчетного
поиска оптимума является «эксперимент» на математической модели.
Задавшись некоторой совокупностью значений независимых пере-
менных, всегда можно путем решения системы расчетных уравнений
вычислить соответствующее значение критерия оптимальности.
Чтобы найти оптимум, не обязательно испытывать все возможные
сочетания значений варьируемых переменных (для этого понадо-
бился бы фантастический объем вычислительной работы); как и при
экспериментальном поиске, здесь должен быть применен один из
методов направленного движения к оптимуму типа метода крутого
восхождения [16]. Поисковые методы редко бывают эффективны.
Кроме того, немаловажно, что ни один поисковый метод не может
дать информации об общей структуре оптимального решения для
ряда сходных задач.
Единый подход к решению широкого класса задач на разыскание
экстремума функции болыпо го конечного числа переменных дает
теория динамического программирования Веллмана [7]. Сущность
этой теории покажем на примере типичной задачи оптимизации,
возникающей в химической технологии. Требуется найти оптималь-
ный режим для последовательности 7V реакторов (или ^стадийного
аппарата), причем на каждой стадии варьируется М независимых
переменных. Пронумеруем реакторы в обратном порядке, так что
первый номер присваивается последнему, а АГ-й — первому по ходу
потока реактору. Состояние потока на выходе га-го реактора обо-
значим индексом к; в соответствии с этим исходное состояние потока
обозначается индексом N + 1 (рис. IX.3). Состояние реагирующего
потока в общем случае описывается некоторым вектором X. Вектор X
часто совпадает с вектором состава С; в более сложных случаях,
однако, компонентами вектора X могут быть, помимо концентраций
ключевых веществ, также и температура потока, давление и пр.
381
Если осуществить оптимальный выбор значений всех MN варьируе-
мых переменных, получаем максимальное значение критерия опти-
мальности, зависящее только от исходного состояния потока:
Вход
+
Выход
Рис. IX.3. Це-
почка последо-
вательно соеди-
ненных реакто-
ров.
|
+
N
max Pjy — (x.v+i) (IX.58)
Выделим из TV-стадийной последовательности
первый по ходу потока реактор и в соответствии
с этим разобьем критерий для TV-стадийного про-
цесса Ря на два слагаемых: критерий для первого
по ходу потока реактора p.v и критерий для остав-
шейся (N — 1)-стадийной последовательности Ру-\.
ру = Ру + ру-г (IX-59)
Очевидно, что режим TV-стадийной последова-
тельности в целом может быть оптимальным только
в том случае, если (TV — 1)-стадийная последова-
тельность работает в режиме, оптимальном относи-
тельно состояния потока, выходящего из TV-го реак-
тора. Действительно, сумма (IX.59) не может стать
максимальной, пока ее слагаемое Py-i не достигнет
максимального значения, зависящего только от со-
стояния потока Ху на выходе из TV-го реактора.
Подставляя (IX.59) в (IX.58) и заменяя, согласно
сказанному, Ру-г на флг-д (Ху), приходим к основ-
ному функциональному уравнению
<PN (xN+1)=max [Pjy+q’jv-i (xN)l (ix.6O)
!
которое представляет собой математическое выра-
жение принципа оптимальности, лежащего в основе
теории динамического программирования и сфор-
мулированного Веллманом: «Оптимальная страте-
гия обладает тем свойством, что, каковы бы ни были
исходное состояние и решение в начальный момент,
последующие решения должны составлять оптималь-
ную стратегию относительно состояния, получаю-
щегося в результате первого решения» [7].
В уравнении (IX.60) максимум достигается варьи-
рованием только параметров, управляющих процес-
сом на первой по ходу потока стадии. Принцип
оптимальности позволяет, таким образом, заменить
задачу одновременного выбора оптимальных значе-
ний MN независимых переменных гораздо более
простой задачей TV-стадийного выбора, на каждой
стадии которого оптимум достигается варьирова-
нием М переменных. Другой отличительной чертой поиска опти-
мума методом динамического программирования является то, что
задача решается не для единственного процесса с какими-то опре-
382
деленными параметрами исходного состояния, как это делается при
использовании метода крутого восхождения, а для совокупности
процессов с различными исходными состояниями. Действительно,
как результат решения получаем зависимость максимального зна-
чения критерия оптимальности от параметров исходного состояния
<p.v (Xn+i)-
Уравнение (IX.60) представляет собой соотношение, позволя-
ющее найти максимальное значение критерия оптимальности для
Лг-стадийной последовательности <p;v, если максимальное значение
критерия для (7V — 1)-стадийной последовательности <p.v-i уже
известно. Этот факт и определяет процедуру поиска оптимума мето-
дом динамического программирования.
Так как выбор оптимальных условий для реакторов, стоящих
в начале последовательности, должен осуществляться с учетом влия-
ния работы этих реакторов на процесс в последующих аппаратах,
а обратное влияние отсутствует, расчет А-стадийной последователь-
ности следует начинать с расчета реактора, последнего по ходу
потока, и вести, оптимизируя цепочки реакторов со все большим
числом членов, подключая к этой цепочке реакторы, все более уда-
ленные от конца последовательности. Начинаем о «последователь-
ности», содержащей ноль реакторов. В такой «последовательности»,
разумеется, никаких превращений не происходит и максимальное
значение критерия оптимальности <р0 (Xi) тождественно равно нулю.
Далее рассматриваем последовательность, состоящую из одного
реактора. Подставляя в (IX.60) N — 1 и <р0= 0, получаем:
<Р! (Х2) = max pi (IX.61)
Вектор Х2 описывает состояние потока, выходящего из реактора 2,
и потому пока неизвестен; поэтому приходится определять оптималь-
ные значения варьируемых параметров и соответствующие им макси-
мальные значения критерия оптимальности <pi (Х2) для некоторой
более или менее широкой совокупности исходных составов и табули-
ровать полученные результаты. После этого можно перейти к рас-
чету двухстадийной последовательности. Пользуясь принципом опти-
мальности (IX.60), находим оптимальные значения параметров,
управляющих процессом в реакторе 2. Варьируя эти параметры,
меняем состояние потока Х2 на выходе из реактора 2; при этом
изменяются как величина р2 (критерий для реактора 2), так и уже
вычисленное максимальное значение критерия оптимальности <fi (Хг)
для реактора 1. Максимизируется сумма этих величин; для реактора
2 оптимальный режим определяется, таким образом, с учетом не
только «локальной пользы», но и влияния работы этого реактора
на дальнейший ход процесса. После того как вычислена функция
Фа (Х3) (в определенной области значений исходных состояний Х3),
можно приступать к расчету трехстадийной последовательности
и т. д., вплоть до любого N.
Описанная общая схема метода динамического программирования,
впервые примененная к расчету химических реакторов Арисом [1,
383
17], является универсальной, но содержит два неприятных момента.
Первый из них — разыскание максимума в уравнении (IX.60),
выполняемое на каждой стадии. Здесь можно воспользоваться
поисковым методом крутого восхождения, что не вызовет больших
затруднений, так как поиск ведется только по малому числу М варьи-
руемых переменных. Второй, еще более неприятный Момент — необхо-
димость вычислять на каждой стадии максимальные значения кри-
терия оптимальности как функции параметров исходного состояния
и табулировать вычисленные величины. Эта чрезвычайно трудоем-
кая задача сильно ограничивает возможности практического при-
менения общей схемы метода динамического программирования
и делает ее подчас менее аффективной, Нем поисковый метод крутого
восхождения. Оба метода являются по своему характеру типично
машинными, так как связаны с многократным использованием
одних и тех же расчетных формул и уравнений, т. е. многократным
Повторением относительно простой программы. Вопрос о том, какой
из методов рационально применить при расчете, должен решаться
в зависимости от конкретных условий задачи. Заранее можно ска-
зать лишь то, что преимущества метода динамического программи-
рования будут сказываться при увеличении числа стадий и при
большей неопределенности исходного состояния потока. Рост числа
компонентов вектора состояния X, затрудняя задачу табулирования,
делает предпочтительным применением метода крутого восхождения.
От недостатков общей схемы метода динамического программи-
рования можно, однако, в значительной мере избавиться, используя
аналитический метод поиска оптимума на каждой стадии. Именно
этот способ будет применен к решению задач оптимизации цепочек
реакторов, рассматриваемых ниже. Отметим, что основные расчет-
ные формулы, которые получим, могут быть выведены не только
С помощью метода динамического программирования, но и на основе
дискретного варианта принципа максимума Понтрягина [18] или
классических вариационных методов.
Цепочка реакторов идеального смешения. При расчете оптималь-
ного режима процесса, протекающего в цепочке реакторов идеаль-
ного смешения, оптимальному выбору подлежат температуры Тп
и времена контакта 8п в каждом из реакторов. Рассмотрим задачу
оптимизации процесса, включающего произвольное число реакций.
Как и выше, примем обратную нумерацию реакторов (см. рис. IX.3).
Очевидно, состав потока в (и + 1)-м реакторе есть одновременно
состав на входе и-го реактора. Материальный баланс га-го реактора
по каждому из ключевых веществ записывается в виде (см. раздел
VII.3):
Сщ-СЬп+1-8пГ1(Сп, Тп) = 0 ' (1 = 1,2. . .. ,К) (IX.62)
Примем следующий критерий оптимальности для TV-стадийной
последовательности:
к N
PN=h u№fl-CbN+1)_x 2 sn (IX.63)
j=l П«1
384
Этот критерий аналогичен критерию (IX.10). Величина Pn может
быть разбита на ряд слагаемых рп, каждое из которых представляет
собой критерий оптимальности для n-го реактора:
N
PN~^1 Рп
п=1
К
Рп=% ч (Cf, „-Cj, л+1) - lSn (IX.64)
3=1
Максимальное значение <pw критерия оптимальности 7<у зависит
только от исходного состава потока С^+1. Используя принцип
оптимальности (IX.60) и выражение для критерия оптимальности
(1Х.(>4), имеем:
4>iv(CN+1)=max
’ к
S ui n+i)~~+
J=i
(IX.65)
, Максимум в уравнении (IX.65) достигается варьированием только
двух параметров — температуры 7jv и времени контакта 5 w в первом
по ходу потока реакторе. Дифференцируя (IX.65) по обоим пара-
метрам, получаем два расчетных уравнения
дф.У-1 \
dCjN ' dSN
d'pN-i \ dC3N __
dC3N 1 9Tn "
(IX.66)
(IX.67)
В эти уравнения входят важные для расчета величины, с кото-
рыми мы уже сталкивались в разделе IX.1 — производные макси-
мального значения критерия оптимальности по концентрациям клю-
чевых веществ. Дифференцируя (IX. 65) по С/, у+1 (i = 1, 2, ..., К),
получаем из рекуррентного соотношения для функций рекуррент-
ные соотношения для производных
__________’V’ / , д'Ру-! \ _
dCi, N+i__7 / дС\tN+l
3=1
(IX.68)
Фдрмулы типа (IX.66)—(IX.68) могут быть, очевидно, найисаны
не только для первого по ходу потока, но и для любого реактора
последовательности. Вводя обозначения
t/n=«/+^<Pn/^n+i (/ = 1, 2,. . . , К, 7i = l, 2,. . . ,ЛГ) (IX.69)
25 Заказ 1441 385
и заменяя индекс N на п, имеем:
Ь п-1
dCjn
dSn
(IX.70)
к
(п = 1, 2, . . , N)
к
dCfn
'' "-1 dTn
О
(п = 1, 2, . . . , N)
(IX.71)
к
/, П-1
dCjn,
n+1
. , К, п = 1, 2, . . . , N)
(IX.72)
(1 = 1, 2, . .
Входящие в формулы (IX.7O)—(IX.72) производные можно полу-
чить непосредственно из системы алгебраических уравнений мате-
риального баланса реактора (IX.62) с помощью правила дифферен-
цирования неявных функций. Если обозначить через Hin левую часть
i-ro уравнения системы (IX.62), выражение для частной производной
любого /-го вещества на выходе из re-го реактора по времени кон-
такта Sn может быть записано в виде:
дС]п
~dS^
QHjn
dSn
(IX.73)
Здесь Д„ — определитель Якоби (якобиан) функций Htn по
переменным С/п, т. е. определитель с К рядами и К столбцами,
элемент которого, стоящий в i-м ряду и /-м столбце, равен частной
производной dHlrJdCjn; Atjn — алгебраическое дополнение эле-
мента dHin/dCjn якобиана Д„, т. е. определитель, получающийся из
Д„ вычеркиванием i-й строки и/-го столбца и умноженный на (—1)‘+/.
Элементы якобиана Д„ вычисляются по формулам
dHin! dCjn = б,/ — S пдг irl/ дС/п (IX. 74)
где — символ Кронекера, принимающий значения
при i /, 6и = 1.
Учитывая, что
dHin/dSn= rin
получаем из (IX.73):
dCjn _ 1 у л .'
dSn SnZit,ntn
i-1
6f/ - О
(IX.75)
(IX.76)
Аналогичным образом определяем и частные производные кон-
центрации Cjn по температуре Тп и входным концентрациям Ci>re+1.
Поскольку
dHinldTn=-r'inSn (IX.77)
386
где штрих обозначает производную по температуре, и
dH(n/dCj,n+i——i>ij (IX.78)
то
i=l
dCjnldCit л+i — AijnJ(IX.80)
Подставляя формулу (IX.76) в (IX.70) и (IX.79) в (IX.71), полу-
чаем расчетные уравнения
К к
У 2 ^/> n—l-^iinrin= (я = 1, 2, ... , ЛГ) (IX-81)
i-1 >1
2 2 и7.«-1ЛО»гг'п = 0 (" = !> 2, . . . , ЛГ) (IX.82)
i-l 3=1
совместным решением которых определяются оптимальные значения
Sn и Тп. Подстановка формулы (IX.80) в (IX.72) дает рекуррентные
соотношения для величин ф,-„:
к
= (i = l, 2, . • . , X; п=1, 2, . . , N) (IX.83)
3=1
Так как в «последовательности», состоящей из нуля реакторов,
вещество не образуется ни при каких условиях, то <р0 (С= 0 и
dtf0/dCi 1 = 0; = Щ 0 = 1, 2...К) (IX.84)
Равенства (IX.84) служат отправной точкой процесса решения.
Задавшись составом реагирующей смеси Ct на выходе последова-
тельности, вычисляем оптимальную' температуру Тj и время кон-
такта 5 i в последнем по ходу потока реакторе по формулам (IX.81),
(IX.82), С2 — из уравнений (IX.62); ф — по формулам (IX.83);
далее рассчитываем S 2 и Tz и т. д. вплоть до любого п = N (рис.
IX.4).
Чтобы прийти к заданному составу С\+1 на входе цепочки
реакторов, необходимо прибегнуть к методу последовательных при-
ближений. *
Если для какого-либо «-го реактора вычисленная оптимальная
температура превышает предельно допустимое значение Т*, то
принимается Тп— Т* и Sn определяется решением уравнения
(IX.81). Как и при выборе ОТП в реакторе идеального вытеснения,
температурный оптимум не достигается, если рост температуры,
ускоряя процесс, не уменьшает его избирательности. Для таких
* Приведенный в тексте вывод расчетных уравнений предложен в работе
[19]. Те же уравнения можно вывести классическим методом [20] или на основе
принципа максимума для дискретных систем [21].
25* 387
процессов оптимальная температура реакции повсюду равна Т*,
и при поиске оптимального режима должно быть выбрано только
время контакта Sn в каждом реакторе последовательности.
В качестве примера рассмотрим простейший процесс, включа-
ющий только одну обратимую или необратимую реакцию [22] *.
Примем продукт реакции за ключевое вещество; соответствующий
Рис. IX.4. Алгоритм оптимизации цепочки реакторов идеального смешения.
коэффициент щ можно принять равным единице. Уравнение мате-
риального баланса и критерий оптимальности записываются в виде:
Сп — Cn+i — Snrn(Cn, Тп) = Ъ (IX.85)
N
PN=^ (Cn-Cn^-XSn) (IX.86)
n-l
Подставляя в формулы (IX.70)—(IX.72) выражения для частных
производных Сп по Сп+1, $п, Тп, определяемые непосредственно
из (IX.85)
дСп 1
dCn+i 1-8пдгп/дСп ’
дСп
дтп- i-Sndm/dCn
получаем расчетные формулы для вычисления оптимальных значе-
ний температуры и времени контакта и рекуррентные соотношения
для величин ф„ = 1 + dqn/dC„+1.
* Этот процесс может быть также рассчитав графическими методами [1.6J.
дСП _ Гп
д8п 1—-SndrnldCn *
8пТп
388
Условие температурного оптимума (IX.71) сводится к виду
г'п = 0; т. е., как и в реакторе идеального вытеснения, температура
реакции повсюду должна локально максимизировать скорость обра-
зования целевого продукта. Согласно этому условию, температура
должна повсюду равняться максимально допустимой при проведении
необратимой или обратимой эндотермической реакции; в обратимой
экзотермической реакции она определяется уравнением (IX.3}
или (IX.5). Оптимальное время контакта равно
Рекуррентное соотношение для величин фя записывается в виде:
(ГХ.89>
Подставляя формулу (IX.88) в (IX.89), находим:
tyre111 ^гп
(IX.90}
Формула (IX.88) для вычисления оптимального времени контакта
приводится теперь к виду:
= 1~дс7 (IX.91)
Так как = 1, формула (IX.91) будет справедлива и для реак-
тора 1, если положить г0 = %. Заметим, что, исключая случаи тор-
можения исходным веществом и автокатализа, дг[дС < 0. При этом
формула (IX.91) дает положительное решение, если выполнены
очевидные неравенства rn > rn_lt > Л. При дг]дС >0 формула
(IX.91) теряет смысл. В этом случае эффективность процесса воз-
растает с увеличением времени контакта Sn и, соответственно, сп
снижением концентрации исходного вещества в реакторе. Поэтому
те реакции, для которых дг/дС > 0 (например, реакции отрицатель-
ного порядка), выгоднее всего проводить в единственном реактора
идеального смешения.
Исходя из формулы (IX.91), нетрудно исследовать характер,
изменения оптимальных времен контакта по ходу потока в изотер-
мической цепочке реакторов. Заменив в уравнении (IX.85) индекс
п чза п — 1 и выразив время контакта в (п — 1)-м реакторе Sn _ t
через С„_ 1 и С„, запишем разность оптимальных времен контакта
в двух соседних реакторах:
5л-Уя_1 = (1
__ rn X /
гп-1 / I дсп
С п-1 — Сп
гп-1
(IX.92}
Преобразуя эту формулу, получаем:
8п—8П-1~
гп гп—1 — (Сп — Сп-1) дгп!дСп
Гп-1дгп/дСп
(IX.93}
38Э
Как это видно из рис. IX.5, числитель в (IX.93) отрицателен,
когда г — выпуклая функция концентрации исходного вещества (а),
и положителен, когда функция г — вогнутая (б) *.
Рассматривая, например, необратимую реакцию, видим, что
оптимальное время контакта и пропорциональные ему размеры
Рис. IX.5. К исследованию формулы (IX.93) hx = гп — гп_х,
= (Сп — Cn-r)
а — г(С) — выпуклая функция; б — г(С) — вогнутая функция.
реакторов возрастают по ходу потока, если порядок реакции превы-
шает единицу. Обратная закономерность действует для реакций
порядком ниже первого. Для реакций первого порядка числитель
Рис. IX.6. Цепочка адиабатических реакторов с промежу-
точными теплообменниками.
в формуле (IX.93) тождественно равен нулю и оптимальные времена
контакта одинаковы на всех стадиях, что согласуется с результатом,
полученным другим методом в разделе VII.3.
Цепочка адиабатических реакторов. TV-стадийный адиабатический
реактор (рис. IX.6) состоит из N реакционных зон и TV — 1 проме-
жуточных теплообменников. Оптимальный режим процесса, проте-
кающего в такой реакторной схеме, достигается варьированием вре-
* Функция называется выпуклой, если ее вторая производная положи-
тельна. Если последняя отрицательна, функция называется вогнутой.
390
мени контакта и начальных температур на каждой из стадий. Опти-
мальное проектирование цепочки адиабатических реакторов идеаль-
ного смешения осуществляется методами, изложенными выше.
Оптимальная действующая температура в каждом из реакторов после-
довательности, рассчитанная по выше приведенным формулам,
может, очевидно, поддерживаться любым возможным способом —
как теплообменом в реакционной зоне, так и с помощью промежут
точных теплообменников. Задача выбора оптимального режима
процесса, протекающего в последовательности адиабатических реакто-
ров идеального вытеснения, требует уже существенно иного матема-
тического аппарата. Такой процесс описывается системой дифферен-
циальных, а не алгебраических уравнений; поэтому здесь невозможно-
получить аналитические выражения для частных производных
максимального значения критерия оптимальности по концентрациям
ключевых веществ и варьируемым параметрам способами, применен-
ными при оптимизации цепочки реакторов идеального смешения,
а следовательно, и использовать ту же схему решения. Ключом
к решению, которое будет здесь изложено, является применение-
математического аппарата, позволяющего вычислять необходимые-
для решения задачи об ОТП значения величин путем интегрирова-
ния надлежащим образом составленных дифференциальных урав-
нений [23].
Рассмотрим процесс, включающий произвольное число реакций.
Химические превращения в реакторе идеального вытеснения описы-
ваются системой уравнений:
Т) (г=1, 2, . . ., К) (IX.94>
При расчете адиабатического реактора эти уравнения должны
быть дополнены уравнением теплового баланса (см. раздел VII.4):
к
4г=2й/г/ <1х-95>
Как и раньше, будем исходить из критерия оптимальности для
стадийной цепочки реакторов (IX.63). Счет реакторов по-прежнему
ведется в направлении, противоположном ходу потока, и индексом
п обозначается состав потока на выходе из n-го реактора. В дальней-
шем будем также отмечать значения переменных на входе в п-й
реактор нижним индексом п и верхним индексом 0.
