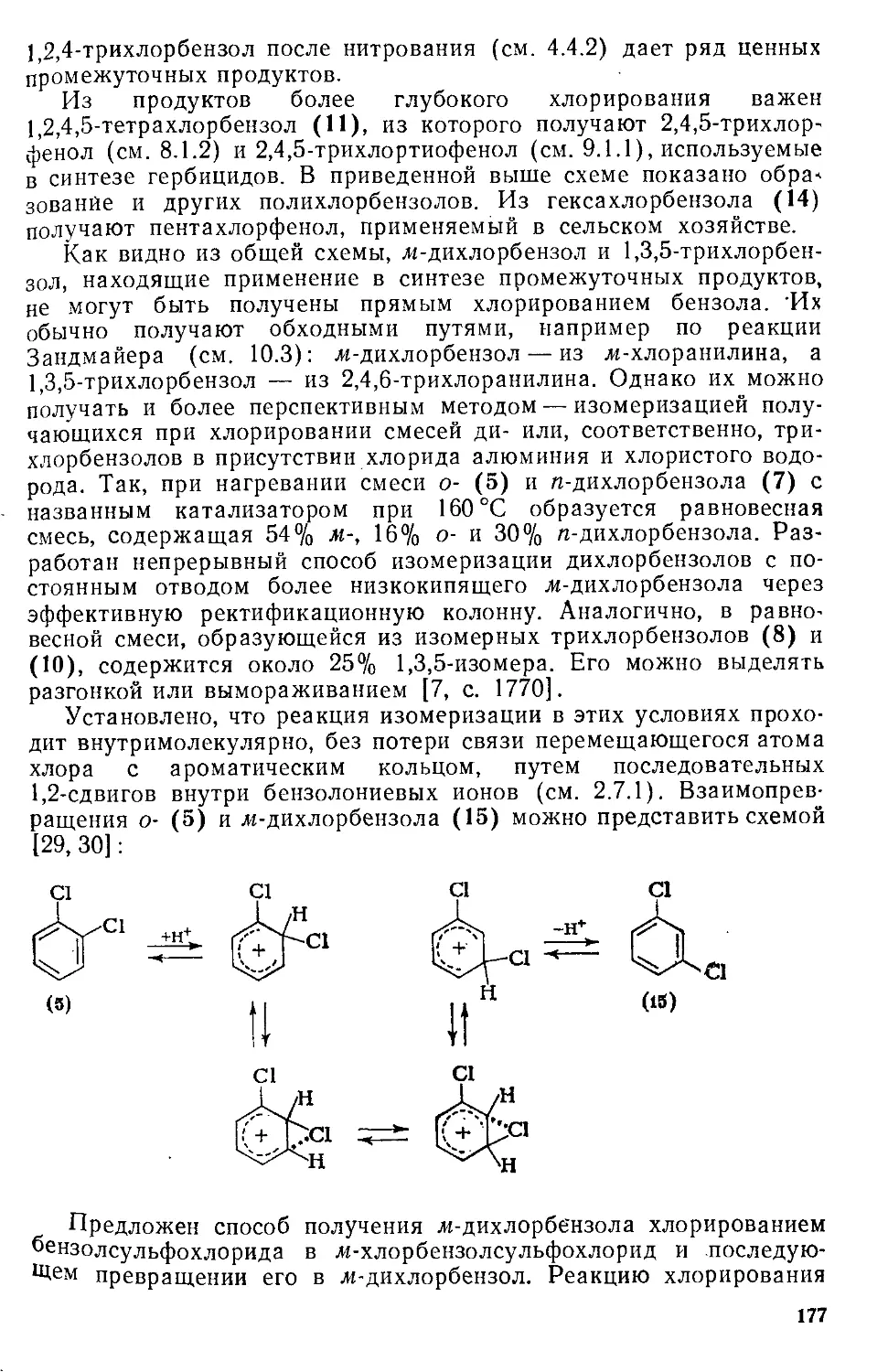
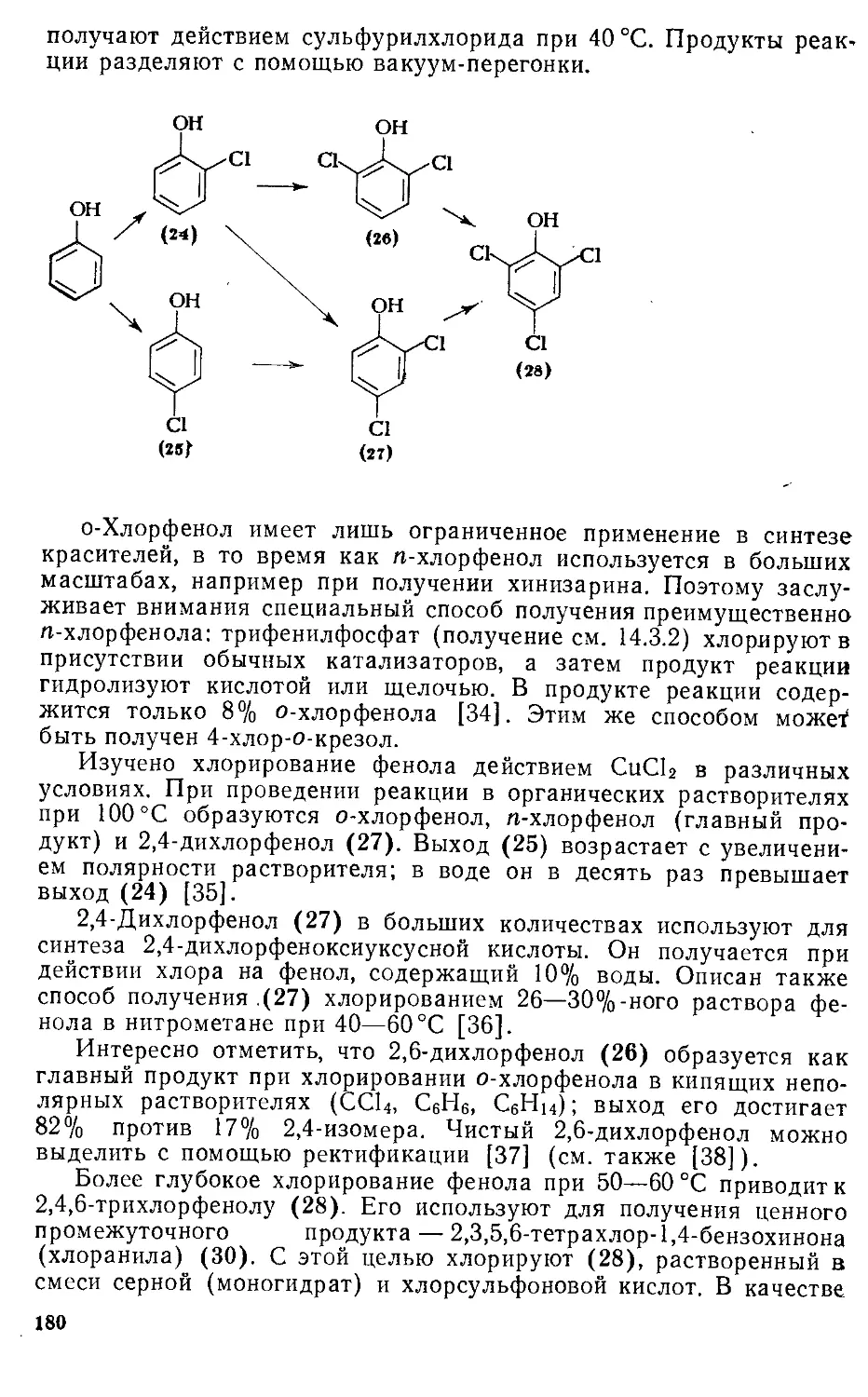
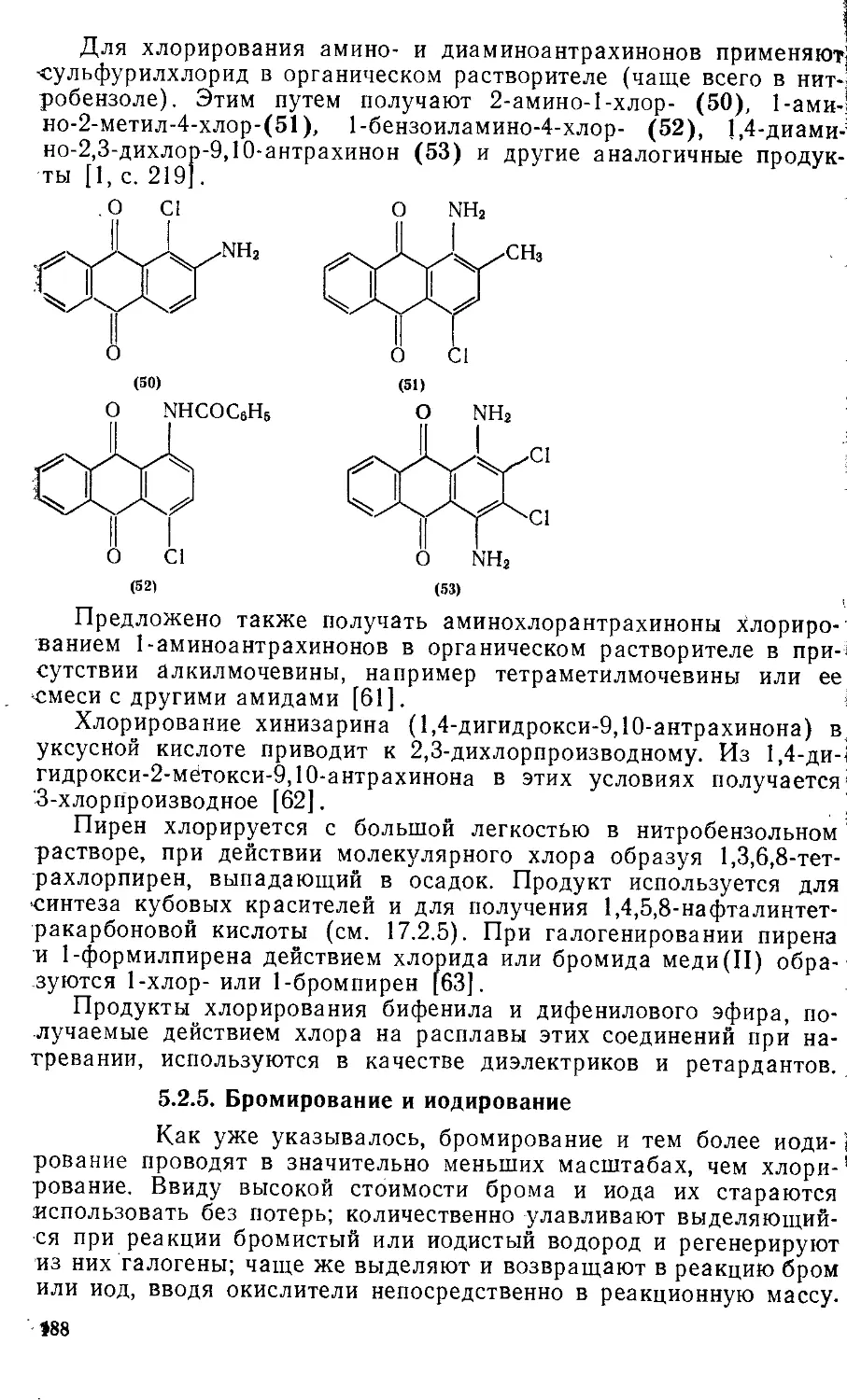
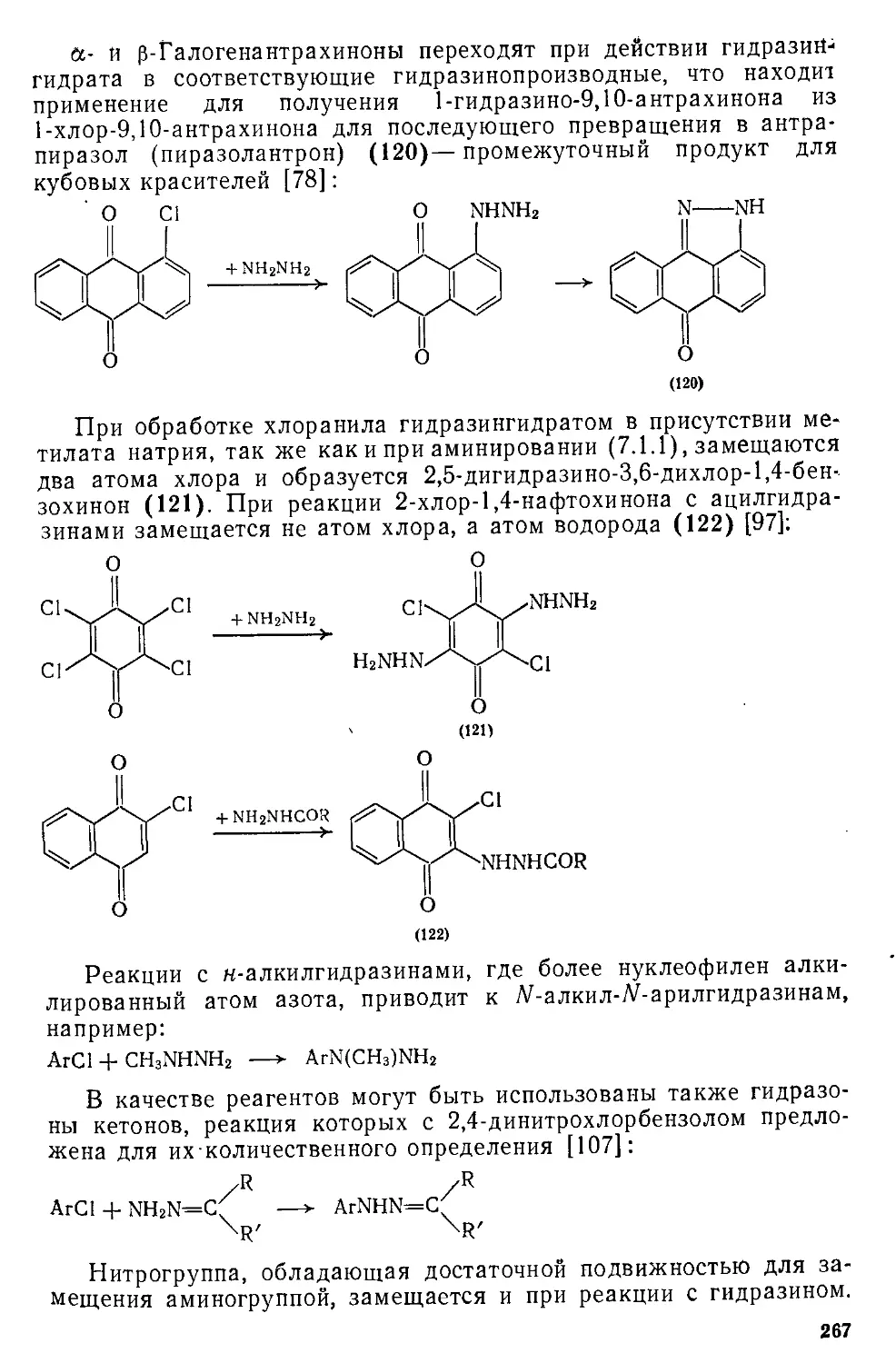
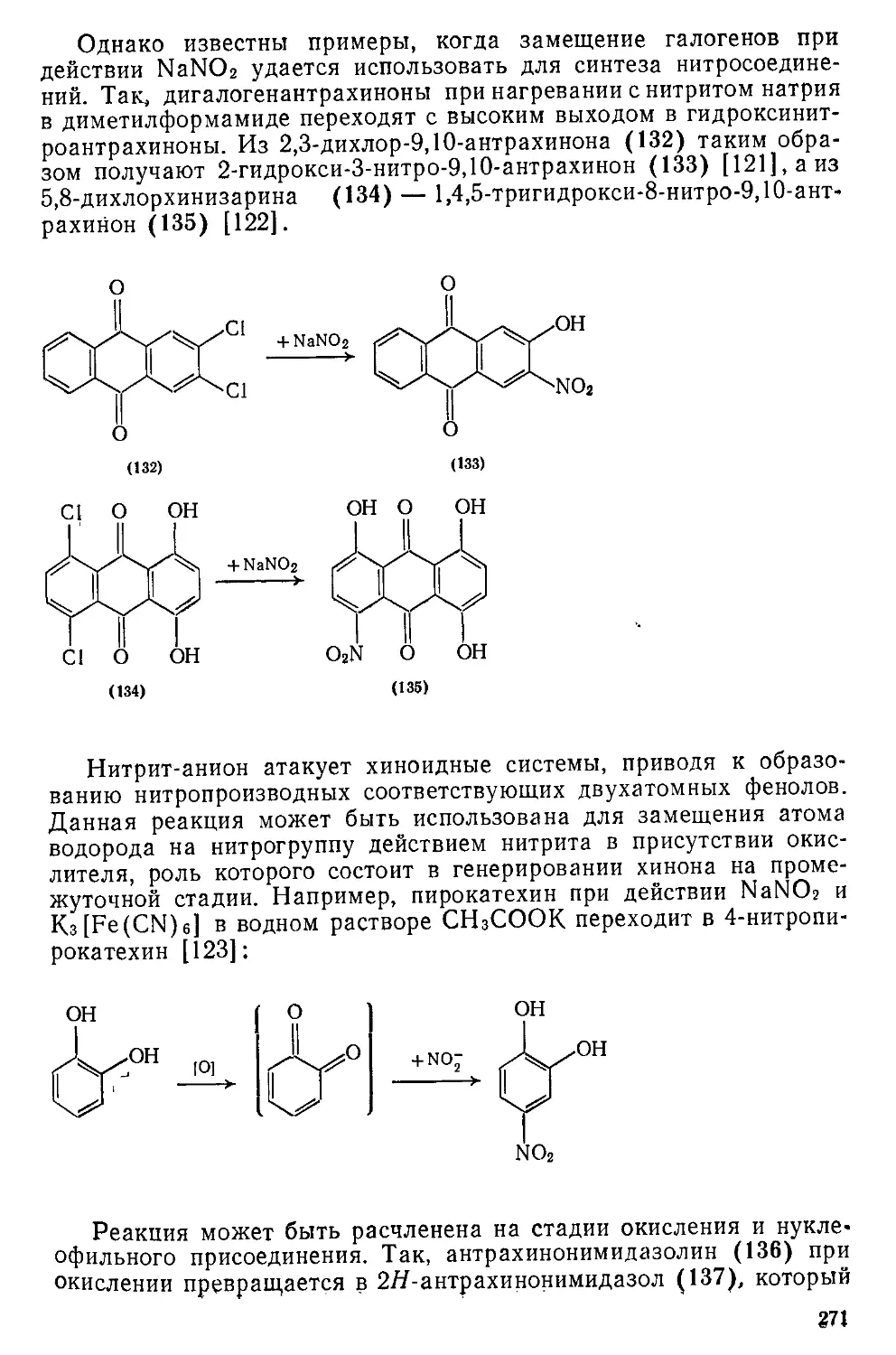
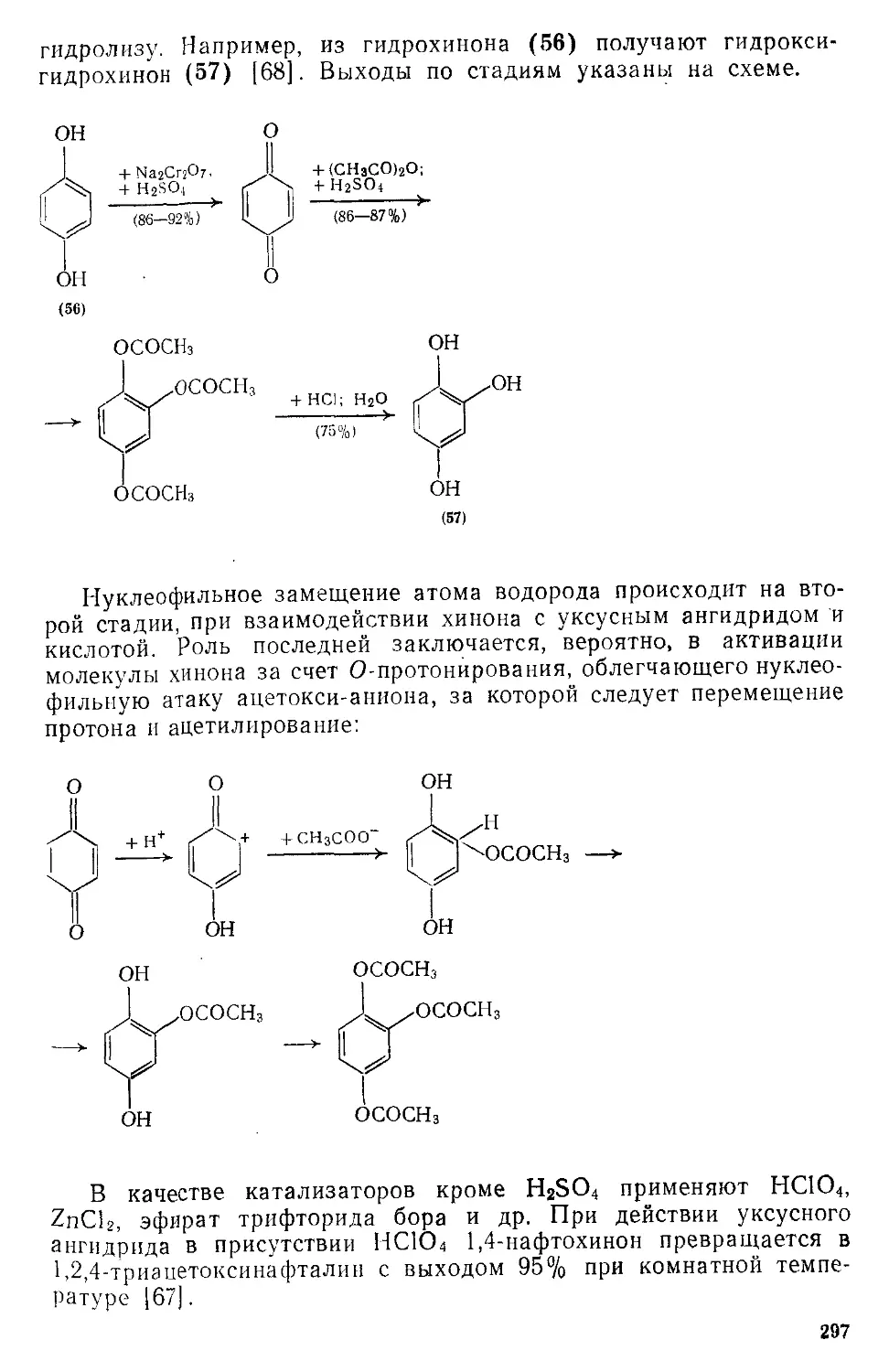
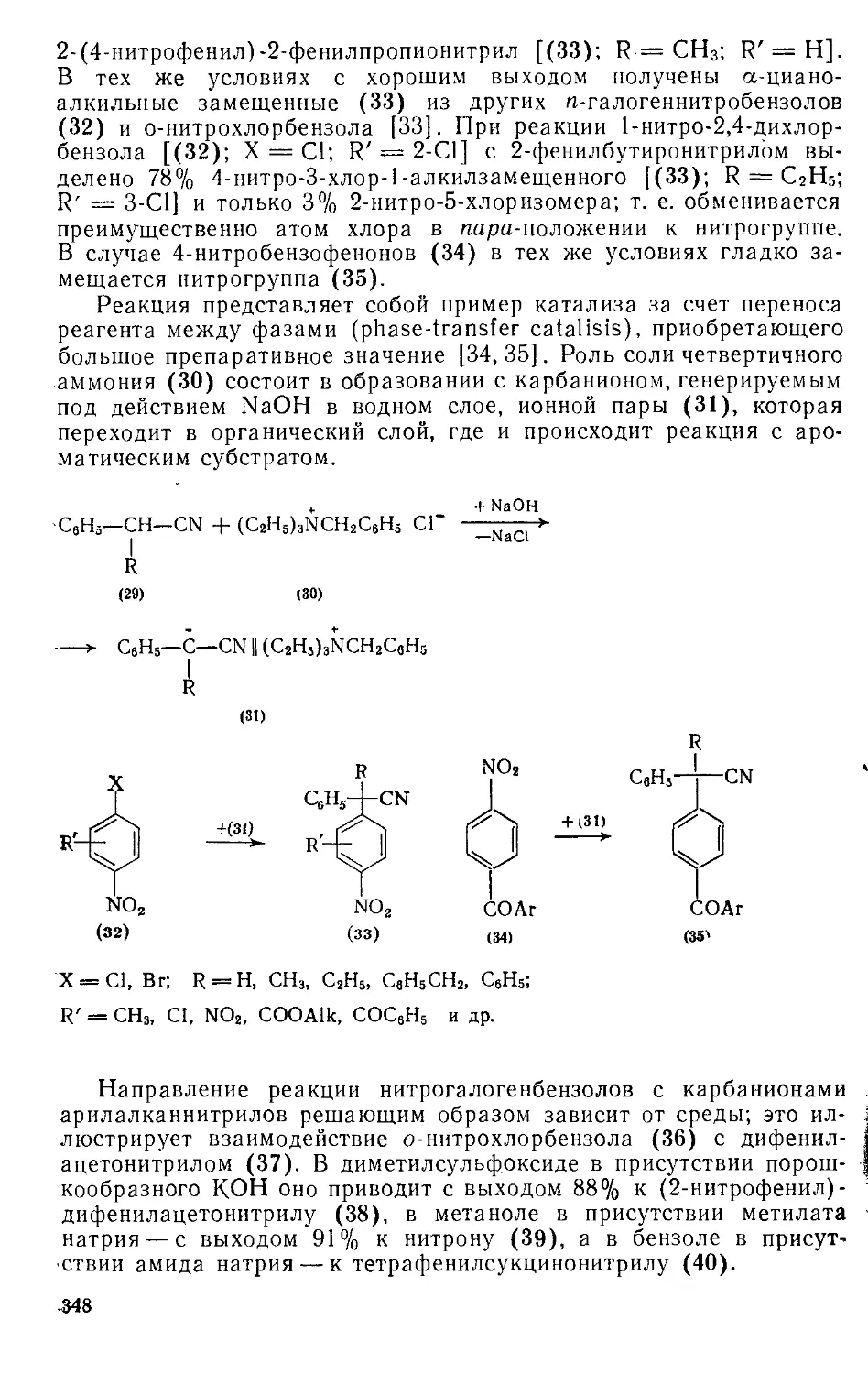
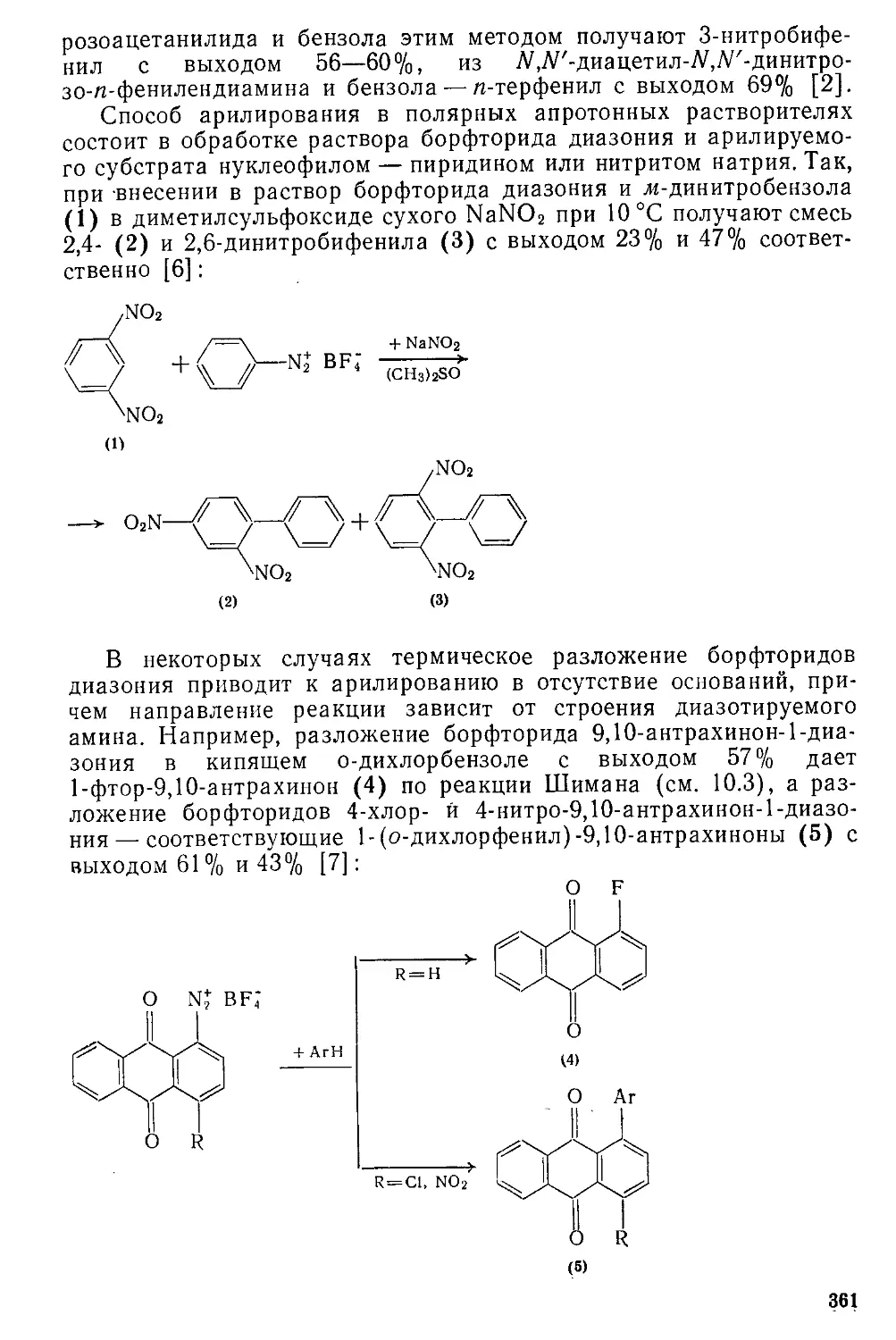
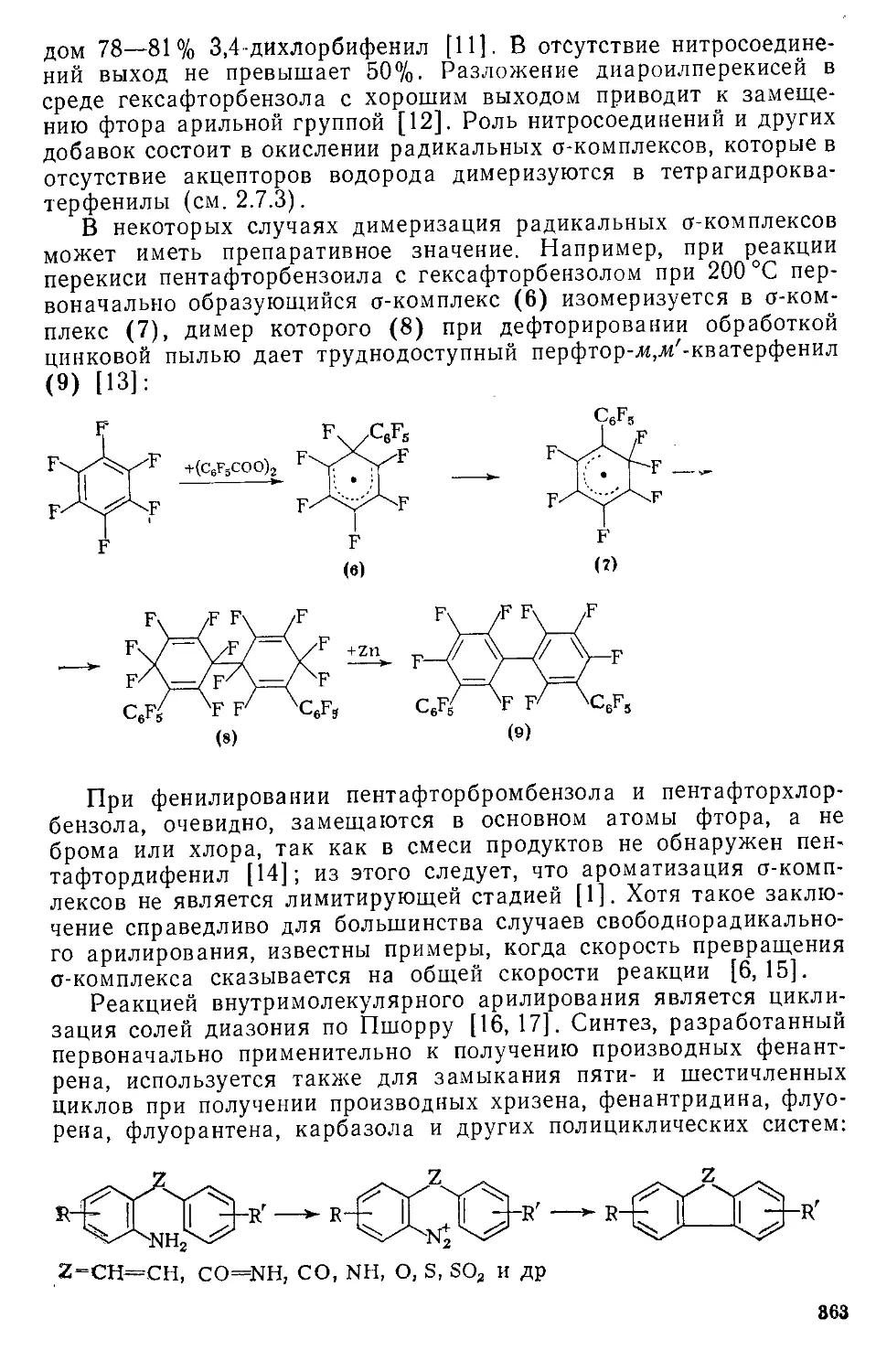
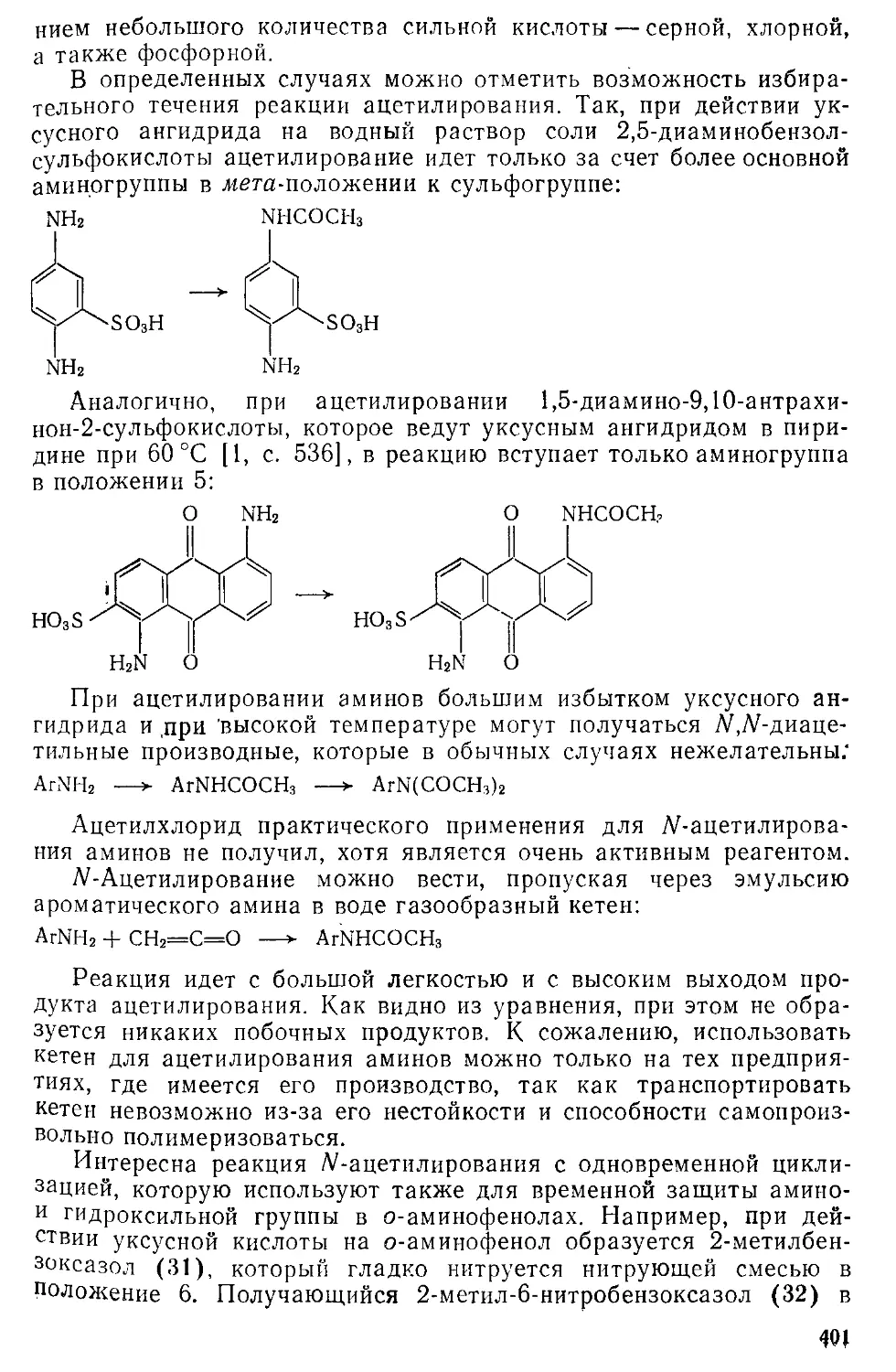
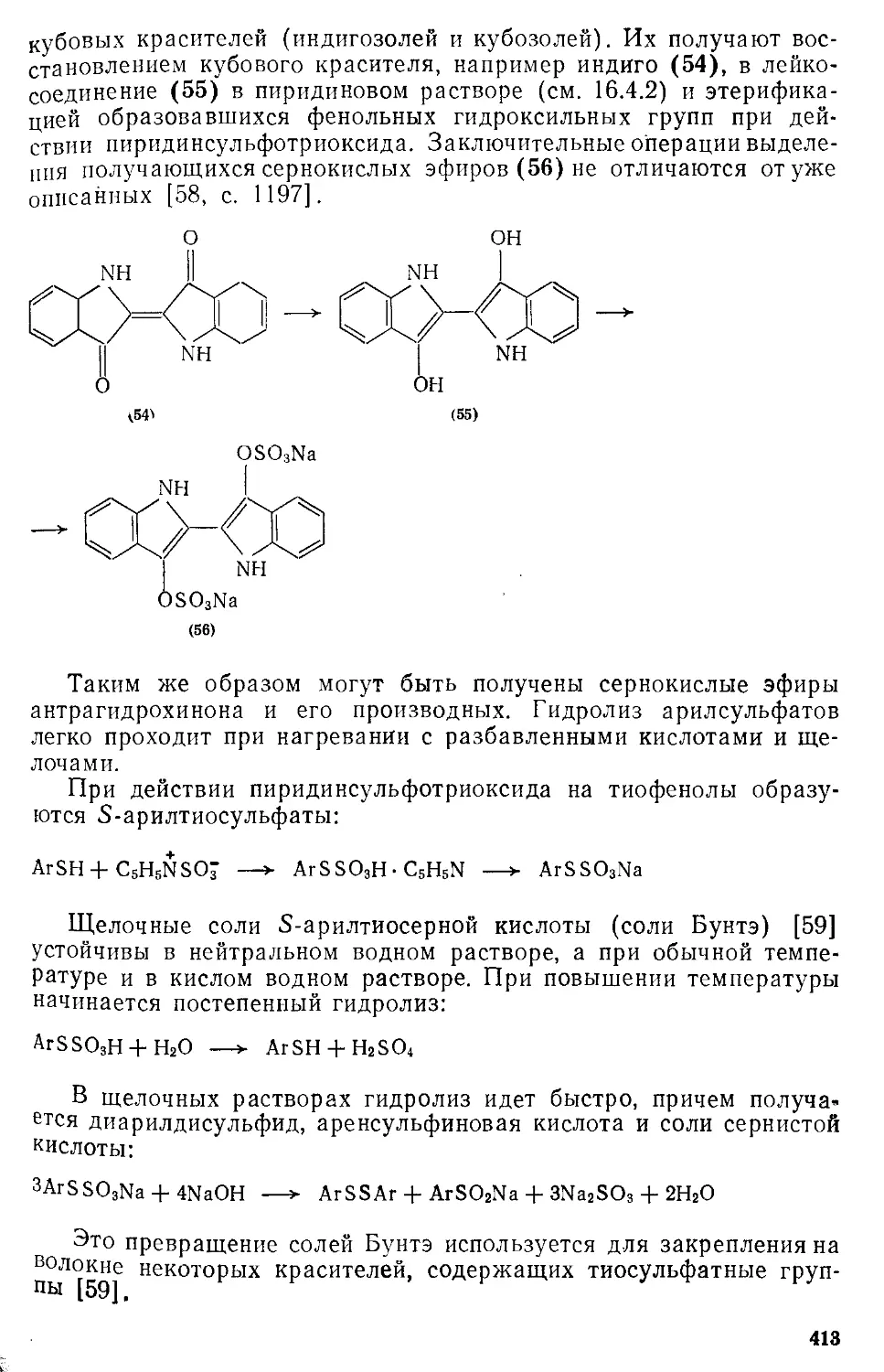
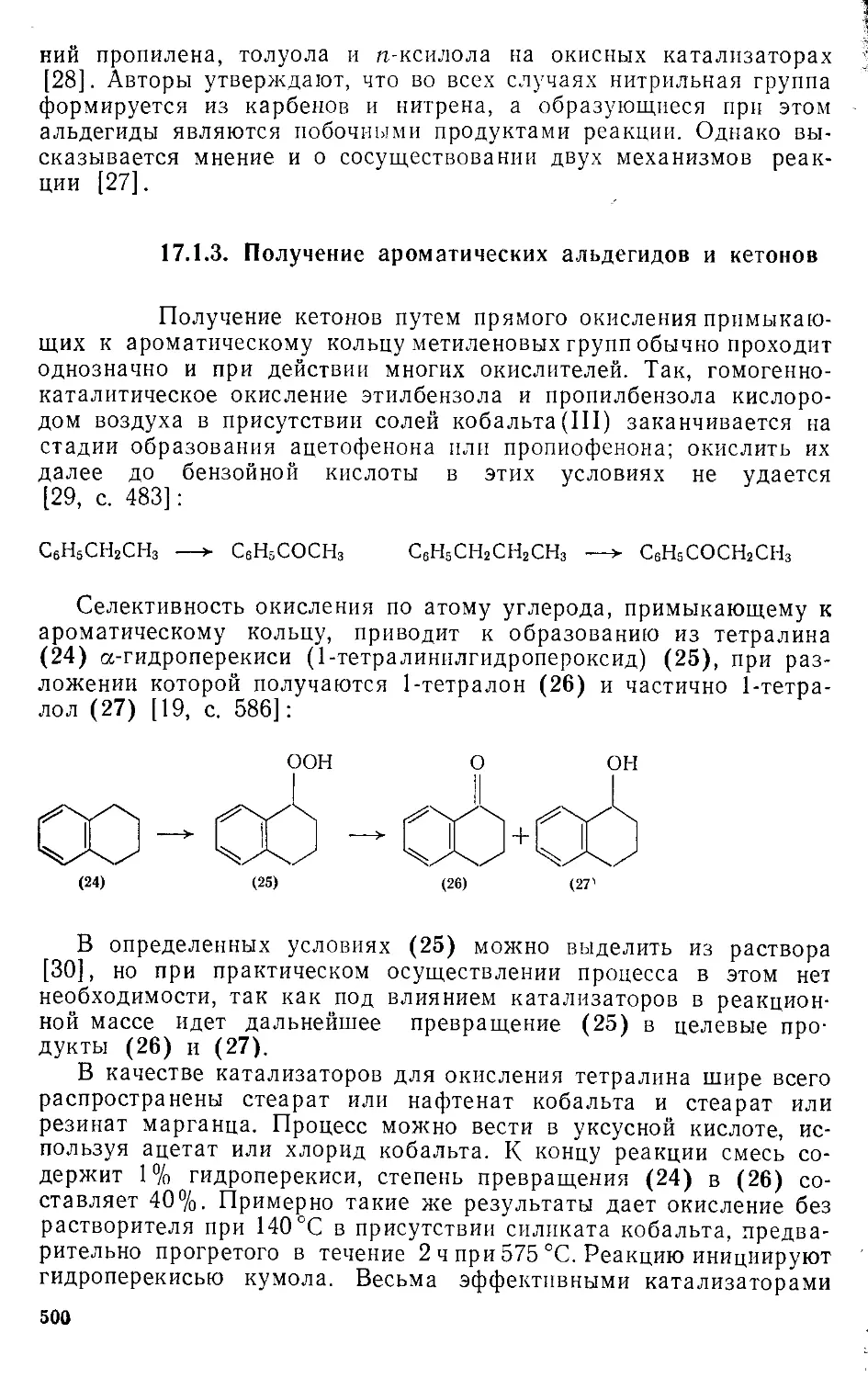
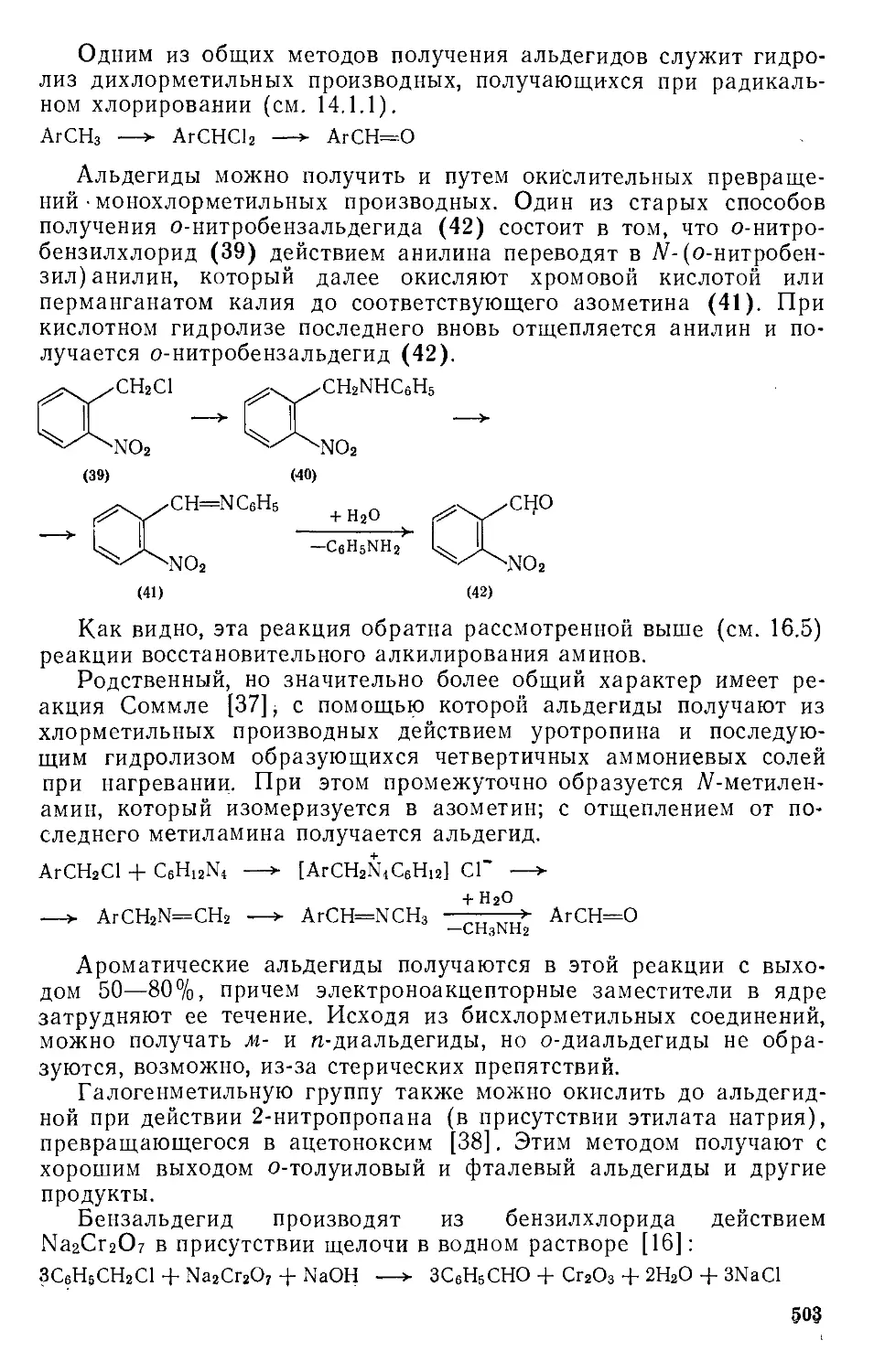
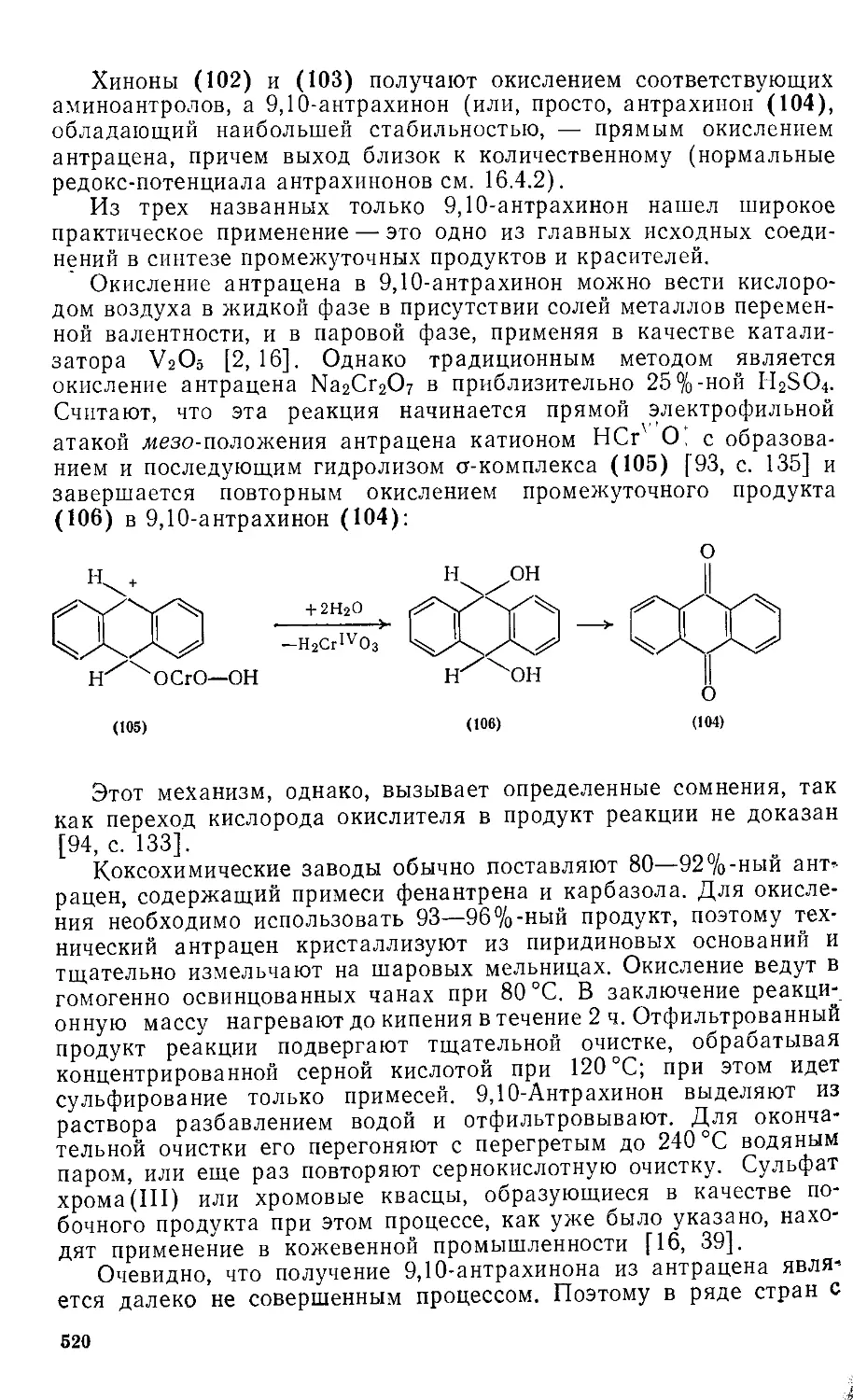
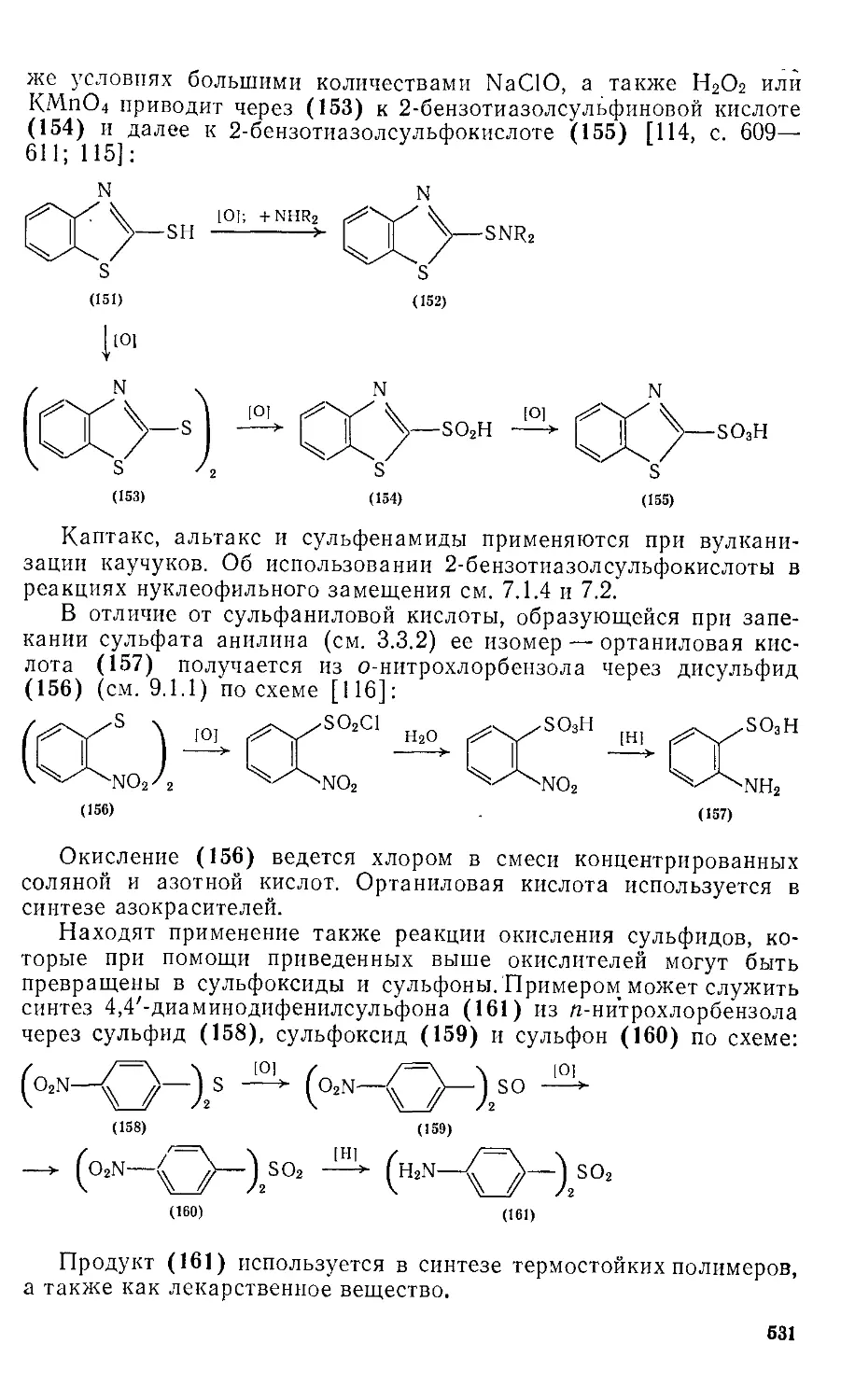
/



































































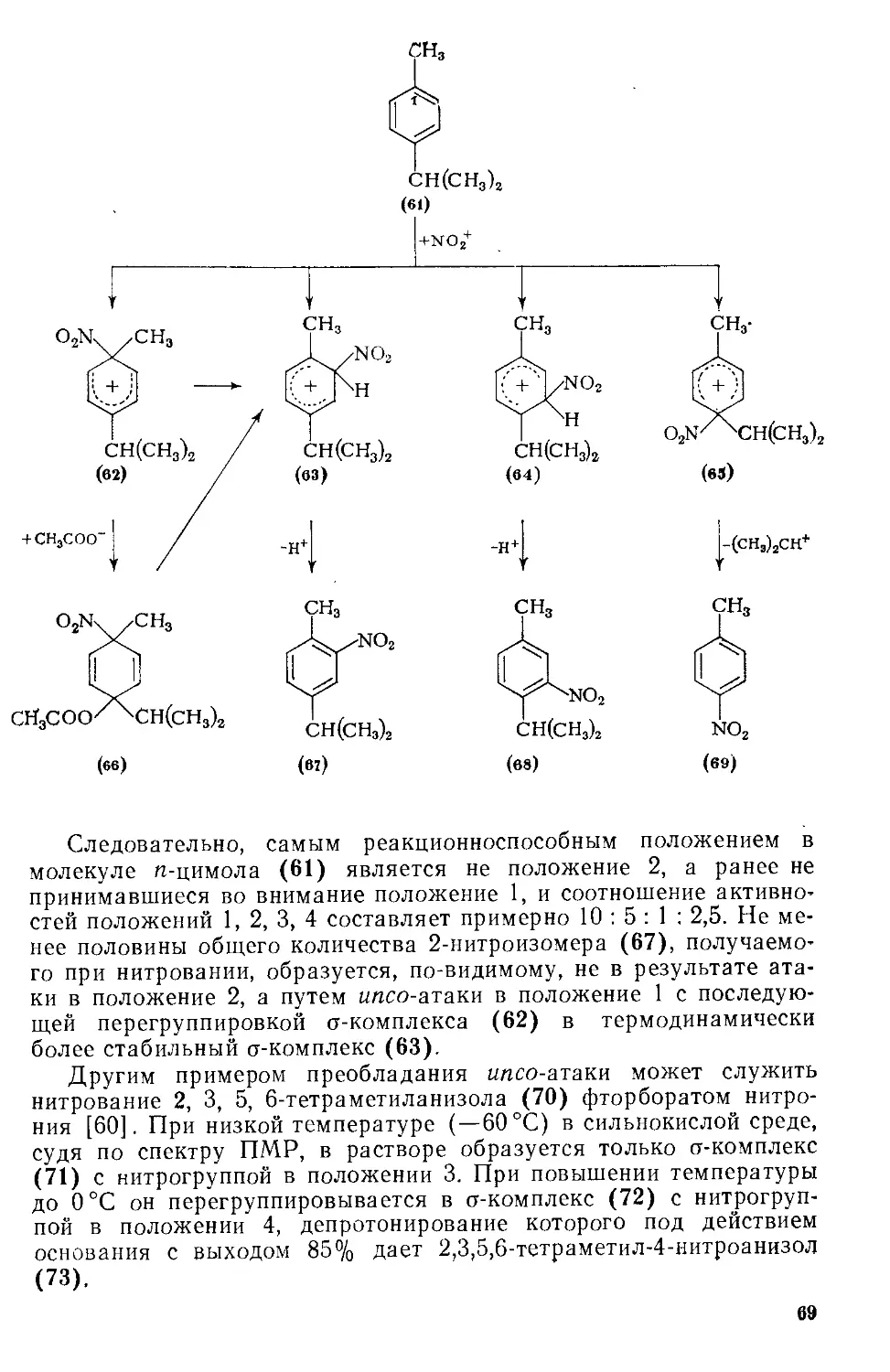





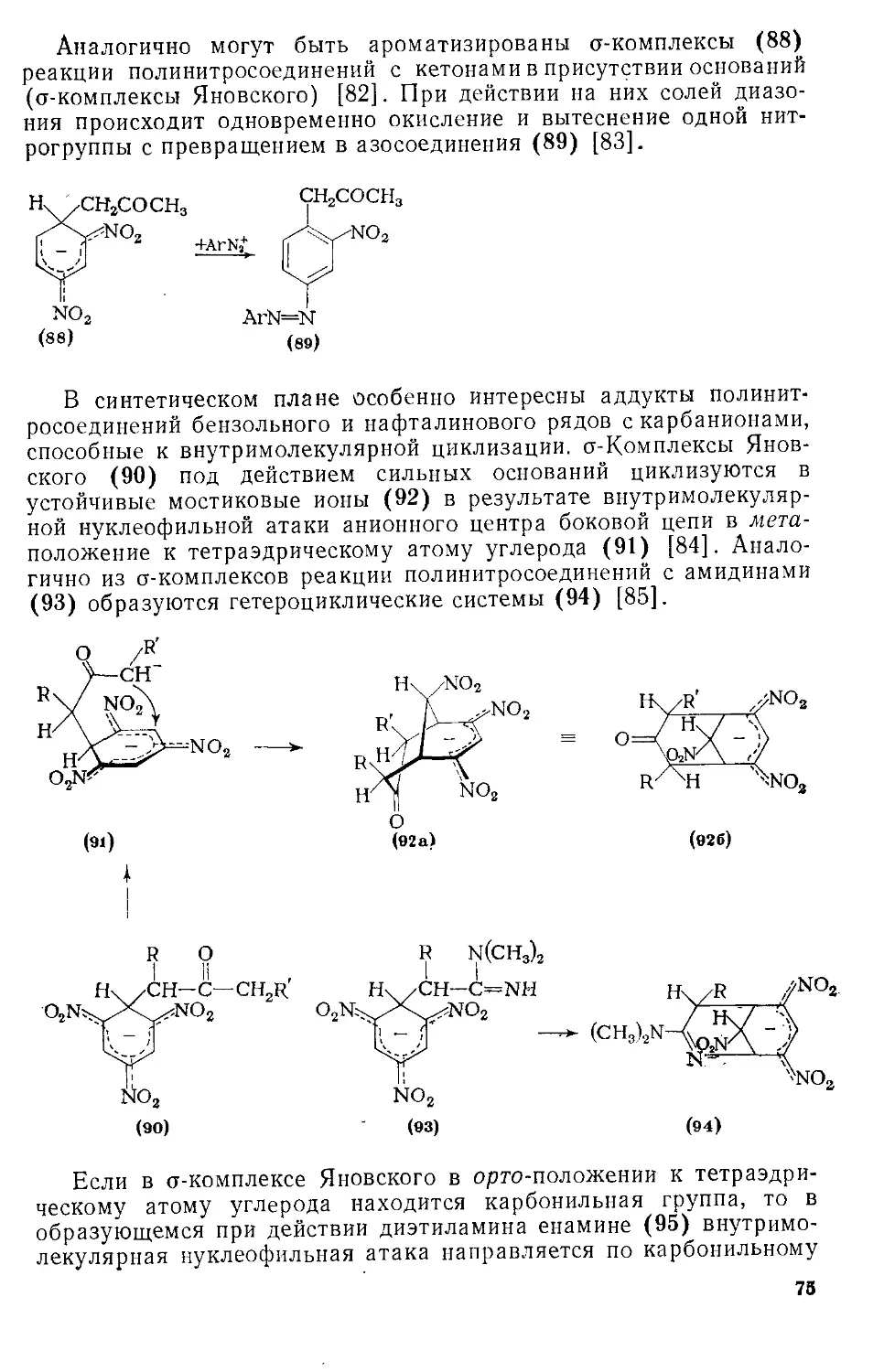



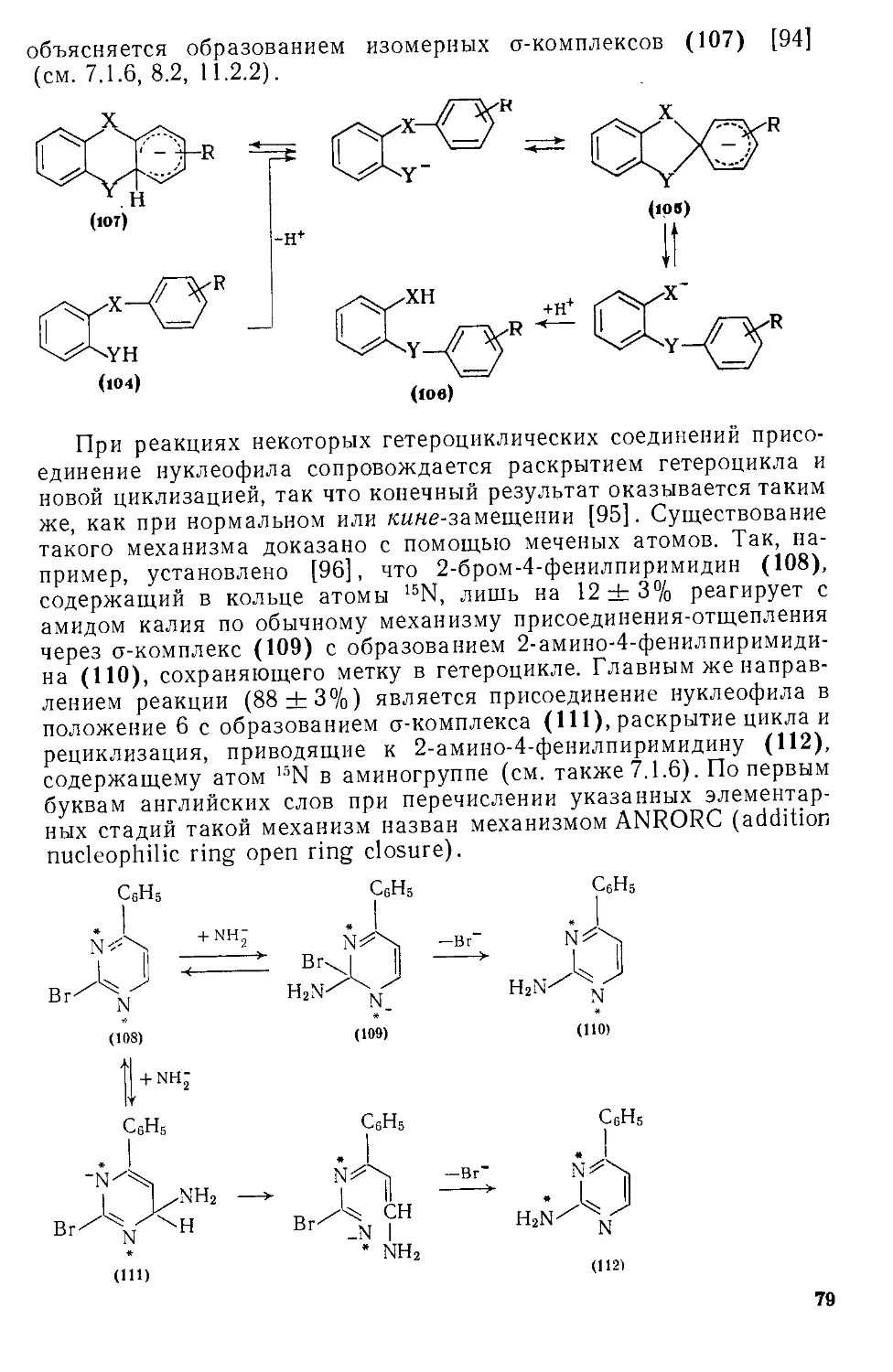













































































































































































































































































































































































































































Автор: Эфрос Л.С. Горелик М.В.
Теги: промышленность красителей производство и применение красителей химия промышленность
Год: 1980




Текст
Л. С. ЭФРОС
М. В. ГОРЕЛИК
химия
И ТЕХНОЛОГИЯ
ПРОМЕЖУТОЧНЫХ
ПРОДУКТОВ
Сканировал: Neptunyi
(Магнитогорск)
Все замечания и пожелания присылать по
адресам:
rhenium@list.ru
nept2006@yandex.ru
Буду рад также высылаемым книгам от вас!
Ленинград
«Химия»
Ленинградское отделение
1980
547 Э • /-h-V
Э94
УДК 667.21.002.62
Сканировал: Neptunyi
(Магнитогорск)
Все замечания и пожелания присылать по адресам:
rhenium@list. ги
nept2006@yandex.ru
Буду рад также высылаемым книгам от вас!
Эфрос Л. С., Горелик М. В.
Э94 Химия и технология промежуточных продуктов.
Л.: Химия, 1979. —544 с., ил.
В книге на основе современных представлений рассмотрены
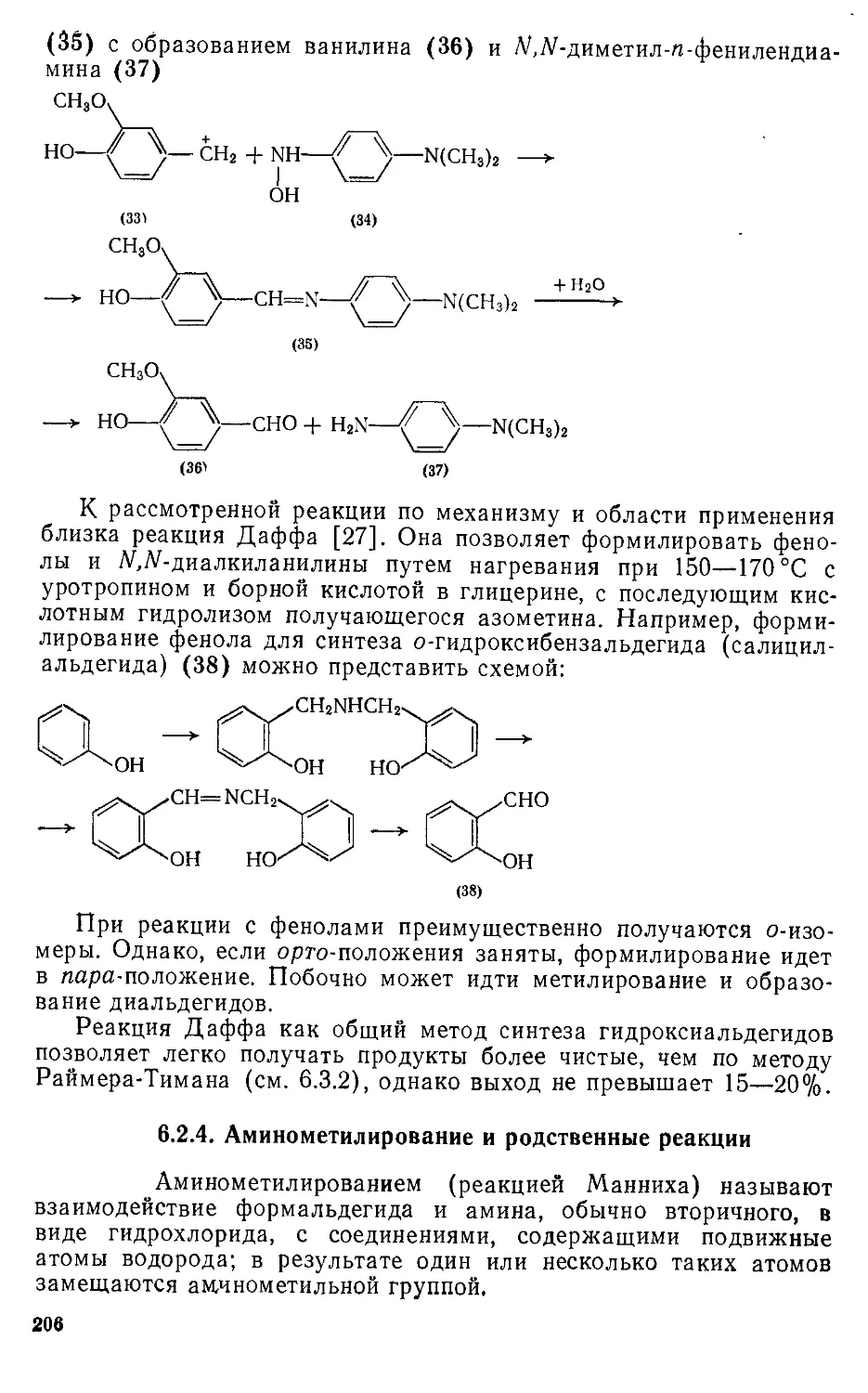
реакции, применяемые в синтезе промежуточных продуктов. Обсуж-
дается электронная структура ароматических соединений, критерии
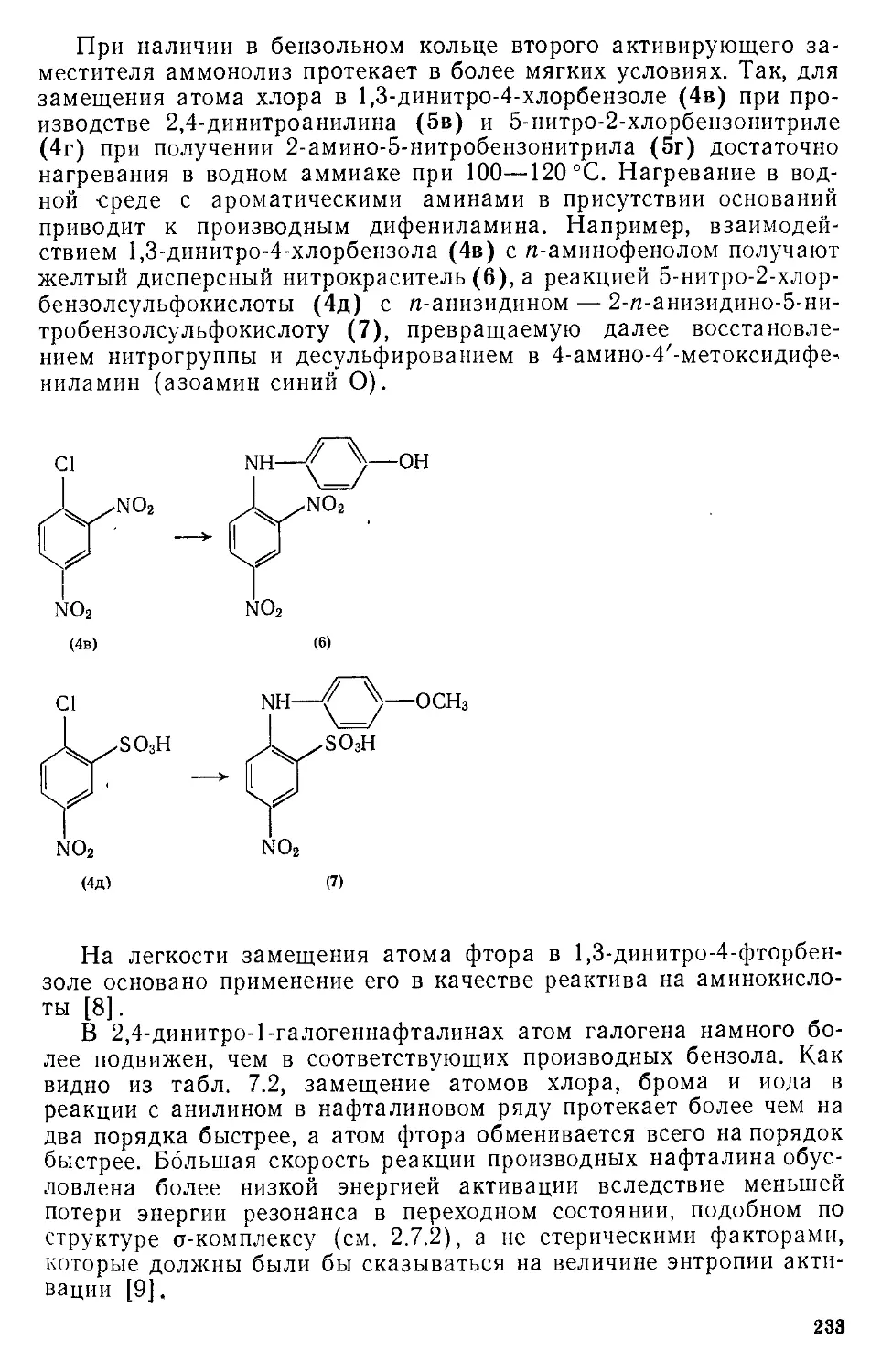
ароматичности, механизмы реакций ароматического замещения, влия-
ние на реакционную способность строения ароматического субстрата,
реагентов и растворителей. Изложены современные данные о применяе-
мых в технических и препаративных целях реакциях электрофильного,
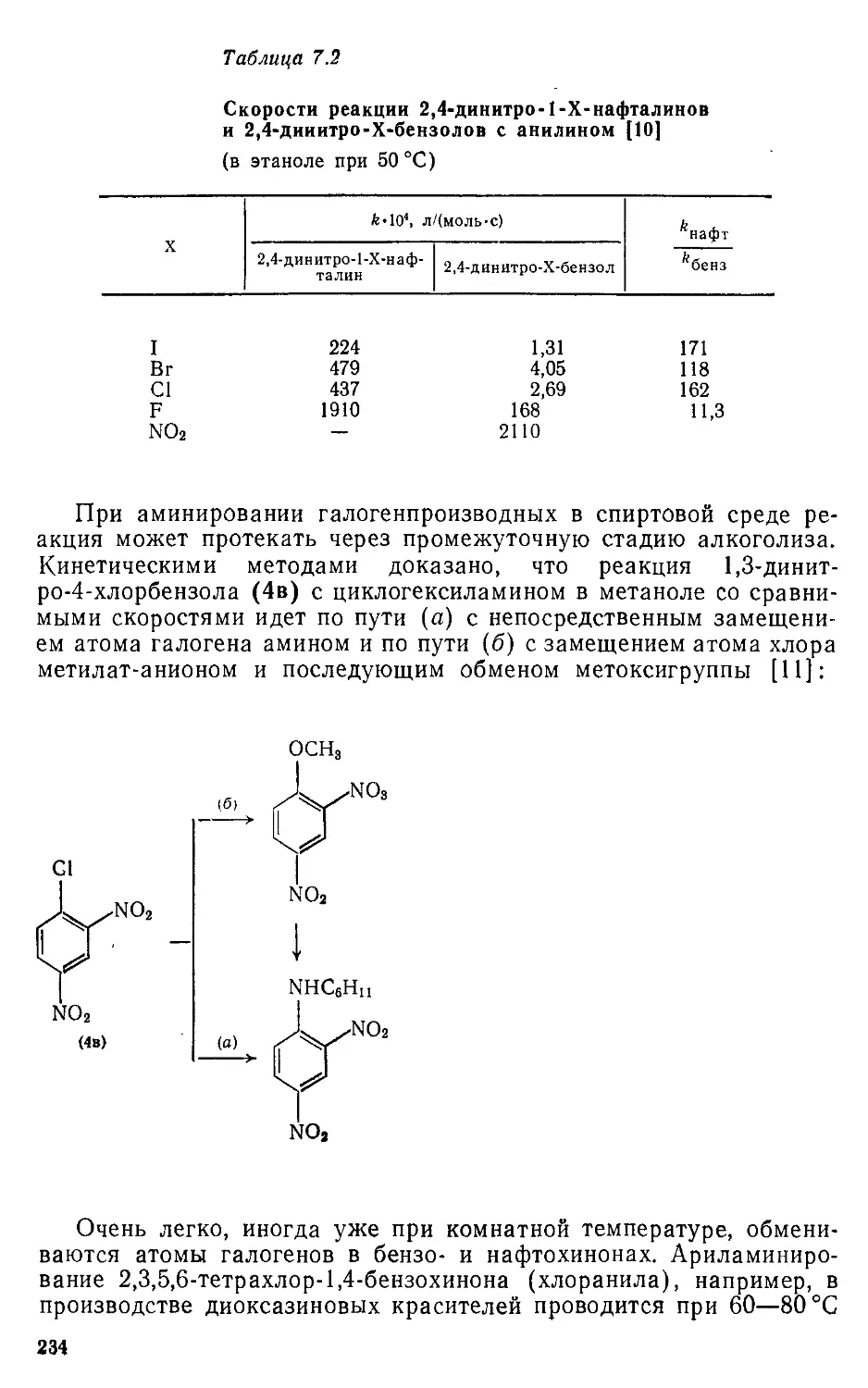
нуклеофильного и свободнорадикального ароматического замещения,
реакциях в боковых цепях, реакциях окисления и восстановления.
Приведены технологические режимы важнейших производств проме-
жуточных продуктов.
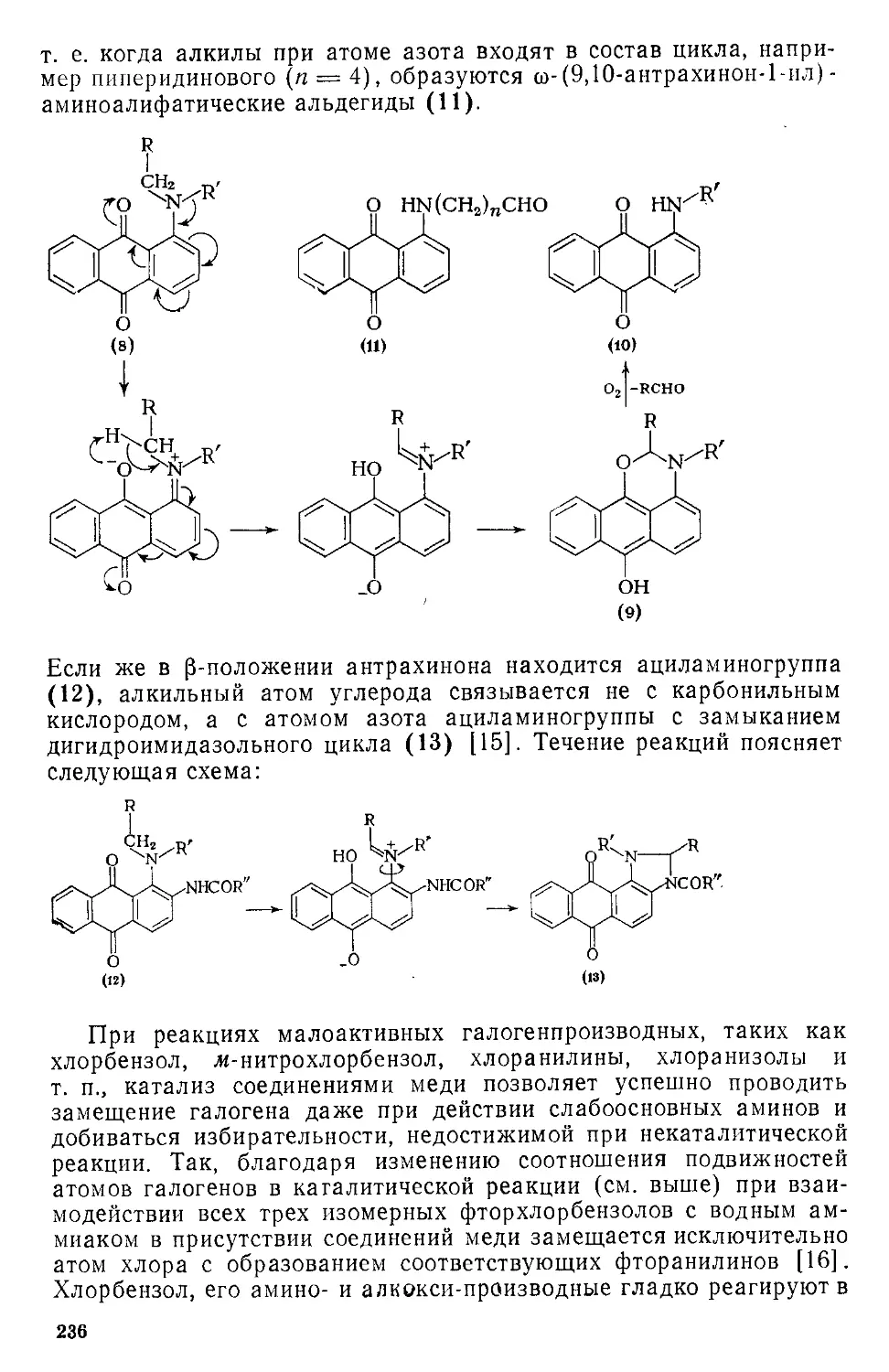
Книга предназначена для инженерно-технических и научных ра«
ботников, занятых в области синтеза ароматических соединений, и мо-
жет быть полезна преподавателям, аспирантам и студентам химико-
технологических вузов и химических факультетов университетов.
544 стр., 10 рис., 13 табл., библиография 1200 названий
547
© Издательство «Химия», 1980
ПРЕДИСЛОВИЕ
Исходными соединениями для промышленного синтеза
красителей, лекарственных препаратов, химических средств защи-
ты растений, взрывчатых веществ, многих мономеров и добавок к
полимерным материалам служат ароматические углеводороды и их
гетероциклические аналоги, получаемые путем переработки горю-
чих ископаемых, главным образом каменного угля и нефти. Чтобы
от сырья перейти к значительно более сложным конечным продук-
там, необходимо осуществить ряд химических превращений, явля-
ющихся промежуточными этапами на пути к целевому соеди-
нению. Разнообразие методов синтеза и дальнейшего исполь-
зования соединений, образующихся на промежуточных этапах,
давно заставили выделить эту область в самостоятельный и
наиболее общий раздел химии ароматических соединений, полу-
чивший интернациональное название химии промежуточных про-
дуктов (Zwischenprodukten в немецком языке, intermediates — в
английском).
Со времени последнего издания классической книги Н. Н. Во-
рожцова «Основы синтеза промежуточных продуктов и красите-
лей» * прошло более 20 лет. За этот период в химии ароматиче-
ских соединений произошел качественный сдвиг, обусловленный
как бурным развитием новых экспериментальных и квантово-
химических методов исследования реакций, так и появлением но-
вых реакций, реагентов и растворителей. Назрела необходимость
рассмотреть основы синтеза промежуточных продуктов с учетом
последних достижений теоретической и препаративной химии аро-
матических соединений.
Хотя ароматические соединения — та область, в которой впер-
вые изучались и устанавливались многие основные закономерности
строения и реакционной способности, до недавних пор эти законо-
мерности недостаточно увязывались с промышленной химией про-
межуточных продуктов и оставались в значительной степени при-
надлежностью теоретической органической химии. Между тем ос-
новная задача химика-практика — уметь управлять химическим
процессом — не может быть решена без понимания того, как
* Ворожцов Н. Н. Основы синтеза промежуточных продуктов и красителей.
Госхимиздат, 1955. 839 с,
I*
3
процесс протекает, а работа химика-теоретика может быть дей-
ствительно плодотворной лишь будучи связана с практическими
задачами. Авторы стремились предоставить читателю возможность
при знакомстве с основами практической химии и технологии про-
межуточных продуктов опираться на современный фундамент тео-
ретической химии ароматических соединений, считая залогом даль-
нейшего развития максимальное слияние этих аспектов.
Специфика промышленности промежуточных продуктов состоит
в огромном разнообразии ассортимента при сравнительно неболь-
шом объеме производства отдельных химикатов. Это обусловлива-
ет с одной стороны разнообразие применяемых реакций и реаген-
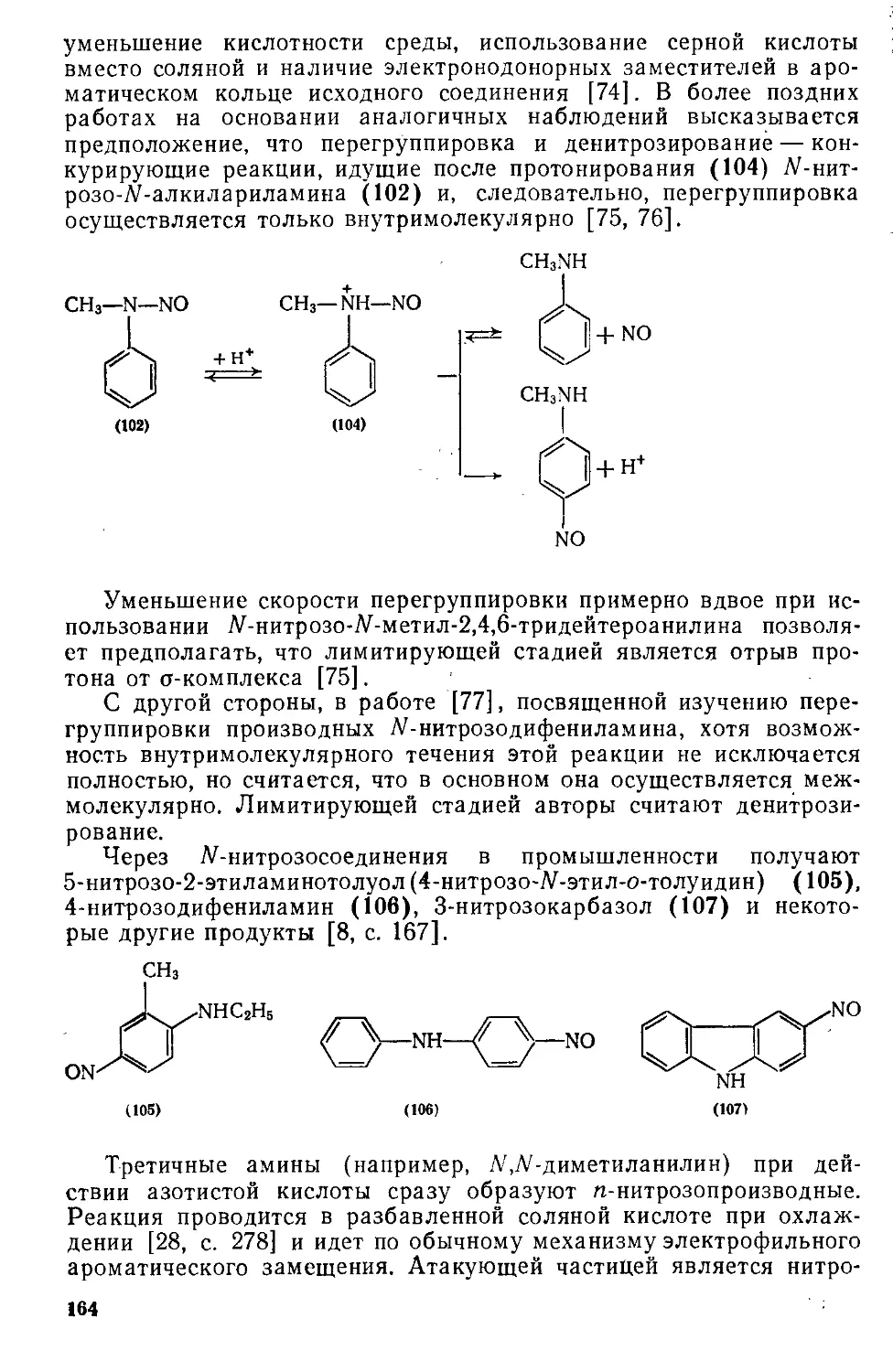
тов, с другой — использование совмещенных или мобильных схем с
унифицированным оборудованием для получения соединений раз-
личного строения. Выбор метода синтеза и параметров его осуще-
ствления является главным в промышленности промежуточных
продуктов, как и других продуктов тонкого органического синтеза.
Типовое оборудование промышленности тонкого органического
синтеза рассматривается в книгах, посвященных процессам и ап-
паратам химической промышленности. Поэтому, касаясь вопросов
конкретного производства, авторы сосредоточили внимание на тех-
нологических режимах и их взаимосвязи с химизмом применяемых
реакций, а не на аппаратурном оформлении. Для иллюстрации
того, как параметры, найденные при изучении химических законо-
мерностей, реализуются в промышленности, в книге приведены
описания технологических режимов производств некоторых на-
иболее важных промежуточных продуктов.
Поскольку объем книги исключает возможность исчерпываю-
щего изложения всего, что известно по рассматриваемым вопро-
сам, с полной библиографией, авторы ограничились выбором важ-
нейшего с их точки зрения материала и ссылками на обзорные
статьи или монографии, оригинальные работы принципиального
характера и последние по времени публикации, в которых можно
найти ссылки на предыдущие исследования.
В книге использованы, по возможности, систематические на-
звания в соответствии с принципами Международной номенклату-
ры органических соединений (ИЮПАК). При этом в сложных
названиях префиксы для обозначения заместителей перечисляются
в порядке русского алфавита. Приведены также укоренившиеся
тривиальные названия, а в отдельных случаях, как синонимы,—
некоторые до сих пор встречающиеся названия по старым номенк-
латурам. Нумерация формул и списки литературы даны по главам.
Части 1, 3 и 4 написаны М. В. Гореликом, части 2, 5 и 6 —
Л. С. Эфросом.
Авторы выражают глубокую благодарность члену-корреспон-
денту АН СССР В. П. Мамаеву за ценные замечания при рецензи-
ровании рукописи и доценту 3. Я. Хавину за большой труд по
научному редактированию. Авторы с благодарностью учтут крити-
ческие замечания и пожелания читателей — они помогут при
дальнейшем совершенствовании книги.
4
СТРОЕНИЕ И СВОЙСТВА
АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Глава 1
АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
1.1. источники ПОЛУЧЕНИЯ
Термин «ароматические соединения» появился в химиче-
ской литературе в первой половине XIX века, когда все соединения
углерода подразделяли на две группы: жирные, или алифатиче-
ские, соединения, названные так потому, что из членов этого ряда
первыми были подробно изучены кислоты, выделенные из природ-
ных жиров, и ароматические соединения, первые представители
которых были выделены из благовонных растительных смол и
бальзамов. В настоящее время главными источниками ароматиче-
ских соединений являются каменноугольная смола, получаемая
при коксовании угля, и нефть [1—3].
Коксование каменного угля проводится с целью получения кок*
са для металлургической промышленности и заключается в обра-
ботке угля при 1000—1250 °C без доступа воздуха с одновременной
отгонкой летучих веществ в виде коксового газа и конденсата,
состоящего из водного аммиака и каменноугольной смолы.
Из коксового газа после охлаждения и обработки промывным
маслом (см. ниже) получают основное количество так называемого
сырого бензола, содержащего бензол и его ближайшие гомологи.
Дополнительную часть сырого бензола извлекают из каменно-
угольной смолы.
Каменноугольная смола представляет собой вязкую темную
жидкость исключительно сложного состава. Считают, что она со-
держит около десяти тысяч соединений, но выделено и идентифи-
цировано около 500. Они составляют до 55% общей массы смолы
12]. Больше всего в смоле содержится нафталина (10%). Далее в
порядке убывания среднего содержания (в %) следуют [1]: Фе-
нантрен 5, флуорантен 3,3, пирен 2,1, аценафтилен 2,0, флуорен 2 0,
хризен 2,0, антрацен 1,8, 2-метилнафталин 1,5, карбазол 1,5, дифе-
ниленоксид 1,0, инден 1,0. Все остальные соединения содержатся в
каменноугольной смоле в количестве менее одного процента, а
ольшая часть из них — в концентрациях, измеряемых сотыми и
тысячными долями процента. Однако масштабы коксохимического
5
производства таковы, что и при таких концентрациях потенциаль-
ные ресурсы этих соединений весьма значительны. Если учесть, что
уже в 1973 г. объем добычи углей для коксования в СССР превы-
шал 170 млн. тонн, а при коксовании 1 гугля может быть получено
до 200 кг сырья для органического синтеза [2], то сотые доли
процента в масштабах страны составляют тысячи тонн в год. На-
пример, при содержании в сыром бензоле 1,3—1,4% тиофена и
выходе сырого бензола около 1% от массы угля потенциал тиофена
только по Украинской ССР достигает 5000 т/год [4]. На практике
лишь немногие соединения извлекаются из каменноугольной смо-
лы для промышленного применения.
Переработка смолы начинается с непрерывной перегонки с от-
бором первичных фракций, которые обычно подразделяют в по-
рядке возрастания температур перегонки [1] на масла: легкое
(0,5—3%), карболовое (2—3%), нафталиновое (10—20%), про-
мывное (7—8%), антраценовое (22—28%) и твердый остаток —
пек (50—55%). Дальнейшая переработка проводится путем ком-
бинирования методов ректификации, кристаллизации и экстракции
с химическими методами (высокотемпературная гидрогенизация,
комплексообразование и др.).
Из легкого масла после обработки раствором едкого натра для
удаления фенолов и экстракции оснований серной кислотой отде-
ляют перегонкой сырой бензол, который объединяют с выделенным
из коксового газа. После гидроочистки и разделения на компонен-
ты получают кроме бензола толуол, о-, м- я-ксилолы, этилбензол,
мезитилен. Экстрактивной ректификацией может быть выделен
также тиофен [4]. Из сернокислотного экстракта обработкой ам-
миаком или щелочью выделяют смесь пиридиновых оснований,
после азеотропного обезвоживания и перегонки которой получают
чистые пиридин, пиколины и более высоко кипящие основания.
Из карболового масла непрерывной экстракцией раствором ед-
кого натра извлекают соли фенолов, которые переводят в свобод-
ные кислоты действием угольного ангидрида и перегоняют, соби-
рая чистые фенол, о-крезол и смесь м- и я-крезолов, которую
разделяют специальными методами. Из высококипящих фракций
могут быть выделены 3,4-, 3,5-диметилфенолы и другие ксиленолы.
Из нафталинового масла кристаллизацией отделяют техниче-
ский нафталин, для дальнейшей очистки которого, главным обра-
зом от примеси тионафтена, достигающей 2%, приходится прибе-
гать к химическим методам или к азеотропной дистилляции.
Промывное масло может быть разделено перегонкой на от-
дельные фракции, которые располагаются по увеличению темпера-
туры перегонки в такой последовательности: метилнафталиновая,
дифенильная, диметилнафталиновая, дифениленоксидная, аценаф-
теновая и флуореновая. Из всех этих фракций указанные состав-
ляющие могут быть получены в чистом виде. В случае необходи-
мости метилнафталиновая фракция может быть превращена
гидродезалкилированием в нафталин, а диметилнафталиновая, со-
держащая 9 из 10 существующих изомеров,—переведена в
6
2 б-диметилнафталин. Из аценафтеновой фракции выделяют аце-
нафтен в количестве, более чем вдвое превышающем его первона-
чальное содержание в смоле, так как при перегонке происходит
гидрирование аценафтилена. Из дифениленоксидной фракции
сравнительно сложным путем извлекают также флуорен. Экстрак-
цией промывного масла или его головных фракций раствором
сернбй кислоты с последующей обработкой аммиаком или ще-
лочью изолируют смесь хинолиновых оснований, из которой удает-
ся выделить чистые хинолин, изохинолин, хинальдин и лепидин.
Антраценовое масло собирают обычно в виде двух фракций.
Охлаждением и центрифугированием первой получают сырой ант-
рацен, содержащий 20—35% антрацена, 30—40% фенантрена и
10—20% карбазола, разделяемых кристаллизацией из растворите-
лей, азеотропной перегонкой, обработкой серной кислотой и други-
ми методами. При экстракции антраценового масла раствором
бисульфита натрия извлекают акридин в виде сернистокислой со-
ли, которую разлагают действием щелочи. Из высококипящей
фракции антраценового масла при повторной перегонке получают
фенил нафталиновую, флуорантеновую, пиреновую, бензофлуоре-
новую и хризеновую фракции, после кристаллизации которых вы-
деляют в чистом виде соответствующие соединения.
Пек представляет собой твердую многокомпонентную смесь вы-
сокомолекулярных ароматических и гетероциклических соедине-
ний, в большинстве своем неустановленного строения. При нагре-
вании для получения остатка с более высокой температурой раз-
мягчения, используемого для получения пекового кокса и для
изготовления электродов, отгоняется пековый дистиллят, который
содержит те же углеводороды, что и вторая фракция антрацено-
вого масла, а кроме того бензофлуорантен, бензопирен и пицен. Из
пека могут быть выделены и наиболее высококипящие соедине-
ния — коронен, нафтохризен, бензопицен.
Длительное время коксохимическая промышленность была мо-
нопольным поставщиком ароматического сырья и до сих пор про-
должает оставаться единственным производителем большинства
полициклических ароматических соединений, хотя из смолы извле-
кается не более 0,1% их общего количества. В производстве бензо-
ла и его гомологов, а также значительной части нафталина на
первое место выдвинулась нефтехимическая промышленность.
В США она дает более 90% бензола, 97% толуола, 99% ксилолов,
более 40% нафталина [3].
Наиболее важным сырьем для получения бензола и его гомо-
логов из нефти являются фракции, образующиеся при термическом
или каталитическом риформинге. При каталитическом риформинге,
осуществляемом на платиновом или более сложном катализаторе
при 500—525 °C, происходят реакции изомеризации нормальных
алканов в изоалканы, алкилциклопентанов в циклогексаны, дегид-
рирование циклогексанов и дегидроциклизация алканов в арома-
тические углеводороды и др. [5, с. 41—77]. Бензол, толуол и
ксилолы могут быть выделены из легких фракций риформинга
7
экстрактивной перегонкой с последующей ректификацией. Фракций
Се, из которых извлекают изомерные ксилолы, содержат в среднем
20—24% о-ксилола, 42—48% л^-ксилола, 1—20% /г-ксилола и 10—
11% этилбензола. Толуол и ксилолы могут быть превращены с
высоким выходом и более дефицитный бензол каталитическим или
термическим гидродезалкилированием. Важным источником полу-
чения бензола и его гомологов кроме продуктов риформинга явля-
ются жидкие продукты пиролиза, образующиеся при кратковре-
менном крекинге нефти с целью получения этилена. На каждые
1000 кг этилена приходится 600—900 кг жидких продуктов с со-
держанием бензола в пределах 20—40%, толуола 15—20%, ксило-
лов 10—15%.
С начала 1960-х годов из нефти получают во все больших
масштабах также и нафталин. Исходными для его производства
служат высококипящие фракции риформинга, газойль каталитиче-
ского крекинга и побочные продукты пиролиза в производстве
этилена. Смеси, содержащие нафталин и его гомологи, подвергают
гидродезалкилированию с последующей очисткой и перегонкой на-
фталина. Согласно прогнозам [3], тенденция к увеличению доли
нефти в производстве ароматического сырья в ближайшие десяти-
летия, по-видимому, сохранится.
1.2. СТРОЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
1.2.1. Ранние представления
Уже для первых полученных ароматических соединений
были обнаружены особенности химического поведения, которые не
укладывались в правила структурной теории, выведенные приме-
нительно к соединениям алифатического ряда. Ароматические со-
единения содержали меньше водорода, чем требовали правила
валентности, и в то же время не обнаруживали характерной для
ненасыщенных соединений способности к реакциям присоединения.
В 1865 г. Кекуле предложил решение, устраняющее это противоре-
чие, допустив, что все ароматические соединения содержат по
крайней мере одно кольцо из шести атомов углерода. Позднее он
дополнил, свою гипотезу предположением о том, что каждый атом
углерода такого кольца, сохраняя по единице сродства, связан с
соседними атомами простой и двойной связями, которые быстро
меняются местами — осциллируют, обусловливая равноценность
атомов углерода (1). Простейшим из соединений ароматического
ряда и его родоначальником был признан бензол, до сих пор
остающийся наиболее ярким примером ароматического соедине-
ния:
О)
8
Вслед за формулой Кекуле (1) для бензола было предложено
несколько десятков разнообразных структур [6]< Из них заслужи-
вают упоминания сегодня так называемая формула Дьюара (2) *,
формула Ладенбурга (3) (призман) и формула Хюккеля (4) (бенз-
вален), поскольку сравнительно недавно, примерно через 100 лет
после того, как эти формулы были выдвинуты на основании чисто
умозрительных соображений, синтезированы соответствующие ре-
альные валентные изомеры бензола [8,9].
Ближе всех из доэлектронных представлений о строении аро-
матических соединений к современным воззрениям гипотеза Тиле,
согласно которой кратные связи имеют остаточные (парциальные)
валентности, определяющие способность сопряженных систем к
реакциям 1,4-присоединения, но взаимно насыщающиеся в бензоле
(5). Однако гипотеза Тиле не могла объяснить неароматичность
циклооктатетраена и неудачи попыток синтеза циклобутадиена,
отчего была вытеснена представлениями о центрических формулах
ароматических соединений (6), дополненными высказанным еще
Бамбергером признанием необходимости наличия непременно ше-
сти «потенциальных валентностей» для появления ароматической
устойчивости. После создания электронной теории валентности это
положение было сформулировано Робинсоном, как необходимость
ароматического секстета электронов, который было предложено
изображать кругом, вписанным в шестиугольник (7). Физическое
обоснование правила секстета для ароматических систем было
найдено позднее при пересмотре электронной теории валентности
с позиций квантовой механики.
7 (5) <6) <7>
1.2.2. Квантовомеханическая трактовка
К настоящему времени физическими методами установ-
лено, что молекула бензола представляет собой плоский правиль-
ный шестиугольник с внутренними углами 120° и расстояниями
* Дьюар никогда не предлагал структуру (2) в качестве альтернативы фор-
муле Кекуле, представления которого он поддерживал, в том числе и в статье,
гДе упоминал о формуле (2) [6, 7].
9
между атомами углерода 1,397 А, т. е. 0,1397 нм (см. 1.3.2). Со-
гласно атомно-орбитальной модели атомы углерода бензольного
кольца рассматриваются как находящиеся в состоянии ^-гибри-
дизации, при котором один s- и два р-электрона из четырех валент-
ных электронов атома углерода находятся на трех равноценных
гибридных орбиталях с копланарным расположением осей под уг-
лом 120°, а один электрон — на негибридизованной р-орбитали,
ориентированной перпендикулярно плоскости остальных. При та-
ком способе рассмотрения считают, что в бензоле за счет сг-пере-
крывания гибридных орбиталей, при котором направления осей
атомных орбиталей и образуемой связи совпадают, формируется
шесть о-связей углерод-углерод и шесть связей углерод-водород,
а за счет «бокового» перекрывания соседних р-орбиталей, заня-
тых электронами с антипараллельными спинами, образуется еди-
ная л-электронная система, которая и обеспечивает дополнитель-
ную энергетическую устойчивость молекулы.
Квантовая механика дает объяснение происхождению этой до-
полнительной энергии стабилизации с помощью молекулярных ор-
биталей (МО) [10, 11]. В простом методе МО, впервые применен-
ном Хюккелем (МОХ), все л-электроны сопряженной системы опи-
сываются как находящиеся на общемолекулярных орбиталях,
охватывающих все атомы, входящие в состав системы. Уровень
энергии электрона на молекулярной орбитали 8/ определяется
уравнением:
е, = 2₽ cos (-^-) (1.1)
где N — число атомов системы; /— квантовое число, которое мо-
жет принимать любое из значений / = 0, ±1, ±2, ...; р—резо-
нансный интеграл, характеризующий перекрывание 2рл-орбиталей
соседних атомов углерода [12].
Уровень энергии электрона на молекулярной орбитали может
быть меньше, равен или больше уровня энергии на атомной орби-
тали, в соответствии с чем молекулярные орбитали называют свя-
зывающими, несвязывающими или разрыхляющими. На каждой
молекулярной орбитали, согласно принципу Паули, может нахо-
диться не более двух электронов с антипараллельными спинами.
В 1931 г. Хюккель предположил, исходя из аналогии с элект-
ронной оболочкой инертных газов, что образование в циклической
системе «замкнутой» л-электронной оболочки, в которой все
л-электроны полностью занимают связывающие орбитали, должно
приводить к повышению устойчивости системы. В общем виде для
моноциклических плоских систем, состоящих из 5/?2-гибридизован-
ных атомов углерода, это условие выполняется при наличии 4п +
4- 2л-электронов (где и — ноль или любое целое число), поскольку
все низшие молекулярные орбитали, как следует из уравнения (1.1),
за исключением орбитали с наименьшей энергией (/=0), являют-
ся дважды вырожденными, т. е. попарно имеют одинаковую энер-
гию, и каждая заселяется двумя электронами,
10
(a)
(б)
(б)
Рис. 1Л. Диаграммы энергетических уровней молекулярных орбиталей (о численном значе-
нии резонансного интеграла |3 см. 1.3.1):
а —бензол (7); б — циклопентадиенид-анион (8); в — циклогептатриенил-катион (9).
Решение уравнения (1.1) для циклов с числом атомов углерода
/V = 5, 6 и 7 указывает на существование для каждого из них трех
связывающих орбиталей, для заполнения которых необходимо
шесть л-электронов (рис. 1.1). Таким образом, правило секстета
электронов, эмпирически найденное для бензоидных систем и но-
сившее первоначально формальный характер, вытекает как част-
ный вывод из теории МОХ. Согласно этой теории, ионы С5Н5 и
С7Н* подобно бензолу должны обладать замкнутыми электронными
оболочками и, следовательно, ароматической стабильностью. Дей-
ствительно, щиклопентадиенид-анион (8), впервые описанный еще
Тиле, известен своей устойчивостью [14], которая служит причиной
высокой кислотности циклопентадиена. Ароматичность циклогеп-
татриенил-катиона (9), названного тропилием, была предсказана
Хюккелем и подтвердилась в результате идентификации и доказа-
тельства электронной структуры тропилийбромида [13].
(7) <8) (9)
Простейшей ароматической системой, содержащей, согласно
правилу Хюккеля, два л-электрона на единственной связывающей
орбитали (п = 0), может быть однозарядный циклопропенил-ка-
тион (10) и двузарядный циклобутенил-дикатион (11). Соли цик-
лопропенил-катиона [15, 16] хорошо изучены и отличаются арома-
тической стабильностью. Соли циклобутенил-дикатиона получены
Для его фенил- и метилзамещенных, охарактеризованных в рас-
творах ЯМР-спектрами, свидетельствующими об ароматичности
[17]. В качестве кислородсодержащего ароматического производ-
ного циклобутенил-дикатиона рассматривается дианион «квадрат-
ной>,кислоты, строение которого может быть представлено форму-
лой (12) [16].
В отличие от циклобутенил-дикатиона (11) незаряженная мо-
лекула циклобутадиена не имеет замкнутой электронной оболочки,
и
так как два л-электрона из четырех находятся неспаренными на
двух несвязывающих орбиталях. Это объясняет, почему, несмотря
на многочисленные попытки, ни одно из производных циклобута-
диена из-за неустойчивости не удалось выделить в свободном
состоянии [18].
(10)
(11)
Аналогичным образом объясняется причина неароматичности
циклооктатетраена, в молекуле которого, если бы она имела строе-
ние правильного плоского восьмиугольника, два электрона из
восьми занимали бы две незаполненные орбитали. Отсутствие вы-
игрыша энергии от делокализации л-электронов приводит к тому,
что циклооктатетраен представляет собой типичный полиен (13) с
некопланарным расположением атомов углерода [19]. В то же
время циклооктатетраенил-дикатион (14), соли метил- и фенил-
производных которого получены окислением соответствующих цик-
лооктатетраенов в среде SbF5 — SO2C1F, является, как следует из
спектров ЯМР, бл-электронной ароматической системой [20]. Цик-
лооктатетраенид-дианион (15), образующийся при двухэлектрон-
ном восстановлении циклооктатетраена, представляет собой пло-
скую ароматическую систему, которая имеет отвечающую правилу
Хюккеля замкнутую оболочку из десяти л-электронов (и = 2)
[21]. Другим примером 10л-электронной моноциклической арома-
тической системы может служит циклононатетраенид-анион (16)
[22]. Обзор, посвященный карбоциклическим ароматическим
ионам, см. [23].
(16)
Убедительные свидетельства в пользу правила Хюккеля полу-
чены при изучении высших моноциклических полиенов CmHm, на-
зываемых аннуленами [24]. При рассмотрении свойств аннуленов
прослеживается четкое различие между [4п + 2] аннуленами и
[4п] аннуленами, из которых только первые являются ароматиче-
скими соединениями. Наглядно это различие можно проиллюстри-
ровать сравнением свойств [18]аннулена (17) и [16] аннулена (18)
при п = 4. В углеводороде (17) все атомы углерода лежат в одной
плоскости, нет альтернирования связей и обнаруживается диамаг-
12
нитный кольцевой ток, тогда как в углеводороде (18) атомы угле-
рода значительно отклоняются от плоскости, наблюдается чередо-
вание связей повышенной и пониженной кратности и отсутствует
кольцевой ток.
(17>
<18>
Таким образом, в ряду моноциклических полиенов теория МОХ
и правило 4и + 2 полностью подтверждается.
1Л2,3. Полициклические соединения
Правило Хюккеля о существовании в циклической систе-
ме замкнутой электронной оболочки при числе л-электро-
нов 4п + 2 выведено и строго применимо только для моноцикличе-
ских соединений. На полициклические конденсированные системы
оно может быть перенесено при допущении [25], что общие для
двух циклов связи не вносят решающих возмущений в л-электрон-
ную структуру по сравнению с соответствующими аннуленами, а
лишь обеспечивают необходимую копланарность. При таком под-
ходе ароматические бициклические углеводороды рассматривают-
ся как электронные аналоги [10]аннулена (циклодекапентаена) с
оболочкой из десяти л-электронов, отвечающей правилу Хюккеля
(ц = 2). Сам [10] аннулен (19) нестабилен [26] из-за некопланар-
ности вследствие стерических препятствий между атомами водо-
рода в положениях 1 и 6, но уже замена двух атомов водорода
метиленовым мостиком приводит к почти плоской молекуле
1,6-метано[10] аннулена (20) [271 или к 1,5-метано[10]аннулену
(21) [28], обладающим ароматическими свойствами. Непосред-
ственная связь между атомами в положениях 1 и 6 или 1 и 5 в
[10] аннулене (19) еще более жестко, чем метиленовый (метано)
мостик, закрепляет плоскую конфигурацию молекулы, создавая
благоприятные условия для образования ароматического децета
электронов в нафталине (22) и азулене (23) соответственно. В то
время как 1,6-метано[10]аннулен (20) и нафталин (22) бесцветны,
1,5-метано[10]аннулен (21) окрашен в оранжевый, а азулен — в
синий цвет. Существенный вклад в электронное строение азулена
13
вносит биполярная структура (236), представляющая собой соче*
тание ядер циклопентаенид-аниона и тропилий-катиона [29].
(19)
(21)
(23б)
Из трициклических систем ароматическими оказываются ана-
логи [14] аннулена, такие как антрацен (24) и фенантрен (25). Сам
[14]аннулен, как и [10]аннулен (19), некопланарен из-за про-
странственных затруднений, но его производные, в которых пло-
ская структура жестко закреплена с помощью мостиков [30], как
в (26), или стяжек [31], как в 10b, 10с-дигидропирене (27), обла-
дают ароматическими свойствами.
(25)
Юс
(27)
Рассмотрение всей совокупности накопленных эксперименталь-
ных данных показывает, что эмпирическое распространение прави-
ла Хюккеля оправдано применительно к любым плоским конден-
сированным системам, у которых нет атомов, общих более чем для
двух циклов [25]. Однако для соединений, имеющих атомы, общие
для трех циклов, правило Хюккеля неприменимо. Для суждения о
наличии или отсутствии в них замкнутой л-электронной оболочки
необходим в каждом случае специальный анализ. Примером аро-
матической системы, не подчиняющейся правилу Хюккеля, может
служить феналенил-катион [32]. Сам фенален (28) неароматичен,
но соответствующий ему феналенил-катион (29) представляет со-
бой ароматическую частицу, имеющую согласно данным расчета по
методу МОХ [33] шесть полностью заполненных связывающих ор-
биталей, образующих замкнутую оболочку с 12 л-электронами,
шесть разрыхляющих орбиталей и одну вакантную несвязываю-
щую орбиталь. В феналенильном радикале (30) и феналенид-
анионе (31) она занята одним или двумя электронами соответ-
ственно, а так как энергия несвязывающей орбитали равна нулю,
14
радикал и анион подобно феналенил-катиону обладают необычно
высокой термодинамической стабильностью.
(28)
(29)
(30)
(31)
Тенденция к образованию систем типа (29) — (31) имеет важ-
ное значение в химии кетонов — феналенона и его бензолога —
бензантрона [34], широко используемого в синтезе красителей.
Другими примерами полициклических ароматических углеводо-
родов с числом л-электронов, не соответствующим формуле Хюк-
келя 4и 4-2, являются пирен (32) (16 электронов) и перилен (33)
(20 электронов).
(32)
(33)
1.2.4. Гетероциклические соединения
Все многообразие ароматических гетероциклических со-
единений может быть формально произведено из карбоцикличе-
ских соединений путем замены одной СН-группы на гетероатом,
способный поставлять в ароматическую систему один электрон,
находящийся на р-орбитали, аксиальной к плоскости цикла, или же
путем замены двух рядом стоящих СН-групп на гетероатом, имею-
щий на подобной орбитали пару электронов.
К гетероатомам и группам, изоэлектронным группе =СН—,
+ + +
относятся =N—, =NR—, =0—, —S—, а также =SR—, =Р— и
некоторые другие, замена которыми СН-группы в молекуле бен-
зола приводит к шестичленным гетероциклам: пиридину (34а),
ионам пиридиния (346), пирилия (34в), тиопирилия (34г), тиабен-
золам (34д) [35], фосфорину (34е) [36] и т. д. соответственно.
Замена двух СН-групп на атомы азота в зависимости от их
взаимного расположения ведет к одному из азинов: пиридазину
(35), пиримидину (36) или пиразину (37), а замена трех
СН-групп — к триазинам (38)—(40). Вместо атомов азота в
15
структурах типа (35) — (40) могут
разные другие гетероатомы Y.
находиться одинаковые или
(38)
Группировку —СН=СН— способны заменить гетероатомы
—NH—, —NR—-, —О—, —S—, —Se—. Такая замена в бензоле
приводит к пятичленным гетероциклам: пирролу (41а), ^заме-
щенным пирролам (416), фурану (41 в), тиофену (41г), селенофену
(41Д) соответственно. Второй гетероатом этого типа не может быть
введен без потери ароматичности, но дальнейшая замена одной или
нескольких СН-групп «пиридиновыми» атомами азота =N— при-
водит к азолам — пятичленным ароматическим соединениям с
двумя (42) и (43), тремя (44)—(47) и более гетероатомами.
(41)
a) X = NH
б) X = NR
в) X—О
Аналогичные замены возможны во всех ароматических поли-
циклических соединениях, причем в числе прочих могут быть заме-
нены и атомы углерода, общие для двух циклов, как например при
переходе от нафталина к индолизину (48). В отдельных случаях
возможна замена атома углерода, общего для трех циклов. Так,
нейтральная молекула (49), содержащая в центре атом бора,
рассматривается как электронный аналог феналенил-катиона (29),
а молекула (50) — как аналог феналенид-аниона (31) [25].
(48)
Поскольку в полициклических системах на гетероатомы могут
быть заменены атомы углерода разных циклов и в разных комби-
нациях, число возможных сочетаний исключительно велико. Число
известных гетероциклических ароматических соединений во много
раз превосходит число карбоциклических.
Хотя введение гетероатома в карбоциклическую ароматическую
систему не нарушает замкнутости электронной оболочки, оно ска-
зывается, и обычно весьма значительно, на распределении элект-
ронной плотности. Характер влияния одного и того же гетероатома
различается в зависимости от того, один или два электрона по-
ставляет он в ароматическую систему. В первом случае — (34) —
гетероатом оказывает на атомы углерода электроноакцепторное
влияние, во втором—(41) — повышает электронную плотность на
них. Различия в электронном состоянии гетероатома сказываются
на кислотно-основных свойствах соединений. Например, пиридин, в
котором неподеленная пара электронов атома азота не взаимодей-
ствует с ароматическим секстетом (она находится на <$р2-гибридной
орбитали в плоскости кольца), обладает ярко выраженными ос-
новными свойствами; пиррол же, в котором неподеленная пара
электронов атома азота (занимающая р-орбиталь) вовлечена в
ароматический секстет, имеет очень слабую основность, но прояв-
ляет кислые свойства. Количественная оценка вклада гетероатомов
в электронную структуру ароматических гетероциклических соеди-
нений представляет собой сложную задачу, не решенную еще
окончательно квантовой химией.
1.2.5. Определение понятия ароматичности
Современные представления о строении ароматических
соединений можно резюмировать следующим определением, сфор-
мулированным Бэджером [37, с. 37]: «Ненасыщенная цикличе-
ская или полициклическая молекула или ион (а также часть моле-
кулы или иона) может рассматриваться как ароматическая, если
все атомы цикла входят в сопряженную систему таким образом,
что в основном состоянии все л-электроны (вступившие в эту си-
стему с атомных орбиталей, ориентированных аксиально к плоско-
сти цикла) находятся на связывающих молекулярных орбиталях
замкнутой (циклической) электронной оболочки».
1.3. КРИТЕРИИ АРОМАТИЧНОСТИ
Согласно приведенному выше определению, ароматич-
ность есть особая структура л-электронной оболочки молекулы,
е. свойство вещества, которое не может быть подвергнуто непо-
и^средственному наблюдению. Поэтому особую важность приобрета-
ет вопрос о выборе критериев, которые бы позволяли судить,
Сселяется ли данное соединение ароматическим, и измерять степень
его ароматичности [38,39]. В качестве критериев в соответствии с
определением должны использоваться показатели, описывающие
основное состояние молекулы. Реакционная способность, кото-
рая была первым и широко применявшимся критерием ароматич-
ности, не удовлетворяет данному требованию, поскольку зависит
как от основного, так и от переходного состояния (см. 2.2). Этого
недостатка лишены физические критерии, которые рассматри-
ваются ниже.
1.3.1. Энергетические критерии
Сама возможность существования ароматических со-
единений обусловлена тем, что образование замкнутой л-электрон-
ной оболочки приводит к повышению термодинамической устойчи-
вости молекулы, т. е. к понижению уровня ее внутренней энергии.
Выигрыш энергии за счет делокализации л-электронов является
необходимым и достаточным условием ароматичности и может
служить надежным критерием при наличии надежных методов
его определения. Сложность использования энергетических крите-
риев заключается как раз в несовершенстве этих методов. Главная
Таблица 1.1
Энергии резонанса (ЭР) и энергии делокализации (ЭД)
Соединение Эмпирическая ЭР, * эВ ЭД на л-элек- трон, ** р ЭР на л-электрон , "1 1 * 1 — МО ССП, 3* эВ МОХ, ** р
Бензол (7) 1,56 0,333 0,145 0,065 Нафталин (22) 2,64 0,368 0,132 0,055 Антрацен (24) 3,62 0,380 0,114 0,047 Фенантрен (25) 3,96 0,389 0,138 0,055 Тетрацен 4,77 0,385 0,101 0,042 Пентацен — ),388 0,091 0,038 Пирен (32) 4,72 0,407 0,131 0,051 Перилен (33) 5,48 0,404 0,131 0,056 Азулен (23) 1,30 0,336 0,017 0,023 Фульфен (51) — 0,244 0,008 -0,002 Циклобутадиен — 0,000 —0,193 —0,268 Бензоциклобутадиен (52) — 0,298 0,054 —0,027 Пентален (53) — 0,307 0,001 —0,018 Пиридин (34а) 1,00 — 0,151 — Пиррол (41а) 0,92 — 0,062 0,039 Фуран (41 в) 0,68 — 0,011 0,007 Тиофен (41г) 0,89 — 0,047 0,032 Хинолин 2,05 — 0,143 — Индол 2,04 — 0,103 0,047 Карбазол 3,42 — 0,127 0,051
* См. [10,40].
** МОХ — метод молекулярных орбиталей Хюккеля, см. [42] и работы тех же авторов
перечисленные в [43].
3* МО ССП —метод молекулярных орбиталей в самосогласованном поле Паризера—- Пар-
ра— Попла, см. [39, 44].
18
Рис. 1.2. Схема соотношения между эмпирической и верти-
кальной ЭР.
трудность состоит в том, что энергию ре-
альной ароматической молекулы приходит-
ся сопоставлять с энергией гипотетической
молекулы, имеющей локализованные про-
стые и двойные связи.
Разность энергий ароматического и со-
ответствующего ему циклического соедине-
ния с фиксированными двойными связями
носит название эмпирической энергии резо-
нанса (ЭР). Ее находят сравнением тепло-
ты сгорания или теплоты гидрирования ароматического соедине-
ния, определяемых экспериментально, с соответствующей величи-
ной для модельного соединения, которая вычисляется по аддитив-
ной схеме как сумма вкладов отдельных связей. Для бензола,
модельным соединением для которого служит 1,3,5-циклогекса-
триен, эмпирическая ЭР составляет около 1,56 эВ, значения для
некоторых других соединений, определенные из теплот сгорания,
приведены в табл. 1.1. Экспериментальное определение теплот
сгорания выполняется в среднем с ошибкой около 0,02 эВ и усту-
пает по точности определению теплот гидрирования, но зато яв-
ляется более универсальным, так как исчерпывающее гидрирова-
ние для многих соединений неосуществимо. Основной источник
ошибок при определении эмпирической ЭР заключается не в несо-
вершенстве методов термохимического эксперимента, а в сложно-
сти учета всех факторов при вычислении энергий атомизации или
гидрирования модельного соединения. Так, в зависимости от ис-
пользования при расчете теплот сгорания усредненных вкладов
связей или вкладов связей с учетом влияния окружения эмпириче-
ская ЭР для нафталина колеблется от 2,90 до 2,64 эВ, а для ан-
трацена от 4,29 до 3,73 эВ [16].
Еще больший элемент неопределенности вносится вследствие
различий геометрических параметров ароматического и модельного
соединений. Для перехода от 1,3,5-циклогексатриена к бензолу
двойные связи должны быть растянуты, а простые — сжаты.
Необходимая для такой деформации энергия по одним оценкам
составляет 1,17 эВ, а по другим —1,60 эВ [10, с. 232]. Для пяти-
и семичленных ароматических соединений кроме энергии деформа-
ции связей необходимо также учитывать энергию напряжения за
счет искажения углов.
Разность энергий реального ароматического соединения и гипо-
тетической модели с локализованными связями, но таким же рас-
положением атомов носит название вертикальной ЭР. Соотношение
между эмпирической и вертикальной ЭР поясняется для случая
бензола на рис. 1.2. Как видно из рисунка, вертикальная ЭР
больше эмпирической на величину энергии деформации, что для
бензола с учетом указанных выше значений составляет 2,73 или
19
4
3,16 эВ. В теоретическом плане эта величина важнее, так как
именно она соответствует энергии делокализации, обеспечивающей
стабильность ароматической молекулы.
Существенная неточность в оценке ЭР возникает из-за допуще-
ния, что энергия простой связи между двумя атомами углерода с
гибридизацией sp3, которая используется при вычислении энергий
модельных соединений, равна энергии связи двух атомов углеро-
да с гибридизацией sp2, существование которых в модельных со-
единениях предполагается. При внесении же поправок на перегиб-
ридизацию атомов углерода в работе [41] для вертикальной ЭР
бензола получено значение всего 0,56 эВ. Аналогичная ситуация
имеет место и в случае вычисления ЭР из теплот гидрогенизации в
результате игнорирования возможной разности энергий связей
С^рг Н и Csp3 Н.
Расчетной величиной, которая должна соответствовать верти-
кальной ЭР, является энергия делокализации (ЭД), представ-
ляющая собой разность между вычисленной методом МОХ л-энер-
гией системы и л-энергией двойных связей, изображаемых в клас-
сической формуле; вклад каждой из них приравнивается к л-энер-
гии молекулы этилена (2(3). Бензол, например, обладает л-энер-
гией, вычисленной методом МОХ, равной 8(3, циклогексатриен —
л-энергией трех молекул этилена, равной 6(3, и, следовательно, ЭД
для бензола составляет 2р. Значения ЭД для некоторых цикличе-
ских углеводородов приведены в табл. 1.1.
Резонансный интеграл р, в единицах которого выражают ЭД,
представляет собой математический параметр, применяемый прй
расчете методом МОХ любой системы, а не специфической мефой
ЭД. Численное значение р зависит от характера корреляции, вы-
бранной для его определения. Из корреляции между ЭД и эмпи-
рической ЭР для бензоидных углеводородов найдено значение
3 « 0,7 эВ [10], для «термодинамического» значения р, свободного
от вкладов энергий деформации и регибридизации о-связей, кото-
рые несет эмпирическая ЭР, предложена величина (3 == 1,29 эВ
[45], а из корреляции между теплотами атомизации и ЭР на
л-электрон, вычисленной методом МОХ (см. ниже), получено
Р = 1,42 эВ [43].
Так как общая ЭД зависит от числа л-электронов системы, то
для сравнения систем с разным числом л-электронов удобнее
пользоваться ЭД на один л-электрон (ее называют также удельной
ЭД). При сравнении значений ЭД на л-электрон, представленных
в табл. 1.1, отчетливо видно несоответствие фактическим данным о
стабильности соединений. Так, по величине ЭД на л-электрон бен-
зол уступает полициклическим углеводородам, в том числе поли-
аценам (антрацен, тетрацен, пентацен), причем у высших полиаце-
нов значения ЭД на л-электрон выше, чем у низших, хотя, судя по
термической устойчивости и свойствам, именно бензол должен
возглавлять ряд ароматичности, а высшие полиацены по арома-
тичности должны уступать низшим (ср. 2.4.2). Бензоциклобута-
диен (52) и пентален (53) по величине ЭД на л-электрон превосхо-
20
дят фульвен (51), хотя из них только последний существует в сво-
бодном состоянии. Исходя из значений ЭД на л-электрон, следо-
вало бы ожидать, что все аннулены, кроме циклобутадиена, а не
только отвечающие правилу Хюккеля являются ароматическими.
(51) (52) (53)
Учитывая неприменимость ЭД в качестве критерия ароматич-
ности и грубые приближения, к которым приходится прибегать при
расчете энергий гипотетических циклических полиенов как объек-
тов сравнения, Дьюар предложил заменить их реальными полиена-
ми с открытой цепью [46]. Для расчета теплот атомизации (теп-
лот, необходимых для фрагментации на отдельные атомы) сопря-
женных циклических систем Дьюар использовал метод МО ССП,
а для расчета теплот атомизации ациклических полиенов — сумми-
рование энергий двойных и простых углерод-углеродных связей,
постоянных для всех классических полиенов [44, 47]. Разность теп-
лот атомизации исследуемого сопряженного соединения и соответ-
ствующего классического полиена называют ЭР Дьюара. Деление
на число л-электронов дает ЭР на л-электрон, обозначаемую в ла-
тинской транскрипции REPE (resonance energy per electron).
Этот параметр гораздо лучше, чем ЭД на л-электрон, согласу-
ется с фактическим материалом (см. табл. 1.1): ЭР на л-электрон,
вычисленная с применением метода МО ССП, имеет наивысшее
значение для бензола, понижается при переходе от низших поли-
аценов к высшим, для фульвена и пенталена приближается к нулю,
а для циклобутадиена приобретает отрицательное значение. Ано-
мальными выглядят эти показатели для бензоциклобутадиена, а
также для азулена и фурана: более высокие и более низкие, чем
следует ожидать, исходя из неудачных попыток синтеза первого и
достаточно высокой стабильности вторых.
Формулируя новый критерий ароматичности, Дьюар отказался
одновременно и от гипотетических циклополиенов в качестве моде-
лей для сравнения, и от метода МОХ для расчета энергий, выска-
зав мнение о нецелесообразности его дальнейшего использования в
органической химии [44]. Однако принципиальным моментом при
переходе от ЭД к ЭР Дьюара является не изменение метода рас-
чета, а смена моделей. Показано [43], что при соотнесении
я-энергии сопряженной циклической системы, рассчитанной мето-
дом МОХ, с л-энергией соответствующего ациклического полиена,
полученной по аддитивной схеме, наблюдается такое же удовле-
творительное согласие с фактическими данными, как и при исполь-
зовании метода МО ССП. Из табл. 1.1 видно, что ЭР на л-электрон,
найденные с применением методов МО ССП и МОХ, в целом
изменяются симбатно, причем в последнем случае значения для
азулена и бензоциклобутадиена более приемлемы.
ЭР на л-электрон представляет собой расчетный энергетический
индекс ароматичности, который позволяет провести грань между
21
ароматическими п прочими соединениями, а также построить еди-
ную шкалу ароматичности. Ароматическими считаются те структу-
ры^ которых циклическая делокализация электронов обеспечивает
выигрыш энергии и для которых ЭР положительна, неаромати-
ческими — те, для которых она равна нулю, и антиароматически-
ми — те, для которых делокализация л-электронов должна вызы-
вать дестабилизацию и ЭР имеет отрицательное значение. Чем
больше выигрыш энергии, т. е. чем больше ЭР на л-электрон, тем
более ароматическим в указанном смысле может считаться соеди-
нение.
Для суждения о надежности этого критерия необходимо знать,
насколько точно принятая методика расчета отражает разность
энергий и насколько ЭР на л-электрон соответствует степени аро-
матичности.
На первый вопрос дает ответ сопоставление вычисленных теп-
лот атомизации с найденными из термохимического эксперимента.
Оно показывает, что ошибка расчета не превышает 0,1%, будучи
близка к ошибке эксперимента, а погрешность при вычислении ЭР
на л-электрон методом МОХ составляет ±0,004(3 [43, 44]. При
наличии термохимических данных ЭР Дьюара может определяться
на экспериментальной основе [48], чему пока препятствует отсут-
ствие достаточных измерений для ациклических полиенов.
Гораздо сложнее с ответом на второй вопрос, который требует
сопоставления с независимыми экспериментальными критериями
ароматичности. Если расчет ЭР на л-электрон дает непрерывную
шкалу, способную, в принципе, охватить все ароматические соеди-
нения, экспериментального количественного критерия с подобной
широтой охвата не существует. Рассматриваемые ниже структур-
ные (см. 1.3.2) и магнитные (см. 1.3.3) критерии, а также реакци-
онная способность (см. 2.2) применимы для количественной оценки
ароматичности лишь в узких пределах. Поэтому сопоставление ЭР
на л-электрон с данными физического и химического эксперимента
ограничено отдельными группами близких по строению соедине-
ний. Так, отличная корреляция отмечена недавно между ЭР на
л-электрон и скоростями образования макроциклических аннуле-
нов при реакции Дильса — Альдера для дегидроаннулено [с] фура-
нов с малеиновым ангидридом [49]. Кроме того, остается неясным,
как сопоставлять количественно степень ароматичности, измеряе-
мую ЭР на л-электрон в единицах энергии, со структурными или
магнитными индексами, поскольку нет никаких оснований ожидать
между ними линейной зависимости.
В условиях отсутствия общего экспериментального критерия
ароматичности становится актуальной задача разработки простых
методов оценки ЭР на л-электрон, не связанных с трудоемкими
расчетами и компьютерами. В этом плане внимание привлекают
формулы ароматических соединений с выделением бензоидных
секстетов. При написании таких формул обозначают кружком —
символом секстета л-электронов (см. 1.2.1)—кольца, в которых
22
подобно структуре Кекуле для бензола могут быть по три сопря-
женных двойных связи, например:
В остальной части молекулы различают кольца, которые спо-
собны «разбавлять» секстет за счет перехода к ним двух л-элект-
ронов (показано стрелкой), как в нафталине (22) и антрацене (24),
кольца с фиксированными двойными связями, как два центральных
кольца в цетрене (54), или «пустые» кольца, как центральное коль-
цо в перилене (33). Такое написание позволяет, как указал Клар
[50], интерпретировать многие свойства ароматических углеводо-
родов, принимая в качестве постулата, что при прочих равных ус-
ловиях соединение тем ароматичнее, чем больше в его формуле
секстетов и чем меньше эти секстеты «разбавлены» смежными
кольцами. Так, фенантрен (25), в формуле которого можно выде-
лить два секстета, более ароматичен, чем антрацен (24), а послед-
ний более ароматичен, чем высшие полиацены, где единственный
секстет сильнее «разбавлен»; эти выводы качественно согласуются
с величинами ЭР на л-электрон (табл. 1.1). Общая ЭР Дьюара
для цетрена (54) и перилена (33) почти вдвое больше, чем для
нафталина (22): 2,69, 2,61 и 1,32 эВ соответственно [44] — как и
можно ожидать, исходя из формул. Кроме того, секстетные фор-
мулы позволяют ввести понятие локальной ароматичности, отно-
сящееся к отдельному кольцу, а не к молекуле в целом; это суще-
ственно для обсуждения влияния структурных факторов на свой-
ства.
В последнее время предпринимаются попытки использовать
секстетные формулы для количественной оценки ароматичности
[51—53]. Этот подход основан на рассмотрении набора всех воз-
можных ковалентных структур типа структур Кекуле и сходен с
применявшимся в структурной теории резонанса [40]. Так, в каче-
стве индекса ароматичности отдельного кольца предложено отно-
шение числа валентных структур, в которых данное кольцо обла-
дает секстетом, к общему числу валентных структур, а в качестве
индекса ароматичности всей молекулы — сумма индексов отдель-
ных колец [52]. Индексы, предложенные разными авторами, в
общем, согласуются друг с другом и, главное, коррелируют со
значениями ЭР на л-электрон, рассчитанными методом МО ССП
или МОХ, не требуя применения ЭВМ. В случае углеводородов с
единственной секстетной формулой для приближенной оценки до-
статочно прохтого подсчета числа секстетов [53]. Так как выделе-
ние изолированных бензоидных секстетов из сопряженной системы
и связывание с ними всей энергии ароматической стабилизации
лишено физического смысла, к использованию секстетных формул
следует, по-видимому, относиться как к полезному формальному
приему, позволяющему быстро оценить ЭР и некоторые свойства
сопряженной молекулы.
Чтобы избежать при определении ЭР сравнения с. моделью,
сильно отличающейся по структуре, заманчиво использовать тер-
модинамические характеристики обратимых химических превра-
щений: кислотно-основных, таутомерных окислительно-восстанови-
тельных, при которых переход ароматического соединения в
неароматическое сопровождается минимальными структурными
изменениями.
Сущность метода можно пояснить на примере определения ЭР
гетероциклических соединений на основании измерений констант
ионизации [39,54]. Известно, что для 0-протонирования ЛЛметил-
пиррола (55) рКа = —5,1, а для p-протонирования неароматиче-
ской модели— 1,4,4-триметил-1,4-дигидропиридина (56) рКа =
= 7,4. Относя разность значений p^a в 12,5 единиц, соответствую-
щую разности энергий в 0,61 эВ, целиком за счет различий ЭР
оснований (55) и (56) и внося поправку в 0,26 эВ на стабилизацию
диенамина (56), обусловленную сопряжением, получают для
А^-метилпиррола (55) величину эмпирической ЭР 0,87 ±0,17 эВ,
что близко полученному для пиррола (41а) из термохимических
измерений (табл. 1.1).
(56)
(56а)
При аналогичных определениях для индола и карбазола на
каждое бензольное кольцо прибавляют 1,56 эВ — эмпирическую
ЭР бензола. Измерение константы pKR+ в реакции R+ +
+ НОН ROH+ Н+ применено при изучении ароматичности
циклопропенильных катионов [55] и катионов кватернизованных
гетероциклов — изохинолиния, тиазолия, бензотиазолия [56].
Из приведенных примеров видно, что хотя определение ЭР из
констант равновесия строится на экспериментальной основе, оно не
свободно от упрощений, несущих в себе элемент неопределенности.
Главными из них являются предположения об адекватности вы-
бранной модели гипотетической молекуле исследуемого соединения
с локализованными связями и об одинаковости изменений энергий
о-связей и энергий сольватации для ароматического соединения и
неароматической модели в изучаемой реакции. Кроме того, метод
требует оценки энергии л-сопряжения в модельном соединении
независимым путем.
В тех случаях, когда обе формы, находящиеся в равновесии,
обладают ароматичностью, использование констант равновесия
?4
дает информацию лишь о разности ЭР. Такая ситуация складыва-
ется при изучении таутомерных превращений. Сопоставление кон-
стант таутомерного равновесия ароматических структур 2-пири-
дон 2-гидроксипиридин (57а) <=>(576) и соответствующих
гидрированных моделей (58а) (586) показывает, что ЭР гид-
роксипиридиновой формы на 0,325 эВ больше, чем пиридоновой
[57].-
(57а) (576)
(58а)
(586)
Аналогично для 2-хинолона получено значение 0,086 эВ, а для
1-изохинолона — 0,190 эВ. Расчет разности ЭР для таутомеров, в
свою очередь, позволяет точно оценить положение таутомерного
равновесия, например при бензоидно-хиноидной таутомерии гидр-
оксиазометинов [58].
Если модельные соединения недоступны, сравнение констант
равновесия близких по строению соединений может дать каче-
ственное указание на тенденцию изменения ароматичности внутри
ряда. Это подтверждается наличием корреляции между значения-
ми р/Са, характеризующими кислотность [10] и основность [59]
полициклических углеводородов, и разностями л-электронных
энергий углеводорода и соответствующего иона, вычисленными
методом МОХ. Хорошая корреляция получена между нормальны-
ми окислительно-восстановительными потенциалами системы хи-
нон-гидрохинон и разностями л-электронных энергий окисленной
и восстановленной форм [10].
1.3.2. Структурные критерии
Как уже отмечалось, ароматические соединения отлича-
ются от неароматических особой геометрией молекулы. Образова-
ние замкнутой л-электронной оболочки обусловливает тенденцию к
копланарному расположению атомов и к выравниванию связей в
ароматическом цикле. Поскольку в настоящее время существуют
надежные экспериментальные методы определения геометрических
параметров молекулы, копланарность и степень выравненное™
связей могут быть использованы в качестве критериев ароматич-
ности.
Для установления молекулярной структуры главными являются
Дифракционные методы [60,61]: дифракция рентгеновских лучей,
электронов и нейтронов; ведущее место принадлежит первому из
этих методов. В последние десятилетия в развитии рентгенострук-
турного анализа произошли сдвиги, качественно изменившие воз-
можности и доступность метода. Внедрение автоматических диф-
рактометров в сочетании с ЭВМ и применение прямых методов
25
определения фаз, не требующих присутствия в молекуле тяжелых!
атомов, настолько расширили круг обследуемых соединений и снич
зили трудоемкость эксперимента, что, по мнению специалистов, в]
ближайшем будущем рентгеноструктурный анализ сможет по до-]
ступности сравняться с ИК- и ЯМР-спектроскопией [62]. В совре-1
менных рентгеноструктурных исследованиях точность определения ;
межатомных расстояний достигает ±0,001—0,003 А (10—30 пм),а?
валентных углов ±0,1 —0,3° [63]. Наиболее серьезным ограниче—'
нием остается необходимость выращивания правильного монокри-1
сталла. Когда вещество обладает достаточной летучестью, приме- 2
няют метод дифракции электронов в парах в сочетании со спект- ’
роскопическими методами. Дифракция нейтронов, при которой :
отражение осуществляется ядрами атомов, а не их электронными
оболочками, позволяет более точно фиксировать положение ато-
мов водорода.
Требование планарности ароматического цикла вытекает из
необходимости параллельности осей атомных р-орбиталей для их
эффективного перекрывания. Как правило, встречающиеся откло^
нения от копланарности атомов в ароматических системах незна-
чительны, хотя в напряженных структурах они могут достигать 5—
20°. На основании этого и данных квантовохимических расчетов
высказано мнение, что и ненапряженные ароматические кольца
обладают определенной гибкостью [64].
В плоских молекулах при наличии ароматичности межатомные
расстояния существенно отличаются от расстояний в соединениях с
локализованными связями *. Длина простой связи между атомами
углерода в зависимости от типа гибридизации составляет:
С5рз—CSP3 1,544А, т. е. 0,1544 нм (в алмазе), 1,533 А (в этане);
С5рз—CSP2 1,504А (в пропилене); CSp2—Csp? 1,466А (в бутадиене),
1,475 (в циклооктатетраене), а длина двойной связи С=С в эти-
лене 1,337 А. В бензоле все углерод-углеродные связи равноценны
и имеют длину 1,397 А — больше двойной, но меньше простой свя-
зи. Однако полная выравненность связей, какая наблюдается в
бензоле, представляет скорее исключение. У подавляющего боль-
шинства карбоциклических ароматических соединений, не говоря
уже о гетероциклических, связи С—С не вполне одинаковы и могут
быть как длиннее, так и короче связи С—С в бензоле. Введение
донорных (D) и акцепторных (А) заместителей в бензольное
кольцо деформирует его так, как показано на схемах, где дугами
отмечены увеличенные углы, а утолщенными линиями — укорочен-
ные связи [63]:
* Ссылки на оригинальную литературу по определению приводимых ниже
значений длин связей см. в обзорах [61, 65].
26
В нафталине (22) связи Са—Ср укорочены до 1,371 А
(0,1371 нм), а остальные удлинены по сравнению с бензолом. То
я<е наблюдается в антрацене (24) и других аценах. В фенантрене
(25) длина связи С9—С10 составляет всего 1,350 А, тогда как длина
связи С10—С10а 1,453 А. В перилене (33) связи, соединяющие на-
фталиновые ядра, по длине (1,471 А) соответствуют простой связи
Csp?—В тех же пределах колеблются длины связей С—С в
гетероароматических соединениях. Связи атом углерода—гетеро-
атом и гетероатом — гетероатом в них также укорочены по срав-
нению с соответствующими простыми связями.
Длины связей в ароматических соединениях могут быть вычис-
лены по эмпирической линейной зависимости между длиной
л-связи d (в А) и ее порядком р, рассчитываемым методами МО,
что дает удовлетворительное совпадение с экспериментом [66]:
d = 1,517 — 0,180р
Распределение длин связей в ароматическом цикле предложено
[67] использовать в качестве количественной характеристики сте-
пени ароматичности путем введения специальных индексов А —
= Д1Д2, где А[ = 1 — 225/м У (1 —drld)\ представляет собой
г
собственно меру выравненное™ п связей, составляющих периметр
молекулы, и имеющих длину dr при средней длине d. Дополни-
тельный индекс А2 = Ц [1 — (hqa/dij)2] учитывает уменьшение
(//)
устойчивости циркуляции л-электронов по кольцу, проявляющееся
в возникновении градиентов заряда \qij/dijy где Д^-/ — разность
л-зарядов на атомах i и /, a dij — расстояние между ними. При
выбранных коэффициентах значение Aj для бензола и значения
А2 для бензоидных углеводородов равны 1. Ниже приводятся
индексы Д1 и результирующие индексы А для некоторых аромати-
ческих систем [67]:
Д1 А А1 А
Бензол 1 1 Пиридин 1,00 0,97
Нафталин 0,90 0,90 Тиофен 0,93 0,67
Антрацен 0,89 0,89 Пиррол 0,91 0,38
Азулен 1,00 0,85 Фуран 0,81 0,06
К индексу /41 близок по физическому смыслу индекс делокали-
зации [68] D = 1 — 2/ц X р — рг, оперирующий не с длинами свя-
г
зей, а с их порядками рг и средним порядком р.
Экспериментально данные о степени фиксации двойных связей
можно получить из значений констант спин-спинового взаимодей-
ствия/ протонов ароматического кольца (59) в ЯМР спектрах [69].
При полной эквивалентности связей отношение констант орто-
взаимодействия /ьс//аь должно быть, как в бензоле, равно 1, а
отношение констант пара- и лета-взаимодействия /ad//ac < 1. При
Полной локализации диеновой группировки, как в соединении (60),
27
/ьс/Ль ~ 0,5, a /ad//ac > 1. Изучение ПМР-спектра 2-метилбен-
зотриазола (61а) показывает, что для него /Ьс//аЬ = 0,781 и
/ad//ac^l, из чего следует, что несмотря на формально орто-
хиноидное строение связи в 2-метилбензотриазоле (61а) в значи-
тельной мере делокализованы, и по степени ароматичности он
превосходит бензо-1,2,5-тиадиазол (616) и тем более
бенз-1,2,5-оксадиазол (61в), для которых Дс/Ль равны соответ-
ственно 0,748 и 0,706 [39].
(60)
a)X=N“CH3
6)X=S
В)Х=О
(61)
Структурные критерии, базирующиеся на прямом измерении
параметров молекулы, дают ценную объективную информацию о
строении, позволяющую различать ароматические и неароматиче-
ские соединения и сравнивать ароматичность близких по структуре
соединений. Однако универсальных количественных критериев,
пригодных для установления на этой основе единой шкалы арома-
тичности, пока не найдено.
1.3.3. Магнитные критерии
При помещении вещества в однородное магнитное поле с
напряженностью Но в системе электронов индуцируются токи, ко-
торые в свою очередь генерируют магнитное поле с напряженно-
стью Н' = //оХг, где — магнитная восприимчивость, отнесенная к
единице объема. Та же величина, отнесенная к единице количества
вещества, %м, измеряется в кубических сантиметрах на моль. Для
диамагнитных веществ, каковыми являются все органические со-
единения, не содержащие неспаренных электронов, индуцирован-
ное магнитное поле направлено противоположно внешнему и мо-
лярная магнитная восприимчивость хм всегда отрицательна. Так
как электронная система органических соединений практически
никогда не обладает сферической симметрией, индуцированное
магнитное поле анизотропно, т. е. изменяет свои характеристики в
зависимости от направления в пространстве. Особенно ярко это
выражено в ароматических соединениях, где помимо эффекта ло-
кальной анизотропии, обусловленной электронными оболочками
отдельных атомов, возникает так называемый «кольцевой ток»
вследствие существования замкнутой системы л-электронов. Для
плоских ароматических молекул, магнитная восприимчивость кото-
рых выражается в прямоугольных координатах в виде суммы:
Хм в ’/з (Хх + %у + Xz)
(1.2)
28
рис. 1*3. Модель кольцевых токов в бензоле.
анизотропия заключается в том,
что магнитная восприимчивость, из-
меренная по оси, перпендикулярной
плоскости ядра, всегда значитель-
но больше, чем' измеренная в
плоскости ядра: Xz » X* ~ Ху
(рис. 1.3). Мерой анизотропии диамагнитной восприимчивости слу-
жит разность между составляющими: Axm=Xz —УзСх* + Ху)-
Поскольку магнитная анизотропия ароматических соединений в
значительной степени определяется наличием замкнутой л-элект-
ронной оболочки, естественна попытка использовать ее в качестве
критерия ароматичности. Прямое измерение магнитной анизотро-
пии сложно, так как требует выращивания монокристалла и пред-
варительного определения в нем ориентации молекул. Более до-
ступно определение магнитной анизотропии, основанное на эффек-
те Зеемана в микроволновых вращательных спектрах [70], но оно
применимо только к соединениям, для которых возможны спект-
ральные измерения в газовой фазе.
Как видно из приведенных в табл. 1.2 примеров, разница в
величинах А%м для ароматических и неароматических (1,3-цикло-
гексадиен) соединений весьма отчетлива, но лишь часть этой раз-
ности (по мнению авторов [71], не более половины) обусловлена
вкладом кольцевого тока, тогда как другая часть должна быть
отнесена за счет локальной анизотропии. Трудности разделения
вкладов кольцевого тока и локальной анизотропии, из которых
только первый свидетельствует о степени ароматичности, в сово-
купности с экспериментальными трудностями ограничивают ис-
пользование анизотропии диамагнитной восприимчивости в каче-
стве критерия ароматичности.
Таблица 1.2
Анизотропия АХМ [71] и экзальтация А [72]
диамагнитной восприимчивости
Соединение А*м А* (—Ю”"6), см3/моль А«( —10 6) на одно кольцо, см3/моль
Бензол 59,7 13,7 13,7
Нафталин 30,5 15,3
Антрацен •— 48,6 16,2
Фенантрен •— 46,2 15,4
Тетрацен 1 66 16,5
Азулен — 29,6
Пиридин — 13,4
Пиррол 42,4 10,2
Фуран 38,7 8,9
Тиофен 50,1 13,0
1,3-Циклогексадиен 7,4 -0,7
29
Проще определить экзальтацию диамагнитной восприимчиво
сти. Делокализация л-электронов в цикле сказывается не только i
увеличении магнитной анизотропии вследствие роста составляю
щей Хг, перпендикулярной плоскости кольца, но и в общем увелц
чении молярной диамагнитной восприимчивости по сравнению <
соединением, имеющим локализованные связи [72]. Это вытекает
из уравнения (1.2), если принять, что % «+ К и, еле
гаром ^неаром
довательно, v « где последний член представ-
аром неаром
ляет собой экзальтацию магнитной восприимчивости ароматиче-
ского соединения Л.
Общая магнитная восприимчивость определяется экспери-
ментально измерением силы, с которой образец диамагнитного
вещества выталкивается из магнитного поля заданной напряжен-
ности. Магнитная восприимчивость модельного соединения с лока-
лизованными связями у' вычисляется как сумма вкладов отдель-
ных структурных элементов молекулы (атомы, связи, группы ато-
мов, электронные пары и т. д.). Экзальтация диамагнитной
восприимчивости Л (см. табл. 1.2) представляет собой разность
этих величин (Л = ум—•%') и рассматривается [72] как непремен-
ное свойство ароматических соединений. Накопленный материал не
противоречит такому заключению, а сравнительная простота опре-
деления, применимость к соединениям различных классов, нечув-
ствительность к влиянию побочных факторов делает экзальтацию
диамагнитной восприимчивости ценным критерием ароматичности.
Его недостатком является необходимость перенесения расчета
магнитной восприимчивости по аддиативной схеме, правомерность
которого показана для реальных неароматических соединении, на
гипотетические, не существующие в действительности модели типа
циклогексатриена. Такой переход всегда таит в себе возможный
источник ошибок, тем более что и для реальных соединений встре-
чаются аномалии. Так, для циклогептатриена и циклопентадиена
получены значения, согласно которым они должны быть отнесены к
ароматическим соединениям (А = 8,1 и 6,5 соответственно). Это
противоречит всем другим данным для этих соединений и потребо-
вало для объяснения привлечения дополнительных гипотез [72].
Если сравнить величины экзальтации диамагнитной восприимчи-
вости на одно кольцо (табл. 1.2), видно, что ароматические угле-
водороды в такой шкале располагаются в прямо противоположной
последовательности, чем в шкале ЭР на л-электрон (табл. 1.1),
согласующейся с совокупностью других данных.
Наиболее простым в выполнении методом исследования арома-
тичности является определение положения сигналов протонов аро-
матического кольца в спектрах ЯМР. Он базируется на упоминав-
шейся выше концепции индуцированных кольцевых токов, генери-
рующих анизотропное магнитное- поле, которое внутри кольца
направлено всегда противоположно внешнему магнитному полю,
но вне кольца, в зависимости от удаления, может быть направлено
30
как по внешнему полю, таки против него (рис. 1.3). Соответственно
влияние кольцевого тока на химический сдвиг зависит от ориента-
ции протона относительно ароматической системы. Сигналы прото-
нов, расположенных внутри ароматического кольца или над ним,
смещаются в сторону более сильного поля, часто даже за сигнал
тетраметилсилана, принятого в шкале б за нуль. Так, внутренние
протоны в [18]аннулене (17) дают сигнал при б =—3,0 м. д.,
протоны метиленового мостика в 1,6-метано [10] аннулене (20) —
при 6 = —0,5 м. д., а внутренние протоны в 10 Ь, 10 с-дигидропи-
рене (27)— при б = —5,49 м. д. В то же время сигналы протонов,
лежащих в плоскости кольца с наружной стороны, смещены в
слабое поле: сигналы протонов бензола проявляются при б =
= 7,27 м. д., сигналы внешних протонов [18]аннулена (17) — при
6 = 9,28 м. д., тогда как сигналы алкеновых протонов 1,3-цикло-
гексадиена — при б = 5,77 м. д. В отличие от [18]аннуленов (17),
в неароматических [16] аннуленах (18) сигналы внутренних и
внешних протонов различаются гораздо меньше: б = —0,32 и
4,8 м. д. соответственно.
Так как согласно изложенной выше концепции смещение сиг-
налов ароматических протонов в слабое поле вызывается кольце-
вым током, а последний обусловливается делокализацией л-элект-
ронов и проявляется в смещении сигналов ароматических протонов,
предложено [731 судить о степени делокализации по значениям
химических сдвигов в спектрах ПМР. Благодаря доступности и
кажущейся простоте интерпретации данные спектров ПМР полу-
чили широкое распространение в качестве критерия ароматично-
сти, причем часто количественное сопоставление ароматичности
делается путем простого сравнения величин химических сдвигов.
