/











































































































































































































































































Текст
Ответственный редактор Л. Н. Павлов. Техн, редактор Ф. Г. Закатов.
Сдана в набор 14/IV 1934 г. Подписана к печати 11/VI 1934 г.
Формат 62Х94>/М. ГХТИ № 519. Инд. Х-31-5-2. Бум. листов 16’/8. Тип. зн. в 1 бум. л. 106.848.
Уполн. Главлита № В—60033. Тираж 3.000—авт. л. 425/,. Заказ М 2611.
2-я тип. ОНТИ им. Евг. Соколовой. Ленинград, пр. Кр. Командиров, 29.
ПРЕДИСЛОВИЕ
Ошибочно думают те, которые пола-
гают, что от лаборатории до завода расстоя-
ние велико. Пример всего мира показывает
противное; всюду прогресс заводских дел
достигается ныне только при помощи людей,
воспитанных в лабораториях или их руко-
водивших. Все новейшие виды заводов суть
не что иное, как увеличенные лаборатории.
Д. И. Менделеев.
... производство обязано науке, ио наука
бесконечно большим обязана производству.
Ф. Энгельс.
В настоящей книге мы стремились дать систематически изло-
женный современный материал по теории и практике синтетиче-
ских превращений, используемых преимущественно в анилино-кра-
сочной промышленности.
Хвтор в 1925 г. опубликовал „Основы синтеза красителей", по-
лучившие признание как пособие для студентов-химиков втузов
и вузов. Приступая к работе над новым изданием, автор осознал
необходимость коренной переработки и расширения объема своей
книги.
„Основы синтеза красителей" составлялись в период 1921—1923 гг.,
в начале восстановительного периода советской промышленности.
Мы не располагали к тому времени сколько-нибудь солидным
опытом в производственном масштабе.
Лабораторное изучение синтеза уже дало некоторые, практи-
чески интересные результаты, но большая часть их оставалась
неосвоенной промышленностью, да и самый путь перехода от лабо-
ратории к производству не был еще достаточно отчетливо намечен.
Реконструктивный период и последующее затем грандиозное
развертывание социалистического строительства создали анилино-
красочную промышленность достаточно мощную для удовлетворе-
ния спроса текстильной промышленности, прочно стоящую на фун-
даменте независимого производства промежуточных продуктов,
освоившую оригинальные приемы работы и смело планирующую
Дальнейшее движение вперед по пути количественного роста и
качественных улучшений продукции.
Инженерно-технические работники красочной промышленности
приобрели за десятилетний промежуток значительный опыт по
з
методике производственной работы и ее рационализации, по пере-
ходу от лабораторных изысканий к проектированию и налажива-
нию новых производств.
Немалое число интересных для промышленности работ совет-
ских ученых и специалистов опубликованы в печати.
Последние четыре года журнал „Анилинокрасочная промышлен-
ность “ публикует большую часть таких работ.
За истекшее десятилетие в научно-технической литературе пере-
довых капиталистических стран Западной Европы и САСШ появи-
лось много интересного материала, частью вполне надежного по
своему значению (большинство научных работ), частью требующего
критического отношения (патенты и сообщения фирм). Этот мате-
риал, отражающий основные тенденции дальнейшего движения
если не производства, заторможенного кризисом, то исследова-
тельской мысли, понятно, также использован нами.
В меру возможности подробно мы пользовались литературой,
включая 1932 г. В процессе подготовки книги к печати мы вклю-
чали в нее показавшиеся нам интересными данные и более позднего
перйода.
Мы хотели в этой книге, освещая современное состояние про-
мышленности органического синтеза в области производных арома-
тического ряда, вывести некоторые общие принципы синтеза, дать
научное освещение установившимся методам промышленной работы,
указать, где это возможно, на перспективы дальнейшего прогресса
производства, подчеркивая границы достигнутого и вскрывая тре-
бующие дальнейшего научного освещения участки производствен-
ной технологии.
Книга планировалась прежде всего как учебное руководство
для студентов-специалистов втузов. Имея однако в виду выявив-
шуюся потребность инженерно-технических работников к система-
тизации новых достижений по основам интересующих их произ-
водств, автор считал для себя обязательным помочь и этому чита-
телю более детальной проработкой отдельных тем.
В тексте более мелким шрифтом отмечены места, которые
можно опустить при первом ознакомлении учащегося с книгой.
В отличие от первой нашей книги мы уделяем достаточно места
не только синтезу промежуточных продуктов, но и синтезу кра-
сителей.
Хотя многие главы книги написаны совершенно заново, но
общая схема, принятая в первом издании, была перенесена и сюда,
как оправдавшая себя с методологической стороны. Таким обра-
зом материал разделен на главы применительно к методам хими-
ческих превращений, но не к видам продуктов.
В отличие от первой нашей книги мы сильно сократили главу
о сырых материалах, учитывая наличие имеющихся на русском и
украинском языках самостоятельных руководств (Вейсгербер,
Орлов, Карпухин). В конце книги введена новая глава о катализе
и контактных реакциях, интерес к которым со стороны производства
все возрастает.
Изложение теории процессов автор стремился построить на
основе схемы о промежуточном образовании продуктов присоеди-
4
нения реагирующих молекул, как подтверждающейся многими фак-
тами и диалектически выдержанной. С этой же точки зрения раз-
вита и теория щелочного плавления сульфокислот.
В изложении технологических вопросов мы стремились, не оста-
навливаясь на частностях, дать главнейшие выводы и обобщения,
могущие быть полезными для понимания принципов производствен-
ной работы по синтезу главнейших промежуточных продуктов и
типов красителей. Учитывая все растущее значение красочной про-
мышленности, как организующей среди других производств органи-
ческого синтеза, мы отмечали попутно значение промежуточных
продуктов красочной промышленности и для многих других отраслей
народного хозяйства.
Кроме студентов мы надеемся заинтересовать книгой работни-
ков производства и исследователей.
Хотелось бы, чтобы первым она была полезна для более пол-
ного освоения процессов производства на научной основе и позна-
комила их с перспективами дальнейшего прогресса технологии.
Автор был бы в высшей степени удовлетворен, если бы исследо-
ватели, работающие для дальнейшего развития нашей красочной
промышленности, получили в результате его труда побуждение
к дальнейшим еще более плодотворным исканиям.
Сводки оригинальной литературы в конце глав помогут заинте-
ресованным книгой читателям более обстоятельно изучить отдельные
вопросы.
В красочной промышленности исследование и производство не-
разрывно связаны, и истекшее десятилетие дало многочисленные
примеры необходимости в дальнейшем культивировать этот тесный
контакт.
Как и в первой нашей книге, мы не стремились давать чисто
рецептурные данные и уделяли лишь строго необходимое внимание
аппаратурно-технологической стороне освещаемых тем.
Работа над книгой заняла много времени (почти два года). Кроме
опубликованных работ нами широко был использован опыт това-
рищей, входящих в коллектив научных руководителей -и сотрудни-
ков Научно-исследовательского института органических полупро-
дуктов и красителей им. К. Ворошилова, сотрудников Рубежан-
ского филиала этого института и заводов Анилобъединения. Работы
наши вместе с рядом сотрудников (по названному институту, Ива-
ново-Вознесенскому политехническому институту и Московскому
химико-техн, институту им. Д. И. Менделеева) помогли нам осве-
тить некоторые положения книги. Некоторая часть материала книги
излагалась нами на лекциях и беседах с инженерно-техническими
работниками заводов. Их вопросы и замечания учитывались при
составлении книги. Таким образом книга фиксирует труд боль-
шого коллектива научных работников и специалистов.
Автор считает приятным долгом выразить благодарность всем
товарищам, помогшим ему в его труде своими замечаниями и сове-
тами, и надеется, что они не откажут ему в указаниях на недо-
статки и упущения этого издания.
Кроме упоминаемых в тексте лиц в работе над книгой мне
много помогли Н. Н. Ворожцов — младший (в корректуре), С. М.
5
Карцев (в изготовлении большей части рисунков), В. В. Козлов
(составлением алфавитного указателя) и дирекция НИОПиК (пре-
доставлением для ознакомления неопубликованных материалов по
исследовательской работе).
Принося всем им на этом месте благодарность, я позволю себе
с особой признательностью отметить участие: редбазы Анилобъеди-
нения, помогшей мне в подготовке книги к изданию, Государствен-
ного химико-технического издательства, давшего книге достойное
внешнее оформление, и Л. Н. Павлова, без деятельной помощи
которого книга имела бы, при спешном ее издании, значительно
больше недостатков.
Н. Ворожцов.
ОГЛАВЛЕНИЕ
Стр.
Предисловие автора..................................................... 3
Сокращенные обозначения................................................. 10
Введение-................•...........•................................ 12
Исходные вещества для синтеза красителей........................... —
Литературные источники •............• . . . 21
Глава I. Общие понятия о превращениях исходных веществ в промежуточные
продукты.............................................................. 22
а) Три основных группы превращений . . . . •................ —
б) Замещения и ориентация заместителей..................... 24
в) Теории ориентирующих влияний при замещениях............. 35
Литературные источники........................................... 42
Глава II. Нитрование н ннтрознрованне................................. 43
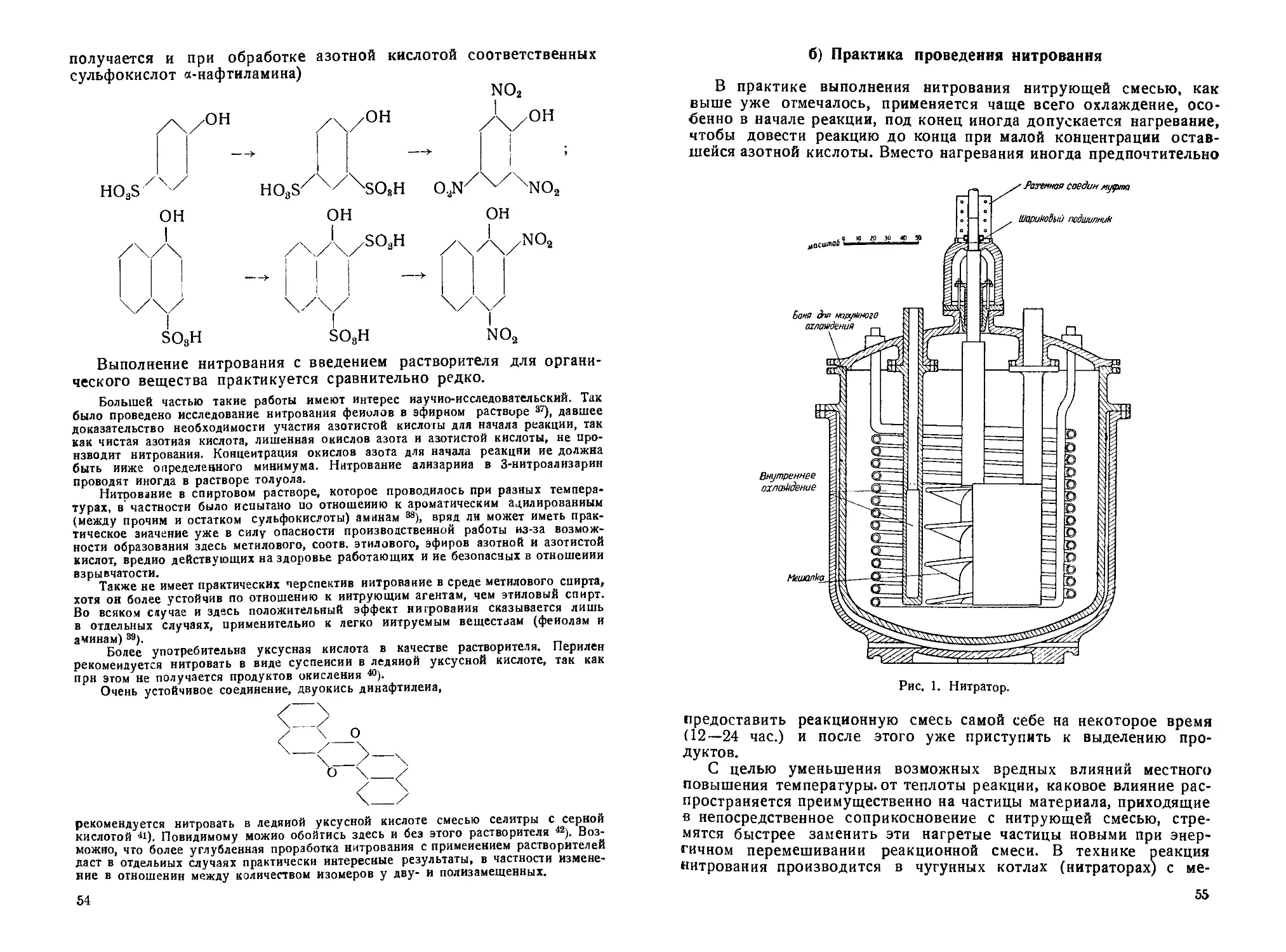
а) Течение, реагенты, факторы ннтроваиия.................... —
б) Практика проведения нитрования.........•................ 55
в) Перспективы дальнейшего прогресса в нитровании.......... 57
г) Выделение и характеристика продуктов нитрования......... 59
д) Ннтрознрованне.......................................... 61
Литературные источники........................................... 69
Глава III. Сульфирование.............................................. 72
а) Течение и факторы сульфирования.............. . . . . —
б) Практика проведения сульфирования........... • , . . . 79
в) Характеристика продуктов сульфирования.................. 87
Литературные источники........................................... 96
Глава IV. Введение галоида (хлора, брома н т. п.)..................... 98
а) Течение и факторы хлорирования бензола................. 101
б) Хлорирование нафталина ................................ 110
в) Хлорирование толуола и других веществ.................. 113
Роданирование .................................................. 120
Литературные источники.....................• •..........• ... 123
Глава V. Образование аминогруппы восстановлением нитрогруппы и других
азотсодержащих групп............................................... 126
а) Восстановление в кислотной среде.......•............... 128
6) Восстановление в нейтральной среде.................... 137
в) Электрохимический метод восстаиовлеиия в кислотной среде . 139
г) Восстановление в щелочной среде (бензидиновая перегруп-
пировка гндразосоединеиий)................................ 140
д) Частичное восстановление полинитросоединеннй........... 146
е) Контактное восстановление водородом . . . . •.......... 153
ж) Характеристика аминосоединеиий........•............... 155
Литературные источники......................................... 162
Глава VI. Превращение сульфогруппы в гидроксильную группу методом
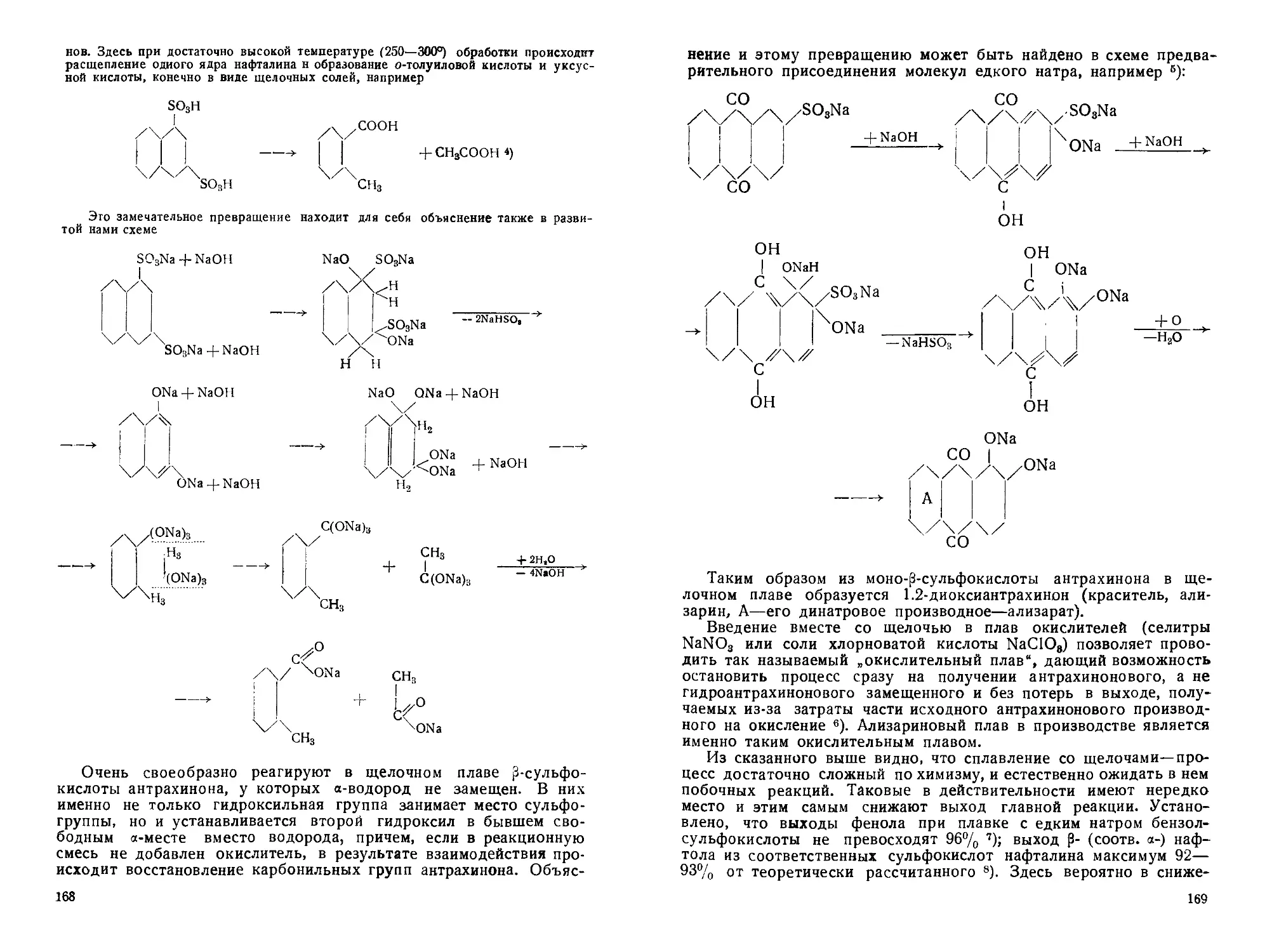
щелочного плавления. Другие превращения сульфогруппы .... 165
а) Характер реакций, протекающих в щелочном плаве сульфо-
кислот. Плав в открытом котле . ......................... 166
б) Автоклавные плавы. Известковые плавы под давлением . . . 172
в) Производственная практика плавления в синтезах фенола,
р-иафтола, резорцина и других оксизамещениых............... 176
г) Характеристика окснсоединеннй . •...................... 182
д) Иные кроме плава методы образования эксигруппы и превра-
щения сульфогруппы........................•................ 188
Литературные источники.......................................... 196
7
Стр.
Глава VII. Обмен галоида (хлора) на другие заместители................ 198
а) Обмен хлора на азотсодержащие группы •................. 201
б) Обмен хлора на кислородсодержащие группы............... 211
в) Обмен хлора на серусодержащие.группы- и на другие заме-
стители ................................................... 227
Литературные источники . ;...................•.................. 231
Глава VIII. Превращение аминосоединений в оксисоединения и обратные
переходы.............................................................. 235
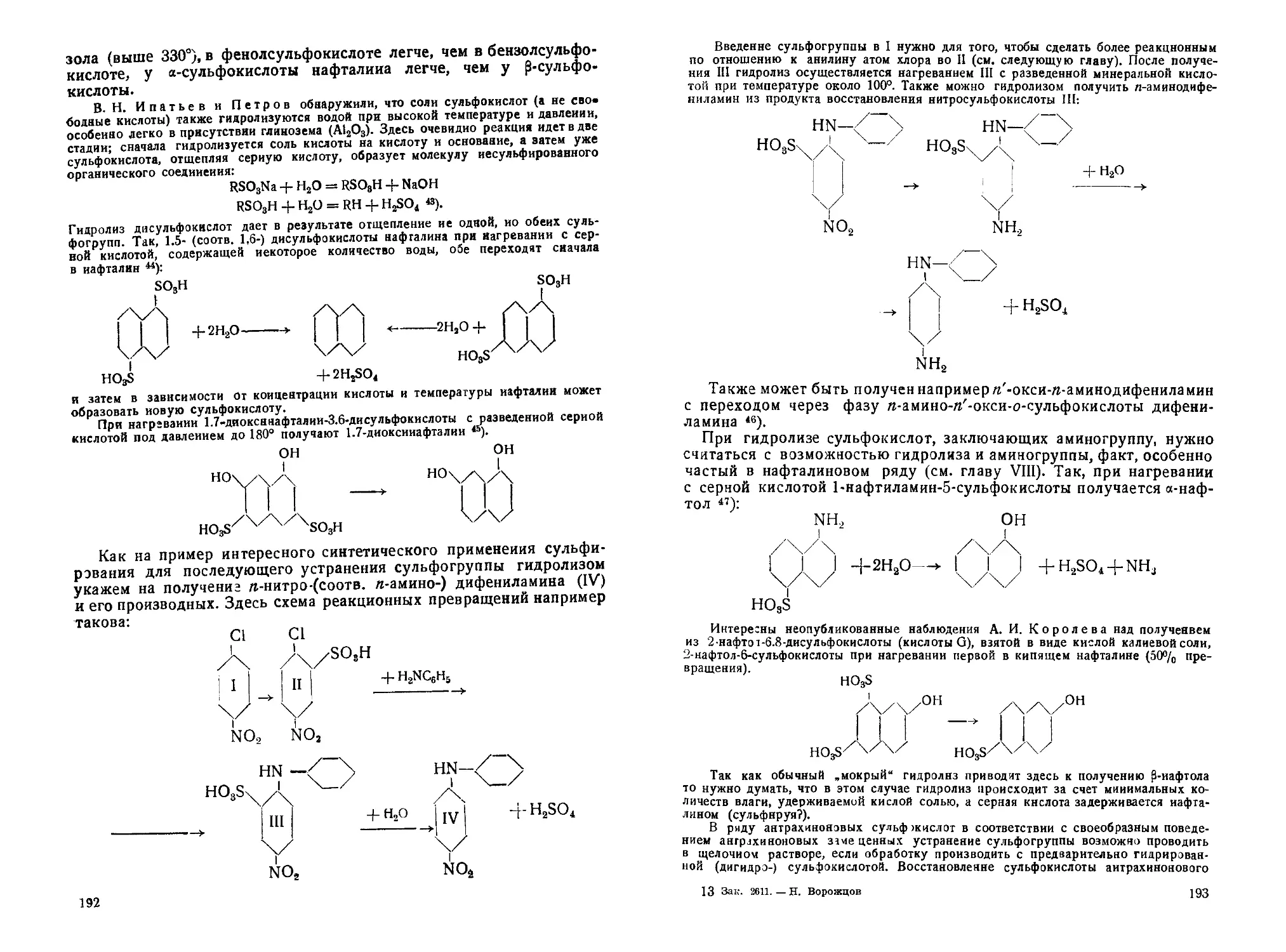
а) Гидролиз аминов с переходом в оксизамещеииые........... 236
б) Аминирование оксисоединений........................... 242
Литературные источники.......................................... 248
Глава IX. Диазотирование и превращения диазогруппы................... 249
а) Методы получения и свойства диазосоединений ............. —
б) Стойкие диазосоедииення . . . •........................ 259
в) Азосочетание........................................... 262
г) Иные, кроме азосочетаиия, превращения диазосоединений . . 269
Литературные источники...............•.......................... 277
Глава X. Араминирование. Образование ариламиновой группы.........• . 280
а) Араминирование в рядах бензола, нафталина, антрахинона . —
б) Характеристика вторичных ароматических аминов.......... 290
Литературные источники.......................................... 292
Глава XI. Алкилирование ............................... • • . . . 293
а) Алкилирование в аминогруппе.......................... —
б) Алкилирование в оксигруппе...................• • ... 311
Литературные источники.......................................... 317
Глава XII. Ацилирование (защнта'реакционной группы).................. 319
а) Ацилирование аминов.................................. —
б) Иные, кроме ацилирования, методы защиты аминогруппы.
Образование азометиновых соединений....................... 334
в) Ацилирование гидроксильных замещенных................... 335
Литературные источники................................• ... 341
Глава XIII. Окисление и осернение...................................... 343
Окисление......................................................... —
а) Образование альдегидной группы окислением............... 344
б) Образование карбоксильной группы окислением............. 352
в) Окисление группы СН2 (соотв. СИ) в карбинольную группу . 356
г) Замещение ароматически связанного водорода гидроксилом . . 358
д) Превращение СН->>СО.................................... 362
е) Превращения R (Rz) СН2-> R(R') СО............• ... 369
ж) Окислительные превращения ароматических соединений
в хиноиды. Интермолекулярные окисления.............• . . —
з) Окисления с изменением углеродного скелета соединений . . 372
и) Окисление не содержащих углерода групп........• ... 378
Осернение.............................................• . . . . 381
Литературные источники.............•............................ 391
Глава XIV. Восстановление соединений с характерными, не заключающими
азота группами . ... ; ........................................ 395
а) Восстановление карбонильных соединений................... —
б) Иные примеры восстановительных реакций ................ 406
Литературные источники...........•.................•..........
Глава XV. Конденсации и перегруппировки............................... 404
А. Конденсации без образования новых циклов.................... 406
а) Реакции с потерей воды (— Н2О)........................... —
б) Конденсации с потерей водорода (дегидрогенизационные
окисления)-............................................... 414
в) Конденсации с отщеплением кислорода (восстановительные
конденсации) . . . . •..................•................. 4.17
г) Реакции с потерей галоидоводорода (НС1) ................. —
д) Реакции отнятия галоида................................ 429
е) Реакции отщепления серы, металла....................... 436
8
Стр.
Б. Конденсации с образованием новых циклов................... . 430
а) Реакции с потерей воды.........................• . . . . —
б) Реакции с отщеплением СО2 и Н2О........................ 440
в) Реакции с потерей хлористого водорода................. 441
г) Реакции с потерей воды и водорода (с окислением)....... 443
д) Реакции с отщеплением С2Н5ОН........................... 449
е) Реакции с потерей аммиака.............................. 450
В. Перегруппировки................................................ 454
Литературные источники............................................. 462
Глава XVI. Катализ и контактные реакции................................. 466
А. Общие определения и теоретические положения..................... —
Б. Катализ в гомогенной системе.................................. 470
В. Катализ в гетерогенной системе............•.................. 480
а) Общие сведения......................; . . ........... —
б) Гидрогенизация, особенно нитросоединений............... 488
в) Дегидрогенизация ...................................... 498
г) Окисление.............................................. 503'
д) Прочие реакции...........................•............. 527
Литературные источники........................................... 530
Предметный указатель............................................. 534
СОКРАЩЕНИЯ
t°njI —Температура плавления.
t°KHn— Температура кипения.
t°3acT. — Температура застывания.
уд. в. — Удельный вес.
S (в формулах иа стр. 94, 95, 160, 161, 195 и 232) обозначает сульфо-
группу SO3H.
А. А. — Aktien-Gesellschaft fiir Anilin-Fabrikation in Berlin.
Ав. n. — Австрийский патент.
Am. n.—Американский патент (США).
Аи. Кр. Пр.—Анилино-Красочная Промышленность (журнал).
Am. Soc. — Journal of the American Chemical Society.
Ann. — Liebig's Annalen der Chemie.
Ann. Ch.-Phys. — Annales de Chimie et de Physique.
В. Б. B. — Badische Anilin und Soda Fabrik in Ludwigshafen a/Rhein.
Bas. — Baseler Chemische Fabrik.
Ber. — Berichte der deutschen chemischen Gesellschaft.
B. D. Co. — British Dyestuff Corporation.
Б. n. — Английский (британский) патент.
Br. A. — British Abstracts.
С. C. — Leopold Cassella und Co. in Frankfurt a/Main.
C. A. — Chemical Abstracts (американский).
Calco — Calco Chemical Company, New Jersey.
CB. — Chemisches Zentralblatt.
Ch. Ztg. — Chemiker Zeitung.
Chem. Met. Eng. — Chemical and Metallurgical Engineering.
Ch. T. Ob. — Chemisch-Technische Obersicht (bei Chemiker-Zeitung).
Ch. Tr. J. — Chemical Trade Journal and Chemical Engineer.
Chim. & Ind. — Chimie et Industrie.
CIBa — Gesellschaff fiir chemische Industrie in Basel.
Cly — Clayton Aniline Co. Ltd.
C. N. M. C. — Compagnie Nationale des Matieres Colorantes et Manu-
facture de Produits Chimiques du Nord Reunies.
C. r. — Comptes rendus de 1’Academie des Sciences,
D. Д. Dingl. Polyt. J. — Dinglers Polytechnisches Journal.
Dow. — Dow Chemical Co., Midland.
DuP. — E. J. Dupont de Nemours et. Co., Delaware.
D. H. — Durand et Huguenin, Soc. Anon., Basel.
F. Ф. F. — Friedlander, Fortschritte der Teerfarbeufabrikation und verwandter
Industrie-Zweige.
Ф. n. — Французский патент (brevet).
G. Г. G. — Joh. Rud. Geigy und Co. in Basel.
Г. и. — Германский патент (D. R. P.).
Gaz. — Gazzetta Chimica Italiana.
Gr. — Chemikalienwerke Griesheim.
Gr. E. — Chemische Fabrik, Griesheim-Elektron.
G. An. W. — General Aniline Works.
Grasselli — Grasselli Dyestuff Corporation, New York.
Griinau — Chemische Fabrik, Griinau; Landshoff und Meyer in Griinau.
H. Helv. — Helvetica Chimica Acta.
Heyden — Chemische Fabrik von Heyden.
10
I. И. IG. — I. G. Farben-Industrie Aktien-Gesellschaft.
ICI. — Imperial Chemical Industries.
Ind. Eng. Ch. — Industrial and Engineering Chemistry.
Изв. И. В. П. И. — Известия Иваново-Вознесенского Политехнического
Института.
J. Ж. Ж. — Журнал Русского Физико-Химического Общества.
Ж. О. X. — Журнал Общей Химии (Химический Журнал сер. А).
Ж. П. X. — Журнал Прикладной Химии.
Ж. X. Пр. — Журнал Химической Промышленности.
Ж. Фарм. И.— Журнал Научного Химико-Фармацевтического И-та ВСНХ.
J. pr. Ch. — Journal fiir praktische Chemie.
J. Soc. D. and Col. — Journal of the Society of Dyers and Colourists.
3. Заявл. Г. п. — Заявление о германской привиллегии (Patent-Anmeldung).
Носят впереди номера инициалы имени заявившего.
К. К. К.—Kalle und Со. in Biebrich a/Rhein.
L. Л. L. — Leonhardt und Co., Fabrik Miilheim.
Lange, S. F. — 011 о Lange, Die Schwefelfarbstoffe, ihre Herstellun^ und
Verwendung, Leipzig 1912.
M. M. M. L. B. — Farbwerke vorm. Meister, Lucius und Briining in Hochst
a/Main.
Mon. — Monatshefte fiir Chemie.
Mon. Scient. — Moniteur Scientifique.
N. H. Nat. An. — National Aniline and Chemical Co.
Newport — Newport Chemical Co.
О. О. O. — K. Oehler, Anilin und Anilinfarbenfabriken in Ofienbach a/Main.
P. П. Pr. Ch. — Fabrique de Produits de chimie organique de Laire a Issy.
R. P. Rec. — Recueil de Travaux Chimiques des Pays-Bas et de la Belgique.
R. G. M. C. — Revue Generale des Matieres Colorantes.
S. С. Ш. Sc. D. — Scottish Dyes Ltd.
Soc.—Journal of the Chemical Society (London).
Soc. Ind. — Journal of the Society of Chemical Industry (Chemistry and
Industry).
St.-Denis — Societe Anonyme des Matieres Colorantes et Produits Chimi-
ques de St.-Denis (Seine).
Cob. п, —Советский патент (авторское свидетельство).
Шв. п. — Швейцарский патент.
Т. t. М. — Chemische Fabriken vorm. Weiler-ter-Meer in Uerdingen a/Rhein.
T. M.— Fabriques des produits chimiques de Thanne et de Mulhouse (Al-
sace).
U. У. Us. Rh. — Societe chimique des Usines du Rhdne.
У. X. Ж. — Украинский Химический Журнал.
V. Ver. Aussig — Verein fiir chemische und metallurgische Produktion in
Aussig (Tschecho-Slovakei).
W. W. — R. Wedekind und Co., G. m. b. H. in Uerdingen a/Rhein.
Wii. — Anilinfabrik, A. Wiilfing.
Z. Zt. angew. — Zeitschrift fiir angewandte Chemie.
Zt. Ch. — Zeitschrift fiir Chemie.
Zt. F. Ch. — Zeitschrift fur Farben- und Textil-Chemie.
Zt. phys. Ch. — Zeitschrift fur physikalische Chemie.
ВВЕДЕНИЕ
ИСХОДНЫЕ ВЕЩЕСТВА ДЛЯ СИНТЕЗА КРАСИТЕЛЕЙ
Синтез красителей, ведется ли он в малых размерах в лаборато-
рии или производится в больших заводских установках, во всех
случаях может быть разделен на отдельные процессы химической
переработки менее сложных химических веществ в более сложные.
Наиболее просто и обще можно представить течение этого синтеза
схемой: сначала превращение органического сырья в сложные со-
единения, не имеющие характера красителей (такие соединения мы
называем промежуточными продуктами), затем превращение про-
межуточных продуктов в красители. Практика организации про-
изводства красителей научила уже давно, что первая стадия —
получение из сырых материалов промежуточных продуктов — явля-
ется вообще значительно более сложной и трудной, чем вторая —
получение красителей из промежуточных продуктов, что поэтому
крайне важно на эту первую стадию обратить особое внимание и
подвергнуть ее специальному изучению.
В этой книге будет представлен систематически материал, касаю-
щийся преимущественно методики проведения синтеза промежуточ-
ных продуктов.
Сырье (исходные материалы) для этого синтеза доставляется
почти исключительно промышленностью, перерабатывающей камен-
ный уголь на кокс с улавливанием газообразных продуктов (коксо-
бензольной промышленностью), и отчасти нефтеперерабатывающей
промышленностью. При изобилии отдельных индивидуальных со-
единений, заключающихся в газообразных и жидких отходах этих
видов промышленности (например в каменноугольной смоле), срав-
нительно небольшая часть интересна для красочной промышлен-
ности в качестве исходных (и иногда вспомогательных) материалов
для синтеза. Эти интересные вещества принадлежат почти исключи-
тельно к соединениям ароматического ряда. Часть этих продуктов —
более легко кипящие углеводороды („сырой бензол")—извлекается
из коксового газа промывным маслом и от этого растворителя
отделяется перегонкой. Другие продукты содержатся в смоле от
коксования и путем первичной разгонки ее собираются в отдель-
ных фракциях. Из последних они выделяются или новой дестилля-
цией или фильтрованием, если выпадают в твердом виде (нафта-
лин, антрацен). Очистка ведется химическим путем (промывка
серной кислотой, иногда раствором щелочи, промывка раствори-
телями) и повторными ректификациями.
Методы их улавливания, извлечения, разделения, очистки и
ректификации не являются предметом нашего-рассмотрения и ра-
12
зобраны в специальных руководствах по переработке смолы. Мы
в дальнейшем только назовем эти исходные материалы и дадим
их краткую характеристику.
Бензол С6Н6, | | , жидкий при обычной температуре углево-
дород, первичный член ароматического ряда соединений; t°K(fn 80,2°;
Лл. 5,5801). Уд. в. (15/4°) 0,87868, при 0° 0,89408, при 50° 0,84539. Тех-
нический бензол заключает всегда некоторое количество посторон-
них веществ, что должно быть учитываемо при его применении.
Наиболее часто встречаемые примеси бензола: сероуглерод CS2,
тиофен C4H4S и неароматические (несульфируемые) углеводороды.
Требования, предъявляемые к чистому каменноугольному бензолу по советско-
му стандарту (ОСТ 463): уд. в. (15°) 0,880—0,885; кипение в пределах 78,5—80,6°.
В пределах 1° должно перегоняться не меиее 95% по объему. Предельное содер-
жание примесей определяется по поглощению брома и по окраске от серной
кислоты.
Бензол находит широкое применение для получения нитросое-
динеиий, сульфокислот и хлоропродуктов.
Толуол С7Н8, С6Н5 • СН3, | |С^3, ближайший гомолог бен-
зола. t°KIin 110,8°, уд. в. (15,2°/4) 0,8702. Примесями технического
толуола могут быть, так же как у бензола, сернистые соединения
(тиотолен, C4H3S • СН3) и жирные углеводороды.
Стандарт (ОСТ 464) ставит требования: уд. в. (15°) 0,870 ± 0,002. Пределы ки-
пения 109—111°. В пределах 1° перегон не меиее 95% по объему. Химические
пробы — как у бензола.
Применяется толуол преимущественно для получения нитропродуктов, затем
для хлоропродуктов и меньше для сульфирования.
Ксилолы, диметилбензолы С6Н4(СН3).2, доставляются промыш-
ленностью в виде смеси трех изомеров орто, пара и. мета с пре-
обладанием мета (60—70% от смеси). Их смесь, в которую
входит немного этилбензола, применяется как технический ксилол.
По стандарту (ОСТ 465) чистый каменноугольный ксилол имеет уд. в. (15°)
0,867 zt 0,002. Пределы кипения 136,5—141,5°. В пределах 4,5° должно перегоняться
не менее 95% по объему.
Температура кипения о-ксилола 144,07°, уд. в.(15°/15) 0,88514; t°KHn п-ксилола
138,23°; t°3acT 13,1°, 1°пл 15—16°, уд. в. (15°/15) 0,8661; t°KIfn .«-ксилола 138,8°; уд. в
(15°/15) 0,8691. Этилбензол С6Н5С2Н5, 1°кип 136°, уд. в. 0,870.
Разделение изомеров можно производить, пользуясь различным
отношением их к серной кислоте. Метаксилол легче других изо-
меров сульфируется (уже при 80°), и сульфокислота его легче
гидролизуется, чем сульфокислоты изомеров (схема CgHgSOjH-f-
+ H2O^C8H10 + H2SO4) 2).
Ввиду трудности получения из технической смеси й-ксилола, имеются предло-
жения синтетического получения его из толуола 3).
Наличие трех изомеров в техническом ксилоле, из которых
каждый в:едет себя различно при химических взаимодействиях,
делает переработку ксилола более затруднительной сравнительно
13
с его низшими гомологами. Ксилол имеет значение почти исклю-
чительно для получения нитропродуктов.
Высшие гомологи бензольных углеводородов не находят при-
менения в виде индивидуальных веществ. Смеси таких соединений
применяются в некоторых областях химической промышленности
в качестве растворителей (например сольвент-нафта с t° , 120—160°
или 160-170°).
Нафталин С10Н8, | | твердый при обычных условиях угле-
водород с двумя конденсированными шестичленными кольцами
в молекуле. Температура плавления 80,1°4), 80,23°5), tKII0 217,96°4), уд.
в. (15°) 1,1517, (79,9°) 0,9777. Технический нафталин заключает всегда
некоторые примеси, присутствие которых изменяет указанные
константы.
По стандарту (ОСТ 277) нафталин кристаллический каменноугольный выпуска-
ется трех сортов: I сорт имеет t°3aCT не ниже 79,6°, II сорт—не ниже 79,3°,
Ill сорт не ниже 79°. При стоянии в течение 1/2 часа над азотной кислотой в эк-
сикаторе I сорт не желтеет, II сорт может дать едва заметное пожелтение. При
нагревании с серной кислотой у I и II сортов получается слабая окраска, сравни-
ваемая по цвету и интенсивности с раствором 0,001 г КМпО4 или 0,01 г КМпО4 +
0,15 г К2Сг2О7 в 1 л. Золы должно быть не более 0,050/0.
Нафталин применяется преимущественно для получения нитро-
продуктов и сульфокислот и отчасти для гидрирования.
Гомологи нафталина не находят себе технического применения.
Н2С —СН2
Аценафтен С12Н10
’ ^пл 95°, t°Kiin 278°, имеет в на-
стоящее время значение для получения так называемого аценафтен-
ОС—СО
хинона
Антрацен С14Н10
, трициклический углеводород
со своеобразной реакционностью водородных атомов во втором
(среднем) кольце, делающей вероятным хиноидную структуру этого
кольца (или допущение прямой углеродной парасвязи в нем).
Получается в технике из антраценового масла каменноугольной
смолы фильтрованием, прессованием и различными химическими
обработками и промывками, удаляющими примеси. Чистый антра-
цен имеет t° 216,1°; t° 339,9°5), 351°; уд. в. 1,242.
ил» < у* - * * -
Технический антрацен с содержанием чистого от 45 — 50%,
до 80 и 95% применяется почти исключительно для окисления
в антрахинон. Примесями технического антрацена являются глав-
ным образом фенантрен, карбазол и метилантрацен наряду с дру-
гими углеводородными и азотсодержащими соединениями. Хими-
ческая оценка технического антрацена имеет назначением исключи-
тельно определить содержание в нем чистого антрацена и произ-
водится с теми или иными видоизменениями по методу Люка:
окислением хромовой кислотой в растворе уксусной кислоты. При
этом антрацен окисляется в стойкий в условиях реакции антрахи-
нон, и примеси переходят или в углекислоту или в легко сульфиру-
емые соединения. По количеству полученного антрахинона судят
о содержании в техническом материале чистого антрацена6).
Фенантрен С14Н10, / \__/ \, изомерный антрацену углево-
дород (t% 100,5°, tKHn 340°; уд. в. 1,182), являющийся его спутни-
ком в сыром антрацене, в отличие от последнего не нашел себе
сколько-нибудь значительного применения в красочной промышлен-
ности. Так как некоторые из производных легко получаемого
фенантренхинона обладают высокими качествами как красители 7),
то вполне возможно, что будут найдены способы технического
использования фенантрена для получения прочных красителей.
Сульфокислоты, оксипроизводные и их замещенные для фенан-
трена во многом похожи на нафталиновые замещенные, поэтому
не исключена возможность их использования по тем путям, кото-
рые характерны для производных нафталина 8).
Другие углеводороды с конденсированными циклами из каменноугольной смолыг
флуорен С13Н10,
пирен
, хризен С18Н12,
СН2
/ \ / \, не имеют пока значения в качестве исходных материалов для
синтеза
Из
Щихся
фенол
красителей, хотя возможно, что окажутся для него интересными.
кислородных соединений ароматического характера, являю-
составными частями каменноугольной смолы, наиболее важны
и его ближайшие гомологи,
/\ /ОН
крезолы.
Фенол С6Н6ОН,
> С.
42,25—43°;
С. 40,9°, уд. в. (21°)
1,0598, t^n 81,2°. Во вполне чистом
состоянии
бесцветен, краснеет
при хранении, особенно на свету. При прибавлении небольших
количеств воды растворяет ее, переходя из кристаллического
в жидкое состояние. 100 г фенола растворяют при 15° 37,4 г воды;
15
100 г воды при 15° растворяют 8,2 г фенола; при 65,3° и выше
фенол и вода смешиваются в любых отношениях.
Применяется в значительных количествах для производства сали-
циловой кислоты, медикаментов (салол), искусственных смол и ла-
ков (бакелит) и в синтезе красителей. Ввиду недостаточных коли-
честв каменноугольного фенола продукт получается в значительных
размерах синтетически из бензола.
Крезолы С6Н4(СН8)ОН получаются в виде смеси трех изомеров
в отношении приблизительно : м : о : п = 40 : 35 : 25. Ортокрезол
с *пл. 30,05°; Г;ип 190,8°; уд. в. (20°/4) 1,0482. Метакрезол жидкий
«с t°„ -4-3 — -4-4°; t°Hn 202,8°, уд. в. (20°/4) 1,0341. Паракрезол — при-
зматические кристаллы, с t°n 36°, t°Hn 202°; уд. в. (20°(4) 1,0347.
Разделение изомерных крезолов, если надо, ведут с помощью
перегонки (отделение о-крезола) и химической переработки, поль-
зуясь различным отношением п- и .и-изомера к серной кислоте и
разницей в способности к гидролизу полученных сульфокислот.
Крезолы находят себе некоторое применение в красочном синтезе
(крезидин из п-крезола, гомологичные салициловой кислоте крезо-
тиновые кислоты) и особенно в производстве искусственных смол.
Из азотсодержащих соединений, получаемых из продуктов коксо-
вания угля, отметим пиридин, хинолин, их гомологи, индол, карба-
зол и акридин. Эти соединения принадлежат к ряду гетероцикли-
ческих соединений, — вместе с углеродными атомами и атом азота
является составной частью их кольца.
Пиридин C5H6N,
> Сш уд. в. (25°) 0,978, почти не имеет
значения как исходный материал для синтеза красителей, но не-
которые его производные имеют применение в синтезе лекарственных
веществ. Вместе с своими гомологами пиридин образует технически
применимую смесь —пиридиновые основания, служащую как раство-
ритель для очистки антрацена от примесей.
Хинолин,
а,
Р-бензопиридин C9H7N
. U. 238°,уд.в.(20°)
1,092, не выделяется из продуктов каменноугольной смолы, а в случае
надобности в нем готовится синтетически. Ядро хинолина, синтети-
чески образованное, входит в состав некоторых интересных краси-
телей антрахинонового ряда. Один из его гомологов — хинальдин
ВД I
N
tK°an. 246—248°, получается также синте-
зе
тически и является исходным веществом в синтезе хинолино-
вого желтого. Хинальдин й другой гомолог хинолина, лепидин,
СНЭ
’ с Lm 258°—260° применяются в синтезе имеющих боль*
N
шую важность при производстве фотографических пластинок кра-
сителей— сенсибилизаторов.
Индол C8H7N,
, Рпл 52,5°, t°KHn 253—254°. Извлечение
ПЛ.
NH
индола из каменноугольной смолы для синтеза красителей пока
не практикуется. Индольное ядро лежит в основе индиго и инди-
гоидных краситилей, причем оно в этих случаях
путем синтеза. Индол находит себе применение
образуется уже
в парфюмерии.
Карбазол C12H9N,
.О
’ пл.
246°, еип.
354—355°, при-
NH
надлежит, как уже было сказано, к спутникам антрацена в ка-
менноугольной смоле. В смоле из донецкого угля содержится при-
близительно на 10 ч. антрацена до 9 ч. карбазола. При очистке
такого антрацена получаются в отходах большие количества кар-
базола. Его значение в красочной промышленности до сих пор не
велико и ограничивается применением в синтезе некоторых кубо-
вых и сернистых (прочных) красителей. Нужно думать, что карбазол
окажется интересным исходным материалом для многих синтезов
как в красочной, так и в других отраслях промышленности. Необ-
ходимо искать для него применений.
Акридин CI8H9N,
. С. 107°. 10кип. 345-346*. До
недавнего времени не выделялся из каменноугольной смолы.
Недавно Виртом предложен способ его выделения в виде N-суль-
фокислоты путем обработки заключающей акридин фракций
бисульфитом®), что делает акридин доступным веществом- Акри-
диновое ядро играет заметную роль в синтезе некоторых основных
красителей, где оно получается в самом процессе синтеза. Воз-
можно, что акридиновые соединения (в том числе очень интересные
лекарственные вещества) будут удобно готовиться из самой* пири-
дина. Химия акридина, как и химии иарбалШ, erne. нештВЬНно
разработана.
2 Зак. 3611. — Н. Ворожцов
17
Количественные отношения, в каких получаются Вышеназванные
продукты при коксовании угля, в достаточной мере постоянны для
каждого сорта угля и системы коксования.
По сообщению проф. М. И. С л а д к о в а, при старой конструкции печей,
при продолжительности коксования 28—30 час., максимальной t° 900° выход сырого
бензола в Донбассе — 0,55—0,60%. или около 6 кг „с 1 т угля, выход бырого бен-
зола в Кемерове (Сибирь) — 0,6—0,65%, или около 6 кг с 1 т угля, выход
сырого бензола на Урале (Кизел)—1,0—1,12%, или около 10—11 кг с 1 т угля.
Состав сырого бензола старых печей.
В среднем для Донбасса и Кемерова:
бензола около 35%.— около 2 кг с 1 т угля
толуола 16—17%— „ 1 „ . 1 „ „
ксилолов 7—8% — „ 0,45 „ „ 1 „
Для Урала (губахнискне печи, кизеловские угли):
бензола около 40—45% — около 4,5 кг с 1 т угля
толуола , 20—25% — . 2—2,4 „ „ 1 „ „
ксилолов „ 12—15% — „ 1,25 „ , 1 „ „
Новые быстроходные печи Донбасса дают сырого бензола 0,8—0,85%, или 8—8,5 кг
с 1 т угля, из него:
бензола около 60% — около 4,8 кг с 1 т угля
толуола „ 8—9%— „ 0,6—0,7 „ „ 1 ,
ксилолов „ 3—4% — „ 0,25 „ „ 1 ,
Соотношение изомерных ксилолов в зависимости от системы коксования в %:
Старые печи Новые печн
Мета-ксилола — 65 — 70 75— 80
Орто-ксилола — около 8 3—5
Пара-ксилола — около 25 около 20
Фенолов (фенола, крезолов и ксиленолов) в смоле старых печей Донбасса
(выход смолы 2,3—2,4%) 0,048—0,050% от угля, или 0,48 кг с 1 т угля, в нх со-
ставе: фенола 20—25%, или 0,12 кг с 1 т угля, крезолов, ксиленолов 80—75%,
или 0,36 кг с 1 т угля.
Соотношение изомерных крезолов приблизительно м : о : п = 45 : 30 : 25.
Фенолов в смоле новых печей Донбасса (выход смолы 26 кг с 1 т угля)
0,025—0,03% от угля, или 0,25—0,3 кг с 1 т угля; из иих фенола 35—40%, или
0,1 кг с 1 т угля; крезолов и ксиленолов 65—60%, или 0,18 кг с 1 т угля.
Соотношение изомерных крезолов приблизительно, м : о : п= 55 : 2$ : 20.
Нафталина со старых печей Донбасса 5—6% от смолы, или 1,2 кг с 1 т угля;
в Кемерове (кузнецкие .угли) около 5% от смолы, или 1,5 кг с 1 т угля; в Губахе
(кизеловские угли) 4—5% от смолы, или 2 кг с 1 т угля.
Нафталина с новых печей Донбасса около 10% от смолы, или около 2,6 кг
elm угля.
Антрацена из смолы старых печей Донбасса около 1% от смолы, нли 0,25 кг
с1тугля; в Кемерове — около 1% от смолы, или около 0,3 кг с 1 т угля; в Губахе
(кизеловские угли) — около 0,75 от смолы, или около 0,5 кг с 1 т угля.
Антрацена с новых печей Донбасса 1,75--2,5% от смолы, или 0,5—0,7 кг с 1 я» угля.
Содержание карбазола по Донбассу в среднем 40% от антрацена, Или 0,1—0,15 кг
elm угля; о содержании карбазола в смоле кузнецких углей точных данных нет,
содержание карбазола в смоле кизеловских углей не свыше 20% от антрацена,
т. е около 0,1 кг с 1 т угля.
В' среднем (по данным Анилобъединения) по коксохимическим установкам
СССР принимают выход смолы в 2% от веса коксуемого угля й ныход „сырого"
бензола в 10 кг с 1 m угля; ректификованного бензола в 4 кг с 1 т угля; толуола
1,4 кг с. 1 т угля; нафталина 8% От смолы, или 1,6 кг с 1 т угля; антрацена
0,5% от смолы, или 0,1 кг с 1 т угля.
Бензола получается таким образом больше, чем толуола, нафта-
18
липа значительно больше, чем других углеводородов с конденси^
рованными циклами.
Само собой понятно, что эти постоянные отношения играют
весьма заметную роль в экономике промышленности. В случае
очень большого запроса со стороны промышленности на какой-
либо из продуктов, получаемый в меньших количествах (таким
например является толуол), усиление его выработки неизбежно
повлекло бы выработку вместе с ним превосходящих его по коли-
честву других продуктов (например бензола, отчасти нафталина),
которые таким образом могли бы сделаться неликвидными. Поэтому
при организации производств, потребляющих большое количество
сырых материалов, всегда предпочтительно выбирать такие исход-
ные материалы, которые не могли бы оказаться дефицитными
при увеличении производства. Этим например обусловливается
то обстоятельство, что производство индиго (в Германии и других
странах Запада) использует в качестве исходного материала не то-
луол, а бензол, хотя методы получения индиго из толуола доста-
точно хорошо разработаны. Некоторые из соединений, заключаю-
щихся в больших количествах в каменноугольной смоле, являются
до сих пор недостаточно ликвидными (таковы например фенантрен,
карбазол), и предстоит большая исследовательская работа, может
быть еще на долгое время, чтобы найти им применение.
С другой стороны, характерной чертой нового периода стано-
вится стремление красочной промышленности использовать обла-
гороженные сырые материалы, которые частично готовятся синтети-
чески. Как пример такого синтеза можно назвать синтез антрахинона.
Последний готовился в довоенное время исключительно из антра-
цена, окислением. Применение в красочной промышленности имеет
главным образом не антрацен, а антрахинон. Экономические со-
ображения (дороговизна извлечения антрацена из смолы и ухуд-
шение, в результате извлечения антрацена, качества смолы для
применения ее в дорожном строительстве) заставили американцев
перейти к синтезу антрахинона, используя как исходные материалы
бензол и нафталин (последний предварительно превращается во фта-
левый ангидрид). Этот метод оказался у них выгоднее старого и стал
постепенно вводиться и на европейских предприятиях (в Англии,
Франции), так что антрахинон становится теперь уже синтетическим
исходным материалом. Таковым же может быть суждено стать
перилену. В известной мере переход к синтезированным, а не прямо
из сырья полученным, исходным материалам можно констатировать
как заметную тенденцию последнего периода красочной промыш-
ленности. Она становится понятной ввиду все большего примене-
ния в практике синтеза сложно построенных исходных веществ.
Антрахинон получается или из антрацена окислением, или синте-
зом. Помимо фталангидридного синтеза, находящего себе примене-
ние и для получения антрахиноновых замещенных fp-метилантрахи-
нона, к3-хлоранитрахинона, 1.4-диоксиантрахииона (хинизарина)
н др.], в последнее время описан метод „диенового" синтеза антра-
хинона и гомологов из хинона (или а-нафтохинона) и углеводородов
ряда бутадиена ,0). Ниже будут указаны условия проведения этих
синтезов.
2*
19
Антрахиион С14Н8О2,
co
, желто-серые ромбические
СО
кристаллы, (’л 284,8°, t’Hn. 376,8°4). В качестве исходного материала
ценится лишь тогда, когда он обладает высокой степенью чи-
стоты, приблизительно 99%- Недопустима примесь метилантрахинона,
возможная при получении из антрацена, не свободного от
метилантрацена. Очищается в случае недостаточной чистоты пере-
кристаллизацией из высококипящих органических растворителей
Или из концентрированной серной кислоты.
Антрахинон применяется как исходный продукт для многих
прочных красителей. Особенно применим для получения нитро-
и сульфопроизводных.
Перилен С^Н
!—\ . Углеводород, получаемый син-
тетически из нафталина или, лучше, из р-нафтола. Желтые кри-
сталлы с 1°л 264—265°. Красные растворы перилена в органических
растворителях показывают синюю флуоресценцию- Периленовое
ядро лежит в основе некоторых особенно прочных красителей,
получаемых из антрахинона (индантреновый темносиний, индан-
треновый яркозеленый). Из перилена довольно просто получаются
подобные прочные красители. В настоящее время в Италии неви-
димому уже из самого перилена производятся красители.
Работа с некоторыми исходными материалами требует соблю-
дения большой осторожности. Бензол и его гомологи представляют
собой жидкости с большой упругостью паров, горючие.
Насыщение воздуха их парами, особенно легко осуществимое
в условиях закрытых помещений, цистерн, приемников, мерников
и т. п., легко может привести к взрывчатой газовой смеси. Взрыв
может последовать от малейшей искры.
Пары бензола и его гомологов при вдыхании могут привести
к обмороку, а при длительном пребывании в такой атмосфере
привести к смерти (например при спуске в аппарат, в котором
был бензол и который не освобожден от его паров). Постоянное
воздействие паров бензола, хотя бы в очень небольших количе-
ствах, отзывается болезненно на нервной системе.
При работе с антраценом, особенно техническим, имеющим
много примесей, надо остерегаться соприкосновения его пыли
с кожей рук, лица. Не говоря уже о легко возникающих, если
такое соприкосновение происходит, кожных заболеваниях экземати-
ческого характера, возможно на основе отравления антраценом
или его примесями и поражение внутренних органов.
Жидкие углеводороды с целью устранения опасности взрывов
и пожара не должны передаваться давлением воздуха (через монте-
20
жюсы). Их можно перемещать или давлением не имеющих свобод-
ного кислорода газов (азот, углекислота) или закачивать насосом
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) J. Masson, СВ 1931, II, 3584.
2) Н. М Кижнер, Г. Г. Вендельштейн, Ж., 57, 1 (1925). Определение
.«-ксилола в техническом ксилоле, Н. Р. Reich el, Ch. Ztg. 55, 744 (1931).
3) Merck, Г. n. 434988, F. XV, 193.
4)1. Timmermans, F. Burriel, CB 1931, D, 875.
5) P. d e В e u 1 e, CB 1931, II, 970.
6) M. А. Ильинский, Ан. Kp. Пр. 1931, № 2—3,4, M. А. И л ь и и,с к и й,
Полупродукты и красители антраценового ряда, Москва, 1932.
7) К. Brass, J. Stadler, Вег. 57. 128 (1924); Г. п. 430631, F. XV, 792;
j. S. Turski, R. Praglerowa, СВ 1929. I, 1692.
8) Werner Lowenstein Wack, Kunz, Ann. 321, 297 (1902); L. F. Fie-
ser, Am. Soc. 51, 940 (1929).
9) Wirt, Г. n. 440771, F. XV, 342.
10) IG. Г. n. 494438, 496393, Б. n. 320375, 324661, CB 1930, II, 807, 609. P. В ейс-
гербер, Химическая переработка каменноугольного дегтя, Москва, 1929.
ГЛАВА I
ОБЩИЕ ПОНЯТИЯ О ПРЕВРАЩЕНИЯХ ИСХОДНЫХ ВЕЩЕСТВ
В ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
а) Три основных группы превращений
Исходные вещества, выделенные из каменноугольной смолы или
полученные синтетически и достаточно очищенные, должны под-
вергнуться часто весьма длительной химической переработке,
чтобы превратиться наконец в годные для фабрикации красителей
промежуточные продукты. Для того чтобы в дальнейшем легко
ориентироваться в сущности этих химических превращений, мы при-
ведем здесь важнейшие из обозначений, отвечающих формулам
строения исходных материалов, которыми мы далее будем пользо-
ваться.
СО
I. Бензол. Все атомы водорода равноценны. Нет изомеров у одно-
замещенных. Для двузамещенных — три изомера: орто (о) —1.2,
или 2.3 или 3.4 и т. д.; мета (м)—1.3 или 2.4 и т. д.; пара (п)—
1.4 или 2.5 и т. д.
У гомологов бензола, например у толуола (II), заместители
водородных атомов могут быть не только в ядре, но и в том
углеводородном радикале (в данйом случае в метиле СН3), кото-
рый составляет так называемую боковую цепь ароматического угле-
водорода. Эти производные называются по тому углеводородному
радикалу, который остается за вычетом заместителя (например
С6Н5СН2 = бензил, С6Н5СН = бензилиден), общее же обозначение
водородов боковой цепи отмечается греческой буквой ш (омега).
III. Нафталин. Среди восьми атомов водорода, способных к за-
мещению. имеется по четыре равноценных .между собой. Это
1.4.5.8 (а-атомы) и 2.3.6.7 (р-атомы); а- и р-атомы между
собой не равноценны. Соответственно с этим возможны изомеры
уже у однозамещенных нафталина (а- и p-изомеры). Изомерия для
двузамещенных более сложна, чем у бензольных производных,
ввиду возможности вхождения второго заместителя не только
в то кольцо, где уже имеется первый заместитель, но и в другое.
При размещении двух заместителей в одном из колец нафталина
22
их можно назвать, так же как в случае бензольных соединений:
о, м и п. При вхождении заместителей в разные ядра изомеры
называются по нумерам атомов С (например 1.5-динитронафта-
лин, 2.6-дисульфокислота и т. д.). Чаще и одноядерные производ-
ные даются только с нумерацией атомов (1.2-нафтиламинсульфо-
кислота, а не о).
IV. Антрахинон имеет 8 атомов водорода, способных к заме-
щению. Как в нафталине, различают по две четверки равноценных
между собой атомов: а (1.4.5.8) и р (2.3.6.7). Возможность
изомерии и нумерация изомеров те же, что и в нафталиновых
соединениях. Химические отношения замещенных антрахинона очень
своеобразны, и многие заместители отличаются повышенной реак-
ционной способностью сравнительно с их поведением при заме-
щении бензольного ядра. Ниже мы встретим некоторые примеры
своеобразия в реакционной способности антрахиноновых производ-
ных, так же как и нафталиновых.
Несмотря на все многообразие методов переработки исходных1
материалов в промежуточные продукты, можно отличить в них
несколько основных групп и в каждой группе по нескольку отдель-
ных химических операций.
Первая и весьма важная по своему значению группа методов
имеет своей задачей введение новых заместителей (субституентов)
в ароматические соединения вместо водородных атомов. Эти ме-
тоды имеют задачей следовательно изменить химическую природу
начальных соединений (в большинстве случаев мало активных угле-
водородов), вводя в них новые реакционные группы, и тем под-
готовить возможность использования второй группы методов.
Эти последние методы, не вводя самостоятельно новых заме-
стителей, берут исходным пунктом уже имеющиеся, т. е. введен-
ные по первым путям, реакционные группы и подвергают их
превращениям в иные замещающие группы часто с повышенной
или пониженной реакционной способностью сравнительно с перво
начальными. Если первую группу мы назовем методами ввода заме-
щающих групп, то вторую будем называть методами превращения
замещающих групп.
Наконец третья, менее значительная, группа методов изменяет
главным образом углеродный скелет органического соединения.
Нужно указать, что если в первой и второй группах можно выде-
лить такие приемы, которые вполне подходят под определение
задач методов, как они выше определены, то все же имеются и
такие, которые одновременно с своей целью осуществляют и цель
другой группы методов, например одновременно с превращением
имеющегося заместителя вводится новый.
В третьей же группе такая коллизия с другими группами
является почти постоянной.
Наиболее употребительными методами переработки начальных
продуктов в промежуточные являются следующие:
1) нитрование и нитрозирование,
2) сульфирование,
3) введение галойда, чаще всего хлорирование,
4) образование аминогруппы восстановлением,
23
5) замена сульфогруппы на гидроксил плавлением со щелочью,
6) обмен хлорного атома на иные группы,
7) замена аминогруппы гидроксилом и замена гидроксила ами-
ногруппой,
8) диазотирование (часто сопровождаемое азосочетанием),
9) алкилирование (гидроксила или аминогруппы),
10) арилирование (главным образом аминогруппы),
11) ацилирование,
12) окисление,
13) реакции конденсации и перегруппировок.
Из названных операций первые три — нитрование, сульфирование, хлорирова-
ние— как-раз подходят в группу методов введения новых заместителей, 4—И (образо-
вание аминогруппы восстановлением, щелочное плавление, превращение хлорного,
соответственно бромного, атома, взаимные превращения амино- и оксигрулп, диазо-
тирование, алкилирование, арилирование, ацилирование) можно включить в группу
методов превращения уже имеющихся заместителей, хотя и могут быть случаи
коллизий. Например при восстановлении нитро- или нитроэогруппы сериистокислыми
солями одновременно с образованием аминогруппы наблюдается часто и вхожде-
ние сульфогруппы в ядро (новое замещение); реакция диазотирования есть метод
превращения аминогруппы, но азосочетание (для пассивно входящего в реакцию
амина или фенола) есть уже способ образования нового заместителя в ядре. Про-
межуточное положение занимает операция окисления, 12, каковая может имёть
применение и в качестве метода превращения заместителя, например в переходе.
СвН6СН3 -> С6Н5СО2Н, и в качестве определяющей вхождение новой гидроксильной
группы в ядро,, например
Y -> ! Y
\Z \/\0H
Эта же реакция окисления имеет значение и как операция, определяющая
в иных случаях изменение скелета соединения (как пример приведем окисление
нафталина во фталевый ангидрид), и в таком случае вместе с реакциями конден-
сации и пр. входит в третью группу.
Первая группа операций наиболее важна для производства, так как она опре-
деляет собой размещение реакционных групп не только в полученном продукте,
но и в тех его производных, которые образуются из него при помощи методов
второй и третьей группы.
Интересно отметить еще одну особенность операций первой группы в Виде
взаимной связи их с одной из операций второй группы. Нитрование и нитрозиро-
вание используются реакцией восстановления для получения аминогруппы. Сульфи-
рование дает материал для реакции сплавления со щелочью (образование оксигруппы).
Хлорирование в огромном большинстве случаев (по крайней мере в технике произ-
водства промежуточных продуктов, а не готовых красителей) имеет задачей не только
ввести хлор как таковой, но путем замещения его получить амино- или оксипроиз-.
водное (6). Таким образом в некоторых из реакций, принадлежащих к двум разным
группам, имеется настолько близкая взаимная зависимость (без сульфогруппы
нельзя образовать во многих случаях гидроксила, а аминогруппа может быть полу-
чена часто только через иитросоединение), что практично будет при специальном
рассмотрении методов получения интересующих нас продуктов учитывать взаимную
связь этих реакций.
б) Замещения и ориентация заместителей
Как уже было выше указано, введенный в ядро заместитель
водородного атома определяет место нахождения будущих реак-
ционных групп. Поэтому чрезвычайно важно установить те пра-
вильности, какие наблюдаются при замещениях, в ароматических
24
циклах^ На весьма большом, числе опытов был» установлено, что
новая замещающая группа становится в ароматическом Ядре в по-
ложении, определяющемся не свойствами ее самой (новой заме-
щающей группы), но указываемом уже имеющимися в ядре заме-
стителями. Реакции, выше нами названные, могут быть причиной
образования таких заместителей: нитрование образует нитро-
группу — NO2, нитрозирование группу нитрозо —NO или изо-
нитрозо “NOH, сульфирование группу сульфо —SO3H, хлориро-
вание (соотв.'бромирование) вводит атом хлора С1 (соотв. брома Вт),
азосочетание сопровождается образованием азогруппы —N —N.
Восстановление, примененное по отношению к группе —NO2, — NO
(или -гNOH) и азогруппе —N--N —, дает аминогруппу —NH2>
щелочное плавление (при реакции с —SO3H), или обработка соеди-
нения с подвижным хлорным атомом щелочью или окисление
водородного атома образуют гидроксильную или оксирруппу
— ОН, диазотирование аминогруппы дает диазониевую группу
N2X, где X — одновалентный анион. Реакциями алкилирования,
арилирования и ацилирования * в применении к амино- или гидро-
ксильной группе производятся алкиламиногруппа —NHAlk, диал-
киламиногруппа— N(Alk)2, ариламиногруппа—NHAr, ацилированная
аминогруппа —NH(Ac), ' алкоксигруппа —OAlk, ацилированная
оксигруппа — ОАс. Если мы к тому же сообщим, что в ядре
исходного продукта может быть заместитель в виде метильной
группы (одной или нескольких) —СН3, что эта последняя может
быть окислением превращена в карбоксильную группу — СООН
или альдегидную группу — СНО, что путем конденсации мы можем
ввести в ароматическое ядро вообще карбонильную группу —СО
и что путем перехода через реакционные диазониевые соединения
можно получить новые заместители (например нитрильную или
цианидную группу — CN и пр.), то мы могли бы притти к выводу
о чрезвычайной сложности вопроса о правильностях при входе
новых заместителей в ароматические ядра. На самом деле, как
мы знаем, вопрос этот значительно упрощается тем, что суще-
ствуют два главные типа замещения.
Именно, наличный в ядре заместитель X направляет (ориенти-
рует) новую, входящую вместо водородного атома в бензольное
ядро, группу или атом Z или таким образом, что она становится
либо в пара- или орто-подожения по отношению к направляющей
группе (причем чаще получается смесь пара- и орто-изомеров)
либо направление новой группы или атома Z от имеющегося в иалич-
* Алкил (Aik) — одновалентный радикал алифатического ряда, например ме-
тил СН3, этил С2Н5. Арил (Аг) — одновалентный радикал ароматического ряда, на-
пример феиил С6Н5, толил С7Н7. Ацил (Ас) — кислотный радикал, сохраняющийся
в ангидридах и хлорангидрндах кислот, например ацетил СН3СО, толуолсульфок-
сил C7H7SO2 и т. п.
25
яости заместителя А таково, что новая группа становится в мета-
положение по отношению к наличной:
Примесь изомеров пара- и орто- в этом случае, так же как примесь
мета-изомеров в случае ориентации первого рода, незначительна.
В применении к соединениям с конденсированными циклами
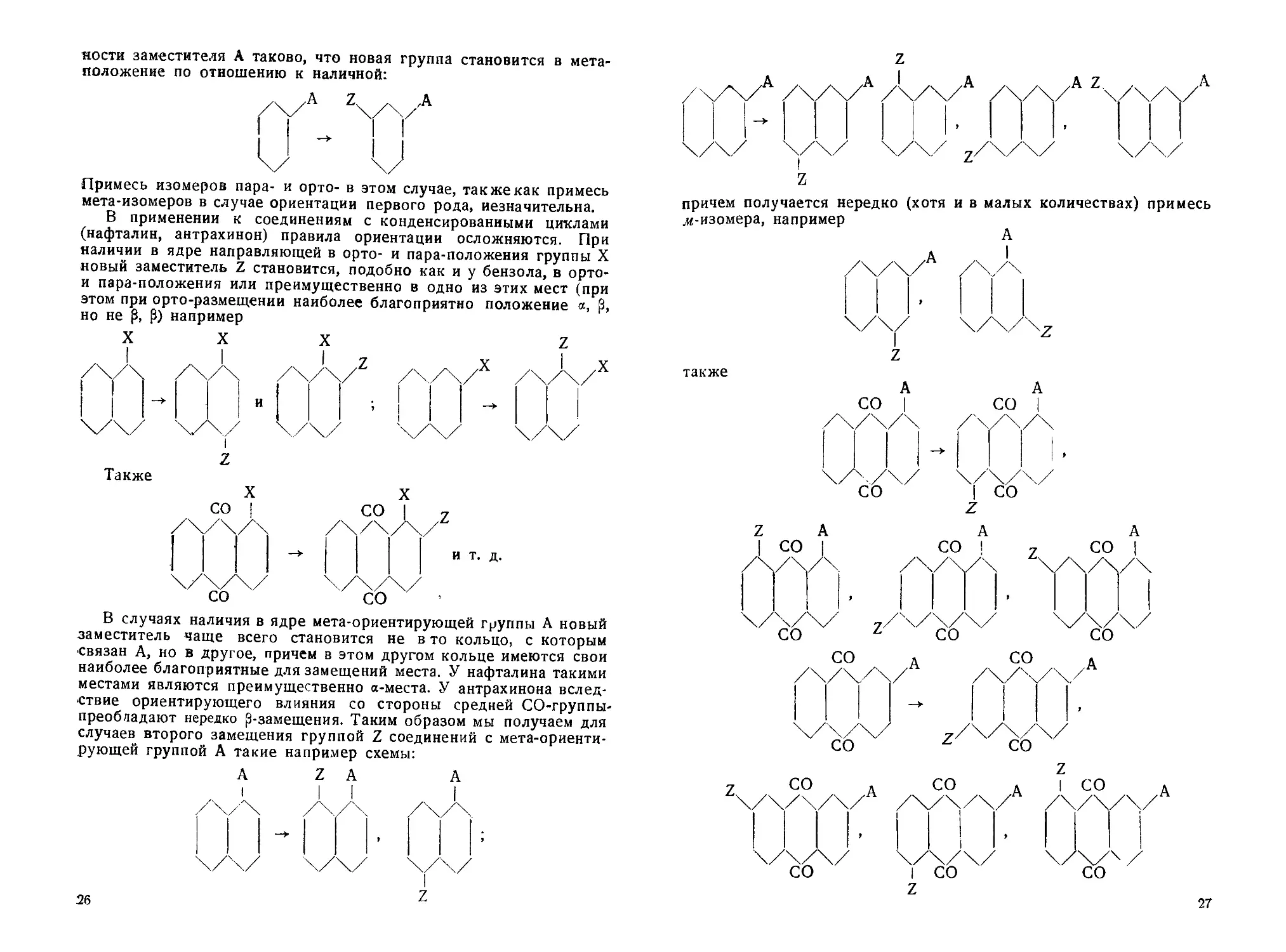
(нафталин, антрахинон) правила ориентации осложняются. При
наличии в ядре направляющей в орто- и пара-положения группы X
новый заместитель Z становится, подобно как и у бензола, в орто-
и пара-положения или преимущественно в одно из этих мест (при
этом при орто-размещении наиболее благоприятно положение а, 0,
но не р, Р) например
В случаях наличия в ядре мета-ориентирующей группы А новый
заместитель чаще всего становится не в то кольцо, с которым
связан А, но в другое, причем в этом другом кольце имеются свои
наиболее благоприятные для замещений места. У нафталина такими
местами являются преимущественно a-места. У антрахинона вслед-
ствие ориентирующего влияния со стороны средней СО-группы-
преобладают нередко ^-замещения. Таким образом мы получаем для
случаев второго замещения группой Z соединений с мета-ориенти-
рующей группой А такие например схемы:
A Z А А
z
z
причем получается нередко (хотя и в малых количествах) примесь
л<-изомера, например
А
также
А А
СО | СО |
27
Мы видим уже по этим схемам, что изомерия в ряду двузаме-
щенных нафталина и антрахинона значительно сложнее, чем в бен-
зольном ряду, и что количество возможных изомеров гораздо больше,
а следовательно и способы выделения и переработки делаются
здесь более сложными х).
Внешние условия (температура, давление и пр.) обычно не
влияют на постоянство типа замещения. Влияние катализаторов
и реакции среды иногда становится заметным, но весьма часто
может быть объяснено изменением структуры самой направляющей
группы, чему примеры мы найдем ниже.
При сопоставлении заместителей бензольного ядра по их напра-
вляющей способности мы получаем следовательно два ряда.
1. Атомы или атомные группы, направляющие в орто- и пара-
III HI III
положения: F, Cl, Br, J, ОН, OAlk, OAc, NH2; NHAlk, N(Alk)2, NHAc,
N-N, CH3, CH2Alk, CH(Alk)2, C(CH3)3, CH2C1, CH2ONO2, CH2SO3H,
CH2NH2, CH2CN, CH2COOH, CH2CH2COOH, CH^CHCOOH,
ch~chno2, c = ccooh, C6Hg.
2. Группы, направляющие в мета-положение: ЫО2, SO3H, СНО,
СН~ЫО2Н (в фенилнитрометане), СООН, СО2А1к, CONH2, COAlk,
СОСООН, = СОН (трифенилкарбинол), CN, СС13, NH3X NH2(Alk)X,
NH(Alk)2X, NH2(Ac)X 2).
Причину разницы этих двух рядов, побуждающих к своеобраз-
ному направлению новых заместителей, мы не знаем. Ниже мы
изложим некоторые гипотезы, имеющие к этому, отношение. Как
правило можно принять, что те группы, в которых между состав-
ляющими их атомами имеется двойная или многократная связь
или имеется атом, могущий легко функционировать в состоянии
высшей валентности, ориентируют в мета-положение *, между тем
как группы, структура которых не заключает многократных связей,
ориентируют в орто- и пара-положения **. В первом ряду заме-
стителей имеется один, именно аминогруппа (и ее алкильные произ-
водные), наиболее характерный атом которого, именно — азотный,
способен переходить из трехвалентного в пятивалентное состояние.
В соответствии с вышеуказанной систематикой нужно ожидать,
что аминогруппа в состоянии ориентировать и в мета-место. В дей-
ствительности аминогруппа проявляет двоякую ориентацию в зави-
симости главным образом от реакции среды: будучи, вообще говоря,
орто- и пара-направляющей, она в сильнокислом растворе (оче-
видно с образованием соединений, отвечающих типу аммония ***,
* Исключение — те группы, где многократная связь имеется между одноимен-
ными атомами, например—СН~СН — , — N — N— и т. п.
Группа SCN повидимому ориентирует в пара- и орто-положення 3).
** Исключение — группы, лишенные многократных связей, но заключающие
(на втором от углерода месте) сильные металлоидно-полярные атомы, так СС13
ориентирует в мета-положение 4).
*** Интересно, что дназониевая группа в сернокислотном растворе, как явствует
из исследований М. И. Сладкова, ориентирует преимущественно в пара-место (не
опубликовано).
28
ориентирует уже в мета-положение. Ниже мы увидим случаи, как
ацилирование аминогруппы делает ориентацию уже однообразной.
Лишенное каких-либо гипотетических предпосылок правило
замещений выставлено недавно двумя английскими исследователями.
Они сформулировали его так:
„Если в бензольном производном (замещенном группой атомов XY)
\ XY
Y стоит в более высокой группе периодической системы, чем X,
или если Y принадлежит той же группе, но атомный вес у Y
ниже, чем у X, тогда второй заместитель (атом или группа), входя-
щий в ядро, вступит в мета-положение к группе XY. Во всех дру-
гих случаях, включая и те, когда вместо сложной группы XY
имеется один атом, второй заместитель становится в орто- и пара-
положения “.
Мета-ориентирующие группы СО2Н, СО, NO2, CN, СС13 покры-
ваются первой частью правила. SO3H— дает пример атомов одной
и той же (6) группы периодической системы, но разных атомных
весов (О < S). Последняя часть правила охватывает такие группы,
как СН3, NH2, ОН, ОСН3 и т. п. В соответствии с этим группы
SCN, SeCN ориентируют в орто- н пара-положения.
Приложение правила к случаю, когда X — Y (например — N — N —,
—С~ С —) также возможно: ближайший к ядру атом не принад-
лежит к низшей группе периодической системы, почему ориента-
ция— не в мета-, но в орто- и пара-положения.
В смешанных группах, например СНаС1, СНС12, СНО, приложе-
ние правила объясняет неотчетливость ориентации. Из хлористого
бензилидена получаются и орто- и пара-замещенные (от ориентирую-
щего влияния СН-группы) и мета-замещенные (от влияния СС12).
Кажущимся исключением из этого правила является нитрозо-
группа NO, так как нитрозобензол при нитровании и бромирова-
нии в растворе сероуглерода или четырехлористого углерода дает
пара-замещенные. Это объясняется тем, что в этих растворителях
нитрозобензол частично находится в бимолекулярной форме. При
замещениях нитрозобензола, растворенного в уксусной кислоте и
находящегося в мономолекулярном состоянии, он дает мета-заме-
щенные 6).
При наличии в бензольном ядре не одного, а двух заместителей
могут быть соединения с согласной или совпадающей ориентацией
заместителей, например мета-двухзамещенное, где оба заместителя
принадлежат к первому ряду (X, Y например NH2 и ОН) или ко
второму ряду (А, В например NO2 и SO3H) (формулы I и II).
Звездочкой <*> помечены места, в которые направляется новый
заместитель
Y\/\/x А\/\/в
29
Таким образом диметилметааминофенол (X=*N(CH3)2, Y = ОН)
фиксирует группу нитрозо (соотв. изонитрозо) при обработке азо-
тистой кислотой и атом С при конденсации с фталевым ангидридом
(образуя краситель — родамин) как-раз в месте совпадающей
ориентации', пара- к N(CH3)2-rpynne и орто- к ОН-группе.
(CH3)2N4/x/
он
(CH^N^/^/O^/^ /N (СН3)2
Также резорцин (X = Y = OH) дает с диазониями два ряда про-
дуктов сочетания: первый с одной и второй с двумя азоарильными
группами. Азогруппа вступает в место 4 (с совпадающей ориента-
цией). При проведении сочетания в сильно щелочной среде стано-
вится все более заметно влияние места 2 (совпадающая орто-
ориентация со стороны ОН-группы).
Изонитрозогруппы при нитрозировании резорцина располагаются
в местах 6 и 2 (также с совпадающей ориентацией):
N = NR
NOH
.и-Крезол при нитровании дает 6-нитро и 4-нитропроизводные
в отношении количеств 2 :3 6)
Н3Ск /ОН н3сх /х/он н3сх/х/он
3 1 3 1
4 6
OgN'7'4/ 4x/XNO2
Следовательно NO2 становится в места с совпадающей ориен-
тацией.
Во второй (II) формуле оба заместителя ориентируют в мета-
положение каждый, и помеченное звездочкой место удовле-
творяет ориентации и того и другого.
30
В согласии с этим м-динитробензол (А = В — NO2) при даль-
нейшем энергичном Нитровании дает 1.3.5-тринитробензол:
O2N\ /X/zNO2 О2К\/, /NO2
NO2
При различно ориентирующих заместителях, принадлежащих
к разным рядам (X, А например ОН и SO3H) при их взаимном
расположении в орто- или пара-местах, налицо также случай сов-
падающей ориентации, например (III и IV):
III
IV
Примером совпадающей ориентации по схеме III может служить
о-нитротолуол (Х = СН3, A = NO2), который при дальнейшем нитро-
вании принимает нитрогруппу в 4- и в 6-положения:
NO2
По схеме IV построена например «-сульфокислота фенола
(Х = ОН, A —SO3H), которая при нитровании, азосочетании и про-
чих реакциях замещения обменивает водород в положении 2
(соотв. 6):
HOgS^^^N —NR
Точно так же «-нитрохлорбензол сульфируется и нитруется
в орто-положение по отношению к атому хлора и в мета-положе-
ние по отношению к NO2-rpynne:
31
Но могут быть такого рода соединения, что заместители, в них
имеющиеся, направляют новые группы в такие места, которые
находятся в неблагоприятном положении по отношению к другому
наличному в ядре заместителю. В таких случаях ориентирующие
влияния конкурируют между собой, и в результате получается
иногда много изомеров, причем преобладают те, которые обусло-
вливаются ориентацией более сильного заместителя. Для наиболее
типичных заместителей относительная энергия определяется местом
в рядах Голлемана, в которых заместители расположены слева
направо по скорости определяемых ими замещений (мерилу энер-
гии по Голлеману):
ш
ОН, NHa, CI, СН3(о-л) иСООН, SO3H, ЫО2(л).
Примеры несовпадающей (несогласной) ориентации:
ш
где X, Y принадлежат к первому ряду (например ОН или NH2),
А — ко второму ряду (например NO2). На формулах видно много
мест, в которые могут войти новые замещающие группы. На опыте
же вопрос решается обычно в пользу ориентации более сильного
заместителя.
Примером несовпадающей ориентации первого рода (I) является
этиловый эфир о-крезола (X = ОС2Н5, Y = СН3), который при нитро-
вании дает последовательно 4-мононитро- и 6.4-ди нитродериваты.
Х/ХОС2Н5
ОС2Н6
Очевидно при этом, что ориентирующее значение метильной группы
(т. е. скорость образования отвечающих ей орто- и пара-производ-
ных) ничтожно по сравнению с таковыми же у группы ОС,Н5.
Подобно этому сам о-крезол при нитровании дает равные количе-
ства 4-нитро- и 6-нитропроизводных ft):
32
Следовательно влияние на ориентацию со стороны СН8-группы
исчезающе мало по сравнению с влиянием более сильной ОН-
труппы.
В виде примера можно также привести о-ацетаннзнднд (Х =
= NHCOCH3, Y —ОСН3), в котором оба заместителя ориентируют
в отдельности в орто- и пара-положения. В результате нх совмест-
ного влияния при нитровании получаются два изомера: приблизи-
тельно 70% 1.2.4- и ЗО°/о 1.2.5-нитропродукта 7):
/./NHCOCHg
O2NZX/,X4OCH3
/\zNHCOCH3
i|
—>
2
N НСО С Н3
ОСН3
В этом случае следовательно конкуренция ориентирующих групп
складывается в пользу образования изомера, отвечающего влиянию
NHCOCHg-группы, но в то же время образуется и нитропро-
дукт, вызванный пара-ориентацией ОСН3-группы, хотя и в мень-
шем количестве.
В качестве соединения с несовпадающей ориентацией двух пара-
замещающих групп (схема II) можно привести, во-первых, л-ацето-
толуидид(Х = ЫНСОСНд, Y = CH3), который при хлорировании дает
только один продукт, именно 2-Хлорацетотолуидид:
NHCOCHg
//ZNHCOCH3
Во-вторых, можно указать на ацет-л-анизидид (Х = NHCOCHg,
Y = OCH3), который при нитровании образует также один лишь
3-нитропродукт 8):
/NHCOCH3
1
o2n
NHCOCHg
Следовательно в обоих последних примерах налицо случай
победы одной из конкурирующих ориентацией. У л-ацетотолун-
Дида наиболее сильной группой оказалась ацетамино-, а не ме-
тильная; в л-ацетанизидиде метоксильная сильнее, чем ацета-
мино-.
л-Крезол дает при нитровании мононитродериват с нитро-
труппой в орто-положении по отношению к ОН: преобладает рриенг
тация со стороны ОН-группы (л). Эфир угольной кислоты и Л-цре-
зола нитруется в орто-положение по отношению к СНз (преобладает
влияние СН3) (б).
3 Зак. 2611. — Н. Ворожцов
33
Таким образом в первом случае более сильная группа-—ОН
сравнительно с СН3, во втором — СН3 сравнительно с О—СО—О.
Наконецкак примернесовпадающей ориентации разно ориентирую-
щих групп (схема III) приведем л-нитроацетанилцд (X = NHCOCH3,
A = NOo). Это вещество при нитровании смесью серной и азотной
кислот дает смесь трех изомеров (Вен дер, Голлем ан), причем
во всех трех оказывается преобладание ориентации ацетамино-
группы:
NO2
O2N^z^/NHCOCH8
O2Nx /Z/NHCOCHs
о2ж ZZ/NHCOCH3
I +
O2NZZZZNHCOCHS
Изомера, отвечающего мета-ориентации нитрогруппьт, не наблю-
далось совсем. Таким образом аминогруппа не только оказывается
в этом случае сильнее нитрогруппы, но ее влияние способно прео-
долеть задерживающее влияние нитрогруппы, которая одна дает
лишь малые количества замещенных в орто- и пара-положения
к ней самой.
На всех разобранных нами примерах и множестве других осу-
ществленных случаях замещений мы усматриваем отчетливо прояв-
ления влияния заместителя на ядро и именно на определенное
место или места в ядре. Очевидно в большинстве случаев заме-
щений углеродные атомы, стоящие в пара- и орто-положениях
34
к заместителю, находятся приблизительно под одинаковым воздей-
ствием со стороны этого заместителя и совсем под иным; чем
стоящие в мета-положении. Чаще всего замещение в мета-местё
(следовательно вызываемое соответственно мета-ориентирующей
группой) проходит в условиях несколько более трудных, чем заме-
щение в орто- и пара-положениях, требуя более высокой темпера-
туры, более высокой концентрации реагентов и т. п. Правда, атом
галоида, принадлежащий к ряду заместителей, ориентирующих
в орто- и пара-положения, в то же' время отличается от своих
аналогов тем, что замещения, вызываемые им, проходят также при
более трудных условиях, чем у соединений (например углеводоро-
дов), лишенных галоида. Естественно, что природа влияния заме-
стителя на ядро, проявляемого в замещениях ароматического ядра,
привлекала и привлекает внимание исследователей.
в) Теории ориентирующих влияний при, замещениях
В последнее время в английской и отчасти в американской лите-
ратуре уделяется очень много места гипотезе о полярных влияниях
со стороны заместителей на атомы ядра через тот атом углерода,
который связан непосредственно с заместителем.
Одним из первых гипотезу о переменной полярности в замеще-
щениях высказал Форлендер 9). Он полагает, что заместители
вносят в ароматическое ядро нарушение в распределении зарядов
углеродных атомов ядра в зависимости от заряда того атома заме-
стителя, который непосредственно связывается с углеродом ядра.
Заместители входят в зависимости от знака этого заряда или как
положительные или как отрицательные радикалы. Если заместитель
состоит из нескольких атомов, та знак его главного атома выясняется
на основании полярности тех атомов, с которыми этот главный
атом связан. Таким образом
4— 4---h 4—-4-4— 4- -
группы: NO2, SO3H, СО2Н, СО, N(Alk)3X * — положительны,
—I- — + —1_ —- +
группы: NH2, ОН, ОСН3, СН3 и т. п. — отрицательны.
По Форлендеру такие группы, как NO2, SO3H, СООН, вхо-
дящие как положительные в бензольное ядро, вызывают отрица-
тельный заряд у связанного с ними атома углерода, и группы NH2
и ОН, входящие как отрицательные, вызывают положительный
заряд у атома С, к которому они присоединены. Эта противопо-
ложность зарядов переносится и на другие атомы ядра, например
+ •— —ь
МП мн
* X—атом галоида или анйон минеральной кислоты.
2*
35
Места наибольших напряжений с отрицательным зарядом С и наиболее способ
ные к замещению Форлеидер отмечает длинными линиями. Замещение пронсхо
дит по преимуществу в этих местах, например
_]—. —I-
мм
Н +
н+
Таким образом правило постоянства типа ориентации по Форлеидеру выра-
жается так: при вхождении второго заместителя в однозамещенное бензола второй
заместитель направляется имеющимся положительным элементом замещающей группы
-4- —
(в Н5С6Е) преимущественно в мета-место, отрицательным элементом (в Н6С6Е) преи-
мущественно в орто- и пара-места. Подвижность галоида в орто- и пара-иитрогалоидо-
замещеииых и малая активность в мета-двузамешенном объясняется противополож-
ностью зарядов углеродов, связанных с галоидом и нитрогруппой в I н II и одина-
ковостью зарядов в III (отрицательные остатки и атомы легче всего обмениваются
если стоят у наиболее сильно положительно заряженного углерода ядра).
Совершенно очевидно, что полярность атомов заместителя имеет
значение в ориентирующем влиянии его на замещение. Если сравнить
ряды углеродсодержащих замещающих групп
СН3(о, п), СН2С1(о, п), СНС12(лг), СС13(л<)
и
CH8(o,n), CHOH(o,n), СНО (л«), СООН(.и).
то перемена типа ориентации оказывается обусловленной измене-
нием полярности атомов, связанных с углеродом (вместо положи-
тельно заряженного водорода становится отрицательно заряженный
хлор или кислород).
Таблица 1
2 положительных заместителя 1.2 1.3 1.4
Имеются заместители . . + (1) + Третий заместитель входит в
NO, no2 5.4.2 —
NO, SO3H —. 5 —
no2 соан 4.5.3 5 3.2
SO3H SO3H — 5.4 —
SO3H СО2Н 4 5 2
со,н СО2Н 3.4 5.4 —
СНО со,н 4.6 — —
36
Форлендером собраны в форме таблиц (см. стр. 36 и ниже),
данные, касающиеся направления третьего заместителя при имею-
щихся двух, — случай вышеназванных совпадающей и несовпадаю-
щей ориентаций.
Таблица 2
2 отрицательных заместителя 1.2 1.3 1.4
Имеются заместители . -(1) Третий заместитель входит в
С1 Cl 4.3 4.2 2*
С1 сн3 4.6 4.6 2.3
С1 NH, 5 4.6 3
С1 ОН 5.3 4.6 3
Вт Вг 4.3 4.2
Вт СН3 4.53 4-2 (?) 2.3
Вг nh2 5 4.6 3.2
Вг он 5.3 6 3
J Вг 4 4 —
J J 4 (?) 4
J nh2 6
J он 5.3 —— 3
J сн3 4-5 (?) — 3
СН3 сн3 4.3 4.2
сн3 NH, 53 6.2.4 3.2
сн3 ОН 5.3 6.4 3
nh2 nh2 — 2.4 —.
NH3 он 5 4.6 (?) 3
ОН он 4 2.4.6 —•
Таблица 3
1 положит, и 1 отрицат. заместители 1.2 1.3 1.4
Имеются заместители . -Н1) NO, no; no; no2 no2 NO, SO3H SO3H SO3H SO3H CO2H CO2H CO2H CO2H CO2H CHO CHO CN CN N(CH3XjX N(CH3)3X Cl Br J CH3 nh2 OH Br CH3 NH.! Orf CI Br CH3 NH, OH Cl OH CH3 Br CH3 Br Третий 3.5 3.5 3.5 5.3 5.3 5.3 5 5 5 5 5.3 5.3 5.3 5 5.3 5 5.3 5 5 5 заместитель 6.2 6 6 (?) 4.6.2 6.4.2 4.2.6 6.4 4 4.6 4.6.2 4.6.2 4.6.2 2.4.6 4.2 4.6 6 4 4 входит в 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
* Введено автором.
37
В существе та же гипотеза Форлендера, выраженная в тер-
минах электронной теории (положительный заряд — потеря элек-
трона, отрицательный — прием электрона), лежит в основе тех
построений, которые приводят сторонники гипотезы „индуцириро-
ванной переменной валентности" 10). Быть может, в будущем раз-
работка этой гипотезы в свете новых данных о строении атома
приведет к возможности предвидения исхода реакции.
Отсылая к оригинальной литературе по этому
предмету, мы укажем здесь иа одну попытку двух
американских химиков Лэтаймера и Портера вы-
вести из этой гипотезы некоторые закономерности
в замещении („полуколичествениый метод опре-
деления полярности групп, связанных с бензоль-
ным кольцом* И).
Структура молекулы бензола выражается сле-
дующей схемой, где • • обозначают электроны,
которые попеременно в количестве 6 или 8 свя-
заны с атомами углерода. Если пара электронов
разделена между двумя атомами, то по предпо-
ложению авторов электроны за исключением слу-
чая с вэдородом квантуются в соответствии с по-
лем между атомами, и действие их зарядов рас-
пределено между атомами в отношении положитель-
ных зарядов их двух ядер.
Так, пара электронов между углеродом и азо-
том рассматривается, как дающая 2 X 4/9 электрон-
ных зарядов углероду и 2Х5/9 электронных зарядов азоту. Основание: поло-
жительный заряд углеродного атома, равный заряду ядра (6), уменьшенному иа
число невалентных электронов (2) = 4, то же для азота 7 — 2 = 5. Общий за-
ряд = 5 + 4 = 9. Распределение зарядов 2 электронов идет в указанном выше
отношении.
В случае связи между водородом и другим атомом авторы допускают, что два
электрона и протон так тесно сближены, что их остаточный заряд равен минус
единице.
Рассчитаем иа этом основании остаточный заряд атома азота анилина. Положи-
тельный заряд ядра атома азота = 5. Из него вычитаем заряд двух неразделенных!
электронов (2), четырех электронов, разделенных с водородом (4 X 1/г>' и ДВУХ< разде-
ленных с углеродом (2Х%). Остаточный заряд равен + 5 — 2—(4 X !/•:>)—(2X79)
= — 0,11.
Н
N :
Н
У нитробензола при таком расчете находим остаточный заряд (положительный заряд
ядра атома 0=6, сумма положительных зарядов ядер N и 0 = 11):
+ 5 - (4 X 71,) - (2 X 6/9) = + 2,07.
: 6 :
\ : N
./ ..
: О :
При аналогичном пересчете большого числа замещающих групп и атомов амЛ
рикаиские авторы получили таблицу остаточных зарядов и ориентирующих влияния
заместителей в бензоле:
3»
Таблица 4
Группы Остат. заряд Главное ориеит. влияние Группы Остат. заряд Главное ориент. влияние
-OR — 0,4 п о —СОС1 + 1,38 м
X (галоид) — 0,27 • —соон + М
-ОН — 0,2 —SO3H + 1,8
-NH, —NHCOR — 0,11 — 0,11 я —NO., —CHRNH2 + 2,1 -t-0,11 я П-'~О
—N—NC6HS — 0,11 » -С=ГСН2 т-0,2
—сн3 0 1
~Cnh2n_|_i 0 о—сн3
—с6н5 0 —N—О + 0,2 я
—сн»соон 0 —СН2Х + 0.27 я
—cr2no, + 0,41 -СНХ, + 0,55 + 0,65 »
-NHOH + 1,0 м —CBr2NO, м
-оно + 1,20 » -сх3 + 0,82 «
—сосн3 + 1,20 я +
—C=N + 1,2 -NH3 + 0,88
—CONИ, + 1,32
-CONHR + 1,32 -NR3 + 0,88 »
Из таблицы, видно, что если остаточный заряд атома, связанного с кольцом,
положителен и достаточно высок, то заместитель ориентирует в мета-положение.
Линия разграничения ие резка, и значения заряда между —{—0,1----------1-0,6 при-
водят к смесям трех изомеров (например СНС12). Этот интересный метод расчета
страдает, естесгвеиио, от того, чго в нем достаточно много допущений, поскольку
мы не знаем количества электронов, участвующих в двойной и одиночной связи.
Авторы этой схемы считают у двойной связи четыре электрона, у единичной два,
в углеродно-кислородной связи (СО) — два электрона, связь С и N в цианидной
группе выражается как : С:: N, а не тремя парами электронов.
В последующем изложении мы будем пользоваться не элек-
тронными, а обычными структурными формулами с учетом того
обстоятельства, что они выражают лишь предельное состояние мо-
лекулы.
Самое течение реакции замещения и многих реакций обмена
заместителей мы склонны объяснить в согласии с развитыми Гол-
леманом и другими взглядами как взаимодействие молекул ис-
ходного вещества и реагента, происходящее при сближении их до
стадии присоединения. Отщепление некоторых элементов из про-
дукта присоединения в виде простой молекулы (воды, галоидово-
дорода и т. п.) приводит к последнему стабильному продукту за-
мещения 12).
Заместители, входящие в ароматическое ядро, делают более
или менее активными двойные связи ароматического ядра и тем
содействуют ускорению образования продукта присоединения,
соотв. и замещения. Подробнее, применительно к отдельным реак-
циям, мы будем иметь возможность развить этот взгляд ниже,
здесь же ограничимся общей схемой. Если в бензольном про-
* Пара- и орто- в хлорировании, мета- в нитровании.
39
изводном имеется заместитель X (I), то в зависимости от хими-
ческой природы заместителя он влияет или на повышение или на
понижение способности присоединения у двойной связи 1 — би.
сопряженной двойной связи 13) 1 — 6 — 5»—4 в местах 1 — 4. Та-
ким образом из имеющихся в бензольном ядре двойных связей,
подвергаются воздействию заместителя или связь 1 — 6 или.
1—6—5—4, дающая эффект в местах 1 — 4, связь же 2—3 остается»
почти вне этого влияния. Если допустить, что наличный в ядре
заместитель X оказывает на первые две связи положительное-
(усиленное в сторону присоединения) влияние, то тогда при взаимо-
действии с активной молекулой реагента YZ могут образоваться
предварительно такие три продукта присоединения молекулы YZ
(II, III, IV):
Z Н
из которых первые два, благодаря названному влиянию замести-
теля X, образуются с гораздо большей скоростью, чем третий, в
потому преобладают в продукте реакции. Эти продукты при-
соединения молекулы YZ, часто нестойкие и не могущие быть
изолированными, отщепляют молекулу YH и переходят в продукты
замещения орто- и пара-
Z
/\ /х z\zx
| Па Ша
\Z z/’'v/
Если действие заместителя А по отношению к тем же системам
1 — би 1—6 — 5 — 4 (т. е. 1—4) отрицательно (понижение спо-
собности присоединения), то тогда из продуктов присоединения
первые два, скорости образования которых будут меньше такой
же для третьего, возникают и в меньшем сравнительно с ним ко-
личестве, и в виде конечного продукта по отщеплении HY мы
получим главным образом мета-замещенное:
z\/A
IVa
40
I
Z
Курт Мейер высказал по поводу явлений замещения взгляд 14), совпадающие
в большой мере с вышеприведенной точкой зрения Голлемана. На примера
соединений с двойной углеродной связью и группами ОН и ОСН3, соединений,,
выбранных и из жирного и из ароматического рядов, ему удалось экспериментально
доказать, что некоторые заместители определенным образом „активируют* двойную,
связь, делая ее особенно способной к реакциям присоединевия и через посредство
отщепления элементов какой-либо простой молекулы от продукта присоединения —
к реакции замещения. К. Мейер считает общим свойством двойных связей всех,
родов — способность активироваться рядом стоящими группами: ОН, ОСН3, NHa,
N(CHg)2- Замещение фенолов, соотв. энолей происходит через стадию начального
присоединения, чем объясняется одновремевно связь между пара- и орто-замеще-
ниями. Группы, ориентирующие в мета-положение, Мейер считает затрудняющими,
активность соседней двойной связи.
А. Е. Чнчнбабин 16), отрицательно отиос$сь к гипотезе многократных угле-
родных связей, принимает, что заместители водорода в бензольном ядре могут
быть более или менее ненасыщенными (склонными к присоединению); таковы
например NH?, ОН. Эта ненасыщенность радикала делает более насыщенвым трех-
атомный (соединенный с тремя атомами) углерод 1, соотв. меиее насыщенными
такие же углероды 2 и 6, бодее насыщеивыми углероды 3 и 5 и всего менее насы-
щенным атом 4. Отсюда можно ожидать наибольшей реакционности у пара- и орто-
атомов. Если заместитель менее, чем водород, иасьщает сродство атома С (на-
пример NQ2), то орто- и пара-атомы являются несколько более насыщенными по»
сравнению с мета-атомами, почему такие заместители ориентируют в мета-положение..
Мы увидим в специальной части примеры активирования
двойной углеродной связи в ароматическом соединении со стороны
некоторых реагентов, применяемых при замещениях (H2SO4, А1С13,
FeCl3 и др.), так что кроме внутримолекулярного влияния на
взаимодействие оказывают влияние и внешние агенты.
Ввиду большого разнообразия тех реакций, которые могут по-
вести к образованию нового заместителя в ароматическом ядре,,
будет ли он образован заменой водородного атома ядра или пре-
вращением уже имеющегося заместителя, иногда возможен ши-
рокий выбор среди тех способов, какими получаются некоторые
из промежуточных продуктов.
Наиболее просто построенные производные (одно- и двузаме-
щенные) могут быть образованы часто многими и разнообразными
путями. Техника выбирает из этих путей те, которые экономически
оправдываются достаточно хорошим выходом продукта, наличием,
сырых материалов, доступностью реагентов, относительной про-
стотой аппаратуры и меньшей сложностью процесса переработки.
Поэтому способ, практически выгодный в одном районе, может
оказаться для другого непригодным. Те же экономические осно-
вания лежат в основе прогресса методики работы. Переход к не-
прерывным методам производства, хотя в иных случаях неизбежна
сопровождается усложнением аппаратурного оформления, открывает
зато возможность получения в единицу времени ббльших количеств-
продуктов и становится поэтому выгодным.
При всех работах по получению промежуточных продуктов и
красителей как в лабораторном масштабе, так и в производстве-
необходимо считаться с тщательным соблюдением всех условий,,
наиболее благоприятных (оптимальных) для данного процесса.
Часто весьма незначительное отклонение от этих
условий приводит к полному нарушению правиль-
ного хода процесса. Температурные пределы, концентрация
и порядок прибавления реагирующих веществ, качество сырых
41
материалов, влияние посторонних химических реагентов, в том
числе и материала, из которого сделана аппаратура, скорость про-
ведения процесса — все это должно строго учитываться при раз-
работке методики, или, как говорят, рецептуры, процесса.
Установление производственной рецептуры требует как правило
продолжительного и очень тщательного изучения процесса в лабо-
ратории для установления всех подробностей его течения в опти-
мальных условиях и отклонений от нормы, вызываемых измене-
ниями отдельных его факторов. При этом необходимо учитывать
и регистрировать все малейшие особенности отдельных стадий
процесса: в случае выделения осадка — скорость его образования,
легкость оседания, отфильтровывания, его объем, отнесенный
к единице веса, скорость и допустимую температуру высушивания:
при перегонках — систему перегонки, температурные пределы и
количество фракций, их состав и т. п.
Лабораторно разработанный рецепт проверяется далее в усло-
виях проведения процесса в аппаратуре, близкой по материалу
к заводской, и в масштабе, в 20—50 раз большем, чем лабора-
торный. Задачей этой проверки на опытно-заводской установке
является совмещение лабораторного опыта и заводских приемов
работы и обнаружение тех неясностей в первоначальном рецепте,
которые не могли быть подмечены в начальной стадии работы.
Само собой понятно, что во всех этих стадиях разработки, так же1
как и при производственной работе, большое значение имеет ана-
литический контроль отдельных стадий процесса, и на эту сторону
вопроса необходимо обращать внимание уже с самого начала.
Понятно также, что производственный рецепт, установленный
после всего этого изучения, требует тщательного выполнения и
не может быть изменен по произволу.
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) Об ориентации у нафталина: V. V е s е 1 У, М. J a k е §, Bull: (4) 33, 954 (1923);
Ю. Залкинд, Ж. О. X., I. (63), 158 (1931). Ср. Н. Н. Ворожцов, Ступени
в синтезе красителей, стр. 94—95, Л., 1926.
2) VorlSnder, Вег. 52, 263 (1919).
3) F. Challenger, A. D. С о 11 i n s, Soc. 125, 1377 (1924), СВ 1924, II, 829.
“) Pauli, J. pr. Ch. (2) 93, 106 (1918).
5) D. L. Hammick, W. S. Illingworth, Soc. 1930, 2358.
6) S. Veibel, Ber. 63, 2074 (1930).
’) Г. n. 98637 (T. M.). F. V, 67.
8) Г. n. 101778 (M. L. B.), F. V, 68.
9) D. VorlSnder, Die Lehre von den innermolekularen Gegensatze und die
Theorie des Benzols, Ber. 52, 263 (1919); H. S. Fry, Zt. phys. Ch. 76, 385,
398. 591 (1911), Am. Soc. 36, 248 (1914).
10) R. Fraser, J. E. Humphries, The Probleme of Substitution in the Ben-
zene Nucleus and the Thomson-Lewis-Langmuir Theorie of Covalence, Chem.
News 1923, 163; Lapworth, Soc. 121, 1391 (1922); J. Stieglitz, Am. Soc.
44, 1299, (1922); R. Kuhn, A. Wasserman n, Uber die Polaritat von Substi-
tuenten am Benzolkern, Helv. 11, 3 (1928,. Электронную интерпретацию
реакций органической химии проводят А. М. Беркеигейм, Я. И. Михайленко
и Б. П. Тронов.
И) W. М. Latimer, С. W. Porter, The Polarities and the orienting Influences
of Sustitution in the Benzene Ring, Am. Soc. 52, 206 (1930).
12) Holleman, Die direkte Einfiihrung von Substituenten in den Benzolkern,
стр. 476, Leipzig, 1910.
I3) J. Thiele, Ann. 306, 94 (1899).
14) к. H. Meyer, Zur Kenntnis der Substitutionsvorg3nge, Ber. 54, 2265 (1921).
15i A. E. Ч и ч и б а б и н, Ж. 43, 1704 (1911).
ГЛАВА II
НИТРОВАНИЕ И НИТРОЗИРОВАНИЕ
а) Течение, реагенты, факторы нитрования
Нитрование (иногда называется нитрацией) — один из наиболее
давно известных методов в практике синтеза промежуточных
продуктов. Датой введения нитрования в круг химических реакций
можно считать 1834 г. — открытие Митчерл ихом нитробензола
при действии азотной кислоты на бензол.
Цель нитрования—замена одного или нескольких водородных
атомов ароматического ядра посредством группы, соотв. групп,
нитро ИО2. Ввиду большого значения нитропроизводных углеводо-
родов (бензола, толуола, нафталина) и многих их замещенных
в практике производства красителей, с одной стороны, и чрезвы-
чайной важности некоторых из полинитросоединений как взрыв-
чатых веществ военного значения (тринитротолуол, динитробензол,
тринитрофенол, динитро- и полинитронафталины, тетранитро-
метиланилин и пр.), с другой, нитрование является процессом, при-
меняемым в очень больших масштабах и достаточно технически
разработанным.
Реагентом нитрования является азотная кислота (в готовом виде
или образуемая в момент реакции—из солей, смешанных ангидридов
и пр.), поэтому общему выражению реакции можно придать вид:
Н:Н-:-нд NO,==RNO2-j Н2О (1)
как реакции отнятия воды (здесь RH — ароматический углево-
дород— или его замещенное, — выменивающий свой водородный
атом на группу NO2).
Нужно иметь однако в виду, что это выражение дает пред-
ставление лишь о результате, а не о течении реакции. Последняя
протекает повидимому всегда через несколько стадий, среди ко-
торых можно различить стадию присоединения и стадию стабили-
зации, последняя уже дает нитропроизводное (ср. стр. 40). Обра-
зование продуктов присоединения сопровождается резкой окраской,
характерно проявляющейся при прибавлении концентрированной
азотной кислоты к углеводородам (красной для бензола)1). Представим
течение реакции с бензолом при участии одной лишь азотной кислоты:
(2)
43
В случае особенно реакционных в смысле присоединения веществ удалось вы-
делить продукты присоединения азотной кислоты. Так, из антрацена получен мезо-
ннтроднгидроантранол, дающий с отщеплением воды (от действвя щелочи) мезо-
ннтроантрацен 2)
Отмечено получение при нитровании нафталина вещества, которому приписы-
вается также строение продукта присоединения 3).
В тех реакциях нитрования, где помимо азотной кислоты принимает участие и
серная кислота, а таконы почти все технически осуществляемые реакции нитро-
вания, можно допустить, что серная кислота играет роль водоотнимающего сред-
ства, ио вероятно помимо этой функции она активно участвует в образовании про-
дукта присоединения. Ввиду ее большой активности по отношению к ароматиче-
скому ядру нужно думать, что первым продуктом, активирующим двойные связи
углеводорода, становится продукт присоединения именно серной кислоты, который
затем либо меняет свою сульфогруппу на нитро (H2SO4 вытесняется азотной кисло-
той; схема Б) либо дает новый продукт с участием и нитро- и сульфогруппы н в
момент стабилизации теряет вместе с нодой и молекулу серной кислоты (схема А).
Вероятность протекания реакции по этой схеме (А или Б) отчасти подтвер-
ждается констатированной на многочисленных примерах склонностью сульфогруппы
уступать место ннтрогруппе, особенно при наличии реактивирующей ядро группы
ОН. Так сульфо- и дисульфокислоты фенола дают при нитронании пикриновую
кислоту.
Далее недавно на производных 4 • 4'-диалкоксндифениламина
AlkO-:
было наблюдено, что окрашенные продукты присоединения серной кислоты обра-
зуются и у тех производных, которые не дают продуктов присоединения с азотной
кислотой, между тем как все соединения, присоединяющие азотную кислоту, при-
соединяют и серную кислоту, следовательно способность серной кислоты давать
продукты присоединения выше в этом случае таковой для азотной кислоты 4).
44
Образование промежуточного соединения [схема (3, I)J делает
вероятным допущение, что стабилизация его из дигидробензоль-
ного в бензольное соединение может итти по двум путям. Пер-
вый, рассмотренный выше, сопровождается отщеплением воды и
приводит к нитрозамещенному. Второй сопряжен с отщеплением
элементов азотистой кислоты, в результате чего получается фенол
(особенно в условиях недостатка водоотнимающего реагента)
Такое направление при реакции с азотной кислотой констати-
ровано особенно в условиях отклонения процесса нитрования (по
температуре, концентрации нитрующего реагента) от нормы s).
Из вышеприведенного общего уравнения нитрования (I) сле-
дует, что при образовании каждой нитрогруппы выделяется одна
молекула воды. Входя в реакционную смесь, вода понижает кон-
центрацию азотной кислоты и мажет довести ее до такой, когда
азотная кислота уже не будет далее нитровать. Вместе с тем раз-
веденная азотная кислота оказывает более окислительное, чем
нитрующее действие на органическое соединение, в отличие от
концентрированной азотной кислоты. С понижением концентрации
азотной кислоты, при нитровании становится заметным выделение
окислов азота, т. е. резче выступает то течение реакции, которое
выше охарактеризовано схемой (4). Для того чтобы избежать
вредного влияния разведения азотной кислоты и сэкономить на
азотной кислоте, прибегают к введению в реакционную массу в
качестве водоотнимающего вещества концентрированной серной
кислоты. Участие ее в реакции делает более вероятным течение
взаимодействия по схеме (2) или, вернее, (3), при этом серная
кислота не только обеспечивает сохранение концентрации нитру-
ющего агента, но и направляет течение процесса.
Смесь азотной и серной кислот, заранее приготовленная,—
наиболее употребительный реагент в нитровании и носит название
нитрующей смеси или нитрующей кислоты. Ее немало-
важное техническое преимущество перед азотной кислотой со-
стоит в том, что она может быть сохраняема в железной аппара-
туре и передаваема по железным трубам, так как не разъедает
этого металла. Количество серной кислоты в нитрующей смеси
рассчитывается по минимальной крепости (общей кислотности)
смеси, при которой еще заметен нитрующий эффект. Если увели-
чить количество азотной кислоты при сохранении того же коли-
чества серной кислоты в правильно составленной нитрующей смеси,
то часть азотной кислоты останется вне взаимодействия. Если уве-
личить количество серной кислоты, это вызовет лишний расход
серной кислоты в отработанной кислоте. На заводах часто при со-
ставлениях нитрующей смеси пользуются отработанной кислотой
предыдущих нитрований, соответственным образом ее подкрепляя
45
добавкой более крепкой серной и по расчету азотной кислоты.
Азотная кислота вводится в смесь примерно в теоретически рас-
считанном количестве с добавкой против расчета в 1—5°/о. Бывают
однако нитрования, в которых избыток HNO8 значительно выше
(10—20%). Таковы например нитрования некоторых полисульфо-
кислот нафталина. Концентрацию серной кислоты в нитрующей
смеси необходимо выбирать в зависимости от большей или мень-
шей реакционности нитруемого вещества и количества нитро-
групп, которое имеется в виду ввести; для получения мононитро-
соединения при прочих равных условиях требуется менее крепкая
кислота, чем для динитросоединения, и для последнего — менее
крепкая, чем для тринитропродукта. Обычно в практике как наи-
более слабая серная кислота применяется купоросное масло (92—
93% H2SO4), далее идет моногидрат (100<>/о-ная H2SO4), олеум
(дымящая серная кислота) с разным содержанием свободного SOs
(10—2О°/о и выше) в зависимости от условий нитрования. В прак-
тике важным фактором при расчете количеств реагентов является
крепость той кислоты, которая остается в результате нитрования
(отработанной, содержащей серную кислоту, воду, окислы азота и
иногда избыток азотной кислоты). Понижение ее крепости вы-
годно с точки зрения экономии серной кислоты, но может, если и
не отразится отрицательно на успехе нитрования, вызвать не-
удобства в работе из-за разъедания кислотой низких концентра-
ций железной аппаратуры. При мононитрациях углеводородов
кончают нитрование при крепости отработанной кислоты в 68 —
72°/о, считая на серную кислоту (примерно отвечает содержанию
H2SO4 4~2НаО); при высших степенях нитрования крепости отрабо-
танной кислоты могут быть выше.
Американец Г рог г н нс, много занимавшийся вопросами нитрования, рекомен-
дует при составлении нитрующей смеси руководствоваться получением в отрабо-
танной кислоте благоприятного отношения между сериой кислотой и водой. Это
отношение он называет отношением водоотиятия 6) серной кислоты * *. Отношение
водоотнятия (ОБ) выражается числом, полученным разделением действительного
количества серной кислоты в иитросмеси на количество всей воды, которая имеется
после проведения нитрования. В последнее число (в знаменатель) входит не только
количество образовавшейся при реакции воды, ио и воды, введенной с нитросмесью
и углеводородом.
Если например смесь состава:
H2SO4 - 6О°/о, HNOg - 320/0, Н2О — 8%
применить для нитрования бензола, то при расчете реакции и молекулярных отно-
шениях
СвН6 + HNO3 = Н,о + C6H5NO2
78 63 18 123
18» 32
на каждые 100 кг иитросмеси (с 32%-ной HNO3) образуется —— = 9,14 кг воды
Do
от реакции. При учете 8 кг воды в самой иитросмеси и 60%-ного содержания
в ней H2SO4
OB-j^-3.51.
*) OB = дегидратирующее значение серной кислоты (dehydrating value of sui-
furic acid).
46
78 • 32
100 кг нашей ннтросмеси отвечает —— = 39,62 кг бензола.
ЬЗ
18 • 100
100 кг бензола отвечает —=-— = 23,08 кг реакционной воды,
/о
В производстве отношения между углеводородом и азотной кислотой заранее
вычисляются, и делается прибавка азотной кислоты (или уменьшение количества
углеводорода) сравнительно с расчетом по теории. Если взять углеводорода 95%,
сравнительно со стехиометрическй требуемым количеством, то 100 кг нитросмеси
95 • 78 • 32
(с 32% HNO3) будут соответствовать —'— — 37,7 кг бензола и 8,71 к&
Do ♦ 100
реакционной воды.
Соответственно этому для практического процесса
ОВ = -^
3,65.
Контроль ОВ имеет большое практическое значение и обеспечивает получение при
нитровании высоких выходов. Отклонение от правильного отношения ОВ приводит
к понижению выхода.
Для получения дивитробензола рекомендуется смесь с большим избытком азот-
ной кислоты (в 10%) и с содержанием 80% H2SO4, 18% HN.O3, 2% Н2О; ОВ = 7,
Для мононитрования хлорбензола — смесь с 71% H2SO4, 18% HNO3 (избыток в 1%),
11% Н2О; ОВ = 4,44. Для мононитроваиия толуола — смесь с 58,7% H2SO4, 23,8%
HNOg (избыток в 1%), 17,5% Н2О; ОВ = 2,42. Для мононитровання нафталина — смесь
с 59,55% H,SO4, 15% HNO3 (избыток в 1%), 24,60% Н2О; ОВ = 2,04.
Кроме смеси азотной и серной кислот в качестве нитрующего
агента могут иметь значение некоторые иные реагенты, о кото-
рых частью будет сказано ниже. Наиболее простой заменой нор-
мальной нитрующей смеси может быть смесь из серной кислоты
и соли азотной кислоты (чаще всего натриевой — чилийской се-
литры). При нагревании такой смеси с нитруемым веществом сер-
ная кислота образует с металлом катиона селитры бисульфат, вы-
деляя азотную кислоту, которая и нитрует:
NaNO3-j-HsSO4 = HNO3%NaHSO4 7).
В качестве отхода нитрования накапливается бисульфат натрия,
не находящий себе большого спроса. Если имеются на заводе де-
нитрационное (для освобождения от остатков HNO3 и окислов
азота) и концентрационное устройства для отработанной кислоты,
то способ нитрования с селитрой не имеет преимуществ.
Применялась также, и в широком масштабе, смесь из аммиач-
ной селитры NH4NO3 и серной кислоты. Судя по наблюдениям
А. В. Степанова, такое нитрование особенно удобно для получе-
ния полинитросоединений, так как реакция протекает гладко и
без осложнений 8). Недостатком является высокая стоимость ам-
миачной селитры, которая частично может быть компенсирована
применением отхода—аммиачной соли серной кислоты — для полу-
чения удобрения.
Интересным, ио не имеющим производственного значения реагентом нитрования
является смешанный ангидрид уксусной и азотной кислот, ацетилннтрат, CH3COONOS.
Он энергично нитрует соединения, не поддающиеся нитрованию обычными кислот-
ными смесями. Образование нитропродукта сопровождается выделением, амаа—ммда
молекулы уксусной кислоты9)
RH + CH3COONO2 = RNO2 + Cl^COOH.
47
При нитровании ацетнлиитратом групп, NO2 становится преимущественно
в орто-положеиие к имеющемуся заместителю. Вместо готового ацетилнитрата (он
готовится взаимодействием уксусного и азотного ангидридов) можно с аналогич-
ными результатами применять смесь концентрированной азотиой кислоты с уксусным
ангидридом10).
Нитрующе действует также смесь уксусного ангидрида (или хлорангидрида
уксусной кислоты) и азотнокислых солей металлов. Применение нитратов тяжелых
металлов (Си. Ре) уже при низкой температуре (ниже 60%) дает быстрое течение
реакции и хорошие выходы интросоедниений у легко осмоляемых в обычных усло-
виях нитрования соедииеинй [амины, фенолы Щ]. Металл нитрата повидимому не
безразличен по отношению к исходу реакции, и перемена металла может повести
к различию в соотношении изомерных нитропродуктов i2). Вероятно нитрующим
агентом здесь оказывается также ацетилнитрат (для нитратов тяжелых металлов) или
диацетнлортоазотная кислота (CH3COO)3N)OH)3.
Предложение нитровать азотнокислыми солями в растворе или суспенсии в без-
водной или высококонцентрированной фтористоводородной кислоте13) конечно не
имеет практического значения.
Также вряд ли можно считать практически интересным применение нитрознл-
сериой кислоты NO2S33H в качестве водоотнимающего средства при нитровании
азотной кислотой за исключением ниже разобранного метода нитрования окислами
азота, где нитрозилсерная кислота является попутно образуемым продуктом взаимо-
действия. Нитрующая смесь с дымящей азотной кислотой проявляет сильное оки-
сляющее действие 14).
Весьма важным фактором при нитровании помимо концентрации
нитрующего агента является температура. Для каждой отдельной
реакции существует максимальная температура, выше которой
нельзя переходить без опасения повредить исходу реакции. Даже
незначительное повышение температуры сверх предельного макси-
мума сопровождается усилением окисляющего действия азотной
кислоты, характеризующегося выделением газообразных окислов
азота, и понижает тем нитрующий эффект.
Один из американских исследователей полагает, что при нитрованиях вообще
теряется заметное количество азотиой кислоты в виде продуктов восстановления:
им при нитровании фенола в пикриновую кислоту открыты в отработанной кислоте
такие окислы азота: NO3, NO, N3O, и констатировано восстановление до азота.
Наиболее сильное газообразование наблюдалось в первую стадию нитрования (на
мононитропродукт). Прибавление железного купороса усилило выделение газов46).
Повышение температуры выше определенного предела как-раз
содействует восстановлению азотной кислоты за счет органического
нитруемого вещества.
Отмеченное в литературе восстановительное влияние электрического тока (иа
аноде) на нитрование, в частности возможность нитрования например нафталина
при применении азотной кислоты пониженной концентрации, повидимому зависит
по преимуществу от повышения при электролизе температуры на аноде вследствие
идущего окисления органического вещества. Поэтому вещества, для которых темпе-
ратурный максимум нитрования высок, могут дать ннтропродукт с хорошим вы-
ходом. Аналогичные результаты получаются при повышении температуры (при той
же концентрации кислоты» и прн чисто химическом нитровании16).
Так как реакция нитрования экзотермична, то, с одной стороны,
необходимо во многих случаях нитрования прибегать к внешнему
охлаждению аппарата, в котором проводится реакция, с другой
стороны, практиковать медленное, порционное смешение нитрую-
щей смеси и нитруемого углеводорода. При введении первой ни-
трогруппы допустимый максимум температуры ниже (следовательно
требуется большее охлаждение), чем при введении следующих
нитрогрупп.
48
Влияние на реакцию нитрования катализаторов систематически
не обследовалось. Наиболее известен своеобразный эффект, вызы-
ваемый присутствием ртути и ртутных солей на смесь аромати-
ческого соединения и азотной кислоты, в результате которого из
углеводородов образуются нитро-, соотв. полинитрофенолы п).
Таким образом из бензола можно получить динитрофенол
и пикриновую кислоту.
По мнению первых исследователей этой реакции Вольфенштейна и Бе-
те р с а образование из бензола пикриновой кислоты происходит по уравнению,
в котором ртуть не участвует:
С6Н6 + 4HNO3 = C6H2(NO2)3OH -f- ЗН2О -f- HNO2.
Реакционное значение прибавки ртути до сих пор не совсем
ясно. Всего вероятнее, ртуть активирует ароматическое соединение,
давая с ним нестойкие, разрушаемые азотной кислотой, продукты
меркурирования RHgX, где X — гидроксил или остаток азотной
кислоты. Некоторые исследователи полагают, что ртуть не является
ни катализатором окисления, ни катализатором нитрования и что
группы N0.3 и ОН вступают одновременно в частицу при разложе-
нии предварительно образовавшегося комплексного соединения
азотнортутной соли с углеводородом 18).
Имеются требующие экспериментальной проверки указания на образование при
реакции днфенилртути (C6H6)2Hg, которая якобы дает 2.4.2'.4'-тетранитродифенил-
ртуть, переходящую от действия азотной кислоты в динитрофенол и азотнокислую
соль ртути!»). Захаров20) видит в ртутн катализатор преимущественно нитрования
при достаточно крепкой азотной кислоте в реакционной смеси, окислительный же
эффект выступает лишь при наличии разведенной азотной кислоты. Роль катализа-
тора согласно этому исследователю сводится к активации бензольного ядра с раз-
рывом двойных связей.
Уместно отметить, что применение нитратов ие ртутн, а иных металлов: Мп, А1,
Си, Zn, дает худшие результаты в отношении получения нитрооксисоединеиий1)9.
Подобный ртути по каталитическому влиянию реагент не был найден также
в РЬ, Ag, As, V, Се, U18).
Подробное обследование прибавки азотнортутной соли Hg(NO3)2 к реакционной
смеси при нитровании различных ооганических соединений 'углеводородов, галоид-
ных производных, фенолов, альдегидов^ при разных составах нитрующей смеси
дало основание английским авторам Файлис и Мак Кайю сделать вывод также
о положительном катализирующем значении прибавки ртути в нитровании. Эта при-
бавка реактивирует органическое вещество н приводит к повышенным выходам
нитросоединеиий. Окислительное течение реакции не идет за счет нитрования и
при поддержании надлежащей температуры вообще не на много превышало идущее
и в отсутствие ртути окисление
Влияние на соотношение изомеров в нитропродуктах присутствия солей ртути
и некоторых других металлов было прослежено и не привело к положительным
результатам применительно к толуолу (в качестве прибавок вводились ртуть и ее
соли, медь и ее соли, никель и его соли, соли металлов: Со, Pb, Zn, Cd, Мп, Bi, U,
соединения Wo, V, борная кислота\ причем во всех случаях получены примерно
одинаковые количества о-нитротолуола, мало отличающиеся от того, которое дает ии-
4 Зак. 3611. — Н. Ворожцов
49
тровавие без прибавок. Нитрование в присутствии ртути нитробензола не отличается
по выходу Jt-динитробензола от проведевного без ртутвой соли.
Нитрование ксилола слабой азотной кислотой в присутствии ртути не обнару-
жило заметного влияния этой прибавки на образование оксипродуктов. Образование
ж-нитрокрезола прн обработке азотной кислотой и ртутью толуола происходит в мень-
шей степени, чем аналогичное превращение бензола в нитрооксипродукты.
Отклонения в поведении антрахинона, соотв. ^-метилантрахинона, при нитрова-
нии без ртути и с прибавкой ртути очевь неотчетливы
Обработка азотной кислотой бензойной кислоты в присутствии азотнокислой
ртути приводит, как и в случае бензола, к образованию оксиполинитропродукта,
в данном случае — 2.4.6-тринитрооксибензойнсй кислоты 23;.
NOa
^/СООН СООН
O2N/Z\/X'NO2
ОН
В связи с вышесказанным кажется неожиданной характеристика, даваемая
Фирц-Давидом действию окиси ртути на азотную кислоту при нитровании:
по нему прибавка окисн ртути устраняет какое-либо окисляющее действие при
образовании нитрата анилина, и при ее участии можно витровать гидрохинон и).
Природа нитруемого вещества имеет очень большое значение
как для хода, так и для результатов нитрования. Часто говорят
о легко нитруемых или трудно нитруемых соединениях, подразуме-
вая под этим, что первые в отличие от вторых требуют менее
энергичных реакционных воздействий (главным образом меньшей
концентрации нитрующего реагента, температурный максимум ле-
жит ниже), чем у вторых. В большинстве случаев скорость проте-
кания реакции у первых больше, чем у вторых.
К сожалению, до сих пор не имеется еще достаточного мате-
риала для полной физико-химической характеристики отдельных
реакций нитрования. Известно лишь, что большое значение для
характера реакции имеют в нитруемом веществе те атомы или
группы, которые замещают водородные атомы ароматического ядра.
Некоторые из них, принадлежащие к ряду ориентирующих в орто-
и пара-положения, реактивируют ядро ароматических соединений и
облегчают вход нитрогруппы, таковы: ОН, ОСН3, СН8, NHAc и
некоторые другие. Другие заместители, главным образом принад-
лежащие к ряду ориентирующих в мета-положения (КО2, SO8H,
СООН, CN), и галоидный атом из ориентирующих в орто- и пара-
положения делают реакцию более затрудненной.
Кинетика реакции нитрования некоторых производных бензола
была прослежена Мартинсеном, причем реакция велась в среде
серной кислоты 25).
Основные выводы работы Мартинсена:
1. Скорость реакции нитрования нитробензола зависит от состава серной кислоты,
служащей средой. Максимального значения скорость достигает нри составе
H2SO4 • 0.7НаО, понижаясь как при понижении, так и при повышении концентрации
серной кислоты против этого предела.
2. Скорость нитрования замещенных бензола находится в сильной зависимости
от характера заместителей. Эти последние могут быть размещены в ряд
NO2 > SO3H > СООН > Cl < CHS < ОСН3 < ОС,Н5 < ОН,
50
причем стоящие вправо от хлора ускоряют реакцию и тем больше, чем далее
вправо они стоят, стоящие же влесо замедляют реакцию все с большей энергией
по мере удаления от хлора.
3. Нитрование фенола в водном растворе — сложная автокаталитическая реак-
ция. Скорость ее возрастает с повышением концентрацви кислоты и уменьшается
с ростом концентрации фенола. Азотистая кислота — сильный положительный ката-
лизатор этой реакции.
Тронов и Бер изучали влияние заместителей на скорость нитровавня за-
мещенных бензола в растворе нитробензола (2 мол.) одним молем HNO3 при ком-
натной температуре 2е), причем пол ученные ими ряды отличаются от вышеприведенных.
Ряд для момента израсходования 5% HNO3:
NO2>CeH5CO>CN>CNCH2>C6H&CH2>CH3>H<C2H5<Br<CH2Ci<Cl<CH3CO
Ряд для момента израсходования 15% HNO3:
... С6Н5СН2 > СН3 > С2Н5 > Н < Вг < С1 < СН2С1 < СН3СО.
Ряд для момента израсходования 25% HNO3:
Cl > Вг > СН3 > Н < С2Н6 <... <СН3СО.
Ряды здесь изображены соответственно вышеприведенному ряду М а р т и в-
с е и а. Центральное место занимает здесь Н, отвечающий незамещенному бензолу.
В этих рядах поражает прежде всего то обстоятельство, что СН3СО-группа, мета-
ориентирующая, является наиболее ускоряющей нитрование.
Любопытны переходы группы по рядам в зависимости от течения нитрования:
С2Н5, замедлявшая вначале, далее ускоряет, затем опять замедляет. Галоиды сначала
ускоряют, потом замедляют. Метил (замедляющий здесь) при 2-кратном, а особенно
3-кратном вхождении сильво ускоряет: о-ксилол нитруется в 1,6—1,9 раз, лг-ксилсл—
в 4,5—4,9, л-ксилол—в 5,7—10,5 раз скорее толуола. Весьма вероятно, что реакци-
онная среда (нитробензол и отсутствие серной кислоты) отразилась здесь на реак-
ционности растворенных в ней производных, может быть и на механизме реакции.
Из отдельных замещенных бензола много внимания было посвя-
щено исследованию нитрования фенола. Фенол принадлежит к наи-
более легко нитруемым веществам и дает мононитропродукты уже
при действии разведенной азотной кислоты.
О каталитическом значения азотистой кислоты при этом было уже упомянуто.
Недавние исследования этого вопроса указывают на необходимость присутствия
азотистой кислоты для гладкого нитрования фенола 2?). Фейбель считает, что
промежуточной фазой при этом является нитрозофенол (см. иитрозированне) в виде
орто- и пара-изомеров, образующийся из продукта присоединения азотистой кислоты
к фенолу и, далее, с окислением переходящий в нитрофенолы (о- и п-).
Выводы его исследования: 1) концентрация азотистой кислоты повидимому не
влияет на отношение количеств о- и п нитрофенолов;
2) увеличение концентрации азотной кислоты благоприятствует образованию
орто-продукта;
3) при концентрациях азотной кислоты в 1—3 N при 25° получаются приблизи-
тельно одинаковые количества орто- и пара-продуктов.
Толуол при нитровании может давать замещение нитрогруппой
как в ядре, так и в метильной группе. При обработке нитрующей
кислотной смесью достаточной крепости получается смесь о- и п-
нитротолуолов с преобладанием орто- и некоторой примесью мета-
изомера. При обработке же азотной кислотой при высокой темпе-
ратуре толуол частично окисляется в бензойную кислоту, а частью
дает замещение в метильной группе и образует фенилнитрометан
С6НбСН2КО22«).
Метод обработки разведенной азотной кислотой при высокой
температуре дал возможность М. И. Коновалову получить ни-
тросоединения парафиновых углеводородов.
4*
51
Для получения феинлннтрометана н феннлинтроэтана C6H6CH(NO2)CH3 Ш о-
рыгнн н Соколова нашли метод более удобный и дающий лучшие выходы,
используя разведение азотной кислоты не водой, а уксусной кислотой; прн этом
отпадает необходимость применения запаянных тр/бок, и можно работать в открытых
сосудах. Прибавка ртутных солей в этой реакции, оказалось, содействует вхожде-
нию иитрогруппы в ядро и не дает ни продуктов окисления, ни замещения в боковой
цепи 2Э).
При -проведении нитрования толуола в условиях повышенной
против нормы температуры (от недостатка охлаждения) наблюда-
лось образование диазо- и даже азосоединений (от действия
азотистой кислоты, соотв. закиси азота, на толуол; ср. ниже нитрози-
рование).
Окисление метильной группы толуола от действия разведенной азотной кислоты
можно представить как переход через фазу образования феинлннтрометана такой
схемой:
C6H5CH3->C6H5CH2NO2->C6H6C(: NOH)OH->C6H5COOH -f- NH2OH,
причем гидроксиламни NH,OH перешел бы в NO нли N2O от азотной нли азоти-
стой кислот з°).
Паранитротуол от действия разведенной азотиой кислоты прн высокой темпе-
ратуре гладко переходит в п-нитробанзойную кислоту, о-нитротолуол дает небольшой
выход (18%) о-ннгробеизойной кислоты и значительно больше пикриновой.
Нитрование близкого к толуолу по строению хлористого бен-
зила С6Н6СН2С1 приводит, как это недавно было проверено, к по-
лучению 52,6% пара-, 32,1% орто-, 15,3% мета-нитропродукта 3l).
Нитрование аминов вследствие влияния очень реакционной амино-
группы может осложниться побочными реакциями с образованием
продуктов окисления, конденсации. Нужно учитывать при этом, что
сама группа NH.2 при образовании солеобразного соединения с со-
ставными частями кислотной нитрующей смеси может частично
v ш
ориентировать в мета- (N) и в орто- и пара-положения (N).
Поэтому для получения однообразно ориентирующего замести-
теля аминогруппу чаще всего стабилизируют или, как говорят,
„защищают", вводя вместо водородного атома ацильную группу,
или иначе (см. ниже ацилирование). Такая стабилизированная амино-
группа меньше активирует ядро и дает меньшее количество изо-
мерных нитросоединений.
Прн нитровании аминов наблюдалось образование (может быть всегда предше-
ствующее включению иитрогруппы в ядро) нитраминов RNHNO2, которые особенно
легко образуются при наличии группы NO2 в мета-положении по отношению к NH2.
Так лт-нитроанилин в крепкой нитрующей смеси (нз 30%-ного олеума н дымящей
азотной кислоты уд. в. 1,52) дает сначала нитрат, а затем, с потерей воды, нитро-
амнн 32).
RNH2HNO3 —> RNHNO2.
Применение крепкой серной кислоты или олеума для раство-
рения амина и последующее нитрование может служить тоже
средством стабилизации аминогруппы, переводя амин в серно-
кислую соль. Ориентация в этом случае — в мета-положение,
в нафталине — в другое ядро88).
При образовании нитропродукта из замещенных ароматических
углеводородов подмечено стремление нитрогруппы во многих, но
не во всех, случаях встать в более близкое положение к имею-
52
щемуся заместителю (орто- для бензольных соединений), хотя бы
главная ориентация была направлена в другое место (мета- для
бензольного ядра).
Так, толуол дает преимущественно о-ннтротолуол (около 60%) (I), нитробензол
прн нитровании образует л-динитробензол, но вместе с тем заметное (7—12%)
количество орто-продукта (11) н меньшее пара-продукта34).
Нитрование сульфокислоты бензола приводит к образованию лт-нитробензол-
сульфокислоты наряду с значительной примесью о-сульфокнслоты (111),
SO3H /x/so3H
+ I I ;
\/\no2
между тем как сульфирование нитробензола доставляет практически только лт-ннтро-
су.иьфокислоту бензола. Нитрование нафталина практически приводит к образо-
ванию лишь а-нитронафталнна (IV), а нитрование последнего продукта к смесн 1.5-и
1.8-дннитронафталинов с заметным количественным преобладанием изомера 1.8
(пери-положение, по близости аналогичное орто-положению).
Также прн нитровании бензальдегида наряду с главным, мета-нитропродуктом,
получается много орто-нитроизомера ®).
Все эти и подобные факты дали повод говорить о „самоориен-
тации нитрогруппы", подразумевая под этим указанное стремление
ориентироваться на наиболее близкое к заместителю место36).
Уместно отметить, что имеются факты, не отвечающие этой пра-
вильности. Так, нитрование фенола приводит к смеси приблизи-
тельно в равных отношениях о- и л-нитрофенолов, при нитровании
хлорбензола получается смесь л- и о нитрохлорбензолов со значи-
тельным преобладанием л-нитрохлорбензола (больше чем 2/з смеси).
На сульфокислоты соединений, имеющих сильно реактиви-
рующую ядро группу ОН, азотная кислота в нитрующей смеси
действует, замещая не только водород ядра, но и вытесняя
нитрогруппой сульфогруппу. Таким образом из сульфо- и дисуль-
фокислоты фенола получается тринитрофенол, из 2-, соотв. 4-,
сульфо- и 2.4-дисульфокислоты а-нафтола — динитронафтол (он же
53
получается и при обработке азотной кислотой соответственных
сульфокислот а-нафтиламина)
NO2
/\/он /\/ОН /\/он
HO3S/X/ HOoS/^/^SOjH
Выполнение нитрования с введением растворителя для органи-
ческого вещества практикуется сравнительно редко.
Большей частью такие работы имеют интерес иаучио-исследовательский. Так
было проведено исследование нитрования фенолов в эфирном растворе 37), давшее
доказательство необходимости участия азотистой кислоты для начала реакции, так
как чистая азотиая кислота, лишенная окислов азота и азотистой кислоты, не про-
изводит нитрования. Концентрация окислов азота для начала реакции ие должна
быть ниже определенного минимума. Нитрование ализарина в 3-нитроализарин
проводят иногда в растворе толуола.
Нитрование в спиртовом растворе, которое проводилось при разных темпера-
турах, в частности было испытано по отношению к ароматическим ацилированным
(между прочим и остатком сульфокислоты) аминам з8), вряд ли может иметь прак-
тическое значение уже в силу опасности производственной работы из-за возмож-
ности образования здесь метилового, соотв. этилового, эфиров азотной и азотистой
кислот, вредно действующих на здоровье работающих и ие безопасных в отношении
взрывчатости.
Также не имеет практических перспектив нитрование в среде метилового спирта,
хотя он более устойчив по отношению к нитрующим агентам, чем этиловый спирт.
Во всяком случае и здесь положительный эффект нитрования сказывается лишь
в отдельных случаях, применительно к легко нитруемым веществам (фенолам и
аминам) м).
Более употребительна уксусная кислота в качестве растворителя. Перилен
рекомендуется нитровать в виде суспеисии в ледяной уксусной кислоте, так как
прн этом не получается продуктов окисления 40),
Очень устойчивое соединение, двуокись динафтилеиа,
О
рекомендуется нитровать в ледяной уксусной кислоте смесью селитры с серной
кислотой 41). Повидимому можно обойтись здесь и без этого растворителя 42). Воз-
можно, что более углубленная проработка нитрования с применением растворителей
даст в отдельных случаях практически интересные результаты, в частности измене-
ние в отношении между количеством изомеров у дву- и полизамещенных.
54
б) Практика проведения нитрования
В практике выполнения нитрования нитрующей смесью, как
выше уже отмечалось, применяется чаще всего охлаждение, осо-
бенно в начале реакции, под конец иногда допускается нагревание,
чтобы довести реакцию до конца при малой концентрации остав-
шейся азотной кислоты. Вместо нагревания иногда предпочтительно
Рис. 1. Нитратор.
предоставить реакционную смесь самой себе на некоторое время
(12—24 час.) и после этого уже приступить к выделению про-
дуктов.
С целью уменьшения возможных вредных влияний местного
повышения температуры, от теплоты реакции, каковое влияние рас-
пространяется преимущественно на частицы материала, приходящие
в непосредственное соприкосновение с нитрующей смесью, стре-
мятся быстрее заменить эти нагретые частицы новыми при энер-
гичном перемешивании реакционной смеси. В технике реакция
нитрования производится в чугунных котлах (нитраторах) с ме-
55
шалками, имеющих двойные стенки (рубашки) для охлаждения
током воды или холодильного рассола, соотв. для нагрева паром;
внутри аппарата установлен также цилиндрический полый или
змеевиковый охладитель, соотв. нагреватель (рис. 1). Следят за ходом
нитрования, анализируя неорганическую часть реакционной массы
иа общую кислотность и содержание азотной кислоты (нитромет-
ром Л у нге). Зная избыток азотной кислоты, взятой против расчета,
легко определить момент, когда вся азотная кислота, могущая
нитровать, уже израсходована.
Выделение нитросоединений, если они жидки при обыкновенной
температуре или плавятся при небольшом нагревании, в производ-
стве осуществляется путем отстаивания всей реакционной смеси
в больших железных разделительных воронках — сепараторах.
Нитропродукт через некоторое время отделяется от разведенной
реакционной водой отработанной кислоты. Последняя по отделении
может быть пущена на денитрацию и концентрацию или путем
подкрепления серной кислотой (или олеумом) и азотной кислотой
вновь войти в состав нитрующей смеси. Если нитрование давт
в результате продукт твердый и не растворимый в холодной отра-
ботанной кислоте, то последнюю можно также отделить от него
(фильтрацией, центрифугированием). Отработанная кислота в этих
случаях всегда может извлечь с собой некоторое количество нитро-
продукта, частично в состоянии раствора. На эту потерю нужно
обращать внимание.
Те процессы нитрования, продукт которых не выделяется из
отработанной кислоты при стоянии (таковы нитропродукты неко-
торых производных аминов, сульфокислот, фенолов), заканчиваются
разведением реакционной смеси при возможно низкой температуре
водой или, лучше, льдом. Такое разведение необходимо проводить
при энергичном размешивании, чтобы избежать местных перегревов,
сопровождающихся обильным выделением окислов азота.
В тех случаях, когда разведением реакционной массы не удается
выделить продукт, иногда пользуются введением в кислотную
смесь (до или после разведения ее) какой-либо легкодоступной
соли, например поваренной, глауберовой, железного купороса и
других. Обычно такое высаливание практикуется по отношению
к нитропроизводным, имеющим кроме нитрогрупп еще кислотные
группы (СО2Н, SO3H). Введение поваренной соли применимо лишь
при отсутствии или очень малой концентрации азотной кислоты
в реакционной массе, так как иначе выделяется хлор, могущий
весьма вредно повлиять на продукты реакции (окислением, охло-
рением).
Нитропроизводные сульфо- и особенно полисульфокислот не-
редко не выделяются после нитрования из реакционной смеси ни
одним из этих методов. Тогда прибегают к способу перехода через
кальциевые соли, который мы рассмотрим ниже (см. сульфирование).
Как новый способ производственного проведения нитрования отметим предло-
женный недавно метод работы в аппарате непрерывного действия ®). Аппарат
состоит в существенной части из нитрующей колонны, разделенной на несколько'
камер (для бензола — 7, для ксилола — больше). Углеводород и нитрующая смесь
подаются непрерывно — первый снизу вверх, вторая — сверху вниз. В камерах нитрую-
щая смесь распределяется мелкими струями системой решеток и сеток. Охлажде-
56
нне свинцовыми змеевиками. Промывка идет также непрерывно в колонне, ннтро-
продукт идет Сверху вниз, вода снизу вверх. Следующая операция — отгонка
непрореагировавшего углеводорода — также осуществлена по принципу непрерыв-
ности.
Этот прием работы является развитием идеи, положенной в основу герм. п. 1915 г.
(К у б е р ш к и). Преимуществом метода, оставляя в стороне трудности аппаратур-
ного оформления, является осуществление работы в противотоке, следовательно
устранение возможности слишком энергичной реакции при соприкосновении массы
углеводорода со свежей нитрующей смесью. Другой принцип — прямого тока —про-
веден в герм. п. 1908 г. (М. L. В.). Углеводород, например бензол, и нитрующая
смесь непрерывно идут каждый из своего сборника и, охлажденные До нужной
температуры, проходят затем ряд (до 4) малых нитраторов с мешалками, соединен-
ных последовательно. Каждый нитратор можно держать при нужной температуре-
Из последнего нитратора реакционная смесь попадает в разделительный цилиндр,
откуда, также непрерывно, выходит сьерху нитробензол, снизу отработанная кислота.
Тот же принцип прямого тока положен в аппарат для проведения нитрования
н подобных реакций герм. п. 1910 г. (tM). В этом вертикальном аппарате, разделен-
ном на несколько камер (сообщающихся через перегородки, попеременно не доходя-
щие то до стенок цилиндрического аппарата, то до вертикального вала), реакционная
смесь подвергается размешиванию в каждой камере пропеллерными мешалками.
Непрерывный метод работы имеет такие большие преимущества перед периоди-
ческим, что при наличии упомянутых решений этого вопроса можно рассчитывать
на практическое осуществление принципа непрерывного нитрования.
в) Перспективы дальнейшего прогресса в нитровании
Ныне вполне своевременно поставить вопрос: каковы перспективы дальнейшего
прогресса в процессе производственного нитрования? До тех пор пока источником
для получения нитрующих агентов была чилийская селитра, нз которой готовилась
азотная кислота и которую в смеси с серной можно было также применить для
получения иитропродуктов, установленные способы можно было делать более удоб-
ными, выгодными, не меняя принципа работы. Теперь же, когда главная масса
азотной кислоты готови|ся из окиси азота NO, получаемой в процессе контактного
окисления аммиака с последующим окислением окиси в двуокись азота, мысль
химиков привлекает возможность использовать для нитрования не ютовую азотную
кислоту, а значительно более доступную и дешевую двуокись азота NO2, ближайший
к ней промежуточный продукт в контактном процессе.
Вопрос о получении нитрозамещенвых действием двуокиси азота на органические
соединения был наиболее обстоятельно обследован Виландом Было установ-
лено, что двуокись азота присоединяется подобно галоидам к алифатической двойной
связи, давая динитропродукты, из которых действием щелочей с отщеплением
азотистой кислоты (в виде соли) получаются мононитрозамещеиные этиленовых
углеводородов:
— НС - СН-> -С — СН—
NO2 NO2 ~hno‘ no2
Из ароматических соединений фенолы реагируют легко с двуокисью азота, давая
иитрофенолы, также относятся и аминопроизводиье. Бензол на холоду не реагирует
с двуокисью азота, при нагревании реакция идет частично в направлении получения
тринитробензола, нитробензола, частично в сторону окисления, сопровождаясь обра-
зованием угольной, щавелевой кислот и тринитрофенола.
Шааршмидт применил при взаимодействии между бензолом (и другими
ароматическими соединениями) и двуокисью азота вспомогательный реагент н виде-
хлористого алюминия А1С13 и хлорного железа FeCl3 !15). Им было установлено, что
в присутствии этих хлористых солей бензол (также толуол и хлорбензол) реаги-
руют очень энергично с двуокисью азота, образуя комплексные соединения, которые
в случае бензола например имеют состав:
2A1C13.3C6H6.3N2O4 и FeCl3.2C6H6.2N2O4.
Вода разлагает эти соединения, причем органический остаток C6H6N2O4, отщепляя
HNO^ переходит в нитробензол. В этом примере, так же как при взаимодействия
NO2 с хлорбензолом, получаются мононитропроизводные с использованием половины
57
всей двуокиси азота, связанной предварительно в промежуточном продукте реакции.
Шааршмидт полагает, что в этих промежуточно-образованных веществах имеется
продукт присоединения к бензолу молекулы тетраокиси азота N2O4, жидкого димера
двуокиси, причем одна азотная группа вступает в виде иитрогруппы, другая —
в виде эфира азотистой кислоты ONO в соответствии с несимметрической структурой
тетраокиси NOONO2.
Их строение поэтому:
ONO—
ft/
i~NO2
соотв.
гн
Н
XONO
Основываясь на соображении, что хлористый алюминий и хлорное железо в опы-
тах Шааршмидта имеют значение активаторов двойных связей ароматического
ядра, Баттге с успехом заменил эти соли более удобным реагентом—серной
кислотой. Прн пропускании NO2 или введении N2O4 в смесь серной кислоты и бен-
зола происходит быстрое и полное поглощение окисла азота с повышением темпе-
ратуры. Углеводород нитруется с потреблением 50% окисла азота, другая половина
которого входит количественно в состав образующейся нитрозилсерной кислоты,
следовательно реакция может быть выражена уравнением:
С6Н6 + 2NO2 + H2SO4 = C6H6NO2 -f- NOOSO3H 4- Н2О.
Аналогично и с такими же выходами протекает реакция с толуолом, м- н
n-ксилолом, хлорбензолом, нитробензолом; антрахинон дает тоже дериват при при-
менении олеума с содержанием 5% SO3 вместо серной кислоты в 77-—100%, какая
применяется при нитровании бензольных и производных ^в). Этим доказана воз-
можность применения двуокиси для целей нитрования, практическая же приложи-
мость этой реакции зависит от того, насколько удобно регенерировать 50% дву-
окиси азота, которые связаны в иитрозилсериой кислоте.
Баттге, изучая различные для этого возможности, останавливается как на наи-
более выгодном на методе анодного окисления отработанной кислоты, заключающей
витрозилсерную кислоту, с применением пористой диафрагмы, причем реакция
окисления дает азотную и серную кислоты по уравнению:
2NSO6H 4- 20 4- 2Н2О = 2HNO3 4- 2H2SO4.
Выход по току был у него 95%, по азотной кислоте 92%.
В американском патенте 1927 г. имеется описание другого метода регенерации
отработанной кислоты, содержащей нитрозилсерную, полученную при нитровании,
аналогичном только что описанному 47) В ием описывается обработка отработанной
кислоты током сернистого газа, причем одновременно достигается выделение окиси,
азота, идущей далее после окисления воздухом в двуокись в процесс нитрования,
и окисление сернистого газа в серную кислоту:
2NSOnH 4- SO2 4- 2Н2О = 3H2SO4 -f- 2NO.
Преимущество этого метода по описанию в том, что в результате регенерации
серная кислота получается более высокой концентрации, чем была в отработанной
кислоте, вследствие того, что реакция потребляет воду.
Процесс нитрования двуокисью азота в присутствии серной кислоты можно
прежде всего представить себе как идущий за счет азотной кислоты, образующейся
вследствие реакции между двуокисью и серной кислотой
2NO2 4- H2SO4 = HNO3 4- NOOSOgH,
и таким образом ои отличается от обычного лишь методом приготовления нитрующей
смесн и тем, что в состав последней входит нитрозилсерная кислота вместе с сеоной.
Одиако на основании своего исследования Баттге развивает иной взгляд на
течение процесса, в котором участвуют три реагента одновременно: углеводород,
двуокись азота (или N2O4) и серная кислота. Основываясь на наблюдении, что бен-
зол с нитрозилсерной кислотой дает цветные молекулярные соединения состава
C6H62NSO5H, и иа структурной аналогии между нитрозилсерной кислотой NOOSO2OH
и тетраокисью азота NOONOg (ннтрозилнитратом), он во взаимодействии, о котором
идет речь, усматривает фазу присоединения 2 молекул NO2 или одной молекулы
58
N20j к бензолу, т. е. промежуточное образование таких же непрочных соединений,
которые получил Шааршмидт при реакции NOa с бензольными соединениями
в присутствии хлористого алюминия. При таком толковании течение реакции может
быть выражено схемой:
С6Н6 4- 2NO2 + H2SO4 —H2SO4+NOOC6H6NO2—- NSO5H 4-C6HbNO2+H2O.
За серной кислотой сохраняется ее значение активатора бензольной молекулы
в направлении реакции присоединения, в данном случае молекул двуокиси азота.
Упомянем здесь, что применение ртути в качестве активатора реакции между дву-
окисью азота и бензолом привело к очень бурной реакции, опасной для повторения,
несмотря на описание герм. пат. о получении в этом случае пикриновой кислоты &).
Применение окислов азота для нитрования выйдет вероятно вскоре на широкий
производственный путь ввиду указанных экономических преимуществ его перед
ныне распространенным способом с азотной кислотой. Важнейшим вопросом для
решения иа ближайшем этапе изучения этой задачи является использование тех
50% окиси азота, которые остаются в нитрозилсерной кислоте или в виде азотистой
кислоты уходят в водном растворе из производства.
Упомянем, что с целью использования окислов азота имелось более давнее
предложение, описанное в герм. пат. 4Э); смесь окислов с воздухом (от окисления
азота в электрической печи) проводят через окись цинка, поглощающую кислые
окислы и отдающую их при нагревании. С парами бензола соединение циика реаги-
рует, переводя его в нитробензол. Толуол, по словам описания, превращается в смесь
из 11% м- и 89% о-нитротолуолов (? НВ). Способ повидимому не нашел техни-
ческого приложения.
г) Выделение и характеристика продуктов нитрования
Полученные тем или другим способом нитросоединения после
своего выделения из реакционной смеси или непосредственно пере-
даются без дальнейшей очистки на последующие операции, например
восстановление в аминосоединения, или (чаще) подвергаются пред-
варительной очистке. Очистка преследует цель: устранение мине-
ральных, главным образом кислотных, примесей, — это достигается
промывкой водой,—и затем освобождение от посторонних органи-
ческих веществ, могущих попасть в сырое нитросоединение из
не вполне однородного сырого материала или оказаться в нем
вследствие отклонения реакции от ее нормального течения (например
вследствие окисления части исходного материала, введения лишней
нитрогруппы и т. п.).
Для нитросоединений, жидких при обыкновенной температуре
и кипящих без разложения, применяется для очистки перегонка.
Для того чтобы понизить температуру кипения, перегонка произ-
водится при разрежении. Нитросоединения, получаемые в виде
смеси двух изомеров (о- и л-нитротолуолы, о- и л-нитрохлор-
бензолы), необходимо разделить на индивидуальные вещества.
Здесь пользуются также перегонкой, разделяя погоны при помощи
колонны на фракции, содержащие преимущественно один из изо-
меров, и подвергая фракции или кристаллизации в надлежащих
температурных условиях, если нужный изомер легко кристалли-
зуется (случай л- и о-хлорнитробензолов, «-нитротолуола), или
ректифицируя фракции в новой перегонке.
Твердые, но легкоплавкие нитро- и полинитросоединения про-
мываются горячей водой в расплавленном состоянии и, если надо,
обрабатываются в таком же виде посредством водных растворов
реагентов для удаления примесей и затем в расплавленном состоянии
выливаются в тару для застывания или, лучше, предварительно
59
получаются в виде более или менее крупных зерен (гранул) выли-
ванием расплавленного продукта тонкой струей в воду при раз-
мешивании.
При получении нитросоединений и их переработке, вообще при
обращении с нитросоединениями, работающим нужно быть осто-
рожными и избегать как попадания нитросоединений на тело, так
и вдыхания их паров и пыли. Могущие выделяться (при нитро-
вании и смешении кислот) окислы азота, „нитрозные газы“, при-
чиняют при вдыхании серьезные поражения дыхательных путей
и могут вызвать очень тяжелые заболевания. Надлежащее обору-
дование с вентиляцией у мест выделения паров, опрятное содер-
жание рабочих помещений с посыпанием мокрыми опилками (и их
сметанием) пола тех помещений, где имеется пыль или легкий
порошок твердых нитросоединений, частая мойка рук и лица и
купание, смена рабочей одежды в случае ее загрязнения нитро-
продуктами, отдельная одежда для работы и дома, принятие пищи
и питья вне рабочего помещения и чистыми руками, устранение
потребления алкоголя работающими в нитроцехах — меры, в значи-
тельной степени обеспечивающие безопасную работу 60).
Нитросоединения, заключающие несколько нитрогрупп, являются
иногда конечным продуктом; таковы вышеупомянутые полинитро-
соединения, применяемые как взрывчатые вещества. Большинство
нитросоединений однако имеют значение как промежуточные про-
дукты, назначаемые для дальнейших превращений — больше всего
в аминосоединения. Некоторые нитросоединения (динитрофенол,
динитротолуол и др.) применяются как исходные для получения сер-
нистых красителей, иные — для получения индулинов и нигрозинов
(нитробензол). Пользуясь окисляющим действием нитрогруппы,
нюросоединения применяют в качестве окислителей при некоторых
реакциях (фуксиновый плав по Купье, получение р-аминоантра-
хинона из р-сульфокислоты антрахинона и т. п.).
Мы знаем, что нитрогруппа оказывает влияние на последующие замещения,
ориентируя новый заместитель в мета-положенне. В некоторых взаимодействиях
ннтросоединений получаются тем не менее орто- (илн пара-) замещенные. Эти
необычайные ориентации нитрогруппы вызываются реагентами, могущими вовлечь
саму интрогруппу в прямое участие (с присоединением н следовательно с измене-
нием ориентирующей группы). Такими реагентами оказываются H2NOH, КОН
и т. п. 61).
Если надо определить присутствие нитрогруппы в соединении,
то качественно ее можно обнаружить иногда по внешним признакам
нитросоединения: оригинальному миндальному запаху, свойственному
весьма многим нитропродуктам, желтому цвету. Лучше (в том слу-
чае, когда соединение не имеет аминогруппы) подвергнуть иссле-
дуемый продукт восстановлению (цинк в уксуснокислом растворе,
SnCl,— в солянокислом) и в полученном продукте восстановления
искать аминогруппу методом диазотирования (см. восстановление).
Метод в случае наличия аминогруппы в нитросоединении не может
быть применен без предварительного удаления через диазореакцию
или приведения в неактивное состояние (например ацилированием)
аминогруппы. Для тех химиков, которым диазопроба является
вполне знакомой, можно и без удаления аминогруппы по оттенкам
60
получаемых с азосоставляющими окрасок сделать сравнение с исход-
ным материалом и вывод о наличии или отсутствии нитрогруппы.
Ди- и полинитропроизводные дают характерные окраски в ацето-
новом растворе от обработки едкими щелочами б2).
Полинитросоединения в отличие от мононитро- разлагаются от
действия горячих растворов щелочей, образуя с металлом щелочи
нитрит. Последний после подкисления массы может быть открыт
по образованию азосоединения (с сульфаниловой кислотой и а-на-
фтиламином Б3).
Нитрозамещенные (моно- и поли-) хорошо характеризуются
точками плавления и кипения.
Зная, что нитрогруппы в веществе присутствуют, их количество
(в чистом продукте) можно определить способом Лимприхта,
именно — обработкой испытуемого вещества раствором хлористого
олова определенного титра и определением в оставшемся растворе
количества непрореагировавшего SnCl2. Реакция протекает по урав-
нению:
RNO2 + 3SnCl2 Ц- 6НС1 = RNH2 4- 3SnCl4 4- 2H2O.
Раствор хлористого олова готовят, растворяя приблизительно 150 г олова
в соляной кислоте; сливают прозрачную жидкость с осадка, добавляют 50 cjks
концентрированной соляной кислоты, водой дополняют до 1 л н определяют тнтр
по 1/10 раствору иода. С этим раствором нагрезают навеску <0,1—0,2 г) иссле-
дуемого материала иг менее х/2 часа в мерной колбе на 100 cat3, при летучих соеди-
нениях— в запаянной трубке 20 см длины и 15 мм диаметра, охлаждают, доводят
объем точно до 100 cat3, встряхивают н отбирают пипеткой 10 cat3. Отобранную
пробу разводят в стакане водой, прибавляют к жидкости раствора соды (90 г
№2СОз + 120г сегнетовой соли в 1 л) до полного растворения образующегося вна-
чале осадка и, разбавив немного водой, титруют 1/10 N раствором иода до остаю-
щегося фиолетового окрашивания крахмального индикатора.
При расчете принимают во внимание, что 1 си3 1j10 N раствора,
иода отвечает 0,0059 г Sn = 0,0007655 г ЫО2 54).
Хорошим методом количественного определения нитрогруппы,
так же как азогруппы, является метод титрования хлористым
титаном 55). Нитросоединения восстанавливаются в кислом растворе
(концентрированная солянтя кислота) посредством горячего титро-
ванного раствора TiCl3, титр которого устанавливается посредством
раствора соли окиси железа, определенного содержания (из железно-
аммиачных квасцов); индикатором служит роданистый калий. Вос-
становление производится в токе СО2 и протекает быстро по урав-
нению :
RNO2 4- 6TiCl з + 6НС1 = RNH2 4- 6TiC4 4- 2H2O.
По окончании процесса восстановления охлаждают и избыток
TiCl8 оттитровывают раствором железных квасцов. Для нитро-
соединений, не растворимых в воде, применяют спиртовые растворы.
Для точных определений вводят поправку на количество кисло-
рода, растворенного в воде, какое количество определяют отдель-
ным опытом.
д) Нитрозирование
Реакция нитрозирования имеет значительно меньшее приме-
нение, чем нитрование. Лишь соединения, заключающие характер-
61
ные группы: ОН (фенольную или энольную), N(Alk)2, NHAr,
иногда NH(Alk), в исключительных случаях NH2-rpynny и также
циклически связанную СО-группу, являются объектом применения
реакции нитрозирования.
Общее уравнение реакции нитрозирования, если не принимать
во внимание перегруппировок внутри молекулы, таково:
RH + HONO = RNO + Н2О.
Реагентом является следовательно азотистая кислота, и эффект
реакции заключается в фиксировании группы нитрозо в ядре дан-
ного соединения с отщеплением молекулы воды, т. е. эффект в зна-
чительной мере напоминает таковой же для реакции нитрования.
В действительности же нитрозирование протекает иначе, чем
можно себе представить по приведенному схематическому уравне-
нию, ибо полученные нитрозопродукты ведут себя так, как если
бы они были производными хинонов. Очевидно реакция эта, про-
ходящая через фазу присоединения (см. выще), дает в результате
продукты с отличным по строению от исходных ядром.
Например
Подобно этому
НО
:ОНН
С1
NO
N:(CH3)2-h2o
^\:N(CH8)t
HON:^
В случае нитрозирования фенолов получаются таким образом
не нитрозофенолы, а оксимы хинонов *, при нитрозировании ал-
килированных аминов — оксимы алкилированных хинониминов.
Поэтому вышеприведенные соединения следует называть изо-
нитрозосоединениями и группу NOH — изонитрозогруппой в отли-
чие от нормальной нитрозогруппы N:O, имеющейся например в
нитрозобензоле CeHBNO.
* При взаимодействии хнвона и гидроксиламина получается хиноноксим, тожде"
ственный получаемому при нитрозировании фенола
^\о ^\noh
+ h2noh —> + н20
0:1 zJ О: । J
62
Возможно тем не менее для некоторых замещенных фенолов
получение отдельно настоящих нитрозо- и изонитрозопроизводных.
Так, при нитрозировавии ж-хлърфенола был получен желто-зеленый 4-ннтрозо-
3-хлорфенол
С1\/\/°Н
перегруппировывающийся от действия соляной кислоты тотчас же и) в соответ-
ственный хиноноксим.
Изонитрозосоединения получаются с размещением группы NOH в пара-месте
по отношению к реактивирующей группе преимущественно, но не исключительно,
ибо наличие примеси орто-соединения констатировано уже н некоторых случаях57).
В отличие от реакции нитрования воздействие азотистой кислоты
не требует всего чаще участия водоотнимающего средства и про-
текает и в водной среде. Фактор концентрации среды не является
следовательно существенным при нитрозировании и интересен
преимущественно ввиду большей или меньшей растворимости про-
дукта реакции.
Значение температуры весьма велико, и повышение ее выше
определенного в каждом случае максимума является весьма неже-
лательным как с точки зрения выходов, так иногда и самого те-
чения реакции, ибо полученные оксимы хинонов являются весьма
реакционноспособными веществами и при высокой температуре
могут претерпеть разложение. Поэтому нитрозирование вообще
ведется при охлаждении реакционной смеси.
При нитрозировании фенолов обычно поступают таким образом,,
что готовят раствор из эквимолекулярных количеств фенола и едкого-
натра и в этот раствор прибавляют азотистокислого натрия (ни-
трита) в количестве, отвечающем одной молекуле NaNO2 на моле-
кулу фенола. Избыток едкого натра против рассчитанного коли-
чества допускается иногда довольно значительный, избыток ни-
трита— незначительный. К смеси при помешивании прибавляют
серную или соляную кислоту с таким расчетом, чтобы ее был»
достаточно не только на нейтрализацию свободной щелочи и вы-
деление из нитрита всей азотистой кислоты, но и на поддержание
в растворе постоянной кислотной реакции, необходимой для вы-
деления нитрозосоединения (хиноноксима) из раствора. В тех слу-
чаях, когда кислотная реакция в образовавшемся осадке неудобна
для последующих операций, прибегают к нейтрализации (точной)
минеральной кислоты аммиаком.
Если в самой молекуле фенола имеется сильная кислотная
группа (например СООН), то тогда достаточно в иных случаях
ввести в реакцию только NaNO2 без прибавления щелочи и
кислоты. Также без щелочи и кислоты обходятся при лишенных
кислотности фенолах, применяя нитриты тяжелых металлов или
смеси солей тяжелых металлов с нитритом натрия. Гидролиз азо-
тистокислой соли тяжелого металла является источником азотистой
кислоты, например
Cu(NO2)24-2H2O-> Cu(OH)2 +2HNO2.
6&
Участие соли тяжелых металлов в реакции образования хиионоксимов пови-
лимэму ие ограничивается лишь передачей элементов азотистой кислоты органи-
ческой молекуле. Здесь возможно комплексообразование. Весьма вероятно, что
известные катионы в нитритах (Си", АГ“) могут быть специфичны для успеха
реакции68).
Для тех изонитрозофенолов, которые далее применяются и реакциях, тре-
бующих водоотнимающего действия серной кислоты (например при получении ин-
дофенола из из шитрозофенола и карбазола), предложен способ нитрозирования
посредством обработки фенолов иитрозилсерной кислотой и последующего выде-
ления азотистой кислоты при разведении смеси водой от реакции
NSO5H + Н2О = HNO2 + H,SO4.
В таком случае изоиитрозосоедииение нет необходимости выделять, а можно при-
менять реакционную смесь прямо дальше 69.
Замечательно, что иитрозироваиие в растворе концентрированной серной кислоты
азотистой кислотой в момент ее образования — от прибавленного нитрита, т. е.
фактически посредством иитрозилсерной кислоты на холоду, позволяет приготовить
нитрозо- или изонитрозосоедииения первичных аминов, которые При обработке
азотистой кислотой в иодиых растворах дают диазосоединения, если эти амины при
свободном парз-месте содержат в орто- и мета-положениях к аминогруппе алкильные
группы или присоединенные в этих же положениях кольца60). Изонитрозосоеди-
нения получаются при этом в виде сернокислых солей, из которых они могут быть
выделены в свободном состоянии посредством обработки щелочными реагентами,
например содой:
NH2 NH
НзС\/\/ННз H3Cw\nh
I I - HON J I I I J- I I I
NOH
Избыток азотистой кислоты при иитрозироваиии может привести к изменению
изонитрозогруппы в диазогруппу. Изоинтрозофенол окисляет при этом азотистую
кислоту в азотиую, переходя сам в витрат дчазония 61). При нитрозироваини (из-
бытком HNO2) некоторых сульфокислот фенола Родионовым и Матве-
евым получен вместо нзэнитрозо- прямо диазопродукт 62).
При приготовлении (изо)нитрозосоедииеиий методом выделения азотистой
кнслоты из ра:твора нитрита вредными оказываются добавочные окислительные
воздействия. Иногда п >этому предпочитают взаимодействие растворов проводить
под уровнем жидкости (вводят трубку с кислотой, соответственно раствором ни-
трита, под жидкость). Повидимому вредное влияние оказывает примеси» азотнокислой
соли в нитрите63).
Из аминосоединений как ингредиенты реакции нитрозирования
в первую очередь интересны третичные жирно-ароматические
амины, общей формулы ArN(Alk)2, причем алкилы необязательно
тождественны. Вторичные жирно-ароматические амины ArNHAlk
дают с азотистой кислотой как правило нитрозосоединения, где
NO-гоуппа связана с азотом (нитрозамины, N-нитрозосоединения)
ArN(NO;Alk. Некоторые из таких вторичных аминов нитрози-
64
руются в ядре, подобно третичным аминам, например монометйл-
о-толуидин дает и-нитрозо-(изонитрозо-)соединение
4- hno2+ нх^
HON:
X
: NHCH8
-т Н2О
^^СНд
(X — анион кислоты).
Очевидно здесь метил, соседний с азотом, принимает участие
в насыщении его сродства.
Первичные амины с азотистой кислотой как правило дают
диазосоединения (см. ниже).
При нитрозировании двуалкилированных аминов обычно в рас-
твор минеральнокислой соли амина, в каковом имеется избыток
минеральной кислоты, прибавляют эквимолекулярное амину коли-
чество нитрита в водном растворе, при помешивании. Выделяю-
щийся осадок минеральнокислой соли оксима алкилированного
хинонимина отделяется от раствора фильтрованием.
Характерной для нитрозосоединений двуалкилированных аминов-
является реакция их с раствором едкой щелочи — при этом ОНИ
разлагаются, отщепляя диалкиламин, и переходят в щелочное со-
единение нитрозофенола, например
/x/N(CH8)2
| +NaOH^
ONXX^
ZO Na
! I HN(CH3)2
ON/Xx//
Таким методом можно получить чистый диметиламин.
Совершенно гак же как с двуалкилированными ароматическими
аминами получаются нитрозопроизводные из вторичных алкилар-
аминов, соответственно ариламинов, и циклических кетонов.
В последних двух случаях образуются часто настоящие нитрозо-
соединения, заключающие характерную NO-, а не NOH-rpynny.
У алкил- (соответственно арил-) нитроза«инов группа NO, стоявшая при атоме
азота, при обработке соединения минеральной кислотой переходит в ядро в пара-
положение по отношению к азоту NH-группы, например
+ HNO,
------)
-Н3О
NO
Карбазол, являющийся также вторичным амином, при нитрозировании, например по-
средством окислов азота, в индифферентном растворителе, дает N-нитрозопроизвод-
ное, которое иследза образованием перегруппировывается в З-(пара) нитрозозамещен-
5 Зак. МП. — Н. Ворожцов 65-
ное, окисляющееся до нитро- от действия окислов азота м). В конечном продукте
можно найти и диннтропродукт:
В последнее время некоюрые из N-нитрозопроизводиых дифениламина при-
обрели большое техническое значение как получающиеся в процессе образования
таких диазопронзводных, которые оказались особенно ценными для так называемых
холодных окрасок. Примером подобных красителей является вариаминовый голубой,
образуемый на волокне и имеющий структуру
\cONHAr
При получении из аминосоединения C2H6OCeH4NHC6H4NH2 дназосоедннения
от действия азотистой кислоты не только диазотируется конечная группа NH2, но
и замещается группой нитрозо NO атом водорода у NH-группы, следовательно
образуется соединение состава: C2H5OC6H,IN(NO)C6H.1N2X (где X — аннон диазо-
группы), которое и входит в сочетание с азосоставляющей. Нитрозогруппу устра-
няют из готового красителя например действием сернистого натрия или сульфитов
в присутствии соединений, способных давать при окислении хиноны или хнно-
нимнны №).
N-нитрозосоединения азокрасителей из дифениламина по описанию германского
патента дают с амино-, окси- и амино-оксисоединениями ароматического ряда (не кра-
сителями) красители неизвестного пока строения 66).
Взаимодействие п-нитрозодифеннламнна с гидроксиламином (в избытке) при-
водит сначала повидимому к образованию диазосоединения, а затем более богатого
азотом соединения:
C6H5NH • C6H,NO + NH2OH C6H6N : С6Н4: N ; N + 2H2O,
CeH5N : C6H4: N2 + NH2OH -> C6HS • NHCeH4N3.
Последнее обладает выдающейся светочувствительностью, почему и названо открыв-
шим его Ангели фотоазидом в7).
Мы видим из всех этих примеров, что нитрозо- или изонитрозо-
группа входит в ядро, где имеется уже активирующий ядро заме-
ститель. Методом прямой обработки азотистой кислотой не удается
образовать нитрозогруппу в незамещенном углеводороде, напри-
66
Промежуточные продукты, получаемые с применением нитрования
и нитрозирования (бензольный ряд).
мер бензоле. Тем не менее нитрозопроизводные углеводородов
известны, но их нитрозогруппа образуется не методом вхождения
ее в незамещенное ядро, а как результат превращения уже имею-
щегося заместителя (NH2-rpynnbi окислением ее с переходом через
6*
67
Промежуточные продукты нафтали-
нового ряда, получаемые иитровнием
ыо?
NO, s s
Азотистая кислота иногда может
быть реагентом нитрования. Так, было
наблюдено, что при действии нитрита
с соляной кислотой в водном растворе
иа диметил-л-толуидии получается с
хорошим выходом иитропродукт,
имеющий строение I; азотная кислота
в одинаковой концентрации с при-
меняемой азотистой ие дает нитропро-
дукта, а при обработке того же ве-
щества нитруощей кислотной смесью
образуется иитрозамещениое со струк-
турой II 69):
no2
kin
NO,
NHOH — см. ниже — окисление).