При выводе расчетных уравнений воспользуемся математиче-
ским аппаратом решения задачи об ОТП в реакторе идеального
вытеснения, осуществляя, однако, выбор оптимальной температуры
не повсюду, а лишь в конечном числе точек. Исходя из (IX.63),
(IX.94), (IX.95) и применяя принцип оптимальности, путем той же-
процедуры, что была использована в разделе IX. 1 при выводе урав-
нения (IX.21), приходим к уравнению первого порядка, в частных
391
производных, которое должно быть выполнено в каждой точке после-
довательности реакторов;
к
2 (t’+Мт) г/-х=0 (IX.96)
i-1
Характеристическими уравнениями (IX.96) служат (IX.94),
(IX.95), а также
К
= - 2 rij </=1- 2.......к) (IX.97)
1=1
, . К
t/=-2(*<+Mt)'-/ (IX.98)
i=i
где r{j — dr{/dCi, r'i = drJdT.
Уравнение (IX.98) отличается от (IX.21) лишь присутствием
частной производной максимального значения критерия оптималь-
ности по температуре 4т — д^/дТ. Действительно, в задаче об ОТП
температура в каждой точке выбирается оптимальной и, следова-
ч тельно, повсюду = 0. В адиабатическом же процессе температуры
могут свободно варьироваться лишь в конечном числе точек — на
входе реакторов. Если температура Т°а на входе в л-й реактор выбрана
оптимальной, то, очевидно, значение производной 4тп равно в этой
точке нулю:
4°Гя=0 (п = 1, 2...N) (IX.99)
Равенства (IX.99) определяют условия оптимального выбора
температур во входных сечениях каждого из реакторов цепочки.
Координаты этих входных сечений, измеряемые текущим временем
контакта t, отсчитываемым от входа в цепочку реакторов, пока не
определены и подлежат оптимальному выбору; при этом, очевидно,
решается поставленная первоначально задача оптимального выбора
времени контакта S в каждом реакторе цепочки. Чтобы осуществить
этот выбор, замечаем, что температура на выходе из любого реактора
уже не влияет на ход процесса и, следовательно,
4 =0 (п = 1, 2, . . . N) (1ХЛ00)
Подстановка (IX.100) в (IX.96) дает условие, которое должно
«быть выполнено в выходном сечении каждого реактора цепочки:
к
(IX.101)
i-i
Если на входе в (и — 1)-й реактор удовлетворено равенство
(IX.99), условие (IX.101) может быть преобразовано к более нагляд-
ной форме. Так как, согласно (IX.99), равенство (IX.101) должно
392
быть выполнено также и на входе в (п — 1)-й реактор и, кроме того,
концентрация ключевых веществ и значения частных производных
максимального значения критерия оптимальности по этим концентра-
циям на выходе из любого и-го реактора и на входе в (л — 1)-й
реактор одинаковы
Czn=c?7-i; ^=^,«-1 (‘=1 2...........К; п=2. 3.......N) (1Х.102>
то можно написать вместо (IX,101):
к
2 п-i) =0 («=2, 3, • . . У) (1Х.103>
i-l
Иначе говоря, величина остается неизменной после опти-
мального изменения температуры в промежуточном теплообменнике.
Равенство (IX.103), очевидно, содержит условие существования
решения в отсутствие температурных ограничений; чтобы (IX.103}
могло быть удовлетворено, функция должна проходить через,
максимум при некоторой температуре, промежуточной между Т®.!
и Тп.
Теперь можем описать процедуру численного решения рассмат-
риваемой задачи. На выходе последовательности выполняются
условия (IX. 25), (IX. 26)
•фГ1=И/ (i= 1, 2 .... К) (IX.104)
к
= Х (IX.105)
i-l
и фТ1 = 0 в силу (IX.100). Задавшись концентрациями ключевых
веществ на выходе последовательности, интегрируем систему уравне-
ний (IX.94), (IX.95), (IX.97), (IX.98), пока не будет выполнено-
условие (IX.99). В результате получаем оптимальную входную
точку реактора 1 и значения всех переменных в этой точке. Условия
(IX.100), (IX.102), (IX.103) дают значения переменных на выходе-
из реактора 2; вновь интегрируем систему расчетных уравнений,
но уже для реактора 2 и т. д.; повторяем это вплоть до п — N.
Чтобы прийти к заданному исходному составу Cjv + 1, приходится
прибегнуть к методу последовательных приближений.
Процедура решения видоизменяется при оптимальном проектиро-
вании процессов, обладающих тем свойством, что функция
при всех условиях возрастает с повышением температуры. Нагрева-
ние, ускоряя такие процессы, ведет к улучшению их избиратель-
ности, и потому их следует проводить при возможно более высокой
температуре. Условие (IX.99) при этом становится уже невыполни-
мым. Вместо него получаем следующие граничные условия:
для экзотермических процессов
Гя=7* (п==1, 2.....N) (IX.106)
для эндотермических процессов
70 = г* (П=1, 2......N) (IX.107)
393
Недействительным становится и равенство (IX.103), но условие
"(IX.101) сохраняет силу.
Расчет эндотермического процесса начинается так же, как расчет
процесса, свободного от температурных ограничений, но интегри-
рование системы (IX.94), (IX.95), (IX.97), (IX.98) для любого п-го
реактора ведется, пока не будет выполнено условие (IX.107). Темпе-
ратура на выходе (п + 1)-го реактора определяется из условия
{IX.101). При расчете экзотермического процесса температура на
выходе последовательности принимается равной Т*, условие (IX.105)
дает при этом соотношение между конечными концентрациями клю-
чевых веществ, ограничивающее на одну степень свободы произволь-
ность их первоначального выбора. Интегрирование упомянутой выше
системы уравнений надо вести до тех пор, пока состав реагирующей
смеси и значения производных не сделаются такими, что при повы-
шении температуры до предела Т* будет выполнено условие (IX.101).
Расчетные уравнения задачи оптимального проектирования це-
почки адиабатических реакторов идеального вытеснения, как и при
расчете оптимального режима реакторов других типов, упрощаются,
если рассматриваемый процесс включает только одну обратимую
или необратимую реакцию и, следовательно, К = 1 [1, 24]. Из про-
цессов с одним ключевым веществом лишь обратимая экзотермиче-
ская реакция обладает температурным оптимумом; росту эффектив-
ности обратимой эндотермической и необратимой реакций благо-
приятствует максимально возможное повышение температуры. При
К = 1 расчетные уравнения (IX.94)—(IX.98) принимают, соответ-
ственно, вид
dC/dt=r(C, Т) (IX.108)
dT/dt = fir(C, Т) (IX.109)
(фс+йфг)г=Х (IX.110)
d^cldt= — (фс + йфг) дг/дС (IX.111)
dtyT]dt= —(фд+Йфу) дг[дТ (IX.112)
где фс = и + dq/d&, а индексы при и, С, г, К опущены.
Выбор ключевого вещества безразличен; для определенности
будем считать, что за ключевое вещество принят целевой продукт;
при этом г > 0 и в экзотермическом процессе Й >• 0. Разделив
уравнение (IX.109) на (IX.108), получаем:
dTjdC = n (IX.ИЗ)
•откуда:
Tn=T<>n+h(Cn-Can) (IX. 114)
Уравнение (IX.114) определяет адиабатический путь процесса,
который изображается в координатах С — Т прямой линией. Урав-
394
нения (IX.Ill) и (IX.112) преобразуются с учетом (IX.110) к виду
d'\'C _ _К_ дг_
dt г ‘ дС
^Фт А, дг
dt
(IX.115)
(IX.116)
Рассмотрим процесс, включающий обратимую экзотермическую
реакцию. В этом процессе на стыке двух последовательных реакто-
ров должно быть выполнено условие (IX.103), которое при К = 1
упрощается и принимает вид:
гп=г&-1 (п = 2, 3......N) (IX.117)
Следовательно, скорость образования целевого продукта на
выходе из и-го и на входе в (и — 1)-й реактор должна быть одинакова.
Это условие впервые было выведено Боресковым [6].
Найдем теперь явный вид условия (IX.99). Интегрируя уравне-
ние (IX.116) от выхода и-го реактора, где выполнено условие (IX.100),
до входа, получаем:
о
Ф°тл=-Ь J-J-~dt=O (IX.118)
in
Умножая и деля подынтегральное выражение этого уравнения
на г = dC/dt, приводим условие оптимальной начальной темпера-
туры в n-м реакторе к виду
сп
С 1 дг
In= J _dc = 0 (IX.119)
где интегрирование ведется вдоль адиабатического пути реакции
(IX.114).
Процедура расчета оптимального режима обратимой экзотерми-
ческой реакции, следовательно, такова. Начав с произвольно выбран-
ной конечной концентрации и температуры, определенной из усло-
вия (IX.105), которое при К = 1 записывается в виде
П = 7.1и (IX.120)
вычисляем интеграл 1Г как функцию нижнего предела, пока не
будет выполнено условие (IX.119). При этом получаем концентра-
цию и температуру на входе в реактор 1; температуру на выходе из
реактора 2 находим из условия (IX.117); точно так же решение
продолжается вплоть до любого п = N.
Процедуру решения можно представить графически в коорди-
натах С — Т (рис. IX.7). Уравнение (IX.120) изображается кривой
1\, тем более близкой к равновесной кривой Ге, чем меньше Z/u.
На кривой 1\ лежат точки, изображающие оптимальное конечное
состояние реагирующей смеси. Кривую 1\, представляющую собой
геометрическое место оптимальных начальных состояний потока
395
Рис. IX.7. Графический расчет опти-
мального режима для обратимой экзо-
термической реакции (стрелки указы-
вают изменение концентрации и темпе-
ратуры в оптимальном процессе, соот-
ветственно исходным состояниям а и Ь).
в реакторе 1, получаем из Гх, проводя из ее различных точек прямо- 1
линейные адиабатические пути реакции с коэффициентом наклона 1
Й-1 до точки, где будет выполнено условие (IX.119). Равенство 1
(IX. 117) дает очевидный способ построения по кривой Г? геометри- s
ческого места оптимальных конечных состояний потока в реакторе 2 ]
Г2. Точно таким же способом построение кривых Г„, может быть |
продолжено вплоть до п = N. j
Все кривые Гп лежат справа, j
а кривые Гйп — слева от кри- j
вой Гт, на которой скорость J
образования целевого продукта J
достигает максимального зна- ]
чения. Максимум скорости про- 1
цесса достигается, таким обра- |
зом, в каждом реакторе после- |
довательности в некоторой I
средней его точке. j
Другим путем ведется расчет I
оптимального режима нербрати- j
мой или обратимой эндотерми- |
ческой реакции. В этих процес- |
сах температура либо на входе, |
либо на выходе реактора должна I
быть фиксирована на верхнем |
пределе Т*. Условие (IX.101) ]
при К = 1 упрощаётся: |
Фспгп = % J
(п = 1, 2, . . N) (IX.121) I
Чтобы раскрыть условие (IX.121), интегрируем уравнение j
(IX.115): j
1
р 1 дг я
’fC>n+l-tCn=fCn-’tCn = ^7^<f' (ix.122) I
0 1
Как и при выводе условия (IX.И9), умножая и деля подынтег- 1
ральное выражение (IX. 122) на r — dCjdt и вводя обозначение 1
1
J (IX.123) 1
Сп Я
где интегрирование ведется вдоль адиабатического пути реакции, Ш
получаем рекуррентное соотношение: м
tC,„+1-%n=^n (IX.124), |
Условие (IX.121) может, следовательно, быть записано в виде: Я
и-к 2 Лп| гЯ=Х (IX.125) 1
т-1 ) Я
I 396
к
Процедура решения для эндотермических процессов показана
на рис. IX.8. В процессе, включающем единственную необратимую
экзотермическую реакцию, существует только одно оптимальное
конечное состояние, определяемое условиями (IX.107) и (IX. 120),
зависящее от параметра %. При этом отпадает надобность в последо-
вательных приближениях.
Другие задачи оптимизации. Рассмотренные здесь примеры
дают представление об основных идеях и методах, лежащих в основе
решения разнообразных за-
Рис. IX.8. Графический расчет оптималь-
ного режима-для эндотермической реакции.
дач оптимизации реакторных
узлов. Можно указать три
направления уточнения и
развития оптимальных расче-
тов. Первое из них — это
анализ различных стадийных
схем. Укажем, например, на
расчет цепочек адиабатиче-
ских реакторов, где охлажде-
ние реагирующей смеси меж-
ду стадиями происходит не
в промежуточных теплооб-
менниках, а путем добавле-
ния холодного сырья или
инертного вещества. Другой
пример — расчет оптималь-
ного трубчатого реактора с
секционированным теплооб-
менником. Второе направление
состоит в уточнении критерия опти-
мальности путем более полного учета затрат на ведение процесса. На-
пример, результаты оптимального расчета цепочки адиабатических ре-
акторов можно уточнить, приняв во внимание расходы на устройство
промежуточных теплообменников. Наконец, третье направление — вы-
бор оптимальных значений других управляющих параметров, помимо
температуры процесса. Так, в работе [25] рассматривается вопрос
об оптимальном профиле давления по длине трубчатого реактора,
а в работе [26] — об оптимальном изменении состава каталитиче-
ской системы. При проектировании стадийных схем, наряду, с опре-
делением оптимального перепада температур между стадиями,
может рассчитываться оптимальное количество свежего реагента,
добавляемого к реагирующей смеси. Вряд ли можно даже перечис-
лить все возможные варианты задач оптимизации; методы их реше-
ния, однако, мало отличаются друг от друга.
При решении всех перечисленных задач оптимизации разыски-
вается оптимальный режим заранее выбранной реакторной схемы,
содержащей заданное число стадий. Эффективность большинства
процессов, оцениваемая с помощью критерия оптимальности (IX.63),
растет с увеличением числа стадий; оптимальное число реакторов
в цепочке можно установить, учтя дополнительные затраты на
397
аппаратуру и регулирование, связанные с увеличением числа стадий.
Сама применяемая схема реакторного узла также должна быть опти-
мальной. Часто этот вопрос удается решить уже на первоначальной
стадии проектирования, исходя либо из опыта проектирования
сходных процессов, либо из теоретических соображений. Примером
последних является сравнение показателей работы реакторов идеаль-
ного смешения, идеального вытеснения и схем, состоящих из после-
довательно соединенных аппаратов обоих типов. Для изотермических
реакторов такое сравнение было проведено в разделе VII.3; анало-
гичным способом оно проводится и для адиабатических аппаратов
[27]. В более сложных случаях точный ответ может дать только
сравнение технико-экономических показателей оптимальных режи-
мов различных технологических схем.
ЛИТЕРАТУРА
1. Р. Арис, Оптимальное проектирование химических реакторов,
ИЛ, 1963.
2. Г. М. Островский, Ю. М. Волин, Методы оптимизации хи-
мических реакторов, Изд. «Химия», 1967.
3. А. И. Бояринов, В. В, К а ф а р о в, Методы оптимизации в хи-
мической технологии, Изд. «Химия», 1969.
4. С. Робертс, Динамическое программирование в процессах хими-
ческой технологии и методы управления, Изд. «Мир», 1965.
5. Р. Арис, Дискретное динамическое программирование, Изд. «Мир»,
1969.
6. Г. К. Боресков, Катализ в производстве серной кислоты, Госхим-
издат, 1956.
7. Р. Веллман, Динамическое программирование, ИЛ, 1960.
8. В. Aris, Chem. Eng. Sci., 12, 243 (1960).
9. S. Katz, Ann. № 4. Acad. Sci., 84, 441 (1960).
10. F. Horn, Chem. Eng. Sci., 14, 77 (1961); Z. Electrochem, 65,209 (1961).
11. И. M. Г e л ь ф а н д, С. В. Ф о м и н, Вариационное исчисление,
Физматгиз, 1961.
12. Л. С. Понтрягин, В. Г. Болтянский, Р. В. Гамкре-
л и д з е, Е. Ф. М ище нк о, Математическая теория оптимальных процес-
сов, Физматгиз, 1961.
13. О. В 11 о us, N. R. Amundson, Chem. Eng. Sci., 5, 81 (1956).
14. Л. М.Письмен, И. И. И о ф ф e, Хим. пром., № 4, 260 (1962).
15. Р. Р 1 a u 1, I. F и с k s, I. J о f f е, W. М i 1 j a w s к i j, Wiss.
Z. der Tech. Hochsch. f. chem. Leuna — Merseburg, 11, № 1, 53 (1969).
16. Д. Дж. Уайлд, Методы поиска экстремума, Изд. «Наука», 1967.
17. R. Aris, Chem. Eng. Sci., 13, 75 (1960); Z. Elekctrochem., 65, 229
(1961).
18. Фан Лянь-цен ь, Вань Чу-сен, Дискретный принцип макси- 1
мума, Изд. «Мир», 1967. 1
19. Л. М. П и с ь м е в, И. И. Иоффе, ДАН СССР, 144, 753 (1962). 1
20. F. Horn, Chem. Eng. Sci., 15, 176 (1961). J
21. S. Katz, Ind. Eng. Chem. Fund., 1, 226 (1962). з
22. Л. M. Письмен, И. И. И о ф ф е, Хим. пром., № 5, 352 (1962). 3
23. Л. М. Письмен, И. И. Иоффе, ДАН СССР, 144, 609 (1962). 1
24. В. Aris, Chem. Eng. Sci., 13, 18 (1960). 1
25. J. G. v a n de V u s s e, H. V о e 11 e r, Chem. Eng. Sci., 14, 90 d
(1961). J
26. D. J. Gunn, W. J. Thomas, Chem. Eng. Sci., 20, 89 (1965). ।
27. A. Cholett, J. Blanchet, Canad J. Chem. Eng., 39, 192 d
(1961); R. Aris, ibid., 40, 87 (1962). |
Сканировал: Neptunyi |
Магнитогорск 1
2008 1
Часть
четвертая
МЕТОДЫ ИССЛЕДОВАНИЯ
КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Глава X _____________________________________________________
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ
ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Х.1. ЦЕЛИ И МЕТОДЫ ЛАБОРАТОРНЫХ ИССЛЕДОВАНИЙ
Многообразие задач, решаемых при лабораторном исследовании
промышленных гетерогенно-каталитических процессов, можно свести
к следующим:
1) первоначальный подбор катализатора;
2) определение приближенно оптимальных условий протекания
процесса;
3) получение данных о кинетике химических и физических
стадий процесса, необходимых для осуществления перехода от лабо-
раторного реактора к промышленному и расчетного выбора опти-
мального режима работы последнего.
Кроме того, в практике исследований по технологическому
катализу значительное место занимает проверка стабильности ката-
лизатора при длительной работе и оценка качества целевого про-
дукта.
Первоначальный подбор катализатора позволяет выявить
катализатор или ряд катализаторов, обладающих хорошей или
удовлетворительной активностью и селективностью по отношению
к исследуемому процессу. Активность катализатора измеряется
количеством исходного вещества, реагирующего в единицу времени
на единице поверхности катализатора. Определенная таким образом
активность катализатора теоретически является самой строгой его
характеристикой; однако для промышленного процесса часто отно-
сят эту величину к единице объема слоя. Пересчет от одной меры
активности катализатора к другой легко выполнить, если известны
удельная поверхность и насыпной вес катализатора.
Более надежной величиной оказывается активность катализа-
тора, отнесенная к единице его веса. Однако, последнюю меру актив-
ности обычно применяют только для процессов с кипящим слоем
катализатора из-за неопределенности объема слоя.
Другая необходимая характеристика катализатора, определяемая
при первоначальном исследовании процесса — его селективность
399
(избирательность), определяемая как отношение скорости образо-
вания целевого продукта к активности катализатора (т. е. скорости,
реагирования исходного вещества). Активность и селективность
катализатора зависят от условий проведения реакции (например,
температуры, концентрации реагентов) и поэтому характеризуют
не только катализатор, но и процесс. Выбирают катализатор
на основании результатов, полученных в оптимальных условиях.
Определение активности и селективности катализатора целе-
сообразнее проводить в отсутствие искажающего влияния процессов
массо- и теплопереноса. Исключение составляют реакции, идущие
в промышленных условиях во внешнедиффузионной области (напри-
мер, окисление аммиака).
Важным условием, необходимым для получения воспроизводимых
экспериментальных данных, является неизменность свойств катали-
затора в течение всей серии опытов для изучения его активности.
Выполнение этого условия достигается выдержкой катализатора
в определенном режиме до получения постоянной активности. Од-
нако к исследованию катализатора с быстро падающей активностью,
например катализаторов крекинга, названное правило неприменимо.
Экспериментальный поиск оптимальных условий — обычный этап,
следующий за первоначальным исследованием катализаторов. До на-
стоящего времени такие эксперименты часто ставят по неверной
методике. Всегда следует помнить, что оптимальные условия, най-
денные опытным путем, относятся лишь к той установке, на которой
проводили эксперимент. Между тем, в ряде случаев при выборе
типа установки для поиска оптимальных условий работы катализа-
тора исходят главным образом из удобства экспериментальной ра-
боты. Но оптимальное условие, найденное на удобной лабораторной
установке, могут не совпадать с наилучшими условиями работы
промышленного реактора.
Экспериментально найденные на лабораторной установке опти-
мальные условия работы катализатора можно непосредственно
перенести на аппарат большого масштаба лишь в том случае, когда
они определены для условий, очень близких к промышленным.
Примером может послужить исследование хода процесса в трубке
промышленных размеров на катализаторе обычной степени дисперс-
ности с целью поиска оптимальных значений температуры реакции,
скорости потока и пр. Эффективный поиск должен осуществляться
с помощью статистических методов направленного движения к опти-
мальному режиму [1, 2]. Этот поиск должен быть более детальным,
чем при первоначальном подборе катализатора. Результаты экспери-
мента, поставленного по такой методике, могут быть непосред-
ственно перенесены на промышленный трубчатый реактор. Это же
имеет место при поиске оптимального режима в жидкофазных про-
точных реакторах идеального смешения.
Метод экспериментального поиска оптимальных условий про-
цесса в реакторе определенного типа дает возможность быстро
получать практически важные результаты, не вникая в механизм
400
процесса. Этот метод обладает определенными достоинствами как
путь оперативного решения промышленных задач, но, разумеется,
не может быть рекомендован как универсальный. Прежде всего,
при экспериментальном поиске оптимальных условий поневоле отка-
зываются от оптимального выбора самого реакционного аппарата,
чем заранее снижают экономические показатели будущего производ-
ства. Далее, путем экспериментального поиска за разумное время
можно найти оптимальный режим варьированием лишь малого
числа независимых переменных. Между тем, достигаемый оптимум,
в общем, тем выше, чем большее число параметров варьировалось
при его поиске.