Такой подход вызывает возражения двоякого рода.
Считают [74], что магнитная анизотропия ароматических со-
единений вообще не связана непосредственно с делокализацией
л-электронов, а целиком обусловлена эффектом локальной анизот-
ропии. Согласно другой точке зрения, разделяемой большим чис-
лом исследователей, концепция кольцевых токов правомерна, но
неправильно увеличение химического сдвига ароматических прото-
нов целиком относить за счет кольцевого тока. Необходимо учиты-
вать также вклад эффекта локальной анизотропии и влияние рас-
пределения электронной плотности. Анализ, проведенный приме-
нительно к ароматическим углеводородам [75], показывает, что
лишь 50% или менее от общего увеличения химического сдвига
может быть отнесено за счет эффекта делокализации, тогда как
остальная часть определяется вкладом локальной анизотропии,
особенно значительным для пространственно затрудненных прото-
нов. Влияние электронной плотности на величину химического
сдвига иллюстрирует сравнение ПМР-спектров циклооктатетраена
(13) и его дианиона (15): положение сигналов протонов в них
почти одинаково, несмотря на то, что дианион (15) представляет
собой ароматическую частицу, а циклооктатетраен (13)—нет. Не-
обходимость учитывать эффект локальной анизотропии и влияние
31
электроотрицательности создает особенно большие трудности при
интерпретации химических сдвигов гетероароматических соедине-
ний, что является причиной противоречий при попытках построить
ряды ароматичности гетероциклов [39].
Очевидно, что простое сравнение значений химических сдвигов
не может дать верного представления о соотношении величин
кольцевых токов и потому не пригодно для количественного сопо-
ставления ароматичности. Отсутствие же надежных методов вы-
деления вклада кольцевых токов заставляет в общем случае рас-
сматривать величину химических сдвигов как качественный крите-
рий, пригодный, скорее, для выявления ароматичности, чем для ее
количественной оценки.
Из параметров, базирующихся на величине химического сдвига,
можно назвать «смещение при разбавлении» А [76] и «смещение
от растворителя» S [77]. Первый из этих параметров представляет
собой отношение
Л = ^ЬЦслед^оед 100о/((
(ДО1 V м)бензола
где Им — молярный объем, а Д61 — смещение сигнала протонов
кольца при разбавлении жидкого ароматического соединения
неполярным растворителем. При применении четыреххлористого
углерода получено [76] для пиридина А = 61 ± 7%, для тиофена
69 ± 1 %, для фурана 42±5%. Параметр S представляет собой
разность химических сдвигов полярной молекулы (ацетонитрила)
и неполярной (циклогексана) в исследуемом.соединении. Для бен-
зола S = 1 м. д., для олефинов — около нуля, а для пиррола, тио-
фена и фурана соответственно 0,82, 0,74 и 0,42 м. д. [77].
Показателем, связанным с магнитной анизотропией, является
также экзальтация вращения плоскости поляризованного света
(эффект Фарадея) образцом прозрачного ароматического веще-
ства, помещенного в магнитное поле, по сравнению с вращением
для гипотетической модели с локализованными связями, вычис-
ленным по инкрементам связей [78]. Все эти параметры примени-
мы только к жидким ароматическим веществам.
1.3.4. Заключение
Рассмотренные критерии опираются на такие признаки
ароматичности, как термодинамическая стабильность, особая гео-
метрия, наличие кольцевого тока. В совокупности они дают экспе-
риментальные свидетельства существования ароматичности как
явления, связанного с особым электронным строением органичен
ских соединений.
Энергетические критерии наиболее универсальны и наиболее
пригодны для построения единой шкалы ароматичности, но соот-
несение их с данными физического и химического эксперимента
затруднительно.
32
Структурные критерии дают объективную картину, целиком
построенную на экспериментальной основе, но определение их
пока еще довольно сложно, и сами по себе они не позволяют по-
строить шкалу ароматичности.
Магнитные критерии легко доступны, но интерпретация их ос-
ложнена необходимостью учитывать влияние многих факторов.
Общая методологическая трудность при использовании указан-
ных критериев состоит в сравнении реального соединения с чисто
умозрительной моделью, имеющей такое же строение, но лишен-
ной ароматичности.
Сегодня нет оснований отказываться от использования какой-
либо из этих групп критериев в пользу других, поскольку при
правильном анализе именно совместное использование их позво-
ляет более надежно проводить сопоставление ароматичности
близких по строению веществ.
Оценка ароматичности имеет важное значение не только для
понимания свойств соединений в основном состоянии, но и для
суждения об их реакционной способности, чему посвящена следу-
ющая глава.
ЛИТЕРАТУРА
1. Коллин Г., Цандер М. — В кн.: Химия синтетических красителей. Т. З/Под
ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия, 1974„
с. 1721 —1739. 2. Привалов В. Е., Литвиненко М. С. — Журн. ВХО, 1976, т. 21,
вып. 3, с. 242—246. 3. Харлампович Г. Д., Чуркин Ю. В. — Там же, 1977, т. 22,
вып. 1, с. 54—61. 4. В кн.: Новые направления химии тиофена/Под ред.
Я. Л. Гольдфарба. М., Наука, 1976, с. 12—13. 5. Теддер Дж., Нехватал А.,
Джубб А. Промышленная органическая химия. Пер. с англ./Под ред. О. В. Кор-
сунского. М., Мир. 1977. 6. Snyder J. Р. — In: Nonbenzenoid Aromatics. V. l./Ed.
J. P. Snyder. N. Y. — London, Academic Press, 1969, p. 1—31. 7. Baker W. — Che-
mistry in Britain, 1965, v. 1, p. 191—197. 8. Болесов И. Г. — Успехи химии, 1968,
т. 37, вып. 9, с. 1567—1576. 9. Liebman J. F., Greenberg А. — Chem. Revs, 1976,
v. 76, № 3, р. 311—365. 10. Стрейтвизер Э. Теория молекулярных орбит для хи-
миков-органиков. Пер. с англ./Под ред. М. Е. Дяткиной. М., Мир, 1965.
11. Дьюар AL, Догерти Р. Теория возмущений молекулярных орбиталей в
органической химии. Пер. с англ./Под ред. Л. А. Яновской. М., Мир, 1977.
12. Лонге-Хиггинс Г. К. — В кн.: Теоретическая органическая химия. Пер. с
англ./Под ред. Р. X. Фрейдлиной. М., ИЛ, 1963, с. 17—30. 13. Деринг У. — Там.
же, с. 50—65. 14. Рыбинская AL И. Корнеева Л. М. — Успехи химии, 1971,
вып. 3, с. 444—461. 15. Дьяконов И. А., Костиков Р. Р. — Там же, 1967, т. 36,
вып. 3, с. 1305—1319. 16. Chandler G. S., Craig D. Р. — In: Rodd’s Chemistry of
Carbon Compounds. V. 3, Part А/Ed. S. Coffey. Amsterdam — London — N. Y.,
Elsevier, 1971, p. 5—44. 17. Olah G. A., Staral J. S.— J. Amer. Chem. Soc., 1976,
v. 98, № 20, p. 6290—6304. 18. Cava M. P., Mitchell M. J. — Cyclobutadiene and
Related Compound. N. Y. — London, Academic Press, 1967, p. 1—87; Lloyd D. —
Intern. Rev. ScL, Org. Chem., Series 1, 1973, v. 3, p. 179—200. 19. Schroder G.
Cyclooctatetraen. Weinheim, Verlag Chemie, 1965; Paquette L. A. — Tetrahedron,
1975, v. 31, № 23, p. 2855—2833. 20. Olah G. A., Staral J. S., Liang G. e.a.—
J. Amer. Chem. Soc., 1977, v. 99, № 10, p. 3349—3354.
21. Katz T. J. — Ibid., I960, v. 82, № 14, p. 3784—3786. 22. Katz T. J., Gar-
rat P. J. — Ibid., 1963, v. 85, № 18, p. 2852—2853; Lalancete E. A., Benson R. E.—
Ibid., p. 2853. 23. Garrat P. J., Sargent M. V. — In: Nonbenzenoid Aromatics..
V. 2/Ed. J. P. Snyder. N. Y. — London, Academic Press, 1971, p. 207—272.
2 Зак. 271 3&
24. Sondheimer F.— Accounts Chem. Res., 1972, v. 5, № 3, p. 81—91. 25. Воль-
пин M. E.— Успехи химии, 1960, т. 29, вып. 3, с. 298—363. 26. Masamune S.,
Hojo Hojo Kiyomi e.a. — J. Amer. Chem. Soc., 1971, v. 93, № 19, p. 4966—4968.
27. Vogel E. — Chimia, 1968, Bd. 22, № 1, S. 21—32. 28. Masamune S.,
Brooks D. W.— Tetrahedron Letters, 1977, № 37, p. 3239—3240. 29. Мочалин В. Б.,
Поршнев Ю. Н.— Успехи химии, 1977, т. 46, вып. 6, с. 1002—1040. 30.- Vogel Е.—
Pure Appl. Chem., 1971, v. 28, № 2—3, p. 355—377.
31. Mitchell R. H., Boekelheide V. — J. Amer. Chem. Soc., 1974, v. 96, № 5,
p. 1547—1557. 32. Reid D. //. — Quart. Revs, 1965, v. 19, № 3, p. 274—302;
Hunig S., Wolff E. — Chimia, 1968, Bd. 22, № 1, S. 33—39. 33. Дяткина M. E.,
Шусторович E. M. — ДАН СССР, 1957, т. 117, № 6, с. 1021 — 1022. 34. Соло-
дарь С. А. — Журн. ВХО, 1976, т. 21, № 3, с. 306—315. 35. Senkier G. FL, Ма-
ryanoff В. Е., Stackhouse J. e.a. — In: Organic Sulphur Chemistry/Ed. C. J. Stir-
ling. London, Butterworth, 1975, p, 157—179. 36. Швецов-Шкловский H. И.,
Бобкова P. Г., Игнатова H. FL и dp. — Успехи химии, 1977, т. 46, вып. 6,
с. 967—1001. 37. Badger G. М. Aromatic Character and Aromacity. Cambridge,
Cambridge University Press, 1969. 38. Agranat I. — Internal. Rev. Sci., Org. Chem,
Series 1, 1973, v. 3, p. 139—178; Agranat L, Barak A.— Ibid., Series 2, 1976, v. 3,
p. 1—24. 39. Cook M. J., Katritzky A. R., Linda P. — Adv. Het. Chem., 1974, v. 17,
p. 257—356. 40. Wheland C. W. — Resonance in Organic Chemistry. N. Y., Wiley,
1955. 325 p.
41. Dewar M. J. S., Schmeising H. N. — Tetrahedron, 1960, v. 11, № 1/2,
p. 96—120. 42. Hess B. A., Schaad L. J.—J. Amer. Chem. Soc., 1971, v. 93, № 10,
p. 2413—2417. 43. Schaad L. J., Hess B. A.—J. Chem. Educ., 1974, v. 51, № 10,
p. 640—643. 44. Dewar M. J. S., de Liano C. — J. Amer. Chem. Soc., 1969, v. 91,
№ 4, p. 789—795. 45. Figeys H, P. — Tetrahedron, 1970, v. 26, № 19, p. 4615—
4620. 46. Dewar M. J. — Ibid., 1966, v. 22, Suppl. 8, p. 75—92. 47. Дьюар M. Тео-
рия молекулярных орбиталей в органической химии. Пер. с англ./Под ред.
М. Е. Дяткиной. М., Мир, 1972, с. 200—246. 48. Baird N. С. — J. Chem. Educ.,
1971, v. 48, № 8, р. 509—514. 49. Hess В. A., Schaad L. J. — Chem. Commun.,
1977, № 8, р. 243—244; Herndon W. C.—Ibid., 1977, № 22, p. 817—818. 5\. Клар Э.
Полициклические углеводороды. T. 1. Пер. с англ., М., Химия, 1971. с. 47—113.
51. Herndon W. С., Ellzey М. L.—J. Amer. Chem. Soc., 1974, v. 96, № 21,
p. 6631—6642. 52. Randic M. — Tetrahedron, 1975, v. 31, № 11/12, p. 1477—1481;
1977, v. 33, № 15, p. 1905—1920. 53. Aihara J. — Bull. Chem. Soc. Japan, 1976,
v. 49, № 5, p. 1429—1430; 1977, v. 50, № 8, p. 2010—2012. 54. Carmody M. P.t
Cook M. J., Dassanaycke N. L. e.a. — Tetrahedron, 1976, v. 32, № 14, p. 1767—1771.
55. Breslow R. — Pure Appl. Chem., 1971, v. 28, № 2—3, p. Ill—130.
56. Cook M. J., Katritzky A. R., Page A. D. e.a. — Tetrahedron, 1976, v. 32, № 14,
p. 1773—1777. 57. Elguero L, Martin C., Katritzky A. R. e.a. — Adv. Het. Chem.,
1976, Suppl. 1. 58. Симкин Б. Я., Брень В. А., Минкин В. И. — ЖОХ, 1977, т. 13,
вып. 8, с. 1710—1722. 59. Бетел Д., Голд В. Карбониевые ионы. Пер. с англ./Под
ред. И. П. Белецкой. М., Мир, 1970, с. 144—151. 60. Molecular Structura by
Diffraction Methods/Eds. G. A. Sim, L. E. Sutton. London, Chem. Soc., V. 1, 1973;
V. 2, 1974; V. 4, 1976.
61. Karie J., Karie 1. L. — Internal. Rev. Sci., Phys. Chem., Series 1, 1973,
v. 11, p. 247—310; Kuchitsu K-— Ibid., 1973, v. 2, p. 203—240. 62. Wheatley P. J.—
Phys. Methods Het. Chem., 1972, v. 5. 598 p. 63. Звонкова 3. В.-—Успехи химии,
1977, т. 46, вып. 5, с. 907—927. 64. Wynberg Н., Nieuwpoort W. С., Jonk-
man Н. Т.— Tetrahedron Letters, 1973, № 46, р. 4623—4628. 65. Herndon W.C. —
J. Amer. Chem. Soc., 1974, v. 96, № 25, p. 7605—7614. 66. Allinger N. L., Spra-
gue J. T. — Ibid., 1973, v. 95, № 12, p. 3893—3907. 67. Julg A. — In: Aromacity,
Psevdo-aromacity, Anti-aromacity. V. 3/Eds. E. D. Bergmann, B. Pullmann. Jerusa-
lem, 1971, p. 383—385. 68. Kemula 117., Krygowski T. M. — Tetrahedron Letters,
1968, № 49, p. 5135—5140. 69. Gunther H. — Ibid., 1967, № 31, p. 2967—2970.
70. Sutter D. H., Flygare W. H. — Topics Curr. Chem., 1976, v. 63, p. 89—196.
34
71. Pochan J. M., Flygare W. H.—J. Amer. Chem. Soc., 1969, v. 91, № 21,
p. 5928—5929. 72. Dauben H. J., Wilson J. D., Laity J. L. — In: Nonbenzenoid
Aromatics. V. 2/Ed. J. P. Snyder N. Y. — London, Academic Press, 1971, p. 167—
206. 73. Elvidge J. A., Jackman L. M.—J. Chem. Soc., 1961, № 3, p. 859—866.
74. Musher J. /.—J. Chem. Phys., 1965, v. 43, № 11, p. 4081—4083; 1967, v. 46,
№ 3, p. 1219—1221. 75. Barfield M., Grant D. M., Ikenberry D. — J. Amer. Chem.
Soc., 1975, v. 97, № 24, p. 6956—6961. 76. Kanekar C. R., Govil G., Khetrapal C. L.
e.a. — Proc. Indian Acad. Sci., 1966, Sect. A, v. 64, № 5, p. 315—320.
77. Anet F. A. L., Schenck G. E. — J. Amer. Chem. Soc., 1971, v. 93, № 2, p. 556—
557. 78. Labarre J. F., Crasnier F.—J. Chem. Phys., 1967, v. 64, № 11—12,
p. 1664—1669.
Глава 2
РЕАКЦИОННАЯ СПОСОБНОСТЬ
АРОМАТИЧЕСКИХ СОЕДИНЕНИИ
2.1. ЭНЕРГЕТИЧЕСКИЕ ХАРАКТЕРИСТИКИ
РЕАКЦИОННОЙ СПОСОБНОСТИ [1—3]
Термодинамически осуществимость любой реакции
контролируется изменением свободной энергии системы AG, кото-
рая состоит из энтальпийной ЛЯ и энтропийной AS составляющих:
\G==\H-T\S (2.1)
Реакция осуществима без подвода энергии извне, если уровень
свободной энергии исходных веществ выше уровня свободной
энергии продуктов реакции. Однако выполнение этого условия еще
не означает, что реакция протекает спонтанно. Для нее всегда
характерна некоторая конечная скорость, которая при обычной
температуре может быть крайне мала и практически равна нулю.
Для объяснения этого используется представление об энергетиче-
ском барьере, который должен быть преодолен в ходе реакции.
Хотя от исходных веществ к продуктам реакции ведет бесчис-
ленное множество путей, обычно рассматривается только тот, ко-
торый в каждый данный момент отвечает минимально возможной
потенциальной энергии системы. Промежуточная структура на пу-
ти реакции, которая имеет наиболее высокий уровень свободной
энергии, называется переходным состоянием или активированным
комплексом. Изменение свободной энергии системы в простейшем
случае представлено для реакции А + В АВ на рис. 2.1, где
под координатой реакции подразумевается свободно выбираемый
параметр, служащий мерой превращения реагентов в продукты
реакции, например межатомное расстояние А---В.
Равновесие между исходными веществами А + В и продуктом
я г) о __ [АВ]
АВ характеризуется константой равновесия л — [А] [В]" и 0ПРеДе‘
ляется разностью их свободных энергий:
ДО ==-/?? 1g К (2.2)
2* 35
Рис. 2.1. Изменение свободной энергии системы
в реакции А + В .•> АВ.
Теория переходного состояния
предполагает наличие бесконечной
последовательности равновесий ме-
жду промежуточными структурами
и, следовательно, наличие равнове-
сия между исходными веществами
А+ В и активированным комплек-
сом АВ^. Определяющая это рав-
новесие разность свободных энер-
гий AG^, называемая свободной энергией активации, и является
тем энергетическим барьером, который должен быть преодолен
для осуществления реакции. Разложение свободной энергии акти-
вации на составляющие:
ДС* ==ДЯ^ - TkS^
(2.3)
из которых энтальпийный член по существу отражает изменение
энергий связей, а энтропийный — изменение упорядоченности си-
стемы, дает возможность глубже понять природу переходного со-
стояния.
Экспериментально значения АЯ^, AS^, AG^ получают путем
измерения скоростей реакции kp при разных температурах из эм-
пирического уравнения Аррениуса:
йр = Ле-Ва/₽Г ' (2Л)
в котором Еа — аррениусовская энергия активации; А — предэкс-
поненциальный фактор.
Между £*а и АЯ^ для реакций в растворе существует соотно-
шение:
ДЯ* = £а~ RT
(2.5)
в котором член RT при комнатной температуре составляет около
2,5 кДж/моль, так что различие между Еа и невелико.
В предположении, что все активированные комплексы распадаются
с одной и той же скоростью, теория переходного состояния дает
для констант скоростей реакции выражение:
kT
р“~ A е
(2.6)
где k—постоянная Больцмана; h — постоянная Планка.
Используя уравнение (2.3) и (2.5), это же выражение можно
записать в форме:
&s*/r
е
е-£а/«г
(2.7)
86
рис. 2.2. Изменение свободной энергии системы
в параллельных реакциях А + В АВ и
А + В <=> А В'
где заключенный в скобки член
эквивалентен предэкспоненциально-
му фактору А в уравнении Арре-
ниуса, зная который рассчитывают
AS+
Наглядное пояснение смысла
разделения свободной энергии ак-
тивации AG^ на две составляющие
дает теория столкновений.
Согласно этой теории, к реакции приводят только те столкновения
молекул, энергия которых превышает критическую величину, рав-
ную АЯ+ при условии благоприятной взаимной ориентации стал-
кивающихся молекул, вероятность чего определяется членом AS^5.
Если вещества А + В могут обратимо реагировать (рис. 2.2) не
только по пути / с превращением в продукт АВ, но и по парал-
лельному пути 2 с превращением в продукт А'В', может сложиться
ситуация, когда реакция, имеющая более низкий энергетический
барьер, приводит к продукту с более высоким уровнем свободной
энергии. В этом случае энергетические профили реакции пересека-
ются.
В начальный период в реагирующей системе преобладает более
быстро образующийся продукт А' В' и соотношение продуктов
определяется отношением свободных энергий активации (кийети-
ческий контроль), но по мере приближения к состоянию термоди-
намического равновесия количество более стабильного продукта
АВ возрастает и при достижении равновесия соотношение продук-
тов определяется отношением изменений свободных энергий (тер-
модинамический контроль). Поскольку в реакциях ароматических
соединений, как правило, возможно образование нескольких изо-
мерных продуктов, вопрос кинетического и термодинамического
контроля реакции имеет важное значение (см. 2.7).
2.2. РЕАКЦИОННАЯ СПОСОБНОСТЬ И АРОМАТИЧНОСТЬ
Как указано выше, фундаментальной величиной,
контролирующей реакционную способность, является свободная
энергия активации AG^, представляющая собой разность между
свободной энергией основного состояния исходных веществ и сво-
бодной энергией переходного состояния. Все ароматические соеди-
нения отличаются повышенной термодинамической устойчивостью
за счет образования замкнутой л-электронной оболочки, т. е. имеют
пониженный уровень свободной энергии. Поэтому при прочих рав-
ных условиях, в частности при неизменности уровня свободной
энергии переходного состояния (АВ+ рис. 2.1), ароматический
субстрат будет тем труднее вступать в реакцию, чем больше сте-
пень его ароматичности. Обнаруженная еще на заре органической
37
химии инертность ароматических производных бензола в сравне-
нии с ненасыщенными алифатическими соединениями является
отражением этого обстоятельства. Тенденция к восстановлению
ароматичности с переходом на наинизший из возможных энерге-
тический уровень обусловливает другую давно отмеченную особен-
ность ароматических соединений — склонность к реакциям заме-
щения, а не присоединения.
Однако нужно еще раз подчеркнуть, что количественная зави-
симость между величиной AG*, определяющей реакционную способ-
ность, и степенью ароматичности, характеризующей основное со-
стояние субстрата, может иметь место только при неизменности
уровня свободной энергии переходного состояния. В общем же
случае энергия переходного состояния может изменяться в столь
широких пределах, что это изменение верхней границы разности
AG* полностью перекроет влияние изменения нижней границы,
зависящей от степени ароматичности. Для иллюстрации рассмот-
рим ряд ароматических соединений: циклопентадиенид натрия (1),
пиррол (2), бензол (3), пиридин (4) и тропилийбромид (5).
(0 (2) (3) (4) (5)
Циклопентадиенид-анион (1) исключительно легко реагирует с
электрофильными агентами, в очень мягких условиях вступая в
реакции галогенирования, алкилирования, азосочетания и др.
При переходе к пирролу (2) и далее к бензолу (3) легкость
электрофильного замещения уменьшается; пиридин (4) уже всту-
пает в реакции такого типа в жестких условиях, а соли тропилия
(5) до сих пор вообще не удалось ввести ни в одну из электро-
фильных реакций. В то же время соли тропилия очень легко взаи-
модействуют с самыми разнообразными нуклеофильными реаген-
тами: водой, алкоголятами и многими другими. Пиридин и тем
более бензол вступают в подобные реакции значительно труднее, а
соли циклопентадиенид-аниона вообще не реагируют с нуклео-
фильными агентами. В представленном выше ряду реакционная
способность по отношению к электрофильным реагентам уменьша-
ется слева направо, а по отношению к нуклеофильным реаген-
там — справа налево без всякой видимой связи со степенью аро-
матичности.
Причина отсутствия корреляции между реакционной способно-
стью и ароматичностью в рассмотренном ряду состоит именно в
том, что энергетический барьер реакции AG* изменяется здесь
главным образом за счет понижения или повышения его верхнего
значения, определяемого энергией переходного состояния, а не
нижнего, отвечающего основному состоянию субстрата и реагента
(рис. 2.1). Природу такого изменения высоты энергетического
барьера удобно пояснить в терминах теории столкновений. Оче-
38
видно, что силы отталкивания, которые необходимо преодолеть
сближающимся частицам, несравненно больше, когда частицы
несут одноименный заряд, как это имеет место в случае электро-
фильной атаки на тропилий-катион, чем когда они разноименно
заряжены, как при электрофильной атаке на циклопентадиенид-
анион. То же относится к легкости нуклеофильной атаки на тропи-
лий-катион и ее затрудненности в случае циклопентадиенид-анио-
на. Гетероциклы пиррола (2) и пиридина (4) занимают промежу-
точное положение соответственно между ионами (1) и (5) и
бензолом (3). П иррол вследствие электронодонорного эффекта
группы NH (см. 1.2.4) имеет избыточную электронную плотность в
кольце и потому легче бензола подвергается электрофильной атаке.
Пиридин вследствие электроноакцепторного влияния атома азота
имеет пониженную электронную плотность в цикле и легче всту-
пает в реакции нуклеофильного замещения.
Приведенный пример одновременно убедительно свидетель-
ствует о неприменимости реакционной способности в качестве кри-
терия ароматичности (см. 1.3) ио том, что данных об ароматич-
ности недостаточно, чтобы судить о реакционной способности
ароматического соединения. Однако без этих данных правильное
понимание реакционной способности и, в особенности, механизмов
реакций также невозможно. Хотя между ароматичностью и сво-
бодной энергией активации нет простой зависимости, электронное
строение субстрата не только определяет нижнюю границу энерге-
тического барьера в реакции, но и накладывает отпечаток на
структуру переходного состояния (см. 2.7 и 2.8). В ряду близких по
строению соединений изменения энергии переходного состояния
реакции и основного состояния субстрата и реагента могут быть
пропорциональны; тогда между изменением энергии резонанса и
энергией активации наблюдается линейная корреляция, как, на-
пример, в случае реакции Дильса — Альдера ароматических угле-
водородов с’малеиновым ангидридом (ссылка [49] к главе 1).
Для изучения реакционной способности сведения об ароматич-
ности соединения недостаточны, но совершенно необходимы. Аро-
матичность является одной из важных характеристик строения
субстрата, влияющих на реакционную способность, наряду с рас-
пределением электронной плотности, стерическими и другими осо-
бенностями.
2.3. КЛАССИФИКАЦИЯ РЕАКЦИЙ
В химической реакции помимо соединений, которые
можно изолировать, могут возникать продукты, малая стабиль-
ность которых не позволяет выделить их из реакционной смеси.
Если нестабильные продукты лежат на пути превращения исход-
ных соединений в конечные, т. е. являются промежуточными, а не
побочными, реакцию можно представить в виде элементарных ста-
дий, каждая из которых характеризуется своей энергией активации
и своим переходным состоянием. Зная расположение атомов
в переходном состоянии, природу взаимодействий, способы разры-
39
ва и образования связей, энергию системы и скорость изменений в
каждый данный момент, можно составить представление о меха-
низме элементарных стадий, а из них — о механизме реакции в
целом.
Сведения об истинных механизмах реакций являлись бы наибо-
лее надежной базой для их классификации. Однако для относи-
тельно простых модельных органических реакций, несмотря на
интенсивные исследования, пока нет полной картины, описываю-
щей все особенности механизма. Тем более неясны механизмы
сложных реакций, применяемых на практике. Помимо эксперимен-
тальных трудностей, связанных с необходимостью наблюдения ла-
бильных промежуточных частиц, определения их строения и роли в
реакции, сложность задачи усугубляется возможностью протекания
одной и той же реакции по разным механизмам в зависимости от
структуры реагентов, природы растворителя, катализатора и дру-
гих условий. Это заставляет проявлять большую осторожность в
перенесении выводов о механизме, сделанных на основании изуче-
ния одной реакционной системы, на другие системы и тем более на
другие реакции. В настоящее время классификация реакций по их
реальным механизмам невозможна ввиду отсутствия достаточных
данных. Поэтому существующая классификация строится на брут-
то-схемах реакций и общих представлениях об образовании связей
[4-7].
По брутто-схемам реакции подразделяются в зависимости от
изменений структурных элементов на реакции замещения, присо-
единения, отщепления и перегруппировки. Выделяют также реакции
внедрения и выбрасывания (экструзии) [4]. Нужно отметить, что
отнесение сложной реакции к тому или другому типу по данному
признаку зависит от степени детализации брутто-схемы. Так, ре-
акция ароматического замещения при разложении на элементар-
ные стадии оказывается совокупностью реакций присоединения и
отщепления (см. 2.7). Реакция перегруппировки может оказаться
комбинацией двух реакций замещения, каждая из которых состо-
ит из реакций присоединения и отщепления.
В зависимости от того, возникает ли новая ковалентная связь в
результате обобщения двух электронов, которые до реакции при-
надлежали разным атомам, или за счет пары электронов, принад-
лежавшей до реакции одному из атомов, различают гомолитиче-
ский (радикальный) и гетеролитический (ионный) тип образования
связи. Такое разделение открывает возможность классификации
реакций на основе природы реагирующих частиц. Поскольку обыч-
но реагирующие частицы условно делят на атакуемую (субстрат)
и атакующую (реагент) , природа которых может быть различна,
классификацию относят к частице реагента.
При гетеролитическом образовании новой связи роль субстрата
и реагента различна, так как один предоставляет пару электронов,
а другой — вакантную орбиталь. Согласно определению Льюиса,
любые частицы, обладающие парой электронов, которая может
принимать участие в образовании новой связи, называются осно-
40
ваниями. а частицы, способные образовать связь с такой парой
электронов,— кислотами. Следовательно, в гетеролитической реак-
ции реагент обязательно является либо кислотой Льюиса, обладая
сродством к электрону, либо основанием, обладая сродством к
ядру. Поскольку термины «кислота» и «основание» имеют более
широкое значение, реагенты в гетеролитических реакциях именуют
соответственно электрофильными и нуклеофильными. Реакции за-
мещения, протекающие под действием электрофильных реагентов,
называются реакциями электрофильного замещения, а протекаю-
щие под действием нуклеофильных реагентов — реакциями нуклео-
фильного замещения.
Гомолитические реакции с участием незаряженных частиц, со-
держащих неспаренные электроны, называют свободнорадикаль-
ными. В особую группу выделяются так называемые реакции с
циклическим переносом электронов (электроциклические), проте-
кающие без участия полярных или свободнорадикальных частиц,
как, например, реакция Дильса — Альдера.
Отнесение реагента к числу электрофильных или нуклеофиль-
ных, как правило, не представляет затруднений, и потому класси-
фикация реакций замещения по типу реагента достаточно одно-
значна. Преимущество такой классификации в том, что она сразу
указывает на характерные особенности реакции, касающиеся ори-
ентации, влияния заместителей, среды и т. д., интерпретируемые
как следствие общности механизма. Однако выводы о механизмах,
базирующиеся на классификации реакций по типам реагентов,
сугубо ориентировочные. Информация о реальных механизмах ре-
акций может быть получена только путем специального экспери-
мента, который, естественно, вносит коррективы в упрощенные
схемы. В последнее время, например, показано, что некоторые ре-
акции, ранее рассматривавшиеся как чисто ионные, протекают че-
рез стадии одноэлектронного переноса с образованием радикалов
(см. 2.7.2). Подобные коррективы, углубляя знания о механизмах
реакций, не противоречат классификации реакций по типам реаген-
тов, если только не пытаться рассматривать ее как классификацию
по механизмам реакций, что не соответствует действительности.
Помимо классификации реакций по брутто-схемам и типам
реагентов используется разделение реакций по механизмам — на
мономолекулярные и бимолекулярные в зависимости от того, одна
или две реагирующие частицы участвуют на стадии, определяющей
скорость реакции. Согласно символике, введенной английской
школой, в обозначении реакции указываются все три признака.
Первым начальной буквой соответствующего английского слова
обозначается тип брутто-схемы (S — substitution, А — addition,
Е — elimination), затем — в индексе — тип реагента (N —
nucleophilic, Е — electrophilic, Н — homolytic или R — radical), по-
том цифрой — тип механизма (1—мономолекулярный, 2 — бимо-
лекулярный), например: SN1, SN2, SE2 и т. д. Специфика реакций
ароматических соединений иногда отражается знаком Аг, напри-
мер SNAr2.
41
2.4. ВЛИЯНИЕ СТРОЕНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
НА РЕАКЦИОННУЮ СПОСОБНОСТЬ
2.4.1. Электронные эффекты заместителей
Влияние заместителей на реакционную способность ин-
терпретируется на базе представлений об электронных и стериче-
ских эффектах. Чисто качественные представления об электронных
эффектах с созданием физической органической химии получили
количественную основу [8—10]. В 1937 г. Гаммет представил за-
висимость констант скоростей некоторых реакций, протекающих по
реакционному центру, примыкающему к бензольному кольцу, в
форме уравнения:
lg£ = lgfe° + Р Ig-^r (2.8)
где k и k°—константы скорости для мета- (лара-)замещенного и
для незамещенного производных, а К и К° — константы диссоциа-
ции для аналогично замещенной и для незамещенной бензойной
кислоты соответственно. Если обозначить lg-^3- = <T, соотношение
(2.8) приобретает вид:
Ig k = 1g k° + pa (2.9)
в котором оно и вошло в органическую химию под названием
уравнения Гаммета.
Параметр п в уравнении Гаммета отражает влияние мета- или
napa-замещенного фенила на величину константы равновесия ре-
акции диссоциации карбоновых кислот в воде при 25 °C, выбран-
ной в качестве стандартной (р = 1). Калибровка шкалы а, в ко-
торой стандартным заместителем (п==0) избран незамещенный
фенил, производится по формуле:
a = Р^що - Р^н2о (2.Ю)
Параметр р в уравнении (2.9) показывает, в какой мере проте-
кание данной реакции зависит от изменения электронной плотности
в реакционном центре по сравнению с реакцией диссоциации со-
ответственно замещенных бензойных кислот. Если уменьшение
электронной плотности благоприятствует реакции, р имеет поло-
жительное значение, если препятствует — отрицательное. Чем
больше скорость или состояние равновесия реакции зависит от
величины электронной плотности в реакционном центре, тем боль-
ше абсолютная величина р, отчего этот параметр называют кон-
стантой чувствительности реакции.
Согласно уравнениям (2.2) и (2.6), логарифмы констант равно-
весия и скоростей реакций при заданной температуре целиком
зависят и линейно связаны с изменениями свободных энергий
42
реакции AG и активации AG^ соответственно. Поэтому легко пока-
зать, что не только разность логарифмов констант (lg k — lgfe°),HO
и разность соответствующих изменений свободных энергий нахо-
дится в линейной зависимости от а-констант заместителей. Таким
образом уравнение (2.9) представляет собой, по сути дела, линей-
ную зависимость между величинами изменений свободных энергий
реакции или активации, являясь отражением принципа линейности
свободных энергий (ЛСЭ).
Еще Гаммет отметил, что, поскольку изменение свободной
энергии состоит из энтальпийной и энтропийной составляющих
[уравнения (2.1) и (2.3)], ЛСЭ может соблюдаться только при
условии постоянства энтропийной составляющей AS или линейной
зависимости между ней и энтальпийной составляющей АЛ. Первое
условие в общем случае не имеет места, а второе, получившее
название изокинетического соотношения, отнюдь не очевидно с
позиций фундаментальных законов термодинамики. Поискам тео-
ретического обоснования ЛСЭ посвящены специальные работы.
Открытие Гамметом принципа ЛСЭ и названного его именем
уравнения (2.9) вызвало огромное количество работ по примене-
нию подобного рода корреляций к исследованию различных хими-
ческих и физических свойств. Вскоре выяснилось, что уравнение
Гаммета даже в пределах мета- и пара-замещенных производных
бензола не всегда соблюдается. Сам Гаммет указал на это обстоя-
тельство, введя для пара-нитрогруппы два значения а: нормальное
(0,78) и завышенное (1,27), предназначенное для применения в
случае таких реакций, как кислотная диссоциация фенолов и ани-
линий-ионов. Использование завышенных значений ст оказалось
необходимым и для других пара-заместителей, обладающих силь-
ным отрицательным резонансным эффектом —R. С помощью этих
констант, обозначаемых в настоящее время а-, оказалось возмож-
ным коррелировать скорости реакций ароматического нуклеофиль-
ного замещения, отчего они получили название нуклеофильных
констант. Аналогично в случае реакций сольволиза замещенных
кумилхлоридов ХСбН4С(СН3)2С1, ионизации три-и диарилкарбино-
лов, а также реакций ароматического электрофильного замещения
заместителям с сильным положительным резонансным эффектом
+/? оказалось необходимым приписать большие отрицательные
значения по сравнению с соответствующими константами Гаммета
(электрофильные константы а+). Численные значения констант о,
<т~ и о+ для некоторых наиболее часто встречающихся заместите-
лей приведены в табл. 2.1.
Однако двойной набор констант для электроноакцепторных
(а, о-) и электронодонорных (а, сг+) пара-заместителей тоже не
обеспечивает универсальности уравнения (2.9). Широкое обследо-
вание показало, что значения ст не группируются около двух
величин — нормальной и экзальтированной, а распределяются в
виде непрерывной шкалы, охватывающей для нитрогруппы, на-
пример, интервал от 0,6 до 1,4. Бесперспективность пути чисто
эмпирического подбора все новых значений ст с ограниченным
43
Таблица 2.1
n-Константы замещенных фенилов*
Заместитель мета- пара-
°м О О м О" п а+ п О О п
н 0,000 0,000 0,000 0,000 0,000 0,000
СНз —0,069 -0,07 —0,170 — -0,311 -0,12
Свн6 0,06 0,005 —0,01 — -0,085 0,04
F 0,337 0,34 0,062 0,02 -0,073 0,21
С1 0,373 0,37 0,227 — 0,114 0,28
Вг 0,391 0,39 0,232 0,26 0,150 0,48
I 0,352 0,34 0,18 — 0,135 0,30
CF3 0,43 0,48 0,54 — 0,612 0,54
ОН 0,121 0,13 -0,37 — -0,853 -0,16
ОСН3 0,115 0,08 -0,268 -0,268 -0,778 -0,15
OCeHs 0,252 — —0,320 — -0,899 0,07
SCH3 0,15 0,12 0,00 — — 0,08
nh2 —0,16 —0,14 -0,66 — -1,111 -0,38
N(CH3)2 -0,05 —0,12 -0,83 — -1,7 -0,44
NHCOCH3 0,21 — 0,00 — —0,249 0,00
COCH3 0,376 0,38 0,502 0,85 0,567 0,49
соон 0,37 0,36 0,45 0,728 0,421 0,46
СООСНз 0,39 0,37 0,31 0,68 0,489 0,46
CN 0,56 0,62 0,660 0,89 0,659 0,72
no2 0,710 0,71 0,778 1,25 0,790 0,81
SO2CH3 0,56 0,70 0,68 1,05 0,747 0,75
О- __ -0,82 —• -0,81 — —1,21
COO’ -0,1 —0,12 0,0 — 0,109 -0,20
Й(СНз)з 0,88 1,04 0,82 — 0,636 0,88
* Значения ам и ап приведены по [8, с. 322—323], остальные—по [1, с. 458 — 459]; отсут-
ствующие в [1] данные—по справочнику [11, с. 166—174].
применением заставила обратиться к анализу физического смысла
используемых констант с позиций представлений об электронных
эффектах заместителей.
Анализ показал, что уравнение Гаммета не соблюдается тогда,
когда между замещенным фенилом и реакционным центром суще-
ствует сильное сопряжение. Непостоянство величин о обусловлено
зависимостью сопряжения от природы реакционного центра.
В реакционных сериях, где реакционный центр обладает +/?-эф-
фектом, при введении лара-заместителя с —/?-эффектом и в сериях,
где реакционный центр обладает —/?-эффектом, при введении ла-
ра-заместителя с -|-/?-эффектом возникает единая сопряженная
система, включающая заместитель, ароматическое кольцо и реак-
ционный центр. Так, при диссоциации иара-(—R)-замещенных
фенолов больший -|-/?-эффект реакционного центра в анионе (7) по
сравнению с кислотой (6) приводит к большей резонансной стаби-
лизации фенолят-иона и сдвигу равновесия в сторону диссоцииро-
ванной формы. При диссоциации же пара-(—R)-замещенных бен-
зойных кислот сопряжение с реакционным центром невозможно,
44
что находит выражение в разности абсолютных значений
ст" и ап для —R-заместителей (табл. 2.1).
Расчленение ст-констант на составляющие, отражающие индук-
ционное и резонансное взаимодействия раздельно, осуществлено
введением шкалы индукционных постоянных замещенных фенилов
ст° (табл. 2.1) с использованием в качестве стандартных процессов
реакций диссоциации замещенных фенилуксусных кислот, гидро-
лиза их эфиров и некоторых других, но с сохранением условий
стандартизации, принятых Гамметом. Чисто индукционное влияние
замещенного фенила, характеризуемое константой ст0, в свою оче-
редь складывается из индукционного эффекта заместителя, пере-
даваемого через фениленовый фрагмент, и резонансного эффекта
заместителя, влияющего на величину электронной плотности на
первом атоме углерода замещенного фенила, но непосредственно
реакционному центру не передающегося. Чтобы подчеркнуть это
обстоятельство, константы ст0 называют константами заместителей,
свободными от прямого полярного сопряжения.
Общее уравнение, учитывающее отдельно индукционное и ре-
зонансное взаимодействие замещенных фенилов с реакционным
центром типа +/?, имеет вид:
lg£ = JgA>° + ра° -ЬрдСТд (2.11)
а с реакционным центром типа —R:
lg*=lg*°-+ ра0 + р+а^ (2.12)
Здесь ст~ = ст" — о° и ст+ = ст+ —ст°; для всех .мета-замещенных
фенилов Стд = ст+ = 0.
Если выразить р+ = гр,уравнение (2.12)преобразуется в урав-
нение, предложенное Юкава и Цуно:
1g & = lgfe° + р [а° + г (о+ — а°)] (2.13)
где г — коэффициент, учитывающий, во сколько раз больше (или
меньше) прямое полярное сопряжение заместителя с реакционным
центром в данной реакционной серии по сравнению со стандарт-
ной (сольволиз кумилхлоридов, г — 1).
Число предложенных индукционных и резонансных ст-кон-
стант заместителей, а тем более способов их обозначения, не
45
исчерпывается указанными выше. Однако уравнения (2.11) и (2.12),
требующие применения двух параметров, в принципе, вполне до-
статочны для описания взаимодействия мета- и пара-заместителей
с реакционным центром в производных бензола. Точность количе-
ственного описания определяется главным образом точностью по-
строения шкалы резонансных постоянных, значения которых за-
метно различаются в разных источниках.
Что касается орто-заместителей, то количественная интерпрета-
ция их влияния пока отсутствует. Уравнение Гаммета не приме-
нимо для этой цели, а попытки модифицировать его путем учета
вклада таких факторов, как стерические затруднения или внутри-
молекулярная водородная связь, не привели к успеху. В ходе этих
попыток предложено не меньше шкал оо, чем насчитывается самих
характеризуемых заместителей (в работе [12] реферировано
23 шкалы), но ни одна из них не может претендовать на универ-
сальность, хотя в отдельных частных случаях соблюдаются про-
стые соотношения. Проблема орто-эффекта, по-видимому, не ис-
черпывается влиянием структурных факторов, а обусловлена раз-
нообразными причинами, в том числе спецификой механизмов
реакций орто-замещенных (см. 2.6).
В ряду гетероциклических соединений для описания эффекта
заместителей широко используются о-константы Гаммета, причем
при размещении заместителя и реакционного центра в положениях
3,5 и 2,4 в пятичленном гетероцикле оперируют о^-константами, а в
положениях 2,5 — ол-константами. Главное внимание при этом
уделяется величинам р, позволяющим оценить проводимость элек-
тронных эффектов в гетероцикле.
Применение корреляционных уравнений в ряду полицикличе-
ских соединений осложняется множественностью вариантов вза-
имного расположения заместителя и реакционного центра. Уже в
случае нафталина даже при исключении из рассмотрения орто- и
перы-положений каждому заместителю соответствует два ряда
констант по пять вариантов в каждом, так как реакционный центр
может находиться либо в а-, либо в p-положении. Для описания
«нафталиновых» констант заместителей вц необходимо указание
двух индексов: i — для положения заместителя в ядре и / — для
положения реакционного центра.
Влияние заместителя на реакционный центр, находящийся в
том же кольце нафталинового ядра, сходно с влиянием в анало-
гичных положениях бензола: о31 и о42 соответствуют ом, а о41 со-
ответствует Оп- Однако величины «нафталиновых» о не следует
непосредственно сравнивать с «бензольными», так как при опреде-
лении первых значение р для реакционной серии соединений на-
фталина условно принимается таким же, как для реакционной
серии производных бензола, что, не изменяя относительных значе-
ний ел/, искажает их абсолютные величины. В смежное кольцо
нафталинового ядра влияние заместителя передается существенно
ослабленным, причем прямое полярное сопряжение может переда-
ваться a-реакционному центру только из положений 5 и 7, а
46
р-реакционному центру — только из положений 6 и 8, отмеченных
звездочками (так называемые «хиногеновые» положения):
Количественная оценка влияния заместителей в многоядерных
соединениях, более сложных, чем нафталин, еще более затрудни-
тельна, и работы в этом направлении весьма ограничены.
2.4.2. Влияние аннелирования
Если для моноциклических ароматических соединений
структурные факторы, определяющие реакционную способность,
исчерпываются характером цикла и влиянием заместителей, для
полициклических соединений важное значение имеет также способ
сочленения (аннелирования) ароматических колец и их число.
Сочленение двух бензольных колец в нафталине порождает
специфические особенности, отличающие нафталин от бензола. Как
уже отмечалось (см. 1.3.2), характерная для бензола выравнен-
ность связей (d = 1,397 А, т. е. 0,1397 нм) в нафталине (8) нару-
шена таким образом, что связи С“ — укорочены до 1,371 А, а
связи —Ср удлинены (1,412 А). Повышенная л-электронная
плотность в области связи Са—нафталинового ядра обеспечи-
вает эффективную передачу электронных влияний а-реакционному
центру из положения 2 и ^-реакционному центру из положения 1.
Понижение л-электронной плотности связи Ср—Се приводит к
тому, что влияние заместителя на реакционный центр из положе-
ния 3 оказывается резко ослабленным и во многих случаях вообще
не играет роли. Классическим примером является реакция азосо-
четания 2-нафтола (9), которая направляется исключительно в
положение 1, а если оно занято, не происходит вовсе.
Присоединение реагента к нафталиновому ядру не ведет, как в
бензоле, к потере всей энергии резонанса, поскольку в молекуле
сохраняется одно бензольное кольцо, и поэтому нафталин легче,
чем бензол, вступает в реакции присоединения, замещения,
47
окисления, восстановления. По той же причине нафтолы легче, чем
фенол, реагируют в оксоформе (10).
При реакциях замещения a-положения в нафталине более ре-
акционноспособны, чем p-положения, вследствие меньшей энергии
активации при образовании а-замещенного ст-комплекса (11) по
сравнению с 0-замещенным ст-комплексом (12); это объясняется,
вероятно, большей возможностью для делокализации заряда: в
первом случае две резонансные структуры содержат ароматиче-
ское кольцо (11), тогда как во втором случае — только одна
(о ст-комплексах см. 2.7).
(и)
(12)
(13)
В то же время заместитель в a-положении нафталина находится
в большей пространственной близости от соседнего атома, находя-
щегося в перм-положении, чем заместитель в p-положении от со-
седнего атома, находящегося в а- или р'-положении (13). Про-
странственные затруднения со стороны пери-расположенного ато-
ма водорода приводят к меньшей термодинамической устойчивости
а-замещенных по сравнению с р-замещенными, а при наличии в
nepu-положении объемистой группировки могут и препятствовать
a-замещению. В результате, когда реакция замещения контроли-
руется кинетическими факторами, она направляется преимуще-
ственно в a-положение, а при термодинамическом контроле —
в p-положение. Примерами могут служить реакции нитрования и
сульфирования. Первая, будучи необратимой, целиком подчиняется
кинетическому контролю и направляется почти исключительно в
a-положения (см. 4.4.3). Вторая сначала, при кинетическом
контроле, приводит к а-нафталинсульфокислоте, а при приближе-
нии к равновесию — к р-нафталинсульфокислоте. Невозможность
сульфирования а-нитронафталина в пери-положение является
следствием стерических препятствий (см. 3.3.2).
Специфические особенности нафталина: ослабление передачи
электронных эффектов через связь С2 — С3, большая активность
a-положений, сильное пространственное влияние пери-заместите-
лей, проявляются и в его гетероаналогах — бензопиридинах (хино-
лин, изохинолин), бензазинах и т. д. Имея ввиду эти особенности,
можно говорить об общем эффекте бензаннелирования к моноцик-
лическим системам.
Присоединение к молекуле нафталина третьего бензольного
кольца возможно двумя способами: линейно и ангулярно. Линей-
ное аннелпрование приводит от нафталина к антрацену, ангуляр-
ное — к фенантрену, свойства которых существенно различаются.
Главное отличие антрацена (14) от нафталина заключается в по-
48
вишенной активности его .мезо-положений, присоединение к
которым не связано с большой потерей энергии резонанса (15).
(14) (15)
За счет мезо-положений антрацен легко вступает в реакцию
Дильса — Альдера в качестве диена, в реакции галогенирования,
восстановления и др. 9-Гидроксиантрацен (9-антрол) (16) суще-
ствует преимущественно в виде оксо-формы (антрон) (17).
(16) (1Т)
Дальнейшее линейное аннелирование приводит к все более
склонным к реакциям мезо-присоединения ароматическим углево-
дородам: тетрацену, пентацену, гексацену. Соответственно снижа-
ется устойчивость гидрокси- и повышается устойчивость оксо-фор-
мы. Тетраценон удается перевести в фенолят-анион действием ще-
лочи, а для пентаценона гидрокси-форма не известна. Указанное
изменение химических свойств в ряду аценов симбатно уменьше-
нию величины ЭР на л-электрон (см. 1.3.1).
Характерной особенностью фенантрена (18) является высокая
реакционная способность связи С9 — С10, близкой по длине к двой-
ной связи (см. 1.3.2), и пространственная затрудненность положе-
ний 4 и 5. Для фенантрена типичны реакции присоединения по
положениям 9, 10, в том числе галогенов, диенов в реакции Диль-
са— Альдера, при окислении и др. (19).
Таким образом, при линейном аннелировании наблюдается
повышенная реакционная способность мезо-положений, при ангу-
лярном — повышенная реакционная способность связи С—С цент-
рального кольца. Если в молекуле сочетаются оба типа аннелиро-
вания, каждый из них накладывает свой отпечаток. Так бенз[а]-
антрацен (20) подобно антрацену присоединяет малеиновый
49
ангидрид в положения 7,12 и подобно фенантрену окисляется
тетраоксидом осмия — реагентом на двойную связь, в 5,6-диол.
Предполагается, что наличие 9,10-фенантреноидной связи, чув-
ствительной к метаболическому окислению (A-область), обуслов-
ливает канцерогенную активность полициклических ароматических
углеводородов. Закономерности аннелирования, характерные для
полициклических углеводородов, в той или иной мере сохраняют
значение применительно к полициклическим гетероароматическим
соединениям.
2.5. ВЛИЯНИЕ СТРОЕНИЯ РЕАГЕНТА
Легкость протекания реакций, естественно, зависит от
строения не только субстрата, но и реагента. Поскольку для аро-
матических соединений наиболее типичны реакции электрофильно-
го и нуклеофильного замещения, вопрос прежде всего сводится к
установлению связи между электрофильностью-нуклеофильностью
реагентов и их строением.
Как уже отмечалось (см. 2.3), все электрофильные реагенты
являются кислотами Льюиса, а все нуклеофильные — основаниями.
Однако между шкалой электрофильности-нуклеофильности с од-
ной стороны и шкалой кислотности-основности с другой нет полной
симбатности. Причина в следующем. Хотя эти шкалы отражают
одно и то же свойство — способность принимать или предоставлять
пару электронов при образовании ковалентной связи — они отно-
сят его к разным партнерам: кислотность и основность характе-
ризуют сродство к протону, а электрофильность и нуклеофиль-
ность — к атому углерода. Как прочность образуемой связи, так
и энергия активации кислотно-основного взаимодействия зависит
от обеих реагирующих частиц. Поэтому нет единого универсаль-
ного показателя, который бы определял кислотность или основ-
ность вообще, независимо от контрагента.
Для характеристики кислотно-основного взаимодействия каж-
дой кислоте А и каждому основанию В помимо параметра, опре-
деляющего их силу S, необходимо приписать второй параметр о,
названный Пирсоном [13] параметром мягкости. С помощью этих
двух параметров константа равновесия кислотно-основной реакции:
А+ В 4=^ АВ, может быть выражена уравнением:
1g К — SASB + оАав (2.14)
Те кислоты и основания, у которых параметр мягкости велик,
получили название мягких, а те, у которых он мал — название
жестких.
Существует несколько эмпирических уравнений, в которых каж-
дый реагент характеризуется двумя параметрами. Наиболее из-
вестно из них уравнение Эдвардса [14]:
ь
= + (2.15)
К
50
где постоянные Н и Еп характеризуют нуклеофил, а постоянные а
и р — электрофил; Н — коэффициент основности нуклеофила, рав-
ный рДа + 1,74; Еп — окислительно-восстановительный коэффи-
циент, равный Е° 4- 2,60. Анализ показывает [15], что член $Н в
уравнении Эдвардса (2.15) может быть сопоставлен с членом SASB,
а член — с членом оАов в уравнении (2.14). Уравнение Эдвард-
са охватывает большое число данных по скорости и равновесию,
но, как и другие подобные уравнения, имеет ограничения, в част-
ности соблюдается не для всех типов электрофилов.
Смысл названий жесткие и мягкие отражает способность ато-
мов кислот и оснований удерживать электроны. Атомы жестких
кислот и оснований имеют высокую электроотрицательность, ма-
лый атомный радиус, малую поляризуемость и удерживают элект-
роны прочно. Атомы мягких кислот и оснований имеют меньшую
электроотрицательность, больший атомный радиус, высокую поля-
ризуемость и удерживают электроны слабее. Классификация неко-
торых кислот и оснований по признаку жесткости и мягкости
приведена в табл. 2.2.
Как видно из таблицы, для элементов первых двух перио-
дов периодической системы жесткой (ж) кислоте соответствует
мягкое (м) основание и наоборот, например: Н+(ж)—Н_(м),
НО“(ж)—• НО+(м), НзС+(ж) —R3C~(m). Для атомов тяжелых
элементов с удаленными от ядра орбиталями и кислота и основание
мягкие, например: I-, I+, RS~, RS+. Молекула ароматического со-
единения является мягкой кислотой (тринитробензол) или мягким
основанием (бензол).
Пирсон в 1963 г. [13] сформулировал простое правило отно-
сительно стабильности и реакционной способности химических
Таблица 2.2
Классификация кислот и оснований*
Жесткие Мягкие Промежуточные
Кислоты
Н+, Li+, Na+, К+, BFa, Hg2+, RS+, I+, Br+, HO+, SO2+, NO+, C6H5+, R3C+
A1C13, RSO2+, ROSO2+, RO+, I2, Br2, C6H3(NO2)3
SO3, RCO+, CO2 хнноны н другие л-кисло-
ты, О, Cl, Br, RO •, ROO .,
карбены
Основания
Н2О, НО’, F", СНзСОО’, R2S, RSH, RS’, Г, SCN~, C6H5NH2, C5H5N, N"
SOf, СГ, ROH, RO', S2O2’, CN', CO, C2H4, Br-, NO’, SO2’
r2o, nh3, rnh2, nh2nh2 C6H6, H*, R3C’, C6H’
* R = Alk, Ar.
51
соединений, названное принципом жестких и мягких кислот и ос- 1
новаций (ЖМКО). Согласно принципу ЖМКО, жесткие кислоты I
предпочтительно связываются с жесткими основаниями, а мягкие^
кислоты — с мягкими основаниями. Принцип ЖМКО [15—17] ;
представляет собой очень общий, но не точный закон. Понятия I
мягкость и жесткость включают совокупность фундаментальных
свойств атомов, молекул и ионов, в том числе электроотрицатель- ’
ность, сродство к электрону, потенциал ионизации, энергия связи, 1
несвязывающие взаимодействия, сольватация и другие, и не могут
быть сведены к какому-либо одному из них. Поэтому физическая
интерпретация этих понятий и их количественное выражение со-
пряжены с трудностями. В то же время именно благодаря своему
феноменологическому характеру и широте охвата принцип ЖМКО
позволяет ориентироваться в самых разнообразных химических
явлениях. Теоретическому обоснованию принципа ЖМКО посвя-
щены специальные работы (см. 2.8).
В свете принципа ЖМКО понятно несовпадение шкал кислот-
ности-основности и электрофильности-нуклеофильности, поскольку
первая отражает сродство к жесткой кислоте Н+ и жесткому
основанию ОН , а вторая — к значительно более мягким кислотам
и основаниям, имеющим в качестве реакционного центра атом
углерода.
В некоторых пределах основность и нуклеофильность изменя-
ются согласованно. Они увеличиваются с переходом от нейтраль-
ных молекул к ионам:
НО' > Н2О, NHJ » NH3
и с уменьшением электроотрицательности ионов:
I I
Н‘ > —С- > N" > —О > СГ
I I
Для кислородсодержащих реагентов основность и нуклеофиль-
ность уменьшаются под влиянием —/-эффекта и увеличиваются
под влиянием -[-/-эффекта:
AlkO' > НО' > АгО' > АсО" > RSO2O"
а для карбанионов возрастают с уменьшением их устойчивости:
III I I
AlkC" > ArC' > С'—CN > С'—СО > С'—NO2
111 I I
Однако при сравнении реагентов с реакционными центрами,
различающимися по жесткости ^мягкости), симбатности в измене-
нии основности и нуклеофильности не наблюдается. Так, слабые, но
мягкие серусодержащие основания (RS~, RSH) значительно пре-
восходят по нуклеофильности сильные, но жесткие кислородсодер-
жащие основания (RO~, ROH). Ряд возрастания нуклеофильности
52
галоген-анионов I > Вг > Cl > F в протонных растворителях
противоположен ряду возрастания их основности в той же среде.
Когда в молекуле нуклеофила возможно взаимодействие с орби-
талями неподеленной пары электронов соседнего атома, мягкость-
реагента и его нуклеофильность увеличиваются, хотя основность-
под влиянием —/-эффекта снижается. Так, реагенты ROO RSS_,
RNHNH'2 более нуклеофильны и менее основны, чем RO , RS ,
RNH2 соответственно. Подобные соотношения можно отметить-
также между кислотностью и электрофильностью.
При наличии в субстрате и (или) реагенте нескольких реакци-
онных центров принцип ЖМКО может быть весьма полезен для
предсказания и объяснения направления реакции [16, 17]. Приве-
дем несколько примеров.
При взаимодействии 2,4-динитрофенил-м-толуолсульфоната.
(21) с жестким основанием — метилат-анионом атака в основном
(88%) направляется по более жесткому реакционному центру
субстрата — сульфонильному атому серы (22), а при взаимодей-
ствии с мягким основанием — тиофенолят-анионом, по более мяг-
кому реакционному центру — ароматическому атому углерода (23>
[18] (см. также 9.2).
(21)
+ СНзО-
-|- M-CH3C6H4SO3CH3
+ CeH6S
-|- M-CH3C6H4SO3
(23)
На жесткость или мягкость реакционного центра существенное-
влияние оказывает характер связанного с ним уходящего атома
или группы. Так, при реакции 2,4-динитро-Х-бензолов (24) с амби-
дентными нуклеофилами атом углерода связывается с более же-
стким реакционным центром реагента, если отщепляемый атом или
группа — жесткое основание, и с более мягким центром, если ухо-
дит мягкое основание. В реакции с нитрит-анионом при X = F
образуется эфир (25), превращающийся далее в фенол (26), при
Х = 1 образуется 1,2,4-тринитробензол (27), а при X = С1, Вг —
смесь обоих продуктов [19]. Реакция с тиоцианат-анионом при
X=NC5H5 приводит в основном к арилизотиоцианату (28), а при
53
X — I — к арилтиоцианату (29), причем отношение скоростей N- и
S-замещения при указанных X, различается более, чем в 105 раз
[20].
Если в ароматическом субстрате имеются две или более потен-
диальные уходящие группы, различающиеся по жесткости, на-
правление атаки зависит от жесткости и мягкости реагента. Так, в
бензогетероцикле (30), содержащем два разных атома галогена,
при действии алкоголята (жесткое основание) замещается атом
фтора (31), тогда как при действии тиолята (мягкое основание) —
атом хлора (32) [21].
(32)
Природа уходящей группы реагента также имеет большое зна-
чение, определяя степень жесткости или мягкости атакующего
центра. Это ярко проявляется при алкилировании амбидентных
соединений [22]. При действии на антрон (33) щелочного раствора
диметилсульфата, уходящая группа в котором — жесткое осно-
вание CH3OSOr, происходит О-метилирование с образованием
'9-метоксиантрацена (34), а при действии метилиодида, где уходя-
54
щая группа — мягкое основание I-, происходит С-метилирование с-
образованием 10-метил- (35) и 10,10-диметилантрона (36) [16]:
+ СН31
+ (CH3O)2SO2
ОСНз
(34)
При метилировании арилсульфинатов (37) действие диметил-
сульфата приводит к О-метилированию (38), а действие метилио-
дида — к S-метилированию (39) [23]:
ArSO2’Na+
(37
О
+ (CH3O)2SO2 II
------------» Ar S—ОСНз
(38)
О
+ СН31 II
-----------► ArS—СН3
(39)
Приведенные примеры показывают, что, руководствуясь прин-
ципом ЖМКО, можно, варьируя строение реагентов и характер
замещаемых групп, воздействовать на скорость и селективность
реакций ароматических соединений.
2.6. ВЛИЯНИЕ РАСТВОРИТЕЛЯ
За исключением отдельных газофазных процессов реак-
ции ароматических соединений проводятся в жидкой фазе, в среде
растворителя, роль которого может играть одно из реагирующих
веществ. Молекулы растворителя взаимодействуют с реагирующи-
ми частицами на всех этапах реакции, образуя вокруг них соль-
ватные оболочки, которые могут изменять свободную энергию и
электронную структуру частиц и, как следствие, скорость, меха-
низм и направление реакции. Фактически в реакции участвуют не
изолированные частицы, которые изображают при написании хи-
мических уравнений, а качественно отличные от них сольватные
комплексы. Без учета влияния среды правильное понимание тече-
ния реакций невозможно.
55.
Принято различать два типа взаимодействий между раствори-
телем и растворенным веществом: неспецифическую и специфиче-
скую сольватацию [24—27]. В первом случае растворитель рас-
сматривается как однородная изотропная среда, характеризуемая
макроскопическими постоянными, такими как диэлектрическая
проницаемость и показатель преломления, во втором учитываются
конкретные особенности строения молекул растворителя: способ-
ность к образованию водородных связей, к донорно-акцепторному
н электростатическому взаимодействию.
По протонодонорной способности при образовании межмолеку-
лярной водородной связи растворители делятся на протонные и
апротонные; последние в свою очередь разделяются по степени
полярности на биполярные и аполярные. Для характеристики по-
лярности используются различные показатели, между которыми
нет строгой симбатности: диэлектрическая постоянная е, диполь-
ный момент ц, спектроскопические параметры (например, энергия
электронного перехода, соответствующего полосе переноса заря-
да в спектре стандартного красителя (40) в данном раствори-
теле —Ет-
(40)
Множественность показателей при отсутствии строгой симбатно-
-сти между ними отражает расплывчатость самого понятия поляр-
ности и отсутствие четкой границы между группами аполярных
и биполярных растворителей. Условно принимают [24], что апо-
лярные апротонные растворители (углеводороды, галогенпроиз-
водные, третичные амины, сероуглерод) имеют е < 15, ц < 2D*,
Ет~ 1304-170 кДж/моль, биполярные апротонные растворители
(У,У-двузамещенные амиды кислот, сульфоксиды, сульфоны,
кетоны, нитрилы, нитропроизводные) — е > 15, ц > 2,5 D,
£т« 1704-200 кДж/моль; протонные растворители (вода, аммиак,
•спирты, карбоновые кислоты, незамещенные амиды) имеют
£т~ 2004-264 кДж/моль. Характеристики некоторых наиболее
распространенных растворителей приведены в табл. 2.3.
Поскольку при донорно-акцепторном взаимодействии с раство-
ренным веществом растворитель выступает либо как кислота, либо
•как основание, для классификации растворителей может быть
привлечен принцип ЖМКО (см. 2.5). В применении к растворам
принцип ЖМКО означает, что жесткие растворители сольватируют
преимущественно жесткие субстраты, а мягкие растворители —
мягкие субстраты, и представляет собой современную формули-
ровку старого правила: «подобное растворяется в подобном».
* 1D = 3,34-10 - 30 Кл-м.
56
Таблица 2.3
Характеристики органических растворителей [24]
Растворитель Т. кип., °C (при 7&0 мм. рт. ст.) е (при 25 °C) ц,, D (в бензоле при 25-30 °C) кДж/моль (при 25 °C)
Гексан 68,8 1,9 0 130,1
Четыреххлористый угле- 76,7 2,2 0 136,0
род
Бензол 80,1 2,3 0 144,3
Диэтнловый эфир 34,6 4,2 1,25 144,7
Диоксан 101,4 2,2 0,4 150,6
Тетрагидрофураи 65,4 7,4 1,7 156,2
Хлорбензол 132 5,6 1,55 157,0
Хлороформ 61,2 4,7 1,1 163,6
Пиридин 115,5 12,3 2,2 168,2
Нитробензол 210,8 34,8 4,0 175,7
Ацетон 56,2 20,7 2,7 176,6
Диметилформамид 153 36,7 3,8 183,2
Сульфолан 283 44,0 4,8 184,1
Диметилсул ьфоксид 189 48,9 3,9 188,2
Ацетонитрил 81,6 37,5 3,5 192,5
Нитрометан 101,1 38,6 3,1 194.2
Этанол 78,3 24,3 1,7 217,2
Метанол 65,0 . 32,6 1,65 232,2
Формамид 210 109,5 3,4 236 8
Вода 100,0 78,5 1,8 264,0
Сольватационные эффекты решающим образом зависят от-
природы сольватируемых частиц, в первую очередь от их заряда.
Анионы, катионы и нейтральные полярные молекулы по-разному
взаимодействуют с растворителями разных классов. Анионы силь-
но сольватируются протонными растворителями за счет образова-
ния водородных связей, причем малые по размеру анионы сольва-
тируются сильнее, чем большие. В биполярных апротонных раство-
рителях анионы сольватируются слабо, но все же в этом случае
большие поляризуемые анионы сольватируются лучше малых. Ка-
тионы плохо сольватируются протонными растворителями, но хо-
рошо сольватируются такими биполярными растворителями, у ко-
торых электронная плотность локализована на атоме кислорода
(диметилформамид, диметилсульфоксид, простые эфиры). Поляр-
ные молекулы, если они не являются сильными донорами или
акцепторами протона при образовании водородных связей, обычно
сильнее сольватируются биполярными апротонными раствори-
телями.
Ионные частицы в растворе ассоциированы не только с окру-
жающими молекулами растворителя, но и с противоионом. Взаимо-
действие противоположно заряженных частиц в растворе не ис-
черпывается двумя крайними ступенями: ковалентным связывани-
ем (АВ) и полной диссоциацией (А++В~). Гетеролитический
разрыв связи А—В может не привести к расхождению ионов,
остающихся в виде контактной (тесной) ионной пары (А+В-), ок-
руженной общей сольватной оболочкой. Создание вокруг одного из
53
ионов собственной сольватной оболочки, вызывая разделение
ионов молекулами растворителя, ослабляет, но не нарушает пол-
ностью взаимодействие между ними. Такая ионная пара (А+ || В~)
носит название сольватно-разделенной или рыхлой. Только когда
каждый из ионов приобретает свою сольватную оболочку, проис-
ходит их полное разделение. Ионные пары влияют на давление
паров, температуру замерзания и электропроводность раствора,
как единое целое, но их спектральные характеристики отличаются
от спектров неионизированной молекулы. Электронные спектры
анионов в контактных ионных парах зависят от радиуса катиона,
приближаясь к спектру свободного аниона с увеличением этого
радиуса. Электронные спектры сольватно-разделенных ионных пар
практически совпадают со спектрами свободных ионов [28]. По
реакционной способности сольватно-разделенные и, тем более,
контактные ионные пары уступают свободным ионам. Таким обра-
зом реакционная способность иона в растворе зависит не только от
его собственной сольватации, но и от сольватации противоиона,
определяющей степень ассоциации ионов.
Реакционная способность иона в растворе тем больше, чем сла-
бее связан он с молекулами растворителя и с противоионом. Оба
эти фактора решающим образом влияют на нуклеофильность ани-
онов по отношению к органическим субстратам. В жестких протон-
ных растворителях сольватация анионов за счет водородных связей
увеличивается с увеличением плотности заряда при уменьшении
размера иона, т. е. с увеличением его жесткости. В мягких бипо-
лярных апротонных растворителях сольватация анионов, вообще
незначительная, увеличивается в обратной последовательности с
увеличением размера иона. В результате нуклеофильность аниона
при переходе от протонного к биполярному апротонному раствори-
телю возрастает, и тем больше, чем более жестким является анион.
Иллюстрацией этого может служить влияние растворителя на ско-
рость нуклеофильного замещения атома галогена в п-нитрогалоге-
нобензоле под действием более жесткого (Из) и более мягкого
(SCN-) аниона (табл. 2.4). При сравнении для уменьшения влия-
ния природы уходящей группы (см. 2.5) в первом случае исполь-
зовано вытеснение жесткого (X=F), а во втором мягкого (Х=1)
основания.
Таблица 2.4
Влияние природы растворителя на скорость реакции
п-нитрогалогенобеизолов с нуклеофильными анионами [26]
n-NO2C6H4X + Y’ —> «-NO2C0H4Y + X'
Растворитель — 1g k [в л/(моль’с) при 25 °C] Растворитель — 1g k [в л/(моль«с) при 25 °C]
Y = N. X=F y=scn, X=I Y = N, X = F Y = SCN, X=I
•СНзОН 7,2 3,8 (CH3)2SO 3,2 2,7
hconh2 6,6 —- (CH3)2CO 2,3 1,4
HCON(CH3)2 2,7 2,1 CH3CN 3,3 2,5
58
Как видно из таблицы, при переходе от протонного растворите-
ля (метанола) к биполярному апротонному (диметилформамиду)
скорость реакции с азид-анионом увеличивается на 4,5 порядка (в
32 000 раз), а скорость реакции с тиоцианат-анионом всего на
1,7 порядка (в 50 раз). В пределах одной группы растворителей,
протонных или биполярных апротонных, скорость реакции изменя-
ется незначительно.
Различия в специфической сольватации анионов в протонных и
биполярных апротонных растворителях приводят к совершенно
различным рядам нуклеофильности анионов. Обычный для про-
тонных растворителей ряд нуклеофильности: I- > SCN~ > Br- >
> Cl- > F~ в биполярных апротонных растворителях полностью
инвертируется: F- > Cl > Br~ > SCN~ > I-. Поскольку различия
вызваны снижением реакционной способности анионов из-за силь-
ной сольватации в протонных растворителях, ряд нуклеофильности
в биполярных апротонных растворителях правильнее отражает
«истинную» нуклеофильность анионов, обусловленную их элект-
ронным строением (см. также 10.1).
«Истинная» нуклеофильность анионов маскируется не только
сольватацией, но и ассоциацией с катионами, что особенно заметно
в слабо ионизирующих апротонных растворителях, таких как аце-
тон, ацетонитрил. Так, нуклеофильная активность галогенид-анио-
нов в ацетоне в реакции бутил-п-бромбензолсульфоната с галоге-
нидами лития изменяется в той же последовательности, что и в
протонных растворителях (1~> Вг~ > СП), а в реакции с галоге-
нидами тетрабутиламмония в противоположной последовательно-
сти, как в диметилформамиде (Cl~> Br > I-):
n-BrCeH4SO2OC4H9 + Х’(М+) —> п-ВгС6Н,50з(м+) + С4Н9Х
Относительные скорости галогенид-анионов при М+ — Li+ и при
М+— N(C4Hg)4 составляют соответственно (стандарт — LiCl):I~
6,2, 3,7; Вг+5,7, 18; С1~ 1,0, 68 [29]. Это объясняется тем, что ма-
ленький катион лития легко объединяется с анионами в ионные
пары, причем тем легче, чем меньше анион, а большой катион те-
трабутиламмония имеет малую склонность к ассоциации и потому
его соли даже в ацетоне практически полностью диссоциированы.
Уменьшение скорости реакции с хлорид-анионом в 68 раз при пере-
ходе от соли тетрабутиламмония к соли лития отражает эффект
образования ионных пар.
Способом разрушения ионных пар является введение специаль-
ных добавок, наппимер краун-эфиров [30], обладающих способно-
стью прочно координироваться с катионом металла, удерживая его
во внутренней сфере (41). Введение комплекса краун-эфира с
фторидом калия [(41), Y~ = F~] делает возможным количествен-
ное замещение атома хлора фторид-анионом в бензоилхлориде и
2,4-динитрохлорбензоле в среде ацетонитрила, в то время как
в отсутствие краун-эфира реакция в тех же условиях практиче-
ски не идет [31]. Нагревание при 90°C комплекса краун-эфира с
59
метилатом калия [(41), Y~ = СН3О“] в о-дихлорбензоле приводит!
к замещению атома хлора метоксигруппой с образованием о-хлор;
анизола (43), хотя в отсутствие краун-эфира о-хлоранизол не обра-
зуется даже в виде следов [32]. Однако в гексаметил фосфортри-]
амиде (гексаметапол) без краун-эфира выход о-хлоранизола со-5
ставляет 78% (см. 8.2).
(41)
(42) (43)
В амбидентных анионах, имеющих два реакционных центра,
направление реакции может изменяться в зависимости от раство-
рителя [33]. Так, при взаимодействии фенолята натрия с алкилга-
.логенидами (бензилхлорид, аллилхлорид) в спиртах, эфирах, ди-,
метилсульфоксиде наблюдается исключительно О-алкилирование,
но в протонных растворителях, экранирующих жесткий оксидный
центр фенолят-аниона вследствие образования прочных водород-
ных связей, происходит также и С-алкилирование (24% в воде, 69%
в феноле, с бензилхлоридом) [34]. При реакции 2-нафтолята на-
трия с бензилбромидом при комнатной температуре в диметил-
формамиде и диметилсульфоксиде алкилирование также направ-
ляется исключительно по атому кислорода, но уже в тетрагидро-
фуране и 1,2-диметоксиэтане наблюдается С-алкилирование, а в
протонных растворителях оно становится преобладающим (84% в
.воде). Алкилирование 2-нафтолят-аниона по атому углерода в
апротонных растворителях тетрагидрофуране и 1,2-диметоксиэтане
объясняют его ассоциацией с катионом натрия в ионные пары;
последние затрудняют О-алкилирование из-за удаленности катио-
на металла от отщепляемого галогенид-аниона в переходном со-
стоянии (44) и способствуют С-алкилированию вследствие сближе-
ния катиона и аниона в переходном состоянии (45) [35].
Из изложенного ясно, что вопрос о нуклеофильности анионов не
может обсуждаться без указания на природу растворителя.
В общем случае оценка активности ионного реагента по скорости
реакции в данном растворителе дает информацию об активности
ассоциата (или ассоциатов), которые образуют реагент с молеку-
•60
лами растворителя и противоионами. Для выяснения собственного
влияния строения частицы реагента требуются специальные мето-
ды. Кроме того, нужно иметь в виду, что природа растворителя
влияет не только на состояние исходных веществ, но и на стабиль-
ность активированных комплексов, .что также изменяет скорость
реакции. Влияние сольватации переходных состояний прослежива-
ется в реакциях между нейтральными полярными молекулами,
сольватация которых меньше влияет на реакционную способность,
чем сольватация ионов.
Согласно качественной теории Хьюза и Ингольда о влиянии
растворителей [36], скорость реакции между незаряженными мо-
лекулами, протекающей через переходное состояние с частичным
разделением зарядов, возрастает с увеличением полярности среды.
В соответствии с этим правилом, реакции ароматического замеще-
ния, протекающие через переходное состояние, подобное по струк-
туре биполярным о-комплексам, ускоряются с увеличением поляр-
ности растворителя. Однако влияние растворителей зависит не
только от их полярности. Наиболее обстоятельно это показано на
примере реакции ароматических галогенпроизводных с аминами
[37]. При близкой полярности растворители тем больше ускоряют
реакцию с пиперидином (46)-*(49), чем больше их основность:
диоксан больше, чем бензол, пиридин больше, чем нитробензол и
т. д. Это объясняют специфической сольватацией за счет образо-
вания водородной связи в о-комплексе (47), облегчающей отрыв
протона от атома азота (общий основной катализ). В значительной
степени влияние основного растворителя зависит от природы заме-
щаемого атома. Увеличение скорости реакции с пиперидином в
ряде растворителей: бензол, этилацетат, метилэтилкетон, ацето-
нитрил, диметилформамид, диметилсульфоксид — составляет при
50 °C для п-нитрофторбензола: 1, 11, 59, 300, 1950, 7200, а для
п-нитрохлорбензола: 1,2, 15, 34, 142, 412 соответственно при отноше-
нии скоростей обмена атомов фтора и хлора в бензоле 24: 1 [38].
Большее влияние основных полярных растворителей В на скорость
замещения атома фтора относят за счет образования более про-
чных водородных связей с сопряженными кислотами ВН+ на ста-
дии отрыва галогенид-аниона (48) — общий кислотный катализ.
Для амфипротных растворителей ZH, обладающих как основным,
так и кислотным характером (например, пиперидин), допускается
возможность одновременного образования водородных связей с
протоном аммониевой группы и с атомом галогена (50) — бифунк-
циональный катализ.
+С5Н,0МН
+в
01
Особый случай представляет собой влияние растворителей на
реакции соединений, содержащих электроноакцепторный замести-
тель в орто-положении к реакционному центру. В неполярной среде
орто-замещенные реагируют значительно быстрее, чем пара-заме-
щенные. В то время как с увеличением полярности растворителя
скорость реакции пара-замещенных возрастает, скорость реакции
орто-замещенных снижается, хотя и в меньшей степени, в резуль-
тате чего соотношение скоростей реакций орто- и пара-замещенных
уменьшается. Так, отношение констант скоростей ko]kn реакции
нитрохлорбензолов с пиперидином в бензоле, метаноле и диметил-
формамиде при 170 °C составляет 64, 2,8 и 1,3 [39]. Большее
активирующее воздействие орто-заместителей объясняют [37] ли-
бо образованием внутримолекулярной водородной связи в пере-
ходном состоянии (51), либо непосредственным электростатиче-
ским взаимодействием соседних противоположно заряженных
групп (52) (внутримолекулярная или «встроенная» сольватация);
уменьшение скорости реакции с увеличением полярности раствори-
теля объясняется нарушением указанных взаимодействий в ре-
зультате «внешней» сольватации молекулами растворителя.
Интерпретация влияния растворителей на переходные состоя-
ния с учетом совокупности электростатических взаимодействий и
образования водородных связей, классифицируемого как проявле-
ние различных вариантов кислотно-основного гомогенного катали-
за, позволяет объяснить широкий круг наблюдений. Однако все это
имеет гипотетический характер ввиду сложности прямого экспери-
ментального обнаружения предполагаемых эффектов.
2.7. ЭЛЕМЕНТАРНЫЕ СТАДИИ РЕАКЦИЙ
АРОМАТИЧЕСКОГО ЗАМЕЩЕНИЯ
Можно считать установленным, что любая реакция аро-
матического замещения представляет собой совокупность двух или
более последовательных элементарных реакций (элементарных
стадий), каждой из которых соответствует свое переходное состоя-
ние и свой конечный продукт. Вопрос о том, из каких элементар-
ных стадий состоит реакция и какова роль отдельных стадий, имеет
фундаментальное значение для понимания природы влияния стро-
ения субстрата, реагента, растворителей и других факторов на
протекание реакции и, следовательно, для управления ею. Будучи
самым важным из вопросов, касающихся механизма реакций, этот
вопрос в то же время и самый сложный. Главная трудность состоит
даже не в том, чтобы идентифицировать возможные лабильные
продукты элементарных стадий, а в том, чтобы определить, яв-
ляются ли они промежуточными или побочными, и если промежу-
точными, то в какой мере определяют течение суммарной реакции.
Ниже этот круг проблем рассматривается применительно к каждо-
му из трех типов реакций ароматического замещения.
2.7.1. Электрофильное замещение
Принято считать [40, 41], что реакции ароматического
электрофильного замещения протекают с участием двух видов ча-
стиц, называемых в соответствии с предполагаемым характером
связывания электрофила л- и о-комплексами.
л-Комплексы построены как молекулярные комплексы донорно-
акцепторного типа [42—44], в которых молекула ароматического
соединения выступает в качестве л-донора, а частица электрофи-
ла — в качестве акцептора (53).
о-Комплексы представляют собой солеобразные соединения, в
которых один из атомов углерода ароматического кольца образует
с частицей электрофила ковалентную о-связь за счет двух элект-
ронов л-системы (54).
(53) (54)
л- и о-Комплексы называют также соответственно внешними и
внутренними [43], подчеркивая этим, что образование первых за-
трагивает лишь периферию молекулы, тогда как образование втО’
рых вызывает перестройку скелета.
Реальность существования комплексов того и другого вида в
общем плане не вызывает сомнений. л-Комплексы ароматических
углеводородов выделены и изучены в кристаллическом состоянии и
в растворах. Кристаллы комплексов бензола с галогенами, имею-
щие состав 1:1, по рентгеноструктурным данным [45] состоят из
цепочек чередующихся молекул донора и акцептора, в которых
молекула галогена (Вг2, С12) расположена перпендикулярно пло-
скости кольца вдоль оси, проходящей через его центр симметрии
(55). В комплексах ароматических углеводородов с органическими
акцепторами (1,3,5-тринитробензол, хлоранил и др.) молекулы до-
нора и акцептора расположены друг над другом, так что центры
колец лежат на одной линии, перпендикулярной плоскости ядра
(56) [42]. Возможна и иная ориентация акцептора относительно
ароматического ядра. В комплексе бензола с перхлоратом серебра
(1:1) катион Ag+ координируется с двумя бензольными ядрами,
63
находясь над и под углерод-углеродными связями 1—2 и 4—|
каждого из них (57) [46].
(S5) (56) (57)
При пропускании в бензол или толуол хлористого водорода в
растворе возникает неокрашенный комплекс (1 : 1), не обладающий
электропроводностью и не обменивающий атомы водорода на дей-
терий при применении DC1 вместо НС1. Это указывает на неионное
строение комплекса (58) и отсутствие в нем ковалентной связи
ароматического соединения с протоном кислоты [47].
о-Комплексы ароматических углеводородов [48—50] легко об-
разуются при действии сильных кислот Льюиса. Эти комплексы
окрашены, их растворы проводят электрический ток, а атомы во-
дорода ароматического кольца легко обмениваются на дейтерий.
Согласно рентгеноструктурным данным [51], в кристалле тетра-
хлоралюмината гептаметилбензолония (59) пять хр2-гибридизован-
ных атома углерода кольца и связанные с ними метильные атомы
углерода лежат в одной плоскости, а кр3-гибридизованный атом
углерода несколько отклонен от нее, и его связи с двумя атомами
углерода метильных групп расположены в перпендикулярной пло-
скости выше и ниже плоскости кольца *.
сн3
(60)
Равновесие реакции протонирования ароматических углеводо-
родов в среде сильных кислот (HF, HSO3F, HF — SbF5 и др.),
например
А + 2HF AH+ + HF2‘
использовано для оценки их основности, варьирующейся в широких
пределах, от рКь = +9,2 для бензола до рКь = —0,4 для гекса-
метилбензола и —5,7 для 9-метилантрацена [52]. Константа ос-
* Межатомные расстояния указаны в ангстремах; 1 А = 0,1 нм.
64
новности Кь служит мерой термодинамической стабильности обра-
зующегося аренониевого катиона AHf.
Особенно ценная информация о строении, образовании и пре-
вращениях аренониевых ионов получена методом ядерного маг-
нитного резонанса [48]. В основном она касается стабильных ионов
типа [(60), X = СНз, NO2, SO3H, С1, Вг], моделирующих о-комп-
лексы разнообразных реакций электрофильного замещения, но в
ряде случаев удалось наблюдать непосредственно ст-комплексы,
содержащие в кольце фрагмент >СНХ.
Факт существования комплексов между ароматическими со-
единениями и электрофильными реагентами послужил основанием
для предположения о том, что л- и о-комплексы являются проме-
жуточными продуктами реакции ароматического электрофильного
замещения, которая таким образом может быть представлена че-
тырьмя последовательными элементарными стадиями: 1 — образо-
вание л-комплекса между исходным соединением и электрофилом;
2 — превращение л-комплекса в о-комплекс; 3 — переход
о-комплекса в л-комплекс между конечным продуктом и уходящей
частицей; 4 — разрушение л-комплекса с элиминированием конеч-
ного продукта:
Такая последовательность вполне вероятна, но само по себе
перечисление вероятных элементарных стадий не представляет
большой ценности, если не установлено, какие именно из них опре-
деляют скорость реакции и каково строение соответствующих пе-
реходных состояний. Согласно постулату Хэммонда [53], переход-
ное состояние каждой элементарной стадии может быть «ранним»
или «поздним», т. е. быть по структуре ближе к исходной или к
конечной молекуле, в зависимости от того, к уровню свободной
энергии которой из них ближе его уровень свободной энергии. Если
скорость реакции лимитируется образованием л-комплексов
(рис. 2.3, кривая /), она протекает через переходное состояние,
близкое по структуре исходному соединению, и активность в реак-
ции замещения должна соответствовать термодинамической
устойчивости л-комплексов. Если скорость лимитируется образова-
нием сг-комплекса и переходное состояние подобно ему по структу-
ре (рис. 2.3, кривая 2), активность должна согласовываться с
термодинамической устойчивостью ст-комплексов. Наконец, если
скорость реакции лимитируется отрывом от ст-комплекса замещае-
мого атома или группы (рис. 2.3, кривая 3), она должна зависеть
от их природы и содействия оснований катионоидному отрыву.
Чтобы оценить роль стадий образования л- и ст-комплексов в
реакциях ароматического электрофильного замещения, константы
3 Зак. 271
65
Рис. 2.3. Схемы возможных энергетических профилей
реакций ароматического электрофильного заме-
щения.
Скорость реакции определяется: 1 — образованием
л-комплекса; 2 — образованием о-комплекса; 3— от-
рывом от 0-комплекса замещаемого атома или группы.
скорости реакций производных бензо-
ла были сопоставлены с константами
равновесия реакций образований л- и
о-комплексов тех же соединений [47].
На рис. 2.4 это сопоставление пред-
ставлено в графической форме для ре-
акции хлорирования метилбензолов в
уксусной кислоте при О °C в сравнении с их константами основности
[в HF, см. уравнение (2.16)] и константами комплексообразова-
ния с НС1 [см. формулу (58)].
Как видно из наклона прямых на рис. 2.4, скорость галогениро-
вания и устойчивость сг-комплексов с введением метильных групп
быстро и пропорционально возрастают, тогда как устойчивость
л-комплексов изменяется незначительно. Малое влияние метиль-
ных групп при образовании л-комплексов естественно, так как в
ароматическое кольцо при этом переносится небольшой положи-
тельный заряд, который делокализуется на л-орбиталях с участием
всех шести атомов углерода, отчего л-комплекс примерно одинако-
во стабилизируется метильными группами, находящимися в раз-
ных положениях. Сильное влияние числа и положения метильных
групп на стабильность о-комплексов обусловлено их участием в
делокализации положительного заряда бензолониевого иона, ко-
торое более эффективно при пара- и орто-расположении по отно-
шению к хр3-гибридизованному атому углерода.
Значительное увеличение скорости с введением метильных
групп в бензольное кольцо присуще, хотя и в меньшей степени, чем
при галогенировании, и другим реакциям электрофильного заме-
щения. Как видно из больших абсолютных величин констант р
(табл. 2.5), реакции ароматического электрофильного замещения
Таблица 2.5
Константы р реакций электрофильного замещения
атома водорода в производных бензола [54]
Реакция Реагент (растворитель, катализатор) р (при 25 °C)
Бромирование Вгг (СНзСООН) — 12,4
Хлорирование С12 (СНзСООН) —8,1
Нитрование НМОз(СНзСООН) -6,4
Ацетилирование СН3СОС1 (дихлорэтан, А1С13) —9,1
Этилирование СгН5Вг (дихлорэтан, GaBr3) —2,4
66
вообще весьма чувствительны к электронным эффектам, а отрица-
тельный знак констант р указывает на электронодефицитность
реакционного центра (см. 2.4.2). Следовательно, во всех этих реак-
циях скорость определяется промежуточным образованием о-ком-
плекса и переходное состояние ближе к о-комплексу по структу-
ре. Промежуточное образование л-комплекса, если оно и имеет
место, представляет собой быструю равновесную стадию, не ока-
зывающую существенного влияния на скорость реакции в
целом.
Правомерность распространения такого вывода на все случаи
электрофильного ароматического замещения служит предметом
дискуссии. Найдены примеры, когда при действии очень активных
электрофилов, в частности при действии солей нитрония в поляр-
ных апротонных растворителях, скорость реакции мало зависит от
наличия в субстрате метильных групп, хотя обычное для электро-
фильных реакций ориентирующее действие метильных групп со-
храняется. По мнению авторов [55], это означает, что скорость
реакции определяется стадией образования л-комплекса (рис. 2.3,
кривая /), а электронные эффекты заместителей сказываются на
следующей, быстрой стадии при конкуренции отдельных положе-
ний ароматического ядра за уже связанный в л-комплексе ион
нитрония. Возражения против такой трактовки состоят в том, что в
столь высокореакционноспособных системах кинетика может
контролироваться не механизмом реакции, а диффузионными про-
цессами: смешением реагентов и вероятностью встречи молекул
реагирующих веществ. Высказано также предположение [50], что
стадией, определяющей скорость реакции, и в этих примерах может
быть образование о-комплексов, которые возникают путем несе-
лективного присоединения активного электрофила в различные
положения со сравнимыми вероятностями, после чего термодина-
мически менее устойчивые о-комплексы быстро перегруппировыва-
ются в более устойчивые.
Так как о-комплексы являются промежуточными продуктами и
наиболее подходящей моделью переходного состояния реакции
ароматического электрофильного замещения, их свойства заслу-
живают самого пристального
внимания. Конечный ароматиче-
ский продукт образуется из о-
комплекса в результате разрыва
ковалентной связи атома углеро-
да кольца с замещаемым атомом
или группой, обычно с атомом во-
дорода, отщепляющимся в виде
Рис. 2.4. Скорости хлорирования метилбеи-
золов в сравнении с устойчивостью a-ком-
плексов (сплошная прямая) и «-комплексов
(пунктир) [5, с. 481].
Цифры на прямой указывают число и положе-
ние метильных групп.
3*
67
протона. Если разрыв связи С—Н осуществляется в стадии, опре-
деляющей скорость реакции, наблюдается изотопный эффект, обус-
ловленный тем, что более тяжелые ядра дейтерия или трития
отщепляются труднее протона: &h/&d(T) > 1. Изучение изотопных
эффектов в реакциях ароматического замещения [56], показало,
что в реакциях нитрования и бромирования с участием электрофи-
ла Вг+ отрыв протона не происходит на стадии, лимитирующей
скорость, а при сульфировании, азосочетании, водородном обмене,
галогенировании в разных условиях разрыв связи С—Н может, по
крайней мере частично, определять скорость процесса. Отсутствие
изотопного эффекта в некоторых реакциях электрофильного заме-
щения водорода подтверждает наличие двух или более элементар-
ных стадий, так как исключает возможность одностадийного син-
хронного механизма, при котором атом водорода неизбежно отры-
вается на лимитирующей стадии.
Важной особенностью о-комплексов является их способность к
изомеризации, которая может происходить внутримолекулярно, за
счет 1,2-перемещений атома водорода или другого заместителя в
кольце, а также путем межмолекулярного переноса протона [48].
Изучение ^-комплексов выявило в ряде случаев существенную роль
таких перегруппировок в механизме электрофильного замещения.
Ранее считалось, что соотношение изомеров, получаемых в реакции
ароматического замещения, отражает соотношение скоростей об-
разования изомерных о-комплексов, то есть априори принималось,
что кинетический и термодинамический контроль реакции совпада-
ют (см. 2.1). Однако в последнее время накапливается все больше
данных, указывающих, что это не всегда так.
Иллюстрацией могут служить работы по нитрованию о- и
п-диалкилбензолов [57,58]. Известно, что нитрование п-цимола
[4-изопропилтолуола (61)] направляется преимущественно в по-
ложение 2. При нитровании (61) фтороборатом нитрония [57]
выделено 85,2% 2-нитро-п-цимола (67), 5,3% 3-нитро-п-цимола
(68) и 9,5% 4-нитротолуола (69); аналогичное соотношение про-
дуктов получено при нитровании п-цимола нитрующей смесью [58].
На этом основании можно считать, что относительная реакционная
способность положений 2,3,4 в «-цимоле (61), равна 16 : 1,3 : 5, если
принять, что изомеры (67) и (68) образуются в результате атаки в
положения 2 и 3 с последующим депротонированием о-комплексов
(63) и (64) соответственно, а 4-нитротолуол (69) — вследствие
unco-атаки * в положение 4 и дезалкилирования о-комплекса (65).
Однако при нитровании п-цимола (61) ацетилнитратом наряду с
примерно теми же количествами 3-нитропроизводного (68) и
4-нитротолуола (69) изолирован всего 41% 2-нитропроизводного
(67) и кроме того выделен 41% циклогексадиена (66), который
легко превращается в 2-нитроизомер (67) в серной кислоте [58].
* Термином unco (ipso) — unco-атака, имсо-замещение — обозначают атаку
или замещение в положение, несущее заместитель, отличный от атома водо-
рода [59].
68
ли
(61)
(бЗ)
l2CH’
Следовательно, самым реакционноспособным положением в
молекуле п-цимола (61) является не положение 2, а ранее не
принимавшиеся во внимание положение 1, и соотношение активно’
стей положений 1, 2, 3, 4 составляет примерно 10 : 5 : 1 : 2,5. Не ме-
нее половины общего количества 2-нитроизомера (67), получаемо-
го при нитровании, образуется, по-видимому, не в результате ата-
ки в положение 2, а путем unco-атаки в положение 1 с последую-
щей перегруппировкой о-комплекса (62) в термодинамически
более стабильный сг-комплекс (63).
Другим примером преобладания unco-атаки может служить
нитрование 2, 3, 5, 6-тетраметиланизола (70) фторборатом нитро-
ния [60]. При низкой температуре (—60 °C) в сильнокислой среде,
судя по спектру ПМР, в растворе образуется только о-комплекс
(71) с нитрогруппой в положении 3. При повышении температуры
до 0 °C он перегруппировывается в о-комплекс (72) с нитрогруп-
пой в положении 4, депротонирование которого под действием
основания с выходом 85% дает 2,3,5,6-тетраметил-4-нитроанизол
(73).
69
осн,
осн.
+NO,
осн.
осн.
NO,
(га)
(го) (71) (гг)
Исследованиям anco-атаки при ароматическом нитровании по-
священ обзор [61].
Случаи несогласованного кинетического и термодинамического
контроля при электрофильном замещении не ограничиваются ре-
акцией нитрования. Так, при протонировании монозамещенных
бензола, содержащих электронодонорные заместители, в сильно-
кислых средах в условиях термодинамического равновесия удается
обнаружить только о-комплексы «-строения, тогда как при изотоп-
ном обмене водорода в сопоставимых количествах образуются ор-
то- и пара-замещенные. Это дает основание считать, что в сильно-
кислых средах первоначально образующийся о-бензолониевый ион
быстро перегруппировывается в термодинамически более устойчи-
вый п-бензолониевый ион [50].
Превращение термодинамически менее стабильных о-комплек-
сов в более стабильные лежит в основе большинства электрофиль-
ных реакций изомеризации ароматических соединений [62]. Одна-
ко известны примеры изомеризации без промежуточного
образования о-комплексов, протекающей по механизму отщепле-
ния-присоединения. Так происходит изомеризация солей аромати-
ческих карбоновых кислот, например, фталата калия (74) в
терефталат калия (76) при высокой температуре в атмосфере
угольного ангидрида. На первой стадии реакции отщепляется мо-
лекула СО2 и возникает карбанион (75а), который перегруппиро-
вывается в более устойчивый карбанион (756), подвергающийся
карбоксилированию
(756)
(76)
Образование термодинамически менее стабильных о-комплек-
сов в условиях кинетического контроля может быть использовано в
синтетических целях путем «улавливания» о-комплекса нуклеофи-
лом, как в вышеприведенном примере, где о-комплекс (62), соеди-
няясь с ацетат-анионом, дает диен (66), который переходит при
термолизе и гидролизе в 2-изопропил-5-метилфенол(тимол), имею-
щий практическое применение [58]. Комплексы фенолов с галоге-
70
нидами алюминия, имеющие строение бензолониевых ионов, спо-
собны сами выступать в качестве электрофильных реагентов, заме-
щая водород в ароматических соединениях [63]. Так, комплекс
2-нафтола с хлоридом алюминия (77) реагирует с бензолом, давая
с выходом 90% 4-фенил-2-тетралон (78):
с6н5
(77) (78)
Комплекс 1-нафтола дает 4-фенил-1-тетралон.
Расчленением реакции ароматического электрофильного заме-
щения на элементарные стадии, соответствующие образованию л-и
ц-комплексов, попытки детализации не исчерпываются. Выдвинуто
предположение, что образованию ц-комплекса предшествует еще
одна элементарная стадия — перенос одного электрона к частице
электрофила с возникновением радикальной пары (79) [64, 65].
(S3) (79) (54)
Эта точка зрения является частью общей концепции, согласно
которой одноэлектронный перенос представляет собой нормальный
акт гетеролитических реакций [64]. Применительно к ароматиче-
скому электрофильному замещению она базируется на исследова-
нии корреляций между реакционной способностью и параметрами,
характеризующими перенос электрона, например, между констан-
тами скоростей азосочетания замещенных катионов арендиазония
с фенолят-анионом и скоростей переноса к ним электрона от тетра-
метил-п-фенилендиамина [66], между относительной реакционной
способностью различных положений ароматических молекул и
распределением спиновой плотности в катион-радикалах ArHt
[67] и т. д. Подобная аргументация относится к обсуждению
вероятности одноэлектронного переноса как одной из стадий реак-
ции электрофильного замещения, но не может служить доказа-
тельством ее реального существования, тем более, что те же кор-
реляции находят объяснение и в рамках чисто гетеролитического
механизма [50].
Прямых экспериментальных подтверждений участия стадии
одноэлектронного переноса в реакции электрофильного ароматиче-
ского замещения пока не найдено. В опытах по нитрованию бензо-
ла фторборатом нитрония не удалось зафиксировать промежуточ-
ного возникновения радикалов с помощью эффекта химической
поляризации ядер [68]. Обнаружение поляризации ядер 15N при
71
взаимодействии фторобората беизолдиазония с фенолятом натрия
не может служить свидетельством одноэлектронного переноса в
реакции азосочетания, как полагали авторы [69], так как в этом
случае должна была бы одновременно происходить поляризация
ядер 13С и >Н фенольного остатка в образующемся л-гидроксиазо-
бензоле, чего не наблюдается в действительности [70]. Зафикси-
рованное присутствие радикальных частиц при взаимодействии,
например, 2,6-ди-трет-бутилфенола с электрофильными реагентами
при низкой температуре [71] еще не значит, что эти частицы лежат
на координате реакции электрофильного замещения. Ввиду отсут-
ствия строгих экспериментальных доказательств, вопрос о роли
одноэлектронного переноса в реакциях ароматического электро-
фильного замещения остается пока открытым.
2.7.2. Нуклеофильное замещение
Известные механизмы реакций ароматического нуклео-
фильного замещения более разнообразны, чем реакций электро-
фильного замещения. Помимо механизма присоединения-отщепле-
ния для некоторых реакций реализуются механизмы отщепления-
присоединения с промежуточным образованием арина или
арилкатиона. Известно также протекание реакций нуклеофильного
замещения с участием радикальных частиц. Каждый механизм
имеет варианты, различающиеся на уровне дальнейшей детализа-
ции.
Наиболее изучен реакционный путь, на первом этапе которого
нуклеофильный реагент присоединяется к ароматическому суб-
страту. Этот путь типичен для замещения в соединениях, содер-
жащих сильные электроноакцепторные заместители, активирую-
щие ароматический субстрат, отчего саму реакцию называют реак-
цией активированного ароматического нуклеофильного замещения
(SnAf). Связывание реагента, как и при электрофильном замеще-
нии, возможно за счет межмолекулярного переноса заряда
(л-комплекс) или образования ковалентной ц-связи (ст-комплекс),
причем ароматический субстрат выступает как акцептор, а реа-
гент — как донор электронов (80).
Молекулярные комплексы с переносом заряда между аромати-
ческими нитросоединениями или хинонами и ароматическими угле-
водородами [см. формулу (56)], ароматическими аминами и други-
ми донорами электронов изучены в кристаллическом состоянии и в
растворах [42,43]. Образование их в условиях ароматического
нуклеофильного замещения дает основание предполагать, что они
участвуют в реакции в качестве промежуточных продуктов [44, 72],
но нет данных для того, чтобы считать эту стадию определяющей
скорость реакции. Ситуация сходна с описанной при обсуждении
вопроса о роли л-комплексов в ароматическом электрофильном
замещении (см. 2.4.1): быстрый равновесный процесс образования
л-комплекса, вполне вероятно, является начальной элементарной
стадией реакции, но не оказывает на нее, по крайней мере в
большинстве случаев, существенного влияния.
72
Анионные о-комплексы, называемые также по именам первых
исследователей комплексами Джексона — Мейзенгеймера [73,74],
надежно охарактеризованы для случаев взаимодействия аромати-
ческих нитросоединений с различными нуклеофилами. Они пред-
ставляют собой окрашенные вещества, стабильность которых обес-
печивается участием в делокализации отрицательного заряда
нескольких электроноакцепторных группировок, что изображают,
как показано в формуле (81), или набором резонансных структур
По рентгеноструктурным данным [75], в комплексе 2,4,6-три-
нитроанизола с метилатом калия (82) все атомы углерода кольца и
атомы азота нитрогрупп лежат в одной плоскости, а два алкок-
сильных атома кислорода — в плоскости, перпендикулярной пло-
скости кольца. Угол а при атоме С1 в плоскости кольца равен
тетраэдрическому—109°, а угол р в перпендикулярной плоско-
сти— 100°. Распределение длин связей в комплексе (82) соответ-
ствует большему вкладу л-хиноидной структуры (81а): связи
С2—С3, С5—С6 и С4—N укорочены по сравнению со связями С3—С4,
С4—С5 и С2—N, С6—N. Интерпретация спектров ПМР показывает,
что в растворах строение о-комплексов в основных чертах такое же,
как в кристаллах *.
К*’
NO,
NO2
(S2)
С помощью спектров ПМР в сочетании со струевыми мето-
дами исследования кинетики быстрых реакций [76] удалось про-
следить за образованием и исчезновением о-комплексов в реакциях
Межатомные расстояния указаны в ангстремах; 1 А = 0,1 нм.
73
ароматических нитросоединений с нуклеофильными реагентами.
Оказалось, что о-комплексы типа (81), содержащие остаток нукле-
офила при замещенном атоме углерода, наблюдаемые в условиях
равновесия и выделенные в свободном состоянии, не всегда являют-
ся первичными аддуктами [77]. При взаимодействии 2,4,6-тринитро-
анизола (83) с метилат-анионом в диметилсульфоксиде первона-
чально в реакционной массе накапливается изомерный о-комплекс
(84), содержащий остаток нуклеофила в незамещенном положе-
нии 3. о-Комплекс (84) затем исчезает, уступая место о-комплексу
(85). Присоединение метилат-аниона обратимо протекает по двум
направлениям: быстро в положение 3 с образованием термодина-
мически менее стабильного о-комплекса (84) и медленнее в поло-
жение 1 с образованием термодинамически более стабильного
о-комплекса (85). Первый преобладает при кинетическом, вто-
рой — при термодинамическом контроле процесса. Аналогичная
картина установлена для других нитропроизводных бензольного,
а также нафталинового и пиридинового ряда [78, 79].
Большую термодинамическую устойчивость 1-о-комплексов ти-
па (85) по сравнению с 3-о-комплексами типа (84), которые назы-
вают также комплексами Сервиса, связывают с уменьшением в
(85) стерического напряжения вследствие вывода алкоксигруппы
из копланарности с орто-расположенными атомами азота нитро-
групп [74]; большую же скорость присоединения нуклеофила в
положение 3 объясняют более благоприятными условиями для
сопряжения в переходном состоянии, соответствующем 3-о-комп-
лексу [78].
Анионные о-комплексы находят применение как исходные ве-
щества в органическом синтезе. Окисление изолированных о-комп-
лексов, содержащих остаток нуклеофила в незамещенном положе-
нии, приводит к продуктам замещения атома водорода [80]. Так,
о-комплексы (86), полученные реакцией ароматических полинит-
росоединений с галоформами [81], при окислении переходят в
полинитробензотригалогениды (87), трудно доступные иным
путем.
74
Аналогично могут быть ароматизированы а-комплексы (88)
реакции полинитросоединений с кетонами в присутствии оснований
(о-комплексы Яновского) [82]. При действии на них солей диазо-
ния происходит одновременно окисление и вытеснение одной нит-
рогруппы с превращением в азосоединения (89) [83].
NO,
(88)
+Аг\У
СН2СОСН3
ArN=N
(89)
В синтетическом плане особенно интересны аддукты полинит-
росоединений бензольного и нафталинового рядов с карбанионами,
способные к внутримолекулярной циклизации. сг-Комплексы Янов-
ского (90) под действием сильных оснований циклизуются в
устойчивые мостиковые ионы (92) в результате внутримолекуляр-
ной нуклеофильной атаки анионного центра боковой цепи в мета-
положение к тетраэдрическому атому углерода (91) [84]. Анало-
гично из a-комплексов реакции полинитросоединений с амидинами
(93) образуются гетероциклические системы (94) [85].
(90)
R n(ch3)2
(93)
(94)
Если в о-комплексе Яновского в орто-положении к тетраэдри-
ческому атому углерода находится карбонильная группа, то в
образующемся при действии диэтиламина енамине (95) внутримо-
лекулярная нуклеофильная атака направляется по карбонильному
75
атому углерода (96) и реакция приводит к производным нафтали-
на (97) [86].
(95) (96) (9Г)
Выделение, установление строения и изучение свойств о-комп-
лексов служит веским аргументом в пользу двухстадийного меха-
низма активированного ароматического нуклеофильного замеще-
ния, на первой стадии которого происходит образование, а на
второй — распад промежуточного о-комплекса (98)->(100). Дан-
ные обширных кинетических исследований [72, 78, 87], включаю-
щие анализ влияния природы заместителей, замещаемого атома
или группы, реагентов, кислотно-основного катализа, растворите-
лей подтверждают, что многие реакции нуклеофильного замеще-
ния протекают по такой схеме.
(99)
В некоторых случаях с применением комбинации спектрофото-
метрии и метода остановленной струи удалось раздельно наблю-
дать в одном процессе последовательные стадии возникновения
о-комплекса и его ароматизации с превращением в продукт заме-
щения [88].
Чувствительность реакций к влиянию заместителей, характери-
зуемая реакционной константой р (см. 2.4.1), мало зависит от
природы реагентов, растворителей, замещаемых атомов или групп
[89]. Увеличение числа и силы электроноакцепторных заместите-
лей ведет к уменьшению реакционной константы вследствие огра-
ниченной донорности бензольного кольца (табл. 2.6). Так, в реак-
ции с метилатом натрия в метаноле при 50 °C константа р в
4-К-замещенных хлорбензолах, равная 6,6, с введением электроно-
акцепторных групп изменяется в такой последовательности: 2-С1
6,3; 2-CF3 5,3; 2-SO2CH3 4,8; 2-NO2 3,6; 2-NO2 и 6-NO2 3,1.
В большинстве случаев подходящей моделью переходного со-
стояния является а-комплекс. Стадией, определяющей скорость
реакции, чаще оказывается образование новой связи с нуклеофи-
лом (рис. 2.5, кривая /), хотя известны случаи, когда скорость
лимитируется разрывом связи с уходящим атомом или группой
76
Таблица 2.6
Константы р реакций нуклеофильного замещения
в производных бензола 4-R-2-NO2CeH3X при 25 °C [90]
X Реагент Растворитель 0 X Реагент Растворитель р
С1 CH3ONa СНзОН 3,9 С1 CH3SNa СН3ОН 3,4
Вг CH3ONa СНзОН 4,1 С1 Пиперидин СН3ОН 3,9
F CHaONa СНзОН 4,0 С1 Пиперидин С6Нб 4,0
I CH3ONa СНзОН 4,2 Вг Пиперидин С2Н5ОН 3,9
С1 NaOH 75% водный 4,3
диокеан
разрыв связи не определяет скорость
(рис. 2,5, кривая 2). То, что
реакции при замещении атома галогена, явствует из значительно
большей легкости замещения атома фтора по сравнению с атомами
хлора и брома, хотя связь С—F разрывается труднее и в алифати-
ческом ряду подвижность фтора наименьшая. Иное положение в
реакциях нуклеофильного замещения атома водорода [80], анио-
ноидный отрыв которого в виде гидрид-иона не выгоден. Гидрид-
ион не образует водородных связей, не сольватируется и до сих пор
не обнаружен в органической химии в качестве кинетически неза-
висимой частицы [91]. Перенос гидрид-иона происходит, вероятно,
всегда в активированном комплексе с участием окислителя, что
эквивалентно переносу двух электронов и протона. Этим объясня-
ется, что анионные о-комплексы, содержащие в геминальном узле
атом водорода, труднее ароматизируются и успех реакции опреде-
ляется возможностью разрыва связи С—Н с удалением гидрид-
иона из a-комплекса под действием окислителя.
Однако при всем обилии накопленного материала нельзя счи-
тать доказанным, что реакции активированного ароматического
замещения всегда протекают по двухстадийному механизму [92,
93]. Далеко не для всех типов ароматических субстратов и нуклео-
фильных реагентов удается обнаружить образование о-комплек-
сов. Они описаны почти исключительно для соединений, имеющих
нитрогруппы [74], и лишь в единичных примерах для производных,
содержащих другие активирующие ~
нений о-комплексы зафиксирова-
ны только в случаях присоедине-
ния нуклеофила к атому углеро-
да, несущему относительно мало-
подвижный атом или группу (Н,
OAlk, ОАг и др.), но не такие
Рис. 2.5. Схемы возможных энергетических
профилей реакций нуклеофильного замеще-
ния:
/ — двухстаднйное замещение, скорость опре'
деляется образованием о-комплекса; 2 —двух-
стаднйное замещение, скорость определяется
разрушением о-комплекса; la, 2а — односта-
дийное «асинхронное» замещение; <3 — односта-
дийное синхронное замещение.
заместители. Для нитросоеди-
Координата реакции
77
типичные для нуклеофильного замещения, легко вытесняемые за-
местители, как атомы галогена или нитрогруппа. Сообщения о
выделении ст-комплексов, содержащих атом галогена в геминаль-
ном узле, оказались ошибочными [89].
Наличие примеров независимости общей скорости замещения от
факторов, определяющих легкость разрыва связи с замещаемой
группой, являясь доводом против строго синхронного одностадий-
ного механизма (рис. 2.5, кривая 3), еще не доказывает присут-
ствия промежуточного продукта, так как энергетический профиль
может описываться кривой, проходящей через точки, соответству-
ющие переходным состояниям двухстадийной реакции, и не иметь
минимума (рис. 2.5, кривые 1а, 2а) [92]. В случае 1а в переходном
состоянии старая связь более ослаблена по сравнению с новой, в
случае 2а более разрыхлена новая связь. Подобные варианты
«асинхронных» одностадийных механизмов реакций можно рас-
сматривать как промежуточные между строго синхронным и двух-
стадийным.
Высказано мнение [87, с. 393], будто правильнее считать, что
механизм ароматического нуклеофильного замещения в большин-
стве случаев описывается схемой, занимающей промежуточное по-
ложение между чисто одностадийной и чисто двухстадийной. Так
как структуры переходных состояний, определяющих скорость ре-
акций, в случае одностадийного (рис. 2.5, кривые 1 и 2) и «асинх-
ронного» одностадийного механизма (рис. 2.5, кривые 1а и 2а)
одинаковы, с точки зрения интерпретации влияния структурных и
других факторов на течение реакции вопрос о выборе между эти-
ми схемами не имеет принципиального значения.
Использование a-комплексов в качестве модели переходного
состояния позволяет объяснить некоторые аномалии нуклеофиль-
ного замещения.
Образование 3-о-комплексов типа (84) делает понятными от-
дельные случаи /снне-замещения, т. е. замещения, при котором
остаток реагента вступает не в то положение, где находилась
уходящая группа (см. 7.2, 8.2). К таким реакциям относится, в
частности, превращение пцра-замещенных нитробензолов (101) в
мета-замещенные бензойные кислоты (ЮЗ) под действием циани-
стого калия (реакция Рихтера), протекающая, по-видимому, через
3-ц-комплекс (102) (см. 11.1.3).
Внутримолекулярное нуклеофильное замещение при перегруп-
пировке Смайлса (104) —(106) протекает через спироциклические
ст-комплексы типа (Ю5), а выделение трициклических продуктов
78
объясняется образованием изомерных о-комплексов (107) [94]
(см. 7.1.6, 8.2, 11.2.2).
При реакциях некоторых гетероциклических соединений присо-
единение нуклеофила сопровождается раскрытием гетероцикла и
новой циклизацией, так что конечный результат оказывается таким
же, как при нормальном или кине-замещении [95]. Существование
такого механизма доказано с помощью меченых атомов. Так, на-
пример, установлено [96], что 2-бром-4-фенилпиримидин (108),
содержащий в кольце атомы 15N, лишь на 12 ± 3% реагирует с
амидом калия по обычному механизму присоединения-отщепления
через сг-комплекс (109) с образованием 2-амино-4-фенилпиримиди-
на (ПО), сохраняющего метку в гетероцикле. Главным же направ-
лением реакции (88 ±3%) является присоединение нуклеофила в
положение 6 с образованием о-комплекса (111), раскрытие цикла и
рециклизация, приводящие к 2-амино-4-фенилпиримидину (112),
содержащему атом 15N в аминогруппе (см. также 7.1.6). По первым
буквам английских слов при перечислении указанных элементар-
ных стадий такой механизм назван механизмом ANRORC (addition
nucleophilic ring open ring closure).
C6H5
(110)
C6H5
(112)
79
По такому же механизму осуществляется обмен местами между
циклическим и экзоциклическим атомами азота в №-замещенных
2-иминопиримидинах под действием щелочей (перегруппировка
Димрота) [97];
+ он-
----->
Нуклеофильное замещение, включающее стадии отщепления-
присоединения, протекает с промежуточным образованием
нейтральных короткоживущих частиц, получивших название де-
гидроаренов или аринов [98—100]. Первый термин подчеркивает,
что частицы представляют собой молекулы ароматических соеди-
нений, лишенные двух соседних атомов водорода, второй — что два
электрона, оставшиеся на двух частично перекрывающихся орби-
талях атомов углерода в плоскости кольца, обеспечивают между
ними дополнительную связь, подобную л-связи в тройной связи
алкинов (ацетиленов).
Наиболее часто аринный механизм встречается при неактиви-
рованном нуклеофильном замещении атомов галогенов под дей-
ствием сильных оснований. Само образование арина является
двухстадийным процессом, на первой стадии которого происходит
отрыв протона из орто-положения к атому галогена, а на второй —
отщепление галогенид-аниона;
Дегидроароматические промежуточные частицы генерированы
из производных бензола, нафталина, фенантрена и различных ге-
тероциклических систем (гетарины [101]).
Скорость образования арина может определяться в зависимо-
сти от среды и природы галогена или стадией депротонирования,
или стадией удаления галогенид-аниона, легкость отрыва которого
уменьшается в ряду Br > Cl > F. В несимметричных ароматиче-
ских соединениях отщепление может приводить к смеси изомерных
аринов, соотношение которых зависит от электронных эффектов
заместителей. На стадии депротонирования лега-замещенных га-
логенбензолов (113) электроноакцепторный заместитель способ-
ствует возникновению аниона (114), а электронодонорный — ани-
она (115). Если скорость образования аринов (116) и (117) опре-
деляется этой стадией, их соотношение соответствует соотношению
анионов (114) и (115), но если скорость определяется отрывом
галогенид-аниона, соотношение аринов может быть иным. Так при
реакции л-бромтолуола [(113), R = СН3, X = Вг] с амидом калия
в жидком аммиаке отрыв галогенид-аниона происходит быстро, и
80
вследствие электронодонорного эффекта метила на стадии депро-
тонирования предпочтительно образуется анион (115), превращаю-
щийся в арин (117). При X = С1, когда наиболее медленной ста-
дией является отрыв галогенид-аниона, из ж-хлортолуола пред-
почтительно образуется арин (116), так как анион (114) легче
отщепляет атом галогена, чем анион (И5). В 3-галогенпиридинах,
где протон легче отщепляется из положения 4, генерируется ис-
ключительно 3,4-дегидропиридин (121), а в 2-галогеннафталинах
благодаря особенностям нафталинового ядра (см. 2.4.2) — исклю-
чительно 1,2-дегидронафталин (122).
Образующиеся арины обладают высокой реакционной способ-
ностью и легко присоединяют различные нуклеофилы. При сим-
метричном строении арина остаток нуклеофила с равной вероятно-
стью вступает в любое из двух положений, связанных тройной
81
связью. Это продемонстрировано опытами с хлорбензолом, мечен-
ным ,4С в положении 1, который при действии амида калия в
жидком аммиаке дает равное количество молекул анилина, содер-
жащих метку в положениях 1 и 2:
knh2
KNH2;NH3
Направление присоединения нуклеофилов к замещенным ари-
нам зависит в основном от индукционного эффекта заместителя,
так как вследствие ортогональности несвязывающих орбиталей
тройной связи с л-орбиталями ароматической системы резонансный
эффект в аринах не играет роли и проявляется только после при-
соединения нуклеофила. В 3-замещенных аринах (116), где заме-
ститель находится вблизи тройной связи, электроноакцепторные
группы, -обладающие —/-эффектом, способствуют нуклеофильной
атаке в лшта-положенце (Н9), а обладающие -(-/-эффектом, — в
орто-положение (118). В 4-замещенных аринах (117) индукцион-
ный эффект сказывается слабее и направляет нуклеофильную ата-
ку соответственно в пара-положение (120) и жета-положение (119).
Так, при аминировании 3-замещенных аринов (116) в жидком
аммиаке в присутствии амида калия соотношение ж-изомер/о-изо-
мер в зависимости от заместителя R, составляет: CN 85 : 15;
F 99 : 1; ОСН3 95 : 5; СН3 45 : 55; N(CH3)2 95 : 5; О" 15 : 85; NH~
10:90 [102], З-Литийарины [(116), R = Li] селективно подвер-
гаются атаке нуклеофила в о-положение (118) [103].
Ориентация нуклеофильного присоединения к арину зависит
также от нуклеофильности реагента и стерических эффектов. Как и
в других реакциях, чем активнее нуклеофил, тем меньше избира-
тельность его атаки. Стерические затруднения возникают из-за
наличия заместителя в орто- или пери-положении к тройной связи и
проявляются тем сильнее, чем больше объем вступающего нуклео-
фила. Так, в 1,2-дегидронафталине атака в положение 1 затрудне-
на наличием в пери-положении атома водорода, отчего при взаи-
модействии с объемистыми аминами, например, с диизопропила-
мином, остаток последнего присоединяется только в положение 2.
Примеры нуклеофильного замещения по аринному механизму
см. 7.1.1, 8.1.2, 8.2.
Нуклеофильное замещение по типу отщепления-присоединения
через арин в некоторых случаях может конкурировать с замеще-
нием по типу присоединения-отщепления. Замещению через арин
способствуют высокая основность нуклеофила, необходимая для
отрыва протона, легкость отрыва замещаемой группы, ее большой
объем, препятствующий присоединению нуклеофила к исходной
молекуле, отсутствие протонодонорных растворителей [104].
Арины и гетарины могут быть генерированы и в отсутствие
нуклеофилов разложением о-диазонийкарбоксилатов (123) или
окислением 1-аминобензотриазолов (124) и введены в разнообраз-
ен
ные реакции, из которых наибольшее препаративное значение име-
ют реакции циклоприсоединения, например:
о-Галогенфенильные анионы, образующиеся из полигалоген-
бензолов под действием оснований в условиях генерирования ари-
нов, могут сами выступать в качестве нуклеофилов, атакуя атом
галогена другой молекулы. Так происходит изомеризация
1,2,4-трибромбензола (125) в 1,3,5-трибромбензол (126) под дей-
ствием анилида калия в жидком аммиаке или трет-бутилата калия
в диметилсульфоксиде. Помимо 1,3,5-трибромбензола (до 60%)
образуются небольшие количества п- и л/-дибромбензолов, 1,2,3,5- и
1,2,4,5-тетрабромбензолов; эта изомеризация названа «танцем га-
логена» [105]:
Арины в данном случае почти не образуются, так как
о-галогенфенильные анионы легче протонируются или отрывают
83
положительно заряженный атом галогена другой молекулы, чем
отщепляют анион брома.
Единственным подробно изученным примером реакции арома-
тического нуклеофильного замещения, протекающей по схеме от-
щепления-присоединения с промежуточным образованием ' арил-
катиона, является реакция разложения солей диазония в отсут-
ствие сильных оснований и восстановителей (см. 8.1.4, 10.3) [104,
106]. Первый порядок реакции, независимость скорости от концен-
трации нуклеофила, постоянство константы скорости при переходе
от Н2О к D2O, указывают, что нуклеофил, в частности вода, не
участвует в стадии, определяющей скорость реакции, и противоре-
чит предположению о механизме присоединения-отщепления; от-
сутствие же обмена водорода на дейтерий в среде D2O исключает
промежуточное образование арина. Малая селективность по отно-
шению к нуклеофилам свидетельствует о высокой активности про-
межуточной частицы, каковой и считают арил-катион (127) ,стадия
генерирования которого определяет скорость всего процесса
(SN1):
Y=OH, Вг, Cl, F
Помимо аринов, арил-анионов и арил-катионов в качестве про-
межуточных частиц в реакциях ароматического нуклеофильного
замещения, протекающих по типу отщепления-присоединения, мо-
гут выступать также арильные радикалы [105]. Первой элемен-
тарной стадией при этом является перенос одного электрона к
молекуле ароматического субстрата с образованием анион-ради-
кала, который затем, отщепляя уходящий атом или группу в виде
аниона, трансформируется в арил-радикал, атакуемый частицей
нуклеофила. Возникающий анион-радикал конечного продукта от-
дает электрон молекуле исходного соединения, завершая цикл и
кладя начало новому звену цепи:
+ е~ -X- + Y- АгХ
АгХ ----> АгХ’. ----> Аг» -----> ArY’» ----> ArY + АгХ •
Аналогичный радикальный механизм SrnI предложен для нук-
леофильного замещения при алифатическом атоме углерода
гг-нитробензилхлорида [107]. Ярким примером такого механизма в
ароматическом ряду может служить реакция о-броманизола (128)
с амидом калия в жидком аммиаке [108]. В отсутствие восстано-
вителей реакция идет через арин (129), приводя (см. выше) к
продукту кыме-замещения— л-анизидину (130). При добавлении
металлического калия, обеспечивающего генерирование радикалов
84
(131), (132), направление реакции полностью изменяется и един*
ственным продуктом оказывается о-анизидин (133)
Известны и другие примеры конкуренции между аринным и
радикальным механизмами ароматического нуклеофильного заме-
щения; они приводят к продуктам различного строения, соотноше-
ние между которыми изменяется от введения инициаторов (ще-
лочной металл) или ингибиторов (тетрафенилгидразин) радикаль-
ных реакций [105] (см. также 10.3 и 11.2).
Донором электрона при генерировании анион-радикалов аро-
матических соединений может служить частица нуклеофила. На-
чиная с работы [109], многими исследователями показано, что
ароматические нитросоединения могут восстанавливаться в анион-
радикалы под действием различных нуклеофильных реагентов.
В свете общей концепции одноэлектронного переноса как нормаль-
ной стадии гетеролитических реакций [64] (см. выше) это дало
основание трактовать образование анион-радикалов как элемен-
тарную стадию ароматического нуклеофильного замещения, пред-
шествующую образованию сг-комплекса [65,89]. Превращение
анион-радикала в анионный о-комплекс ArXY- изображается или
нецепной схемой, как рекомбинация с радикалом, возникающим в
результате переноса электрона от нуклеофила [НО]:
АгХ + Y" —> АгХ"» + Y • —> ArXY"
или как цепной путь, где анион-радикал присоединяет нуклеофил
с образованием дианион-радикала ArXY; , который окисляется мо-
лекулой исходного соединения [111]:
+ Y" + Y" АгХ
АгХ ----> АгХ"» ----► ArXY2"» ----> ArXY" + АгХ"»
причем допускается, что стадия переноса электрона и рекомбина-
ции с радикалом могут происходить «в клетке», то есть практи-
чески одновременно.
Представление об анион-радикалах как промежуточных части-
цах подтверждается наличием максимума на кинетических кривых
85
их накопления и расходования, ингибированием реакции замеще-
ния при добавлении более сильных, чем исходное нитросоедине-
ние, акцепторов электронов (тетрацианоэтилен, 1,4-бензохинон) и
ускорением ее в присутствии кислорода (см. также 8.1,3, 8.2).
Методом электрохимического моделирования продемонстриро-
вано, что не анион-радикал, а дианион исходного нитросоединения,
возникающий при переносе второго электрона от нуклеофила к
анион-радикалу, может являться реакционноспособной частицей
при реакции ж-динитробензола с ацетоном и щелочью, приводящей
к о-комплексу Яновского (88) или при реакции замещения атома
хлора в и-нитрохлорбензоле под действием тиофенолятов, которая
минует стадию о-комплекса [112].
Полученные данные показывают, что реакции нуклеофильного
замещения могут протекать с участием радикальных частиц, воз-
никающих в результате одноэлектронного переноса. Однако из
этого не следует, что стадия одноэлектронного переноса обяза-
тельна для реакций ароматического нуклеофильного замещения.
Образование анион-радикалов, как и о-комплексов, удалось за-
фиксировать почти исключительно при реакциях ароматических
соединений, содержащих нитрогруппу, Кроме того, само по себе
появление анион-радикалов еще не означает, что они участвуют в
реакции, так как может объясняться побочными процессами. Что-
бы выяснить, лежит ли стадия одноэлектронного переноса на ко-
ординате реакции, необходимы в каждом случае эксперименталь-
ные доказательства.
Особое место занимают реакции ароматического нуклеофиль-
ного фотозамещения [113]. Протекая под действием тех же реа-
гентов, но при фотовозбуждении субстрата, они подчиняются иным
правилам ориентации, противоположным тем, которые наблюда-
ются при «термических» реакциях: электроноакцепторные замести-
тели направляют вступающие группы в мета-, а электронодонор-
ные — в орто- и пара-положения. Такая ориентация определяется
специфическим механизмом реакций. Например, при нуклеофиль-
ном фотозамещении в ароматических метоксипроизводных первой
химической стадией является фотоионизация субстрата в низшем
триплетном состоянии с образованием катион-радикала, взаимо-
действующего далее с нуклеофилом, атака которого направляется
по месту наименьшей электронной плотности в катион-радикале.
2.7.3. Свободнорадикальное замещение
Ароматическое свободнорадикальное замещение проис-
ходит под действием радикалов, возникающих при гомолитическом
расщеплении двухэлектронной связи в соединениях — источниках
радикалов. Число типов радикальных частиц, атака которых может
приводить к замещению в ароматическом ядре, ограничено по
сравнению с числом известных электрофильных и нуклеофильных
реагентов. Обычно используются углеродные (арильные, алкиль-
ные, ацильные), или кислородные (гидроксильные, ацилоксиль-
86
ные) нейтральные радикалы, а также азотные (аммониевые) ка-
тион-радикалы (см. главы 12 и 13).
Ароматическое свободнорадикальное замещение' представляет
собой нецепную реакцию, протекающую по схеме присоединения-
отщепления [114,115]. На первой стадии радикал присоединяется
к молекуле ароматического соединения с образованием радикаль-
ного а-комплекса (134), который на второй стадии взаимодейству-
ет с таким же или другим радикалом, отдавая атом водорода и
превращаясь в продукт замещения.
Одностадийный механизм с синхронным связыванием радикала
и отрывом атома водорода исключается, так как при арилировании
и алкилировании нитросоединений совершенно не наблюдается
восстановления нитрогрупп, как этого следовало бы ожидать при
выделении атомарного водорода.
Радикальные ц-комплексы типа (134) нельзя выделить подобно
катионным ц-комплексам при электрофильном или анионным
о-комплексам при нуклеофильном замещении. Однако побочные
реакции, наблюдаемые при арилировании, алкилировании и других
свободнорадикальных реакциях, не оставляют сомнения, что
ц-комплексы, имеющие строение циклогексадиенильных радикалов
(135), действительно образуются, поскольку изолированные про-
дукты не могут иметь иных предшественников. Так, при фенилиро-
вании бензола перекисью бензоила наряду с бифенилом выделены
тетрагидро-п-кватерфенил (136) и дигидробифенил (137), образу-
ющиеся соответственно при димеризации и диспропорционирова-
нии фенилциклогексадиенильного радикала (135).
с6н5Х/н
(135)
/W
(/35/
W \=J 41
(137)
C6HS— C6HS
Скорость реакции определяется стадией присоединения ра-
дикала к ароматическому субстрату. Об этом свидетельствует от-
сутствие заметного изотопного эффекта при арилировании бензола
87
Таблица 2.7
Факторы общей и парциальных скоростей фенилирования
однозамещенных бензола С6Н5Х [116]
X Общая относи- тельная скорость Факторы парциальных скоростей
о- м- п-
и 1 1 1 1
no2 2,94 5,50 0,86 4,90
СНз 2,58 4,70 1,24 3,55
трет-С^Нд 1,09 0,70 1,64 1,81
С1 2,20 3,90 1,65 2,12
Вг 1,90 3,05 1,70 1,92
О СНз 2,71 5,6 1,23 2,31
и уменьшение подвижности атомов галогена в ряду: F > I > Вг >
> О при замещении их фенильным или циклогексильным радика-
лом в гексагалогенбензолах, указывающее, что разрыв связи с
уходящим атомом происходит не на лимитирующей стадии.
Сравнение скоростей свободнорадикального замещения произ-
водных бензола, содержащих различные заместители, показывает
что скорость реакции при введении заместителя увеличивается
независимо от его электронной природы (табл. 2.7). Так, нитро-
бензол и анизол оба фенилируются примерно в три раза быстрее
бензола. Вне зависимости от природы заместителя в субстрате
радикальная атака всегда направляется преимущественно в орто- и
«ара-положения, причем атака в орто-положение преобладает.
Подобное влияние объясняют способностью как электронодонор-
ных так и электроноакцепторных групп к делокализации неспа-
ренного электрона на атомах заместителя, находящегося в орто- и
«ара-положении к реакционному центру. Преобладание орто-атаки
пока не находит убедительного объяснения. Замещению в орто-
положение препятствуют лишь пространственные затруднения,
возникающие при наличии в субстрате объемистого заместителя,
например трет-бутильной группы (табл. 2.7).
Влияние полярных эффектов становится заметным при одно-
временном варьировании заместителей в субстрате и атакующем
арильном радикале. Так, «-нитрофенильный радикал медленнее
реагирует с нитробензолом, чем с анизолом, и дает больше
л-изомера, тогда как «-толильный радикал, напротив, быстрее
реагирует с нитробензолом, чем с анизолом и дает меньше
л-изомера [114]. Заместители в орто-положении к атому радикала,
несущему неспаренный электрон, снижают общую скорость реак-
ции замещения и количество о-изомера, по-видимому, вследствие
пространственных затруднений.
Скорости замещения в лета-положение хорошо коррелируют с
а.я-константами заместителей в соответствии с уравнением [117]:
k
(2.16)
К М
88
Это означает, что полярность радикалов определяется в основ-
ном индукционным эффектом. n-Арилированию отвечает уравне-
ние
lg-^- = pa„+Cre (2.17)
где член Сп отражает вклад эффекта делокализации неспаренного
электрона в переходном состоянии при /г-замещении.
Ниже приведены реакционные константы р для некоторых
арильных радикалов (при 20 °C), характеризующие их полярность
[И7]:
n-NO2C6H4* -0,81 с6н5. 0,05
но. -0,41 * п-СНзС6Н4 • 0,03
п-С1С5Н4• -0,27 fz-CH3OC6H4 • 0,09
* Из данных работы [118].
Как видно из знака и величины констант р, /г-нитрофенильный
радикал обладает ярко выраженным электрофильным, а фениль-
ный и /1-толильный — слабо нуклеофильным характером. Наиболь-
шее ускорение реакции арилирования достигается при противопо-
ложных электронных эффектах заместителей в субстрате и атаку-
ющем радикале. Гидроксильный радикал по электрофильности
занимает промежуточное положение между n-нитрофенильным и
n-хлорфенильным радикалами. Наиболее электрофильны аммоние-
вые катион-радикалы, являющиеся атакующими частицами при
гомолитическом аминировании, которое направляется исключи-
тельно в пара- и орто-положения к электронодонорному замести-
телю [119] (см. также 13.2).
Интересно, что в некоторых свободнорадикальных реакциях,
так же как в реакциях электрофильного и нуклеофильного заме-
щения, начальной, видимо, является стадия одноэлектронного пе-
реноса [114]. В частности, при действии перекиси бензоила на
производные бензола, содержащие электронодонорные заместите-
ли, протекает реакция бензоилоксилирования, тогда как при нали-
чии электроноакцепторных заместителей — только реакция фени-
лирования. Это объясняют переносом электрона ароматического
субстрата, например 1,4-диметоксибензола (138) к бензоилоксира-
дикалу CgHgCOO* с образованием катион-радикала (139), кото-
рый, будучи более активным, чем нейтральная молекула, рекомби-
нируется с бензоилоксирадикалом до распада последнего на фе-
нильный радикал и угольный ангидрид, превращаясь в продукт
замещения (140).
ОСНз ОСН3 ОСНз
(138) (139) (140)
89
Введение электроноакцепторных заместителей повышает окис-
лительно-восстановительный потенциал субстрата и делает пере-
нос электрона невозможным.
2.8. КВАНТОВОМЕХАНИЧЕСКОЕ ОПИСАНИЕ
РЕАКЦИОННОЙ СПОСОБНОСТИ
В настоящее время общепризнанной основой квантово-
механического описания свойств органических соединений стал ме-
тод молекулярных орбиталей (МО). Он не только является наи-
более распространенным расчетным методом квантовой химии, но
именно в его терминах обсуждаются результаты эксперименталь-
ных исследований физических свойств, строения и реакционной
способности молекул [120—122].
Для реализации молекулярных расчетов обычно используется
приближенное представление МО в виде линейной комбинации
атомных орбиталей (ЛКАО) всех атомов молекулы с привлечением
для решений уравнений самосогласованного поля (ССП). Основ-
ные вычислительные трудности метода МО ЛКАО ССП, развитого
Хартри, Фоком и Рутаном, связаны с необходимостью расчета
чрезвычайно большого числа молекулярных интегралов, пропорци-
онального приблизительного и4, где п — общее число базисных
атомных орбиталей (АО). Эти трудности возрастают при учете
конфигурационных взаимодействий: чтобы вычисленная полная
энергия удовлетворительно соответствовала опытным оценкам,
необходимо учитывать сотни и тысячи конфигураций. Поэтому
подавляющая часть расчетов по методу МО ЛКАО ССП выполня-
ется с применением приближенных неэмпирических и различных
полуэмпирических подходов.
Приближенные неэмпирические (ab initio) методы МО
ЛКАО ССП строятся на минимальном базисе (наборе) АО,
включающем только АО, заполненные в основном состоянии ато-
мов молекулы. Для упрощения расчетов молекулярных интегралов
используется представление АО в виде линейных комбинаций
гауссовых функций.
В полуэмпирических методах отдельные интегралы не вычис-
ляют теоретически, а задают как исходные параметры, определяе-
мые из опытных данных. Существует множество различных систем
параметров, предложенных разными авторами, и результаты рас-
чета существенно зависят от их выбора. Для сокращения числа не-
обходимых интегралов и упрощения методов расчета вводятся раз-
личные приближения. Широкое распространение получил принцип
пренебрежения дифференциальным перекрыванием (ПДП), обо-
значаемый в английской транскрипции буквами NDO (neglect of
differential overlap) с добавлением впереди букв, указывающих
на разновидность метода: CNDO (complete — полное пренебреже-
ние, INDO (intermediate — частичное пренебрежение), MINDO
(modification — модификация метода 1NDO) и т. д. В приближе-
нии NDO (ПДП) количество оставшихся интегралов становится
90
пропорциональным п2, а не я4. К ароматическим системам приме-
нимы полуэмпирические методы Паризера—Парра—Попла (ППП)
и расширенный метод Хюккеля. В л-электронном приближении,
когда не учитываются в явном виде не только взаимодействия
между внутренними электронами, но и валентными о-электронами,
расчет сводится к чисто эмпирическим методам.
Полуэмпирические методы гораздо проще любых неэмпириче-
ских и при удачном выборе параметров лучше согласуются с
экспериментом, поскольку эмпирические значения интегралов
неявно учитывают искажения АО в молекуле и отчасти вбирают в
себя ошибки, вносимые принятым приближением. Однако, если
приближенные неэмпирические методы позволяют проследить, как
изменяются при уточнении расчетной схемы различные вычислен-
ные величины, то в полуэмпирических расчетах с неконтролируе-
мыми приближениями и теоретически необоснованным выбором
параметров такой анализ невозможен. Кроме того, в рамках одно-
го полуэмлирического метода не удается описать всю совокупность
молекулярных свойств; каждый метод имеет свою область приме-
нения и в зависимости от рассчитываемых свойств и от типов
молекул требует различных наборов параметров. Процедура под-
гонки параметров обеспечивает возможность достичь хороших
совпадений с экспериментальными данными, но зато снижает
предсказательную силу полуэмпирических методов.
Для квантовомеханической оценки реакционной способности
соединения необходимо рассчитать скорость его реакции с за-
данными реагентами, для чего, в соответствии с теорией переход-
ного состояния, достаточно найти энергию активации реакции.
В общем случае для этого необходимо вычислить поверхность
потенциальной энергии реакционной системы, определить на этой
поверхности профиль реакции, найти точку «перевала», отвечаю-
щую переходному состоянию, и, зная из расчета энергию послед-
него и энергию исходных реагентов, по разности получить энергию
активации. Строгое решение этой задачи для сложных реакций
пока практически невозможно. При изучении сложных реакций
приходится иметь дело с многомерными поверхностями, так как
энергия N атомных ядер зависит от всех внутренних координат,
число которых М = 3N — 6. Чтобы рассчитать геометрическую
структуру переходного состояния в реакционной системе с большим
числом степеней свободы, нужно перебрать десятки тысяч пробных
структур, о вычислительных трудностях расчета каждой из кото-
рых говорилось выше. Поэтому приходится выбирать: либо вычис-
лить с достаточной точностью энергию нескольких выбранных то-
чек, не имея гарантии, что одна из них действительно соответствует
точке «перевала», либо приближенно рассчитать поверхность по-
тенциальной энергии, чтобы получить представление о ее рельефе,
пожертвовав при этом надежностью вычисления энергий [121].
Практически при расчете энергии переходного состояния не
учитывают всех внутренних степеней свободы, принимая, что ато-
мы, непосредственно не примыкающие к реакционному центру, не
91
меняют координат. При выборе конфигурации переходного состоя-
ния не прибегают к широкому варьированию, а руководствуются
знаниями, полученными экспериментальным путем, заранее закла-
дывая таким образом представление о типе и пути реакции. При
расчете энергии активации как разности между общей энергией
переходного состояния и исходных реагентов существует возмож-
ность существенных ошибок, связанных с необходимостью точно
определять малую разность между большими величинами, рассчи-
тываемыми приближенными методами. Чтобы избежать этого,
принимают, что переходное состояние по большинству параметров
остается близким к исходным соединениям и ведут расчет, учиты-
вая изменения одной-двух связей, энергия которых составляет
незначительную часть общей энергии системы.
Дальнейшее упрощение подхода состоит в том, что реакцию
данного соединения не исследуют, а сравнивают с аналогичной
реакцией другого соединения, выбранного в качестве стандартного.
Полагая, что обе реакции протекают через переходные состояния
одинаковой структуры и зная из эксперимента скорость стандарт-
ной реакции k°, методами МО вычисляют разность энергий акти-
вации Л-Е*— Л-Е* и, применяя уравнение (2.2), находят кон-
станту скорости исследуемой реакции:
lg* = lg fe°+ Р RT
(2.18)
При таком подходе нет необходимости обязательно вычислять
энергии активации. Достаточно воспользоваться какой-либо вели-
чиной, связанной с энергией активации линейной зависимостью,
которая в неявном виде будет отражаться эмпирическими кон-
стантами. Тогда уравнение (2.18) приобретает вид:
lg k = А + BW
(2.19)
где А и В — эмпирические константы, a W—индекс реакционной
способности, вычисляемый методом МО.
Выражение (2.19) представляет собой форму записи соотноше-
ния линейности свободных энергий (ЛСЭ)—см. 2.4.1. Именно
применение различных расчетных индексов реакционной способно-
сти, базирующееся на принципе ЛСЭ, является до сих пор глав-
ным способом оценки реакционной способности ароматических со-
единений методами квантовой механики.
Все применяемые в настоящее время индексы реакционной спо-
собности ароматических соединений [123, 124] можно разделить на
две группы. К первой относятся индексы, полученные расчетом
изолированной молекулы ароматического субстрата, ко второй —
полученные расчетом возможной структуры активированного или
промежуточного комплекса. В приближении изолированной моле-
кулы в качестве индексов реакционной способности положения
используется индекс свободной валентности Fr, л-электронная
плотность qr, собственная поляризуемость атома лгг, граничная
92
электронная плотность С”, сверхделокализуемость Sr и др.
В приближении промежуточного комплекса индексами реакционной
способности служат главным образом энергии локализации Lr.
Различные индексы реакционной способности соответствуют
различным моделям переходных состояний и движущих сил реак-
ции. Индексы первой группы исходят из предположения о «ран-
нем» переходном состоянии, близком по структуре и положению на
энергетическом профиле реакции к исходной молекуле. Индекс
свободной валентности Fr является современным видоизменением
представлений Тиле об остаточном сродстве (см. 1.2.1). Чем боль-
ше степень участия атома г в образовании л-связей с соседними
атомами ароматической системы, тем меньше его индекс свободной
валентности и его способность связываться с атакующим реаген-
том. Использование л-электронной плотности qr, рассчитываемой
суммированием вкладов всех заполненных МО, адекватно пред-
ставлению об определяющем значении электростатического взаи-
модействия между субстратом и реагентом, благодаря которому
электрофильная атака легче направляется на атомы с наибольшей,
а нуклеофильная — с наименьшей электронной плотностью. Индекс
собственной поляризуемости пгг отражает легкость изменения
суммарной л-электронной плотности на атакуемом атоме под вли-
янием реагента. Чем больше индекс лгг атома г, тем легче в это
положение должны идти реакции как электрофильного, так и
нуклеофильного замещения.
Граничная электронная плотность учитывает распределение
электронной плотности только на граничных орбиталях: на высшей
занятой молекулярной орбитали (ВЗМО) при электрофильном
замещении и на низшей свободной молекулярной орбитали
(НСМО) после переноса на нее двух электронов при нуклеофиль-
ном замещении. Мерой граничной электронной плотности положе-
ния является коэффициент С™, отражающий вклад атомной орби-
тали атома г в граничную молекулярную орбиталь т. Считается,
что электрофильное и нуклеофильное замещение протекают по
месту наибольшей электронной плотности на соответствующей гра-
ничной орбитали. При свободнорадикальном замещении и ВЗМО,
и НСМО рассматриваются как граничные орбитали [125]. По-
скольку граничная электронная плотность пригодна только для
рассмотрения ориентации в данной молекуле, для выявления отно-
сительной реакционной способности различных систем введен ин-
декс, названный сверхделокализуемостью Sr. При формулировке
этого индекса использована теория возмущений [126] в примене-
нии к модели, в которой вступающая группа образует слабую
л-связь с атомом г, а л-система в целом не изменяется. К индексам
теории граничных орбиталей [125] близки другие индексы, осно-
ванные на представлении о переходном состоянии, как комплексе с
переносом заряда, например Z-фактор [123]. Обсуждавшиеся в
связи с концепцией одноэлектронного переноса (см. 2.7.1) корре-
ляции между относительной реакционной способностью различных
положений ароматических молекул и распределением спиновой
93
плотности в катион-радикалах фактически эквивалентны использо-
ванию индексов типа Sr [50].
Индексы второй группы, к которым относятся энергии локали-
зации Lr, исходят из представления о том, что переходное состоя-
ние при ароматическом замещении по структуре и положению на
энергетическом профиле реакции близко к промежуточному о-ком-
плексу, т. е. является «поздним». Энергией локализации называют
изменение энергии л-электронного взаимодействия при переходе от
исходной ароматической молекулы к о-комплексу. Энергия лока-
лизации для электрофильного замещения L? характеризует за-
трату энергии на локализацию в положении г пары электронов
л-системы, энергия локализации для свободнорадикального заме-
щения — затрату энергии на локализацию в положении г одного
электрона л-системы, энергия локализации для нуклеофильного
замещения Lr — затрату энергии на распределение всех электро-
нов л-системы в поле остальных атомов без участия положения г.
Сопоставление различных индексов реакционной способности
с экспериментальными данными для незамещенных бензоидных
углеводородов показывает, что все индексы, кроме qr, всегда рав-
ного в данном случае единице при расчете простым методом моле-
кулярных орбиталей Хюккеля (МОХ), в большей или меньшей
степени пригодны для описания реакционной способности с по-
мощью линейных уравнений типа (2.19) и линейно связаны между
собой. Включение в ароматическую систему гетероатомов и введе-
ние заместителей модифицирует волновые функции, нарушает рав-
номерность распределения л-электронной плотности (ф-УМ) и
изменяет соотношения между индексами реакционной способности.
В результате одни из них сохраняют применимость, тогда как
другие ее утрачивают. Отвлекаясь от возможных ошибок, связан-
ных с трудностью учета влияния гетероатомов и приближенностью
методов расчета, следует указать на принципиальные обстоятель-
ства, которые могут повлечь за собой неприменимость тех или
других индексов.
Использование индексов реакционной способности на основе
соотношений ЛСЭ типа 2.19 предполагает соблюдение «правила
непересечения» энергетических профилей подобных реакций, со-
гласно которому энергия, необходимая для достижения любой
точки на координате реакций пропорциональна отношению их
де{ л с'11 л
энергий активации, т. е. —-у
/X £ 2
Д£[‘ ДЯ^
—П~ ’ как показано на
рисунке 2.6, а. Однако нет никаких причин считать «правило непе-
ресечения» общей закономерностью. Выше уже рассматривались
случаи, когда профили реакций пересекаются, что для обратимых
реакций ведет к несогласованности кинетического и термодинами-
ческого контроля (см. 2.1 и 2.7). Пересечение энергетических про-
филей приводит к нарушению пропорциональности между энерги-
ями на разных участках пути реакции. Если профили пересекаются
на нисходящей ветви кривых, более стабильному о-комплексу со-
94
Рис. 2.6. Возможное расположение энергетических
профилей реакций.
ответствует больший активацион-
ный барьер и «работают» только
индексы . в приближении изолиро-
ванной молекулы, предполагающие
«ранние» переходные состояния
(рис. 2.6, б). Если профили пересе-
каются на восходящей ветви кри-
вых,большей энергии, необходимой
для движения по координате реак-
ции в начальный период, соответ-
ствует меньшая энергия активации
и «работают» только индексы в
приближении локализации, предпо-
лагающие «поздние» переходные
состояния (рис. 2.6, в). Если же
профили пересекаются дважды, не
пригодны обе группы индексов и
единственно приемлемым является f
собственно приближение активиро-
ванного комплекса.
Причина возможного пересече-
ния энергетических профилей за-
ключается не только в различном
влиянии на разных участках пути
реакции структурных факторов, но координата реакции
и сольватационных эффектов. Кван-
товохимические расчеты индексов реакционной способности отно-
сятся, по существу, к газовой фазе и переносятся на реакции в
растворах в допущении, что изменения энергий сольватации срав-
ниваемых систем одинаковы на всем реакционном пути. Сложность
и многообразие сольватационных взаимодействий (см. 2.6) застав-
ляет думать, что и это правило часто не соблюдается.
По-видимому, простой универсальный индекс реакционной спо-
собности вообще не может быть найден. Более перспективен под-
ход, учитывающий разную природу факторов, определяющих ве-
личины энергий активации. Одним из примеров такого подхода
может служить обобщенный индекс реакционной способности ге-
тероароматических систем в реакциях электрофильного замещения
[122]:
lg = A AL+ + В Д/
(2.20)
где А и В — эмпирические коэффициенты; AL? и А/ — соответ-
ственно разности энергий локализации и потенциалов ионизации
сравниваемых молекул.
95
Молекула Переходное Молекула Молекула Переходное
донора состояние акцептора донора состояние
Молекула
акцептора
Рис. 2.7. Схема энергетических уровней молекулярных орбиталей реагирующих частиц.
а — зарядно-контролируемая реакция; б — орбитальио-контролируемая реакция.
Общий подход к задаче взаимодействия двух реагирующих
частиц в рамках теории возмущений основан на разделении сил
взаимодействия на электростатические и ковалентные [127, 128].
Энергия взаимодействия молекул донора (нуклеофила) R и ак-
цептора (электрофила) S с образованием химической связи между
атомами г и s этих молекул выражается суммой двух составляю-
щих— электростатической и ковалентной;
(2.21)
где qr и qs — общие заряды атомов; drs— расстояние между ни-
ми; е — диэлектрическая постоянная растворителя; сТ и с"—гра-
ничные электронные плотности; Aflrs — изменение резонансного
интеграла при взаимодействии орбиталей атомов г и $ на расстоя-
нии drs', {Em — Е’п) — разность энергий граничных орбиталей.
Электростатическая составляющая тем больше, чем больше
заряды связываемых атомов, а ковалентная — чем меньше раз-
ность энергий граничных орбиталей реагирующих молекул, т. е.
разность энергий ВЗМО донора и НСМО акцептора. Реакции, при
которых главный вклад в энергию взаимодействия вносит электро-
статическая составляющая, названы зарядно-контролируемыми, а
реакции, при которых главный вклад принадлежит ковалентной
составляющей — орбитально-контролируемыми.
Обобщенный подход на базе уравнения (2.21) служит теорети-
ческим обоснованием принципа ЖМКО [15] —см. также 2.5. Же-
сткие основания (доноры) имеют низко лежащую ВЗМО, а жест-
кие кислоты (акцепторы) —высоко лежащую НСМО. В результате
между уровнями энергий граничных орбиталей жесткого основа-
ния и жесткой кислоты существует большой разрыв (рис. 2.7, а) и
ковалентная составляющая в уравнении (2.21) из-за большой ве-
личины знаменателя {Е*т — Е*п) не играет существенной роли.
Взаимодействия типа жесткий — жесткий являются зарядно-
контролируемыми. Мягкие основания имеют высоко лежащую
ВЗМО, а мягкие кислоты — низко лежащую НСМО, вследствие
чего уровни энергий оказываются почти вырожденными
(рис. 2.7,6) и ковалентная составляющая в уравнении (2.21) при-
обретает доминирующее значение. Взаимодействия типа Мягкий —
мягкий орбитально-контролируемые. При Е^ — Е'п «з 0 уравнение
(2.21) для орбитально-контролируемой реакции приобретает вид:
ЬЕ ж 2c"c"prs (2.22)
Понятия «жесткость» и «мягкость» определяют, склонен ли
реагент реагировать преимущественно по зарядно-контролируемо-
му или орбитально-контролируемому пути. Анализ выражения
(2.21) показывает, что только взаимодействия между донором и
акцептором, стремящимися реагировать по одному и тому же пути,,
зарядно-контролируемому или орбитально-контролируемому, энер-
гетически выгодны, что и составляет сущность принципа ЖМКО.
Обобщенный подход делает понятными физический смысл и
область применения граничной электронной плотности как индекса
реакционной способности (см. выше). В незамещенных бензоидных,
углеводородах, где заряды атомов qr равноценны, ориентация
замещения целиком определяется орбитально-контролируемой со-
ставляющей, зависящей от граничных электронных плотностей,
которые и дают верное представление о направлении реакций.
В замещенных и гетероциклических производных, где заряды ато-
мов различны, граничные электронные плотности мало пригодны,
так как не учитывают влияния на ориентацию зарядно-контроли-
руемых факторов.
Поскольку ориентация в незамещенных бензоидных углеводо-
родах полностью контролируется орбитальными взаимодействия-
ми, селективность реагента в этих случаях зависит только от спо-
собности реагировать по орбитально-контролируемому пути и тем
больше, чем больше эта способность. При неравномерном распре-
делении электронной плотности в ароматической молекуле значе-
ние приобретает также величина заряда на атакующем атоме реа-
гента. Так, в толуоле, согласно расчетам [129], положение 2 имеет
большую общую л-электронную плотность, а положение 4—
большую граничную электронную плотность. Поэтому следует
ожидать, что положение 2 будет предпочтительнее для атаки за-
рядно-контролируемыми реагентами (нитрование, хлорирование),
а положение 4 — для атаки орбитально-контролируемыми реаген-
тами (бромирование, меркурирование). Справедливость такого за-
ключения подтверждается экспериментальными данными. В част-
ности, соотношение о-изомер/п-изомер при алкилировании и аци-
лировании толуола возрастает с введением в молекулу реагента
электроноакцепторных заместителей, увеличивающих заряд 6 +на
атакующем атоме, и снижается с введением электронодонорных
заместителей, уменьшающих этот заряд. Субстратная селектив-
ность, измеряемая соотношением констант скоростей &толуола/&бензола
и зависящая в основном от орбитально-контролируемого взаимо-
действия, изменяется в противоположном направлении [55]. Те же
объяснения можно дать в терминах принципа ЖМКО, но разделе-
ние сил взаимодействия на зарядно-контролируемые и орбитально-
4 Зак. 271
97
контролируемые благодаря не только качественному, но и количе-
ственному подходу позволяет улавливать более тонкие особенности
реакционной способности.
Учет орбитально-контролируемых факторов позволил предло-
жить унифицированный подход к вопросу о роли одноэлектронного
переноса в реакциях гетеролитнческого ароматического замещения
[130]. Необходимым условием переноса электрона при нуклео-
фильном замещении является сближение уровней ВЗМО реагента
и НСМО ароматического субстрата. Поскольку между этими
уровнями, как правило, существует значительный разрыв, истин-
ный механизм реакции должен включать взаимодействие, которое
повышало бы уровень ВЗМО нуклеофила до уровня низшей раз-
рыхляющей орбитали ароматической системы. Таким взаимодей-
ствием не может быть образование комплекса с переносом заряда,
стабилизирующее ВЗМО, но может быть, как показано расчетом
[130], атака нуклеофила Y: на связь С—X с возникновением
трехцентрового переходного состояния (141). Вырождение уровней
в результате сближения нуклеофила с атакуемой связью делает
возможным перенос электрона от нуклеофила к ароматическому
субстрату с образованием пары ион-радикалов, которая далее
рекомбинируется в сг-комплекс (142) или выходит в объем. Если
при сближении вырождение уровней не наступает, реакция проте-
кает без одноэлектронного переноса непосредственно через стадию
образования сг-комплекса.
Вероятность выхода ион-радикалов в объем тем больше, чем
больше межмолекулярное расстояние, на котором происходит пе-
ренос электрона, а вырождение уровней наступает тем раньше, чем
выше находится невозмущенная ВЗМО нуклеофила и чем ниже
НСМО ароматического соединения. Следовательно, одноэлектрон-
ного переноса следует ожидать при реакциях между высокоактив-
ными нуклеофилами, имеющими низкий потенциал ионизации, и
ароматическими субстратами, обладающими высокой электроно-
акцепторной способностью. Этим условиям отвечают реакции аро-
матических нитросоединений и хинонов с сильными нуклеофилами,
для которых пока только и обнаружено возникновение анион-
радикалов (см. 2.7.2). Аналогичный подход правомерен примени-
тельно к реакциям электрофильного замещения.