Нитрозоуглеводороды и их за-
мещенные малостойкие соеди-
нения, но имеющие некоторый
технический интерес для получе-
ния только что упомянутых ами-
нозамещенных дифениламина 68).
/X/N(CH3)2 z /N(CH3)2
II I 1 I
H3CZZZ H3CZZZZNO,
o2nx/. zN(CH3)2
M
H3CZ z
Для качественного опре-
деления нитрозогруппы в сое-
динении обычно пользуются
либермановской реак-
цией, нагревая испытуемое ве-
щество с избытком фенола. По
охлаждении прибавляют к сме-
си немного концентрированной
серной кислоты, разбавляют
водой и при охлаждении де-
лают жидкость щелочной при-
бавлением едкого кали. При
наличии в соединении нитро-
зо- (изонитрозо-) группы жид-
кость окрашивается в интен-
сивно синий цвет.
Для количественного опре-
деления нитрозо-, так же как
изонитрозо-, нитро- и других,
способных к восстановлению
в NH.2, групп можно применить
метод Кауфлера, состоящий
в нагревании вещества с соля-
ной кислотой и оловом (в
избытке, чтобы реакция не шла
по уравнению SnCl34-2HCl =
= SnCl4+H2).
Зная по азотометру, сколько водо-
рода (собирать над КОН) выделяется
68
в холостом опыте, и зная, сколько
его получено прн опыте с веществом,
можно рассчитать, сколько его потреб-
лено иа восстановление характерной
группы. Прибор до опыта наполня-
ется СО2, и водород после опыта ею
же вытесняется. Навеска вбрасывается
в прибор без проникновения туда воз-
духа. К олову для лучшего раство-
рения добавляется некоторое (во всех
опытах одинаковое) количество очень
разведенного раствора PtCl4.
Нередко изонитрозосоеди-
нения аналитически характери-
зуются, как и нитросоедине-
ния, в виде полученных из них
аминопроизводных путем обра-
ботки их избытком цинковой
пыли в присутствии соляной
или уксусной кислоты. Амино-
группа затем определяется s
методом количественного диа-
зотирования (см. главу V.)
В предварительном выделении
аминосоединения при этом нет
необходимости. Можно при-
менять для диазотирования
отделенный от избытка цинка
кислый раствор.
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
') Ср. М. Battegay, BulJ. (4),
43, 103 (1928).
2) J. Meisenheimer, Вег. 33.
3547 (1900), Meisenheimer, Con-
ner a d е. Ann. 330, 133 (1904).
3) G. Oddo, CB 1925. II, 1598.
4) J. A 1 p h e п, CB 1932, I, 675.
5) H. E. Armstrong, E. C. Ros-
siter, Ber. 24, Ref. 720 (1891), Ё. N o-
e 11 i n g, S. F о r e 1, Ber. 18,2670 (1885)
Noelting, B. Pick, Ber. 21, 3158
(1888), Battegay, 1. c. 128
6) P. H. Groggins, Chem.Met.
Eng., 1928, ннтир. no Ch.-Ztg 1929, 68,
G г о g g i n s, Anilin and its Derivatives,
New York, 1924, 72.
7) Hanp. Livingston, Kyri-
d e s. Am. n. 1638045 (получение нит-
робензола), Am. n. 1586253(иитрохлор-
бензол).
8) А. В. С т e п а н о в. Ж. 50, 309
(1918), Ж. П. X. 1, 195 (1924).
») А. Р i с t е t, Г. п. 202201 F.
IX, 70, A. Pictet, К h о t i n s k у,
Ber. 40, 1163 (1907).
Промежуточные продукты антрахино-
нового ряда, получаемые путем нитро-
вания
69
1») Witt, U ter шапп, Вег. 39, 3901 (1906).
п) J. В. Menke, СВ 1925, I, 1066, II, 153, Б. п. 235698. заявл. Г. п. М. 83728,
F. XV, 199.
12) G. Bacharach, СВ 1927, И, 810, В а с h а г а й h, Р е i t z, СВ 1931, II, 233.
A. Pictet, Р. Genequand, Вег. 35, 2526 (1902).
is) К. Fredenhagen, Г. п. 529538, СВ 1931, 11, 1924.
М) Heinemann, Б. п. 102216, Н. Wolf, Г. п. 310772, F. XIII, 1104, P.S. Varma.
D. A. Kulkarni, СВ 19’5, I, 1302.
15) F. О. R i с е, СВ 1921, I, 530.
1«) F. Fichter, Pluss, Helv. 15, 236 (1932), Н. Trill er, Г.п. 100417,F. V, 159.
i’) W oilfens te in, Boters, Г. n. 194883, F. IX, 140, Ber. 46, 586 (1913), cp.
L. Vi gn on, Bull. (4) 27, 547 (1920).
1Й) T. L. D a v i s, Worral, Dreke, Helmkamp, Young, CB 1921, 111, 404,
Davis. Am. n. 1417368, CB 1923, IV, 922.
I») L. Des vergnes, Chirrt. & Ind. 22, 451 (1929) CB 1929, II, 2730.
20) А. И. 3 a x a p о в, Ж. X. П. 8, 30 (1931).
а) Phyllis, Me Kie, Soc. Ind. 46, 261T, (1927).
22) Holderman n, Ber. 39, 1250 (1906), H. A. X о л e во, И. И. Э й т и и г о н,
Ж. П. X. 5, 612 (1932).
23)Wolffenstein, Paar, Ber. 46, 589 (1913).
м) Н. Е. Fierz-David, The Sulphonation and Nitration of Naphthalene, Soc. Ind.
1923, 424 T.
25) H. Martinsen, Zt. phys. Chem. 50, 385 (1905), 52, 605 (1907). По нитрова-
нию в уксусном ангидриде см. J. Р. W i b a u t, Rec. 34, 241; СВ 1915, II, 653.
26) Б. В. Тронов, Г. Л. Бер, Ж. 62, 2337 (1930).
27) S. Veibel, Вег. 63, 1577 (1930), А. И. К а р та ш е в, Ж. 62, 2129(1930). Там
же библиография предмета.
26) М. И. Коновалов, Вег. 28, 1861 (1895), Ж. 31. 254 (1899). Также Г. и.
239953 (Us. Rh) F. X, 158, J. Humphrey, CB 1918, I, 824.
22) П. П. HI о p ы г и н, А. И. Соколова, Ж. 62, 673 (1930).
6°) P. As ken as у, Eldd, Trogus, Ann. 461, 109 (1928), Seydel, Am. n.
1576999.
я) T. v. d. Linden, CB 1932, 1, 215.
32) E. Macciotta, CB 1930, II, 1068; E. Bamberger, Ber. 30, 1253 (1897).
зз) G. T. Morgan, Jones, Soc. Ind. 1923, 341 T.
З4) O. Wyler, Zur Kenntnis der isomeren Dinitrobenzole, Helv. 15, 23 (1932), cp.
Linden, Helv. 15, 591 (1932).
зз) Hodgson, Beard, Soc. Ind. 45, 91 T (1926)
36) Obermiller, Die orientierende Einfliisse und der Benzolkern, Leipzig, 1909.
3’) A. Klemenc, Eki, CB 1919, I, 827.
36) F. Re verdin, Rec. 48, 838 (1929), Helv. 12, 1053 (1929), Г. n. 164130 (A),
F. VIII, 107.
38) E. Pl azek, CB 1931, 1, 1427.
б®) В e n z а, Б. n. 260568, CB 1927, II, 506.
61) 1G., Б. n. 317304, CB 1930, 1, 2011.
62) 1G„ Г. n. 497809, CB 1930, 11. 986.
63) О. К r e b s, Die Nitrierung der Kohlenwasserstoffe in der stetig betriebenen
Nitrierungsanlage, Teer & Bitumen, 1929, 393; К Kuberschky, Г. n. 287799
(F. Xll, 109); MLB, Г. n. 201623 (F. IX, 105); t. M. Г. n. 228544, CB 1910, 11, 1693,
cp. Ullmann, Enz. d. techn. Chemie, 1 Aufl., 11, 374—375. R. Norris Shreve,
Equipment for Nitration and Sulfonation Ind. Eng. Chem., 24, 1344 (1932).
66) H. Wieland, Ann. 328, 154 (1903), Wieland, Stenzl, Ber.40,4828(1907),
Ann. 360, 299 (1908), Wieland, Ber. 54, 1776 (1921). Сравни H. А. Д е м ь я н о в и
К. В. Сидоренко Ж. 41, 832 (1909).
б5) A. Schaarschmidt, Вег. 57, 2065 (1924).
66) М. В a 11 е g а у, La nitration 4 1’hypoazotite, Bull. (4) 43,109 (1928), В a 11 e g ay,
Rasumejew, Б. n. 262097, CB 1927, 11. 2352, L. A. Pi nek, Am. Soc.49, 2536 (1927),
CB 1928, 1, 27, Г. n. 310772 (H. Wolf) F. X11I, 1104, A. Schaarschmidt,
E. Smo 11 a, Ber. 57, 32 (1924), A. Schaarschmidt, Balzerkewicz, Gante,
Ber. 58, 499 (1925), также 65). м. И. Богданов, Нитрование окислами азота
Ан. Кр. Пр. 3 № 4, 133 (1933).
47) G. В. Taylor, A. S. Richardson (DuP). Am. п. 1640737 (1927).
66) Н. Wieland, Ber. 54, 1777 (1921), ср. W о 1 f f е п s t е i п, Boters, Г.П.
214045, F. IX, 142.
70
«) Grflnau, Г. п. 207170, F. IX, 107.
®°) См. Alice Hamilton, Industrial Poisoning in Making Coal-Tar. Dyes and
Dye-Intermediates, Washington (U.S. Dep. of Labor) 1921, 18; К. E gl i - E. Riist,
Несчастные случаи при химических работах, НХТИ, Ленинград, 1926.
si) Meisenheimer, Вег. 53, 358 (1920), A. Wohl. Г. п. 116790, F. VI, 113.
И) Ср. Desvergnes, СВ 1932, 1, 710..
53) Р. К. В о s е, СВ 1932, 1. 260.
54) Ср. С о 1 vег, Ргi dеа их, Soc. Ind. 1917, 480; О. Wallerius, СВ 1928,.
11, 32а.
55) Knecht, Hibbert, Ber. 36, 166, 1554 (1903), 38, 3318 (1905), 40, 3819
(1907), Koi th of f, Robinson, Rec. 45, 109 (1926). Об источниках ошибок при
определении группы NO2 по методу Лимприхта и о видоизменении его с при-
бавлением двуаммиачно-лимовной соли вместо сегнетовой см. D. F 1 о rentin, Н. Van-
der be rghe, Bull. (4) 27, 158 (1920).
5в) H. H. Hodgson, CB 1931, II, 2319, H odg son, Moore, CB 1924, 1,307.
5?) W. Lesnianski, CB 1923, IV, 497, Veibel 2’).
58) Ср. В. Г. Гу л инов, Ж. X. П. 5, 225 (1928), 6, 214 (1929).
53) Siderfin, Tallantyre, Shannan, Galbraith Б. n. 203060, Soc. Ind.
1923, 1064A, также Gas Ligth & Coke Co (London) Ф. n. 566874, CB 1926, 11, 1903.
60) 1G„ Г. n. 519729, CB 1931, П, 1755.
6») A. Morel, P. Sisley, Bull. 41, 1217 (1927), Hepp, Ber. 10, 1654 (1877).
62) W. M. Rodionow, W. K- Matweew, Ber. 57, 1711 (1924).
63) Cp. Knecht, J. H. Platt, CB. 1926, 1. 359.
61) O. F i s c h e r, Hepp, Ber. 19, 299 (1886), 20, 1247 (1887), M. Ильинский,
Б. M а к с о p о в, Ж. X. П. 1928, 469.
65) 1G., Ф. п. 38059 С. А. 25, 5573 (1931).
66) Ю„ Г. и. 535671, СВ 1932, I, 137.
6’) A. A n g е 1 i, А. Р i е г о n i, Atti ас. Lincei, Anno 320 (5) 32 (1923).
«в) Напр. 1G., Г. п. 499824, 511526, 512722, СВ 1930, И, 982, 1931, И, 1492,
см. ниже — араминирование.
69) Н. Н. Hodgson, A. Karshaw, Soc. 1930, 277, СВ 1930,1,3032. О нитро-
вании хлорбензола в 24-динитрохлорбензол, L. Desvergnes, Chim. & Ind. 26,.
1271 (1931). О нитровании фенола на дннитро- и тринитрофенол, L. Des Verg-
ne s, Le 2.4-dinitrophenol, fabrication, proprietes et emplois, Chim. & Ind. 27, 527
(1932), ib. 26, 507 (1931).
ГЛАВА III
СУЛЬФИРОВАНИЕ
Сульфирование (нередко называемое также „сульфурацией")
имеет назначением образоватьв исходном соединении характерную
группу — SO3H, так называемую сульфогруппу. Чаще всего сульфо-
группа, образуется замещением водорода в ядре ароматического
соединения, хотя имеются и другие методы ее возникновения.
О них будет сказано в своем месте.
Сульфирование — реакционный процесс, имеющий значение и
применение в производстве промежуточных продуктов не меньшее,
чем нитрование. Сульфирование применяется как к соединениям,
имеющим определенные реакционные, следовательно ориентирую-
щие группы, так к углеводородам и наконец к готовым красите-
лям,— в последнем случае—с целью увеличения растворимости
соединений, иногда — с целью сообщить более кислотный характер
красителю.
Как химический метод, дающий определенный производственный эффект, суль-
фирование известно уже более 100 лет. Так, еще в конце XVIII века сульфирова-
нием естественного индиго приготовлялся саксонский синий — дисульфокислота
индиго. Получение индивидуальных сульфокислот ароматических углеводородов
относится к 20-м г. XIX столетия. В 1819 г. Бранд (Brande) наблюдал образова-
ния нового соединения из нафталина от действия серной кислоты, в 1826 г. Фара-
дей получил в нечистом состоянии две изомерных сульфокислоты нафталина.
а) Течение и факторы сульфирования
Сульфирующим агентом служит прежде всего серная кислота
различных концентраций (в технике чаще всего купоросное масло
с содержанием 92—93% H2SO4 или моногидрат, содержащий 100%
H2SO4) и олеум, или дымящая серная кислота, содержащая различ-
ные количества SO3. Наиболее часто находят себе применения
такие сорта олеума, которые достаточно концентрированы в отно-
шении SO3 и в то же время не затвердевают при обычной темпе-
ратуре. Таковы олеумы, содержащие до 25% SO3 и около 65%
SОз. Их удобно транспортировать и отмерять, между тем как
олеум например с содержанием 40% SO3, застывший в кристаллах,
требует длительного разогрева для превращения его в жидкость.
Далее сульфирующими агентами служат хлорсульфоновая кис-
лота (монохлорангидрид серной кислоты) C1SO3H, некоторые веще-
ства, могущие выделять серный ангидрид при реакциях, и — реже —
сернистая кислота в виде солей (при одновременном восстановле-
нии заместителя, уже имеющегося) и иногда кислые соли серной
кислоты.
72
Для наиболее употребительных агентов сульфирования общее
выражение процесса таково;
RH + HOSO3H = RSO3H + H2O. (1)
При избытке.серной кислоты реакция сульфирования протекает, как реакция
первого порядка, при соотношениях между реагентами, близких к эквимолекуляр-
ным,— как реакция второго порядка.
Константа скорости этой реакции изменяется при прочных равных условиях
в зависимости, от концентрации сериой кислоты, и она тем больше, чем выше .эта
концентрация. Константа скорости убывает с ходом сульфирования в связи с нара-
станием количества реакционной воды.
Температурный коэфициент реакции сульфирования (возрастание константы
скорости на каждые 10°) равен 2—‘/>/2 (для сульфирования л-нитротолуол^).
Константа скорости нитрования приблизительно в 1000 раз более, чем у сульфи-
рования, и температурный коэфициент для нитрования выше, именно 3 1).
Выражение процесса (1) дает собственно его результат. Реакция
и в этом случае идет через стадию присоединения молекулы сер-
ной кислоты к молекуле ароматического вещества, вначале веро-
ятно без участия атомных связей, затем с активированием двойной
связи ароматического ядра. Есть много оснований полагать, что
серная кислота активирует ароматическое ядро более энергично,
чем другие агенты реакции замещений. Применительно к реакции
с бензолом ход сульфирования можно представить таким образом:
4-но
so3H
Подобный продукт присоединения становится уже стабильным в случае наличия
в циклическом углеводороде изолированной двойной связи. ”
дает при обработке серной кислотой и уксусным ангидридом
генсаиолсульфокислоту (II) (Фризе) 2).
Н2
Так, циклогексен (1)
количественно цикло-
но
Н.
н/'
, "С™
2\/\SO3H
н2 н2 3
Недавно на примере нитробензола доказана возможность
н,.
SO3H
н.
Недавно на примере нитробензола доказана возможность образования вполне
определенного соединения с сериой кислотой у однозамещениыхбевзола. Получен-
ное соединение отвечает до составу формуле C6H&NO2H2SOi и, вероятно, построено
по вышеприведенной схеме 3).
Выделяющаяся при реакции вода постепенно понижает концен-
трацию сульфирующего агента и в некоторой мере содействует
протеканию обратной реакции гидролиза, ниже более подробно
разобранной:
RSO3H -b Н2О -> RH 4- H2SO4.
В реакционной смеси устанавливается равновесие между прямой
и обратной реакциями (в пользу первой из них). При этом ско-
рость прямой реакции (сульфирования) постепенно снижается по
мере разведения серной кислоты выделяющейся в процессе водой.
н
73-
При определенном для каждого сульфирования разведении,
соотв. концентрации, серной кислоты скорость сульфирования ста-
новится настолько малой, что при этом можно считаться практи-
чески с остановкой сульфирования. Чрезвычайно длительное взаимо-
действие сульфируемого вещества и серной кислоты предельной
концентрации может повидимому сопровождаться, хотя и незначи-
тельным, эффектом сульфирования (И. С. Иоффе).
Предельная концентрация серной кислоты, при которой она
практически уже не сульфирует исходное вещество, называется
(Гюйо, Курго) к данного сульфирования и выражается в процен-
тах SO3 4. (Содержание SO3 в моногидрате равно 81,6%)- кон-
центрация практически несульфирующей серной кислоты, различна
для сульфирований различных органических веществ.
По Kyp.ro, те для моиосульфироваиия бензола 66,4, для моиосульфнроваиня
нафталина (при 55—60°) — около 56 и (при 160°) около 52, для дисульфироваиия
нафталина (при 10°)<82, (при 80—90°) 80,8 и (при 160°) 66,5. Нитробензол,
л-иитротолуол имеют те для моносульфирования близкий к 82. Мы видим иа этих
примерах, что в некоторых случаях, как в двух последних, концентрация практи-
чески иесульфирутощей кислоты те достигает и даже превосходит концентрацию
моиотидрата. В таких случаях, естественно, требуется применение еще более кон-
центрированного агента сульфирования, каким является олеум, содержащий свобод-
ный серный ангидрид. Серная кислота с концентрацией те, остающаяся и процессе
сульфирования за счет разведения более крепкой кислоты реакционной водой,
является уже не работающей более составной частью реакционной смеси, так на-
зываемой отбросной кисчотой. При малой разнице между концентрацией исходного
сульфирующего агента и те (например случай моиосульфироваиия бензола моно-
гидратом с концентрацией 81,6% SO3, те = 66,4%) количество отбросной кислоты
может быть очень значительным.
Вообще же обязательность образования серной кислоты „нерабо-
тающей" концентрации те делает избыток сульфирующего агента про-
тив рассчитанного по молярным отношением часто весьма большим.
Если требуется х кг серной кислоты с концентрацией а процентов SO8 для
моиосульфироваиия килограммолекулы органического вещества RH, то 1 моль
SO3 [ = 80] из нашей кислоты будет израсходован на образование сульфогруппы,
а остальная часть, т. е. х—80 войдет в состав отбросной кислоты, по условию имею-
щей концентрацию к.
Выразив это уравнением, получим:
Л 100 = 80 + - 80) j00
откуда
_ 80(100 —те)
а — те ’
т. е. чем выше концентрация а исходной серной кислоты, чем больше знаменатель, тем
меньшее количество х кислоты может быть применено. В практике одиако прихо-
дится считаться с необходимостью предела в концентрации исходного сульфирую-
щего агента ввиду возможйости вторичных реакций при применении слишком кон-
центрированных сульфирующих средств: образования полисульфокислот, сульфонов
и т. д. Возможно также образование сульфокислот с различным положением сульфо-
группы в зависимости от концентрации сульфирующего агента.
Мы увидим ниже, какими средствами пользуются для умень-
шения количества отбросной серной кислоты концентрации те.
О^ень важным фактором сульфирования является температура.
Помимо того, что при переходе к температуре, более высокой,
чем допустимый для реакции предел, можно осложнить процесс 74
74
(2)
нежелательными изменениями — окислением органического веще-
ства, конденсациями, образованием сульфонов — сохранение тем-
пературы в строго определенных для каждого процесса пределах
важно потому, что в зависимости от температуры весьма часто
сульфогруппа фиксируется различными атомами ядра. Так, фенол
при сульфировании серной кислотой на холоду дает преимуще-
ственно о-сульфокислоту, при сульфировании же при температуре
водяной бани «-сульфокислоту, причем в пара-продукт превра-
щается и образовавшаяся сначала о-сульфокислота.
ОН
ОН
SO3H HO3S-
Образование сульфокислот нафталина дает очень много приме-
ров влияния температуры на положение сульфогруппы. Наиболее
простой случай — получение моносульфокислот нафталина. Сульфи-
рование нафталина моногидратом при низких температурах (35—60°)
дает в качестве главного продукта а-сульфокислоту; та же
реакционная смесь при высокой температуре (160°) образует пре-
имущественно ^сульфокислоту. Если сначала получить «-сульфо-
кислоту (сульфированием при низкой температуре) и затем, не
выделяя продукта, нагреть все до 160°, то по мере хода нагрева-
ния будет заметно уменьшение количества а-сульфокислоты и по-
степенное возрастание количества [3- изомера Б). Здесь, как и во
многих аналогичных случаях сульфирования, имеются равновесные
системы из нескольких компонент, и каждой температуре при до-
статочной выдержке отвечают строго определенные соотношения
между количеством отдельных компонент (здесь a-и р-сульфо-
кислот). Почти всегда вызываемый повышением температуры сдвиг
равновесия в пользу одного продукта объясняется участием обра-
зующейся при сульфировании воды, которая, несмотря на присут-
ствие избыточной серной кислоты в реакционной массе, вызывает
отщепление сульфогруппы (в виде молекулы серной кислоты) от
молекулы сульфокислоты, образовавшейся при более низкой тем-
пературе (гидролиз сульфокислоты), и регенерирует первоначалшое
вещество, которое переходит при повышенной температуре в друг
гой сульфоизомер с более устойчивой по отношению к гидролизу
сульфогруппой, например
SO8H
/\/\
+H2so4 !
при 35—60 |
с повыше-
нием темпе-
ратуры
4-h2so4
при 160°
H,SO4
при 160°
\/\/
/\/\/SO3H
75
Кроме гидролиза в смеси могут иметь место и необратимы6
реакции изомеризации.
Аналогичные равновесия в сульфурационном процессе с участием гидролити-
чески действующей воды констатированы были при сульфировании а-нафтиламина 6)
и во многих других случаях. В этих процессах следовательно перемещение («стран-
ствие") сульфогруппы происходит в связи с временным образованием в смеси не-
сульфированиого продукта.
Интересен пример перехода сульфогруппы у 1-нафтиламин-8-сульфокислоты
при нагревании ее долгое время при 75—80° с концентрированной серной кислотой
в 4-место (с образованием нафтионовой кислоты) 7). Здесь вероятно также имеет
место гидролиз SO3H у менее стойкой формы (1.8) и сульфирование в более стой-
кий изомер. Любопытно, что по опытам автора и К. А. Грибова при обработке
сульфокислот а-нафтиламина разведенной серной кислотой (3—5%) при температуре
до 180° сульфогруппа в 8-месте оказывается более устойчивой, чем в 4-месте.
Констатировано и в технике используется в широких размерах
иное перемещение группы SO3H. Именно, многие соли первичных
аминов и серной кислоты при нагревании сухими до высокой тем-
пературы (около 180°) переходят в сульфокислоты аминов, главным
образом пара-ряда. Такой метод приготовления сульфокислот пер-
вичных аминов, впервые испытанный на получении из сульфата
анилина сульфаниловой кислоты и из сульфата нафтиламина наф-
тионовой кислоты, носит в технике названия метода „запекания".
/y/NH2- H2SO4
; —н2о
NH2 • H2SO4
Промежуточно при подобном сульфировании аминов из кислого сульфата ве"
роятно получается соответственная сульфаминовая кислота, которая далее перегруп’
пировывается в сульфокислоту амина. Для фенилсульфаминовой кислоты (1) Бам-
бергером констатированы два перехода от действия концентрированной серной
кислоты: при более низкой температуре в о-сульфокислоту анилина, при высокой
температуре (180—190°) в сульфаниловую кислоту 8).
NH2- H2SOj
-Н2б
76
р-нафтиламнн при запекании с сериой кислотой дает 2-амияо-6-сульфокислоту нафтил-
амииа (2.6 конфигурация у нафталина во многом напоминает размещение пара)
zNH2 • H2SO4 /X/X/NH,
I | ; | I |
\/\/
Аналогично 1.8 - нафтилендиаминсульфат дает 1.8 - иафтилендиамин - 4 - сульфо-
кислоту 9). а-Амииоантрахинонсульфат при нагревании до 240— 245° дает 1-амнно-
антрахиноно-2-сульфокнслоту. В данном случае предложено применять серную
кислоту в виде кислых солей (бисульфатов) i°).
Переход остатка сульфокислоты от азота аминогруппы в ядро
является частным случаем общей тенденции аминосоединений, имею-
щих у азота электроотрицательный радикал (ОН, С1), уступать его
ядру.
Имеется случай перехода сульфогруппы, связанный с повышением температуры,
который трудно объяснить гидролизом, так как он происходит в безводной среде.
Именно, нагревание сухой натриевой соли нафтионовой кислоты и кипящем нафта-
лине (218°) или самой по себе приводит к образованию изомерной 1-нафтиламии-2-
сульфокислоты 1Х)
nh2 nh2
/\ \ z\A/SOsNa
UJ LU
I
SO3Na
Объяснения этой странной и не дающей количественного выхода реакции, которая
аналогична превращению 1-нафтол-4-сульфокислоты в 1-иафтол-2-сульфокислоту 12),
пока не имеется, хотя Эрдманом высказан взгляд, что она также представляет
собой случай гидролитического превращения п).
Есть еще один фактор, влияющий на положение сульфогруппы
при некоторых случаях сульфирования, именно — участие катали-
з 1торов.
Вопрос о значении катализаторов в процессе сульфирования во
всей его широте еще не подвергался систематическому изучению.
Довольно случайно было открыто окисляющее действие серной
кислоты на нафталин при высокой температуре в присутствии
ртути. Это наблюдение Е. Зап пера (Е. Sapper) было использо-
вано в течение некоторого времени для фабрикации фталевой ки-
слоты из нафталина 13).
М. А. Ильинским в 1891 г. 14) и позже Р. Шмидтом 1Б)
было открыто значение ртути как катализатора при сульфировании
антрахинона. Сульфируя антрахинон в отсутствии ртути, получают
^-моносульфокислоту:
77
между тем в присутствии малйх количеств ртутной соли сульфо-
группа закрепляется в а-месте:
SO8H
СО СО |
/\/\/\ /\/\/\
Этот чрезвычайно интересный факт каталитического влияния
ртути на положение сульфогруппы в антрахиноне привлек внима-
ние исследователей в позднейшее время. Можно считать доказан-
ным, что а-сульфокислота образуется только в присутствии ртути
и что нагревание чистой а-сульфокислоты с серной кислотой до
180° не приводит к превращению ее в [3-кислоту. Если же в смеси
с серной кислотой и а-кислотой имеется ртуть, то тогда в резуль-
тате нагревания получается р-кислота. Очевидно в этих условиях
присутствие катализатора содействует гидролизу а-кислоты (кото-
рый без катализатора не происходит), затем избыток серной ки-
слоты образует уже более стойкую р-кислоту (А. Мейер).
Вероятно образование ртутно-органического соединения антрахинона в присут-
ствии серной кислоты содействует вхождению сульфо1руппы в a-мест о, ибо в остат-
ках от сульфирования со ртутью можно констатировать всегда ртутвс-срганическое
соединение (с иенонизнроваииой ртутью). Предположительно течение сульфирования
проходит через стадии:
С14Н8О2 + HgSO4^ (CUH7O2) • HgSO4H (а)
С14Н7О2 • HgSO4H 4- SO3-> C14H7O3SO3H + HgSO4.
Скорость образования а-сульфокислоты пропорциональна (для малых количеств
ртути) концентрации HgSO4 1е).
Имеется возражение против допущения образования ртутно-органического
соединения и утверждение, что окись ртути образует с группой СО антрахинона
соединение со связью через кислород. Такая группа может иначе ориентировать,
чем СО 17).
Недавно в патенте 1G заявлено о значении ртути (в виде HgSO4) как. напра-
вляющего катализатора для получения 4-сульфофталевой кислоты из фталевого
ангидрида. Л а у е р в нижецитированной работе не подтвердил данных этого
патента, установив, что при нормально проведенном сульфировании в присутствии
ртути первым продуктом является 3-сульфофталевая кислота, переходящая далее
в 3.5-дисульфокислоту 18). Ртутный катализатор предложен также при сульфиро-
вании соединений пиридинового ряда 19)<
При сульфировании некоторых антрахиноновых производных констатировано, что
ртуть и селен являются катализаторами окисления, между тем соли ванадия — уско-
рителями собственно сульфирования 20).
Морман исследовал влияние катализаторов на равновесие между м- и л-ди-
сульфокислотамн бензола при нагревании до 240—250° и констатировал наиболь-
шее значение ртути для сдвига равновесия в пользу пара-продукта (без катализа*
тора отношение изомеров л:л«=1,3:98, с ртутью — л: л =1:2).
Недавно Л а у е р о м установлено было, что своеобразное поведение ртути при
сульфировании антрахинона, проявляющееся в смещении положения сульфогруппы,—
лишь частный случай влияния этого катализатора при сульфированиях соединений,
имеющих в ядре заместители, ориентирующие в мета место. Лауер доказал, что
действие серной кислоты, ие содержащей воды (не слабее моногидрата), на ртутно-
органические ароматические соединения типа RHgX, где X — неорганический анион,
сопровождается образованием сульфокислоты, содержащей SOgH у того атома угле-
рода, с которым был связан атом ртути. Действие серной кислоты, содержащей
78
воду, на такое ртутно-органическое соединение проявляется в его разложении,
и если в последием случае имеет место сульфирование, то сульфогруппа стано-
вится в то место ядра, которое ориентируется со стороны заместителя (а ие
Hg-атома).
Известно, что прн меркурироваини ароматических соединений Обработкой их.
например растворами ртутиых солей остаток, содержащий ртуть, устанавливается
в о-л-положение к имеющемуся заместителю, независимо от его природы (т. е. от
того, принадлежит ли он к о-л- или .«-ориентирующим).
Вследствие этого можно ожидать разницы в ориентации между сульфированием
в присутствии и в отсутствии ртутн для тех лишь соединений, где имеется ^-ориен-
тирующий заместитель. Такая разница и была установлена Л а у е р о м при сульфиро-
ваниях с ртутью и без ртути нитробензола, бензойной кислоты н сульфокислоты
бензола (заместители: NO2, СООН, SO3H), причем обнаружено, что сульфирование
со ртутью дает смесь изомеров с пониженным против нормы содержанием .«-сульфо-
кислоты и значительно повышенным л- 1илч л- и о-) сульфокислоты. Понятно, что
сульфирование в присутствии ртути таких соединений, у которых заместители при-
надлежат к л-о-ориентирующим, ие дает изменений в содержании изомеров срав-
нительно с сульфированием без ртути.
Сульфирование анилина в присутствии соли ртути при 180° протекает бурно
с обугливанием органического вещества и выделением сернистого газа. При
125—130° наблюдается также раскисление серной кислоты и образование неболь-
ших количеств бензидина (за счет окисления анилина) 22)
2CeH5NH2 4- О = H2N • С6Н4 С6Н4 • NH2 + HLO.
В недавнее время отмечено некоторое влияние иодного катализатора па реак-
цию сульфирования в смысле облегчения реакции 32).
Ускорение реакции сульфирования со стороны катализаторов изучалось иедс-
статочио. Имеются наблюдения, что катализаторы сульфирования бензола можно
разделить иа 2 группы: слабые (Си, Hg, V, Сг, К, Li, Na), и более сильные (V-f- Na
в определенных отношениях). Повиднмому элементы первой группы периодической
системы ускоряют образование дисульфокислот, другие катализаторы кроме V -ф- Na
благоприятствуют моиосульфироваиию 24).
б) Практика проведения сульфирования
Выбор сульфирующею агента не является случайным, но дол-
жен быть обоснован для каждого отдельного случая с учетом
максимального выхода желаемого продукта и минимальных отхо-
дов отработанной кислоты.
Для сокращения потери серной кислоты при сульфировании бензола в бензол-
сульфокислоту и при последующих обработках в Америке практикуется непрерыв-
ный способ сульфирования (D е nn is - В ul I), основанный иа использовании замет-
ной растворимости сульфокислоты бензола в бензоле (2 — 3%) при экстрагировании
последним сульфурациониой смеси. Бензол идет навстречу движению сульфосмеси
в батарее из четырех экстракторов; аппарат, содержащий больший процент сульфо-
кислоты, служит сульфуратором. Отработанная серная кислота в 70— 77% H2SO4
отходит практически лишенная сульфокислоты. Сульфокислота вымывается из бен-
зольного раствора водой и почти ие дает примесн сульфата при нейтрализации 25)-
Своеобразным сульфирующим агентом являются полисульфаты
общей формулы Me*H3 (SO4)2 *, получаемые нагреванием бисульфата,
например NaHSO4, с серной кислотой (не обязательно 100%-ной,
необходимо только нагревание для удаления воды).
По описанию Ламбертса 2в) сульфирование при помощи
полисульфатов протекает более гладко, без побочных реакций
и с лучшими выходами, чем при работе с одной серной кислотой.
Температура плавления полисульфата NaH3(SO4)2 лежит между
* Me1— одновалентный металл щелочной группы.
7»
95—100°, почему работа с ними возможна и при низких темпера-
турах. Это обстоятельство позволяет готовить по этому методу
и моносульфокислоты. В некоторых случаях применения полисуль-
фата (для полисульфокислот) обнаружилось преимущество калие-
вого полисульфата пред натриевым в отношении выходов и каче-
ства сульфокислот. Возможно, что при применении полисульфатов
играет известную роль вышеуказанное каталитическое влияние
ионов К или Na. В этом случае при обработке продукта сульфиро-
вания известковым молоком получаются прямо щелочные соли
сульфокислот:
RSO3H • KaHSO4 + Са(ОН)2 -> RSO3Na + CaSO4 + 2Н2О.
Полисульфаты находят применение в дисульфировании р-наф-
тола27).
Хлорсульфоновая кислота в качестве агента сульфирования
интересна главным образом как дающая возможность получить не
самое сульфокислоту, но ее хлорангидрид, сульфохлорид, общей
формулы RSO2C1.
Примение хлорсульфоновой кислоты дает возможность напра-
влять реакцию двояко. При взаимодействии молекулярных коли-
честв сульфируемого соединения и хлорсульфоновой кислоты
в первую очередь образуется сульфохлорид:
RH -J- HOSO2C1 = RSO2C1 -I- Н2О.
Образовавшаяся вода, действуя на хлорсульфоновую кислоту,
разлагает ее по уравнению:
HOSO2C1 4- Н2О = HOSO2OH + НС1.
Полученная таким образом серная кислота может сульфировать,
причем образующаяся вода опять разлагает оставшуюся хлорсуль-
фоновую кислоту. В результате реакции получается значительное
количество сульфокислоты и малое — сульфохлорида, как будто
реакция протекала по уравнению:
RH + C1SO2OH = RSOsH 4- НС1.
Поэтому для того чтобы направить реакцию сульфирования
хлорсульфоновой кислотой в сторону получения сульфохлорида,
требуется применять весьма большой избыток хлорсульфоновой
кислоты (4—5-кратный).
Хлорсульфоновая кислота является тем не менее весьма инте-
ресным сульфирующим агентом тогда, когда получаемые с ее
помощью сульфохлориды легко выделяются или разделяются,
особенно при сульфировании толуола.
Предложено с целью уменьшения расхода хлорсульфоновой кислоты применять
ее для сульфирования в количествах, лишь немногим превышающих молекулярные,
добавляя зато в качестве водотнимающего средства олеум и используя вместо
выделенной хлорсульфоновой кислоты техническую смесь из соляной кислоты или
ее солей и олеума й).
Реакция хлорсульфоновой кислоты с углеводородами в применении к бензолу,
толуолу, ксилолам, дифенолу, нафталану была изуиена Поллаком и сотрудни-
ками29^
80
Метиловый эфир хл^рсульфоновой кислоты, C1SOsCH3, имеющий значение
в газовой войне вайантит (vaillantite) реагирует не полно с бензолом, толуолом,
хлорбензолом, давая как главный продукт эфир сульфокислоты RSO3CH3. Выход
уменьшается от вторичной реакции: RSO3CH3 4- НС1 = RSO3H -|-СНзС130—
В недавнее время изучено сульфирующее действие фторсульфоиовой кислоты
FSOgH. Подобно хлорсульфоиовой кислоте, она дает с углеводородами и их заме-
щенными сульфокислоты и сульфофториды RSO2F. Остаток SO2F становится всегда
иа то же место, что и SO2C1. Между группами 8О2С1 и SO2F существуют отноше-
ния взаимного обмена при действии хлор-(соотв. фтор-) сульфоновой кислоты 32).
Сульфофториды бензола можно получить из сульфохлоридов нагреванием их с вод-
ным раствором нейтральной соли фтористоводородной кислоты (например KF) 33).
Вряд ли можно рассчитывать иа введение в практику фторсульфоновой кислоты,
хотя некоторые свойства сульфофторидов [стойкость к гидролизу — их можно пере-
гонять с водяным паром, — стойкость по отношению к восстановителям, дающая
возможность получать из нитросульфофторидов аминосульфофториды 32)J делают их
интересными объектами лабораторного исследования.
Полный хлорангидрид серной кислоты, хлористый сульфурил SO2C12 в качестве
сульфирующего агента практически не имеет значения. Мы встретимся с этим сое-
динением в главе о хлорировании.
В качестве побочного продукта сульфирования образуются суль-
фоны, общей формулы RSO2R. Их образование происходит или из
двух молекул сульфокислоты по уравнению
2RSO3H = RSOaR + H2SO4 3i)
или (более вероятно) из молекулы сульфокислоты и несульфиро-
ванного вещества с выделением молекулы воды
RSOsH 4- HR = RSO2R 4- Н2О.
Содействуют образованию сульфонов: значительная концентра-
ция сульфирующего (водоотнимающего) агента при наличии суль-
фокислоты и непросульфироцанного вещества и высокая темпера-
тура.
При сульфированиях более затрудненных, где прибегают к про-
должительному и энергичному нагреванию реакционной смеси,
нужно считаться с возможностью нежелательного образования суль-
фонов и стремиться свести к минимуму их количество ЗБ).
Сульфоны — соединения, нерастворимые в холодной воде, раство-
римые в некоторых органических растворителях (спирте, эфире)
и в водных растворах сульфокислот и их солей.
Некоторые производные сульфонов по-
лучают значение для красочной промышлен-
ности, как например амины типа А и подоб- । ।
ныеему[1-амино-4-диалкил-(диаралкил-) ами- А |
но и т. п.] сульфоновые дериваты, в каче- м/\/\сл /-----\ гы
стве диазосоставляющей при холодном кра- (CH;!)2N 4SO2 —, ,— СН8
шении 36).
Выше было отмечено, что теневой стороной процесса сульфи-
рования является накопление значительных масс отбросной кис-
лоты с предельной концентрацией тс, вообще довольно высокой.
Потери здесь неизбежны, так как в отличие от того, что имеет
место при нитровании, отбросная кислота сульфирования уходит
с производства в такой форме, что не может быть легко сконцен-
трирована (или в виде солей или будучи слишком разведенной).
Так как снижение концентрации тс по сказанному практически не-
возможно, то задача уменьшения потерь должна быть понята как
6 Зак. 2611. — Н. Ворожцов
81
сведение до минимума количества отбросной щислоты, а это значит
что разрешение нужно искать в устранении воды из самой реакцион-
ной смеси и в приближении количества серной кислоты к теорети-
чески требуемому количеству.
Одним из таких способов является применение для сульфиро-
вания не серной кислоты, а серного ангидрида SO3 самого по себе
или в виде соединений, могущих его выделять.
Если обратиться к вышеприведенному уравнению (2) (стр. 74), то видно, что при
прогрессивном возрастании «(концентрации SO3 в применяемой кислоте) х — количе-
ство кислоты уменьшается и при «=100 (т. е. при применении серного ангидрида
х = 80; таким образом для сульфирования молекулы органического вещества тре-
буется молекула SO3).
Сам ангидрид может быть применен только в отдельных исключительных слу-
чаях. Так, удалось, насыщая расплавленный фталевый ангидрид серным ангидридом,
получить сразу чистый 4-сульфофталевый ангидрид 37).
Действие парообразного SO3 на антрахинон при 150—170° приводит к образо-
ванию главным образом ^-моносульфокислоты 38). Реакция с серным ангидридом
протекает здесь медленно и в результате является собственно реакцией присоеди-
нения (без потери каких-либо составных частей реагирующих молекул):
RH -J- SO3 = RSO3H.
Вода при реакции не должна образоваться, следовательно нет и отработанной
кислоты.
Судя по описанию испытавших этот метод сульфирования Вальдмава и
Ш венка, процесс (для антрахинона н фталевого ангидрида) протекает крайве
медленно сравнительно с обычными методами. Не является ли это доказательством
той акгнвирующен роли, какую играет молекула H2SO4, а не SO3 прн сульфирова-
ниях с олеумом? Серная кислота вероятно легче вступает в реакцию присоединения.
Серный ангидрид очень энергично реагирует со многими орга-
ническими веществами, и сульфирование свободным SO3 может при-
вести к осложнениям реакции вследствие поднятия температуры,
обугливания, образования сульфонов и т. п. Поэтому более удоб-
ным способом кажется применение SO3 в виде его соединений,
особенно с пиридином и подобными ему основаниями.
Действующим веществом является здесь ангидрон-сульфо-
кислота пиридиния (соотв. его гомологов и аналогов), получаемая
действием SO3 на пиридин в присутствии безводного растворителя
(CC1J
4-so3= I !
I
N N\
I /°
O2SZ
Соединение это довольно стойко, сохраняется на воздухе без изменения, устой-
чиво при нагревании до 200° и не быстро реагирует с водой прн обыкновенной
температуре.
Ангидрон-сульфокислота отдает SO3 органической (и неорга-
нической) молекуле НХ по схеме:
CBHBN • SO34-HX CBHBN • HO3SX 39).
При сульфировании нафталина ангидро-М-сульфокислотой пиридиния наблюда-
лось интересная разница с обычным сульфированием в том, что при повышенной
82
температуре (до 170°) получалось относительно больше а-изомера, а при понижении
(до 140°) — относительно больше fi-изомера 40).
Сульфирование антрацена ангидро-К-сульфокислотой пиридиния в среде пири-
динкеросин при 165—175° дает продукт, состоящий почти вацело из а-моно-
сульфокислоты антрацена, между тем как сульфирование антрацена олеумом или
хлорсульфоновой кислотой в среде уксусной кислоты (при 95°) приводит к смеси
почти равных количеств а-и 0-моиосульфокислот. В области сульфирования антра-
цена до сих пор много неясного 41).
Ниже, особенно в главе об ацилировании, мы встретим много примеров приме-
нения ангидро-К-сульфокнслоты пиридиния и подобных ей веществ для введения
остатка серной кислоты в кислородные, азотные и серу содержащие группы.
Ангидро-^-сульфокислота пиридиния может быть получена из
пиридина не только воздействием SO3, но и олеума, а также хлор-
сульфоновой кислоты и хлористого сульфурила 42).
Имеется предложение получать эту н хинолиниевую ангидро-К-сульфокислоТу
действием пиросульфатов щелочных металлов (К252О7, Na2S2O7) на соответственное
основание при нагревании (до 120°) 43).
Наконец можно, казалось бы, применить серный ангидрид
в растворах таких веществ, с которыми он не реагирует при тем-
пературе реакции. Такой метод сульфирования ряда веществ
(в большинстве углеводородов) серным ангидридом в хлорофор-
менном растворе был испытан Курто и сотрудниками 44) и дал
удовлетворительные результаты с выходами, близкими к количе-
ственным при получении моносульфокислот. При дальнейшем суль-
фировании моносульфокислоты нафталина обнаружено образование
продукта присоединения SO3 к молекуле дисульфокислоты, которое
снижало выхода дисульфокислоты.
При применении серной кислоты в качестве сульфирующего
средства возможно также освободиться от реакционной воды
(устранить отработанную кислоту с концентрацией л). Наиболее
разработанным методом является увод воды с парами вещества,
подвергающегося сульфированию. Сульфируя например бензол,
можно вводить в реакцию серную кислоту с концентрацией ниже л.
При нагревании до кипения бензола его пары уносят воду, подни-
мают концентрацию кислоты до начала сульфирования, а затем,
уводя постепенно реакционную воду, дают возможность использо-
вать до конца серную кислоту. Практично, разумеется, бензол,
сконденсированный из пара, по отделении его от воды вводить
обратно в реакционный сосуд 45).
Сульфирование нафталина в парах (220°) было испытано в Аме-
рике и дало преимущественно рр-дисульфокислоты (2.7 главным
образом) 46).
Можно также устранять воду, образующуюся при реакции суль-
фирования, сообщая сульфурационный прибор с вакуумом. Вода
в виде пара уходит (вместе с частью органического вещества,
если оно летуче) из нагретого прибора при понижении в нем да-
вления. Этим способом можно например из моносульфокислоты
бензола получить хорошие выхода дисульфокислоты 47). Также
хорошо этот способ проявил себя при получении 0-сульфокислоты
нафталина 48).
Можно наконец вводить в реакционную смесь добавочное не-
сульфирующееся вещество с невысокой температурой кипения,
6*
83
задачей которого являлось бы увлечение со своими парами воды,
образующейся при реакции. Этот способ, понятно, особенно удо-
бен по отношению к сульфируемым веществам с высокой темпе-
ратурой кипения 49). Вспомогательной жидкостью может быть
четыреххлористый углерод, легкие нефтяные углеводороды, угле-
кислый газ и т. п. Сравнительно со способом, основанным на при-
менении разрежения, это видоизменение особых преимуществ не
имеет.
При проведении процесса сульфирования обычно в состав реак-
ционной смеси входят только два компонента: сульфирующий
агент и сульфируемое вещество. В некоторых частных случаях одна-
ко сульфируемое вещество вводится в состоянии раствора в каком-
либо неизменяемом в процессе растворителе. Это облегчает взаимо-
действие, если сульфируемый продукт сам не легко образует
в условиях реакции гомогенную смесь с сульфирующим агентом,
особенно если взаимодействие должно протекать быстро. Как при-
мер такого ведения процесса можно привести сульфирование
^-нафтола на 1-сульфокислоту (оксикислоту Тобиаса), которое
осуществляется обработкой олеумом или хлорсульфоновой кисло-
той р-нафтола в растворе частью в суспенсии нитробензола или
о-нитротолуола 50). Нитроуглеводороды здесь конечно удобнее,
чем ранее рекомендованный сероуглерод, хотя их токсичность зна-
чительно понижает преимущества их применения.
Имеются предложения сульфировать первичные амины в рас-
творе тетрахлорэтана хлорсульфоновой кислотой 6I) или серным
ангидридом 62), при этом будто бы сульфогруппа становится лишь
в орто-положение к аминогруппе.
Сульфирование аминов, особенно тех, которые можно сульфировать методом
запекания (анилин, толуидины, ксилидины, хлоранилнны, а- и р-нафталамины), пред-
ложено проводить в смесях с высококипящими иидиферентными по отношению
к серной кислоте и к амину органическими растворителями (хлорбензол, сольвент-
нафта, керосин, трансформаторное масло). Здесь мы имеем разновидность только-
что отмеченного метода, так как с парами растворителя удаляется реакционная
вода 53).
Уксусная кислота с добавкой уксусного ангидрида в качестве среды при суль-
фировании фенолов и их эфиров дает количественные выходы сульфокислот (фе-
нола, анизола, пирокатехина, гваякола, анетола) 51)-
Прибавление пористого вещества в реакционную среду рекомендовано было
давно как ускоряющее процесс (повидимому от увеличения поверхности). Таким
веществом, пористым и химически безразличным, является инфузорная земля (ки-
зельгур) к/.
В практике реакция сульфирования выполняется в металличе-
ских, чаще всего — в чугунных аппаратах, называемых сульфура-
торами. (Вихельгаус считает чугун неподходящим материалом
аппаратуры для сульфирования с олеумом при высокой темпера-
туре.) Сульфураторы снабжены мешалкой, паровой рубашкой для
нагревания и змеевиком или трубопроводом к рубашке для охла-
ждения водой. За границей практикуется газовый нагрев голых
сульфураторов и охлаждение их брызгом из дырчатой трубы, коль-
цом обнимающей верх сульфуратора. Сульфуратор кроме прочей
арматуры непременно имеет гильзу для термометра, входящую
в реакционную массу.
Порядок прибавления компонентов реакционной смеси может
84
Рис. 2. Сульфуратор.
быть различен в зависимости от условий и назначения данного
сульфирования. Температурные условия и свойства исходного ма-
териала больше всего играют роль при определении приемов вве-
дения составных частей смеси. Твердые исходные материалы, если
они вводятся нерасплавленными, должны быть хорошо измель-
чены, чтобы реакция протекала одинаково скоро со всеми пор-
циями сульфируемого вещества.
В иных сульфированиях, особенно требующих повышенной тем-
пературы, серную кислоту прибавляют к жидкому (расплавлен-
ному) веществу.
При обычном сульфировании
количество серной кислоты рас-
считывается по входящему во
взаимодействие избытку SO3 или
H2SO4 свыше ее количества, ос-
тающегося в отработанной ки-
слоте предельной концентрации.
Общее молекулярное количество
серной кислоты (также хлор-
сульфоновой кислоты), расходуе-
мое на сульфирование, может
быть высоким: 2—5-кратным от
стехиометрических отношений.
Полисульфокислоты получа-
ют обработкой исходного про-
дукта или сразу всем потреб-
ным количеством серной кисло-
ты, или методом ступенчато-
го сульфирования. Последний
характеризуется прибавлением
сульфирующего агента в несколь-
ко приемов, при этом для каж-
дой „ступени сульфирования"
(ввода новой сульфогруппы) вы-
бираются надлежащая концен-
трация сульфирующего агента и
температура взаимодействия.
По окончании прибавления
всего количества температуру,
при размешивании, держат на
желаемой высоте. От тщательности соблюдения всех условий (как
в отношении концентрации и относительной чистоты от посторон-
них примесей сульфирующего агента и исходного материала, так
и сохранения ровной повсюду темперагуры реакционной массы —
здесь играет роль целесообразно сконструированная мешалка) за-
висит успех сульфирования и последующих операций выделения
сульфокислот.
За сульфированием следят, отбирая пробы от смеси и испыты-
вая растворимость их в воде, соответственно в растворе соды,
поваренной соли и т. п. Определяется иногда количество свобод-
ной серной кислоты в смеси. Введение одной сульфогруппы ска-
85
зывается на проявлениях растворимости у продукта. При сульфи-
ровании бензола взаимодействие производится на практике в за-
крытых сульфураторах и за концом сульфирования следят по
спаду давления в аппарате (от понижения количества бензола
в паровой фазе).
Для выделения продуктов сульфирования реакционную смесь
подвергают разведению (холодной водой или льдом) или нейтра-
лизации. Часто практикуется также метод высаливания. Выбор
метода выделения зависит больше всего от свойств продукта.
Моносульфокислоты аминов малорастворимы в разведенных
кислотах и выпадают в осадок при разведении водой реакционной
смеси. Они растворимы в виде щелочных солей и потому очи-
щаются переводом в раствор действием соды, фильтрованием от
примесей и последующим подкислением.
Внесение реакционной массы в раствор поваренной соли содей-
ствует образованию натриевой соли сульфокислоты, которая менее
растворима в растворе соли и может выпасть при этом. Иногда
при этом полезна предварительная, хотя бы частичная, нейтрали-
зация кислого раствора содой. Таков случай выделения р-сульфо-
кислоты нафталина. Практикуется иногда высаливание хлористым
калием. Некоторые сульфокислоты образуют характерные, мало-
растворимые в кислой жидкости соли с металлами высших групп
периодической системы (Mg, Fe). Их удобно тогда выделять в виде
таких солей.
Так, .и-сульфокислота нитробензола выделяется по прибавлении железного
купороса в виде железной соли «в), 4.8-днсульфокнслота 2-нитронафталина —
в виде магниевой соли от введения Mg- в кислую смесь, также в виде солей цинка,
марганца, кобальта или никеля при прибавлении окнслов или солей этих металлов
в разведенную после реакции нитрования смесь 67).
К сожалению, систематического исследования растворимости
солей сульфокислот и их характерных реакций не производилось,
или по крайней мере такие данные не опубликованы. Возможно,
что при систематическом изучении найдутся еще удобные соче-
тания металла и сульфокислоты, которые дадут метод выделения
некоторых сульфокислот.
Иногда после полной нейтрализации содой раствор натриевой
соли сульфокислоты подвергается сгущению с выделением пред-
варительно сульфата натрия и с последующим выпадением натрие-
вой соли сульфокислоты (пример—моносульфокислота бензола).
Имеется мало практическое предложение производить без разведения реакцион-
ной смеси водой обработку поваренной солью при нагревании, что якобы дает воз-
можность улавливать хлористый водород и разделить массу на два слоя: соль суль-
фокислоты, внизу — бисульфат натрия ю).
В тех случаях сульфирования, когда продукт не выделяется ни
одним из вышеуказанных приемов в практически заметных коли-
чествах (полисульфокислоты, особенно при наличии нитрогруппы),
кислую жидкость нейтрализуют прибавлением избытка мела (или
извести). Кальцевые соли большинства сульфокислот легкораство-
римы, между тем как серная кислота переходит в осадок в виде гипса:
2RSO3H 4- nH.SO4 4-(n 4- 1)СаСО3
= (RSO3)2Ca 4- nCaSO4 4" пН2О 4 (и 4~ 1) СО2.
86
Мел необходимо добавлять в тонкоизмельченном состоянии (лучше всего оса-
жденный в виде сметаиообразной взвеси в воде), причем практично вести нейтра-
лизацию при температуре около 60° с сохранением до конца слабокислотной реак-
ции. При этом получается наиболее легко фильтруемый и промываемый осадок гипса.
Отделив гипс (на нутче) и промыв его, получают прозрачный
раствор кальциевых солей сульфокислот, из которых можно вы-
делить сгущением как кальциевые, так и натриевые соли; послед-
ние— с предварительной обработкой содой и отделением выпав-
шего мела. В заводском масштабе, когда раствор кальциевых солей
стремятся получать не чрезмерно разведенным, утилизируют по-
следние промывные воды для смешения с мелом или разведения
сульфурационной смеси. Этот метод известкования, наиболее упо-
требительный в технике получения полисульфокислот, устраняет
отбросную кислоту в виде гипса, которого большие количества на-
капливаются поэтому на заводах.
Нейтрализация мелом практикуется по отношению главным
образом к полисульфокислотам. Для моносульфокислот вполне
практичным оказывается метод нейтрализации посредством суль-
фита натрия, отхода при получении фенолов из сульфокислот.
Нейтрализация происходит с выделением сернистого ангидрида,
который находит себе обычно тут же применение для выделения
фенола из фенолята:
H2SO4 + Na2SO3 = Na2SO4 -|- Н2О + SO2,
2 RSO3H — Na2SO3 = 2RSO3Na4-H2O4-SO2 Б9).
в) Характеристика продуктов сульфирования
Помимо выделения сульфокислот практика требует также мето-
дов разделения отдельных сульфокислот. Реакционная смесь после
сульфурации крайне редко может содержать только одну сульфо-
кислоту. При сульфированиях нафталина и антрахинона примеси
изомерных и более сульфированных продуктов неизбежны. Опре-
деление состава сульфурационной смеси в отношени отдельных
ингредиентов становится иногда трудной задачей. Определение
различных изомеров сульфокислот основывается на различной их
реакционности в одинаковых условиях: различие в растворимости
солей, различная скорость гидролиза и пр.
Определение различных степеней сульфирования (моно- и дисульфокнслот)
можно производить например по методу Г а с л е р а 60). Метод основан на объемном
или весовом определении нужного для нейтрализации смеси сульфокислот количе-
ства соды (например после предварительного перевода серной кислоты в BaSO4
и сульфокислот в Ba-соли обработкой сульфосмеси посредством ВаСО3). На осно-
вании стехиометрических отношений (1,11, III) можно рассчитать отношение сульфо-
кислот (моно- и ди-, нли ди- н три-) по израсходному количеству соды из двух
уравнений с двумя неизвестными. Таким образом для примера сульфирования на-
фталина имеем расчетные уравнения.
C10H7SO3H 4- V3Na2CO3 = C10H,SO3Na +i/2 (CO3 + H2O) (I)
C10H6 (SO3H)3 4- Na3CO3 = C10H6 (SO3Na)3 + CO3 + H3O (II)
C10H5 (SO3H)3 + 3/2Na3CO3 = C10H5 (SO3Na)3 + з/2(СО2 + H2O) (HI)
При расчете на г-мол. прореагировавшего нафталина моносульфокислота тре-
бует 53 г (R, мол.), дисульфокнслота —106 1 (1 мол.) и трнсульфокислота 159 г
(3/2 мол.) Na2CO3.
87
Назвав х и у количества (в г) соды, нужные для нейтрализацви ди- и моно-
сульфокислот в смеси (из 1 г-мол. нафталина) и т—найденное по опыту количе-
ство соды, получаем уравнения:
х Ч-у == т,
х + 2у = 106,
откуаа можно рассчитать х и у. Подобно этому, количества ди- и трисульфокислот
находятся по разрешении уравнений:
х z — т,
%х + г = 159.
Большинство методов разделения сульфокислот основано на
использовании различной растворимости сульфокислот или их
солей в определенных условиях: разведения, концентрации серной
кислоты и пр.
Так, из кислотной смеси после сульфирования толуола на моиосульфокислоту
можно выделить n-сульфокислоту, прибавив в смесь такое количество воды (соот-
ветственно льда), чтобы серная кислота имела концентрацию 66—71% H2SOP
Здесь выпадает трудиорастворимое кристаллическое соединение имеющее состав:
2CH3CeH4SO3H H2SO4 + Н2О. Ортосульфокнслота толуола в свою очередь трудно-
растворима в сериой кислоте 45 — 55%, поэтому может быть выделена дальнейшим
разведением фильтрата после выпадения пара-изомера (В и х е л ь га у с).
1.5-дисульфокислота нафталина может быть отделена от изомеров, сопрово-
ждающих ее в сульфурационной смеси, после доведения концентрации сериой
кислоты до 60% — 1.5-изомер в этих условиях выпадает.
Здесь важно принять во внимание, что весьма небольшие прнмеси (загрязнения)
посторонних веществ отзываются крайне резко иа повышении растворимости суль-
фокислот и затрудняют выпадение их. Часто практикуется разделение сульфокислот
не после сульфирования непосредственно, а после следующих обработок, приводя-
щих к образованию других замещений (например в виде аминосульфокислот). Поль-
зуются также образованием смешанных солей полисульфокислот, например солей
Са и Na, кислых солей и т. п. 61).
Несмотря на огромную практическую важность сульфокислот,
они принадлежат к наименее изученным соединениям органической
химии, что объясняется отчасти трудностью их характеристики.
В самом деле, сульфокислоты — вещества, не имеющие определен-
ной температуры кипения. За исключением отдельных индиви-
дов у них нет характерной температуры плавления. Характери-
зуются сульфокислоты чаще всего в виде металлических солей
(содержание кристаллизационной воды, растворимость) или солей
с органическими основаниями. Для образования соли с ароматиче-
скими аминами смешивают раствор сульфокислоты, по возмож-
ности свободный от анионов, могущих осадить амин, рассчитанным
количеством растворенной в воде солянокислой соли амина. Оса-
ждается солеобразное соединение, которое часто имеет характер-
ную температуру плавления 62).
Взамен неприложимого к сульфокислотам определения точки
плавления Обермиллер рекомендует определение „числа рас-
творимости" для солей сульфокислот, т. е. определение удельного
веса их насыщенного водного раствора при обыкновенной темпе-
ратуре. Если имеется смесь нескольких солей, то при перекристал-
лизации получают маточный раствор, удельный вес которого чаще
всего значительно выше, чем число растворимости одной из ком-
понент. Ерли же при перекристаллизации соли удельный вес ма-
точного раствора остается постоянным, то можно считать соль
88
чистой. Такое установление чисел растворимости таким образом
не менее полезно, чем определение точек плавления 03).
Сульфокислоты — сильные кислоты, могущие отнимать катион
у таких минеральных солей, как хлористый натрий, сульфат и т.п.
Для более точной характеристики строения сульфокислот поль-
зуются соединениями с измененной сульфогруппой. Такими соеди-
нениями в первую очередь являются сульфохлориды и сульфон-
амиды. Кроме прямого образования сульфохлоридов введением
группы SO2C1 в ядро при обработке органического вещества хлор-
сульфоновой кислотой можно образовать сульфохлориды и из
сульфокислот, замещением гидроксила в сульфогруппе хлорным
атомом. Употребителен метод перевода сульфокислоты в щелочную
соль и обработки последней пятихлористым фосфором в неболь-
шом избытке против требуемого по реакции количества:
RSO3Na 4- РС16 = RSO2C1 + POC1S + NaCl.
Обращает внимание в недавней американской работе, что при действи пяти-
бромистого фосфора на соли арилсульфокислот часто проявляется восстановитель-
ный эффект (от действия РВг3 иа сульфобромиды) и наряду или вместе с сульфо-
бромидами образуются отвечающие сульфокислотам дисульфиды. Относительные
количества сульфобромида и дисульфида зависят от природы арильного остатка,
связанного с сульфогруппой 61). Реакции можно передать рядом уравнений:
RSOgNa + PBr5 = RSO2Br NaBr + POBr3.
PBr8^PBr3-|-Br2>
2RSO2Br + 5PBr3 = RSSR + 4POBr3 + PBr5.
Сульфохлориды углеводородов (и многие нитросульфохлориды)
хорошо изучены, имеют определенные температуры плавления и
могут дать достаточные указания о природе исходной сульфокислоты.
Сульфохлориды отличаются от хлорангидридов карбоновых
кислот относительно большей стойкостью по отношению к гидро-
лизу, и превращение их водой в сульфокислоты по уравнению:
RSO2C1 + Н2О = RSO3H НС1
требует обычно повышенной температуры. Возможно, что предва-
рительно имеет место образование продукта присоединения
RSO2CI. Н2О 6В).
Сульфохлоридная группа SO2C1 может быть образована из сульфогруппы при
обработке сульфокислот хлорсульфоновой кислотой на холоду. Таким образом из
аминонафтолсульфокислот получаются их сульфохлориды, иногда с дополнительным
сульфированием 66), например
89
Затем сульфокислоты при взаимодействии на холоду с хлоридами серы нли при
обработке серой и хлором в присутствии ниднферентного растворителя, например
четыреххлористого углерода, также переходит в сульфохлорнды 87). Сульфохлориды
можно перевести в сульфииовые кислоты при гидролизе в присутствии восстано-
вляющнх веществ, например взаимодействием хлористого олова и соляной кислоты88).
Взаимодействие сульфохлоридов с сульфитом натрия (в присутствии соды) приводит
к образованию натриевых солей сульфиновых кислот. Реакция протекает по схеме:
RSO2C1 4-Na2SO3-> RSO2SO3Na 4-NaCl
RSO2SO3Na 4- Na2CO3 -> RSO2Na 4- Na2SO4 4- CO2 69).
Сульфохлориды углеводородов (бензола, толуола) находят себе
применение для получения алкильных эфиров сульфокислот, которые
в свою очередь имеют интерес как алкилирующие средства (см. алки-
лирование), При взаимодействии с аммиаком, соотв. аминами, сули-
фохлориды легко переходят в амиды сульфокислот (сульфонамиды):
RSO2CH-NH3-^ RSO2NH2 и, соотв., RSO.,C1+H,NR' -> RSO2NHR'
Последние соединения, которые легко индивидуализировать,
имеют также значение для характеристики сульфокислот. С дру-
гой стороны, реакция сульфохлоридов с аминами находит приме-
нение для характеристики и разделения аминов первичных, вто-
ричных и третичных из их смеси в процессе получения алкили-
рованных аминов.
Группа NH2 н сульфонамидах прн обработке нх хлорной известью обменивает
водород на атом хлора; нз щелочных солей таких охлоренных сульфонамидов известна
/С1
натриевая соль л-толуолсульфохлорамида CI-^CeHiSOjNif • ЗН2О нлн, вероятнее,
^Na
CH3C8H4S (: О) = N — Cl • ЗН2О, так называемый актнвнн. Он применяется довольно
(4ыа
широко за границей в качестве средства, отделяющаго хлор для дезинфекцион-
ных целей 7в),
Некоторые производные сульфонамидов интересны как азосоставляющне. Имея
сродство к животным волокнам, они, будучи нанесены на этн волокна, образуют прн
обработке диазосоеднненнямн азокрасители в самом волокне,—процесс, похожий на
образование так называемых холодных окрасок на хлопчатобумажном товаре71 .
Описаны нижеприведенные производные сульфонамидов, интересные как азо-
составляющне:
•ОН
NHO2S-
-сн3
ZOH ____
I f NHO2S-Z V-CH3 .
H3CZZ/ ZCOHN-Z Z
90
—N-N
! । и
co c-ch3
so2nh2 CH2
Интересно, что производные сульфонамидов сходного строения предложены
как средства, защищающие шерсть от моли, например72)
Сложные арилсульфонарнламино-пронзводные, где остатками —SO2NH— свя-
заны по нескольку ароматических (многоядерных) радикалов, обладают дубящими
свойствами. Таков например
-SO2N11
Z\Zs°3Na
SO2NH нт. п.’З)
- so2nh
91
Алкильные (илн аралкильные) эфиры сульфокислот при термическом разложении
(t°>200°) лают сульфокислоту с теоретическим выходом, алкил (илн аралкил) дает
отвечающий ему ненасыщенвый (этиленовый) углеводород74).
Фенольные эфиры ароматических сульфокислот при нагревании с хлоридами
металлов, например А1С13 или ZnCl2 в присутствии ннднферентных растворителей
(например нитробензола) или без них. превращаются по описанию патента с хоро-
шим выходом в оксндиарилсульфоны 7б), например ___
. /SO2OC6H5 SO2- \-ОН
II -II-:
н3с/\/ н3с/
Сульфокислоты находят себе применение кроме производства
фенольных соединений и красителей еще в приготовлении синте-
тических дубителей 76) и в производстве смачивающих, эмульгирую-
щих и моющих средств.
В последней из названных групп продуктов имеют значение
преимущественно сульфокислоты нафталина, в который одновре-
менно или ранее сульфирования вводится или алкильный (пропил,
бутил) или аралкильный (бензил) остаток или остатки.
Наиболее употребительные в практике производства сульфо-
кислоты фенолов, аминов, аминонафтолов сравнительно легко отли-
чаются друг от друга по их реакциям, описанным и в обшей, и
в патентной литературе (образование диазосоединений, азокраси-
телей, цвета водных растворов, отношение к реагентам и т. п.).
Для изучения строения сульфокислот, особенно в ряду нафта-
лина, применяют реакцию замены сульфогруппы атомом хлора77).
Эта реакция осуществляется нагреванием (до 150—160°) пятихло-
ристого фосфора с сульфокислотой. Сульфогруппа, переходя вре-
менно в сульфохлоридную группу, отщепляется в виде хлористого
тионила. Например
C]0H7SO8Cl + РС15 = С10Н7С1 + SOCla 4- РОС18.
Для сульфокислот антрахинона практикуется другой метод об-
мена сульфогруппы на атом хлора (см. хлорирование).
Качественно присутствие сульфогруппы в неизвестном соеди-
нении (если оно не карбоновая кислота) может быть определено
по проявлению соединением кислотных свойств: растворимости
(соотв. образованию солей) в растворах углекислых щелочей, по
осаждению из щелочных растворов минеральными кислотами, при
этом часто в виде кислых солей.
Технические продукты весьма часто сильно загрязнены мине-
ральными солями, например гипсом и глауберовой солью. Так как
количественное определение сульфогрупп сводится к определению
серы, то примесь сульфатов, сульфитов и пр. должна быть исклю-
чена или точно установлена предварительно. Очищение произво-
дят многократной перекристаллизацией вещества из воды, из
спирта или иного подходящего растворителя.
92
Количественно сульфогруппы в таких очищенных соединениях
можно определить по способу Мессингера окислением в ще-
лочном растворе марганцевокислого калия (или в солянокислом
растворе бихромата). Метод пригоден для определения серы в неле-
тучих серусодержащих соединениях.
Навеску соединения (сульфокислоты) переводят в колбу на 500 см3 4, куда при-
бавляют 1,5—2 г КМпО4 н 0,5 г чистого КОН. В горло колбы вставляют обратный
холодильник. Через верхнее отверстие холодильника вводят в колбу 25—30 см3
воды и нагревают 2—3 часа. По охлаждении жидкости, которая должна быть еще
красной, через холодильник постепенно приливают ковцентрированной соляной
кислоты и по окончании выделения газов нагревают до тех пор, пока жидкость
станет прозрачной. Содержимое колбы переводят затем в стакан н осаждают серную
кислоту хлористым барием.
Реакция сульфирования служит главным, но не единственным,
методом получения сульфокислот. Сульфокислоты могут быть
образованы и путем превращения иного заместителя.
Из таких методов отметим:
1. Обмен подвижного атома хлора на сульфогруппу действием сульфита ще-
лочного металла:
RC1 + Na2SO3 = RSOgNa + Na Cl.
2. Обмен нитрогруппы на сульфогруппу действием сульфита щелочного металла
(имеет значение преимущественно в ряду антрахиноновых производных).78).
4- Na2SO3
— NaNO,
3. Обмен аминогруппы, переведенной в диазогруппу на остаток SC(: S)OC2H5
(кСантогенатную группу) и омяление последней (до SH) с последующим окисле-
нием 79)
RNH, -» RN2X -» RSC(: S)OC2H5 -» RSO3H
(подробнее ниже в главе о диазотировании).
4. В особенно реакционноспособном ядре антрахинона имеются примеры ввода
сульфогруппы, происходящего в водной среде, именно путем обработки соответ-
ственного производного сульфитом натрия в присутствии перекиси марганца (окис-
литель) и борной кислоты 80), например
Здесь можно себе представить течение реакции, как имеющее промежуточную
фазу образование хинона (I), который, присоединяя элементы соли сернистой кис-
лоты, восстанавливается до гидрохинона (хиннзариндисульфокислоты) (11).
О вноде сульфогруппы обработкой ннтросоединений кислыми солями сернистой
кислоты см. главу V.
93
Промежуточные продукты бензольного ряда, Получаемые путем
сульфирования
Промежуточные продукты анграхинового ряда, получаемые путем
сульфирования
94
Промежуточные продукты нафталинового ряда, получаемые путем
сульфировавня
95
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
Н. Wichelhaus, Sulfurieren, Alkalischmelze der Sulfosauren, Esterifizieren,
Leipzig, 1911.
R. Norris Shreve, Equipment for Nitration and Sulfonation, Ind. Eng. Chem.
24, 1344 (1932).
l) Martinsen, Zt. phys. Ch. 1908, I, 713.
2) H. Friese, Ber. 64, 2101 (1931).
3) A. J. Masson, Soc. 1931, 3200, Cherbuliez, Helv., 6, 281 (1923).
4) Guyot, Chitn. & Ind. 1919, tl, 879, Ch. Courtot, Le re de la Sulfonation,
R. G. M. C. 33, 177 (1929). Courtot, H on net. C. r. 182, 855. Б. П. Федоров,
Ан.-Кр. Пр. 2, 2, 1, 1932. И. С. Иоффе, Ж. О. X. 3 (65), 437, 505, 1933; Ан.-Кр.
Пр. 3. 7, 296, 1933. Н. Н. Ворожи о в младщ. Ан.-Кр. Пр. 4, 84, 1934.
5) Р. С. J. Euwes, La sulfonation de la naphthaline, Examen quantitatif, Rec. 28,
298 (1909).
6) H. Erdmann, Ann. 275, 192 (1893).
') A. Wahl, G. V e r m e у 1 e n, C. r. 184, 334 (1927).
8) E. Bamberger, E. Hindermann, Ber. 30, 654 (1897); Bamberger,
J. Kunz, Ber. 30, 2274 (1897).
9) Liebmann, заяв. Г. n. L 3205, F. I, 420. Bischoff, Ber. 23, 1914
(1890); By, Г. n. 216076, F. X, 187.
to) IC1., Б. n. 299279, 311977. IG. Г. n. 484997, Sandoz, Г. n. 530135.
u) Г. n. 72833 (By), F. Ill, 427, 56563 (Grinau) F. 11, 269. Ср. H. Erdmann,
Ann. 275, 225, 1893.
to) By, Г. n. 237396, F. X, 177.
13) В., Г. n. 91202, F. IV, 164. R. E. Schmidt, Ber. 37, 66 (1904) примечание.
to) M. A. I Ij inski, Ber. 33, 4194 (1903).
to) R. E. Schmidt, Ber. 37, 66 (1904). Cp. G. W. Ciough, CB. 1923, 1, 317.
16) A. Cop pens. Rec. 44, 937 (1925), The Influence of Mercury on the Sulpho-
nation of Anthraquinone, J. P. W i b a u t, Helv. 10, 380 (1927).
”) A. Meyer, C. r. 183,519 (1926), 184, 609 (1927). Cp. Martinet, Roux,
C. r. 172, 385 (1921). H. E. Fierz-David, Helv. 10, 197, 429 (1927).
18) IG., Г. n. 500914, Am. n. 1745025 (G. An. W.); Cp. Waldmann, E. Schwenk,
CB. 1931, tl, 1278. Ср. К Lauer, J. pr. Ch (3) 138, 81, (1933).
19) IG., Ф. n. 685062, CB. 1930, II, 2576. Г. n. 544036, CB. 1932, I, 1298.
to) F. Th urn ml er, Г. n. 214156. F. IX, «70.
a) G. Mo hr man n, Ann. 410, 373 (1915); CB. 1916, I, 50.
to) Holderman n, Ber. 39, 1250 (1906).
^Heinemann, Б. n. 12260 (1915). J. Soc. D & Col, 1916, Cp. Auger,
Vary, CB. 1922, I, 493. также Annual Reports of the Progress of Chemistry for 1920
(XVII), 76
24) Ambler, Cotton, Ind. Eng. Chem. 12, 968 (1920).
2B) A. E. Pet er kin, Synthetic Phenol, ind. Eng. Chem. 10,738 (1918), LM. Den-
nis, Am. n. 1247499, 1260852, H. Bull (The Barrett Co), Am. n. 1212612, 1211923,
1227894, 1228414, 1229593.
to) E. Lamberts, Г. n. 113784, F. VI, 62.
to) И. И. Воронцов, Ан. Kp. Пр. 1931, № 6, 14.
to) Г. п. 385049, t М, F. XIV, 386.
to) Mon. 55, 358 (1930), СВ. 1930. II, 567.
зо at) f г ё г е j а с q u е, С. г. 183, 607 (1926).
за) Steinkopf, J. pr. Ch. (2) 117, 1 (1927).
33) W. Davies, Dick. СВ. 1931, 11, 2865.
31) F о u q u e, Lacroix, CB. 1923, I, 1120.
;’5) О п >лучении сульфонов, H. Meyer, Ann. 433, 327 (19231.
36) Ci ba, Ф. n. 697742, CB. 1931, II, 3273.
ЗА W a 1 d m a n n, Schwenk, 1. c., Cp. Am. n. 1321994 (The Barrett Co); Ciba,
Шв. и. 150612, CB. 1932, II, 123. Cp. Nat. An., Am. n. 1676211, CB. 1928, 11, .066.
to) Schwenk, CB. 1932, 1, 388.
39) Baumgarten, Ber. 59, 1166, 1976 (1926), Г. n. 499571, CB. 1930,11, 986.
10 j M. В a 11 e g a y, Schneider (Kern), Bull. (4) 41, 1491 (1927).
41) Battegay, Brandt, Bull. (4) 33, 1667 (1923).
to) Baumgarten, Г. n. 514821, Ch. T. Ob. 1931, 182.
96
«) Sc. D., Г. п. 535147, CB. 1931, П, 3395, Б. п. 317735, СВ. 1929, II, 2734.
**) Courtot, 1. с. 181.
4S) D. Тугег, Ам. п. 1210725, Guyot, 1. с., Н. Meyer, 1. с.,Harvey, Steg-
man. CB. 1924, II, 1787.
**) Ambler, Gibbs, Ind. Eng. Chem. 12, 968 (1920).
«) The Barrett Co, Шв. n. 90292, 90298.
48) Опыты И. Я. Гришина и Спрыскова (не опубликовано).
49) Н. Meyer. 1. с., L. Gay, М. A u m 6 г a s, М i о n, Chim. & Ind. 19,387 (1928),
Ф. п. 619439, Ch. Т. Ub., 1927, 174; ср. СВ. 1928,11, 1198, А. А. С п р ы с к о в, Ж. X.
Пр. 8, № 20, 41 (1931).
5°) Например Ам. п. 1716082 (DuP).
61) BD Со. Г. п. 392460, F. XIV, 405.
52) Newport, Ам. п. 1794861, СВ. 1931, I. 3610.
И) IC1, Б. п. 354201, Ch. Tr. J. 89, 463 (1931), Ф. п. 718678, Ind. Chim. 19, 219,
262 (1932), Г. п. 549136, СВ. 1932, II, 443. W. Huber. Helv. 15, 1372 (1932).
м) Н. Friesе, Вег. 64, 2109 (1931).
«б) G. Wen dt, Г. п. 71556, 75455, F. III, 19.
5®) Fiегz-David, Schlitter, Waldmann, Helv. 12, 663 (1929).
м) Am. п. 1756837 (N. Cotton 1930), 1836204 (DuP). CB. 1932, I, 1830. Cp.
И. И. Ворэяцов, Ан.-Кр. Пр. 1931 № 2—3, 41.
M) Miersch, Г. п. 199959 F. IX. 101, 201971, F. IX, 102.
й) P. W. Uh Imann, Г. п. 229537, F. X, 112.
1923в0 A Hasler, Uber die Disufurierung des Naphthalins, Diss.. Zfirich,
61) Например А. А. К у p о ч к и н, Ж. О. X. I (63), 689(1931), М. L. Crossley,
G. S. Simpson (Calco), Ам. п. 1701259 (1929).
«2) Н. Erdmann, Вег. 32, 3186 (1899), W. Konig. Haller, J. pr. Ch. 101,
38 (1920/21), A. G. P er k i и, Sewell, Soc. Ind. 1923, 27 T, F о rs t er, Keyworth,
Soc. Ind. 43, 299 T (1924), Forster, Watson, Soc. Ind. 1927, 224 T, R. B. Fоr-
s t e r, T. H. Hanson, R. Watson, Soc. Ind. 47, T 155, Forster, D. H. Mosby,
Soc. Ind. 47, T 157; CB. 1928, II, 768. J. A. Ambler, Ind. Eng. Chem., 12, 108
(1920).
к*) J. О ber mi 11 er, Zt. angew. Ch. 27, 37 (1914).
6I) A. H. Kohlhase, Am. Soc. 54. 2441 (1932).
65) E. Berger, Olivier, CB. 1927, II, 1818.
ee) IG, Б. n. 326226, CB. 1930, II, 469; Г. n. 532399, CB. 1932, II, 775.
й) H ey de n, Г. n. 499052, CB. 1930, II, 1444.
«в) M. Claasz, Ann. 380, 303 (1911).
в») В I o m s t r a n d, Ber. 3, 965 (1870), L i m p r i c h t, Ber. 25, 3477 (1892), E r d-
mann, Silvern, Ann. 275, 305 (1893), Krishna, Dass, CB. 1928,1,555.
’°) Hey den, Г. n. 422076, F.XV, 274, 514094. IG., Г. n. 514802, Ch. T. Ub. 1931,
175, By, Б. n. 241579, 241580, CB. 1927, II, 977, 1084; Esseff Австр. .n 107722, CB.
1928, II, 2752.
!I) E. Schwenk, Reich n er, Knob. Am. n. 1718882.
ra) IG., Г. n. 506988, CB. 1930, II, 3220.
ra) IG., Б. n. 353046, CB. 1931, II, 3710.
74) Z. Feldi, Ber. 60, 656 (1927).
75) Heyden, Г. n. 532403; CB.1931, II, 3264.
76) См. работы У крайней. Ин-та прикл. физ.-химнн по еннтанам (Я. П. Берк-
ман). Г. п. ничииая с F XI, 187 и след. тома.
”) Armstrong, Wynne, СВ. 1897, II, 551.
’8) R. Е. Schmidt, Вег. 37, 66 (1904). Г. п. (By) 164292, F. VIII, 231.
те) Leuckardt, J. pr. Ch. 41,188 (1890), Ср. Pollak, Deutsche г, Krauss,
CB. 1927, II, 1021.
80) IG., Б. n. 324120, CB. 1930, I, 3489.
7 Зак. 2611. — H. Ворожцов
97
ГЛАВА IV
ВВЕДЕНИЕ ГАЛОИДА
(хлора, брома и т. п.)
Введением галоида, или галогенированием, называется вообще
реакция замены одного или нескольких атомов водорода органи-
ческих,— в интересующих нас случаях,— ароматических соединений
иа атом или атомы галоида. В применении к участвующим в ре-
акции галоидам и получающимся продуктам замещения, реакцион-
ный процесс принимает частные наименования хлорирования, бро-
мирования и т. д. Хотя все элементы этой группы, начиная с фтора,
могут при разных условиях давать продукты замещения водорода
в ароматических соединениях, тем не меиее только хлорирование
имеет преимущественное значение в практике получения большин-
ства технически интересных галоидопроизводных. Это объясняется
и техническими и экономическими причинами.
Сам фтор реагируете органическими веществами слишком энергично, для того
чтобы можно было управлять реакцией в сторону получения однородных продуктов
замешевня, поэтому фтористые соединения ароматических веществ получают обход-
ным путем, главным образом нз аминов через диазореакцню, или вытеснением хлора
действием активных соединений фтора (SbFg)1).
Принимая во внимание большую активность фтора и затруднения подобрать
такие материалы для постройки аппаратуры, которые были бы достаточно стойки
против фюра, и сравнительно малую доступность, а следовательно и дороговизну
фтора, становится понятным, почему фторирование не имеет технического значения.
Бром и иод менее распространены в природных соединениях,
чем хлор, они дороже хлора, и поэтому, хотя методы работы с ними
и не являются затруднительными, все же бромирование и особенно*
иодирование применяются по сравнению с хлорированием редко,
главным образом по отношению к готовым красителям или же для
получения особенно ценных промежуточных продуктов.
Введение галоида в ароматические углеводороды вместо водо-
рода сделалось известным как метод лабораторный в конце 40-2
и начале 50-х годов прошлого столетия. Позднее метод хлориро-
вания стал заводским процессом, сначала в применении к толуолу
для получения продуктов охлорения СН3-группы и через них —
бензальдегида (80-е годы). Значительно позднее техника заинтере-
совалась получением хлоропродуктов с замещением галоидом водо-
родных атомов ароматического ядра (главным образом хлорбензола).
Этот период соответствовал упрочению производства щелочей эле-
ктролизом хлористых солей, когда хлор стал отходом производства.
Хлорирование ароматических соединений в ядре вошло в практику
в самом конце XIX столетия и начале XX.
93
В конце главы описано несколько примерен введения в качестве заместителя
аналога галоидов, родана SCN, который не только по ионным реакциям в солях,
но и по методу введения в ароматические вещества очень близок к галоидам.
Что касается хлорирования, то его преимущественное значение
в технике понятно; хлор — вещество очень распространенное в при-
роде (хлористый натрий) и получаемое в свободном состоянии со-
временной химической промышленностью в огромных количествах.
Развитие электрохимических производств уже в конце прошлого
столетия и особенно в начале XX заставило искать применений для
образующегося при электролизе поваренной соли хлорг.» произ-
водство которого возрастало более интенсивно, чем могла его потре-
бить неорганическая химическая промышленность.
Со времени мировой войны и появления химических боевых
средств (отравляющих газов) в современной военной технике, сам
хлор и многие его производные приобретают и серьезное военное
значение. Вполне понятно поэтому интерес капиталистических стран
к расширению производства хлора и нахождению для него новых
путей использования в промышленности с целью сохранения по-
тенциальных запасов его на случай войны.
Среда других новых областей применения хлор-газа и жидкого
хлора (беление текстильных волокон, бумажной и древесной массы,
очистка питьевых вод) утилизация хлора для получения аромати-
ческих хлорозамещенных заняла довольно заметное место. Вместе
с тем оказалось возможным и в некоторых случаях практически вы-
годным получать из хлоропроизводных замещенные обменом хлора
на иные реакционные группы, и в хлорировании был найден замет-
ный конкурент двум вышеописанным методам ввода заместителя
в ядро, именно сульфированию и нитрованию (см. главу VII).
Наиболее .интересными в настоящее время исходными материалами для проведе-
ния хлорирования являются бензол н толуол, н с недавнего времени привлекает
внимание нафталин. Далее, некоторые хлоропродукты готовятся из антрахинона н его
замещенных, так же как и из иных исходных материалов. В частности реакция хлори-
рования имеет приложение, как уже сказано, ие только к промежуточным продук-
там, но н к гораздо более сложно построенным готовым красителям.
Самый употребительный агент хлорирования — сам хлор, чаще
всего применяемый газообразным. В отдельных случаях для полу-
чения хлорозамещенных применяются соединения или смеси со-
единений, выделяющие хлор в условиях реакции, например гипо-
хлориты, как NaClO, соляная кислота вместе с солями хлорноватой
кислоты (НС1 NaC10s), хлористый сульфурил (SO2C12).
Если представить себе процесс хлорирования по отношению к
углеводороду с участием только хлора, то, не говоря о самом те-
чении реакции, мы получаем такое общее выражение для ее резуль-
тата:
RH-j-CI2 = RCl + HCl.
При этом в бензоле (RH = C6HBH) замещение хлором может
иметь место только по отношению к водородному атому ядра,
между тем как у гомологов бензола, и в первую очередь у толуола,
оно может произойти и в ядре и в боковой цепи, следовательно
при однократном замещении дать начало либо соединению С1С6Н4СН3
7#
99
либо С3НБСН2С1. Оказывается, эти два направления вступающего
в соединение галоида (в ядро и боковую цепь) требуют для про-
ведения значительно различных реакционных условий, Именно, за-
мещение углеводорода в ядре обусловливается главным образом
участием в реакционной смеси некоторых простых веществ или их
галоидных соединений, которые оказываются, как говорят, пере-
носчиками галоида, или катализаторами хлорирования в ядре. Хлори-
рование углеводородов, имеющих в качестве заместителя метильные
группы, например хлорирование толуола, без катализатора при под-
ходящих условиях (преимущественно со стороны температуры) дает
хлорозамещенные (<в) в метильной группе: С6Н3СН2С1, С3НБСНС12,
С6НБСС13.
Таким образом обнаруживается, что катализатор нужен для
хлорирования ядра, между тем как боковая цепь при замещении
водородных атомов хлором не нуждается в каталитическом со-
действии (ниже мы увидим, что описаны некоторые прибавки, якобы
ускоряющие реакцию и здесь, но вещества, рекомендуемые для по-
следнего рода процессов хлорирования,—иной природы, чем в пер-
вых процессах).
Как катализаторы хлорирования в ядре испытаны и применяются
прежде всего: из металлов—железо, из металлоидов—иод. Железо —
наиболее употребительный в широком промышленном масштабе
катализатор. Наилучшая форма применения железа как такового —
возможно мелкое раздробление. Галоидные соединения железа и
других металлов оказываются очень реакционноспособными ката-
лизаторами, и не подлежит сомнению, что активность железа, взя-
того для хлорирования в виде металла, начинается лишь с того
момента, когда оно под действием хлора переходит в хлористые
соединения (FeCl2, Fe.Cl3).
Сильным катализатором хлорирования является хлористый алюминий, часто
настолько энергично действующий, что его применение не дает возможности упра-
влять реакцией. Описаны применения также металлического алюминия и амальгамы
алюминия. Хлористое олово (SnCl2) и хлористая и хлорная сурьма (SbCl3, SbCl8)
также извзстны как хорошие катализаторы. Железный катализатор предложено
применять и ввите готового хлорного железа. Как активный катализатор рекомен-
довано недокисное хлористое железо, получаемое из смеси равных количеств вос-
становленного водородом железа и хлорного железа 2). Аналогично действует само
железо и хлорное железо, прибавленные в отдельности в реакционную массу з). Очень
активные каталитические свойства обнаруживает также смешанный катализатор:
железо с малым количеством иода (например I: Fe = 1:10)4). Хлористые соедине-
ния других металлических элементов: меди, свинца — невидимому содействуют
также вхождению хлора в ядро. Что касается металлоидных соединений, то хлориды
фосфора предложены и применяются в качестве помогающих реакции охлорення
боковой цепи (С.Ч3-группы) в толуоле, но их роль в этом случае нуждается в ис-
следовательском освещении.
Вышгназванные катализаторы или активаторы хлорирования не
все одинаковы по своему значению и влиянию на процесс. При их
выборе и применении нужно принимать во внимание их свойства
в отношении реакционной смеси: так, самым удобным для примене-
ния был бы такой катализатор, который растворяется в хлорируе-
мом веществе или образует с ним однородную смесь, — этим веро-
ятно и обусловливается большая активность иода. Далее, нужно
считаться с индивидуальными качествами каждого отдельного ка-
тализатора, имея в виду, что хлорирование—процесс, в котором
можно подметить отдельные стадии и в течение которого протекают
обычно не одна, а несколько реакций; как выяснено будет ниже,
от катализаторов можно ожидать различного поведения в отдель-
ные моменты процесса, что может в результате отразиться так или
иначе на количестве и качестве продуктов реакции и методах их
дальнейшей обработки.
Для того чтобы выяснить, в чем сущность действия катализа-
торов хлорирования, рассмотрим пример хлорирования бензола.
а) Течение и факторы хлорирования бензола
Общее выражение результата реакции для этого частного слу-
чая имеет вид:
С6Н6+С12 = СвН6С1 + НС1, (1)
если продуктом реакции оказывается одно хлорозамещенное,
монохлорбензол. В уравнение реакции, дающее начальное и конеч-
ное состояния реакционной смеси, катализатор не входит. Между
тем если бензол обрабатывать хлором при полном отсутствии по-
сторонних веществ, особенно металлов, например в стекле, то ре-
акция принимает иное течение, особенно отчетливо проявляющееся
при солнечном освещении: бензол присоединяет молекулы хлора
одну за другой, ие выделяя хлороводорода. В качестве конечного
продукта такой реакции выделен продукт присоединения шести
атомов (трех молекул) хлора: бензолгексахлорид С6Н6С16. Его по-
лучение может быть выражено уравнением:
С6Н6 + ЗС12 = С6Н6С1в.
Вне сомнения, присоединение хлора к бензолу происходит по-
степенно, по одной молекуле, переходя, может быть и с разной
скоростью, через три фазы:
С1 Н
СвН6С14-ЬС1а =
Н С1
С1
Н
С1 н
Таким образом в отсутствии катализаторов бензол ведет себя
по отношению к хлору так же (хотя в общем с меньшей актив-
ин
ностью), как этиленовые углеводороды, присоединяющие по месту
двойной связи молекулы галоида. В вед ние в реакционную смесь
катализатора направляет реакцию согласно уравнению (1) или, что
вероятнее, не уменьшая значения реакции присоединения, содей-
ствует отщеплению от продуктов присоединения элементов, обра-
зующих молекулы хлороводорода. В самом деле, если представить,
что молекула бензолдихлорида теряет хлористый водород, то по-
лучается молекула хлорбензола:
^\/н ^С|
^С1___
/Н -на
V\r.
Для бензолгексахлорида известно, что его нагревание с щелоч-
ными агентами дает начало 1.2.4-трихлорбензолу:
НС1 С1
НС! НС! ^\/ С1
—знсТ
НС1\/ НС!
НС! С/^
По представлению Меервейна продукты присоединения галоида по месту
двойных связей ароматического углерода построены таким образом, что один из ато-
мов галоида находится в нормальной связи с насыщенным атомом углерода, второй же
связан координационно (следовательно солеобра ;но) во внешней сфере5; например
для продукта присоединения брома к бензолу строение будет таково:
Вт
Эти первичные продукты присоединения галоида до сих пор
не изолированы.
В течении реакции хлорирования мы сталкиваемся с теми же
явлениями, которые отмечены нами выше для нитрования и суль-
фирования, т. е. с замещением, проходящим через стадию при-
соединения к ароматическому ядру элементов реакционной моле-
кулы. Ввиду того что прибавки, катализаторов, хотя бы и не
в значительных количествах, вызывают направление реакции в сто-
рону замещения водородного атома хлором, естественно допустить,
что значение катализирующего агента — в такого же рода активации
молекулы бензола, какое вызывается уже рассмотренными реаген-
тами — серной и азотной кислотами.
Что аналогия здесь ие кажущаяся, а более глубокая, доказано возможностью
получения хлорозамещенных бензола без участия металлических катализаторов или
галоидо-металлнческнх соединений, но с участием серной и азотной кислот.
102
Так, Баттгэ получил хорошие выхоли хлорбензола и дихлорбензола при хлори-
ровании бензола с серной кислотой (93%-ной), взятой в небольших количествах,
и наблюдал не меньшую легкость хлорирования, чем при применении хлорного
железа или хлорвстого алюминия, даже работая при низкой температуре. Лишь по-
нижение температуры ниже 0° благоприятствовало образованию продуктов присо-
единения 6.
Имеются также указания в патентной литературе на возможность успешного
получения хлоро-(соотв. бромо-)замещенных трудно галогенируемого антрахинона
в растворах серной кислоты (моногидрата или олеума) без добавки катализатора 6).
С другой стороны, Ионеску удалось прохлорировать и ранее Датта прокоти-
ровать бензол с добавкой азотной кислоты, причем первый нз названных нашел
кроме хлорбензола в продуктах реакции нитробензол и нитрохлорбензол 7). Наи-
более вероятная схема течения реакции для последнего случая такова:
Что касается нитробензола, то он получается очевидно из бензола, непро-
реагировавшего с хлором, пройдя через продукт присоединения азотной кислоты,
как выше было рассмотрено.
Подобно этому н хлорирование прн участии серной кислоты протекает ве-
роятно по схеме:
Н, /ОН
Основываясь на этих фактах, мы вправе высказать положение,
что роль употребительных металлических и солеобразных катали-
заторов состоит в такой же активации молекулы бензода со сто-
роны галоидных солей [в сторону образования продуктов при-
соединения с временным переходом в ди-(соотв. тетра-) гидро-
бензольную молекулу и с последующим образованием вновь
бензольного соединения], какая активация характерна со стороны
серной и азотной кислот. Мы находим подтверждение для такого
утверждения и в том обстоятельстве, что хлористый алюминий
оказывается особенно сильным активатором хлорирования; между
тем выше были приведены уже примеры активирования им нитро-
вания ароматических углеводородов окислами азота.
Не исключена возможность, что некоторые катализаторы (такова повидимому
медь), не активируя или слабо активируя реакцию присоединения галоида, проте-
кающую с небольшой скоростью сама по себе, энергично влияют на отщепление
галоидного водорода.
Железо и другие металлы, применяемые в качестве активаторов,
очевидно переходят при хлорировании в соответствующие хлористые
соединения. Но хлорное железо FeCl3 по отношению к молекулам
ЮЗ
ароматических углеводородов ведет себя во многом подобно хло-
ристому алюминию (см. главу XV).
Пфейфер и Вициигер видят промежуточную стадию хлорирования при
участии „передатчика галоида" (активатора) в комплексных галогеногалоидных
солях, в состав которых входит и передатчик галоида. Например для случая бро-
мвроваиия бензола в вижеприведенисй соли, где А — передатчик галоида, а*
указывает место ионизации молекулы8. Эти комплексные соли распадаются далее
по схеме:
/\/ВГ
| 1 + НВг + А
А. Воль полагает, что так называемые „передатчики галонда" при галогени-
ровании ароматических соединений действуют ускоряюше не иа присоедивевне
галоида, но на последующую стабилизацию (в галоидозамещенные). „Посредством
связывания галоидов, присоединенвых главной валевтносню к ядру, например
с хлорным железом и т. п., происходят комплексы, представляющие ионы более
сильных кислот, и из них тот легче отщепляет (галоид), который встречает наиболее
подвижной водород у соседнего углеродного атома промежуточно-образованного
продукта (присоединения); последний не имеет более бензольного характера, и для
него имеют значение известные правила об ослаблении водородного атома в али-
фатических соединениях",—отсюда объясняется получение веожидавных метагалоидо-
замещеннй бензольных производных9.
Количество катализаторов, необходимых для проведения хло-
рирования, очень незначительно. Для проведения процесса доста-
точно прибавки 1% от веса бензола железных опилок или стружек,
0,1% иода, 0,5% хлорного железа, 0,1°/о алюминия10). Хлорирование
бензола для получения монохлорбензола было обстоятельно про-
слежено Бурь оном11), из работы которого мы заимствуем неко-
торые приводимые ниже выводы.
При хлорировании бензола, если проводить его пропусканием
хлора в бензол в присутствии катализатора (в технике часто при-
меняется хлорное железо, образуемое в самом процессе из загру-
женного в аппарат с бензолом железа), только в первый, непро-
должительный период процесс идет в направлении образования
хлорбензола по уравнению (1).
Когда в смеси имеется уже некоторое количество молекул хлор-
бензола, то хлорирование распространяется и на него по уравне-
нию (2):|
С6НбС1 + С12=С6Н4С12 + НС1 (2)
(образуется преимущественно п- и о-дихлорбензол).
Если в смеси имеется только 1% хлорбензола, то количество бензола, превра-
щаемого по уравнению (1), в 842 раза больше, чем колвчество хлорбензола, реаги-
рующего далее по уравнению (2). Если же хлорбензола 74,1%, эти количества
становятся приблизительно равными, следовательно с возрастанием концентрации
образующегося хлорбензола в смеси при продолжающемся хлорировании его вы-
ходы относительно падают.
Нужно заметить, что наиболее практически интересным продук-
том хлорирования бензола является именно хлорбензол, и потому
выходы дихлорозамещенных и высших продуктов хлорирования
104
(полихлоридов), образующихся при накоплениии дихлоридов в смеси,
например таким образом:
c6h4ci24- ci2=с6н8с13+на,
при всех условиях желательно свести к минимально возможным,
Бурьон выяснил, что порядок реакции как (I1, так и (2) равен 1 (реакции
моиомолекулярны). Конставты реакции (1) и (2) k н k' находятся из диферевциаль-
ных уравнений, определяющих скорости:
^- = й'С1(х1 —х),
где а — начальная концентрация бензола, xt — концевтрапвя бензола, превращевного
в хлорбензол, х — концентрация л-дихлорбензола (преобладающего среди дихлсри-
дов), Cj — концентрация хлора порядка п для обеих реакций.
k'
Отношение констант этих двух реакций-^- — К, определимое из уравнения
dx _ k' xt — х _ Xi — x
t/Xj ~ k a —Xt ~ a —xt
и по интегрировании
(1 -10g = log f1 +11+•
и — x a — Xt J
остается постоянным в ряде проведенных нм опытов, где определвлись величины
разницы концентраций а — х, Xj — х и кс вцевтрации х.
Величина К для температуры 25° равна в среднем 0,118. Отсюда и вытекают
приведенные выше выводы о значении концентрации хлорбензола в смесв для
образования дихлорида. В самом деле, если #=0,118, то для одной и той же кон-
центрации и одного времени часть молекул бензола, превращенных в мовохлор-
беизол, в Усив — 8,5 раз больше, чем часть молекул хлорбензола, превращающихся
в дихлорбевзил, но при возрастании концентрации мснохлор бензола сравнительно
с концентрацией бензола отношение абсолютных количеств превращаемых по реак-
циям (1) и (2) веществ может измениться в пользу преобладания дихлорнда.
Повышение температуры при хлорировании влияет на повыше-
ние выхода. Нужно заметить однако, что с повышением темпера-
туры количество n-дихлорбензола в смеси растет быстрее количе-
ства хлорбензола.
Вышеприведенное отношение констант К, бывшее при 18° равным 0,107, воз-
росло до 0,123 при 35°. Приложив уравнение Вант Гоффа, Бурьон определил,
что в пределах обычной температуры для повышения температуры иа 10° темпе-
ратурный коэфициент скорости превращения монохлорбензола в дихлорбензол
превышает таковой для превращения бензола в монохлорбевзол примерно на 8,5%,
Наблюдения производственного процесса подтверждают эти
выводы; так, при повышенной температуре хлорирование бензола
всегда дает ненормально высокое содержание полихлоридов в про-
дуктах.
Очень вероятно, что природа употребляемого прн хлорировании катализатора
ие остается без влияния ва отношенве скорости реакций (1) и (2). Этот вопрос
мало освещен в литературе в приложение к получению хлорбензола. Иначе, как
увидим ниже, он обстоит в отвошении хлорирования нафталина. Сложный катализатор
из железа с хлорным железом, взятых в равных весовых частях, судя по патентному
описанвю, дает более низкое содержание полихлоридов, чем отдельно примененные
его составные части у. Железо с иодом (1 и 0,1% от веса хлорируемого продукта)
105
в применении к хлорированию бензольных производных значительно ускоряет про-
цесс, что особенно заметно при хлорировании м=ло реакционных нитропроизводных.
Иод с железом, примененный как катализатор реакции хлорирования бензола (при
температуре 20—70°), благоприятствует образованию высших продуктов замещения
(наблюдается сдвиг отношения молекул С6Н5С1: СвН4С12 с 1 :0,С6 до 1:0,25) при
•общем ускорении хлорирования. Эффективность этого катализатора видна из того
обстоятельства, что при хлорировании толуола (при 50° и выше) наблюдается при
ускорении процесса противодействие входу хлора в бокбвую цепь даже при повы-
шении температуры до температуры кипения толуола4).
В опытах автора совместно с В. М. Григорьевым обнаружено очень свое-
образное влияние меди прн хлорировании бензола. В присутствии меди при обыч-
ной температуре поглощение хлора бензолом идет медленно и не сопровождается
образованием продуктов замещения, а только дает продукты присоединения хлора
к бензолу. Введение вместе с медью небольших количеств железа ускоряет реакцию,
направляет ее в сторону образования продуктов замещения, и поиохлорбензол среди
них является преобладающим.
Корчи иски й приводит список элементарных веществ, хлористые н броми-
стые соединения которых в качестве катализаторе в применялись для галогенирова-
ния бензола (Au, Al, Ga, In, Tl, Zr, Sn, Sb, Bi, Те, J, Fe, Mo, (J, P), н установил
полезный эффект W и WCI6. Он усматривает некоторую связь между каталитиче-
скими свойствами и атомным весом элемента 1а).
Хлорирование бензола в паровой фазе при освещении протекает через несколько
этапов, где вначале можно подметить образование хлорбензола и в качестве глав-
ного конечного продукта — бензолгексахлорида13).
Всегда в процессах хлорирования стремятся устранить из реак-
ционной среды воду и пользуются поэтому сухими материалами
(бензолом и хлором). Это стремление вполне понятно, так как
образующийся при взаимодействии хлористый водород, растворяясь
вводе реакционной массы, очень быстро разрушает металлическую
аппаратуру (абсорбер) и самый железный катализатор.
Некоторые американские исследователи находят тем не меиее, что полное от-
сутствие воды дает ие то направление процессу, который останавливается в случае
применения вполне сухих хлора и бензола на стадии образования продуктов при-
соединения. По нх мнению очень малое количество воды (вряд ли устранимое
в обычном техническом процессе осушки) является активатором процесса м).
Выводы из вышеприведенного обзора для практики получения
хлорбензола: нужно работать с возможным исключением воды
в реакционной среде, по возможности поддерживая постоянную
и не превосходящую невысокий максимум (20—25°) температуру,
нужно стремиться к тому, чтобы в реакционной среде не накапли-
валось много хлорбензола, ибо в противном случае будет усилено
образование полихлоридов.
В производственной практике хлориррвание бензола произво-
дится в цилиндрических довольно значительного диаметра метал-
лических (чугунных) аппаратах (абсорберах), которые заполняются
обрезками железа, служащего катализатором. После заполнения
абсорберов бензолом подают хлор через отверстия трубы, приво-
дящей хлор в низ аппарата.
Выделяющийся при реакции хлористый водород отводится
сверху аппарата по трубопроводу и идет на поглощение водой
для получения соляной кислоты. Хлористый водород уносит с собой
заметное количество паров бензола, соотв. хлорбензола, которые,
конденсируясь, загрязняют соляную кислоту.
Недавний американской патент фирмы Dow Со (1932 г.) описывает прием осво-
бождения газа от паров углеводорода: предварительную промывку отходящего
106
газа в системе из дзух скрубберов, из которых первый ваполиеи охлоренной жид-
костью, как она получается после обработки бензола хлором, второй — жидкими
полихлоридами (преимущественно о-дихлорбензолом). Второй скруббер охлаждается
приблизительно до 0э. При такой системе промытый в скрубберах газ не заключает
паров углеводорода и является достаточно чистым для поглощения иодой15).
С целью избежать далеко идущего, как говорят, „глубокого",
хлорирования, т. е. образования больших количеств полихлоридов,
процесс останавливают, далеко не до конца используя бензол. За про-
цессом хлорирова-
ния следят по по-
вышению удельного
веса реакционной
смеси. В опытах с ма-
лыми количествами
контролируют ход
реакции по привесу
массы или по расхо-
ду хлора. Останав-
ливают процесс на
достижении массой
удельного веса 1,00
или немного выше
(1,04). При благопри-
ятном течении реак-
ции этот момент от-
вечает содержанию
40 — 45% бензола,
около 50% хлор-
бензола и 8—10°/о
полихлоридов.
Полученную смесь
продуктов хлориро-
вания („охлоренный
бензол") подвергают
затем обработке ще-
лочным агентами (со-
дой, едким натром)
для нейтрализации•
содержащегося всме-
си хлористого БО-
РИС. 3. Абсорбер.
дорода, разложения
унесенных растворимых соединений железа (FeCl3), разрушения
могущих сохраниться комплексных соединений железных солей
с органическими веществами, так же как разрушения тех органи-
ческих хлоропроизводных, которые при дальнейшем нагревании
могли бы выделить хлористый водород.
Как выяснилось из опытов автора и К. А. М р о с т. комплексные соединения
жСлезвых солей с продуктами охлорения разлагаются только при нагревании
с крепкими щелочными растворами.
Далее, после осушения массы, если это вызывается необходи-
мостью, она идет на разгонку через фракционные колонны, отде-
10Г
лающие одно- или двукратной перегонкой бензол от хлорбензола
и последний от полихлоридов. Разгонку более высококипящих
фракций предпочительно вести под разрежением.
Такой способ работы не является совершенным. В объемистых аппаратах трудно
достичь ранкомерного распределения катализатора, также трудно ныдержать опре-
деленный температурный режим. Наружное охлаждение абсорбера не дает уверен-
ности, что внутри него температура не перейдет допустимые границы (н следова-
тельно не создадутся условия, благоприятствующие образованию полихлоридов).
Большим неудобством является также необходимость перерыва процесса прн исполь-
зовании всего около половины бензола нз-за опасения получить большой остаток
полихлоридов.
Поэтому a priori представляется целесообразным найти такую систему работы,
которая устраняла бы эти недостатки. Нам представляется вероятным, что такая
система — в проведении хлорнронання непрерывным потоком в более узких
колонных аппаратах с устранением образующегося хлорбензола тотчас по возник-
новении из сферы реакции. В других производствах имеются подобные процессы
и аппараты, и вероятно можно и для хлорбензольного производства найти по их
примеру практическое решение задачи16).
Имеются предложения получать хлоропроизводные бензола, используя иные
принципы работы, но практический их интерес невелик.
Так, в патентах немецкой фирмы указывается на метод хлорирования бензола,
используя хлор в момент образования прн контактном окислении хлористого водо-
рода: смесь паров бензола, хлористого водорода и воздуха (или кислорода) пропу-
скают при температуре свыше 300° через подходящие катализаторы окисления,
например через хлорную медь, осажденную на пемзе.
Другой, также немецкий, патент дает прн этом указания на более низкие пре-
делы температурного воздействия (170—200°) с прнмеьением смешанного ката-
лизатора, состояще) о из медной соли и соли другого металла (Со, Ni, Мп, Fe, Ст)
на пористом носителе, например геле кремневой кислоты п).
Хлорирование бензола в парах при отсутствии катализаторов
за исключением стенок кварцевой трубки, в которых проводился
процесс, изучалось М э с о н о м и сотрудниками при температуре
в пределах 250—400° и выше. Температура, необходимая для пол-
ного хлорирования, зависит от диаметра реакционной трубки. При
хлорировании при температуре свыше 400° получается хлорбензол
и дихлорбензолы, причем расходуется до 95°/о бензола и хлора.
При избытке хлора констатировано образование трихлорбензола.
Влияние изменения молекулярных соотношений реагентов видно
из таблицы (для диаметра трубки около 10 мм при температуре
450° и объемной скорости 50 (число объемов газа, проходящего
через единицу объема нагретой трубки при температуре реакции
в 1 мин.).
Молекулярные отношения С1д: свнв Количество % бензола, пре- вращенного С1 в % к взя- тому Отношение ны- ходов CgHjCl и CgH4c£
н CgHgCl в СвН4С1
1 : 1,1 44,7 15,2 нет 2,9
1 : 1,6 43,3 6,8 нет 6,4
1 : 2,2 34,3 3,7 0,3 9,3
1 : 3,4 24,0 2,2 0,5 10,9
Прн увеличении общего количества израсходованного бензола содержание
дихлорбензола, незначительное вначале, возрастает нее больше, начиная приблизи-
тельно с 60% расхода бензола, и при 1ООо/о расхода бензола достигает максимума
(и 80%), тогда как количество хлорбензола прн этом отвечает 26% превращения.
108
В Этом случае повышение температуры паровой фазы действует подобно катализи-
рую пей реакцию прибавке хлорида металла: продукт присоединения хлора оказы-
вается нестойким при этих термических условиях и разлагается, образуя продукт
замещения.
Присутствие катализаторов, применимых в жидкой фазе, способствует хлори-
рованию в парах, в частности хлорное железо содействует „полнхлорированню**—
увеличению содержания дихлорбензола и появлению трвхлорбензола и>.
Хлорирование бензола пропусканием паров бензола вместе с хлором в кислоро-
дом (с добавками разбавляющих газэв) через катализаторы (уголь—хлориды Си, Fe,
Tl, щелочноземельных металлов и редких земель) прн ЗОЭ—660° приводит по сло-
вам австрийского патента (Gremli) к получению хлорбензола18).
С другой стороны, имеются указания, что медь в процессе хлорирования
(в парах) действует иначе, чем железо или алюминий, давая преимущественно выс-
шие продукты присоединения (в особенности CgHgClJ и как продукт распада по
следнего 1.2.4-трнхлорбензол19).
Изучалось также хлорирование бензола в момент образования хлора при элек-
тролизе соляной кислоты. Было установлено, что выход хлорбензола и полихлоридов
растет с повышением температуры и зависит от скорости размешивания. Электро-
химический метод хлорирования при прочих равных условиях благоприятствует
бэлее глубокому хлорированию м).
Отметим, хотя и не имеющий практического значения, способ хлорноования,
основанный на фотохимическом окислении соляной кислоты в присутствии антра-
хинона, катализатора фотореакцин: бензол, 0,5% антрахвиоиа н 0,5% Fe2Os вместе
с разведенной соляной кислотой — н нижнем слое — образует прн освещении в ат-
мосфере кислорода монохлорбеизол 21).
Хлорбензол — продукт большого значения, являясь исходным
материалом для получеиия«-нитрохлорбензола,соотв. «-нитрофенола,
о-нитроанизола, соотв. дианизидина, «-нитранилина, динитрофенола,
сырого материала для сернистого черного. В ближайшем будущем
хлорбензол сделается несомненно сырьем для получения фенола
(каким он уже стал в Америке) и может быть анилина. «-Ди-
хлорбензол применяется как ннсектисид в деле защиты растений
от насекомых. Он и о-дихлорбензол могут быть применены для
синтеза соответственных двух- и полизамещенных: гидрохинона,
пирокатехина, их эфиров, хлоранилинов, хлораминофенов, их алкиль-
ных эфиров, фенилендиаминов и т. п.
Действие хлора и вообще галоида на такие бензольные про-
изводные, которые заключают реактивирующие группы: ОН или
NH2, обычно не требуют катализатора для получения галоидоза-
мещенных. Здесь ядро ароматического соединения достаточно акти-
вировано и не нуждается в добавочном воздействии. Тем не менее
получение полихлорфенолов (тетра, пента замещенных) из менее
охлоренных фенолов рекомендуется производить в присутствии
катализатора (FeCl3) и лучше — в подходящих органических рас-
творителях22).
Влияние определенных заместителей при низших ступенях замещения галоидом
приписывается тому обстоятельству, что „позитивнрующне" группы (ОН, NH2,
также OAlk) способствуют образованию положительного иона из органической
молекулы, который входит в сэсгав промежуточно-образуемого продукта присоеди-
нения. По вышеприведенному взгляду Пфейфера и Вицингера тнп этих
комплексных продуктов, полученных как первичные прн бромировании, таков (I):
^\/Х
Вг3
4- Вг2 + НВт
^/\Вг
109
X—„позитнвнрующая" группа,*—место ионообразовання у молекулы. Так как
тдкое комплексообразование идет или у той лвойвой связи, где имеется замещающая
группа, нлн по сопряженной системе в 4-м месте, то здесь возможно образование
только орто- и пара-замещенных 23). Для преимущественного вхождения галоидного
атома в фенолы в орто- или пара-место имеет повндимому наибольшее значение
температура. Повышение ее содействует орто-орнентации.
Активирующие группы, как ОН, NH2, в ядре соединения, под-
вергаемого воздействию хлора, нередко нужно предварительно
привести в неактивное состояние, например посредством ацилиро-
вания. Это важно для точного ориентирования галоида и для
устранения побочных реакций.
Наличие свободной аминогруппы может благоприятствовать образованию арома-
тических хлораминов RNC12, перегруппировывающихся (под влиянием Н-ионов)
в хлорированные ароматические амины 24).
Способ хлорирования лс-ксилидина в растворе серной кислоты является оче-
видно частным примером ннактивирования аминогруппы с переходом амина в соль
серной кислоты (подобно как и при ннтрованнн аминов в избытке серной кислоты).
В результате получаются мета- (по отношению к NH2) хлорозамещенвые 25):
Здесь преимущество метода в том, что сульфат 3.5-днхлорозамещенного не-
растворим в реакционной смеси и легко выделяется из нее.
б) Хлорирование нафталина
Хлорирование нафталина до сих пор не вошло в широкую
заводскую практику для получения массовых продуктов такого
большого значения, какое имеет хлорбензол. Есть однако полное
основание ожидать, что процесс хлорирования, надлежаще про-
работанный, может дать и в приложении к нафталину интересные
практические результаты, особенно для получения а-хлорнафталина
С1
и высокохлорированных производных.
Нафталин по отношению к хлору ведет себя несколько отлично
от бензола. Он сравнительно легко поглощает хлор, что объясняется
его структурой из двух колец, из которых одно может проявлять
себя как циклоолефиновое (ненасыщенное), если второе становится
вполне ароматическим 2в).
В нафталине значительно отчетливее, чем в бензеле, проявляется стремление
к образованию продуктов присоединения. Присоединение хлора особенно легко
протекает лишь в одном вз шестнчлениых колец нафталиновой молекулы. Про-
дукты присоединения, теряя элементы хлороводорода, переходят в хлорозамещенные
ПО
нафталина. Образование а-хлорнафталина проходят таким образом также стадию
присоединен ня, например:
НС1 С1
Дихлорнафталнны получаются из нафталннтетрахлорида:
Продукты присоединения хлора к нафталину легко образуются- при проведении,
реакции прн пониженной температуре. С повышением температуры онн отщепляют
в большей своей части хлористый водород, переходя в хлопозамещенные. Терми-
ческая неустойчивость хлоридов наф|алнна делает возможным получение хлор-
нафталнна без участия катализатора, ио при нагревании. Тем не менее не вся масса
хлоридов переходит в хлорозамещевные и прн повышении температуры; часть нх
остается в продукте, выделяя хлорородород при дальнейшей перегонке, если не
устранить возможность его образования целесообразно проведенной обработкой
щелочными агентами.
Хлорирование нафталина путем введения хлора в расплавленный
или кипящий нафталин впервые практиковалось Рымаренко,
причем главным продуктом реакции был а-хлорнафталин. В не-
давнее время опубликовано несколько исследований Ферреро
и сотрудников и работа советских химиков Траубенберга
и Вассермана 27), посвященные вопросу получения а-хлорнафта-
лина. В этих работах имеются данные относительно хлорирования
нафталина как такового в расплавленном состоянии и в паровой,
фазе, и особенно подробно освещен процесс хлорирования нафта-
лина, растворенного в органических растворителях.
Ферреро и Вуненбургер установили, что нафталин в состоянии пара
с 1,5 мол. хлора дает оптимальный выход ахлорнафталниа прн 350° (без катали-
затора) в 85% иа прореа!ировавшнй нафталин (59% на взятый в реакцию). При
применении в еачестве катализатора иола (0,5%) выхода выше: 94% на вошедший
в реакцию н 70% на весь нафталин. Хлорное железо облегчает образование высших
продуктов замещения; применение иода встречает затруднения в его большой лету-
чести, так как он отгоняется из контактной трубки при температуре реакции..
Пемза и древесный уголь активируют и моно- и полнхлорнрованне. Иод заметно
затрудняет образование полнхлорозамешенных; несмотря на высокую температуру,
выход полихлорозамешенкых н> же 5%. Способ хлорирования в паровой фазе пред-
ставляет большие трудное.и прн осуществлении в заводских размерах и вряд ли
будет конкурентоспособен по отношению к хлорированию в жидкой фазе.
Ферреро н Фельма и установили, чго расплавленный нафталин прн про-
пускании через вето при 126° тока хлора в отсутствии катализатора образует
а-хлорвафталнн с выходом около 80%, считая на вошедший в реакцию нафталин,
причем высших продуктов замещения образуется до 20%. В присутствии иода
(%—%% ст веса CioHg) выход а-хлорнафталнна достигает 84%, выход полиза-
мещеииых— около 1..%.
При применении для хлорирования растворов нафталина в органических раство-
111
рителях (наиболее благоприятное отношение по весу растворителей н нафталина—2:1)
удалось установить, что химическая природа растворителя имеет весьма большое
значение для результата про iecca. Так, растворители неактивные или очень мало
активные по отношению к хлору, каковы охлоренные углеводороды жирного ряда
(СС14, CgHGIg, CjHaClj), дают неважные результаты: выхода хлорнафталина — ниже
получаемых при хлорировании одного нафталина, реакция замедлена даже и при
повышении температуры.
Растворители, могущие сами реагировать с хлором, можно разделить на две
группы.
В первую входят такие растворители, которые хлорируются в условиях реакции
труднее, чем нафталин, даже в присутствии иодного катализатора, и потому защи-
щаются нафталином от действия хлора. В этой группе особенно интересны бензол
и хлорбензол из ароматических соединений и уксусная кислота из жирных. Они
активируют реакцию и дают высокие выхода а-хлорнафталина: применение хлорбен-
зола при 126° дало 92% а-хлорнафгалина, считая на вошедший в реакцию нафталин.
Вторую группу составляют растворители, сами хлорирующиеся легче, чем нафта-
лин. Таковы например толуол и декагидронафтална. Эти растворители, реагируя
в первую очередь с хлором, тем защищают нафталин от хлорирования. Понятно
само собой, что такие растворители непригодны для получения хлорнафталина. Были
испытаны и оказались мало пригодными в качестве растворителей для хлорирова-
ния нафталина этиловый спирт, этиловый эфир, уксусно-этиловый эфир, о- и л-ди-
хлорбеизолы и нитробензол.
Повышенная температура благоприятствует получению монохлорозамещенного.
Повышение температуры имеет однако границу (ниже 160°), выше которой итти не
следует.
Количество растворителей определяется двумя пределами. Если растворителя
мало, то усиливается полихлорирование, что наблюдается и при хлорировании без
растворителя. Если растворителя слишком много, то наблюдается постепенное хло-
рирование растворителя, в то же время растет и количество полнхлорозамещенных.
Оптимальное соотношение между весом растворителя н нафталина было определено
опытным путем и указано выше.
Для получения лучших выходов монохлорозамещенного выгоднее останавливать
хлорирование при затрате хлора несколько ниже требуемого теоретическим расче-
том (моль Ci2 на моль С10Н8< Йз испытанных Ферреро и сотрудниками катали-
заторов иод (%—%% от веса нафталина) благоприятствует монохлорнрованню, хлор-
ное железо (%%) содействует образованию полнхлорозамещенных. Невидимому для
получения а-хлориафгалина выгоднее обходиться совсем без катализатора, чем при-
менять хлорное железо. Применение смеси железа и иода (%%: V^/o) не изменило
поведения железного катализатора.
М. И. Сладко виМ. П. Гер применяли в хлорировании нафталина в каче-
стве катализатора хлористую сурьму (SbCi3), также мелкораздробленную металличе-
скую сурьму причем наряду с хлорнафталином получали значительное количество
•(до 20%) высшнх хлорозамещенных. Преимуществом этого катализатора является
его хорошая растворимость в реакционной смеси и то обстоятельство, что после
щелочной нейтралнзацнн сырой продукт прн перегонке не отделяет хлористого
водорода, между тем как полученный с иодом при прочих равных условиях выде-
ляет НС1 в значительных количествах. Вообще устранение выделения хлористого
водорода при термической переработке сырых продуктов хлорирования часто пред-
ставляет трудности.
Недавняя работа двух японских химиков, изучавших хлорирование нафталина
при разных температурах в присутствии восстановленной меди, делает вероятным
такое течение реакции, при котором медь (в виде своего хлорного соединения)
содействует в первую очередь образованию продуктов присоединения. Из последних
при термическом разложении, следовательно прн более высокой температуре, обра-
зуются продукты замещения 28).
Механизм реакции для этого катализатора объясняется исследователями такой
схемой:
Си + хС12 = Си • хС12; Си • хС12 + С10Н8 = Си • хС12С10Н8 = Си (х — 2п) С12 +
+ Сю^8С12п (продукт присоединения) С^НуС!^^ (продукт замещения) -f- НС1
Судя по недавним патентным заявкам иностранных фирм, за границей про-
является интерес к изучению высших продуктов хлорирования нафталина &). Хло-
рирование идет или с катализаторами в две стадии: до монохлорнафталнна (85—100°)
112
и до полихлорнафталинов (140—150°) или при непрерывном пропускании хлора
через ряд связанных между собой закрытых сосудов при пониженном давлении.
Можно повидимому и при температуре ниже 150° получить достаточную степень
охлорения. Продукт представляет воскообразную массу. В качестве катализаторов при
таком хлорировании упомянуты железо, сурьма, фосфор, сера, иод и их соединения.
а-Хлорнафталин кроме возможных его применений для полу-
чения иных замещенных нафталина с обменом хлора имеет значе-
ние как составная часть антидетонирующей прибавки к моторному
бензолу. Полихлориды находят себе применение в производстве
электроизолирующих материалов, пластических масс, огнестойких
покрытий и смачивающих веществ.
в) Хлорирование толуола и других соединений
Задача хлорирования толуола своеобразна, так так в этом углево-
дороде практически интересно заместить хлором водородные атомы
не в ядре, а в метильной группе и получить таким образом после-
довательно производные: C4H5CHjCl (хлористый бензил), С6Н5СНСГ2
(хлооистый бензилиден) и СвН5СС13 (бензотрихлорид или фенилхло-
роформ).
Вхождение хлора в ядро одновременно с замещением водорода
метильной группы должно быть по возможности устранено. Поэтому
при охлорении толуола тщательно избегают присутствия тек ката-
лизаторов, которые применяются при хлорировании бензола и ко-
торые благоприятствуют замещению в ядре, ибо при их участии
получаются вместо только-что названных продуктов о- и га-хлорто-
луолы С1С6Н4СН3. Таким образом возможно в зависимости от вы-
бранных условий направить реакции в ту или другую сторону по
схеме:
ClCfiH4CH3 <- С6Н5СН3 С8НбСН2С1.
Значение отдельных факторов прн реакции взаимодействия галонда и толуола
было обстоятельно выяснено Голлеманом и сотрудниками, из ннх главным
образом ф а н-д е р - Л а а н о м 30) иа примере бромирования толуола. Ими изу-
чалось значение катализатора, температуры, освещения и концентрации брома.
Было констатировано, что в полной темноте и без катализатора при высокой
температуре (выше 83°) бром реагирует исключительно в метильной группе, между
тем как при температуре 25° (реакция идет в течение долгого времени — многих дней)
бромистый беизнл получается всего лишь в количестве 10,6% от всей массы бромо-
продуктов. Из бромтолуолов орто-изомер образуется в меиьших сравнительно с пара-
изомером количествах, но относительное содержание орто-изомера в смеси увели
чивается с повышением температуры.
Под влиянием света реакция бромирования идет очень быстро, и в результате
даже на рассеянном свете образуется исключительно бромистый бензнл.
Как катализатор интересные результаты дало при этом бромное железо FeBr3.
Примесь уже 0,002 мол. FeBr3 на 1 мол. брома приводит, несмотря на сравнительно
высокую температуру (50°), в полной темноте только к образованию бромтолуодов
при полном исчезновении из продукта реакции бромистого бензила. Таким образом
влияние катализатора в этом случае успешно конкурирует с ориентирующим галоид
в метильную группу влиянием повышения температуры.
Повидимому влияние катализатора также преобладает над действием освещения,
само по се.бе благоприятствующим для замещения в СН3-группе.
Значение концентрации галоида выяснилось в ряде опытов бромировании зна-
чительных избытков толуола при 50° и 25°. Оказалось именно, что при увелнчении-
изЗытка толуола возрастает количество бромистого бензила в продуктах реакции.
Работы по хлорированию толуола в общем подтверждают пра-
вильности, установленные при бромировании.
8 Зак. 2611__Н. Ворожцов
113
Производилось сравнительное хлорирование толуола прн осве-
щении и в темноте, в отсутствии и в присутствии катализатора
(FeCl3, J) при очень низкой (—80°) и при высокой температурах
(105—110°) 31). Высокая температура при отсутствии катализатора
как в темноте, так и при освещении приводит к замещению только
в боковой цепи толуола, не затрагивая ядра, но при солнечном
освещении реакция проходит при этом приблизительно в два раза
скорее, чем в темноте.
Прн очень низкой температуре хлор н толуол в темноте практически не реаги-
руют; освещение же в этих условиях вызывает отчетливее взаимодействие. Расчет
выхода по квантам выясняет, что каждому абсорбированному кваиту при —80°
отвечает 25 реагирующих молекул хлора.
Хлорирование толуола в присутствии катализаторов замещения
в ядре изучалось А. Валем и сотрудниками 32). Действие таких
катализаторов успешно конкурирует с влиянием высокой темпера-
туры в пользу замещения в ядре. Алюминий оказывается слишком
энергичным катализатором, делающим реакцию настолько бурной,
что ею трудно управлять. Повышение температуры в присутствии
железа не изменяет получаемого с этим катализатором вообще
соотношения между изомерными хлоропродуктами (в среднем 58%
орто- и 42°/о пара-хлортолуола). Присутствие значительных коли-
честв хлористого свинца при температуре, близкой к 100°, благо-
приятствует ориентации хлора в ядро. Так как в производстве
хлорирование толуола проводится обычно в свинцовой аппара-
туре, то нужно тщательно избегать условий (главным образом
присутствия воды), могущих привести к образованию хлористого
свинца, чтобы не получить нежелательных примесей к продуктам.
Хлорирование толуола для получения хлористого бензила и даль-
нейших замещенных в метильной группе рекомендуется проводить
с добавкой хлористых соединений фосфора (иногдасеры и селена)38),
катализирующих будто бы эту реакцию. Смысл такой прибавки не
вполне ясен. С одной стороны, хлориды фосфора не влияют на
замещение в ядре, но опыты Бе pre л я 31)> сравнившего скорость
образования хлористого бензила при высокой температуре в темноте
и при освещении, с участием и в отсутствии пятихлористого фос-
фора, не обнаружили совсем ускоряющего влияния такой прибавки.
В производственной практике пользуются для хлорирования
толуола аппаратами из свинца, который должен быть свободен от
примесей, могущих катализировать замещение в ядре (Sb, Fe, Sn,
As, Си) при повышенной температуре (100—105°), близкой к темпе-
ратуре кипения толуола, и иногда пользуются в качестве стимули-
рующего реакцию средства освещением например светом ртутной
лампы, богатым ультрафиолетовыми лучами.
По Фрей иду з3) хлорирование толуола производится в железных гомогеино-
освинцованных аппаратах. Для получения в продукте преимущественно хлористого
бензилидена в 567 кг толуола (Д-5 кг РС13) вводят 1 С00 кг хлора при температуре
90—135° в течение 32 час. Получающаяся в конце смесь имеет уд. в. 1,2853 (32,2° Боме)
при 17° и заключает около 1 000 кг хлористого бензилидена. Такой продукт прямо
идет на гидролиз для получения бензальдегида и бензойной кислоты.
При достижении надлежащей степени охлорения освобождаются
продуванием воздухом и промыванием водой от растворенного хло-
ристого водорода (соотв. вымывают фосфорные соединения) и дроб-
ей
ной перегонкой разделяют продукты с разным содержанием хлора34).
При фракционировании нужно всячески устранять влагу и сопри-
косновение с железом или его соединениями со стороны продуктов
во избежание могущих быть потерь из-за образования продуктов
конденсации.
В одном американском патенте за) описана ниая система охлорення толуола:
в толуол добавляется сода (V2 веса толуола), и прн 60—70° прн хорошем размеши-
вании вводится хлор. По утверждению патента такое хлорирование не опасно в смысле
влияния катализаторов замещения в ядре (Fe), предположительно, здесь хлорирует
образовавшаяся НС1О (нз NaClO + СО2+ Н2О). Вряд ли такой способ хлорирования
в густой массе может оказаться практически удобным.
В недавней английской работе установлено, что хлорноватистая кислота (НС1О),
реагируя с толуолом, образует продукты замещения хлором как в ядре, так и в боко-
вой цепи. Полное отсутствие света содействует замещению в ядре. Свет, содержа-
щий большое количество инфракрасных лучей, благоприятствует замещению в боко-
вой цепи, ультрафиолетовые лучи разлагают НСЮ. Разбавленные растворы НСЮ
(до 1/7V) дают замещение в ядре. При образовании НСЮ из соли (NaClO) и слабой
кислоты (борной, угольной) наступает хлорирование в ядре, между тем как сильные
кислоты содействуют хлорированию в боковой цепи. Повышение температуры за
40° приводит к хлорированию в цепи. Избыток реагента не изменяет отношений
между замещением в ядре и цепи. Соли железа и
меди вызывают замещение в ядре. Там же указывается
иа возможность применения в качестве хлорирующего
агента эфиров хлорноватистой кислоты, например V
(СН3)3С—ООД. \
. - , X J I ««ч»_
Интересен способ хлорирования толуола
в паровой фазе при освещении, испытанный
в лабораторном масштабе Н. Д. Зелин- V. )
с ким. Подвергая кипению толуол и проводя
пары через дефлегматор с наполнителями,
кончающийся шарообразным расширителем Ц
с обратным холодильником и вводной труб- Щ
кой для хлора, в паровом пространстве этого
шара всегда можно иметь только толуольные Щ
пары, так как выше кипящий хлористый бен- \
зил конденсируется и стекает по дефлегма- \ 8В
тору в нижний сосуд с толуолом. Таким r-'X
образом в лабораторном масштабе можно /д \
прохлорировать толуол практически до конца ?=4
и получить продукт без высших хлорозаме- Vi у
щенных. Подобный прибор впервые приме-
нен был Н. Й. Курсановым для хлори- Рис. 4. прибор для
рования циклогенсана в парах, а затем и для хлорирования толуола,
хлорирования толуола.
Так же на использовании освещения основан способ, описанный
в недавнем американском патенте. Хлорирование ведется непре
рывным процессом. Горячий толуол входит сверху в башню из
стекла Pyrex, заполненную стеклянными кольцами Рашига (башня
длиной 10 м, диаметром 150 мм) и встречает ток хлор-газа, иду-
щего снизу. Башня освещается или солнцем или лампами с ультра-
фиолетовым излучением. При соответственном регулировании ско-
ростей тока хлора и толуола и температуры получается смесь,
содержащая до 57°/0 хлористого бензила с толуолом, практически
не имеющая примеси хлорозамещенных в ядре 87).
8*
115
М э с о н при хлорировании толуола в парах (прн высокой температуре) полу-
чил максимальный выход хлористого бензила (по отношению к взятому толуолу)
в 44,4% по отношению к израсходованному 70,3% от теоретически рассчитанного 18).
Хлоропроизводные толуола с замещением в метильной группе
имеют большое значение в красочной и некоторых других отраслях
химической промышленности.
Хлористый бензил С6Н5СН2С1 применяется в широких раз-
мерах для введения бензильного остатка (С6Н6СН2) в органические
соединения: для получения бензиловых эфиров, интересных для.
лакокрасочной промышленности, бензилцеллулозы (для искусствен-
ного волокна и пластических масс), бензилированных ароматических
аминов, например этилбензиланилина, некоторых смачивающих и
эмульгирующих веществ. Хлористый бензил имеет значение и для
производства химических боевых средств (например бензилциа-
нида). Хлористый бензилиден С6Н6СНС12 больше всего
интересен для производства бензойного альдегида. Бензотри-
хлорид С6Н5СС13—для производства бензойной кислоты и ее
хлорангидрида, хлористого бензоила С6Н5СОС1, который в свою
очередь служит наиболее употребительным в технике средством для
ввода бензоильного остатка С6Н5СО в органические соединения.
Некоторые кубовые красители антрахинонового ряда—аминобен-
зоильные производные.
Помимо этих имеют значение некоторые хлоропроизводные толу-
ола с хлором в ядре, особенно о-хлортолуольные производные для
производства о-хлорбензойного альдегида.
Непосредственное хлорирование антрахинона и его производ-
ных обработкой хлором — сравнительно мало применяемый в тех-
нике прием. Тем не менее, судя по описаниям патентов, хлориро-
вание и бромирование удается сравнительно легко, если применять
раствор антрахинона, соотв. его производных, например сульфо-
кислот, в серной кислоте разных концентраций (от моногидрата до
80°/о-ного олеума) с участием или в отсутствии иода. Например:
СО СКЛ tj Cl СО С ГЛ 1J
/\ /\ /\ /йизп
Cl3
в растворе H2SO4 160'
СО С1 со
Во многом похожий на антрахинон по стойкости и реакционным особенностям
фталевый ангидрид также дает полигалоидопронззодные при обработке галоидом в
растворе серной кислоты 6)
Так
СО
С1
I со
С1
СК 1 S?
о----------------1---------
в олеуме с 25»;О SO3, 40—60'
о
со
С1
I со
С1
со
со
I I
С|/Х%0
116
Применение олеума в качестве растворителя при галогенировании в этом н по-
добных Примерах полезно ие только для активирования реакции, но н потому,
что SO3 связывает выделяющуюся галоидоводородиую кислоту, в случае хлориро-
вания иапрнмер образуя хлорсульфоновую кислоту:
SO3+HC1 = HO3SC1;
последняя может быть в большей части отделена от реакционной массы отгонной.
Хлорирование многоядериых ароматических углеводородов (типа дифенила)
может быть проведено подобно хлорированию нафталина в растворе органических
растворителей (например бензола) в присутствии катализаторов (Fe, FeCl3, AlClg, J
и т. п.), причем вначале (до образования дихлоропроизводного) при низкой темпера-
туре (около 0°) охлорения растворителя не наблюдается з®).
Обработка галоидом сложно построенных красителей требует
всегда участия растворителя. В качестве такового берут неактив-
ный по отношению к галоидам нитробензол (случай бромирования
индиго) или крепкие кислородсодержащие кислоты (чаще олеум,
также НС1О4, Н3РО4) — примеры иодирования антрахиноновых и
индигоидных красителей зэ). Находит себе применение также и
спирт как растворитель: таково бромирование и иодирование
флуоресцерина 40).‘
Метиловый спирт испытан как растворитель при хлорировании ароматических
соединений (аминов и фенолов). Никаких преимуществ применение этого раствори-
теля не имеет4!)-
Кроме воздействия молекулярного хлора имеются иные методы
введения этого галоида в молекулу ароматических соединений.
Выше мы видели уже некоторые примеры хлорирования, прово-
димого за счет хлора в момент его образования из соляной
кислоты (в результате электролиза или контактного окисления).
Интересным хлорирующим агентом, хотя и более дорогим, чем хлор, является
хлористый сульфурил SO2C12. Его воздействие на многие органические вещества
сопровождается их хлорированием по реакции:
RH + SO2Cl2-> RC14- SO2-f- НС1.
Газообразные побочные продукты реакции (SO2 и НС1) могут быть улавлеиы
и использованы 42).
Способ такого хлорирования, естественно, может найти применение лишь в
исключительных случаях, применительно например к получению определенных по
положению хлорозамещенных, также в ряду антрахиноновых производных и примени-
тельно к алкилнафталииам 43).
Недавно метод обработки хлористым сульфурилом был испытан в применении к
получению хлорозамещенных толуола, причем выяснено было каталитическое влия-
ние хлористых соединений большого числа металлов и металлоидов. Хлориды: Sb,
Bi, Fe, Mo, Al, Те, Se благоприятствуют замещению в ядре. Хлориды: Р, As, Мп,
Вт благоприятствуют замещению в метильной группе. Хлористая сера SC12 ускоряет
оба вида замещения. Хлористый алюминий дает возможность провести исчерпыва-
ющее хлорирование ядра до пентахлортолуола. Любопытна подмеченная здесь зави-
симость характера каталитического воздействия от атомного веса элементов одной
и той же группы периодической системы 44).
В ряду антрахиноновых производных нередко пользуются свое-
образным методом введения хлора в ядро, именно—обработкой
соединений в растворе или суспенсии в соляной кислоте солями
хлорноватой кислоты. Реакции должно бы отвечать уравнение:
3RH + NaC10s + ЗНС1 = 3RC1 + NaCl + ЗН2О,
но соли хлорноватой кислоты (хлората) расходуется значительно
117
больше, чем отвечает этому уравнению «). Часто эта обработка
применяется для замены сульфогруппы в антрахиноновом ядре
атомом хлора и имеет значение для определения структуры антра-
хиноновых сульфокислот, например
SO3H
СО I
/\/\/\
I NaC103+HCl
+ Na9SO4
Несмотря на крайне энергичную обработку (крепкая кислота,
окислительная смесь, кипячение), антрахиноновые производные вы-
держивают ее, не разрушаясь. Можно заместить сульфогруппу
хлором и прямо обработкой сульфокислоты в среде соляной кис-
лоты хлором 48).
Интересно отметить, что аналогичная обработка, примененная к 4-сульфофтале-
вому ангидриду, приводит к получению также хлорозамещенного фталевой кислоты 47).
Cl^/^/COOH
I I
I I
\соон
Воздействие хлора в щелочной среде наилучше используется
в реакции ароматических соединений с солями хлорноватистой
кислоты — гипохлоритами 48).
Реакция выражается уравнением:
RH + NaClO = RC1 + NaOH.
Реакция имеет значение применительно к оксипроизводным бен-
зольного, нафталинового и антрахинонового рядов, причем замеще-
нию хлором подвергается преимущественно атом водорода, стоящий
в орто-положении к гидроксилу.
Обработка водным раствором гипохлорита проводится или на холоду илн при
температуре до 30—40° н даже до кипения раствора. При работе с легко окисля-
ющимися соединениями илн такими, в которых имеются легко окисляющиеся группы,
необходимо учесть возможность окислительного влияния гипохлорита по реакции:
NaClO = О + NaCl.
Хлорирование, также как н бромирование, обработкой галоидом в водном рас-
творе проходит вероятно с промежуточным образованием хлорноватистой, соотв.
бромноватистой, кислоты, например
С1, + Н2О = СЮН -f- НС1.
Но агентами галогенирования в этом случае служат не эти кислоты, а галоид в мо-
мент выделения, так как ие содержащая брома бромноватнстая кислота НВгО бро-
нирует в 1 000 раз медленнее, чем бромная вода 4в).
Недавнее исследование Дм. Тищенко бромирования н хлорирования арома-
тических соединений в водном растворе соды позволяет сделать выводы, согласные
с этим.
Как „мягкое" бромнрующее средство указаны дибромбромгидраты пи-
ридина н хинолина, например C5H5N • HBrBr2, в среде уксусной кислоты 5°). Также
118
чожно указать и на упомянутые выше хлорамины, которые в присутствии соляной
кислоты передают хлор от азота заместителя в ядро Б1). Испытано было также хло-
рирование за счет хлора, связанного с азотом, не принадлежащим молекуле хлори-
руемого вещества, например посредством дихлормочевииы СО (NHCI)2 (хлорирова-
ние фенола), днхлорамнда л-толуолсульфокн слоты СН3С5Н48О2МС12 (днхлорамнна
Т) в среде уксусной кислоты с добавлением соляной кислоты (хлорирование азо-
фенолов) 52); в хлорировании толуола как хлорирующий агент применялся также
не замещенный органически хлорамин (NClj; готовится взаимодействием NH4C1 и
хлора) 83).
Выше была отмечена возможность (особенно в водной среде) течения взаимо-
действия между галоидом и органическими соединениями в направлении окисле-
ния. Иногда галогенирование происходит вместе с окислением, и в результате по-
лучаются галогенированные продукты окисления.
Прежде продуктам обработки избытком галоида фенолов илн галондофеиолов
приписывалась структура так наз. кетогалогенидов (например кетобромндов) (И).
Ныне доказано, что такие продукты построены как эфиры бромноватистой (соотв.
хлорноватистой) кислоты (I) к.
Вт Вт
Хлорирование ^-нафтола приводит, по Цинке, к днхлоркетонафталнну 66)
С1 С12
Действие хлора на анилин в неводном растворителе поиводит к получению сим-
метрического трихлоранилнна; в водной среде получаются помимо того окрашенные
продукты, которые легче получаются прн взаимодействии хлора с кипящим анили-
ном. Это сложные продукты окисления — нндулиновые красители вероятной струк-
туры
CBH5HNx^xJ4x/x/NHCeH5
I
СУЬ
Хлорирование первичных аминов в виде солянокислых солей прн полном рсклю-
ченни влаги в растворе или суспенсни в индиферентиом растворителе дает воз-
можность получить азотсодержащие высокоохлоренные продукты, где частично
илн полностью двойные связи ядра насыщены н превратились в одиночные. Так,
анилиновая соль в виде суспенсин в хлорбензоле дает соединение
С12
HCI:/\:NC1
НС1
Подобные соединения дают а- н fi-нафтиламины, а- и “-амииоантрахиноны 53).
Третичные ароматические амины с свободным орто-, соотв. пара-местом по отно-
119
шению к аминогруппе легко хлорируются в водном растворе в форме своих ми-
неральнокислотных солей (Кал и шер, Келлер). Например68)
/ч^/N (СН3)2 • НС1 /\/N (СНз)? ’ ЧС1
I I 1
Н3с/ Н3С/Х/ \ci
Хлорирование и бромирование п-аминобевзоил-о-бензойной кислоты удается в
растворе концентрированной серной кислоты при действии хлора (соотв. брома м),
например
'Ч/'ХСООН \/\сООН
Прн хлорировании или бромировании соединений, могущих легко перейти
н хиноны, получается хлор- (соотв. бром-) анил, тетрахлор- (соотв.тетрабром-) хинон,
например нз изонитрозофенола
С1
jA: NOH ci, Cl^\ : О
в кисл. среде ;
0:U O:UC'
Ci
Роданирование
Имеется метод введения более дорогих галоидов, брома и иода,
в органические соединения, использующий вытеснение их хлором
из неорганических соединений. Смесь, отвечающая по содержанию
NaBrO8-|- 5NaBr (или таким же соединениям иода) при обработке
их раствора хлором отдает весь свой бром, соотв. иод, органиче-
скому веществу, например
6RH + NaBrO3 + 5NaBr + ЗС12 = 6RBr -f- 6NaCl + 3H2O.
Раствор бромата н бромида в соответственных отношениях молекул получается
при прибавлении брома к раствору шелочн при нагревании:
6NaOH + 3Br2 = 5NaBr -f- NaBrO3 -J- 3H2O.
Трудно судить, как широка область применения этого метода,
но он интересен, так как использует весь ценный галоид, а не
половину всего взятого количества, как при обычном бромирова-
нии 61).
Правда, при бромировании флуоресцеина с 1,елью получения
эозина, которое проводится в спиртовом растворе непосредственно
бромом, расход брома сокращается также вдвое путем прибавле-
ния соли хлорноватой кислоты (например КС1О3), которая и выте-
сняет бром из образовавшейся бромистоводородной кислоты, пере-
водя его в ядро органического вещества:
3RH + ЗНВг 4- КС1О3 = КС1 + ЗН2О 4- 3RBr в2).
Похожим способом вытеснения можно вызвать реакцию между
достаточно реакционным органическим веществом и роданом (SCN)a
120
в момент его образования из неорганических солей роданисто-
водородной кислоты. Подобно галоидам, этот комплексный псевдо-
галоид входит в качестве заместителя водорода в органическую
молекулу. В наиболее простой форме это взаимодействие можно
осуществить, растворив роданид в воде, введя в водный раствор
роданируемое соединение (фенол, соль амина и т. п.) и прибавив,
туда галоид (бром или хлор), например по схеме:
2NaSCN 4- Br2 = 2NaBr+ (SCN)2,
RH 4- (SCN)2 RSCN 4- HSCN.
.1ри правильном манипулировании, сохранении избытка роданида, реакция между
галоидом и органическим веществом практически не имеет места; также можно
устранить, поддерживая достаточно кислотную реакцию среды, н гидролиз родана,
ведущий к образованию роданистоводородной, серной и синильной кислот-.
3(SCN)2 + 4Н2О = 5HSCN + H2SO4 + HCN.
В качестве кислот, в среде которых реакция протекает, предложены минеральные
кислоты, уксусная и муравьиная в3).
Имеются предложения проводить роданирование в растворе метилового спирта,,
насыщенного предварительно солью галоидоводородной кислоты, галоид который
применяется для вытеснения родана (NaCl при хлоре, NaBr прн броме). В послед-
нем случае выхода получаются выше, и в частности устраняются побочные про-
дукты — тиазоловые производные, образующиеся при замещении роданом в орто-
месте к аминогруппе м), например
Роданирование имеет применение по отношению к соедине-
ниям, имеющим реактивирующие амино- или оксигруппы, к кото-
рым в пара- (или если оно занято) и в орто-место становится
группа SCN. Таким образом получены например соединения:
Применяются также н нерастворимые соли роданистоводородиой кислоты,
в частности Pb(SCN)2 или Hg(SCN)j в растворе органического растворителя, насы-
121
шейного соответственным влектролитом (NaCl, NaBr). Как растворители рекомен-
дуются: метиловый спирт, этиловый спирт, четыреххлористый углерод, сероуглерод,
ацетон, уксуснометиловый вфир “).
Наконец видоизменением метода является взаимодействие не между галоидом
и роданидом, но между родаиндом [Pb(SCN)2] и хлористым сульфурилом в рас-
творе четыреххлористого углерода. Промежуточно вероятно образуется роданистый
сульфурил, который и роданирует, подобно тому как хлористый сульфурил хло-
рирует в®),
Pb(SCN)2 + SO2C12 = РЬС12 + SO2(SCN)3,
RH + SO2(SCN)2 =» RSCN 4- SO2 + HSCN.
Роданозамещенмые—новые еще для техники продукты, воз-
можно могущие оказаться интересными для промышленности фарма-
цевтических средств, флотационных реагентов и может быть и для
красочной.
Хлоро- и бромозамещенные углеводородов имеют характерные
для них температуры кипения и плавления. В том случае(довольно
редком в практике), когда желают качественно определить хлор
или бром, пользуются известной пробой Бейльштейна: накали-
ванием в ушке платиновой проволоки кусочка прокаленной окиси
меди с весьма малым количеством вещества, причем при наличии
галоида в испытуемом соединении бесцветное пламя горелки окра-
шивается в зеленый или зеленовато-голубой цвет.
Количественно галоид определяется или
Jno методу Ка риуса (нагреранием в за-
паянной трубке с крепкой азотной кисло-
той в присутствии AgNO3) или по сухому
способу Либиха — прокаливанием с чи-
стой известью, причем органически связан»
ный хлор переходит в хлористые соли.
Ряс. 5. Прибор из стекла
Ругех для количественного
определения галоида.
Эти способы количественного определения гало-
ида дают удовлетворительные результаты во всех
случаях. Иные упрощенные способы пригодны только
применительно к определенным соединениям и типам
соединений. Таков способ А. В. Степанова (нагре-
вание с металлическим натрием и абсолютным этило-
вым спиртом и последующее определение галоида
в подкисленном азотной кислотой растворе), который
с ароматическими хлоропроизводными не всегда дает
удовлетворительные результаты 87).
Интересен как удобный, но требует
изучения способ Томпсона и Окедла6*),
испытанный в нескольких опытах с поло-
жительным результатом К. Г. Мизучем.
Ниже приводится рисунок прибора, материал м
которого является стекло Ругех. я
В колбу вводится навеска галоидопроизводного
и 0,5 г CuSO4. В коническую колбу, служащую для
поглощения хлора, наливают 100 см3 10%-ного рас-
твора едкого натра, содержащего 1 г As2O3. После
того как аппарат собран и вода пущена в холодиль-
ник, осторожно приливают в колбу с навеской, через
воронку, 20 вм3 олеума с 20—30%SO3, дают постоять
некоторое время без нагревания, затем кипятят
122
30 минут, стараясь не допустить сильного выделения SO3, Затем приливают смесь
и? 10 г персульфата калия и 20 см* 95%-ной сериой кислоты. Прилив еще 15 см3
95%-ной серной кислоты, кипятят до позеленения раствора в нагретом состоянии.
Затем крайне осторожно приливают 50 см* воды и, выпустив воду из холодильника,
концентрированный раствор марганцевокислого калия (до красного окрашивания).
Смесь кипятят 5—10 минут.
Содержимое поглотительной склянки переводят в стакан, подкисляют азотиой
кислотой и определяют С1 или объемным путем поФольгарту или весовым
путем.
Промежуточные продукты, получаемые методом
хлорировании
(*мета]+ полихлориды.
+ паталврийы.
CI
а
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
») IG, Б. п. 361097, СВ 1932, 1, 1445.
2) L. Rabinowitz, Ам. п. 1189736 (1916).
3) Fahlberg-List, Г. п. 219242, F. IX, 1179.
4) L. Grin га их, Helv. 12, 921 (1929).
5) М е е г w е i п, Zt. angew. Ch. 38, 816 (1925).
e) Batt egay, Bull. [4] 43. 120 (1928); Ф. n. 641102, CB 1928, II, 1718; Г. n.
216071 (B), F. IX, 677, 107721, F. V, 302, ср. Г. n. (By) 228901, F. X, 578; па полу-
чение галогенированных фтвлевых кислот см. N. J u v а 11 а, Г п. 50177, F. Н, 93,
Villiger, Ber. 42, 3538 (1909).
123
i) М. V. Jon esc u, CB 1927, 11, 243, Datta, Am. Soc. 39, 435, CB 1918, Ir
188, Varma, Kulkarnl, CB 1927, I, 1433, Varma, Narayan, CB 1928, I, 489.
Ср. E. de Barry-Barnett, Cook, Rec. 43, 262, 1924. Bull. (4) 36, 1328, (1924).
«) Pfeiffer, Witzinger, Ann. 461, 132 (1928).
9) A. Wohl, Ber. 64, 1382 (1931), прим. 6.
ю) J. Meunier, Bull. (4) 27, 833 (1920).
U) F. Bourion, C. r. 170, 1319, 933, 990, 1115, 1181 (1920).
l2) К or c z i n s к i Bull. (4) 29, 283 (1921).
ls) Lane (jr), Noyes (jr), Am. Soc. 54, 161 (1932).
14) Lowy, Frank, Trans. Am. Erectrochem. Soc. 43, 107 (1923).
15) M. E. P u t m a n (Dow. Co), Am. n. 1858521 (1932).
ie) Cp. Co. Prod. Chim. d’Alais et de la Camargue, Г. n. 410529, F. XV, 78;
L. Marckwald, Г. n. 142939, F. VH, 46.
17) Б. n. 208155 (MLB), CB 1924, 1, 2822, 1G, Г. n. 487596, F. Raschig, Г. n.
539176 CB 1932 1 2642.
18) Mason, Smale, Norman, Wheeler, Soc. 1931,3150; E. Grem 1 i, Ab. n.
108424, CB 1928, I, 1229.
I9) S h o-e T e i, S h i g e r u К о m a t s u, CB 1928, I, 2370.
20) C. W. Croco, Lowy, CB 1927, 1, 421, также 4
2i) Nozicka, Г. n. 526195, CB 1931, 11, 2931. О фотохимической реакции,
в газовой фазе между бензолом и хлором см. 13.
22) IG, Г. п. 527393, СВ 1931, II, 2785.
23) См. 8, также F. G. S о р е г, G. F. S m i th, Soc. 1926, 1582.
24) Г. n. 240038 (A), F. X, 178,94410 (L ev i n ste i n), F. IV, 73; S. Goldschmidt,
L. Strohmenger, Ber. 55, 2450 (1928).
25) G. An. W„ Am. n. 1730729.
26) Ср. H. H. Ворожцов, Химические особенности нафталинового ядра. Изве-
стия Ив.-Возн. пол. ин-та № 6, Woroshtzow, Bull. (4) 35, 996 (1924).
27) Рымаренко, Ж. 8, 141 (1876); Р. Ferrero, Wunenburger, Helv. 11,
416 (1928), Ferrero, Fehlmann, Helv. 11, 763, 12, 583 (1929), J. pr. Ch (2) 122,
340 (1929), Ferrero, Corbez, Helv. 13, 1009 (1930); Шв. n. 134089 (F. F.),
CB 1930, 1, 2165; Traubenberg, Wasserman n, J. pr. Ch. (2) 120, 177
(1928).
28) Sh. Komatsu, Sb. Tanaka, С. A. 26, 722 (1932).
2”) ICI, Б. n. 357743, CB 1932,1, 1440, IG, Б. n. 324774, CB 1930,1, 3486, Halowax,
Corp. Am. n. 1564044, CB 1926, I, 1717. B. D. Co, Б. n. 291849, CB 1928, 11, 2286.
°°) Holleman, Drrekte Einitihrung, 84 и след.
si) Book, Eggert, Zt. f. Elektrochem. 29, 512 (1923), Ber.59, 1192 (1926), Ber-
gel, Ber. 59, 153 (1926).
82) A. Wahl, Normand, Vermeylen, Bull. (4) 31, 570 (1922).
83) R. Freund, Die Fabrikation von Benzaldehyd und BenzoesSure im Grossbe-
trieb, Ch. Ztg. 51, 803 (1927); H. Erdmann, Ann. 279, 148 (1893); L. Kyrides (Nat.
An.) Am. n. 1733268.
8i) Dow. Co, Am. n. 1804458, CB 1931, II, 497.
35) Ch. C. Loomis (Sem. Solvay Co), Am. n. 1384909.
Зв) B. Clark Hr), C. A. 26, 1591 (1932), из Chem. News 143, 265(1931).
37) H. Д. Зелинский, Б. В. Мак cop о в, Ж. X. Пр. 1, № 5-6, 4 (1925);
Н. И. Курсанов, Ж. 38, 1304 (1906); М. М. Кацнельсон, Приготовление синте-
тических химико-фармацевтических препаратов, Москва, 1915, 99, 100; Solvay Proc.
Со (Conklin), Am. п. 1828858, 1828859, СВ, 1932, I, 1575; Ан.-кр. пр. 1932 № 2, 56.
38) Dow. Со, Ам. п. 1835754, СВ 1932, 1, 1440.
з») 1G, Б. п. 327702, СВ 1939, II, 632.
«) Ср. Pyro-Pack Prod. Со, Ам. п. 1733776, СВ 1930, II, 805.
'!) Plaiek, СВ 1931, I, 1427.
42) Ср. Peratoner, Condorelli, Gazz, 28, 1 226 (1898).
4») Г. п. 158951 (В), F. VIII, 277, 146796, F. VII, 46, 160102, F. VIII, 86, 162394
(Wohl) Е. VIII, 86, IG, Б. п. 263844, Ch. Tr. J. 1927,272.
«) О. Silberrad, Ch. A. Silberrad, Parke, Soc. 127, 1724 (1925), Silberrad,
Baake, Roberts & Co, Б. n. 259329, Br.A.B. 1926, 1029.
«) Cp. W. —Ильинский, Г. n. 167743, F. VIII, 273, 172300, F.VII1, 274, 202770,.
F. IX, 684, 189937, F. IX, 685.
«) By, Г. n. 205195, F. IX, 673, H. Schilling, Ber. 46, 1066 (1913).
«) H. Waldmann, E. Schwenk, Ann. 487, 287 (1931), CB 1931, II, 1278.
«) Ср. Г. n. 167458, F. VIII 164, 168824 (K), F. VIII, 165, 152175, F. VII, 167,.
124
153194 (W.—Ильинский), F. VII, 169, Ber. 16, 1749 (1883), IG. Ф. u. 669388, CB
1930, 1, 3240.
«) H. Baines, CB 1923, I, 746, A. W. Francis, CB 1926, I, 353. Дм. Тищенко,
Ж. 60, 153 (1928).
ю) Rosenmund, Kuhnheim, Ber. 56, 1262 (1923).
5l) Например: Orton, Bradfield, CB 1927, П, 686, Bradfield, CB 1928, I,
2499.
к) Лнюшерстов, Ж. 61, 1019 (1929), L. Hunter, R. S. Barnes, CB 1928,
II, 1762.
63) Stoll, CB 1929, II, 35.
61) Kohn, Rosenfeld, CB 1926,1,2332.
бб)Сукневнч и Будницкий (Ssuknewitsch, Budnitzky) J. pr. Ch. 138, 18,
1933, CB. 1933, II, 2002.
66) Zin eke, Ber. 21, 3378, 3540 (1888).
57) Brown, Cumming, CB 1927, I, 757.
D. H., Г. n. 400254, F. XIV, 381.
б9) С..Г. n. 453427, G, Kalischer, Keller Am. n. 1777266(1930). E.T. Howell
(DuP), Am. n. 1832211 (1931).
e°) Holliday & Co, Б. n. 274700, CB 1927, II, 2227.
61) Zmaczinski (Змачинский), Ber. 59, 710 (1926).
62) Heumann, Die Anilinfarben und ihre Fabrikation, Braunschweig, 1888, I, 473.
63) H. P. Kaufmann, Oering, Ber. 59, 187 (1926), IG, Г. n. 484360; Dienske,
CB 1927, I, 2410.
6«) IG, Г. n. 491222, CB 1930, I, 2631, Ф. n. 620799, CB 1929, I, 2697, 35025, CB
1930, 1, 1696.
65) IG, Г. n. 493025, CB 1930, II, 1283.
66) IG, r. n. 439604, F. XV, 278, Spengler (A), Am. n. 1594697.
67) По сравнению различных методов определения галоида в органических сое-
динениях см. К. В. Алексеевский, Я- С. Пиказин, Ж. П. Х.З, №2, 273 (1930).
б®) J. Thompson, Oakadle, Am. Soc. 5?, 1195(1930), Willard, Thompson
Am. Soc. 52, 1893 (1930) см. Ж. X. Up. 8, 533 (1913).
ГЛАВА V
ОБРАЗОВАНИЕ АМИНОГРУППЫ ВОССТАНОВЛЕНИЕМ
НИТРОГРУППЫ И ДРУГИХ АЗОТСОДЕРЖАЩИХ ГРУПП
Образование аминогруппы (NH2) путем восстановления азотсо-
держащих групп: нитро-(NO2), изонитрозо- (NOH), нитрозо-(NO) и
азогруппы (N:N), принадлежит к числу реакций превращения уже
имеющихся заместителей. Следовательно при этом процессе не
может быть, как правило, затронут какой-либо из водородных ато-
мов, связанных с углеродным ядром (хотя замещение посторонними
группами и может иногда иметь место в качестве сопутствующего
реакции восстановления). Место аминогруппы в ядре таким обра-
зом уже предуказано наличной в нем группой, подвергающейся
превращению. Ввиду того что наиболее часто в практике получе-
ния промежуточных продуктов эта реакция применяется по отно-
шению к нитрогруппе, ниже мы остановимся более подробно на
процессе восстановления нитросоединений. Это — настолько важ-
ная реакция, что синтез нитросоединения весьма часто является
лишь этапом на пути получения аминосоединения.
Учитывая разницу в молекулярном составе нитро- и аминосоеди-
нений, реакцию проще всего представить как воздействие водо-
рода на нитросоединение и дать ей такое общее выражение:
RNO2 + 6H = RNHa+2H2O. (1)
Однако такое выражение отвечает только частным случаям реак-
ции, при которых используется молекулярный водород в катали-
тических процессах или водород в момент выделения в чисто хи-
мических, в большинстве же процессов восстановления, имеющих
практический интерес, водород не вводится в реакционную среду.
Поэтому в общем виде процесс правильнее представить как си-
стему реакций, в которых участвует нитросоединение, дающее
свой кислород (окисляющее), и другие ингредиенты реакционной
среды, принимающие этот кислород (окисляющиеся). Если при те-
чении этих реакций останавливаться только на этой стадии, то
можно получить в продуктах реакции соединения с заместителями,
менее богатыми кислородом, чем нитрогруппа [RNO, RN(O): NR],
или совсем, лишенные кислорода (RN=:NR). Если же кроме того
реакционная среда делает возможным проявление реакционности
со стороны водородных соединений (например воды, когда она
вовлечена в окислительный процесс с расщеплением молекулы на
Н и ОН), тогда продуктами реакции восстановления оказываются
и имеющие при азоте водород (с кислородом или без него):
RNH • ОН, RNH • HNR, RNH2.
126
При таком понимании процесса реакция восстановления нитро-
соединений требует вещества, способного окисляться за счет кис-
лорода нитрогруппы (непосредственно или через посредство дру-
гих ингредиентов реакционной среды). Эти окисляющиеся вещества
по отношению к нитросоединениям оказываются „восстановителями".
Восстановителями могут быть очень многие вещества, как простые
(элементы), так и соединения, в том числе и органические, поэтому
литература дает богатый фактический материал по проведению
этой реакции с разными восстановителями. Техническая практика
пользуется однако сравнительно небольшим ассортиментом восста-
новителей, наиболее доступных по цене и удобных для практиче-
ской работы, и особенно широко применяет в качестве таковых
металлы, из них главным образом железо.
Рассматривая огромный материал методов проведения реакции
восстановления нитросоединений в водйых растворах (или суспен-
сиях), можно вывести руководящее заключение, что течение реак-
ции и ее результат в большой степени определяются реакцией
водного раствора, т. е. концентрацией водородных и гидроксиль-
ных ионов в нем. Проводимые по этому признаку в одинаковых
условиях восстановления дают приблизительно одинаковые резуль-
таты, хотя индивидуальные особенности нитросоединений необхо-
димо должны быть учитываемы, ибо структура ядра, связанного
с нитрогруппой, положение последней по отношению к другим
заместителям, реакционный характер замещающих радикалов мо-
гут вызвать и вызывают иногда серьезные изменения в ходе и
результате процесса.
По признаку относительной кислотности или щелочности реак-
ционной среды можно разделить процессы восстановления на три
основные группы: 1) такие процессы, которые проводятся в при-
сутствии кислотных агентов, 2) такие, которые используют прак-
тически нейтральную среду, и 3) проводимые в щелочной среде.
Надо сказать, что, поскольку известно автору, вопрос о концентра-
ции водородных ионов в водных растворах, применяемых при вос-
становительных процессах, вопрос о влиянии анионов различных
кислот не освещались сколько-нибудь систематично научным ис-
следованием, так же как и относительная способность к восста-
новлению отдельных восстановителей (окислительно-восстанови-
тельный потенциал процесса). Об этом приходится пожалеть:
исследования в этом направлении могли бы быть очень плодотвор-
ными и разрешить много недоуменных вопросов практики.
Эффект действия железа в растворе хлорида щелочного металла
(получение амина) не тождественен эффекту действия цинка в-рас-
творе хлористого аммония (получение p-арил гидроксиламина);,
возникает вопрос: чем вызывается эта разница?
Самый механизм реакции восстановления в гетерогенной системе с участием
твердою восстановителя (Fe, Zn и т. п.) нуждается в выяснении. Бесспорно, реак-
ция здесь идет иа поверхности, разделяющей твердую фазу от жидкой. Не только
размеры поверхности (крупность зерна металла), но и ее состояние имеют большое
значение для начала и скорости восстановления; этому мы найдем примеры осо-
бенно в щелочном методе восстановления. Однако составление хотя бы схемати-
ческой картины для взаимодействия между атомами металла, Н’-или ОН'-иоиами и
носителем иитрогруппы—дело будущего, и здесь положение вопроса ие более, если
127
не менее, ясно, чем в механизме гетерогенно-газового катализа. Частичное прибли-
жение к разрешению вопроса в частном случае дает Принс х).
Проведение восстановительного процесса может быть далее
чисто химическим и электрохимическим, если восстановление осу-
ществляется на катоде электролитической ванны. В восстанови-
тельном процессе, проводимом в жидкой фазе, могут принимать
участие катализаторы, содействующие например проявлению реак-
ционности молекулярным водородом. Наконец восстановление
может быть осуществлено в паровой фазе водородом при содей-
ствии катализаторов, но последний вид каталитического восста-
новления мы рассмотрим в отдельной главе в конце книги.
а) Восстановление в кислотной среде
Наиболее употребительным материалом для кислотного восста-
новления служит железо (чугунные стружки, опилки) в присут-
ствии соляной кислоты. Кислотный способ вообще характерен
тем, что дает сразу конечный продукт восстановления, т. е. ами-
носоединение. Казалось бы, что уравнение, отвечающее такому
восстановлению, должно быть таково:
RNO2 4- 3Fe + 6НС1 = RNH, + 3FeCl2 + 2НаО. (2)
На самом же деле стехиометрические отношения реагентов
согласные с этим уравнением, применяются лишь в исключитель-
ных случаях для цели наилучшего выделения получающихся про-
дуктов2), и соляная кислота в огромном большинстве практически
интересных процессов применяется в значительно меньших коли-
чествах, чем это требует уравнение (2). Больше всего кислота
нужна в начальный период (до прибавления нитросоединения) для
возбуждения реакционной способности (как говорят, „для протрав-
ления") железа3).
Всего 2,5—3% рассчитанного по уравнению (2) количества со-
ляной кислоты, при достаточном запасе железа, уже производят
нужный эффект. Это обстоятельство, позволяя экономно расхо-
довать соляную кислоту, является весьма удобным для техники.
Так как в результате реакции из железа образуется закись-
окись Fe3O4, то течение ее очевидно проходит через процесс оки-
сления железа за счет кислорода группы NO2.
Наряду с взаимодействием между железом и соляной кислотой
по уравнению:
Fe-f- 2НС1 = H2%FeC!a (3)
приходится допустить окисление образовавшегося хлористого же-
леза в основную соль, согласно уравнению:
2FeCl2 % О % Н2О = 2FeCl2OH, (4)
причем очевидно атом кислорода, требуемый уравнением (4), при-
надлежит нитрогруппе. Основная соль восстанавливается далее
железом вновь в хлористую соль, окисляя железо до закись-окиси:
8FeCl2OH 4- 3Fe = 8FeCl2 4- Fe3O4 % 4H2O. (5)
128
При таком толковании в реакции наиболее важным фактором
является не соляная кислота, а железо, хлористое же железо
является лишь передатчиком кислорода от нитрогруппы железу.
Действительно, железо в присутствии лишь FeCl2 реагирует с нитро-
бензолом, причем стехиометрические отношения реакции отвечают
приблизительно схеме
4C6H5NO24-9Fe + 4Н2О--------> 4CeH5NH2 + 3Fe3O4. (6)
(в присут-
ствии FeCl2)
В тех случаях, когда есть осиоваиия опасаться или освобождения ацилирован-
ной аминогруппы, имеющейся в исходном нитросоединении, или слишком большой
подвижности образовавшейся аминогруппы в присутствии солиной кислоты или ее
солей, видоизменяют этот распростраиеннейший способ, употребляя не соляную
кислоту, а уксусную, также в количестве меиьшем, чем требует после пересчета
стехиометрия реакции (2) (5—ЛОУо теоретического).
И в том и в другом случае реакцию проводят в присутствии
большого количества воды и при повышенной температуре,
обычно около 100°. Ввиду того что в реакционной массе имеется
большое количество твердого железа, требуется основательное
перемешивание.
Можно применять вместо соляной кислоты серную кислоту также при соот-
ветственном уменьшении требуемого уравнением (2) количества. Один американ-
ский патент описывает также применение кислых сульфатов, например бисуль-
фата NaHSO4, получаемого как отход при фабрикации азотной кислоты по Вален-
ти и е р у 4).
Роль соляной или других кислот при этом методе восстановле-
ния сводится повидимому к активированию, возбуждению начала
реакции. После растворения поверхностного слоя чугунной
стружки в растворе находятся хлорные соединения железа. Есте-
ственно спросить: нельзя ли осуществить процесс восстановления
железом в отсутствии свободной кислоты?
Выше мы указывали, что в присутствии хлористого железа
восстановление проходит с тем же конечным эффектом.
Семь лет назад американские химики Лайонс и Смит изу-
чали вопрос о восстановлении нитросоединений посредством железа
(чугунных опилок) в присутствии водных растворов хлористого
натрия н хлорного железа FeCl3 на примерах нитробензола,
«-нитротолуола, .и-динитробензола, «-нитрофенола и 2.4.6-три-
нитрофенола, причем установили, что во всех этих примерах про-
цесс восстановления проходит, давая в результате амины (с обра-
зованием лишь моноаминопроизводных из ди-, соотв. три-, нитроза-
мещенных6).
При этом достаточно, чтобы в растворе было очень небольшое количество этих-
солей, именно 1,672—2,675 г FeCl3 или 1,523—2,9 г NaCl на 100 г нитросоединения,
т. е. 1/160—1/100 количества СГ, рассчитанного по отношению 3Fe:6HCl.
При концентрации СГ в виде FeCl3 или NaCl от 0,0188—0,035 г в 1 см3
раствора получены выходы соответствующих аминов в 95—100% от теоретически
рассчитанных из следующих соединений:
C6H6NO2, zi-CH3C6H4NO2 и .и-С6Н4 (NO2)-2-
При концентрации от 0,009—0,006 г СИ получены теоретические выходы ами-
нов нз n-HOC6H4NO2, соотв. из пикриновой кислоты.
9 Зак. 2611. — Н. Ворожцов
129
Чугунные опилки должны быть возможно мелкими, однообраз-
ными по размеру. Наилучшие результаты дали опилки, просеян-
ные через сито с 80 отверстиями на 1 си2, более грубые (просеян-
ные через сито с 40, 20, 13 отверстиями на 1 си2) оказались ме-
нее деятельными.
Хлористый натрий при прочих равных условиях обладает 84%
от активности FeCl3.
Нитрофенол и пикриновая кислота восстанавливаются железом
в присутствии названных солей гораздо легче, чем нитробензол,
нитротолуол и .и-динитробензол. Пикриновая кислота восстанавли-
вается одним железом в водном растворе в динитроаминофенол.
Таким образом гидроксил в орто- или пара-месте к нитрогруппе
сильно уменьшает сопротивление нитрогруппы восстановлению.
Еще ранее этого исследования Воль наблюдал, что нитросое-
динения восстанавливаются железом в растворе хлористого каль-
ция 6).
Если в процессе восстановления, проводимом в растворе хлор-
ного железа, легко усмотреть соответствие вышеприведенной
схеме (4) (5) (6), ибо в водном растворе FeCl3 легко может давать
вследствие гидролиза основную соль:
FeCl3 + Н2О = FeCl2(OH) -f- НС1,
то восстановление в растворе поваренной соли и других прочных
к гидролизу хлористых солей металлов, не переходящих в иные
степени окисления, требует иных объяснений. По Волю здесь
восстановителем служит мелкораздробленное влажное железо, и
основная реакция восстановления выражается так: например
C6H5NO2 + 2Fe -f- 4Н2О = С6Н5МН2 + 2Fe(OH)s.
Вторичная реакция железа с гидратом его окиси приводит к об-
разованию закиси-окиси железа:
8Fe(OH)3 + Fe = 3Fe3O4 + 12Н2О.
Легко видеть, что результирующее основное расчетное уравне-
ние (6) остается при этом тем же.
Способ восстановления в растворах хлористых солей (в частно-
сти NaCl) особенно практичен при работе с нитрофенолами и их
эфирами, где он нередко дает повышение выхода сравнительно
с чисто кислотным методом работы.