Кинетические исследования. Наиболее эффективный, хотя и
самый трудный путь экспериментального исследования состоит
в раздельном количественном изучении всех разнородных явлений
(например, кинетики химических превращений, переноса массы
и тепла, движения потока), взаимодействие которых определяет
закономерности реального каталитического процесса. Эксперимент
при этом должен быть поставлен либо в таких условиях, когда
исключено действие всех факторов, кроме исследуемого, либо когда
методами математического анализа может быть выявлено влияние
каждого исследуемого фактора. Результатом такого исследования
является построение математической модели, на основании которой
может быть осуществлен расчетный выбор оптимального режима
процесса.
Многочисленные методы исследования кинетики гетерогенно-
каталитических реакций могут быть разделены на несколько групп,
некоторые из них взаимно перекрываются. Прежде всего, различают
динамические и статические методы, в зависимости от того, является
ли реактор проточным или нет. В свою очередь, динамические методы
могут быть проточными и проточно-циркуляционными. Другим
важным принципом классификации кинетических методов исследова-
ния является математическая характеристика величин, получаемых
в результате эксперимента. Если при проведении опыта непосред-
ственно определяется скорость реакции, метод называют дифферен-
циальным, если же определяется количество вещества, прореагиро-
вавшего за какой-то период времени или на каком-то участке реак-
тора, то метод называют интегральным (поскольку полученные
величины являются интегралом от скорости реакции по времени
или длине слоя катализатора). Наконец, в зависимости от постоян-
ства температуры опыта или вдоль слоя катализатора различают
изотермические и неизотермические эксперименты.
Всякий статический метод, очевидно, является интегральным,
так как в нем может быть измерено только изменение концентрации
вещества за какой-либо период времени, причем условия процесса
в течение этого периода не могут оставаться постоянными., Проточ-
ные интегральные и дифференциальные реакторы представляют собой
ни что иное, как реакторы соответственно идеального вытеснения
и смешения (см. главу VII). Дифференциальные реакторы идеального
26 Заказ 1441
401
вытеснения — это. проточные аппараты, работающие при очень
малых степенях превращения. В проточном реакторе идеального
смешения концентрации реагентов и температура повсюду одинаковы
и скорость образования любого вещества, отнесенная к единице
объема зоны реакции, равна разности между стационарной выход-
ной и исходной концентрациями этого вещества, деленной на сред-
нее время контакта (см. раздел VII.3). Математическая обработка
экспериментальных данных, полученных на дифференциальном реак-
торе, значительно проще, что является важным преимуществом
аппаратов этого типа по сравнению с интегральными. Преимуще-
ством же интегральных методов является большая скорость исследо-
вания, поскольку эти методы позволяют в одном опыте получить
кйнетическую кривую, содержащую гораздо больше информации,
чем измерение скорости реакции при определенных условиях, про-
водимое в дифференциальных реакторах. Это преимущество может
оказаться решающим по мере развития методов машинной обработки
экспериментальных данных.
Преимущество статических методов (немаловажное в практиче-
ской работе) — отсутствие дозирующих устройств и связанных
с ними ошибок дозировки, а также надежность при определении
неанализируемых продуктов по уравнению материального баланса^
Для реакций, происходящих между газообразными продуктами,
статический метод обычно применяют в циркуляционном варианте,
когда газ непрерывно прокачивается через катализатор, циркулируя
в системе.
Проточные интегральные реакторы, обычно заполненные ката-
лизатором трубки, аналогичны аппаратам, применяемым в промыш-
ленности, и по условиям своей работы близки к ним. Это имеет суще-
ственное значение в прикладных исследованиях, когда кроме чисто
химических и расчетных данных необходимо выявить технологи-
ческие особенности процесса, получить образцы целевого продукта,
сведения о длительности работы катализатора и качества целевого
продукта и т. п. Поэтому стадия модельной установки с проточным
реактором является практически необходимой в разработке промыш-
ленных гетерогенно-каталитических процессов. Целесообразно
использовать эти реакторй для получения данных по кинетике,
необходимых для расчета и проектирования промышленных реакто-
ров. При применении современной машинной вычислительной тех-
ники постановка опытов на проточных интегральных реакторах
может дать большой объем информации, позволяющий составить
математическое описание процесса с большой степенью надежности
и тем самым решить задачу перехода от лабораторного или пилот-
ного реактора к промышленному любой схемы и конструкции, в том
числе и к оптимальному.
Особую разновидность проточных реакторов представляют собой
дифференциальные проточно-циркуляционные установки, в которых
поток в реакторе перемешивается или часть потока циркулирует
по контуру, включающему в себя реактор и циркуляционный насос.
402
Помимо простоты математической обработки результатов опыта,
такие реакторы обладают тем существенным преимуществом, что
вследствие больших скоростей циркулирующего потока или примене-
ния специальных турбулизаторов можно устранить внешнедиффу-
зионное торможение. Конструкция аппаратов позволяет применять
мелкодисперсный катализатор, тем самым ликвидируя и внутри-
диффузионные помехи. Поэтому аппараты такого типа нашли широ-
кое применение для детального изучения химической кинетики
гетер огенно-ката литических процессов.
В последнее время все большее распространение получает им-
пульсный метод исследования гетерогенно-каталитических реак-
ций [3]. Он имеет существенные преимущества в отношении быстроты
эксперимента и малого количества реагентов для исследования.
Однако ввиду нестационарности условий опыта и математической
сложности явлений в эксперименте этот метод пригоден только для
исследования кинетики простейших реакций. Гораздо в большей
степени преимущества этого метода реализуются в исследованиях
по подбору катализаторов.
При исследовании кинетики реакций весьма важен вопрос о вы-
боре контролируемого параметра. В простых газо-жидкостных
процессах, в которых хорошо изучены направления химических
превращений (например, реакции гидрирования непредельных соеди-
нений или восстановления нитросоединений водородом), контроли-
руемым параметром может служить давление. Процесс в этом случае
проводят статически в изохорических условиях, а скорости реакций
измеряют по скорости изменения давления в системе. Математиче-
ская обработка полученных результатов достаточно проста. Для
сравнительно простых реакций можно применять адиабатический
метод исследования кинетики [4—6], когда контролируемым пара-
метром является только температура. Метод основан на определении
скорости разогрева (охлаждения) адиабатического реактора и при-
меним для сильно экзотермических (или эндотермических) реакций.
Для его использования нужно знать тепловые эффекты реакций
и теплоемкости реагентов и продуктов. Надо, однако, иметь в виду,
что при применении чисто адиабатического метода всегда есть опас-
ность непредвиденного изменения направления реакции по мере
повышения температуры, что сразу затрудняет расшифровку полу-
ченных данных. Гораздо большую перспективу имеет применение
для исследования каталитических процессов метода неизотермиче-
ского эксперимента, где наряду с анализом веществ производится
замер профиля температуры nd длине слоя катализатора или по
ходу опыта.
Наиболее существенными параметрами для контроля за ходом
реакции являются концентрации реагентов и продуктов. При этом
всюду, где это возможно, следует вести полный анализ реагирующей
смеси с расчетом материального баланса. Такой метод контроля
эксперимента существенно необходим в прикладных кинетических
исследованиях.
26*
403
В кинетических установках обязательным условием получения
правильных результатов является либо поддержание постоянной
температуры по всему объему катализатора, ли(эо точный замер
температурного поля в этом объеме. Далее необходимы стабилизация
давления в системе, стабилизация исходной концентрации реагентов
и точное определение времени реакции, т. е. для динамических
систем — скорости реагирующего потока. Поэтому эксперименталь-
ная техника исследований кинетики процессов гетерогенного ката-
лиза в значительной степени связана с разработкой надежных систем
термостатирования, замера температур, дозировки газа и жидкости,
стабилизации давления. Мы не будем касаться здесь методов иссле-
дования физических процессов, протекающих в каталитических
реакторах, а также общих вопросов экспериментальной техники,
связанных с исследованиями каталитических процессов, поскольку
по этим вопросам имеются специальные монографии [7—9].
Х.2. ИНТЕГРАЛЬНЫЕ РЕАКТОРЫ
ДЛЯ ИССЛЕДОВАНИЯ ГАЗОФАЗНЫХ РЕАКЦИЙ
На основании изложенного выше различают конструктивно два
типа лабораторных интегральных реакторов: 1) статические, т. е.
непроточные; 2) динамические, т. е. проточные. Математически опи-
сание обеих конструкций идентично при замене координаты времени
на координату длины слоя катализатора.
В прикладных исследованиях кинетики целесообразно применять
только циркуляционные статические установки. Основное преиму-
щество статических систем — отсутствие дозирующих устройств,
недостаточно точная работа которых, как показали обследования,
является, наряду с неточностями анализов, основным источником
ошибок в исследованиях кинетики процессов. Замкнутость системы
и возможность обеспечить высокую точность в определении коли-
чества вводимых реагентов позволяет сводить баланс вещества по
разности, что очень существенно при изучении сложных реакций,
где не все компоненты удается аналитически определять. В стати-
ческих установках за один опыт снимается полная кинетическая
кривая реакции при заданных начальных условиях с практически
неограниченным числом точек, что значительно ускоряет весь про-
цесс исследования.
Применение циркуляции также имеет свои достоинства:
1) весь слой катализатора работает при одинаковых значениях
кинетических переменных;
2) принудительная циркуляция дает возможность применять
катализатор мелкого зернения и тем исключить внутридиффузионные
явления;
3) вариацией циркуляции можно устранить внешнедиффузионное
торможение и установить его границы;
4) возможно проводить исследования с удалением продуктов
реакции из циркуляционной зоны.
404
Главный недостаток статических систем, в том числе и циркуля-
ционных, заключается в том, что они могут применяться только
к исследованию таких катализаторов, стационарный состав которых
либо не зависит от реакционной среды, либо устанавливается со
скоростью, сравнимой со скоростью реакции. Если же время уста-
новления стационарного состояния' катализатора сравнимо со вре-
менем эксперимента, то результаты исследований будут отражать
нестационарность свойств катализатора и вследствие этого их будет
трудно интерпретировать. Если, однако, время установления ста-
ционарного состояния катализатора велико по сравнению с длитель-
ностью опыта, то можно приближенно говорить о квазистационарном
состоянии катализатора и правильно интерпретировать результаты
исследований. Решить вопрос о пригодности статического метода
исследований можно лишь на основе предварительных качественных
исследований катализатора или. по теоретическим соображениям.
При проектировании статических циркуляционных установок
кроме общих вопросов, связанных с прочностью, обогревом и дру-
гими, следует учитывать соблюдение следующих условий:
1. Скорость циркулирующего газа в системе должна обеспечи-
вать малую (порядка 1—5 отн. %) разность концентраций компонен- '
тов до и после реактора. Если подсчитанное из предварительных
опытов на проточном реакторе время превращения примерно на
1—5% составляет /мин, то скорость циркуляции можно оценить
по формуле
где рцир — объемный расход газа, циркулирующего в системе;
Vp — объем реактора (катализатора); е — порозность слоя ката-
лизатора.
2. Объем системы с учетом буферной емкости должен обеспечи-
вать длительность эксперимента £эксп, приемлемую с точки зрения
отбора проб и (или) выполнения анализов
Усист еГР
— ^эксп
где S — время контакта для заданного превращения по предвари-
тельным опытам в интегральном проточном реакторе; Усист — объем
системы. ~~ t
3. Общий объем проб, изымаемых из системы, не должен превы-
шать примерно 5% от объема системы в пересчете на соответствующее
состояние.
4. Во избежание заметного искажения со стороны объемных
реакций «мертвый» объем е высокими температурами должен быть
сведен к минимуму.
В статических установках реакции проводят не в изобарических,
а в изохорических условиях, что нужно учитывать при обработке
405
результатов опытов, и поэтому вносить для реакций с изменяющимся
числом молей соответствующие поправки по уравнениям главы II.
На рис. Х.1 дана схема циркуляционной установки для изуче-
ния кинетики газофазных каталитических реакций под давлением
до 100 ат [10]. Через штуцер 3 шприцем либо другим способом
подается заданное количество жидкого реагента, который тут же
испаряется; реакционный газ дозируется по парциальному давле-
нию. Приготовленную реакционную паро-газовую смесь прокачи-
вают циркуляционным на-
сосом через слой катали-'
затора, расположенного в
трубчатом изотермическом
реакторе 1. Через опреде-
ленные промежутки време-
ни небольшая часть цирку-
лирующей реакционной
смеси отбирается через
калиброванные камеры 4
и 5 на анализ.
Установки низкого дав-
ления работают по ана-
логичной схеме и могут
быть снабжены ловушками
для конденсации продук-
тов реакции.
Интегральные реакторы
могут представлять собою
стеклянные, кварцевые
или металлические трубки,
снабженные обогревом раз-
личной конструкции. Од-
К иронагпогрифу
Рис. Х.1. Схема кинетической циркуляцион-
ной установки для изучения кинетики газо-
фазных реакций под давлением до 100 ат'.
1 — реактор; 2 — устройство для ввода реагентов;
з — штуцер; 4 и s — калиброванные камеры; в —
циркуляционный насос; 7 — термостат.
нако такие реакторы из-за нечеткого температурного профиля
обычно мало пригодны для получения данных о процессе. Кроме
того, в них трудно учесть влияние осевого смешения и пристеноч-
ного эффекта на поток газа.
Исследование кинетики изотермических процессов, не осложнен-
ных внешней диффузией, в простейших интегральных реакторах,
видимо, не связано со значительными ошибками, если диаметр
зерен катализатора це превышает 1/i0—1/i0 диаметра трубки, а длина
слоя и скорость потока таковы, что исключено заметное влияние
продольного перемешивания. Практически приходится, однако,
брать диаметр зерна равным примерно 1/6—х/8 диаметра трубки.
Независимость хода процесса от внешнедиффузионных и гидродина-
мических факторов может быть проконтролирована сравнением ре-
зультатов, полученных при одинаковых временах контакта, но
с различными линейными скоростями потока газа. Более надеж-
ные данные могут быть получены в интегральных реакторах, спе-
циально предназначенных для кинетических исследований.
406
Темкин и Кулькова [11] предложили конструкцию трубчатого
проточного реактора, предназначенного для исследования кинетики
на таблетированных или гранулированных катализаторах (рис. Х.2).
Разделение таблеток катализатора металлическими цилиндрами
или шариками, близко прилегающими к стенке.трубки, обеспечивает
хорошую изотермичность потока газа без осевого перемешивания.
Рис. Х.2. Элемент реакционной трубки
интегрального реактора системы Тем-
кина — КулЬковой:
1 —стальные шары; 2 — направляющие прово-
лочки; з — зерна катализатора; 4 — реакци-
онная трубка.
Рис. Х.З. Интегральный реактор по
системе Кикотя:
1 — штуцер; 2 — пластинка пористого
стекла; з — реакционная трубка; 4 — кор-
пус бани; 5 — место для отбора пробы;
в — штуцер для ввода смеси; 7 — штуцер
для вывода смеси.
На рис. Х.З показан реактор, предложенный Кикотем, по прин-
ципу совпадающий с реактором Ройтера и Корнейчука для окисле-
ния нафталина [12]. Он представляет собою свернутый в цилиндр
плоский змеевик, помещенный в баню с псевдоожиженным песком.
В точках перегиба впаиваются диафрагмы и пробоотборники. Такая
конструкция дает возможность в одном опыте получить ряд точек,
в данном случае пять, на кинетической кривой. Изотермичность
процесса, кроме хорошего теплоотвода, обеспечивается малым
диаметром трубки.
При моделировании каталитических установок часто применяют
трубчатые реакторы. \
Для того чтобы на таком реакторе одновременно с подбором
режимных условий можно было бы получать представительную
информацию о кинетике процесса, реактор должен быть снабжен
по высоте реакционной трубки несколькими пробоотборниками.
Кроме того, в реакторе должно хорошо измеряться температурное
407
поле, хотя бы по оси. Это дает возможность получать надежные дан-
ные при неизотермических экспериментах.
Измерение температурного поля может производиться либо
рядом специальных очень тонких термопар, либо при отсутствии
таковых — движущейся термопарой, один из вариантов которой
приведен на рис. Х.4. Принцип замера температур движущейся
Рис. Х.4. Схема установки дви-
жущейся термопары:
1 — спай термопары; 2 — контакт-
ная трубка; з — капилляр для
термопары; 4 — блок; 5— тросики;
в — барабан, закрепленный на валу
реверсивного двигателя; 7 — за-
писывающий потенциометр; 8 —
груз.
термопарой состоит в том, что спай
термопары медленно движется вдоль
слоя катализатора и показания термо-
пары непрерывно записываются потен-
циометром. Скорость движения спая
устанавливают с учетом инерционности
замера температуры, практически 1 —
^2 м/ч. Пренебрежение остальными
ошибками дает меньшую погрешность,
чем погрешность принятого приближе-
ния о постоянстве температуры цо се-
чению трубки.
Ход кинетических исследований на
интегральных реакторах сводится к
следующему. Пусть в результате опыта
измерены концентрации реагентов С и
температура реагирующей смеси Т в по-
следовательные момен!ы времени т или
в последовательных сечениях реактора
идеального вытеснения с координатами
I, измеряемыми временем, за которое
поток проходит расстояние от входа до
данного сечения. Таким образом полу-
чают интегральные кривые исследуе-
мого процесса. Кривые изменения
концентраций представляют собой ре-
шение кинетических уравнений процесса
= п (Х.1)
записанных для каждого из ключевых веществ при известной инте-
гральной температурной кривой.
Задача исследования кинетики процесса состоит в определении
зависимости скоростей образования ключевых веществ гс (или ско-
ростей реакции) от концентраций реагентов и температуры. Кине-
тика становится известной, когда найдены такие функции гг, что ’
интегральные кривые уравнения (Х.1) достаточно мало отличаются
от кривых изменения концентраций реагентов во времени (или по
длине реактора), найденных экспериментально. Подробнее вбпросы
расшифровки экспериментальных кинетических данных изложены
в главе XI.
408
Х.З. ДИФФЕРЕНЦИАЛЬНЫЕ РЕАКТОРЫ
ДЛЯ ИССЛЕДОВАНИЯ ГАЗОВЫХ РЕАКЦИЙ
Дифференциальные реакторы, работающие по принципу малых
степеней превращения, конструктивно и по экспериментальной
технике не отличаются от проточных интегральных |реакторов,
поэтому не будем на них останавливаться. Прочие варианты диф-
ференциальных реакторов снабжены приспособлениями, обеспечи-,
вающими одинаковые условия работы всего слоя катализатора по
всем кинетическим параметрам, в том числе и по концентрациям.
Рис. Х.5. Проточно-циркуляционная установка окисления бензола
в малеиновый ангидрид:
J —1 реометр на подаче воздуха; 2 —,шприц-дозатор; з — испаритель бен-
зола; 4 — реактор; 5 — блок-термостат реактора; в — клапанная система;
7 — контрольный пробоотборник; 8 — циркуляционный насос; 9 — щ>омы-
валки с водой; 19—пружина.
Такие типы реакторов часто называют безградиентными. По сути,
как указывалось, они являются реакторами идеального смешения
или приближаются к ним. С конструктивной точки зрения, кинети-
ческие установки с безградиентными реакторами делятся на две
основные группы: проточные циркуляционные и проточные смеше-
ния. Кроме того, конструкция каждой группы установок зависит от
назначения: для работы под обычным или повышенным давлением.
Темкин положил начало широкому применению проточно-цирку-
ляционного метода для изучения кинетики гетерогенно-каталити-
ческих-процессов [131. На рис. Х.5 представлена схема установки
такого типа для окисления бензола в малеиновой ангидрид. Общий
принцип работы установки ясен из рис. Х.5. Вся циркуляционная
система: реактор, насос с клапанной коробкой и коммуникации — на-
ходится в термостате или имеет специальный обогрев. Температура
409
в термостате и коммуникациях должна быть выше точки росы
реагирующей смеси.
В проточно-циркуляционных установках для прокачки реагиру-
ющей смеси часто используют стеклянные плунжерные циркуля-
ционные насосы. Поршень насоса приводится в возвратно-поступа-
тельное движение с помощью соленоида. Прерывистое магнитное
поле соленоида создается посредством релейной схемы. Магнитные
плунжерные насосы не всегда удобны в применении, в частности
при сильно экзотермических реакциях, когда требуется создавать
большую циркуляцию газа, чтобы избежать неоднородного темпера-
турного поля в реакторе. Поэтому наряду с этими насосами при-
меняют и другие конструкции, например, сильфонные или диафраг-
менные насосы, приводимые в движение от электродвигателя [14,15].
Весьма целесообразно включать в схему центробежные газодувки
высокой производительности. Здесь, однако, надо исключить утечки
газа через сальники на оси ротора насоса.
Для исключения влияния на ход реакции продуктов превраще-
ния в циркуляционный контур проточно-циркуляционных устано-
вок включают какого-либо рода конденсаторы или поглотители,
селективно извлекающие все или некоторые продукты реакции из
газовой смеси.
Схема проточно-циркуляционных установок повышенного давле-
ния может не отличаться от описанной схемы низкого давления
и аналогична схеме периодической циркуляционной установки
высокого давления (при отсутствии буферной емкости). Конечно,
в конструктивном отношении имеются особенности, например силь-
фонное уплотнение вентилей для гарантии от утечки, насосы с маг-
нитным приводом без смазки и т. п.
Наряду с приточно-циркуляционными реакторами в качестве
дифференциальных реакторов применяют проточные реакторы сме-
шения. Одним из наиболее простых конструктивных вариантов
таких реакторов являются реакторы с виброкипящим слоем [16].
В этом случае смешение потока происходит за счет перемешивания
катализатора,' находящегося в реакторе в виде порошка, под влия-
нием механического вибратора с частотой 50 гц и более. На рис. Х.6
приведена удобная конструкция простейшего типа. Реактор (стек-
лянный или металлический) соединен с металлическими капиллярами,
на которых он и подвешен. Воздействие вибратора с малой амплиту-
дой передается на петлю одного из капилляров. Такой реактор
можно использовать и при работах в импульсном режиме. Тогда
в штуцер 7 с силиконовой пробкой шприцем вводят реагент, а выход
реактора соединяют непосредственно с хромотографом.