ЛИТЕРАТУРА
1. Гаммет Л. Основы физической органической химии. Пер. с англ./Под ред.
Л. С. Эфроса. М., Мир, 1972. 2. Эмануэль Н. М., Кнорре Д. Г. Курс химической
кинетики. Изд. 3-е, М., Высшая школа, 1974. 3. Джилкрист Т., Сторр Р. Орга-
98
нические реакции и орбитальная симметрия. Пер. с англ./ Под ред. Б. Л. Дят-
кина. М., Мир, 1976. 4. Матье Ж-, Панико Р. Курс теоретических основ органи-
ческой химии. Пер. с франц./Под ред. Л. А. Яновской. М., Мир, 1975. 5. Бек-
кер Г. Введение в электронную теорию органических реакций. Пер. с нем. М.,
Мир, 1977. 6. Сайкс П. Механизмы реакций в органической химии. Изд. 2-е.
Пер. с англ./Под ред. Я- М. Варшавского. М., Химия, 1973. 7. Бреслоу Р. Меха-
низмы органических реакций. Пер. с англ./Под ред. В. Н. Сеткиной. М., Мир,
1968. 8. Пальм В. А. Основы количественной теории органических реакций.
Изд. 2-е. Л., Химия, 1977. 9. Жданов Ю. А., Минкин В. И. Корреляционный
анализ в органической химии. Ростов, Изд-во Ростовского ун-та, 1966. 470 с.
10. Джонсон К. Уравнение Гаммета. Пер. с англ. М., Мир, 1977.
11. Гордон А., Форд Р. Спутник химика. М., Мир, 1976. 12. Charton М.—
Progress Phys. Org. Chem., 1971, v. 8, p. 235—317. 13. Pearson R. G.—J. Amer.
Chem. Soc., 1963, v. 85, № 22, p. 3533—3539. 14. Edwards J. 0.—Ibid., 1954, v. 76,
№ 6, p. 1540—1547. 15. Пирсон P. Дж.— Успехи химии, 1971, т. 40, вып. 7,
с. 1259—1282. 16. Но Т. L.— Chem. Revs., 1975, v. 75, № 1, р. 1—20. 17. НоТ. L.—
Hard and Soft Acids and Bases Principle in Organic Chemistry N. Y.— London,
Academic Press. 1977. 18. Bunnett J. F., Bussett J. У.— J. Amer. Chem. Soc.,
1959, v. 81, № 9, p. 2104—2109. 19. Rosenblatt D. H., Dennis W. H., Goodin RD.—
Ibid., 1973, v. 95, № 7, p. 2133—2136. 20. Giles D. E., Parker A. J.—Austr. J.
Chem., 1973, v. 26, № 2, p. 273—279.
21. Davies J. H.. Haddock E., Kirby P. e.a.— J. Chem. Soc., 1971C, № 16,
p. 2843—2846. 22. Реутов О. А., Курц А. Л. — Успехи химии, 1977, т. 46, вып. 11,
с. 1964—1994. 23. Meek J. S., Fowler J. S.— J. Org. Chem., 1968, v. 33, № 9,
3422—3424. 24. Райхардт X. Растворители в органической химии. Пер. с нем./Под
ред. Л. С. Эфроса. Л., Химия, 1973. 150 с. 25. Паркер А. Д. — Успехи химии,
1963, т. 32, вып. 10, с. 1270—1295; в кн.: Успехи органической химии. Т. 5. Пер.
с англ./Под ред. И. Л. Кнунянца. М., Мир, 1968, с. 5—50. 26. Паркер А. Д.—
Успехи химии, 1971, т. 40, вып. 12, с. 2203—2249. 27. Dack М. R. J. — In: Solu-
tions and Solubilities. Part 2/Ed. M. R. J. Dack. N. Y., Wiley, 1976, p. 95—157.
28. Белецкая И. П., Соловьянов А. А. — Жури. ВХО, 1977, т. 22, вып. 3, с. 286—
300. 29. Winstein S., Savedoff L. G. Smith S. G. e.a. — Tetrahedron Letters, 1960,
№ 9, p. 24—30. 30. Педерсен К. Д., Френсдорф X. К.—Успехи химии, 1973, т. 42,
вып. 3, с. 492—510.
31. Liotta С. L., Harris Н. Р.—J. Amer. Chem Soc., 1974, v. 96, № 7,
p. 2250—2252. 32. Sam D. J., Simmons H. E. — Ibid., 1974, v. 96, № 7, p. 2252—
2253. 33. Шевелев C. A. — Успехи химии, 1970, т. 39, вып. 10, 1773—1800.
34. Komblum N., Berrigan P. J., le Noble IF J. — J Amer. Chem., Soc., 1963,
v. 85, № 2, p. 1141—1147. 35. Komblum N., Setzler R., Halerfield P. — Ibid., 1963,
v. 85, № 8, p. 1148—1154. 36. Ингольд К. К. Теоретические основы органической
химии. Пер. с англ./Под ред. И. П. Белецкой. М., Мир, 1973, с. 379—390.
37. Росс С. Д. — В кн.: Современные проблемы физической органической химии.
Пер. с англ./Под ред. М. Е. Вольпина. М., Мир, 1967, с. 33—68. 38. Suhr Н.—
Chem. Вег., 1964, Bd. 97, № 12, S. 3277—3283. 39. Шейн С. М., Данилова Н. К. —
ЖОХ, 1968, т. 4, вып. 11, с. 1940—1947. 40. Norman R. О. C.t Taylor R. Electro-
filic Substitution in Benzenoid Compounds. Amsterdam, Elsevier, 1965. 343 p.
41. de la Mare P. B. D. — In: Rodd’s Chemistry of Carbon Compounds. 2nd ed.
V. ЗА/Ed. S. Coffey. Amsterdam, Elsevier, 1971, p. 45—88. 42. Эндрюс Л., Ки-
фер P. Молекулярные комплексы в органической химии. Пер. с англ./Под ред.
И. И. Моисеева. М., Мир, 1967, 207 с. 43. Mulliken R. S., Person IT. В. Molecular
Complexes. New York, Wiley Interscience, 1969. 498 p. 44. Сергеев Г. Б. — Жури.
ВХО 1974 т. 19, вып. 3, с. 285—294. 45. Hassel О., Stromme К. О. — Acta chem.
Scan’d., 1958, v. 12, № 5, p. 1146; 1959, v. 13, № 9, p. 1781—1786. 46. Smith H. G„
Rundle R. E. — J. Amer. Chem. Soc., 1958, v. 80, № 19, p. 5075—5080.
47. Brown H. C., Brady J. D. — Ibid., 1952, v. 74, № 14, p. 3570—3582. 48. Kon-
тюг В. A. — В кн.: Современные проблемы химии карбониевых понов/Под ред.
В. А. Коптюга. Новосибирск, Наука, 1975, с. 5—178. 49. Brouwer D. М.,
4*
99
Mackor E. L., MacLean C. — In: Carbonium Ions. V. 2. N Y„ Wiley Intercience,
1970, p. 837—897. 50. Коптюг В. A.— Журн. ВХО, 1976, т. 21, вып. 3, с. 247—255.
51. Baenziger N. С., Nelson A. D. — J. Amer. Chem. Soc., 1968, v. 90, № 24,
p. 6602—6607. 52. Перкампус Г. Г. — В кн.: Новые проблемы физической .орга-
нической химии. Пер. с англ./Под ред. И. П. Белецкой. М., Мир, 1969, с. 257—
337. 53. Hammond G. S. — J. Amer. Chem., Soc., 1955, v. 77, № 2, p. 334—338.
54. Справочник химика. Изд. 2-е. Т. 3. Л., Химия, 1965, с. 938—951, 968—993.
55.0/пА G. А. — Accounts Chem. Res., 1971, v. 4, Ns 7, p. 240—248. 56. Мелан-
дер Л. Изотопные эффекты в скоростях реакций. Пер. с англ./Под ред. С. 3. Ро-
гинского. М., Мир, 1964. 57. Olah G. A., Kuhn S. L. — J. Amer. Chem. Soc., 1964,
v. 86, Ns 6, p. 1067—1070. 58. Hahn R. C., Strack D. L. — Ibid. 1974 v. 96 N° 13,
p. 4335—4337; Galley M. Г., Hahn R. C. — Ibid., 1974, v. 96, Ns 13, p. 4337—4339.
59. Perrin C. L., Skinner G. A. — Ibid., 1971, v. 93, № 14, p. 3389—3394. 60. Кам-
ший Л. П., Маматюк В. И., Коптюг В. А. — ЖОХ, 1977, т. 13, вып 4,
с. 810—817.
61. Moodie R. В., Schofield К- — Accounts Chem. Res., 1976, v. 9, Ns 8,
p. 287—292. 62. Коптюг В. A. — Изомеризация ароматических соединений. Ново-
сибирск, Изд-во СО АН СССР, 1963. 63. Коптюг В. А. — Журн. ВХО, 1976, т. 21,
вып. 3, с. 315—322. 64. Билевич К. А., Охлобыстин О. Ю. — Успехи химии, 1968,
т. 37, вып. 12, с. 2162—2191; Охлобыстин О. Ю. — Перенос электрона в орга-
нических реакциях. Ростов-на-Дону, Изд-во Ростовского ун-та, 1974. 65. Тод-
рес 3. В. — Успехи химии, 1978, т. 47, вып. 2, с. 260—288. 66. Медведев Б. Я-,
Полякова Л. Я., Билевич К. А. и др. — Теорет. эксп. химия, 1972, т. 8, вып. 2,
с. 256—259. 67. Epiotis N. D.—J. Amer. Chem. Soc., 1973, v, 95, Ns 10, p. 3188—
3193. 68. Beletskaya I. P., Rykov S. V., Buchachenko A. L.— Org. Magnet. Reso-
nance, 1973, v. 5, Ns 12, p. 595—597. 69. Bubnov N. N., Medvedev B. Ya., Polyako-
va L. A. e. a. — Ibid., 1973, v. 5, Ns 9, p. 437—439. 70. Бучаченко Л. A. — Химиче-
ская поляризация электронов и ядер. М., Наука, 1974, с. 167.
71. Походенко В. Д., Хижный В. А., Кошечко В. Г. и др. — ЖОХ, 1975,
т. 11, вып. 9, с. 1873—1876. 72. Miller J. Aromatic Nucleophilic Substitution. Am-
sterdam, Elsevier, 1968. 408 p. 73. Холл T., Порански Ч„ мл. — В кн.: Химия
нитро- и нитрозогруппы. Т. 2/Под ред. Г. Фойера. Пер. с англ, под ред.
В. А. Тартаковского. М., Мир, 1973, с. 254—296. 74. Штраус М.— В кн.: Методы
и достижения в физико-органической химии. Пер. с англ./Под ред. И. П. Бе-
лецкой. М., Мир, 1973, с. 422—544. 75. Ueda Н., Sakabe N., Tanaka J. е. а.—
Bull. Chem. Soc. Japan, 1968, v. 41, Ns 12, p. 2866—2871. 76. Соловьянов A. A.,
Белецкая И. П. — Журн. ВХО, 1974, т. 19, вып. 3, с. 275—284. 77. Servis К-L.—
J. Amer. Chem. Soc., 1965, v. 87, Ns 23, p. 5495—5497; 1967, v. 89, Ns 6, p. 1508—
1514. 78. Bernaskoni C. F. — In: Internat. Rev. Sci., Org. Chem., Series 1, 1973,
v. 3, p. 33—64. 79. Fyfe C. A., Cocivera M., Damji S. W. H. — J. Amer. Chem. Soc.,
1975, v. 97, Ns 20, p. 5707—5713. 80. Чупахин О. H., Постовский И. Я. — Успехи
химии, 1976, т. 45, вып. 5, с. 908—937.
81. Шейн С. М., Хмелинская А. Д.— ЖОХ, 1972, т. 8, вып. 6, с. 1237—1242.
82. Калинкин М. И., Парнес 3. Н., Пузанова В. Е. и др. — Там же, 1973, т. 9,
вып. 11, с. 2354—2359. 83. Каминский А. Я-, Гитис С. С., Багал И. Л. и др.—
Там же, 1977, т. 13, вып. 4, с. 803—805. 84. Strauss М. J. — Accounts Chem. Res.,
1974, v. 7, Ns 5, p. 181—188. 85. Bard R. R., Strauss M. J. — J. Org. Chem., 1976,
v. 41, Ns 14, p. 2421—2428. 86. Alpha S. R. — Ibid., 1973, v. 38, Ns 18, p. 3136—
3139. 87. Де Бур T. Дж., Диркс И. П. — В кн.: Химия нитро- и нитрозогруппы.
Т. 1/Под ред. Г. Фойера. Пер. с англ, под ред. С. С. Новикова. М., Мир, 1972,
с. 371—466. 88. Orvik J. A., Bunnett J. F. — J. Amer. Chem. Soc., 1970, v. 92, №8,
p. 2417—2427; Gaboriaud R., Schaal R. — Bull. chim. France, 1969, Ns 8, p. 2683—
2690. 89. Шейн С. M. — Журн. ВХО, 1976, т. 21, вып. 3, с. 256—266.
90. Шейн С. М„ Козорез Л. А.— Реакц. способность орг. соедин., 1966, т. 3,
вып. 4, с. 45—90.
91. Парнес 3. Н., Курганов Д. Н. Реакции гпдрпдного перемещения в орга-
нической химии. М., Наука, 1969. 92. Баннет Д. Ф.— В кн.; Теоретическая
100
органическая химия. Пер. с англ./Под ред. Р. X. Фрейдлиной. М., ИЛ, 1963,
с. 181—199. 93. Мамаев В. П., Загуляева О. А., Шейн С. М. — Химия гетероцикл,
соед., 1973, вып. 6, 723—734. 94. Дрозд В. И. — Жури. ВХО, 1976, т. 21, вып. 3,
с. 266—273. 95. Crampton М. R. — In: Organic Reaction Mechanisms 1973. Lon-
don, Wiley, 1973, p. 225—242; Organic Reaction Mechanisms, 1974. London. Wiley,
1976, p. 249—267. 96. Kroon A. P., van der Plas H. C. — Res. Trav. chim, 1973,
c. 92, № 9/10, p. 1020—1026. 97. Brown D. J. — In: Mechanisms of Molecular
Migrations. V. 1/Ed. B. S. Thyagarajan. London, Wiley, 1968, p. 209—245.
98. Hoffmann R. IF. Dehydrobenzene and Cycloalkynes. N. Y. — London, Academic
Press, 1967. 99. Gilchrist T. L., Rees C. W. Carbenes, Nitrenes and Arynes. N. Y.,
Appleton-Century-Crofts, 1969, p. 42—54, 103—119. 100. Хофманн P. B. — В кн.:
Успехи химии ацетиленовых соединений/Под ред. Г. Г. Вийе. Пер. с англ, под
ред. В. Ф. Кучерова. М., Химия, 1973, с. 141—204.
101. ден-Хертог X., ван-дер-Плас X.—Там же, с. 205—254.
102. de Graaf G. В. R., den Hertog H. J., Megler IF. C. — Tetrahedron Letters,
1965, № 15, p. 963—968. 103. Стоянович Ф. M., Маракаткина M. A. — Изв. AH
СССР, сер. хим., 1978, № 1, c. 142—150. 104. Pietra F. — Quart. Rev., 1969, v. 23,
№ 4, p. 504—521. 105. Bunnett J. F. — Accounts Chem. Res., 1972, v. 5, № 4,
p. 139—147. 106. Swain C. G., Sheats J. E., Harbison К G. — J. Amer. Chem. Soc.,
1975, v. 97, № 4, p. 783—790. 107. Russel G. A., Norris R. K.— In: Organic Reac-
tive Intermediates/Ed. S. P. McManus. N. Y. — London, Academic Press, 1973,
p. 423—448. 108. Kim J. K., Bunnett J. F.— J. Amer. Chem. Soc., 1970, v. 92, № 25,
p. 7464—7466. 109. Miller R. E.t Wynne-Jones IF. F. K- — Nature, 1960, v. 186,
№ 15, p. 149—150. ПО. Блюменфельд Л. А., Брюховецкая Л. B.t Фомин Г. В.
и др. — ЖФХ, 1970, т. 44, вып. 4, с. 931—944.
111. Шейн С. М., Брюховецкая Л. В., Иванова Т. М.— Изв. АИ СССР, сер.
хим., 1973, вып. 7, с. 1594—1601. 112. Сосонкин И. М, Каминский А. Я-, Ги-
тис С. С. и др. — ЖОХ, 1973, т. 9, вып. 7, с. 1470—1476; Сосонкин И. М., По-
лынникова Т. К-, Каминский А. Я. и др. — Там же, 1975, т. 11, вып. 1, с. 115—
121. 113. Havinga Е., Cornelisse J. — Chem. Revs., 1975, v. 75, № 4, p. 353—388;
den Heifer J., Shadid О. B., Cornekisse J. e.a.—Tetrahedron, 1977, v. 33, № 7,
p. 779—786. 114. Нонхибел Д., Уолтон Дж. Химия свободных радикалов. Пер. с
англ./Под ред. И. П. Белецкой. М., Мир, 1977, с. 441—493. 115. Perkins М. J.—
In: Free Radicals. V. 2/Ed. J. К. Koshi. N. Y. — London, Wiley, 1973, p. 231—271.
116. Davies D. I., Hey D. H., Summers B. — J. Chem. Soc. (c), 1971, № 15,
p. 2681—2684. 117. Ito R., Migita T., Morikawa N. e.a. — Tetrahedron, 1965, v. 21,
№ 5, p. 955—961. 118. Anbar /И., Meyerstein D., Neta P. — J. Phys. Chem., 1966,
v. 70, № 8, p. 2660—2662. 119. Minisci F. — Synthesis, 1973, Ns 1, p. 1—24; To-
pics Curr. Chem., 1976, v. 62, p. 1—48. 120. Дяткина M. E. Основы теории моле-
кулярных орбиталей. М., Наука, 1975. 190 с.
121. Базилевский AL В. — Журн. ВХО, 1977, т. 22, вып. 3, с. 245—251.
122. Абронин И. А., Жидомиров Г. М. — Химия гетероцикл, соед., 1977, вып. 1,
с. 3—22. 123. Стрейтвизер Э. Теория молекулярных орбит для химиков-органи-
ков. Пер. с англ./Под ред. М. Е. Дяткнной. М., Мир, 1965, с. 291—336.
124. Ридд Д. — В кн.: Физические методы в химии гетероциклических соедине-
ний/Под ред. А. Р. Катрицкого. Пер. с англ, под ред. Л. С. Эфроса. М. — Л.,
Химия, 1966, с. 125—175. 125. Fukui К- — Theory of Orientation and Stereoselec-
tion. Berlin/ Springer-Verlag, 1975. 134 p. 126. Дьюар M., Догерти P. Теория
возмущения молекулярных орбиталей в органической химии. Пер. с англ./Под
ред. Л. А. Яновской. М., Мир, 1977. 695 с. 127. Klopman G. — J. Amer. Chem.
Soc., 1968, v. 90, Ns 2, p. 223—234. 128. Клопман Г. — В кн.: Реакционная спо-
собность и пути реакций/Под ред. Г. Клопмана. Пер. с англ. М., Мир, 1977,
с. 63—174. 129. Dewar М. I. S., Klopman G. — J. Amer. Chem. Soc., 1967, v. 89,
Ns 13, p. 3089—3098. 130. Дьяченко А. И., Иоффе А. И. — Изв. АН СССР, cep.
хим., 1976, № 5, c. 1160—1163.
Часть 2
РЕАКЦИИ АРОМАТИЧЕСКОГО
ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ
Глава 3
СУЛЬФИРОВАНИЕ
3.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О РЕАКЦИИ
СУЛЬФИРОВАНИЯ И СУЛЬФИРУЮЩИЕ АГЕНТЫ
Сульфированием (по международной номенклатуре —
сульфонированием) называют взаимодействие органического со-
единения с концентрированной серной кислотой и некоторыми ее
производными, в результате которого водород при одном или
нескольких атомах углерода замещается сульфогруппами и обра-
зуются сульфокислоты (сульфоновые кислоты):
ArH + HOSO3H ArSO3H + Н2О
Будучи одной из важнейших реакций органического синтеза,
сульфирование широко используется с целью химической перера-
ботки ароматических углеводородов в промежуточные продукты
различного строения, а также для придания конечным продуктам
синтеза — красителям, физиологически активным соединениям,
поверхностно-активным и текстильно-вспомогательным веществам
и др.— кислотных свойств и растворимости в воде.
Для осуществления реакции чаще всего применяют 92—
94%-ную серную кислоту (купоросное масло), 98—100%-ную (мо-
ногидрат) и жидкий при обычной температуре олеум (дымящую
серную кислоту), содержащий 20—25% или 60—65% SO3, раство-
ренного в моногидрате. Сульфирующие агенты иной концентрации
обычно готовят, смешивая перечисленные продукты в рассчитанном
соотношении. Олеум 30—55%-ной и более чем 65%-ной концентра-
ции при обычной температуре является твердым продуктом. Он
неудобен в обращении и поэтому не производится.
В ряде случаев пользуются также монохлорангидридом серной
кислоты — хлорсульфоновой кислотой CISO3H, которую получают
из серного ангидрида и хлористого водорода, а также самим
серным ангидридом (иногда в растворителях).
Для сульфирования высокореакционноспособных соединений,
нестабильных в кислой среде, иногда применяют продукты присое-
динения серного ангидрида к пиридину или диоксану (в избытке
102
этих растворителей) — пиридинсульфотриоксид и диоксансульфо-
триоксид, имеющие строение биполярных ангидрооснований:
Из сделанного перечисления видно, что основным сульфирую-
щим агентом служит SO3, все остальные продукты можно рас-
сматривать как получающиеся из него при действии соответствую-
щих нуклеофилов. В соответствии с этим именно серный ангидрид
является наиболее активным реагентом сульфирования. Его взаи-
модействие с ароматическим соединением проходит по следующей
простой схеме:
АгН + SO3 —> ArSO3H
Однако, как будет видно из дальнейшего, возможность прямого
использования серного ангидрида для сульфирования резко огра-
ничивается неоднозначностью направления протекания реакции и
чрезмерной активностью этого реагента, а также необходимостью
применения растворителей.
Как показали кинетические исследования, скорость сульфиро-
вания чрезвычайно сильно зависит от концентрации серной кисло-
ты. Например, при уменьшении ее от 100 до 99,5% скорость суль-
фирования хлорбензола падает на несколько порядков [1]. Если
при реакции образуется вода, разбавляющая серную кислоту, ско-
рость сульфирования может уменьшиться вплоть до полного его
прекращения. В связи с этим количество сульфирующего агента,
необходимого для проведения реакции, рассчитывают не по стехи-
ометрическому соотношению, а по концентрации серной кислоты,
которая должна получиться после завершения процесса сульфиро-
вания. Эта эмпирически найденная оптимальная концентрация
отработанной серной кислоты неодинакова для различных соеди-
нений и зависит от их реакционной способности: при прочих равных
условиях она может быть тем ниже, чем легче ароматическое
соединение вступает в электрофильную реакцию сульфирования.
Предельную концентрацию отработанной серной кислоты, вы-
раженную в процентах SO3, называют л сульфирования. Даже для
одного и того же соединения она не строго постоянна: несколько
падает с повышением температуры проведения реакции и зависит
от исходного соотношения реагентов. Поэтому л сульфирования
нельзя использовать для строгой оценки реакционной способности
соединений, однако для практических задач этой величиной поль-
зуются часто.
Так, для моносульфирования бензола л составляет 66,4, а для
моносульфирования нафталина — 56 при 55—60 и 52 при 160 °C;
для дисульфирования нафталина 82 при 10, 80,8 при 80—90 и
66,5 при 160 °C. Для нитробензола и л-нптротолуола л « 82 [2].
103
Как видно из этих примеров, для полного сульфирования некото-
рых соединений необходимо, чтобы концентрация отработанной
кислоты находилась на уровне моногидрата (в нем концентрация
SO3 81,6%). В таких случаях обычно используют олеум, что позво-
ляет резко сократить количество отработанной серной кислоты и
получающихся из нее после выделения продуктов сульфирования
кислых промышленных отходов.
Другой важнейшей особенностью реакции сульфирования яв-
ляется ее обратимость. Почти все сульфокислоты способны гидро-
лизоваться в кислой среде с образованием соответствующего угле-
водорода и серной кислоты по схеме:
ArSO3H + Н2О ---> ArH + H2SO4
н+
Если при сульфировании в реакционной массе содержится хотя
бы немного воды, наряду с прямой реакцией, может проходить и
эта обратная реакция десульфирования. В результате устанавли-
вается равновесие
ArH + H2SO4 4=* ArSO3H + H2O
положение которого зависит от соотношения скоростей прямой и
обратной реакции. Константы скорости сульфирования и десуль-
фирования, по-видимому, в равной или близкой степени зависят от
строения исходного ароматического соединения, так как экспери-
ментально доказано: чем быстрее соединение сульфируется, тем
легче десульфируется образовавшаяся сульфокислота [3]. Дело в
том, что обратная реакция, так же как и прямая, является реакцией
электрофильного ароматического замещения и обе они подчиняют-
ся общим закономерностям.
Скорость прямой реакции должна возрастать с увеличением
концентрации сульфирующего агента, а скорость десульфирова-
ния— с увеличением содержания воды в реакционной массе. Ско-
рость сульфирования максимальна в начале процесса и падает к
его завершению, в то время как скорость десульфирования начи-
нает играть заметную роль только в конце реакции. По-видимому,
приведенные выше значения величин п сульфирования примерно
соответствуют тому моменту реакции, когда скорости сульфирова-
ния и десульфирования сближаются и устанавливается равновесие.
Так как состав продуктов сульфирования в состоянии равнове-
сия— при термодинамическом контроле реакции — может сильно
отличаться от первоначального, полученного при кинетическом
контроле (см. 2.1), обратимость реакции сульфирования имеет
большое практическое значение. Варьируя концентрацию сульфи-
рующего агента и температуру, во многих случаях удается регули-
ровать скорости прямой и обратной реакций и таким образом
направлять течение сульфирования в сторону целевого продукта
необходимого строения.
104
Так, при сульфировании фенола (1) до моносульфокислот
главными продуктами реакции являются о- и п-гидроксибензол-
сульфокислоты:
Электронодонорный заместитель — гидроксильная группа — в
большей степени активирует замещение в орто-положение (&0 >
>kn), и при низкой температуре и кинетическом контроле в суль-
фомассе преобладает о-гидроксибензолсульфокислота. По мере те-
чения реакции, особенно при повышении температуры до 100 °C,
концентрация о-изомера уменьшается и накапливается га-изомер,
становящийся при термодинамическом контроле главной составной
частью продуктов моносульфирования. Объясняется это тем, что
скорость десульфирования о-изомера больше, чем n-изомера. Ког-
да в сульфомассе накапливается реакционная вода и при повы-
шенной температуре создаются благоприятные условия для гидро-
литических процессов, о-изомер превращается в фенол, который
далее медленно сульфируется в более стабильную п-гидроксибен-
золсульфокислоту [3]. Такого рода изомеризация сульфокислот
широко используется в синтезе промежуточных продуктов, особен-
но нафталинового ряда.
Значительно меньшее значение имеют реакции так называемой
термической изомеризации сульфокислот. Они проходят в усло-
виях, исключающих возможность течения реакции десульфирова-
ния, при простом нагревании солей сульфокислот в подходящем
теплоносителе или без него. Например [4], нагревание натриевой
соли 4-амино-1-нафталинсульфокислоты (нафтионовой кислоты)
(2) при 200—250 °C, лучше всего в кипящем хинолине, приводит к
получению соли 1-амино-2-нафталинсульфокислоты (3);
(2) (3)
Сводку таких реакций и обсуждение возможного их механизма
можно найти в монографии [5].
3.2. МЕХАНИЗМ РЕАКЦИИ СУЛЬФИРОВАНИЯ
Вопрос о строении электрофильной частицы, получаю-
щейся из серной кислоты или ее производных и являющейся реа-
гентом реакции сульфирования, до сих пор строго не решен. Целе-
сообразно поэтому вначале несколько подробнее рассмотреть
строение уже упомянутых сульфирующих агентов [1].
Серный ангидрид в парах преимущественно мономерен, а в виде
жидкости имеет структуру циклического шестичленного тримера в
форме кресла, находящегося в равновесии с небольшим количе-
ством мономера. В разбавленных растворах в жидком SO2, в СС14,
СС12=СС12, SO2C12 он преимущественно мономерен, но присут-
ствует и тример, количество которого растет с увеличением кон-
центрации раствора.
Молекула SO3 плоская, атомы кислорода эквивалентны. Вслед-
ствие разницы в электроотрицательности серы и кислорода, связи
между их атомами поляризованы в сторону кислорода. В соответ-
ствии с этими физическими данными молекула SO3 является очень
сильной кислотой Льюиса и активно образует комплексы с перено-
сом заряда даже с очень слабыми основаниями, такими, например,
как ароматические углеводороды. С более сильными основаниями
образование комплексов проходит очень энергично, с большим
тепловым эффектом. По данным диэлькометрического титрования
и калориметрии были определены дипольные моменты и степени
переноса заряда б, константы диссоциации и энтальпии образова-
ния АЯ комплексов SO3 с эфирами, триалкилфосфатами, аминами,
диоксаном и др. Оказалось, что между параметрами АЯ и б, а
также между АЯ и энтропией образования этих комплексов су-
ществует взаимосвязь по корреляционным уравнениям, характер-
ным для комплексов с переносом заряда [6].
Механизм сульфирования комплексными соединениями серного
ангидрида в значительной степени зависит от полярности и степени
диссоциации последних. В случае сильно диссоциированных комп-
лексов, например, как упомянутый выше комплекс с диоксаном,
сульфирование проходит фактически за счет свободного SO3.
В случае значительно более прочных комплексов с пиридином [3]
или с триалкилфосфатами [6] в качестве электрофильной частицы
выступают уже сами эти комплексы.
Серную кислоту многие исследователи также склонны рассмат-
ривать как очень прочный комплекс H2O-SO3. Кроме этого, мето-
дами криоскопии зарегистрированы соединения H2O-2SO3 (пиро-
серная кислота — H2S2O7), 2H2O-SO3, 3H2O-SO3, 4H2O-SO3,
5H2O-SO3 и 7H2O-SO3.
Серная кислота 100%-ной концентрации имеет диэлектрическую
постоянную s = 100 (при 25 °C) и Гамметовскую кислотность
Я° = —12,20 [1]. Она на 0,2—0,3% ионизирована по следующим
уравнениям автопротолиза:
2H2so4 Hssot + Hso;
106
2h2so4 +=> so3 + h3o+ + hso;
3H2SO4 H2S2O7 + H3O+ + HSO7
Таким образом, автопротолиз вызывает образование активных
частиц сульфирования H3SO4, SO3 и H2S2O7. К ним, вероятно,
следует добавить и катион SO3H+, легко получающийся при прото-
нировании SO3, особенно в олеуме.
При добавлении воды к 100%-ной H2SO4 начинается ее иони-
зация по уравнению:
H2O + H2SO4 h3o++hso4-
Увеличение концентрации ионов Н3О+ и HSO4 вызывает со-
ответствующее уменьшение количества активных частиц сульфиро-
вания, образующихся при автопротолизе, благодаря чему происхо-
дит резкое уменьшение скорости реакции.
Длительное время считалось, что основным сульфирующим ре-
агентом во всех случаях является SO3. Этот же вывод сделан в
работе [7], где помимо традиционных химических и физико-хими-
ческих методов для обработки сложных систем кинетических
уравнений'использовано математическое моделирование. Кинети-
ческие исследования последних лет, однако, приводят к выводу, что
в реакции сульфирования участвуют два реагента. В более кон-
центрированной серной кислоте это—пиросерная кислота H2S2O7
(H2SO4-SO3), а в более разбавленной —ион Н3 SOt(HaO+• SO3)
Предполагается, что наличие двух активных частиц сульфирования
позволяет объяснить изменение соотношений скоростей образова-
ния отдельных изомеров, наблюдающееся при изменении концент-
рации сульфирующего агента. Катион H3SO4 — менее активный и
более селективный сульфирующий реагент, чем H2S2O7. Поэтому
реакции с его участием (в более разбавленной серной кислоте)
идут более избирательно [8].
Представление о том, что активным реагентом сульфирования
является не SO3, а значительно более объемные частицы
H2S2O7 и H3SO4 хорошо согласуются с давно известными фактами
существования при реакции сульфирования стерических затрудне-
ний. В отличие от нитрогруппы, сульфогруппа обычно не становится
в орто- или иери-положение к нитро- или сульфогруппе (правило
Армстронга — Вина), как и в любое другое стерически затруднен-
ное положение (см., однако, сульфирование метаниловой кисло-
ты— 3.3.2). Соответствующие соединения, однако, могут быть лег-
ко получены при нуклеофильном введении сульфогруппы, так как
атакующей частицей здесь является анион SO3 .имеющий неболь-
шой объем. Этим путем удалось получить даже бензолгексасуль-
фокислоту, оказавшуюся весьма устойчивым соединением
(см. 9.3).
Вне зависимости от действительного строения активного реа-
гента сульфирования (на схеме он условно изображен в виде
107
SO3H+), механизм сульфирования ароматических соединений в
настоящее время представляют следующим образом
(4) (5) (6)
Первоначально в результате взаимодействия ароматического
соединения с сульфирующим реагентом обратимо образуется по-
ложительно заряженный сг-комплекс (5). Дальнейшее его превра-
щение в сульфокислоту (6) происходит после отрыва протона ос-
нованием, например HSO4. Вторая стадия реакции в абсолютном
большинстве случаев также является обратимой, но при очень
высокой кислотности среды и недостатке основания, необходимого
для связывания протона, может сильно замедляться, становясь
«узким местом» реакции. В соответствии с этим при сульфировании
галогенбензолов моногидратом или олеумом наблюдается кинети-
ческий изотопный эффект. Если же для сульфирования использо-
вать 96%-ную или менее концентрированную серную кислоту, то
кинетический изотопный эффект отсутствует — лимитирующей ста-
дией реакции становится образование п-комплекса [8—10].
Высокий изотопный эффект наблюдается при сульфировании
9-П-антрацена в диоксане действием SO3 при 40 °C [9].
Как видно из изложенного, разница между энергиями актива-
ции первой и второй элементарной стадии при сульфировании
должна быть небольшой и может меняться в зависимости от усло-
вий.
Реакция десульфирования, которую в рассматриваемом случае
называют также реакцией протодесульфирования, может прохо-
дить как сопутствующая процессу сульфирования или же в рас-
творах сильных кислот, например соляной,— как независимая.
В соляной кислоте скорость этой реакции возрастает с увеличением
кислотности среды [Н]. В реакции, по мнению Серфонтайна [1],
участвует не сама сульфокислота, а ее анион. Реакция начинается с
протонирования атома углерода, несущего сульфогруппу; образу-
ющийся сг-комплекс (5) затем превращается в углеводород по
приведенной ранее схеме. Течение реакции в обратную сторону
здесь обеспечивается водой, которая немедленно превращает от-
щепившуюся активную частицу сульфирующего реагента в мало-
реакционноспособную серную кислоту.
3.3. ПРАКТИКА СУЛЬФИРОВАНИЯ
Реакция сульфирования протекает сравнительно мед-
ленно. Поэтому, хотя она экзотермична (тепловой эффект 126—
168 МДж/моль), ее чаще всего проводят при нагревании. Реакци-
онная масса в первый период бывает гетерогенна, и поэтому очень
важно эффективное перемешивание.
108
В производственных условиях сульфирование проводят в типо-
вых чугунных или стальных аппаратах — сульфураторах — со сфе-
рическим днищем и крышкой. Течение реакции контролируют по
изменению общей кислотности среды, содержанию в- сульфомассе
серной кислоты и сульфокислот, а окончание реакции — по исчез-
новению исходного соединения (в случае получения моносульфо-
кислот—проба на растворимость в воде).
Предложен также метод контроля процесса сульфирования,
основанный на растворении пробы в органическом растворителе,
например ацетоне или спирте, с последующим определением степе-
ни сульфирования по интенсивности флюоресценции [12].
После окончания реакции сульфомассу передавливают или вы-
ливают в воду, полученный раствор частично нейтрализуют и вы-
саливают натриевую соль сульфокислоты, добавляя к раствору
сульфат, хлорид или сульфит натрия. Если натриевые соли суль-
фокислот хорошо растворимы и плохо высаливаются, их выделяют
из раствора в виде труднорастворимых калиевых, магниевых, же-
лезных, медных или каких-либо других солей. Иногда, однако, и
этот способ оказывается непригодным. В таком случае водный
раствор, содержащий серную кислоту и сульфокислоту, полностью
нейтрализуют известью или мелом. При этом сульфат кальция
выпадает в осадок, который отделяют от водного раствора. Выпа-
ривая последний, получают продукт сульфирования в виде каль-
циевой соли сульфокислоты.
3.3.1. Сульфирование бензола и его гомологов
Бензол — одно из очень немногих соединений, при суль-
фировании которых в первой стадии образуется только один про-
дукт — моносульфокислота. Но по мере течения процесса, умень-
шения концентрации сульфирующего агента и накопления бензол-
сульфокислоты (6) она может вступить в реакцию с избытком
бензола, образуя дифенилсульфон (7):
(в)
Этому побочному направлению реакции особенно благоприят-
ствуют условия, создающиеся при сульфировании бензола серным
ангидридом или высокопроцентным олеумом, взятыми с очень
небольшим избытком, когда при небольшой концентрации сульфи-
рующего агента воды в сульфомассе практически нет. Наиболее
вероятно в этих условиях, что активной частицей реакции является
сульфонильный катион (8), образующийся по схеме:
/А—SO3H + 2H2SO4 ч=±; —SO2+ + H3O+ + 2HSO4-
(8)
109
Один из наиболее эффективных методов подавления этой реак-
ции — введение в сульфомассу сульфата натрия, образующего би-
сульфат-ион HSO4- Последний смещает приведенное выше равно-
весие влево пропорционально квадрату своей концентрации и тем
самым резко уменьшает содержание в сульфомассе сульфонильно-
го катиона (8) [8]. Для ингибирования образования сульфонов в
реакционную смесь вводят также кислородсодержащие соедине-
ния (кетоны до Се, простые эфиры, сложные эфиры, спирты до С6 и
низшие карбоновые кислоты или Н3РО4, диоксан) [13], алифати-
ческие амины С16 — Cis в алкильных остатках, их соли, амиды Ci —
Cig, алканоламиды, алканоламины, аминокарбоновые кислоты, ор-
ганические или неорганические соли аммония, ароматические или
гетероциклические амины [14] и неорганические сульфиты [15].
При избытке сульфирующего агента и надлежащей его концен-
трации образующаяся на первом этапе бензолсульфокислота мо-
жет сульфироваться дальше до дисульфокислот, хотя эти реакции
идут на несколько порядков медленнее, чем первичное сульфиро-
вание бензола. В соответствии с ориентирующим влиянием элект-
роноакцепторной сульфогруппы, с наибольшей скоростью при этом
образуется ж-бензолдисульфокислота. Сульфирование в пара-по-
ложение идет значительно медленнее; орто-положение из-за сте-
рических причин при реакции не затрагивается. Поэтому при суль-
фировании бензолсульфокислоты (6) высокопроцентным олеумом
образуется только ж-бензолдисульфокислота (9), не содержащая
даже примеси n-изомера. Однако, при длительном нагревании (9)
в условиях, благоприятствующих гидролитическому процессу де-
сульфирования (90%-ная H2SO4, 235 °C) получается равновесная
смесь, содержащая около 66% м- и 34% п-бензолдисульфокислоты
(10) [16]. Интересно отметить, что это превращение было пред-
сказано еще за 25 лет до эксперимента [3].
SO3H SO3H SO3H
SO3H
(9) (6) (10)
Дальнейшее сульфирование (9) до 1,3,5-бензолтрисульфокис-
лоты удается провести только при нагревании с 66%-ным олеу-
мом при 250 °C в присутствии ртути [17].
Бензолсульфокислота получается при сульфировании бензола
92—94%-ной H2SO4 или моногидратом при нагревании. Так как эта
сульфокислота до сих пор производится в больших масштабах для
последующего превращения в фенол при щелочном плавлении
(см. 8.1.1), весьма интенсивно разрабатывались наиболее рацио-
нальные пути ее получения. В ряде патентов описано сульфирова-
ние бензола до моносульфокислоты с помощью серного ангидрида
110
в жидком сернистом ангидриде, а также без этого растворителя с
применением веществ, препятствующих полимеризации серного
ангидрида. В обоих случаях рекомендуется использовать добавки,
подавляющие реакцию образования сульфона (7). Один из непре-
рывных методов сульфирования бензола основан на заметной рас-
творимости бензолсульфокислоты в бензоле, что позволяет, пропу-
ская жидкий бензол противотоком через серную кислоту, полно-
стью извлекать из нее продукт сульфирования. В дальнейшем
бензолсульфокислота из бензольного раствора извлекается водой,
а бензол после сушки возвращается в цикл [18, с. 34].
Наименьший расход серной кислоты и наилучшие экономиче-
ские показатели достигаются при сульфировании бензола «в па-
рах», и этот способ наиболее часто используется в технике. Пары
бензола, перегретые до 150—160 °C и взятые в 7—8-кратном из-
бытке, барботируют через нагретую до той же температуры серную
кислоту. При этом бензол частично сульфируется, а частично уле-
тает, унося с собой пары воды и укрепляя таким образом серную
кислоту, концентрация которой постоянно сохраняется на уровне
90% —вполне достаточном для эффективного течения реакции.
Улетающий бензол после отделения воды возвращается в цикл.
При желании можно полностью использовать всю взятую серную
кислоту для сульфирования. Однако обычно процесс заканчивают,
когда в сульфомассе содержится еще 4—6% H2SO4, так как при
более низкой концентрации начинают с большой скоростью идти
побочные реакции, главным образом образование дифенилсульфо-
на. При указанных оптимальных условиях сульфомасса после от-
дувки бензола имеет следующий состав (в %):
Бензолсульфокислота 88,5—91,5
лг-Бензолдисульфокислота 0,4—0,6
Свободная серная кислота 3,0—3,5
Нерастворимые примеси 1,3
в том числе дифенилсульфон 1,0
Свободный бензол 0,2
Выход бензолсульфокислоты достигает 96—98% от теоретиче-
ского, в расчете на бензол. Сульфомассу нейтрализуют суспензией
карбоната или сульфита натрия в воде и без отделения обра-
зующегося при этом сульфата натрия [18, с. 35] передают на
щелочное плавление, при котором получается фенол (см. 8.1.1).
Газообразный сернистый ангидрид, образующийся при нейтрали-
зации сульфомассы сульфитом натрия, улавливается и по трубам
передается в отделение щелочного плавления, где используется
для выделения фенола из щелочного раствора. Этим достигается
значительная экономия расхода нейтрализующих агентов в произ-
водстве фенола. Недостатком этого приема является необходи-
мость использования материалов, стойких к коррозии, а также
повышенные требования к герметичности оборудования — все это
удорожает производство. Поэтому сульфит натрия для нейтрали-
зации используется не всегда.
111
Сульфирование бензола в промышленности осуществляется
следующим образом [18, с. 51]. Бензол подают в стальной испа-
ритель типа «труба в трубе». По внутренним трубам испарителя
проходит бензол, по внешним — греющий пар давлением 0,9 МПа.
В испарителе бензол нагревается, испаряется и перегревается до
150—160 °C. Образующиеся пары с давлением 0,15—0,18 МПа по-
ступают в чугунный сульфуратор со стальной рубашкой, куда
подается греющий пар с давлением 0,9 МПа. В сульфуратор пред-
варительно загружают 94—96%-ную серную кислоту. Пары бензо-
ла поступают в кислоту через чугунный или термосилидовый бар-
ботер. Избыток бензола вместе с водяными парами направляется
через брызгоуловитель в свинцовый змеевиковый конденсатор, от-
куда попадает в отстойник, где при 30—35 °C отделяется вода.
Далее бензол нейтрализуется, проходя через раствор щелочи, и
возвращается на сульфирование.
В сульфураторе в течение первых двух часов работы процесс в
целом имеет явно выраженный экзотермический характер. Теплота
отводится парами бензола, которые в начале процесса имеют более
низкую температуру, чем реакционная масса. На этом этапе тем-
пературу паров бензола поддерживают не выше 150 °C, чтобы
сульфомасса не перегревалась выше 160—170 °C. В последующий
период выделяющейся теплоты недостаточно для компенсации
теплопотерь, поэтому сульфуратор необходимо обогревать паром.
После завершения реакции сульфомасса передается в обогревае-
мый сборник, а оттуда — в обогреваемую чугунную колонну, рабо-
тающую под вакуумом. Здесь идет десорбция бензола, которого
первоначально в сульфомассе содержится до 4%. Пары бензола
задерживаются в конденсаторе и в абсорбере, заполненном актив-
ным углем и после очистки возвращаются в процесс. Сульфомассу,
содержащую не более 0,2% бензола, нейтрализуют суспензией
сульфита натрия в воде. Реакция ведется в стальном, футеро-
ванном кислотоупорной плиткой аппарате, снабженном фаолити-
рованной мешалкой. Аппарат соединен с футерованной колонной, в
которой газообразный сернистый ангидрид отделяется от раствора
соли сульфокислоты, стекающего в нижнюю часть колонны. Про-
тивотоком стекающему раствору пропускают водяной пар для вы-
деления из массы остатка сернистого ангидрида, который исполь-
зуется при получении фенола. Реакция нейтрализации непрерывно
контролируется с помощью pH-метра. Образующийся раствор со-
держит 47—51% бензолсульфоната натрия, 0,2% сульфита натрия,
3—5% сульфата натрия, 1—2% хлорида натрия, 0,7% сульфонов,
0,5% соли л/-бензолдисульфокислоты и 40—45% воды. Он непо-
средственно используется для щелочного плавления. Выход
продукта — 95%.
л-Бензолдисульфокислота, которая используется преимуще-
ственно для получения резорцина при щелочном плавлении
(см. 8.1.1), производится в значительно меньших масштабах. Так
как при прямом сульфировании бензола олеумом до л-бензолди-
сульфокислоты получается значительное количество сульфонов.
112
обычно процесс ведут в два этапа—вначале сульфируют бензол
до моносульфокислоты, а затем, после укрепления сульфомассы
олеумом,— до дисульфокислоты. Так как соли ж-бензолдисульфо-
кислоты хорошо растворимы в воде и не высаливаются, продукт
получают после полного осаждения серной кислоты в виде суль-
фата кальция (гипса) с помощью выпаривания [17]. Описано так-
же получение ж-бензолдисульфокислоты пропусканием паров бен-
зола и избытка серной кислоты (слоем толщиной 0,5—3 мм)
в течение 60 с через испаритель при 200—240 °C и давлении
10 мм рт. ст. Избыток серной кислоты удаляют, вводя в реакцион-
ную смесь при 150—180 °C пары бензола [19].
В промышленности двухстадийное сульфирование бензола ве-
дут следующим образом [18, с. 96]. Сначала бензол сульфируют
на моносульфокислоту 100%-ной серной кислотой при 50—80 °C,
затем — на дисульфокислоту 65%-ным олеумом при 30—80 °C. На
27,5 кг бензола берут 71 кг моногидрата в первой стадии и 98 кг
65%-ного олеума во второй. Выдержка во второй стадии при 80 °C
составляет не более часа.
Сульфомассу передавливают в аппарат, содержащий раствор
сульфата натрия, и нейтрализуют гидроксидом кальция (известко-
вым молоком) или карбонатом кальция (мелом). Раствор кальци-
евой соли дисульфокислоты отфильтровывают от сульфата кальция
на вакуумных нутч-фильтрах. В фильтрат загружают карбонат
натрия и отделяют выпавший осадок карбоната кальция на
фильтр-прессе. Остатки л-бензолдисульфоната натрия вымывают
из осадка горячей водой, раствор соли выпаривают и сушат на
вальцевой сушилке. Получают 100 кг сухого продукта, содержа-
щего 85% соли л-бензолдисульфокислоты, что соответствует
85%-ному выходу.
В описанном методе расход серной кислоты составляет 5,4 моль
на 1 моль бензола. Значительно лучше этот показатель — всего
3,4 моль на моль бензола — в разработанном в СССР процессе
одностадийного сульфирования бензола 65%-ным олеумом в при-
сутствии сульфата натрия, подавляющего побочную реакцию об-
разования сульфонов (см. выше). Как и в предыдущем способе,
сульфомассу нейтрализуют мелом, а гипс отделяют на нутч-фильт-
ре. Раствор кальциевой соли л-бензолдисульфокислоты обрабаты-
вают карбонатом натрия, осадок карбоната кальция отфильтровы-
вают, но не промывают, а возвращают в процесс на нейтрализацию
сульфомассы. После фильтрации раствор лт-бензолдисульфоната
натрия выпаривают до 60%-ной концентрации и в этом виде ис-
пользуют для щелочного плавления.
Толуол сульфируется легче бензола, причем вначале образуется
смесь о- и n-изомеров толуолсульфокислоты, которая при нагрева-
нии в серной кислоте, содержащей воду, превращается в основном
в n-изомер с небольшой примесью .и-изомера. Описан способ полу-
чения чистой n-толуолсульфокислоты, состоящий в проведении
процесса при 150 °C и промывке продукта реакции толуолом, со-
держащим этанол [20]. Найдены условия, в которых количество
НЗ
образующегося л-изомера можно довести до 19% [21] и даже до
50%, продлив время реакции и нагревая массу до 180—200 °C [22].
Изучено влияние растворителей на ориентацию при сульфировании
толуола серной кислотой и серным ангидридом [23—25].
Изучалось также сульфирование о-ксилола серной кислотой
различных концентраций в пределах 0—150 °C. Определена зави-
симость количества 3-изомера о-ксилолсульфокислоты от темпера-
туры, концентрации кислоты и степени превращения о-ксилола.
При сульфировании двукратным избытком 100% -ной H2SO4 сум-
марный выход 3- и 4-о-ксилолсульфокислот при 100 °C достигает
99% на взятый о-ксилол [26].
Большое промышленное значение имеют так называемые суль-
фонолы RGjHiSOsNa — поверхностно-активные вещества. Их полу-
чают сульфированием длинноцепочечных алкилбензолов, чаще все-
го додецилбензола (алкил С12) и керилбензола (алкилы Сю—Сю) —
см. 6.1.3 — и последующей нейтрализацией образующихся алкил-
бензолсульфокислот. Сульфирующим агентом может служить
25%-ный олеум или газообразный серный ангидрид, разбавленный
инертным газом, например азотом. Однако наилучшие результаты
достигаются при использовании 15%-ного раствора SO3 в жидком
сернистом ангидриде; такой раствор обычно получают на месте
применения путем дозированного окисления сернистого ангидрида
[27, с. 396]. Непрерывное сульфирование алкилбензолов этим
агентом можно вести с большой скоростью, так как теплота реак-
ции очень эффективно отводится кипящим сернистым ангидридом.
Таким способом возможно получать до 200 т сульфокислот на
1 м3 объема реактора в час [18, с. 107].
Аппарат для сульфирования алкилбензолов представляет ци-
линдрический сосуд с нижним сливом, в центре которого распола-
гается стакан с конусной мешалкой. Алкилбензол и сульфирующий
агент из двух мерников непрерывно подаются в центр стакана, где
смешиваются при помощи конусной мешалки, засасывающей смесь
и отбрасывающей ее на стенки аппарата. Реакция быстро доходит
до конца, и сульфомасса, содержащая до 5% растворенного SO2,
стекает в теплообменник для полного удаления сернистого ангид-
рида. Последний конденсируется в холодильниках и возвращается
в цикл. В дальнейшем сульфомасса поступает на нейтрализацию,
которая ведется непрерывным способом путем прибавления
25%-ного раствора NaOH до pH 7,5—8,0. Полученный 50%-ный
раствор используется как таковой или после добавления наполни-
телей подвергается сушке в распылительной сушилке.
3.3.2. Сульфирование производных углеводородов
ряда бензола
Хлорбензол примерно в 10 раз медленнее реагирует с
сульфирующим агентом, чем бензол. При сульфировании его моно-
гидратом или олеумом при 80—100 °C образуется п-хлорбензол-
сульфокислота, почти не содержащая о-изомера [17]. Продукт
1И
Вопросу об активном реагенте нитрования и механизме реак-
ций в уксусной кислоте и уксусном ангидриде посвящены работы
[17, 18], выполненные с привлечением метода МО и кинетических
методов.
В практическом отношении большое значение имеет нитрование
высокореакционноспособных ароматических соединений разбав-
ленной азотной кислотой при нагревании. Естественно, что в этих
условиях в реакционной массе не могут содержаться нитрацидий-
и нитроний-катионы и даже недиссоциированная азотная кислота.
В этом случае доказано, что реакция идет под влиянием четы-
рехокиси азота N2O4, первоначально образующейся за счет восста-
новления азотной кислоты нитруемым соединением или примесями
[19]. Четырехокись азота (3) может распадаться, давая радикаль-
ную двуокись азота или же нитрозоний-катион и нитрат-анион:
_>)
2NO2 ч=± O=NON :₽=> NO++NOs
V)
(3)
Так как разбавленной азотной кислотой способны нитроваться
лишь ароматические соединения, которые особенно легко реагиру-
ют с электрофильными реагентами: фенолы, амины, диметоксипро-
изводные и другие, то высказывается предположение, что реаген-
том в такой реакции является катион NO+. При этом вначале при
его действии образуется нитрозосоединение, которое окисляется
азотной кислотой в нитропроизводное:
-Н+ + HNO3
АгН + NO+ ---> ArNO -------► ArNO2 + HNO2
Однако это предположение не согласуется с некоторыми экспе-
риментальными наблюдениями, показывающими, в частности, что
разбавленная азотная кислота нитрует многие соединения, не спо-
собные вступать в реакцию нитрозирования. Более вероятно, что
реагентом при нитровании разбавленной азотной кислотой являет-
ся сама четырехокись азота:
Многие исследователи предполагают также, что реакция нит-
рования разбавленной азотной кислотой имеет радикальный ха-
рактер и реагентом в ней служит двуокись азота, представляющая
собой свободный радикал:
J87
Радикальная реакция, подобно рассмотренным электрофиль-
ным, имеет двухстадийный механизм. Промежуточно образую-
щийся о-комплекс, содержащий делокализованный неспаренный
электрон, превращается в конечный продукт реакции при действии
еще одной радикальной частицы, отрывающей атом водорода
(см. 2.7.3).
Таким образом, вопрос о механизме и реагенте реакции нитро-
вания в разбавленной азотной кислоте в настоящее время еще до
конца не выяснен. Известно лишь, что в отсутствие четырехокиси
азота эта реакция не идет. Вместе с тем, начавшись, она доходит
до конца, так как получающаяся азотистая кислота при любом из
обсужденных механизмов реагирует с азотной кислотой, регенери-
руя четырехокись азота, необходимую для нитрования:
HNO2 + HNO3 —> N2O4 + H2O
Обзору исследований по механизму реакций нитрования посвя-
щена работа [20].
Описано нитрование нитратом титана(1У) Ti(NO3)4. Этот агент
легко нитрует ароматические соединения. С бензолом реакция за-
канчивается за 10 с, а с толуолом — еще быстрее. Нитробензол и
хлорбензол реагируют медленнее, а ж-динитробензол вообще не
нитруется. Соотношение о-, м-, и п-изомеров близко к соотношению
изомеров, получающихся при нитровании смесью азотной и серной
кислот,— это свидетельствует, что Ti(NO3)4 действует как электро-
фильный реагент [21].
4.3. ОСОБЕННОСТИ ОРИЕНТАЦИИ ПРИ НИТРОВАНИИ
При нитровании ориентация вступающей нитрогруппы
существенно отличается от ориентации сульфогруппы при сульфи-
ровании. Это связано с малым объемом нитрующих частиц и, как
следствие, с их сравнительно малой чувствительностью к стериче-
ским препятствиям; имеет значение также высокая электрофиль-
ность катиона NO2, которая в сочетании с необратимостью про-
цесса приводит к снижению селективности направления реакции.
Так, при нитровании алкилбензолов в уксусном ангидриде при
0°C получаются следующие соотношения изомеров:
o-NO2 .«-NO, n-NO
Толуол 58,5 4,4 37,1
Этилбензол 45,0 6,5 48,5
Изопропилбензол 30,0 7,7 62,3
трет-Бутилбензол 15,8 11,5 72,7
С увеличением объема алкильной группы постепенно уменьша-
ется количество о-изомеров, а п- и .«-изомеров становится больше.
Последнее особенно интересно, так как свидетельствует о падении
селективности в этом ряду веществ.
На ориентацию при реакции нитрования особенно большое вли-
яние оказывает стабильность переходного состояния и промежу-
138
обычно не выделяют из сульфомассы и тут же нитруют, получая
З-нитро-4-хлорбензолсульфокислоту, которая применяется в синте-
зе промежуточных продуктов.
Нитробензол, «-нитротолуол, n-нитрохлорбензол и некоторые
другие продукты, содержащие нитрогруппу, сульфируют олеумом,
так как в этих случаях величина л сульфирования равна 82. Про-
цессы эти небезопасны и при нарушении технологии могут приво-
дить к взрывам. Температура ни в коем случае не должна превы-
шать 100—110 °C, а нитросоединение загружают непременно в
олеум, а не наоборот. Такой порядок загрузки уменьшает возмож-
ность накопления в реакционной массе нерастворившегося в серной
кислоте нитропродукта. Существенным условием является также
непрерывное и эффективное перемешивание.
При сульфировании нитробензола олеумом получается ж-нит-
робензолсульфокислота [17,28], Ее используют главным образом
для получения метаниловой кислоты (лг-аминобензолсульфокисло-
та), из которой в свою очередь получают лг-аминофенол и серию его
производных. Реакцию ведут с одно- двухкратным по массе коли-
чеством 65%-ного олеума при 80—105 °C. Конец реакции определя-
ется по отсутствию в пробе нитробензола. Сульфомассу выдавли-
вают из аппарата в раствор сульфата натрия при 80—85 °C, ох-
лаждают до 20—25 °C и отделяют выпавшую натриевую соль
ж-нитробензолсульфокислоты на центрифуге. Выход продукта —
65%. Если продукт далее используется для восстановления, его не
выделяют из раствора, но с помощью гидроксида кальция полно-
стью осаждают сульфат-ион в виде сульфата кальция, а последний
отфильтровывают при 80 °C. Побочным продуктом сульфирования
является 3,3'-динитродифенилсульфон.
Сульфирование «-нитрохлорбензола дает 5-нитро-2-хлорбен-
золсульфокислоту, которая применяется для получения производ-
ных дифениламина и 2-амино-5-нитробензолсульфокислоты.
«-Нитрохлорбензол при 80 °C растворяют в 1,3 части 20%-ного
олеума, добавляют 1 часть 65%-ного олеума и сульфируют при
107—110 °C. После окончания реакции массу охлаждают до 40 °C и
выдавливают ее в воду при температуре не выше 80 °C. Продукт
высаливают из раствора сульфатом натрия, который добавляют до
достижения плотности 1,33—1,36. Суспензию охлаждают и фильт-
руют на барабанном нутч-фильтре, промывая раствором хлорида
натрия. Выход — 93%. [18, с. 188].
«-Нитротолуол сульфируют трехкратным по массе количеством
18—25%-ного олеума при 60 °C. По окончании реакции сульфомас-
су выливают в такое количество воды, чтобы получился примерно
40%-ный раствор серной кислоты, в котором продукт практически
нерастворим. Получаемая таким образом 4-нитро-2-толуолсульфо-
кислота используется преимущественно для окисления в 4,4'-ди-
нитро-2,2'-стильбендисульфокислоту (см. 14.1.2). Предложено
также получать 4-нитро-2-толуолсульфокислоту в несколько ста-
дий контактированием расплавленного «-нитротолуола со смесью
115
не менее 50% (об.) газообразного серного ангидрида и инертного
газа при 90—150 °C [29].
Хлорангидриды сульфокислот — сульфохлориды (сульфонил-
хлориды) обычно синтезируют, сульфируя ароматические соедине-
ния хлорсульфоновой кислотой. При этом на первом этапе реакции
образуется сульфокислота:
ArH+CISOaH —> ArSO3H + HCl
Эта стадия реакции необратима, так как вода в реакционной
массе отсутствует. Получившаяся сульфокислота взаимодействует
с хлорсульфоновой кислотой далее по равновесной реакции [30],
образуя сульфохлорид:
ArSO3H + CISO3H ч=> ArSO2Cl + H2SO4
Чтобы сместить это равновесие вправо, необходим избыток
хлорсульфоновой кислоты. Поэтому обычно для сульфирования
берут не менее 4—5 моль хлорсульфоновой кислоты на 1 моль
исходного соединения. После проведения реакции сульфомассу
осторожно выливают на колотый лед или в охлажденную воду.
Продукт реакции отделяют фильтрованием, или, если он жидкий,
расслаиванием. Дальнейшую очистку жидких сульфохлоридов про-
водят вымораживанием и вакуум-перегонкой.
Действием хлорсульфоновой кислоты в промышленности полу-
чают бензолсульфохлорид, о- и п-толуолсульфохлорид, 5-(хлор-
сульфонил) салициловую кислоту, и-ацетиламинобензолсульфохло-
рид и ряд других продуктов. Сульфохлориды получаются также
при действии пятихлористого фосфора на сульфокислоты и их соли.
Их используют в качестве ацилирующих агентов для получения
различных сульфамидов (сульфониламидов), а также для восста-
новления в сульфиновые кислоты и тиофенолы.
Спектрофотометрическим методом изучена кинетика хлорсуль-
фирования бензола и толуола и превращения бензолсульфокисло-
ты в сульфохлорид в 1,2-дихлорэтане при 30—75 °C. Высказано
предположение, что реакция хлорсульфирования ароматических
углеводородов протекает по электрофильному механизму с уча-
стием ионной пары SO2C1+ || SO3CH и образованием промежуточ-
ного циклического комплекса [31].
Сульфированием ароматических аминосоединений получают
аминосульфокислоты. Аминогруппа является одним из сильнейших
электронодонорных заместителей и ускоряет течение реакции
сульфирования в пара- и орто-положения. Однако, при действии
серной кислоты или олеума аминогруппа протонируется, причем
равновесие почти полностью смещается вправо:
ArNH2 + H2SO4 :?=> ArNHl + HSO;
Протонированная аминогруппа (аммониевая) является уже
сильным электроноакцепторным заместителем, минимально тормо-
зящим реакцию сульфирования в л-гето-положепие. Поэтому при
прямом сульфировании аминов практически неизбежно образова-
на
йие изомеров, которые, как правило, с трудом разделяются.
В связи с этим амины сульфируют непосредственно обычно лишь в
тех случаях, когда наличие изомерных продуктов не мешает даль-
нейшему использованию аминосульфокислот. Для получения же
чистых индивидуальных продуктов пользуются обходными мето-
дами или применяют так называемую «реакцию запекания».
Реакция запекания позволяет получать чистые сульфокислоты
многих 'первичных и вторичных ароматических аминов, а также
гетероциклических соединений, содержащих группу NH в кольце,
например производных бензимидазола [32], при минимальной за-
трате серной кислоты.
Эту реакцию осуществляют путем длительного нагревания за-
ранее приготовленных кислых сульфатов (бисульфатов) аромати-
ческих аминов при определенной температуре, обычно не превы-
шающей 200 °C. Раньше сухие бисульфаты аминов нагревали на
металлических противнях в специальных печах при заданной тем-
пературе, иногда под вакуумом. В настоящее время в промышлен-
ности бисульфаты аминов нагревают в высококипящих органиче-
ских растворителях, например в полихлоридах бензола. Это обес-
печивает значительно более равномерный обогрев и исключает
подгорание твердого продукта.
При нагревании вначале с отщеплением воды образуется арил-
сульфаминовая кислота, которая далее изомеризуется с переходом
сульфогруппы от азота в орто- или пара-положение ароматическо-
го кольца, в зависимости от температуры, при которой ведется
процесс. Например, бисульфат анилина (11) при этом через
Af-фенилсульфаминовую кислоту (12) образует сульфаниловую
кислоту (п-аминобензолсульфокислоту) (13) по схеме:
NHJSO.JF NHSO3H NHa
SO3H
(11) (121 (13)
В чугунный аппарат с мешалкой и рубашкой, обогреваемый
высококипящим органическим теплоносителем, загружают анилин,
четырех- пятикратное количество растворителя — технического
о-дихлорбензола и эквивалентное количество концентрированной
серной кислоты. Температуру в аппарате постепенно повышают до
175 °C. При этом отгоняется азеотропная смесь воды и растворите-
ля, содержащая около 0,25 взятого количества о-дихлорбензола.
Основную часть последнего отгоняют из аппарата под разреже-
нием, после чего при 70—80 °C приливают воду и нейтрализуют
раствор щелочью. Остатки о-дихлорбензола и не вступивший в
реакцию анилин отгоняют острым паром. Полученный раствор
117
обрабатывают активным углем, подкисляют соляной кислотой,
охлаждают и отфильтровывают выпавшую сульфаниловую кисло-
ту [18, с. 189].
Аналогично из о-толуидина получается 2-амино-5-толуолсуль-
фокислота, а из п-толуидина— 4-амино-З-толуолсульфокислота.
В нафталиновом ряду этот метод используется для промышленного
получения нафтионовой кислоты из 1-нафтиламина, а в антрахино-
новом— для синтеза 1 -амино-9,10-антрахинон-2-сульфокислоты из
1-ам ино-9,10-антрахинона.
Описано влияние сульфата ртути(II) на ориентацию сульфо-
группы при сульфировании диметиланилина олеумом. При этом
сокращается продолжительность реакции и образуются сульфо-
смеси с повышенным содержанием лг-сульфокислоты диметилани-
лина и пониженным содержанием о- и п-изомеров [33].
ж-Аминобензолсульфокислота (метаниловая кислота) при дей-
ствии олеума и умеренной температуре дает смесь 2-ами-
но- 1,4-бензолдисульфокислоты, 5-амино-1,2,4-бензолтрисульфо-
кислоты и 4-амино-1,2,3,5-бензолтетрасульфокислоты:
Степень полисульфирования растет с концентрацией олеума, и
соотношение продуктов контролируется термодинамически. Таким
образом, под влиянием аминогруппы замещение идет с наруше-.
нием правила Армстронга — Вина (см. 3.2) в орто- и пара-положе-
ния к сульфогруппе. Высказывается предположение, что сульфи-
рованию в ядро предшествует образование арилсульфаминовой
кислоты, которая сульфируется в непротонированной форме. В вод-
ной серной кислоте 4-амино-1,2,3,5-бензолтетрасульфокислота лег-
ко десульфируется до 5-амино-1,2,4-бензолтрисульфокислоты [34].
3.3.3. Сульфирование нафталина и его производных
Основные пути первичной переработки нафталина — это
окисление его во фталевый ангидрид и реакция сульфирования,
служащая первым этапом на пути получения целой серии важней-
ших промежуточных продуктов.
Нафталин значительно легче бензола вступает в реакцию суль-
фирования. Скорость сульфирования нафталина в 79%-ной
118
H2SO4 при 25 °C в 80 раз больше скорости сульфирования бензола,
а в 83,4 %-ной — в 43 раза [3].
SO3H
(25)
Как видно из этой схемы, продуктами моносульфирования
нафталина (14) могут быть 1- и 2-нафталинсульфокислоты (15) и
(16). Как сообщалось ранее, ос-положения нафталина активнее
[3-положений и соотношение скоростей образования 1- и 2-сульфо-
кислоты меняется от 5,9 до 4,1 с увеличением концентрации серной
кислоты от 75 до 95% при 25 °C. При постоянней 95%-ной концент-
рации серной кислоты это отношение уменьшается от 5,2 при 0,5°C
до 3,3 при 70 °C [35]. Примерно в таком же отношении находятся
и скорости десульфирования этих сульфокислот.
Сульфирование нафталина на 1-сульфокислоту следует вести,
создавая условия, в которых первично образующаяся 1-сульфо-
кислота не успевала бы гидролизоваться — это обеспечивает удов-
летворительный выход. Наоборот, в условиях, благоприятствую-
щих гидролитическим процессам, главным продуктом реакции
становится 2-изомер. Обычно сульфирование нафталина на 1-суль-
фокислоту ведут моногидратом при 60 °C. При этом в сульфомассе
содержится до 20% 2-изомера. Фирц-Давид описывал также суль-
фирование нафталина моногидратом при 0°С. В этих условиях
скорость образования 2-изомера уменьшается и содержание его в
сульфомассе не превышает 4% [17]. В литературе описано мно-
жество других приемов, позволяющих уменьшить количество
2-изомера при сульфировании нафталина на 1-сульфокислоту:
119
сульфирование действием SO3 или C1SO3H, в органических раство- =
рителях или в жидком SO2, при низкой температуре и т. д. [8,36]. -
Из-за примеси 2-изомера 1-нафталинсульфокислота лишь огра-
ниченно используется для синтеза 1-нафтола (последний поэтому
получают из 1-нитронафталина через 1-нафтиламин или из тетра-
лина— см. гл. 17), но находит применение в синтезе ряда других
промежуточных продуктов. Среди них важнейшие — 5- и 8-нит-
ро-1-нафталинсульфокислоты, 1,5-нафталиндисульфокислота (17) и
1,3,5-нафталинтрисульфокислота (22).
Сульфирование нафталина на 2-сульфокислоту ведут в услови-
ях, максимально благоприятствующих течению реакции десульфи-
рования, которой в первую очередь подвергается ненужная здесь
1-сульфокислота: используется купоросное масло при 160—165 °C и
длительная выдержка. Тем не менее в сульфомассе остается до
10% 1-изомера, который удается в значительной степени гидроли-
зовать до нафталина.
В промышленном производстве получение 2-нафталинсульфо-
кислоты (16) проводят в чугунном аппарате вместительностью до
5000 л, обогреваемом паром и снабженном якорной мешалкой.
В аппарат по обогреваемым трубам заливают расплавленный наф-
талин и при 125—158°С постепенно вводят 96%-ную H2SO4—
приблизительно 1,1 моль на 1 моль нафталина. Поскольку около
9% нафталина не вступает в реакцию, избыток серной кислоты
составляет примерно 20%. После 3-часовой выдержки при 160 °C
сульфомассу передают в другой аппарат, где производится гидро-
лиз побочно образующейся 1-сульфокислоты (ее содержится 4%).
Этот аппарат представляет собой чугунный котел, футерованный
двумя слоями кислотоупорной плитки, с якорной мешалкой.
К сульфомассе добавляют 0,4 моль воды на 1 моль взятого нафта-
лина и выдерживают 1 ч при 140—150 °C. По окончании этой
операции сульфомасса содержит 8,5% серной кислоты, 65%
2-сульфокислоты и 2% 1-сульфокислоты, а также около 9% нафта-
лина. Для удаления последнего сульфомассу передают в отдувоч-
ный аппарат, представляющий собой стальной футерованный котел
с мешалкой и медным барботером для подачи пара. В него пред-
варительно помещают раствор сульфата натрия с плотностью 1,2 в
количестве 70 л на 1 кмоль взятого нафталина, в который и сливают
сульфомассу. Через барботер пропускают водяной пар сдавлением
0,3—0,4 МПа до прекращения отгонки нафталина. В реакционной
массе остается не более 0,2% нафталина. Водяной пар, содержа-
щий нафталин, попадает в конденсаторы и специальные ловушки, в
которых отогнанный нафталин отделяется от воды и возвращается
в процесс.
Нейтрализуют сульфомассу раствором сульфита натрия в за-
крытом аппарате, футерованном кислотоупорной плиткой. После
проверки полноты нейтрализации и плотности раствора выделив-
шуюся натрцевую соль 2-нафталинсульфокислоты отфильтровыва-
ют на полуавтоматических горизонтальных центрифугах и с влаж-
ностью 15—20% передают в отделение щелочного плавления; вы-
120
ход продукта — 85—86%. Образующийся при нейтрализации суль-
фомассы сернистый ангидрид (газ) по трубам передается в
аппараты для подкисления щелочных растворов 2-нафтола, полу-
чающихся при щелочном плавлении 2-нафталинсульфокислоты
[18, с. 125].
2-нафталинсульфокислота используется в очень больших коли-
чествах, для щелочного плавления в 2-нафтол (см. 8.1.1) и для
получения А^-фенил-2-нафтиламина (неозона Д — см. 7.1.5), исполь-
зуемого в промышленности в качестве стабилизатора полимеров.
Меньшие ее количества подвергаются дальнейшему сульфирова-
нию или нитрованию, при которых получаются 5- и 8-нитро-2-наф-
талинсульфокислоты. Из них после восстановления получают
используемые в синтезе азокрасителей кислоты Клеве: 5-амино-
2-нафталинсульфокислоту (26) и 8-амино-2-нафталинсульфокис-
лоту (27).
NH2
nh2
7-кислота Клеве
(27)
SO3H
6-кислота Клеве
(26)
Дальнейшее сульфирование моносульфокислот нафталина про-
ходит преимущественно за счет второго, незамещенного бензоль-
ного кольца, так как на него в меньшей степени распространяется
электроноакцепторное влияние сульфогруппы. Поэтому из 1-на-
фталинсульфокислоты (15) получаются 1,5-, 1,6- и 1,7-нафталинди-
сульфокислоты (17), (18) и (19); в положение 8 сульфирование не
идет из-за стерических препятствий. При этом сульфирование в
положения 5 (a-положение) и 6 (нехиногеновое положение) дол-
жно идти заметно скорее, чем в положение 7 (хиногеновое и
р-положение). Соответственно дальнейшее сульфирование 2-на-
фталинсульфокислоты (16) идет в положения 5, 6, 7 и 8, из кото-
рых по понятным причинам наиболее активно положение 5, а наи-
менее активно — 6.
В условиях, при которых возможно течение гидролитических
процессов, десульфированию в первую очередь подвергаются ди-
сульфокислоты, содержащие сульфогруппы в сс-положениях. По-
этому для получения с удовлетворительным выходом 1,5-нафта-
линдисульфокислоты (17) реакцию сульфирования надо вести ак-
тивным сульфирующим агентом — олеумом при сравнительно
низкой температуре. Для накопления в сульфомассе 2,7-нафталин-
дисульфокислоты (20) и особенно 2,6-нафталиндисульфокислоты
(21) применяют 92—94%-ную H2SO4 и реакцию ведут длительное
время при 160—170 °C.
Таким образом, при сульфировании нафталина может быть
получено пять дисульфокислот (17) — (21). В практическом отно-
шении из них важнейшей является (17), из которой синтезируют
5-гидрокси-1 -нафталинсульфокислоту (1 -нафтол-5-сульфокислоту),
121
1,5-дигидроксинафталин, 3- и 4-нитро-1,5-нафталиндисульфокисло-
ты, при восстановлении и дальнейших превращениях образующие т
ряд ценных промежуточных продуктов. Остальные используются в
значительно меньших масштабах. Например, из (20) получают
7-гидрокси-2-нафталинсульфокпслоту (2-нафтол-7-сульфокислоту).
Получение 1,5-нафталиндисульфокислоты (17) в промышленно-
сти ведут следующим образом [18, с. 170]. Вначале нафталин
сульфируют 98%-ной H2SO4 при 40—55 °C до 1-сульфокислоты
(15), а затем сульфомассу укрепляют 65%-ным олеумом и ведут
реакцию при температуре не выше 35 °C. На 1 кг нафталина за-
гружают 2,3—2,4 кг H2SO4 и 1,7 кг 65%-ного олеума. Концентра-
ция отработанной кислоты составляет 74—76% Готовую сульфо-
массу выливают в воду так, чтобы серная кислота разбавилась до
30—32 %-ной концентрации и при 60—65 °C добавляют хлорид нат-
рия. Выпавшую соль дисульфокислоты отделяют в горизонтальной
гуммированной центрифуге при 25—30 °C. При реакции получается
значительная примесь изомерных продуктов — в основном 1,6-на-
фталиндисульфокислоты (18), большая часть которой (около 7%)
остается в фильтрате.
Для получения трисульфокислот нафталина приходится ис-
пользовать олеум, и поэтому гидролитические процессы на этом
этапе исключаются. Из этих продуктов практически важная
1,3,6-нафталинтрисульфокислота (23) не Может сульфироваться
далее и является конечным продуктом реакции, а две другие —
1,3,5- и 1,3,7-нафталинтрисульфокислота (22) и (24) — образуют
1,3,5,7-нафталинтетрасульфокислоту (25).
Важнейшая из трисульфокислот (23) используется для нитро-
вания и последующих превращений в 4-амино-5-гидрокси-2,7-на-
фталиндисульфокислоту (Аш-кислота), находящую широкое при-
менение в синтезе азокрасителей. Возможно несколько путей для
получения (23) с минимальным количеством примесей [3]. Тради-
ционно ее получают, последовательно сульфируя нафталин сначала
на 2-сульфокислоту (16), затем на 1,6-дисульфокислоту (18) и,
наконец, на 1,3,6-трисульфокислоту (23) [17].
В заводской практике сульфирование нафталина на (23) про-
водят в две стадии [18, с. 158]. На 1000 кг нафталина загружают
1300 кг 100%-ной H2SO4, смесь нагревают с размешиванием при
80—85 °C и затем повышают температуру до 145 °C (первая ста-
дия). При этом образуется 2-нафталинсульфокислота (16) с при-
месью дисульфокислот. После часовой выдержки сульфомассу ох-
лаждают до 80—85 °C, добавляют еще 1000 кг моногидрата, охла-
ждают до 40—60 °C, загружают 2600—3000 кг 65%-ного олеума
и нагревают до 145—160 °C (вторая стадия). По окончании вы-
держки массу разбавляют водой и направляют на нитрование.
Практически достигнутый выход целевого продукта в производ-
ственных условиях составляет около 65%.
Из двух других нафталинтрисульфокислот 1,3,5-изомер (22)
используется для синтеза 4-амино-5-гидрокси-1,7-нафталиндисуль-
фокислоты (8-амино-1-нафтол-3,5-дисульфокислоты; К-кислоты).
122
Его получают ступенчатым сульфированием нафталина через
1,5-дисульфокислоту (17). 900 кг нафталина и 2880 кг 65%-ного
олеума загружают одновременно в течение 32 ч в 1575 кг моно-
гидрата при 30—35 °C. Смесь нагревают 1 ч при 50 °C, затем 1 ч
при 70 °C, наконец, 7 ч при 90 °C. При реакции образуется также
некоторое количество (23) [4].
1,3,7-нафталинтрисульфокислота (24) и 1,3,5,7-нафталинтетра-
сульфок-ислота (25) практически почти не используются.
Нафталинтрисульфокислоты (22), (23) и (24) способны изоме-
ризоваться, превращаясь друг в друга. В присутствии 100%-ной
H2SO4 и HgSOi при 182 °C этот процесс проходит вдвое быстрее,
чем в отсутствие ртути. При 234 °C уже через час достигается
состояние, близкое к равновесию с содержанием 54% 1,3,6-нафта-
линтрисульфокислоты (23) [37].
Изучено сульфирование 2-метилнафталина действием 93 %-ной
H2SO4 при различных температурах. Анализ образующейся реак-
ционной смеси проводят химическим путем: сульфокислоты пере-
водят в натриевые соли, затем в нитрилы (см. 11.1.2); последующей
реакцией нитрилов с CH3MgI и гидролизом получают кетоны, ко-
торые определяют методом ГЖХ. Отмечено образование всех воз-
можных семи моносульфокислот 2-метилнафталина [38].
Сульфирование 2-нафтола является одним из наиболее важных
для практики и сложных процессов. Строение и взаимосвязь полу-
чающихся при этом главных продуктов представлена на схеме
он
so3n
123
В молекуле 2-нафтола (28) из четырех хиногеновых по отноше-
нию к гидроксилу положений — 1,3,6,8 — наиболее активно в реак-
циях электрофильного замещения положение 1; заметно менее
активны положения 8 и 6; положение 3 активируется гидроксиль-
ной группой лишь в очень слабой степени (см. 2.4.2). В соответ-
ствии с этим при сульфировании с максимальной скоростью обра-
зуется 2-гидрокси-1-нафталинсульфокислота (2-нафтол-1-сульфо-
кислота; оксикислота Тобиаса) (29). Ее десульфирование идет
настолько легко, что получить этот практически важный продукт
удается лишь проводя реакцию в условиях, когда гидролитические
процессы затруднены (низкая температура, высокая концентрация
сульфирующего агента, короткое время проведения реакции).
Иногда с этой же целью в сульфомассу добавляют борную кислоту,
образующую с (29) циклический эфир. Интересный вариант полу-
чения чистой (29) путем сульфирования 2-нафтола хлорсульфоно-
вой кислотой в нитробензоле при температуре не выше 5 °C описан
в [17].
В промышленности оксикислоту Тобиаса получают, действуя на
1000 кг 2-нафтола 2300—2400 кг 98%-ной H2SO4 в интервале от
—10 до —1 °C. Важным условием является эффективное размеши-
вание вязкой реакционной массы, для того чтобы весь 2-нафтол
вступил в реакцию. Остатки последнего в конечном продукте недо-
пустимы, так как при последующем аминировании может полу-
чаться канцерогенный 2-нафтиламин. После окончания реакции
сульфомассу выдавливают при 20—30 °C в раствор хлорида калия
с плотностью 1,17, выпавший осадок отделяют на центрифуге и
промывают тем же раствором. Выход продукта составляет 60—•
62,5%. Побочные продукты — изомерные сульфокислоты 2-нафто-
ла — переходят в фильтрат и промывные воды.
Главное назначение оксикислоты Тобиаса (29) — превращение
ее в 2-амино-1-нафталинсульфокислоту (кислоту Тобиаса) (39) с
помощью реакции Бухерера (см. 7.1.5).
При сульфировании 2-нафтола 92—94 %-ной H2SO4 или моно-
гидратом и нагревании до 90—100 °C кислота (29) практически не
образуется. В этих условиях получается 6-гидрокси-2-нафталин-
сульфокислота (2-нафтол-6-сульфокислота; кислота Шеффера)
(30) и 7-гидрокси-1-нафталинсульфокислота (2-нафтол-8-сульфо-
кислота; кроцеиновая кислота) (31), содержащие небольшую при-
месь З-гидрокси-2-нафталинсульфокислоты (2-нафтол-З-сульфо-
кислоты) (32) и дисульфокислот. Отделение используемой в тех-
нике кислоты Шеффера от менее важной кроцеиновой кислоты
представляет известные трудности. Поэтому кислоту Шеффера ча-
ще получают в более жестких условиях, при повышенной темпера-
туре, способствующей почти полному гидролитическому расщепле-
нию кроцеиновой кислоты, но приводящей к накоплению заметных
количеств З-гидрокси-2,7-нафталиндисульфокислоты (2-нафтол-
3,6-дисульфокислота; Р-кислота) (35), имеющей самостоятельное
значение. Разделение этих продуктов сульфирования, отличаю-
щихся по числу сульфогрупп, осуществляется значительно легче.
124
Для получения такой смеси сульфирование 2-нафтола ведут сер-
ной кислотой при ПО—120 °C в течение нескольких часов.
Из дисульфокислот особенно важна 7-гидрокси-1,3-нафталин-
дисульфокислота (2-нафтол-6,8-дисульфокислота; Г-кислота) (34),
являющаяся исходным продуктом для синтеза 6-амино-4-гидро-
кси-2-нафталинсульфокислоты (Гамма-кислоты — см. 7.1.5). Г-кис-
лота получается сульфированием 2-нафтола моногидратом или
слабым олеумом при 70—80 °C, она образуется в смеси с заметным
количеством кислоты Шеффера (30) и Р-кислоты (35). Для пре-
имущественного получения последней сульфирование 2-нафтола
ведут моногидратом при более высокой температуре— 120—125 °C
и более длительно — до 18 ч [17].
При промышленном сульфировании 2-нафтола на Г-кислоту (с
попутным получением Р-кислоты) 2-нафтол загружают в равное
по массе количество 98%-ной H2SO4 при 20—25 °C и выдерживают
при 40—45 °C. В этих условиях образуются кислоты (30) и (31).
При 50 °C в сульфомассу вводят 20%-ный олеум (примерно 2 кг на
1 кг 2-нафтола) и выдерживают при 60 °C. Сульфомассу передав-
ливают при 85 °C в воду и добавляют суспензию хлорида калия в
его насыщенном растворе. После охлаждения до 35 °C и кристал-
лизации дикалиевой соли Г-кислоты (34) ее отфильтровывают на
центрифуге. В фильтрат вводят хлорид натрия до содержания
около 11% и отделяют на центрифуге выпавшую динатриевую соль
Р-кислоты (35). Выход Г-соли достигает 63%, Р-соли—13—14%
[18, с. 180]. К недостаткам описанного процесса сульфирования
относится образование весьма густой сульфомассы, которая требу-
ет мощной мешалки. Однако разбавление массы дополнительным
количеством сульфирующего агента ведет к увеличению выхода
Р-кислоты и соответствующему падению выхода Г-кислоты. Чтобы
облегчить передавливание сульфомассы после сульфирования, ее
иногда разбавляют водой.
Сульфирование кислоты Тобиаса (39) ведут большим количе-
ством олеума при 20—30 °C; в результате образуется преимуще-
ственно 2-амино-1,5-нафталиндисульфокислота (40); при дальней-
125
шем повышении температуры она превращается в 6-амино-
1,3,5-нафталинтрисульфокислоту (41). После завершения про-
цесса сульфомассу разбавляют водой и нагревают. При этом идет
гидролитическое отщепление сульфогруппы в положении 5 и обра-
зуется 6-амино-1,3-нафталиндисульфокислота (42). Из нее при
щелочном плавлении получают практически очень важную 7-ами-
но-4-гидрокси-2-нафталинсульфокислоту (6-амино-1-нафтол-3-
сульфокислота; И-кислота) (43) —см. 8.1.1.
Промышленное сульфирование кислоты Тобиаса (39) проводят
следующим образом. Продукт, полученный после аминирования
оксикислоты Тобиаса (29) и содержащий около 10% воды, раство-
ряют в 20%-ном олеуме (0,85 кг на 1,5 кг продукта), добавляют
2 кг 65%-ного олеума и сульфируют при температуре не выше
35 °C. Содержание свободного серного ангидрида в сульфомассе
должно составлять не менее 12%. После небольшой выдержки
сульфомассу нагревают до 45—100 °C, и в этих условиях образует-
ся трисульфокислота (41). Сульфомассу охлаждают до 75 °C и
передавливают в аппарат для гидролиза, куда предварительно
загружают раствор сульфата натрия с плотностью 1,345. Реакци-
онную массу нагревают при 105 °C. После охлаждения и кристал-
лизации кислой натриевой соли дисульфокислоты (42) ее отфильт-
ровывают на барабанном вакуум-фильтре; выход — около 75%
[18]. Эту соль направляют на щелочное плавление.
При сульфировании 1,8-нафталинсультама (44) 96—100%-ной
H2SO4 и 20%-ным олеумом при 0 и 25°C образуются моно- и ди-
сульфокислоты. Преобладающим продуктом реакции является
1,8-нафталинсультам-5,7-дисульфокислота (45) [39].
SO3H
(44) (45)
3.3.4. Сульфирование антрахинона и его производных
9,10-Антрахинон — третья по значимости после бензола
и нафталина ароматическая структура, используемая для построе-
ния молекул органических красителей. Его молекулу (46) можно
рассматривать как состоящую из двух ароматических бензольных
колец, жестко связанных двумя изолирующими эти кольца карбо-
нильными группами. Межатомные расстояния в этих кольцах та-
кие же, как в бензоле.
Сульфирование является главным методом первичной химиче-
ской переработки антрахинона. Благодаря влиянию двух карбо-
нильных групп, проявляющих электроноакцепторные свойства, он
вступает в эту реакцию только при нагревании с олеумом и суль-
фогруппы становятся исключительно в |3-положения. Так как бен-
зольные кольца антрахинона в значительной степени автономны,
126
введенный в одно из них электроноакцепторный заместитель
(SO3H) почти не влияет на реакционную способность второго бен-
зольного кольца и получившаяся в первой стадии 9,10-антрахи-
нон-2-сульфокислота (47) в тех же условиях и с близкой скоростью
сульфируется далее до 9,10-антрахинон-2,6- и 2,7-дисульфокислот
(48) и (49):
о
(49)
Их удается довольно легко разделить в виде калиевых солей,
имеющих разную растворимость в воде.
Длительное время были неясны причины, по которым антрахи-
нон сульфируется в p-положение, тогда как реакции нитрования и
галогенирования в основном идут в ос-положения. В настоящее
время считают, что причиной этого являются неодинаковые разме-
ры активных частиц, атакующих антрахинон при электрофильном
замещении. Пространственные затруднения мешают подходу объ-
емистой сольватированной частицы сульфирующего реагента к
a-положению антрахинона, экранированному кислородом карбо-
нильной группы (см. также 3.2).
В 1891 г. М. А. Ильинский обнаружил, что направление реакции
сульфирования изменяется в присутствии солей ртути (II): суль-
фогруппы вступают исключительно в сс-положения 9,10-антрахино-
на и схема сульфирования может быть представлена следующим
образом:
О SO3H
(52)
127
М. А. Ильинский предполагал, что образованию 9,10-антрахи-
Ион-1-сульфокислоты (50) и 1,5- и 1,8-дисульфокислот (51) и (52)
предшествует меркурирование в ос-положения антрахинона. Взаи-
модействие сс-меркурпроизводных с олеумом приводит к сульфо-
кислотам и сульфату ртути(П), который вновь вступает в реакцию
с антрахиноном. В настоящее время эти предположения подтвер-
ждены прямым меркурированием антрахинона в полифосфорной
кислоте, моделирующей олеум, идущим исключительно в ос-поло-
жения [40].
Комбинируя реакцию сульфирования в присутствии солей рту-
ти (II) и без них, можно получить еще две дисульфокислоты
9,10-антрахинона — 1,6- и 1,7-, а также две изомерные тетрасуль-
фокислоты (53) и (54).
(53)
Катализаторами сульфирования антрахинона в ос-положение
являются также соли таллия (III) [41] и палладия (II).
При получении моносульфокислот 9,10-антрахинона реакцию
сульфирования обрывают еще тогда, когда около 50% антрахинона
в эту реакцию не вступило и скорость образования дисульфокислот
сравнительно невелика. В дальнейшем, после разбавления реакци-
онной массы водой, антрахинон отделяют от продуктов сульфиро-
вания и возвращают в процесс. Такое проведение реакции называ-
ется сульфированием с «обратным» антрахиноном.
Важнейшей в практическом отношении является 9,10-антрахи-
нон-1-сульфокислота (50), используемая для синтеза Камино- и
1-хлор-9,10-антрахинона, а также и других продуктов. Существует
несколько способов ее получения [18, с. 196].
По одному из способов в обычный чугунный сульфуратор,
снабженный паровой рубашкой и мешалкой, вносят 400 масс. ч.
20%-ного олеума и 4 масс. ч. оксида или сульфата ртути(П) и
208 масс. ч. антрахинона. Перемешивают при 50 °C до полного рас-
творения, затем нагревают в течение часа до 130—135 °C и далее в
течение 2 ч прибавляют 50 масс. ч. 60%-ного олеума, после чего
выдерживают при той же температуре еще 1 ч. Сульфомассу раз-
бавляют водой и при 70—80 °C отфильтровывают на центрифуге
антрахинон, не вступивший в реакцию. Его промывают до
нейтральной реакции, сушат и возвращают на сульфирование.
К фильтрату и промывным водам добавляют сульфат натрия, ох-
лаждают до 30 °C и выпавшую Na-соль 9,10-антрахинон-1-сульфо-
кислоты отфильтровывают на центрифуге, промывая осадок рас-
128
твором сульфата натрия. Выход продукта — около 55%, считая на
взятый в реакцию антрахинон [17].
1,2-Дигидрокси-9,10-антрахинон (ализарин) (55) сульфируется
серной кислотой или моногидратом исключительно в активируемое
гидроксильной группой положение 3 (56). При использовании оле-
ума вторая сульфогруппа вступает в положение 6 или 7 (57). Если
сульфомассу несколько разбавить водой и нагреть, то гидролити-
ческому отщеплению подвергается сульфогруппа в положении 3
1-Амино-9,10-антрахинон превращают в сульфокислоту (60) ре-
акцией запекания его бисульфата (59) или хлорсульфоната, лучше
в среде полихлоридов бензола (см. также 3.3.1).
(59)
(80)
Получающаяся 1-амино-9,10-антрахинон-2-сульфокислота (60)
имеет очень большое значение.
3.3.5. Сульфирование антрацена
Как видно из 3.3.4, сульфирование 9,10-антрахинона,
особенно для получения наиболее важной 9,10-антрахинон-1-суль-
фокислоты, представляет определенные трудности. Использование
солей ртути в этом процессе повышает токсичность промышленных
стоков, а следы ртути сохраняются не только в продуктах сульфи-
рования, но и в соединениях, образующихся на последующих ста-
диях их переработки. Поэтому еще в прошлом веке были начаты
исследования по сульфированию антрацена, выделяемого из
5 Зак. 271
129
тяжелой фракции каменноугольной смолы (см. 1.1), с целью даль-
нейшего окисления образующихся антраценсульфокислот в антра-
хинонсульфокислоты.
Антрацен (61) вступает в реакцию сульфирования значительно
легче нафталина и несравненно легче антрахинона. Наиболее ак-
тивны в его молекуле лгезо-положения 9 и 10 (ср. 2.4.2), затем
следуют а-положения — 1,4,5,8 и р-положения — 2,3,6,7. Сульфо-
группа, введенная в одно из этих положений, мало влияет на
реакционную способность других колец, и поэтому антрацен обла-
дает повышенной способностью к образованию дисульфокислот.
В старых работах утверждалось, что при сульфировании ант-
рацена концентрированной серной кислотой при 20 °C образуются
1,5- и 1,8-антрацендисульфокислоты, а при действии 67%-ной
H2SO4 при 130 °C — 2,6- и 2,7-антрацендисульфокислоты, а также
2-антраценсульфокислота. Однако, как показали более поздние
исследования, выход этих продуктов в указанных условиях невелик
из-за образования большого количества 9,9'-биантрила и соеди-
нений полимерного строения [42].
Более интересные результаты получаются при проведении ре-
акции в органических растворителях, применение которых позво-
ляет резко уменьшить течение окислительных процессов. Так, суль-
фирование (61) хлорсульфоновой кислотой в диоксане приводит
главным образом к 9-антраценсульфокислоте (62) [9]. Сульфокис-
лота эта лабильна и подвергается десульфированию, в результате
чего в реакционной массе постепенно накапливаются 1-антрацен-
сульфокислота (63), а затем и 2-антраценсульфокислота (64), из
которых могут получаться и дисульфокислоты. Побочно при реак-
ции получается 9-хлорантрацен (65) и Э^'-биантрил (66); послед-
ний, по-видимому, через промежуточное равновесное протонирова-
ние антрацена и образование соответствующего радикала. Сум-
марно взаимосвязь продуктов получающихся при сульфировании
антрацена может быть представлена схемой (где А— антрил):
1-А—-SO3H
(вз)
2-А—
(64)
9-А—9-А
(ее)
9-Ar-Cl
(65)
9-A—SO3H
(62)
Соотношение этих продуктов зависит от условий проведения
реакции и существенно меняется в различных растворителях: хло-
роформе, диоксане, смеси пиридина с изоалканом [42].
Изучалось также сульфирование антрацена серной кислотой в
ацетилхлориде и хлорсульфоновой кислотой в ледяной уксусной
130
кислоте. В обоих случаях при реакции получаются 1- и 2-антра-
ценсульфокислоты, смесь дисульфокислот и небольшие количества
9-хлорантрацена и его сульфокислоты. С повышением температуры
сульфирования возрастает образование дисульфокислот. Высказы-
вается предположение, что активным агентом сульфирования в
этих условиях является ацетилсульфат CH3COOSO3H [43].
Предложен технический способ получения 1-антраценсульфо-
кислоты (63), образующейся при сульфировании антрацена
100%-ной H2SO4 в алкановой кислоте или еехлорангидриде (выход
62%). Продукт можно окислить азотной кислотой в 9,10-антрахи-
нон-1 -сульфокислоту (50) [44]. Тот же продукт можно получить,
сульфируя антрацен хлорсульфоновой кислотой в алкановых кис-
лотах Се — Сд при 50 °C и окисляя затем 1-антраценсульфокислоту
водной азотной кислотой без выделения из реакционной смеси.
9 10-Антрахинон-1-сульфокислота при этом образуется с выходом
45,7—54% [45].
О выделении антраценсульфокислот, образующихся при дей-
ствии серной кислоты на антрацен в растворе уксусной кислоты и
уксусного ангидрида, см. [46].
ЛИТЕРАТУРА
1. Cerfontain Н. Mechanistic Aspects in Aromatic Sulfonation and Desulfona-
tion. New York, Intersciens Publishers, 1968, p 1—201. 2. Ворожцов H. H. Основы
синтеза промежуточных продуктов и красителей. 4-е изд. М,' Госхимиздат, 1955,
с. 60—118. 3. Иоффе И. С. Сульфирование органических веществ. Л., Изд-во
ВММА, 1944. 4. Доналдсон Н. Химия и технология соединений нафталинового
ряда. Пер. с англ./Под ред. А. И. Королева. М, Госхимиздат, 1963, с. 125—443,
5. Коптюг В. А. Изомеризация ароматических соединений. Новосибирск, Изд-во
СО АН СССР, 1963, с. 116—152. 6. Мельник А. И., Пассет Б. В. — В кн.: Труды
VII Международного конгресса по ПАВ. Тезисы. М., 1976, секция А, докл. 151.
7. Холодное В. А., Матвеев А. В., Пассет Б, В. — В кн.: Математическое моде-
лирование в науке и технике. Вып. 2.4.3/Под ред. И. Н. Таганова. Л., ЛТИ
им. Ленсовета, 1975, с. 31—42. 8. Ворожцов Н. Н., мл. — В кн.: Химия синтети-
ческих красителей. Т. 3/Под ред. К. Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса. Л., Химия, 1974, с. 1740—1750. 9. Koeberg-Telder A., Cerfon-
tain И. — Rec. trav. chim., 1972, v. 91, № 1, p. 21—32. 10. Cerfontain H., Koeberg-
Telder A., Ris A. e.a. — J. Chem. Soc. Perkin 2, 1975, № 9, p. 970—973.
11. Левин И. А., Кухтин B. A. — ЖОХ, 1957, t. 27, c. 3067. 12. Замышев-
ская H. И., Канушкин А. В., Крикун С. А. и др.—Авт. свид. СССР 363903,
1973. 13. Пат. ФРГ 2019250, 1971; Chem. Abstr, 1971, v. 74, 99639С. 14. Пат.
ФРГ 2019527, 1970; Chem. Abstr, 1971, v. 74, 31652. 15. Пат. США 3789067, 1974;
Chem. Abstr, 1974, v. 80, 82402. 16. Старков С. П., Спрысков А. А.— ЖОХ, 1957,
т. 27, вып. 11, с. 3067—3071. 17. Фирц-Давид Г. Э., Бланже Л. Основные про-
цессы синтеза красителей. Пер. с нем./Под ред. С. В. Богданова и И. В. Фоди-
мана. М, ИЛ, 1957. 18. Беркман Б. Е. Сульфирование и щелочное плавление в
промышленности органического синтеза. М, Госхимиздат, 1960. 19. Бельг, пат.
698821, 1971; РЖ Хим. 1972, ЗН184П. 20. Пат. ПНР 59938, 1970.
21. Broyles A. R., Eckert R. Е. — Ind. a. Eng. Chem. Process Des. a. Develop,
1973, v. 12, № 3, p. 295—300. 22. Яп. пат. 70/28976, 1970; Chem. Abstr., 1971,
v. 74, 12828. 23. Качурин О. И., Зарайский А. П. — Укр. хим. жури, 1973, т. 39,
№ 1, с. [2—16. 24. Cerfontain Н., Koeberg-Telder A, van Kuipers Е. — J. Chem.
Soc. Perkin 2, 1972, № 14, p. 2091—2093. 25. Van Albada M. P., Cerfontain H„
Koeberg-Telder A. — Rec. trav. chim, 1972, v. 91, № 1, p. 33—36. 26. Гнедин Б.Г.,
5*
131
Спрысков А. А., Чудинова Г. П. — Изв. вузов. Химия и хим. технол., 1974,
т. 17, № 3, с. 388—391. 27. Лебедев Н. Н. Химйя и технология основного орга-
нического и нефтехимического синтеза. М., Химия, 1975. 28. Франц, пат. 1544998,
1968; Chem. Abstr., 1970, v. 72, 21490. 29. Пат. США 3840591, 1974; Chem.
Abstr., 1975, v. 82 3988. 30. Спрысков А. А., Кузьмина Ю. А. — ЖОХ, 1951, т. 21,
вып. 4, с. 714—719, 1951, т. 21, вып. 10, с. 1887—1891, 1953, т. 23, вып. 9,
с. 1536—1539.
31. Чижик С. М., Пассет Б. В. — ЖОрХ, 1975, т. 11, вып. 8, с. 1649—1651.
32. Эфрос Л. С. — ЖОХ, 1953, т. 23, вып. 5, с. 842—843. 33. Хе.гевин Р. И.—
ЖПХ, 1972, т. 45, вып. 7, с. 1557—1558. 34. Maarsen Р К-, Bregtnan R., Cerf on-
tain Н. — J. Chem. Soc., Perkin 2, 1977, № 14, p. 1863—1868. 35. Cerfontaln H.,
Telder A. — Rec. trav. chim., 1967, v. 86, № 5, p. 527—536. 36. Пассет Б. В.,
Рачковская H. В.— ЖПХ, 1971. т. 44, вып. 12, с. 2761—2763. 37. Караваев Б. И.,
Егоров В. Е. — Тр. Ивановск. хим.-технол. ин-та, 1969, № 11, с. 49—52.
38. Core Р. Н., Siddtquei A. S —J. Chem. Soc. Perkin 1, 1972, № 18, р. 2344—
2346. 39. Спрысков А. А., Козлов В. А., Захаров А. Г. — Изв. вузов. Химия и
хим. технол., 1975, т. 18, вып. 12, с. 1868—1872. 40. Докунихин Н. С. — Журн.
ВХО, 1976, т. 21, № 3, с. 292—298.
41. Гаева Л. А., Докунихин Н. С. — В кн.: Химия антрахинона/Под ред.
А. И. Королева. М., Химия, 1969, с. 57—65. 42. Morley J О.—J. Chem. Soc. Per-
kin 2. 1976, № 13, р. 1554—1560. 43. Morley 1. О. — Ibid., 1976, № 13, р. 1560—
1564. 44. Пат. ФРГ 2600332, 1976; Chem. Abstr., 1976, v. 85, 94145. 45. Пат. ФРГ
2600363, 1976; Chem. Abstr., 1976, v. 85, 94146. 46. Пат. ЧССР, 159084, 1975;
Chem. Abstr., 1976, v. 84, 164536.
Глава 4
НИТРОВАНИЕ И НИТРОЗИРОВАНИЕ
4.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О РЕАКЦИИ НИТРОВАНИЯ
И НИТРУЮЩИЕ АГЕНТЫ
Нитрованием принято называть взаимодействие органи-
ческого соединения с азотной кислотой и некоторыми ее производ-
ными, в результате которого водород при одном или нескольких
атомах углерода замещается нитрогруппами и образуются нитро-
соединения:
ArH + HONO2 —► ArNO2 + H2O
Нитрование, как и сульфирование, является важнейшей реак-
цией органического синтеза и имеет широкое применение в про-
мышленности. Нитросоединения могут являться целевыми продук-
тами органического синтеза, например, взрывчатыми веществами
или красителями, но чаще используются как промежуточные про-
дукты в цепи химических превращений исходных ароматических
углеводородов, например, для синтеза практически очень важных
ароматических аминов.
Для проведения реакции нитрования промышленность выпу-
скает 60—63%-ную (плотность около 1,40) и 98—100%-ную дымя-
132
щую азотную кислоту (плотность 1,50—1,52); из них в случае
надобности готовят кислоты другой концентрации. Однако нитро-
вание одной азотной кислотой проводят сравнительно редко; чаще
для реакции применяют смеси азотной и серной кислот, так назы-
ваемые нитрующие смеси, количество азотной кислоты в которых
близко к теоретически необходимому.
Вода, образующаяся при реакции, разбавляет азотную кислоту
или нитрующую смесь и быстро уменьшает скорость процесса.
Поэтому при нитровании, как и при сульфировании, количество
азотной кислоты (если нитрование ведется только ею) или серной
кислоты в нитрующей смеси рассчитывают по концентрации отра-
ботанной кислоты, получающейся после завершения реакции.
Практически важным преимуществом нитрующих смесей перед
просто азотной кислотой является также то, что их можно хранить
в железной аппаратуре и передавать по железным трубам, так как
они не вызывают коррозии этого металла. Этим же свойством
обладает так называемый меланж, представляющий собой кон-
центрированную азотную кислоту, содержащую 7,5—9% H2SO4 и
до 4,5% воды. Меланж значительно удобнее транспортировать, чем
азотную кислоту, и в промышленности его часто используют
вместо азотной кислоты для приготовления нитрующих смесей.
Иногда для нитрования вместо концентрированной азотной
кислоты используют ее соли — нитраты натрия или аммония. Рас-
творяясь в серной кислоте, эти соли выделяют азотную кислоту,
которая и нитрует органическое соединение:
NaNO3 + H2SO4 —> NaHSO4 + HNO3
За последние годы наметилась тенденция замены при нитрова-
нии концентрированной серной кислоты другими кислотными аген-
тами, легче поддающимися регенерации. Один из вариантов —
использование в этих процессах нерастворимых обезвоженных
смол, содержащих свободные сульфогруппы (например, полимеров
стиролсульфокислот) [1,2]. Реакция нитрования проходит на по-
верхности этих смол, по-видимому, через промежуточное образо-
вание нитроний-катионов (см. 4.2), Вместо смол используют также
каталитические количества серной или фосфорной кислот на носи-
теле (А120з, AI2O3 + Si О?) [ 3].
Другой вариант — использование очень сильных перфторал-
кансульфокислот, например CF3SO3H [4], или их смесей с P2Os
для связывания выделяющейся воды. Во втором случае реакцию
ведут в среде инертного органического растворителя, например
метилендихлорида или дихлорэтана [5].
Иногда нитрование высокоактивных ароматических соединений
ведут, действуя на них 10—40%-ной азотной кислотой, обычно при
нагревании. В этих случаях азотную кислоту берут со значитель-
ным избытком против теоретического количества. Реакционноспо-
собные соединения нитруют также азотной кислотой в среде орга-
нических растворителей. Особенно часто используют ледяную
133
уксусную кислоту и уксусный ангидрид. Последний с азотной кис-
лотой образует ацетилнитрат:
(СН3СО)2О-f-HONO2 —> CH3COONO2-f-СНзСООН
Ацетилнитрат является активным нитрующим агентом, но
взрывоопасен. Поэтому его используют лишь в особых случаях.
В качестве высокоселективного нитрующего агента предлагает-
ся использовать нитрат алюминия Al(NO3)3-9Н2О в уксусном ан-
гидриде или уксусной кислоте [6], а также нитраты других двух- и
трехвалентных металлов. Кинетическими методами показано, что в
определенных условиях наиболее активны нитраты цинка и меди, а
наименее — нитраты алюминия и железа. Отмечается сильное
влияние иона металла на соотношение изомеров, образующихся
при нитровании салицилового альдегида [7].
Для нитрования алифатических соединений часто применяют
четырехокись азота N2O4 (димер двуокиси NO2; жидкость, кипя-
щая при 21,4 °C; хранится в баллонах) [8,9]. Промышленного
применения для нитрования ароматических соединений она пока
не получила.
В отличие от реакции сульфирования, реакция нитрования
необратима и идет с большой скоростью. Поэтому, хотя тепловой
эффект нитрования близок к тепловому эффекту сульфирования
(151 —159 МДж/моль при вступлении одной нитрогруппы плюс
теплота разбавления серной кислоты водой, образующейся при
реакции), выделение тепла при нитровании происходит быстро и
может сопровождаться значительным разогревом реакционной
массы. Между тем для каждого случая существует оптимальная
температура, при которой следует вести нитрование; при более
низкой температуре скорость реакции резко уменьшается, а при
более высокой настолько возрастает, что может приобрести взрыв-
ной характер. Поэтому обычно нитрующий агент вводят в реакци-
онную массу постепенно по мере его расходования, а процесс ведут
в аппаратах с эффективным отводом тепла и постоянным
контролем температуры. Так как нитрование чаще всего идет в
гетерогенной среде, важным условием является постоянное и хоро-
шее перемешивание.
4.2. МЕХАНИЗМ РЕАКЦИИ НИТРОВАНИЯ
Нитрование концентрированной азотной кислотой и нит-
рующими смесями является классическим и наиболее изученным
примером реакций электрофильного замещения.
Азотная кислота в водных растворах ионизирована:
HNO3-|-H2O =?=*: NO3 + НзО+
Однако уже в 75%-ной азотной кислоте такая ионизация прак-
тически не происходит. Азотная кислота при этой концентрации
существует в виде неионизированных молекул HNO3 и неионного
134
гидрата состава HNO3-H2O. В очень небольшой степени осуществ-
ляется также ионизация по уравнению
3HNO3 < НзО+ -)-• NO2 -р 2NO3
приводящая к образованию наиболее активного электрофильного
реагента нитроний-катиона NO2. Роль этого равновесия возрастает
по мере уменьшения содержания воды, и в 100%-ной азотной
кислоте концентрация NO2 составляет уже около 1%.
В нитрующих смесях взаимодействие серной и азотной кислот
может быть представлено уравнением, в котором азотная кислота
играет роль основания:
2H2SO4 -|- HNO3 < > 2HSO4 Н3О+ NOj
Если количество воды в смеси невелико, а концентрация азот-
ной кислоты не превышает 15%, то приведенное равновесие сильно
смещено вправо. Азотная кислота в такой смеси почти полностью
превращается в катион NO% Разбавление нитрующей смеси водой
приводит к уменьшению концентрации NO% так как серная кислота
в этих условиях взаимодействует не с азотной кислотой, а с водой,
являющейся значительно более сильным основанием. Образование
NOJ из азотной кислоты доказано как физическими и физико-
химическими методами, так и выделением солей этого катиона с
анионами ряда кислот. Некоторые из этих солей, например
NO2BF4, используются не только при изучении механизмов реак-
ций, но и в препаративных целях [10].
Опубликованные данные о равновесиях в растворах азотной
кислоты и в ее смесях с серной кислотой и результаты обширных
кинетических исследований показывают, что электрофильной ча-
стицей с участием которой идет реакция нитрования, в этих усло-
виях является катион NO2 [11, с. 270]. Уменьшение концентрации
этих активных частиц при разбавлении нитрующей смеси водой
хорошо объясняет причину уменьшения скорости нитрования.
Согласно общепринятым представлениям, нитрование аромати-
ческих соединений с участием нитроний-катиона проходит по
схеме:
Образование ^-комплексов — ионный бимолекулярный процесс,
зависящий как от реакционной способности ароматического соеди-
нения, так и от сольватационных эффектов среды. В рассматрива-
емом случае скорость образования о-комплексов % слишком вели-
ка, а скорость обратной реакции k_\ слишком мала, чтобы обрат-
ная реакция могла влиять на суммарную скорость реакции. Полу-
чившийся о-комплекс является сильной кислотой и быстро, со ско-
ростью k2, отдает свой протон основанию. Поэтому данный этап
135
реакции не может определять ее суммарную скорость. В отличие от
сульфирования, при нитровании k2 всегда значительно больше
и кинетический изотопный эффект не наблюдается. Единственное
исключение отмечено при нитровании 1,3,5-три-трет-бутилбензола
[12]: пространственные препятствия делают реакцию образования
о-комплекса из этого соединения обратимой. Увеличение /г_) вы-
зывает появление кинетического изотопного эффекта.
Как уже указывалось (см. 2.7.1), предшественником ст-комп-
лекса является л-комплекс. Имеются данные в пользу того, что в
некоторых случаях скорость образования л-комплексов может ли-
митировать общую скорость реакции нитрования [13]. Обсужда-
ется также вопрос об окислительно-восстановительном характере
реакции нитрования и о существовании одноэлектронного переноса
при ее течении. Однако прямых доказательств существования
этого процесса пока нет [14].
Выше указывалось, что влияние концентрации серной кислоты
на скорость нитрования связано со степенью превращения азотной
кислоты в катион NOz- Однако серная кислота служит также и
растворителем, в котором образуются сильно полярные л- и
о-комплексы. Уже давно отмечалось, что при нитровании азотной
кислотой в серной кислоте и в олеуме скорость нитрования мини-
мальна в 100%-ной серной кислоте. При разбавлении серной кис-
лоты скорость нитрования заметно возрастает и при некоторой
концентрации, лежащей в пределах 90—95%, достигает максиму-
ма. В том же направлении действует добавление к 100%-ной
H2SO4 ряда солей и пиридина. Скорость нитрования возрастает
также при переходе от моногидрата к олеуму различных концент-
раций и этим широко пользуются при нитровании соединений с
малой реакционной способностью. Такого рода явления объясняют
тем, что в соответствии с общей теорией влияния растворителя
на скорость реакции скорость нитрования должна увеличиваться с
уменьшением диэлектрической постоянной среды. Последняя мак-
симальна для 100%-ной H2SO4 и падает от добавления воды, солей
или серного ангидрида [15]. Другое объяснение, опирающееся на
представление о большем или меньшем протонировании субстра-
та, затрудняющем реакцию [13], кажется менее убедительным.
Хотя при нитровании концентрированной азотной кислотой и
смесями с серной кислотой участие катиона NO2 строго доказано,
нет оснований полагать, что во всех случаях нитрования активной
частицей является именно этот катион (см., например, [16]). Вы-
сказываются предположения о возможности участия в этой реак-
ции нитрацидий-катиона (1) и недиссоциированной азотной кисло-
ты, а при нитровании в уксусной кислоте — протонированного аце-
тилнитрата (2), получающегося по схеме:
/ОН /О—no2
h2ono? + СНзС^ —-> сн3с^ + н2о
•'О чо+—н
(1) 12)
136
Точного о-комплекса. Например, возможна циклическая координа-
ция иона NO2 за счет его взаимодействия с электроотрицательным
атомом орто-расположенных групп —СНО, —СООН, —NO2,
- SO3H и др. [22, с. 174]:'
NO2
+
Это приводит к образованию значительных количеств о-нитро-
производных, несмотря на то, что перечисленные группы имеют
электроноакцепторный характер. Ниже показан выход (в %) изо-
меров, получающихся при нитровании некоторых замещенных бен-
золов:
СНО СООН СООС2Н5 CN NO2
Введение в лета-положение электронодонорной метоксигруппы
резко увеличивает количество продуктов орто-замещения:
СНО NO2 no2
ОСОСНз
При нитровании нафталина (4) большая стабильность о-комп-
лексов приводит к образованию почти исключительно 1-нитропро-
изводного (5). Дальнейшее нитрование последнего идет также за
счет сс-положений в другом кольце с преимущественным образова-
нием 1,8-динитропроизводного (за счет циклокоординации иона
NO2). Примерно так же идет и нитрование 1-нафталинсульфокис-
лоты (6).
96-97
' (4) (5) (6)
Как видно, стерические препятствия здесь не оказывают замет-
ного влияния на ориентацию.
Нитрование 2-нафталинсульфокислоты (7) идет также в поло-
жения 5 и 8, причем получаются близкие выходы обоих продук-
тов.
139
1,5-Нафталиндисульфокислота (8) нитруется в положения 3 и 4,
причем с увеличением концентрации отработанной кислоты возра-
стает выход 3-нитропроизводного и при нитровании в олеуме он
становится единственным продуктом [23]. Этот своеобразный эф-
фект среды на ориентацию вступающей нитрогруппы по-видимому
связан с процессами протонирования субстрата, затрудняющими
циклокоординацию иона NO*. 1,3,6-Нафталинтрисульфокислота
(9) нитруется в положение 8.
(9)
Интересное влияние среды на ориентацию прослеживается при
нитровании 1,5-динитронафталина [24]. При проведении реакции с
нитрующей смесью из него образуется 94% 1,4,5-тринитропроиз-
водного и только 6% 1,3,5-изомера (10). Очевидно, в этом случае
большое влияние на ориентацию оказывает циклокоординация
иона NO2 и повышенная стабильность ст-комплекса, образующегося
за счет сс-положения. Соотношение продуктов нитрования сильно
меняется при использовании вместо нитрующей смеси одной
70%-ной азотной кислоты: получается 42% 1,4,5- и 58% 1,3,5-три-
нитронафталина (11).
(10)
Причину этих различий видят в изменении строения атакующей
частицы: в смесях с серной кислотой это нитроний-катион NO2,
в 70%-ной азотной кислоте — нитрацидий-катион H2ONO2 — реа-
гент, менее способный к координации с нитрогруппой и более се-
лективный [13] (см. также 4.4.3).
9,10-Антрахинон нитруется преимущественно за счет а-положе-
ний, что объясняется дополнительной стабилизацией переходного
состояния за счет координации ионаЬЮг с карбонильными атома-
ми кислорода. Дальнейшее нитрование 1-нитро-9,10-антрахинона
идет практически с такой же скоростью и приводит к образованию
смеси, содержащей равные количества 1,5- и 1,8-динитро-9,10-ант-
рахинонов (12). 9,10-Антрахинон-1- и 2-сульфокислоты (13) и (14)
140
нитруются в «-положений незамещенного бензольного кольца, да-
вая эквимолекулярные смеси изомерных нитросульфокислот
(см. также 4.4.4).
(12) (13) (14)
Об unco-атаке и изомеризации о-комплексов см. стр. 68.
4.4. ПРАКТИКА НИТРОВАНИЯ
В промышленности процессы нитрования, в зависимости
от объема производства, ведут периодическим или непрерывным
методами, как правило с использованием нитрующих смесей. При
периодическом методе применяют стальные котлы — нитраторы —
с большой поверхностью теплообмена в виде рубашек, змеевиков
или полых цилиндров, в которые подается вода или холодильный
рассол. Нитратор обязательно снабжается хорошо работающей
мешалкой, термопарой для непрерывной регистрации температуры
и автоматическим устройством, закрывающим подачу нитрующего
агента при прекращении размешивания массы или ее перегреве.
Особенное значение при нитровании имеет эффективный массо- и
теплообмен, для которого необходимо полное смешение, так как
реакционная масса чаще всего состоит из двух слоев — кислотного
и органического. Азотная кислота из постепенно добавляемого
нитрующего агента распределяется между этими слоями и боль-
шей частью находится в органическом слое; однако реакция идет
преимущественно в кислотном слое и с большой скоростью [15].
В настоящее время нитрование толуола, бензола, хлорбензо-
ла — наиболее крупнотоннажные процессы — ведутся преимуще-
ственно непрерывными методами. Описаны разнообразные системы
аппаратов для осуществления этих процессов [15], а также ните-
вой реактор с микродозированием для реакции нитрования [25] и
ленточный реактор, рекомендуемый для применения в промыш-
ленном масштабе [26]. Предложено автоматическое регулирова-
ние процесса по температуре и давлению в аппарате [27].
Выделение жидких или низкоплавких продуктов нитрования,
отслаивающихся от отработанной кислоты, ведется в сепараторах
периодического или непрерывного действия. Получившиеся нит-
ропродукты освобождают от остатков кислот, промывая их водой,
и нейтрализующим агентом, после чего для очистки подвергают
вакуум-перегонке или кристаллизации. Если при нитровании полу-
чаются смеси изомерных соединений, для разделения их использу-
ются оба эти метода.
141
Отделенную от продуктов нитрования отработанную кислоту
подвергают очистке от органических примесей и остатков азотной
кислоты. Часто для этого отработанную кислоту промывают ис-
ходным соединением для нитрования. После этой операции его
возвращают в цикл нитрования, а очищенную отработанную кис-
лоту укрепляют до необходимой концентрации серной кислотой
или олеумом и также вновь используют в процессе.
В тех случаях, когда продукты нитрования высокоплавки или
не отделяются от отработанной кислоты, реакционную массу вы-
ливают в воду. Если продукты выпадают в осадок, их отделяют
фильтрованием, а если они остаются в растворе (обычно так ведут
себя нитросульфокислоты), их высаливают, добавляя сульфат
натрия или иные соли.
Для выделения нитросульфокислот часто приходится перево-
дить их в кальциевые соли (см. 3.3). Это трудоемкая операция, и
поэтому иногда предпочитают без предварительного выделения
нитросульфокислот восстанавливать их растворы до аминосульфо-
кислот, которые значительно легче выделяются из растворов
[28, с. 163].
При нитровании обязательному анализу подлежит нитрующая
смесь, в которой определяют содержание азотной и азотистой
кислот и отдельно содержание азотистой кислоты, а также содер-
жание серной кислоты.
Качество нитропродуктов определяют по температуре плав-
ления, температуре застывания, плотности, а нитросульфокис-
лоты восстанавливают в аминосульфокислоты и титруют нитри-
том натрия.
4.4.1. Нитрование бензола и его гомологов
Бензол при действии нитрующей смеси, содержащей
0,97—1,01 моль азотной кислоты на моль соединения при 40—50 °C
гладко превращается в нитробензол (15); концентрация отрабо-
танной серной кислоты должна быть не ниже 69%. Дальнейшее
нитрование нитробензола идет со скоростью в 105 —107 раз мень-
шей [15] и поэтому требует значительно более жестких условий:
температура до 90 °C, концентрация отработанной кислоты 86%.
При этом образуется л-динитробензол (16), содержащий 9% о- и
1% п-изомера.
NO2 ь
(15) (
Дальнейшее нитров
JO2
^^''X'NO2
л-динитробензола требует настолько
жестких условий и идет с таким плохим выходом 1,3,5-тринитро-
142
бензола, что получение последнего этим методом считается неце-
лесообразным.
Производство нитробензола — крупнотоннажное и осущест-
вляется непрерывным методом. Нитрующий агент берут с неболь-
шим недостатком, чтобы азотная кислота использовалась полно-
стью и в продукте не было даже следов динитросоединений. Не-
большие количества бензола легко отделяются при перегонке [29].
Основное назначение нитробензола — производство анилина.
Непрерывное нитрование бензола проводится в нитраторе с
охлаждающей рубашкой и внутренними змеевиками, куда посту-
пает холодная вода. Аппарат оборудован мощным перемешиваю-
щим устройством. В нитратор непрерывно поступают бензол, нит-
рующая смесь и отработанная кислота. Подача бензола и нитро-
смеси автоматически регулируется по заданному соотношению, а
количество отработанной кислоты—по температуре в нитраторе.
Реакционная масса из верхней части аппарата перекачивается в
два расположенных последовательно спиральных холодильника,
где завершается реакция нитрования и температура реакционной
массы снижается до 30 °C. Затем в сепараторе непрерывного дей-
ствия нитробензол отделяется от отработанной кислоты. Сырой
бензол нейтрализуют аммиачной водой или раствором соды, после
чего перегоняют. Отработанная кислота содержит до 2,2% раство-
ренного нитробензола и около 0,5% азотной кислоты. Поэтому ее
подвергают двухступенчатой экстракции полуторным по массе ко-
личеством бензола, в результате чего нитробензол полностью из-
влекается, а азотная кислота почти полностью расходуется на
нитрование бензола. Бензол отработанной кислотой не задержи-
вается и после экстракции используется для нитрования. Отрабо-
танная кислота идет на регенерацию или используется для полу-
чения суперфосфата [30].
м-Динитробензол (16) используется в значительно меньших
масштабах, и поэтому его получают преимущественно периодиче-
скими методами. Нитрование нитробензола ведут при 90°C, ис-
пользуя небольшой (до 10%) избыток азотной кислоты. После
отделения сырого продукта нитрования от отработанной кислоты
его подвергают очистке путем обработки раствором сульфита на-
трия [28]. При этом примеси о- (17) и п-динитробензолов (18) в
результате нуклеофильного замещения одной из нитрогрупп суль-
фогруппой (см. 4.3) превращаются в соли соответствующих нитро-
бензолсульфокислот и переходят в раствор. Для окончательной
очистки продукт (16) подвергают гранулированию.
(W)
(18)
Т43
Используют л-динитробензол в основном для производства
л-нитроанилина и л-фенилендиамина; потребность в последнем за
недавние годы резко возросла в связи с применением его для
получения термостойких полимеров и волокон.
Толуол (19) нитруется на мононитропроизводные непрерывным
методом в условиях, близких к условиям нитрования бензола. При
этом образуется 55—57% о- (21), 38—42% п- (22) и 3—4%
.и-изомера (20). Соотношение изомеров мало меняется с изменени-
ем условий проведения реакции нитрования. В связи с большой
потребностью в н-нитротолуоле (22) и сравнительно малой в
о-изомере (21) описано [31,32] несколько способов повышения
выхода (22). Один из них основан на нитровании толуола в при-
сутствии пористых материалов (японская кислая глина, обрабо-
танная соляной кислотой, силикагель, активный уголь). Преобла-
дание н-изомера, по-видимому, связано со стерическими затрудне-
ниями орто-замещения в мелких порах катализатора [33].
(22) (24)
Разделение изомеров основано на различии температур кипе-
ния и застывания. Его проводят ректификацией в вакууме с отбо-
ром дистиллята, содержащего о-нитротолуол, и с последующей
кристаллизацией п-нитротолуола из кубового остатка в трубчатых
кристаллизаторах. Остающуюся эвтектическую смесь, содержа-
щую до 20% л-изомера, подвергают повторной ректификации. При
этом получают дистиллят, содержащий около 80% л-изомера. Для
окончательной очистки дистиллят охлаждают, и из него выкри-
сталлизовывается чистый л-нитротолуол.
Дальнейшее нитрование смеси нитротолуолов проводят в более
жестких условиях (температура 75—80 °C, концентрация отрабо-
144
тайной кислоты 80%). При реакции образуется главным образом
2,4-динитротолуол (24), содержащий значительную примесь
2,6-динитротолуола (23) и в гораздо меньших количествах другие
динитротолуолы, получающиеся преимущественно из лг-нитротолу-
ола (20). Чистый 2,4-динитротолуол выделяют из смеси кристал-
лизацией из расплава.
Дйнитротолуол с содержанием до 90% 2,4-изомера можно по-
лучить прямым нитрованием толуола смесью азотной и серной
кислоты при температуре ниже 0 °C (лучше от—35 до—5 °C) и при
молекулярном соотношении толуола и азотной кислоты 1 : (2ч-10)
в присутствии небольшого количества воды. Реакцию осущест-
вляют добавлением толуола к охлажденной нитрующей смеси.
Реакция протекает в течение нескольких секунд с почти количе-
ственным выходом. Динитротолуол извлекают растворителем, на-
пример метилендихлоридом, и очищают промыванием водой [34].
Главный продукт дальнейшего нитрования — 2,4,6-тринитрото-
луол (25) используется как взрывчатое вещество (тротил, тол).
Основное назначение изомерных нитротолуолов — получение
соответствующих толуидинов, из которых наибольшее значение
имеет п-толуидин. Кроме того, п-нитротолуол в больших количе-
ствах используется для производства n-нитробензойной кислоты,
4,4'-динитро-2,2'-стильбендисульфокислоты, п-нитробензальдегида
и ряда других продуктов.
Из чистого 2,4-динитротолуола получают главным образом
2,4-толилендиамин, используемый в синтезе красителей, а техниче-
ская смесь 2,4- и 2,6-динитротолуолов, получающаяся при нитрова-
нии, после восстановления в больших количествах перерабатыва-
ется в толилендиизоцианаты, используемые в химии полимеров.
Изомерные ксилолы подвергаются нитрованию в небольших
масштабах. Важнейшими из получающихся продуктов являются
4-нитро-лг-ксилол (26), 4-нитро-о-ксилол (27) и 2-нитро-п-ксилол
(28), используемые для получения соответствующих ксилидинов.
no2
сн3
<2в)
Соотношение 3-нитро-о-ксилола и 4-нитро-о-ксилола (27) зави-
сит от концентрации серной кислоты, в которой ведется нитрование
о-ксилола. При 25°C в интервале концентраций H2SO4 от 50 до 70%
оно повышается от 0,45 до 1,5; в H2SO4 с концентрацией выше 70%
оно становится постоянным и равным 1,5. На основании установ-
ленной зависимости скорости реакции от кислотности раствора
считают, что в 70%-ной H2SO4 электрофильной атакующей части-
цей является катион NO] [35].
145
Изучена кинетика нитрования 2,4- и 4,6-динитро-лг-ксилола
азотной кислотой различной концентрации. Установлено, что, ве-
роятно в связи со стерическими препятствиями, 4,6-динит-
po-.w-ксилол нитруется в 3—5 раз медленнее 2,4-динитро-л-ксило-
ла [36].
Разработан способ получения нитромезитилена (2-нит-
ро-1,3,5-триметилбензола) (29а) нитрованием мезитилена (29)
смесью азотной и серной кислот в присутствии ПАВ [37].
(29)
(29а)
Восстановлением нитромезитилена получают мезидин (2-ами-
но-1,3,5-триметилбензол), имеющий большое значение в синтезе
антрахиноновых красителей.
4.4.2. Нитрование производных углеводородов
ряда бензола
Большое значение в промышленности имеет нитрование
хлорбензола.
(32)
(38)
Благодаря —/-эффекту атома хлора, хлорбензол вступает в эту
реакцию примерно в 7—8 раз медленнее, чем бензол. В зависимо-
сти от условий при реакции образуется 33—37% о- (31), 63—67%
п- (32) и около 1,6% лг-нитрохлорбензола (30). Из них особенно
146
широко используется «-изомер, служащий исходным продуктом
для синтеза «-нитроанилина, «-нитрофенола и многих других со-
единений. Применение о-нитрохлорбензола имеет значительно бо-
лее ограниченный характер. Поэтому ведутся поиски условий нит-
рования, при которых получается большее количество «-изомера.
В известных пределах оно является функцией температуры и
концентрации отработанной серной кислоты С0Тр. Так, при
Corp = 70% и повышении температуры нитрования от 50 до ПО °C
выход о-нитрохлорбензола возрастает с 32,3 до 36,1%. Повышение
Corp до 75% при ПО °C увеличивает образование о-изомера до
37,4% [30]. Таким образом, увеличение жесткости условий нитро-
вания снижает его селективность. Напротив того, добавление в
нитрующую смесь уксусной кислоты позволяет повысить содержа-
ние «-нитрохлорбензола до 74% [38]. Другие данные по этому
вопросу см. [15]. О получении лг-нитрохлорбензола см. 5.2.2.
Нитрование хлорбензола на мононитрохлорбензолы осуществ-
ляется по непрерывному способу в системе из двух аппаратов
нитрующей смесью, содержащей 35% HNO3, 53% H2SO4 и 12% во-
ды. Отработанная кислота содержит 70—71% H2SO4 и 0,7—1,3%
HNO3. Сырье дозируют в первый реактор, где поддерживается
температура 55—60 °C и протекает основная часть процесса. Реак-
ция завершается во втором аппарате при 70—75°C. Реакционная
масса, вытекающая из нитратора, попадает в отстойник, где слой
нитрохлорбензолов отделяется от отработанной кислоты. Органи-
ческий слой промывают горячей водой и 1%-ным раствором карбо-
ната натрия (соды). Из отработанной кислоты хлорбензолом
экстрагируют при 60—65 °C растворенные нитрохлорбензолы. Од-
новременно идет частичное нитрование хлорбензола, благодаря
которому утилизируется остаточная азотная кислота. Хлорбен-
зольный экстракт направляется на нитрование, а отработанная
кислота — на регенерацию.
Промытая смесь нитрохлорбензолов подвергается осушке путем
испарения воды в вакууме, после чего поступает на разделение.
Для этого она при 90 °C закачивается в трубчатые кристаллиза-
торы, в межтрубном пространстве которых циркулирует холодная
вода. При охлаждении до 13—14 °C из нее кристаллизуется «-нит-
рохлорбензол. После удаления жидкой эвтектической смеси, в
межтрубное пространство подают горячую воду и таким образом
чистый «-нитрохлорбензол расплавляют и сливают. Отделенную
эвтектическую смесь, содержащую 28—34% «-изомера, подвергают
перегонке под вакуумом; «-изомер отгоняют с заметной примесью
о-нитрохлорбензола и таким образом содержание последнего в
кубовой жидкости доводят до 80%. При кристаллизации из нее
выделяется чистый о-нитрохлорбензол. Эвтектическую смесь воз-
вращают в цикл.
Дальнейшее нитрование хлорбензола требует значительно бо-
лее жестких условий — концентрации отработанной серной кисло-
ты 82—85% и избытка азотной кислоты до 10% против теоретиче-
ски необходимого [28,30]. В результате реакции получается
147
1,3-динитро-4-хлорбензол (35) с заметным содержанием 1,3-ди-
нитро-2-хлорбензола (34) и очень небольшим количеством 1,2-ди-
нитро-4-хлорбензола (33), образующегося преимущественно из
л-гштрохлорбензола (30). Примеси (33) и (34) легко удалить,
промывая (35) органическими растворителями. Продукт в боль-
ших количествах используется для получения 2,4-динитроанилина,
в синтезе красителя сернистого черного, для производства 2,4-ди-
нитрофенола и ряда других соединений.
Получение 1,3-динитро-4-хлорбензола (35) ведется непрерыв-
ным методом в каскаде из двух нитраторов. В первый непрерывно
дозируются реагенты, температура в нем поддерживается в преде-
лах 55—60 °C; температура во втором реакторе 90—100 °C. Далее
реакционная масса перетекает в сепаратор, где продукты нитрова-
ния отделяются от отработанной кислоты. Последнюю в насосе-
смесителе обрабатывают хлорбензолом, извлекающим нитропро-
дукты и частично азотную кислоту, после чего хлорбензол
используют для нитрования, а отработанная кислота идет на реге-
нерацию. Динитрохлорбензол в расплавленном виде промывают
водой и раствором соды и собирают в хранилище [30].
Исчерпывающее нитрование хлорбензола дает 1,3,5-тринит-
ро-2-хлорбензол (36), применяемый главным образом для получе-
ния пикриновой кислоты.
При нитровании о-дихлорбензола образуется смесь 1-нит-
ро-2,3-дихлорбензола (37) и 1-нитро-3,4-дихлорбензола (38), из
которых практическое значение имеет только (38). Его удается
выделить, обрабатывая смесь 7—13%-ным олеумом, который суль-
фирует только (37) [39].
NO2
no2
(38)
Практическое применение имеют также продукты мононитро-
вания п-дихлорбензола — 1-нитро-2,5-дихлорбензол (39) [27,
с. 100] и 1,2,4-трихлорбензола — 1-нитро-2,4,5-трихлорбензол (40).
NO2
o2n-
Cl
(39)
Cl
(40)
О получении изомерных нитрохлорбензолов см. также 5.2.2.
Фенол очень легко вступает в реакцию нитрования. Так, о- и
'«нитрофенолы могут быть получены действием разбавленной
азотной кислоты, хотя их традиционно получают из о- и п-нитро-
хлорбензолов (см. 8.2). Предложен промышленный способ нитрова-
ния фенола и его производных, обеспечивающий высокий выход
п-нитрризомера. Это достигается подбором температуры реакции и
соотношением HNO3, HNOa и H2SO4 в нитрующей смеси [40].
С концентрированной азотной кислотой фенол реагирует черес-
чур энергично, поэтому для получения 2,4,6-тринитрофенола (пик-
риновой кислоты) (42) фенол вначале сульфируют с таким расче-
том, чтобы образовалась преимущественно 4-гидрокси-1,3-бензол-
дисульфокислота (41). После этого сульфомассу несколько
разбавляют водой и ведут нитрование. При этом сульфогруппы
подвергаются электрофильному вытеснению нитрогруппами (нит-
родесульфирование) и одновременно третья нитрогруппа вступает
в свободное орто-положение [28, с. 136].
Впрочем, этот метод получения пикриновой кислоты в настоя-
щее время утрачивает свое значение.
Практическое значение имеет 4-гидрокси-З-нитробензолсульфо-
кислота (43), которая получается путем последовательного суль-
фирования и нитрования фенола:
Нитрование салициловой (о-гидроксибензойной) кислоты в
мягких условиях приводит к 5-нитросалициловой (2-гидро-
кси-5-нитробензойной) кислоте (44), используемой при синтезе
красителей:
а С ООН
ОН
ОгМч^^/ОООН
Y Т
^/^он
<44)
149
В отличие от этого при нитровании n-гидроксибензойной кисло-
ты в обычных условиях происходит нитродекарбоксилирование с
образованием п-нитрофенола (45):
Однако при действии на n-гидроксибензойную кислоту 25—
35%-ной HNO3 при 20—40 °C в присутствии каталитических коли-
честв нитритов щелочных металлов получается 4-гидрокси-З-нит-
робензойная кислота (46) [41].
n-Гидроксибензанилид хорошо нитруется при нагревании с
разбавленной азотной кислотой, образуя 4-гидрокси-З-нитробенза-
нилид (47), применяемый в синтезе красителей:
(47)
В таких же условиях ведется нитрование диметилового и ди-
этилового эфиров гидрохинона; образуются диметиловый (48) и
диэтиловый (49) эфиры 2-нитрогидрохинона:
(48)
ОС2Н5
(49)
При нитровании бензальдегида нитрующей смесью получается
л-нитробензальдегид (50), содержащий до 20% о-нитробензаль-
дегида (51). Его очищают с помощью бисульфита натрия, который
реагирует с (51) значительно легче, чем с (50).
(SOI (81)
150
Бензотрифторид (52) и «-хлорбензотрифторид (53) нитруются
исключительно в жета-положение к трифторметильной группе [42]:
(53)
Бензолсульфокислоту обычно не нитруют, потому что при этом
получаются смеси изомеров. Так, при нитровании бензолсульфона-
та натрия нитрующей смесью с понижением температуры от 20 до
—50 °C содержание ж-нитробензолсульфокислоты увеличивается с
68,5 до 83,9%, а содержание о- и «-изомеров уменьшается соответ-
ственно с 12,4 до 2,6% и с 19 до 14%. В качестве побочного продук-
та реакции обнаружена стифниновая кислота (2,4,6-тринитрорезор-
цин), получающаяся через ж-гидроксибензолсульфокислоту [43].
Изучалось также дезактивирующее влияние сульфогруппы на
бензольное кольцо, передающееся благодаря индуктивному эф-
фекту через одну, две и три группы СН2 в СбН5(СН2)п5О3Н (где
« = 04-3). Расчет парциальных факторов показывает, что даже
при трех группах СН2 преобладает нитрование в жета-положе-
ние [44].
При нитровании ж-хлорбензолсульфохлорида с помощью азот-
ной кислоты или ее смеси с серной кислотой или олеумом атакуется
преимущественно «-положение к хлору и в качестве главного про-
дукта образуется 2-нитро-5-хлорбензолсульфохлорид [45].
Нитрование аминов протекает очень легко и с образованием
смеси изомеров. Поэтому перед нитрованием их подвергают аци-
лированию (см. 14.2.3), чаще всего муравьиной или уксусной кис-
лотой. Ацилирование ослабляет активирующее действие амино-
группы и устраняет возможность образования ж-изомеров. Нитро-
вание Л/-ацильных производных аминов ведут нитрующей смесью
(иногда с добавлением уксусной кислоты) при низкой температуре,
а выделение — разбавлением реакционной массы водой. Ацильную
группу в полученном продукте в дальнейшем удаляют, нагревая
его с разбавленной щелочью или кислотой.
В случае нитрования «-толуидина 4-амино-З-нитротолуол (55)
может быть получен через А-формил-«-толуидин (54):
Т51
Этим же путем в промышленности получают 2-амино-5-нитро-
толуол (56), 2-метокси-4-нитроанилин (57) и 4-метокси-2-нитро-
анилин (58).
(56) (57) (58)
Нитрование ароматических аминов проводят также в сильно-
кислых средах, в которых практически весь амин находится в виде
соли.
Аминогруппа является сильным электронодонором
(см. табл. 2.1) и ускоряет реакцию нитрования в 106 раз. Получа-
ющаяся из нее аммониевая группа— сильный электроноакцептор,
уменьшающий скорость этой реакции в 106 раз [11, с. 258]. Таким
образом, разница в скоростях нитрования амина и его соли со-
ставляет около двенадцати порядков. Поэтому, если даже равно-
+
весне ArNH2 ArNH3 смещено в сторону соли так, что кон-
центрация амина в растворе составляет 10-12 от концентрации со-
ли, продукт нитрования амина (n-изомер) может получаться во
вполне заметных количествах.
Существует и иная точка зрения на происхождение п-изоме-
ра — будто он получается исключительно за счет нитрования про-
тонированного амина, а не амина-основания. Утверждают, что по-
следнее присутствует в таком незначительном количестве, что даже
если замещение происходит при каждом столкновении с нитроний-
катионом, его скорость вносит исчезающе малый вклад в наблю-
даемую скорость процесса. Подтверждением этой точки зрения
может служить нитрование соли триметилфениламмония, при ко-
тором получается 89% м- и 11% п-нитропроизводного [46].
Однако это категорическое суждение не согласуется с данными
по нитрованию У,У-диметиланилина (59) нитрующей смесью,
при котором выход n-нитропроизводного значительно выше. Реак-
ция проводится в концентрированной серной кислоте, взятой в
количестве 9 моль на моль (59) и при этом образуется 63—70%
А,А-диметил-.и-нитроанилина (61) и 30—37% У,У-диметил-п-нит-
роанилина (62) [47]. Увеличение количества и концентрации сер-
ной кислоты приводит к некоторому уменьшению количества
образующегося n-изомера. Поэтому, по-видимому, правильнее счи-
тать, что п-изомер (62) здесь может образоваться двумя путя-
ми — из основания (59) и из соли (60).
152
быстро
(59\
(60)
(61) (62)
Таким образом, при нитровании солей аминов почти неизбежно
получаются продукты изомерного строения. Поэтому этот прием
нитрования аминов имеет ограниченное применение. Обычно им
пользуются для соединений, в которых лара-положение к амино-
группе занято слабым электронодонорным заместителем. Напри-
мер, в промышленности этим путем получают 4-амино-2-нитро-
толоул (63) и 4-метокси-З-нитроанилин (64).
(63) (64)
4.4.3. Нитрование нафталина и его производных
Нафталин (4) вступает в реакцию нитрования значи-
тельно легче, чем бензол.
NO2
(67)
Для получения 1-нитронафталина (5) реакцию проводят, ис-
пользуя близкое к теоретическому количество азотной кислоты в
водной серной кислоте при 50—60 °C.
153
Реакцию ведут в питраторе из хромоникелевой стали, оборудо-
ванном мешалкой, рубашкой и змеевиком для охлаждения. Из-
мельченный нафталин суспендируют в 1,7—3-кратном количестве
61—65%-ной H2SO4 при 30—40 °C, после чего постепенно добавля-
ют нитрующую смесь, содержащую 28—42% HNO3, 42—57%
H2SO4 и 15—16% воды. Избыток азотной кислоты 1—3% от теоре-
тического. В конце нитрования температура поддерживается в
пределах 52—62 °C. После отделения продукта от отработанной
кислоты его несколько раз промывают горячей водой и раствором
соды, в который уходит получающийся побочный продукт —
2,4-динитро-1-нафтол. Иногда для удаления остатков нафталина
используется перегонка с паром. После такой очистки расплавлен-
ный 1-нитронафталин выливают в холодную воду, где он застывает
мелкими гранулами. Температура плавления продукта 52—52,5 °C
(чистый продукт плавится при 61 °C), в нем содержится 4—5%
2-нитронафталина (65) и до 3% динитронафталинов. В таком виде
его используют для восстановления в 1-нафтиламин [30].
Для того чтобы не размалывать нафталин перед реакцией,
предлагается [48] нитровать его расплавленным при температуре
не выше 95 °C; дальнейшее повышение температуры увеличивает
выход 2-нитронафталина (65) [8].
Присутствие (65) крайне нежелательно, так как при восстанов-
лении он образует канцерогенный 2-нафтиламин, опасный для ра-
ботающих [49, с. 292]. Как видно из предыдущего (65), сопут-
ствующий 1-нитронафталину (5), не удаляется из продукта нитро-
вания. При нитровании нафталина в уксусном ангидриде выход
2-изомера возрастает до 10%. Напротив, при нитровании азотной
кислотой в ацетонитриле получается только 1-нитронафталин.
Предлагается также нитровать нафталин четырехокисью азота
при 180 °C [15].
Дальнейшее нитрование 1-нитронафталина (5) приводит к об-
разованию смеси, содержащей около 7з 1,5- (66) и 2/з 1,8-динит-
ронафталина (67), наряду с небольшим количеством продуктов
изомерного строения. Разделение изомеров может осуществляться
кристаллизацией из органических растворителей, а также обра-
боткой восстановителями (1,5-изомер восстанавливается легче,
чем 1,8-), или сульфитами (1,8-изомер образует растворимые в
воде сульфокислоты, а 1,5- не вступает в реакцию) [30; 50, с. 202].
Один из способов промышленного получения динитронафтали-
нов состоит в следующем [50]. В нитрующую смесь, приготовлен-
ную из 1850 кг 98%-ной H2SO4, 1220 кг 53%-ной HNO3 и 50 кг
98%-ной HNO3, загружают при 30 °C чистый нафталин (530 кг)
пятью порциями в течение 21 ч. После загрузки четвертой порции
температура 37—38, после пятой 44 °C. Нагревают до 80 °C в тече-
ние 4 ч, выдерживают 1 ч и передавливают в 7000 л воды. При
30 °C осадок фильтруют, промывают и сушат при 70—80 °C. Выход
технического продукта 80% (состав: 20% 1,5-динитронафталина,
53% 1,8-изомера и 27% смол). Для разделения изомеров 1000 кг
технического продукта нагревают с дихлорэтаном (4180 л) и ведут
154
отгонку азеотропа до полного удаления воды. Затем нагревают с
обратным холодильником до растворения осадка, быстро охлаж-
дают до 60 и далее медленно до 50 °C. При этой температуре
отфильтровывают 1,5-динитронафталин и промывают его несколько
раз дихлорэтаном (выход 230 кг). Из маточных растворов после
отгонки части дихлорэтана и охлаждения кристаллизуется
1,8-динитронафталин (выход 525 кг).
Динитронафталины можно получать, проводя реакцию нитро-
вания в полифосфорной кислоте [51]. Смесь, в которой преоблада-
ет 1,5-динитронафталин, предложено получать нитрованием в
CCh в присутствии ионообменной смолы, а также в виде раствора
в ацетонитриле [15].
1,5- и 1,8-Динитронафталины используются для восстановления
в соответствующие нафтилендиамины, а 1,5-изомер, кроме того,
для синтеза 5,8-дигидрокси-1,4-нафтохинона (нафтазарина —
см. 16.6.2).
Продукты более глубокого нитрования — три- и тетранитро-
нафталины — применения в синтезе промежуточных продуктов не
нашли.
Практически очень важны продукты, образующиеся при нитро-
вании многочисленных моно-, ди- и тринафталинсульфокислот. Их
обычно получают, проводя нитрование непосредственно после
сульфирования нафталина в необходимых условиях, без предвари-
тельного выделения сульфокислот, что удешевляет производствен-
ный процесс. Так как нитросульфокислоты непосредственно и в
виде солей хорошо растворимы в воде, их обычно не выделяют из
растворов. Реакционную массу разбавляют водой и подвергают
денитрации. Для этого ее нагревают некоторое время до полного
удаления азотной кислоты и окислов азота. Затем, нейтрализуя
жидкость известью или мелом, удаляют серную кислоту. Нит-
росульфокислоты — обычно в виде магниевых солей — восстанав-
ливают до аминонафталинсульфокислот. Последние значительно
хуже растворимы и в определенных условиях выделяются из рас-
твора (см. [28]).
Естественно, что при таких синтезах образуется значительное
количество изомерных соединений, загрязняющих целевые про-
дукты (см., например, [15, с. 1760]). Их удается отделить лишь на
этапе заключительной очистки, к которой должны предъявляться
поэтому достаточно высокие требования.
Ориентационные эффекты при нитровании нафталинсульфокис-
лот уже обсуждались ранее (см. 4.3). Следует отметить также,
что увеличение числа сульфогрупп в молекуле нафталина требует
проведения реакции в более жестких условиях: концентрация от-
работанной серной кислоты при нитровании моносульфокислот
Должна быть около 70%, дисульфокислот — 78% и трисульфокис-
лот—82%. Кроме того, если при нитровании моносульфокислот
нитрующий агент берется в теоретических количествах или даже с
очень небольшим недостатком, то в случае трисульфокислот его
следует вводить с 10—20%-ным избытком.
155
При нитровании 1-нафталинсульфокислоты
смесь, содержащая 30—40% 5-нитро- (68) и
ро-1-нафталинсульфокислоты (69).
(6) получается,
60—70% 8-нит-,
SO3H SOaH NO2 SO3H
NO2
(68)
(6)
(69)
Эту смесь разделяют после восстановления в соответствующие ;
аминонафталинсульфокислоты, из которых особенно важна 8-ами- '
но-1-нафталинсульфокислота (1-нафтиламин-8-сульфокислота; пе-
рикислота), используемая для синтеза красителей и промежуточ- i
ных продуктов (ср. 10.3). 1
Из 2-нафталинсульфокислоты (7) при нитровании образуется 1
смесь 5-нитро- (70) и 8-нитро-2-нафталинсульфокислоты (71). Ее
можно разделять, используя плохую растворимость в воде барие-
вой соли изомера (70) [15].
no2
no2
(70) (7) (71)
При восстановлении (70) и (71) получаются соответствующие
аминонафталинсульфокислоты, называемые кислотами Клеве.
В виде смеси и в индивидуальном виде их, как уже указано,
применяют в синтезе азокрасителей (ср. 3.3.2).
Нитрование 8-хлор-1-нафталинсульфокислоты смесью HNO3
(4,8%), H2SO4 (85%) и воды (10,2%) приводит к 8-хлор-7-нитро-
и 8-хлор-5-нитро-1-нафталинсульфокислоте. В небольшом количе-
стве выделена 8-хлор-5,7-динитро-1-нафталинсульфокислота [52].
При нитровании 1,5-нафталиндисульфокислоты (8) образуются
3-нитро- (72) и 4-нитро-1,5-нафталиндисульфокислота (73), разде-
ление которых можно осуществить как до, так и после восстанов-
ления.
156
Из этих продуктов особенно важен (73). Он используется для
синтеза 1-амино-8-гидрокси-2,4-нафталиндисульфокислоты (8-ами-
но-1-нафтол-5,7-дисульфокислота; Чикаго СС-кислота; 2С-кис-
лота).
При нитровании 1,6-нафталиндисульфокислоты (74) получается
в основном 8-нитро-1,6-нафталиндисульфокислота (75) с примесью
ее 3-нйтроизомера (76):
Ниже указаны стрелками положения, в которые вступает нит-
рогруппа при моно- и динитровании 2,6- и 2,7-нафталиндисульфо-
кислот:
Важнейшее практическое значение имеют 8-нитро-1,3,6- и
8-нитро-1,3,5-нафталинтрисульфокислота (77) и (78), получаемые
соответственно нитрованием 1,3,6- и 1,3,5-нафталинтрисульфокис-
лоты.
(77)
Кислота (77), будучи предварительно восстановлена в 8-ами-
но-1,3,6-нафталинтрисульфокислоту (Т-кислота), служит исход-
ным продуктом для синтеза 4-амино-5-гидрокси-2,7-нафталинди-
сульфокислоты (Аш-кислота; ср. 8.1.1). Кислота (78) применяется
Для получения 4-амино-5-гидрокси-1,7-нафталиндисульфокислоты
(К-кислота; ср. 3.3.3) [28].
157
1-Нафтол, подобно фенолу (см. 4.4.2), нитруют после предвари-
тельного сульфирования до 4-гидрокси-1,3-нафталиндисульфокис-
лоты (79) или до 4-гидрокси-1,3,6-нафталинтрисульфокислоты (80)
(79) (80)
В зависимости от этого при последующем нитродесульфирова-
нии получают 2,4-динитро-1-нафтол (81) или 8-гидрокси-5,7-динит-
ро-2-нафталинсульфоки слоту (2,4-динитро-1-нафтол-7-сульфокис-
лота; нафтоловый желтый) (82).
(81)
4.4.4. Нитрование антрахинона и его производных
Нитрование 9,10-антрахинона обычно ведут нитрующими
смесями при повышенной температуре. При этом вначале обра-
зуется1-нитро-9,10-антрахинон (83), который с близкой скоростью
нитруется далее до 1,5- и 1,8-динитро-9,10-антрахинонов (84) и
(85) в близком соотношении:
При реакции получается некоторое количество примесей, со-
держащих нитрогруппу в положении 2. Они отделяются, например
158
при промывке органическими растворителями, а смесь динитро-
продуктов непосредственно используется в синтезах или предвари-
тельно подвергается разделению кристаллизацией.'
Чистый 1-нитро-9,10-антрахинон (83), пригодный для восста-
новления в практически очень важный 1-амино-9,10-антрахинон
до недавнего времени получить прямым нитрованием не удавалось
[53]. При нитровании антрахинона большим избытком дымящей
азотной кислоты получается до 90% (83) [54—58], но примеси
динитропроизводных и 2-нитроизомера отделяются с большим тру-
дом [59]. Предложено получать чистый (83) нитрованием антра-
хинона в 85%-ной H2SO4 (20 : 1) при температуре от —10 до
-[-35 °C; выпадающий при разбавлении реакционной массы водой
продукт перегоняют под вакуумом. Таким образом удается полу-
чить 99%-ный 1-нитро-9,10-антрахинон с достаточно высоким выхо-
дом [60]. Он может быть получен с чистотой 95% без примеси
динитроантрахинонов нагреванием технического продукта нитро-
вания с водными растворами сульфитов [61, 62].
Динитроантрахиноны (84) и (85) получают нитрованием в при-
сутствии индифферентных органических растворителей [63], а
также при 0—50 °C нитрующей смесью [64].
2-Метил-9,10-антрахинон при нитровании образует с хорошим
выходом практически важный 2-метил-1-нитро-9,10-антрахинон
(86):
5,8-Диметил-1-нитро-9,10-антрахинон получают нитрованием 1,4-
Диметил-9,10-антрахинона нитрующей смесью, содержащей четы-
рехкратное количество азотной кислоты, при 20—25 °C [65].
Изучено нитрование 1-метокси-9,10-антрахинона нитрующей
смесью при различной концентрации серной кислоты и разной тем-
пературе. В результате реакции образуются 1-метокси-4-нитро- и
1-метокси-2-нитро-9,10-антрахиноны. На изомерный состав влияет
протонизация 1-метокси-9,10-антрахинона. Положение 2 в прото-
нированной форме менее реакционноспособно, чем в непротониро-
ванной. Поэтому при концентрации H2SO4 98,34% и 20 °C получа-
' ется чистый 4-нитроизомер. В серной кислоте высокой концентра-
ции происходит одновременное нитрование протонированной и
непротонированной формы [66].
1,5-Дигидрокси-9,10-антрахинон (антраруфин) вначале сульфи-
руют серной кислотой до 2,6-дисульфокислоты (87), а затем под-
вергают нитрованию. Получается 1,5-дигидрокси-4,8-динитро-
159
9,10-антрахинон-2,6-дисульфокислота (88), применяемая в произ-
водстве кислотных и дисперсных красителей:
О - ОН о он
он о он о
(87)
no2 о он
он о no2
(88)
При нитровании 1,2-дигидрокси-9,10-антрахинона нитрогруппа,
в зависимости от условий, вступает в положение 3 или 4 [1].
Интересны особенности нитрования хлорантрахинонов. Атом
хлора, как известно, обладает значительным —/-эффектом и в
обычных случаях заметно уменьшает скорость реакции нитрования
(ср. 4.4.2). Поэтому, например, нитрование 1-хлорнафталина в
основном идет в незамещенное кольцо. Иначе проявляет себя как
заместитель хлор в молекуле антрахинона: 1- и 2-хлор-9,10-антра-
хиноны и даже 2,3-дихлор-9,10-антрахинон нитруются в «-положе-
ния того кольца, где находятся атомы хлора, с образованием
соответственно 1-нитро-4-хлор- (89), 1 -нитро-2-хлор- (70) и 1-нит-
ро-2,3-дихлор-9,10-антрахинона (91):
(91)
Вероятно это объясняется тем, что на фоне сильного электроно-
акцепторного действия двух карбонильных групп, хлор выступает
преимущественно как электронодонорный заместитель.
4.4.5. Нитрование других полициклических соединений
Нитрование бифенила в серной кислоте приводит к о- и
n-нитропроизводным в соотношении opTolnapa от 3,1 до 3,7 [67].
Дифеновая (2,2'-бифенилдикарбоновая) кислота, образующая-
ся при окислении фенантрена, при нитровании нитрующей смесью
дает 4,4',6,6'-тетранитродифеновую кислоту (92) [68]. Продукты
160
восстановления и циклизации последней, например 2,7-диамино-
4,5,9,10-тетрагидро-5,10-диазапирен-4,9-дион (93), могут найти при-
менение в синтезе прямых азокрасителей вместо канцерогенного
бензидина.