Немецкие фабрики в последнее время обращают внимание не только на полу-
чение аминов в процессе восстановления, но и на качество остающегося железного
осадка (шлама), состоящего нз окислов железа, заменяя полностью или частично
соляную кислоту хлоридами (Fe, щелочных и щелочноземельных металлов, А1, Сг,
Се, Th, металлов редких земель, Zn); иногда прн употреблении негндролизнруемых
хлоридов, добавляя соляную кислоту, они получают закись-окись железа в виде
темноокрашенного порошка (с оттенками краено-коричневыми, фиолетовым до чер-
ного), который особенно пригоден для получения минеральной железной краски
(мумии, сурика).
Хорошего осадка соединений железа можно достичь и проведением чисто
кислотного восстановления прн расчете например количества соляной кислоты
в отношении 0,6 мол. НС1:1—1,5 мол. C6H5NO2 с 5 грамматомами Fe при сохра-
нении минимальной концентрации соляной кислоты в 6° Боме и при температуре
в 100°’).
130
При восстановлении нитро- и нитрозосоедивений железом иногда оказывается
полезным прибавлять к водной жидкости некоторое количество медной соли (CuSO4,
CuCl2). Образующаяся железно-медная пара является очевидно более активным
восстановителем. В втом ваправлении работы ведутся А. Е. П о р а й-К о ш и ц е м8).
Упомянутые выше Л а й он с н Смит не констатировали ускорения реакции
с железом прн прибавлении хлористых солей ртутн или меди.
Подобно тому как работа с железом в растворе, не заключающем свободной
кислоты, а только содержащем хлористые соли, приводит, как мы видели, к вос-
становлению одной лишь нитрогруппы в полинитросоединениях (к частичному вос-
становлению полинитросоединений), такой же эффект может быть достигнут приме-
нением железа с весьма малым количеством кислоты (соляной, уксусной или сер-
ной), а также очень малым количеством воды. При соблюдении этих условий из
ле-динитробензола можно получить лг-ннтранилнн9).
II -> II
Если принять во внимание уравнения:
RNO2 + 2Fe + Н2О = RNH2 + Fe2O3,
RNO2 + 2Fe + 3H2O = RNH2 4- Fe2O(OH)4,
то количество воды, допустимое для такого восстановления, не должно превышать
двойного против теоретически рассчитанного и должно прибавляться не сразу,
а постепенно.
Обращает на себя внимание еше одна разновидность кислотного восстановле-
ния с железом, дающая в результате применения, как и вышеприведенный способ,
частичное восстановление одной ннтрогруппы в полнннтросоединениях. Мы хотим
указать на использование реакции между железом и водным раствором сернистой
кислоты10).
Даже при избытке железа и долгом пропускании SO2 реакция останавливается
на восстановлении лишь одной NO2-
Техника выполненние реакции такова.
Нитропродукт смешивается с железными стружками и водою, и в эту смесь при
нагревании пропускается SO2, пока не растворится почти все железо. Полученное
нитроаминосоединенне остается в зависимости от своих свойств (вполне или ча-
стично) в растворе или остается нерастворенным. Из реакционной смеси нитро-
амин выделяется непосредственно или отфильтровывается по удалении SO2 (вдува-
нием воздухом) нлн наконец, после выпаривания реакционной смеси досуха, извле-
кается подходящим растворителем.
От только-что приведенного способа Вильфинга новый способ отличается
большей уверенностью работы при обычных, но не исключительных условиях.
Из других кислотных восстановителей некоторое техническое
значение имеет цинк с соляной или уксусной кислотой. Он при-
меняется в такой среде главным образом для восстановления не
нитросоединений, а других восстанавляющихся с образованием
аминогруппы веществ.
Хлористое олово в растворе соляной кислоты является мало
применимым в технике, по дороговизне своей, восстановителем
и может быть употребляемо только при условии регенерации олова
для получения особенно дорогих препаратов (при восстановлении
азокрасителей) или, так же как и цинк, в кислой среде в аналити-
ческой лабораторной практике.
При восстановлении нитросоединений хлористым оловом в солянокислом
растворе наблюдается образование хлорозамешенных аминов (С1 в орто- или пара-
положении по отношению к NH>), что объясняется промежуточным образованием
хлораминов, перегруппировывающихся далее в хлорозамещениые, например:
CeH5NO2-> C6H5NHOH 4- НС1-» CeH5NHC] -» С1СвН^Н2И).
9
131
Восстановление посредством SnCl2 при 75° применялось для определения отно-
сительной скорости восстановления нитросоединений. Ннтрозамещенные, имеющие
группу NO2 в пара-положенни к другому заместителю (за исключением трех ннтро-
бензоивых "кислот), восстанавливаются скорее мета-, последние — скорее орго-про-
нзводиых. Хлор-, бром-, иод- нитробензолы восстанавливаются приблизительно
с одинаковой скоростью. Увеличение количества кислоты в растворе SnCl2 увели-
чивает скорость реакции, прибавление воды уменьшает ее12).
Весьма своеобразным восстановителем из слабокислотных
является сернистая кислота, применяемая главным образом в виде
своих кислых солей (бисульфитов). Наряду с образованием амино-
группы при обработке бисульфитом происходит сульфирование
ядра, если к этому нет препятствий в строении молекулы.
В качестве продуктов, образующихся промежуточно при этой
реакции, констатированы сульфаминовые кислоты с характерной
группой NHSO3H. Например:
/NO2 /NHSO8Na
С6Н/ 3NaHSO3 -> C6hZ 4- 2NaHSO41S)
XCO,C2H5 XCO2C2H5
и
/NO, /NH2
CqHZ ' + 3NaHS0s->C6HZ C02H + 2NaHSO4 “).
xCOaH xSO3Na
Здесь проявляется одно из свойств сульфогруппы, связанной
через азот, — ее стремление переходить из внешней цепи в ядро.
Этот способ восстановления с одновременным сульфированием
имеет известный технический интерес и помимо нитросоединений
используется в ряду нитрозопроизводных. Восстановление проте-
кает обычно при кипячении в присутствии избытка бисульфита и
дает для бензольных производных почти исключительно пара-
сульфокислоты аминов (если пара-место к N свободно). Как про-
дукты кислотного гидролиза сульфаминовых кислот получаются и
несульфированные амины:
RNHSO3H + Н2О -> RNH2 -4- H2SO4.
Интересно, что не только кислые, но и средние соли сернистой
кислоты производят восстанавляющее действие на нитросоеди-
нения. Раствор сульфита натрия (Na2SO3) до 90° не действует на
нитробензол при суспендировании последнего. При кипячении
раствора Na2SOg (2 мол.) с 1 мол. CGH5NO2 происходит реакция
с образованием 1-амино-4-фенол-2-сульфокислоты повидимому
вследствие двух реакций:
C0HsNO2Z2Na2SO3+H2O = C6H4(NHOH)SO3Na + Na2SO4 -f- NaOH,
CeH4(NHOH)SO3H -> HO • C6H3(NH2)SO3H .
Образуются при этом и иные продукты, лишенные азота вслед-
ствие побочных реакций 15).
Свободный сернистый ангидрид реагирует с аминосоедииениями, образуя про-
дукты присоедииення. Различную реакционность п- и о-толуидина с SO2 предло-
жено использовать для разделения смеси изомерных аминов, из которой таким
образом можно выделить с хорошим выходом соединения n-толуидина состава
2C-,H.)N • SO2 • Н,0 1В).
132
В недавнем германском патенте описывается применение для восстановления
ннтросоединеннй в кислотной среде (уксусная кислота) восстановителя, имеющего
значение главным образом для превращения кубовых красителей в лейкосоеди-
иения и для последней цели применяемого в присутствии щелочи. Речь идет о
гидросульфите Na2S2O4, восстановительная способность которого основана иа легкой
окисляемости его в производные сернистой кислоты (пиросульфит) >;):
Na2S2O4 + О = Na2S2O5
Однако метод этот не имеет никакой практической ценности, сопровождаясь
большим расходом ценного гидросульфита и давая в результате ве только одни
амины, ио и их сульфокислоты, очевидно в результате действия образующихся
кислых солей сернистой кислоты на иитросоедивеиия:
Na2S2O5 + Н2О = 2NaHSO3.
Из кислотных восстановительных агентов своеобразно и, как говорят, „мягко"
действует на азотсодержащие группы иодистый водород, своеобразие которого
проявляется в том, что он атакует по преимуществу азогруппу, не трогая нитро-
группы (стоящей ие в орто-положении к N:N-rpynne). Таким образом из нитроазо-
краснтелей можно получить нитроамины. Возможно при этом применение не го-
тового йодистого водорода, а смесей, его образующих, например SO2 -)- J
Р J — в присутствии ВОДЫ.
Иодистый водород в присутствии избытка SO2 и минеральной кислоты
является хорошим восстановителем азосоедииений до гидразосоедииений, которые
затем перегруппировываются в бензидивовые производные (Бодеиштейи)18).
Этот метод работы вследствие дороговизны реагента вряд ли может рассчитывать
на сколько-нибудь серьезный технический интерес.
Метод катодного восстановления при электролизе, при условии
применения содержащего кислоту (невысокой концентрации) ката-
лита, приводит к получению из нитросоединений аминов, но электро-
химический метод в этом случае, вследствие более высокой стои-
мости, уступает по своему значению чисто химическим методам1Э).
Методом электрохимического восстановления в кислотных растворах пытались
пользоваться для выяснения реакционности отдельных нитрофенолов, отсчитывая
разницу между всем количеством водорода, образующимся на катоде, и тем коли-
чеством, которое выделяется как не вошедшее в реакцию. Опыты не дали вполне
точных результатов, так как нередко образовывались продукты неполного восста-
новления (окрашенные вещества) и восстановление полинитрофенолов шло неравно-
мерно. В отношении легкости восстановления первое место занимает о-интрофенол,
затем я-нитрофенол, о-я-дииитрофеиол, наименее легко—пикриновая кислота (по-
следняя— приблизительно вдвое труднее о-нитрофенола в равных условиях). Пнкра-
мнновая кислота не обнаруживает неровностей при восстановлении, какие показы-
вает пикриновая кислота20).
Из всех перечисленных видоизменений кислотного метода вос-
становления несомненно наибольшее значение и почти исключи-
тельное применение в практике имеет метод работы с железом.
Последнее применяется в виде чугунных стружек, какими они
получаются на машиностроительных заводах.
Выяснено в процессе работы, что не каждый чугун дает одинаковые результаты
в качестве материала для восстановительных процессов. Вопрос о наилучшем сорте
нз нескольких образцов решается лучше всего параллельными пробами в восстано-
влении ннтросоединеннй, проведенными в строго одинаковых условиях. Стружки
должны быть возможно однородны, лишены посторонних включений (от которых,
если они есть, следует избавиться сортировкой и просеиванием), свободны от
ржавчины и однообразного размера. Чем мельче стружки, тем скорее и полнее
проходит, прн однваковых прочих условиях, восстановление. Поэтому например на
некоторых американских заводах предпочитают размалывать стружки до состояния
мелкого порошка н пропускать их через сито.
133
Метод работы с железом в слабокислотной среде применяется
для получения аминов, производимых в массовых количествах,
каковы анилин, а-нафтиламии, толуидины; он применяется также
для получения некоторых диаминов, например лг-фенилендиамина (1),
ж-толуилендиамина (II) из соответственных динитропроизводных:
o.2nx/x/no2 h2nx/x/nh2
Нужно сказать, что в этих последних случаях остается недостаточно изученным
процесс восстановления: если, что вероятно, не обе нитрогруппы сразу сменяются на
аминогруппы, а имеет место промежуточное, хотя бы и быстротечное, образование
аминоиитросоединений, то не правильнее ли было бы процесс проводить в две
стадии, подбирая для каждой наиболее благоприятные условия (иапример с точки
зрения концентрации Н’-иопов в растворе, температуры и т. д.).
Этот же метод восстановления применяется также для получе-
ния большого числа нафталиновых аминосульфокислот, главным
образом из нитросульфокислот а-нафтиламина, образуемых в свою
очередь нитрованием сульфо- и полисульфокислот нафталина. Не-
обходимо отметить еще раз, что каждый отдельный нитропродукт
имеет свои особенности при восстановлении. Появление всякого
рода побочных продуктов и понижение в выходе главного про-
дукта обнаруживает такие индивидуальные особенности.
Наиболее часто встречаемое осложнение в процессе восстановления в кислотной
среде — это остановка на промежуточной фазе, могущей дать изменение структуры
полученного в результате продукта. Если промежуточно образуется например
арилгидроксиламин (RNHOH) (вследствие неполного восстановления из-за недо-
статка железа или малой его активности), то это соединение может сохраниться
в готовом продукте, понизив его качества, может в условиях кислотной обработки
перегруппироваться в оксиаминосоедннение, например
/\/NH0H /X/NHj
j j Н*-ионы | j
\Z
и опять-таки загрязнить продукт и снизить выход главного.
Тщательным подбором наиболее благоприятных условий прове-
дения восстановления и соблюдением их при проведении процесса
можно добиться постоянно одинаково удовлетворительных резуль-
татов.
В производственном масштабе восстановление нитросоединений
проводится в чугунных цилиндрических аппаратах (редукторах),
обычно выложенных внутри кислотоупорным материалом (керами-
ковыми плитками на кислотоупорной, свинцово-глицериновой за-
134
мазке). Аппарат снабжен чугунной же мешалкой, тяжелой и проч-
ной, для того чтобы она могла справиться с разгребанием тяжелой
массы металлической мелочи и илистого осадка. Кроме того у верх^
ней части редуктора имеется сообщение с обратным холодильником.
По окончании процесса восстановления, после нейтрализации рас-
твора щелочью или известью, амин выделяют различными методами
в зависимости от его свойств. Так, анилин снимают в большей
Рис. 6. Схема редукционной установки.
/—Редуктор. 2—Холодильник трубчатый. 3 — Холодильник змеевиковый. Холодильник для легки
углеводородов. 5—Отводная труба. 6—Труба для загрузки чугунных стружек. 7—Шибер (задвижка)
части посредством сифона, дав отстояться его слою над водным рас-
твором, к которому для увеличения удельного веса прибавляют
поваренной соли, а затем отгоняют с паром остаток. Твердый при
обычной температуре а-нафтиламин или извлекается из подщело-
ченной массы подходящими растворителями (ксилолом, сольвент-
нафтой и т. п.) и выделяется отгонкой растворителя, либо отгоняет-
ся из твердой массы, заключающей кроме него все железные
остатки, посредством перегретого пара.
Летучие с водяным паром амины частично растворяются в воде,
и в производстве является весьма существенным вопросом извлече-
ние из такой воды амина. Прежде для извлечения практиковалась
135
или система сложных отгонок или применение такой заключающей
амин воды для питания парового котла, откуда пар отбирается
в восстановительное отделение. Позже предложен практичный спо-
соб извлечения амина из водного раствора, именно — экстрагиро-
вание последнего нитросоединением, служащим для получения
амина. Двукратного размешивания с нитросоединением (если надо —
при повышенной температуре) достаточно, чтобы извлечь весь
амин. Нитросоединение вместе с растворенным в нем амином идет
на восстановление 21).
Если дело идет о сульфокислотах аминов (например нафтила-
миновых сульфокислотах), не летучих и не перегоняющихся с па-
ром, то приходится, осадив металлические соединения щелочью
(содой или едким натром), подкислить фильтрат от гидроокисей
минеральной кислотой (или уксусной), причем весьма часто и вы-
падает аминосульфокислота. Иногда для выделения требуется
высаливание раствора (поваренной, глауберовой солью, хлористым
калием и пр.) (см. выше — выделение сульфокислот). Бывают тем
не менее случаи, когда амин (особенно часто полиамин) упорно не
выделяется ни одним из этих способов. Тогда его выделяют в виде
соли с какой-либо минеральной кислотой. Многие амины (также
и полиамины), будучи хорошо растворимы в разведенной соляной
кислоте (в виде солянокислой соли), в то же время труднораство-
римы в той же, только концентрированной, кислоте. Следовательно
прибавлением к возможно малому объему раствора хлористоводо-
родного амина избытка крепкой соляной кислоты можно такой
амин выделить в виде соли.
Для приготовления особо чистых препаратов аминов, например применяемых
как фотографические проявители, пользуются иногда раствором амииа в органи-
ческом растиорителе, не смешивающемся с водой (бензол, бензин, хлорбензол и
т. п.), и, пропуская через него ток сухого хлористого водорода, осаждают чистую
солядокислую соль амина.
Дробным осаждением Смесей аминов при использовании этого же приема можно
отделить амины друг от друга в виде солянокислой соли. Указывается иа возмож-
ность разделения смеси толуидинов, толуидина от анилина и т. п.
Некоторые полиамины, хорошо растворимые в виде солянокислой
соли, дают труднорастворимую сернокислую Соль. Таков например
бензидин, и его потому выделяют после перегруппировки гидразо-
бензола в виде сульфата. Отделив мннеральнокислую соль амина,
разлагают ее содой или едкой щелочью, если необходимо получить
свободное основание, и соответственной обработкой (кристалли-
зацией, перегонкой) очищают амин.
Сгущением, по возможности свободных от минеральных соеди-
нений, водных растворов аминов с высокой температурой кипе-
ния и не летучих с водяным паром, причем сгущение для пони-
жения температуры раствора лучше вести под разрежением, можно
выделить такие амины (например лг-диамины бензольного ряда,
л-аминофенол и т. п.) в свободном состоянии иногда в виде кри-
сталлогидрата (лг-фенилендиамнн). При выделении или после него-
применяется, если надо, дополнительная очистка: перерастворение,
обработка сернистой кислотой и ее солями и т. д., с целью устра-
нить например продукты окисления и другие загрязнения особо
чувствительных аминов. Перегонку аминов для окончательной
136
очистки производят в вакууме; может быть иногда полезна была
бы кондестилляция. Сушат — в вакууме, избегая повышения
температуры свыше некоторого допустимого предела.
Практичным кажется предложенный И. Я. Гришиным способ перегонки
а-нафтиламцна в парах керосина: из керосиноиого раствора он выпадает сразу
в состоявии высокой чистоты. Метод перегонки трудиокнпящнх веществ с органи-
ческими растворителями—новый метод, который может найти себе повидимому
более широкое применение, чем только и названном примере. Он дает хорошие
результаты и в перегонке 13-иафтола.
Метод такой перегонки назван Рассовым и Шульцки »кондестнлляцией“
(совместная перегонка), причем ими приведены и те условия, которым должен удо-
влетиорять растворитель для того, чтобы быть пригодным для коидестилляции: 1) он
не должен или может лишь в незначительной мере растворять подлежащее пере-
гонке вещество, 2) упругость его паров должна быть возможно близка к упругости
пара перегоняемого продукта, 3) молекулярный вес по крайней мере не выше, чем
у перегоняемого вещества, 4) он ие должен быть дорогим.
Указанные авторы получили хорошие результаты при коидестилляции «-нитро-
фенола с фракцией нефтяных погонов (с t°Kan 240°/760 мм}, одновременно освобож-
дая продукт от смолистых веществ 23).
Сульфокислоты аминов, будучи образованы восстановлением
нитросульфокислот, выделяются способами, характерными для
сульфокислот: подкислением или выделением в виде солей и т. п.
б) Восстановление в нейтральной среде
Восстановление, проведенное с железом в растворе нейтраль-
ных солей (при повышенной температуре), как мы видели выше,
заканчивается получением аминосоедннений нли нитроаминов из
полиннтросоединеиий. Нейтральное восстановление на холоду
с применением некоторых иных, кроме железа, металлов, особенно
цинка, дает возможность задержать процесс на образовании проме-
жуточного продукта неполного оводороживания нитрогруппы в ис-
ходном материале, именно образовании p-арилгидроксиламина. Так,
нитробензол при обработке цинковой пылью на холоду в водйо-
спиртовом растворе хлористого аммония образует фенилгидроксил-
амнн 24).
CeH5NO2+ 2Zn -НН2О = C6H6NHOH -|- 2Zn(OH)2-L Н2О.
Интересно отметить, что хлористый аммоний из всех испытанных
хлористых солей (щелочных и щелочноземельных металлов) дает
наилучший выход фенилгндроксиламина.
Сравнивая концентрацию водородных ионов параллельно и растворах хлористого
аммония и хлористого кальция, в которых велось восстановление нитробензола цин-
ковой пылью, Бранд установил причину благоприятного илияния раствора с хло-
ристым аммонием в его меньшей щелочности. Значение pH для раствора от вос-
становления нитробензола цинковой пылью с хлористым кальцием — 11- 11,75, то же
с хлористым аммонием—7,6—8,4.
Таким образом смесь из водного спирта, хлористого кальция и цинковой пыли
после прибавления нитросоединения не остается нейтральной, а по мере хода реак-
ции становится щелочной. Щелочная же реакция при восстановлении содействует
преимущественному направлению реакции в сторону образования сложных продуктов
преиращения с группами N(O): N, N: N, NH • NH, о чем ниже сказано более подробно.
Явления, приводящие к проявлению щелочной реакции, Бранд передает схе-
мами:
RNO2 + 2Н" + 2ОН' + Zn -> RNO + Н2О + 2ОН' + Zn" (а
RNO + 2Н’ + 2ОН' + Zn RNHOH + 2ОН' + Zn", (б)
137
из (а), и (б):
RNO2 + 4Н’ + 40Н' + 2Zn -> RNHOH + Н20 + 40Н' -f- 2Zn" (в)
2Zn" + 40Н' £ 2Zn(OH)2. (г)
Устранение щелочности с целью повышения выходов р-арил-
гидроксиламина возможно производить просто введением в реак-
ционную смесь кислоты (уксусной, щавелевой или минеральной).
При таком видоизменении восстановление в растворе хлористого
кальция дает хорошие выхода и при применении динитро- (соотв.
хлорнитро-) производных в качестве исходных материалов. Количест-
во кислоты — около 0,5 эквивалента по отношению к нитросоеди-
ненню. При этих условиях можно работать и при повышенной
температуре, на кипу 25).
Цинк при восстановлении с хлористым аммонием входит вероятно в состав
комплексных солей, например [Zn(NH3)2]Cl2, [Zn(NH3)4]Cl2 и т. п., и повидимому в со-
став промежуточно образующихся органических, содержащих цннк, соединений,
разлагающихся о г действия кипящего спирта с выделением двойной соли состава
2NH4C1 • 5Zn(OH)2 26).
При дальнейшем действии восстановителя на ^-арилгидроксил-
амин можно получить продукты более полного восстановления
иитрогруппы. Например p-фенилгидроксиламнн переходит при этом
преимущественно в анилин:
CflH3NHOH +Zn + Н2О = C6H5NH24-Zn(OH)2.
Восстановление n-нитротолуола до того, как получится конечный продукт—
амин, дает еще промежуточно гндразотолуол CH3C6H4NH • HNC6H4CH3.
Кроме цинка с раствором хлористого аммония или хлористого кальция, неко-
торые другие восстановители образуют (хотя не всегда с хорошим выходом) гидро-
ксиламиновое замещенное из ннтросоединення: амальгама цника в присутствии
сернокислого алюминия (а) или алюминиевая амальгама с безводными растворителями
(эфир, бензол, нефтяные углеводороды) с добавкой необходимого по расчету коли-
чества воды (б), сернистый аммоний на холоду (в) и сульфгндрат аммония с ней-
трализующим этилуксусиым эфиром (г), также применялся алюминий в порошке
в растворе NH4C1 при сильном охлаждении (д) %7) Гидроксиламииовые производные
антрахинона получаются из соответственных ннтрозамещеиных антрахинона при
восстановлении их посредством щелочного раствора закиси олова (из SnCl2 н NaOH)28).
З-Арилгидроксиламины очень реакционноспособные вещества.
В частности для них характерна неустойчивость по отношению
к минеральным кислотам, воздействие которых превращает гидро-
ксиламиновые замещенные в оксиаминодвузамещенные (и-амино-
фено.ты) 29), например:
y^yNHOH Z\/NH2
минеральная ,
кислота j j
Эта перегруппировка, с одной стороны, с другой—возможность
получения из [3-арилгидроксиламиновых производных соответствен-
ных нитрозосоединений: RNHOH -> RNO (см. ниже гл. XIII) при-
влекают интерес к арилгидроксиламиновым соединениям.
Получение л-аминооксисоединений не только в бензольном, но
и в других рядах ароматических соединений, в том числе и в антра-
хиноновом (где гидроксиламиновые замещенные довольно стойки),
138
осуществляется нередко с переходом через арилгидроксиламины,
выделяемые при этом не во всех случаях.
Замечательно, что методом электрохимического восстановления в растворе
нейтральных электролитов (Na2SO4, MgSOJ из нитробензола можно получить иитро-
зобензол 30 . Повидимому образование последнего — вторичная реакция, связаииая
с окислением иа аноде предварительно образовавшегося фенилгидрокснламина:
Характерен метод восстановления нитросоедниеиий цинковой пылью в растворе
концентрированной серной кислоты.
При действии цинковой ныли на нитробензол в концентрированной серной
кислоте (температура 50—80°) получается п-аминофенол, прн восстановлеиин
.и-ннтробензойной кислоты - п-аминосалнциловая кислота, из дииитроавтрахииона—
диаминодиоксиантрахннон 31). В конечном счете следовательно реакция сопровож-
дается не только образованием аминогруппы, но и вхождением (в «-положение
к аминогруппе) гидроксильной группы. Этот оригинальный результат восстанови-
тельного процесса объясняется образованием в качестве промежуточного продукта
^-арилгидрокснламииа, который в момент образования под влиянием концентриро-
ванной кислоты перегруппировывается в амииооксисоединение. Значит, для нитро-
бензола например схема реакции такова:
4- 2Н.
- Н2О
в присутствии
КОНЦ, кислоты
в) Электрохимический метод восстановления в кислотной среде.
Растворяя нитробензол в довольно крепкой серной кислоте
и подвергая его при электролизе катодному восстановлению, полу-
чают в качестве главного продукта также л-аминофенол, очевидно
как результат двух последовательно идущих реакций: восстано-
вления до фенилгидрокснламина и перегруппировки последнего
в оксиаминопроизводное. Этот метод получения п-аминофенола
имеет бесспорно практический интерес и при наличии дешевой
энергии может быть вполне рентабелен. Образование анилина прн
этом всегда наблюдается 32).
Практиковавшееся вначале введение концеитрированной серной кислоты
в качестве среды для реакции постепенно заменялось применением значительно
разведенной кислоты, в которой нитробензол не растворяется, а суспендируется
при энергичном размешивании. Применение катодов из неблагородных металлов
снижает выход амииофенола за счет увеличенвя выхода анилина.
Применение катодов из неблагородвых металлов в присутствии других металлов
(Pb, Bi, Си, Ni и т. п.) или угольных катодов в присутствии некоторых металлов
дает возможность повысить выход свыше 50% от теоретического ю). Удачным
поввдимому разрешением вопроса является метод, описанный в германском патенте
(1927 г.), основанный на применении разведенной серной кислоты (30° Боме) с вве-
дением в ваниу коллоида (клея), замедляющего образование порошковатого металла,
который содействует восстановлению до амина. В описании указывается иа посте-
пенное добавление нитросоединеиия к католиту по мере хода восстановления.
Анод — свинцовый, катод — медный. При этом можно значительно повысить плот-
ность тока (до 6—20 амп/дм2), что при катодах из неблагородных металлов было
невозможно без понижения выхода по току. Электролиз идет при размешивании при
температуре 80—90°. Выхода аминофенола около 65%, считая на нитробензол.
139
Другие исследователи применяли иные условия: катоды из серебра, никеля, 50%-ная
серная кислота, температура опыта 25—60е с более низкой плотностью тока
(3,4 амп/дм2). Выход по току 60—62%, выход по веществу 60—65% :!4).
г) Восстановление в щелочной среде (бензидиновая нерегруп
пировка гидразосоединений)
Применение для восстановительного процесса щелочной среды
делает возможным получение продуктов иного строения, чем до
сих пор упоминавшиеся, именно азоокси-, азо- и гидразопроизводных,
которые своим возникновением обязаны участию уже не одной,
а двух молекул нитросоединения и получаются через превращение
его характерной группы с промежуточным образованием нестой-
ких продуктов 35).
RNO3 £^2-> RNO, (1)
(нитрозосоедннеиие)
RNO., RNHOH, (2)
—rigO
(арилщдрок'силамин)
RNO4-HOHNR ztho-* RN —NR> (3)
О
(азоксисоедннение
RN=NR RN = NR. (4)
/S —HgU
~ (азосоедпнеиие)
При дополнительном восстановлении азокси- и азосоединеиий
образуются гидразосоединения.
RN=NR RNH — HNR RN=NR.
Q ‘
Гидразосоединение может быть восстановлено с образованием
двух молекул моноамина:
RNH • HNR+ Н2-------> 2RNH.,,
может быть окислено в азопроизводное (легко в щелочной среде):
RNH —HNR + O RN = NR.
Последняя реакция, весьма вероятно, является, наряду с восста-
новлением азоксисоединений, причиной образования азосоедииений
при щелочном восстановлении нитрозамещенных.
Наконец гидразопродукт в кислой среде (водный раствор мине-
ральной кислоты) претерпевает своеобразную перегруппировку
в я, я'-диамин— типа бензидина, например
Н— / \-NH—HN—/ Н-------->
——> h2n—/ V-nh2.
140
В качестве конечных продуктов процесса, начинающегося вос-
становлением нитросоединений в щелочной среде, может оказаться
таким образом или моноамин, образование которого в данном
случае нежелательно, так как его легче получить кислотным вос-
становлением, или диамин. Обычно в технике щелочное восстано-
вление имеет целью получение я, я'-диаминобифениловых про-
изводных или бензидиновых оснований.
Среди перечисленных выше продуктов восстановления продукты
реакции (1) и (2) нестойки и ие могут быть изолированы в про-
цессе восстановления, и первыми удобными для наблюдения веще-
ствами являются поэтому в щелочном восстановлении азокси- и
азосоединения (3) и (4), которые получаются обыкновенно вместе,
чаще с преобладанием азоксипродукта.
Тем не менее, без сомнения, азоксисоединения образуются через
фазы (1) и (2) вероятно по схеме:
RN:O RNOH . RN:O
+ RNHOH RNOH 2 RN
и известно, что особенности во внутреннем строении ароматического
остатка, связанного с нитрогруппой, могут отзываться на увели-
чении или уменьшении скорости образования азоксисоединения из
этих составлящих 36). Метильная группа в орто- или параположе-
нии к азоту понижает эту скорость.
В качестве щелочных восстановителей предложено очень много
комбинаций чисто химического характера, не говоря об электро-
химическом способе. Из них наиболее интересными технически
являются цинковая пыль с раствором едкого натра и железо
с таковым же раствором 37). Последний восстановитель интересен
потому, что он позволяет повидимому проходить через все сту-
пени восстановления.
Кроме этих предложены в качестве восстановляющих средств для получения
из ннтросоединеннй азокси- и азопродуктов соли мышьяковистой кислоты в щелоч-
ном растворе з®), свинец с раствором едкого натра ®*), недокись свинца *°), магний
в метилоспиртовом растворе вместе с водным раствором хлористого аммония, где
очевидно необходимая щелочность поддерживается за счет выделяющегося аммиака —
способ не имеет конечно технического значения 41),— суспенсия каменного угля
в щелочи, суспеисня древесных опилок и других целлюлозных материалов в щелоч-
ном растворе 42), сернистые соединения (сульфиды) тяжелых металлов с едким
натром 43), сернистые щелочи в крепких растворах едких щелочей м), метилат натрия,
лучше в присутствии органических оснований [пиридина, хинолина, вторичных или
третичных жирноароматических аминов45)], гидразосоединеиия в щелочном растворе46),
причем в этом случае реакция протекает по такой схеме, например
2RNO2 + 3CeH5NH • HNCeH5 —> RN(O): NR + 3CeH5N : NCeH5 + 3H2O.
Гидразобензол взят в качестве восстановителя как простейший представитель
класса гндразосоедниеннй. Арнл R в нитросоедииении может и не быть фенилом С6Н3.
Легкая окисляемость гидразобензола до азобензола (и возможность превраще-
ния азобензола обратно в гидразо) может быть использована для получения пере-
киси водорода. При действии кислорода на бензольный раствор гидразобензола
может (Тыть получена с почти количественным выходом высокопроцентная (94%)
перекись водорода по реакции:
CeH5NH • HNCgH5 + О2 = CeH5N : NCeH3 + Н2О2 «).
Щелочное восстановление нитросоединений в азо-, азокси-
141
(соотв. гидразо) соединения производится при повышенной темпе-
ратуре и при размешивании.
Многочисленность патентных заявок на различные воспроизве-
дения процесса щелочного восстановления не доказывает, что все
патентованные способы практиковались и практикуются в технике.
В настоящее время производственное значение осталось за методами
восстановления посредством цинка, железа и более недавним методом
работы с амальгамой натрия, какой она получается при электро-
лизе хлористого натрия с ртутным катодом. Последний способ
восстановления, входящий в цикл процесса получения щелочи,
представляет большие технические преимущества 48).
Для технически наиболее важных процессов восстановления
трех нитросоединений: нитробензола, о-нитротолуола и о-нитро-
анизола C6H4(0CHg)NO2 техника восстановления по цинковому
методу и для нитробензола по железному изучена 49). Ввиду того
что исходные материалы жидки при обыкновенной уже темпера-
туре, щелочь же применяется в водном растворе, в котором
нерастворимы ни исходные нитросоединения, ни образуемые твер-
дые продукты, а металл —восстановитель и продукты его окисления
образуют осадок, необходимо применять очень энергичное разме-
шивание для того, чтобы все нужные для реакции вещества могли
достаточно совершенно приходить в соприкосновение друг с дру-
гом. Практикуется иногда прием введения в реакционную сферу
органического растворителя, индиферентного к участвующим в про-
цессе веществам (спирт, бензол, толуол, ксилол, сольвентнафта,
жидкие полихлориды бензола, нефтяные погоны) для того, чтобы
сделать реакционную смесь более подвижной и перевести в раствор
продукты реакции [азокси- и азо- и особенно гидразосоединения 60 ].
При методе с цинком однако можно обойтись и без раствори-
теля; при восстановлении с железом приходится к нему прибегать.
Выяснено, что выгодно проводить процесс восстановления нитро-
соединений в гидразо-, используя различные условия для отдель-
ных стадий или фаз процесса. Таким образом для стадии
2RNO-2----> RN(O): NR (соотв. RN = NR)
выгодно применять более высокую температуру и более высокую
концентрацию щелочи, чем для стадии
RN(O) = NR (соотв. RN~ NR)------> RNH —HNR.
Значение разницы в концентрациях щелочной жидкости особенно
проявляется при восстановлении о-нитротолуола и о-нитроанизола,
возможно в связи с вышеотмеченным понижающим влиянием со
стороны в орто-положении к NO2 стоящей СН3- (соотв. ОСН3-)
группы на скорость образования азоксисоединения из первичных
продуктов восстановления.
При соблюдении всех оптимальных для каждой стадии условий
протекания реакции, образование первичного амина, как продукта
далеко идущего восстановления, хотя и имеет место, но не пре-
восходит некоторых допустимых пределов.
По относительной легкости проведения процесса восстановле-
ния, соответствующего более узким пределам отклонений от опти-
142
мальных концентраций щелочи и температуры, для названных
выше исходных нитросоединений получается такой ряд:
нитробензол, о-нитротолуол, о-нитроанизол
(ряд уменьшающейся легкости).
Конечным продуктом собственно восстановительного процесса
является гидразосоединение, которое нужно не само по себе, но
для получения из него диамина ряда дифенила посредством так
называемой бензидиновой перегруппировки.
Гидразосоединения RNH—HNR обладают основными свойствами лишь в очень
малой степени. Хлористоводородные соли они могут давать при условии солеобр. -
зования в отсутствии воды: или бесцветные одиокислотные соли в неводных раство-
рителях (в эфире и т. п.) при пропускании сухого хлористого водорода, или двукис-
лотные соли зеленые или фиолетовые при действии сухого хлористого водорода на
твердые гидразосоединения И).
В присутствии воды соли гидразосоединения не образуются.
Под влиянием минеральной кислоты — серной, соляной или смеси
обеих (например смеси серной кислоты с водным раствором хло-
ристого натрия) — при поддержании надлежащих температурных
условий гидразо- перегруппировывается в более основное вещество,
имеющее две аминогруппы, связанные с ядром дифенила. Главный
продукт такой перегруппировки п, п'-диамин (I), наряду с которым
образуется некоторое количество п, о-диамина (дифенилина (II) •—
при восстановлении нитробензола] не имеющего технического зна-
чения) например:
/ NH • HN—>------------------->
NH2
--> H.,N — Z NHa + Z \-Z >-NH2
/ t \ / n \ '
Б. П. Opeл кин и его сотрудники м) полагают, что возможно образование
малого количества продуктов .семидиновой” (полубеизидиновой) перегруппировки
(соединений строения RNHRNH2) за счет предварительного образования двукислот-
иой соли гидразосоединения, распадающейся в воде на феиилхлорамии и амин,
сочетающихся в семидии (III), например
однокислотная соль
двукислотная соль
—> МНС] -)-/ nh2 —>
NH — NH2 -}- НС1
III z
Аналогично бензидину перегруппировкой соответственных гид-
143
разосоединений получаются технически ценные диамины ряда дифе-
нила: толидин (IV) и дианизидин (V).
/СН3 /х /СН3 Н3С\ НзС\ /СНз
2| | _, | I | | >-<—>-NH’
\/\NO3 IV
/X/OCH3 /\zOCH3 H3COXZ. H3CO\ zOCH3
2 | | —II | | —
\/\no2 \/Z'x-NH----------HN/\/ V
Все эти три диамина имеют большое техническое значение
в синтезе азокрасителей, применяемых для непосредственного кра-
шения хлопчатобумажных изделий.
Получение из азосоедииений диаминов типа бензидина удается провести с при-
менением кроме минеральной и арилсульфиновых кислот (RSO2H), действующих
восстановительно, причем в этом случае одна из аминогрупп диамина связана
с кислотным арилсульфоновым остатком, иапринер:
\ /—N - N-<Z> + H02S*~\ /-снз --%
—> HjjN-/___\-NHOgS-/ \-СН3 “)
Оригинально ведут себя при восстановлении нитроанилины, нитрофенолы,
алкильные эфиры последних. Именно, в то время как о- и л-нитрофеиолы (могущие
образэвать соответственные хииоиды) дают и при щелочном восстановлении амнно-
феиолы, о- и л-нитранилины дают диамииы, лт-ннтрофенол, наоборот, образует прн
атом азоксисоединение (н азо-). Так же как лг-ннтрофенол, относятся к восстанов-
лению алкильные эфиры всех трех ннтрофенолов (соответственно невозможности
образовывать хиноид) 53).
Аналогичные взаиморазличия наблюдаются при восстановлении оксиазокраси-
телей и их алкильных эфиров. Последние имеют тенденцию к переходу в гидразо-
соединеиня, первые — к распадению иа два аминосоединения.
Щелочное восстановление применительно к о-ннтрофенолу с целью получения
дифениловых оснований возможно при использовании сернокислотного эфира
о-нитрофенола м) таким образом:
/\/N°* /\/NH-----------------------------HN\/\ HS0
I I Zn-|-NaOH, aq I I II H S°
\ z\ ” \ J\ /\ z лед
x z ^OSOsNa 'z OSO;1Na NaO3SOz
H2Nz/z Z\ZNH2
Illi гидролиз
NaO^SO^/ \/''4OSO8Na
H2N\/z zz/NH2
144
В качестве восстановителя, применимого в условиях щелочной среды, в част-
ности для получения азоксисоединеиий, может применяться также глюкоза. Вероятно
процесс идет с окислением ее карбонильной группы. Таким образом при нагрева-
нии в щелочном (NaOH) растворе глюкозы при 60—70° сложного нитропроизвод-
ного со структурой (1)
/к / /ОН
\-NO2
\/\/Z\cOHN-/ \-N : N—/ \-NHOC/\-/\/
\--/ q n \----/
получают азоксисоединение (II), имеющее значение как азосоставляющая (один из
нафтолов типа AS) при холодном крашенин. Вместо глюкозы в этом случае можно
применять мышьяковистую кислоту, формальдегид, сервистый натрий и цинк Б5).
Восстановление нитросоединений в щелочной среде с получе-
нием азо-,азокси- и гидразосоединений возможно и при посредстве
электролиза. Средой выбирают водно-спиртовые растворы едких
щелочей, растворы слабощелочных солей органических кислот,
щелочные растворы окисей тяжелых металлов. Катод и анод раз-
деляют диафрагмой иди ведут электролиз без диафрагмы Б6).
Электрохимические методы имеют известные преимущества
перед цинковым и железным методами восстановления прежде
всего в отсутствии добавочных реагентов. Возможно, что при
подходящей экономической конъюнктуре и чисто электрохимиче-
ский метод восстановления нитросоединений в щелочной среде
окажется выгодным для применения в практике и выдержит кон-
куренцию с методом использования амальгамы натрия, получаемой
при электролизе поваренной соли, о котором было упомянуто
выше. В последнем случае восстановительный процесс не требует
специальной затраты тока, а проходит как побочный процесс при
главном — выщелачивании амальгамы.
В производственной обстановке восстановление нитросоедине-
ний (цинком или железом) в щелочной среде — растворе едкого
натра — осуществляется в чугунных аппаратах, снабженных мощными
мешалками, при соблюдении оптимальных условий в отношении
температуры и концентрации щелочи для каждой из стадий про-
цесса. Готовое гидразосоединение остается или в растворе орга-
нического растворителя, если он применяется, или выпадает
в твердом виде, отделяется от жидкости и требует в таком случае
отделения от увлеченных им цинка и цинковых соединений, что
достигается чаще всего осторожной обработкой разведенной
кислотой. Перегруппировка в бензициновое основание произво-
дится в отдельном аппарате, сделанном из материала, достаточно
прочного к действию кислоты. Бензидин легко выделяется в виде
практически нерастворимого сульфата, дианизидин — в виде соля-
нокислой соли. Свободные основания выделяются из солей дей-
ствием щелочных агентов. Для получения совершенно чистых осно-
ваний их перегоняют в ваккумме.
Ю Зак. 2611. — Н. Ворожцов
145
В производстве нужно внимательно относиться к качеству вос-
становительного агента, так как и цинк и железо обладают разной
активностью в зависимости от не совсем точно определяемых
аналитически свойств их поверхности. У цинка констатирована
возможность наступления пассивного состояния, задерживающего
дальнейшее движение восстановительного процесса. Наблюдают
иногда и явление неожиданных температурных „толчков11 — не-
объяснимого внезапного повышения температуры (особенно часто
в случае цинка, содержащего примесь серы). С этими явлениями
борются успешно предварительной обработкой цинковой пыли
водным раствором щелочи и подачей цинка в виде пасты с этим
раствором. При цинковом методе восстановления существенно
необходимо использовать цинковые отходы для получения техни-
чески ценных продуктов — белил, литопона и т. п. — или регене-
рировать цинк.
Возможно проводить процесс восстановления, используя два
восстановителя, например доводя его до стадии азосоединения
посредством железа, и восстановляя азо- в гидразосоединение
цинком. При этом значительно сокращается (на 4/5) расход ценного
цинка.
При всех прочих равных условиях железный способ имеет
преимущество в дешевизне материала, но зато при нем нельзя
обойтись без растворителя, что усложняет процесс.
д) Частичное восстановление полинитросоединений
Среди восстановителей, применяемых в щелочной среде, особое
место занимают сернистые щелочные соли (Na.2S, NaHS,
(NH1)2S, Na2S2 и т. п.), особенно благодаря их специфическому
действию при так называемом частичном восстановлении поли-
нитросоединений. Именно когда в ди- или полинитросоединении
требуется восстановить одну лишь нитрогруппу или восстановить
нитрогруппу в нитроазокрасителе, не разрушая азогруппы, тогда
чаще всего пользуются этими „мягкими" восстановителями.
Своеобразное действие растворов сернистых щелочных солей
перед кислотным восстановлением обусловливается повидимому
тем обстоятельством, что имеющаяся хотя бы очень слабая щелочная
реакция раствора (некоторая минимальная концентрация ОН'-ионов)
затрудняет переход образовавшегося нитроаминосоединения в рас-
твор, почему оно и избегает дальнейшего действия восстановителя,
полинитросоединение же легче растворимо в этой среде.
Между тем при кислотном восстановлении отношение раство-
римости обратное. Образовавшаяся аминогруппа вызывает усиленную
растворимость соединения при малой даже концентрации водо-
родных ионов, и если имеются другие восстановляющиеся группы,
они могут быть также легко вовлечены во взаимодействие с вос-
становителем.
Но при взаимодействии нитросоединений с растворами серни-
стых солей щелочных металлов нужно учитывать и иное направ-
ление реакции, чем получение нитро- (соотв. полинитро-) аминов.
Растворы этих солей сами имеют щелочную реакцию. При наличии
146
же щелочи в среде, протекающий через определенные выше стадии
процесс восстановления может привести к получению значительных
количеств нитроазоксисоединений, так как образованию из арил-
гидроксиламинов и нитрозопроизводных азоксисоединений, как
выше было уже отмечено, благоприятствует достаточно щелочная
среда. Получение азоксисоединений здесь нежелательно, как умень-
шающее выход главного продукта реакции. Различные полинитро-
соединения имеют разную чувствительность к концентрации гид-
роксильных ионов, во всяком случае для каждого из исходных
материалов следует определить предел допустимой максимальной
концентрации.
В технике имеется значительный выбор восстановителей этого типа с разной
щелочностью. Характеристика солен натрия, интересных технически, представлена
в виде таблицы, заимствованной у Бранда:
Соли Концентрация ОН'-ионов в I/IOTV растворе Гидролиз в 1/10 N растворе
Сернистый натрий Na3S 0,0396 86,4%
Двусерннстый натрий Na2S2 0.0298 64,6%
Трехсерннстый натрий Na9S3 0 016R 37 6°/л
Четырехсернистый натрий Na2S4 0,0059 11,8%
Пятисерннстый натрий Na2S5 0,00285 5,7%
Сульфгидрат натрия NaHS 0,00015 0,15%
Сернистый аммоний (NH4)2S и сульфгидрат аммония (NH4)HS имеют концен-
трацию ОН'-нонов в 1/10 N растворе ниже 0,0015 (концентрации ОН'-ионои
в 1/10 N растворе аммиака), следовательно вообще очень низкую концентрацию
ОН'-нонов 67).
Таким образом при прочих равных условиях сернистый аммоний, а особенно
сульфгидрат аммония, создают среду, наименее благоприятную для направления
реакции в сторону образования азоксисоединеиий.
Сернистый аммоний исторически интересен тем, что был первым
по времени восстановителем, пользуясь которым профессор Н и-
колай Николаевич Зинин превратил в первый раз аромати-
ческое нитросоединение в амин (1842 г.).
В практике производства тем не менее чаще всего пользуются
сернистыми соединениями натрия, более дешевыми и доступными.
Реакция между нитросоединениями и сернистым натрием про-
текает по уравнению:
4RNO2 -Ь 6Na2S + 7Н2О = 4RNH2 -J- 3Na2S2O3 6NaOH, (1)
т. е. к той щелочи, которая получается при гидролизе сернистого
натрия прибавляется еще образующаяся при реакции щелочь
в количестве, равномолекулярном с взятым сернистым натрием.
Немногие процессы восстановления могут быть удачными при
этих условиях, поэтому гораздо практичнее работать в среде,
менее щелочной. Этому последнему условию отвечают методы,
применяющие растворы многосернистых соединений (полисуль-
фидов) натрия (начиная от двусернистого Na2S2), получаемые
10*
147
растворением рассчитанного количества серы в растворе сернистого
натрия, или пользующиеся сульфгидратом натрия.
Выше мы видели, что концентрация ОН^-ионов и гидролиз
в водной среде в полисульфидах и в сульфгидрате натрия значи-
тельно ниже, чем соответственно у сернистого натрия. Если же
мы примем во внимание уравнения, выражающие ход реакции, то
убедимся, что при их применении самый восстановительный про-
цесс не сопровождается накоплением свободной щелочи.
В самом деле:
RNO2 + Na2S2 + Н2О = RNH, + Na2S2O8, (2)
RNO2 + Na,S6 4- H2O = RNH2 + Na2S2O3 + 3S, (3)
4RNO2 + 6NaSH4-H2O = 4RNH24-3Na2S2O3; (4)
все три уравнения [(2), (3), (4)] в отличие от (1) не имеют в правой
части щелочных соединений. Поэтому применение таких восстано-
вителей, как полисульфиды или сульфгидрат натрия, вполне целе-
сообразно. Из полисульфидов наиболее употребителен дисульфид
Na2S2 как не дающий при реакции выделения серы, выпадение
которой, при применении высших полисульфидов, создает некоторые
затруднения при изолировании и очистке продукта восстановления.
Сульфгидрат натрия является еще более удобным, по своей малой
щелочности, восстановителем, и применение его дает положитель-
ный эффект.
Можно понижать щелочность реакционной массы даже при применении сер-
нистого натрия, вводя в реакционную смесь вещества, могущие нейтрализовать
свободную образующуюся щелочь. Бранд применял в своих лабораторных опытах
уксусноэтиловый эфир, омыляюшнйся от действия свободной щелочи, причем
уксусная кислота образовывала соль с едким натром, и получал хороший выход
.и-нитранилина из динитробензола. А. А. Разумеев с успехом испытывал при-
менение бикарбоната натрия, связывающего едкий натр с образованием соды м).
Можно также прибавлять к раствору сернистой щелочи растворимую аммиачную
соль, например сульфат, и проводить реакцию практически посредством сернистого
аммония.
Реакция частичного восстановления полинитросоединений посред-
ством сернистых щелочей протекает при нагревании, обычно при
невысоких температурах (40—60°), реже—на кипу. Количество
восстановителя определяется по расчету, с избытком против теории
в несколько процентов. Метод имеет применение для получения
дг-нитранилина, пикраминовой кислоты 59)
O2N
O,n/\/\nO2
no2
А /ОН
>
O2n/\/\nH2
и ряда других нитроаминосоединений и для восстановления нитро-
группы в нитроазокрасителях с целью превратить азокраситель
в диазосоставляющую, например:
148
При проведении этой реакции нужно иметь в виду, что азогруппа
в красителе может быть затронута восстановлением. При доста-
точно долгом кипячении азокрасителя с избытком полисульфида
можно нацело расщепить краситель с образованием двух амино-
соединений, например:
3NaO3S—N~N—> + 2Na2S3-f-
-j-6H2O — 3NaOsS—/ >-NH2-J-
-4-3H2N-/ >4- 2Na2S.2O3 + 2S 60).
Окси- и аминоазокрасители при восстановлении вообще не задер-
живаются на стадии гидразо (в отличие от алкильных эфиров
оксиазокрасителей), а дают сразу два амина 61).
Работы химиков за границей освещают за последние годы некоторые новые
направления, касающиеся восстановления с помощью сернистых соединений
металлов.
Так, сернистый аммоний, там где он является отходом или малоценным сырьем,
может найти применение для восстановления нитросоединений, нерастворимых
в воде, при работе в закрытых аппаратах, выложенных внутри алюминием. На
1 мол. нитросоединения нужно 2 мол. (NH4)2S и около 2 мол. водного раствора
аммиака. Работа идет при 100— 115° и давлении в 7 —8 ат. В реакционную смесь
прибавляются и сернистые соединения тяжелых металлов, например FeS. Метод
имеет значение для восстановления мононитросоединеиий в2).
Полного восстановления ннтро- н полинитросоединений можно достичь, приме-
няя легкорастворимые сульфиды, полисульфиды или соли серноватистой кислоты
в количестве, достаточном для восстановления нитрогруппы (или групп), если про-
водить реакцию под давлением водорода или окиси углерода или смеси обоих
газов, причем в реакционной смеси непрерывно происходит восстановление гипо-
сульфита или полисульфида в сульфид, а этот последний по мере образования пре-
вращает нитросоединения в амины. Таким образом практически восстановление идет
за счет водорода или окиси углерода. В качестве катализаторов реакции полезна
прибавка веществ с сильно развитой поверхностью, каковы гель кремневой кислоты
и т. п., а также прибавки тяжелых металлов, например железа, нх окислов, гидро-
окисей, сульфидов, карбонатов и т. п. Реакция протекает при 150— 180° н давлении
100 ат ч выше. Таким образом иа 24 мол. нитробензола достаточно всего 1 мол.
сернистого аммония и около 2/3 мол. FeS, для того чтобы получить количественные
выходы анилина. Преимущество метода — в том, что в качестве отхода при работе
с сернистым аммонием получается раствор сульфата аммония — ценного как
удобрение.
149
Восстановления сульфокислот иитросоединений можно достичь пропусканием
тока газа, заключающего сероводород и аммиак (соотв. 3 г и 0,5 г в 1 ж3) в вод-
ный раствор нитросульфокислоты в присутствии пористого угля ®з).
Описывается также применение в качестве щелочного восстановителя извести
с серой [Са(ОН),S] с прибавкой солн Fe" как катализатора. Восстановление при-
меняется к нитросоедниеииям, ие имеющим гидроксила в ядре. Реакции отвечает
уравнение:
2RNO2 + 3S + ЗСа(ОН)2 = 2RNH2 + 3CaSO3 + Н2О «).
Возможно и применение сульфидов тяжелых металлов самих по себе для полу-
чения эффекта частичного восстановления полиннтросоедииений. Так, прибавив
к водному раствору железного купороса водный раствор сернистого натрия и раз-
мешав полученный осадок FeS с водным раствором динитрофеиолята (2.4), после
нагревания в течение 1/2 — 2 час. прн 40 — 80° получают 2-амнно-4-интрофенол65).
/\/ONa
O2n/\/\n о, O2N Z\/\n н2
Подобный эффект производит свежеосажденный сернистый марганец MnS в ней-
тральной илн щелочной суспеисии 66). Сернистый марганец повидимому даже в не-
больших прибавках при обычном восстаиовлеинн сернистыми щелочами оказывает
благоприятный ускоряющий эффект (неопубликованное наблюдение В, К. Матвеева).
Растворы сернистых щелочей применяются как удобный восста-
новитель при получении аминофенолов, соотв. их замещенных, из
нитрозофенолов и их замещенных. Так как полученные при этом
восстановлении продукты, обладая одновременно и амино- и окси-
группами, растворимы и в кислотных и в щелочных жидкостях, то,
работая в концентрированных растворах сернистой щелочи, удается
повысить выходы и получить более чистый продукт прибавлением
к реакционной смеси во время или после восстановления раствори-
мой аммиачной соли, чем создается аммиачно-щелочная среда,
содействующая выделению аминофенолов из смеси при достаточ-
ной их концентрации. Такой же метод восстановления в концен-
трированном растворе сернистого натрия приложим и к нитро-
фенолам за исключением л-аминдофенола. Реакция проходит при
40—60°
Выделение аминофенолов можно в отдельных случаях прово-
дить осторожной нейтрализацией подходящей, не сильной кисло-
той, например углекислотой, соотв. двууглекислой солью, также
сернистой кислотой и т. п.
Восстановление сернистыми щелочами нитрогруппы в амино-
группу находит применение в отношении превращения антрахино-
новых нитрозамещенных (при наличии других заместителей и без
него) в аминопродукты 68).
Любопытна своеобразная избирательность сернистых щелочей по отношению
к соединениям, имеющим лишь одну иитрогруппу. Так, при обработке технического
нитротолуола, заключающего орто- и пара-нзомеры, растворами сернистых щело-
чей восстаиовляется преимущественно пара-продукт, иа чем основан н способ раз-
деления этой смеси изомерных интро- (соотв. амино-) производных69).
Практически важно далее применение растворимых сернистых
металлов для восстановления всех нитрогрупп в полинитросоеди-
нениях тогда, когда кислотное восстановление мало пригодно
ввиду затруднений как с его проведением, так и особенно с изо-
150
лированием нелетучцх полиаминосоединений. Чтобы избежать обра-
зования вследствие гидролиза свободной щелочи, которая может
действовать на нитросоединения разрушительно и в силу меньшей
растворимости в щелочах нитроаминосоединений, сравнительно
с полинитросоединениями, обусловливать плохие выходы и низкое
качество продуктов восстановления, оказалось в этих случаях
практичным не нагревать исходный материал с водными растворами
сернистых металлов, но сплавлять с твердыми сернистыми соеди-
нениями при температуре, не превосходящей 110°; наиболее подхо-
дящей солью оказался кристаллический сернистый натрий, отве-
чающий формуле Na2S.9H2O. Сама реакция протекает уже по иной
схеме, чем вышеприведенная (1), именно:
RNO2 + Na2S + H2O = RNH24-Na2SO8. (5)
Таким образом например n'-динитродифениламин легко переходит
в л-диаминодифениламин:
Этим же методом можно получить с хорошим выходом амино-
салициловую кислоту из нитросалициловой ’!0):
Процесс проводится при температуре в 105 —110° при разме-
шивании в аппарате с обратным холодильником.
Пиридин, как органическое основание и хороший растворитель Для многих
антросоедииеннй, нерастворимых в воде и спирте, также оказывается пригодным
как среда и реагент восстановительного процесса в присутствии сероводорода (или
сернистого аммония). Температура от 0 до 100°. Сероводород и его соли действуют
здесь как частичные восстановители полннитросоедниеиий. Иногда полезна при-
бавка пиперидина 71)-
В германском патенте описан метод получения амино-, иитроамино- и диамино-
аитрахниоиов с использованием в качестве восстановителей интропроизводиых гид-
рированных оснований хинолинового ряда, например тетрагндрохннолина. Последние
окисляются прн этом в хинолиновые производные и могут быть после отделения
вновь гидрированы и введены в процесс, так что прн восстановлении расходуется
собственно водород. Например
151
Восстановление протекает при невысокой температуре и без катализаторов.
Изменением условий (устранение уксусной кислоты, понижение температуры) можно
получить дигидроксиламии, с введением пиридина—диамииоантрахиион72).
Из неорганических восстановителей, имеющих характер осно-
ваний, следует упомянуть о соединениях закиси железа. Гидрат
закиси железа, лучше —в момент его образования из соли закиси и
раствора щелочи, или углекислые или уксуснокислые соли закиси
железа восстанавливают нитросоединения в амины уже при обы-
кновенной температуре. Удобным оказалось такое восстановление
нестойких к кислотам сернокислотных эфиров нитронафтолов и
т. п. соединений, например
NO2 nh2
/X/\ZOSO3Na /ZzZ/OSO3Na
проводить, взбалтывая их растворы с суспендированным мелом
или углекислым барием и обрабатывая затем при 70 — 75° раство-
ром железного купороса '33).
Осаждение гидрата закиси железа щелочью из железного купороса, находя-
щегося н совместном растворе с моиоазокрасителем, приводит к восстановлению
этого последнего по месту азогруппы (неопубликованное наблюдение В. К. М а-
т в е е в а).
Продукты воздействия карбонильных соединений железа [например Fe(CO)3],
получивших в германской технике большое значение в качестве прибавок к мо-
торному топливу для устранения стука в моторах, обладают также восстановитель-
ными свойствами по отношению к иитросоединениям. В зависимости от количества
щелочи можно при этом получить как амии, так и азокси-, азо-, и гидразосоедине-
иия 74).
Из восстановителей, употребляемых в щелочном растворе, не-
обходимо отметить гидросульфит Na2S2O4, который, правда, не
имеет значения для практики производственного восстановления
нитросоединений, но находит большое применение в лаборатории
для восстановления особенно азокрасителей, при выяснении их
строения и природы их составляющих. Работа ведется в нейтраль-
ном или слабощелочном водном или спиртовом растворе 7&). Восста-
новительное действие гидросульфита основано на легком проте-
кании его окисления в водных растворах по двум направлениям —
преимущественно:
Na2S2O44- О24- Н2О = NaHSO3 4- NaHSO4
и в меньшей степени:
Na.2S,O4 4- О 4- Н2О = 2NaHSOs.
Восстановительная способность гидросульфита используется
особенно для получения продуктов восстановления (лейкопродук-
тов) кубовых красителей.
Некоторые аналитики предпочитают при восстановлении азокра-
сителей для выяснения их состава пользоваться хлористым оловом
с соляной кислотой.
152
Препаративное значение для получения амииов из азосоединений имеют более
дешевые восстановители. В американском патенте описан способ получения аминов
из алкилированных оксназосоединеиий, например
CeH6N —N—ОС2Н6 ----------* CeH5NH24-H2N-<^___\-ОС2Н3,
нагреванием с железом в присутствии уксуснокислого железа и органического
растворителя 7в). Железо И цинк наиболее пригодные восстановители азокрасителей
с целью получения из них аминов для дальнейшего синтеза.
О восстановлении нитрозофенолов (хиноноксимов)уже выше было
сказано. Помимо сернистого натрия и вообще сернистых соединений
щелочных металлов здесь имеют значение кислые соли сернистой ки-
слоты, специально для получения солей сульфокислот аминофенолов,,
например: NOH NH2
/\/\;о Z\/\/0H
SO3Na
Применяется также железо в виде чугунной стружки с кислотой
или поваренной солью. В отношении восстановления пара-нитро-
зозамещенных диалкилированных аминов (оксимов, соотв. хинон-
иминов) метод восстановления железом (если надо в присутствии
меди) дает удовлетворительные результаты.
е) Контактное восстановление водородом
Каталитическое восстановление в жидкой фазе (в растворах)
до сих пор не занимает среди производственных процессов замет-
ного места, какое например уже заняло гидрирование жировых ве-
ществ и некоторых ароматических производных (нафталин, фенолы).
Об одном аналогичном приеме работы было уже упомянуто: о получении амн-
ион из иитросоединеиий прн обработке водородом или окисью углерода исходных
материалов в присутствии солей сероводорода [(NH4)2S] и некоторых катализато-
ров. В этом процессе водород действует иа иитросоеднненне не непосредственно, а
через сернистый аммоний, который он регенерирует из соли серноватистой кислоты.
Восстановление водородом обычно при повышенном давлеини удается провести
в растворах органических растворителей (спирт, гликоль, бензол и т. п.) в присут-
ствии подходящих катализаторов. Наиболее применим н лабораторной практике
палладий н коллоидных растворах или осажденный иа животном угле. Пользуясь
этим катализатором, благодаря большой его способности окклюдировать водород
чаше обходятся без повышения давления. Работа с этим катализатором приводит к
трем типам продуктов в зависимости от господствующих при опыте условий: в ней-
тральном растворе получают из нитробензола p-фенилгидроксиламин, в щелочном
растворе азоксибеизол и гидразобензол. Концентрированная серная кислота в каче-
стве среды содействует образованию амииофенолов 7?).
При этом восстановлении можно пользоваться не молекулярным водородом, а
образующимся например из гидразина NH2 — NH2 прн действии едких щелочей
(с разложением этого вещества на азот и водород): при нагревании в спиртовом
растворе едкого кали с палладием, осажденным на углекислом кальции, из нитро-
бензола количественно получен азоксибензол; с ббльшим количеством катализатора
и восстановителя — гидразобензол и анилин 78).
Никелевый катализатор (реже — Со, Fe, Си) при каталити-
ческом восстановлении в жидкой фазе действует слабее, чем пал-
153
ладий. Восстановление достигается при повышении температуры
иногда до 100э, в иных случаях значительно выше. Никелевый ка-
тализатор готовится осаждением разными приемами гидрата окиси
никеля или окиси никеля на подходящем носителе (например геле
кремневой кислоты, пемзе, угле) и последующим прокаливанием
сухого осадка в токе водорода. Получение аминосоединений удается
при подходящих условиях не только из нитро-, но и из нитрозо-
(изонитрозо-)азо-и азоксисоединений.Растворитель чаще всего спирт.
Возможно получение диаминов из динитросоединений 79), например
В настоящее время нет еще оснований видеть в способе ката-
литического восстановления в жидкой фазе заметного конкурента
установившихся ранее способов получения аминов из нитросоеди-
нений и т. п. При доступности и дешевизне водорода способ мо-
жет в частных случаях быть полезным для производства отдель-
ных продуктов немассового потребления. При сокращении продол-
жительности восстановительного процесса каталитический метод
может стать практически очень интересным с возможностью пере-
хода к непрерывному процессу.
В Италии при низкой стоимости электролитического водорода
он используется для восстановления нитросульфокислот в амино-
сульфокислоты в водных растворах, в частности для получения
1-нафтиламин-3.6.8-трисульфокислоты — промежуточной стадии в
образовании кислоты Н 80).
О каталитическом восстановлении нитросоединений в паровой
фазе сказано ниже в своем месте.
Технически интересные превращения нитросоединений методом
восстановления приводим в виде следующей схемы:
R,NH — HNR
2R1NHOH
2R1NHSO3Na
!
H2NR2 -R2NH2 (л. n')
2H2NRaSO3Na (л)
154
Здесь Rj— одновалентный радикал, например фенил, R2 — двувалентный радикал
отвечающий первому, например феиилен СвН4.
Схема получения аминосоединений из нитрозосоединений н
азокрасителей:
Rx------►RxNH.a
ar2no-------> ar2nh2 <-----r2a
Здесь А обозначает ауксохромовую группу.
ж) Характеристика аминосоединений.
Амины и их замещенные занимают крайне важное место среди
промежуточных продуктов. Прежде всего аминопроизводные нужны
для производства азокрасителей: не менее 50% от веса индиви-
дуальных азокрасителей принадлежит аминам, входящим в их состав
в виде так называемых диазосоставляющих. Удельный же вес азо-
красителей среди всех органических красителей очень велик, так
как они составляют около 40% всей массы красителей.
Амины играют самую существенную роль в производстве
основных красителей всех рядов и особенно в производстве основ-
ных триарилметановых и хинониминовых. Нет почти ни одного
класса красителей, в котором производство не было бы основано
на применении аминов либо в виде исходного материала, либо
использованных для промежуточной обработки.
Амины в значительных количествах потребляются текстильной
промышленностью для окислительных окрасок (черный анилин),
для проявления холодных крашений (основания по (3-нафтолу, по
нафтолам типа AS).
Многие из аминопроизводных (полиамины, аминооксисоединения,
их эфиры и пр.) стали уже давно признанными красильными мате-
риалами для мехов, давая темные тона разных оттенков путем
окислительного крашения (урсолы).
Амины же являются весьма существенной составляющей в син-
тезе крайне необходимых для резиновой промышленности ускори-
телей и антиоксидантов (противостарителей), препаратов, содей-
ствующих ускорению вулканизации резины и препятствующих
преждевременному износу — старению — резиновых изделий. В Аме-
рике большая часть производимого анилина идет на нужды рези-
новой промышленности.
Аминопроизводные ароматического ряда, имеющие еще некото-
рые заместители, очень ценятся как фотографические проявители
или применяются для приготовления последних (амидол, глицин,
родинал, метол).
Некоторые аминосоединения интересны как исходный материал
для синтеза важных фармацевтических препаратов (анилин или
л-аминофенол для диаминодиоксиарсенобензола, о-анизидин для
гваяколовых препаратов, фенэтидин для фенацетина и т. п.).
Наконец некоторые аминопроизводные важны как исходные
материалы для продуктов, нужных для целей обороны государства.
155
При производстве аминосоединений и при обращении с ними
необходимо помнить о вредностях, связанных с производством.
Те амины, которые не имеют сульфогруппы в своем составе, за-
служивают С этой стороны наибольшего внимания. Анилин как
самый употребительный в разных производствах амин особенно
известен своими токсическими свойствами. Но и другие амины,
как жидкие (о-толуидин, о-анизидин), так и твердые (а-нафтиламин),
также и полиамины (.и-фенилендиамин) и аминооксисоединения
далеко не безвредные вещества. В рабочие помещения, где произ-
водятся или перерабатываются амины, пары последних не должны
проникать, так как вдыхание их вредно отзывается на кровенос-
ной системе. Не должно допускать попадания аминов или нх
растворов на кожу работающих; в случае загрязнения кожи амином
необходимо освободить это место от одежды и тщательно промыть
теплой водой (подкисленной), а затем с мылом. Одежду, облитую
амином, необходимо сейчас же сменить и не надевать до устранения
с нее загрязнения. Чаще мыть руки в рабочих помещениях, следить за
чистотой у аппаратов и в помещении, не брать пищи невымытыми
руками, не употреблять спиртных напитков не только во время
работы, но и вне ее, — вот основные правила личной гигиены,
которые при соблюдении общих правил охраны труда в рабочих
помещениях делают достаточно безопасной работу по производству
и переработке аминов.
Аминогруппа, ароматически связанная, может быть определена
качественно помощью диазопробы: к водному раствору соединения,
в котором ее предположительно можно ожидать встретить, при-
бавляют избыток минеральной кислоты (до кислой реакции по бу-
мажке Конго*) и азотистокислого натрия (нитрита) в таком количестве,
чтобы присутствие азотистой кислоты в свободном состоянии можно
было только констатировать по иодокрахмальной бумажке, но не
было бы большого избытка HNO2. Прибавление нитрита ведется
при возможно низкой температуре (0—5°). Полученный таким
образом раствор соли диазония (если она образовалась из амино-
группы) обрабатывается уксуснокислым натрием до исчезновения
минеральнокислой (на Конго) реакции и затем на холоду прили-
вается к содовому раствору соли R (натриевая соль 3.6-дисульфо-
кислоты ^-нафтола) или щелочному раствору резорцина. В случае,
если аминогруппа присутствовала в соединении и дала диазоние-
вую соль, — при сочетании этой последней с солью R или резор-
цином наблюдается образование яркой и интенсивной окраски
азокрасителя. Отсутствие окраски служит указанием на отсутствие
аминогруппы в исследуемом соединении. Следует тщательно из-
бегать при этой пробе избытка кислоты в растворе после слива-
ния с солью R или резорцином, так как в таких условиях при
малейшем избытке азотистой кислоты вместо сочетания (в азокра-
ситель) происходит реакция нитрозирования нафтолдисульфо-
кислоты, или ж-диоксибензола, в результате которой не полу-
* Реактивную бумагу Конго получают пропитыванием чистой фильтровальной
бумаги 0,5—1%-иым раствором красного Конго (из бис-дназотнрованиого бензи-
дина и нафтионовой кислоты) и высушиванием. От мниеральвых кислот бумажка
синеет, в щелочах и органических кислотах остается красной.
156
чается уже характерной окраски оксиазосоединения, но заметно
позеленение раствора. Эта диазопроба имеет в ароматическом
ряду весьма большое значение для характеристики аминов как „ди-
азосоставляюш.их“ красителей. Некоторые трудно диазотируемые
амины требуют иногда специальной обработки для перевода в диа-
зоний (см. ниже о диазотировании). Некоторые полиамино- (соотв.
аминоокси-) производные орто- и пара-рядов реагируют с азотистой
кислотой, окисляясь в хиноны, и не образуют поэтому диазосое-
нений при применении обычной методики диазотирования.
Количественное определение аминогруппы в сое-
динении основано на использовании той же способности к переходу
аминов в диазосоединение под влиянием азотистой кислоты.
Известно, что реакция между первичным ароматическим амином
и азотистой кислотой протекает количественно по уравнению
RNH2 • НС14- HNO2 = 2Н2О + RN2C1.
Следовательно по количеству израсходованной азотистой кислоты
(соотв» ее соли) можно судить о содержании первичных аминогрупп
в соединении, если оно нормально диазотируется.
Для определения применяется раствор нитрита определенного титра (0,1 N).
Для установки титра нитрита пользуются кристаллическим сульфаииловокислым
натрием CeH4(NH2)SO3Na.2H2O, который легко получить в совершенно чистом
состоянии. Готовят приблизительно 0,1 N раствора этой соли (1 мол. = 231) и, беря
по 20—25 см3 раствора с добавлением достаточного количества соляной кислоты
(иа 1 мол. соли минимально 3 мол. кислоты), титруют при охлаждении помощью
раствора нитрита. Индикатором конца диазотирования является иодокрахмальная
бумажка, обнаруживающая посинением малейший избыток HNO2- Установив титр
нитрита по сульфаииловокислому натрию (1 мол. сульфаниловой'" кислоты отвечает
1 мол. нитрита), можно пользоваться этим раствором для количественных определений
амииосоедииений. Необходимо лишь всегда следить за достаточной кислотностью
титруемого раствора, так как при недостаточно кислой среде происходит образова-
ние амииоазокрасителей и определение значительно затрудняется. Вторая предосто-
рожность, которую важно всегда иметь в виду при этом определении, это — охла-
ждение раствора, дабы избежать неверных результатов от потери азотистой
кислоты. В остальном проба очень удобна и проста и дает хорошие результаты
в применении к большинству амииосоедииений. Воздействие азотистой кислоты
на первичную аминогруппу с некоторыми аминами протекает быстро, с другими
требует более или меиее продолжительного времени для завершения реакции
диазотирования. Это необходимо иметь в виду при определении конца реакции.
Некоторые амииосоединеиия, как вышеуказанные орто- и пара-диамино-, соотв.
аминоокси-, соединения, ие могут быть точно определены этим методом, так как
с ними реакция диазотирования или совсем ие идет или осложняется реакцией
окисления. Мета-диамины часто даюг амииоазо-(соотв. аминодисазо-) красители, что
затрудняет определение их этим методом.
Ниже (см. определение ОН-группы) указан другой метод анализа амииосоеди
нений по азосочетанию.
Для многих аминов характерны их цветные реакции 81)-
Имеются методы прямого ввода аминогруппы в ароматическое ядро, минуя
промежуточную стадию иитросоедииеиия. Своеобразная рекация, принадлежащая
по вышесказанному к первой группе процессов (образование новых заместителей
вместо водородного атома), протекает между гидроксиламином и некоторыми арома-
тическими ди- и полииитросоединеииями в спиртово-щелочной среде 82). Реакция,
общее уравнение которой
RH + HONH2 = RNH2 + Н2О,
имеет большой теоретический интерес и может приобрести и техническое значение,
если дальнейшая разработка создаст для нее удобвую форму примеиеиия и воз-
можность управления процессом.
157
И. Т. Турский предлагает метод введения аминогруппы в ароматические
соединения обработкой их гидроксиламииом и концентрированной серной кислотой.
При этом, в зависимости от большей или меньшей легкости сульфирования соеди-
нения, получаются или аминозамещенные или сульфокислоты аминов. Катализато-
ром является FeSO4 в3).
К этому же типу реакций принадлежит реакция взаимодействия между некото-
рыми ароматическими соединениями (нафтолами, сульфокислотами нафтолов) и
натрий-амидом NaNH2
Метод, интересный пока с теоретической стороны, для ввода аминогруппы
непосредственно в ароматическое ядро, описан в германском патенте: это действие
азотнстоводородной кислоты N3H на исходное органическое соединение в присут-
ствии концентрированной минеральной кислоты. Так, из бензола можно получить
анилин ®)
z\ .NH.,
II 60° I I '
+ N3H----------------------+ + N2
' I H2SO, или H3PO4 I I
Выхода в описании однако далеки от теоретических, и сам способ не может пре-
тендовать на практическую ценность.
Для нитросоедннений превращения в аминосоединения, соотв. в промежу-
точные стадии восстановления (гидроксиламиновое, азокси-, азо-, гидразосоединения),
не являются единственно возможными методами превращения реакционной нитро-
группы. Не такое общее, как восстановление, значение имеют иные процессы,
применяемые к ннтросоединениям, где нитрогруппа находится в особо реакционных
условиях. Таким образом например одна нитрогруппа в о- и л-динитробензолах
при нагревании с растворами щелочей замещается на гидроксил, и в результате
из Диннтросоедннеиия получается нитрофенол, например:
/X/Zno.,
| | 4-2NaOH------->
\z\no2
Z\/0Na
I | J- NaNO, + H2O
\-/\no2
Эту обработку можно применять для освобождения сырого л/-динитробензола от
сопровождающих его орто- и пара-нзомеров. Практичнее повидимому здесь оказы-
вается метод обработки динитробензола раствором сульфита натрия, причем о- и п-
изомеры переходят в растворимые соединения (О. М. Голос енко).
Особенно своеобразно реакционна нитрогруппа, замещающая
водород в антрахиноне. Здесь возможны многочисленные превраще-
ния нитросоединений с обменом нитрогруппы как на заместители,
заключающие азот (без восстановления ее), так и на безазотные
заместители. Так, нагревание нитроантрахиноновых производных
с аминами, как жирными, так и ароматическими (не третичными;
у азота амина должен быть по крайней мере один свободный водо-
родный атом), приводит к образованию алкил-, соотв. диалкил-,
соотв. ариламинозамещенных антрахинона 86):
NO2
СО |
/\/\/\
+ HN(CH3)2->
N(CH3)2
СО |
/\/\/\
4-hno2
Нагревание нитроантрахиноновых замещенных с водным пири-
дином (с обратным холодильником или под давлением), такая же
158
обработка известковым молоком или сплавление при нагревании
до 170 — 180° с кислым уксуснокислым калием является методом
замены нитрогруппы на гидроксил 87):
Обработка щелочами в растворе метилового спирта (или мети-
латом щелочного металла) открывает путь для получения из нитро-
метоксизамещенных антрахинона; при этом из динитро- можно
получить, в зависимости от их строения и метода обработки, ди-
метоксильные и нитрометоксильные производные антрахинона 88)
NO2
Ц-NaOCHg--->
NO2
СО |
/\/\/\
4-NaNO2
Обработка фенолятом щелочного металла переводит нитроантра-
хинон в арилоксисоединение 69)
NO2
СО I
/\/\/\
I +NaOC6HB
ОС6Н5
СО I
/\/\/\
+ NaNO2.
Кипячение с раствором сульфита щелочного металла вводит
сульфогруппу вместо нитрогруппы т):
NO2
СО |
SO3Na
СО |
/\/\/\
+ NaNO2.
Наконец обработка хлором в высококипящем растворителе дает
замещение нитрогруппы на атом хлора 9I).
Эту замечательную подвижность нитрогруппы в антрахиноне,
имеющую аналогию в подвижности и других заместителей (напри-
мер сульфогруппы), следует отнести на счет особенного реакти-
вирующего влияния со стороны антрахинонового ядра.
159
Промежуточные продукты, получаемые методом восстановления
А. Кислотное восстановление
NCH
S = SO3H
7—Анилин. 2—Толуидин (пара-). 3—Толуидин (орто-). 4— Анизнднн (орто-). 5— Аминофенол (пара).
6—Метаниловая кислота. 7—л-Аминоацетаннлнд. 5—п-Нитооанилин. .9—.и-Фенилеидиамии. Ю—лг-То-
лунлендиамнн. 11 — Диамнностильбенднсульфокнслота. 12 — п-Толуилендиамин. 13 — «-Нафтиламин-
7-7—1.5-нафтнламинсульфокнслота. 7<5— 1.8-нафтиламиисульфокнслота (перн-). 16—1.6-нафтиламиисуль-
фокислота (кислота Клеве). 17—1.7-нафтиламннсульфокислота (кислота Клеве). 18— 1.4-ацетаминонаф-
тиламин-6-(7-) сульфокислота (днаминогеновая кислота). /9—!-нафтиламнн-4.8-дисульфокислота (амино-
кислота S). 29— 2-нафтилямин-4.8-днсульфокнслота (аминокислота С). 21—1-нафтидамнн-3.8-дисульфо-
кнслота (аминокислота Е). 22— 1-нафтиламин-3.6-днсульфокнслота (кислота Фрейнда). 23— 1-нафтиламин-
3.6.8-трисульфокиелота (кислота Т). 24—1.2-аминонафтол-4-сульфокислота.
160
Промежуточные продукты, получаемые методом восстановления .
Ц Зак. 2611. — Н. Ворожцов
161
Весьма замечательно, что фталевый ангидрид (1), принадлежащий к бензольным
замещенным, имеющий общую с антрахиноном (И) схему двух орто-размещенных
СО-групп, в своих нитропродуктах повторяет некоторые реакционные особенности,
характерные для нитроантрахиноновых замещенных
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ.
1) Н. J. Prins, Rec. 42, 942 (1923), СВ 1924, И, 2091.
s) Н. Pomeranz, Г. п. 269542 (F. XI, 149).
3) A. Bechamp, Ann. Chim. Phys. (3) 42,186 (1854); Brimmeyr первый
предложил восстановление с малым количеством соляной кислоты; Р. Н. Grog gins;
Anilne and its derivatives. New. York, 1924.
4) C. W. Davis (DuP), Am. n. 1663476.
5) Lyons, Smith, Ber. 60, 173 (1927), Cp. A. Bretniitz, A. Pens a, CB
1927, 11, 243.
в) A. Wohl, Ber. 27. 1432, 1815 (1894); O. N. Witt, Chem. Industrie 10, 218-
(1887). Ср. E. v. Meyer: Mu spratt; Encyklopad. Handbuch der technisch. Chemie,
4 Aufl., Braunschweig, 1888, 942; P. N. Raikow, Zt. angew. Ch. 29, 196, 239; CB
1916, II, 315, 380.
’) IG. Г. n. 463773, 464561, Б. n. 263376, CB 1928, II, 1488, Г. n. 515758, CB
1931, I, 1827, 516999, CB 1931, II, 2932, 518929, Ch. T, Ob. 1931, № 76/77, 183,
Б. n. 274562, Ch. Tr, J. 81, 329, (1927), Am. n. 1793941, CB 1931, 1, 2936; cp. IG,
Г. ri. 551254, 551255, CB 1932, 11, 621. 1G. Б. n. 279283, CB 1930, I, 2479.
8) Cp. Hertel, Ши. n. 118720, CB 1927, II, 869.
9) W tilling, Г. и. 67018 (F. ill, 47).
10) H. Pomeranz, Г. n. 289454 (F. XII, 117).
И) В 1 a n k s m a, Proc. Ak. Wetensk. Amsterdam, 24, II, 1906, cp. Rec. (4) 49,
148 (1929).
12) J, R. S a m p ey, CB 1930, I, 1612.
13) H. Weil, Ber, 55, 132, 2533 (1922), Г. n. 147552 (F. VII, 58).
К) T. Walter, MLB, Г. n. 109487 (F. V, 70).
15) S eye wets, Vi gat, Bull. (4), 32, 684 (1922); ср. P i r i a, Ann. 78, 31 (1851),
A. W. Forsberg, Ber. 20, Ref. 107 (1887), S a n d m e у e г, Г. n. 65236 (F. Ill, 56),
71368 (G) (F. Ill, 57), также E. Erdmann, H. Erdmann, Г. n. 64908 (F. Ill, 40),
R. Nietzki, Halbach, Ber. 29, 2448 (1896), Nietzki, Т.п. 86097 (F. IV, 90),
E. Bamberger, Ber. 30, 654 (1897), F. Raschig, Schwefel- & Stickstoffstudien,
Leipzig, 1924, 2k5, Hunter, Sprung, CB 1931, I, 3464.
16) W. Blythe & Co Ltd. Ф. n. 720029, CB 1932, I, 2996.
1’) H. В u c h e г e г, Г. n. 423029 (F. XV, 223).
18) H. В 4 r t, Г. n. 258059 (F. XI, 152); M. Bodenstein, Г.п. 172569 (F. VIII, 118).
19) Ch. F. Bohringer & Sohne, Г. n. 117007 (F. VI, 69); О tin WSser,
Г. n. 235955 (F. X, 130); A. K. Doolittle, Trans. Am. Electrochem. Soc. 45, 43,
CB 1924, I, 1918.
г») K. Elbs, J. pr. Ch. (2) 43, 39, (189J).
2i) tM. Г. ci. 282531 (F. XII, 116).
22) IG. Б. n. 328210, CB 1930, II, 1134.
28) B. Rassow, H. S c h u 11 z k y, Zt. angew. Ch. 44, 669 (1931).
24) A. Wohl, Г. n. 84138 (F. IV, 44), 84891 (F. IV, 46); К, Г. n. 89978
(F. IV, 47); E. Bamberger, Ber. 27, 1347, 1549 (1894), A. Wohl, Ber. 27, 1432
(1894),
25) R. Brand, A. Modersohn, J. pr. Ch. (2) 120, 160 (1928), здесь пол-
ная библиография предмета; К. Brand, F. S t а с h е, J. pr. Ch. (2) 133, 355
(1932), I G. Г. n. 488947, CB 1930, I, 2008, cp. CB 1929, I, 236.
26) W. M. Cumming, G. S. Ferrier, CB 1931, II, 187.
27) a) E. Bamberger, M. Knecht, Ber. 29, 864 (1896); б) H. W is lice-
nus. Ber. 29, 894 (1896), G. Austerweil, Г. n. 453428, CB 1928, 1, 2307.-
в) R. W i 11 s t a 11 e r, Kubli, Ber. 41, 1936 (1908); О. В a u d i s c h, H. Rom,
162
Ber, 49, 203 (1916); г) К. Brand, J. pr. Ch. (2) 74, 449 (1906), Д) A. Ch. Ray,
S. D u 11, CB 1928, 1, 2370.
28) L. Gattermajin, R. Schmidt, Ber. 29, 2934 <1896), F. IV, 265.
29) A. Wohl, Г. n. 83433 (F. IV, 53).
so) Dieffenbach, Г. n. 192519 (F. IX, 111).
si) M. L. В., Г. n. 96853 (F. V, 58).
32) K. Brand, Die elektrochemische Reduktion organischer Nitrokorper und ver-
wandter Verbindungen, Ahrens Sammlung chem. & chem.-techn. VortrSge 13, 51 (1908),
Г. n. 75260 с добавлениями (By) (F. Ill, 53); F. Darmstadt er, Г.’п. 150800’
154086 (F. VII, 90, 91); L. Gattermann, Ber., 26, 1844 (1893).
зз) CIBa, Г. n. 295841 (F. XIII, 255).
31) G r ii n a u, Г. n. 437002 (F. XV, 226), Б. n. 254204, Ф. n. 609631, CB 1927,
I, 85; F. M. Brigham, H. S. Lukens, CB 1932, I, 1364, cp. D. Caeser, CB
1927, 11, 2499.
35) Cp. F. Haber, Zt. f. Electrochemie 4.506 (1898), F. Haber, C. Schmidt
Zt. Phys. Ch. 32, 271 (1900).
3») K. Brand, J. Mahr, CB 1931, 11,2145; E. Bamberger, A. Rising,
CB 1901, II, 333; Bamberger, Renauld, Ber. 30, 2278 (1897).
37) Г. n. 138496 (tM.) (F. VI, 1290); С. Д. Топорков, H. И. Амиантов,
Ан.-Кр. Пр. 1931, № 6, 3.
38) H. Loes пег, Г. п. 77563 (F. IV, 42).
39) A. Wohl, Г. п. 81129 (F. IV, 43).
4°) Deutsch. Gold & Silber-Scheide Anstalt, Г. п. 486598, CB 1930, I, 1536.
41) L. Z e c h m e i s t e r, P. R о in, Г. n. 446867 (F. XV, 227).
42) By, Г. n. 210806 (F. IX, 115); Gr. Г. n. 225245 (F. IX, 1180).
43) By, Г. n. 204653 (F. IX, 114).
44) M. L В., Г. n. 216246 (F. IX, 116), ср. значение Na2S2 у Blanksma, Rec. 20,
141 (1901).
43) H. S. Fry, P. E. В о w m a n, Am. Soc. 52,1531 (1930). Cp. Suter, Dains,
CB 1928, И, 2645.
46) D i ef f e n b a c h, Г. n. 197714 (F. IX, 112).
47) J. H. Walton, G. W. Filson, Am. Soc. 54, 3228 (1932).
48) G. Pellegrini, Giorn. Chim. ind. applic. 8, 175 (1926), G. Poma,
G. Pelligrini, Г. n. 410180 (F. XV, 216).
49) В. А. Измаильский, В. H. К о л п e н с к и й, Ж. X. Пр. I, № 4, 31
(1925), Измаильский Г. И. Арбузов, Ж. X. Пр. I, № 5—6, 35 (1925),
Измаильский, Е. П. Рустанович, П. Т. Шпунтенок, Ан.-Кр. Пр. 1932,
№ 3, 10; Топорков, Амиантон см. 37), В. О. Лукашевич, Получение
гидразоанизола и дианизидина (не опубликовано).
30) Ср. Nat. An., Ам. п. 1633123, 1644484, СВ 1929, I, 1613, 1718373, СВ 1930, I,
2010.
зг) Б. П. Орел кин, А. Т. Рыскальчук, М. А. А й з и к о в и ч, Ж. О. X.,
I (63), 698 (1931), Н. Wieland, Вег. 45, 492, примечание 2.
52) IG. Г. п. 530823, СВ 1931, II, 3398.
53) Ср. Bamberger, Вег. 28, 250 (1895); Wislicenus, Kaufmann,
Ber. 28, 1326 (1895); Grandmougin, Ber. 39, 3562 (1906); Elbs, Zt. f. Elektro-
chemie 7, 133. 141 (1900).
54) I G. Г. n. 488611, CB 1930, I, 1698.
55) I G. Шв. n. 117474, CB 1927, I, 1743.
56) Ср. Г. n. 127727 (F. VI, 78), 141535 (F. VII, 57) (MLB); 116467, 122046 (F. VI,
76, 77) (Lob); 79731 (F. IV, 63); (E. Straub); 100233,100234 (F. V,62, 63) (W filling);
121899, 121900 (F. VI, 74, 75) (By).
57) K. Bran d, Uber die Sulfide des Natriums als partielles Reduktionsmitte’l
fur die Polynitroverbindungen, J. pr. Ch. (2), 74, 449 (1906); V. Vesely,
L. K- Chud ozi 1 о w, CB 1926, 1, 86.
а8) См. И. И. Воронцов, Ж. X. Пр. 7, 2145 (1930).
59) Cobenzl, Ch. Ztg 1913, 299,
60) И. И. Воронцов, Ан.-Кр. Пр. 1931, №9—10, 43.
61) J. В. Conant, М. F. Pratt, Am. Soc. 48, 2468 (1926).
6S) IG. Г. n. 449405 (F. XV, 220).
63) Grasse Hi, Dyest. Co, Am. n. 1662421, IG. Г. n. 458088, CB 1928, I. 2539;
ср. Г. n. (By) 388185 (F. XIV, 394).
M) Nat. An. Am. n. 1765367, CB 1930. II, 1442.
6Б) Nat. An. Am. n. 1689014, CB 1929 I, 1615.
11*
163
ев) Gen. Anil. Works, Am. n. 1765660, CB 1930, II, 1442.
87) A. E. P о r a i - К о s c h i t z, Г. n. 463519, Пора й-К о ш и ц, Сов. п. 19627, СВ
1931. II, 3393.
68) By, г. п. 135561 (F. VI, 298), Lauth, С. г. 137, 662 (1903); Beisler,
J о n е s, СВ 1923, I, 929.
в®) С1у, Г. п. 92991 (F. IV, 32).
70) В. И. Мииаен, Б. П. Федоров, С. М. Карцев, Ан.-Кр. Пр. 1931,
№ 8, 1, также неопубликованные опыты С. М. Карцева.
’1) О. L. Brady, J. N. Е. Day, С. V. Reinolds, СВ 1930, I, 42.
72) IG, Г. п. 473871, СВ 1929. II, 96.
’3) Fino w, Н. Muller, Г. п. 418497 (F. XV, 224); G. N. Burkhard.
Н. Wood, СВ 1929, I, 1565,
74) IG, Г. п. 441179, СВ 1927, I, 2135.
75) Gran dm о u gin, J. pr. Ch. (2) 76, 125 (1907), Ber. 39, 2494, 3929 (1906).
76) Dow, Am. n. 1722417, см. также во).
77) K. Brand, Steiner, Ber. 55, 875 (1922); Parrett, Lowy, Am. Soc.
1926, 778; R. Adams, F. L. Cohen, O. W. Rees, CB 1927, II, 60.
78) M. Busch, H. Schulz, Ber, 62, 1458 (1929).
’9) W. E. Bradt, CB 1931, II, 987; IG. Ф. n. 621434, CB 1927, II, 975,
Newport, Am. n. 1589936, CB 1926, II, 1192.
8°) G. Poma. G. Pellegrini, Б. n. 190114, CB, 1923 II, 962; IG. Б. n.
540327, CB 1932, I, 1154.
81) Cp. G. Grunow, Bericht tiber die Fortschritte, d. analyt. Chemie, Ill, Zt.
analyt. Ch. 86, 262 (1931).
82) Ср. заявл. Г. п. M. 28239 (Meisenheimer, Patzig) (F. VIII, 95),также
Ber. 39, 2533 (1906).
®) J. F. Turski, Г. n. 287756 (F. XII, 120). Cp. J. Kniatowna, Bull. (4) 35,
207 (1924), CB .924, I, 2695.
si) Sachs, Г. n. 173522, 181333 (F. VIII, 172, 173), Ber. 39, 3006 (1906).
85) Knoll & Co, Г. n. 427858 (F. XV, 221); ср. Б. n. 307798, CB 1930, I, 1369.
®) By, Г. n. 136777. 136778 (F. VI. 374, 381), 126542 (F. VI, 300).
87) By, Г. n. 145238 (F. VII, 188), 158891 (F. VIII, 253); E. Schwenk, CB
1922, 1, 866.
®) M.L.B. Г. n. 75054 (F. Ill, 268), 77818 (F. IV, 304), 145188 (F. VII, 189), 158278
(F. VIII, 265), 167699 (F. VIII, 266).
88) By, Г. n. 158531 (F. VIII, 241).
®) R. E. Schmidt, Ber. 37, 66 (1904), Г. n. 164272 (By) (F, VIII, 231).
»1) В., Г. n. 252578 (F. XI, 545).
®) ICI, Б. n. 356609, Chem. Age 1932, 56.
ГЛАВА VI
ПРЕВРАЩЕНИЕ СУЛЬФОГРУППЫ В ГИДРОКСИЛЬНУЮ ГРУППУ
МЕТОДОМ ЩЕЛОЧНОГО ПЛАВЛЕНИЯ. ДРУГИЕ ПРЕВРАЩЕНИЯ
СУЛЬФОГРУППЫ.
Одно из наиболее широко известных превращений аромати-
ческих сульфокислот—это их переход в гидроксильные произ-
водные— чаще всего фенолы. Такое превращение осуществляется
в наиболее простой и давно установленной форме действием
расплавленной безводной горячей гидроокиси щелочного металла
(чаще NaOH; иногда КОН) на щелочные соли сульфокислот.
Превращение сульфогруппы в гидроксильную группу в этом процессе было
открыто в 1864 г. Дюзаром (Dusart) и независимо от него и почти одновременно
В горцем (Wurz) во Фрайции и Кекулен Германии 1). Довольно скоро оно
вошло в обиход широкой заводской практики, чему в особенности благоприятствовало
большое значение новой реакции, получиншей назнаняе „щелочного плавления*
или „сплавления со щелочами* в возникшем в 70-х годах в Германии производстве
синтетического ализарина и производстве р-нафтола.
Основное структурно е отличие этого превращения от рассмот-
ренных в предшествовавшей главе то, что, в то время как при
восстановлении азотных групп (NO2 и других) при перемене
заместителя сохраняется в связи с углеродом ядра тот же атом
азота, который был и в исходном заместителе, здесь в новом
заместителе уже отсутствует атом серы. Можно сказать, что ще-
лочное плавление сульфокислот дает один из примеров вытеснения
новым заместителем прежнего.
В качестве минерального реагента в этой реакции наиболее
употребителен уже вследствие своей большей дешевизны едкий
натр. Едкое кали находит применение тогда, когда едкий натр
оказывается недостаточно реакционным. В некоторых случаях
пользуются смесью из едкого кали с едким натром, удобной
благодаря тому, что такая смесь имеет более низкую температуру
плавления, чем каждая из ее составных частей.
Процесс сплавления солей сульфокислот со щелочами имеет
большое производственное значение, служа методом получения
ароматических оксизамещенных и в первую очередь фенолов.
Обработка горячей плавленной щелочью применяется не только для взаимодей-
ствия с солями сульфокислот, ио и н реакциях с некоторыми другими промежу-
точными продуктами, содействуя тогда образованию новых гетероциклических
соединений, иногда красителей. В этих применениях едкая щеаочь является уже
„конденсирующим средством*, н механизм ее действия, в основе аналогичный
такому же в рассматриваемой нами здесь реакции, будет ниже разобран по-
дробнее.
165
а) Характер реакций, протекающих в щелочном плаве сульфо-
кислот. Плав в открытом котле
Обычно уравнение реакции между солью сульфокислоты и гидро-
окисью щелочного металла в процессе сплавления со щелочами
(для едкого натра) выражается так:
RSO3Na + 2NaOH = RONa + Na2SO3 + H2O. (1)
Однако нужно заметить, что течение процесса гораздо сложнее,
чем может выразить это уравнение, регистрирующее лишь резуль-
тат главного процесса: в последнем имеет место не только простое
вытеснение гидроксильной группой сульфогруппы. Едкая щелочь
в расплавленном, безводном состоянии — реагент очень энергичного
действия. В этих условиях, как обнаружено рядом исследований
действия ее на органические вещества, имеющие ненасыщенные
группы, у молекул щелочи наблюдается наклонность к присоеди-
нению по месту многократных связей, причем едкий натр например
реагирует как составленный из частей ЫаО...Н.
Таким путем возможно например получение продуктов окисления ненасыщен-
ных углеводородов. Так, ацетилен переходит при пропускании через расплавленную
смесь едкого кали и едкого натра при 220° в уксуснокислый натрий очевидно
по схеме:
NaO Н
НС = СН4-2ЫгЮН -> NaO— С— С—Н + Na°g—» (NaO)3C-CH3
/ “орто-ацетат натрия
н н
(NaOJgC—СН3 Н-Н2О -» О:С—CH3 + «NaOH 2).
NaO
Сплавление фенола с шестикратным количеством едкого натра, как установлено
было Бартом и Шредером, сопровождается выделением водорода и дает
в качестве главных уловленных продуктов реакции: резорцин, пирокатехин и фло-
роглюцин вместе с малым количеством дифенильного окснпронзводного. Естествен-
нее всего течение реакции представить н здесь проходящим через стадию при-
соединения остатков едкого натра, например для резорцина и пирокатехина:
ONa
I
f^l
+ NaOH j
ONa
+ NaOH
ONa
ONa
A. E. Чичибабнным установлено для хинолина, что действие едкого калн
при 200—225° сопровождается превращением хинолина в карбостнрил с количе-
ственным отщеплением водорода очевидно по схеме:
166
/\/А
+ КОН
OK
NH
+ н2 »).
Ниже мы увидим еще довольно показательные примеры подобного действия едкой
щелочи на антрахиноновые производные.
По сказанному выше представляется наиболее вероятным, что
и в случае сплавления солей сульфокислот со щелочами как пер-
вая фаза реакции наступает присоединение элементов едкой ще-
лочи к ароматическому ядру, и лишь в результате стабилизации
продукта при отщеплении молекулы сульфита получается напри-
мер фенолят. Схема образования фенолята, в развитие общего
выражения (1), поэтому такова:
X\/SO3Na ^\<ONa
+ NaOH -> )
ч/ ч/<:н
X\X°Na
4-NaHSO3;
к/
NaHSO3 + NaOH = Na2SO3 + H2O.
Такое понимание процесса уясняет некоторые особенности его
течения в отклоняющихся от нормы примерах. Нужно заметить,
что при сплавлении со щелочами моносульфокислот бензольного
и нафталинового рядов получаются всегда, хотя и не с количе-
ственным выходом, одноатомные фенолы, заключающие группу ОН
в том же положении, какое занимала сульфогруппа. Следова-
тельно здесь вероятнее всего допустить присоединение остатка
ONa к тому же атому углерода, с которым была связана сульфо-
группа.
Имеются однако примеры отклоненна от этой правильности. Так л-днсульфо-
кнслота бензола прн сплавлении со щелочами дает не пара- (соотв. орто-) днокси-
бензол, ио мета-диоксибензол (резорцин), например:
/\/SO3Na NaO^^x/ONa
| | + 4NaOH —> | | +2Na2SO3 + 2H2O.
NaO3SZ\/
Здесь своеобразный исход реакции можно объяснить лишь допущением при-
соединения элементов молекулы едкой щелочи таким образом, что одна молекула
щелочи отдает остаток ONa атому углерода, связанному с сульфогруппой, а другая
молекула соседнему по отношению к сульфогруппе атому, например:
/-\^^/SO3Na X\z/SO3Na ^x/ONa
| | -J- 2NaOH > H I ^ONa _ 2NaHSO, * | !
NaO3S-/\/' NaOgS^XZ^Hs V
A I
H ONa ONa
Своеобразно же происходит взаимодействие с расплавленной щелочью у ж-дн-
судьфокнслот нафталина, соотв. у ж-сульфокнслот окси- и соотв. амннонафтали-
167
нов. Здесь при достаточно высокой температуре (250—300°) обработки происходит
расщепление одного ядра нафталина н образование о-толуиловой кислоты и уксус-
ной кислоты, конечно в виде щелочных солей, например
SO3H
I
+ CHgCOOH *)
Это замечательное превращение находит для себя объяснение также в разви-
той нами схеме
SO3Na + NaOH
SO3Na +NaOH
NaO SO3Na
/\Х<н
H
.SO3Na
\Z\Z^ONa
H H
— 2NaHSO,
ONa + NaOH
Z\Z\
ONa + NaOH
NaO ONa + NaOH
+ NaOH
/\^(ONa)3
Г"
I (ONa)3
CH3
I
C(ONa)3
4- 2H,O
- 4NaOH
/\/ \0Na
i
I
z Xch3
CH3
k°
\QNa
Очень своеобразно реагируют в щелочном плаве р-сульфо-
кислоты антрахинона, у которых а-водород не замещен. В них
именно не только гидроксильная группа занимает место сульфо-
группы, но и устанавливается второй гидроксил в бывшем сво-
бодным a-месте вместо водорода, причем, если в реакционную
смесь не добавлен окислитель, в результате взаимодействия про-
исходит восстановление карбонильных групп антрахинона. Объяс-
168
некие и этому превращению может быть найдено в схеме предва-
рительного присоединения молекул едкого натра, например б):
/\/\/\/S°3Na
+ NaOH
Z\/\^\/S°3Na
| X ONa +Na0H >
\/VV
C
OH
OH
| ONaH
/\Д\Х/ада
^ONa
OH
— NaHSO3
OH
I ONa
z\zC\\/V0Na
\Z\Z\Z
c
OH
ONa
ONa
Таким образом из моно-р-сульфокислоты антрахинона в ще-
лочном плаве образуется 1.2-диоксиантрахинон (краситель, али-
зарин, А—его динатровое производное—ализарат).
Введение вместе со щелочью в плав окислителей (селитры
NaNO3 или соли хлорноватой кислоты NaC108) позволяет прово-
дить так называемый „окислительный плав“, дающий возможность
остановить процесс сразу на получении антрахинонового, а не
гидроантрахинонового замещенного и без потерь в выходе, полу-
чаемых из-за затраты части исходного антрахинонового производ-
ного на окисление 6). Ализариновый плав в производстве является
именно таким окислительным плавом.
Из сказанного выше видно, что сплавление со щелочами—про-
цесс достаточно сложный по химизму, и естественно ожидать в нем
побочных реакций. Таковые в действительности имеют нередко
место и этим самым снижают выход главной реакции. Устано-
влено, что выходы фенола при плавке с едким натром бензол-
сульфокислоты не превосходят 96% 7); выход р- (соотв. а-) наф-
тола из соответственных сульфокислот нафталина максимум 92—
93% от теоретически рассчитанного 8). Здесь вероятно в сниже-
169
нии выхода играет роль побочная реакция превращения продукта
присоединения в углеводород и сульфат, например для нафтола
Ч-NaOH
SO3Na
SO3Na
— Na2SO4 ~*
Нельзя сказать с уверенностью, происходит ли побочная реак-
ция за счет именно этого продукта присоединения NaOH или
такого, где ONa присоединен к соседнему а-атому
NaO Н
SO3Na \/ SO3Na
Ввиду особенной структуры продукта присоединения и нали-
чия в нем ненасыщенных групп можно ожидать, что он легко
будет отзываться на различные реакционные воздействия, в част-
ности на окислительные, в процессе плавки.
Относительная стойкость отдельных сульфокислот ароматического ряда в про-
цессе щелочного плава не была подвергнута, насколько нам известно, сравнительному
исследованию. Известно из недавней американской работы, что сульфокислоты
жирвого ряда различно стойки по отношению к нагреванию с раствором (4/V) едкого
натра до 315—375°. Наиболее стойки метилсульфокислота, бензилсульфокислота.
Вычисленные теплоты активации сульфокислот с остатками СН3-, С6НБСН2-, С6НБ-
соответствеино таковы: 54 700, 62 500, 29300 е).
В рассматриваемом нами превращении вообще участвуют два
главных вещества: соль сульфокислоты (сульфонат) и едкая ще-
лочь. Для успеха реакции необходимо, чтобы и та и другая были
по возможности чисты и не заключали примесей. Соль сульфо-
кислоты должна быть по возможности свободна от минеральных
солей, таких, как поваренная соль или сульфат натрия, тем более
она не должна обладать кислой реакцией. Наличие значительного
количества минеральных солей вследствие их нерастворимости
в расплавленной щелочи является причиной образование комков
в плаве, понижает его подвижность и делает поэтому возможным
и местные перегревы всей массы плава, что при высокой темпера-
туре стенок реакционного сосуда (внутри него температура под-
держивается часто выше 300°) может повести к подгоранию и даже
настоящему горению всей массы.
Кислотность же назначенной к плавлению сульфосоли, вообще
зависящая от присутствия свободной минеральной кислоты,
является невыгодной двояко: во-первых, она приводит к беспо-
лезной затрате излишнего количества щелочи из той, которая
участвует в качестве реагента при щелочном плавлении, и, во-вто-
170
рых, в результате нейтрализации этой свободной кислоты щело-
чью образуется добавочное количество минеральных солей, кото-
рые, как только-что сказано, вредны как примесь к плаву. Однако
незначительное количество минеральных солей (приблизительно до
10% по весу сульфоната) не может отозваться вредно на успехе
процесса при условии хорошего размешивания плава.
Что касается едкой щелочи, считается важным, чтобы она не
заключала заметных количеств хлоратов (СЮ'), могущих сохра-
ниться как примесь при электролитическом получении едкого
натра, соотв. едкого кали, из хлористых солей. Считается, что
ввиду большой реакционности фенолов по отношению к окисли-
телям примесь хлората в плаве может повести к осложнениям
(вплоть до взрыва) в условиях проведения щелочного плавления.
Нам кажется тем не менее, что, для того чтобы быть уверенным в правильно-
сти этого мнения, следовало бы провести лабораторное исследование в направле-
нии выяснения максимально допустимого количества хлората в плаве и тех отли-
чий от нормального течения, которые эта примесь вызывает. Впрочем обычно
полученная электролизом, упаренная и сплавленная твердая щелочь заключает очень
небольшую примесь хлората и не вызывает осложнений при применении.
Согласно уравнению (1) в плаве должно быть 2 мол. едкой
щелочи на каждую сульфогруппу сульфоната. Это — теоретически
мыслимый предел, к которому и стараются приблизиться в произ-
водственной практике. В лаборатории, при малых масштабах ра-
боты, редко удается провести плав с близким к теоретически
требуемому количеством щелочи, здесь применяют для достиже-
ния равномерной консистенции плава 3 мол. щелочи и даже выше.
В производстве, где экономические соображения имеют существен-
ное значение, необходимо стремиться к предельно малому расходу
щелочи. Успех здесь возможен при условии хорошего качества
сульфоната, рационально сконструированного аппарата и тща-
тельного наблюдения за ходом процесса.
В некоторых примерах щелочного плавления даже при всех
прочих благоприятных условиях необходим некоторый избыток
щелочи сверх теоретически рассчитанного для сохранения выхода
на достаточном уровне.
Для того чтобы создать для плава большую подвижность при
температуре более низкой, чем отвечающая плавлению безводной
щелочи (едкий натр плавится при 318°, едкое кали при 360°),
к щелочи добавляют некоторое количество воды (5—10%) и, полу-
чив жидкий плав при нагревании до более низкой температуры,
чем с безводной щелочью (с едким натром например при 275—
280°), начинают вносить сульфонат в плав при размешивании.
Вода постепенно выпаривается, но плав, раз став подвижным,
уже легко размешивается, тем более, что выделяющиеся пары
воды, как ранее введенной, так и образующейся при реакции,
также несколько размешивают густую массу.
Сульфонат вводят постепенно, порциями, или в виде сухой соли
или в виде насыщенного водного раствора, как например при
,.жидком плаве“ для получения фенола. Наблюдено, что введение
раствора сульфоната вместо твердой соли, которая во всяком слу-
чае должна быть в состоянии возможно мелкого порошка, имеет
171
известные преимущества в смысле большей „гладкости" протека-
ния процесса, отсутствии вспенивания и пыления. Возможно, что
и здесь быстрое образование пара в массе содействует поддержа-
нию однородности ее состава i°).
По окончании введения всего сульфоната, причем температура
поддерживается обычно не на самом высоком уровне, установлен-
ном заранее для процесса, продолжая размешивание, реакционный
аппарат подогревают некоторое время (’/8 часа—1 час), доводя
температуру до нужной максимальной (например 310—320°). После
этого плав готов; он состоит чаще всего из некоторого избытка
свободной щелочи, сульфита и фенолята, может быть части остав-
шегося продукта присоединения (см. стр. 167), который окон-
чательно превращается в фенолят уже при обработке плава
водой.
б) Автоклавные плавы. Известковые плавы под давлением
Современная практика дала уже много примеров перехода от
такого „открытого" способа сплавления со щелочами к „закры-
тому" или „автоклавному" методу. Вода, по крайней мере когда
ее масса не велика, не вредит реакции, между тем присутствие
воды позволяет поддерживать щелочь, так же как фенолят и суль-
фит, в растворе. Следовательно если применением закрытого аппа-
рата (автоклава), выдерживающего достаточное давление, можно-
добиться задержания воды в массе при температуре, значительно
выше температуры ее кипения, то нужную для реакции темпера-
туру можно получить с сохранением хорошей подвижности и го-
могенности реакционной массы при расходе щелочи более близ-
ком к рассчитанному по теории количеству.
Автоклавный метод работы чаще всего применяется в тех слу-
чаях, когда в полисульфонате, т. е. соли полисульфокислоты,
нужно заменить на гидроксил одну, а не все сульфогруппы, и где
плавление в открытом аппарате при слишком энергичном воздей-
ствии могло бы повести к получению неоднородного продукта.
Таковы например превращения:
SO3Na ONa
NaO3S
NaOgS
172
Также проводится в автоклаве „окислительный" ализариновый
плав р-сульфокислоты антрахинона (в ализарин), о котором выше
было уже упомянуто.
Возможно, что и в случае одиночно стоящей сульфогруппы
работа в автоклаве окажется не менее, а может быть и более
удобной, чем работа в открытом аппарате.
В частности Фр. Вилльсон и К. Мейер установили, что при
нагревании до 300° уже с 10%-ным раствором едкого натра моно-
сульфокислоты нафталина очень гладко переходят в соответствен-
ные нафтолы, очень чистые и свободные от продуктов окисле-
ния п).
В бензольном ряду названные исследователи наблюдали не-
сколько меньшую наклонность сульфогруппы замещаться гидро-
ксилом от действия разведенной щелочи при высокой температуре
и давлении, причем из наблюдаемых ими фактов они выводят
заключение, что при этих гидролизах конкурируют две реакции:
расщепление сульфокислоты на углеводород и сульфат, с одной
стороны, и с другой — превращение в фенол и сульфит. В боль-
шинстве процессов становится заметным лишь второе течение
реакции (особенно с сульфокислотами нафталина), но например п-
и о-сульфокислоты фенола превращались при их опытах в чистый
фенол (первое течение реакции):
ОН
+ NaOH
Na2SO4
NaO^4^
4-NaOH
— Na2SO4
При применении разбавленной щелочи нельзя считать исклю-
ченной и возможность чисто гидролизующего ее влияния в силу
ионного расщепления молекулы, например Na ... ОН, и соответ-
ственно этому следующего протекания реакции:
+ NaHSO4;
NaHSO4 + NaOH = Na2SO4 H,O.
С подобным превращением сульфокислоты в углеводород и
сульфат мы уже встретились как с побочной реакцией при обыч-
ном получении фенольного производного (нафтола). Неизвестно,
имеет ли место при гидролизе влияние со стороны металла стенок
автоклава.
173
В новой патежной литературе имеются указания на возможность использова-
ния автоклавного метода работы в применении к получению фенола нз сульфр-
кнслоты бензола. Так, американцы ГэлнБрнттон, о работе которых по полу-
чению фенола из хлорбензола будет подробно сказано ниже, заявили недавно
патент на получение фенола нз натриевой солн бензолсульфокислоты нагреванием
ее в течение % часа до 300—375° в автоклаве вместе с водными растворами едкого
натра илн едкого кали или гидроокиси щелочноземельного металла, лучше в при-
сутствии окнси меди, причем в смесь, подобно как и прн превращении хлорбен-
зола в фенол, добавляют 10% дифенилового эфира, собирающегося в этом же
количестве после реакции 12).
Также в закрытом аппарате, но в присутствии фенола и инднферентного газа
и при пониженном давлении (!) рекомендует вести плавку бензолмоносульфоната
другой американский патент 13).
Разновидностью автоклавных плавов являются так называемые
„известковые плавы под давлением" (Kalkdruckschmelzen).
Здесь в качестве щелочного агента применяется известковое
молоко, вместе с которым нагревается известковая соль сульфо-
кислоты. Исключительная пока область применения известковых
плавов —получение оксипроизводных антрахинонового ряда с за-
меной только сульфогруппы на гидроксил, но без вступления
новой оксигруппы в свободное от сульфогруппы место. Этот ме-
тод работы был введен М. А. Ильинским и описан в патентах
немецких красочных заводов в Гехсте 14).
Отличие в поведении гидроокиси щелочноземельного металла
сравнительно с щелочной гидроокисью весьма вероятно обусло-
вливается не только разной степенью щелочности (концентрации
ОН-ионов), но и возможностью для едкой щелочи, как мы видели,
своеобразно окислительно реагировать, между тем как щелочно-
земельная гидроокись реагирует предположительно лишь как
гидролизующий агент, т. е. содействует отнятию элементов сер-
нистой кислоты при происходящем от действия воды гидролизе
Следовательно схема течения реакции например с р-сульфокисло-
той антрахинона здесь была бы такова:
+ CaSO3+H.2O.
Подобно этому также гидролитическим лишь влиянием обусловливается благо-
приятный эффект применения гидроокисей щелочноземельных металлов при полу-
чении диоксипроизводных из п- и о-дихлорбензолов (см. следующую главу).
174
Известковые плавы под давлением употребительны
в следующих технически интересных превращениях 15):
например
Тогда когда исходным материалом для реакции берется не кальциевая, а натрие-
вая (нли калиевая; соль сульфокислоты антрахинона, даже н прн применении ще-
лочноземельной гидроокиси не исключается образование нежелательных вторичных
продуктов обработки: при взаимодействии гидроокиси кальция с образовавшимся
сульфитом щелочного металла образуется едкая щелочь, которая содействует выше-
указанному образованию продуктов восстановления антрахиноновых производных.
Для того чтобы воспрепятствовать этим осложнениям процесса, предложено
прибавлять к смеси хлористый кальций и окислитель, причем, несмотря на присут-
ствие последнего, прн известковой обработке имеет место только обмен сульфа-
группы на гндрокснл, но не образование добавочного водного остатка 1е).
Относительная легкость осуществления процесса сплавления со
щелочами зависит в значительной мере от большей или меньшей
подвижности (лабильности) сульфогруппы или нескольких сульфо-
групп в соединении и характеризуется прежде всего предельной
температурой превращения соответственной сульфогруппы. В бен-
зольном ряду сульфогруппа сравнительно мало подвижна, темпе-
ратура реакции весьма высока, свыше 300°. В нафталиновом ряду
а-сульфокислоты замещаются с большей легкостью, — сульфо-
группа лабильна,—температура плава для замещения сульфогруппы
сравнительно низка; р-сульфокислоты нафталинового ряда обла-
дают менее лабильной сульфогруппон. Температура реакции дохо-
дит до 300° и выше. Зная это, можно выбирать для случаев поли-
сульфокислот нафталина такую температуру плава, чтобы замести-
лась только одна из нескольких сульфогрупп гидроксилом. При
замещении нескольких сульфогрупп, даже стоящих в благоприят-
ном положении, температуру приходится несколько поднимать.
Таким образом не только сульфокислоты нафталина, но и сульфо-
кислоты нафтиламинов, соотв. нафтолов, оказываются излюблен-
ным материалом для получения аминонафтолов, соотв. полиокси-
дериватов нафталина и их сульфокислот, причем в руках знающего
экспериментатора управление ходом реакции совершается с боль-
шой легкостью, главным образом регулированием температуры
175
плава, часгью концентрации щелочи и, как выше было сказано,
выбором подходящей для реакции щелочи. В некоторых случаях
сплавления нафтиламиновых сульфокислот необходимо учитывать
возможность гидролиза аминогруппы с отщеплением ее в виде NH3
и замещением на ОН. Особенно легко подвижна NH2-rpynna в 4-м
положении к сульфогруппе; аминогруппа, стоящая в 8-м (пери)
месте по отношеню к а-сульфогруппе, обычно не легко сменяется
на ОН при обработке щелочью 17). Обработка методом щелочного
плавления нитросульфокислот не практикуется. Повидимому при
ней можно ожидать весьма сложного течения реакции с образова-
нием продуктов окисления за счет нитрогруппы.
в) Производственная практика плавления в синтезах фенола,
^-нафтола, резорцина и других полизамещенных
В производстве сплавление со щелочами производится, как
сказано уже, или в открытых котлах, или в автоклавах. Первые—
чугунные, обычно слегка конические, невысокие котлы с округ-
ленным дном и энергично работающей мешалкой, по возможности
близко отвечающей форме стенок и дня и оставляющей минималь-
ный йромежуток до стенок, так чтобы на стенках и на дне не
задерживалось значительного слоя плава, могущего подвергнуться
перегреву. Сверху во время работы котлы обязательно должны
быть закрыты крышкой с отверстиями для введения загрузки
сульфоната и взятия пробы плава в различные моменты процесса.
Котел обогревается посредством или печного или газового нагрева.
Обогрев производят также при помощи металлической или
масляной бани. За границей в ходу чугунные котлы с залитыми
в чугунных стенках стальными трубами, по которым циркулирует
перегретая вода (система обогрева Фредеркинга).
Автоклавы (см. рис. 8,. стр. 230) для проведения сплавления со
щелочами бывают или вертикальными или горизонтальными. Исклю-
чительно последняя система применяется для ализаринового плава.
Здесь применяют вполне цилиндрический, гладкий внутри (без
заклепок) стальной или железный котел с мешалкой по оси его.
Лопасти мешалки почти касаются стенок котла, что облегчает
соскребание со стенок котла густой и вязкой массы, а следова-
тельно и перемешивание. Очень ответственной частью аппарата
является сальник при месте входа оси мешалки в крышку авто-
клава. Большинство современных автоклавов делаются стальными.
Ниже приведем несколько примеров наиболее широко приме-
няемых в технике щелочных плавлений и именно связанных
с производством фенола, fi-нафтола и резорцина.
С целью понижения расходных коэфициентов материалов
в производстве, получение одноатомных фенолов комбинируют
вместе с производством исходной сульфокислоты таким образом,
чтобы отходы одного цеха служили вспомогательными материа-
лами для другого.
Таким образом для нейтрализации сульфомассы, соотв. выса-
ливания из нее сульфокислой соли, можно пользоваться сульфитом
натрия, получающимся в таком же молекулярном количестве, как
376
и фенолят при плаве, а выделяющийся при действии на сульфит
кислой сульфомассы сернистый ангидрид
(2RSO3H 4- Na2SO3 = 2RSO3Na + Н2О -f- SO2)
применять как кислотный агент для разложения щелочного фено-
лята и выделения из него фенола.
Плавка моносульфоната бензола на фенол была обстоятельно
изучена американцами Родсом и сотрудниками7).
Рнс. 7. Котел для щелочного плавления.
При прибавлеини сульфоиата к плавленному едкому натру прн 300° сначала
не заметно реакционного воздействия, а затем наступает быстрая реакция со вспени-
ванием от выделяющихся паров воды., что согласуется с развитым выше взглядом
на двухфазное протекание реакции. Прн температурах в 300 и 320° н 30 мин.
выдержки (после прибавления всего сульфоната) оптимальный выход в 95 и 96%
от теории фенола получен прн 50%-ном избытке щелочи против расчета; при 350“
достаточно для этого уже 15%-ного избытка щелочи и 15 мин. выдержки, т. е.
Продолжительность плава уменьшается по мере повышения температуры. Более
длительное нагревание после достижения максимума превращения приводит к сни-
жению выхода.
На выходах не отражается заметно присутствие сульфата, если его не более
чем 10% от сульфоната.
Если под крышку котла во время плавки пропускать водяной пар, то последний
создает предохраняющую От окисления атмосферу над плавом. При пропускании
12 Зак. 2611. — Н. Ворожцов 177
место «ара воздуха наблюдалось побурение плава н даже сильное обугливание
при повышенной температуре плава. В этой случае в продукте появляются белее
высококипящие сравнительно с фенолом соединения, главным образом дифенолы:
ОН ОН
получающиеся в результате окисления.
При более жестких условиях плава процесс окисления может нттн дальше
образования дифенолов с выделением углекислого газа, частью нейтрализующегося
едкой щелочью. Отсюда вывод для производственной работы: необходимо котел для
плавления держать закрытым, чтобы образующийся при плавлении водяной пар мог
собираться под крышкой и тем мешать развитию окислительных процессов за счет
кислорода могущего проникнуть воздуха. Размешивание должно быть достаточным,
иначе возможно появление в массе таких участков, где сульфонат будет преобла-
дать над щелочью. В подобных условиях наблюдается появление в продукте дифе-
нилового эфира, очевидно вследствие реакции:
CeH5ONa 4- NaO3SC6H5 = C6H5OCgH5 + Na3SO3
и некоторого количества тиофенола CrHsSH.
Образование тиофенола происходит вследствие перегревания непрореагнроваи-
шего сульфоната, почему важно прибавление его вести постепенно и избегать ком-
ков. Возможно, что восстановление сульфогруппы в сульфгидрильную SH облегчается
действием иа сульфонат горячих железных стеиок 18).
Присутствие тнофенола из-за его резкого и неприятного запаха даже при малой
примеси его к фенолу сильно портит продукт. Прибавление к щелочи сульфоната
в растворе, о чем ранее было упомянуто, затрудняет образование тиофеиола.
Контроль взаимодействия при щелочном плавлении очень важен
для наблюдения течения процесса. Обычно для этого достаточно
определения свободной щелочи и фенола. Последний определяется
в виде трибромфенола или как таковой или титрованием раствором
соли бромноватой кислоты (КВгО3) в кислом растворе, заключаю-
щем бромистую соль (КВг)19), или титрованием иодом (образование
трииодфенола).
Плав, полученный после конца операции, еще в жидком состоя-
нии вносится в воду в аппарате, носящем название гасителя, где
после некоторого охлаждения можно наблюдать разделение на два
слоя: внизу насыщенный раствор сульфита натрия, вверху — фено-
лят. Эти слои могут быть разделены. Сульфит, после того как он
кристаллизуется, может быть употреблен, как сказано, для ней-
трализации кислот сульфомассы, фенолят же подкислен, до выде-
ления фенода в свободном состоянии, образующимся сернистым
ангидридом.
Выделенный фенол очищается перегонкой. Водный слой содер-
жит в растворе фенол и может быть использован.
По одному американскому патенту (1931 г.) фенол из кислого раствора выду-
вается паром, идущим навстречу раствора через башню, наполненную кольцами
Раш ига. Улавливается фенол раствором шелочн, откуда выделяется посредством
подкисления раствора фенолята сернистым газом.
В патентной литературе можно иайти указания на отличающиеся от описанных
выше составы реакционной смеси для сплавления сульфокислот со щелочами. Так,
по американскому патенту 1924 г. применяют для сплавление с сульфонатами коли-
чества щелочи большие, чем необходимо по расчету реакции. По растворении
плава и достаточном количестве воды выпадают минеральные соли (NagSOg), в раствор
178
переходят фенолят и избыток щелочи. Этот растррр после сгущения пускается вновь
в плав вместо свежей щелочи, причем процесс повторяется до использования всей
свободной щелочи. Способ предлсвкен для получения фенола, нафтола и резорцина,
во не имеет никаких преимуществ перед обычным, обладая в то же время суще-
ственным недостатком, как связанный с лишним расходом тема на сгущение щелоч-
ного раствора и как могущий привести к образованию примесей в продукте из-за
более продолжительного нагревания готового фенолята с сульфонатом 2°).
По другому амервканскому натенту (1931 г.) песле нормального сплавления
беизолсульфоната натрия с едким натром при 300—340® прибавляют свободного
фенола очевидно для связывания свободной шелочи плава, причем якобы в таких
условиях плав резче делится иа слои сульфита и фенолята. Способ вряд ли может
иметь технический интерес 21).
Более обоснован с химической стороны прием сплавления с вве-
дением в плав индиферентного растворителя, например высоко-
кипящих фракций нефтяных углеводородов (330—360°), и нагрева-
нием плава в открытом аппарате — с обратным холодильником.
Индиферентный растворитель мешает возникновению окислительных
процессов, содействует лучшему размешиванию и регулированию
температуры реакционной массы.
Применение нейтрального растворителя запатентовано вместе с этим в приме-
нении к ряду других процессов, где используется действие щелочи при высокой
температуре (превращение n-дихлорбеизола с амидом натрия в диамин, плавка
Р-амнноаитрахинона иа иидантрен) &).
В частном случае получения фенолов из сульфокислот. защити-
тельный эффект против окисления может быть достигнут и без
применения вового ингредиента смеси в виде растворителя, как
выше уже было сказано.
Действие прибавки окислителя, именно перекиси свинца РЬО2, при щелочной
плавке сульфокислот бензольных производных было испытано Гребен Краф-
том 23). Сульфокислоты гомологов бензола дают при этом карбоновые кислоты
(сульфокислоты толуола — бензойную), лишенные сульфо- (соотн. оксн-) группы.
Между тем как сульфобензойиые кислоты при обычной щелочной плавке переходит
в оксибензойиые кислоты, крезолы при окислительной плавке (РЬО^ образуют
также оксибензойвые кислоты,—окислительная плавка сульфокислот гомологе? бен-
зола приводит только к карбоновым кислотам. Сплавление со щелочью например
о-сульфокислоты бензилового спирта
СН2ОН
\(
^^SOgH
при 220—240® дает бензойную кислоту. Очевидно отщепляющий сульфогруплу
процесс, подобный рассмотренному в начале этой главы, обусловливается напра-
вляющим влиянием образующегося сперва от окисления группы СН3 заместителя
СН2ОН, Интересно отметить, что Гребе и Крафт получили прн реакции в усло-
виях окислительного плава (КОН РЪО2) из о-крезола (при 210—220°) с количе-
ственным почти выходом салициловую кислоту. Прн такой же плавке фенола (250°)
обнаружено небольшое количество салициловой кислоты. Очевидно в последнем
случае имело место окислительное расщевленне, наблюденное позже Родсом и
сотрудниками (см. выше); образующийся при этом углекислый газ реагировал
с частью еще не затронутого окислением фенолята, образуя салициловую кислоту.
Плавка нафталин-р-сульфоната со щелочью в общем похожа
на плавку сульфоната бензола. Чаше всего сульфонат вводится су-
хим, следовательно нужно, чтобы он был возможно более мелко
раздроблен. Также вредно отзывается превосходящая допустимый
12*
179
предел примесь минеральных солей. Так как р-сульфонат полу-
чается в твердом состоянии, а не в растворе, как сульфонат бен-
зола, то следовательно нужно обращать внимание на достаточную
его промывку перед пуском в плав. Выше было упомянуто, что
выход при плаве сульфокислоты нафталина, достижимый в опти-
мальных условиях, составляет 92—93°/о от теории, следовательно
заметная часть сульфокислоты в процессе плава претерпевает иное
превращение (см. выше).
Контроль за плавом приблизительно такой же, как и при фе-
нольной плавке. Определяется избыток свободной щелочи и наф-
тол; последний обычно иодометрически. Готовый р-нафтол харак-
теризуется методом азосочетания (о нем см. ниже).
Окислительные влияния ввиду более реакционного характера нафталинового
ядра здесь могут быть еще более значительными, чем при работе с бензольным
производным. Повидимому, как вытекает из опытов автора, Н. Е. Благове-
щенского н Е. К. Безнер, имеет известное значение у материал плавиль-
ного котла: в железном аппарате выходы fl-нафтола меньше, чем в чугунном; потеря
относится на счет образования труднэрастворимых продуктов (от окисления, соотв.
конденсации).
Разложение нафтолята практичнее всего производить подкис-
лением сернистым газом.
Промытый от минеральных солей р-нафтол обычно очищается
окончательно посредством перегонки в вакууме или с перегретым
паром. Первый способ практичнее при условии достаточного осво-
бождения сырого продукта от минеральных примесей, могущих
обусловить при высокой температуре в перегонном аппарате обра-
зование из р-нафтола продуктов ангидризации, например динаф-
тильного эфира24). Возможно, что и в этом случае окажется прак-
тически интересным метод кондестилляции с нефтяным маслом,
разработанный И. Я- Гришиным.
а-Нафтол готовится обычно нз а-нафтнламина, а не из а-сульфокислоты наф-
талина. В американской практике повидимому имеет место применение н метода
щелочного плавления. Так как а-сульфокислоту трудно получить без примеси
fl-сульфокислоты нафталина, то сырой а-нафгол содержит значительное количество
fl-нзомера (до 20%). В одном американском патенте предложено отделять ^-нафтол
в этом случае кратковременным иагреваннем сырого продукта с известковым моло-
ком. При этот fl-нафтол дает нерастворимое соединение с известью, а в фильтрат
переходит известковое соединение а-нафтола, подкислением которого получают
чистый а-иафтол25).
Плавка .и-дисульфокнслоты бензола на резорцин была
изучена Филлипсом и Гиббсом26).
Оптимальные услозия из нх работы: продолжительность плава 2 часа при 310°
н соотношении 14—16 мол. едкого натра на 1 мол. днсульфоната беизола обусло-
вливают выход резорцина в 63—65%. Присутствие воды в плаве понижает выход.
В производстве этот пример сплавления со щелочью оказы-
вается одним из наиболее трудных по выполнению ввиду чрезвы-
чайной реакционности продукта реакции — резорцина и необходи-
мости, связанной с этим, по возможности быстро выводить из
сферы высокой температуры готовый продукт. Повидимому в этом
и подобных случаях (например получении 1.5-диоксинафталина из
1.5-дисульфокислоты нафталина) выгоднее проводить процесс по
возможности с малыми количествами материала, работать в не-
180
больших аппаратах. Как на предельное выражение тенденции
к уменьшению размера плавильного котла укажем, что на неко-
торых заграничных фабриках достаточно однородная смесь из
дисульфоната бензола и едкой щелочи загружается в чугунные
трубки, имеющие дно на одном конце и плотную крышку на
другом, и эти трубки помещаются в печь, где поддерживается
нужная для реакции температура. Это — способ „запекания" плава.
Здесь благодаря незначительной толщине нагреваемого слоя массы
устраняется необходимость в размешивании.
Подобный только-что описанному прием сплавления приводится
в американском патенте (1928 г). Бензолдисульфонат натрия в вод-
ном растворе смешивается с водным же раствором едкого натра,
которого берут с 15 — 25%-ным избытком против требуемого
количества (4 мол.). Смесь выпаривается затем в плоском чане
(глубина слоя раствора вначале 15 см), и полученный твердый
слой (около 4 см толщины) подвергается дальнейшему нагреванию
в металлической бане до нужной температуры (в металлической
бане поддерживается температура в 375 — 400°) 27). Наличие боль-
шого открытого сверху слоя, где могут иметь место окислитель-
ные процессы, не дает преимущества этому способу сравнительно
с применением трубок.
Запекание в тонком слое с пропуском сверху водяного пара является практич-
ным видонзмененнем этого метода (Н. Г. Лаптев).
Резорцин в отличие от фенола и р-нафтола очень хорошо рас-
творим в воде и водных растворах солей. Поэтому его приходится
извлекать после „гашения" плава из водного раствора с примене-
нием растворителей. Наиболее употребительным растворителем
резорцина в производстве является амиловый спирт, в лаборато-
рии— эфир.
Современная тенденция к работе непрерывным процессом
в связи с возможностью во многих случаях щелочного плавления
применять водные растворы щелочи делает вероятным, что непре-
рывный процесс щелочного плавления со временем сделается упо-
требительным приемом. Очевидно это будет связано с введением
в работу трубчатых систем, выдерживающих давление изнутри,
по которым будет циркулировать нагретая щелочная смесь. В аме-
риканском патенте (1917 г.) уже описана подобная схема работы28).
Сульфокислоты аминов, в частности полисульфокислоты
нафтиламино в, могут быть подвергнуты щелочному плавлению
для получения соединений с окси- и аминогруппой, если надо—
с сохранением некоторых сульфогрупп, или в открытых аппаратах
или н автоклавах. Выбор при прочих равных условиях зависит от
нужной температуры, относительной реакционности сульфо- и под-
вижности аминогруппы.
При наличии особенно подвижной перисульфогруппы при сплавлении амнно-
нафталннсульфокислот с целью избежать гидролиза а-ПН2-группы американский
патент рекомендует проведение плава под давлением аммиака29).
Замечательно ведут себя прн реакции со щелочами арилсульфаминовые кислоты
строения RNHSOjH (о них см. ниже в главе об ацилировании). Будучи очень
нестойкими по отношению к кислотам, от действия которых оин расщепляются гидро-
литически иа RNHa и H2SO4, они выдерживают, сохраняя связь атомов N и S, обра-
ботку щелочами н даже щелочное плавление.
181
Подобно сулвфаминовым кисдотам выдерживают без расщепления реакцию
щелочного плава сульфонамидные производные ароматических углеводородов. Таким
образом возможно по описанию патента например превращение (действием едкого
кали при 140—160°)
HOsS NHOaSCgHa НО NHO^SCgHg
HOsS^/k^SOaH HOgS^/L^SOsH
Выделение аминооксисоединений из плава, если они заключают
сульфогруппу или сульфогруппы, удается чаще подкислением или
высаливанием. При отсутствии же в соединении сульфогрупп выде-
ление связано нередко с применением растворителя. А. Е. Порай-
Кошиц предлагает облегчить выделение, вводя в водный раствор
плава соль аммония [(NH4)2SO4 или NH4CO2CHS]31). В слабоаммиач-
ных растворах аминооксисоединения менее растворимы, чем в рас-
творах щелочей.
г) Характеристика оксисоединений
В преобладающем числе случаев щелочное плавление дает нача-
ло оксигруппе, которая способна реактивировать азосочетание —
образование при взаимодействии с диазониями характерных окра-
шенных оксиазосоединений, т. е., иначе говоря, сплавление со
щелочами образует „азосоставляющие" красителей. На этом свой-
стве основан общий способ определения, как качественного, так
и количественного, гидроксильной группы. Если в соединении нет
других реактивирующих азосочетание групп [например NH2, NHAr,
N(Alk)2 и пр. у аминонафтолов и их замещенных], то тогда каче-
ственное определение сводится к наблюдению, появляется ли интен-
сивная окраска, обычно в оранжевый до красного тон, при слива-
нии раствора испытуемого соединения и соды с раствором доста-
точно удобной для приготовления диазониевой соли. В качестве
таких солей пользуются или хлористым фенилдиазонием (из ани-
лина) или хлористым n-ннтрофенилдиазонием (из п-нитроанилина).
Последний предпочитают ввиду его относительной стойкости.
Раствор диазониавой соли не должен заключать свободной мине-
ральной кислоты, исключаемой прибавлением уксуснокислого
натрия, так чтобы при смачивании Конго-бумажки употребляемым
раствором она оставалась красной. Для количественного анализа
ароматических оксисоединений применяют раствор хлористого
n-нитрофенилдиазония определенного титра. Для приготовления
его можно пользоваться таким приемом:
14 г п-нитроанилина растворить при нагревании в 25 см? кон-
центрированной соляной кислоты (приблизительно 2,5 мол. НС1
на 1 мол. амина) и 200 см? воды. Вылить раствор в размолотый
на мелкие куски лед. По охлаждении в раствор с осадком хло-
ристоводородной соли всыпать 7 г 100°/о-ного сухого нитрита нат-
рия NaNOa или соответственно большее количество нитрита, менее
высокопроцентного. Всыпание производить быстро при энергичном
размешивании жидкости, которую по окончании реакции (осветление,
исчезновение осадка) поставить добавлением воды на 1 л и про-
182
фильтровать. Раствор сохранять »а холоду в месте, закрытом от
прямого света. При этих условиях титр может считаться постоян-
ным в течение дяя.
Титр диазораствора устанавливается по химически чистому
р-иафтолу с t° пл. 1223. Отвесив 1,44 г р-нафтола ( = 0<01 мол.),
растворяют эту навеску в воде с прибавлением необходимого коли-
чества едкого натра, доводя раствор до 100 см3 ( = 0,1 N). Беря
от этого раствора по 20 см3, разводят его в стакане 600 — 700 см3
воды и слабо подкисляют уксусной (или соляной) кислотой, при-
бавляют 5 — 10 г уксуснокислого или двууглекислого натрия и при-
ливают из бюретки раствор соли диаэония, пока „проба на вытек*
перестанет давать ясные результаты.
Для производства пробы берут стеклянной палочкой каплю рас-
твора с взмученным в ней осадком азокрасителя и помещают ее
на фильтровальную бумагу: в середине образуется красное пятно
от нерастворимого красителя, около которого кругом заметен
достаточно широкий ореол — вытек бесцветного или чуть окрашен-
ного раствора. В этом растворе может быть избыток либо диазо-
составляющей, в данном случае n-нитрофенилдиазония, либо азосо-
ставляющей, т. е. здесь р-нафтола. Если мы возьмем далее чистой
палочкой капельку содового раствора соли R, иногда раствора кис-
лоты Н, дающей белее темное окрашивание, или раствор быстро
реагирующего резорцина и опустим ее на нашу бумажку на сухое
место вблизи вытека, то, растекаясь, эта капля дойдет до первого
вытека, и, если там был избыток диазония, он, реагируя с солью R,
соотв. другими азосоставляющими, даст красный мениск от образо-
вавшегося азокрасителя. Если эта проба указала на отсутствие
избытка диазония, то следует испытать, нет ли избытка нафтола.
Для этого капелька раствора диазония наносится вблизи первого
вытека, и, если окраска в месте соприкосновения первого и нового
вытеков не образуется, значит р-нафтол не в избытке. При установ-
лении титра для большей верности в случае даже отрицательных
результатов пробы на вытек необходимо увериться в действитель-
ности конца реакции. Поэтому в пробирку отфильтровывают
2—3 см3 титруемой жидкости, делят фильтрат на две части и одну
обрабатывают диазораствором, а другую нейтральным раствором
соли R (в обоих случаях, понятно, прибавление идет каплями).
Если и та и другая пробы при этом не окрасятся, это служит при-
знаком конца реакции. В противном случае будет видно, избыто»
какого реагента имеет место.
При титровании нафтола солью диазония краситель обычно быстро коагули-
рует и препятствует этим сочетанию большей части адсорбированного осадком наф-
тола. Титрование затягивается надолго и не дает точных результатов, если ие при-
менять очень разведенного раствора нафтола или же не подогревать слегка рас-
твор с нафтолом после прибавления избытка диазония. Применение защитных колло-
идов (гуммиарабик, свежеприготовленный раствор желатины) помогает получить
хорошие результаты. Титрование ведут в небольшом объеме воды (100 —150 см-1
на 0,3 — 0,4 г нафтола). Титрование заканчивается в 15 — 20 мин. Избыток диазо-
ния легко и точно определится на бумажке (с высаливанием и лучше по реакции
с вытеком кислоты Н) (сообщение А. И. Королева).
С таким титрованным раствором диазония и ведут количествен-
ные определения оксисоедииений. Определенная навеска вещества
183
растворяется в таком количестве воды, чтобы образовать прнбли-
знтельно 0,1 — 0,05 7V раствор. От этого раствора берут для титро-
вания равные объемные части (20 — 25 см3), прибавляя к титру-
емому раствору двууглекислый натрий (3 — 5 г), и если образу-
емый краситель легко растворим, то поваренной соли перед взя-
тием пробы на вытек. Она производится по уже указанному выше
способу. Если краситель растворим настолько, что трудно полу-
чить бесцветный вытек обычным способом, иногда на бумажку
насыпают слой порошкообразной соли и на него капают уже ис-
следуемый раствор. Высаливание доканчивается тогда в порах
бумаги, и вытек становится бесцветным. Полезно применять бумаж-
ку, пропитанную раствором хлористого аммония и высушенную
(А. И. Короле в).
Применение диазо-п-нитроанилина недопустимо в случаях рез-
кой щелочной реакции в среде, где производится азосочетание:
даже в присутствии соды, не говоря уже об едком натре, это
дназосоединение претерпевает изменения (см. главу IX), н опреде-
ления становятся неточными.
Так как исследуемые окснсоединения имеют в большинстве
случаев назначение быть азосоставляющимн, то такой способ, кото-
рым можно количественно определить в нем гидроксильную группу,
является очень удобным для техники. Предпочтительно данные
анализа выражать, записывая молекулярный вес соединения, так
как технические продукты всегда более нли менее загрязнены
минеральными солями и количество молекул в одной и той же
навеске различных партий продукта может сильно колебаться.
Поэтому, указав, что молекулярный вес данной сульфокислоты
р-нафтола равен, положим, 290, мы сообщаем, что необходимо
взять 290 г этого вещества, чтобы при сочетании с 1 мол. амина
получить одну молекулу оксназокрасителя, безотносительно, явля-
ется ли эта сульфокислота в нашей пробе в свободном состоянии
нли в виде соли и какой именно соли.
Наконец тогда, когда анализу на содержание окснгруппы под-
вергается соединение, уже имеющее аминогруппу или группу из
ее замещенных, благоприятствующих реакционности соединения
по отношению к диазониям, например когда анализу подвергаются
моно-нли днсульфокислоты аминонафтолов, положим, кислота Н,
то определение гидроксила по азосочетанию, проведенное по выше-
описанному, может иногда возбуждать сомнения в точности резуль-
татов.
ОН NH2
I I
*7 2*
HO3SZ v v XSO3H
Наряду с образованием окснмоноазокрасителя (азогруппа в месте 7)
можно ожидать получения также днсазокрасителя (азогруппы в ме-
стах 7 н 2) и амнномоноазокраснтеля (азогруппа в месте 2).
184
Опытами установлено тем не менее, что сочетание в щелочной
среде, обусловленное реакционностью гидроксила, протекает с зна-
чительно большей скоростью, чем сочетание, вызываемое влиянием
аминогруппы (последнее в кислой среде), поэтому метод „щелоч-
ного* сочетания применяется уверенно для большинства определе-
ний амннонафтолсульфокислот, видоизменяясь иногда в отношении
условий.
Если в окснаминосоеднненнях подобного рода важно опреде-
лить аминогруппу, то это делается или методом диазотирования
или методом азосочетания в кислой среде. Практика титрования
совершенно такова же, как и в предшествовавшем случае, только
к титруемому раствору не прибавляют бикарбоната илн уксусно-
кислого натрия, но добавляют ацетат вместе с уксусной кислотой
(вес ацетата: вес уксусной кислоты — 1 :4) нлн в иных случаях при
наличия особенно реактивирующих азосочетание оксигрупп под-
кисляют минеральной кислотой до слабокислой реакции по бумажке
Конго (пример: кислота Н). При проведении титрования в кислой среде
азосочетанне останавливается на стадии амнномонодзокраснтеля.
Проба на вытек, уточненная, если понадобится, пробой фильтрата,
обнаруживает конец реакции. Полученный аминомоноазокраситель
может быть переведен, если в нем имеется ОН-группа, в днсазокрасн-
тель сочетанием с дназоннем в слабощелочной среде. Ввиду труд-
ности, работая с сильно окрашенными веществами, проследить ана-
литически точно конец реакции, обычно эта проба как метод опреде-
ления и не применяется. Если тем не менее необходимо, хотя бы н без
гарантий большой точности, провести количественно реакцию образо-
вания днсазокраснтеля, то полученный моноазокраситель, растворен-
ный в воде с прибавкой соды, подвергается обработке раствором
соли диазоння определенного содержания. Для прослеживания
процесса азосочетания берут пробу раствора до прибавления дназо-
ння и отбирают пробы по мере прибавления 1li0, 2/j0, и т. д. рас-
считанного количества диазония. Эти пробы высаливаются пова-
ренной солью и исследуются пробой на вытек, причем в первых
порциях только посредством обработки вытека минеральной кис-
лотой н щелочью, чтобы получить отчетливое представление об
изменениях цвета, как моноазо-, так и смеси моноазо- с дисазокра-
сителем. Прибавление последней порцнн днсазораствора требует
большой осторожности и сопровождается частыми пробами с об-
работкой вытека солью R, дабы избежать избытка непрореагнро-
вавшего диазоння. В практике производства красителей предпочи-
тают скорее прибавить немного меньше диазоння, чем больше, про-
тив требуемого количества.
Кроме диазония из n-ннтроанилина в лабораториях применяют
еще дназонин из анилина и n-толундина. Эти диазонии готовятся
не таким быстрым прибавлением солн азотистой кислоты к под-
кисленному раствору солн амина, как первый. Растворы диазоннев
менее прочны, чем n-ннтранилнновый, н часто нх титр изменяется
уже через 3 — 4 часа стояния, но зато они имеют перед первым
диазонием то преимущество, что менее чувствительны по отноше-
нию к щелочным растворам н потому применяются там, где диазо-
нз n-ннтранилина не дает удовлетворительных результатов. Дназо-
185
инй из анилина применяется например для определения 2-амино-5-
й#фтол-7-сульфокислоты (И кислоты), диазо- из n-толуидина для
определения оксигруппы в кислоте Н
По стойкости диазониев можно распределить амины в такой ряд:
п-нитранилин > п-толуидин > анилин 32).
При титровании бюретку с диазораствором лучше помещать
в более широкую стеклянную трубку, наполненную чистой ледяной
водой, к которой полезно прибавить немного раствора сафранина
(красного красителя) для предохранения от разложения светом.
Фенолы, аминофенолы и их производные, в особенности
сульфокислоты, имеют большое значение в синтезе красящих ве-
ществ. Простейший по строению фенол С6Н6ОН применяется
в производстве некоторых азокрасителей, служит исходным мате-
риалом для получения салициловой кислоты и аминофенольных
производных, для некоторых искусственных смол и пластических
материалов (бакелит) и некоторых фармацевтических препаратов.
р-Нафтол — очень важный промежуточный продукт в синтезе
азокрасителей, так же как и получаемые из него сульфокислоты и
особенно одна из его карбоновых кислот, именно 3-карбоновая
кислота (р-окси-3-нафтойная кислота) — исходный материал для
получения применяемого для холодного крашения нафтола AS и
его многочисленных аналогов. Производство р-нафтиламина и полу-
чаемых из него дисульфокислот 5.7 и 6.8 и производство двух
ценных сульфокислот аминонафтолов (7 и И) также пользуются
р-нафтолом как исходным веществом. Некоторые другие соедине-
ния, например применяемый как краситель нитрозо-р-нафтол,
некоторые оксазиновые красители и важный для протравных азо-
красителей промежуточный продукт, 1-амино-2-нафтол-4-сульфо-
кислота, получаются из р-нафтола. Кроме того р-нафтольные произ-
водные находят себе-применение в медицине.
Резорцин применяется как азосоставляющая в производстве
азокрасителей, в синтезе некоторых фталеиновых красителей (эозин),
применяется в медицине и в крашении мехов.
Многочисленные сульфокислоты а-нафтола и аминонафтольные
сульфокислоты служат почти исключительно для синтеза азокраси-
телей. Назовем из них как наиболее употребительные
ОН
HO3S
1-нафтол-5-сульфокислота (азуриновая кислота)
186
HO8S он
I ! Готовится без применения
метода щелочного плавления
из а-яафгйламин-3.8-дисуль-
фокиелоты (см. гл. Vlttf.
SO3H
1-нафтол-3.8-днсульфокнслота с-(эпснлон)-кислота
1-амняо-8-нафтол-4-сульфокнслота (S-кислота) (Кислота Чикаго S)
НО
HOgS^/X/
2-амино-3-нафтол-6-сульфокислота (т-кислота)
HO3S\/\/\zHH2
ОН
2-амино-5-нафтол-7-сульфокислота (Изо-гамма- илн И-кислота)
НО ЫН2
/\/\
III
HO3SZ\'/VXSO3H
1-амино-8-нафтол-3-6-днсульфокислота (Н-кнслота)
НО NHa
I 1 2
HOgS^./4/
SO3H
1-амиио-8-нафтол-4-6-дисульфокислота (К-кислота)
НО NH2
A/\zSO3H
SO8H
1-амино-8-вафтол-2-4-дйСульфокислота (2S- или Чикаго 25-кислота)
187
В ряду сульфокислот антрахинона щелочное плавление, по ска-
занному выше, дает или красители типа ализарина (с введением до-
бавочного гидроксила в свободном месте) или, при использовании
известкового плава под давлением, соединения, играющие роль про-
межуточных продуктов при образовании более сложно построен-
ных красителей.
д) Иные кроме плава методы образования оксигруппы и превра-
щения сульфогруппы
Гидроксильная группа входит в ароматическое ядро, как выше
было подробно выяснено, в результате замещения сульфогруппы.
Кроме этого наиболее широко применимого способа имеются для
частных случаев другие способы, например превращения МН2-группы,
обмен Cl-атома на ОН. Об этих методах сказано ниже, в своем
месте. Независимо от них существуют, правда, не все имеющие
значение для практики способы непосредственного ввода ОН-группы
в ароматическое ядро. Таковыми являются прежде всего электро-
химический метод окисления ароматических производных аз), ката-
литическое окисление углеводородов в парах 84) и затем метод
нагревания со щелочами соединений, заключающих реактивирую-
щую окислительную группу. Примеры такого окисления уже были
отмечены выше, при описании применения метода щелочного пла-
вления к ^-сульфокислотам антрахинона.
Здесь отметим, что обработка порошкообразными щелочами нитрссседннений
приводит к образованию иитрофенолов ®). Так, из нитробензола получается 0-нитро-
фенол, из нитронафталина—1.2-нитроиафтол
В этих последних примерах, где наряду с нвтрофенолами получаются азокси-
соединения (из 5 мол. нитросоедннения образуется 3 мол. нитрофенола и 1 мол.
азоксисоединения), вероятный первый продукт (I) всздейтвия щелочи на ннтрссос-
динение, например
ОН
подвергается окислению с устранением водорода со стороны питрогруппы самой
реагирующей молекулы.
Имеются уже литературные указания на возможность такого же окислительного
эффекта извне (кислород воздуха при содействии каталитически действующих оки-
слов элементов V и VI групп периодической системы).
Таким образом при нагревании до 320—400° бензола с 20%-ным раствором
едкого натра в присутствии окислов например U, V, Mo, W получен фенолят нат-
рия38). И здесь в виде первой фазы вероятно образование продукта присоединения типв
A A4U
+ NaOH
\Н
ХН
188
При действии воздуха в ирису гствви катализаторов происходит окисление во-
дорода в воду и стабилизация продукта в фенолят.
Сплавление со щелочами с целью получения гидроксильных со-
единений—одно из самых важных применений ароматических суль-
фокислот в технике производства промежуточных продуктов. Име-
ются тем не менее и другие приемы использования сульфокислот.
Прежде всего кажется вероятным ожидать превращения сульфо-
кислоты с заменой сульфогруппы на сульфгидрильную группу SH
при обработке сульфокислоты не едким натром, а его тиоаиалогом
(сульфгидратом), причем уравнение реакции было бы подобно (I)
RSO3Na + 2NaSH = RSNa Na2SO3 -f- H2S.
Опыты такого взаимодействия при высокой температуре и да-
влении с В-нафталинсульфокислотой привели частично к получению
Р-тионафтола, частично к получению смеси его дисульфида с р-наф-
толом. Сульфокислота и дисульфокислота бензола или не реагиро-
вали в автоклаве с сульфгидратом натрия или при более высокой
температуре образовывали сложные молекулы сернистых красите-
лей 87).
В ограниченном пока круге производных антрахинонового ряда
сульфокислоты оказываются способными к иному замещению своей
сульфогруппы, именно на аминогруппу. Для этого сульфокислота
или ее соль подвергаются нагреванию до высокой температуры
с водно-аммиачным раствором, что сопровождается, понятно, и вы-
соким давлением вследствие большой упругости пара аммиака.
Процесс, происходящий в этом случае, недостаточно освещен
исследованием. Возможно, что и тут взаимодействие начинается
с присоединения элементов аммиака к сульфокислоте, например:
и в последующей стадии стабилизация соединения в аминоантра-
хинон происходит при отщеплении элементов сернистой кислоты
ОН
Такое толкование реакции находит себе подтверждение в том,
что особенно успешно замещение сульфогруппы на аминогруппу
происходит в присутствии окислителей, добавленных в реакцион-
189
ную смесь, например нитросульфокислот, мышьяковой кислоты,
соли азотной кислоты (NH4NO3) и т. п. яв).
В согласии с таким навиванием хода реакции находится и описываемы* в не-
давнем немецком затейте фдог образования диаминоантрахинона ив р-амиярдятраде-
нома прн нагревании его с концентрированным аммиаком в закрытом аппарате
в присутствии окислителя (например NaNOa). Здесь реакция повидимому Проходит-
черрз стадию, подобную получению ализарина при плаве ^-сульфокислоты антрахи-
нона со щелочью 8»):
ОН Н NHa
Описываемое в некоторых патентах введение в реакционную
смесь соли щелочноземельного металла, преследующее цель связы-
вания освобождающейся сернистой кислоты и вывода ее из про-
цесса в виде нерастворимой соли, не дает таких выходов, как при-
менение окислителя 40).
Сульфогруппа в сульфокислотах может быть замещена группой CN (цианидной,
илн иитрильиой) при иагреваиии соли сульфокислоты с циаиистым калием. Метод
для получения нитрилом ароматических карбоновых кислот в общем уступает ме-
тоду образования их через диазосоедииеиия (см. ниже). Тем не менее в отдельных
приложениях ои может оказаться практичным. Так, в недавнем немецком патенте
описывается получение нитрилов из производных 1.4-диаминоантрахииои-2-сульфо-
кислоты нагреванием солей сульфокислот с раствором KCN -иод давлением до 119—
115° как метод образования соответствующих нитрилов 41):
При разборе процессов сульфирования мы имели возможность
познакомиться с явлениями перехода сульфогруппы с одного места
молекулы в другое обычно при повышении температуры во время
самого сульфирования, причем для большинства примеров можно
считать доказанным, что переходу сульфогруппы предшествует
предварительный гидролиз сульфозамещенного с образованием про-
межуточного несульфированного соединения и с установлением На-
конец более устойчивой при белее высокой температуре формы
сульфокислоты с иным, отличным от исходного, положением суль-
фогруппы.
Не останавливаясь на этом месте на явлениях „странствия0 суль-
фогруппы при сульфировании, мы рассмотрим здесь некоторые при-
меры замены сульфогруппы водородным атомом, т. е. устранения
сульфогруппы как заместителя, причем это превращение обусло-
вливается таким же гидролитическим влиянием, как и отмеченные
нами выше ,странствия“ сульфогруппы.
В практике получения промежуточных продуктов иногда встре-
чается необходимость использовать введение сульфогруппы в ядро
190
ароматического соединения для временного сообщения ему особен-
ной реакционности, чаще всего повышения или понижения актив-
ности определенных участков молекулы ароматического производ-
ного. После использования преимуществ, предоставляемых таким
замещением сульфогрунпой, в дальнейших реакциях ее желательно,
устранить, что обычно и осуществляется нагреванием до опреде-
ленной температуры сульфированного продукта с разведенной до>
нужной концентрации минеральной кислотой (серной или соляной).
Реакция здесь протекает, как гидролитическая, т. е. по урав-
нению
RSO3H Н2О = RH + H2SO4.
Понятно, что отщепляющаяся серная кислота не должна уже
сульфировать это соединение, т. е. необходимо или достаточное
разведение водой реакционной смеси или участие в ней такого
агента, который мог бы удержать серную кислоту от реакционного
воздействия.
Гидролиз ароматических сульфокислот под влиянием минераль-
ных кислот рассматривается как каталитический процесс: катализа-
тором служит здесь Н-ион. Тем не менее и не диссоциированная
на ионы часть молекул кислоты принимает участие в гидролизе,
так как более концентрированная кислота обусловливает более
скорый гидролиз 42).
Самая схема воздействия воды может быть похожа на такую же
при участии щелочи, т. е. и здесь имеется стадия присоединения,
например
SO3H HO3S ОН
Гидролиз сульфокислот производится или нагреванием их, ил»
их солей, с разведенными кислотами в закрытых аппаратах—авто-
клавах, в лаборатории — в запаянных трубках, или пропусканием пе-
регретого водяного пара через смесь сульфокислоты с серной кис-
лотой, или наконец кипячением в открытых аппаратах с обратным
холодильником с такой концентрации серной кислотой, темпера-
тура кипения главной массы которой отвечала бы желаемой темпе-
ратуре гидролиза. Из кислот, входящих как составная часть ре-
акционной смеси при гидролизе, имеют значение прежде всего сер-
ная кислота, соляная—в лабораторной преимущественно работе—
и фосфорная кислоты.
Способ устранения сульфогруппы гидролизом имеет значение
в анализе соединений и в синтезе, как увидим ниже, для получе-
ния некоторых промежуточных продуктов. Сила, с какой удержи-
вается сульфогруппа, у углерода различна для разных соединений
и зависит главным образом от структуры тех соединений, в кото-
рые входит сульфогруппа, и от положения в них сульфогруппы.
Так, сульфогруппа в моносульфокислоте бензола отщепляется легче
(при более низкой температуре, 170°), чем у дисульфокислоты бен-
191
зола (выше 330°), в фенолсульфокислоте легче, чем в бензолсульфо-
кислоте, у а-сульфокислоты нафталина легче, чем у р-сульфо-
кислоты.
В. Н. Ипатьев и Петров обнаружили, что соли сульфокислот (а не сво«
бодные кислоты) также гидролизуются водой при высокой температуре и давлении,
особенно легко в присутствии глинозема (А12О3). Здесь очевидно реакция идет в две
стадии; сначала гидролизуется соль кислоты на кислоту и основание, а затем уже
сульфокислота, отщепляя сериую кислоту, образует молекулу иесульфироваиного
органического соединения:
RSO3Na -f- Н2О = RSO8H -)- NaOH
RSO3H + Н2О = RH + H2SO4 «).
Гидролиз дисульфокислот дает в результате отщепление ие одной, ио обеих суль-
фогрупп. Так, 1.5- (соотв. 1,6-) дисульфокислоты нафталина при нагревании с сер-
ной кислотой, содержащей некоторое количество воды, обе переходят сначала
в нафталин м):
и затем в зависимости От концентрации кислоты и температуры нафталин может
образовать новую сульфокислоту.
При нагревании 1.7-диоксннафталии-3.6-дисульфокислоты с разведенной сериой
кислотой под давлением до 180° получают 1.7-диоксииафталии ®).
ОН
Как на пример интересного синтетического применения сульфи-
рования для последующего устранения сульфогруппы гидролизом
укажем на получение д-нитро-(соотв. я-амино-) дифениламина (IV)
и его производных. Здесь схема реакционных превращений например
такова:
192
Введение сульфогруппы в I нужно для того, чтобы сделать более реакционным
по отношению к анилину атом хлора во II (см. следующую главу). После получе-
ния III гидролиз осуществляется нагреванием III с разведенной минеральной кисло-
той при температуре около 100°. Также можно гидролизом получить и-аминодифе-
ниламин из продукта восстановления нитросульфокислоты III:
Также может быть получен например я'-окси-я-аминодифениламин
с переходом через фазу я-амино-я'-окси-о-сульфокислоты дифени-
ламина 46).
При гидролизе сульфокислот, заключающих аминогруппу, нужно
считаться с возможностью гидролиза и аминогруппы, факт, особенно
частый в нафталиновом ряду (см. главу VIII). Так, при нагревании
с серной кислотой 1-нафтиламин-5-сульфокислоты получается «-наф-
тол |17):
Ч-2Н2О—>
+ h2so4+nh3
Интересны неопубликованные наблюдения А. И. Королева над полученвем
из 2-нафто 1-6.8-дисульфокислоты (кислоты G), взятой в виде кислой калиевой соли,
2-нафтол-6-сульфокислоты при нагревании первой в кипящем нафталине (50% пре-
вращения).
HO3S
Так как обычный „мокрый” гидролиз приводит здесь к получению ^-нафтола
то нужно думать, что в этом случае гидролиз происходит за счет минимальных ко-
личеств влаги, удерживаемой кислой солью, а серная кислота задерживается нафта-
лином (сульфируя?).
В ряду антрахиноновых сульфокислот в соответствии с своеобразным поведе-
нием ангрзхиноновых ззме ценных устранение сульфогруппы возможно проводить
в щелочном растворе, если обработку производить с предварительно гидрирован-
ной (дигидро-) сульфокислотой. Восстановление сульфокислоты антрахинонового
13 Зак. 2611.—Н. Ворожцов
193
ряда в слабокислотном, нейтральном или слабощелочном растворе приводит к со-
единениям, которые Р Э. Шмидт называет дигидросоединениями. Эти соединения
легко отщепляют свою сульфогруппу в виде сернистой, но не серной кислоты
в щелочном растворе (NaOH, NH4OH, Na2CO3) при обыкновевиой температуре.
Реакцию можно объяснить двоякой схемой (1 и II), например
или
+ Н2О + SO2
Реакция легко проходит с сульфокислотами, заключающими еще амино-и окси-
группы.
Можно дигидросоедииения сульфокислот лишить сульфогруппы и посредством
нагревания с крепкой серной кислотой 48).
Применение восстановления к растворам сульфокислот приводит нередко к устра-
нению сульфогруппы не только в антрахиноновом ряду, ио и в нафталиновом и бен-
зольном.
Так, метод обработки растворов сульфокислот или их солей посредством амаль-
гамы натрия рекомендован Фридлендером и Л у х т о м для устранения осо-
бенно подвижных a-стоящих сульфогрупп в нафталиновых производных, например
для перевода 49):
SO3H
Нагреванием водно-щелочного раствора полисульфокислот нафталиновых про-
изводных (амиио-, окси- или аминоокси-замешенных) с Цинковой пылью можно до-
стичь того же эффекта замещения а-сульфогруппы на водородный атсм по схеме:
RSO3H -ь Н2 -> RH 4- Н2О 4- SO2.
Таковы например превращения 60):
HO3S NH,
ЛЛ'
HOgS/X/xjxSOgH
nh2
/\А
J I
HO3S/\/\/\SOgH
194
13*
195
Применение амальгамы натрия, но не в готовом виде, а образуемой при элек-
тролизе щелочного раствора сульфокислоты при ртутном катоде, описывается в не-
мецком патенте 51). Интересно отметить, что в этом случае наблюдается отщепление
не непременно а-сгоящих суяьфэгрупп, но и ^-стоящих в зависимости от положе-
ния отщепляемой группы в отношении заместителя. Констатируется и возможность
замены водородом сульфогруппы в полисульфокислотах бензольного ряда. Среди
многочисленных примеров такого метода превращения укажем:
Недавняя работа японских химиков устанавливает, что при электролитическом
отщеплении сульфогруппы из замещенных сульфокислот (преимущественно в бен-
зольном ряду) на легкость отщепления имеет наибольшее влияние характер и относи-
тельное (к сульфогруппе) положение заместителя. Оно облегчается наличием кар-
боксильной группы и затрудняется аминогруппой Из сульфобензойных кислот орто-
н пара-кислоты легче восстанавливаются, чем мета-кислота. Из аминосульфокислот,
наоборот, мета-производные восстанавливаются легче всего, между тем как пара-
изомеры повидимому наиболее прочны м).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) Ср. A. Wahi, Bull (4) 41, 1431 (1927).
2) Н. Feuchter, Ober Alkalischmelzen, Ch. Ztg.38, 273 (1914). Ср. H. S. Fry,
E. L. Schulze, H. Weilkamp, Am. Soc. 46, 2268, CB 1924, 11, 2828.
3) L. В a r t h, J. S c h r e d e r, Ber. 12, 417 (1879); A. E. Ч и ч и б а б и н, Ж. 55, 7
(1924). О поведении в плавлении дисульфокислот бензола н сульфокислот
фенола ср. Barth, Sehnhofer, Ber. 8, 1483 (1875), 9, 969(1876).
4) К, Г. п. 79028 (F. IV, 147).
5) Аналогично, как для плава (З-аминоантрахинона, см. Е. Schwenk, Ch. Ztg.
52, 45, 62 (1928).
6) Ср. A. W ahi, Le centenaire de la decouverte de I’alizarine, Bull. (4) 41. 1417
(1927).
7) F. H. Rhodes, D. W. Jayne jr., F. H. Bivins, The phenol fusion, Ind. Eng.
Chem. 19, 804 (1927).
s) H. Fierz, Helv. 3, 318 (1920).
9) T. C. Wagner, E. E. Reid, Am. Soc. 53. 3407 (1931).
10) A. G. Peterkln, The synthetic phenol, Ind. Eng. Chem. 10, 738 (1918).
u) FrancisWilson, Kurt H. Meyer, Ber. 47, 3160 < 1914).
Hale, Britton (Dow), Am. n. 1789071, CB 1931, 11,1348.
13) A. Hempel, Am. n. 1820274, QB 1931, II, 2933.
H) M. А. Ильинский (11 j i n s k i), Ber. 36, 4198 (1903); R. E. Schmidt, Ber.
37, 66, (1904); By заявл. Г. n. F. 17474 (F. VII, 188); M. L. В., Г. n. 148875
(F. VII 190).
ls) S. Aoyaraa, J. Morita, CB 1932, 1, 2467 [в последнем случае улучшение
выхода дает применение Ва(ОН)2 и натриевой соли дисульфокислоты].
16) W.-Ильинский, Г. п. 195874 (F. IX, 683).
17) R. v. Mural t, Ober die Einwirkung von Natriumhydroxyd auf die a-Naphthyi-
aminsulfosSuren. Diss. Zfirich 1919; Fierz, 1. c.
196
18) Capelli, Gazz. 48, II, 107 (1918), CB 1919, I, 722.
i«) Cp. R. Scott, Ind. Eng. Chem. Anal. Ed. 3, 67 (1931).
20) Elko Chem. Co, Am. n. 1817444, CB 1931, II, 3042; R. P о ci us, Am. n. 1430184,
CB 1924, II, 1277.
2i) A. Hempel, Am. n. 1808722, CB 1931, II, 1348.
22) V. R. Kokatnur, Am. n. 1667480 (1928).
аз) C. Graebe, H. Kraft, Ober Oxydationsschmelzen, Ber. 39, 794, 2507(1906).
2J) W. M. Rodiono-w, S. J. Manzow, Soc. Ind. 42, 509 T (1923).
25) A. B. Davis (Pennsylvania Coal Prod. Co), Am. n. 1717009 (1929).
гв) M. Ph у 11 ips, H. D. Gibbs, The fusion of sodium benzene-m-disulfonate with
sodium hydroxide for the production of resorcinol, Ind. Eng. Chem. 12, 857
(1920); cp. Bindschedler, Busch, Mon. Scient. 20, 1169 (1878); De-
gener, J. pr. Ch. (2) 20, 300 (1879); Miihlhauser, Dingl. Polyt. J. 263,
154 (1887).
2’) Ch. R. Downs (Weiss & Downs), Am. n. 1658230, CB 1929, II, 96.
28) А у 1 s w о r t h, Am. n. 1213142.
29) 1. Gubelman n, Tinkler (Newport), Am. n. 1573056.
30) I. G„ Г. n. 497243.
31) A. E. Порай-Кошиц, Сов. n. 19628, CB 1931, II, 3393.
32) Ср. T. Callan, Analytical control-in the dyestuff-industry, Soc. Ind. 1923,828
33) Hanp. Fr. Fichter, Fr. А с к e r m a n n, Helv. 2, 583 (1919) и последующие
работы Fichter (Helv), ср. главу XV.
34) I. G., Г. n. 501467, CB 1930, II, 1771.
35) A. Wohl, Г. n. 116790. (F. VI, 113).
36) W. J. Hale (Dow), Am. n. 1595299, CB 1926, II, 2116.
37) C. Schwalbe, Ber. 39,3102 (1906) Cp. Hale, E. Britton, Ind. Eng. Chem.
20, 114 (1928).
38) В., Г. n. 256515 (F. XI, 551); CIBa Г. n. 391073 (F. XIV, 847); A. Sander
Г. n. 484432, Ch. T. Ob. 1930, 47; Lauer, J. pr. Ch. 135, 7 (1932).
39) IG., Г. n. 523523. CB 1931, II, 2388.
4°) M. L. В., Г. n. 267212 (F. XI, 552).
«) Merz, Miihlhauser, Ber. 3, 710 (1870);IG„ Г.п. 536998, CB 1932, II 1975.
42) Сабатье, Катализ в органической химии. Ленинград 1932, 21. Tolle ns,
Wigan d, Ann. 265, 317 (1891). Ср. I. M. Crafts, Ber. 34, 1350 (1901).
43) W. Ipatiew, A. Petrow, Ber 59, 1737 (1926).
44) D. L. L у n c h, J. T. Scanlan, Ind. Eng. Chem. 19, 1010 (1927), также Ind
Eng. Chem. 19, 417 (1927),
45) IG., Г. n. 535080.
46) F. Ullman n, R. D ah men, Ber. 41, 3744 (1908); F. U 11 m a n n, K. J u n-
gel, Ber. 42, 1077 (1909); F. U 11 m a n п, Г. n. 193351 (F. IX, 133), 193448 (F.
IX, 129).
4') Г. Вен дел ь штейн, P. M. Шпинель, Ан.-Кр. Пр. 1932, № 4, 16
48) R. E. Schmidt, Am. n. 1782747 (1930); IG. Б. n. 274558, CB 1928, II, 1623.
49) P. Friedlander, Lucht, Ber. 26, 3028 (1893).
50) К., Г. n. 233931 (F. X, 184).
51) By, Г. n. 255724 (F. XI, 217), 248527 (F. X, 186).
52) M. M a t s u i, G. S a k u r a d o, CB 1932, II, 2173
ГЛАВА VII
ОБМЕН ГАЛОИДА (ХЛОРА) НА ДРУГИЕ ЗАМЕСТИТЕЛИ
До сравнительно недавнего времени считалось установленным
существенное различие между галоидом, связанным с ароматиче-
ским ядром, и галоидом в соединении с углеродом алифатической
цепи — в том, что в первом случае галоид неспособен к обмену
на другие заместители, между тем как алифатически связанный
галоид подвижен и легко выменивается в многочисленных реак-
циях.
С накоплением экспериментального материала обнаружился
целый ряд исключений из этого правила о „неподвижности" аро-
матически закрепленного галоида, особенно когда последний на-
ходится под благоприятным влиянием особых реактивирующих,
иногда называвшихся негативирующими, групп, напр. NO2, СООН
и др. Когда же аппаратурная техника дала возможность работать
при сильно повышенных давлениях и очень высокой температуре
в подходящих автоклавах, ароматические хлорозамещенные начали
вести себя совершенно так же, как вели бы себя в соответствую-
щей реакции алифатические соединения галоидов, и разница между
свойствами „ароматического" и „алифатического" хлора осталась
только как количественная (применение более высокой темпера-
туры при реакциях с ароматическими хлоропроизводными), но не
принципиальная.
Ныне можно уже утверждать, что реакция хлорирования в ог-
ромном большинстве случаев ее применения в красочном произ-
водстве имеет в виду не зафиксировать хлор раз навсегда, но
использовать хлоропроизводные как с ароматической, так и с али-
фатической связью галоида для получения иных, свободных от
хлора, замещенных.
Курт Мейер х), основываясь на своей гипотезе о значении двойной угле-
родной связи для реакционности ароматических соединений, видит в крепости
галоида, связанного с ароматическим ядром, относительную меру ненасыщенности
двойной связи, вызываемой большей илн меньшей активностью заместителя. Так, им
обнаружено, что при обработке бромопроизводных алкогольным раствором NaOH
(при 180°) количество отщепляемого галоида понижается в порядке соединений:
бромбензол, л-бромтолуол, л-броманизол, n-бромфенол, л-бромдиметиланилин.
Следовательно, чем активнее (по отношению к двойной связи) замещающая
л-вздород группа, тем крепче связан галоид. Группы, затрудняющие активность
двойной связи (например NO,), делают галоид связанным слабее.
В ряду галоидов относительная легкость их отщепления, характеризующая силу
их связи с углеродным атомом, изменяется обычно в порядке:
J>Br> С1>Е
Фгоросоедннения наименее реакционны, но и в них фтор может сделаться под-
вижным под влиянием реактивирующих групп 2).
198
Относительная подвижность галоида в различных галоидозамещенных зависит
от многих факторов, в том числе и от характера реагента, содействующего вовле-
чению галонда в реакцию. Мы увидим ниже несколько примеров неожиданных
отклонений от общей схемы.
Необходимо поэтому индивидуализировать понятие „подвиж-
ности галоида" указанием на характер реакции, применительно
к которой изучается относительная реакционность галоидного
атома.
Кроме нитрогруппы подвижность галоида в орто- и пара-местах
обусловливают также группы: CN, COR (R может быть и водоро-
дом), СООН, SO3H и в несколько более слабой степени азогруппа:
N = NR з).
Если эти активирующие группы стоят не в орто- или пара-,
но в мета-положении к галоиду, то их влияния на подвижность
галоида чаще, но не всегда незаметно.
Особенно велико активирующее влияние на галоид со стороны
этого характера групп, если их не одна, а две или три в ядре и
все они расположены в орто- и пара-положении к галоиду. Поэ-
тому например способность к обмену хлора у 2.4-динитрохлор-
бензола (I) превосходит такую же у о- и л-иитрохлорбензолов:
Можно было бы ожидать, на основании вышеприведенного ряда реакционности
К. Мейера, что влияние галоидного атома на подвижность другого галоидного
атома в ароматическом ядре проявится в направлении понижения реакционности
галонда, стоящего в орто- и пара-месте, так как галоидный атом принадлежит
к орто-пара-ориентирующим заместителям, т. е. активирует двойную связь (выше,
правда, упомянуто, что в реакцию нитрования галоидозамещенные вступают не
легче, но труднее незамещенных углеводородов). Этот вопрос недостаточно еще
освещен точными наблюдениями, но известно, что в полигалоидопроизводных не-
которые положения галоида благоприятны для его обмена. Так, с метилатом натрия
наиболее легко, при 183°, реагируют .и-дихлорбензол, симм. трихлорбензол и
1.2.4.6-гетрахлорбензол. Пентахлорбензол и гексахлорбензол реагируют медлен-
нее, чем тетрахлорбензол (может быть от меньшей растворимости в реакционной
среде); о-дихлорбензол скорее реагирует, чем пара-изомер, тетрахлорбензол рядо-
вой (1.2.3.4) скорее, чем симметричный (1.2.4.5) 4).