Из проточных реакторов смешения широкое распространение
получили реакторы конструкции Корнейчука [12, 17]. В простей-
шем варианте этого реактора (без циркуляционного насоса) переме-
шивание газов и поддержание изотермического режима осущест-
вляется возвратно-поступательным движением поршня. Более совер-
шенным оказывается реактор с внутренней циркуляцией (рис. Х.7).
410
Благодаря размещению одного клапана в поршне, а другого в суже-
нии трубки реактора газы циркулируют в одном направлении.
Скорость циркуляции газа зависит от высоты подъема поршня
и частоты переключения электромагнитной катушки. Реакторы
изготавливают из стекла, а поршни — из стекла, металла или дру-
гих химически стойких материалов.
(
Рис. Х.6. Реактор с виброкипя-
щем слоем:
1 — реактор; 2 — металлический ка-
пилляр; з — гильза термопары; 4 —
печь; 5 — слой псевдоожиженного
песка; в — вибрирующее устройство;
7 — штуцер для ввода пробы при ра-
боте в импульсном режиме.
Рис. Х.7. Безградиептпый реак-
тор с внутренней циркуляцией:
1 — чехол термопары; 2 — катализа-
тор; 3 — змеевик для отбора проб;
4 — корпус; 5 — клапаны; в — пор-
шень; 7 — индукционная катушка.
К достоинствам реакторов Корнейчука следует отнести их ком-
пактность, относительную простоту, возможность работать без
термостата. Недостаток заключается в большом мертвом объеме
горячего пространства, что затрудняет или даже делает невозможным
изучение гетерогенно-каталитических реакций с заметно текущими
параллельными или последовательными объемными реакциями.
Кроме того, Интенсивность циркуляции газов в этих реакторах
не всегда достаточна и не подается контролю. Последнее замечание
касается многих моделей кинетических реакторов с циркуляцией.
До последнего времени этот момент недооценивался, однако, как
доказал опыт, без учета линейных скоростей потока можно
411
пропустить переход реакции во внешнюю диффузионную область,
что происходит чаще, чем ранее предполагалось.
Иногда пригодна модель безградиентного реактора с враща-
ющейся магнитной мешалкой (рис. Х.8). Реактор состоит из цельно-
Рир. Х.8. Безградиентный реактор
вращающейся магнитной мешалкой: ,
1 — железный стержень; 2 — фторопластовый
подпятник; 3 — фторопластовая втулка; 4 —
паянного стеклянного корпуса,
в который помещена мешалка,
представляющая собой стеклян-
ную трубку, к верхнему концу
ее прикреплен запаянный в стек-
ло железный стержень, а к ниж-
нему — П-образные перемеши-
вающие лопасти. Для избежания
с
ный реактор с внутренним контуром-
циркуляции для работы под высо-
ким давлением:
I — сетка с катализатором; 2 — диффузор;
3 — турбинка; 4 — ротор; 5 — статор;
в — электрообогрев.
корпус реактора; 5 — мешалка; в — столик
для катализатора; 7 — термопары; 3 — резерв-
ный штуцер.
вибрации и биения лопастей о стенки реакционной камеры в корпус
реактора помещен подшипник скольжения из фторопласта, а на
Т-образный спай трубки со стержнем насаживают конический
подпятник 2. В реакционной камере реактора имеется перфориро-
ванный столик для зерен катализатора, в который впаяна термо-
пара. Реактор помещают под электромотором с закрепленным на оси
постоянным магнитом,, с помощью которого вращается мешалка
и перемешиваются газы в реакционной камере. В описанном реак-
торе стационарное состояние устанавливается в течение нескольких
412
минут, он мал по габаритам и весьма прост в обслуживании. К недо-
статкам его следует отнести большой горячий объем и очень малую
загрузку катализатора.
Для высоких давлений весьма удобны реакторы с внутренним кон-
туром циркуляции и магнитным приводом. Один из конструктивных
вариантов такого реактора описан в работе [18]. Реактор (рис. Х.9)
представляет собою автоклав, внутри которого установлен диф-
фузор для направления потока газа с вмонтированной для зерен
катализатора сеткой. Над диффузором расположены крылья-отра-
жатели потока. В нижней части реактора расположен ротор, на
котором укреплено колесо турбинки, прокачивающей газ через диф-
фузор. Статор, представляющий собою катушку с вращающимся
магнитным полем, надет на внешнюю сторону выступающей вниз
гильзы автоклава; где расположен ротор. Конструкция испытана
в работе при' 500° С и 300 ат. Эта конструкция отличается компакт-
ностью, относительной простотой, надежностью. К недостаткам
этой конструкции можно отнести большой горячий объем, отсутствие
контроля циркуляции газов, невозможность вывода продуктов
реакции из циркуляционного цикла.
Все изложенные варианты безградиентных проточно-циркуля-
ционных и проточных реакторов обладают преимуществами в отно-
шении исключения диффузионных, теплообменных и гидравлических
помех, которые были перечислены для статических циркуляционных
установок. Все эти реакторы, что весьма существенно, работают
в условиях стационарного состояния катализатора. Общий их недо-
статок — необходимость применения дозирующих устройств и отно-
сительно большая длительность эксперимента.
Некоторые основы расчета кинетических проточно-циркулйцион-
ных реакторов, а, следовательно, и реакторов с внутренним контуром
циркуляции даны в работе [19].
Соотношение между основными параметрами установки рассчи-
тывают из уравнения реактора идеального смешения
g = fc7KaTC»:i (1^*)П / (Х.2)
где G — подаЧа реагента; к — константа скорости реакции, отнесен-
ная к единице объема катализатора; VKaT — объем катализатора;
Со — начальная концентрация реагента; п — порядок реакции;
х — степень превращения.
Кратность циркуляции на установке, т. е. отношение объемного
расхода циркулирующего потока к расходу входящего через систему
потока, определяется, с одной стороны, точностью анализа и сте-
пенью превращения реагента и выражается формулой
nSs&r-l (Х.З)
(где £ — величина, обратная точности анализа (в долях) при
определении начальных концентраций, £ «=* 102—103), а, с другой
413
стороны, зависит от теплового эффекта реакции, который не должен
создавать градиент температур в реакторе более АГ, т. е.
Т] Cq'&X— 1
(Х.4)
Рис. Х.10. Номограмма для
определения величины цирку-
ляции.
мое отклонение величины
где 'fl' = /гр/урАГ; hp — теплота реак-
ции.
На рис. Х.10 приведена номограмма
для определения ц по уравнению (Х.4).
В зависимости от того, какая из вели-
чин, t, или 'ОСо, больше, необходимо
пользоваться .либо уравнением (Х.З),
либо (Х.4).
Время установления выхода уста-
новки на. новый стационарный режим
приближенно определяют по уравнению
<ст 1----х 1 Да:
Г“7----Л— 1п ---------
Т 1 + (« — 1) X X
(Х.5)
где %' — среднее время пребывания,
определяемое как отношение полного
объема установки к объемной скорости
подачи газа, пересчитанной на условия
реакции; Ах — разность степеней пре-
вращения при начальном и конечном
стационарных состояниях; х — допусти-
конверсии от ее значения в идеальном
стационарном режиме, достигаемом при fCT -> оо.
Х.4. ЛАБОРАТОРНЫЕ РЕАКТОРЫ %
ДЛЯ ИССЛЕДОВАНИЯ ЖИДКОФАЗНЫХ РЕАКЦИЙ
Жидкофазные лабораторные реакторы обладают рядом отличий
от газофазных, поэтому их целесообразно рассмотреть особо. Устрой-
ство аппаратов мало меняется от того, проводятся ли в них чисто
жидкофазные или газо-жидкофазные реакции с твердым катализа-
тором. Последний тип реакций, к которому относятся жидкофазное
гидрирование, восстановление водородом, жидкофазное окисление
молекулярным кислородом в настоящее время более распространен
в технике, чем первый, 'к которому принадлежат реакции алкили-
рования, дегидратации и этерификации.
Аппараты для газо-жидкофазных реакций практически всегда
выполняются для работы под давлением. По вопросу о технике
лабораторных работ при высоком давлении имеется специальная
литература [20]. Поэтому, остановимся только на вопросах, особо
актуальных при исследовании гетерогенных каталитических реак-
ций, не рассматривая конструктивные детали.
414
Конструктивно лабораторные, аппараты для жидкофазных реак-
ций, работающие под давлением, можно разделить на. автоклавы
и аппараты колонного или трубчатого типа. Первые можно применять
и как статические реакторы, и (при нали-
чии специальных штуцеров) как динами-
ческие безградиентные аппараты для ис-
следования кинетики реакций. Все ва-
рианты автоклавов должны предусматри-
вать хорошее перемешивание реакционной
массы. Аппараты второго типа представ-
ляют собой интегральные реакторы, по-
добные описанным выше для газофазных
процессов.
Наиболее надежным типом лаборатор-
ных автоклавов являются различные ва-
рианты бессальниковых аппаратов. Один
из самых распространенных вариантов
такого аппарата — автоклав с внутрен-
ним контуром циркуляции системы Виш-
невского [21] представлен на рис. Х.11.
Особенность аппарата — экранирование
статора асинхронного электродвигателя
от реакционного пространства немагнит-
ным материалом, что позволяет отказаться
от сальникового уплотнения вала ме-
шалки, так как ротор двигателя находится
под реакционным давлением. Турбинная
мешалка обеспечивает эффективное переме-
шивание реакционной смеси. Аппарат мо-
жет быть использован и в проточных си-
стемах. При работе с гранулированным
катализатором в аппаратах конструкции
Вишневского внизу диффузора устанав-
ливают редкую сетку, на которую поме-
щают зерна катализатора. За кинетикой
реакций в таких аппаратах наиболее
целесообразно следить, отбирая пробы
через определенные промежутки времени.
В случае, когда катализатор находится
в аппарате в суспендированном состоянии,
погруженный в реакционную массу конец
пробоотборника должен быть снабжен
фильтром, изготовленным из тонкой сетки
Рис. Х.11. Реактор с внеш-
ним статором конструкции
Вишневского емкостью
0,5 л:
1 — ротор; 2 — обмотка стато-
ра; 3 — корпус реактора; 4 —
винтовой насос; 5 — направля-
ющий стакан.
или из керамики, или металлокерамики.
Все же отбор проб из автоклава в течение опыта иногда вызывает
трудности из-за забивки пробоотборника или непредставительности
проб в случае многофазных систем (несколько несмешивающихся
жидкостей). Тогда приходится для. определения каждой точки на
415
кинетической кривой проводить специальный опыт, что занимает
много времени и создает значительные ошибки при быстрых реак-
циях. В этих случаях гораздо эффективнее для исследования кине-
тики применять ампульную методику, которая заключается в том,
что ряд ампул помещают в общий термостат и затем ампулы выни-
мают последовательно через заданные интервалы времени, охлаждают
и их содержимое анализируют. Интервал времени при этом состав-
ляет несколько минут и поэтому данные
1
I
Рис. Х.12. Ампула с маг-
нитной возвратно-поступа-
тельной мешалкой:
1 — запорная игла; 2 — уплот-
няющая головка; з — корпус;
4 — катушка-соленоид; 5 —
плунжер; 6 — лопасти.
получаются для относительно малых вре-
мен реакции. Для работы под давлением
предпочтительно работать с металличе-
скими ампулами, которые фактически
представляют собою микроавтоклавы. При
особой необходимости можно работать и
со стеклянными ампулами (до давления
100 ат), которые коваром сварены с ме-
таллическими запорными устройствами.
Конструкция таких ампул дана в работе
120].
На рис. Х.12 приведена очень удобная
конструкция металлической ампулы ем-
костью 30 мл с магнитной возвратно-по-
ступательной мешалкой, в которой исклю-
чены холодные выступающие части запор-
ных приспособлений и связанные с этим
ошибки из-за наличия мертвого объема с
пониженной температурой [22]. В верх-
ней части ампулы находится уплотняющая
головка 2 с размещенной в ней частью
вентиля с запорной иглой 1. Другая часть
вентиля расположена в специальном вы-
носном устройстве, служащем для заполне-
ния ампулы газом до давления опыта. При
необходимости работать с постоянной по-
ддачей газа головку с иглой заменют на
головку с приваренным капилляром. Мешалка состоит из плун-
жера 5, выполненного из железа Армко и, если нужно, защищен-
ного коррозионностойким покрытием, и лопастей 6. Всю ампулу
вставляют в катушку соленоида, помещенную в жидкость термо-
стата. При числе переключений соленоида 2—3 в 1 сек обеспе-
чивается весьма интенсивное перемешивание содержимого ампулы.
В некоторых случаях проточные реакторы при исследовании
жидкофазных реакций обладают преимуществами в сравнении со
статическими, например при исследовании реакций с суспендирован-
ным катализатором. В этом случае в установках с безградиентным
реактором не применяют внешнего контура для циркуляции жид-
кости, а используют принудительное перемешивание. На рис. Х.13
приведена схема установки с проточным безградиентным реактором,
416
применявшейся для изучения кинетики жидкофазного восстановле-
ния нитросоединений водородом на суспендированном катализаторе
при давлениях порядка 150 ат. В основу ее положен реактор с маг-
нитной возвратно-поступательной мешалкой, что, однако, необяза-
тельно. Особенностью реактора является укрепленная в дне на
Рис. Х.13. Безградиентный жидкофазный реактор:
1 — насос-дозатор; 2 — мешалка; 3 — пористая пластина.
замазке фильтрующая керамическая пластинка 5, которая препят-
ствует удалению катализатора из аппарата.
Необходимость устройства газового циркуляционного контура
для стабилизации состава газовой фазы определяется характером
реакции. Если продукты реакции не попадают в газовую фазу, как,
например, в процессах гидрирования, то надобность в таком контуре
отпадает. В других случаях, например при Окислении в жидкой
фазе воздухом, когда продукты реакции выделяются в газовую
фазу, наличие циркуляционного контура по газу может оказаться
желательным. Однако из-за сложности его осуществления лучше
27 Заказ 1441 417
заменять циркуляцию газа приготовлением искусственных газовых
смесей.
Проточные интегральные лабораторные реакторы для жидко-
фазных реакций представляют собой простые трубки из стекла или
металла. При работе с высокими давлениями конструктивно при-
ходится вносить значительные усложнения, которые подробно изло-
жены в работе [20].
Лабораторные газо-жидкофазные проточные реакторы выполняют
в виде колонок, где в качестве насадки применяют таблетки катали-
затора. Конструкция их в основном зависит от давления, при кото-
ром проводится процесс, и не представляет ничего оригинального
по сравнению с конструкциями реакторов для жидкофазных реакций.
При перенесении результатов исследований кинетики для газо-
жидкостных или многофазных жидкостных систем из статических
интегральных или из дифференциальных реакторов на промышлен-
ные аппараты надо учитывать поправки на различие в газосодержа-
нии, связанные со скоростями подъема пузырей или капель. По-
скольку гидродинамика в лабораторных колонных реакторах резко
отличается от таковой в промышленных аппаратах, применение
интегральных проточных реакторов при исследовании кинетики
большей частью не создает преимуществ перед статическими.
Х.5. ЛАБОРАТОРНЫЕ АППАРАТЫ
ДЛЯ ИССЛЕДОВАНИЯ ПРОЦЕССОВ В КИПЯЩЕМ
И ДВИЖУЩЕМСЯ СЛОЯХ КАТАЛИЗАТОРА
В процессах с движущимся катализатором, а особенно в кипя-
щем слое, влияние гидродинамических факторов еще сильнее, чем
в процессах с неподвижным слоем катализатора. Поэтому при их
разработке целесообразнее всего исследовать различные стороны
процесса отдельно. Активность и стабильность катализатора и кине-
тику химических превращений наиболее удобно исследовать на
проточно-циркуляционных или других кинетических установках.
Истираемость катализатора, скорость падения активности, условия
регенерации следует изучать в специальных условиях. Целесообразно
отдельно исследовать гидродинамические характеристики аппарата.
Однако практически последнее редко удается осуществить полностью
и с достаточной надежностью и поэтому пока часто нельзя обойтись
без предварительного моделирования процесса в целом в лаборатор-
ных условиях. Поскольку при этом целью является фактически
исследование не катализатора, а аппарата, то лабораторную аппара-
туру желательно выполнять в наибольших возможных размерах,
чтобы устранить влияние стенок и других особенностей малых
аппаратов.
Опыт показывает, что аппараты с кипящим слоем катализатора
диаметром менее 100—120 мм не дают надежных результатов для
разработки промышленных процессов, и применение их в лаборатор-
ной практике, в общем, нецелесообразно. Исключения возможны,
418
По A A
Рис. X.14. Стеклянный ре-
актор для работы с кипя-
щим слоем катализатора:
1 — реакционная часть колонки;
2 — сепарационная часть колон-
ки; з — отдувочное сопло;
4 — керамические фильтры; 5 —
газоотводящий коллектор; 6 —
влектрообмотка; t — катализа-
тор; 8 — загрузочное отверстие;
9 — отверстие для термопары;
1 о — пластинка из пористой ке-
рамики или пористого стекла.
Рис. Х.15. Стальной реактор с фильтрами для
работы с кипящим слоем катализатора (диаметр
рабочей части 50 мм\.
1 — штуцер для ввода воздуха на отдувку; 2 — распре-
делительный кран; 3 — керамические фильтры; 4 — сепа-
рационная часть колонки; 5 — реакционная часть ко-
лонки; в — распределительная решетка; 7 — гильзы для
термопар.
27*
когда требуется приближенно оценить показатели процесса и возмож-
ность его осуществления или найти относительные показатели работы
катализатора для кипящего слоя (например, при контроле произ-
водства катализатора), а также при
Рис. Х.16. Металлический пи-
лотный реактор для работы с
кипящим слоем катализатора
без фильтров:
1 — уловитель катализатора; 2 —
сепарационная часть колонки; 3 —
провальные тарелки; 4 — реакцион-
ная часть колонки; 5 — распреде-
лительная решетка; в — рубашка
для охлаждения; 7 — предохрани-
тельная мембрана.
первоначальном исследовании процес-
сов с циркулирующим кипящим слоем
катализатора. В последнем случае вза-
имосвязь работы реактора и регенера-
тора может быть определена и на уста-
новке малого масштаба. Те же сообра-
жения, в общем, применимы и к
аппаратам с' движущимся катализа-
тором.
Лабораторные аппараты для иссле-
дования катализатора в кипящем слое
представляют собой уменьшенную ко-
пию промышленных аппаратов, но имеют
обычно значительно большее отношение
высоты к диаметру. Аппараты лучше
всего изготавливать из металла, хотя
имеется опыт работы и со стеклянным
реактором [23] (рис. Х.14). В послед-
нем случае колонка представляет собой
трубку из молибденового или другого
тугоплавкого стекла диаметром 30—
40 мм и высотой до 1 м с расширением
наверху. В нижнюю часть колонки вма-
зана опорная пластинка 10. В верхней
части колонки в расширении располо-
жены фильтры из пористых керами-
ческих гильз.
На рис. Х.15 представлена сталь-
ная лабораторная колонка диаметром
50 мм для исследования реакций в ки-
пящем слое катализатора. Кроме мате-
риала и конструктивных деталей уст-
ройств для отдувки фильтров^ она прин-
ципиально ничем не отличается от стек-
лянной колонки РИС; Х.14.
На рис. Х.16 приведена лабора-
торная реакционная колонка с диамет-
ром рабочей части 150 мм. В отличие
от предыдущих, колонка не имеет
фильтров, снабжена распределитель-
ными провальными тарелками и предназначена для работы с относи-
тельно крупным катализатором (диаметром —1 мм). Схема лабора-
торной установки, состоящей из реакторд и регенератора, принци-
пиально не отличается от производственной.
420
Рис. Х.17. Лабораторная установка
с движущимся шариковым катализа-
тором:
1 — газовые часы; 2 — приемник; 3 —
конденсатор; 4 — блок дифференциальных
манометров; 5 — смотровой фонарь; в —
контрольная коробка; 7 — труба для ка-
тализатора; 3 — бункер для катализатора;
9 верхний затвор; 10 — подогреватель
воздуха для эрлифта; 11 — эрлифт; 12 —
реактор; 13 — средний затвор; 14 — реге-
нератор; 15 — подогреватель воздуха для
регенерации; 1в и 18— реометры; 17—
баллоне азотом; 19 —дозатор катализа-
тора; 20 — нижний затвор; 21 — насос-до-
затор сырья; 22 — бюретка для сырья;
2з — конденсатор газоанализатора; 24 —
газоанализатор.
СО
Сырье
Газ крекинга
Воздух
Устройство лабораторной установки каталитического крепинга
с движущимся шариковым катализатором понятно из рис. Х.17.
Следует подчеркнуть, что установка таких размеров пригодна только
для изучения основных параметров процесса, но не дает возмож-
ности отработать конструкцию аппаратуры.
ЛИТЕРАТУРА
1. В. В. Н а л и м о в, Н. А. Чернова, Статистические методы
планирования экстремального эксперимента, Изд. «Наука», 1966.
2. , Ю. П. Адлер, Введение в планирование эксперимента, Изд. «Метал-
лургия», 1969.
3. С. 3. Р о г и н с к и й, М. И. Я н о в с к и й, А. Д. В е р м а н, Ос-
новы применения хромотографии в катализе, Изд. «Наука», 1972.
4. Gordon, Trans. Faraday Soc., 44, № 4, 196 (1948).
5. H. К a g о m, M. В о nue m a у, Bull. soc. chim. France, 68, № 5,
931 (1961).
6. M. Zarhiewski, Roczn. Chem., 30, 999 (1956).
7. M. Э. А э p о в, О. M. T о д e с, Гидравлические и тепловые основы
работы аппаратов со стационарным и кипящим зернистым слоем. Изд. «Химия»,
1968.
8. В. А. Осипова, Экспериментальные исследования процессов те-
плообмена, Изд. «Энергия», 1964.
9. Я. П и н к а в а, Лабораторная техника непрерывных процессов,
ИЛ, 1961.
10. Н. А. А р о н о в и др., Кинетика и катализ, 11, № 4, 1048 (1970).