НООС NO2
o2n с ooh
о
(92) (93)
Пирен при нитровании разбавленной азотной кислотой дает
1-нитропирен. Образующийся при восстановлении последнего
1-пиренамин может применяться в синтезе диоксазиновых краси-
телей. Такое же назначение имеют 3-нитрокарбазол (94) и 3-нит-
ро-9-этилкарбазол (95), получающиеся при нитровании разбавлен-
ной азотной кислотой карбазола и его JV-этильного производного.
Можно также получить (94) нитрованием в присутствии уксусной
кислоты и уксусного ангидрида при 30 °C [69]. Последовательным
сульфированием и нитрованием из карбазола получаю1?
1,3,6,8-тетранитрокарбазол (96) [8].
(96)
Нитрование флуорена в присутствии полифосфорной кис-
лоты приводит к смеси 2,5-, 2,7-динитро- и 2,4,7-тринитрофлуо-
ренов [51].
Описано нитрование перилена действием азотной кислоты
в Диоксане: в результате получается смесь 1- и 3-нитропери-
ленов [70].
4.5. НИТРОЗИРОВАНИЕ
Реакция нитрозирования, при которой атом водорода
ароматического кольца замещается остатком азотистой кислоты,
имеет значительно менее общий характер, чем реакция нитрования;
6 Зак. 271 161
в нее могут вступать лишь ароматические соединения, содержащие
наиболее сильные электронодонорные заместители — гидроксиль-
ные или аминогруппы. Реакции эти большей частью проводят в
водной среде, причем источником азотистой кислоты служит нит-
рит натрия в кислой среде.
Кинетические исследования показывают, что атакующей части-
цей — реагентом при этих реакциях — может служить нитрозоний-
катион NO+, нитрозилхлорид С1—NO, азотистый ангидрид N2O3
или неионизированная азотистая кислота [3]:
HONO + Н+ H2O + NO+
NO+ + СГ Cl—NO
NO+ + NOJ ^=±: ON—ONO (N2O3)
NOJ + H+ HO—NO
При проведении реакции в соляной кислоте основным электро-
фильным реагентом, по-видимому, является нитрозилхлорид.
Устойчивость этого соединения невелика, и поэтому его концентра-
ция в растворе сравнительно низка. В бромистоводородной кислоте
реагентом оказывается значительно более устойчивый нитрозил-
бромид, в связи с чем реакция нитрозирования протекает с боль-
шей скоростью.
Использование разбавленной серной кислоты приводит к обра-
зованию азотистого ангидрида, который в этих реакциях является
менее активным реагентом. Недаром в рассматриваемых условиях
реакция имеет третий порядок — первый по амину и второй по
азотистой кислоте.
Наиболее активен нитрозоний-катион, который в заметных ко-
личествах образуется лишь в концентрированной серной кислоте из
нитрозилсерной кислоты:
HOSO2ONO ^=±: HOSO2O' + NO+
Иногда для нитрозирования используются также алкиловые
эфиры азотистой кислоты, чаще всего бутил- и амилнитриты. Ре-
акция с ними проводится в среде органического растворителя.
4.5.1. Нитрозирование аминов
Первичные ароматические амины при действии азоти-
стой кислоты в солянокислом растворе быстро нитрозируются по
аминогруппе и получающийся Af-нитрозамин тотчас же превраща-
ется в соль арендиазония (см. 15.2). Однако, если в используемом
амине (97) свободно и достаточно активно пара-положение, a
реакция проводится в концентрированной серной кислоте, при дей-
ствии нитрозилсерной кислоты, первоначально получающийся
Af-нитрозамин (98) может перегруппироваться в С-нитрозопроиз-
водное п-нитрозоариламин (99). Соединения этого типа, по-види-
162
мому, существуют преимущественно в таутомерной форме хинон-
иминоксимов (100) [8, с. 168].
(99) (100)
Высказывается также предположение [71], что 4-нитро-
зо-л«-толуидин (99) при этой реакции образуется не путем пере-
группировки, а в результате прямого нитрозирования в кольцо, так
как в концентрированной серной кислоте аминогруппа полностью
превращена в аммониевую. Однако такое течение реакции пред-
ставляется маловероятным, так как соединения с аммониевой
группой неспособны нитрозироваться (см. также 4.4.2).
Вторичные амины (101) реагируют с азотистой кислотой в
разбавленной соляной кислоте по схеме:
СНз—NH СНз—N—NO СН3—NH
NO
(101) (102) (103)
Реакция проводится при охлаждении, и получающийся jV-нит-
розо-АДалкилариламин (102) [72] выделяется в виде масла или
твердого осадка. При дальнейшей обработке разбавленной кисло-
той или раствором НС1 в спирте, продукт типа (102) перегруппи-
ровывается в п-нитрозо-ЛАалкилариламин (103). Считалось, что
эта перегруппировка имеет межмолекулярный характер, т. е. идет
через стадии отщепления нитрозогруппы от атома азота и нового
ее вступления в пара-положение арильного кольца. Поэтому, если в
кислый раствор ЛАнитрозамина (102), подвергаемого перегруппи-
ровке, ввести соединения, более активно взаимодействующие с
азотистой кислотой, например мочевину или дифениламин, то в
результате реакции вместо n-нитрозоариламина (ЮЗ) образуется
исходный /V-алкилариламин (101).
Однако при изучении перегруппировки лгета-замещенных
ЛАнитрозо-ААалкиланилинов в присутствии серной кислоты и боль-
шого количества мочевины в продуктах реакции обнаруживается
заметное количество соответствующих п-нитрозо-ААалкиланилинов
[73]. Это свидетельствует о частично внутримолекулярном проте-
кании реакции. Внутримолекулярному превращению способствует
6* 163
уменьшение кислотности среды, использование серной кислоты
вместо соляной и наличие электронодонорных заместителей в аро-
матическом кольце исходного соединения [74]. В более поздних
работах на основании аналогичных наблюдений высказывается
предположение, что перегруппировка и денитрозированиё — кон-
курирующие реакции, идущие после протонирования (Ю4) У-нит-
розо-У-алкилариламина (102) и, следовательно, перегруппировка
осуществляется только внутримолекулярно [75, 76].
СНз—N—NO
(102)
Уменьшение скорости перегруппировки примерно вдвое при ис-
пользовании У-нитрозо-У-метил-2,4,6-тридейтероанилина позволя-
ет предполагать, что лимитирующей стадией является отрыв про-
тона от 0-комплекса [75].
С другой стороны, в работе [77], посвященной изучению пере-
группировки производных У-нитрозодифениламина, хотя возмож-
ность внутримолекулярного течения этой реакции не исключается
полностью, но считается, что в основном она осуществляется меж-
молекулярно. Лимитирующей стадией авторы считают денитрози-
рование.
Через У-нитрозосоединения в промышленности получают
5-нитрозо-2-этиламинотолуол (4-нитрозо-У-этил-о-толуидин) (105),
4-нитрозодифениламин (106), 3-нитрозокарбазол (107) и некото-
рые другие продукты [8, с. 167].
(105)
(106)
(107)
Третичные амины (например, А7,А-диметиланилин) при дей-
ствии азотистой кислоты сразу образуют п-нитрозопроизводные.
Реакция проводится в разбавленной соляной кислоте при охлаж-
дении [28, с. 278] и идет по обычному механизму электрофильного
ароматического замещения. Атакующей частицей является нитро-
164
зилхлорид. Продукт реакции (108) выделяется в виде имеющего
хиноидное строение гидрохлорида интенсивного желтого цвета:
(108)
4.5.2. Нитрозирование фенолов и енолов
Нитрозирование растворимых в воде фенолов можно
проводить в подкисленных водных растворах при постепенном
добавлении раствора соли азотистой кислоты. Нерастворимые со-
единения гладко нитрозируются в уксусной кислоте [78]. В произ-
водственных условиях эту реакцию чаще всего проводят постепен-
но, при охлаждении, добавляя серную или соляную кислоту к
водно-щелочному раствору фенола, содержащему соль азотистой
кислоты, взятую в количестве, близком к эквимолекулярному
[28, с. 178; 79].
Продукты, образующиеся при нитрозировании фенолов, иден-
тичны хиноноксимам (Ю9), получающимся при действии гидро-
ксиламина на соответствующий хинон:
ОН
+ nh2oh
(109)
(109)
Многочисленными, в том числе спектральными, исследованиями
показано, что таутомерное равновесие нитрозофенол
хиноноксим (Ю9) в большинстве случаев сильно смещено в
сторону хиноноксимной формы. В химических реакциях (109) так-
же ведут себя двойственно; например, при окислении они легко
образуют соответствующие нитрофенолы, а при действии гидро-
ксиламина превращаются в хинондиоксимы:
лев
В практическом отношении из этих соединений наиболее инте-
ресны п-нитрозофенол (109), 2,4-динитрозорезорцин (ПО), 1-нит-
розо-2-нафтол (111) и 6-гидрокси-5-нитрозо-2-нафталинсульфокис-
лота (1-нитрозо-2-нафтол-6-сульфокислота) (112).
N—ОН
N—ОН
(112)
Образование при нитрозировании резорцина 2,4- (ПО), а не
4,6-динитрозопроизводного объясняется следующей схемой тече-
ния этой реакции:
В реакцию нитрозирования могут вступать также многие гете-
роциклические фенолы и кетоны, способные к кето-енольной тау-
томерии. Наиболее важный из таких кетонов З-метил-1-фенил-
5-пиразолон и ряд его производных нитрозируются в положение 4
с образованием продукта, способного к образованию нескольких
таутомерных форм:
Обзор по реакции нитрозирования соединений, содержащих
реакционноспособные метильные, метиленовые и метиновые группы
см. [80].
166
ЛИТЕРАТУРА
1. Катео Т., Nishimura S., Manabe О.— J. Chem. Soc. Jap. Chem. a. Ing.
Chem. 1974, № 1, p. 122—126; Катео T., Manabe 0.— Ibid., p. 127—130. 2. Пат.
США 2948759, 1960; Chem. Abstr., 1961, v. 55, 3544D. 3. Пат. ФРГ 2408664, 1974;
Chem. Abstr., 1975, v. 82, 3955. 4. Пат. США 3714272, 1973; Chem. Abstr., 1973,
v. 78, 97303. 5. Пат. ФРГ 2249373, 1974; Chem. Abstr., 1974, v. 81, 13324. 6. Raja-
mohan K-, Rao S. N. V.— Indian J. Chem. 1973, v. 11, № 10, p. 1076—1077.
7. Fukunaga K., Kimura M.— J. Chem. Soc. Jap. Chem. a. Ind Chem., 1973,
№ 7, p. 1306—1313. 8. Ворожцов H. H. Основы синтеза промежуточных продук-
тов и красителей. Изд. 4-е. М., Госхимиздат, 1955, с. 119—170. 9. Топчиев А. В.
Нитрование углеводородов и других органических соединений. Изд. 2-е. М„
Изд-во АН СССР, 1956. 488 с. 10. Olah G. A., Olah J. A., Overhuk N. А. — J.
Org. Chem.. 1965, v. 30, Xs 10, p. 3373—3376.
11. Ингольд К. Теоретические основы органической химии. Изд. 2-е. Пер. с
англ./Под ред. И. П. Белецкой. М., /Чир, 1973. 12. Myhre Р. С., Beug М.—J.
Amer. Chem. Soc., 1966, v. 88, № 7, p. 1569—1570. 13. Уивер У. — В кн.: Химия
нитро- и нитрозогрупп./Под ред. Г. Фойера. Пер. с англ. М., Мир, 1973, т. 2,
с. 5—43. 14. Коптюг В. А.— Журн. ВХО, 1976, т. 21, Ns 3, с. 247—255. 15. Во-
рожцов Н. Н., мл. — В кн.; Химия синтетических красителей. Т. 3/Под ред.
К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия, 1974,
с. 1751—1763. 16. Ridd J. Н. — In: Studies on Chemical Structure and Reactivity/
Ed. J. H. Ridd. New York, Wiley, 1966. 290 p. 17. Hartshorn S. R., Moodie R. B.,
Schofield K. e.a.—J. Chem. Soc. (B), 1971, Ns 12, p. 2447—2453. 18. Hart-
shorn S. R., Hogett J. G., Moodie R. B. e.a.— J. Chem. Soc. (B), 1971, № 12,
p. 2461—2469. 19. Титов А. И.— Успехи химии, 1952, т. 21, вып. 8, с. 881—913;
Баллод А. П., Штерн В. Я. — Там же, 1976, т. 45, вып. 8, с. 1428—1460.
20. Hartshorn S. R., Schofield К.— In: Progress in Organic Chemistry/Ed. W. Car-
ruthers and Sutherland. London, Butterworths, 1973, v. 8, p. 278—313.
21. Amos D. W., Baines D. A., Flewett G. W.— Tetrahedron Letters, 1973,
Ns 34, p. 3191—3194. 22. Хэммонд Г., Хаусорон M. — В кн.: Пространственные
эффекты в органической химии/ Под ред. М. С. Ньюмена. Пер. с англ, под ред.
А. Н. Несмеянова. М., ИЛ, 1960. 23. Пат. ФРГ 1176648, 1964; Chem. Abstr.,
1964, v. 61, 13257Е; см. также [8, с. 158]. 24. Ward Е. R., Johnson С. D.,
Day L. А. — J. Chem. Soc., 1959, № 2, р. 487—493. Szabo М., Meszaros L.,
Baumann Z. — Acta phys. et chem., Szeged., 1974, Bd. 20, № 1—2, S 177—181.
26. Szabo M., Meszaros L., Urbanies Z. — Ibid., S. 171—175. 27. Егоров С. B.,
Ивантеев В. П. Авт. свид. СССР 306868, 1971; Егоров С. В., Лобанов Н. В.,
Обновленский П. А. и др. Авт. свид. СССР 332852, 1972. 28. Фирц-Давид Г. Э.,
Бланже Л. Основные процессы синтеза красителей. Пер. с нем./Под ред.
С. В. Богданова и И. В. Фодимана. М., ИЛ, 1957. 29. Пат. США 3928457, 1975;
Chem. Abstr., 1976, v. 84, 104999. 30. Беркман Б. Е. Промышленный синтез аро-
матических нитросоединений и аминов. М. Химия, 1964.
31. Vorozhtsov N. N. jr. — In: Aromatic Compounds/Ed. by H. Zollinger. Lon-
don, Butterworths; Baltimore, Univ. Park press, 1973, p. 319—320. 32. Ma-
nabe О., Катео T. — Dyestuffs and Chemicals (Tokyo), 1974, v. 19, № 6,
p. 154—162. 33. Nishino ]., Moriyama H. e.a. — J. Synth. Org. Chem. Japan, 1976,
v. 34, № 4, p. 232—235. 34. Пат. США 3708546, 1973; Chem. Abstr., 1973, v. 78,
58008. 35. Coombes R. G., Russel L. W. — J. Chem. Soc. (B), 1971, № 12, p. 2443—
2447. 36. Лейтман Я. И., Заславец Е. Ф., Илюшин М. А. — ЖПХ, 1973, т. 46,
вып. 2, с. 366—370. 37. Смурова Р. Е., Казанцева А. Е. Авт. свид. СССР 451689,
1975. 38. Пат. США 3180900, 1965; Chem. Abstr., 1965, v. 63, 1734Е. Пат. США
3253045, 1966; Chem. Abstr., 1966, v. 65, 5400A. 39. Пат. США 2883435, 1959;
Chem. Abstr., 1959, v. 53, 17054. 40. Англ. пат. 1098717, 1968; Chem. Abstr., 1968,
v. 69, 10221.
41. Пат. ФРГ 2316495, 1974; Chem. Abstr., 1975, v. 82, 31139. 42. Порай-Ко-
ишц A. E., Порай-Кошиц Б. А., Эфрос Л. С. и др. — ЖПХ, 1955, t. 28, вып. 9,
c. 969—975. 43. Барвинская И. К., Спрысков А. А. — Изв. вузов. Химия и хим.
167
технол., 1970, т. 13, вып. 6, с. 802—804. 44. Moodie R. В., Schofield К., loshi-
da T.—J. Chem. Soc. Perkin 2, 1975, Ws 7, p. 788—792. 45. Пат. ФРГ, 2321332,
1974; Chem. Abstr, 1975, v. 82, 100087. 46. Ridd G. H., Utley G. H. P. — Proc.
Chem. Soc., 1964, № 1, p. 24—25. 47. Фитч Г. — В кн.: Синтезы органических
препаратов. Сб. 4. Пер. с англ. М, ИЛ, 1953, с. 374—376. 48. Франц, пат.
1352613, 1964; Chem. Abstr., 1964, v. 61, 626Е. 49. Вредные вещества в промыш-
ленности. Изд. 7-е. Т. 2./Под общ. ред. И. В. Лазарева и Э. Н. Левиной. Л.,
Химия, 1976. 50. Доналдсон Н. Химия и технология соединений нафталинового
ряда. Пер. с англ./Под ред. А. И. Королева. М, Госхимиздат, 1963.
51. Козлов Н. С., Клейн А. Г., Пунина М. Ш. — Изв. вузов. Химия и хим.
технол., 1974, т. 17, вып. 7, с. 1043—1046. 52. Станкевич Г. С., Лисицын В. И.—
Тр. МХТИ им. Д. И. Менделеева, 1975, вып. 86, с. 30—31. 53. Моисеева 3. 3. —
В кн.: Химия антрахинона/Под ред. А. И. Королева. М, Химия, 1969, с. 70—79;
Пат. ФРГ 2039822, 1972; Chem. Abstr., 1972, v. 76, 140338. 54. Пат. ФРГ
2162538, 1972; Chem. Abstr., 1972, v. 77, 114132; пат. ФРГ 2227340, 1972; Chem.
Abstr., 1973, v. 78, 58147. 55. Пат. ФРГ 2241627, 1974; Chem. Abstr., 1974, v. 80,
145908. 56. Пат. ФРГ 2253016, 1974; Chem. Abstr., 1974, v. 81, 25463. 57. Пат.
ФРГ 2301735, 1974; Chem. Abstr., 1974, v. 81, 135817; Плакидип В. Л., Мо-
лот Л. М. и др. Авт. свид. СССР 394357, 1974. 58. Пат. ФРГ 2252013, 1973;
Chem. Abstr., 1973, v. 79, 31740. 59. Докунихин Н. С. — Журн. ВХО, 1976, т. 21,
№ 3, с. 292—298. 60. Пат. ФРГ 2461648, 1976; Chem. Abstr., 1976, v. 85, 94144;
пат. ФРГ 2256644, 1974; Chem. Abstr., 1974, v. 81, 77744.
61. Пат. ФРГ 2206960, 1972; Chem. Abstr., 1972, v. 77, 164318. 62. Пат. ФРГ
2248990, 1974; Chem. Abstr., 1974, v. 81, 25464. 63. Яп. пат. 7652165, 1976; Chem.
Abstr., 1976, v. 85, 94148; Пат. ФРГ 2346317, 1975; Chem. Abstr. 1975 v. 83,
96868. 64. Яп. пат. 76—86644, 1976; РЖ Хим, 1976, т. 24, Н201П. 65. Пат. ФРГ
2200185, 1972; Chem. Abstr, 1972, v. 77, 166169. 66. Adamek M., Remes M.~
Chem. prum, 1974, Bd. 24, № 11, S. 556—559. 67. Coombes R. O., Golding J. G.—
Tetrahedron Letters, 1976, № 10, p. 771—774. 68. Мигачев Г. И., Андриев-
ский А. М., Докунихин Н. С. — Химия гетероцикл, соед. 1975, № 12, с. 1699—
1700. 69. Pielichowski J., Puszynski А. — Monatsh. Chem, 1974, Bd. 105, № 4,
S. 772—774. 70. Looker J. J. —J. Org. Chem. 1972, v. 37, № 21, p. 3379—3381.
71. Zollinger H.— Chimia, 1968, Bd. 22, № 1, S. 9—20. 72. Хартман 3.,
Ролл Л. — В кн.: Синтезы органических препаратов. Сб. 2. Пер. с англ. М, ИЛ,
1949, с. 372. 73. Беляев Е. К)., Никуличева Т. И., Порай-Коишц Б. А.—ЖОрХ,
1969, т. 5, вып. 12. с. 2204—2207. 74. Беляев Е. К>., Никуличева Т. И., Сели-
на Л.' П. — Там же, 1974, т. 10, вып. 6, с. 1198—1201. 75. Morgan Т. D. В.,
Williams D. L. H. — J. Chem. Soc, Perkin 2, 1972, № 1, p. 74—78. 76. Mor-
gan T. D. B., Williams D. L. H., Wilson J. A. — Ibid, 1973, № 4, p. 1343—1349;
Williams D. L. H. — Tetrahedron, 1975, v. 31, p. 1343—1349. 77. Кампель Л. П.,
Наумова И. И., Стрепихеев Ю. А. и др. — ЖОХ, 1975, т. 45, вып. 12. с. 2734—
2739. 78. Порай-Кошиц А. Е., Эфрос Л. С. — ЖОХ, 1947, т. 17, вып. 10. с. 1801 —
1806. 79. Марвел, Портер — В кн.: Синтезы органических препаратов. Сб. 1.
Под общ. ред. Гильмана. Пер. с англ. М, ИЛ, 1949, с. 300—301. 80. Пау-
стер О. — В кн.: Органические реакции. Сб. 7. Пер. с англ. М, ИЛ, 1956,
с. 409—475.
Глава 5
ГАЛОГЕНИРОВАНИЕ В АРОМАТИЧЕСКОЕ
КОЛЬЦО
В промышленном синтезе соединений ароматического
ряда введение атомов галогенов электрофильным замещением на-
ходит обширное применение не только как метод первичной пере-
работки исходного сырья, но и на более поздних этапах получения.
168
промежуточных продуктов и красителей. В крупных масштабах
используется практически только хлорирование; бромирование
применяется значительно реже и в небольших по объему процессах,
а иодирование — лишь в отдельных случаях. Это объясняется пре-
жде всего значительно более высокой стоимостью брома и особенно
иода по сравнению с хлором, но также и уменьшением реакционной
способности в ряду С1 > Вг > I.
Что касается реакции фторирования, то слишком высокая ак-
тивность фтора (тепловой эффект реакции 428 МДж/моль) делает
ее трудно осуществимой. Поэтому фторпроизводные обычно пред-
почитают получать обходными путями — нуклеофильным замеще-
нием атомов хлора, диазониевой группы и др. (см. гл. 10).
5.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О РЕАКЦИИ, РЕАГЕНТАХ
И МЕХАНИЗМАХ
Наиболее распространенными галогенирующими агента-
ми являются хлор, бром и иод. Значительно реже для хлорирова-
ния применяются гипохлориты, соляная кислота с окислителями
(кислород воздуха, хлорат натрия и др.), сульфурилхлорид SO2C12,
хлорамины, а для бромирования и иодирования — бромиды и
иодиды в присутствии окислителей (чаще всего хлора).
В присутствии катализаторов или растворителей, способных
поляризовать молекулу галогена, последний замещает атомы во-
дорода ароматического кольца:
АгН + С12 —► ArCl + НС1
В отсутствие катализаторов при нагревании и действии света
галоген замещает атомы водорода алифатических боковых цепей
(см. 14.1.1):
ftv
АгСНз4-С1г ---► АгСН2С1 4-НС1
Если же боковых цепей нет, то в тех же условиях может идти
присоединение галогена к ароматическому кольцу:
С6Н6 + ЗС12 С6Н6С1в
Галогенирование в ароматическое кольцо идет со значительно
большей скоростью, чем другие процессы. Поэтому обычно в при-
сутствии катализаторов образуются только продукты этой реак-
ции.
В свою очередь, галогенирование в боковую цепь идет значи-
тельно быстрее, чем реакция присоединения галогена к ароматиче-
скому кольцу. Таким образом, различия в скоростях и условиях
проведения реакций позволяют осуществлять галогенирование од-
нозначно в избранном направлении.
В качестве катализаторов галогенирования в ароматическое
кольцо чаще всего используют хлорид железа (III) (хлорное же-
лезо, получается в самом процессе из железа и хлора), иод, серная
169
кислота; аналогично действуют хлориды алюминия, титана (IV)
(четыреххлористый титан), сурьмы(III) (треххлористая сурьма) и
ряд других соединений [1]. В последнее время предложено прово-
дить бромирование и иодирование с использованием органических
и неорганических солей таллия [2—5]. Все эти катализаторы
необходимы для получения из галогенов электрофильных частиц,
которое, возможно, идет по схеме:
FeCh + Ch —> Cl++FeCl4’
H2SO4 + C12 —> C1+ + HC1 + HSO4"
При использовании иода вначале образуется хлориод, который
взаимодействует с хлором, давая хлор-катион:
IC1 + С12 —> 1С13 1С12 + С1+
Впрочем, прямых доказательств того, что при этих реакциях
образуется в виде кинетически независимой частицы катион гало-
гена, нет. Более того, высказывается мнение [6], что свободный
хлор-катион, согласно правилу Хунда, должен подобно кислороду
находиться в триплетном состоянии с двумя одиночными элект-
ронами, имеющими параллельные спины на разных орбиталях.
Такой катион не является координативно ненасыщенным и не мо-
жет быть непосредственным реагентом электрофильного замеще-
ния. Для того чтобы реакция стала возможной, необходима
Энергетически невыгодная перестройка электронной оболочки хлор-
катиона, при которой все электроны спарены, а одна из Зр-орбита-
лей свободна. Поэтому более вероятно [7, с. 1763], что действие
катализаторов сводится лишь к поляризации молекул галогена с
образованием электрофильных частиц следующего типа:
«+
С1—С1-” катализатор
Подобную же поляризацию молекулы галогена, но в меньшей
степени, могут вызывать растворители, обладающие большой диэ-
лектрической постоянной, например нитробензол, нитрометан, аце-
тонитрил, уксусная кислота и др.
Механизм реакции каталитического хлорирования можно пред-
ставить схемой:
+
+ НС1 + FeCl3
Вслед за этапом, на котором образуется о-комплекс, следует
обычно более быстрый этап отрыва протона ионом FeClJ с обра-
зованием хлористого водорода и регенерацией катализатора [7].
О таком соотношении скоростей свидетельствует отсутствие кине-
170
тического изотопного эффекта. Однако в других случаях соотно-
шение этих этапов может изменяться.
Так, при бромировании соли 7-гидрокси-1,3-нафталиндисульфо-
кислоты (Г-кислоты) [8] молекулярным бромом или бромновати-
стой кислотой быстро образуется ст-комплекс (1), который медлен-
но превращается в конечный продукт реакции (2)
При бромировании молекулярным бромом в уксусной кислоте
З.б-ди-трет-бутилфенола [9] удается выделить промежуточное со-
единение (3), превращение которого в конечный продукт (4) ката-
лизируется кислотой и основаниями:
Интересно отметить, что такое, катализируемое кислотой, пре-
вращение в тяжелой воде идет в три раза быстрее, чем в обычной.
Отрицательный кинетический изотопный эффект, очевидно, объяс-
няется быстрым протонированием промежуточного продукта по
карбонильному кислороду [10, с. 444].
Высокая устойчивость промежуточных соединений, образую-
щихся при галогенировании фенолов, уже давно отмечается в ли-
тературе. От «нормальных» о-комплексов они отличаются отсут-
ствием заряда вследствие отщепления протона от гидроксильной
гРуппы. Частичная нейтрализация положительного заряда в
171
a-комплексе может осуществляться и при наличии других электро-
нодонорных групп, например в случае Л^Л'-диметиланилина:
Такая стабилизация a-комплекса увеличивает скорость его об-
разования и, казалось бы, должна уменьшать скорость его пре-
вращения в конечный продукт галогенирования. Однако кинетиче-
ский изотопный эффект обнаруживается только при иодировании
М,М-диметиланилина; при хлорировании и бромировании он отсут-
ствует [10].
Описаны реакции хлорирования и бромирования аминобензо-
лов, в ходе которых образуются a-комплексы, иногда стабильные
при комнатной температуре [11]. Влияние изменения полярности
растворителя на скорость хлорирования ароматических соединений
в системе вода — уксусная кислота проявляется в увеличении ско-
рости реакции с ростом содержания воды [12].
Изучена кинетика бромирования бензола и толуола в жидком
SO2 при —9 °C. По сравнению с обычным некаталитическим бро-
мированием реакция в жидком SO2 ускоряется и процесс высоко-
селективен. В случае бромирования толуола образуется 88,5% п-,
11,5% о- и 0,1% лт-изомера [13].
Как уже указано, кроме галогенов реагентами галогенирования
в ароматическое кольцо являются также гипогалогениты, галоге-
намины, галогенамиды и другие соединения, содержащие положи-
тельно поляризованный галоген:
6+ б+ +6
НО—Cl RNH—Cl RSO2NH—С1
Механизм их действия принципиально не отличается от уже
рассмотренного. Однако использование этих реагентов позволяет
проводить реакцию галогенирования в водной среде, что имеет
практическое значение для синтеза и открывает широкие возмож-
ности для кинетических исследований. Последними установлено
наличие катализа этих реакций ионами водорода [14].
В ряде случаев кинетика реакции при использовании хлорнова-
тистой (и бромноватистой) кислоты выражается уравнением:
скорость = k [АгН] [СЮН] [Н+]
Из этого следует, что реагентом галогенирования в этих усло-
виях являются СЮНг или СГ. Более подробное обсуждение этих
вопросов см. [6]. Обзор применения хлор- и бромаминов для
галогенирования см. [15].
Сульфурилхлорид SO2C12 — мягкий и удобный в применении
хлорирующий агент, нашел довольно широкое использование в
172
промышленности для введения хлора в ароматические соединения,
обладающие высокой реакционной способностью [16]. С бензолом
в обычных условиях он не взаимодействует. Реакции с
SO2CI2 обычно проводят в среде органических растворителей (на-
пример, в нитробензоле) на холоду или при легком нагревании:
дгН -Ь SO2CI2 —► ArCl -р SO2 -р НС1
Хлорирование ароматических соединений хлористым сульфури-
лом ускоряется под действием света и в присутствии перекисей,
способных инициировать образование свободных радикалов. Эти
данные свидетельствуют в пользу радикального механизма рас-
сматриваемой реакции. Вместе с тем, добавление в SO2CI2 катали-
тических количеств однохлористой серы S2CI2 и хлорида алюминия
приводит к мощному перхлорирующему реагенту, по-видимому,
электрофильного действия, позволяющему легко получать, напри-
мер, перхлорфенантррн и перхлортолуол [17].
Хлорирование ряда соединений может быть проведено с по-
мощью хлорида меди(II). Например, мезитилен при кипячении с
СиС12 превращается в 2-хлормезитилен с выходом 90%. В анало-
гичных условиях n-ксилол образует 2-хлор-п-ксилол, только с
20%-ным выходом. Предполагают, что реакция идет через образо-
вание катион-радикалов [18]:
+ C11CI2 • , + Сис12
АгН --------->• АгН+ ---------> ArCl
—С1“, —CuCl — CuCl, —Н+
Фенол и его гомологи можно хлорировать с хорошим выходом,
пропуская кислород через их растворы в соляной кислоте, содер-
жащей эквимолекулярное количество хлорида меди(II). Основным
продуктом является n-хлорфенол. Для хлорирования аминов целе-
сообразно брать 2 моль СиСЬ на 1 моль амина и проводить
реакцию в возможно более концентрированной соляной кислоте.
Хлор и здесь вступает преимущественно в «-положение к амино-
или диметиламиногруппе [19].
Предложен имеющий промышленное значение процесс монохло-
рирования в кольцо соединений, содержащих электронодонорную
группу, например фенолов и ароматических аминов, взаимодей-
ствием их с СиСЬ в водном солянокислом растворе, с введением
газообразного хлора для окисления образующегося CuCl в СиС12;
Достигаются хороший выход и высокая селективность [20]. Можно
также хлорировать фенолы и ароматические амины в кольцо с
получением преимущественно /г-производных, используя хлориды
не только меди, но и других элементов I, II и III групп [20].
Взаимодействием ароматических соединений с солями меди и до-
нором иода (12 или любые иодиды металлов и неметаллов I—VIII
гРупп) получают арилиодиды [21]. Описан простой метод направ-
ленного галогенирования алкилароматических соединений в кольцо
в присутствии оксида кремния [22]. Хлорирование с помощью
хлоридов палладия, железа, титана и других металлов см. [23].
173
5.2. ПРАКТИКА ГАЛОГЕНИРОВАНИЯ
5.2.1. Хлорирование бензола и его гомологов
Среди множества используемых в промышленности ре-
акций хлорирования в ароматическое кольцо хлорирование бензо-
ла имеет наибольшее значение. Как правило, его проводят, ис-
пользуя в качестве катализатора хлорид железа (III), получаю-
щийся при взаимодействии хлора и металлического железа,
загружаемого в аппарат в виде обрезков листового железа,
железных колец и т. д. Образующийся безводный FeClg раство-
ряется в бензоле и продуктах его хлорирования и благодаря
этому реакция протекает как гомогенно-каталитическая
[1, с. 181]. Очень важно применять сухие бензол и хлор, так как
гидратированный FeCl3 нерастворим в органической фазе и реак-
ция может приобрести менее выгодный гетерогенно-каталитиче-
ский характер. Кроме того, хлористый водород, образующийся при
реакции, с водой дает соляную кислоту, агрессивно действующую
на металлы.
В отличие от нитро- и сульфогрупп атом хлора в бензольном
кольце уменьшает скорость дальнейшего электрофильного хлори-
рования очень мало — всего в 7—8 раз. Поэтому при хлорирова-
нии бензола реакция образования хлорбензола (6) в чистом виде
идет только в самом начале процесса.
174
По мере образования хлорбензола начинается дальнейшее хло-
рирование его, причем скорость процесса увеличивается с повыше-
нием концентрации хлорбензола. Благодаря этому при хлорирова-
нии бензола неизбежно получаются полихлориды, количество
которых быстро возрастает с увеличением глубины процесса. По-
этому реакцию приходится вести с «обратным» бензолом, обрывая
ее задолго до полного израсходования бензола. Например, при пе-
риодическом способе производства реакцию обычно заканчивают,
когда в смеси содержится 30—40% хлорбензола, 10—15% поли-
хлоридов и около 50% бензола. После нейтрализации реакционной
массы эти вещества легко разделяются при перегонке.
Так как объем производства хлорбензола, широко используе-
мого в качестве промежуточного продукта и растворителя, очень
велик, получать его в аппаратах периодического действия эконо-
мически невыгодно, и в настоящее время почти во всех странах
применяются непрерывные способы хлорирования, аппаратурное
оформление которых может быть весьма разнообразным [24, 25].
Определенные трудности при осуществлении непрерывного процес-
са вызывает необходимость отвода выделяющейся при реакции
теплоты — 32 МДж/моль [7, с. 1766]. Видимо, наиболее интересен
метод непрерывного хлорирования, при котором выделяющаяся
теплота полностью расходуется на нагревание исходных веществ
до кипения и на испарение части бензола и хлорбензола [24].
Использование непрерывных методов позволяет уменьшить глуби-
ну хлорирования и тем самым значительно сократить образование
полихлорпроизводных, находящих лишь ограниченное применение.
Например, при упомянутом выше способе поддерживают темпера-
туру 76—83 °C; в реакционной массе остается до 65% бензола и
образуется 3,5—4% полихлоридов от получившегося хлорбензола.
В связи с этим преимуществом интенсивно изучается непрерывное
хлорирование и других ароматических соединений [26].
Непрерывное хлорирование бензола обычно проводят в хлора-
торах, представляющих собой трубу, заполненную перемешанными
стальными и керамическими кольцами размером 25 X 25 мм.
В верхней части хлоратора имеется сепарационный объем, не за-
полненный насадкой. Бензол и хлор подают прямотоком в нижнюю
часть аппарата. Подачу реагентов регулируют таким образом,
чтобы полностью использовать хлор, и чтобы температура в аппа-
рате поддерживалась на уровне 76—83 °C. Пары бензола и, в
небольшой степени, хлорбензола вместе с выделяющимся НС1 по-
падают в теплообменник, конденсируются, отделяются от НС1 и
после осушки возвращаются в процесс. Реакционная масса, выте-
кающая из верхней части аппарата, поступает на непрерывную
ректификацию, при которой хлорбензол и полихлориды отделяются
от бензола, возвращающегося в цикл [24].
Для получения хлорбензола в промышленности используется
также парофазный метод так называемого окислительного хлори-
рования. Реакцию осуществляют, пропуская пары бензола и хло-
ристый водород при 200—250 °C над катализатором, содержащим
175
медь(II). В присутствии кислорода воздуха идет окисление НС1 и
одновременное хлорирование бензола по суммарному уравнению:
С6Н6 + НС1 + 0,5О2 —> С6Н5С1 + Н2О
Степень превращения бензола за проход около 10% и выход
хлорбензола 90% от теоретического [24]. Хлорбензол, получаемый
этим методом, используют для парофазного гидролиза в фенол
(см. 8.1.2). Одно из преимуществ этого метода заключается в
практическом отсутствии полихлоридов, а основной его недоста-
ток— трудность подбора материалов для аппаратуры, которая
должна работать в среде хлористого водорода и воды при высокой
температуре.
Обзор материалов о каталитическом окислительном хлорирова-
нии органических соединений различных классов см. [27].
Определенный интерес вызывает также парофазное получение
хлорбензола из бензола и хлора. В отсутствие катализаторов эта
реакция начинается только при температуре выше 260 °C и, по-
видимому, идет по радикальному механизму. Добавление в каче-
стве инициатора перхлордиметилсульфона (ССЬЬВОг, который,
распадаясь, образует SO2 и радикалы *СС1з, позволяет снизить
температуру реакции до 180 °C и получать только продукты заме-
щения, не содержащие даже следов продуктов присоединения
хлора и диарилов. В этих условиях бензол хлорируется в 7 раз
быстрее, чем хлорбензол [28].
Основные количества хлорбензола, вырабатываемого промыш-
ленностью, расходуются на производство фенола (см. 8.1.2) и на
получение нитро- и динитрохлорбензолов (см. 4.4.2). В меньших
масштабах он применяется во многих других синтезах и в качестве
растворителя.
При дальнейшем хлорировании хлорбензола в присутствии
FeCh вначале образуются о- и п-дихлорбензолы (5) и (7), обычно
в соотношении 30 и 70%. Количество n-изомера можно несколько
увеличить, добавляя при хлорировании сернистые соединения
[7, с. 1766]. Из смеси n-изомер может быть выделен с помощью
кристаллизации. Остающаяся эвтектика (так называемый техни-
ческий о-дихлорбензол) содержит около 70% о-изомера и часто
используется в качестве растворителя. Чистый о-дихлорбензол из
нее получается путем ректификации. Оба продукта используются
для нитрования (см. 4.4.2), а также в некоторых других реакциях;
n-изомер, кроме того, в качестве инсектицида (антимоль). о-Дих-
лорбензол значительно легче n-изомера вступает в реакции элект-
рофильного замещения, и на этом основано несколько методов
удаления его из практически более важного п-изомера [1, с. 197].
Впрочем, промышленного применения эти методы не получили.
При дальнейшем хлорировании из о-дихлорбензола получается
смесь 1,2,3-(8) и 1,2,4-трихлорбензола (10), а из и-дихлорбензо-
ла — только (Ю). 1,2,3-Трихлорбензол сульфируют и затем ис-
пользуют в синтезе одного из тиоиндигоидных красителей, а
176
1,2,4-трихлорбензол после нитрования (см. 4.4.2) дает ряд ценных
промежуточных продуктов.
Из продуктов более глубокого хлорирования важен
1,2,4,5-тетрахлорбензол (11), из которого получают 2,4,5-трихлор-
фенол (см. 8.1.2) и 2,4,5-трихлортиофенол (см. 9.1.1), используемые
в синтезе гербицидов. В приведенной выше схеме показано обра*
зованйе и других полихлорбензолов. Из гексахлорбензола (14)
получают пентахлорфенол, применяемый в сельском хозяйстве.
Как видно из общей схемы, ж-дихлорбензол и 1,3,5-трихлорбен-
зол, находящие применение в синтезе промежуточных продуктов,
не могут быть получены прямым хлорированием бензола. ’Их
обычно получают обходными путями, например по реакции
Зандмайера (см. 10.3): лг-дихлорбензол— из .м-хлоранилина, а
1,3,5-трихлорбензол — из 2,4,6-трихлоранилина. Однако их можно
получать и более перспективным методом — изомеризацией полу-
чающихся при хлорировании смесей ди- или, соответственно, три-
хлорбензолов в присутствии хлорида алюминия и хлористого водо-
рода. Так, при нагревании смеси о- (5) и л-дихлорбензола (7) с
названным катализатором при 160 °C образуется равновесная
смесь, содержащая 54% м-, 16% о- и 30% л-дихлорбензола. Раз-
работан непрерывный способ изомеризации дихлорбензолов с по-
стоянным отводом более низкокипящего л«-дихлорбензола через
эффективную ректификационную колонну. Аналогично, в равно-
весной смеси, образующейся из изомерных трихлорбензолов (8) и
(10), содержится около 25% 1,3,5-изомера. Его можно выделять
разгонкой или вымораживанием [7, с. 1770].
Установлено, что реакция изомеризации в этих условиях прохо-
дит внутримолекулярно, без потери связи перемещающегося атома
хлора с ароматическим кольцом, путем последовательных
1,2-сдвигов внутри бензолониевых ионов (см. 2.7.1). Взаимопрев-
ращения о- (5) и ж-дихлорбензола (15) можно представить схемой
(29, 30]:
Предложен способ получения л-дихлорбе'нзола хлорированием
бензолсульфохлорида в ж-хлорбензолсульфохлорид и последую-
щем превращении его в л-дихлорбензол. Реакцию хлорирования
177
проводят в среде разбавителей (CCI4, C2H2CI4, С1гС=СС12, уксус*
ной кислоте, петролейном эфире) в присутствии катализаторов
Фриделя — Крафтса [31].
Хлорирование толуола в кольцо ведут в значительно меньших
масштабах и обычно периодическим способом. В качестве катали-
затора здесь используются хлориды железа(III) или сурьмы(III).
Применение SbCK дает меньшее количество продуктов осмоления и
более выгодное соотношение изомеров. В этом случае необходимо
использовать освинцованные аппараты [1, с. 201].
Последовательное образование важнейших продуктов, получа-
емых при хлорировании толуола можйо представить схемой:
На первом этапе хлорирования толуола образуется смесь о-
(16) и п-хлортолуола (17) в приблизительном соотношении 1:1,
которую разделяют с помощью вакуум-перегонки. о-Хлортолуол
используется для синтеза о-хлорбензальдегида, о-хлорбензойной
кислоты и некоторых других соединений. n-Хлортолуол находит
значительно большее применение для получения п-хлорбензальде-
гида, ц-хлорбензотрихлорида, n-хлорбензойной кислоты и других
продуктов, а также для дальнейшего хлорирования в кольцо.
В присутствии FeCl3 при 50°C в результате дальнейшего хло-
рирования (16) и (17) получается практически важный 2,4-ди-
хлортолуол (20), содержащий около 25% 3,4-дихлортолуола (21).
178
Последний отделяют вакуум-перегонкой. Из 2,4-дихлортолуола в
дальнейшем получают 2,4-ДИХлорбензотрихлорид и 2,4-дихлорбен-
зойную кислоту.
Могут быть получены также 2,5- (19) и 2,6-дихлортолуол (18).
Более глубокое хлорирование на схеме не показано полностью.
Из n-хлортолуола, например, образуются смеси, содержащие че-
тыре трихлор- (с преобладанием 2,3,6- и 2,4,5-изомеров), два тетра-
хлор- и пентахлортолуол. Наблюдаемое на отдельных стадиях рас-
пределение изомеров удовлетворительно согласуется с рассчитан-
ным по аддитивной схеме с учетом парциальных скоростей заме-
щения [32].
Из продуктов, образующихся при хлорировании ксилолов, наи-
большее значение имеет 2,5-дихлор-л-ксилол (22). Его получают с
примесью 2,3-изомера (23) и очищают кристаллизацией из изо-
пропилового спирта. Он служит исходным продуктом для получе-
ния 2,5-дихлортерефталевой кислоты.
Более чисто хлорирование м- и n-ксилолов в присутствии
FeCl3 проходит в среде ледяной уксусной кислоты. В этих условиях
в молекулу ксилолов вступает не более двух атомов хлора.
Хлорирование n-ксилола до 2,3,5,6-тетрахлор-п-ксилола реко-
мендуется вести в СС14 с FeCl3 в качестве катализатора [7,
с. 1769]. Образующийся продукт используется для синтеза тетра-
хлортерефталевой кислоты.
о-Ксилол при хлорировании в присутствии железа дает смесь,
содержащую 3-хлор- и 4-хлор-о-ксилол [33].
5.2.2. Хлорирование производных углеводородов
ряда бензола
Фенол подвергается хлорированию с большой скоростью
без катализаторов. Монохлорпроизводные — о-хлорфенол (24)
(преимущественно) и п-хлорфенол (25)—получаются уже при
Действии на фенол водного раствора гипохлорита натрия. В завод-
ской практике смесь (24) и (25), в которой преобладает (25),
179
получают действием сульфурилхлорида при 40 °C. Продукты реак-
ции разделяют с помощью вакуум-перегонки.
о-Хлорфенол имеет лишь ограниченное применение в синтезе
красителей, в то время как n-хлорфенол используется в больших
масштабах, например при получении хинизарина. Поэтому заслу-
живает внимания специальный способ получения преимущественно
n-хлорфенола: трифенилфосфат (получение см. 14.3.2) хлорируют в
присутствии обычных катализаторов, а затем продукт реакции
гидролизуют кислотой или щелочью. В продукте реакции содер-
жится только 8% о-хлорфенола [34]. Этим же способом может
быть получен 4-хлор-о-крезол.
Изучено хлорирование фенола действием C11CI2 в различных
условиях. При проведении реакции в органических растворителях
при 100 °C образуются о-хлорфенол, n-хлорфенол (главный про-
дукт) и 2,4-дихлорфенол (27). Выход (25) возрастает с увеличени-
ем полярности растворителя; в воде он в десять раз превышает
выход (24) [35].
2,4-Дихлорфенол (27) в больших количествах используют для
синтеза 2,4-дихлорфеноксиуксусной кислоты. Он получается при
действии хлора на фенол, содержащий 10% воды. Описан также
способ получения .(27) хлорированием 26—30%-ного раствора фе-
нола в нитрометане при 40—60°С [36].
Интересно отметить, что 2,6-дихлорфенол (26) образуется как
главный продукт при хлорировании о-хлорфенола в кипящих непо-
лярных растворителях (СС14, СбН6, C6Hi4); выход его достигает
82% против 17% 2,4-изомера. Чистый 2,6-дихлорфенол можно
выделить с помощью ректификации [37] (см. также [38]).
Более глубокое хлорирование фенола при 50—60 °C приводит к
2,4,6-трихлорфенолу (28). Его используют для получения ценного
промежуточного продукта — 2,3,5,6-тетрахлор-1,4-бензохинона
(хлоранила) (30). С этой целью хлорируют (28), растворенный в
смеси серной (моногидрат) и хлорсульфоновой кислот. В качестве
180
промежуточного продукта реакции здесь образуется пер-
хлор-2,5-циклогексадиенон (29) [39].
(28) (29) (30)
Наиболее удобным методом получения монохлорпроизводных-
о-, м- и n-крезолов, этилового эфира n-гидроксибензойной кислоты,,
диметиловых эфиров гидрохинона и резорцина является хлориро-
вание этих соединений действием сульфурилхлорида SO2C12 [U
с. 212].
Анилин реагирует с хлором так же легко, как и фенол. Напри-
мер, действие хлора на анилин в неводном органическом раство-
рителе приводит к получению 2,4,6-трихлоранилина. Проведение-
этой реакции в водной среде сопровождается образованием окра-
шенных продуктов и смол.
Для получения моно- и дихлорпроизводных первичных арома-
тических аминов эти соединения вначале формилируют или ацети-
лируют по аминогруппе, а затем ведут хлорирование, обычно в
органическом растворителе (хлорбензол, нитробензол, диметил-
формамид). Отмечается, что в диметилформамиде хлорирование
идет значительно быстрее, чем в других растворителях. Первич-
ными продуктами хлорирования в этих случаях являются А-хлор-
производные, которые затем под влиянием хлористого водорода
перегруппировываются в п- и о-хлорпроизводные (перегруппировка.
Ортона). Перегруппировка имеет межмолекулярный характер-
[6, с. 739].
Этот путь синтеза используют, например, при получении-
4-хлор-о-толуидина (31), применяемого для холодного крашения:.
Предложен способ получения 5-хлор-о-толуидина и
3-хлор-о-толуидина хлорированием о-толуидина в 90—100%-ной
H2SO4 при молекулярном соотношении о-толуидин : H2SO4 —
= 1 : 4 4- 1 : 8 и С12: о-толуидин ^1:1 [40].
Нитропроизводные аминов гладко хлорируются без защиты
аминогруппы в водном растворе. Например, 4-нитро-2,6-дихлор-
181
анилин (32) получается при приливании раствора хлората натрия
к суспензии n-нитроанилина в соляной кислоте; 2,4-динитро-6-хлор-
анилин (33) образуется при действии хлора на водную суспензию
2,4-динитроанилина в присутствии FeCl3:
nh2
no2
(32)
no2 no2
(33)
Для получения 4-нитро-2-хлоранилина предложено действовать
на n-нитроанилин соляной кислотой в присутствии Н2О2 [41].
4-Нитро-2-хлор-МАДдиэтиланилин, применяемый в синтезе диа-
зотипных материалов [42], получают хлорированием п-нит-
ро-У,А^-диэтиланилина сульфурилхлоридом в органическом рас-
творителе.
Производные бензола, содержащие только электроноакцептор-
ные заместители, хлорируются в более жестких условиях — при
нагревании в присутствии катализаторов. Например, нитробензол
хлорируется в присутствии FeCl3 с образованием м-нитрохлорбен-
зола. Реакцию приходится вести при сравнительно невысокой тем-
пературе (35—40 °C), так как нагревание способствует замещению
нитрогруппы хлором. Побочно образуются изомерные моно- и ди-
хлорнитробензолы, которые отделяют ректификацией.
лг-Нитрохлорбензол используется главным образом для получе-
ния м-хлоранилина, применяемого в холодном крашении.
Описан непрерывный способ получения м-нитрохлорбензола
хлорированием нитробензола в каскаде из четырех реакторов.
Нитробензол в смеси с катализатором, содержащим иод, непре-
рывно подают в первый реактор с одновременной подачей хлора.
Реакционная смесь из последнего реактора поступает в колонну
фракционирования, откуда отогнанный непрореагировавший нит-
робензол и 70—80% иода возвращаются в начало процесса; сырой
л-нитрохлорбензол очищают ректификацией. В качестве катализа-
тора используют FeCl3 или SbCl3 с 12 [43].
Упомянутая выше побочная реакция замещения нитрогруппы
хлором может иметь и самостоятельное значение. Так, при пропу-
скании хлора через расплавленный м-динитробензол при 220 °C в
отсутствие катализаторов и воды с хорошим выходом образуется
м-дихлорбензол:
NO2 Cl
182
Процесс приходится осуществлять в стеклянной аппаратуре
[1, с. 215], (см. также [44]). Полагают, что реакции этого типа
идут по радикальному механизму.
При хлорировании «-нитротолуола в присутствии FeCl3 получа-
ется 4-нитро-2-хлортолуол. Для получения 4-нитро-2,6-дихлортолу-
ола хлорирование «-нитротолуола рекомендуется вести в присут-
ствии SbCh. Побочно образующиеся при этом нитротри- и нитро-
тетрахлорпроизводные толуола превращают в целевой продукт,
действуя порошком меди в смеси хлорбензола с уксусной кисло-
той [45].
Для получения смеси 1-нитро-2,5- и 1-нитро-2,3-дихлорбензолов
хлорируют о-нитрохлорбензол в присутствии сульфида железа(II)
[46].
О получении изомерных нитрохлорбензолов см. также 4.4.2.
Большое практическое значение имеют продукты, образующие-
ся при хлорировании фталевой кислоты и фталевого ангидрида,,
используемые в синтезе антрахиноновых красителей и фталоциа-
нинов [47, с. 224].
Фталевая кислота легко хлорируется в водном слабощелочном
растворе, образуя вначале 4-хлорфталевую (34), а затем 4,5-ди-
хлорфталевую (35) кислоту с примесью изомеров, которые, одна-
ко, легко отделяются [48]:
^.COONa
^^^^-COONa
COONa
COONa
(34) (35)
4,5-Дихлорфталевую кислоту можно получить также хлорируя
фталевый ангидрид в паровой фазе над хлоридом марганца и
оксидом алюминия при 400 °C [49].
При хлорировании фталевого ангидрида в олеуме получается
преимущественно 3,6-дихлорфталевый а'нгидрид (36), а более глу-
бокое проведение реакции дает тетрахлорфталевый ангидрид.
(37).
При увеличении длительности процесса происходит декарбок-
силирование и образуется гексахлорбензол [1, с. 212].
Необходимость значительно более жестких условий проведения
реакции хлорирования со фталевым ангидридом объясняется более
сильным электроноакцепторным действием ангидридной группы..
18$
Изучалось также хлорирование азобензола, которое идет пре-
имущественно в орто-положения к азогруппе. Газообразный хлор
пропускают в раствор азобензола с каталитическим количеством
PdCU в смеси воды с органическими растворителями (например, со
‘смешивающимися с водой простыми эфирами). Получается смесь,
-содержащая 2-хлоразобензол, 2,6-дихлоразобензол, 2,2'-дихлор-
азобензол и 2,6,2'-трихлоразобензол, из которой выделяют 2,2'-ди-
хлоразобензол. В отсутствие PdCl2 в аналогичных условиях обра-
зуется смесь 4-хлоразобензола и 4,4'-дихлоразобензола [50].
5.2.3. Хлорирование нафталина и его производных
Нафталин хлорируется значительно легче, чем бензол,
так что последний может даже быть использован как относительно
инертный растворитель при этой реакции. Чаще, однако, раствори-
телем служат полихлориды бензола или же ведут реакцию без
растворителей, пропуская хлор в расплавленный нафталин. В ка-
честве катализатора рекомендуются иод или FeCl3.
Главным продуктом монохлорирования является 1-хлорнафта-
лин (38), наряду с которым в обычных условиях получается 9—
10% 2-хлорнафталина (39) и полихлорпроизводные:
С1
(38) (39)
В чугунном аппарате на 8000 л расплавляют 6000 кг нафталина
и при перемешивании прибавляют 15 кг железного катализатора.
’Хлор пропускают при ПО—120 °C до достижения плотности жид-
кости 1,2 (при 20 °C). Содержимое аппарата продувают воздухом в
течение 2 ч и нейтрализуют 50—80 кг порошка NaOH до полного
отсутствия НС1. Продукт реакции (16 500 кг) перегоняют в вакууме
(при 25 мм рт.ст.), собирая следующие фракции: а) предваритель-
ный погон (нафталин, 2500 кг), б) промежуточная фракция
(2000 кг; 240—260 °C), в) 1-хлорнафталин (7000 кг, 250—262 °C),
т) промежуточная фракция (3000 кг, 262—280 °C), д) дихлорнаф-
талины (1700 кг). Фракции «а» и «д» возвращают в аппарат для
хлорирования, «б» и «г» снова подвергают перегонке, присоединяя
к следующей партии продукта хлорирования [51, с. 176].
Получаемый 1-хлорнафталин содержит примесь 2-хлорпроиз-
водного, полное отделение которого является трудной задачей [1,
<с. 203].
Практически интересна смесь полихлорнафталинов, образую-
щаяся при более глубоком хлорировании. Она имеет воскоподоб-
ную консистенцию и применяется в качестве диэлектрика в элект-
ротехнической промышленности (галовакс). Кроме этого, ее ис-
пользуют для пропитки тканей и бумаги с целью придания им
-огнестойкости.
184
Полихлорнафталины получают как периодическим, так if
непрерывным хлорированием в присутствии катализаторов. Темпе-
ратуру к концу процесса повышают до 160 °C, ход реакции
контролируют по плотности и температуре застывания продукта.
После нейтрализации продукт подвергают фракционированию под
вакуумом. Основная часть получающихся этим способом соедине-
ний содержит 3—4 атома хлора. Исчерпывающее хлорирование
нафталина требует повышения температуры до 200 °C. Образую-
щийся октахлорнафталин (перхлорнафталин) (40) при дальней-
шем хлорировании и повышении температуры до 220—230 °C пре-
вращается с выделением
риндан (41) [51].
четыреххлористого углерода в перхло-
С1
С1
С1
|си /С1
С1
+ СС14
С1
С1/ЧС1
С1
(41)
2-Нафтол и его производные, не содержащие заместителей в
положении 1, легко и избирательно хлорируются в это положение-
при действии гипохлоритов в водно-щелочной среде или при дей-
ствии сульфурилхлорида в органических растворителях.
1-Нафтол в аналогичных условиях образует 2,4-дихлор-1-на-
фтол (42), который при дальнейшем хлорировании и гидролизе
превращается в 2,3-дихлор-1,4-нафтохинон (43):
(42) (43)
2,3-Дихлор-1,4-нафтохинон применяют в синтезе красителей
[52, с. 17] и в качестве инсектицида. В промышленности его обычно
предпочитают получать хлорированием 1,4-нафтохинона (44). Ре-
акцию рекомендуют [53] проводить в дихлорэтане при 75-—80 °C с
добавлением 10% масс, мочевины или диметилмочевины. Выход
продукта—86%. При отсутствии 1,4-нафтохинона продукт (43)>
обычно получают хлорированием нафтионовой кислоты (45):
О
nh2
SO,H
(«)
(44)
18&
В последнем случае азот аминогруппы выделяется из реак-
ционной массы в виде взрывоопасного хлористого азота NC13 и
это вызывает необходимость принятия специальных мер предосто-
рожности.
5.2.4. Хлорирование антрахинона, его производных
и других полициклических соединений
9,10- Антрахинон можно хлорировать в среде олеума.
Выделяющийся при реакции хлористый водород образует хлор-
сульфоновую кислоту, значительная часть которой может быть
отогнана из реакционной массы. Таким образом получают
1,4,5,8-тетрахлор-9,10-антрахинон (46) [54]:
(46)
Останавливать процесс на более низких стадиях хлорирования
нецелесообразно, так как получающиеся смеси моно-, ди- и три-
хлорпроизводных разделяются с большим трудом. Поэтому прак-
тически важные 1-хлор-, 1,5- и 1,8-дихлор-9,10-антрахиноны полу-
чают хлорированием соответствующих сульфокислот, при котором
•сульфогруппа замещается хлором. Процесс проводят, постепенно
добавляя раствор хлората натрия NaC103 к водному раствору ант-
рахинонсульфокислоты, подкисленному соляной кислотой, при на-
гревании [55, с. 212]. Например:
(47)
(48)
Реакция протекает очень гладко, с хорошим выходом, но при
заводском осуществлении она неудобна, так как идет медленно, в
больших объемах и тормозится солями некоторых тяжелых метал-
лов, особенно марганца. Изучение механизма этой реакции пока-
зало, что она ускоряется при действии света и инициаторов, т. е.
имеет радикальный характер [56]. Это позволило предложить
новую технологию, при которой водный раствор соли антрахинон-
сульфокислоты хлорируют молекулярным хлором, постепенно до-
бавляя в ходе процесса небольшие количества раствора инициато-
186
ра, например хлорида аммония. Последний с молекулярным хло-
ром образует хлористый азот, термически нестойкий выше 80 °C?
NH4C1 + 3C12 —> 4НС1 + NC13 —> O,5N2 + 3C1.
Цепной радикальный характер реакции получения хлорантра-
хинона АгС1 из антрахинонсульфокислоты ArSO3H может быть
представлен следующими уравнениями [57];
ArSOj + С!. —> ArCl + SO;.
SO3. + С12 —> СЫ-8ОзСГ
so3cr + он- —> sor + cr + н+
Соли аммония как инициаторы этих реакций можно использо-
вать и при проведении процесса не с молекулярным хлором, а с
хлоратами. Так, хлорзамещенные 9,10-антрахиноны, пригодные
для получения прочных красителей, получают нагреванием
9,10-антрахинонсульфокислот с хлоратами в 5—25%-ной
H2SO4 при 98 °C, с постепенным добавлением раствора хлорида,
нитрата или сульфата аммония, содержащего хлорид натрия и
соляную кислоту. В качестве исходных используют, например:
9,10-антрахинон-1-сульфокислоту, -1,5- или -1,8-дисульфокислоту,
или 5-нитро-9,10-антрахинон-1-сульфокислоту [58].
Интересно отметить, что при фотоинициировании реакция заме-
щения хлором сульфогруппы в 9,10-антрахинон-1-сульфокислоте
(47) количественно проходит в водном растворе хлорида натрия
на холоду [59].
При обработке озоном водного сернокислого раствора натрие-
вой соли 9,10-антрахинон-1-сульфокислоты (47) в присутствии хло-
рида натрия образуется 1-хлор-9,10-антрахинон (48) с выходом
87%.
Скорость реакции и выход продукта увеличивается при введе-
нии каталитических добавок сульфата марганца, практически пол-
ностью предотвращающих разрушение (47) озоном. В водном рас-
творе и в отсутствие серной кислоты (47) преимущественно под-
вергается озонолизу, и 1-хлор-9,10-антрахинон не образуется [60].
Замещение хлором сульфогрупп в p-положении 9,10-антрахино-
на идет значительно труднее и препаративного значения не имеет.
2-Метил-1-хлор-9,10-антрахинон (49) получается прямым хлориро-
ванием 2-метил-9,10-антрахинона в серной кислоте или в олеуме в
присутствии каталитических количеств иода [1, с. 211]:
О
И»)
187
Для хлорирования амино- и диаминоантрахинонов применяют!
оульфурилхлорид в органическом растворителе (чаще всего в нит-,
робензоле). Этим путем получают 2-амино-1-хлор- (50), 1-ами-'
но-2-метил-4-хлор-(51), 1-бензоиламино-4-хлор- (52), 1,4-диами-
но-2,3-дихлор-9,10-антрахинон (53) и другие аналогичные продук-
ты [1, с. 219].
(51)
Предложено также получать аминохлорантрахиноны хлориро-
ванием 1-аминоантрахинонов в органическом растворителе в при-;
сутствии алкилмочевины, например тетраметилмочевины или ее
•смеси с другими амидами [61].
Хлорирование хинизарина (1,4-дигидрокси-9,10-антрахинона) в,
уксусной кислоте приводит к 2,3-дихлорпроизводному. Из 1,4-ди-!
гидрокси-2-мётокси-9,10-антрахинона в этих условиях получается1
‘3-хлорпроизводное [62].
Пирен хлорируется с большой легкостью в нитробензольном
растворе, при действии молекулярного хлора образуя 1,3,6,8-тет-
рахлорпирен, выпадающий в осадок. Продукт используется для
•синтеза кубовых красителей и для получения 1,4,5,8-нафталинтет-
ракарбоновой кислоты (см. 17.2.5). При галогенировании пирена
и 1-формилпирена действием хлорида или бромида меди(П) обра-
зуются 1-хлор- или 1-бромпирен [63].
Продукты хлорирования бифенила и дифенилового эфира, по-
лучаемые действием хлора на расплавы этих соединений при на-
гревании, используются в качестве диэлектриков и ретардантов.
5.2.5. Бромирование и иодирование
Как уже указывалось, бромирование и тем более иоди-!
рование проводят в значительно меньших масштабах, чем хлори-1
рование. Ввиду высокой стоимости брома и иода их стараются
использовать без потерь; количественно улавливают выделяющий-
ся при реакции бромистый или иодистый водород и регенерируют
из них галогены; чаще же выделяют и возвращают в реакцию бром
или иод, вводя окислители непосредственно в реакционную массу.
Л88
В промышленности в небольших масштабах ведут бромирова-
ние бензола на бромбензол, мезитилена на 2-броммезитилен и
1,3,5-триэтилбензола на 2-бром-1,3,5-триэтилбензол [64, с. 89; 65].
Получаемые продукты могут быть использованы в синтезе антра-
хиноновых красителей. Выделяющийся при реакции бромистый
водород улавливают и в дальнейшем превращают в бром.
Бромирование и иодирование о-ксилола в присутствии железа
в смеси уксусной, серной и азотной кислот приводит к смеси 3- и
4-бром- или -иод-о-ксилолов; иодопроизводные получаются в соот-
ношении 16:84 [33].
Для получения о-бромфенола раствор фенола в дибромэтане
или дибромпропане и в их аналогах обрабатывают бромом и затем
отгоняют растворитель [66].
Бромирование терефталевой кислоты или ее хлорангидрида до
2,5-дибромпроизводного ведут при 40—55 °C в хлорсульфоновой
или фторсульфоновой кислоте, применяя серный ангидрид и иод в
качестве катализаторов [67].
Фталевый ангидрид иодируют также в хлорсульфоновой
кислоте в присутствии серы (как переносчика иода) избытком
иода. В результате реакции образуется тетраиодфталевый ан-
гидрид [68].
Полибромбифенил, содержащий 8—10 атомов брома, получают
действием брома на раствор бифенила в метилендибромиде при
20—100 °C в присутствии катализаторов А1С13, А1Вг3, FeCl3,
FeBr3 или их смеси [69].
Для получения 5-бромизатина (54), применяемого в синтезе
тиоиндигоидов, к суспензии изатина в разбавленной соляной кис-
лоте приливают солянокислый раствор брома, а затем для полного
использования брома в реакционную массу пропускают хлор [70,
с. 39]:
Этим же приемом пользуются при получении 1-бром-4-метил-
амино-9,10-антрахинона (55) из 1-метиламино-9,10-антрахинона
[70] и 1-амино-2,4-дибром-9,10-антрахинона (56) из 1-ами-
189
Бромирование 1-метиламино-9,10-антрахинона можно вести i
также в пиридине [71] ив нитробензоле [72]. Утверждают, что в *
нитробензоле выход (55) составляет до 94,5%, в то время как в
водной среде только 70,5%.
В качестве среды для бромирования 1-амино-9,10-антрахинона;
на 2,4-дибромпроизводное (56) рекомендуют смесь соляной кисло-
ты и хлорбензола. Перед проведением реакции 1-амино-9,10-ант-
рахинон длительное время размешивают в этой смеси до образо-
вания однородной суспензии; затем добавляют бром и гипохлорит.
Выход продукта 97% [73].
Для синтеза технически важной 1-амино-4-бром-9,10-антрахи-
нон-2-сульфокислоты (бромаминовая кислота) (58) к раствору
1 -амино-9,10-антрахинон-2-сульфокислоты (57) в воде или в слабой
кислоте добавляют раствор брома в щелочи или в соляной кислоте
и одновременно пропускают хлор. Очень чистая бромаминовая
кислота может быть получена при бромировании (57) в нейтраль-
ной среде в присутствии ацетата натрия [74].
Побочно при этой реакции, особенно при повышении темпера-
туры, может получаться уже упомянутый 1-амино-2,4-дибром-
9,10-антрахинон (56) (см. также [75]). Бромаминовую кислоту
производят в довольно больших масштабах.
При бромировании бензантрона в водно-солянокислой суспен-
зии получается 3-бромбензантрон [1, с. 220 и сл.]. Для полного
использования брома в реакционную массу добавляют раствор
гипохлорита натрия.
Диангидрид 3,4,9,10-перилентетракарбоновой кислоты брони-
руют в олеуме, в хлорсульфоновой или в серной кислоте [76].
Реакции бромирования подвергаются не только промежуточные
продукты, но и многие красители, особенно кубовые, так как вве-
дение брома углубляет их цвет, а во многих случаях и повышает
их прочностные характеристики. Эти реакции обычно проводят в
концентрированной серной кислоте. Типичным примером является
получение 5,5', 7,7'-тетраброминдиго (60):
190
Бромирование индиго (59) ведут в 98—99%-ной H2SO4 поло-
винным от представленного в уравнении количеством брома, так
как серная кислота окисляет образующийся бромистый водород до
брома, возвращая его в реакцию:
H2SO4 + 2НВг —> 2Н2О + SO2 + Вг2
Суммарное уравнение реакции приобретает следующий вид:
С^НщХгОг -f- 2Вгг -Ь 2H2SO4 —>- СцзНеМгОгВ^ -|- 4НгО Ч- 2SO2
(59) (60)
Выделяющаяся при реакции вода разбавляет серную кислоту и
ослабляет ее окисляющее действие. Поэтому в ходе процесса сер-
ную кислоту постоянно укрепляют олеумом, сохраняя в пределах
указанной выше концентрации. Превышение этой концентрации
недопустимо, так как вызывает сульфирование красителя.
Для регенерации брома непосредственно в реакционной массе
можно использовать также нитробензол, сульфурилхлорид и неко-
торые другие реагенты. Следует однако отметить, что такая реге-
нерация брома безусловно выгодна только при сравнительно
небольшом объеме производства. Когда же мощность производ-
ства значительна, например в производстве бромаминовой кисло-
ты, технически более целесообразно выносить регенерацию брома
из бромистого водорода в самостоятельный производственный
цикл. Это значительно упрощает и улучшает технологию получе-
ния основного продукта.
Дибромвиолантрон получают бромированием виолантрона
бромом в хлорсульфоновой кислоте в присутствии катализатора
S2CI2 [77].
Реакцию иодирования практически применяют только для вве-
дения иода в молекулы нескольких готовых красителей.
ЛИТЕРАТУРА
1. Ворожцов Н. Н. Основы синтеза промежуточных продуктов и красите-
лей. Изд. 4-е. М., Госхимиздат, 1955, с. 179—230. 2. Uemura S., Sohma К.,
Okano М. e.a. — Bull. Chem. Soc. Jap., 1971, v. 44, № 9, p. 2490—2495. 3. McKil-
lop, Bromley D., Taylor E. C. — J. Org. Chem., 1972, v. 37, № 1, p. 88—92.
4. Erickson K. L„ Barowsky H. W.— Chem. Commun., 1971, № 24, p. 1596—1597.
5. Ischikawa N., Sekiya A. — Bull. Chem. Soc. Jap., 1974, v. 47, № 7, p. 1680—
1682. 6. Ингольд К. Теоретические основы органической химии. Изд. 2-е. Пер. с
англ./Под ред. И. П. Белецкой. М., Мир, 1973, с. 285—291. 7. Ворожцов Н. Н.,
мл. — В кн.: Химия синтетических красителей. Т. З/Под ред. К. Венкатарамана.
Пер. с англ, под ред. Л. С. Эфроса. Л., Химия, 1974. 8. Christen М., Zollin-
ger Н. — Helv. chim. acta, 1962, v. 45, № 6, p. 2057—2066; 2066—2077. 9. Володь-
кин А. А., Ершов В. В. — Изв. АН СССР; Сер. хим., 1962, № 11, с. 2022—2031;
Ершов В. В., Никифоров Г. А., Володькин А. А. — Пространственно-затруднен-
ные фенолы. М., Химия, 1972, с. 177. 10. Берлинер Ж.— В кн.: Современные
проблемы физической органической химии. Пер. с англ./Под ред. М. Е. Воль-
пина. М„ Мир, 1967.
191
11. Menzel P. Effenberger F. — Angew. Chem., 1972, B. 84, Ns 19, S. 954—955.
12. Kalachandra S., Ganesan R. — Curr. Sci. (India), 1972, v. 41, № 18, p. 677—
678. 13. Conselier J.-P.— Bull. Soc. chim. France, 1971, Ns 5, p. 1785—1788.
14. Каняев H. П., Шилов E. A. — ДАН СССР, 1939, т. 24, № 9, с. 891—893;
De la Mare P. B. D., Ketley A. D., Vernon C. A. — J. Chem. Soc., 1954, p. 1290—
1297. 15. Kovacic P., Lowery M. K-, Field K. — Chem. Rev., 1970, v. .70, № 6,
p. 639—665. 16. Физер Л., Физер M. Реагенты для органического синтеза, Т. 3.
М., Мир, 1970, с. 292; Brown Н. С. — Ind. Eng. Chem., 1944, v. 36, № 9, p. 785—
791. 17. Ballester M, Molinet C, Castaner J. — J. Amer. Chem. Soc., 1960, v. 82,
Ns 16, p. 4254—4258. 18. Cummings C. A., Millner D. J. — J. Chem. Soc. (C),
1971, p. 1571—1572. 19. Crockner H. P., Walster R. — J. Chem. Soc. (C), 1970,
№ 14, p. 1982—1986. 20. Пат. ФРГ 2337621, 1974; Chem. Abstr, 1974, v. 80,
145730.
21. Baird W. C. jr., Surridge I. H. — S. Org. Chem., 1970, v. 35, Ns 10,
p. 3436—3442. 22. Yaroslavsky C. — Tetrahedron Letters, 1974, Ns 38, p. 3395—3396.
23. Франц, пат. 2008869, 1970; Chem. Abstr, 1970, v. 73, 45091; Vorozhtscov N.N.
jr. — In: Aromatic Compounds/Ed. H. Zollinger, London, 1973, p. 324. 24. Берк-
ман Б. E. — Промышленный синтез хлорбензола. M, Госхимиздат, 1957. 25. Яп.
пат. 69—13133, 1969; Chem. Abstr, 1969, v. 71, 112599. 26. Пат. Нидерл. 6508531,
Chem. Abstr, 1966, v. 65, 551; пат. ВНР 154184, 1967, Chem. Abstr, 1968, v. 69,
27007. 27. Allen J. A.. Clark A. I. — Rev. Pure a. Appl. Chem, 1971, v. 21, Sept,
p. 145—166; Суворов Б. В., Букейханов H. Р. Окислительные реакции в органи-
ческом синтезе. М, Химия, 1978, с. 173—183. 28. Dorrepaal IP, Louw Р. — Rec.
trav. chim, 1971, v. 90, Ns 7, p. 700—704. 29. Коптюг В. А., Исаев И. С.,
Ерыкалов Ю. Г. и др. — ЖОрХ, 1965, т. 1, вып. 12, с. 2081—2083. 30. Коп-
тюг В. А. — Изомеризация ароматических соединений. Новосибирск, Изд-во СО
АН СССР, 1963. 176 с.
31. Пат. ФРГ 2326414, 1974; Chem. Abstr, 1975, v. 82, 155715. 32. Алиха-
нов П. П., Сакадынский К. И. — ЖОрХ, 1976, т. 12, вып. 9, с. 1931 —1935.
33. Меировиц И., Мазер И., Вальтер С. и др. — Изв. АН ЛатвССР, Сер. хим,
1970, № 5, с. 591—594. 34. Sumimoto Chem. Со. Авт. свид. СССР 285634, 1970.
35. Crocker Н. Р., Walser R. — J. Chem. Soc. (С), 1970, № 14, р. 1982—1986.
36. Пат. ЧССР 149803, 1973; Chem. Abstr, 1973, v. 79, 146197. 37. Зубарев С. В.
и др. Авт. свид. СССР 252319, 1970. 38. Симонов Е. Д., Бабинцева В. Н., Икри-
на М. А. и др. Авт. свид СССР 197547, 1967. 39. Шилов Е. А., Я сникав А. А. —
ЖФ.Х, 1954, т. 28, вып. 9, с. 1680—1681. 40. Пат. США 3890388, 1975; Chem.
Abstr, 1975, v. 83, 78827.
41. Islam А. М., Hassan Е. A., Hannout I. В. е.а. — J. Chem. (UAR), Cairo,
1970, v. 13, Ns 3, р. 297—308. 42. Динабург М. С. Светочувствительные диазо-
соединения и их применение. М. — Л, Химия, 1964. 43. Пат. ФРГ 2156285, 1973;
Chem. Abstr, 1973, v. 79, 31654. 44. Пат. ФРГ 2256167, 1974; Chem. Abstr, 1974,
v. 81, 37375. 45. Пат США 3423475, 1969; Chem. Abstr, 1969, v. 70, 87266.
46. Бабин E. П., Гейд Ю. П., Сергеев Е. В. и др. Авт. свид. СССР 363679, 1973.
47. Бутс Г. — В кн.: Химия синтетических красителей. Т. 5/Под ред. К. Венка-
тарамана. Пер. с англ, под ред. Л. С. Эфроса. Л, Химия, 1967. 48. Ayling Е. Е.—
J. Chem. Soc, 1929, Part I, p. 253—256. 49. Brintzinger H., Orth H. — Monatsh.
Chem, 1954, Bd. 85, S. 1017. 50. Fahey D. R.—J. Organometal. Chem, 1971,
v. 27, Ns 2, p. 283—292; Пат. США 3676421; Chem. Abstr, 1972, v. 77, 101118.
51. Доналдсон H. Химия и технология соединений нафталинового ряда.
Пер. с англ./Под ред. А. И. Королева. М, Госхимиздат. 1963. 52. Тилак Б. А.—
В кн.: Химия синтетических красителей. Т. 5/Под ред. К. Венкатарамана. Пер.
с англ, под ред. Л. С. Эфроса. Л, Химия, 1977. 53. Яп. пат. 7668554, 1976;
Chem. Abstr, 1976, v. 85, 94136. 54. Пат. США 2378745, 1945; Chem. Abstr, 1946,
v. 40, 365. 55. Фирц-Давид Г. Э., Бланже Л. — Основные процессы синтеза
красйтелей. Пер. с ием./Под ред. С. В. Богданова и И. В. Фодимана. М, ИЛ,
1957 . 56. Докунихин Н. С., Егорова Л. М. — Хим. наука и пром, 1957, т. 2,
Ns 1, с. 132. 57. Докунихин Н. С. — Журн. ВХО, 1976, т. 21, Ns 3, с. 292—298.
192
58. Пат. США 3378572, 1968; 59. Ельцов А. В., Эфрос Л. С., Студзинский О. Н.
Авт. свид. СССР 295746, 1971. 60. Галстян Г. А., Якоби В. А, Ристер Н. А.
и др. — ЖПХ, 1976, т. 49, № 6, с. 1330—1333.
61. Франц пат. 2114433, 1972; Chem. Abstr., 1973, v. 78, 59789. 62. Sar-
gent M. V., Smith D. — Tetrahedron Letters, 1970, № 24, p. 2065—2068. 63. Mon-
naim A. D., Wolf M. E., Saavedra L. e.a. — Tetrahedron Letters, 1973, № 17,
p. 1491—1494. 64. Шоенауер В., Бенгуерел Ф., Бенц Дж. — В кн.; Химия синте-
тических красителей. Т. 5./Под ред. К. Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса. Л., Химия, 1977. 65. Смит Л. — В кн.; Синтезы органических пре-
паратов. Сб. 2. Пер. с англ. М, ИЛ, 1949, с. 118—119. 66. Пат. ФРГ 2005259,
1971; Chem. Abstr., 1971, v. 75, 129523. 67. Пат. США 3894079, 1975; Chem.
Abstr., 1975, v. 83, 113992. 68. Hashem A. 1. — J. AppL Chem. a. Biotechnol., 1972,
v. 22, № 10, p. 1041—1042. 69. Пат. США 3833674, 1974; Chem. Abstr, 1974,
v. 81, 169275. 70. Николенко Л. H. Лабораторный практикум по промежуточным
продуктам и красителям. Изд. 2-е. М, Высшая школа, 1965.
71. Вильсон К. — В кн.: Синтезы органических препаратов. Сб. 4. Пер. с
англ. М, ИЛ, 1953, с. 300—301. 72. Франц, пат. 1432211, 1966; Chem. Abstr.,
1966, v. 65, 12149D. 73. Рашевская С. Т. и др. Авт. свид. СССР 196763, 1967.
74. Пат. США 3428659, 1969; Chem. Abstr, 1969, v. 70, 106269. 75. Пат. ФРГ
2303746, 1975; РЖ Хим., 1976, 10Н248П. 76. Роговик В. Н., Ставинчук В. Г.,
Денисова Е. В. Авт. свид. СССР 273192, 1970. 77. Плакидин В. Л., Мамот А. М.,
Дуйко Н. А. Авт. свид. СССР 408942, 1974.
Глава 6
ВВЕДЕНИЕ В АРОМАТИЧЕСКОЕ КОЛЬЦО
УГЛЕРОДСОДЕРЖАЩИХ ГРУПП
В этой главе рассмотрена обширная группа важных в
практическом отношении реакций электрофильного замещения, с
помощью которых в ароматические соединения вводят алифатиче-
ские цепи, получают ароматические спирты, альдегиды, кетоны и
кислоты. При всем разнообразии исходных веществ, условий про-
ведения и деталей механизмов в этих реакциях есть общая, объ-
единяющая черта; атакующим реагентом является карбкатион или
катионоидная частица, в которой имеется атом углерода с частич-
но незаполненной электронной оболочкой. Роль катализаторов при
этих реакциях в большинстве случаев состоит в том, что они со-
действуют образованию таких активных реагентов в концентра-
циях, достаточных для успешного протекания процесса.
Реакции введения углеродсодержащих групп в значительно
большей степени, чем рассмотренные ранее реакции сульфирова-
ния, нитрования и галогенирования, чувствительны к влиянию за-
местителей в субстрате. Электронодонорные заместители резко
облегчают их, в то время как сильные электроноакцепторные, на-
пример нитрогруппа, как правило полностью исключают возмож-
ность проведения процессов. Константа р этих реакций имеет от-
рицательный знак (см. 2.7.1).
7 Зак. 271
193
6.1. РЕАКЦИИ С ГАЛОГЕНАЛКИЛАМИ,
НЕПРЕДЕЛЬНЫМИ СОЕДИНЕНИЯМИ И СПИРТАМИ
6.1.1. Общие представления об алкилировании
Для алкилирования ароматических соединений могут
быть использованы галогеналкилы, этиленовые углеводороды и
спирты.
В первых двух случаях алкилирование ведут в присутствии
катализаторов Фриделя — Крафтса [1], представляющих собой
безводные галогениды ряда металлов, например, А1С13, А1Вг3,
FeCl3, существующие в виде димеров, а также BF3, TiCh, SnCK,
ZnCl2 и некоторые другие. Общим структурным признаком этих
солей является незаполненность электронной оболочки их метал-
лов; благодаря этому они очень активно координируются с соеди-
нениями, содержащими л-электроны или неподеленные электрон-
ные пары.
Наиболее активен из перечисленных катализаторов бромид
алюминия. Широкое применение в лабораторной практике и про-
мышленности имеет несколько менее активный безводный хлорид
алюминия, получаемый действием газообразного хлора на алюми-
ниевые стружки, и значительно менее активный безводный хлорид
цинка, который готовят обезвоживанием этой гигроскопической
соли при нагревании.
Взаимодействие галогеналкилов с хлоридом алюминия идет по
схеме:
RX + А1С13 ч=ь R-X.-.A1C13 ч=Ь R++A1C13X'
В настоящее время считают, что частицей, атакующей и алки-
лирующей ароматическое кольцо, является алкил-катион (свобод-
ный или в виде ионной пары), хотя пока еще не подтверждено,
может ли образование такого иона быть стадией, контролирующей
скорость алкилирования [2].
Тенденция галогенов к комплексообразованию с катализатора-
ми Фриделя — Крафтса падает с увеличением их атомного объема
от фтора к иоду, и галогеналкилы по алкилирующей способности
образуют ряд:
RF > RC1 > RBr > RI
Поэтому алкилиодиды для алкилирования обычно не применя-
ются. Состояние приведенного выше равновесия и концентрация
ионов или ионных пар зависят также от стабильности образую-
щихся карбкатионов, которая возрастает с увеличением степени
делокализации их положительного заряда в ряду:
CHJ < CH3CHj < (СН3)2СН+ < (СН3)3С+
Еще стабильнее ионы, положительный заряд которых делока-
лизуется в ароматических кольцах:
CsH5CHt < (С6Н5)2СН+ < (С6Н5)3С+
194
Стабильность трифенилметил-катиона так велика, что для об-
разования его из соответствующего хлорида достаточно действия
сольватирующего растворителя.
Образование карбкатионов из этиленовых углеводородов при
действии катализаторов Фриделя — Крафтса требует так называе-
мых сокатализаторов, которыми являются протонные кислоты, на-
пример НС1. Реакция может быть представлена следующим обра-
зом:
СН2 = СН2 + НС1 + А1С1з —> СН3СН^ + А1С17
При алкилировании ароматических соединений спиртами вме-
сто катализаторов Фриделя — Крафтса часто используют сильные
протонные кислоты, например фосфорную, пирофосфорную, сер-
ную, хлорсульфоновую, олеум. Карбкатионы из спирта и этих кис-
лот образуются путем следующих равновесных реакций:
ROH + HX ч=>: ROH^X’ R+ + H2O + X’
Дальнейшее взаимодействие карбкатионов с ароматическими
соединениями идет по общей схеме реакций электрофильного за-
мещения; с А1С13 это можно представить следующим образом
(см. 2.7.1):
Для получения конечного продукта алкилирования от образую-
щегося л-комплекса необходимо оторвать протон. Так как исход-
ный углеводород менее основен, чем продукт алкилирования, его
избыток выполнить это не может. Реакцию завершают, разлагая
получившийся комплекс водой. Таким образом, катализатор не
регенерируется, и его приходится брать в количестве, эквивалент-
ном количеству алкилируемого соединения.
Реакционная способность бензольного кольца при алкилирова-
нии по Фриделю — Крафтсу качественно подобна проявляемой при
сульфировании и нитровании. Так, толуол активнее бензола в
2,2 раза, этилбензол — в 1,8, а изопропилбензол — только в
1,4 раза. Хлорбензол алкилируется примерно в десять раз медлен-
нее бензола. Однако различие между активностью орто-, мета- и
пара-положений при этих реакциях выражено слабее (получается
больше ж-изомера), чем при других. Возможно, это объясняется
тем, что скорость реакции алкилирования более зависит от кон-
центрации карбкатиона, чем от скорости его взаимодействия с
субстратом [2].
Высказывается также предположение, что продукты лета-за-
мещения частично образуются в результате изомеризации перво-
начально получившихся соединений [3]. Поэтому во всех случаях,
7*
195
когда реакцию алкилирования проводят с участием А1С13, ее стре-
мятся вести при возможно более низкой температуре, так как на-
гревание может ускорить эти нежелательные процессы.
6.1.2. Изомеризация алкильных производных
Реакции изомеризации алкиларенов при сравнительно
невысоких температурах имеют внутримолекулярный характер, но
в более жестких условиях могут протекать и межмолекулярно.
В присутствии таких сильных кислот, как НС1 + А1С13, арома-
тические соединения образуют соли, катионы которых представля-
ют собой бензолониевые ионы (см. 2.7.1). В том случае, когда эти
ионы содержат алкильную группу при зр3-гибридизированном ато-
ме углерода, связь ее с ароматическим кольцом ослабевает и она
может внутримолекулярно переместиться к соседнему атому угле-
рода ароматического кольца. Например, взаимопревращения
п- (1) им- (2) ксилолов можно представить схемой:
Кинетические характеристики такого 1,2-сдвига алкильной
группы определяются степенью электронного дефицита в карбони-
евом центре аренониевого иона и поэтому хорошо коррелируются с
величинами химического сдвига в ЯМР-спектре 13С [4]. Для кси-
лолов в результате перемещений группы СНз устанавливается рав-
новесие между всеми тремя изомерами, преобладает наиболее
термодинамически устойчивый лг-изомер. В зависимости от темпе-
ратуры смесь содержит 60—52% л-ксилола, 24—23% п-ксилола и
16—25% о-ксилола [5, 6].
По-видимому, по такому же механизму идет изомеризация
ксилолов на алюмосиликатных катализаторах в паровой фазе при
500 °C — этот процесс имеет большое практическое значение для
получения дешевого ц-ксилола, используемого для окисления в
терефталевую кислоту (см. 17.1.1). п-Ксилол хорошо выделяется из
полученной смеси изомеров при охлаждении до —50 ч----70 °C, а
оставшуюся жидкую эвтектическую смесь вновь подвергают изо-
меризации на катализаторе для накопления п-изомера [7, с. 90].
В более жестких условиях реакция изомеризации алкилбензо-
лов приобретает межмолекулярный характер, в результате чего из
ксилолов могут получаться смеси, содержащие три-, тетра-, пента-
и даже гексаметилбензол, наряду с эквивалентными количествами
толуола и бензола. Этот процесс также используется в промыш-
ленности, в частности для получения бензола, дефицит которого
иногда ощущается [8].
19в
К таким же превращениям способны алкилнафталины. Напри-
мер, из 1-метилнафталина (3) образуется смесь 1- и 2-метилнаф-
талина (4) с преимущественным содержанием (4), как термодина-
мически более устойчивого. В более жестких условиях образуются
изомерные диметилнафталины, из которых более термодинамиче-
ски стабилен 2,6-диметилнафталин (5). Его применяют для окис-
ления в 2,6-нафталиндикарбоновую кислоту, используемую в син-
тезе термостойких полимеров.
СН3
6.1.3. Алкилирование углеводородов
Важнейшими продуктами, получаемыми реакцией алки-
лирования, являются этилбензол и изопропилбензол (кумол). Их
получают сотнями тысяч тонн при действии на бензол этилена или,
соответственно, пропилена в присутствии А1С13 и НС1.
С2н5
сн2=сн2
<--------
СН(СН8)2
СН2=СНСНз
--------->
(7)
Так как этилбензол (6) и кумол (7) алкилируются быстрее
бензола (см. 6.1.1), глубину алкилирования в этих процессах под-
держивают на уровне 10%. Тем не менее наряду с целевыми
продуктами образуются заметные количества диалкилпроизвод-
ных (преимущественно м- и п-изомеров), которые отделяются при
ректификации.
Алкилирование бензола ведут преимущественно в аппаратах
непрерывного действия. Они представляют собой эмалированные
или футерованные графитовой плиткой многосекционные колонны.
Три секции снабжены рубашками для отвода теплоты, основное
количество которой, однако, отводится с парами кипящего бензола.
Хлорид алюминия вводится в аппараты в виде заранее приготов-
ленного раствора, содержащего 10—12% А1С1з, 50—60% бензола и
25—30% соответствующего диалкилбензола, так как в бензоле
А1С1з растворяется плохо. Для образования хлористого водорода,
который является сокатализатором (см. 6.1.1), в раствор добав-
ляют воду (2% от массы А1С13). В нижнюю часть колонны через
распылитель подается алкен [9, с. 115].
Этилбензол используется главным образом для синтеза стиро-
ла. Из изопропилбензола получают а-метилстирол и фенол
(см. 17.2.3). Побочно образующиеся диэтилбензолы превращают
197
в дивинилбензолы, используемые в химии полимеров, а диизопро-
пилбензолы могут быть применены для синтеза гидрохинона и
резорцина (см. 17.2.3).
О прямом получении лг- и n-диизопропилбензолов из бензола
и пропилена см. [10].
Большое значение имеет получение алкилбензолов, содержа-
щих достаточно длинную и, по возможности, неразветвленную
алифатическую цепь. Бензол алкилируют продуктом, образую-
щимся при хлорировании керосина (монохлоркеросин, керилхло-
рид). Получающийся при этом продукт известен под названием
керилбензол; с бензольным кольцом связаны алкилы Сю—С^.
Для алкилирования могут быть использованы 1-алкены (а-олефи-
ны) с неразветвленной цепью, получаемые крекингом парафина
или синтезом на катализаторах Циглера. При алкилировании бен-
зола они образуют смесь втор-алкилбензолов. Длинноцепочечные
алкилбензолы служат промежуточными продуктами для синтеза
поверхностноактивных веществ типа сульфонолов, получаемых
сульфированием (см. 3.2.1).
В условиях, аналогичных алкилированию бензола, но в значи-
тельно меньших масштабах алкилируют толуол, нафталин и дру-
гие ароматические углеводороды [7].
Недостатком описанных процессов алкилирования является
безвозвратное расходование А1С1з, остающегося в промышленных
стоках после разложения водой реакционной массы. В связи с этим
ведутся поиски путей проведения реакций алкилирования в виде
гетерогенных процессов на поверхности твердого катализатора.
Заслуживают внимания гетерогенный катализ твердыми суперкис-
лотами; в качестве таких используют [11] хлорид и бромид алю-
миния, интерполированные на графите, и перфторированные поли-
мерные сульфокислоты (Nafion-H). Реакции с ними проводят в
псевдоожиженном слое при 125—210 °C. Интерполированные
А1С13 и А1Вг3 вначале дают хорошие результаты, но быстро теряют
активность, так как удаляются с графита. Значительно лучше и
стабильнее проявляет свою каталитическую активность полимер-
ная перфторированная сульфокислота. В качестве алкилирующих
агентов в этом случае можно использовать не только этиленовые
углеводороды, но и спирты.
6.1.4. С-Алкилирование фенолов
Фенолы легко вступают в реакции С-алкилирования при
действии алкенов и катализе сильными кислотами. Первоначально
считалось, что эти реакции проходят через промежуточное образо-
вание алкиловых эфиров фенолов, которые в дальнейшем пере-
группировываются с перемещением алкильной группы в аромати-
ческое кольцо:
С6Н5ОН —CSH5OR —> RC6H4OH
В настоящее время, однако, доказано, что основной путь этих
реакций — прямое алкилирование ароматического кольца, активи-
198
роьанйого сильным электронодойорным заместителем (гидро-
ксильной группой). Подобно сульфированию, алкилирование фе-
нолов — обратимый процесс, и это позволяет при необходимости
накопить в реакционной массе термодинамически более устойчивый
изомер. Так, при алкилировании фенола вначале, в условиях кине-
тического контроля, образуется смесь о- и «-изомера в близких
количествах:
ОН он он
R
Увеличение длительности реакции и некоторое повышение тем-
пературы позволяют приблизиться к равновесию, при котором пре-
обладает более стабильный п-изомер. Соответственно, из двух
образующихся диалкилфенолов 2,4-изомер термодинамически бо-
лее устойчив, чем 2,6-изомер.
В качестве катализатора при этой реакции чаще всего исполь-
зуют концентрированную серную кислоту в количестве 3—10%
(масс.). В зависимости от активности алкена реакцию ведут при
50—110 °C. После завершения процесса и нейтрализации кислоты
продукт отделяют и очищают перегонкой под вакуумом. В послед-
нее время в качестве катализатора стали использовать также
нерастворимую в воде катионообменную смолу КУ-2; она легко
отделяется от реакционной массы фильтрованием. При этом спосо-
бе, однако, требуется более длительное проведение реакции и более
высокая температура [7, с. 316].
В ' синтезе' полимеров широко используют п-трет-бутилфенол
(8), получаемый из фенола и изобутилена. 2,6-Ди-трет-бу-
тил-п-крезол(ионол), получаемый тем же способом из п-крезола
и изобутилена, применяется для стабилизации полимеров.
Продукты алкилирования фенола 2-метил-1-бутеном (изоами-
леном) (9) и диизобутиленом (10) могут быть использованы для
синтеза кислотных антрахиноновых красителей [12].
СНз НО——с—сн3 но— СНз (8) (t СНз НО— /"Д—С—СН2—С(СНз)з \=/ 1 СНз (10) СНз %—с—С2Н5 СНз 1)
199
n-Алкилфенолы, такие, как (10), и фенолы с более длинной
алкильной группой, например Ci2, применяют для получения
неионогенных ПАВ; для этого их подвергают реакции полиоксиэ-
тилирования по гидроксильной группе (см. 14.3.1).
Весьма интересно каталитическое действие фенолята алюминия
А1(ОСбН5)з, который избирательно направляет алкилирование фе-
нола в орто-положение, даже если лара-положение свободно. Так,
при алкилировании изобутиленом получаются о-трет-бутилфенол
(11) и 2,6-ди-трет-бутилфенол (12).
ОН
(СНз)зС
\Х/С(СНз)з
и
(И)
(12)
Механизм этой реакции до конца не выяснен, но можно пола-
гать, что первоначально алкилирование идет в орто-положение
одного из активированных бензольных колец фенолята алюминия;
затем это кольцо отщепляется в виде о-алкилфенола, вытесняемого
более кислым неалкилированным фенолом. Реакция имеет боль-
шое практическое значение для синтеза стерически затрудненных
фенолов, используемых в качестве стабилизаторов полимеров про-
тив термоокислительной деструкции [13].
6.1.5. C-Алкилирование аминов
При С-алкилировании аминов, в отличие от фенолов,
четко прослеживаются две стадии реакции: алкилирование по
аминогруппе (А-алкилирование) и перегруппировка с перемеще-
нием алкильной группы от азота в ароматическое кольцо (пере-
группировка Гофмана — Марциуса; о механизме см. [2, с. 745—
747]). Первая стадия идет в значительно более мягких условиях,
чем вторая.
Так, синтез п-бутиланилина (14), используемого для получения
антрахиноновых красителей, ведут в две стадии [14]. Вначале
получают А-бутиланилин (13): на анилин действуют бутилхлори-
дом или бутиловым спиртом в присутствии кислот или алкоголя-
тов. Выделив (13), его подвергают перегруппировке при нагрева-
нии до 200—250 °C в присутствии катализаторов ZnCl2 или СаС12
NH2 nhch2ch2ch2ch3 nh2
СН2СН2СН2СНз
(13) (14)
Следует отметить, что при перегруппировке не происходит изо«
меризации алкильной группы. Это, однако, не служит доказатель-
200
ством внутримолекулярного механизма миграции алкила, так как
может быть объяснено тем, что скорость превращения первичного
+ +
бутил-катиона во вторичный СН3СН2СН2СН2 —> СН3СН2СНСН3
меньше, чем скорость реакции алкилирования пара-положения.
Совершенно аналогично из о-толуидина и 1-бутанола получают
4-бутил-2-метиланилин, а из анилина и триизобутилена — и-доде-
циланилин. Эти продукты также могут быть использованы в син-
тезе красителей.
Для тех же целей из 2,4-ксилидина (2,4-диметиланилина), его
гидрохлорида и метанола при 270 °C под давлением получают
мезидин (2,4,6-триметиланилин) (15), а из 2,4-ксилидина и его
гидрохлорида в присутствии металлического алюминия при дей-
ствии этилена — 2,4-диметил-б-этиланилин (16) [14].
nh2 . nh2
СНз СНз
(15) (16)
В отличие от первичных, третичные амины могут алкилировать-
ся сразу в ароматическое кольцо. Так, при действии на
Д1,ДСдиметиланилин бис (п-диметиламинофенил) метанола (гидро-
ла Михлера) (17) в присутствии минеральной кислоты легко полу-
чается трис(п-диметиламинофенил)метан (18):
[(CH3)2N——]cHOH + f)—N(CH3)2 -+
(17)
—> £(ch3)2n——]сн
(18)
Окисление (18) дает краситель кристаллический фиолетовый.
Образующийся из (17) ион (19) очень стабилен, так как поло-
жительный заряд в нем сильно делокализован. Поэтому как алки-
лирующий реагент он очень мало активен. Чтобы увеличить его
реакционную способность, процесс ведут в среде концентрирован-
ной серной кислоты, которая протонирует диметиламиногруппы и
таким образом уменьшает делокализацию положительного заряда
(20).
(19)
(CH3)2NH——CH—\^/—NH(CH3)2
(20)
201
В этих условиях гидролом Михлера (17) удается алкилировать
многие малоактивные соединения. Например, из 2,7-нафталинди-
сульфокислоты получают соединение
окисление которого дает краситель кислотный зеленый Ж
[15, с. 275].
6.2. РЕАКЦИИ С АЛЬДЕГИДАМИ И КЕТОНАМИ
6.2.1. Получение ди- и триарилметановых производных
Для введения в ароматические соединения углеродсо-
держащих групп широко применяют альдегиды, кетоны и их про-
изводные. Таким образом могут быть синтезированы разнообраз-
ные производные диарил- и триарилметана. В качестве катализа-
торов при этом используют сильные кислоты; они протонируют
кислород карбонильной группы, увеличивая положительный заряд
на ее атоме углерода:
R—СН=О + Н+ [R—СН=6—Н ч-> R—СН—ОН]
Образующиеся катионы взаимодействуют по уже приведенной
схеме электрофильного замещения, и получаются соответствующие
ароматические спирты. Однако под влиянием того же катализато-
ра образующийся спирт (21) легко отщепляет гидроксил; возника-
ющий новый карбкатион (22) с большой скоростью реагирует еще
с одной молекулой ароматического соединения.
(22'
(23)
Взаимодействие #,#-диметиланилина с бензальдегидом идет в
присутствии каталитических количеств НС1 или ZnCl2. В результа-
те образуется бис(м-диметиламинофенил)фенилметан (24)—лей-
кооснование малахитового зеленого [15, с. 275]:
СН
2
(И)
202
Из фенола при действии на него ацетона получают, например
4,4/-изопропилидендифенол [2,2-бис(м-гидроксифенил) пропан; ди-
фенилолпропан; диан; бисфенол А] (25) [7];
СН3
С=О
I
СНз
СН3
СНз
(25)
Бисфенол А используется в больших количествах в производ-
стве эпоксидных смол.
Взаимодействие ароматических соединений с альдегидами и
кетонами полностью подчиняется уже рассмотренным закономер-
ностям реакций электрофильного ароматического замещения. Вли-
яние строения альдегида или кетона на его активность в этих
реакциях изучалось кинетическими методами [16]. Показано, что
реакция AJV-диметиланилина с бензальдегидом и его производны-
ми при катализе бензолсульфокислотой идет тем медленнее, чем
больше в бензальдегиде электронодонорность заместителя, кото-
рая уменьшается в ряду NO2 > Cl > Н > СН3 > ОСН3>
>N(CH3)2. Следовательно, чем выше электронный дефицит на
атоме углерода карбонильной группы, тем более активным элект-
рофилом является альдегид или кетон.
Кинетические исследования подтверждают также, что первая
стадия реакции — образование спирта (21)— идет значительно мед-
леннее второй, при которой получается продукт взаимодействия
(23). Это объясняется высокой реакционной способностью карб-
катиона (22), который быстрее, чем исходный альдегид или кетон,
взаимодействует со второй молекулой ароматического соединения.
Поэтому остановить реакцию на первом этапе в обычных условиях
не удается. Если, однако, в реакционную массу ввести какое-
нибудь соединение, способное реагировать с карбкатионом (22)
быстрее, чем исходное ароматическое соединение, то направление
реакции можно изменить. На этом приеме основано несколько
практически важных реакций, рассматриваемых ниже.
6.2.2. Реакция хлорметилирования
При действии формальдегида и хлористого водорода в
ароматическое соединение можно ввести одну или несколько хлор-
метильных групп [17, 18]:
АгН + НСНО + НС1 —> АгСН2С1 + Н2О
Реакцию обычно осуществляют, пропуская при небольшом на-
гревании НС1 в смесь ароматического соединения с раствором
формальдегида или триоксиметилена (параформальдегида) в при-
сутствии ZnCl2, H2SO4 и др. Вместо формальдегида или триокси-
метилена можно использовать дихлордиметиловый эфир
203
С1СН2ОСН2С1. С малоактивными соединениями реакцию прово-
дят в безводной среде в присутствии А1С1з.
При хлорметилировании бензола получается бензилхлорид (о
синтезе его хлорированием толуола см. 14.1.1). Механизм реакции
можно представить схемой [18]:
сн2о + Н+ СН2ОН
+ -н +
СвНв + сн2он ----► С6Н6СН2ОН
С8Н5СН2ОН 4- Н+ ч=> СбН5СН2 + Н2О
СбН5СН2 + СГ —> С8Н5СН2С1
Таким образом, успех реакции определяется тем, что бензил-
катион реагирует с хлорид-анионом быстрее, чем с избытком бен-
зола. Если в реакции использовать ароматическое соединение зна-
чительно более реакционноспособное, чем бензол, например анилин
или фенол, то вместо хлорметильных производных образуются
только производные диарилметана. Впрочем, и при реакции с бен-
золом побочно получается дифенилметан, количество которого воз-
растает с повышением температуры [7, с. 669].
Для хлорметилирования бензола обычно используют формалин
и в качестве катализатора ZnCl2. Последний вводят в виде кон-
центрированного раствора в соляной кислоте и во время реакции,
которую ведут при температуре не выше 50—60 °C, барботируют
НС1. Так как бензилхлорид алкилируется почти с такой же скоро-
стью, как и исходный бензол, при реакции образуется о- и, в
большей степени, n-бисхлорметильные производные. Чтобы умень-
шить образование этих соединений, глубину превращения взятого
в реакцию бензола поддерживают в пределах не более 20—25%.
В реакцию хлорметилирования можно вводить толуол и ксило-
лы [19]; получающиеся соединения могут быть использованы для
окисления в терефталевую, тримеллитовую и пиромеллитовую кис-
лоты (см. 17.1,1). Их применяют в синтезе термостойких полиме-
ров и волокон.
При хлорметилировании n-нитротолуола образуется 4-нит-
ро-2-(хлорметил)толуол (26) [20, с. 688]; при восстановлении из
него получается 3,4-ксилидин (3,4-диметиланилин) (27), приме-
няемый в синтезе витаминов.
(26) (27)
204
Хлорметилирование нафталина дает главным образом ^(хлор-
метил) нафталин (28), который при дальнейшем хлорметилирова-
нии образует смесь 1,4- (29) и 1,5-бисхлорметилнафталина (30)
[21]:
СН2С1 СН2С1 СН2С1
СН2С1 СН2С1
(28)
(29) (30)
Реакция хлорметилирования нашла также применение и в син-
тезе красителей. Фталоцианин меди вступает в эту реакцию при
действии дихлорметилового эфира или триоксиметилена в расплаве
А1С1з [22]. Позднее выяснилось, что эту реакцию можно вести,
используя триоксиметилен в смеси серной и хлорсульфоновой кис-
лот [23] или с дихлордиметиловым эфиром в растворе только
серной кислоты [24]. Этими методами можно ввести в молекулу
фталоцианина до восьми хлорметильных групп. Полученные про-
дукты перерабатывают в ценные красители. К реакции хлормети-
лирования способны многие кубовые красители и пигменты.
6.2.3. Получение альдегидов реакцией с формальдегидом
Некоторые труднодоступные другими методами альде-
гиды, например ванилин (4-гидрокси-З-метоксибензальдегид) (36)
и n-диметиламинобензальдегид, получают в промышленности при
совместном действии формальдегида и А^ААдиметил-п-нитрозоани-
лина на гваякол (о-метоксифенол) [25, с. 254] или, соответствен-
но, на АЦУ-диметиланилин [26] в минеральнокислой среде.
Считают [20], что эти реакции проходят таким образом, что,
например, в случае ванилина, вначале гваякол (31) реагирует с
формальдегидом, образуя соответствующий (ванилиловый) спирт
(32);
(31>
CH3Ov<
+ сн2о —> НО-(Л—СН2ОН
(32)
Образующийся из (32) ванилил-катион (33) захватывается
м-диметиламинофенилгидроксиламином (34), получающимся в ре-
акционной массе в результате восстановления формальдегидом
А^ААдиметил-и-нитрозоанилина; при этом образуется соответству-
ющий азометин (35); этот продукт может быть легко выделен из
реакционной массы. Заключительным этапом является гидролиз
205
(35) с образованием ванилина (36) и MAf-диметил-и-фенилендиа-
мина (37)
(35)
(36>
(37)
К рассмотренной реакции по механизму и области применения
близка реакция Даффа [27]. Она позволяет формилировать фено-
лы и JV.lV-диалкиланилины путем нагревания при 150—170 °C с
уротропином и борной кислотой в глицерине, с последующим кис-
лотным гидролизом получающегося азометина. Например, форми-
лирование фенола для синтеза о-гидроксибензальдегида (салицил-
альдегида) (38) можно представить схемой:
(38)
При реакции с фенолами преимущественно получаются о-изо-
меры. Однако, если орто-положения заняты, формилирование идет
в пара-положение. Побочно может идти метилирование и образо-
вание диальдегидов.
Реакция Даффа как общий метод синтеза гидроксиальдегидов
позволяет легко получать продукты более чистые, чем по методу
Раймера-Тимана (см. 6.3.2), однако выход не превышает 15—20%.
6.2.4. Аминометилирование и родственные реакции
Аминометилированием (реакцией Манниха) называют
взаимодействие формальдегида и амина, обычно вторичного, в
виде гидрохлорида, с соединениями, содержащими подвижные
атомы водорода; в результате один или несколько таких атомов
замещаются анчнометильной группой.
206
С ацетофеноном реакция Манниха проходит по метильной
группе:
СвН5СОСНз + сн2о + NHR3 —> C6H5COCH2CH2NR2 + Н2О
Промежуточным продуктом является М-гидроксиметильное
(метилольное) производное амина, образующееся при взаимодей-
ствии формальдегида с амином:
CH2O + HNR2 —> HOCHjNRj
В кислой среде оно превращается в карбкатион, стабилизиро-
ванный смещением неподеленной пары электронов азота:
HOCH2NR2 + Н+ —> Йн2-4ш2 + Н2О
Благодаря этой стабилизации такой карбкатион в реакциях
электрофильного ароматического замещения сравнительно мало
активен. В обычных условиях реакция Манниха хорошо идет толь-
ко с такими ароматическими соединениями как фуран, тиофен,
пиррол, некоторые производные пиразола, индол и другие гетеро-
циклы, а также фенолы бензольного, нафталинового и гетероцик-
лического ряда и некоторые подобные соединения [28].
Например с n-крезолом реакция проходит путем последова-
тельного вступления в орто-положения к фенольному гидроксилу
одной (39) и двух (40) аминометильных групп:
СП. CU СН.
(39)
(40)
В молекулу фенола могут вступить три таких группы.
С бензолом, нафталином, их гомологами и производными менее
активными, чем фенолы, реакция Манниха в обычных условиях не
идет. Благодаря этому до недавнего времени в синтезе промежу-
точных продуктов и красителей эту реакцию использовали сравни-
тельно редко [29].
Оказалось, однако, что если в реакцию вместо амина ввести
амид или имид карбоновой кислоты или сульфокислоты, например
хлорацетамид, бензамид, фталимид, бензолсульфамид или же лак-
тамы 2-пирролидон, 2-пиридон и др., то образующиеся из них
гидроксиметильные (метилольные) производные в концентриро-
ванной серной кислоте проявляют высокую реакционную способ-
ность; они амидометилируют даже сравнительно мало активные
соединения, например, производные антрахинона, а также соеди-
нения, содержащие нитрогруппу и др. [30].
?07
Так, М-гидроксиметил-2-пирролидон (42) в концентрированной
серной кислоте реагирует с п-нитротолуолом (41) по схеме:
Образующийся продукт (43) после восстановления нитрогруппы
используется в синтезе красителей [31].
М-Гидроксиметилбензамид (45) реагирует в тех же условиях с
4-нитровератролом (44); причем бензамидометильная группа всту-
пает в положение 5. Это доказывается тем, что продукт (46) после
восстановления при действии хлорокиси фосфора превращается в
производное хиназолина (47) [32].
Реакцией амидометилирования пользуются не только для полу-
чения промежуточных продуктов, но и для химической переработки
готовых красителей и пигментов, например производных антрахи-
нона [14], фталоцианинов меди и кобальта [33].
Многие М-гидроксиметильные производные амидов легко обра-
зуются при взаимодействии формальдегида и амида кислоты в
присутствии каталитических количеств КзСОз.'
C6H5CONH2 + СН2О —> C6H5CONHCH2OH
В ряде случаев эти соединения получаются непосредственно в
реакционной массе, куда добавляют амид и формалин. Иногда для
реакции вместо амида кислоты вводят нитрил [30].
В серной кислоте М-гидроксиметильные производные амидов
+
дают карбкатионы RCONHCH2. Положительный заряд в них ком-
пенсируется влиянием амидного азота лишь в очень небольшой
степени, особенно если учесть возможность протонирования карбо-
нильного кислорода в этих условиях. Этим и объясняется высокая
реакционная способность таких карбкатионов как амидоалкили-
рующих реагентов.
Другим родственным реагентом, с помощью которого можно
вводить в ароматические соединения аминометильную группу, яв-
ляется М-хлорметилфталимид (48). В реакции используются ката-
лизаторы Фриделя — Крафтса, обычно ZnCK или А1С1з [34]. Ами-
?08
нометилирование бензола, например, проходит следующим об-
разом:
(50)
От полученного А^-бензилфталимида (49) остаток фталевой
кислоты может быть отщеплен путем гидролиза, вначале в щелоч-
ной, а затем в кислой среде с образованием бензиламина (50).
Эта реакция также находит применение в синтезе фталоциани-
новых красителей [32], хотя препаративно она менее удобна, чем
с амидами.
Близкой по характеру является и реакция сульфометилирова-
ния. Она протекает при действии на ароматические соединения с
высокой реакционной способностью бисульфитного производного
формальдегида. С 2-нафтолом, например, реакция идет с образо-
ванием соли 2-гидрокси-1-нафтилметансульфокислоты (51) [20]:
+ HOCH2SO3Na
CH2SO3Na
(5П
6.3. РЕАКЦИИ С ПРОИЗВОДНЫМИ КАРБОНОВЫХ кислот
6.3.1. Общие представления о реакции ацилирования
Введение в ароматические соединения углеродсодержа-
щих групп осуществляется также с помощью карбоновых кислот и
их производных — галогенангидридов, ангидридов, эфиров, ами-
дов и нитрилов — применяемых в реакциях ацилирования. Их
сравнительная реакционная способность, так же как и реакционная
способность рассмотренных выше альдегидов (см. 6.2.1), зависит
от степени поляризации группы С=О (или C=N) и, следователь-
но, от величины положительного заряда, возникающего на ее атоме
углерода. Влияние других заместителей, связанных с этим атомом,
изменяющее величину его положительного заряда, различно.
В соответствии с этим по реакционной способности карбоновые
кислоты и их производные образуют ряд:
RCOHal > (RCO)aO > RCOOH > RCOOCH3 > RCOO'
209
Обычно реакции ацилирования ароматических соединений про-
водятся с использованием катализаторов Фриделя — Крафтса, ча-
ще всего А1С13, и этим напоминают уже рассмотренные реакции
алкилирования. Однако по своей реакционной способности галоген-
ангидриды кислот образуют ряд, обратный ряду галогеналкилов
(ср. 6.1.1):
RCOF < RCOC1 < RCOBr < RCOI
Это свидетельствует о том, что активация галогенангидридов
хлоридом алюминия осуществляется по иному механизму, чем
активация галогеналкилов. Он, очевидно, состоит в присоединении
хлорида алюминия к карбонильному кислороду с последующим
отщеплением галогена и образованием ионной пары:
R—С=О + А1С13 R—С=О—A1C1J :?=£ R—СО+|| А1С17
I I
Cl С1
Кинетическими исследованиями установлено, что именно ад-
дукт А1С1з с хлорангидридом кислоты в биполярной или ионной
форме, а не ацил-катион, получающийся при диссоциации ионной
пары, образует о-комплекс при реакции ароматического ацилиро-
вания. Вместе с тем, сходство в кинетике и строении продуктов,
образующихся при реакции ацетилирования по Фриделю — Крафт-
су и при той же реакции с использованием заранее приготовлен-
ных ацетилгексафторантимоната и ацетилгексахлорантимоната
CH3CO+||SbF6 и СН3СО+|| SbCK [2, с. 304—307], позволяют утвер-
ждать, что этот аддукт имеет строение ионной пары.
Дальнейшее взаимодействие аддукта с ароматическим соеди-
нением идет по уже рассмотренной для реакции алкилирования
схеме (см. 6.1.1).
Однако между течением реакций алкилирования и ацилирова-
ния по Фриделю — Крафтсу наблюдаются и существенные разли-
чия. Они прежде всего определяются значительно меньшей ак-
тивностью ацилирующего реагента, благодаря чему реакция с
ним заметно селективнее и чувствительнее к влиянию имеющихся
заместителей. Так, если толуол при алкилировании вступает в
реакцию со скоростью в 5—6 раз большей, чем бензол, то скорости
ацилирования этих аренов различаются в ПО—150 раз. Константа
реакции р в уравнении Гаммета для алкилирования обычно имеет
величину от —2 до —3, а для ацилирования она достигает вели-
чины —9 и более (см. 2.4.1). Это свидетельствует о более позднем
переходном состоянии при ацилировании (см. 2.7.1).
В результате ацилирования в ароматическое кольцо вступает
остаток с карбонильной группой, являющейся сильным электроно-
акцептором. Поэтому реакция ацилирования отличается четкой
стадийностью. Основность карбонильной группы настолько велика,
31Q
что введенный в реакцию катализатор Всегда остается связанным
с этой группой:
C6He + R—С=О—A1C1J —► С5Н5—С=б—A1C1J + НС1
I I
Cl R
Продукт реакции обычно затем выделяется при разложении
реакционной массы водой. Благодаря этому для осуществления
реакции необходимо не менее чем стехиометрическое количество
катализатора, и реакция суммарно имеет третий порядок:
p = fe[RCOCl][C6H6][AlCJ3]
При ацетилировании бензола заранее приготовленными аце-
тилгексафторантимонатом и ацетилгексахлорантимонатом наблю-
дается небольшой кинетический изотопный эффект: скорость реак-
ции с гексадейтеробензолом в 2,2 раза меньше, чем с бензолом. Из
этого делается вывод [35], что скорость реакции может частично
определяться стадией отщепления протона от о-комплекса.
Реакцию ацилирования обычно проводят в среде органического
растворителя; чаще всего это избыток ароматического соединения
или же другое соединение, обладающее меньшей реакционной
способностью, чем вводимое в реакцию ацилирования. Безводный
А1С13 сильно гигроскопичен и полностью теряет активность, связы-
ваясь с водой. Поэтому все реагенты и растворители перед исполь-
зованием сушат, а для защиты от влаги воздуха все воздушные
линии аппарата должны быть снабжены поглотителями. Контроль
процесса обычно ведут по скорости выделения НС1, для этого его
поглощают щелочью. После завершения реакции массу выливают в
подкисленную воду, чтобы соли алюминия остались в растворе.
Органический слой отделяют и целевой продукт выделяют из него,
отгоняя растворитель с водяным паром или же, после осушивания,
фракционной вакуум-перегонкой.
6.3.2. Получение кетонов
В промышленности промежуточных продуктов метод
ацилирования широко используется для синтеза жирноароматиче-
ских и ароматических кетонов. Так, из бензола и ацетилхлорида
получается ацетофенон:
С5Н6 + СН3СОС1 —> С5Н5СОСН3 + НС1
Вместо ацетилхлорида можно применять уксусный ангидрид:
С5Н5 + (СН3СО)2О —> CSH5COCH3 + СНзСООН
В этом случае, однако, необходимо брать 2 моля катализатора,
так как образующаяся уксусная кислота безвозвратно потребляет
один его моль на солеобразование.
Аналогично получают пропиофенон и кетоны с более длинной
цепью.
211
В реакцию с ароматическими соединениями можно вводить
также ангидриды двухосновных кислот [36], например янтарный,
малеиновый и др.
Н2(Х \
свнв + । ;о —> с6н5сосн2сн2соон
Н2С\ /
с
Таким способом получают ароматические оксокарбоновые кис-
лоты (кетокислоты), которые часто используют для последующей
циклизации.
При взаимодействии бензола и бензоилхлорида получается
бензофенон:
С6Нв + С6Н5СОС1 СвН5СОСвН5 + НС1
Его можно синтезировать из бензола и карбонилхлорида (фос-
гена) в присутствии А1С13. Реакцию целесообразно вести под дав-
лением:
2СвН6 + СОС12 СвН5СОСвН5 + 2НС1
Практически бензофенон удобнее получать из бензола и четы-
реххлористого углерода [37].
Для ацилирования по Фриделю — Крафтсу в промышленности
часто используют фталевый ангидрид. При взаимодействии с бен-
золом, хлорбензолом, толуолом и другими продуктами он образует
ряд производных бензофенона, содержащих карбоксильную груп-
пу в орто-положении — получаются различные о-бензоилбензой-
ные кислоты (52)-
О О
(R = H, СНз, С1)
Катализатор AICI3 в эти реакции вводится из расчета 2—
2,5 моль на 1 моль фталевого ангидрида. Растворителем служит
избыток ацилируемого соединения [20, с. 714].
Из соединений типа (52) при циклизации под действием
100%-ной H2SO4 легко получаются антрахинон и ряд его производ-
ных, важных для синтеза красителей.
Сравнительно меньшее значение имеют аналогичные продукты,
получающиеся из бензола и его производных и диангидрида пиро-
212
меллитовой (1,2,4,5-бензолтетракарбоновой) кислоты (53), став-
шего теперь вполне доступным продуктом.
(53) (54)
Циклизация образующихся соединений типа 2,5-дибензоилте-
рефталевой кислоты (54) приводит к получению производных пен-
тацендихинона [38, с. 409], еще не получивших практического
применения.
Практически интересный продукт — 4-ацетилбифенил (55) об-
разуется при ацетилировании бифенила ацетилхлоридом в нитро-
бензоле в присутствии А1С1з. Его используют для окисления в
4-бифенилкарбоновую кислоту. Аналогично из 4-нитробифенила
получают 4-ацетил-4'-нитробифенил (56) [39], при окислении обра-
зующий 4'-нитро-4-бифенилкарбоновую кислоту.
СОСН3
(55)
—СО СН3
(56)
Таким образом, нитрогруппа в соседнем кольце не препятствует
течению реакции ацетилирования.
Ацетилирование нафталина всегда приводит к смеси 1- и
2-ацетильных производных. Соотношение изомеров более всего за-
висит от примененного растворителя, но также и от температуры и
количества введенных А1С13 и ацетилхлорида. Например, в дихлор-
этане выход 1-ацетилнафталина (57) составляет 98%, в то время
как в нитробензоле получается до 70% 2-ацетилнафталина (58).
Если реакцию в дихлорэтане вести с избытком ацетилхлорида
(2 моль на 1 моль А1С1з, то образуется (58) с выходом 60%
[40, с. 536—539].
СОСНз
(57* (58)
Единого объяснения этим наблюдениям нет. Вероятно различ-
ная ориентация при реакции связана с изменяющимся строением
атакующей частицы (см. также 2.6).
Бензоилирование нафталина ведут действием бензоилхлорида в
присутствии А1С13 обычно в среде полихлорбензолов. Реакция идет
213
В «-Положения, Но стадиям — вначале образуется 1-бензоилнафта-
лин (59), который несколько медленнее реагирует еще с одним
молем бензоилхлорида, образуя смесь, содержащую около 80%
1,5-(60) и 20% 1,8-дибензоилнафталина (61), Побочный продукт
(61) легко отделяется от (60) обработкой хлорбензолом.
СОСбН5 СбН5СО СОСбН6
СОСбН5
(60) (61)
Из этих кетонов практически важны (59) и (60). Из первого при
циклизации получается бензантрон, а из второго—дибензпирен-
хинон.
Из аценафтена при действии ацетилхлорида образуется 5-аце-
тилаценафтен (62), служащий главным образом для окисления
в 1,4,5-нафталинтрикарбоновую кислоту [41, с.191]. При бензоили-
ровании получается 5-бензоилаценафтен (63), используемый для
синтеза кубовых красителей.
(62)
СОСбНб
(63)
Ароматические соединения, содержащие сильные электронодо-
норные заместители, могут вступать в реакцию ацилирования и без
А1С13, иногда в очень мягких условиях. Так, 4,4/-бисдиметиламино-
бензофенон (кетон Михлера) (65) получается из /У,ЛСдиметилани-
лина и карбонилхлорида (фосгена) в две стадии:
(CH3)2N—^2/+СОС12 —> (CH3)2N——СОС1+НС1
164)
(CH3)2V 0-СОС1 + —N(CH3)2 —>
—> (CH3)2N —СО——N(CH3)2 + НС1
,65)
Первая стадия — образование 4-диметиламинобензоилхлорида
(64) идет уже при комнатной температуре и не нуждается в ката-
214
лизаторах. Вторая стадия требует несколько более жестких усло-
вий— нагревания до 100 °C в автоклаве и прибавления плавленого
ZnCU [20, с. 705]. В таких же условиях получают 4,4'-бисдиэтил-
аминобензофенон.
Резорцин ацилируется в положение 4 при нагревании с безвод-
ными алифатическими кислотами до 125—135 °C в присутствии
ZnCl2:
НО. , НО.
СНз(СН2)5СООН + —ОН —> СН3(СН2)5СО—(Л—он
В аналогичных условиях реагируют флороглюцин и пирогал-
лол.
Ацилирование фенолов ароматическими кислотами обычно про-
водят нагреванием в концентрированной серной кислоте.
6.3.3. Реакция формилирования
Использование муравьиной кислоты и ряда ее произ-
водных для реакции ацилирования позволяет вводить в аромати-
ческие соединения формильную группу и получать альдегиды.
Однако применить для этой цели хлорангидрид муравьиной
кислоты (формилхлорид) невозможно, так как он нестоек и сразу
же разлагается по реакции:
НСОС1 —> СО4-НС1
Поэтому Гаттерман предложил получать альдегиды, пропуская
в смесь ароматического соединения и А1С1з безводные окись угле-
рода и хлористый водород:
С6Нв + СО+НС1 —> СвН5СНО + НС1
При реакции действительно образуется бензальдегид, однако с
ничтожным выходом. Использование дополнительного катализато-
ра— хлорида меди(1)—позволило значительно увеличить при
этой реакции выход многих альдегидов (реакция Гаттермана —
Коха, см. [42, 43]), но не бензальдегида. Основная причина —
нерастворимость А1С13 в бензоле. Это препятствие удалось преодо-
леть, используя вместо хлорида алюминия его бромид или при-
меняя в качестве растворителя нитробензол. Смесь окиси углерода
с хлористым водородом получают, приливая хлорсульфоновую
кислоту к муравьиной кислоте:
CISO3H4-HCOOH —> H2SO4 + CO + HC1
Техническое решение задачи было найдено проведением реак-
ции арена с СО и НС1 под давлением. В автоклаве при 6—7 МПа и
?15
20—60 °C формилирование бензола, его гомологов, производных
нафталина и более сложных соединений идет, как правило, с
выходом не менее 90% от теоретического. При этом необязательно
применять сокатализаторы. В реакцию не вступают фенолы. Из
побочных процессов наблюдается изомеризация с перемещением
алкильных групп, очевидно благодаря каталитическому действию
А1С1з + НС1 (см. 6.1.2). Реакция является важным техническим
методом получения ароматических альдегидов.
Высокий выход продуктов в описанных условиях объясняется
тем, что давление смещает равновесие СО 4- НС1 4=^ НСОС1
вправо. Формилхлорид, как и другие хлорангидриды кислот
(см. 6.3.1), взаимодействует с хлористым алюминием, образуя
ионную пару НС=О+1| AICU, которая и является активным реаген-
том. Благоприятное влияние добавки CuCl, вероятно, объясняется
тем, что при этом увеличивается концентрация формил-катионов за
счет реакции СиСО+ с протоном.
Введение альдегидной группы в фенолы и их эфиры удается при
помощи реакции, также предложенной Гаттерманом [44, 45], — в
ней используются безводные цианистый водород и хлористый во-
дород с катализаторами Фриделя — Крафтса. Например, при об-
работке фенола HCN и НС1 в эфире в присутствии ZnCK из
раствора выделяется гидрохлорид л-гидроксибензилиденамина, ко-
торый далее гидролизуют водной кислотой и получают п-гидро-
ксибензальдегид:
Эфиры фенолов и некоторые другие фенолы реагируют в этих
условиях с трудом. Поэтому для реакции с ними вместо
ZnCl2 пользуются AICI3.
Считают, что при этой реакции НС1 присоединяется к HCN с
образованием формимидоилхлорида (66); последний реагирует еще
с одной молекулой HCN и полученный продукт (67) дает с А1С13
комплекс (68), взаимодействующий с ароматическим соедине-
нием; образуется сложный имин (69), который далее гидролизуют
до альдегида:
+ HC=N AICI3
HC==N + HC1 —> C1CH=NH ---------> C1CH=NCH=NH ------>•
(66) (67)
+ ArH +H2O
—> C1CH=NCH=NH • А1С13 -------> ArCH=NCH=NH • AICI3 ------->
—HC1
(68) (69)
—> ArCH=O + 2NH3 + HCOOH
216
Вместо HCN можно использовать Zn(CN)2. Необходимый реа-
гент при этом образуется в реакционной массе при пропускании
НС1. При таком проведении реакции выход альдегида несколько
возрастает.
Ценным методом, позволяющим вводить в ароматические со-
единения альдегидную группу, является реакция Вильсмайера [46;
47, с. 798]. В качестве реагентов используются полностью заме-
щенные амиды муравьиной кислоты, чаще всего М.АГдиметилформ-
амид и АГметил-АГ-фенилформамид, а в качестве катализатора,
активирующего эти соединения — хлорокись фосфора, реже —
карбонилхлорид (фосген). Катализатор взаимодействует с ами-
дом, образуя карбкатион (70), который, в свою очередь, реагирует
с ароматическим соединением.
(CH3)2NCH=O + РОС13 —*- (СН3)2Ы-СН—о—РС13О“—
—*• (СН3У,Н—СИ—о—РОС12 + сг
(то)
АгН + СИ—О—РОСЫ- сг —> Ar—СН—О—РОС12 + НС1
I I
N(CH3)2 N(CH3)2
(70) (71)
Гидролиз продукта (71) приводит к получению альдегида, ди-
метиламина, соляной и фосфорной кислот. Карбкатион (70) срав-
нительно мало активен, так как его эффективный положительный
заряд компенсируется неподеленной парой электронов соседнего
атома азота. По реакционной способности этот ион близок к обра-
зующемуся при реакции аминометилирования (см. 6.2.4).
С помощью реакции Вильсмайера, однако, нельзя ввести аль-
дегидную группу в бензол, нафталин, но антрацен легко формили-
руется за счет лтезо-положения. В реакцию вступают также произ-
водные бензола с сильными электронодонорными заместителями;
например, А^АГдиметиланилин образует по этой реакции
п-диметиламинобензальдегид с выходом 80—84% [48]. По этому
методу формилируются также многие гетероциклические соедине-
ния — фуран, тиофен, некоторые пиразолы, а также соединения с
подвижными атомами водорода.
Интересно отметить, что при осторожном гидролизе первичных
продуктов типа (71), образующихся при реакции Вильсмайера, в
ряде случаев удается остановиться на стадии получения достаточ-
но устойчивых диметиламинометиленовых производных [49]. Так,
исходя из 3-метил-1-фенил-5-пиразолона, можно получить димети-
ламинометиленовое производное (72), которое в кислой среде гид-
ролизуется далее доо-гидроксиальдегида (74) через промежуточное
217
образование таутомера (73) с гидроксиметиленовой группой:
н3с н3с СН—N(CH3)2
N N
С5Н5 С5Н5
(72)
Н3С СН—он
I
свн5
(73)
н3с сно
N
I
свн5
(74)
При низких температурах приведенное равновесие смещено в
сторону альдегидной группы (74) [50].
При реакции с М-замещенными сукцинимидами с хорошим
выходом образуются 3,4-диформил-2,5-дихлорпирролы [51]:
Несколько особняком в рассматриваемой группе реакций стоит
синтез Раймера — Тимана, с помощью которого из фенолов и хло-
роформа в водно-щелочной среде получаются гидроксиальдегиды
преимущественно орто-ряда [52]. В этом случае первичное взаи-
модействие хлороформа со щелочью приводит к образованию ди-
хлоркарбена:
НСС1з + ОН' —> н2о + сг + * СС12
Дихлоркарбен, подобно рассмотренным выше карбкатионам,
содержит атом углерода с координативно-незаполненной элект-
ронной оболочкой (6 электронов), но, в отличие от карбкатионов,
не несет заряда. Взаимодействие дихлоркарбена с фенолят-ионом,
очевидно, проходит по схеме:
О” О О’ О’
Хотя в этой реакции выход альдегидов обычно менее 50% от
теоретического, она находит практическое применение, так как
218
очень проста в осуществлении. Особенно гладко и со значительно
лучшими выходами она идет с 2-нафтолом и соответствующими
сульфо- и дисульфокислотами, например:
HO3S
HO3S
СНО
6.3.4. Реакция карбоксилирования фенолов
Угольный ангидрид — очень слабый электрофил, не спо-
собный реагировать с ароматическими углеводородами. Однако
реакционная способность ароматического кольца фенолов в виде
фенолятов настолько велика, что их можно ввести в реакцию с СО2,
в результате которой образуются практически очень важные гид-
роксиаренкарбоновые кислоты (реакция Кольбе — Шмитта).
Например, для получения салициловой (о-гидроксибензойной)
кислоты безводный фенолят натрия нагревают в автоклаве при
180 °C и вводят туда под давлением СО2. Реакцию считают закон-
ченной, когда прекращается поглощение СО2 [20]. При этом с
хорошим выходом образуется натриевая соль салициловой кисло-
ты. В аналогичных условиях из фенолята калия, а также из фено-
лятов рубидия и цезия получаются калиевая, рубидиевая или
цезиевая соли n-гидроксибензойной кислоты.
Влияние катиона металла на направление реакции карбоксили-
рования в настоящее время объясняют следующим образом. Фено-
лят натрия образует о-комплекс, стабилизированный образованием
хелатной связи через натрий; этот о-комплекс далее обычным
путем превращается в конечный продукт реакции:
ON а
+ С°2
Калий и более тяжелые металлы — несравненно более слабые
хелатообразователи, чем натрий. Поэтому о-комплексы фенолятов
этих металлов за счет орто-положения менее стабильны, чем в
случае фенолята натрия, и атака СО2 идет в более активированное
гидроксильной группой лара-положение;
219
Оказалось также, что безводный салицилат калия при нагрева-
нии в открытом сосуде при 220 °C образует калиевую фенолят-соль
n-гидроксибензойной кислоты и фенол:
ОН ОК ОН
COOK
Так же ведут себя салицилаты рубидия и таллия. Со значи-
тельно лучшим выходом, достигающим при 250°C 90—93%, идет
перегруппировка калиевой фенолят-соли салициловой кислоты в
фенолят-соль n-гидроксибензойной кислоты. Эту реакцию исполь-
зуют как промышленный метод получения п-гидроксибензойной
кислоты. Фенол при изомеризации не образуется [5, с. 153].
Интересно, что при нагревании мононатриевой соли п-гидро-
ксибензойной кислоты до 280—290°C она частично, с образованием
фенола, превращается в фенолят-соль салициловой кислоты:
Таким образом, изомеризация гидроксибензойных кислот обра-
тима и направление ее зависит только от свойств взятого катиона.
Аналогичные явления наблюдаются и в случае карбоксилиро-
вания 1- и 2-нафтолов.
Карбоксилирование безводного 1-нафтолята натрия при 120—
140 °C под небольшим давлением СОг приводит к получению с
выходом 82—83% практически важной 1-гидрокси-2-нафтойной
кислоты (75) [40], используемой при получении реактивов для
цветной фотографии:
При карбоксилировании 2-нафтолятов в зависимости от метал-
ла и температурного режима процесса карбоксильная группа мо-
жет вводиться в положения 1, 3 и 6 с образованием солей, соответ»
220
ствующих гидроксинафтойных (нафтолкарбоновых) кислот. Так,
2-гидрокси-1-нафтойная кислота (76) с наилучшим выходом,
достигающим 87% от теоретического, получается при карбоксили-
ровании 2-нафтолята калия (а не натрия, как можно было бы
ожидать) при 130 °C. Проведение той же реакции при 230—240 °C
дает с 50%-ным выходом 6-гидрокси-2-нафтойную кислоту (77),
содержащую 12% примеси З-гидрокси-2-нафтойной кислоты (78):
COOK
(76)
Технически наиболее важную кислоту (78) получают с выходом
72% (с учетом регенерации взятого в реакцию 2-нафтола) путем
карбоксилирования строго безводного 2-нафтолята натрия при
260 °C [40].
Так как реакционная масса получается очень вязкой, автоклав
снабжают мощной пятилопастной мешалкой, а между лопастями
устанавливают неподвижные пластины. Нагрев паром осущест-
вляется с помощью змеевиков, расположенных снаружи. В аппа-
рат загружают нагретый до 100 °C 50%-ный раствор едкого натра
(840 кг), затем 3000 кг расплавленного 2-нафтола. Аппарат закры-
вают и нагревают до 182 °C, после чего давление медленно спуска-
ют. При этом температура несколько понижается несмотря на
непрерывное нагревание. Высушивание продолжают при нагрева-
нии и удалении пара вакуум-насосом, пока температура не подни-
мется до 195 °C, а давление снизится до 40 мм рт. ст. Последние
следы влаги удаляют при 220—230 °C и остаточном давлении 15 мм
рт. ст. Далее вводят СО2 с такой скоростью, чтобы за 2 ч давление
поднялось до 0,45 МПа и поддерживалось на этом уровне 8—10 ч;
температура в аппарате 260 °C. Затем давление спускают и в ваку-
уме при остаточном давлении 15 мм рт. ст. и 230 °C отгоняют около
800 кг 2-нафтола. Операции карбоксилирования и последующей
отгонки 2-нафтола повторяют еще два раза, получая еще 310 и
150 кг регенерированного 2-нафтола. Далее реакционную массу
растворяют в воде и передавливают в аппарат для нейтрализации.
После дополнительного разбавления раствора до плотности 1,045—
1,052 его осторожно нейтрализуют соляной кислотой для осажде-
ния 2-нафтола, доводят объем раствора до 40 000 л (pH 6,8) и
отделяют осадок на фильтр-прессе. Раствор З-гидрокси-2-нафтой-
ной кислоты подкисляют соляной кислотой и центрифугируют,
221
после чего сушат в тарельчатой сушилке. Выход продукта —
1392 кг. В нем содержится 1,2% 2-нафтола.
Гидроксинафтойные кислоты способны изомеризоваться. Так,
натриевая фенолят-соль З-гидрокси-2-нафтойной кислоты может
быть получена нагреванием безводной натриевой фенолят-соли
2-гидрокси-1-нафтойной кислоты при 240 °C:
COONa
ONa
COONa
Изучение механизма изомеризации гидроксиаренкарбоновых
кислот показывает, что такие реакции имеют межмолекулярный
характер.
Обычно считают, что миграция карбоксилатной группы в солях
таких кислот является результатом предварительного распада соли
на фенолят и угольный ангидрид. Повторное их взаимодействие
приводит к образованию устойчивой в условиях реакции изомерной
гидроксиаренкарбоновой кислоты:
Образование СО2 при изомеризации убедительно доказано
многими методами, в том числе и с помощью меченого углерода в
СО2, который в дальнейшем оказывается в продуктах реакции. Тем
не менее, в открытых сосудах изомеризация проходит гладко, без
уменьшения выхода продуктов. Это явление объясняют большой
скоростью изомеризации, благодаря которой СО2 не успевает вый-
ти из реакционной массы. Если, однако, реакцию вести под глубо-
ким разрежением, то значительная часть СО2 может быть уловлена
[5, с. 153].
Рассматриваемый механизм не объясняет, почему при исполь-
зовании фенолят-солей гидроксиаренкарбоновых кислот выход
продуктов, получаемых при изомеризации, значительно возра-
стает.
Более вероятным поэтому представляется следующий меха-
низм. Декарбоксилирование фенолят-солей приводит к образова-
нию дианионов с карбанионным центром в ароматическом кольце,
который может менять свое положение с соответствующим пере-
мещением атомов водорода. Например, в случае изомеризации
фенолят-соли салициловой кислоты такой дианион (79) превра-
222
щается в дианион (80). После захвата СОг образуется конечный
продукт изомеризации.
(79) (80)
Такой механизм изомеризации в случае однометаллических
солей гидроксиаренкарбоновых кислот может реализоваться лишь
частично, поскольку обладающий очень высокой основностью кар-
банионный центр должен быстро нейтрализоваться водородом фе-
нольного гидроксила. Подобным же действием обладает водород
воды, если она присутствует в реакционной массе. Именно поэтому
для успеха реакции воду необходимо тщательно удалять.
Представленные выше анионы в действительности существуют в
виде ионных пар с катионами металлов; последние благодаря
этому и могут оказывать свое влияние на направление реакции
изомеризации.
Карбоксилированию подвергаются и более сложные фенолы,
например 2-антрол, из которого получают З-гидрокси-2-антрацен-
карбоновую кислоту (81). Этим же способом можно получать
З-гидрокси-2-дифениленоксидкарбоновую (82) и З-гидрокси-2-кар-
базолкарбоновую кислоту (83).
(81)
ОН
СООН
ОН
СООН
ОН
СООН
I
н
(83)
В отличие от перечисленных соединений резорцин карбоксили-
руется в очень мягких условиях при пропускании СОг в его водно-
щелочной раствор. При этом с хорошим выходом образуется
2,4-дигидроксибензойная (резорциловая) кислота (84):
СООН
ОН
(84)
Кроме фенолов в эту реакцию с успехом можно ввести и ами-
нофенолы. Среди получаемых из них продуктов наиболее важна
223
n-аминосалициловая кислота (85), образующаяся при карбоксили-
ровании л-аминофенола:
(85)
Под названием ПАСК ее в больших количествах используют
как лекарственное вещество.
Амины, преимущественно в виде А-ацетильных производных,
также могут вступать в реакцию карбоксилирования [53]. Однако
выход продуктов этой реакции невелик, и она пока что практиче-
ского применения не получила.
6.3.5. Диспропорционирование и изомеризация солей
карбоновых кислот
Впервые еще в 1873 г. было открыто необычное превра-
щение бензоата калия при нагревании до 400 °C в смесь солей
бензолдикарбоновых кислот и бензол. Лишь спустя 80 лет эта
реакция привлекла к себе пристальное внимание многих исследо-
вателей, так как выяснилось, что с ее помощью можно получать
терефталевую кислоту исходя из толуола, производство которого в
ряде стран превышает потребность [5, с. 158; 54].
Превращение бензоата калия в терефталат (86) идет по урав-
нению:
Реакцию ведут при нагревании до 350—450 °C под давлением
СО2, в присутствии ряда катализаторов, среди которых, по-види-
мому, наиболее эффективен иодид кадмия. Выход терефталевой
кислоты и бензола близок к теоретическому [55].
Интересно влияние катионов взятых солей на протекание реак-
ции. Бензоат лития диспропорционируется труднее, чем бензоат
калия, а бензоаты рубидия и цезия — заметно легче. Бензоаты
стронция и бария образуют при реакции соответствующие соли
фталевой (87), а не терефталевой кислоты. Бензоат кальция дает
смесь солей всех трех бензолдикарбоновых кислот.
СООч
/Ва +
СОСУ
(87)
224
При нагревании rt-хлорбензоата калия в атмосфере СО2 при
360 °C с катализатором фторидом кадмия получается соль 4-хлор-
изофталевой кислоты (88):
Диспропорционированием солей 1- и 2-нафтойных кислот при
400 °C с фторидом кадмия получается соль 2,6-нафталиндикарбо-
новой кислоты (89):
соок
(89)
Реакция распространяется также на гетероароматические со-
единения, Так, калиевые соли всех трех изомерных пиридинкарбо-
новых кислот образуют при диспропорционировании соль 2,5-пи-
ридиндикарбоновой кислоты (90):
Аналогично из калиевой соли 2-хинолинкарбоновой кислоты
образуется соль 2,4-хинолиндикарбоновой кислоты (91):
COOK
(91)
Еще большее практическое значение имеют открытые в 1955 г.
[56] реакции изомеризации солей дикарбоновых кислот, проходя-
щие в сходных условиях и позволяющие, например, из солей
8 Зак, 271
225
фталевой и изофталевой кислот получать соль терефталевой кис-
лоты (86):
COOK
COOK
КООС
COOK
COOK
(86)
Температура процесса 400—450 °C. Существенно отсутствие во-
ды и свободных кислот. Реакцию ведут под давлением СОг до
5 МПа в присутствии фторида или иодида кадмия. Выход тере-
фталевой кислоты из фталевой достигает 95%. Изофталевая кисло-
та изомеризуется значительно труднее и с худшим выходом [55].
Изомеризацией можно получить и ряд других кислот. Так,
дикалиевая соль дифеновой (2,2'-бифенилдикарбоновой) кислоты
(92) при 380 °C в сходных условиях превращается в соль
4,4/-бифенилдикарбоновой кислоты (93):
(92) (93)
Дикалиевая соль нафталевой (1,8-нафталиндикарбоновой) кис-
лоты (94) в присутствии фторида кадмия при 425 °C за 4 ч дает
соль 2,6-нафталиндикарбоновой кислоты (89) с выходом 62%:
КООС соок
(94)
коос
(89)
Такие же превращения претерпевают соль 2,3-пиридиндикарбо-
новой кислоты, образующая соль 2,5-пиридиндикарбоновой кисло-
ты (90), и соль 2,3-пиразиндикарбоновой кислоты, изомеризующа-
яся в соль 2,5-пиразиндикарбоновой кислоты.
Механизм реакции диспропорционирования и изомеризации
солей ароматических карбоновых кислот, по всей вероятности,
близок к рассмотренному механизму реакции Кольбе — Шмитта
(см. 6.3.4). Родственность этих реакций отмечена многими иссле-
дователями [57].
Например, в случае диспропорционирования соли бензойной
кислоты вначале идет процесс декарбоксилирования и, с выделе-
нием СО2, образуется ионная пара (96). При отсутствии в реакци-
онной массе воды или кислот анион СеНГ в этой паре нейтрализу-
220
ется, отрывая, как более «кислый», атом водорода из пара-положе'
ния бензоат-аниона. При этом получаются бензол и металлоргани-
ческое соединение (97), с дианионом, содержащим карбанионный
центр в пара-положении ароматического кольца. Заключительным
этапом реакции является взаимодействие металлорганического со-
единения (97) и СО2 с образованием терефталата калия (86):
СОСГ К
к+ гоос-
сосг к'
СОО’ к + со.
Так как реакции отщепления СО2 с образованием металлорга-
нического соединения и взаимодействия этого соединения с
СО2 обратимы, общее направление рассматриваемого превращения
должно определяться положением наиболее «кислого» атома во-
дорода в ароматическом кольце аниона исходной соли. Например,
в бензоатанионе это водород в пара- и орто-, но не в лета-положе-
нии. В случае пиридинкарбоновых кислот активно прежде всего
положение 2. Образование при изомеризации этих кислот
2,5-пиридиндикарбоновой кислоты (90), очевидно, проходит через
стадию, в которой получается 2-пиридинкарбоновая кислота; в ней
«кислый» характер приобретает водород в положении 5.
В свою очередь, общая скорость химического превращения
по-видимому, должна определяться скоростями установления рав-
новесия бензоат-анион металлорганическое соединение. Сле-
дует полагать, что соли кадмия катализируют именно эти стадии
процесса. Упомянутые выше фенолят-соли салициловой и п-гидро-
ксибензойной кислоты (см. 6.3.4) легче всего дают такие равнове-
сия. Фенолят-соли именно этих кислот рекомендуются «в качестве
доноров карбоксилатных групп» [58] при получении терефталевой
кислоты из бензойной. Реакцию с бензоатом калия проводят под
давлением СО2 при 400 °C в присутствии солей кадмия, и терефта-
левая кислота получается с выходом 80%. Авторы полагают, что
процесс протекает при этом с предварительным образованием
4-гидроксиизофталевой кислоты, которая передает карбоксильную
группу бензоату [58].
Однако с точки зрения рассматриваемого механизма следует
полагать, что при декарбоксилировании фенолят-соли салициловой
кислоты образуется металлорганическое соединение (98); оно
нейтрализуется путем захвата более «кислого» водорода из
«-положения бензоат-аниона, превращаясь в фенолят; при этом
8*
227
образуется новое металлорганическое соединение (99). Последнее
с СО2 дает терефталевую кислоту.
Из сказанного следует, что фенолят-соль салициловой кислоты
в этой реакции является источником образования металлорганиче-
ского соединения, а не донором карбоксилатной группы.
Таким образом, рассматриваемые реакции карбоксилирования
являются реакциями электрофильного ароматического замещения,
в переходном состоянии которых участвует не о-комплекс, а кар-
банион (см. 2.7.1). Реакции эти в настоящее время усиленно изу-
чаются и, по-видимому, получат большое распространение.
ЛИТЕРАТУРА
1. Friedel-Krafts and Related Reactions. V. 1—4/Ed. G. A. Olah. N. Y., Lon-
don, 1963—1965. 2. Ингольд К. Теоретические основы органической химии, Пер.
с англ./Под ред. И. П. Белецкой. Изд. 2-е. М„ Мир, 1973, с. 298—304.
3. Прайс Ч. — В кн.: Органические реакции. Сб. 3. Пер. с англ. М., ИЛ, 1951,
с. 7—87. 4. Бородкин Г. И., Шакиров М. М., Шубин В. Г. и др. — ЖОрХ, 1978,
т. 14, № 2, с. 321—331. 5. Коптюг В. А. Изомеризация ароматических соедине-
ний. Новосибирск, Изд. СО АН СССР, 1963, с. 5—56. 6. Коптюг В. А. — В кн.:
Современные проблемы' химии карбониевых ионов/Под ред. В. А. Коптюга. Но-
восибирск, Наука, 1975, с. 103—142. 7. Лебедев И. Н. Химия и технология
основного органического и нефтехимического синтеза. Изд. 2-е. М., Химия, 1975.
8. Сулимов А. Д. Производство ароматических углеводородов из нефтяного
сырья. М., Химия, 1975, с. 150—208. 9. Паушкин Я. М., Адельсон С. В., Вишня-
кова Т. И. Технология нефтехимического синтеза. Ч. 1. М., Химия, 1973. 10. Пав-
лов Г. П., Беленький М. С. и др. — Хим. пром., 1975, № 12, с. 892—894.
11. Olah G. A., Kaspi L, Bukala J. — J. Organic Chem, 1977, v, 42, № 26,
p. 4187—4191. 12. Франц, пат. 1478768, 1967, Chem. Abstr., 1968, v. 68, 96800;
1478769, 1967, Chem. Abstr., 1967, v. 67, 109626. 13. Ершов В. В., Никифоров Г'. А.,
Володькин А. А. Пространственно-затрудненные фенолы. М., Химия, 1972.
14. Шоенауер В., Бенгуерел Ф., Бенц Дж. — В кн.: Химия синтетических краси-
телей. Т. 5/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л.,
Химия, 1977, с. 69—102. 15. Фирц-Давид Г. Э., Бланже Л. Основные процессы
синтеза красителей. Пер. с нем./Под ред. С. В. Богданова и И. В. Фодимана.
М„ ИЛ, 1957. 16. Pratt Е. F., Green L. О. — J. Amer. Chem. Soc., 1953, v. 75,
228
№ 2, р. 275—278. 17. Фюзон Рейнгольд К., Мак-Кивер К- Г- — В кн.: Органиче-
ские реакции. Сб. 1. Пер. с англ. М, ИЛ, 1948, с. 84—115. 18. Беленький Л. И.,
Волькенштейн Ю. Б., Карманова И. Б. — Успехи химии, 1977, т. 46, вып. 9,
с. 1698—1719. 19. In: Methoden der organischen Chemie (Houben-Weyl). 4 Aufl.
Bd. 5/3. Stuttgart, Georg Thime Verlag, 1959, S. 1001. 20. Ворожцов И. H. Осно-
вы синтеза промежуточных продуктов и красителей. Изд. 4-е. М, Госхимиздат,
1955.
21. Груммитт О., Бэк А. — В кн.: Синтезы органических препаратов. Сб. 3.
Пер', с англ. М, ИЛ, 1952, с. 481—483. 22. Пат. США 2435307, 1948; Chem.
Abstr., 1948, v. 42, 3187. 23. Пат. ФРГ 843726, 1952; Chem. Abstr, 1958, v. 52,
9218А. 24. Франц, пат. 1403353, 1965; Chem. Abstr, 1965, v. 63, 15026C. 25. Об-
щий практикум по органической химии. Пер. с нем./Под ред. А. И. Коста. М,
Мир, 1965. 26. Адамс Р„ Колеман — В кн.: Синтезы органических препара-
тов. Сб. 1/Под общ. ред. Гильмана. Пер. с англ. М, ИЛ, 1949, с. 193—196.
27. Анжиал С. Дж. — В кн.: Органические реакции. Сб. 8. Пер. с англ. М, ИЛ,
1956, с. 263—287. 28. Hellmann И., Opitz G. a-Aminoalkilierting. Darstellung und
Eigenschaften der Kondensationprodukte H-acider Stoffe mit Carbonyl-verbindun-
gen und Aminen. Weinheim, Chemie, 1960. 29. Бэр Д. P. — В кн.: Химия синте-
тических красителей. Т. 4/Под ред. К. Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса. Л., Химия, 1975, с. 167—172. 30. Цаугг Г., Мартин В. — В кн.:
Органические реакции. Сб. 14. Пер. с англ. М, Мир, 1967, с. 65—286.
31. Пат. США 2427527, 1947; Chem. Abstr, 1948, v. 42, 1431. 32. Downes А. М.,
Lions F. — J. Amer. Chem. Soc, 1950, v. 72, № 7, p. 3053—3056. 33. Бутс Г.—
В кн.: Химия синтетических красителей. Т. 5/Под ред. К. Венкатарамана. Пер.
с англ, под ред. Л. С. Эфроса. Л, Химия, 1977, с. 233. 34. Герм. пат. 442774;
Friedlender, Bd. 15, S. 1700. 35. Olah G. A. et al. — J. Amer. Chem. Soc, 1964,
v. 86, № 11, p. 2198—2202. 36. Берлинер Э. — В кн.: Органические реакции. Сб. 5.
Пер. с англ. М„ ИЛ, 1951, с. 195—270. 37. Марвел, Сперри — В кн.; Синтезы
органических препаратов. Сб. 1. Пер. с англ. М, ИЛ, 1949, с. 99—102.
38. Клар Э. Полициклические углеводороды. Т. 1. Пер. с англ. М, Химия. 1971.
39. Mowry D. Т., Renoll М., Huber W. F. — J Amer. Chem. Soc, 1946, v. 68,
p. 1648—1654. 40. Доналдсон H. Химия и технология соединений нафталинового
ряда. Пер. с англ./Под ред. А. И. Королева. М, Госхимиздат, 1963.
41. Красовицкий Б. 44, Болотин Б. 44. Органические люминофоры. Л, Хи-
мия, 1976. 42. Краунз Н. — В кн.: Органические реакции. Сб. 5. Пер. с англ. М,
ИЛ, 1951, с. 271—283. 43. Трюс, У. Э. — В кн.: Органические реакции. Сб. 9.
Пер. с англ. М, ИЛ, 1959, с. 45—81. 44. Мачинская И. В. — В кн.: Реакции и
методы исследования органических соединений. Сб. 7./Под ред. В. М. Родионова,
Б. А. Казанского и др. М, Госхимиздат, 1958, с. 277—306. 45. Физер Л., Фи-
зер М. Реагенты для органического синтеза. Пер. с англ./Под ред. И. Л. Кну-
нянца и Р. Г. Костяновского. М, Мир, 1971, т. 4, с. 187; т. 5, с. 430. 46. Grund-
mann С., Dean J. М. — Angew. Chem, 1965, Bd. 77, № 21, S. 966—967. 47. Вей-
ганд-Хильгетаг. Методы эксперимента в органической химии. Пер. с нем./Под
ред. Н. Н. Суворова. М, Химия, 1968. 48. Кампень Э., Арчер У. — В кн.: Син-
тезы органических препаратов. Сб. 5. Пер. с англ. М, ИЛ, 1954, с. 22—24.
49. Порай-Кошиц Б. А, Квитко И. Я.— ЖОХ, 1962, т. 32, вып. 12, с. 4050—4056.
50. Курковская Л. Н., Шапетько Н. И., Витвицкая А. С. и др. — ЖОрХ, 1977,
т. 13. вып. 6, с. 1203—1209.
51. Квитко И. Я-, Панфилова Е. А. — ХГС, 1973, № 4, с. 507. 52. Wyn-
berg Н. — Chem. Rev, 1960, v. 60, № 2, p. 169—184. 53. Кизбер А. И., Глаго-
лева А. С. — ЖОХ, 1953, т. 23, вып. 6, с. 1028—1034. 54. Ворожцов Н. Н. мл.—
В кн. Химия синтетических красителей, Т. З/Под ред. К. Венкатарамана. Пер. с
англ, под ред. Л. С. Эфроса. Л, Химия, 1974, с. 1806. 55. Ichikawa V., Ta-
keuchi Y. — Hydrocarb. Proc, 1972, v. 51, № 11, p. 103—108. 56. Пат. ФРГ
936036, 1955; Chem. Abstr, 1958, v. 52, 20065A. 57. Королев A. И. — В кн. [40],
c. 501. 58. Пат. США 2904587, 1959, Chem. Abstr, 1960, v. 54, 1448H.
229
Часть 3
РЕАКЦИИ АРОМАТИЧЕСКОГО
НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
Глава 7
РЕАКЦИИ С АЗОТОСОДЕРЖАЩИМИ
НУКЛЕОФИЛЬНЫМИ РЕАГЕНТАМИ
7.1. ВВЕДЕНИЕ АМИНОГРУПП
Реакция нуклеофильного аминирования является одной
из важнейших в химии промежуточных продуктов. Это определяет-
ся большим практическим значением получаемых таким образом
ароматических аминосоединений и широкими препаративными
возможностями реакции. Для введения незамещенных и замещен-
ных аминогрупп в качестве реагентов используются аммиак, пер-
вичные и вторичные амины, их соли со щелочными металлами,
ацильные производные, мочевина, гидроксиламин. Вытесняться из
ароматического кольца при аминировании могут самые разнооб-
разные атомы и группы: галогены, нитро-, алкокси-, арилокси-,
ацилокси-, гидрокси-, сульфо-, алкил- и арилсульфонильные, ал-
кил- и арилтио-, амино- и аммониевые группы, атомы водорода
Таблица 7.1
Относительные скорости реакции нуклеофильного замещения
для п-нитро-Х-бензолов [1]
F 412 OC6H4NO2-n 2,9
С1 * 1 ОСОСНз 0,06
Вг 1,17 OSO2C6H4CH3-n 5,5
I 0,26 SO2C6H5 0,0088
no2 8,7 8О2СбН4МО2-п 0,11
ОСНз 0,0017 SC6H5 0,001
OCHaCgHj 0,0011 SC6H4NO2-n 1,15
ОС6Н5 0,030 CN 0,001
При Х = С1 А=66-10“8 л/(моль-с).