Таким образом для реакционности галонда благоприятны повидимому мета- и
орто-положення в полигалоидопроизводных по отношению к другому галоиду.
Галоид, в частном случае хлор, связанный не непосредственно
с ядром ароматического соединения, но находящийся в боковой
цепи в виде группы СН2С1, СНС12, СС13, ведет себя, как и следует
ожидать, более активно сравнительно с ароматически связанным
галоидом, хотя на его активность в этом случае может быть ока-
зано влияние со стороны заместителей ядра. Отметим, что здесь
наибольшее влияние проявляет активирующая группа (NO2), стоя-
щая не в пара- и орто-, но в мета-положении к углеродному за-
местителю, несущему хлор.
199
Выразив схематически строение замещенного хлористого бензила ХС6Н4СН2С),
где X — заместители, перечисленные ниже, в том числе и водородный атом, можно
получить такой ряд изучеиых О л н в ье заместителей по силе их влияния 3):
л-СН3 > о-СН3 > л«-СН3 > Н > л-С1 > л-Вг > о-С1 > о-Вг > .и-С1
> >и-Вг > .u-NO2 o-NO2 > n-NO2.
Общее выражение течения реакции обмена хлора (галоида) на
другие заместители таково:
RC14-HX — RXH-HC1. (1)
Из этого видно, что реакция сопровождается уходом одной
молекулы хлористого (галоидо-) водорода. Ясно также, что ре-
акция будет облегчена, если имеется возможность выделяющийся
хлористый водород вывести из сферы взаимодействия, хотя бы
связав его в виде соли. В некоторых частных применениях реакции
эта нейтрализация хлористого водорода происходит сама по себе за
счет щелочного металла (Me'), замещающего водород. В таких слу-
чаях уравнение реакции (1) приобретает вид:
RCl-j-Me'X = Me'CI-|- RX. (2)
Как пример такого взаимодействия по уравнению (2) можно привести полу-
чение .и-иитробеизальдегид-л-сульфокислоты 6)
/Х/С1 +Na2SO3 /X/SO3Na
—NaCl
В тех же случаях, когда уравнение (1) выражает схему течения
реакции, необходимо прибегать к связывающим кислоту (следова-
тельно щелочным) средствам, каковыми могут быть или соли сла-
бых кислот— ацетаты (натрия), карбонаты — или гидроокиси ще-
лочноземельных металлов, аммиак, органические основания и т. п„
Можно с достаточным основанием предполагать, что общее уравнение (1) выра-
жает лишь начальное и конечное состояния реакционной массы и что в ходе реак-
ции существует момент присоединения реагирующей молекулы ХН (соотв. ХМе')
к галоидопроизводиому. Такие продукты могут быть очень нестойки и, распадаясь
с выделением галондоводородиой кислоты, дают конечный продукт замещения хлора
иа группу или атом X. Например:
Следовательно здесь предполагается такое же течение процесса, какое мы считаем
наиболее вероятным при щелочном плавлении сульфокислот.
В некоторых взаимодействиях, протекающих с обменом галоида, новейшие
исследования подтверждают приемлемость такого взгляда на ход реакции 7).
Наиболее важными превращениями атома галоида являются
замещения его посредством азотсодержащих групп, в том
числе и NH-2-группы, посредством кислородсодержащих
труп, между прочим и ОН-группы, и посредством серусодер-
жащих групп.
200
а) Обмен хлора на азотсодержащие группы
Общее выражение первого рода превращений таково:
RC1 + HNR'R" = RNR'R" + НС1, (3)
причем R' и R" могут быть в частном случае водородными ато-
мами.
Необходимо различать при этом те взаимодействия, при кото-
рых в исходном галоидозамещенном соединении галоид связан
в алифатической цепи и которые протекают в сравнительно легких
условиях (низкая температура, отсутствие побудителей реакции),
как например получение бензиланилина из хлористого бензила и
анилина 8)
/\/СН2—
i II---------► ; I
U + U ~HCJ L U
и особенно интересного его орто-нитрозамещенного.
В тех же случаях, когда галоид в исходном продукте связан
непосредственно с ароматическим ядром и не находится под влия-
нием сильных реактивирующих групп, взаимодействие протекает
при высокой температуре и весьма часто требует присутствия
катализатора, каковым является медь, или в виде мелкораздроб-
ленного металла, окисла, например Си2О, или в виде солей (CuSO4,
CuJ, CuCl2, CuCl).
Бесспорно наибольшего внимания в этом ряду превращений
заслуживает получение анилина из хлорбензола, принявшее не-
давно уже производственные формы и масштаб в Америке, так же
как получение л-нитроанилина из л-нитрохлорбензола, практи-
ковавшееся в Западной Европе уже в более ранний период и поста-
вленное в СССР.
Вышеприведенное уравнение (3) для первого случая принимает
такой вид:
С6Н5С1 + NHS = C6H5NH2 + НС1,
а так как хлористый водород связывается избыточным аммиаком,
то выражение общего баланса реакции таково:
C6H6C1 + 2NH8 = C6H6NH2 + NH4C1. (4)
Американским химиком Квиком, а затем Гелем и Брит-
тоном при этом взаимодействии, протекающем при высокой тем-
пературе и большом давлении, было обнаружено особое значение
медных катализаторов, особенно солей закиси меди. Введение ме-
таллической меди, при наличии соли закиси, еще более усиливает
действие катализатора.
Можно с успехом применять н соли окиси меди (Си”), если ввести металли-
ческую медь в таком количестве, чтобы нацело вся Си" перешла в Си'. Выяснено,
что если в реакционную смесь нз 1 мел. хлорбензола н водного аммиака, содер-
жащую 0,1—0,2 мол. Си2С12, прибавить металлической меди, то реакция ускоряется
настолько, что практически полное превращение хлорбензола в анилин может быть.
201
достигнуто в течение нескольких часов прн 200°; при несколько же более высокой
температуре полное превращение требует менее часа времени. Реакция совер-
шается в медных трубчатых аппаратах или автоклавах или в аппаратах, выложенных
или покрытых внутри медью.
На 1 мол. хлорбензола требуется около 5 мол. водного 25—
30°/о-ного аммиака и 0,1 мол. Си2С12. Температура 150—250°. Час-
тично реакция течет по другому направлению, и вследствие гидро-
лиза (ОН'-ионы) происходит образование фенола:
CeH5Cl + H2O + NH8 = C6H6OH + NH4Cl. (5)
По описанию патента, при обыкновенных условиях отношение фенола к ани-
лину 1:20 (наделе несколько выше). При повышении концентрации аммиака
можно достичь отношения 1:50, но авторы не считают практичным итти к этому
пределу. Вместе с тем получается незначительная примесь дифениламина.
Хлористые (галоидные) соли других кроме меди металлов, в том числе NH4C1,
замедляют каталитический эффект медных солей. Накопляющийся по реакции (4)
хлористый аммоний все более тормозит каталитическое влияние, делая невозмож-
ным полное превращение. Присутствие меди однако содействует преодолению
этого тормозящего эффекта со стороны хлористого аммония. Именно медь в при-
сутствии аммиака соединяется с кислородом (из воздуха), давая закись меди Си2О,
а последняя, реагируя по схеме
СщО + 2NH4C1 == 2CuCl -L 2NHs + H2O,
устраняет накопляющийся хлористый аммоний и увеличивает концентрацию
соли Си*, которая, по вышесказанному, является положительным катализатором глав-
ной реакции.
Если воздуха в системе нет, того же действия на хлористый аммоний можно
добиться введением малых количеств (0,2 мол.) Сп2О. Максимально необходимое
количество Си2О для разложения всего хлористого аммония 0,5 мол. на 1 мол.
С6Н5С1 8).
Замечательно, что недавно в патенте той же фирмы, которая ранее отмечала
вредное влияние на процесс обмена хлора на аминогруппу хлористых солей иных
кроме меди металлов, появляется указание на состав реакционной смеси с участием
кроме аммиака еще хлористого кальция (или ZnCl2 или AgCl) и гидроокиси каль-
ция (или NaOH).
При этом отмечена, правда, длительность процесса (в 35 час.). Кажется сомни-
тельным польза неведения гидроокисей щелочноземельных или щелочных металлов:
повышение концентрации ОН-ионов при прочих равных условиях будет сдвигать
процесс в сторону гидролиза, и следовательно получаться оудет не анилин, но
фенол (Ам. и. 1840760).
Дальнейшие патенты сообщают некоторые дополнительные сведения, касаю-
щиеся главным образом медных катализаторов. Так, взамен части и всей закиси
меди, соотв. закисномедной соли, можно применять с успехом медиые соединения
неопределенного состава, получаемые в водном слое после обработки реакционной
массы щелочью для удаления связанного аммиака: соединения (вероятно гидраты
окислов) оседают в виде илистой массы, которую можно применять вместо катали-
затора в реакции. Хорошо действуют как добавочные катализаторы восстановитель-
ного характера при наличии главного катализатора — производного закнсн меди
(например Си2С12) — непрочно связанные соединения меди, могущие в условиях
реакции освобождать металлическую медь, как например: недокись меди, получаемая
при сохранения из первичной нестойкой четвертной закиси меди Си4О (последняя
получается действием восстановителей в щелочном растворе на медные соли), нит-
рид меди Cu3N, готовящийся нагреванием с аммиаком свежеприготовленной закиси
меди до 240—260° 1°).
Применяемые при этом давления порядка 60—100 ат.
С целью уменьшения расхода на образование дифениламина из хлорбензола по
реакции
СвНлн2 4- с1с6н. = c6h5nhc6h5 + на (6)
(хлористый водорот конечно связывается при этом аммиаком реакционной смеси),
новыми патентами рекомендуется заранее вводить в реакционную смесь нужное
202
количество дифениламина, которое соответствует получаемому в равновесном со-
стоянии после реакции. Польза такой прибавки должна быть проверена: не уч-
тена ли в этом патенте просто аналогия процессоз получения анилина и фенола,
причем в последнем названном процессе вполне убедительно доказана выгода при-
бавления дифенилового эфира как ингредиента, входящего в реакционную смесь.
Прибавление едкого натра, для повышения концентрации аммиака (путем раз-
ложения NHjCl), рекомендуемое в этих последних патентах, представляется опас-
ным a priori как могущее с усилением концентрации ОН-ионов содействовать
образованию фенола по схеме (5) и).
Изменяя исходное хлоропроиззодное, можно аналогично проводимым про-
цессом (по описанию патентов) получать различные ароматические амины: так, ди-
хлорбензолы даюг фенилендиамины (I), хлорголуолы — толуидины (II), хлорнафта-
лип — наФтиламин (III), хлэрантрлхинон — аминоантрахинон (IV):
I QHjCl, -> СвЩЫНз),; II С1 • С8Н4 • СН3 -> H,N • C8Hj • СН3; III С10Н7С1 -> C--H7NH2
NH,
Последнее из указанных превращений, именно ,3-хлор- (соотв
бром- ) антрахинона в ^-аминоантрахинон, недавно было подробно
изучено Гроггинсом с сотрудниками, причем установлено, что
наиболее легко и полно реакция превращения протекает именно
у бромозамещенного. На этом превращении подробно изучалось
реактивирующее значение медных катализаторов (Cu2Cl2, Си.2О).
Без их участия реакция хотя и идет, но дает превращение немно-
гим выше ЗО°/о сравнительно с тем, что получается в присутствии
катализаторов. Заметный катализирующий эффект дает значитель-
ная примесь нитробензола (без меди) в равных количествах с броман-
трахиноном.
Прибавление окислителей (солей НСЮ, НС1О3, НС1О4) благо-
приятствует замещению аминогруппой атома хлора в р-хлоран-
трахинонб.
Относительно причины каталитического влияния медных солей
в этом и подобных превращениях Гроггинс высказывает мнение,
что оно связано с проявлением электролитической активности.
Комплексные ионы, как купраммониевый [Cu(NH3),]+, облег-
чают галоидопроизводному контакт с большим количеством частиц
аммиака.
Для случая презращения бромантрахинона в аминоанграхинон количества мед-
ных соединений, достаточные для превращения : 0,1 — 0,2 мол. Си2С12 и 0,2 мол.
Си2О на 1 мол. исходного галоидопрэизводного- С повышением температуры
реакция идет скорее. Превращение за 2 часа при 180° приблизительно эквивалентно
такому же за 15 час. при 150° при прочих равных условиях 12).
В немецком журнале мы находим описание производства ценного (3-аминоан-
трахинона из |3-хлорантрахннона [нагревание до 220° с 15-кратным количеством
аммиака (уд. в. раствора 0,9) вместе с порошком меди]; выход указан как почти
теоретический 13).
Химики английской фабрики, известной своими достижениями
в области производных антрахинона (Scottish Dyes Ltd), находят
практичным готовить а-аминоантрахинон не из а-сульфокислоты,
как это обычно принято, но из а-хлорантрахинона, указывая, что
процесс протекает полнее и с более хорошим выходом. Присут-
203
ствие медных солей, характер которых они подробно не описы-
вают, благоприятно влияет на реакцию, которая проходит в авто-
клаве с 26%-ным аммиаком (700 г на 1С0 г хлоропродукта, 12 ча-
сов при 170°)14):
Cl NH2
/\/сО\/\ /\/СО
Близкие к антрахиноновым производные о-бензоилбевзойной кислоты, вклю-
чающие хлор в благоприятном положении к реактивирующей СО-группе, особенно
при наличии второй активирующей группы (например NO2), также могут быть
превращены в аминозамещенные, например
/\/ со \/\ /\/ со \/\
j | | | +2NH, | I | j -J-NHjCl
\/Ч'СО2Н \/\сО2Н \/XNH,
/\/ со \/\/N^
-pHNXa j | j | 4- X2NH. MCI
^/XCO2H \/\nX2
(X—ьодородный атом, алкильная или арильная группа)
Первый процесс требует нагревания с водным аммиаком в присутствии медной
соли (Си”) н автоклаве, второй удается провести с аммиаком при нагревании
смеси на водяной бане. Применение в последнем случае кислотных амидов, соотв.
сульфонамидов RSO2NH2, требует повышенной температуры, участия щелочного
агента и медного катализатора 15).
В американских патентах указывается на возможность получе-
ния хлорозамещенных анилинов из ди-, соотв. полихлорозамешен-
ных бензола при обработке их аммиаком в неводной среде (напри-
мер в спиртовом растворе). Аммиак берется в количестве 3 мол.
на 1 атом обмениваемого хлора. Как катализатор применяется
опять хлористая медь Си2С12. Соображения о значении металличе-
ской меди и пр. остаются в силе. По этому способу возможно сле-
довательно осуществление таких например превращений:
Получение п-фенилендиамина из /г-дихлорбензола описано
в ам. пат. 1919 г. Оно проводится посредством аммиака (избы-
ток 10—20% от теоретического количества) в присутствии соеди-
нения меди как катализатора.
Редкшюннаа жидкость, заключающая катализатор, готовится растворением мед-
ного купороса в горячей воде и рививмиваиием с водным раствором (28%-иым)
204
аммиака до почти полного растворения всего. Паэа-дихлорбеизол смешивается
с этой жидкостью и нагревается во вращающемся автоклаве при 195—200° в те-
чение 14—16 часов 16).
Дальнейшая разработка этого способа сделала возможным на-
правление обмена атома хлора в хлорбензоле не на аминогруппу,
а на ариламиногруппу, причем используется как главная реакция (6),
которая при получении аминозамещенных была бы нежелательной.
Большая часть процессов, ведущих к образованию группы NHC8H5
и т. п. ариламиногрупп, разобрана в главе X, здесь же мы ограни-
чимся лишь теми процессами, которые связаны с заменой атома С1
на группу NHR.
Направлению реакции в сторону образования дифениламина
из хлорбензола содействует понижение количества аммиака
(1—1,5 мол. NH3 на 1 мол. хлорбензола), более высокая темпера-
тура (до 250°) и большая продолжительность взаимодействия.
Катализаторы — те же, что и в ранее описанных процессах (Си2О
и более бедные кислородом окислы). Прибавление едкого натра
для разложения NH4C1 и снижения тем расхода аммиака, рекомен-
дованное в патенте, вероятно будет сопровождаться образованием
больших количеств фенола ”).
Совсем иным путем к разрешению пэследнего вопроса о превращении хлоро-
замещеияого в направлении
RC1 -> RNHR',
где R и R'— ароматические углеводородные радикалы, подходит патент швейцар-
ской фабрики в Базеле 18).
Вводящие группу NHR' ароматические амины переводятся предварительно
в соединения со щелочными металлами I группы, например с Na, и эти соедине-
ния уже легко реагируют с хлорозамещеиными при участии или в отсутствии
таких катализаторов, как медь и соединения меди. При таком методе работы легко
получаются диариламины с замещающими группами, например СН3 —, в одном или
обоих из ядер, несимметрические диариламины и т. п.
Таким способом можно получить например о, о-дитолиламин по схеме:
\/\nh2
нагрев 175—190'
в присутствии Си
и прибавл. Na
в автоклаве
24 часа
/Х/СИз
\/\NHNa
нагрев в 250—ЗОО1
Без практического опроооваиия трудно сказать, имеет ли этот способ значи-
тельные преимущества сравнительно с американским.
Отметим потытки получать амияосоединения из хлорозамещен-
ных действием аммиака на пары, например хлорбензола, в присут-
ствии катализ (тора — мелкораздробленного никеля при температуре
свыше 30Ээ. При этом получается немного анилина вместе с бен-
золом. Опыты с окисью тория дали отрицательные 19).
205
В превращениях рассматриваемого нами типа
RCl^RNXy
по всей вероятности обмену галоида предшествует образование
продукта присоединения, и вероятно роль катализаторов — ускоре-
ние и участие в этом образовании, хотя изолировать такие продукты
и не удалось до сих пор. Вероятно взаимодействие протекает по
такой например схеме:
Н Н
| ! +NH.CI
В строении промежуточно-образуюшегося продукта присоединения вероятно
участвует и катализатор, что не указано на нашей схеме.
Если активность хлора в незамещенном ароматическом ядре невы-
сока, то она становится значительно большей при благоприятном
влиянии некоторых замещающих групп, например NO2, SO3H, СО2Н,
СНО (в орто- и пара-положениях к галоиду). При этом активность
галоида, особенно при наличии не одной, а двух или трех реактиви-
рующих галоид групп в соответственном положении, может быть
так повышена, что по реакционности хлора такое соединение упо-
добляется алифатическим галоидопроизводным. Из наиболее инте-
ресных технически превращений здесь в первую очередь должно
назвать получение n-нитроанилина из п-нитрохлорбензола:
+ 2NH3
+ NH.C1
Реакция осуществляется нагреванием п-хлорнитробензола с вод-
ным, по возможности высокой концентрации, раствором аммиака
в стальных автоклавах с мешалками (см. рис. 8, стр. 230) в тече-
ние 8—16 час. до температуры 170—190° при давлении 30—40 ат.
Количество аммиака: 7—8, иногда 10 мол. NH3 на 1 мол. хлорнит-
ропроизводного. В давнем германском патенте указан даже более
значительный избыток NH3 20). Концентрация аммиачного раствора
существенно важна для успеха превращения: чем концентрирован-
нее аммиак, тем скорее и полнее при прочих равных условиях про-
текает обмен хлора на аминогруппу. Более разведенный аммиак
открывает большую возможность направлению реакции частично
в сторону обмена хлора на гидроксил. Катализаторы — медиые
соли — содействуют ускорению реакции.
Вопрос о их значении и роли недостаточно освещен в литературе, и выгод-
ность их применения оспаривается. Указывается — как на отрицательные стороны
применения катализаторов в виде соединений закиси меди — на возможность восста-
новительного процесса по отношению к ннтрогруппе, на то, что медные соединения,
увлекаемые п-нитроаиилином, значительно изменяют оттенок красителя (особенно
206
пара красного), приготовляемого из нитроанилина, — возражение существенное, но
освобождение от следов меди в процессе последующей очистки л-нитроанилина—
задача вполне разрешимая. Между тем если сразить условия работы прн полу-
чении анилина из хлорбензола и n-нитроанилина из n-хлорнитробензола, то о' ра-
щает внимание то обстоятельство, что во втором случае, где имеется благоприятно
поставленная л-ннтрогруппа, процесс требует почти вдвое больше относительных
количеств аммиака, чем в первом, где применяются непременно катализаторы.
Прн реакциии п- н о-хлорнптробензола с аммиаком в спиртовом растворе
констатировано ускоряющее действие прибавления небольших количеств иоднстого
калия или натрия. Превращение при прочих равных условиях протекает гораздо
полнее с этой прибавкой, которая, возможно, содействует промежуточному образо-
ванию вместо хлоро- иодозамещенного. При введении медного порошка в этом
случае наблюдалось странным образом снижение выхода как в смеси, где была
иодистая соль, так и без нее 21).
Аналогичный получению п-нитроанилина способ с применением катализа-
тора (СнС12) дает возможность приготовить сульфаниловую кислоту из л-хлорбен-
золсульфокислоты:
/\/С1 /\/NHa
| | NH„ 170^ | |
HO-.s// HO3s/X/
Способ вряд ли можег прн каких-либо условиях оказаться выгоднее употребитель-
ного метода получения сульфаниловой кислоты сульфированием анилина—запе-
канием 2а).
В немецком патенте 1919 г. описан способ получения n-нитроанилина из п-
хлорнитробензола с участием всего 6 мол. NH3, причем взаимодействие происхо-
дит в безводной среде в растворе этиленгликоля (отношение весовых количеств
хлорннтробензола н гликоля около 1 : 2и). Указывается 140—150° как температура
взаимодействия и 5 час. продолжительность 23). Вряд лн этот метод, требующий
расхода этиленгликоля вместо воды, может оказаться выгодным.
Хлорнафталин до сих пор еще не вошел в производство как
исходный материал для реакций обмена галоида, и мы мало знаем
о подвижности его хлора, но галоид в 1-хлор-4-нитронафталине
повидимому подвижен не менее, чем в л-хлорнитробензоле, и реакция
может быть проведена при тех же условиях, что и получение /г-ни-
троанилина ‘2*)- п-о- Динитрохлорбензол, п-нитрохлорбензол-о-суль-
фокислота, 2.4-динитрохлорнафталин и т. п. замещенные с двумя
благоприятно расположенными реактивирующими галоид группами
реагируют еще легче с аммиаком, чем мононитрогалоидозамещен-
ные, например:
/Х/С1
O2NZ Х Z 4 SO3H
2Q7
Ввиду сравнительной легкости обмена галоида на аминогруппу в 2-4-ди-
япгрэ-1-хлэрбгнзате, получечие дияитроанилина можно осуществить, сплавляя
его при 24Э° с 2,4 мол. мочевины без применения автоклава. При этом образуется
цитнуровая кислота. Можно представить эго превращение как переходящее через
фазы 25):
/C1 + H2N
со
02N/'/xNO2 NH;
i
O2N'/\'/\nO,
/X/NH2 ho4^n\ zoh
-----> | + HCON; 3HCON =
О21\Т/\4\ХО2
OH
Альдегиды с подвижным под влиянием CHO, соотв. NOa-, группы галоидом
при обработке их амидами арилсульфокислот в присутствии связывающего кислоту
средства дают замещение азотсодержащей группой на месте галоида. Такие заме-
щенные легко переходят в аминоальдегиды. Вместо амидов сульфокислот (сульфон-
амидов) можно применять сульфамид SO,(NH2)2, амиды карбоновых кислот (бен-
замид, фтал амид). Обработка происходит также в присутствии растворителей
(нитробензол, амиловый спирт, циклогексанол) и катализаторов: меди и ее солей 26).
Например:
OsN^/^/CHO
170-190°
(К*СОЯ, Си-ЬСи3С1л)
О2Хх /ч /СНО O2N /х /СНО
60—8(Р X у
X /X H,SO495»'0 . I
\/j>NH NIL,
O,S . С;Н7
В соединениях, имеющих несколько хлорных атомов и одну
реактивирующую, например нитрогруппу, при действии аммиака
вступает в реакцию тот лишь атом хлора, который находится в со-
ответственно благоприятном положении к этой реактивирующей
группе. Таким образом из нитро-га-дихлорбензола получается хлор-
нитроанилин
Z\/C1 /\/NH>
! i - I I ;
C1//'x/\nO2 C|//\/\nO.2
также
/\ /С1
Cl/4' /X'SO3H ClZ/Z/sCLH
208 3 3
так как галоид, находящийся в мета-положении к нитрогруппе, ею
не активируется 27).
Обмен хлора (галоида) на группу NHR, где R— ароматический
радикал, имеет довольно распространенное применение в практике
получения промежуточных продуктов и самих красителей (в антра-
хиноновом ряду).
Выше мы рассмотрели уже такой обмен применительно к ис-
пользованию хлорозамещенных без реактивирующих групп. Реакти-
вирующие группы в соответственном положении к галоиду, по-
нятно, повышают его подвижность.
Несколько работ посвящено освещению меры влияния различ-
ных структурных факторов на реакционность галоида и амино-
группы в подобных превращениях 2S).
Реакция например между хлорнитробензолом и анилином протекает согласно
уравнению
Cl-C6HrNO2 + 2CeH6NH2 = C6H6NH3HC14- МО2-С6Н^НСвН5.
Прн этом мононитрогалоидопроизводные бензола реагируют с анилином мед-
ленно, 2.4-динитро-1-хлорбензол со свободным анилином при 50° реагирует быстро,
с анилиновыми солями медленно, 2.4.6-тринитрохлорбензол—чрезвычайно быстро
с свободным анилином и скоро —с анилиновыми солями.
Реакционность галоида в нитрогалоидопроизводных бензола изменяется согласно
ряду:
Br>Cl>J.
Реакционная способность галоидэнитробензолов по отношению к пиперидин^-
изменяется совершенно своеобразно применительно к изменению положения и ха-
рактера галоида. Так, в то время как хлор в орто-положении по отношению к NO2
значительно активнее п-С1, иод по активности почти не отличается, стоит ли он
в орто- или пара-месте, бром занимает промежуточное положение. Распределение
пиперидина между двумя эквимолекулярными количествами о-галоидиитробензолов
идет по такому ряду:
С1;Вг = 0,6:1; Br:J = 1,5:1; J: Cl =1:5,4;
между тем как для 1-галонд-2.4-динитробензолов ряд распределений таков:
С1: Вг = 1,75:1; Вг: J == 1; 2,22; J : Cl = 1 : 6,83.
Очень большое значение имеют для хода и полноты реакции структурные
особенности входящего во взаимодействие амина.
Монометиланилин C6H5NHCH3 реагирует всегда медленнее, чем анилин. Заме-
щение в анилине атомов ядра сопровождается в большинстве случаев уменьшением
реакционности аминогруппы во взаимодействии с нитро- и полинитрохлорбензолом.
При этом реакционность аминогруппы внутри ряда замещенных посредством
групп NO2, SOgH, СО2Н, Cl, Вг, J возрастает в порядке: о <п<м. При прочих
равных условиях с возрастанием атомного веса замещающего галоида все более
затрудняется реакционность аминогруппы. SOgH-группа сильнее затрудняет реак-
ционность, чем СООН. Иные заместителя, как то: ОН, ОС2Н5, СН3 и NH» в амине,
не позволяют сделать точного разграничения. Замещение гидроксилом и OAlk-груп-
пой пара-места к NH2 сообщает последней ббльшую реакционность сравнительно
с незамещенным анилином; о- н n-толуидины реагируют медленнее соответственных
аминофенолов, .и-толуидин, наоборот,— быстрее лг-аминофенола. Аминогруппа
наименее других заместителей затрудняет реакционность первой аминогруппы;
таким образом лг-фенилендиамин по реакционности равен анилину, п-фенилендиамин
его превосходит в этом отношении.
Три заместителя последнего ряда — ОН, СН3 и NH2 — прн замещении по отно-
шению к реакционной аминогруппе ускоряют реакцию в порядке
о <" м <7 п.
14 Зак. 2611. — Н. Ворожцов
209
Чем более реакционен галоид, например чем больше реактиви-
рующих групп на него влияет, тем с большей легкостью протекает
замена атома галоида (хлора), при температуре, не превышающей
температуру кипения компонентов реакционной смеси (главным
образом амина), без катализаторов, с участием или без участия
отнимающих кислоту средств (в последнем случае с кислотой свя-
зывается избыток амина, который почти всегда имеет место при
реакции).
Отметим как интересное практически превращение этого рода —
получение п-нитродифениламин-о-сульфокислоты:
/\/С1
| + H2N<^__>
180-185°
СаО для
отнят. НС1
также
уже прн кипячении в водном растворе
с Са(ОН)2
При превращении в отсутствии воды полезной добавкой (рас-
творителем) оказывается глицерин.
В подобных превращениях галоидозамещенных ароматического
ряда, изучавшихся Ульманом, отмечена благоприятствующая,
роль меди как катализатора.
Недавно американскими учеными Вестоном н Адкинсом обнаружено
что в зависимости от условий реакции влияние медн, соотв. ее солей, может быть
различным. Каталитически действует по нх мнению соединение, даваемое медью
с амином (в нх случае N-ацетил-п-толуидином) в присутствии воздуха. Раствори-
тель в таких случаях понижает концентрацию действительного катализатора н при-
водит к уменьшению выхода29).
В патенте немецкой фирмы приводится ряд продуктов присоединения аромати-
ческого амина к 1-хлор-2.4-днннтробензолу, из которых более нли менее легко
образуются диариламины с отщеплением НС1. Здесь констатированы повидимому
те промежуточные стадии обмена Cl-атома на ариламиногруппу, о которых было
сказано выше:
O2N
+H,NCSH3
O2N,^^NOOH O2NX//XyNO,
/С1 - нС1
^/\nhc6h5 х/\хнс6н5
Среди хлорозамещенных производных нафталина в рассматриваемом нами пре-
вращении оказался интересным 1-хлор- (соотв. бром-) р-нафтол, дающий с первич-
210
пымн ароматическими аминами, например с анилином, арнламннопронзводные
6-нафтола, например
Cl NHC6H6
/\/V0H z-x/'\/0H
I I | + H,NC,HS -» III +HCI-
В превращении используют избыток амина, можно пользоваться подходящими
растворителями (в случае например твердого н трудноплавкого амина), возможны
и добавки катализаторов (Zn, Sn, Fe, Pb, Cu)3°).
Аналогичные рассмотренным выше превращения хлора (галоида),
связанного с антрахиноном, используются для получения кубовых
красителей или промежуточных продуктов, наиболее близких
к последним. При взаимодействиях, проводимых при высокой
температуре, часто в присутствии высококипящих растворителей
(нафталин, трихлорбензол) оказывает большую помощь прибавка
меди или как таковой в виде тонкого порошка или в виде солей
закиси (Си2С12). Почти всегда прибегают к связывающим свободную
минеральную кислоту средствам (ацетат натрия), так как применяе-
мые аминосоединения не обладают для этого достаточной основ-
ностью31). Примеры:
СО
H2N NH
Оранжевый алголевый
Подобные этим двум, превращения хлоро- (бромо-) замещенных
антрахинона используются во многих случаях для получения ку-
бовых красителей рядов полиантрахинониламинов и антрахи-
нонакридонов.
б) Обмен хлора на кислородсодержащие группы
Обмен хлора на кислородсодержащие группы — процесс, приме-
няемый для синтеза не особенно большого числа промежуточных
продуктов, но в то же время крайне важный, так как некоторые
из продуктов такого превращения сделались практически доступ-
ными именно благодаря ему.
14* 211
Блестяще разработанный американскими химиками метод полу-
чения фенола из хлорбензола является наибольшим достижением
в этой группе превращений и обещает при благоприятной эконо-
мической конъюнктуре, главным образом при доступности хлора,
следовательно при дешевой электрической энергии, стать серьез-
ным конкурентом классического метода фабрикации фенола через
сульфокислоту бензола. В Америке по данным энциклопедии тех-
нической химии Ульмана уже теперь около 7О°/о фенола произво-
дятся этим путем32). Нет сомнения, что в промышленности СССР
с ростом электрификации этот способ получения фенола будет также
преобладающим. Из разновидностей реакций обмена хлора на
кислородзаключающие группы практически интересны две: обмен
на гидроксил и на оксиалкильную группу. Общий вид таких пре-
вращений можно выразить схемой:
RCl-f-HOX -+ ROX4-HC1, (7)
где X—водородный атом или алкильная группа.
Тот вид превращений, где в результате вместо галоида обра-
зуется гидроксильный остаток, может быть назван гидролизом
хлорозамещенного.
И здесь очень вероятно, что в течение реакции имеет место образование про-
дукта из двух реагирующих молекул, например
который далее, отщепляя молекулу хлористого водорода, нейтрализуемого свобод-
ной щелочью среды, переходит в фенолят:
Применительно к бензольному производному, имеющему только
один заместитель — галоидный атом, в частном случае к хлорбен-
золу, реакция гидролиза водными растворами щелочей разрабаты-
валась достаточно долго, но практические результаты, лежащие
в основании современного производства фенола, получены только
во второй половине двадцатых годов нынешнего столетия, главным
образом благодаря работам уже упоминавшегося ранее Гела и
БриттонаЗз).
Ранее, в 1914 г., гидролиз хлорбензола разведенными щелочами
при высокой температуре (до 300°) и соответственно высоком
давлении изучался К. Мейером и Бергиусом, констатиро-
вавшими, что реакция протекает при этих условиях гладко, осо-
бенно при добавке полухлористой меди. Получается чистый феиол,
главной примесью его, убывающей при повыщении температуры
реакции, является дифениловый эфир СвН5ОС6Н5 34).
212
Два последних исследователя, испытав гидролиз хлорбензола
раствором едкого натра, сравнили действие других оснований при
температуре 300° и установили, что известь (известковое молоки)
практически не гидролизует хлорбензола, раствор соды гидролизует
слабо (35% фенола и 13,7% дифенилового эфира), раствор буры
действует немного слабее соды.
Работами американцев было установлено, что минимальная тем-
пература, нужная для превращения хлорбензола в фенол, лежит
около 300 (295°), но для ускорения реакции выгоднее работать
при высшей температуре (340°). Концентрация раствора щелочи при
этом, дающая достаточную скорость гидролиза и в то же время мини-
мум побочных продуктов (смолы), лежит в пределах 6 — 10°/о. Молеку-
лярное отношение между едким натром и хлорбензолом 2iji—
в соответствии с уравнением
С6Н6С14- 2NaOH == C6H6ONa + NaCl + НаО. (8)
Реакция с химической стороны является именно гидролизом,
она мономолекулярна (подобно гидролизу сложного эфира водой
в присутствии Н‘).
Присутствие меди (медная стенка аппарата) содействует уско-
рению реакции, что становится особенно заметным при более низких
температурах (при 300°, 2х/2 мол. NaOH, 10 час. нагрева — 39%
превращения в стальном автоклаве и 92% в медном). Медь как мате-
риал автоклава имеет то преимущество что не изменяется или мало
изменяется от действия ингредиентов реакционной смеси. Медные
соли (в стальном автоклаве) также действуют каталитически.
Нужно добавить, что медь, в меньшей степени чем железо, под-
вергается окислительному воздействию со стороны горячей щелочи,
поэтому в медных аппаратах выделение водорода, действующего
как на исходные материалы, так и на продукты реакции гидроге-
низующе, происходит в меньшей степени, чем в железных.
Во вэех опытах гидролиза хлорбензола водным раствором едкого
натра констатировано образование дифенилового эфира в коли-
чествах, пропорциональных количеству получаемого фенола, обычно
около Ю°/о на взятый хлорбензол.
Введя заранее в смесь щелочи и хлорбензола дифениловый
эфир в том отношении, в каком он получается, американские
химики убедились, что в продуктах процесса его остается столько
же, как и получалось прежде, т. е. не более того количества,
какое было введено. Если же ввести его в смесь в большем коли-
честве, чем он обычно получается, то в результате реакции часть
его превращается в фенол. Здесь имеется следовательно равновесие
в процессе. Дифениловый эфир образуется вследствие взаимодей-
ствия между хлорбензолом и уже накопившимся фенолятом натрия:
С6НВС1 % NaOC6H5 = СеН6ОСбНб % NaCJ.
Но образовавшийся дифениловый эфир гидролизуется под дей-
ствием фенолята с образованием фенола (этот процесс не требует
катализатора и идет так же быстро и полно в стальном автоклаве,
как в медном):
С6Н5ОС6Н5 % Н2О = 2С6Н5ОН.
2 В
С другой стороны, фенол при высокой температуре в присутствии
окислов металлов (стенок аппарата) частично переходит, с потерей
воды, в дифениловой эфир:
2С6НЬОН = С6Н6ОС6Н5 + Н2О.
Таким образом после израсходования большей части хлорбен-
зола в реакционной системе устанавливается равновесие:
2С6Н5ОН С6Н6ОС6Н5 4- Н2О,
которое может быть сдвинуто в зависимости от большей или мень-
шей концентрации гидроксильных ионов в системе.
Соли сильных оснований и слабых кислот (например сода),
дающие достаточную концентрацию гидроксильных ионов, в при-
сутствии катализатора (меди) также гидролизуют хлорбензол, причем
в таком видоизменении процесса выделяющаяся при реакции со-
ляная кислота переводит среднюю соль в кислую, например в слу-
чае применения соды реакция выразится так:
С8Н5С1 + Na2CO3 4- Н2О = С6НбОН -j-NaCl + NaHCO3,
и фенол будет уже не в виде фенолята, а в свободном состоянии
в реакционной смеси. Водные растворы солей сильных оснований
со слабыми кислотами содействуют гидролизу дифенилового эфира,
хотя и в меньшей мере, чем едкий натр или фенолят. При доста-
точно хорошем смешении реакционной массы и достаточно высо-
кой температуре, время, потребное для превращения хлорбензола,
измеряется всего несколькими минутами, а потому этот процесс
легко осуществить в виде непрерывного процесса, протекающего
в трубчатой системе (медных или выложенных медью стальных
труб), где с одной стороны непрерывно накачивается смесь хлор-
бензола, раствора едкого натра и дифенилового эфира, а с дру-
гой— выходит смесь раствора фенолята и дифенилового эфира,
разделяемая и перерабатываемая далее.
Побочные реакции процесса дают некоторое количество про-
дуктов, в большинстве высококипящих и потому скопляющихся
в фенольной смоле. Наиболее заметный из этих побочных продук-
тов (до 25% всей смолы) — оксидифенил С6НБС6Н4ОН (преиму-
щественно пара-изомер). Образование его протекает, возможно, по
двум путям (I из хлордифенила, И из фенола)
I. С6Н6С1 % С6НБС1 = С6Н6 • С6Н4С1 % НС1
С6НБ - С6Н4С1 + Н2О == С6Н3 • С6Н4ОН % НС1
II. Фенол реагирует с хлорбензолом в своей тавтомерной форме,
например ____
Н2: / : О
214
Последний путь менее вероятен, чем первый, так как фенол в реакционной
смеси связан со щелочным металлом в виде фенолята, следовательно представлен
в энольной форме. Более правдоподобное видоизменение последней схемы основан»
на допущении, что в образовании оксидифенилов принимает участие вышеупомя-
нутый продукт присоединения едкого натра к хлорбензолу, например
"V^ONa
^ZX-ONa
HQ
Вышеописанное является химическим основанием самого совер-
шенного из современных методов производства фенола из хлор-
бензола с применением высокого давления, значительно превы-
шающего 100 ат.
Отметим указание в американском патенте на способ работы, при котором
применяют сравнительно небольшое давлевие (до 20 ат), но который не дает зато
превращения всего хлорбензола: смесь хлорбензола и едкого натра нагревается
в автоклаве под давлением 14—20 ат до 300° (? Н. В.). Реакционная смесь через вентиль
спускается в камеру с атмосферным давлением; фенолят при этом отделяется, непро-
реагировавший хлорбензол, дифениловый эфир и вода ввовь входят в процесс35).
Имеется метод гидролиза хлорбензола в фенол, протекающий
без повышения давления и основанный на действии на пары хлор-
бензола паров воды при высокой температуре и участии катали-
тически действующих поверхностью агентов36).
Реакция, проходящая здесь в отсутствии щелочного агента,
может быть выражена уравнением:
с6н5с14- н2о = с6н5он + на.
Водяной пар вводится в избытке, с которым уходит и хлористый
водород.
Патенты немецкого концерна описывают применение в качестве
катализатора силикагеля, самого по себе или в присутствии метал-
лов. Однократный пропуск смеси паров хлорбензола и воды не
дает в результате полного превращения хлорбензола.
Хлорбензол превращается в пар в парообразователе, где он
нагревается вместе с раствором едкого натра, смесь паров хлор-
бензола и воды проходит через горячие контактные трубки, напол-
ненные активным гелем кремневой кислоты, пары, отходящие из
контактного аппарата, проходят вновь в тот же щелочной раствор,
и не вошедший в реакцию хлорбензол повторно увлекается парами
воды в контактную трубку.
Неопубликованные опыты Г. Н. Г у л и н о в а, изучавшего этот процесс, опре-
деляют благоприятную для превращения температуру при применении фарфоровой
трубки в пределах 410—445°. Вместе с фенолом получается некоторое количество
дифенилового эфира. Медь без силикагеля дает очень малые выхода фенола.
Опыты д-ра Мигге при работе в кварцевой трубсе при температуре 520—
600° указывают иа значение иных катализаторов этого процесса, среди иих — олова
(металлического -f- Sn”), магния и иных. Процент превращения — до 54% при одном
проходе через трубку. При большем проценте превращения хлорбензола наблю-
дается образование заметных количеств бензола. Отношение реагентов: на 1 мол.
хлорбензола—3 мол. НаО.
215
Американский патент Dow Chemical Со описывает применение в качестве ката-
лизатора окиси меди, осажденной на пемзе при температуре 450°. В смесь паров
хлорбензола и воды добавляется в количестве 0,2% (от веса хлорбензола) дифени-
ловый эфир (очевидно с целью затормозить его образование из фенола и хлорбен-
зола). Превращение хлорбензола не полное.
Американский патент Federal Phosphorus Со пользуется катализатором, содер-
жащим силикагель. Температура реакции 700° (!). Выход до 90% фенола.
Французский патент фирмы Прожиль описывает как отличительную составную-
часть своих катализаторов фосфорную кислоту или ее соли, осажденные на пори-
стых телах. Температура реакции 350—700°.
Французский патент Р а ш и г а основан на использовании других катализа-
торов: глинозема, не содержащего железа, или не содержащих железа соединений
других элементов I—IV групп периодической системы, или также свободных от
железа естественных силикатов.
Фирма Дрейфус в своем французском патенте указывает на главный ката-
лизатор — хлористую медь на пемзе, температура реакции 320—380°. Названы
вместе с тем в качестве катализаторов — из металлов: Си, Ag, Fe, Со, Ni, Zn, Sn,
окислы металлов: Сг, Мп, V, Се, Be, Са, далее СаСО3, CuCl, CuCl2 и ВаС12. Реакцию
можно проводить, разводя смесь паров воздухом, азотом или углекислотой.
Из этих разноречивых указаний можно вывести, что наивыгод-
нейшее решение задачи контактного гидролиза хлорбензола в парах
нельзя считать найденным. Возможно, что здесь при переходе
к производственному масштабу встретится много затруднений со
стороны подбора материала для аппаратуры, могущей выдержать
при высокой температуре, совместное корродирующее влияние водя-
ного пара и хлористого водорода. Ввиду того что при этом спо-
собе применение едкого натра не исключено, экономичность его
сравнительно с методом высокого давления—под сомнением.
Отметим тем не менее недавний американский патент на применение гидролиза
в парах к полихлорозамещенным бензола и некоторым хлорозамещенным с другими
заместителями. По патенту взаимодействие с водяным паром при температурах
в 550—850° над пористыми катализаторами (силикагель, боксит, магнезит, окиси
Zn, Th или Ti) приводит к получению диоксибензолов (любого изомера) из соот-
ветственного изомера дихлорозамещенных (также из трихлорбензола получают
пирогаллол или флороглюцин); из нитрохлорбензола — нитрофенол. При количестве
воды, меньшем эквивалентного, получают обмен не всех атомов хлора на гидро-
ксил и следовательно как продукт хлорфеиолы37).
Превращение галоидбензола в дифениловый эфир
С6Н5Х+ Ме'ОС6НБ С6Н5ОС6Н5 + Ме'Х
где X — атом галоида, а Me' — атом металла щелочной группы,
изучалось Ульманом, который констатировал благоприятное
для исхода реакции влияние прибавок небольших количеств меди
(в виде порошка). Реакционность галоидов изменяется в порядке:
J < Вг< С1. Реакция с хлорбензолом и фенолятом калия с прибавкой
меди (0,1 г Си на 1 мол. С6Н.,С1) проходит при температуре 200°
в течение 7 час. Этим методом, используя полигалоидбензолы
и феноляты одноатомных фенолов (но не моногалоидбензол и фе-
ноляты полиатомных фенолов), можно получить фениловые эфиры
полиатомных фенолов, например:
/X
С6Н4 / J- 2Ме'ОС8НБ С6Н4(ОС6Н6)2 + 2Ме'Х.
ХХ
216
Возможно в непрерывном процессе, как описанный выше для
фенола, проводить получение дифенилового эфира из хлорбензола
с одновременным получением уменьшенных количеств фенола, поль-
зуясь упомянутым выше равновесием между фенолом и дифениловым
эфиром в реакционной смеси. При этом нужно иметь в виду, что
понижение концентрации гидроксильных ионов (щелочи вводится
5—15°/о, считая в молекулярном отношении к хлорбензолу) задер-
живает гидролиз дифенилового эфира и следовательно содействует
его повышенному выходу38). Дифениловый эфир имеет практический
интерес для парфюмерной промышленности, производства пласти-
ческих масс и может быть полезен как передатчик тепла в тепло-
обменных аппаратах для высоких температур.
Вопрос об обмене хлора на гидроксил в полихлорозамещенных
бензола, так же как в хлорфенолах, данные литературы не вполне
разъясняют.
Получение хлорфенолов из п- и о-дихлорбензолов предложено
осуществлять действием едких щелочей (NaOH) в среде метило-
вого спирта.
Например n-дихлорбензол с 2,5 частями метилового спирта и 4,5 мол. NaOH
дает после 40 часов нагревания до 190—195° в железном автоклаве 90%-ный выход
n-хлорфенола а9):
/\/С1 /\/0№
I | 4-2NaOH= | | -4-NaCl+H2O.
С1/\/ СГ4/
Так же ведет себя и о-дихлорбеизол.
А. И. Киприяиов и Дашевский получили выход в 74,6% о-хлорфенола
нз о-дихлорбензола при нагревании последнего с метиловоспиртовым раствором
3 мол. NaOH, i/70 ч. (по весу дихлорозамещенного) меди в течение 2,5 час. до 225°
в железном автоклаве (не опубликовано).
С другой стороны, такой же и даже более полный обмен хлора был достигнут
введением медного катализатора без обязательного устранения воды. Например
при 21-час. нагревании до 200° о-дихлорбензола с 4-кратным количеством разведен-
ного на Via водой этилового спирта и 21/3 мол. КОН (на 1 мол. дихлорозамещенного)
в автоклаве с пластинкой меди получают по описанию немецкого патента пирока-
техин:
Здесь подчеркивается благоприятное влияние меди, весьма вероятное на осно-
вании известных уже нам фактов в аналогичном превращении хлорбензола. Не
лишена вероятия возможность, что железо, а может быть и другие металлы при
обработке в щелочной среде могут содействовать отщеплению хлора с замещением
его водородным атомом, о чем подробнее будет сказано ниже.
Подобно этому, но уже с полным устранением из реакционной среды спирта
и при работе с водными растворами едких или углекислых щелочей в медных или
серебряных автоклавах (в последнем случае с добавкой медной соли) можно по
описанию патентов получить из хлорфенолов диоксипроизводные (соотв. пирокатехин
и гидрохинон) 40).
Тем не менее обработка концентрированными растворами щелочей в приме-
нении к днхлорозамещенным (и хлорфенолам) может повести, подобно тому как
указывалось в предыдущей главе, к образованию гидроксильного остатка не у того
атома углерода, какой был связан с хлором. При сплавлеиин n-хлорфенола со
щелочами получается в большей или меньшей мере резорцин41).
217
Поэтому более практичным с этой стороны кажется описанное в весколькнх
германских патентах применение в качестве веществ, содействующих гидролизу н
отнятию кислоты, окисей нли гидроокисей щелочноземельных металлов (Са, Ba, Sr).
Эти реагенты содействуют только обмену хлора иа гидроксил н позволяют
мягко, постепенно проводить реакцию как с хлорокси- так и с дихлорозамещен-
ными. Прибавление меди (иногда иодистых солей) дает полезный эффект .Реакция
проводится в аппарате с медными стенками в водной или водио-спиртовой среде,
пределы температуры 170—240° в зависимости от реакционности галоида (у хлор-
фенолов ои подвижнее, чем у дихлорбензола) 42).
Имеющееся в литературе указание (Schwyzer) на возможность получения
пирокатехина в гомогеиио-освинцованном железном автоклаве при нагревании
o-ClCgH^OH с гидроокисью бария в воде, кажется сомнительным и требует поверки.
Обмен хлора на гидроксильную группу в хлорнафталине недо-
статочно изучен. Имеются противоречия в литературе, касающиеся
возможности гладко провести это превращение а-хлорнафта-
лина 34), 39). Во всяком случае вопрос заслуживает более тщатель-
ного изучения.
Обмен хлора в антрахиноне и в данном случае проходит срав-
нительно легко. Нагреванием с концентрированным раствором ще-
лочи из p-хлорантрахинона можно получить ализарин 43). Следо-
вательно здесь наряду с обменом хлора происходит такое же
образование гидроксила в a-положении, какое характерно для
щелочной плавки ^-сульфокислоты:
ОН
ONa
СО | ONa
Превращение хлоропроизводиого в оксизамещенное может быть осуществлено
во время самого синтеза антрахинонового замещенного. Так, при конденсации
фталевого ангидрида (1) с л-хлорфенолом (в среде концентрированной серной
218
кислоты в присутствии борной кислоты) получается не 1.4-хлороксиантрахинон
как следовало бы ожидать, но хинизарнн (II) «):
При более низкой температуре можно получать и оксихлоропродукты ®). Ход про-
цесса гидролиза хлорозамешенного в кислотной смеси требует выяснения. Стран-
ным образом получение хииизарина якобы удается (с пониженными выходами) прн
применении не п-, а о-хлорфенола.
При применении о-хлорфенола нужно ожидать при конденсации с фталевым
ангидридом получения 1.2-замещенного, следовательно ализарина:
ОН
S? X /ОН
СО
который действительно н получен с выходом в 75% Танака (с сериой н борной
кислотами при 255°) 46).
Метод получения хииизарина н его замещенных таким методом конденсации,
использующим хлорфенолы, в последнее время интересует многие заграничные пред-
приятия 47).
Наличие в бензольном ядре кроме галоида еще иных реакти-
вирующих галоид групп, стоящих к галоиду в благоприятном по-
ложении, естественно, повышает реакционность галоида и облег-
чает превращение хлоропроизводного в оксипроизводное. Гидролиз
п- или о-хлорнитробензола в соответственные нитрофенолы вод-
ными щелочами происходит, правда, в закрытых аппаратах (под
давлением), но температура, для этого требуемая, невысока
(110—130°), давление — 3—5 ат.
Повышение температуры значительно ускоряет реакцию гидро-
лиза. 2.4-Динитрохлорбензол дает динитрофенол при нагрева-
нии с водным раствором едкого натра или даже соды уже при
обычном давлении. Мы видим, как повышается реакционность
галоида в ряду
/\/С1
При получении этим методом нитро-(соотв. динитро-) фенолов
необходимо считаться с возможностью побочных реакций, возник-
новение которых может быть обусловлено помимо прочих факто-
ров реакции и материалом стенок реакционного аппарата.
Нитрогруппа, особенно стоящая в орто-месте по отношению к галоиду, обла-
аает достаточной подвижностью и при щелочной обработке может легко отще-
219
пляться, связываясь в виде соли азотистой кислоты. Большая концентрация щелочи,
перегревы, содействуют такой нежелательной реакции. Проведение гидролиза о-ии-
трохлорбензола в присутствии меди не позволяет при прочих равных условиях по-
лучить о-нитрофенол, что удается сделать, используя стальную аппаратуру (не-
опубликованные опыты Э. С. Л е в и н а и Ш. М. И т т е н б е р г a)
Отмечено ускоряющее влияние ультрафиолетового облучения на обмен хлора
под действием щелочных агентов в хлоро- (соотв. бромо-) замещенных бензола (при
участии медных катализаторов). Облучение по мнению Розенмунда возбуждает
молекулу, особенно активно относящуюся в этом случае к каталитическим воздей-
ствиям 49).
Нитрофенолы и особенно динитрофенол — очень важные про-
межуточные продукты; первые — для получения соответственных
аминосоединений, нужных для красочной, меховой и фотографи-
ческой промышленности, 2.4-динитрофенол — как промежуточный
продукт для получения сернистого черного и пикриновой кислоты»
нитро-о-аминофенола и 2.4-диаминофенола, служащего фото-
проявителем (амидол).
Реактивирующее влияние со стороны групп NO,, SO3H, СООН
на гидролиз хлорозамещенных ряда бензола при благоприятном их
размещении по отношению к атому хлора определяется рядом
NO2 > SO3H > СООН.
При этом влияние этих групп измеряется величинами весьма различного по-
рядка. Так, для 2.4-двузамещенного хлорбензола это отношение приблизительно
таково: NO2: SO3H : СООН = 1:1/70 000: 1/200 000.
Для п - замещенного 1-хлор-2-нитробензола ряд SO3H : СООН : Н = 16 :4,4:1;
здесь содействует проявлению реакционности прежде всего o-NO2-rpynna и за-
тем стоящая в пара-положенин — SO3H нли СООН в°).
4-Хлор-о-бензоилбензойная кислота и ее 3-нитропроизводное, интересные
как промежуточные продукты в получении антрахиноновых замешенных, также
легко превращаются при нагревании с раствором щелочи (первая в автоклаве)
в соответственные оксизамешенные и), например
z\/C0\z\zN°2 z\zC0\z\zN°2
N Н — II II
'Z^COaH^/^Cl \/\с02н\/\0Н
Как очень практически интересные превращения галоидопроиз-
водных в кислородсодержащие замещенные необходимо отметить
получение из хлорозамещенных алкильных эфиров фенолов.
Общее выражение реакции (7) при этом получает такой вид:
RC14- HOAlk = ROAlk -f~ НС1.
Так как реакция ведется всегда в присутствии щелочного агента,
например едкого натра, то соляная кислота связывается им, и
в уравнение реакции входят дополнительные члены:
RC14- HOAlk 4-NaOH = ROAlk 4- NaCI 4- H2O. (8)
Этот вид превращений в производстве имеет значение для о- и
и-нитрохлорбензолов (и нитродихлорбензолов) с целью получе-
ния из них алкильных эфиров соответственных нитрофенолов. Ал-
килы, входящие в состав продукта, — или метил или этил, а ре-
агентом, HOAlk (8), является или метиловый или этиловый спирт.
Несмотря на кажущуюся простоту реакции и удобные условия
220
ее выполнения — в однородной среде, спиртовом растворе, в ко-
тором растворимы и исходный материал и реагент и органический
продукт реакции, это превращение тем не менее принадлежит
к числу очень деликатных и требующих очень тщательного наблю-
дения для того, чтобы не быть осложненным крайне нежелатель-
ными побочными реакциями. В самом деле, исходным материалом
является соединение, заключающее кроме достаточно подвижного
галоида нитрогруппу. Последняя в щелочной среде может под-
вергнуться восстановительному воздействию со стороны спирта
(не исключается и катализирующее влияние металла стенок реак-
ционного аппарата), — результатом может оказаться более или
менее значительная примесь к главному продукту продуктов вос-
становления, преимущественно азоксисоединения, и далее амина,
например
/\/С1 /\ /ОС.,Н- С1\/\ /\/С1
-» j + +•
О
/\/G1
"Ь
h2n/\/
Восстановительное действие метилата натрия на нитробензол
обнаружено давно 52). Работа с этиловым спиртом требует не ме-
нее, если не более, осторожности в этом отношении. Поэтому
здесь внимание ведущих процесс должно быть направлено на устра-
нение побочных реакций, в которых участвует нитрогруппа исход-
ного материала, и доведение до минимума возможного гидролиза
хлорнитрозамещенного от действия щелочи и воды, образующейся
при реакции:
O3NC6H4C1 + 2NaOH = O2NC6H4ONa -f-NaCl + H2O.
Факторами, влияющими иа образование азоксисоединений,
являются повышенная температура реакции, примесь альдегида
в применяемом спирте, повышение концентрации щелочи в реак-
ционной смеси. Количество нитрофенола в продукте реакции рас-
тет с повышением температуры и концентрации щелочного рас-
твора. Индивидуальная природа самого нитрохлоропроизводного,
число и положение нитрогрупп также конечно много значат в исходе
реакции Б3).
Так, интересно, что действие алкоголятов высокомолекулярных спиртов на п- и
м - нитрохлорбеизолы дает начало азокснсоедннениям и аминам, между тем о-ии-
трохлорбензол образует с теми же алкоголятами замещенные аминокислоты 51), на-
пример
о-С1 • C6H4NO2 + RCH2CH2OH-> o-Cl • C6H4NHCH(R)COOH.
122
Реакция нитрохлоропроизводнык со спиртовым раствором щелочи
протекает при нагревании в открытом аппарате, например с обрат-
ным холодильником, но для более быстрого ее проведения в произ-
водстве пользуются закрытыми аппаратами (автоклавами). Нагре-
вают смесь до температуры, не на много превышающей темпера-
туру кипения соответственного спирта при атмосферном давлении.
Состав и порядок прибавления щелочи может быть различен в за-
висимости от оптимальных условий проведения реакции. Ниже мы
укажем некоторые из приемов, которые описаны в патентной и
научной литературе.
Нитроаннзолы O2N • С6Н4 • ОСН3 получаются при нагревании до 105—110° ии-
трохлорбеизолов с нодным метиловым спиртом (80—90%-вым) и щелочью в тече-
ние 4—5 час., продукт промывается горячей водой (очевидно для устранения нитро-
фенола). Тот же эффект получается при многочасовом (60—65 час.) кипячении смеси
с обратным холодильником, причем конденсирующиеся в холодильнике пары захва-
тывают при обратном токе щелочь (КОН или NaOH) и уносят ее в реакционную
смесь, ... поддерживается постоянная н невысокая концентрация щелочи. Ранее
появления этого американского патента такой же принцип работы был с успехом
испытан Филипповым и Сериковым55).
л-Нитрохлорбензол превращается в л-нитрофенетол п - O2N • С6Н4 • ОС2НБ при
нагревании сильно разведенного (3—5% содержания нитрохлорбензола) раствора
исходного продукта в спирту, содержащем 15% воды, до 90—120° с количеством
щелочи ие выше эквивалентного хлоросоединению w).
Слабую концентрацию ОН-ионов предлагается осуществить в реакционной смеси
(с участием порошка меди) введением в качестве щелочного агента избытка карбо-
натов щелочных или щелочноземельных металлов или таких гидроокисей металлов,
которые не обладают щелочной реакцией [Zn(OH)2]. Возможно применение водного,
спирта 57).
Участие своеобразных, содержащих медь, катализаторов обеспечивает по опи-
санию германского патента количественный выход п - нитрофенетола при нагрева-
нии до 100° нитрохлорбензола со спиртовым раствором едкого натра (около 50%
избытка). Катализатор — комплексное соединение меди (Си-) и глицерина или дру-
гого полиатомного спирта
Наконец с целью устранить восстановительный процесс американская фирма
применяет введение в реакционную смесь кислорода или воздуха под давлением
(или введение иных окисляюших средств), причем этот метод дает якобы возмож-
ность работать в сравнительно очень концентрированвом растворе при 100° с л-ни-
трохлорбензолом и при 105—110° с о - нитрохлорбензолом 5Э).
Из алкильных эфиров нитрофенолов наибольший практический
интерес представляют о-нитроанизол (I) и п-нитрофене-
тол (11), затем 2.4-динитроанизол (III) и я-х л о р-о-н и т р о-
анизол (IV).
I
zxzoch3 ZZZOCHS
III IV
Cl/^/^NOs,
Первый служит исходным материалом для о-анизидина, кото-
рый помимо своего значения как диазосоставляющей красителей
находит большое применение в промышленности фармацевтических
продуктов (для получения гваякола и его препаратов). Для кра-
сочной промышленности о-нитроанизол особенно важен как исход-
ное нитросоединение в синтезе дианизидина (см. восстановление);
n-нитрофенетол при восстановлении дает амин—п-фенетидин,
222
применяемый для синтеза известного медицинского средства фена-
цетина (n-ацетаминофенетола). Амины, отвечающие двум послед-
ним нитроэфирам (III и IV), применяются как диазотирующиеся
основания в холодном крашении по нафтолам типа AS.
Эфиры фенолов с другими, кроме NO2, реактивирующими группами не встре-
чают пока большого интереса. Реактивирующее значение отдельных групп при
одинаковом их размещении относительно галоида во взаимодействви с метилатом
натрия изменяется в порядке:
NO2 > CN > SO3H > СООН > СН2ОН.
Группы ОН, ОСН3, SH, NHg не реактивируют атома хлора, стоящего в орто-
положенин 4).
л-Хлор-о-нитробензоилбензойная кислота может быть переведена в мети-
ловый, соотв. этиловый, эфир л-оксипроизводного нагреванием ее в очень мягких
условиях (40—60°) с метиловым, соотв. этиловым, спиртом и щелочью в количестве
1—1,5 мол. на 1 мол. хлорнитробензоилбензойной кислоты 6“), например
/Х/СО\/\/ СО \/\/NO3
| | | | 4-СН3ОН + NaOH = I I J j +NaC14-H2O
\/'\cO2Na\/z\ci ^^COaNa^/^OCHs
Нитро- и динитропроизводные галоидоняфталина практически
до сих пор не применяются в производстве.
Интересно отметить, что хлорный атом в 1-хлор-2.4-дини-
тронафталине (II) значительно подвижнее при взаимодействии
с алкоголятами натрия, чем хлор в 2.4-динитрохлорбензоле (I).
Константы скоростей превращения с CH3ONa относятся (для 25°) как к\'. Л„ =
= 1:13,9 и (для 0°) Ai:Air= 1 : 30,8; с C2H6ONa: для 25° = 1: 8,9; для 0°
kx: Лп = 1:24,9.
Таким образом второй углеродный цикл нафталина влияет как
реактивирующая группа в орто-месте [например в пикрилхло-
риде (III)] 61)
С1 С1 С1
Y/N°2 /X/!X//NO2 02NxJx/N0.2
I II III
Y XYZ Y
no.2 no2 no2
В тех соединениях, где хлор связан с алифатической
боковой цепью, он является наиболее подвижным и может
быть легко замещен на гидроксильную или иную кислородсодер-
жащую группу. Наиболее интересным примером таких галоидных
соединений являются производные толуола, особенно хлористый
бензилиден С6Н5СНС1а и бензотрихлорид С6НБСС13 как
дающие бензальдегид и бензойную кислоту и хлорангидрид послед-
ней— хлористый бензоил.
Реакция замещения хлорных атомов для получения двух пер-
вых из названных продуктов производится действием гидроокиси
223
кальция при нагревании с известковым молоком, причем очевидно
предварительно происходит замещение хлора на гидроксил и затем
уже образование карбонильной, соотв. карбоксильной, группы 62):
С6Н6СНС12
С6Н3СН(ОН)9-------->
— Н2о
С6Н6СС13-------► С6Н5С(ОН)3------------►
- Н2О
Гидролиз проходит удовлетворительно и в присутствии иных
гидроокисей, связывающих образующуюся соляную кислоту, на-
пример Zn(OH)2. Могут найти применение в аналогичных гидроли-
зах и карбонаты щелочных и щелочноземельных металлов (Па2СО3,
СаСО3).
При этой реакции гидролиза по описанию германского патента
может оказывать значительное влияние в смысле ускорения тече-
ния процесса и облегчения его условий (более низкая температура)
прибавление железных солей и самого железа 63) и без участия
щелочных агентов.
Так как железо влияет очень активно на другое направление
реакции ад-хлорозамещенных толуола—-отнятие хлороводорода без
гидролиза с возникновением продуктов конденсации 64), например
дифенилметанового ряда, то очевидно здесь должны быть подо-
браны специальные условия для осуществления гидролиза.
Для некоторых замещенных (хлористого бензилидена и бензогрихлорида) за-
мена хлора кислородом может быть произведена нагреванием с крепкой серной
кислотой 63).
Я. Я. Макаров-Землянский предлагает для гидролиза хлористого бен-
зилидена применять борную кислоту, переходящую в метаборную:
С6Н6СНС12 4- Н3ВО3 = С6Н6СНО + 2НС1 + НВЭ2.
Реакция проходит при 130°. Выход бензальдегида 85—90% 66).
Прежде для получения бензальдегида применялся хлористый
бензил, причем обработка велась в присутствии окисляющих
средств (азотнокислого свинца), так как один обмен хлора на
гидроксил приводит к образованию бензилового спирта:
CeHsCH2Cl-----► С6Н5СН2ОН.
Недавно П. П. Щзрыгиным предложено применять для получения бензаль-
дегида также окислительный гидролиз хлористого бензила прн пропускании его
паров вместе с воздухом и перегретым паром через катализатор окисления (V2OS
на пемзе) при 360—330°. Образующийся при этом вначале бензиловый спирт окис-
ляется далее в бензальдегид и отчасти в бензойную кислоту 67). Вряд ли этот спо-
соб может оказаться более выгодаым сравнительно с обычным методом гидролиза
хлористого бензилидена.
224
Хлористый бензил имеет ныне значение fi качестве „бензили-
рующего" (алкилирующего) средства для аминосоединений и для
получения бензиловых эфиров карбоновых кислот, каковые эфиры
входят в состав некоторых современных лаков для покрывных
красок и может быть для искусственных смол м).
Получение из хлористого бензила бензилового спирта,
если таковой нужен, может быть достигнуто одним из способов гидро-
лиза, практикуемых при получении бензальдегида из хлористого
бензилидена. Повидимому слабоосновные гидроокиси, например
Zn(OH)2 или карбонаты щелочноземельных металлов (СаСО8), ока-
зываются при этом пригодными 68).
Б енза л ь д е г и д — продукт важный в синтезе многих основ-
ных и кислотных красителей триарилметанового ряда; он же
находит спрос в парфюмерной промышленности.
Бензойная кислота как таковая для красочной промышлен-
ности не имеет серьезного значения, но важна для производства
натриевой соли, применяемой в медицине и в консервном деле. Способ
производства бензойной кислоты параллельно с получением бензаль-
дегида— наиболее старый из синтетических. В последнее время
он имеет сильного конкурента в методе получения ее из фталевой
кислоты, каковая в виде ангидрида готовится окислением нафта-
лина. Этот последний метод дает бензойную кислоту, абсолютно
свободную от хлорозамещенных, чего нелегко достигнуть при ста-
ром методе. Окислением толуола также получается вполне чистая
бензойная кислота.
Для красочной промышленности важно иметь средство ввести
преимущественно в аминосоединения, радикал бензойной кислоты,
бензоил С6Н5СО—. Бензоиламинозамещенные антрахинонового
ряда — частью сами уже красители кубового ряда, как некоторые
желтые, красные (дибензоилдиаминоантрахинон), фиолетовый ди-
бензоилдиамино-диоксиантрахинон и др.; среди промежуточных
продуктов имеются также в нафталиновом по преимуществу ряду
ценные бензоиламинозамещенные, например бензоил-И-кислота:
ho3sx/4/x/nhcoc6h5
он
Сама бензойная кислота для введения бензоила мало пригодна,
и предпочтительно пользуются ее хлорангидридом — хлористым
бензоилом С6Н6СОС1 (см. главу XII).
Вместо того чтобы готовить этот последний из бензойной кис-
лоты, выгоднее оказалось проводить гидролиз исходного для бен-
зойной кислоты вещества, бензотрихлорида, останавливаясь
на стадии образования именно хлористого бензоила, т. е. осущест-
влять реакцию:
С6Н6СС13 4- Н2О = С6Н5СОС14- 2НС1.
15 Зак. МП. —Н. Ворожцов 225
Предложено довольно много способов такого частичного
гидролиза. Вода добавляется постепенно, в строго рассчитан-
ном по реакционному уравнению количестве (если надо, с неболь-
шим избытком в 5—1О°/о), к нагреваемой до 100° смеси бензотри-
хлорида с катализатором (безводное хлорное железо, хлористый
цинк, серная кислота). Реакция обнаруживается по выделению
хлористого водорода. Под конец реакция оживляется подогрева-
нием при размешивании. Количество катализатора 0,1 — 1% от
веса бензотрихлорида. Гидролизом уже образовавшийся хлористый
бензоил можно перевести в бензойную кислоту:
СГ)Н5СОС1 + Н2О == С6Н6СО2Н -4- НС1.
Но последняя при наличии избытка бензотрихлорида постепенно
реагирует с ним, давая вновь хлористый бензоил:
С6Н5СООН + С6Н3СС13 = 2С6Н5СОС1 + НС1.
Эту последнюю реакцию можно проводить при более высокой
температуре.
Подобным гидролизом замещенных бензотрихлорида могут быть
приготовлены замещенные хлористого бензоила 69).
Катализаторами этого гидролиза вероятно могут быть и другие
соли и кислоты кроме перечисленных. При проведении процесса
и при последующей очистке перегонкой хлористого бензоила
нужно выбирать материал аппаратов, не могущий проявить ката-
литических влияний и достаточно стойкий против коррозии. По
описаниям патентов — подходящий материал или свинец или ке-
рамика.
По американскому патенту 1932 г. можно получать хлористый бензоил как
промежуточный этап для синтеза бензойной кислоты [катализаторы гидролиза:
ZnCl2, А1С13, Zn (пыль), Fe], пользуясь далее превращением хлораигидрида в эфир
бензойной кислоты — при нагревании его со спиртом — и последующим гидролизом
сложного эфира. Таким образом удается получить вполне чистую бензойную кис-
лоту, соотв. ее соль:
+ Н.О + СН3ОН + NaOH
С6Н5СС13-----> С6Н6СОС1---------> С6Н3СООСН3--------> C6H3COONa
- 2НС1 - НС1 — СН3ОН
Можно проводить реакцию получения хлористого бензоила из бензотрихлорида,
пользуясь не водой, а другими соединениями, могущими дать водород и кислород.
Тогда вместе с хлористым водородом получаются иные соединения. Действием
спирта иа бензотрихлорнд получают наряду с хлористым бензоилом хлористый
этил:
с6н3са3 с2н-он = свн5соа 4- с2н3а 4- на
Лучше эту реакцию вести в присутствии хлорного железа. При действии амидов
оргаинческих кислот или аммониевых солей последних, в качестве второго про-
дукта реакции получается нитрил кислоты, взятой в виде амида или соли, например
С6Н3СС13 4- CH3CONH2 = С6Н6СОС1 4- CHSCN 4- 2НС1
2С6Н5СС13 4- RCOONH4 = 2С6Н3СОС1 4- RCN 4- 4НС1
С6Н3СС13 4- RCOONH4 = С6Н3СОС1 4- RCN 4- 2НС1 4- Н2О
Полезно и здесь прибавление катализаторов (ZnC!.,, FeCl3, А1С13, H2SO4, Н3РО4),
так как они позволяют снизить температуру реакции до 100° 72).
Само собой разумеется, что замена простого гидролиза реакцией с введением
нового органического компонента может оправдываться лишь экономическими вы-
годами такого комплексного направления реакции, например потребностью в допол-
226
ните.чьном продукте (нитриле, хлористом этиле) в количествах, приблизительно
эквивалентных потребности в хлористом бензоиле.
Гидролизом бензотрихлорида в присутствии подходящих соеди-
нений металлов (соли, окислы: Zn, Си, Fe, Al, Sn) количеством
воды, отвечающим стехиометрии уравнения:
2С6Н5СС13 + ЗН2О = (С6Н5СО)2О + 6НС1,
получается ангидрид бензойной кислоты, который, так же как и
хлористый бензоил, но менее выгодно, может служить для внесе-
ния остатка С6Н5СО в амино-, окси- и другие соединения 73).
Хлорный атом хлористого бензоила и других хлорангидридов, обмениваясь на
водород под действием водорода в присутствии катализатора (коллоидальный Pd),
дает альдегид и)
RCOC1 + Н2 = RCOH + НС1.
С другой стороны, действием хлора на бензальдегид или смеси, его содержа-
щие, можно получить хлористый беизоил 73).
в) Обмен хлора на серусодержащие группы и на другие
заместители
Обмен хлора на содержащие серу группы, по пре-
имуществу на SH, —S—S —, соотв. S— и SO3H, происходит по
общей для всех этих превращений схеме с выделением хлористого
водорода, для связывания которого прибегают к применению ще-
лочных агентов. Очень часто в этих превращениях применяют
солеобразные соединения с содержащим серу анионом, катионом же
служит атом щелочного металла, который и связывает соляную
кислоту. Применение катализаторов здесь далеко не так широко
испытано и используется, как в двух рассмотренных выше типах
превращений.
Получение тиофенолов в общем имеет много сходства с полу-
чением фенолов.
Так, хлорбензол в водной среде обрабатывается 2 мол. сульф-
гидрата натрия NaSH в автоклаве, в котором пространство над
жидкостью заполнено сероводородом, при 250°, и дает тиофенол
вместе с малым количеством (3%) фенола и 10% дифеннлсульфида
C6HS•S • С6Н5. Главной реакции отвечает уравнение:
CSH5C1 + 2NaSH = C6H5SNa + NaCl % H2S,
совершенно подобное уравнению (8) получения фенола. Аналогично,
как в фенольном процессе, и здесь можно парализовать образо-
вание новых количеств дифеннлсульфида (аналога дифенилового
эфира), прибавив к реакционной смеси его в соответственных
отношениях 76).
Действием в водном растворе сернистого натрия Na2S (2,5 мол.) на о- н я - ни-
трохлорбензолы при нагревании можно получить о- и я - аминотиофенолы. Вероятно
реакция проходит через несколько стадий, например
O2N • СвН4 • С1 4- Na2S —> O2N • CSH4 • SNa NaCl,
O2N • CeH4 • SNa + Na2S + вода —> H2N • C6H4 • SH 77).
Действием сульфгидрата натрия NaHS иа хлористый бензил при 75— 80° полу-
чается бензиловый меркаптан
СвН5СН2С1 + NaHS —> C6H6CH3SH + NaCl
15*
227
Похоже на гидролиз хлорбензола в паровой фазе протекает взаимодействие между
хлорозамещенными бензольного ряда и сероводородом при высокой температуре
(около 700°). Катализаторами служат окись нли карбонат магния, окись тория, хло-
ристые соли металлов, стойкие к нагреванию и сероводороду. Реакция выражается
уравнением
RC1 + H2S = RSH + НС1 та).
Тиофенол ы — еще мало употребительные в практике вещества,
возможно их вовлечение в синтез некоторых кубовых красителей,
заключающих серу, или ускорителей вулканизации резины, воз-
можно, что они найдут себе применение в качестве агентов фло-
тации при обогащении руд и т. п.
Пара- и орто-нитрохлорбензолы реагируют с сернистыми ще-
лочами легко уже при невысоких сравнительно температурах.
Применение двусернистого натрия дает возможность получить ди-
сульфиды нитросоединений, например
2 O,N • С6Н4С1 + Na2S2 = O2N • С6Н4 • S • S • C6H4NO2 + 2NaCl so).
Действием метилмеркаптана CH3SH иа спиртово-щелочной раствор о-нитро-
хлорбензола получается о-иитрофенилметилсульфид 81):
/\/N°2
I
\/\sch3
Сами тиофеиольные производные могут быть использованы как реагенты для
введения содержащих серу радикалов вместо галоида, например:
Взаимодействие здесь проходит при 145—150° (растворитель — амиловый спирт)
в присутствии поташа 82).
Обмен галоида на сульфогруппу особенно интересен в приме-
нении к хлоропроизводным с галоидом, активированным благо-
приятно размещенными альдегидными СНО-группами.
В то время как стоящие в пара- или орто-положении по отно-
шению к СНО атомы хлора способны к замещению на сульфо-
группу при обработке солью сернистой кислоты, мета-расположен-
ный галоид остается вне взаимодействия. Поэтому возможно полу-
чить как хлорсульфокислоты бензальдегида, так и дисульфокислоты
его 83). Так,
/^/СНО
+ NaaSO3
— NaCl
^/^SOaNa
228
/Х/СНО ,^/СНО
2Na2SO3
- 2NaCl
NaO3S/^/^SOgNa
Cl-^/^/CHO Cl^/^/CHO
Na2SO3
' -NaCl
^/^SOgNa
Различные нитро- и полинитрозамещенные хлор- (соотв. бром-) бензола были
испытаны в реакции со средним сульфитом, и изучены скорости образования со-
ответственных нитро- и полинитросульфокислот. Обнаружено, что хлоро- и бромо-
производные реагируют приблизительно с одинаковой скоростью. Неожиданно было,
что о-, м- и я-нитрохлорбензолы приблизительно одинаково реакционноспособны.
Ряд активности галоида для них даже: л«>о>я. В ряду нитробромбензолов:
ii/jt/O. Здесь таким образом резкое отклонение от правильностей активности
в других видах превращений с обменом галоида. 2.4-дииитрохлорбеизол реагирует
в 2000 раз скорее, чем моноиитропродукт, между тем как 2.4.6-тринитрохлорбеизол
только в 25 раз скорее дииитропродукта 84).
Приведенным выше не ограничиваются все возможности пре-
вращения хлорного атома на иные атомы или группы, интересные
технически.
Отметим прежде всего, что имеются методы замены галоида,
и в том числе хлора, водородным атомом. Обычно это сопряжено
с участием восстановительно действующих агентов.
Так, из хлор- (соотв. бром-) фенолов можио получить феиолы действием водо-
рода в присутствии катализатора, заключающего никель илн другие металлы (Со,
Си, Мп) s5).
Действием железа в щелочном растворе прн высокой температуре также можно
из хлорфеиола получить фенол, например 86)
нох/ч/сн3 нох/х/сн3
\Z\ci х/
Также восстановительно действует и закись меди при температуре 300° и выше
в щелочном растворе 87). Из я-броманилин-о-сульфокислоты можно устранить
бромный атом обработкой цинковой пылью88).
1.3-Дихлор-2-аминоантрахииои может быть переведен в З-хлор-2-аминоаитрахиион
действием восстановителей (Na2S2O4, цинк, глюкоза) в щелочной среде при на-
гревании 89):
С1
Галоид в кубовых красителях устраняется обработкой гидразином NH2—NH2
в присутствии меди или подобного катализатора 90).
Возможна также замена хлора на нитрильную (цианидную)
группу. При действии на ароматические галоидопроизводные циа-
229
нида металла в присутствии каталитически действующих броми-
стых солей меди, никеля и кобальта или самих металлов в состоянии
тонкого раздробления получаются соответствующие галоидопроиз-
водным нитрилы, например
300°
AgCN + 5% (NiBr2 + CuBr)
Рис. 8. Автоклав.
Также из о-хлортолуола [Zn(CN)2-j- CuBr] получается нитрил о-толуиловой
кислоты, а из а-бромнафталина нитрил а-нафтойной кислоты 91).
а-Хлорантрахиноновые замещенные могут быть переведены в нитрилы нагре—
230
ваиием до 230—240° с цианистой медью (в эквивалентных обмениваемому хлору
количествах) и цианистым бензилом или другим нитрилом, причем роль нитрилов
(применяемых в 3-5-кратных количествах сравнительно с хлорозамещениым) неясна.
Пример э2):
Нужно отметить также попытку ввести в бензольное ядро вместо хлора кар-
боксильную группу путем пропускания паров хлорозамешенного, например хлор-
бензола, вместе с окисью углерода и водяным паром над катализаторами (соли
металлов или сами металлы: Ni, Си). Полного превращения за один пропуск не
удается достигнуть. Предполагается промежуточное образование хлорангидрида
кислоты RC1 —> RCOCI. Вряд ли этот метод может быть технически интересным 93).
Довольно мало до сих пор данных относительно реакционности
галоида в нафталиновых производных, но имеющиеся материалы
позволяют сделать вывод, что галоид как в а-, так и в р-месте
нафталинового ядра приблизительно так же, но не более, реакцио-
нен в превращениях, связанных с обменом на азот-, кислород-
и вероятно серусодержащие группы сравнительно с галоидом в бен-
зольных соединениях 94).
Аппаратура, применяемая в превращениях, частично указана
выше. Наиболее характерные аппараты здесь — закрытые аппа-
раты высокого давления (рис. 8), иногда (для непрерывного про-
цесса) — трубчатой системы 95).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) К. Н. Meyer, Вег. 54, 2265 (1921).
2) G. S с h i е m а п п, W. Ro s е 1 i u s, Ber. 64, 1332 (1931).
3) W. В о r s c h e, J. E x s s, Ber. 56, 2357 (1923); W. В о rs c h e, H. Feske, Ber.
61, 690 (1928).
4)T. deCrauw, Rec. 50, 780 (1931); A. F. Holleman, Rec. 35, 1 (1915).
5) S. C. J. Olivier, Rec. 42, 775 (1923), Soc. A. 123, i, 197 (1923).
6) H. Erdmann, Г. n. 61843 (F. Ill, 65).
7) См. H. Lindemann, A. Pabst, Ann. 462, 24, CB 1928, II, 349; Bonsche,
Ber. 56, 1488 (1923), Ann. 386, 356 (1912).
ч) Описание производства бензилаиилина см. Ch. Tr. J. 1925, 610.
^William J. Hale, Joseph W. Britton (Dow). Am. n. 1607824, Re
17280 (Apr. 1929). A. J. Quick, Am. Soc. 42, 1033 (1920). Более ранний (1908)
Г. п. (А) 204951 (F. IX, 116), ср. К- W. R о s е n m un d, Е. S t r u c k, Ber. 52,
1749 (1919) W. H. Williams (Dow), Am. n. 1840760, CB 1932, I, 3498.
10) J. W. Britton, W. H. Williams (Dow), Am. n. 1726170, 1726172, 1726173;
W. J. Hale (Dow), Am. n. 1764869.
u) Dow, Am. n. 1775360. 1804466, CB 1931, II, 1195. Также Г. п. (A) 202170, 204848
(F. IX, 118), 205415 (F. IX, 120).
12) P. H. Groggins, H.P. Newton, Ind. Eng. Chem. 21, 369 (1929); P. H. G г о g-
g i n s, A. J. S t i r t о п, H. P. N e w t о n, Ind. Eng. Chem. 23, 893 (1931); Grog-
gins, Stir ton, lb. 25, 42, 46, 169, 274, 277 (1933).
13) Anonymos, Metallborse 15, 2332 (1926).
u) J. Thomas, A. H. Davies, Am. n. 1657420.
lo) J. G u b e 1 m a n n (Newport), Am. n. 1654290,. R. A d a m s, J. M. D a v i d s о n,
I. Gubelmann, Am. n. 1614584, R. Adams, W. W. Moyer, Am. n. 1720751.
1B) W. J. Hale, G. H. Cheney (Dow), Am. п. 1729775; W. M. Grosvenor,
J. Miller, Am. n. 1445637, (1919 r.), CB 1925. II, 1800.
231
Промежуточные продукты, получаемые из галоидопроиэводных обменом
галоида на иные заместители
А. Обмен на азотные группы.
Б. Обмен на кислородные группы.
В. Обмен на содержащие серу группы.
1 — Анилин. 2 — л-Яитроанилия. 3 — 2.4-Динитроанилин. 4— л-Нитроанилин-о-сульфокислота..
«5—л-Хлор-о-нитроанилин. 6 — л-Хлоранилин-о-сульфокислота. 7 — л-Фенилендиамин. 8— Дифе-
ниламин. у — л-Нитродифениламин-о-сульфокислота. 10 — (З-Амнноантрахинон. 11 — Фенол.
12 — л-Ннтрофенол. 13 — о-Ннтрофенол. 14 — 2.4-Динитрофенол. 15 — Дифениловый эфир (ди-
фенилоксид). 16— о-Хлорфенол. 17— Пирокатехин. 18 — о-Нитроанизол. 1У— л-Нитрофенэтол.
20— 2.4-Динитроанизол. 21 — л-Хлор-о-нитроанизол. 22 — Тиофенол. 23 — Дифенилсульфид.
24 — о-Сульфокислота бензальдегида. 25 — 2.4-Днсульфокислота бензальдегида.
232
W) W. J. Hale (Dow), Am. n. 1804466 (1931).
18) CIBa, Б. n. 250819, Brit. Ch. Abs't. B. 528 (1926), CB 1927, 1 804
и) J. P. Wibant, CB 1916, II, 380.
20) С1 у, Г. n. 148749 (F. VII, 66).
2i) A. Wohl, Ber. 39, 1951 Cl906).
22) А., Г. n. 205150 (F. IX, 117).
23) O. Matter, Г. n. 375793 (F. XIV, 403).
2i) Cm. 22). О подвижности галоида в бромннтронафталииах — Ю. С. Залкннл
Ж. О. X. 1 (63), 151 (1931).
25) К. Н. Т. Pfister, Ам. п. 1752998.
2в) IG., Б. п. 339699, СВ 1931, II, 1925.
2?) А., Г. п. 202564 (F. IX, 119), А. И. Киприанов, М. М. Дашевский.
Получение хлорнитроанилинов из п- и о-дихлорбензолов — У. X. Ж 5
H.-Техн. ч. 241 (1930).
28) А. Н. Rhein lander, Soc. 1923, Т. 123, В. Linke, Ber. 56, 848 (1923);
А. В г е wi п, Е. Е. Т u г п е г, Soc. 1928, 332, СВ 1928,1,2081; Franzen,
Bockhauer, Ber. 53, 1174 (1920).
29) F. U11 m a n п , R. Dahmen, Ber. 41,3744 (1908); W. L e 4 n i a n s k i, CB
1929, I, 3105; P. E. W e s t о п, H. A d k i n s, Am. Soc. 50, 859; CB 1928, 1, 2380;
IG. (B. Malenkovic), Г. n. 459548, CB 1928,11, 1617.
30) A. Wahl, R. Lantz, Bull. (4) 33, 93 (1923), R. G. M. C. 28, 33 (1923);
St. Denis, Г. n. 365367 (F. XIV, 468), CB 1932, I, 2464.
si) F. U1 Iman n, Ann. 381, 1 (1911); Г. n. 221853 (F. X. 703), 237236 (A) (F. X,
708); Г. n. 174699 (By) (F. VIII, 365), 237546 (By) (F. X, 711).
32)Ullmann, EnzyklopSdie der technischen Chemie, 2 Anil. 8 (1931), 338,
J. В e b i e, Chem. Met. Eng. 37, 473 (1930).
33) W. J. Hale, Edgar C. Britton, Ind. Eng. Chem. 20 114(1928), Am. n. 1607618-
1737841, 1756110 (Dow), Aylsworth, Am. n. 1213142 (1917). Также M. И.
Ушаков, H. Д. 3 e л и н с к и й, Ж. П. X. 5, 362 (4932). H. Н. Ворожцов
мл., А. Г. О ш у е в, Ан. Кр. Пр. 1933 № 6, 224. Н. Н. Ворожцов мл.,
А. Г. О ш у е в, Сов. пат. 28219.
34) К. Н. Meyer, F. Bergius, Ber. 47, 3155 (1914).
35) Dow, Am. n. 1821800, CB 1931, II, 3042.
36) IG., Б. n. 288308, Chem. Age. 1928, 555, Бельг, n. 350287, CB 1929, I, 2234;
Г. n. 487310, (1927). Federal Phosphorus Co, Am. n. 1735327, CB 1930, I, 2009,
Hale, Britton (Dow), Am. n. 1806798, CB 1931, II, 1348; F. Raschig,
Ф. n. 698341, CB 1931, 11,1491. H. Dreyfus, Ф. n. 709184, CB 1931,11,2933;
Progil, Ф. n. 720721, CB 1932, 11, 616.
3?) S. J. Lloyd, A. M. Kennedy. Am. n. 1849844, CB 1932, 1, 2994.
38) F. U11 m a n п, P. Sponagel, Ann, 350, 83 (1906), также Ullmann,
C. Wagner, Ann, 355, 359 (1907); W. J. Hale (Dow) Am. n. 1744961.
39) Chem. Werke Ichendorf, Г. n. 281175 (F. XII, 155).
4°) В б h r i n g e r & S 6 h п e, Г. n. 284533 (F. XII, 157), BShringer & S 6 h n e,
Г. n. 269544 (F XI, 160).
41) A. Faust, Ber. 6, 1022 (1873).
42) By, Г. n. 249939 (F. X, 1330), BShringer & SShne, Г. n. 286266
(F. XII, 158), 288116 (F. XII, 159); Scfrwyzer, Ch. Ztg 34, 818 (1930).
43) J. Thomas, H. W. Here wo rd (Sc. D.). Am. n. 1744815, C. A. 24, 1394
(1930), ср. M. Phillips, Am. Soc. 49, 473 (1927); Sc. D„ Б. n. 246529, CB
1926, II, 2948.
44) By, Г. n. 255031 (F. XI, 588); H. H. Reynolds, L. A. В i g e 1 о w, A Study
of the preparation of quinizarine, Am. Soc. 48, 420 (1926), также Б n. 245584
(Unit. Aik. Co, Dodd, S p r e n t), Б. n. 248874 (те же).
43) Б. п. 234533 (В. D. L. Thomas, Hooley), Chem. Age, March. 3, 1926,.
Dyestuff Monthly Suppl. 24.
46) Tanaka, Ch. T. Ub. 1928 2/4, 6 no Proc. Imp. Akad. Tokio.
47) Например Newport, Am. n. 1790915; J. W. Or el up. Am. n. 1790510, CB-
1932,1, 2238; Newport, Am. n. 1790932, CB 1932, 1, 2239.
4e) А. В. Попов, У. X. Ж. 5, .H.-Техн, ч. 105 (1930), А. И. Киприанов,
П. И. Михайленко, У. X. Ж. 5, H.-Техн. ч. 225 (1930), BOhringer &
Sdhne, Г. п. 288116 (F. XII, 159). По гидролизу 2.4-динитрохлорбензола
в диннтрофенол см. L. D е s v е г g п е s, Chim. & Ind. 26, 507 (1931).
43) К. W. Rosenmund, К. Luxat, W. Tiedemann, Ber 56, 1950 (1923)..
M) W. Davies, E. E. Wood, Soc. 1928, 1122, CB 1928, 11, 238.
233
5i) I. Gubelfflann, J. Welland, O. Stallmann (Newport), Am. n. 1654287,
1654289.
53) К 1 i n g e r, Ber, 15, 866 (1882); H. S. F r y, J. L. Cameron, CB 1927, I, 2721.
53) Blom. Helv. 4, 1029 (1921), L. C. Raiford, J. C. Colbert, CB 1926, II,
2894; Richardson, Soc. 129, 522 (1926).
5i) С. M. Suter, F. B. D a i n s, CB 1928, II, 2645; F. B. D a i n s, V. О. К e п у о п,
CB 1931, II, 986.
65) DuP., Am. n. 1578943, CB 1926, I, 3363, Nat. Ап. Б. n. 239320, CB 1926, II, 1335,
О. P. Филиппов, С. А. Сериков, Сообщ. Научно-Техн. раб. республ.,
1920, II, 115.
56) W. Lew cock & The Caz Light & Cocke Co, Б. n. 204591, Soc. Ind. 1923,1169.
«) O. Matter, Г. n. 386618 (F. XIV, 425).
58) Ver. Aussig, Г. n. 453429.
53) L. S. Pratt, E. H. Weitz, W. L. Mills (DuP.), Am. n. 1619368.
w) I. Gubelmann, H. J. Weiland, O. Stallmann (Newport), Am. n. 1665541.
si) H. W. Talen, Rec. 47, 329; CB 1928, I, 1768.
63) R. Freund, Die Fabrication von Benzaldehyd und BenzoesMure im Grossbe-
trieb. Ch. Ztg 1927, 803.
63) p. Schultze, Г.п. 82927 (F. IV, 143).
«В Ср. С. H. У шаков, К о н, Ж. П. X. 3, 69 (1930).
65) Us. Rh. Г. п. 98433 (F. V, 41).
ее) Сов. п. з. св. 39261 (1929).
6’) Сов. п. з. св. 33783 (1929), И. П. Ш о р ы г и н, И К и з б е р, Е. Смолья-
нинова, Ж. П. X. 2, 149 (1929).
б») С h. F a b г. Pott Г. п. 484662.
«9) Г. п. (В) 331696 (F. XIII, 272); A. G е о г g е, Ам. п. 1557154, Р. F. Banghame
& Sc. D. Б. п. 308231 (1927), А. И. Киприанов (не опубликовано);
В. D. Со Б. п. 293924, СВ 1929, I, 1614.
70) A. George (Monsanto), Ам. п. 1866849 (1932).
’1) Ver. Aussig, Г. и. 472422, СВ 1929 I, 2823.
12) 1G., Б. п. 323948, СВ 1930, I, 2630.
та) В. D. Со, Б. п. 280373, СВ 1928, I, 1460.
,4) К. W. Ros enm und, Вег. 51, 585 (1918).
73) IC.L, Б. п. 310910, СВ 1929, II, 1217. .
’в) Dow Ам. п. 1825662, СВ 1931, II, 3264. Н. Н. Ворожцов мл. С. П. Ми-
ценгеидлер, Доклады Акад. Наук СССР, 1933, № 6.
”) R. Lantz (St.-Denis), Ф. п. 714682, СВ 1932, I, 1828.
78) Nat. Ап., Ам. п. 1842414, СВ 1932, I, 1829.
79) Н е у d е п, Г. п. 497570, СВ 1930, II, 622.
st) Wohlfahrt, J. pr. Ch. (2) 66, 553 (1902); В 1 а пk sтa, Rec. 19, 111 (1900);
Н. Н. В о р о ж и о в, В. В. Козлов, Ж. О. X., II, 940; Good-Jear Tire &
Rubber Со; Ам. п. 1669630 (1928).
st) Н. Н. Hodgson, F. W. Handley, Soc. Ind. 1927, 435 T.
®) F. U 11 m а п п, E. Knecht, Ber. 44, 3125 (1911); H. H. Hodgson, W. Ro-
senberg, Soc. 1930, 180, CB 1930, I, 1774.
ss) Г. n. 88952 (F. IV, 133), 98321 (F. V, 207), 91818 (F. IV, 187) (G).
st) M. M. Sprung, Am. Soc. 52, 1650(1930). По обмену галоида на содержащие
серу группы в присутствии меди: К. W. R о s е п m u п d, Н. Harms, Вет. 53,
2226 (1920); Rosenmund, Вег. 54, 438 (1921).
85) Rhein. Kampf er Fabrik, Г. п. 432802 (F. XV, 235).
8в) F. R a s c h i g, Г. n. 396454 (F. XIV, 423).
87) H. H. В о p о ж ц о в-мл. и П. В. Ка р л а ш, Доклады Акад. Наук СССР
1933. № 5.
88) Н. Kreis, Ann. 286, 377 (1898); Bas., Г. п. 84141 (F. IV, 90).
®9) Sc. D., Б. п. 299333, СВ, 1929, 1, 1615.
«°) IG., Ф. п. 699492, СВ 1931, I, 3517.
91) Roessler & Hasslacher, Chem. Со, Ам. п. 1702711, СВ 1929, I, 2824;
К. W. Rosenmund, Е. Struck, Вег. 52, 1749 (1919).
®2) К. S с h i г m а с h е г, L. Z u t р h е п, Ам. п. 1728216.
93) Н. Dieterle, W. Es с he п b a ch, СВ 1927,11, 2986; Г. п. 537610, СВ
1932, I, 1155.
91) Loevenich, Loeser, Ber. 60, 320 (1927).
®3) Р. Н. G г о g g i п s, R. Н а 11 е г, Chem. Met. Eng 37, 693 (1930), СВ 1931, I, 2094.
ГЛАВА VIII
ПРЕВРАЩЕНИЯ АМИНОСОЕДИНЕНИЙ В ОКСИСОЕДИНЕНИЯ
И ОБРАТНЫЕ ПЕРЕХОДЫ
Значение аминосоединений и оксисоединений в синтезе кра-
сящих веществ так велико, что очень немногие синтезы проходят
без участия представителей хотя бы одного из этих двух классов
соединений. Обычно или амин или гидроксильное соединение
в какой-либо из стадий синтеза применяются как важная состав-
ляющая новых более сложных молекул.
Аминогруппа, соотв. ее производные, например алкиламино-,
диалкиламино-, ариламиногруппы, так же как оксигруппа, оказы-
вают, как выше уже неоднократно было упомянуто, очень суще-
ственное влияние на повышение реакционности ароматического
ядра, заключающего эти заместители. Включение новых замещающих
атомов или групп протекает у амино- (соотв. окси-) соединений
с значительно большей легкостью, чем у соединений, свободных
от этих групп. Стойкость по отношению например к окисляющим
воздействиям, характерная для бензола, в значительной мере ослаб-
ляется уже в анилине и феноле и еще более у двузамещенных
амино- (соотв. окси-) группами. Можно сказать поэтому, что амино-
и оксигруппы являются заместителями, характерно усиливающими
реакционность ароматических соединений.
К сказанному необходимо добавить, что участие аминогруппы
(и ее N-замещенных), так же как оксигруппы, в молекуле краси-
теля оказывает очень существенное влияние на его оттенок и на
красильные свойства. Эти две группц являются, как говорят,
типичными „ауксохромами" (усиливающими цветность). Таким обра-
зом значение амино- и оксигрупп в синтезе красителей трудно
переоценить.
Будучи сходными по своему влиянию на несущее их аромати-
ческое ядро, эти два заместителя могут получаться нередко один
на месте другого. Практика пользуется такими переходами амино-
соединений в оксизамещенные и обратно — оксизамещенных в амино-
соединения, особенно там, где обычные приемы получения окси-
соединений например через сульфокислоты или аминосоединений
через нитрозамещенные оказываются почему-либо мало удобными,
например дающими практически недостаточные выходы или боль-
шое количество изомеров и т. п.
Мы можем следовательно представить себе такую схему пре-
вращений для нитросоединений, соотв. для сульфокислот, с пере-
ходом их в амино- и оксисоединения:
RNO2 RNH2 ROH RSO3H.
235
Главное в этой схеме — превращение аминов в оксисоединения
и обратный переход оксисоединений в амины—может быть с успе-
хом осуществлено только применительно к соединениям опре-
деленной структуры.
а) Гидролиз аминов с переходом в оксизамещенные
Превращение аминов в оксизамещенные проще всего предста-
вить как пример гидролитического взаимодействия по уравнению
RNH24-H2O = ROH + NH3. (1)
Уравнение (1) является выражением лишь начального и конеч-
ного состояний реакционной смеси, и течение процесса вообще
проходит через промежуточные, иногда довольно сложные, стадии.
Гидролиз аминогруппы в гидроксил может быть выполнен раз-
личными методами. Самым общим методом, пригодным для гидро-
лиза первичной, ароматически связанной аминогруппы, является
метод перехода через диазосоединения: амин подвергается пред-
варительно диазотированию, и полученный продукт, соль диазония,
при кипячении с разведенной минеральной кислотой (серной) выде-
ляет азот и превращается в гидроксильное соединение с ОН-группой
вместо диазониевой группы. Схема такого превращения выражается
уравнениями
RNH2 + HNO2 + H2SO4 = RN2HSO4 4- 2H2O,
RN2HSO4 + H2O = ROH + N2 + H2SO4.
Подробности о диазотировании и превращениях диазогруппы —
в следующей главе. Здесь укажем лишь, что замещение группы
диазо гидроксилом протекает вообще довольно легко (хотя и не
с количественным выходом) при кипячении диазосоединений в кислот-
ном растворе. Нередко, особенно в применении к малореакцион-
ным диазосоединениям, полезно участие каталитически действую-
щих солей, например медного купороса. Такая прибавка описана
в немецком патенте на способ превращения о-аминофенола в пиро-
катехин ]):
y^yNH2 z^/N2X /\/ОН
\/\он \/\эН \/\оН
Менее общим методом гидролиза аминогруппы и применимым
лишь к аминосоединениям с особенно реакционной по положению
аминогруппой является обработка первичных аминов непосред-
ственно водными растворами щелочей или кислот.
Кипячением с щелочными растворами например удается, хотя и медленно,
заместить аминогруппу гидроксилом в 2.4-дииитроавилиие, я-иитроаиилине.
Более употребителен метод прямого гидролиза в применении
к а-нафтиламиновым замещенным и самому а-нафтиламину. Хотя
для некоторых сульфокислот а-нафтиламина, соотв. а-амино-8-наф-
236
толов, гидролиз удается и при действии щелочных водных раство-
ров, но гораздо удобнее его проводить действием разведенных
кислот. Как выяснили наши с К. А. Грибовым опыты, амино-
группа в нафтил аминовых и аминонафтольных сульфокислотах
практически полностью гидролизуется при нагревании соответствен-
ной содержащей аминогруппу сульфокислоты в течение 1—2 час.
с 3%-ным раствором серной кислоты при 180—190°. Конечно реак-
цию нужно проводить, при этом в автоклаве 2).
Таким образом удаются например гидролизы
HO3S NH2
\/\/\so3h
HO3S ОН
/\/\
\/\/\so8h
он
HO3s/\/\/\sO8H
HO nh2
HO3s/W\sO3H
HO OH
HOgS/^/^/ \SO3H
Интересно отметить, что в этих случаях сульфогруппы в р-поло-
жении и 8-(пери-)положении к аминогруппе оказываются невовле-
ченными в реакцию, между тем как сульфогруппы в 4.5-(а-)местах
по отношению к аминогруппе в вышеприведенных условиях не-
стойки и при нагревании с разведенной серной кислотой отщеп-
ляются в виде молекулы серной кислоты (см. главу VI, например3)
237
Температура гидролиза при этом может быть снижена за счет увеличения его
длительности, как выяснили наши с Е. Н. Юрыгиной опыты применительно
к получению хромотроповой кислоты (1.8-диоксинафталин-3.6-дисульфокислоты) из
кислоты Н (1-амино-8-нафтол-3.6-дисульфокислоты) 4).
Незамещенный а-нафтиламин также легко превращается при
нагревании с разведенной серной кислотой в а-нафтол. Этим пре-
вращением практически пользуются для получения в производстве
а-нафтола, так как получение последнего через сульфокислоту
менее выгодно уже прежде всего потому, что а-нафтиламин почти
свободен от примеси p-изомера, чего нельзя сказать про а-сульфо-
кислоту нафталина.
Наши опыты с А. В. Гуторко выяснили условия протекания процесса гид-
ролиза а-нафтиламина при различных температурах н концентрациях растворов
серной кислоты 6).
Хотя промежуточные соединения, образующиеся при гидролизе нафтиламиновых
соединений растворами серной кислоты, и не изолированы, тем не менее пред-
ставляется вероятным, особенно учитывая своеобразную реакционность нафтали-
новых замещенных, что здесь замена аминогруппы гидроксилом проходит через
стадию присоединения элементен воды, например
NH2 НО NH2 ОН
причем участие в реакционной среде серной кислоты облегчает стабилизацию про-
межуточио-образующегося соединения н нафтол, содействуя отнятию аммиака
н виде сернокислой соли аммония. При таком допущении понятно, что гидролиз
аминогруппы действием разведенной щелочи проходит через ту же стадию с уходом
аммиака из реакциоииой сферы в виде газа, и понятво, что в условиях обработки
в закрытом сосуде (автоклаве) аммиак не уходит из реакционной среды вследствие
растворения и тормозит гидролитическое превращение.
При всех прочих равных условиях гидролиз в кислотной среде
для нафтиламиновых производных проходит легче, чем в щелоч-
ной среде 6).
Подобно нафталиновым аминопроизводным а(5)-аминоаценафтен переходит
н оксипроизводное при действии водных растворов минеральных кислот или ки-
слотнореагирующих солей (ZnCla, AICI3) при высокой температуре (200-—225°) '):
Н2С—СН2 Н2С—СН2
I I
nh2 он
238
Среди гидролизующих аминогруппу агентов имеется нашедший
себе особенное применение в нафталиновом ряду и дающий исклю-
чительно хороший эффект, именно—сернистая кислота и ее кислые
соли. Применение бисульфитов дает возможность работать в откры-
тых сосудах без повышенного давления при температуре до 100°-
Как выяснилось из исследований автора, этот реактив специфичен
и гидролизует легко те аминосоединения, которые можно интер-
претировать как реагирующие в двух тавтомерных формах.
Промежуточные соединения, заключающие остатки сернистой кислоты, полу-
чающиеся при гидролизе, близки по строению к бисульфитным соединениям
кетонов 8):
Образование сернистокислых эфирон, отвечающих нафтолам, например
/\ //\/zOSO2Na
\J\Z
каконое строение приписывает этим промежуточным соединениям Бухерер9)
мало правдоподобно.
Эти промежуточно-образующиеся соединения (некоторые из них удалось изоли-
лировать) таким образом аналогичны по строению тем, которые мы считаем вероят-
ными в ниде промежуточной фазы процесса щелочного плавления сульфокислот.
Полная схема превращений в этом процессе например такова:
+ Н2О + so
H2N SO3H
+ Н2О
-NHs*
Н^ЧозН
Практически этот процесс выполняется помощью обработки
соответственного амина бисульфитом натрия, как он получается
в технике, т. е. в виде раствора 35—38° Боме.
239
Такой раствор готовят насыщением сернистым газом раствора
соды или едкого натра, причем достигают полного превращения
вначале образующейся средней соли Na2SO3 в кислую NaHSO3.
Хорошей пробой полноты превращения н этом случае может служить проба
с формальдегидом: смешиваются ранные объемы раствора и продажного форма-
лина, н н смесь прибавляют каплю фенолфталеина, — в случае образонаиия бисуль-
фита растнор останется бесцветным.
Применение бисульфита требует избытка его н количестве 3—4 и более молекул.
Обработка сводится лишь к кипячению с обратным холодильником, иногда при
небольшом превышении давления сверх атмосферного (10—15 мм ртутного столба).
Необходимо наблюдать за исчезнонеиием взятого амина иапример по диазопробе,
если был взят моноамии. Закончив реакцию, полученное оксисоединение выделяют,
разложив полученный промежуточный продукт щелочью [NaOH, Na2CO3, Са(ОН)2],
прокипятив до полного удаления аммиака, подкислив затем минеральной кислотой
и удалин из кислотной жидкости сернистый газ кипячением. Продукт выпадает
по охлаждении или высаливании. Иногда можно не прибегать к обработке щелочью,
довольствуясь лишь кислотной обработкой, получая при этом н некоторых случаях
продукты присоединения сернистой кислоты к оксипроизводному.
Реакция обработки кислыми солями сернистой кислоты, как
выше сказано, имеет значение преимущественно в нафталиновом
ряду. В бензольном оказываются способными к ней лишь мета-
диамиио- (соотв. мета-оксиамино-) производные.
В нафталиновом ряду бисульфитная обработка в применении
к аминам дает фенолы в обоих случаях (для а- и р-производных),
когда аминогруппа является единственным заместителем.
Во многих нафтиламиновых сульфокислотах также удается
бисульфитный гидролиз. Не поддаются такой обработке те лишь
сульфокислоты нафтиламинов, у которых сульфогруппа стоит
в мета-положении к аминогруппе, следовательно производные р-наф-
тиламина с сульфогруппой в 4-м месте и производные «-нафтил-
амина с сульфогруппой в 3-м месте. Кроме того из а-нафтилами-
новых сульфокислот нереакционны имеющие сульфогруппу во
2-м месте. Между тем расположение сульфогруппы в 1-м месте
по отношению к ,3-стоящей \'Н2-группе или в 4-м месте по отно-
шению к «-расположенной аминогруппе делает заместитель очень
способным к гидролизу.
Наиболее известен производственный пример приложения би-
сульфитного метода гидролиза — получение из нафтионовой кислоты
так называемой кислоты Н ев и л я-В и н т е р а (1-нафтол-4 сульфо-
кислоты)
NH2
I
Тормозящее влияние сульфогруппы, стоящей в мета-положении, и некоторые
другие факты реакционности бисульфитных соединений делают вероятным допу-
щение С. В. Богданова, что одной из реагирующих форм н бисульфитной
240
реакции является соединение с лабильной связью сульфогруппы с мета- (по отно-
шению к NH, или ОН) расположенным углеродным атомом 10), например
NH3
Азокрасители с нафтолом или нафтиламином в качестве азо-
составляющей, также нитрозонафтолы с соответственным положе-
нием азо- (NzrN) и нитрозо- (изонитрозо-) групп по отношению
к амино- (соотв. окси-) группам ведут себя в реакции с кислыми
солями сернистой кислоты совершенно подобно сульфокислотам
нафтиламинов, соотв. сульфокислотам нафтолов, образуя в резуль-
тате взаимодействия продукты присоединения кислой соли сер-
нистой кислоты к нафтольным соединениям. Таким образом реак-
ционными оказываются I, II [X = N —NR, NO, (:NOH), SO3H] и не-
реакционными— III u)
NHo(OH)
NH.,(OH)
III
Аминогруппа (а-) в антрахиноне, соединения которого, как не-
раз уже было упомянуто, отличаются своеобразными химическими
отношениями, гидролизуется легко при условии введения в реак-
цию не самого антрахинонового производного, а продукта восста-
новления (лейкосоединения), в котором характерные СО-группы
превращены в С • ОН. Гидролиз осуществляется или при действии
кислотных агентов или при кипячении со щелочами; в последнем
случае вместе со щелочью вводится и восстановительный реагент
(гидросульфит, Na2S2O4) 12).
Таким образом возможно например превращение
16 Зак. 2611.—И. Ворожцов
кипячение с NaOH -J-Na^Qj
ОН
| он
/\/с\/\
\Z\Z\/
с I
I он
он
241
б) Аминирование оксисоединений
Превращения гидроксила в аминогруппу, или так
называемое аминирование, имеет большое значение для полу-
чения тех аминосоединений, которые не могут быть приготовлены
через нитростадию и для которых аналогично построенные соеди-
нения с оксигруппой легче доступны.
Проще всего представить себе общее выражение этого взаимо-
действия в виде
ROH -Ь NH3 = RNH2 + Н2О,
не говоря о подробностях протекания процесса.
Отсюда видно, что процесс нуждается в аммиаке или отдающих
аммиак веществах в качестве так называемых аминирующих аген-
тов. Ввиду легкости улетучивания аммиака в большинстве случаев
приходится проводить процесс в закрытых сосудах при повышен-
ном давлении.
Аминирующими агентами служат, во-первых, сам аммиак как
таковой или в виде водного, реже — спиртового раствора, во-вто-
рых, соединения аммиака с хлористым кальцием и хлористым
цинком, получаемые насыщением газообразным аммиаке м соответ-
ственных солей, и, в-третьих, целый ряд аммиачных легко диссо-
циирующих солей, каковы хлористый аммоний NH4C1, уксусно-
кислый аммоний NH4O2CCH3, углекислый аммоний (NH4)2CO3 и
особенно — сернистокислый аммоний (NH4)2SO3, также как двойная
аммонийнонатриевая соль сернистой кислоты.
Бринером и его сотрудниками доказано, что аммиак образует уже при
низкой температуре нестойкие продукты присоединения с одно- и двуатомными
фенолами и их замещенными. В этих продуктах соотношение между количеством
молекул фенола и аммиака различно для разных фенолов. Так, аммониакат р-наф-
тола содержит % молекулы NH3 на 1 молекулу нафтола, состав резорцинового
производного — средний между моно- и диаммониакатом, о-витрофенол дает моно-
и триаммониакат, ж-нитрофенол — только моноаммониакат 13).
Фенолы замещают свой гидроксил аминогруппой с различной
легкостью.
В то время как обыкновенный фенол С6Н5ОН при обработке аммиачным соеди-
нением хлористого цивка (ZnCl2-2NH3) и хлористым аммонием при 330° дает
выход в 44% анилина наряду с 6% непрореагировавшего фенола и ЗЬ% дифенил-
амина 14), флороглюцин переходит просто при настаивании с аммиаком в 5-амнно-
резорцин 15):
ОН ОН
I I
П —* П
Но/^/^ОН Hq/^/^NH
Для вовлечения во взаимодействие ф. нолов бензольного ряда с аммиаком
(и аминами), по английскому патенту, рекомендуется применение хлоридов метал-
лов, могущих образовывать больше чем один хлорид (Fe, Со, Мп, Си, Sn и их
смеси) при температуре в 350—450° 16).
В английском же патенте немецкой фирмы указывается на применение дегид-
ратирующего катализатора [А1(ОН)3] в этих реакциях, проводимых при 400—4Ж J7).
Недавно Морганом и Пратом было констатировано, что хлористый
242
аммоний проявляет свойства хорошо действующего агента аминирования, особенно
выгодного для получения таких аминов, которые трудно получаются обычвым
путем нитрования и восстановления. Нагревая например л-крезол с половинным
по весу количеством хлористого аммония (2 часа до 350°, давление 50—60 ат), они
получили при общем превращении в 35% смесь оснований, состоящую из раввых
количеств лг-толуидива и ди-л-толиламина, следовательно превращение крезола
идет приблизительно так же, как и фенола 18):
Наибольшая реакционность и здесь проявляется у тех произ-
водных, которые могут реагировать в двух тавтомерных формах
(кето- и энольной).
Действительно, нитрозофенол (тавтомер — хиноноксим) уже при
нагревании на водяной бане с растворами аммиачных солей пре-
вращается в нитрозоанилин 19):
Так же легко реагирует с NH3 1.2-нитрозонафтол 20), нитрозо-
нафтолсульфокислота 21).
Понятно само собой, что практически эта реакция замены ОН
на NH2 применяется в сравнительно узком круге объектов и осо-
бенное значение она приобрела в нафталиновом ряду.
Получение p.-нафтилами на и его весьма ценных сульфокислот,
так же как сульфокислот р-аминонафтолов, ввиду практической
невозможности приготовить соответственные p-нитродериваты до-
стигалось и достигается с предварительным получением соответ-
ственных оксипроизводных и последующим их аминированием.
Само аминирование нафтолов и их замещенных производилось
прежде по преимуществу нагреванием с аммиаком, лучше в при-
сутствии хлористого кальция 22).
Б. Н. Меншутквн и Н. Бутков исследовали вопрос о получении нафтил-
аминон из нафтолов и аммиака при посредстве хлористого, соотв. бромистого,
цинка. Оказалось, что ZnCl2 и ZnBr2 не растворяются в нафтолах. С нафтилами-
нами же эти соли образуют кристаллические соединения: ZnCl2 и а-нафтиламин—
ZnCl2.2C]0H7NH2; ZnC)2 н fJ-нафтиламин — ZnCl2.2C10H7NH2; ZnBr2 и а-нафтнламин —
ZnBr2.4C10H7NH2; ZnBr2 и ₽-нафтиламин — ZnBr2.2C]0H7NH2; fi-нафтол и fi-нафтил-
амин' образуют соединение 2С10Н,ОН -J- C10H7NH2.
Наиболее действительный катализатор аминирования нафтолов СаС12 не обра-
зует повидимому соединения с исходными органическими веществами и продуктами;
а-нафтиламин из а-нафтола получается с ZnCl2 (соединение с 4ZnCl2) менее скоро,
чем при тех же условиях (З-нафтиламин (соединение с 2ZnCl3). Соединения продуктов
с катализатором таким образом затрудняют реакцию. Получение р-нафтиламина
затрудняется еще образованием продукта присоединения (3-нафтола и [3-нафтиламииа.
Повидимому способ работы с хлористым кальцием или цинком
имеет значение и теперь для получения аминосоединения, отве-
16*
243
чающего очень интересной практически 2-окси-З-нафтойной ки-
слоте, исходному продукту для многочисленных „нафтолов* типа AS:
/\/\/°Н
Так, американский патент 1927 г. описывает получение 2-амино-З-нафтойной
кислоты путем нагревания оксинафтойиой кислоты в автоклаве с избытком водного
раствора аммиака в присутствии хлористого цинка или хлористого кальция при
температуре свыше 200° 23).
Патенты немецкой фнрмы указывают иа применение здесь смеси хлористого
аммония с углекислым цинком или окисью циика. Реакция осуществляется в ша-
ровой мельнице просто нагреванием смеси или в токе газообразного аммиака.
Вследствие возможности взаимодействия между основным цинковым соединением
и хлористым аммонием, например
ZnO + 2NH4CI = ZnCl2 J- 2NH3 + Н2О,
с химической стороны в этом случае нет разницы от только-что описанного про*
цесса 21).
В одном американском патенте для этого же превращения опи-
сывается применение вместе с аммиаком соли железа (Fe") или
железных стружек; с последними необходимо введение в реакцию
растворимого хлорида или соли с другим анионом, могущей пере-
вести железо в ионное состояние. Температура реакции 200—210°.
Роль железа в самой реакции аминирования здесь не разъяснена,
но известно, что оно образует нерастворимое кристаллическое
соединение с продуктом реакции вероятного строения 2б)
z\/\/N\
>Fe
Может быть образование этого соединения, выпадающего из
реакционного раствора и тем выходящего из сферы реакции, облег-
чает дальнейшее продвижение взаимодействия в сторону образо-
вания аминопродукта и обеспечивает повышение его выхода.
Производные вышеупомянутой З-оксн-З-нафтойной кислоты, имеющие еще одну
группу ОН, при взаимодействии с аммиаком, а также его производными типа NH2R,
где R — алкильная или арильная группа, характеризуются повышенной реакцион-
ностью со стороны одной только оксигруппы. Так, при нагревании с аммиаком,
соотв. cNH,?, 2.6-диокси-З-нафтойной кислоты получается производное Р-окси-
Р"-аминонафтойной кислоты 26) по схеме
/\/\/°Н /\/\/°Н
III - HI
R = H, Aik, Ar.
Среди производных строения А имеются повидимому входящие в состав зеленых
азокрасителей, пригодных для холодного крашения.
244
Здесь уместно отметить, что весьма похожий по своим свойствам на а-нафтсл-
3-окситиоиафтен (1) при нагревааии с водным рас’вором аммиака до 180° образует
почти колнчествевно ди-(тьонафтенил-3-)-амин (II), между тем как в:греваиие его
до низшей температуры (135°) с большим избытком сильно концентрированного
(5С%-ного) раствора аммиака является методом получения Замино-тисвафтева (III) -7).
Более употребителен и выгоден способ аминирования окси-
соединений нафталина посредством нагревания с сульфитом аммония
и аммиаком.
Он разработан Бухерером и Баденской анилиновой и содовой
фабрикой 28). Реакция в этом процессе оказывается обратной реак-
ции гидролиза аминогруппы. Это можно выразить схемой:
Не исключена возможность возникновения как фазы реакцион-
ного превращения р-нафтольных, соотв. р-нафтиламиновых, произ-
водных промежуточного соединения с прямой связью углерода
и сульфогруппы (для ^-соединений в 4-м месте)10), нарример
4- h3O-4-So2
Н SOsH
Границы применимости сульфитной реакции аминирования
в нафталиновом ряду те же, что и для бисульфитной реакции гид-
ролиза. Сульфогруппа в мега-положении к гидроксилу защищает
последний от аминирования, так же как стоящая в орто-положении
245
по отношению к а-гидроксилу сульфогруппа. Это обстоятельство
делает возможным вести реакцию аминирования диоксисоединений
с уверенностью, что в результате получится не диамино-, но амино-
оксисоединение, если один из гидроксилов находится под неблаго-
приятным влиянием сульфогруппы. Таким образом
ОН
HO3s/\/\/
В то же время сульфогруппа, стоящая в 1-м месте по отношению
к гидроксилу во 2-м месте (р), так же как сульфогруппа, находя-
щаяся в 4-м положении по отношению к гидроксилу в 1-м месте (а),
сообщают гидроксильным группам большую реакционноспособность
во взаимодействии с сульфитом аммония, чем она проявляется со
стороны гидроксилов, не находящихся под таким благоприятным
влиянием сульфогруппы. Таким образом
ОН ОН
переходит в
ОН NH2
О возможности получения нафтиламинов из нафтола действием аммиака
в паровой фазе будет сказано ниже, в главе о каталитических процессах.
В бензольном ряду сульфитная реакция аминирования имеет весьма ограни-
ченное значение только для днокси- н аминооксидериватов (мета-).
Необходимо отметить при этом, что если вместо аммиака и
аммиачного сульфита применить моно- или диалкиламин, соотв. их
сернистокислые соли, то в результате получатся моно- (соотв. ди-)
алкилированные амины )29.
Аналогичным путем возможно введение в нафталиновое ядро остатка ш-амиио-
алкиламина, например NH2 — СН2 — СНа — NH2, причем кроме диамина в реакцион-
ную среду вводится кислая соль сернистой кислоты (натрия нли калня), а в каче-
стве нафтольной составляющей реакционной массы применяются цо патенту окси-
246
нафтойные кислоты (1.2, 2.3, 2.6). Таким образом процесс включения аминоалкил-
аминового радикала сопровождается отщеплением карбоксила, например
ZX/\/0H + н.» - сн,- СН, - nh. zx/x/NHICH^NH,
| | j(+H8so,)ill J
\/\/\ссюн \/\Z
Реакция происходит при нагревании до 90—100°, следовательно в открытом
аппарате з°).
Также возможно введение остатка гидразина H3N — NH2 вместо гидроксильной
группы в нафтолы н в резорцин при участии солей сернистой кислоты 31).
Оксихинолиновые производные подобно нафталину аминируются при обработке
их сернистокислым аммонием 32), например
Также действием сернистокислого аммония можно заместить аминогруппой
остаток ОН в 1-оксикарбазоле зз).
NH I
NH2
На этих примерах мы видим, как велика область применения
сульфитной реакции аминирования и соответственно бисульфитной
реакции гидролиза.
Практика выполнения всех видов реакции аминирования, как сказано выше,
требует обычно повышенного давления. Сульфитная реакция по сравнению с дру-
гими способами работы протекает при низших температурах и давлениях. Ее
проводят в технике в закрытых железных, чугунных или эмалированных котлах
с мешалками. Значительного избытка против теоретически потребного количества
аминирующего агента не применяют. По окончании реакции отгоняют избыточный
аммиак и отделяют выделившийся продукт, если он не растворим в воде. Если же
аминопронзводное растворимо в слабощелочных жидкостях (сульфокислоты наф-
тиламинов, аминонафтолов), то подкисляют полученную жидкость, что обычно ведет
к выделению продукта аминирования. Иногда необходимо бывает н высаливание
(см. выше, выделение сульфокислот).
Аминирование оксиантрахиноновых производных, так же как и
оксипроизводных нафтохинона, осуществляется легче всего в при-
менении к восстановленным (лейко-) продуктам. Нагревание этих
последних с аммиаком или аминами, частью даже не при высокой
температуре, лучше при участии щелочей в реакционной массе,
приводит к замещению оксигруппы аминогруппой, например 84)
ОН ОН
ОН ОН
247
OH nh2
\/
I I
он nh.2
окисление
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
Us. Rh. Г. п. 167211 (F. VIII, 128).
2) Н. Н. Ворожцов и К. А. Грибов. Заяв. свид. СССР 19397 (1927),
30277 (1928).
3) Ю. Г. В е н д е л ь ш те й н, Р. М. Шпинель, Ан.-Кр. Пр. 1932, № 4, «16.
Н. Н. Ворожцов, Е. Н. Ю р ы г и н а, Ан. Кр. Пр. 3 № 10, 453 (1933).
6) Н. Н. Ворожцов и А. В. Г у т о р к о, О гидролизе а-нафтиламина (готов,
к печати).
в) Ср. Г. п. 74879 MLB. (F. III, 422), 69190, Г. п. 68721 (F, III, 464—5) (By),
67062. 75153 (F. Ill, 466) (С).
’) IG., Г. п. 517264, СВ 1931, II, 1757.
8) Ворожцов Н. Н., О реакции между кислым сернистокислым натрием н
азотокрасящими веществами, Москва, 1916, 13, Ж. 47, 1669, (1915), Bull (4) 35, 996
(1924), Известия И. В. П. И. № 6, Ж. 61, 483 (1929), Вег. 62, 57 (1929).
9) Bucherer, J. pr. Ch. 69, 49 (1904).
10) С. В. Богданов, Ж. О. X. 2 (64), 9 (1932).
и) Ворожцов, 1. с., Н. Н. Ворожцов и С. В. Богданов, Ж. 61, 497
(1929).
12) IG., Г. п. 436526 (F. XV, 661) MLB, Г. п. 207668 (F. IX, 699), 148792,
(F. VII, 186).
13) Е. В г i п е г, О. Agathon, Helv. 10, 770 (1927), Е. В г i n е г, А. М о г f, Helv.
11, 926 (1928).
И) Merz, Mil 11 er, Ber. 19, 2906 (1886).
15) Pollak, Mon. 14, 419 (1893).
16) ICI. Б. n. 355715; Ch. Tr. J. 89, 543 (1931).
ii) IG., Б. n. 368373; Br. A. B. 1932, 591.
is) G. T. Morgan, D. D. Pratt, Soc. Ind., 51, 283 T, 1932.
1») O. Fischer, Hepp, Ber. 20, 2475 (1887), 21, 684 (1888).
2°) M. А. Ильнискнй, Ber. 19, 343 (1886).
21) К., Г. n. 601,20 (F. Ill, 808).
22) Merz, Weith, Ber. 13, 1300 (1880), 14, 2343 (1881), Benz, Ber. 16, 8, (1883).
23) R. Tobi er (CIBa), Am. n. 1629894, IG, Б. n. 282450, Chem. Age, Dyest. Monthly
Suppl. 20, 15 (1929). Cp. R. Mohlau, Ber. 28, 3096 (1895).
21) IG., Ф. n. 672305, CB 1930, I, 3486, Ср. Б. n. 284998, CB 1929, II, 1472.
25) E. Hotz, V. Lanz, Am, it. 1690785 (1928).
26) IG., Ф. n. 670462, CB 1930, I, 1538.
27) W. К о n i g, G. H a m p r e c h t, Ber. 63, 1546 (1930).
28) Bucherer, J. pr. Ch. (2) 69, 49 ^1904).
2») В., Г. n. 121683 (F. VI, 192).
з°) W. Duisberg, W. Hen tri ch. L. Z e h, J. Huismann, Am. n. 1633929
(1927), IG., Г. n. 468811, CB 1929, I, 3146; Г. n. 467627.
ai) H. Franzen, J. pr. Ch. (2) 76, 205 (1907). Franzen W. Deib e 1, J. pr.
Ch. (2) 78, 157 (1908). H. Wieland, D. Juchum, J. Maier, Ber. 64, 2513 (1931).
й) N. N. W о г о s h t row, J. M. К о g a n, Ber. 65, 142 (1932).
33) IG., Б. n. 316962, CB 1929, II, 2732.
34) B„ D. Co, Б. n. 268891, CB 1929,1,145. D re y f u s, Ф. n. 667415, CB 1930; I, 596;
Brit. Celanese Ltd, Б. n. 310784 (1928).
248
ГЛАВА IX
ДИАЗОТИРОВАНИЕ И ПРЕВРАЩЕНИЯ ДИАЗОГРУППЫ
а) Методы получения и свойства диазосоединений
Реакция диазотирования имеет большое значение в производстве
азокрасящих веществ и обычно рассматривается вместе с этим
классом красителей.
Но имея в виду, что, с одной стороны, диазосоединения, хотя
и не долговечные, все же несомненные промежуточные продукты
фабрикации красителей и что, с другой, многие простейшие азо-
красящие вещества применяются в технике почти исключительно
как исходные материалы для получения иных, более интересных
по окраске и свойствам соединений, мы в дальнейшем подвергнем
обозрению вышеназванную реакцию.
Продукты диазотирования носят названия диазосоединений.
В зависимости от последующих взаимодействий с составными частями
реакционной среды диазосоединения могут проявляться в различных
изомерных и стереоизомерных формах. Наиболее часто получаются
диазосоединения в форме солей диазония, где из двух атомов
азота реакционной группы Ы2 один трехвалентен, другой пяти-
валентен.
Реакция диазотирования применяется к первичным аминам и
требует участия одной молекулы азотистой кислоты и какой-либо
минеральной кислоты для образования соли диазония. Так как
в качестве источника азотистой кислоты в технике применяется
исключительно азотистокислый натрий, „нитрит** NaNO?, то общее
выражение реакции принимает такой вид:
RNH2 -j- МаМО2 + 2НХ = R • N2 • X -j- NaX -f- 2Н2О,
где X—одновалентный анион, например С1, х/2 SO4 и т. п.
Реакция сопровождается образованием чрезвычайно способных
к превращениям солей диазониев R — N —X. На один моль первич-
Ы
ного амина требуется и расходуется 1 моль нитрита. Что касается
минеральной кислоты, то ее количество обычно превышает теоре-
тически рассчитанное, и вместо двух молей кислоты (одноосновной)
употребляют 2i/4, 2lj2 и 3—4 молей. Необходимость избытка кислоты
обусловливается возможностью, при недостаточно кислой реакции
смеси, осложнения реакции диазотирования последующими реак-
циями образования диазоаминосоединений или азосочетания с уча-
249
стием в них свободного амина, еще находящегося в реакционной
смеси:
R(N = N)X4-H2NR------->R -N— N — HN — R
—HX диазоаминосоединеиие
R(NsN)X+H • R, • NH.,-------> R • N =zN • R2 • NH2.
—HX аминоазосоединение
Стойкость раствора диазо обычно выше в присутствии большего
избытка минеральной кислоты.
Те амины, которые дают особенно реакционноспособные диазо-
нии, исами очень склонны к азосочетанию, например а-нафтиламин,
л<-фенилендиамин, нуждаются в большем избытке минеральной
кислоты при диазотировании.
Типически реакция диазотирования проводится таким путем: амин растворяется
в минеральной (обычно—соляной) кислоте с водой, и к полученному раствору соли
амина при охлаждении (около 0э) прибавляют, размешивая, раствор соли азотистой
кислоты. Конец диазотирования узнается пробой капли раствора на иодо-крахмаль-
ной бумажке (фильтровальная бумажка, смоченная раствором йодистого калия в жид-
ком крахмальном клейстере и высушенная в темном месте с чистым воздухом);
(отношение количеств крахмала к KJ = 5:1, воды 500 ч.).
Эта реактивная бумажка окрашивается в темносиний цвет от выделившегося
действием азотистой кислоты иода.
Когда диазотируют сульфокислоты аминов, нерастворимые в ми-
неральных кислотах, то их сначала растворяют в эквивалентном
количестве щелочи и затем осаждают при охлаждении вливанием
избытка кислоты (с расчетом на добавочно необходимую молекулу
кислоты для нейтрализации щелочи). При такой обработке амино-
сульфокислота выпадает в мелкораздробленном виде, удобном для
диазотирования.
С целью сделать невозможными вышеназванные последующие
за диазотированием реакции азосочетания практикуются иногда
отклонения от типического диазотирования.
Так, амин (легче сульфокислоты аминов) переводится в раствор
с нитритом, и этот раствор, слабощелочной или нейтральный,
приливается к охлажденной льдом соляной кислоте. В таких усло-
виях свободный амин находится все время до своего полного
превращения в диазоний в присутствии очень большого избытка
минеральной кислоты, т. е. в условиях, затрудняющих азосоче-
тание.
Таков же смысл второго видоизменения хода диазотирования:
амин вводится, иногда в виде свободного основания, в охлажденный
льдом раствор азотистой кислоты, приготовленный из нитрита и
•соляной кислоты. Возможно низкая температура содействует сохран-
ности этого последнего раствора.
Ввиду большой способности к химическим взаимодействиям
получаемых диазосоединений, особенно при повышенной темпера-
туре, диазотирование как общее правило проводится при охлаж-
дении (0—5°)-
Тем не менее есть исключения из этого правила, и многие амины
выдерживают более высокую температуру при диазотировании.
250
Таковы особенно сульфокислоты аминов. Так, диаминостильбенди-
сульфокислоту
— СН —СН—<
•SO3H
диазотируют при 39°. Большинство сульфокислот нафтиламинов
может быть диазотировано при такой же или комнатной темпера-
туре. Диазонии, производные дифениламина ’) и антрахинона, могут
быть даже нагреваемы без разложения.
Реакция диазотирования протекает с различной скоростью в за-
висимости от свойств применяемого амина. Обычно скорость диазо-
тирования находится в прямой функциональной зависимости от
растворимости амина или его соли в данных условиях процесса.
Так, амины бензольного ряда типа анилина и его гомологов, кото-
рых соли с минеральными кислотами хорошо растворимы в водных
растворах, диазотируются легко. Сульфат бензидина растворим
трудно, и диазотирование этого основания в виде сернокислой соли
требует продолжительного времени. Сульфокислоты аминов бен-
зольного и нафталинового рядов в большинстве трудно растворимы.
Поэтому например нафтионовая кислота
NH2
SO3H
требует для полного диазотирования продолжительной обработки
азотистой кислотой. На энергичное размешивание при диазотирова-
нии таких нерастворимых аминов необходимо обращать внимание.
Скорость диазотирования различных аминов изучалась особенно Тассильи г).
Она изменяется в зависимости от природы диазотируемого амина. Работа произво-
дилась в присутствии 5 мол. соляной кислоты. Реакция диазотирования—бимолеку-
лярная реакция.
Скорость диазотирования различных аминов такова.
Анилин диазотируется практически в течение 1 часа, максимум через 5 час.
о-Толуидин диазотируется практически в течение 1,5 час.
л-Толуидин » „ „ » 5 час.
.«-Ксилидин быстро диазотируется вначале (45 мин.), затем медленно до 3 ч.45 мии.
л-Аиизидии—постепенно до 3 час.
о-Анизидин—быстро, реакция заканчивается в 15 мин.
Все три изомерных нитроанилина диазотируются сполна в 15 мин.
Толндин диазотируется в 15 мин.
Диазотирование сульфаниловой кислоты идет скорее в более концентрированных
растворах. Избыток нитрита при прочих равных условиях ускоряет, недостаток —
замедляет реакцию.
Е. И. Шпитальский и В. В. Монбланова установили, что при диазоти-
ровании анилина в солянокислом растворе скорость реакции несравненно больше,
чем при прочих равных условиях в эквивалентном растворе серной кислоты. Повы-
251
шение концентрации соляной кислоты до 9 эквивалентов кислоты на 1 эквивалент
анилина не влияет заметно на скорость, повышение же концентрации кислоты до
13 эквивалентов увеличивает скорость, дальнейшее возрастание концентрации кислоты
уже не влияет на скорость реакции. В одинаковой степени скорость диазотирования
солянокислого раствора анилина повышается при действии хлористых солей, откуда
вытекает возможность каталитического влияния хлор-иона.
Диазотирование сернокислых растворов анилина протекает прн достаточном
избытке серной кислоты одинаково медленно, почти независимо от ее концентрации
и сильно ускоряется хлор-ионами.
Присутствие серной кислоты сильно замедляет диазотирование анилина в соля-
нокислых растворах, но присутствие в одинаковых условиях эквивалентного коли-
чества сернокислых солей такого замедляющего действия не оказывает. Присут-
ствие сернокислых солей калия и аммония значительно ускоряет диазотирование
сернокислых растворов анилина 3).
Г. Г. Be н д е л ь ш т е й н, сравнивая отношение к диазотированию различных
изомерных сульфокислот а-нафтнламииа, констатировал, что введение сульфогруппы
в молекулу а-нафтиламииа понижает устойчивость NH2 к действию азотистой кислоты
и повышает Прочность диазосоединений.
Изомерные сульфокислоты нафтиламина можно разделить на три группы:
1) Изомеры 1.3, 1.6 и 1.7, имеющие близкие между собой константы скорости
диазотирования, скорости разложения диазосоединеиий и сочетания.
2) Изомеры 1.5 и 1.8, более устойчивые к действию азотистой кислоты и
дающие менее прочные диазосоединения.
3) Изомеры 1.4 и 1.2, менее устойчивье (сравнительно с 1 группой) по отно-
шению к азотистой кислоте и дающие более прочные диазосоединения 4).
Стойкость растворов диазосоединений была испытана Кэном и Николом,
а также Тассильи. Последним установлено, что хлористый февилдиазоний (QH^NgO)
в растворе при 0° не разлагается еще через 43 часа.
Диазо из о-толуидина при 0° не разлагается в течение 24 час.
„ , л-толундина при 15° практически стойко в течение 96 час.
„ „ о- и л-анизидинов не разлагаются заметно даже при 60°.
, „ л-иитроанилина выдерживает 2-часовое нагревание до 60°, но разла-
гается при 80°. Диазо из о- и лг-иитроанилинов менее стойки уже при 66°.
Бисдиазо из толидина стойко в течение 20 час. при 18°, но не выдерживает
нагревания.
Диазо из сульфаниловой кислоты при 18° стойко в течение 23 час. 2).
Кэн и Никол установили, что разложение диазосоединений
в водном растворе — мономолекулярная реакция, которой отвечает
обычное уравнение
А
А — х
где С—константа реакции.
Сравнивая полученные для разных температур значения константы С для неко-
торых диазо бензольного ряда, эти исследователи нашли, что у трех диазотолуолов
стойкость отличается между собой и сравнителгно с таковой у диазобензола: орто-
и мета-соединения менее устойчивы, чем диазобензсл, между тем как диазо из
л-толуидина гораздо более устойчиво, чем диазо из анилина. Введение сулгфо- и
нитрогруппы в высокой степени повышает устойчивость диазо. Диазо из сульфанило-
вой кислоты во много раз более устойчиво, чем диазо /z-толуидива, а все три диазо
изомерных нитранилинов устойчивее диазотированной сульфаниловой кислоты. Среди
диазо из нитранилинов самый устойчивый—орто-изомер, затем мета-и наконец пара-
изомер. Влияние положения нитрогруппы иа устойчивость диазо совегшевно противо-
положно действию метильной группы. Диазо из л-аединоацетанилида в 6,5 раз более
устойчиво, чем диазосульфаниловая кислота.
Для диазо из нафталиновых производных, диазоссединений, растворимых в усло-
виях проведения реакции, исследователи вывели следующую таблицу относительных
скоростей разложения при 60°.
252
Диазосоли из Относитель- ная скорость разложения Устойчивость
1. Анилина 2. а-нафгиламина . • 64,9 11,9 В 5,4 раза устойчивее 1, менее
3. „ ........ 14,9 устойчив, чем диазо из « толуи- дина. В 4,3 раза устойчивее 1, менее
4. 1-нафтиламин-8-сульфокислоты 6,1 устойчив, чем диазо из «-толуи- дина. Устойч. 1 и диазо из «-толуидина.
5. 2-нафтиламин-6-сульфокислоты 6,9
6. 2-нафгиламиН-7-сульфокислоты 5,9
7. 2-нафтиламин-6,8-Дисульфоки- слоты • .... 8. 2-иафтиламин -3,6-дисульфоки- слоты 3,2 1 Устойч. диазо из сульфаниловой кислоты. Устойчив, диазо из сульфаниловой кислоты.
Нерастворимые в воде диазосоединения сульфокислот нафтил-
аминов разлагаются по иному закону. Их скорость разложения выра-
жается формулой (реакции нулевого порядка):
где х—объем азота, выделившийся в промежуток времени t (в ми-
нутах).
Часто разложение осложняется вторичной реакцией сочетания
диазо с образовавшимся нафтольным производным в азокраситель.
Тем не менее до известного момента диазо и нафтольное произ-
водные могут находиться в смеси без сочетания. Вероятно это За-
висит от недостаточной диссоциации диазосоли (особенно в при-
сутствии минеральной кислоты—см. ниже) б).
Необходимо отметить работы японского химика Ямомото по скорости разло-
жения хлористого фенилдиазония в водном растворе. В частности разложение, вызы-
ваемое действием солнечного света, как можно ожидать, усиливается в соответствии
с силой света. Прибавка восстановителей, например Na3S2Os и NaHSO3, к водному
раствору диазосоединения для раскисления имеющегося в нитрите нитрата по
реакциям
2NaNO3 Н- Na,S2O5 = 2NaNO2 4- Na2S2O7,
NaNO3 + NaHSO3 = NaNO3 + NaHSO4
несколько повышает стойкость диазораствора в течение первых часов по пригото-
влении, что подтверждено иа примере диазо из а-нафгиламина П. П. Викторовым.
Из вышеприведенного понятно, почему для аналитических опре-
делений азосочетанием предпочтительно применяют диазо из п-
толуидина, а не его изомеров, так же как диазо из нитроанилинов.
В тех случаях, когда (пример — нафтионовая кислота) и амин и
диазосоединение трудно растворимы, по внешним признакам конец
диазотирования узнать невозможно. Но если занейтрализовать
избыток минеральной кислоты, прибавив соли уксусной кислоты
или бикарбоната, то образование азокрасителя (см. азосочетание)
дает доказательство присутствия непрореагировавшего амина.
253
Эта проба может быть применена в том лишь случае, когда
в диазотируемом соединении нет заместителя, могущего вызвать
независимо от аминогруппы азосочетание. Аминонафтольные суль-
фокислоты (правда, в большинстве хорошо растворимые) могут
давать интрамолекулярное азосочетание в очень разведенной кислоте
или слабой щелочи с образованием окрашенных продуктов, напри-
мер
ОН
ОН
НО35'/ч/Х/
Амины, в которых имеются сильно активные электроотрицатель-
ные группы, понижающие основность аминогруппы, могут быть
иногда с успехом диазотированы в концентрированной серной
кислоте нитритом или в концентрированной азотной кислоте с при-
бавлением раскисляющей азотную кислоту до азотистой пиросер-
нистокалиевой соли K2S2O5 6). Уравнение реакции образования
азотистой кислоты таково:
K2S2O6 + 2HNO3 = K2S2O7 + 2HNO2.
Слабоосновные амины диазотируются также раствором нитрита
в моногидрате, будучи растворены на холоду в ледяной уксусной
кислоте. Моногидрат (для NaNO2) и уксусная кислота (для раство-
рения амина) берутся в одинаковых количествах.
2.4.6-Тринитроанилин можно диазотировать и без уксусной
кислоты в концентрированной серной кислоте или моногидрате.
Добиться диазотирования трудно диазотируемых аминов можно,
по опыту Кришна и сотрудников, растворением амина в неболь-
шом количестве пиридина и, после введения туда же концентриро-
ванного раствора нитрита, вливанием полученной смеси в раз-
бавленную соляную кислоту 7).
Наиболее совершенным, хотя и не отличающимся простотой,
методом диазотирования аминов с значительным числом нитро-
или иных отрицательных групп в молекуле является путь перевода
амина в сульфаминовую кислоту (с группой NHSO3H) и обработки
последней азотистой кислотой. Для этого диазотируемый амин
вносят в предварительно приготовленную смесь пиридина (в избытке)
и хлорсульфоновой кислоты, заключающую по вышесказанному
(см. гл. III) ангидро-Ы-сульфокислоту пиридиния при 35—40° или
выше, и через некоторое время добавляют туда такое количество
едкого натра, чтобы образовать натриевую соль сульфаминовой
кислоты, отгоняют пиридин с паром и полученную соль, размешав
ее с водой, обрабатывают концентрированной соляной кислотой
и рассчитанным количеством нитрита при невысокой темпера-
туре 8).
-254
Реакции здесь могут быть выражены следующими уравнениями:
C6H6N + C1SO3H = C5H6NSO3 4- НС1,
C6H6NSO3+RNH2 = RNHSO3H4-C5H5N,
RNHSO3H 4-NaOH = RNHSO3Na4-H2O,
RNHSO3Na 4- 3HC14- NaNO2 4- H2O=RN2C14- 2NaCl 4-H2SO44- 2H2O.
Этим способом можно легко получить диазосоединения из поли-
хлоранилинов, пикраминовой кислоты, аминоазобензола, нитро- и
полихлорнафтиламинов, 3-аминокарбазола и т. п.
Очень трудно протекает диазотирование аминоантрахинонов. Здесь для успеха
диазотирования нужно пользоваться предварительным раствсревием амина в крепкой
серной кислоте и, после добавления в раствор эквивалентного амину количества
нитрита, выливанием массы в холодную воду, следовательно разложением ннтро-
зилсерной кислоты.
Другой метод диазотирования состоит в использовании для диазотирования
двусернокислотного эфира аминопроизводного, отвечающего антрагидро.хинону,
например
OSO3Na
OSO3Na
(см. ацилирование, глава XII). При этом лучше, если аминогруппа предварительно
переведена в сульфаминовую группу.
Имея в виду, что сернокислотный эфир гидролизуется от свободной азотистой
кислоты, выгоднее вводить сразу весь нитрит и постепенно прибавлять эквивалент-
ное количество соляной кислоты 9).
Применение концентрированной соляной кислоты оказывается
необходимым при диазотировании л<-диаминов для противодей-
ствия очень настойчивому стремлению полученных моно- и бис-
диазониев к сочетанию с непрореагировавшим еще диамином в азо-
красители, В этом и аналогичных примерах получения активных
диазониев (например из n-нитранилина) рекомендуется вести реакцию
возможно быстро 10).
Очень интересные для получения своеобразных азокрасителей
«-диамины и особенно п-аминофенолы бензольного и нафталино-
вого рядов не поддаются обычному диазотированию, так как азо-
тистая кислота окисляет их в хиноны и хиноиды.
Поэтому диазотировать их аминогруппы можно лишь посте-
пенно одну за другой, переходя или через стадию п-нитроамина
с последующим (за закреплением в азокрасителе группы — N : N—)
восстановлением нитрогруппы
o2n/ Озм/4'/ o2n/4//
255
Zx//N : NR /\/N : NR
H2N"/X/ cinX4^
или диазотируя n-аминоациларилид и отщепляя ацильную группу
после фиксирования группы—N :N— (азосочетанием) п)
NH, NHCOCHg NHCOCH3 NHCOCH3
nhcoch8 nh2 n2ci
гидролиз
N : NR N : NR N : NR
Посредством предварительного перевода в сульфаминовые кис-
лоты можно без затруднения провести одностороннее диазотиро-
вание и диаминов, в том числе и п- и о-диаминов, с получением
в результате сульфаминовых кислот диазосоединений. Метод по-
лучения полисульфаминовых кислот практически не отличается от
вышеуказанного для моносульфаминовых кислот. Диазотирование
производится посредством рассчитанного на одну аминогруппу
количества нитрита.
Таким образом возможны например превращения
/^/NH.;, NHSO3H Z\/N2
h2n/X// ho3shn//Xx// Z/X//
nhso2—о
причем полученная диазосульфаминовая кислота может быть
употреблена для сочетания. В американском патенте описаны при-
меры аналогичных превращений для .и-толуилендиамина, о-фенилен-
диаиина, бензидина, 1.5- и 1.8-нафтилендиамина 12).
Такие разноядерные n-диамины, как бензидин, диазотируются обычным приемом
сразу в обоих ядрах, и односторонне диазотированный бензидин можно получить
довольно длительным взаимодействием бас-диазосоединения бензидина, так назы-
ваемого хлористого дифенилтетразония (который правильнее было бы назвать хло-
256
ристым дифеиил-бас-диазоиием) и свободного бензидина. Обмен диазоииевой группы
на аминовую дает в результате хлористый л-амииодифеиилдиазоний 13):
cin2—/ / ^>-N2Cl
HgN-Z V NH2
НС1 \----z х---z НС1
Необходимо отметить, что о-диамины при диазотировании дают
не диазонии, но путем внутримолекулярного сочетания образуют
своеобразные циклические диазоаминосоединения, так называемые
азоиминосоединения, или азимины. Например
+ HNO,
—2НгО
Промежуточным соединением очевидно должно быть
Так же реагируют с азотистой кислотой и пери-диамины нафта-
линового ряда, образующие при этом пери-азимины типа
N
N^H
Азимины подобно диазоаминосоединениям — кристаллические
вещества, окрашенные в желтый цвет.
Применяя превращение в дисульфаминовую кислоту, можно как
уже было упомянуто, просто получать односторонне диазотиро-
ванные бензидин и о-диамины.
В триамиие такого строения
H2N^ /CH.,
Н,С-<~ >-NH2
h2nz \сн3
диазотируются лишь две аминогруппы. Повидимому бензольное ядро не может
нести более двух диаэогрупп н).
17 Зак. 2611. — Н. Ворожцов 257
Пара- и орто-аминофенолы как бензольного, так и особеннонаф-
талинового ряда, как было сказано, не поддаются обычным приемам
диазотирования. Тем не менее среди них имеются такие, которые
находят себе большое применение в качестве диазосоставляющих,
как например 1.2-аминонафтол-4-сульфокислота
NH2
/\А/ОН
SO3H
имеющая значение для получения черных хромируемых красителей.
Подобные соединения можно диазотировать, или применяя вместо
минеральной кислоты органическую, например уксусную, щавелевую,
в большом избытке (8—9 мол.) или проводя диазотирование в от-
сутствии минеральной кислоты, но в присутствии некоторых солей
тяжелых металлов (Си, Zn, Fe) или солей щелочных или щелочно-
земельных металлов 1б).
Наиболее употребителен прием диазотирования с введением
медной соли, например CuSO4 или СиС12, причем в результате
взаимодействия ее с нитритом натрия образуется нитрит меди,
который и производит диазотирование (см. нитрозирование).
Кроме аминонафтольных производных этим путем можно диа-
зотировать аминокрезол, аминотимол, аминоксиленол, а также и-ами-
норезорцин. Для этого предложены кроме меди и другие ме-
таллы (Zn, Ni, SnIV)16).
Получаемые из аминофенолов, соотв. их сульфокислот, диазо-
соединения не заключают аниона минеральной кислоты и имеют
строение или диазооксисоединений (I) или хинондиазидов (II)
N=N
I
SOsH
Химизм такого диазотирования нуждается в разъяснении.
Для большинства применений как в технике приготовления
промежуточных продуктов, так и самых красителей дназосоеди-
нения применяются в растворенном виде и тотчас же по приго-
товлении. Некоторые сульфокислоты (нафтил-) аминов и амины
антрахинона дают мало растворимые в воде диазосоединения, вы-
падающие в осадок.
258
б) Стойкие диазосоедииеиия
В некотором, сравнительно ограниченном, числе случаев при-
менения аминов в качестве диазосоставляющей для так называе-
мого холодного крашения и печати (по ^-нафтольному плюсу
или по нафтолам типа AS) диазосоединения заготовляют в твердом
виде или в виде пасты.
Получение таких стойких к хранению диазосоединений осно-
вано на некоторых приемах лишения их реакционности. Наиболее
простым и грубым приемом является введение в диазораствор
большого количества минеральных солей тяжелых или щелочных
металлов с последующим высушиванием всей смеси в вакууме до
тех пор, пока она будет застывать в твердую массу при обычной
температуре (применимы NaHSO4, ZnCl2, алюминиевые квасцы).
Двойные соли с солями тяжелых металлов (Zn, Cd) также в не-
которых случаях выделяются и без выпаривания как малореак-
ционные в твердом виде вещества 17).
При изучении стойких диазосоединений много внимания уде-
лено внесению анионов, могущих с положительным остатком диа-
зония образовать труднорастворимую соль.
Таково например предложение немецкого патента вводить кислоту с малой
pH (в частности борную кислоту) при устранении свободной азотистой кислоты
мочевиной и высаливать соль диазония нейтральными солями (NaCl). Так по-
лучают стойкие диазо из несульфированных амннопроизводных дифениламина18).
Несколько патентов заявлено на получение стойких препаратов диазониев по-
средством прибавления к диазорастворам борофтористоводороднсй кислоты BHF4
и некоторых других комплексных кислот: HaTiF«, H3A1F6, H2SnF6, H2SnCl6 • 6H,O,
HSbF4, HSbF6, HSbBr6 • 3H2O, H2ZnF4.
В этих случаях изолируются борофториды, соотв. соли иных комплексных солей
и диазониев 19).
Фторосульфонаты диазониев обладают малой способностью к вспышке и взрыву 20)
Большое применение в группе методов введения нового аниона
в соль диазония имеет обработка дназорастворов ароматическими
сульфокислотами. Уже малая примесь сульфокислоты стабилизи-
рует диазосоединение.
Диазониевые соли сульфокислот по малой растворимости по-
хожи на большинство диазониев из сульфокислот ароматических
аминов. В этих последних диазосоединениях солеобразование про-
исходит интрамолекулярно (внутри молекулы), например в п сульфо-
фенилдиазонии (из сульфаниловой кислоты)
so2-----О
Из сульфокислот, имеющих интерес для стабилизации диазо-
соединений, следует упомянуть: «-сульфокислоту хлорбензола *21),
сульфокислоты «-нитротолуола и динитротолуола22), 1.5 и 1.6-дисуль-
фокислоты нафталина, смесь три- и тетрасульфокнслот нафталина 23).
Имеется затем довольно широко применяемый способ перевода
17*
259
диазосоединения с изменением его структуры в иную, нереакцион-
ную форму, из которой тем не менее можно при определенных
условиях получить вновь реакционное соединение.
Диазосоединение, полученное в кислом водном растворе, какой
в результате получается при диазотировании амина, построено ана-
логично аммониевым (с одним пятивалентным атомом азота) со-
лям, отсюда их название — соли диазониев:
R —N2 —Cl = R — N = N
Cl
Диазониевые соединения с простым минеральным анионом об-
ладают большой реакционностью по отношению многих превра-
щений, в том числе и азосочетания, и поэтому не могут быть
сохраняемы ни в водных растворах, ни в твердом состоянии, если
они получены такими: твердые соли диазония очень легко раз-
лагаются со взрывом при нагревании, трении и пр.
Действие щелочей на соли диазония приводит к образованию
соединений с измененной реакционной группой, заключающей уже
два трехвалентных атома азота, где гидроксил, заменяющий мине-
ральный анион, функционирует как слабокислотная группа, реагируя
с катионом щелочи. Эти новые соединения, так называемые диа-
зотаты (т. е. щелочные соли кислотной формы диазо), проявляются
в двух стереоизомерных формах син- и анти- (изо-), из которых первая
сохраняет реакционность диазония, вторая же менее реакционна.
Изомеризация синформы в анти- происходит при более высокой
температуре. Превращения можно представить схемой:
R — N — Cl R — N
HI +2NaOH -> || +NaC14- Н2О
N NaO —N
сннднлзотат
R — N R —N
II II
NaO—N N • ONa
антидиазотат
(или изодиазотат)
Неактивной форме диазотата придавалось прежде строение струк-
турного изомера RN • Na(NO) — соли нитрозамина. Для свободных
нормальных диазотатов Ан гели высказывается в пользу такого
строения RNO • NH (иминонитроарил). Неактивным (изо- или анти-)
диазотатам щелочей присвоено в технике неправильное название
нитрозаминов. Антидиазотаты (нитрозамины) при подкислении пере-
ходят в активную синформу дназотата- и в соль диазония
R —N + нх R —N + нх R —N—X
II ------------> II--------------> III
N —ONa -Nax НО —N -н2О N
Антидиазотат в виде щелочной соли выпадает при обработке
раствора соли диазония концентрированной щелочью при повы-
260
шении температуры. Обработку удобно проводить в две стадии:
сначала при низкой температуре (около 5°) до образования синди-
азотата и затем применяя очень концентрированную щелочь (КОН)
и высокую температуру (100—105°) до образования антидиазотата 24).
Такие препараты стойких диазо-, носящие название азофоров,
азогенов, нитрозаминов (нитрозаминовый красный) и пр., в большом
употреблении в текстильной промышленности для получения „хо-
лодных" окрасок, особенно в ситцепечатании.
Разные диазо в зависимости от своего строения и заместителей
относятся с различной легкостью к превращению в антиформу
диазотата.
Паранитранилин, который легко дает антидиазосоль, приме-
няется, как мы видели выше, для количественного определения
помощью азосочетания окси-групп в ароматических соединениях.
Отсюда понятна необходимость избегать избытка щелочи, хотя
бы и в виде соды, при реакции сочетания с диазонием из л-нитр-
анилина, так как такой избыток, содействуя переходу части ди-
азония в неактивную форму, приведет к неверным результатам
анализа.
Наконец можно закрепить диазосоединение в виде более проч-
ного соединения, переведя его в такое диазоаминосоединение, ко-
торое само не в состоянии быть превращенным в аминоазосоеди-
нение, иначе говоря, пассивная аминосоставляюшая которого не
в состоянии функционировать, как говорят, в виде азосоставляющей
Диазоаминосоединения при достаточной концентрации Н-ионов
в среде претерпевают распад на диазосоединение и амин; последний
же не может по вышесказанному дать начало азокрасителю, сле-
довательно если при разложении диазоаминосоединения в реак-
ционной среде имеется посторонняя, достаточно активная азосо-
ставляющая, то она и будет связана с азогруппой. Для удобства
отделения выходящего из реакции амина (от диазоаминосоединения)
предпочтительно его иметь растворимым, например обладающим
сульфо- или карбоксильной группой. Так как диазоаминосоеди-
нения несравненно более стойки, чем диазо-, то такой метод по-
лучения стойких диазо- имеет большое значение, особенно в ситце-
печатании в комбинации с нафтолами типа AS в виде так на-
зываемых рапид-красок. Здесь схема превращений диазо такова:
RN2X + R' — NH — R" -> R — N : N — N(R'R")
(R'— ароматический остаток с сульфо- (соотв. карбоксильной) группой, R"’ —алкил,
арил илй Н)
N: NR
/\/\/°Н /\/\/°Н
R-N : N-N(R'R") + | | j | | | -j-HNR'R"
'^/’V^CONHRx \/\/\cONHR,
Стойкие диазо, построенные по схеме диазоаминосоединений, по-
лучили название „рапидогенов".
261
Из аминов общей формулы: HNR'R" могут быть названы этйл-
о-толуидиисульфокислота
СН3
сульфоантраниловая кислота
СООН
и др.25).
Уместно отметить здесь, что в некоторых соединениях удается образовать
диазогруппу (без амино-) прямым замещением: так, действием азотистой кислоты
иа антрахинон в присутствии окиси ртути получают 1.4-оксидиазоннй антрахи-
нона Я). Повидимому то же происходит иногда и в нафталиновом ряду, например
с 1-ннтро-7(6)-сульфокислотой нафталина 27) (см. иитрознрование).
в) Азосочетание
Реакция азосочетания, как уже было отмечено, дает начало
азокрасителям. Многие продукты ее оказываются интересными не
только как красители, но и как исходные соединения для полу-
чения иных промежуточных, часто не окрашенных соединений.
Восстановлением сравнительно простых моноазокрасителей гото-
вится целый ряд парадиаминов, смеси диаминов с моноаминами
(для сафранина) и пр. Ниже кратко остановимся на реакции
азосочетания 28).
Азосочетание происходит между активным диазосоединением,
следовательно солью илн гидратом диазония или синдиазотатом, и
теми циклическими соединениями, которые обладают соответст-
венными замещающими группами, становящимися ауксохромными
группами образующегося азокрасителя. Такими группами являются
NH2, NHAlk, NHAr, N(Alk)2, NHSOaAr *, ОН и в некоторых слу-
чаях OAlk 29).
* NHSO2Ar-rpynna являлась до недавнего времени единственной в своем роде
ацилированной аминогруппой, сообщающей ядру достаточную активность в азосоче-
тании (О. Witt). Такой же активностью к азосочетанню повидимому обладают и
сульфаминовые кислоты типа RNHSO3H.
W. KOnig и К. КбЫег констатировали в некоторых случаях активность
к азосочетанию и у ацетилированных соотв. бензоилированных) нафтиламииов.
Энергия азосочетаиия, правда, в этих случаях очень невелика 30). Таким образом
понятия о защите аминогруппы от азосочетания путем ацилирования должно пре-
терпеть некоторое ограничение. Согласно новым исследованиям К. Г. Майера
262
Мы будем называть амин, дающий активное диазосоединение,
диазосоставляющей реакции азосочетания, а второй ком-
понент (амин, фенол, энол, например в метилфенилпиразолоне
Н3С
СН
N
n-c6h5
или ацетоуксусном анилиде СН3 — С(ОН) = СН — CONHC6H5), пас-
сивно участвующий в реакции, — ее азосоставляющей.
Конечным продуктом реакции азосочетания является аминоазо-
(соотв. оксиазо-) краситель.
Азосочетание, всего вероятнее, протекает через стадию при-
соединения молекулы диазосоединения к азосоставляющей с по-
следующей стабилизацией отщеплением воды или галоидоводо-
родной кислоты и т. п. Например
/Ч/Н N : \R ^/N:NR
+ ; НО\ : —н2сГ
ОН НО/НО/!^/
В некоторых случаях сочетания с фенолами констатировано образование ди-
азоокснсоедниений с связью — N : N — О —, особенно прн наличии в азососта-
вляющей кроме ОН таких групп, как NO2, СООН, близко стоящей SO8H. Присоеди-
нение в таких случаях происходит по иной схеме, чем только-что упомянутая.
Прн сочетании диазосоединеннй с фенолами (среда близка к нейтральной или
слабощелочная) можно ожидать гидролиза соли диазония в гидрат диазония с по-
следующей перегруппировкой в синдназогидрат по схеме (Эг. Мелау):
R - N — х R-N-OH R — N
ill ill п
N N НО —N
гидрат синдназо-
диазония гидрат
При сочетании с аминами (более кислотная реакция) активна соль диазония.
Ангидиазотаты сочетают обычно при понижении щелочности среды иапример
прибавкой MgCI2 и др. Активен к сочетанию вероятно дназогидрат, образующийся
например по схеме
R —N nH2O R-N
Il ;=± II
N —ONa nNaOH N —ОН
антндиаэогидрат
о значении так называемой активной двойной связи для азосочетання уже угле-
водороды циклические, как мезитилен (I)
НзСу,/.^/,СН3
н
I
СН3
или ненасыщенные, бутадиен (СН2 — СН — СН — СН2, его гомологи) дают сочета
ния с п-ннтрофеннлдиазонием в настоящие азосоедннення. В циклических угле-
водородах при этом имеют значение в качестве реактивирующей группы (подоб-
ной NH2, ОН и пр.) метилы, накопление которых в подходящих размещениях
делает двойные связи особенно активными к азосочетанию.
263
При сочетаниях диазосоединений с аминами, промежуточными
соединениями могут быть диазоаминосоединения, разлагающиеся
от кислот на диазониевую соль и амин. Диазоаминосоединения
при диазотировании и азосочетании преимущественно образуются
при недостаточно минеральнокислотной реакции среды и наличии
избытка свободного ароматического (первичного или вторичного)
амина. Их превращение в аминоазосоединения от действия соли
первичного амина (при повышенной температуре), которое истол-
ковывалось прежде как вторичный процесс (перегруппировка)
Н • R —NH —N : NRj->H2N —R —N : NRb
иыне К. Майер рассматривает как первичный процесс, вызываемый
конкуренцией относительно азогруппы (— NzrrN—) между имино-
группой NH, с которой азогруппа уже связана, и ядром первич-
ного амина (его активной двойной связью по Майеру), введен-
ного вновь в реакцию и тяготеющего также к азогруппе 31).
Общее выражение реакции азосочетания таково:
R — N— Х-(-Н — R" — Y = R —N : N —R" —Y-j-HX,
где R" — двувалентный ароматический радикал, например фени-
лен С6Н4, нафтилен С10Н6 и т. п., a Y — реактивирующая (ауксо-
хромная) группа, одна из вышеприведенных.
Если обратить внимание на диазоний, то можно притти к вы-
воду, что азосочетание есть один из способов превращения его
характерной группы — N2X в ^R^Y. Если же учесть изменения
второй составляющей азосочетания (амина или фенола), то можно
констатировать образование совершенно новой группы R — N~N —
на месте водородного атома ядра этой составляющей. Таким об-
разом реакция азосочетания должна рассматриваться, в зависи-
мости от того, за судьбой какого из ингредиентов этой реакции
мы следим, или как реакция первого рода (ввод новой группы)
или как реакция второго рода (превращение существующей группы).
Закономерности вхождения новой азогруппы ( —N —N—) в
ядро, заключающее ауксохром, определяется ориентирующим влия-
нием названных заместителей, каковые, как известно, направляют
новые заместители в пара- или орто-положения. Азогруппа стано-
вится преимущественно в пара-положение к ауксохрому. Чаще
всего наряду с пара- получается и незначительное количество орто-
азопродукта. Если пара-место занято или (в нафталиновом ряду)
находится под неблагоприятным влиянием сульфогруппы, стоящей
в орто- или пери-положении, то тогда азогруппа становится в орто-
положение к ауксохрому. Мета-амино-, соответственно мета-оксиазо-
соединения не могут быть получены прямым азосочетанием и об-
разуются лишь обходным путем.
При наличии заместителей и в пара- и в орто-местах по от-
ношению к активирующей группе (ОН, NH, и т. п.) сочетания во-
обще не происходит. При некоторых азосоставляющих, содержащих
в пара- или в единственно способном к сочетанию орто-месте до-
статочно подвижные замещающие группы (СООН, SO3H, CH2R),
264
азосочетаиие все-таки совершается, но с вытеснением азогруппой
другого заместителя.
Таким образом например а-карбоновая кислота ^-нафтола (I)
дает р-нафтольный краситель. Так же ведет себя 1-сульфокислота
р-нафтола (II), дающая как первый продукт взаимодействия диазо-
оксисоединение
СООН
N:NR
+ RN2x
-НХ
Динафтилметановые соединения, соответствующие арилидам ^-окси-3-нафтойной
кислоты (III) прн сочетании с диазониями разрывают связь углеродных атомов
в месте, куда входит азогруппа 32)
- RNHOC^
ОН
: CH2-)-R'-N2-X
В. В. Шарвин и Кальянов наблюдали, что реакция диазо из сульфанило-
вой кислоты и тетраметилдиаминодифеинлметана (IV), соотв. диметнламино-л-бен-
зойной кислоты (V), приводит к образованию л-днметиламиноазобензол-л-сульфо-
кислоты (гелиантнна)
/\/N2
^-М(СН3)2 + 2 j |
SO2^_ О
(Н3С)2М-
zZ\/Z^2 / . //NICHj 2
-> (h3c)3n-^2/-N:N~\2/_’s°3H j ! + M
SO^—O OCZ X//
OH
в первом случае с отщеплением формальдегида, во втором — углекислоты.
Примеры полного отказа в сочетании со стороны таких соединений, как ди-
метнлсульфаииловая кислота, диметиламииодифенилметан, тетраметилбензидин, ди-
метил ф-нафтиламин, указывают на препятствие со стороны диметиламииогруппы —
N(CH3).2 вхождению в орто-положение к ней азогруппы, причем это препятствие
более значительное, чем в равных условиях со стороны незамещенной амино-
группы. Это препятствие можно отнести к так называемым „пространственным затруд-
нениям" (К е р м а н и, В. М ай е р) ®).
265
В зависимости от природы азосоставляющей (амин, фенол) ре*
акция азосочетания проходит в определенной среде. Обычно
сочетание с аминами проводят в отчетливо кислотной среде: раз-
веденная соляная кислота, уксусная кислота; сочетание с фенолами
протекает иногда достаточно хорошо в слабокислотной среде,
обычно же лучше в нейтральной или слабощелочной (уксусная
кислота с уксуснокислыми солями, бикарбонат, гидроокись кальция,
водно-аммиачный раствор, сода, реже — едкий натр) 34). Избыток
щелочи во всяком случае может, как сказано выше, повести к по-
тере части диазосоедииения с переходом его в неактивное соеди-
нение. В тех случаях, когда азосоставляющая заключает два раз-
личных ауксохрома (NH2- и ОН-группы), из которых каждый
независимо может образовывать азокрасители (случай двуядер-
ных замещенных нафталина), начинают азосочетание в кислой
среде, благоприятной для образования аминоазокрасителя и, устра-
нив с образованием последнего влияние ЫН2-группы, проводят вто-
рое азосочетание на оксиазокраситель в слабощелочной среде.
Таковы например ступени реакции образования дисазокрасителей
нз кислоты Н
ОН NH2 ОН NH,
НОзБ/^/^/'ЧОзН HOgS/^/^/^SOsH
ОН NH2
HOgS/^^/^SOgH
В тех случаях, когда в азосочетание вступают тождественные
диазо- и азосоставляющие, часто не заготовляют заранее соли
диазония, но прибавляют рассчитанное на половину всего имею-
щегося амина количество нитрита к подкисленной соли амина и,
промешав достаточно массу, ожидают выкристаллизовывания соли
образовавшегося аминоазосоединения. При взаимодействии сульфо-
кислот диазониев с первичными ароматическими аминами часто
наблюдают своеобразный обмен между диазо- и аминогруппами
(ср. выше односторонне диазотированный бензидин).
Так, при действии диазотированной сульфаниловой кислоты на
анилин наблюдают течение реакции по двум направлениям: (I и II):
266
Наряду с этим, благодаря вышеназванному перемещению групп,
происходит образование фенилдиазония и сульфаниловой кислоты,
и диазоний реагирует с свободным анилином, образуя несульфи-
рованный аминоазобензол
В последнее нремя было найдено, что применение арилсульфоиовых произ-
водных первичных аминов, например толуолсульфанилида, в слабощелочном рас-
творе дает гарантию вполне однообразного течения азосочетаиия и первом напра-
влении ®).
По американскому патенту, а-нафтилсульфамииовая кислота (I) прн сочетании
дает лишь исключительно пара-азопродукт и ие образует орто-изомера ®®)
I
NHSOgH
При азосочетаниях различных диазо- и азосоставляющих обычно
прибавляют охлажденный раствор соли диазония в охлажденный
же раствор азосоставляющей, каковому раствору прибавлением
кислоты или щелочи сообщена нужная реакция, и, энергично раз-
мешивая, доводят азосочетание до конца. Бывают азосичетания,
проводимые при высокой температуре, это — случаи стойких и
мало реакционных диазосоединений, таковы например диазоокиси
нафталина 37), для которых указывается оптимальная температура
сочетания 40°.
Азосочетание протекает с различной скоростью в зависимости
главным образом от свойств азосоставляющей. Повидимому орто-
азогруппа образуется труднее, чем в пара-месте. Иные азосоче-
тания в технике требуют многих часов и даже дней (например
с нафтионовой кислотой).
Природу реакции азосочетаиия точными методами изучал ряд исследователей®8).
Г. Гольдшмидт и сотрудники установили, что при сочетании солянокислой
соли третичного амииа с диазобензолсульфокнслотой [например в гелнантнн
(CH3)2N — CeH4 —N:N —CeH4SO3H] реагирует освобожденное нз солн гидро-
лизом оснонаине с диазо как таковым. При этом избыток соляной кислоты умень-
шает скорость сочетания. Концентрация солянокислой соли и дназосульфокнслоты
не влияет на скорость реакции. Реакция между третичным амином и дназобензол-
сульфокислотой протекает тем быстрее, чем слабее применяемая кислота среды.
Прн образовании оксиазосоедииений нз фенолов и диазосоединеннй в щелочном
растворе в реакцию вступают освобождающиеся (вследствие гидролиза фенолята)
части фенола и снндиазосоединение; избыток щелочи при этом замедляет реакцию.
Требуемое для взанмодейстния время тем больше, чем концеитрированиее растворы
фенола и диазо.
267
В недавней американской работе Ко и а нт а и Питерсона по изучению
реакции сочетания исследователи изучали значение концентрации водородных ионов pH
во взаимодействии между фенолами и диазо, имея в виду, что обынно при таком
сочетании кислотность поддерживается ниже определенного значения прибавкой
соли слабой кислоты (например уксусной, угольной). Исследователи установили так же,
как ранее Гольдшмидт, бимолекулярный порядок реакции, отвечающей при
равномолекулярных количествах реагентов уравнению
ftC=4l^z’ (1>
где С—концентрация реагентов, Z—прореагировавшая часть во время t SR).
Отношение между k и pH выражается прямолинейной функцией, что дает воз-
можность вести расчеты, сравнимые для различных азо- и диазосоставляюших прн
таком pH, при котором k — 10 (logA = 1).
Это значение pH авторы считают характеристикой для азосочетания и называют
его „значением сочетания" (coupling value).
„Значение сочетания* данной пары составляющих определяется как природой
дназо-, так и азосоставляющих. Авторы нашли возможным установить константы
для аминов и фенольных соединений, так что сумма их определяет значение соче-
тания „з. с." для данной пары. Приводим ниже их таблицу, где А и В—кон-
станты для дназо- (и соотв. для азо-) составляющих.
Азосоставляющие Диазосоставляющие
сульф- анилов. к-та /?-бром- анилии анилин «-толуи- дин о-анизи- дин
1-нафгол-4-сульфонаг В — 3,5. А = 0 „з. с.“ 3,5 о , II о | А = 1,1 ,з. с.“ 4,6 А = 1,5 .3. с.' 5,0 А = 2,1 „з. с.“ 5,6
2-нафгол-3.6-дисульфонат В = 4,2 4,2 4,2 5,3 5,7 6,3
1-нафтол-3,8-дисульфонат В = 5,1 5,1 5,1 6,2 6,7 7,3
2нафтол-6.8-дисульфонат В = 6,4 6,4 6,4 7,5 7,9
и-фенолсульфонат В = 8,3 . . 8,3 — — — —
В пределах ошибки опыта вычисленные „з. с.“ совпадают с рассчитанными сум-
мированием А и В. Для и нитранилнна А = 2, для салициловой кислоты В=8
и для резорцина В = 3,4.
Сочетание, по мнению американских исследователей, проходит в две стадии:
сначала — гидролиз диазония (в сиидиазосоединение) — энергично протекающая
обратимая реакция:
Аг — NZTN — X + OH“^Ar-N — N — OH-f-X-
и вторая — собственно сочетание с остатком фенола Р:
Аг • N — N • ОНHP —> Аг — N — N — Р-f-Н2О,
относительно медленно идущая и необратимая. Относительная энергия второй
стадии зависит от концентрации Аг — N = N — ОН, т. е. является функцией pH
буферного раствора.
В работе двух итальянских исследователей установлено, что при сочетании 1.8-ами-
нонафтол-З.б-дисульфокислоты (Н) с диазониями до стадии моноаминоазосоедине-
ния в кислотной среде выход моноазо возрастает в зависимости от силы кислоты
(ненормально в ряду кислот ведет себя фосфорная кислота) 39).