11. М. И. Темкин, Н. В. Кулькова, Кинетика и катализ, 10,
№ 2, 461 (1969).
12. В. А. Ройт ер и др., Каталитическое окисление нафталина, Изд.
АН УССР, Киев, 1963.
13. М. И. Т е м к и н, С. Л. К и п е р м а н, Л. И. Лукьянова,
ДАН СССР, 74, № 4, 763 (1950).
14. Ю. В. Суздальницкая, К. И. Матвеев, Кинетика и ката-
лиз, 5, № 1, 194 (1964).
15. И. И. И о ф ф е, Г. В. Д е р г у н о в а, ЖФХ, 41, № 2, 511 (1967).
16. Г. П. К о р н е й ч у к, В. Г. В ы с о ч е н к о, сб. «Катализ и ката-
лизаторы», Изд. «Наукова думка», Киев, 1971, стр. 103; В. Л. Щ у к и н,
•С. А. Веньеминов, Кинет, и катализ, 12, № 2, 533 (1971).
17. Г. П. Корнейчук, сб. «Катализ и катализаторы», т. 4, Изд.
«Наукова думка», Киев, 1968, стр. 151.
18. В. М. Г а р а н и н, У. М. К у р к ч и, X. М. Миначев, Кине-
тика и катализ, 8, № 3, 701 (1967).
19. A. Luft, A. Herbert z, Chem. Ing. Techn., 62, № 11, 363 (1964).
20. Д. С. Цикли с, Техника физико-химических исследований при вы-
соких и сверхвысоких давлениях, Изд. «Химия», 1965.
21. Н. Е. В и ш я е в с к и й, Н. П. Г л у х а н о в, И. С. К о в а л е в,
Аппаратура высокого давления с герметическим приводом, Машгиз, 1960.
22. И. И. И о ф ф е, Н. А. А р о н о в, А. А. X а р ч е н к о, Кинетика
и катализ, 12, № 2, 531 (1971).
23. А. Ф. Г р и г о р о в, Ю. Ф. Г о л ы н е ц, И. И. Иоффе, Зав.
лаб., 23, № 3, 370 (1957).
Глава XI
МЕТОДЫ ПОСТРОЕНИЯ
МАТЕМАТИЧЕСКИХ МОДЕЛЕЙ
КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
XI.1. ПРИНЦИПЫ ПОСТРОЕНИЯ МОДЕЛЕЙ
При разработке математических моделей химических реакторов;
часто лимитирующей (в смысле времени и надежности) становится
стадия раскрытия составляющих этих моделей, характеризующих
химическую реакцию. Актуальность проблемы, внимание к ней
исследователей, специфичность используемых методов выдвинули
этот комплекс вопросов в самостоятельный раздел инженерной
химии.
Разработка кинетической составляющей математических моделей
состоит из ряда этапов: теоретический анализ химизма процесса
с целью выбора возможных вариантов кинетической схемы; проведе-
ние экспериментов на кинетических или укрупненных установках;,
оценка параметров математического описания по полученным экспе-
риментальным данным; оценка доверительных областей параметров;
оценка принятых гипотез о механизме реакций и планирование
дополнительных экспериментов для уменьшения доверительной
области параметров и выбора механизма, адекватно описывающего
процесс в исследованной области режимных параметров. Описанная
процедура является итеративной, так как не всегда удается получить-
однозначный ответ об адекватности единственной модели из всех
выдвинутых априори после первой серии экспериментов. Процесс
отбраковки неадекватных моделей продолжается до тех пор, пока не
останется единственная модель, не противоречащая всей совокуп-
ности' экспериментальных данных.
Общих принципов выбора той или иной кинетической модели
реакции нет, он осуществляется на основе довольно грубой пред-
варительной теоретической оценки зависимостей и отчасти на ин-
туиции исследователя. Однако не следует преувеличивать трудности
этого этапа, так как последующая математическая обработка дает
возможность отобрать из ряда предполагаемых моделей наиболее
правдоподобную [1].
Этапы проведения экспериментов и их обработки тесно связаны;
между собой: выбор аппаратуры и методики исследований опреде-
ляет методы обработки экспериментальных данных; в свою очередь,,
наличие или отсутствие в распоряжении исследователя средств,
вычислительной техники и математического обеспечения может
иногда обусловить выбор того или иного метода кинетических иссле-
дований. Отсутствие до недавнего времени эффективных алгоритмов
обработки данных объясняет, в основном, появление и широкое
использование в последнее время безградиентных методов
423
исследования кинетики химических процессов. Математическаямодель
•безградиентного реактора представляет собой систему алгебраических
уравнений, как правило, линейных или достаточно легко линеари-
зуемых при принятии некоторых упрощающих предположений.
Задача обработки экспериментальных данных тогда решается с по-
мощью классического аппарата метода наименьших квадратов или
методов линейного программирования. Разработка общих принципов
линеаризации сложных нелинейных моделей [2] и успешное разви-
тие поисковых методов определения экстремума функции многих
переменных [3, 41 позволили создать эффективные алгоритмы об-
работки экспериментальных данных, полученныхирактически в лю-
бых условиях проведения процесса — при наличии градиентов кон-
центраций и температур по времени, длине и радиусу слоя катали-
затора, внешней и внутренней диффузии и т. д. В связи с этим выбор
типа установки должен определяться, в основном, спецификой ис-
следуемого процесса и имеющейся в распоряжении исследователя
экспериментальной базой. При этом следует учитывать, что резуль-
таты, полученные на безградиентных реакторах, имеют менее
наглядный характер по сравнению с данными, получаемыми с ин-
тегральных реакторов, и их кинетическая трактовка иногда затруд-
нительна.
XI.2. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ
ПРОСТЫХ КИНЕТИЧЕСКИХ ЗАВИСИМОСТЕЙ
Рассмотрим задачу обработки экспериментальных данных при
разработке математических моделей реакций, описываемых простей^
шими кинетическими схемами. Фактически под этим понимаются
достаточно простые методы определения порядков единичной реак-
ции и констант ее скорости, не требующие применения вычислитель-
ной техники. Такие методы могут оказаться полезными не только
для простых, но и для сложных реакций, когда исследуется брутто-
реакция превращения исходного вещества или интересным является
формальная кинетика образования целевого продукта. Поэтому для
целостности изложения напомним эти методы, хотя некоторые из
них достаточно подробно описаны в ряде монографий, например [5].
Для быстрой, но приближенной оценки порядка реакции по од-
ному компоненту можно применять способ сравнения времен рав-
ных превращений, основанный на соотношении
\ И) /
(XI.1) -
где t{ — время, необходимое для превращения /-й доли исходного
реагента; п — порядок реакции; Сц — исходная концентрация реа-
гента в опыте.
424
Другим приемом для обработки выражений степенного типа
является способ логарифмирования уравнения скорости реакции,
приводящий к выражению
In | rJl = ln /сЦ-nln С (XI. 2}
которое дает возможность методом наименьших квадратов (МНК)
определить к и п из серии опытов. Значения г определяются либо
непосредственно в дифференциальных установках, либо графическим
или численным дифференцированием данных интегральных реак-
торов.
В несколько более сложных случаях строят графики в коорди-
натах t — kf (С), где / (С) — вероятные формы функциональных за-
висимостей. Выбирается такая функция /, при которой экспери-
ментальные точки ложатся наиболее блцзко к прямой линии, наклон
которой соответствует значению к.— v
Энергия активации реакции легко определяется из графика Ар-
рениуса в координатах In к — 1/Т или из выражения:
Т\~
Для простых реакций, однако, можно определить приближенно
энергию активации и без знания константы скорости реакции. Ме-
тод сводится к следующему. Для любого порядка реакции имеем;
гт, \ _к(Т1)
ТТг Л-const к
Из формулы (XI.4) и уравнения
E = RTZ
следует, что
dink
дТ
(XI.4)
(Х1-5>
(XI. 6),
где х — степень превращения.
Подставляя в (XI.6) вместо производной отношение конечных
разностей, полученных из опытных данных при х — const, можно
рассчитать энергию активации реакции.
Эти методы пригодны для обработки экспериментальных данных,
полученных в изотермических условиях (при отсутствии градиентов
температуры по длине слоя катализатора и радиусу трубки). Про-
ведение экспериментов в неизотермических (адиабатических, в част-
ном случае) условиях приводит к усложнению методов обработки.
Один из этих методов заключается в дифференцировании экспе-
риментальных концентрационных кривых. Если подставить получен-
ные таким образом значения скорости реакций в нескольких точках
по длине слоя катализатора (т. е. при различных температурах)
425.
IB дифференциальное уравнение, описывающее процесс, то получится
•система алгебраических уравнений, содержащая неизвестные кине-
тические параметры — предэкспонент и энергию активации:
^|;=Zexp(_w)/(C)l/(7=1,2...........т) (XL7>
Эта система приводится логарифмированием к линейному отно-
сительно In Z и Е виду.
О степени превращения исходного компонента при исследовании
единственной или брутто-реакции, протекающей с тепловым эффек-
том, можно судить по тепловыделению или теплопоглощению в ходе
процесса. Так, при проведении экзотермической реакции в адиабати-
ческих условиях изменение температуры процесса пропорционально
изменению концентрации исходного вещества или продукта
h
Д2’=-р—|ДС| (XI.8)
тде h — тепловой эффект реакции; Ср — удельная теплоемкость реак-
ционной смеси.
Используя (XI.8), можно получить концентрационный профиль
и с помощью изложенного выше метода найти кинетические пара-
метры.
Случай мономолекулярной реакции любого порядка рассматри-
вается в работе [6]. Путем несложного преобразования"система диф-
ференциальных уравнений, включающая уравнении скорости реак-
ции и уравнение теплового баланса, приводится к виду:
( CD V-l
~^Z -=£ (Гмакс-7’)»ехр(-Е/Я7’)
\ fl /
(XI.9)
Разделив выражение (XI.9) на (Гмзкс
уовав, получаем:
Т)п и прологарифми-
ln (dT/dt)
(Тмакс-Т)п
_1_
т
(XI.10)
Значения dT/dt получаются графическим или чисЛЬнным диф-
ференцированием измеренного температурного профиля. При пра-
вильно подобранном порядке реакции п экспериментальные данные *
. (dT/dt) 1
ложатся на прямую в координатах In г- ——, —
V1 макс —1 ) -К-*
Из этого графика определяются значения Е и Z.
Следует отметить, что изложенные методы носят приближенный
характер, так как используют графическое дифференцирование экс-
периментальных кривых, что вносит значительные погрешности
в результаты решения.
426.
XI.3. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ
СЛОЖНЫХ КИНЕТИЧЕСКИХ ЗАВИСИМОСТЕЙ
(ЛИНЕЙНЫЕ ЗАДАЧИ)
Если исследуемая реакция является сложной и протекает как
ряд параллельных и последовательных превращений, представля-
ющих собой отдельные стадии всего химического процесса, и, если
все параметры, включая порядки реакций, неизвестны, то расшиф-
ровка кинетической схемы процессов и определение значений кине-
тических параметров является сложной задачей. Этой проблеме
в настоящее время посвящено много работ [2, 7, 8]. Здесь рассмотрим
некоторые наиболее, на наш взгляд, существенные и близкие к пред-
мету книги методы решения указанного типа задач. Последним можно
дать наименование «обратные задачи химической кинетики», по-
скольку в них по известному решению, найденному экспериментально,
должны быть определены структура и параметры уравнений кине-
тической модели.
Если зависимость скорости реакции от концентраций реагентов Ct
представить в форме
г=кПс^с (xi.il)
i
то задача расчета сведется к вычислению константы скорости к
и порядков а,. Специфика гетерогенно-каталитических процессов
состоит в том, что скорость каждой реакции может зависеть не
только от концентраций исходных веществ, но и от концентраций
продуктов реакции и веществ, в данной реакции не участвующих.
Если кинетическое уравнение представлено в виде (XI.11), то по-
рядки реакции по веществам, тормозящим процесс, будут отрица-
тельными. В.процессах гетерогенного катализа всегда можно ожи-
дать появления в выражении (XI.11) концентраций всех веществ,
присутствующих в реагирующей смеси и, следовательно, произведе-
ние в правой части (XI. 11) всегда будет содержать большое число
сомножителей.
Логарифмируя формулу (XI.ll), получаем:
In г = In к-f-ai 1н Ci (XI.12)
При обработке экспериментальных данных, полученных на ин-
тегральных проточных реакторах в «идеальных» условиях, т. е.
при отсутствии градиентов температуры и продольного перемеши-
вания, математическая модель представляет собой систему кинети-
ческих дифференциальных уравнений материального баланса, ли-
нейных относительно входящих в них неизвестных констант ско-
рости реакций
(XI.13)
at
427
где /х (Л, С) — вектор-функция, компоненты которой — функции,
линейные относительно констант скорости реакций.
Систему (XI. 13) можно привести к системе линейных алгебраи-
ческих уравнений одним из следующих двух способов.
Если Численно продифференцировать в нескольких точках tt
экспериментальные зависимости С (t) и подставить полученные зна-
чения скорости изменения концентраций реагирующих компонен-
тов в (XI.13), то получится линейная система, аналогичная модели
дифференциального реактора.
Интегрированием система (XI. 13) приводится к виду:
/ *i \
С(г)-С0 = /2 fc, J Cdt (XI.14)
\ о /
В этой линейной системе коэффициенты при константах скорости
реакций находятся численным интегрированием табличных экспе-
риментальных данных С (t) (например, методом Симпсона) при не-
скольких значениях верхнего предела интегрирования.
Сравнение рассмотренных методов сведения системы дифферен-
циальных уравнений к системе алгебраических, проведенное на
модельных типовых реакциях с использованием математического
эксперимента, показало, что дифференцирование вносит значитель-
ные погрешности в результаты обработки по сравнению с интегри-
рованием. Надежность расшифровки и точность расчета констант
для рассмотренных случаев обработки кинетических изотермических..
данных при использовании метода интегрирования не хуже, чем при
применении для исследований кинетики безградиентных реакто-
ров [7].
Таким образом, независимо от типа кинетической установки
и методики исследования, задача определения константы путем об-
работки экспериментальных данных, полученных в идеальных усло-
виях, сводится при известных порядках реакций к решению системы
линейных алгебраических уравнений вида (XI.12) или (XI.14), кото-
рую можно записать в общем матричном виде
у = Ах - (XI.15)
где х — вектор неизвестных параметров — констант скорости реак-
ции; у — вектор значений левых частей уравнений (XI.12), (XI.13)
или (XI.14); А — матрица коэффициентов при неизвестных пара-
метрах размером п X (го X ц) (здесь п — число параметров (столб-
цов матрицы; т — число реагирующих компонентов; р — число
пределов интегрирования).
Вектор оценок для констант к находится методом наименьших
квадратов (МНК):
а: = (Лг4)_14гу (XI.16)
428
Формула (XI.16) справедлива для частного случая, когда ковариа-
ционная матрица свободных членов у диагональная и их среднеквад-
ратичные отклонения одинаковы. Если эти условия не соблюдаются,
то МНК-оценки определятся в виде
x = [ATDl(y)A] 1ATD~1(y)y
(XI.17)
где D (у) — ковариационная матрица (матрица моментов) свобод-
ны^ членов, элементы которой
( о? при i = i
I cov (У1, yf) при i =/= j
В случае, когда концентрации компонентов измеряются с раз-
ными, но независимыми погрешностями, матрицу Z)-1 в (XI. 17)
можно заменить диагональной матрицей весовых коэффициентов:
<J2
W(y) =
(XI.18)
Однако при применении МНК в ряде случаев может наблюдаться
значительная чувствительность определяемого набора констант к из-
менениям экспериментальных данных. Значения отдельных констант
могут выходить за" пределы допустимых (например, принимать даже
отрицательные значения).
Задача определения неизвестных параметров кинетических урав-
нений по экспериментальным данным станет, по-видимому, более
корректной, если при ее решении будет, кроме соотношений вида
(XI.11)—(XI.14), учитываться и другая информация о кинетике
процесса и о константах, например, положительность величин к
и а1 и их приближенное равенство стехиометрическим коэффициен-
там для реакций низких порядков, отрицательность порядков по ве-
ществам, тормозящим процесс, и т. д. /
Задачи такого рода решаются в рамках хорошо разработанных
методов математического программирования. Покажем это для
задачи определения константы к и порядков реакции а, гетерогенно-
каталитической реакции при зависимости логарифма скорости реак-
ции от концентраций вида (XI.12). Допустим, известны следующие
факты об определяемых параметрах:
In к г; а, 0 (г = 1, 2, , . т^); а; О (г = mj-|- 1, . . ., т)
(XI, 19)
Тогда задачу определения параметров к и формулируют сле-
дующим образом. Задана система линейных равенств
t\j(k, a)=lnfc + 2 MnQ/ —1пг/
l—l
(где а = (ап аа, . . ., ат}) •
(XI.20)
429
и система линейных неравенств (ограничений):
In А — z SsO;
ai 2s О,
2s о, а.„ ., о, . . а с; о
* mi * Ш1+1 ’ » т
(XI.21)
Индекс / обозначает экспериментальные точки (/ = 1,2, . . ., N).
Требуется определить минимум выпуклой кусочно-линейной
функции:
N
V (к, а)= 2 «)1 (XI.22)
з=1
Используя метод минимизации суммы модулей, предложенный
Зуховицким, сводим задачу минимизации (XI.22) к задаче линей-
ного программирования [9]. Полагая | т], (к, а) | sg у,, приходим
к минимизации
N
^=2^- (Х1-23>
j=i
при ограничениях:
т
У,— In к— 2 In C/y + lnr/^ О
3=i ~ (XI.24)
г// + 1п к+ 2 ln Cij~~1пг/ 0 -
1 = 1
In к — z ~s О, J/-1 2s 0, ami ~:s 0, —ami_|_i₽= 0, — ат О, У} 2s О
(7 = 1, 2, . . ., N)
Эта задача решается симплекс-методом. Прежняя при исполь-
зовании МНК формулировка задачи (минимизация суммы квадратов
отклонений логарифмов расчетных значений скоростей реакций от
экспериментально определенных) изменена на новую, соответству-
ющую минимизации суммы модулей отклонений логарифмов рас-
четных значений скоростей от экспериментальных.
XI.4. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ
СЛОЖНЫХ КИНЕТИЧЕСКИХ ЗАВИСИМОСТЕЙ
(НЕЛИНЕЙНЫЕ ЗАДАЧИ)
^Рассмотрим основные типы нелинейности сложных кинетических
моделей. В общем случае скорости реакций можно записать в виде:
r=F(T, С) (XI.25)
Функция F для реакции, протекающей по гомогенному или
квазигомогенному механизму, имеет вид:
Р = (XI.26)
1=1
430
Для лангмюровской кинетики скорость реакции определяется
выражением
7И '
fc П c“z
Г =--------—-----т----- (XI-27)
/ т \2 ai
1 + 2 b‘Ci Г1
\ г=1 /
где к — эффективная константа скорости реакции.
Зависимость константы скорости реакции от температуры обычно
принимается в виде уравнения Аррениуса:
/с(Т)=2еХр(-А;
(XI.28)
Неизвестные кинетические параметры (за исключением предэкс-
поненциальных множителей Z): энергии активации Е, порядки реак-
ций аг, адсорбционные коэффициенты bt — входят в выражения
(XI.26)—(XI. 28) нелинейно. Прямое применение изложенных в раз-
деле XI.3 методов в этом случае невозможно. Для решения задачи
оценки параметров нелинейных моделей возможны два подхода:
1) линеаризация модели относительно параметров и сведение ее
к системе линейных алгебраических уравнений;
2) применение поисковых методов минимизации критерия эффек-
тивности оценки констант.
Подробное рассмотрение второго подхода не входит в задачу
настоящей книги, так как этот вопрос полно освещен в некоторых
монографиях [3, 41. Поэтому остановимся лишь на методах линеари-
зации.
Писаренко и Погорелов предложили общий метод линеаризации
кинетических уравнений сложных реакций, основанный на много-
кратном дифференцировании скорости реакции по определяющим
переменным [2].
Рассмотрим систему N реакций типа
<хА+РВ+... ► pD + vE-|---- (IX.29)
с неизвестными в общем случае порядками реакций. Модель этой
системы имеет вид
dC- т
—^Lri = Zexp ( — J] Cvtil (Z = l, 2, . . ., A) (XI.30)
7 i=l
Продифференцируем левую и правую части этого выражения по
некоторым параметрам Ц; (/ = 1, 2, . . ., т + 1), смысл которых
будет объяснен ниже. Здесь примем только, что величины скорости
реакций г1г концентраций и температуры Т являются функциями
431
этих параметров и дифференцируем по ним необходимое число раз.
Тогда получим
^-г,ехр(-ВДДГ)^.^-П CZ'*+
г-1
m т
+ ХггеХр(-л///?п-^--4т£-П c*«z=
т
El дТ , X? 47 дС> , , , '
RT2‘ dfij Z1 Ci ' дц{ U — 1’ • i (m+l)) (XI.31)
i-l
или в матричной форме
где
Ut=riWPi
W=
'fl dCj )
I Ci ' дщ J
. ((™+1)Хт)
1 дТ Г
RT2 ' ди/ J
((m+l)Xl) .
(XI-32)
(XI.33)
Искомые параметры — порядки реакции va я энергия актива-
ции Et — входят в систему (XI.32) линейным образом. Поэтому
вектор параметров Р( может быть найден как z
Pz=(rzlF)-lI7z (XI.34)
Таким образом, задача сводится к нахождению значений элемен-
тов матриц Ut и W хотя бы для одной точки (тп + 2)-мерного про-
странства, образованного переменными plf . . ., p,m+1 и t, для ко-
торой матрица W будет невырожденной.
В качестве первых тп параметров Ц; (j = 1, 2, . . ., тп) могут
быть выбраны следующие величины: тп значений концентраций ком-
понентов С( в некоторый момент времени, в том числе и начальные
значения; тп любых соотношений бу (j = 1, 2, . . ., тп) между С, (i =
= 1, 2, . . ., тп), из которых может быть однозначно, определена
величина каждой из С,; то же, но в отношении значений начальных
концентраций Ci0.