230
и др. Способность этих атомов й групп подвергаться замещению
варьируется в широких пределах в зависимости от прочности связи
с ядром и электроотрицательности элиминируемого атома, поляри-
зуемости, специфической сольватации и т. д. (см. гл. 2). Некоторое
представление об относительной подвижности уходящих групп да-
ет сопоставление скоростей реакций n-нитро-Х-бензолов с пипери-
дином в диметилсульфоксиде (табл. 7.1).
Как видно из этой таблицы, скорости реакции с изменением
природы замещаемой группы изменяются более чем на пять по-
рядков. Однако и сравнительно мало подвижные группы также с
успехом используются в реакции аминирования, поскольку малая
подвижность может быть компенсирована повышением температу-
ры, применением более активных аминирующих агентов, наличием
в субстрате активирующих заместителей. Выбор вытесняемой
группы определяется осуществимостью реакции в приемлемых ус-
ловиях и доступностью исходных соединений.
7.1.1. Замещение атомов галогенов
Замещение галогенов наиболее распространено, так как
они обладают высокой подвижностью (см. табл. 7.1) и могут быть
легко введены в ароматическое ядро (см. гл. 5 и 10). Замещение
галогена, активированного влиянием электроноакцепторных групп,
проводится непосредственно обработкой аминирующим агентом в
отсутствие катализатора. В случаях, когда галоген недостаточно
подвижен, применяется катализ соединениями меди.
В некаталитической реакции фтор значительно превосходит по
скорости замещения хлор и бром, которые, в свою очередь, заме-
щаются легче, чем иод. Это объясняется определяющей ролью ста-
дии присоединения нуклеофила с образованием о-комплекса
(см. 2.7.2), а также общим кислотным катализом, осуществляв»
мым реагентом и растворителем и способствующим анионоидному
отрыву (см. 2.6).
В каталитической реакции [2] скорость замещения изменяется
в зависимости от природы атома галогена в противоположной
последовательности: I >> Br >> Cl >> F. Катализаторами могут
служить соединения меди разной валентности. Константа чувстви-
тельности каталитической реакции (р = 0,54-0,6) почти на порядок
меньше, чем некаталитической (ср. табл. 2.9). Малая чувствитель-
ность каталитической реакции к влиянию заместителей, находя-
щихся в пара- и лега-положениях, сочетается с сильным ускоряю-
щим эффектом о-заместителей, таких как карбокси-, азо-, гидро-
кси-, нитрогруппа и др. Представления о механизме каталитиче-
ской реакции в большинстве работ базируется на предположении
[3] о координации соединения меди как кислоты Льюиса по заме-
щаемому атому галогена, например:
ArCl + [Cu(NH3)2]+ [ArCl • Cu(NH3)2]+ ----► ArNH2 + [Cu(NH3)2]+
U)
231
Согласно другим точкам зрения, атом меди координируется с
л-системой ароматического ядра или образует медьорганическое
соединение, внедряясь между атомами галогена и углерода. Пред-
ставление о протекании реакции через комплекс типа (1) лучше
согласуется с наблюдаемыми закономерностями. Аномально боль-
шой эффект о-заместителя объясняется его участием в координа-
ции с атомом меди с замыканием хелатного цикла, а увеличение
подвижности атомов галогенов — уменьшением их электроотрица-
тельности и увеличением поляризуемости, которые облегчают при-
соединение к катализатору. На примере реакции 1-бром-9,10-ант-
рахинона с 2-аминоэтанолом показано [4], что соединения меди
(II) сами обладают низкой каталитической активностью, но уве-
личивают каталитическую активность соединений меди (I). Раз-
рыв связей в комплексе (1) на пути к продукту замещения может
происходить по синхронной, ионной или радикальной схеме [5], из
которых предпочтение отдается последней [2, 6].
Важнейшим применением некаталитического замещения гало-
гена в бензольном ряду являются процессы получения нитроани-
линов и их производных.
a) R = H
б) R=C1
в) R=NO2
г) R = CN
д) R —SO3H
(4) (5)
Аммонолиз о- и н-нитрохлорбензолов (2) и (4а) для получения
соответствующих нитроанилинов (3) и (5а) проводятся в среде
водного аммиака в автоклавах при температуре до 200 °C или
непрерывным методом в трубчатом реакторе при более высокой
температуре [7].
Так, при производстве н-нитроанилина (5а) в стальной авто-
клав вместимостью 3000 л загружают 1700 л 40%-ного родного
раствора NH3 (35 кмоль) и 640 кг (4 кмоль) расплавленного
п-нитрохлорбензола (4а), нагревают смесь до 170—190 °C и разме-
шивают при этой температуре 7—10 ч. По окончании реакции
спускают давление, поглощая аммиак водой, осадок п-нитроани-
лина отделяют на центрифуге, промывают 1000 л воды и отжима-
ют; выход 94%. Из фильтрата после добавления раствора NaOH
отгоняют аммиак, который улавливают водой и возвращают в
цикл.
ж-Нитрохлорбензол в тех же условиях в реакцию не вступает.
Различная подвижность атомов галогена в мета- и в пара-положе-
нии к нитрогруппе делает возможным избирательное замещение
одного из них в 1-нитро-3,4-дихлорбензоле (46) при производстве
4-нитро-2-хлоранилина (56).
232
При наличии в бензольном кольце второго активирующего за-
местителя аммонолиз протекает в более мягких условиях. Так, для
замещения атома хлора в 1,3-динитро-4-хлорбензоле (4в) при про-
изводстве 2,4-динитроанилина (5в) и 5-нитро-2-хлорбензонитриле
(4г) при получении 2-амино-5-нитробензонитрила (5г) достаточно
нагревания в водном аммиаке при 100—120 °C. Нагревание в вод-
ной среде с ароматическими аминами в присутствии оснований
приводит к производным дифениламина. Например, взаимодей-
ствием 1,3-динитро-4-хлорбензола (4в) с «-аминофенолом получают
желтый дисперсный нитрокраситель (6), а реакцией 5-нитро-2-хлор-
бензолсульфокислоты (4д) с п-анизидином — 2-«-анизидино-5-ни-
тробензолсульфокислоту (7), превращаемую далее восстановле-
нием нитрогруппы и десульфированием в 4-амино-4'-метоксидифе-
ниламин (азоамин синий О).
На легкости замещения атома фтора в 1,3-динитро-4-фторбен-
золе основано применение его в качестве реактива на аминокисло-
ты [8].
В 2,4-динитро-1-галогеннафталинах атом галогена намного бо-
лее подвижен, чем в соответствующих производных бензола. Как
видно из табл. 7.2, замещение атомов хлора, брома и иода в
реакции с анилином в нафталиновом ряду протекает более чем на
два порядка быстрее, а атом фтора обменивается всего на порядок
быстрее. Большая скорость реакции производных нафталина обус-
ловлена более низкой энергией активации вследствие меньшей
потери энергии резонанса в переходном состоянии, подобном по
структуре a-комплексу (см. 2.7.2), а не стерическими факторами,
которые должны были бы сказываться на величине энтропии акти-
вации [9].
233
Таблица 7.2
Скорости реакции 2,4-динитро-1-Х-нафталинов
и 2,4-дииитро-Х-бензолов с анилином [10]
(в этаноле при 50 °C)
X fe-104, л/(моль-с) ^нафт
2,4-динитро-1-Х-наф- талин 2,4-ДИнитро-Х-бензол ^бенз
I 224 1,31 171
Вг 479 4,05 118
С1 437 2,69 162
F 1910 168 Н,
no2 — 2110
При аминировании галогенпроизводных в спиртовой среде ре-
акция может протекать через промежуточную стадию алкоголиза.
Кинетическими методами доказано, что реакция 1,3-динит-
ро-4-хлорбензола (4в) с циклогексиламином в метаноле со сравни-
мыми скоростями идет по пути (а) с непосредственным замещени-
ем атома галогена амином и по пути (б) с замещением атома хлора
метилат-анионом и последующим обменом метоксигруппы [11];
Очень легко, иногда уже при комнатной температуре, обмени-
ваются атомы галогенов в бензо- и нафтохинонах. Ариламиниро-
вание 2,3,5,6-тетрахлор-1,4-бензохинона (хлоранила), например, в
производстве диоксазиновых красителей проводится при 60—80 °C
234
связывания выделяющегося НС1:
в присутствии оснований для
В производных 9,10-антрахинона, где заместители находятся в
ароматическом, а не в хиноидном кольце, способность атомов га-
логенов к замещению на аминогруппы сравнимы с таковой в нит-
рогалогенбензолах. 2-Амино-9,10-антрахинон получают в промыш-
ленности аммонолизом 2-хлор-9,10-антрахинона в условиях, близ-
ких к применяемым при производстве о- и п-нитроанилинов [7]. На
1 кмоль 2-хлор-9,10-антрахинона в реакцию вводят 17,5 кмоль
Ь1Нз и 0,06 кмоль CuSO4. Процесс ведут при 204—206 °C в течение
10 ч при давлении 4,5 МПа, выход 90%. Атомы галогена в
a-положении антрахинонового ядра более подвижны. Отношение
скоростей аминирования а- и 0-хлорантрахинонов пиперидином в
бензоле при 50 °C равно 440, а фторантрахинонов — 2250 [12], что
в 8—9 раз больше, чем отношение о-изомер/и-изомер для нитрога-
логенбензолов (см. 2.6). Это свидетельствует о более сильном
взаимодействии нуклеофила с карбонильной группой антрахинона
в переходном состоянии при a-замещении по сравнению с взаимо-
действием с нитрогруппой при реакции о-нитрогалогенбензолов.
С увеличением полярности растворителя отношение а-изомер/0-
изомер для хлор- и фторантрахинонов намного уменьшается (до
3,6 и 5 соответственно в диметилформамиде), так же как отноше-
ние о-изомер/п-изомер для нитрогалогенбензолов. При аминирова-
нии полигалогенантрахинонов это приводит к изменению ориента-
ции замещения при переходе от неполярных растворителей к более
полярным. Так, при реакции 1,2,3,4-тетрафтор-9,10-антрахинона с
пиперидином в бензоле при 80 °C образуется 86% 1-пипериди-
но-2,3,4-трифтор-9,10-антрахинона, а в диметилсульфоксиде — 82%
2-пиперидино-1,3,4-трифтор-9,10-антрахинона [13].
Реакция между а-галоген-9,10-антрахинонами и вторичными
аминами при нагревании в полярной среде осложняется дезалки-
лированием с превращением диалкиламинопроизводных (8) в мо-
ноалкилпроизводные (10). В результате исследования этого пре-
вращения найдено [14], что дезалкилирование протекает через
стадию замыкания связи между ближайшим к азоту атомом угле-
рода алкила и карбонильным кислородом с образованием дигид-
роантраоксазинов (9); последние были изолированы в виде аце-
тильных производных. Окисление антраоксазинов (9) кислородом
воздуха приводит к раскрытию оксазинового кольца и элиминиро-
ванию алкильного остатка в виде альдегида, Когда R, R'— (СН2)П,
235
т. е. когда алкилы при атоме азота входят в состав цикла, напри-
мер пиперидинового (п = 4), образуются со-(9,10-антрахинон-1-ил) -
аминоалифатические альдегиды (11).
Если же в p-положении антрахинона находится ациламиногруппа
(12), алкильный атом углерода связывается не с карбонильным
кислородом, а с атомом азота ациламиногруппы с замыканием
дигидроимидазольного цикла (13) [15]. Течение реакций поясняет
следующая схема:
R
(12) (13)
При реакциях малоактивных галогенпроизводных, таких как
хлорбензол, л-нитрохлорбензол, хлоранилины, хлоранизолы и
т. п„ катализ соединениями меди позволяет успешно проводить
замещение галогена даже при действии слабоосновных аминов и
добиваться избирательности, недостижимой при некаталитической
реакции. Так, благодаря изменению соотношения подвижностей
атомов галогенов в каталитической реакции (см. выше) при взаи-
модействии всех трех изомерных фторхлорбензолов с водным ам-
миаком в присутствии соединений меди замещается исключительно
атом хлора с образованием соответствующих фторанилинов [16].
Хлорбензол, его амино- и алкокси-производные гладко реагируют в
236
присутствии солей меди в кипящем нитробензоле с такими слабо-
основными аминами, как аминоантрахиноны [17], давая арилами-
ноантрахиноны с более высоким выходом, чем удается достигнуть
реакцией галогенантрахинонов с соответствующими производными
анилина.
При каталитическом аминировании галогенантрахинонов выхо-
ды часто оказываются низкими из-за побочных процессов; главным
является замещение галогена атомом водорода [18]. Оно происхо-
дит в результате одноэлектронного восстановления галогенантра-
хинона до анион-радикала атомом Си1 катализатора или другим
восстановителем с последующим отщеплением галоген-аниона и
отрывом нейтральным семихинонным радикалом атома водорода
от молекулы растворителя [4]. Катализ соединениями меди при-
меняется для замещения а-атома галогена на ариламиногруппу в
1,4-аминогалогенпроизводных при получении кислотных, дисперс-
ных и активных красителей: в бромаминовой кислоте (14а) (ее
аминируют в водной среде); в А-метил-1-амино-4-бром-9,10-антра-
хиноне (146) и в 1 -амино-2,4-дибром-9,10-антрахиноне (14в) (с
двумя последними реакцию проводят в среде амина или в органи-
ческом растворителе).
(14)
(15)
a) R=H, Х= SO3H
б) R = СНз, Х=Н
в) R = H, Х = Вг
Добавка третичных аминов ингибирует замещение атомов хло-
ра в присутствии солей меди в 1-алкиламино-4-хлорантрахинонах;
благодаря этому последние удается получить в чистом виде с
хорошим выходом алкиламинированием 1,4-дихлорантрахинонов
[19]. а-Атом галогена в 1-амино-4-хлор-9,10-антрахиноне и 1-ами-
но-2,4-дибром-9,10-антрахиноне (14в) замещается уже при ком-
натной температуре в среде анилина с выходом 85—88% в присут-
ствии А1С1з [20].
Помимо аммиака и аминов в качестве аминирующих агентов
применяют их ацильные производные. При введении остатков ам-
миака и низших алкиламинов применение амидов позволяет избе-
жать работы с автоклавами. В присутствии оснований ацильные
производные аммиака и первичных аминов способны ионизиро-
ваться, генерируя А-анионы, являющиеся более сильными нуклео-
филами. Реакция с ацильными производными вторичных аминов
протекает, по-видимому, путем непосредственной нуклеофильной
атаки амидного азота на ароматический атом углерода с отщепле-
нием ацильного остатка.
Производные хлорбензола, содержащие не менее двух доста-
точно сильных электроноакцепторных заместителей, превращаются
237
в соответствующие производные анилина при нагревании в рас-
плавленной мочевине до 180 °C [21]. Так же может быть введена
аминогруппа в другие соединения, имеющие подвижный атом га-
логена. о- и n-Нитрохлорбензолы не реагируют с мочевиной при
нагревании до 250 °C.
Для введения диметиламиногруппы используется взаимодей-
ствие с диметилформамидом или гексаметилфосфортриамидом
(гексаметапол). Кипячение в диметилформамиде нитрогалоген-
бензолов (16) с добавкой CuSO4 приводит с выводом до 80% к
соответствующим А/,./У-диметилнитроанилинам (17) [22]. 2-Хлор-
бензимидазолы в этих условиях превращаются в 2-диметиламино-
бензимидазолы в отсутствии катализатора [23]. При нагревании в
гексаметаполе при 210—230 °C без давления /г-нитрогалогенбензо-
лы переходят в ЛС./У-диметил-4-нитроанилины (17) с умеренным
выходом [24].
(«)
+(ch3)2ncho _
или[(СН3)2И]3РО
N(CH3)S
no2
(17)
Нагревание 1-хлор-9,10-антрахинона с соответствующими
/V-замещенными формамидами и ацетамидами в кипящем нитро-
бензоле или в самом амиде без катализатора дает с выходом 97—
99% (V-метил-, А/Д-диметил-, А/,А(-диэтил-1-амино-9,10-антрахино-
ны; однако выход А-фенил- и А,-метил-А/-фенил-1-амино-9,10-ант-
рахинонов не превышает 30% [25].
R = H, СН3
R', R" = Н, СНз, С2Н5, С6Н5
В ряду 9,10-антрахинона большое распространение получило
введение аминогрупп действием n-толуол- или п-бензолсульфами-
да. Реакцию обычно проводят в присутствии соединения меди и
карбоната или ацетата щелочного металла в среде органического
растворителя; выделенный jV-антрахинонилсульфамид затем гидро-
лизуют действием серной кислоты. В бромаминовой кислоте (14а)
замещение осуществляется в водном растворе в присутствии
небольших количеств CuSO4. В реакцию могут вводиться также
(V-алкил- и АКарил-n-толуолсульфамиды [26].
Аминирующий агент может быть переведен в более активную
А-анионную форму не только ионизацией ацилированного амина,
но и непосредственно действием на амин щелочного металла или
238
его амида. Так, при действии на анилин амида натрия протекает
обменная реакция с выделением аммиака:
C6H6NH2 + NaNH2 —> C6H6NHNa + NH3
Образовавшийся анилид натрия способен замещать неактиви-
рованный атом галогена в галогенбензолах в сравнительно мягких
условиях. Например, при нагревании анилида натрия с п-хлорани-
золом при 90—100 °C с хорошим выходом образуется 4-метоксиди-
фениламйн [27, с. 76].
При аминировании действием амидов щелочных металлов часто
наблюдается кмне-замещение вследствие протекания реакции по
аринному механизму (см. 2.7.2). Так, взаимодействие с амидом
натрия о-фторхлорбензола в жидком аммиаке приводит исключи-
тельно к лг-фторанилину [16]. Реакция 1-метокси-2-хлорнафталина
(18) с амидом калия в жидком аммиаке предложена как препара-
тивный метод синтеза 4-метокси-2-нафтиламина (19) [28]:
ОСНз ОСНз
(18) (19)
7.1.2. Замещение нитрогруппы
Нитрогруппа по способности замещаться подобна фтору.
По подвижности они превосходят все остальные атомы и группы.
В реакциях 4-нитро-Х-бензолов с пиперидином в диметилсульфок-
сиде нитрогруппа по скорости замещения уступает атому фтора, а в
реакциях 2,4- и 2,6-динитро-Х-бензолов с анилином в этаноле пре-
восходит его (ср. табл. 7.1 и 7.2). Неактивированная нитрогруппа в
нитробензоле не подвергается нуклеофильному замещению, но ре-
акция становится возможной при наличии в п- или во-положении к
нитрогруппе электроноакцепторного заместителя. Так, в о-нитро-
бензонитрилах (20) нитрогруппа легко обменивается при действии
различных нуклеофильных реагентов в диметилформамиде, в том
числе при действии метил- или диметиламина [29]:
R = NO2, ОСНз
Если в молекуле кроме нитрогруппы присутствуют другие спо-
собные вытесняться группировки, направление реакции зависит от
взаимного расположения заместителей. Так, в о-нитросульфоне
(21) замещению подвергается нитрогруппа; в 2,4-динитросульфоне
239
(22), где обе нитрогруппы активируют сульфоновую группу RSO2,
но не друг друга, замещается эта группа [27].
(21)
(23)
В о- и м-нитрогалогенбензолах, как уже отмечалось выше,
(см. 7.1.1), вследствие активирующего действия нитрогруппы за-
мещается только атом галогена. Но если сама нитрогруппа в нит-
рогалогенбензолах находится под активирующим электроноакцеп-
торным влиянием, она замещается в первую очередь; например, в
1,2-динитро-4-хлорбензоле (23) при аммонолизе замещается нит-
рогруппа в положении 2 [27]. При ослаблении активирующего
воздействия нитрогруппы на атом галогена из-за пространствен-
ных затруднений, нарушающих ее сопряжение с ароматическим
кольцом, замещение нитрогруппы становится предпочтительней; в
результате, например, в нитропентахлорбензоле (24) нитрогруппа
замещается легче, чем любой атом хлора.
При взаимодействии 2,3-динитронафталина с пиперидином ко-
личественно образуется З-нитро-1-пиперидинонафталин. В данном
случае кыне-замещение протекает по механизму присоединения —
отщепления (S.vAr) в результате атаки нуклеофила в а-положение
(см. также. 7.2) [30]:
Высокой подвижностью обладает нитрогруппа в а-положении
антрахинона и это находит техническое применение. Скорость за-
мещения нитрогруппы в реакции 1-Х-антрахинонов с пиперидином
в бензоле при 50 °C в 12 раз больше скорости замещения атома
хлора [31]. В патентной литературе имеется много предложений по
оформлению процесса получения 1-аминоантрахинонов из 1-нитро-
антрахинонов, например аммонолиз в присутствии хлорбензола
или в ацетонитриле [32], реакция с мочевиной [33], взаимодей-
ствие с алкиламинами в тетраметилсульфоне [34] и т. п. Анало-
гично могут быть получены 1,5-диаминопроизводные из 1,5-динит -
роантрахинонов. Промышленное значение при получении красите-
лей имеет замещение нитрогрупп в дигидроксидинитроантрахино-
нах. При нагревании 1,5-дигидрокси-4,8-динитро- (26) и
1,8-дигидрокси-4,5-динитро-9,10-антрахинона (29) с ароматически-
ми аминами в обоих случаях замещается одна нитрогруппа (25),
?4Q
(28); в присутствии же борной кислоты в первом случае замеща-
ются обе нитрогруппы (27), а во втором — гидрокси- и нитрогруппа
в положениях 1 и 4 (30) [35]. Причины различий течения реакций
кроются, по-видимому, в различии строения комплексов с борной
кислотой.
(26)
+ ArNH2
ОН о он
+ ArNHa
Н3ВО3
(29)
+ ArNH2
Н3ВО,
(30)
При совместном присутствии атома галогена и нитрогруппы в
1-нитро-4-хлор-9,10-антрахиноне замещение протекает одновремен-
но в двух направлениях; соотношение их зависит от природы амина
и условий реакции [18]. При взаимодействии с алифатическими
аминами при 50—60 °C замещается преимущественно нитрогруппа
(74%, с бутиламином в этаноле), а с ароматическими аминами при
96—98°C — атом галогена (56%, в анилине).
7.1.3. Замещение амино- и
аммониевых групп
Аминогруппы могут быть не только введены, но и вытес-
нены в реакциях нуклеофильного замещения. Обмен одной ами-
ногруппы на другую применяется на практике для получения ди-
ариламинов взаимодействием первичных ароматических аминов
241
между собой [36,37]. Реакцию обычно проводят в присутствии
кислого катализатора (соляная кислота, фосфорная кислота, хло-
рид фосфора(III), ароматические сульфокислоты). Нагревание
анилина с его гидрохлоридом в автоклаве при 230 °C приводит к
дифениламину и хлориду аммония:
C6H5NH2 + C6H5NHt СГ —> (C6H5)2NH2 + NH4C1
При проведении реакции в присутствии лишь каталитических
количеств кислоты выделяется свободный аммиак, который непре-
рывно выводят из автоклава. В промышленном масштабе реализо-
ван парофазный процесс получения дифениламина из анилина на
гетерогенном алюмосиликатном катализаторе.
Особенно широко замещение аминогруппы на ариламиногруппу
используется в ряду нафталина. Нагреванием 1-нафтиламина с
анилином или его производными в присутствии кислоты или ката-
литических количеств иода получают 1М-арил-1-нафтиламины, при-
меняемые в качестве антиоксидантов:
NH2
+ ArNH2
NHAr
Ариламинирование аминонафталинсульфокислот осуществляют
нагреванием с избытком амина без давления. Кислотный катализ
может осуществляться имеющимися в соединениях сульфогруппа-
ми, но добавление минеральной кислоты часто позволяет снизить
температуру реакции и повысить выход. Таким путем из 8-ами-
но-Гнафталинсульфокислоты (перикислота) получают практиче-
ски важные М-фенил- и М-п-толилперикислоты:
Аг — CgHs, м-СНдСбН4
При кислотно-катализируемом ариламинировании аминонафта-
линсульфокислот может происходить замещение не только амино-,
но и сульфогруппы, например в случае 8-амино-1,6-нафталинсуль-
фокислоты (1-нафтиламин-3,8-дисульфокислота; амино-е-кислота):
+ CeH5NH2
--------->
HO3S nhc6h5
nhc6h5
Такое течение реакции указывает, по-видимому, на протониро-
вание нафтиламинов с образованием не только аммониевых, но и
аренониевых ионов (см. 7,1.5), в которых сульфогруппа обладает
повышенной подвижностью.
243
Если аминонафталинсульфокислота содержит способную заме-
щаться гидроксигруппу, реакцию для избирательного обмена ами-
ногруппы ведут с небольшим избытком ариламина и с добавлением
воды под давлением, например:
Обмен аминогруппы осуществляют также в присутствии би-
сульфита натрия в условиях реакции Бухерера (см. 7.1.5 и 8.1.3).
Так, А-фенил-6-амино-4-гидрокси-2-нафталинсульфокислоту (фенил-
Гамма-кислота) (31) получают кипячением 6-амино-4-гидро-
кси-2-нафталинсульфокислоты (Гамма-кислота) в смеси анилина с
водным раствором бисульфита натрия:
ОН
+ свн5ын2
--------->
+ NaHSO3
Примером переаминирования в полициклических соединениях
может служить образование 6-алкиламино-2-ариламиноантрапи-
ридинов (33) при взаимодействии 2,6-ди (ариламино) антрапириди-
нов (32) с алкиламинами [38]:
(32)
+ rnh2
(33)
Аммониевая группа благодаря своей высокой электроотрица-
тельности обладает значительно большей подвижностью, чем ами-
ногруппа. Препаративное значение приобрели синтезы с примене-
нием солей А-арилпиридиния [39], способных в зависимости от
условий и строения реагировать с аминами по двум направлениям:
с раскрытием пиридинового цикла и образованием производного
глутаконового альдегида (реакция Цинке — Кенига) или с заме-
щением пиридиниевой группы. Так, хлорид А-(2,4-динитрофенил)-
пиридиния (34), легко получаемый взаимодействием 1,3-динитро-
4-хлорбензола с пиридином, при нагревании в анилине превраща-
ется в дианил глутаконового альдегида (35) и 2,4-динитроанилин,
243
а при нагревании с гидрохлоридом анилина— в 2,4-динитродифе-
ниламин (36):
(36)
Оба превращения можно использовать для введения амино-
групп в ароматическую молекулу. Расщепление Цинке—Кенига
оказалось удобным методом получения из пиридиниевой соли (37)
1-амино-З-метилантрапиридона (38), являющегося люминофором
244
Нуклеофильное замещение пиридиниевой группы служит мето-
дом получения 4-аминопиридинов из дихлорида N- (4-пиридил)пи-
ридиния (39), образующегося при реакции пиридина с тионилхло-
ридом. При взаимодействии с водным аммиаком, алифатическими
аминами или с гидрохлоридами ароматических аминов дихлорид
(39) переходит в соответствующие 4-аминопиридины (40) [41]:
(39) (40)
7.1.4. Замещение сульфогруппы
Замещение сульфогруппы аминогруппой в ароматиче-
ских соединениях привлекательно ввиду доступности ароматиче-
ских сульфокислот (см. гл. 3). Оно, однако, имеет ограниченное
применение из-за недостаточной подвижности сульфогруппы, не
активированной влиянием электроноакцепторных группировок.
Введение же сульфогрупп в орто- и пара-положения к электроно-
акцепторным заместителям прямым сульфированием невозможно.
Бензол- и нафталинсульфокислоты, как правило, не обменивают
сульфогруппу при действии аммиака и аминов, но реагируют с
амидами щелочных металлов. Выходы первичных аминов при
сплавлении солей сульфокислот с амидом натрия невысоки [42].
Лучше протекает реакция с замещенными амидами щелочных ме-
таллов в среде соответствующего амина. Так, при кипячении
2-нафталинсульфоната натрия (42) в пиперидине с двухкратным
избытком амида натрия реакция с образующимся пиперидидом
натрия приводит к М (2-нафтил)пиперидину (41) с выходом 71%
[43]. Недавно найдено, что подобным образом при действии на
1- и 2-нафталинсульфонаты ариламидов щелочных металлов с вы-
соким выходом удается получать диариламины (43) [44]. Метод
может иметь преимущества по сравнению с синтезом М-арил-2-наф-
тиламинов (43) ариламинированием 2-нафтола (см. 7.1.5), по-
скольку сам 2-нафтол получают из 2-нафталинсульфокислоты
(см. 8.1.1).
245
В нафталиновом ряду сульфогруппа способна замещаться при
нагревании под давлением с водным аммиаком и хлоридом аммо-
ния, если она находится в мета-положении по отношению к ами-
ногруппе [42]. Например, 4-амино-2,7-нафталиндисульфокислота в
этих условиях превращается в 5,7-диамино-2-нафталинсульфокис-
лоту:
NH2
NHa
+ NH3
SO3H
HO3S
•nh2
HO3S
Замещение сульфогруппы остатком анилина при ариламини-
ровании 4-амино-2-нафталинсульфокислот отмечалось выше (см.
7.1.3).
Главным применением реакции замещения сульфогруппы ами-
ногруппой в промышленности являются производства Гамино- и
/V-метил-!-амино-9,10-аптрахинона из 9,10-антрахинон-1-сульфо-
кислоты и 1,5-диаминоантрахинона из 9,10-антрахинон-1,5-дисуль-
фокислоты.
NH2
+ NH3
о
Реакция проводится в автоклаве в водном растворе аммиака в
присутствии окислителя — соли мышьяковой кислоты или м-нит-
робензолсульфокислоты [7]. Окислитель необходим для разруше-
ния отщепляющегося сульфита, который, если его не окислить,
внедряется в ядро, приводя к образованию значительных коли-
честв аминоантрахинонсульфокислот [45].
При производстве 1-амино-9,10-антрахинона смесь, содержа-
щую 25%-ный водный раствор NH3 (7,05 кмоль), 1,40 кмоль калие-
вой соли 9,10-антрахинон-1-сульфокислоты, 0,55 кмоль натриевой
соли м-нитробензолсульфокислоты и 125 кг (NH4)2SO4, нагревают
в автоклаве вместимостью 3000 л при 180 °C и давлении до 2,3 МПа
в течение 14 ч; выход 76%.
Аналогичным образом могут быть получены 2-амино- и
2,6 (2,7)-диамино-9,10-антрахиноны из соответствующих р-сульфо-
и рф'-антрахинондисульфокислот. Аммонолиз 9,10-антрахинон-1-
сульфокислоты и -1,5-дисульфокислоты предложено осуществлять
непрерывным методом в трубчатом реакторе при более высокой
температуре [46, 47].
Большой подвижностью обладает сульфогруппа, находящаяся
в хиноидном кольце и в некоторых гетероциклах. Высокая актив-
ность 1,2-нафтохинон-4-сульфокислоты (44) по отношению к ами-
нам позволяет использовать ее для идентификации аминокислот и
протеинов [48]. Сульфогруппа в 2-бензотиазолсульфокислоте (45)
246
замещается на аминогруппу при кратковременном нагревании с
водным раствором аммиака [49].
О
SO3H
(44) (43)
Замещение других серусодержащих групп в ароматических со-
единениях имеет ограниченное препаративное значение. Метилтио-
и фенилтиогруппы в бензольном кольце, содержащем в положени-
ях 2 и 4 относительно этих групп две нитрогруппы, не обмениваются
на аминогруппу при сплавлении с мочевиной в отличие от анало-
гично расположенных атома хлора, метокси- и феноксигрупп [21].
Метилсульфонильная группа в метил(1-нафтил)- и метил(2-на-
фтил)сульфоне может быть замещена остатком амина при кипяче-
нии в пиперидине с амидом натрия [50]. Внутримолекулярное
замещение арилсульфонильной группы ариламиногруппой в о-ами-
но-о'-нитродиарилсульфонах при перегруппировке Смайлса
(см. также 7.1.6) с высоким выходом приводит к 2'-нитро-2-дифе-
ниламинсульфиновым кислотам, например:
он-
7.1.5. Замещение гидроксильной группы
Нуклеофильное замещение гидроксигруппы осуществля-
ется в условиях, исключающих ее ионизацию с отщеплением про-
тона, поскольку отрицательный заряд в фенолят-анионе препят-
ствует нуклеофильной атаке. Реакцию обычно проводят в присут-
ствии кислот, которые не только подавляют такую ионизацию, но и
играют роль катализатора. Известно, что ароматические гидрокси-
соединения и их эфиры при действии кислот протонируются по
двум центрам: по атому кислорода и по атому углерода аромати-
ческого кольца, главным образом в пара-положении, с образова-
нием соответственно оксониевого (46) и гидрокси (алкокси) арено-
ниевого (47) ионов [51]:
(46)
R-H.A1K
247
Сущность кислотного катализа состоит в генерировании аре-
нониевых ионов типа (47), которые значительно легче подвергают-
ся нуклеофильной атаке, чем непротонированная молекула. Обра-
зование аренониевых ионов наряду с аммониевыми не исключено
также и при протонировании ароматических аминов. Существова-
ние таких ионов показано спектроскопическими методами в случае
протонирования 1,3,5-триаминобензолов. Возможно, что аминоаре-
нониевые ионы участвуют как активные частицы в реакциях нук-
леофильного замещения аминогрупп в присутствии кислот
(см. 7.1.3).
Другим обстоятельством, определяющим легкость обмена гид-
роксигруппы в фенолах, является их способность к фенол-цикло-
гексадиеноновой таутомерии (см. [52]). Для самого фенола тауто-
мерное равновесие полностью сдвинуто в сторону гидроксиформы,
но с увеличением числа гидроксигрупп и аннелированных арома-
тических циклов (см. 2.4.2) устойчивость оксо-форм возрастает.
Так как нуклеофильная атака атома углерода карбонильной
группы в оксо-форме должна протекать легче, чем атома углерода,
несущего гидроксигруппу в феноле, легкость нуклеофильного за-
мещения гидроксигруппы связывают со склонностью гидроксисо-
единения переходить в оксо-форму.
О сравнительной подвижности гидроксигрупп в фенолах можно
судить по данным об изотопном обмене кислорода 18О в водных
растворах соляной кислоты (табл. 7.3). Последовательность про-
исходящих при этом превращений отражается следующей схемой:
Как видно из табл. 7.3, по скорости обмена гидроксигрупп
нафтолы и двухатомные фенолы резорцин и гидрохинон превосхо-
дят фенол и крезолы. о-Нитро- и о-аминофенол не вступают в
реакцию; это обусловливается, очевидно, прочной внутримолеку-
лярной водородной связью. В пирокатехине, где водородная связь
слабее, обмен затруднен, а существующая в о-бромфеноле водо-
родная связь заметным образом не сказывается. Медленный обмен
гидроксигрупп в n-аминофеноле по сравнению с лг-аминофенолом
объясняется, вероятно, протонированием по атому азота амино-
группы, затрудняющим протонирование в ядро. Большая же
248
Таблица 7.3
Изотопный обмен атома кислорода в фенолах [53]
(в 10 н. НС1 в НгО, обогащенной 18О)
Изотопный обмен, %
Фенолы ПО °C, 24 ч 180 °C, 6 ч 180 °C, 24 ч
Фенол 0 33,3 93,0
Пирокатехин 0 31,5 —
Резорцин 30,0 57,9 —
Г идрохинон 16,7 55,6 —
1-Нафтол 41,5 59,7 61,1
2-Нафтол 35,1 63,2 66,7
о-Нитрофенол — 0 0
м-Нитрофенол — 7,2 8,7
п-Нитрофенол — 43,5 37,7
о-Аминофенол — 0 0
л-Аминофенол — 38,9 40,0
и-Аминофенол — 7,4 13,0
о-Крезол — 10,5 31,6
л-Крезол — 17,5 43,9
п-Крезол — 10,5 43,9
о-Бромфенол — 22,2 40,7
га-Бромфенол — 17,5 43,9
скорость обмена в n-нитрофеноле по сравнению с фенолом и
л1-нитрофенолом обусловлена, по-видимому, иным механизмом ре-
акции, при котором нуклеофильной атаке подвергается непротони-
рованная молекула n-нитрофенола; это становится возможным
благодаря активирующему влиянию п-нитрогруппы.
В реакции обмена, данные о которой представлены в табл. 7.3, в
качестве нуклеофила выступает вода, но эти же данные ввиду
общности механизма могут быть применены к реакциям с другими
нуклеофилами в кислой среде, в том числе к реакциям с аминами.
Действительно, сведения об активности гидроксисоединений в ре-
акции аминирования в условиях кислотного катализа [54] согла-
суются с рядом активности, вытекающим из табл. 7.3.
Аминирование моногидроксибензолов, не содержащих активи-
рующих заместителей, требует очень жестких условий и проходит с
малоудовлетворительными результатами. Например, нагревание
ж-крезола с NH4C1 при 350 °C под давлением в течение 2 ч приво-
дит к смеси равных количеств .«-толуидина и ди-.и-толиламина с
общим превращением всего 35% [36]. Для получения анилина из
фенола и толуидинов из крезолов разработаны гетерогеннокатали-
тические методы. При пропускании паров фенола или крезола и
аммиака над катализатором при 425 °C выход анилина составляет
88%, а выход м- и n-толуидинов соответственно 95% и 92% [55].
Нитрофенолы и нафтолы вступают в реакцию легче, но также
при высоких температурах. 4-Метил-2-нитрофенол (48а) превра-
щается в 4-метил-2-питроанилин (49а) при нагревании с водным
NH3 и NH4C1 при 140 °C в течение 30 ч с выходом 83% [56],
249
й 2-метил-4,6-динитрофенол (486) в 2-метил-4,6-динитроанилин
(496) в присутствии толуола при 160 °C в течение 1 ч с выходом
90% [57]. Для перевода З-гидрокси-2-нафтойной кислоты (50) в
З-амино-2-нафтойную кислоту (51) ее нагревают с водным NH3
и ZnCl2 в автоклаве 36 ч при 195 °C (выход 66—70%) [58].
a) R = CH3, R' = H;
б) R = NO2, R' = СНз
Имеющие большое техническое значение Л/'-арилнафтиламины
получают в производстве нагреванием 1- и 2-нафтола с арилами-
нами при температуре выше 200 °C в присутствии кислого катали-
затора. Так, ЛГ-фенил-2-нафтиламин — наиболее многотоннажный
продукт этой группы — получали в производстве нагреванием сме-
си 1940 кг расплавленного 2-нафтола, 1460 кг анилина и 20 кг
96%-ной H2SO4 за 5,5 ч от 170 до 260 °C с одновременной отгонкой
воды, после чего остаток перегоняли в вакууме и продукт чешуи-
ровали; выход около 95% [59, с. 231].
Нитрозофенолы, способные существовать в хиноноксимной
форме, обменивают гидроксигруппу при более низких температу-
рах. п-Нитрозофенол (52) при нагревании с NH3 и NH4C1 в ацето-
нитриле при 115 °C в течение 2,5 ч превращается в п-нитрозоанилин
(53) с выходом 88% [60], а при обработке анилином в пиридине в
присутствии BF3 при 0—35 °C — в 4-нитрозодифениламин (54) с
выходом 62% [61].
NH2
NO
(54)
250
Примерно в тех же условиях обменивают гидроксигруппу на
аминогруппу 1-нитрозо-2-нафтол и нитрозонафтолсульфокислоты
[36].
Резорцин благодаря способности переходить в оксо-форму мо-
жет обменивать одну гидроксигруппу в отсутствие катализатора.
Пропускание NH3 в расплавленный резорцин при температуре око-
ло 160°С дает л-аминофенол (55а) с выходом 85% и степенью
превращения около 30% [62], а нагревание с диметиламином при
290°C — WJV-диметил-л-аминофенол (556) с выходом 66% [63].
В присутствии борной кислоты реакция с этаноламином проходит
при 170 °C в течение 5 ч с выходом /7-(2-гидроксиэтил)-лг-аминофе-
нола (55в) 87% [64]. Для замещения гидроксигруппы в пирокате-
хине его 48 ч нагревают с анилином и ZnCl2 в ксилоле при 180—
195 °C; получается Л^-фенил-о-аминофенол (56) с выходом 59%
'[65].
(55) (56)
a) R = R' = H; б) R = R' = СН3; в) R = С2Н4ОН, R' = H
Очень легко обменивается гидроксигруппа в тригидроксибензо-
лах. Для замещения одной гидроксигруппы в флороглюцине до-
статочно обработки водным NH3 при комнатной температуре. При
действии метил- и диметиламина в воде с высоким выходом обра-
зуются соответствующие производные 3,5-диаминфенола (57), тог-
да как с диметилформамидом в водном растворе получаются про-
изводные 5-аминорезорцина (58) [66]. В жестких условиях, при
нагревании с диметиламином при 220 °C под давлением флороглю-
цин обменивает все три гидроксильные группы, превращаясь с
выходом 69% в 1,3,5-трис(диметиламино) бензол (59) [67].
(59)
В гетероциклических гидроксисоединениях, таутомерное равнове-
сие которых практически целиком сдвинуто в сторону оксо-формы,
251
диметиламиногруппа может быть введена нагреванием в гекса-
метаполе. Так, 2-хинолон (60) в результате нагревания в гекса-
метаполе при 230 °C в течение 4 ч образует 2-диметиламинохинолин
(61) с выходом 79% [68]. Считают, что реакция заключается в
непосредственной нуклеофильной атаке амидного атома, гексаме-
тапола на атом углерода группы СО с последующим разрывом
связи N—Р.
(60)
+ [(CH3)2N]3PO
(6D
Важным способом получения аминов является аминирование
гндроксисоединений в присутствии солей сернистой кислоты (ре-
акция Бухерера) [69—71]. Реакция позволяет превращать гидро-
ксипроизводные, главным образом нафталинового ряда, в первич-
ные, вторичные и третичные амины с высоким выходом в условиях,
когда при отсутствии соли сернистой кислоты гидроксисоединение
не изменяется. Так, взаимодействие 2-нафтола с ароматическими
аминами и бисульфитом натрия дает возможность получать
М-арил-2-нафтиламины в водной среде при 110—120 °C, тогда как в
отсутствие бисульфита требуется нагревание до 260 °C с отгонкой
воды (см. выше). Для получения первичных аминов гидроксисо-
единение нагревают в водном растворе NH3 и сульфита аммония,
для получения вторичных и третичных аминов помимо аминирую-
щего агента вводят сульфит натрия или аммония.
Особенно большое промышленное значение имеет реакция Бу-
херера для получения |3-аминонафталинсульфокислот, так как их
производство сульфированием 2-нафтиламина прекращено ввиду
канцерогенности последнего. В настоящее время р-аминонафта-
линсульфокислоты получают сульфированием 2-нафтола с после-
дующей заменой гидроксигруппы на аминогруппу. Так, 7-ами-
но-1,3-нафталиндисульфокислоту (63) — промежуточный продукт1
в производстве важной азосоставляющей для красителей — 6-ами-
но-4-гидрокси-2-нафталинсульфокислоты (7-амино-1-нафтол-3-
сульфокислота; Гамма-кислота) (67) получают нагреванием 7-гид-
рокси-1,3-нафталиндисульфокислоты (Г-кислота) (62) с водным
NH3 и (МНДзБОз при 185 °C; 2-амино-1-нафталинсульфокислоту
(2-нафтиламин-1-сульфокислота; кислота Тобиаса) (65) — про-
межуточный продукт в производстве Гамма-кислоты и 7-ами-
но-4-гидрокси-2-нафталинсульфокислоты (И-кислота) — готовят
взаимодействием 2-гидрокси-1 -нафталинсульфокислоты (оксикис-
лота Тобиаса) (64) с аммиаком и сульфитом аммония при 150°С4
SO3H SO3H
(62) (63)
252
SO3H
(64)
SO3H
(65)
В. производстве кислоты (65) суспензию 2,60 кмоль калиевой
соли кислоты (64) объемом 3100 л, 3,23 кмоль (NFh^SOs и
1,25 кмоль NH3 нагревают при 150°С в стальном автоклаве при
давлении до 1 МПа в течение 4—5 ч. Реакционную массу разбав-
ляют 1700 л воды, размешивают с 50 кг активного угля, фильтруют
и раствор при 50—60 °C подкисляют 35%-ной H2SO4 (около
5,6 кмоль). Продукт отфильтровывают, промывают водой и отжи-
мают, получая 75%-ную пасту кислоты (65); выход 82%.
Для получения кислоты (63) по реакции Бухерера предложена
непрерывная технологическая схема [7].
Эмпирически установлены некоторые закономерности влияния
взаимного расположения сульфо- и гидроксигруппы на протекание
реакции. Замещению гидроксигруппы препятствует сульфогруппа,
находящаяся в .«-положении, т. е. в З-гидрокси-1-нафталинсульфо-
кислоте и в 4-гидрокси-2-нафталинсульфокислоте, а также суль-
фогруппа в 1-гидрокси-2-нафталинсульфокислоте. Способствует
замещению сульфогруппа в 4-гидрокси-1-нафталинсульфокислоте
и в 2-гидрокси-1-нафталинсульфокислоте. Различная подвижность
гидроксигруппы в зависимости от положения сульфогрупп позво-
ляет осуществлять с помощью реакции Бухерера избирательное
аминирование полигидроксипроизводных. Так, аминирование
4,6-дигидрокси-2-нафталинсульфокислоты (66) приводит исключи-
тельно к образованию Гамма-кислоты (67), а аминирование
4,5-дигидрокси-1-нафталинсульфокислоты (68)—только к 4-амино-
5-гидрокси-1-нафталинсульфокислоте (69).
Фениламинирование Гамма-кислоты (67) затрагивает только
аминогруппу, но не находящуюся в .«-положении к сульфогруппе
гидроксигруппу (образуется фенил-Гамма-кислота — см. 7.1.3).
253
Реакция Бухерера применима также к гидроксипроизводным
антрацена, фенантрена и к гетероциклическим соединениям, со-
держащим гидроксигруппу в аннелированном бензольном кольце,
например к 6-гидрокси- и 8-гидроксихинолину. В бензольном ряду
она описана для производных резорцина и л-аминофенола.’
Роль солей сернистой кислоты в реакции Бухерера была пред-
метом интенсивных исследований. Длительное время считалось, что
бисульфит присоединяется к группе СО оксо-формы нафтола с
образованием а-гидроксисульфокислот типа (70), которые и обме-
нивают гидроксигруппу на остаток амина. Получение и установле-
ние структуры продукта присоединения бисульфита к 1-нафтолу
[72] и 1-антролу [73], а затем получение бисульфитного соедине-
ния 2-нафтола из 2-нафтолкарбоновых кислот [74] положило конец
многолетней дискуссии о строении бисульфитных соединений. Вы-
деленные аддукты нафтолов с бисульфитом имеют строение солен
л/-тетралонсульфокислот — 4-оксо-2-тетралинсульфокислоты (71)
и З-оксо-1-тетралинсульфокислоты (72).
ОН
SO3Na
(70)
На основании этого факта для реакции Бухерера принята схе-
ма, выдвинутая Богдановым еще в 1932 г. [75]. Согласно ей,
аминированию подвергаются ля-тетралонсульфокислоты, после че-
го образовавшиеся енамины стабилизируются путем отщепления
бисульфита. Обратная реакция приводит от аминов к гидроксисо-
единениям. Для 1-нафтола схема имеет вид:
ОН о
(71)
—NaHSO3
NRR'
Следует отметить, что несмотря на установление строения би-
сульфитных соединений нафтолов предложенная схема реакции
254
Бухерера носит гипотетический характер. Нельзя йе согласиться С
мнением [76, с. 1788] о том, что выделение бисульфитных соеди-
нений (71) и (72), как и вообще выделение при реакции какого-
либо соединения, само по себе не может служить доказательством
того, что это соединение является промежуточным продуктом. Для
выяснения истинного механизма реакции Бухерера необходимы
специальные исследования.
В ряду 9,10-антрахинона замещение гидроксигруппы находит
основное применение для получения разнообразных 1,4-диамино- и
1-амино-4-гидрокси-9,10-антрахинонов из хинизарина и его произ-
водных. Для этого хинизарин (73) восстанавливают в лейкосоеди-
нение, которое, как доказано с помощью ПМР-спектров [77], су-
ществует не в виде 1,4,9,10-антратетрола, а в более стабильной
таутомерной форме 2,3-дигидро-9,10-дигидрокси-1,4-антрахинона
(74). В последнем две оксогруппы постадийно аминируются амми-
аком, первичным алкил- или ариламином и образовавшиеся лейко-
соединения аминопроизводных (75) и (76) окисляются кислородом
воздуха или другим окислителем в амино-9,10-антрахиноны (77)
и (78). В отсутствие восстановителя происходит замещение не гид-
роксигрупп, а атома водорода в положении 2 хинизарина
(см. 7.1.6).
(77)
Восстановление хинизарина (73) в лейкосоединение (74) осу-
ществляют дитионитом («гидросульфитом») натрия Na2o204 или
255
цинковой пылью. Первый способ чаще применяют, если лейкосо-
единение выделяют и затем вводят в реакцию с аминами, второй —
когда восстановление совмещают с реакцией аминирования.
Нередко восстанавливают лишь часть хинизарина, тогда как
остальная часть переходит в лейкосоединение за счет окисления
образующейся лейкоформы аминогидрокси- или диаминоантрахи-
нона. Например, 1-гидрокси-4-(п-толиламино)-9,10-антрахинон—
промежуточный продукт для фиолетового кислотного красителя —
получают взаимодействием хинизарина с n-толуидином в кипящем
этаноле в присутствии соляной кислоты и менее 0,5 моль цинковой
пыли на 1 моль хинизарина. При проведении реакции в среде
n-толуидина с добавкой борной кислоты при 100 °C образуется
1,4-ди(п-толиламино)-9,10-антрахинон — промежуточный продукт
для зеленого кислотного красителя [78]. Реакция лейкохинизарина
(74) с этилендиамином сопровождается внутримолекулярным за-
мещением атома водорода в положении 2 с замыканием тетрагид-
ропиразинового цикла [79].
Замещение гидроксигруппы в ядре антрахинона без перевода в
лейко-форму применяют для получения 1-амино-2,4-дигидрок-
си-9,10-антрахинона (81) из пурпурина (80) при действии аммиака,
а также 1-гидрокси-2,4-ди(фениламино)-9,10-антрахинона (79) при
взаимодействии с анилином в присутствии борной кислоты:
(81)
7.1.6. Замещение алкокси-, арилокси- и ацилоксигрупп
По сравнению с замещением гидроксигрупп нуклео-
фильное замещение алкокси-, арилокси- и ацилоксигрупп имеет
меньшее значение. Из них алкоксигруппы наименее подвижны
(см. табл. 7.1), однако при синтезах замещение их используется
чаще. Это связано с большей доступностью алкоксисоединений по
сравнению с арилоксисоединениями и с большей устойчивостью их
256
к гидролитическому расщеплению по сравнению с ацилоксисоедй-
нениями. Алкилирование гидроксигруппы может облегчать переход
от гидроксисоединения к амину. Так, реакция м-нитрозофеиола
(52) с анилином проходит в пиридине в присутствии BF3 с посред-
ственным выходом (см. выше), тогда как алкиловые эфиры п-нит-
розофенола (82) превращаются в 4-нитрозодифениламин (54) в
спирте в присутствии кислоты при 25—50 °C с выходом, близким к
количественному [80].
Замещение гидроксигруппы алкиламиногруппой в 1-гидро-
кси-4-ариламиноантрахинонах (83) не удается осуществить, не за-
трагивая ариламиногруппы из-за побочной реакции переаминиро-
вания, но после метилирования — в соединениях (84)— метокси-
группа гладко обменивается на остаток алкиламина (85) в среде
кетонного растворителя [81]:
(83) (84) (85)
Метокси- и феноксигруппа в производных бензола, так же как
атом хлора, замещаются на аминогруппу при сплавлении с моче-
виной, если в орто- и пара-положении к ним имеются сильные
электроноакцепторные заместители [21]. Замещение метоксигруп
пы в нитриле 1-метокси-4-нитро-2-нафтойной кислоты проходит
при нагревании с водным аммиаком в этаноле при 70 °C [82];
NO»
NO2
9 Зак, 271
267
Очень легко взаимодействуют с нуклеофильными реагентами
О-арилсульфонильные производные фенолов. Соотношение скоро-
стей реакции с пиперидином в диметилсульфоксиде для 1-меток-
си-4-нитробензола, n-нитродифенилового эфира и и-нитрофенило-
вого эфира n-толуолсульфокислоты составляет 1:17,6:3230 (ср.
табл. 7.1). Однако практическое использование арилсульфонил-
оксигрупп в качестве уходящих в реакциях аминирования затруд-
нено из-за того, что нуклеофильная атака амина идет в двух на-
правлениях: по ароматическому атому углерода (а) и по атому
серы (б) (см. 2.5).
Например, взаимодействие 2,4-динитрофенилового эфира п-то-
луолсульфокислоты (86) с пиперидином в метаноле при 0 °C лишь
на 55% протекает в направлении (а) с разрывом связи С—О и
образованием ариламина (87); на 45% реакция идет в направлении
(б) с разрывом связи S—О и образованием сульфамида (88).
В 60%-ном водном диоксане при 40 °C реакция на 90% идет в
направлении (б) [83].
Реакцией внутримолекулярного аминирования является пере-
группировка Смайлса [84], при которой происходит замещение
алкокси- или арилоксигруппы, а также алкил (арил) сульфонильной
и других групп аминогруппой, находящейся в той же молекуле
(см. также 7.1.4). В частности, 2-амино-2',4'-динитродифениловые
эфиры (89) при кратковременной обработке смесью уксусной кис-
лоты и пиридина (4:1) при 50 °C с количественным выходом
переходят в 2-гидрокси-2',4'-динитродифениламины (90) [84];
1-амино-2-арилокси-4-гидрокси-9,10-антрахиноны (91) при нагре-
вании с КОН в диметилсульфоксиде превращаются в 1-арилами-
258
но-2,4-дигидрокси-9,10-антрахиноны (92) [85]. Реакция протекает
через спироциклические ст-комплексы (см. 2.7.2).
(89)
(92)
Интересен пример двойной перегруппировки Смайлса гидро-
ксиалкиламидов нитробензолсульфокислот, приводящей к элими-
нированию фрагмента — SO2 — из молекулы [86]. Так, при
20-минутном кипячении в 6% водном растворе NaOH Д/-(2-гидро-
ксиэтил)-4-нитробензолсульфамид (93) с выходом 96% превраща-
ется в Л^-(2-гидроксиэтил)-4-нитроанилин (94). На первой стадии в
результате перегруппировки Смайлса сульфамидная группа внут-
римолекулярно замещается алкоксигруппой, после чего гидроли-
тически отщепляет сульфит и при второй перегруппировке алкок-
сигруппа замещается аминогруппой:
Н2С-СН2
МО2
9*
259
7.1.7. Замещение атома водорода
С практической точки зрения нуклеофильное замещение
водорода особенно привлекательно, так как избавляет от необхо-
димости сначала с помощью электрофильных реакций вводить
вместо атома водорода подходящий заместитель и только потом,
иногда после дополнительных операций, вытеснять его действием
нуклеофильного реагента. Сравнительно малое распространение
реакций нуклеофильного замещения водорода объясняется труд-
ностью удаления гидрид-иона Н_. В отличие от других уходящих
атомов и групп гидрид-ион не способен к анионной стабилизации
(см. 2.7.2). Он не известен в органической химии как кинетически
независимая частица, и следует полагать, что перенос его всегда
происходит с участием акцептора электронов; им может служить
специально вводимый окислитель, сам субстрат или отщепляемая
вместе с гидрид-ионом частица.
Замещение водорода аминогруппой возможно в ароматических
и гетероароматических соединениях, содержащих электроноакцеп-
торные атомы и группы. Особое место занимают высокотемпера-
турные гетерогеннокаталитические методы получения аминов из
бензола или его гомологов и аммиака, например, получение анили-
на на никелевом катализаторе [87].
Аминирующими агентами при нуклеофильном замещении водо-
рода в активированных ароматических соединениях служат гидро-
ксиламин в присутствии щелочей и амиды щелочных металлов.
Первый применяют главным образом для аминирования аромати-
ческих нитропроизводных, вторые — для аминирования азотсодер-
жащих гетероциклов по Чичибабину. В хинонах и некоторых
полициклических кетонах замещение водорода может происходить
непосредственно при действии аминов.
Аминирование нитросоединений гидроксиламином в щелочной
среде позволяет получать с удовлетворительным выходом первич-
ные амины бензольного и нафталинового рядов [27]. Для протека-
ния реакции в бензольном ряду необходимо наличие не менее двух
нитрогрупп или нитрогруппы и других электроноакцепторов; в ряду
нафталина достаточно присутствия одной нитрогруппы. Реакцию
проводят в этиловом или метиловом спирте при нагревании с
гидрохлоридом гидроксиламина и гидроксидом щелочного метал-
ла; затем разбавляют реакционную смесь водой. Аминогруппа
вступает в орто- или пара-положение к нитрогруппе.
/«-Динитробензол и его производные (95) при действии гидро-
ксиламина и щелочи образуют смесь 1-амино-2,4-динитро- (98) и
1,3-диамино-2,4-динитрозамещенных (99). В случае алкоксисоеди-
нений (956) выделено также гидроксиламинозамещенное (ЮО).
Соотношение продуктов реакции зависит от природы заместителя
R, количества реагентов и других условий, меняя которые удается
направить реакцию в сторону преимущественного образования од-
ного из продуктов. Так, 1,3-диамино-2,4-динитробензол (99а) по-
260
лучен из Л!-динитробензола (95а) с выходом 90% [27], а моноами-
нозамещенные (98) из алкокси-2,4-динитропроизводных (956) с
выходом 55—60% и из 2,4-динитротиопроизводных (95в) с выхо-
дом 80—90 % [88].
Реакция заключается в нуклеофильной атаке W-аниона, генери-
руемого из гидроксиламина при действии щелочного агента, с
последующим разложением ст-комплекса при обработке водой.
В случае ^-динитробензолов (95) присоединяются две частицы
нуклеофила с образованием дианионных ст-комплексов типа (96) и
(97). Превращение в моноаминосоединение (98) или в диамино-
соединение (99) зависит от того, разлагается ли ст-комплекс (96)
при гидролизе с отщеплением молекулы воды и гидроксиламина
или с отщеплением двух молекул воды. Гидроксиламинопроизвод-
ное (ЮО) возникает в результате замещения алкоксигруппы
остатком гидроксиламина с промежуточным образованием ст-ком-
плекса (97). Опытами с полностью дейтерированным Л1-динитро-
бензолом показано [89], что атом водорода ядра входит в вводи-
мую аминогруппу. Следовательно, о-комплекс стабилизируется не
путем удаления гидрид-иона из молекулы, а путем внутримолеку-
лярного переноса его к атому азота с отрывом от последнего гид-
роксид-аниона. Именно этим обстоятельством объясняется, по-ви-
димому, легкость протекания реакции.
Амины (98) аммонолизом количественно переводятся в диамин
(Ю1), являющийся промежуточным продуктом для получения
1,2,5,6-тетраминобензола — ценного мономера для термостойких
волокон [88].
Взаимодействие динитрила 5-нитроизофталевой кислоты с гид-
роксиламином в щелочной среде предложено для синтеза 2,6-ди-
Циано-4-нитроанилина — промежуточного продукта для дисперс-
ных красителей [90]. Тем же методом из 1-нитронафталина с
261
выходом 55—60% получают 4-нитро-1-нафтиламин [91], а из
2-нитронафталина — 2-нитро-1-нафтиламин.
Подобно реагируют гетероциклические аналоги нитронафтали-
нов, например 4- и 5-нитробензо-1,2,5-тиа- и -селенадиазолы (102)
[92] или полициклические нитросоединения — 9-нитрофенантрен,
no2
Замещение водорода аминогруппой в гетероциклах по Чичиба-
бину [94] используется в промышленности для получения 2-ами-
нопиридина и 2,6-диаминопиридина и применимо к соединениям
хинолина, изохинолина, фенантридина, перимидина, бензимидазо-
ла, бензотиазола и некоторых других TV-гетероциклов. По легкости
аминирования они располагаются в ряд 1-метилбензимидазол >
> изохинолин > 1-метилперимидин > бензо [f] хинолин > пири-
дин акридин [95]. Аминогруппа вступает в a-положение к пи-
ридиновому азоту и только если эти положения заняты — с боль-
шим трудом в у-положение. Процесс сопровождается выделением
водорода и аммиака. Его проводят обычно при нагревании гете-
роциклического соединения с амидом натрия при температуре
выше 100°C в углеводороде (толуол, ксилол, декалин и др.), ди-
метиланилине или в эфирном растворителе (диоксан, анизол) и
обрабатывают реакционную смесь водой.
Реакция протекает через промежуточное образование ст-комп-
лексов типа (ЮЗ), присутствие которых доказано с помощью спек-
тров ПМР [96]. Стадия образования ст-комплекса оказывает на
262
скорость реакции лимитирующее влияние. Активность гетероцикла
зависит от его основности, определяющей сорбцию соединения на
поверхности NaNH2 за счет ион-дипольного взаимодействия между
ионом Na+ и пиридиновым азотом; от величины положительного
заряда 6+ на а-атоме углерода; от поляризуемости связи C=N,
обеспечивающей делокализацию отрицательного заряда, приноси-
мого ионом NH2. Ингибирование реакции в присутствии кисло-
рода, азобензола, нитробензола дает основание для предположе-
ния о существовании радикальной стадии одноэлектронного
переноса от амид-аниона к субстрату, предшествующей образова-
нию ст-комплекса [95] (см. 2.7.2).
Разрушение ст-комплекса происходит, вероятно, с участием би-
функционального катализатора, роль которого играет натриевая
соль гетариламина. Катализатор отрывает протон от атома азота
группы NH2, передавая его через шестичленное циклическое пере-
ходное состояние (Ю4) на соединение с гидрид-ионом, в результате
чего и элиминируется молекула водорода [95]. Изучение аминиро-
вания фенил-1,3,5-триазина по Чичибабину при действии K15NH2 в
жидком 15NH3 показало, что в образовавшемся 2-амино-4-фе-
нил-1,3,5-триазине (105) 55% меченого атома находится в гетеро-
цикле и, следовательно, реакция на 55% идет по механизму
ANRORC [97] (см. 2.7.2).
Иногда в условиях близких к условиям реакции Чичибабина
удается осуществить введение алкил- или ариламиногруппы. Так,
при действии пиперидина и NaNH2 1-нитронафталин переходит в
1- (4-нитро-1-нафтил) пиперидин [37].
Замещение атома водорода непосредственно при действии ами-
нов типично для хинонов, имеющих доступное для атаки хиноидное
кольцо [98]. Эта реакция состоит в нуклеофильной атаке амино-
азота по атому углерода ненасыщенной связи хинона и окислении
образующегося аддукта молекулой исходного хинона; последний
при этом восстанавливается в гидрохинон и затем регенерируется
при действии кислорода воздуха. Суммарное превращение
263
соответствует замещению атома водорода с участием окислителя,
например:
В некоторых случаях в Подобную реакцию вступают и произ-
водные 9,10-антрахинона, хотя у них оба крайние кольца аромати-
ческие. При нагревании 2-гидрокси-9,10-антрахинона (106) в авто-
клаве е водным ЫНз с выходом около 70% образуется 1-ами-
но-2-гидрокси-9,10-антрахинон, а при аналогичной обработке
1-гидрокси-9,10-антрахинона (107) — небольшое количество 2-ами-
но-1-гидрокси-9,10-антрахинона [45]. При действии на 1-гидро-
кси-9,10-антрахинон вторичных аминов в присутствии кислорода
воздуха остаток амина вступает в положение 2 или 4 уже при
комнатной температуре. Аминирование хинизарина (73) вторичны-
ми и первичными аминами направляется в положение 2, приводя к
2-амино-1,4-дигидрокси-9,10-антрахинонам [99]. Реакция значитель-
но облегчается при образовании хелатных комплексов хинизарина
с соединениями борной кислоты [100]. Направление аминирования
указано в формулах стрелками:
В мягких условиях способны также обменивать атом водорода
на остаток амина некоторые ангулярно конденсированные гетеро-
циклические производные 9,10-антрахинона (108), (109), (110)
[101] и п^рм-конденсированные производные антрона — 3-мети-
лантрапиридон (111) [102], керамидонин (112) [103].
264
о
Z = O, S, Se, NR
Изучение реакции Af-замещенных антрахинонтриазолов (113) с
пиперидином позволило выявить различное влияние структурных
факторов на легкость замещения атомов водорода и галогена
[104]. Из трех изомерных Af-метилантрахинонтриазолов (113а-в)
при замещении атома хлора (X = С1) наиболее активен М'-метил-
антрахинонтриазол (113а), тогда как при замещении водорода
(X = Н) он наиболее инертен. Введение заместителей в орто-поло-
жение фенильного кольца №-фенилантрахинонтриазола (113г) не-
зависимо от их электронной природы ускоряет замещение хлора
и замедляет замещение водорода. Эти различия отражают, по-
видимому, различие механизмов, заключающееся в том, что обра-
зующийся на первой стадии о-комплекс при замещении гало-
гена (Х = С1) распадается с отрывом аниона (114)->(116), а при
замещении водорода (Х = Н)—с отрывом протона (115)->(117)
и переносом пары электронов в ядро. При замещении галогена
главное значение имеют факторы, способствующие образованию
о-комплекса, а при замещении водорода — способствующие от-
щеплению протона.
Таким образом, во всех рассмотренных случаях замещения
водорода аминогруппой гидрид-ион фактически не является ухо-
дящей частицей, и подобные реакции могут лишь формально счи-
таться реакциями с вытеснением гидрид-иона. Это положение
265
сохраняет силу и применительно к нуклеофильному замещению
водорода другими группами, которое рассматривается в после-
дующих главах.
7.2. ВВЕДЕНИЕ ГИДРАЗИНОГРУПП
Гидразин, алкил- и арилгидразины являются более
сильными нуклеофилами, чем соответственно аммиак, алкил- и
ариламины (см. 2.5). Поэтому большинство реакций нуклеофиль-
ного замещения, которые могут быть реализованы для введения
аминогрупп (см. 7.1), пригодны и для введения гидразиногрупп.
Ограничения связаны главным образом с восстановительными
свойствами гидразинов, вызывающими нежелательные побочные
реакции. Меньшее распространение реакции введения гидразино-
групп по сравнению с реакцией аминирования обусловлено мень-
шим значением ароматических гидразинопроизводных и меньшей
доступностью замещенных гидразинов как реагентов по сравнению
с аминами.
Замещение в активированных ароматических соединениях про-
водят обычно нагреванием с гидразингидратом в спирте, пиридине
или другом растворителе или в среде самого гидразингидрата
[105]. При использовании солей гидразина реакцию ведут в при-
сутствии оснований. Так, кипячение 1,3-динитро-4-хлорбензола (4в)
с сульфатом гидразина и ацетатом калия в этаноле служит мето-
дом получения 2,4-динитрофенилгидразина (выход 81—85%)
[106]. Последний образуется также при нагревании с гидразин-
гидратом 1-метокси-2,4-динитробензола (2,4-динитроанизола)
[54]:
(4в)
+ NH2NH2
-------->
+ NH2NH2
<--------
Взаимодействие гидразингидрата с 5-нитро-2-хлорбензонитри-
лом (4г) приводит к 2-гидразино-5-нитробензонитрилу (118), ко-
торый далее циклизуется в З-амино-5-нитроиндазол (119) — про-
межуточный продукт для катионных красителей:
+ nh2nh2
-------->
no2 no2
(4г) (J18) (1|9)
?6₽
а- и р-Галогенантрахиноны переходят при действии гидразин-
гидрата в соответствующие гидразинопроизводные, что находит
применение для получения 1-гидразино-9,10-антрахинона из
1-хлор-9,10-антрахинона для последующего превращения в антра-
пиразол (пиразолантрон) (120)—промежуточный продукт для
кубовых красителей [78]:
При обработке хлоранила гидразингидратом в присутствии ме-
тилата натрия, так же как и при аминировании (7.1.1), замещаются
два атома хлора и образуется 2,5-дигидразино-3,6-дихлор-1,4-бен-
зохинон (121). При реакции 2-хлор-1,4-нафтохинона с ацилгидра-
зинами замещается не атом хлора, а атом водорода (122) [97];
Реакции с н-алкилгидразинами, где более нуклеофилен алки-
лированный атом азота, приводит к Л^-алкил-А/-арилгидразинам,
например:
ArCl + CH3NHNH2 —> ArN(CH3)NH2
В качестве реагентов могут быть использованы также гидразо-
ны кетонов, реакция которых с 2,4-динитрохлорбензолом предло-
жена для их количественного определения [107]:
ZR /R
ArCl + NH2N=C^ —> ArNHN=C^
XR' \R'
Нитрогруппа, обладающая достаточной подвижностью для за-
мещения аминогруппой, замещается и при реакции с гидразином.
267
Например, в 3,4,5-тринитротолуоле (123) легко замещается про-
странственно затрудненная нитрогруппа в положении 4. При дей-
ствии гидразингидрата на 2,3-динитронафталин, как и при дей-
ствии амина (см. 7.1.2), с высоким выходом образуется продукт
киме-замещения— З-нитро-1-нафтилгидразин (124) [108].
NO2
СН8
NHNH2
no2
(123)
(124)
Гидроксигруппа может быть замещена гидразиногруппой при
длительном кипячении в гидразингидрате, как, например, в случае
2-антрола [109], или в условиях реакции Бухерера [36, 37].
Для введения гидразиногруппы в неактивированные к нуклео-
фильному замещению ароматические соединения необходимо ис-
пользовать гидразиды щелочных металлов в безводном гидразине
[ПО]. В этих условиях фторбензол и n-фтортолуол с выходом 95—
96% превращаются в фенил- и n-толилгидразин, а бензол- и
2-нафталинсульфокислота — в фенил- и 2-нафтилгидразин со-
ответственно.
Взаимодействие с гидразинами применяется для синтеза гете-
роциклических азосоединений, получить которые диазотированием
и азосочетанием невозможно, например арилазотриазинов. Реак-
ция хлор-1,3,5-триазинов (125) с гидразингидратом и последующая
конденсация с а-дикетонами приводит к гидроксиазосоединениям
(126) [111]; реакция с арилгидразинами с последующим окисле-
нием— карилазосоединениям (127) [112].
Бензазолы общей формулы (128) легко обменивают замести-
тели X в положении 2 при действии гидразингидрата. Так,
268
2-аминобензотиазолы (128) при 1 — S, X = NH2 дают 2-гидрази-
нобензотиазолы (129) с выходом 82—93% при нагревании с гид-
разингидратом и гидрохлоридом гидразина в этиленгликоле при
140°C [ИЗ]. Еще легче осуществляется замещение в кватернизо-
ванных соединениях (130) с образованием гидразинов (131). Соли
2-амино- и 2-диметиламино-./У-метилбензотиазолия (130)npnZ=S
превращаются в гидразоны (131) кипячением с гидразином в
водном растворе [114, 115]. 2-Гидразоно-А^-метилбензотиазол по-
лучают из 2-меркаптобензотиазола (каптакса) ?/,.$-диметилирова-
нием с последующим замещением метилтиогруппы в кватернизо-
ванном соединении.
(129}
(130) (131)
Z=O,S,NH,NCH3; Х=С1, SO3H, SCH3, NH2,N(CH3)2
2-Гидразинобензазолы (129) и 2-гидразоно-А(-алкилбензазолы
(131) служат промежуточными продуктами для синтеза моноазо-
красителей методом окислительного сочетания.
7.3. ВВЕДЕНИЕ НИТРО- И АЗИДОГРУППЫ
Нуклеофильное замещение действием нитрит-аниона
NO 2 позволяет вводить нитрогруппу в такие положения ароматиче-
ской молекулы, куда она не может быть введена электрофильной
реакцией нитрования, а именно в орто- или пара-положение к
электроноакцепторному заместителю, в частности к другой нитро-
группе. Нуклеофильное замещение нитрит-анионом используется
для перевода аминогруппы в нитрогруппу через стадии диазотиро-
вания и последующего обмена диазониевой группы или, реже, для
непосредственного обмена атома галогена, и иногда водорода.
Замещение диазониевой группы реакцией с NO2 проводят в
нейтральной или щелочной среде как с катализаторами — соеди-
нениями меди, так и без них [116]. Для удаления кислоты соль
диазония осаждают в виде сульфата, борфторида, кобальтинитри-
та или же раствор диазосоединения нейтрализуют после диазо-
тирования. Хорошие результаты при получении о- и п-динитро-
бензолов из соответствующих нитроанилинов (выход 97%) дает
269
приливание раствора соли диазония к раствору, содержащему из-
быток NaNO2 и NaHCO3 [117].
Изучение взаимодействия выделенных борфторидов нитробен-
золдиазония с водными растворами NaNO2 в отсутствие катали-
затора [118] показало, что реакция имеет суммарный третий по-
рядок: первый — по соли диазония и второй — по нитриту. Это
объяснено последовательностью двух бимолекулярных реакций, в
первой из которых соль бензолдиазония присоединяет нуклеофил с
образованием нестойкого бензолдиазонитрита, далее взаимодей-
ствующего со вторым нитрит-анионом по типу SnAf:
Замещение диазониевой группы нитрит-анионом в присутствии
катализатора идет по иному механизму, типичному для реакции
Зандмайера (см. 10.3.) Данный способ дает лучшие результаты в
ряду нафталина и при отсутствии в солях бензолдиазония сильных
электроноакцепторных заместителей. Для получения 1,4-динитро-
нафталина из 4-нитро-1-нафтиламина раствор осажденного и тща-
тельно отмытого от кислоты сульфата 4-нитро-1-нафталиндиазония
вливают в раствор нитрита натрия в присутствии сульфитов меди
(выход 52—60%) [119].
Нитрит-анион, будучи достаточно сильным нуклеофилом, спо-
собен замещать атомы галогенов, активированные влиянием элек-
троноакцепторных групп. Практическое использование этой реак-
ции затруднено амбидентным характером нитрит-аниона, который
может атаковать ароматическое кольцо как атомом азота, так и
атомом кислорода. Соотношение N- и О-атаки зависит от замести-
теля в субстрате, природы уходящей группы и растворителя
(см. 2.5 и 2.6). В случае О-атаки (а) образующиеся нитрозоэфиры
гидролитически разрушаются до фенолов. В случае A-атаки (б)
вступившая нитрогруппа может вытесняться при О-атаке другого
нитрит-аниона (в). В результате в обоих случаях конечными про-
дуктами оказываются гидроксисоединения (см. 8.1) в соответствии
со схемой [120]:
(в) -
<-ArX + 2 NOj q, ..,=£ ArNO2 + X + ЫОг -i
(а) (в)
1-------► [ArONO] + NO; --------------1
J
ArO + N2O3
270
Однако известны примеры, когда замещение галогенов при
действии NaNO2 удается использовать для синтеза нитросоедине-
ний. Так, дигалогенантрахиноны при нагревании с нитритом натрия
в диметилформамиде переходят с высоким выходом в гидроксинит-
роантрахиноны. Из 2,3-дихлор-9,10-антрахинона (132) таким обра-
зом получают 2-гидрокси-3-нитро-9,10-антрахинон (133) [121], а из
5,8-дихлорхинизарина (134)-1,4,5 -тригидрокси-8-нитро-9,10-ант-
рахинон (135) [122].
(132) (133)
(134) (135)
Нитрит-анион атакует хиноидные системы, приводя к образо-
ванию нитропроизводных соответствующих двухатомных фенолов.
Данная реакция может быть использована для замещения атома
водорода на нитрогруппу действием нитрита в присутствии окис-
лителя, роль которого состоит в генерировании хинона на проме-
жуточной стадии. Например, пирокатехин при действии NaNO2 и
Ks[Fe(CN)e] в водном растворе СНзСООК переходит в 4-нитропи-
рокатехин [123]:
он о он
no2
Реакция может быть расчленена на стадии окисления и нукле-
офильного присоединения. Так, антрахинонимидазолин (136) при
окислении превращается в 2Д-антрахинонимидазол (137), который
271
при обработке нитритом натрия в диметилформамиде дает 4-нит-
роантрахинонимидазолин (138) [124]:
Азид-анион N3 по нуклеофильности близок нитрит-аниону
[120] и так же, как последний, может вводиться замещением
диазониевой группы или атомов галогена, нитрогруппы и др. [125].
Ароматические азидосоединения находят применение в качестве
светочувствительных материалов [126] и промежуточных продук-
тов в некоторых синтезах. Для замещения диазониевой группы на
азидную нет необходимости, как при замещении нитрогруппой, в
удалении кислоты после диазотирования. о-Нитрофенилазид (139),
например, получают с выходом 94—97% добавлением раствора
азида натрия в солянокислый раствор диазотированного о-нитроа-
нилина [127]. Азид (139) термическим разложением переводят в
бензофуроксан (бензо-1,2,5-оксадиазол-А-оксид) (140), служащий
исходным соединением для разнообразных синтезов [128].
(139) (140)
Для замещения галогенов или нитрогруппы в активированных
ароматических соединениях достаточно обработки азидом натрия в
спирте или, лучше, в биполярном апротонном растворителе. Обмен
нитрогруппы на азидогруппу в 2-нитро-6-хлорбензонитриле, на-
пример, протекает в диметилформамиде при 25 °C с выходом
74% [29].
ЛИТЕРАТУРА
1. Suhr //. — Chem. Вег., 1964, Bd. 97, № 12, S. 3268—3276. 2. Шейн С. М„
Литвак В. В.—Журн. ВХО, 1976, т. 21, вып. 3, с. 274—279. 3. Ворожцов Н. Н.,
мл., Кобелев В. А. — ЖОХ, 1939, т. 9, вып. 17, с. 1569—1576. 4. Aral S., Hida М.,
Yamagishi Т.— Bull. Chem. Soc. Japan, 1978, v. 51, № 1, p. 277—282. 5. Ba-
con R. G. R., Hill H. A. 0. — Quart. Rev., 1965, v. 19, N 2, p. 95—125.
6. Tuong T. D., Hida M.— J. Chem. Soc. Perkin 2, 1974, № 6, p. 676—682.
7. Беркман Б. E. Промышленный синтез ароматических нитросоединений и ами-
нов. М., Химия, 1964, с. 233—241, 256—273 . 8. Sanger F.— Biochem. J., 1945,
v. 39, р. 507—517. 9 Krauss R. C., Reinheimer J. D. — Can. J. Chem., 1967, v. 45,
№ 1, p. 77—79. 10. Elias D. H. D., Parker R. E. — J. Chem. Soc., 1962, № 7,
p. 2616—2624; Parker R. E., Read T. 0. — Ibid., 1962, № 8, p. 3149—3153.
11. Nudelman N. S., Garrido D.—J. Chem. Soc. Perkin 2, 1976, № 11,
p. 1256—1260. 12. Штерншис M. В., Шейн С. M. — ЖОрХ, 1971, т. 7, вып. 8,
с. 1707—1711. 13. Фокин Е. П., Лоскутов В. А. — ЖОХ, 1967, т. 38, вып. 8,
с. 1884—1888. 14. Фокин Е. П., Русских В. В. — ЖОрХ, 1966, т. 2, вып. 5,
с. 907—912, 912—916. 15. Lynch J., Meth-Cohn О. J. — Chem. Soc. Perkin 1, 1973,
№ 9, p. 920—926. 16. Ворожцов H. H., мл., Якобсон Г. Г., Рубина Т. Д.— ДАН
СССР, 1959, т. 127, № 6, с. 1225—1227. 17. Красносельская М. И., Гудзен-
ко В. //. — Журн. ВХО, 1971, т. 16, № 1, с. 116—117; ЖОрХ, 1972, т. 8, № 11,
с. 2379—2382. 18. Курдюмова Т. Н. — В кн.: Химия антрахинона/Под ред.
А. И. Королева. М., Химия, 1969, с. 94—104. 19. Франц, пат. 2008169, 1970;
Chem. Abstrs., 1971, v. 74, 3439. 20. Philip G., Sunthankar S. — Chem. Ind., 1975,
№ 10, p. 433.
21. Лаврищев В. А., Плакидин В. Л., Кретов А. Е. — Изв. вузов. Химия
и хим. технол., 1960, № 1, с. 127—129; ЖОХ, 1960, т. 30, вып. 9, с. 3064—3072.
22. Wakae М., Hamano К — Bull. Chem. Soc. Japan, 1963, v. 36, № 2, p. 230;
Deorha D. S., Sharma H. L.—J. Indian Chem. Soc., 1963, v. 40, № 9, p. 819—820.
23. Josef L., Albert A. H. — J. Heterocycl. Chem., 1966, v. 3, № 1, p. 107—108.
24. Pedersen E. D.t Perregard J., Lawesson S.-O. — Tetrahedron, 1973, v. 29,
№ 24, p. 4211—4213. 25. Lord W. M., Peters A. T. — Chem. Ind.. 1973, № 5,
p. 227—228. 26. Докунихин H. С., Шейн С. M., Богуславская И. Л. Журн. ВХО,
1965, т. 10, № 5, с. 596—597. 27. In: Methoden der Organischen Chemie (Houben-
Weyl). Bd. 11/1. Stuttgart, Georg Thieme Verlag, 1957, S. 24—267. 28. Smith-
wick E. L., Shuman R. T. — Synthesis, 1974, № 8, p. 582. 29. Beck J. R., Sob-
szak R. L., Suhr R. G. e.a. — J. Org. Chem., 1974, v. 39, № 13, p. 1839—1845.
30. Guanti G., Thea S., Dell’Erba C. — Tetrahedron Letters, 1976, № 6, p. 461—462.
31. Штерншис M. В., Якушева Г. А., Шейн С. M. — ЖОрХ, 1972, т. 8, вып. 1,
с. 173—177. 32. Яп. пат. 76/32552, Chem. Abstr., 1976, v. 85, 94143; Пат. ФРГ
2409542, 1975, Chem. Abstr., 1975, v. 83, 181116. 33. Пат. ФРГ 2211411, 1972;
Chem. Abstr., 1973, v. 78, 17637. 34. Пат. ФРГ 2541663, 1976; Chem. Abstr., 1976,
v. 85, 48269. 35. Пат. ФРГ 1277475, 1969; РЖ Хим., 1970, 9Н242. 36. Ворож-
цов Н. Н. Основы синтеза промежуточных продуктов и красителей. М., Госхим-
издат, 1955, с. 400—418, 466—482. 37. Вейганд-Хильгетаг. Методы эксперимента
в органической химии. Пер. с нем./Под ред. Н. Н. Суворова. М., Химия, 1968,
с. 502—505. 38. Попов С. И., Курдюмова Т. Н., Докунихин Н. С. — Химия гете-
роцикл. соед., 1968, № 5, с. 869—872. 39. Krohnke F. — Angew. Chem., 1963,
Bd. 75, N 7, S. 317—329. 40. Казанков M. В., Пуца Г. И., Мухина Л. Л. — Химия
гетероцикл, соед., 1972, вып. 12, с. 1651 —1655.
41. Thomas К., Jerchel D. — Angew. Chem., 1958, Bd. 70. № 24, S. 719—737.
42. Сьютер Ч. Химия органичесскнх соединений серы. Ч. 2. Пер. с англ/Под
?73
ред. Н. Н. Мельникова. М., ИЛ, 1951, с. 247—248. 43. Bunnett J. F., Brother-
ton Т. К., Williamson S. М. — In: Organic Syntheses Coll. v. 5, N. Y., Wiley,
1973 p. 816—818. 44. Шейн С. M., Русов В. П., Соколенко В. И. и др., Авт. свид.
СССР 517581; БИ, 1976, № 22. 45. Докунихин Н. С.— Жури. ВХО, 1976, т. 21,
вып. 3, с. 292—298. 46. Беркман Б. Е., Гишплинг М. Я-, Гофмейстер К. К- и др.
Авт. свид. СССР 143696; БИ, 1961, № 1. 47. Англ. пат. 1141655, 1969; Chem.
Abstr., 1969, v. 70, 87386. 48. Matsushima A., Hachimori У., Inada У. e.a.—
J. Bio’chem., 1967, v. 61, № 3, p. 328—336. 49. Эфрос Л. С., Давиденков Л. P.—
ЖОХ, 1951, t. 21 вып. 11, c. 2046—2050. 50. Brotherton T. K-, Bunnett J. F.—
Chem. Ind., 1957, № 3, p. 80.
5t. Коптюг В. A. — В кн.: Современные проблемы химии карбониевых ионов.
Новосибирск, Наука, 1975, с. 6—178. 52. Ершов В. В., Володькин А. А. — Успе-
хи химии, 1966, т. 35, вып. 11, с. 1953—1985. 53. Оае S., Kiritani R., Tagaki W.—
Bull. Chem. Soc. Japan, 1966, v. 39, № 9, p. 1961—1967. 54. Wedemeyer K--F.—
In: Methoden der Organischen Chemie (Houben-Weyl): Bd. 6/lc. Stuttgart, Georg
Tieme Verlag, 1976, S. 1140—1186. 55. Бельг, пат. 635927, 1964; Chem. Abstr.,
1964 v. 61 13237. 56. Пат. США 2970171, 1961; Chem. Abstr., 1961, v. 55, 15416.
57. Пат. США 3658906, 1972; Chem. Abstr., 1972, v. 77, 48040. 58. Аллен 4„.
Белл A. — В кн.: Синтезы органических препаратов. Сб. 3. Пер. с англ. М., ИЛ,
1952, с. 51—54. 59. Доналдсон Н. Химия и технология соединений нафталино-
вого ряда. Пер. с англ, под ред. А. И. Королева. М., Госхимиздат, 1963. 60. Пат.
США 3338966, 1967; Chem. Abstr., 1968, v. 68, 77955.
61. Пат. США 3340302, 1967; Chem. Abstr., 1968, v. 68, 12681. 62. Пат. ФРГ
2140786, 1973; Chem. Abstr., 1973, v. 78, 124270. 63. Effenberger R, Prossel G.,
Auer E. e.a. — Chem. Ber., 1970, Bd. 103, № 5, S. 1456—1462. 64. Англ. пат.
1190274, 1971; Chem. Abstr., 1972, v. 76, 45927. 65. Schmitt J., Suguet M„
Calet G. e.a. — Bull. Chem. France, 1962, № 3, p. 444—455. 66. Petrzilka T.,
Lusardi W. G. — Helv. chim. acta, 1973. Bd. 56, № 1, S. 510—518. 67. Effenber-
ger R, Niess R. — Chem. Ber., 1968, Bd. 101, № 11, S. 3787—3793. 68. Peder-
sen E. B., Lawesson S.-O. — Tetrahedron, 1974, v. 30, № 7, p. 875—878.
69. Дрейк H. — В кн.: Органические реакции. Сб. 1. Пер. с англ. М., ИЛ, 1948,
с. 133—162. 70. Тодрес-Селектор 3. В. — В кн.: Сульфирование солями серни-
стой кислоты. М., Химия, 1965, с. 52—89.
71. Seeboth Н.— Angew. Chem., 1967, Bd. 79, № 8, S. 329—340. 72. Богда-
нов С. В., Карандашова Н. Н. — ЖОХ, 1955, т. 25, вып. 6, с. 1152—1156;
Богданов С. В. — Там же, 1958, т. 28, вып. 5, с. 1324—1327, 1328—1332. 73. Бог-
данов С. В., Горелик М. В.— Там же, 1959, т. 29, вып. 1, с. 136—139.
74. Rieche A., Seeboth Н.— Lieb. Ann. Chem., 1960, Bd. 638, S. 76—81. 75. Бог-
данов С. В. — ЖОХ, 1932, т. 2, вып. 1, с. 9—22. 76. Ворожцов Н. Н., мл.—
В кн.: Химия синтетических красителей. Т. З/Под ред. К- Венкатарамана. Пер.
с англ, под ред. Л. С. Эфроса. Л., Химия, 1974, с. 1740—1816. 77. Bloom S. М.,
Hutton R. F.— Tetrahedron Letters, 1963, № 28, р. 1193—1997. 78. Степанов Б. И.
Введение в химию и технологию органических красителей. Изд. 2-е. М., Химия,
1977, с. 180—189. 79. Greenhalgh С. W., Hughes N. J. — Chem. Soc. (С), 1968,
№ 10, р. 1284—1288. 80. Hays J. Т., Yong Н. L., Espy H. H. — J. Org. Chem.,
1967, v. 32, № 1, p. 158—162.
81. Англ. пат. 807241, 1959; Chem. Abstr., 1960, v. 54, 1873. 82. Горелик M.B.,
Ломзакова В. И. — ЖОрХ, 1978, т. 14, вып. 5, с. 1051 —1055. 83. В unpett J. R,
Bassett I. У. —J. Org. Chem., 1962, v. 27, № 7, p. 2345—2348. 84. Truce W. E.,
Kreider E. M., Brand W. W. — In: Organic Reactions. V. 18, N. Y., Wiley, 1970,
p. 99—215. 85. Пат. ФРГ 2019427, 1971; Chem. Abstr., 1972, v. 76, 101224.
86. Kleb K. G. — Angew. Chem., 1968, Bd. 80, № 7, S. 284—285; Knipe A. C„
Lound-Keast J. — J. Chem. Soc. Perkin 2, 1976, № 14, p. 1741—1748. 87. Пат. ФРГ
2460212, 1975, Chem. Abstr.. 1975, v. 83, 163797; 2460233, 1975, Chem. Abstr., 1975,
v. 83, 178538. 88. Грудцын Ю. Ш, Лакомова H. А., Гитис С. С. — ЖОрХ, 1975,
т. 11, вып. 3, с. 638—643; Лакомова Н. А. Автореф. канд. дисс., Л., 1975.
89. Гитис С. С., Глаз А. И., Григорьев В. В. и др. — ЖОрХ, 1967, т. 3, вып. 9.
с. 1617—1620. 90. Англ. пат. 1127085, 1968;
91. Прайс Ч., Воонг Синг-ту. — В кн.: Синтезы органических препаратов.
Сб. 4. Пер. с англ. М., ИЛ, 1953, с. 376—377. 92. Brizzi С., Dal Monte D., San-
dri E. — Ann. Chim. (Rome), 1964, v. 54, № 5, p. 476—485. 93. Barton J. W.,
Grunham A. R., Whitaker К. E.— J. Chem. Soc. (c), 1971, № 8, p. 1384—1386.
94. Пожарский А. Ф., Симонов A. M. Аминирование гетероциклов по Чичибаби-
ну. Ростов-на-Доиу, Изд-во Ростовского ун-та, 1971. 95. Новиков В. И., Пожар-
ский А. Ф., Доронькин В. Н. — Химия гетероцикл, соед., 1976, № 2, с. 244—252.
96. Zoltewicz J. A., Helmick L. S., Oestreich Т. М. е. а. —J. Org. Chem., 1973,
v. 38, № 10, р. 1947—1949. 97. Simig Gy., van der Plas H. C. — Rec. trav. chim.
Pays-Bas, 1976, v. 95, № 5, p. 125—126. 98. Grundmann Ch.— In: Methoden der
Organischen Chemie (Houben-Weyl.) Bd. 7/3a. Stuttgart, Georg Thieme Verlag,
1977 S. 404—533. 99. Фокин E. П., Русских В. В., Русских С. А. и др.—ЖПХ,
1971’, т. 44, вып. 10, с. 2271—2276. 100. Курдюмова Т. Н. — ЖОрХ, 1968, т. 4,
вып. 9, с. 1683—1684.
101. Горелик М. В. — В кн.: Химия антрахинона/Под ред. А. И. Королева.
М., Химия, 1969, с. 5—56. 102. Казанков М. В., Уфимцев В. Н. — ЖОХ, 1964,
т. 34, вып. 12, с. 4124—4125. 103. Эктова Л. В., Шишкина Р. П., Фокин Е. П.—
Изв. СО АН СССР, сер. хим. наук, 1975, № 4, вып. 2, с. 133—138. 104. Горе-
лик М. В., Хараш М. С. — ЖОрХ, 1976, т. 12, вып. 4, с. 864—871. 105. Eders Е.—
In: Methoden der organischen Chemie (Houben-Weyl). Bd. 10/2. Stuttgart, Georg
Thieme Verlag, 1967, S. 234—287. 106. Аллен К. — В кн.: Синтезы органических
препаратов. Сб. 2. Пер. с англ. М., ИЛ, 1949. с. 229—230. 107. Китаев Ю. П.,
Бузыкин Б. И. Гидразоны. М., Наука, 1974, с. 124—125. 108. Пат. США 3294838,
1966; Chem. Abstr., 1967, v. 66, 55277. 109. Болотин Б. М.— Методы получения
химических реактивов и препаратов, 1967, № 15, с. 13. ПО. Kauffman Т,—
Angew. Chem., 1964, Bd. 76, № 5, S. 206—217; Burkhardt W., Kauffman T.—
Ibid., 1967, Bd. 79, № I, S. 57.
111. Горелик M. В., Гладышева T. X., Шилова Г. И. и др. Авт. свид. СССР
410069; БИ, 1974, № I. 112. Пат. ФРГ 1132270, 1962; Chem. Abstr., 1963, v. 58,
4673. 113. Barnett C. J., Smirz J. C.—Org. Prep. Proc. Intern., 1974, v. 6, № 4,
p. 179—182; Пат. ФРГ 2350875, 1974; Chem. Abstr., 1974, v. 81, 13492. 114. Уфим-
цев В. H., Аринич Л. В., Собчинская С. А. и др. Авт. свид. СССР 334832; БИ,
1973, № 22. 115. Горелик М. В., Аринич Л. В. Авт. свид. СССР 649708; БИ,
1979, № 8. 116. Уивер У. — В кн.: Химия нитро- и иитрозогруппы. Т. 2/Под ред.
Г. Фойера. Пер. с англ, под ред. В. А. Тартаковского. М., Мир, 1973, с. 5—43.
117. Ward Е. R., Johnson С. D., Hawkins J. G. — J. Chem. Soc., 1960, № 2,
р. 894—896. 118. Багал Л. И., Певзнер М. С., Фролов А. Н. — ЖОрХ, 1969, т. 5,
вып. 10, с. 1820—1828. 119. Ходгсон X., Махадеван А., Уорд Э. — В кн.: Синтезы
органических препаратов. Сб. 4. Пер. с англ. М., ИЛ, 1953,с. 206—208. 120. Brox-
ton Т. J. Muir D. М., Parker A. J. — J. Org. Chem., 1975, v. 40, № 14, р. 2037—
2042; Ibid., № 22, р.3230—3233.
121. Пат. США 2587093, 1952; Chem. Abstr., 1952, v. 46, 8679. 122. Пат. ФРГ
2227766, 1973; Chem. Abstr., 1973, v. 78, 99054. 123. Wanzlick H.-W. — Angew.
Chem., 1964, Bd. 76, № 8, S. 313—320. 124. Горелик M. В., Хан Иp Геон,— ЖОрХ,
1978, т. 14, вып. 2, с. 414—423. 125. Biffin М. Е. С., Miller J., Paul D. В.— In:
The Chemistry of the Azido Group/Ed. S. Patai. London, Interscience Publishers,
1971, p. 57—190. 126. Динабург M. С. Светочувствительные диазосоединения и
их применение. М., Химия, 1964, с. 181—187. 127. Смит П., Бойер Дж.—В кн.:
Синтезы органических препаратов. Сб. 4. М., ИЛ, 1953, с. 395—397. 128. Ley К.,
Seng F. — Synthesis, 1975, № 7, р. 415—422.
275
Глава 8
РЕАКЦИИ С КИСЛОРОДСОДЕРЖАЩИМИ
НУКЛЕОФИЛЬНЫМИ РЕАГЕНТАМИ
8.1. ВВЕДЕНИЕ ГИДРОКСИЛЬНОЙ ГРУППЫ
Реакции замещения различных групп или атомов под
действием нуклеофилов, имеющих в качестве реакционного центра
атом кислорода, являются наиболее широко применимым методом
введения гидроксильной группы в ароматические соединения [1].
Большая универсальность нуклеофильного замещения по сравне-
нию с другими методами гидроксилирования: свободнорадикаль-
ным замещением (см. 13.1) и окислением (см. 17.2.1)—обуслов-
лена разнообразием доступных О-нуклеофилов и уходящих атомов
и групп. При нуклеофильном гидроксилировании реагентами могут
служить вода, гидроксиды щелочных и щелочноземельных метал-
лов, алкоголяты, соли карбоновых кислот, нитриты и некоторые
другие. Обмениваться на гидроксильную группу могут почти все
атомы и группы, способные участвовать в реакциях нуклеофиль-
ного замещения. Наибольшее применение находит замещение
сульфогруппы, галогенов, а также нитро-, амино- и диазо-
ниевой группы.
8.1.1. Замещение сульфогруппы
Из многих известных реакций обмена сульфогруппы замещение
гидроксигруппой — самая важная в практическом отношении. Ре-
акция протекает при действии щелочей на соли сульфокислот с
образованием фенолята и сульфита:
ArSO3Na + 2NaOH —> ArONa + Na2SO3 + Н2О
Процесс проводят или при атмосферном давлении, в среде
расплавленного гидроксида щелочного металла или при повышен-
ном давлении при нагревании с водным раствором гидроксида в
автоклаве [2]. Оба варианта в литературе принято называть ще-
лочным плавлением. Из щелочей обычно применяют более дешевый
гидроксид натрия; гидроксид калия используют в специальных
случаях в качестве более активного реагента. Для понижения
температуры при плавлении с твердыми щелочами применяют
смесь NaOH и КОН. Иногда, главным образом в ряду антрахино-
на, замещение сульфогруппы ведут при нагревании в водной сус-
пензии Са(ОН)2 в автоклавах. Температура реакции варьируется
от 100—150 до 300—350 °C, а в непрерывных процессах — до 400 °C
и выше.
Исследованием щелочного плавления бензосульфокислоты, ме-
ченной атомом углерода Ь14С, установлено, что гидроксильная
276
группа вступает к тому же атому углерода, у которого находилась
сульфогруппа [3], и, следовательно, аринный механизм, сопро-
вождающийся кине-замещением, в данной реакции не играет роли.
Это подтверждается также образованием только n-крезола из
n-толуолсульфокислоты и отсутствием резорцина в продуктах ще-
лочного плавления 1,2- и 1,4-бензолдисульфокислот [4J. С примене-
нием NaOH, обогащенного 18О, показано, что в ароматическое
кольцо внедряется атом кислорода, принадлежащий ранее гидро-
ксид-аниону, а не сульфогруппе [3, 5].
При изучении кинетики замещения сульфогруппы в бензол-
сульфокислоте и нафталинсульфокислотах в водных растворах
NaOH под давлением для большинства сульфокислот порядок
реакции был найден равным 2, но для бензолсульфокислоты, для
6- и 7-гидрокси-2-нафталинсульфокислот он ближе к 3 (первый по
сульфокислоте, второй — по щелочи) [6]. Оказалось, что порядок
реакции зависит от концентрации щелочи и наблюдаемая констан-
та скорости Ан описывается линейной зависимостью основно-ката-
лизируемых реакций:
ka = kz + kz [В]
где [В]—концентрация щелочи, а А2 и А3— некаталитическая и
каталитическая константы скорости реакции соответственно [7].
Предполагается, что на первой стадии реакции гидроксид-анион
присоединяется к сульфонат-аниону с образованием о-комплекса,
который далее превращается в фенолят-анион как по каталитиче-
скому, так и по некаталитическому пути:
Для бензолсульфоната и 2-нафталинсульфоната каталитиче-
ские константы скорости А3 практически одинаковы, и большая
реакционная способность 2-нафталинсульфоната целиком опреде-
ляется большей скоростью реакции по некаталитическому пути А2.
Отношение констант А3/А2, характеризующее вклад каталитическо-
го пути, для бензолсульфокислоты в 7 раз больше, чем для
2-нафталинсульфокислоты, вследствие чего реакция первой из них
более чувствительна к изменению концентрации щелочи.
Условия проведения реакции зависят от подвижности сульфо-
группы и свойств образующегося гидроксисоединения. Наименее
подвижна сульфогруппа в бензольном ряду; ее подвижность боль-
ше в нафталиновом, еще больше — в антрахиноновом. Щелочное
плавление бензолсульфокислоты в производстве фенола и 2-на-
фтал' чсульфокислоты в производстве 2-нафтола ведут при атмоС’
Ферном давлении в среде NaOH при температуре 320 °C и выше.
277
Так, при производстве 2-нафтола в чугунный котел вносят около
2500 кг 85—90% расплавленного NaOH (55 кмоль), приготовлен-
ного упариванием 42%-ного водного раствора, а при 280—310 °C
добавляют 24 кмоль натриевой соли 2-нафталинсульфокислоты,
вводят водяной пар для защиты плава от кислорода воздуха,
размешивают при 325—335 °C 1,5—2 ч и через нижний штуцер
сливают плав в 6500 л воды. Раствор нафтолята натрия подкисля-
ют пропусканием SO2 и слои разделяют в стальной воронке при
98—100 °C; нижний слой раствора Na2SO3 направляют на стадию
нейтрализации после сульфирования; верхний слой 2-нафтола два-
жды промывают водой при 98—100°С, продукт обезвоживают,
дистиллируют и чешуируют на барабанном кристаллизаторе; вы-
ход 87% [2].
1-Нафталинсульфокислота реагирует при 300 °C примерно в
3 раза быстрее 2-нафталинсульфокислоты, но не применяется для
получения 1-нафтола из-за примеси в техническом 1-сульфонате
2-изомера [2,6].
Введение второй сульфогруппы сильно ускоряет (в 20—30 раз),
а введение гидроксильных групп замедляет реакцию. Благодаря
этому из ди- и трисульфокислот можно получать гидроксиарен-
сульфокислоты. Избирательное замещение одной из сульфогрупп
имеет большое значение для получения гидрокси- и аминогидрок-
синафталинсульфокислот. Эта реакция является заключительной
стадией в производствах таких важных азосоставляющих, как
Аш-кислота, И-кислота, Гамма-кислота. Аш-кислоту (4-амино-5-
гидрокси-2,7-нафталиндисульфокислота или 8-амино-1-нафтол-3,6-
дисульфокислота) (2) получают, исходя из 8-амино-1,3,6-нафталин-
трисульфокислоты (Гнафтиламин-3,6,8-трисульфокислота; Т-кис-
лота) (1):
ОН NH2
(О (2)
Реакции проводят в закрытых аппаратах при атмосферном или
повышенном давлении при температурах ниже 200 °C и строгом
соблюдении температурного режима [2]. При производстве Аш-
кислоты (2) в стальной автоклав на 5200 л вносят 24,0 кмоль
NaOH в виде 70%-ного водного раствора и затем около 4200 кг
нагретого до 90 °C водного раствора, содержащего 3,0 кмоль на-
триевой соли Т-кислоты. Смесь нагревают 4 ч от 178 до 184 °C при
давлении до 0,9 МПа, спускают давление и переносят плав в
7200 л 17 %-ной H2SO4, размешивают 4 ч при 85—90 °C, фильтруют
при 50°C, промывают водой и отжимают. Выход натриевой соли
Аш-кислоты 75%.
278
Введение двух гидроксигрупп требует более жестких условий.
В производстве резорцина при взаимодействии динатриевой соли
ж-бензолдисульфокислоты с NaOH при атмосферном давлении
первая сульфогруппа замещается при температуре ниже 300 °C, а
вторая — после нагревания при 330—340 °C в течение 8—И ч. За-
местить сульфогруппу в n-гидроксибензолсульфокислоте вообще не
удается. Это соединение в условиях щелочного плавления относи-
тельно устойчиво и лишь медленно превращается в иные кроме
гидрохинона продукты. 4-Гидрокси-1,3-бензолдисульфокислота в
результате плавления с NaOH при 330—350 °C превращается с
выходом 80% в 3,4-дигидроксибензолсульфокислоту, обменивая
только сульфогруппу в орто-положении к гидроксилу [8].
2,3-Дигидроксинафталин с хорошим выходом образуется из 3-гид-
рокси-2-нафталинсульфокислоты [9]
Побочными реакциями при щелочном плавлении сульфокислот
являются замена сульфогруппы на атом водорода, расщепление
ароматического ядра, образование димеров и тримеров, замещение
атома водорода гидроксигруппой. Все эти реакции происходят в
результате дальнейших превращений первоначально образующих-
ся гидроксисоединений. Кроме того, гидроксисоединения очень
легко окисляются в щелочной среде при высокой температуре;
поэтому в процессе плавления необходимо защищать реакционную
смесь от кислорода воздуха.
Замещение водорода гидроксигруппой при щелочном плавле-
нии характерно для антрахинонсульфокислот. Взаимодействие
9,10-антрахинон-2-сульфокислоты с NaOH при 190—200 °C в при-
сутствии окислителя КЙОз служит одним из промышленных мето-
дов получения 1,2-дигидрокси-9,10-антрахинона (ализарина) (см.
8.1.5). Применение водной суспензии Са(ОН)а (известкового мо-
лока) позволяет избежать замещения водорода. Сообщается, что
вместо известкового молока можно использовать разбавленные
(3—6%) водные растворы NaOH при 150 °C [10, 11]. В частности,
из 9,10-антрахинон-2,6- и 2,7-дисульфокислот получены соответ-
ствующие дигидрокси-9,10-антрахиноны [10]. Однако при реакции
а-антрахинонсульфокислот в этих условиях образуется сложная
смесь продуктов вследствие вступления сульфо- и гидроксигрупп в
3-положения. После 18-часового нагревания 9,10-антрахи-
нон-1,8-дисульфокислоты с разбавленным раствором NaOH при
145 °C идентифицированы кроме 1,8-дигидрокси-9,10-антрахинона
(17%) и 8-гидрокси-9,10-антрахинон-1-сульфокислоты (20%) еще
шесть основных продуктов, в том числе 1,2,8-тригидрок-
си-9,10-антрахинон (21 %), 7,8-дигидрокси-9,10-антрахинон-1-суль-
фокислота (10%), 8-гидрокси-9,10-антрахинон-1,7-дисульфокисло-
та (9%), 1,8-дигидрокси-9,10-антрахинон-2-сульфокислота (6%)
и ДР- [12].
Сульфогруппа в 1,2-нафтохинон-4-сульфокислоте, 2-бензотиа-
зол- и 2-бензимидазолсульфокислотах, 2- и 4-пиримидинсульфокис-
лотах и подобных соединениях замещается гидроксигруппой уже
?7?
при кипячении в водных растворах в присутствии кислоты или
щелочи.
В связи с ростом потребления важнейших гидроксисоединений
разработаны непрерывные технологические схемы получения фе-
нола, резорцина, 2-нафтола [2, 13] в трубчатых реакторах под
высоким давлением. Перспективы внедрения таких установок за-
висят от конкурентоспособности щелочного плавления в ряду дру-
гих промышленных методов получения указанных продуктов. Для
производства фенола, например, в настоящее время используется
шесть промышленных методов, выбор между которыми требует
учета многих факторов, включая местные условия [14]. Серьезным
недостатком метода щелочного плавления для создания на его
основе многотоннажных производств является большое количество
отходов, затрудняющее охрану окружающей среды.
Алкил- и арилсульфонильная группа также замещаются на
гидроксигруппу при действии щелочей. Расщепление диарилсуль-
фонов при щелочном плавлении обычно дает смесь фенола и аро-
матического углеводорода. Изучение плавления дифенилсульфона,
меченного 1-14С, с КОН при 300 °C и опыты со щелочью, обогащен-
ной 18О, исключают, как и для сульфокислот, аринный механизм, но
полученные результаты не позволяют сделать окончательный вы-
бор между механизмом с атакой гидроксид-аниона на атом угле-
рода и на атом серы [15]. В первом случае (а) первоначально
образуются фенолят и бензолсульфинат, превращающийся при
действии щелочи в бензол; во втором (б) — бензол и бензолсуль-
фонат, который далее переходит в фенолят:
Выделение с хорошим выходом гидроксибензоилбензолсульфи-
новых кислот (4) при расщеплении циклических сульфонов (3)
действием NaOH в кипящем водно-диоксановом растворе [16]
может служить аргументом в пользу протекания реакции по пути
(а). Расщепление сульфонов (3) особенно легко происходит при
R = NOa, останавливаясь на стадии сульфиновой кислоты (4); но
при R = Н, Cl и др. за расщеплением следует внутримолекулярное
замещение с образованием ксантонов (5).
О
(5)
8.1.2. Замещение атомов галогенов
Замещение атомов галогенов на кислородсодержащие
группы, как и замещение на аминогруппы, может проводиться без
катализатора и в присутствии соединений меди. Помимо щелочей
при замещении галогенов используются все другие нуклеофильные
реагенты, пригодные для введения гидроксигруппы: вода, алкого-
ляты, карбонаты, соли карбоновых кислот, нитриты, соли оксимов и
т. д., но главное значение сохраняют гидроксиды щелочных ме-
таллов.
Ароматические галогенпроизводные, не содержащие активиру-
ющих электроноакцепторных атомов и групп, обменивают атом
галогена в очень жестких условиях. При нагревании паров хлор-
бензола и воды при 500 °C фенол не образуется, при 600 появля-
ются лишь небольшие его количества, но происходит обугливание.
Однако при пропускании паров хлорбензола и воды над специаль-
ными катализаторами при 400—500 °C выход фенола составляет
около 90%. В производстве получение фенола по этому методу
комбинируют с получением хлорбензола из бензола, хлористого
водорода и кислорода (окислительное хлорирование)—(см. 5.2.1):
С6Н5С1 + Н2О —> С6Н5ОН + НС1
С6н6 + НС1 + 0,5О2 —> СбН5С1 + Н2О
Промышленный метод получения фенола из хлорбензола при
действии щелочей состоит в нагревании с 15%-ным раствором
NaOH (2,2—2,5 моль на 1 моль хлорбензола) при 360—390 °C и
28 МПа в аппаратах непрерывного действия с добавкой дифени-
лового эфира 15—20% от массы хлорбензола [1]. В этих условиях,
как доказано опытами с хлорбензолом-1-14С [17], реакция идет
281
преимущественно по аринному механизму (см. 2.7.2). Дегидробен-
зол, возникающий в результате отрыва протона и последующего
отщепления хлорид-аниона, присоединяя гидроксид-анион, пре-1
вращается в фенолят, а взаимодействуя с фенолят-анионом,— в
дифениловый эфир, анионы 2- и 4-фенилфенолов: ;
+ он-
------>
+ С6Н5О- +СвН5О-, н2о
Дальнейшие реакции с участием дегидробензола приводят к
феноксибифенилам, 2,6-дифенилфенолу и другим побочным про- ’
дуктам. Дифениловый эфир необратимо гидролизуется в фенол с :
такой скоростью, что при введении в хлорбензол 15—20% дифени- ,
левого эфира количества образовавшегося и гидролизовавшегося |
эфира оказываются примерно равными, что и используется в прак-
тике (см. выше).
Аринный механизм подтверждается также образованием смеси
м- и о-крезолов из о-хлортолуола (в соотношении около 6 : 4 при
350 °C), м- и n-крезолов из n-хлортолуола, смеси 1- и 2-нафтолов
как из 1-, так и из 2-хлорнафталина. При снижении температуры и
концентрации NaOH доля реакции, протекающей по аринному ме-
ханизму, уменьшается, уступая место механизму присоединения-
отщепления SnAf. Уже при 340 °C в 14"%-ном растворе NaOH
превращение хлорбензола в фенол на 16% идет по этому пути, а
при 300°C в 10%-ном и еще более слабых растворах — целиком по
механизму SnAf [6].
В присутствии катализатора (меди, ее оксидов или солей)
аринный механизм подавляется за счет ускорения прямого заме-
щения галогена. Природа влияния соединений меди в реакциях
гидроксилирования такая же, как в реакциях аминирования
(см. 7.1.1). Применение соединений меди при взаимодействии га-
логенпроизводных со щелочами позволяет получать без примеси
изомеров 2-нафтол из 2-хлорнафталина, n-крезол из п-хлортолуо-
ла, 3-гидроксипирен из 3-бромпирена [18], 3-гидроксихинолин из
3-бромхинолина [19].
Роль катализа соединениями меди ярко проявляется при полу-
чении двух- и трехатомных фенолов, в отсутствие катализатора
протекающем по типу кпне-замещения. Так, нагревание о-хлорфе-
нолята натрия с порошкообразным КОН в о-дихлорбензоле при ]
282
178 °C приводит к смеси резорцина и пирокатехина в соотношении
1,6 : 1 [20]; нагревание же о-хлорфенола в водном растворе NaOH
при 190 °C с добавкой CuSO4 дает пирокатехин с выходом 90—95%
[21]. При взаимодействии n-хлорфенола с 20%-ным NaOH при
265 °C образуется смесь двухатомных фенолов с выходом гидрохи-
нона всего 3% и резорцина — 6,7%, тогда как в присутствии
CuSO4 при 175 °C получают гидрохинон с выходом 75% [!]•
Иногда протекание реакции по пути кпне-замещения представ-
ляет препаративную ценность. Например, 5-метилрезорцин (орсин)
можно с удовлетворительным выходом получать при действии
КОН на 4- и 6-хлор- или -бром-я-крезолы [22], тогда как 5-хлор-
или -бром-м-крезолы в качестве исходных продуктов трудно до-
ступны. Аналогично из 4-хлоррезорцина и 2,6-дихлорфенола можно
получать флороглюцин (выход 66% и 46% соответственно) [23]:
Для получения галогенфенолов из ди- и полигалогенбензолов
реакцию с гидроксидами щелочных металлов обычно проводят в
спиртовой среде. При этом первоначально происходит замещение
галогена на алкоксигруппу, а затем образовавшийся эфир фенола
расщепляется в результате атаки алкоголят-аниона на алифатиче-
ский атом углерода с выделением диалкилового эфира [24]; на-
пример, таким путем из 1,2,4,5-тетрахлорбензола через 1-меток-
си-2,4,5-трихлорбензол (6) получается 2,4,5-трихлорфенол (7);
+ CH3ONa
-------->
+ CH3ONa
-(СН3)2О
ОН
283
Реакция в спиртово-щелочной среде позволяет более избира-
тельно и в более мягких условиях провести обмен одного из атомов
галогена на гидроксигруппу. Так, при действии NaOH в метаноле
при 130 °C 1,2,4-трихлорбензол превращается в 2,5-дихлорфенол
(8) с выходом 93%, а в водной среде при 250 °C — в смесь (8) с
2,4- и 3,4-дихлорфенолами в соотношении 3,9: 1 : 1,4 [1]:
(8)
При тщательном выборе параметров в некоторых случаях про-
цесс удается провести строго селективно и в водной среде. Нагре-
вание 1,2,3,5-тетрахлорбензола с 4—8%-ным водным раствором
NaOH при 210 °C дает 2,3,5-трихлорфенол (9) с выходом 98%
[25]:
Полихлорфенолы из полихлорбензолов производятся в про-
мышленном масштабе для получения пестицидов, например
2,4,5-трихлорфеноксиуксусной кислоты и ее эфиров из 2,4,5-три-
хлорфенола (7). Опасной побочной реакцией в этих производствах
является образование дибензо-1,4-диоксинов, обладающих исклю-
чительно высокой токсичностью [26]; в частности 2,3,6,7-тетра-
хлордибензо-1,4-диоксин (10) образуется из 2,4,5-трихлорфено-
ла (7).
(7) (Ю)
Образования дибензодиоксинов можно избежать при тщатель-
ном контроле температуры, исключающем ее повышение, исполь-
зовании разбавленных растворов и значительного избытка щелочи
(не менее 3,5 моль на 1 моль галогенпроизводного).
284
Введение в молекулу галогенпроизводного электроноакцеп-
торных заместителей в орто- и пара-положения увеличивает лег-
кость обмена галогена на гидроксил в некаталитич'еской реакции, а
введение в орто-положение таких хелатообразующих групп как
карбокси- и азогруппа сильно увеличивает его подвижность в ре-
акции, катализируемой соединениями меди.
При производстве о-нитрофенола в стальной котел вместимо-
стью 5000 л вносят 3500 л водного раствора, содержащего
8,95 кмоль NaOH, и затем 3,60 кмоль о-нитрохлорбензола; созда-
ют давление воздуха 0,25—0,3 МПа и нагревают при 145—150 °C
около 8 ч (давление до 1 МПа). По окончании реакции охлажден-
ный раствор подкисляют 40%-ной H2SO4 до pH 4,5, нижний слой
отделяют и дважды промывают порциями по 2000—3000 л воды;
выход о-нитрофенола 72%.
Роль кислорода воздуха в реакции обмена на гидроксил атома
галогена, активированного нитрогруппой в орто- и пара-положе-
нии, весьма существенна, но пока не вполне выяснена. Предполо-
жительно ее связывают с протеканием реакции через стадию об-
разования анион-радикалов (см. 2.7.2). Согласно патенту [27],
n-нитрофенол можно получить с выходом, близким к количествен-
ному, нагреванием n-нитрохлорбензола при 160 °C в автоклаве в
2—4%-ном водном растворе NaOH при непрерывном продувании
воздухом.
Накопление электроноакцепторных групп приводит к способно-
сти галогена гидролитически замещаться в отсутствие щелочей.
Так, пентацианохлорбензол переходит в пентацианофенол уже при
нагревании в водном диоксане или диметоксиэтане [28].
При наличии в молекуле трифторметильных групп под действи-
ем алкоголятов или спиртовой щелочи вслед за обменом галогена в
ароматическом кольце гидролизуются атомы фтора трифторме-
тильной группы, находящейся в орто- или пара-положении к
фенольному гидроксилу. п-Хлорбензотрифторид (11) при 180 °C
превращается в n-гидроксибензойную кислоту (12), а 1,4-бис(три-
фторметил) -2-хлорбензол — в 2-гидрокси-4-трифторметилбензой-
ную кислоту. Последняя представляет практический интерес, и
другим путем получить ее трудно [29]. Гидролиз трифторметиль-
ной группы не происходит при реакции нитротрифторметилгало-
генбензолов с твердым NaOH в диметилсульфоксиде при 20—25 °C
[30]. В этих условиях З-нитро-4-хлорбензотрифторид (13) превра-
щается в 2-нитро-4-трифторметилфенол (14) с выходом 96%.
С1 ОН
(13) (14)
285
Если в ароматическом кольце одновременно присутствуют га-
логен и сульфогруппа, первый в условиях щелочного плавления
замещается быстрее. Это позволяет получать гидроксибензолсуль-
фокислоты из хлорбензолсульфокислот, например, при нагревании
2,4-дихлорбензолсульфокислоты (15) при 200—222°C с 50%-ным
водным NaOH в автоклаве с выходом 96% образуется 2-гидро-
кси-4-хлорбензолсульфокислота (16), десульфированием которой
при кислотном гидролизе получают 3-хлорфенол (17) [31].
+ NaOH
SO3H
Cl
+ H2O
НС1
(15) (16) (17)
Галогенкарбоновые кислоты при щелочном плавлении могут
декарбоксилироваться. 2,4-Дихлоризофталевая кислота, например,
при действии КОН при 200 °C в присутствии Си2О дает резорцин
(97%), а 2,5-дихлортерефталевая кислота — гидрохинон (76%)
[32]. 2-Хлорбензойная кислота при действии NaOH при 200°С
превращается в смесь м-гидроксибензойной и салициловой кислот
в соотношении 2 : 1 [33]. Подобных осложнений не возникает, если
замещение галогена в орто- и иерм-галогенкарбоновых кислотах
проводить в водных растворах третичных или вторичных аминов в
присутствии соединений меди. Кипячение 1- и З-хлор-2-нафтойной и
8-хлор-1-нафтойной кислот [34] в водном растворе пиперидина или
морфолина приводит с выходом 75—85% к соответствующим гид-
роксикарбоновым кислотам. В тех же условиях гидроксильная
группа замещает галоген и в о-галогенфенилуксусных кислотах
'[35].
Атомы галогена, обладающие высокой подвижностью, замеща-
ются гидроксигруппой при действии не только щелочей, но и других
О-нуклеофилов. Как уже отмечалось (см. 7.3), при действии
NaNO2 вследствие его амбидентности атомы галогена обменивают-
ся не только на нитро-, но и на гидроксильную группу:
ArCl + 2NaNO2 —> ArONa + NaCl + N2O3
При реакции 1,3-динитро-4-хлорбензола и З-нитро-4-хлорбензо-
трифторида (13) с NaNO2 в диметилсульфоксиде при 100 °C обра-
зуются с выходом >95% 2,4-динитрофенол и 2-нитро-4-трифторме-
тилфенол (14) соответственно. Реакция же З-нитро-4-хлортолуола
(18) дает с выходами 70 и 30% 2-нитро-п-крезол (19) и 6-нит-
286
ро-м-крезол (20); последний возникает вследствие замещения at6-
ма хлора нитрогруппой при А/-атаке и последующего замещения
орто-расположенной нитрогруппы при О-атаке нитрит-аниона
[36].
ONO ОН
СН3 СНз СНз
(20)
В аналогичных условиях из 4-хлорфталодинитрила получают
4-гидроксифталодинитрил (60%), из пентахлорбензонитрила —
4-гидрокситетрахлорбензонитрил (89%) [37]. Замещение на гид-
роксигруппу представляет промежуточную стадию при синтезе ди-
ариловых эфиров под действием NaNO2 (см. 8.2).
В диметилсульфоксиде подвижный атом галогена замещается
на гидроксигруппу при действии солей альдоксимов с образовани-
ем О-ариловых эфиров, расщепляющихся при атаке второй моле-
кулы соли альдоксима на фенол и нитрил [38]:
“ON=CHCeH5 -ON=CHCeH5
ArCl --------> ArON=CHC6H5 --------> ArO‘+ C6H5CN
Когда реакция проводится в диметилсульфоксиде, сам раство-
ритель может выступать в качестве О-нуклеофила. При нагревании
в нем 1,3,5-тринитро-2-хлорбензол в течение 2 ч при 100 °C с выхо-
дом 90% переходит в пикриновую кислоту, а 1,3-динитро-4-хлор-
бензол при 190°С — в смесь 2,4-динитрофенола (34%) с другими
продуктами. При добавлении ацетата или бензоата натрия выход
2,4-динитрофенола повышается до 78—80% [39]. Считают, что
реакция с диметилсульфоксидом идет через стадию сульфониевых
о-комплексов типа (21), а в присутствии карбоксилат-анионов,
являющихся более сильными нуклеофилами,— через ацилоксиза-
мещенные (22), расщепляющиеся при О-атаке диметилсульфокси-
да на ацильный атом углерода.
287
Замещение на гидроксильную группу при действии солей кар-
боновых кислот характерно для соединений с подвижным атомом
галогена. Так, пентафторбензонитрил, легко обменивающий атом
фтора в положении 4 в реакциях с различными нуклеофилами,
переходит в 4-гидрокситетрафторбензонитрил при нагревании с
бензоатом натрия в А^-метилпирролидоне (56%) или без раствори-
теля (88%) [40].
В 5-хлорантрахинонпиридине (23) атом галогена замещается
на гидроксильную группу не только при действии солей карбоновых
кислот в биполярных апротонных растворителях, но и при нагре-
вании в самих карбоновых кислотах, например в уксусной.
В реакцию вступает, по-видимому, протонированная форма соеди-
нения (23), обладающего высокой основностью вследствие тенден-
ции протонированной формы к образованию внутримолекулярной
водородной связи. Образующееся 5-гидроксисоединение существу-
ет в стабилизированной водородной связью таутомерной форме
(24) [41].
(24)
2f8
В ряду антрахинона галоген в a-положении замещается гидро-
ксильной группой при нагревании с Н3ВО3 в концентрированной
серной кислоте или слабом олеуме при 120—160 °C. Из 1-ами-
но-2,4-дибром- или -дихлор-9,10-антрахинона таким способом полу-
чают 1-амино-2-галоген-4-гидрокси-9,10-антрахиноны. При произ-
водстве хинизарина взаимодействием фталевого ангидрида с
п-хлорфенолом в H2SO4 с добавкой Н3ВО3 атом хлора замещается
гидроксильной группой в процессе конденсации. Реакция, ве-
роятно, идет через промежуточное образование циклических борно-
кислых эфиров, гидролизующихся при выливании реакционной
смеси в воду.
8.1.3. Замещение нитрогруппы
Активированная нитрогруппа замещается гидроксиль-
ной группой при действии тех же нуклеофильных реагентов, кото-
рые приводят к аналогичному замещению атома хлора; по подвиж-
ности в одинаково активированных соединениях нитрогруппа
превосходит атом хлора (ср. 7.1.2). Тем не менее фенолы реже
получают замещением нитрогруппы, чем замещением галогенов,
из-за малой доступности соединений, содержащих активирован-
ную нитрогруппу.
о-Динитробензол, и-динитробензол и 1,2-динитронафталин пре-
вращаются в о-нитрофенол, n-нитрофенол и 2-нитро-1-нафтол со-
ответственно при кипячении в разбавленном водном растворе
NaOH [1]. Побочно образуются продукты восстановления: азо- и
азоксисоединения.
Изучение взаимодействия о- и п-динитробензолов с NaOH в
диметилсульфоксиде [42,43] показало, что предшественником
продукта замещения — нитрофенолята — является анион-радикал
динитросоединения:
+он“
С помощью видимых и ЭПР спектров и метода «спиновой
ловушки» (2-метил-2-нитрозопропан) показано образование анион-
радикала динитробензола, радикалов ОН- и радикалов, генериро-
ванных из растворителя (СН3- в диметилсульфоксиде), а кинети-
ческими измерениями подтверждено, что анион-радикал переходит
в конечный продукт при действии гидроксид-аниона. Относительно
того, как именно осуществляется последнее превращение, пока
высказываются различные предположения (см. также 2.7.2).
Намного легче обменивают нитрогруппу 1,2,3- и 1,2,4-тринитро-
бензол, а 1,2,3,5-тетранитробензол и гексанитробензол чувстви-
тельны уже к действию влаги [44]. В сильной степени акти-
вирует нитрогруппу орто-расположенная диазониевая группа, от-
чего при диазотировании, например, 2-нитро-1-нафтиламинов (25)
10 Зак. 271 2 89
нитрогруппа гидролитически замещается в кислой среде с образо-
ванием гидроксинафталиндиазосоединений (26).
(25) (28)
В биполярных апротонных растворителях замещение нитро-
группы гладко протекает при отсутствии сильных электроноакцеп-
торов. Так, 4-нитробензофенон переходит в 4-гидроксибензофенон
при нагревании в 1 н. растворе NaOH в диметилсульфоксиде при
200 °C [45]. 4-Нитробензонитрил с высоким выходом (94%) пре-
вращается в 4-гидроксибензонитрил при действии 2 моль соли
бензальдоксима в диметилсульфоксиде при 25 °C; так же реаги-
руют этил-4-нитробензоат и некоторые другие соединения [38].
Практическое значение приобретает замещение а-нитрогруппы
в антрахинонах. При действии на 1-нитро- и 1,5-динитро-9,10-ан-
трахинон 15—20%-ного водного раствора КОН в тетраметиленсуль-
фоне при 100—105 °C с выходом более 95% образуется 1-гидрокси-
и 1,5-дигидрокси-9,10-антрахинон соответственно [46]. Обработка
КОН, К2СО3 или СаО в диметилформамиде, АЛметилпирролидоне,
тетраметиленсульфоне при 130—180 °C позволяет селективно заме-
стить гидроксильной одну нитрогруппу в 1,5- и 1,8-динит-
ро-9,10-антрахиноне [47].
8.1.4. Замещение амино- и диазониевой групп
Замещение аминогрупп гидроксилом стехиометрически
представляет собой реакцию гидролиза, обратную аммонолизу и
аминолизу гидроксисоединений:
ArNRRz + H2O ArOH + RR/NH2
Гидролиз аминов в кислой среде и в присутствии солей серни-
стой кислоты, действительно, является следствием смещения в
обратную сторону равновесного превращения фенолов в амины.
Так как прямая и обратная реакции идут по одному и тому же
пути, все сказанное о характере влияния катализатора и других
факторах в разделе об аминировании гидроксисоединений
(см. 7.1.5) полностью применимо к гидроксилированию аминов
водными растворами кислот и бисульфита натрия. Помимо того
амины способны гидролизоваться в щелочной среде, когда фенол
уже не может превращаться в амин, так как находится в ионизи-
рованной форме, отрицательный заряд которой препятствует нук-
леофильной атаке.
290
В условиях кислотного гидролиза амины бензольного ряда, не
содержащие электроноакцепторных групп, переходят в соответ-
ствующие фенолы с большим трудом. После нагревания анилина в
41%-ной фосфорной кислоте при 280 °C в течение 25 ч получают
77% фенола, 8% дифениламина и 10% непрореагировавшего ани-
лина [1]. Значительно легче реагируют диамины и аминофенолы.
Гидролиз лгфенилендиамина в разбавленной серной [48] или фос-
форной [49] кислоте при температуре около 220 °C дает резорцине
выходом до 98%. Еще легче протекает гидролиз 1,3,5-триамино-
бензола, который превращается в флороглюцин при кипячении его
гидрохлорида в воде; выход 90%. Это служит промышленным
методом получения флороглюцина [50].
В нафталиновом ряду аминогруппа замещается гидроксигруп-
пой легче, чем в бензольном. 1-Нафтиламин превращается в
1-нафтол при нагревании его солей в серной и другими кислотами в
воде при 200 °C с выходом порядка 95% [51]. Примерно в тех же
условиях в разбавленной серной или фосфорной кислоте из 5- и
8-аминохинолинов образуются соответствующие гидроксихиноли-
ны (78—90%) [52].
Гидролиз аминогруппы в нафталиновом ядре находит прак-
тическое применение для получения а-гидроксинафталинсульфо-
кислот, например 4,5-дигидрокси-2,7-нафталиндисульфокислоты
(хромотроповая кислота) (27) из Аш-кислоты (2); 4-гидрокси-2,7-
нафталиндисульфокислоты (оксикислота Фрейнда) (28) из 4-ами-
но-2,7-нафталиндисульфокислоты (кислота Фрейнда); 8-гидро-
кси-1,6-нафталиндисульфокислоты (е-кислота) (29) из 8-ами-
но-1,6-нафталиндисульфокислоты (амино-е-кислота).
Сульфогруппа в положении 1 при кислотном гидролизе ами-
ногруппы в 4- и 5-амино-1-нафталинсульфокислотах может отщеп-
ляться, замещаясь на водород.
Производные 1,4-диамино- и 1-амино-4-гидрокси-9,10-антрахи-
нонов превращаются в лейкохинизарин как в кислой, так и в
щелочной среде в присутствии восстановителя. Эта реакция гидро-
лиза, обратная реакции аминирования лейкохинизарина (см. 7.1.5),
применяется для установления строения антрахиноновых краси-
телей.
Обмен аминогрупп на гидроксильные по реакции Бухерера осу-
ществляется кипячением амина в водном растворе NaHSO3. Влия-
ние взаимного положения амино- и сульфогрупп в нафталиновом
Ядре в данной реакции такое же, как при аминировании гидрокси-
нафталинсульфокислот (см. 7.1.5). Промышленным приложением
10*
291
бисульфитного метода гидролиза является получение 4-гидро-
кси-1-нафталинсульфокислоты (кислота Невиля — Винтера) из
4-амино-1-нафталинсульфокислоты (нафтионовая кислота):
NH2 ОН
SO3H SO3H
Смесь 8,0 кмоль натриевой соли нафтионовой кислоты, 1325 л
40%-ного раствора NaHSOs и 460 кг жидкого SO2 нагревают при
95 °C в течение 24 ч и вносят в 6000 л известкового молока
(1300 кг СаО); повышают температуру за 12 ч до 107 °C, поглощая
выделяющийся аммиак водой; осадок CaSO3 отфильтровывают,
промывают водой, из фильтрата осаждают Са2+ добавлением 60 кг
ЫагСОз и снова фильтруют, получая раствор натриевой соли
4-гидрокси-1-нафталинсульфокислоты; выход 90% (см. ссылку [59]
к гл. 7, с. 332).
В лабораторной практике обмен аминогруппы по реакции Бу-
херера используется также для синтеза гидроксипроизводных ан-
трацена, фенантрена и бензогетероциклов [1,51].
К гидролизу в щелочной среде способны амины, содержащие
сильные электроноакцепторные заместители. Так, 3-ацетилами-
но-4-бром-2-нитробензойная кислота (30) [53] и 5-ацетилами-
но-2-нитро-4-этилбензойная кислота (32) [54] дают соответствую-
щие ж-гидроксибензойные кислоты (31) и (33) при длительном
кипячении в 20—25%-ном растворе КОН с выходом 84—90%’
(30' (31)
(32) (33)
Известным примером щелочного гидролиза является превра-
щение /У.М-диметил-4-нитрозоанилина (34) в п-нитрозофенол (35),
282
служившее в прошлом промышленным методом получения диме-
тиламина:
(34)
(34)
о
Л
+ (CH3)2NH
II
NOH
(35)
Аминогруппа, активированная влиянием нитрогруппы в нафта-
линовом ядре, также замещается гидроксигруппой при кипячении в
водном щелочном растворе. Из 2-ацетиламино-1-нитронафталина
таким образом получают I-нитро-2-нафтол с выходом 88—89%
[55]. В аминонафталинсульфокислотах при действии щелочей мо-
гут замещаться одновременно как амино-, так и сульфогруппа.
Например, 8-амино-1,3,6-нафталинтрисульфокислота (Т-кислота)
(1) при нагревании в концентрированном растворе NaOH до 230 °C
превращается в хромотроповую кислоту (27).
Универсальным методом перевода аминогруппы в гидроксиль-
ную является диазотирование амина с последующим разложением
соли диазония в кислой среде:
ArNH2 —>• ArN2 —> ArOH + N2
В отсутствие восстановителей замена диазониевой группы на
гидроксигруппу протекает по механизму SnI с промежуточным
образованием арил-катиона (см. 2.7.2); в присутствии соединений
меди замещение идет по механизму, подобному механизму реак-
ции Зандмайера, с участием радикалов (см. 10.3). Общая методика
получения фенолов разложением солей диазония состоит в посте-
пенном добавлении раствора диазосоединения, не содержащего
избытка азотистой кислоты, в кипящий водный раствор H2SO4.
Образующийся фенол может быть отогнан с водяным паром или
экстрагирован органическим растворителем. Выход .и-нитрофенола
из лг-нитроанилина в этих условиях составляет 81—86% [56]; вы-
ход м- и /г-крезолов из толуидинов — 91% и 89% соответственно
[57]. Для промышленного получения о-метоксифенола (гваякола)
из о-анизидина предложена непрерывная схема с разложением
соли диазония в присутствии сульфата меди и экстракцией про-
дукта о-дихлорбензолом [58]. Применения НС1 рекомендуется из-
бегать, так как при этом возможна побочная реакция замещения
диазониевой группы атомом хлора.
При разложении диазосоединения в среде уксусной кислоты
или уксусного ангидрида диазониевая группа замещается ацет-
оксигруппой. Так, ацетоксиантрахиноны образуются при диазоти-
ровании аминоантрахинонов в уксусной кислоте и последующем
нагревании с выходом 41—71% [59].
293
Разложение солей диазония может сопровождаться, в зависи-
мости от их строения, окислительно-восстановительными реакция-
ми, циклизацией и др. Например, при диазотировании 8-нит-
ро-1-нафтиламина и разложении диазосоединения (36) образуется
не 8-нитро-1-нафтол (37), как предполагалось первоначально, а
8-гидрокси-1,4-нафтохинон-1-оксим (38) [60]. Кипячение раствора
диазотированной 8-амино-1-нафталинсульфокислоты с почти коли-
чественным выходом приводит к 1,8-нафталинсультону (39) [61].
(39)
(38)
Разложение производных о-метилбензолдиазония сопровожда-
ется образованием индазолов, которые нередко являются главны-
ми продуктами.
8.1.5. Замещение атома водорода
Еще в прошлом веке показано, что нитробензол при
нагревании с безводным КОН образует о-нитрофенол, одновре-
менно восстанавливаясь до азоксибензола; 1,3,5-тринитробензол
превращается в пикриновую кислоту при действии щелочного рас-
твора гексациано-(III) феррата калия. Эти реакции были одними из
первых примеров нуклеофильного замещения атома водорода, в
которых гидрид-ион отщепляется с участием окислителя.
Трудности удаления гидрид-иона, обсуждавшиеся в связи с
реакцией аминирования (см. 7.1.6), ограничивают возможности
прямого гидроксилирования при действии О-нуклеофилов. Препа-
ративное значение имеет замещение атома водорода гидрокси-
группой под действием щелочей в W-гетероциклах и, в отдельных
случаях, в 9,10-антрахинонах и полициклических кетонах, а также
ацетоксилирование хинонов по Тиле — Винтеру.
Гидроксилирование W-гетероциклов проводят сплавлением с
большим избытком безводной щелочи при высокой температуре.
Реакция идет с выделением водорода, подобно аминированию по
294
Чичибабину (см. 7.1.6), и направляется в a-положение к пириди-
новому азоту. Несмотря на жесткие условия гидроксисоединения
при этом удается получать с выходом до 90% и более. Для дости-
жения стабильных результатов важно тщательно обезвоживать
щелочь. Для этого ее предварительно нагревают выше температуры
реакции или добавляют к ней Na2O, связывающий следы влаги.
После «прокаливания» смеси КОН и NaOH при 475 °C плавление в
ней при 240 °C изохинолина (40) дает с выходом 98% 1-изохинолон
(41), а плавление хинолина (42)—с выходом 70% 2-хинолон (43)
при проведении опытов в укрупненных масштабах [62]:
О
(40) (41) (42) (43)
Помимо пиридинов, хинолинов, изохинолинов, бензохинолинов,
фенантридинов в реакцию гидроксилирования вступают бензими-
дазолы, нафтимидазолы и перимидины [63]. Так, 1-метилбензими-
дазол (44) переходит в 1-метил-2-бензимидазолон (45) с выходом
90% при сплавлении с КОН при 230—250 °C в течение 20 мин;
1-метилперимидин (46) превращается в 1-метил-2-перимидон (47)
с тем же выходом при 160—170 °C в течение 5 мин:
(46) (47)
Гидроксигруппа может быть введена в положение 2 хинолино-
вого ядра нагреванием в 40%-ном растворе NaOH при 300 °C
3-гидроксихинолина (48), который перегруппировывается при этом
в 2-хинолон (43) с выходом 88% [64]:
(48)
(43)
295
Замещение атома водорода гидроксигруппой в а-положении
9,10-антрахинона используется в производстве 1,2-дигидро-
кси-9,10-антрахинона (ализарина) (53) из 2-хлор-9,10-антрахинона
(49) или 9,10-антрахинон-2-сульфокислоты (см. также 8.1.1). При
получении (53) из (49) реакцию проводят в 40 %-ном водном рас-
творе NaOH при 235 °C в течение 3 ч в присутствии KNO3 в ка-
честве окислителя. Предполагается, что образующийся вначале
анион 2-гидрокси-9,10-антрахинона (50), подвергаясь нуклеофиль-
ной атаке гидроксид-аниона, превращается в лейкоализарат (51),
который окисляется в ализарат (52) и переходит в ализарин (53)
при подкислении.
При действии КОН и КС1О3 на бензантрон (54) аналогичная
реакция приводит к смеси 4- и 6-гидроксибензантрона [65]. При
кипячении 3-метилантрапиридона (55) в растворе NaOH в водном
диоксане образуется 1-гидрокси-З-метилантрапиридон [66].
Бензо- и нафтохиноны реагируют со щелочами сложно, с обра-
зованием смесей продуктов. Намного более гладко протекает за-
мещение атома водорода при ацетоксилировании хинонов по реак-
ции Тиле—Винтера [67], применяемой для введения гидрокси-
группы в несколько этапов. Соответствующий фенол или гидрохинон
окисляют в хинон, который в уксусном ангидриде в присутствии
кислоты превращается в триацетоксипроизводное, подвергаемое
296
гидролизу. Например, из гидрохинона (56) получают гидрокси-
гидрохинон (57) [68]. Выходы по стадиям указаны на схеме.
(56)
+ ШгСпО?.
+ H2SO4
---------—>
(86—92%)
+ (СНзСО)гО;
4- H2SO4
------------>
(86—87%)
ОСОСНз
+ НС1; Н2О
------------>.
(75%)
(57)
Нуклеофильное замещение атома водорода происходит на вто-
рой стадии, при взаимодействии хинона с уксусным ангидридом и
кислотой. Роль последней заключается, вероятно, в активации
молекулы хинона за счет О-протонирования, облегчающего нуклео-
фильную атаку ацетокси-аниона, за которой следует перемещение
протона и ацетилирование:
В качестве катализаторов кроме H2SO4 применяют НСЮ4,
ZnCl2, эфират трифторида бора и др. При действии уксусного
ангидрида в присутствии НС1О4 1,4-нафтохинон превращается в
1,2,4-триацетоксинафталин с выходом 95% при комнатной темпе-
ратуре [67].
297
8.2. ВВЕДЕНИЕ АЛКОКСИ- И АРИЛОКСИГРУПП
Введение алкокси- и арилоксигрупп осуществляется
действием алкоголятов и фенолятов щелочных металлов на гало-
ген- и нитроароматические соединения. В отдельных случаях ис-
пользуется замещение других групп: диазониевой, аммониевой,
эфирной (OAlk, ОАг) и т. д.
Замещение галогенов проводят как в отсутствие, так и в при-
сутствии катализаторов — соединений меди. При алкоксилирова-
нии в отсутствие катализатора образующееся алкоксизамещенное
может взаимодействовать с избытком алкоголята, расщепляясь на
диалкиловый эфир и фенол (см. 8.1.2). Расщепление наиболее
легко проходит при реакции с метилатом, медленнее с этилатом и
еще медленнее с алкоголятами высших спиртов. Чтобы предотвра-
тить расщепление, нужно вести процесс при возможно более низ-
кой температуре и избегать избытка алкоголята. Таким образом
при реакции 1,2,4-трихлорбензола (58) в диметилсульфоксиде при
100—110 °C с 1 моль метилата натрия получают 1-меток-
си-2,5-дихлорбензол (59) с выходом 75%, а при избытке метила-
та — 2,5-дихлорфенол (60) [69]:
(58) (59) (60)
Использование биполярных апротонных растворителей, таких
как диметилсульфоксид, диметилформамид, гексаметилфосфортри-
амид (гексаметапол), в которых алкоголят-анионы обладают зна-
чительно большей нуклеофильной активностью, чем в протонных
растворителях (см. 2.6), позволяет проводить реакцию не только в
более мягких условиях, но и замещать атом галогена в соединени-
ях, которые не реагируют с алкоголятами в иных средах. Так, при
нагревании о-дихлорбензола с небольшим избытком метилата на-
трия в гексаметаполе при 90 °C в течение 20 ч получают о-мето-
ксихлорбензол (о-хлоранизол) с выходом 78% [70]. В среде
о-дихлорбензола такое превращение удается осуществить только
при действии комплекса метилата калия с краун-эфиром (см. 2.6).
Аналогичным образом в гексаметаполе из лг-дихлорбензола полу-
чен л-ьметоксихлорбензол (лг-хлоранизол) (87%), из хлорбензола
при 120 °C — метоксибензол (анизол) (50%), из 1-хлорнафтали-
на—1-метоксинафталин (54%) [70]. Отсутствие изомеров, про-
дуктов восстановительного дегалогенирования и нечувствитель-
ность к ингибиторам радикальных реакций (п-динитробензол)
свидетельствует в пользу бимолекулярного механизма SnAt по
сравнению с аринпым SnI или радикальным SrnI (см. 2.7.2).
298
При действии более сильного основания, чем метилат-анион,—
трет-бутилата калия в диметилсульфоксиде — замещение неакти-
вированного атома галогена протекает по аринному механизму
[71, 72]. При обработке бромбензола смесью трет-бутилата калия и
трет-бутилового спирта в диметилсульфоксиде получают трет-бу-
токсибензол (42—46%) [71], а из 1- и 2-бромнафталпна — в каж-
дом -случае смесь 1- и 2-трет-бутоксинафталинов [72]; реакция
2-фторнафталина не сопровождается изомеризацией.
Замещение галогена на алкоксигруппу в соединениях, содер-
жащих активирующие электроноакцепторные заместители, имеют
промышленное значение для производства о- и п-метоксинитро-
бензола (о- и п-нитроанизол) (61), (62), п-нитроэтоксибензола
(и-нитрофенетол) (63), 1-метокси-2,4-динитробензола (2,4-динит-
роанизол) (64), 1,5- и 1,8-диметокси-9,10-антрахинона из соответ-
ствующих хлорпроизводных.
(61) (62) (63) (64)
Процесс обычно проводят в среде соответствующего спирта в ав-
токлаве с добавкой концентрированного водного раствора гидро-
ксида щелочного металла, под действием которого спирт переходит
в алкоголят.
При производстве о-нитроанизола (61) в стальной автоклав
вносят смесь 2400 л (60 кмоль) метанола, 6,5 кмоль NaOH в виде
52%-ного раствора и 5,2 кмоль о-нитрохлорбензола; создают дав-
ление воздуха 0,15—0,16 МПа, нагревают до 97—99 °C и разме-
шивают при этой температуре 5—7 ч. По охлаждении раствор
нейтрализуют 30%-ной H2SO4 до остаточной щелочности 0,5 г/л
NaOH, метанол отгоняют, приливают около 1000 л воды, органи-
ческий слой отделяют, фильтруют и промывают водой до отсут-
ствия о-нитрофенолята натрия и NaOH. После этого продукт под-
вергают в вакууме отгонке с водяным паром и обезвоживают в
вакууме; выход о-нитроанизола 72%.
Имеются специальные рекомендации проводить реакции алко-
ксилирования под давлением кислорода или воздуха [51]
(ср. 8.1.2 о роли кислорода при гидроксилировании). Побочно
образуются продукты восстановления нитрогруппы, преимуще-
ственно азоксисоединения.
В активированных соединениях галоген замещается в отсут-
ствие катализатора также и на арилоксигруппу. Простое нагрева-
ние 1,3-динитро-4-хлорбензола или, еще легче, 1,3-динитро-4-фтор-
бензола с фенолом приводит к 2,4-динитродифениловому эфиру;
это используют для идентификации фенолов [73]. Столь же легко
299
обменивается в динитропроизводных пиридиниевая группа, напри-
мер [74]:
+ АгОН
При перегруппировке Смайлса в водном щелочном растворе с
выходом, близким к количественному, происходит внутримолеку-
лярное арилоксилирование (см. ссылку [84] к гл. 7):
В производстве антрахиноновых красителей находит примене-
ние замещение на арилоксигруппу атома галогена в а- и в
p-положении антрахинонового ядра. Например, таким путем полу-
чают 1,5-дифенокси-9,10-антрахинон из 1,5-дихлор-9,10-антрахино-
на и 1-амино-2-арилокси-4-гидрокси-9,10-антра.хиноны (66) из
1-амино-2-бром-4-гидрокси-9,10-антрахинона (65) действием со-
ответствующего фенолята в феноле. Показано, что реакцию бром-
производного (65) с фенолятом можно проводить не только в среде
фенола, но и в водной среде с выходом феноксипроизводного 90%
[75]. При обработке (66) фенолятом, например гидрохинона, одна
арилоксигруппа вытесняет другую (67) [76].
В большинстве случаев диариловые эфиры получают в присут-
ствии меди или ее соединений (эфирная конденсация Ульмана
800
[77]). Так, нагревание n-крезолятов Na пли К с п-бромтолуолом
при 230—240 °C с добавкой порошка меди дает 4,4'-диметилдифе-
ниловый эфир [73]; а феноляты [78], n-крезоляты и 3,4-ксилено-
ляты [79] с п-нитрохлорбензолом при 180 °C образуют соответ-
ствующие 4-нитродифениловые эфиры (выход 80—85%), Вместо
фенолятов в реакцию можно вводить фенол и К2СО3 [73]. Особенно
эффективен этот прием в случае двухатомных фенолов, феноляты
которых осмоляются при проведении конденсации в обычных усло-
виях. Этого удается частично избежать, предварительно приготав-
ливая сухие феноляты в инертных растворителях [80].Применение
К2СО3 исключает эту стадию и позволяет получать ди- и полиэфи-
ры в среде пиридина с более высоким выходом [81 ]. Например, так
получают 1,3-дифеноксибензол из бромбензола и резорцина в при-
сутствии иодида меди(1) с выходом 84%:
Растворитель имеет важное значение, поскольку выступает, по-
видимому, в роли сокатализатора, участвуя в образовании ком-
плекса с медью. Пиридин является одним из лучших растворите-
лей [80].
Особой подвижностью в каталитической реакции обладают
атомы галогена в орто- и иери-положении к о'-гидроксиарилазо-
группе. Атомы хлора в 1-(2,6-дихлорфенилазо)-2-нафтоле (68)
[82] с высоким выходом замещаются на алкокси- и арилоксигруп-
пу при нагревании с соответствующим алкоголятом или феноля-
том в диоксане или другом растворителе. Аналогично реагируют
1-(2-хлорфенилазо)-2-нафтол [82], а также 8-(2-хлорфенилазо)-1-
нафтол [83].
+ RO"
C11SO4
R = Alk, Аг
(69)
301
Атомы иода и брома, как характерно для каталитической реак-
ции (см. 7.1.1), замещаются легче, а атомы фтора труднее:
I > Вг > Cl » F
Легкость замещения атома галогена в о-гидроксиазосоединени-
ях объясняется его координацией с атомом меди в комплексе типа
(69) [82]. В отсутствие о-гидроксигруппы в азосоединении подвиж-
ность атома галогена в каталитической реакции значительно
ниже [84].
Обмен галогена или нитрогруппы на арилоксигруппу — одна из
стадий получения симметричных диариловых эфиров взаимодей-
ствием активированных ароматических соединений с нитритом ще-
лочного металла. Фенолят-анион генерируется в результате
О-атаки нитрит-аниона (см. 8.1.2) и реагирует затем с исходным
соединением:
+ NOI + Noj + АгХ
АгХ ------>• ArONO —->• ArO* ----------> Ar2O
—n2o3
X= Cl, NO2
Хлорбензолы, содержащие в орто- или пара-положении элект-
роноакцепторный заместитель, при нагревании с NaNO2 и Ыа2СОз
(для связывания N2O3) в воде при 100—150 °C переходят в со-
ответствующие диариловые эфиры с выходом до 90% [85].
А-Метил-4-нитрофталимид превращается в диариловый эфир с
выходом 65% при кипячении с KNO2 в диметилформамиде [86].
При действии n-крезолята натрия в диметилформамиде при 25 °C
А-метил-4-нитрофталимид быстро и количественно переходит в
А/-метил-4-(п-толилокси)фталимид [87]. Спектральными методами
(ЯМР *Н и 13С, ЭПР, УФ) зафиксировано образование анионра-
дикала нитросоединения, но не установлено, предшествует оно или
сопутствует нуклеофильному замещению.
Алкоксигруппа может быть успешно введена замещением нит-
рогруппы. Так как алкоголят-анионы (метилат, этилат)—более
сильные нуклеофилы, чем гидроксид-анион, соединения, способные
обменивать нитрогруппу на гидроксильную (см. 8.1.3), обменивают
ее и на алкоксильную, причем в более мягких условиях, особенно в
биполярных апротонных растворителях. 4-Нитро- и 4,4'-динитро-
бензофенон при взаимодействии с метилатом пли этилатом натрия
в диметилформамиде, диметилсульфоксиде, гексаметаполе при
20°C за 24 ч количественно переходят в соответствующие 4-алкок-
си- и 4,4/-диалкоксибензофеноны. Замещение гидроксильной груп-
пой при действии NaOH не завершается и через 100 ч.
В 2,2/-дибром-4,4'-динитробензофеноне при реакции с алкоголята-
ми замещаются обе нитрогруппы (90%), но не атомы брома [45].
При обработке метилатом натрия в гексаметаполе 2,6-динит-
ро-4-трифторметилбензонитрил (706) обменивает одну нитро-
группу при 0 °C за 5 мин (83%) и обе — за 30 мин (78%);
303
2,6-динитробензонитрил (70а) превращается
водное (71а) при 65°C с выходом 81% [88].
в диметоксипроиз-
CN
CN
(70)
a) R=H
б) R = CF3
(71)
В достаточно активированных нитросоединениях замещение
на алкоксигруппу можно проводить в спиртовой среде. а-Мет-
окси-9,10-антрахиноны получают нагреванием 1-нитро-, 1,5- и
1,8-динитро-9,10-антрахинонов в метаноле с безводным К2СО3 [89]
или в присутствии оснований при пропускании воздуха [90]. При
нагревании в метаноле с метилатом натрия при 100—ПО °C
1,3-динитронафталин (72) с выходом 43% переходит в 1-метокси-
3-нитронафталин (73) [91]. Примечательно, что то же самое
соединение (73) образуется с выходом 94,6—96,8% при обработке
2,3-динитронафталина (74) избытком метилата натрия в метаноле
при 40-45 °C [92].
Можно полагать, что кине-замещение в данном случае связано
с возникновением изомерного ст-комплекса при атаке метилат-
анионом атома углерода, не несущего заместителя '(см. 2.7.2).
Подобный механизм кпне-замещения предполагается, например,
для превращения 4-нитробензофуразана (4-нитробензо-1,2,5-окса-
диазол) (75) в 5-метокси-4-хлорбензофуразан (76) при действии
щелочного раствора гипохлорита натрия в метаноле (67%) [93]-.
(75)
303
Справедливость такого предположения подтверждена кинети-
ческими исследованиями с помощью струевой методики, показав-
шими, что присоединение метилат-аниона к 4-нитро-7-Х-бензофу-
разанам (78) в метаноле значительно быстрее идет в положении
5 с образованием о-комплексов (77), чем в положении 7 с образо-
ванием термодинамически более стабильных о-комплексов (79),
приводящих к продукту «нормального» замещения (80) [94].
Алкоксизамещенные могут быть получены разложением солей
диазония в спирте в присутствии кислоты. Обработка спиртом,
главным образом этанолом, применяется как метод восстанови-
тельного замещения диазониевой группы атомом водорода
(см. 15.4.3), при котором алкоксипроизводные являются побочны-
ми продуктами. Однако часто, особенно при разложении в метано-
ле, алкоксипроизводные образуются в значительных количествах
[95]. Один из препаративных способов перевода аминов в метокси-
производные состоит в добавлении алкилнитрита в нагретый до
50—60 °C раствор амина в метаноле (выход около 70%) [73].
Подробное исследование разложения солей R-бензолдиазония
(81) в метаноле [96] показало, что замещение N) метоксигруппой
(82) и атомом водорода (83)—две независимые параллельные
реакции, первая из которых идет по ионному, а вторая — по ради-
кальному механизму.
Радикальная реакция подавляется в атмосфере кислорода и в
присутствии некоторых акцепторов радикалов (2-метил-2-нитро-
зопропан, FeSO4, HNO2) и ускоряется в присутствии инициаторов
(а-фенилазотрифенилметан). Так, термолиз борофторида /г-бром-
бензолдиазония [(81), R = п-Br] в метаноле при 65 °C в атмо-
сфере азота дает 18% п-броманизола и 74% бромбензола, в атмо-
сфере кислорода — 72 и 4°/о, а в присутствии а-фенилазотри-
фенилметана — 0 и 99% соответственно. Образование арильных
804
радикалов в атмосфере азота подтверждается идентификацией
побочных продуктов (4,4'-дибромбифенил), выделением п-бромиод-
бензола в присутствии иодбензола и др.
Для ионного механизма предложен путь с промежуточным
генерированием арил-катионов (см. 2.7.2):
СНзОН
ArNj > Аг+ 4- N2 -------► АгОСНз + Н+
Для радикального механизма — цепная схема:
Аг • + СН3ОН —> АгН + • СН2ОН
• СН2ОН + ArN2 —> ArN=N • + СН2О + Н+
ArN=N • —> Аг • + N2
с зарождением цепи, предположительно, через стадию ковалент-
ных диазоэфиров:
ArNj + СН3ОН —> ArN=N—ОСН3 —> Ar • + N2 + CH3O •
При R=Ji-Br, n-Cl, лг-Cl, л-ОСН3, как и при R = n-Br, тер-
молиз солей диазония (81) в атмосфере азота идет преимуще-
ственно по радикальному механизму с образованием R-аренов
(83), а в атмосфере кислорода — по ионному механизму с образо-
ванием метоксиаренов (82). При R = Н, п-СН3, лг-СН3, Л4-ОСН3 в
той и другой атмосфере преобладает ионный механизм, а при
R = n-NO2 — радикальный. Знание механизма и природы влияния
различных факторов позволяет, как и в других случаях, созна-
тельно выбирать условия проведения процесса.