Значение физико-химической характеристики среды, в которой
происходит сочетание, таким образом вне сомнения, и исследова-
268
ния в этом направлении могут очень облегчить целесообразное
проведение синтеза азокрасителей.
Азосочетание чаще всего, но не всегда, необходимо заканчивать
при значительном избытке азосоставляющей, дабы не было в про-
дукте свободного диазосоединения, которое своими вторичными
превращениями может сильно осложнить реакцию, увеличив коли-
чество посторонних и иногда трудно отделимых продуктов. За хо-
дом азосочетания следят, беря описанные пробы на вытек (см.
определение аминогруппы), и добавляют новую порцию диазосое-
динения тогда лишь, когда эта проба дает доказательство отсутст-
вия в реакционной смеси свободного диазония.
Продукт азосочетания или выделяется вследствие его малой
растворимости в реакционной смеси, или его выделяют помощью
высаливания, иногда подкислением.
Тиофенолы с диазониями не образуют азосоединений, но продукты замещения
с вытеснением азота. Хнноны реагируют с диазониямн также с потерей азота.
г) Иные, кроме азосочетания, превращения диазосоединений
Кроме азосочетания диазосоединения применяются для весьма
большого круга превращений характерной групп ы—N2X,
частью с сохранением атомов азота, частично же при отщеплении
всего азота.
Прежде всего заслуживает упоминания реакция восстано-
вления. Обработка диазониевой соли посредством подходящих
восстановителей, например хлористым оловом в крепкой соляной
кислоте (2—2’/г мол. SnCl2 на 1 мол. амина), дает начало образова-
нию гидразиновой группы 40)
R^-N2 —Cl — R — NH — NH2.
Наиболее употребителен иной способ (Э. Фишер) получения
ароматических гидразинов. Взаимодействие соли диазония с избыт-
ком средней натриевой соли сернистой кислоты Na2SO3 приводит
к образованию диазоарилсульфоновокислой соли натрия. Восста-
новление этой последней цинком дает арилгидразиносульфокислую
соль. Гидролитическое расщепление действием горячей соляной
кислоты устраняет сульфогруппу, связанную с азотом.
Ход реакции при этом схематически таков:
+ н,
R — N — X-J-Na2SO3---->R —N —N —SO3Na —
III — NaX
N
+ Н2О 4- HC1
--------► R —NH —NHSO3Na--------------->
—NaHSO4
--------► RNH —NH2HC1
Этот способ находит себе широкое применение при приготов-
лении ароматических гидразинов. Иногда восстановление диазо-
сульфоновокислой соли производят не цинком, но помощью сер-
нистой кислоты, прибавив к кислой жидкости еще эквимолекуляр-
ное количество сульфита натрия 41).
269
Лучшие выходы нитрофенилгцдразииов получают при применении вместо нат-
риевой соли сернистой кислоты сульфита аммония (NH4)2SO3 ^).
Арилгидразины — весьма реакционноспособные соединения и
находят себе применение в практике производства промежуточных
продуктов (производных пиразолона).
При окислении (например посредством CuSO4, FeCl3) они переходят, с потерей
обоих атомов азота, в углеводород, отвечающий арилу 4d):
R — NH — NH2 + О — RH + N2 + H2O.
Этой реакцией пользуются для устранения группы —NH — NHa, соотв. — NH3,
в тех случаях, когда необходимо выяснить структуру связанного с аминогруппой
арила. Такое же значение имеет другая восстановительная реакция, практикуемая
по отношению к диазогруппе, которая одним приемом устраняет ее.
Если соль диазония, полученную лучше всего в твердом виде,
обработать большим количеством спирта на кипу44), то с выделе-
нием азота диазогруппа замещается водородом, спирт же окис-
ляется в альдегид:
R — N — X 4- С2Н5ОН — RH 4- N2 + НХ Ц- СН3СОН.
Ill
N
Такое замещение водородом может сопровождаться, и даже отодвигаться на
второе место, образованием алкильных эфиров соответствующего арила45):
RN2C1 -J- С3Н5ОН = ROC2H5 4- N2 + НС1.
Часто можно передвинуть реакцию в направлении оводорожения арила при-
бавлением щелочных агентов, например алкоголята натрия, едкого натра, поташа
или цинковой пыли46), порошка меди или алюминия.
Восстановление диазосоединения на этот раз в щелочном растворе, полученном
из хлористого олова 47), дает возможность также сразу заместить водородом диазо-
группу:
RN = NOH Н2 —> RH 4- N2 4- Н2О.
Арилгидразины легко характеризуются своей большой окисляе-
мостью: они восстановляют раствор Фелинга уже на холоду. При
окислении окисью ртути переходят в диазосоединения
RNH • NH2 • НХ 4- 0.2 — RN2X 4- 2Н2О,
которые могут быть характеризованы своими красочными реакциями.
С альдегидами и кетонами (обычно в присутствии уксусной кис-
лоты) арилгидразины дают продукты водоотщепления, гидразоны
>С—N-NHR.
Количественно гидразины определяются иодометрически, так как от действия
иода они разлагаются с выделением азота:
RNH • NHa 4- 4J = RJ 4- Ns 4- 3HJ.
Для сульфокислот арилгидразинов практичным методом анализа является окис-
ление их медиым купоросом в растворе соляной кислоты. Измеряется объем выде-
ленного азота. Реакция, представляемая уравнением
RNH • NH2 4- 2CuSO4 4- Н20 = RH 4- Cu2O 4- 2H2SO4 4-
повидимому идет и в ином направлении: в сторону получения хлорозамещенньх
углеводородов 48).
Превращения диазосоединений без устранения азота возможны еще в сторону
образования азидов, соединений с группой N3. При обработке диазосоединений
в водном растворе, слабокислом или аммиачнощелочном, посредством галоидами-
270
нов (например хлорамина, водный раствор которого получается при смешении рав-
ных количеств N растворов аммиака и гипохлорита иатрия) или хлораминовых со
единений сульфокислот (например RSOgNCl Na, — см. стр. 90) получаются с хоро*
шими выходами арилазиды 49):
RN2X + NH2C1 — RN3 + НС1 + HX,
RN2X + R'SOjNCINa — RN3 -J- NaCl -J- R'SO2X.
Азиды, производные азотистоводородной кислоты HNs, обладают большой
реакционностью и могут быть использованы для получения иных продуктов. Так,
при действии крепких кислородных кислот в присутствии растворителей (уксусной
кислоты ит. п.) они образуют аминофеиолы “°):
Иные, кроме восстановительного, превращения диазогруппы со-
пряжены с устранением азота реакционной группы. Принадлежа
к ряду наиболее реакционноспособных органических веществ, ди-
азосоединения дают возможность очень большого числа замещений
своей характерной группы на другие атомы или группы. Возможно
замещение диазогруппы на ОН, OAlk, OAr, Cl, Вг, J, F, CN, SH,
SAr, SCN, SO2H, NO2, наконец возможно смыкание углеводородных
ядер диазосоединения. Эти превращения, имея большое препаратив-
ное и теоретическое значение, сравнительно мало используются про-
изводственной практикой и по сравнению с азосочетанием играют
очень невидную роль в применении диазосоединений. Мы остано-
вимся только на важнейших из них.
Замене диазогруппы гидроксилом отвечает уравнение
rn2x 4- н2о = roh 4- нх 4- ы2.
Превращение выполняется посредством диазотирования (в растворе
серной кислоты) амина и последующего кипячения кислого вод-
ного раствора диазо. Мы уже упоминали об этом методе в преды-
дущей главе. Там же было указано на то, что при этом в не-
которых случаях выгодно прибавлять в реакционную смесь мед-
ную соль.
Присутствие галоидоводородных кислот при разложении диазо
необходимо избегать, так как от них можно ожидать превращения,
хотя бы частично, в галоидозамещенные.
Интересно отметить, что имеются указания на повышение выходов, если кроме
серной кислоты в растворе присутствуют фосфорная кислота или ее соли, напри-
мер трииатрийфосфат 51). Возможно, что в разных случаях могут оказаться полез-
ными и другие анионы.
Для устранения могущих образоваться при разложении диазо окислов азота
может оказать услугу прибавление мочевины52). С целью изолирования из реакци-
онной среды образующегося фенола предложено введение в нее индиферентных,
не смешивающихся с водок растворителей фенолов (ароматические углеводороды,
их хлорозамещенные, эфиры, сложные эфиры и т. п.)53).
Нередко диазораствор заготовляется отдельно и вводится постепенно в нагре-
тую серную кислоту надлежащей концентрации. Для того чтобы избежать при этом
нежелательных побочных реакций, например от нагрева диазо ранее попадания
в слой серной кислоты или задержки его в среде кислой пены, практично приме-
нять испытанное Е. Б. Ашкинази приспособление (рис. 9). Раствор диазо охлаж-
дается холодильником в реакционном аппарате до входа во взаимодействие-
271
Попадая во внутреннюю трубку аппарата, порция диазо выталкивается образую-
щимся азотом в более глубокий слой кислоты. Применение непрерывного отгона
с паром для летучих с ним продуктов или слоя растворителя над кислотой позво-
ляет быстро выводить продукт нз зоны реакции.
Метод превращения диазо в фенолы имеет применение для по-
лучения пирокатехина, его метилового эфира — гваякола52), лт-окси-
бензальдегида54).
Разложение диазосоединений в водных растворах в сторону об-
разования фенолов побуждается не только повышением темпера-
туры, но и действием света.
В частности это обстоятель-
Рис. 9. Прибор для проведения превраще-
ния дназосоединений в фенолы.
ство используется при приго-
товлении так называемых ди-
азокопий с чертежей вместо
употребительных прежде „си-
нек". Диазо наносится на по-
верхность бумаги вместе с азо-
составляющей— активным фе-
нолом (например резорцином)
в среде, не дающей возмож-
ности сочетания этих соста-
вляющих в краситель. При ко-
пировании чертежа (кальки)
на таком слое в местах, где
свет проникает, диазо разла-
гается, под черными линиями
диазо сохраняется. Если после
экспозиции создать условия,
облегчающие сочетание, — об-
работкой светочувствительно-
го слоя парами или раствором
аммиака и т. п. —то краситель
образуется лишь в тех местах,
где сохранилось диазо, следо-
вательно колия воспроизведет
цветными линиями черные ли-
нии чертежа. Этот метод по-
лучения светочувствительных
слоев получил очень большое
значение в практике копиро-
вания чертежей 55).
Получение галоидозамещеннык из дназосоединений предполагает обязательное
участие в реакционной среде галоид-иона. Недавно было исследовано разложение
дназобензола в водном растворе и установлено, что скорость разложения CeH5NaCl
зависит от концентрации соляной кислоты в растворе, причем реакция происходит
как с молекулами HCI, так н (в меньшей степени) с ионами СИ. Присутствие нейтраль-
ных хлористых солей (NaCl, КО) повышает выход хлорбензола; вероятно недиссо-
циированная молекула C6H5N2C1 сразу переходит в С6Н5С1 и N2. Наряду с этим проис-
ходит распад ионов C6H5n + — от воды в СвН5ОН и от соляной кислоты в СвН5С1м).
Замена диазогруппы хлором (или бромом) по Зандмейеру
совершается при участии в реакции с галоидной солью диазония
272
закисной соли меди (Си2С12 или Cu2Br2). Например:
RN2C1 — RC1 + N2.
Зандмейер считает процесс переходящим через промежуточ-
ную фазу — присоединения молекулы Си2С12 к диазо:
R —N------N — С1
R — N2C14-Cu2C12- I I -RC1 + N2 + Cu2C12«).
CuCl CuCl
По Гаттерману для этого превращения можно применять свежеосажденный
порошок медн, прибавляя его к раствору в галоидоводородной кислоте соли ди-
азоння з8).
При соблюдении таких молекулярных отношений между реа-
гентами-— галоидоводородная кислота: соль Си-: амин = 10: 1:1—
оказалось практически одинаковым, проводить ли взаимодействие
на холоду или при нагревании59).
Родановая группа SCN входит на место дназогруппы при обработке диазо солью
роданистоводородной кислоты в присутствии катализаторов. Были испытаны хлори-
стые соединения ряда металлов и свежеосажденный порошок меди в этом послед-
нем превращении, причем обнаружено, что металлы с атомным весом 55,84—63,57
наиболее активны. Наилучший выход в 80% получен с хлорным железом (активны:
Fe, Си, Со, Ni). По предположению Корчннского атомный вес элемента илн его
кратное играет какую-то роль в проявлении каталитической активности в этом пре-
вращения60).
Замену дназогруппы хлором предложено проводить при посредстве облучения
хлористоводородной соли диазония в првсутствин хлористого цинка н хлористого
аммония, например светом ртутной лампы61). Прн мало реакционных дназо полу-
чают иногда замену диазо бромом, переходя через стадию пербромнда.
Недавно Швехненом предложено замещать хлором н бромом N2-rpynny в
мало реакционных диазо нагреванием сухих комплексных соединений соли диазоння
с галоидными соединениями ртути (HgBr2, HgCl2), осажденных соответственной ртут-
ной солью из водного раствора диазониевой солн. Осадок, высушенный промывкой
ацетоном или спиртом с эфиром, смешивается с двойным илн большим количест-
вом галоиднощелочной соли (КВт, КС1), н смесь, помещенная в тугоплавкую трубку,
подвергается нагреванию. Реакция идет спокойно. Выщелачивая после нагревания
растворимые соли водой, получают хорошие выходы галоидозамещениого62).
Замена иодом чаще всего выполняется введением в раствор диазо растворимой
соли иодистоводородной кислоты (KJ). Замещение фтором— через предварительное
получение борофторидов диазония, например добавлением к дназо борофторнсто-
водородной солн (NaBF4) и нагреванием полученного борофторнда диазония до вы-
сокой температуры в3).
Аналогично вводу галоида вместо дназогруппы протекает вве-
дение цианидной (нитрильной) группы CN путем взаимодействия
по Зандмейеру диазо с щелочным раствором цианистой закиси
меди Cu2(CN)264).
По патентам немецкой фирмы расход медной соли и цианистого калия при
этом взаимодействии можно уменьшить вдвое, применяя диамминоцианиды щелочных
металлов. Способ получения этих последних комплексных солей и ход взаимодей-
ствия выражается уравнением:
CuSO4 + NH3 4KCN = K2CuNH3(CN)4 + K2SO4,
K,CuNH3(CN)4 + 2RN2CI = 2RCN + 2KC1 + NH3 + CN + CuCN °5).
Замещение посредством сульфгидрильной группы, SH, произво-
дится по методу Лейкардта66) действием на соль диазония ксан-
тогеновокислой соли при нагревании и омылении спиртовой ще-
18 Зак. 2611. — Н. Ворожцов
273
лочью образовавшегося вначале арильного эфира этилксантогено-
вой кислоты
RN2X + KS • CS • ОС2Н5 = RS•CS• ОС2Н5 + КХ Н- N2,
RS • CS • ОС2Н5 4- 2Н2О = RSH + СОа+ H2S+ С2Н5ОН.
Эта реакция открывает путь для получения как тиофенолов,
так и сульфокислот с таким положением SH-(cootb. SOsH-) группы,
в которое невозможно ввести атом серы методом сульфирования.
В отдельных случаях можно обойтись без этой, сравнительно
дорогой, обработки и(взаимодействием двусернистого натрия Na2S2
с солью диазония) приготовить тиофенол более простым способом:
2RN9X4-Na,S2-------► RN : N —S—S-N : NR-------->
- 2NaX —2N.?
+ H2
----> R-S—S-R--------- 2RSH
Способ нельзя считать вполне безопасным, так как при нем
возможно получение сильно взрывчатых промежуточно образую-
щихся сернистых производных диазосоединений. Тем не менее он
применяется при получении тиосалициловой кислоты из антрани-
ловой:
/x/N2X /\/SH
II -II — II
Х''/'ХСО2Н ^-/^СОаН
Отметим, что недавно опубликованы патенты немецкой фирмы иа применение
реакции взаимодействия с полисульфидами щелочных металлов диазо (не заклю-
чающих гидроксильных групп) в щелочном растворе для получения дисульфидов,
переходящих при восстановлении в меркаптаны, и на аналогичную обработку диазо-
нафталиисульфокислот 6”).
Тиофенолы, как мы видели выше, не сочетаются с диазониями: они реагируют
с ними с образованием диазотиоэфиров, которые при повышении температуры (70°)
выделяют азот и дают ароматические сульфиды, например
CcH3SNa + CcH-N2C1--► CeH5N2SCcH5 -> CcH3SC6H, + N,.
—NaCl
Введение порошка меди позволяет понизить температуру реакции. Взаимодей-
ствие иногда необъяснимым образом идет в сторону образования не сульфида,
а дисульфида. Вместо допущения здесь разложения диазотиоэфира, например
С6Н5К>8СсН3, проще объяснить это окислением тиофенола
2C6H3SH + CoH5N2C1 -> CeH5 • SS • С6Н3 + N2 + С6Нс + НС1 «*).
Не только меркаптаны ароматического ряда, ио и жирные тиоспирты дают невиди-
мому аналогичное превращение в сульфиды. Так, обработка диазосоединений тио-
гликолевой кислотой HS-CH2COOH ведет к образованию арилтиогликолевых
кислот 69).
Замещение диазогруппы арильным радикалом, т. е. образование углеродной
связи между ароматическими радикалами, несущими остатки диазо, происходит
по Гаттерману при обработке диазосое шнеиия в подходящем растворителе
(спирт, уксусный ангидрид) порошком меди
2RN2X + Си = R — R + 2N, 4- СиХ2.
274
Структура диазосоедииения имеет большое значение при этом превращении.
Найдено, что присутствие нитрогруппы облегчает сцепление двух ароматических
остатков, другие заместители затрудняют 71).
Практично оказалось применять в качестве реагента аммиачный раствор закиси
медн в отношении 1 мол. Сп2О иа 1 мол. диазо. Реакция протекает в аммиачно-
щелочной среде н при обыкновенной температуре. Таким образом готовят например
по немецкому патенту дииафтилдикарбоновые кислоты, например
или соответственные производные аценафтена 27), например
Интересно, что углеродный заместитель вместо диазогруппы может возникнуть
при реакции диазо с хинонами (в водно-спиртовом растворе). С уходом азота обра-
зуются арильные производные хинонов 73) например
I I + RN2C1
0А^
(+CH3COONa)
Замещение остатком — SO2H с переходом диазония в сульфи-
новую кислоту происходит при восстановлении сульфата диазония
в сульфит при действии газообразного сернистого ангидрида и обра-
боткой полученного таким образом сульфита диазония порошком
меди 74).
Все вышеперечисленные превращения диазониев происходят
при соответственных для каждого температурных условиях. Ре-
акции сопровождаются выделением элементарного азота, следо-
вательно сильным вспениванием. Метод выделения продукта реак-
ции сильно зависит от его свойств, определяющихся характером
образовавшейся реакционной группы и уже присутствующими за-
местителями.
Несмотря на большую реакционность, а следовательно малую
стойкость диазосоединений, при известных условиях и определен-
ном строении они способны претерпеть без заметного разложения
реакции замещения в ядре. Больше других здесь применяется,
а потому и освещена, реакция нитрования.
В бензольном ядре нитрогруппа ориентируется со стороны
диазогруппы по преимуществу в пара- и орто-места 75).
Получаемые посредством диазотирования 1.2-аминонафтольных
производных диазоокиси легко выносят нитрование 76) и сульфи-
рование 77); последнее осуществляется посредством олеума. При
нитровании диазо из 1.2-аминонафтол-4-сульфокислоты получается
18*
275
ценная составляющая черных хромовых красителей 6-нитро-1-диазо-
2-окси-4-сульфокислота нафталина 78):
N—N
/\/\/°
SO3H
Хлорирование этого же диазо приводит к получению хлорозамещенного, причем
хлор становится также в место 6.
Диазониевая группа в ароматическом, особенно нафталиновом, ядре производит
своеобразное реактивирующее действие на заместители, стоящие в орто-месте,
именно — сообщая им способность сравнительно легко обмениваться на гидроксил.
Например ’9)
С1 ОН
N=N
-f-NajCOg
—Na.SO3—СО.
Аналогично превращение диазоазосоедииений бензольного ряда:
/NO2
HO3S—/ \-N=N—/>—N,C1 ->
,он
-> HO3S-/ N=N—N,CI8»)
Диазосоединения в случае необходимости их аналитического
определения чаще всего определяются методом азосочетания, опи-
санным в главе V. В тех случаях, когда азосочетание затрудни-
тельно и не протекает быстро с достаточной полнотой (таково на-
пример диазо из 1-амино-2-нафтол-4-сульфокислоты), можно поль-
зоваться определением выделяющегося при разложении диазосоеди-
нения азота.
С этой целью берут навеску (0,3—0,7 г) диазосоедииения (вероятно можно при-
менять и достаточно концентрированные растворы их) в пробирку, закрытую проб-
кой с 3 отверстиями, через которые проходят: 1) трубка, согнутая под прямым углом
и расходящаяся сверху иа две, каждая с краном: одна, соединенная с источником COg,
276
другая с насосом, 2) воронка с краном, 3) трубка, кончающаяся недалеко от пробки
и соединенная с градуированным цилиндром для газа, наполненным 50%-иым рас-
твором кон.
После вытеснения воздуха углекислотой создают вакуум и через воронку при-
ливают 25 ел3 концентрированной соляной кислоты и пускают вновь ток СО2, на-
гревая дно пробирки на песчаной бане. Отсчитав количество азота и приведя его
«нормальным условиям, можно рассчитать по нему процентное содержание диазо81).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
A. Hantzsch und G. Reddelien, Die Diazoverbindungen, Berlin, 1921.
1) Jkuta, Ann. 243, 281 (1888).
2) E. Tassilly, Le facteur de la diazotation, Bull. (4), 27, 19 (1920).
3) E. И. Шни та л ьс к и й, В. В. М о н бл а нов а, Кинетические явления при
реакции диазотирования анилина, Труды Научно-Исслед. Химич. Ин-та 1 МГУ,
1925, 44.
4) Г. Г. В е и д е л ь ш т е й н, Ж. 59, 146 (1927).
5) J. С. Cain, F. N i с о 11, Soc. 81,1412 Т (1902), 83, 206,470 (1903). Ср. J. С.С a i п,
Soc. 83, 688 (1903). Е. Yam от oto, J. Soc. Chem. Ind. Japan 32, 279 В,208 В,352В
(1929), 33, 177 В (1930).
П. П. Викторов, Ж. П. X. 4, 777 (1931). Ср. Knecht, Platt., J. Soc. D. Col.
1925, № 8.
6) Witt, Ber. 42, 2953 (1909), заявл. Г. n. W. 28063 (F. IX, 302), cp. Elion, CB
1923, I, Ю21, Fuchs, CB 1923, III, 552, также, Am. n. 1460708, Soc. Ind. 1923,
A. 922.
7) M i s s 1 i n, Helv. 3, 626. S. Krishna, R. L. В a t.h i a C. A. 25, 2981 (1931).
8) Cm. IG., Г. n. 462149 (1928).
9) T. Maki, J. Soc. Chem. Ind. Japan. 34, 514 (1931). Г. n. 537021,CB 1932,1,140
Ch. Age 26, 22 (1932).
10) Epstein, Г. n. 103660 (F. V, 634), Tauber, W a 1 d e n, Ber. 30,2901 (1897).
И) Ср. С, Г. n. 74177 (F. Ill, 499), 78831 (F. IV, 732).
12) K. & W. Schirmacher (G. An. W.), Am. n. 1750057 (1930), IG. Г.n. 473217,
Б. n. 269582, CB 1929, II, 659.
13) Ta uber, Ber. 27, 2627 (1894).
14) Morgan, Davies Soc. 123, 228 (1923), CB 1923, I, 1218.
15) t. M., Г. n. 155083 (F. VII, 405). G., Г. n. 171024 (F. VIII, 640,) 172446,
(F. VIII, 646), К, Г. n. 175593 (F. VIII, 648), 178621 (F. VIII,649), 178936 (F. VIII, 650),
В, Г. n. 189179 (F. VIII, 654).
16) Rhein. Kampfer. Fa b r ik. Г. n. 431513 (F. XV, 215). H. H. Ворожцов
и A. M. Го p ь ко в, Ж. О. X., II. 421 (1932).
17) IG., Г. п. 454894 (1928), К- Ф. п. 657852,СВ 1930,1, 2008, S а п d oz, Ф. п. 708822,
СВ 1932, I, 587.
18) IG., Г. п. 515205, СВ 1931, I, 2114.
19) IG., Б. п. 316691, Ф. п. 657041, СВ 1930, I, 2008, Б. п. 317355, СВ 1930,1, 892,
Ф. п. 673576, СВ 1930, II, 1281, 672466, СВ 1930, П, 1443.
20) IG., Б. п. 321737, СВ 1930, II, 1141.
21) IG., Г. п. 484905 (1926).
22) Опыты А. Е. Пора й-К о ш и ц а (не опубликованы).
23) IG. Г. п. 430621 (1925), Б. п. 269212, СВ 1928, I, 1714, F. Keller,
К. Schnitzspahn, Ам. п. 1717453 (Grasselli) (1929), 1744903 (G. An. W.).
СВ 1930,1, 3610, О. Staehlin (G Ап. W.), 1744193 (1930), IG. Г. п. 438743,
(F. XV, 574).
24) С. Schraube, С. Schmidt, Вег. 27, 514 (1894).
IG. Б. п. 307965, СВ 1930, 1, 2007, Ам. п. 1724062, СВ 1930, 1, 3723.
О строении норм, диазогидратов: А. А п g е 1 i, Вег. 63, 1977 (1930),
25) Ю„ Б. п. 320824, СВ 1930, II, 1287. Ср. G. М. Norman, Soc. 115,673(1919),
Bull. 26, 474, IG. Г. п. 500437, 502334, 513209, Ch. Т. Ob. 1931, 80, 182.
26) By, Г. п. 161954 (F. VIII, 252).
27) Fierz-David, Soc. Ind., 1923, 423 T.
28) Cm. Dimroth, Ber. 40, 2404, 4460 (1907), 41, 4012 (1908); К. H. Meyer,
Ann. 380, 212 (1911), 398, 49 (1913), Ber. 52, 1472 (1919): К. v. Auwers, Ber. 47,
1275 (1914); Karrer, Ber. 48, 1398 (1915), 50, 1479 (1917); Chataway, Hill,
CB 1923, 1, 747.
277
29) Ср. К- Н. Meyer, Lenhardt, Ann. 393, 66 (1913); Ворожцов, Хими-
ческие особенности нафталинового ядра, Иваново-Возиесенск (Известия И.-В. Полит.
Ии-та № 6); Eg. Mohlau, Cb.-Ztg. 56, 31, 305 (1932).
30) Witt, Ber. 27, 2370 (1894); W. К 0 n i g, K. Kohler, CB 1921, III, 316.
31) К. H. Meyer, Ber. 54, 2267 (1921). Ср. E. Rosenhauer, H. Unger,
Ber. 61, 392 (1928), Rosenhauer, Ber. 63. 1056 (1930), 64, 1438 (1931).
32) Brass, Sommer, Ber. 61, 993 (1928). Cp. R. Mohlau, E. Strohbach,
Ber. 33, 804, 1900.
33) W. W. S c h a r w i n, К a 1 j a n о w, Ber. 41, 2056 (1908).
34) Ср. H. Hagenbach, Traugott Sandmeyer’s Forschungen und Erfindungen,
Helv 6, 159 (1923).
35) См. А., Г. n. 217935, (F. IX, 302) M. В a 11 e g а у, P. S c h n e i d e r, Chim. & Ind.
21, 497 (1929), CB 1930 I, 1618.
36) Heusner, Simon, Am. n. 1532627, CB 1925, II, 1230; By, Ф. n. 585664,
CB 1921, I, 2662.
37) К., Г. n. 188645, (F. VIII, 676).
38) H. Goldschmidt, A. Merz, Dinamische Untersuchungen uber die Bildung
der Azofarbstolfe, Ber. 30, 970 (1897), F. Buss, Ber. 30, 2075 (1897); E. Buikle,
Ber. 32, 355 (1899); К e p p e 1 e r, Ber. 33, 893 (1900); H. Keller, Ber. 35,3534 (1902).
J. B. Conant, W. D. Peterson, Am. Soc. 52, 1220 (1930).
39) G. R. Levi, Delponte, CB 19’9, II, 165.
40) V. Meyer, L e с c o, Ber. 16, 2976 (1883), 17, 572 (1884).
41) Schering, Г. n. 62004 (F. Ill, 50).
42) W. Davies, CB 1922, III, 1081.
43) А. В а у e r-H a 11 e r, Ber. 18, 90 (1885).
44) Griess, Ann. 137, 69 (1886), E. & O. Fischer, Ann. 194, 270 (1878).
45) J. Remsen, Ber. 18, 65 (1885), Hantzsch., Jochem, Ber. 34, 3337 (1901).
46) Winston, CB 1904, I, 809.
47) P. Friedlander, Ber. 22, 587 (1889).
48) E. v. Meyer, J. pr. Ch., 36, 115 (1887); Ю. Г. В e н д e л ь ш т е й и, Ан.-Кр.
Пр. 1932, № 1, 37.
49) Rhein Kampfer FabriK, Г. п. 456857 (1925).
50) Rhein. Kampfer F a b г i к, Г. п. 462153, CB 1929, I, 2234.
51) J. Gubelmann, H. Weiland, O. Stallmann (Newport), Am. n. 1623949
(1927).
52) A. Corbellini, Debenedetti, CB 1929, II, 2046.
53) IG., Г. n. 497412, CB 1930, II, 983, С r a w f о r d, W i 11 s о n, Б. n. 274960 Br. A. B.
1927, 772.
54) F. H. Kranz (Nat. An.), Am. n. 1715417 (1929).
55) J. Schmidt, W. Maier, Ber. 64, 767 (1931), CB 1932, I, 1230.
O. Ruff, V. Stein, Ber. 34, 1668, CB 1901, II, 351.
A. S e у e w e t z. Chim. & Ind. 20, 218, 1928; A. Seyewetz, D. Mounier,
C. r. 188, 953, CB 1928, I, 2578. И. M. Коган, А. Пнщулина, К. Анкиновнч,
Ан.-Кр. Пр. 1932, № 11, 34.
56) J. S. Р. Blumberger, Rec. 49, 259 (1930), СВ 1930, I, 3298.
57) San dm е у er, Ber. 17, 1633, 2650 (1884), 23, 1880 (1890), ср. La villa Lo-
rens. Ch. Ztg. 1912, 898,
58) L. Ga Hermann, Ber. 23, 1218 (1890), 25, 1091 (1892).
59) Fry, Gratt, Am. Soc. 1926, 710.
60) A. Korczinski, Bull. (4) 29, 283 (1921), A. Korczinski, Mrozinski,
V i e 1 a n, CB 1920, III, 505.
61) R. Kuhn, E. E i c h e n b e r g e г, Шв. n. 141750, C. A. 25, 2153 (1931).
62) Le Fevre, Turner, Soc. 1928, 967.
H. W. Schwechten, Ber. 65, 1605 (1932).
63) Balz, Schiemann, Ber. 60, 1186 (1927), Le Fevre, Turner, Soc.
1930, 1158.
64) См. »), ср. H. T. Clarke, R. R. Read, Am. Soc. 43, 1001, CB 1924,1,2877.
65) IG., Г. n. 524187, CB 1931, II, 632, Б. n. 326149, CB 1930,11. 307.
66) Leuckardt, J. pr. Ch. 41, 179 (1890).
67) IG., Б. n. 279136, CB 1929, I, 2693, IG. Г. n. 433103 (F. XV, 325).
68) Ziegler, Ber. 23, 24S9 (1890), G. E. H i 1 b e rt. T. B. J о h n s о n. Am. Soc.
51, 1526, CB 1929, II, 303.
69) F. VIII, 470.
70) G a 11 e r m a n n, Ber. 34, 3328 (1901).
278
71) Brydowna, CB 1928, I, 2820.
72) С., Г. n. 445390 (F. XV, 300), IG. Г. n. 485787.
73) G. An. W., Am. n. 1735432, CB 1930, II, 137.
74) Gattermann, Ber. 32, 1137 (1899), By, Г. n. 97286 (F. V, 202).
75) I. J. Rinkes, CB 1927, II, 1471. Неопубликованные опыты M. И. С л а fl-
ко в а. Несогласные данные у Schoutissen, СВ 1928, I, 803, См. MLB, Г. и. 224387
(F. X, 787).
76) G., Г. п. 164655 (F. VIII, 647), К., Г. п. 176619 (F. VIII, 653).
77) К-, Г. п. 176618, 176620 (F. VIII, 651, 652).
78) Р. R u gg 1 i, F. Knapp, E. Merz, A. Zimmermann, Helv. 12,1034 (1929).
79) В., Г. n. 145906, 148881, 148882 (F. VII, 400—402) 156440, 160536, 162009
(F. VIII, 656-659).
80) Hagen ba ch, см. 3t,', 1. c. 157.
81) L. Less a, A. Mossin i, Chim. & Ind. 28, 121 (1932).
ГЛАВА X
АРАМИНИРОВАНИЕ. ОБРАЗОВАНИЕ АРИЛАМИНОВОЙ ГРУППЫ
а) Араминирование в рядах бензола, нафталина, антрахинона
Получение вторичных, чисто ароматических аминов типа RNHR'
где R и R'— ароматические углеводородные радикалы, происходит
путем превращения характерной группы у ароматического амино-
замещенного по схеме RNHa -> RNHR' или путем изменения ре-
акционной группы в окси-(фенольных) соединениях: ROH-> RNHR'.
Возможны также и практически используются, как уже было отме-
чено выше (см. главу VII), переходы от хлоро-(галоидо-)замещен-
ных к вторичным аминам: RC1 -> RNHR'. Наконец в исключитель-
ных случаях остаток NHR' вступает на место сульфогруппы.
Группу NHAr мы будем называть ариламиногруппой, в частном
случае C6H5NH — фениламино-, СН3 • С6Н4 • NH — толиламино-,
CioH7NH — нафтиламиногруппой и т. п. Отсюда за процессом ее
введения остается название ариламинирования или араминиро-
вания.
Общее выражение процесса араминирования
RY 4- H2NR' = RNHR' + Н Y,
где Y — реакционный атом или группа, претерпевающая превра-
щение: Cl, NH2, ОН, SO8H; HY следовательно — НС1, NHS, Н2О, H2SO3.
Взаимодействие требует, с одной стороны, участия вещества, под-
вергающегося араминированию (амина, фенола и пр.), с другой —
реагента, образующего ариламиновую группу. Такой реагент на-
ходим в виде первичного амииа.
Мы не будем здесь останавливаться на характеристике превра-
щения во вторичный ароматический амин хлоро- и вообще галоидо-
производных, как уже приведенной выше (гл. VII), н рассмотрим
преимущественно превращения первичных аминов и фенолов, со-
отв. гидроксильных производных. При замещении аминогруппы на
ариламиногруппу (RNH2 -► RNHR'), которое можно назвать арили-
рованием аминогруппы, чаще всего в реакцию достаточно ввести
исходное аминосоединение и араминирующий амин в избытке
(2—3-кратном), причем часть его в виде солянокислой соли: соляная
кислота нужна для связывания освобождающегося при реакции
аммиака
RNH24- R'NH2 -> RNHR' 4- NH3.
Иногда при араминировании, в особенности красителей, вводят в реакционную
смесь небольшое количество бензойной кислоты. В недавнем американском патенте
указывается на применение сульфаниловой кислоты (0,2% по весу ингредиентов)
280
в качестве катализатора при получении из л-фенетидина и а-иафтиламииа л-этокси-
фенил-а-нафтиламина
При получении дифениламина из анилина по американскому патенту 1921 г.
в качестве кислоты, связывающей анилин в соль, применяется беизолсульфокислота.
Нагревание анилина с сульфокислотой бензола производится в открытых аппаратах
при слабом кипении. В смесь добавляется время от времени свежий, свободный
от аммиака, анилин. После 72-часового нагревания температура постепенно повы-
шается с отгоном непрореагировавшего анилина. Температура поддерживается
ие выше 220—230°. Беизолсульфокислота при этом не разлагается J).
Взаимодействие в таких случаях происходит иногда в закрытом
сосуде (автоклаве) из кислотоупорного материала прн высокой
температуре и соответственно повышенном давлении, иногда при
кипячении с обратным холодильником. Взаимодействие с гидро-
ксильными замещенными (фенолами) происходит с освобождением
воды
ROH -J- H2NR' = RNHR' + Н2О.
При работе в закрытом аппарате предпочитают иногда для свя-
зывания воды прибавление гигроскопических солей, например хло-
ристого кальция или хлористого цинка.
Интересно, что из аминокрезола структуры (I) при нагревании
его (в присутствии соляной кислоты) с аминами, не заключающими групп SO3H,
СО2Н, NOS, до 210—215° получаются, по словам патента немецкой фирмы, только
ариламинопроизводные типа
NHR'
НО/\'/Х'СН8
несмотря на замедляющее влияние на реакционность NH2 со стороны стоящей
в орто-месте СН8-группы, и гидроксил не вовлекается в реакцию 2).
Нагреванием с анилином (соотв. другим ароматическим аминам) и соляной
кислотой с обратным холодильником 3.5-диоксибензойной кислоты (II)
CONHCjHg
CONHC6H5
СООН
I
получают вместе с карбарилидом этой кислоты (III) также и 3-оксидиариламин-
5-карбоновую- кислоту в виде арилида (IV). Реакцию араминирования в этом и по-
добных случаях можно проводить в присутствии хлористого циика 8).
Высокая температура кипения ароматических аминов позволяет
работать в открытых сосудах с обратным холодильником, нагревая
281
реакционную смесь не выше температуры, нужной для кипения
амина. В таких случаях прибегают к большим (до 10 мол. и выше)
количествам амина. Избыток араминирующего амина вообще может
быть только полезен, если его легко удается извлечь после завер-
шения реакции, лишь бы не было опасности вовлечения во взаимо-
действие иной, кроме затрагиваемой, реакционной группы. Если, как
в примере 1.8-аминонафтолсульфокислот 4)
ОН NH2
SO3H
или
ОН NH2
HOgS/^/^/^SOgH
имеются две реакционные группы, NH2 и ОН, которые можно заме-
ненить на ариламиновую, то для того, чтобы получить однород-
ный продукт замещения лишь аминогруппы, например
ОН NHC6H&
SO3H
нужно работать в условиях, исключающих проявление активности
со стороны гидроксила.
Оказалось полезным в этом случае работать в присутствии большого избытка
воды (равновесие в пользу водоотиятия делается мало вероятным), под давлением,
со сравнительно небольшим избытком амииа. Если вести процесс обычным путем, без
воды, в присутствии амииа и соли амииа, то, по словам германского патента, наблю-
дается осмоление.
Применение водных растворов оказывается весьма полезным для чистоты про-
дукта и в условиях, исключающих вторичные реакции в).
При описанном ходе реакции следят за исчезновением исход-
ного араминируемого вещества по диазо- или азореакции, отмыв
предварительно разведенной кислотой или отгоняя с паром из под-
щелоченной пробы первичный амин, служащий переносчиком арил-
аминовой группы. Достигнув желаемого результата, реакционную
282
смесь или делают щелочной, прибавив соды, и отгоняют с паром
первичный амин, или делают смесь слабокислотной для растворе-
ния того же амина, если его избыток невелик. Полученные арил-
аминосоединения (вторичные амины) обладают невысокой раство-
римостью в разведенных минеральных кислотах и могут быть по-
этому отделены от непрореагировавшего первичного амина. Если
продукт реакции растворим в растворе соды (сульфокислоты ди-
ариламинов), то последующим подкислением выделяют его из рас-
твора в свободном состоянии или в виде кислой соли.
Сульфокислоты с ариламиногруппой нередко получаются непо-
средственно после выделения в крайне загрязненном продуктами
окисления амина состоянии, например фенил-(соотв. толил-) а-нафтил-
амин-8-сульфокислота. В таком случае их очищают, переводя
в магнезиальные или кальциевые соли, причем осадки отходящих
минеральных соединений (MgCO3, CaSOJ захватывают большую
часть примесей.
При араминировании нафталиновых производных наблюдается
некоторое своеобразие в поведении сульфогруппы.
Именно при нагревании (190—200°) с первичными ариламинами
сульфокислот нафтолов * получаются с водоотнятием сначала
сульфокислоты арилированных нафтиламинов, которые при даль-
нейшем нагревании постепенно, с отщеплением сульфогруппы, пе-
реходят в арилнафтиламины 6):
/\/\/°Н
+ c6h5nh.
-NHC6H5
— н,о
HO8SZ 4
/NHCgH5
+ н,о
- H2SO4
Применяя ту же реакцию к сульфокислотам нафтиламинов, по-
лучают, с отщеплением NH3, лишь сульфокислоты арилированных
нафтиламинов при более легких условиях взаимодействия 7).
Для сульфокислот нафтиламииа, у которых сульфогруппы стоят в положениях
3, 4 или 5, рекомендуется при этом не переходить предел температуры в 140°.
При нагревании 1-иафтиламнн-4.8-дисульфокислоты с ариламииом до 180—
190° происходит замещение водорода аминогруппы арилом и отщепление одной
лишь пара- (к NH2- ) стоящей сульфогруппы 8):
HO3S NH2 HO3S nhc6h5
+c6h5nh2
so3h
* За исключением .и-сульфокислот нафтолов (о иих сказано ниже).
283
Наконец при нагревании с первичными ароматическими ами-
нами .^-сульфокислот нафтиламинов, соотв. нафтолов а- и ₽-, без-
различно, имеют ли место в этом случае моно- или полисульфо-
кислоты, наблюдается не только замещение амино- (соотв. окси-)
группы на ариламиновую, но и вторичный процесс — отщепление
сульфогруппы (в виде сернистой кислоты) и вхождение на ее
место второй ариламиновой группы 9):
HO3S NH2
+ 2C6H5NH2
------------>
— NH3 — H2SO3
HO8S NHC6H6
I I
В качестве первого продукта реакции иногда наблюдается образование соеди-
нений с одной ариламиновой группой и незатронутой лг-сульфогруппой, како-
вые соединения при дальнейшем нагревании с ариламином отщепляют SO2 и пере-
ходят в замещенные .и-нафтилендиаминовые производные.
При обработке производных диоксинафталина ароматическими
аминами наблюдается избирательное действие со стороны одной
из двух оксигрупп, именно находящейся в особо благоприятном
положении для замены на араминовую группу. Такой пример
имеется в 2.6-диоксинафталин-З-карбоновой кислоте (1)
/\/\/°Н
I
НО^4 /\/\сООН
/\/\/он
!п ।
RNH/\/\/\coOH
R — арильный остаток,
которая при нагревании с избытком анилина или его производных
до сравнительно невысокой температуры в 170° переходит в 6-арил-
амино-2-нафтол-З-карбоновую кислоту (П)‘.
Нужно отметить, что нагревание этой диоксикислоты с аммиаком в автоклаве
приводит к замещению на NH2 гидроксила также в 6 месте.
6-Арил-амино-З-оксинафталинкарбоновые кислоты возбуждают большой инте-
рес в качестве азосоставляющих (разновидность нафтолов типа AS), так как
наличие добавочной ауксохромной группы RNH — в частице азокрасителя ведет
к углублению цвета его до зеленых оттенков 10).
В ряду антрахиноновых производных, где замещения в а-месте,
как известно, совершаются с большой легкостью, образование
ариламиновой группы на месте ОН или NH2, стоящей в этом актив-
ном a-положении, требует менее высокой температуры, чем р-заме-
щения [иногда участия вспомогательных веществ, например В2О3 31),
СН8СООН 12)], для своего завершения 13).
Легче всего такие араминирования окси- (соотв. амино-) произ-
водных антрахинона происходят при употреблении продуктов
с оводороженной карбонильной группой (лейкопроизводных).
Прямой ввод ариламиногруппы в благоприятное положение
(а-) антрахинонового ядра может быть осуществлен нагреванием
284
соответствующих производных вместе с первичными ароматиче-
скими аминами в присутствии слабых окислителей (воздуха) и
с металлическими соединениями ариламинов или с металл-ами-
дами 14).
Полученные ариламиносоединения антрахинона являются частью
готовыми красителями, частью их лейкосоединениями. Наиболее
известно превращение этого рода в применении к хинизарину (I),
лучше его лейкопроизводному (II), с целью получения моно- (III)
и ди-л-толиламинопроизводного (IV), из которых последнее после
введения сульфогрупп в остатки толуидина и окисления превра-
щается в зеленый кислотный краситель:
6н NHC6H4 • сн3
2Н МНС6Н4СН3
с ।
IV
\/\сА/
nhc6h4ch3
При превращении хииизарина в ариламииопроизводное подмечен (в присут-
ствии борной кислоты) интересный факт влияния на полноту превращения метал-
лов (материала аппаратуры). Именно, при работе в железном аппарате превраще-
ние идет преимущественно до моноариламинозамещенного (Ill), между тем в нике-
левом — до диариламинопроизводного (IV) 16).
Из всего вышесказанного об интересующем нас сейчас про-
цессе получения промежуточных продуктов можно сделать вывод,
что процесс требует участия двух веществ: одного — принимающего
ариламиновую группу (амин, фенол, соотв. их замещенные), дру-
гого— образующего эту группу (первичный ариламин) и затем при-
сутствия некоторого количества минеральной (бдльшею частью со-
ляной) или органической (бдльшею частью бензойной) кислоты.
Эти кислоты, иногда прибавляемые в меньших количествах, чем
следует для связывания всего амина в соль, отчасти участвуют
в процессе активно, устраняя образование вторичных продуктов
взаимодействия, частью действуют каталитически.
Нет ли других катализаторов данной реакции?
Киевенагель, изучивший каталитическое действие иода при подобных
реакциях, получил в этом случае удовлетворительные результаты 1е). Тем не менее
иод — слишком дорогой реактив для техники, и вдобавок его применение не дает
285
преимуществ в смысле заметного сокращения времени или понижения температуры
реакции.
Одна нз немецких фабрик защищает в недавнем патенте применение гидро-
снликата, активированного кислотами (Tonsil), каолина, глнн, белиль-
ных земель и т. п. прн синтезе вторичных ароматических аминов. Реакция требует
продолжительного нагревания до высокой температуры (350°) в автоклаве, смесн
первичных аминов илн смесн амина н фенола. Здесь возможно осуществление
таких например реакций:
/ NHa-l-HO-/ \-NH-/ >|~Н2О
Наряду с последней — н такой:
2 ОН -> <f О-/ >+Н2О
Выходы повидимому невысоки, и метод вряд лн найдет применение в произ-
водстве 17).
Совсем иное техническое значение имеет другой катализатор —
соли сернистой кислоты. Выше мы видели уже их значение для
взаимных превращений групп OH^NH2.
В настоящем процессе применение солей сернистой кислоты
позволяет работать при невысоких температурах и без применения
повышенного давления 18).
Операция проводится нагреванием смеси из араминируемого
вещества, кислой (NaHSO3) или средней [(NH4)2SO3] соли сернистой
кислоты и первичного амина на водяной бане при энергичном раз-
мешивании до полного превращения исходного вещества в ара-
минопродукт. При изучении реакции обнаружилось, что она имеет
ограниченную сферу применения. Именно, она идет превосходно
в нафталиновом ряду для получения ^-арилнафтиламиновых заме-
щенных, но неприменима к соединениям бензольного ряда;
а-нафтольные сульфокислоты не дают в этом случае замещения
гидроксила на группу NHR.
Значение особенностей строения, констатированное при рас-
смотрении сульфитной реакции нафтолов и нафтиламинов, играет
роль и здесь: сульфогруппа, стоящая в мета-положении по отно-
шению к ОН- или NH2-rpynne нафталинового производного, пре-
пятствует замещению ауксохрома на ариламиновую группу. Сле-
довательно при взаимодействии 2.5-диоксинафталин-7-сульфо-
кислоты и сернистокислого анилина нет основания ожидать заме-
щения обоих гидроксилов, но лишь одного, находящегося в ядре,
свободном от сульфогруппы, стоящей в мета-положении:
286
Из аминов, переносчиков группы RNH, активны в реакции
с участием солей сернистой кислоты анилин, толуидины и ксили-
дины, n-фенетидин, п- и л«-сульфокислоты анилина, особенно же
реакционноспособны я-аминофенол и я-фенилендиамин 19), мало
реакционны бензидин и а-нафтиламин.
Предложены в немецком патенте также тиосульфокнслоты n-диаминов, напри-
мер
H2N-/\/'\sSO3H
в качестве активного амнна прн этом методе работы 20).
Третичные амины (например диметиланнлнн), находясь в реакционной смеси,
действуют при этой сульфитной реакции как катализатор, увеличивая выходы
ариламннопродуктов до теоретических.
Этот способ образования группы RNH — помощью сериистокнслых солей нахо-
дит себе объяснение в допущении промежуточного образования продуктов присо-
единения элементов сернистой кислоты к иафтольиому, соотв. нафтилэмииовому,
производному. На строение таких продуктов, как кетопронзводиых нлн хниои-
дов, было указано выше в главе VIII. Очевидно эти продукты присоедниеиия
значительно реакционны по отношению к амниам, давая с ннмн замещеииые через
промежуточную фазу продукта присоедниеиия, например
Н3 Н3
Н2 Н2
H/^SOgNa H^^-SOgNa
2-Окси-З-нафтойная кислота очень реакционна по отношению к сернисто-
кислым аминам 21). Возможно, что эта реакционность обусловливается перегруппи-
ровкой в кетосоединенне
Hs
Х-/\/\сООН ^/^^СООН
Повидимому и по отношению к реакции с аминами в сульфит-
ном процессе, как и по отношению к обмену NH2!z;OH, наиболее
отзывчивы те соединения, которые могут реагировать в двух фор-
мах: кетоновой и энольной 22).
Названные выше приемы позволяют получать большинство упо-
требительных ариламинопроизводных: дифениламина и некоторых
из его замещенных, фенилированных, соотв. толилированных, наф-
287
тиламинов, соотв. их сульфокислот, и арилированных аминонаф-
толсульфокислот.
Большая часть этих производных имеет значение в красочной
промышленности как азосоставляющие, таковы дифениламин (I),
фенил- и п-толил-а-нафтиламин-пери-сульфокислоты (II, III), фе-
нил-И- (IV) и фенил-7-кислоты (V)
ОН
HO3SX/X/X/NHC6H6 .
также динафтиламиновое производное И-кислоты (VI)
Дифениламин вследствие способности образовывать соединения
с азотистой кислотой, свойственной вторичным аминам, приме-
няется в больших размерах как „флегматизатор" для предохране-
ния от саморазложения бездымного пороха, в массе своей являю-
щегося эфиром азотной кислоты.
Некоторые из этих производных применяются в качестве про-
тивостарителей (антиоксидантов) в резиновом производстве; таков
например фенил-р-нафтиламин
•NHC6H6
(неозон)
Араминирование антрахиноновых производных и главным обра-
зом аминоантрахинонов по недавнему немецкому патенту можно
легко проводить, применяя первичные ароматические амины вместе
288
с их металлическими соединениями, металларилидами (например
C6Hr,NHNa и т. п.), или вместе с амидами металлов, например
NaNH2. Реакция проходит вероятно через хиноидную форму, на-
пример для аминоантрахинона (I)
ONa
| NH
nh2
СО
со
СО I
NHC6H5
NHCsH-,
СО I
СО I
NHCeH3
к которой присоединяется остаток ариламина, входя уже в ядро.
Таким образом возможно осуществить вышеприведенное превраще-
ние. Одинаково реакционны и (3-аминозамещенные. Иногда по-
лезно при взаимодействии прибегать к окисляющему действию
воздуха 23). Ариламиноантрахиноновые замещенные — промежуточ-
ные продукты ценных кислотных красителей; таковы вышеупо-
мянутые производные 1.4-диоксиаитрахинона
ОН
NHC6H4 СН
NHC6H4 • СН3
nhc6h4 • сн3
сульфокислоты которых —фиолетовый и зеленый красители.
Недавно описан новый способ получения вторичных ароматических аминов,
имеющих в пара-положеиии к NH еще первичную аминогруппу. Такие пара-амино-
производные дифениламина н его замещенных приобрели в последнее время
большой интерес, как диазосоставляющие для так называемых нариамииовых
рапнд-красителей при холодном крашении и печати (см. ннтрозирова-
н и е).
Способ нх получения — взаимодействие равномолекуляриых количеств пара-
замещенного нитрозобензола и орто-замещенного, но с свободным пара-местом
нитрозобензола и последующее восстановление первичного продукта взаимодей-
ствия. Первая реакция выполняется в среде концентрированной серной кислоты
19 Зак. 2611. — Н. Ворожцов
259
в присутствии уксусной при пониженной температуре; восстановление же — по-
средством раствора сернистого натрия 24). Например
Na2S
—NO—> первичный продукт —>
—> С1~\ NH-
\сн3
Не выяснен ход первого взаш’оденствия н характер цветного промежуточного
соединения, где очевидно имеет уже место связь между атомом азота, принадле-
жавшим пара-нитрозозамещенному, и парауглеродом орто-нитрозосоединения.
Отметим еще способ получения замещенных дифениламина из N-ариларимино-
ZOR'
арильных эфиров RCZ . Нагреванием последние переводятся в ацильные сое-
XNR"
динения соответственных диариламинов: RCON(R') (R"), которые при омылении
спиртовой щелочью образуют дпариламины R'NHR" к). Иминоэфиры получаются
легко из соответственных аминов и фенолов через анилидиминохлориды кислот.
Схема получения такова:
R'NHot^^'RCONHR' RCC1:NR' RC(OR"): NR'
—> RCONHR'R" R'NHR".
Арилирование оксигруппы, т. e. образование арилокси-
группы RO, — операция, осуществимая различными путями, но
в производстве промежуточных продуктов не используемая. Осу-
ществить эту реакцию возможно действием водоотнимающих
средств при высокой температуре на арилируемое оксисоединение
и фенол, дающий OR-группу (см. гл. XVI).
б) Характеристика вторичных ароматических аминов
Отличить качественно ариламиновую группу среди других заме-
стителей ароматического ядра можно по некоторым реакциям, ко-
торые характерны не только для этой специально группы, но
вообще для вторичных аминов, безразлично, будет ли азот такого
амина связан с двумя ароматическими радикалами или с одним
жирным, другим ароматическим. В технике наиболее удобны реак-
ции нитрозирования и азосочетания.
Если в соединении пет первичной аминогруппы (отсутствие диазореакции) и
нет оксигруппы и в то же время соединение реагирует с азотистой кислотой, обра-
зуя обычно желтые продукты, не дающие с фенолами азокрасителей; если прн
этом данное соединение само по себе в слабокислотной среде с диазониями обра-
зует азокрасители, то можно подозревать присутствие вторичной (или третичной)
аминогруппы.
Продукты нитрозореакции для вторичных аминов, так назы-
R к
ваемые нитрозамины pN • NO обнаруживаются по либерма-
RZ
но в с кой реакции (см. нитрозирование). Устранение нитрозо-
группы, связанной с азотом, следовательно получение из нитроз-
амииов вторичных аминов, возможно при обработке такими сла-
быми восстановителями, как сероводород (в среде разведенной
290
серной кислоты) или сернистая кислота (при нагревании в водном
растворе) 2С). Под влиянием спиртовой соляной кислоты (для не-
растворимых в воде нитрозаминов) ‘47) или под влиянием концен-
трированного водного раствора соляной кислоты нитрозамины пре-
терпевают перегруппировку в n-нитрозозамещенные аминов:
R\ R\
>N-NO -> /NH
H—R/(l) ON—R/ (1)
(4) (4)
Последние отличаются от нитрозаминов более глубокой окрас-
кой (зеленовато-голубой). Нитрозогруппа становится в ароматиче-
ском ядре в пара-месте к центральному атому азота. В том слу-
чае, когда с этим азотом связаны, с одной стороны, бензольное,
с другой — нафталиновое ядро и у обоих пара-места не заняты,
нитрозогруппа предпочтительно образуется в нафталиновом ядре 28).
По некоторым отношениям /г-нитрозозамещенных можно опре-
делить их строение и соответственно строение исходных вторич-
ных аминов.
Так n-нитрозооснования при действии едких шелочей разлагаются на первич-
ный амин и нитрозофенол
R
.... \Nfl _> RNH2 4- ON • R4 • ОН
ON —
По полученному амину RNH2 судят о характере того арила, который участво-
вал в образовании ариламиновой труппы. Такое же разложение с еше большей
легкостью производится действием разведенной серной кислоты для нитрозоосно-
ва иий нафталинового ряда
Некоторые нитрозооснования от действия избытка азотистой кислоты легко
переходят в соли диазония 29), например
CcH5NHC6H4NO -J- H2SO4 + 3HNO2 = C6H6NHCeH4-N,-HSO4 + 2HNO3-|- H2O.
Бамбергером констатировано, что нитрозобенгол CcH.-NO под действием
окиси азота и, возможно, азотистой кислоты переходит в нитрат диазония (см. также
нитроз и рование) 30)
RNO -)-2NO-> RN0NO3.
Нужно заметить, что вторичные амины, производные (3-нафтиламина, дают
нитрозамины. которые под влиянием спиртовой (или водной) сольной кислоты
переходят в ннтрозоосновайия. Нитрозогруппа этих последних становится в орто-
(я-) место но отношению к аминовому азоту. В тех случаях, когда такому пере-
мещению препятствуют структурные причины (пери-стоящая сульфогруппа), внт-
розогруппа переходят в пара-место бензольного ядра 3i).
Нитрозамины, где один из углеродных радикалов принадлежит антрахинону,
а другой — фенил, также при перегруппировке в кислой среде удерживают иитрозо-
группу в фенильном ядре, причем действием окислителей, в том числе и азотистой
кислоты, можно получить прямо нитрозамещенные зз), например
СО СО
в ледяной уксусн. кислоте
+ NaNO2
ON-N—
CH:.
CO I
NH-
NO,
-CH.
19*
291
Зная природу ариламиновой группы, определить ее количественно, т. е. опре-
делить молекулярный вес соединения, где она находится, можно путем той же
реакции иитрознровання. При этом сама обстановка и выполнение опыта таковы же,
как при определении аминогруппы методом диазотирования.
Иногда удобнее пользоваться методом количественного азосочетания с диазо-
ииами для определений вторичных ароматических аминов.
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
О DuP., Ам. п. 1840576, СВ 1932, 1,3499.
A. Lachmann, Ам. п. 1549136, СВ 1925, II, 2296.
2) 1G., Ф. п. 697764, СВ 1931, II, 1492.
3) 1G., Б. п. 355114, СВ 1931, II, 3663.
*) By, Г. п. 181929 (F. VIII, 169).
') К., Г. п. 170630, (F. VIII, 168).
в) А., Г. п. 38424 (F. 1, 417).
’) By, Г. п. 70349 (F. III, 513).
») А., Г. п. 158923 (F. VIII, 167).
By, Г. п. 75296 (F. III, 500), 76414, 77866, 78854 (F. IV, 594—596).
1") В. Неуп (G. Ап. W.), Ам. п. 1754’90 (1930).
И) By, Г. п. 86150 (F. IV, 308).
12) By, Г. п. 93224 (F. IV, 320).
1-!) By, Г. п. 91152 (F. 1V, 318) IG., 1. i . 537023, СВ 1932, I, 1006.
И) В., Ф. п. 526686 (1920).
’5) Un. Aik. Со, Б. п. 248874. СВ 1928, И, 2290.
10) К п о е v е п a g е 1. J. рг. С1>. 89, I (1914).
17) R h е i п Kampfer Fa, Г. п. 530735. СВ 1331, II, 3515.
is) Bucherer, J. pr. Ch. 71, 433 (1905), 75, 249 (1907); В., Г. п. 122570 (F. VI,
194).
И) Например IG, Бп. 282111, СВ 1930, 1,3361.
2») 1G., Г. П. 485435 (1926).
‘-1) Bucherer, 1. с.
22) Ворожцов, Химич, особенности нафталинового ядра. ср. С. В. Богда-
нов, Ж. О. X. 11 (64) 9 (1932).
W. Кб nig, Haller, J. pr. Ch. 101, 43 (1920J21), P. Friedlander, Ber. 54,
620 (1921).
») В., Г. n. 360530 (F. XIV, 854).
2i) 1G„ Г. n. 499824, CB 1930, II, 982, 511526, 512722, CB 1931, II, 1492.
2b) A. W. Chapman, Soc. 1929, 569.
26) А., Г. n. 377589 (F. XIV, 399).
21) O. Fischer, Hepp., Ber. 19, 2991 (1886), 20, 1247 (1887).
28) Wacker, Ann. 243, 306 (1888).
2») M. Jkuta, Ann. 243, 281 (1888), Jager, Ber. 8, 894 (1875),
3°) E. Bamberger, Ber. 51, 634 (1918).
:1i) В., Г. n. 205414 (F. IX, 186).
32) IG., Г. n. 443126 (F. XV. 664).
292
ГЛАВА XI
АЛКИЛИРОВАНИЕ
Название „алкилирование" в общем смысле покрывает собой
всякое вхождение алкила на место водорода в органическое соеди-
нение. Ниже мы будем принимать этот термин в ограничительном
смысле, имея в виду лишь замену алкилом водорода в амино- или
оксигруппе (реже в группе сульфгидрильной SH). В таком пони-
мании процесс алкилирования принадлежит к реакциям 2-го рода
(замена существующего заместителя новым). Метод алкилирования
при этом имеет значение в применении к красителям и в применении
к получению промежуточных продуктов.
а) Алкилирование в аминогруппе
Общее выражение реакций алкилирования аминогруппы таково:
RNH2 -Н XAlk = RNHAlk + НХ,
RNHAlk 4- XAlk = RN (Alk)2 XH,
где X — одновалентный радикал, связанный с алкилом (С1, Вг, ОН
и т. д.), R — арил, Aik — алкильная группа. Из этого выражения
видно, что реакция может иметь две ступени. Для первой хара-
ктерно образование вторичного (жирно-ароматического) амина
RNHAlk, для второй — образование третичного амина RN(Alk)2.
По этим двум ступеням и проходит обычно реакция, задержи-
ваясь на каждой из них таким образом, что даже в случае избытка
алкилирующего средства AlkX все-таки в продуктах реакции необ-
ходимо искать вместе с третичным и вторичный и иногда даже
первичный амин.
Полученные алкилированные амины интересны с той стороны,
что влияние алкил- и диалкиламиногруппы на ароматическое ядро
остается во всяком случае не меньшим, чем оно было со стороны
аминогруппы, и даже скорее усиливается. Мы можем это видеть
по подвижности п-водородного атома в третичных аминах в раз-
ного рода реакциях конденсации и замещений (см. например полу-
чение кетона Михлера из диметиланилина и фосгена, нитрози-
рование диалкиланилина) и по легкости, с какой такие амины
входят в реакцию азосочетания.
Наиболее употребительными алкилами, с которыми приходится
иметь дело в технике производства промежуточных продуктов,
являются: метил СН8—, этил С2НБ—, бензил С6НБСН2— (последний
е возможными заместителями в ароматическом радикале), реже —
такие производные метила и этила, как —СН2СООН, соотв.
293
— СН,-СН,ОН, и т. п. Применение бензила для алкилирования
аминогруппы часто имеет основанием использовать этот аралкил
(алкил с ароматическим, замещающим Н, радикалом), как носитель
сульфогруппы с целью например сделать полученный путем алки-
лирования продукт растворимым в воде.
Менее изучено применение бозее сложных алкилов, например пропила С>Н7.
бутила CjHg, амила С-,Нц, также гидроароматических радикалов, как циклогексил
СН2 ' СН2
С6НЦ— : — НС\ \сн2. Не исключена возможность их практического при-
СГЦ СН2
менения в будущем.
Как алкилирующие средства, передатчики алкилов (практически
этила и метила), находят себе применение в этом процессе прежде
всего смеси соответствующего алкилу алкоголя с мине-
ральной кислотой. Нагревая например анилин в виде соли
соляной кислоты или бромистоводородной кислоты1) (также иодисто-
водородной кислоты) или серной кислоты с метиловым спиртом
до достаточно высокой температуры (200—210°, в случае бромисто-
водородной кислоты на 50° ниже), переводят первичный амин во
вторичный и третичный, которых количественные отношения в смеси
зависят от относительного избытка спирта. Происходит ли в авто-
клаве реакция только с отнятием воды, например
C6H5NH2^-CH3OH------------> CeH3NHCH3 (1)
-н2о
CGH-NH2-h-2CH3OH--------------► C6H5N(CH3), (2)
— 2Н,0
или предварительно образуются из спирта и кислоты алкильные
соединения (галоидалкилы в присутствии НС1 или НВг, алкилсер-
ные кислоты с H2SO4) и последние реагенты алкилируют, отщепляя
галоидоводород или серную кислоту, т. е. проходит ли в таком
случае взаимодействие через этапы:
СН3ОН4-НС1-------------► СН8С1 (3)
— н2о
CGH5NH9-t-ClCHs----------► C6H3NHCH3 (4)
— HCl
с уверенностью сказать невозможно. Против первого предположе-
ния говорит необходимость участия сильной минеральной кислоты
и возможность алкилирования аминосоединений непосредственно
алкильными соединениями, которые могли бы здесь образоваться
например по уравнению (3). Правда, такое алкилирование (галоид-
алкилами и пр.) протекает успешно в присутствии связывающих
свободную кислоту агентов, но в данном случае амин и исходный
и алкилированный может быть таким агентом.
Не в пользу второго толкования реакции говорят, между про-
чим, опыты Кнолля и К02), получающих моно-и диалкиларамины
нагреванием ариламина со спиртом в присутствии малых количеств
иода. Тут с трудом можно допустить предварительное образование
294
йодистого алкила (за счет чего раскислится ОН), и повидимому иод
является лишь катализатором реакции, отвечающей уравнениям
(1) и (2).
По недавним американским патентам получение диметиланилина производится
нагреванием анилина с метиловым спиртом и небольшим количеством бромистого
метила в течение 12—20 час. в железном автоклаве до 230—240°.
Реакции, протекающие при этом, таковы:
C6H5NH2 + 2СН3ОН + СН3Вг —► C6H5N (СН3)3Вг + 2Н.,О,
C8H5N (СН3)3 Вт —> C6Hr,N(СН3)2 + СН3Вг.
Таким образом бромистый метил регенерируется из бромистого триметилфенил-
аммоиия, который все-таки частично сохраняется в готовом продукте. Подобно
этому можно получить и диметиловые производные других ароматических аминов3).
Способ другого американского патента—вводить в аминогруппу ароматических
аминов низшие алкилы обработкой их галоидалкиламн в присутствии щелочи — не
представляет преимуществ по сравнению с обычным4).
Природа галоидоводородной кислоты весьма существенно отра-
жается не только на энергии течения алкилирования (выше при-
ведены указания на различие оптимальных температур), но и на
результате.
Этиловый спирт и солянокислый анилин образуют диэтил-
анилин и моноэтиланилин. Применение соли бромистоводородной
кислоты обеспечивает образование преимущественно диэтиланилина.
При этилировании с участием этилового спирта применяется
лишь галоидоводородная кислота, но не серная, так как последняя
может вызвать образование из спирта этилена (С2Н5ОН == С2Н4-(-
— Н2О) и соответственно поднятие давления в аппарате выше допу-
стимого предела.
При прочих равных условиях применение серной кислоты
имеет большое преимущество перед применением соляной в воз-
можности в первом случае пользоваться металлической (стальной)
аппаратурой без защитного покрытия ее изнутри кислотоупорной
эмалью. Эмалирование автоклава или чаще его вкладыша и крышки
необходимо при работе с солянокислой солью анилина.
Поэтому было бы весьма ценно для производства выработать
приемы безопасной работы с использованием серной кислоты не
только для получения диметиланилина.
М. К. Беззубец и С. А. Лившиц, изучая этилирование анилина (в виде
солянокислой соли) этиловым спиртом при 185—190°, наблюдали зависимость между
избытком этилирующего агента и выходами моно- н диэтилпроизводного. Эту зави-
симость можно представить в виде диаграммы (см. рис. 10).
Таким образом с увеличением количества этилового спирта в смесн растет выход
третичного амина за счет уменьшения количества вторичного. При нагревании
в автоклаве (180—200°) солянокислого анилина с метиловым спиртом английскими
химиками был наблюден наибольший выход монометиланнлина в 58% 5).
В пользу понимания реакции между спиртом и ариламином как реакции водо-
отнятия говорит также получение алкильных производных ариламинов при пропу-
скании парообразно'й смеси амина и спирта (этилового или метилового) через ката-
лизатор (А12О3, ThO2. ZrO2) при 350—450°.
Подобно этому при нагревании анилина и метилового спирта до 230° с гидратом
кремневой кислоты (Tonsil) получается диметиланилин 6).
Алкилирование аминогруппы может быть достигнуто, как ука-
зано выше, и обработкой амина посредством галоидного алкила.
Между тем как первый способ имеет значение по преимуществу
295
для введения таких алкилов, отвечающие которым спирты — мети-
ловый, этиловый — доступнее и дешевле галоидных замещенных,
этот последний способ интересен для фиксирования таких алкилов,
спирты которых не более доступны сравнительно с галоидозаме-
щенными и благодаря высокому молекулярному весу менее реак-
ционноспособны в рассматриваемом процессе, ибо алкилирующая
способность спирта падает с возрастанием молекулярного веса.
Таковы из алкилов С6Н5-СН2 —, —СН2СООН и т. п. Урав-
нение реакции в этом случае аналогично уравнению (4). Присут-
ствие в реакционной смеси таких веществ, которые бы связывали
освобождающуюся кислоту, оказывается полезным для ускорения
реакции и для ее течения в одном лишь направлении. Связываю-
щим кислоту агентом может быть, как сказано, амин, имеющийся
в реакционной смеси, чаще же более или менее щелочные соеди-
нения, которые прибавляют для взаимодействия с кислотой: едкий
натр, сода, известь, углекислый кальций, гидроокиси тяжелых ме-
таллов и т. п. Температура реакции чаще не превышает 100°, почему
можно работать с обратным холодильником, если галоидный алкил
(С6Н5СН2С1, ClCH2COOh) кипит выше 100°. С низкокипящими
галоидными алкилами (С2Н3С1) работают в закрытых сосудах —
автоклавах. Количество алкилирующего галоидного соединения
рассчитывается в отношении не более, чем моль на моль алкили-
руемого амина, щелочной агент также берется в эквивалентном
к галоидалкилу количестве.
При получении алкилараминов первым методом (амин, спирт,
кислота) количественные отношения изменяются в пользу спирта,
избыток которого против теории может быть до 20% и выше,
особенно тогда, когда имеют в виду получение диалкилараминов.
296
Количество кислоты или эквивалентно количеству амина, или —
обычно — меньше. Комбинирование обоих способов работы приме-
няется иногда для получения диалкилараминов с различными алки-
лами. Таким образом полученный при этилировании по первому
способу мсноэтиланилин при обработке хлористым бензилом дает
бензилэтилаиилин. Метод особенно удобен для применения к смеси
вторичного и третичного аминов, полученной после этилирования
анилина, так как дает возможность легче разделить перегонкой
диэтиланилин от бензилэтиланилина, чем это возможно по отноше-
нию к смеси моно- и диэтиланилина. Нейтрализующим соляную
кислоту веществом здесь служит едкая щелочь или сода. Суммар-
ное выражение процессов таково:
C6H5NH,+ C2H5OH -------C6H5NHC2H6,
C6H6NHC9H54-CrH5CH2C1 — нс,-> C6HbN(C2H5)CH2C6H5.
О получении бензиланилина C6H5NHCH2CBH5 см. главу VII.
С целью получения моноэтиланилина,свободного от третичного
амина, можно пользоваться хлористым этилом, применяя его для
обработки не самого анилина, но соли щелочного металла арил-
сульфонанилида типа ArSOoN (Na) С6Н5, например полученного из
бензолсульфохлорида и анилина. Полученный после этилирования
Х-этил-Й-фениларилсульфонамид гидролизуется кипячением с раз-
веденной минеральной кислотой в этиланилин и арилсульфокис-
лоту 7).
Схема реакций в этом случае:
NH,
Na
+ NaOH
------->
- С2Н6С1
- NaCl
Этилирование хлористым этилом анилина в присутствии извести
(50% избытка С2Н5С1 сверх рассчитанного по теории количества.
125°, 10—12 ат, 12 час.) дает преимущественно диэтиланилин (58%)
при минимальном количестве непрореагировавшего анилина8).
297
Введение бензильного остатка в аминогруппу /г-сульфокнслоты аиилнна, сульфа-
ниловой кислоты, в присутствии щелочных агентов (соды, едкого натра) приводит
к образованию моиобензнльного производного HO3S C6H(NH- CH2Ct>H3 наряду
с дибензильным н даже соответственным четырехзамещенным аммонийным продук-
том OgS • Cgf-^N (CH2Cf.H3)3. Продукт в виде натриевой соли находит себе при-
менение как смщнвающее вещество в печати кубовыми красителями под названием
„солюционной соли'.
Замещенные в ядре проилводные хлористого бензила, типа ХСиН4СН2С1, где
X — С1, СООН и т. и., получаемые действием формальдегида и соляной кислоты на
соответственные бензольные производные (см. ниже, конденсация), предложено при-
менять для получения соединений типа например C(;H3N (СН3) СН2СвН4Х. Обра-
ботка аминов хлорбензильным производным происходит при невысокой температуре
(около 35°)9).
Своеобразный галоидный алкил с карбоксильной группой, моно-
хлоруксусная кислота С1СН2СООН, находит широкое применение
для получения замещенных глицинов RNHCH2CO2H, особенно инте-
ресных для производства индиго и индигоидных красителей. Моно-
хлоруксусная кислота применяется в водном растворе, реакции
с ней проводятся в открытых сосудах с обратным холодильником.
Нейтрализующим освобождающуюся соляную кислоту агентом
является или сам амин (взятый в большом избытке)10) или, чаще,
минеральные основания, прибавляемые в эквивалентном соляной
кислоте количестве.
Необходимо отметить, что в качестве побочных продуктов прн действии хлор-
уксусной кислоты на арамин, с одной стороны, можно ожидать образования ар-
амида арнлглицина
RNHCH2CONHR,
с другой — образование дналкнльного производного
RN(CH2COOH)2.
Предложено для устранения образования арамида прибавление нейтрализующих
окисей (СаО илн MgO)11) нли водной закиси железа 12). Последняя прибавка
является повидимому в этом случае наиболее полезной.
В некоторых исключительных случаях реакция с монохлоруксус-
ной кислотой имеет целью введение не группы СН,СООН, но ме-
тила СН3, образующегося из первого заместителя с. потерей дву-
окиси углерода. Процесс алкилирования проходит тогда через две
необходимые ступени:
RNH2 + C1CH.,COoH ---------> RNHCH,CO.,H
— HCI
RNHCH0CO0H------------> RNHCHS
- CO2
Вторая фаза процесса13) вызывается нагреванием полученного
продукта до более или менее высокой температуры.
Этот способ введения метила особенно интересен в применении к получению
л-моно-Н-метиламннофенола (И)
/\/NHCH2CO2H NHCH3
111 -co^ ! 111
HO/Z\/Z HO ''
служащего фотографическим проявителем (метол).
298
Последние американские патенты по этому предмету описывают применение
крезола как среды для нагревания п-оксифенилгдицниа (I) до 165—180° для отще-
пления СО2 14)-
Из галоидных алкнлов отметим еще этиленхлоргндрнн С1СН2-СН2ОН, служа-
щий для введения остатка — СН2 • СН2ОН. Замещение в последнем гидроксила по-
средством эфнрной „сульфатогрунпы* O3OSH открывает возможность получения
новых кислотных красителей (сульфатных) 1;’)-
Введение этанольного остатка в анилин с переходом его в окси-
этиланилин достигается действием этиленхлоргидрина на анилин
С1СНЙСН.2ОН
/-X//NHCH2CH.,OH
Продукт при щелочном плавлении переходит в индиго 1й).
Введение этанольного остатка в аминогруппу, например у амннонафтолсульфо-
кислот, можно производить обработкой нх щелочных солей посредством окнси
этилена (СН2)2О 17;. например
Реже применяются в практике алкилирования аминогруппы
алкильные эфиры серной кислоты, например диме-
тилсульфат (CH3)2SO4 == CH8OSO2OCHs, соли алкилсерной
кислоты, например метилсернокислый натрий CH3OSO3Na, и
алкильные эфиры арилсульфоиовых кислот. Из по-
следних наибольшее техническое значение имеют эфиры л-толуол-
сульфокислоты, например л-СНа С6Н4 • SO.,OCH3, для которых
исходный материал, n-толуолсульфохлорид, доступен как значитель-
ный по количеству побочный продукт сахаринового производства.
Схемы реакции во всех случаях похожи, как схемы омыления
кислотных эфиров, например
RNH2 4- CH3OSO.,OCH3 = RNHCH3 + CH3OSO_.OH, (5)
RNH, 4- CH3OSO3Na = RNHCH3+NaHSO„ (6)
RNH., 4- AlkOSO.,R' = RNHAlk 4- R'SO3H, (7)
RNH., 4- 2AlkOSO2R' = RN(Alk)., 2R'SO3H. (8)
Алкилсерные соли из этих агентов действуют нанмеиее энергично. Днметил-
сульфат, по Ульману 18), прн взаимодействии с двумя молекулами первичного
амина (в водной суспенсни прн встряхивании) дает вторичное основание и метил-
сернокислую соль первичного амнна по уравнению (5). При нагревании первичного
амина с днметилсульфатом до более высокой температуры можно по произволу напра-
влять реакцию в сторону образования преимущественно моно- и дналкнльных про-
изводных. Выходы продуктов алкилирования изменчивы: они выше у гомологов
анилина, чем у самого анилина. Третичные основания в неводной среде (например
эфирном или бензольном растворе) действием того же алкилирующего средства
переводятся в четвертичные аммониевые солн.
Диметилсульфат прн обработке им натриевых солей арилсульфоиовых кислот
превращает последние в метиловые эфиры.
299
В одном из немецких патентов описывается применение метилсериокнслого
натрия CH3OSO3Na для метилирования п амииофенола в аминогруппе. Для этого
нужно нагревать с обратным холодильником л-аминофенол и эквимолекулярное
количество метилсериокнслого натрия в водном растворе в течение 6—8 часов 18).
В. М. Родионов н В. Е. Введенский испытали обработку метиловым
эфиром п-толуолсульфокислоты в применении к а-(соотв. р-)нафтиламину и обнару-
жили, что оптимальная температура взаимодействия для обоих лежит в пределах
155—160е. При избытке метилирующего агента получаются преимущественно диме-
тильиые производные. ?-Нафтиламии метилируется несколько труднее а-изомера.
Диметил-а-иафтиламин при нагревании с эквимолекулярным количеством метило-
вого эфира толуолсульфокислоты дает количественно метил-п-толуолсульфонат
лиметил нафтиламина
C10H7N(CH3)3 OSO2C6H4 • CHS,
пригодный для алкилирования подобно соединениям типа R—N(OH)=Alk;i (см- ниже) 2°).
Недавно Слотта и Франке приготовили эфиры п-толуолсульфокис-
лоты с высшими спиртами, начиная от пропилового, и испытали их в каче-
стве алкнлируюших агентов применительно к соединениям, заключающим группы
NH2, ОН и SH. При нагревании до 110—125° амина и эфира сульфокислоты в отно-
шениях молекул амии :эфнр = 2 :1 получаются с хорошими выходами моноалкили-
рованиые амнны. Лишняя молекула амина служит в этом случае для связывания
в соль п-толуолсульфокислоты, получающейся по уравнению (7). При нагрева-
нии смеси амина и эфира толуолсульфокислоты в молекулярных отношениях
амин : эфир — 1:2 до более высокой температуры и с добавкой едкого кали в ко-
личестве, эквивалентном сложному эфиру, образуется диалкилированный амин.
Алкилирование группы SH у тнофеиолов производится также при нагревании
тиофенола и сложного эфира (в эквимолекулярных отношениях) с добавкой едкого
кали.
Введение алкила в трудноалкнлируемые аминосоединения, например антрахино-
нового ряда, предложено в одном английском патенте осуществлять, применяя для
этого сульфаминовые кислоты, отвечающие этим аминам. Их преимущество заклю-
чается в растворимости в воде и стойкости по отношению к щелочным агентам.
Таким образом например из {3-аминоантрахинона (1) действием ангидро-М-сульфо-
кислоты пиридиния готовится сначала антрахинон-^-сульфамииовокислая соль (11),
которая затем обрабатывается при 50° диметилсульфатом, и полученная сульф-
аминовокислая соль метилированного амииа (111) гидролизуется кипячением с мине-
ральной кислотой в [-i-метиламиноантрахинон (IV):
+ (CH3)2SJ,
Способ алкилирования первичного амина во вторичный с пред-
варительным введением сульфогруппы в связь с азотом приложим
и к просто построенным аминам, например к переводу анилина
в монометиланилин:
NHCHS
300
и принципиально очень напоминает ранее описанный (см. стр. 297)
способ Ульмана, где „защитой" аминогруппы является сульфо-
арильная группа 21).
При „защите" аминогруппы со стороны сульфогруппы может
получиться таким образом только моноалкильное производное.
Подобно как при алкилировании фенолов, в этом случае из двух
метильных групп диметилсульфата в реакцию (при низкой темпера-
туре) вступает лишь одна, другая же остается связанной в виде
метилсернокислой соли [см. уравнение (5)].
В американском патенте (Lapworth) предлагается с целью получения только
вторичных аминов из пара-замещенных первичных (1) пользоваться альдегидными
соединениями последних (Шиффовыми основаниями) (11) (см. след, главу) и обра-
боткой таких соединений диалкилсульфатом или алкильным эфиром сульфокислоты
в бензольном растворе и последующим гидролизом получать продукт моиоалкилн-
рования (IV), например 2i)
гидролиз
(HCI)
ь ОСН • С6Н3
В американской химической литературе описан метод получения только вто-
ричных аминов без примесн третичных, использующий в качестве алкилирующего
средства алкоголяты алюминия (А1кО)3А1. Последние соединения готовятся дей-
ствием амальгамы алюминия на безводные спирты. Этилат алюминия плавится
прн 140° и перегоняется при 200° при давлении 6 —8 мм. Реакция этилирования
здесь требует энергичного нагревания в закрытых аппаратах при температуре
в 275—350°. Реакция протекает повидимому по схеме например
(С,Н-О)3А1 4- 3C6H5NH2-> 3CeH5NHC2H5 + А1(ОН)3.
Выхода моноалкнлированного амина очень высокие. Примесью является первич-
ный амин. Повидимому такой способ получения нторичиых жирно-ароматических
аминов применяется в американской практике 23).
Воздействием метиловых эфиров сульфокислот (фенолов) третичные амины мо-
гут быть превращены в соли четвертичных аммониевых оснований 23).
Кроме вышеперечисленных приемов введения только одного
алкила в аминогруппу обращает внимание еще один, именно исполь-
зующий в качестве промежуточных стадий реакции альдегидные
соединения аминов, где альдегиды имеют то же число углеродных
атомов, что и во входящем в связь с азотом алкиле.
Наиболее широкую известность этот метод получил в отноше-
нии введения метильной группы (и некоторых ее производных)
при участии формальдегида.
301
Формальдегид (в водном растворе) легко образует с ароматиче-
скими аминами ангидросоединения:
RNH2 + OCH„---------> RN : СН2; (а)
-Н2О
последние могут быть восстановлены (цинковой пылью или акти-
вированным алюминием) в метилированные ариламины 24):
+ Н2
RN : СН2------> RNHCH3.
Здесь повидимому имеется метод получения лишь вторичных
метилированных аминов без примеси третичного, особенно инте-
ресный для приготовления монометил-/г-аминофенола, имеющего
применение как фотографический проявитель, (метол) (ср. франц,
пат. фабрики Griinau).
Однако при восстановительном процессе нередко имеет место гидролиз ангидро-
основания, почему в продукте восстановления имеется иногда довольно много пер-
вичного амина.
Восстановление по патенту немецкой фирмы (1924) осуществляется каталитиче-
ским путем: обработкой водородом в спиртовом растворе в присутствии металличе-
ских катализаторов (палладий на угле); по патенту Риделя — действием активи-
рованного алюминия в метиловоспиртовом растворе. Полученное по вышесказан-
ному ангидроформальдегидное соединение амина может быть переведено обработ-
кой синильной кислотой в нитрил кислоты, причем группа CN станет у углерода
метиленовой группы, т. е. получится ш-цианметиларамин 25).
RN:CH24-HCN —> RNHCH2CN.
Такие же нитрилы могут быть получены быстрее и проще, если
вместо формальдегида обрабатывать амин бисульфитным соедине-
нием формальдегида, имеющим строение
НО • СН2 • SOsNa,
причем в результате водоотнятия получается продукт замещения
в метилированном амине водорода метила на группу SOsH, ш-суль-
фокислота метилированного амина
RNH24-HOCH2SO3Na----------> RNHCH,SO3Na. (б)
— н2о
Полученное соединение легко обменивает свою сульфогруппу
на цианидную при обработке солью синильной кислоты
RNHCH2SO3Na -f- NaCN = RNHCH2CN -f- Na2SO3 2«).
Реакция в этом измененном виде применима и к вторичным аминам, так как из
сравнения уравнений (а) и (б) явствует, что при использовании бнсульфитного
соединения формальдегида достаточно наличия всего лишь одного водородного
атома в аминогруппе для того, чтобы могло образоваться алкильное соединение 27д
Полученные вышеописанными приемами ш-цианметиларамины
могут быть гидролизом переведены в соответственные карбоновые
кислоты
RNHCH2CN------> RNHCHoCOOH
и образовать таким образом замещенные глицины.
Получение нитрила фенилглицина, так же как и получение са-
мой карбоновой кислоты, является важнейшим этапом в имеющих
302
практическое значение синтезах индиго. Большинство европейских
производителей индиго пользуется методом получения фенилгли-
цина из хлоруксусной кислоты и анилина. В Америке кроме этого
практикуется и нитрильный метод.
Предложения алкилировать ароматические амнны через их соединения с жир-
ными альдегидами (высшими гомологами формальдегида) — Шиффовы основания—
посредством их восстановления—многочисленны. Вряд ли однако онн могут рас-
считывать на практический успех, так как реакция восстановления не протекает так
гладко, как это желательно.
Из описанных преимущественно в немецких патентах приемов восстановления
альдегидных соединений аминов назовем применение цинка в присутствии едкого
натра или разведенной серной кислоты или уксусной кислоты; медный купорос
активирует цинк 28). Также предложено восстановление цинком в присутствии вод-
ной сернистой кислоты 29, магнием н метиловым спиртом з°) илн методом катали-
тического гидрирования (под давлением около 5 ат) в присутствии катализаторов
группы железа (Ni, Со), содержащих окислы или соли тяжелых металлов 31).
Наибольшего внимаиня заслуживает метод Валлаха, практикующего приме-
нение смеси из амииа, альдегида и муравьиной кислоты, играющей очевидно роль
восстановителя для альдегидного соединения. Таким образом получен например
диамиланилнн из валерианового альдегида, муравьинокислого анилина и муравьи-
ной кислоты. Повидимому можно вводит в смесь в качестве восстановителей альде-
гида и иные органические вещества, способные окисляться, например изопропило-
вый спирт 3'-).
Выше было упомянуто уже о каталитическом методе получения
вторичных и третичных аминов из первичных аминов и спиртов
алифатического ряда.
Этот метод привлекает исследователей своей простотой, хотя
повидимому не имеется еще достаточных оснований для перевода
его в производственный масштаб. Отметим, что получение моно-
и диэтиланилина по французскому патенту немецкой фирмы
удается при пропускании паров ингредиентов (анилина и этилового
спирта) через никель, осажденный на пемзе.
Гюйо и Фурнье получали циклогексиланилин при нагревании смеси
циклогексанола и анилина с небольшим количеством никеля в автоклаве до 170°.
Выражение результата реакции:
C6HUOH -j- CgHsNHa = C6H-NHC8Hn + Н3О (1)
нелогично с тем, которое отвечает опытам Мэль. В то время как там катализа-
тор — окислы металлов, здесь катализирует металл (никель может быть заменен
Со, Си, Fe, но Ni особенно благоприятен). Исследователи полагают, что процесс
здесь более сложен, чем выражение его результата (I), и что металл дегидрогенизи-
рует спирт в альдегид или кетон, который конденсируется с амином в альдимин
илн кетимни, принимающий водород от первой фазы реакции, иапример 33)
+ R'nh2 +н2
RCHoOH —> RCHO-----------> RCH : NR'--------> RCH,NHR'.
- н. — Н2О
Метиловый эфир (СН3)2О является метилирующим средством, если пропускать-
его пары вместе с парами вторичного илн первичного амина черезокнеиые катали-
заторы (Ai2O3, ZrO2, ThO2, TiO2) при температуре 270 — 350°. В этом случае можно
применять н смесь" метилового спирта с метиловым эфиром м).
Обработка ароматических аминов, в частности диаминов акридина, посредством
бисульфита и альдегидов (не одного только формальдегида) является способом обра-
зования соотв. алкнл-ы-сульфокислот аминов 3;’).
Отметим далее способ образования алкиламиновой группы вне-
сением не только алкила, но и азота этой группы извне.
303
Сульфитная реакция, описанная в главе о превращениях
NH.j^OH, дает к этому возможность. Алкилнафтиламины могут
быть образованы нагреванием производных нафтиламииов, а также
и нафтолов с раствором средней сернистокислой соли
соответственного алифатического амина в присут-
ствии свободного алкиламина. Таким образом возможно
осуществить превращение
(NH,CH3)2H2SO3
nh2ch8
Очевидно реакция проходит по тем схемам, которые развиты
выше (см. главу VIII).
В антрахиноновом ряду введение алкиламиновой, например метил-
аминовой, группы вместо иных групп, чаще всего вместо сульфо-
группы, осуществляется простым нагреванием исходного материала
с алкиламином, лучше — в присутствии окислителя, например
NHCH.,
СО |
Оксипроизводные антрахинонового и нафтохинонового рядов легче
поддаются превращению в алкиламинозамещенные при обработке
их алкиламином, если их применять в виде продуктов восстановле-
ния (гидрохинонового типа). Так, при нагревании лейкопроизводных,
получаемых действием например гидросульфита Na2S2O4 в щелоч-
ной среде на такие полиоксиантрахиноны, как хинизарин (1.4-ди-
оксиантрахинон), 1.4.5-триоксиантрахинон, 1.4,5.8-тетраоксиантрахи-
нон, ализарин бордо (1.2.5.8-тетраоксиаитрахинон), диаминоантра-
руфин (1.5-диамино-4.8-диоксиантрахинон), диаминохризазин (1.8-
диамино-4.5-диоксиантрахинон), получаются замещения метиламино-
выми группами преимущественно в 1.4 местах. Образованные таким
образом лейкопродукты действием окислителя, например нагрева-
нием в нитробензоле, переводятся в замещенные антрахинона,
имеющие значение как красители для ацетатного шелка, например36)
ОН ОН
I СО
ОН
ОН
+ Na,SA
------->
2CH3NH..
304
----->
окисл.
HNCH3 ОН
i 9? А /он
| со
HNCH3
Отметим попутно, что в патентной литературе имеются указания на примеры
замены аминогруппы гуанидиновой
/NH,
RNH3 —> RNH—CZ
'NH
каковая замена, аналогично как при получении гуанидина из аммиачных солей, и
здесь происходит действием цианамида или дициандиамида на аминогруппу. При-
меры даиы применительно к оксинафтиламнновым производным с свободным к NH3
орто- или пара-положением 37).
Противоположная алкилированию аминов задача может быть
поставлена как освобождение аминогруппы от алкильных групп,
замещающих ее атомы водорода. Чаще всего речь может итти об
устранении одного из двух имеющихся алкилов у третичного
жирно-ароматического амина. Такое устранение алкила, например
образование монометиланилина из диметиланилина (с выходом в 55С/О),
можно проводить нагреванием диметиланилина с солянокислым
анилином до 180° 38).
Предложено также осуществлять получение вторичного амина из третичного,
нагревая его солянокислую соль, например C6H6N (СН3)2 • НС1, до 170 — 180° при
медленном приливании уксусного ангидрида. При реакции, по описанию, выделяется
сначала хлористый метил, а затем уксусная кислота. В продукте остается ацетиль-
ное производное моиоалкиламина, например
C6H5N (СН3)2НС1 + (СН3СО)2О -> C6H6N (СН3) СОСН3 + СН3СО2Н 4- СН3С1 39).
Описано также как дающее возможность устранить один алкил из третичного амина
нагревание около 200° третичного амина с органическими кислотами (гидрокоричной,
бензойной, пальмитиновой, уксусной), — во всех случаях получается моноалкилани-
лид соответствующей кислоты в выходах, во всяком случае незначительных (10 — 30%).
Отщепление алкильной группы действием бромистого циана BrCN (бромангид-
рида циановой кислоты), протекающее по схеме
R2-N + BrCN =
R‘\ /Вг
r2-n<
R3\
>NCN + Rj.Br,
R/
принадлежит к этому же классу процессов и позволило установить ряд прочности
связи алкилов с азотом: С6Н5СН2, СН3, С2Н5, С3Н;, — высшие алкилы, С6Н5 40). Бен-
зильный остаток, всего слабее связанный с азотом, может быть действительно устра-
нен уже просто гидрированием. Обработка водородом в присутствии палладия и угля
таких аминов, которые имеют в связи с азотом бензил, независимо от того, вторич-
ный или третичный этот амин, приводит к отщеплению бензила, гидрированного до
толуола, например
CgHaN (CHg) СН2С6Н5 + Н2 C6H5NHCH3 + С6Н6СН3 ").
Наиболее часто в технике получения алкилированных аминов
в продукте не получается вполне индивидуального вещества, а смесь
20 Зак. 2611. — Н. Ворожцов
305
различных степеней алкилирования вместе с исходным первичным
амином. Нужно уметь как определить в этой смеси отдель-
ные составн нечаст и, так и располагать технически пригодным
способом разделения смеси на ингредиенты.
Первичные, вторичные и третичные амины разно относятся
к действию азотистой кислоты: азотистая кислота в присутствии
минеральной кислоты переводит первичные амины в соли диазония
(1), вторичные — в нитрозамины (II), третичные — в соли п-нитрозо-
оснований (III):
dktui "г HNOg -|- НХ р. v ...
RNH^^-^J------->R —N —X, (I)
N
RNHAlk o2 -> R (Alk)N • NO, (II)
H X
/ » T / ft 11 X HN Oo —J— H X . T / X. Я 1 1 X
< ^N(Alk),-7-——--------->ON-\ V-N(Alk)2zi
z^\ x
Z1H0N;/ \;N(Alk)2. (Ill)
Соли диазония и соли нитрозооснований растворимы в воде,
между тем как нитрозамины в воде нерастворимы и могут быть из
нее извлечены например эфиром.
В недавней работе А. Нелюбиной изучены скорости взаимодействия между
азотистой кислотой н аминами: первичными, вторичными и третичными. Константы
скоростей для реакций I, И, III значительно различаются между собой, как это пока-
зано в приводимой таблице
Реакция (при 0°) Константа
I Диазотирование анилина.......................... 3,3- 10“2
И Ннтрознрованне метиланилина . . •................ 10 • 10—2
„ „ этиланилииа.................... 1,34-Ю-2
III „ диметиланилина.................... 0,78 • 10~2
„ „ днэтиланилина.................... 0,72-10-2
Большая скорость нитрозирования вторичного амина сравни-
тельно с третичным дает возможность пользоваться методом нитро-
зирования для определения первого в присутствии второго. Одно-
временно диазотируется первичный амин, который должен быть
определен в отдельной пробе 42).
Удобный способ анализа, особенно для характеристики смеси
метилированных и этилированных аминов, предложили Ревердэн
и Ла-Гарп43). В нем диазопробой с последующим сочетанием
определяется первичный амин, вторичный амин определяется по
расходу уксусного ангидрида на ацетилирование его и первичного
амина. Определение выполняется таким образом:
1. Определение первичного амина. Навеску в 7 — 8 г испытуемой
смеси аминов растворяют в 28 — 30 см3 соляной кислоты и разводят водой до 100 с.п3.
306
Одновременно приготовляется титрованный раствор соли R, содержащий в 1 л такое
ее количество, которое эквивалентно приблизительно 10 г ,8-нафтола. Отбирают
питеткой 10 с.и3 раствора солянокислых солей аминов, разводят водой и льдом
и прибавляют такое количество нитрита, которое понадобилось бы израсходовать,
если бы смесь состояла только нз одного анилина, и полученную смесь постепенно
приливают в отмеренный объем раствора соли R, к которому добавлен избыток
соды. Образующийся азокраситель осаждается прибавкой поваренной соли, отфиль-
тровывается, и фильтрат испытывается уже описанным методом на избыток диазо-,
соотв. соли R. Повторными опытами устанавливается точно объем раствора соли R,
необходимый для связывания диазо в 10 с.и3 раствора смеси солей аминов. Так как
раствор соли R предварительно титруется раствором диазо из анилина, то установ-
ление израсходованного объема раствора соли R позволяет установить, сколько
диазобензола вступило с ним в соединение, следовательно и количество анилина.
1а. Нелюбиной испытан иной метод определения первичного амина в смеси
с вторичным и третичным, основанный на применении работы Цинке.
Первичный амин в смеси аминов реагирует в растворе уксусноэтилового эфира
с хлористым пикрилом:
RNH2 + C1C6H,(NO,)3 RNHC6H.2(NO2\ + НС1,
выделяя эквивалентное количество хлористого водорода. СИ оттитровывается по
Фольгарту 43).
2. Определение вторичного амина. К 1 г смеси оснований быстро
прибавляют 2 г уксусного ангидрида, дают смеси постоять V, часа при комнат-
ной температуре, вливают затем 50 сл<3 воды и нагревают 3/4 часа на водяной бане.
Избыток не вошедшего в реакцию с первичным н вторичным амином уксусного
ангидрида, согласно уравнениям
RNH2 4-(СН3СО)2О = RNHCOCH3 + СНзСОоН, (1)
RNHAlk -г (СН3СО)2О = RN(Alk)COCH3 4- СН3СО2Н (2)
превращенный водой в уксусную кислоту, оттитровывается вместе с уксусной
кислотой, образовавшейся при реакциях (1) и (2), 1 N раствором NaOH (с фенол-
фталеином). При обычной температуре уксусный ангидрид с первичным амином
дает только моноацетилпродукт по реакции (1) и не дает диацетнлзамещенного типа
RN(COCH3)2, какой получается при продолжительном нагревании амина с избытком
ангидрида. Количество вошедшей в реакцию уксусной кислоты, уменьшенное на то.
которое нужно по уравнению (1) для связывания количества анилина, ранее опре-
деленного, дает основания для расчета количества вторичного амина. Третичное осно-
вание определяется по разности между всем количеством амннов и суммой вторич-
ного и третичного.
В тех случаях, когда имеется только двойная смесь аминов,
например только первичный и вторичный или только вторичный
и третичный, можно пользоваться методом Л а з и е р а и Адкинса2-).
Титрованием такой смеси аминов 0,5 N соляной кислотой (с бумажкой Конго
в качестве индикатора) можно рассчитать эквивалент нейтрализации N этой смеси,
т. е. средний молекулярный вес.
Если N— эквивалент нейтрализации, х — число молекул основания с меньшим
молекулярным весом А, а у — число молекул другого основания с молекулярным
весом В, то
д?_ Ах +
х+у
Если Р — процентное содержание одного основания, то 100 — Р — содержание другого
Р 100 — Р
ЯХ = ~А ’ а У^-В-
для Р, получаем
. Подставляя значения х н у в уравнение (а) и решая его
lOOA(fi-TV)
Г N(B-A) ’
Первичный амин обнаруживается в смесн с третичным таким образом, что техни-
ческий продукт растворяют в эфире и прибавляют осторожно разведенную серную
кислоту до тех пор, пока продолжает осаждаться сернокислая соль первичного амииа.
Осадок можно затем отфильтровать и взвесить.
20*
307
Характерные указания относительно того, состоит ли техниче-
ский продукт из диалкилированного лишь амина или заключает
примесь моноалкцлзамещенного, дает проба с уксусным ангидри-
дом. В то время как третичные амины смешиваются с уксусным
ангидридом без повышения температуры и даже показывают харак-
терное понижение температуры на V20 ПРИ смешивании равных
объемов реагентов одной температуры, вторичные и первичные
амины относятся при этом иначе.
Как пример можно привести пробу Гофмана41) на содержание моиометил-
анилииа в техническом диметиланилиие: 4 см3 сухого диметилаиилина смешивают
с равным объемом уксусного ангидрида; предварительно обе жидкости должны
иметь одинаковую (комнатную) температуру. Если термометр, опущенный в смесь,
обнаруживает повышение температуры, то имеется моиометиланнлин, и именно иа
каждый один градус повышения можно считать наличность приблизительно в V2V0
моиометиланилниа.
Нитрозамины, отвечающие вторичным аминам (II), могут быть
превращены обратно во вторичные амины или восстановлением
(оловом с соляной кислотой) или действием концентрированной
соляной кислоты. При пользовании вторым приемом получаются
иногда смеси, из которых трудно изолировать вторичный амин.
Предложен поэтому иной метод устранения NO-группы, именно —
прибавление мочевины или тиомочевины к раствору нитрозамина
в воде серная кислота при температуре 50—100°. Мочевины
берется избыток против молекулярного отношения. Изонитрозо-
соединения с NOH-группой в ядре при этом не изменяются45).
Третичные амины могут быть определены и методом азосоче-
тания, так как с диазониями они легко реагируют, образуя гладко
азокрасители.
Нитрозосоедииеиия, получаемые из третичных аминов, могут оказывать услугу
при определении структуры последних; будучи нагреты со щелочами, иитрозоосио-
ваиия разлагаются иа вторичные чисто жирные амины и на иитрозофенолы (см. гл. П,
нитрозироваиие).
По строению иитрозофенола и диалкиламнна можно составить заключение
о строении самого ннтрозоосноваиия, а следовательно и исходного третичного
амина. Эта реакция может быть использована для получения вторичного жирного
амина, например диметиламииа из иитрозодиметилаиилица.
Разделение смеси аминов разных степеней алкилирования в тех-
нике может быть осуществлено различными способами. В практике
чаще всего имеется необходимость в разделении не всех трех типов,
а только двух, так как проводить реакцию стараются таким образом,
чтобы получался преимущественно желательный продукт, и поэтому
например первичного амина при получении третичного в смеси
остается немного. Поэтому уже простой ректификацией удается
приготовить технический продукт, заключающий практически не
более двух компонент: чаще — третичный амин вместе со вто-
ричным.
Разделение таких смесей практично осуществлять действием
кислотных ангидридов или хлорацгидридов, используя реакцион-
ность незамещенного алкилом водорода аминогруппы по отношению
к кислотным агентам (см. след, главу).
Также можно пользоваться хлорсульфоновой кислотой, обрабаты-
вая ею сухую смесь аминов: первичные и вторичные переходят при
ЗС8
этом в сульфаминовые кислоты, третичный остается без изменений
RNH2 + C1SO3H = RNHSO3H + НС1,
RNHAlk + C1SO3H = RN(Alk)SO3H + HC1.
По прибавлении щелочи, переводящей сульфаминовые кислоты
в соли, третичный амин может быть отогнан с паром, а соли суль-
фамивовых кислот первичного и вторичного амина разложены
добавлением минеральной кислоты и гидролизованы обратно в амины:
RNHSOsH 4- Н2О = RNH2 4- H2SO4.
Для успеха такого разделения в смеси не должно быть больше 15% вторичного
амина46).
Можно для образования сульфамииовых кислот вторичных аминов применять
серный ангидрид в присутствии веществ, связывающих кислоту 47). Фосген, хлораи-
гидрид угольной кислоты СОС1а также пригоден как реагент для разделения вторич-
ных и третичных аминов. Для этого смесь моно- и дналкиларамииов обрабатывают
фосгеном в присутствии воды при температуре ниже 15°. Если третичного амина
недостаточно для связывания выделяющейся кислоты (НС1), то во время обработки
фосгеном прибавляют постепенно щелочи. Хлористоводородная соль третичного
амина растворяется в разведенной соляной кислоте, а соединение, получаемое из
вторичного амина, отфильтровывается. Реакция фосгена с вторичным амином выра-
жается такой схемой:
RN(Alk)H 4- СОС12-> R(Alk): N • COCI -I- НС1.
Полученное соединение или подвергается гидролизу или конденсируется сновой
молекулой вторичного амниа до карбарилнда 48):
R(Alk)NCOCl 4- HN(Alk, R -> ОС [N(Alk)R]2.
Известен метод, применяющий хлорангидрид сульфокислоты,
например п-толуолсульфохлорид.
Обработка смеси аминов эквивалентным количеством сульфо-
хлорида (и раствором едкого натра) переводит анилин и вторичный
амин в сульфонарилиды (I) и (II), из которых только отвечающий
первичному амину (I) продукт растворим в щелочи и может быть
поэтому отделен от нерастворимого сульфонарилида (II). Третичный
амин при этом не вступает в реакцию и отгоняется с паром:
1 C6H5NH(O2)SC6H4CH3 II CeH6N(C2H5)(O2)SC6H4CH3
Имеются предложения разделять смесь вторичного и третичного аминов посредством
фталевого ангидрида, переводящего моиоалкилированиый амии в соответственную
фталаминовую кислоту:
R(Alk)NH 4- С6Н4 (СО)2О -> R(Alk)NCOC6H4COOH.
Обработку фталевым ангидридом смеси аминов можно проводить в бензольном
растворе при размешивании. Разведенной щелочью извлекается соль фталаминовой
кислоты, и по отгоике растворителя получается свободное третичное основание49).
Практичнее работать в отсутствии постороннего растворителя, прибавляя фта-
левый ангидрид при обычной температуре и при размешивании в количестве, отве-
чающем содержанию вторичного амина в смеси. Прибавкой щелочи извлекают соли
фталамивовой (и фталевой) кислот. Третичный амин получается сразу в товарном
виде (заключает максимально 0,1% вторичного амина). Фталаминовая соль гидроли-
зуется по уравнению
R(Alk)NCOC6H4COONa NaOH = R(Alk)NH + CeH4(CO2Na)2
действием 40%-ного едкого иатра при высокой температуре, например 180 — 200°,
в закрытом аппарате (10—15 ат). Отдуванием с паром получают почти колнчест-
309
венный выход вторичного амина. Подкислением концентрированной серной кислотой
и кристализацаей можно регенерировать фталевую кислоту 5Э).
Моио- н диметнланилин разделяют также, пользуясь различной растворимостью
щавелевокислых солей 31}.
Для разделения смеси вторичного и первичного аминов предложена обработка
смеси 50%-ным раствором хлористого цинка, причем осаждается труднорастворимое
соединение, отвечающее составу RNH2ZnCl3 (скорее — RNH2 • НС1 • ZnCl3), вторичный
амин в присутствии воды ие образует соединения с хлористым цинком и может
быть извлечен подходящим растворителем, например петролейным эфиром 32).
Алкилированные амины имеют важное значение для получения
красителей, преимущественно ряда триарилметановых основных
и некоторых кислотных. Введение в амин алкильной группы
усиливает „ауксохромное* действие, сообщая красителю более
„глубокий* оттенок и повышая его основные свойства. Вместе
с этим алкилированные амины находят себе применение в синтезе
хинониминовых красителей, оксазинов, в том числе галлоцианиновых,
тиазинов и некоторых азокрасителей основного характера. Наиболее
важны третичные диалкилированные амины. Бензильный остаток
часто вводится в амин для дальнейшего сульфирования амина или
его производного, так как сульфогруппа легко входит в фениль-
ный радикал бензила. Сульфокислоты бензилированных аминов, на-
пример этилбензиланилннсульфокислота CsH5N(C2H5)CH2C6H4SO3H,
играют заметную роль в синтезе кислотных трифеннлметановых
красителей.
Вторичные амины имеют более ограниченную область примене-
ния в синтезе красителей. Напомним, что диметнланилин, а лучше —
монометиланилин, является исходным материалом для получения
тетранитрометиланилина (тетрила) — взрывчатого вещества для за-
палов C8H2(NO,)SN(NO3)CH3.
Введение алкила практикуется как в применении к незамещен-
ным, первичным аминам (например анилину), так и к их замещен-
ным, например сульфокислотам. Алкилированные в аминогруппе
сульфокислоты аминов нужны для получения алкиламинофенолов
(особенно мета-ряда).
При получении из анилина и его гомологов алкилированных
аминов при участии галоидалкилов или при нагревании амина со
спиртом и соляной кислотой возможно — при определенных усло-
виях^— образование таких производных аминов, где алкильная группа
закрепляется не у азота, а у углерода ядра. Переход алкила в ами-
нах от азота к ядру был подмечен Гофманом и Марциусом
еще в 1871 г.
При нагревании ароматических аминов с метиловым спиртом и соляной кислотой
в автоклаве до 200 — 250° наблюдается постепенное метилирование ядра, причем СН3
становится преимущественно в пара- и орто-места к азоту. Прн нагревании смеси
до более высокой температуры (250 — 300°) становится более заметной мета-ориеи-
тация; так, из о-толуидина (I) получается мезидин (II) и некоторое количество изо-
дуридина (III):
112 llg
/х zNH., ' .NH, Н,С A /NH,
III I II 1 1 III I
НзС-^/^СНз
Попытки полного метилирования ядра(диметилмезидин,соляная кислота, метиловый
спирт) дали ие пентаметиланилин, но пентаметилфенол 33).
310
Переход алкила от азота в ядро, например
-NHCHg
-NH,
легко осуществляется при участии галоидных солей металлов (Со, Zn, Cd, Мп).
Реакцию можно было бы объяснить схемой с допущением предварительного гидро-
лиза галоидной соли металла, например
2C6HjNHAlk —±^gk±H-° 2CeH3NHAlk. НС1 2 Aik • C6H4NH2• HC1.
Но, как оказалось, оиа протекает в отсутствии воды, и следовательно ее можно
рассматривать как изомеризацию алкиланилина 51).
Тем ие менее недавний американский патент описывает получение гомологов
анилина нагреванием бромистоводородных, соотв. иодистоводородных, солей, алки-
лированных в азоте аминометилбензолов до 230—350° 53), например
СН, Н3С СН,
/\/ \/\/
II ->11
NHCHg • HJ NH2
Подобные реакции протекают не однообразно, н ими трудно
управлять, так что всегда можно ожидать в продуктах сложной
смеси различных аминов.
Получение из углеводородов таких амииов, где аминогруппа связана ие
с углеродом ядра, а с углеродом боковой цепи (например CeH5CH2NH2), будет
рассмотрено в соответственном месте (см. конденсации).
б) Алкилирование в оксигруппе
Алкилирование фенольного гидроксила — превра-
щение, имеющее малое значение для получения промежуточных
продуктов,особенно после введения в практику получения алкокси-
производных через хлорозамещенные. Большее применение этот
метод находит в ряду красителей с целью сделать красители менее
чувствительными по отношению к щелочным агентам.
Общее выражение алкилирования гидроксильных замещенных:
ROH-L-AlkX—^ROAlk, (1)
где X—чаще всего атом галоида (Cl, Вг), остаток серной кислоты
или арилсульфокислоты. Для связывания выделяющейся кислоты
чаще всего фенол применяется в виде щелочного соединения —
фенолята. Во многих случаях алкилирования пользуются избытком
щелочи в реакционной среде.
Практически для алкилирования гидроксильных соединений
находят себе применение только метил СН3 —, этил С2Н5— и окси-
этил НОСН2СН2—.
Метилирование чаще всего осуществляется при помощи мети-
ловых эфиров серной кислоты: диметилсульфата (CH3O)2SO2 и
311
метилсернокислой соли, например CH3OSO3Na и метилового эфира
арилсульфокислоты, например и-толуолсульфокислоты.
Для введения этила пользуются хлористым (или бромистым)
этилом С2Н5С1 или этиловым эфиром арилсульфокислоты. Ди-
этилсульфат (C2H5O)2SO2 менее доступен и потому дороже.
Оксиэтильную группу вводят при посредстве хлористого произ-
водного, этиленхлоргидрина НОСН2СН2С1.
Реакция метилирования диметилсульфатом, так же как и
в случае аминов, протекает в две стадии:
RONa + CH3OSO2OCH3 = ROCH3 + CH3OSO2ONa, (2)
RONa CH3OSO2ONa = ROCH3 + Na2SO4. (3)
Первая стадия (2) отличается легкостью и гладкостью взаимо-
действия и обычно заканчивается уже при температуре значительно
ниже 100°, между тем как вторая стадия (3), с участием кислого
эфира серной кислоты, требует большей энергии и обычно прово-
дится при такой температуре, когда нужно пользоваться уже
закрытыми аппаратами.
Недавно американец Льюис с сотрудниками установили, что прн метилиро-
вании фенола вторая стадия проходит довольно полно и при работе с обратным
холодильником, если не вводить большого избытка воды. По их наблюдениям
можно использовать на 90% метиловые группы в диметилсульфате, применяя на
2 мол. фенола 1 мол. диметилсульфата в присутствии 3 мол. едкого натра и 2 мол.
воды и проводя взаимодействие в течение 5 час. при 100° 56).
По исследованию Ходжсона и Никсона метилирование диметилсульфатом
с водной щелочью дает хорошие результаты в применении к крезолам (70—80%-нын
выход), удовлетворительные с фенолом (65%) н нитрофенолами (45—65°/о), и не
идет совсем с хлорнитрофенолами и нитрокрезолами. Такие соединения можно
метилировать в ксилольном растворе, применяя углекислый калий как отнимающее
кислоту средство. Повидимому в некоторых взаимодействиях требуется большая
ионизация фенолята: может быть реакция начинается с присоединения диметил-
сульфата к аниону фенола
/ /СН3 \ —
CgH5O—O3SOCH3
Фрейдеиберг рекомендует при алкилировании фенолов, когда исходные
материалы ценны и нестойки, пользоваться не свободными фенолами, а их ацетиль-
ными соединениями (это особенно важно для таких фенольных соединений, которые
легко могут реагировать в тавтомерной кетоформе; таков например флороглюцин)
НО ОН СН2
YY осАсо
Н2С\/СН,
| со '
он
ибо ацетильное соединение стабилизирует энольную форму. Метилирование лучше
всего проводить, приливая заранее избыток диметилсульфата к ацетильному соеди-
нению фенола и затем постепенно, по мере развития реакции, вводя щелочь57).
Диметилсульфатом пользуются при метилировании нитро-п-кре-
зола (I) в метиловый эфир n-нитрокрезола (II). Последний является
312
промежуточным этапом при получении ценного для синтеза проч-
ных азокрасителей крезидина (III):
он осн3 JX/NO2 Д/NO, I 1 -ч II1 OCHg XIX/NH2 ► jm 1
CHS CHS CH3
По недавнему немецкому патенту также помощью диметилсульфата или мети-
лового эфира арилсульфокислоты в присутствии кетона (например ацетона) и
щелочных средств (сода, щелочи, гидроокиси щелочноземельных металлов в водных
растворах; метилируют амино-₽-оксиантрахиноны, например
Получаемые таким образом продукты частью являются красителями для ацетатного
шелка 58).
Полные эфиры сернистой кислоты, например (CH3O)SO, приготовляемые обра-
боткой спиртов посредством хлористого тионила
SOC12 + 2AlkOH -> [CISOOAlk] -> AlkO • SO • OAlk,
могут быть также применяемы для алкилирования фенолов. Взаимодействие про-
ходит между фенолятом и сернистокислым эфиром в среде индиферентного раство-
рителя по возможности с высокой температурой кипения.
Этот метод представляется значительно менее удобным и общим в сравнении
с применением эфиров серной кислоты59).
Алкилсернокнслые соли, в частности CH3OSO3K, можно применять прн высокой
температуре в открытых аппаратах, заменяя воду глицерином. Фан Эрп получал
алкильные эфиры нитрофенолов, нагревая сухую смесь калиевого нитрофенолята
с калиевой солью алкилсерной кислоты и безводным глицерином (по весу в коли-
честве х/з—от фенолята) до 180— 200°60).
Обработка диметилсульфатом хиноноксимов (изоиитрозофенолов) в присутствии
водной щелочи приводит к образованию метиловых эфиров хиноноксимов, напри-
мер 61)
,-^\noh Z\noch3
Эфиры арилсульфоновых кислот, например СН3 • С6Н4 • SO2OC2H5>
очень удобные агенты для введения в гидроксил алкильной группы-
Их приготовление при наличии доступного сульфохлорида чрез-
вычайно просто, а применение легко и проходит гладко м). Их
употребление позволяет легко вводить в связь с кислородом и
высшие алкилы с более чем двумя углеродными атомами (С л о та,
Франке).
Прн помощи эфиров арилсульфоновых кислот с тяжелыми алкилами (например
бутил-, бензил-) можно ввести алкилы при сравнительно низкой температуре не
только в группу ОН, но и в ядро соединения, например из динитробензола и бен-
зилового эфира толуолсульфокислоты образуется бензилдиннтробензол 63).
Реакции алкилирования фенольных соединений посредством
эфиров арилсульфоновых кислот отвечает общее уравнение
RONa 4- R,SO2OAlk = ROAlk RjSOoONa. (4)
313
Нагревания на водяной бане с обратным холодильником смеси из
соответственного арилсульфонового (чаще п-толилсульфонового)
эфира и фенолята достаточно для того, чтобы перевести фенол
в алкильное производное.
При введении более тяжелых алкилов (начиная с С3) оказалось практичным
работать в алкогольной среде с едким кали (в колвчествах, немногим меньше экви-
молекулярного отношения). При таких условиях например из пирокатехина полу-
чается сначала моноэфир с небольшой примесью диалкильного эфира.
Освобождающаяся в виде соли по уравнению (4) арилсульфокислота — ценный
отход и должна быть регенерирована (через сульфохлорид) в эфир или найти
какой-либо другой путь применения для того, чтобы метод был рентабельным.
Этильная группа вводится в гидроксил, особенно в готовые
красители, с целью устранения реакционности гидроксила в отно-
шении щелочей и с целью сделать окраски прочными к стирке.
Этилирование осуществляется обработкой таких гидроксильных
соединений хлористым этилом. Обработка ввиду летучести этого
реагента происходит в закрытых аппаратах в присутствии щелочи
и обычно в спиртовой среде. Как на пример такой реакции можно
указать на превращение яркого желтого (I) в хризофенин (II):
(НО—/ N : N —^>—СН : )
~ I —
По сравнению с другими этилирующими агентами хлористый
этил имеет преимущество дешевизны и, хотя и требует для избе-
жания его потери соответствующей специальной аппаратуры,
широко применяется.
В последнее время в производстве промежуточных продуктов и красителей
наблюдается интерес к введеиню оксиэтильиого остатка в оксигруппу, причем это
введение обеспечивает возможность реакционности гидроксила в этом алифатическом
радикале в направлении перевода его в эфирное соединение с серной кислотой.
Получаемые кислые эфиросоли (11) обладают большей растворимостью сравнительно
С исходными продуктами (I). Схема получения таких продуктов такова:
ROH -J- С1СН,СН90Н ROC.H.OH ROC,H4OSO3K.
’ ' 1 'II
Пример такого окснэтилирования (этанолирования):
ОН ОСН2СН9ОН
A/n0> А/А
I | 4-С1СН,СН.,ОН-> | I
\/ “ \/
I I
сн3 сн3
Реакция протекает при иагреваиии с обратным холодильником смеси эквимоле-
кулярных количеств реагентов; в качестве щелочного средства применяется едкий
натр в водном растворе (также в эквивалентном хлоргидрину количестве).
Реакция может быть воспроизведена и с гидроксильными соединениями нафта-
лина, антрацена61).
Подобно окснэтильному остатку могут быть введены помощью соответственного
хлористого производного иные производные этнла, например диэтиламиноэтил
314
I I
^/'СНоС.Н-
(C2H3j2N • C2H4 — , Метод применяется для производства некоторых фармацевти-
ческих препаратов65).
Необходимо отметить, что обработка фенолятов галоидными алкилами приводит
к алкоксильным соединениям лишь в тех случаях, если реакционной средой служит
вода, спирты (метиловый или этиловый) или ацетон. В среде, не содействующих
диссоциации растворителей (бензол, толуол), н при применении галоидал-
килов с особенно реакционным галоидом (аллил СН2 'СН—СН,—, циинамил
СвН3СН=СН—СН,—, беизил С6Н5СН2—, беизгидрил (С6Н5)2СН—) получаются не
алкильные эфиры фенолов, а фенолы, замещенные в орто-месте к гидроксилу алкиль-
ной группой, например66)
/^yONa /^/ONa
I I + С6Н5СН2С1
в толуоле
при кипении
Для некоторых гидроксильных групп в нафталиновом и антра-
ценовом рядах и для некоторых полиоксибензольных производных
оказывается возможным иной способ алкилирования: не щелочной,
но кислотный. Нагревание таких гидроксильных соединений со
спиртом и минеральной кислотой приводит к образованию алкокси-
замещенных. Значит, в этом случае ароматически связанный ги-
дроксил Подобен по реакционности гидроксилу спиртов, в присут-
ствии кислот образующих со спиртом же простые эфиры. Соеди-
нения, способные к такому алкилированию, обычно характери-
зуются склонностью реагировать в двух формах, отвечающих ке-
тоновой и энольной структуре (как нафтолы). Поэтому можно ска-
зать, что такой метод алкилирования приложим к гидроксилам не
чисто ароматически связанным, но энольным.
В некоторых особенных случаях алкилирования применяются в качестве алки-
лирующего средства четвертичные трналкилариламмонии. Реакция протекает по
схеме
ROH 4- Аг - N = (AIk)3 ROAlk -|- Ar — N : (А1 к)2 -|- Н.2О6").
ОН
Реакция имеет отношение по преимуществу к оксигруппе, не требует приме-
нения повышенного давления и совсем не затрагивает вторичных и третичных
аминогрупп. Но реактив дорог, и поэтому применение его имеет очень ограничен-
ную область.
Алкилоксисоединения менее растворимы, чем исходные гидрок-
сильные (фенольные) производные и потому могут быть от них
отделены и отличены. Если алкилирование применяют по отноше-
нию к окрашенным или красящим соединениям, то цвет при этом,
как выражаются, „повышается", т. е. становится более близким к
желтому. Химическая активность в некоторых случаях, например
у эфиров фенола, значительно понижена: эфиры фенола не сочетаются
с диазониями, нелегко бромируются, не нитрозируются. В других
случаях при алкилировании активность сохраняется, если даже не
усиливается. Так, в нафтольных эфирах, в эфирах резорцина,
флороглюцина не наблюдается заметного ослабления активности
в вышеназванных замещениях. Продукты алкилирования в гидро-
ксиле часто можно узнать по внешнему виду, запаху, нераство-
римости в щелочах (последнее при отсутствии в соединении
карбоксильных или сульфоновых групп) и т. п.
315
Если потребовалось бы определить алкнлоксигруппу и она принадлежала бы
к числу „активных* по вышесказанному, то определение не представляло бы
никаких трудностей прн употреблении уже известных нам реакций.
Вообще же алкилоксигруппу определяют по методу Ц е й з е л я 88). Метод со-
стоит в переводе метила или этила алкокснльной группы кипячением с иоднсто-
водородной кислотой в иодистый алкил и определении иода в иодистом серебре,
полученном взаимодействием образовавшегося летучего йодистого алкила и спир-
тового раствора азотнокислого серебра. По количеству йодистого серебра рассчиты-
вают количество алкокснльных групп в соединении.
Редко в препаративных интересах, чаще в интересах аналити-
ческих или с целью определения структуры, бывает желательно
заменить имеющуюся алкоксильную группу обратно гидроксилом,
т. е. перейти от фенольных эфиров к свободным фенолам. Этот
процесс обезалкилирования производится действием
иодистоводородной (соотв. бромистоводородной) кислоты, причем
сущность реакции заключается в замене водородом алкильной
группы у фенола, алкил же соединяется с галоидом:
ROCHS 4 HBr = ROH + СН3Вг.
При этом желательно создать прибавлением индиферентных
для данного процесса растворителей среду, в которой бы наилучше
реагировали составные части смеси, т. е. среду гомогенную69).
Весьма активным веществом для обезалкилирования фенольных
эфиров является также хлористый алюминий70).
Наконец имеются предложения применять для данной цели галоидоводородвые
солн ароматических оснований71). В частности для обезметилирования фенольных
эфиров рекомендуется нагревать 1 мол. эфира с 2—3 мол. хлористоводородного
анилина (илн его гомолога) до сплавления (180—230е). Реакция заканчивается
в 1/з~1 час. Горячий сплав выливают в крепкую соляную кислоту. Течение реак-
ции очень сложно.
Описан случай устранения метильной группы из эфирной связи с фенильным
радикалом нагреванием в течение нескольких минут с хлористым тиовилом72).
Освобождение группы: ROAlk-> ROH для частного случая этилового или метило-
вого эфира я-аминофенола или его N-алкилпроизводного, по описанию советской
заявки немецкой фирмы, производится нагреванием с 70—75%-ной серной кислотой
до 160° с обратным холодильником. Прн этом алкил отщепляется в виде спирта.
Любопытно, что прн этом не наблюдается (судя по описанию) сульфирования и что
связь N—Aik (например N— СН2.СН2ОН) не нарушается 7з).
В некоторых алкильных эфирах, например в нитранизолах, обезалкилирования
удается достигнуть нагреванием эфира с пиридином н пиперидином и небольшим
количеством воды 71).
При изучении обезалкнлирующего влияния хлористого водорода (1Б°/0-ная
соляная кислота, 130°) на алкильные эфиры фенолов констатировано, что мета-
замещенные фенольные эфиры реагируют всего медленнее, пара-замешенные—всего
скорее, орто-замещенные лежат посредине. Кислотные группы Вг, Cl, NO2, также
СН3, но не СОСН3, повышают стойкость эфира. Этиловые эфиры разлагаются
медленнее метиловых. Увеличение отрицательных групп в частице повышает стой-
кость эфира, аминогруппа ослабляет. Метиловый эфир а-нафтола менее стоек, чем
его тетрагндропроизводное.
Алкилирование соединений,содержащих сульфгидрильную
группу (SH), имеет сравнительно ограниченный круг приложений
в технике. Водород сульфгидрила очень подвижен и легко заме-
щается на алкил теми же приемами, что и водород гидроксила.
Наиболее часто встречается необходимость введения остатка
— СН2СООН в связь с серой с целью получения производных
арилтиогликолевой кислоты RSCH2COOH, употребительных проме-
316
жуточных продуктов в синтезе индигоидных красителей. Для этого
тиофенольное производное обрабатывается при нагревании в вод-
ном растворе щелочи монохлоруксусной кислотой, например75)
/\/SH /^/SCHoCOOH
| I -;-С1СН2СООН^ I |
V^cooh \/\:ooh
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
i) W. Stadei, Г. п. 21241 (F. I, 21).
2) К п о 11 & Co, Г. п. 250236 (F. XI, 156).
3) Dow, Am. n. 1794057, 1793993, CB 1931, I, 3288.
i) Ch. B. Chatfield, Am. n. 1429714, CB 1924, II, 1271.
5) P. F. Frankland, F. Challenger, N. A. Nicols, Soc. 115, 198 (1919).
в) A. M a 1 h 1 e. Ф. n. 23891, CB 1922, IV, 760; M a 11 h e, de G о d о п, C. r. 166,
467, 564 (1918), Bull. (4) 29, 106 (1921), C.r. 172, 1417(1921), Bull. (4) 30, 1035(1921);
Rhein, Kampfer Fa, Ф. n. 681049,CB 1930, II, 1132, Б. n. 319205,CB 1930, 1,736.
') Cp. Hi ns berg, Ann. 265, 178 (1891); Fr. Ullmann, Ann. 327, 110 (1903).
Опыты H. M. К и ж и e p, К. А. С л а в н н о н (не опубликовано).
8) М. К. Б е ззу б ец, С. А. Лившиц (не опубликовано).
8) 1G-, Г. п. 494803, СВ 1930, II, 466.
1») Ср. В., Г. п. 169358 (F. VIII, 389).
U) Wohl, Blank, Г. п. 167698 (F. VIII, 388).
12) В., Г. п. 177491 (F. VIII, 390).
И) Ср. М. L.B., Г. п. 255304 (F. XI, 171).
1‘) W. Т. Mac L eester, Ам. п. 1844926, A. W. М. D i с k 1 п s, Ам. п. 1844844, СВ
1932, I, 2895.
I5) Green, Saunders В. D. Со, Б. п. 181750, СВ 1923, II, 191; Green,
Saunders, R. G. M. С. 28, 81 (1923); 1G., Г. п. 487788, СВ 1930, I, 1537.
16) В., Г. п. 163043 (F. VIII, 391).
17) Например IG., Г. п. 500439, СВ 1930, II, 1142.
18) Fr. Ullman п, Ann. 327, 104 (Ullmann, Wenner) 129 (1903). Z. Fold!,
CB 1923, III, 519, Lower Manfg. Co, Am. n. 1550749 (1927).
19) K- F r i с к e г, Г. n. 449047 (F. XV, 208).
-ч) В. M. Родионов, В. E. Введенский, Ж. X. Пр. 7, II (1930). К. Н-
Slotta, W. Franke, Darstellung und Verwendung hoherer Ester der p-tohiolsul-
fonsaure, Ber. 63, 678 (1930). Там же — библиография вопроса. Ср. Zoltan Fold!,
СВ 1923 III 519.
2') Sc. D., Б. n. 319805, CB 1930,1,596; A. Lap worth, Am. n. 1431470, CB 1924,
II, 1276.
22) W. Lazier, H. Adkins, Am. Soc. 46, 741 (1924), cp. Ind. Eng. Chem., New
Ed. 1931, 129 (The Mattin Laboratories).
23) L. J. S i m о n, M. Frere ja cqu e, C. r. 178, 944, CB 1924, I, 2267.
21) См. T. Sandmey er (G), Г. n. 75854 (F. Ill, 22), Morgan, B. D. Co, Шв. n.
91563, CB 1922, IV, 837; E. M e г с к, Г. n. 437975 (F. XV, 208); J. D. Riedel, Г. n.
406533 (F. XIV, 429); Gru паи, Ф. n. 667551, CB 1930, I, 436.
й) В, Г. n. 157346, 157617 (F. VIII, 396, 398).
26) Г.п. 157909, 157910 (Bucherer) (F. VII, 777, 779), 156760, 181723 (B)
(F. VIII, 400, 393).
27) Cp. MLB, Am. n. 1426348, CB 1923, IV, 592, Б. n. 164002, CB 1921, IV, 708,
IG, Г. n. 476643, 476663, добавление к Г. п. 421505 (MLB) (F. XV, 1470).
28) Ср. О.,Г. п. 75854 (F. Ill, 22), Г. п. 157710, 157840 (Bucherer) (F. VII,779
780). Cp. Bucherer, G г о 1 6 e, Ber. 39,986 (1906); Knudsen, Г. n. 143197 (F. VII,
24); Morgan B. D. Co 1. c.; G. L о c k e r m а п п, Г. n. 491856, CB 1930, II, 982,
503113, CB 1930, II, 3850; Goldschmidt, Ch. Ztg 28, 1229.
29) tM, Г. n. 376013 (F. XIV, 398).
3°)L. Zechmeister, J. Truka, Ber. 63, 2883 (1930).
«) J. D. R1 e d e 1, AG, Г. n. 423132 (F. XV, 206).
32) Wallach, Ann. 343, 54 (1905); CB 1906, I, 355; R. Leuckart, Ber. 18,
317
2342 (1885); Leuckart, Bach, Ber. 19, 2128 (1886)By, Г. n. 287802, 291222 (F.X11,-
800 — 802).
зз) IQ, Ф. n. 669824, CB 1930,1,1697. Аналогично: Co Alai s, Froge, Camargue,
Б. n. 317079, CB 1930,1, 1536; A. Guyot, M. Fournier; C. r. 189, 927 (1929),
Bull. (4), 47, 203 (1930), CB 1930, 1, 674, 2730.
З4) A. Mackert, Б. n. 275377, Br. A. B. 1926, 772.
35) A. Q. Green, Б. n. 293617, CB 1929, I, 1615.
Зв) B. D. Co (W. W. T a t u m), Б. n. 268891, 270779, CB 1930,1, 145; H. Dreyfus,
Ф. n. 667415, CB 1930, I, 596; Brit. Celanese Ltd. Б. n. 316989, CB 1929, II, 2829.
Ср. В., Г. n. 265515 (F. XI, 551).
s’) К., Г. n. 530398, CB 1931, II, 3400.
38) Frankland, Challenger, Nicols cm. 5).
39) Sc hi It, Шв. n. 117162, 1927, II, 866.
40) J. v. Braun, K. Weissbach, Ber. 63, 488 (1930); J. Braun, Ber. 35, 1279
(1902). Cp. Staedel, Ber. 19, 1947 (1886).
4i) E. Merck, Г.п. 432151 (F. XV, 200\
42) Нелюбина, Ан. Kp. Пр. 4, № 2, 120 (1934).
43) Fr. R е v е г d i и, d е la Harpe, Ber. 22, 1004 (1889). Нелюбина, Ан.
Кр. Пр. 3, № 8, 355 (1933).
41) A. W. Hofmann, Ber. 10, 591 (1887).
W. G. Macmillan, T. H. Reade, Soc. 1929, 585.
4B) B. D. Co, R о d d, E v e r a 11, Б. n. 270930, Br. А. В. 1927, 622.
«) lG.,r. n. 513679, Ch. T. Ub. 1931, № 73/73, 175.
4S) B. D. Co, Everatt, Rodd, Б. n. 273923, Br. A. B. 1927, 648.
«) Silesia, Б. n. 280877, CB 1929, I, 1046.
“) IG. Г. n. 523603, 526881, CB 1931, II, 3545, 3546.
51) L. Desvergnes, Chim. & Ind. 24, 785 (1930), CB 1931, I, 1274.
32) W. J. Hickinbottom, Soc. 1930, 992 CB 1930, 11, 1218.
И) D. H. H e y, CB 1931, II, 1696.
З4) W. J. H i c k i n b 0 11 о m, Soc. 1927, 64, CB 1927, I, 1950, H i c k i и b ot t 0 m,
A. C. W a i n e, G. H. P r e s t о n, CB 1930, II, 2638. Ср. A. M. Hofmann, Mar-
ti u s, Ber. 4, 742 (1871), A. W. Hofmann, Ber. 5, 704, 720 (1872), Howard,
Derick, Am. Soc. 46, 166 (1924).
33) Nat. An. (Minnis), Am. n. 1844518, CB 1932. II, 122.
зв) H. F. Lewis, S. Shaffers, W. T r i e s c h m a п n, H. С 0 g a n, Ind. Eng.
Chem. 22, 34 (1930).
5;) H. H. Hodgson, J. Nixon, CB 1930, II, 3139; Haworth, Lap worth,
CB 1924, I, 1660.
K. Freudenberg, Ann. 433, 230 (1923), CB 1924, I, 2190.
IG., Г. n. 549285, CB 1932, II, 447.
59) W. Voss, E. Blanke, Ann. 485, 258, CB 1931, I, 2603.
ю) Van Erp, Ber. 56, 217 (1923).
«i) S. Veibel, M. H. Sim esen, Ber. 63, 2478 (1930).
82) См. В. А. Измаильский, Б. A. P а з о p e н о в, Ж. 52, 359 (1920); см. (18), ('-°).
83) Z. Fol di, Б. п. 319273. СВ 1930, I, 744.
«4) 1G., Г. п. 443340, СВ 1928, I, 3113.
85) Н. Н a h 1 (Winthrop Chem. Со), Ам. п. 1725136.
88) L. Ciaisen, Zt. angew. Ch. 36, 478 (1923), Г. п. 412169 (F. XV, 267).
67) BOhringer & S о h п е, Г. п. 247180 (F. X, 1215). В. М. Родионов и
Каневская, Ж. Фарм. Ин-та 1921, 46, А. Е. Пора й-К о ш и ц, Ю. А. А у ш к а п,
Ж. 43, 673 (1911).
«8) Zeisel, Mon. 6, 986 (1885), 7, 406(1886); ср. Ki гр al, В u h 11, Ber. 47, 108',
(1914), Mon. 36, 853, (1915).
в9) Cp. D. M. Birosei, Am. Soc. 52, 1944 (1930', CB 1930, II, 722.
™) Ср. E. Merck, Г. n. 444587 (F. XV, 1521).
71) A. KI emen c, Ber. 49, 1371 (1916), CB 1916, II, 174.
и) H. Q. Rule, E. Brets ch er, Soc. 1927, 925, CB 1927, II, 566.
73) IG., Сов. заяв. свид. 30009.
74) R. S. Cahn, CB 1931, II, 842.
7Ь) К., Г. n. 192075 (F. VIII, 1371), 181658 (F. VIII, 477\ Заяв. Г. п. К. 34994
(F. IX, 542). Г.п. 240118 F. X, 510). Ср. Р. F г i е d 1 а п d е г, N. Woro.shtzow,
Ann. 288, 1 (1912).
318
ГЛАВА Xll
АЦИЛИРОВАНИЕ
(защита реакционной группы)
Ацилированием мы будем называть в дальнейшем введение
ацила (остатка кислород заключающей минеральной кислоты,
карбоновой или сульфокислоты, получаемого отнятием ОН от ча-
стицы кислоты) в характерную группу NH2 или ОН соединения
вместо водородного атома. Таким образом ацилами могут быть:
для серной кислоты (HOSOoOH) остатки: SO2OH (сульфонил) и S02
(сульфон); для угольной кислоты (НОСООН) — остатки СООН (кар-
боксил) и СО(карбонил); для муравьиной кислоты (НСООН)—остаток
НСО (формил); для уксусной кислоты (СН3СООН) — остаток СН3СО
(ацетил); для бензойной кислоты (С8Н5СООН) — остаток С6Н5СО
(бензоил) и т. п.
Ацилирование имеет назначением изменить химический харак-
тер заместителя и тем самым сделать возможным такие превра-
щения несущего заместитель соединения, которые при свободной
ной амино- или оксигруппе недостижимы или удаются лишь с
большими потерями. Так как группы NH2 и ОН принадлежат
к активнейшим заместителям, в сильнейшей степени влияющим
на органическое ядро, с которым они соединены, и так как
ацилирование уменьшает в заметной мере активность этих групп,
делая ароматическое ядро более устойчивым против многих воз-
действий (например окисления), то принято говорить, что аци-
лирование „защищает" аминогруппу, соотв. оксигруппу. В тех
случаях, когда представляется необходимым выявить превраще-
нием ароматический характер ядра и когда есть основание ожи-
дать, что заместитель (NH2 или ОН) может повлечь добавочные
изменения при превращении, прибегают к ацилированию или дру-
гим средствам химической защиты замещающей группы.
а) Ацилирование аминов
В предыдущей главе мы видели уже несколько примеров целесо-
образности ацилирования аминогруппы с целью вовлечь в реакцию
алкилирования лишь один атом водорода у азота и получить только
вторичные амины.
Выше, в главе о нитровании, мы видели примеры необходимости
такой защиты. Азотная кислота, нитрующая углеводородное ядро
соединения, может при наличии свободной аминогруппы в значи-
тельной мере содействовать его окислению. Чтобы избежать обра-
зования побочных продуктов, всегда понижающих выход и каче-
ство главных, мы стабилизуем аминогруппу (ацилированием, пре-
319
вращением в азометиновые соединения или прибавлением избытка
серной кислоты). Так же может обстоять дело при реакциях оки-
сления аминосоединений с боковой алифатической цепью, при хло-
рировании и пр. Реакция ацилирования аминогруппы схематически
выражается уравнением
RNH2-4-X- Ас-----> RNHAc,
-нх
где Х=ОН, С1, ОАс и т. п.
В высшей степени вероятно, что реакция ацилирования начинается с присоеди-
нения молекулы ацилирующего агента к молекуле амина (у азота) и протекает по
такой схеме:
RNH, + ХАс—> RNH.Z----------> RNHAc.
хАс -нх
Ацилирующими агентами могут быть, во-первых, кислоты в со-
стоянии более или менее высокой концентрации, во-вторых, анги-
дриды кислот, в-третьих, хлорангидриды, включая сюда и сульфо-
хлориды. О применении для ацилирования аминогруппы остатка
серной кислоты (сульфогруппы) мы упоминали в главе об алкили-
ровании и диазотировании. И там и тут сульфаминовые кислоты
общей формулы RNHSO3H оказываются очень полезными как поз-
воляющие вовлечь аминосоединение в односторонне протекающую
реакцию.
Лучший способ получения сульфаминовых кислот из аминов — это воздействие
ангидро-Хт-сульфокислоты пиридиния (см. стр. 82) C3H3N • SO3, или заранее
образованной или образующейся в процессе реакции. В последнем случае целесо-
образно брать амии в растворе или суспенсин в пиридине (также хинолине, иногда
можно брать и третичный жирио-ароматический амин, например диметиланилин)
н воздействовать на смесь хлорсульфоиовой кислотой (или олеумом). Образующаяся
сульфаминовая кислота обработкой водным раствором соды (или другим щелочным
агентом) переводится в соль, которая может быть, если надо, выделена выпарива-
нием или, чаще, может быть применена непосредственно в растворе.
Ход реакций при ацилировании в таких случаях следующий:
RNH2 4- C5H3N + C1SO3H —► [RNH2 4- C3H3NSO3] —> RNHSO3H • C3H3N,
(пиридиновая соль
сульфаминовой кислоты)
2RNHSO3H • C3H6N + Na2CO3 —> 2RNHSO3Na + CO, + 2C3H5N + H2O.
Следует учитывать свойство ароматических сульфаминовых
кислот, отвечающих первичным аминам, легко гидролитически рас-
падаться на амин и серную кислоту под влиянием уже слабых
растворов кислот
RNHSOsH + Н2О = RNH, + H2SO4,
между тем как по отношению к щелочам они в высшей степени
стойки и могут вообще выдерживать даже сплавление со щело-
чами без нарушения связи между азотом и серой.
Можно пользоваться также хлорсульфоновой кислотой без третичного осно-
вания, вводя ее постепенно во взаимодействие с тройным против требуемого коли-
чеством ароматического амина в растворе в хлороформе или ином инертном рас-
творителе при охлаждении. Тогда избыток амина принимает на себя функцию ней-
трализации выделяющегося хлористого водорода, какую роль в первом способе
играет третичное основание (пиридин н т. п.). Реакция во втором случае выра-
жается уравнением (для анилина):
3C6H5NH2 4- CISO3H = CeH3NHSO3H • CeH5NH2 4- C6H3NH2 • HC1.
320
Для выделения соли феиилсульфаминовой кислоты здесь нужно освободиться
не только от хлористых солей, но и от избыточного амина, что во всяком случае
является более трудной задачей, чем устранение пиридина, берущегося в избытке
по первому способу 1).
Применение фторсульфоновой кислоты FSO3H для получения ароматических
сульфаминовых кислот не имеет никаких преимуществ перед пользованием хлор-
сульфоновой кислотой 2).
Образование сульфаминовых кислот становится все более ши-
роко применяемым в практике приемом, который дает во многих
случаях возможность совместить свойства исходного аминосоеди-
нения в новом производном с повышенной растворимостью в воде.
Таким образом возможно получить например продукты типа окси-
сульфаминовых кислот, действуя хлорсульфоновой кислотой в при-
сутствии пиридина (или других подобных органических оснований)
на аминонафтолы, аминоарилпиразолоны, аминоарилиды ацето-
уксусной кислоты или аминоарилиды 2.3-оксинафтойной кислоты.
Во всех этих случаях наблюдается преимущественная реакцион-
ность у аминогруппы сравнительно с имеющейся в соединении
оксигруппой. Также приобретают интерес сульфаминовые кислоты
антрахинонового ряда. Арилсульфаминовые кислоты 3) интересны
еще и потому, что их характерная —NHSOgH-группа обладает
достаточной способностью к активированию ароматического ядра
в реакции азосочетания, подобно тому как это делает амино-
сульфарильная группа — NHSO.2R (см. главу IX, стр. 262). Так
как сульфаминовые соли в то же время, как указано, растворимы
в воде, что свойственно далеко не всем сочетающим аминопроиз-
водным, то они интересны и как азосоставляющие. Имея в виду,
что сульфогруппа легко может быть отщеплена от азота амино-
группы, например нагреванием с разведенной серной или соляной
кислотой, и что сама реакция сочетания протекает гладко в напра-
влении пара-места, без побочных реакций, такое применение суль-
фаминовых кислот в водно-содовом растворе может оказать боль-
шую пользу например при получении промежуточных соединений
в синтезе вторичных дисазокрасителей. Как пример приведем 4)
,N.,C1 / NHSO3Na
/\/ - I 3
гидролиз
(MCI)
SO3Na
^SO3Na
—NHSO3Na
21 Зак. 2611. — II. Ворожцов
321
Сульфаминовые кислоты в свободном состоянии невыделимы из растворов
вследствие гидролиза. Оии вероятно образуются в качестве промежуточной стадии
при сульфировании аминов методом „запекавия“.
Ацилирование серной кислотой с полным использованием последней до сульф-
арилида типа RNHSO2NHR не имеет практического значения.
Построенный по такой схеме сульфанилид получен из анилина, взятого в эфир-
ном растворе, действием иа него хлористого сульфурила, также растворенного
в эфире. Без растворителя хлористый сульфурил действует на анилин преимуще-
ственно хлорирующе, здесь же реакция протекает по схеме:
2C6H5NH2 + SO2C12 = (C6H5NH)2SO2 + 2НС1.
Сульфанилид — соединение прочное, не разлагающееся заметно ни от щелочей
ни от кислот, со щелочами дает соли, например C12H10N2SO2Na2. В содовом растворе
сочетает с диазоииями, образуя красители, обладающие сродством к бумажному
волокну.
В настоящее время из неорганических кислот имеет значение
для ацилирования еще угольная кислота. Передатчиком ацила
здесь служит полный хлорангидрид, фосген, хлорокись углерода,
которая легко выменивает свои атомы хлора на остатки амино-
групп, сцепляя посредством группы СО два остатка амина
2RNH2 + СОС12 = RNH • СО • NHR -ф- 2НС1.
Часто обработка с фосгеном имеет целью закрепить в соеди-
нении СО-группу надолго. Реакция, как видно из уравнения, про-
ходит с выделением хлористого водорода, поэтому для успеха ее
необходим или избыток ацилируемого амина, если он достаточно
основен, или введение щелочного агента, например соды или едкого
натра (например при ацилировании аминонафтолов). Этот способ
ацилирования имеет значение для получения особенно интересных
в нафталиновом ряду замещенных мочевины. Их представителем
является производное известной аминонафтолсульфокислоты И (алая
кислота):
HO3SX — СО — HNX
I I
OH OH
Такое же ацилирование помощью фосгена практикуется и по
отношению к готовым азокрасителям, имеющим аминогруппу, для
связывания двух частиц азокрасителя помощью СО-группы. Такие
„фосгенированные" азокрасители проявляют большую устойчивость
в окрасках по отношению к окислительным воздействиям и влия-
нию солнечного света. Обычно, имея дело с азокрасителями в виде
сульфокислот, пользуются водными содово-щелочными растворами
при обработке фосгеном.
В недавнем немецком патенте для получения производных мочевины из амино-
азобензола указывается метод с взмучиванием мелкоистертого аминоазобеизола
в сухом нитробензоле и внесение взвеси в раствор фосгена в нитробензоле же в).
В предыдущей главе мы встретились с возможностью приме-
нения обработки фосгеном для разделения вторичных и третич-
ных аминов.
322
Фосген — сильно ядовитый газ, и работа с ним требует соблю-
дения мер предосторожности с целью избежать проникновения
его в рабочее помещение.
Аналогичные карбарилидам, замещенным мочевины, производные
тиоугольной кислоты, следовательно замещенные тиомочевины,
тиокарбарилиды общей формулы
RNHC( : S)NHR
имеют практическую важность как в производстве красителей, так
и особенно в промышленности ускорителей вулканизации резины.
Для получения наиболее простого и часто применяемого пред-
ставителя этого класса соединений дифенилтиомочевины, тиокарб-
анилида (C6H5NH)2CS, пользуются взаимодействием сероугле-
рода CS2 (сернистого аналога угольного ангидрида) на анилин,
причем реакция протекает с выделением сероводорода
CS2 + 2H2NCgH3 = H2S -j- SC(HNC6H5)2.
Дифенилтиомочевина служит исходным продуктом в синтезе
индиго по Зандмейеру и в синтезе изатина.
Применение ее и ее замещенных (в фенильных остатках) для
получения ускорителей вулканизации резины основано прежде
всего на превращении ее, действием аммиака с добавкой отнимаю-
щих сероводород окислов (РЬО), в арилзамещенные гуанидина
(C6H5NH)2CS 4- NH3 = (C6H5NH),C : NH 4- H2S,
I II "
а эти последние, например дифенилгуанидин (II), уже служат уско-
рителями вулканизации.
При получении дифеиилгуанидииа (II) из дифенилтиомочевины (I) взаимодей-
ствие, весьма вероятно, происходит с предварительным отщеплением из замещен-
ной тиомочевины сероводорода и образованием промежуточно очень реакционного
карбодифенилдиимида (III), который уже, присоединяя аммиак, образует дифенил-
гуанидин 6)
-г хн, c6h6nh4
(C6H5NH)2CS----> >С------------> >С : NH
I H2s C6H5N/ CgHjNH/
III II
Воздействие сероуглерода и серы на анилин при высокой тем-
пературе приводит к образованию более сложного продукта с до-
бавочным кольцом тиазола
именно меркаптобензотиазола (IV) или так называемого каптакса,
очень ценящегося как ускоритель вулканизации 7). Его получение
протекает по общей схеме
znh24-cs24-s /\/n^
I 1 ----> I iv 1 c • sh 4- H2S
\Z
21*
323
Работа с сероуглеродом требует соблюдения мер безопасности
ввиду, с одной стороны, вредного влияния вдыхания его паров на
здоровье работающих, с другой — чрезвычайной опасности его
в пожарном отношении. Будучи крайне летуч (t°KHn 46°) и в то же
время горюч, сероуглерод с воздухом легко образует взрывчатые
смеси.
Из ацилов, отвечающих одноосновным кислотам жирного ряда,
наиболее употребительны остатки муравьиной кислоты
(формил) и уксусной кислоты (ацетил).
Муравьиная кислота, приготовляемая теперь в больших количествах синтети-
чески в виде своей натриевой соли из едкого натра и окиси углерода, имеет пре-
имущества меньшего молекулярного веса и следовательно меньших расходных
коэфипиеитов при одинаковом действии. В большинстве ацилирований формильный
остаток удерживается в связи с атомом азота менее прочно, чем ацетил, н это
обстоятельство надо учитывать при последующих обработках формилированных
аминов (например при нитроваини и т. п.).
Формилирование свободных аминов без кислотных групп осу-
ществляется нагреванием с обратным холодильником амина с избыт-
ком муравьиной кислоты по уравнению
RNH2 4- НСООН = RNHCOH 4- Н2О.
Если для формилирования берется минеральная соль амина, то в реакционную
смесь вводится более чем необходимое количество муравьиноиатриевои солн, коли-
чество же свободной муравьиной кислоты может быть взято меньшее, чем требует
молекулярное отношение, так как от действия минеральной кислоты, связанной
с амином, будет освобождаться соответственно свободная муравьиная кислота из
ее соли 8), например
/NHaHCl
| |Z + NaOoCH--->
HOZ'Z
zx/NHa ZZ/NHCOH
---> | j + HCO2H + NaCl------> ||
hozZ/Z---------------------------hoZZ/Z
При формилированни сульфокислот аминов практично также прибавлять в реак-
ционную смесь муравьинонатриевую соль для нейтрализации свободных кислотных
групп, иногда для лучшего выделения готового продукта.
В последнее время имеется много предложений проводить формилирование,
обходясь без употребления готовой муравьиной кислоты, а используя ее амид
(формамнд) HCONH2, который может быть получен из окисн углерода н аммиака
по немецкому патенту К. Мейера, причем таким же способом в присутствии
воды получается аммонийномуравьииая соль, формиат аммония HCC^NH^ также
предложенная в качестве формилирующего агента 9).
Формамид при действии на амины выделяет аммиак:
RNH2 + HCONH2 —> RNHCOH -f- NHS.
Ацилирование при посредстве формамида удается провести полно и совершение
при условии быстрого удаления аммиака из сферы реакции. Поэтому взаимодей-
ствие производится при сильно пониженном давлении (10 — 30 мм) при сравни-
тельно низкой температуре (70°) в присутствии катализирующе действующих
веществ (вода, алкоголяты. муравьиная кислота). Можно работать без вакуума, про-
пуская через реакционную смесь иидиферентный газ (азот), и тогда применять
более высокую температуру (100 — 110°) 10).
324
По сравнению с этим методом работы кажется мало практичным способ, опи-
санный в американском патенте, применяющий не формамид, но его сульфат
HCONH2 • H2SO4. Реакция проходит здесь также при невысокой температуре (85°):
RNH2 + HCONHa • HaSO4 -> RNHCOH + NH4HSO4.
По патенту, возможно применение способа н к иным ацилированиям,
при использовании сульфатов иных ациламидов, например CH3CONH2 • H2SO4,
CeH5CONH2• H2SO4. Недостатком метода является связывание серной кислотой
аммиака, следовательно, с одной стороны, затруднительность регенерирования
аммиака и с другой — большие осложнения при извлечении органического продукта
из смеси п).
Введение формильного остатка в амины, особенно в аиилни, можно, по опи-
санию патента, производить действием окиси углерода при высокой температуре
(200°) и давлении (200 ат) на амины в присутствии мураньиной кислоты илн ее
производных нли таких веществ, которые могут превратиться в условиях взаимо-
действия с окисью углерода в производные муравьиной кислоты (например едкая
щелочь, щелочь и метиловый спирт и т. п.) 12).
Вышеупомянутый формиат аммония при нагревании с аромати-
ческими аминами (анилином и гомологами) до 100 — 200° реаги-
рует, отщепляя аммиак и давая сначала муравьинокислые соли
аминов, которые при более высокой температуре переходят в фор-
мильные производные с отщеплением воды:
RNH2+HCO.2NH4 -> RNH2-HCO2H + NHs RNHCOH % Н2О.
Ввиду большей простоты получения формиата аммония срав-
нительно с муравьиной кислотой —этот способ заслуживает вни-
мания 13).
Для введения ацетила СН8СО в ароматические амины чаще
всего пользуются уксусной кислотой. Уксусная кислота в технике
может оказаться в различных концентрациях: 100%-ная (ледяная)
уксусная кислота, 80%-ная и ниже до 50—30%. Наиболее легко
и удобно реакцию ацетилирования проводить с 100%-ной кислотой,
так как в таких условиях имеет меньшее значение обратное тече-
ние реакции с распадением ацилпродукта на амин и кислоту:
RNH2 + HOCOCHS = RNHCOCH3 % ДО.
Процессу ацетилирования обычно предшествует образование солн из амина
и уксусной кислоты. Выделение воды нз образовавшегося ацетата дает в резуль-
тате ацильный продукт. Этот ступенчатый ход процесса позволяет пользоваться
при нем более слабой, чем 100%-иая, уксусной кислотой. В самом деле, кипяче-
нием 80%-ной кислоты и амина можно получить ацетат, частью находящийся
в растворе, дальнейшим нагреванием можно удалить избыток воды, в виде
слабой кислоты, и затем сильным нагреванием уже сухого ацетата отщепить
и конституционную воду, получив ацильный продукт. Имеются предложения при-
менять более слабую (например 50%-ную) кислоту, пользуясь для реакции работой
при повышенном давлении 14).
Избыток ацетилирующего агента (уксусная кислота) необходим для того, чтобы
проацетилнровать весь амин. По Н. А. Меншуткину, при соотношении: 1 мол.
анилина -j- 2 мол. уксусной кислоты достигается предел взаимодействия в 96,88%
прореагировавшего амина; 1 мол. анилина-)-4 мол. уксусной кислоты — предел
в 99,8% 15).
Прибавка ииднферентныхрастворителей, например кснлола, при ацетилировании
уксусной кислотой не может быть признана целесообразной. Увеличение выхода
при этом практически незаметно, а разделение растворителя от кислотного слои
требует добавочной аппаратуры и труда 16).
При ацетилировании сульфокислот амвнов полезно применять введение в реак-
ционную смесь вместе с уксусной кислотой соли уксусной кислоты, например
ацетата натрия. При этом, как выяснилось из опытов автора и А. И. Титова
325
над ацетилированием 1-нафтиламин-6-сульфокислоты (кислота Клеве), это выгодно
потому, что сульфокислота взаимодействует, подобно как было отмечено выше
при формилированин, с уксусионатриевой солью, освобождая уксусную ки-
слоту, которая и производит по преимуществу ацетилирующий эффект, например
C10H6(NH2)SO3H 4- CII3CO2Na = C10HB(NH2)SO3Na + СН3СО,Н,
C10H6(NH2)SO3Na 4- CHSCO2H = C10H6(NHCOCH3)SOsNa -j- H3O,
и следовательно реакция идет в значительной мере не за счет свободной уксусной
кислоты, а за счет ацетата. Вместе с тем при этом реакция протекает скорее,
она может проходить достаточно полно даже при применении 80—90%-ной кислоты.
Полноте реакции содействует устранение из реакционной сферы соли ацетил-
продукта вследствие высаливающего эффекта ацетата натрия.
Ацетилирование уксусной кислотой в больших размерах лучше
всего производить в алюминиевой аппаратуре.
Уксусный ангидрид (СН3СО)2О — энергичный ацетилирующий
агент; действию его на амины отвечает уравнение
RNH2 4- (СН3СО)2О = RNHCOCH3 4- СН3СООН.
В заграничной практике, где стоимость 100%-ной уксусной кислоты и ее
ангидрида мало разнятся одна от другой, уксусный ангидрид применяется для ацети-
лирования довольно широко. Сульфокислоты аминов, соотв. аминофенолов, для этого
переводятся с содой в водный раствор, и раствор, подогретый до 50—70°, быстро
обрабатывается уксусным ангидридом, взятым в некотором избытке против требуемого
теорией количества (30—50%). Быстрое прибавление уксусного ангидрида является
существенным для успеха ацетилирования, так как наряду с прямым использованием
ангидрида на соединение с аминогруппой в водном растворе, особенно достаточно
щелочном, имеет место и гидролитический распад ангидрида, бесполезный для
ацетилирования:
(СН3СО)2О 4- Н,О = 2СН3СООН.
Поэтому важно сохранение высокой концентрации ангидрида при уменьшенной
вследствие ацетилирования концентрации амииосоедннения и при возможно малом
количестве водного раствора.
Такое видоизменение ацетилирования может проводиться и в деревянных чанах
и в эмалированной посуде 17).
В предыдущей главе мы видели пример ацетилирования не-
сульфированных аминов при действии уксусного ангидрида на
холоду.
Обработка уксусным ангидридом арилсульфамидов (при нагревании, иногда
в присутствии серной кислоты) приводит к замещению аминогруппы амида аце-
тилом, например
л-СНдСбН48О,КН2 4- (СН3СО)2О = «-CHsCjjH^SOjNHCOCHg -f- СН3СООН.
Полученные N-ацетиларилсульфамиды служат прибавками к ацетилцеллюлозе 18).
Хлористый ацетил СН3СОС1, хлорангидрид уксусной кислоты,
ввиду своей относительной дороговизны не находит применения
в обычных для техники ацетилированиях аминов. По энергии
своего действия он похож на уксусный ангидрид.
В случае, если такие энергичные переносчики ацила, как хлористый ацетил
или уксусный ангидрид, взяты в большем избытке, а температура поднята значи-
тельно выше требуемой, не исключается возможность получения диацетильного
продукта 19):
RNHCOCH34- С1СОСН3-> RN(COCH3)2 4- НС1.
Упомянем, что как новый ацетилирующий агент для амииосоедииений пред-
ложен кетен СН9 : СО, реагирующий преимущественно с аминогруппой и позво-
326
яяющий поэтому проводить частичное ацетилирование аминофенолов. Реакция
с газообразным кетеном протекает например таким образом:
п-НО • С6Н4 • NH2 + СН2 : СО В СПИРТ1:,ВОМ > Л.НО • С6Н4 •NH•СО• СН3 «).
растворе
В настоящее время кетен нельзя рассматривать как реагент, так же доступный,
как вышеупомянутые агенты ацилирования.
Своеобразным ацилирующим агентом оказывается щавелевая
кислота (СООН)2. Ввиду того что эта кислота — двухосновная,
она может давать два ряда ацилированных продуктов: 1) с одной
замещенной и одной свободной карбоксильной группой, так назы-
ваемые замещенные оксаминовые кислоты, и 2) продукты
с замещением в обоих карбоксилах, так называемые оксарилиды.
Примерами таких соединений являются СПНГ) • NH• СО • СООН—фенил-
оксаминовая кислота и С6Н5 • NH • СО • СО • HN • С6Н5 — оксанилид.
Особенно интересны н наиболее легко получаются соединения первого рода.
Получаются оин при взаимодействии между несколько большим, чем эквивалентное,
количеством щавелевой кислоты н амином при нагревании. Щавелевая кислота
•может быть безводной, может быть кристаллической (4-2 молекулы Н.2О) и даже
в некоторых случаях в водном растворе. Нагревание требуется вести до темпера-
туры не очень высокой: максимум 130—140°, при этом реакция становится заметной
по обильному выделению воды. Интересны же оксаминовые кислоты, благодаря
тому обстоятельству, что присутствие свободной карбоксильной группы делает их
легкорастворимыми, особенно в виде щелочных солей, и поэтому удобными для
многочисленных применений, где требуется растворимость в воде.
При обработке щавелевой кислотой диаминов ацилиоование
особенно легко происходит в одной из двух аминогрупп. Так, при
взаимодействии уже водного раствора щавелевой кислоты на
ж-фенилендиамин образуется ж-аминофенилоксаминовая кислота
H2Nx /у /NH2-r НООС • СООН H2N~X zNH • СО • СООН
+ Н2О
Также нагревание диаминоаитрахиноиов с щавелевой кислотой и водой до
105—110° приводит к образованию лишь монооксаминовой кислоты (для 1.5-, 1.4-,
1.8-диаминоантрахннонов, днаминоантраруфина и т. п.) 21).
Оксарилиды получаются при более высокой температуре взаимо-
действия амина с щавелевой кислотой, при сплавлении ингредиен-
тов 22):
2RNH2 4- (НООС)2 = RNH СО•СО• HNR + 2Н2О.
Для получения оксарилилов проще, во избежание побочных реакций, пользо-
ваться хлорангидридом щавелевой кислоты С1ОС-СОС1, хлористым оксалилом
2RNH2 4- (СЮС)2 = (RNHCO)2 4- 2НС1.
1.1'-диметил-2.2'-оксалиламинобензол (I) при конденсации в при-
сутствии алкоголята натрия дает я.я'-дииндил (II), из которого
легко образуется индиго23)
327
Вероятно получение оксарилидов с помощью хлористого оксалила проходит
через стадию образования хлораигидрида оксамииовой кислоты
RNH2 + С1СО • СОС1 RNH • СО • СО • Cl RNHCO • COHNR,
ио на этой стадии ие задерживается. Если же пользоваться аминами не в виде
свободных оснований, а в виде солянокислых солей, реакция с энергичным отде-
лением хлористого водорода происходит уже при обычной температуре или при
охлаждении и гладко дает хлораигидрид арилоксаминовой кислоты. Избыток хло-
ристого оксалила может быть различен. Иидифереитиые растворители вводятся
в смесь, если в этом представляется необходимость 21).
Бензоил С6Н6СО и некоторые его замещенные (хлор-, нитро-)
имеют наибольшее значение среди ацилов ароматического ряда..
Введение бензоила в ароматические соединения чаще всего произ-
водится действием хлористого бензоила СбН5СОС1.
Введение бензоила в аминогруппу преследует обычно не пре-
ходящую цель использования вызываемой ацилированием стабили-
зации аминогруппы, как это имеет место в отношении ацетилиро-
вания и формилирования, но цель сохранения бензоила или его
замещенного в готовом продукте. Таково например бензоили-
рование аминонафтолсульфокислот. Введение нитробензоильного-
остатка в 2-нафтиламин-5-окси-7-сульфокислоту (кислоту И) со-
действует образованию новой азосоставляющей со способной диазо-
тироваться аминогруппой:
HOgS^/^/^/NHCO—/ N02
ОН он
HOgS^/^/. NHOC-^ V-NH.,
328
HO3SX/X/X/NHOC- /
H5CcN : N/X
OH
Также обрабатывая дихлор хлористым бензоилом 1-амино-8-на-
фтол-4.6-дисульфокислоту (кислоту К), образуют азосоставляющую,
особенно пригодную для синтеза лаковых азокрасителей.
Введение лг-нитро-п-метилбензоильного остатка в 1-нафтил-
амин-4.6.8-трисульфокислоту дает начало синтезу ценного меди-
камента против сонной болезни — г е р м а н и н а
СО
NH
СО—NH SO3Na
I I
X.ZZZZSOsNa
I d
NaOsS
NaOsS
A/\/S0“Na
I I
CO—NH SOsNa
в структуре которого участвуют и л/-аминобензоильные остатки "’)-
Наиболее серьезное значение в химии красителей бензоилиро-
вание имеет по отношению к аминоантрахиноновым производным,
где введение бензоила в аминогруппу часто обусловливает пере-
ход от промежуточного продукта к кубовому красителю. 1.5-ди-
бензоилдиаминоантрахинон (I) — желтый кубовый краситель; 1-мето-
кси-4-бензоиламиноантрахинон (II) — алый кубовый; 1.5-диокси-
4.8-дибензоилдиаминоантрахинон (III) — фиолетовый кубовый кра-
ОСН3
СО I
/\/\/\
II
\/\/\/
СО |
НЫОССсН3
hnoccsh5
СО |
/\/\/\
I I
| со
H-CgCONH
329
h5c6conh он
I co I
z\/\/\
III
I CO I
OH hnoccsh5
Таким образом вхождение бензоильной группы имеет известное
значение и для проявления цвета и для красильных свойств этих
красителей.
При бензоилировании хлористым бензоилом в волной среде, например сульфо-
кислот амиионафтолов, для связывания образующейся соляной кислоты (могущей
повести к гидролизу бензоиламииосоедииеиия) практично прибавлять соду ' или,
лучше, ацетат натрия 26).
При образовании бензоиламинопроизводных антрахинона работают в безводной
среде, например с высококипящими растворителями (хлорбензол, нитробензол);
газообразный хлористый водород удаляется при этом из сферы реакции сам.
Выгодно применять избыток ацилирующего средства (3—5 мол. С6Н5СОС1 на
каждую NHg-rpynny) и проводить взаимодействие при 100°, а ие выше 27).
Вместо применения готового хлористого бензоила можно также пользоваться
для ацилирования исходным для него продуктом, бензотрихлоридом, который при
нагревании с бензойной кислотой в высококипящем растворителе (нитробензол,
нитрохлорбензолы, дихлорбеизолы), лучше н присутствии подходящего активатора
(ZnCL), образует хлористый бензонл или бензойный ангидрид, например
С6НБС13 + С6Н5СООН = 2С6Н3СОС1 + HCI,
СбН5С13+ 2С6Н3СООН = QHjCOCI + (С6Н3СО)2О + 2НС1,
С6Н3С13 + ЗС6НБСООН = 2(С6НБСО)2О + 3HCI.
Такой раствор может быть непосредственно применен для бензоилирования й).
Бензойный ангидрид (СеН3СО)2О, примененный в надлежащем количестве
в среде растворителя (нитробензол, полихлориды бензола), дает возможность гото-
вить трудно синтезируемые иным путем моиобензоильиые производные диамиио-
антрахиионов 29), например
NHa NHCOC6H5
СО | СО I
<YY> YAY
1111+ (CGH5CO)2O -> I I I I + C6H3COOH
\/\/\/
I CO I co
H,N H2N
Подобно бензоильному остатку надолго вводится в амины
сложный ацил, отвечающий ацетоуксусной кислоте СН3СОСН2СООН,
который следует назвать ацетоацетилом СН3СОСН.?СО—. Полученные
с ним арилиды общей формулы CH3COCH2COHNR имеют спе-
циальное применение в качестве азосоставляющих, где активное
участие в сочетании принимает как-раз остаток ацила. Полученные
азопродукты (хорошие красители для лаков) отвечают по строе-
нию формуле:
CH.QO-CH-CO-NHR СН3 • С(ОН)=С—СО—NHR
| , СООТВ. I
NsrNR' N~NR'
330
Примером таких красителей является ганза желтый 5G из анилида ацетоуксусной
кислоты и диазо из о-нитроаиилииа з°). Введение ацииа в аминогруппу здесь
происходит в результате взаимодействия ацетоуксусного эфира CHgCOCHjCOOCaHs,
соотв. СН3С(ОН): CHCOOCjHj, с ароматическим амином. Взаимодействие лучше
идет при нагревании в среде иидифереитиого растворителя (ксилол, хлорбензол) sl),
например
СН3СОСН2СООС2Н5 +
СН3СОСН2СО • HN.
+ С2Н5ОН
Совершенно так же готовятся N-ацетоацетилпроизводные из
аминоазопроизводных или диаминов, — в последнем случае двузаме-
щенные 32), например
/\/NHCOCH2COCH3
CH3COCH2COHn/\/
Также надолго вводится в амины остаток карбоновой кислоты
f-нафтола, чаще всего 2.3-оксинафтойной кислоты (I) (в послед-
нее время предложены и другие оксикарбоновые кислоты не только
нафталинового ряда). Образующиеся в результате ацилирования
арилиды оксинафтойной кислоты служат очень важными промежу-
точными продуктами в качестве азосоставляющих для образования
с диазосоединениями непосредственно на волокне так называемых
холодных, или ледяных окрасок. Представителем таких соединений
является так называемый нафтол AS, анилид (3-оксинафтойной
кислоты (II)
ОН
СООН
Применение вместо анилина других аминов, например ^-нитро-
анилина, n-анизидина, я-нафтиламина, р-нафтиламина и т. п., при-
водит к образованию других „нафтолов" этого типа (соотв.: наф-
толов ASBS, ASRL, ASBO, ASWS), имеющих каждый значение
в практике ситцепечатания для получения ярких и прочных окра-
сок, соотв. узоров. Техническая номенклатура часто не считается
с химическим строением, и находящие себе одинаковое применение
с только-что названными вышеупомянутые производные ацето-
331
уксусной кислоты также называются „нафтолами типа AS“, на-
пример
CH3COCH2COHN— / ^-NHOCCH,COCHS—
/ \ —нафтол ASG.
Н3С СН3
Для получения нафтолов типа AS ^-оксинафтойная кислота и
амин в подходящем растворителе (толуол, хлорбензол) обрабаты-
ваются треххлористым фосфором для отнятия воды. Реакция при
повышенной температуре проходит обычно гладко33).
Своеобразная ацильная группа циануровой кислоты (I) в последнее время не
раз была испытана в качестве заместителя водорода в аминогруппе в азо- н дназо-
составляющих азокрасителей. Наиболее употребительным методом введения циану-
рового остатка в молекулу служит реакция между хлористым цнануром (II) и
имеющими аминогруппу промежуточными продуктами (производными амннонафтола,
феннлендиамнна н т. п.). Как результат такой реакции может быть получен либо
продукт, где циануровая группа только одним из своих углеродов связана с служащим
для образования азокрасителя соединением (например III), либо продукт с исполь-
зованием двух углеродных атомов цианурового ядра (например IV), либо продукт,
где три атома углерода циануровой кислоты сцеплены через атомы азота с арома-
тическими аминозамещеннымн (например V):
СОН
I 1
нос!^ 1'СОН
N
СС1
к/А
11
cicL. Jlcci
N
СС1
HO3S/Z\/'X/Z\SO3H HOaS/X/X/ SO:1H
Ацилирование производится в водном растворе или суспенсни с участием сла-
бощелочных агентов.
332
Такие циан/ровыс производные применяются для синтеза преимущественно
суб.тантивных азокрасителей, особенно ценных по оттенку и прочности 84).
По отношенвю к аминоантрахнноновым производным подобный метод ацилирования
хлористым циануром также оказался интересным. Среди полученных продуктов
имеются ценные по свойствам кубовые красители. Реакция производится в растворе
или суспенсии нитробензола при повышенной температуре вплоть до температуры
кипения нитробензола. Химическая природа продуктов такого взаимодействии
в смысле сохранности в них цианурового остатка не установлена а"’).
О применении ароматических сульфохлоридов к ацилиро-
ванию аминов было неоднократно уже упомянуто.
Так как наиболее употребительные сульфохлориды обладают
невысокой температурой плавления, то при наличии жидкого или
легкоплавкого амина можно работать при сравнительно низкой
температуре в гомогенной среде, — в этом большое преимущество
этого способа ацилирования. Иногда взаимодействие происходит
в присутствии воды. Ацилирование помощью сульфохлорида со-
провождается отделением хлористого водорода:
RNH2 Ц- C1SO2R' = RNHO2SR' + НС1.
Для ее связывания очень практично, по Витту, применять
сплавленный уксуснокислый натрий. Соляная кислота выделяет
из него свободную уксусную кислоту, и эта последняя участвует
в смеси в качестве растворителя, поддерживающего однородность
смеси.
Обычно ацилирование помощью сульфохлорида производится на водяной бане,
причем берут небольшой избыток амина (1—5%) против теоретически требуемого
количества и за реакцией следят по исчезновению характерного запаха сульфо-
хлорида. Достигнув этого момента, отгоняют с паром амин и выделяют нелетучий
сульфонарнлнд в кристаллическом состоянии. Нелетучие амины могут быть отде-
лены переводом в растворимые солн, например соли соляной кислоты. В. А. Соло-
нина указал, что при взбалтывании первичных аминов с арилсульфохлорндами
(в большом избытке) и с едким натром (в малом избытке) наряду с нормальными
моноарнлсульфонарилндами образуются в малых количествах диацнлпродукты м).
Арилсульфонарилиды типа RNHO2SR' легко сочетают, как мы
видели выше, с диазониями, образуя n-азокрасители. Уже на этом
примере мы видим, что природа ацила далеко не безразлична для
характера продуктов ацилирования.
В действительности наблюдается большая избирательность в по-
следующих реакциях ацилированных аминов.
Л а у е р недавно установил, что чем выше константа диссоциации кислоты,
ацил которой связан с атомом азота аминогруппы, тем более пара-орнентацня
преобладает (над о- н м-) при нитровании соответственного аннлида н тем более
превалирует ориентация 1.4 (перед 1.2) прн нитровании а-аминоантрахнноиа
л ориентация 2.1 (предпочтительно перед 2.3) при нитровании {3 - амнноантрахн-
нона эт).