В качестве (тп + 1)-го параметра могут быть использованы: Т —
температура внутри реакционной зоны при изотермических усло-
виях проведения процесса; Тб„ — температура блока нагрева (охла-
ждения) реактора при условии постоянства во времени температур-
ного поля блока при неизотермических условиях проведения про-
цесса; То — начальная температура реакционной смеси; Т„ — тем-
пература любой точки блока при условии сложного температурного
поля.
В принципе систему типа (XI.32) можйо написать для N реакций
и для любой модели, в том числе и для такой существенно нелиней-
ной, как лангмюровская модель реакции, проводимой в неизотерми-
ческих условиях. Однако в этом случае в матрицу U входят вторые
производные от скорости реакции по параметрам ц,-.
432
Если бы были известны точно значения всех элементов матриц U
и W, входящих в расчетные выражения типа (XL34), то можно
было бы получить точные значения всех искомых параметров для
любой формы моделей реакций и реакторов и любых условий прове-
дения процесса. Но так как значения этих элементов зависят от зна-
чений параметров, заранее неизвестных, то даже при условии, что
точно известна форма математической модели, невозможно вычис-
лить все производные, входящие в указанные расчетные выражения.
Поэтому значения производных определяются экспериментальным
путем, для чего должен быть проведен специальный эксперимент.
Если эксперимент проводится по специальному факторному плану,
то оказывается возможным написать сравнительно простые расчет-
ные выражения для элементов матриц U и W. Некоторым недостат-
ком рассмотренного метода следует считать необходимость проведе-
ния зксперимента по специальному плану, т. е. невозможность
обработки «непланированных» экспериментальных данных. Более
существенным недостатком является необходимость эксперименталь-
ного определения первых или даже вторых производных от скорости
реакций, что в случае проведения экспериментов в интегральном
реакторе фактически означает определение вторых и третьих смешан-
ных производных от концентраций. Как отмечалось выше, даже одно-
кратное дифференцирование экспериментальных данных вносит зна-
чительные ошибки в результаты обработки. При определении же
производных высших порядков эти ошибки существенно возрастают.
К сожалению, авторы слабо иллюстрируют возможность метода
на конкретных численных примерах с анализом погрешностей оценки
кинетических констант, поэтому вопрос о корректности применения
метода остается неясным.
Гуревичем [10] предложен аппроксимационный метод расчета ки- -
нетических параметров по данным пассивного эксперимента для
процессов, которые адекватно описываются системой дифференциаль-
ных уравнений вида
-§- = У VijZ0jexp(-Ej/RT) П С8 (i = l, 2, .... m) (XI.35)
jfiii s^Nl
где Mt — множество реакций, в которых участвует i-e вещество;
v,y — стехиометрический коэффициент i-ro вещества в j-й реакции,
взятый с соответствующим знаком; zoj — предэкспонента константы
скорости j-й реакции; Е/ — энергия активации, j-й реакции; Cs —
концентрация 5-го вещества; nSj — порядок j-й реакции по 5-му
веществу; N] — множество веществ, являющихся реагентами j-й
реакции. z
Искомыми параметрами являются предэкспонеяты, энергии акти-
вации и порядки реакций. Суть алгоритма удобно изложить на более
простом примере, а именно, для процесса, протекающего в изотерми-
ческом реакторе интегрального типа, причем когда скорость каждой
28 Заказ 1441 1 ' 433
простой реакции зависит от концентрации только одного вещества.
Система (XI.35) преобразуется к виду
2 V‘^CSS5’ (i = l>2, (XI.36)
3 t ме
здесь к/ — константа скорости j-й реакции.
Функция
у (С, П) = сп (XI.37)
аппроксимируется во всей области изменения переменных С и га*.
Предварительно с помощью сжатия по координате С и сдвига по п
исходная область изменения этих величин преобразуется в такую,
аппроксимация в которой является в некотором смысле оптимальной.
Формулы преобразования к новым переменным С' и п'
С = цС'; п = ге' + у (XI.38)
где р, и 7 — коэффициенты сжатия и сдвига.
Функция у (С, п') — (С')п' аппроксимируется функцией:
г (С , п ) = а (С ) + 6 (С ) f (n') (XI.39)
При известном виде функции/(га') коэффициенты а и Ъ для каждого
конкретного С найдутся из условия минимизации интеграла
%
(?=J [(C')n -a(C')-&(C')/(n')]Mn' (XI.40)
х п1
где га' и га' — верхний и нижний пределы изменения га'.
Сама же функция / (га') определяется из условия минимизации
интеграла
S=J J [(С')п-a(C')-b(C)f(n')Pdn'dC' (XI.41)
с; п;
где С' и С" — пределы изменения переменной С'; а (С') и Ъ (С')
находятся так, как указано выше.
Для реального нахождения функции / (га') она раскладывается
в ряд по полиномам, ортонормированным на отрезке [га(, гаД. Раз-
ложение обрывается на полиноме третьей степени. Коэффициенты
разложения подсчитываются с помощью условия минимизации ин-
тегралов (XI.40) и (XI.41).
Далее вводятся новые переменные и и и по формулам:
(XI.42)
v = uf (п')
* Индексы реакции и вещества здесь и в дальнейшем опускаем для удобства
записи.
434
Подставив в (XL36) вместо C"Sj’ функцию z с помощью числен-
ного интегрирования правых частей системы (XI.36) по временным
отрезкам (по длине реактора) можно получить переопределенную
систему линейных алгебраических уравнений относительно и vf
(j = 1, 2, . . TV), где N — число реакций. (Далее снова вводим
индексы). Величины Uj и Vj находятся линейным МНК, или, если
на искомые параметры наложены линейные связи, — симплекс-
методом, как неизвестные в задаче линейного программирования.
Затем, используя формулы (XI.42) и (XI.38), легко найти порядки
реакций и константы скоростей. Заметим, что оптимальная область
аппроксимации обладает тем свойством, что уравнения третьей сте-
пени / («') = const имеют только один вещественный корень. Эта
область находится в пределах: для С от 0,4 до 2,0, для п' от 0 до 2.
В приведенном примере искомыми величинами являлись кон-
станты скорости и порядки реакций. Если же искомыми неизвест-
ными являются энергии активации, то аппроксимация проводится
аналогичным способом. Изложение велось, кроме того, в предполо-
жении, что в каждом слагаемом системы (XI.36) имеется по два
неизвестных параметра. В случае, если в слагаемых системы до трех
неизвестных параметров, применяется следующий прием: фикси-
руется часть параметров из тех, которые входят в показатели сте-
пени (к примеру часть порядков реакций или часть энергий актива-
ции) таким образом, что оставшиеся параметры (основная часть)
находятся изложенным выше способом. Используя найденные зна-
чения параметров, аналогично можно вычислить параметры первой
группы, затем снова второй и т. д.
К примеру, в системе реакций
А+В C + D
C + D^^a+B
D E+F
первую группу составляют параметры п2 и га4 (их первоначально
фиксируют), вторую — остальные. Практика показывает, что для
полного решения такой задачи достаточно двух-трех итераций.
XI.5. ПОСТРОЕНИЕ МАТЕМАТИЧЕСКИХ МОДЕЛЕЙ
ПО НЕИЗОТЕРМИЧЕСКИМ ЭКСПЕРИМЕНТАЛЬНЫМ ДАННЫМ
Нелинейность математической модели, обусловленная неизотер-
мическими условиями проведения экспериментов, является наибо-
лее часто встречающимся на практике случаем и требует отдельного
рассмотрения. Прежде чем приступить к изложению методов об-
работки неизотермических экспериментальных данных, рассмотрим
вопрос о принципиальной возможности раздельной оценки по таким
данным предэкспоненциальных множителей и энергий активации.
28*
435
При исследовании реакций с высокими тепловыми эффектами в
реакторах с неподвижным слоем твердого катализатора не всегда
удаётся поддерживать строгое постоянство температуры по длине
или радиусу трубки. Перепады температур могут достигать при этом
40—50° С. Такая же разница температур используется и при про-
ведении изотермических экспериментов для обеспечения раздельной
оценки предэкспонентов и энергий активации. Поэтому, казалось бы,
неизотермический эксперимент, так же как и изотермический, позво-
лит оценить раздельно кинетические константы.
Для того чтобы решить этот вопрос, сравним два эксперимента —
изотермический и адиабатический. Примем, что адиабатический экс-
перимент проводится при одной начальной температуре Тд, а изо-
термический при двух 7\ и Т2, так что разность между ними АТ =
= Тг—Т2 равна адиабатическому перепаду температур. Будем счи-
тать, что конструкция реактора обеспечивает отбор проб в m точках,
равномерно распределенных по длине слоя катализатора. Сравнение
будем проводить на примере единственной мономолекулярной реак-
ции типа А -> В. Тогда для изотермических условий можно написать
систему дифференциальных кинетических уравнений:
— — — exp^«0
Й [А(2)1 ( J
(А <!>](«)
£) lA'»> W
(XI.43)
Здесь к0 = In z, В — EfR, а индексы (1) и (2) указывают на опыт
с температурами 7\ и Т2, соответственно.
Интегрируя уравнения системы (XI.43) от t = 0 до нескольких
верхних пределов получаем:
IA(1Jh-Pol = - exp J [А(1>] (t) dt
о
—lA<J = —exp^fco —JlA(2)](0<W (i= 1, 2, . . ., m) (XI.44)
о
или после преобразований
In =fc0— Д/Г1 (XI.45)
j‘ [Ao,] (t)dt _
о
In IAol — =k0-B/T2
о
436
что можно в компактной матричной форме записать так
1=Рязх (XI.46)
где I — вектор-столбец свободных членов; х — вектор-столбец оце-
ниваемых параметров к0 и В; Риз — матрица коэффициентов раз-
мерностью 2т X 2:
г 1 1 : 1 . 1 1
рт г из 1 1 1 1 1 i (XI.47)
L ” Л : ’ ' Taj
тп 1 пг
Для адиабатического эксперимента можно по аналогии записать:
[Al] —[Ао] = — J exV(k0-B/T(t))[A](t)dt '
.............°........................ (XI.48)
[Am]-(A0] = —J exp(k0-B/r(t))IA)(t)dt
О •
г
Пользуясь теоремой о среднем, так как функция [A] («) не знако-
переменная, имеем
[Аг] —[Ао] —— ехр (Ао—В/Тср,) J [A] (t) dt
о
....................•.............. (XI.49)
[Ат]~[Ао] = — ехр (к0-В/Тсрт) j [A] (t) dt
о .
где'Тер = Т (tcpi-), 0 < «ср / ti-
Полученную таким образом систему можно привести к виду
(XI. 46). Транспонированная матрица коэффициентов системы (XL46)
в этом случае имеет вид:
1 1 • • • 11
Рт -
гад —
1_______1_
ГСР1 rcp>
1
т
1 Сртп J
(XI.50)
Составим квадратную (2 X 2) матрицу D вида
(XI.51)
Мерой обусловленности системы нормальных уравнений может
служить величина: Аад = | D |.
437
Найдем определитель матрицы D:
Представим Тср[ в следующем виде:
Гер / = + АЛ = 71 (1 + = 71 (1+az)
Тогда
1
l + az
(XI.52)
(XI.53)
(XI.54)
Раскладывая функцию
чиваясь первыми двумя членами,
ряд Тейлора
имеем
(az 1)
и ограни-
- откуда, после преобразований, получаем:
ЛГад-|Я|=~
(XI.56)
Легко показать, что для изотермического зксперимента
Л^из —
тДуз
“тГ
(XI.57)
В случае адиабатических экспериментальных данных, как указы-
валось выше, Гх< Гср/< Тогда Тср1 можно представить как
(XI.58)
7ср» — 71 + A7yz
Здесь yz < 1.
Подставляя формулу (XI.58) в (XI.56) и преобразовывая, имеем:
(XI.59)
Отношение
из
(XI.60)
можно определить как степень информативности адиабатического
эксперимента по сравнению с изотермическим. Так как у < 1, то
Ф < 1.
438
Численные расчеты для различного количества эксперименталь-
ных точек тп, дают следующие значения:
тп . • .......................... 2 3 .6
Ф....................... . . 0,04 0,07 0,13
Отсюда видно, что информативность адиабатического экспери-
мента даже в сравнительно простом случае единственной реакции
по крайней мере на порядок ниже, чем изотермического.
В случае же сложного механизма, когда имеет место сильная кор-
реляция между отдельными параметрами, обработка единственного
адиабатического эксперимента может дать совершенно неправильные
результаты.
Легко показать, что при проведении серии адиабатических экспе-
риментов при нескольких начальных температурах, их информатив-
ность несколько выше, чем информативность изотермических экспе-
риментов, проведенных при тех же температурах.
Для обработки полученных при нескольких начальных темпера-
турах экспериментальных данных можно использовать один из
описанных в разделе -XI.4 методов оценки параметров нелинейной
модели.
Рассмотрим еще один метод оценки параметров, использующий
тот факт, что поверхность квадратичного критерия оценки йараме-
тров является почти вырожденной (имеет вид «оврага»), а сами
оценки предэкспонентов и энергий активации сильно попарно кор-
релированы между собой. Уравнение регрессии имеет вид:
k0-$-aBt=b (XI.61)
Если обрабатывается изотермический эксперимент, проведенный
при одной температуре, то поверхность критерия строго вырождена.
Параметры fc0 и В связаны между собой уравнением:
к0-1/Т„аВ = 1пк(Т) (XI.62)
Сопоставление уравнений (XI.61) и (XI.62) показывает, что можно подобрать такую матрицу размера 2 X тп
Г 1 1 ... 1 "1
рт = гив 1 1 Т экв экв 1 экв - (XI.63)
что квадратичный функционал
(Ряах-1) (XI.64)
будет удовлетворительно аппроксимировать в некоторой окрестности
минимума аналогичный функционал Fafl, являющийся критерием
оценки при обработке адиабатического эксперимента.
439
Будем в дальнейшем называть параметр Т,кв, входящий в мат-
рицу Ри3, температурой эквивалентного изотермического экспери-
мента или эквивалентной температурой. Тогда параметр кжв, полу- •
чающийся из выражения (XI.62) подстановкой в него эквивалент- .
ной температуры, можно назвать эквивалентной константой скорости. :
Из условия наилучшей аппроксимации функционала Fatk функ- j
ционалом FB3 вытекает, что функционал
‘k
(^»кв> ^экв)=2| I (^Чад—из)2 (XI.65)
i. h О
имеет минимум по эквивалентным параметрам.
Здесь С*ад — расчетные значения концентраций i-ro компонента,
соответствующие минимуму функционала Fan; Cilf3 — расчетные значе-
ния концентраций в эквивалентном изотермическом эксперименте,
являющиеся функциями эквивалентных параметров /сэкв и Тэкв;
к — индекс интервала интегрирования.
До сих пор рассматривали случай единственной реакции типа
А -> В. В случае сложного механизма реакций оказывается невоз-
можным подобрать единственное значение ТЭКВ таким образом, чтобы
одновременно получался минимум выражения (XI.65) для всех ста-
дий реакции. Тогда имеет смысл ввести понятие эквивалентной тем-
пературы q-й стадии, так чтобы выполнялось условие:
t
^д(^кИв%. экв <?) = min S f [^*ад из (^экв q, ^эквд))2^ (XI.66)
i. k О
Трудно дать физическое толкование понятия эквивалентной тем-
пературы g-й стадии реакции, так как невозможно поддерживать
одновременно в одной точке пространства q различных температур.
Поэтому будем понимать эквивалентную температуру в строгом
смысле как некоторый формальный параметр, определяемый регрес-
сионным коэффициентом уравнения (XI.61). На практике же ока-
зывается, что значения ТЭКВ9 очень близки между собой и отли-
чаются не более, чем на 2—3 град, что соизмеримо с погрешностями
эксперимента.
Общность приведенных выше рассуждений сохраняется и в слу- /
чае проведения реакций в аппаратах с внешним теплообменом, с той
лишь разницей, что значения средних температур в выражениях
(XI.49)—(XI.60) более близки между собой, поэтому степень инфор-
мативности такого эксперимента оказывается еще ниже, чем адиаба-
тического, т. е. поверхность квадратичной формы (XI.64) еще более
приближается к вырожденной. Последнее объясняется тем, что
концентрации компонентов в неизотермическом и эквивалентном
изотермическом экспериментах незначимо различны при наличии
погрешностей измерения, т. е. СнеиЭ <=« Сэкв из. '
Алгоритм метода эквивалентных параметров можно представить
следующим образом. -
440
Пусть математическая модель процесса задана системой дифферен-
циальных уравнений материального и теплового балансов
/ [А (Т), С]; /= (ilt - • fN)T; =fN+i [А (Г), С] (X1.67)
с начальными условиями: при t — О, С — Со, Т = Т0.
Для простоты дальнейшего изложения примем, что реакции моно-
молекулярны и йротекают по первому порядку, т. е.
N п
л=2 2 <XL68)
i-i «=1
где
1, если в q-й реакции l-е вещество превращается в i-e;
q 0, если в q-й реакции не участвует ни l-е, ни i-e вещество;
V/I —1, если i-e вещество в результате q-й реакции превра-
. щается в i-e;
п — количество реакций.
Отметим, что алгоритм не меняется и для более общего случая
N
л=2
Ь« /-1
(XI.69)
при известных порядках реакций nj{.
Зависимость константы скорости от температуры представим
в виде
А9(7) = ДА9ехр(А09--^-) (XI.70)
где Д&в — некоторый поправочный множитель, равный 1, если
значения параметров kOq и Вq минимизируют функционал FaK.
Система концентрационных уравнений из (XI.67) после инте-
грирования по нескольким пределам t принимает вид:
t
Ci (Ц) -• Ci о = 2 v?i J71 e*p (ft0 4 - ) Cl (t) dt (1 = 1,2,..., N)
q, I 0
(XI.71)
Положим в системе (XI.71) все kqq и Bq равными нулю. Тогда
система (XI.71) сводится к линейной системе, такой же, как и в слу-
чае изотермического эксперимента, и Д/са = кэкъд находятся ме-
тодом наименьших квадратов. Все дальнейшее изложение проведем
в предположении, что эквивалентные температуры Тэкв д известны,
а методы их определения обсудим отдельно.
После нахождения решения систем (XI.71), написанных для не-
скольких температурных профилей Тр (t) (р = 1,-2, . . ., Р), кпд
и Bq определяются решением систем уравнений для каждой q-и
441
стадии реакции так же, как и в случае изотермического экспери-
мента:
1
lnAfc9P = fc09— ----Bq (p=i, 2, . Р) (XI.72)
1 экв qP
Исключив из системы (XI.72) неизвестную kQq, получаем
1-----—\вч (р = 2,...,Р) (XI.73)
\ 1 ЭКВ qp 1 экв q 1 /
или в матричной форме
Lq = MqBq (XI.74)
где Lq — (Р — 1)-мерный вектор-столбец свободных членов;
Mq — (Р — 1)-мерный вектор-столбец коэффициентов при един-
ственной переменной Bq.
Решая систему (XI.76) методом наименьших квадратов, находим
р-1
2 lqimql
—
* 2
1=1
(XI.75)
'.Oq при значении Bq — О,
константы скорости:'
(Р=1, 2, . .., Р) (XI.76)
где lqi и mql — компоненты векторов Lq и Mq.
Полученные значения требуют уточнения, для чего вводится ите-
ративная процедура. Рассмотрим порядок действий на /-й итерации.
Перед /-й итерацией уже определены векторы констант к0 и В.
Их значения подставляются в систему (XI.71). Решением этой си-
стемы находится точка на прямой регрессии (XI.61). Полученная
точка и угол arctg Тзкв вполне задают положение прямой. Следо-
вательно, можно определить значение ~
являющееся логарифмом эквивалентной
В1
1п ^экв qp = ln &kqp + k0 q
Подставляя уравнение (XI.76) в выражение константы скорости,
получаем после преобразований:
В} . Bi+l
1пД4 + Ц3-^- = ^-1^- (Р=1, 2, ..., Р) (XI.77)
4 •* экв qp •* экв qp
Введем новые переменные:
= ~к0 q
м^в^-Bi
Тогда систему (XI.77) можно записать в матричном виде
(XI.78)
&q — ®qzq
(XI.79)
442
где Sg — вектор-столбец свободных членов с элементами In
©о — матрица размером 2 X Р, аналогичная (XI.63); z. = (Afcj,
ДВ')Т.
Несколько более простое решение для найдем, если исключим
из системы (XI.79) переменную А
p-i
ЛВ9=-^1------- (XI-80)
i-1
где lQi и ntj имеют тот же смысл, что в выражении (XI.75), которое
непосредственно вытекает из (XI.80) при j = 0.
Выражение (XI.80) может оказаться полезным, так как его легче
анализировать при рассмотрении сходимости процесса решения.
Проанализируем, какое влияние могут оказать Тэкм на резуль-
таты решения. Предположим, что в выражении (XI.80) вместо век-
тора М используется вектор М с элементами р, отличными от
~—. Тогда получающиеся значения АВ, будут отличаться от значе-
экв р А
ний Ь-В’д. Если АВ, > 2АВ£, то итеративный процесс оценки кон-
стант — расходящийся, если АВ^ < АВ^ < 2АВ^, то процесс сходится
колебательно, и при АВ^ < АВ£ — монотонно. При фиксированном
векторе L, определенном решением системы (XI.71), значение АВ,
полностью определено заданием вектора М. Из сказанного следует,
что характером процесса решения (в том числе и в отношении его
сходимости) можно управлять, изменяя вектор М.' Заметим, что
при этом степень свободы выбора элементов этого вектора велика,
так как одно и то же значение АВ(, обеспечивается не единственным
вектором М, т. е. точное знание вектора М не обязательно для реше-
ния. Схему управления сходимостью можно представить следующим
образом: при расходящемся или колебательно сходящемся решении
следует увеличивать величину | 1VPM | [знаменатель в формуле
(XI.80)], при сходящемся монотонно — уменьшать. Это может быть
достигнуто соответствующим изменением одного или нескольких
элементов вектора М.
В качестве начального приближения вектора М, рбеспечива-
ющего сходимость процесса решения, можно выбрать вектор, соста-
вленный из обратных значений начальных температур. Если при
этом сходимость будет медленная, то необходимо менять элементы
этого вектора в соответствии с приведенной выше схемой. Хорошие
результаты дает также использование вместо эквивалентных тем-
ператур среднеинтегральных температур по времени:
s
J т (г) dt
7’ср = ^---- (XI-81)
г о
443
Так как время решения системы линейных алгебраических урав
нений на ЭВМ невелико, то количество итераций при решении задачи
обработки экспериментальных данных не имеет существенного зна-
чения, поэтому единственное требование, предъявляемое к вектору М,
заключается в обеспечении сходимости.