ЛИТЕРАТУРА
1. Wedermeyer K.-F.— In: Methoden der Organischen Chemie (Houben-Weyl).
Bd. 6/lc. Stuttgart, Georg Thieme Verlag, 1976, S. 146—312. 2. Беркман Б. E.—
Сульфирование и щелочное плавление в промышленности органического синтеза.
М., Госхимиздат, 1960. 3. Оае S., Furukawa N., Kise М. е. а. — Bull. Chem. Soc.
Japan, 1966, v. 39, № 6, p. 1212—1216. 4. Buzbee L. R.—S. Org. Chem., 1966,
v. 31, № 10, p. 3289—3292. 5. Маколкин И. 4. —ЖОХ, 1942, т. 12, вып. 7—8,
с. 356—360. 6. Ворожцов Н. И. мл. — В кн.: Химия синтетических красителей.
Т. 3/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия,
1974, с. 1740—1816. 7. Шейн С. М.. Русов В. П. — ЖОрХ, 1975, т. И, вып. 1,
с. 96—99. 8. Япон. пат. 70/21501; Chem. Abstr., 1970, v. 76, 76874. 9. Пат. США
3309402, 1967; Chem. Abstr., 1967, v. 67, 81999. 10. Шейн С. М., Игнатов В. А.,
Ворожцов И. Н., мл.*.— ЖПХ, 1966, т. 39, вып. 8, с. 1867—1869.
11. Шейн С. М., Пирогова П. /1., Ярцева Р. В. и др. Авт. свпд. СССР
230187; БИ, 1968, № 34. 12. Morley 1. O.—i. Chem. Soc. Perkin 2, 1973, № 12.
р. 1626—1632. 13. Русов В. П., Шейн С. А4. — Изв. СО АН СССР, сер. хим.
наук, 1974, № 4, вып. 2, с. 138—142; Русов В. П., Ливанов В. А., Шейн С. М.—
Там же, с. 143—149. 14. Теддер Дж., Нехватал А., Джубб А. — Промышленная
органическая химия. Пер. с англ./Под ред. О. В. Корсунского. М., Мир., 1977,
с. 198—205. 15. Оае S.. Furukawa N. — Bull. Chem. Soc. Japan, 1966, v. 39, № 10,
p. 2257—2260; Furukawa N., Oae S. — Ibid., 1968, v. 41, № 4, p. 949—953.
16. Bennett O. F., Bouchard M. J., Malloy R. e.a. — J. Org. Chem., 1972, v, 37,
№ 9, p. 1356—1359. 17. Bottini A. T., Roberts J. D. — J, Amer. Chem. Soc., 1957,
v. 79, № 6, p. 1458—1462. 18. Gumprecht W. H. — In: Organic Syntheses. Coll,
v. 5. N. Y., Wiley, 1973, p. 632—635. 19. Дюмаев К- M., Попова Е. П. Авт. свид.
305
СССР 438649; БИ, 1974, № 29. 20. Пат. ФРГ 2042569, 1972; Chem. Abstr., 1972,
v. 76, 140170.
21. Пат. ФРГ 2119676, 1971; Chem. Abstr., 1972, v. 76, 33957; Пат. ФРГ
2237808, 1973; Chem. Abstr., 1974, v. 80, 14732. 22. Англ. пат. 1149447, 1969;
Chem Abstr 1969, v 71, 70296; Biller E., Carmichael W. M., Richter C. — Chem.
Ind., 1973 № 14, p. 685—687. 23. Пат. ФРГ 2231005, 1973; Chem. Abstr., 1973,
v. 78, 84014, 24. Шейн С. M., Игнатов В. А, —ЖОХ, 1962, т. 32, вып. 10,
с 3220—3222. 25. Скибинская М. Б., Шаров В. Г., Сергеев Е. В. — ЖПХ, 1972,
т 45 вып. 3, с. 702—703. 26. Kende A. S., Wade 1. 1., Ridge D. e.a.— J. Org.
Chem’., 1974, v. 39, № 7, p. 931—937. 27. Пат. США 3283011, 1966; Chem. Abstr.,
1967, v. 66, 2340. 28. Friedrich K-, Oeckl S — Chem. Ber., 1973, Bd. 106, № 7,
S. 2361—2365. 29. Шейн С. M., Красносельская M. Л. — ЖОХ, 1965, t. 35,
вып. 11, c. 1952—1955. 30. Jacobs R. L. — J. Org. Chem., 1971, v. 36, № 1.
p. 242—243.
31. Пат. США 2835707 1958; Chem. Abstr,, 1958, v. 52, 16295. 32. Пат. США
3760008, 3760009, 1973; Chem. Abstr., 1974, v. 80, 14714. 33. Яп. пат. 73/00696,
1973; Chem. Abstr., 1973, v. 78, 124296. 34. Лисицын В. И., Диденко Л. А. —
ЖОрХ, 1969, т. 2, вып. 6, с. 1063—1066; Там же, 1968, т. 4, вып. 7, с. 1225—
1227. 35. Лисицын В. И., Луговская Е. К. — Там же, 1971, т. 7, вып. 12,
с 2567—2568 36. Broxton Т. j. Muir D. At., Parker A. J. — J. Org. Chem., 1975,
v. 40. № 14, p. 2037—2042. 37. Пат. ФРГ 1933525, 1971; Chem. Abstr., 1971, v. 74,
111780. 38. Knudsen. R. D., Snuder H. R. — J. Org. Chem., 1974, v. 39, № 23,
p. 3343—3346; Knudsen R. D., Morrice A. G., Snyder H. R. Ibid., 1975, v. 40, Ns 20,
p' 2878—2883. 39. Biffin M. E. C.. Paul D. B. — Austr. J. Chem., 1974, v. 27, № 4,
p 777—788. 40. Birchall J. M., Haseldine R. N., Jones M. E.— J. Chem. Soc. (C),
1971, № 7, p. 1343—1348.
41. Горелик M. В., Евстратова M. И., Михайлова T. А. и др. — ЖОрХ, 1970,
т. 6, вып. 6, с. 1271 —1277. 42. Билькис И. И., Шейн С. М. — ЖОрХ, 1974, т. 10,
вып. 5, с. 1126—1128; Bilkis I. I., Shein S. М. — Tetrahedron, 1975, v. 31, № 8,
p. 969—971. 43. Abe T., Ikegami Y. — Bull. Chem. Soc. Japan, 1976, v. 49, № 11,
p. 3227—3231; Ibid., 1978, v. 51, № 1, p. 196—200. 44. Urbanski T. — Chemistry
and Technology of Explosives. Oxford, Pergamon Press, 1964, p. 258—259. 45. Gor-
vin J. H. — Chem. Ind., 1967, № 36, p. 1525—1526. 46. Пат. ФРГ 2537798, 1976;
Chem. Abstr., 1976, v. 84, 166268. 47. Пат. ФРГ 2254199, 1974; Chem. Abstr., 1974,
v 81 105107. 48. Пат. ФРГ 1808389, 1969; Chem. Abstr., 1969, v. 71, 123934.
49, Пат. США 3462497, 1969; Chem. Abstr., 1970, v. 72, 3216. 50. Kastens M. L„
Kaplan J. F. — Ind. Eng. Chem,, 1950, v. 42, p. 402—413; Англ. пат. 1106088,
1968, Chem. Abstr., 1968, v. 69, 35731.
51. Ворожцов П. П. Основы синтеза промежуточных продуктов и красите-
лей. М„ Госхимиздат, 1955, с. 371—375, 399—404. 52. Пат. ФРГ 2352976, 1974;
Chem, Abstr., 1974, v. 81, 91374. 53. Иванов В. А., Глибин Е. Н., Гинзбург О. Ф,—
ЖОрХ, 1972’, т. 8, вып. 9, с. 1891 — 1894, 54. Brockmann Н., Seela F.—Chem.
Ber., 1971, Bd. 104, № 9, p. 2751—2771. 55. Хартманн В., Байерс Дж., Дик-
ки Дж. — В кн.: Синтезы органических препаратов. Сб. 2. Пер. с англ. М., ИЛ,
1949, с. 379—380. 56. Манске Р. — Там же. Сб. 1, с. 313—315. 57. Lain-
booy J. Р. — J. Amer. Chem. Soc., 1950, v. 72, Ns 11, p. 5327—5328. 58. Пат. ФРГ
1148236, 1963; Chem. Abstr., 1963, v. 59, 11338. 59. Козлов В. В., Белов Б. И,—
ЖОХ 1959, т. 29, № 10, с. 3450—3455. 60. Morrison D. С., Heinritz D. W.—
J. Org. Chem., 1962, v. 27, № 6, p. 2229—2231.
61. Silcox С. M., Zuckerman J. J. — J. Organometal. Chem., 1966, v. 5, Ns 5,
p. 483—486. 62. Vandewalle J. J. M., de Ruiter E., Reimlinger H. e. a. — Chem.
Ber., 1975, Bd. 108, № 12, S. 3898—3899. 63. Кашпаров И. С., Пожарский А. Ф. —
Химия гетероцикл, соед., 1971, № 1, с. 124—128. 64. Дюмаев К. М., Попо-
ва Е. П. — Там же, 1973, № 6, с. 859. 65. Bradley W., Jadhav G. V. — J. Chern.
Soc., 1937, p. 1791 — 1792. 66. Казанков M. В., Уфимцев В. Л. — Химия гетеро-
цикл. соед., 1966, Ns 2, с. 315—316. 67. McOmie J. F. W., Blatchly J. М. — In:
Organic Reactions. V. 19. N. Y., Wiley, 1972, p. 199—277. 68. Влие E. — В кн.:
306
Синтезы органических препаратов. Сб. 1. Пер. с англ. М., ИЛ, 1949, с. 396 - 397,
463—465; Препаративная органическая химия. Пер. с польского/Под ред.
Н. С. Вульфсона. М., Госхимиздат, 1959, с. 574—576. 69. Пат. ФРГ 2210254,
1973; Chem. Abstrs., 1973, v. 79, 136781. 70. Shaw J. E., Kunerth D. C., Swan-
son S. B. — J. Org. Chem., 1976, v. 41, № 41, p. 732—733.
71. Sahyun M. R. V., Cram D. J. — In: Organic Syntheses. Coll. v. 5. N. Y„
Wiley, 1973, p. 926—928. 72. Bradshaw J. S., Hales R. H. — J. Org. Chem., 1971,
v. 36, № 2, p. 318—322; Bradshaw J. S., Chen E. У., Hales R. H. e.a. — Ibid..
1972, v. 37, № 12, p. 2051—2052. 73. Meerwein H. — In: Methoden der Organischen
Chemie (Houben-Weyl). Bd. 6/3. Stuttgart, Georg Thieme Verlag, 1965, S. 75—90.
74. Англ. пат. 805761. 1958; Chem. Abstr., 1959, v. 53, 8074. 75. Штейнберг Я. Б.,
Ткаченко С. С. — ЖПХ, 1976, т. 49, вып. 4, с. 904—905. 76. Яп. пат. 71/32596;
Chem. Abstr., 1972, v. 76, 60945. 77. Мороз А. А., Шварцберг М. С. — Успехи хи-
мии, 1974, т. 43, вып. 8, с. 1443—1461. 78. Брюстер Р., Тренинг Т. — В кн.: Син
тезы органических препаратов. Сб. 2. Пер. с англ. М., ИЛ, 1949, с. 370—371.
79. Миронов Г. С., Устинов В. А., Фарберов М. И.—ЖОрХ, 1973, т. 9, вып. 1,
с. 128—132. 80. Williams A. L.. Kinney R. Е., Bridger R. Е. — J. Org. Chem. 1967,
v. 32, № 8, р. 2501—2505.
81. Шейн С. М.. Литвак В. В. — Изв. СО АН СССР, сер. хим. наук, 1973,
№ 2, вып. I, с. 104—107; Литвак В. В., Маматюк В. И., Шейн С. М.—Там же,
1973, № 4, вып. 2, с. 100—103. 82. Степанов Б. И., Андреева М. А. — ЖОХ, 1960,
т. 30, вып. 8, с. 2748—2754; Степанов Б. И. — Там же, 1962, т. 32, вып. 11,
с. 3741—3744. 83. Степанов Б. И., Аринич Л. Н. — Там же, 1959, т. 29, вып. 9,
с. 3052—3054. 84. Rolling Р. V. — J Org. Chem., 1975 v. 40 № 17, р. 2421 —
2425. 85. Пат. ФРГ 2547037, 1977; РЖХим, 1978, 6Н179П. 86. Markezich R. L.,
Zamek О. S. — J. Org. Chem., 1977, v. 42, № 21, p. 3431—3436. 87. Relles H. M„
Johnson D. S., Manello 1. S. J. Amer. Chem. Soc., 1977, v. 99, № 20, p. 6677—6686.
88. Beck J. R., Sobczak R. L., Suhr R. G. e.a. — J. Org. Chem., 1974, v. 39, № 13,
p. 1839—1845. 89. Пат. ФРГ 2314696, 1974; Chem. Abstr., 1975, v. 82 45063.
90. Пат. ФРГ 2607036, 1976; Chem. Abstr., 1976, v. 85, 161884.
91. Hanker J. S., Katzoff L. Aronson L. D. e.a. — J. Org. Chem. 1965, v. 30,
№ 6, 1779—1781. 92. Morrison D. C. — Ibid., 1962, v. 27, № 1, p. 296—297. 93.
Mallori F. B., Varimbi S. P.—Ibid., 1963, v. 28, № 6, p. 1656—1662. 94. Di
Nunno L., Florio S., Todesco P. E. — J. Chem. Soc. Perkin, 2, 1975, № 14, p. 1469—
1472. 95. Корнблюм H.—В кн.. Органические реакции. Сб. 2. Пер. с англ. М.,
ИЛ, 1950, с. 285—361. 96. Broxton Т. J., Bunnett J. F., Paik С. Н,-—J. Org. Chem.,
1977, v. 42, № 4, р. 643—649; Bunnett J. F., Yijima C. — Ibid., p. 639—643.
Глава 9
РЕАКЦИИ С СЕРУСОДЕРЖАЩИМИ
НУКЛЕОФИЛЬНЫМИ РЕАГЕНТАМИ
Нуклеофильные реагенты, реакционным центром в кото-
рых является атом серы, в зависимости от валентного состояния
этого атома подразделяются на две группы: содержащие двухва-
лентный (двухкоординационный) атом серы и содержащие четы-
рехвалентный (трехкоординационный) атом серы. Первые исполь-
зуются для получения соединений с двухкоординационной серой:
тиолов, сульфидов, тиоцианатозамещенных, вторые — для получе-
ния соединений с четырехкоординационной серой: сульфокислот и
сульфонов.
807
9.1. ВВЕДЕНИЕ МЕРКЛПТОГРУППЫ
Получение тиолов непосредственным введением меркап-
тогруппы возможно только при действии сероводорода или его
солей. Все остальные методы [1,2] предусматривают введение
серусодержащих групп, которые превращаются в меркаптогруппу
на последующих стадиях: при гидролизе, восстановлении, расщеп-
лении основаниями.
S-Нуклеофилы, используемые для введения таких групп, весьма
разнообразны. К ним относятся ди- и полисульфиды металлов
M2Sn, тиомочевина NH2—CS—NH2, ксантогенаты RO—CS—S~,
тиокарбонаты RO—CO—S", тиосульфаты S2Oj" тиоцианаты SCN",
тиоляты RS" и О-ариловые эфиры тиоугольной ArO—CS—OAr и
тиокарбаминовой ArO—CS—NR2 кислот, перегруппировывающиеся
в соответствующие S-ариловые эфиры путем внутримолекулярного
нуклеофильного замещения. Вытесняемыми группами при межмо-
лекулярном замещении служат обычно атомы галогенов и диазо-
ниевая группа. Упомянутые внутримолекулярные перегруппировки
позволяют заменять гидроксигруппу на.меркаптогруппу, а окисле-
ние ароматического соединения в хиноидное и последующее вза-
имодействие с S-нуклеофилом дают возможность вводить мер-
каптогруппу вместо атома водорода.
9.1.1. Замещение атомов галогенов
Замещение галогенов меркаптогруппой при отсутствии
активирующих электроотрицательных заместителей требует очень
жестких условий. Так, нагревание хлорбензола с избытком суль-
фида натрия (1 :2,5) в АСметилпирролидоне при 300 °C приводит к
смеси тиофенола и дифенилсульфида с выходом 53 и 26% соответ-
ственно; в тех же условиях образуется только дифенилсульфид
(87%) при соотношении реагентов 1 : 0,5 [3]. Предложено получать
тиофенол пропусканием паров хлорбензола и сероводорода при
температуре около 400 °C над катализатором, содержащим суль-
фид металла на активном угле [4].
В полихлорбензолах возможно замещение хлора меркапто-
группой действием гидросульфида натрия или калия в этиленгли-
коле, метаноле, жидком аммиаке. Так, гексахлорбензол при обра-
ботке NaHS в жидком аммиаке под повышенным давлением при
80 °C в течение 10 ч превращается в пентахлортиофенол с выходом
96%. В тех же условиях замещается атом хлора в положении 3
пентахлорбензола (86%), в положении 2 1,2,3,4-тетрахлорбензола
(98%), труднее — в 1,2,4,5-тетрахлорбензоле (34%), 1,2,3-трихлор-
бензоле (34%) и 1,3,5-трихлорбензоле (17%), а я-дихлорбензол не
реагирует с NaHS даже при 125 °C в присутствии ацетата меди (II)
[5]. При нагревании пентахлорпиридина с KHS в этиленгликоле
может быть получен 4-меркаптотетрахлорпиридин с выходом до
98% [6]. 2,5-Дибромтерефталевая кислота переходит в 2,5-димер-
каптотерефталевую кислоту при нагревании с KHS и порошком
меди [7] (см. также 9.1.3).
308
При реакции полихлорпропзводных бензонитрила с NaHS в
жидком аммиаке предпочтительно замещается хлор в пара-поло-
женин, а при реакции полихлорпроизводных нитробензола — в ор-
то-положении [1]. При действии на о- и п-нитрохлорбензолы суль-
фида натрия в водной среде наряду с замещением хлора наблю-
дается частичное восстановление нитрогруппы. Для получения
о-аминотиофенолов стадии замещения и восстановления совмеща-
ют, проводя процесс при кипячении в водном растворе дисульфида
натрия [8]. При применении Na2S2 в этаноле из о-нитрохлорбензо-
ла с выходом 58—66% получают ди(2-нитрофенил)дисульфид [9],
который можно восстановить в о-нитротиофенол, не затрагивая
нитрогруппы.
R=H, Cl, Вг
2-Амино-1-хлор-9,10-антрахинон в подобных условиях перехо-
дит в 2-амино-1-меркапто-9,10-антрахинон. Соединения, содержа-
щие галоген в цикле пиримидина, пиридазина и других азотсодер-
жащих гетероциклах легко обменивают его на меркаптогруппу при
действии NaHS или Na2S.
Соединения, содержащие активированный атом галогена, спо-
собны реагировать с тиомочевиной, образуя изотиурониевые соли,
которые гидролизуются с образованием тиолов в щелочной среде:
zNH2
ЛгХ + S=C(NH2)2 —> ArSC' + X’ —-> ArSH + (NH2)2CO
4nh2
Этот метод широко применяется в ряду гетероциклов с
несколькими атомами азота [1], но малоэффективен для нитрога-
логенбензолов. Выход изотиурониевой соли при взаимодействии
1,3-динитро-4-хлорбензола (2,4-динитрохлорбензола) с тетраметил-
тпомочевиной составляет всего 20% [Ю]. Очевидно, подвижность
хлора в 1,3-динитро-4-хлорбензоле недостаточна для обмена при
действии такого слабого нуклеофила, как тиомочевина, значитель-
но уступающая по нуклеофильности не только тиолят-анионам, но
н метилатаниону. Реакция 1,3-динитро-4-хлорбензола с тиомочеви-
ной в метаноле при 50 °C идет в 20 400 раз медленнее, чем с мети-
латом натрия, тогда как реакция с тиофенолятом натрия в 250 раз
быстрее [11].
309
Тиопианат-анион— более сильный нуклеофил, чем тиомочеви-
на, хотя тоже уступает метилат-аниону. Взаимодействие 1,3-ди-
нитро-4-хлорбензола с тиоцианатом аммония в водной среде слу-
жит промышленным методом получения 2,4-динитрофенилтиоциа-
ната (2), применяемого в качестве пестицида («нирит») [12]. На
направление реакции с тиоцианат-анионом ввиду его амбидентно-
сти существенное влияние оказывает характер уходящей группы
[13], определяющий степень жесткости (мягкости) реакционного
центра (см. 2.5). Если при реакции 1,3-динитро-4-хлорбензола [(1),
X == С1] с тиоцианатом калия в ацетоне при О °C образуется с
выходом 98% тиоцианатозамещенное (2), то при реакции 2,4-ди-
нитрофенилпиридиния [(1), X = NC5H5] получается, с выходом бо-
лее 98%, изотиоцианатозамещенное — 2,4-динитрофенилизотиоци-
анат (3); при реакции же 2,4-динитродифенилового эфира [(1),
X = ОСеНб] получается 69% тиоцианатозамещенного (2) и 31%
изотиоцианатозамещенного (3) [И-
Восстановление тиоцианатозамещенных или расщепление при
действии щелочей приводит к получению арентиолов [1]. Действи-
ем восстановителей арентиолы могут быть получены также из
сульфидов после введения нуклеофильным замещением алкилтио-
или арилтиогруппы (см. 9.2).
9.1.2. Замещение диазониевой группы
Замещение диазониевой группы при действии S-нуклео-
филов представляет собой более универсальный метод, чем заме-
щение галогенов, так как не требует наличия в молекуле активи-
рующих заместителей. Любое производное анилина или другого
диазотируемого амина может быть переведено в арентиол взаимо-
действием соли диазония с дисульфидом или полисульфидом, ксан-
тогенатом, тиоцианатом металла или с тиомочевиной и последую-
310
щим гидролизом или восстановлением образовавшегося S-заме-
щенного. Ограничения обусловлены побочными реакциями, в част-
ности обменом заместителей (атома галогена, нитрогруппы) в орто-
и пара-положении к диазониевой группе, а также взрывоопасно-
стью реакционных смесей при взаимодействии с полисульфидами
и особенно с ксантогенатами [14].
Реакции с полисульфидами щелочных металлов M2Sn, где
п 2, проводят во избежание взрыва при постепенном добавле-
нии раствора соли диазония к нагретому до 45—60 °C водному
раствору полисульфида. Образующийся диарилполисульфид пере-
водят в тиол кипячением в растворе NaOH [15] или восстановле-
нием [16, 17]:
+ M2s„ - N2
ArN] ------> (ArN2)2Sra -> Ar2Sra —> ArSH
В некоторых случаях процесс вполне безопасен и его можно
проводить при низкой температуре. Так, взаимодействие диазоти-
рованной антраниловой (о-аминобензойной) кислоты с дисульфи-
дом натрия ведут при температуре не выше 5 °C, получая
2,2'-дитиодибензойную кислоту, которую восстанавливают далее в
2-меркаптобензойную кислоту [18], применяемую в производстве
тиоиндигоидных красителей.
При взаимодействии соли диазония с алкилксантогенатами ще-
лочных металлов сначала образуются арендиазоксантогенаты, ко-
торые разлагаются с выделением азота, превращаясь в О-ал-
кил-5-арилдитиокарбонаты. Последние гидролизуют в щелочной
среде или восстанавливают в арентиолы:
-n2
ArN2 + ’S—CS—OR —> ArN=N—S—CS—OR ---------►
—>- ArS—CS—OR —> ArSH
Алкилксантогенаты получают внесением сероуглерода в рас-
твор щелочи в спирте [14, с. 627]:
ROH+CS2 + KOH —> RO—CS—SK+H2O
Для получения лг-тиокрезола раствор диазотированного ,м-то-
луидина постепенно приливают в нагретый до 40—45 °C водный
раствор этилксаитогената калия, после чего арилдитиокарбонат
извлекают и нагревают с КОН в этаноле; выход 63—75% [19].
Подчеркивается, что процесс взрывоопасен из-за возможности на-
копления и затем спонтанного разложения арендиазоксантогената.
Ни в коем случае нельзя смешивать растворы диазония и этил-
ксантогената при низкой температуре и потом нагревать. Разложе-
ние этилксаитогената диазония значительно ускоряется и гладко
протекает уже при 0 °C в присутствии каталитических количеств
ионов Ni2+, что делает процесс более безопасным [20]. Затрудне-
ния возникают также на стадии гидролиза, возможно, из-за легкой
окисляемости арентполятов. При получении n-тиокрезола и
2,6-диметилтиофенола по упомянутой выше методике с гидролизом
Действием щелочи выходы не превышали 39—49%, но повысились
811
до 84—89% при переводе арилтиокарбонатов в тиолы восстанов-
лением алюмогидридом лития [21].
Борфториды диазония в водном растворе реагируют с тиомоче-
виной, образуя S-арилтиурониевые соли, которые без выделения
гидролизуют в бикарбонатной среде, получая тиофенолы с выхо-
дом 25—50% в расчете на борфторид диазония [22]. В случае
n-хлорбензолдиазония в результате замещения атома хлора полу-
чают 1,4-димеркаптобензол, так же как из диазотированного
и-фенилендиамина. По другой методике, в раствор диазония непо-
средственно после диазотирования вносят тетраметилтиомочевину
и хлорид меди(П), затем выделяют перхлорат изотиурония и под-
вергают его гидролизу в растворе КОН [10]:
zNR2
ArNH2 —> ArNj —> ArSCZ + —> ArSH
4NR2 R=H, CH3
Взаимодействие солей диазония с тиоцианатом меди в условиях
реакции Зандмайера является общим методом введения тиоциана-
тогруппы. При реакции о- и п-галогенбензолдиазониев помимо
диазониевой группы тиоцианатогруппой может замещаться атом
галогена. Например, при обработке раствора З-нитро-4-хлор- или
4-бром-З-нитробензолдиазония тиоцианатами калия и меди обра-
зуется смесь приблизительно равных количеств 4-галогено-З-нит-
рофенилтиоцианата и 2-нитро-1,4-фенилендитиоцианата [1]:
Х = С1, Вг
Как уже указано выше (см. 9.1.1), арилтиоцианаты могут быть
превращены в тиолы действием щелочей или восстановителей.
В отдельных случаях тиоцианатогруппу вводят вместо аминогруп-
пы в красители и другие химикаты. Так, монодиазотированием
1,4-диамино-5-нитро-9,10-антрахинона и кипячением раствора вы-
деленного сульфата диазония с тиоцианатом калия в воде полу-
чают краситель 1-амино-5-нитро-4-тиоцианато-9,10-антрахинон [23].
9.1.3. Замещение гидроксильной группы
Методы превращения гидроксильной группы в меркап-
тогруппу основаны на термических перегруппировках О-ариловых
эфиров тиокарбоновых кислот в S-ариловые эфиры:
ArO—CS—R —> ArS—СО—R
Известны перегруппировки соединений, в которых R = ОАг,
NAlk2, Аг и Aik, SAr и SAlk. Препаративное значение для получе-
312
ния тиофенолов имеют только две первые: перегруппировка 0,0'-
диарилтиокарбонатов (4) в О,5-диарилтиокарбонаты (5) (перегруп-
пировка Шенберга [24]) и перегруппировка О-арил-ММ-диалкил-
тиокарбаматов (6) в S-арил-^А^-диалкилтиокарбаматы (7) (пере-
группировка Ньюмена-Куарта) [25, 26].
S О II II —> ArO—С—ОАг —> ArS—С—ОАг —> ArSH + СО2 + АгОН (4) (5) (8)
АгО'1 — S О II II —> ArO—С—NAlk2 —> ArS—С—NAlk2 —> ArSH + СО2 + AlksNH (6> (7) (8)
О,О'-Диарилтиокарбонаты (4) получают ацилированием фе-
нолов тиофосгеном (тиокарбонилдихлоридом) CSC12 (см. [27]),
О-арил-МАГдиалкилтиокарбаматы (6)— ацилированием ЛАЛАдиал-
килтиокарбамоилхлоридами (10) [28]. Последние, в свою очередь,
с высоким выходом приготавливают хлорированием промышленно
доступных тетраалкилтиурамдисульфидов (9): тиурама Д (Aik =
= СНз) или тиурама Е (Aik = С2Н5)—в инертной среде (СС14,
бензол) или без растворителя [29]:
? ? + ci2
Alk2N—С—S—S—-С—NAlk2 -------► Alk2N—(Г + 2S
\ci
(9) (10)
Перегруппировки проводят при нагревании без растворителя
или в высококипящем инертном растворителе при температуре
170—300 °C и выше в зависимости от строения арила. Перегруппи-
ровку О-арилтиокарбаматов (6) предложено проводить также в
газовой фазе пропусканием смеси тиокарбамата и разбавителя
(толуол) через кварцевую трубку, нагретую приблизительно до
400 °C [25]. Выходы при перегруппировке Шенберга составляют
50—80%, при перегруппировке Ньюмена — Куарта— 70—95% от
теоретического.
Гидролиз получаемых в результате перегруппировок S-арило-
вых эфиров (5), (7) в тиофенолы (8) осуществляют нагреванием с
водным раствором NaOH или КОН, иногда с добавкой органиче-
ского растворителя; выходы достигают количественных.
В препаративном отношении перегруппировка Ньюмена-Куарта
предпочтительней, поскольку использует для ацилирования более
доступные ЛСА-диалкилдитиокарбамоилхлориды (10), а не тио-
фосген, дает более высокие выходы и обеспечивает полное превра-
щение фенола, тогда как при перегруппировке Шенберга по сте-
хиометрии реакции половина исходного фенола возвращается не-
измененной.
313
2-Тионафтол (2-меркаптонафталин) получают указанным мето-
дом из 2-нафтола ацилированием A',.'V диметилтиокарбамоилхло-
ридом, перегруппировкой и гидролизом с общим выходом около
52% [28]. 3,5-Диметокситиофенол синтезируют из диметилового
эфира флороглюцина с выходом 40,5% [30]. Преимущества метода
таковы, что иногда оказывается целесообразным предпринять син-
тез фенола из галогенпроизводного, чтобы перевести его затем в
тиофенол перегруппировкой А^М-диалкилтиокарбамата вместо то-
го, чтобы непосредственно замещать атом галогена. Так, обследо-
вание различных методов получения 2,5-димеркаптотерефталевой
кислоты из 2,5-дибромтерефталевой кислоты показало, что шести-
стадийный синтез с общим выходом 36%, включающий перевод в
2,5-дигидрокситерефталат и перегруппировку Ньюмена — Куарта,
более эффективен, чем упомянутое ранее одностадийное превра-
щение при действии KHS и меди (см. 9.1.1), которое дает с
выходом 60—90% трудноочищаемый продукт [7].
Перегруппировки облегчаются при наличии в ароматическом
кольце электроноакцепторных заместителей, отчего при пиролизе
несимметричных О.О'-диарилтиокарбонатов АгО—CS—ОАг'(Пб)
с атомом серы всегда связывается арил, обладающий большей
электроноакцепторностью. 2- и 4-Нитрозамещенные О-фе-
нил-ААМ-диметилтиокарбаматы (На) перегруппировываются в
S-фенилтиокарбаматы (13а) с выходом более 90% при 170—180 °C,
а 3-нитро- и 2-метоксизамещенные — при 235 и 280 °C соответствен-
но [26]. Скорости перегруппировок коррелируют с о~-константами
заместителей R' в арильном остатке при реакционной константе
р == 1,83 для перегруппировки тиокарбаматов (Па) и р = 1,06 для
перегруппировки тиокарбонатов (116) (в дифенилоксиде при
200 °C) [31].
(и)
(12)
R
(13)
a)R=N(CH3)2
6)R=OC6HS
Влияние заместителей не оставляет сомнений в нуклеофильном
характере замещения атомом серы, а первый кинетический поря-
док реакции и отсутствие обмена арилами при перегруппировке
несимметричных тиокарбонатов АгО — CS — ОАг' или смесей
О-арилтиокарбаматов, содержащих разные арилы, указывает, что
реакции внутримолекулярны. Предполагается, что они протекают
через четырехчленное циклическое промежуточное состояние,
близкое по структуре к биполярному спиро-о-комплексу (12)
[31]. Это предположение вытекает из постулата Хэммонда, по-
скольку ввиду большой энергии активации перегруппировок пере-
ходное состояние должно лежать на координате реакции ближе к
продукту элементарной стадии, чем к исходному соединению
(см. 2.7.2).
314
9.1.4. Замещение атома водорода
Меркаптогруппа может быть введена в незамещенное
положение окислением ароматического дигидрокси-, аминогидрок-
си- или диаминосоединения в соответствующую хиноидную систему
с последующим замещением атома водорода действием S-нуклео-
фила (тиомочевина, тиосульфат-аниоп), сопровождающегося внут-
римолекулярным восстановлением и заключительной трансформа-
цией вступившей группировки в меркаптогруппу. Таким образом,
окисляя пирокатехин или 4-метилпирокатехин (14) гексациа-
но-(Ш)ферратом калия в водном растворе ацетата натрия при
комнатной температуре в присутствии тиомочевины, с выходом
80—90% получают S-арилтиурониевые соли (15), которые перево-
дят нагреванием в растворе NaOH в меркаптосоединения (16).
Последние выделены в виде триацетильных производных с выхо-
дом 81—86%.
Аналогично окислением фенотиазина хлоридом железа(III) в
соль фенотиазиния, обработкой тиомочевиной и гидролизом полу-
чают 2-меркаптофенотиазин [32].
Взаимодействием п-бензохинонов (17) с тиосульфатом нат-
рия уксусной кислоте получают сульфотиогидрохиноны (18), ко-
торые далее восстанавливают в 2-меркаптогидрохиноны (19)
[14, с. 556].
315
Подобным образом окислением п-фенилендиамина в п-хиион-
диимин в присутствии тиосульфата натрия с последующим восста-
новительным расщеплением сульфотиогрупп можно получить мер-
капто-п-фенилендиамины [1].
9.2. ВВЕДЕНИЕ АЛКИЛТИО- И АРИЛТИОГРУПП
Введение алкилтио- и арилтиогрупп осуществляется
действием соответствующих тиолят-анионов с замещением атомов
галогенов, нитро-, сульфо-, алкилсульфонильных и некоторых дру-
гих групп и действием тиолов на хиноны с замещением атома
водорода.
Алкил- и арилтиолят-анионы AlkS-, ArS-, будучи слабыми, но
мягкими основаниями, являются сильными нуклеофилами (см.
2.5). Специфические особенности тиолят-анионов как нуклеофилов
обусловлены именно их «мягкостью», подразумевающей высокую
поляризуемость при малой энергии удерживания электронов на
ВЗМО. Реакции с тиолят-анионами очень чувствительны к харак-
теру реакционного центра и его окружения, включая природу
уходящей группы и заместителей в ароматическом кольце. Чем
более мягкое основание представляет собой отщепляемый анион X ,
тем легче протекает замещение под действием тиолят-аниона. При
переходе от X = F к Х = 1 в 2,4-динитро-Х-бензолах скорость
реакции с тиофенолятом натрия в метаноле увеличивается в 285 раз
больше, чем скорость реакции с жестким основанием — метилатом
натрия [33]. Тиофенолят-анион C6H5S~ обладает большей поляри-
зуемостью, чем метантиолят-анион CH3S_, вследствие чего при
X = I реакция быстрее идет с тиофенолятом, а при X = F — с
метантиолятом [2]. При реакции 1,3-динитро-4-трифторметилсуль-
фонилбензола (X = SO2CF3) с тиофенолятом натрия в метаноле
замещается трифторметилсульфонильная группа (96%), тогда как
при реакции с метилатом натрия и пиперидином — в основном
нитрогруппа в «-положении к трифторметилсульфонильной группе
[34]:
+ сн3о~
+ CeH5S-
Влияние электроноакцепторных заместителей на скорость за-
мещения при действии тиофенолят-аниона не описывается уравне-
нием Гаммета с применением о~-констант [35], как в реакциях с
алкоголятами и другими нуклеофилами (см. 2.4.1 и 2.7.2). Так,
316
реакция замещенных хлорбензолов с тиофенолятом натрия в мета-
ноле протекает быстрее при наличии в «ара-положении более сла-
бого электроноакцептора — нитрогруппы, чем более сильного —
трифторметилсульфонильной группы. По-видимому, определяю-
щим фактором при реакции с тиофенолятом является не столько
электроноакцепторная способность заместителя, сколько его вос-
приимчивость к поляризующему действию нуклеофила из-за боль-
шего вклада прямого полярного сопряжения заместителя с реак-
ционным центром по сравнению со стандартными реакционными
сериями [34, 35].
Замещение активированного атома галогена на алкил(арил)тио-
группу проводят обычно при нагревании с тиолом и гидроксидом
щелочного металла в протонном или полярном апротонном раство-
рителе. Нитрогруппы при этом могут восстанавливаться с образо-
ванием азоксисоединений, чего удается избежать, если вести реак-
цию при избытке свободного тиола, добавляя щелочь постепенно.
Постепенное внесение раствора КОН в раствор п-нитрохлорбензо-
ла и метан- или этантиола в метаноле или диметилформамиде с
последующим нагреванием дает возможность получать сульфиды с
выходом 92—95% [36]:
R = H CF3
R' = CH3, С6Н5
В активированных полигалогенпроизводных при действии тио-
лята можно заместить несколько атомов галогена. Например, ки-
пячение тетрафторфталодинитрила с тиофенолятом натрия в
метаноле в течение 15 мин приводит с выходом 62% к 4,5-ди(фенил-
тио)-3,6-дифторфталодинитрилу наряду с 8% тетра(фенилтио)-
фталодинитрила, а кипячение тетрахлорфталодинитрила с тиофе-
нолятом натрия в водном ацетоне в течение 30 мин — к
тетра (фенилтио) фталодинитрилу с выходом 76% [37].Добавлени-
ем водного раствора NaOH к раствору 1,4-диамино-2,3-ди-
хлор-9,10-антрахинона и арентнола в диметилформамиде при 70°С
осуществляют замену обоих атомов хлора на арилтиогруппы [38].
В некоторых случаях аналогично удается заместить неактиви-
рованные атомы галогена. Например, нагревание 1- и 2-бром- или
фторнафталинов с 1-бутантиолом или 2-метил-2-пропантиолом в
диметилсульфоксиде при ПО °C приводит к соответствующим бу-
тилтионафталинам с выходом 50—96% [39].
Неактивированные атомы галогена в бензольном ряду обмени-
ваются на алкил- или арилтиогруппу при действии тиолятов меди.
Как и в других катализируемых соединениями меди реакциях
обмена атома галогена (см. 7.1.1), легче всего в этих условиях
обменивается атом иода, труднее — бром и намного труднее --
хлор. Нагревание бромбензолов с этан- и 1-бутантиолятом или
317
тиофенолятом меди в хинолине при 200 °C в течение 4—5 ч дает с
хорошим выходом соответствующие сульфиды, например 1,4-ди-
(этилтио)бензол из 1,4-дибромбензола с выходом 96% [40]. Данный
метод использован для введения трифторметилтиогруппы в бен-
зольное и пиридиновое кольцо замещением атома иода действием
трифторметантиолята меди в Af-метилпирролидоне, хинолине, ди-
метилформамиде [41]. Иногда соли меди вводят в реакцию с
тиолом в каталитических количествах, как например при получении
красителя 1-амино-4-(2-бензотиазолилтио)-9,10-антрахинона реак-
цией 1-амино-4-бром-9,10-антрахинона с 2-меркаптобензотиазолом
(каптакс) в кипящем бутаноле в присутствии К2СО3, CuCl и по-
рошка меди [42]. То же самое соединение можно получать заме-
щением нитрогруппы в 1-амшю-4-пи'гро-9,10-аитрахиноне при дей-
ствии 2-меркаптобензотиазола и К2СО3 в диметилформамиде [43].
Тиолят-анионы могут образовываться непосредственно в про-
цессе синтеза сульфидов. На этом основано получение диарил-
сульфидов взаимодействием арилгалогенидов с сульфидом натрия
[14, с. 584], на первой стадии которого образуется арентиолят,
реагирующий со второй молекулой арилгалогенида:
—NaX —NaX
2ArX + Na2S ------► ArSNa-f-ArX -------► Ar2S
Алкантиолы могут быть генерированы в реакционной среде
разложением S-алкилтиурониевых солей. Так, 1-алкилтио-9,10-ан-
трахиноны получают, с хорошим выходом, кипячением с S-алкил-
тиурониевыми солями 1-хлор-9,10-антрахинонов в спирте [44] или
9,10-антрахинон-1-сульфонатов в воде [45] в присутствии избытка
щелочи.
В сильно активированных соединениях возможно вытеснение
одной алкил (арил) тиогруппы другой, как в 2-метилтио- или
2-этилтио-4-пиримидонах при действии алкан- и арентиолятов
[46].
Вытеснение арентиолят-анионов при нуклеофильной атаке ме-
тилата приводит к расщеплению 2,4-динитродифенилсульфидов в
кипящем метанольном растворе КОН и образованию тиофенолов
с выходом 76—80% [47]:
+ СН30-
о- и w-Хиноны присоединяют алкан- и арентиолы в кислой среде,
превращаясь соответственно в 2- и 4-алкилтио- или -арилтиозаме-
318
Щенные арендиолы, легко окисляемые в замещенные хиноны [48].
Подобным же образом реагируют некоторые хиноидные полицик-
лические системы, например 3-феноксазиноны (20), при действии
на которые тиолов замещается атом водорода в положениях 1 или
2 хиноидного кольца или в положении 7 бензоидного кольца, в
обоих случаях с одновременным восстановлением гетероцикла. Ес-
ли атака в хиноидное кольцо невозможна из-за 1,2-бензаннелиро-
вания (21), замещение направляется только в бензоидное кольцо
[49].
9.3. ВВЕДЕНИЕ СУЛЬФОГРУППЫ, АЛКИЛ-
И АРИЛСУЛЬФОНИЛЬНЫХ ГРУПП
Сульфогруппы вводятся при действии солей сернистой
кислоты, а алкил- и арилсульфонильные — соответственно, солей
алкан- и аренсульфиновых кислот, нуклеофильным замещением
атома галогена или нитрогруппы, а также атома водорода. Суль-
фогруппа, кроме того, может быть введена замещением диазоние-
вой группы при действии сернистого ангидрида.
Обмен галогена и нитрогруппы практикуется при наличии в
молекуле активирующих заместителей. Реакция с сульфитом по-
зволяет получать сульфокислоты, которые нельзя синтезировать
электрофильным сульфированием (гл. 3). При нуклеофильном за-
мещении сульфогруппа вступает в пара- или орто-положение к
электроноакцепторному заместителю, в том числе в орто-положе-
ние к другой сульфогруппе, что обычно исключено при сульфиро-
вании из-за стерических факторов даже при благоприятном элек-
тронном влиянии других заместителей. Возможности метода хоро-
шо иллюстрирует синтез ароматических полисульфокислот. Нагре-
вание 1,4-динитротетрахлорбензола (22)— продукта нитрования
1,2,4,5-тетрахлорбензола — с сульфитом натрия в водном диоксане
без катализатора приводит к натриевой соли тетрахлор-1,4-бензол-
Дисульфокислоты (23), а в присутствии солей меди — с выходом
319
75% к натриевой соли бензолгексасульфокислоты (25) [50]. Ана-
логично из пентахлорпиридина синтезирована пиридинпентасуль-
фокислота [51].
(24)
Замещением атомов хлора и нитрогруппы в производных
9,10-антрахинона действием Йа250з удалось получить многие но-
вые антрахинон-сульфокислоты: 1,2-, 1,4- и 2,3-дисульфокислоты,
1,2,3- и 1,4,5-трисульфокислоты, 1,2,3,4- и 1,4,5,8-тетрасульфокисло-
ты и др. [52]. Комбинацией сульфирования, хлорирования и обра-
ботки сульфитом получают 1-амино-9,10-антрахинон-2,5,8-три-
и 2,4,5,8-тетрасульфокислоты [53].
Соединения, в которых атомы галогенов и нитрогруппы способ-
ны обмениваться при реакции с сульфитом, при действии арен-
сульфинатов образуют соответствующие сульфоны. Так, (22) при
нагревании в водном диоксане с бензолсульфинатом натрия с
выходом 85% переходит в 1,4-ди(фенилсульфонил)тетрахлорбен-
зол (24); последний превращается в (25), при обработке сульфи-
том в присутствии соли меди. Нагреванием п-нитрохлорбензола с
п-ацетиламинобензолсульфинатом натрия в смеси этиленгликоля и
его метиловых эфиров при 142 °C получают 4-ацетиламино-4'-нит-
родифенилсульфон (51%) [54]. При реакции 1,8-динитро-9,10-ант-
рахинона с этансульфинатом натрия в диметилформамиде при
120—130°С получается 1,8-ди(этилсульфонил)-9,10-антрахинон
(74%) [55]. Реакцией 2-хлор-9,10-антрахинона с п-толуолсульфи-
натом натрия в кипящем диметилформамиде готовят 2-(п-толил-
сульфонил)-9,10-антрахинон (85—90%) [56].
Метод введения сульфо- и хлорсульфонильных групп замеще-
нием диазонневой группы состоит в добавлении раствора хлорида
320
диазония, приготовленного диазотированием амина в соляной кис-
лоте, в насыщенный раствор SO2 в ледяной уксусной кислоте в
присутствии CuCh- При этом из анилина, его производных, со-
держащих в кольце галогены, метильные, нитро-, метокси- и кар-
боксильные группы, а также из нафтиламинов с выходом 70—80%
получают соответствующие сульфохлориды, из анилин- и 1-нафтил-
аминсудьфокислот — дисульфокислоты [57]. Если замещение ди-
азониевой группы проводить в присутствии восстановителя—ме-
ди, оксида меди(1) или соли железа (II), продуктом реакции яв-
ляются сульфиновые кислоты. В частности, пропусканием SO2 в
сернокислый раствор диазотированного 2,4-динитроанилина в при-
сутствии FeSO4 с выходом 74% получают 2,4-динитробензолсуль-
финовую кислоту [57].
Для введения сульфогруппы в незамещенное положение может
быть использована обсуждавшаяся ранее в связи с реакцией Бу-
херера (см. 7.1.5) способность нафтолов и некоторых других гид-
роксисоединений присоединять бисульфит натрия. Ароматизация
аддуктов с бисульфитом позволяет вводить сульфогруппу в мета-
положение к гидроксигруппе, чего невозможно достигнуть элект-
рофильным замещением. Бисульфитное соединение 1-нафтола —
.и-тетралонсульфокислота (26) превращается в 4-гидрокси-2-на-
фталинсульфокислоту (27) при бромировании в положение 2 с
последующим дегидробромированием при действии небольших ко-
личеств гидразина или его производного [58]:
(27)
Подобный метод оказался пригодным и для введения сульфо-
группы в молекулу 9,10-антрахинона. При внесении NaHSO3 в рас-
твбр натриевой соли 1-гидроксиантрагидрохинона (1,9,10-антра-
центриол), полученного восстановлением 1-гидрокси-9,10-антрахи-
нона (28) дитионитом натрия в щелочной среде, образуется
бисульфитное производное (29), которое при обработке бромом
или хлором в водном растворе с высоким выходом превращается
непосредственно в 4-гидрокси-9,10-антрахинон-2-сульфокислоту
11 Зак. 271 321
(30). Аналогично из 1,5-дигидрокси-9,10-антрахинона получена
4,8-дигидрокси-9,10-антрахинон-2,6-дисульфокислота [59].
В препаративном отношении представляют интерес превраще-
ния бисульфитного соединения 1-нитрозо-2-нафтола [60, 61], име-
ющего строение производного 3,4-дигидро-1,2-нафтохинона —
4-гйдроксиимино-3-оксо-1-тетралинсульфокислоты (31). При под-
кислении раствора соединения (31), приготовленного обработкой
1-нитрозо-2-нафтола избытком NaHSO3, образуется 4-амино-З-гид-
рокси-1-нафталинсульфокислота (1-амино-2-нафтол-4-сульфокис-
лота) (32), что служит промышленным методом ее получения.
В суспензию 1-нитрозо-2-нафтола, приготовленную нитрозировани-
ем 4 кмоль 2-нафтола, приливают 2400 кг 40%-ного раствора
NaHSO3. К раствору бисульфитного соединения (31) добавляют
500 кг NaCl и 700 кг 78%-ной H2SO4 (разогрев до 50—52°C),
оставляют на 36—40 ч, после чего продукт отфильтровывают и
промывают водой; выход кислоты (32) 80—81% (ссылка [59] к
гл. 7).
При нагревании с гидроксиламином в солянокислом рас-
творе соединение (31) переходит в диоксим (33), который далее
гидролизуется и окисляется гидроксиламином в 4-гидрокси-З-
нитрозо-1-нафталинсульфокислоту (34), восстанавливаемую в
3-амино-4-гидрокси-1 -нафталинсульфокислоту (2-амино-1-нафтол-4-
сульфокислоту) (35). В уксуснокислой среде диоксим (33) устой-
чив и может быть без выделения с высоким выходом окислен
азотной кислотой в 4,5-дигидронафтофуроксан-5-сульфокислоту
(36). Последняя в щелочной среде претерпевает перегруппировку,
открытую Богдановым [62], превращаясь в растворе ЫагСОз в
4-амино-3-нитро-1-нафталинсульфокислоту (37), а в растворе
NaOH — в 1,2-нафтохинондиоксим-4-сульфокислоту (38). Анало-
гичные превращения реализованы на основе замещенных 1-нитро-
зо-2-нафтолов, а также в ряду антрацена и фенантрена [61].
322
Сульфит- и сульфинат-анионы легко присоединяются к хинонам
с последующим внутримолекулярным переносом пары электронов в
структурах типа (40) и отщеплением — присоединением протона,,
что приводит к замещению атома водорода с одновременным
восстановлением хиноидной группировки. Так, при обработке
1,4-нафтохинона (39) сульфитом натрия или солью алкан- или
аренсульфиновой кислоты с высоким выходом образуются соответ-
ственно 1,4-нафтогидрохинон-2-сульфокислота (41), 2-алкилсуль-
фонил- и 2-арилсульфонил-1,4-нафтогидрохиноны. 1,2-Нафтохинон
в тех же условиях образует соответствующие 4-замещенные
1,2-нафтогидрохинона. Подобным же образом реагируют другие
пара- и орто-хиноны [48]. Заместители в хиноидном кольце оказы-
вают значительное влияние на строение образующихся продуктов.
1,4-Нафтохинон-2-сульфокислота (42), присоединяя бисульфит, пе-
реходит в 2,3-дисульфокислоту (43), существующую в отличие от
моносульфокислоты (41) не в^ароматической 1,4-дигидроксиформе,
а в 2,3-дигидро-1,4-диоксоформе [63].
11*
323
Взаимодействие 2-метил-1,4-нафтохинона (44) с бисульфитом
приводит к смеси 3-метил-1,4-нафтогидрохинон-2-сульфокислоты
(45) и 2,3-дигидро-2-метил- 1,4-нафтохинон-2-сульфокислоты
(46)- аналога витамина К, обладающего антигеморрагнческим
действием.
О
О
(46)
Замещение атома водорода при действии солей сернистой кис-
лоты и алкан- или аренсульфиновых кислот возможно и в некото-
рых гетероциклических производных 9,10-антрахинона, содержа-
щих ангулярно конденсированный гетероцикл. Например, 9,10-ан-
трахинон-1,2,5-тиадиазол (47) при кипячении в водном растворе
NaHSO3 в присутствии пиридина с выходом около 90% переходит в
гидрохинон-4-сульфокислоту (48), окисляемую в хинон-4-сульфо-
кислоту (49), а в уксусной кислоте присоединяет бензолсульфино-
вые кислоты с образованием 4-фенилсульфонилзамещенных гидро-
хинонов [64].
Взаимодействие гетероциклических хиноидных систем с алкан-
или аренсульфиновыми кислотами также приводит к замещению
атома водорода с одновременным восстановлением хиноидной
группировки; так реагирует, например, бензо[а]феноксазин-9-он
[65]:
324
Способность хиноидных систем реагировать с S-нуклеофилами
может быть использована для замещения атома водорода в гете-
роароматических соединениях, способных окисляться в хиноидные.
Так, алкил- и арилсульфонильные группы могут быть введены в
2,2-диалкилзамещенные бензимидазолины (50) окислением их в
2/7-бензимидазолы (51), которые взаимодействуют с сульфиновы-
ми кислотами с образованием 5-К5О2-замещенных бензимидазо-
линов (52). Повторение операций приводит к 4,5-дизамещенным
(54). Поскольку бензимидазолины легко получаются конденсацией
о-фенилендиамина с кетонами и легко гидролизуются в кислой
среде, реакции 2/7-бензимидазолов с нуклеофилами могут служить
для получения замещенных о-фенилендиаминов типа (53) и (55).
Стадии окисления и взаимодействия с нуклеофилом могут быть
совмещены, а замещенные бензимидазолины — введены во многие
реакции, характерные для о-фенилендиаминов, без предваритель-
ного гидролиза [66].
Окисление фенотиазина в присутствии аренсульфиновых кислот
позволяет ввести арилсульфонильную группу в положение 3 за
счет промежуточного образования фенотиазиний-катиона, присое-
диняющего сульфиновую кислоту [67] :
NH
32S
ЛИТЕРАТУРА
1. Wardell J. L. — In: The Chemistry of the Thiol Group. Part 1/Ed. S. Patai.
London, Wiley, 1974, p. 163—269. 2. Peach M. E. — Ibid. Part 2, p. 721—784.
3. Пат. США 3374274, 1968; Chem. Abstr., 1968, v. 69, 51832. 4. Яп, пат.
70/19046; Chem. Abstr., 1970, v. 73, 55812. 5. Takikawa У.— Kogyo Kagaku Zasshi,
1967, v. 70, № 8, p. 1384—1388; Chem. Abstr., 1968, v. 68, 59210. 6. Ager E.,
Jddon B., Suschitzky H.— J. Chem. Soc. (C), 1970, № 1, p. 193—197. 7. Field L„
Engelgardi P. P. — J. Org. Chem,, 1970, v. 35, № 11, p. 3647—3655. 8. Jain S. K.,
Chandra D., Mital R. L. — Chem. Ind., 1969, № 29, p. 989—990. 9. Богерт M. T.,
Сталл A. — В кн.: Синтезы органических препаратов. Сб. 1. Пер. с англ. М., ИЛ,
1949, с. 200—201. 10. Kessler Н., Kalinowski H.-О., v. Chamier С. — Lieb. Ann.
Chem., 1969, Bd. 727, v. 228—230.
11. Miller J., Yeung H. W.—J. Chem. Soc. Perkin 2, 1972, № 11, p. 1553—
1557. 12. Мельников H. H. Химия и технология пестицидов. М., Химия, 1974,
с. 418—419. 13. Giles D. Е., Parker A, J.— Austr, J. Chem., 1973, v. 26, № 2,
р. 273—299. 14. Вейганд-Хильгетаг. Методы эксперимента в органической химии.
Пер. с нем./Под ред. Н. Н. Суворова. М., Химия, 1968, с. 599—602. 15. Оксен-
гедлер Г. М., Герасименко Ю. Е. — ЖОХ, 1957, т. 27, вып. 12, с. 3214—3217. 16.
Rundel W. — Chem. Вег., 1968, Bd. 101, № 8, S. 2956—2962. 17. Crampton M. R —
J. Chem. Soc. (B), 1971, № 11, p. 2112—2116. 18. Аллен К., Мак-Кей Д. — В кн.:
Синтезы органических препаратов. Сб. 2. Пер. с англ. М., ИЛ, 1949, с. 455—
458. 19. Тарбелл Д„ Фукушима Д. — Там же, Сб. 4. М., ИЛ, 1953, с. 469—471.
20. In: Organic Syntheses. Coll. v. 5, N. Y., Wiley, 1973, p. 1050.
21. Campaigne E., Osborn S. W. — J. Org. Chem., 1957, v. 22, № 5, p. 561 —
562. 22. Копылова Б. В., Хасанова M. Н., Фрейдлина Р. X. — Изв. АН СССР,
сер. хим., 1970, № 3, с. 633—636. 23. Пат. США 3113952, 1963; РЖ Хим., 1965,
11Н195П. 24. Schonberg A., v. Vargha L., Paul W. — Lieb. Ann. Chem., 1930,
Bd. 483, S. 107. 25. Kwart H., Evans E. R. — J. Org. Chem., 1966, v. 31, № 2,
p. 410—413, 26. Newman M. S., Karnes H. A. — Ibid., 1966, v. 31, № 12, p. 3980—
3984. 27. Titles //. — In: The Chemistry of Organic Sulfur Compounds. V. 2/
Ed. N. Kharasch, C. Y. Meyers. Oxford, Pergamon Press, 1966, p. 311—336.
28. Newman M. S., Hertzel F. W.— In: Organic Syntheses. V. 51. N. Y„ Wiley,
1971, p. 139—142. 29. Госхорн P., Левис У. мл., Джаул Э. и др.—В кн.: Син-
тезы органических препаратов. Сб. 7. Пер. с англ. М., ИЛ, 1956, с. 33—35.
30. Wolfers Н., Kraatz U., Korte /'. — Synthesis, 1975, № 1, р. 43—44.
31. Miyazaki К. — Tetrahedron Letters, 1968, № 23, p. 2793—2798; Kaji A.,
Araki У., Miyazaki K. — Bull. Soc. Chem. Japan, 1971, v. 44, № 5, p. 1393—1399.
32. Daneke J., Jahnke U., Pankow B. e.a. — Tetrahedron Letters, 1970, № 15,
p. 1271—1272. 33. Bunnett J. F. — J. Amer. Chem. Soc., 1957, v. 79, № 22, p. 5969—
5974. 34. Бойко В. H., Щупан Г. М., Ягупольский Л. М.—ЖОрХ, 1975, т. II,
вып. 8, с. 1713—1718. 35. Солодова К. В., Кузнецов А. Е., Шейн С. М. — Там же,
1972, т. 8, вып. 3, с. 574—582. 36. Gold Е. Н., Piotrowski V., Weiner Ben Zion. —
J. Org. Chem., 1977, v. 42, № 3, p. 554—556. 37. Birchall J. M., Haszeldine R. N.,
Morley J. O. — J. Chem. Soc. (C), 1970, № 3, p. 456—462. 38. Пат. США 3656880,
1972; Chem. Abstr., 1972, v. 77, 76671. 39. Bradshaw J. S., Soth J. A. Hales R. H.—
J. Org. Chem., 1972, v. 37, № 15, p. 2381—2383. 40. Adams R., Ferretti A. — J.
Amer. Chem. Soc., 1959, v. 81, № 18, p. 4927—4931.
41. Yagupolski L. M., Kondratenko N. V., Sainbur V. R. — Synthesis, 1975,
№ 11, p. 721—723. 42. Нидерл. заявка 6411507, 1965; Chem. Abstr., 1965, v. 63,
13457. 43. Пат. ПНР 73000, 1974; Chem. Abstr., 1975, v. 83, 44725. 44. Panico R.,
Pouchot O. — Bull. Soc. chim. France, 1965, № 6, p. 1648—1652. 45. Русских В. В.,
Фокин Е. П. — Изв. СО АН СССР, сер. хим. наук, 1968, № 12, вып. 5, с. 97—98.
46. Хейфец Г. М., Хромов-Борисов Н. В., Гаврилова Л. А. — ЖОрХ, 1971, т. 7,
вып. 1, с. 199—207. 47. Kharasch N., Swidler R. — J. Org. Chem., 1954, v. 19, № 10,
p. 1704—1707. 48. Grundmann Ch. — In: Methoden der Organischen Chemie (Hou-
ben-Weyl). Bd. 7/3a. Stuttgart, Georg Thieme Verlag, 1977, S. 599—623. 49. Паш-
326
кевич К. И., Афанасьева Г. Б., Постовский И. Я- — Химия гетероцикл, соед.,
1971, № 6, с. 746—749. 50. Докунихин Н. С., Гаева Л. А., Мезенцева Г. А.—
ДАН СССР, 1972, т. 206, № 3, с. 624—626.
51. Мощицкий С. Д., Залесский Г. А., Кухарь В. П. и др.—ЖОрХ, 1975,
т. 11, вып. 5, с. 1134. 52. Докунихин Н. С.— Журн. ВХО, 1976, т. 21, вып. 3,
с. 292—298. 53. Нидерл. заявка 6409875, 1965; Chem. Abstr., 1965, v. 63, 1756.
54. Ферри К., Бэк Дж., Балтцли Р. — В кн.: Синтезы органических препаратов.
Сб. 3. Пер. с англ. М, ИЛ, 1952, с. 160—162. 55. Франц, пат. 1402324, 1965;
Chem. Abstr., 1965, v. 63, 15021. 56. Пат. ФРГ 1644578, 1971; РЖ Хим, 1972,
19Н174. 57. Meerwein Н., Dlttmar С., Gollinger R. e.a.—Chem. Ber, 1957, Bd. 90,
№ 6, S. 841—852. 58. Rieche A., Seeboth H.— Lieb. Ann. Chem, 1960, Bd. 638,
S. 101—110. 59. Горелик M. В., Шибряева Л. C. — ЖОрХ, 1976, т. 12, вып. 6,
с. 1327—1332. 60. Тодрес-Селектор 3. В. — В кн.: Сульфирование солями серни-
стой кислоты. М, Химия, 1965, с. 52—89.
61. Горелик М. В. — Там же, с. 90—119. 62. Богданов С. В., Короле-
ва И. Н.— ЖОХ, 1953, т. 23, вып. 10, с. 1761 —1764; 1954, т. 24, вып. 11,
с. 1994—1998. 63. Asahi Y. — Chem. Pharm. Bull. (Tokyo), 1963, v. 11, p. 813—
814. 64. Горелик M. В., Ланцман С. Б. — Химия гетероцикл, соед, 1968, Ns 3,
с. 453—458. 65. Афанасьева Г. Б., Постовский И. Я. — ЖОХ, 1964, т. 34, вып. 12,
с. 3893—3898. 66. Горелик М. В., Гладышева Т. X.— ЖОрХ, 1977, т. 13, вып. 9,
с. 1974—1981. 67. Карпищенко Л. С., Просяник А. В., Бурмистров С. И. — Хи-
мия гетероцикл, соед, 1977, № 5, с. 616—618.
Глава 10
РЕАКЦИИ С АНИОНАМИ ГАЛОГЕНОВ
Введение галогенов с помощью реакций нуклеофильного
замещения осуществляется при действии галогеноводородных кис-
лот, их солей со щелочными и переходными (медь и др.) металла-
ми, солей комплексов галогеноводородов с кислотами Льюиса
(NaBF4 и др.), галогенангидридов неорганических кислот (РООз,
SOCh) и т. д. Подвергаться замещению могут атомы другого
галогена, нитрогруппа, гидроксильная и диазониевая группы; за-
мещение последней позволяет перейти от любого диазотируемого
амина к галогенпроизводному. Механизмы реакций нуклеофильно-
го замещения при действии галогенид-анионов весьма разнообраз-
ны. Они могут протекать не только по типу SnAt через элементар-
ную стадию образования о-комплекса, но, при замещении
диазониевой группы, также через стадию образования арил-катио-
на (SnI) или радикальных частиц.
10.1. ЗАМЕЩЕНИЕ АТОМОВ ГАЛОГЕНОВ И НИТРОГРУППЫ
Замещение атомов галогенов и нитрогрупп при действии
галогенид-анионов относится к реакциям типа SnAc, в которых
определяющей скорость стадией обычно является присоединение
нуклеофила (см. 2.7.2). Характеристика активности галогенид-ани-
онов, как нуклеофилов, может быть дана на основании сопостав-
ления констант скоростей реакций замещения в галогендинитро-
бензолах с нитрогруппами в орто- и пара-положениях к галогену.
327
Таблица 10.1
Относительная нуклеофильность анионов Y” в реакции
с 1-иод-2,4-динитробензолом по сравнению с анионом СГ [1]
Анион Y lg k (y-)-lg *(СГ)‘ Анион Y“ lg k (y-)-ig k (cr)’
в метаноле в диметил- формамнде в метаноле в диметил- формамиде
СГ 0 0 127J- 2,5 -3,3
F' >0 >0 СН3О” 7,7 6,5
В г- 1,3 -1,3 c6H6s- п,о 9,4
* lg k (Cl ) = —8,86 в метаноле, — 2,53 в диметилформамиде [при 50°С; выражено в
л/(моль-с)[.
В табл. 10.1 представлены разности логарифмов констант скоро-
стей реакций анионов Y" и аниона С1~ с 1-иод-2,4-динитробензолом в
двух различных растворителях: метаноле и диметилформамиде.
Как следует из таблицы, анионы галогенов по нуклеофильности
значительно уступают таким нуклеофилам, как и метилат- или, тем
более, тиофенолят-анионы. Нуклеофильность галогенид-анионов по
отношению друг к другу зависит от природы растворителя.
В протонной среде (метанол) она уменьшается в ряду I- > Вг~>
> Cl- > F~, в полярной апротонной среде (диметилформамид) —
в противоположном порядке. Эта последовательность сохраняется
и когда уходящей группой является не I, a F— в аналогично по-
строенном динитрофторбензоле [1].
Инверсия ряда относительной нуклеофильности галогенид-ани-
онов при перемене растворителя обусловлена большим снижением
реакционной способности анионов, являющихся жесткими основа-
ниями (F , С1 ), в протонной среде из-за сольватации с образова-
нием водородных связей (см. 2.5 и 2.6). Однако даже реакция с
наименее сольватируемым иодид-анионом быстрее протекает в ди-
метилформамиде, чем в метаноле (lg k равен соответственно —5,8 и
—6,4; по данным таблицы). Поэтому замещение атомов галогенов
и нитрогрупп при взаимодействии с галогенидами щелочных ме-
таллов предпочтительно проводить в апротонной среде. Так,
1-иод-2,4-динитробензол получают 15-минутным кипячением
1,3-динитро-4-хлорбензола с KI в диметилформамиде с выходом
65—71% [2]. Хлорпентацианобензол превращается в иодпентациа-
нобензол при размешивании с KI в ацетоне при 20 °C в течение
1 ч с выходом 90% [3].
Обмен галогенов (Вг, I) проводят также каталитически, дей-
ствуя галогенидом меди, который служит одновременно и реаген-
том, и катализатором. Атомы брома и иода в галогеннитробензолах
обмениваются на атомы хлора при реакции с CuCl в уксусной
кислоте [4]. При действии CuCl в кипящем диметилформамиде
328
моно-, ди- и трихлортиофены с хорошим выходом синтезированы
из более доступных бромтиофенов [5].
В среде концентрированных галогеноводородных кислот заме-
щение может облегчаться вследствие протонирования ароматиче-
ского субстрата. При нагревании с концентрированной соляной
кислотой в уксусной кислоте 2,3-, 2,5-, 3,4-динитроанилины количе-
ственно обменивают нитрогруппу, находящуюся в орто- или пара-
положении к протонируемой аминогруппе, на атом хлора и диазо-
тируются далее выделяющейся азотистой кислотой, превращаясь в
хлориды соответствующих нитрохлорбензолдиазониев. Так же ве-
дут себя 1-нитро-2-нафтиламин, 2-нитро-, 2,4-, 3,4-динитро-1-на-
фтиламины; 2,4- и 2,6-динитроанилины образуют смесь хлоридов
дихлор- и динитробензолдиазониев [6]. Нитрогруппа замещается
атомом брома при кипячении 3-нитро-1,2,4-триазол-5-онов (1) в
20%-ной НВг, а при кипячении в концентрированной НС1 нитро- и
бромпроизводные (1), (2) переходят в хлорпроизводные (3) с
выходом 90—95% [7]:
+ НС1
+ НС1
Хотя замещением атома галогена и нитрогруппы могут быть
введены атомы всех галогенов, главное препаративное значение
имеет введение этим путем атомов фтора. Реакцию проводят
обычно нагреванием хлор- или нитропроизводного с KF без рас-
творителя или в полярном апротонном растворителе. Активность
фторида в сильной степени зависит от катиона щелочного металла,
уменьшаясь в ряду CsF > RbF > KF > NaF > LiF [8, 9].
Без растворителя процесс проводят при нагревании хлорпроиз-
водного с безводным KF при 200—500 °C. Выбор температуры
зависит от подвижности атомов хлора и от желаемой степени
замещения в полихлорпроизводных. При наличии сильных активи-
рующих заместителей атом хлора обменивается уже при 190—
200 °C. В этих условиях при действии небольшого избытка KF в
течение 7—8 ч из 1,3-динитро-4-хлорбензола с выходом 92% по-
лучен 1,3-динитро-4-фторбензол, из 1,3-динитро-4,6-дихлорбензо-
ла — 1,3-динитро-4,6-дифторбензол (87%) [9], из 3,5-динитро-2-
хлорбензотрифторида —- 3,5-динитро-2-фторбензотрифторид (85%)
329
[10]. При несколько более высокой температуре (220—230 °C) и
большей длительности процесса замещаются атомы хлора в
1-хлор- (88%), 2-хлор-9,10-антрахиноне (53%) [10], все четыре
атома хлора в 1,2,3,4-тетрахлор-9,10-антрахиноне (92%) [11].
В гораздо более жестких условиях удается заместить атомы хлора
в полихлорпроизводных, не содержащих сильных электроноакцеп-
торных заместителей [12]. Так, нагреванием гексахлорбензола с
KF в автоклаве при 510 °C в течение 20 ч получают смесь гекса-
фторбензола (выход 35—37%) и пентафторхлорбензола (36—40%)
[13]. Для замещения всех атомов хлора на фтор в три-
хлор-1,3,5-триазине в качестве катализатора вводят триоксид сурь-
мы Sb2O3 и проводят процесс в автоклаве при 320—350 °C, полу-
чая трифтор-1,3,5-триазин с выходом 94% [14].
Варьируя температуру, из хлорпроизводных можно получать
продукты частичного замещения, причем вступающие атомы фтора
ориентируют последующее замещение преимущественно в мета-
положение. При реакции пентахлорбензонитрила (5) с KF при
300 °C образуется с выходом 64% 2,4,6-трифтор-3,5-дихлорбензо-
нитрил (6), а при 350 °C — с выходом 70% пентафторбензонитрил
При взаимодействии тетрахлорфталевого ангидрида (7) с KF
при 300 °C замещение атомов хлора сопровождается своеобразной
циклизацией через стадию дилактона (8); в результате с выходом
40—45% выделен октафтор-9,10-антрахинон (9) [16]:
330
В качестве ристворителей для введения атомов фтора дей-
ствием KF применяются диметилформамид, диметилсульфоксид,
А-метилпирролидон, сульфолан и др. Проведение реакции в рас-
творителе позволяет снизить температуру, сократить длительность,
а иногда и осуществить замещение в соединениях, не реагирующих
при сплавлении с KF. Так, о- и n-нитрохлорбензолы не изменяются
при нагревании с KF без растворителя при 190—200 °C [9], тогда
как в диметилсульфоксиде n-нитрохлорбензол переходит в п-нит-
рофторбензол при 185—195 °C с выходом 72% [17], а
1-бром-2-нитро-3-хлорбензол при кипячении в диметилформамиде
(150°С)—в 1-бром-2-нитро-3-фторбензол с выходом 76% [18].
Для замещения атомов хлора в производных антрахинона предло-
жен высококипящий растворитель— дифенилсульфон, применение
которого позволяет сократить длительность нагревания по сравне-
нию с процессом без растворителя с 25 до 4 ч при получении
1-фтор-9,10-антрахинона и с 48 до 74 при получении
2-фтор-9,10-антрахинона [19].
Нитрогруппа, обладающая при одинаковом активирующем
влиянии со стороны других заместителей значительно большей
подвижностью, чем атом хлора (см. 7.1.2), может быть замещена в
среде органического растворителя в слабо активированных соеди-
нениях. Так, л«-динитробензол превращается в л«-нитрофторбензол
при нагревании с KF в А-метилпирролидоне или гексаметилфос-
фортриамиде (гексаметапол) при 180 °C за 48 ч с выходом 40—
45%, а 2-нитропиридин — в 2-фторпиридин при 160 °C за 24 ч с
выходом 55—60% [20]. 3- и 4-Фторфталевые ангидриды можно
получить из соответствующих нитрофталевых ангидридов с выхо-
дом 56—66% при кратковременном (30—75 мин) нагревании с KF
без растворителя при 180—195 °C или в диметилсульфоксиде при
80 °C [21].
10.2. ЗАМЕЩЕНИЕ ГИДРОКСИЛЬНОЙ ГРУППЫ
Превращение ароматических и гетероциклических гид-
роксисоединений в галогенпроизводные осуществляется при
действии галогенангидридов неорганических кислот: фосфор-
ной (РООз, РС15, РОВгз, РВг5), сернистой (SOCU), угольной
(СОС12, COC1F) и триарилгалогенфосфонийгалогенидов [Аг3РСГ] СГ,
[Аг3РВг] Вг-. Реакции заключаются в ацилировании гидроксиль-
ной группы с последующим замещением анионом галогена по ме-
ханизму SnAt или при термическом разложении эфира. Реакции
первого типа реализуются в соединениях, где гидроксильная груп-
па находится под сильным электроакцепторным влиянием: в ди- и
тринитрофенолах и в гидроксигетероциклах. Термическое разло-
жение эфиров позволяет замещать гидроксильную группу в неак-
тивированных соединениях.
Замещение гидроксильной группы в нитрофенолах проводят
обычно нагреванием с галогеноангидридом неорганической кислоты
331
в органическом растворителе или без него в присутствии ката-
лизатора— третичного амина или диметилформамида. Так,
2,4-динитрофенол при нагревании с избытком оксохлорида фосфо-
ра (V) РОС13 в присутствии пиридина, М,М-диметиланилина,
диметилформамида превращается с. выходом 72—87% в
1,3-динитро-4-хлорбензол, а 4,6-динитрорезорцин — с выходом
91—97% в 1,3-динитро-4,6-дихлорбензол [22]. При кипячении в
РОС1з или в РОВгз в присутствии пиридина гидроксибензолпента-
карбонитрил переходит соответственно в хлор- или бромбензол-
пентакарбонитрил с выходом 90—95% [3]. 4-Алкил-2,6-динитрофе-
нол ы (Юа) переводят в хлорзамещенные (Па) кипячением с тио-
нилхлоридом в смеси гексана и диметилформамида (93%) [23];
метиловый эфир 4-гидрокси-3,5-динитробензойной кислоты (106)
может быть превращен в хлорзамещенное (116) кипячением с
тионилхлоридом в CCU в присутствии каталитических количеств
диметилформамида (95—96%) [24]; метилтиодинитро- и метилди-
нитрофенолы (10в) переходят в хлорзамещенные (Пв) в тех же
условиях при действии фосгена (до 97%) [25]. В тринитрофеноле
замещение может протекать и в отсутствие катализатора.
a) R = NO2, R' = CH(CH3)2, С(СНз)э
б) R = NO2, R^COOCHs
R = COOCH3, R' = NO2
в) R==NO2, R' = CH3, SCH3
(Ю)
Действием галогенангидридов фосфорной кислоты переводят в
галогенпроизводные гетероциклические оксосоединения, потенци-
ально способные существовать в гидроксиформе; при этом введе-
ние катализаторов не обязательно. Так, нагреванием с РОС13 из
4-метил-2-хинолона получают 2-хлорлепидин (89—92%) [26], из
9-акридона — 9-хлоракридин (около 100%) [27], из 3-индазолона
в присутствии пиридина — 3-хлориндазол (68—74%) [28]. Неред-
ко применяют смесь оксогалогенида и галогенида фосфора (V)
[29].
Метод замещения гидроксигруппы на атом хлора или брома в
неактивированных фенолах при действии галогенидов фосфора (V)
состоит в синтезе тетра (арилокси) фосфонийгалогенидов, которые
при термолизе распадаются на арилгалогенид и триарилфосфат:
4АгОН + РУ5 ----> [(АгО)4Р+] Y —> ArY + (АгО)3РО
Y = С1, Вг
Первую стадию проводят при нагревании фенола и галогенида
фосфора (V) при 140—160, вторую — при 250—300 °C. Этим мето-
дом из фенола, .м-крезола, 1- и 2-нафтола, о-гидроксибифенила
получены хлорпроизводные с выходом 92—95%, бромпроизвод-
332
ные — с выходом 70—93% [30]. Недостатком метода является
большой расход реагентов, поскольку 3Д фенола и 4/s связанногос
фосфором галогена не используется по стехиометрии реакции.
Более экономично применение триарилгалогенфосфонийгалоге-
нидов — аддуктов триарилфосфина, например трифенилфосфина с
галогенами. При взаимодействии с фенолами они образуют трифе-
нил (арилокси) фосфонийгалогениды (12), термически расщепляю-
щиеся на арилгалогенид и трифенилфосфиноксид:
(СвН5)3Р + У2 —> [(С6Н5)3РY] Y-
+ -ну +
АгОН + [(СвН5)зРУ] Y' --> [АгОР(СвН5)3] Y" —> ArY + (С6Н5)3РО
(12)
Y = С1, Вг
Присоединение галогена к трифенилфосфину проводят в орга-
ническом растворителе при охлаждении, после чего вносят фенол,
отгоняют растворитель, а эфир (12) подвергают термическому
разложению. Так, действием (СеНа^РСП на фенол, о- ии-крезолы,
1- и 2-нафтолы в СС14 и термолизом при 220 °C с выходом 60—85%
получают соответствующие хлорпроизводные [31], а действием
(СбН5)3РВг2 в ацетонитриле — бромпроизводные с выходом 60—
90% [32,33]. Эфиры нитрофенолов разлагаются легче, превраща-
ясь в нитрохлорбензолы при 140—160 °C. При получении 2-бром-
нафталина из 2-нафтола разложение эфира рекомендуется вести
при температуре в бане до 340 °C (70—78%) [34]. Синтезированы
также бромпроизводные, исходя из 2-пиридона, 3-гидроксипири-
дина, 2-хинолона.
Пиролиз фторкарбонатов фенолов, получаемых ацилированием
фенолов карбонилфторидхлоридом, дает возможность заменять
гидроксигруппу на атом фтора:
—на
ArOH-4-COFCl ----> ArOCOF —> ArF + СО2
Карбонилфторидхлорид готовят взаимодействием фосгена
(карбонилдихлорида) с AsFs, SbF5, SiF4. Ацилирование ведут под
давлением при 35—50 °C с практически количественным выходом
фторкарбонатов. Пропускание паров фторкарбоната фенола в сме-
си с азотом над платиновой сеткой, нагретой до 700—750°C, дает
фторбензол с выходом 90—93% при высокой степени превращения
[35]. Также получен фторбензол из тиофенола (78%). Выходы
фторзамещенных из фтор- и бромфенолов и 1-нафтола заметно
ниже (25—55%). С небольшим выходом удалось осуществить пре-
вращение ди(фторкарбонатов) резорцина и гидрохинона в один
прием в соответствующие дифторбензолы.
Отсутствие изомеров исключает радикальный механизм пере-
группировки, а ее протекание в газовой фазе при столь высоких
температурах делает невероятным межмолекулярное гетеролити-
ческое замещение. Очевидно, реакция представляет собой внутри-
молекулярное нуклеофильное замещение с четырехцентровым
333
переходным состоянием [35], подобное перегруппировкам Шен-
берга и Ньюмена — Куарта (см. 9.1.3);
10.3. ЗАМЕЩЕНИЕ ДИАЗОНИЕВОЙ ГРУППЫ
Обмен диазониевой группы на атом галогена [36—38]
может осуществляться при действии галоген-анионов без катали-
затора, в присутствии галогенидов меди(1) (реакция Зандмайера)
и термическим разложением солей диазония (реакция Шимана и
примыкающие к ней). Названные методы различаются как усло-
виями проведения, так и механизмами реакций.
Замещение в отсутствие катализатора может_ протекать по ме-
ханизму SnI при действии анионов Y~ = F , Cl , Вг" (см. ссылку
[106] к гл. 2):
-n2 . +Y-
ArN2 ----> Аг+ ---► ArY
или по механизму SrN1 при действии иодид-аниона [39]:
—N2 4-1
ArNj + Г ----► Аг- + V2I2 -> Ari
В обоих случаях не исключена возможность механизма SnAf.
Препаративное значение имеет синтез иодзамещенных, который
осуществляют простым смешением раствора диазотированного
амина с водным раствором КТ. Например, диазотированием ани-
лина в НС1, добавлением раствора КТ и нагреванием получают
иодбензол (выход 74—76%) [40], а диазотированием о-бромани-
лина в H2SO4 и приливанием раствора КТ — о-бромиодбензол
(выход 72—83%) [41]. Диазотирование аминов в безводной HF
добавлением сухого NaNO2 позволяет получать некоторые фторза-
мещенные бензола с высоким выходом, но из-за агрессивности HF
метод сложен в оформлении и не всегда дает хорошие результаты
([38]. В отдельных случаях амины можно перевести в хлорпроиз-
водные диазотированием в концентрированной НС1 без катализа-
тора. Так, из 2-аминопиримидина диазотированием при низкой
температуре получают с выходом 26—27% 2-хлорпиримидин [42].
Введение атомов хлора и брома по реакции Зандмайера осуще-
ствляется в присутствии галогенидов меди(1). Скорость реакции
прямо пропорциональна концентрации диазоний-катионов и Си+ и
обратно пропорциональна квадрату концентрации галогенид-анио-
на [43]. Считают, что лимитирующей стадией является образова-
ние лабильного комплекса между диазоний-катионом и галоге-
нидом меди(1), который затем быстро распадается с переносом
334
электрона от иона Си+ к диазоний-катиону, элиминированием азо-
та и отрывом арильным радикалом атома галогена от иона Си2+
[44, 45], например:
. 4. ~N2
ArNj + CuCl —> [ArN=N «—CuICl]+ -----►
—> [Ar- + (CuiiCl)+] —> ArCl + Cu+
Превращение комплекса диазосоединения с галогенидом меди
происходит, очевидно, в клетке, вследствие чего количество биари-
лов, образующихся путем рекомбинации арильных радикалов при
их выходе в объем, невелико. Другими побочными продуктами
являются фенолы. Скорость образования побочных продуктов пря-
мо пропорциональна концентрации ионов Си+ и обратно пропорци-
ональна четвертой степени концентрации ионов С1 . Поэтому с
увеличением концентрации ионов С1~ скорость основной реакции
уменьшается не так быстро, как побочных; это используется для
подавления последних. Снижение скорости основной реакции объ-
ясняют связыванием галогенида меди(1) в комплексный анион;
CuCl 4- 2СГ —> CuCl;
Введение электроноакцепторных заместителей увеличивает, а
электронодонорных — уменьшает скорость основной реакции; это
рассматривается как аргумент в пользу механизма с переносом
электрона к диазоний-катиону в медьсодержащем комплексе, по-
скольку при его распаде с образованием арил-катиона влияние
заместителей было бы противоположным. На основании сравнения
выходов хлорпроизводного и фенола при варьировании заместите-
лей в реакции Зандмайера (катализатор FeCh) высказано пред-
положение, что реакция идет с переносом электрона при электро-
ноакцепторных заместителях и с образованием арил-катиона при
электронодонорных [46]. В целом, в механизме реакции Зандмайе-
ра остается много неясного.
Эту реакцию обычно проводят, добавляя раствор соли диазония
в раствор галогенида меди(1) в соответствующей галогеноводо-
родной кислоте. Хлорид или бромид меди(1) готовят обработкой
раствора сульфата меди(II) и хлорида или бромида натрия суль-
фитом, бисульфитом или пиросульфитом натрия. Так, о- и п-хлор-
толуолы получают, приливая солянокислый раствор диазотирован-
ного о- или n-толуидина в охлажденный до О °C раствор CuCl в
НС1 с последующим постепенным повышением температуры и за-
ключительной отгонкой хлортолуола с водяным паром (выход
70—79%) [47]. При получении лг-нитрохлорбензола из м-нитроани-
лина растворы смешивают при 25—30 °C (68—71%) [48], а при по-
лучении о- и м-бромхлорбензолов из соответствующих хлоранили-
нов, м-дибромбензола из лг-броманилина, о-броманизола из м-ани-
зидина раствор соли диазония, приготовленный диазотированием
амина в НВг, постепенно прибавляют к кипящему раствору CuBr
в НВг (выход 80—95%) [49]. Диазосоединения из слабоосновных
335
ароматических и гетероциклических аминов готовят диазотирова-
нием в концентрированной серной или фосфорной кислоте (см. 15.1)
и затем вносят в раствор галогенида меди(1) [36, 37]. В приведен-
ных примерах на 1 моль диазотируемого амина вводят не менее
1 моль CuCl [47,48] и 0,55 моль СиВг [49], но в других случаях
количество галогенида меди может составлять 0,1—0,2 моль [45].
Вместо галогенида можно применять порошок меди, который при-
бавляют после диазотирования в галогеноводородной кислоте и
нагревают, как например при получении о-бромтолуола (выход
43—47%) [50]. В производстве 8-хлор-1-нафталинсульфокислоты,
промежуточного продукта для тиоиндигоидных красителей, соля-
нокислый раствор диазотированной 8-амино-1-нафталинсульфо-
кислоты (перикислота) добавляют к раствору CuCl (0,37 моль на
1 моль амина) и NaCl в НС1 при 5—35 °C (выход 81%):
Хорошие результаты дает иногда совмещение стадий диазоти-
рования и замещения диазониевой группы при внесении NaNO? в
смесь амина и раствора галогенида меди в галогеноводородной
кислоте. Таким образом из 3-аминопиридинов удалось получить
3-хлор- и 3-бромпиридины с выходом 74—87% против 16—30% при
разделении стадий [51]. С хорошим выходом (60—97%) получены
хлор- и бромпроизводные бифенила при реакции выделенных бор-
фторидов диазония с галогенидами меди в диметилсульфоксиде
[52].
Термическое разложение борфторидов диазония по Шиману
[38, 53, 54] служит одним из основных методов введения фтора в
ароматическое ядро. Борфториды диазония, отличающиеся малой
растворимостью в воде и высокой стабильностью, получают добав-
лением борфтористоводородной кислоты или ее солей в процессе
или после диазотирования амина в минимальном количестве воды.
Осадок борфторида диазония отделяют, промывают, тщательно
высушивают и нагревают до разложения, при котором соль диазо-
ния превращается в арилфторид с элиминированием азота и три-
фторида бора:
-n2
ArNH2 —>• ArN) BF4" ---> ArF + BF3
Выход и температура разложения зависят от строения исход-
ного амина. В общем случае, чем ниже температура разложения,
тем более гладко протекает реакция. Присутствие влаги в борфто-
риде диазония, как правило, оказывает отрицательное влияние,
приводя к неуправляемому течению процесса с образованием
большого количества смол. Наличие функциональных групп повы-
шает температуру разложения и снижает выход. Наиболее высо-
336
кой температуры требует разложение нитрозамещенных, протека-
ющее бурно и с низким выходом. Для более равномерного хода
реакции в таких случаях борфторид диазония смешивают с песком
или другим инертным наполнителем. Разложение борфторидов
диазония проводят также в среде органических растворителей,
чаще всего в углеводородах. Выходы фторпроизводных в расчете
на борфторид диазония достигают 80—90%. Так, нагреванием бор-
фторида п-этоксикарбонилбензолдиазония и последующим гидро-
лизом получают п-фторбензойную кислоту с выходом 84—89%
[55], из борфторида бисдиазотированного бензидина — 4,4'-ди-
фторбифепил с выходом 80—81,5% [56], разложением борфторида
3-хинолиндиазония в толуоле— 3-фторхинолин с выходом 91,5%
[38]. Однако поскольку выходы борфторидов из-за потерь при
выделении сравнительно невысоки, общий выход в приведенных
примерах составляет 54—73%. В некоторых случаях удается повы-
сить выход, применяя соли диазония с другими фторсодержащими
комплексными анионами: гексафторфосфатом PFe, гексафторанти-
монатом SbFs, гексафторарсенатом AsFs. Так, гексафторфосфат
о-бромбензолдиазония выделяют с выходом 94—97% и превраща-
ют при нагревании в минеральном масле при 167—170 °C в
о-бромфторбензол с выходом 78%, тогда как при переходе через
борфторид диазония общий выход составлял 37% [57]. Гексафтор-
антимонаты бензолдиазониев, содержащих электроноакцепторные
группы (NO2, СООН), превращаются во фторпроизводные с бо-
лее высоким выходом, чем борфториды. При термолизе гекса-
фторантимоната и борфторида о-нитробензолдиазония, например,
выход о-нитрофторбензола равен соответственно 40% и 9—17%
]58].
Для реакции Шимана наиболее вероятен гетероциклический тип
распада солей диазония. При разложении борфторида бензолдиа-
зония в нитробензоле или алкилбензоате выделены только лг-нит-
робифенил или алкил-л!-бифенилкарбоксилат соответственно, что
свидетельствует об электрофильной атаке промежуточно образую-
щегося арил-катиона [59]. Изучение термолиза серии борфторидов
бензолдиазониев в резонаторе ЭПР-спектрометра показало, что
наибольшее число радикальных частиц возникает при разложении
п-нитрозамещенного, дающего наименьший выход арилфторида;
из этого сделано заключение о превращении радикалов в побочные
продукты [60]. Существует мнение [61], что разложение борфто-
рида бензолдиазония в 2,2,2-трифторэтаноле, приводящее к смеси
фторбензола и 2,2,2-трифторэтоксибензола (в соотношении около
1 : 2) с общим выходом 96,8%, протекает в обоих направлениях по
гетеролитическому механизму.
Помимо термического разложения сухих борфторидов диазо-
ния по Шиману описано их разложение в водной или ацетоновой
среде в присутствии порошка меди или CuCl. В частности, обра-
боткой раствора диазосоединения борфтористоводородной кисло-
той и порошком меди при комнатной температуре из W-аце-
тил-п-фенилендиамина получен А-ацетил-п-фторанилин с выходом
337
82%, а из 2-амино-9,10-антрахинона — 2-фтор-9,10-антрахинон с
выходом 86% вместо 41% при термическом разложении [62].
Подобным образом выполняется реакция Шимана с целью
замены аминогруппы на атом галогена при термическом разложе-
нии комплексных солей диазония с галогенидами ртути(II), (реак-
ция Швехтена). Этим способом вводят атомы хлора и брома в тех
случаях, когда реакция Зандмайера дает низкие выходы, главным
образом в ряду нафталина, фенантрена, бифенила:
+ HgY2 -Ы2
ArNj Y -------> ArN2 HgYj ----> ArY 4- HgY2
Y = Cl, Br
После диазотирования амина к раствору диазосоединения до-
бавляют растворы HgCl? и КС1 или HgBr2 и КВг, затем осадок
тригалогенмеркуроата диазония отделяют, высушивают и разла-
гают при нагревании в смеси с КС1 или КВг; выходы арилгалоге-
нидов составляют 60—80% [36,37].
Препаративное значение для получения бромпроизводных гете-
роциклов имеет разложение пербромидов диазония, образующихся
при действии брома на бромид диазония и разлагаемых в присут-
ствии воды, нагреванием в этаноле или в уксусной кислоте:
4-Вг2 —N2
RNJ Вг’ ----> RN2 BrJ -----> RBr + Br2
Так, диазотированием 2-аминопиридина в НВг в присутствии
избытка брома и разложением пербромида добавлением раствора
NaOH получают 2-бромпиридин с выходом 86—92% [63].
ЛИТЕРАТУРА
1. Broxton Т. J., Muir D. М., Parker A. J.— J. Org. Chem., 1975, v. 40, № 22,
р. 3230—3233. 2. Банкет Дж. Ф., Коннер Р. М. — В кн.: Синтезы органических
препаратов. Сб. 12. Перев. с англ. М., Мир, 1964, с. 47—49. 3. Friedrich К.,
Oeckl S. — Chem. Вег., 1973, Bd. 106, № 7, S. 2361—2365. 4. Liedholm В.—Acta
chem. Scand. 1971, v. 25, № 1, p. 106—112, 113—117. 5. Conde S., Corral C., Mad-
ronero R. e.a. — Synthesis, 1976, № 6, p. 412—413. 6. Sihlbom L. — Acta chem.
Scand., 1953, v. 7, № 8, p. 1197—1206; 1954, v. 8, № 9, p. 1709—1719. 7. Kro-
ger C.-F., Mietchem R., Frank H. e. a. — Chem. Ber., 1969, Bd. 102, № 3, S. 755—
766. 8. Ворожцов H. H., мл., Якобсон Г. Г. — ЖОХ, 1957, т. 27, вып. 6, с. 1672—
1676. 9. Ворожцов Н. Н., мл., Якобсон Г. Г. — Хим. наука и пром., 1958, т. 3,
№ 3, с. 403. 10. Пат. ФРГ 1018042, 1958; РЖ Хим., 1959, 75717.
11. Якобсон Г. Г., Штейнгарц В. Д., Костина Н. Г. и др. — ЖОХ, 1966,
т. 36, вып. 1, с. 142—145. 12. Yakobson G. G., Vlasov V. М. — Synthesis, 1976,
№ 10, р. 652—672. 13. Синтезы фторорганических соединений/Под ред. И. Л. Кну-
нянца, Г. Г. Якобсона. М., Химия, 1973, с. 130—132. 14. Forche Е.— In: Metho- i
den der Organischen Chemie (Houben-Weyl). Bd. 5/3. Stuttgart, Georg Thieme •[
Verlag, 1962, S. 160—166. 15. Birchall J. M., Haszeldine R. N., Jones M. E. —
J. Chem. Soc. (C), 1971, № 7, p. 1341—1342. 16. Vorozhtsov N. N., Yakobson G. G.,
Odinokov V. N. — Tetrahedron Letters, 1965, № 49, p. 4473—4475. 17. Finger G. C.,
Kruse C. W. — J. Amer. Chem. Soc., 1956, v. 78, № 23, p. 6034—6037. 18. Lied-
holm B. — Acta chem. Scand. 1975, v. B29, № 9, p. 981—982. 19. Докунихин H.C.,
338
Колоколов Б. Н., Салов Б. В. Авт. свид. СССР 164615, 1964; БИ, 1964, № 16.
20. Bartoli G., Latrofa A., Naso F. e.a. — J. Chem. Soc. Perkin 1, 1972, № 21,
p. 2671.
21. Ishikawa N., Tanabe T., Hayashi D. — Bull. Chem. Soc. Japan, 1975, v. 48,
№ 1, p. 359—360. 22. Збарский В. Л., Шутов Г. М., Жилин В. Ф. и др.— ЖОрХ,
1971, т. 7, вып. 2, с. 310—314. 23. Tao Е. V. Р., Christie С. F. — Org. Prep. Proc.
Intern., 1972, v. 4, № 2, p. 73—74; Пат. США 3665042, 1972; Chem. Abstr., 1972,
v. 77, 61528. 24. Matsumoto I. — J. Pharm. Soc. Japan, 1965, v. 85, № 6, p. 544—
546; Chem. Abstr., 1965, v. 63, 6898. 25. Пат. США 3657355, 1972; Chem. Abstr.,
1972, v. 77, 48039. 26. Кэзлоу К., Лауер Б. — В кн.; Синтезы органических пре-
паратов. Сб. 3. Пер. с англ. М., ИЛ, 1952, с. 480—481. 27. Альберт А., Рит-
чи 5.— Там же, с. 36—39. 28. Стефенсон Э. — Там же. Сб. 4. 1953, с. 262—263.
29. Вейганд-Хильгетаг. Методы эксперимента в органической химии. Пер. с нем./
Под ред. Н. Н. Суворова. М., Химия, 1968, с. 229—231. 30. Нефедов О. М.,
Левкое Я. Л., Петров А. Д. — ДАН СССР, 1960, т. 133, № 4, с. 855—858.
31. Hoffmann Н., Horner L., Wippel Н. G. e.a. — Chem. Вег., 1962, Bd. 95,
№ 2, С. 523—527. 32. Wiley G. A., Hershkowitz R. L., Rein В. M. e. a. — J. Amer.
Chem. Soc., 1964, v. 86, № 5, p. 964—965. 33. Schaefer J. P., Higgins J.—J. Org.
Chem., 1967, v. 32, № 5, p. 1607—1608. 34. Schaefer J. P., Higgins J., She-
noy P. K. — In: Organic Syntheses. Coll. v. 5. N. Y., Wiley, 1973, p. 142—145.
35. Christe К. O., Pavlath A. E.— J. Org. Chem., 1965, v. 30, № 9, p. 3170—3173;
№ 12, p. 4104—4107; 1966, v. 31, № 2, p. 559—561. 36. Stroh R. — In: Methoden
der organischen Chemie (Houben-Weyl). Bd. 5/3. Stuttgart, Georg Thieme Verlag,
1962, S. 843—852. 37. Roederig A.—Ibid., Bd. 5/4, 1961, S. 438—451. 38. For-
che E.— Ibid., Bd. 5/3, 1962, S. 213—231. 39. Kumar R., Singh P. R. — Tetrahedron
Letters, 1972, № 7, p. 613—616. 40. Люкас X., Кеннеди E. — В кн.; Синтезы
органических препаратов. Сб. 2. Пер. с англ. М., ИЛ, 1949, с. 278—280.
41. Хиней Ч., Миллар И. Т. — Там же. Сб. 12.- 1964, с. 152—154. 42. Ка-
ган И.,'Минин Р., Овербеджер Ч. — Там же. Сб. 7. 1956, с. 67—68. 43. Pfeil Е.—
Angew. Chem., 1953, Bd. 65, № 6, S. 155—158; Cowdrey W. A., Davies D. S.—
Quart. Revs., 1952, v. 6, № 3, p. 358—379. 44. Laing I. G. — In: Rodd’s Chemistry
of Carbon Compounds. 2-nd ed. V. 3, part С/Ed. O. Coffey. Amsterdam, Elsevier,
1973, p. 63—74. 45. Hertel H. — In: Ullmanns Enzyklopadie der technischen Che-
mie. 4-te Aufl. Bd. 10. Weinheim, Verlag Chemie, 1975, S. 109—132. 46. Naka-
tani F. — Tetrahedron Letters, 1970, № 51, p. 4455—4458. 47. Марвелл Ч., Мак
Эльвен С. — В кн.: Синтезы органических препаратов. Сб. 1. Пер. с англ. М.,
ИЛ, 1949, с. 491—493. 48. Хартман В., Брезен М.— Там же, с. 485—487.
49. Хартвелл Дж.—Там же. Сб. 3. 1952, с. 467—469. 50. Бигелов Л.—Там же.
Сб. 1. 1949, с. 136—137.
51. Talik Т., Talik Z., Ban-Oganowska Н. — Synthesis, 1974, № 4, р. 293—294.
52. Kobayashi М., Yamada E., Matsui M. e.a. — Org. Prep. Proc. Intern., I960,
v. 1, № 4, p. 221—224. 53. Роэ A. — В кн.: Органические реакции. Сб. 5. Пер.
с англ. М., ИЛ, 1951, с. 155—194. 54. Сушицкий Н. — В кн.: Успехи химии фто-
ра. Т. 3—4. Пер. с англ./Под ред. А. П. Сергеева. М., Химия, 1970, с. 335—373.
55. Шиман Дж., Винкельмюллер В. — В кн.: Синтезы органических препаратов.
Сб. 2. Пер. с англ. М., ИЛ, 1949, с. 534—537. 56. Там же, с. 244—247. 57. Ru-
therford К. G., Redmond W. — In: Organic Syntheses. Coll. v. 5. N. Y., Wiley,
1973, p. 133—136. 58. Sellers C., Suschitzky H. — J. Chem. Soc. (C), 1968, № 18,
p. 2317—2319. 59. Nesmeyanov A. N., Makarova L. G., Tolstaya T. P. — Tetra-
hedron, 1957, v. 1, № 1/2, p. 145—157. 60. Альсинг T. K-, Болдырев A. Г.—
ЖОрХ, 1970, т. 6, вып. 3, c. 627.
61. Burri P., Loewenschuss H., Zollinger H. — Helv. chim. acta, 1974, v. 57,
№ 2, p. 395—402. 62. Bergmann E. D., Bentov M. — J. Org. Chem., 1954, v. 19,
№ 10, p. 1594—1599; Bergmann E. D., Berkovic S., Ikan R. — J. Amer. Chem. Soc.,
1956, v. 78, N 23, p. 6037—6039. 63. Аллен Ч., Тиртл Дж. — В кн.: Синтезы орга-
нических препаратов. Сб. 4. Пер. с англ. М., ИЛ, 1953, с. 94—96.
Глава 11
РЕАКЦИИ С УГЛЕРОДСОДЕРЖАЩИМИ
НУКЛЕОФИЛЬНЫМИ РЕАГЕНТАМИ
11.1. ВВЕДЕНИЕ ЦИАНОГРУППЫ
11.1.1. Замещение диазониевой группы
Замена диазониевой группы на цианогруппу представ-
ляет собой частный случай реакции Зандмайера, рассмотренной в
предыдущей главе применительно к введению атомов галогенов
(см. 10.3). Раствор диазосоединения обрабатывают раствором,
содержащим комплексную соль цианида меди(1) и цианида натрия
или калия, например:
—n2
ArN2Cl + Na[Cu(CN)2) -> ArCN + CuCN + NaCl
Раствор комплексной соли обычно готовят, смешивая раствор
цианида щелочного металла и суспензию хлорида меди(1) при их
мольном соотношении (2н-3,5): 1. Раствор диазотированного ами-
на нейтрализуют для предотвращения выделения цианистого во-
дорода (синильная кислота), смешивают при охлаждении с рас-
твором комплексной соли меди, взятой с небольшим избытком, и,
если нужно, нагревают, о- и n-Толунитрилы получают этим
методом из соответствующих толуидинов с выходом 64—70% [1].
Описано применение Ni(CN)2 вместо CuCN [2].
Введение цианогруппы по Зандмайеру находит применение в
промышленном синтезе о-цианофенилтиогликолевых кислот — про-
межуточных продуктов для тиоиндигоидных красителей, 8-циано-
нафталин-1-сульфокислоты — промежуточного продукта для кубо-
вых красителей ряда антантрона [3] и др. После отделения циа-
нозамещенных растворы обрабатывают полисульфидом натрия,
который превращает цианиды в безвредные тиоцианаты и оса-
ждает сульфид меди.
11.1.2. Замещение атомов галогенов и сульфогруппы
Важным способом введения цианогруппы является за-
мещение галогенов при действии CuCN (реакция Розенмунда —
Брауна). По способности к замещению атомы галогенов распола-
гаются в обычной для каталитической реакции последователь-
ности: Cl < Br < I (см. 7.1.1). Реакцию можно проводить без
растворителя, в пиридине и в полярных апротонных растворителях.
В частности, нагреванием 9-бромфенантрена с небольшим избыт-
ком CuCN при 260 °C в течение 6 ч с выходом 87% получают
9-фенантренкарбонитрил [4]. Нагревание смеси 1-бромнафталина,
340
CuCN и пиридина при 215—225 °C в течение 15 ч дает 1-нафта-
линкарбонитрил (нитрил 1-нафтойной кислоты) с выходом 82—
90%. Чтобы достигнуть того же результата, исходя из 1-хлорнаф-
талина, необходимо нагревание при 245—250 °C в течение 24 ч [5].
Применение полярных апротонных растворителей позволяет зна-
чительно ускорить реакцию. Для превращения 1-бромнафталина в
1-нафталинкарбонитрил достаточно 3-часового кипячения в (V-ме-
тилпирролидоне [6], для превращения 2-хлорнафталина в 2-на-
фталинкарбонитрил (нитрил 2-нафтойной кислоты) — нагревания в
гексаметилфосфортриамиде в течение 3 ч при 230—240°C (96%)
[7].
Метод применим для синтеза полицианозамещенных. Нагрева-
нием 2,4,6-трибромезитилена и CuCN в пиридине при 205 °C полу-
чен мезитилен-2,4,6-трикарбонитрил (56%) [8], взаимодействием
1,3-дииодтетраметилбензола с CuCN в гексаметилфосфортриамиде
при 90—100 °C — тетраметилизофталонитрил (73%) [9]. При ре-
акциях о-дигалогенпроизводных образующиеся о-динитрилы пре-
вращаются во фталоцианины меди. Этого превращения удается
избежать, если проводить процесс в кипящем диметилформамиде,
непременно при больших разбавлениях [10]. В этих условиях из
замещенных о-дибромбензолов с выходом 40—83% получены со-
ответствующие фталонитрилы.
При синтезе гексацианобензола (бензолгексакарбонитрил) ис-
пользовано цианирование в отсутствие солей меди, для чего атомы
хлора в трихлор-1,3,5-трицианобензоле (трихлор-1,3,5-бензолкар-
бонитрил) замещены атомами фтора, которые обмениваются на
цианогруппу при действии Ca(CN)2 в диметилформамиде при 20—
25-°С [11]. Обработкой KCN в водном метаноле осуществляют
замещение двух орто-расположенных атомов галогена в тетрага-
логен-1,4-бензохинонах и в 2,3-дигалоген-1,4-нафтохинонах, причем
избыток цианида восстанавливает цианозамещенный хинон в гид-
рохинон [12]:
X = Cl, Br, I
Техническое значение имеет замещение галогенов на циано-
группу при получении 1,4-диамино-2,3-дициано-9,10-антрахинона
(1,4-диамино-9,10-антрахинон-2,3-дикарбонитрил) (2), применяемо-
го в производстве синих дисперсных красителей [13]. Например,
динитрил (2) можно получить, действуя цианидом щелочного ме-
талла в присутствии соли меди (II) и сульфита на комплекс
341
1,4-диамино-2,3-дибром-9,10-антрахинона (1) с борной кислотой
При наличии в орто-положении к галогену нитро- или азогруп-
пы легкость замещения по Розенмунду — Брауну увеличивается.
Так, при реакции 1-нитро-2,3-дихлорбензола и 1,2-дибром-З-нитро-
бензола с CuCN в диметилформамиде обменивается в первую
очередь атом хлора в орто-положении к нитрогруппе [2, 15]. Обмен
галогена в о-галогеназобензолах используется в промышленности
при производстве дисперсных красителей. Реакцию проводят в
диметилформамиде или метилцеллозольве в присутствии пиридина
[16] ив водной среде [17]. Считают, что реакция протекает через
стадию комплекса с CuCN, являющегося лабильным промежуточ-
ным продуктом в реакциях каталитического замещения галогена
(см. также 7.1.1):
ArX + [CuL3]+ CN’ —»•
X
Ai\ )CuL2
CN
—»• ArCN + [CuL3]+ X"
L = лиганд; X = галоген
Роль о-заместителя в ароматическом кольце, по-видимому, сво-
дится к участию в образовании комплекса с CuCN. Предположение
о промежуточном возникновении медьорганических соединений
при каталитическом замещении галогенов не получило подтверж-
дения [18].
Сульфогруппа обменивается на цианогруппу при сплавлении
арилсульфонатов с цианидами или гексациано-(II)ферратами ще-
лочных металлов при температуре порядка 400 °C:
ArSO3Na + NaCN —> ArCN + Na2SO3
Этим методом из цианонафталинсульфокислот синтезированы с
выходом 8—76% все 10 изомерных дицианонафталинов (нафта-
линкарбонитрилы) [19]. Примером применения реакции в гетеро-
циклическом ряду может служить синтез нитрила 3-пиридинкар-
боновой — никотиновой кислоты (3-цианопиридин) нагреванием
3-пиридинсульфоната натрия и цианида натрия при 340—400 °C;
выход 46% [2]. В менее жестких условиях возможно замещение
сульфогрупп цианогруппами в 1,4-диамино-9,10-антрахинон-2,3-ди-
сульфокислоте; получается динитрил (2), но выход его ниже, чем
при синтезе замещением галогена [14].
342
11.1.3. Замещение атома водорода
Водород замещается цианогруппой в ароматических
нитросоединениях, хинонах и азотсодержащих гетероциклах. При
действии цианидов щелочных металлов на нитросоединения циа-
ногруппа вступает в орто-положение к нитрогруппе, которая,'буду-
чи таким образом активирована, сама может подвергаться затем
нуклеофильному замещению. Так, при обработке л-динитробензо-
ла (3) в метаноле водным раствором KCN при 40 °C и последующей
выдержке 2—3 дня при комнатной температуре получают с выхо-
дом 22—23% 2-метокси-6-нитробензонитрил (4) [20]:
При взаимодействии нитросоединений (5) с KCN в диметил-
сульфоксиде при 100 °C вслед за замещением водорода нитрогруп-
па обменивается на гидроксигруппу и с выходом 55—60% образу-
ются о-гидроксибензонитрилы (6) [21]. Окислителем на стадии
ароматизации о-комплекса служит нитросоединение, превращаю-
щееся в смесь азо- и азоксисоединений.
+ KCN '
(CHshSO
R = СОС6Н5, SO2C6H5, CN
(5)
(6)
При нагревании нитросоединений (7) с KCN в протонодонорных
растворителях (водном этаноле, метилцеллозольве и др.) атака
цианид-аниона в орто-положение к нитрогруппе приводит к эли-
минированию ее и образованию бензойных кислот (11) (реакция
Рихтера):
R=H, AIK, Hal, 8О3Н и др.
343
Выходы в реакции Рихтера невысоки, но она имеет определен-
ное препаративное значение, позволяя синтезировать карбоновые
кислоты, труднодоступные иным путем. Так, кипячением раствора
2-бром-4-нитротолуола (12) с KCN в водном метилцеллозольве и
подкислением получают с выходом около 7% 2-бром-л-толуиловую
кислоту (13) [22]:
(12) (13)
При изучении механизма реакции Рихтера выяснилось, что в
процессе превращения выделяется газообразный азот в количестве
примерно эквивалентном количеству карбоновой кислоты. Опыта-
ми с n-нитрохлорбензолом, меченным 15N, установлено, что один из
двух атомов газообразного азота ранее принадлежал нитрогруппе,
а опытами в CgHgOD + D2O показано, что на место нитрогруппы
вступает атом водорода, принадлежавший растворителю. Воз-
можность образования газообразного азота при взаимодействии
аммиака с нитрит-анионом, если бы последний отщеплялся в реак-
ции, исключается, так как при проведении реакции в присутствии
15NH3 газообразный азот остается немеченным [23]. Применение
смеси этанола с водой, обогащенной 18О, показывает, что один из
атомов кислорода карбоксильной группы переходит из растворите-
ля, а второй—из нитрогруппы [24]. Среди продуктов реакции не
найдено соединений, предшественниками которых могли бы быть
арины. Совокупность изложенных данных согласуется со схемой
[23]; согласно ей, изомеризация ст-комплексов (8) приводит к
«-нитрозобензамидам (9), которые циклизуются в 3-индазолоны
(10), распадающиеся с элиминированием азота. Вероятность тако-
го механизма подтверждается независимым синтезом 3-индазоло-
на, который мгновенно превращается в бензойную кислоту с выде-
лением азота при внесении в раствор KCN в водном этаноле [25].
Хиноны могут реагировать с цианидами с образованием циано-
замещенных гидрохинонов. При действии на 1,4-бензохинон (14)
цианида калия в водном изопропиловом спирте в присутствии
H2SO4 с выходом близким к количественному выделены в эквимо-
лярном соотношении 3,6-дигидроксифталонитрил (15) и гидрохи-
нон (16) [12]. При обработке 2-арил-1,4-нафтохинонов цианидом
натрия в смеси диоксан — этанол — вода при комнатной темпера-
туре и подкислении с выходом 64—68% получены 3-арил-1,4-нафто-
хинон-2-карбонитрилы [26].
О ОН ОН
<(14) (15) (16)
344
Методом введения цианогруппы в азотсодержащие гетероцик-
лы служит взаимодействие солей Л'-алкоксипроизводных, получае-
мых алкилированием гетероциклических АГокисей (оксидов) с ци-
анидами щелочных металлов. Реакция состоит в присоединении
цианид-аниона в а- или у-положение гетероцикла с последующим
отщеплением протона и алкоголят-аниона. Процесс проводят в-
воде без нагревания, получая цианозамещенные с высоким выхо-
дом. Метилируя хинолин-1-оксид (17) диметилсульфатом и обра-
батывая метилсульфат 1-метоксихинолиния (18) цианидом полу-
чают с выходом 93°/о 2-хинолин карбонитрил(нитрил хинальдино-
вой кислоты) (19); из 2-и.зохинолиноксида— 1-изохинолинкарбо-
нитрил (95%); из 1-пиридиноксида — 32% 2-пирндинкарбонитрила
(нитрил пиколиновой кислоты) и 49% 4-пиридинкарбонитрила
(нитрил изоникотиновой кислоты); из 2-пиколин-1-оксида получено
40—46% 6-метил-2-пиридинкарбонитрила (нитрил 6-метилпиколи-
новой кислоты) [27].
При взаимодействии азотсодержащих гетероциклов, в которых
имеется связь —C = N—, с ацилхлоридом и цианидом калия легко
образуются аддукты типа (20)—так называемые соединения
Райссерта. При гидролизе из них образуются амид карбоновой
кислоты (22) и альдегид (23), соответствующий взятому ацилхло-
риду. Гидролиз соединений Райссерта в кислой среде протекает с
выходами близкими к количественным и является препаративным
методом синтеза как гетероциклических карбоксамидов и кислот,
так и альдегидов из ацилхлоридов [28, 29]. При получении альде-
гидов обычно используют хинолин или изохинолин.
(21)
(22)
(23)
345
Исследованиями с помощью спектров ЯМР установлено, что в
кислой среде соединения Райссерта существуют в виде конденси-
рованных производных 5-аминооксазолия типа (21), которые и
подвергаются гидролитическому расщеплению [30].
11.2. ВВЕДЕНИЕ АЛКИЛЬНЫХ ГРУПП
11.2.1. Замещение атомов галогенов и других групп
Галогены способны обмениваться на алкильные группы
при действии карбанионов в присутствии солей меди, а также без
.катализаторов, в двухфазных системах в присутствии четвертичных
-аммониевых оснований и в присутствии инициаторов радикальных
реакций.
Катализ солями меди эффективен почти исключительно в реак-
циях карбанионов с орто- и пери-галогенкарбоновыми кислотами
[31]. В качестве нуклеофилов используются карбанионы, образую-
щиеся под действием алкоголятов на р-дикарбонильные соедине-
ния малоновый или ацетоуксусный эфир, ацетилацетон, бензоил-
ацетоп, 1,3-циклогександион. Катализаторами служат металличе-
ская медц соединения меди(1) и (II), растворителем, как
правило,— сухой этанол; в пиридине, ацетонитриле, диметилсуль-
•фоксиде, обычно используемых при катализируемом медью заме-
щении галогенов (см. 7.1.1, 8.2, 11.1.2), реакция не идет. Получае-
мые диоксоалкильные производные в процессе реакции или при
дополнительной обработке могут подвергаться кислотному рас-
щеплению, превращаясь в оксоалкильные замещенные. Так, кипя-
чение эквимолярных количеств о-бромбензойной кислоты (24) и
бензоилацетона в этаноле с двукратным избытком этилата натрия
и порошком меди приводит с выходом 57% к 2-(бензоилметил)-
бензойной кислоте (25). В трет-бутиловом спирте с трет-бутила-
том калия выход повышается до 75%. Из 8-бром-1-нафтойной
кислоты (26) в этаноле с выходом 77% получают 8-(1-бензоилаце-
тонил)-1-нафтойную кислоту (27), которая переходит в 8-ацето-
нил-1-нафтойную кислоту (28) при кипячении в водном растворе
NaOH.
+ СвН5СОСН2СОСНз
С2Н5О"; Си
- С ООН
СНСОС6Н5
I
СОСНз
—СНзСООН
соон
СН2СОСвН5
(25)
846
+ с6н5сосн2сосн3
C2H5O~; Си
C6HSCO
+ NaOH
--------->
—С6Н5СООН
(27)
no2-
Так же, но с меньшим выходом, реагируют 2-бром-1-нафтойная-
и З-бром-2-нафтойная, З-бром-2-пиридинкарбоновая (3-бромпико-
линовая) и 10-бром-9-фенантренкарбоновая кислоты. о-Хлорбен-
зойные кислоты в реакцию не вступают, а о-иодбензойная дает
выход ниже, чем о-бромбензойная. Если карбоксильная группа
находится в мета- или нпрп-положении к брому или если в орто-
положении к нему находится другой заместитель — ацетильная,
формильная, карбоксиметильная, гидроксиметильная, гидроксиль-
ная, цианогруппа, — замещения не происходит. Жесткие требова-
ния к строению ароматического субстрата связывают с определяю-
щим влиянием геометрии молекулы на координацию атома галоге-
на с ионом меди в медной соли карбоновой кислоты [31].
Другой катализируемой медью реакцией обмена галогена на
алкильную группу является взаимодействие арилгалогенида с ал-
килгалогенидом, родственное, по-видимому, получению диарилов
по реакции Ульмана (см. 11.3). Например, нагревание п-бромнит-
робензола с пентафторэтилиодидом в диметилформамиде в при-
сутствии металлической меди при 100—130 °C приводит к п-нитро-
пентафторэтилбензолу [32]:
/==\ +c2f5i /==\
NO2-^J>—C2F5
>—В г
галогеннитропроизводные способны обменивать
замещенную алкильную группу и в отсутствие
Реакция n-галогеннитробензолов с а-фенилал-
Ароматические
хлор или бром на
соединений меди,
каннитрилами (29) протекает в диметилсульфоксиде в присутствии
избытка твердого КОН или NaH или в двухфазной системе, состо-
ящей из органического растворителя (бензол, ацетонитрил) и кон-
центрированного водного раствора NaOH в присутствии каталити-
ческих количеств соли четвертичного аммония, обычно бензилтри-
этиламмонийхлорида (30). В результате размешивания в такой
системе (3—4 ч при 40—50 °C) эквимолярных количеств п-нитро-
хлорбензола и 2-фенилпропионитрила с выходом 92% получают
347
2-(4-нитрофенил)-2-фенилпропионитрил [(33); R — СН3; R' = H],
В тех же условиях с хорошим выходом получены а-циано-
алкильные замещенные (33) из других п-галогеннитробензолов
(32) и о-нитрохлорбензола [33]. При реакции 1-нитро-2,4-дихлор-
бензола [(32); X = Cl; R' = 2-С1] с 2-фенилбутиронитрилом вы-
делено 78% 4-нитро-3-хлор-1-алкилзамещенного [(33); R = С2Н5;
R' = 3-С1] и только 3% 2-нитро-5-хлоризомера; т. е. обменивается
преимущественно атом хлора в пара-положении к нитрогруппе.
В случае 4-нитробензофенонов (34) в тех же условиях гладко за-
мещается нитрогруппа (35).
Реакция представляет собой пример катализа за счет переноса
реагента между фазами (phase-transfer catalisis), приобретающего
большое препаративное значение [34,35]. Роль соли четвертичного
аммония (30) состоит в образовании с карбанионом, генерируемым
под действием NaOH в водном слое, ионной пары (31), которая
переходит в органический слой, где и происходит реакция с аро-
матическим субстратом.
+ +NaOH
-CeH5—CH-CN + (C2H5)3NCH2CeH5 СГ >
R
(29) (30)
С6Н5—С—CN || (C2H5)3NCH2CeH5
I
R
Х = С1, Вг; R = H, СН3, С2Н5, СвН5СН2, С6Н5;
R' = СН3, CI, NO2, COOAlk, СОС6Н5 и др.
Направление реакции нитрогалогенбензолов с карбанионами
арилалканнитрилов решающим образом зависит от среды; это ил-
люстрирует взаимодействие о-нитрохлорбензола (36) с дифенил-
ацетонитрилом (37). В диметилсульфоксиде в присутствии порош-
кообразного КОН оно приводит с выходом 88% к (2-нитрофенил)-
дифенилацетонитрилу (38), в метаноле в присутствии метилата
натрия — с выходом 91% к нитрону (39), а в бензоле в присут-
ствии амида натрия — к тетрафенилсукцинонитрилу (40).
348
+ (C3H5)2CHCN
(Зв) (37)
CN
CN
(40)
Для объяснения различий в направлении реакций в диметил-
сульфоксиде и в метаноле выдвинуто предположение, что в первом
случае реакция находится под термодинамическим, а во втором —
под кинетическим контролем (см. 2.7.2). При кинетическом
контроле быстрее образующийся 3-ст-комплекс, возникающий в ре-
зультате атаки в незамещенное положение о-нитрохлорбензола
(36) необратимо превращается в (4-нитрозо-З-хлорфенил) дифенил-
ацетонитрил (на схеме не указан), который взаимодействует со
второй частицей карбаниона, давая соединение (39). При термо-
динамическом контроле дальнейшие превращения претерпевает
преобладающий в условиях равновесия более стабильный 1-о-ком-
плекс, распадающийся с отщеплением хлораниона, что приводит к
соединению (38). Образование сукцинонитрила (40) рассматрива-
ется как результат побочной реакции одноэлектронного переноса
от карбаниона к нитросоединению с последующей димеризацией
дифенилцианометильных радикалов [33].
Активированная нитрогруппа легко обменивается на остаток
СН-кислоты в полярных апротонных растворителях. Замещенные
нитробензолы взаимодействуют с литиевой солью 2-нитропропана
в гексаметилфосфортриамиде при 25 °C, давая соответствующие
а-нитрокумолы с выходом 75—82% [36]:
R \—NO2 + (CH3)2CNO2 Li+ —->• R —C(CH3)2
\=/ \=/ |
NO2
R = CN, C3H5SO2, С3Н5СО, NO2
Описанные выше реакции нитросоединений с карбанионами
представляют собой примеры активированного нуклеофильного
349
замещения типа SnAt2. В неактивированных ароматических соеди-
нениях атомы галогенов и другие уходящие группы могут быть
замещены в реакциях типа SrnI. Метод состоит в генерировании
анион-радикала ароматического соединения АгХ *, который, рас-
падается на анион уходящей группы X- и арильный радикал Аг*,
атакуемый нуклеофилом Y :
+ е + Y + АгХ
АгХ ----> АгХ". —> Аг * + X" ----> ArY"* ----> ArY + ^rX".
Реакции проводят в жидком аммиаке, применяя для генериро-
вания анионов амид калия, а для генерирования арильных анион-
радикалов щелочной металл (находящийся в жидком аммиаке в
основном в виде катионов и сольватированных электронов), или
фотооблучение. В качестве уходящих групп помимо галогенов об-
следованы триметиламмониевая, фенокси-, триэтилфосфокси- и
фенилтиогруппы, в качестве нуклеофилов — анионы из алифатиче-
ских кетонов, диенов и циклических СН-кислот (инден, флуорен)
;[37,38].
При взаимодействии с нитрилами образующиеся а-цианоал-
килзамещенные (41) в присутствии избытка щелочного металла
могут подвергаться децианированию, превращаясь в алкилзаме-
щенные (42),
(41) (42)
В=Н,СН3,СН3СН2, СН3СН2СН2, С6Н5 и др.
Хотя общие выходы алкилбензолов не превышают 50%, по
мнению авторов [38], метод представляет препаративную цен-
ность, так как в отличие от электрофильного С-алкилирования
(см. 6.1.3) ориентация вступающего алкильного остатка определя-
ется исключительно положением уходящей группы, а не влиянием
других заместителей, и сама алкильная цепь не претерпевает пере-
группировок, что исключает образование изомеров. Кроме того,
исчерпывающее А-алкилирование аминов и О-ацилирование фено-
лов с последующим обменом триалкиламмониевой или ацилокси-
группы на алкильную позволяет перейти от аминов и фенолов к
соответствующим С-алкилзамещенным. Реакции SRNl-алкилирова-
ния возможны также при фотовозбуждении, как показано на при-
мере взаимодействия енолятов кетонов с галогензамещенными
бензолами и пиридинами [39].
11.2.2. Замещение атома водорода
Ранее уже отмечалось (см. 2.7.2), что нуклеофильная
атака карбаниона в незамещенное положение приводит к стабиль-
ным анионным ст-комплексам, которые могут быть окислены в
продукты С-замещения. Непосредственный обмен атома водорода
350
на алкильную группу возможен при действии С-нуклеофилов, спо-
собных после присоединения отщеплять анионный фрагмент, обес-
печивая тем ароматизацию ст-комплекса. К таким нуклеофильным
реагентам относятся карбанионы, генерируемые при действии на
диметилсульфоксид, диметилсульфон, метилфенилсульфон трет-
бутилата калия или гидрида натрия. При их реакциях с ароматиче-
скими соединениями вслед за образованием о-комплекса происхо-
дит разрыв связи С—S с отщеплением метилсульфинильного или
метилсульфонильного аниона и внутримолекулярным переносом
гидрид-иона из геминального узла к метиленовой группе
(ср. 7.1.6 — ароматизация о-комплекса при аминировании гидро-
ксиламином) :
< ,, +сн3зосн2 SOCH3 -chjSo-
/~н " ~~^7“СНз
При обработке раствором трет-бутилата калия или гидрида
натрия в диметилсульфоксидё при 55—70 °C полициклические аро-
матические углеводороды и гетероциклы подвергаются метилиро-
ванию [40—42]. Антрацен в зависимости от избытка реагента и
длительности обработки переходит в 9-метилантрацен (74%) или
9,10-диметилантрацен (до 96%); фенантрен превращается в
9-метилфенантрен (93%), акридин — в 9-метилакридин, бензокса-
зол — в 2-метилбензоксазол. Хинолин, изохинолин, фенантридин
почти количественно дают 4-метилхинолин, 1-метилизохинолин и
6-метилфенантридин соответственно [41]. Бензол, пиридин, фена-
зин в реакцию не вступают. После нагревания нафталина в течение
92 ч с выходом 15% получена смесь, состоящая на 96% из
1-метил- и на 4% из 2-метилнафталина [40]. Метилирование фе-
нилсульфонилметанид-анионом C6H5SO2CHJ проводилось обработ-
кой метилфенилсульфоном и гидридом натрия в гекса метил фос-
фортриамиде (гексаметапол) [42].
Нуклеофильное метилирование нитросоединений протекает при
действии диметилсульфоксонийметилида, возникающего при обра-
ботке соли триметилсульфоксония основанием. Отщепляемой груп-
пой здесь является молекула диметилсульфоксида:
+(ch3)2sochJ ?уСП2 ДО(СН3)2 _(cH3)tSO^ <
- Ан " у СНз
При внесении нитробензола в раствор диметилсульфоксонийме-
танида в диметилсульфоксиде с выходом до 67% на прореагиро-
вавший нитробензол образуется смесь о- и n-нитротолуолов со
значительным преобладанием о-изомера [(104-15): 1]. Метилиро-
вание изомерных нитрохлорбензолов, нитротолуолов, нитроанизо-
лов, 1-нитронафталина также направляется преимущественно в
орто-положение к нитрогруппе [43, 44].
Замещение водорода в гетероароматических соединениях легко
протекает при действии алкилирующих или ацилирующих агентов
331
на их TV-окиси (Диоксиды) с последующей обработкой четвертичной
соли С-нуклеофилом. Ароматизация о-комплексов осуществляется
путем отрыва алкокси- или ацилокси-аниона и протона. Так, при
добавлении раствора Na2CO3 или NaOH к водному раствору метил-
сульфата 1-метоксихинолиния (18) (см. 11.1.3) и ацетона с вы-
ходом 88% получают 2-ацетонилхинолин (43) [45], а при обра-
ботке хинолин-1-оксида (17) этиловым эфиром цианоуксусной кис-
лоты и уксусным ангидридом с выходом 83% 2-[ (этоксикарбонил-) -
цианометил] хинолин (45) [46]. В последнем случае, по-видимому,
первоначально образуется ацетат 1-ацетоксихинолиния (44), кото-
рый присоединяет карбанион, возникающий при отщеплении
протона от метиленовой компоненты под действием ацетат-аниона.
+ ncch2cooc2h5
+ (СН3СО)2О
СНзСООН
+ N
| СНзСОО’
-СНзСОО
"'-СНСООС2Н5
I
CN
(45)
Нуклеофильное орто-алкилирование ароматических соединений
возможно путем внутримолекулярного замещения типа перегруп-
пировки Смайлса и Соммле. Диарилсульфон (46) под действием
бутиллития в эфире или трет-бутилата калия в диметилсульфокси-
де ионизируется по о-метильной группе (47) и затем через спиро-
циклический о-комплекс (48) перегруппировывается в бензилзаме-
щенную аренсульфиновую кислоту (49) (выход 98%) [47]. В отли-
чие от других случаев перегруппировки Смайлса (см. 7.1.4, 7.1.5,
8.2) в данном примере внутримолекулярному нуклеофильному
замещению подвергается неактивированное ароматическое кольцо.
При перегруппировке Соммле соли триалкилбензиламмония
под действием оснований переходят в о-алкилбензилзамещенные
соединения [48]. Например, иодид бензилтриметиламмония (50)
352
почти количественно превращается в ЛГ,У-диметил (о-метилбеН-
зил)амин (51) при обработке амидом натрия в жидком аммиаке:
ch2n(ch3)3
I
+NaNH2
5K.NH3
СН
ch2n(ch3)2
(50)
(51)
Подобное превращение лежит в основе препаративного метода
о-алкилирования ароматических аминов, включающего перегруп-
пировку азасульфониевых солей. Процесс о-метилирования состоит
в Дехлорировании производного анилина (52а) или N-ал кил анили-
на (526) гипохлоритом кальция или трет-бутилгипохлоритом, за-
мещении атома хлора в /V-хлорамине (53) обработкой диметил-
сульфидом, перегруппировке сульфониевой соли (54) в при-
сутствии метилата натрия в о-(метилтиометил)анилин (55) и
десульфуризации с превращением в о-метиланилин (56) никелем
Ренея [49]. Первые три стадии выполняются без выделения проме-
жуточных соединений при температуре ниже О °C в неполярном орга-
ническом растворителе (метилендихлорид, пентан, толуол). Выход
о-метилтиометильных замещенных (55) составляет 50—75% в рас-
чете на исходный амин (52), и достигает по стадии собственно
С-алкилирования 90—95%. Применение циклических сульфидов
тетрагидротиофена и тетрагидротиопирана позволяет ввести в
о-положение н-бутильную и н-пентильную группу соответственно
(57)—>(58).
+Са(С1О)2
(55)
a)R=H; 6)R=CH3,C(CH3)3; b)R“CH3CO
R'=C1, CH3, OCH3> COOC2Hs и др.
4
12 Зак. 271
353
В отличие от электрофильного С-алкилирования ароматических
аминов (см. 6.1.5) данный способ обеспечивает отсутствие м- и
«-изомеров и перегруппировок алкильных групп. Показана приме-
нимость метода к ацетанилидам (52в) [50] и к гетероциклическим
аминам на примере орто-алкилирования 2-аминопиридинов- [51].
11.3. ВВЕДЕНИЕ АРИЛЬНЫХ ГРУПП
Арильные группы могут быть введены в ароматическое
кольцо свободнорадикальным арилированием (см. 12.1), реакцией
Ульмана и нуклеофильным замещением при действии на активи-
рованный субстрат л-избыточных молекул.
Синтез биарилов по Ульману [52,53] состоит в обработке
арилгалогенидов порошком меди:
+ Си
2АгХ ---> Ar—Ar + 2СиХ X = Cl, Br, I
Активность арилгалогенидов возрастает в ряду: Cl < Br < I.
Введение электроноакцепторных заместителей (NO2, СН3СО), осо-
бенно в орто-положение к атому галогена, способствует реакции.
Процесс проводят, нагревая арилгалогенид или его раствор с
3—10-кратным избытком порошка меди. В качестве растворителя
используют нитробензол, алифатические и алициклические углево-
дороды, пиридин, хинолин и другие, но наиболее часто диметил-
формамид. Последний позволяет снизить температуру, сократить
избыток меди, подавить побочную реакцию дегалогенирования.
Промежуточные продукты для кубовых красителей и пигментов
М'-биантрахинонилы (61) получают взаимодействием 1-хлор-
9,10-антрахинонов и 2-ациламино-1-хлор-9,10-антрахинонов (60) с
порошком меди в диметилформамиде при 80—120 °C [54]:
R=H, СН8,
2-NHCOCH31
2-NHCOOC2Hsh др.
Помимо синтеза симметричных биарилов реакция Ульмана
применяется для получения несимметричных биарилов, полиарилов
и циклических соединений. Образование симметричных биарилов
облегчается, когда один из арилгалогенидов более реакционноспо-
собен, чем другой, причем реакция между активным и неактивным
354
арилгалогенидом протекает легче, чем между двумя молекулами
активного арилгалогенида:
Замыкание новой связи С—С по реакции Ульмана применяет-
ся для построения четырехчленных (бифенилены), пятичленных
(флуореноны и их гетероаналоги), шестичленных (трифенилены),
восьмичленных (тетрафенилены) и еще больших углеродных цик-
лов. Линейные полиарилены получают, вводя в реакцию смесь
моно- и дигалогенпроизводного.
Принятый механизм реакции Ульмана [53] включает промежу-
точное образование арилмедного соединения, которое, будучи ста-
билизировано в виде сольватного комплекса, выступает в качестве
нуклеофила по отношению к молекуле арилгалогенида:
+ Си + Аг X
АгХ ----> АгСи -----> Аг—Аг
Исследование роли арилмедных соединений в реакции Ульмана
привело к методу получения несимметричных биарилов с раздель-
ным приготовлением медьорганического соединения и введением
его во взаимодействие с галогенпроизводным. В некоторых случаях
такой метод дает результаты, недостижимые классическим путем.
Например, взаимодействием 1,8-дииод-9,10-антрахинона с литий-
дифенилкупратом в тетрагидрофуране и последующим окислением
кислородом воздуха получен 1,8-дифенил-8,10-антрахинон; выход
42% [55]:
Соединения, обладающие высокой активностью по отношению к
нуклеофильным реагентам: нитросоединения, хиноны, азотсодер-
жащие гетероциклы — способны арилироваться при действии ари-
ламинов, фенолов, их эфиров, л-избыточных гетероциклов. Ди- и
тринитрогалогенбензолы обменивают галоген на арильную группу
уже при кипячении с 1,3-бис(диалкиламино)бензолами в этаноле.
При сливании растворов метил-3,5-динитро-2-хлорбензоата (62) и
1,3-дипирролидинобензола (63) сначала выпадает комплекс с
переносом заряда, который затем растворяется, и после 4 часов
12* 355
кипячения с выходом 84% получают производное бифенила (64)
[56]
(62) (63)
(64)
1,4-Бензохиноны (65) реагируют с фенолами, анизолом,
Л\М-диметиланилином в присутствии кислот, образуя арилгидро-
хиноны (66), выделенные с выходами менее 50% [57]. Нуклео-
фильной атаке подвергается, по-видимому, протонированная моле-
кула хинона, выступающая по отношению к арилирующему агенту
в роли электрофила, подобного участвующим в реакциях ацилиро-
вания по Фриделю — Крафтсу (см. 6.3.1).
+ с6н5х
[Н+] ’
X = ОН, ОСНз, N(CH3)2
R = СОСНз, СООСНз
(65)
(66)
Промышленное значение для производства дисперсных краси-
телей имеет арилирование 4,8-диамино-1,5-дигидрокси-9,10-антра-
хинон-2,6-дисульфокислоты (67), которая при действии ароматиче-
ских С-нуклеофилов (фенол, анизол и др.) в H2SO4 в присутствии
Н3ВО3 переходит в моносульфокислоту (68), подвергаемую затем
восстановительному десульфированию. В реакцию вступает, оче-
видно, молекула хинона (67), активированная комплексообразова-
нием с Н3ВО3 и протонированием по аминогруппам. Как показано
спектрами ЯМР и другими данными [58], арилирование направ-
356
ляется в незанятое сульфогруппой положение 3 и представляет
собой случай кине-замещения.
Азотсодержащие гетероциклы, активированные протонировани-
ем или A-замещением, присоединяют ариламины, фенолы и
л-избыточные гетероциклы, превращаясь в дигидропроизводные,
которые могут быть ароматизированы при действии окислителя
[59, 60]. Особенно легко протекает такая реакция с солями акри-
диния (69), которые превращаются в соли 9-аминоарилакридиния
(70) с выходами 80—90%.
(69)
(70)
Из других гетероциклических катионов в реакцию вступают
катионы хинолиния и некоторых бензазинов, а из незаряженных
соединений — акридин и оксобензазины; 2-хиноксалон, 2-хиназо-
лон, 3-циннолон [61].
ЛИТЕРАТУРА
1. Кларк Г., Рид Р. — В кн.: Синтезы органических препаратов. Сб. 1. Пер.
с англ. М., ИЛ, 1949, с. 391—394. 2. Friedrich К., Wallenfels К- — In: The Che-
mistry of the Cyanogroup/Ed. Z. Pappoport. London, Wiley, 1970, p. 67—122.
3. Венкатараман К. Химия синтетических красителей. Т. 2. Перев. с англ./Под
ред. Н. С. Вульфсона. Л., Госхимиздат, 1957, с. 1097, 1175—1176. 4. Каллен Дж.,
Дорнфельд К-, Кальман Дж. — В кн,: Синтезы органических препаратов. Сб. 4.
357
Пер. с англ. М., ИЛ, 1953, с. 555—556. 5. Ньюмен М. — Там же, Сб. 3. 1952,
с 344—346. 6. Newman М. S, Boden И. J.—Org. Chem., 1961, v. 26, № 7,
р. 2525. 7. House Н. О., Fischer W. F. — Ibid., 1969. v. 34, № 11, p. 3626—3627.
8. Weis C. D. — Ibid., 1962, v. 27, № 8, p. 2964—29 65.9. Suzuki H., Hanafusa T.~
Synthesis, 1974, № 1, p. 53—55. 10. Ковшов E. И., Соловьева Л. И., Михайлен-
ко С. А. и др. —Журн. ВХО, 1976, т. 21, вып. 4, с. 465—466.
11. Friedrich К., Oeckl S. — Chem. Вег., 1970, Bd 103, № 12, S. 3951.
12. Wallenfels К., Bachmann G., Hoffmann D. e.a. — Tetrahedron, 1965, v. 21,
№ 9, p. 2239—2256. 13. Стрела Дж. M. — В кн.: Химия синтетических красителей.
Т. 3/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л, Химия,
1974, с. 2053—2057. 14. Яп. пат. 74/17643; Chem. Abstr., 1975, v. 82, 32456.
15. Михайленко С. А., Лукьянец Е. А. Анилинокрас. пром., 1975, вып. 3, с. 3—10.
16. Пат. ФРГ 2364205, 1975, Chem. Abstr., 1975, v. 83, 133374; Пат. США
3821195, 1974, Chem. Abstr., 1975, v. 83, 61646. 17. Пат. ФРГ 2134896, 1973,
Chem. Abstr., 1973, v. 78, 148953; 2310745, 1974, Chem Abstr., 1974, v. 82, 45018.
18. Van Koten G., Jastrzebski J., Noltes J. G. — Ibid., 1976, № 3, p. 223—226.
19. Доналдсон H. Химия и технология соединений нафталинового ряда. Пер.
с англ, под ред. А. И. Королева. М, Госхимиздат, 1963, с. 482—490. 20. Рас-
селл А., Теббенс В. — В кн.: Синтезы органических препаратов. Сб. 3. Пер. с
англ. М, ИЛ, 1952, с. 201—203.
21. Gorvin J. Н. — Chem. Commiin., 1971, № 18, р. 1120—1121. 22. Бан-
нет Дж., Раухут М. — В кн.: Синтезы органических препаратов. Сб. 10. Пер. с
англ. М, ИЛ, 1960, с. 14—16. 23. Rosenblum М.—J. Amer. Chem. Soc., 1960,
v. 82, № 14, р. 3796—3798. 24. Samuel D. — J. Chem. Soc., 1960, № 3, p. 1318—
1320. 25. Ullman E. F., Bartkus E. A. — Chem. Ind., 1962, № 2, p. 93—94. 26. Гри-
нев A. H., Клягина А. И., Терентьев А. ПТ—ЖОХ, 1959, т. 29, вып. 8, с. 2773—
2711. 27. Feely IP. £., Evanega G., Beavers E. M. — In: Organic Syntheses. Coll,
v. 5. N. Y, Wiley, 1973, p. 269—273. 28. Мозеттиг Э. — В кн.: Органические реак-
ции. Сб. 8. Пер. с англ. М., ИЛ, 1956, с. 288—332. 29. Popp F. D. — In: Advances
in Heterocyclic Chemistry. V. 9. N. Y, London, Academic Press, 1968, p. 1—25.
30. McEwen IP. E., Mineo I. C., Shen У. H. — J. Amer. Chem. Soc., 1971, v. 93,
№ 18, p. 4479—4484; Cook M. J., Katritzky A. R., Page A. D. — Ibid., 1977, v. 99,
№ 1, p. 165—169.
31. Cirigottis K. A., Ritchie E., Taylor IP. C. — Austr. J. Chem., 1974, v 27,
Ns 10, p. 2209—2228. 32. Франц, пат. 2301511, 1976; Chem. Abstr., 1977, v. 85.
33. Makosza M., Jagusztyn-Gror.howska M., Ludwikow M. e.a. — Tetrahedron, 1974,
v. 30, Ns 20, p. 3723—3735. 34. Dockx A —Synthesis, 1973, Ns 8, p. 441—456;
Dehmlow E. V. — Angew. Chem. intern. Ed., 1974, v. 13, Ns 3, p. 170—179. 35. Ma-
коша M.—Успехи химии, 1977, т. 46, вып. 12, с. 2174—2202. 36. Kornblum N.,
Cheng L., Kerber R. С. e.a. — J. Org. Chem., 1976, v. 41, № 9 ,p. 1560—1564.
37. Rossi R. A., Bunnett J. F. — J. Amer. Chem. Soc., 1972, v. 94, Ns 2, p. 683—
684; J. Org. Chem., 1973, v. 38, № 17, p. 3020—3025; Rossi R. A., de Rossi R. H„
Lopez A. F. — J. Amer. Chem. Soc., 1976. v. 98, № 5, p. 1252—1257. 38. Bun-
nett J. F., Gloor B. F. — J. Org. Chem., 1973, v. 38, № 24, p. 4156—4163. 39. Bun-
nett J. F., Sundberg J. E. — Ibid., 1976, v. 41, Ns 10, p. 1702—1706; Komin A. P.,
Wolfe J. F. — Ibid, 1977, v. 42, Ns 14, p. 2481—2486. 40. Argabright R. A., Hof-
mann J. E., Schriesheim A. — Ibid, 1965, v. 30, Ns 9, p. 3233—3235.
41. Russel G. A., Weiner S. A. — Ibid, 1966, v. 31, Ns 1, p. 248—251. 42. No-
zaki N., Yamamoto У, Nisimura T. — Tetrahedron Letters, 1968, № 44, p. 4625—
4626. 43. Metzger H., Konig H., Seelert K. — Ibid, 1964, Ns 15, p. 867—868.
44. Traynelis V. J., McSweeney J. V. — J. Org. Chem, 1966, v. 31, № 1, p. 243—
247. 45. Okamoto T., Takayama H. — Chem. Pharm. Bull. (Tokyo), 1963, v. 11,
p. 514—517. 46. Хамана M. — Химия гетероцикл, соед, 1973, Ns 9, с. 1155—1171.
47. Truce W. E., Kreider E. M., Brand IP. W. — In: Organic Reactions. V. 18,
N. Y, Wiley, 1970, p. 99—215. 48. Pine S. H. — Ibid, p. 403—464. 49. Gas-
sman P. G., Gruetzmacher G. D. — J. Amer. Chem. Soc, 1973, v. 95, № 2,
p. 588—589; 1974, v. 96, № 17, p. 5487—5495. 50. Claus P. R., Schwarz H. A.,
358
Rieder W. e.a.— Phosphorus and Sulfur, 1976, v. 1, № 1, p. 11—18; Gassman P.G.,
Balchunis R. J. — Tetrahedron Letters, 1977, № 26, p. 2235—2238,
51. Gassman P. G., Huang С. T. — Ibid., 1973, № 13, p. 4453—4455. 52. Po-
щаев M., Отрощенко О. С., Садыков А. С. — Успехи химии, 1972, т. 41, вып 12,
с. 2198—2223. 53. Fanta Р. Е. — Synthesis, 1974, № 1, р 9—21 54. Пат. ФРГ
2200127, 1973; Chem. Abstr., 1973, v. 79, 106144; 2250107, 1973; Chem. Abstr.,
1973, v. 79, 20312. 55. House H. O., Don Koepsell G., Campbell W. J.—J. Org.
Chem,, 1972, v. 37, № 7, p. 1003—1011. 56. Eff enberger F., Nagel f\., Agster W.—
Angew. Chem., 1971, Bd. 83, № 16, S. 619—620. 57. Kuser P., Inderbitzin M,
Brauchli J. e.a. — Helv. chim. acta, 1971, Bd. 54, № 4, S. 980—995. 58. Clark M.C.,
Marley T. J., Lowe P. A.—J. Appl. Chem. Biotechnol., 1974, v. 24, № 6, p 343—
348; Adam. L-М., Hindermann P., Winkler T. — Helv. chim. acta, 1976, Bd. 59, № 8,
S. 2999—3012. 59. Шейнкман A. Ал—Химия гетероцикл, соед., 1974, № 1, p. 3—
18. 60. Чупахин О. Н., Постовский И. Я. — Успехи химии, 1976, т. 45, вып. 5,
с. 908—937.
61. Постовский И. Я., Чупахин О. Н., Пиличева Т. Л. и др. — ДАН СССР,
1973, т. 212, № 5, с. 1125—1127; Е. О. Сидоров, Чупахин О. Н. — ЖОрХ, 1978,
т. 14, вып. 1, с. 134—140.
Сканировал: Neptunyi
(Магнитогорск)
Все замечания и пожелания присылать по
адресам:
rhenium@list.ru
nept2006@yandex.ru
Буду рад также высылаемым книгам от вас!
Часть 4
РЕАКЦИИ АРОМАТИЧЕСКОГО
СВОБОДНОРАДИКАЛЬНОГО ЗАМЕЩЕНИЯ
Глава 12
РЕАКЦИИ С УГЛЕРОДНЫМИ РАДИКАЛАМИ
12.1. ВВЕДЕНИЕ АРИЛЬНЫХ ГРУПП
Арилирование ароматических соединений нейтральными
арильными радикалами представляет собой наиболее полно изу-
ченную реакцию свободнорадикального ароматического замеще-
ния. Как уже отмечалось (см. 2.7.3), такие реакции протекают по
двухстадийному механизму с промежуточным образованием ради-
кального a-комплекса и его последующей ароматизацией и на-
правляются независимо от характера заместителя в ароматическом
кольце субстрата в орто- и пара-положения.
Источниками арильных радикалов при свободнорадикальном
замещении служат диазосоединения и родственные им произ-
водные (ТУ-ацетил-ТУ-нитрозоариламины, триазены, арилазоалка-
ны) или диароилперекиси [1].
Арилирование диазосоединениями (реакция Гомберга — Бах-
мана) [2] проводят, добавляя раствор основания NaOH или
CHsCOONa в гетерогенную смесь водного раствора соли диазония
и жидкого арилируемого соединения. Так, постепенным добавле-
нием раствора NaOH в смесь раствора хлорида п-бромбензолдиа-
зония с бензолом при 5 °C получают 4-бромбифенил; выход 34—
35% [3]. Гомогенности реакционной смеси и повышения выхода
можно достигнуть диазотированием ароматического амина алкил-
нитритом непосредственно в среде арилируемого соединения.
В частности, нагревание н-хлоранилина с пентил нитритом в бензо-
ле приводит к 4-хлорбифенилу; выход 73% [4]. Иногда соль диа-
зония обработкой алифатическим амином переводят в триазен,
который затем разлагают в ароматическом углеводороде. Триазен
из диазотированного лг-нитроаннлина и диметиламина при разло-
жении в кипящем бензоле в присутствии п-толуолсульфокислоты
превращается в 3-нитробифенил с выходом около 40% [5].
При арилировании ?У-ацетйл-?У-нитрозоаминами ацетилирован-
ный амин jV-нитрозируют в уксусной кислоте или уксусном ангид-
риде, выделяют нитрозопроизводное разбавлением водой и обра-
батывают жидким ароматическим соединением. Из 3-нитро-?У-нит-
360
розоацетанилида и бензола этим методом получают 3-нитробифе-
нил с выходом 56—60%, из А^'-диа цетил-jV,Л'7-динитро-
зо-/г-фенилендиамина и бензола — /г-терфенил с выходом 69% [2].
Способ арилирования в полярных апротонных растворителях
состоит в обработке раствора борфторида диазония и арилируемо-
го субстрата нуклеофилом — пиридином или нитритом натрия. Так,
при внесении в раствор борфторида диазония и м-динитробензола
(1) в диметилсульфоксиде сухого NaNO2 при 10 °C получают смесь
2,4- (2) и 2,6-динитробифенила (3) с выходом 23% и 47% соответ-
ственно [6]:
В некоторых случаях термическое разложение борфторидов
диазония приводит к арилированию в отсутствие оснований, при-
чем направление реакции зависит от строения диазотируемого
амина. Например, разложение борфторида 9,10-антрахинон-1-диа-
зония в кипящем о-дихлорбензоле с выходом 57% дает
1-фтор-9,10-антрахинон (4) по реакции Шимана (см. 10.3), а раз-
ложение борфторидов 4-хлор- й 4-нитро-9,10-антрахинон-1-диазо-
ния — соответствующие 1-(о-дихлорфенил)-9,10-антрахиноны (5) с
выходом 61 % и 43% [7]:
О F
(5)
361
Арильные радикалы могут быть генерированы также одноэлек-
тронным восстановлением катионов арендиазония металлами
(цинком, медью) и электролитически:
ArNj ----► ArNj* —► Ar* + N2
или термическим разложением арилазотрифенилметанов при
80°С:
ArN=NC(C6H5)3 —► Аг • + N2 + (С6Н5)3С •
Механизм образования арильных радикалов при взаимодей-
ствии диазоний-катионов с О-нуклеофилами (ОН', СНзСОО ,
NO2 и др.) заключается, очевидно, в промежуточном образовании
арендиазооксидов (окисей), которые затем гомолитически распа-
даются на радикалы ArN2O* и ArN2*; последний из этих радикалов
элиминирует азот, давая арил-радикал, а первый выступает в
качестве окислителя при отрыве атома водорода от радикального
о-комплекса [8]:
+ RO” + RO" + ArN2
ArNj ------> ArN=NOR --------> ArN=NO" ------->
.—> ArN2O*
—> ArN=N—О—N=NAr —
1—> ArN2* —> A1-. + N2
Аналогичная схема предложена для разложения А-ацетил-А-
нитрозоариламинов, которые первоначально перегруппировываются
в диазоацетаты, находящиеся в равновесии с диазоний-катионами:
ArN(NO)COCH3 —> ArN=NOCOCH3 ArN) + СН3СОО'
Альтернативный механизм, включающий гомолитический рас-
пад диазоэфиров
ArN=NOR —> Ar* + N2 + RO*
исключается отсутствием ацетоксилирования радикалами
СНзСОО* при разложении А-нитрозоацетанилидов [1] и отсут-
ствием эффекта химической поляризации ядер (ХПЯ) на продук-
тах превращения нуклеофилов RO~ [9]. Возникновение радикалов
ArN=NO* при разложении А-нитрозоацетанилидов подтвержде-
но идентификацией сигнала ЭПР при проведении реакции в резо-
наторе ЭПР-спектрометра [10].
Арилирование диароилперекисями (пероксидами) проводят на-
греванием последних в среде ароматического соединения. При тер-
молизе диароилперекиси гомолитически распадаются на ароил-
оксирадикалы, которые затем отщепляют диоксид углерчда, пре-
вращаясь в арил-радикалы, атакующие ароматический субстрат:
-со2
(АгСОО)г —> 2А1-СОО • --> 2Аг *
В результате 40-часового кипячения перекиси 3,4-дихлорбензо-
ила в бензоле в присутствия лг-дщщтробензола получают с выхо-
86?
дом 78—81% 3,4дихлорбифенил [11]. В отсутствие нитросоедине-
ний выход не превышает 50%. Разложение диароилперекисей в
среде гексафторбензола с хорошим выходом приводит к замеще-
нию фтора арильной группой [12]. Роль нитросоединений и других
добавок состоит в окислении радикальных о-комплексов, которые в
отсутствие акцепторов водорода димеризуются в тетрагидроква-
терфенилы (см. 2.7.3).
В некоторых случаях димеризация радикальных о-комплексов
может иметь препаративное значение. Например, при реакции
перекиси пентафторбензоила с гексафторбензолом при 200 °C пер-
воначально образующийся о-комплекс (6) изомеризуется в а-ком-
плекс (7), димер которого (8) при дефторировании обработкой
цинковой пылью дает труднодоступный перфтор-лцлг'-кватерфенил
(9) [13]:
При фенилировании пентафторбромбензола и пентафторхлор-
бензола, очевидно, замещаются в основном атомы фтора, а не
брома или хлора, так как в смеси продуктов не обнаружен пен-
тафтордифенил [14]; из этого следует, что ароматизация о-комп-
лексов не является лимитирующей стадией [1]. Хотя такое заклю-
чение справедливо для большинства случаев свободнорадикально-
го арилирования, известны примеры, когда скорость превращения
a-комплекса сказывается на общей скорости реакции [6, 15].
Реакцией внутримолекулярного арилирования является цикли-
зация солей диазония по Пшорру [16, 17]. Синтез, разработанный
первоначально применительно к получению производных фенант-
рена, используется также для замыкания пяти- и шестичленных
циклов при получении производных хризена, фенантридина, флуо-
рена, флуорантена, карбазола и других полициклических систем:
Z-CH—CH, CO=NH, СО, NH, О, S, SO2 и др
863
Синтез заключается в диазотировании амина, содержащего
аминогруппу в орто-положении к мостику Z, который соединяет
арильные кольца, и разложении соли диазония, обычно в присут-
ствии порошка меди. При получении фенантренов предложено
проводить циклизацию, обрабатывая соль диазония, приготовлен-
ную диазотированием изопентилнитритом в ацетоне или диметил-
формамиде, иодидом натрия [18]. При замыкании пятичленных
циклов иногда нагревают соль диазония в кислом растворе без
восстановителя.
Роль меди, иодида натрия или другого восстановителя состоит в
генерировании арильных радикалов путем переноса одного элект-
рона к соли диазония. Свободнорадикальный характер реакции
подтверждается-, малым влиянием заместителей в арилируемом
кольце на выход продуктов циклизации; арилированием бензола
диазотированным 2-амино-транс-стильбеном в условиях, в которых
диазотированный 2-амино-цис-стильбен дает фенантрен; выделе-
нием димерного продукта при реакции соли диазония из
MAf-замещенного о-аминобензанилида (10). Обработка последней
порошком меди или иодидом натрия приводит помимо фенантри-
дона (12) к димеру (14), который при пиролизе почти количе-
ственно переходит в фенантридон (12). Образование димера (14)
можно объяснить только рекомбинацией радикальных спиро-о-
комплексов (13), находящихся в равновесии с арильными радика-
лами (11) [19].
Циклизация с образованием пятичленных колец при нагревании
соли диазония в кислом растворе в отсутствие восстановителя
протекает по гетеролитическому механизму с промежуточным
образованием карбкатионов (ср. 2.7.2, 8.1.4). При термическом
разложении диазотированных 2/-метил-2-бифениламинов в раз-
бавленной H2SO4 арилирование направляется в метильную группу,
приводя к производным флуорена; так образуется, например, три-
864
метилфлуорен (15) с выходом 36% [20]:
Ni СН0 СН2
(15)
К реакциям свободнорадикального арилирования ароматиче-
ских соединений примыкает реакция арилирования ненасыщенных
алифатических соединений солями диазония (реакция Меервейна)
[21,22]. Она протекает с участием тех же атакующих частиц—
арильных радикалов, но по характеру субстрата классифицируется
(см. 2.3) как реакция алифатического свободнорадикального за-
мещения. Арильная группа вступает к атому углерода с кратной
связью, находящемуся в [J-положении к активирующей электроно-
акцепторной группе Z, с замещением атома водорода или присо-
единением аниона соли диазония:
zR/
ArNj СГ + R—сн=с; ---------->- АгС=С—Z или ArCH—СС1—Z
' I II
R' R R'
Источником арильных радикалов является одноэлектронное
восстановление диазоний-катиона, которое, как полагают, протека-
ет через стадию образования комплекса с ионом меди и арилируе-
мым соединением, что сближает эту реакцию по механизму с
реакцией Зандмайера (см. 10.3). Допускают, что при арилировании
хинонов в переносе электронов участвуют семихиноны (см. 16.4.2).
Реакцию обычно проводят, смешивая раствор арилируемого
соединения в воде или органическом растворителе (ацетон, акри-
лонитрил, диметилсульфоксид) с частично нейтрализованным рас-
твором соли арилдиазония и СиС12. При взаимодействии 1,3-бута-
диена с диазотированным /г-нитроанилином в этих условиях
образуется 1 -(тг-нитрофенил) -4-хлор-2-бутен (16), который дегид-
рогалогенируют метанольным раствором КОН, и получают
1-(/г-нитрофенил)-1,3-бутадиен (17) с выходом 57—61% [23].