При ацилировании первичных ароматических аминов исчезнове-
ние диазореакции последних предоставляет удобный метод контроля
конца реакции.
Большинство ацилированных аминов получается не для долго-
временного существования, но для преходящего использования их
в реакциях замещения с тем, чтобы далее освободить аминогруппу
от защищающего его ацила. Такой гидролиз, или так называемое
„омыление11, ацилированных аминов производится или действием
333
щелочей или действием кислот. Кипячение с едким натром (в не-
большом избытке против количества, требуемого для нейтрали-
зации ацила в соль) освобождает аминогруппу (например при
получении n-нитроанилина). В других случаях (пример — арилсуль-
фоновые дериваты) полезно прибегать к омылякпцему действию
серной кислоты и смесь разводить немного водой. Развившаяся
при этом теплота часто бывает достаточна, чтобы вызвать распа-
дение ацильного соединения3»), Иногда требуется добавочное
нагревание. Соляная кислота может оказать также существенную
помощь при гидролизе ациламинов. Нагревание с соляной кислотой
(после отделения ацила в виде свободной кислоты) влечет образо-
вание солянокислой соли свободного амина. В продукте гидролиза
может быть определена природа ацила, если это нужно, по соли
соответствующей ему кислоты.
б) Иные, кроме ацилирования, методы защиты аминогруппы.
Образование азометиновых соединений
Защитить аминогруппу возможно и иными способами кроме
ацилирования. Особенно часто практикуется способ, основанный
на применении альдегидов, использующий образование так назы-
ваемых Шиффовых оснований, или азометиновых соедине-
ний *, в которых атом азота соединен двойной связью с углеродом
метиновой группы:
RNH2 + О : С • Rj --, > RN : CHRr
н
При этом из алифатических альдегидов наиболее употребителен
формальдегид СН2О, из ароматических — бензальдегид СсН5СОН.
Формальдегид с первичными ароматическими аминами дает так
называемые ангидроформальдегидарамины типа RN~CH2, соеди-
нения, которые легко полимеризуются (вероятно в тример). Их
значение особенно видно из легкости их перехода через п-амино-
бензиланилин (и его гомологи) в диаминодиарилметаны (при
нагревании с первичными аминами) и на легкости получения при
этом переходе трифенилметановых красителей (см. главу XV).
Ангидроформальдегидамины получаются при размешивании пер-
вичного амина с водным раствором формальдегида (продажным
формалином). Последнего берут в небольшом избытке против
теории. Продукт взаимодействия (полимер) получается в виде
нерастворимого осадка, который можно отделить фильтрованием.
Бензальдегидные соединения (бензилиденарамины) получаются
обычно также без участия каких-либо реактивирующих или отни-
мающих воду средств. Если амин применен в виде соли минераль-
ной кислоты, то необходимо прибавление нейтрализующего агента,
например соды, уксуснокислого натрия и пр. Иногда взаимо-
действие побуждается слабым нагреванием. Образовавшееся азо-
метиновое соединение отделяют от жидкости фильтрованием.
* С характерной группой — С—N— азометиновой, аналогичной —N—N— азо-
группе.
334
Реакция образования таких соединений весьма чувствительна,
так что может служить как для изолирования первичных аминов
посредством альдегидов, так и для выделения альдегидов. Азоме-
тиновые соединения могут быть, после доведения до конца тех.
превращений, ради которых производилась защита аминогруппы,
разложены на свои составляющие —амин и бензальдегид — простым
кипячением с разведенной кислотой. Бензальдегид отгоняется при
этом с водяным паром.
Азометиновые соединения являются производными аминов
с хорошо защищенной аминогруппой. Они ориентируют (подобно
ациламинам) при замещениях в пара и орто (преимущественно
в пара) и не дают реакции азосочетания с диазониями.
Азометиновые соединения в практике синтеза промежуточных
продуктов интересны, с одной стороны, как этап в получении
амина, в частности вторичного амина, чему примеры мы уже
видели в предшествовавшей главе. В двух следующих мы встре-
тимся еще с азометиновыми производными как стадиями на пути
к синтезу альдегидов.
Шиффовы основания из алифатических альдегидов или их
производных имеют большое значение для резиновой промышлен-
ности, служа частью ускорителями вулканизации, частью типич-
ными „противостарителями", т. е. веществами, задерживающими
преждевременный износ (от окисления, действия света, тепла)
резиновых изделий. Из таких веществ назовем продукт взаимо-
действия альдоля [СН3СН(ОН)СН2 • СНО] с я-нафтиламином (аль-
нафт).
в) Ацилирование гидроксильных замещенных
Почти все ацилирующие средства, находящие себе применение
в технике ацилирования аминов, употребляются и для ввода
ацила в гидроксильную группу. Общее выражение процесса
в этом случае таково:
ROH ХАс---------> ROAc.
—нх
В зависимости от избранного ацилирующего средства отходящее
при реакции вещество (НХ) может быть водою, хлористым водо-
родом и т. п., причем очень важно для успеха реакции использо-
вание мер к побуждению его отщепления прибавлением в реакцион-
ную смесь соответствующих реагентов. В тех случаях, когда как
передатчики ацилов применяются органические кислоты (НХ = HgO),
для связывания воды употребляют такие агенты, как РС13, РОС13.
В иных случаях вводят ангидрид соответственной кислоты или
обезвоженную щелочную соль этой же кислоты (Na-ацетат). Тем-
пература реакции колеблется в пределах от немногим превышаю-
щей комнатную до температуры кипения реакционной смеси. При
употреблении в качестве ацилирующего средства хл оран гидридов
(и сульфохлоридов) прибегают к нейтрализующим минеральную
кислоту, следовательно более или менее щелочным, агентам для
ускорения и завершения реакции39).
335
Таким агентом может быть раствор едкого иатра, находящий себе применение
ири а шлировании гидроксильных соединений (бензоилом, сульфонарнлом, карбо-
нилом). особенно в тех случаях, когда фенольное производное нерастворимо
в органических растворителях (сульфокислоты фенолов, нафтолов и пр.). Едкий
натр берется в количестве, эквивалентном фенолу. Поташ К>СО3 или сода Na2CO3,
оба в сухом виде, удобны для применения и при органических растворителях.
Фенол в бензольном растворе в присутствии избытка поташа дает с хлористым
бензоилом бензойный эфир фенола40;:
С8Н5ОН + С1ОСС6Н5 = С6Н5ООСС6Н5 + НС1.
В водном растворе в присутствии щелочи хлорангилрнды кислот различно
относятся к ацилированию. Наилучшие выходы ацилированного продукта дает хло-
ристый бензоил, ниже — бензолсульфохлорид и еще ниже — хлористый ацетил.
Уксусный ангидрид дает лучшие выходы, чем хлористый ацетил. Понижение тем-
пературы благоприятствует выходу. Фенольный гидроксил менее активен, чем
спиртовый. Применение КОН вместо NaOH содействует повышению выхода4!).
Ацетилирование посредством уксусного ангидрида фенольных
гидроксилов можно производить в водно-спиртовом растворе при
употреблении небольшого избытка щелочи и охлаждении вбрасы-
ванием льда. Холодной водой уксусный ангидрид гидролизуется
в весьма малой степени. Таким образом удается в фенолах и поли-
оксизамещенных бензола добиться полного замещения всех гидро-
ксилов ацетоксигруппами4’2).
Исследование ацетилирования ^-нафтола уксусным ангидридом в растворе
уксусной кислоты с различной концентрацией водородных ионов обнаружило, что
скорость ацилирования зависит от кислотности или основности среды. Максимальная
скорость РН<НАс> = —4,5, минимальная — приблизительно при pH (НА£)= 2,0 .
При значениях между 2,0 и 5,0 скорость вновь возрастает, следовательно про-
цесс катализируется как кислотами, так и щелочами 4:1).
Влияние заместителей в феноле иа реакционность гидроксила при ацилирова-
нии несомненно. Испытание галоидозамещениых фенолов в реакции с бромистым
ацетилом в среде уксусиоэтилового эфира определило такой ряд по реакционности:
С6Н5ОН, л-С1С6Н4ОН, п-Вг • С6Н4 • ОН, л«-С1 • С6Н4.ОН, .w-J • С6Н4 • ОН,
ж-Вг.СвН4.ОН, (2)Вг(4)СН3.С6Н3. ОН, о-С1 • С6Н4 • ОН, о-Вг • С6Н4 • ОН,
(2.4.6)-С13.С6Н2.ОН«).
Характер введенного в гидроксил ацильного остатка имеет большое значение
не только для крепости ацилированного продукта, ио и для последующих замещений.
Так, в феноле крепче удерживается остаток карбэтоксн COOC^Hg (вводимый по-
средством С1СООС2Н5), чем ацетил. При воздействии х.юрсульфоиовой кислоты иа
карбэтоксифеиол
/XzOCOOC2H5
ч
можно получить моиосульфэхлорид (л), между тем как из феннлацетата — только
дисульфохлориды i3).
При ацилировании фенолов посредством хлораигидридов ароматических и али-
фатических кислот очень полезной оказывается прибавка весьма небольших коли-
честв каталитически действующих неорганических хлористых соединений, как то:
олова (SnCl4), титана (TiCl4), свинца (РЬС14) или слабее действующих хлоридов
As, Sb, Р. Хлораигидрид применяется при этом в некотором избытке против моле-
кулярного отношения. Реакция протекает полно уже без внешнего нагревания. Для
больших количеств рекомендуется разбавлять смесь иидифереитиыми раствори-
телями: CS2, СНС13, СС14 46).
Удобным средством для отнятия минеральной кислоты является пиридин, кото-
рый имеет значение и как растворитель47). Можно растворить фенол в 5 — 10-
кратном количестве пиридина и прибавить при охлаждении постепенно хлораигид-
рид кислоты. При этом часто оседает солянокислый пиридин. Через 6 — 8 час
336
реакционную массу понемногу выливают в холодную разведенную серную кислоту,
причем продукт аапмгровиния выпадает в виде масла или в твердом состоянии.
Применение пиридина позволяет часто приготовить продукты частичного аци-
лирования полиоксипроизяодиых. Пиридиновый метод позволяв» ацилировать фенолы
не только в нейтральной, но и и кислой среде, например бекзоилировать хлористым
бензоилом в растиоре ледяной уксусной кислоты.
Пиридин действует не только как нейтрализующее минеральную кислоту
средство, ио и как посредник при передаче ацила, образуя реакционные продукты-
присоединения, например
/И
QHsN-b СЮСС6Н5+ C5H6N<
ХОСС6Н5
которые разлагаются прн взаимодействии с фенолом:
/С1 /С1
СбНзЫ/ + HOR -> ОДЖ + ROOCCeH3
Ч)СС6Н6 н
Так как ацилированные фенолы, в отличие от незамещенных,
не реагируют с диазониями, то в реакции азосочетаиия можно
найти средство различения продукта ацилирования от исходного
материала.
Методику ацилирования фенолов посредством арилсульфохлоридов описымет
Георгеску: взбалтывание на холоду слабощелочного раствора фенола с посте-
пенно прибавляемым сульфохлоридом при сохранении щелочной реакции. Он реко-
мендует избегать алкогольных растворов, так как спирт в присутствии щелочи
реагирует с сульфохлоридом легче, чем фенол 48).
Ориентация ацилированных фенолов при замещениях остается
орто-пара.
Ацалирования одной из реакционных труппу оксиамииов (NH2
либо ОН) достигают частичным исключением одной из замещающих групп.
Реакция с альдегидами парализует NH^ переводя амии в азометииовое соеди-
нение, при этом ОН-группа может быть свободно ацилирована (бензоилироваиа).
Только NH2 можно ацилировать посредством полнсульфидов кислот которые
не затрагивают ОН-группы:
(С6Н5СО)2 S2 + 2RNH, = 2CeH3CONHR + H2S -f- S.
Чтобы получить ацилированной одну лишь ОН-группу, можно также ацилиро-
вать сначала обе: и ОН и NH3 и затем действием РС13 и спирта отпять ацил от
азота *9).
Ацильная группа (от органического ацила) крепче удерживается кислородом,
чем азотом.
Введение неорганического ацила, и в частности остатка серной
кислоты, в гидроксильные соединения имеет малое техническое
значение в применении к самим фенолам.
Известно, что фенольные эфиры (кислые) серной кислоты в виде солей (ф*М-
сернокислые соли) получаются после взаимодействия хлорсульфоновой кислоты
с фенолом в нейтральном растворителе (CS2) в присутствии третичного амииа
(отнимающего соляную кислоту); смесь делают затем щелочной (например приба-
влением соды):
ROH + CISO3H ROSOgH + HCI,
2ROSO3H + Na,CO3 2ROSO3Na + H2O 4- CO2.
Вместо хлорсульфоиовой кислоты можно применять пиросульфат KgS2O7 с третич-
ным амином.
Порядок введения ингредиентов такой: к смеси хлорсульфоиовой кислоты и
диметиланилина в. сероуглероде, подогретой до 35°, прибавляют фенол или его
замещенное; раствор затем подщелачивают и арилсупьфат изолируют из водиого
22 Зак. 2811. — Н. Ворожцов 337
слоя. Соли сернокислых эфиров витрофенолов могут быть восстановлены осторожно
в аминосоединении без потери сернокислотной группы (например FeSO4 в суспен-
сии CaCOg или BaCOs)M).
Из недавних работ по исследованию фенольных эфиров сернистой кислоты,
получаемых действием хлористого тионила на феноляты, отметим работу Баттге5!).
В последнее время синтез кислых эфиров серной кислоты прак-
тикуется в отношении не имеющих фенольного характера лейко-
соединений индигоидных красителей, также кубовых красителей
антрахинонового ряда. Получаемые соли этих эфиров, называемые
индигозолями, представляют большой интерес как закреплен-
ная форма энолей лейкосоединений этих красителей.
Способ получения таких индигозолей практически не отличается
от известных уже нам приемов: обработка лейкосоединений кра-
сителя (II), по возможности в отсутствии влажности, хлорсуль-
фоновой кислотой в среде пиридина (или диметиланилина или ани-
лина), если надо — с добавкой растворителя (например хлорбен-
зола), и перевод полученного кислого эфира в соль прибавлением
достаточного количества щелочи. Органическое основание, нужное
для связывания хлористого водорода в первой стадии взаимо-
действия, отделяется от подщелоченной массы отгонкой с паром.
Структура таких продуктов видна на индигозоле О (IV) из индиго (I):
Вместо хлорсульфоновой кислоты можно применять ее эфиры, например
ClSOaOCgHj, получая предварительно средние эфиры серной кислоты, которые
далее уже переводятся в соли кислых эфиров с предварительным отщеплением прн
омылении алкильной группы. Хлорсульфоновая кислота может быть также заме-
нена олеумом.
Есть указания английского патента, что при получении индигозолей благоприят-
ным оказывается присутствие в реакционной среде некоторых иеществ, как то:
фталевого нли янтарного ангидридов или имидов, фосгена, хлоругольиого эфира
(CICOOC^Hg), хлористого ацетила или бензоила, л-толуолсульфохлорида и других
хлорангидридов органических кислот 62).
Вероятно подобные по строению эфиры, содержащие остаток фосфорной,
соотв. алкилфосфорной, кислоты, получаются при обработке кубовых красителей
галоидангидридами алкнлфосфориой кислоты или хлорокисью фосфора в при-
сутствии металла (Zn) и третичного основанияю).
338
Полученные соли сернокислых эфиров лейкосоединений оказа-
лись особенно интересными для ситцепечатания, так как они дают
возможность легко закреплять кубовые красители действием оки-
слительных и гидролизующих агентов 64).
Индигозоли — название целого класса этих препаратов, их мар-
ки и иногда указание на цвет объясняют, из какого красителя они
получены.
Энольяые эфиры серной кислоты кубовых красителей могут быть использованы
и для дальнейшего синтеза. Если в молекуле кубового красителя имеется аминогруп-
па, то его сернокислотный эфир может быть диазотирован и применен в качестве
диазосоставляющей для получения азокрасителей, например
OS03K OSO3K OSO9K OSO3K
В последнее время появился интерес к получению эфиров сер-
ной кислоты энолей, отвечающих некрасящим антрахиноновых произ-
водных. Примеры диазотирования таких эфиров, соответствующих
аминоантрахинонам, мы уже встречали в главе о диазотировании.
В отличие от приготовления индигозолей здесь можно в одну
операцию производить и восстановление группы СО антрахинона
до С • ОН и этерификацию энола. Роль восстановителя играет
здесь мелкораздробленный металл, например медь. Ацилирующим
средством служит лучше всего уже упомянутая ранее ангидро-N-
сульфокислота пиридиния, средой — пиридин.
Таким образом например ацетил-^-аминоантрахинон нагревают в пиридине с до-
бавкой аигидрб-Г4-сульфокислоты пиридиния (из пиридина и хлорсульфоновой кис-
лоты) до 90°, прибавляют порошка меди и после часового нагревания разводят во-
дой, подщелачивают едким натром и пиридин отгоняют с паром. Последующее
кипячение щелочного раствора приводит к отщеплению ацетильной группы. Схема
превращений здесь такова в“):
ОН
Illi -Jill 1 он OSO3Na 1 A/WMmc“* r ' * Illi ” \Z\/\Z ' 1 OSO3Na 22 * ZNHCOCH3 OSO3Na 1 /xy\Q/NH2 4//'c l OSO3Na
339
Подобные сернокислотные эфиры антрагидрохиноновых произ-
водных вероятно могут быть применены ко многим превращениям
антрахиноновых замещенных.
При обработке хлорсульфоиовой кислотой в присутствии третичного амина
(пиридина и т. п.) оксизамещенных антрахинона в отсутствии восстановителя полу-
чаются сернокислотные эфиры преимущественно {3-оксиантрахиноиовых производ-
ных 66).
Например ализарин (I) образует такой эфир (II):
ОН
ОН
Эти эфиры интересны для применения красителей в крашении шерсти.
Образование солей кислых эфиров серной кислоты находит себе применение
в случае о-оксиазокрасителей для повышения их растворимости, причем получен-
ные эфиры можно подвергать некоторым реакциям для изменения их заместителей
без потери группы серной кислоты Б7).
Ацилирование остатком серной кислоты возможно не только
в приложении к амино- и оксигруппам, но и к сульфгидрильной
группе SH с получением в этом случае арилтиосерных кислот:
RSH - RSSO3H.
Ацилирующим агентом здесь служит также ангидро-М-сульфо-
кислота пиридиния б8).
Реакция по отношению к тиофенолу идет по уравнению
C6H5SH 4-C5H6NSOs^C6H5SSO8H • C5H5N.
Щелочные соли фенилтиосерной кислоты в нейтральном водном растворе устой-
чивы. При обычной температуре феиилтиосерная кислота также устойчива и в кис-
лом растворе. При повышении температуры наступает постепенно разложение гидро-
литическое:
CeH6SSO3H + Н2О = CeH5SH + H2SO4.
В щелочных растворах разложение наступает быстро с образованием дифенил-
дисульфида, бензолсульфиновой кислоты и сернистой кислоты.
Арилтиосериые кислоты, замещенные в арильном остатке окси- (соотв. амиио-)
группами, получаются при взаимодействии хинонов, соотв. хннонимииов, с серно-
ватистокислой солью в присутствии окислителя, например при синтезе метиленового
голубого (см. следующую главу).
Ацилированные амино- (соотв. окси-) производные иногда нуж-
даются в аналитической характеристике ацила, связанного с харак-
терным их заместителем и в количественном его определении.
И то и другое выполняется сначала гидролитическим расщеплением
ацилпродукта, например действием щелочей или кислот в водном
растворе, с последующим отделением водного раствора образо-
вавшейся кислоты или ее соли и применением тех реакций, кото-
рые характерны для аниона данной кислоты, соотв. титрованием
кислоты щелочью. Если кислота летуча, то ее изолируют отгон-
кой из водного раствора.
Фрейдеиберг рекомендует для определения ацетильной группы нагревать
содержащее ацетил соединение в спиртовом растворе с толуолсульфокислотой,
отгонять образовавшийся уксусноэтиловый эфир и титровать уксусную кислоту
после щелочного омыления эфира. 59).
340
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) J. Wagner, Ober Sulfamintauren der aromatischen Reihe, Ber. 19, 1157 (1886):
W. Tra u be, Ber. 23, 1653 (1890), 24, 360 (1891).
2) W. Trau be, Г. n. 317668 (F. XVIII, 199), Ber. 52, 1292 (1919).
3) IG, Б. n. 328032, CB 1930, 11, 805; cp. IG, Б. n. 370459 Br. A. B. 1932, 763.
Сульфаминовые кислоты антрахннового ряда; D. Н., Шв. и. 114914, СВ 1927, 1, 817.
4) К. Heusner, М. Simon (G. An. W.), Ам. и. 1725076(1929).
Б) 1G, Г. и. 493689, СВ 1930, II, 306.
6) Н. С. Дроздов, Ж. X. Пр. 9. № 7 44 (1932).
7) См. В. Sebrell, Ам. и. 1604199. М. Н. У е д и и о в, Об ускорителях вулка-
низации резины. Аи. Кр. Пр. 1932, № 3, 21.
8) By, Г. и. 49075 (F. II, 528).
9) К. Н. Meyer, Г. и. 390798, 392409 (F. XIV, 240, 241), ср. 1G. заявл. Г. п.
В 115369 (F. XV, 117).
1°) IG, Г. и. 449112 (F. XV, 234).
u) R. В. Т г u s I е г (R о е s s 1 е г & Hasslacher Chem. Со), Ам. п. 1656252
(1928),
12) IG, Г. и. 445774 (F< XV, 400).
13) 1G, Г. п. 462303, СВ 1929, I, 2233.
И) WMJ, Matheson & Со, Г. и. 98070 (F. V., 752)
13) N. A. Mens ch utkin, Цег. 15, 2502 (1882).
I6) Ср. Е. Sakeliar ios, Ber. 60, 218 (1927).
п) См. Г. Е. Фир ц-Д а в и д, Производство органических красок, Гиз 1927, 143.
I8) IG, Г. п. 466519, СВ 1929, 1, 3143.
1Э) Ср. Кау, Вег. 26, 2853 (1893), Ulffers, Jansen, Ber. 27, 93(1894). L. C. Rai-
ford; R. Taft, H. Lankelma, Am. Soc. 46, 2051 (CB 1924, II, 2331).
2°) M. Bergmann, Г. n. 453577 (1927).
2i) BDCo, Б. n. 250883, CB 1929,1, 145.
22) См. например A. A. Levine, L. Wehmhoff, Am. Soc. 51, 1243 (1929).
23) W. Madelung, Ber. 45, 1128, 3521 (1912), Г. n. 262327 (F. XI, 278).
a4) IG, Г, n. 463140 (1928), J. Haller (Gr as sel 1 i), Am. n. 1685698 (1928).
26) E. F о u r n e a u, J. T r e f о u e 1 (Poulenc F г ё r e s), Г. n. 427857 (F. XV,
1434). Cp. J. E. В al a ban, H. King, Soc. 1927, 3068; CB 1<)28, I, 1036.
26) Например CIBa, Г. n. 170045 (F. VIII, 174), By, Г. n. 252159 (F. XI, 225).
27) Sc. D„ Б. n. 324311, CB 1930, II, 143.
-8) BDGo, Б. n. 293924, CB 1929, I, 1614. Cp. R a b c e w ic z-Z u b k о w s k i, CB
1929, II, 2766.
29) BDCo, Г. n. 462053 (1928).
3°) H. E. Fierz-David, E. Ziegler, Helv. 11, 776 (1928); CB 1928, II, 2365.
si) By, Г. n. 256621 (F. XI, 159).
32) Gr.-E., Г. n. 409949 (F. XV, 203), IG (A. Zitscher, R. Schmidt), Am. n.
1634090 (1927).
33) См. A. E. Вольхин, К вопросу получения нафтолов AC, АС-БС, АС-БО.
Ан. Кр. Пр. 1933, 1, 11.
31) CIBa, Г. п. 436179 (F. XV, 531).
зз) СЛВа, Г. п. 390201, 399485 (F. XIV, 820, 878, 880).
36) В. А. Солонина, Ж. 29, 405 (1897), 31, 640(1899).
з’) К. Lauer, J. pr. Ch. [2] 137, 161 (1933).
38) В., Г. п. 145191 (F. VH, 130).
в») Baumann, Вег. 19, 3219 (1886).
®) Claisen, Вег. 27, 3182 (1894).
Щ F. A. Menalda, Rec. 49, 967 (1930); ср. J. F. М. Caudri, СВ 1929, II,
2802.
«) F. D. Chattaway, Soc. 1931, 2495; СВ 1931, II, 2980.
«) J. В. Conant, G. М. Bramann, Am. Soc. 50, 2305, СВ 1928, II, 1545.
44) Н. L. Busse it, СВ 1931, II, 3464; 1930, II, 3020; ср. Raiford и сотр., I. с.
<*5) Е. Gebauer-Fiillnegg, F. Meissner, СВ 1928, II, 1322; Gebauer-
Ftillnegg, J. Smith-Reese, CB 1928, II, 2459.
«) H. Reihlen, Г. n. 463518, CB 1929, I, 2236.
47> Einhorn, Holland, Ann. 301, 95 (1898).
48) Georgescu, Ber. 24, 416 (1891).
49) Bergmann, Ulpts, Camacho, Ber.55, 2796 (1922).
341
ю) G. N. Burkhardt, Lapworth, Soc. 129, 684 (1926); Burkhardt,
H. Wood, Soc. 1929, 141, CB 1929, I, 1565.
B1) M. Batt egay, L. Dani veil e, CB 1931, I, 2605.
Ba) M. Bader, Ch. Sunder, DH, Г. n. 424981, 430548, 435787, 436176, DH
428241 (F. XV, 592—596); Morton Sundour Fabrics, Б. n. 278399, CB 1928, I,
852
B3) Sc. D. (Wylam, Hariss-Thomas), Б. n. 260647, CB 1927, T, 1232.
B1) DH, Г. n. 418487, 431501, 431250, 433146. 432726, 441371 (F. XV, 915-921).
м) Sc. D. (F a i r w a t h e r, J. T h о m a s), Б. n. 312243, CB 1929, II, 2830.
E. Geb a u e r-F till n eg g, J. Ei s n e r, CB 1928, II, 601. IG (Mi eg, Hei-
denreich), Г. n. 491424, CB 1930, I. 3487.
B7) IG, Б. n. 282107, CB 1928, I, 1719.
58) R. Baumgarten, Ber. 63, 1330 (1930).
M) K. Freudenberg, Ann. 433, 230 (1923), CB 1924, I, 2189.
ГЛАВА XIII
ОКИСЛЕНИЕ И ОСЕРНЕНИЕ
ОКИСЛЕНИЕ
Метод окисления — один из весьма распространенных и приме-
няемых в технике как для производства промежуточных продуктов,
так и для получения из них готовых красителей. Окисление в широ-
ком понимании обнимает, во-первых, те химические превращения,
которые связаны с введением в соединения кислорода в виде напри-
мер новых кислородсодержащих заместителей и, во-вторых, такие
превращения, где при помощи воздействия кислорода от органи-
ческого соединения только отнимается водород (в виде воды).
В настоящей главе будут рассмотрены лишь процессы окисле-
ния, подходящие под первое из двух вышеназванных определений,
т. е. дающие в результате обогащение кислородом исходных соеди-
нений, частично и процессы, сопровождающиеся потерей водорода
(дегидрогенизацией). Процессы же второго рода, где роль кисло-
рода или отдающего кислород агента (окислителя) заключается
лишь в уменьшении содержания водорода в соединении, найдут
себе место в следующей главе, поскольку при этом меняется угле-
родный скелет соединения.
В отличие от всех вышеописанных методов получения проме-
жуточных продуктов окисление не может найти себе общего
выражения в формуле, так как случаи его приложения, равно
как и реагенты, чрезвычайно разнообразны. Всякое окисление
связано с сообщением исходному материалу кислорода непосред-
ственно или через посредство третьего вещества — окислителя. В не-
которых окислениях кислород участвует в образовании новой моле-
кулы, входя в состав новой содержащей кислород группы без
потери молекулой водородного или иного атома, в других — обра-
зование кислородного соединения сопровождается потерей водо-
родных атомов. Имеются наконец и такие примеры окисления, где
взаимодействию сопутствует отщепление углеродных атомов исход-
ного соединения, в виде например молекулы СО2 и т. п., причем
в таких реакциях изменяется уже и углеродный скелет соединения.
Существует большой выбор веществ, могущих функциониро-
вать в качестве окислителей. Наиболее доступен и дешев из них
конечно кислород воздуха. Тем не менее большая часть самых
интересных примеров его применения в практике окисления будет
рассмотрена ниже как процессы контактного (каталитического)
окисления, протекающие в паровой фазе.
Часто применяемыми сложными окислителями служат неорга-
нические и иногда органические кислородсодержащие соединения
343
(из последних — особенно часто нитросоединения), могущие в усло-
виях реакции передать свой кислород окисляемому веществу.
В некоторых реакциях окисления пользуются неорганическими
соединениями высшей степени валентности (например CuCI2> FeCl8,
SnClt), каковые, переходя в низшие степени окисления, содей-
ствуют отнятию водорода от окисляемой молекулы.
Для того чтобы уверенно проводить реакции окисления, необ-
ходимо познакомиться со специфическими качествами отдельных
окислителей, так как в этой группе реагентов в значительной мере
проявляются избирательные свойства: есть окислители особенно
пригодные для получения определенного эффекта реакции при
определенном положении заместителей в окисляемом соединении
и не дающие этого эффекта при иной структуре молекулы, есть
окислители .мягкие" и окислители энергичные и т. п.
Во многих реакциях окисления выяснилась серьезная роль ката-
литических агентов, в некоторых примерах окисления значение
катализаторов не выяснено, но несомненно имеет место. Тут еще
предстоит много работы исследователю.
Окисление в большинстве случаев производят в водной среде,
весьма редко пользуясь органическими растворителями. Иногда
реакцию проводят при употреблении и воды и органического, не
смешивающегося с водой, растворителя. Это важно например для
изолирования от дальнейшего окисления уже образованного про-
дукта; переходя из водного раствора, продукт избегает переокисле-
ния. Расчет стехиометрических количеств реагентов для окисления
удобнее всего вести, имея в виду степень раскисления окислителя
и пользуясь формулой исходного и конечного соединения.
Ниже будут прослежены процессы окисления по характеру
изменений, вызываемых в исходном материале. Вначале поста-
влена наиболее значительная по количеству примеров группа пре-
вращений гомологов бензола и их замещенных в альдегиды и кар-
боновые кислоты окислением группы СН3 в СНО и СООН.
а) Образование альдегидной группы окислением
Превращения СН3 -> СНО имеют значение для получения альде-
гидов непосредственно из метилзамещенных бензола. Уравнение
реакции, отнесенной к кислороду:
RCH3 + О2 = Н2О + RCHO.
Альдегиды имеют выдающееся значение среди промежуточных
продуктов, служа посредствующими соединениями для получения
ценных триарилметановых замещенных. Поэтому получению альде-
гидов всегда было посвящено много исследований. Мы видели уже
выше, что возможно получить бензойный альдегид из ш-дихлорозаме-
щенного (хлор в метильной группе) обменом хлора на гидроксиль-
ную группу. Этот метод требует предварительного хлорирования,
разделения от изомеров, низших и высших степеней охлорения,
и поэтому способ прямого окисления представляется теоретически
гораздо более удобным.
Среди ароматических альдегидов имеют значение как сам бен-
344
зальдегид, так и различные его замещенные (о-нитробензальдегид—
для индиго, о-сульфо-, о-хлорбеизальдегид, .и-оксибензальдегид—
для красителей ряда триарилметана). При этом необходимо отме-
тить, что у альдегидной группы—СНО — ориентирующая способ-
ность (мета-) при замещениях совсем иная, чем у группы СН3
(орто-, пара-), следовательно во многих случаях является интерес-
ным получать замещенные бензальдегиды не превращениями гото-
вого бензальдегида, но образовывать альдегидную группу в соеди-
нении с имеющимися уже заместителями.
Среди окислителей, применяющихся для превращения метильной
группы в альдегидную, прежде всего необходимо назвать кисло-
родные соединения марганца. Перекись марганца МпО2 применяется
главным образом в виде регенерированной перекиси марганца
(какой она получается из марганцовокислого калия КМпОх в резуль-
тате применения этой соли для окислительных процессов в щелоч-
ной или нейтральной среде). Применяется она в избытке по отно-
шению к окисляемому соединению в присутствии водной серной
кислоты, или с избытком кислоты, но сама в требуемом коли-
честве ’).
Последний способ дает хорошие результаты при окислении гомологов толуола,
но представляет трудности при окислении о-нитротолуола. Сульфокислоты углево-
дородов с метильной группой окисляют в олеуме, причем при избытке SO3 даже
очень большое против теории количество окислителя ие дает переокисления до
карбоксильной группы, но лишь альдегидную группу СНО. Температура во всех
таких окислениях ие высока. С олеумом работают при охлаждении до 0—10°.
С водной серной кислотой — при 20—40°. Очень полезными окислителями оказа-
лись электролитически приготовляемые сернокислые соли марганца, отвечающие или
трехвалеитиому марганцу, например Mn2(SO4)3 • (NH^SOi2), или перекиси марганца 3).
Наконец предложено применять перекись марганца с разведенной серной кисло-
той при очень высокой температуре до 165е в автоклавах при давлении 10 ат.
В этих условиях окисляется о-нитротолуол в о-иитробензальдегид 4). Предложение
окислить о- и л-иитротолуол в альдегид нагреванием с перекисью марганца 5) дает
слишком мало шансов получить достаточные для технического использования вы-
ходы альдегида.
Повидимому также невыгоден способ окисления с примеиеиием никелевой или
кобальтовой окисей с добавочной регенерацией окиси металла из закиси посред-
ством прибавления хлорноватистокислой соли ®).
Интересно, что по неопубликованным наблюдениям Н. М. Киж-
нера перекись марганца не регенерированная, а природная дает
меньшие выхода альдегида, но больше продуктов сцепления двух
ядер (типа стильбена, диарилэтана и пр.).
Двуокись церия в серной кислоте при 90° оказывается достаточно энергичным
окислителем для получения альдегида из метилового замещенного.
Своеобразно действие хромовой кислоты (СгО3) на углеводо-
роды с метильными группами, если обработку вести в присутствии
уксусного ангидрида и серной кислоты при 0—10°.
Окисляемое вещество растворяют в смеси уксусного ангидрида и концентрирован-
ной серной кислоты при низкой температуре и при энергичном размешивании окисляют,
внося постепенно твердый хромовый ангидрид. При этом оседает вязкая масса
хромсульфата, и требуется прибавление уксусной кислоты, чтобы не задерживать
работы мешалки. По окончании реакции массу выливают на лед. Как продукт реак-
ции в таком случае получают диацетат, отвечающий орто-форме альдегида 7).
345
Таким образом реагирует например о-нитротолуол
,СН: (ОСОСН3)2
'2
Метод требует большого расхода хромовой кислоты, которая частью окисляет
и уксусный ангидрид. Поэтому практическое значение его мало. При обработке
гидролизующими агентами, с потерей ацильных групп, получают соответствующие
альдегиды.
Если в этом последнем случае предохранителем от слишком далекого окисления
углеродного заместителя является уксусный ангидрид, ацилирующий орто-форму
альдегида, то опыты Зинькова доказывают, что можно в известной степени
ослабить окислительный эффект хромовой кислоты, выводя вместе с избытком толуола
образовавшийся уже бензальдегид чисто механически. При непрерывном прохождении
толуола через аппарат хромовая кислота, образуемая из двухромовокислого калия,
приливаемого в водном растворе к сериой кислоте, находящейся под слоем толуола,
осуществляет две окислительных реакции: превращение толуола в бензальдегид
и в бензойную кислоту согласно уравнениям:
ЗСвН3СН3 + 2К2Сг2О7 + 8H,SO4 = ЗС^СНО + 2K2SO4 -f- 2Cr2(SO4)3 + 11Н2О,
ЗС6Н3СН3 + ЗК2Сг2О7 4- 12H2SO4 = ЗСвН3СООН + 3K2SO4 + ЗСг2(ЗО4)з 4- 15Н.,О.
Количества бензальдегида и бензойной кислоты в продуктах окисления прибли-
зительно одинаковы 8).
Методика непрерывного проведения процесса в предлагаемой форме требует
еще большой работы по выяснению практической пригодности этого метода.
Предложен автоклавный метод окисления боковой цепи в гомологах бензола
с образованием альдегидной и карбоксильной групп. Так, толуол нагреванием в авто-
клаве с водой и кислородом (до 235—240J при 50—60 ат} в присутствии катали-
затора (Fe2O.s-H3O) переводится в бензальдегид 4* (немного) бензойную кислоту.
Подобно окиси железа могут действовать и окислы иных тяжелых металлов: Cr, V,
Си, Се, U 8).
Получение о-нитробензальдегида из о-нитротолуола ввиду лег-
кости перехода от него к индиго (по методу Бейера и Древ-
сен а) было издавна интересной задачей, и, хотя из приведенных
прямых методов окисления не оказалось ни одного, который безу-
словно был бы технически удобен для получения этого производ-
ного, выяснилась возможность выгодных способов его фабрикации
методами непрямого окисления.
Назовем, во-первых, способ перехода через изоиитрозосоедннение действием
амилнитрита в спиртово-щелочной среде. Изонитрозосоедииеиие (альдоксим) обычной
кислотной обработкой превращается в альдегид 10):
/\/СН3 /-i-oNOCsH,,ч /\ /СН— N-ONa /СНО
II ( -f-NaOCjHj ) || кислотный гидролиз ( ,
\/\nO2 \ZXNO2
Во-вторых, о-нитрохлористый бензил (из о-нитротолуола) пере-
водят воздействием анилина в о-нитробензиланилин, который
окисляют (марганцевокислой солью или хромовой кислотой)
в бензилиденовое (азометиновое) соединение и это последнее обра-
боткой кислот разделяют на альдегид и соль анилина J1).
346
+ H.NAr
— HC1
//\/CH = NAr
1 1 гидролиз
I +ArNHg
Третий способ основан на образовании ртутного соединения о-иитротолуола
при нагревании его с окисью ртути в щелочной среде. Обработка ртутного соеди-
нения посредством азотистой кислоты переводит его в дииитрит, который при под-
кислении превращается с потерей ртути в нитробеизальдоксим и в иитробеизаль-
дегид 12):
/х/сн3 /x/CHHg2O ^XHCHgNO^
11 -I I 41
^^NO, \/xNO2 Xz/xNO3
Наконец по четвертому способу о-иитробензальдегид получают из о-нитрото-
луола, предварительно переведя его в о-иитрофеиилиитрометаи (выхода низки;
нитрование разведенной азотиой кислотой при ПО—120°). Окислением последнего
посредством маргаицевокислого калия в нейтральной среде переводят соединение
в нитробензальдегид 13):
/4/ZCH2NO, /\yCHO
II 41 -и
^/'4NO2 \/\чО2 '4'/\nO2
Окисление метильной группы в альдегидную в zz-иитротолуоле происходит за
счет внутримолекулярного окисления кислородом нитрогруппы (интрамолекулярное
окисление в щелочной среде при участии серы). Одновременно нитрогруппа восста-
навливается до аминогруппы:
ух/сн3 /х/сно
Подобное превращение возможно осуществить и в дымящей сериой кислоте 14).
Образование альдегидной группы, вместе с карбоксильной окислением метила,
замещающего водородный атом в сложных конденсированных системах типа бен-
зантрона и т. п.. предложено проводить при помощи селенистой и селеновой кислот
H2SeO3 и H2SeO4, которые восстанавливаются при этом до селена. Так, 2-метилбен-
заитрон при нагревании с селенистой кислотой и водой в автоклаве до 230—240°
переходит в беизаитрои-2-альдегид 1в).
Для частного случая окисления метильной группы в 2.4.6-трииитротолуоле (I),
где метил находится очевидно в особо активном состоянии благодаря сосредото-
ченному влиянию трех иитрогрупп в орто- и пара-положениях, можно пользоваться
347
с успехом своеобразным окислителем в виде нитрозодиметнланилина в присутствии
пиридина 16).
Взаимодействие проходит через стадию азометинового основания (П)
NO2
/\/СНз
I 1 I + 0N~\ /-N<CH3)2
o2n/\/\no2
no2
1 ,CH —N — CeHj — N(CH3)2
+ Hso
нагрев с HC1
NO,
A/CH°
> | | + H2N — CeH4 — N(CH3)2
O2N/Z\'/X'NO2
Выше было уже указано на практическое значение для получе-
ния бензальдегида предварительного хлорирования толуола в хло-
ристый бензилиден. Метод превращения обменом хлора на гидро-
ксил и окислительный метод могут быть совмещены 17).
Смесь хлористого бензилидена и хлористого бензила (техническая смесь, полу-
ченная при хлорировании толуола до удельного веса 1,175) обрабатывается с обрат-
ным холодильником в водной среде посредством порошковатой перекиси марганца
(моль иа моль хлорвстого бензила) до превращения всех хлоридов в альдегид. По-
видимому этот метод оказался технически непригодным.
Окисление СН2С1 -► СНО предложено производить действием
бихромата щелочного металла с добавлением едкой или углекислой
щелочи в водном растворе. Уравнение реакции таково:
3RCH2C1 + Na2Cr2O7 + NaOH = 3RCHO + 3NaCl Cr2O8 4- 2H2O.
Аналогичный способ предложен для получения альдегидов
вообще из соединений, в которых имеется группа СН2С1. Странным
образом в качестве агента в этом процессе, где требуется и заме-
нить хлор на кислородную группу и окислить соединение, предло-
жен гексаметилентетрамин C6N4H12, получаемый взаимодействием
формальдегида и аммиака 18).
Необходимо отметить затем способы каталитического окисления
метильной группы в карбонильную при действии на пары углево-
дорода в смеси с воздухом или иным кислородсодержащим газом
определенных соединений некоторых металлов при достаточно вы-
сокой температуре. О них ниже будет сказано более подробно.
Полученный тем или иным способом ароматический альдегид
обычно сопровождается примесью непрореагировавшего углеводо-
рода, соотв. замещенного; иногда к нему могут быть примешаны
продукты дальнейшего окисления. Очищают альдегид от примесей
во-первых (если он летуч) отгонкой с водяным паром, во-вторых,
348
обработкой раствором кислого сернистокислого натрия (бисуль-
фита) NaHSO8, в каковом растворе альдегид на холоду раство-
ряется и образует при этом с бисульфитом хорошо кристаллизую-
щее соединение типа RCH(OH)SO8Na.
Такое соединение разлагается от действия даже слабых щело-
чей (соды) и кислот, регенерируя альдегид.
В воде растворимы не только соединения альдегидов с кислыми
солями сернистой кислоты, но и соединения их с самой сернистой
кислотой. Этим обстоятельством также можно пользоваться для
выделения альдегидов из продуктов реакции 19).
Способность к взаимодействию с бисульфитом различна у различных альдегидов.
Этим пользуются для отделения .и-иитробензальдегида, получаемого нитрованием
бензальдегида, от примешанного к нему о-нитроизомера. Обработкой смеси иитро-
беизальдегидов таким количеством раствора бисульфита, которое отвечает прибли-
зительно количеству орто-изомера, удаляют в раствор преимущественно его, а не
Jt-нитробензальдегид (неопубликованные опыты Н. М. К и ж н е р а и Н. Г. Л а п-
т е в а).
Легкость взаимодействия альдегидов с первичными аминами,
уже упомянутая в главе об ацилировании, может также оказаться
полезной для выделения альдегидов. Для этого необходимо разме-
шать смесь, заключающую альдегид, с раствором соли щелочно-
земельного металла некоторых сульфоновых или карбоновых
кислот первичных аминов, причем выделяется азометиновое соеди-
нение в кристаллическом виде. Это соединение, путем горячего
гидролиза без участия кислоты (с паром), разлагается на альдегид
и кислоту амина 20).
Для альдегидов также характерны гладкие реакции с гидро-
ксиламином (образование альдоксима RCH:NOH), с фенилгидра-
зином (образование фенилгидразона RCH :N • NHC6H5) и неко-
торые цветные реакции.
Выше было приведено, каким образом получают бисульфитиые соединения аль-
дегидов. Добавим, что эти соединения, будучи трудиорастворимыми в избытке
бисульфита, в то же время часто становятся легче растворимыми и труднее выде-
лимыми при большом избытке свободной сернистой кислоты, каковой поэтому
следует избегать в растворе бисульфита.
Образование альдоксима производят воздействием на 1 мол. альдегида водного
раствора солянокислого гидроксиламина (1 мол.) и соды (,/2 мол.) на холоду. Если
альдегид нерастворим в воде, то работают в водно-спиртовом или метиловоепирто-
вом растворе. С такими альдегидами, которые легко окисляются (бензальдегид),
образование оксима проводят в сосуде, иаполиениом углекислотой.
Для получения фенилгидразона применяют вполне чистый фенилгидразвн и
проводят реакцию лучше всего в уксусиокислотиом (слабом) растворе. Вещество
растворяют в воде или спирте и обрабатывают свежеприготовленной смесью вз рав-
ных объемов феиилгидразииа и 50%-ной уксусной кислоты, разведя все тройным
объемом воды. Реакция часто проходит гладко уже иа холоду, иногда легче при
нагревании иа водяной бане. При взаимодействии с феии л гидразином вредно отзы-
вается присутствие в реакционной смеси свободных минеральных кислот и особенно
азотистой кислоты. Последняя должна быть разрушена предварительным прибавле-
нием мочевины.
В качестве хорошего реагента иа карбонильную группу (в альдегидах и ке-
тонах) предложен 2.4-дииитрофенилгидразии, легко получаемый из 2.4-дииитро-
хлорбеизола н гидрата гидразина 2I).
Реакция с метадиамииами характерна, как и вышеописанные, для ароматиче-
ских альдегидов (и карбонильных соединений вообще). Для ее производства при-
готовляют водный или (лучше) спиртовый 0,5—1%-иый раствор солянокислой соли
метадиамииа (например .и-фенилеидиамина) и приливают несколько кубич. санти-
метров этого раствора к спиртовому или водному раствору исследуемого вещества.
340
Через несколько минут наступает реакция, проявляющаяся в интенсивной зелено-
ватой флуоресценции раствора, достигающей самое большее через 2 часа макси-
мума ннтеисивиостн. Прибавка щелочи затемняет эту цветную реакцию, последую-
щее подкисление опять ее выявляет.
Очень чувствительна реакция с диазосульфаннловой кислотой. Чистую кристал-
лическую дназосульфаниловую кислоту N2CeH4SO3 растворяют приблизительно
в 60 частях холодной воды с прибавлением небольшого количества едкого натра,
затем вводят вещество, смешанное с разведенной щелочью, и малое количество
амальгамы натрия. При наличии альдегида через 10—20 мин. образуется красно-
фиолетовая, похожая на чисто фуксиновую, окраска. Это—вполне типичная реак-
ция альдегидов, отличающая их от других карбонильных соединений, и в то же
время это — очень чувствительная реакция, позволяющая открывать например
бензальдегид при разведении 1: 3 000.
Если требуется определить количественно альдегид, то можно пользоваться
следующим объемным методом Риппера 22).
Готовят приблизительно 1/2%*ный по возможности водный раствор испытуемого
альдегида, 25 см3 этого раствора вливают в колбочку (на 150 см3), где уже налито
50 гм3 раствора бисульфита калия (содержащего 12 г вещества в 1 л). Колбочку
плотно закупоривают и оставляют иа у4 часа. В течение этого времени определяют
(по у10 N раствору иода) титр 50 см3 раствора бисульфита калия. Затем оттитровы-
вают этим же У10 У раствором иода количество несвязанной сернистой кислоты
в растворе альдегида. Разница между расходом иода в первом и втором титровании
дает содержание связанной сернистой кислоты, соотв. содержание альдегида,
в 25 см3 раствора альдегида. Метод дает хорошие результаты для альдегидов, кото-
рые растворимы в воде или могут быть переведены в водный раствор малым количе-
ством спирта. Сколько-нибудь значительное количество спирта (например более 5%)
в растворе уже вредно отражается на чувствительности иодо-крахмальиой реакции.
Другой метод, позволяющий определять количественно альдегид (нли другое
карбонильное соединение, реагирующее с гидроксиламином), выработан Петрен-
ко-Криченко и Лордкипанидзе и основан на наблюдении, что оксимы в
разведенных растворах ие соединяются с кислотами, почему избыток гидроксила-
мииа, остающийся при получении оксима, можно титровать кислотами. К алкогольному
раствору карбонильного соединения прибавляют свежеприготовленный раствор
сернокислого гидроксиламина и эквивалент гидроокиси бария. Определения произ-
водятся таким образом, чтобы при сливании жидкостей получился раствор в 50%-
иом алкоголе приблизительной у100 N концентрации 23). Индикатором служит мети-
ловый оранжевый. Существуют еще иные методы определения альдегида: по раз-
ложению избытка фенилгидразииа окислителем и йодометрический.
Окисление арилкарбинолов в альдегиды, следовательно пре-
вращения RCH2OH-> RCHO, используются в значительно мень-
шей мере, чем окисление метильной группы в альдегидную.
Общее выражение реакции таково: RCH2OH4-O = RCHO-f-H2O.
Непрямой способ окисления дает возможность готовить окси- и
аминоальдегиды. Как описано в следующей главе, формальдегид
с ароматическими аминами и фенолами дает амино- или окси-
производные ароматических спиртов (соотв. их ангидридопроиз-
водные). Если после образования или в процессе образования
таких спиртов прибавить к реакционной смеси сульфокислоту
ароматического гидроксиламина, то получается продукт дегидра-
тации, переходящий в ангидро- (азометиновое) соединение, легко
распадающееся при действии разведенных щелочей или кислот
на альдегид к амин 24):
(CH3)2Nx/x
+ H(HO)N—/~\-СН3-------------->
^/^СНгОН ^SOsH
350
(CH8)jN.
(CH3)2N4/x
гидролиз
—----> ----------------------------->
/ /—СН3
^SOgH
(CH8)3NX/X
-----> -+ h2n-<^2/--ch3
Ч/ХчСНО XSOgH
Из ш аралкиламиновых соединений RCH2NH2 возможно получение альдегидов
также непрямым методом окисления. При взаимодействии с хлорозамещенным, обла-
дающим подвижным галоидом, из аралкиламина (I) образуется соответствующее
замещенное типа беизиланилина (И), окислением которого, как выше было уже
сказано при о-нитробензальдегиде, в азометииовое соединение (III) и гидролизом!
последнего получается альдегидное соединение, например
SO3H
I
ОСН3
Н0\Л
| | И SOgH
X'/4CH2-NH-/ - no2
°СНз SO3H
Н0\А /'
| I + no2
X/Z\cho
(ванилин^
Кроме ш-аралкиламинов с гидроксильной группой исходные соединения могут
быть и с другими замещающими, в том числе с углеводородными радикалами
Способ получения аралкиламинов (ш-амииов) рассмотрен ниже в гл. XV.
Из многочисленных способов получения альдегидных соедине-
ний практическое значение имеют немногие, и для бензальдегида
в частности метод превращения хлористого бензилидена гидроли-
зом сохранил до сих пор свою важность.
351
б) Образование карбоксильной группы окислением
Превращения СН8 -► СООН имеют назначением превратить
метильную группу в карбоксильную. Общее выражение реакции:
RCH3 + 30 = RCO2H 4- Н20.
Карбоксильная группа является последним предельным окисли-
тельным превращением метильной группы (до отщепления угле-
родного заместителя от ароматического ядра), и для получения
карбоксила можно пользоваться более сильными окислителями.
Наиболее употребительные окислители для этого рода превращений,
это — хромовая кислота, азотная кислота и марганцовокислый калий.
Если в соединении имеется несколько боковых цепей, то азот-
ная кислота вызывает чаще всего лишь частичное окисление, произ-
водя иногда и нитрующее действие. Хромовая кислота (вместе
с серной) окисляет вполне, причем результат окисления зависит
и от взаимного положения боковых цепей. Именно орто-изомеры
хромовой кислотой или не окисляются или вполне разрушаются,
при работе же с марганцовокислым калием окисление в этом
случае протекает удачно. Боковые цепи в пара-положении легче
окисляются, чем в мета-положении. Если требуется окислить лишь
одну боковую цепь, то и на ее окисляемость имеют также влияние
другие заместители. Отрицательная группа, стоящая в орто-месте
к метилу, повидимому затрудняет окисление посредством хромовой
кислоты. Наиболее пригодны для окисления таких орто-замещен-
ных соединений повидимому марганцовокислый калий26), так же
как железосинеродистый калий K8Fe(CN)6 27), мало употребитель-
ный в других случаях.
В редких случаях применяется в качестве окислителя хромовый ангидрид СгО3;
при этом рассчитанное количество его растворяется в ледяной уксусной кислоте,
и этим раствором обрабатывают при нагревании до кипения растворенное в уксус-
ной же кислоте окисляемое вещество. Гораздо чаще пользуются так называемой
хромовой смесью (40 ч. двухромовокислого калия или, еще лучше, такой же соли
натрия и 55 ч. концентрированной сериой кислоты, которая иногда разводится
двойным объемом воды). С помощью некоторых искусственных приемов удается
при работе с хромовой кислотой получить карбоновую кислоту, избегая дальней-
шего окисления с разрушением соединения (окисление до СО2), например пропуская
пары окисляемого вещества через нагретую хромовую смесь, причем карбоновая
кислота, собираясь в плавающем сверху слое углеводорода, избегает дальнейшего
окисления ®). Из работы Каминского выяснилось любопытное влияние при-
бавки к хромовой смеси небольшого количества соляной кислоты: при сравнительно
низкой температуре (40°) метильная группа в ароматических углеводородах окис-
ляется почти количественно в карбоксильную. Возможно, что в сказанных условиях
окислительно действует хлористый хромил СгО2С12 в момент своего образования м).
Более энергичное взаимодействие между хромовой кислотой н серной обеспечи-
вается повидимому прибавлением адсорбирующего пористого материала (кизель-
гура, каолина) и хорошим перемешиванием смеси 30).
Азотная кислота (концентрированная) для избежания нитрующего эффекта раз-
водится до подходящей концентрации (например 1 : 4). Вообще же оиа — мало
удобный окислитель, применяемый только там, где трудно без нее обойтись. Разве-
денная азотная кислота, как указано выше (см. гл. И), действует при высокой
температуре, нитруя боковую цепь, а не ароматическое ядро. Такой продукт пови-
димому в дальнейшем легко окисляется. Пример мы видели уже при получении
альдегидов. Заксе получил бензойную кислоту, нагревая толуол с азотиой кисло-
той (22°/0-ной) до 120—150° в автоклаве. Очевидно здесь процесс окисления про-
ходит через фазу фенилнитрометана 31).
352
Подобен stow способ американского патента, использующий применение азот-
иой кислоты (в 35% HNO3) с добавкой перекиси марганца. Нагревание в автоклаве
ведется долговременно при температуре 70—НО032).
Не говоря о дороговизне специальной аппаратуры (из хромоникелевой стали),
способ ие имеет никаких преимуществ перед другими Методами окисления приме-
нительно к получению бензойной кислоты.
В растворе разведенной азотной кислоты (20%-ной) был испытан электрохими-
ческий метод окисления между платиновыми электродами, давший положительный
результат по отношению к л-бромтолуолу с выходом по току в 115,75% (что делает
вероятным образование промежуточно ВгСвН4СН2ОН и ВгС6Н4СНО, окисляющихся
уже за счет азотной кислоты). Из о-иитротолуола" в этих условиях окисления было
получено максимум 4,3% нитробензойной кислоты 33).
Четырехокись азота N2O4 окисляет толуол в бензойную кислоту. Так же реаги-
руют с N2O4 и нитротолуолы %.
Марганцовокислый калий КМпО4 (перманганат) и марганцовисто-
кислый калий К2МпО4 (манганат) — превосходные и часто упо-
требляемые окислители для образования карбоксильных групп.
Мы отметили выше значение перманганата для окисления орто-
замещенных соединений Зб).
Для окисления перманганат применяется в водном растворе,
причем в тех случаях, когда выделяющаяся при реакции щелочь
(2KMnO4-j-H2O = 2KOH-f-2MnO2-(-3O) может оказывать вредное
влияние, например омыляя ацильный продукт, прибавляют серно-
кислого магния как легко диссоциирующую соль, которая связы-
вает свободное основание.
В тех случаях, когда нужно окислить метильную группу в амииосоедииеиии,
аминогруппу предварительно ацилируют. Таким образом получают из о-толуидина
антраниловую кислоту зв)
^z^CHg /х^/СООН /^/СООН
\/\nh2 \/\мНСОСН3 \/\nhCOCH3 \^\nH2
В этом примере прибавление MgSO4 к реакционной жидкости оказало решаю-
щее влияние на выходы, которые поднялись с 30% (в слабом уксусиокислотиом рас-
творе) до 75°/0.
При окислении вторичных аминов защиту реакционной группы проводят нли
ацилированием илн иитрозироваиием. Таким образом о-толилглиции окисляют
в о-феиилглициикарбоиовую кислоту, переходя через промежуточные соединения
/\/СН3 /\/СН3
I I или I I
X/XN—СОСН3 \/Xn—NO
I I
СН2СО2Н СН2СО2Н
Удивительно, что при окислении группа нитрозо остается незатронутой 37).
При окислении соединений кислотного характера применяют
щелочные растворы перманганата или раствора манганата.
Окисление гомологов фенола марганцовокислым калием может
повести к нежелательным добавочным реакциям вплоть до распада
циклического соединения. Чтобы избежать этого, пользуются
методом „защиты" оксигруппы. Защищают ее или ацилируя или
алкилируя.
23 Зак. 2611. — Н. Ворожцов
353
Так, нагреванием пиросульфата калия Кг52О7 до 60—70° с горячим концентриро-
ванным раствором n-крезола последний переводят в калиевую соль крезолсериой
кислоты. Этот эфир растворяют в малом количестве воды с прибавлением едкого
кали и окисляют при нагревании иа водяной бане 4%-иым раствором КМпО4;
л-оксибеизойиая кислота образуется с теоретическим выходом з®).
Алкильные эфиры фенолов относятся к окислению перманганатом ие активнее
гомологов бензола 39). Для окисления метильных групп в фенолах без ацилирования
можно пользоваться в качестве окислителей окислами тяжелых металлов, например
СиО. Нагревание с таким окислом до достаточно высокой температуры переводит
о-крезол в салициловую кислоту 40).
Марганцовокислый калий, как сказано, является очень удобным
средством для окисления боковой углеродной цепи до карбоксиль-
ной группы. Он широко применяется для этой цели в лаборатор-
ной практике. В производстве его применение ограничивается
необходимостью использовать образующуюся перекись марганца.
Удешевлением этого метода окисления является предлагаемый немецким патен-
том способ использования маргаицово-(или марганцовисто-) кислой соли только
как прибавки к окислительно действующему раствору гипохлорита NaClO (или
гипобромита). Окисление проводится в щелочной среде. Получающаяся при окисле-
нии толуола бензойная кислота, по описанию патента, ие содержит хлора 41).
Недавно французской фирмой был заявлен в Германии патент, использующий
в качестве окислителя метильной группы уже известный окислитель МпО2 в среде
серной кислоты. Мы только-что упоминали этот прием окисления применительно
к получению альдегида. Оказывается, если вести процесс при температуре свыше
40° (преимущественно при 60—70°), то в продукте окисления преобладает бензойная
кислота. Расчет процесса ведется применительно к реакциям:
ЗМпО2 + 3H2SO4 = 3MnSO4 + ЗН2О + 30,
СвНвСН3 4- 30 = СвНвСО2Н + Н2О.
Выходы приведены в патенте: из прореагировавшего толуола (50%) получается
иа 100% бензойной кислоты 8% бензальдегида. Вряд ли этот способ окисления
может оказаться интересным для практики получения бензойной кислоты, так же
как и бензальдегида, вследствие затруднений с большой массой кислотных отходов,
заключающих марганцовые соединения 43).
Ароматические иитросоединеиия оказываются мало подходящими окислителями
метильной группы в бензольных углеводородах и производят окисление лишь кар-
бинольной группы СН2ОН н щелочной среде, понятно — с одиоиремеииым восста-
новлением иитрогруппы в азо- (соотв. азокси-) группу 43).
Между тем окисления метилзамешениого в карбоновую кислоту в сложных
коидеисироваииых системах, например в метилбензантроие, возможно проводить
при нагревании с едким кали и нитробензолом до высокой температуры (140—150°)44)-
Например
354
Интересующее нас окисление СН3 -> СООН,так же как и другие окислительные
превращения, могут быть осуществлены помощью электрохимического метода
окисления с участием в нем соли марганца, служащей передатчиком кислорода 45).
Эта соль в анодном пространстве окисляется до высших степеней окисления (от
окиси до перекиси) и передает н момент своего образования кислород окисляю-
щемуся веществу, раскнсляясь сама до начального состояния с тем, чтоб начать
вновь тот же цикл изменений. Повидимому такой способ работы во многих случаях
практически очень удобен.
Окисление метила до карбоксильной группы может быть достигнуто и без
участия внешнего окислителя, если внутри молекулы соединения имеется такая
богатая кислородом группа, как NO2 46). В присутствии спиртового или водного
едкого кали без притока кислорода извне о-иитротолуол превращается в заметных
количествах в антраниловую кислоту (из 25 г о-нитротолуола, 25 г КОН, 12,5 г воды
5 час. кипячения в атмосфере водорода дали 5,47 г антраниловой кислоты):
Интрамолекулярная реакция о- и л-нитротолуолов может привести прн полном,
как здесь, изменении СН3 и к образованию азо- (соотв. азокси-) кислоты, например
/СО2Н НО2Сх
c5-n=n-<z>
При частичном окислении можно получить таким образом динитродибеизил,
дииитро- и дииитрозостильбеи 47).
Воздействие активных лучей (света) чаще всего содействует окислительному
процессу как при интрамолекулярном, так и иитермолекулярном его течении.
Изиестио, что под влиянием освещения о-иитробеизальдегид переходит в о-иитро-
зобеизойиую кислоту:
ух^/СНО /х^/СООН
и II
\NO
Работами ряда исследователей доказано, что фотоокисление толуола кисло-
родом в бензойную кислоту—процесс очень медленный. Если же освещению под-
вергается толуол в присутствии антрахинона (и воды), то окисление идет с заметной
скоростью. Применяя известковую воду под толуолом, можно получить раствор
бензоата кальция. Прибавление очень небольших количеств некоторых солей
[CuSO4, Bi(NOs)2] или активированного угля, также силикагеля, благоприятствует
ускорению фотоокислеиия 48).
Возможно получить карбоксильную группу за счет окисления
альдегидной группы.
Известно так называемое превращение Канницаро, харак-
терное для ароматических альдегидов: под влиянием щелочи они
превращаются в смесь соответственного алкоголя (восстановление)
и карбоновой кислоты, например
2С8Н5СНО4-КОН -► С6Н5СНвОН4-СвН5СООК.
Пользуясь добавлением подходящего окислителя, в том числе
и кислорода, чаще с участием катализаторов (окислов металлов
Fe, Ni, Со или металлов Ag, Pt, Ni, Си), можно в щелочной среде
окислить бензальдегид количественно в бензойную кислоту 49).
Кроме окисления метильной и карбонильной групп известны
иные способы образования карбоксильной группы. Так, предвари-
23*
355
тельным хлорированием метила можно получить трихлорозамещен-
ное, например бензотрихлорид из толуола:
СвН5СН3 - СвН6СС18 - С6НВСООН
и гидролитической обработкой в присутствии извести или ката-
литически действующих соединений железа перевести трихлоро-
замещенное в соответственную карбоновую кислоту. Такой метод
был уже разобран в главе VII.
Можно также, не доводя хлорирования до получения трихлоро-
замещенного, подвергнуть окислению менее охлоренные продукты
с получением карбоксильной группы. Подходящий для этой цели
окислитель — хлорная известь. Нагревание с раствором хлорной
извести окисляет смесь хлористого бензила и хлористого бензили-
дена в бензойную кислоту 50).
Иные, не окислительные методы образования карбоновых кислот
не входят в задачу этой главы. Образование кислот путем обмена
хлорных атомов на кислород рассмотрено в главе о превращениях
хлорного атома. Получение нитрилов кислот, которые могут быть
действием гидролизующих агентов переведены в кислоты, частично
затронуто было в главе об алкилировании. Здесь мы остановимся
еще на вопросе о выделении кислот из реакционной смеси после
их получения и об определениях их химически.
В большинстве случаев карбоновые кислоты менее растворимы
в воде, чем их щелочные соли. Поэтому их можно выделять, под-
кисляя щелочную жидкость после реакции и отфильтровывая
выпавший осадок свободной кислоты. Чтоб отделить кислоту от
некислотных примесей, можно перевести ее в щелочную, например
натриевую, соль, обработав раствором соды или едкого натра и,
по отделении от нерастворимого, из фильтрата выделить по выше-
сказанному кислоту. Некоторые кислоты можно очищать перегон-
кой самих по себе или с водяным паром, все—перекристаллиза-
цией из воды или органических растворителей.
Характерно для карбоксильной группы образование солей
со щелочами и иногда очень типичных солей тяжелых металлов.
Способность образовывать кислотные эфиры в растворе спирта
при пропускании хлороводорода может быть также учтена как
качественная реакция карбоновых кислот. Количественно кислоты
определяются посредством титрования ’/ю N раствором щелочи
NaOH, КОН, Ва(ОН)2 в присутствии фенолфталеина как индикатора.
Можно также приготовить серебряную соль (обменным разложе-
нием щелочной соли кислоты и азотнокислого серебра) или медную
и после сжигания и прокаливания определить обычным способом
металл.
в) Окисление группы СН2 (соотв. СН) в карбинольную группу
Окисление группы, связывающей арильные радикалы в ди-(соотв.
три-) арилметаиовых замещенных, практикуется преимущественно
для получения основных и кислотных красителей типа триарил-
метана.
При этом окисление дифенилметановых производных R—СН2—R',
где R, R' — фенильные замещенные, приводит к образованию дифе-
356
ниловых замещенных метилового спирта (карбинола), называемых
п
бензгидрольными замещенными р,>СНОН и служащих употреби-
тельными промежуточными веществами в дальнейшем синтезе
триарилметановых красителей. Замещенные аминогруппами в ари-
лах производные триарилкарбинола R(R')(R2)C • ОН, например
[(СН3)^С6Н4]3СОН-соединения— весьма близкие генетически к три-
арилметановым красителям, в которые они переходят уже при
подкислении. Исходные соединения с неизмененным водородом при
метановом углероде носят названия лейкопродуктов по отношению
к только-что названным красителям.
Получение бензгидрольных замещенных по общему уравнению
R(R1)CH24~O = R(Ri)CHOH производится легче всего обработкой
замещенного диарилметана в сернокислотном растворе (также и
в соляной и уксусной кислоте) перекисью свинца. Если, как всего
чаще встречается в практике, приходится окислять производные
л-диаминозамещенного диарилметана, то эффект окисления узнается
по интенсивному голубому окрашиванию жидкости в результате
образования хиноидной соли, отвечающей ангидропроизводному
бензгидрола, например
(H3C)2N—\-СН2—/ N(CH3)2 —
(H3C)2N—
\-N(CH3)2 -4^
----> (H8C)2N-/
\-CH—N(CH3)2
\so4
Перекись свинца—излюбленный окислитель для получения из лейкосоединений
самих красителей, особенно в ряду производных дифеиил- и трифенилмета на ьг)
Здесь образованию красителя, как сказано, предшествует получение сс ответствен-
ного бесцветного гидрола, который затем переходит в окрашенную соль—краситель:
R(Ri)(R2)CH---> R(R1)(R2)C • ОН.
Перекись свинца применяется и в этом случае в присутствии разведенной серной
(иногда соляной) кислоты, чаше без подогревания. Окисление триарилметаиа стремя
нитрогруппами в соответственный трииитрогидрол было осушествлено братьями
Фишер действием хромовой кислоты в растворе ледяной уксусной кислоты.
Так же окисляется сам трифенилметаи в трифеиилкарбииол Бг).
Водородный атом, связанный с углеродом, входящим в арома-
тическое кольцо, может претерпеть различные окислительные
превращения. Он может быть замещен гидроксилом по схеме:
\сн + о-------->\сон (1)
или же на месте группы СН может возникнуть карбонильная
группа, причем в этом последнем случае распределение связей
357
в ядре существенно изменяется в сторону образования хиноидного
соединения
СН СН
\СН--------> >С: О (2)
СН “СН
Чаще образование карбонильной группы имеет место не путем
замены водородного атома кислородом, но вследствие замены
на атом кислорода эквивалентной водороду группы (NH2 или ОН),
особенно склонной к реакциям.
г) Замещение ароматически связанного водорода гидроксилом
Замещение ароматически связанного водорода
гидроксилом по схеме (1)—превращение, исключительно свой-
ственное производным антрахинонового и отчасти нафталинового
рядов и протекающее при действии концентрированной и дымящей
серной кислоты вместе с окислителями или без них. Реакция имеет
значение для получения ценных красителей и некоторых проме-
жуточных продуктов.
Антрахиноновые производные легко выносят без изменений
(вроде сульфирования) растворение в крепкой серной кислоте.
Вместе с тем прибавление к раствору антрахинонового производ-
ного в сериой кислоте перекиси марганца вызывает замену водо-
родного атома гидроксилом б3).
Синтез полигидроксилированных производных антрахинона
посредством обработки олеумом или окисления перекисью марганца
в среде серной кислоты получил по имени изобретателей, разра-
батывавших этот синтез на немецких фабриках, название реакции
Бона и Шмидта (R. Bohn, R. Е. Schmidt).
Таким образом путем обработки более низко гидроксилиро-
ванных продуктов получают три-, тетра-, пента-, гексаоксиантра-
хиноны, имеющие значение как красители. Температура при такой
обработке бывает различной, от 25—30° и до 180°, в зависимости
от желаемой степени гидроксилирования и природы окисляемого
продукта. Бывают процессы, кончающиеся образованием новых
хиноидных циклов с переходом в антрадихиноны.
Количество окислителя и температура влияют иа степень окисления. В присут-
ствии дымящей серной кислоты с высокой концентрацией SO3 оказывается воз-
можным гидроксилирование аитрахиноиовых продуктов при относительно низкой
температуре и без добавления окислителей.
Так, ализарин (I) переходит при обработке 10-кратным количеством олеума
(70% SO3) в течение 24—48 час. при 35—40° в продукт, выделяющий при обработке
щелочами, подкислении и нагревании тетраоксидериват антрахинона (1П). Как первый
продукт взаимодействия сериой кислоты и ализарина образуется повидимому средний
эфир тетраоксиаитрахииона (И) м):
358
Сернокислотные эфиры оксиаитрахиноновых производных омы-
ляются нагреванием с кислотами или растворением в щелочах и
последующим подкислением кислотой при нагревании. Промежу-
точно при действии щелочи образуются соли кислых эфиров,
например
(HO)2CuH4O2<o>SO2 (НО)2С14Н4О2<°^°зК
Далее выяснилась возможность аналогичных вышеописанным
превращений антрахиноновых производных без участия дымящей
серной кислоты, но лишь с концентрированной серной кислотой
в присутствии борной кислоты. При этом борная кислота, образуя
эфиры с гидроксильными антрахиноновыми замещенными, делает
окисляемое соединение устойчивым. Иногда прибавление борной
кислоты полезно и в обработке с участием окислителя (МпО2) В6).
Введение в практику окисления антрахиноновых производных,
проводимого в растворе серной кислоты, прибавки борной кислоты,
следовательно образование в реакционной среде эфиров, например
типа эфиров метаборной кислоты ROB:О, оказалось существенно
важным для успеха многих процессов окисления.
Эфиры борной кислоты характеризуются по их цвету и флуоресценции. Они
легко омыляются. Этерификация бориой кислотой гидроксила, сообщая реакцион-
ность определенным местам соединения в процессе окисления (например ализарина
в пурпурин или в 1.2.3.4-тетраоксиантрахиион) и в иных замещениях, имеет пови-
димому значение и в процессе конденсаций оксиаитрахииоиоиых производных из
бензольных производных. Так, при синтезе хииизарина из л-хлорфеиола и фталевого
ангидрида в качестве конденсирующего средства применяется серная кислота
вместе с борной.
Для получения из антрахинона 1.4-диоксипроизводиого (хииизарина) давний
германский патент указывает путь нагревания антрахинона с концентрированной
сериой кислотой в присутствии борной кислоты и ртутных солей. Недавний фран-
цузский патент развивает эту же идею, предлагая регулировать течение реакции
путем прибавления различных количеств ртути (0,5—4 г иа 1 000 г антрахинона) №).
Обработка дымящей серной кислотой, применимая к динитро-
замещенным антрахинона, дает начало сложной реакции, в резуль-
тате которой образуются также полигидроксильные замещенные
антрахинона. Особенно эффективно действует олеум, в который
прибавлена сера в количестве одного атома S на молекулу SO3.
Здесь, как и в описанном далее превращении 1.5-динитронаф-
талина, из серы и серного ангидрида получается нестойкое, легко
разлагающееся синевато-зеленое соединение S2O3 (недокись серы).
1.5- динитронафталин под влиянием нагревания до невысокой
(40—45°) температуры с олеумом (7—10°/о SO3) и серой переходит
в 1.4-аминоокси-а-нафтохинонимин
NO2 nh2
I О |
I NH I
no2 oh
359
каковой далее переходит (при кипячении разведенного кислотного
раствора) в ценный краситель—нафтазарин. Здесь, в окислительном
процессе, принимают таким образом участие ннтрогруппы (процесс
идет следовательно интрамолекулярно), так как констатированы
промежуточно-образующиеся такие продукты 57):
NO ОН NO
! I I
i
no2 он no он
Так же относится к нагреванию с дымящей серной кислотой н 1.8-диннтро-
нафталнн, давая 1-нитро-8-ннтрозо-4-нафтол. Наклонность ннтрогруппы окислять
прн нагревании с олеумом пара-стоящий водород затрудняет приготовление нз
полиннтроиафталннов полннитросульфокнслот. Прн сульфировании мононнтронаф-
талииа (олеум, лучше—хлорсульфоновая кислота, которая повидимому ие вызывает
описанных превращений) необходимо избегать высокой температуры.
Интрамолекулярное окисление может дать интересные результаты н при иной
обработке нитросоединеннй. Выше были уже упомянуты примеры превращения
ннтросоединений в оксннитросоединеиня при умеренном нагревании с порошко-
образным едким кали. Для нитробензола реакция была прослежена в больших раз-
мерах, и оказалось—отвечает уравнению в®):
5СвН5МО2 = 3C6H4(NO2)OH 4- С6НВ — N : N — С6Н3
О
Пока еще не имеется общего метода окислительного введения
гидроксильной группы на место водородного атома любого арома-
тического ядра. Овладение таким методом упростило бы и уде-
шевило получение фенольных производных, устранив необходи-
мость например предварительного получения сульфокислот или
хлорозамещенных. Разрешение этой задачи для некоторых частных
случаев в антрахиноновом и нафталиновом рядах описаны выше.
Ниже мы укажем на иные методы и попытки получения оксизаме-
щенных прямым окислением, частью они будут описаны в главе
о контактных реакциях.
Известен и практически интересен способ получения ализарина
из антрахинона при обработке последнего в щелочной среде в при-
сутствии сульфита окислительным агентом (солью азотной кислоты).
При этом превращении, весьма возможно, едкая щелочь прини-
мает участие не только в качестве среды, но и как реагент, содей-
ствуя вхождению гидроксила в виде ONa, как это рассмотрено
в главе VI °9): СО /\/\/\ 4-NaOH - \АА/ со /\/A\/0Na „ \н -|~NaOH> \/\/\/ С 1 он
360
он
I Н ONa ONa
4)Na +° .
2H2O
Опыты, проведенные M. А. Ильинским, A. H. Николаевой
и А. И. Перельман, указали, что в этом случае весьма суще-
ственно прибавление извести для связывания образующегося ализа-
рина в виде ализарата кальция и что сульфит натрия может быть
заменен с выгодой сернистым натрием.
Также в щелочной среде бензол был переведен в фенол при нагревании до
высокой температуры, причем окислительное воздействие производил кислород
воздуха прн содействии катализаторов-окислов некоторых металлов V н VI групп
периодической системы. Вряд ли в той форме, какая описана в американском
патенте 60) способ этот может претендовать на практическое использование, ио
несомненно он интересен как дающий отправную точку для дальнейших исканий
практического решения задачи.
Электрохимическому окислению ароматических углеводородов
посвящено много работ швейцарского ученого Фихтера с сотруд-
никами, выяснивших, что анодный кислород является одним из
наиболее сильных окислителей. При помощи него окисляется даже
бензол, но продуктов непосредственного окисления, каковым для
бензола Фихтер считает фенол, не удается изолировать, и они
окисляются дальше до щавелевой, муравьиной кислоты, углекислоты
и окиси углерода. При работе без диафрагмы может быть изоли-
рован гидрохинон, получающийся вследствие восстановления хинона
на катоде. Из фенола также электрохимическим методом окисления
можно получить гидрохинон. Толуол в растворе ацетона и водной
серной кислоты окисляется на аноде в бензальдегид и бензойную
кислоту, из которых изолируется лишь бензальдегид, вследствие
малой растворимости, между тем как бензойная кислота окисляется
тотчас дальше, в СО2 и воду.
При анодном окислении изомерных ксилолов обнаружено много
продуктов различного направления окислительного воздействия.
При работе с свинцовым (с осадком РЬО2) анодом и оловянным
катодом с диафрагмой и без нее в среде разведенной серной
кислоты, в продуктах реакции преобладают образованные окисле-
нием метильной группы соответственного ксилола толуиловый
альдегид (I) и изомерная фталевая кислота (П), наблюдаются также
хиноны, образованные через посредствующую стадию фенола (кси-
ленола) (III), с отщеплением метильной группы (IV) или с ее сохра-
нением, но перегруппировкой; в последнем случае промежуточно
образуется диметилхинол (VI), перегруппировывающийся в диме-
тилгидрохинон (VII). Окислительной конденсацией ксиленола (де-
гидрогенизацией) получается и дифенол (диксиленол) (V).
361
Схема окислительных превращений для .«-ксилола например такова:
С6Н4(СНО)ЧСН3)з -> С6Н4(СО2Н)1(СН3)з СвНДСОгН)1;3
При окислении на аноде (с осадком РЬО2) в серной кислоте фенола констатиро-
вано образование гидрохинона и пирокатехина, которые окисляются далее в окси-
хинон 61).
Нафталин дает при анодном окислении а-нафтол в качестве одного из первых
продуктов окисления. Сам а-нафтол прн анодном окислении (РЬО2,1О°/о-ная H2SO4)
дает в зависимости от плотности тока -или а-нафтохинои и фталевую кислоту, или
а-нафтогидрохинон вместе с аа'-динафтолом 62).
В настоящее время электрохимический метод окисления бензола и его гомоло-
гов не может рассчвтывать на практическую приложимость для получения гидро-
ксильных замещенных.
О. ГО. Магидсон и Н. Преображенский, окисляя фенол 2%-ной пере-
кисью водорода в водном растворе в присутствии железного купороса обнаружили
образование заметных количеств диоксипроизводных (пирокатехина и гидрохи-
нона) 63). Е. Гольдгаммерв продуктах окисления фенола нашла следы гидро-
хинона и в значительных количествах пирокатехин и пирогаллол 61).
д) Превращения > СН -> > СО.
Второе из возможных при окислении превращений группы СН,
именно превращение в СО-группу (см. выше), находит большое
применение для получения некоторых хинонов и главным образом
технически важного антрахинона.
Хиноны и ряд производных, имеющих строение хинонов, так
называемые хиноиды, интересны для красочной промышленности
как аналогичные по структуре некоторым красящим веществам, ка-
ковы хинониминовые красители, или как дающие представление
об одной из возможных реакционных форм красителей, например
в ряду азокрасителей, нитро- и нитрозокрасителей. Кроме того не-
замещенные хиноны в последнее время становятся интересными
объектами синтеза кубовых красителей.
Простейший хинон бензольного ряда — л-хинон (I) и нафтохи-
ноны «-(II) и Р-(Ш)
О О
362
о
готовятся практически окислением не углеводородов, а их заме-
щенных.
Прямое окисление бензола в хинон посредством анодного кислорода при элек-
тролизе французским исследователям удалось проводить с 65%-ным выходом, пере-
водя в катодном пространстве хинон в гидрохинон.
Электролиз проводился в среде, содержащей 25% серной кислоты, 33% уксус-
ной кислоты и бензол, в присутствии 1—1,5% мелкого порошка сернокислого
свинца. Электроды—свинцовые, разделенные диафрагмой. Сильная мешалка эмуль-
гирует анолит. Температура 24°. По достижении содержания хииона в бензоле в 1%
он тотчас переводится в катодное пространство, для превращения в гидрохинон.
Повышение температуры анолита содействует быстрому снижению выхода хи-
нона ю).
При окислении бензола перекисью свинца в среде 30%-ной серной кислоты по
описанию недавнего немецкого патента также получается хинон №).
Обычно при производстве хинона пользуются анилином как
исходным материалом. Окислителем служит хромовая кислота —
из двухромовокислой соли с серной кислотой. Окислительный про-
цесс можно представить наиболее просто соответственно уравне-
ниям:
3C6H5NH2 + 2Na2Cr2O7 + 8H2SO4 =
=30 :С6Н4: NH -1- 2Na2SO4 + 2Cr2(SOJ3 +11H2O. (1)
30: CGH4: NH -f- 3H2O = 30: C6H4:0 + 3NH3.
Однако выход хинона не превосходит 88 — 89% от теорети-
чески рассчитанного, и исследование Виллыптеттера объясняет
это обстоятельство. Именно, при окислении анилина хромовой ки-
слотой образуется сначала известный в технике крашения анили-
новый черный, который после перехода через ряд промежуточных
стадий имеет строение
и при последнем этапе окисления и гидролиза образует хинон лишь
на счет своих га-двузамещенных бензольных ядер, т. е. на счет 7/8
из своих бензольных остатков 67).
Введение бихромата в раствор анилина в серной кислоте прак-
тикуется при обычной температуре в два приема. Общее коли-
чество бихромата на 50°/о превосходит рассчитанное по урав-
нению (1).
Недавно в немецком патенте описан прием окисления анилина в хинон, при ко-
тором расход бихромата снижен до % с заменой остальной части перекисью мар-
ганца. Окисление анилина проводится также в растворе серной кислоты, в который
прибавляется сначала перекись марганца (2 части) и затем 1 часть бнхромата. Выход
хиноиа по патенту 90% «в).
Получение отходов синтеза, заключающих одновременно соли марганца и хрома—
слабая сторона такого процесса, так как для таких отходов труднее найти какое-
либо применение нли возможность регенерации, чем для отходов несмешанных
солей.
Недавний немецкий патент описывает способ окисления анилина в хинон по-
мощью хромовой кислоты, проводимый в растворе не серной, а разведенной азотиой
кислоты. Способу этому приписывается то преимущество, что ои позволяет легко
регенерировать хромовую кислоту путем окисления солей окиси хрома за счет
363
азотной кислоты и окислов азота, выделяющихся при концентрировании отработан-
ной минеральной кислоты. Если учесть одиако, что соли окиси хрома сами по себе
имеют большое техническое значение и получаются во всяком случае раскислением
хромовокислых солей, то вряд ли можно рассчитывать на внедрение в широкую
практику этого способа окисления, предложенного для получения и автрахинона,
особенно приняв во внимание необходимость сложных установок по улавливанию
и регенерации в азотную кислоту выделяющихся оквслов азота 6»),
Наиболее простой по химизму процесс получения хинонов, в том
числе и бензохинона, — окисление с потерей водорода соответ-
ствующих гидрохинонов, если они доступны
/\ /ОН
/\/ //\; о
— н2 О:
В качестве окислителей употребительны хромовая кислота (хро-
мовая смесь), хлорное железо, перекись марганца с серной кисло-
той. В тех случаях, когда хинон мало устойчив в водной среде,
можно пользоваться для окисления средой безводного раствори-
теля (бензол, его гомологи и т. п.) и как окислителем — перекисью
свинца, окисью серебра. Такая система окисления дает возмож-
ность получения и хиноноподобных (хиноидных) соединений, ка-
ковы хинонимины, например O:C6H4:NH и т. п.
Образование хиноидных соединений из соединений типа гидро-
хинонов (хотя бы один или два остатка ОН были в последних за-
мещены аминогруппами или замещенными аминогруппами или угле-
родными группами), употребительно в практике получения краси-
телей из так называемых лейкосоединений красителей. Так, окисле-
ние лейкоиндоанилина (и-окси-п'-амино-л-метилдифениламина) (I)
приводит к индоанилину (II).
Подобны окислительные процессы образования и других более
сложных хинониминовых красителей.
Четырехлорозамещеиное хинона—хлоранил получается воздействием хлорирую-
щих и одновременно окисляющих агентов на достаточно реакционные бензольные
замещенные, особенно же n-диаминозамещеиные. Так, его можно приготовить из п- ни-
троанилина по схеме ’°);
I I --------*
' I КСЮз
o2n/\/ ‘НС1>
С1
------>
SnCl2
364
Cl
Cl
В недавнем английском патенте описан способ получения хлоранила (соотв. бром-
аиила) при помощи обработки хлором (соотв. бромом) нитрозофенола (т. е. хиион-
оксима) в кислом растворе при повышенной температуре п).
Хинон применяется для получения гидрохинона — интересного
как фотографический проявитель и как исходный материал для
синтеза некоторых промежуточных продуктов (хинизарин).
С ароматическими аминами хинон и его замещенные реагируют,
давая арилидохиноны, из которых некоторые сами по себе обла-
дают свойствами кубовых красителей или могут быть превращены
в ценные сернистые красители.
Из двух одноядерных нафтохинонов а-нафтохинон получается
с выходом около 5О°/о как продукт окисления нафталина хромовой
кислотой в растворе уксусной кислоты при обычной темпера-
туре 72).
Лучшим методом его получения является однако окисление
1.4-аминонафтола или 1.4-нафтилендиамина, получаемых как про-
дукты восстановительного расщепления а-нафтольных (соотв. а-наф-
тиламинойых) азокрасителей, например:
N: NC6H4SO3H
/\/'\
------->
SnCl,
(НС1)
+ H2NC6H4SO3H------>
окисле-
ние
Окисление лучше проводить посредством хромовой кислоты.
Подобным же путем р-нафтохинон образуется при окислении
1.2-аминонафтола, получаемого как продукт восстановительного
расщепления р-нафтольного азокрасителя или восстановления ни-
трозо-р-нафтола.
Антрацен вследствие своеобразной реакционности его мезоато-
мов (9.10) легко переходит при всякого рода окислительных воз-
действиях в антрахинон.
+ 30
-н2о
365
Наиболее употребительным и интересным технически приемом
окисления является применение хромовой кислоты в растворе сер-
ной кислоты (приблизительно 50%-ной).
Реакция окисления отвечает уравнению
CJ4H10-}-Na8Cr2O7-}-4H2SO4=CuH8O2-]-Cr2(SO4)34-Na2SO44-5H2O. (1)
Антрацен, применяемый для окисления, содержит различное ко-
личество примесей, оставшихся в нем после предварительного вы-
деления из антраценового масла и очистки.
В большей мере эти примеси состоят из карбазола и изомер-
ного антрацену фенантрена. В практике советской промышленности
имело место окисление 80%-ного и 45%-ного антрацена. Разу-
меется само собой, что наиболее выгодно применять антра-
цен наиболее чистый, и в западноевропейской промышленности
окислению подвергается антрацен с содержанием свыше 95% чи-
стого. Примеси антрацена вызывают добавочную трату окисли-
теля, затрудняют очистку полученного сырого продукта окисления
и загрязняют ценный отход — раствор солей хрома, не говоря
о ряде технологических затруднений, вызываемых ими, в виде по-
нижения скорости фильтрования в различных стадиях фабрика-
ции и т. п.
Подготовка антрацена к окислению имеет целью придать сырому продукту наи-
более мелкую структуру. Это достигается нли перегонкой сырого антрацена с пере-
гретым водяным паром, или иногда растиранием его в шаровой мельнице.
Для окисления измельченный антрацен переводится в чан с вод-
ным раствором двухромовокислого натрия (с избытком против тео-
ретически рассчитанного и тем большим, чем менее чист антрацен).
При повышении температуры до 90—100° в смесь прибавляют
50°/о-ной серной кислоты и поддерживают в течение 1—2 часа эту
температуру смеси. Сырой продукт окисления отделяют от рас-
твора, содержащего сульфат хрома, промывают водой и, если надо—
под конец содовым раствором, собирают и высушивают осадок,
содержащий антрахинон в количестве, отвечающем окисленному
антрацену. Отделение антрахинона от примесей в сыром продукте
основано на значительно большей стойкости антрахинона сравни-
тельно с примесями при обработке концентрированной серной ки-
слотой при температуре 100—105°. При этих условиях (серной
кислоты берут в 2—2% раза больше по весу сырого антрахинона)
примеси антрахинона переходят в растворимые сульфокислоты
и могут быть отделены от неизмененного антрахинона фильтрова-
нием, причем надо учитывать необходимость получения антрахи-
нона в таком кристаллическом состоянии, чтоб фильтрование не
затруднялось (обычно: медленное прибавление воды, медленное
понижение температуры). Промывка водой и содовым раствором
позволяет получить антрахинон достаточно чистый от примесей
(96—95°/о-ный) и пригодный для многих технических применений.
Если требуется антрахинон еще более чистый, то полученный про-
дукт можно перекристаллизовать из органических растворителей
(например хлорбензол, смесь крезолов) или из горячей серной
кислоты. Как метод окончательной очистки применяется и пере-
гонка антрахинона с перегретым паром или сублимация.
366
При получении антрахинона этим методом остается ценный
отход в виде раствора сульфата хрома, который идет или как та-
ковой на кожевенные фабрики для хромового дубления, или пере-
рабатывается на гидрат окиси хрома, соотв. другие соли хрома
(ацетат, бисульфит, формиат), находящие применение в текстиль-
ной промышленности 78).
При окислении очень чистого антрацена (96— 97%-ного) хромовой кислотой хро-
мовые отходы можно подвергать регенерации путем электролиза (анодным окисле-
нием), что н практикуется местами за границей.
Регенерация возможна, но вероятно невыгодна при окислении в растворе раз-
веденной азотной кислоты. Регенерация хромовой кислоты в прежнее время за
границей проводилась осаждением окиси хрома известью и прокаливанием обра-
зующегося осадка в муфельных печах.
Окисление антрацена до антрахинона возможно и другими методами. Гофман
описывает любопытный прием окисления азотнокислыми н хлорноватокислыми со-
лями в присутствии большого избытка сернокислого или хлористого магния. При-
меняются содержащие кристаллизационную воду магниевые соли (в 3—5 кратном ко-
личестве от ннтратов илн хлоратов). Процесс окисления главным образом проходит
при 120—150°, затем масса нагревается до 210° и даже до 290—300° (до возгонки
антрахинона) 74).
В водной суспенсии посредством раствора хлорноватистонатриевой соли при
добавке следов OsO4 окисление проходит уже при обыкновенной температуре 75).
Суспендированный в растворе хлората железа (также хрома, никеля, кобальта,
марганца) или в таких растворах, в которых хлораты могут образоваться (например
смеси NaClO3 и FeCIg), антрацен окисляется в антрахинон прн нагревании раствора
до кипения 76).
В растворах или суспеисиях в органических растворителях окисление антра-
цена удается посредством азотной кислоты при поддержании в течение нескольких
часов температуры 15—35°. Аитрахинои по словам патента получается в состоянии
высокой чистоты, азотиую кислоту частично можно регенерировать. Возможно при-
менение катализаторов—ртутных солей Т!).
Действием на антрацен окислов азота при 100—200° можно также произвести
окисление углеводорода в антрахинон. Антрацен выгоднее применять в тесной смеси
с разводящими веществами, например пемзой или порошком асбеста и окислов ме-
таллов, нейтрализующих азотную кислоту та).
Окисление антрацена окислами азота в растворах или суспенсиях в органиче-
ских растворителях особенно хорошие выходы дает прн участии ртутных солей.
Температура взаимодействия 100—110° ге).
В немецких патентах имеются указания на метод окисления антрацена посред-
ством солей Fe’" в нейтральных или кислых растворах 8°) н окисления в щелочных
растворах или суспенсиях прн нагревании с кислородом (нли иоздухом) в присут-
ствии таких например катализаторов, как окись меди м).
Электрохимический метод окисления антрацена в антрахинон
был описан 35 лет назад в немецком патенте. В настоящее время
имеется уже значительная литература по этому способу, в том
числе и исследования советских химиков Флоренского и Ме-
телкина 82).
Наилучшим приемом для такого окисления, по опытам послед-
них названных авторов, оказывается проведение электролиза в среде
серной кислоты с концентрацией в 35%. К раздробленному антра-
цену перед суспендированием добавляется небольшое количество
ацетона и уксусной кислоты. Окислителем является собственно
анодный кислород, но с целью более полного его использования
необходимо участие катализаторов — переносчиков кислорода—
в виде солей таких металлов, которые легко меняют свою валент-
ность под влиянием окислителей и восстановителей. Из таких ока-
зался вполне пригодным двухромовокислый калий или натрий
367
в 3%-ной концентрации. Температура электролита—около 1ОСР,
и электролит необходимо энергично размешивать. Анодом служит
чистый свинец, покрытый слоем перекиси свинца, катод жела-
тельно иметь из металла с малым водородным перенапряжением
и не разъедаемого серной кислотой. Удобна форма электродов
в виде коаксиальных цилиндров с анодом внутри; электроды отде-
ляются один от другого пористой кислотоупорной диафрагмой.
Плотность тока на аноде 10 А^дм^. Рабочее напряжение 3—5 V.
Интересно, что при меньшей плотности тока на аноде (1,2 А/йм2)
при катализаторе Се’" получается в качестве продукта окисления
не антрахинон, а антрагидрохинсгн. Очевидно реакция идет в две
фазы:
Н ОН
Окислительный процесс на аноде можно скомбинировать с вос-
становительным на катоде например для восстановления нитросо-
единений, чем конечно значительно повышается эффективность
всего электролитического процесса 83).
Антрахинон имеет исключительно важное значение в практике
производства особо прочных сортов красителей как протравных, так
и кислотных и кубовых. Окислительный метод получения не является
единственным, имеющим практический интерес. Ниже мы опишем
метод синтетического образования антрахинона (см. главу XV).
Окисление изомерного антрацену фенантрена привлекло несра-
вненно меньше внимания, чем окисление антрацена. Больше всего
это объясняется отмеченным уже малым до настоящего времени
практическим значением фенантрена. Окисление фенантрена прежде
всего приводит к фенантренхинону- (9.10).
СО СО
/ 9 ю\ / \
/»
7
\б 5
Окисление хромовой кислотой, так же как в случае антрацена,
дает фенантренхинон. Окисление хромовой кислотой, по крайней
мере в растворе уксусной кислоты, протекает труднее, чем с антра-
ценом. Имеются описания способов окисления фенантрена посред-
ством солей марганца (электролитически окисленных), посредством
хлорноватокислого натрия и уксусной кислоты в присутствии со-
лей рутения и электролитического окисления в присутствии соеди-
нений церия 84).
Очистка фенантреихинона легко осуществляется посредством получения его
кристаллического бисульфитного соединения C14H8O2-NaHSOa. 2НаО, регенерирую-
щего от действия гидролизующих агентов (щелочей и кислот) фенантренхинон.
368
Недавней работой итальянских химиков установлено, что обработка фенантрена
перекисью водорода в растворе уксусной кислоты дает фенантренхинон. Подоб-
ная же обработка антрацена дает антрахинон и немного 9.9'-днантрона
СО
е) Превращения R(R')CH2 -> R(R')CO
Этн превращения имеют значение для получения из аценафтена
так называемого аценафтенхинона (собств. дикетона):
Такое окисление выполняется действием хромовой кислоты
в растворе уксусной и сопровождается образованием ангидрида
нафталевой кислоты 88):
НО2С СО2Н
Аценафтенхинон дает с бисульфитом кристаллическое соеди-
нение состава С12Н6О2 • NaHSO3 • 2Н2О.
ж) Окислительные превращения ароматических соединений
в хиноиды. Интермолекулярные окисления
Среди продуктов, получаемых методом окисления, имеется
группа таких производных, которые, сохранив углеродный скелет
исходного соединения, отличаются от него перераспределением
двойных связей по типу хинона. В то же время такие соединения
отличаются от вышеупомянутых хинонов отсутствием кислорода
в одной из двух карбонильных групп. Их характерные группы —
замещенные карбонила в виде C~NH, C~NC1 и т. п. Такие
соединения носят частные названия хинониминов, хинонмонохло-
риминов, хинондихлордииминов и пр., объединяясь в общем наиме-
новании „хиноидов*1. Они имеют значение в качестве обычно не-
стойких и весьма реакционных промежуточных продуктов для по-
лучения главным образом азиновых и азониевых красителей. Хи-
24 Зак. 2611. — Н. Ворожцов
369
нонхлоримины и хинондихлордиимины готовят из «-аминофенолов,
соотв. n-диаминов, одновременным окислением и хлорированием.
Наиболее употребительным окислителем, понятно, является хлор,
который вводится во взаимодействие или в виде газа или, чаще,
в виде солей кислородных кислот хлора, например гипохлорита
натрия NaClO, или в виде дешевой хлорной извести СаС12О.
Зная количество активного хлора в таком окислителе, стехио-
метрию процесса можно рассчитать по уравнению, иапример
H,N • R • NH2 + 6Cl = C1N: R: NCI -f- 4HC1.
В. Г. Шапошниковым было предложено применять при получении нафто-
хинондихлордиимииа вместо хлориой извести хлориоватонатриевую соль, причем
реакция идет по схеме 87)
H2N• R • NH2 + 6НС1 + NaClO3-> ON: R: NCI + NaCl 4-3H,O + 4HC1.
Этот способ ие может быть применен в тех случаях, когда энергичное хлори-
рование влечет за собой замещение хлором водородных атомов самого ядра.
Хиноидные же соединения, именно—замещенные ароматиче-
ским остатком в N хинонимины, могут быть получены совместным
окислением двух ароматических веществ, из которых одно заклю-
чает аминогруппу, а другое реактивирующую ОН- или NH2- (или ее
замещенную) группу. Получающиеся таким образом окрашенные
индофенолы, соотв. индамины, не имеют как конечные продукты
самодовлеющего значения, но очень интересны как промежуточные
соединения при получении весьма ценных красителей, больше
всего сернистых и отчасти тиазиновых, оксазиновых и азониевых.
Формула строения простейшего индофенола (окисление л-фе-
нилендиамина с фенолом или л-аминофенола с анилином):
О : С6Н4 : N • C6H4NH2 *
и индамина (окисление л-фенилендиамина с анилином)
HN: С6Н4: N • C6H4NH2.
Окислительный синтез индофенола может быть осуществлен или в щелочном
растворе действием соли хлорноватистой кислоты, причем соль отдает свой атом
кислорода, иапример
NaOCl-> NaCl 4-0,
или в щелочном же растворе действием красной кровяной соли K3Fe(CN)e, пере-
ходящей в желтую соль K4Fe(CN)e, например
H2NC6H4NH2 4- / ОК 4- ЗКОН 4- 4K3Fe(CN)6) =
\nhcoch3
= H2NC6HiN:<^ ^>:0 4-4K4Fe(CN)e 4-ЗН2О.
\nhcoch3
Окисление может быть проводимо и в кислой среде действием хромовой кис-
лоты (серная кислота и бихромат). Так как хиноиимииовые производные типа
индофенола (и индамина) мало стойки вообще, особенно же по отношению к раз-
веденным минеральным кислотам (в достаточно крепкой серной кислоте иидофе-
* Точнее было бы назвать это соединение индоанилииом, так как собственно
ивдофенол отвечает формуле О: С6Н4: N • СоН4ОН.
370
нолы можно получать с хорошими выходами), то их, имея в виду их назначение
в производстве сернистых красителей, переводят тотчас после приготовления
в лейкопроизводные обработкой сернистым натрием.
Эти лейкосоединеиия являются производными диариламина, например
ZCH3 ZCH3
/—NH2 + НО-^ >.NH\ >-Nh3
Имеются наконец методы производства
окислителя.
Хинонхлоримид, например (II),
индофенолов без участия внешнего
получаемый из n-аминофеиола (I) окислением его в кислом растворе хлорноватистой
кислотой,—в щелочной среде легко сочетается например с фенолом, нафтолом и их
замещенными в индофенолы
О:</ \:NCI + НZ:N'\ /~он
Работами Гиббса и сотрудников установлено, что этот метод обеспечивает полу-
чение наиболее чистых индофенолов, могущих быть примененными в качестве
индикаторов при электропотеициометрическом исследовании окислительно-восста-
новительных процессов 88).
Другим методом образования индофенолов без участия внеш-
него окислителя пользуются, применяя хинонокснмы (соотв. оксимы
хинониминов). При этом окисление происходит за счет кислорода
нитрозо- (соотв. изонитрозо-) группы одного из ингредиентов
реакции. Реакционная смесь состоит обычно из нитрозофенола
(хиноноксима) и аминосоединения со свободным пара-местом (по
отношению к азоту).
Реакция протекает например по схеме:
\ О) (4)
О:< > :N • ОН + Н • R • NH • Rt —
X —/ —
\ (1) (4)
---->0:^ / : N • R • NH • Rx (1)
где R — двувалентный ароматический радикал типа фенилена, a Rt —
алкил, арил или водородный атом.
Выдающееся значение приобрели получаемые сказанным спосо-
бом индофенолы из карбазола, соотв. его замещенных, и л-нитро-
зофенола. Карбазол реагирует при этом как вторичный амин аро-
матического ряда и дает индофенол
NH
24*
371
при подходящей обработке в сернистой плавке дающий весьма
ценный кубовый краситель — гидроновый синий, первый по вре-
мени появления из гидроновых красителей. Их синтез привлек
внимание химиков к карбазолу как важному исходному материалу
для получения прочных красителей.
Схема получения индофенола из нитрозофенола (1) указывает на необходи-
мость при этом водоотнятня. Соответственно этому реакция проводится или в до-
статочно крепкой серной кислоте (70—100%-ной) или в ледяной уксусной кислоте
в присутствии крепкой серной кислоты. Температура реакции невысока, 20—30э
По окончании реакции продукт изолируют выливанием смеси на лед и фильтро-
ванием.
з) Окисления с изменением углеродного скелета соединений
На первом месте среди названных превращений должны быть
поставлены такие, которые сопровождаются разрушением имею-
щегося углеродного кольца и образованием из его углеродных
атомов новых заместителей ароматического ядра. Речь идет глав-
ным образом о нафталине и других системах с конденсирован-
ными ароматическими циклами.
Выше мы видели примеры таких систем, как аценафтен (I) и
фенантрен (IV)
НО2С
СО2Н
из которых первый переходит при окислении в аценафтенхинон (II)
и в двуосновную нафталевую кислоту (III), между тем как фенан-
трен образует сначала фенантренхиной (V) и при дальнейшем
окислении дифеновую кислоту (VI). Нагревание со щелочью фе-
нантренхинона приводит (с вероятным промежуточным образо-
ванием соли дифеновой кислоты) к отщеплению карбоксильных
групп и к количественному превращению в дифенил 8Э) (VII):
ОС СО
+ 2NaOH
NaOOC
COONa
+ 2NaOH
\ + 2Na2CO3
372
Незамещенный нафталин при окислении в подходящих усло-
виях дает два главных конечных продукта: фталевую и фталоно-
вую кислоты, согласно схеме:
.СОСООН
' 47 +80
-СО2-Н2О
Х//^СООН
СН
+ 90
— 2СО2 — Н20
/^/СООН
1
\/\соон
Первый улавливаемый продукт окисления нафталина, как упо-
мянуто выше, — нафтохинон, который при дальнейшем действии
окислителя дает вышеназванные продукты уже с разрушением не-
сущего кислород углеродного кольца, например
СО
+ 50
-со2
/х/СОСООН
^/^СООН
Окисление нафталина во фталевую кислоту сыграло существен-
ную роль в первом технически осуществленном в больших разме-
рах синтезе индиго (на Баденской анилиновой и содовой фабрике),
так как фталевая кислота сравнительно легко превращается во
фталимид
и далее в антраниловую кислоту
/у/СООН
\/\nh2
(см. ниже), являющуюся исходным продуктом для получения фе-
нилглицин-о-карбоновой кислоты и индиго.
Технически удобным и выгодным методом окисления, откры-
тым случайно Заппером 8°), оказалось нагревание нафталина
с моногидратом серной кислоты до высокой температуры (250—
300°) в присутствии ртути или сернокислой ртути. В этих усло-
виях серная кислота раскисляется до сернистой, нафталин же пе-
реходит во фталевую, частью — в сульфофталевую, кислоту. Окис-
ление нафталина идет при температуре выше 200° и без ртути,
за счет серной кислоты, но присутствие ртути значительно уско-
ряет процесс.
373
По наблюдениям Мильбауэра и Немца серная кислота в процессах окис-
ления алифатических соединений раскисляется нацело до SO2. Ароматические угле-
водороды сгорают с Н25О.,, тем более, чем более боковых групп, содержащих
углерод, находятся у бензольного ядра, так что можно составить ряды возрастания
окислительного воздействия:
1) нафталин — аценафтен — антрацен,
2) дифенил — флуорен — фенантрен 91).
Наиболее существенно для повышения выхода фталевой кис-
слоты— быстрое удаление ее или ее ангидрида из сферы взаимо-
действия 9'2).
Этот метод получения фталевой кислоты имеет в настоящее
время лишь историческое значение, будучи вполне вытесненным
способом каталитического окисления нафталина воздухом (см.
главу XVI). Заслуживает быть отмеченным факт, что практика
окисления нафталина серной кислотой, потребляя огромное коли-
чество серной кислоты (9 мол. на 1 мол. нафталина), освобождала
соответственные массы сернистого ангидрида. Необходимость их
утилизации вызвала быстрое освоение контактного производства
серной кислоты. Таким образом развитие органическо-химического
производства (индиго через фталевый ангидрид) отразилось на пе-
реходе основного из неорганических производств на высшую сту-
пень. Примеры такого взаимодействия двух отраслей производств
в их росте и развитии не редки в истории химической техники.
Окисление нафталина посредством хромовой кислоты или перманганата в кис-
лом растворе оказывается слишком энергичным для того, чтобы получать хорошие
выходы фталевой кислоты, и часто приводит к сгоранию всей молекулы нафта-
лина в СО2 и Н2О. Окисление же перманганатом (КМпО4) или манганатом (КгМпО^)
без подкисления, т. е. фактически в щелочной среде, каковая образуется в усло-
виях окислительного процесса при распаде молекулы окислителя, дает смесь из
фталоновой и фталевой кислот с преобладанием фталоиовой 93). Интересно, что при
окислении тетрагидроиафталина, в зависимости от реакции среды, получается либо
фталевая (КМпО4 в щелочной среде) либо фталоновая кислота (в кислой среде) 91).
Подобное течение окисления возможно при участии гипохлорита с примесью пер-
манганата (один гипохлорит ие окисляет) 93).
Замещенные нафталина, нафтолы, нафтиламины, сульфо- и
нитропроизводные, также фенантрен и антрацен дают при нагре-
вании с концентрированной или дымящей серной кислотой в при-
сутствии ртути фталевую и сульфофталевые кислоты в6). Иные
методы окисления нафталиновых замещенных дают однако другие
продукты.
Так, при обработке а-нитронафталииа в щелочной среде с раствором марган-
цовокислого калия получается фталоновая кислота. При обработке того же нитро-
продукта хромовой кислотой наблюдалось образование нитрофталевой кислоты 97).
При нагревании сухой смеси из нафтола, едкого натра, и окиси меди обра-
зуется смесь бензойной и фталевой кислот. Такая же смесь получается при нагре-
вании при высокой температуре и давлении иитронафталина с едким натром в при-
сутствии окиси меди 98). В этом последнем случае сначала происходит окисление
водорода того же ядра, где стоит нитрогруппа, с образованием смеси двух нитро-
нафтолов (1.2 н 1.4), и эти последние окисляются затем в бензойную и фталевую
кислоты. В этой стадии окисления очевидно принимают участие иитрогруппы, так
как среди продуктов реакции констатирован аммиак.
Интересные продукты получаются при окислении р-нафтола и
некоторых его производных.
При обработке ^-нафтола (1) в щелочной среде марганцовокислым калием про-
дуктом окисления оказывается в первую очередь о-карбоксикоричная кислота (11),
374
затем в сильнощелочной среде при действии окислителя образующая с {3-нафтолом
продукт окислительной конденсации: 2’-карбокси-4-фенил-5.6-бензокумарин (III) "):
,^/СООН
zk z z°H z СООН 7 г
/\/Х/ /\/
Замечательное окислительное расщепление в щелочном растворе нитрозо-₽-наф-
тола (I), вызываемое обработкой ароматическим сульфохлоридом, описывает гер-
манский патент 1923 г., именно, превращение в о-цианкоричную кислоту [моди-
фикацию с Спл. 137° (II)] 10°):
NOH
При дальнейшем нагревания щелочного раствора цианидная группа гидролизуется
до карбоксильной, и образуется упомянутая выше о-карбоксикоричная кислота.
Подобные описанным выше окислительные превращения с разрушением цикла
известны и в антрахиноновом ряду.
Таковы например окисления диоксиантрахиноиов (1.2- и 1.4-), производимые
железосинеродистой солью [K3Pe(CN)e] в щелочном растворе 101):
Отметим возможность образования из 1.4-диоксиантрахинона (хинизарииа)
при иагреваиии со слабыми щелочами (раствором соды, лучше с пиперидином)
также путем окисления, но с конденсацией 1.4.1'.4'-тетраокси-2.2'-диантрахино-
нила 102):
ОН ОН ОН
СО | СО | | СО
ОН ОН ОН
375
Насколько различно могут относиться к одинаковым химическим воздействиям
близкие по строению производные, видно из того, что пурпурин (I), обладающий
только одной лишней гидроксильной группой по сравнению с хинизарином и али-
зарином, распадается при окислении K3Fe(CN)e в щелочном растворе по месту
среднего ядра. Продуктами являются уже не конденсированные ароматические
производные 103):
Нарушение углеродного скелета соединения при окислении
может происходить и без размыкания кольца, но с уменьшением
числа углеродных атомов соединения. При этом для убыли угле-
рода имеются различные возможности, например переход части
его атомов в углекислый газ при разрыве двойной связи в боко-
вой цепи и образование по месту ее новых кислородсодержащих
соединений. При этом если двойная связь в исходном соединении
делила молекулу на две равнозначные половины (пример — стиль-
беновые соединения), то в результате окисления образуются две
молекулы с половинным против исходного содержанием углерода.
Хорошим окислителем для образования кислородсодержащих за-
местителей на месте бывшей двойной связи является соль марган-
цевой кислоты (КМпО4) без прибавления кислоты или с прибавкой
щелочи.
Известная Гофмановская реакция образования первичных
аминов из кислотных амидов — также один из примеров окисления
с потерей углерода в виде СО2 и в применении к фталимиду
(гидратация которого дает моноамид фталевой кислоты — фтала-
миновую кислоту) открывает верный путь для производства техни-
чески важной (для индигоидных красителей) антраниловой кислоты.
Окислителем при этом служит соль хлорноватистой (или бромно-
ватистой) кислоты в щелочном растворе (избыток щелочи — суще-
ственный фактор для получения хороших выходов).
Раствор натриевой соли хлорноватистой кислоты (гипохлорита) готовят, пропу-
ская рассчитанное количество хлора в 10%-ный раствор едкого натра прн охлаж-
дении водой. Правильно приготовленный раствор заключает в 100 г до 5,5 г де-
ятельного хлора, отвечающего 5,78 г NaClO или 1,24 г кислорода 1м). Количество
деятельного хлора эквивалентно количеству кислорода хлорноватистокислой соли.
Определяется содержание деятельного хлора объемным путем или по окислению
мышьяковистой кислоты или иодометрически (по количеству выделившегося иода).
Для сохранности раствора гипохлорита необходимо присутствие хотя бы незначи-
тельного избытка свободной щелочи, например в отношении
2NaOH + Cl2 -J-1/4 NaOH.
376
Схема реакции образования антраниловой кислоты такова:
+ Н2О -f- Cl2 + 4NaOH
----> -------------.----->
^/^СООН
z\/NHa
4- Na2CO3 + 2NaCl + 2Н,0
\/\соон
Промежуточно образуются при этом продукты охлорения амида,
которые претерпевают перегруппировку, сопровождающуюся отще-
плением СО2:
ONa
/С1 |
zCONH2 ,CON< /C:NC1
/ \/ /\/ \Na / X/
'Z/'^COONa \/\3OONa
^/^COONa
Cl
/N: C (ONa) yx ,N: C(ONa)2
+H2O
+H3o—Na2CO3
^/^COONa ^/^COONa
/4yNH2
^/^COONa
При отсутствии избытка щелочи происходит превращение без выделения уголь-
ной кислоты и с образованием нзатовой кислоты 103). Именно, при взаимодействии^
эквимолекулярных количеств фталимида, едкого натра и гипохлорита получают
натриевую соль изатовой кислоты(1). По Гребе и Ростовцеву 106), течение
реакции здесь таково:
т
+ NaOH + NaOCl
,con/C1
\ ^Na
xCOONa
+н.о
----->
NT л
------->
— NaCl
zNa
—co
СбН<\ I
j\co—о
377
Потеря углерода в виде углекислоты может иметь место у не-
которых продуктов окисления (карбоновых кислот) уже без уча-
стия добавочных окислительных воздействий. Наиболее интересно
в этом отношении превращение фталевой кислоты в бензойную,
так как оно приводит к практическому решению задачу получения
чистой бензойной кислоты.
Нагревание солей щелочноземельных металлов и фталевой
кислоты, в частности кальциевых солей, самих по себе или с до-
бавкой воды, под давлением, дает возможность получения бензой-
ной из фталевой кислоты
/^/СООН /^/СООН
I — СО2
\/Ч\СООН
Присутствие щелочных соединений в последнем случае, так же
как наличие катализаторов, содействующих отщеплению СО2
(соединения А1 или элементов 6-й группы периодической си-
стемы Сг и W), благоприятствует такому превращению.
Так, по описанию английского патента нагреванием расплавленного фталевого
ангидрида (100 ч.) в присутствии фталата хрома (3 ч.) и фталата натрия (2,35 ч.)
до 240° с введением (50—20 ч.) водяного пара осуществляется желаемое превра-
щение, и отгоняется с паром сначала непрореагировавшая фталевая кислота (соотв.
ангидрид) и затем бензойная кислота i°7).
По американскому патенту нагревание кальциевой соли фталевой кислоты
вместе с гидроокисью кальция осуществляется в трубчатой системе с червячным
внутренним транспортером при температуре 440 —450° в непрерывном процессе.
Французский патент (Г о л ь д ш м и д т а) описы вает нагревание до той же температуры
фталевого ангидрида с известковым молоком периодическим процессом i08).
Непрерывный процесс получения бензойной кислоты описывает в своем па-
тенте и немецкая фирма: пропускание паров фталевого ангидрида вместе с водя-
ным паром при 380—400° через контактный слой, содержащий катализаторы отщеп-
пления СО2, например NagCOg, СаСО3, окиси, гидроокиси и силикаты разных эле-
ментов, например:
Zn, Cd, Pb, Bi, Si, Al, Ti, Fe, Ni 109).
Получаемая одним из этих способов бензойная кислота подвергается очистке
разными приемами 110).
и) Окисление не содержащих углерода групп
Среди названных превращений наибольшее теоретическое и
практическое значение имеют окисления азотсодержащих групп.
Окисление анилина и других ароматических аминов может быть
выполнено таким образом, чтобы, сохранив азот заместителя, по-
лучить (вместо амино-) нитрозогруппу N: О. Необходимым для этого
окислителем является кислота Каро ш) H0-SO2-OOH, получае-
мая действием надсернокислых солей, например: (NH4)2S2O8 на
концентрированную серную кислоту при низкой температуре.
Промежуточно образуются арилгидроксиламины 112)
RNH2 -+ RNHOH RNO.
Кислота Каро (I) — производное перекиси водорода, так же как н надсерная
кислота (II)
1. НО • SO2 • ООН II. HOSO2 • О • О • SOaOH.
378
Ее образование из солн надсерной кислоты действием серной кислоты обусловли-
вается более легким гидролизом образовавшейся вначале свободной надсерной
«ислоты:
HSO3- О • О • SO3H 4- Н2О = НО • SO3 • ООН -J- H2SO4.
Соли кислоты Каро очень легко разлагаются. Окислительное действие ее
усиливается при применении в нейтральном или щелочном растворе. Окислитель-
ный эффект обусловливается освобождением одного атома кислорода на 1 мол.
кислоты Каро. Для превращения например анилина в нитрозобензол требуется
участие двух атомов кислорода;
С(;Н6НН2 -j- 20 = C6H3NO + Н20.
Практически берется иногда избыток окислителя (10—20%)
против рассчитанного количества. Избыток окислителя иногда мо-
жет привести к дальнейшему окислению нитрозо- в нитрогруппу.
Система обработки такова: сначала заготовляется раствор свободной кислоты
Каро взаимодействием соли надсерной кислоты и избытка серной кислоты при
размешиваиин и при отсутствии повышения температуры, выдерживают смесь
в продолжение некоторого времени (1 час), выливают затем в двойное-тройное ко-
личество ледяной воды, нейтрализуют содой, слегка подкисляют затем иногда
уксусной кислотой и смешивают с раствором или эмульсией амина в воде (рас-
твор для анилина например 3:150). При этом тотчас наступает превращение амина
в нитрозозамещенное, которое отделяется и очищается перегонкой с паром 113).
Окислением кислотой Каро или надсерной кислотой, получае-
мой непосредственно обработкой водного раствора соли надсерной
кислоты посредством серной кислоты, можно при достаточном
избытке окислителя получить из амина нитросоединение. Превра-
щение происходит при нагревании в среде разведенной серной
кислоты. Таким путем без всяких посторонних добавок удается
перевести p-аминоантрахинон и его замещенные в р-нитропроизвод-
яые антрахинона. Амины бензольного ряда лучше окисляются
в присутствии азотносеребряной соли как катализатора 1и).
Интересно отметить, что обработка аминов нейтрализованным водным раствором
кислоты Каро в присутствии алкильного эфира азотистой кислоты (например амил-
нитрита) и щелочи (например соды) приводит к образованию нитрозоарилгидро-
ксиламинов RN(NO)OH, легко изолируемых в виде своих солей, например комплекс-
ных солей тяжелых металлов.
Те же соединения могут быть получены из соответствующих нитропроизводных
восстановлением ннтросоединеннй цинком в присутствии алкильных эфиров азо-
тистой кислоты в щелочном или аммиачном растворе ш).
Арилгидроксиламины RNHOH окисляются в нитрозосоединения
различными окислителями. Наиболее употребительны хромовая
кислота (двухромовокислая соль в водном растворе серной кислоты
при охлаждении), хлорное железо, железосинеродистый калий
(в щелочном растворе; практичный метод получения сульфокислот
арилгидроксиламинов) Пб).
Нитрозо- и изонитрозосоединения (например нитрозофенолы)
могут быть окислены в нитросоединения или посредством красной
кровяной соли K3Fe(CN)6 в щелочном растворе или помощью раз-
веденной азотной кислоты.
Арилгидразины окисляющим действием медной соли серной ки-
слоты при нагревании превращаются в ароматические углеводоро-
ды (соотв. их замещенные)117):
RNHNH2 4- 2CuSO4% Н2О = RH 4- Cu2O 4- N3 4- 2H2SO4.
379
Этот так называемый Бейеровский метод устранения азота из гидразина
является средством для перехода от амииа к отвечающему ему углеводороду через
диазосоедииеиие и гидразин и является дополнением к указанному в своем месте
восстановительному способу замещения диазоииевой группы непосредственно водо-
родом. Оба оии служат преимущественно аналитическим целям, — для определения
строения полизамещениых.
Окисление серусодержащих заместителей касается больше всего
тиофенолов и дисульфидов.
Тиофенолы RSH крайне легко уже под влиянием кислорода воз-
духа в щелочном растворе подвергаются окислению в дисульфиды:
2RSH + О = RS • SR + Н2О.
Очень гладко такое окисление осуществляется при действии
иода в растворе йодистого калия.
Окисление тиофенолов в сульфогалогениды (RSO2X, где X—
галоидный атом) предложено в недавнем немецком патенте дости-
гать посредством обработки тиофенола галоидом в присутствии
воды, но в индиферентном растворителе (для галоида), например
СС14. Реакции отвечает уравнение 118)
RSH4-3X, 4- 2Н2О = RSO2X + 5НХ.
Дисульфиды RS • SR при нагревании с азотной кислотой уме-
ренной крепости или (особенно дисульфиды арилсульфокислот)
при обработке марганцовокислой солью в щелочном растворе пе-
реходят в сульфокислоты:
RS • SR-> 2RSO3H.
Так как дисульфидная группа может быть образована и окисле-
нием сульфидрильной SH, а эта последняя может возникнуть-
в результате превращения диазогруппы, то таким способом можна
сменить последовательными превращениями аминогруппу на суль-
фогруппу:
2RNH2 -> 2RN2X -> 2RSH -> RSSR -> 2RSOsH.
С другой стороны, возможно образование дисульфидной группы
вследствие обмена подвижного атома галоида (действие Na2S2):
2RC1-+RSSR.
При окислении дисульфидов нужно считаться с возможностью наличия обна-
руженных в одном случае автором и В. В. Козловым своеобразных форм ди-
сульфида, дающих при окислении ие сульфокислоту, а сульфон 119):
О
II
R — S — R-> R — S — R.
II II
S О
Окисление сульфиновых кислоте сульфокислоты достигается по-
средством обработки их марганцовокислой солью в щелочном рас-
творе:
RSO2H -+ RSO3H.
Обработка хлором раствора щелочных солей сульфиновых кислот
переводит их в сульфохлориды:
RSO.Na 4- Cl2-> RSO,C1 4- NaCl 12°).
380
Так как образование ароматических сульфиновых кислот, так
же как образование тиофенолов, легко достигается превращением
диазониевой (следовательно и амино-) группы, то в таком окисле-
нии имеется метод для введения сульфогруппы в такие места аро-
матического ядра, которые при обычном сульфировании не зани-
маются сульфогруппой, например для образования п.п- или
о.о-дисульфокислот.
ОСЕРНЕНИЕ 121)
Сера — элемент VI группы периодической системы, как и кис-
лород, ближайший его аналог. Во многих своих неорганических
и органических соединениях сера функционирует подобно кисло-
роду (H2S— Н2О; NaOH — NaSH, CS2— СО2, Na2CO8 — Na,CS3,
CeH5SH — С6Н5ОН), сообщая, естественно, сернистым продуктам
своеобразные свойства в силу более электроотрицательного (кислот-
ного) сравнительно с кислородом характера.
Подобно тому как кислород и соответственно кислород-отдающие
соединения вызывают окисления или путем уменьшения количества
водорода в соединении или путем сообщения ему заключающих
кислород заместителей, точно так же и сера при подходящих тем-
пературных условиях может отнимать водород (действуя дегидро-
генизирующе), связывая его в сероводород, или (и иногда одно-
временно с первым направлением) вызывать возникновение новых
серузаключающих заместителей (SH, SS и т. п.) или содержащих
серу циклов.
Такие взаимодействия серы или серупередающих агентов
(Na2Sx, S2C12, HSSO3H и т. п.), которые сопровождаются вхожде-
нием серы в молекулу продукта, мы будем называть реакциями
осернения.
Подобные процессы имеют большое значение в технике полу-
чения главным образом сернистых красителей, некоторых проме-
жуточных продуктов, при получении тиазиновых красителей и
индигоидных. Продукты воздействия осерняющих агентов далеко
не всегда и везде достаточно изучены вследствие трудности выде-
ления их в чистом виде и возможности одновременного протека-
ния реакции осернения в разных направлениях.
Молекулярная сера при нагревании с органическими вещества-
ми энергично с ними реагирует, причем процесс сопровождается
обычно выделением сероводорода. В продуктах, как уже было
указано, трудно определимых и не индивидуальных, можно конста-
тировать наличие серы, связанной в органической молекуле. Так
как сера плавится при температуре около 119°, то реакцию между
серой и органическими веществами проводят при температурах
не ниже 120°, а иногда значительно выше (200—250—280°). Полу-
ченные в результате такого плава, например из диаминов, динитро-
соединений продукты—аморфные вещества, нерастворимые в воде,
но переходящие в раствор от действия сернистого натрия и обла-
дающие свойствами сернистых красителей.
Выяснено, что нагревание с серой ароматических аминов и фе-
нолов приводит прежде всего к замещению серой орто-места ядра
по отношению к реакционной группе NH2 или ОН. Таким образом
381
анилин при сплавлении с серой образует так называемый тиоани-
лин (I);
Вероятно промежуточно образуется о-аминотиофенол (II), две моле-
кулы которого сцепляются атомом серы с выделением серово-
дорода
11 +
Точно так же ведет себя и п-толуидин при нагревании до 140°
с серой (и окисью свинца, прибавляемого в качестве связываю-
щего H2S реагента).
Вслед за образованием таких простых сернистых соединений
при дальнейшем действии серы, например при повышении темпе-
ратуры плава, получаются продукты более сложного строения, на
которых мы остановимся ниже.
Часто употребительны в практике как осерняющие средства
полисульфиды натрия различного состава Na2Sx (от Na2S2 до
Na2S5 и с еще более высоким содержанием серы). Полисульфиды,
растворы которых приготовляются растворением серы в концен-
трированных растворах сернистого натрия (Na2S), особенно часто
применяются для получения сернистых красителей черных, синих,
зеленых и красноватых методом „мокрого" плава при проведении
процесса в водной среде при температурах кипения раствора со-
лей, т. е. в пределах 105—115°. Исходными материалами для таких
плавов служат полинитро-(обычно динитро-) соединения, производ-
ные дифениламина (лейкоиндамины и лейкоиндофенолы) и иногда
готовые красители азинового ряда.
Полисульфид реагирует в плаве в трех направлениях: с одной
стороны, он действует как восстановитель на иитросоединения
(переводя нитрогруппы в амино-) и на могущие образоваться хино-
идные соединения, и в этом направлении преимущественно реакци-
онным должен быть сернистый натрий Na2S или двусернистый
Na2S2, с другой, вследствие лабильности связи серы в полисуль-
фиде Na2Sx, часть ее выделяется в свободном, особенно активном,
состоянии по мере ухода в реакцию части Na2S. Эта возникающая
в процессе сера оказывает свой осерняющий эффект на органичес-
кую молекулу, направляясь прежде всего в орто-место по отноше-
нию к имеющемуся, чаще всего азотному атому, связывающему
два ароматических остатка.
382
Очень вероятно, как предполагает И. Л. Хмельницкая, что
именно в момент распада полисульфида в процессе его окисления
особо активная сера входит в состав молекулы продукта.
Наконец третья возможность участия полисульфида в процессе
осернения — это включение сульфгидрильных групп (SH, соотв.
SNa), где сера только односторонне связывается с углеродом. Та-
кое включение повидимому чаще всего имеет место в тех положе-
ниях ароматического ядра, которые могут быть в процессе плава
особенно реакционными вследствие превращения органической мо-
лекулы (от действия серы) в хиноидное состояние.
Поясним примерами развитые выше положения.
4-нитро-4'-оксидифениламин (I) может претерпеть предвари-
тельное превращение:
+ Na2S2 4- Н2О
-f- Na2S2O3
Выделяющаяся избыточная сера производит далее осерняющий
эффект
н
Такой путь осернения нитрооксидифениламина не прослежен экс-
периментально, но некоторые его этапы достаточно достоверны.
Осернение, как указано выше, протекает часто по разным напра-
влениям. Для тех соединений, которые имеют структуру дифе-
ниламиновых производных, как I и II, этот процесс почти всегда
имеет этап образования циклического производного с кольцом тиа-
зина (V). Возможности же включения сульфгидрильных групп
383
(IV, VI) повидимому могут быть значительно шире, чем однократное
только вступление. Судя по недавно опубликованным исследова-
ниям Бернаскоии 1а2), в соединении, полученном в результате
осернения полисульфидом натрия индофенола строения
/\ Z\ZN\Z4
атомы серы располагаются в таком порядке:
S—
Нужно принять во внимание, что при плаве с полисульфи-
дом известную роль в осернении может выполнять и тиосульфат,
образующийся от окисления полисульфида, — ниже мы увидим при-
меры такого осерняющего эффекта солей серноватистой кислоты, —
что могущий образоваться сульфгидрат натрия (Na2S-f-H2S =
= 2NaHS) может также принять участие в осернении, соотв. вос-
становлении, например
-f-NaSH
/\ Z\ZNH\Z\/SNa
4x/Z\nh//4zz\s/\zz\oh
что наконец амино- и аминооксисоединения, полученные например
из нитросоединений, взятых как сырые материалы, могут в плаве
образовывать, с потерей аммиака, соотв. воды, дифениламиновые
соединения
RNH2 4- H2NR -> RNHR + NH3; R'NH2 + HOR" R'NHR" + H2O.
Мы видим таким образом, как причудливо запутана химиче-
ская картина полисульфидного плава, и отсюда становится по-
нятным, почему в этом вопросе еще так много неясности. Странным
может показаться в самом деле, что для сернистого черного из
384
динитрофенола, фабрикуемого ежегодно во всех культурных стра-
нах в количестве многих миллионов килограммов, до сих пор еще
не установлена сколько-нибудь правдоподобная формула строения,
так же как и вероятный ход реакции его получения, при котором
отщепляется 1 молекула NHS на 3 молекулы динитрофенола (Вейн-
берг).
Мы уверены теперь в наличии тиазинового кольца во многих
тех соединениях, где дифениламиновое производное берется исход-
ным материалом»в процессе осернения, мы считаем вероятным вхо-
ждение групп SH в молекулу хиноида одним из вышеописанных
путей, причем атомы серы в этих группах в продукте, выделяемом
из плава, обычно сцепляются вследствие окисления в дисульфидные
группы — SS —, переходящие от восстановительного действия Na2S
при обработке его водным раствором вновь в сульфгидрильные.
Понятно на основании сказанного в главе VII, что имеющиеся
в молекуле исходного материала атомы хлора при обработке поли-
сульфидом можно сменить также на группы SH, соотв. — SS —.
Более „мягко" действующий агент осернения — соли сернова-
тистой (тиосерной) кислоты, например Na2S2O8, (NaS • SO8Na). При-
менение тиосульфатного метода осернения особенно известно в син-
тезе тиазиновых красителей, например метиленового голубого
(Бернтсен)123). Остаток тиосерной кислоты — тиосульфогруппа
SSO3H — легко присоединяется, при восстановлении, к хиноидным
соединениям, например
//\^NH /\/NH2
-t- HSSO3H I
(CH8)2N^^ (CH8)2N/Z\/\sSO3H
Такая реакция облегчается выделением свободной тиосерной
кислоты из соли применением например одновременно раствора
гипосульфита натрия и раствора сернокислого алюминия, причем
образовавшийся гипосульфит алюминия вследствие диссоциации
реагирует как свободная тиосерная кислота; также можно под-
кислить раствор гипосульфита например уксусной кислотой.
При достаточном избытке соли тиосерной кислоты, соотв. до-
статочном количестве окислителя для окисления в хиноид арома-
тического производного, можно ввести в ядро больше чем одну
тиосульфогруппу и получить ди- и даже тетратиосульфокислоты,
соли которых изолируются в кристаллическом состоянии, например
получить из n-фенилендиамина дитиосульфокислоту:
HOgSS^/^ zNH2
H2n/\/\sSO8H
25 Зак. 2611. — Н. Ворожцов
385
Полученные тиосульфокислоты достаточно легко при нагревании
в водном растворе теряют остаток сульфо (в виде сернистой кис-
лоты H2SO3), причем атом серы связывается с ароматическим ядром,
замещая его водородный атом. Таким образом получают тиази-
новые красители из тиосульфокислот индаминов, причем обычно
для связывания сернистой кислоты в дитионат прибавляют в смесь
окислитель. Примером такого превращения служит синтез метиле-
нового голубого:
(CH3)2N^X^XS \/\n(CH3)2
Cl SO3H
(CH3)2N/^X/X'/\n(CH8).,
s
Подобно вышеописанному получаются тиосульфокислоты из хинонов (1), на-
пример
/\ /О /х /ОН /Х /О
I I + HSSO3H -> || -> | 1 |
Ho/^/^SSOaH O^\^\sSO3H
которые, взаимодействуя с аминами, образуют соответствующие арилидохиноиы (II)
I I П
O^\^\SSO3H
при действии кислотных конденсирующих средств переходящие, с замыканием
цикла, также в (гидро) тиазиновые производные (III), имеющие значение в качестве
темнокоричневых сернистых красителей для шерсти.
386
В литературе имеются указания на возможность применения тиосульфата для
получения продуктов осернения более далеко идущего, чем при образовании тио-
сульфокислот. Нагревание с кислыми растворами исходных продуктов (диаминов,
нитрозофеиолов и т. п.) при разложении тиосульфата, например по уравнению
Na2S2O8 + H2SO4 = Na2SO4 -f- SO2 -f- S + H2O,
может содействовать осернению продукта за счет выделяющейся серы.
Нагревание водного тиосульфата до высокой температуры, например 160—165°
(под давлением), при его разложении (с выделением сероводорода) иа сульфит, сульфат
и серу также может служить способом осернения ш).
Так же интересна, как осерняющий агент, хлористая сера S2C12
Ее действие по отношению к ароматическим аминам со свободным
пара- и с одним свободным орто-местом (к NH2) приводит к об-
разованию хлористых арилентиазтиониевых соединений (I) и часто
к входу хлора в пара-место (к NH2). Так, из о-толуидина полу-
чается соединение (I) 125).
С1
В реакцию вводятся солянокислые соли аминов, хлористая сера берется всегда
в избытке (не менее 3 мол.) по отношению к амину. Реакция проходит при на-
гревании (иногда в присутствии нейтрального по отношению к S2C12 растворителя),
сопровождаясь выделением хлористого водорода. Температура реакции (чаще 50—
120°, иногда ниже) зависит от реакционности аминосоединеиия. Получаемые соеди-
нения, очищаемые без участия воды (разведением и промыванием жидкими угле-
водородами), легко кристаллизуются и обладают большой реакционной способностью.
В тех из них, которые в пара-месте к азоту несут атом хлора, этот последний
особенно склонен к обмену, например на азотные группы, и уже при растворении
в анилине они дают замещенные, иапример
С1 С1
При действии восстановителей (Na2S2O4) тиазтиониевые соединения образуют
аминомеркаптаны
X
находящие себе применение в получении тиоиндигоидных красителей и т. п.
Упомянутые уже выше методы введения остатка родана в ароматическое ядро
(см. главу IV), например обработка роданистым свинцом вместе с хлористым сульфу-
25*
387
рилом, применяются иногда как начальная фаза осернения, так как роданопродукты
прн взаимодействии с двусерннстым натрием дают тнофенольные производные,
например некоторые сернистые красители 126).
Действие серы как таковой на ароматические амины рассмот-
рено в начале этого раздела. При повышении температуры воз-
действия серы на n-толуидин (до 175—185°) в результате полу-
чаются соединения, заключающие серу в циклической связи—в тиа-
золовом кольце (I) 127)
+ 4S
-3H2S
-Ж + Н,(С—
---II
Образующееся соединение здесь называется дегидротио-п-толу-
идином (II). В зависимости от большего или меньшего избытка
серы и степени нагрева получают соединения с несколькими тиа-
золовыми циклами, например
каковые вероятно имеют место в составе молекул сернистых кра-
сителей желтых, оранжевых и коричневых, образуемых методом
„запекания" (безводного плава).
Дегидротиотолуидин играет некоторую роль в получении азо-
красителей как диазосоставляющая их. Соединения с несколькими
тиазоловыми кольцами вышеуказанного типа (III) входят в состав
основания примулина (субстантивного для хлопка красителя, полу-
чаемого сульфированием основания).
Наклонность к образованию тиазоловых колец резко проявляется
у гомологов анилина (толуидина), у ацилированных остатком орга-
нической кислоты аминов, например
Атом азота и углеродный атом тиазолового кольца могут не принадлежать мо-
лекуле одного вещества, н все-такн кольчатое соединение образуется в результате
сплавления с серой. Например при нагревании с серой 2 мол. дегндротнотолунднна
388
н1 мол. бензидина образуется желтый краситель, который имеет вероятно стрсе^
вве (IV):
Среди различных сернистых соединений, имеющих значение как
красители или как промежуточные продукты для сернистых, инди-
гоидных и тиазиновых красителей, в недавнее сравнительно время
(с 1920 г.) вызвали большой интерес продукты осернения фенола
и его замещенных. Ранее в колористической практике было уста-
новлено, что сернистые красители, нанесенные на хлопчатобумаж-
ное волокно, являются хорошей „протравой" для основных кра-
сителей: на окрашенном сернистым красителем волокне легко
закрепляются основные красители, сами не окрашивающие хлопок;
при этом цвета того и другого красителя, смешиваясь, дают в ре-
зультате смешанные тона. С целью испытать бесцветные серни-
стые соединения на их способность закреплять основные красители
было испытано (Тауссом и Гюнтером) осернение фенола и
его замещенных, и полученные в результате продукты, сами бес-
цветные, оказались обладающими сродством к хлопчатобумажному
волокну и хорошими закрепителями на нем основных красителей.
Они вошли в продажу в Германии под названием „катанолов".
Н. В. Филиппов разработал в Иванове метод приготовления
подобногоже соединения,получившего название „закрепителя Т“ 128).
По тому в другому способу осернение ведется в водном растворе, причем
к фенолу добавляется эквивалентное фенолу колнчестио едкого натра (или немногим
меньше); серы же добавляется в количестве от 1,5—2,5 эквивалентов по отношению
к фенолу. Нагревание с обратным холодильником ведется 24—30 часов.
389
А. А. Разумеев предлагает иной метод работы без воды для получения ана-
логичного препарата ,таннола“, причем берет едкого натра в количестве 0,2 экви-
валента по фенолу, а серы 2,1 эквивалента 129).
Американский патент Таусса описывает получение подобного препарата нагре-
ванием фенола с серой и 0,01—0,03 мол, натриевых солей слабых кислот (NaOCOCHo,
NaOCOH, Na2S2O4, Na2SO3, NaNO2) до 180—210°.
Реакция протекает через промежуточное образование диокси-
дифенилдисульфида
Л/ОННО д /д/ОННОд/д
вероятно из
который сам не обладает свойствами катанола и лишь при даль-
нейшем осернении дает желательный продукт.
Филиппов и Воронков считают для закрепителя Т вероятным такое
строение
где х вероятно = 2. Исследования Липатова подтверждают такое предположение,
так как медная соль продукта отвечает составу
Роль едкого натра, который может быть заменен Na2S или NaaCO3,
Разумеев считает каталитической. Возможно действительно,
что, образуя хотя бы в малых количествах молекулу фенолята,
едкий натр активирует тем самым бензольное ядро к дальнейшим
присоединениям или участвует в образовании хотя бы весьма
малых количеств продуктов воздействия щелочи на бензольное
ядро того вида, который мы усматривали уже в главе VI:
ONa
Н
390
Вполне возможно, что такой продукт будет очень реакционно-
способен по отношению к сере.
Дальнейшее уплотнение молекулы диоксидифенилдисульфида
возможно в двух направлениях. С одной стороны, под влиянием
дегидрогенизирующего действия серы может возникнуть новая
углеродная связь между бензольными ядрами, например
С другой стороны (и параллельно с первым течением процесса
уплотнения), вероятно возникновение связи отдельных молекул
диоксидифенилдисульфида посредством атомов серы, по орто-
месту к группе НО, т. е. так:
Во всяком случае химизм получения бесцветных сернистых про-
изводных фенола заслуживает внимательного изучения, так же
как и вообще химизм интересных процессов осернения аромати-
ческих соединений.
В последнее время описано получение тиопроизводных фенолов
осернением последних в присутствии щелочных солей таких кис-
лот, которые заключают в анионе металлы, осаждающиеся от
сероводорода или сернистого аммония. Такими солями могут быть
станнаты, антимонаты, алюминаты и т. п. Также описано приме-
нение молибденовых соединений при осернении 13°).
Обработка соединениями олова уже готовых тиопроизводных
фенолов самих по себе или в присутствии фенолов дает продукты,
резервирующие при крашении шерсть 131).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) Us. Rh, Г. п. 101221, 107722 (F, V, 95, 96).
a) Lang, Г. п. 189178 (F. VIII. 1357).
3) В., Г. п. 175295 (F. VIII, 152).
391
4) В., Г. п. 179589 (F. VIII, 151).
5) Заявлен. Г. п. В. 25232 (F. VI, 123).
в) В., Г. п. 127388 (F. VI, 122).
’) By. Г. п. 121788 (F. VI, 123), Tiele, Winter, Ann. 311, 353 (1900).
8) 3. Е. 3 и н ь к о в, С. Е. Л и в ш н ц, Ж. Пр. X. 5, 604 (1932).
9) 1G, Ф. п. 678826, СВ 1930, 1, 3831.
Ю) MLB, Г. п. 107095 (F. V, 128).
И) MLB, Г. п. 91503, 92084, 93539, (F. IV, 129—132), 103859, (F. V, 122).
12) MLB, A. Reissert, Г. п. 186881, К., Г. п. 199147 (F. IX, 162, 163),
13) Us. Rh, Г. п. 239953, 246381, 237358, 246659 (F. X, 158—160).
11) Sandmeyer (G), Г. п. 86874, 87255 (F. IV, 136, 137).
15) IG, Б. п. 347743, СВ 1931, II. 2930; 1G, Г. п. 557249, СВ 1932, II, 2376.
is) St. S ё с а г е a n и, Вег. 64, 837 (1931).
17) Н. Schmid t, Г. п. 20909 (F. I, 23).
is) G. Blanc, Г. п. 347583 (F. Х111, 1107).
Pr. Ch., Г. п. 268786 (F. XI, 197).
19) Gr.-E., Г. п. 154499 (F. VII, 111).
го) Heyden, Г. п. 124229 (F VI, 134).
21) О. L. Brady, G. V. Elsmie, СВ 1926, I, 2610.
22) Ripper, Mon. 21, 1079 (1900).
2а) Петренко-Крнченко и Канчев, Вег. 34, 1702 (1901); Ж. 33, ТГ&
(1906).
21) Sandmeyer, Г. п. 103578, 105103, 105105, 105798 (G) (F. V, 101—109);
Н. Hagenbach, Helv. 6, 155 (1923).
25) IG, Б. п. 250955, Вг. А. В 1927, 572.
26) F a h 1 b е г g, L i s t, Ber, 21, 243 (1838).
27) Noyes, Ber. 16, 52; 2296 (1883).
28) Ch. F. Buckau, Г. n. 261775 (F. XI, 207).
29) Каминский, Ж. Фарм. Ин-та, 1921, № 2.
зо) Е. Е i с h w а 1 d, Ph. Fr. H a r d t, Г. n. 360528 (F. XIV, 439).
3i) Sachse, Г. n. 216091 (F. X, 166).
32) P. Seydel, Am. n. 1576999, CB 1926, I, 3631.
33) J. F. С о n n, A. L о w y, CB 1926, II, 2789.
3i) L. W. В a s s, T. B. Johnson, Am. Soc. 46, 456; CB 1924, I, 1924,
35) См. также Monnet, Reverdin, Noelting, Ber. 12, 443 (1879)
1 2
(CgHJCHgXNOg)C6H4(CO2H)(NO2)]; Ullmann, Am. Soc. 16, 533 (1894);
14 14
Michel, Norton, Ber. 10, 580 (1877) [С6Н4С1(СО2Н), C6H4(NO2)(CO2H)].
36) В., Г. n. 94629 (F. IV, 146).
37) By, Г. n. 102893 (F. V, 402); D. Vorliinder, R. S c h i 11 i n g (B), 121287
(F. VI, 540).
38) Heymann, Ko nigs, Ber. 19, 705 (1886).
39) Cp. Oppenheim, Pfaft, Ber. 8, 887 (1875).
4°) Friedlander, Low-Beer, Г. n. 170230.
4i) Ch. Werke Grenzach, Г. n. 377990 (F. XIV, 441).
42) Etab I. Poulenc Freres, Заявл. Г. п. E. 25065 (F. XIV, 444).
43) L.T. Smith, R. E. Lyons, Am. Soc. 48, 3165; CB 1927, I, 1001.
44) IG, Шв. n. 130696, CB 1929, II, 218; Gen. An. W., Am. n. 1740771, CB 1930,
I, 1539.
46) BOhringer & Sohne, Г. n. 117129 (F. VI. 110).
46) Binz, Zt. angew. Ch. 1900, 385.
47) O. F i s c h e r, H e p p, Ber. 26, 223 (1893).
48) C. J. Kothari, H. E. Watson, CB 1931, II, 2425; A. E k e r t, Б. n. 182487,
CB 1923, 11, 1029; E. N оziсkа, Г. n. 526195, CB 1931, II, 2931, Ver. ch. F.
Kr. Heller & Co, Ф. n. 702183, CB 1931, II, 1190.
49) G. Blanc, Ф. n. 586383, CB 1927, J, 806; IG, Г. n. 506438, CB 1930, 11,3850.
M) Jessnitzer, Г. n. 236489 (F. X, 165), см. также 1J).
6i) Например G„ Г. n. 88085 (F. IV, 219), ClBa, Г. n. 71370 (F. Ill, 106).
52) E. & O. Fischer, Ann. 194, 256 (1878).
33) Hanp. By, Г. n. 68114, 68123 (F. Ill, 220—222), MLB, Г. п. 111919 (F. V. 270).
См. многочисленные Г. n., В. и By под рубрикой Anthracenfarbstoffe, F. Ши
сл. томы.
392
54) By, Г. п. 60855, 63693 (F. Ill, 198, 201).
55) By. Г. п. 81481 (F. IV, 272), 421235 (F. XV, 662).
56 By, Г. n, 161954 (F, VIII, 252); Dreyfus, Ф. n. 705161, CB 1931, II, 3274.
Cp. F. V, 239.
57) P. F ri e d 1 a n d e r, S c h e г z e r, CB 1900, 1, 409; Friedlander, Ber, 32,
3528 (1899).
®) A. Wohl, Ber. 32, 3486 (1899); H. F. L e w i s, G. W. T h i e s s e n, C. A, 20
2316 (1926).
59) By Г. n. 241806 (F. X, 594).
во) Dow (W. J. Hale), Am. n. 1595299, CB 1926, II, 2116.
6i) Fr. Fichter, CB 1924, II, 2832; Fichter, J. Meyer, Helv. 8, 74 (1925); CB
1925, I, 1591; Fichter, M. Rin d e r s p a c h er, Helv. 9, Ю97 (1926, CB
1927, I, 999; Helv. 10, 4U (1927;, CB 1927, 1, 1374.
Fr. Fichter, F. Ackermann, Helv. 2, 583 (1919); Bull. 28, 768 (1920).
62) Kashi chi Ono, J. Chem. Soc. Japan 42, 559 (1921;; Bull. 32, 1591 (19z2).
63)0 Магидсон, H. Преображенский, Труды Научн. хим.-фарм.
ин-та вып. 16, 65 (1926), СВ 1928, 1, 35.
64) Н. G о 1 d h a m m е г, СВ 1928, II, 650.
б5) A. Seyewetz, G. М i о d о п, СВ 1923, 111, 304; Ср. Т. Kempf, Г. п.
117251 (F. VI, 109).
66) IQ Г. п. 533128, СВ 1931, II, 3042.
67) R.W i 1 Is ta 11 er, St. D о г о g i, Ber. 42, 2147 (1909).
68) Byk-Guldenwerke, Г. n. 369354 (F. XIV, 432).
б9) В б h r i n g e r & S о h п e, Г. n. 420444, CB 1926, 1, 2247.
’°) O. N, Witt, S. T о e c h e - M i 111 e r, Ber. 36, 4390 (1903).
71) Holiday & Co C. Shaw, Б. n. 274700, CB 1927, 11, 2227.
72) О. Мнллер, Ж. 16, 414 (1884); Grove, Ann. 167, 357 (1873),
И) P. К. Э й x м а и, О производстве антрахинона в СССР. Ан.-Кр. Пр. 1932,
№ 1, 21. Подробнее о способах получения антрахинона, Н о u b е n, Das
Anthracen und die Anthrachinone, Lpz, 1929, 209-213.-
74) К Hofmann, Г. n. 277733 (F. Xll, 919), Holman n, Quoos, Schnei-
der, Ber, 47, 1991 (1914).
75) Hofmann; Ritter, Ber. 47, 2238 (1914).
76) MLB, Г, n. 273318, 273319 (Xll, ч0/, 408).
77) Gr.-E., Г. n. 283213, 284083, 284084 (F. XII, 404-406).
’8) G г й n а и, Г. n. 234289 (F. X, 574), 254710, MLB, Г. n. 256623 (F. XI, 540):
Ср., Г. n. 215335 (F. IX, 668).
’9) В., Г. n. 268049 (L. XI, 541), Gr.-E., Г. n. 284179 (F. Xll, 407).
80) К. H. Meyer, Заявл. Г. n. M. 46377 (F. XI, 539).
81) MLB, Г. n. 292681 (F. XIII, 384).
82) Fr. Darmstadt er, Г. n. 109012 (F. V, 664), By, Г. n. 252759 (F. XI, 538);
П. А. Флоренский, К. T. Метелкин, Об электрокаталитическом окис-
лении антрацена в антрахинон, Ан.-Кр. Пр. 1931. № 8, 9, № 9/10, 7. Там же
литература вопроса. Ср. Г. п. 172654 (F. VIII, По).
83) Tratscher, Ам. о. 1397562 (1922).
84) С. Graebe, Ann. 167, 131 (1873); R. Anschiitz, G. Schultz, Ann. 196, 38.
(1879); Lang, Г. n. 189178 cm. 2); В. Г. n. 275518 (F. Xll, 37), MLB, 1. n.
152063 (F. Vli, 105).
85) G. C h a r r i e г, A. M о g g i, CB 1928, 1, 701, C h a r r i e r G. В. С r i p p a, ibid.
86) C. Graebe, E. Gfeller, Ber. 25, 652 (1892); Ann. 276, 1 (1893).
87) В. Г. Шапошников, Исследования из области азиновых и аэониевых
красящих веществ, Киев 1904.
88) н. G. Gibbs, W. L. Hall, W. М. Clark, Studies on Oxydation-Reduc-
tion, XIII Public Health Report (supf. N 69), Wachington 1928; Division,
о f C h e m i s t r y, Hygiene Laboratory; Studies on Oxydation-Reduction I—X.
Washington 1928; G. Heller, Ann. 392 16 (1912); CB 1912, 11, 1544;
A. Hirsch, Ber. 13, 1903 (1880), R. Mohlau, Ber. 1<ч 2843 (1883).
8») C. Graebe, Ann. 167, 131 (1873).
9°) Cm. Bru nek, Ber. 33, Sonderheft, LXXX (1900)
S1) Mi lb an er, N e mec, J. pr. Ch. (2) 99, 93 (1919)
9a) Winteler, Ch. Zig. 1908, 602. 1 A
83) Tscherniak, Г. n. 79693, 86914 (F. IV 162 163)
94) J. v. В r a u n, Ber. 56, 2332 (1923). ’
85) Ch. W e r k e Grenzach, AG, f. n. 377990 (F. XIV, 441).
393
96) В., Г. п, 91202 (F. IV, 164).
°7) J. Н. Gardner, Am. Soc. 49, 1831; СВ 1927; II, 1268.
98) Bas., Г. n. 138790, 140099 (F. VII, 112, 114).
99) Ehrlich, Benedikt, Mon. 9, 527 (1888); O. Dischendorfer, W. D a n-
ziger, Mon. 48, 315. CB 1927, II, 1839.
к») t. M„ Г. n. 411955 (F. XV, 263); Cp. Werner, Piguet, Ber. 37, 4310(1904),
Ш) R. Scholl, A. Zincke, Ber. 51, 1419 (1919), 52, 1142 (1919); R. Scholl,
Ber. 52, 1829 (1919); R. Scholl, P. Dahl, Fr. Hansgirg, Ber. 56,
2548 (1923).
юз) R. E. Schmidt, B. S t e i n, C. Bamberger, Ber. 63, 300 (1930).
юз) R. Scholl, P. Dahl, Ber. 57, 80 (1924).
Ю4) C. G r a e b e, Ber. 35. 2753 (1902).
105) MLB, Г. n. 127138 (F. VI, 156).
ioo)Graebe, Rostowzeff, Ber. 35, 2748 (1902).
Ют) Monsanto, Ch. W.. Б. n. 341902, CB 1931, I, 3722.
108) S e 1 d e n Co, Am. n. 1727102, CB 1930, II, 307, T. Go 1 d s c h m i d t, AG., Ф. n.
692521, CB 1931, I. 1170.
109) IG, Г. n. 445565 (F. XV, 396).
HO) Cp. IG. Г. n. 315892, CB 1930, I, 740; Б. n. 329375, 329389, CB 1930, II, 1614;
Ф. n. 670642, CB 1930, I, 1221; Г. n. 523115. CB 1931, II, 632; Monsanto,
Ch. W„ Am. n. 1755242, Ch. T. Ub. 1931, № 13/15.
Ш) H. Caro, Zt. angew. Ch. 1898, 845; A. В a ey er, V il 1 i g e r, Ber. 32, 3628
(1899); D’Ans, Friedrich, Ber. 43, 1880 (1910).
11®) E. Bamberger, Tschirner. Ber. 32, 1672 (1899).
из) В., Г. n. 110575 (F. V, 45', A. Baeyer, E. Knorr, Ber. 35, 3034 (1902);
Bamberger, Htibner, Ber. 36, 3803, 3827 (1903); Freundler, Seve-
stre, C. r. 147, 981 (1908), CB 1909, I, 69.
ui) E. К о p e t s c h n i, Г. n. 363930, (F. XIV, 850); Witt, Kopetschni, Ber.
45, 1134 (1912); Meisenheimer, Hesse, Ber. 52, 1162 (1919).
us) О. В a u d i s c h, Г. n. 227659 (F. X, 125).
U6) Bamberger, Ber. 27,1555 (1894), 28. 1221 (1895); Bamberger, Pyman,
Ber. 36, 2701 (1903); Wacker, Ber. 36, 668 (1902).
«’) Cm. Haller, (Bayer) Ber. 18, 90, 92 (1885).
ив) He у den, Г. n. 550685, CB 1932, 11, 1511.
11») H. H. Ворожцов и В. В. Козлов, Ж. О. X. II (64), 947 (1932).
19°) G a 11 е г m а п п, Вег. 32, 1136 (1899); Ullmann, Lehner, Ber. 38, 732
(1905).
i21) О. Lange, Die Schwefelfarbstoffe, Lpz. 1912; A. v. Weinberg, Neuere
Forschungen auf dem Gebiete der schwefelhaltigen organischen Verbindungen,
Ber. 63, A 117 (1930); A. W., La constitution des colorants sulfures. Rev. Gen.
M. C. 36, 161 (1932); И. Хмельницкая, Кинетика процесса образования
сернистого черного, Ан.-Кр. Пр. 1931, № 11—12,22; И. Хмельницкая,
В. В е р х ов с к а я, О химизме процесса осернения ароматических соеди-
нений, Ан.-Кр. Пр. 1932, № 1, 31.
122) Е. Bernasconi, Helv. 15, 287 (1932).
i23) Bernthsen, Ber. 16, 2903 (1883). Ann. 251, 1 (1889); В., Г. n. 45839 (F. Il,
144); MLB, Г. n 46805 (F. II, 152),
121) Cly, Г. n. 106030 (F. V, 469); Vidal, 116351 (F. VI, 745); Sandoz, Г. n.
136016 (F. VI, 735).
125) R. Herz (С). Г. n. 350590, 367344, 367345, 370854 (F. XIV, 908-915). Заявл.
Г. п. С. 33548, С. 33559 (F. XV, 231).
12») 1G, Б. п. 289241, СВ 1923, П, 496.
’2’) М. Т. Bogert, L. Smidth, Am. Soc. 50, 428, CB 1928, I, 1851.
128) A. Thauss, A. Gunther (By), Г. n. 400242 (F. XIV, 1069); H. В. Фи-
липпов, К вопросу получения суррогатов таннина, Изв. Текст. Пром.
1926, 1, № 8, 26; Н. В. Ф и л и п п о в, Б. С. Воронков, Изв. Текст. Пром.
1926, № 6, 7; С. М. Липатов, Изв. Текст. Пром, и Торг. 1929, № 7—8,
76 (514).
129) А. А. Разумеев, Таниол, Изв. Текст. Пром, и Торг. 1929, № 12, 70 (872).
А. Т a u s s (Gen. An. W.), Ам. п. 1757400 (1930), 1С1, Б. п. 370497, СВ 1932,
П, 781.
’зо) Sandoz, Ф. п. 39838, 40088, СВ 1932, II, 780.
131) 1G, Б. п. 360378, СВ 1932, 1, 743; Б. п. 370458, СВ 1932, II, 780.
394
ГЛАВА XIV
ВОССТАНОВЛЕНИЕ СОЕДИНЕНИЙ С ХАРАКТЕРНЫМИ, НЕ
ЗАКЛЮЧАЮЩИМИ АЗОТА, ГРУППАМИ
Выше были рассмотрены многообразные превращения, насту-
пающие при восстановительной обработке соединений с группами
NO2, N: N, N2X и другими содержащими азот группами (см.
главы V, IX).
Метод восстановления находит себе применение и по отношению
к таким соединениям, которые содержат иные характерные группы
независимо от того, входит ли в состав молекулы таких соединений
азот или не входит.
а) Восстановление карбонильных соединений
Особенное значение метод восстановления имеет по отношению
к карбонильным соединениям (с группой СО), причем карбонильная
группа может быть или в виде кетоновой R • СО • R' или циклически
связанной, например в ангидридах двуосновных кислот
/СО\
R2< >0
xcoz
в ангидридных производных замещенных кетокислот, например
СО
NH
или в хинонах и хиноидах.
Восстановление подобных соединений практикуется как для
синтеза промежуточных продуктов и красителей, так нередко при-
меняется и по отношению к готовым красителям с целью привести
их в иную реакционную форму, иногда уже в практике самого
крашения (лейкосоединения кубовых красителей).
Ароматические кетоны, например бензофенон, переводятся в соот-
ветствующие вторичные спирты — бензгидролы действием цинковой
пыли в щелочном (иногда аммиачном) концентрированном растворе
(обычно в водно-спиртовом). Цинк применяется в значительном
избытке против количества, рассчитанного по реакции
RCOR' 4- Zn + 2Н2О = RCH(OH)R' + Zn(OH), i).
395
Кроме этилового спирта для проведения реакции при более высокой
температуре применяется амиловый спирт.
Лабораторным методом может служить во многих случаях вос-
становление посредством натрия в спиртовом растворе, а также
посредством амальгамы натрия 2). Нужно указать, что более энер-
гичное восстановление (в том числе и работа с амальгамой натрия),
особенно же кислотное восстановление, переводит карбинолы в ли-
шенные кислорода диарилметановые производвые:
R(R')CO - R(R')CHOH - R(R')CH2.
Третичные ароматические карбинолы, получаемые при обработке
щелочью триарилметановых красителей, переходят в триарилмета-
новые замещенные уже при обработке цинком в присутствии
кислоты.
Таким путем образуются так называемые лейкооснования кра-
сителей этого класса:
RRiR2COH -> RR^CH.
Восстановление самих красителей триарилметанового ряда в лей-
кооснования можно проводить также цинком в кислом растворе,
лучше же гидросульфитом в спиртовом растворе (предпочтительно
с добавкой небольшого количества цинка) 3). При обработке гидро-
сульфитом красителей этого типа (I) в воднощелочном растворе
получаются в растворах или в кристаллическом виде соли триарил-
метановых сульфинокислот (П) или изомерных им кислых эфиров
сульфоксиловой кислоты (и карбинолов) например
С1
[(CH8)8NC6HJ2C: : N(CH8)2-^[(CHS)2NC6HJ2C • <“>-N(CH3)2 ->
I И |
SO2Na
(или OSONa)
ZS^51(CHs)>N-C,HJ, ; СН
которые при нагревании с избытком щелочи, отщепляя кислотный
остаток в виде сульфита, образуют лейкооснование красителя (III)4).
Восстановление карбонильной группы практикуется в примене-
нии к индиго и подобным ему индигоидным красителям для пе-
реведения их в растворимое в щелочном растворе энольное соеди-
нение. Такие растворы носят название „куба" соответственного
красителя. Для индиго (I) схема процесса такова:
396
Соединение (II), получаемое в результате восстановления, назы-
вают белым или лейко-индиго.
В практике приготовления кубов для крашения применялись многие восстано-
вительно-действующие агенты: анаэробное брожение углеводов, железный купорос
с известью, цинковая пыль в щелочном растворе. Наиболее же употребительный
в настоящее время восстановитель — гидросульфит натрия.
Применяя этот восстановитель в виде готовой соли Na2S2O4 или
образуя соль гидросернистой кислоты взаимодействием сернистой
кислоты и цинковой пыли в самой реакционной смеси, можно, поль-
зуясь спиртом как средой, получить лейкосоединения индигоидных
красителей в твердому состоянии. В производстве их освобождают
от спирта и сушат в вакууме 5).
Оказалось возможным получение лейкосоединений из индиго и
других кубовых красителей путем обработки их водородом в ще-
лочной среде в присутствии восстановленного никеля или палла-
диевой черни 6).
Также интересным методом получения лейкосоединений кубовых
красителей, особенно содержащих нитрогруппы, является обработка
их в среде из третичного основания (например пиридина) посред-
ством сернистой и муравьиной кислот. При этом NO2-rpynna
остается без изменения 7).
Также посредством восстановления гидросульфитом, обычно
в щелочной среде, приготовляются куба из антрахиноновых кубо-
вых красителей для целей крашения ими.
Твердые, сухие лейкосоединения кубовых красителей служат
исходными материалами при получении вышеупомянутых (глава XII)
индигозолей— кислых эфиров серной кислоты.
Изатин (I) — вещество, инею цее интерес как промежуточный продукт при син-
тезе некоторых индигоидных красителей, и историческое значение в установлении
структуры индиго,—под влиянием восстановительных агентов обнаруживает наклон-
ность к превращению прежде всего в группе СО, непосредственно связанной с бен-
зольным ядром. Таким образом из него образуются сначала диоксиидол (И) и затем
оксиидол (III)
Превращение изатина в диоксиндол было впервые осуществлено Байером
и Кио пом при действии амальгамы натрия иа изатин в водной суспе.исии. Ныне
имеется более удобный способ его получения — кипячением изатина с водным рас-
твором гидросульфита натрия.
В щелочной среде диоксиндол далее ие восстанавливается. При восстановлении
диоксиндола также амальгамой натрия, но избегая образования сильнощелочной среды,
например проводя процесс в среде разведенной соляной или серной кислоты илн
s растворе бикарбоната натрия, насыщаемом во все время процесса углекислотой,
получают оксиндол 8).
Восстановление СО-группы в хинонах в С —ОН, т. е. превра-
щение хинонов в гидрохиноны, производится чаще всего действием
сернистой кислоты на хинон в водной суспенснн. Промежуточно
397
образуется хингидрон (I), который
к действию сернистой кислоты, чем
более стоек по отношению
хинон:
Поэтому предложено заканчивать восстановление хингидрона с уча-
стием металлического железа или соли закиси железа в присут-
ствии окиси или карбонатов магния или других щелочноземель-
ных металлов 9).
Хиноны различного строения по-разному относятся к восстановительному дей-
ствию сернистой кислоты, иоднстоводородной кислоты и фенилгидразина. В зависи-
мости от относительной легкости перехода хиноидной структуры в чисто аромати-
ческую (у гидрохинонов) наиболее легко поддается восстановительному воздействию
беизохииои, затем следуют иафтохииоиы, некоторые многоядерные хиноны, например
аитаитрои, и наиболее трудно переходит в гидрохинон антрахинон, являющийся
скорее дикетоиом, чем хиионом. В частности феиилгндразин, который более или
менее легко восстанавливает беизохинои, его производные и иафтохииоиы ие изме-
няют антрахинона и его производных 10).
Восстановление антрахинона, в зависимости от условий, дает на-
чало различным продуктам, частью находящимся в состоянии кето-
энольной тавтомерии одни к другим.
Схему восстановительных превращений антрахинона можно
представить в следующем виде:
ОН
\/\/\/
I
ОН
СН
ОН
Восстановление в антрагидрохинон (I) впервые было произведено
Гребе и Либерманом цинковой пылью в среде 50°/0-ного
раствора едкого натра. Позднее был предложен (Гранмуженом)
способ восстановления гидросульфитом в щелочном же растворе п).
Аитрагидрохииои (эиольная) форма (I) является если ие единственно существую-
щей, то во всяком случае подавляющей количественно другую, тавтомериую, форму
оксиантрона (II). Последняя отличается от первой нерастворимостью в щелочи, от-
сутствием флуоресценции и стойкостью по отношению к кислороду воздуха. Окси-
398
антрои, полученный из (мезо) бромантроиа гидролизом, энолизируется в антрагидро-
хинои действием кипящего спиртового раствора щелочи
При действии на оксиантрои (ио ие аитрагидрохинои) цинка и уксусной кис-
лоты получается антранол (111), тавтомерный антрону (IV).
Из последней названной пары тавтомеров каждый может существовать в твердом
состоянии, в растворах же устанавливается равновесие большей частью с преоблада-
нием кетоформы (IV). Присутствие следов соляной кислоты ускоряет кетизацию.
Характер растворителя существенен для преобладания кето- (соотв. энольиой) формы
(уксусная кислота и особенно пиридин благоприятствуют энолвзации, хлороформ
и ацетон — кетизации) 13).
Антрон интересен практически как промежуточный этап в полу-
чении ценного бензантрона. Он был получен впервые Либерма-
ном умеренным восстановлением антрахинона иодистоводородной
кислотой или оловом и соляной кислотой в растворе ледяной
уксусной кислоты. Как наиболее употребительный способ его по-
лучения из антрахинона применяется восстановление последнего
посредством стружек меди или алюминиевой бронзы в концентри-
рованной серной кислоте при 30—40°. При этом первой стадией
реакции является повидимому образование оксиантрона, который
далее восстанавливается в антрон. Имеются указания, что приме-
нение в качестве восстановителя железа и соляной кислоты или
железа с хлористым железом в водной суспенсии или в среде
уксусной кислоты дает лучшие результаты по выходу антрона 14).
Подобно антрахинону восстанавливаются и антрахиноновые
производные, причем наиболее употребительным для получения
гидрохиноновых производных восстановителем служит гидро-
сульфит.
Восстановление обеих СО-групп в хинонах до СН или СН2 практикуется пре-
имущественно при исследованиях структуры хиноиов и хиноидных соединений для
определения углеводорода или оводороженного гетероциклического соединения,
лежащего в основе таких соединений. Наиболее известен исторически метод
Байера — нагревание вещества, смешанного с большим избытком цинковой пыли,
в трубке или реторте. Таким путем Гребе и Либерман получили антрацен
из ализарина, раскрыв впервые природу этого красителя и тем наметив путь к его
синтезу. Из антрахинона также получается, понятно, антрацен,причем путь восстано-
вления вероятно проходит по начерчевиой выше схеме, включая и дигидроантраиол (I):
СН2 СН
/\/\/\ Z\Z\Z\
I
ОН
Практически метод восстановления антрахиноновых производных
до антраценовых может применяться лишь для синтеза тех соеди-
нений, которые непосредственно из антрацена не получаются или
получаются с трудом. Такова например р-сульфокислота антрацена
СО
Z\Z\/\/S°3H /\/\Z\/S°3H
\z\z\z \z\z\/
со
399
Восстановление осуществляется действием иодистоводородной
кислоты с фосфором или цинком в аммиачном растворе 15).
Хинониминовые производные (индофенолы, индоанилины), о ко-
торых шла речь в предыдущей главе, переводятся в свои гидро-
хиноновые производные (лейкоиндофенолы) преимущественно путем
обработки раствором сернистого натрия. Так как весьма часто
индофенолы играют роль промежуточных продуктов в синтезе
сернистых коасителей, то обработка сернистым натрием подгото-
вляет среду для осернения.
Упомянем еще, что СО-группа в аигидондах двуосновных кислот, например
фталевой кислоты, может быть восстановлена в СН2-группу. Для фталевого анги-
дрида (I) путь превращений начинался переходом ангидрида во фталимид (П) (дей-
ствием сухого аммиака на расплавленный фталевый ангидрид), далее шло восста-
новление фталимида оловом и соляной кислотой во фталнмидии (41), превращение
фталимиднна действием азотистой кислоты в нитрозосоедииеиие (IV) и расщепление
последнего с образованием фталида (V):
Описан также способ превращения фталимида во фталид непосредственно дей-
ствием цинковой пыли и раствора едкого натра иа холоду с последующим нагре-
ванием для удаления аммиака и кипячением после подкисления соляной кислотой.
Ход превращений здесь таков:
/\/\ /\/\ /\ /СН,ОН
I I >4 I I
« Х/\СООН
Наконец недавно описан в немецком патенте способ восстановления самого
фталевого ангидрида и его замещенных действием водорода в присутствии катали-
затора гидрирования (никеля) в среде подходящего растворителя 16).
Редко встречаемый в практике получения промежуточных продуктов пример
перехода карбоксильной группы в альдегидную становится интересным в приме-
нении к получению ароматических оксиальдегидов, в частности ценного исходного
для душистых веществ салицилового альдегида. Этот переход
достигается обработкой соли кислоты в водном растворе в присутствии борной
кислоты амальгамой натрия (или магния). Такой процесс может быть проведен и
электрохимическим путем (прн ртутном катоде). Выделение альдегида облегчается
введением ароматического амина (п-толундииа), дающего с альдегидом бензилиде-
новое соедниеине 17).
б) Иные примеры восстановительных реакций
Практика присоединения вэдорода к ненасыщенным участкам
молекулы ароматических соединений (к двойным связям) с пере-
ходом их в циклические насыщенные соединения менее интересует
400
собственно красочное производство, но имеет значение для полу-
чения некоторых соединений, важных для народного хозяйства
(растворителей для лако-красочной промышленности, жидкого го-
рючего и т. п.). Наиболее употребительны каталитические методы
гидрирования в жидкой фазе под давлением с применением мелко-
раздробленных металлов как катализаторов (особенно никеля или
его окислов, смеси никеля с медью и т. п.).
Не останавливаясь здесь более подробно на этих вопросах,
укажем лишь, что техническое значение получили продукты гидри-
рования нафталина (I): тетрагидронафталин (тетралин) (II) и дека-
гидронафталин (декалин) (III)
также продукт гидрирования фенола, соотв. .«-крезола — цикло-
гексанол (IV), соотв. метилциклогексанол
Н2
н/^н • он
IV
н2Х/н2
н2
В практике приготовления нитролаков имеют большое значение
и отвечающие названным циклическим спиртам кетоны: цикло-
гексанон (V) и метилциклогексанон (VI) 18):
Н3СН
Работам по гидрированию ароматических соединений под давле-
нием в присутствии катализаторов начало положено исследованиями
В. H. Ипатьева.
Некоторые ароматические соедииеиия прн восстаиовлеини претерпевают изме-
нения в самом углеродном скелете. Так например ₽Р-дииафтол-аа-метаи распадается
прн обработке цинком в щелочном растворе на р-иафгол н а-метнл-р-иафтол
гн
^6 Зак. 2611. — Н. Ворожцов
401
В иных случаях по месту бывшей СО-группы возникает углеродная связь
между двумя углеводородными остатками. Подобные превращения рассмотрены
будут ниже.
Восстановление в применении к группам, содержащим серу,
частично было рассмотрено в главе о сульфировании. Восстано-
вление сульфокислот до сульфиновых кислот легче всего прово-
дится через стадию сульфохлорида. Помимо указанных выше спо-
собов восстановления
RSO2C1 -+ RSO2H
имеют применение цинковая пыль (рассчитанное количество) в спир-
товом растворе на холоду, также сернистый натрий при нагревании.
Описаны и другие методы восстановления, в том числе и электро-
химический 20).
Для получения тиофенолов из сульфокислот пользуются в ка-
честве исходного материала также сульфохлоридом. Восстановление
RSO2C1 -+ RSH
проводится посредством цинковой пыли и серной (или концентри-
рованной соляной) кислоты. Можно применять и олово с соляной
кислотой 21). Из сернистых соединений довольно часто в практике
получения промежуточных продуктов встречаются дисульфиды
RSSR, например при обработке диазосоединений дисульфидом
натрия. Их восстановительный переход в тиофенолы осущест-
вляется при посредстве солей сероводорода. Нагревание со спиртовым
раствором сернистого натрия вызывает образование тиофенола.
Можно восстановить дисульфиды нитропроизводных, не затрагивая
восстановлением нитрогруппы: при этом употребляют сульфгидрат
натрия (NaSH) в количестве % мол. на 1 мол. дисульфида в из-
бытке свободной щелочи:
4RSSR -h ЗНаО -f-2NaSH = 8RSH + Na2S2O3.
Можно пользоваться и сернистым натрием в эквивалентном ко-
личестве, имея в виду равновесия:
NaOH + NaSH Na2S -j- Н2О.
Восстановителями дисульфидов до тиофенолов могут быть,
далее, железо или цинк в кислотной среде, также глюкоза в ще-
лочном растворе (на 1 мол. дисульфида 1 мол. глюкозы и 3—4 мол.
NaOH)22).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1) Elbs, J. pr. Ch. (2) 33, 183 (1886); В., Г. п. 27032 (F. I, 75).
2)M6hlau, Klopfer, Ber. 32, 2148 (1899); Mohlau, Heinze, Ber. 35,
360 (1902); KI ages, Allendorf, Ber. 31, 998 (1898).
3) O. Fischer, Fritzen, J. pr. Ch. 14, 79, 562 (1909); CB 1909, II, 530.
*) Wieland, Ber. 52, 880 (1919);
Scheuing, Berliner, Ber. 56, 1586 (1923).
5) B„ Г. n. 204568 (F. IX, 621).
6) А. В г о c h e t, Г. n. 325562 (F. Х1П, 460); Зелинский, Борисов, Вег.