Изложенный метод можно проиллюстрировать примером, в кото-
ром использовалась модельная реакция
2—>С3
Система дифференциальных уравнений, соответствующая этой
схеме, имеет вид:
1
dCi /,
T = -exp
Cl
dC2
exp
Д1
т
i —exp
Cj —exp (fcea—C2 (XI.82)
“dt =exP (fc03-
dCi । A-ffg
1F+~C7
dT __ ЫЦ
dt Cp
Здесь кН — тепловой эффект реакции.
Последний член уравнения теплового баланса учитывает внеш-
ний теплообмен; Твя — температура внешней среды.
Для построения математического эксперимента были выбраны
следующие начальные условия: при 1—0; С10 = 1; С20 = C3Q =
= С40 = 0. «Эксперимент» проводился при двух начальных темпе-
ратурах: Г01 = 453° К и Г02 = 473° К. Принятые «истинные» зна-
чения кинетических констант даны в табл. XL1. На расчетные зна-
чения концентраций и температур накладывалась случайная нор-
мально распределенная ошибка с о = 0,05.
Для сравнительной оценки точности определения констант по
данным, полученным в неизотермических и изотермических условиях,
описанным выше способом были получены изотермические «экспери- ;
Результаты обработки «экспериментальных» данных,
полученных в неизотермических (при начальном .
приближении ко=О и В = 0) и изотермических условиях
dC3 । ДЯ4
~dt~'~C^~
tZ*' p
Таблица XI.1
Стадия реакции «Истинные» значения кинетических констант Неиаотермические ' условия (получено расчетом) Изотермические " условия (получено расчетом)
fe0 В А. В А. в
1 19,605 10 000 22,953 11642 21,954 10 779
2 13,554 7 500 12,972 7 216 13,558 7 501
3 22,920 12 500 20,899 11493 22,957 12 517
444
ментальные» данные при температурах изотермического эксперимента
Т г = 453° К и Т\ = 473° К, т. е. примерно в равных сиеизотерми-
ческим экспериментом условиях.
Результаты обработки обеих серий «экспериментальных» данных,
представленные в табл. XI. 1, показывают, что в случае неизо-
термического эксперимента точность оценки кинетических кон-
стант в этом случае сравнима с точностью при обработке методом
наименьших квадратов изотермического эксперимента. Некоторые
различия между полученными и «истинными» значениями констант
(порядка 10—15%) могут быть объяснены тем, что при определении
коэффициентов системы (XI.71) численным интегрированием функ-
ции вида exp ~ 5^)) с использованием эксперименталь-
ных значений Q (t) имеют место большие погрешности, чем в случае
изотермического эксперимента.
XI.6. КОРРЕКТНОСТЬ ОБРАТНЫХ ЗАДАЧ
ХИМИЧЕСКОЙ КИНЕТИКИ
И ПЛАНИРОВАНИЕ ЭКСПЕРИМЕНТА
Как было показано выше, задача определения параметров кине-
тических моделей часто сводится к решению переопределенной си-
стемы линейных алгебраических уравнений (XI.15) методом наимень-
ших квадратов. Оценка искомого вектора х получается минимиза-
цией квадратичного функционала
F = (y-Ax)TW(y—Ax) - (XI.83)
которому соответствует квадратичная форма (однородный квадратич-
ный функционал):
Ф=хтАтАх (XI-84)
Эта оценка единственна, если только ранг матрицы АТА равен
числу искомых параметров, т. е. размерности вектора х. Квадратич-
ная форма Ф определяет характер поверхности F, а свободные члены
квадратичного функционала, содержащие вектор свободных членов
уравнения (ХГ;15), определяют лишь ее положение. Следовательно,
корректность постановки задачи, т. е. обусловленность системы
(XI.15), может быть полностью охарактеризована свойствами фор-
мы Ф, т. е. обусловленностью матрицы АТА.
Ковариационная матрица оценок (матрица моментов) имеет вид
= <ХЕ85)
где пг — число уравнений в системе (XI. 15); п — число определяе-
мых параметров.
Недиагональные элементы D (х) указывают на степень корреля-
ции между оценками параметров, а ее определитель является в из-
вестном смысле обобщенной характеристикой достоверности всех
445
оценок (объема, гиперэллипсоида доверительной области). Методы
планирования экспериментов, по-существу, сводятся к минимизации
этого определителя, либо других характеристик той же матрицы,
как, например, максимального собственного значения или следа.
На практике, однако, обусловленность матрицы АТА часто пло-
хая, что может привести к бессмысленным результатам при определе-
нии вектора х и даже ковариационной матрицы D (ж). Это обстоятель-
ство указывает на необходимость привлечения средств линейной
алгебры для предварительного анализа экспериментальных данных.
Неожиданные трудности, скрывающиеся в сравнительно неслож-
ных расчетах при обработке экспериментальных дацных химиче-
ской кинетики, ярко проявляются даже в простейшем случае ана-
лиза системы мономолекулярных реакций, которыми и ограничимся,
хотя предлагаемый нами подход применим и в более сложных задачах.
Может оказаться, что коэффициенты при неизвестных в системе
уравнений (XI. 15) не являются независимыми величинами и даже
очень большой экспериментальный материал не гарантирует доста-
. точной обусловленности матрицы А, т. е. четко выраженной линей-
ной независимости ее столбцов; в предельном случае ее ранг может
оказаться меньше числа оцениваемы? параметров, т. е. имеет место
избыточность принятой модели по отношению к имеющимся экспери-
ментальным данным. Последняя может быть объяснена либо недо-
статочностью экспериментальных данных, либо избыточностью при-
нятого механизма реакций относительно истинного.
На стадии обработки экспериментальных данных, следовательно,
прежде всего необходимо вычислить ранг матрицы А. Формально он
равен числу линейно независимых столбцов и при точных вычисле-
ниях обычно совпадает с рангом нормальной матрицы АТА. На са-
мом деле, однако, элементы АТА суть скалярные произведения типа
>т
2 aik аИ> т- е- являются суммами многих, резко различных по своей
i-i
величине чисел разных знаков, поэтому «линейная независимость»
ее столбцов может оказаться результатом ошибок округления, сле-
довательно, предпочтительно рассматривать непосредственно матри-
цу А. Для определения ранга прямоугольной матрицы наиболее
целесообразно пользоваться немодифицированным методом ортого-
нализации Шмидта [11]; при этом полным перебором из каждого
столбца А вычитаем его проекцию на все остальные столбцы, в ре-
зультате получаем группы векторов-столбцов, линейная комбина-
ция которых дает вектор с нормой, не превосходящей е ]/т (где е —
погрешность опыта), т. е. которые могут быть рассмотрены как ли-
нейно зависимые. Число оставшихся независимых столбцов назовем
8 — рангом матрицы А. Сам процесс состоит из п основных шагов,
каждый i-й из которых состоит из (п — i) шагов, /-й из которых за-
ключается в ортогонализации у-го столбца по отношению к предше-
ствующим (п — i — j) столбцам, начиная с (п — г)-го. Если в ре-
зультате получается вектор, незначимо (с точностью до 8) отличный
446
от нулевого, то одна линейная зависимость уже найдена, но процесс
должен быть продолжен для выявления остальных зависимостей;
Этот метод вычисления ранга дает важную информацию и о при-
чине вырожденности А; номера коррелированных столбцов (вместе
с соответствующими коэффициентами корреляции) указывают на
реакции, относительно которых экспериментальный материал недо-
статочно информативен. Эта информация позволяет либо спланиро-
вать дополнительные эксперименты, восполняющие указанные про-
белы, либо, если исчерпаны все возможности варьирования режим-
ных параметров (с учетом ограничений), принять менее избыточную
гипотезу о механизме реакции.
Поясним это более подробно. Допустим, что система кинетиче-
ских уравнений имеет вид
at
где К — кинетическая матрица размером N X N (здесь N — число
компонентов, участвующих в процессе).
Недиагональные элементы ktj матрицы К — константы скорости
реакций превращения j-ro вещества в г-е, а диагональные ее эле-
менты ки выражаются суммой:
N
ка — У, kji
i=i
i + i
В случае работы на установке интегрального типа начальный
вектор состава можно выразить как линейную комбинацию собствен-
N
пых векторов qt кинетической матрицы К в виде С (0) = 2a»ft-
i=i
Через некоторое время t такое разложение уже имеет вид С (t) =
n -л t
= ‘ ft, рДе К — соответствующие собственные значения ма-
/=1
трицы К, о которых известно, что все они вещественны и неположи-
тельны. Если в систему имеются быстрые стадии, либо некоторые
во всех рассматриваемых опытах малы, то ||а(е~х**д,-1| следо-
вательно, среди уравнений системы (XI. 15) может вовсе не быть п
линейно независимых в вышеуказанном смысле. В дополнительных
опытах следует стремиться к тому, чтобы высокие значения соответ-
ствующих аг компенсировали малость е-Л/*<7г. Упрощение модели
для устранения избыточности приводит к исчезновению соответству-
ющего аг или Хг.
Если же среди столбцов матрицы А не найдутся линейно зависи-
мые, то в первом приближении можно считать, что задача поста-
влена корректно, т. е. имеет единственное решение. Проведенный
анализ, однако, еще не свидетельствует о хорошей обусловленности
нормальной матрицы АТА, поэтому при оценке параметров жела-
тельно избежать получения последней в явном виде. Для этого можно
использовать следующий прием 111].
447
Матрица А разлагается на произведение ортонормированной
матрицы Q и верхней треугольной U. Тогда, принимая обозначение
z — Ux, имеем Qz = у, т. е.
z = QTy
(XI.86)
x = U~lQTz
Имея оценки для х (т. е. и для элементов матрицы К), можно проин-
тегрировать систему кинетических уравнений. В результате получаем
концентрационные кривые, которые сопоставляются с эксперимен-
тальными. Резкое несоответствие, заключающееся в существенном
различии априорно известной дисперсии ошибок измерений о2 от ее
выборочной оценки о2 = очевидно, указывает либо на крайнюю
недостаточность экспериментальных данных, либо на неадекватность
выбранной модели. Если о2 о2, проводим математический экспери-
мент; повторим вычисление ранга Л и оценки х на основании расчет-
ных кривых, т. е. подвергаем анализу систему, не завуалированную
случайными погрешностями опыта. Окончательную оценку достовер-
ности результатов получим в виде сопоставления двух наборов
искомых констант — элементов вектора х, соответствующих реаль-
ному и математическому экспериментам соответственно.
Изложенный метод оценки обусловленности системы предпола-
гает линейность либо возможность легкой линеаризации модели.
Если же линеаризация приводит к большим ошибкам, то предпочти-
тельнее для оценки параметров использовать поисковые методы
минимизации функции нескольких переменных. При этом в про-
цессе поиска получается обширная информация о поверхности кри-
терия оценки, которую можно использовать для непосредственного
вычисления матриц корреляции параметров. Так, в работе [12] пред-
лагается поисковый метод, основанный на вычислении коэффициен-
тов регрессии оцениваемых параметров. Покажем, как можно ис-
пользовать матрицу коэффициентов регрессии для нахождения кор-
реляционной и ковариационной матриц. Из матрицы коэффициентов
регрессии образуем матрицу вида
“ 1
— 021
_ — 0П 1
— 012 • • • — Р1П~
1 • • • — 02П
— Рл 2 " • • • 1 _
(XI.87)
которая несет достаточную информацию о поверхности критерия
оценки F вблизи минимума. Пусть ковариационная матрица (матрица
моментов) имеет вид:
D(x) =
Х12
Х12 • . • Мл
А.22 • • • Мл
(XI.88)
~ Mi
_ .... Хпп—
Сканировал: NeptUliyi
Магнитогорск
2008
448
Здесь
(о? при t^k
= { , . .
[COv(ZjZfc)
— коэффициент корреляции.
Известно [13], что,
о . A,fe
Лп
(XI.89)
при t =^= к
где Л с индексами — элементы матрицы Л — D-1 (х) — матрицы,
обратной матрице моментов.
Проделаем следующие преобразования:
которую назовем матрицей относительных ковариаций.
где D' с индексами — элементы матрицы D'.
29 Заказ 1441
449
Используя диагональные элементы матрицы 0 \ можно получить
сводный коэффициент корреляции
который является мерой корреляции между xt и остальными п — 1
параметрами.
Из матрицы (XI.87) получается коэффициент корреляции пара-
метров Xi и xk по отношению к параметрам Xj {j =£ к, i)
----77=== = I'iWlT (7 l, к) (XI.95)
1 V ^ii^kk
Отметим, что при n = 2 по определению
Ф12 е== гр (tj) ф (г2) S ф12/0 = /р12р21 (XI.96)
что можно легко проверить, проделав соответствующие преобразова-
ния над матрицей 0 размером 2x2.
Прежде чем перейти к изложению применения описанных выше
методов проверки обусловленности систем для планирования иссле-
дований кинетики, сделаем некоторые замечания о самой постановке
задачи планирования. Обычно эта задача формулируется как требо-
вание минимизации числа экспериментов для оценки параметров
с заданной точностью. Однако, на наш взгляд, такая постановка
задачи не совсем верна.
Во-первых, сложность большинства задач современной химиче-
ской кинетики приводит к их слабой обусловленности даже при
наилучшем выборе условий проведения экспериментов. «Экономия»
нескольких опытов только ухудшает и без того слабую обусловлен-
ность.
Во-вторых, «себестоимость» экспериментальной точки опре-
деляется в основном не суммарным количеством таких точек, а об-
щими затратами времени на эксперимент, слагающегося из времени
на пуск и освоение установки, отладку методик анализа, подготовку
сырья, профилактическое обслуживание установки, первичную обра-
ботку экспериментальных данных и т. д., а также затратами на
строительство установки. Поэтому традиционную постановку задачи
планирования экспериментов следует несколько изменить, а именно:
требуется найти такие условия проведения экспериментов при за-
данных ограничениях на варьируемые переменные, которые обеспе-
чили бы максимальную точность оценки констант модели.
Обобщая изложенное выше, можно следующим образом предста-
вить методику проведения исследований при разработке математи-
ческой модели сложного каталитического процесса.
На первом этапе исследований проводятся эксперименты при
произвольно выбранных условиях. 'Начальные температуры выби-
раются из условия максимальной надежности раздельной оценки
450
к
предэкспонентов и энергий активации, т. е. таким образом, чтобы
разность между наибольшей и наименьшей начальными температу-
рами была максимальной в пределах допустимой области варьиро-
вания входных температур. Ограничения по температурам могут
быть обусловлены, например, стабильностью катализатора или устой-
чивостью процесса (верхняя граница) и требованием достижения
за приемлемое время контактирования такой степени превращения
реагирующих компонентов, которая могла бы быть измерена с до-
статочно малой относительной погрешностью (нижняя граница).
Полученные экспериментальные данные используются для нахо-
ждения предварительных оценок параметров модели, которые ис-
пользуются для анализа обусловленности системы, определения кор-
реляционных зависимостей параметров и построения плана допол-
нительного эксперимента. С использованием найденных оценок
определяются расчетные значения концентраций компонентов, и на-
ходится матрица А. Отметим, что матрица А может быть построена
и на основании априорных значений параметров модели, если тако-
вые имеются. Так как точную оценку погрешности е найти трудно,
а известна только достаточно широкая область, в которой может
быть заключено ее значение, то следует определить е-ранг матрицы
(Q (е)) как целочисленную функцию от е в указанной области. Если
окажется, что при некотором е матрица А содержит попарно зависи-
мые с точностью до е столбцы, то зто означает, что имеются попарно
коррелированные между собой параметры. Если коэффициенты ли-
нейной зависимости соизмеримы друг с другом, то все параметры
коррелированы и не могут быть достаточно надежно оценены раз-
дельно. В первом случае необходимо изменить начальные концен-
трации тех компонентов, которые существенно входят в линейно за-
висимые с точностью до е столбцы; во втором — для надежной
оценки параметров желательно изменить начальные концентрации
всех компонентов. Наилучшие условия можно подобрать, максими-
зируя максимальную величину е, при которой еще сохраняется
D (е) — п.
Если для оценки параметров модели используется поисковый
метод, то для построения плана дополнительного эксперимента
удобно использовать анализ корреляционных зависимостей пара-
метров. При этом для уменьшения коэффициента корреляции ф№
следует изменить начальные условия тех компонентов, совместно
с которыми г-й и /с-й параметры входят в выражение скорости реак-
ции. Наилучший план дополнительного эксперимента находится
из условия одновременной минимизации максимального коэффи-
циента корреляции и максимальной величины отношения стг/стй.
Полученные при новых условиях проведения процесса экспери-
ментальные данные обрабатываются совместно с данными, получен-
ными в предварительных экспериментах, для нахождения уточнен-
ных оценок параметров. Последние используются для построения
плана следующего уточняющего эксперимента до тех пор, пока не
выполнится одно из следующих условий:
29*
451
a) e — ранг матрицы равен п при любом е из области его задания; -
б) за две последовательных итерации не получено значительного
изменения коэффициентов корреляции и величин Oi/ak;
в) принятая модель противоречит экспериментальным данным,
полученным при использовании новой матрицы планирования.
В последнем случае следует выдвинуть новую гипотезу о меха-
низме процесса.
ЛИТЕРАТУРА
1. Р, Schneider, М. Kraus, Chem. Eisty, 55, 1358 (1961).
2. В. Н. Писаренко, А. Г. П огоре лов, Планирование кине-
тических исследований, Изд. «Наука», 1969.
3. Д. Дж. У а й л ь д, Методы поиска экстремума, Изд. «Наука», 1967.
4. X. Розенброк, С. Стори, Вычислительные методы для инже-
неров-химиков, Изд. «Мир», 1968.
5. С. Л. К и п е р м а н, Введение в кинетику гетерогенных каталитиче-
ских реакций, Изд. «Наука», 1964.
6. Г. И. Лихтенштейн, Ю. М. С и в е р г и н, А. А. Б е р л и н,
\ Теорет. и эксперимент, хим., 1, 690 (1965).
7. Е. Ш. Фук с, Автореф. канд. дисс., ВНИИНефтехим, 1967—
8. В. М е z a k i, J. В. К i 11 г е 11, Ind. Eng. Chem., 59, 63 (1967).
9. С. H. 3 у x о в и ц к и й, Л. И. А в д е е в а, Линейное и выпуклое
программирование, Изд. «Наука», 1964.
10. Б. Ю. Г у р е в и ч, И. И. И о ф ф е, И. С. Фукс, ЖФХ, 45,^
№ 11, 1253 (1971).
11. Дж. X. Уилкинсон, Алгебраическая проблема собственных зна-
яений, Изд. «Наука», 1970.
12. А. П. Д о р о х о в, И. И. И о ф ф е, НТС ЦНИИТЭНефтехим «Неф-
тепереработка и нефтехимия», № 3, 1971.
13. Г. К о р н, Т. К о р н, Справочник по математике для научных ра-
ботников и инженеров, Изд. «Наука», 1968.
УКАЗАТЕЛЬ
Автоклав 415
бессальниковый 274, 415
с внутренним контуром циркуляции
Вишневского 415
Адсорбционный центр 23
Адсорбция
активированная 14
каталитических «ядов» 87, 147
равновесие 92
скорость 84
теория Рогинского 18—20
тепловой эффект 159
теплота 20, 24, 82, 83, 159, 160
физическая 14
химическая 14, 15
энтропийный аффект 159
яда 146
Активированный комплекс 10, 11
Активность
катализатора 28, 31, 147, 152, 158,
199, 263, 316, 399, 400, 418
Активные центры 12, 13, 160, 194
Алюмосиликаты 35, 39, 47, 177
Алюмотитанаты 35
Аниониты 35, 38
Асбест 187
Блокировка катализаторов 52
Вихри турбулентные 204, 216
Возмущение
скорость затухания 352
стационарного режима 336, 337
Вольфраматы 35
«Вулканообразные кривые» 31
Гептан 43 ..
Гидрирование каталитическое 42
Гидроформинг 272
Гистерезис 116
Глины 186, 201 '
Гранулятор тарельчатый 181
Графит 201
Динамическое программирование 370,
371, 381
Динас 187
Диффузия 140 сл.
в массе полимера 39
внутренняя 39, 263 1
в порах ионита 39, 98
в сплошной фазе 305
коэффициент 42, 99, 103, 122, 208,
211, 212, 225, 233, 260, 305, 314
молекулярная 99, 102, 103, 207,
211, 225
поверхностная 48
поперечная, коэффициент 219, 221,
290
продольная, коэффициент 219, 229,
234, 305
турбулентная 211
эффективный коэффициент 146, 211,
222, 223, 239, 289, 293, 302, 310,
. 360
Дихотомия 169
«Зажигание» реакции 117
Закоксовывание катализаторов 52,149
Закон
Аррениуса 256, 330, 425
Генри 16, 19, 81, 82, 312
Максвелла 99
Фика 98, 103
«Затухание» реакции 118
Зауглероживание катализатора 52
453
Избирательность катализатора
Изотерма
адсорбции 20, 24, 85, 86
адсорбции Лангмюра 16, 17, 19,
81, 92
Генри 17
Зельдовича — Рогинского 20
Фрейндлиха 20
Ингибиторы твердые 170
Индекс реакционности 163
Иониты 38, 39
Истираемость катализатора 418
Карборунд 187
Катализ
гетерогенный ионный 33, 38, 53
гомогенный ионный 33
кислотно-основной 34
на ионообменных смолах 38
на полупроводниках 26
«электронный» 13, 25
Катализатор(ы)
л-активирующие 155, 156
а-активирующие 155, 156
активная составляющая 200
активность 28, 31, 147, 158, 199,
263, 291, 316, 399, 400, 418
алюмосиликатные 36, 37, 178,
199
«армированные» 198
Бага 186, 197
гетерогенные
кислотно-основные 35, 152
комплексообразующие 152, 153
металлы 152, 157
подбор 151
производство 176
электронные полупроводники
152, 154
гидроформинга 183
гранулирование 181, 273
дегидрирование 183
дезактивация 268
железо-хромовые, производство
179, 180
закоксование 52, 149
зауглероживание 52
зернистый слой 203 сл.