NT СГ
f J + СН2—СН—-СН=СН2 —->
Ч. CUC12
no2
СН2—СН=СН—СН2С1 сн2=сн—сн=сн2
no2
(16)
no2
(17)
365
Реакцией бисульфата 9,10-антрахинон-1-диазония (18) с
1,1-дихлорэтиленом в присутствии CuCl в метаноле с последующим
алкоголизом при нагревании получают с высоким выходом (до
98%) метиловый эфир 9,10-антрахинон-1-илуксусной кислоты
(19) — промежуточный продукт для производства синего кубового
красителя [24]:
+ СН2=СС12
------------>
CuCl; СН3ОН
Арилирование хинонов можно проводить в отсутствие солей
меди. Так, добавлением солянокислого раствора диазотированного
о-хлоранилина к суспензии 1,4-бензохинона в водном растворе
NaHCO3 при 5—8 °C получают 2-(о-хлорфенил)-1,4-бензохинон с
выходом 90% [21].
12.2. ВВЕДЕНИЕ АЛКИЛЬНЫХ И ДРУГИХ ГРУПП
Реакция свободнорадикального алкилирования по меха-
низму подобна реакции арилирования. Алкил-радикал присоеди-
няется к атому углерода ароматического кольца, после чего обра-
зующийся радикальный о-комплекс может отщеплять атом водо-
рода, диспропорционировать или димеризоваться (см. 2.7.3). Для
того чтобы преобладала реакция замещения, необходимо присут-
ствие радикалов, способных отрывать от сг-комплекса атом водо-
рода. При алкилировании эту роль выполняет часть алкил-радика-
лов или избыток окислителя, применяемого для их генерирования.
Источники алкил-радикалов весьма разнообразны. К важней-
шим из них относятся [25, 26]:
а. Разложение диацилперекисей и тетраацетата свинца:
*/2(RCOO)2 —> RCOO. —> r» + co2
(CH3COO)4Pb —>- 2СН3 • + 2СО2 + (CH3COO)2Pb
б. Разложение диалкилперекисей:
'/2(СН3)3СООС(СНз)з —► (СН3)3СО. —> СН3 • + (СН3)2СО
в. Отрыв атома водорода от алкана другим радикалом. Так
получают, например, циклогексил- и бензил-радикалы при разло-
жении ди-трет-бутилперекиси в среде циклогексана или толуола
соответственно:
RH + 72(СНз)3СООС(СН3)з —> R . + (СНз)зСОН
366
Этим же методом могут быть генерированы гидроксиалкил-
радикалы из спиртов и алкокси- или алкилендиоксирадикалы из
эфиров, например:
+ (СНз)зСООН
. Г- 2 +
+ Fe
+ (СНз)зСОН
г. Разложение алкилгидроперекисей, а-гидроксигидропереки-
сей, а-алкоксигидроперекисей и оксазиридинов при одноэлектрон-
ном восстановлении солями железа (II):
(СН3)3СО • —► СНз • + (СНз)гСО
+ Ре2 +
(СНз)зСООН --------
| +СН3ОН
—> • СН2(СН2)4СООН
0/ОСН3 + ре2+ .----. /ОСНз
( ----> \ А —* • СН2(СН2)4СООСН3
V)OH '--' 'О •
(20)
(21)
д. Окислительное декарбоксилирование кислот тетраацетатом
свинца или персульфатом аммония, катализируемое ионами
серебра:
RCOOH +72S2O82' ---> 2R • + СО2+ Н++SO24-
Ag+
Последний метод найден особенно ценным в препаративных
целях.
Изучение влияния заместителей в бензольном кольце на изо-
мерный состав продуктов алкилирования показывает, что отноше-
ние jn-изомер/о-изомер снижается в порядке: циклогексил > ме-
тил > фенил при наличии в кольце электронодонорных заместите-
лей и в обратном порядке — при наличии электроноакцепторных
заместителей. Следовательно, указанный ряд соответствует умень-
шению нуклеофильности радикалов. Поскольку фенилрадикал сам
является слабым нуклеофилом (см. 2.7.3), нуклеофильность метил-
и, тем более, циклогексил-радикалов должна быть явно выражена.
Действительно, для реакции фенилирования р = 0,05, для цикло-
гексилирования р= 1,10, тогда как для фенилэтинилирования
367
р=—1,56 [1]. Нуклеофильность радикального углеродного цент-
ра зависит от энергии удерживания неспаренного электрона и тем
меньше, чем больше s-характер орбитали, на которой он находится:
sp3(алкил) > sp2 (фенил) > sp (этинил). В соответствии с уве-
личением s-характера орбитали нуклеофильность циклоалкильных
радикалов уменьшается при переходе от циклогексила к цикло-
пропилу, который по нуклеофильности близок фенильному радика-
лу [27].
Явно выраженная нуклеофильность алкильных радикалов об-
условливает пути синтетического использования свободноради-
кального алкилирования [25,28]. Для синтеза карбоциклических
ароматических соединений оно не имеет практического значения,
поскольку приводит к сложным смесям изомеров и уступает дру-
гим методам введения алкильных групп (см. 6.1 и 11.2). Зато в
ряду гетероароматических соединений, обладающих в протониро-
ванной форме высокой электронодефицитностью и высокой реак-
ционной способностью по отношению к нуклеофильным реагентам,
свободнорадикальное алкилирование оказалось ценным синтетиче-
ским методом.
Роль протонирования азотсодержащего гетероцикла может
быть проиллюстрирована на примере метилирования хинолина ди-
трет-бутилперекисью. В отсутствие кислоты метилирование на-
правляется во все возможные положения: 2 (в смеси содержится
10,2% изомера), 4 (24,2%), 5 (16,1%), 8 (30,8%), остальные
(18,7%); в уксусной кислоте преобладают 2- и 4-изомеры; 2
(33,6%), 4 (38,7%), 5(8,8%), 8 (13,2%), остальные (5,7%), а в
присутствии соляной кислоты метилирование направляется исклю-
чительно в 2- и 4-положения: 2 (49,0%), 4 (50,0%), остальные
составляют около 1 % [29].
Высокая селективность наряду с хорошими выходами и просто-
той эксперимента может сделать свободнорадикальное алкилиро-
вание в гетероциклическом ряду таким же важным синтетическим
методом, как электрофильное алкилирование в ряду карбоцикли-
ческих ароматических соединений. В ряду азотсодержащих гете-
роциклов электрофильное алкилирование мало применимо и дол-
жно приводить к совершенно другой ориентации. Свободноради-
кальное алкилирование не сопровождается изомеризацией внутри
алкильных групп даже при введении неопентильного или цикло-
пропильного остатка, как это наблюдается при электрофильном
замещении.
Препаративные возможности метода продемонстрированы син-
тезами производных пиридина, хинолина, изохинолина, хинокса-
лина, акридина и бензотиазола [25,26]. Так, при добавлении к
раствору хинолина в смеси СНзОН и H2SO4 раствора 1-метокси-
циклогексилгидроперекиси (20) и затем FeSO4 при 20—30 °C с
количественным выходом получают смесь, состоящую из 53,8%
2-(5-метоксикарбонилпентил) хинолина (22) и 46,2% 4-изомера
(23). Пр имерно в таком же соотношении количественно образуют-
ся 2- и 4-[5-(метилкарбамоил)пентил] хинолины (24) и (25) при
368
одновременном внесении водного раствора FeSO4 и 2-мс-
тил-3,3-пентаметиленоксазиридина (21) в раствор хинолина в
20%-ной H2SO4.
СН2(СН2)4СООСН3
(24)
(25)
Обработка раствора 2-метилхинолина и пропионовой кислоты в
водной серной кислоте персульфатом аммония в присутствии
А§ЫОзпри70оС количественно приводит к 2-метил-4-этилхинолину
(26). При кипячении 2-метилхинолина в водном метаноле с
(NH4)2S20s и H2SO4 с выходом 86% выделен 4-гидроксиме-
тил-2-метилхинолин (27), а при нагревании в 50%-ной H2SO4 с
(NH4)2S2O8 и диоксаном — 4-диоксанил-2-метилхинолин (28) с
выходом 74%.
Из примеров введения алкильных групп в карбоциклические
соединения с помощью свободнорадикального замещения при син-
тетических исследованиях следует упомянуть метилирование хино-
нов и ароматических нитропроизводных тетраацетатом свинца в
уксусной кислоте [30].
869
Помимо алкильных и замещенных алкильных групп свободно-
радикальное замещение позволяет ввести в ароматические азотсо-
держащие гетероциклы ацильные, карбамоильные [25,26] и кар-
боксильные [31] группы. Ацильные радикалы получают отрывом
атома водорода от альдегидов при действии перекисей:
+ Fe2+
RCHO + (СНз)зСООН ----->• RCO . + (СН3)3СОН
карбамоильные — окислением формамида или диметилформами-
да:
—н»
R2NCHO ----> R2NCO • R = Н, СН3
карбоксильные — восстановительным разложением эфиров а-гид-
рокси-а-гидропероксикарбоновых кислот, получаемых действием
перекиси водорода на а-оксокарбоновые кислоты:
? „ НОЧ /ООН 2+
II + Н2О2 \/ + Fe
R'— С—COOR ------► R'—С—COOR -------->
н+
НО\ /О •
—> R—С—COOR —> R'COOH + • COOR
При одновременном добавлении раствора FeSO4 и трет-бутил-
гидроперекиси в смесь альдегида, H2SO4 и бензотиазола, хинолина
или хиноксалина с выходом 45—80% получают соответствующие
2-ацильные производные этих гетероциклов, например (29). При
внесении трет-бутилгидроперекиси и FeSCh в раствор хиноксалина
и H2SO4 в формамиде с выходом 71—82% выделяют 2-хиноксалин-
карбоксамид (30). В этих же условиях из бензотиазола получается
2-бензотиазолкарбоксамид. При взаимодействии эфира а-гидро-
кси-а-гидропероксикарбоновой кислоты, приготовленной из этило-
вого эфира пировиноградной кислоты, с хиноксалином в H2SO4 в
присутствии FeSO4 с выходом 70% образуется этиловый эфир
2-хиноксалинкарбоновой кислоты (31).
(29)
(31)
|.conh2
(30)
Можно полагать, что введение различных групп действием на
протонированные гетероароматические основания углеродных ра-
дикалов получит дальнейшее развитие.
370
ЛИТЕРАТУРА
1. Нонхибел Дж., Уолтон Дж. Химия свободных радикалов. Пер. с англ./
Под ред. И. П. Белецкой. М., Мир, 1977, с. 441—493. 2. Бахман В. Е., Гоф-
манн Р. А.—В кн.: Органические реакции. Сб. 2. Пер. с англ. М., ИЛ, 1950,
с. 244—284. 3. Гомберг М., Бахман В. Е. — В кн.: Синтезы органических препа-
ратов. Сб. 1. Пер. с англ. М., ИЛ, 1949, с. 106—108. 4. Cadogan J. I. G. — J.
Chem. Soc., 1962, № 10, р. 4257—4258. 5. Кэзлоу К., Сэммерс Р. — В кн.: Синтезы
органических препаратов. Сб. 5. Пер. с англ. М., ИЛ, 1954, с. 46—49. 6. Ko-
bayashi М., Minato Н., Kobori N. e.a. — Bull. Chem. Soc. Japan, 1970, v. 43, № 4,
p. 1131—1135. 7. Valkanas G., Hopff H.— J. Org. Chem., 1962, v. 27, № 10,
p. 2431—3682; 1964, v. 29, № 2, p. 489—490. 8. Rilchardt C., Merz E. — Tetra-
hedron Letters, 1964, № 36, p. 2431—2436; Rilchardt C., Freudenberg B. — Ibid.,
1964, № 48, p. 3623—3628. 9. Душкин А. В., Лешина T. В., Шуваева О. И.
и dp.--ЖОрХ, 1977, т. 13, вып. 6, с. 1231—1236. 10. Cadogan J. I. G.—Accounts
Chem Res., 1974, v. 4, № 5, p. 186—189.
11. Hey D. H., Perkins M. J. — In: Organic Syntheses. Coll. v. 5, N. Y.,
Wiley, 1973, p. 51—54. 12. Bolton R., Williams G. H. — In: Advances in Free-
Radical Chemistry. V. 5. London, Elek Science, 1975, p. 1—25. 13. Власова Л. В.,
Кобрина Л. С., Якобсон Г. Г. — ЖОрХ, 1974, т. 10, вып. 4, с. 787—791. 14. Old-
ham Р. Н., Williams G. Н., Wilson В. А. — J. Chem. Soc. (В), 1970, № 7, р. 1346—
1349. 15. Kamigata N., Minato Н.. Kobayashi М. — Bull. Chem. Soc. Japan, 1972,
v. 45, № 7, p. 2042—2046. 16. Де-Тар Д. Л. — В кн.: Органические реакции.
Сб. 9. Пер. с англ. М., ИЛ, 1959, с. 529—600. 17. Abramovitch R. А. — In: Advan-
ces in Free-Radical Chemistry. V. 2. London — N. Y., Logos Press-Academic Press,
1967, p. 87—138. 18. Chauncy B., Gellert E. — Austr. J. Chem. 1969, v. 22, № 5,
p. 993—995; 1970, v. 23, № 12, p. 2503—2516. 19. Hey D. H., Jones G. H., Per-
kins M. J. — Chem. Comm., 1970, № 21, p. 1438—1439. 20. Puskas I., Fields E. K.—
J. Org. Chem., 1968, v. 33, № 11, p. 4237—4242.
21. Рондестведт X. C. — В кн.: Органические реакции. Сб. 11. Пер. с англ.
М., Мир, 1965, с. 199—266. 22. Rondestvedt С. S. — In: Organic Reactions. V. 24.
N. Y., Wiley, 1976, p. 225—259. 23. Ponn Г., Койнер Ю. — В кн.: Синтезы орга-
нических препаратов. Сб. 4. Пер. с англ. М., ИЛ, 1953, с. 380—382. 24. Пат.
ФРГ 2323543 (1973); Chem. Abstr., 1974, v. 80, 61068. 25. Minisci F. — Synthesis,
1973, № 1, p. 1—24; Topics Curr. Chem. 1976, v. 62, p. 1—48. 26. Minisci F„
Porta O. — In: Advances in Heterocyclic Chemistry. V. 16. N. Y. — London, Aca-
demic Press, 1974, p. 123—180. 27. Shono T., Nishiguchi I. — Tetrahedron, 1974,
v. 30, № 14, p. 2183—2190. 28. Citterio A., Minisci F., Porta O. e.a. — J. Amer.
Chem. Soc., 1977, v. 99, № 24, p. 7960—7968. 29. Bass К. C., Nababsing P. — J.
Chem. Soc. (C), 1970, № 16, p. 2169—2172. 30. Физер Л., Физер M. Реагенты для
органического синтеза. Т. 3. Пер. с англ. М., Мир, 1970, с. 235—236.
31. Bernardi R., Caronna Т., Galli R. e.a.—Tetrahedron Letters, 1973, Ns 9,
p. 645—648.
Глава 13
РЕАКЦИИ С ГЕТЕРОАТОМНЫМИ РАДИКАЛАМИ
13.1. ВВЕДЕНИЕ ГИДРОКСИЛЬНОЙ
И АЦИЛОКСИЛЬНЫХ ГРУПП
Свободные радикалы, имеющие неспаренный электрон
на атоме кислорода, получают, исходя из перекисных соединений.
Источником гидроксильных радикалов служит одноэлектронное
восстановление перекиси водорода ионами Fe2+ или других метал-
лов [1];
H2O2 + M«+ —> НО-4-ОН’4-M(rt+1)+ Mra+ = Fe2+, Ti3+, Cu+, Co2+
371
источником ацилоксильных радикалов — разложение диацилпере-
кисей и тетраацилоатов свинца:
(RCOO)2 —> 2RCOO.
(RCOO)4Pb —> RCOO • + (RCOO)3Pb •
При действии реактива Фентона (Н2О2 + FeSCM бензол пре-
вращается в фенол и бифенил по схеме [2]:
(4)
(2)
Образование гидроксициклогексадиенильных радикалов типа
(1) подтверждено регистрацией спектров ЭПР [3]. При окислении
ионами Fe3+ с последующей отдачей протона радикал (1) пере-
ходит в фенол, а при димеризации — в 1,Г,4,4'-тетрагидробифе-
нил-4,4'-диол (2), который дегидратируется в бифенил. Выход
фенола повышается с увеличением концентрации ионов Fe3+ или
при введении других окислителей (О2, Си2+) и снижается при до-
бавлении фторидов, которые связывают ионы Fe3+, препятствуя
окислению [4].
Гидроксильные радикалы обладают электрофильными свой-
ствами, как это следует из отрицательной величины константы
р = —0,41 в уравнении Гаммета (см. 2.7.3). При гидроксилирова-
нии толуола реактивом Фентона образуется смесь о-, м- и
n-крезолов в соотношении 71 : 5 : 24, а также 2,2'-диметилбифенил
и продукты реакции по метильной группе (дибензил и др.) [5].
К последним приводит фрагментация циклогексадиенильных ра-
дикалов, содержание которых в реакционной смеси возрастает с
увеличением кислотности [4]:
В случае нитробензола доля лега-гидроксилирования увеличи-
вается — соотношение орто!мета1пара составляет 24:30:46 [3].
372
При гидроксилировании нафталина 1- и 2-нафтол образуются в
соотношении примерно 3:1 [6]. Окисление ароматических соеди-
нений перекисью водорода в присутствии FeSO4 предложено' для
получения гидроксипроизводных [7,8]. Большее техническое зна-
чение особенно для совместного получения пирокатехина и гидро-
хинона из фенола [9], имеет кислотно-катализируемое окисление
перекисью водорода, протекающее в отличие от рассматриваемых
реакций по ионному механизму (см. ссылку [1] к гл. 8).
Другой путь свободнорадикального введения гидроксильной
группы заключается в генерировании арильных радикалов из солей
диазония (см. 12.1) и окислении их ионами Си2+ [10]:
Си+ Си2+
ArNj ---> Аг- —АгОН
H2V
В отличие от превращения солей диазония в гидроксисоедине-
ния по ионному механизму (см. 8.1.4), требующего длительного
нагревания в сильнокислой среде, данная реакция протекает в
течение нескольких минут при комнатной температуре с выходом
80—95%. Недостаток метода—-необходимость применения боль-
ших (15—50-кратных) избытков окислителя Cu(NO3)2 и больших
объемов растворов; достоинства — мягкие условия и возможность
получать гидроксисоединения из аминов, которые иначе не могут
быть переведены в фенолы через соли диазония с удовлетвори-
тельными результатами. Например, разложение З-нитро-2-толуол-
диазония (4) в кипящей H2SO4 дает 46% 7-нитро-Ш-индазола (3)
и только 10% 6-нитро-о-крезола (5), тогда как фторборат диазония
(4) в нейтральном водном растворе при обработке Cu(NO3)2 и
Си2О при комнатной температуре превращается в 6-нитро-о-крезол
(5) с выходом 80% (74% в расчете на амин). Соль диазония из
2-аминобензофенона (7) при нагревании в H2SO4 переходит в
9-флуоренон (6) с выходом 65—68%, а при действии соединений
меди — в 2-гидроксибензофенон (8) с выходом 88% [10].
373
Введение ацилоксигрупп под действием диацилперекисей [11]
протекает, как и другие реакции свободнорадикального замеще-
ния, с присоединением на первой стадии радикала к ароматиче-
скому кольцу и последующим окислением а-комплекса:
Р*+СОг
OCOR
Конкурирующая реакция представляет собой диссоциацию
ацилоксильных радикалов на алкильные или арильные и диоксид
углерода. Сдвиг в сторону ацилоксилирования зависит от легкости
присоединения ацилоксильного радикала и скорости окисления
о-комплекса.
При разложении диароилперекисей в бензоле главным направ-
лением является арилирование (см. 12.1), но при взаимодействии
перекиси бензоила с нафталином с выходом 55% образуется смесь
1- и 2-нафтилбензоатов в соотношении примерно 7:3 [11]. Осо-
бенно высока степень бензоилоксилирования в случае метоксипро-
изводных бензола. При реакции анизола с перекисью бензоила
образуется смесь, содержащая 82% о- и 18% п-метоксифенилбен-
зоата без примеси лг-изомера. При реакции 4-метиланизола (9а) и
1,4-диметоксибензола (96) выделяют 5-метил-2-метоксифенилбен-
зоат (Юа) и 2,5-диметоксифенилбензоат (106) соответственно,
причем образования биарилов совершенно не наблюдается [12].
ОСН3
]f^| + (С6Н6СОО)2
Г
ОСНз
ОСОС6Н5
R
a) R = СНз
б) R = ОСНз
(9) (10)
Особенности ацилоксилирования метоксипроизводных связыва-
ют с механизмом, включающим стадию одноэлектронного перено-
са, предшествующую образованию радикального о-комплекса
(см. 2.7.3). По такому же пути, возможно, протекает ацилирование
анизола тетраацетатом свинца в присутствии инициаторов ради-
кальных реакций. Реакционная способность анизола в реакции
ацетоксилирования в 200 раз больше, чем у бензола, тогда как в
реакции метилирования — всего в 1,7 раза [13]. Это различие
объясняют тем, что при ацетоксилировании атакуется катион-ра-
374
дикал анизола, а при метилировании (см. 12.2) — нейтральная мо-
лекула:
Реакция ацилоксилирования катализируется солями меди(II),
способствующими окислению радикального a-комплекса. Так, то-
луол при действии на него диизопропилпероксидикарбоната
(СНз) 2СНОСО—00 — СООСН(СН3)2 в присутствии СиС12 в аце-
тонитриле при 60 °C с выходом 79% переходит в смесь, содер-
жащую 57% изопропил-о-толил-, 15% изопропил-м-толил- и 28%
изопропил-п-толилкарбоната СН3С6Н4ОСООСН(СНз)2; в тех же
условиях нафталин с выходом 89% дает смесь 92% изопропил-1-
нафтил- и 8% изопропил-2-нафтилкарбоната [14].
13.2. ВВЕДЕНИЕ АМИНОГРУПП
В качестве источника /V-радикалов, применяемых для
введения в ароматическое кольцо аминогрупп, служит одноэлект-
ронное восстановление солями металлов /V-хлораминов или гидро-
ксиламина и гидроксиламин-О-сульфокислоты [15, 16]. Восстанов-
ление в зависимости от кислотности среды может приводить к
нейтральным аминным радикалам или к их протонированной фор-
ме— аммониевым катион-радикалам. Наибольший интерес в син-
тетическом плане представляет замещение при действии аммоние-
вых катион-радикалов, генерируемых из алкилхлораминов в силь-
нокислой среде:
R2NHCI + Мп+ —> R2NH • + М<п+1)++ СГ Mn+ = Fe2+, Ti3+, Ci?, Сг2+
//-Хлорамины, получаемые хлорированием соответствующих
аминов [17], легкодоступны и достаточно устойчивы. Их одноэлек-
тронное восстановление и взаимодействие радикалов с ароматиче-
ским субстратом протекает с высокой скоростью при комнат-
ной и более низких температурах. Протонирование радикала и об-
разующегося аминозамещенного определяет высокую селектив-
ность, исключая возможность вступления второй аминогруппы и
375
обусловливая при наличии сильных электронодоноров только орто-,
пара-ориентацию. Предполагают два пути ароматизации радикаль-
ного ц-комплекса — под действием хлорамина и под действием
иона металла:
Второй путь вероятнее при наличии в ароматическом кольце
электроноакцепторных заместителей, при более высоком окисли-
тельно-восстановительном потенциале пары М2+ ( > и более
— е~
низкой концентрации хлорамина.
Реакцию обычно проводят, добавляя кристаллический
FeSO4 или соль другого металла в смесь ароматического соедине-
ния, хлорамина и H2SO4 при комнатной температуре [16]. В этих
условиях при действии эквимолярного количества М-хлордимети-
ламина в смеси серной и уксусной кислоты (85 : 15) из бензола с
выходом 79% получают А(,Л^-диметила нилин (100% в расчете на
прореагировавший бензол), из нафталина — с выходом 68%
МА^-диметил-1-нафтиламин, содержащий незначительную примесь
2-изомера. При действии /V-хлормоноалкиламинов аминозамещен-
ные образуются с более низкими выходами. Диалкиламинирование
бензола и нафталина можно осуществить также при термическом
разложении А(-хлордиалкиламинов в H2SO4 [18,19], но при этом
образуется значительное количество хлорпроизводных. В случае
соединений, имеющих сильные электронодонорные заместители
термическое разложение неприемлемо, так как приводит только к
хлорированию и сульфированию.
Фенол, анизол, Af-ацетиланилин удается перевести в аминоза-
мещенные с выходом 65—98% в присутствии FeSO4 при комнатной
температуре. Во всех этих случаях преобладает пара-замещение,
хотя отношение о-изомер/п-изомер зависит от условий, возрастая
с увеличением кислотности среды. Сильные электроноакцепторные
заместители NO2, CN, NR3, СООН, SO3H полностью дезактивируют
ароматическое кольцо, препятствуя аминированию. В бицикличе-
ских соединениях, содержащих электроноакцепторную группу в
одном из колец, аминирование направляется в незамещенное
кольцо. Так 4-нитробифенил, 4-бифенилиламин и 4-бифенилсуль-
фокислота при взаимодействии с AZ-хлордиметнламином с выходом
376
86—90% превращаются в соответствующие 4'-диметиламинопроиз-
водные, а 1-хлорнафталин — с выходом 90% в смесь, состоящую из
76% АААСдиметил-5-хлор-1-нафтиламина и 21% ЛГ.ЛСдиме-
тил-8-хлор-1-нафтиламина.
При реакции алкилбензолов наряду с аминированием в кольцо
происходит хлорирование в a-положение боковой цепи, доля кото-
рого возрастает с увеличением алкильного остатка при атоме азота
хлорамина. Это следствие пространственных препятствий амини-
рованию и уменьшения концентрации серной кислоты. При обра-
ботке AZ-хлордиметиламином и FeSO4 в 100%-ной H2SO4 толуол на
95,4% аминируется в кольцо и только на 4,6% хлорируется в
метильную группу, тогда как в 100%-ной СН3СООН образуется
только бензилхлорид. Получаемая смесь А?,АСдиметилтолуидинов
содержит около 9% о-, 53% м- и 38% «-изомера. Подобная ориен-
тация сильно отличается от наблюдаемой в реакциях электро-
фильного замещения, что не согласуется с представлением об
электрофильном характере атакующего аммониевого катион-ради-
кала. Для объяснения преимущественного лгета-замещения выдви-
нута гипотеза о нечувствительности свободнорадикального амини-
рования к эффекту гиперконъюгации алкильных групп, обуслов-
ливающему их орто-, «ара-ориентирующее влияние [20].
Внутримолекулярное аминирование в jV-хлор(3-фенилпропил)-
аминах (11) позволяет гладко получать тетрагидрохинолины
(12), но замыкание пятичленного цикла в АСхлор(2-фенилэтил)-
аминах (13) с превращением в индолины (14) идет с низким вы-
ходом.
(Н) (12)
(13) (14)
В то время как аминирование ЛСхлораминами наиболее при-
годно для введения диалкиламиногруппы, применение гидроксил-
амина и гидроксиламин-О-сульфокислоты позволяет ввести сво-
бодную аминогруппу. Соединения, содержащие электронодонорные
группы, при действии этих реагентов в присутствии катионов Fe2+
или TF+ в мягких условиях количественно превращаются в произ-
водные анилина, но выход в расчете на аминирующий агент не
превышает 40%. Ориентация при аминировании гидроксиламином
и гидроксиламин-(9-сульфокислотой существенно различается. Так,
обработка 4-метиланизола (9а) гидроксиламином в метаноле при
комнатной температуре в присутствии FeSO4 дает 88% 2-аминоза-
мещенного (15) и 12% 3-аминозамещенного (16), а обработка
гидроксиламин-О-сульфокислотой соответственно 48% и 52%.
377
(Яа) (15) (16)
При реакции анизола с гидроксиламином в присутствии
TiCl3 образуется 63% о- и 37% n-анизидина, а при реакции с
гидроксиламин-О-сульфокислотой соответственно 4 и 96%. Пред-
полагают, что причина разной ориентации может состоять в разной
природе атакующего радикала, который в случае гидроксиламина
представляет собой нейтральную, а в случае гидроксила-
мин-О-сульфокислоты— протонированную частицу [16]:
NH2OH-|-M'!+ —> • NH2 + MOHrt+
NH3OSOJ + Mrt+ —> • NH3 + M(rt+’>+ + SO]'
Mrt+ = Fe2+, Ti3+
Гидроксиламин-О-сульфокислота, существующая в виде бипо-
лярного иона, может выступать по отношению к сильным нуклео-
филам в качестве электрофильного аминирующего агента [21] и, в
частности, широко применяется для электрофильного iV-аминиро-
вания ароматических азотсодержащих гетероциклов.
При реакциях слабо активированных ароматических соедине-
ний с гидроксиламином и гидроксиламин-О-сульфокислотой наря-
ду с аминозамещенным образуются продукты димеризации ради-
кальных a-комплексов. При взаимодействии мезитилена (17) с
гидроксиламином и TiCU в атмосфере азота выделено 50% мези-
дина (19) и 34% 1,1',3,3',5,5'-гексаметил-1,Г,4,4'-тетрагидробифе-
нил-4,4'-диамина (20). В присутствии кислорода димер (20) не
образуется в результате участия кислорода в окислении а-комп-
лекса (18).
378
К такого же типа процессам относится, вероятно, давно извест-
ное аминирование гидроксиламином в концентрированной
H2SO4 при нагревании выше 100 °C (реакция Турского) [22]. Ис-
следование продуктов, полученных при аминировании 9,10-антра-
хинона и 9,10-антрахинон-2-сульфокислоты в H2SO4 при 130 °C в
присутствии FeSCh, обнаружило наряду с 1- и 2-амино-9,10-антра-
хинонами димерные амины типа (21) с сочленением ядер по 1,1'-,
1,2'-, 2,2'- и 2,4'-положениям [23].
ЛИТЕРАТУРА
1. Денисов Е. Т., Метелица Д. И. — Успехи химии, 1968, т. 37, вып. 9,
с 1547—1566. 2. Smith J. R. L., Norman R. О. С.—J. Chem. Soc., 1963, № 5,
р. 2897—2905. 3. Jef coate C. R. E., Norman R. О. C. — J. Chem. Soc. (B), 1968,
№ 1, p. 48—53. 4. Jef coate C. R. E., Smith J. R. L., Norman R. О. C.— Ibid., 1969,
№ 8, p. 1013—1018. 5. Walling C., Johnson R. A. — J. Amer. Chem. Soc., 1975,
v. 97 № 2, p. 363—367. 6. Boyland E.y Sims P. — J. Chem. Soc., 1953, № 10,
p 2966—2972. 7. Нидерл. пат. 6613970, 1967; Chem. Abstr., 1968, v. 68, 12689.
8 Пат. ФРГ 2162589, 1972; Chem. Abstr., 1972, v. 77, 114026. 9. Пат. ФРГ
2138735, 1973; Chem. Abstr., 1973, v. 78, 135870. 10. Cohen T., Dietz A. G„
Miser J. R. — J. Org. Chem., 1977, v. 42, № 12, p. 2053—2058.
11. Rawlinson D. J., Sosnowsky G. — Synthesis, 1972, № 1, p. 1. 12. Da-
vies D. I., Hey D. H., Williams G. H.— J. Chem, Soc. (C), 1970, № 19, p. 2653—
2660. 13. 'McClelland R. A., Norman R. О. C., Thomas С. B. — J. Chem. Soc. Per-
kin 1, 1972, № 4, p. 562—570. 14. Kovacic P., Reid C. G., Kurz M. E. — J. Org.
Chem., 1969, v. 34, № 11, p. 3302—3308. 15. Sosnovsky G., Rawlinson D. J.—
In: Advances in Free-Radical Chemistry. V. 4. London, Logos Press, 1972, p. 203—
284. 16. Minisci F. — Synthesis, 1973, № 1, p. 1—24; Topics Curr. Chem., 1976,
v. 62 p. 1—48. 17. Kovacic P.y Lowery M. L., Field K. W. — Chem. Revs., 1970,
v. 70’ № 6, p. 639—665. 18. Bock H., Kompa K-L. — Chem. Ber., 1966, Bd. 99,
№ 4, S. 1347—1356. 19. Нидерл. пат. 6614947, 1966; Chem. Abstr., 1968, v. 68,
68642. 20. Clerici A., Menisci F., Perchinunno M. e.a.—J. Chem. Soc. Perkin 2,
1974, № 4, p. 416—419.
21. Шмитц Э. — Успехи химии, 1976, т. 45, вып. 1, с. 54—70. 22. Препара-
тивная органическая химия. Пер. с англ, под ред. Н. С. Вульфсона. М., Госхим-
издат, 1959, с. 283—286. 23. Yoshida Z., Matumoto Т., Oda Р. — Kogyo Kagaku
Zasshi, 1964, v. 67, № 1, p. 67—69, 70—72; Chem. Abstr., 1964, v. 61, 1810.
Часть 5
РЕАКЦИИ В ЗАМЕЩАЮЩИХ ГРУППАХ
АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Глава 14
ЗАМЕЩЕНИЕ ВОДОРОДА В БОКОВЫХ ЦЕПЯХ
И В ФУНКЦИОНАЛЬНЫХ ГРУППАХ
В химии промежуточных продуктов большое значение
имеют реакции, при которых замещаются атомы водорода боковых
цепей и других заместителей, например групп СН3, NH2, ОН, SH и
других. Механизмы этих реакций иные, чем у рассмотренных ранее
реакций ароматического замещения.
Вместе с тем, эти реакции отличаются от реакций алифатиче-
ских соединений, потому что благодаря сопряжению боковые цепи,
так же как и другие замещающие группы, передают часть своей
электронной плотности в ароматическое кольцо, что увеличивает их
кислотность, и они приобретают специфический характер. По той
же причине промежуточные частицы, образующиеся в ходе таких
реакций — радикалы, катионы или анионы — отличаются повы-
шенной стабильностью.
14.1. ЗАМЕЩЕНИЕ В АЛКИЛЬНЫХ ГРУППАХ
Замещение в алкильных группах может проходить по
гомолитическому или по ионному механизму.
В первом случае начальным этапом реакции является отрыв от
алкильной группы атома водорода, осуществляемый реагентом,
содержащим неспаренный электрон; при этом образуется радикал
бензильного типа, стабильность которого определяется степенью
делокализации одиночного электрона в ароматическом кольце:
CeH6CH3 + R« C6H5CH2 + RH
Именно поэтому при наличии в боковой цепи нескольких атомов
углерода водород отрывается в первую очередь в а-положении,
т. е. от углерода, непосредственно связанного с ароматическим
кольцом.
Обычно стадия образования радикала лимитирует общую ско-
рость реакции. Дальнейшие его превращения в зависимости от
380
условий быстро приводят к образованию продуктов замещения или
димеризации:
СбНзСНг + R • —> C6H5CH2R
2С6Н5СН2 —> С6Н5СН2СН2С6Н5
Реакции электрофильного замещения в алкильных группах на-
чинаются с отрыва протона от атома углерода, связанного с аро-
матическим кольцом каким-либо основанием:
C6H5CH2Cn + B’ —> C6H5CHCN+BH
Образовавшийся карбанион далее взаимодействует с электро-
фильным реагентом, например с альдегидом или с галогеналкилом;
возникает первичный продукт взаимодействия, который иногда до
окончательной стабилизации испытывает еще ряд превращений:
C6H5CHCN + СН3СН2Вг —► C6H5CH(CN)CH2CH3 + Вг'
Когда образование карбаниона идет с трудом, концентрация
его мала и он высокоактивен, лимитирующей стадией реакции
является именно этот этап. Обычно при таких реакциях наблюда-
ется кинетический изотопный эффект и их скорость не зависит от
концентрации электрофила — имеет первый порядок.
Если же карбанион образуется легко, так как его отрицатель-
ный заряд сильно делокализован, то он сравнительно мало реак-
ционноспособен и лимитирующей стадией реакции становится ско-
рость взаимодействия карбаниона с электрофильным реагентом.
Реакция имеет второй порядок и не сопровождается кинетическим
изотопным эффектом.
14.1.1. Свободнорадикальное галогенирование
в алкильных группах
В отличие от рассмотренных в гл. 5 реакций галогениро-
вания в ароматическое кольцо, которые идут по механизму элект-
рофильного ароматического замещения, галогенирование в боко-
вую цепь является свободнорадикальной реакцией. Эта реакция
тормозится кислородом и ускоряется при действии инициаторов,
при нагревании и действии света — факторов, которые могут вы-
звать диссоциацию молекулы галогена на атомы:
Cl—Cl 4-=t 2С1.
В данном случае образующиеся атомы служат активными ча-
стицами, необходимыми для ведения процесса, который может
идти по схеме:
С6Н5СНз + С1. —> С6Н5СН2+НС1
Сен6сн2 + С12 —> С6Н5СН2С1 + С1 •
881
Как видно, в результате второй реакции происходит регенера-
ция атомарного хлора; благодаря этому цикл превращений может
повторяться до тех пор, пока активная частица — атомарный хлор
или бензильный радикал — не израсходуется в каком-либо необ-
ратимом побочном процессе: превращении атомарного хлора в
молекулярный (2С1 • —> С12), димеризации органических ради-
калов (2С6Н5СН2- —* С6Н5СН2СН2С6Н5), взаимодействии орга-
нического радикала с атомарным хлором (С6Н5СН2 • + С1 • —>
С6Н5СН2С1). Экспериментально показано, что при хлорировании
толуола в метильную группу каждая активная частица, содержа-
щая неспаренный электрон, может участвовать во многих тысячах
актов химических превращений до своей «гибели» по одной из
перечисленных реакций [1, с. 206].
Важным этапом таких цепных реакций является начальный
процесс, в котором зарождаются активные частицы. При ради-
кальном хлорировании ароматических соединений, обычно проис-
ходящем в жидкой фазе, зарождение активных частиц может идти
за счет термической диссоциации молекулярного хлора с участием
стенок аппарата. Однако термическое инициирование реакции не
всегда достаточно эффективно. Значительно сильнее действует свет
кварцевой лампы, кванты которого имеют высокую энергию и вы-
зывают распад молекулярного хлора на атомы. С повышением
температуры квантовый выход при фотохимическом хлорировании
повышается. Так, в случае хлорирования толуола при —80 °C каж-
дый поглощенный квант вызывает вступление в реакцию 25 моле-
кул хлора, а при 20°C квантовый выход достигает 8-Ю4 [2].
Весьма эффективна химическая активация реакции с помощью
инициаторов, чаще всего перекиси бензоила или 2,2'-азобисизобу-
тиронитрила, которые при нагревании в реакционной массе распа-
даются с образованием радикалов:
(С6Н5СОО)2 —> 2С6Н5СОО « —> 2С6Н5 • + 2СО2
СНз СНз СНз
I I I
NC—C—N=N—С—CN —> 2NC—C« + N2
I I I
СНз СНз СНз
Следует отметить, что химическое инициирование не часто ис-
пользуется при хлорировании ароматических соединений, более
всего из-за взрывоопасности названных инициаторов. В качестве
невзрывоопасного инициатора при хлорировании толуола рекомен-
дуется диацетильное производное сх,о/-дигидроксиперекиси хлора-
ля [СН3СООСН(СС13)]2О2, позволяющее вести реакцию при 60 °C и
получать бензилхлорид с выходом 90% [3]. При хлорировании в
метильную группу толуола, n-ксилола рекомендуются также нит-
раты спиртов — метилнитрат, тетранитропентаэритрит и др. [4].
При хлорировании в боковую цепь в реакционную массу до-
вольно часто добавляют РС13, ускоряющий течение реакции и
уменьшающий образование побочных продуктов [2]. Считают, что
382
каталитическим действием в этом случае обладает благодаря спо-
собности координироваться с молекулярным хлором хлорид фос-
фора (V), образующийся из РС13 и хлора [5]. Продукт координа-
ции далее диссоциирует с образованием атомарного хлора:
РС15 + С12
PCh + Ch + Cl.
Как уже указано (см. 14.1), хлорирование в боковую цепь
направляется преимущественно в a-положение. Так, при взаимо-
действии пропилбензола с хлором под влиянием света и при 25 °C в
основном образуется а-хлорпропилбензол. Меньше получается 0-и
еще меньше у-замещенного. Введение CS2 значительно повышает
селективность реакции — получается 81,5% а-замещенного. Одна-
ко при освещении и 160 °C начинает преобладать вступление хлора
в 0-положение — образуется 48,5% 0-хлорпропилбензола [6].
Казалось бы, что влияние заместителей в ароматическом кольце
должно мало сказываться на скоростях радикальных реакций в
боковой цепи, лимитирующей стадией которых является отрыв
атомарного водорода. В действительности же оно проявляется
вполне отчетливо в виде корреляционных зависимостей с отрица-
тельным значением константы реакции р. По мнению большинства
исследователей, причина полярных влияний заместителей лежит в
разделении зарядов в переходном состоянии для отрыва атома
водорода; однако предлагаются и другие объяснения этих эффек-
тов [7]. Например, при радикальном бромировании в боковую
цепь серии замещенных в ядре этилбензолов получена корреляция
с о+-константами, в которой р = —0,69 [8]. Получены корреляции
скоростей реакции с ст-константами заместителей при радикальном
хлорировании замещенных в ядре этилбензолов, толуолов и ди-
фенилметанов сульфурилхлоридом (SO2C12) [9, 10]. При радикаль-
ном хлорировании той же серии соединений фенилхлориодоний-
хлоридом CeHsICK найдено очень высокое значение р = —2,76.
Хлорирующим агентом в этих реакциях является селективный
фенилхлориодонильный радикал С6Н51С1* [11].
Интересно, что при переходе к реакциям с изопропилбензолом,
возрастание стерических препятствий вызывает уменьшение кон-
станты реакций р радикального хлорирования и бромирования,
очевидно, как следствие уменьшения роли полярного влияния за-
местителей [12].
С другой стороны, влияние атома хлора, уже введенного в
алифатическую цепь, сравнительно невелико и лишь в небольшой
степени затрудняет дальнейшее замещение водорода. Поэтому,
например, при хлорировании толуола последовательно проходят
следующие стадии:
С6Н5СН3 —> С6Н5СН2С1 —> С6Н5СНС12 —> СеН5СС13
383
Следует отметить, что все продукты, представленные в этой
цепи превращений, практически интересны. Бензилхлорид исполь-
зуется в больших количествах как алкилирующий агент (см. 6.1.1)
и для других целей. Его получают также с помощью реакции
хлорметилирования (см. 6.2.2). Бензилидендихлорид (бензальхло-
рид) при гидролизе образует бензальдегид. Бензотрихлорид при-
меняется для получения бензоилхлорида, образующегося при гид-
ролизе или при реакции с бензойной кислотой:
С6НГ,СС13 + С6Н5СООН —> 2С6Н5СОС1 + НС1
Бензотрихлорид используют также для превращения в бензо-
трифторид действием безводного HF при нагревании под давле-
нием:
CeH5CCl3 + HF —> C6H5CF3 + 3HC1
Для того, чтобы реакцию хлорирования в боковую цепь остано-
вить на нужной стадии, процесс контролируют по ГЖХ и плотно-
сти реакционной массы, которая растет с увеличением глубины
хлорирования.
В производстве, кроме толуола, хлорированию в боковую цепь
подвергают хлортолуолы (преимущественно п-, м- и 2,6-дихлорто-
луол), а также м- и n-ксилолы. Из последних получают главным
образом со-гексахлорпроизводные, используемые большей частью
для синтеза хлорангидридов терефталевой и изофталевой кислот.
Продукты исчерпывающего хлорирования в боковую цепь хлорто-
луолов используют в основном для перевода в хлорбензотрифто-
риды, подвергаемые далее нитрованию. Хлорирование 2,6-дихлор-
толуола заканчивается на стадии 2,6-дихлорбензилидендихлорида,
из которого получают 2,6-дихлорбензальдегид [13, с. 144].
Для хлорирования в боковую цепь применяются аппараты как
периодического, так и непрерывного действия, иногда снабженные
ртутными лампами для инициирования реакции. Хлор подается
через барботер в нижнюю часть аппарата. Хлорирование чаще
всего ведут при температуре, близкой к температуре кипения ре-
акционной смеси. При исчерпывающем хлорировании в реакцион-
ную смесь почти всегда добавляют хлорид фосфора(III) или ини-
циаторы, например 2,2'-азобисизобутиронитрил.
Так как электрофильное хлорирование в ядро идет на несколько
порядков быстрее свободнорадикального (см. 5.1), принимаются
особые меры, чтобы в реакционную массу не попадали вещества,
способные катализировать электрофильное хлорирование. Для
этого в производственной практике пользуются аппаратами, по-
крытыми эмалью или свинцом, не содержащими даже примесей
сурьмы, железа, олова, мышьяка, меди. Исходные продукты перед
реакцией подвергаются сушке и перегонке, а от хлора, поступаю-
щего в аппарат, отфильтровывают следы ржавчины. Для связыва-
ния следов железа в реакционную массу иногда добавляют урот-
ропин.
384
Имеются данные о том, что реакция хлорирования и бромиро-
вания ксилолов в боковую цепь при инициировании УФ-светом
может быть значительно ускорена добавлением 0,01—5%
уУ,Л^-диметилформамида, МД-диметилацетамида и аналогичных
соединений [14]. Это же относится и к хлорированию толуола
[15].
Радикальное бромирование алкилароматических соединений
может проходить в аналогичных условиях, хотя практического
применения почти не получило. Благодаря большому объему реа-
гента и меньшей его реакционной способности при бромировании
гораздо легче, чем при хлорировании, остановить реакцию на ста-
дии образования бензилиденовых производных, при гидролизе ко-
торых затем получаются альдегиды. Так, терефталевый альдегид
(1) с хорошим выходом может быть получен по схеме [16]:
СНз СНВГ2 СНО
СНз СНВг2 СНО
(1)
Радикальное иодирование не применяется. Для получения бен-
зилиодидов соответствующие хлорпроизводные вводятся в реак-
цию нуклеофильного замещения при действии иодида натрия в
ацетоновом растворе:
С6Н5СН2С1 + Nal —> С6Н5СН21 + NaCl
14.1.2. Получение производных дибензила и стильбена
Производные дибензила (2), а из них и производные
стильбена (3) могут образоваться при димеризации бензильных
радикалов:
—н2
2С6Н5СН2. —> с6н5сн2—сн2с6н5 ------> С6Н5СН=СНС6Н5
(2) (3)
Для получения бензильных радикалов обычно применяются
специфические реагенты окисляющего действия, способные отры-
вать атомарный водород от метильной группы,— такие, как орга-
нические перекиси, персульфат аммония, перекись натрия, диоксид
свинца, гипохлорит натрия. Соответствующие реакции проводятся
в Щелочной среде. В последние годы для этой же цели используют
кислород воздуха в водно-щелочной среде в присутствии эмульга-
торов и нафтенатов кобальта или марганца в качестве катализа-
тора. При проведении этих реакций полезен нагрев до 50—60 °C.
13 Зак. 271 385
В химии промежуточных продуктов особенно важна 4,4'-динит-
ростильбен-2,2'-дисульфокислота (4), получаемая из 4-нитро-2-то-
луолсульфокислоты при действии кислорода воздуха в щелочной
среде (ср. также [13, с. 150]):
В очень мягких условиях при действии перекисей на 2,6-ди-
трет-бутил-п-крезсл, используемый как стабилизатор полимеров
(ионол — получение см. 6.1.4), получается соответствующий ди-
бензил-4,4'-диол (5), а затем стильбенхинон (6) [17, с. 146]:
СНз
(СНз)зС
НО---
(СНз)зС
(СНз)3С
НО—
(СН3)3С
Специфическим методом, с помощью сплавления с серой м- и
n-толуиловых кислот при 260—280 °C получаются 3,3'- и 4,4'-стиль-
бендикарбоновые кислоты [1, с. 699].
Рассматривая димеризацию радикалов бензильного типа как
метод получения дибензила и его производных, необходимо иметь
в виду возможность проявления их двойственной реакционной
способности, связанной с делокализацией неспаренного электрона
в пара- и в меньшей степени в орто-положениях бензольного
кольца:
386
При стерической затрудненности радикального центра такие
радикалы могут образовать не димеры дибензильного типа, а
соединения с циклогексадиеновой структурой. Например, димер
трифенилметильного радикала представляет собой не гексафе-
нилэтан, как еще недавно думали, а 6-дифенилметилен-З-трифенил-
метил-1,4-циклогексадиен:
/==\ /н . /=\ /н
(CgHs)2C=. >• + С(СвН5)з < > (СбНз)2С=, «
ХС(С6Н5)з
Такого рода процессы имеют, по-видимому, довольно большое
распространение [18].
14.1.3. Электрофильное замещение в алкильных группах
Выше уже указывалось, что первым этапом реакций
электрофильного замещения в алкильных группах является иони-
зация соединения с образованием карбаниона под влиянием како-
го-либо соединения, способного оторвать протон:
АгСНз + СНзО" ч=* АгСЩ + СН3ОН
Соединения, способные в условиях эксперимента образовать
такой карбанион, вступают в реакцию с электрофилом. Традици-
онно они получили название соединений с подвижными атомами
водорода. Если карбанион не образуется, то реакция электрофиль-
ного замещения не идет.
На подвижность водорода в алкильных группах большое влия-
ние оказывают структурные факторы. Так, электроноакцепторные
заместители в сопряженных положениях оттягивают электронную
плотность от атома углерода, при котором находится алкильная
группа, и тем самым делают более подвижными атомы водорода,
а соединение — более склонным к. ионизации. Для качественной
оценки подвижности водорода было предложено несколько тестов
в виде электрофильных реакций, требующих все более и более
высокой способности соединений к ионизации [19].
За последние полтора десятилетия накоплен большой фактиче-
ский материал, позволяющий значительно строже оценивать реак-
ционную способность и подвижность атомов водорода, которая,
как ясно из всего изложенного, представляет собой проявление
СН-кислотности соединений. Большая часть этих данных получена
путем изучения кинетики основного дейтерообмена [20].
Реакция основного дейтерообмена в сущности является про-
стейшей реакцией электрофильного замещения в алкильных груп-
пах. Доказано, что она протекает по карбанионному механизму,
медленной стадией которого является отрыв протона основанием:
+RO~ + ROD
АгСН3 АгСЩ ArCH2D
Следовательно, скорость дейтерообмена может служить мерой
СН-кислотности соединений.
13“
Прямым подтверждением этого является соблюдение линейной
зависимости между логарифмом констант скорости дейтерообмена
и величиной Р^аН, характеризующей кислотно-основное равнове-
сие сходно построенных алкилароматических соединений в прото-
фильных средах:
lg k = 14,26 - 0,84рК^н
Это соотношение выдерживается в диапазоне изменения
ДрД™^ 15.
Ниже приведены значения логарифмов констант скорости дей-
терообмена и величины термодинамической кислотности метиль-
ных групп некоторых ароматических соединений [20]:
- Ig k (при 25 °C) „СН Р*а
Толуол — 36
Дифенилметан —- 28,6
Трифенилметан — 27,2
Флуорен — 20,5
2-Метилпиридин 8,2 (27)
З-Метилпиридин 11,0 (28)
4-Метилпиридин 6,3 (25)
2-Метилхинолин 5,4 (23)
З-Метилхинолии (9,5) (28)
4-Метилхинолин 5,1 22,2
6-Метилхинолин Н,1 (30)
8-Метилхинолин 11,8 (31)
1 -Метилизохинолин 5,7 25,0
2-Метилхиноксалин 2,7 (20)
9-Метилфенантридин 5,1 22,8
9-Метилакридин 4,3 21,1
2-Метилбензотиазол 3,5 —
1,2-Диметилбензимидазол 5,6 —
Примечание. В скобки заключены вычисленные значения.
Для алкилароматических соединений р/(аН может изменяться в
очень широких пределах: от 36 для толуола (для метана 42) и до
7—8 для четвертичных солей 2- и 4-метилпиридина. При действии
щелочей последние образуют электронейтральные метиленовые ос-
нования, причем некоторые из них настолько стабильны, что мо-
гут быть получены в химически чистом виде:
Скорость образования метиленовых оснований велика и не ли-
митирует суммарной скорости химического превращения
(см. 14.1).
383
Термодинамическая кислотность позволяет количественно ха-
рактеризовать подвижность атомов водорода и в некоторых пре-
делах прогнозировать реакционную способность соединений. Эти
пределы ограничены возможностями проведения конкретной реак-
ции в условиях, наиболее благоприятных для ионизации соедине-
ния, и зависят, кроме того, от стерических факторов, содействую-
щих или препятствующих подходу электрофильного реагента к
карбанионному центру соединения.
Соединения с подвижными атомами водорода способны ко
многим электрофильным реакциям. Например, их можно алкили-
ровать галогеналкилами и непредельными соединениями. Реакция
с акрилонитрилом идет по следующей схеме:
АгСНз + CH2=CHCN —> ArCH2CH2CH2CN
Процесс катализируется сильными основаниями. Многие соеди-
нения вступают в реакцию Манниха см. 6.2.4). Соединения с
повышенной подвижностью атомов водорода нитрозируются в бо-
ковую цепь при действии азотистой кислоты:
АгСНз + HONO —> ArCH2NO ArCH=NOH
Для таких соединений характерно также взаимодействие с
нитрозосоединениями и образование азометинов, при гидролизе
которых получаются альдегиды [19]. Например, из 2,4-динитро-
толуола этим путем через азометин (7) получается 2,4-динитро-
бензальдегид (8) с выходом 38% [21]:
Некоторые соединения с подвижными атомами водорода в бо-
ковых цепях вступают в реакции азосочетания (см. 15.4.1).
Наибольшее практическое значение в синтезе промежуточных
продуктов находит реакция алкилароматических соединений с
альдегидами, в результате которой получаются разнообразные
стирильные производные:
Отщепление воды, как правило, проходит быстро и не лимити-
рует суммарной скорости реакции. При проведении этих реакций
обычно используют катализ основаниями, добавляя в реакционную
массу триэтиламин или пиперидин. В случае необходимости при-
меняют также водоотнимающие агенты, например уксусный
389
ангидрид. Условия проведения реакции в основном определяются
подвижностью атомов водорода взятого соединения.
Как было указано, толуол к этой реакции не способен, но уже
re-нитротолуол реагирует по метильной группе с бензальдегидом и
его производными [22].
Другим способом повышения подвижности атомов водорода
метильной группы является временное введение в нее электроно-
акцепторных заместителей. Например, фенилуксусная кислота и ее
производные гладко реагируют с альдегидами в присутствии три-
этиламина и уксусного ангидрида
2CsH5CH2 +ОСН——сн0 —* CSH5C=CH——СН=СС6Н5
СООН СООН СООН
Полученные продукты в дальнейшем могут быть декарбоксили-
рованы [23].
Реакция с альдегидами нашла особенно большое применение в
гетероаромэтическом ряду: с ее помощью получают мероцианино-
вые и цианиновые красители [24].
Для успешного проведения реакций с соединениями, имеющими
малую СН-кислотность, крайне желательна сильно щелочная сре-
да, благоприятствующая образованию карбанионов. Первые
эксперименты в этом направлении проводились на примере взаи-
модействия ге-толилфенилсульфона (9) с бензальдегидом (10) в
диметилсульфоксиде в присутствии трет-бутилата калия; в резуль-
тате был получен 4-стильбенилфенилсульфон (11), правда, с
небольшим выходом [25]:
C6H5SO2C6H4CH3 + CSH6CHO —> CsH5SO2C6H4CH==CHCsH5 + Н2О
(9) (10) (11)
Предполагая, что причиной низкого выхода стильбенового со-
единения является образующаяся при реакции вода, авторы заме-
нили бензальдегид Д'-бензилиденанилином (12) и, проводя реак-
цию в диметилформамиде с трет-бутилатом калия, за 15 мин при
комнатной температуре получили тот же продукт (11) с выходом
84% [26]:
C6H5SO2C6H4CH3 + CsH6CH=NC6H5 —> CsH5SO2C6H4CH=CHCsH5 + c6h5nh2
(9) (12) (11)
Если вместо трет-бутилата калия использовать метилат натрия,
то реакция останавливается на стадии образования продукта при-
соединения карбаниона к Д-бензилиденанилину:
C6H5SO2C6H4CH2CHC6H5
I
NHC6HS
В дальнейшем эта реакция была детально изучена и под на-
званием апил-синтеза распространена на очень большое число
разнообразных гетероароматических и ароматических соединений,
содержащих боковые метильные группы. Оказалось, что наилуч-
390
шим растворителем для ее проведения является диметилформа-
мид. В диметилацетамиде, гексаметилфосфортрйамиде и диметил-
сульфоксиде реакция идет значительно хуже. трет-Бутилат калия в
большинстве случаев может быть заменен порошком КОН. Вопре-
ки первоначальным предположениям, небольшие количества воды
не мешают реакции.
Для проведения анил-синтеза большое значение имеет темпе-
ратура. Она должна быть возможно более низкой, так как ее
повышение вызывает протекание побочных реакций и гидролиз
растворителя. Анил-синтез следует вести в токе азота, чтобы пре-
дотвратить реакции окисления, которые могут идти за счет кисло-
рода воздуха [27]. При недостатке азометина (12) наряду с
образованием стильбеновых соединений получаются продукты ти-
па (13), построенные из двух арилметильных остатков на один
остаток альдегида:
2АгСН3 -f- C6H5CH=NC6H5 —> (ArCH2)2CHC6H5 + C0HBNH2
(12) (13)
С помощью анил-синтеза с удовлетворительным выходом могут
быть получены стирильные производные из толуола, изомерных
ксилолов, три- и тетраметилбензолов, метил- и диметилнафтали-
нов, метилбифенилов и терфенилов, метильных производных
стильбена, толана, дифенилбутадиена и т.п. [28]. В эту реакцию
вступают метильные производные гетероциклов, такие как
2-(n-толил) бензоксазол, -бензотриазол, -5-фенилоксазол,
2,5-ди(п-толил) оксадиазол и многие другие [29].
Таким образом, анил-синтез является общей реакцией получе-
ния стирильных производных из метилароматических соединений.
Применительно к последним анил-синтез удобнее, чем реакция
Виттига, исходными соединениями в которой служат алкил- и
аралкилгалогениды и фосфорорганические соединения [30].
Успешное течение реакции анил-синтеза определяется высокой
щелочностью среды, благодаря которой алкилароматическое со-
единение дает максимально возможную концентрацию карбанио-
на, активно взаимодействующего с азометином. Это происходит
несмотря на то, что азометин является более слабым электрофиль-
ным реагентом, чем альдегид (связь C = N менее поляризована,
чем связь С—О в альдегиде).
Активность альдегидов как электрофильных реагентов в зави-
симости от их строения уже рассматривалась (см. 6.2.1).
14.2. ЗАМЕЩЕНИЕ В АМИНОГРУППАХ
Реакции замещения в аминогруппах весьма разнообраз-
ны, но, как правило, проходят по единому механизму; лимитирую-
щей стадией его является присоединение катиона или катионоид-
ной частицы к неподеленной паре электронов атома азота с обра-
зованием аммониевого иона:
ArNH2 + x+ —> ArNHsX
391
Поэтому чем больше основность амина, тем легче он вступает в
реакции электрофильного замещения, в результате которых полу-
чаются алкильные производные, азометины или ацильные произ-
водные. Так как ароматические амины вследствие эффекта сопря-
жения на 5—6 порядков менее основны, чем алифатические, ука-
занные реакции с ними протекают значительно медленнее.
14.2.1. Л/-Алкилирование
Af-Алкилирование аминов проходит по общей схеме:
ArNH2 —> ArNHR —> ArNR2 —> ArNR3
На промежуточных этапах реакции образуются вторичные и
третичные амины, а на завершающем — соли полностью замещен-
ного (четвертичного) аммония. В общем случае, когда алкил R не
вызывает больших пространственных препятствий для дальнейше-
го течения реакции, скорости отдельных ее этапов мало различа-
ются. Поэтому в результате алкилирования образуются смеси про-
дуктов. Из них соли триалкилариламмония легко выделить благо-
даря их растворимости в воде или же превратить в третичные
амины при действии щелочи и нагревании под давлением:
ArNR3 + OH- > ArNR2 + ROH
1OU*- loo V
Наибольшие трудности связаны с получением вторичных жир-
ноароматических аминов; в обычных условиях они неизбежно об-
разуются в смеси с первичными и третичными аминами. Разделе-
ние таких смесей часто затруднительно, так как их компоненты
имеют близкие температуры кипения.
В качестве алкилирующих агентов применяются непредельные
соединения и спирты (в присутствии кислот), алкилгалогениды,
алкиловые эфиры серной кислоты и сульфокислот.
В промышленности алкилирование ароматических аминов ме-
тиловым, этиловым, бутиловым и другими спиртами ведется в
автоклавах при 180—220°C в присутствии минеральных кислот
(НО или H2SO4) в количестве 0,06—0,3 моль на 1 моль амина.
Для превращения в третичные амины образовавшихся четвертич-
ных солей по приведенной выше реакции, полученный алкилат
обычно сильно подщелачивают, добавляя 30%-ный NaOH и вновь
нагревают под давлением при 160—180 °C в течение нескольких
часов.
Кислотный катализ рассматриваемых реакций осуществляется
путем протонирования спирта и образования алкоксониевого иона,
который далее может диссоциировать с образованием алкил-кати-
она и воды
ROH + H* ч=ь ROH2 R+ + H2O
Алкил-катион далее взаимодействует с амином как показано
выше,
392
В зависимости от условий и от строения спирта реакция с
амином может идти и без предварительной диссоциации алкоксо-
ниевого иона путем синхронного разрыва старой связи R—О и
образования новой связи N—R.
c6h5nh2 + roh2 [C6H5NH2-.-R.- OH2] —> C6H5NH2R + H2O
Реакция алкилирования аминов в условиях кислотного катали-
за обратима. Так, нагревание /V-метиланилина в присутствии кис-
лоты приводит в результате равновесной реакции к образованию
М,М-диметиланилина и анилина:
2C6H5NHCH3 C6H5NH2 + C6H5N(CH3)2
Возникновение этого равновесия объясняется тем, что аммоние-
вый ион соли алкилированного амина, например СбН5ЫН(СН3)2,
подобно изображенному выше алкоксониевому иону способен ал-
килировать ароматический амин.
Вместо соляной или серной кислот при алкилировании спиртами
можно с успехом пользоваться хлоридом фосфора(III), который
добавляют в количестве 0,8—0,9% от взятого амина. Использова-
ние этого катализатора позволяет значительно сократить время
реакции и получить вторичные и третичные амины с хорошим вы-
ходом. Этот катализатор используется для получения диметилани-
лина непрерывным методом.
Л7-Бутиланилин получается из анилина и бутилового спирта,
взятого с 15%-ным избытком против теории при использовании
0,2% РС1з [1, с. 488]. Механизм каталитического действия по-
следнего не выяснен. Полагают, что он образует со спиртом на
первом этапе алкилфосфиты, но не исключена также возможность
образования фосфазосоединений.
Алкилирование ароматических аминов спиртами может прово-
диться и в виде гетерогенно-каталитического процесса в паровой
фазе над соответствующим катализатором, например А120з
[1, с. 488].
В промышленности алкилирование спиртами применяется для
получения М-алкил- и М,М-диалкиланилинов (алкилы — СНз, С2Н5,
К-С4Н9 и др.).
Широко используется в производстве промежуточных продук-
тов и А-алкилирование алкилгалогенидами. С ароматическими
аминами они взаимодействуют в сравнительно очень мягких усло-
виях. Скорость реакции обычно описывается уравнением второго
порядка
г = й [RCI] [ArNH2]
и сильно зависит от свойств среды, в которой процесс протекает.
Повышение полярности среды увеличивает скорость реакции, ко-
торая осуществляется по синхронному механизму:
C6H5NH2 + RCl [C6H5NH2--R-C1] —> CeH5NH2R+Cr
393
Наибольшее применение для алкилирования находят бензил-
хлорид, хлоруксусная кислота, этиленхлоргидрин, реже — эпихлор-
гидрин и окись этилена.
При использовании бензилхлорида для алкилирования анилина
и некоторых его производных, благодаря стерическим затрудне-
ниям второе бензилирование идет значительно медленнее первого:
CeH5NH2 —> C6H5NHCH2C6H5 —C8H5N(CH2C6H5)2
Тем не менее при получении А-бензиланилина на 2 моль ани-
лина берут 1 моль бензилхлорида [31]. Выделяющийся хлористый
водород связывается избытком анилина:
2C6H5NH2 + С6Н5СН2С1 —> CeH5NHCH2CeH5 + C6H5NH3 СГ
Аналогично бензилируются вторичные амины — А-метилани-
лин, А-этиланилин [13, с. 123] и Л'-этил-.и-толуидин, образуя со-
ответственно третичные амины (14), (15) и (16), применяющиеся в
синтезе трифенилметановых красителей. При бензилировании
N,N-диметиланилина получается хлорид бензилдиметилфенилам-
мония (17), известный под названием лейкотроп и используемый в
качестве бензилирующего агента для лейкосоединений некоторых
кубовых красителей в щелочной среде.
CsHsNCHjCgHs CsHsNCHjCsHs -M-CHaCebLiNCHgCeHs
I I I
СНз C2H5 C2H5
(14) (15) (16)
(CH3)2NCH2C6H5 СГ
C6H5
(17)
Взаимодействие анилина с хлоруксусной кислотой проводят в
водной среде, применяя нейтрализующие агенты, так что фактиче-
ски алкилирующим средством служит хлорацетат-анион. Целевым
в этой реакции является продукт моноалкилирования — А-фенил-
аминоуксусная кислота (фенилглицин) (18), используемая для син-
теза индиго. Однако при реакции с большой легкостью образуется
и не находящее применения диалкильное производное — А-фенил-
иминодиуксусная кислота (19):
CeH5NH2 —> C6H5NHCH2COOH —CeH5N(CH2COOH)2
(18) (IB)
Чтобы избежать образования (19), реакцию ведут в присут-
ствии солей железа (II), которые образуют с (18) нерастворимую в
воде соль, выпадающую в осадок и поэтому не вступающую в
повторную реакцию с алкилирующим агентом.
В реакцию с хлоруксусной кислотой вводят также п-аминофе-
нол. Чтобы не проалкилировать гидроксильную группу, реакцию
ведут в кислой среде. Полученную А-(n-гидроксифенил) аминоук-
Сусную кислоту (20) используют в качестве проявителя (глицин);
894
декарбоксилированием из нее получают п-метиламинофенол (21),
представляющий собой более ценный проявитель (метол):
но——NHa —* но—\~У—МНСН2СООН —>-
__ (20)
—> НО——ННСНз
(21)
Хлоргидрин этиленгликоля используют для алкилирования
многих первичных и вторичных аминов с целью получения моно- и
бис-2-гидроксиэтильных производных, используемых в синтезе
дисперсных красителей [32]. Реакция идет по схеме:
C6HSNH2 + С1СН2СН2ОН —> C6H5NHCH2CH2OH + НС1
Выделяющаяся кислота связывается избытком амина или
нейтрализующим агентом.
Чаще, однако, для синтеза тех же продуктов применяют окись
этилена, легко реагирующую с аминами в присутствии небольших
количеств воды. С первичными аминами реакция идет в два этапа,
причем второе алкилирование с такой же скоростью, как и первое:
О
ArNH2 + Н2С^——СН2 —> ArNHCH2CH2OH
О
ArNHCH2CH2OH + Н2б—СН2 —> ArN(CH2CH2OH)2
Поэтому для получения моноалкильного производного реакцию
ведут с большим недостатком окиси этилена. Продукт реакции
отделяют разгонкой, используя большую разницу в температурах
кипения. Для получения бис-2-гидроксиэтильных производных, на-
против того, применяют небольшой избыток окиси этилена.
В промышленности реакции гидроксиэтилирования окисью эти-
лена подвергают анилин, N-метил-, 7У-этил- и /У-бутиланилины,
лг-толуидин и Л;'-этил-,и-толуидин, а также некоторые другие про-
дукты. Окись этилена дает с воздухом взрывчатые смеси в очень
широком диапазоне концентраций. Поэтому аппараты, в которых
ведутся реакции, до введения окиси этилена необходимо тщательно
продувать азотом.
В ограниченном числе случаев для алкилирования аминов при-
меняют также эпихлоргидрин. Последний реагирует с ароматиче-
скими аминами по окисному циклу, а не за счет атома хлора.
Однако при действии щелочи окисное кольцо вновь образуется в
результате дегидрохлорирования:
О ОН
. / \ I +он-
ArNH2 + H2C--СНСН2С1 —-> ArNHCH2CHCH2Cl --------►
О
—> ArNHCH2CH—СН2
895
Эпихлоргидрин используют для алкилирования некоторых ами-
нов антрахинонового ряда и аминов, применяемых в синтезе ак-
тивных красителей [33].
Алкилирование ароматических аминов ведут также с помощью
акрилонитрила. Реакция катализируется кислотами и щелочами:
ArNH2 + CH2=CHCN —> ArNHCH2CH2CN
Например, с анилином процесс идет при нагревании до 120—
140°C в присутствии уксусной кислоты. При более высокой темпе-
ратуре образуется смесь продуктов моно- и дицианоэтилирования:
C6H5NH2 —> C6H5NHCH2CH2CN —> C6H5N(CH2CH2CN)2
В сходных условиях в реакцию вступают и другие амины —-
А-этиланилин, м- и п-хлоранилины, .w-толуидип и др. [34]. Полу-
чающиеся продукты находят применение в синтезе дисперсных
[32] и катионных [35] красителей.
О трудности выделения вторичных жирноароматических ами-
нов из смесей с первичными и третичными аминами, образующихся
при алкилировании ароматических аминов в обычных условиях,
уже говорилось. Разработаны методы, позволяющие синтезировать
чистые вторичные жирноароматические амины, необходимые в
производстве промежуточных продуктов и красителей.
По наиболее общему методу Ульмана — Кижнера первичный
амин подвергают действию бензол- или толуолсульфохлорида и
получившееся А-аренсульфонильное производное, в котором водо-
род при азоте проявляет «кислые» свойства, алкилируют в щелоч-
ной среде; затем аренсульфонильную группу отщепляют гидроли-
зом в кислой среде:
+ RX
ArNH2 —> ArNHSO2Ar' —> ArNSO2Ar' -----> ArN(R)SO2Ar' —> ArNHR
Вторичные амины иногда получают через А-арилсульфамино-
вые кислоты, образующиеся при действии пиридинсульфотриокси-
да на ароматические амины (см. 3.1). Обработка солей А-арил-
сульфаминовых кислот диметилсульфатом и последующий гидро-
лиз (ср. 14.2.3) приводят к получению вторичных аминов:
ArNH2 ArNHSOaNa —> ArNSO3Na —> ArNHCH3
I
СНз
Иногда вторичные амины получают также через А-ацильные
производные [1, с. 496].
Другим общим и находящим большое практическое применение
методом получения вторичных аминов является восстановительное
алкилирование [36]. Сущность его состоит втом, что амин вводят в
реакцию с альдегидом или кетоном, и образующийся азометин (22)
гидрируют над катализатором, например над никелем Ренея или
палладием на угле (подробно см. 14.2.2 и 16.5).
396
14.2.2. Реакции первичных аминов с альдегидами
Первичные ароматические амины легко взаимодейству-
ют с альдегидами, образуя азометины (основания Шиффа) (22):
ArNH2 + CHR —> ГArNH2CHRl —> ArNHCHR —> ArN=CHR
11 1 ।
о L o' J он
(22)
Кинетическими исследованиями показано, что реакции образо-
вания и гидролиза азометиков в водных растворах протекают по
двухстадийному механизму через аминоспирт, о чем свидетель-
ствует наличие максимума на кривой pH — скорость реакции.
В кислых средах скорость реакции определяется стадией образо-
вания аминоспирта, в нейтральных — дегидратацией [37].
На активность ароматических альдегидов при образовании
анилов — азометинов (22), в которых R = арил,— большое влия-
ние оказывают заместители в их ароматическом кольце: электро-
ноакцепторные заместители, увеличивающие положительный за-
ряд на углероде карбонильной группы, повышают их реакционную
способность, а электронодокорные — понижают (ср. 6.2). Однако
из этой зависимости выпадают о-гидроксиальдегиды, которые вза-
имодействуют с аминами быстрее всего, по-видимому, потому, что
водородная связь в о-гидроксиальдегиде усиливает поляризацию
карбонильной группы и увеличивает положительный заряд карбо-
нильного углерода.
Естественно, что на скорость образования азометинов влияет и
основность аминов — чем она больше, тем легче идет реакция.
Однако, значительные поправки могут вносить стерические факто-
ры.
При нагревании азометинов с аминами, амины с большей ос-
новностью вытесняют менее основные. При этом устанавливаются
равновесия, положение которых определяется соотношением рАь
использованных аминов. Промежуточным продуктом обменных
реакций является соединение (23), образующееся путем присоеди-
нения амина по двойной связи азометина [38]:
Ar'NFR
ArN=CHR + Ar'NH2 ^CHR Ar'N=CHR + ArNH2
ArNH/
*22) (23) (24)
Равновесие можно полностью сдвинуть в одну из сторон, уда-
ляя один из аминов из сферы реакции, например отгонкой в ваку-
уме. Аналогичным способом можно произвести и замену альдегид-
ной компоненты азометина [39]:
RCH = NAr + R'CH=O 4=fc R'CH=NAr + RCH=O
Азометины могут подвергаться обменной реакции и при взаи-
модействии друг с другом [40].
397
Все азометины легко гидролизуются в минеральнокислых сре-
дах, причем реакция идет уже на холоду и начинается с протони-
рования атома азота.
Образование азометинов используют для временной защиты
аминогрупп и в качестве промежуточных стадий органического
синтеза (см. 6.2.3, 14.1.3, 14.2.3 и др.). В отдельных случаях азоме-
тины находят и самостоятельное применение в качестве красителей
и люминофоров [41, с. 60]. Среди последних особенно важны
анилы салицилового (25) и 2-гидрокси-1-нафтойного (26) альдеги-
дов:
(26)
14.2.3. /V-Ацилирование
M-Ацилирование аминов — часто используемая реакция.
Ее применяют как для временной защиты аминогруппы в соедине-
ниях, подвергаемых сульфированию, нитрованию или галогениро-
ванию [42, с. 235], так и для получения соединений, обладающих
определенными практически важными свойствами. В первом слу-
чае в реакцию чаще всего вводят муравьиную или уксусную кис-
лоту и ацетангидрид, во втором — самые разнообразные карбоно-
вые кислоты и их производные — обычно ангидриды и хлорангид-
риды.
Механизм реакции М-ацилирования аминов может быть пред-
ставлен следующей схемой:
ОН
I
ArNH2 + С—R
ОН п
+ I
ArNH2—С—R
I
О’ -I
ArNH—С—R + Н2О
Стадией, лимитирующей суммарную скорость реакции, являет-
ся образование промежуточного продукта, который, как видно из
схемы, получается путем внедрения неподеленной пары электронов
атома азота аминогруппы в электронный пробел углерода карбок-
сильной группы. Электронодонорные заместители в ароматическом
кольце амина увеличивают, а электроноакцепторные уменьшают
скорость реакции ацилирования — константа реакции р в уравне-
нии Гаммета имеет отрицательный знак. Чем больше основность
амина, тем легче он вступает в реакцию ацилирования.
Противоположное влияние на скорость реакции оказывают за-
местители в остатке карбоновой кислоты R. Здесь электроноакцеп-
торные заместители увеличивают, а электронодонорные — умень-
398
шают скорость ацилирования, и константа р имеет положительное
значение. Отсюда следует, что чем сильнее выражен электронный
пробел на углероде карбоксильной группы, тем более активна
кислота или ее производное как ацилирующий агент (ср. 6.3).
В соответствии с этим может быть представлен следующий ряд
ацилирующих агентов по их сравнительной реакционной способно-
сти:
RCOC1 > RCOOCOR > RCOOCeH5 > RCOOH > RCOOCH3 >
> RCONH2 > RCOO"
Анион карбоновой кислоты практически уже не способен аци-
лировать ароматические амины.
Выбор реагента для А-ацилирования зависит от реакционной
способности амина, условий, в которых можно проводить реакцию,
доступности и стоимости ацилирующего агента.
Для временной защиты аминогруппы часто используют реак-
цию А-формилирования. Обычно ее проводят, нагревая амин с
избытком муравьиной кислоты;
ArNH2 + НСООН ArNHCHO + Н2О
Реакция обратима, но образующаяся вода отгоняется с избыт-
ком муравьиной кислоты, и благодаря этому процесс доходит до
конца. Отгонку заканчивают в вакууме при 150 °C. Если для фор-
милирования используют гидрохлориды аминов или амины, содер-
жащие сульфогруппы, в реакционную массу добавляют формиат
натрия для нейтрализации этих соединений [1, с. 525].
В отдельных случаях, когда формилирование амина надо про-
вести в мягких условиях и с хорошим выходом, пользуются сме-
шанным муравьино-уксусным ангидридом [43], который получают
непосредственно перед реакцией, добавляя уксусный ангидрид к
избытку муравьиной кислоты:
НСООН + СН3СООСОСН3 :?=* СНзСООСНО + СНзСООН
В смешанном ангидриде формильный остаток значительно бо-
лее активен, чем ацетильный, и поэтому реакция с амином идет
однозначно в сторону образования А-формильного производного:
СНзСООСНО + ArNH2 —ArNHCHO + СН3СООН
При проведении этой реакции можно использовать обычную
85%-ную муравьиную кислоту, связывая воду необходимым коли-
чеством уксусного ангидрида [44].
Для временной защиты аминогрупп особое значение имеет ре-
акция JV-ацетилирования:
ArNH2 4- СНзСООН ArNHCOCHa + Н2О
Реакция обратима, поэтому обычно предпочитают использовать
98—100%-ную уксусную кислоту, которую берут с небольшим из-
бытком (10—15%) против теоретического количества. После
нескольких часов нагревания при кипении избыток кислоты вместе
с образовавшейся водой отгоняют от продукта реакции.
399
При ацетилировании аминосульфокислот, например сульфани-
ловой или нафтионовой, перед проведением реакции кислую функ-
цию необходимо нейтрализовать; с этой целью к уксусной кислоте
можно добавить необходимое количество безводного ацетата на-
трия [1, с. 525].
Чаще, однако, для ацетилирования таких аминов и еще более
слабоосновных соединений используют уксусный ангидрид, обла-
дающий значительно большей реакционной способностью, чем ук-
сусная кислота. Его широко используют для ацетилирования рас-
творимых в воде в виде солей аминосульфокислот, аминокарбоно-
вых кислот, а также растворимых в воде диаминов, аминофенолов
и других соединений. Реакции проводят, постепенно прибавляя к
водным растворам названных соединений уксусный ангидрид в
количестве, несколько превышающем теоретически необходимое.
Реакция хорошо идет уже при комнатной температуре. Таким об-
разом, например из 7-амино-4-гидрокси-2-нафталинсульфокислоты
(И-кислота) получают Х-ацетил-И-кислоту (27):
>27)
Процесс ведут в следующих условиях [45, с. 415]. К слабокис-
лому водному раствору натриевой соли И-кислоты приливают при
20—30 °C уксусный ангидрид, прибавляют NaOH и Na2CO3, нагре-
вают 2 ч при 95 °C для отщепления О-ацетильной группы, нейтра-
лизуют соляной кислотой и (27) высаливают; выход 85%.
Аналогично получают Af-ацетилсульфаниловую кислоту (28),
А-ацетил-Аш-кислоту (29) [13, с. 237], А-ацетил-Гамма-кислоту
(30) и многие другие соединения.
NHCOCH3
SO3H
(28)
HO3S
ОН NHCOCH3
(29)
(30)
Уксусным ангидридом в водной суспензии или эмульсии можно
ацетилировать и многие нерастворимые в воде амины, если только
они не слишком мало реакционноспособны.
Ацетилирование низкоосновных аминов, таких как 2,4-динитро-
аиилин или аминоантрахиноны, ведут уксусным ангидридом в ук-
сусной кислоте, в пиридине, а иногда и просто’ при нагревании с
избытком уксусного ангидрида. Реакция катализируется добавле-
но
нием небольшого количества сильной кислоты — серной, хлорной,
а также фосфорной.
В определенных случаях можно отметить возможность избира-
тельного течения реакции ацетилирования. Так, при действии ук-
сусного ангидрида на водный раствор соли 2,5-диаминобензол-
сульфокислоты ацетилирование идет только за счет более основной
аминогруппы в лгетп-положении к сульфогруппе:
NH2 NHCOCH3
NH2 NH2
Аналогично, при ацетилировании 1,5-диамино-9,10-антрахи-
нон-2-сульфокислоты, которое ведут уксусным ангидридом в пири-
дине при 60 °C [1, с. 536], в реакцию вступает только аминогруппа
в положении 5:
При ацетилировании аминов большим избытком уксусного ан-
гидрида и при 'высокой температуре могут получаться А/Д-диаце-
тильные производные, которые в обычных случаях нежелательны."
ArNH2 —> АгШСОСНз —► ArN(COCH3)2
Ацетилхлорид практического применения для ^ацетилирова-
ния аминов не получил, хотя является очень активным реагентом.
А-Ацетилирование можно вести, пропуская через эмульсию
ароматического амина в воде газообразный кетен:
ArNH2 + СН2=С=О —> ArNHCOCH3
Реакция идет с большой легкостью и с высоким выходом про-
дукта ацетилирования. Как видно из уравнения, при этом не обра-
зуется никаких побочных продуктов. К сожалению, использовать
кетен для ацетилирования аминов можно только на тех предприя-
тиях, где имеется его производство, так как транспортировать
кетен невозможно из-за его нестойкости и способности самопроиз-
вольно полимеризоваться.
Интересна реакция М-ацетилирования с одновременной цикли-
зацией, которую используют также для временной защиты амино-
и гидроксильной группы в о-аминофенолах. Например, при дей-
ствии уксусной кислоты на о-аминофенол образуется 2-метилбен-
зоксазол (31), который гладко нитруется нитрующей смесью в
положение 6. Получающийся 2-метил-6-нитробензоксазол (32) в
401
дальнейшем при обработке раствором щелочи, взятой в количестве,
близком к теоретически необходимому, при 70—75 °C отщепляет
остаток уксусной кислоты и получается 2-амино-5-нитрофенол
(33), используемый в синтезе красителей [46].
Аналогичная реакция с о-фенилендиамином приводит к 2-ме-
тилбензимидазолу. Однако это соединение вполне устойчиво к
гидролизу и в щелочной и в кислой среде, и отщепить от него
уксусную кислоту в обычных условиях невозможно.
A-Формильные или А-ацетильные производные аминов легко
гидролизуются при нагревании с разбавленным раствором щелочи
или сильной кислоты. Для этого в заводской практике применяют
нагревание при 70—100 °C с 5—10%-ным раствором NaOH, взятым
с небольшим избытком против теоретического количества. Щелоч-
ной гидролиз проходит очень гладко, причем формильные произ-
водные гидролизуются значительно быстрее ацетильных. Особенно
легко, уже на холоду, гидролизуются трифторацетильные про-
изводные аминов ArNHCOCF3. А-Бензоиламины, напротив того,
гидролизуются значительно медленнее.
Эти важные для практики закономерности легко объясняются
механизмом щелочного гидролиза, медленной стадией которого
является внедрение гидроксид-аниона в электронный пробел кар-
бонильного углерода:
О
II +ОН"
ArNH—С—R ------►
ОН -]
ArNH—С—R
О’ -
—► ArNH2 + RCOO"
К реакциям собственно А-ацилирования аминов примыкают
реакции образования А-сульфонильных производных ароматиче-
ских аминов — А-арилсульфаминовых кислот ArNHSOsH и А-бен-
золсульфонил- и А-толуолсульфониламинов ArNHSO2CeH4X (где
Х = Н или СНз). Эти реакции уже были рассмотрены в связи с
получением вторичных аминов (см. 14.2.1). Их механизм сходен с
механизмом реакций А-ацилирования. А-Ареисульфонильные про-
изводные аминов также используются и для временной защиты
аминогруппы и как самостоятельные продукты. Однако область
применения таких производных ароматических аминов значитель-
но шире благодаря их высокой стойкости к гидролизу в щелочных
402
средах, т. е._в условиях^ в которых они образуют соответствующие
анионы: ArNSOi и ArNSCbQPhX. В щелочных средах эти соеди-
нения гладко вступают в реакцию азосочетания с диазосоединени-
ями (см. 15.4.1).
Д/-Аренсульфонильные производные аминов часто вводят в ре-
акции нитрования с последующим восстановлением нитросоедине-
ний до аминов, причем защищенные аминогруппы сохраняются.
При этом TV-толуолсульфонил- и TV-бензолсульфониламины обра-
зуют максимальное количество п-нитропроизводных.
А-Арилсульфаминовые кислоты легко гидролизуются при на-
гревании подкисленных водных растворов с образованием серной
кислоты:
ArNHSO3H Н2О —> ArNH2 -р H2SO4
В отличие от них, М-аренсульфонильные производные аминов
обычно стойки к действию разбавленных водных растворов кислот.
Отщепление аренсульфонильной группы легко проходит при
непродолжительном нагревании в серной кислоте, содержащей
небольшое (до 25%) количество воды.
Кроме использования для временной защиты аминогрупп, ре-
акции ацилирования широко применяются в синтезе промежуточ-
ных продуктов и красителей, в молекулах которых сохраняются
введенные ацильные группы. С такой целью применяют иные аци-
лирующие агенты, большей частью хлорангидриды карбоновых и
сульфокислот.
Простейший ацилирующий агент этого назначения — фосген
(карбонилхлорид). Его вводят в реакцию с разнообразными аро-
матическими аминами, в том числе и с красителями, содержащими
свободные аминогруппы:
2ArNH2 + COCI2 —> ArNHCONHAr + 2НС1
Реакцию большей частью ведут в водном растворе, пропуская
через него газообразный фосген. Для нейтрализации образующе-
гося НС1 используют ЫагСОз или NaOH. Этим способом из
И-кислоты получают так называемую алую кислоту (7,7'-уреилен-
бис-4-гидрокси-2-нафталинсульфокислота) (34) [45], а из 2,5-
диаминобензолсульфокислоты — 5,5'-уреилендиортаниловую кис-
лоту (35). Обе они используются в синтезе красителей.
(35)
403
Для получения несимметричных производных мочевины в каче-
стве ацилирующих агентов применяют хлорангидрпды А’-арилкар-
баминовых кислот (36) или, чаще, арилизоцианаты (37). Их полу-
чают взаимодействием фосгена и ароматических аминов, раство-
ренных в толуоле, хлорбензоле или в полихлорбензолах по
суммарной реакции:
—на —НС1
ArNH2 + СОС12 --------> ArNHCOCl ---------> ArN=C=O
нагрев.
(36) (37)
Процесс обычно ведут в две стадии. Вначале к раствору фосге-
на в хлорбензоле при О °C медленно при перемешивании добавляют
двойное количество амина. При этом одновременно с хлорангид-
ридом ДАарилкарбаминовой кислоты (36) образуется соль амина
ArNH2-HCl, которая выделяется в осадок. Вторую стадию ведут
при нагревании до 150—200 °C и оставшуюся часть фосгена пропу-
скают в виде газа в реакционную смесь до тех пор, пока осадок
соли амина полностью не перейдет в раствор. Выделяющийся хло-
ристый водород очищают от фосгена, пропуская через раствори-
тель, и улавливают водой. Получающийся арилизоцианат (37)
отделяют от растворителя перегонкой [47, с. 248].
Арилизоцианаты легко взаимодействуют с аминами в воде или
в органических растворителях и образуют несимметричные произ-
водные мочевины. Например, с 4,4'-диамино-2,2'-стильбендисуль-
фокислотой-реакция идет в водной среде:
Получающаяся 4,4'-ди (3-фенилуреидо) -2,2'-стильбендисульфо-
кислота используется в качестве оптического отбеливателя.
Для ацилирования красителей, содержащих первичную ами-
ногруппу, находит применение хлорангидрид фумаровой кислоты
[48]. Его получают, нагревая малеиновый ангидрид с дихлоран-
гидридом фталевой кислоты в присутствии каталитических коли-
честв ZnCl2. Продукт выделяют перегонкой под вакуумом [49].
Хлорангидрид фумаровой кислоты прибавляют к водному рас-
твору амина; взаимодействие идет по схеме:
2ArNH2 + С1СОСН=СНСОС1 —> ArNHCOCH=CHCONHAr + 2НС1
Из хлорангидридов ароматических карбоновых кислот наи-
большее применение для ацилирования находит бензоилхлорид, а
также п-нитробензоилхлорид. Используются, но в значительно
меньших масштабах хлорангидриды .11- и «-толуиловой кислот,
л-хлорбензойной кислоты, 4-бифенилкарбоновой кислоты. Хлоран-
404
гидриды двухосновных кислот: изофталевой и терефталевой,
2,5-дихлортерефталевой, 4',4"-азобпс-4-бпфенилкарбоновой — ис-
пользуются для синтеза термостойких полимеров [50, с. 393].
Ацилирование ароматических аминов бензоилхлоридом и его
производными чаще всего ведут в водном растворе в присутствии
нейтрализующих средств путем постепенного добавления реагента
при перемешивании.
В случае малоосновных или плохо растворимых в воде аминов
реакцию ведут в органических растворителях—в хлорбензоле,
о-дихлорбензоле или нитробензоле, обычно при нагревании в пре-
делах 50—150 °C. В этих условиях образующийся хлористый водо-
род не связывается амином, а выделяется из реакционной массы.
В водных растворах бензоилируют разнообразные сульфокис-
лоты и аминогидроксинафталинсульфокислоты. Получаются цен-
ные для синтеза азокрасителей соединения, в частности А-бензоил-
Аш-кислота (38), ДАбензоил-И-кислота (39), ДАбензоил-Гамма-
кислота (40) и др. [45].
ОН NHCOCSH5
HO3S
(38)
Ацилирование н-нитробензоилхлоридом позволяет в дальней-
шем восстановить нитрогруппу и либо повторить реакцию ацили-
рования полученного аминосоединения, либо использовать его для
синтеза азокрасителей.
В органических растворителях ацилируют разнообразные ами-
нонитросоединения. Например, бензоилирование 5-метокси-4-нит-
ро-о-толуидина (41) и последующее восстановление нитрогруппы в
его /V-бензоильном производном (42) приводит к азоамину фиоле-
товому (43), применяемому при ледяном крашении:
СН3 СН3 1 СНз
Jx/NH2 J\zNHCOC6H5 1^NHCOCSH5
1 1 - Г ¥ — II
O2N ОСНз (41) h2n^ ОСНз (42) (!сн3 (43)
405
Аналогичным путем получают 4-нитро-Л^-(«-нитробензоил)-ани-
лин (44) и 2,4 -динитро-А-(n-нитробензоил)анилин (45);
NO2
O2N—(
NHCO
(44)
(45)
Среди разнообразных соединений, получаемых путем ацилиро-
вания ароматических аминов, особое место принадлежит азосо-
ставляющим для холодного крашения, так называемым азотолам
(нафтолам AS). В качестве ацилирующих агентов при их получе-
нии используют о-гидроксикарбоновые кислоты, преимущественно
З-гидрокси-2-нафталинкарбоновую (получение см. 6.3.4), а для
синтеза азотолов желтых тонов — ацетоуксусный эфир или дике-
тен. Реакцию ацилирования аминов З-гидрокси-2-нафталинкарбо-
новой кислотой (46) и другими о-гидроксикарбоновыми кислотами
проводят по единой методике. Она состоит в нагревании эквимоле-
кулярных количеств амина и кислоты в толуоле, хлорбензоле или
полихлорбензолах и добавлении в раствор хлорида фосфора (III) в
количестве, несколько превышающем теоретически необходимое.
Когда заканчивается выделение НС1, реакционную массу нейтра-
лизуют содой и отгоняют растворитель с водяным паром. Выде-
лившийся ариламид (47) отфильтровывают и обрабатывают рас-
твором соды, чтобы очистить от не вступившей в реакцию
о-гидроксикарбоновой кислоты [45, с. 516].
(46)
+ 3ArNH2 + РС13
(47)
ОН
CONHAr
+ Р(ОН)3 + ЗНС1
О роли, которую в этой реакции играет РС1з, нет единого мне-
ния. Высказывается предположение, что он превращает о-гидро-
ксикарбоновую кислоту в хлорангидрид, который далее взаимо--
действует с амином. По другой гипотезе, РС1з вначале взаимодей*!
ствует с аминами, образуя фосфазосоединения:
2ArNH2 + РС13 —► ArN=PNHAr + ЗНС1
Последние взаимодействуют с карбоновой кислотой и дают
ариламид:
ArN=PNHAr + 2Аг'СООН —> 2Ar'CONHAr + НРО2
406
Имеются доводы в пользу обоих предположений [1, с. 556].
Для получения ?/-ацетоацетильных производных аминов ис-
пользуется специфический ацилирующий агент — дикетен. Его по-
лучают путем димеризации кетена в ацетоновом растворе и отде-
ляют перегонкой:
СН2=С + СН2
IP II
сн2=с—сн2
О с=о
о—с=о
Реакция дикетена с аминами идет по уравнению:
СН2=С—СН2
О—с=о + ArNH2 —► CH3COCH2CONHAr
Чаще всего ее проводят, прибавляя дикетен к раствору, суспен-
зии или эмульсии амина в воде; выход продуктов близок к теоре-
тическому. Лишь в случае слабоосновных аминов, например
1-амино- или 1-метиламино-9,10-антрахиноыа, реакцию целесооб-
разно вести в органических растворителях — бензоле, хлорбензоле
или ацетоне [51].
Ариламиды ацетоуксусной кислоты — удобные исходные со-
единения для синтеза ариламидов других оксокарбоновых кислот.
Так, ариламиды бензоилуксусной кислоты рекомендуется получать
действием бензоилхлорида на водно-щелочной раствор Фацето-
ацетильного производного с последующей обработкой промежуточ-
ного образующегося ариламида ацетилбензоилуксусной кислоты
аммиаком в водном растворе; при этом отщепляется ацетильный
остаток:
С6Н5ССк
CH3COCH2CONHAr + С6Н5СОС1 —> ^CHCONHAr —>
СНзСО/
—> CeH6COCH2CONHAr
Реакция имеет общий характер и позволяет получать из ацето-
ацетильных производных самые разнообразные ариламиды оксо-
карбоновых кислот [1, с. 562].
Ариламиды ацетоуксусной и З-гидрокси-2-нафталинкарбоновой
кислоты используются не только в качестве азотолов, но и приме-
няются в больших количествах в синтезе пигментов для лакокра-
сочной промышленности [52].
14.3. ЗАМЕЩЕНИЕ В ГИДРОКСИ- И МЕРКАПТОГРУППАХ
В неионизированном виде фенолы и тиофенолы значи-
тельно менее нуклеофильны, чем амины. Поэтому реакции алкили-
рования и ацилирования этих соединений предпочитают проводить
в Щелочных растворах — в условиях образования фенолят-анионов,
Обладающих высокой реакционной способностью.
407
14.3.1. О- и S-Алкилирование
Для О-алкилирования фенолов применяются галогенал-
килы, алкиловые эфиры серной кислоты и сульфокислот, а также
некоторые четвертичные аммониевые соединения, например лей-
котроп— хлорид бензилдиметилфениламмония (ср. 14.2.1). Фено-
лят-анионы обычно реагируют с этими соединениями по синхрон-
ному Sn2 механизму
АгО' + СНзХ —> [АгО ... СНз ... X] —> АгОСНз + X'
или же по механизму, при котором медленной стадией является
образование алкил-катиона (механизм SN1).
Для О-метилирования можно использовать метилхлорид, одна-
ко процесс в этом случае приходится вести под давлением. Поэтому
часто предпочитают пользоваться диметилсульфатом или ме-
тил-н-толуолсульфонатом — н-СНзС6Н45ОзСНз.
С диметилсульфатом взаимодействие протекает в две стадии;
ArONa -|- (CH3O)2SO2 —> АгОСНз -Ь NaOSO2OCHg
ArONa + CH3OSO2ONa —> АгОСНз + Na2SO4
В первой стадии реакция идет с большой легкостью уже при
температуре значительно ниже 100 °C, в то время как во второй, где
алкилирующим агентом служит соль метилсерной кислоты, требу-
ются более жесткие условия.
Диметилсульфат используют при получении метилового эфира
лг-крезола, диметилового эфира гидрохинона, а также крезидина
(метилового эфира 2-амино-м-крезола); синтез последнего идет по
схеме:
Из резорцина действием диметилсульфата получают моно- и
диметиловый эфиры:
Монометиловый эфир (лг-метоксифенол) применяется в каче,
стве азосоставляющей в диазотипных бумагах и кальках. Для того,:
чтобы получить его с хорошим выходом, реакцию метилирования^
ведут в присутствии не смешивающегося с водой органического
растворителя и при pH ^9 4-10. В этих условиях образующийся'
403
монометиловый эфир резорцина переходит в органическую фазу и
благодаря этому не вступает в повторную реакцию алкилирова-
ния.
Для этилирования и введения более сложных алкилов обычно
используют соответствующие галогениды или алкилсульфонаты.
В качестве алкилирующего агента для фенолов часто использу-
ют хлоруксусную кислоту. Реакция проходит очень гладко при
нагревании водного раствора фенолята с натриевой солью этой
кислоты. Например:
OCH2COONa + NaCl
Получающаяся 2,4-дихлорфеноксиуксусная кислота, ряд ее
аналогов и сложных эфиров находят обширное применение в каче-
стве ядохимикатов для сельского хозяйства.
В производстве промежуточных продуктов применяется и реак-
ция гидроксиэтилирования фенольных ОН-групп. Реагентами слу-
жат хлоргидрин этиленгликоля, а также окись этилена. С хлор-
гидрином берется эквимолекулярное соотношение реагентов и ще-
лочи.
При гидроксиэтилировании окисью этилена вводят небольшое
количество фенолята щелочного металла, который катализирует
реакцию. Скорость этой реакции пропорциональна концентрации
фенолята. Процесс идет по схеме:
О
/ \ +н+
АгО’ + Н2С--СН2 —> АгОСН2СН2О' ------> АгОСН2СН2ОН
При недостатке фенола и избытке окиси этилена образовав-
шийся сначала алкоксил-анион не успевает нейтрализоваться фе-
нолом и реагирует с окисью этилена—-идет реакция полиоксиэти-
лирования с образованием 7—10 и более 0-оксиэтильных звеньев.
Получающиеся продукты являются ценными неионогенными по-
верхностно-активными веществами (ОП-7, ОП-Ю и др.). Их обычно
получают [47, с. 351], нагревая n-алкилфенолы с окисью этилена
при 150—180°C в присутствии 0,3% NaOH или CHsONa. Реакция
идет по схеме:
КС6Н4ОН + и(СН2—-СН2) —> RC6H4O(CH2CH2O)n-iCH2CH2OH
Определенные затруднения возникают при необходимости из-
бирательного алкилирования гидроксильной группы в соединении,
содержащем также аминогруппу. В ряде случаев их удается пре-
одолеть, проводя реакцию в щелочной среде, так как обычно фе-
нолят-анион более нуклеофилен, чем аминогруппа. В других
случаях, когда в соединении более активна аминогруппа, этого
409
оказывается недостаточно. В таких соединениях перед О-алкили-
рованием аминогруппу предварительно защищают ацильной груп-
пой:
-NH—
—NHCOCH3 —>
NH-
ОН
--NHCOCHa
ОСН3
О реакциях, дающих возможность получать алкиловые эфиры
нафтолов, антролов, резорцина, флороглюцина и некоторых других
при действии спиртов в присутствии кислот по механизму нуклео-
фильного ароматического замещения см. 8.2.
В сравнительно редких случаях О-алкилирование используется
как способ временной защиты фенольного гидроксила. При этом
возникает вопрос о возможности последующего снятия алкильной
группы — реакции дезалкилирования. Оно достигается более или
менее длительным нагреванием эфиров в сильнокислых средах — в
бромистоводородной кислоте или ее смесях с уксусной кислотой, с
безводными гидрохлоридами или гидробромидами пиридина или
же с А1С1з в присутствии НС1 [53, с. 361].
Дезалкилирование проходит через промежуточное образование
оксониевых солей по схеме:
АгОСНз .. t HBr..> АгОНСНз Вг’ —> АгОН + СН3Вг
Так как скорость таких реакций лимитируется стадией распада
оксониевой соли с образованием карбкатиона, она зависит от ‘
строения алкильной группы. Труднее всего дезалкилируются мети- i
ловые эфиры, а легче всего — бензиловые и особенно трифенилме-
тиловые. Гидролиз последних проходит очень быстро при обработ-
ке раствором НВг в уксусной кислоте уже при 10 °C [54, с. 51].
Гваякол, например, чтобы получить из него пирокатехин, кипя- 1
тят 6—7 ч с 48%-ной НВг. Непрореагировавший гваякол, отгоняю- •
щийся при этом с водяным паром, отделяют в отстойнике и воз- )
вращают в реактор. Выход пирокатехина 69—72% [55]. ।
Тиофенолы по реакционной способности напоминают уже рас-
смотренные фенолы. S-Алкилирование их обычно проводят в тех ]
же условиях, что и О-алкилирование фенолов, т. е. в водно-щелоч- I
ной среде. При этом тиофеноляты, более нуклеофильные, чем фе- j
ноляты, легко вступают в реакцию. 1
Наиболее часто для S-алкилирования тиофенолов прибегают к 1
хлоруксусной кислоте. Например, из тиосалициловой (2-меркапто- ]
бензойной) кислоты (48) при этом образуется 5-(о-карбоксифе- i
пил) — тиогликолевая кислота (49), применяющаяся при синтезе |
тиоиндигоидных красителей: ]
O/SNa ^^^SCHjCOONa I
+ClCH2COONa —> Г + NaCl j
^COONa ^-"^COONa 1
(48) . (49) I
410
Для тех же целей аналогичным способом получают 5-(2-ме-
гиЛ.4-хлорфенил)тиогликолевую (50) и S- (2-нафтил)тиогликоле-
вую (51) кислоты и другие продукты.
Интересно отметить, что благодаря высокой нуклеофильности
тиофенолятная группа может быть избирательно проалкилирована
при наличии в соединении аминогруппы. Так получают в частности
5-(2-амино-5-хлорфенил)тиогликолевую (52) и S-(2-амино-З-ме-
тил-5-хлорфенил)тиогликолевую (53) кислоты.
(52)
NH2
SCH2COOH
Продукт алкилирования 2,4,5-трихлортиофенола хлоруксусной
кислотой используется в качестве ядохимиката.
S-Алкильные производные в щелочной среде могут быть полу-
чены алкилированием не только тиофенолов, но и легко образую-
щихся из них при окислении дисульфидов. Реакция идет по урав-
нению:
2ArSSAr + 4ClCH2COONa + 4NaOH —>
—> 3ArSCH2COONa + ArSO2CH2COONa + 4NaCl + 2H2O
Обзор методов расщепления тиоэфиров см. [56].
14.3.2. О- и S-Ацилирование
Для О-ацилирования фенолов чаще всего используют
хлорангидриды и ангидриды кислот, обычно в водно-щелочном
растворе. Так как гидроксид-анионы катализируют гидролиз
сложных эфиров, реакцию ведут на холоду, постепенно добавляя
ацилирующий агент (реакция Шоттен — Баумана) [57]:
АгО- + RCOC1 —> ArOCOR + СГ
ArO' + (RCO)2O —> ArOCOR + RCOO"
Кроме водно-щелочной среды можно вести О-ацилирование
фенолов в среде пиридина. Хороший растворитель, он, кроме того,
катализирует реакцию: вызывает частичную ионизацию фенола и
образует с ангидридами и хлорангидридами кислот очень реакци-
онноспособные продукты присоединения типа хлорида АГ-бензоил-
пиридиния:
csH6N + С1СОС6Н5 —> C5H5NCOCsHs СГ
411
I
J
s
Последние реагируют с фенолят-анионами: [
C5H5NCOC8H5 + ArO' —> ArOCOC8H5 + C5H5N
По сравнению с водно-щелочными растворами преимущество
пиридина еще и в том, что в этой среде не может идти гидролиз;
сложных эфиров.
При ацилировании аминофенолов или аминонафтолов прежде
всего идет А-ацилирование, т. е. реакция за счет более нуклео-
фильной аминогруппы, и гидроксильная группа не затрагивается
(ср. 14.2.3). Но если реакцию проводить в сильнощелочной среде,:
когда фенольный гидроксил ионизирован, в первую очередь идет'
О-ацилирование за счет гидроксильной группы. Этот путь избирав
тельного ацилирования по гидроксильной группе находит разнооб-,
разное практическое применение. !
Аналогично, при действии бензолсульфохлорида на 4-ами-]
но-5-гидрокси-2,7-нафталиндисульфокислоту (Аш-кислота) в вод-1
ном сильнощелочном растворе на холоду получается 4-ами-]
но-5-бензолсульфонилокси-2,7-нафталиндисульфокислота, исполь-'
зуемая в качестве диазосоставляющей при синтезе красителей
[см. 15.1, формула (13)]. ]
Обратную реакцию — гидролиз сложных эфиров карбоновых]
кислот и сульфокислот, проводят, нагревая их с 5—10%-ным вод-]
ным раствором щелочи, или, реже, с разбавленными кислотами.]
Из эфиров фенолов с неорганическими кислотами известны И]
используются в технике эфиры фосфористой и фосфорной кислот.)
Их получают, действуя на фенолы соответственно хлоридом фос-.
фора(Ш) или оксохлоридом фосфора(V) в присутствии основа-;
ний:
ЗСвН5ОН + РС13 (С8Н6О)3Р + ЗНС1 (
ЗСвН5ОН+РОС13 —> (С8Н8О)3РО + ЗНС1 !
Триарилфосфиты и триарилфосфаты находят применение в ка-;
честве растворителей, пластификаторов, а также как ядохимикаты]
в сельском хозяйстве. Их начинают также использовать и в синтезе:
промежуточных продуктов (см., например, 5.2.2). 1
Значительно большее значение при получении промежуточных,
продуктов имеют эфиры серной кислоты — арилсульфаты. Их]
обычно получают, действуя на фенолы продуктом присоединения!
серного ангидрида к пиридину — пиридинсульфотриоксидом (по-|
лучение см. 3.1). Реакцию ведут в избытке пиридина: ;
ArOH + C5H5NSO3 —> ArOSO3 C5H5NH —> ArOSO3Na + C8H5N
Получающийся при этом арилсульфат пиридиния растворяют в,
воде, переводят в арилсульфат натрия, добавляя необходимое КО'
личество соды и высаливают из раствора хлоридом натрия.
Важнейшее применение арилсульфатов в практике промежу-.
точных продуктов и красителей — синтез водорастворимых форм;
412
кубовых красителей (индигозолей и кубозолей). Их получают вос-
становлением кубового красителя, например индиго (54), в лейко-
соединение (55) в пиридиновом растворе (см. 16.4.2) и этерифика-
цией образовавшихся фенольных гидроксильных групп при дей-
ствии пиридинсульфотриоксида. Заключительные операции выделе-
ния получающихся сернокислых эфиров (56) не отличаются от уже
описанных [58, с. 1197].
(56)
Таким же образом могут быть получены сернокислые эфиры
антрагидрохинона и его производных. Гидролиз арилсульфатов
легко проходит при нагревании с разбавленными кислотами и ще-
лочами.
При действии пиридинсульфотриоксида на тиофенолы образу-
ются S-арилтиосульфаты:
ArSH+ C5H5NSO7 —> ArSSO3H • C5H5N —> ArSSO3Na
Щелочные соли S-арилтиосерной кислоты (соли Бунтэ) [59]
устойчивы в нейтральном водном растворе, а при обычной темпе-
ратуре и в кислом водном растворе. При повышении температуры
начинается постепенный гидролиз:
ArSSO3H + Н2О —> ArSH + H2SO4
В щелочных растворах гидролиз идет быстро, причем получа-
ется диарилдисульфид, аренсульфиновая кислота и соли сернистой
кислоты:
3ArSSO3Na + 4NaOH —> ArSSAr + ArSO2Na + 3Na2SO3 + 2H2O
Это превращение солей Бунтэ используется для закрепления на
пьЩБЭ]6 некотоРых красителей, содержащих тиосульфатные груп-
413
ЛИТЕРАТУРА
1. Ворожцов И. Н. Основы синтеза промежуточных продуктов и красителей.
Изд. 4-е. М., Госхимиздат, 1955. 2. Ворожцов И. И. мл. — В кн.: Химия синте-
тических красителей. Т. 3/Под ред. К. Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса — Л., Химия, 1974, с. 1763. 3. Козлов Е. М. и др. Авт. свид. СССР
213783, 1968. 4. Андрухов Н. А. и др. Авт. свид. СССР 273186, 1970. 5. Фрид-
ланд С. В., Чернокальский Б. Д.—Успехи химии 1978, т. 47, вып. 8, с. 1397—
1413. 6. Vorozhtsov N. N. jr. — In: Aromatic Compounds. Ed. H. Zollinger, London,
Butterworths; Baltimore, Univ. Park press, 1973, p. 325. 7. Спирин Ю. Л.—
Успехи химии, 1969, т. 38, вып. 7, с. 1201—1222; Zavitsas A. A., Pinto J. А.—
J. Amer. Chem. Soc, 1972, v. 94, № 21, p. 7390—7396.,8. Huang R. L., Lee K. H.~
J. Chem. Soc. (C), 1966, № 10, p. 932—935. 9. Lee К. H., Teo T. 0. — J. Chem.
Soc. Perkin 2, 1973, № 6, p. 683—693; Chem. Communs, 1970, № 14, p. 860—861.
10. Lee К. H, — J. Chem. Soc. Perkin 2, 1973, № 6, p. 693—696.
11. Днепровский А. С, Касаточкин A. H., Темникова T. И. — ЖОрХ, 1974,
т. 10, вып. 10. c. 2136—2139. 12. Днепровский А. С., Касаточкин А. //. — Там
же, 1975, т. 11, вып. 6. с. 1350. 13. Фирц-Давид Г. Э., Бланже Л. Основные
процессы синтеза красителей. Пер. с нем./Под ред. С. В. Богданова и И. В. Фо-
димана. М., ИЛ, 1957. 14. Пат. ФРГ 2303115, 1973, Chem. Abstr., 1973, v. 79,
104907. 15. Момот В. Л., Бондарь Ю. Р. и др. Авт. свид. СССР 449025, 1975.
16. Снелл Дж., Вайсбергер А. — В кн.: Синтезы органических препаратов. Сб. 3.
Пер. с англ. М, ИЛ, 1952, с. 397—399. 17. Хавкине Э. Дж. Э. Органические
перекиси, их получение и реакции. Пер. с англ./Под ред. Л. С. Эфроса. Л., Хи-
мия, 1964. 18. Иоффе Б. В., Эфрос Л. С. — Реакционная способность органиче-
ских соединений, 1972, т. 9, вып. 4, с. 1165—1186. 19. Порай-Кошиц А. Е. Из-
бранные труды. М., Изд-во АН СССР, 1949, с. 186—261. 20. Зацепина Н. Н.,
Тупицын И. Ф. — Химия гетероцикл, соед., 1974, № 12, с. 1587—1612.
21. Беннет Дж., Белл Е. — В кн.: Синтезы органических препаратов. Сб. 2.
Пер. с англ. М„ ИЛ, 1949, с. 224. 22. Пат. США 3514495, 1970; Chem. Abstr.,
1970, v. 73, 35025. 23. Шубина Л. В., Малкее Л. Я.— ЖОрХ, 1965, т. 1, вып. 5,
с. 896—898. 24. Фикен Г. — В кн.: Химия синтетических красителей. Т. 4/Под
ред. К- Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия, 1975,
т. 4, с. 207. 25. Russel G. A., Becker Н. D. — J. Amer. Chem. Soc., 1963, v. 85,
№ 21, p 3406—3410. 26. Becker H. D. — J. Org. Chem., 1964, v. 29, № 10, p. 2891—
2894. 27. Франц, пат. 1506629, 1967; Chem. Abstr., 1969, v. 70, 38906. 28. Sieg-
rist A. E.. Liechti P., Meyer H. R. e.a. — Helv. chim. acta, 1969, v. 52, № 8,
p. 2521—2554. 29. Siegrist A. E. — Ibid., 1967, v. 50, № 3, p. 906—957; Sieg-
rist A. E. e.a. — Ibid., 1978, v. 61, № 11, p. 129—170. 30. Маеркер A. — В кн.:
Органические реакции. Сб. 14. Пер. с англ. М., Мир., 1967, с. 287—530.
31. Вильсон Ф. Г., Уилер — В кн.: Синтезы органических препаратов. Сб. 1.
Пер. с англ. М., ИЛ, 1949, с. 85—87. 32. Стрела Дж. М. — В кн.: Химия синтети-
ческих красителей. Т. 3/ Под ред. К. Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса. Л., Химия, 1974, с. 2029. 33. Зигель Э. — Там же. Т. 6. Л., Хи-
мия, 1977, с. 16; Эфрос Л. С., Томчин А. Б. — ЖОХ, 1963, т. 33, вып. 7, с. 2321—
2327. 34. Терентьев А. П., Кост А. Н. — В кн.: Реакции и методы исследования
органических соединений. Кн. 2/Под ред. С. С. Наметкина, В. М. Родионова,
Н. И. Мельникова. М. — Л., Госхимиздат, 1952, с. 47—208. 35. Бэр Д. Р. —
В кн.: Химия синтетических красителей. Т. 4/Под ред. К- Венкатарамана. Пер.
с англ, под ред. Л. С. Эфроса. Л., Химия, 1975, с. 163—204. 36. Эмерсон В.—
В кн.: Органические реакции. Сб. 5. Пер. с англ. М., ИЛ, 1951, с. 347—432.
37. Cordes Е. И., Jencks W. Р. — J. Amer. Chem. Soc., 1962, v. 84, № 5, p. 832—
837; 1963, v. 85, № 18, p. 2843—2848. 38. Ремизов А. Л., Порай-Кошиц Б. A.—
В кн.: Сборник статей по общей химии. Т. 2. Л., Изд-во АН СССР, 1953,
с. 1570—1597. 39. Пат. США 2513996, 1950; Chem. Abstr., 1950, v. 44, 8361. Пат.
США 2765340, 1956; Chem. Abstr., 1957, v. 51, 5826. 40. Toth G„ Pinter I., Mes-
smer A. — Tetrahedron Letters, 1974, № 9, p. 735—738.
414
41. Красовицкий Б. М., Болотин Б. М. Органические люминофоры. Л., Хи-
мия, 1976. 42. Хискин Р. О., Рао В. Р., Роудс В. Дж. — В кн.: Защитные группы
в органической химии/Под ред. Дж. Мак Оми. Пер. с англ. М., Мир, 1976,
с 235. 43. Gibson Н. W.—Chem. Rev., 1969, v. 69, № 5, р. 673—692.
44. Закс Э. Р., Стрелец Т. Р., Эфрос Л. С. — Химия гетероцикл, соед., 1970, № 10,
с. 1387—1392. 45. Доналдсон Н. Химия и технология соединений нафталинового
ряда. Пер. с англ./Под ред. А. И. Королева. М„ Госхимиздат, 1963. 46. FIAT,
v. 1016, р. 12. 47. Лебедев Н. Н. Химия и технология основного органического
и* нефтехимического синтеза. Изд. 2-е. М., Химия, 1975. 48. Стид К. В. — В кн.:
Химия синтетических красителей. Т. З/Под ред. К. Венкатарамана. Пер. с англ,
под ред- Л. С. Эфроса. Л., Химия, 1974, с. 1924. 49. Киридес Л. — В кн.: Син-
тезы органических препаратов. Сб. 3. Пер. с англ. ЙЛ, 1952, с. 464—467.
50. Брилл У., Бейкер Ж. — В кн.: Мономеры для поликонденсации. Пер. с англ,
под ред. В. В. Коршака. М., Мир, 1976, с. 393.
51. Перекалин В. В., Соколова Т. А. — Успехи химии, 1956, т. 25, № 11,
с. 1351—1372. 52. Ленуар Дж. — В кн.: Химия синтетических красителей. Т. 5/
Под ред. К- Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия,
1977, с. 274. 53. Вейганд-Хильгетаг. Методы эксперимента в органической химии.
Пер. с нем./Под ред. Н. Н. Суворова. М„ Химия, 1968. 54. Бартон Дж. У.—
В кн.: Защитные группы в органической химии/Под ред. Дж. Мак Оми. Пер.
с англ. М„ Мир, 1976, с. 51. 55. Кларк, Тейлор — В кн.: Синтезы органических
препаратов. Сб. 1. Пер. с англ. М., ИЛ, 1949, с. 347. 56. Хаслам Е. — В кн.: За-
щитные группы в органической химии/Под ред. Дж. Мак Оми. Пер. с англ. М.,
Мир, 1976, с. 153. 57. In: Methoden der organischen Chemie (Houben-Weyl).
Stuttgart, Georg Thieme Verlag, 1952, Bd. 8, S. 545; 1958, Bd. 11/2, S. 478.
58. Венкатараман К. Химия синтетических красителей. Т. 2. Пер. с англ./Под
ред. Н. С. Вульфсона. Л., Госхимиздат, 1957, с. 1197. 59. Weston S. D. — In:
The Chemistry of Synthetic Dyes. V. 7/Ed. K. Venkataraman. N. Y. — London,
Academic press, 1974, p. 35.
Глава 15
ДИАЗОТИРОВАНИЕ.
СТРОЕНИЕ И РЕАКЦИИ ДИАЗОСОЕДИНЕНИИ
Выше были рассмотрены реакции ацилирования аминов
различными кислотами или их производными и свойства образую-
щихся А/-ацильных производных (14.2.3). Рассматривались также
реакции взаимодействия вторичных и третичных аминов с азоти-
стой кислотой, приводящие к получению N- и С-нитрозопроизвод-
ных (см. 4.5.1), сходные с реакциями ацилирования. Однако при
взаимодействии первичных аминов с азотистой кислотой обра-
зуются, как это впервые обнаружил Петер Грисс, не /V-нитрозо-
производные, а диазосоединения, химия которых играет очень боль-
шую роль в синтезе промежуточных продуктов и красителей.
Диазосоединения могут вступать в многочисленные и разнооб-
разные реакции. Они могут проявлять себя как электрофильные
реагенты в реакциях азосочетания, в качестве ароматических суб-
стратов в реакциях нуклеофильного замещения и как источники
Радикалов в реакциях ароматического свободнорадикального за-
мещения. Наконец, диазосоединениям свойственны своеобразные
415
фотохимические превращения, Ё настоящей главе рассматривают-
ся способы получения и строение диазосоединений, а также основ-
ные особенности их реакционной способности с целью дать общие
основы химии диазосоединений в той мере, в какой она связана с
синтезом промежуточных продуктов.
15.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ
О РЕАКЦИИ ДИАЗОТИРОВАНИЯ
Обычно реакцию диазотирования проводят, приливая
раствор нитрита натрия к амину, растворенному в водной, чаще
всего соляной, кислоте, при размешивании и низкой температуре:
ArNH2 + NaNO2 + 2НС1 —> ArN2Cl + NaCl + 2Н2О
Количество НС1, как видно из приведенного уравнения, должно
составлять два эквивалента. Фактически кислоту надо брать с
небольшим избытком в 0,2—1 эквивалент с тем, чтобы в конце
реакции pH реакционной массы находился в пределах 0,5—1,5. При
недостаточной кислотности среды реакция диазотирования не про-
ходит до конца, главным образом из-за двух побочных реакций, в
которых участвует образовавшееся диазосоединение и еще не ус-
певший вступить в эту реакцию исходный амин — образование
диазоаминосоединения ArNH—N=N—Аг, а также аминоазосо-
единения Аг — N=N—АгМНг. Избыток кислоты тормозит тече-
ние этих побочных реакций и одновременно несколько увеличивает
скорость основной реакции.
Что касается нитрита натрия, то его применяют в строго рас-
считанном по теории количестве, следя за тем, чтобы к концу
реакции в растворе обнаруживался очень небольшой избыток азо-
тистой кислоты. Его устанавливают с помощью иодкрахмальной
бумажки, или, что надежнее, с помощью красителя метанилового
желтого (1) или так называемого сульфонреактива (2) [1, с. 12],
дающего с азотистой кислотой сине-зеленую окраску.
(2)
О)
В настоящее время, однако, предпочитают осуществлять непре-
рывный контроль реакции диазотирования по содержанию в реак-
ционной массе азотистой кислоты, измеряя окислительно-восстано-
вительный потенциал системы редоксметром [2].
После окончания реакции небольшой избыток азотистой кисло-
ты, сохраняющийся в реакционной массе, уничтожают, добавляя
мочевину или сульфаминовую кислоту, быстро реагирующие с азо-
тистой кислотой:
H2NCONH2 + 2HNO2 —> СО2 4-2N2 4-ЗН2О
H2NSO2OH 4- HNO2 —> H2SO4 4-N2 4-Н2О
416
Заметный избыток этих реагентов нежелателен, так как они
способны медленно реагировать и с диазосоединениями, превра-
щая их в исходные амины:
ArN2Cl +H2NSO3H + H2O —> ArNH2 + N2 + H2SO4 + НС!
В большинстве случаев диазосоединения неустойчивы и их раз-
ложение ускоряется при повышении температуры. Диазотирова-
ние— экзотермическая реакция с тепловым эффектом около
135 кДж/моль (например, для n-хлоранилина эта величина со-
ставляет 117 кДж/моль) [3, с. 219]. Именно поэтому диазотиро-
вание ведут с охлаждением (чаще всего при 0—10 °C), непосред-
ственно добавляя лед в реакционную массу или с помощью зме-
евиков, через которые циркулирует охлажденный рассол. Лишь в
отдельных случаях при получении термостабильных диазосоедине-
ний, например из п-амино-А^,А^-диметиланилина (М,А/-диметил-п-
фенилендиамин) и его аналогов, для успешного проведения реак-
ции температуру поднимают до 40—50 °C и выше.
При диазотировании в водной среде (основной способ проведе-
ния реакции) большое значение имеет растворимость исходного
амина в разбавленной кислоте или возможность получения его
мелкодисперсных взвесей. Затруднения, возникающие в связи с
этим, например при диазотировании п- и о-нитроанилинов, успешно
преодолеваются тщательным измельчением продуктов перед реак-
цией или длительным (иногда в течение суток) размешиванием
кислых водных суспензий. Используются и другие приемы, которые
варьируют в зависимости от свойств взятого амина [3, с. 219].
Например, при диазотировании о- и n-нитроанилинов, образую-
щих очень активные диазосоединения, способные образовывать
диазоаминосоединения даже в сильнокислой среде, рекомендуется
быстро прилить весь раствор NaNO2, необходимый для реакции.
При диазотировании аминов, дающих менее активные диазосоеди-
нения, раствор NaNO2 приливают постепенно, так как при этом
Удобнее поддерживать постоянную температуру.
В случае особенно плохо растворимых аминосульфокислот и
некоторых аминоазокрасителей, содержащих сульфогруппы, ис-
пользуется иной порядок проведения диазотирования: водно-ще-
лочной раствор амина, содержащий необходимое количество
NaNO2, медленно вливают в охлажденный раствор соляной или
серной кислоты при хорошем перемешивании.
Диазотирование слабоосновных аминов, таких как 2,4-динитро-
анилин, 4-нитро-2,6-дихлоранилин, аминоантрахиноны, не удается
провести в водной среде. Их диазотируют в концентрированной
серной кислоте действием нитрозилсульфата. Последний получают
растворением сухого измельченного нитрита натрия в концентри-
рованной серной кислоте при нагревании:
2H2SO4 4- NaNO2 —> ONOSO3H + NaHSO4 + Н2О
К полученному раствору нитрозилсульфата при 15—25 °C мед-
, ленно присыпают измельченный амин при перемешивании. После
14 Злк. 2'1
417
окончания реакции раствор выливают на лед и фильтруют от
осадка амина, не вступившего в реакцию диазотирования.
Для диазотирования используют также твердый нитрозилсуль-
фат, который получают, пропуская SO2 в дымящую азотную кис-
лоту при охлаждении до полного затвердевания всей реакционной
смеси. Продукт отмывают на фильтре небольшим количеством
уксусной кислоты, а затем СС14; выход 70% [4].
Многие о- и га-амннофенолы бензольного и нафталинового ряда,
их сульфокислоты и другие производные нельзя диазотировать ни
одним из описанных методов, так как в кислой среде они окисля-
ются азотистой кислотой до соответствующих хинонов. Однако
диазотирование этих соединений в виде их гидрохлоридов или если
они содержат хотя бы одну свободную сульфогруппу, проходит
очень гладко при добавлении к их растворам NaNO2 в отсутствие
дополнительного количества кислоты и в присутствии либо эквива-
лентного количества ZnCl2, либо небольшого количества соли меди,
например медного купороса [3, с. 219].
Этим путем в технике диазотируют 4-амино-3-гидрокси-1-на-
фталинсульфокислоту (1-амино-2-нафтол-4-сульфокислота) (3)
для последующего использования в синтезе ценных металлсодер-
жащих азокрасителей, а также 6-амино-5-гидрокси-1-нафталин-
сульфокислоту (2-амино-1-нафтол-5-сульфокислота) (4), диазосое-
динение из которой применяется в светочувствительных составах
для радиоэлектроники, так называемых фоторезистах [5, е. 196].
ОН
SO3H
(4)
Продукты, образующиеся при диазотировании о-аминонафто-
лов типа (3) или (4), отличаются исключительной для рассматри-
ваемого класса соединений термостойкостью. Многие из них можно
сульфировать, нитровать, кристаллизовать из горячей воды без
разложения. Это их свойство объясняется тем, что они имеют
строение соответствующих нафтохинондиазидов. Так, при диазо-
тировании (3) получается 1,2-нафтохинон-1-диазид-4-сульфокисло-
та (5), а при диазотировании (4) — 1,2-нафтохинон-2-диа-
зид-5-сульфокислота (6) (ср. 15.4.4).
N=N
SO3H
(5)
О
SO3H
(6)
418
Затруднения возникают и при диазотировании о- и пери-, а
также и-диаминов. Из о-диаминов образуются очень стабильные и
неспособные вступать в реакцию азосочетания производные бен-
зотриазолов (7) или нафтотриазолов (8).
Аналогично из гаери-диаминов получаются соединения (9), со-
держащие триазиновое кольцо:
N
(9)
Бисдиазосоединения из о-диаминов удается получить при дей-
ствии нитрозилсерной кислоты в уксусной кислоте.
Диазотирование n-диаминов в обычных условиях невозможно
из-за окисления, при котором образуются хиноны. Реакцию удает-
ся провести в концентрированной серной кислоте, но практического
применения она не имеет. Для диазотирования, однако, часто ис-
пользуют производные А-ацил-м-фенилендиамина, вступающие в
реакцию как обычные первичные амины.
Осложнения возникают и при диазотировании ж-фенилендиами-
на. Здесь они связаны с тем, что образовавшееся диазосоединение
быстро вступает в реакцию азосочетания с ж-фенилендиамином,
еще не успевшим продиазотироваться. В результате получаются
сложные смеси продуктов. Успешно провести диазотирование
ж-фенилендиамина можно, добавляя его раствор к заранее приго-
товленному раствору соляной кислоты, содержащей необходимое
количество азотистой кислоты, образовавшейся в этом же растворе
из добавленного NaNO2.
Подобные осложнения при диазотировании свойственны не
только ж-фенилендиамину, но и всем аминам, обладающим повы-
шенной способностью к реакции азосочетания. Ярким примером
таких соединений являются 4-амино- и 5-аминобензо-1,2,5-тиадиа-
золы (Ю) и (11), которые удается диазотировать только в описан-
ных выше условиях [7].
Nh2
N N
НО) (11)
14*
419
Аналогичная тенденция наблюдается и при диазотировании 1-и
2-нафтиламинов. Ее удается преодолеть, используя повышенную
кислотность среды при реакции и возможно более быстрым добав-
лением всего необходимого раствора NaNC>2.
В случае некоторых аминонафтолов, например, при диазотиро-
вании 4-амино-5-гидрокси-2,7-нафталиндисульфокислоты (Аш-кис-
лота) (12) во избежание реакции самосочетания действием бен-
золсульфохлорида (см. 14.3.2) временно защищают фенольный,
гидроксил (13), а затем уже получают диазосоединение (14):
(14)
C6H5SO2O NH2
SO3H
(13)
15.2. МЕХАНИЗМ ОБРАЗОВАНИЯ
И СТРОЕНИЕ ДИАЗОСОЕДИНЕНИЙ
Итак, реакция диазотирования обычно проходит легко и
осуществляется в сравнительно простых условиях. Однако, как
справедливо утверждал Б. А. Порай-Кошиц, диазотирование ами-
нов является одной из сложнейших реакций в органической химии
[ 1, с. И]. Многолетние дискуссии по этому вопросу только срав-
нительно недавно завершились исчерпывающими работами К. Ин-
гольда и его школы [6, с. 512; 8].
Активными реагентами при диазотировании в водной среде
могут быть неионизированная азотистая кислота или продукт ее
протонирования — нитрозацидий-катион:
no; + н3о+ hno2 + н2о
hno2 + h3o+ h2no; + h2o
Нитрозацидий-катион является очень активным реагентом ре-
акции диазотирования, но кинетические данные показывают, что он
реагирует с амином приблизительно на порядок медленнее, чем с
неорганическими анионами, которые могут присутствовать в вод-
ном растворе. В результате образуются новые активные реагенты:
h2no; + no; on-ono+h2o
h2no; + сг +=> on—ci + н2о
H2NOJ + Br' <=* ON—Вг + Н2О
h2no; + hso; on—oso3h + h2o
420
Таким образом, реакция диазотирования в водных средах почти
всегда проходит под влиянием нескольких реагентов. Кинетические
уравнения этих реакций поэтому могут включать два и более
членов и имеют сложный порядок. Доля участия перечисленных
реагентов в реакции диазотирования определяется их концентра-
цией, зависящей от состояния приведенных выше равновесий, и их
активностью, которая падает в следующем ряду:
ON—OHJ > ON—Cl > ON—Br > ON—ONO > ON—OSO3 > ON—OH
Кинетические исследования позволили установить, что в водной
хлорной кислоте, анион которой не способен образовать ковалент-
ное соединение с нитрозогруппой, главным реагентом диазотиро-
вания является азотистый ангидрид ON—ONO. В разбавлен-
ной соляной кислоте таким реагентом выступает нитрозилхлорид
ON — Cl, а в бромистоводородной— нитрозилбромид ON — Вг.
В разбавленной серной кислоте (до 0,3 н.) основным диазотирую-
щим реагентом является азотистый ангидрид, а нитрозилсульфат в
виде ON—OSO3 или ON — OSO3H — в этих условиях существен-
ного значения не имеет. Наиболее активный реагент диазотирова-
ния — нитрозоний-катион NO+ в водных кислотах не образуется и в
реакциях не участвует. Он образуется только при проведении ре-
акции в концентрированной серной, кислоте в результате диссоциа-
ции нитрозилсульфата:
ON—OSO3H NO+ + HSO4'
Эти выводы хорошо согласуются с обсуждавшимися в
15.1 особенностями реакции диазотирования в различных средах.
Реакция в соляной кислоте всегда идет быстрее, чем в водной
серной кислоте, потому что нитрозилхлорид как реагент значи-
тельно активнее азотистого ангидрида. Ускорение реакции диазо-
тирования некоторых аминов в соляной кислоте при добавлении
бромидов (особенно часто — при титриметрическом определении
аминов) является результатом концентрированного эффекта: нит-
розилбромид на несколько порядков термодинамически более ста-
билен, чем нитрозилхлорид, и поэтому равновесная реакция его
образования сильно смещена вправо.
Наконец, приведенные данные позволяют объяснить, почему
диазотирование слабоосновных аминов необходимо проводить в
концентрированной серной кислоте: только в этих условиях обра-
зуется наиболее активный, способный с ними взаимодействовать,
диазотирующий реагент — нитрозоний-катион.
Реакция диазотирования проходит в сильнокислой среде, в
которой протолитическое равновесие сильно смещено вправо:
ArNH2 + H3O+ =₽=>: ArNHj + Н2О
Тем не менее диазотированию подвергается амин в виде осно-
Вания (15). Лимитирует суммарную скорость реакции, как это
421
предполагал еще в 1900 г. Бамбергер, стадия образования арил-
нитрозаммония (16).
ArNH2 + ON—X ArNH2—NO + X’
(15)
(16)
Получившаяся соль быстро депротонируется и далее, проходя
ряд быстрых превращений — нитрозоамин (17)—диазогидроксид
(диазогидрат) (18) — образует соль диазония (19);
ArNH2—NO + Н2О ч=> ArNH—NO + Н3О
(16)
(17)
ArNH—NO
Ar—N=N—ОН
+ Н3о
ч------
(17) (18)
Ar—N=N + 2Н2О
(10)
Продукты диазотирования ароматических аминов — довольно
лабильные и весьма реакционноспособные соли диазония — полно-
стью ионизированы в водных растворах и имеют строение:
+ в+
дг—х
Положительный заряд в их катионах в основном локализован
на атоме азота, связанном с ароматическим кольцом, но вследствие
индуктивного эффекта и показанного в формуле эффекта сопря-
жения частично передается и на смежный атом азота. Частичный
положительный заряд на последнем возрастает под влиянием
электроноакцепторных заместителей в ароматическом кольце и
уменьшается под действием электронодонорных заместителей, что
существенно сказывается на реакционной способности катиона.
Рентгеноструктурный анализ кристаллов солей диазония пока-
зывает, что диазоний-катион представляет собой плоскую, систему
с линейно расположенными атомами азота. Анион располагается
по одну сторону от этой плоскости, на расстоянии 3,2 А (0,32 нм)
от диазониевой группы, причем чуть ближе к крайнему атому
азота, чем к азоту, непосредственно связанному с ароматическим
кольцом [9]. Это свидетельствует о том, что оба атома азота несут
положительный заряд. Это же подтверждается квантовохимиче-
ским расчетом бензолдиазоний-катиона по методу ССП ППП [10]:
оказывается, положительный заряд на крайнем атоме азота до-
вольно велик и лишь вдвое меньше, чем на азоте, связанном с
бензольным кольцом. Порядок связи азот — азот тем не менее
довольно высок и составляет 2,8826.
Расчет показывает также, что диазониевая группа сильно вы-
тягивает электронную плотность из бензольного кольца, особенно
из лара-положения, положительный заряд которого составляет
422
0,1581. Индукционные константы о0 для n-нитро- и диазониевой
группы равны соответственно 0,89 и 1,56 [1].' Таким образом,
диазониевая группа — очень сильный электроноакцептор, по силе
почти вдвое превосходящий нитрогруппу.
При добавлении щелочи к раствору соли диазония гидроксид-
анион медленно связывается и образуется диазогидроксид (18):
+ ki
Ar—+ он 5=^ Ar—N=N—ОН
(19) (18)
С образованием этого соединения длительное время связыва-
лись многочисленные реакции дпазосоединений, в том числе и ре-
акция азосочетания. В действительности оказалось, что это не так
(см. 15.4.1). В отличие от диазоний-катиона (19), который является
кислотой Льюиса, диазогидроксид (18) является протонной кисло-
той средней силы и быстро реагирует с еще одним гидроксид-
анионом, образуя диазотат-анион (20):
^2
Ar—N=N—ОН + ОН" 4=^ Ar—N=N—О + Н2О
(18) (20)
Благодаря этому обстоятельству концентрация (18) в растворе
минимальна, и его не удается обнаружить никакими физическими
методами. Многие исследователи даже считают, что такое соеди-
нение не существует, так как реакция диазоний-катиона идет как
бы сразу с двумя гидроксид-анионами [11, с. 50].
Ar—N==N4-2OH~ —> Ar—N=N—О" + Н2О
(19) (20)
Тем не менее, используя данные потенциометрического титро-
вания и применяя специальную методику, удалось рассчитать кон-
станту кислотности диазоний-катиона, которая для различных со-
единений находится в пределах 10-10—10-13, и диазогидроксида
Для тех же соединений — Ю-0 —10-10.
Значит, при кислотности среды, соответствующей области пере-
хода (19) в (20), т. е. при pH = + р^2 , (18) должен присут-
ствовать в соизмеримом количестве с этими двумя формами — 7%
при 0 и 22% при 20 °C, независимо от характера заместителя в
ароматическом кольце. Повышение ионной силы раствора увеличи-
вает кислотность и основную диссоциацию диазогидроксида, в
связи с чем резко уменьшает его содержание [12].
Подкисление раствора (20) приводит к смещению реакции
влево и быстрому образованию (19). Однако, если раствор (20)
оставить на некоторое время в щелочной среде или нагреть со
423
щелочью, то он превращается в более стабильную соль того же со-1
става; последняя при подкислении лишь очень медленно переходит|
в соль диазония, причем эта реакция идет через промежуточное]
образование нитрозамина. Ганч, открывший это явление,, объяснил ]
его тем, что образующийся вначале диазотат-анион имеет сан- ?
строение (21), а более стабильный продукт его изомеризации —
а«ти-строение (22):
Ar—N Ar—N ,н+ Ar—NH ]
II — II I ।
О—N N—О' N=O |
(21) (22) (17) j
£
i
Получающийся при подкислении антп-диазотата нитрозамин \
(17) в ряде случаев может быть выделен, но, оставаясь в кислой]
среде, он медленно через (23) изомеризуется в соль диазония (19). j
Интересно отметить, что реакция эта, по-видимому, проходит че-1
рез те же этапы, что и диазотирование: ;
+ н+ + + !
ArNH—N=O -------> ArNH=N—ОН —> ArtesN + Н2О
(17) (23) (19)
Медленной стадией этой реакции, вероятно, является процесс:
отщепления воды от (23) [1, с. 38].
Скорость изомеризации сик-диазотата (21) в а«ты-диазотат|
(22) очень сильно зависит от влияния заместителей в ароматиче-j
ском кольце. Электроноакцепторные заместители облегчают тече-j
ние этих превращений. Так, превращение п-нитробензолдиазоний-5
катиона в соответствующий ант и-диазотат проходит очень быстро в j
водно-щелочной среде. Возможность такой изомеризации необхо-;
димо учитывать при проведении реакции азосочетания (см. 15.4.1).]
В то же время для превращения п-метоксибензолдиазоний-катионаЛ
в соответствующий антц-диазотат необходимо нагревать соль диа-]
зония с концентрированным раствором щелочи при 130—140 °С.й|
анти-Диазотаты — стабильные соединения, неспособные встуЧ
пать в реакцию азосочетания. Их используют в технике как источ-j
ник получения солей диазония — при действии кислот или света.]
Описанные выше протолитические равновесия и свойства раз-1
личных форм диазосоединений установлены путем детального ис-|
следования соединений преимущественно бензольного ряда. Как]
видно из приведенных ниже данных [13], эти свойства могут!
существенно изменяться при переходе к диазосоединениям гетеро-!
ароматического ряда. |
Так, при диазотировании 2-амино-1-метилбензимидазола (24м
бутилнитритом или нитрозилсульфатом образуется М-нитрозамим
424
(25), существующий в иминной форме (26), в стабилизацию кото-
рой вносит значительный вклад цвиттерионная структура (27).
(24) (25)
(26) (27)
(29)
(28)
М-Нитрозамин (25) плохо растворим в воде и в органических
растворителях и вполне устойчив. В сильнокислой среде, например
в концентрированной серной кислоте, он вначале образует диазо-
гидроксид (28), а затем соль двузарядного диазоний-катиона
(29)—гетероциклического ряда. Такие соли обладают исключи-
тельно высокой активностью в реакции азосочетания (см. 15.4.1).
При диазотировании 1-аминофеназина (30) образуется 1,2-фе-
назинхинон-1-диазид (31), очень мало активный в реакциях азосо-
четания [14]:
(30)
(31)
О строении и реакционной способности других гетероцикличе-
ских диазосоединений см. [15].
425
15.3. СТОЙКИЕ ФОРМЫ ДИАЗОСОЕДИНЕНИЙ
Диазониевые соли сравнительно редко выделяют в сво-
бодном виде и в большинстве случаев их перерабатывают в виде
растворов или суспензий в воде. Чтобы выделить соль .диазония
диазотирование обычно ведут в спиртовом растворе при действии
алкилнитритов. Получаемые таким образом диазоний-хлориды в
большинстве неустойчивы и опасны в обращении, так как могут
взрывать.
Оказалось, однако, что с некоторыми другими противоионами,
например с BF$, ZnCll', SnCle', с анионами ароматических
сульфокислот, диазоний-катионы образуют соли, обладающие зна-
чительно более высокой стабильностью как в растворах, так и в
твердом состоянии [И, с. 61]. Это позволило начать промышлен-
ный выпуск стойких солей диазосоединений, главным образом для
применения в текстильной промышленности при ледяном крашении
[16, с. 752; 17]. Стойкие соли диазосоединений используют также
для изготовления диазотипных бумаг и калек.
Для получения стойких солей диазосоединений (по принятой в
СССР номенклатуре — диазолей) ароматические амины диазоти-
руют в возможно более концентрированном растворе, отфильтро-
вывают раствор от загрязнений и осаждают соль диазония добав-
лением ZnCk и NaCl или какого-нибудь другого реагента: HBF4,
SnCl4 или 1,5-нафталиндисульфокислоты. Осадок отфильтровыва-
ют, смешивают с безводными Na2SO4 или A12(SO4)3 и, если нужно,
досушивают в вакууме при температуре не выше 50 °C. Обычно в
выпускаемых диазолях содержится не более 20—25% соли диазо-
ния, остальное представляет собой наполнитель.
Из числа сульфокислот для стабилизации диазосоединений
обычно используют 1,5-нафталиндисульфокислоту. Борофтористо-
водородную кислоту HFB4, которая является превосходным ста-
билизатором солей диазосоединений, в чистом виде используют
редко, так как она дает недостаточно хорошо растворимые про-
дукты.
Почему противоионы оказывают на диазосоединения стабили-
зирующее действие? Этот вопрос давно обсуждается в литературе
[8]. Противоионы не образуют ковалентных соединений с диазо-
пий-катионом, так как в ПК-спектрах стойких солей всегда обна-
руживается характерная полоса N = N. Высказывают предполо-
жения, что стабилизация связана с образованием донорно-акцеп-
торных комплексов анионов с ароматическим кольцом диазосо-
единения. Вероятность такого взаимодействия подтверждается
данными приведенных выше рентгеноструктурных исследований
(см. 15.2). Образование КПЗ лишь незначительно уменьшает
электрофильность солей диазония и скорость реакции азосочета-
ния [18].
Кроме диазолей, способных вступать в реакцию азосочетания и
поэтому называемых активными формами стойких диазосоедине-
ннй, промышленность выпускает пассивные, не способные в обыч-
426
пых условиях сочетаться формы диазосоединений. Для того, чтобы
пассивная форма перешла в активную, ее раствор необходимо
нагреть со слабой! кислотой — уксусной или муравьиной, а иногда
и без кислоты.
К пассивным формам диазосоединений относятся арендиазо-
сульфонаты натрия (32), образующиеся при действии на растворы
солей диазония смеси сульфита и бисульфита натрия:
Ar—N^N + SO3Na“ —> Ar—N=N—SO3Na
(19) (32)
и арил (арилдиазо) сульфоны (33), получающиеся из диазониевых
солей и солей аренсульфиновых кислот:
Ar—N=N +’O2S—Аг' —> Ar—N=N—SO2—Аг'
(19) (33)
Соединения (32) и (33) не сочетаются с азосоставляющими, но
при нагревании, действии света или окислителей ковалентная связь
N—S разрывается и образуются соли диазония, способные всту-
пать в азосочетание.
Другим типом пассивных диазосоединений являются уже рас-
смотренные антц-диазотаты (22). Если в бензольном кольце соли
диазония имеется нитрогруппа, они легко образуются при вли-
вании раствора диазосоединения в 25%-ный раствор NaOH.
анти-Диазотат выделяют из раствора высаливанием, отфильтро-
вывают и смешивают с безводным ацетатом натрия. Этим путем в
промышленности получают анти-диазотат из о-нитроанилина (ни-
трозамин оранжевый О).
В настоящее время, однако, диазосульфонаты иантц-диазотаты
почти полностью утратили свое значение и заменены третьей фор-
мой пассивных диазосоединений — диазоаминосоединениями (ди-
азаминами) .
Диазоаминосоединения (34) образуются при взаимодействии
солей диазония (19) с первичными и вторичными ароматическими
(а также жирными) аминами в нейтральной или слабощелочной
среде:
Ar—N=N + H2NAr' + ОН’ —> Ar—N=N—NH—Аг' + Н2О
(19) (34)
При действии кислот диазоаминосоединения расщепляются с
образованием диазосоединения и амина, и на этом основан прин-
цип их применения. Важно, чтобы амин, использованный для такой
стабилизации соли диазония, сам не был бы способен вступать в
реакцию азосочетания. Необходимо также, чтобы диазоаминосое-
динение было растворимо в воде, и поэтому амин-стабилизатор
должен содержать сульфо- или карбоксильные группы.
Диазоаминосоединения, образованные из солей диазония и
первичных аминов, способны к таутомерии:
Ar—NH—N=N—Аг' Аг—N=N—NH—Аг'
427
При этом из двух таутомеров всегда более устойчив тот, в
котором группа —NH— связана с более электроноакцепторным
кольцом. Поэтому, например, при расщеплении кислотой диазо-
аминосоединения, полученного из соли и-нитробензолдиазония и
анилина, образуется п-нитроанилин и бензолдиазоний-катйон:
Это явление получило название «переноса» диазогруппы. Его
иногда используют для синтеза диазосоединений, которые трудно
получить иными способами.
Однако при получении диазоаминосоединений как пассивных
форм диазосоединений перенос диазогруппы от исходного диазо-
соединения на амин-стабилизатор является недопустимым. Для
того чтобы перенос диазогруппы был невозможен, в молекулу
амина-стабилизатора вводят электроноакцепторные заместители
или вместо первичных аминов применяют вторичные, образующие
диазоаминосоединения, не способные к таутомерии. В качестве
аминов-стабилизаторов чаще всего используют 5-сульфоантрани-
ловую кислоту (35), М-метил-5-сульфоантраниловую кислоту (36),
УУ-(о-карбоксифенил)глицин (37), а также саркозин (38) и метил-
таурин (39).
CH3NHCH2COOH
(38)
CH3NHCH2CH2SO3H
(39)
Диазоаминосоединения, используемые в качестве пассивных
форм диазосоединений (диазамины), получают следующим обра-
зом. Раствор диазотированного амина медленно приливают к со-
дово-щелочному раствору амина-стабилизатора, взятого с неболь-
шим избытком от теоретического количества. К концу реакции
раствор должен сохранять слабощелочную реакцию и в нем не
должна обнаруживаться способная к азосочетанию соль диазония.
Диазамин выделяют высаливанием, фильтруют и высушивают в
вакууме при 70—80 °C.
15.4. РЕАКЦИИ ДИАЗОСОЕДИНЕНИЙ
15.4.1. Азосочетание
Важнейшей реакцией диазосоединений является реак-
ция азосочетания, лежащая в основе получения азокрасителей —
обширнейшего класса органических красителей. В настоящем раз-
428
деле эта реакция рассмотрена лишь в общих чертах, как один из
методов синтеза промежуточных продуктов.
Азосочетание — типичная реакция электрофильного ароматиче-
ского замещения, причем из всех рассмотренных выше форм диа-
зосоединений в качестве активного реагента этой реакции высту-
пает только арендиазоний-катион [19].
Б.ензолдиазоний-катион, так же как и абсолютное большинство
используемых в практике его производных, сравнительно слабый
электрофил и вступает в реакцию азосочетания только с фенолами
(в слабощелочной среде) и с ароматическими аминами, т. е. с
соединениями наиболее сильно активированными для реакций
электрофильного замещения. Уже с метиловыми эфирами фенолов
и У-ацетильными производными аминов эти реакции не идут.
Вместе с тем электрофильность бензилдиазоний-катиона сильно
зависит от влияния заместителей в ароматическом кольце — воз-
растает под влиянием электроноакцепторных заместителей и
уменьшается под влиянием электронодонорных. Наблюдается хо-
рошая корреляция скоростей реакции азосочетания с о-константа-
ми заместителей в уравнении Гаммета, причем константа реакции
р имеет положительный знак. Так, n-нитробензолдиазоний сочета-
ется с фенолами в 1300 раз быстрее бензолдиазония, а п-метокси-
бензолдиазоний — в 10 раз медленнее [6, с. 308].
Увеличение электрофильности диазоний-катиона путем усиле-
ния электроноакцепторности связанного с ним ароматического
кольца позволяет расширить область применения реакции азосоче-
тания. Так, 2,4-динитробензолдиазоний способен сочетаться с ани-
золом и фенетолом, а 2,4,6-тринитробензолдиазоний реагирует да-
же с мезитиленом, образуя 2,4,6-триметил-2/,4',6/-тринитроазобен-
зол:
Однако и это не является пределом: в гетероциклическом ряду
получены соли еще более электрофильных диазоний-катионов (29),
способные сочетаться с толуолом и бензолом [20]. Хотя диазосое-
динения подобного рода практического значения не приобрели, они
важны как доказательство общего характера реакции азосочета-
ния.
Азосочетание идет по бимолекулярному механизму через про-
межуточное образование о-комплекса, превращающегося в конеч-
ный продукт при действии основания:
+ /N=N—Аг' + в
ArH + Ar'Nj =?=*: Аг ---> Ar—N=N—Ar'+ ВН*
Обычно вторая стадия реакции — отрыв протона основанием —
Не лимитирует ее скорость. Но в тех случаях, когда в орто- или
429
перг>положениях к месту сочетания находится сульфогруппа, по-
является значительный кинетический изотопный эффект и реакция
катализируется основаниями. Таким образом, в определенных слу-
чаях отрыв протона становится лимитирующей стадией реакции
азосочетания [11, с. 175].
От других реакций электрофильного ароматического замещения
реакция азосочетания отличается высокой селективностью, что
связано со слабой электрофильностью диазоний-катиона и, со-
ответственно, близостью переходного состояния к структуре
о-комплекса (см. 2.7.1). При проведении реакции в неводных сре-
дах ее скорость сильно зависит от свойств растворителя. Так,
константа скорости азосочетания с ¥,¥-диметиланнлином в нитро-
метане при проведении той же реакции в АД¥-дпметнлацетамиде
возрастает на три порядка [21].
Процесс азосочетания обычно проводят медленным приливани-
ем раствора диазосоединения к охлажденному и забуференному
раствору амина или фенола при размешивании. Контролируют
значение pH раствора: обычно при сочетании с аминами оно нахо-
дится в пределах 3—5, а с фенолами 8—10. Реакцию стремятся
провести таким образом, чтобы в конце раствор не содержал ни
соли диазония, ни избытка азосоставляющих — амина или фенола.
Растворимые в воде продукты азосочетания выделяют из раствора
высаливанием.
С большой легкостью идет восстановление азосоединений; про-
исходит разрыв азосвязи и образуется смесь аминов (см. 16,7);
Аг—N=N—Аг' —> Ar\'H2 + Ar'NFh
Поэтому в синтезе промежуточных продуктов реакцию азосо-
четания нередко используют как промежуточный этап для избира-
тельного введения в пара- и орто-положение к амино- или гидро-
ксильной группе аминогруппы. Например, 6-амино-5-гидрокси-
1-нафталинсульфокислоту (2-амино-1-нафтол-5-сульфокислота)
(41) получают в промышленности из 5-гидрокси-1-нафталинсуль-
фокислоты (40) по схеме [22, с. 419]:
О получении (41) восстановлением азосоединения см. также
16.7.
Диазосоединение, используемое в подобных реакциях, подбира-
ют с таким расчетом, чтобы амин (42), образующийся при восста-
430
новлении, можно было легко отделить от целевого продукта (41).
Например, если целевой продукт содержит сульфогруппы и, сле-
довательно, растворим в воде, для реакции следует использовать
дпазосоединение, не содержащее сульфогрупп, и т. д.
Уже было упомянуто, что реакция азосочетания отличается
высокой селективностью, отчего ориентация при этой реакции осо-
бенно сильно зависит от структурных и стерических факторов и
часто существенно отличается от ориентации при других реакциях
электрофильного ароматического замещения [16, с. 464].
Азосочетание с фенолом и его производными в отличие от
нитрования и сульфирования идет почти исключительно в пара-
положение к гидроксилу. Если пара-положение занято, как напри-
мер, в п-крезоле, сочетание идет в орто-положение. Однако о- и
и-нитрофенолы не способны вступать в реакцию азосочетания.
С двумя молекулами диазосоединения фенол может давать дис-
азосоединения; третье азосочетание идет с большим трудом и лишь
частично.
Салициловая кислота сочетается менее активно, чем фенол,
вследствие участия ее фенольного гидроксила в образовании водо-
родной связи. Тем не менее для технического применения скорость
азосочетания салициловой кислоты достаточна, и эта реакция ши-
роко используется в практике. Она идет исключительно в пара-
положение к гидроксилу.
Резорцин вступает в азосочетание с большой легкостью, причем
с первой молекулой соли диазония реакция идет преимущественно
(приблизительно на 90%) в положение 4 — образуется азосоеди-
нение (43). Со второй молекулой диазосоединения (43) реагирует
избирательно — в положение 2 и образуются дисазосоединения
(44). Это объясняют возникновением в (43) внутримолекулярной
водородной связи
(43)
N=N—Аг
Пирокатехин сочетается крайне медленно и не находит приме-
нения в этой реакции. Гидрохинон вызывает разложение диазосо-
единений, сопровождающееся окислением его в 1,4-бензохинон,
выделением азота и арилированием образующегося хинона.
Ароматические амины ряда бензола (анилины) сочетаются с
диазосоединениями в нейтральной, а чаще — в слабокислой среде.
С повышением кислотности скорость азосочетания уменьшается,
так как образуется соль амина, не способная к этой реакции.
С другой стороны, нейтральная среда благоприятствует взаимодей-
ствию диазоний-катионов и анилинов с образованием диазоамино-
соединений (45), которые изомеризуются в аминоазосоединения
431
(46) лишь в кислой среде, через промежуточное расщепление с
образованием диазоний-катиона (см. 15,3).
CsH5Nt + C6H5NH2 CsH5NH—N=N—CsH5 CSH5—N=N—C6H4NH2
(45) (46) .
Поэтому сочетание анилинов предпочитают вести в слабокислой
среде; таким образом во многих случаях удается провести реакцию
без промежуточного получения диазоаминопроизводного. Особен-
но легко это удается, когда в качестве азосоставляющих берут
л-фенилендиамин, 2,5-диметоксианилин, л-толуидин, 6-меток-
си-ж-толуидин (крезидин) и др. 4-Аминоазобензол и о-аминоазо-
толуол (4-амино-2',3-диметилазобензол) получают только через
перегруппировку диазоаминосоединений.
Вторичные амины, например дифениламин, и третичные амины,
например ДСА-диалкиланилины, гладко сочетаются с диазосоеди-
нениями в пара-положение.
л-Нитроанилин, .м-хлоранилин, метаниловая кислота и анало-
гичные соединения с электроноакцепторными заместителями в ре-
акцию азосочетания не вступают. Не могут быть введены в эту
реакцию также о- и п-фенилендиамины и о- и п-аминофенолы —
при действии диазосоединений они окисляются.
Ряд особенностей отличает азосочетание с нафтолами и нафтил-
аминами. 1-Нафтол и 1-нафтиламин сочетаются преимущественно
в положение 4. Однако направление реакции азосочетания 1-на-
фтола, по-видимому, зависит от pH среды и-еще более от активно-
сти соли диазония. С наименее активными солями диазония, име-
ющими строение хинондиазидов (см. 15.1), сочетание 1-нафтола
идет почти исключительно в положение 2, и получающиеся про-
дукты находят широкое практическое применение для получения
металлсодержащих красителей.
Если 1-нафтол или 1-нафтиламин содержат сульфогруппу в
положении 3, 4 или 5, сочетание также идет, как правило, в
положение 2. Объясняют это тем, что сульфогруппа в положении
3 в значительно большей степени дезактивирует положение 4, чем
положение 2, так как связь С3—С4 имеет повышенный порядок
(см. 2.4.2). Влияние сульфогруппы в положении 5 объясняют ее
стерическим действием, затрудняющим азосочетание в положение
4. Тем не менее, с диазосоединением из n-нитроанилина, обладаю-
щим высокой активностью, 4-гидрокси-2-нафталинсульфокислота
(1-нафтол-З-сульфокислота) и некоторые ее производные сочета-
ются преимущественно в положение 4 [11, с. 471]. Таким образом,
ориентационные эффекты и их причины в рассматриваемых случа-
ях нуждаются в дальнейших исследованиях.
2-Нафтол и 2 нафтиламин сочетаются только в положение
1, так как о-комплекс, образующийся за счет этого положения,
отличается исключительно высокой стабильностью (этот вопрос уже
обсуждался — см. 1.2.3). Если положение 1 в 2-нафтоле занято
метильной группой (47а), то реакция азосочетания останавливает-
ся на стадии образования о-комплекса (48а).
432
Если же в положении 1 в 2-нафтоле находятся более подви-
жные группы (476), например галоген, сульфогруппа, альдегидная,
гидроксиметильная или ацетильная группа, а также некоторые
другие, более сложные, реакция азосочетания проходит через
^-комплекс (486) с вытеснением этих групп в виде соответствую-
щих катионов Х+ и образованием азосоединения (49). Впервые это
было показано на примере вытеснения галогенов, катионы которых
захватывались тиосульфатом [23].