56, 402 (1923).
7) IG, Г. n. 548820, CB 1932, II, 1977, Ф. n. 721034, CB 1932, II, 1527.
8) A. Baeyer, A. A. Knop, Ann. 140, 1 (1866), O. Marschalk, Ber. 45,
583 (1912).
402
9) Nietzki, Ann. 215, 127 (1883); Em de (S ch eгi ng), Г. n. 352982, 354401,
380503 (F. XIV, 430, 431).
1°) Wil Is tatter, Parnas, Ber. 40, 1406 (1907); D i m г о t h, H i 1 с к e n, Ber.
54, 3053 (1921); Scholl, Ber. 44, 2371 (1911).
ii) Graebe, Liebermann, Ann. 160, 126 (1871); К. H. Meyer, Sander,
Ann. 420, 121 (1920); CB 1920, III, 138; Grandmougin, J. pr. Ch. (2), 76,
137 (1907); Ber. 39, 3563 (1906).
12) К. H. Meyer, Ann. 379, 44 (1911); CB 1911, I, 886.
13) К. H. Meyer, Ann. 379, 37 (1911); К. H. Meyer, A. Sander, 396, 140
(1913); CB 1911, I, 886, 1913, J, 1921.
il) Liebermann, Gembel, Ber. 20, 1854 (1887); К. H. Meyer, Ann. 379,
55 (1911); CB 1911, I, 886. By, Г. n. 201542 (F. IX, 682), A. E ckert, R. Pol-
lak, Mon. 38, 11 (1917). Gr.-E. 249124 (F. X, 575).
15) В a e у e r, Ann. 140, 295 (1866); Graebe, Liebermann, Ann. Spl. 7, 287
(1870); Liebermann, Hdrmann, Ber. 12, 589 (1879); C. Liebermann
A. Bischof, Ber. 13, 47 (1880).
16) Graebe, Ber. 17, 2548 (1884);
K-, Г. n. 267596 (F. XI, 197);
MLB 364414 (F. XIV, 454).
П) H. Weil, Ber. 41, 4147 (1908); Г. n. 196239 (F. IX, 158).
18) Sc h rd ter, (Tetralin G. m. b. H.), Г. n. 299013, 324861 (F.X1I1, 307, 302).
K. Wimmer, Г. n. 300052 (F. XIII, 301); S ch rd ter, Ann. 426, 1 (1922);
CB 1922, I, 556.
Ипатьев, Яковлев, Ракитин, Ber. 41, 996 (1908); Ипатьев, Ber. 42,
2092 (1909).
19) Fries, Hubner, Ber. 39, 435 (1906), MLB, Г. n. 161450 (F. VIII, 163).
ЗД) BDCo, Б. n. 245865, CB 1928, 1, 2288; MLB, Г. n. 224019 (F. X, 115); F 1 c h t e r,
Bernoulli, Ber. 42, 4308 (1909); Fichter, Thamm, Ber. 43, 3032 (1910).
21) E d. Bourgeois, Ber. 28, 2319 (1895); Zincke, Brune, Ber. 44, 188
(1911).
22) MLB, Г. n. 228868 (F. X, 152).
С., Г. n. 193290 (F. VIII, 1369); К., Г. n. 205450 (F. IX, 541).
Claasz, Ber. 45, 2424 (1912); Lecher, Simon, Ber. 55, 2427 (1922).
26*
ГЛАВА XV
КОНДЕНСАЦИИ И ПЕРЕГРУППИРОВКИ
Название конденсация не является таким определенным, как на-
пример окисление, нитрование и пр. Оно часто прилагается к про-
цессам самого различного характера как по природе участвующих
в химической игре соединений, так и по структурным превраще-
ниям, вызываемым ими. Разумея под конденсацией всякого рода
более тесное (плотное, dense) сближение прежде раздельно стоящих
атомов и частиц, нередко говорят о конденсациях там, где по су-
ществу имеются основания к более определенной номенклатуре.
В патентной и научной литературе, случается, называют конден-
сацией реакцию обмена галоида на арил-амино-(соотв. алкил-амино-)
группу, реакцию замены такими же группами гидроксила, иногда
даже алкилирование аминогруппы. Можно найти под названием
конденсации и этерификацию и окисление интермолекулярное и
ангидризацию (CONH2->CN) и т. д.
Такая расплывчатость определения может повести к ряцу недо-
разумений. Мы попытаемся поэтому по возможности сузить понятие
конденсации и будем называть этим именем лишь такого рода
химические процессы, отличием которых служит появление в про-
дукте новой связи между двумя углеродными атомами, каковой
связи не существовало в исходном (или исходных) соединении.
Возникновение новой связи углерода с азотом, кислородом, серой
и т. д. или возникновение связи углерода с углеродом через азот,
кислород и пр. не будут подходить под это определение. Короче
говоря, конденсация непременно сопровождается изменением в про-
дукте углеродного скелета исходного соединения. При этом реак-
ция конденсации происходит при отщеплении из реагирующего
вещества (веществ) элементов, образующих (обыкновенно легкие)
частицы: Н2, О>, Н2О, НС1 и пр. Сказанное выше определяет раз-
личие конденсации от таких процессов, как некоторые виды окис-
ления с изменением скелета: нафталин во фталевую кислоту, ре-
акции присоединения (циангидриновый синтез), полимеризации
(ацетилен в бензол).
Ниже под названием конденсации мы будем понимать такого
рода реакции, в которых при отщеплении некоторых элементов,
входящих в состав исходных органических веществ, образуются
продукты, характеризующиеся появлением прежде не существо-
вавшей связи углерода с углеродом. При этом факт отщепления
не является самым характерным сравнительно с фактом возникно-
вения новой углеродной связи, как мы увидим ниже на примерах
синтезов бензоилбензойных кислот. В этом отношении конденсации
404
имеют много общего с ниже рассмотренными перегруппировками.
Такое определение, суживая круг явлений, подлежащих рассмотре-
нию, оказывается все же достаточно широким по количеству обни-
маемых им отдельных примеров. Нет никакой надежды в кратком
обзоре исчерпывающе изложить обильный материал по этому во-
просу. Также нет возможности дать общие формулы этих процессов.
Эти последние имеют значение лишь для некоторых групп явлений;
далеко не во всех случаях имеется достаточно данных для соста-
вления картины течения всего процесса в целом.
Некоторые конденсации протекают без участия посторонних ве-
ществ,— для их выполнения достаточно лишь привести в сопри-
косновение при подходящих условиях температуры исходные ма-
териалы.
Чаще приходится пользоваться для успеха конденсации приба-
влением посторонних веществ, так называемых конденсирующих
средств, число которых очень значительно: неорганические кислоты,
щелочи, соли, восстановители и окислители, некоторые органиче-
ские вещества играют роль конденсирующих средств.
Цель прибавления конденсирующего средства — ускорить ре-
акцию между органическими молекулами. Нередки примеры, когда
конденсирующее средство само вступает во взаимодействие с ис-
ходными материалами или промежуточно образуемыми соедине-
ниями, делая их более реакционноспособными к дальнейшим пре-
вращениям. Поэтому многие конденсирующие средства являются
типичными катализаторами реакций (см. следующую главу).
Конденсирующие средства не имеют такой специфичности, как
сульфирующий или нитрующий и т. п. агенты, и могут найти
многообразное применение в других реакциях. Мы видим в конден-
сирующем средстве реагент, могущий удалить или побудить к вы-
делению уходящую молекулу (таково конденсирующее значение
окислителей для удаления водорода в виде Н2О, восстановителей
для удаления кислорода в виде Н2О, ZnO и пр.), или реагент,
образующий с одним из ингредиентов реакции соединение,
обладающее большей реакционной способностью по отношению ко
второму ингредиенту, чем молекула самого первого ингредиента.
Образование промежуточных соединений может быть доказано,
правда, далеко не часто.
Что касается отдельных конденсирующих средств, то некоторые из них имеют
ясно выраженную специфичность, другие ее не имеют. Так, более илн менее креп-
кую серную кислоту можно считать водоотнимающим (дегидратационным) средством
по преимуществу (хотя она же иногда отнимает аммиак). Таким же свойством от-
нимать воду обладает соляная кислота (также отнимает и NH3), фосфорный анги-
дрид Р2ОВ и хлорокись фосфора POCI3, щавелевая кислота, уксусиокалиевая и
уксуснонатрневая соли (также отщепляют НС1), хлорное олово SnCl4. Хлористый
цинк содействует иногда отделению воды, иногда хлороводорода, иногда аммиака.
Едкий натр содействует также отщеплению воды, в иных случаях прн одновремен-
ной реакции восстановления.
Как отнимающие галоидоводород средства известны прежде всего хлористый
алюминий А1С13, хлористый циик ZnCl^ в некоторых случаях металлы алюмввий и
цинк, причем возможно, что вначале их воздействия образуются минимальные коли-
чества хлористых солей, которые и производят требуемый эффект. Углекислые соли
щелочных металлов, К2СО3 и Na2CO3, также заслуживают упоминания. Хлористая
медь Си2С12 находит себе применение, в смеси с хлористым алюминием, хлорное
железо FeClg иногда оказывается полезным, так же как хлористый алюминий.
405
В качестве отнимающих галоид средств выступают металлы: натрий, его амаль-
гама, медь (вместе с тем отнимает серу), серебро и цинк.
Если не упоминать об окислителях и осерняющих агентах и восстановителях,
которые по вышесказанному в результате своего прямого воздействия производят
и конденсации, то этим можно закончить перечисление наиболее типичных конден-
сирующих средств.
Необходимо наряду с этим всегда иметь в виду, что реакции
конденсации зачастую проявляют весьма отчетливую избиратель-
ность в отношении конденсирующих средств, и для одного и того
же эффекта, скажем, — отнятия воды, может быть при одном составе
ингредиентов применен лишь один какой-либо реагент из водоот-
нимающих средств и никакой другой.
Органические молекулы, участвующие в конденсации, могут
быть все одинаковы, и в таком случае конденсация происходит или
внутри каждой молекулы (интрамолекулярная конденсация) или
между двумя (и более) молекулами одного и того же или различ-
ных соединений (интермолекулярная конденсация). Далее, конден-
сации могут давать начало соединениям нециклического (неколь-
чатого) строения или образовывать циклические соединения.
Синтез а-цафтола по Фиттигу и Эрдману1) нагреваниемфеиилизокротоио-
вой кислоты—пример интрамолекулярной циклической конденсации:
СООН
\сн2
\/\Лн
сн
+ Н2О
Получение по Лорану2) стильбена из бензилового тиоэфира—конденсация интра-
молекулярная нециклическая:
C6HSCH2 • S • H2CCgHB-> С6НВСН ~ СНС6НВ 4- Н^.
Синтез дифенилэтаиа3) из хлористого бензила помощью металлического натрия—
нециклическая интермолекуляриая конденсация однородных молекул:
СсН5СН2С1 с6нБсн2
4-2 Na -> 4-2 NaCl.
СсН3СН2С1 СвНвСН2
При последующем нашем кратком рассмотрении наиболее типиче-
ских примеров конденсаций мы будем систематизировать их по
отделяющимся при этих процессах, в большинстве неорганическим,
молекулам и будем говорить о конденсациях с потерей Н2О,
с потерей НС1 и пр.
Конденсации имеют очень большое значение как в практике
получения промежуточных продуктов, так и самих красителей.
Образование новых углеродных цепей, колец характерно для мно-
гих реакций получения красителей (например антрахиноновых кубо-
вых).
А. КОНДЕНСАЦИИ БЕЗ ОБРАЗОВАНИЯ НОВЫХ ЦИКЛОВ
а) Реакции с потерей воды (—Н2О)
Для этого вида конденсаций наибольшее значение в качестве
исходных материалов имеют карбонильные соединения, из них
преимущественно альдегиды.
406
Из жирных альдегидов чрезвычайно часто и широко приме-
няется формальдегид в виде технически известного своего водного
40%-ного раствора (формалина).
Механизм отнятия воды от формальдегида и другого (или дру-
гих) органического соединения реакционной смеси выясняется, если
формальдегид СН2О рассматривать в гидратированной форме (gem-
уОН
гликолевой) СН24 . Мы усматриваем тогда два возможных тече-
'ОН
ния конденсации: с отнятием одной гидроксильной группы от муравь-
иного альдегида (а), или с отнятием от него двух гидроксилов (б).
Наиболее типичны по вышесказанному такие схемы процессов
конденсации с формальдегидом:
/ОН
СН2< 4-HR------>R-CH2-OH
'ОН -н2о
(а)
/ОН
сн2<
2\он
HR
HRi
—2Н2О
(б)
При этом в схеме (б) возможна тождественность обоих радикалов.
Конечные продукты реакций в (а) и в (б) могут претерпеть после-
дующие превращения, если в молекуле имеется еще реакционный
водородный атом, соотв. ОН-группа.
Конденсирующими средствами при реакциях с формальдегидом
служат щелочные жидкости (растворы едких и углекислых щело-
чей и т. п.) или (чаще) минеральные или органические кислоты.
Конденсация протекает обыкновенно в водном, часто—разведенном
растворе. Концентрация ионов ОН', соотв. Н’, может быть очень не-
значительной. Их роль чаще всего может быть объяснена катализом.
Формальдегидные конденсации интересны для получения окси-
бензиловых, соотв. аминобензиловых, спиртов и их замещенных,
и аминобензиланилиновых производных и получаемых из них
диаминодиарилметановых и для синтеза динафтолметановых произ-
водных и т. п.
Исходными материалами кроме формальдегида служат таким
Образом соединения с активными ОН- или ЫН2-группами. Приме-
нение углеводородов, например нафталина, вызывает необходимость
конденсировать СН2О [или его производные, например метилаль
СН2(ОСН3)2] в безводной среде посредством энергичных водоотни-
мающих средств (HaSOj*).
Формальдегид, сульфит и ароматические оксисоединения легко образуют ш-суль-
фометилпроизводиые, подобно тому как бисульфитиое соединение формальдегида
может служить для введения ш-сульфометильиого остатка в аминогруппу (см. гл. XI),
например
407
Интересен факт, что эти а>-сульфометильные производные океисоедииений при
нагревании со щелочными растворами фенолов или их производных замешают свою
<о-сульфогруппу иа оксиарильный остаток, например
Подобные соедииеиня после введения сульфогруппы обладают дубящими свой-
ствами 5).
Своеобразная формальдегидная конденсация, в которой участвует
кроме формальдегида, с одной стороны,—амин или фенол (в том
числе и многоатомный), с другой — ароматическое гидроксиламино-
замещенное, дает начало амино-(соотв. окси-) производным арома-
тических альдегидов. Способ такого синтеза приложим к получению
широкого круга альдегидных производных Реакция начинается
вероятно стадией образования окси-, соотв. амино-, спирта, который
затем окисляется в альдегид, соотв. в его азометиновое производное,
например
гидролиз
--------->
(Chs)2nx/x
^/\сно + H2N'/'4'/\sO8H
Конденсация протекает в нейтральной или кислой среде, замеще-
ние альдегидной группой направляется преимущественно в пара-
место к амино- (соотв. окси-) группе. Применение арилгидроксил-
амина в готовом виде не обязательно. Можно пользоваться отве-
чающими ему нитросоединениями и, вводя в реакционную смесь
подходящий восстановитель или пользуясь катодным восстановле-
нием, использовать гидроксиламиновое замещенное в момент его
образования е).
В качестве окислителей промежуточно-образующегося аромати-
ческого алкоголя в дальнейшем развитии процесса оказались при-
годными ароматические нитрозо- (и соотв. изонитрозо-) производные,
408
например нитрозодиметиланилин. Реакция проводится в присут-
ствии минеральной (соляной) кислоты и, если надо, органического
растворителя (спирта). Нитрозосоединение переходит при этом
в амин, образующий с альдегидом также азометиновое соединение.
Этот способ, так же как и вышеупомянутый, имеет значение для
синтеза некоторых трудно получаемых иначе альдегидов, например
ванилина (II) из гваякола (I) 7):
/\/ОН /\/°н
I ---> II
ОНС/\/\оСН8
Взаимодействие незамещенных в азоте ароматических аминов
с формальдегидом, как упомянуто было в главе XII, приводит
к образованию ангидроформальдегидных соединений типа
RN = CH2.
При проведении реакции в присутствии минеральной (соляной)
кислоты получаются продукты конденсации, строения ангидро-
л-аминоароматических спиртов, например
C6H4(NH)CH2.
I___I
Реакция протекает через фазу включения остатка формальдегида
(СН2) в непосредственную связь с азотом аминогруппы и с после-
дующим переходом N-замещенного (I), легче при повышенной
температуре, в вышеупомянутые ангидропроизводные (II) 8), на-
пример
/^/NHaHCl
+ СН2О
Образование таких производных на пути синтеза например
диаминодифенилметановых производных (из анилина и формальде-
гида) вполне вероятно, так как например л-аминобензиловый спирт
дает при нагревании с солянокислым анилином до 80° диаминоди-
фенилметан (I). Также возможно получение и несимметрических
диарилметановых производных (II) 9):
409
Диарилметановые производные—конечные продукты превраще-
ния по схеме (<?), имеющие, как это чаще всего бывает, строение
диаминозамещенных, например
и т. п., играют большую роль в синтезе триарилметановых краси-
телей.
Превращения ангидроформальдегиданилина в дифенилметановое
производное идет через ступень образования га-аминобензилани-
лина (III) — при взаимодействии на холоду ангидроформальдегидани-
лина с солянокислым анилином
/NH2HC1
C6H6NH2HCl + CH2=NCaH5^C6HZ (III)
\CH2NHC6H6
Нагревание в присутствии кислоты с анилиновой солью или
с солью другого ароматического амина превращает аминобензил-
анилин (с отщеплением анилина) в замещенное дифенилметана,
например
/СНз
H2N—CH2NH— Z / + \ nh2^
-+ h2n—\-сн2—/ nh2 4- /nh2
\снз
Подобным образом из аминобензильных производных гомологов
анилина можно получить различные симметрично и несимметрично
построенные диаминодифенилметановые основания, имеющие зна-
чение при синтезе трифенилметановых красителей 10).
Формальдегидная конденсация может повидимому иметь значение для образова-
ния хлористого бензила и его замещенных в ядре. Ароматические углеводороды при
пропускании хлористого водорода, после того как в них суспендирован триоксиме-
тилен (или 4О°/о-ный формалин) в присутствии ZnCl2 или H2SO4, замещают один из
своих водородных атомов на группу СН2С1. Эта интересная реакция протекает
вероятно по схеме (для бензола):
2С6Н6 + О(СН2С1 )2-> 2С6Н3СН2С1 + Н2О
с предварительным образоианием из формальдегида н соляной кислоты дихлордн-
метилового эфира.
410
Также реагируют с формалином, соляной кислотой в присутствии конденсирую-
щих средств (ZnCl2, А1С13> SnClj, FeClg) и гомологи бензола. Описан метод полу-
чения этим путем о- и л-ксилилхлоридов из толуола, которые по восстановлении
(химическим,электролитическим или каталитическим методом) дают о- и п-ксилолы
например
Аналогичен метод для синтеза толуола Н).
Формальдегидная конденсация в применении к фенолам имеет
большое практическое значение как служащая для получения цен-
ных продуктов — пластических масс типа так называемых бакели-
тов 12). В их строении без сомнения имеется связь СН2-группы
с ароматическими ядрами фенола.
Конденсация фенола и их замещенных, например л-хлорфенола в кислой среде
с формальдегидом, приводит к образованию соединений типа полиоксиди- или
триарилметановых соединений, например
Продукты сульфирования таких соединений предлагаются как средство защиты от
моли шерстяных изделий 13).
Конденсации ароматических альдегидов имеют также два типич-
ных течения по схемам:
RCH(OH)24-H3CR1------> RCH — CH2R,-----> RCHzCHR!
-н2о | -н2о (в)
ОН
/R2 /Ra
RCH(OH)2 4- HR.,--► R-CH • OH 4- HR3----> RCH< (г)
-H2O -H2O \R3
В схеме (в) образование промежуточно альдольного соединения
не всегда имеет шансы осуществиться.
Наиболее известны из таких конденсаций с участием ароматиче-
ских альдегидов синтез Бейера и Древсена, имеющий значение
в ряду индиговых красителей, и Перкиновская реакция полу-
чения ненасыщенных ароматических кислот.
И та и другая реакции принадлежат к классу (в). Первая реакция
происходит между о-нитробензальдегидом и ацетоном. Конденси-
рующим средством служит едкий натр в водном растворе. Проме-
жуточно образуется альдольный продукт, который может быть изо-
лирован.
411
Схема реакции:
хх /СНО
СН(ОН)СН2СОСН8
Подобно этому реагирует с о-нитробензальдегидом уксусный
альдегид, пировиноградная кислота 14).
Весьма важная реакция Перкина характерна для ароматиче-
ских альдегидов, каковые при высокой температуре обрабатываются
ангидридом жирной кислоты и натриевой ее солью.
Роль ангидрида и соли кислоты в этой реакции оценивается различно. Исследо-
ватели первоначально считали более вероятным, что ангидрид иужеи лить для водо-
отиятия в конденсации, а реакционной молекулой оказывается лить соль жирной
кислоты. Более поздние работы разъясняют важное значение ангидрида кислоты
и возможность замены соли другими веществами, ие имеющими ничего общего
с карбоновыми кислотами и играющими, как и соль кислоты, роль катализаторов
в этом процессе конденсации. Такими катализаторами являются третичные основа-
ния: пиридин, хинолии и т. п., также щелочнореагирующне соли, например потат.
Значение катализаторов—в эиолизации ангидрида кислоты (I), соотв. продукта при-
соединения (И). Таким образом синтез иапример коричной кислоты проходит че-
рез стадии:
СН3СО.
>0
СНдССУ
СНдСОх I
>0
СН,—с<
2 \он
+ С6Н6СО
I
н
СН3-СОх II
>0
сн2-с<
I I хон
С6Н5СО н
iCHg-CO О
I /01ч (! /он
с6н6с±:сн/ Х'н
-СНдСООН^
......0\ /ОН '
с6н5с=сн/ \н
о
---> CgHsCH—сн-с—он
412
При недостаточной энолизации продукта присоедниеиия возможна иная схема
отщепления кислоты с образованием альдегида:
/СНСОСНз ZO
с6н5сосн2с^-- рн-------> с6нвсосн2с<^н+сн3соон.
Выходы различны в зависимости главным образом от степени энолизации ангидридов
кислот 15).
Реакции типа (г) имеют значение для получения замещенных
триарилметановых дериватов. Кроме ароматического альдегида
в реакции участвуют соединения с подвижным [например под влия-
нием пара-М(А1к)2-группы] водородным атомом. Конденсирующими
средствами служат хлористый цинк, соляная кислота, серная кислота,
ее кислые соли, безводная щавелевая кислота и т. п. кислые, типич-
ные водоотнимающие реагенты.
Типичным примером такого взаимодействия может служить
получение лейкосоединения зеленого малахитового из бензальдегида
и двух молекул диметиланилина
СеЩСНО + г/ N(CH3)2------------->С6Н5СН
\-----/ -Н2о
-N(CHS)J
h
Карбонильная группа кетоиов, подобно альдегидной, вступает в реакцию водо-
отиятия с достаточно активным водородом ароматического ядра. Таким образом из
ацетона и анилина получается л.л'-диамииодифеиилдиметилметаи 16).
СН3 СН3
/---\ 1 /---К /---\ I /---ч
у СО < >—NH, -> H2N-< С—< >-NH2
\--- । ----/ \---/ , х----'
сн3 сн3
Та же в конечном результате реакция водоотиятия, ио проходящая через стадию
образования активных соединений щелочных металлов и 4.4'-тетраалкилдиамииодиа-
рилкетоиов (I) и стадию образования карбинола (II) приводит к получению триарил-
замещеииых этиленов 17), иапример
/Na
[(CH3)2NC6H4]2: СО-----> [(CH^NC.HJ, : С<^ + Н3ССвН5------>
ОН
I
-----► [(CH3)2NC6HJ2: С • CHaCeHs----> [(CH3)2N • C6HJ2: С: CHC^
Промежуточное образование диарилкарбинолов (бензгидролов)
не всегда достигается. Но в отдельных случаях оно имеет место 18).
Готовые бензгидролы имеют наряду с альдегидами значение для
реакции конденсации с водоотнятием. Взаимодействие и в этом
случае обусловливается наличием подвижного водорода во втором
ингредиенте реакционной смеси.
Самое реакцию можно рассматривать при этом как последнюю
фазу реакции типа (г). Конденсирующими средствами являются раз-
веденные соляная или серная кислоты. Эти последние конденсации,
как и таковые по (г), имеют значение для получения лейкосоеди-
нений триарилметановых красителей.
413
Так называемый бензгидрол Мих лер а, тетраметилдиаминоди-
фенилкарбинол (I), получаемый окислением продукта конденсации
диметиланилина и формальдегида, при конденсации его со многими
представителями ароматических веществ, имеющих достаточно
подвижной атом водорода, образует также лейкосоединения (II),
например
(CH3)2N-< )-СН-<
1 4
1 ОН
\-N(CH8)2 + /\-N(CH8)a->
---> СН; [/^-N(CH3)2 ]3 + Н2О
Спирты жирного ряда с ароматическими углеводородами или их
замещенными (например сульфокислотами) часто вступают в реак-
ции конденсации с водоотнятием, давая гомологи углеводородов,
соотв. их замещенных. Особенно легко это удается в применении
к нафталину. Водоотнимающим средством является чаще всего
хлорсульфоновая кислота или олеум, которые вероятно дают сна-
чала со спиртом алкилсерную кислоту AlkOSOaH и сульфируют
углеводород. Наиболее известны полученные таким образом пропил-,
бутил-, амил-замещенные нафталина, сульфокислоты которых прак-
тически интересны как эмульгирующие и смачивающие вещества,
находящие большой спрос в текстильной промышленности (леонил,
некая) 19).
Отметим еще как конденсацию, происходящую с водоотнятием
(при участии хлористого цинка), образование хинофталона (II),
основания хинолинового желтого, при взаимодействии фталевого
ангидрида с хинальдином (I)
СО
возможно с предварительным образованием изохинофталона (III).
б) Конденсации с потерей водорода (дегидрогенизационные оки-
сления)
Окислительное удаление водородного атома с одновременным
насыщением свободного углеродного сродства углеродным же
414
атомом—весьма частый пример конденсаций при получении как
красителей, так и промежуточных продуктов. Достаточно вспомнить
(пара-) фуксиновый плав:
>-СН3 4- О2 + 2
—2Н2О
В качестве окисляющих, следовательно конденсирующих, веществ
в этом случае могут быть применены многие реагенты: хлорное
олово, азотнортутные соли (закиси или окиси), чаще всего нитро-
бензол (в присутствии Fe или FeCl3) и мышьяковая кислота.
В окислительных конденсациях в триарилметаиовые производные центральный,
углерод метана образуется или из метильной группы бензольных замещенных (как
только- что показано); или из простых метановых соединений, или из центрального
атома производного дифенилметаиа, например в процессе образования основания
нового фуксива
h2nx/4
3 | I-j-CH3OSO3Na20
H3CZX/
СН + NaHSO4 + 2 Н2О
в процессе образования кислотного фиолетового
HO3S • С6Н4 • СН2—N • С6Н4 —
I
С2Н5
:СН2+О +
2
___\-N(CH3)2
HO8S • С6Н4СН2—N • С6Н4 -
Сан5 2
CH-CeH4.N(CH3)2
Окисление не останавливается на образовании трехзамещенного
метана, но переходит за эту ступень, давая карбинольное соедине-
ние, соотв. его ангидро-соль, — краситель.
Упомянем далее примеры сцепления двух бензольных циклов
при окислении боковой метильной группы с образованием дифе-
нилэтановых (дибензильных) или дифенилэтиленовых (стильбеновых)
дериватов
RCH3 R—СН2
RCH3+° ~"а° R—СН.2
(1)
RCH3 RCH
+ °2 ^2Н О II <2)
RCH3 RCH
Наилучшим окислителем для обеих реакций является хлорноватистоиатриевая
соль NaClO, ио можно применять и перекись натрия, персульфат аммония (NH4)2S2O8,
перекись свинца или электрохимическое окисление в щелочной ванне. Для получе-
ния дибензильных соединений требуется ие слишком высокая температура и больший
избыток NaOH, для стильбеновых—более высокая температура, меньшее количе-
ство NaOH и избыток NaClO против теоретического количества.
415
Из стильбеновых замещенных особенно важное значение имеет ди-
нитро- (соотв. диамино-) дисульфокислота стильбена такого строения:
о2ы-/ СН—НС—S—ИО2
(H2N) х---< >---z (NH2)
\so3h HO3SZ
Диаминокислота может быть получена путем интрамолекуляр-
ного окисления с раскислением нитрогруппы до нитрозо- (или
азокиси-) группы. Такое самоокисление происходит при нагревании
л-нитро-о-сульфокислоты толуола (I) с раствором едкого натра.
/^/CHs
I
Сцепление двух бензольных (или нафталиновых) циклов непосредственно угле-
родной связью требует чаще окисления в кислотной среде.
Окислителями могут служить: перекись марганца с разведенной H2SO4 (о преиму-
ществах естественной МпО2 см. выше в главе об окислении), перекись свинца
в сернокислотном растворе хромовая кислота и ее кислые соли, марганцовокалиевая
соль в сернокислотном растворе, также хлорное железо.
Легче всего такие конденсации протекают между ароматическими ядрами, обла-
дающими реактивирующими (ОН, МН2) группами, с образованием производных дифе-
нила, динафгила и пр. Очевидно в таких соединениях одни водородный атом,
находящийся в благоприятном положении по отношению к характерной группе,
особенно склонен к отщеплению.
Примером такой окислительной (собственно— дегидрогеиизациониой) конденса-
ции может служить образование тетраметил-М-бензидина из диметилаиилииа; оки-
слитель—перекись свинца в среде серной кислоты 21).
Таково же получение красного конго из аиилиндиазоиафтионовой кислоты и
подобные окисления моиоазокрасителей в бензидиновые дисазокрасители: окисли-
тель—МпО2 в среде сериой кислоты
I
SO3H
1- (соотв. 2-) нафтолы от действия окисной соли железа (FeCl3) или окисей железа,
меди, перекиси марганца переходят в соотв. динафтолы (значение окисей просле-
жено для ^-нафтола я);
'ОН 'ОН но/
416
Иногда конденсации этого рода вызываются действием окислителей в щелочной
среде. Таков например синтез динитродиоксидифеиила из о-нитрофенола: окисли-
тель—КМпО4 в щелочной среде ,8*).
Окисление фенолов бензольного ряда в среде разведенной (6—10%-ной) серной
кислоты посредством кислородных соединений марганца (КМпО4, МпО2 и т. п.)
приводит при достаточном избытке окислителя к образованию продуктов дегидро-
генизационной конденсации, типа полиоксиполифеиилов, простейшим представи-
телем которых является например л.л'-дноксиднфенил:
В продуктах может иметься более длинная цепь гидроксилированных ароматических
ядер. Такие продукты обладают интересным свойством закреплять иа растительных
волокнах основные красители подобно вышеописанным сернистым производным
фенолов ®).
Дегидрогенизационные конденсации без окислителя с участием
каталитических агентов, в частности хлористого алюминия, рассмо-
трены в следующей главе.
в) Реакции с отщеплением кислорода (восстановительные конден-
сации)
Цинковая пыль в безводной среде при нагревании с некоторыми заключающими
СО-группу соединениями отнимает их кислород и создает таким образом двойную
связь между углеродами. Так получается дифталил
О—С—С —О
/ I I \
ОС-Н4Св С6Н4-СО
прн нагревании фталевого ангидрида с цинковой пылью. Такой же эффект обнаружи-
вают и некоторые другие восстановители: олово и соляная кислота, медь и серная
кислота и т. п.
Не забудем, что при кислотном восстановлении кетонов (R)2CO могут по-
лучиться пинаколины
(R)3=C- COR,
а при щелочном восстановлении пинаконы
(R)2=C-C-(R)2
I I
ОН ОН
все также продукты конденсации.
г) Реакции с потерей галоидоводорода (НС1)
Эти реакции принадлежат к числу наиболее важных в ряду
конденсаций и открывают дорогу приготовлению многих весьма
интересных соединений.
Всего легче отнятие галоидоводорода происходит при наличии
в реакционной смеси двух условий: подвижного галоида у одной
составной части смеси и реактивированного подходяще расположен-
ной группой водородного атома у другой.
Подвижной галоид, как известно, характерен для жирных гало-
идных соединений или таких соединений ароматического ряда
(аралкильных), где галоид связан не непосредственно с ядром, но
через боковую алифатическую группу; также легко подвижен он у
галоидоангидридов кислот. Такие вещества и берутся чаще всего
27 Зак. 2611. — Н. Ворожцов
417
для синтезов, проводимых просто при нагревании реакционной
смеси или при добавлении более или менее основных (отни-
мающих кислоту) средств.
Известей синтез оксиальдегидов по Реймеру и ТимануM):
фенолы в растворе едкой щелочи обрабатываются при повышен-
ной температуре хлороформом, — в результате образуются о- и п-
оксиальдегиды.
Общее уравнение этого процесса таково (для незамещенного фе-
нола):
/ОЫа
С6Н5ОН СНС13 4- 4NaOH = С6Н*\СНО + 3NaCl + ЗН2° •
Действием галондоалкилов на нафталин в присутствии галоидных солей метал-
лов и связывающих кислоту веществ получают алкилнафилины, например
CjoHs + ClCHg —* С1он7сн3.
Конденсирующими средствами могут быть ZnCl2, FeCI3; отнимающими кислоту —
ZnO, MgO2’).
Нагревание нафталина с этиленом -|- хлористый водород в присутствии таких
конденсирующих средств, как А1С13 или FeCl3, приводит к образованию этил-
(соотв. полиэтил-) нафталина. Ход реакции здесь вероятно таков
СН2:СН2 —СН3-СН2С1 С1ОН7С2НВ.
В недавнем германском патенте описана конденсация с отня-
тием галоидоводорода, приводящая к образованию аралкиламинов
типа Ат • СН2 • МН2.
Ароматические углеводороды обрабатываются ш-галоидметил-
фталимидом, и полученные продукты расщепляются затем посред-
ством обработки щелочами и кислотами. Конденсирующим средством
является здесь хлористый цинк. Например
/со\
с6н6+С1сн2 • \с6н4
/с°\
>С6НВСН2Ы< >С6Н4.
ХССИ
Полученное производное фталимида при действии водных ще-
лочных растворов на холоду переходит в соль N-замещенной фтал-
аминовой кислоты, например
C6H5CH2N
C6H4 + NaOH
C6H5CH2NHCOC6H4CO2Na
и далее — при обработке разведенной кислотой:
CeH5CH2NHCOC6H4CO2Na 2НС1+Н2О -+
-> C6H5CH2NH2HC1 + С6Н4 (СО2Н)2 4- NaCL
Таким образом можно ввести co-аминометильную группу в бензол,
нафталин (до трех групп в a-положение), в витрацея89).
418
При замещении при посредстве галоидозамешеиного углеводородный остатком
водородного атома ядра фенола или амнна, следовательно при отщеплений галоидо-
водорода, часто повидимому имеет место предварительное образование замещенных
в N-(соотв. О-) атомах и последующая перегруппировка, часто под влиянием хлори-
стого цинка, в С-замещешюе, например30)
С6НВОН + С1С(С6Н6)8 тГнсГ* С6НвОС(СвНв)8 -ёйсц--* (СвНв)3: С • C^OH.
Тетраметилдиаминобензофенон (кетон Мих л ера) (П) получается
с отнятием элементов хлористого водорода из фосгена СОС12 и диме-
тиланилина, при этом связывающим кислоту средством является
избыток диметиланилина; реакция идет в две фазы31):
СОСХ4-2 N(CH3)2->
N(CH3)2 - НС1;
С1СО—Z
-N(CH8)2^
->(CH3)2N—II
\-N(CH3),+
+ С6Н6М(СНз)2.НС1.
Следовательно на 1 мол. фосгена нужно брать не менее 4 мол.
диметиланилина. Обе фазы реакции идут при подходящей для
каждой температуре и без прибавления посторонних веществ (вторая
фаза при нагревании на водяной бане в автоклаве), но оказалось,
зто прибавление хлористого алюминия во второй стадии процесса
тначительно ускоряет процесс и позволяет вести его при низшей
чемпературе. В А1С13 имеется таким образом средство, конденси-
рующее и действующее каталитически. Нужно указать, что хло-
ристый алюминий, прибавленный к диметиланилнну в первой стадии
процесса, содействует образованию (вероятно, с промежуточным
образованием кетона М и х л е р а) фиолетового кристаллического ®2)
Образовавшийся в дайной реакции промежуточный продукт — хлорангидрид
диметил-д-аминобеизойиой кислоты (I) можно закрепить в виде стойкого соедииеиия,
иапрнмер анилида, прибавлением 1 мол. анилина в смесь, полученную при 20° из
4 мол. диметиланилина и 1 мол. фосгена. Для этого требуется кратковременное
нагревание до 50—60° и последующее удаление избытка диметиланилина в виде рас-
твора солянокислой соли 83).
(CH3)2N-Z У~СОС1 + H3NC8H5 (CH3)2N-/__________COHNCeHB
Кетон Михлера и подобные ему диарилкетоны обычно конден-
сируются с вторичным или третичным ароматическими аминами,
содержащими активный атом водорода в пара-положении к азоту.
Кетон или предварительно обрабатывается хлорными соединениями
фосфора (РОС13, РС13) для получения хлорида кетона, или соответ-
ственное галоидное соединение фосфора вводится в смесь кетона
27*
419
с амином. Таким образом получаются интересные триарилметановые
красители, например
[(CHS)2N—]3СО (pcigT
NHC6HS
голубой Виктория В34).
В недавнем английском патенте описан иной метод конденсации производных
бензофенона в триарнлметановые красители, основанный на промежуточном образо-
вании из кетонов в растворе ароматического углеводорода, действием металлического
натрия, натриевых соединений кетонов (I), которые затем реагируют с ароматическим
галондопронзводным, отщепляя галоид (например хлор) и образуя триарилметановые
соединения. Последние при подкислении переходят в красители. Например
, ,ONa
•kC\Na
—NaCl
Cl-NfCHsb
малахитовый зеленый
Этим методом может быть использован большой круг галондо- (в частности
хлоро-) производных; это ие только хлорированные углеводороды; но и галоидо-
производные их замещенных. Замещение ядра группами, реакционноспособными по
отношению к натрию (NH2, NO2), препятствует реакции35).
Особого внимания как конденсирующее средство в реакциях,
сопровождающихся выделением галоидоводорода, заслуживает хло-
ристый алюминий (см. также следующую главу). Реакции с участием
хлористого алюминия носят название реакций Фриделя и
Крафтса по имени первых исследователей, выработавших этот
метод синтеза ароматических производных86).
420
Реакции находят себе применение прежде всего для получения
углеводородов (1) и (2) и кетонов (3), например
С6Н6+ВгСаН5 -> С6Н5 • С2Н6 + HBr; (1)
ЗС6Нв + СНС13 -> НС (С6Н6)8 + ЗНС1; (2)
СеНв4-С1СОСН8->С6Н5СОСН3 + НС1 (3)
и т. п.
Несмотря на практическое значение таких синтезов и очень
большое число исследований, до сих пор не освещен достаточно
ход превращений в реакциях Фриделя и Крафтса. Несом-
ненно одно: хлористый алюминий в большей или меньшей мере
играет роль катализатора в этих реакциях, хотя во многих из них
его количества отвечают стехиометрическим по отношению к одному
из органических ингредиентов.
С одной стороны, известны продукты присоединения хлористого алюминия
к галондоангидридам кислот (также и к кетоиам), образующиеся в стехиометри-
ческих количествах, типа RCOC1 • А1С13, RCOC6HB • А1С13. Такие продукты, тем ие менее,
поскольку они достаточно стойки, не влияют положительно на ускорение реакции.
Добавление уже весьма малых количеств хлористого алюминия сверх потребного
по стехиометрии количества вызывает значительное ускорение реакции. Следова-
тельно образование продуктов присоединения А1С13 является фактически отравле-
нием катализатора.
С другой стороны, известны факты, что конденсирующая роль хлористого
алюминия сохраняется за ним в тех взаимодействиях, где он не образует продук-
тов присоединения с реагентами, как например в превращении
С6НВСН2С1 + С6Н6->С6Н5СН2С6НВ + НС1,
причем прибавление в эту реакционную смесь нвтробеизолз, образующего продукт
присоединения CgHgNOj • А1С13, дает лучший выход для вышеприведенной реакции,
причем нитробензол не вступает в реакцию с органическим хлоропроизводным а).
Влияние хлористого алюминия на химическое взаимодействие
своеобразно. Безекен усматривает это влияние в деформации
(dislocation) молекул. Такие деформированные молекулы оказыва-
ются более активными в реакциях.
Шааршмидт усматривает роль хлористого алюминия, а также
FeCls в активировании ароматического углеводорода и в одновре-
менном ослаблении связей галоидоангидрида или галоидалкила.
По нему, А1С13 проявляет активность побочных валентностей угле-
водорода, причем одновременно ставшие активными главные валент-
ности углеводорода присоединяют молекулу галоидоангидрида,
или галоидалкила (RX). Первичный комплекс оказывается постро-
енным по типу
/К
С13А1::::::С6Н6/
= ХХ
Если этот первичный комплекс устойчив при внутримолекуляр-
ном перераспределении валентностей, то А1С13 связывается в сте-
хиометрическом отношении, и наблюдается молекулярное проте-
кание синтеза.
При отщеплении в неустойчивых комплексах, по месту присое-
динения главной валентностью, галоидоводорода (НХ) и обратном
421
возникновении двойной связи ароматического углеводорода, реге-
нерируется в свободном состоянии А1С13, и процесс течет как
каталитический.
Различные вторичные взаимодействия между продуктами реак-
ции и хлористым алюминием могут при первичном каталитическом
процессе потребовать стехиометрических количеств хлористого
алюминия, например когда продукты реакции (кетоны) образуют
стойкие продукты присоединения А1С13. Заместители в бензольном
ядре, взаимодействующие с А1С13, препятствуют реакции ®8). Схему
присоединения А1С13 к реагентам принимают так или иначе и
другие исследователи реакций Фриделя и Крафтса.
По Шретеру первым этапом реакции является присоединение хлористого
алюминия и углеводорода (бензола), дающее в результате перегруппировки особо
склонное к обмену атомов металлоорганическое соединение (I)
С6Нв + А1С13 СвНв-А1С13 -> СвН5.А1С13 {” I
При взаимодействии галоидалкила, например СН3С1, это последнее соединение
обменивает свой активный водородный атом на метил и с отделением хлористого
алюминия переходит в толуол
С6Н3 А1С12{£[ + СН3С1 -> СвН5А1С12{£[Нз + HCI
QHsAIC^ |^Нз-СвН3СН3 + AlClg.
Допуская большую склонность в промежуточном соединении (I) к обмену водород-
ного атома на алкильные радикалы, можно объяснить этим и проявляющееся, наря-
ду с конденсирующим, деструктивное действие хлористого алюминия, приводящее
к уменьшению боковых цепей в ароматических углеводородах.
Таким образом полученное из толуола, сообразно вышеуказанной схеме, проме-
жуточное соединение (II), встречаясь со второй молекулой толуола, обменивает
метка второго бензольного ядра на водород, и таким путем вместе с ксилолом
(конденсация) образуется бензол (деструкция):
СН3СвН4А1С12[Н + СН3 ^CH3C3HjA1CI2 {^Нз + свНв
П 1 Cen3
СН3С6Н4А1С12 [СН3->СН3С6Н4СН3 + А1С13 30).
Даугерти усматривает в первичных продуктах присоединения хлористого
алюминия как к бензолу, так и к галоидалкилу (RX) комплексные молекулы, поля-
ризующиеся и даже диссоциирующие на ионы.
Для реакций он приводит такие схемы:
RX + А1СЦ г* RX • AlCl3 F R+ — (ХА1С13)~
CeHe + AICI3 £ C3He • А1С13 £ (С6Н5А1С13) ~-Н+
(СвН5А1С13Г-Н + + R+-(XA1CI3)~ $ (CjHjAlClg)-—R+ + Н+-(ХА1С(3)-
(C6H5AIC13)--r+ £ C6H5RAIC13 £ C6H5R+ A1C13
н+— (XAlCIs)” F HXA1C13 F HX 4- A1C13.
Обмен ионами iF и R“^ между комплексами приводит к вхождению алкила.
Так как этапы реакции обратимы, то можно ожидать как конденсирующего, так и
деструктивного действия хлористого алюминия.
Образование комплексов он объясняет октетно-электронной схемой:
:С1: :С1: :С1:
R:X: + А1:С1: ->R:X:A1:CI: £ R+— :X:Al:Cl:
-Cl: :CU
422
Тронов и сотрудники в реакции хлористого алюминия с ароматическим
ядром усматривают также стадию комплексообразования, в котором принимают
участие, с одиой стороны, углеродные атомы бензольного ядра, с другой — атом
алюминия в AlClg. Обусловленное этим ослаблевие электронной связи между ато-
мами С и Н содействует отрыву водородного атома **).
Недавние исследования Воля и Вертипороха подводят
более солидное физико-химическое основание под гипотезу про'
межуточных комплексов, содержащих галоидные соли алюминия 41).
Названные исследователи наблюдали, что тройная система:
С6Нв, С2Н5Вг, А1Вг3, хорошо проводит ток, между тем как раствор
бромистого алюминия в бензоле тока не проводит, а раствор бро-
мистого алюминия в бромистом этиле проводит его слабо.
Связь реагирующих веществ обусловливается образованием
сольвата псевдосоли алюминия, например Al(AlBrJ3 и галоидопро-
изводного, например С2Н3Вг, и переходом этой слабо проводящей
соли в сильно проводящую путем внедрения в нее ненасыщенного
или ароматического углеводорода.
Для тройного соединения из А1Вг3, С2Н3Вг, С0(С2Н5)е принимается
формула
[А1(С2НбВг)п(С18Н30)4] • [А1Вг4]3.
Такое объединение компонентов в одну комплексную молекулу —
первое условие взаимодействия. Ослабление первоначально имев-
шихся связей в органическом галоидопроизводном (энергия акти-
вирования уменьшается на энергию присоединения) является вто-
рым реакционным условием, и выделение галоидоводорода стаби-
лизирует молекулу. Воль и Вертипорох усматривают большую
аналогию в таком действии галоидных соединений алюминия
с другими реакциями замещения ароматических углеводородов,
например нитрованием.
Вспомним, что уже около 30 лет назад Густавсон нашел,
что бензол, хлористый этил и хлористый алюминий образуют жел-
тый маслообразный продукт состава С6Н3(С2Н6)3 • А12С1в, „фермент"
Г уставсона, обладающий каталитическими свойствами. Этот про-
дукт присоединяет различные ароматические углеводороды, которые
в таком состоянии легко реагируют без введения новых порций
А1С13 с галоидалкилами при выделении хлороводорода.
Бензол в первоначальном комплексе при этом лишь постепенно
замещается алкилами, переходя например в двойное соединение
Сб(С2Н3)е • А1С13, обладающее каталитическими свойствами перво-
начального комплекса 42).
Принцип метода Г уставсона использован в английском патенте, предлагающем
пользоваться тяжелым маслом — продуктом реакции между бензолом, галоидалкилом
и хлористым алюминием — для взаимодействия дальнейших порций бензола с гало-
идалкилом, оживляя каталитическую активность первоначального продукта посте-
пенным прибавлением порошка алюминия 43).
Применяя хлористый алюминий в реакциях с жидкими ингре-
диентами, иногда не применяют добавочных растворителей, так
как для поддержания подвижности реакционной среды достаточно
введения избытка реагирующего углеводорода. Нередко однако
вводят в смесь добавочный растворитель; естественно, он должен
быть достаточно индиферентным по отношению к реакции с хло-
ристым алюминием .Чаще всего применяется сернистый углерод,
имеющий преимущество в низкой температуре кипения и потому
регулирующий температуру взаимодействия. Мало реакционный
нитробензол в комбинации с хлористым алюминием удобен как
содействующий большему проявлению активности со стороны
ароматического ядра (см. следующую главу). Применяются также
и легкие нефтяные погоны. Различные растворители отзываются
не всегда одинаково на реакционности углеводородов 44).
Порядок смешения составных частей реакционной массы бывает
различен: или к А1С18 в растворителе прибавляют раствор органи-
ческих составных частей или в готовую органическую смесь вво-
дят постепенно порошок хлористого алюминия.
Важным фактором реакции является температура, каковую
опасно держать высокой, особенно вначале. Поэтому стараются
вести реакцию осторожно и, если надо, при внешнем охлаждении.
Лишь заканчивают реакцию при повышенной температуре.
Аналогичным, но ослабленным конденсирующим свойством обладает безводное
хлорное железо. Описано применеине такового в стадии его образования (нз пирита
и хлористого водорода), правда, по отношению к особенно реакционному хлоропро-
нзводиому — хлористому бензилу 46).
Испытание смесей нз А1С13 н FeCls дало в большинстве реакций Фриделя и
Крафтса прямую пропорциональность выходов содержанию в смесн хлористого
алюминия, особенно в присутствии сернистого углерода как растворителя. Для
реакций с ангидридами кислот, где конденсирующее средство не играет роли отни-
мающего хлористый водород агента (см. ниже), максимум выходов в большинстве
случаев отвечает содержанию 50 молекулярных процентов FeCl3 в смеси FeCl3 н А1С1348).
Для реакций типа Фриделя н Крафтса, проходящих между газообразными
нлн парообразными веществами, возможно, по немецкому патенту, применение вместо
готового хлористого алюминия металлического алюминия в присутствии хлористого
водорода нлн алюминия, активированного прибавкой небольшого количества хлорис-
того алюминия или кратким нагреванием алюминия в токе хлора или хлороводорода.
Вместо чистого алюминия здесь можно применять его сплавы, иапример нз 30% Zn
и 70% А1. Если хлороводород выделяется во время реакции, то нет необходимости
все время его вводить в смесь.
Таким образом возможно получение в трубке, содержащей алюминий (не-
много А1С13), при температуре 50—60°, нз фосгена и бензола — беизофенсва:
2СвН6 + COC12->C6HSCOC6H5 + 2НС1 «).
Заметим, что хлористый алюминий конденсирует смесь олефиновых углеводо-
родов с ароматическими в их гомологи.
Описано применение вместо хлористого алюминия также галоидных соединений
бора, например BF3 48).
Реакции этого типа имеют большое значение и могут быть
распространены на большое число ароматических производных.
При наличии гидроксила в соединении, реагирующем с галоидо-
ангидридом и т. п., можно ожидать прежде всего связывания кис-
лотного остатка кислородом ОН-группы, но в присутствии хло-
ристого алюминия имеет место чаще перегруппировка такого
О-эфира в оксикетон (см. ниже).
Подобно этому, из фенола с хлористым алюминием и арнлсульфохлоридом
получается оксидиарнлсульфон, например
z\/OH /\/СНз /\/он
II II 120—130° I I
II |1 <А1С1з) * | | ___
+ ClOaS/^/ 'ч'/'\5О2—/ ^>-СН3
424
При 170—180° получают пара«изомер оксисульфона48). Применение нитробензола
в качестве растворителя позволяет повидимому обойтись без защиты оксигруппы
в фенолах при действии например хлорангидрндов кислот (Behn).
Что касается галоидопраизводных, то тут возможно большое
разнообразие, и наряду с готовыми галоидалкилами, галоидоан-
гидридами оказалось возможным употреблять и такие смеси, где
еще нет, но где может образоваться галоидозамещенное. Пропу-
ская в смесь бензола с хлористым алюминием НС1 и SOa, получают
бензолсульфиновую кислоту C6H6SO2H 50); может быть, в этом слу-
чае промежуточно образуется монохлорангидрид сернистой кис-
лоты НО—SO—Cl.
Из этих последних синтезов очень интересны и важны те, ко-
торые позволяют образовать в углеводороде (соотв. в его заме-
щенном) альдегидную группу.
Если основываться на вышеприведенной схеме (3), то легко
понять, что получения альдегидной группы в связи с ароматиче-
ским ядром можно ожидать, если ввести в реакцию хлорид С1СОН,
т. е. хлорангидрид муравьиной кислоты. Он не получен, ибо раз-
лагается при образовании на СО и НС1.
Если в ароматический углеводород в присутствии хлористого
алюминия пропускать смесь газов СО и НС1, то реакция получе-
ния альдегида идет, но с ничтожными выходами. Вводя в смесь
добавочный катализатор в виде хлористой меди Си2С12, делающей
очевидно активной окись углерода, Гаттерман и Кох достигли
весьма успешных результатов, делающих доступным техническое
использование этого метода для получения альдегидов б1). Реакция
хорошо проходит с гомологами бензола, и СНО-группа становится
в пара-положение к боковой цепи. Реакцию проводят при посто-
янном размешивании и при надлежащей температуре, колеблющейся
в отдельных случаях между комнатной и 50°.
Окиси углерода пропускают приблизительно в 2 раза больше чем НС1. Взаимо-
действие продолжают в течение иесколькнх часов. Масса в большинстве случаев
в конце реакции загустевает, так что мешалка двигается с трудом илн совсем
останавливается. Реакционную смесь выливают иа лед н очищают обычным способом
(отгонка с паром и очистка альдегида через бисульфнтное соединение).
Способ в описанной форме неприложим к получению бензаль"
дегида, с одной стороны, с другой — к получению альдегидофено"
лов (вида НО • R • СНО) и алкоксиарилальдегидов (типа AlkORCHO),
так как в бензол, так же как в фенолы и в эфиры фенолов, по
этому методу ввести альдегидную группу не удается.
Для бензальдегида А. Н. Реформатскому62) удалось удов-
летворительно разрешить проблему, заменив нерастворимый
в бензоле хлористый алюминий растворимой бромистой солью,
оставив в остальном обстановку процесса прежней. Выход бенз-
альдегида достигает 9О°/о теории.
Кихлер и Буфф рекомендуют применение вместе с тем н ноднстого алюми-
ния, также растворимого в бензоле. Хлористая медь может быть замевеиа броми-
стой нлн просто металлической медью, каковая в течение процесса образует за счет
галоидонодорода реактивирующую соль.
Новый патент немецкой фирмы обходит затруднения прн синтезе бензальдегида
из бензола, пользуясь введением и реакционную смесь ннтроуглеводородов, в ча-
стности нитробензола. Этим достигается переведение хлористого алюминия (соотв.
425
его продуктов присоединения) в раствор и выход бензальдегида в 90°/в по израс-
ходованному бензолу й).
Другой патент описывает метод получения бензальдегида из бензола действием
окиси углерода при повышенном давлении (например в 60 ат} в присутствии хло-
ристого алюминия, активированного посредством TjCl< (1%) и иногда посредством
FeCl3. ° Температура взаимодействия, проводимого во вращающемся автоклаве,
Ввести альдегидную группу в фенолы и фенольные эфиры уда-
лось Гаттерману 55) новым видоизменением реакции Фриделя
и Крафтса, Основываясь на тавтомерной форме синильной кис-
лоты HN : С и на возможности образования из нее и хлористого
водорода хлорангидрида иминомуравьиной кислоты
HN~C —С1
I
Н
который, подобно хлорангидридам кислот, вообще при воздействии
на ароматические соединения в присутствии хлористого алюминия,
казалось, должен давать иминопроизводные альдегидов (альди-
мины), например
HgCOC6H6 + С1 — СН ~ NH----------> Н3СОС6Н4 — СН — NH,
—НС1
Гаттерман применил смесь цианистоводородной кислоты и хло-
ристого водорода, воздействуя ими на фенольные эфиры и фенолы,
и получил с удовлетворительными выходами альдимины, перехо-
дящие при нагревании с разведенными кислотами в альдегиды
RCH~NH-}-H2O -> rch-o + nh3.
Синильную кислоту (безводную) применяют или в жидком виде или в виде
газа. В первом случае в смесь, где уже имеется А1С13 и, если надо, растворитель,
например бензол, пропускают (вначале при наружном охлаждении) хлористый водо-
род; во втором — оба газа вводят одновременно. Реакционную массу выливают
по окончании взаимодействия в воду, затем подкисляют соляной кислотой и отго-
няют с паром летучие части смеси. Если альдегид летуч, то его извлекают из погона
обычными приемами, если он не летуч, то выделяют из остатка после отгона
с паром.
Американские исследователи предлагают упрощение этого синтеза с применением
вместо сииильиой кислоты циавистого цинка Zn(CN)2 (1,5 мол. по отношению
к фенолу в эфирном или бензольном растворе). Сухой хлористый водород пропус-
кают при размешивании в присутствии 1,5 мол. А1С13 =®).
Пользуясь вместо цианистоводородной кислоты нитрилами кислот и реакцион-
ными ароматическими соединениями в качестве второго ингредиента, можно расши-
рить синтез Гаттермаиа иа получение кетонов м).
При действии бромистого циана и хлористого водорода на многоатомные фено-
лы образуются полиоксиальдегиды. Процесс проходит через несколько стадий 58),
например
BrC = N + HCl н- BrCH~NCl
/ОН .ОН
/---/-------------------------------\ гидролиз
НО-< > + BrCH“NCl-------------> НО-< >—CH=NCI----------->
\----/ — НВг \----/
/ОН
------> HO-Z \-CHO
426
Альдегидная группа встает преимущественно в пара-место по отношению к ОН-
Если пара-место занято, то в бензольном ряду о-оксиальдегиды получаются с пло-
хими выходами. Для алкильных эфиров феволов ориентация остается той же (преи-
мущественно пара), ио прн занятом пара-месте легко получаются орто-продукты.
Обычно входит лишь одна карбонильная группа в ядро, даже у полиокси- (соотв.
полиалкокси-) замещенных.
В некоторых случаях хлористый алюминий для этого видоизменения реакции
может быть заменен хлористым цивком.
В недавнее время опубликован ряд патентов немецкой фирмы,
описывающих новый конденсационный метод введения альдегидной
группы в ароматические соединения, в том числе и углеводороды.
Реагентом, вносящим карбонильный заместитель, служит здесь
не окись углерода или цианистоводородная кислота, но остаток
муравьиной кислоты, формил, находящийся в связи с азотом, чаще
всего в виде формильных производных вторичных жирно-арома-
тических аминов, например в виде формилмонометиланилнна
C3H3N(COH)CH3. Конденсирующим средством служат преимуще-
ственно хлорные соединения фосфора (например POCU), также
названы хлористые соединения серы и хлористый алюминий ®).
Взаимодействие между формилметиланилииом и хлорокисью фосфора прохо-
дит через стадию образования продукта присоединения
C8H5N(CH3)CH(CI)OPOCl2 (I).
Этот продукт, так же как полученное из него обработкой пятихлористым фос-
фором дихлоропроизводиое (II),
C6HSN(CH3)CH(CI)OPOC12 + РС15 -> CBH6N(CH3)CHC1, (П) + 2РОС13,
реагирует например с третичными жирио-ароматическими аминами с образованием
сначала продуктов коидеисации (III) с отщеплением НС1, соотв. НОРОС12 с заме
«цепнем пара-места к атому N третичного амииа, например
C8H6N(CH3)CHC12 + C6H-N(CHs)2 -> C6H6N(CH3)CHCI • C6H4N(CH3)2 (II1) + HC1.
Этот продукт (111) реагирует с 1 мол. (II)
СбН5К(СН3)СНС1 • C8H4N(CH3)2 + C6H6N(CH3)CHCI2 -> CI2HCC6H4N(CH3)2 (IV) +
+ C8Hb(CH3)N • CH: N(CH3)(C8H3)Cl.
Гидролиз соединения (IV) переводит его в «-амииоальдегид м).
Для метода возможен довольно широкий круг применений в ря-
дах углеводородов, особенно с отличным по реакционности атомом
водорода (антрацен), оксизамещенных (нафтолы), их О-эфиров,
гетероциклических соединений (пиридин, нафтостирил, карбазол).
В циклические соединения, содержащие кислород, можно вводить группу СНО
при посредстве формамида (HCONHJ, а ие только формильных соединений вторич-
ных аминов, причем конденсирующим средством служит хлористый алюминий или
глинозем. Таким образом из ^-нафтола посредством HCONH2 в присутствии А1С13
(130—140°) можно получить Jl-окси-а-иафтальдегид в1).
Приведенные выше конденсации в присутствии хлористого
алюминия связаны преимущественно с выделением молекулы гало-
идоводорода. Тем не менее имеются и иного типа конденсации,
вызываемые участием хлористого алюминия. Дегидрогенизационные
конденсации, вызываемые им, рассмотрены в следующей главе.
Здесь же мы остановимся на реакциях водоотнятия и перегруп-
пировках.
427
Как выше уже отмечалось, при действии этиленовых углеводородов (пропилен,
бутилен) на нафталин в присутствии хлористого алюминия образуются алкилиро-
ванные производные нафталина, например тетраизопропилиафталин:
С10Н8 + 4СН3 • СН: СН2 -> С10Н4(СН(СН3)2]4 «).
Из сравнительно простых интермолекулярных перегруппировок
или конденсаций с присоединением, производимых в присутствии
хлористого алюминия в реакционной среде, необходимо отметить
прежде всего синтезы ароматических карбоновых кислот из угле-
водородов и их замещенных, с одной стороны, и из углекислого
газа — с другой. Они проводятся при высоких давлениях в не-
сколько десятков атмосфер и при повышенной температуре. Наряду
с карбоновыми кислотами получаются и ароматические кетоны.
Таким образом возможны превращения, например
СвН6+СО2-С6Н5СООН + С6НвСОСеН6 и т. д.
В применении к амино- и окси-производным ароматических углеводородов этим
методом возможно, в зависимости от применяемого давления (60—90 ат), темпера-
туры (100—150°) и конденсирующего средства (А1С13, ZnCl2, FeCl3), преимущественно
получать нли карбоновые кислоты или кетоны вз).
Практически весьма интересны синтезы ароматических
кетокислот из фталевого ангидрида и ароматических углеводо-
родов в присутствии хлористого алюминия, например образование
о-б е нз о и л б енз о й н о й кислоты (I)
В выражении этой реакции не видно отщепления каких-либо
атомов из реагирующих молекул, а только иное структурное раз-
мещение с образованием новой углеродной связи между бензоль-
ным ядром и углеродным атомом, принадлежавшим карбоксильной
группе. Это можно рассматривать как конденсацию двух молекул
в одну по схеме присоединения или интрамолекулярную перегруп-
пировку. Однако наблюдения над течением такой и подобных ре-
акций убеждают в большей сложности этого процесса, чем это
выражает приведенная простая схема.
Прежде всего количество хлористого алюминия, требуемого
для процесса, находится в стехиометрическом отношении с коли-
чеством фталевого ангидрида, затем при взаимодействии фталевого
ангидрида, бензола и хлористого алюминия выделяется хлористый
водород, и в результате взаимодействия получается продукт ком-
плексного характера, заключающий алюминий и о-бензоилбензой-
ную кислоту. Все эти обстоятельства делают вероятным предвари-
тельное образование из бензола, ангидрида и А12С16 промежуточ-
ного продукта присоединения, отщепляющего затем хлористый
428
водород с переходом в комплексное соединение о-бензоилбензой-
ной кислоты 6‘):
СвН4(СО)2О+А12С16+С6Н6 С6Н4(СО)2О • А12С16 • С6Нв
/СОСеН5
СвН5 • СО • СвН4СОО • А12С15 + НС1 -► сен4<
хсоон
Так же как бензол, во взаимодействие с фталевым ангидридом
можно вовлечь аналогичным образом толуол, нафталин, хлорбен-
зол и т. п.
Применение жидких и невысококипящих ингредиентов (бензол, его гомологи,
хлорбензол) позволяет обходиться без введения растворителя в смесь. Применение
твердого углеводорода, например иафталииа для синтеза нафтоилбеизойиой ки-
слоты, требует уже растворителей. В этом случае можно с успехом применить бен-
зол и толуол, менее активные, чем нафталин, по отношению к хлористому алюми-
нию углеводороды, роль которых в данном случае сводится к образованию предва-
рительно активного комплекса с хлористым алюминием н фталевым ангидридом.
В этот комплекс внедряется затем иафтални, вытесняя бензол и образуя затем
обычным порядком иафтоилбеизойиую кислоту вБ).
В этом примере наблюдается такая же конкуренция между менее и более
активными йигредиеитами смеси, какую мы видели на примере хлорирования
нафталина в растворе бензола, соотв. хлорбензола (см. главу IV).
Бензоилбензойная кислота, ее гомологи и аналоги, получаемые
аналогичным синтезом с фталевым ангидридом, служат важными
исходными материалами для получения антрахинона и его произ-
водных синтетическим путем. Отметим из них наиболее важные:
I—производное бензола, II—толуола, III—хлорбензола, IV—нафталина. О превраще-
нии нх в антрахиноновые производные сказано ниже.
д) Реакции отнятия галоида
В этих реакциях существенную роль как конденсирующее средство играет
металл; наиболее употребительный—медь (в состоянии мелкого раздробления—вос-
становленная медь), также металлический натрий. Чтобы сделать галоид, ароматиче-
ски связанный, достаточно подвижным для взаимодействия с металлом, необходимо
присутствие подходяще расположенной реактивирующей группы, ианример NO»ю),
н высокая температура реакции, 200—300°. Из галоидов наиболее подвижен J, наи-
менее—С1. Легко удается реакция в антрахиноновом ряду, где галоид в а-месте
активируется со стороны СО-группы.
Галоид В боковой цепи легко подвергается отщеплению посредством обра-
429
ботки натрием. Так, при кипячении хлористого бензила с Na гладко получают ди-
беизил, дифенилэтаи и):
2СвН5СН2С1 + 2Na = CeH6CH2CH2CeHs + 2NaCl.
Восстановительные влияния (присутствие воды или гидроксильных производ-
ных) препятствуют правильному ходу реакции, содействуя замене галоида водо-
родом.
е) Реакции отщепления серы, металла
Выбором подходящего конденсирующего средства можио вызвать отщепление
и иных атомов или групп.
Для удаления серы пользуются, так же как и для удаления галоида, металлом,
чаще всего медью. При нагревании с заключающими серу соединениями в отсут-
ствии кислорода медь связывает серу, вызывая соединение углеродных атомов,
иапример ®8)
Отнятие металла с образованием углеродной связи в исключительном случае
ацетиленового замещенного производится посредством окисления красной кровяной
солью K3Fe(CN)e в щелочном растворе. По этому способу был получен Бейером
имевший значение для синтеза иидиго дииитродифеиилдиацетилеи:
2C6H4(NO2)C=C • Си QH4(NO2)C==C-C = C(NO2)CeH4.
В следующем разделе об образовании конденсацией циклических соединений
нз соединений с открытой цепью приведены примеры отщепления элементов амми-
ака с образованием индоловых и гидракридииовых соединений.
Б. КОНДЕНСАЦИИ С ОБРАЗОВАНИЕМ НОВЫХ ЦИКЛОВ
а) Реакции с потерей воды
Очень важное место среди этих конденсаций занимают синтезы
антрахинона и его производных.
Прежде всего следует остановиться на интрамолекулярной
конденсации вышеупомянутой о-бензоилбензойной кислоты в ан-
трахинон
СО
/Х/СОХ/Х /\/\/\
Реакция проходит количественно при нагревании исходной
кетокислоты с концентрированной серной кислотой и является
завершительным этапом нового метода получения антрахинона из
фталевого ангидрида и бензола. В Америке этот метод является
единственно применяемым. В Западной Европе—во Франции, Ан
глии и Италии—он также вошел в практику 69).
Подобно этому, из о-(4-толуил)-бензойной кислоты (I) получается
2-метилантрахинон (II), нз о-(4-хлор)-бензоилбензойной кислоты (III)—
430
^-хлорантрахинон (IV), из о-нафтоилбензойной кислоты (V) —
нафтантрахинон (VI) (1.2-бензантрахинон).
СО
/\/\/\
---► | VI
I
Замечательно, что при действии фталевого ангидрида иа Р-нафтол и его произ-
водные в присутствии хлористого алюминия (прн температуре не свыше 150°) по-
лучаются ие нафтантрахиноиовые производные, ио сначала повидимому производ-
ные 1.8-фталоилиафталина (I), например 2-окси-1.8-фталоилиафталии (II)
и только при нагревании с серной кислотой (75—96°) из этих продуктов образу-
ются 1.2-беизаитрахииоиовые производные. При нагревании со щелочными рас-
творами в присутствии окислителя [KsFe(CN)e] образуется 3-В2-оксибеизантрои(Ш),
без окислителя 4-оксибеизантрон (IV) 70).
Антрахиноновые замещенные, особенно оксизамещенные, могут
быть синтезированы из фталевого ангидрида без промежуточной
стадии в виде ароилбензойной кислоты * при использовании со-
ответственного метода конденсации.
Известно, что фенолы, соотв. диоксипроизводные, дают при
дегидратационной конденсации с фталевым ангидридом под влия-
нием типично водоотнимающих средств (ZnCl^, SnCl4) преимуще-
* Ароил—ацил, отвечающий ароматической одиоосиовиой кислоте.
431
ственно фталеины с вовлечением в конденсацию лишь одного
углеродного атома карбоксильной группы и двух ядер фенола,
например
Получение из фенолов окси- (соотв. полиокси-) бензоилбензой-
ных кислот возможно при участии хлористого алюминия. Получен-
ные продукты обычно не замыкаются в антрахиноновые окси-
производные от действия серной кислоты, вероятно вследствие
сульфирующего эффекта.
Применяя при температуре свыше 150° (180—240°) хлористый
алюминий, можно получить за один прием из фталевого ангидрида
и фенолов (в том числе и многоатомных), нафтолов, оксиантраце-
нов— оксиантрахиноны и нх производные. Хлористый алюминий
функционирует при этом уже как водоотнимающее средство 71)-
При этом отношения количеств хлористого алюминия н фталевого ангидрида
меньше, чем прн синтезе бензоилбензойных кислот (мол. отношения ангидрида: AlClg
приблиз. = ®/1 — 5/s); фталевый ангидрид вводится в многократном избытке по
сравнению с фенолом. Таким образом возможно например получение аитрагаллола
из пирогаллола и фталевого ангидрида:
Применяя смесь хлористого алюминия и хлористого натрия
(в отношении 5:1), можно получить оксиантрахинон при нагрева-
нии смеси до 200—230° 78), например
С1 ОН
Ангидрид янтарной кислоты оказывается также реакционноспособным к кон-
денсациям в дикарбонильные производные под влиянием сплацления (160—220э)
с хлористым алюминием (илн смесью А1С13 NaCl) вместе с фенолами или п-ди-
оксисоединеииями 7i).
Такого же типа конденсация фталевого ангидрида с гидрохи-
ноном проходит при содействии серной кислоты вместе с борной
кислотой.
432
Смесь хлористого алюминия и хлористого натрия служит кон-
денсационным средством при водоотнятии с получением бензан-
троновых производных из производных 2-окси-З-нафтойной ки-
слоты. Таким образом например из бензоил-О-эфира 2.3-оксинаф-
тойной кислоты при нагревании его с AlCla-|-NaCl до 140—150°
получают 4-оксибензантрон-З-карбоновую кислоту, причем предва-
рительно происходит перегруппировка* исходного эфира в 1-бен-
зоил-2-окси-З-нафтойную кислоту 75):
СООН
’,Х-СО//\-/ХСООН
он
со ।
он
Типично дегидратационной конденсацией, как и получение антрахинона из беи-
зонлбензойной кислоты, является синтез антантрова (II) из 1.1-дниафтил-2.2'-ди-
карбоновой кислоты (I) илн из 1.1-дииафтил-8.8'-дикарбоиовой кислоты (III)
Здесь имеют значение кислотные конденсирующие средства, главным образом
концентрированная серная кислота 7в). Антантрон прввадлежит к кубовым краси-
телям, производным пирена.
В последнее время кроме фталангидридного синтеза антрахино-
новых производных заслуживает внимания новый путь конденса-
ций, исходными продуктами для которых служат хиноны и угле-
водороды ряда бутадиена. Эти синтезы, сближающие химию кра-
сителей с производством синтетического каучука, для стабилиза-
ции своих продуктов нуждаются в отщеплении водорода, отннмае-
* См. ниже—перегруппировка Фрнса.
28 Зак. 2611. — Н. Ворожцов
433
мого окислителем в виде воды. Таким путем возможно получение
нафтохинона, антрахинона и их гомологов.
Хинон при нагревании иапример с 1.3-бута диеном (с добавлением щелочи или
без нее) дает продукт присоединения, путем перегруппировки при обработке
в спиртовой среде соляной кислотой или при нагревании до 170—175°, переходя»
щий в дигидро-5.8-диоксинафталии-1.4. Последний хлорным железом окисляется
в 5.8-дигидронафтохинон и избытком FeCl3 — через фазу 1.4-диоксииафталина в наф-
тохинон. Схема реакций такова:
Аналогично могут быть получены антрахинон и его гомологи. Так, при действии
изопрена на хинои в присутствии 5%-ного раствора КОН и при последующей
обработке воздухом получен 2.7- и 2.6-диметилантрахинон:
СН2
II
Н3С-С
сн +
II
сн2
с-сн3
I
сн
II
сн2
4- 2.6-диметил-
антрахинон
Реакция проходит также через ряд промежуточных стадий: образование про-
дукта присоединения, переходящего например в тетрагидродиоксиантрацен (I),
окисляющийся в тетрагидроантрахинон (II), тавтомеризирующийся в дигидроди-
оксиантрацен (III), например
ОН ОН
Реакция между диолефиновыми углеводородами и хинонами ускоряется прибавле-
нием катализаторов, например соединениями металлов Си, Со, Ni, Fe. Как окисли-
тели имеют значение перекись водорода, пербораты, марганцевокислый калий.
Подобные синтезы могут иметь исходным материалом не бензохинон, но нафтохи-
нон и его производные 77):
434
Довольно близкую связь с такими методами имеют работы Дильса и Аль-
дера по так называемому диеновому синтезу конденсированных систем те).
Образование важного для производства прочных кубовых кра-
сителей бензантрона из антрахинона и глицерина действием серной
кислоты является примером сложной дегидратацнонной и дегидро-
генизационной конденсации.
Сам антрахинон как исходный материал для синтеза уступает
по своему значению продукту, получаемому его восстановлением—
антранолу или его тавтомеру антрону:
ОН
который приготовляется обычно в первой фазе синтеза бензан-
трона, например при обработке антрахинона в среде серной ки-
слоты медными стружками или цинковой пылью при 35°.
Как известно, глицерин при действии крепкой серной кислоты
при повышенной температуре теряет воду, переходя в ненасыщен-
ный альдегид — акролеин:
НОСН2—СН(ОН) —СН2ОН "2Н СН2 = СН —СНО
Таким образом в реакционной смеси встречаются антранол (соотв.
антрон) и акролеин.
Их превращение в бензантрон можно представить проходящим
по одному из двух путей, из которых в первом принимается реак-
ционной формой антранол, во втором — антрон 79):
28*
435
II
Первый путь предусматривает тип альдольной конденсации
акролеина с участием карбонильной его группы; второй, более
вероятный,—присоединение акролеина к мезо-атому углерода ан-
трона ненасыщенным атомом альдегида с образованием 0-антро-
нилпропионового альдегида. Серная кислота является в последую-
щем водоотнимающим агентом, она же, если нет в среде антрахи-
нона, окисляет выделяющийся водород в воду, раскисляясь сама
до сернистой кислоты. Реакция конденсации в бензантрон опти-
мально происходит в среде 85%-ной серной кислоты при темпе-
ратуре 105—108°.
Важное место средн конденсаций с водоотнятием занимают
реакции получения индоксила (I) из фенил-(соотв. арил-)глицнна
или так называемые индоксиловые плавы, открывающие путь син-
теза индиго из анилина, а не из антраниловой кислоты.
Общую схему реакции с фенилглицнном можно выразить
схемой:
он
НОСО
\:н2
\Z\nh/
Реакция отщепления элементов воды происходит прн высокой
температуре в расплавленной щелочной массе. Удовлетворитель-
ные выходы получаются при применении специальных конденси-
рующих средств, особенно натрнй-амнда NaNHg80). Среда, в кото-
436
рой производят реакцию, состоит из легкоплавкой смеси эквимо-
лекулярных количеств едкого натра с едким кали; прибавляют
иногда цианистого калия KCN. Натрий-амид нет необходимости
готовить заранее, но можно к расплавленной щелочи прибавить
металлического натрия и затем, медленно пропуская сухой аммиак,
вводить фенилглицин. При последнем видоизменении получается
более полное использование аммиака и более хорошие выходы про-
дукта 81).
Сплавление с натрий-амидом фенилглицина — наиболее употре-
бительный метод получения индоксила, а следовательно и индиго.
Введение этого конденсирующего средства в свое время решило
вопрос о методе фабрикации индиго из анилина, так как дало
возможность получать выходы, не меньшие, чем при первом из
осуществленных в практике (на Баденской фабрике) методе из
антраниловой кислоты.
Предложено также в качестве конденсирующих средств добавлять к плаву ще-
лочные илн щелочно-земельные металлы 82', их водородные соединения83), нитриды
щелочно-земельных металлов м), карбиды металлов ®), анилиды щелочных металлов
вместе с гидроокисью и всегда с самвм щелочным металлом 86). При этом необхо-
димо, чтобы состав щелочного плава обеспечивал этим реагентам растворение
соотв. равномерное распределение в массе и следовательно более полное взаимо-
действие с ароматическими продуктами, и чтобы температура плава не была слиш-
ком высока. Предпочтительно работать в отсутствии воздуха или в разреженном
пространстве. Образование индоксила имеет место во всех случаях, во обычно его
не выделяют, но перерабатывают разведенный водой плав дальнейшим окислением
воздухом непосредственно в индиго
Предложено проводить индоксиловый плав, защищая массу от окисления слоем
углеводородов, например нефтяных продуктов и т. п. 87.
Вышеприведенная схема реакции вероятно не отражает всей сложности тече
ния процесса сплавления.
Имея в виду, что плав происходит в присутствии натрия или амида натрия,
Фейхтер представляет его проходящим через такие этапы:
NH ч
СН2
I
/С=О
NaO/
+ Na + NaOH
—Н
—NaOH
NH
fYY*'
--!c=(ONa)2
-NaOH
437
Следовательно считать этот процесс дегидратационной конденсацией можно лишь
формально на основании состава исходного и конечного продуктов ®®).
Превращение окснэтиланилина, одного из возможных исходных материалов для
синтеза нндиго (см. гл. XI), в нндиго—еще более сложный процесс так как прн
сплавленни со щелочью непосредственным продуктом явлиется днгидронндол:
В этом процессе очевидно одновременно с дегидрогенизацией днгндронндола
или самого оксиэтиланнлина происходит окисление, например
Тиоаналог индоксила, так называемый 3-окситионафтен (II)
\/\sch2cooh
можно получить из фенилтиогликолевой кислоты (III), причем
в этом случае применяются кислотные конденсирующие средства:
серная кислота или хлорсульфоновая кислота 8Э). Недавно предло-
жена и фторсульфоновая кислота Конденсация с помощью сер-
ной кислоты (моногидрата или купоросного масла) происходит при
температуре водяной бани, между тем как хлорсульфоновая ки-
слота конденсирует уже при комнатной температуре, а иногда
требуется при этом и охлаждение.
При взаимодействии арилтноглнколевых кислот с C1SO3H оказалось, что выходы
красителей (соотв. тиоиндоксила) существенно зависят от температуры, быстроты
прибавлении реагента н общей продолжительности взаимодействия. Во многих слу-
чаях прибавление безразличных к хлорсульфоиовой кислоте нитросоединеннй (нитро-
бензол, хлорвитробеизол, диннтрохлорбензол) создает наиболее благоприятные усло-
вия для выхода продуктов. Окситнонафтен, подобно индоксилу, не изолируется,
а перерабатывается непосредственно в кубовый краситель (тионнднго н его про-
изводные).
438
Интересный пример конденсации с образованием одновременно двух нвдольвых
циклов дает Маделунг, получивший динндил (IV) из оксал-о-толуидина:
исходный материал для своеобразного синтеза индиго и). Конденсирующим веще*
ством является здесь сухой алкоголя! (амнлат) ватрия.
Шестнчленный углеродно-азотный цикл образуется прн нагревавии со щелочью
(КОН 4- СаО) апетантраинловокалневой соли. Образуется двокснпроизводное хино-
лина, 7-оксикарбостирвл 92) (V):
Это соединение интересно тем, что его изонитрозопроизводное при кипячении
с соляной кислотой переходит в изатин (VI), имеющий важное значение в синтезе
индигоидных красителей:
СО СО СО
Среди конденсаций с водоотнятием необходимо отметить еще
синтезы акридонов из диариламин о-карбоновых кислот. Акридоны
антрахинона являются довольно интересными кубовыми красите-
лями. Водоотнимающими средствами служат здесь хлористые со-
единения фосфора или концентрированная сериая кислота.
Лешнянскнй нашел, что лучшим средством коидевсацнн является хлорокнсь
фосфора, избыток которой можно отогнать после реакции. Пример такой реакции93):
Подобным образом в индиферентных растворителях получаются
антрахинонакридоны, например
439
и т. п.
При этом водоотнятие происходит или посредством хлоридов фос-
фора или посредством ангидро-М-сульфокислоты пиридиния или
просто вследствие нагревания растворителя (нитробензол, поли-
хлориды бензола и т. щ) до высокой температуры 94).
Крепкая серная кислота при 200° конденсирует хиноидиантраниловую кислоту
в хииондиакридон, как это наблюдено В. В. Ш а р в и н ы м 96):
Аналогично получаются тиоксантоны. Тиоксаитоны антрахинонового ряда (I) —
также кубовые красители, например M)
— н2о
б) Реакции с отщеплением СО2 и Н2О
Из этих реакций отметим те, которые имеют отношение к по»
лучению индоксила из о-карбоновой кислоты фенилглицина. Спла-
вление со щелочью (едкое кали или смесь его с едким натром)
при высокой температуре (200° и выше) — употребительнейший спо-
соб такой обработки. При этом реакция проходит через фазу обра-
зования индоксилкарбоновой кислоты (соотв. ее соли), которая, при
продувании через ее раствор воздуха, отщепляет СО2, переходя
в индоксил:
^z^zCOOH
-Н2О
^/^NHCHaCOOH
-СО2
440
сон
NH
Описанный щелочной метод конденсации для устранения окисле-
ния выгодно проводить с вполне безводными материалами и при
сильном разрежении воздуха. В свое время эта реакция была одним
из этапов первого практически осуществленного метода производ-
ства индиго. Ныне он уступил место индоксиловому плаву из фе-
нилглицина. Вне сомнения, и здесь течение процесса сплавления
сложнее, чем выражает эта схема.
В качестве кислотного средства для конденсации можно пользоватьсяуксусным
ангидридом, дающим также индоксил (в виде ацетильного производного) &).
Что касается получения сернистого аналога индоксила — окснтионафтена — из со-
ответственной дикарбоновой кислоты, фенилтиоглнкол-о-карбоновой кислоты (VII),
то н в этом случае конденсация может быть вызвана щелочным плавлением, причем
получается карбоновая кислота окситионафтена (VIIIj
/^/СООН
i VII j
сн2 • соон
- н2о
которая может отщепить далее СО2 и перейти в окситионафтеи, илн реакция про
водится лишь нагревавием дикарбововой кислоты выше температуры ее плавления
с удалением и СО2 н Н2О
/ЧуСООН
• сн2 • соон
— со2 —н,о-
СОН
Промежуточно и в этом случае вероятно образуется окситиоиафтенкарбоновая ки-
слота.
в) Реакции с потерей хлористого водорода
В этой группе реакций отметим, во-первых,те, которые откры-
вают путь к получению производных изатина из хлорангидридов
оксаминовых кислот, например
441
Cl-co
—NH•CO
Взаимодействие происходит в среде нитробензола в присутствии
хлористого алюминия “).
Также при посредстве хлористого алюминия можно получить добавочный угле-
родный цикл в пери-местах аценафтена с хлорангидридом малоновой кислоты (или
ее нитрилом)
HO2C-Z' \-СО2Н
НО2С-<^ у—СО2Н
Такие полициклические соединения переходят при окислении в нафталии-1.4.5.8-
тетракарбоновую кислоту (I), приобретающую интерес для синтеза красителей ").
При наличии особенно подвижного водородного атома, например в гидроксиле,
возможны такие превращения галоидозамещенных, которые хотя и не дают новой
связи между углеродами, но сопровождаются образованием новых гетероциклов.
Реакции эти являются развитием известных методов обмена галоида на кислородные
и тому подобные группы. Примеры 10°):
— 2НС1
Получение изовиолантрона из хлорбензантрона являет пример
такого же отщепления НС1 посредством щелочного агента (едкого
442
кали) (например в амиловом спирте при 120—140°) с образованием
нового углеродного цикла
г) Реакции с потерей воды и водорода (с окислением)
Некоторые из подобных реакций были упомянуты нами выше:
таковы диеновые синтезы антрахиноновых производных, получение
бензантрона из антрона.
В этом месте мы более подробно остановимся на этого типа
реакциях, причем вместе с тем рассмотрим и некоторые уже не де-
гидратационные, но дегидрогенизационные (окислительные) кон-
денсации.
Синтез хинолина по Скраупу—одна из наиболее употреби-
тельных реакций этого рода. Необходимые ингредиенты этого син-
теза— анилин (или иной ароматический амин), глицерин, серная
кислота. Взаимодействие вызывается нагреванием смеси с окисли-
телем; в качестве окислителя при типичном синтезе с анилином
применяют нитробензол, при других аминах — отвечающие им
нитросоединения, либо мышьяковую кислоту. Ясно, что процесс
довольно сложен и наряду с конденсацией в нем имеет место
и просто водоотнятие (с переходом глицерина в акролеин)
Серная кислота необходима как водоотнимающее, а следовательно
конденсирующее средство во всех фазах реакции, окислитель же—
в последней фазе.
Концентрированной серной кислоты берется приблизительно тройное количество
по отношению к амину и глицерина — столько же, сколько серной кислоты, нитро-
соединения (обычно отвечающего взятому амину —для аминофенола, следовательно
нитрофенол и т. п.) — в половинном приблизительно количестве против амина. На-
гревание первое время приходится вести с осторожностью ввиду часто бурного
характера реакции.
443
Можно умерить течение реакции путем введения и смесь с глицерином кри-
сталлического железного купороса. Кроме значения этой соли как участвующей
в передаче кислорода (от иитросоединеиия) возможно, что ее кристаллизационная
вода, разводя сервую кислоту, также содействует умереиию энергии реакции 101).
В некоторых синтезах хинолиновых производвых, по Скраупу, необходвмо
для повышения выхода продукта понижать концентрацию серной кислоты прибавле-
нием иоды.
Продукт реакции выделяют, разведя реакционную массу водой н освободившись»
напрвмер отгонкой с паром, от ивтросоединения. Хинолиновые оснонания не растно-
римы в воде и щелочах, чем и пользуются для их выделения.
Метод имеет весьма общее значение и нашел себе применение
в широком кругу аминосоединений. Этим методом пользуются
не только для получения хинолинов, его гомологов, окси- и про-
чих замещенных, но и для образования хинолинового цикла у го-
товых уже красителей, „хинолинизируя“ их. Таким образом напри-
мер готовят из аминоализарина (I) ализариновый голубой (II):
Заметим, что факт получения синего красителя (II) из 3-нитро-
ализарина, глицерина и серной кислоты был открыт Прюдомом
и структура его выяснена Гребе ранее, чем выработан Скрау-
пом общий метод синтеза хинолина. Синтез по Скраупу, или
„скраупирование", применяется во многих случаях для получения
многообразных замещенных хинолина 102).
Интересный как исходный материал для желтого хинолинового
гомолог хинолина, хинальдин (а-метилхинолин) готовится из ани-
лина и параацетальдегида (или альдоля) в присутствии концентриро-
ванной соляной кислоты.
Взаимодействие начинается вероятно с образования из альдоля
ненасыщенного кротонового альдегида, который с анилином дает
ариламиноальдегид, далее переходящий в циклическое соединение,
подобно как при синтезе хинолина:
/ОН
2СН3СНО -> CH3—CH—CH2—СНО -+ сн3—сн=сн—сно
осн
сн
II
сн
сн3
осн
\сн2
—н.,о
/СН • сн3
NH
444
Водород не выделяется и свободном состоянии, но расходуется на гидрирова-
ние хинолиновых производных, так что н реакциовиой смеси имеются как негидри-
роваиные, так и тетрагидрированные продукты. Продукты выделяются, по англий-
скому патенту, через двойную соль с хлористым цинком. Выходы чистого продукта
далеко отстают от теории.
Американский патент описывает метод получения хинальдина из солянокислого
анилина, кротонового альдегвда н нитробензола. Побочные продукты выделяют
и ииде нерастворимых иитросоедииений обработкой нитритом кислой смеси при
охлаждевии 1<в).
Очень важный в практике получения кубовых красителей антра-
хинонового ряда метод сплавления со щелочами можно уложить
в схему дегидрогенизационной конденсации. Таково например по-
лучение индантренового синего RS из р-аминоантрахинона:
Э. Ш в е н к очень основательно рассматривает этот процесс, как
протекающий через ряд последовательных стадий.
Несколько отклоняясь от схемы Швенка и сторону принятия
промежуточных продуктов присоединения едкого кали к хиноидной
форме аминоантрахинона и тем сближая эту реакционную схему
с рассмотренной выше в главе VI, мы получаем такое выражение
хода процесса:
ОН
СО
С
/\/Ч/Ч
445
он
он
он
о
он
, \/y\/\NH
но °н
HN\/\^ С \у\
н/\он
Отсюда вытекает, между прочим, констатированная опытом не-
возможность получить выход индантрена более 50°/о от теорети-
чески рассчитанного по количеству p-аминоантрахинона, так как
предварительно образованный тетрагидроиндантрен (I) под влия-
нием щелочи претерпевает двоякое расщепление (подобно реакции
Канниццаро в случае альдегидов): в продукт окисления, дигидро-
индантрен (II), являющийся лейкоиндантреном и переходящий в по-
следний при окислении, и продукт восстановления (III), переходя-
щий с потерей воды в 1.2.2'.Г-антрагидрохинонантранолазин (IV),
446
уже ие переходящий в индаитрен. Можно ли найти методы превра-
щения последнего в индантрен, остается пока нерешенным вопросом.
Подобна этой, по Швенку, схема превращения бензантрона в дибен-
зантронил и затем в виолантрои (индантреновый темносиний ВО):
ОН ОН
Получаемый в качестве первичного продукта дигидродибензантро-
нил (V) в последующей дегидрогенизационной конденсации пере-
ходит в дигидровиолантрои (VI). Возможно, что и здесь с послед-
ним продуктом идет, подобная вышеназванной, окислительно-вос-
становительная реакция с переходом половины вещества в продукт,
не имеющий уже свойств кубового красителя *. Такая схема ще-
лочных плавлений антрахиноновых производных повидимому при-
ложима во многих случаях 1М).
Заслуживают внимания окислительно-дегидрогенизациоввые превращения р.р'-
бинафтола (см. выше).
Бинафтол (I) под влиянием некоторых окислителей (например Ag2O в бензоль-
ном растворе) переходит в оксибинафтиленовую окись (II), которая быстро с по-
терей водородного атома (например при окислении посредством K3Fe(CN)e в щелоч-
ном растворе) превращается далее в дегидрооксибивафтиленовую окись (III). Послед-
нее соединение, которое растворяется в органических растворителях, окрашивая нх
в красновато-фиолетовый цвет, диссоциирует на свсбоднь е радикалы (IV, V).
Более энергичные воздействия окислителя превращают сразу бинафтол в ди-
окись динафтилена (VI).
Недавно английскими исследователями установлено влияние прибавки различ-
ных окислов (V2O5, CaO, WO3) на характер продуктов окисления ^-нафтола. При
нагревании с СаО не выше 310° получают только ₽.р'-бинафтсл, при 340° только
* Опыты А. М. Лукина доказывают, что выход виолантрона может быть выше
предельной для схемы Швейка величины в 5С% от теории. Поэтому, по крайней
мере для виолантроновой плавки, схема эта требует пересмотра.
447
окись изоди нафтилена (VII). При нагревании с V2O3 fi-бинафтол образует окись
ди нафтилена (VII) 1м).
Соединения I—III, возможно, образуются в качестве побочных продуктов при
ненормально проводимой плавке нафталии-р-сульфокислого натрия на ^-нафтол.
Продолжительное нагревание плава в открытом сосуде без предохранительного слоя
пара сверху, присутствие окислов железа в плаве, — все условия, благоприятствую-
щие такому течению процесса. Возможны потери (3-нафтола, при перегонке его для
очистки, в результате возникновения таких же. окислительных реакций. Благоприят-
ствующим нежелательным побочным реакциям фактором в этом случае может быть
продолжительное нагревание до высокой температуры, особенно при содержании
в нафтоле окисных соединений тяжелых металлов (железа). Не в этом ли лежат
причины известного смолообразования прн перегонке ^-нафтола?
Диокись дннафтилена (VI) специально интересна как резко окрашенный в жел-
тый цвет продукт, получаемый сравнительно простым дегидрогеиизационным оки-
слением из ^.р'-бинафтола.
Таким образом нагревание р.З'-бинафтола с избытком окиси серебра в индифе-
реитном растворителе служит методом получения дюкиси дан 1фтилеиа. Значительно
практичнее метод нагревания р.р'-бинафтола с окисью меди до 290—300э или кипя-
чения с окисью меди или перекисями тяжелых металлов (МпО2, РЬО2) в нитробеи-
зольном растворе. Сам нитробензол и другие нитросоединеиия могут служить оки-
слителями рЗ'-бинафтола в присутствии поташа или других щелочных безводных
агентов при продолжительном кипячении в них его растворов we).
448
Диокнсь динафтилена, получаемая по вышесказанному достаточно легко и с хо-
рошим выходом, заинтересовала уже заграничных исследователей и предприятия
ввиду возможности получения из нее красящих веществ, хинонов и т. п. Не ли-
шено вероятия, что ее использование приведет к получению новых типов краси-
телей.
д) Реакции с отщеплением С2Н3ОН
Металлический натрий, этилат натрия, натрий-амид — употребительные конден-
сирующие средства в вышеназванных реакциях в алифатическом ряду, когда идет
речь о получении -1.3-дикетонов нли (3-кетокарбоновых кислот (ацетоуксусный
эфир).
В практике получения промежуточных продуктов конденсации
с отщеплением спирта имеют значение при приготовлении пиразо-
лонов, т. е. ароматических замещенных такого соединения
ОС
HN<fs
It
сн2
сн
из гидразоновых производных, отвечающих p-кетокарбоновым эфи-
рам. Конденсация таких соединений в пиразолоновые производ-
ные идет самопроизвольно, без конденсирующих средств, легче
при нагревании. Исходными материалами являются, с одной
стороны, арилгидразины, с другой — эфиры р-кегокарбоновых
кислот или а-дикетодикарбоновых кислот, например диоксивинной
кислоты.
Примеры:
а)
СООС2Н6
СО
| 4- H2N • NHC6H4SO3H
СН2
СООС2Н5
щавелевоуксусный эфир
-Н3О
СООС2Н5
С—N • N Н
сн2
СО ОС2Й5
C6H4SO3H
-------->.
-QHjOH
СООС2Н5
I
С—N
Н2Сч
ОС^
NC6H4SO8H
1-сульфофенил-З-карб-
этокси-5-пиразолон
29 Зак. 2611. — Н. Ворожцов
449
б)
сн8
со
I 4-h2n-nhc6h5
сн2
СООС2Н6
ацетоуксусный эфир
-Н2О
сн8
С—N • N
сн2
С6Н
СОЮС2Н6 ;
-С2НБОН~*
Н
СН3
I
C=N
I
Н2С
1
ОС—
NC6H6
1-феиил-З-метил-
5-пиразолон
Таким же путем готовится сульфокислота последнего 1-л-суль-
фофенил-З-метил-5-пиразолон (из л-сульфокислоты фенилгидразина
и ацетоуксусного эфира).
Пиразолоиовые производные способны к сочетанию с диазо-
соединениями, причем к сочетанию они относятся подобно фейолам
и реагируют преимущественно в энольной форме, например
Н8С • С N
нс/ + XN2R
HOC NC6H5
N
H8C • С
RN: N • C<^
HOC
NCeH6
Пиразолоновая азосоставляющая в азокрасителе сообщает послед-
нему обычно большую прочность по отношению к свету.
1-Феиил-Зметил-5-пиразолон наиболее употребительное в технике производное
пиразолоиа. Ои служит исходным материалом для азокрасителей и некоторых
лекарственных веществ (антипирин, пирамидон).
е) Реакции с потерей аммиака
Чисто углеродные коидеисации такого рода имеют пока второстепенное зна-
чение в технике промежуточных продуктов. Из иих отметим синтез индольных про-
изводных, по Э. Фишеру, нагреванием фенилгидразонов альдегидов и кетонов
с подходящими конденсирующими средствами (хлористый цинк или соляная ки-
слота).
За счет одного из атомов азота гидразона отщепляется при этом молекула
аммиака, и образуется индольное замещенное.
Следует остановиться на одной конденсации производных ин-
дола, практически заканчивающейся отщеплением аммиака, хотя
и проходящей предварительно через иные стадии. Мы говорим
о синтезе изатина по Зандмейеру 1<п)-
450
При нагревании соли гидроксиламина с солью анилина и хлорал-
гидратом (I) в водном растворе происходят последовательно ре-
акции, кончающиеся образованием изонитрозоацетанилида (II):
CC18CH(OH)24-H2NOH -> CClgCHtNOH (I)
CCI8CH: NOH4-C6H5NH2+H2O -> 3HCl-f-C6H6NH • COCH: NOH (II)
Вместо Готового гидроксиламина можно пользоваться раствором его сульфата, как
он получается из дисульфокислоты гндроксиламнна, готовящейся обработкой нитрита
натрия раствором бисульфита (2 мол.), последующим подкислением серной кислотой и
нагреванием полученного раствора до кипения. В процессе получения изоиитрозоацет-
аиилида полезно для высаливания продукта вводить в водвый раствор сернокислый
натрий.
Изонитрозоацетацнлид при нагревании до 60—75° с концентри-
рованной серной кислотой дает с отщеплением аммиака изатин:
Способ имеет общее значение по отношению получения заме-
щенных в ядре изатинов (галоидом, алкилом, алкокси-, карбоксиль-
ной группой) н замещенных в атоме азота (алкилом, аралкилом).
Изатин и его замещенные имеют значение для синтеза кубовых
индигоидных красителей.
Здесь же необходимо упомянуть о довольно важной конден-
сации, сопровождающейся потерей аммиака, с той оговоркой, что
последняя потеря не обусловливает возникновения углеродной связи.
Мы говорим о синтезе гидракридиновых соединений, окислением
которых получаются акридиновые красители.
Если мы проследим этот синтез с начала, то усмотрим в нем на-
ряду с отщеплением NH3 отщепление и других молекул (Н2О,
иногда RNH2 и пр.), т. е. конденсации, уже рассмотренные в пре-
дыдущей главе. Например:
(CH8)2Nx/x/NH2 H2Nx/^/N(CH8)2
29*
451
Конденсирующие средства для первой фазы — минеральная ки-
слота (Н'-ионы) (также щелочь :ОН'-ионы), для второй (отщепле-
ние NH3) — соляная кислота при высокой температуре. Аналогично
протекает несимметрический синтез:
452
Полученные гидракридиновые. производные при окислении в ки-
слой среде переходят в акридиновые красители.
Относительная легкость отщепления молекулы аммиака в ди-
аминопроизводных в значительной мере зависит от относительной
близости двух аминогрупп и соответственно легкости замыкания
цикла атомом азота. В предыдущих примерах обе аминогруппы
в дифенилметановых соединениях стоят в орто-местах к централь-
ному атому углерода. Подобная близость аминогрупп и склонность
образовывать пятичленный гетероцикл имеется в о.о'-диаминоди-
фениле и его производных, переходящих при обработке кислыми
агентами в карбазол и его замещенные. При пользовании разведен-
ными кислотами и нагревании до высокой температуры здесь
можно провести и гидролиз сульфогруппы. Например
+Н2О(2%НС1 или H2SO4)
(200®)(—NHS—H2SO4)
(100°)
-NH3
Своеобразная конденсация в нафтокарбазольные замещенные
происходит в результате обработки нафтиламиновых или нафтоль-
ных замещенных фенилгидразином. и бисульфитом и последующей
кислотной обработки. Отдельные ступени реакции здесь не вполне
ясны, но на основании наших сведений о бисульфитной реакции
мы можем дать им предположительно такое обьяснение, например
+NaHSO3
HOgS
H2N^ SO8Na
п2 + h2n • nhc6h5
1.2-нафтокарбазол-7-сульфо-
кислота ш)
45
Подобные сульфокислоты нафтокарбазолов, получаемые методом
бисульфитной реакции, в последнее время приобрели известное
техническое значение для получения из них путем щелочного
плавления соответственных оксинафтокарбазолов, применяемых
в качестве азосоставляющих для получения с диазосоединениями
красителей прочных и глубоких оттенков при сочетании на во-
локне 11?).
Возможен синтез карбоновых кислот оксинафтокарбазолов из замещенной в 5-
или 8-месте посредством NH2 или ОН 2.3-оксииафтойной кислоты при обработке ее
бисульфитом и арилгидразином. Таким образом получена иапример 7.8-бенз ока р ба-
зол-3'-окси-2’-карбоновая кислота ш)
ОН
1 /СООН
Z3'\ /
W \5/
Циклические конденсации под действием осерняющих агентов
рассмотрены выше в гл. XIII в разделе „Осернение".
В. ПЕРЕГРУППИРОВКИ
В широком смысле перегруппировкой называется такое превра-
щение реакционной молекулы, которое связано лишь с изменением
структуры соединения, но не сопряжено ни с потерей, ни с при-
обретением им новых (качественно и количественно) атомов.
Подобные перегруппировки мы встречали уже многократно
в предыдущем изложении. Напомним например перегруппировку
арилгидроксиламинов в аминооксисоединения, N - нитрозаминов
в нитрозозамещенные, перегруппировки в ряду сульфокислот наф-
талина. Можно указать также на примеры странствия галоидного
атома от азота в аминогруппе и ацетиламиногруппе в ядро 11S).
Если ограничиться лишь такими перегруппировками, которые
изменяют углеродный скелет органического соединения, то трудно
провести резкую разделительную линию между перегруппировкой
и конденсацией: перегруппировка в этом смысле — это интрамоле-
кулярная конденсация без потери каких-либо атомов реагирующей
молекулы. Пример подобной, только интермолекулярной, конден-
сации представляет синтез о-бензоилбензойной кислоты из фтале-
вого ангидрида и бензола: формально, если не учитывать роли
хлористого алюминия, здесь получается новый продукт того же
состава, что и сумма реагирующих молекул. Некоторые из пере-
группировок в указанном ограничительном понимании были уже
освещены в предшествовавшем изложении.
Такова например бензидиновая перегруппировка гидразосоеди-
нений, происходящая под влиянием минеральной кислоты и веду-
щая в результате к образованию п .га'-диаминозамещенных ди-
фенила:
СвН3 • NH • HN • С6Н3 -> H2N • С6Н4 • С6Н4 • NH2.
454
Такая же перегруппировка используется в цепи превращений, кончающихся
образованием диамииобеизаитронового производного. Схема этих превращений
такова:
\/\соон
Н'-иоиы
NH ------>
Исходное азосоедииеиие готовится из амииоазокрасителя (диазосоставляющая—
ж-амииобензойная кислота, азосоставляющая — а-нафтиламии) путем устранения NHa
после диазотирования и кипячения со спиртом.
Этим путем возможно получение и замещенных диамииобензантрона при при-
менении иных, чем вышеприведенные, компонентов, лишь бы была возможна бен-
зидиновая перегруппировка, т. е. были бы свободны месга помеченные *иб.
При описании методов алкилирования указывалось на возмож-
ность перегруппировки вторичных алкилараминов в первичные
амины, гомологичные исходному первичному амину, например
Интересным и имеющим большое значение для практики при-
мером перегруппировки является получение карбоновых ки-
слот фенолов действием углекислоты на феноляты щелочных
металлов при высокой температуре и давлении. При этом в первой
фазе реакции происходит внедрение элементов двуокиси углерода
455
в замещающую группу ароматического радикала с образованием
соли фенилового эфира угольной кислоты:
/x/OCOONa
Этот последний под действием избытка СО2 при высокой тем-
пературе (120—140°) и давлении претерпевает перегруппировку
в соль оксикислоты
/4/OCOONa Z4/COONa
Таким образом готовится салициловая кислота 117).
Автоклав для получения салициловой кислоты изображен на рис. 11.
При нагревании фенолята с углекислотой при обычном давле-
нии, течение реакции иное — используется лишь половинное коли-
чество наличного фенола (синтез Кольбе):
2C6H5ONa + СО2 = C6H4(ONa)COONa+C6H5OH.
Замечательно, с точки зрения влияния отдельных факторов на течение реакции, то
обстоятельство, что, в то время как натриевый фенолят при этой перегруппировке
дает о-оксикарбоновую (салициловую) кислоту, фенолят калия образует п-оксикар-
боиовую, ие имеющую такого технического зиачевия, как первая.
Описанный метод имеет значение для получения оксикарбоно-
вых кислот не только бензольного, но и нафталинового ряда. Тем-
пературный фактор имеет при синтезе карбоновых кислот нафто-
лов большое значение. При 120—140° 0-нафтолят натрия дает
с СО2 под давлением нестойкую 2.1-нафтолкарбоновую кислоту
(t°njI 157°). При нагревании той же реакционной смеси до 200—
250° образуется 2.3-оксинафтойная кислота
С°пл. 216°), имеющая большое значение в практике получения не-
растворимых азокрасителей.
В литературе имеются указания иа различные видоизменения метода получе-
ния 2.3-оксинафтойиой кислоты. Немецкий патент описывает перегруппировку
с введением в смесь р-нафтола в свободном состоянии, причем он при высокой
температуре плава служит растворителем нафтолята.
Американский патент фирмы Du Pont de Nemours & Co рекомендует
проводить перегруппировку при давлении углекислоты, превышающем некоторое
предельное давление. Последнее соответствует давлению СОг, необходимому для
456
предупреждения разложения промежуточно образующейся соли изомерной 2.1-ок-
синафтойной кислоты (II) при нагревании ее до соответствующей каждому пределу
давления температуры. В патенте указано давление в 33 ат при
температуре 225°.
С другой стороны, Швейк центр внимания в перегруппировке
перемещает иа температуру, ие интересуясь совершенно давлением-
ifli <Ri.
.. с О Ю 23 30 40 50
Масштаб: , ।,
Рис. 11. Автоклав для получения салициловой кислоты.
Весьма замечательно, что при подобной обработке ₽-нафтолята калия констати-
ровано образование не только 2.3-оксинафтойиой кислоты, но также 2.6-оксииаф-
тойиой кислоты (IV):
COONa
/\/\/0Н
М 1
\/\//'4'COONa
1,в)
М I
соок
КООС'
457
Э. Швенк устанавливает, что в то время как нагревание натриевой солн р-иаф-
тилугольного эфира (I) до 80—100° разлагает ее с выделением углекислоты, на-
гревание ее же в запаяивой трубке до 120—130° приводит к образованию натрие-
вой соли 2-иафюл-1-карбововой кислоты (II).
При нагревании вполне сухой соли (II) в запаянной трубке до 250—260° или
свыше образуется 2.3-оксннафтойиокислая соль (111).
Подобные перегруппировки известны для калиевой соли салициловой кислоты,
которая при нагревании до 210—220° дает двукалиевую соль п-оксибеизойной
кислоты и фенол
/Чх/СО2К /ХуСО3К
2 | | -> I | + С6Н5ОН
\/'хОН КСУ4/
Перегруппировки при нагревании натриевой соли салициловой кислоты не
происходит, при этом соль только превращается в двунатриевую соль и фенол,
согласно реакциям
CeH4(OH)CO2Na-> C6H5ONa + СО2
CeH4(OH)CO2Na + CeHjONa -> C6H4(ONa)CO2Na + CeH5OH.
Опыты Швеи к а подтверждают также, что при перегруппи-
ровках 2.1-оксинафтойной соли в 2.3-оксинафтойную имеет место
предварительное отщепление углекислоты и образование нафто-
лята. Такая подвижность СО2 обусловливается вероятно переходом
при высокой температуре реакционной формы соли карбоновой
кислоты (I) в фенолятную (II)
COONa
в которой не отяжеленная катионом металла карбоксильная группа
легко отделяется при нагревании.
Соль 2.3-оксикарбоновой кислоты уже стойка при нагревании.
Калиевая соль ее также не перегруппировывается при повышении
температуры. Вероятно превращение в 2.6-оксикарбоновую кис-
лоту (IV) р-нафтолята калия проходит не через стадию 2.3-кислоты,
ио параллельно с ней.
Из этих карбоновых кислот р-нафтола изомер 2.3 имеет на-
ибольшее практическое значение ввиду того, что ее арилиды
общей формулы
X/Xx/\CONHR
где R —арильный остаток, получили большое распространение
в ситцепечатной промышленности в качестве азосоставляющих для
образуемых на волокне так называемых холодных, или ледяных,
458
окрасок, ярких и достаточно прочных. Полученные арилиды, так
называемые нафтолы типа AS, различаются, в зависимости от ха-
рактера арила R, по своим колористическим свойствам (нафтол AS —
производное анилина, нафтол ASBS—.и-нитранилина, нафтол
ASRL — га-анизидина и т. п.).
Введение в практику нафтолов типа AS дало текстильной про-
мышленности новые простые методы получения ярких и прочных
окрасок, вызвало подъем изобретательства для получения новых
диазосоставляющих для нафтолов, тем более, что некоторые из
полученных с нафтолами типа AS непосредственно на волокне азо-
красителей выдерживают в отношении прочности конкуренцию
таких прочных красителей, как индиго и ализарин.
В последние годы интерес к получению оксикарбоновых кислот
и их арилидов все растет.
В диоксипроизводные нафталина возможно ввести две карбоновых группы, на-
пример
Запатентовано получение 3-карбоновой кислоты 6-метилмеркапто- (и соотв.
6-алкокси-) 2-оксинафталнна
\/\/\сО2Н
действием углекислоты на щелочные соли 6-метилмеркапто- (соотв. 6-алкокси-)
₽-нафтола при давлении 50—60 ат и температуре 260—280° 12°).
Констатированы нревращения аминофенолов в оксикарбоновые кислоты при
обработке их щелочного соединения углекислотой при повышенной температуре (I)
Так же реагируют 4-оксидифениламин (II) и л-оксидифениламииовые производ-
ные (III). Таким же путем получены карбоновые кислоты вз хлор-л-крезола н
1.4-дихлорфенола
Описаны арилиды различных карбоновых кислот производных оксидиарилами-
иов и оксикарбазола 122_).
459
2.3-окснантраценкарбоновая кислота получена из калиевого 2-антролата при
действии на него, при 260° и 60 ат давления, углекислоты илн прн продолжитель-
ном нагревании 2-антрола с поташом при 220—230° и 80 ат:
/\/\/\/0Н
1111
2.3-окснантраценкарбоновая кислота интересна как азосоставляющая для зеленых
красителей, образуемых на волокне.
Замечательно, что щелочные производные тиофенолов реагируют с углекислым
газом так же, как и производные фенолов, требуя повидимому лишь более энер-
гичного нагревания и высокого давления. Так, сухой тнофенолят натрия (1), насы-
щенный углекислым газом при давлении в 30—50 ат при 150—190°, превращается
в феннлтноуглекнслый натрий (II). Последний прн 250— 270° и давлении СО2 в
50—70 ат превращается в натриевую соль тносалнцнловой кислоты:
|ш|
COONa
При занятом орто-месте СО2 становится в пара-положение к SH. Калиевый
тнофенолят прн 330—350° и давлении СО3 в 80—90 ат превращается в дифенил-
дисульфнд-4.4-дикарбоновую кислоту 124)
Такой метод может вероятно найти применение для синтеза тиокарбоновых
кислот, интересных как исходный материал прн получении индигоидных красите-
лей, производных тионафтена, если соответственные тнофенолы станут легко до-
ступными, например прн получении нх из хлоропроизводных.
В ряду фенольных производных любопытна перегруппировка, вызываемая дей-
ствием хлористого алюминия илн хлористого цинка у ацилированных фенолов:
последние перегруппировываются при этом в п- (соотв. о-) оксикетоны. Например
ОСОСН3
Эта так называемая перегруппировка Ф р н с а в случаях гомологичных фено-
лов сопровождается иногда переходом алкила на другое место, например
Реакцию с хлористым алюминием производят при нагревании смеси до темпе-
ратуры выше 100°. Повидимому эта перегруппировка не сопровождаетси предва-
рительным омылением эфира и последующим новым замещением водорода ациль-
ной группой 126).
Огметнм наконец интересную перегруппировку в нафталиновом ряду, наблю-
денную Роун сотрудниками. Перегруппировка сопровождается интрамолекулярной
конденсацией прн раскрытии нафталинового кольца. Речь идет о производных
2.1-нафтолсульфокислоты (Тобиаса) н некоторых дназосоединений ^в).
460
Прн действии диазосоедниеинн из п-нитраиилииа, также л-ннтранилииа и не-
которых других аминов на 2-нафтол-1-сульфокнслоту образуется в первую очередь
продукт, отвечающий дназосульфонату (I), который от действия раствора соды
перегруппировывается в натриевую соль п-иитробензол-1газо-₽-нафтохиион-1-суль-
фонат (II). Последний при нагревании с раствором едкого натра без выделения
азота перегруппировывается, с раскрытием кольца (III) н замыканием его в гете-
роцикл, в 3- (n-нитрофеннл-) 1.3-дигидрофталазнн-4-уксусную-1-сульфокислоту (IV):
Этим собственно кончается схема перегруппировки. Замечательна здесь лег-
кость раскрытия шестичленного углеродного кольца и образования нового также
шестнчленного гетероцикла. Путем последовательных обработок можно превратить
полученную сульфокислоту (IV) в соответственное окснсоедивение (V) (гидролиз
кислотой), в продукт восстановления (VI), нагреванием последнего с разведенной
минеральной кислотой, в п-амино-З-феннлфталазон-4 (VII) и восстановительной об-
работкой последнего (цинк н соляная кислота) — перевести его в п-амино-Ы-фенил-
фталнмнднн (VIII)
I
снасоон
Очень интересна также перегруппировка в нафталиновом ряду, установленная
С. В. Богдановым. Действие солянокислого гидроксиламина на бисульфитиое
соединение нитрозо-₽-иафтола (I) приводит к образованию 2-нитрозо-1-нафтол-4-суль-
фокнслоты (III) в виде аммонийной соли. Здесь следовательно атомы азота и ки-
461
слорода меняются местами. Промежуточной фазой вероятно является здесь днокснм
соответственного дикетона (II)
Продукт восстановления III — 2-амнно-1-нафтол-4-сульфокнслота (IV)
ОН
I
SO3H
может быть интересным исходным материалом как дназосоставляющая — в частности
для приготовления диазокопнй с чертежей 127).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
*) Fit tig, Erdmann, Ber. 16, 43 (1883).
2) Laurent (Traite de Chitnie organique p. Ch. Gerhard, Paris 1856, III, 1856).
3) Comey, Ber. 23, 1115 (1890).
i) Grabowcki, Ber. 7, 1605 (1874).
5) MLB, Г. n. 426434 (F. XV, 1292), By, Г. Ц. 87335 (F. IV, 97).
6) G., Г. n. 103578, 105103, 105105, 105798 (F. V, 101—109).
7) А.,заявл. Г. п. H. 73706; Us Rh.,заявл. Г. п. S. 52610 (F. XIV, 437); S. Berg-
mann, Ch. Ztg. 51, 700 (1927).
8) К, Г. n. 95184 (F. IV, 52), 95600, 96851, 96852, 97710 (F. V, 91—94).
9) К., Г. n. 96762 (F. V, 77).
1°) MLB, Г. n. 107718 (F. V, 78); Сравн. H. King.Soc. 117, 988 (1920); CB 1920,
1П, 767.
U) G. Blanc, Bull. (4) 33, 313 (1923), Ворожцов, Юрыгнна, Ж. О. X.
(63), 49 (1930).
Е. Tschunkur (Gen. An. W.), Ам. n. 1727682 (1929); IG, Г. n. 509149, CB
1931, I, 360.
Ср. — для л-бром-хлорнстого бензила. R. Que let. Bull. 41, 329 (1927).
Аналогично также: IG., Б. n. 347892, 347887; CB 1932, I, 2927.
i2) Cp.—Ullmann, Enzykldpgdie der techn. Chem. 1 Aufl., II, 129.
13) IG, Г. n. 530219, 535151, 535152, 536551, 539182, 540208, 542067, 542068,
537768, 541629, 544293; CB 1932, I, 3072—3015.
Ср. — Ворожцов, Юрыгнна, 1. с.
И) J. Tanasescu, A. Georgescu, Bull. (4) 51, 254 (1932); Ср. Ваеует,
Drew sen, Ber. 15, 2856 (188'2), 16, 2205 (1883), также A. Janson, Г. n. 112400
(F. V, 406).
») P. Kalnin, Helv. 11, 977; CB 1929, II, 562; cp. Fittig, Ber. 14, 1824,
(1881); 16, 1436 (1883), Michel, Ber. 34, 918 (1901).
16) Homo Ik a, Am. n. 1591384, CA. 20, 3697 (1926).
W) BDCo, Б. n. 289571, CB 1929, I, 1614; ср. E. H. Rodd, F. W. Lin ch, Soc.
1927, 2174, 2179, CB 1927, II, 2392.
18) Ср. К., Г. n. 45806 (F. II, 26), 73147 (F. Ill, 85), 119461 (F. VI, 228).
i«) IG., Б. n. 253118, 269155, 277277, CB 1928, I, 849; Г. n. 449114, CB 1927, II,
2118; Ф. n. 624843, CB 1928, I, 850; Г. n. 476906, 478332, CB 1930, 1, 3610. A. V e r I e y,
Ф. n. 632124, CB 1928, I. 1708. Ср. H. Meyer, K. Bernhauer, Mon. 53/54, 721
(1929), CB 1930, I, 355.
462
О циклогексилнафтолах (из циклогексанола и нафтолов в присутствии ZnCL)
см. В. Alberti, Ann. 450, 304; СВ 1927, I, П60.
») Green, Wahl, Ber. 30, 3097 (1897).
21) Mi chi er, Pattinson, Ber. 17, 115 (1884\
22) В., Г. n. 84893, 87976; 88595 (F. IV, 846—850).
2») Дианин, Ж. 6, 183 (1874), 14, 130, 281 (1882); Ber. 6, 1252 (1873), 7, 125
(1874), 8, 166 (1875); Walder, Ber. 15,2166 (1882); A. M. Clifford, Am. n. 1717093
(1929), Опыты автора и И. С. Травкина.
24) Дианин, Ж. 23, 507 (1891).
25) IG, Г. п. 432113 (F. XV, 946).
2в) Reimer, Tiemann, Вег. 9, 824, 1268 (1876). Ср. Dоw., Ам. п. 1839526, СВ
1932, I, 1829.
27) IG, Б. п. 260604, СВ 1928, I, 2308.
28) 1G, Ф. и. 628440; СВ 1928, 1, 2309. Ср. t. М., Г. п. 417170 (F. XV., 72).
28) IG, Г. п. 442774 (F. XY, 1700).
з°) J. v. А1 р h е n, Rec. 46, 287; СВ 1927, II, 562; L. С1 a i s е п. Вег. 45, 317
(1912); Г. п. 268099 (F. XI, 181). См. ниже о перегруппировке Фриса.
31) См. Е. А. Шилов, Дементьев, Изв. И. В. П. И. № 3, 194.
33) В., Г. п. 26016 (F. 1 78).
33) MLB, Г. п. 44238. W i n t h е г, Patente d. organ. Chemie, 1, 565.
3*) В., Г. п. 27789 (F. I, 80).
BDCo, Rodd. L inch, Б. n. 272321, no Chem. Age. 1927 Dyest. Monthly
Suppl. 85. Cp. Rodd, Linch, 1. c.
36) Cp. A. A. Ashdown, Earliest Hystory of the Friedel-Crafts Reaction,.
Ind. Eng. Chem. 19, 1063 (1927). Г. Кренцлейн, Хлористый алюминий в органи-
ческой химии, Усп. Хим. I, 427 (1932).
37) J. Boeseken, Rec. 45, 458 (1926); СВ 1926, II, 1925; 2-ше Conseil de Chimie,
Paris, 1926, 449.
S. C. J.-Olivier, Rec. 45. 817 (1926), CB 1927, I, 268.
38) A. Schaarschmidt, Zt. angew. Ch. 37, 286 (1924).
39) G. Schroeter, Ber. 57, 1990 (1924). Ср. B. Leone, CB 1928, I, 2174.
®) G. Dougherty, Am. Soc. 51, 576 (1929); CB 1929, I, 1796. Б. В. Тронов,
Л. В. Ладыгина, И. М. Карпенко, Ж. О. X. I (63), 911 (1931).
«•) A. Wohl, Е. W е г t у р о г о с h, Вег. 64, 1357 (1931).
42) Г. Густа в со и, Ж. 10, 390 (1878). 16, 95(1884); СВ 1903, I, 1333; 1905,1, 1379,
«) Р. С. Rus chen, Б. п. 259507, Вг. А. В. 1926, 1028.
44) Ср. Behn, Г. п. 95901 (F. V. 143); Chopin, Bull. (4) 35, 610; СВ 1S24,
II, 625.
«) J. A. Smythe, Soc. 121, 1270 (1922); СВ 1922, III, 1369; Nencki, Ber. 32,.
2414 (1889); Perrier, Ber. 33, 816 (1900).
«) W. A. Riddel, C. R. Nolle r, Am. Soc. 54, 290 (1932), 52, 436 (1930).
47) В., Г. n. 403507 (F. XIV, 438).
«) F. Hofmann, C. Wulff, Б. n. 307802, CB 1929, II, 2101.
«) Hey den, Г. n. 555409, CB 1932, 11, 1692.
50) Knoevenagel. Kenner, Ber. 41, 3315 (1908).
51) Ga 11 er m a n n, Koch, Ber. 30, 1632 (1897). Об аппаратуре см. N. Scha-
piro, Ch. Ztg. 50. 438; CB 1926, II, 554.
52) Ж. 33, 154 (1901); Cp. Kiichler & Butt, Г. n. 126421 (F. VI, 120).
W) Gelsenkirc.hner Bergwerks AG. & Fr. S ch fit z, Г. n. 403489 (F. XIV, 435).
M) IG, Б. it. 334009; CB 1930, II, 3850.
») Gat terma nn, Ber. 31, 1149 (1898), 32, 278 (1899).
s®) R. Adams, J. Levine, Am. Soc. 45, 2373; CB 1924, I, 1186; R. Adams,
E. Montgomery, Am. Soc. 45, 1518; CB 1924, II, 1189.
57) K. Hoes ch, Ber, 48, 1122 (1915); 50, 462 (1917); H ouben, W. Fischer,
Ber. 60, 1759 (1927).
«*) P. Karrer, Helv. 2, 89 (1919).
W) IG. Г. n. 514415, CB 1931, I. 1361; 519444, CB 1931, II, 3394; Ф. n. 648069,
CB 1929,1, 2825; G. An. W. (К a 1 i s c h e r, S c h e у e r), Am. n. 1763557 (1930); Gras-
selli Co, Am. n. 1717567 (1929).
«>) Vilsmeier, H a a k, Ber. 60, 119 (1927).
si) IG, Г. n. 519806, CB 1931, II, 3394.
*2) IG, Чехо-Слов. n. 34979, CB 1932, II, 2531.
«) IG, Б. гь 307223, CB 1929, I, 3144; К. H. Meyer, H. H о pf f (IG), Am.
n. 1866717 (1932).
463
G. T. Morgan, D. D. Pratt, Б. n. 353464, CB 1931, II, 3266,
6i) G. Heller, K. Schiilke, Ber. 41, 3627 (1908).
65; q. Heller, Г. n. 193631 (F. IX, 669); Heller, Schiilke, I. c.; L. Gresly,
Ann. 234, 234, Ber. 19 Ref. 686 (1886).
ев) и 11 m a n n, Ber. 34, 2176 (1901).
6?) Comey, Ber. 23, 1115 (1890).
68) Ris, Ber. 19 2243 (1886).
69; G. Heller, Zt. angew. Ch. 19, 669 (1906); H. Lauth, Zt. angew. Ch. 39,
653 (1926).
To) IG, Г. n. 555081, CB 1932, II, 2243. В прогиворечие Г. n. (By) 298345 (F. XIII, 390).
IG, Г. п. 555938, CB 1932, II, 2532.
’i) By, Г. n. 298345 (F. XIII, 390).
to,73) H. Waldmann, J. pr. Ch. (2) 126,250(1930); CB 1930, II, 1076; cp. Zahn,
Ochwat, Ann. 462, 72 (1928), CB 1928, II, 352.
«) IG, Г. n. 454762 (1926).
’5) IG, Г. n. 508109, CB 1930, II, 3650; cp. IG, Б. n. 248791, CB 1927, II, 336.
76) L. Kalb, Г. n. 280787 (F. XII, 498), Ber, 47, 1724 (1914).
77) IG Б. n. 324661, Г. n. 494438, 496393, Б. n. 320375; CB 1930, II, 807; Б. n.
341553, CB 1931, II, 497; Ф. n. 37684; CB 1930, II, 124; Шв. n. 143258, CB 1931. 1,
2937; Г. n. 554878, CB 1932, II, 1836.
78) O. Diels, K- Alder, Ann. 486, 191; CB 1931, 11, 435-437.
IG (Kai is ch er, Scheyer), Am. n. 1721560 (1929).
To) O. Bally, R. S c h о 11, Ber. 44. 1656 (1911); M e e r w e i n, J. p r. Ch. (2) 97,
225 (1918).
A. M. Лукин, Ан.-Кр. Пр. 1931, № 11/12, 14; Benzanthrones, Chim. & Ind.
1929 (227).
80) Deutsche Gold &Silbe r-S c h e i d e-A nstalt, vorm Rdssler in
Frankfurt a/M, Г. n. 137955 (F. VI, 567).
si) To же, Г. п. 180394 (F. VIII, 408),
88) MLB, Г. п. 163039 (F. VIII, 404).
83) MLB, Г. п. 166974 (F. VIII, 405).
31) MLB, Г. n. 166213 (F. VIII, 406).
85) MLB, Г. n. 166214 (F. VIII, 407).
80) ClBa, заявл. Г. n. G. 31241 (F. X, 344).
87) D о w Co, Am. n. 1564218, CB 1926, I, 1717.
88) H. Feuchter, Ch. Ztg. 38, 273 (1914).
в») См. например K„ Г. n. 245633, 243087 (F. X. 498, 504), cp. Friedlander,
Woroshtzow, Ann. 388, 15 (1912).
90) IG, Г. n. 464087 (1928)
91) Madelung, Ann. 405, 61 (1914).
92) В., Г, n. 117167 (F. VI, 1224).
93) W. Lesnianski, CB 1929, I, 3105.
9i) Cm. Ullmann, Г. n. 221883 (F. X 703); В., Г. n. 237236 (F. X, 708). Cp. IG,
Г. n. 450921 (1927); Sc. D., Б. n. 333191 (1929).
9d) B. B. HI a p в н н, Ж. 47, 1260 (1915).
98) В., Г. n. 243750 (F. X, 726).
»’) Cp. Friedlander, RGMC 1900, 1.
98) IQ, Г. n. 448946 (F XV, 615); G. An. W., Am. n. 1856210, C. A. 26,3522 (1932).
9>) IG (A. Eckert). Г. n. 439511; CB 1927, 1, 1527; Ф. n. 704634; CB 1931, II,
3267; cp. IG., Шв. n. 134084; CB 1930, I, 1372; IG, Б. n. 359201, 363044, CB 1932,
II, 1514.
i°°) By, Г. n. 223367 (F. X, 155); W. (Ильинский), Г. п. 257832 (F. XI, 650).
101) См. Organic Synteses, New York, 1922, II, 79 (Adams, Glarke).
юз) Skraup, Mon. 1, 316 (1880). 2, 139 (1881).
403) Dobner, Miller, Ber. 16, 2465 (1883), 17, 1648 (1884). BDCo, Б. n. 276156;
CB 1928, II, 2286; Nat. An., Am. n. 1752492; CB 1930, II, 139.
4°4) E. Schwenk, Ober Alkalischmelzen der Anthrachinonderivaten, Ch. Ztg. 52,
45 (1928).
I05) Pummerer, Frankfurter, Ber. 47, 1472 (1914).
G. R. Cl emo. R. Spence, Soc. 1928, 2811; CB 1929, I, 651; Cl emo,
LG. Cockburn, Spence, Soc. 1931, 1265; CB 1931, II, 716.
10e) IG, Г. n. 454700 (1927); К., Г. n. 462152 (1928); IG, Г. n. 459363 (1928).
407) T. Sandmeyer, Helv. 2, 234 (1919); G., Г. n. 113848 (F. VI, 573); Cp.
Organ. Syntheses, V, New York 1925, 71.
464
««) Leonhardt, Г. п. 59179 (F. Ill, 290).
10!)) Oehler, Г. п. 43714 (F. II, 104).
НО) MLB, Г. п. 107626 (F. V, 378).
1П) IG, Г. п. 542422; СВ 1932, II, 1516.
1I2) Н. Т. Ви с here г, W. Zimmermann, J. pr. Ch. (2) 103, 277 (1921).
нз) Ср. IG, Б. п. 275326 (1927); Г. п. 533470; СВ 1932, II, 1075.
lw) 1G. Г. п. 553627; СВ 1932, II, 1516.
и5) В1 а п к s m a, Rec. 22, 290 (1903); Cornelia Ch. J. F о n t е i n, Rec. 47,
685; CB 1928, II, 4.
i‘e) С., Г. n. 426317 (F. X'V, 716).
117) Cp. Davies, Zt. Phys. Ch. 134, 57; CB 1928, 11, 3. Об очистке cp. Mon-
santo, Б. п. 353921; СВ 1931, II, 3157.
и8) Е. Schwenk, Ober die Enwlrkung von Kohlensaure auf 2-Naphtolnatrium,
Ch. Ztg. 53, 297, 333 (1929). Там же, библиография вопроса.
Ver. A u s s i g, Г. п. 485274 (F. XVI, 491). IG, Г. n. 436524 (F. XV, 298); By, Г. n.
423034 (F. XV, 295); DuP., Am. n. 1648839 (1927).
и») 1G, Г. n. 554235, CB 1932, II, 1970.
120) IG, Г. n. 548823; CB 1932, II, 777; Г. n. 555006, CB 1932, II, 1701.
121) 1G, Г. n. 525654; CB 1931, II, 1927; Г. n. 526394; Б. n. 336394; CB 1931, II,
1928; IG, Г. n. 554645, CB 1932, II, 1702; IG, Г. n. 536997, 537453; CB 1932, II, 1079.
122) IG, Г. n. 528581; CB 1931, 11, 2785; Ф. n. 39249; CB 1932, I, 587; Г. n. 531480,
CB 1931, II, 3264.
123) ]G, Г. n. 553538, CB 1932, II, 1526.
121) Heyden, Г. n. 514507, CB 1931, 11, 1350.
нз) K. Fries, G. Fink, Ber. 41, 4276 (1908); Fries, Pfaffendorf, Ber. 43)
215, прим. (1910); Fries, Ber. 54, 709 (1921).
K. Auwers, H. Bund esmanh, F. Wieners, Ann. 447, 162, CB 1926, 1, 3470.
S. Skraup, K- Poller, Ber. 57, 2033 (1924).
K. Auwers, Ber. 61, 416 (1928).
K. Auwers, W. M a u s s, Ann. 460, 240, CB 1928, I, 1853.
В. И. Минаев, Ж. 58, 729 (1926).
12в) F. M. R о w e, E. L e v i п, Д. Ch. Burns, J. St. H. Davies, W. Tepper,
Soc. 1926, 690; Rowe, Levin, Soc. 1928, 2550; Rоwe, Himmat, Levin, Soc.
1928 2556
Ц С.’в. Богданов, Ж. О. X. 2 (64), 9, 770 (1932'.
30 Зак. 2611. — И. Ворожцов
ГЛАВА XVI
КАТАЛИЗ И КОНТАКТНЫЕ РЕАКЦИИ
А. ОБЩИЕ ОПРЕДЕЛЕНИЯ И ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
В изложенном выше обозрении главнейших химических процес-
сов, применяемых для синтеза промежуточных продуктов и краси-
телей, мы нередко встречались с понятием катализа и катализато-
ров. В последующем мы несколько подробнее остановимся на вы-
яснении тех явлений, которые принято называть каталитическими,
рассмотрим их техническое значение в интересующем нас круге
химических превращений, их разновидности и практику их исполь-
зования. Такое выделение этой темы тем более необходимо, что
каталитические явления играют весьма важную роль в технически
применяемых реакциях, что некоторые виды катализируемых пре-
вращений, вошедшие в практику сравнительно недавно, быстро за-
воевали для себя широкое признание, и потому наконец, что изуче-
ние теории каталитических явлений позволяет глубоко осветить
ход химических реакций.
Катализом называется явление ускорения, иногда и вызывания,
реакции без притока энергии извне, обусловливаемое присутствием
в реакционной системе некоторого вещества или некоторых ве-
ществ в количествах, иногда очень малых и не находящихся в сте-
хиометрических отношениях с реагирующими веществами системы.
Вещество (или вещества), применяемое для ускорения данной реак-
ции, называется ее катализатором. Количество катализатора и его
состав не изменяются в течение реакции. Катализатор не входит
в уравнение катализируемой реакции. В течение реакции в катали-
заторе имеют место иногда физические изменения (изменение по-
верхности, вида кристаллов и т. п.), и этим обусловливаются нередко
различия в катализирующей способности отдельных катализаторов,
зависящие от продолжительности их службы.
Катализаторы пригодны для определенных лишь реакций, как
говорят, они специфичны. Таким образом имеются например катали-
заторы гидрирования, катализаторы окисления, катализаторы водо-
отнятия и т. п., причем катализаторы одного типа могут быть со-
вершенно неактивными в другом роде реакций. Есть катализаторы
с более широким кругом активности, так же как и обнимающие
более узкий круг только одного рода реакций. Активность ката-
лизатора прежде всего функция его химического состава (при кон-
тактных реакциях помимо этого играют большую роль и характер
и размеры поверхности).
Антагонистом положительного катализатора, ускоряющего, как
466
сказано, реакцию, является отрицательный катализатор, замедляющий
ее. Отрицательные катализаторы иногда называют антикатализато-
рами.
Есть примеры каталитических явлений, где катализатором ре-
акции оказывается или одно из реагирующих веществ или продукт
реакции. Такие реакции носят название автокаталитических.
В зависимости от того, представляет ли катализатор вместе
с реагирующими веществами однофазную или многофазную систему,
различают гомогенный и гетерогенный катализ.
Гомогенный катализ может различаться по агрегатному со-
стоянию химической системы. В практике интересующего нас круга
вопросов наиболее часто встречаются примеры гомогенного ката-
лиза в жидкой среде.
Из разновидностей гетерогенного катализа наибольший интерес
для наших целей представляют примеры применения твердого ката-
лизатора в жидкой среде и твердого катализатора в газовой или
паровой фазе. В гомогенном катализе участвует в реакции вся
масса катализатора, и скорость реакции пропорциональна концен-
трации катализатора. В гетерогенном же катализе скорость реакции
не находится в прямой пропорциональности массе катализатора, но
зависит от величины его поверхности, его физических и химических
свойств. Гетерогенный катализ представляет огромный интерес для
химической промышленности вообще и для производства промежу-
точных продуктов в частности. Выше мы видели уже примеры
этому, а в последующем будем иметь возможность разобрать еще
некоторые. Тем не менее, несмотря на большое число исследований
в этой области, она остается еще очень слабо теоретически осве-
щенной, и достижения в ней завоевывались по преимуществу пу-
тем эмпирических проб и поисков, но не приложением научной
теории.
Сложность проблемы изучения гетерогенной реакции А. В. Раковский харак-
теризует так: „В гетерогенной реакции мы различаем объемную массу фазы и по-
верхность раздела фаз (или поверхность стенок сосуда). Реакция протекает нли
начинается там, где вещества различных фаз впервые встречаются, т. е. иа поверх-
ности раздела; отсюда видна доминирующая роль последней в гетерогенных ре-
акциях. Очевидно, что вещества должны подойти к этой поверхности, а продукты
реакции отойти от нее, следовательно в общем случае явление диффузии н меры
к ее ускорению имеют большое значение для хода реакции. Если реакция происхо-
дит иа поверхности или в тонком поверхностном слое, то казалось бы, что основные
законы кинетики должны прилагаться к концентрациям в этом Слое, а ие к объемным
концентрациям в фазе. Но с точки зрения конечных результатов только последние
представляют интерес, поэтому приходится нли обходить вопрос о конпентрацнях на
поверхности раздела или искать связь между поверхностными и объемными концен-
трациями'.
Для уяснения роли катализатора в реакциях мы вспомним неко-
торые основные положения химической кинетики.
Известно, что скорость химической реакции пропорциональна действующим мас-
сам или концентрациям реагирующих веществ. Скорости реакции определяются как
de
производные концентрации по времени, например -—ц~
В зависимости от числа молекул, вступающих в реакцию, реакции различаются
как реакции первого порядка, илн мономолекулярные, реакции второго порядка, или
бимолекулярные, реакции третьего порядка, или тримолекулярные, н т. д.
В мономолекулярных реакциях (А -> А') имеет место разложение одного вида
30* 467
молекул. Число молекул dN, измененных в единицу времени, пропорционально
числу N всех наличных молекул, т. е.
dN м
~-dT = kN-
Приводим формулу к концентрации (деля на объем):
de к dc к
— —rr- — kc нли —-----= kdt.
dt с
Интегрируя, получаем
— In с = kt-}- const.
При t — 0 н концентрации cOl — In с0 = const. Отсюда
— 1п с -4- in с0 = In -^2- — kt-, I = In ,
и с k с
или, пользуясь показательной функцией,
c = c<^-kt.
.Средняя продолжительность жизни“ превращающихся молекул — При
, 1
t — т — , с = Cq₽, т. е. концентрация уменьшается за t в е раз.
Величина k носит названия константы скорости реакции н является постоянной
при одной и той же температуре. Размерность k обратна времени •
Уравнение, определяющее скорость, может быть дано в иной форме, если при-
нять за а — количество молей реагирующего вещества, во время t = 0 и за х—ко-
личество превращенных молей его во время t.
Следовательно количество молей исходного вещества ко времени t равно а — х,
н скорость реакции, пропорциональная действующей массе а — х,
В бимолекулярной реакции (Л -|-В -> А' + В') при начальных концентрациях
действующих веществ СА — а и Св = b уравнение скорости
dx
= kCACB = k(a — x)(b — X).
После интегрирования получаем
При условии равномолекулярных отношений между А и В, т. е. при а = Ь, уравне-
ние скорости принимает вид
-^- = k(a-x)\
н по интегрировании получаем
kt == --------------------------------.
(а — х)а
Размерность константы скорости бимолекулярной реакции обратна концентрации
н времени. Размерность константы скорости полнмолекулярной реакции определяется
концентрацией в степени 1 — п, где п — порядок реакции, и обратна времени
L/г] = [с]1~ге [£]—\ где размерность [с] = [Af] [L3] ~1
Порядок реакции устанавливается на основании постоянства значений k в урав-
нениях скорости реакции прн подстановке в них определенных опытом значений пе-
ременных.
468
В химической системе возможно нередко протекание не одной,
а нескольких реакций. Получаемое в результате изменение оказы-
вается суммой всех отдельных реакций.
Из таких сложных реакций отметим прежде всего параллельные
и последовательные (консекутивные) реакции.
Параллельными реакции называются тогда, когда реагиру-
ющие вещества взаимодействуют не в одном, а в нескольких направле-
ниях. Примером параллельных реакций могут служить столь час-
тые получения нескольких изомеров при замещениях ароматических
производных.
Среди параллельных реакций интересны „сопряженные" реакции,
где участие в смеси двух не реагирующих друг с другом веществ
третьего реакционноспособного по отношению к одному из них
вызывает проявление реакционности (как говорят, индуцирует ре-
акционноспособность) и другого вещества, без этого неактивного.
Последовательными (консекутивными) реакциями называ-
ются такие, где в течение реакционного процесса имеются промежу-
точные стадии. Таковы реакции синтеза полизамещенных арома-
тических производных (с одним и тем же заместителем). Например
/\ /\/n°2 O2N\^/X/NOa
j 4-nHNO3 -> | I и t. n.
Кинетика многих из рассмотренных нами выше реакций арома-
тических соединений прослежена еще недостаточно. Мы отмечали
уже не раз тот факт, что многие реакции нельзя рассматривать как
односторонние, т. е. идущие до конца. Реакция, начавшись с боль-
шой скоростью в направлении образования определенных веществ,
под конец замедляется и в дальнейшем может сопровождаться
обратной реакцией разложения продуктов реакции. Пример: суль-
фирование и гидролиз сульфокислот в серной кислоте от образо-
вавшейся воды.
Отношение констант скоростей прямой и обратной реакций опре-
деляет постоянную равновесия.
Известно, что скорость химических реакций находится в прямой
зависимости от температуры. Обычно при повышении температуры
на 10° скорость гомогенной реакции увеличивается в 2—3 раза.
Определяя из опыта константы скорости реакции для различных
(постоянных для каждого ряда опытов) температур, можно опреде-
лить „температурный коэфициент реакции” как отношение констант
при температурах 10° и t°, т. е. как At+10„/AfO. Изменение по-
стоянной ke температурой определяется уравнением Аррениуса
(Ван т-Г о ф ф а)
d In k А
dT ~ RT2 ’
где R— газовая постоянная, Т—абсолютная температура, А — так
называемая теплота или энергия активирования.
469
Последнее понятие уясняется на основе представления, что мо-
лекулы, находящиеся в реакционной смеси, обладают различной
энергией и что только столкновение молекул, обладающих некото-
рой энергией выше среднего значения, или так называемых актив-
ных молекул, приводит к реакции. Величина энергии для молекул
данной химической системы, превышение которой характерно для
активных молекул, носит название энергии активирования и выра-
жается в калориях на грамм-моль. Эту энергию, характерную для
реакции, мы можем таким образом вычислить из опытов изменения
скорости реакции (а следовательно k) с температурой. Зависи-
мость между постоянной скорости и энергией активирования (£)
выражается часто другой формулой:
k = Ce RT,
где С — коэфициент, зависящий от природы компонентов реакции
и скорости молекул.
Б. КАТАЛИЗ В ГОМОГЕННОЙ СИСТЕМЕ
Роль катализатора в гомогенной среде для большинства изучен-
ных примеров объясняется химическим его воздействием на реаги-
рующее вещество (или вещества) с возникновением при его участии
промежуточных стадий главной реакции. Ускорение реакции при
посредстве катализатора К при этом находит объяснение в том, что
энергии активирования промежуточной реакции (II), так же как
энергия активирования реакции образования из промежуточного
этапа последнего продукта реакции (III), ниже, чем энергия активи-
рования прямой реакции между двумя реагирующими молекулами (I)
(I) А-\-В -+ АВ-,
(II) А-\-К-^> АК\
(III) АК-^-В АВ + К.
Мы встречали уже неоднократно в ранее изложенном материале
примеры участия в жидкой системе катализаторов и в большинстве
находили объяснение их значения в протекании реакции через про-
межуточные этапы.
Напомним роль серной киолоты в процессе нитрования, особенно окислами азота,
и ее роль в процессе хлорирования бензола по Батт г е. И там и тут мы усматри-
ваем в серной кислоте такой активатор, т. е. катализатор реакции, который вовлекает
молекулы бензола в предварительное взаимодействие с ним самим (образуя с бензо-
лом нестойкие промежуточные соединения), и затем образовавшиеся продукты при-
соединения, под воздействием двуокиси азота, соотв. хлора, дают уже замещенные
бензола.
Прибавкой глицерина к толуолу значительно ускоряется процесс хлорирования
в боковой цепи углеводорода. Факт, установленный наблюдениями Н. Ф. Силина
и В. С. Калининой, заслуживает изучения для выяснения роли глицерина и вообще
многоатомных спиртов в образовании как главных, так и промежуточных
продуктов реакции.
Уместно здесь отметить своеобразный взгляд Г. Е. Армстронга
на течение каталитических реакций в гомогенной среде. Термин
„катализ" он применяет лишь к реакциям, проходящим на поверх-
470
ности твердых тел и ускоряющихся влиянием этих поверхностей.
Он рассматривает все реакции как электролитический процесс. Ре-
акция между неэлектролитами является непрямым процессом, вызы-
ваемым присутствием электролита, действующего на реагирующие
вещества с образованием промежуточных стадий. Электролит, для
которого этот исследователь предлагает название „детерминант",
имеет способность соединять реагирующие молекулы в электропро-
водящий комплекс. Общий вид реакционной схемы по Армстронгу
такой:
A2 + s84-52 АВ-\-АВ-\-&,
где еЗ— электролит детерминант1).
Этот взгляд не противоречит известным до сего времени фактам
катализа в гомогенной среде, которые мы встречали в этой книге.
Явление своеобразного сульфирования антрахинона при участии
ртутных солей — также один из примеров гомогенного катализа.
Без участия ртути сульфирование приводит к замещению р-атома
антрахинона, но уже участие в реакционной смеси малейших следов
ртутной соли делает заметным образование а-сульфокислоты.
Как было указано выше, здесь вероятно имеет место предвари-
тельное участие в реакции ртути с образованием ртутно-органи-
ческого соединения, которое в дальнейшем процессе переходит
в а-сульфокислоту. Это явление замечательно еще и потому, что
направление a-замещения становится заметным только в присут-
ствии ртути, следовательно катализатор не только ускоряет, но вы-
зывает реакцию. М. А. Ильинским, впервые наблюдавшим этот
катализ, был найден и отрицательный катализатор, именно хлори-
стый натрий, введение которого в реакционную смесь парализует
катализирующее влияние ртути. Требует выяснения вопрос, имеет ли
здесь место ионный катализ (со стороны иона ртути) или, что более
вероятно, катализ молекулярного характера, так же как и вопрос
о химическом влиянии антикатализатора.
Последние работы Ла у ера подтверждают вполне положения
Ильинскогои дают определения энергии активирования дляр-суль-
фирования антрахинона: E(W/^ = 8 220 кал.-, Е^^ — 14 220 кал.
Это одна из немногих энергетических характеристик изучаемых
нами реакций2).
В процессе восстановления нитросоединений легко усматри-
вается каталитическое влияние ионного характера. Здесь мы будем
говорить только о реакциии восстановления в гомогенной среде
без участия металлических восстановителей, как железо и т. п., хотя
и в фактах гетерогенного восстановления, которые будут рассмо-
трены ниже, каталитическое влияние со стороны ионов имеет место.
В своем месте было изложено различие между так называемым
кислотным и щелочным методом восстановления нитросоединений
и на основании работ Бранда сделан вывод о влиянии щелоч-
ности раствора (концентрации ОН'-ионов) на выход азоксисоедине-
ния при применении растворов сернистых щелочей в качестве вос-
становителей.
Представляется наиболее вероятным в этом случае каталити-
ческое влияние ОН'-ионов видеть в воздействии их на всю моле-
471
кулу нитросоединения, толкающем ее в направлении образования
ациформы ннтрогруппы, которая при воздействии водорода или
отдающих водород реагентов легко может перейти в нитрозо- и
гидроксиламиновые замещенные, дающие в результате своего взаимо-
действия азоксисоединение
В главе VI мы уже рассмотрели процесс гидролиза сульфокис-
лот в водных растворах кислот, где легко усматривается каталити-
ческий процесс в гомогенной системе с катализирующим влиянием
водород-иона.
Влияние катализаторов в процессе щелочного плавления сульфо-
кислот еще не достаточно прослежено, но оно вне сомнения имеет
место особенно при окислительном течении сплавления.
При обмене хлора в хлоропроизводных изучение влияния ката-
лизаторов только и позволило сделать эти реакции производ-
ственно интересными. Для обмена хлора на аминогруппу характер-
ным катализатором являются медные соединения, отвечающие за-
киси меди. В реакционной смеси проявляется недостаточно изу-
ченное каталитическое влияние со стороны комплексных купри- и
купро-аммониевых ионов.
В обмене хлорного атома на гидроксил (получение фенола из
хлорбензола) играют роль также заключающие медь катализаторы и
вместе гидроксил-ионы. Медные стенки аппарата — катализатор ге-
терогенный, быть может, частично работающий в направлении обра-
зования содержащих медь соединений, гомогенно катализирующих
реакцию. Присутствие железа (в молекулярном или ионном состоя-
нии) благоприятствует сдвигу реакции в направлении конденсации
в дифенильные производные.
Катализирующее значение сернистой кислоты и ее солей в взаимо-
превращениях амино- и оксипроизводных разобрано подробно
в своем месте. В этой группе реакций имеется молекулярный ката-
лиз и образование промежуточных соединений в их ходе во мно-
гих случаях подтверждается опытами их выделения.
Диазосоединения и их превращения являются благодарным объ-
ектом исследования каталитических влияний. Напомним факты ди-
азотирования аминофенолов (аминонафтолов) с участием нитритов
меди и других тяжелых металлов, превращения диазосоединений
472
в нитрилы, галоидозамещенные, сульфиновые кислоты, нитросоеди-
нения, в которых играет роль тот или другой катализатор, чаще
всего медь или ее соединения. Химизм таких воздействий далеко
не везде еще ясен.
Не развивая здесь подробнее, упомянем лишь, что в процессах
алкилирования, так же как ацилирования, имеется много примеров
каталитических влияний. Практически серная или соляная кислота,
входящая как ингредиент реакционной смеси при алкилировании
аминов спиртами, — является катализатором реакции. При ацили-
ровании фенолов и аминов ангидридами кислот часто б&вает очень
полезным в смысле большего выхода продукта и ускорения ре-
акции прибавление весьма малых количеств крепкой серной кис-
лоты как катализатора реакции.
В главе об окислении мы рассмотрели значительное количество
примеров окисления органических веществ при участии окислителей.
Нужно сказать, что ни одной реакции из тех, для которых катали-
тические влияния были замечены, не было посвящено так много
работ, как окислению. Это вполне понятно ввиду значения про-
цессов окисления в биологии и большой их важности для научного
понимания многих химических явлений.
Многие органические вещества, в том числе большое количе-
ство ароматических соединений: фенолы, полиоксизамещенные,
альдегиды и т. п., принадлежат к числу „самоокисляющихся",
точнее — легко окисляющихся свободным кислородом. Основы для
понимания таких процессов окисления положены в теории А. Н. Баха
и Энглера, и в последнее время эта теория была очень широка
освещена исследованиями Муре и Дюфресса3).
Согласно этой теории, окисление тех соединений, которых
структура и наличие определенных функциональных групп делает
способными к окислению их свободным кислородом, протекает
через стадию промежуточного образования нестойких и очень
реакционноспособных перекисей с „активным" кислородом. Такие
перекиси для соединения А будем обозначать А [О2]. По Муре
и Д ю ф ре с с у, такая первая ступень соединения А с молекулой кисло-
рода образуется без потери энергии или падения потенциала,
но часто с повышением потенциала. Обладая различными уровнями
энергии, не все молекулы А и О2 участвуют в образовании такой
„первичной" перекиси, но только те немногие из них, которых
энергетический уровень выше энергии активирования (complement
critique d’energie). При этом первичная перекись образуется за счет
энергии окружающей массы без передачи системе какой-либо
внешней энергии. Молекулы первичной перекиси А [О2], если они
не диссоциируют на составные части, в дальнейшем эволюциони-
руют в направлении стойких форм, где кислород уже не имеет
тех свойств, которые отличают первичные перекиси, где он пере-
ходит в неактивное состояние.
Могут быть различные виды такого превращения первичной
перекиси. Перекись сама может разложиться с образованием новых
продуктов окисления: такой пример представляет окисление кете-
нов ввиду особо ненасыщенной их природы
RR'C : С : О4-О2~- RR'C : СО [О2]—RR'C : О + СО2.
473
Далее перекись может оказать окисляющее влияние на окружаю-
щие ее молекулы как исходного для перекиси вещества, так и
иных веществ, имеющихся в реакционной системе. Схема первого
превращения такова
Д [О2] 4- А —► 2Д [О] —* 2ЛО — (устойчивая форма)
неустойчиво
Окисление бензальдегида служит примером такого превращения:
2С6Н5СНО Ч- О2 — С6Н3СНО [О2] + С6Н5СНО —
— 2С6Н5СНО [О] — 2С6Н5СООН.
При наличии в системе молекул вещества В, отличных от исход-
ного вещества А, кислород перекиси Л[О2] может распределиться
между Л и В, например
Л[О2]4-5^Л [O]4-5[OJ —Л0 + 50.
нестойкие стойкие
В такой сопряженной реакции окисления окисление В в ВО
называется индуцированной реакцией, А — автоокислителем, а В—
акцептором.
Может быть и иной вид эволюции первичной перекиси, когда
Л[О2] отдает весь свой кислород В, например
Л [О2] 4- В -» Л 4- В [О2] -» ВО2 (стойкое)
или
Л [О2] 4- 25-» Л 4- 25 [О] -» 250 (стойкие).
В последних случаях имеется типично каталитическое окисле-
ние 5 через посредство самоокисляющегося вещества Л.
Образно можно представить себе, по Муре, явления, описан-
ные выше, в виде схемы вытекания жидкости через сифон из выше-
стоящего приемника в нижний. Такая схема приложима вообще
к химической реакции (рис. 12).
Жидкость не может вытекать до тех пор, пока она как смесь 4 + О2 не достиг-
нет некоторого предела потенциальной энергии (энергии активирования), измеряе-
мого высотой столба в малой ветви сифона. Эта часть явления отвечает образованию
первичной перекиси А [02]. Последующее вытекание по длинной трубке сифона
отвечает дальнейшим превращениям перекиси в стойкие продукты окисления.
На диаграмме рядом с фигурой дана кривая изменений энергетического уровня
для молекул в разные моменты реакции. Средний уровень энергии молекул системы
отвечает Е}. Реагирующие молекулы достигают уровня Е2, и затем уровень пони-
жается в стойких продуктах окисления до £3.
Катализатор в системе, как мы ниже увидим, может благоприятствовать ходу
реакции снижением необходимого высшего уровня Е2 до более низкого.
Первая стадия явления самоокисления, т. е. образование первич-
ной перекиси, может поддаваться каталитическим воздействиям
как положительным, так и отрицательным. Если катализатор С
образует с самоокисляющимся веществом соединение, энергия
активирования которого ниже, чем потребно для одного А, при
соединении его с молекулой 02, то понятно, что в единицу вре-
мени большая часть молекул А будет реакционноспособна. В таком
случае будем иметь
А 4- С -» АС + 02 — А С [02] -» А02 4- С.
474
Катализатор С после регенерации вновь войдет во взаимодействие. Подобного
типа реакции имеют место особенно в щелочных средах (например окисление пиро-
галлола н оксизамещенных). Щелочь здесь является катализатором типа С.
Очень интересны явления отрицательного катализа в случаях
самоокисления, где катализатор мешает нормальному образованию
первичной перекиси. Такие катализаторы Му ре и Дд) ф р е с с назы-
вают „антиокислителями" в отличие от „прОокислителей"— поло-
жительных катализаторов окисления (как С в вышеприведенной
схеме). Как правило „антиокислители" сами принадлежат к числу
соединений, легко взаимодействующих с кислородом.
Сиапепа A+Oj
Стойкий npoqi/кт
окисления
Рис. 12. Схема окисления (по Муре).
Фенолы и полиоксипроизводные, например пирокатехин н гидрохинон, пиро-
галлол, нафтолы, оказываются для многих реакций окисления хорошими анти-
окислителями, такими же являются иод, неорганические галоидные соли (преиму-
щественно нодистые н менее бромистые), гидронодиды органических оснований,
иоднстые алкилы, нодистые четырехзамешенные аммонии, йодоформ, четырехноди-
стый углерод, сера, полуторасернистый фосфор P4S3, неорганические сульфиды,
амины, нитрилы, амиды, карбамиды, уретаны, некоторые красители, неоргаинческне
соединения фосфора, мышьяк, сурьма, висмут, ванадий, бор, кремний, олово, свв-
нец. В качестве самоокисляющихся веществ были испытаны ненасыщенные угле-
водороды, сложные органические соединения (каучук, жиры), сульфит натрия, раз-
личные классы альдегидов и т. п.
Достаточно очень малых количеств антиокислителя, чтобы обнаружить замедле-
ние в процессе окисления таких веществ. Так Viooooo части серы или Vaoooo гидро-
хинон! по весу бензальдегида уже достаточно для точного обнаружения замедления
окисления; '/дао серы или x/iooo гидрохинона—для практически полной остановки
процесса.
Способность замедлять окисление свободным кислородом при-
суща веществам, могущим окисляться. Эта способность расшифро-
вывается Муре как катализ реакции, обратной образованию пер-
вичной перекиси А [О2], т. е. ее распада. Таким образом перекись
А [О2] окисляет антиокислитель В в перекись В [О], между тем
как сама превращается при этом в также нестойкую другую
перекись Д[О]. Два перекисных соединения А [О] и В [О] взаимно
разрушаются с регенерацией молекул А, В и О2 в их первона-
чальном состоянии.
475
Схема превращений такова:
д_|_Оа->Д[Ог]; Д[О2Ц-В->Д[О] + В[О];
Л[О] + В[О]->Л+В4-О2.
Возможно, что имеет место и прямое окисление антиокислителя
например по схеме:
Д-|-О2->-Д [О.-]; ВОа-> В [О2]; А [О2] 4-В [О2] -> Д-]-В4-О2.
При таком непрямом дезактивировании — молекулы А и О2, вошед-
шие в реакцию как активные молекулы, возвращаются в систему
в неактивном состоянии. При небольшом числе активных молекул
в реакционной среде понятно, что достаточно крайне незначитель-
ных количеств антиокислителя, чтобы задержать ход окисления.
Явления отрицательного катализа при окислении в высшей
степени интересны и в приложении к интересующим нас пробле-
мам. Ими объясняются в известной мере применения многих орга-
нических соединений типа замешенных аминов для предохранения
от старения резиновых изделий. Такие „противостарители" произ-
водятся красочной промышленностью из ее промежуточных про-
дуктов. Сохранение без изменений альдегидов, даже таких мало-
устойчивых, как фурфурол, удается теперь без затруднений
посредством введения в него подходящего антиоксиданта. Вероятно
возможно найти подходящие антиокислители для сохранения фено-
лов от изменений на воздухе, также каки для аминов. Обработка
анилина железом, покрытым слоем сернистого железа, испытана
как средство против изменений анилина от потемнения при хра-
нении [П. К. БуличЗЬ18]- Не имеет ли здесь место также действие
антиокислителя в виде следов сернистого железа? Наконец воз-
можно теоретически найти надлежащие антиоксиданты для краси-
телей, особенно легко изменяющихся при окислении, и тем самым
сделать прочными окраски красителями, которые сами по себе
непрочны. Практически давно известно, что на разных волокнистых
материалах, соотв. субстратах, и при разных подготовках (протра-
вах) одни и те же красители дают окраски разной степени проч-
ности.
Таким образом катализатор может содействовать разложению
активной первичной перекиси на составные части через посредство
взаимодействия образующихся вторично перекисей А [О], В [О]
с выделением всего кислорода. Но катализатор может содейство-
вать и образованию стойких окисных соединений—продуктов
эволюции первичной перекиси — при условии, если В [О] реагирует
не с молекулами А [О], а с веществом А, количество молекул
которого неизмеримо больше количества молекул первичной
перекиси.
В таком случае ход превращений передает схема:
А + [О,] -> А [Оа]; А [О2] + В -> А [О] 4- В [О];
В[О]+Д->В-]-ДО (устойчиво)
В случае перехода В [О] в стойкую форму ВО катализатор
не регенерируется и постепенно теряется для катализа.
476
Схема всех превращений молекулы А с участием катализаторов
С и В передается Муре таким образом:
Осаждение кубовых красителей из растворов их лейкосоеди-
нений и сернистых красителей из растворов их в сернистых ще-
лочах можно рассматривать как окисление в гомогенной среде,
катализируемое действием щелочных растворов.
Нужно указать еще на оставленный метод окислительного син-
теза фталевого ангидрида из нафталина действием серной кислоты
в присутствии ртути, метод, имевший большое историческое зна-
чение в разработке первого технически осуществленного синтеза
индиго. Здесь ртуть — катализатор этой реакции. Весьма вероятно,
что реакция проходит через стадию промежуточного ртутного соеди-
нения, так же как продуктов присоединения серной кислоты, рас-
падающихся, возможно также не без участия ртути, с выделением
сернистой кислоты, например
Z\/\/S°3H
I
I /Н
\/\/xSO3H
но^н
--------->
— 2H,SOs
Гидроксилированные циклы нафталина, естественно, окисляются
далее еще легче, чем незамещенные.
477
Приведенная схема основана на известных и выше упоминав-
шихся фактах присоединения серной кислоты к сульфируемым
соединениям в первой фазе реакции, на фактах выделения серни-
стой кислоты в процессах сульфирования, особенно заметных при
повышении температуры. Окисляющее действие серной кислоты,
проявляющееся в некоторой лишь степени в обычных условиях
сульфирования, при участии ртути значительно усиливается.
Подобное этому же явлению находим в ряде вышеупомянутых
(см. главу XIII) примеров введения гидроксильной группы в антра-
хиноновое ядро действием олеума. В этих многочисленных при-
мерах констатировано также раскисление серной кислоты до серни-
стой и катализирующая роль ртути и селена при этих окислениях.
В какой фазе таких сложных реакций участие катализатора эффек-
тивно,— трудно установить. Имея в виду, что многие из реакций
окисления антрахиноновых замещенных в полиоксипроизводные
протекают при сравнительно невысокой температуре, часто при
40—50°, можно считать вероятным, что эти прибавки катализируют
стадию распада продукта присоединения серной кислоты с выде-
лением сернистой.
В числе реакций конденсации имеется немало примеров воз-
действий каталитического характера. Таковы например многие
альдегидные конденсации, проходящие в кислотной и щелочной
среде. Ионный катализ со стороны Н’ или ОН' здесь проявляется
с очевидностью. Особенно замечательны в этом отношении реак-
ции с участием хлористого алюминия. Участвуя в ряде химических
превращений, известных под общим названием реакций Фриделя
и Крафтса, хлористый алюминий в некоторых группах процес-
сов участвует как реагент, требуемый в стехиометрических коли-
чествах,— таково большинство синтезов кетонов и кетонокислот,—
в других — достаточно минимального его количества для успеха
реакции. К последнему типу процессов принадлежат например
конденсации с введением алкила в ароматическое ядро.
Вышеупомянутые недавние исследования Воля н Вертипороха устано-
вили, что в тройной системе — бензол, бромистый этил н хлористый алюминий —
смеси попарно взятых ингредиентов не проводят тока, между тем как тройная
смесь его проводит (см. главу XV). На основании этого авторы допускают суще-
ствование промежуточного тройного собдннення из этих ингредиентов, построенного
по типу сольвата, например псевдосоли А1 [А1С14]3 с галоидопроизводными н после-
дующий переход этой соли в сильно электропроводящую соль с включением в нее
ароматического углеводорода 4), например
[Al(C2H5Br)n(C6H6)m] [А1Вг4]3.
Безекен на основании наблюдений своих и Оливье дер-
жится того взгляда, что хлористый алюминий (и вообще катали-
затор), образуя продукт присоединения (напр. типа RCOC1A1C13),
теряет в значительной мере свою активность (парализуется)
и что введение весьма незначительного избытка А1С13 сверх
связанного в продукте присоединения приводит к внезапному уско-
рению реакции. Он полагает, что объяснение каталитического дей-
ствия нужно искать не в образовании этих продуктов присоеди-
нения, но в изменении состояния молекул органического вещества
478
в момент взаимодействия с катализатором. Он называет это со-
стояние деформацией молекулы б).
Только что описанные факты, характеризующие каталитическую
активность хлористого алюминия, хорошо согласуются с выше-
упомянутой теорией Г. Армстронга, касающейся роли катали-
затора (детерминанта — электролита) в гомогенной системе.
Для понимания хода реакций в газовой и жидкой среде боль-
шое значение имеет теория „цепных" реакций, развитая особенно
полно Н. Н. Семеновым. Начало цепной реакции заключается
в активировании одной из имеющихся в системе разновидностей
молекул вплоть до ее диссоциации. Начало может быть положено
воздействием лучистой энергии, местным нагреванием и т. п.
Образовавшиеся атомы (или радикалы) реагируют с другого вида
молекулой, освобождая ее атом или радикал, который продолжает
цепь. Например
Л2-^Л+Л; Л + Д2->Лб + б; В+Л2 = Л54-Л...
Такие цепи, на концах которых — активные атомы, могут быть
разной длины. Обрыв их, т. е. соединение концевых атомов в моле-
кулу (типа АВ, Л2 или В2) в объеме гомогенной системы — редкое
явление, но может произойти на стенке сосуда, соотв. поверхности
твердого тела, или при встрече с посторонними молекулами
с потерей активными атомами их энергии.
Энергия активирования для ослабления связей молекулы доста-
вляется при экзотермических процессах теплотой реакции. Получив
запас энергии, столкнувшиеся молекулы становятся источниками
новых цепей. Введение катализатора понижает необходимую энер-
гию активирования, но катализатор не обязательно принимает
участие в создании дальнейших звеньев цепи, и реакция, начав-
шись, дальше может итти сама, если она экзотермична.
Выше, в главе IV, мы имели случай познакомиться с реакцией,
где действие лучистой энергии содействует определенному химиче-
скому эффекту,— мы говорим о реакции между хлором и толуолом.
Освещение при этом направляет реакцию в сторону введения хлора
в метильную группу. Из цитированной в своем месте работы Бука
и Эггерта следует, что каждому абсорбированному кванту (Av)
энергии при —80° отвечает 25 реакционных молекул С12. Это обстоя-
тельство объясняется образованием реакционных цепей такого
например вида:
ci24-Av-^ci4-ci,
С1 + с5н5сн3 СбНбСН2С1 4- н,
h4-ci2-^hci4-ci,
С1 + С6Н5СН3 С5Н5СН2С1+ Н и т. д.
В окислительных реакциях с достаточным основанием
можно предполагать наличия цепного их протекания. Для фото-
химических окислений (например бензальдегида) цепной механизм
можно считать доказанным, для термических весьма вероятным.
479
В. КАТАЛИЗ В ГЕТЕРОГЕННОЙ СИСТЕМЕ
а) Общие сведения
Гетерогенный катализ, особенно в газовой фазе, в настоящее
время привлекает внимание очень широкого круга выдающихся
исследователей. Хотя его теоретическое обоснование требует еще
многосторонней разработки, приложение гетерогенного катализа
дало уже практические результаты огромной значимости. Напом-
ним о синтезе аммиака из азота и водорода и синтезе метанола и
высших спиртов из окиси углерода и водорода. Период по пре-
имуществу эмпирических поисков наилучшего состава и вида ката-
лизатора сменяется ныне углубленным изучением теории этих
явлений, и чисто научные изыскания оплодотворяют производ-
ственную практику.
На гетерогенном катализе с очевидностью обнаруживается зна-
чение специфичности катализаторов, влияния химического со-
става и развития поверхности катализатора.
Известно, что одни катализаторы преимущественно пригодны
для введения водорода, например в органические соединения (г и д-
рогенизация, восстановление) и они же нужны для отщепле-
ния водорода, например при переходе гидроароматических соеди-
нений в ароматические. Таковы металлы платиновой группы, никель,
кобальт, железо, медь. Меньшую роль играют другие металлы, на-
пример серебро, кадмий, олово. Палладий и платина дают особенно
выдающийся эффект в гидрогенизации, так как их препараты можно
применять с успехом для обработки водородом в жидкой среде
и при обыкновенной температуре.
Окислы металлов (никеля, железа, алюминия) также хорошо
катализируют реакцию гидрогенизации, особенно при повышенном
давлении водорода (В. Н. И п а т ь е в).
Н. Д. Зелинский применил в качестве катализатора металлы
платиновой группы, особенно палладий на угле, никель, окислы
никеля и алюминия и указал условия дегидрогенизации гидро-
ароматических соединений, а следовательно пути получения арома-
тических соединений из природных углеводородов нефти.
Реакции окисления по отношению к органическим вещест-
вам катализируются преимущественно окислами и солями элемен-
тов с переменной валентностью. Находят применение также сме-
шанные катализаторы, иногда с добавлением так называемых
промотеров. Наиболее известны в этой группе катализаторов
окислы ванадия, молибдена, вольфрама, железа, марганца, урана,
свинца, меди, серебра, кобальта и никеля. Описаны катализаторы
типа солеобразных соединений, где кроме металлического окисла
имеется характерный анион (кремневой кислоты, фосфорной,бор-
ной кислоты), так же как такие, где перечисленные выше окислы
выполняют кислотную функцию (ванадаты).
Для реакций водоотнятия (дегидратации) особенно
пригодны окислы алюминия, кремния, циркона, тория, хрома и
вольфрама. Дегидратация спиртов и получение из них этиленовых
углеводородов на глиноземе (А12О3) была изучена В. Н. Ипать-
480
евым и вошла в заводскую практику. Некоторые из окислов при
переходе в соли например с фосфорной и серной кислотой сохра-
няют, а иногда повышают каталитическую активность.
Иногда катализаторы этой группы, например окись тория,
применяются и для гидратации, т. е. присоединения элементов воды
к дегидратированному соединению.
Реакция хлорирования, которую мы рассмотрели ранее как
проводимую в гетерогенной системе (с твердым катализатором),
нуждается в катализаторах с характером элементов или хлористых
солей.
Так как при катализе в гетерогенной системе реакция разви-
вается на поверхности раздела двух фаз, например твердой и жид-
кой или твердой и газообразной, то, понятно, кроме химического
состава катализатора, развитие и характер поверхности имеют
самое важное значение для успеха реакции. Такие реакции, про-
ходящие на поверхности твердого тела — катализатора, часто
называются контактными реакциями.
Соответственно этому, методика приготовления катализаторов,
определяющая и состав его и вид поверхности, имеет большую
важность для каталитических реакций. Очень существенно соблю-
дение в точности тех приемов приготовления катализаторов,
которые дали лучшие результаты в предварительных пробах.
В жидкой системе выгодно применять катализаторы в коллоид-
ном состоянии для наиболее равномерного распределения их в сис-
теме. Твердые катализаторы для газообразной системы применя-
ются в виде порошка, твердых плотных или пористых гранул и
кусков или нанесенными на носитель, например пемзу, асбест,
глинозем, гель кремневой кислоты и т. п. Подробности методов
приготовления катализаторов описаны преимущественно в патентах
и иногда в научной оригинальной литературе. Большое значение
имеют сложные катализаторы, где при химически разнородном
составе достигается в сумме больший эффект, чем при участии
только одной действующей составной части. В состав такого слож-
ного катализатора могут входить две и более составных части
в определенных весовых отношениях для достижения максималь-
ного полезного эффекта. Особенно отчетливо значение сложных
катализаторов выявлено на процессе получения аммиака из азото-
водородной смеси. Повидимому цеолитовые катализаторы, при-
меняемые для окисления углеводородов, принадлежат к такому
же типу.
Посторонние вещества, включенные в состав основной массы
катализатора, могут быть вредными, безразличными и полезными
для проявления его каталитической активности.
Вредные вещества или „каталитические яды" могут пра-
ктически привести к нулю эффект действия катализатора. Таким
ядом оказываются например сера и серосодержащие соединения
для многих катализаторов гидрогенизации: так как сера может
заключаться в составе реагирующих веществ, если они тщательно
не освобождены от сернистых соединений (случай, не редкий
в практике применения ароматических углеводородов и их произ-
водных), то, накапливаясь в ходе процесса на катализаторе, она
31 Зак. 2611.—Н. Ворожцов
481
может его практически отравить, понизив ниже допустимого пре-
дела его активность.
Яды так же специфичны, как и катализаторы, т. е. для каждого
катализатора ядовиты определенные вещества. Важно для успеш-
ного пользования катализатором знать специфичные его яды, так
как при этом можно или устранять их внесение на катализа-
тор или восстанавливать соответственными приемами активность
катализатора, освобождая его от ядов последующей обработ-
кой.
Безразличные вещества, не изменяющие катализатор, сами никак
не влияющие на реакцию, в которой имеет применение катализатор,
нередко употребляются при получении катализатора в качестве
носителя или подкладки. Такими веществами являются иногда
асбест, пемза, различные силикаты, гель кремневой кислоты, уголь,
гранулы металлов и т. п. Цель их введения в практику — увеличе-
ние поверхности катализатора при той же массе, создание более
теплопроводящего катализатора и т, п.
Полезными прибавками к катализатору (промот ерами,
активаторами) являются такие, которые, будучи сами по себе
неспособными функционировать как катализатор реакции, в то же
время усиливают действие катализатора. Промотеры по реакцион-
ности и по составу обычно сильно отличаются от катализатора.
Определенного состава промотеры специфичны для определен-
ных катализаторов. Главные элементы, входящие в состав промотера,
принадлежат к иным группам периодической системы, чем элементы
катализатора.
Наиболее прочно обоснованным и принятым в настоящее время
воззрением на сущность тех процессов, которые происходят при
реакции на поверхности твердого катализатора в газовой среде,
является так называемая адсорбционная теория.
Согласно этой теории молекулы газообразного вещества удер-
живаются на поверхности катализатора, образуя облекающий ее
одномолекулярный (по толщине) слой. Образование этого слоя
адсорбированных молекул происходит при участии сил валентности
катализатора (главных и побочных), таким образом известное срод-
ство между молекулами газа и катализатора должно иметь место.
Такая адсорбция, связанная с перестройкой электронных орбит
в атомах, связана и с изменением химических свойств газовых
молекул. Весьма вероятно, что в зависимости от своего химического
состава, соотв. полярности отдельных атомов или радикалов, моле-
кулы определенным образом ориентированы к поверхности катали-
затора. Реакция, протекающая на поверхности катализатора, может
итти различными путями: 1) между молекулами или атомами,
адсорбированными на соседних участках поверхности; 2) между
молекулами адсорбционного слоя и атомами находящегося под
ним твердого адсорбента и наконец 3) при столкновении молекул
газа, находящегося в данном объеме, с уже адсорбированными
молекулами или атомами (Лангмюр).