истираемость 418
кобальтсодержащие 28
коллоидные 176
механическая прочность 200
модифицированные 44, 45
на носителях 45, 46, 176, 182,
184, 185
никелевый 185, 309
окисления аммиака 185
органические 176
454
Катализатор(ы)
осажденные 176, 179
отравление 12, 52, 140, 146, 315
первоначальный подбор 399
пларленые 176, 185
платиновый 47, 184
полупроводниковый 25, 26
пористый 108, 109
природные 176, 186
промотирование 12
промышленный 44, 151—171, 173
сл.
процессов крекинга нефти 186
регенерация 54, 293, 299
1?енея 185, 186
селективность (избирательность)
158, 399
скелетные 176, 185, 186, 197
сложные 44
смешанные 45
с падающей активностью 319
с переменной активностью 294
стабильность структура 199
старение 294
структура 173
поверхности 193, 194
пор 174, 175, 193, 195
таблетирование 181, 200, 201
термическая, дезактивация 200
утомление 294, 315
фазовая структура (фазовый со-
став) 50, 193, 194
химическая активность 151
хромсодержащие, производство 184
цеолитные 12, 39, 176
эффективность 188
Каталитические системы 46, 169—
171
Каталитическое гидрирование 42
Катиониты 38
Керамика 174
Кизельгур 35, 185, 187
Кинетика
лангмюровская 81
реакций в потоке 74
сложных реакций 87—96
Кипящий слой 198, 269—270, 310 сл.,
316
Кислота(ы)
апротонные 33
бренстедовские 34, 35
кремнефосфорная 185
льюисовские 34, 35
протонные 33
Кнудсеновская область 100, 101
Коагуляция в капле 177, 178, 198
Кольца Рашига ,198
л-Комплекс 153, 154
Комплекс активированный 10, И
«Комплексные соединения» 153
Корреляции линейные 158
Коррозия каталитическая 199
Корунд (а-окись алюминия) 187
Коэффициент
адсорбционный 16—18, 81, 87
асимметрии 224, 228, 229
вязкости
динамической 303, 305
кинематической 305
диффузии 42, 43, 122, 208, 211,
212, 233, 260, 314
в сплошной фазе 305
кнудсеновский 100, 101
молекулярной 99, 103, 225
поперечной 219, 221, 290
продольной 219, 228, 229, 234,
, 239—241, 305
турбулентной 211
эффективной 146, 222, 223, 239,
289, 293, 302, 310, 360
корреляции 449—452
массопередачи 103, 104, 106, 114,
249, 250, 260, 312
межфазного переноса 314
падения активности, эффективный
295, 296
поперечной теплопроводности 222,
290
продольной теплопроводности 222,
246, 247
регрессии 448
семиинвариантный 206
температуропроводности пористого
зерна 360
теплопередачи 104, 250
теплопереноса 260
теплопроводности 102, 105, 289
торможения реакции 286
эффективности ионита 38
Крекинг каталитический 149
Критерий
оптимальности 189—190, 365, 369,
370, 373, 383, 385, 388, 391,
397
Рауса — Гурвица 335
Ксерогели 174
«Маршруты реакций» 90
Массопередача
внешняя 102
в порах 98
к внешней поверхности 262
коэффициент 103, 104, 106, 114,
249, 250, 312, 315
Массоперенос коэффициент 260
Материальный баланс 275
реактора идеального смешения
275
реакции 130, 147
Матрица
кинетическая 71
констант скорости 70, 72
корреляционная 449
коэффициент регрессии 448
оценок, ковариационная (матрица;
моментов) 445, 448
стехиометрических коэффициентов
66, 67, 90, 91
Машины пропиточные 183
Металлы
механизм каталитических реакций,
22
Метод
аппроксимационного расчета кине-
тических параметров Гуревича
433
Белммана 370, 371, 381
вмазывания 181
дифференциально изотопный 18
дифференцирования эксперимен-
тальных концентрационных кри-
вых 425
кинетических исследований
динамический 401
дифференциальный 401
изотермический эксперимент
401
интегральный 401
неизотермический эксперимент-
403
проточно-циркуляционный 401
проточный 401
статический 401, 402
линеаризации кинетических урав-
нений Писаренко и Погорелова.
431
наименьших квадратов (МНК)
428—430
ортогонализации Шмидта 446
«пептизации» 196
Понтрягина 371
статистического прогнозирования
каталитических свойств 164
Мешалка
магнитная, возвратно-поступатель-
ная 416 ’
турбинная 415
Микроавтоклав 416
Моделирование математическое
каталитических, реакций 423 сл.
по неизотермическим эксперимен-
тальным данным 435—445
Модель
дискретная ячеистая 220, 221, 223,
227
диффузионная 207, 212, 219, 222,.
227, 230, 232, 233
«двойная» 101
химических реакторов 208
455.
Модель
Тернера — Ариса 220
ячеек идеального смешения 220
М одификаторы
структурные 45
фазовые 45, 51
Модуль Тиле 107, 109, 111, 122 —
124, 126, 130', 149, 358
Муллит 187
Насос стеклянный плунжерный, цир-
куляционный 410
Никель Ренея 186
Носитель гранулированный, пропит-
ка 182, 183
Носители для катализаторов 187
Область
внешнедиффузионная 112, 144
внутридиффузионная 112, 121, 128,
144
внутрифазнокинетическая 308
диффузионная 308
кинетическая 308
межфазнодиффузионная 308
Окисление 26, 27, 48
Окись
алюминия 47, 187, 196
циркония 187
Оператор Лапласа 121, 122
Определитель Якоби (якобиан) 386
Оптимальное проектирование реак-
торов 365 сл.
Оптимальные условия
процессов в реакторе 400
работы катализатора 400
Оптимальный температурный профиль
(ОТП) 365 сл., 380, 387
Оптимизация стадийных схем 380 сл.
«Открытая система» 303
Отравление катализаторов 52, 140,
146
Параметрическая чувствительность
реакторов 339
Пемза 187, 200
Перемешивание
гидродинамическое 218 -
поперечное 234
принудительное 204
продольное 234
Планирование эксперимента 445
П олупроводники
механизм каталитического действия
22
теория зонная 29.
П отенциа л окислите льн о-в осстанови-
тельный 28
Преобразование Лапласа 205, 210
Принцип
геометрического соответствия Ба-
ландина 32
линейных соотношений свободной
энергии (ЛССЭ) 158
Паулинга 152
энергетического соответствия Ба-
ландина 30, 158
Промотирование 44
Процессы жидкофазные 273, 274
Равновеси с адсорбционное 84, 85, 92,
94
Размер частиц 198
«Распознавание» катализатора 166
Расчет(ы)
квантово-химические 163
лабораторного изотермического ко-
лонного реактора гидрирования
додецена 309
реакторов
емкостных 313
идеального смешения 276
реакции изотермической 130
режима регенерации 300
Реактор(ы)
автотермические с внутренним теп-
лообменом 353—358
адиабатические 262, 264, 284 , 286,
390, 397
емкостные 264
с внешним теплообменом, устой-
чивость 344 сл.
безградиентный 409, 411, 412, 417,
424
газо-жидкофазный 418
двухфазная модель 291
дифференциальный 409
для газофазных процессов
с кипящим слоем катализатора
269, 273
с неподвижным слоем катали-
затора 264—268, 283
для окисления' нафталина Рей-
тера и Корнейчука 407
емкостные, расчет 313
жидкофазный лабораторный 414—
418
идеального вытеснения 391
выбор оптимального темпера-
турного профиля 387
дифференциальный 401
с однофазным потоком 282 сл.
идеального смешения 274, 334, 384,
389
материальный баланс 275
тепловой баланс 275
устойчивость 278, 324 сл.
456
Реактор(ы)
изотермический 254—256, 309
интегральные лабораторные 404—
408, 418 -
Кикотя 407
статический 404
Темкина — Кульковой 407
математическое моделирование 260
сл.
оптимальное проектирование 365 сл.
полочный 265, 266
промышленный, основные требова-
ния 261
проточно-циркуляционный 402, 409,
410, 412
с виброкипящим слоем 411
с внутренним теплообменом 287, 342
с движущимся катализатором лабо-
раторный 420
с движущимся слоем 318
с двухфазным потоком 301
с кипящим слоем 310 сл.
емкостные (несёкционирован-
ные) 311
модели 311
нециркулирующим 270
пилотный 421
секционированные 311
стеклянный 419
циркулирующим 271, 272
с неподвижным зернистым слоем
катализатора 213
с однофазным потоком 284
с радиальным распределением газа
265
с теплообменом в реакционной зоне
(внутренним теплообменом) 262
с теплоотводом из зоны реакции 267
трубчатые 267, 268, 336 сл.
лабораторный 407
с секционированным теплооб-
менником 397
цилиндрический, двумерная модель
290
Реакция(и)
акцепторные 25»
в потоке, кинетика 74
донорные 25
«зажигание» 117
«затухание» 118
изотермическая 121
изотермические, расчет 130 сл., 329
каталитические
в жидкой фазе 41
лабораторные исследования 399
сл.
математическое моделирование
423 сл.
механизм 22
на металлах 22
Реакция(и)
каталитические
на полупроводниковых катали-
заторах 26
определение скорости 61, 62
стационарные, теория Хориути —
Темкина 87
Фаворского 153
Фишера — Тропша 37
эволюционные 94
Режим(ы)
внешнедиффузионный 111, 112
внешнекинетический 111, 112
внутридиффузионный 109, 110, 112,
113, 128, 145, 147
внутрикинетический 109, НО
гидродинамический 212
идеального вытеснения 212, 213
идеального смешения 212, 213
неполного смешения 213
работы реактора изотермический 254,
255, 256
регенерации, расчет 300
стационарный 229, 248 сл., 344,
350
экзотермической реакции 248
Связь
«прочная» 23
«слабая» 23
Селективность
во виешнедифф у знойном режиме 145
во внутридиффузионном режиме 145
катализаторов 158
Силикагель 47, 177, 185, 187
Силикат циркония (циркон) 187
Системы "
каталитические 46
с сосредоточенными параметрами
325
Скорость
адсорбции 84
внутренней диффузии 263
потока в зернистом слое 215—218
псевдоожижения, критическая 313
реакции
во виепшедиффузнойной области
113 сл.
во внутридиффузионном режиме
128, 147
• гетерогенной 79
зависимость от скорости потока
112
зависимость от температуры 112
тепловыделения 67
Слой зернистый
гидродинамика 213 сл,
«живое сечение» 214
механизм теплопередачи 241
перенос тепла 222
457
Слой зернистый
продольное перемешивание 223
процессы переноса 213
скорость потока 215—218
с неправильной упаковкой 234
стационарные режимы процессов
248
структура упаковки 214
теплопроводность в стационарном
режиме 241
Смолы ионообменные 186
Спектр турбулентных пульсаций 216
Станнаты 35
Стекло жидкое 179, 181
Сульфаты 35
Сульфоуголь 186
Таблетирование порошков 179, 200,
201
Тальк 201
«Твердая фосфорная кислота» 35,
185
«Твердая ячейка» 241
Температурный оптимум 389, 394
Теория
адсорбции
Лангмюра 82
Рогинского 18
зонная полупроводников 29
мультиплетная Баландина 33
«пересыщения» Рогинского 25, 194
прецептрона 164
промежуточных ионов Гарднера 163
промежуточных соединений 12, 26,
30
распознавания 164, 165
стационарных реакций Хориути —
Темкина 87
столкновений 62
цепных процессов Боденштейна 88
Тепловой баланс реакции 115, 130,
147, 275
Тепловой эффект адсорбции 159
Тепловыделение скорость 67
Теплопередача
в зернистом слое 241
внешняя 102
в порах 98
коэффициент 104, 250
Теплоперенос, коэффициент 260
Теплопроводность
зернистого слоя 241
молекулярная, коэффициент 105
поперечная 222, 243, 290-
по твердой фаэе 241
продольная 245
Теплота адсорбции 20, 24, 82, 83,
159, 160
Термопара движущаяся 408
Торможение Д
внешнедиффузионное 113 |
реакций реагентами 106 |
Точки ветвления 358 |
1
Уголь активный 187 Л
У равнение /
Аррениуса 62, 86, 118, 129, 342, Ч
367, 377 ' 4
Бренстеда 28, 35, 36 |
Гаммета — Тафта 158, 160, 164 %
Диффузии конвективной 103, 211, i
239, 244 2
изменения активности катализатора
295
кинетические, интегрирование 63
Лангмюра 160
материального баланса 277, 300,
302, 312, 316, 334, 388
Поляни — Воеводского — Семенова
158
Поляни — Семенова 28
реактора идеального смешения
413
Рогинского 161
Тафта 160
теплового баланса 275, 277, 300,
334
Френкеля 43
Эргана 193
Уровень Ферми 23—25, 49
У стойчивость
реакции на пористом зерне катали-
затора 358—363
системы реактор — теплообменник
350
условия 360
Фактор
формы 109, 110
эффективности 109, 110, 111, 126,
127, 130
Фильтры 270
Форма -частиц 198
Формула
Кэлдербенка и Моо — Юнга 310
Эргана 290
Фосфаты 35
Функция
Бесселя 110, 237
Гамильтона (гамильтониан) 371 —
374, 377
кислотности Гаммета 36, 161, 162
макрораспределения 235
распределения
времени пребывания в реакторе
209
коэффициент асимметрии 228
Сканировал: NeptUliyi
Магнитогорск
2008
458
Хемосорбция 14, 15, 27, 154
кислорода 26
«прочная» 22
реагента 26
«слабая» 22
Число
Рейнольдса 104, 105, 216, 219,
222, 223, 225, 227, 229, 241, 290,
293
Шервуда 104
Шмидта 104, 310
Центр(ы)
адсорбционный 23
активные 12, 13
кислотные, бренстедовские 41
льюисовские 41
Цеолиты
(молекулярные сита) 174, 186
механизм действия 39
получение 186, 187
Циклогексан 43
Циклон 270
Число
Био 107
Нуссельта 104, 105, 250
Пекле 209, 211, 213, 219, 220, 228,
231, 240, 245, 283
Прандтля 104, 105, 222, 250, 293
Электрофильтры 270
«Элемент потока» 207, 212
Энергетический барьер 30, 31
Энергия активации 9, 10, 29, 31, 34,
81, 82, 83, 86, 113, 146, 250, 285, 367
Энтропийный эффект адсорбции 159
Энтропия активации 11
Эффект компенсационный 11, 12, 159
«Яды» каталитические 52—57, 86
адсорбционный коэффициент 87
адсорбция 87, 146, 147
скорость адсорбции 146
Ячейка
идеального смешения 210, 220
«надкритическая» 251
«подкритическая» 251
с застойными зонами 225
ОГЛАВЛЕНИЕ
Предисловие к 2-му изданию........................................ 3
Из предисловия к 1-му изданию.................................... 4
Введение ..........................................................5
ЧАСТЬ ПЕРВАЯ
ОСНОВЫ ТЕОРИЙ ГЕТЕРОГЕННОГО КАТАЛИЗА
Глава I. Теория каталитического действия.......................... 9
1.1. Общие представления о катализе........................ 9
1.2. Адсорбция ............................................ 14
1.3. Электронные представления в катализе.................. 21
1.4. Промежуточные соединения, энергетические и геометриче-
ские фактори я катализе................................... 30
1.5. Ионный катализ на поверхности......................... 33
1.6. Цеолитные катализаторы ............................... 39
1.7. Особенности гетерогенно-каталитических реакций в жидкой
фазе ..................................................... 41
1.8. Сложные катализаторы и каталитические системы . . . . 44
1.9. Взаимодействие катализатора и реакционной среды....... 49
1.10. Отравление катализаторов............................. 52
Литература ..................................'............. 57
Глава II. Кинетика гетерогенно-каталитических реакций ........... 61
II.1. Скорость реакции.........’..........'................ 61
11.2. Кинетика сложных реакций............................. 65
П.З. Кинетика реакций в потоке............................ 74
II.4. Особенности кинетики гетерогецно-каталитических реакций 79
II.5. Кинетика сложных гетерогенно-каталитических реакций .. 87
Литература ................................................ 97
Глава III. Макрокинетика каталитических реакций ......... 98
II 1.1. Законы тепло- и массопереноса..................... 98
III.2. Области протекания реакции . ..................... 106
III.3. Внешне диффузионное торможение и разогрев внешней по-
верхности катализатора........................_.......... 113
III.4. Внутридиффузионное торможение и внутренний разогрев
1 катализатора........................................... 121
II 1.5. Расчет экзотермической реакции с учетом внешнего и внут-
реннего тепло- и массопереноса........................... 130
III.6. Селективность сложных реакций и отравление катализа-
тора при диффузионном торможении процесса................ 140
Литература .............................................. 149
460
ЧАСТЬ ВТОРАЯ
ОСНОВЫ СИНТЕЗА ПРОМЫШЛЕННЫХ КАТАЛИЗАТОРОВ
Глава IV. Методы подбора химически активных катализаторов... 151
IV.1. Качественные принципы подбора катализаторов...... 152
IV.2. Количественные методы прогнозирования активности и се-
лективности катализаторов............................... 158
IV.3. Подбор каталитических систем..................... 169
Литерат ура ....................................... 171
Глава V. Основы технологии промышленных катализаторов....... 173
V.I. Характеристика н структура промышленных катализаторов 173
V.2. Краткие сведения о технологии производства катализаторов 176
V.3. Оценка эффективности и оптимизация промышленных катали-
заторов •............................................. 187
V-4. Методы управления структурой и формой катализаторов . . 193
Литература ............................................ 201
ЧАСТЬ ТРЕТЬЯ >
ОСНОВА РАЗРАБОТКИ И ОПТИМИЗАЦИИ
„ КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
Глава VI. Зернистый слой катализатора........................ 203
VI .1. Распределение времени пребывания в реактору..... 203
VI. 2. Гидродинамика и процессы переноса в зернистом слое . . . 213
VI.3 . Продольное перемешивание в зернистом слое....... 223
VI .4. Продольное и поперечное перемешивания в зернистом слое
с неправильной упаковкой................................ 234
V I.5. Теплопроводность зернистого слоя в стационарном режиме 241
VI .6. Стационарные режимы. процессов в зернистом слое .... 248
Литература ......................................". . . 257
Глава VII. Расчет реакторов по математической модели........ 260
VII .1. Принципы математического моделирования каталитических
реакторов ...................................... ...... 260
VII. 2. Промышленное осуществление гетерогенно-каталитических
реакций ............................................... 261
VII .3. Реакторы идеального смешения с . однофазным потоком
реагентов ................. . . ..................... 274
VI I.4. Реакторы идеального вытеснения с однофазным потоком 282
VII .5. Усложненные модели для реакторов с однофазным потоком 288
VI 1.6. Катализаторы с переменной активностью и регенерация
катализаторов.......................................... 294
VII.7 . Реакторы с двухфазным потоком (для жидкофазных реакций) 301
VII .8. Реакторы с кипящим слоем...................... 310
VI 1.9. Реакторы с движущимся слоем ....'.............. 318
Литера т у р а ...................................... 322
Глава VIII. Устойчивость каталитических реакторов............ 324
VII I.1. Устойчивость реакторов идеального смешения... 324
VIII .2. Параметрическая чувствительность трубчатых реакторов 336
VIII. 3. Стационарные режимы и устойчивость адиабатических
реакторов с внешним теплообменом...................... 344
VIII .4. Автотермические реакторы с внутренним теплообменом 353.,
VII I.5. Устойчивость реакции на пористом зерне катализатора 358
Литература............................................. 363
461
Глава IX. Оптимальное проектирование реакторов .... ...... 365
IX.1 . Оптимальный температурный профиль............. 365
IX.2. Оптимизация стадийных схем..................... 380
Литература .......................................... 398
ЧАСТЬ ЧЕТВЕРТАЯ
МЕТОДЫ ИССЛЕДОВАНИЯ КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Глава X. Лабораторные исследования гетерогенно-каталитических реак-
ций ............................................. 399
Х.1. Цели и методы лабораторных исследований . ........ 399
Х.2. Интегральные реакторы для исследования газофазных реак-
ций ................................................... 404
Х.З . Дифференциальные реакторы для исследования газофазных
реакций ............................................... 409
Х.4. Лабораторные реакторы для исследования жидкофазных
реакций ............................................... 414
Х.5. Лабораторные аппараты для исследования процессов в кипя-
щем и движущемся слоях катализатора.................... 418
Литература ........................................... 422
Глава XI. Методы построения математических моделей каталитических
реакций ..................................................... 423
XI.1. Принципы построения моделей....................... 423
XI.2. Определение параметров простых кинетических зависимостей 424
, XI .3. Определение параметров сложных кинетических зависимостей
* (линейные задачи)................................. 427
XI.4. Определение параметров сложных кинетических зависимо-
стей (нелинейные задачи) .............................. 430
XI.5. Построение математических моделей по данным неизотер-
мического эксперимента ................................. 435
XI.6. Корректность обратных задач химической кинетики и плани-
рование эксперимента ................................... 445
Литература ............................................. 452
Сканировал: Neptlinyi
Магнитогорск
2008
ИОСИФ ИСАЕВИЧ ИОФФЕ
ЛЕОНИД МИХАЙЛОВИЧ ПИСЬМЕН
ИНЖЕНЕРНАЯ ХИМИЯ
ГЕТЕРОГЕННОГО КАТАЛИЗА
Ленинградское отделение издательства «Химия»
Невский пр., 28
Редакторы: 3. И. Грива, Ю. М. Левин
Техн, редактор Ф. Т. Черкасская
Корректор Б. Н. Тамаркина
Переплет художника Л. А. Яценко
Сдано в набор 31/XII 1971 г. Подписано в печать 3/V 1972 г. М-12676.
Формат 60 X 90*/ie. Печ. л. 29. Уч.-изд. л. 30,87. Бумага № 2.
Тираж 4200 экз. Цена 3 р. 33 к. Заказ 1441.
Ленинградская типография № 6 Главполиграфпрома Комитета по печати
при Совете Министров СССР. Московский пр., 91.