Адсорбированная молекула может отделиться от поверхности —
это явление называется десорбцией. Оно обратно адсорбции—
сцеплению молекул с поверхностью. Столкновение молекул из
482
объема газа вне поверхности с адсорбированными на поверхности
приводит к реакции в случае, если они активны.
По Лангмюру большая или меньшая степень покрытия поверх-
ности твердого вещества (адсорбента) молекулами газа зависит от
средней продолжительности пребывания газовых молекул на поверх-
ности или от промежутка времени, протекающего между конден-
сацией газовой молекулы на поверхности адсорбента и ее испаре-
нием.
При постоянной температуре и однородной поверхности скорость конденсации
молекул пропорциональна величине свободной поверхности, еще не покрытой моле-
кулами газа (1 — с) (с—часть поверхности, покрытой газом), н давлению р.
Таким образом
г»1 — kJ (1 — с).
Скорость испарения молекул т»2 пропорциональна части поверхности, покрытой
газовыми молекулами, т. е.
ц2 = /?„т.
При наступлении адсорбционного равновесия эти скорости равны, значит
kiP (1 — c) = k2<s,
откуда часть поверхности, покрытая молекулами газа, определяется как
_ К Р
k* 4- '
Приняв b = упрощаем:
- _ Ър
“ ~ i и- Ьр
При незначительной адсорбции и малых давлениях (Ьр мало по сравнению
с единицей) получаем
= = Ьр,
т. е. при этих условиях имеется линейная зависимость между величиной поверх-
ности, покрытой молекулами газа, и давлением его.
Для высоких давлений (Ьр велико по сравнению с единицей)
= = 1,
т. е. величина поверхности, покрытой молекулами газа, не зависит от давления: она
насыщена. В части изотермы адсорбции между этими крайними значениями зависи-
мость а от р может быть выражена как
с Р Л
где zz> 1.
Скорость реакции на однородной поверхности пропорциональна части поверхности,
покрытой адсорбированным газом. При допущении, что реагирует только газ
и что продукты реакции не адсорбируются катализатором, для трех выше разобран-
ных случаев мы получаем три уравнения скорости. Отвечающее малой адсорбции
(а = Ьр)
— — kz— kbp = к^р, (1)
f 1 \
для частичной адсорбции ( = — р п I
— = ftP ~ (2)
31*
483
и для адсорбции, соответствующей насыщению поверхности, (а = 1)
dp
— dF = const- (3)
Уравнение (1) отвечает мономолекулярной реакции, уравнение (3) отвечает реак -
ции нулевого порядка — скорость реакции постоянна, независима от количества
адсорбированного газа, вошедшего в реакцию. Уравнение (2) отвечает установлен-
ной опытом для адсорбции формуле Фрейндлиха.
Изучение явлений адсорбции некоторых газов на поверхности
твердых тел позволило установить два вида адсорбции; иногда
называют их первичной и вторичной адсорбцией, иногда
говорят о неспецифическом, или физическом, типе адсорбции и о
специфическом, или химическом, типе.
Работы последнего времени устанавливают, что адсорбция при
низких температурах носит неспецифический характер: газ адсор-
бируется в молекулярном состоянии, равновесие адсорбции насту-
пает быстро, причем большая часть молекул газа, ударяющихся о
поверхность, ею удерживается.
При переходе к более высоким температурам обнаруживается
медленно протекающзя специфическая адсорбция, при которой мо-
гут быть поглощены значительно большие количества газа, нежели
при адсорбции в области низких температур. Скорость специфи-
ческой адсорбции связана с температурой зависимостью типа по-
казательной функции, так что имеется достаточно оснований для
суждения об энергии активирования адсорбции для данного газа и
данной поверхности адсорбента, подобно тому, как говорят об
энергии активирования химических реакций.
Исследования Тэйлора и Вильямсона установили, что
величина энергии активирования адсорбционного процесса меняется
в отдельные периоды процесса адсорбции в зависимости от того,
какая часть поверхности участвует в процессе. Существует таким
образом два типа специфической адсорбции: с высокой и с низкой
энергией активирования 6).
Различные места поверхности катализатора обладают различной
активностью при образовании адсорбционного слоя. Более актив-
ные места (активные центры) легко удерживают газ, — ско-
рость процесса велика. Скорость адсорбции уменьшается по мере
увеличения части поверхности, покрытой газом.
При измерении скорости адсорбции водорода на смеси окиси марганца с окисью
хрома при температурах 100 и 132° можно было вычислить энергию активирова-
ния процесса по уравнению
din v Е
~dT RE1’
причем оказалось, что иа наиболее активной части поверхности энергия активиро-
вания Е = 5 900 кал, между тем как для второго периода замедленной адсорбции
энергия активирования равна 8 500 кал.
Неоднородность структуры активных поверхностей таким обра-
зом подтверждается. Скорость адсорбционного процесса может быть
_________________________________£_
рассчитана по выражению е RT. Отношение скоростей первого
и второго типов адсорбции позволяет судить об относительной
484
энергии активных центров. Введевие промотеров в состав катали-
затора во многих случаях приводит к увеличению скорости, соотв.
понижению энергии активирования, адсорбционного процесса.
Причины активаторного действия промотеров могут
быть различны. Возможно, что некоторые промотеры содействуют
образованию определенных размеров и структуры частиц катали-
затора, например приданию катализатору особенной тонкозерни-
стой структуры. Также промотеры могут содействовать сохране-
нию структуры катализатора, противодействуя его спеканию при
нагревании. Промотеры, благодаря большей своей адсорбционной
способности по отношению к реагирующим продуктам, могут благо-
приятствовать подводу их к каталитически активным частям по-
верхности, также могут способствовать удалению продуктов реак-
ции, если их способность адсорбировать продукты реакции неве-
лика. Возможны вместе с тем и химические взаимные влияния
катализатора и активатора, повышающие их отдельно взятую
эффективность.
Примешивание определенных газообразных веществ к реак-
ционвой смеси своим действием на катализатор может, как это
констатировано на ряде примеров, также повысить активность
катализатора. Очевидно роль этих газов сходна с таковой же
твердых промотеров.
Явления отравления к а та л и з а то р о в противоположны
положительному влиянию промотеров в смешанвых катализаторах.
Они обнаруживаются ослаблевием каталитически активных веществ
вследствие перехода их от действия „контактных ядов“ в такое
состояние или соединения, которые больше не способствуют нор-
мальному течению катализируемого процесса. Действие контактного
яда проявляется внешне в постепенном понижении выхода глав-
ной реакции и может повести к полному парализованию катали-
затора. Иногда уже минимальное количество отравляющих при-
месей в составе газовой смеси может вызвать остановку катали-
зируемого процесса.
Иногда не только посторонние вещества, во и продукты реак-
ции могут вызвать явления отравления. Действие контактных ядов
направляется по преимуществу на активные центры поверх-
ности. Активные центры с разными значениями энергии активиро-
вания адсорбции ивогда относятся различно к отравляющему
эффекту. Эффект отравлевия таким образом связан с выключением
более активных центров с передачей их функций центрам с иным
энергетическим уроввем 7). Различают отравления обратимые, т. е.
исчезающие при постепенном удалении задерживающих реакцию
веществ, и необратимые отравления, окончательно парализующие
катализатор. Своевременное распознавание и устранение действия
контактного яда обеспечивает часто удачу технического способа,
основавного на примевевии катализатора.
Подобно отравлению, вредвым для каталитических реакций
является разложение или „старение" катализаторов. Это не-
желательное явление сопровождается изменением формы (микро-
структуры) твердых катализаторов, с уменьшением, вплоть
до полного исчезвсвевия, числа активных центров поверхвости.
485
При прочих равных условиях катализатор тем ценнее, чем он более
долговечен, т. е. стоек в условиях контактного процесса. Пере-
грев катализатора может повести к вредному спеканию его поверх-
ности, т. е. такой ее деформации, которая связана с ослаблением
характерной зернистости, и тем привести также к ослаблению
действия катализатора.
А. А. Баландин излагает очень интересную схему гетерогенных каталити-
ческих реакций, дающую достаточно ясное представление о роли и характере
«активных центров* в катализе и достаточно оправданную экспериментом в прило-
жении к дегидрогеиизациониым каталитическим реакциям. По нему, определенные
активные точки поверхности катализатора ориентируют к себе требующие активи-
рования химические связи и тем облегчают протекание реакции. Активные точки
притягивают к себе некоторые атомы адсорбированной молекулы. При этом моле-
кула может быть притянута активными точками разного рода за различные свои
атомы. Если при этом силы притяжения больше сопротивлений, удерживающих
атомы в молекуле, то молекула может быть разорвана. Если при этом к каждой из
двух активных точек притянуто по дза атома, связанных с остатками молекулы, то
результатом разрыва может быть образование новых двух молекул. Совокупности
таких активных точек мультиплетов на поверхности катализатора характери-
зуются специфичностью притяжения. Схемы каталитических превращений, напри-
мер этилового спирта, по гипотезе мультиплетов могут быть изображены так:
Н
• I
Т СН, — СН., п СН3 —С—О
1 I • I “ 11 II
I.....Г... "Г I
н он н•н
Активные притягивающие центры изображены точками около них поставлены
части молекулы, атомы которых к ним притягиваются и которые образуют своим
соединением новые молекулы. Разрывающиеся связи первоначальной молекулы
перечеркнуты.
Из схемы понятно, что превращение I, где притянуты к одному активному
if! j? группа СН2 —СН2, к другому ОН и Н должно дать в результате этилен и
воду. Превращение II (к одному центру притянуты С и О, к другому Н и Н) дает
альдегид и водород.
Мультиплеты характеризуются также особой конфигурацией, связавной с осо-
бенностями кристаллической решетки поверхности катализатора. На примере дегидро-
генизации циклогексана кубической решеткой платины можно считать доказан-
ным наличие общих элементов симметрии органической молекулы и катализатора.
Мультиплеты, собственно активные центры, по Б а л а н д и и у, не наиболее рыхло
расположенные атомы (по Т а й л о р у), но кристаллические зародыши—центры
кристаллизации, оставшиеся при приготовлении катализатора. Их сохранность за-
висит от температуры приготовления. Они уничтожаются при нагревании вслед-
ствие рекристаллизации, они могут оставаться, не разрастаясь, адсорбируясь на
носителях.
Влияние промотеров объясняется при этом тем, что привнесение посторонних
атомов создает особые центры, могущие притягивать определенные части молекул.
Если мультиплет АВ нуждается в точках А и В, но они не одинаково часто рас-
пределены, то примесь может образовать недостающие 8).
Гипотеза мультиплетов объясняет отсутствие полного параллелизма между адсорб-
цией и катализом.
Для характеристики катализатора и химического превращения,
в котором он участвует, имеет значение время соприкосновения,
нужное для данного превращения, под каковым понимается сред-
ний промежуток времени, в течение которого единица объема, про-
пускаемого через контактное пространство газа, находится внутри
свободного пространства, определяемого как разность между объе-
мом контактного пространства и собственным объемом катализатора.
486
В практике каталитическую способность данной системы выра-
жают объемной скоростью, т. е. объемом реагирующего газа
или пара, измеренного при нормальных температуре и давлении, при-
ходящим в контакт с одним объемом катализатора в течение часа.
По мере возрастания от нуля объемной скорости, превращение
в продукты (выход в процентах от теории) от равновесного его
значения начинает снижаться, так как превращение может вскоре
сделаться малым из-за уменьшенного времени контакта реагентов
с катализатором. В это же время производительность
(выход в единицу времени), т. е. произведение объемной скорости
и превращения, возрастает от низшего значения, проходит через
максимум и затем понижается. Когда объемная скорость низка,
превращение может быть близко или равно определяемому равно-
весием, и получаются только низкие выходы. Часто бывает, что
возрастание объемной скорости сначала заметно не отражается на
превращении, и выход возрастает приблизительно пропорционально
количеству обрабатываемого материала в единицу времени. Затем,
если реакция не обладает слишком большой скоростью, при воз-
растании скорости прохождения реагентов через катализатор пре-
вращение может убывать. Превращение может убывать при этом
в большей мере, чем пропорционально возрастанию объемной ско-
рости. В таком случае выход начинает убывать тем более, чем
более материала обрабатывается в единицу времени. Очевидно,
если скорость реакции не бесконечно велика, превращение при-
ближается к нулю по мере того, как объемная скорость прибли-
жается к большему значению, и следовательно выход приближается
к нулю в этих условиях. В производственных операциях, стремясь
получить высокие выходы, применяют высокие объемные скорости,
хотя превращение при этом ниже того, какое отвечает равно-
весию (Марек, Ган).
Адсорбция газовых молекул поверхностью катализатора или не-
которыми местами поверхности—явление, несомненно играющее
роль при гетерогенном катализе, но она одна не объясняет всего
химического процесса. Адсорбция—лишь ступень к дальнейшему
взаимодействию. Адсорбированные молекулы газа, склонные к хи-
мическому взаимодействию, могут быть различно активными. Ад-
сорбированные молекулы, обладающие предельной энергией акти-
вирования, вступают во взаимодействие на поверхности катализа-
тора. Опытом определено для некоторых реакций, что энергия
активирования реакций, проведенных без катализатора, выше, чем
с участием катализатора. Таким образом можно полагать, что
вообще роль катализатора в гетерогенных реакциях заключается
в понижении энергии активирования.
Практическое использование гетерогенного катализа для синтеза
промежуточных продуктов ароматического ряда идет пока не-
сколько позади применения контактных реакций для получения
неорганических продуктов (аммиак, азотная кислота, серный ангид-
рид) и некоторых синтезов алифатических соединений (метанол,
хлоропроизводные метана, этилена, уксусная кислота из ацетилена).
Тем не менее имеется ряд производств, где гетерогенный катализ
уже применяется или с успехом может быть применен.
487
б) Гидрогенизация, особенно нитросоединений
Очень актуальной задачей для применения катализа является
гидрогенизация, особенно нитросоединений для пре-
вращения их в амины. Воздействием водорода здесь можно ожи-
дать наиболее простого химически разрешения задачи восстанов-
ления:
RNO2 + ЗН2 = RNH2 + 2Н2О.
Возможность такого метода восстановления была доказана в са-
мом начале настоящего столетия работами Сабатье и Санде-
ра н, причем ими же было доказано, что мелкораздробленные
металлы, например получаемые восстановлением их окислов при
нагревании в струе водорода, оказываются эффективными катали-
заторами этой реакции.
При этом никель оказался слишком энергичным катализатором, так как он
при температуре выше 200° не только превращает например нитробензол з анилин,
но н переводит его с гидрированием ядра в циклогексиламищ
C0H5NO2-> C0H5NH2-> C0HuNHs
Прн температуре свыше 250° происходит уже отщепление аммиака
C6H6NO2 + 4Н,_ = CeH6 + NH3 4- 2Н2О.
Прн еще более высоких температурах (свыше 300°) это направление реакции
еше более заметно, причем происходит уже распад и бензольного ядра с образо-
ванием метана
C6H6NO2 4- 13Н2 = 6СН4 4- NH3 4- 2Н3О.
Платина с сильно развитой поверхностью (чернь, губка, платинированный
асбест) обладает, по наблюдениям этих исследователей, слабой активностью, и, если
нет большого избытка водорода, восстановление не бывает полным; в результате
образуется некоторое количество гидразобензола.
Медь, по наблюдениям Сабатье иСандеран — наиболее подхо-
дящий катализатор для восстановления нитросоединений в амины,
так как ее действие распространяется только на NO2-rpynny, не
затрагивая ароматического ядра. Нитробензол превращается в ани-
лин, начиная с температуры в 230°. Между 300 и 400° реакция
проходит быстро, и при избытке водорода получаются выхода
до 98°/о анилина, содержащего лишь следы азобензола. Металл,
по их словам, может служить долго. Водород может быть заме-
нен водяным газом (смесь СО и Н2, при этом окись углерода уча-
ствует также в восстановлении, переходя сама в углекислый газ)8).
В немецком патенте Сандеран указаны и другие катализаторы:
Со, Fe. Последующая разработка задачи в патентах основана на
этих наблюдениях с использованием частью сложных катализато-
ров и промотеров. Содержащие медь катализаторы во всяком слу-
чае оказались легко отравляемыми, особенно сернистыми соеди-
нениями, которые должны быть тщательно удаляемы как из
нитросоединений (производные тиофена), так и из восстановляю-
щего газа.
Серебро и золото, осажденные в мелкораздробленном состоянии на пемзе или
асбесте в отдельности и в смеси, катализируют, по описанию германского патента,
получение аннлина нз смесн паров нитробензола и водорода или водородсодер-
жащего газа в пределах 230 — 250°. Указывается на возможность применения
в смеси с этими металлами и меди10).
488
К металлам, уже частью отмеченным работами Сабатье, воз-
вращаются позднейшие немецкие патенты.
Патент 1912 г. (W. ter Meer) описывает каталитическое восстановление нитро-
соединений посредством водорода или водяного газа в амины с участием катали-
затора: закиси или закиси-окиси железа в мелкораздробленном состоянии на асбесте.
Температура взаимодействия паров нитробензола и водорода не приводится, но
отмечается, что продолжительность восстановления 3—5 час., т. е. процесс идет
далеко не быстро и).
Патенты Баденской анилиновой и содовой фабрики (О. Шмидт)
преимущественно описывают применение соединений меди большей
частью в виде сложных (смешанных) катализаторов12).
В патентах отмечается прежде всего благоприятная роль про-
мотеров (активаторов) в работе медного катализатора. Такими
промотерами названы щелочные соединения, окислы (MgO, A12OS),
другие кроме меди тяжелые металлы или их соединения (Fe,
Ag, Zn).
Пример приготовления такого сложного катализатора: 130 ч. пемзы замеши-
вают с смесью нз 20 ч. 40%-ного раствора силиката натрия, 24,3 ч. углекислой
меди, 2,7 ч. углекислого цинка и некоторого количества воды. Массу вводят впечь
и восстанавливают водородом при 180—220°. После восстановления катализатор
пригоден для восстановления нитробензола избытком водорода в анилин (при 200°).
В позднейших из этих патентов обращено внимание на освобож-
дение от контактных ядов, главным образом от соединений серы,
примешанных к нитросоединениям. Кроме собственно восстанови-
тельно действующей части, катализатор поэтому должен заключать
окиси или карбонаты щелочноземельных металлов или алюминия
или соединения редкоземельных элементов. Сернистые соедине-
ния всех этих элементов характеризуются большой теплотой
образования, соотв. разложения. Как катализаторы восстановления
названы: Си, Ag, Au, Fe, Zn, Pb, Sn; промотеры: окислы Сг, Мп,
щелочи и силикаты щелочей.
По одному из названных патентов, можно разобщить два кон-
тактных слоя, над которыми последовательно проходит смесь
водорода и нитробензола. Первый имеет задачу освобождение от
ядов: он состоит из ВаСО3, нанесенного на пемзу при помощи си-
ликата (температура контакта 200°); задача второго — собственно
гидрогенизация, и состав его практически не отличается от выше-
названных.
При восстановлении нитросоединеннй газами, содержащими окись углерода,
возникает опасность отравляющего действия газа на катализатор, если
даже омывающие катализатор газы тщательно освобождены от сернистых соедине-
ний. Ядовитой примесью оказываются карбонилы металлов, особенно железа (от
взаимодействия СО и мелкораздробленных металлов). От них предложено осво-
бождаться пропусканием газа через активный уголь. В последующем затем проходе
газа необходимо устранять возможность соприкосновения его с железом (в газо-
проводах, контактном пространстве) ы).
Отдельно медь и серебро как катализаторы восстановления
нитробензола в анилин были недавно обследованы двумя итальян-
скими химиками14).
Выход сильно зависит от температуры. Для меди оптимальный
выход лежит при температурах не выше 350° (при 400° обнару-
живается образование бензола и аммиака), для серебра оптимум
489
400°. При увеличении количества катализатора выход быстро
растет, достигая затем предельного значения. При более низких
температурах (250—300°) повышение количества катализатора со-
действует большему повышению выхода, чем при более высоких
(300—350°). Влияние количества водорода возрастает с темпера-
турой и количеством катализатора. Активность катализаторов
постоянна. Серебряный катализатор при 380° дает чистый анилин,
между тем как анилин, полученный на меди, всегда окрашен сле-
дами азобензола.
Работа американцев Броуна н Генке подтверждает наблюдения Сабатье,
что медь, восстановленная из Си(ОН)2 и имеющая кристаллическую структуру, бо-
лее активна в восстановительном катализе, чем полученная из Cu(NO3)2. Благо
приятную температуру восстановления эти исследователи определяют в 260°. Медь
нанесенная на асбест, более активна, чем отложенная на пемзе или совсем без но-
сителя. При быстром ходе нитробензола медь на асбесте быстрее теряет свою
активность. Следы железа на катализаторе из азотномедной соли затормаживают
ослабление деятельности катализатора15).
Медь, осажденная на пемзе из Cu(NO3)2 обжиганием и восстановлением во-
дородом при 325°, была испытана как катализатор восстановления нитрофенолов:
о-нитрофенол восстанавливается водородом с медью прн 265°, пара-изомер— так же,
но медленнее. Нитроанилины восстанавливаются в диамины (мета-изомер при
270—310°). Никель в этом случае менее активен16).
Очень активные катализаторы гидрогенизации, содержащие металлы Ni, Со, Си,
получаются по американскому патенту 1931 г. при выделении таких металлов из
водного раствора их солей в присутствии аммиака (или органических оснований)
посредством другого более электроположительного металла (Al, Zn, Fe). Выделение
происходит при температуре ниже температуры кипения раствора. Это выделение
иыгодно производить на носителе (инфузорная земля, гель кремневой кислоты,
уголь). Выделяющийся вместе с металлом окисел электроположительного металла
играет роль промотера. Такие катализаторы имеют значение при работе и в паро-
вой и в жидкой фазе (например при восстановлении нитросоединений)17). По не-
мецкому патенту, нитросоединение, например нитробензол, увлекается парами воды и
вместе с ними смешивается с водородом. При этом восстановление с участием
активного катализатора (никель) происходит при более низкой температуре, и про-
дукты поверхностью катализатора не удерживаются. Указана температура контакта
(предварительный нагрев) в 120018).
Американскими исследователями и преимущественно упомяну-
тыми Броуном и Генке изучалась гидрогенизация нитросое-
динений с участием различных металлических катализаторов.
При этом обнаружена значительная специфичность металличе-
ских катализаторов; некоторые из них (Pb, Bi, Т1) дают преиму-
щественно с хорошим выходом азосоединения, другие (Au, Sb, Сг,
Мп) дают малые выходы последних, между тем например Ni, Си,
Ag дают исключительно амины. Авторы усматривают некоторую
связь между атомным весом металла и его свойствами как ката-
лизатора.
В американском патенте этих же исследователей описывается применение как
катализаторов Pb, Bi, Т1 для получения азокси-, азо-, гндразо-и аминосоедннений.
По словам патента эти катализаторы имеют преимущество перед Ni, Со, Fe, Си,
Pt, Os, Ir. Сомнение возбуждает это утверждение в отношении N1 и Си. При про-
пускании паров нитробензола с избытком водорода иад мелкораздробленным свин-
цом (из РЬО н Н2) при 290° получают 78% анилина. 21% азобензола и примесь
азокси- и гидразобензола. При таких же условиях свинец, полученный из аморф-
ного глета, дает 24^ азобензола ( +азокси и гидразо) и 53,6% анилина.
Примененный в качестве катализатора В1, полученный осаждением Bi(NO3)3
водным аммиаком и восстановлением водородом при 250 — 350°, дает при 230°:
92% азобензола (-f- азокси % гидразо) и 4,4% анилина; при 240°: 89,2% азобен-
490
зола и 9,1% анилина; при 300°: 29,5% азобензола и 65,2% аннлнна. Носителями
служат’, асбест, пемза, А12О3, MgSO4 или металлы, не дающие сплавов с катализа-
торами 19).
Свинцовые катализаторы испытаны, и при получении анилина из нитробензола.
Катализаторы готовились или переводом PbfNO^ в РЬСО3 и обжиганием послед-
него при 430° до сурика илн прямым обжиганием Pb(NO3)2 при 430°. Полученные
образцы сурика РЬО после восстановления их водородом испытывали на катали-
тическую активность прн восстановлении нитробензола водородом в анилин. Ката-
лизатор, полученный из Pb(NO3)2, обнаружил длительную активность и давал 97%
выхода анилина. Активность катализатора, ослабевающая от действия одного во-
дорода, восстанавливается скорее в железных трубках, чем в стеклянных20).
Очень деятельным катализатором восстановления нитробензола в анилин оказа-
лось олово. Катализатор получается осаждением из раствора хлористого олова
посредством соды гидрата закиси и его восстановлением. Иногда перед восстановле-
нием окисел снова предварительно окисляют. В последнем случае предпочтительно
при окислении применять более низкую температуру. Повышение температуры
при катализе содействует понижению активности. В стеклянных трубках оптималь-
ные пределы температуры катализатора 275 — 294°. При работе в железных труб-
ках ниже (230°)21).
Таллий как катализатор восстановления нитробензола, полученный в мелкораз-
дробленном состоянии на асбесте восстановлением Т12О при 260°, дает в быстром
токе водорода до 84% азобензола вместе с анилином. Повышение температуры
снижает выход.
Кадмий оказался очень хорошим катализатором превращения нитробензола
в анилин. Испытай был кадмий, полученный восстановлением щавелевокислого
кадмия (Н2 при 390°), и оказался более активным, чем приготовленный прокалнва
нием щавелевокислой соли при 280° и последующим восстановлением. Кадмий из
Cd(OH)2, обожженного при 250° и затем восстановленного, более активен, чем полу-
ченный при обжигании гидрата при 350°. Кадмий на асбесте давал лучшие резуль-
таты. Наибольшие выхода анилина при 319°. Количество водорода, необходимое
для полного превращения нитробензола в анилин, 514% от теоретически рассчитан-
ного. Любопытно, что при повышении температуры свыше 350°, когда кадмий уже
плавится, каталитическое действие кадмия на асбесте еще велико22).
Как выше было уже указано, возможно применение для вос-
становления не только водорода, но и водяного газа. Наконец
применяется смесь окиси углерода с водяным паром, причем
возможно, что ранее восстановления нитросоединения имеет место
образование водорода по реакции: CO-f-H2O = CO24-H2.
Сложные металлосодержащие катализаторы для контактного восстановления
в парообразной и жидкой фазе описывает в своем патенте американская фирма
The Selden Со ->3).
Патент в части описания катализаторов перечисляет так много составных ча-
стей, что трудно сказать, какие из перечисленных в нем ингредиентов действи-
тельно нужны для приготовления практически ценных катализаторов. Катализаторы
составлены нз силикатов типа цеолитов (например Ме2О • А12О3 • nS102)—обмениваю-
щих основания или не обменивающих основания соединений, вместе с каталитически
действующими соединениями. Цеолиты готовятся действием силиката на соль метал-
ла. Как природные силикаты со способностью к обмену оснований приведены не-
фелин, полевой шпат, лейцит.
Силикаты типа цеолитов приготовляются в смеси с носителями, например ин-
фузорной землей, силикагелем и многими другими, часть которых никак не может
быть названа только носителями (каковы порошки металлов, солн, вольфраматы,
ванадаты, хроматы и другие соединения металлов: Си, Fe, Ni, Со, Al, Ti). В со-
став катализаторов входят далее стабилизаторы: соединения щелочных и щелочно-
земельных металлов, также соединения металлов 5-й и 6-й групп и солеобразные
соединения, заключающие поостой или комплексный кислотный радикал, включаю-
щий элементы: Сг, V, W, U, Мо, Мп, Та, Nb, Sb, Se, Те, Р, Bi, Sn, Cl, Pt, В. Сое-
динения, обменивающие основания (силикаты), выщелачиваются минеральными
кислотами для удаления обмениваемых щелочей. Из веществ, входящих в основную
часть обменивающего основания силиката, названы соединения: Си, Ag, Au, Bi,
Be, Zn, Cd, В, Al, редких земель, Ti, Zr, Sn, Pb, Th, Nb, Sb, Ta, Cr, Mo, W,
491
U, V, Мп, Fe, Ni, Со, Pt, Pd. Еще большим количеством элементов может быть за-
менена щелочь в соединении, обменивающем основание.
Из сказанного видно, как трудно найти зерно истины в таксм запутанном спи
санин. К сожалению, подобный патент, не разъяснякщий, но затемняющий вопрос,
не является одиноким среди других патентов по каталитическим процессам.
Более простую комбинацию кремнекислоты и окислов тяжелых
металлов, в частности окиси меди, дает американский патент
1927 г. 2*).
Силикагель, пропитанный окислами тяжелых металлов, оказы-
вается более энергичным катализатором восстановления, чем один
силикагель. Количества окислов металлов могут быть невысоки.
Гидраты окислов в реакционном процессе переходят в окислы и
в свободные металлы. Восстановление нитробензола над силика-
гелем с медью протекает при 200°. При работе с высококипящими
нитросоединениями рекомендуется применять разрежевие в системе
или вводить в процесс нитропродукты помощью перегретого пара.
Как выше было уже упомянуто (глава V), восстановляюшим га-
зом для получения аминов из нитросоединений может быть не
водород, но сероводород. Необходимо при этом участие угля (дре-
весного или животного), в порах которого и протекает реакция по
общему уравнению25):
RNO2 + 3H2S = RNH2 + 2Н2О 3S.
Метод интересен по преимуществу для использования бедных
сероводородом газов, которые нуждаются в освобождении от сер-
нистых соединений. Таков например коксовый газ, содержащий
10 г H2S в 1 л3, или генераторный газ с 3 г H2S в 1 л/8. Их осво-
бождение от серы таким путем могло бы стать в то же время
средством получения ценных полупродуктов. Практически метод
не может быть назван удобным, хотя бы по одному тому, что
сера, осевшая в порах угля, останавливает его каталитическую ак-
тивность, и требуется ее извлечение, например экстракцией, для
восстановления активности угля. Полезным оказалось введение
небольших количеств аммиачного газа для ускорения процесса.
Процесс восстановления экзотермичен и не требует внешнего
нагрева. Метод применим и в жидкой фазе, например к раство-
рам сульфокислот нитросоединений.
Несмотря на большое количество работ и патентов, касаю-
щихся контактного восстановления водородом нитросоединений
в паровой фазе, этот метод работы повидимому до сих пор не во-
шел еще в широкую заводскую практику. Вероятно главной при-
чиной является незначительная продолжительность службы катали-
заторов, выводимых отравлениями из работы. Это неприятное
обстоятельство не дает возможности установить непрерывный
процесс работы, который для производства массовых продуктов,
естественно, всегда предпочтительнее периодического. Более прак-
тически интересным оказывается метод восстановления нитросоеди-
нений водородом с твердым катализатором не в газовой, но в жидкой
среде. Этот метод в практике западноевропейской промышлен-
ности доведен уже до производственного применения.
Так как один из ингредиентов реакции (водород) газообразен,
492
то, с целью увеличить его концентрацию в системе, в таких про-
цессах прибегают к работе под давлением водорода свыше атмо-
сферного, применяя соответствующие автоклавы. Само собой
понятно, что процесс восстановления протекает на поверхности
катализатора, которая должна быть по возможности сильно раз-
вита,— следовательно важно, чтобы катализатор был по возмож-
ности мелко раздроблен и был, если не в состоянии коллоида, то
нанесен на носитель с большой поверхностью (пемза, инфузорная
земля и т. п.). Явления диффузии в этом случае играют заметную
роль в обновлении слоя, окружающего катализатор непрореагиро-
вавшими молекулами, и уводе из него продуктов реакции, следо-
вательно при процессе должно быть обеспечено хорошее переме-
шивание жидкости, газа и катализатора.
Наиболее испытанные в практике катализаторы такого восста-
новления нитросоединений — это никель (также окислы) для за-
водских процессов и палладий для лабораторных целей.
К сказанному в главе о восстановлении нитросоединений в амины
(глава V) добавим указания американских авторов, проводивших
восстановление нитро- и полинитрозамещенных бензольного и нафта-
линового рядов с никелевым катализатором в атмосфере водорода
при давлении 15 — 47,5 ат. Итальянец Пома для водной среды
дает указания на давление 5 —10 ат. Подходящими растворителями
названы спирт, бензол, причем они имеют преимущество перед
этиленгликолем. При давлении в 33 ат и при 215° достигается
количественный выход аминов. Оптимальные результаты дости-
гаются при количестве катализатора, отвечающем 0,52 г Ni на
каждый грамм восстанавливаемого продукта, т. е. очень значи-
тельном 26).
Подобно этому можно восстановить в присутствии никеля азо-
бензол (соотв. азоксибензол) в спиртовом растворе при 100° под
давлением водорода в гидразобензол. Повышение температуры
(до 126°) количественно переводит азобензол в анилин. Можно поль-
зоваться окислами или солями никеля, кобальта и прибавлять
окислы других металлов (Fe, Си, Мп)27).
Замечательно, что при гидрогенизации нитросоединений в при-
сутствии никелевого катализатора и раствора едкого натра полу-
чаются последовательно азокси-, азо- и гидразосоединения и амины;
таким образом и при этом методе влияние ОН-иона проявляет себя,
как и при восстановлении без молекулярного водорода метал-
лами в водной среде28).
Подобно как при превращении нитросоедииеиий в амины, метод гидрогениза-
ции в присутствии катализаторов в жидкой фазе может быть применен с успехом
и дня восстановительных превращений в приложении к несодержащим азота группам,
например при восстановлении индиго и индигоидных красителей в лейкосоедиие-
ния. Реакция проводится в присутствии раствора щелочи w).
При пользовании никелевыми и иными металлическими катали-
заторами гидрогенизации выгодно применять носители с возможно
более развитой поверхностью. Английский патент германской фирмы
предлагает для этой цели кислородные соединения кремния более
низкой степени окисления, чем SiO2 (например оксидисилин Si2OH2,
силоксен Si6O3H6).
493
Например раствор уксуснокислого никеля нагревают с оксидисилином и
занейтрализовав уксусную кислоту, промывают осадок. С таким катализатором
нитробензол в присутствии воды, при перемешивании, 30 — 40 ат давления
водорода и 60 — 80°, переходит в анилин. С подобным же катализатором, заклю-
чающим медь, а-нитронафталин в спиртовом растворе при 80—100е и 40 ат можно
перевести в а-нафтиламин. Для приготовления катализаторов никель можно полно-
стью или отчасти заменить другими металлами: Zn, Си, Pd, Pt, Au, Sn, Ag, Co, Pb,
Th, Bi 3C).
[Оксидисилин Si2(OH)H получается обработкой силицилида кальция в водно-
спиртовой среде посредством соляной кислоты на холоду, лучше в атмосфере
углекислого газа. Ои обладает сильно восстановительными свойствами.]
Упомянутый в главе V способ восстановления нитросоединений
в водной среде с участием недостаточного для полного взаимодей-
ствия количества растворимого сульфида в атмосфере водорода
или окиси углерода также связан с применением твердых катали-
заторов, например геля кремневой кислоты, окиси алюминия,
а также тяжелых металлов, их сульфидов, окислов и т. п.
Французский патент немецкой фирмы защищает способ, который
в сущности является просто гидрогенизацией в жидкой фазе нит-
росоединений в присутствии катализаторов (металлы 2—8-й групп
периодической системы, особенно тяжелые металлы этих групп,
далее Си, Ag, Аи и соединения этих групп, как таковые или на
носителях). По словам патента ни нитросоединения, ни водород не
требуют при этом очистки от сернистых соединений, причем эти,
обычно ядовитые для катализаторов, примеси регулируют действие
катализатора так, что реакция не идет далее образования амино-
группы, т. е. не имеет места ни гидрогенизация ядра, ни отщепление
NH3.
Это новое направление в гидрогенизационном катализе, утили-
зирующее обычно вредно действующие смеси, заслуживает боль-
шого внимания.
Реакция начинается уже при 100° и значительно ускоряется более тесным сопри-
косновением между газом, жидкостью н катализатором. Восстановление проходит
в железных автоклавах при температуре 150 — 200° под давлением водорода 140 —
200 ат. К чистым нитросоединениям добавляют при этом некоторое количество
серы (1%) или вводят с водородом сероводород. Из катализаторов названы железо
(в порошке или губчатом виде) и никель на искусственном цеолите. В последнем
случае катализатор находится в выложенной изнутри алюминием трубке, и нитро-
бензол в атмосфере водорода (4- NH3 4- H2S 4- Н2О) пропускается над его слоем 31).
Гидрогенизация для получения вторичных аминов испытывалась Мель(МайЬе)
и по отношению к основаниям Шиффа (типа RCH“NR'). Никелевый катализатор
при 220° дает с водородом и основанием нормальную реакцию:
RCHzzNR' + Н2 -» RCH,—NHR',
наряду с этим идет и побочная 32):
RCH—NR' 4- 2Н, -» RCH3 4- R'NH2.
Восстановление нитросоединений в присутствии воды, малого
количества кислоты и железа (чугунных стружек), подробно рас-
смотренное выше (глава V), по существу является реакцией, про-
текающей на поверхности железа. Гидрогенизация в этом случае
происходит не за счет молекулярного водорода, введенного вместе
с нитросоединением, но посредством водородных атомов воды, при
ионном ее расщеплении. Можно, в развитие схемы Воля, предста-
494
вить процесс гидрогенизации нитросоединения проходящим через
такие например фазы:
rnCo+h-‘ ё :oH'+Fe-*RN:O+H^o+Fe(°H)2>
RN : О + £ ; ; • ОН' Fe _> RN < + Н2О + ре(0Н)2,
rn <+н:::: он'+еЖ -> ™н2+2Ре(он)3.
Ввиду участия железа непосредственно в реакции с переходом
его в окислы взаимодействие не может быть рассматриваемо как
катализируемое железом; железо здесь — один из ингредиентов
сложной, по меньшей мере двухфазной, химической системы и участ-
ник последовательно развивающихся взаимодействий. Тем не менее,
наблюдаются в этом процессе определенно каталитические влия-
ния. Прежде всего само железо обнаруживает разную степень
активности, вероятно вызываемую или весьма малыми примесями
посторонних веществ или особенностями его макро-и микрострук-
туры. Причины этих антикаталитических, тормозящих влияний тре-
буют научного объяснения. Затем, как выше было уже установлено,
весьма существенным фактором в процессе восстановления желе-
зом оказывается присутствие электролитов в водном растворе.
Присутствие ионов, как анионов (чаще всего СГ), так и катионов
(Fe"’, Fe", Na’, АГ", Са” и др.), ускоряет процесс или содействует
его своеобразному течению, главным образом в направлении полу-
чения особой формы окислов железа. Примесь медной соли в про-
цессе восстановления железом ускоряет реакцию (А. Е. Порай-
Кошиц). Ввиду более низкого положения меди сравнительно
с железом в ряду напряжений последнее явление может найти себе
объяснение в образовании медно-железной пары на поверхности
железа, более активно содействующей ионному распаду воды. Вли-
яние же таких электролитов, как NaCl и т. п., на процесс желез-
ного восстановления нуждается в объяснении.
Метод восстановления цинком, особенно в щелочной среде,
употребительный для получения гидразосоединений, использует
реакцию, развивающуюся на поверхности металла. Значение катали-
тических влияний гидроксил-ионов по всей вероятности сводится
к активированию ациформы нитросоединений, как выше было уже
отмечено.
Гидрогенизация, приводящая к оводорожмванию ароматических
соединений и превращению их в гидроароматически е, может
быть интересна в приложении, к фенолам и нафталину. Выше
(глава XIV) эти примеры были уже упомянуты.
Гидрогенизация фенолов в паровой фазе над восстановленным
никелем изучалась уже Сабатье и Сандераном аз).
Ими констатировано, что гидрогенизация фенола при 180° приводит к получе-
нию циклогексанола С6Н31ОН с небольшой примесью фенола (5—10%) и еще мень-
шими количествами циклогексанона СвН]0О и циклогексена С8НЗП. Пропустив второй
раз смесь, кипящую в пределах 155—165°, через никель при 150— 1ТО°, получают
полное превращение фенола и циклогексанова в циклогексанол. Подобно этому
495
технически ценный лг-крезол превращается двойной гидрогенизацией на никеле
(200—220 и 180°) в метилциклогексанол. Гидрогенизация над никелем паров фенола
прн температуре 250 — 300° приводит преимущественно к бензолу
C^HjOH -J- Н3-»С8Н6 -j- Н2О
и прн дальнейшем повышении температуры — к метану. Диокснпронзводные бен-
зола прн 250° реагируют с водородом на никеле аналогично: последовательно пре-
вращаясь сначала в фенол, затем в бензол.
Замечательно, что, по германскому патенту 1923 г., можно из
гомологов толуола, в том числе прежде всего из крезолов, получить
с выделением метана фенол, соотв. низшие гомологи из высших,
пропуская их пары с водородом через активированные катализа-
торы гидрирования (Ni, Си, Ag с промотерами). Реакция по отно-
шению например к крезолу выражается уравнением:
С6Н4(СН8)ОН + Н2 = С6НВОН + СН4.
Катализатор готовится из хромовокислого никеля, осажденного например на
пемзе, восстановлением его в токе водорода при 800°. Смесь паров, состоящая на-
пример нз 85 объемных процентов о-крезола и 15 объемных процентов водорода,
проведенная при 400° через катализатор, дает с хорошим выходом фенол si).
Насколько специфично действие катализаторов при одинаковом
казалось бы составе реакционной смеси, обнаруживают например
исследования Фр. Фишера с сотрудниками над восстановлением
фенолов в паровой фазе водородом в углеводороды с катализа-
торами при обыкновенном давлении. лг-Крезол над окисью молиб-
дена уже при 330° образует толуол, при повышении температуры
до 360 — 380° восстановление идет наиболее полно:
С6Н4(СН8)ОН + Н2 -> С6НВСН3 + Н2О.
Наиболее активный катализатор — нз нитрата, получаемого растворением молиб-
дена в азотной кислоте. Окись молибдена мало чувствительна к контактным ядам
и по утверждениям исследователей не отравляется серой. Катализатор оживляется
окислением на воздухе прн невысокой температуре. Повышение давления до
10 — 20 ст ускоряет восстановление, но в этом интервале давления образуются
уже гидроароматические соединения з6).
Пропусканием смеси крезолов (трикрезола) в парах, в смеси
с водородом, через железную трубку, покрытую изнутри слоем
сернистого железа (обработкой сероводородом), при 750° можно
превратить их в бензол:
С6Н4(СН3)ОН + 2Н3->С6Н6-Ь СН4 + Н2О «в).
Стадников, Гаврилов и Виноградов восстанавливали
крезол в паровой фазе активным углем, нанесенным на мелкораз-
дробленное железо37).
Асбестом, пропитанным раствором азотнокислого железа, обработанным после
отжимания 10%-ным раствором аммиака, промытым, высушенным и разделенным
иа волокна, заполнялась реакционная трубка. Она нагревалась до 460 — 470° при
пропускании в течение 3—4 час. водорода для восстановления железа. После этого
температура поднималась до 480 — 490°, и в трубку при медленном токе водорода
вводился по каплям n-крезол, распадающийся при этом на водород, окись угле-
рода н углерод, оседающий на железе. После введения некоторого количества кре
зола в газе появляется СО2, и из трубки выделяется толуол. Не прерывая впуска
крезола, температуру печи постепенно снижают до 430° н получают в хороших,
выходах толуол. В этом примере восстановление идет однако не за счет водорода
496
но за счет углерода, осевшего на железе, так как после вытеснения водорода нз
трубки азотом реакция прн 430° приводит к тем же результатам. Контактными
ядами оказываются сернистые соединения. Таким образом реакция предположи-
тельно может быть выражена уравнением
2СН3С6Н4ОН + С = 2С6Н5СН3 + СО,.
Гидрогенизация нафталина водородом в паровой фазе над нике-
лем по Сабатье дает при 200° тетрагидронафталин, при 175° —
декагидронафталин. Нафтолы, а и р, переходят после двух после-
довательных гидрогенизаций (а — при 170 и 135°, р — при 170
и 150°) в соответственные декагидронафтолы.
Антрацен, подобно нафталину, гидрируется при более высоких
температурах менее полно, чем при менее высоких (в тетрагидро-
при 260, в октогидро- при 200°). Подобно этому относится к гидро-
генизации и фенантрен 38).
В широкую производственную практику вошел метод гидриро-
вания ароматических колец с катализатором при высоких давлениях
водорода, разработанный впервые В. Н. Ипатьевым39).
Работа производится в стальном трубчатом автоклаве — бомбе.
Катализатором служат чаще всего окислы никеля (NiO , Ni2O3),
температура 200 — 250°, давление 250 — 300 ат. Гидрогенизируемое
вещество вместе с катализатором вводится в автоклав, туда же
накачивается водород до давления например 100 ат, и по закрытии
автоклава при нагревании его давление повышается. Таким образом
гидрогенизация фенола при 245° в течение 14 час. дала превраще-
ние исходного материала в циклогексанол с некоторым количеством
циклогексана.
Практика заграничной промышленности использует этот метод
работы, применяя несколько более мягкие условия. Так, во фран-
цузском журнале указывается на давление водорода в 15 — 25 ат
и нагревание до 150° в течение нескольких часов при размешивании
для превращения фенола в циклогексанол. Катализатор — никель
в количестве 1/35 по весу фенола 40).
Патент немецкой фирмы дает указания также на более низкую температуру
гидрогенизации. Для превращения фенола в циклогексанол применяется катализатор
из 45 ч. окнси никеля, 45 ч. окиси кобальта и 10 ч. окисн медн. Отношение между
весом фенола н катализатора — 2:1. Температура взаимодействия 190—215°, дав-
ление 25 ат. Продолжительность взаимодействия 1% часа. Катализатор применяется
многократно 41).
Применение такого же метода каталитической гидрогенизации
с никелевым катализатором в случае а-нафтола при 130° дало смесь
из 85°/0 ас-тетрагидро- (I) и 15% ar-тетрагидронафтолов (II)
НО Н ОН
и в случае р-нафтола (150°): 75% ас-тетрагидро- и 25% аг-тетраги-
дропродукта 42).
Эти результаты в отношении а-нафтола совершенно отличны от
32 Зак. Э611. — H. Ворожцов
497
полученных Бамбергером при восстановлении «-нафтола натрием
в амиловом спирте, при чем не образуется ас-тетрагидродериват.
Методика Ипатьева применяется и при гидрогенизации нафта-
лина в тетрагидро- и декагидронафталины (тетралин и декалин)
в западноевропейской промышленности. Для сохранения активности
катализаторов здесь необходимо тщательно устранять серу из сое-
динений, входящих в реакционную смесь. Так как даже лучшие
сорта технического нафталина всегда содержат сернистые соеди-
нения (до 0,25°/о S в виде главным образом тионафтена), то его
очистка необходима. Это обстоятельство усложняет и удорожает
производство гидрированных продуктов, тем более, что наиболее
эффективная очистка требует участия металлического натрия.
Катализаторами служат преимущественно металлы: Ni, Со, щелоч-
ные и щелочноземельные металлы, смеси Си4 Ni-f-Co; окислы:
NiO, CuO, MnO, ThO2, смеси NiO-4CuO, MnOJ-ThO2. Гидрогени-
зацию проводят при давлении в 25 ат и температуре 120—140°.
Если нафталин для более равномерного нагрева разбавлен тетра-
лином, то температуру держат в 160—180°. По переходе всего
нафталина в тетралин начинается гидрирование до декагидропроиз-
водного. При температуре свыше 250° декалин отщепляет водород,
переходя в тетралин43).
Неизвестно, найдет ли тетралин себе применение как исходный
углеводород в красочной промышленности. Его ароматическое ядро
относится к замещениям как орто двузамещенное бензольное.
Гидрирование по Ипатьеву удается также и по отношению к более сложным
конденсированным системам: антрацену, фенантрену, флуорену.
Гидрогенизация антрахинона в присутствии никеля (при 160—170') легко при-
водит к получению антранола. Работа в растворе декалина неудобна, так как легко
перейти эту ступень ,4).
ОН
СО
Z\/\zz\
в) Дегидрогенизация
Гидрогенизации как присоединению водорода по месту двойных
углеродных связей в системе: ароматическое соединение — водород,
отвечает обратное превращение — дегидрогенизация: отщепление
водорода от гидрированного продукта с переходом его обратно
в ароматическое производное. Обычно равновесие в пользу дегидро-
генизации устанавливается при более высокой температуре, чем
нужно для гидрогенизации. Мы выше видели это уже на примере
превращения декалина в тетралин. Подобно этому циклогексанол
на никеле при 360° почти нацело превращается в фенол 45):
С8НПОН -> С6Н5ОН4-ЗН2.
Применение специальных катализаторов благоприятствует еще более протеканию
реакции дегидрогенизации гидроароматических соединений. В приложении к угле-
водородам это в высокой степени важно для превращения гидроароматических
углеводородов нефти в ароматические. Эта проблема изучается Н. Д. Зелинским,
и полученные им результаты по применению платины и палладия (в виде черни),
а также смешанного никелево-глиноземного катализатора практически весьма
интересны. Весьма замечательно то обстоятельство, что разработанный им метод
дегидрогенизации дает возможность превращения в ароматические углеводороды
только гидроароматических колец, но не 5- н 7-члениых или таких шестичлеиных,
которые не могут быть превращены в ароматические 46).
При описании реакций конденсации (гл. XV) были приведены
такие, где образование новой углеродной связи происходит через,
отщепление водорода. В некоторых случаях водородные атомы
могут быть отняты путем действия кислорода или серы на орга-
нические соединения и связаны в виде воды, соотв. сероводорода.
Имеются однако факты отнятия водорода при образовании новых
углеродных связей, где реакция возбуждается каталитическими
воздействиями и где водородные атомы связываются лишь в моле-
кулу водорода. Здесь следовательно имеет место тоже дегидроге-
низация своеобразного характера, которой должно предшествовать
некоторое ослабление, разрыхление связи между водородным ато-
мом и углеводородным остатком, при благоприятных к тому усло-
виях могущее закончиться полным разрывом этой связи с устано-
влением новой углеродной:
2RH -> 2R...H -> 2R--------1-2Н---> R — R + H2.
Такого типа реакции распада органической молекулы под влия-
нием высокой температуры называют пиролизом. Разрыхлению
углеродо-водородных связей прежде всего содействует повышение
температуры до достаточно высокого предела, — этим например
пользуются при крекинге нефтяных углеводородов. С другой сто-
роны, действие такого катализатора, как хлористый алюминий, осо-
бенно активного по отношению к углеродо-водородной связи, может
дать тот же результат и при более низкой температуре. Не исклю-
чается возможность участия и других катализаторов.
В наиболее простом виде подобная дегидрогенизационная кон-
денсация имеется в превращении бензола в дифенил. Практика
американской промышленности использует в широком размере
метод, описанный в швейцарском патенте, для получения дифе-
нила, имеющего большое значение в качестве теплопередающей
среды для обогрева при высоких температурах 47).
Бензол прогоняется через баню с расплавленным свинцом, причем он испа-
ряется н подогревается до максимальной температуры в 600— 650°, прн которой еще
не имеет места образование дифенила, и затем прогоняется через вторую свинцовую
баню, нагретую до 750—800°, в которой уже идет реакция с образованием значи-
тельных количеств дифенила. Пары, выходящие из реакционной бани, быстро охла-
ждаются и конденсируются, водород отводится в газгольдер. Конденсат дает при
перегонке дифенил. Выход дифенила 10—12%, считая на пропущенный бензол.
Реакции отвечает уравнение
2СВН6 = С6Н5- С„НЙ + Н2.
Исследование влияния температуры, давления и скорости тока бензола при получе-
нии дифенила выяснило, что оптимальная температура 740°, давление приблизи-
тельно 1 ат. степень превращения падает с возрастанием скорости, причем большее
падение констатировано при атмосферном давлении, чем при более высоком. Израс-
ходованный бензол превращается в дифенил на 92-98% 48).
В работе американцев Л оу и Джемса описывается ниой метод дегидроге-
низации бензола — пропускание его паров над нагретой до слабокрасного каления
нихромовой проволокой 49).
32*
499
Уже чисто контактные методы дегидрогенизации бензола опи-
сывает в своих патентах немецкая фирма. В ее французском патенте
указывается на пропуск паров бензола, соотв. других ароматических
углеводородов, не имеющих цепей больше, чем в один атом угле-
рода, при 500—800° через катализаторы, содержащие минимум
один трудновосстанавливающийся и трудноплавкий окисел, по-
крываемые иногда графитированным углеродом. Работу можно
проводить в присутствии инертного газа.
Окись магния, обработанная при 700° ацетиленом для образования графитового
слоя, является катализатором для реакции превращения бензола в дифенил: при
775° однократный пропуск дает около 10% дифенила. Нафталин с 0,2% по объему
водяного пара при пропуске над катализатором из WO3-|-A12O3 образует смесь из
-динафтила и ап'-динафтила с преобладанием первого.
В немецком патенте этой же фирмы описывается иной прием работы и другие
катализаторы (карбонаты). Бензол нагревается до с0°, увлекается током углекислого
газа, затем пары проводятся через смесь из MgCO3 + Li2CO3 при 500°. В про-
дукте реакции дифенил (I) вместе с 20% дифенилбензола (11)
(I) С6Н3 • С6Нб (II) С6Н5 • С6Н( • С6Н5 (III) С6Н5 • СН2 • С6Н5.
Подобно этому толуол сСО2, пропущенный через катализатор К2СО3 • H2O-(-ThO2-|-CoO
на пемзе дает дифенилметан (III) и смесь других углеводородов 5°).
Дегидрогенизационные конденсации могут сопровождаться при
подходящих для этого условиях изменением структуры соединений
и образованием новых циклов (чисто углеродных и гетероциклов).
Так метил-о-толуидин при 300—330° над никелем в присутствии водорода пре-
вращается на 6% в индол (1)
С большим выходом (24%) получается из диметил-о-толуидина N-метилиндол
(II) 61).
По недавнему немецкому патенту, подобную дегидрогенизацию вместе уже
с окислением водорода возможно осуществить для получения индолов не только
из производных о-толуидина, но и в юбще из ацилированных аминов (если алкил
минимум С2Н3) при ие замещенном алкилом орто-меще к N. Для достижения этого
нужно пары алкилированных аминов проводить в присутствии окисляю цего вещества
(воздух, пары иитросоединений) через пористые катализаторы: гель кремневой
кислоты, активированный фосфорной кислотой уголь, лучше — с добавкой катализа-
торов окисления (окислы Се, V, Мп, Си, Со). Температура контакта лежит в преде-
лах 300—400°. Таким образом возможно например получение N-этилиндола из ди-
этиланилина 5-)
Возможно, что реакция в этом случае более сложна, чем это передает уравнение,
и в ней имеет место зредварительное окисление этильной группы в ацетильную,
так как примерно те же реакционные условия нужны для осуществления ниже
рассмотренного превращения ацилированных аминов в индолы с дегидратацией.
500
Хлористый алюминий, с некоторыми из применений которого
в конденсациях органических молекул (с отщеплением галоидово
дорода) мы познакомились в предыдущей главе и (отчасти во II и IV)
вообще приводит молекулы органических соединений в состояние
с более ослабленными связями между углеродом и водородом.
Таким образом, действуя на ароматические углеводороды, хлори-
стый алюминий содействует их расщеплению на водородные атомы
и ненасыщенные радикалы, из которых последние могут соединиться
в молекулу с большим числом ароматических ядер, чем было в на-
чальном продукте. Вследствие благоприятного влияния катализа-
тора эта реакция дегидрогенизации проходит при более низкой
температуре, чем вышеуказанные конденсации, например в свинцо-
вой бане.
Таким образом из бензола при действии хлористого алюминия
образуется дифенил, из нафталина р,3'-динафтил, из этилового эфира
а-нафтола (I) в нитробензольном растворе — 4.4'-диэтокси-1.1'-ди-
нафтил (II) 53).
В последнем из названных превращений следует обратить вни-
мание на роль нитробензола. Хлористый алюминий при высокой
температуре содействует, как мы видели в главе XI, отрыву алкила
от кислорода (в гидроксиле). Между тем с нитробензолом реакция
протекает уже при обыкновенной или даже более низкой темпе-
ратуре и идет только в сторону образования диалкоксидинаф-
тила (II):
Шолль усматривает в этом явлении совместное активирующее по
отношению а-водородного атома нафтильных остатков действие и
нитробензола и хлористого алюминия. Вместе с тем нитробензол
устраняет из сферы реакции образующийся водород, восстановляясь
сам. Активирующее влияние на определенные участки ароматиче-
ского ядра со стороны нитросоединений Шолль видит в молеку-
лярных соединениях нитросоединений с углеводородами, где нитро-
группа „насыщает побочные валентности углерода в ароматическохм
ядре и делает подвижным ароматически связанный водородный
атом".
В практике получения некоторых сложных производных антра-
хинона хлористый алюминий нашел себе применение как средство
для дегидрогенизационной конденсации. Так, из ^-аминоантрахинона
нагреванием его до 250—280° с хлористым алюминием образуется
продукт дегидрогенизационно-дегидратационной конденсации —
501
ценный желтый кубовый краситель, флавантрен (I)84). Реакция про-
текает через ступени:
СО СО
Лучшим, чем хлористый алюминий, вспомогательным катализато-
ром является однако здесь пятихлористая сурьма SbCl3, применяю-
щаяся в растворе нитробензола 55).
Другим примером аналогичного применения хлористого алюминия (в пиридино-
вой среде) может служить превращение продукта (II), заключающего и фенаитрен-
хиноиовое и антрахиноновые ядра в производное карбазола (III) нагреванием до
150—180° •«):
ОС СО
502
Особенно легко удаются при действии хлористого алюминия
такие замыкания внутренних циклов с потерей водорода в углево-
дородных производных с конденсированными циклическими систе-
мами (нафталин и т. п.). Эти реакции, изученные III о л л е м, проте-
кают при сравнительно невысокой температуре (80—180°).
Таким путем из а-нафтилфенилкетона (I) может быть получен
бензантрон (II) 57), из аа'-динафтила (III) — перилен (IV), крайне инте-
ресный углеводород, находящийся в составе наиболее прочных
кубовых красителей антрахинонового ряда.
Действие хлористого бензоила на бензантрон в присутствии
хлористого алюминия приводит последовательно сначала по реакции
Фриделя и Крафтса к фенилбензантронилкетону (V), а затем
с отщеплением водорода, который практично устранять при окисле-
нии воздухом, — необходим избыток хлористого алюминия для свя-
зывания воды, — к 3.4.8.9-дибензпирен-5.10-хинону (VI) — прочному
желтому кубовому красителю 5Ч):
г) Окисление
Большого внимания заслуживает задача окисления аромати-
ческих соединений кислородом воздуха с применением катализато-
ров. В практическом приложении она ставится преимущественно как
окисление ароматических углеводородов. Согласно сказанному в гла-
503
ве XIII, в зависимости от наличия боковой цепи (группа СН3) в угле-
водороде, целью превращения может быть либо получение альдегид-
ной (СНО), либо карбоксильной (СООН) группы у углеводородного
ядра: в том случае, когда исходным материалом оказывается не
имеющий боковых цепей углеводород, окислительным превраще-
ниям подвергается в первую очередь ароматически связанный водо-
род, заменяясь или гидроксилом или — с перестройкой ядра-—ато-
мом кислорода, входящим в состав карбонильной группы хинона.
Наконец последним, но далеко не самым маловажным типом окисли-
тельных превращений углеводородов являются такие, в которых
нарушается сам углеродный скелет ароматического соединения.
Выше были уже разобраны явления самоокисления в гомогенной среде. Подоб-
ные явления возможны и в гетерогенной среде. Известно, что водородистый палла-
дий в водном растворе, где он сам жадно реагирует с кислородом, вызывает энер-
гичные окисления: не говоря об окислении неорганических веществ (например
аммиака в азотную кислоту), констатировано превращение бензола в февол н то-
луола в бензойную кислоту. Здесь очевидно имеют место упомянутые выше сопря-
женные реакции 59).
Наиболее интересно проведение окислений углеводородов в па-
ровой фазе при высокой температуре при наличии катализаторов.
Химически это — реакции неполного сгорания углеводородов, с оста-
новкой процесса на получении определенных промежуточных про-
дуктов на пути полного окисления: всего углерода в СО2 и всего
водорода в воду 60).
Полное окисление ароматических углеводородов при помощи воз-
духа требует определенной температуры. Эту температуру можно
снизить с применением катализаторов. Такова например платина,
с которой смесь паров бензола с воздухом дает продукты полного
окисления при пониженной температуре, если сравнить с той, кото-
рая требуется для сгорания бензола в парах без катализатора.
Однако реакция происходит слишком бурно, и уловить промежу-
точные стадии окисления при этом не удается. Лишь подбор соот-
ветственных катализаторов и определение для каждого из них
наиболее благоприятных температурных пределов процесса, так же
как и иных факторов реакции, дало возможность создать произ-
водственно приемлемые формы окисления.
Первым наиболее известным фактом каталитического окисления
углеводорода, имевшим самые серьезные производственно-техниче-
ские последствия, было одновременное открытие Вол ем в Герма-
нии и Гиббсом и Конновером в Америке (в сентябре 1916 г.)
возможности превращения нафталина в паровой фазе с воздухом
во фталевый ангидрид при пропускании смеси паров через ката-
лизатор, содержащий пятиокись ванадия 61).
Изобретателями и многими другими исследователями были
испытаны затем этот и другие катализаторы в различных комби-
нациях с иными составными частями контактного слоя, и, хотя эту
работу нельзя считать ни в какой степени законченной, к настоя-
щему времени известен список наиболее подходящих катализаторов
окисления ароматических углеводоров; это по преимуществу —
окислы металлов 5-й и 6-й групп периодической системы: ванадия,
вольфрама, молибдена, урана или смеси их окислов, также иногда
504
более сложные смеси с участием окислов или соединений иных:
тяжелых металлов: Сг, Си, Sn и т. п.
Приемы для приготовления катализаторов, количество и при-
рода входящих в состав катализаторов веществ могут быть весьма
разнообразны — в зависимости от природы окисляемого продукта,,
равно и степени окисления. Учитывая то обстоятельство, что все
реакции окисления экзотермичны и что для каждой степени окис-
ления имеется своя оптимальная температура, выше которой пере-
ходить нельзя без опасности нарушения наиболее благоприятного
для нее теплового режима, необходимо при всяком катализаторе
обеспечить достаточно полный отвод выделяющегося при окислении
тепла, чтобы не получалось „застоев тепла" на катализаторе,,
а следовательно окисление не шло бы дальше нужной фазы. Это*
обстоятельство приводит как к обязательным условиям, во-первых,
к применению для помещения катализатора трубок неширокого
сечения, чтобы передача тепла совершалась по возможности не
через большой слой катализатора и в разных точках его сечения
не было бы слишком разительной разницы температур, и, во-вторых,
к преимущественному использованию контактных слоев, обладаю-
щих большой теплопроводностью, почему в практику входят
такие например инертные в окислительном процессе носители, как
гранулированный алюминий.
По расчету американца Геффа при окислении нафталина до фталевого ангид-
рида кислородом воздуха (в четырехкратном количестве по отношению к теореа
тнчески требуемому) в совершенно изолированном пространстве температур-
смеси, а значит и катализатора, поднялась бы приблизительно на 594—616°. Если
представить, что прн этих условиях будет поступать в контактную трубку по-
догретая до 250° смесь паров с воздухом, то температура поднимется до крас-
ного каления, и начнется полное окисление уже фталевого ангидрида до углекис-
лоты и воды не только на поверхности катализатора, ио во всей массе,, причем, по-
нятно, и катализатор, спекаясь от жары, потеряет свою каталитическую актива
ность в2).
По этим основаниям отвод тепла от контактных трубок является
крайне важной задачей для поддержания нужного теплового режима
процесса окисления. Практичнее с этой стороны пользоваться для:
окисления не чистым кислородом, но воздухом, так как балластный
азот будет уносить с собой избыток тепла. Применяемые для
окисления количества воздуха чаще всего значительно превышают
рассчитанные по стехиометрии. Контактные трубки делаются из
материала, достаточно дешевого, индиферентного в окислительном
процессе и теплопроводного, стойкого механически. Таким мате-
риалом оказывается обыкновенная сталь, хотя и имеются указания
в патентной литературе на преимущество трубок из нержавеющей,
хромоникелевой стали, никеля и даже серебра. При всех прочих
равных условиях имеют преимущество трубки не круглого, но
прямоугольного сечения: более развитая поверхность, преимущества
размещения в агрегаты, равномерность слоя катализатора. Но
и круглого сечения трубки при небольшом диаметре оказываются
достаточно удобными.
Снаружи контактные трубки соприкасаются со средой, которая
принимает на себя тепло реакции и выравнивает температуру.
В практике такой средой служит обычно свинец в расплавленном
505
состоянии, омывающий все трубки системы, обеспечивающий нагрев
системы до нужной для начала реакции окисления температуры
и отбирающий затем тепло от трубок.
В американской литературе отмечаются преимущества жидкой
бани с постоянной, отвечающей температуре реакции температурой
кипения. В качестве наполнителя такой бани указывается например
ртуть или смесь ртути с кадмием, причем давление азота, под
которым идет кипение, обеспечивает поддержание температуры на
желаемом уровне. Указывается также сера как материал для напол-
нения бани с контактными трубками. Ниже в своем месте мы при-
ведем схему такого аппарата. При прочих равных условиях однако
сопряженные с применением ртути расходы и опасности для здо-
ровья работающих несомненно заставляют, где можно, избегать
применения этого материала.
Работа с окислением углеводорода кислородом воздуха доста-
точно безопасна при правильном ходе процесса. Тем не менее
в заграничной технической литературе зарегистрированы отдельные
случаи взрывов, весьма тяжелых по своим последствиям, на уста-
новках например фталевого ангидрида. Эти обстоятельства необ-
ходимо иметь в виду и, тщательно соблюдая, с одной стороны,
нормальные условия для проведения процесса, не забывать при
конструировании аппаратуры о необходимых на случай взрывов пре-
дохранительных приспособлениях; например устройстве слабых
мест — диафрагм из мягкого металла (при входе и выходе газов из
контактного аппарата конвертора), которые, открывшись в случае
быстрого повышения давления, могли бы дать выход газам.
Окисление метильной группы в ароматических углево-
дородах изучалось преимущественно применительно к получению
бензальдегида из толуола. Американский исследователь Кревер,
оценивая каталитическую активность окислов металлов в этом
процессе, выделяет, как наиболее энергичный, пятиокись ванадия,
которая окисляет толуол часто с распадом ядра. Не указывая
условий окисления, он утверждает, что толуол с V2O5 дает бензаль-
дегид, бензойную кислоту, малеиновую кислоту и продукты полного
сгорания в количествах соотв. 1:2,47:1,45: 1,35, между тем как
окислы Mo, U, W, Та и Сг дают только бензальдегид с весьма
малым количеством продуктов дальнейшего окисления.
Кревер делит окислы иа следующие классы в зависимости от их активности
при одинаковых условиях проведения процесса.
1. Относительно высокая продукция бензальдегида и относительно низкий эффект
полного сгорания. Окислы Та, W, Zr, Мо.
2. Относительно высокая продукция альдегида и относительно высокий эффект
полного сгорания. Окислы Мп, Си, Ni, Cr, Th, U.
3. Относительно низкая продукция бензальдегида н относительно высокий эффект
полного сгорания. Окислы Со и Се.
4. Относительно низкая продукция бензальдегида и низкий эффект полного сго-
рания. Окнслы Ti, Bi, Sn.
Смеси окислов, по утверждению Кре вера, дают более благоприятные результаты
в смысле выходов бензальдегида; так, смесь из 88,5% UO3 и 11,5% Мо03 дает при
выходе в 6,44% бензальдегида только 1,1% продуктов полного сгорания, между
тем как для одного 1Ю3 отношение выходов 6,5:39,7, а для МоО3 1:1.
Пятиокись ванадия, благоприятствуя более полному окисли-
тельному эффекту гомологов бензола, приводит к образованию пре-
506
имущественно карбоновых кислот, соотв. дикарбоновых кислот
(при орто-положении двух СН3), и дает заметное количество малеи-
новой кислоты.
Кревер готовит катализаторы осаждением соли металла, соотв.
солей металлов (иногда лучше солей органических кислот), на асбесте,
пемзе и тому подобных носителях, или осаждением на носителях
окислов из их суспенсии и прокаливанием до образования из солей
соответствующих окислов.
Оптимальное отношение толуола н воздуха по весу 1:14, т. е. близкое к теории
при смешанном урано-молнбденовом катализаторе (лучше вместе с промотером СиО),
температура немногим выше 500°. Вообще же пределы температур Кревер уста-
навливает между 300 — 700° в зависимости от окисляемого углеводорода, скорости
прохода паро-воздушной смесн, давления и соотношения между количествами кис-
лорода и углеводорода га).
Немецкий патент 1927 г. описывает применение для получения
бензальдегида из толуола смешанных катализаторов по типу ката-
лизаторов, указанных Вол ем в его патентах, например смесь
молибдатов меди и урана на подходящем носителе.
Температура реакции в пределах 400 — 500°, в паро-воздушной смеси, получен-
ной продуванием воздуха через толуол, нагретый до 60—70°, содержится 400 —
800 г толуола на 1 м':; воздух добавляется по мере потребления кислорода. Окис-
ление идет, как н всегда с толуолом, не нацело, почему в патенте описывается
проведение кругового процесса с насыщением (добавочно, в промывателях) толуои-
лом нескондеисировавшихся паров н воздуха и с введением нх вновь в контактный
аппарат 6‘).
Мэкстед исследовал в окислении толуола и других углеводо-
родов катализаторы типа солей ванадиевой кислоты, особенно вана-
даты олова и висмута, упомянуты также соли РЬ и Со.
Исследования Мэкстеда очень полно освещают значение от-
дельных факторов каталитического окисления углеводородов.
С
и
Рис. 13. Прибор для каталитического окисления углеводородов (по Мэкстеду).
Применявшийся им прибор (рис. 13) позволяет отсчитывать скорость тока воа-
духа, увлекающего пары толуола (первичный воздух), и тока воздуха, вводимого
в смесь перед соприкосновением с катализатором (вторичный воздух). Измерения
производились реометрами А н С. Толуол находится в склянке В, подобной про-
мывной, находящейся в бане (нагретой до 40° при окислении толуола). Склянка В
(карбюратор) сообщается с контактной U-образиой трубкой из кварца, внутреннего
диаметра около 4 мм. Объем (кажущийся), занимаемый катализатором, 10 смл.
507
Катализатор состоит из осажденного и высушенного ванадата олова, после раз-
дробления разделенного на гранулы приблизительно в 3 мм линейного размера.
Вес катализатора 9—10 г. Катализатор трудно отравляется и может долго приме-
няться. Трубка с катализатором помещена в баню D из расплавленной эвтектической
смеси нитратов натрия и калия. Контактная трубка своим выходным отверстием
сообщается с конденсатором Е—суженной посредине пробиркой, на запаянном
конце которой выдуто небольшое отверстие. В ней собирается продукт окисления —
в опытах Мэкстеда—кристаллы бензойной кислоты.
Мэкстед выражает скорости газовых потоков в литрах в час на 1 л объема
катализатора; выхода в процентах по весу сырого материала, прошедшего через
катализатор. Выходом по времени на объем он называет количество граммов про-
дукта в час, отнесенное к 1 см3 объема катализатора. Выходы в процентах выво-
дятся на основании разницы в весе карбюратора до и после опыта, следовательно
допускают потерю непрореагировавшего толуола. Температурой катализатора счи-
талась температура бани, в которую погружена контактная трубка.
Для характеристики полученных данных приведем выдержку
из таблицы его опытов при температуре контакта — 290°.
Таблица 1.
Опыт Объемная скорость первичного воздуха Объемная скорость вторичного воздуха Выход и °/0 бензойной кислоты Выход по времени на объем
1 150 700 32,5 0,010
2 200 500 50,1 0,018
3 200 500 57,1 0,028
4 200 500 49,0 0,017
5 200 500 57,1 0,018
Среднее• • • • 53,3 0,020
6 200 700 41,6 0,0125
13 300 1 500 44,6 0,0145
Нанлучшие результаты (50 — 57°/0 выхода) получены при объемной скорости
в 200 первичного и 500 вторичного воздуха. При повышении скорости свыше опти-
мального предела много толуола проходит без окисления. При малой скорости,
с другой стороны, происходит сгорание части образовавшейся бензойной кислоты
до СО2 и HjO.
Работая прн более низкой температуре и изменяя условия, возможно получать
и бензальдегид. Замещенные толуола подобным образом окисляются с образованием
кислот на ванадате олова прн 270—300° ®).
Катализатор, заключающий окислы железа и ванадия, описан
в английском патенте 1929 г. Он готовится распределением в асбесте
метаванадата аммония и окислов железа [при обработке FeS04
аммиаком (отношение количеств NH4VO3:FeSO4 = 30:107)]. Слегка
промытую массу формуют в палочки, сушат и превращают в неболь-
шие куски. Этот катализатор пригоден для окисления толуола уже
при 290° 66).
Смешанный катализатор, состоящий из окислов металлов различных групп
периодической системы с преобладанием основного окисла, особенно пригодный
для проведения гидрогенизаций (например превращений: СО Н2 -> СН3ОН;
СН^СН, -» СН3 • СН3), может оказаться полезным и для окисления толуола в бензаль-
дегид. Так, катализатор из 3 ч. ZnO и 1 ч. СгО3 при пропускании над ним смеси
508
воздуха в паров толуола при 450 — 500° благоприятствует образованию бензаль-
дегида и бензойной кислоты W).
Исследованию различных смешанных катализаторов, заключаю-
щих окислы ванадия, молибдена и марганца, в окислении толуола
посвящена работа М. И. Кузнецова и сотрудников 68).
Были испытаны: основной манганат циика, ванадат висмута, метаванадат олоиа,
окислы: V2O5 на пемзе и на асбесте, МоО3 на пемзе и на асбесте. Воздух проду-
вался со скоростью 8 — 10 л в час при 60 — 80° и насыщался парами толуола (1 г
толуола в 1,6 — 2 л воздуха) и затем проводился через катализатор. Наибольший
эффект окисления обнаружен у катализаторов: МоО3 на асбесте (9,2 — 55,7% окислен-
ного толуола), ванадата висмута на пемзе (20,5 — 32,0%), Sn(VO3)4 (16,8 — 31,6%),
при этом МоО3 на пемзе дает наилучшие отношения между бензальдегидом и бен-
зойной кислотой [(RCHO : RGOOH = (68,3 — 87,8%): (1,5 — 8,0%)]. В этом полученные
результаты согласуются с данными К р е в е р а. Однако в противоречие с послед-
ним V2O3 при относительно небольшом проценте окисленного толуола дал в про-
дуктах окисления также преимущественно бензальдегцд, а не бензойную кислоту.
С. В. Богданов и Н. С. Лезнов получили при контактном
окислении толуола достаточно высокие выходы бензальдегида для
возможности практического использования этого метода. Катали-
затором у них служит смесь V2OS и Мо03, осажденная на метал-
лическом инертном носителе (NiCr). Предварительно катализатор
подвергается восстановительной обработке (например цинком и соля-
ной кислотой). Быстрая „приспособляемость" катализатора вероятно
находится в связи с понижением степени валентности окислов
металла, служащего катализатором. Возможно, что промотером
являются и хлористые соединения.
Отметим, что еще в 1908 г. Е. И. Орловым было констати-
ровано образование бензальдегида при контактном окислении
толуола воздухом над окисью меди при 300 — 320°. Выход рав-
нялся 1 —1,5°/0 на весь введенный в реакцию толуол и около
2,4 — 4% на прореагировавший толуол 70).
При окислении паров толуола и других углеводородов, в том числе антрацена,
над катализаторами—окнслами элементов 5-й и 6-й групп периодической сиссемы
(V2O5, МоО3) — можно пользоваться при более высокой температуре вместо воздуха
углекнслотой. Углекислота удобна как не дающая опасности взрыва с парами
углеводорода. Окисляя углеводород, она раскисляется до окиси углерода. Продук-
том окисления толуола, по утверждению немецкого патента, является бензаль-
дегид в9).
Синтез бензальдегида и бензойной кислоты путем контактного
окисления толуола интересен для получения этих продуктов, не
содержащих совершенно примесей хлорозамещенных, какие могут
быть в полученных из хлористого бензилидена, соотв. бензотри-
хлорида, соединениях. Чистые от органических хлоросоединений
продукты, особенно бензальдегид, нужны например для фабрикации
душистых веществ. Повидимому возможно проводить хлорирование
и обмен хлора в <о-хлорозамещенных толуола настолько совер-
шенно, что бензальдегид и бензойная кислоты практически не
заключают аналитически определяемых количеств хлора. Недостат-
ком способа контактного окисления толуола оказывается невоз-
можность получить сколько-нибудь полное окисление толуола
в один проход и связанная с этим необходимость улавливания
непрореагировавшего толуола и, по отделении от него продуктов
окисления, введения толуола повторно в контактный аппарат, сле-
509
довательно необходимость создания более громоздкого с производ-
ственной стороны кругового процесса.
На основании опытов окисления в жидкой среде гомологов бензола кислородом
без катализатора, не давших возможности открыть в продуктах окисления спирта
АтСН2ОН и определивших тормозящее влияние воды в этом процессе, Стнфенс
полагает, что процесс окисления здесь проходит последовательно через стадии обра-
тимых реакций, заканчивающиеся необратимым превращением „ненасыщенного
остатка" в карбонильное соединение (альдегид, кетон). Схема Стифенса такова:
Углеводород -Н О» комплекс
Н,О+ „ненасыщенный остаток" -* альдегид.
Понятно при этом, что повышение количества воды может сдвигать равновесие
в последней обратимой реакции не в пользу образования „ненасыщенного остатка ,
т. е. тормозить полезную реакцию.
Комплекс, предполагаемый Стифе псом, можно представить согласно Муре
переходящим например для толуола из состояния активной перекиси СсН5СНз(Ог]
в состояние настоящего перекисного соединения C6H5CH3'Og, кислород в котором
реагирует далее с водородом, образуя ненасыщенный радикал, например С6Н5СН<^
стабилизирующийся в альдегид 71)- Повидимому подобные же стадии нужно пред-
полагать и прн течении реакции на катализаторе.
Контактное окисление бензилового спирта до бензальдегида — случай практи-
чески мало интересный — возможно при пропускании смесн егр паров с воздухом
(12% беизилового спирта) при 250 — 300е через катализатор, состоящий нз носителя
(шамот, железо, пемза) и фосфорной кислоты и).
При окислении кислородом бензилового спирта в бензойный альдегид в жидкой
среде (в растворе толуола и его гомологов) из испытанных металлических катализа-
торов оказалось преимущественно активной медь 73).
О возможности контактного окисления хлористого бензила
в бензойный альдегид было сказано выше, в главе VII.
Окисление бензальдегида в бензойную кислоту—процесс само-
произвольно, хотя и медленно, протекающий уже при стоянии бен-
зальдегида на воздухе. Блан нашел, что этот процесс ускоряется
от добавления катализаторов, каковы металлы: Ag, Pt, Ni, Си, и
окислы и соли металлов Ni, Со, Си. Носителем катализатора слу-
жили песок, инфузорная земля, уголь, безводные соли натрия. Тем-
пература поднимается сама при продувании воздуха до 100° и
должна поддерживаться на этом уровне внешним охлаждением 74).
Окисление бензальдегида в паровой фазе над ванадатом олова изучалось Мэк-
с те дом, констатировавшим, что окисление идет практически при той же темпера-
туре, которая характерна и для окисления толуола в бензойную кислоту. Таким
образом оптимальная температура образования продукта окисления зависит в боль-
шей степени от качества катализатора, чем от природы исходного материала. Ма-
ксимальный выход бензойной кислоты из бензальдегида не превосходил выхода
кислоты прн получении ее непосредственно из толуола. Можно было бы ожидать,
что катализатор в этом окислении будет более продуктивен, чем при окислении
бол^е трудно окисляемого толуола. На самом же деле максимальный выход по
времени на объем — величина того же порядка, что н при окислении толуола. Сле-
довательно ограничительный фактор присущ самому катализатору (относительная
скорость адсорбции и десорбции), а не реакции. Оптимальная температура катали-
затора (300°) выше, чем прн окислении толуола, вероятно в зависимости от отно-
сительно невысокой экзотермнчностн окисления бензальдегида 75).
Окисление бензола представляет интерес с двух сторон.
Во-первых, как теоретически мыслимый метод получения таким
путем фенола, во-вторых, как метод получения малеиновой кис-
лоты. Оставляя рассмотрение последнего превращения до дальней-
шего, остановимся на первом.
510
Превращение бензола в фенол непосредственным окислением
могло бы крайне упростить задачу получения фенола, так как
устраняло бы ряд промежуточных стадий получения сульфокислоты,
соотв. хлорбензола. Выше, в главе VI, было уже упомянуто о попытке
разрешения этой задачи методом щелочного плавления с участием
катализаторов окисления — окислов металлов 6-й и 7-й групп пери-
одической системы. Бибб в американском патенте 1923 г. описы-
вает метод гомогенного окисления бензола в паровой фазе при
пропускании паров бензола с воздухом или кислородом с добав-
лением небольшого количества окислов азота (1°/о от паро-воздуш-
ной смеси) через кварцевую трубку, нагретую до 700°. Соотношение
между парами бензола и воздуха 1 :3. Выделяющийся при охлажде-
нии фенол содержит в небольших количествах примеси нафталина,
дифенила, окиси дифенилена и др. 76).
Немецкая фирма в своем патенте описывает окисление бензола
в фенол в гетерогенно-паровой фазе при пропускании паров бензола
в смеси с воздухом над катализаторами, представляющими собой
окисные соединения металлов, которые на кривой атомных объемов
находятся на минимумах больших периодов или вблизи минимумов.
Подходящими катализаторами таким образом оказываются окисли
Cr, Mo, W, U, V, Nb, Та, Ti, происходящие от этих металлов
кислоты или соли, например молибдат меди, ванадат серебра,
вольфрамат цинка. К окислам возможна добавка других металлов —
Fe, Си, Ag— или окислов А12О8, СаО или карбонатов, фосфатов и
других солей. Воздух предпочтительно разбавить (до содержания
1 — 5 объемных процентов кислорода) индиферентными газами или
парами [азот, СО2, водяной пар, малые количества водорода (!)].
Температура дается в пределах 400—500°.
Примеры приготовления катализаторов:
1. WO(OH)4 замешивается в тесто с раствором (NH4)2CrO4, н масса отсасывается.
После сушкн масса нагревается в течение часа до 700—800°, по охлаждении замеши-
вается с малым количеством воды, наносится на кусочкн чистой пемзы и высуши-
вается. Через такой катализатор пропускают при 400—500° круговым процессом
смесь паров бензола н разведенного азотом воздуха. Выход фенола около 750/0,
считая на прореагировавший бензол.
2. Тщательно размешивают на шаровой мельнице основную углекислую соль
медн, Н2МоО4, WO(OH)4 и 1Ю3, нагревают порошок до 350°, формуют в зериа
надлежащего размера и обрабатывают их в контактной печн воздухом сначала 5 час.
при 360°. Затем при 350-37о° пропускают над катализатором смесь паров бензола,
водяного пара и разведенного азотом кислорода круговым током со скоростью
большей, чем 3 л3 в час. Выход фенола на прореагировавший бензол до 88%.
3. Подобно вышесказанному, прн пропускании круговым током при 350—380°
паров хлорбензола с воздухом -f- азот через катализатор из CuO, МоО3 WO3, UO3
получают преимущественно смесь о- и n-хлорфенолов и немного фенола 77).
Опытная проверка патента в нашей лаборатории И. С. Трав-
киным подтвердила образование фенола в описанных условиях,
но в таких минимальных количествах при одном проходе через
контакт, что становится сомнительной практическая осуществи-
мость такого процесса, особенно если принять во внимание рас-
ход щелочи на вымывание фенола 78).
Окисление связанного в ароматическом цикле водорода
в карбонильный кислород с образованием хинонов, соотв. дике-
тонов, имеет значение преимущественно для получения антрахинона
511.
и фенантренхинона, хотя образование хинонов при контактном
окислении кислородом констатировано и по отношению к другим
исходным продуктам, например при окислении нафталина.
Окисление антрацена в парах кислородом воздуха возможно
при участии уже вышеупомянутых катализаторов, главным образом
окислов ванадия. Ввиду особой реакционности мезоатомов антра-
цена и сравнительно большой стойкости антрахинона к окислению,
кажется понятной осуществимость такого окисления. Смесь паров
антрацена и нагретого воздуха проходит над контактным слоем
с подходящими окислами, и полученный антрахинон конденсируется
из пара в конденсаторах.
Повидимому первым, указавшим на возможность получения антрахянона прн
окислении антрацена воздухом в паровой фазе, был И. Вальтер. Применявшийся
им катализатор состоял нз окислов ванадия на асбесте, покрывавших платиновую
проволоку, слой катализатора тонок (толщина асбестнрованной проволоки), темпе-
ратура на контакте свыше 500°. Выход малый и).
А. Вол ем разработан более практически приемлемый тип
катализатора и способ работы 80).
Им описаны катализаторы характера кислотных окислов, могу-
щих менять свою степень окисления от действия кислорода при
температуре ниже красного каления: V2O5, МоО3, урановая кислота
в форме желтого ураната, калия, хромовая кислота в виде двухро-
мовокислых солей. Катализаторы этого типа он получает распре-
деленными на пемзе, иногда с предварительным осаждением соот-
ветственных солей на угле. Например раствор молибденовокислого
аммония с углем осаждается на пемзе, прокаливается затем при
300—400° в токе воздуха, пока сгорит уголь. Такой катализатор
окисляет антрацен в паро-воздушной смеси при 350°.
Оказалось далее, что интересные результаты дает применение
не одних кислотных окислов или отвечающих им щелочных солей
кислот, но солей этих кислот с тяжелыми металлами, так как
в этих условиях можно окислить антрацен при более низкой
температуре. Как кислота, особенно применимая в этом случае,
названа ванадиевая кислота (далее — хромовая, молибденовая, ура-
новая, оловяная и даже мышьяковая), как металлы оснований —
Си, Ag, Pb, Tl, Pt, Се, Ni, Со.
Пример приготовления такого катализатора: 1 мол. ванадиевой кислоты (V2O5)
растворяется в 6 эквивал. щелочи и осаждается 3 мол. раствора сульфата меди,
зеленовато-желтый осадок (I) промывается н отделяется от жидкости отсасыванием.
С другой стороны, приблизительно пятидесятнкратным количеством (от применен-
ной) ванадиевой кислоты пропитывают подходящего размера зерна пемзы с помощью
гуммиарабика, сушат, затем распределяют осадок (I) равномерно на зернах посред-
ством механического встряхивания н нагревают осторожно в токе кислорода или
воздуха до исчезновения органического вещества. Такой катализатор благоприят-
ствует окислению кислородом антрацена в антрахинон уже прн 180—190°. Выгоднее
для постоянства работы этого катализатора (для регенерации его поверхности)
пользоваться более высоким давлением кислорода.
Ванадат серебра на железе дает прн 280° хорошие результаты окисления пара
антрацена при невысокой ковцентрацни и при достаточно большой скорости воздуха.
Воздух, соотв. кислород, пропускается через нагретый приблизительно до 200°
антрацен для образования паро-воздушной смеси.
Об аппаратах для производственного осуществления такого
окисления сказано ниже. Они подобны применяемым для получения
фталевого ангидрида.
512
Близки по принципу к описанным в патентах Воля приемам
методы Гиббса и Коновера и Даунса81).
Последний указывает температуру реакции в 375°, первые—в 500°.
Основательно проработан вопрос о контактном окислении антра-
цена Сенсэменом и Нельсоном, стремившимся определить
все основные факторы реакции 82).
Они пользовались для этого аппаратом (рис. 14), где карбюратор А, загружен-
ный антраценом, нагревали в электрическом нагревателе (на схеме не показано).
Воздух вводился двумя трубками: I—для первичного воздуха, 2 — для вторичного
Температура паро-воздушной смесн определялась ртутным термометром 3. Хорошо
изолированный снаружи карбюратор соединен с реакционной камерой В посредством
шлнфа. Последняя, также шлифом, соединена другим своим концом с конденсатором
для паров антрахинона С, сделанным из укороченной кьельдалевской колбы с отвер-
стием посередине. Реакционная камера-трубка окружена муфтой электропечи, темпе-
ратура в трубке регистрируется электропирометром. Конденсатор тщательно изоли-
рован и может быть обогреваем посредством электрического тока. Катализатор
(V2O5) в реакционной трубке применялся нанесенным на пемзу. Кусочки пемзы
Рис. 14. Прибор для контактного окисления антрацена.
с нанесенным слоем (полученным погружением пемзы в тесто VaO5 с водой) нагре-
вают затем на паяльной лампе до плавления пятиокисн ванадия. Другой вид
носителя — кружки нз прессованного асбеста с отверстиями, на которые таким же
путем наносится катализатор. Этн кружки вводились в стеклянную трубку н затем
оплавлялись. Третий носитель — наружная поверхность стеклянной трубки (20—25 см
длины, внутренний диаметр 1,5 см). На поверхности трубки плавится пг.тяокись
ванадия и вводится вместе с трубкой (внутри запаивается термопара пирометра)
в реакционную камеру, так что остается небольшое только свободное пространство
(около 6—7 мм) у сгенкн реакционной камеры. Лучшие результаты дает как-раз
последнего типа нэснтель (выход 78,7—81,1% теории). Пемза (59%) н асбест
дают более низкие выхода (64,6—68,4%) при оптямальных условиях: температура
в реакционной камере 400—425°, суммарный ток воздуха 300 см3 в 1 мин. при
концентрации антрацена в паро-воздушной смесн 0,22—0,32 г антрацена в 1 000 см8
воздуха. Скорости прохода воздуха через карбюратор 70—125 см® в 1 мни.
Как катализатор окисления антрацена пятиокись ванадия по
наблюдениям этих авторов требует некоторого „периода приспо-
собления* для проявления своей максимальной активности. Повы-
шение температуры реакции за предел 425’ обычно снижает выход
антрахинона вследствие очевидно более далеко идущего окисления
уже готового антрахинона, между тем свежий катализатор даже
при 500’ не дает полного окисления антрацена, и в продукте
имеется неизмененный антрацен. После нескольких окислений ката-
лизатор „прирабатывается", цвет его становится зеленовато-синим,
и он начинает работать нормально. Сплавленный катализатор дает
лучшие результаты, чем применяемый в виде порошка. Авторы
склонны к допущению химического участия катализатора в про-
цессе окисления с временным переходом в низшие окислы ванадия
и с последующим окислением вновь. Схема реакций на поверхности
катализатора по ним такова:
VaOj+C^Hio —► низшие окислы V-f-С14Н8Оа 4-НаО.
Низшие окислы V-J-O2 —* V2O6.
33 Зак. 2611. — H. Ворожцов
513
В последнее время Ю. Г. Шерман достиг ценных результа
тов при окислении технического антрацена (содержащего около
60% чистого антрацена) в антрахинон, пользуясь в качестве ката-
лизатора пятиокисью ванадия. Схема его установки в основных
чертах близка к схеме получения фталевого ангидрида (стр. 522):
подогретый воздух проходит над расплавленным антраценом, увле-
кает его пары и, смешиваясь с „вторичным" воздухом, проходит
в контактный аппарат, подобный описанному ниже для фталевого
ангидрида. Антрахинон собирается в конденсаторах за конверто-
Рис. 15. Кривые давления паров на-
фталина (1\ аценафтена (2), фенант-
рена (3), антрацена (4), кат базола (5)
и антрахинона (6).
ром. Примеси антрацена и малая
часть его самого сгорает, образуя
частично фталевый ангидрид, являю-
щийся единственной примесью такого
антрахинона, легко удаляемой про-
мывкой.
Ниже приведены кривые давлений паров
определенных для нафталина, антрацена, фе-
нантрена и др., которые важно учитывать при
определениях концентраций различных паро-
воздушных смесей при различных температу-
рах (рис. 15) 83).
Значительно более сложные по
составу катализаторы, заключающие
окислы ванадия и других элемен-
тов, описаны Егером в его мно-
гочисленных патентах. Основу ка-
тализатора составляет вещество, об-
менивающее основание, типа цеолита
(см. выше), но необязательно про-
изводное кремнекислоты 84).
Получают их обменным разложением раствогимых силикатов щелочей с солями
тяжелых металлов, также взаимодействием алюминатов с солями пиых металлов
и т. п., вводя в них некоторые другие составные части, в том числе и соли.щелоч-
ных металлов. Автор считает, что специфические катализаторы окисления можво
.стабилизировать* введением в нпх соединений щелочных металлов, щелочноземель-
ных и иных с трудно восстанавливаемыми окислами, утверждая, что такие прибавки,
умеряют активность специфических катализаторов.
По его утверждениям, можно добиться с некоторыми сложными катализаторами
значительного избирательного эффекта в процессе окисления, причем например будут
затронуты окислением в парах технического антрацена лишь карбазол, ио антрацен
останется неизмененным. Помимо стабилизатора в составе катализатора важную
роль играют ,промотеры стабилизатора*. Это компоненты, имеющие характер актив-
ного, по не высокоспецифичного катализатора. Его участие .повидимому смягчает
я активирует действие наличного стабилизатора, и таким образом проявляется
высокоэффективное избирательное действие катализатора*.
Пример такого катализатора: 8,7 г окиси железа и 8 г титава осаждаются
щелочью из растворов солей металлов Окислы взвешиваются в 100 г воды с добав-
лением 14,2 объемн. процентов 10 раствора КОН (стабилизатор). Сусненсию
наносят на 200—250ч. по объему зерен пемзы (величиной с горошину) и обжигают
при 400-500°. При пропускании через такой катализатор паров, полученных из
30%-иого технического антрацена, содержащего 22% карбазола в смеси с воздухом
(отношение по весу 1 :30), при 380° получают антрацен с содержанием лишь 3,5%
карбазола, при 400°—1,4% карбазола и при 4:0—440°—антрацен совсем без
карбазола. Здесь специфическим катализатором является окись железа, окись же
титана — .промотером стабилизатора*.
514
соответственно построенный цеолит или подобное ему обмени-
вающее основание вещество может, по словам автора, вызвать еще
более разительный эффект селективной очистки.
Для окисления антрацена в антрахинон, которое можно прово-
дить в одной аппаратуре с избирательным окислением примесей
(от фенантрена таким образом освободиться не удается), пригоден
содержащий V2O5 катализатор, приготовленный таким например
образом:
200 г раствора калиевого силиката (33° Боме) развести 1600 воды и прибавить
70 г Celite earth (ипфузориой земли? Н В.). Восстановить 18 г V2O6 в воде посред-
ством сернистого газа и перевести в соответственный вввадит калия. Смешать этот
раствор с суспенсией силиката, обработать смесь таким количеством раствора серно-
кислого марганца, чтобы опабыла нейтральной пли слабошелочвой по фенолфталеину.
Перед употреблением обжечь массу при 400—500° в токе сернистого газа (с 7°/0-иым
содержанием SO2).
Эти сложные смеси соединений, выдвигаемые Егером как особо
активные катализаторы с избирательной способностью производить
окисления только примесей антрацена, подлежат настоящей научной
проверке для выяснения полезности и роли отдельных составных
частей, хотя бы и носящих „научные" названия „стабилизаторов",
„промотеров стабилизации". Рецептура Егера кажется попыткой
эмпирических поисков среди чрезвычайно большого числа комби-
наций большого числа элементов.
Окисление антрацена в антрахинон в парах над контактом, содержащим фосфор-
ную кислоту (3% антрацена в смеси, ЗаО—400°), описано в примере упомянутого
германского патента ®).
Окисление в парах воздухом фенантрена в феиантрепхиноп удается, по описанию
американского патента Льюиса и Гиббса, также при помощи пятиокисн
вавадив (также окислов Mo, W, U). Оптимальная температура 400° ®).
Каталитическое окисление антрацена в жидкой фазе кислородом
воздуха под давлением описано в германском патенте: суспенсия
антрацена в водном растворе аммиака или пиридина нагревается
20 час. до 170—175°. Катализаторами окисления служат соединения
металлов (Си, Ni, Со, РЬ) ет).
Окисление также за счет кислорода воздуха, служащего для
регенерации солей закиси железа в соли окиси при нагревании
антрацена с солями окиси железа, предлагает К. Мейер ев). Ката-
лизаторами названы соли меди и нитрит натрия.
Окисление аценафтена (I) контактным путем удается, согласно
патентам немецкой фирмы, с применением ванадиевых катализа-
торов. В состав продуктов окисления входят кроме ацеиафтенхинона
(IV) аценафтилен (И), биаценафтилидендион (III), нафталевая альде-
гидокислота (V) и нафталевая кислота (VI):
/ \-сн, / \-СН < \-С=С-/ \ со
\/ I -> )----------/ tl -> Ъ----<6 I 1 >—\ -* /---------ч I
/ 1 \-СН, / II >-сн < >-со ос-< > < IV >-со
\----/ \----/ \----/ III X-------/ х-------/
I
/ СООН / V-соон
\. X---------------б (и ангидрид)
V \-CHO VI Х-СООН
33*
515
При отношении между аценафтеном и воздухом в паро-воздушной смеси: 2—Зг
ацепафтепа на 10—60 л воздуха, при 310—330° получаются в значительных коли-
чествах вышеупомянутые низшие продукты окисления (И —V), между тем при
понижения концентрации аценафтена в смеси (1 г иа 30—200 л воздуха) и повы-
шении температуры (330—360°) получается с хорошим выходом нафталевая кислота.
В этом и аналогичных примерах контактного окисления (для
нафталина например) в патенте отмечено преимущество последова-
тельного применения в аппарате контактных масс разной актив-
ности. Таким образом первый слой катализатора дегидрировал бы
аценафтен до аценафтилена, а второй при подходящих температур-
ных условиях превращал последний в нафталевую кислоту 89).
Нафталевая кислота интересна как исходный материал для
превращения в тетракарбоновую кислоту нафталина (1.4.5.8), при-
меняющуюся в синтезе кубовых красителей.
На примере окисления аценафтена в нафталевую кислоту, так же
как в окислении фенантренхинона в дифеновую кислоту (частично
образуемую при получении хинона из фенантрена), окисления
сопровождаются нарушением углеродного скелета исходных соеди-
нений.
Значительно большую важность имеют, среди таких процессов
окисления, соответственные превращения бензола в малеиновую
кислоту и особенно нафталина во фталевый ангидрид.
Последнее из названных превращений лежит в основе широко
применяемого в Западной Европе и Америке способа производства
фталевого ангидрида. Экспериментально метод был выработан, как
выше упомянуто, одновременно и независимо друг от друга Во л ем
в Германии и Гиббсом в Америке в 1916г. В производство он был
введен ранее, чем во всех других странах, в Америке, и уже в 1919 г.
полученный каталитическим окислением нафталина дешевый фтале-
вый ангидрид был там в продаже. Фталевый ангидрид с введением
нового метода получения становится крайне широко и много-
образно потребляемым продуктом для синтеза антрахинона и
антрахиноновых производных, синтеза фталеиновых красителей,
производства бензойной кислоты; главная же сфера его применения—
это лакокрасочная промышленность и производство пластических
масс (эфиры фталевой кислоты, продукты конденсации с глице-
рином). Производство фталевого ангидрида в 1929 г. в Америке
дало наибольшее количество продукта — около 4 155 т при цене
в 16,3 цента за англ. фунт.
Принципиально метод получения не отличается от вышеописан-
ных. Нафталин, нагретый выше точки плавления, омывается в закры-
том карбюраторе протекающим воздухом, пары нафталина увле-
каются воздухом и вместе с ним или с добавкой вторичного
воздуха входят в контактный аппарат (конвертор), где соприка-
саются с слоем катализатора и окисляются на его поверхности.
Продукты окисления выходят вместе с избытком воздуха из кон-
вертора в охладительные аппараты — конденсаторы, где фталевый
ангидрид осаждается в кристаллическом виде и накапливается до
удаления из конденсаторов:
Наиболее пригодным катализатором в этом процессе оказалась
пятиокись ванадия. Испытаны и иногда рекомендованы различные
516
более сложные соединения и смеси, заключающие окислы ванадия.
Превосходство окиси ванадия перед другими катализаторами дока-
зано исследованиями Гиббса и Коновера90).
Продуктом, уловимым первоначально при окислении нафталина,
является нафтохинон, он мало стоек в условиях реакционного про-
цесса; образующийся затем фталевый ангидрид более стоек и
составляет при правильном ведении процесса главную массу про-
дукта. В качестве примеси возможна малеиновая кислота, бензой-
ная кислота, нафтолы, акриловая кислота. Главнейшие этапы полу-
чения фталевого ангидрида контактным путем из нафталина можно
представить следующей схемой (по Мареку и Ган):
О
С*к)Н8 О2 —> C10H8[OJ
(1)
со
Н-бо->
О4-2СО2 + Н2О
(2)
/^/СООН
^/^соон
(3)
(4)
^уСООН
\/\сООН
(5)
о
517
(6)
Прн подходящих температуре и времени контакта можно получить
хорошие выходы нафтохинона. По Ма ре к у и Г а н, с катализатором,
состоящим из пемзы, на которую нанесено 10% пятиокиси ванадия,
можно получить до 25% от теоретического выхода нафтохинона.
Условия опыта в этом случае были следующие: температура свинцовой бани
418°, отношение воздуха 6,5-кратиое по отношению к теоретически необходимому
для полного сгорания, и продолжительность контакта в 0,13 сек. Даже при условиях,
благоприятных для образования фталевого ангидрида, т. е. при меньшем содержании
воздуха в паро-воздушной смеси и большей продолжительности контакта, нафто-
хнноя будет появляться в продукте, если температура будет снижаться или время
контактирования станет меньше.
Для лабораторных исследований этого процесса по Гиббсу
можно пользоваться просто сконструированным аппаратом (рнс. 16).
Рис. 16. Прибор для изучения процесса окисления нафталина.
Воздух, выходящий с известным давлением (от воздушного насоса, газометра
или нз бомбы), проходит через реометр 1 н идет в карбюратор 2, представляющий
горизонтальный сосуд из медной жести с впаянными вверху входной и выходной
трубками. В карбюраторе находится нафталин, заполняя его приблизительно до по-
ловины высоты. Карбюратор стоит в масляной бане 3, нагретой до надлежащей темпера-
туры (например 105°). Воздух, пройдя через насыщенное парами нафталина простран-
ство карбюратора, увлекает их. Паро-воздушная смесь проходит затем через трубку из
тугоплавкого стекла 5, лежащую в стальной муфте б, пронизывающей корытообразную
железную баню 4, наполненную свинцом. Газовая горелка 9 позволяет нагревать свинец
в бане до плавления и до нужной температуры. Термометры 7 в железных муфтах
или соотв. пирометры показывают температуру двух точек свинцовой бани. Пары
продуктов окисления охлаждаются в стеклянных конденсационных трубках 8, фтале-
ный ангидрид и другие твердые продукты оседают иа внутренних стенках трубок.
Марек описывает иную систему прибора для лабораторного изу-
чения процесса окисления нафталина. Схема прибора изображена
иа рис. 17 и 18.
Пятиокись ванадия применяется нанесенной на пемзе (Воль)
или просто в виде твердой, сплавленной и дробленной на куски
518
окиси (Гиббс). Температура реакции, приведенная в патенте Водя
(370°), кажется слишком низкой для окисления всего нафталина,
и пределы 400—450° (Гиббс) — более целесоответственны. Кон-
центрация нафталина в паро-воздушной смеси вообще довольно
низка. Воль указывает сравнительно весьма небольшой избыток
Воздуз
J — реометр,
2 — подогреватель воздуха,
3 — предварительный карбюратор,
4 — масляная баня.
5 — трубка к манометру.
6 — масляный термостат,
7 — карбюраторы.
3 — подогреватель для паровоздушной
смеси,
9 — подогреватель для инертного газа*
Рис. 17.
Рис. 18. Прибор для изучения про-
цесса окисления нафталина
воздуха против теоретически требуемого количества (около 20—
30%), между тем в практике пользуются значительно большим
(свыше 100%-иого) избытком воздуха, учитывая, что избыток воз-
духа уносит с собой часть теплоты, выделяющейся при реакции, и
тем стабилизирует тепловой режим конвертора91)- По весу коли-
чество воздуха превосходит
количество нафталина в 7—20
раз.
Марек и Ган приводят
в своей книге кривые, ха-
рактеризующие соотношения
выходов фталевого ангидрида
с количеством воздуха в паро-
воздушной смеси и со време-
нем контактирования (рис. 19
и 20).
В недавнем немецком патенте указаны оптимальные условия окисления: 170 г
нафталина (в смеси с воздухом) на 1 л контактного пространства в час 92). Марек
приводит как благоприятное отношение воздуха к нафталину 6,5-Кратный избыток
его сравнительно с теоретически необходимым количеством для полного окисления
нафталина в СОг и Н2О.
Кроме пятиокиси ванадия испытывались другие катализаторы этого процесса
окисления. Даунс в своем патенте описывает применение глинозема при темпера-
туре 350—700° (преимущественно 450°). Однако выход фталевого ангидрида, указы-
ваемый им, крайне мал, к тому же он устанавливает получение около 50% нафто-
хинона. Во всяком случае этот катализатор ие имеет значения9J). Коновер и
Гиббс в своей работе нашли, что при 40(У° А12О3 дает едва заметное превращение
нафталина во фталевый ангидрид (0,6%)9t)*
519
Смеси окислов металлов при разной каталитической актииности отдельных ком-
понентов рекомендуются Крэвером как дающие лучшие результаты в смысле
выхода полезной реакции—получения фталевого ангидрида—и задержки после-
дующих стадий — образования бензохинона, малеиновой кислоты и углекислоты.
Рис. 19. Кривые, характеризующие, выходы фталевого ангидрида.
Рис. 20. Кривые, характеризующие выходы фталевого ангидрида.
Им названы двойные смеси (например 65% V8O5-|-35% МоОз), тройные (65% VaO6-|-
-J-30% МоО3 -|- 5% МпО2)96). Однако наши опыты с В. О. Лукашевичем не
обнаружили преимущества таких смесей перед одной пятиокисью ванадия. Выхода
фталевого ангидрида ниже полученных с одной V8O5, необходимая же темпера-
тура—выше.
Мэкстедом был обстоятельно исследован в качестве ката-
лизатора окисления нафталина кислородом воздуха в паровой фазе
520
ванадат олова, проявивший большую активность. Для него обна-
ружена сравнительно низкая температура реакции (280°). При этом
оптимальные результаты отвечали таким факторам (ср. выше}.
Объемная скорость первичного воздуха 200, вторичного 2 100; выход фталевого
ангидрида 84,3%, выход по времени на объем 0,041. Во всех опытах Мэкстеда
последний фактор—выход по времени на объем—для окисления нафталина значи-
тельно выше, чем при прочих равных условиях для окисления например толуола.
Катализатор начинает проявлять свою активность при таком же приблизительно
уровне температуры, что и при окислениях других углеводородов. Повышение
скорости пропуска воздуха содействует повышению выхода.
Ваиадат висмута требует для окисления более высокой (приблизительно на
100°) температуры, чем соединение олова 96). Метаванадат кобальта, по опытам К о-
новера и Гиббса, менее активен сравнительно с V2O5.
Еще более сложные смеси в качестве катализаторов описаны
Егером87). Они во многом напоминают предлагаемые им же для
окисления антрацена и его примесей. Егер полагает, что для избе-
жания далеко идущих реакций окисления и с целью сообщения
достаточной активности катализатору для вовлечения всего нафта-
лина во взаимодействие нужны катализаторы, в которых активные
элементы находятся в форме двух сложных цеолитов (алюмо-
силикатов). Каталитические элементы входят в состав этих сильно-
пористых цеолитов в четырех различных формах: или входя в ядро
цеолита в необмениваемой форме, или в виде одного из способ-
ных к обмену катионов цеолита, или в виде аниона, могущего
образовать с цеолитом солеобразное соединение, или наконец вхо-
дить в состав растворителя цеолита. Приготовление таких катали-
заторов с образованием сложных цеолитов описано в оригинале
патента. Оно заканчивается подкислением образовавшейся массы
(серной кислотой, пропуском сернистого газа и т. п.). По описа-
ниям патента, катализаторы содействуют получению превосходных
выходов фталевого ангидрида. Катализаторы этой группы имеют
тот недостаток, что они хрупки, легко распыляются. Хотя некото-
рые из них, по нашим с В. О. Лукашевичем опытам, позволили
получить выходы фталевого ангидрида не ниже, чем с V2O5, но во
всяком случае не превышающие. Вопрос о применимости сложных
катализаторов в этом процессе следовало бы подвергнуть основа-
тельному экспериментальному обследованию.
Все вышеприведенные комбинированные катализаторы имеют
своей задачей (которую, может быть, невсегда правильно выпол-
няют) торможение слишком далеко заходящего окисления, оста-
новку реакции на стадии фталевого ангидрида. Этой же цели
немецкая фирма имеет в виду добиться иным путем: двухфазным
окислением нафталина последовательно над двумя контактными
слоями, из которых первый (например V2O5, нанесенная на зерно
металла) активен уже при 320°, а второй (например V2OR, смешан-
ная с инфузорной землей) при более высокой: 350—410°. Темпера-
тура первой фазы окисления 320°; реакция начинается здесь и
заканчивается во второй фазе при 410Од8).
Форма нанесения катализатора на металлические поверхности,
по вышесказанному, имеет большие преимущества для поддержа-
ния теплового режима реакции на определенном оптимальном
уровне. Немецкий патент чехо-словацкой фирмы защищает приме-
521
некие поверхностей из сплавов алюминия с металлами, окислы
которых служат катализаторами99).
Схема производства фталевого ангидрида по методу Гиббса и
Коновера дана ниже (рис. 21).
Z — карбюратор: at паровая рубашка; наф-
талин; смотровое стекло.
2 — реометр — измеритель скорости воздуха.
3 — конвертор: вводная коробка для паро-
воздушной смеси; выходная коробка;
е3 свинцовая баня.
4 — горелки газовые.
5 — конденсаторы»
6 — смотровые окошки.
Рис. 21. Схема производства фталевого ангидрида.
Нафталин поступает в карбюратор-испаритель*/, цилиндрический, обогреваемый
паром сосуд, где он поддерживается в расплавленном состоянии прн нужной тем-
Рис. 22. Кривые давления паров составных частей смеси,
полученной прн окислении нафталина.
ратора, пройдя предварительно по изогнутой трубе, погруженной в нафталин, и
будучи нм подогрет. Паро-воздушная смесь идет далее в реакционный аппарат—кон-
вертор 3, где сначала проходит по средней нустой стальной трубе, погруженной, как и
соседине трубки, заполненные катализатором, в баню на расплавленного снинца. Выйдя
522
мз трубы, паро-воздушная смесь попадает в головную распределительную коробку
конвертора, а оттуда распределяется по открывающимся в коробку контактным
трубкам (стальные^ диаметром 22 мм, 2,5 м длины), находящимся, благодаря окру-
жению расплавленным свинцом, прн одинаковых температурных условиях. В труб-
ках происходит окислительное взаимодействие, причем тепло реакции отбирается
свинцовой баней. Баня доводится до плавления свинца и нужной темпера гуры
(обычно в пределах 410—440°) посредством газовых горелок 4 под конвертором.
Во время процесса подогревание газом илн совсем не нужно нли нужно в мини-
мальном размере, так как теплота, воспринимаемая от реакцнн свинцом, является
илн совершенно достаточной илн нуждающейся в незначительном только пополне-
нии для покрытия потерь на лучеиспускание и пр. Продукты окисления вместе
с избытком воздуха проходят сначала в сборную выходную коробку конвертора и
оттуда по выходной трубе идут в охлаждаемые воздухом железные конденсаторы 5,
где из паров оседает фталевый ангидрид н отчасти сопутствующие ему примеси
(обычно немного нафталина и нафтохииона). Фталевый ангидрид удаляется из кон-
денсаторов через задвижки внизу. Катализатор, почти полностью заполняющий
контактные трубки, — кусочки сплавленной пятиокисн ванадия. В такой системе
получают в среднем выход фталевого ангидрида в 69—70% от теории, т. е. 80 ч.
на 100 ч. взятого нафталина.
Выше приведены заимствованные у Марека и Ган кривые да-
вления паров" составных частей смеси, полученной при окислении
нафталина (рис. 22).
Описаны и имеют применение
иные системы конверторов. Наи-
более интересная по идее, но в то
же время дорогостоящая система—
с баней, саморегулирующей тем-
пературу процесса. Жидкость, за-
полняющая баню, может быть на-
пример ртуть, кипящая под дав-
лением азота при температуре
свыше 400°, или сплав ртути с кад-
мием.
или пар
Рис. 23. Система конвертора с баней.
Схема одного элемента такой бани
представлена на рис. 23. Смесь воздуха и
паров нафталина идет в верхнюю часть трубки, внизу которой, примерно до уровня
жидкости, лежит катализатор. Верхняя часть трубки служит теплообменником,
в котором паро-воздушная смесь воспринимает температуру паров кипящей жид-
кости. Подогретая смесь идет затем через катализатор, Продукты окисления и
избыточный воздух уходят через нижвюю часть трубки в конденсационную систему.
Выделившееся прн реакции тепло идет от поверхности катализатора через стенки
трубки и нагревает жидкость, поддерживая ее в состоянии кипения. Пары кипящей
жидкости из верхней части бани конденсируются в холодильнике и, спускаясь
обратно в баню, поддерживают ее уровень постоянным. В практике конверторы
такого типа с вертикальными стальными трубками делают на очень большое число
узких трубок (1,6—1,9 см в свету)—до 900—1 200 в одном конверторе. Длина трубок
около 90 см. Узкие трубки имеют большое преимущество для удержания реакции
на предельном уровне, так как в них легче осуществляется передача тепла от
катализатора стенкам. Предпочтительно для использования пространства и пере-
дачи тепла применять трубки ие круглого, а прямоугольного сечения. Такой кои-
неотор работает автоматически. Несмотря иа большие преимущества такой системы
конвертора, помимо его дороговизны и сложиостн монтирования он представляет
значительную опасность для работающих, из-за возможности проникания ртутн,
жидкой и в парах, и рабочее помещение н связанных с этим отравлений
Английский химический концерн в недавнем патенте устанавли-
вает преимущество применения такой аппаратуры (трубок для
проведения окисления), материал которой был бы стоек в усло-
523
виях реакции по отношению к окислигельным воздействиям и
не образовывал бы стойких фталатов. Такими материалами явля-
ются металлы Ni, Cr', Ag, Al, сплавы Ni-Cr-Fe, Cr-Fe, Si-F6, Cr-W-Cu.
В примере патента указано применение никелевой контактной
трубки при 490° ш).
Повидимому применение стальных трубок для пррведения в них
контактного окисления не отзывается вредно на исходе реакции.
По Грину, стенки реакционной камеры покрываются каталитически неактивным,
слоем, например порошком асбеста или силикатом натрия. Внутри камера разде*
лена на части полками с отверстиями, покрытыми кусочками пемзы с нанесенной
на ней пятиокис.ью ванадия. В промежутках между полками установлены мелкие
отверстия для пульверизирования воды внутрь камеры. Тотчас переходя в пар,,
вода служит для отвода тепла реакции и для регулирования температуры про-
цесс#,’ устраняя перегрев поверхности катализатора и возможный переход за жела-
тельную стадию окисления 1°8).
Так как теплота,-выделяемая при окислении нафталина во фта-
левый ангидрид, очень значительна, но ниже теплового эффекта
окисления нафталина например в малеиновый ангидрид и тем
более в углекислоту и воду, то понятно, почему важно задерживать
более далеко заходящую реакцию окисления: увеличение общего'
количества тепла содействует все большей активности катализа-
ора с повышением значения реакций более полного окисления.
Раз начавшийся переход за желательную стадию окисления акти-
вирует своей теплотой как-раз окисление до более далеких пре-
делов.
Фталевый ангидрид от контактйото окисления получается в за-
висимости от системы работы более или менее чистым. При пра-
вильном режиме- работы конвертора и конденсаторов главная
масса получаемого фталевого ангидрида может итти для большей
части применений без предварительной очистки. Тем не менее
известная часть продукта оказывается загрязненной примесями:
нафталином, частью хинонами. Гиббс и Ко-новер среди примесей
нашли также бензойную кислоту, нафтолы, фталевый альдегид;
Кревер указывает на бенздхинон и малеиновый ангидрид.
Хиноны сообщают желтый цв&т фталевому ангидриду. Образо-
вание нафтохинона характерно для более низкой температуры
конвертора, чем этого требует образование фталевого ангидрида.
При пуске в работу свежего контактного слоя вначале получается
большое количество нафтохинона. Характерным признаком не полно
проходящего окисления служит кроме желтого цвета продукта
еще резкий, очень раздражающий слизистые оболочки носа и глаз,
запах паров, затрудняющий работу у*конвертора и конденсаторов.
При нормальном режиме конвертора этот запах заметен, но не
так интенсивен. Очистку загрязненного фталевого ангидрида прак-
тически выполняют различными путями.
Можно, подвергнув сырой продукт нагреванию, увлечь пары воздухом я, про-
ведя через конденсационную систему, собрать более чистую фракцию возгона
в первых камерах (Г и б б с). Предложено также растворять фталевый ангидрид
в неводных растворителях (например СС14) и, обесцветив. раствор например углем,
кристаллизовать из него фталевый ангидрид (К о н о в ё р, Г и б б с, 1919)—способ,
заведомо мало практичный вследствие дороговизны н громоздкости!®).
Предложена также очистка путем нагревания сырого продукта с триоксимети-
леном, органическими высокомолекулярными полиосиовными кислотами, иеоргани-
524
ческими конденсирующими средствами и последующей перегонкой. Вполне понятно,
что метод очистки может быть выбран лишь после основательного обследования
сортов сырого продукта, подлежащего очистке, на .количество и характер его
примесей101).
Оригинальный прием очистки фталевого ангидрида еще до конденсация его из
паров описывает американский патент двух немецких химиков: пропуск паров про-
дуктов окисления непосредственно за контактным аппаратом при температуре
-около 200° через твердые тела с* сильно развитой поверхностью (зерна пемзЫ,
черепки глиняного товара, гель кремнекислоты, активированный уголь и т. -п.).
Иногда для усиления активности этих адсорбентов можно пропитать их до употреб-
ления растворами солей илн оснований или покрыть их солимн или окнсламн
металлов. Так как, выполняя свою очистительную функцию по отношению к парам
продуктов, очиститель теряет часть своей активности, то предлагается пользоваться
нм при постоянной смене новыми слоями уже отработавших105).
В главе XIII было уже упомянуто о получении из фталевого
ангидрида бензойной кислоты контактным путем: пропусканием
<меси паров фталевого ангидрида с водяным паром и азотом через
СаСО3 (при 400°) или через А12О3 (390—400°) или через Fe4O8 С до-
бавкой других активирующих окислов металлов (330—360°). П8сте-
ленным охлаждением смеси паров можно отделить бензбйную
кислоту от фталевого ангидрида.
Превращение проходит очевидно по схеме:
+н2о
С6Н4(СО)2О-----► СбН4(СООН)2-----► С6Н5СООН
-со2
Каталитическое декарбоксилирование фталевого ангидрида в бен-
зойную кислоту, по патенту Коновера, идет с выходом в 90%
при применении приблизительно 50 ч. водяного пара (перегретого
до 200°) и 1 ч. паров фталевого ангидрида и проведении смеси
паров в реакционную камеру с окисью цинка (или СиО или А12О3),
нанесенной на инертный носитель. Для достижения лучших выхо-
дов необходим вообще значительный избыток водяного пара про-
тив требуемого теорией количества106).
Недавний немецкий патент описывает еще более замечательнее
превращение фталевой кислоты в бензальдегид. Для этого нужны
два последовательных изменения: отщепление СО2 с переходом
в бензойную кислоту и восстановление СО2Н в СНО:
/х^/СООН
-СО2
^/^соон
Ток водорода, насыщенного при 215° парами фталевого ангидрида, проводится
над катализатором: висмут и окись меди с прибавкой окиси цинка и небольшой
количества окиси магния. Катализатор перед употреблением восстанавливается
водородом. Температура каталитического воздействия 350э. При скорости тока
газовой смесн в 1 л3 в час на 1 л катализатора более 80% нсего фталевого анги-
дрида переходит в бензальдегид, 1% остается без изменения, и остаток переходит
в бензол107).
В новых патентах предлагается последовательно проводить
более сложные превращения контактным путем108).
526
Так, смесь паров нафталина н воздуха проводится через V2O6 прн 400s
а затем смесь паров идет при 380° над катализатором декарбоксилирования (CdO,
ZnO, Ai2O3), в результате последнего получается бензол, который над третьим
контактным слоем (V2O5, 410°) может быть окислен, как ниже будет изложено,
в малеиновый ангидрид. Еще горячая смесь паров, если в вей иет избытка воздуха,
может быть гидрогенизирована водородом (Ni нли Со) с образованием янтарной
кислоты. Эта цепь превращений может быть передана схемой:
/х^/СООН
СО к!
| I 1*11 /° /\ сн-со сн,-соон
w 'Ч I - II >ч
\/ СН-СО СН2-СООН
Контактное окисление а-ннтронафталина воздухом в паровой фазе над пяти-
оки сью ванадия на пем е при 320—370° приводит к образованию фталимида
с хорошим выходом. Окислением затрагивается следовательно то ядро нафталина,
которое несет группу NO^i09):
В этом превращении обнаруживается с несомненностью, как активны водород-
ные атомы углеводородов в процессах контактных окислений. По всей вероятности
нх активность проявляется не только по отношению к кислороду связанному (как
в этом примере в нитрогруппе), но и по отношению к активированному молеку-
лярному кислороду.
Окисление бензола воздухом в паровой фазе может, как
было уже упомянуто, пройти с разрушением углеродного ядра.
Как и в случае нафталина, первой улавливаемой стадией окисле-
ния здесь является образование хинона (I), который далее уже
образует малеиновую кислоту (II):
СН—СООН
С6Не-|-О2-► СбН6[О2]-► I -> || II
О\ J СН-СООН
Реакция требует участия 9 атомов кислорода:
СН—СООН
С6Н6-]-9О = || + Н2О 4- 2СО2.
СН-СООН
Практика применяет небольшой избыток против этого теоретиче-
ского расчета (бензол: воздух =1 : 12,1 —13). Указание немецкого
патента американской фирмы (The Barret Со) на равные весовые
части бензола и воздуха вряд ли отвечает принятым в производ-
стве отношениям.
Катализатором служит по преимуществу пятиокись ванадия.
Предложены также сложные катализаторы: смешанные окислы
526
(V, Mo; Mo, U; V, Mo, U) на носителе: пемзе или металле (Al, Си,
Ni, Ag и т. п.)110).
Очистка малеиновой кислоты от примесей (бензохинона и др.)
производится путем добавления органического растворителя (арома-
тического углеводорода) и перегонки, причем малеиновый ангидрид
перегоняется уже после отхода примесей.
Малеиновый ангидрид имеет в настоящее время меиыпее зна-
чение, чем фталевый. Он находит для себя применение в произ-
водстве пластических масс, для получения янтарной и яблочной
кислот.
д) Прочие реакции
В главе IV были упомянуты неоднократно новые способы полу-
чения хлорбензола посредством пропускания паров бензола
в смеси с хлористым водородом (соотв. хлором) и воздухом при
температуре в пределах 200—300° над контактным слоем, содержа-
щим преимущественно соединения меди. Здесь имеет место прежде
всего катализ окислительной реакции:
2НС14-О->Н2О + С1а,
и выделяющийся хлор реагирует с бензолом, находящимся в паро-
образном состоянии, при участии того или иного катализатора или
без каталитических воздействий. В примере упомянутого там же
австрийского патента смесь хлора, например с бензолом н воздухом,
пропускается при 300 —650° над катализаторами (уголь или хло-
риды Си, Fe, Т1, щелочных или щелочноземельных металлов)11г).
Насколько полно может быть использован при этом хлор и
насколько выгодно распределяются в массе продуктов монохлор-
бензол и полихлориды, неизвестно.
В типичном хлорировании бензола в жидкой фазе с участием
хлорного железа или металлического железа остаются невыяснен-
ными как промежуточные фазы реакции и соответственно с этим
моменты воздействия катализатора, так и вопрос, протекает ли
Здесь катализ в гетерогенной или гомогенной системе (вследствие
растворимости хлоридов железа, быть может с образованием комп-
лексных соединений в среде бензола и хлорбензола).
Гетерогенные превращения хл оро заме ще н н ы х и
значения в них каталитических воздействий, особенно со стороны
соединений меди, были достаточно подробно разобраны в главе VII.
При этом превращения в паровой фазе над поверхностью катали-
заторов, которых многочисленные примеры были в своем месте
приведены, яаляются реакциями гидролиза, соотв. аминолиза, на-
пример 1,а)
сбнаС14- н2о -+ с6н5он 4- НС1,
С6Н5С14- NHS -+ C6H5NH2 + НС1.
Реакции, связанные с дегидратацией, мы встречали а ряде
примеров в предыдущем. Напомним например аминирование фено-
лов, соотв. нафтолов
ROH 4- NH3 -> RNH2 4- Н2О.
527
Бринер и Ферреро установили, что при взаимодействии
аммиака и фенолов предварительно получаются продукты при-
соединения аммиака к фенолам, а при пропускании паров фенолов
с аммиаком над глиноземом, типичным катализатором дегидратации,
получаются с заметными выходами аминозамещенные, причем
констатировано, что со свежим катализатором выходы амино-
производных ниже (40—43%), чем с уже работавшим (51—55%).
Температура реакции 420°. Смесь фенола и аммиака дает максимум
12% анилина. Более положительные результаты получены приме-
нительно к трем изомерам крезола и резорцину 113).
Американские химики, изучавшие дегидратацию смеси паров
Р-нафтола с аммиаком над глиноземом при 430—450°, достигли
очень хороших результатов: 95,5%-ного выхода р-нафтиламина с не-
большой примесью рр'-динафтиламина ш).
Окись тория оказывается в этих превращениях менее хорошим
катализатором. Дегидратация фенола сама по себе и с участием
окиси тория изучалась Сабатье, Мэль и Бринером с со-
трудниками. Бринер установиллюбопытныйфакт, что без катали-
затора фенол при 450° может быть полностью дегидратирован
в дифениловый эфир:
2С6Н5ОН =2 Н2О + СеН5ОС6Н5,
между тем как при той же температуре, в присутствии окиси
тория равновесию в статической системе отвечает всего 62% пре-
вращения. Таким образом присутствие катализатора ускоряет
наступление равновесия, которое однако менее благоприятно для
выхода, чем без катализатора. Такое необычное вмешательство
катализатора в равновесную систему может быть объяснено лишь
проявлением химического сродства катализатора к реагирующим
веществам и появлению таким образом в системе новых компо-
нентов, которых не было без катализатора. Возможно, что здесь
оправдывается предположение Сабатье о роли образования то-
ратов в этой дегидратации 115).
При использовании ароматических аминов, а не фенолов деги-
дратация может иметь место в смеси их с спиртами. Таким обра-
зом возможно например протекание взаимодействий:
C6H5NH2 4- HOCHg -+ CeH6NHCHs 4- Н2О;
CeH6NHCHs + НОСН8 -+ CSH6N(CH3)2 4- Н2О.
В главе XI уже было упомянуто о такого рода дегидратации в
паровой фазе с целью получения алкилированных аминов. Катали-
заторами являются преимущественно ThO2, A12OS, SiO2. Температура
реакции различна при различных катализаторах и изменяется в
пределах 300—430°. Активность А12О3 проявляется при более низкой
температуре, чем у SiO2 (силикагель)116).
Позднейшим — более полным — исследованием этих процессов
предстоит установить, насколько и в каких случаях экономически
выгодна каталитическая дегидратация сравнительно с употреби-
тельными методами дегидратации смеси амина со спиртом с уча-
стием кислот (H2SO4, НС1) или иными методами алкилирования в
жидкой фазе без катализаторов.
528
По немецкому патенту, дегидратация в паровой фазе смеси фта-
левого ангидрида с бензолом при пропускании паров в токе
сухого азота над активированным гелем кремнекислоты при 440°
приводит к превращению смеси на 25°/0 в антрахинон. Пары фта-
левого ангидрида сами по себе также образуют при этих условиях
антрахинон
СО
+ 2СО2.
В патенте указывается на возможность использования других ката*
лизаторов, например заключающих РЬО или Li2CO3 на пемзе.
Вероятно последние катализаторы типа декарбоксилирующих
нужны для протекания реакции по второй схеме 117).
Это интересное превращение, могущее лечь в основу нового
синтеза антрахинона, заслуживает внимательного изучения для
выяснения как ступеней его течения, так и наилучших условий
образования антрахинона.
Отметим еще среди каталитических реакций дегидратирования
синтез индолов из таких производных о-толуидина, которые заклю-
чают связанную с азотом ацильную (органическую) группу. Схема
превращения выражается так:
Пары исходных ацилированных аминов проводятся над пористыми активными
веществами (активный гель кремнекислоты; стекловидный глинозем нли активиро-
ванный уголь), к которым могут быть прибавлены катализирующие водоотнятие
соединения, например соли фосфорной или борной кислоты. Температура взаимо-
действия 380—400°. Предпочтительно вести работу прн пониженн м давлении 118).
Также с дегидратацией протекает недавно описанный в патентах немецкой
фирмы синтез сложных конденсированных углеводородов, исходящий нз аромати-
ческих илн смешанных ароматическн-алнфатнческих кетонов, в которых в орто-
положении к СО-группе стоит СН3- нлн СН2-грунпа. Превращение осуществляется
пропусканием паров исходных кетонов над пористыми массами (уголь, сили-
кагель, глинозем). Иногда полезно присутствие добавочных катализаторов (Си, Fe,
Се, их соли). Протекающие при этом реакции дают продукты циклнчесю й кон-
денсации не всегда шестичленной, а также и 5-членной (типа индена), Температура
взаимодействия 380—420°. Иногда применяется введение в реакционную среду
34 Зак. 2611. — Н. Ворожцов.
529
воздуха для разведения паров кетонов. Давление Варьирует в зависимости от
условии ог долей атмосферного до превышаю него атмосферное ч»), Примеры таких
взаимодействий:
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
А. В. Раковский, Химическая кинетика и катализ, Госхимтехнздат, Москва —
Ленинград 1932.
Райдил н Тэйлор, Катализ в теории и практике, Госхимтехнздат, 1933.
Э. 3 а у т е р, Гетерогеииый катализ, ОНТВУ, „Кокс и химия", Харьков 1932.
В. Франкенбургер и Ф. Дюрр, Катализ, «Успехи химии' 1, 76 (1932).
Ю. С. Тэйлор, Химические рескции на поверхностях, „Успехи химии' 1,
576 (1932).
X. Брюкнер, Каталитические реакции в химнкс-органической промышленности,
Госхим1ехиздат, Ленинград 1932.
Р. Sabatier, La Catalyse еп chimie organique, Paris et Liege 1920; русск. перевод:
Сабатье, Катализ в органической химии, Госхимтехнздат, Ленинград 1932.
В. В. Ч е л и н ц е в, Контактно-каталитнческне процессы в области органических
соединений и их приложение к технике, Химтехиздат, Леиинград 1927.
Г. К р е и ц л е й н, Хлористый алюминий в органической химии, „Успехи химии',
1, 421 (1932).
L. F. Marek and D. А. Н a h п, The Catalytic oxidation of organic compounds in
the vapor phase, New York 1932.
A. Mittasch, Bemerkungeu zur Katalyse, Ber. 59, 13 (1926).
Deuxifeme Conseil de chimie, Structure et activit6 chimique, Paris, 1926.
Доклады: A. J о b, M. E r i c et K. R i d ea 1, E.- F. Armstrong et P. H i 1 d i t c h,
Ch. Moureu et Ch. Dulraisse, H. E. Armstrong.
i) Armstrong, 2-me Conseil de Chimie, 612.
2) Ильинский, см. Houben, Das Anthraren und die Anthrachinone, Lpz.,
1929,295, K- Lauer, J. pr. Ch. (2) 135, 164 (1932).
3) A. Bach; Ber. 43, 366 (1910), 46, 3327 (1913), Bach, Nikolajew, Ber. 64,
2769 (1931), Ch. Moureu, Ch. Dufraisse, 2-me Conseil de Chimie (1. c.),
524, Soc. Ind. 1928.
a bis) п. К. Б у л и ч, Аи.-Кр. Пр. 3 № 9, 405 (1933). Ср. A. Weissberger,
Е. Strasser, J. pr. Ch. [2], 135,209 (19324
4) Wohl, Wertyporoch, Ber. 64, 1357 (1931).
5( Boeseken, 2-me Conseil de Chimie, 449.
6) H. S. Taylor, Am. Soc. 53, 578 (1931); Taylor, Williamson, Am. Soc. 53,
813 (1931).
7) И. E. А д а д у p о в, Заводская лаборатория, 1932, № 2, 53.
8) А. А. Баландин, К теории гетерогенных каталитических реакций, Ж. €1,
909 (1929).
Он же. Опыт естественной классификации каталитических реакций в органи-
ческой химии, Ж. О. X. П. (644 166 (1932).
®) J. В. S е п d е г е п s, Н. A. D’A п d г о g u е de S ё г i ё g е, Н. R. de С h е f d е b i с п,
Г. п. 139457 (F. VH, 55).
1°) В., Г. п. 263396 (F. XI, 146).
Ч) См. Г. п. 273322 (F. ХП, 113).
530
13) В., Г. и. 282568, 283449 (F. ХП, 113, 114), 328339, 331393 (F. XIII, 230, 231), 15,
Г. п. 436820 IF. XV. 391), 462641 (F. XVI, 6«2).
13) 10 (О. Schmidt, A. Johansen), Г. п. 429102 (F. XV, 392).
14) О. В. Semeria, М. Mil one, СВ 1928, If, 2380.
») О. W. Brown, С. О. Henke, СВ 1923, I, 1116.
16) О. W. Brown, L. L. Carrik, Am. Soc. 41, 436 (1919), no Bull. [4] 26,
244 (1919).
)7l К. Marx, К. Bro der sen. Am. n. 1830705 (1931).
'в) MLB, Г. n. 282492 (F. XII, 115).
"») Brown, Henke, CB 1923, I, 1572; 1924, I, 1478, Am. n. 1451489; CB 1925,
II, I860.
-’°) F. A. M a d e n w a 1 d, Henke, Brown, CB 1927, II, 779.
21) Brown. Henke, CB 1924, I. 728, Am. п. 1459969, CB 1925. И, 1800.
22) Brown, Broters, Etzel, CB 1928, I, 2046; Hartmann, Brown, CB 1931,
II, 956.
33) Selden Co (A. O. Jaeger), Б. n. 304640; CB 1929, II, 485.
2I) M. Kahn. R. May er (Or as sell!), Am. n. 1639186; IO, Б. n. 260186; CB 1927,
II, 1088.
25) By, Г. n. 388185 (F. XIV, 394).
2*) Brown, Etzel, Henke, CB 1928, I, 2691; О. P о m a. G. Pellegrini, Ф. n.
559730. 559732; 0. Poma, Ф. n. 560318, CB 1926, I, 497.
2?) Brown, Henke (Newport), Am. n. 1589936; CB 1926,1', 1192; cp. Tetralin,
Г. n. 406064 (F. X V, 3951.
28) Brochet, C. r. 158, 135 (1914); CB 1907, JI, 600.
28) Brochet. C. r. 160, 306 (1915), cm. Sabatier, Catalyse, 231.
я») IG. Б. n. 301577, CB 1929, II, 93; cp. Kautsky, Herzberg, CB 1924, II, 2234.
Kautsky, CB 1921. Ill, 767.
a') IG, Б. n. 295824, 297212; CB 1929, I, 3143; Г. n. 467638, CB 1929, 1, 3142; ср. Г. n.
458088. CB 1928, I, 2039.
32) M. Mailhe, Bull. (4) 29, 106 (1921).
зз) Sabatier, Catalyse, 138, 174.
si) В., Г. n. 420393 (F. XV, 387); ср. о катализаторах В., Г. п. 307580 (F. XI1,174),
408811 (F. XIV, 52).
зз) По W. G. Rum bo Id, Chem. Age 26, 215 (1932).
Зв) Fr. Fischer, Г. n. 431479 (F. XV,393).
351 Стадников, Гаврилов, Виноградов, Ber. 58, 2428 (1925).
38) Sabatier, Catalyse. 182—183; ср. H. Д. Зелинский, Ber. 56, 1723 (1923).
39) 14 патьев, Ж. 36, 786, 813 (1904)
40) M. Donau, Pev. d. la Parfumerie, 1926; Ind. Chim. 1926) 551, cm. A. Brochet,
CB 1923, III, 748.
4t) J. D. R i e d e 1, Г. n. 444665 (F. XV, 394).
42; Brochet, Cornubert. Bull. (4) 31, 1280(1922).
«) H. Briickner. Ch. Ztg 55, 401 (1931'; ср. 11 u о К о g e s h i r a, CB 1932, I, 1358.
W) J. Braun, O. Bayer, Ber. 58, 2675 (1925).
45) Sabatier, Catalyse, 247.
4*) Зелинский, К омарев ск и й, Вег. 57, 667 (1924); Зелинский, Турова-
Поллак, Вег. 58, 1298 (1925).
4?) Federal Phosphorus Со., Шв. п. 140707, СВ 1931, 1, 160, Ф. п. 667840’
СВ 1931. II, 3060.
48) A. Hixson, L. Т. Work и сотр., Ind. Eng. Chem., Anaiyt. Ed. 3, 289 (1931).
4*) С. H. Lowe, C. James, CB 1924, 1, 1664.
«>) IG, Ф. n. 707431, CB 1931, II, 3265; cp. IO, Б. n. 369613, CB 1932, II, 1925;
Г. n. 555079, CB 1932, II, 1835; Б. n. 356189, Chem. Age 1932, 33.
Ы) Carrasco, Pad о a, CB 1906, II, 683; 1907, i, 571.
51) IG (N i с о d e m u s), Г. n. 446544 (F. XV. 406).
53) Friedel. Crafts, C. r. 100 692 no Ber. 18, R. 336 (1885); A. Homer, CB
1907, II. 600; S c h о 11, S e e r, Ber. 55, 332 (1922).
54) В., Г. n. 136015 (F. VI, 417).
55) В., Г. n. 138119 F. VI', 228).
58) IQ. Г. n. 446932 (F. XV, 700).
5’) S ch oil, Seer, Ann. 394, 111 (1912); Scholl, Seer, Weitzenbock, Ber.
43, 2202 <1910); cp Zin eke, Funke, Ber. 58, 2222 (1925; C. N. M. С., Б. n.
208720, CB 1924, 1, 1714.
MLB, Г. n. 423720 (F. XV, 733), cp. S c h о 11, Neumann, Ber. 55, 118 (1922).
34*
531
M) Sabatier, Catalyse, 52, Hoppe-Seyler, Ber. 12, 1551 (1879).
e°) L. F. Marek, Methods & Apparatus for oxidation of hydrocarbons, Ind. Eng.
Chem. 24, 1103 (1932). C. R. Downs, Каталитическое окисление органических
соединений в паровой фазе, Ж. X. Пр. II, 965 (1926).
И) По поводу формального приоритета Воля на 3 дня см. Ind. Eng. Chem., New.
Ed. 10, 55 (1932) „Phthalic anhydrid patent suit filed".
M) W. J. Huff, The thermal probleme in organic contact catalysis, Trans. Am. El-
Chem. Soc. 36, 167 (1919).
«3) A. E. Craver, Am. n. 1636853, 1636855 (1927); The Barret Co, Am. n. 1638854,
CB 1928 I 1231
IG, Г. n. 446912 (F. XV, 390) cp. A. Wohl, Г. n. 347610 (F. XIV, 836).
®) E. B. Maxted, Soc. Ind. 47, 101 T (1928); Maxted, Dunsby, Soc. 1928, 1439,
CB 1928, I!, 647; Maxted, Coke, Б. n. 237688, CB 1928, 1712.
««) Gas Light & Coke Co, Б. n. 331535, CB 1930, II, 3081.
и) В (Mittasch, Pier, Winkler), Г. n. 415686 (F. XV, 355).
M) M. И. Кузнецов, Степаненко н сотр., У. X. Ж 4, Технич. ч. 153 (1929);
СВ 1930, I, 3548.
«9) В., Г. п. 408184 (Е. XIV, 185).
») Е. И. О р л о в, Ж. 40, 656 (1908).
я) Н. N. Stephens, Am. Soc. 50, 2523 (1928).
’2) В., Г. п. 419861 (F. XV, 389).
73) Rosenmund, Zeitsche, Ber. 54, 1092 (1921).
«) G. В 1 а п с, Ф. п. 586383, СВ 1927, I, 806.
?6) Maxted, lb. 102 Т.
?6) С. Н. Bibb, Am. n. 1547725, СВ 1925, II, 2297.
”) IG, Г. n. 501467, СВ 1930, П, 1771; Ф. n. 686650, С. А. 25, 710 (1931).
58) Ср- J. Bebie, Chem. Met. Eng. 37, 473 (1930).
ra) J. Wa Iter, Г. n. 168291 (F. VIII,29).
so) A. Wohl, Г. n. 347610, 349089 (F. XIV, 836-838).
• 1) H. D. Gibbs, C. Conover, Am. n. 1417367. Ch. R. Downs, Am. n. 1374721
(1921).
ю) С. E. Senseman, O. A. Nelson, Catalytic oxidation of anthracene to anthra-
quinone, Ind. Eng. Chem. 15, 521 (1923).
®) Nelson, S e n s e m a n, Ind. Eng. Chem. 14, 58 (1922).
«) A. O. Jaeger, Ind. Eng. Chem. 20, 1330 (1928).
«) В., Г. n. 419861 (F. XV, 389), ср. Б. n. 199886, Soc. Ind. 863 A.
») H. F. Lewis, H. D. Gibbs, Am. n. 128843 (1918).
* 7) MLB, Г. n. 292681 (F. XIII, 384).
®) К. H. Meyer, заявл. Г. п. M. 46377 (F. XI, 539).
* ») IG, Г. п. 428088, 441163 (F. XV, 394, 350);The Barret Co, Am. n. 1439500, CB
1924, II, 2823.
®o) Gibbs. Conover, Ind. Eng. Chem. 11, 1031 (1919), 12, 1707 (1920), 14, 120 (1922).
я) A. Wohl, Г. n. 379822 (F. XiV,450), G i b b s, С о n о v e r, Am. n. 1284888 (1918)
1285117 (1918).
M) Gewerkschaft M. Stinnes, Г. n. 553408, CB 1932, II, 1366.
«>) Ch. Downs (Barret Co).Am. n. 1374722 (1921).
91) C. Conovet, H. D. Gibbs, Ind, Eng. Chem. 14, 120 (1922).
8s) А. E. Craver (Barret Co), Am. n. 1489741 (1924).
80) Maxted, 1. c.
82) A. O. Jaeger (Selden Co), Am. n. 1692126 (1928).
*») IG., Г. n. 441163 (F. XV, 350).
») Ver. Aussig, Г. n. 478192, CB 1929, II. 3251.
leo) Marek, 1. c., F. A. Canon Ch. A. Andrews, Am. n. 1689860 (1928).
101) ICI, Б. n. 351185, CB 1931, Ц. 2387.
'«a) A. G. G r e e n, Б. n. 249973, CB 1927, II, 2229.
103) Conover, Gibbs, Am. n. 1301388 11919).
1W) Conover, Nat. An., 1817304, CB 1931, I, 290.
los) J. В г о d e, A. Johansen (Grasselli), Am. n. 1693913 (1928).
los) С. С о n о v e г (M о n s a n t o), Am. n. 1645180 (1927).
M7) IG, Г. n. 526482, CB 1931, Ц, 770.
I»3) IG, Ф. n. 623807, Б. n. 262101, 291326, CB 1929, I, 2470; Б. n. 268775, CB 1928,
I, 1818.
I®») A. G. Green, S. J. Green (B. D. Co), Г. n. 394849 (F. XIV, 452).
ив) The Barrett Co,Г. n. 365894 (F. XIV, 294); A, Craver, Am. n. 1637857 (1927).
532
Hl) MLB, Б. n. 208155, CB 1924, I, 2822; IG, Г. n. 487596; F. Rasch i g,T. n. 539176,
CB 1932, I, 2642; E. G rem 11, Ab. n. 108424, CB 1928, 1, 1229.
«») IG, B.n. 288308, CB 1930, II, 1772; Бельг, n. 350287, CB 1929, I, 2234; Г. n,
487310 (1927); Federal Phosphorus Co, Am. n. 1735327, CB 1930,1, 2009;
Hale, Britton (D о w), Am. n. 1806768, CB, 1931, II, 1348; F. Raschig,
Ф. n. 698341, CB 1931 II, 1491; H. Dreyfus, Ф. n. 709184, CB 1931, II, 29 33;
Progil, Ф. n. 720721, CB 1932, II, 616; S. J. Lloyd, Kennedy, Am. n.
1849844, CB 1932, I, 2994; J. P. W i b a u t, CB 1916, II, 380, Ber. 50, 541 (1917).
пз) E. Brine r, P. Ferrero, E. de tuserna, Helv. 7, 282 (1924); A. Ferrero,
Recherches sur I’amination catalytique des phenols... Thfese, GGnfeve 1925.
™) Howald, Lowy, Ind. Eng.Chem. 15, 397.
115) Sabatier, Ma i 1 h e, C. r. 151, 492 (1920;; Brine r, Brow-Stale t, Paillard,
Helv. 15, 619 (1932).
и») Mallhe, de God an, C. r. 166, 467, 564 (1918); Smolensk!, CB 1923, III,
204; Brown, Reid, Am. Soc. 46, 1836, CB 1924, II, 1681.
П7) IG, Г. n. 533465.
118) IG, Г. n. 458383, CB 1928, II, 1386
1M) IG, Б. n. 251270, 253911, CB 1928, II, 1820.
ПРЕДМЕТНЫЙ указатель
Абсорбер 107
Азимины см. Азоиминосоединения
Азиновые красители 370
Азобензол 141, 490
Азоиминосоединения 257
Азокрасители 148, 184, 241
Азоксибензол 153, 490
Азохсисоединения 140, 145, 152, 221
Азометиновые соединения 334
Азониевые красители 370
Азосоединения 140, 145, 152
Азосоставляющне 182, 263
Азосочетание 180, 184, 262, 266
Азофоры 261
Азуриновая кислота 186
Акридин 322
Акридины антрахинона 442
Акролеин 443
Алая кислота 322
Ализарин 169, 171, 219, 340
Ализарин бордо 304
Ализарин голубой 444
Ализариновый плав 169
Алкил 25, 293, 296
Алкиламино- (и диалкиламино-) груп-
па 304
Алкиламиноантрахиноны 158
Алкилирование 293
Алкилирование фенольного гидрокси-
ла 34
Алкоксигруппа 315
Алкилоксиазосоединения 153
Алкильные эфиры сульфокислот 92
— — фенолов 220
Альдегиды 208
Альдоль см. Ацетальдегид
Альнафт 335
Амидол 220
Амил 294
Амиловый спирт 208, 396
Аминирование 292
Амины 155
Аминоазобензол 255, 267
Аминоазокраснтели 184
Аминоантрахиноны 119
Амино-а-антрахинонсульфат 77
Аминоантрахинон-2-сульфокислота 60
77
Аминоантрахинон (а) 203
Аминоантрахинон (•) 60, 203, 445
л-Аминоациларилид 256
Амино- а -аценафтен 238
534
Амино-я-бензоил-о-бензойная кислота
120
л-Аминобензиловый спирт 409
л-Аминобензиланилин 410
Аминогруппа образование 126
— определение 69, 156, 157
— превращения 176, 235
Аминокарбазол (3) 255
Аминокрезол 256, 281
Аминоксиленол 258
Аминонафтолдисульфокислота (1.8.
.3.6) 187, 208
— (1.8.2.4) 187
— (1 .8.4.6) 187, 329
Аминонафтолсульфокислота (1.2.4)
186, 258, 275
— (1.8.4) 187, 282
— (2.5.7) 186, 187
— (2.8.6) 187
Аминоокси (1,4)-а-нафтохинонимин 359
zi-Амино-я'-окси-о-сульфокислота ди-
фениламина 193
п-Аминооксисоединения 139, 182
Амино-fi-оксиантрахиноны 313
Аминорезорцин (5) 242, 258
Амино-п-салициловая кислота 139, 15г
Аминосульфокислота нафталина
(2.6) 77
Аминотимол >258
Амино- ₽-тионафтен 245
Аминотиофенол 382
Аминотиофенолы (о и л) 227
Амино-м-фенилоксаминовая кислота
327
Амино-п-фенилфталазон (3) 461
Амино-л-фенетедин 222
Аминофенол (о) 220
— (л) 136, 139, 155, 287
Аминофенолсульфокислота (1 .4.2) 132
Ангидроформальдегиданилин 410
Ангидро-1У-сульфокислота пиридиния
82, 254, 300, 320, 339, 440
Анизидин (о) 155, 251
— (л) 251, 331
Анилин 79, 134, 153, 155, 182, 185, 201,
242, 251, 281, 287, 294, 363, 370, 409,
490
Антиокислители 475
Антиоксиданты 155, 288
Анилид оксинафтойной кислоты 331
Антидиазотаты 260, 263
Антрагидрохинон 368, 398
Антрагаллол 432
Антранол 399, 435
Антрон 399, 435
Антраниловая кислота 274, 353, 373,
377, 437
Антрахинон 19, 20, 23, 77, 116, 365, 398,
429 513 529
Антрацен 14,16, 314, 366, 399,427, 497, 513
Антраценовое масло 14
Араминирование 280
— амнноантрахинонсв 288
Ариламиногруппа 280
6-Ариламнно-2-нафтол-3-карбоновая
кислота 284
Ариламиносоединения антрахинона 158
Арилирование оксигруппы 290
Арилгидразины 270, 379
Арилгидроксиламины 137, 140, 379
Арилсульфонариламинопроизводные 91,
333
Асфальт 16
Ауксохромная группа 245, 262
Аценафтен 14, 275, 369, 372, 515
Аценафтенхинон 14, 369, 372, 515
Аценафтилен 515
Ацетанизидид (о) 33
Ацетотолуидид (п) 33
Ацетилнитрат 48
Ацетальдегид 335, 412, 444
Ацетон 315, 361, 411
Ацетаминофенетол 223
Ацетоуксусная кислота 330
Ацетоуксусный эфир 331, 449
Ацил 25, 319
Ацилирование 319, 331
— аминогруппы 319
Ацетил-р-аминоантрахинон 339
Бейлыптейна проба 122
Бензальдегид 116, 229, 225, 335, 425,507
Бензантрон 447
Бензантрахинон (1.2) 431
Бензидин 140, 145, 256, 287
Бензидиновая перегруппировка 140,143
Бензил 22, 293
Бензил хлористый см. Хлористый бен-
зил
Бензиловый спирт 224
Бензиланилин 201, 297
Бензиловый меркаптан 227
Бензилиден 22
Бензоил 328
Бензоил хлористый см. Хлористый
бензоил
Бензоил-И-кислота 225
Бензоилирование 330
Бензоилбензойная кислота (о) 428
Бензойный ангидрид 330
Бензойная кислота 51, 116. 179 223
225, 280, 352, 378, 509, 525
Бензоилоксинафтойная кислота (1.2.3)
443
Бензгидролы 395
Бензодихлорид 102
Бензол 13, 22, 83, 101, 142, 153, 315,
361, 424, 499
Бензолгексахлорид 102
Бензокарбазолоксикарбоновая кислота
(7.8.3.2) 224
Бензолсульфокислота 281
Бензотрихлорнд 112, 116, 223
Бензофенон 395, 424
Бензохинон 364, 434, 524
Биэценафтилидендион 515
Бннафтол (РФ') 447
Бисульфитная реакция 240, 245
Бромантрахинон (8) 203
Броманизол (л) 198
п-Броманилин-о-сульфокислота 229
Бромбензол 198
Бромдиметиланилин (п) 198
Бромистый бензил 113
Бромнафталин (а) 230
Бромтолуол 198
Бромфенол (л) 198
Бутил 294
Ванилин 409
Виолантрон 447
Восстановление 126
Вторичные амйны 290
Галогенирование 98
Гамма-кислота 187
Гаттермана реакция 274
Гидрогенизация 488, 494
Гваякол 272, 409
Гексаметилентетрамин 348
Гексахлорбензол 199
Гетерогенный катализ 467, 480
Гидразобензол 141, 490
Гидразосоединения 140, 145, 152
Гидразотолуол 138
Гидроксил 185, 188
Гидроксиламин 66, 157, 349
Гидролиз аминогруппы 104, 236, 245
— сульфогруппы 75, 190
— хлорзамещеиных 213, 224
Гидросульфит 133, 152, 304, 398
Гидрохинон 50, 361, 397, 432
Гипохлориты 118
Гликоль 152
Глицерин 222, 313, 443
Голлемана ряд 32
Гомогенный катализ 467, 470
Гофмановская реакция 376
Дегидротиотолуидин 388
Декагидронафталин 401, 497
Декалин см. Декагидронафталин
Диазоаминосоединеиия 250
Диазосоставляющие 148, 263
Диазотирование 249, 255
Диалкоксидифениламин (4.4') 44
Диалкиламиногруппа см. Алкиламино-
группа
Диаминоантрахинонсульфокислота
(1.4.2) 190
Диаминоантраруфин см. Диаминоди-
оксиантрахинон (1 .5.4.8)
535
Диаминоантрахинон 152
Диаминобифениловые производные 141
Диаминодиоксиантрахинон 139
Диаминодиоксиарсенобензол 155
Диаминодифениламин (п. д') 151
Днаминодиоксиантрахинон (1.5.4.8)
304
Днаминодиоксиантрахинон (1.8.4.5)
304
Диаминодифенилметан 409, 419
Диаминохризазин см. Диамннодиокси-
антрахинон (1.8.4.5)
Диаминофенол 220
Днанизндин 144, 145
Диантрен (9.9') 369
Диарилметановые замещенные 356
Дибензопирен-(3.4.8.9) -хинон (5.10)
503
Дигидроантранол 399
Дигидровиолантрон 447
Дигидродибензантронил 447
Дигидроксиламин 152
Дигидронафтохинон (5.8) 434
Дииндил (а.а' ) 327
п-Диметиламиноазобензол-л-сульфо-
кислота 265
Диметилметааминофенол 30
Диметиланилин 305, 320, 416
Диметилантрахинон (2.6 и 2.7) 434
Диметил-р-нафтиламин 265
1,Г-Диметил-2,2'-оксалиламинобензол
327
Диметилсульфаниловая кислота 265
Диметилсульфат 299, 311
Диметилхинон 361
Динафтиламин (₽•£') 528
1,1'-Динафтил-8,8'-дикарбоновая кисло-
та 433
₽.₽'-Динафтол-а.а'-метан 401
Динитроанизол (2.4) 222
Динитроантрахинон 139
Динитробензол (м) 31, 129, 131
— (о) 158
(п) 158
Динитродифениламнн (п.п'~) 151
Динитронафталин (1.5) 23, 359
— (1.8) 360
Динитро-4-оксидифениламин 133
Динитротолуолсульфокислота 259
Диннтрофенолы (о и п) 49, 220, 349
Динитрохлорбензол (2.4) 199, 207,209,
229
Диоксиантрахинон (1.2) 169, 375
— (1.4) 19, 304, 375
Диоксибензол (п ио) 167
— (м) 156, 167
Диоксибензойная кислота (3.5) 281
Диоксивинная кислота 449
1,5-Диокси-4,8-дибензоилдиаминоантра-
хинон 329
Диоксидинафтилен 449
Диоксидифенилдисульфид 390
Диоксидифенил (л.д') 417
Диоксинафталин (1.5) 180
— (1.7) 192
— (1.4) 434
Диоксинафталиндисульфокислота
. (1.7.3.6) 238
- - (1 .8.3.6) 238
Диоксинафталинкарбоновая кислота
(2.6.3) 284
Диоксинафталннсульфокислота (2.5.7)
286
Дисульфокислота бензола (м) 180
--- (и) 167
---(о) 167
— нафталина (1.5) 180, 192, 259
---(1.6) 192, 259
— стильбена 416
Дитолиламин 205, 243
Дифенил 499
Дифениламин 192, 202, 204, 242, 281
Дифенилгуанидин 323
Дифенилдисульфид-4.4'-дикарбоновая
кислота 460
Дифениловый эфир 212, 214
Дифенилсульфид 227
Дифенилтиомочевина 321
Дифенилэтан 406
Дифенол 178, 361
1,3-Дихлор-2-аминоантрахинон 229
Дихлорбензол (Я) 104, 174, 204
— (о) 104, 174, 217
— (м) 199
Дихлоркетонафталин 119
Дихлорнафталин 111
Дихлорфенол (1.4) 459
Диэтиланилин 297
4,4'-Диэтокси-1,1'-динафтил 501
Закрепитель Т 389
Заместителей ориентации см. Ориента-
ция заместителей
Замещающие группы 25
Зандмейера реакция 273
Запекание 76, 181, 388
Значение сочетания 208
Изатин 397, 450
Известковый плав 174
Изо-гамма кислота см. И-кислота
Изодуридин 310
Изонитрозогруппа 30, 62
И-кислота 186, 187, 328
Индоанилины 400
Индантрен 447
— синий RS 445
— темносиний ВО 447
Индиго 17, 298, 321, 327, 338, 373, 396
Индигозоли 338, 397
Индигоидные красители 117, 298, 389
Индоанилины 364
Индоксил 441
Индол 17
Индофенолы 370, 400
Индулиповые красители 119
Иодирование 117
536
Каменный уголь 141
Канницаро превращение 355
Каптакс см. Меркаптобензтиазол
Карбазол 15, 17, 366, 372, 427
Карбоксифенилбензокумарнн (2.4.5.6)
375
Карбоксикоричная кислота (о) 374
Кариуса метод 122
Катализаторы хлорирования 112, 201,
226
— восстановления 153
Катализ гомогенный см. Гомогенный
катализ
— гетерогенный см. Гетерогенный ка-
талнз
Каталитические яды 481, 485
Катанолы 389
Кауфлера метод 69
Кетон Михлера 419
Кислота Клеве см. 1-Нафтиламин-6-
сульфокислота
— К 187, 329
- G 193
— Каро 378
— Н 154, 186, 187, 238, 266
= •( см. Гамма-кислота
— 2S см. Чикаго 2S кислота
Кислотное восстановление 128
Контактное восстановление 153
Крезидин 16, 313
Крезол (о) 15, 32, 179
- (п) 15
— (и) 15, 30, 243, 401, 496
Крезотиновые кислоты 16
Ксиленол 361
Ксилидин (и) 251
Ксилол (о и Л) 13, 142, 331, 411
— (и) 13, 362
Кубовые красители 329, 440
Лейкардта реакция 273
Лепидин 17
Либермана реакция 69, 290
Либиха способ 122
Малеиновая кислота 527
Малеиновый ингидрид 524
Мезидин 310
Меркаптобензотиазол 323
Метил 22, 220, 293, 311
Метил-а-нафтол-р 401
Метиламиноанграхинон ф) 300
Метилантрацен 15
Метилантрахиноны 19, 430
Метиленовый голубой 385
Метилмеркаптан 228
Метиловый спирт 220, 223, 295, 310
— эфир 303
----а-нафтола 316
------ п-нитрокрезола 312
Метиловый эфир хлорсульфоновой
кислоты 81
Метилхинолин (а) 444
Метилциклогексанол 401
Метилциклогексанон 401
Метол 302
1-Метокси-4-бензонламиноантрахинои
329
п-Моно-М-метиламинофенол 298
Моногидрат серной кислоты 46, 72, 438
Монометиланилии 300, 305
Монохлоруксусная кислота 317
Моноэтиланилии 297
Мягкие восстановители 146
Нафтазарин 360
Нафталевая кислота 515
Нафталин 14, 22, 77, 83, ПО, 153, 192,
193, 211, 314, 404, 429, 497
Нафталинтетракарбоновая кислота
(1.4.5.8) 440, 516
Нафталинтетрахлорид 111
Нафтиламии (а) 61, 76, 119, 134, 180,
190, 236, 287, 331
Нафтиламии (₽) 77, 119, 186, 190, 273,
331, 528
Нафтиламиндисульфокислота (1. 4.8)
283
Нафтиламиноксисульфокислота (2.5.7)
328
Нафтиламинсулькофислота (1.8) 76
— (1.6) 326
— (1 : 5) 193
— (1.2) 23, 77
Нафтиламинтрисульфокислота (1.3.6.
.8) 154
— (1 . 4.6.8) 329
Нафтилендиамин (1 .4) 365
— (1.5) 256
— (1.8) 77, 256
Нафтилендиаминсульфокислота (1.8.4)
77
о-Нафтилсульфаминовая кислота 267
а-Нафтилфенилкетон 503
Нафтионовая кислота 77, 240, 251
Нафтоилбеизойная кислота (о) 431
Нафтолкарбоновая кислота (2.1) 265,
456
---(2.3) 456, 457, 458
---(2.6) 457
Нафтол (а) 169, 180, 193, 406, 497
Нафтол (₽) 84. 119, 137, 169, 176, 180,
183, 186, 189, 243, 336, 374, 401, 416,
497
Нафтол AS 186, 331
Нафтол типа AS 186, 227, 244, 259, 261,
284, 331, 459
Нафтолдисульфокислота (2.6.8) 193
— (1.3.8) 187
Нафтокарбазолсульфокислота (1.2.7)
453
---(2.3) 456, 457, 458
Нафтолсульфокислота (1 . 5) 180
— (1.2) 77
— (1.4) 77. 240
— (2.1) 460
— (2.6) 193
587
Нафтохинон (а) 362, 434
— (?) 362
Нафтохннондихлордиимин 370
Невиль-Винтера кислота 240
Неозон см. Фенилнафтиламин
Нейтрализация сульфокислот 86
Нефтяные погоны 142
Нейтральное восстановление 137
Нитратор 55
Нитрильная группа 229
Нитрил я -нафтойной кислоты 230
Нитрил о-толуиловой кислоты 230
Нитроазокрасители 148
Нитроанизол (о) 142, 222
Нитроанилнн (о и п) 144, 182, 201, 206,
364
— (и) 131, 148, 331
Нитроацетанилид (и) 34
Нитробензальдегид (о) 345, 411
— (м) 349
м-Нитробензаль дегид-л-сульфокислота
200
Нитробензол 129, 139, 142, 153, 188,
208, 425, 440, 489
Нитробензолсульфокислота (и) 86
Нитробензойная кислота (о) 355
----(л) 139
Нитрование 43, 57
л-Нитродифениламин-о-сульфоКислота
210
Нитрозамины 260, 291, 308
Нитрозодиметиланилин 409
Нитрозирование 61
Нитрозо- ?-нафтол 186
Нитрозобензол 62
Нитрозогруппа 30, 62
Нитрозодифениламин (л) 66
Ннтрозонафтолсульфокислота (2 . 1 . 41
461
Нитрозофенолы 153, 365
4-Нитрозо-З-хлорфенол 63
6-Нитро-1-диазо-2-окси-4-сульфокисло-
та нафталина 276
Нитрокрезол (л) 312
Нитронафтол (1.2) 188
Нитронитрозонафтол (1.8.4) 360
Ннтросалициловая кислота (л) 151
Нитротолуол (л) 51, 129, 138
-- (о) 51, 142, 346
Нитротолуолсульфокислота 259. 416
4-Нитро-4'-оксидифениламин 383
Нитрофенетол (л) 222
Нитрохлорбензол (л) 53, 199, 201, 206.
227
— (о) 53, 199, 227
л-Нитрохлорбензол-о-сульфокислота
207
о-Нитрофенилметилсульфид 228
Нитрофенол (и) 129, 130, 144
— (о) 133, 144, 188, 417
— (л) 133, 144, 243
Нитрующая смесь 45
Окисление 343
538
Окнсь динафтилена 448
— этилена 299
Оксаминовые кислоты 327
л-Окси-л-аминодифениламин 193
Оксиамнносоединения 185
л-Окси-л-амино-л'-метил дифениламин
314
Р-Окси-Р'-аминонафтойная кислота 244
Оксиантрон 399
Оксиантраценкарбоновая кислота (2.3)
460
Оксибензальдегид (и) 272, 345
Оксибензантронкарбоновая кислота
(4.3) 433
Оксибензойная кислота (л) 458
Оксидиариламинкарбоновая кислота
(3.5) 281
Оксикарбазол 247
Т-Оксикарбостирил 441
Оксинафтойная кислота (2.3) 244, 287,
493, 458
----(2.6) 456
?-Окси-я-нафтальдегид 427
Окситионафтен (3) 245, 438, 441
2-Окси-1,8-фталоилнафталин 431
Окси-тобиаса кислота 84
Оксихннон 362
Оксиэтнлирование 314
Оксиэтил 311
Олеум 46, 72, 117, 275, 320 , 345, 358,
415
Определение галоидов 122
— аминов 306
— ннтрогруппы 61
— оксисоединений 182
Ориентация заместителей 25, 28, 37
Осернение 381
Пентаметиланилин 310
Пентаметилфенол 310
Пентахлорбензол 199
Нерилен 54
Пикраминовая кислота 148, 255
Пикриновая кислота 49, 130, 133, 220
Пиперидин 151, 209
Пирен 15
Пиридин 16, 82, 152, 320, 336, 427
Пиридиновые основания 16
Пировиноградная кислота 412
Пирогаллол 362, 432
Пирокатехин 166, 272, 362
к (пи) сульфирования 74
Подвижность галоида 199
Полисульфиды натрия 147, 384
Полихлориды бензола 142, 440
— нафталина 113
Полупродукты кислотного восстано-
вления 160
— нейтрального восстановления 161
- щелочного плавления 195
Пропил 294
Противостарители 155, 288, 335
Разложение диазосоедннений 252
Рапидогены 261
Растворители при хлорировании 112
Редиягар 135
Р«®рцин Ж), 156, 166, 176, 180, 186
Рщеоль 156, 183
Родамин 30
Роданирование 120
Салициловая кислота 16, 179
Семиднновая перегруппировка 143
Серная кислота 72
Сернистая кислота 132, 239
Сернистый ангидрид 132
Сернистый аммоний 147
Сернистые красители 220, 365, 386
Сернистые щелочи 146
Серный ангидрид 82
Сернокислый эфир о-иитрофенола 144
Сероуглерод 13
Сиидиазотаты 260, 263
Синтетические дубители 92
Скорость диазотирования 25
Скорость разложения диазосоединеиий
253
Скраупнрование 444
Смола каменноугольная 14
Соль R см. R соль
Сольвентнафта 14, 142
Спирт этиловый 142, 153, 220, 224, 295
Сплавление со щелочами см. Щелоч-
ное плавление
Сульфамнновые кислоты 132, 254, 309,
320
Сульфаниловая кислота 61, 76, 252, 259,
265, 280
о-Сульфобензальдегнд 345
Сульфгидрат аммония 147
Сульфгидрильная группа 178, 189, 273,
316, 380
Сульфоантраннловая кислота 262
Сульфогруппа 93, 178, 189
P-Сульфокислота антрахинона 77, 168,
171
Сульфокислота антрацена (Р) 399
— (а) 399
Сульфокислота нафталина (а) 75, 175
----(Р) 75, 83, 175, 238
Сульфокислота фенола (о и п) 31
Сульфокислоты ксилолов 13
Сульфонамиды 90
Сульфоны 81
Сульфофталевая кислота (Р) 78, 373
4-Сульфофта левый ангидрид 118
Сульфохлоридная группа 84
Сутьфохлорнды углеводородов 89
Сульфуратор 85
Сульфурил хлористый 81, 99, 117
л-Сульфофеинл-З-метнл-5-пирозолон
450
Таннол 390
Тетрагидронафталин 401, 497
Тетрагидрохннолнн 151
Тетралин см. Тетрагндронафталин
Тетраметилбензидин 265
Тетрамйтилдиамияовенаофежщ 419
Тетраметилдиаминодифенил метан 255
1>4,1\4'-тетраокси-2,2'-диантрахииоиил
Тетраннтрометиланилин 310
Тетраоксиантрахиион (1.4.5.8) 304
— (1.2.5.8) 304
— (1.2.3.4) 359
Тетрахлорбензол (1.2.4.6) 199
— (1.2.3.4) 199
— (1.2.4.5) 199
Тиазиновые производные 384
Тиоиндиго 323
Р-Тиоиафтол 189
Тиосалнцнловая кислота 274
Тиотолен 13
Тиофеи 13, 488
Тнофенол 178, 288, 274, 340
Толидин 144, 251
Толил 25
Толуидин (о) 132, 134, 156, 251, 287,
О1П ОКО QQ7
— (п)’132,’134, 185, 186, 251, 287, 400
— (и) 242
Толуилендиамин (аг) 134, 256
о-4-Толунл-бензойная кислота 430
Толуол 13, 51, 113, 142, 315, 411, 427,
507
п-Толуолсульфохлорид 299, 309
Толуолсульфокислота (п) 311
Толуиловая кислота (о) 168
Трибромфенол 178
Тринитробензол 31
Тринитрооксибензойная кислота (2.4.
.6) 50
Тринитротолуол (2.4.6) 347
Триннтрохлорбензол (2.4.6) 209, 229
Трннитрофенол (2.4.6) 129
Триокснантрахинон (1.4.5) 304
Трихлорбензол (1.2.4) 102, 199, 211
Трифеннлметановые красители 310
Уксусный альдегид см. Ацетальдегид
Уксусный ангидрид 48, 305, 326, 335,
439
Уксусная кислота 54, 168, 258, 268
Уксусноэтиловый эфир 307, 335
Ускорители вулканизации 228, 323, 335
Урсолы 155
Фенантрен 15, 366, 497, 515
Фенацетин 15, 155
Фенантренхинон 368
Фенетедин 155, 281, 287
Фенил 25
Фенилгндразин 349
Фенилгидразон 349
Фенилглицин 303, 436
Фенилгндроксиламин (Р) 177, 157
Фенил-гамма-кислота 288
Феннлглицннкарбоновая кислота (о)
302, 373, 440
Фенилендиамнн (п) 204, 287, 349, 370,
385
539
WbWMB mw ж ж
Феявл-И-кислота 288
Фенилизокротоновая кислота 406
Фенил-а-иафтиламин-8-сульфокислота
283
Фенил-?-нафтиламии 288
Фенилнитрометан 352
Фенилсульфаминовая кислота 76
Фенилтиогликоль-о-карбоиовая кисло-
та 441
Фенол 15, 63, 169, 176, 186, 212, 214,
229, 312, 335, 361, 456
Фенольные эфиры сульфокислот 92
Фенолят натрия 167, 456
Флавантреи 502
Флегматизаторы 288
Флороглюцин 242, 312
Флуорен 13, 498
Флуоресцеин 117, 120
Форлендера гипотеза 35
Формальдегид 301, 407
Формилированне 324
Фосгенирование 322
Фриделя и Крафтса реакция 420, 478
Фталеиновые красители 186
Фталевый ангидрид 30, 219, 309, 414,
429, 506, 518
Фталевая кислота 77, 361. 373, 378, 404
Фталоновая кислота 373
Фторсульфоиовая кислота 81, 321, 438
Фуксиновый плав 60, 415
Фурфурол 476
Хинальдин 16, 414, 444
Хингидрон 398
Хинизарин 19, 219, 304
Хиноиды 144
Хинолии 16, 166, 320, 443
Хинон (л) 362, 397, 526
4-Хлор-о-беизоилбензойиая кислота 220
223, 430
П-Хлор-О-нитроанизол 222
Хлораминоантрахинон (3.2) 229
Хлорантрахинон (а) 203
—(?) 19, 203, 431
Хлорацетотолуидид 33
Хлорбензальдегнд (о) 345
Хлорбензол 101, 104, 201, 227, 331, 429
Хлорбензолсульфокислота (л) 259
Хлординитронафталин (1.2.4) 203
Хлористая сера 117
Хлористый алюминий 58
— ацетил 326
Хлористый бензил 52, 112, 116, 199,
201, 225, 348, 406
— л-амннодифенилдиазоинй 257
— бензилиден 112, 116, 223, 348
— бензоил 116, 223, 225
— дифенилтетраазоний 256
—- фльфурид см. суЖфурм’"7^!ИК-
стый
Хлористый фенилдиазоний 182, 252
Хлористый тиоиил 313
Хлористый этил 312, 314
Хлорнафталин (а) Ш
Хлорнитробеизол (1.2) 220
Хлорннтронафталин (1.4) 207
Хлороксиантрахинон (1.4) 219
Хлорсульфоновая кислота 72, 80, 308.
320, 336, 360, 415, 438
Хлортолуолы 112
Хлорфенол (л) 219, 229
— (о) 217, 219
Хризен 15
Хромотроповая кислота 238
о-Цианкоричная кислота 375
Циануровая кислота 332
Циклогексан 111
Циклогексанол 208, 401, 497
Циклогексанолсульфокислота 73
Циклогексанон 401
Циклогексен 73
Цнклогексил 294
Циклогексиламин 303
Частичное восстановление 146
Чикаго-28-кислота 187
Число растворимости 88
Шиффовы основания 334
Щавелевая кислота 327, 413
Щелочное восстановление 140, 145
Щелочное плавление 165, 171
Электролитическое отщепление 196
Электронная структура атома 38
Электрохимическое восстановление ЭД
139 145
— окисление 188, 361, 367
— хлорирование 109
Энергия активирования 469
Эозин 186
Эпсилон кислота 187
Этил 25, 220, 293, 311
М-Этил-Й-фениларилсульфонамид 297
Этил-о-толуидннсульфокислота 262
Этилбензиланилин 116
Этилбензиланилинсульфокислота 3W
Этилбензол 13
Этиленхлоргидрин 299, 312
Этиловый спирт см. Спирт этиловый
Этиловый эфир-о-крезола 32
Этилсульфат 312
и -Этоксифенил- а -иа фтиламии 281
Н. Н. ВОРОЖЦОВ
ОСНОВЫ СИНТЕЗА
ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ
И КРАСИТЕЛЕЙ
Допущено в качестве учебного пособия
к изданию в 1934 г. Главным Управле-
нием Учебных Заведений НКТП СССР
нктп
СССР
ГОСУДАРСТВЕННОЕ ХИМИКО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ОНТИ * МОСКВА - ЛЕНИНГРАД * 1934