N=N—Аг
(47) (48) (49)
а) X = СН3; б) X = Hal, SO3H, СНО, СН2ОН, СОСН3 и др.
Следует отметить, что вытеснение диазоний-катионом заме-
стителей, находящихся в положениях, активированных гидр-
оксильной или аминогруппой, — достаточно общее явление и много-
численные примеры такого рода реакций описаны в литературе
[И, с. 179; 24, с. 433].
Аминонафтолы, содержащие амино- и гидроксильную группы в
разных кольцах нафталиновой группировки, в зависимости от сре-
ды, в которой проводится реакция, могут сочетаться с образовани-
ем различных продуктов. Азосочетание в кислой среде идет под
ориентирующим влиянием аминогруппы, а в щелочной — гидро-
ксила. При последовательном проведении реакций (в порядке,
указанном над стрелками в приведенных ниже формулах) можно
получать дисазосоединения.
Так, 8-амино-1-нафтол (50) сочетается в положения 4 и 5;
4-амино-5-гидрокси-2,7-нафталиндисульфокислота (Аш-кислота)
(51) благодаря влиянию сульфогрупп — в положения 3 и 6, а
4-амино-5-гидрокси-1-нафталинсульфокислота (8-амино-1-нафтол-
5-сульфокислота; Чикаго-С-кислота; С-кислота) (52)—также в по-
ложения 3 и 6.
НО nh2
(51)
SO3H
(52)
433
7-Амино-4-гидрокси-2-нафталинсульфокислота (И-кислота)
(53) сочетается в положения 8 и 3, а изомерная ей 6-ами-
но-4-гидрокси-2-нафталинсульфокислота (Гамма-кислота) (54) —
в положения 3 или 5. Дисазосоединение из (54) получить не уда-
ется, по-видимому, вследствие водородной связи, которая блоки-
рует гидроксил в моноазосоединении (55).
(55)
По этой же причине не дает дисазосоединений 8-амино-4-гид-
рокси-2-нафталинсульфокислота (5-амиио-1-нафтол-3-сульфокисло-
та; М-кислота) (56); азосочетание ограничивается образованием
моноазосоединения (57).
(56) (57)
Интересны реакции азосочетания солей диазония с соединения-
ми, содержащими подвижные атомы водорода, т. е.
условиях реакции образовывать соответствующий ;
(14.1.3). Важнейшими в практическом отношении су
способными
карбанш
этого типа являются p-дикарбонильные соединения и
производные р-кетокислот. Такие соединения, как
1,3-
(58) [25], ацетоуксусный эфир (59), амиды н ариламиды ацетоук-
сусной кислоты (60) (азотолы, см. 14.2.3), в щелочной среде соче*
таются с диазосоединениями по метиленовой группе, образуя сме-
шанные (жирноароматические) азосоединения:
(58)
СН3СОСН2СООС2Н5 CH3COCH2CONHAr
(59) (60)
К азосочетанию (в положение 4) способен З-метил-1-фе-
нпл-5-пиразолон (61) и многие его производные, имеющие большое
практическое значение. Получающиеся продукты во всех случаях
имеют хинонгидразонное строение (62) [26, с. 1899]:
N—NH—Аг
Н3С
С6Н5
(61)
Н3С
. / 0
N
N
С6н5
(62)
Изучение кинетики и механизма азосочетания солей диазония с
ацетоацетанилидом показывает, что скорость реакции пропорцио-
нальна концентрациям карбаниона и диазоний-катиона, т. е. имеет
второй порядок. В кислых буферных средах, когда концентрация
карбаниона мала, скорость реакции не зависит от концентрации
диазоний-катиона и идет по первому порядку, так как лимитирую-
щей ее стадией становится образование карбаниона [27].
15.4.2. Особенности механизма
нуклеофильного замещения диазониевой группы
Как известно, диазосоединения используют для разно-
образных реакций нуклеофильного ароматического замещения,
идущих с выделением молекулярного азота:
ArN) + Y~ —> ArY + N2 Y = OH, OCH3, Hal, CN, N3 и др.
Применение и частные механизмы таких реакций обсуждены в
^•3, 8.1.4, 9.1,2 и 10.3, Поэтому в настоящем разделе будут рас-
смотрены только некоторые общие особенности механизма этих
Реакций, связанные со строением и свойствами диазосоединений.
Некаталитические процессы нуклеофильного замещения с же-
сткими основаниями — гидроксид-анионом, анионами фтора и хло-
Ра^проходят при нагревании по механизму SnI, в котором медлен-
ной стадией является разложение диазоний-катиона и образование
очень реакционноспособного арил-катиона:
Аг< —> Дг+ + Nj
435
Арил-катион дальше быстро реагирует с нуклеофильной части
цей, образуя продукт замещения.
Значительно сложнее проходят реакции замещения диазогруп
пы на мягкие основания, а также каталитические реакции в при
сутствии солей меди(1) (реакции Зандмайера — см. 10.3). В эти:
случаях кинетическими методами доказан бимолекулярный харак
тер реакций. В связи с тем, что в качестве катализаторов част<
используются соли металлов в их низшей степени окисления, ;
также свободные металлы (реакция Гаттермана), способные вое
станавливать диазоний-катион, в литературе утвердилось мнение
согласно которому определяющей в этих реакциях является стади!
переноса электрона. Такой механизм, распространенный и н;
некаталитические процессы, предусматривает одноэлектронно
восстановление арендиазоний-катиона до неустойчивого радикала
отщепляющего азот с образованием арильного радикала, взаимо
действующего
Например,
предлагается
ный перенос:
Аг—NJ + Г —
с нуклеофилом или его окисленной формой,
для реакции соли арендиазония с анионом
[28] следующая схема, включающая одноэле:
Аг—Г
Аг + Г
Аг—i" +Ar—NJ —> Ar—I + Ar—N2
Механизм подтверждается тем, что из бензолдиазония в ка
честве продуктов реакции получаются иодбензол (45%), молеку
лярный иод (10%), бензол (30%) и формальдегид (15%); послед
ний образуется из метанола, служащего растворителем.
Следует отметить также попытки интерпретировать с позици
одноэлектронного переноса и реакцию азосочетания [29; 30, с. 1 If
31], хотя серьезные аргументы, подтверждающие такой взгляд, w
приведены.
Механизм реакций с одноэлектронным переносом подвергну
справедливой критике, как исходя из общих соображений [32], та!
и применительно к рассматриваемым реакциям диазосоединени!
[33]. Было указано, например, что в ряде реакций замещени!
диазогруппы в качестве катализаторов успешно используете
медь(П), являющаяся окислителем, а не восстановителем [34],
также соли металлов постоянной валентности (хлорид цинка [35]
Таким образом, в механизме реакций нуклеофильного замещени
диазониевой группы остается еще много неясного.
Недавно предложен новый подход к механизму нуклеофилый
го замещения диазогруппы — без представления об одноэлектро!
ном переносе [33]. Взаимодействие диазоний-катиона с основание
(Y~) трактуется в виде термодинамического цикла (63), состою
щего из трех процессов: ионной моляризации (а), окислительис
восстановительной димеризации (б) и радикального диспропорЦй
436
онирования (s). Аналогичным циклом (64) уже давно объясняю!
известное свойство раствора иодистоводородной кислоты неизмен-
но содержать определенную концентрацию молекулярного иода:
n^ArNj + Y
'[L i/2(ArN2)2
=?=t ArN2Y^n
+ i/2Y2L=Lf
(5)
Y"’+ r
L^>/2H2 +
(63)
(64)
В термодинамический цикл (63) входят два ковалентных диа-
зопродукта: продукт присоединения основания ArN2Y и димер
диазосоединения (ArN2)2, из них первый является промежуточным
продуктом реакций нуклеофильного замещения, а второй служит
основным источником получения арильных радикалов. Предпочти-
тельное направление процесса в сторону образования ковалентного
продукта присоединения или димера однозначно обусловливается
кислотно-основными свойствами реагентов и их окислительно-вос-
становительными характеристиками и поддается расчету. Медь и
ее соли, участвуя в реакции, образуют комплексы с ковалентными
диазоформами ArN2Y и сдвигают приведенное выше равновесие в
сторону этих форм, уменьшая образование димеров диазосоедине-
ний и радикальный распад.
Что касается строения ковалентных продуктов ArN2Y, то счита-
ют [36], что они являются лабильными син-диазосоединениями
(65), стабилизирующимися в виде циклического комплекса (66) с
участием соли металла (MY). В дальнейшем этот комплекс по
синхронному пути превращается в продукт нуклеофильного заме-
щения (67).
(65) (66)
(67)
Для реакций нуклеофильного замещения, протекающих без ка-
тализатора, стабилизация с««-диазосоединения может осуществ-
ляться за счет специфики нуклеофила. Так, в литературе имеются
Указания [37], что при взаимодействии с азид-анионом, стабили-
зация связана с образованием циклического пентазена (68). Ста-
билизация ковалентного бензолдиазоиодида (69) происходит бла-
годаря координации его с находящимися в растворе анионами
иода.
(69)
437
Образование молекулярного иода в рассмотренной выше реак4
ции бензолдиазония с анионами иода свидетельствует о значив
тельном вкладе окислительно-восстановительного взаимодействия;
обусловленного высоким восстановительным потенциалом I-; обра-
зование бензола и формальдегида при проведении этой реакции в
среде метанола связывают с гомолитическим разложением димера
диазосоединения:
(Ar—N2)2 —> 2Аг • + 2N2
2АГ.4-СН3ОН •—> 2АгН + СН2О
Таким образом, изложенная трактовка реакции нуклеофильно-,
го замещения диазогруппы основанием по схеме (63) предусмат-
ривает только гетеролитический путь развития реакции с конку-
ренцией двух механизмов: мономолекулярного— через арил-катиоц
и бимолекулярного — через промежуточное образование кова*
лентного диазосоединения ArN2Y. Вклад мономолекулярного ме*
ханизма при прочих равных условиях тем больше, чем меньше
константа образования ArN2Y и чем более разбавлены реагирую-
щие растворы.
В противоположность представлениям об одноэлектронном пе-
реносе, такая концепция связывает образование радикальных ча-
стиц со вторичными явлениями, обусловленными необратимыц
разложением ковалентных димеров диазосоединений. ;
15.4.3. Свободнорадикальные реакции диазосоединений
При практическом осуществлении реакций свободнора*
дикального арилирования (см. 12.1) диазосоединения — наиболее
распространенный источник получения арильных радикалов. i
Уже указывалось (см. 15.4.2), что разложение диазосоединений
может приводить к образованию арильных катионов и арильны^
радикалов. Первые образуются в кислой среде, препятствующей
образованию ковалентных форм диазосоединений, тогда как обра^
зование вторых, наоборот, требует избытка основания, сдвигающей
го равновесие в сторону ковалентных продуктов. В связи с этим|
уместно напомнить (см. 15.2), что концентрация диазогидроксид^
достигает максимальной величины при определеннном значений
pH, и именно при таком значении кислотности среды наблюдаете!
наиболее интенсивное разложение диазосоединений, протекающее
с образованием высокомолекулярных смол [38]. Это можно свЯ
зать с гомолитической диссоциацией диазогидроксида:
Аг—N=N—ОН —> Аг • 4- ОН • 4- N2 —> диазосмолы
В неводных средах взаимодействие солей диазония с гидре
ксид-анионом или метилат-анионом приводит к образованию арев
диазооксидов Аг—N=N—О—N —N—Аг [39], при гомолитическо
диссоциации которых также получаются радикалы (см. 12.1). Та
ким же образом в неводных средах распадаются через промежУ
точное образование диазоацетатов У-нитрозоацетанилиды: .
ArN(NO)COCH3 —> [Ar—N=N—ОСОСН3] —> Аг . 4-N2 4-- ОСОСНз
-
438 1
Следует отметить, однако, что диазоацетаты полностью иони-
зированы даже в неполярных средах [40,41]. Арильные радикалы
можно получать также из арил (арилдиазо) сульфонов (33) [42] и
при термолизе диазоаминосоединений (34) и смешанных азосое-
динений (70).
Ar_N=N—SO2—АГ Ar—N=N—NH—АГ Ar—N=N—Aik
(33) (34) (70)
Важным источником получения арильных радикалов является
восстановление солей диазония. В качестве восстановителей ис-
пользуют медь, цинк, хлорид титана (III). При подборе восстано-
вителей следует учитывать их потенциал, который должен быть
ниже следующего за одноэлектронным потенциалом диазоний-ка-
тиона. В ином случае в присутствии протонов образуются арил-
гидразины (см. 16.8):
ArN2 + 4е~ + ЗН+ —> ArNHNH2
В связи с этим весьма удобным способом генерирования ариль-
ных радикалов следует признать электрохимическое восстановле-
ние солей диазония при заданном потенциале [43].
Дальнейшая судьба арильных радикалов, обладающих высокой
реакционной способностью, зависит от условий проведения реак-
ции. Они могут отрывать атом водорода от растворителя, образуя
углеводороды, арилировать растворитель (см. 12.1) или образовы-
вать биарилы при обязательном присутствии солей меди(1), бла-
годаря координирующим свойствам которой две радикальные ча-
стицы рекомбинируют друг с другом.
Способность диазотированных аминов при нагревании со спир-
том превращаться в соответствующий углеводород была открыта
еще Гриссом. Это происходит по реакции:
ArNJ + С2Н5ОН —> ArH + N2 + Н+ + СНзСНО
Значительно позднее было обнаружено, что наряду с таким гомо-
литическим превращением идет параллельная реакция гетеролити-
ческого характера по механизму SnI (см. 8.2), в результате кото-
рой образуются арилэтиловые эфиры:
ArNJ 4- С2Н5ОН —> АгОС2Н5 + N2 + Н+
Соотношение продуктов, образующихся при параллельных ре-
акциях, сильно зависит от строения диазосоединений и от pH сре-
ДЫ- Так, в минерально-кислой среде из диазотированного анилина
образуется фенетол с выходом до 85—90%, в то время как в
ацетатном буферном растворе получается бензол с почти количе-
ственным выходом [44]. В отличие от этого диазотированный
4'Нитро-л-толуидин при нагревании со спиртом в сильнокислой
сРеде образует ж-нитротолуол с 70%-ным выходом [45]. Таким
00Разом, реакция замещения диазониевой группы водородом об-
легчается под влиянием электроноакцепторных заместителей в
439
ароматическом кольце. С другой стороны, течение этой радикал!
ной реакции подавляется действием кислорода воздуха и ингиб!
торов свободных радикалов, например трет-бутилнитрита. В связ
с этим процесс рекомендуется вести в атмосфере азота (см. 8.2'
Вместо этилового спирта можно использовать этиленгликол]
тетрагидрофуран, а также некоторые восстановители. Из после;
них особенно хорошо влияет на течение реакции соль фосфорновг
тистой кислоты — гипофосфит натрия NaH2PO2, а в некоторы
случаях глюкоза в щелочном растворе [24, с. 449].
Реакцию замены диазониевой группы водородом использую
в технике для получения .и-нитротолуола из 2-нитро-я-толуидин
[45], 1,3,5-трихлор- и 1,3,5-трибромбензола из 2,4,6-трихлор-
2,4,6-триброманилина [46], З-гидрокси-1-нафталинсульфокислот;
(2-нафтол-4-сульфокислота) из 4-амино-3-гидрокси-1-нафталш
сульфокислоты (1-амино-2-нафтол-4-сульфокислота) [22, с. 338]
некоторых других продуктов. .
Для получения симметричных биарилов путем рекомбинаци
арильных радикалов, образующихся из диазосоединений, после;
ние разлагают при действии порошка меди в подходящем раствс
рителе, например в спирте или в уксусном ангидриде. Можн
также пользоваться солями меди(1) в жидком аммиак
[24, с. 456]:
2Ar—Nj X" + Си
Ar—Ar + 2N2 + CuX2
В практике эту реакцию обычно ведут с водно-аммиачны;
раствором оксида меди(1) Cu(NH3)2OH в расчете 1 моль на мол
диазосоединения. Роль аммиака при этой реакции может заклк
чаться в связывании Си2+ в медноаммиачный ион [Cu(NH3)4]^
благодаря этому тормозится образование продуктов нуклеофил!
ного замещения.
Пример практически важной реакции этого типа — получени
1,1'-бинафтил-8,8'-дикарбоновой (8,8'-би-1-нафтойной) кислот!
(71а), применяемой в синтезе кубовых красителей:
N,+ СООН
h2n СООН
(71)
Для проведения процесса 172 кг 8-амино-1-нафтойной кислот
(71) растворяют в 2000 л воды и 220 л 27%-ного NaOH и внося
70 кг NaNO2. Полученный раствор медленно выливают на сме<
1600 кг льда и 450 кг НС1. По окончании диазотирования раство
нейтрализуют раствором карбоната натрия. Отдельно готовят ра<
твор купрохлорида натрия NaCuCls, содержащий НО кг меди-
440
2000 л воды, 800 кг льда, 120 кг бикарбоната натрия и 340 л
25%-ного аммиака. Поддерживая температуру 15—20 °C, быстро
приливают раствор диазосоединения и в конце прибавляют еще
100 л аммиака. Смесь фильтруют и кислоту (71а) осаждают из
фильтрата прибавлением серной кислоты. Выход 132 кг, что СО'
ставляет 83,9% от теоретического [22, с. 106].
Интересно отметить, что стерическая затрудненность не пре-
пятствует течению димеризации,
15.4.4. Фотохимические реакции диазосоединений
Все диазосоединения обладают светочувствительностью,
и это необходимо учитывать при получении, хранении растворов и
твердых диазопродуктов и при их переработке.
Способность диазосоединений разрушаться под действием све-
та нашла обширное практическое применение для осуществления
фотопроцессов без использования серебра, а в некоторых случах и
как препаративный метод синтеза труднодоступных соединений
[5]. Поэтому целесообразно рассмотреть фотохимические превра-
щения диазосоединений несколько подробнее.
Как уже указано (см. 15.2) арендиазосульфонаты (32), обра-
зующиеся при взаимодействии солей диазония с сульфитом, явля-
ются одной из пассивных форм диазосоединений и не способны
вступать в реакцию азосочетания. При действии света они диссо-
циируют с образованием диазоний-катиона и сульфит-аниона, бла-
годаря чему приобретают способность к азосочетанию. Считают,
что это превращение идет через фотостереоизомеризацию аренди-
азосульфонатов, имеющих анты-строение, в лабильную сым-форму,
которая сразу же диссоциирует с образованием соли диазония:
Ar—N h Ar—N
II II —>
N—SO3Na NaO3S—N
Ar—N=N SO3Na*
K таким же превращениям способны арил (арилдиазо) сульфо-
ны (33) и арендиазоцианиды Ar—N=N—CN.
Действие света на соли диазония приводит к распаду диазоний-
катионов по гетеролитическому механизму с образованием азота и
очень активных арил-катионов, так же как при нагревании.
В зависимости от среды, в которой проводится эта реакция, арил-
катионы могут превращаться в фенолы, арилалкиловые эфиры,
галогенпроизводные или даже восстанавливаться в соответствую-
щий углеводород (ср. 15.4.2 и 15.4.3). Однако особенностью всех
этих фотохимических реакций нуклеофильного замещения диазо-
ниевой группы, по сравнению с рассматривавшимися раньше «тем-
новыми» реакциями, является влияние заместителей. Течение фо-
тохимических реакций ускоряется при наличии в ароматическом
кольце арендиазония электронодонорных заместителей и тормо-
зится электроноакцепторными. Эта закономерность характерна не
i, 441
ь
только для реакций нуклеофильного замещения диазониевой груп-1
пы, но имеет общий характер [47]. Для диазосоединений ее можно"
было бы иллюстрировать величинами квантовых выходов реакций,
характеризующих отношение числа молекул, вступивших в реак-
цию, к числу поглощенных квантов света. Так, квантовый выход
фоторазложения соли 4-диметиламинобензолдиазония (72а) бли-
зок к теоретическому и составляет 0,95—0,99, в то время как
квантовый выход фоторазложения соли 4-сульфобензолдиазония
(73) только 0,07—0,10 [5]. Аналогичные результаты получены при
изучении кинетики фоторазложения диазосоединений в пленках из
ацетата целлюлозы [48],
Хотя причина этой закономерности до сих пор не выяснена, она
уже давно находит практическое применение в диазотипии, где
применяются термостойкие и обладающие высокой светочувстви-
тельностью диазосоединения, содержащие сильные электронодо-
норные заместители в пара-положении к диазониевой группе, на-
пример соли 4-диметиламино- (72а) и 4-диэтиламинобензолдиазо-
ния (726), 4 -морфолинобензолдиазония (74) и др.
(72) (73) (74)
a) R = СН3; б) R = С2Н5
Квантовый выход фоторазложения таких солей диазония велик
и не зависит от природы растворителей. Предполагается, что реак-
ция идет через образование синглетного возбужденного состояния
[49]. Преимуществом диазосоединений этого типа является также
и то, что они имеют интенсивный максимум в спектре поглощения,
лежащий на границе УФ и видимой части спектра.
Диазотипные материалы бывают одно- и двухкомпонентные.
В первом случае на бумагу или пленку наносят только диазосое-
динение, чаще всего в виде соли с хлоридом цинка и некоторые
вспомогательные вещества. После засветки ртутной лампой такие
материалы проявляются путем нанесения раствора азосоставляю-
щей и нейтрализующих агентов.
В двухкомпонентных материалах в состав светочувствительного]
слоя входит, кроме диазосоединения, также и азосоставляющая. ]
Чтобы они не сочетались друг с другом, в слой добавляют кисло- ]
ты — чаще всего лимонную и фосфорную, а также вспомогатель- ]
ныевещества. После засветки ртутной лампой эти материалы про-’
являют действием паров аммиака. ?
Весьма интересны реакции, проходящие при действии света на]
диазосоединения, образующиеся из о-аминофенолов и имеющие]
строение хинондиазидов (см. 15.1). В ходе фотохимических пре-]
вращений таких диазосоединений идет перегруппировка, в резуль- ]
тате которой образуются производные циклопентадиенкарбоновой
кислоты (реакция Зюса) [50,51]. Изучение механизма реакции;
показывает, что в этом случае от хинондиазида (75) под влиянием
442
света отщепляется молекула азота и в результате образуется кар-
бен (76), быстро изомеризующийся в производное кетена (77).
Присоединяя воду, оно образует конечный продукт реакции
1-инденкарбоновую кислоту (78).
Реакция имеет общий характер, идет с высоким выходом и дает
возможность получать многие труднодоступные соединения. Так,
недавно этим методом из хинондиазида антрапиридона (79) уда-
лось получить ранее неизвестную антрапирролкарбоновую кислоту
(80), декарбоксилирование которой приводит к антрапирролу (81)
[52]:
(79)
(80)
(81)
Реакция Зюса находит широкое применение и в светочувстви-
тельных составах для получения рельефных изображений, так
называемых фоторезистах [5]. Так как квантовый выход фотопре-
вращения 1,2-нафтохинон-2-диазида значительно выше, чем 1,2-на-
фтохинон-1-диазида, в светочувствительных составах обычно ис-
пользуют производные первого — например, 1,2-нафтохинон-2-диа-
зид-5-сульфокислоту (6) (см. 15.1). Действием хлорсульфоновой
кислоты ее превращают в соответствующий хлорангидрид (82) и
Далее им этерифицируют высокомолекулярный фенол:
(83)
(в)
443
Получающийся продукт (83) вводят в светочувствительный со-
став. Такой эфир гидрофобек и нерастворим в щелочных средах, на
при действии света он образует соответствующее производное
1-инденкарбоновой кислоты (78), которое легко растворяется в!
слабощелочных растворах. На этом и основан принцип действия]
светочувствительных составов, находящих применение в полигра-]
фии и радиоэлектронике. 1
ЛИТЕРАТУРА
1. Порай-Кощиц Б. А. Азокрасители. Л., Химия, 1972. 2. Пат. ФРГ 960205,
1955, Chem. Abstr., 1959, v. 53, 17055; Пат. США 2950178, 1960, Chem. Abstr.^
1960, v. 54, 23491С. 3. Фирц-Давид Г. Э., Бланже Л. Основные процессы синтеза;
красителей. Пер. с нем./Под ред. С. В. Богданова и И. В. Фодимана. М„ ИЛ,'
1957. 4. Piercey М. R., Ward Е. R. — J. Chem. Soc., 1962, р. 3841—3844. 5. Дина-:
бург М. С. Светочувствительные диазосоединения и их применение. Л., Химия/
1964. 6. Ингольд К. Теоретические основы органической химии. Изд. 2-е. Пер. с:
англ./Под ред. И. П. Белецкой. М., Мир, 1973. 7. Эфрос Л. С., Левит Р. М, —1
ЖОХ, 1955, т. 25, вып. 1, с. 199—207. 8. Zollinger Н. — Chimia, 1968, Bd. 22,;
№ 1, S 9—20. 9. Полынова Т. Н., Бокий Н. Г., Порай-Кошиц М. Л.—Журн.!
структ. химии, 1965, т. 6, № 6, с. 878—887; Ramming Chr., Tjornhom Т. — Acta'
chim Scand. 1968, v. 22, № 9. p. 2934—2942. 10. Трейгер В. M., Багал И. Л.,
Порай-Кошиц Б. Л. — ЖОХ, 1970, т. 6, № 12, с. 2535—2538.
11. Цоллингер Г, Химия азокрасителей. Пер. с нем./ Под ред. Б. А. Порай-;
Кошица. Л., Госхимиздат, 1960. 12. Кетлинский В. А., Багал И. Л., Порай-Ко->
шиц Б. А. — Реакц. способн. орган, соедин., 1971, т. 8, вып. 2, с. 463—475,:
с. 475—483. 13. Симонов А. М., Колодяжная С. Н., Подладчикова Л. Н. — Хи-:
мня гетероцикл, соед., 1974, № 5, с. 689—692. 14. Olson Е. S, — Heterocycl.:
Chem., 1977, v. 14, р. 1255—1256. 15. Butler R. N. — Chem. Revs., 1975, v. 75, №2,::
p. 241—257. 16. Венкатараман К. Химия синтетических красителей. Т. 1. Пер. с;
аигл./Под ред. Б. А. Порай-Кошица. Л., Госхимиздат, 1956. 17. Kirst W., Neu-i
mann W. — Angew. Chem., 1954, Bd. 66, № 15, S. 429—434. 18. Koller S., Zollin-i
ger H. — Helv. chim. acta, 1970, Bd. 53, № 1, S. 78—89. 19. Wistar R„ Bart--
lett F. D. — J. Amer. Chem. Soc., 1941, v. 63, № 2, p. 413—417; Порай-Ко-,
шиц Б. А., Грачев И. В. — ЖОХ, 1946, т. 16, вып. 4—5, с. 571—582. 20. Сима-
нов А. М., Колодяжная С. Н.— ЖОрХ, 1967, т. 3, с. 1146—1147. '
21. Багал И. Я., Скворцов С. А., Ельцов А. В. — ЖОрХ, 1978, т. 14, № 2,
с. 361—373. 22. Доналдсон Н. Химия и технология соединений нафталииовогс
ряда. Пер. с англ./Под ред. А. И. Королева. М., Госхимиздат, 1963. 23. Иоф-
фе и. С.— ЖОХ, 1937, т. 7, № 20—21, с. 2637—2638. 24. Ворожцов Н. И,
Основы синтеза промежуточных продуктов и красителей. Изд. 4-е. М., Госхим-
издат, 1955. 25. Сахаре Л, Ю., Гудриниеце Э. Ю., Лаукманис В. А. — ЖОХ,
1976 т. 12 вып. 7, с. 1455—1458; Сахаре Л. Ю., Гудриниеце Э. Ю., Тихона-,
ва А. М. — Изв. АН ЛатвССР, сер. хим., 1976, № 1, с. 57—60. 26. Стид К. В.-
В ки.: Химия синтетических красителей. Т. 3/Под ред. К. Венкатарамана. Пер;
с англ, под ред. Л. С. Эфроса. Л., Химия, 1974, с. 1899. 27. Machacek Y. et al.—
Collect. Czech, chem. Commun., 1970, v. 35, № 3, p. 844—856. 28. Kumar R-i
Singh P. R. — Tetrahedron Letters, 1972, № 7, p. 613—616. 29. Bargon J., Sei-
fert K. G. — Ibid., 1974, Ks 26, p. 2265—2268. 30. Бучаченко Л. А. Химическая
поляризация электронов и ядер. М., Наука, 1974.
31. Охлобыстин О. Ю. Перенос электрона в органических реакциях. Ростов
н/Д, Изд-во Рост, ун-та, 1974. 32. Белецкая И. П. — ЖОрХ, 1976, т. 12, № 9,
с. 2045—2047. 33. Багал И. Л., Ельцов А. В., Степанов Н. Д. — ЖОрХ, 1977;
т. 13, № 1, с. 22—30. 34. Contardi—Ann. chim. appl., 1923, v. 7, p. 13—23
35. Sawanishi Naomi, Takino, Yoshio — J. Pharm. Soc. Jap., 1976, v. 96, № ?
p. 214—218. 36. Багал, И. Л., Ельцов А. В. — В кн.; Реакционная способности
444
ароматических соединений/Под ред. Б. И. Степанова. М., Изд-во МХТИ
им Менделеева, 1978, с. 28. 37. Ritchie Calvin D., Wright D. J. —J Amer.
Chem Soc., 1971, v. 93, № 10, p. 2429—2432. 38. Matzka M„ Sdgner Z., Chrna-
tal V. e.a. — Chemicky prumysl, 1969, v. 19, № 3, p. 106—109. 39. Степанов H. Д.,
Багал И. Л., Сосонкин. И. М. и др. — ЖОрХ, 1975, т. И, № 11, с. 2330—2336.
40. Казицина Л. А., Дзечиленко Н. 5. — ДАН СССР, 1970, т. 192, № 3, с. 570—
571.
41.' Riichardt Ch„ Tan Chuan Cheng. — Chem. Ber. 1970, Bd. 103, № 6,
S 1774—1783. 42. Kojina Minoru, Minato Hiroshi Kobayashi Michio, — Bull
Chem. Soc. Jap., 1972, v. 45, № 7, p. 2032—2035. 43. Gadallah F. F., Elof-
son R. M. — J. Org. Chem., 1969, v. 34, № 11, p. 3335—3338. 44. Корнблюм H.—
В кн.: Органические реакции. Сб. 2. Пер. с англ. М., ИЛ, 1950, с. 285—361.
45. Кларк, Тейлор — В кн.: Синтезы органических препаратов. Сб. 1. Пер. с
англ. М., ИЛ, 1949, с. 310. 46. Колеман Дж., Талбот Б. — Там же. М., ИЛ, 1949,
с. 467. 47. Ельцов А. В., Фролов А. Н., Кульбицкая О. Н.—ЖОрХ, 1972, т. 8,
№ 1, с. 76—84; 1973, т. 9, вып. И, с. 2320—2331. 48. Barraclough R. e.a.—
J. Soc. Dyers and Colour, 1972, v. 88, № 1, p. 22—25. 49. Mizianty Michael F.
e.a. — Photogr. Sci. and Eng. (USA), 1971, v. 15, № 3—4, p. 331—333. 50. Sus 0.,
Munder J., Steppan H. — Angew. Chem., 1962, Bd. 74, № 24, S. 985—988.
51. Родина Л, Л., Коробицина И. К- — Успехи химии, 1967, т. 36, вып. 4,
с. 611—635. 52. Казанков М. В., Макшанова Н. Я.— Журн. ВХО, 1974, т. 19,
№ 4, с. 461—462.
Сканировал: Neptunyi
(Магнитогорск)
Все замечания и пожелания присылать по
адресам:
rhenium@list.ru
nept2006@yandex.ru
Буду рад также высылаемым книгам от вас!
Часть 6 1
РЕАКЦИИ ВОССТАНОВЛЕНИЯ I
И ОКИСЛЕНИЯ ’
АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ i
Глава 16 J
ВОССТАНОВЛЕНИЕ АРОМАТИЧЕСКИХ КОЛЕЦ
И ФУНКЦИОНАЛЬНЫХ ГРУПП
16.1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ
ОБ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ РЕАКЦИЯХ
Реакции восстановления и окисления органических со- ]
единений, так же как и неорганических, характеризуются процес- (
сом передачи электронов; восстанавливаясь, соединение приобре-!
тает, а окисляясь — отдает электроны. Вместе с тем круг органи- ]
ческих окислительно-восстановительных реакций очерчен гораздо]
менее четко, чем в неорганической химии. j
Так, в неорганической химии для характеристики окислительно-]
восстановительных реакций принято представление о степени|
окисления, которая выражается числом переданных или смещен-]
ных электронов (электронных пар). Однако степень окисления — ]
понятие формальное, оно не характеризует истинное электронное)
состояние атома углерода и не связано с механизмом химического
превращения. Даже в модификации этого понятия, предлагаемом
французскими исследователями [1, с. 22], единственное его на-]
значение — расчет стехиометрических отношений химических реак-
ций. Поэтому в органической химии понятием о степени окисления
предпочитают не пользоваться. 1
По установившейся в органической химии традиции, реакциями]
окисления считают такие химические процессы, в которых органи-]
ческое соединение отдает электроны окислителю и при этом в нем]
увеличивается число связей С—О (N—О, S—О и т. п.) или умень-
шается число связей С—Н (N—Н, S—Н и т. п.), соответственно,;
реакции восстановления, когда органическое соединение приобре-)
тает электроны, должны характеризоваться уменьшением числа;
связей С—О или увеличением числа связей С—Н.
Реакции окисления — восстановления могут иметь весьма раз-
нообразные и часто очень сложные многостадийные механизмы.
При этом механизмы многих важных реакций еще не исследованы
и о них можно высказывать лишь предположения.
16.2. МЕХАНИЗМЫ РЕАКЦИЙ ВОССТАНОВЛЕНИЯ
Для восстановления ароматических соединений могут
быть использованы практически все обычные неорганические вое*
становители, но наиболее часто применяются водород в присут*
446
1
ствии катализаторов, Металлы и некоторые соли металлов пере-
менной валентности — железо, цинк, олово, хлорид олова(П), на-
трий и соединения серы — соли сероводородной кислоты — суль-
фид Na2S и гидросульфид NaHS, дитионистой кислоты — дитионит
Na2$2O4 (иногда неправильно называемый «гидросульфитом»),
сернистой кислоты — сульфит Na2SO3 и гидросульфит NaHSO3
(бисульфит) и др. Приобретают значение также смешанные гид-
риды металлов — алюмогидрид лития LiAlH4, боргидрид натрия
NaBH4 и др.
Для простейших случаев восстановления возможны по крайней
мере четыре варианта протекания реакций: с передачей электронов,
с передачей гидрид-иона, с передачей пары электронов от нуклео-
фильного реагента и восстановление водородом на катализаторе.
16.2.1. Восстановление с передачей электронов
Этот механизм наиболее вероятен, когда восстановите-
лями служат металлы и соли металлов переменной валентности
(SnCl2, FeSO4 и др.). Органическое соединение вначале получает
электроны от восстановителя и образует соответствующие анион-
радикалы и дианионы. Последние отрывают протоны от раствори-
теля (например, от спирта или воды), превращаясь в конечные
продукты восстановления. Естественно, что расчленить этапы та-
кого восстановления можно только в том случае, когда реакция с
металлами проводится в апротонной среде — в эфире, тетрагидро-
фуране и т. п.
При таком восстановлении электрон металла переходит на
нижнюю вакантную молекулярную орбиталь соединения, и даль-
нейшая судьба образовавшейся амбидентной частицы зависит от
распределения в ней электронной плотности и от условий проведе-
ния реакции [2]. Так, при восстановлении карбонильных соедине-
ний, например бензофенона (1), натрием вначале образуется со-
ответствующий анион-радикал (металл-кетил), отрицательный за-
ряд которого локализован на атоме кислорода, а неспаренный
электрон делокализован в дифенилметильном остатке (2).
Нейтрализация анионного центра приводит к возникновению
Радикалов (3), которые димеризуются с образованием бензопина-
кона (4);
(С6Н5)2С=О —> (С6Н5)2С—О’ —>
0) (2)
ОН ОН
Z • I I
—* (СсН5)2С—ОН —> (С6Н5)2С—С(С6Н5)2
(3) (4)
Действие еще одного эквивалента натрия на анион-радикал (2)
Приводит к образованию дианиона (5), один из анионных центров
Оторого делокализован в дифенилметильном остатке. Амбидент-
н°сть дианиона подтверждается тем, что при алкилировании
447
трет-бутилхлоридом, подход которого к алифатическому атому
углерода стерически затруднен, алкильный радикал количествен-
но вступает в пара-положение одного из бензольных колец с обра-
зованием (6). Протонирование дианиона (5), однако, дает бенз-
гидрол (7):
(С6Н5)2С—О’ —>
(2)
ОН
+(СН3'зСС1 /=
(CeHshC—О’ ----------> (СН3)3С—(
н/
(5) (6)
;=С—С6Н5
(С6Н5)2СН—ОН
(7)
восстановления рассматриваемого типа наиболее!
Из реакций
изученной является реакция Бёрча — восстановление ароматиче-'
ских соединений щелочными металлами в жидком аммиаке или;
аминах [3]. Применительно к бензолу (8) механизм этой реакции]
представляется следующим образом: j
Сольватированный электрон, образовавшийся при растворении?
щелочного металла в жидком аммиаке, присоединяется к арома-|
тическому соединению,—’Образуется анион-радикал (9). В зависи-|
мости от стабильности последнего, возрастающей в ряду
бензол < нафталин < антрацен < фенантрен
положение равновесия этой реакции различно. Для бензола оно
сильно смещено влево. Для того чтобы сдвинуть его вправо, необ-'
ходимо выводить анион-радикал (9) из равновесия протонирова-;
нием, которое приводит к радикалу (10). Источником протонов в;
классическом варианте реакции Бёрча служат спирты, но может;
быть также и значительно менее «кислый» аммиак. Анион-радикал;
(9) может также связать еще один электрон, образуя дианиоИ:
(11), протонирование которого дает карбанион (12), а затем цик-
логексадиен (13). К тем же продуктам приводит присоединение
электрона к радикалу (10) и последующее протонирование. По-,
скольку атака протоном контролируется зарядом, следует пола-
гать, что местом присоединения второго протона будет углерод С
максимальной электронной плотностью. По этой причине из бензо<
ла и нафталина получаются 1,4-, а из антрацена и фенантрена —;
9,10-дигидропроизводные. ;
448
Если в соединении имеется электронодонорный заместитель D,
при восстановлении, как показывают расчет и опыт, образуются
производные 1,4-дигпдробензола (14), содержащие заместитель
или заместители при двойных связях:
Соединения же с электроноакцепторными группами А, такими
как —СООН, —СООСНз, при восстановлении образуют производ-
ные 1,4-дигидробензола (15), в которых эти заместители находятся
в положении 1 [4]. Таким образом, при восстановлении аромати-
ческих соединений по механизму, при котором первым этапом
является приобретение электрона и образование анион-радикала
или дианиона, новые электроны заполняют нижнюю вакантную
молекулярную орбиталь соединения. Дальнейшее протонирование
анион-радикалов и дианионов идет по атомам с максимальной
электронной плотностью. Этот же механизм характерен для реак-
ции электрохимического восстановления [5].
По-видимому, такой же механизм имеют реакции восстановле-
ния цинком, оловом, алюминием, железом — эти восстановители
значительно чаще применяют в производстве промежуточных про-
дуктов, чем щелочные металлы. Так, для восстановления полицик-
лических кетонов, хинонов, нитро-, нитрозо- и азосоединений при-
меняют железо, цинк, олово и хлорид олова (II). При этом строение
конечных продуктов во многих случаях зависит от того, в каких
условиях ведется реакция (см,, например, 16.4.2 и 16.6). В завод-
ской практике для исчерпывающего восстановления нитросоедине-
ний широко используют железо (чугунная стружка) в присутствии
электролитов.
16.2.2. Восстановление с передачей гидрид-иона
Этот вариант реакции восстановления предполагает пе-
ремещение от восстановителя к органическому соединению водо-
рода с двумя электронами — гидрид-иона Н~. Такой механизм ста-
новится возможным, когда восстановителями служат гидриды ме-
таллов, муравьиная кислота, изопропилат алюминия, органические
производные силана типа R3S1H, а также некоторые органические
соединения, при отщеплении гидрид-иона от которых образуются
ароматически стабилизированные соединения или ионы. Из них в
Качестве доноров гидрид-ионов чаще всего используются смешан-
ные гидриды металлов LiAlH4, NaBH4 и КВН4, отдающие четыре
иона Н~. Способность отдавать гидрид-ионы наиболее сильна у
L1A1H4, значительно слабее у КВН4 и NaBH4. Реакционную спо-
собность этих гидридов можно уменьшить, превратив их в
15 За к. 271 449
алкоголяты типа 1лА1Н(ОС2Н5)з, обладающие большей селектив-
ностью.
Реакция восстановления может проходить по типу либо нукле-
офильного замещения, либо нуклеофильного присоединения гид-
рид-иона, перемещающегося ,От донора в реакционном комплексе
[6]. В обоих случаях, однако, восстанавливаемое соединение дол-
жно в условиях реакции образовать электронодефицитный центр,
достаточно активный для того, чтобы оторвать гидрид-ион от вос-
становителя.
Так, при действии LiAlH« на соединения с подвижным атомом
галогена идет нуклеофильное замещение последнего гидрид-
ионом:
I
С—В г
С— + Вг-
н/ ^-Аг
Нуклеофильному замещению гидрид-ионом может подвергать-
ся также остаток n-толуолсульфокислоты (тозил) при действии
LiAlH4 на соответствующие эфиры:
С—О—Ts
I
С— + О—Ts
Гидриды металлов очень легко восстанавливают альдегиды и
кетоны до соответствующих спиртов:
:С=О
+н~
/СН— о
-±2^- ^сн—он
Донором гидрид-ионов в этой реакции могут служить также
алюминиевые алкоголяты вторичных спиртов, чаще всего изопро-
пилат алюминия (реакция Меервейна — Понндорфа — Верлея)
[7]. Перенос гидрид-иона осуществляется через промежуточное
шестицентровое переходное состояние:
Н3С\ /СНз
/с\
о7 он
I Ч /R
О к
Н
, I /R
[(СН3)гСНО]2 А1\ /С (
о к
Карбоновые кислоты, их эфиры и хлорангидриды восстанавли-
ваются гидридами металлов в две стадии, образуя спирты:
сн—о — сн=о -AU—сн2—о —снгон
450
Нитрилы и амиды кислот, равно как и азометины, при действии
гидридов металлов превращаются в соответствующие первичные
или вторичные амины:
RCH=N—R' RCH2—N"—R' RCH2NH—R'
Гидриды металлов, однако, не восстанавливают слабо поляри-
зованные двойные углерод-углеродные связи, не способные под-
вергаться атаке нуклеофильными реагентами, так как не содержат
электронодефицитного центра.
Благодаря различию механизмов реакций восстановления с
передачей электронов и реакцией, с передачей гидрид-иона, про-
дуктами этих реакций часто могут быть соединения различного
строения.
16.2.3. Восстановление с передачей пары электронов
от нуклеофильного агента
Только что рассмотренные реакции восстановления пу-
тем передачи гидрид-иона являются простейшей моделью более
сложных по механизму реакций, идущих при действии ряда нук-
леофильных реагентов на соединения, обладающие электрофиль-
ными центрами. Нуклеофильный реагент на первом этапе передает
свою пару электронов в совместное обладание атакуемому соеди-
нению, а на втором этапе отрывается без этой пары электронов; при
этом он замещается протоном из среды, в которой проводится
взаимодействие. Таким образом, восстанавливаемое соединение
получает вначале пару электронов, а затем протон. Следует пола-
гать, что восстановление упомянутыми выше соединениями серы
(см. 16.2) и другими соединениями проходит именно по такому
механизму. Реакции эти находят большое практическое примене-
ние, хотя известен механизм лишь очень немногих из них.
Так, восстановление солей арендиазония до арилгидразинов
обычно ведут действием солей сернистой кислоты. Первой стадией
этой реакции является взаимодействие соли арендиазония с суль-
фитом натрия, в результате которого получается ковалентное
соединение — арендиазосульфонат (16). Последний присоединяет
молекулу бисульфита по азогруппе, образуя АГарилгидразоди-
сульфонат (17). Одна из сульфогрупп в этом продукте отщепляется
в виде серной кислоты уже на холоду — образуется АГ-арилгидра-
зиносульфонат (18). Гидролитическое отщепление второй сульфо-
группы происходит лишь при нагревании в кислой среде, и полу-
чается арилгидразин (19) [8, с. 447].
+ SO3Na“ + SO3Na“; + Н +
ArN2 ---------> Ar—N=N—SO3Na ---------------►
(16)
SO3Na
I + H2O +HC1
—> ArNNHSO3Na --------->- ArNHNHSO3Na ----> ArNHNH2 • HC1
нагр.
(17) (18) (19)
15*
451
Суммарно эта реакция представляется уравнением:
ArN? + 2NaHSO3 + 2Н2О —> ArNHNH2 + 2NaHSO4 + Н+
(19)
16.2.4. Восстановление водородом
Этот вариант восстановления должен был бы предпола-
гать присоединение к органическому соединению ядер водорода с
одним электроном — атомарного водорода. Близкая картина на-
блюдается в условиях каталитического гидрирования, при котором
органическое соединение и водород, адсорбированные поверхно-
стью катализатора, вступают в тесный контакт друг с другом.
Водород в присутствии главным образом переходных металлов
Pd, Pt, Rh, Ru, Ni, а также никель-хромовых, никель-медных и
других комплексных катализаторов может присоединяться к крат-
ным связям С—С, С—О, C=N, N=O, N—N и т. п. Кроме того,
может идти гидрогенолиз связей С—Hal, С—OTs и др. Эффектив-
ность катализатора зависит от способа его приготовления, от его
удельной поверхности, от носителя, от свойств и pH реакционной
среды [9].
При гетерогенном катализе в присутствии переходных метал-
лов важную роль играет хемосорбция реагентов на активных цент-
рах катализатора (Kat), при которой за счет электронных взаимо-
действий с участием катализатора ослабляются или полностью
разрываются некоторые химические связи в адсорбированной мо-
лекуле. Например, физическими методами показано, что в случае
сорбции водорода металлами связь атомов в его молекуле ослабе-
вает, а затем и полностью разрывается по схеме:
+ Kat
Kat + Н2 Kat—Н2 KatH—Н *=* 2KatH
Насыщенные углеводороды сорбируются в меньшей степени, но
для них также возможна диссоциация по связи С—Н, проходящая,
однако, при повышенной температуре:
+ Kat
Kat + СНзСНз KatH—СН2СН3 KatH + KatCH2CH3
Непредельные и ароматические соединения обладают высокой
способностью к сорбции, которая протекает главным образом за
счет частичного или полного раскрытия л-связи:
Kat + СН2=СН2 + Kat Kat—СН2=СН2—Kat KatCH2CH2Kat
Дальнейшее взаимодействие протекает между двумя хемосор-
бированными молекулами, находящимися на соседних активных
центрах поверхности катализатора:
KatCH2 + KatH СН3
| —► | +4Kat
KatCH2 + KatH CH3
Продукт гидрирования обладает меньшей сорбционной способ-
ностью и вытесняется с активных центров катализатора новой
молекулой олефина или ароматического соединения.
452
Таким же образом гидрируются альдегиды и кетоны до спир-
тов, нитрилы, амиды и азометины до аминов, нитро- и нитрозосое-
динения до аминов. Все эти процессы имеют очень широкое прак-
тическое применение.
Как видно, восстановление водородом на катализаторе являет-
ся гомолитическим процессом. Существенно также, что эта реакция
обратима. Прямая реакция—гидрирование — всегда экзотермич-
на, и ее тепловой эффект, например, для превращения нитробензо-
ла в анилин достигает 435 кДж/моль. Реакция идет с уменьшени-
ем объема. Поэтому ее проводят при сравнительно невысоких тем-
пературах и под давлением. Напротив того, дегидрирование ведут
при сравнительно высокой температуре и часто при атмосферном
давлении или даже в вакууме (см. 17.1.5).
16.3. ВОССТАНОВЛЕНИЕ АРОМАТИЧЕСКИХ КОЛЕЦ
Основным методом восстановления ароматических угле-
водородов в том числе и с кратными углерод-углеродными связями
в боковых цепях является каталитическое гидрирование. При этом
ароматические кольца, стабилизированные энергией резонанса
(см. 1.3) восстанавливаются в более жестких условиях, чем крат-
ные связи боковых цепей. Это позволяет избирательно восстано-
вить кратную связь, не затрагивая ароматическую часть молекулы.
Для гидрирования кратной связи С=С или С=С в качестве
катализаторов могут быть использованы платиновая чернь, ске-
летный никелевый катализатор (никель Ренея), никель на носите-
лях, медь, смешанные оксидные катализаторы (медь-хромитный и
цинк-хромитный) и др. В промышленной практике обычно приме-
няют металлический никель и никель, осажденный на оксиде алю-
миния, оксиде хрома или других носителях [10, с. 597]. Гидриро-
вание протекает очень гладко и с высоким выходом. Скорость
гидрирования падает в зависимости от увеличения степени экра-
нирования двойной связи заместителями. Диеновые углеводороды
гидрируются быстрее углеводородов с одной связью С=С.
Катализаторами гидрирования ароматических колец могут
быть все металлы VIII группы, но в промышленности применяют
главным образом никель, нанесенный на оксид хрома (III), С этим
катализатором реакцию ведут при 120—200 °C под давлением 1 —
5 МПа, которое необходимо для увеличения степени конверсии.
Ароматические углеводороды необходимо предварительно очищать
от сернистых соединений, отравляющих катализатор. Можно ис-
пользовать также катализатор из оксидов и сульфидов никеля и
вольфрама, нечувствительный к сернистым соединениям, однако
этот катализатор требует значительно более жестких условий про-
ведения процесса — 320—360 °C и давления 30 МПа.
Гидрирование бензола (8) проходит три этапа:
l^jl АНЛ ff^jl
медл. 11 U быстро I^J. быстро
(8) (20) (21) (22)
453
Однако первая стадия реакции, идущая вследствие разрушения
ароматического секстета электронов с поглощением тепла
(—23,8 кДж/моль) и значительно медленнее, чем экзотермические
вторая и третья стадии, не позволяет остановить процесс на про-
межуточных продуктах 1,4-циклогексадиене (20) и циклогексене
(21)-в результате гидрирования образуется только циклогексан
(22). Последний в больших масштабах используют для производ-
ства циклогексанона, капролактама и адипиновой кислоты.
Гидрирование нафталина (23) также представляет собой цепь
из пяти 'последовательных реакций присоединения водорода, но
остановить процесс удается только на одной — стадии получения
тетралина (1,2,3,4-тетрагидронафталина) (25). Медленными ста-
диями являются присоединение первой и третьей молекул Н2.
В результате разрушаются соответственно ароматический децет
электронов (24) и ароматический секстет электронов с образова-
нием (26). При дальнейшем присоединении водорода через (27)
получается конечный продукт — декалин (декагидронафталин)
(28).
Тетралин и декалин получают гидрированием нафталина с
никелевым катализатором, первый в паровой, второй — в жидкой
фазе. Тетралин используется в синтезе 1-нафтола (см. 17.1.3 и
17.1.5) и в качестве растворителя, декалин — преимущественно в
качестве растворителя.
Гидрирование фенола (29) на никелевом катализаторе при
130—150°C и давлении 0,5—2 МПа дает циклогексанол (30),
важный промежуточный продукт для синтеза циклогексанона,
адипиновой кислоты и капролактама. Среди побочных продуктов
этой реакции циклогексанон (31), выход которого растет с повы-
шением температуры и уменьшением давления. Образование его
можно объяснить реакцией равновесного дегидрирования цикло-
гексанола. Однако считают более вероятным [10, с. 611], что гид-
рирование фенола (29) идет ступенчато через енольную форму
(32) циклогексанона, которая частично гидрируется в циклогекса-
нол (30), а частично изомеризуется в кетонную форму — циклогек-
санон (31). Другими побочными продуктами гидрирования фенола
являются циклогексен (21), образующийся путем дегидратации
циклогексанола, и циклогексан (22). Гидрирование фенола над тем
454
же катализатором при 250—300 °C приводит к образованию бен-
зола (8).
1-Нафтол и 2-нафтол при гидрировании над никелевым катали-
затором образуют соответствующие смеси алициклических и аро-
матических гидроксипроизводных тетралина — ац-тетралолов,
т. е. 1-тетралола (33) и 2-тетралола (35) и ар-тетралолов, т. е.
5-тетралола (34) и 6-тетралола (36). В этих смесях преобладают
бщ-тетралолы.
На других никелевых и кобальтовых катализаторах эта реакция
идет при 200 °C под давлением 5—20 МПа. В значительно более
мягких условиях ее удается провести на черни родия [11].
Используя специфические катализаторы и проводя реакцию в
мягких условиях, при гидрировании фенолов и нафтолов можно
получить соответствующие циклические кетоны [12]. Так, из фено-
ла над палладием при 50°C образуется циклогексанон, из 2-наф-
тола при комнатной температуре над родием на угле — 2-тетра-
лон, а из резорцина при комнатной температуре в водно-щелочной
среде над родием на АЬОз—1,3-циклогександион.
Анилин гидрируется с образованием циклогексилам ина (38) в
присутствии никеля Ренея при давлении 9,5 МПа и температуре
30—150 °C. В верхнем пределе указанного интервала температур
образуется также дициклогексиламин с выходом до 25%. Добавка
к исходному анилину дициклогексиламина устраняет образование
последнего и позволяет увеличить выход циклогексиламина. Про-
межуточным продуктом при гидрировании анилина является цик-
логексанонимин (37) [8, с. 677]:
NH2 NH NH2
6 - 6 - 6
(37) (38)
465
Аналогично идет гидрирование изомерных толуидинов. Полу-
чающийся из «-толуидина 4-метилциклогексиламин (39) использу-
ется для синтеза 4-метилциклогексанона (40):
Гидрирование кольца ароматических карбоновых кислот во
многом напоминает гидрирование соответствующих углеводородов.
Реакцию ведут на никелевом катализаторе при 160—200 °C под
давлением водорода; она идет труднее, чем гидрирование углево-
дорода или соответствующего фенола.
Из бензойной кислоты при этом получается циклогексанкарбо-
новая кислота, используемая в одном из вариантов синтеза капро-
лактайа:
+ зн2
СаН6СООН ----> С6НцСООН
м- и n-Аминобензойные кислоты при гидрировании над рутени-
ем в водном этаноле образуют бициклические лактамы [13].
Все три изомерные фталевые кислоты при гидрировании пре-
вращаются в соответствующие гексагидрофталевые (циклогексан-
дикарбоновые) кислоты, которые в небольших количествах приме-
няют для производства полимеров:
+ зн2
С6Н4(СООН)2 ---> CSH10(COOH)2
При гидрировании нафтойных кислот водород присоединяется в
первую очередь к ароматическому кольцу, не содержащему заме-
стителя. Из З-гидрокси-2-нафтойной кислоты (41) при этом полу-
чается 3-гидрокси-5,6,7,8-тетрагидро-2-нафтойная (или 7-гидро-
кси-6-тетралинкарбоновая) кислота (42), находящая некоторое
применение в синтезе красителей [8, с. 677]:
(41) (42)
Из приведенных примеров следует, что при гидрировании аро-
матических колец карбоксильная группа замедляет реакцию, а
гидроксильная ее ускоряет.
Совершенно иные закономерности отмечаются при восстановле-
нии ароматических колец по Бёрчу (см. 16.2.1) и при действии
амальгам щелочных металлов [14, с. 78]. При этих родственных
реакциях карбоксильная группа облегчает восстановление, в то
456
время как гидроксильная замедляет. Так, восстановление 8-гидро-
кси-1-нафтойной кислоты (43) идет избирательно в сторону обра-
зования 1,4-дигидропроизводного (44) [15]:
НО СООН НО СООН
(43) (44)
16.4. ВОССТАНОВЛЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ,
СОДЕРЖАЩИХ УГЛЕРОД-КИСЛОРОДНЫЕ СВЯЗИ
В производстве промежуточных продуктов и красителей
из этих реакций особенно большое значение имеет восстановление
соединений с карбонильными группами •— кетонов, альдегидов, хи-
нонов и кубовых красителей.
16.4.1. Восстановление кетонов и альдегидов
Ароматические кетоны и альдегиды легко восстанавли-
ваются до соответствующих спиртов и гораздо труднее — до со-
ответствующих углеводородов:
+ 2Н +2Н
Ar—С—R ------► Ar— CH—R ----> Ar—СН2—R
II I
О ОН
В качестве восстановителей чаще всего применяют металлы:
цинк, олово, железо, амальгаму натрия или цинка—в щелочной
или в кислой среде. Восстановление можно также вести алюмо-
гидридом лития (см. 16.2) или водородом над никелем Ренея и
другими катализаторами.
Альдегиды в сравнении с кетонами восстанавливаются заметно
легче, но восстановление кетонов имеет значительно большее
практическое значение.
Так, практически важные бензгидрол (дифенилметанол) и его
производные, например гидрол Михлера (бис-п-диметиламинофе-
нилметанол), которые применяют в синтезе триарилметановых
красителей, получают восстановлением бензофенона (дифенилке-
тона) и его производных. Чаще всего действуют цинковой пылью
в воднощелочном или аммиачном растворе, к которому для повы-
шения растворимости продукта добавляют спирт. Цинк применяют
в значительном избытке против количества, рассчитанного по
уравнению [8, с. 667]:
Ar—СО—Ar + Zn + 2Н2О —> Ar—СНОН—Ar + Zn(OH)2
Для исчерпывающего восстановления кетонной группы реакцию
желательно вести в кислой среде; это благоприятствует отрыву
гидроксильной группы гидрола и образованию карбкатиона, легко
восстанавливающегося до углеводорода. Такие условия создаются
457
при восстановлении кетонов с использованием амальгамы цинка и
соляной кислоты (реакция Клемменсена) [16]; в результате груп-
па С=О сразу восстанавливается до СНг:
С6Н5СОСН2СН2СН3 —>• С6Н5СН2СН2СН2СНз
Кетоны, в которых карбонильная группа непосредственно свя-
зана с ароматическим кольцом, легко восстанавливаются до угле-
водородов также при действии сплава никеля с алюминием в
водно-щелочном растворе [17].
Производные о-бензилбензойной кислоты, необходимые для
синтеза замещенных антрона, получают из соответствующих
о-бензоилбензойных кислот действием цинковой пыли в водно-
аммиачном растворе [8, с. 667]. Например, из о- (п-хлорбензоил)
бензойной кислоты (45) получается о-(и-хлорбензид)бензойная
кислота (46).
СООН С1 СООН С1
Особенно легко диссоциируют с образованием карбкатионов
триарилметанолы (триарилкарбинолы) и их производные, являю-
щиеся карбинольными основаниями триарилметановых красителей.
Поэтому они легко могут быть восстановлены цинком в кислой
среде до лейкосоединений триарилметановых красителей, т. е. со-
ответствующих производных триарилметана:
+ Н+ +2е~; +Н +
АгзСОН -----+ Аг3С+ ---------->- Аг3СН
Восстановить триарилметановые красители в лейкосоединения
можно и действием дитионита натрия в водно-спиртовом растворе.
Интересно, что если эту реакцию вести в водно-щелочной среде, то
удается выделить промежуточный продукт, представляющий собой
соль триарилметансульфиновой кислоты (47), которая при нагре-
вании с избытком щелочи отщепляет кислотный остаток в виде
сульфита и превращается в лейкосоединение триарилметанового
красителя (48) [8, с. 668]:
(CH3)2N-
С+ -------->• (CH3)2N---f \------ CSO2Na
(CH3)2N-----6 >---JCH + Na2SO3
(48)
Таким образом, восстановление дитионитом проходит через
нуклеофильное присоединение реагента и последующий его отрыв,
в соответствии с одним из уже обсуждавшихся механизмов
(см. 16.2.3).
458
16.4.2. Восстановление хинонов и кубовых красителей
Практически важны обратимые реакции восстановления
хинонов в соответствующие двухатомные фенолы (гидрохиноны).
На примере 1,4-бензохинона (49) и гидрохинона (50) их можно
представить схемой:
(49) (50) (51)
Промежуточными продуктами одноэлектронного восстановле-
ния хинонов или окисления гидрохинонов являются анион-радика-
лы типа (51) — их называют семихинонами. Димеризация семихи-
нонов приводит к соединениям, имеющим строение перекисей.
Взаимодействие хинонов с соответствующими гидрохинонами
приводит к образованию стабильных и интенсивно окрашенных
хингидронов, представляющих собой комплексы переноса заряда.
Молекулы хинона и гидрохинона в этих комплексах располагаются
в двух параллельных плоскостях друг над другом [18].
Способность хинона восстанавливаться или, что то же, его
окислительная способность оценивается с помощью нормального
редокс-потенциала, составляющего для 1,4-бензохинона в воде
0,715 В. Высокий редокс-потенциал хинона связан с тем, что пре-
вращение его в гидрохинон приводит к выигрышу энергии сопря-
жения: непредельно-кольчатая система (энергия сопряжения
12,5—17 кДж/моль [19, с. 98]) превращается в ароматическую.
о-Хиноны имеют более высокий потенциал, чем изомерные «-хино-
ны; аннелирование бензольных и гетероароматических колец при-
водит к снижению редокс-потенциалов; тот же эффект дает
наличие электронодонорных заместителей. Величина редокс-потен-
циала хорошо коррелирует с энергией низшей свободной молеку-
лярной орбитали соединения [20, с. 188].
Ниже приведены хинонов [21, с. 47]: нормальные редокс-потенциалы некоторых
Е’, мВ Е°, мВ
1,2-Бензохинон 783 1,4-Бензохинон 715 (696) 1,2-Нафтохинон 576 1,4-Нафтохинон 484 2,6-Нафтохинон 758 1,2-Нафтохинон-4-сульфокис- 630 лота 2-Метил-1,4-нафтохинон 422 2-Амино-1,4-нафтохинон 274 2Тидрокси-1,4-нафтохинон 362 2,3-Дихлор-1,4-нафтохинон 499 1,2-Антрахинон 1,4-Антрахинон 9,10-Антрахинон 1,2-Фенантренхинон 1,4-Фенаитренхинон 3,4-Фен антрен хинон 9,10-Фен антренхинон 1,6-Пиренхинон 1,8-Пиренхинон 4,5-Пиренхинон 490 401 154 660 523 621 416 612 514 424
459
Константа приведенного выше равновесия хинон=^гидрохинон
пропорциональна квадрату концентрации ионов водорода. Благо-
даря этому редокс-потенциал чувствителен к pH водного раствора:
изменение величины pH на единицу меняет потенциал на 0,059 В.
В производстве промежуточных продуктов гидрохинон (хинол)
(50) обычно получают путем восстановления 1,4-бензохинона (49),
образующегося при окислении анилина. Реакция может быть осу-
ществлена действием сернистой кислоты на 1,4-бензохинон в вод-
ной суспензии. Промежуточно образующийся хингидрон труднее
восстанавливается сернистой кислотой, и реакцию заканчивают,
добавляя в раствор металлическое железо при 70—80 °C. Практи-
чески восстановление 1,4-бензохинона чаще ведут порошкообраз-
ным железом при уже указанной температуре. Гидрохинон может
быть получен также путем каталитического восстановления водо-
родом на никеле Ренея и электрохимически в среде водной серной
кислоты [8, с. 671].
Практическое значение имеют и продукты, образующиеся при
восстановлении 9,10-антрахинона и его производных по общей
схеме:
О ОН О
(55) (56) (57)
Восстановление 9,10-антрахинона в антрагидрохинон (9,10-ант-
радиол, антрахинол) (53) легко проходит при действии цинка в
щелочной или аммиачной среде, но чаще для этого пользуются
дитионитом натрия Na2S2O4 в присутствии щелочи. Фактически при
этом образуется не антрагидрохинон, а его моно- или дианион,
имеющие в водном растворе интенсивный желто-коричневый цвет.
При подкислении этого раствора он обесцвечивается — образуется
антрагидрохинон (53), медленно изомеризующийся в 10-гидр-
окси-9-антрон (54). Этот продукт отличается от антрагидрохинона
отсутствием флуоресценции и нерастворимостью в щелочах на хо-
лоду. Лишь при нагревании со спиртовым раствором щелочи (54)
медленно переходит в раствор, образуя дианион антрагидро-
хинона.
В отличие от антрагидрохинона, 10-гидрокси-9-антрон (54) лег-
ко восстанавливается цинком и уксусной кислотой, а также дитио-
нитом натрия (при недостатке щелочи) до 9-антрола (55); послед-
460
ний в кислой среде изомеризуется в таутомерный ему 9-антрон
(56). Продукты (55) и (56) могут быть выделены, в индивидуаль-
ном виде, но в растворах устанавливается их таутомерное равно-
весие, состояние которого зависит от использованного растворите-
ля: полярные растворители — уксусная кислота, пиридин — благо-
приятствуют образованию енольной формы (55), а ацетон и
хлороформ вызывают накопление кетонной формы (56). При даль-
нейшем восстановлении (56), которое может быть осуществлено
нагреванием с цинковой пылью в водном аммиаке, получается
антрацен (57). Эта реакция представляет практический интерес
для получения таких производных антрацена, которые не могут
быть синтезированы непосредственно из него. Этим путем, напри-
мер, можно получить 1- и 2-антраценсульфокислоты [22].
Практически важны 9-антрон и некоторые его производные,
применяемые в качестве промежуточных продуктов. Сам 9-антрон
в лабораторной практике лучше всего получается действием на
9,10-антрахинон оловом и соляной кислотой в уксуснокислом
растворе. 1-Хлор-9-антрон (59) получают восстановлением
1-хлор-9,10-антрахинона (58) порошком алюминия в 95%-ной
H2SO4 [23]:
О С1
(58)
(59)
В производстве кислотных антрахиноновых красителей широко
используется продукт восстановления 1,4-дигидрокси-9,10-антра-
хинона (хинизарина) (60)—лейкохинизарин. Его получают при
действии дитионита натрия в присутствии соды. Лейкохинизарин
значительно стабильнее описанных выше антрагидрохинонов и
имеет строение 2,3-дигидро-9,10-дигидрокси-1,4-антрахинона (61):
О ОН он-о
С&--С65
II I I II
о он он-о
(60) (61)
Аналогичное строение имеют и продукты восстановления
1,4,5,8-тетрагидрокси-9,10-антрахинона и 1,4-диамино-9,10-антрахи-
нона (ср. 7.1.5). В стабилизации такого рода структур, содержа-
щих непредельно-кольчатую группировку, большую роль играют
внутримолекулярные водородные связи — см. (61).
Восстановление кубовых красителей в щелочной среде исполь-
зуется для перевода этих нерастворимых в воде соединений в
461
раствор перед крашением. В качестве восстановителя обычно ис-
пользуется дитионит натрия. Например, восстановление индиго
(62) в лейкоиндиго (63) идет по схеме:
(62)
(63)
Количество дитионита натрия, необходимое для осуществления
реакции с таким красителем (Кр), может быть рассчитано по урав-
нению:
2Кр + Na2S2O4 + 2NaOH + 2Н2О —> 2КрН2 + Na2SO3+N Na2SO4
Аналогично проводится восстановление и более сложных кра-
сителей. Например, индантрон (64) восстанавливается в две ста-
(65)
(66)
Вначале образуется лейкосоединение (65), щелочной раствор
которого имеет синий цвет. Более глубокое восстановление дает
раствор коричневого цвета, содержащий анион соединения (66)
[24].
Для восстановления кубовых красителей может быть использо-
ван водород в присутствии никеля Ренея, а также металлы, чаще
всего порошок железа. Восстановление железом в сухом пиридине
462
Л
используется для получения сернокислых эфиров лейкосоединений
кубовых красителей—кубозолей [25]. В этом случае лейкосоеди-
нения не выделяют, а сразу же после образования подвергают
ацилированию пиридинсульфотриоксидом (см. также 14.3.2),
В дальнейшем пиридиниевую соль сернокислого эфира лейкосо-
единения в водно-щелочной среде превращают в натриевую.
Окислительно-восстановительные свойства кубовых красителей,
так же как и хинонов, количественно оцениваются по величине
нормальных редокс-потенциалов [26].
16.4.3. Восстановление фенолов
Наиболее жестких условий требует восстановление фе-
нолов, когда нужно заместить группу ОН атомом водорода и
получить ароматический углеводород. Такого рода превращения
бывают необходимы для идентификации соединений неизвестного
строения, для синтеза ароматических углеводородов сложного
строения, а в технике — для обесфеноливания продуктов деструк-
тивного гидрирования различных смол, образующихся при пироге-
нетической переработке твердых топлив.
В последнем случае это достигается путем гидрирования над
селективными катализаторами, например над сульфидом кобальта,
при 200 °C и давлении 10—20 МПа или над сульфидами молибдена
и вольфрама при 450—480 °C и 8 МПА [10, с. 611]. Этим методом,
однако, нельзя получать сложные и зачастую лабильные аромати-
ческие соединения, которые в столь жестких условиях неизбежно
подвергаются деструкции и изомеризации.
Исторически первым предложенным для этого методом была
сухая перегонка с цинковой пылью, с помощью которой из ализа-
рина был получен антрацен. Более совершенным методом является
нагревание соединения с концентрированной иодистоводородной
кислотой под давлением при 150—200 °C. Однако при этом прохо-
дит и частичное гидрирование ароматических колец — получаю-
щиеся продукты нуждаются в последующем дегидрировании.
Удобнее метод, дающий значительно лучший выход продукта и
заключающийся в сплавлении фенола с цинком и хлоридом цинка
[27, с. 164].
Самый мягкий способ удаления из ароматических соединений
фенольных гидроксильных групп — это каталитическое гидрирова-
ние аренсульфонатов или сульфатов этих соединений над никелем
Ренея в водном растворе [28].
16.5. ВОССТАНОВЛЕНИЕ АЗОМЕТИНОВ,
АМИДОВ И НИТРИЛОВ
Как уже было сказано (см. 14.2.2), азометины (67) легко
образуются из альдегидов или кетонов и первичных аминов.
Обычно их не выделяют из реакционной массы и подвергают
гидрированию на никеле при 80—150 °C и 0,2—2 МПа. Такой метод
463
восстановительного алкилирования позволяет получать чистые^
вторичные амины (68) [29].
—Н2О + Н2
R—СН=О + Н2.\’Аг ---> R—CH=NAr -----> R—СН2—NHAr
(67) (68)
В основной химической промышленности этот метод используют i
и для синтеза первичных аминов из альдегидов или кетонов и!
аммиака. Для хорошего выхода необходим по крайней мере деся-
тикратный избыток NH3, циркулирующего в системе, иначе наряду
с первичным амином образуются значительные количества вторич-
ного.
Перспективнее, однако, иной способ получения аминов, при
котором вместо альдегида исходным является более доступный
соответствующий спирт и процесс проводится над катализатором,
ускоряющим реакции гидрирования и дегидрирования, например
над никелем при 200—250 °C идавлении!—2 МПа. Для частичного
подавления дегидрирования в массу добавляют водород. Первой
стадией процесса является дегидрирование спирта:
-н2
R—СН2ОН------> R—СН=О
Образующийся альдегид затем реагирует с амином и по приве-
денной выше схеме дает азометин (67), в результате гидрирования
которого получается вторичный амин (68). Суммарно же реакция
представляется как процесс А-алкилирования первичного амина
спиртом (см. также 14.2.1) [10, с. 613].
Этим путем из циклогексанола и анилина в автоклаве над
никелем на пемзе при 170 °C получается А-циклогексиланилин:
С6НцОН+C6H5NH2 —> CsHnNHCeHs + Н2О
При пропускании паров анилина с 1,5 моль СН3ОН и 2,5 моль
Н2 над дегидрирующим катализатором при 250 °C образуется
смесь, содержащая 96,1% А-метиланилина, 2,8% А',А-диметилани-'
липа и 1,1% анилина. Аналогично можно получать А-этиланилин
[8, с. 489].
Гидрированием амидов карбоновых кислот могут быть получе-
ны амины:
RCONH2 RCH2NH2
Реакцию проводят над катализаторами гидрирующего типа,
например над никелем, при 250 °C и давлении 1—2 А4Па. Обычно,
однако, исходные амиды получают одновременно с их гидрирова- .
нием, вводя в реакцию карбоновую кислоту и аммиак:
RCOOH + NH3 ——> RCONHs
—Н2О
Если целевой продукт — первичный амин, процесс ведут с
10-кратным избытком аммиака. Это делается для того, чтобы пре-
464
дотвратить реакцию ацилирования образующегося первичного
амина, восстановление продукта которой дает вторичный амин:
+ Н2
RCH2NH2 + RCOOH ——-> RCH2NHCH2R
—2Н2О
Этот метод используется для синтеза высших аминов из со-
ответствующих карбоновых кислот. Например, октадециламин по-
лучают из стеариновой кислоты:
+ н2
С17Н35СООН + NH3 * c,8h37nh2
—ri2U
Продукт применяется в производстве поверхностно-активных
веществ, а также в синтезе некоторых красителей.
Гидрирование нитрилов также является удобным методом син-
теза аминов, если только исходные нитрилы доступны (см. 17.1.2).
Реакция проходит в присутствии гидрирующих катализаторов
обычного типа под давлением через промежуточное образование
альдиминов:
+ Н2 + Н2
RC=N -----> RCH—NH ------> RCH2NH2
Альдимин при этом может реагировать с конечным продуктом,
образуя азометин, гидрирование которого приводит к получению
вторичного амина:
RCH2NH2 + HN=CHR «=> RCH2N=CHR + NH3
+ Н2
RCH2N=CHR -----> RCH2NHCH2R
Добавление аммиака подавляет этот процесс, так как сдвигает
равновесие реакции образования азометина влево. Таким образом,
при гидрировании нитрила можно получать достаточно чистый
первичный амин.
При гидрировании динитрила адипиновой кислоты важнейшим
продуктом является гексаметилендиамин, используемый для син-
теза полимера и волокна анид:
+ 4Н2
NC(CH2)tCN -----> H2N(CH2)6NH2
Побочным продуктом этой реакции является циклический гек-
саметиленимин (CH2)eNH, за последние годы находящий примене-
ние в синтезе промежуточных продуктов и красителей.
Ограниченное применение находит также лг-ксилилендиамин,
образующийся при гидрировании динитрила изофталевой кис-
лоты:
CN
CH2NH2
CH,NH,
465
16.6. ВОССТАНОВЛЕНИЕ АРОМАТИЧЕСКИХ
НИТРОСОЕДИНЕНИЙ
Среди реакций восстановления функциональных групп
важнейшей является открытая еще в 1842 г. Н. Н. Зининым реак-
ция восстановления нитросоединений. Она дает возможность полу-
чать самые разнообразные первичные ароматические амины:
ArNO2 —> ArNH2
В промышленности для восстановления нитросоединений ис-
пользуют железо, цинк, сульфиды щелочных металлов, дитионит
натрия NasSsCh; применяется также электрохимическое и катали-
тическое восстановление. Каталитическое восстановление водоро-
дом — наиболее перспективный и прогрессивный метод получения
ароматических аминов, но в технике по разным причинам, которые
отчасти станут ясными из дальнейшего изложения, сохраняются и
консервативные методы химического восстановления, хотя они и
связаны с образованием отходов производства и токсичных сточ-
ных вод.
Восстановление нитросоединений в первичные амины представ-
ляет собой ступенчатый процесс и проходит через следующие ста-
дии:
ArNO2 —> ArNO —> ArNHOH —> ArNH2
(69) (70)
На стадии образования нитрозосоединений (69) при обычном
проведении процесса остановиться нельзя, так как они имеют вы-
сокий окислительный потенциал и легко восстанавливаются далее
до (V-арилгидроксиламинов (70).
Изучение механизма этих реакций [30] показало, что при вос-
становлении нитросоединений натрием или калием в апротонных
растворителях первичный продукт реакции—анион-радикал (71)
образуется путем передачи одного электрона от металла к нитро-
соединению:
Ar-N^° Ar^Is/°
Vo ч(>
(71)
Отрицательный заряд анион-радикала распределен на кисло-
родных атомах, а неспаренный электрон делокализован в арома-
тическом кольце. Такой же анион-радикал может быть получен при
электрохимическом восстановлении нитросоединений на катоде.
Катион металла связан с анион-радикалом в тесную ионную пару.
В результате действия второго атома металла анион-радикал
(71) превращается в дианион (72), который в определенных усло-
виях может быть выделен из раствора в виде двуметаллической
соли, комплексно связанной с молекулой нитросоединення
466
ArNO2Na2-ArNO2. При гидролизе дианион образует нптрозосое-
динение (69):
. /О +е~ Л® + 2Н+ /ОН1 —Н20
ArNz ------* ArNz ----—>• ArNz I -----> ArNO
\o_ L N)hJ
(71) (72) (69)
Дальнейшее восстановление (69) также проходит через стадии
образования анион-радикала (73) и дианиона (74), при гидролизе
которого получается А-арилгидроксиламин (70):
+ е « + е — + 2Н
ArNO ----► ArN—О' -------> ArN—О' ----► ArNHOH
(69) (73) (74) (70)
Как видно, рассматриваемые здесь стадии процесса восстанов-
ления нитросоединений металлами [30] принципиально ничем не
отличаются от стадий восстановления карбонильных соединений
(см. 16.2.1). Что касается завершающего восстановления А-арил-
гидроксиламинов (70) в амины, идущего, в отличие от восстанов-
ления спиртов, с большой легкостью, то следует полагать, что эта
реакция протекает с гетеролитическим разрывом связи N — ОН
(75) +(76), скорее всего после протонирования по атому азота
(70)+ (75):
+ н+ + —ОН“ + +2е~
ArNHOH -----► ArNH2OH ------> [ArNH2] ----> ArNH2
(70) (75) (76)
В щелочной среде нитрозосоединения (69) быстро реагируют с
А-арилгидроксиламинами (70), образуя азоксисоединения (77), а
с аминами — азосоединения (78). Затем (77) и (78) могут после-
довательно восстанавливаться в гидразосоединения (79), а по-
следние в амины:
+ ArNHOH
---------->
(70)
ArNO —
(691
+ ArNH2
---------->
О
ArN=NAr
(77)
ArN=NAr —> ArNH—NHAr —> 2ArNH2
(78) (79)
Азоксисоединения (77) могут получаться и при взаимодействии
А-арилгидроксиламинов (70) с исходными нитросоединениями по
реакции [30]:
3ArNHOH + ArNO2 —> 2ArNO=NAr + ЗН2О
(70) (77)
С учетом рассмотренной выше схемы восстановления нитросое-
динений не исключено также что образование азокси- и азосоеди-
нений происходит путем димеризации анион-радикалов или ради-
калов.
467
Если восстановление нитросоедннений в амин идет по такойj
усложненной схеме, уменьшается его скорость и заметно снижает-
ся выход целевого продукта. Поэтому обычно восстановление аро- :
матических нитросоедннений до аминов стараются не проводить в
щелочной среде.
Приведенные данные показывают, что в зависимости от усло-
вий: количества и свойств использованного восстановителя, а так-
же pH среды, в которой проводится реакция,— при восстановлении
нитросоедннений могут быть получены различные продукты. Почти
все приведенные в схемах соединения вырабатываются промыш-
ленностью и имеют практическое значение.
16.6.1. Получение аминов
Основным методом получения первичных ароматических
аминов и диаминов служит каталитическое восстановление нитро- ‘
соединений водородом в паровой или в жидкой фазе.
Восстановление нитросоединений до аминов может сопровож-
даться гидрированием ароматического кольца (см. 16.3); это осо-
бенно характерно при катализе платиной, палладием и никелем.
Поэтому при проведении процесса в паровой фазе наиболее часто
используют медь на носителях. Катализатор получают нанесением
карбоната меди(II) из суспензии в растворе силиката натрия на
пемзу и восстановлением водородом.
При восстановлении нитробензола, последний из резервуара
проходит через экстрактор для извлечения получающегося анили-
на из воды, которая образуется в результате реакции, затем в
испаритель и, наконец, после смешения с водородом, в два соеди-
ненных последовательно контактных аппарата. Реакция начинает-
ся в первом аппарате при 170 °C. При выходе из второго аппарата .
температура смеси доходит до 350—370 °C. Для отвода тепла ре-
акции применяется большой избыток водорода (50 моль на 1 моль
нитробензола), возвращаемого в процесс после отделения продук-
тов реакции. Выход анилина достигает 98% от теоретического
[8, с. 269].
Хорошие результаты дает Ni-катализатор, комбинированный с ,
оксидами ванадия и алюминия — выход анилина достигает 97—
99% от теоретического. Катализатор работает в относительно ши- i
роком интервале температур (240—300 °C) и легко регенерируется j
при окислении воздухом. Описано также производство анилина с
использованием подвижного слоя катализатора (медь на силика-
геле) . Метод позволяет легко отводить выделяющееся при реакции
тепло. Для этого применяют контактные аппараты с межтрубным
пространством, заполненным органическим теплоносителем.
В других случаях для отвода тепла используется кипящая вода, --
дающая перегретый пар.
Если в исходных соединениях присутствуют каталитические I
яды, например сернистые соединения, гидрирование можно вести J
на сульфидах никеля и молибдена при 300—350 °C под давлением j
468
20—30 МПа, что значительно усложняет аппаратуру. Поэтому
обычно предпочитают проводить дополнительную очистку продук-
тов от сернистых соединений и пользоваться медным катализато-
ром.
Каталитическим гидрированием нитроксилолов в паровой фазе
с медно-никелевым катализатором на носителе в промышленности
получают ксилидины [31, с. 198—200].
Жидкофазное гидрирование ароматических нитросоединений
универсальнее парофазного, так как позволяет восстанавливать не
только высококипящие нитросоединения, но и неплавкие и нелету-
чие, например нитроаренсульфокислоты. Кроме того, реакцию
можно проводить в мягких температурных условиях, что благо-
приятно сказывается на выходе и качестве целевых продуктов.
При жидкофазном гидрировании через жидкую реакционную
массу, находящуюся в контакте с твердым катализатором, барбо-
тируют водород. Для увеличения скорости процесса его почти
всегда ведут под давлением водорода. Исходные нитросоединения
вводят в реакцию в виде растворов в органических растворителях
или в воде, а иногда в виде водных эмульсий. Это облегчает отвод
тепла реакции и выделение продуктов восстановления, которые
необходимо отделять от катализатора. Свойства растворителя су-
щественно влияют на скорость процесса гидрирования. Обычно она
максимальна в спиртах и в воде и резко падает, если раствори-
тель— ароматическое соединение, диметилформамид и другие. Это
объясняется сорбцией таких растворителей активными центрами
катализатора.
Наиболее употребительные катализаторы для жидкофазного
гидрирования — никель Ренея и палладий на угле. В производ-
ственных условиях, однако, удобнее работать с катализатором в
виде гранул, достаточно крупных, чтобы их не уносили потоки
жидкости или газа. В этом случае исключается необходимость
фильтрования продуктов восстановления и легче организуется
непрерывный процесс.
Важнейшим условием успешного проведения этих реакций яв-
ляется возможно более энергичное перемешивание, необходимое
Для наилучшего контакта газообразного водорода, раствора нит-
росоединения и твердого катализатора. Для этого прибегают к
специальным размешивающим устройствам или усиленному бар-
ботажу водорода, взятого со значительным избытком, с последую-
щей его рециркуляцией.
Жидкофазное гидрирование позволяет восстановить до амина
любой нитропродукт и широко используется в технике, особенно
в крупнотоннажном производстве аминов, таких как толуидины,
л-фенилендиамин (из л-динитробензола) [32], л-толилендиамин
(из смеси 2,4- и 2,6-динитротолуолов) и др.
Один из важнейших процессов — каталитическое восстановле-
ние смеси динитротолуолов, образующейся при нитровании, в смесь
толилендиаминов, применяемую для приготовления толилендиизо-
Цианата. Реакцию ведут в метанольном растворе при 100—170 °C и
469
давлении 0,5—10 МПа с применением никеля Ренея. Предложено'
также реакцию вести в водной эмульсии с палладием на угле, а
также в водно-спиртовом растворе с тем же или платиновым ката-
лизатором [30]. Изучение кинетики реакции показало, что скорость
реакции лимитируется диффузией водорода в жидкости к поверх-
ности катализатора [33].
Было показано также, что анилин можно получить при гомо-
геннокаталитическом восстановлении нитробензола или азобензола
при действии водорода в диметилформамиде в присутствии рас-
творимого комплексного катализатора (CsH.sN^RhCla-NaBin [34].
Гомогеннокаталитическое восстановление нитробензола можно
провести также с помощью Н2 и СО в присутствии карбонилов
рутения Ru3(CO)i2 и Ru(CO)s. Кроме анилина при этом получает-
ся АЖ-дифенилмочевина [35].
Обзор патентной литературы по каталитическому восстановле-
нию нитросоединений см. [30, 36].
Многие ароматические нитросоединения можно гладко восста-
навливать каталитически над никелем Ренея в спиртовом и водно-
спиртовом растворе, используя вместо водорода гидразин, который
постепенно добавляют при перемешивании. Этот метод, в частно-
сти, рекомендуется для восстановления З.З'-динитробензидина,
4,4'-диамино-3,3/-динитродифенилметана и аналогичных продуктов
до соответствующих тетраминопроизводных, используемых в син-
тезе термостойких полимеров [37].
Вместо водорода можно вводить также муравьиную кислоту
или ее соли. Например, 8-нитро-1,3,6-нафталинтрисульфокислоту
восстанавливают при кипении водной муравьиной кислотой или ее
солями в присутствии палладия на угле. Выход продукта — 98%
[38].
Наряду с контактно-каталитическим методом в технике про-
должают широко пользоваться способом восстановления нитросо-
единений до аминов чугунными стружками в водном растворе
электролита [39; 8, с. 234]. Таким электролитом служит хлорид
железа(III), вводимый извне или же получающийся в реакционной
массе при добавлении небольших количеств соляной кислоты.
Иногда вместо этого, в воду добавляют небольшие количества
NaCl или NH4CI. По некоторым патентным данным [30], рекомен-
дуется также добавлять пиридин. Восстановление ведут в цилинд-
рических аппаратах (редукторах), выложенных изнутри кислото-[
упорной плиткой. Аппарат снабжается прочной чугунной мешал-J
кой, необходимой для размешивания тяжелого осадка, состоящего.]
из чугунных стружек и окислов железа. Поскольку восстановление |
нитросоединений сопровождается значительным тепловым эффек-j
том, чугунные стружки и нитросоединение загружают в аппарату
по частям. Л
Об окончании восстановления можно судить по исчезновению!
нитросоединения или по концентрации образовавшегося амина,|
которую определяют титрованием раствором NaNO2. Чугунные!
стружки при этом превращаются в смесь FeO и Ре20з; и при-I
470 1
специально подобранных условиях в присутствии А1С13 чугунные
стружки могут образовать Fe3O4 — оксид железа (II, III) (магне-
тит), находящий применение в качестве желтого пигмента. Урав-
нение этой реакции обычно представляется следующим образом:
дАгЫОз + 3Fe + 4Н2О —> 4ArNH2 + 3Fe3O4
После нейтрализации реакционной массы продукт восстановле-
ния выделяют различными способами в зависимости от его свойств
(сифонирование, экстракция, перегонка с водяным паром, кри-
сталлизация или высаливание).
Этот способ, в течение многих десятилетий применявшийся для
восстановления большинства нитросоединений, даже в форме
непрерывного процесса постепенно утрачивает свое значение из-за
большого количества сточных вод, содержащих токсичные амины и
нитросоединения, громоздкости аппаратуры и трудоемкости; его
заменяют описанным выше каталитическим восстановлением.
В настоящее время чугунные стружки используют для восстанов-
ления нитробензол- и нитронафталинсульфокислот и других соеди-
нений, которые производятся в сравнительно небольших масшта-
бах.
Восстановление нитросоединений в амины с применением цин-
ка, олова или хлорида олова (II) используют очень редко; даже в
лабораторной практике также предпочитают каталитическое вос-
становление в жидкой фазе.
Вследствие токсичности образующихся сточных вод и отсут-
ствия больших преимуществ за последнее время значительно со-
кратилось применение в качестве восстановителей нитросоединений
в амины сульфидов металлов. Их, однако, продолжают применять
для восстановления о-нитроанилина в о-фенилендиамин, о- и
n-нитрофенолов в о- и и-аминофенолы и в некоторых других синте-
зах. Особенно часто восстановление сульфидами металлов исполь-
зуют в антрахиноновом ряду, например, для восстановления 1,5- и
1,8-динитро-9,10-антрахинонов в соответствующие 1,5- и 1,8-диами-
но-9,10-антрахиноны. Таким же путем из 1,5-дигидрокси-4,8-динит-
ро-9,10-антрахинон-2,6-дисульфокислоты (78) получают ализарин-
сафирол (79):
(78)
(79>
Реакцию проводят нагреванием нитросоединения с водным
раствором Na2S при интенсивном перемешивании.
471
Во всех перечисленных выше случаях восстановление сульфц|
дами с успехом может быть заменено каталитическим восстанов!
лением в жидкой фазе. Однако сульфиды металлов пока чтд
незаменимы для целей частичного восстановления нитрогрупп $
полинитропроизводных, например при получении лг-нитроанилина
(81) из л-г-динитробензола (80) или пикраминовой кислоты (83) из
пикриновой кислоты (82), а также для избирательного восстанов-'
ления нитрогруппы в азокрасителях, например при получении ди-
аминоазокрасителя (84а) из (84):
(82)
При восстановлении нитросоединений сульфидом натрия реак-
ция идет по суммарному уравнению:
4ArNO2+6Na2S + 7Н2О —> 4ArNH2 + 3Na2S2O3 + 6NaOH
При этом оказалось, что реакция идет ступенчато [40]. В первой
стадии сульфид натрия переходит в тетрасульфид, что сопровож-
дается образованием большого количества щелочи:
АгГУО2 4~ 4Na2S 4- 4Н2О —> ArNH2 4- Na2S, 4- 6NaOH . .
Щелочность несколько уменьшается во второй стадии:
ArNO24-3Na2S44-6Na0H + 2H20 —► 5ArNH2 4- 6Na2S2O3
Высокая щелочность среды, особенно в первой стадии процесса,!
приводит к образованию азокси- и азосоединений и значительней
ухудшает качество и выход амина. Поэтому вместо сульфида на*!
трия в последнее время для восстановления применяют дисульфид^
реагирующий с нитросоединением по уравнению: 1
ArNO2 4- Na2S2 4- Н2О —> ArNH2 4- Na2S2O3 1
Еще более удобен гидросульфид натрия, в процессе восстанов-|
ления которым также не образуется щелочь: |
4ArNO2 4- 6NaSH + Н2О —> 4ArNH2 4- 3Na2S?O3 ]
472 1
При восстановлении дисульфидом натрия активной восстанав-
ливающей частицей является двухзарядный анион .Si’. Этот же
анион получается в первой стадии восстановления нитросоединения
гидросульфидом натрия. Будучи более сильным восстановителем,
чем исходный анион SH~, он восстанавливает нитросоединение до
соответствующего амина [41].
Влияние заместителей в ароматическом кольце на восстановле-
ние нитросоединений дисульфидом натрия хорошо описывается
уравнением Гаммета, причем константа реакции р =+3,55. Столь
большая чувствительность реакции к природе заместителя, вероят-
но, связана с двойным зарядом восстанавливающей частицы. По-
ложительный знак и большая величина р делают понятным, почему
сульфиды с успехом применяются для частичного восстановления
нитросоединений. Например, л-динитробензол должен восстанав-
ливаться в 103 раз быстрее, чем л-аминонитробеизол [42].
Сообщается, что спиртовый раствор гидросульфида натрия яв-
ляется наилучшим реагентом для частичного восстановления нит-
рогрупп в производных динитробифенила [43].
Частичное восстановление нитрогрупп в 2,4-динитроанилине с
преимущественным образованием 4-нитро-о-фенилендиамина, а
также избирательное восстановление нитрогруппы в азокрасителях
может быть проведено также с применением в качестве восстано-
вителя резорцина в растворе бикарбоната натрия [44].
16.6.2. Получение Af-арилгидроксиламинов
и их перегруппировка в аминофенолы
Восстановление нитросоединений можно задержать на
стадии образования /V-арилгидроксиламинов (70), если вести ре-
акцию при низкой температуре и поддерживать pH раствора около
нейтральной точки. Такие условия создаются, например, при вос-
становлении нитробензола в /V-фенилгидроксиламип (85) с цинко-
вой пылью в водно-спиртовом растворе и с использованием в каче-
стве электролита хлорида аммония [45]:
CeH5NO2 + 2Zn + ЗН2О —> C6H5NHOH + 2Zn(OH)2
(85)
В этих условиях pH раствора по окончании восстановления
составляет 7,6—8,4; если же в качестве электролита используется
СаС12, получающийся раствор имеет pH 11—11,75. Между тем
Щелочная среда при синтезе арилгидроксиламинов недопустима,
так как ионы ОН- катализируют реакции образования азокси- и
азосоединений. В связи с этим разработаны и другие методики
получения М-арилгидроксиламинов, в которых образующуюся при
восстановлении щелочь нейтрализуют добавлением кислоты или
Же весь процесс ведут в слабокислой, например в уксуснокислой,
среде [8, с. 240].
МАрилгидроксиламины обладают высокой реакционной спо-
собностью. Они являются слабыми основаниями и с минеральными
473
кислотами дают соли. Легко окисляются, образуя нитрозосоединё*
ния:
ArNHOH —> ArNO
Окисление идет с очень большой легкостью; /V-арилгидроксила-
мины — настолько сильные восстановители, что дают реакцию се-
ребряного зеркала и взаимодействуют с фелинговой жидкостью.
Окисление N-арилгидроксиламинов является основным методом
получения многих нитрозосоединений.
В нейтральной и слабокислой среде ЛАарилгидроксиламины
восстанавливаются до аминов.
Характерно также диспропорционирование М-арилгидроксила-
минов с образованием азоксисоединения (77) и амина; реакция
катализируется солями железа и меди:
3ArNHOH —> ArNO = NAr + ArNH2 + 2Н2О
(70)
(77)
При действии разбавленной серной кислоты ААарилгидрокси-
ламины подвергаются перегруппировке в аминофенолы с перехо-
дом гидроксильной группы в пара-положение ароматического
кольца. Из /V-фенилгидроксиламииа (85) при этом получается
п-аминофенол (86) с хорошим выходом; реакция идет по уравне-
нию первого порядка [46, с. 750]:
NHOH —> НО—:
-nh2
(85) (86)
Препаративно эту реакцию осуществляют, не выделяя N-фе-
нилгидроксиламин, промежуточно образующийся при восстанов-
лении нитробензола. Для этого нитробензол восстанавливают в
сильнокислом растворе и получают непосредственно п-аминофенол.
Интересно, что в этиловом спирте образуется п-фенетидин [47],
что свидетельствует о межмолекулярном механизме перегруппи-
ровки. ;
При восстановлении нитробензола водородом на платиновом.;
катализаторе в безводном метанольном растворе с добавко^
1,4 моль 60%-ного олеума и 0,26 моль диметилсульфоксида
1 моль нитросоединения при 50—100 °C образуется n-анизидин О
выходом 98%. При проведении этой реакции в среде безводного
этанола получается n-фенетидин с выходом только 50%. В отсут-|
ствие диметилсульфоксида выходы этих продуктов значительно
ниже и образуется анилин. Предполагают, что диметилсульфоксиЯ
сольватирует фенилгидроксиламин и облегчает его удаление с пбя
верхности катализатора [48]. 1
474
Рассматриваемые реакции используют в промышленности
для получения нафтазарина — 5,8-дигидрокси-1,4-нафтохинона из
1,5-динитронафталина [49, с. 459]; процесс идет по схеме:
Реакцию обычно ведут в концентрированной серной кислоте, к
которой добавляют раствор серы в 40%-ном олеуме, а гидролиз,
приводящий к образованию конечного продукта (после разбавле-
ния водой) — при 60 °C.
При действии на А-фенилгидрокСиламин (85) азотистой кисло-
ты образуется А-нитрозо-А-фенилгидроксиламин (87). Это соеди-
нение дает с ионами Си2+ и Fe2+ комплексные соединения и приме-
няется в аналитической химии (купферон). Оно взаимодействуете
металлами в таутомерной форме А-оксида диазогидроксида (88).
hno2
С8Н5—NH --—CeH5—N—N=O =?=* С6Н5—N=N—ОН
I -НгО I ।
ОН он о
(85) (87) (88)
16.6.3. Получение азо- и гидразосоединений.
Бензидиновая перегруппировка
Выше было указано, что при восстановлении нитросоединений в
присутствии щелочей могут быть получены азокси- (77), азо- (78)
и гидразосоединения (79) (см. 16.6).
Азосоединения, образующиеся при восстановлении нитросоеди-
нений, сами по себе имеют сравнительно ограниченное применение.
Так, в промышленности из n-нитробензойной кислоты получают
азобензол-4,4'-дикарбоновую кислоту (89), применяемую в
небольших количествах для синтеза полимеров и некоторых кра-
сителей. Восстановление ведут цинком в водном аммиаке. Анало-
гично восстановлением 4'-нитро-4-бифенилкарбоновой кислоты
475
глюкозой в щелочной среде [50] получают 4,4'-азобис-4-бифенил-
карбоновую кислоту (90), используемую в синтезе красителей.
Главное же применение азосоединений — восстановление до
гидразосоединений, перегруппировкой которых получают произ-
водные бензидина, имеющие большое значение в синтезе азокра-
сителей. Из нитробензола постадийно могут быть таким образом
получены азобензол (91), гидразобензол (92) и бензидин
(4,4'-бифенилдиамин) (93):
(93)
Можно, минуя стадию выделения азосоединений, получать гид-
разосоединения одностадийно, непосредственно из нитросоедине-
ний. Именно этим путем их чаще всего получают в промышленно-
сти.
Гидразосоединения — лабильные вещества основного характе-
ра, легко автокаталитически окисляющиеся кислородом воздуха до
азосоединений:
Ar—NH—NH—Аг + О2 —► Ar—N=N—Аг + Н2О2
Они способны также диспропорционировать с образованием
азосоединения и амина:
2Ar—NH—NH—Аг —> Ar—N=N—Ar + 2ArNH2
Важнейшим для практики свойством гидразосоединений, одна-
ко, является способность под влиянием кислот, обычно соляной или
серной, подвергаться упомянутому выше превращению в произ-
водные бензидина — так называемой бензидиновой перегруппи-
ровке. Впервые такая перегруппировка была осуществлена в 1845 г.
Н. Н. Зининым превращением гидразобензола в бензидин под
действием серной кислоты. В настоящее время этим термином
обычно объединяют ряд перегруппировок, в результате которых из
гидразосоединений могут образоваться производные не только
476
бензидина (93), но также о-бензидина (2,2/-бифенилдиамина)
(94), дифенилина (2,4'-бифенилдиамина) (95), а также-о-семидина
(А-фенил-о-фенилендиамина) (96) и д-семидина (ААфенил-/1-фени-
лендиамина) (97).
(96) (97)
Не все эти продукты образуются в ходе какой-либо одной ре-
акции, но в различных случаях перегруппировки их удалось выде-
лить. Например, при перегруппировке самого гидразобензола (92)
получается смесь, содержащая 70% бензидина (93) и 30% дифе-
нилина (95). Другие продукты можно обнаружить только путем
хроматографирования. При низкой температуре в безводной среде
получается также о-бензидин (94) [51]. Перегруппировка других
соединений бензольного ряда дает значительное количество неко-
торых о-бензидинов, но нередко образуются и о-семидины, дифе-
нилины и n-семидины. В отличие от этого, в нафталиновом ряду
выделены продукты типа о-бензидинов, но дифенилины как основ-
ные продукты не образуются.
В ряде случаев перегруппировка идет с вытеснением имеюще-
гося заместителя. При этом легче всего вытесняются сульфо- и
карбоксильная группы, труднее — хлор и ацетоксигруппа, еще
труднее — алкоксильная. Амино- и алкильные группы не вытесня-
ются, и перегруппировка идет в свободные орто- или /шра-положе-
ния. Интересно, что сульфо- и карбоксильная группы при вытесне-
нии образуют серную и угольную кислоты, т. е. отрываются без
пары электронов, в то время как хлор и ацетоксигруппа уходят с
парой электронов в виде анионов. Это свидетельствует о том, что
при перегруппировке реакционный центр в кольце может быть
заряжен как положительно, так и отрицательно [46, с. 756].
Например, при перегруппировке 4-гидразобензолкарбоновой
кислоты получается с хорошим выходом бензидин и выделяется
Угольный ангидрид:
NH—NH
—СООН —> h2n-
—nh2 + со2
В отличие от этого при перегруппировке 4-ацетоксигидразобен-
зола бензидин получается с малым выходом. Основным продуктом
477
Механизм бензидиновой перегруппировки изучается уже более
70 лет. Итоги многочисленных исследований исчерпывающе осве-
щены [46]. Уже давно и вполне надежно доказано, что эта пере-
группировка идет строго внутримолекулярно.
Длительное время дискуссировался вопрос, какие соли гидра-
зобензолов подвергаются перегруппировке — однопротонные или
двухпротонные. Для перегруппировки самого гидразобензола (92)
кинетическими методами показано, что скорость реакции пропор-
циональна квадрату концентрации протонов в растворе, т. е. что
перегруппировывается двухпротонная соль (99):
+ н+ + + н+ + +
c6h5nh—nhc6h5 -----> CeH5NH2—NHCeHs -----> c6h5nh2—nh2c6h5
(92) (98) (99)
Эти кинетические данные подкреплены получением однопро-
тонной соли (98), которая превращается в бензидин только при
дальнейшей обработке кислотой [52]. Однако более поздними,
преимущественно кинетическими исследованиями показано, что в
случае других гидразобензолов реакция может иметь дробный
порядок и даже первый порядок по концентрации протонов. Это
значит, что в зависимости от строения исходного гидразобензола
перегруппировываться могут как однопротонная, так и двухпро-
тонная соли. Была обнаружена и наиболее интересная третья воз-
можность протекания этой реакции — без промежуточного прото-
нирования. Так, 1,1'-гидразонафталин при нагревании в спиртовой
среде образует 1,Г-бинафтил-4,4'-диамин (100) и 2,2'-бинаф-
тил-1,Г-диамин (101), а 2,2'-гидразонафталин в этих же условиях
превращается в 1,Г-бинафтил-2,2'-диамин (102) [53].
NH2 NH2
non
478
Побочно в результате циклизации (101) и (102) образуются
дибензокарбазолы. Эти реакции проходят быстрее в спиртах, чем в
апротонных растворителях. Некатализируемые кислотами пере-
группировки сохраняют внутримолекулярный характер и поляр-
ность переходного состояния [54, 55].
Предполагается, что во всех трех вариантах механизма бензи-
диновой перегруппировки переходное состояние образуется из та-
кой конформации молекулы гидразосоединения, в которой сбли-
жаются реагирующие друг с другом центры, например пара-поло-
жения:
+ б+
H2N......nh2
6“
Расстояния между реагирующими центрами в такой конформа-
ции, как показывает расчет, составляет всего 1,5 А (0,15 нм) и по-
чти не отличается по длине от бифенильной связи [46]. Далее по
полярному механизму идет образование новой углерод-углеродной
связи с синхронным разрывом связи азот — азот.
При промышленном получении бензидина и его производных
для восстановления нитросоединений в гидразопроизводные чаще
всего используют цинковую пыль в щелочной среде или амальгаму
натрия. Последний способ обладает большими техническими пре-
имуществами и особенно часто применяется для получения бензи-
дина [56].
Восстановление нитросоединений до гидразопроизводных дей-
ствием цинковой пыли в щелочной среде протекает по суммарному
уравнению:
2ArNO2 + 5Zn + Н2О —> ArNH—NHAr + 5ZnO
Цинковую пыль обычно берут с 10—15%-ным избытком, едкий
натр, не входящий в приведенное уравнение реакции,— в количе-
стве 0,1—0,2 моль на 1 моль нитропродукта. Так как среда полу-
чается гетерогенной, реакционную массу необходимо эффективно
размешивать. Для того, чтобы реакционная масса была более
подвижной и продукты реакции переходили в раствор, в качестве
растворителя часто применяют хлорбензол, сольвент-нафту, этанол
и др.
В сольвент-нафте реакция восстановления нитробензола идет
при кипении растворителя и гидразобензол полностью переходит в
раствор, что позволяет легко отделить оксид цинка. Раствор гид-
разобензола в сольвент-нафте далее обрабатывают соляной кисло-
той, происходит перегруппировка и образующаяся соль бензидина
переходит в водный раствор; затем бензидин может быть выделен
479
в виде гидрохлорида, основания или очень трудно растворимого "
сульфата (57, с. 114].
Разработан и осуществлен в промышленном масштабе непре-
рывный метод восстановления нитробензола до гидрозобепзола
цинковой пылью в щелочной среде в каскаде из трех аппаратов
(31, с. 198—200].
Восстановление нитробензола в гидразобензол при помощи
амальгамы натрия в производственных условиях проводят следу-
ющим образом [56]. Амальгама получается электролизом хлорида
натрия с ртутным катодом и содержит 0,2—0,3% натрия [14]. Из
электролизера она поступает в никелевый реактор, снабженный
охлаждающим змеевиком, в котором находится нитробензол в во-
дно-спиртовом щелочном растворе. Процесс идет по уравнению;
2CeH5NO2 + 10Na(Hg)„ + 6Н2О —> C6H5NH—МНС6Н5 + lONaOH + lOreHg
Гидразобензол получается в виде суспензии и его отделяют
фильтрованием. В маточном растворе содержится некоторое коли-
чество азоОиизола (около 'А от количества гидразобензола); его
повторно восстанавливают. Полученный гидразобензол подверга-
ют бензидиновой перегруппировке действием соляной кислоты
(охлаждение ведется водой со льдом).
Из реактора разбавленная амальгама вытекает в аппарат, где
она подвергается дополнительному разложению водой до образо-
вания 50%-ной щелочи. Затем, ртуть возвращается в электролизер.
Описано также непрерывное трехстадийное восстановление
нитросоединений амальгамами натрия или калия [58].
Разработан электрохимический метод получения гидразобензо-
ла и 2,2'-гидразоанизола (2,2/-диметоксигидразобензол). Восста-
новление ведется в электрохимической ячейке на 1000 А. Католи-
том служит 8%-ный раствор NaOH, содержащий 1% РЬО, ано-
лит— 30%-ный раствор NaOH [59].
В промышленности аналогичными способами получают
З.З'-диметилбензидин (о-толидин) (103) из о-нитротолуола и
3,3'-диметоксибензидин (о-дианизидин) (104) из о-нитроанизола.
Эти производные бензидина в больших количествах используют в
синтезе азокрасителей.
(103)
Меньшее значение имеют 2,2/-бензидиндисульфокислота (105),
3,3'-бензидиндикарбоновая кислота (106) и 3,3'-дихлорбензидин
(Ю7), получаемые исходя, соответственно, из л-нитробензолсуль-
фокислоты, о нитробензойной кислоты и о-нитрохлорбензола. )
480 j
З,3'-Дихлорбензидин применяют, главным образом, для производ-
ства азопигментов [60, с. 274].
(106)
Бензидин и многие его производные (о-дианизидин, о-толидин,
З.З'-дихлорбензидин и др.) сильно канцерогенны и вызывают опу-
холи мочевого пузыря. В связи с этим производство их ежегодно
сокращается. Многочисленные красители и пигменты на основе
этих продуктов заменяются новыми марками. Полностью запре-
щено использование бензидина и его производных в текстильной
промышленности [61, с. 316].
Выше был упомянут способ восстановления нитросоединений в
щелочной среде до азо- и гидразосоединений с помощью альдеги-
дов, например глюкозы. Оказалось, что эти реакции идут значи-
тельно лучше в присутствии хинонов, которые промотируют вос-
становление. В частности, в присутствии 1,4-нафтохинона эту реак-
цию удается провести, применяя в качестве восстановителя
различные гликоли. Последние при этом окисляются в кислоты.
Так, из этиленгликоля образуются щавелевая и гликолевая кисло-
ты [62].
При восстановлении ароматических нитросоединений боргидри-
дом натрия с хорошим выходом получаются азоксисоединения,
которые далее образуют смеси азосоединений и аминов. Реакция
проводится в диметилсульфоксиде или в сульфолане [63].
16.7. ВОССТАНОВЛЕНИЕ НИТРОЗО- И АЗОСОЕДИНЕНИЙ
Нитрозо- и азосоединения используют как исходные
продукты для восстановления в амины, если получить их — и при-
том с необходимым строением — путем реакции нитрозирования
(см. 4.5) или азосочетания (см. 15.4.1) легче, чем соответствующие
нитросоединения. Так как обе эти реакции обычно гладко проходят
только в орто- или пара-положения к сильным электронодонорным
группам, восстановлением нитрозо- и азогрупп обычно пользуются
Для синтеза о- и п-арилендиаминов, а также о- или п-аминофено-
лов.
В качестве восстановителей при этих реакциях в практике при-
меняют железо, цинк, Na2S, NaHSO3, Na2S2O4, а в лабораторных
16 Зак, 271
481
синтезах также хлорид олова (II) в соляной кислоте. Универсала ]
ним методом восстановления нитрозо- и азосоединений является ’
каталитическое гидрирование в жидкой фазе на никеле Ренея; в
качестве растворителя при этом применяют воду или этанол.
Восстановление нитрозосоединений и азокрасителей, содержа-
щих в орто- или пара-положениях сильные электронодонорные
группы, проходит сразу до аминов:
ArNO —> ArNH2
ArN=NAr' —> ArNH2 + Ar'NHa
Остановиться на промежуточных стадиях — У-арилгидроксилам’и-
нов в первом или гидразосоединений во втором не удается
[8, с. 263].
В технике этим методом получают серию производных
А^-диалкил-п-фенилендиамина из соответствующих У,У-диал-
кил-п-нитрозоанилинов, образующихся при нитрозировании
У,У-диалкиланилинов:
Восстановление ведут каталитическим гидрированием в спир-
товом растворе при нормальной температуре, или железом в при-
сутствии электролитов. Продукты используются для получения
светочувствительных диазосоединений (см. 15.4.4).
Из п-нитрозофенола (108) под действием Na2S получают
n-аминофенол (109), используемый в качестве промежуточного
продукта и в фотографии:
(108) (108) (109)
Из 2-нафтола и ряда соответствующих ему гидроксинафталин-
сульфокислот нитрозированием получают соответствующие 1-нит-
розо-2-нафтолы, при восстановлении которых получаются произ-
водные 1-амино-2-нафтола, находящие применение в синтезе кра- )
сителей. Например, 5-амино-6-гидрокси-2-нафталинсульфокислоту
(1-амино-2-нафтол-6-сульфокислота) (112) в технике получают
нитрозированием 6-гидрокси-2-нафталинсульфокислоты (ПО) с |
482 I
последующим восстановлением нитрозопроизводного (111) цинко-
вой пылью в серной кислоте [49, с. 425]:
Важнейшей среди этих продуктов является 4-амино-З-гидро-
кси-1-нафталинсульфокислота (1-амино-2-нафтол-4-сульфокнсло-
та); она получается непосредственно из 1-нитрозо-2-нафтола при
действии бисульфита натрия [49, с. 421]. О механизме этой реак-
ции и условия синтеза см. 9.3.
Изомерную 3-амино-4-гидрокси-1-нафталинсульфокислоту
(2-амино-1-нафтол-4-сульфокислота) получают нитрозированием
4-гидрокси-1-нафталинсульфокислоты и восстановлением нитро-
зопроизводного бисульфитом [49, с. 418].
Практически более важную 6-амино-5-гидрокси-1-нафталин-
сульфокислоту (2-амино-1-нафтол-5-сульфокислота) (116), приме-
няемую в синтезе красителей и в производстве светочувствитель-
ных материалов (см. 15.1), синтезируют, исходя из 5-гидро-
кси-1-нафталинсульфокислоты (114). В этом случае предпочитают
вводить аминогруппу с помощью реакции азосочетания, которое
идет значительно более избирательно в положение 6, чем нитрози-
рование. Получающийся азокраситель (115) восстанавливают
действием дитионита натрия [49, с. 419]:
(114) (115) (116)
Восстановление азокрасителей используют не только для син-
тетических целей. В текстильной промышленности эту реакцию
применяют для получения так называемых восстановительных вы-
травок на тканях. Восстановителем служит ронгалит (формальде-
гидсульфоксилат натрия) и процесс ведется в условиях запарива-
ния при 100 °C,
16* 483
В лабораторной практике восстановление азокрасителей при- ]
меняется как метод установления их строения [64]. При этом 1
особенно удобно действие хлорида олова(II) или каталитическое !
гидрирование.
16.8. ВОССТАНОВЛЕНИЕ ДИАЗОСОЕДИНЕНИЙ
Восстановление диазосоединений с замещением диазо-
ниевой группы водородом уже обсуждалось в 15.4.3. Поэтому в
настоящем разделе будет рассмотрена только реакция восстанов-
ления диазосоединений с образованием арилгидразинов, являю-
щаяся важнейшим методом получения этих соединений.
В данном случае восстановление идет по гетеролитическому
механизму, по-видимому, в две стадии:
+ + 2е~; +н+ + 2е~; + 2Н+
Аг—N^N --------->• Ar—N=NH ---------> Аг—NH—NH2
Хорошие результаты дает восстановление солей диазония хло-
ридом олова (II) с соляной кислотой:
ArNsN СГ + 2SnCl2 + 4НС1 —> ArNHNHa • НС1 + 2SnCl4
Практически хлорид олова берут с небольшим избытком против
теории.
В заводских условиях значительно удобнее проводить эту ре-
акцию, используя в качестве восстановителя соли сернистой кисло-
ты [65], окисляющиеся до солей серной кислоты. Механизм реак-
ции уже рассмотрен в 16.2.3. Было приведено суммарное уравнение
образования арилгидразинов (19). Однако фактически в результа-
те взаимодействия вначале образуются М'-арилгидразиносульфо-
наты (18), гидролиз которых протекает в условиях нагревания с
соляной кислотой, и тогда получаются арилгидразины (19).
Этим путем в промышленности получают кроме фенилгидрази-
на ряд его производных, например п-гидразинобензолсульфокис-
лоту, 2-хлор- и 2,6-дихлорфенилгидразины и другие продукты.
Среди них отметим 4,4'-дигидразино-2,2'-стильбендисульфокис-
лоту
H2NNH—— СН=СН—АА—NHNH2
\so3h ho3s/
Основное назначение таких производных фенилгидразина—
синтез производных пиразолона, применяемых при промышленном
получении красителей'и лекарственных средств.
Арилгидразины — сильные восстановители и уже на холоду .
реагируют с фелинговым раствором. Окисление арилгидразинов, а *
также АГ-арилгидразиносульфокислот проходит очень гладко с[
образованием диазосоединений. |
484 -]
1
16.9. ВОССТАНОВЛЕНИЕ ГРУПП, СОДЕРЖАЩИХ СЕРУ
Сульфогруппа, так же как и карбоксильная, восстанав-
ливается с большим трудом. Поэтому аренсульфокислоты чаще
всего переводят в аренсульфохлориды, обрабатывая их хлоридом
фосфора (V) или хлорсульфоновой кислотой. Аренсульфохлориды
восстанавливаются ступенчато, образуя аренсульфиновые кислоты,
а затем'тиофенолы:
ArSO3H —> ArSO2Cl —> ArSO2H —-> ArSH
Для получения аренсульфиновых кислот применяют сульфит
натрия, цинковую пыль в стехиометрических количествах в водном
или спиртовом растворе на холоду и некоторые другие восстанови-
тели [66].
Из аренсульфиновых кислот, получаемых таким образом, прак-
тически важны n-амино- и дг-аминобензолсульфиновые, 4-ами-
но-3-метоксибензолсульфиновая и некоторые другие кислоты. Вза-
имодействием их с этиленхлоргидрином в водно-щелочной среде
получают арил-2-гидроксиэтилсульфоны, используемые в синтезе
активных красителей.
Тиофенолы получают, восстанавливая аренсульфохлориды цин-
ковой пылью в серной или концентрированной соляной кислоте
[67, с. 606]. Таким путем из 2-нафталинсульфохлорида получают
2-тионафтол, который далее S-алкилируют хлоруксусной кислотой
(см. 14.3.1) и полученную S-(2-нафтил)тиогликолевую кислоту
применяют для синтеза тиоиндигоидного красителя. Аналогично
тем же путем из 8-хлор-1-нафталинсульфохлорида (117) получают
8-хлор-1-тионафтол (118), а из него 5-(8-хлор-1 -нафтил)тиоглико-
левую кислоту (И9):
(117) (118) (119)
Тиофенолы окисляются с большой легкостью даже кислородом
воздуха, образуя диарилдисульфиды:
2Ar—SH —> ArS—SАг
Последние в ряде случаев получаются и непосредственно, на-
пример при действии Na2S2 на диазосоединения (см. 9.1.2). Вос-
становительное превращение диарилдисульфидов в тиофенолы мо-
жет быть осуществлено реакцией с сульфидом или гидросульфи-
Дом натрия, причем осторожное проведение реакции позволяет не
затронуть при этом, если она имеется, нитрогруппу [8, с. 679].
В качестве восстановителей дисульфидов могут быть использо-
ваны, кроме того, железо или цинк в кислой среде, а также глюко-
За в щелочной среде.
485
При действии восстановителей на ароматические сульфокисло-1
ты и сульфамиды может идти замещение сульфогруппы водоро-1
дом — восстановительное десульфирование. В отличие от реакции |
протодесульфирования (см. 3.1), проходящей в сильнокислых сре- ]
дах, эта реакция идет в щелочной или близкой к нейтральной
среде, причем отщепляющаяся сульфогруппа уходит с парой элек-
тронов в виде сульфит-аниона:
+ 2е
ArSO3Na + Н2О ---> АгН + SO3Na' + ОН-
Реакция восстановительного десульфирования известна уже
давно и находит разнообразное применение в синтезе промежу-
точных продуктов, главным образом для удаления сульфогрупп из
«-положений нафталиновых производных, в то время как сульфо-
группы в P-положениях этой реакцией практически не затрагивают-
ся [8, с. 328]. В качестве восстановителей используются амальга-
ма натрия или цинковая пыль в щелочной среде.
Например, этим путем из 7-амино-1,3-нафталиндисульфокисло-
ты (120) получается 6-амино-2-нафталинсульфокислота (121), а из
6-амино-1,3-нафталиндисульфокислоты (122)— 7-амино-2-нафталин-
сульфокислота (123):
HO3S
(120) (121)
(122>
(123)
В аналогичных условиях из 8-амино-1,3,6-нафталинтрисульфо-
кислоты (124) образуется 4-амино-2,7-нафталиндисульфокислота
(125); выход 93% [68].
(124)
(125)
Названные продукты применяются в синтезе азокрасителей.
Были проведены обстоятельные исследования условий, области
применения и механизма реакции восстановительного десульфиро-
вания соединений нафталинового ряда. Используя цинковую пыль
и сульфат меди (II) в нейтральных растворах или цинковую пыль и ,
486
хлорид олова (II) в присутствии щелочи, удалось значительно
улучшить выход уже известных продуктов и распространить реак-
цию на новые объекты. Так, разработаны новые способы получения
4-амино-2,7-нафталиндисульфокислоты (125), 4-гидрокси-2,7-на-
фталиндисульфокислоты, 4-а мино-2-нафталинсульфокислоты,
7-гидрокси-2-нафталинсульфокислоты, 7-амино-2-нафталинсульфо-
кислоты (123), 4-гидрокси-2-нафталинсульфокислоты и другие
[69,70].
Исследование механизма этой реакции показало, что она идете
промежуточным восстановлением ароматического кольца нафта-
лина и последующим быстрым отщеплением от него сульфогруппы
в виде соли сернистой кислоты:
SOaNa SOaNa
В том случае, когда сульфогруппа находится в (5-положении,
отщепление соли сернистой кислоты идет с трудом и реакция
заканчивается на стадии образования производного дигидронаф-
талина, которое в определенных условиях удается выделить [71].
Реакция восстановительного десульфирования может прохо-
дить и с соединениями ряда бензола; показано, что в таких случаях
она облегчается влиянием электроноакцепторной карбоксильной
группы в орто- или пара-положениях и тормозится так же распо-
ложенной аминогруппой. Эти закономерности находятся в согла-
сии с влиянием заместителей на восстановление ароматического
кольца (см. 16.2.1 и 16.3).
В антрахиновом ряду восстановительное десульфирование про-
ходит особенно легко. Так, разнообразные производные 1-амино-
или 1-гидрокси-9,10-антрахинон-2-сульфокислот отщепляют суль-
фогруппу при обработке их водных или водно-спиртовых растворов
дитионитом натрия:
О NH2 О nh2
ЛИТЕРАТУРА
1. Матье Ж-, Паника Р. — Курс теоретических основ органической химии.
Пер. с франц./Под ред. Л. А. Яновской. М.; Мир, 1975. 2. Иоффе Д. В., Мосто-
ва М. И. — Успехи химии, 1973, т. 42, вып. 1, с. 102—120. 3. Ахрем А. А., Ре-
шетова И. Г., Титов Ю. Т. — В кн.: Реакции и методы исследования органиче-
ских соединений/Под ред. Б. А. Казанского, И. Л. Кнунянца и др. М., Химия,
1969, т. 20, с. 7—127. 4. Журкович И. К., Иоффе Д. В. — ЖОрХ, 1976, т. 12,
487
Вып. 5, с. 980—984. 5. Майрановский В. Г. Журн. ВХО, 1977, т. 22, № 3,1
с. 334—347. 6. Гейлорд Н. Восстановление комплексными гидридами металлов. 1
Пер. с англ./Под ред. Н. К. Кочеткова. М., ИЛ, 1959. 7. Уайлдс А. Л. — В кн.: |
Органические реакции. Сб. 2. Пер. с англ. А1., ИЛ, 1950, с. 194—243. 8. Ворож-‘.
цов И. Н. Основы синтеза промежуточных продуктов и красителей..Изд. 4-е. М.,
Госхимиздат, 1955. 9. Balandin А. А. — In: Advances in Catalysis. V. 19/Ed.
D. D. Eley, H. Pines, P. B. Weifi, N. Y. — London, Academic Press, 1969, -
p. 1—210. 10. Лебедев H. H. Химия и технология основного органического и
нефтехимического синтеза. Изд. 2-е. М., Химия, 1975.
11. Сокольский Д. В., Омаркулов Т. О., Нургожаев К. X. ЖОрХ, 1978,
т. 14, № 2, с. 334—339. 12. Augustine R. L. — In: Catalysis Rev., Science and
Engineerings. V. 13/Ed. H. Heinemann, J. J. Carberry, M. Dekker. New York — Ba-
sel, 1977, p. 285. 13. Augustine R. L.t Vag L. — J. Org. Chem., 1975, v. 40; p. 1074—
1082. 14. Смирнов В. А. Восстановление амальгамами. Л., Химия, 1970. 15. Пржи-
ягловская Н. Л1., Мондодоев Г. Т., Белов В. Н.—ЖОХ, 1961, т. 31, вып. 10,
с. 3375—3378. 16. Buchanan J. G. St. С., Woodgate Р. D.— Quart. Rev. 1969,
v. 23, № 4, р. 522—536. 17. Papa D., Schwenk E., Whitman B. — J. Org. Chem.,
1942, v. 7, № 6, p. 587—590. 18. Foster R., Foreman M. J. — In: The Chemistry
of the Quinonoid Compounds. Part 1/Ed. S. Patai London, Wiley, 1975, p. 304.
19. Wheland G. W. Resonanse in Organic Chemistry. N. Y., Wiley, 1955. 20. Май-
рановский С. Г., Страдынь Я. П„ Безуглый В. Д. Полярография в органиче-
ской химии. Л., Химия, 1975.
21. Gleicher G. J.— In: The Chemistry of the Quinonoid Compounds. Part 1/Ed.
S. Patai. London, Wiley, 1975, p. 18. 22. Методы получения реактивов и препа-
ратов, вып. 15, М, Изд. ИРЕА, 1966, с. 102. 23. FIAT, v. 1313, р. 105. 24. Ля-
шенко В. Д., Кирзнер Н. А. — ЖОХ, 1946, т. 16, вып. 4—5. с. 583—592. 25. BIOS,
V. 960, р. I; v. 1493, р. 60. 26. Чурсина Л. А., Артым М. И., Андросов В. Ф.—
Изв. вузов. Технол. текст, пром., 1973, № 3, с. 88—93. 27. Клар Э. Полицик-
лические углеводороды. Т. 1. Пер. с англ. М., Химия, 1971. 28. Венкатара-
ман К-, Айер В. Н. — В кн.: Химия синтетических красителей. Т. 5/Под ред.
К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л., Химия, 1977, с. 120.
29. Эмерсон В.—В кн.: Органические реакции. Сб. 5. Пер. с англ. М., ИЛ,
1951, с. 347—432. 30. Ворожцов Н. Н., мл. — В кн.: Химия синтетических кра-
сителей. Т. 3/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфро-
са.— Л., Химия, 1974, с. 1176.
31. Беркман Б. Е. Промышленный синтез ароматических нитросоединений
и аминов. М., Химия, 1964. 32. Albright L. F., Van Munster F. H., Forman J. G.—
Chem. Eng. (New Jork), 1967, v. 74, № 23, p. 251—259. 33. Dovell F. S., Fergus-
son W. E., Greenfeld H.—-Ind. Eng. Chem. Prod. Res. Develop., 1970, v. 9, № 2,
p. 224—229. 34. Jardine I. McQuillln F. J.—Chem. Commun., 1970, № 11, p. 626.
35. L’Eplattenier F., Matthys P., Calderazzo F.— Inorganic Chem., 1970, v. 9, №2,
p. 342—345. 36. Vorozhtsov N. N. jr- — In: Aromatic Compounds/Ed. H. Zollinger,
London, Butterwoths; Baltimore, Univ. Park Press, 1973, v. 3, p. 326. 37. Kop-
шак В. В. и др. Авт. свид. СССР 218898, 1968. 38. Пат. ФРГ 2434466, 1975;
Chem. Abstr., 1975, v. 82, 172614. 39. Лукашевич В. О. — Усаохн химии, 1948,
т. 17, вып. 6, с. 692—709. 40. Рашевская С.—ЖОХ, 1940, т. 10, вып. 12,
с. 1089—1093.
41. Соре О. J., Brown R. К. — Сап. J. Chem., 1962, v. 40, № 12, р. 2317—2328.
42. Hojo М., Takagl У., Ogata У. — J. Amer. Chem. Soc , 1960, v. 82, № 10, '
p. 2459—2462. 43. Idoux J. P. — J. Chem. Soc. C, 1970, p. 435—437. 44. Kvapil J-,
Viba Z., Allan Z.— J. Collect. Czech. Chem. Commun., 1970, v. 35, № 1, p. 366— <
370. 45. Камм О. — В кн.: Синтезы органических препаратов. Сб. 1. Пер. с англ.
№., ИЛ, 1949, с. 432—434. 46. Ингольд К. Теоретические основы органической «
химии. Изд. 2-е. Пер. с англ./Под ред. И. П. Белецкой. М., Мир, 1973. 47. Яп. -j
пат. 175973, 1950; Chem. Abstr., 1951, v. 45, 7146. 48. Ценюга В. С., Падежи- '
на Н. А., Овчинников П. Н. и др. — ЖОрХ, 1978, т. 14, № 2, с. 337—343. 4
488
49. Доналдсон Н. Химия и технология соединений нафталинового ряда. Пер.
с англ./Под ред. А. И. Королева. М., Госхимиздат, 1963. 50. FIAT, v. 1313, № 2,
р. 179.
51. Лукашевич В. О., Кролик Л. Г. — ДАН СССР, 1948, т. 63, № 5, с. 543—
544. 52. Кролик Л. Г., Лукашевич В. О. — ДАН СССР, 1953, т. 93, № 4, с. 663—
666. 53. Кролик Л, Г., Лукашевич В. О. — ДАН СССР, 1949, т. 65, № 1, с. 37—
40. 54. Лукашевич В. О., Кролик Л. Г, — ДАН СССР, 1962, т. 147, № 5,
с 1090—1093. 55. Banthorpe D. V., Hughes Е. D, — J. Chem. Soc., 1964, № 9,
р. 2849—2854; р. 2860—2864 . 56. Funke W. — Chem.-Ing. Techn., 1963, Bd. 35, № 5.
S. 336—340. 57. Фирц-Давид Г. Э., Бланже Л. Основные процессы синтеза кра-
сителей. Пер. 'с нем./Под ред. С. В. Богданова и И.В. Фодимана. М., ИЛ, 1957,
с. 114. 58. Пат. ФРГ 1004190, 1957; Chem. Abstr., 1960, v. 54, 414С; Пат. ФРГ
1005525, 1957; Chem. Abstr., 1960, v. 54, 414H; Пат. ФРГ 1012305, 1957; Chem.
Abstr., 1960, v. 54, 414. 59. Balakrishnan T. D. e.a. — Chem.-Ing. Techn., 1969,
Bd. 41, № 13, S. 776—777. 60. Ленуар И. — В кн.: Химия синтетических краси-
телей. Т. 5/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л.,
Химия, 1977, с. 274.
61. В кн.: Вредные вещества в промышленности. Т. 2. Изд. 7-е/Под ред.
Н. В. Лазарева и Э. Н. Левиной. Л., Химия, 1976, с. 316. 62. Нидерл. пат.
6604118, 1966; Chem. Abstr,, 1967. v. 66, 65277. 63. Hutchins R. О. e.a.—J. Org.
Chem., 1971, v. 36, № 6, p. 803—806. 64. Analitical Chemistry of Synthetic Dyes/
Ed. K. Venkataraman. London, Wiley, 1977. 65. Колеман — В кн.: Синтезы орга-
нических препаратов. Сб. 1. Пер. с англ. М., ИЛ, 1949, с. 429. 66. Truce W. Е.,
Murphy А. М. — Chem. Rev., 1951, v. 48, № 1, р. 69—124. 67. Вейганд-Хильгетаг.
Методы эксперимента в органической химии. Пер. с нем./Под ред. Н. Н. Суво-
рова. М., Химия, 1968. 68. Острожинская Г. И. — Анилинокрас. пром., 1935, т. 5,
с. 138—140. 69. Лемин Н. Н, Авт. свид. СССР 236480, 1968; 249396, 1969;
274117, 1970; 297275, 1970. 70. Лемин Н. Н., Карандашева Н. Н. Авт. свид.
СССР 296410, 1970; 296411, 1970.
71. Лемин Н. Н., Ямщиков А. В. — ЖОрХ, 1972, т. 8, вып. 8, с. 1672—1677.
Глава 17
ОКИСЛЕНИЕ БОКОВЫХ ЦЕПЕЙ,
АРОМАТИЧЕСКИХ КОЛЕЦ
И ФУНКЦИОНАЛЬНЫХ ГРУПП
Как уже говорилось (см. 16.1), реакции окисления — это про-
цессы, в которых органическое соединение отдает электроны окис-
лителю и при этом в нем увеличивается число связей С—О (N—О,
S —О и т. п.) или уменьшается число связей С—Н (N—Н, S—Н и
т. п.). Более строгое определение этим реакциям в органической
химии дать, к сожалению, невозможно.
Практическое значение реакций окисления в промышленности
органического синтеза, в том числе и в производстве промежуточ-
ных продуктов исключительно велико, так как они представляют
собой один из основных методов переработки ароматического
сырья и получения его из нефти (см. 1.1). В этой главе рассматри-
ваются только те процессы окисления, которые непосредственно
связаны с химией промежуточных продуктов.
489
В крупнотоннажных процессах в качестве окислителя исполь-
зуют кислород воздуха с применением катализаторов. Реакции
проходят в жидкой или паровой фазе и имеют цепной радикальный
механизм. Реакции каталитического дегидрирования, которые так-
же следует отнести к реакциям окисления, могут идти без внешне-
го окислителя, внутримолекулярно, с образованием водорода:
С6Н,2 •—> СвНв + ЗНг
Механизм дегидрирования противоположен механизму реакций
гидрирования (см. 16.2.4). На специальных катализаторах реакции
дегидрирования можно проводить и в присутствии кислорода
(окислительное дегидрирование). При этом образуется не водород,
а вода.
В некаталитических реакциях чаще всего применяют следую-
щие окислители: бихромат натрия Na2Cr2O7, перманганат калия
КМпО4, оксид марганца (IV) МпО2, оксид свинца (IV) РЬО2, азот-
ная кислота, пероксид водорода Н2О2 и надкислоты. Реакции с
этими окислителями могут протекать с отрывом пары электронов,
т. е. по ионному механизму или же с отрывом одного электрона —
по радикальному механизму.
В молекулах органических соединений наиболее чувствительны
к окислению положения, имеющие наибольшую электронную плот-
ность,— те, что подвергаются атаке электрофильных реагентов.
Окисление органических соединений можно проводить и элект-
рохимическими методами [1].
17.1. ОКИСЛЕНИЕ БОКОВЫХ ЦЕПЕЙ
При окислении алкилароматических соединений дей-
ствие реагента в первую очередь направляется к атому углерода,
примыкающему к ароматическому кольцу. В результате этих реак-
ций могут быть получены перекиси, спирты и фенолы, альдегиды и
кетоны, карбоновые кислоты, нитрилы и непредельные соединения.
17.1.1. Получение ароматических карбоновых кислот
Окисление боковых цепей — наиболее общий и класси-
ческий метод получения аренкарбоновых кислот. Предпочтительно
исходить из метиларенов, так как на окисление более сложных
боковых алкильных групп требуется больше окислителя и реакция
идет медленнее, по усложненной схеме. Лишь когда необходимое
метильное производное трудно доступно, окислению подвергают
ацетильную или иную группу, которую вводят в ароматическое
соединение, например, с помощью реакции Фриделя — Крафтса
(см. 6.1 и 6.3).
Окисление метиларенов до карбоновых кислот проходит по схе-
ме:
АгСНз —> АгСН2ОН —> АгСН=О —> АгСООН
490
Однако, так как труднее всего идет первая стадия, на проме-
жуточных удается остановиться лишь при соблюдении особых ус-
ловий.
Для окисления метильной группы в карбоксильную применяют
Na2Cr2O7 с H2SO4, КМпО4, МпО2 с H2SO4, разбавленную HNO3
при нагревании, но наиболее прогрессивным является окисление
кислородом воздуха в жидкой фазе в присутствии катализатора,
чаще всего нафтената или резината кобальта(III).
Окисление кислородом воздуха начинается со стадии иници-
ирования; от метильной группы ароматического соединения отры-
вается атом водорода и образуется радикал бензильного типа (1):
АгСНз —> АгСН2- + Н»
О)
Этот процесс катализируется солью металла переменной валент-
ности, которая превращает атом водорода в протон с переходом
металла в низшую степень окисления. В ароматическом ряду ре-
акция идет особенно гладко потому, что металл переменной валент-
ности окисляет ароматическое соединение, захватывая электрон с
верхней заполненной молекулярной орбитали; в результате перво-
начально образуется катион-радикал и уже он, отдавая протон
какому-либо основанию, превращается в бензильный радикал;
+в“
-вн
Радикал (1) далее реагирует с кислородом воздуха, превра-
щаясь в пероксирадикал (2), достаточно активный для того, что-
бы с исходным соединением образовать первичную гидроперекись
(3) и новый радикал (1):
АгСН2« + О2 —> АгСН2ОО-
(О (2)
АгСН2ОО . +АгСНз —> АгСН2ООН + АгСН2-
(2) (3) (1)
Таким образом, в результате второй стадии регенерируется
стартовый радикал (1) и цикл превращений может многократно
повторяться; реакция имеет цепной характер.
Однако это первичное образование радикалов (1) при окисле-
нии имеет значение лишь для сокращения индукционного периода
реакции, тогда как в развившемся процессе новые радикалы обра-
зуются путем более быстрых реакций вырожденного разветвления
цепи. При умеренной температуре жидкофазных процессов они
происходят путем разложения гидроперекисей (3), а в дальнейшем
491
и надкислот, которое ускоряется металлами переменной валентно-
сти:
2АгСН2ООН —> АгСН2О • + АгСН2ОО • + Н2О
(3) (4) (2)
АгСН2ООН + Со2+ —> АгСН2О • + ОН* + Со3+
43) (4)
Потеря радикалов, могущая вызвать обрыв цепи при жидко-
фазном окислении, происходит, главным образом, путем диспро-
порционирования наименее активных перекисных радикалов по
схеме:
2АгСН2ОО • —> молекулярные продукты
Разложение первичных гидроперекисей (3), образующихся при
окислении метиларенов, идет почти исключительно в сторону обра-
зования альдегидов по следующей автокаталитической реакции:
+ он.
АгСН2ООН —> АгСН=О + ОН •
—‘Г12О
(3)
Однако альдегиды при жидкофазном окислении обычно не на-
капливаются, так как быстро превращаются далее в ацильные (5)
и ацилпероксильные (6) радикалы и надкислоту (7):
О О
II II
АгС • + О2 —>- АгСОО •
(5) (6)
О О О
II II II
АгСОО . + АгСН=О —> АгСООН + АгС.
(6) (7) (3)
В свою очередь, надкислота (7) вступает во взаимодействие с
альдегидом, образуя по равновесной реакции а-гидроксиперекись
(8), в присутствии катализатора диспропорционирующую с обра-
зованием двух молекул карбоновой кислоты:
ОО О ОН
II II II I
АгСООН + СНАг ^=±: АгСООСНАг —> 2АгС00Н
(7) (8)
Столь сложный путь окисления метильной группы в карбо-
ксильную при окислении кислородом воздуха [2] обычно не ме-
шает получению карбоновых кислот с высоким выходом.
Рассмотренный здесь механизм инициирования реакций жид-
кофазного окисления углеводородов солями металлов переменной
валентности излагается во многих монографиях и учебниках и,
казалось бы, является общепризнанным. Однако за последние
годы накопился ряд фактов, не укладывающихся в рамки этого
механизма. Так, установлено, что не только соли металлов пере-
менной валентности, но исами металлы в мелко диспергированном
виде (размер частиц 25 А, т. е. 2,5 нм), весьма эффективно ини-
циируют реакции окисления [3]. Такое же действие оказывают
492
соли многих металлов постоянной валентности, например натрия,
калия, кальция, бария, цинка, кадмия и др. Они; хотя и не могут
окислять органические соединения, отнимая у них электроны, про-
являют высокий каталитический эффект, часто превосходящий
каталитическое действие солей металлов переменной валентно-
сти [4].
В ' связи с изложенным высказываются предположения, что
механизм каталитического действия солей металлов при гомоген-
ном жидкофазном окислении принципиально не отличается от ме-
ханизма действия катализаторов гетерогенного парофазного окис-
ления (см. 17.2.5) и состоит в активации молекул кислорода
[5, с. 45]. Предполагается, что активация происходит путем
включения молекул кислорода в координационную сферу металла
6+ в-
с образованием частиц М • О2, взаимодействующих с углеводо-
родом:
б+ 6-
М • О2 + RH —-> М + но2. + R •
Степень окисления металла при этом не меняется. В случае
комплексных соединений кобальта образование частиц
III *-
Со • О2 и CoL2(B)O2 доказано спектральными методами [6].
Процессы окисления, периодические или непрерывные, ведут в
аппаратах колонного типа, в которых окисляющееся соединение
стекает сверху вниз по тарелкам, а воздух подается в нижнюю
часть. Теплота реакции отводится с помощью внутренних змееви-
ковых теплообменников или выносных холодильников. Так как
получающиеся карбоновые кислоты обладают корродирующим
действием, аппараты изготовляют из специальных сортов стали,
алюминия, иногда титана. Процесс окисления ведут при 120—
200°C под давлением 0,2—1 МПа с катализатором (0,05—0,2%) до
определенной конверсии, при которой скорость окисления начинает
падать. Образовавшуюся карбоновую кислоту отделяют от исход-
ного продукта, который возвращают на окисление.
Скорость реакции окисления метильных групп по причинам,
которые уже изложены, коррелируют с п-константами заместите-
лей в ароматическом кольце, причем электроноакцепторные заме-
стители замедляют реакцию. Этим объясняется четкая стадийность
окисления ди- и триметилбензолов, наблюдаемая при окислении в
описанных условиях:
Образующиеся из ксилолов м- или п-толупловые кислоты окис-
ляются далее до практически ценных изофталевой и терефталевой
493
кислот лишь в значительно более жестких условиях и с плохим
выходом, по-видимому, не только из-за дезактивирующего влияния
карбоксильных групп, но и потому, что толуиловым кислотам и
особенно конечным дикарбоновым кислотам свойственны высокие
температуры плавления и плохая растворимость в углеводородах;
превращение толуиловых кислот в метиловые эфиры позволяет
гладко довести процесс до конца — окислить вторую метильную
группу [2]. Повторным метилированием получают, например, ди-
метилтерефталат, используемый в очень больших количествах для
производства волокна терилен (лавсан):
СНзСбН4СООН —> СН3С6Н4СООСН3 —> НООСС6Н4СООСН3 —>
—> СН3ООСС6Н4СООСНз
Но окисление м- и n-ксилолов можно провести и одностадийно,
если реакцию вести в растворителе, чаще всего в уксусной кислоте,
используя в качестве катализатора не только соль кобальта, но еще
и ацетальдегид [7] или бромиды натрия и калия. Механизм дей-
ствия таких смешанных катализаторов до конца не выяснен.
Предполагается, что с бромидами в ходе реакции образуется эле-
ментарный бром, инициирующий образование радикалов по обыч-
ному механизму.
Одностадийное окисление м- и n-ксилолов реализовано в про-
мышленности и успешно конкурирует с двухстадийным. Окисление
проводят воздухом в уксуснокислом растворе при 125—200 °C и
давлении до 4 МПа в барботажных колоннах. Катализаторами
служат ацетат кобальта и ацетальдегид. Продукты реакции —
дикарбоновые кислоты — выпадают из раствора и отделяются
фильтрованием. Уксусную кислоту после регенерации и отделения
воды возвращают в процесс. Выход терефталевой кислоты состав-
ляет 98% от теоретического [7].
В промышленности осуществляется также одностадийное окис-
ление n-ксилола при 195—205 °C кислородом воздуха в присут-
ствии 0,45% (масс.) солей кобальта, марганца и молибдата аммо-
ния с добавкой 0,45% (масс.) бромида натрия; выход продукта
90% [8].
Все эти методы приобрели большое значение и для окисления
других соединений, например псевдокумола в тримеллитовую кис-
лоту (9) и дурола в пиромеллитовую кислоту (Ю) [9] , хотя по-
следнюю предпочитают получать контактным окислением в паро-
вой фазе над ванадиевым ангидридом V2O5 [10].
Нзсх^ х .СН3 НООСх.х.СООН X — х х ^^СНз ^^соон (9)
H3CY х.СНз НООСх.х/СООН X — ТТ
НзС-^5 ^^СНз ноосх^"^соон (10)
494
Обе кислоты находят применение для получения термостойких
полимеров и волокон.
Окисление о-ксилола до фталевой кислоты (11), образующей
летучий и термостойкий фталевый ангидрид (12), обычно ведут в
паровой фазе с помощью кислорода воздуха и катализатора V2O5
иногда с различными добавками.
СООН
СООН
(И) (12)
В промышленности фталевый ангидрид до сих пор получают в
основном контактно-каталитическим окислением нафталина в па-
ровой фазе (см. 17.2.5). Процесс получения фталевого ангидрида
из о-ксилола за последние годы подвергался усиленной разработке
в связи с тем, что нефтехимический о-ксилол вполне доступен, а
окисление его теоретически выгоднее окисления нафталина [2, 12].
На практике, однако, исходя из о-ксилола, трудно достичь выходов
фталевого ангидрида выше 60—70% от теоретического, в то время
как при окислении нафталина выход составляет 85—90%.
Для окисления о-ксилола используются как низкотемператур-
ный, так и высокотемпературный режимы [13]. В первом случае
работают с закрепленным слоем катализатора при 350—360 °C и
продолжительности контакта 1 с, причем лучшие результаты до-
стигаются при добавлении в реакционную смесь SO2. Во втором
реакцию ведут при 420—450 °C и времени контакта 0,1 с. Условия
процесса с применяемым в промышленности кипящим слоем ката-
лизатора не опубликованы.
Разрабатывается также окисление о-ксилола во фталевый ан-
гидрид в жидкой фазе с солями кобальта (III) и бромидами [14].
Реакция идет постадийно, с промежуточным образованием о-толу-
иловой кислоты, которую этерифицируют метиловым спиртом и
эфир затем окисляют; в результате образуется фталевая кислота и
ее ангидрид. Авторы утверждают, что этот процесс позволяет
создавать установки большой единичной мощности (до
64000 т/год) и способен конкурировать с парофазным окислени-
ем.
Наряду с каталитическим окислением кислородом воздуха в
промышленности продолжают использовать окислительные реак-
ции, идущие при нагревании с 40—60%-ной HNO3 под давлением
[И]. При условии улавливания окислов азота и регенерации азот-
ной кислоты эти реакции достаточно эффективны, но производство
терефталевой кислоты этим способом [15] быстро теряет свое зна-
чение. Метод, однако, дает хорошие результаты при получении
поликарбоновых кислот, например при окислении гексаметилбен-
зола в бензолгексакарбоновую кислоту и в некоторых других ре-
акциях [11].
495
Каталитическим окислением кислородом воздуха постепенно
заменяют процессы, в которых еще недавно применялись неорга-
нические окислители. Уже сейчас таким путем получают, например,
n-нитробензойную кислоту из и-нитротолуола [11] и более слож-
ные продукты, в частности поликарбоновую кислоту (23) из (22).
При сравнительно небольших объемах производства и в лабо-
раторной практике для окисления метильных групп в карбоксиль-
ные до сих пор пользуются и такими распространенными неорга-
ническими окислителями, как бихромат натрия и перманганат ка-
лия.
Бихромат натрия применяют в виде раствора в серной кислоте
(хромовая смесь). Он значительно лучше растворим, чем калиевая
соль (К2СГ2О7). Концентрацию H2SO4 подбирают в зависимости от
необходимого температурного режима процесса. Отходы произ-
водства, содержащие соли хрома (III), можно использовать в ко-
жевенной промышленности. Этот метод используют для получения
о- и n-нитробензойной, 2,4-дихлорбензойной, о-сульфамоилбензой-
ной кислоты, производных 9,10-антрахинон-2-карбоновой кислоты
и др. [16, с. 588]. Реакции идут по уравнению:
АгСН3 + Na2Cr2O7 + 4H2SO4 —> ArCOOH + Cr2(SO4)3 + Na2SO4 + 5H2O
Интересно, что при действии хромовой смесью на метилнафта-
лины идет окисление ароматического кольца и вместо соответству-
ющих нафталинкарбоновых кислот получаются производные на-
фтохинона:
О
Окислить метильные группы в метилнафталинах можно, ис-
пользуя водный раствор бихромата без кислоты при нагревании в
автоклаве при 250 °C. Нафталинкарбоновые кислоты получаются
при этом с хорошим выходом [17]. Обзор современных методов
получения 2,6-нафталиндикарбоновой кислоты см. [18].
Перманганат калия добавляют в твердом виде или в форме
водного раствора к раствору, суспензии или эмульсии окисляемого
соединения в воде. Перманганат при этом восстанавливается до
диоксида марганца, выпадающего в осадок, который отделяют и
используют для других окислительных реакций. При реакции об-
разуется щелочь, благодаря чему карбоновая кислота переходит в
раствор:
АгСНз + 2КМпО4 —-> АгСООК + 2МпО2 + КОН + Н2О
Когда окисляемое соединение кроме метильной группы содер-
жит еще амино- или гидроксильную группу, их защищают ацили-
рованием, так как иначе может пойти окисление ароматического
кольца. В этом случае щелочь, образующаяся при восстановлении
496
перманганата, нежелательна: она катализирует гидролитическое
отщепление защитных ацильных групп. Чтобы воспрепятствовать
этому в раствор добавляют сульфат магния, образующий со ще-
лочью очень малоосновный гидроксид магния. Так получают, .на-
пример, «-аминобензойную кислоту из п-толуидина:
СНз СНз СООН СООН
NH2 NHCOCH3 NHCOCH3 nh2
Другие примеры технического использования КМпСЧ для полу-
чения карбоновых кислот см.. [16]. Для этой же цели можно
использовать и МпО2 с разбавленной H2SO4.
Аренкарбоновые кислоты можно получать окислением не только
боковых метильных групп, но и других алкильных заместителей.
Так, при окислении аценафтена (13) с хорошим выходом образу-
ется 1,8-нафталиндикарбоновая (нафталевая) кислота (17), самая
доступная из нафталиндикарбоновых кислот. Ее получают, окис-
ляя аценафтен кислородом воздуха в уксусной кислоте в присут-
ствии солей кобальта (III), а также контактно-каталитически в па-
ровой фазе. Можно окислять аценафтен также раствором Na2Cr20r
в серной или уксусной кислоте, а также нейтральным раствором
Na2Cr2O? при 260 °C под высоким давлением по непрерывному
способу [19]. Реакция проходит через стадии образования аце-
нафтенона (14), аценафтенхинона (15) и нафталальдегидовой кис-
лоты (16):
(13) (14) (15)
ОСН СООН НООС СООН
(16) (17)
Нафталевую кислоту (17) используют в синтезе промежуточ-
ных продуктов, красителей и люминофоров, а также для изомери-
зации в 2,6-нафталиндикарбоновую кислоту (см. 6.3.5).
Практически важным продуктом окисления аценафтена явля-
ется также дикетон, известный под тривиальным названием аце-
нафтенхинон (15). Остановить процесс окисления на стадии по-
лучения этого продукта довольно трудно, так как он легко
превращается далее. Можно, однако, получать аценафтенхинон, на-
пример, окисляя аценафтен действием хромового ангидрида СгОз
497
в ледяной уксусной кислоте, но при этом попутно образуется зна-
чительное количество нафталевой кислоты (17). Поэтому аценаф-
тенхинон целесообразнее получать, действуя на аценафтен этил-
нитритом в спиртовом растворе соляной кислоты. Образующийся
вначале аценафтенхинондиоксим (18) превращается затем в аце-
нафтенхинон действием формальдегида в водном растворе H2SO4:
Продукт очищают, переводя его в растворимое в воде бисуль-
фитное соединение, разрушающееся при действии соды [16].
Интересно, что при нагревании с 30%-ным раствором КОН
аценафтенхинон с почти количественным выходом образует на-
фталальдегидовую кислоту (16) [19, с. 487].
1,4,5-нафталинтрикарбоновую кислоту (21), которую применя-
ют для синтеза люминофоров [20, с. 191], до недавних пор полу-
чали окислением 5-ацетилаценафтена (19) в две стадии: вначале
Na2Cr2O7 в уксусной кислоте до 4-ацетилнафталевой кислоты
(20), а затем NaClO в водно-щелочной среде в целевой продукт
(21).
(20)
(19)
(21)
Оказалось, что это превращение можно провести и в одну
технологическую стадию, если вести реакцию с Na2Cr2O7 в уксус-
ной кислоте в присутствии солей железа [21].
Применяемую в синтезе кубогенов 1,Г-бинафтил-4,4',5,5/,8,8'-
гексакарбоновую кислоту (23) получают каталитическим окисле-
нием кислородом воздуха б.б'-диацетил-б.б'-биаценафтенила (22)
[22]:
(22)
(23)
В общем случае, однако, окисление ацетильных групп в кар-
боксильные предпочитают вести действием гипохлоритов или ги-
побромйтов (солей хлорноватистой НСЮ и бромноватистой НВгО
кислот) в водной среде. Этот метод используется в технике для
получения 4-бифенилкарбоновой и 4'-нитро-4-бифенилкарбоновой
кислот из соответствующих ацетильных производных [16]. Реак-
ции, по-видимому, идут с промежуточным хлорированием или бро-
мированием метильных групп и с последующим отрывом их в виде
хлороформа или бромоформа:
АгСОСНз —> АгСОССЦ —> АгСООН + СНС1Э
17.1.2. Получение ароматических нитрилов
(окислительный аммонолиз)
Окислительным аммонолизом называют одностадийный
синтез нитрилов и некоторых других азотсодержащих соединений
путем сопряженного окисления кислородом соответствующего уг-
леводорода и аммиака [23]. Реакцию обычно проводят в газовой
фазе с гетерогенными катализаторами из оксидов переходных ме-
таллов, чаще всего ванадия, молибдена, титана, при 500—550 °C.
Процессу способствуют пары воды, галогены, органические гало-
генпроизводные и серусодержащие соединения.
Этим путем из толуола или этилбензола получается бензонит-
рил с выходом более 90%, [24], из о-ксилола—фталодинитрил с
выходом 80% [25]. Последний используется в синтезе фталоциа-
нинов. Этот же продукт может быть получен из нафталина [26]. Из
м- и n-ксилолов образуются изофтало- и терефталодинитрилы,
которые можно гидролизовать в дикарбоновые кислоты или вос-
станавливать в ксилилендиамины, применяемые в химии полиме-
ров. Описано также получение ряда других ароматических про-
дуктов, в том числе пиридинкарбонитрилов и пиридиндикарбони-
трилов [27].
Существует две гипотезы относительно механизма этих практи-
чески очень важных реакций. Одна из них основана на предполо-
жении, что в присутствии молекулярного кислорода может проте-
кать каталитическое дегидрирование углеводородов с образовани-
ем карбенов, а аммиак в тех же условиях может дать нитрен.
Рекомбинация этих частиц приводит к альдиминам, которые при
дальнейшем дегидрировании дают нитрилы:
+ О2 ••
АгСНз ----> АгСН —
—> ArCH=NH —> ArC^N
Согласно другой точке зрения, альдимины образуются из аль-
дегидов или радикальных продуктов, возникающих при окислении
углеводорода и аммиака. Справедливость первой гипотезы недавно
получила подтверждение при изучении окислительных превраще-
499
ний пропилена, толуола и n-ксилола на окисных катализаторах
[28]. Авторы утверждают, что во всех случаях нитрильная группа
формируется из карбенов и нитрена, а образующиеся при этом
альдегиды являются побочными продуктами реакции. Однако вы-
сказывается мнение и о сосуществовании двух механизмов реак-
ции [27].
17.1.3. Получение ароматических альдегидов и кетонов
Получение кетонов путем прямого окисления примыкаю-
щих к ароматическому кольцу метиленовых групп обычно проходит
однозначно и при действии многих окислителей. Так, гомогенно-
каталитическое окисление этилбензола и пропилбензола кислоро-
дом воздуха в присутствии солей кобальта(III) заканчивается на
стадии образования ацетофенона или пропиофенона; окислить их
далее до бензойной кислоты в этих условиях не удается
[29, с. 483]:
СбН5СН2СН3 —> С6Н5СОСН3 С6Н5СН2СН2СН3 —> С6Н5СОСН2СН3
Селективность окисления по атому углерода, примыкающему к
ароматическому кольцу, приводит к образованию из тетралина
(24) «-гидроперекиси (1-тетралинилгидропероксид) (25), при раз-
ложении которой получаются 1-тетралон (26) и частично 1-тетра-
лол (27) [19, с. 586]:
В определенных условиях (25) можно выделить из раствора
[30], но при практическом осуществлении процесса в этом нет
необходимости, так как под влиянием катализаторов в реакцион-
ной массе идет дальнейшее превращение (25) в целевые про-
дукты (26) и (27).
В качестве катализаторов для окисления тетралина шире всего
распространены стеарат или нафтенат кобальта и стеарат или
резинат марганца. Процесс можно вести в уксусной кислоте, ис-
пользуя ацетат или хлорид кобальта. К концу реакции смесь со-
держит 1% гидроперекиси, степень превращения (24) в (26) со-
ставляет 40%. Примерно такие же результаты дает окисление без
растворителя при 140°C в присутствии силиката кобальта, предва-
рительно прогретого в течение 2 ч при 575 °C. Реакцию инициируют
гидроперекисью кумола. Весьма эффективными катализаторами
500
для окисления тетралина служат соли или комплексные соедине-
ния рутения и осмия [31]. Изучена также кинетика жидкофазного
окисления тетралина [32].
Продукты окисления (26) и (27) имеют близкие температуры
кипения, и поэтому разделять их приходится преимущественно
химическими методами; например, легко проходит дегидратация
1-тетралола (27) в 1,2-дигидронафталин (28) [33]:
ОН
(27) (28)
Однако при использовании 1-тетралона и 1-тетралола по основ-
ному назначению — каталитическом дегидрировании до 1-нафтола
(см. 17.1.5) •—можно употреблять и неразделенную смесь.
Окисление тетралина кислородом воздуха в присутствии ката-
лизатора проводят в аппаратах непрерывного действия при 100—•
150 °C [31]. Образующаяся смесь продуктов реакции и исходного
тетралина стекает в ректификационную колонну непрерывного
действия, работающую под вакуумом, в которой идет их разделе-
ние. Тетралин возвращается в цикл, а 1-тетралон и 1-тетралол
поступают на дегидрирование.
Общим методом получения ароматических альдегидов является
окисление метильных групп ароматических соединений. Но хотя
при окислении метиларенов кислородом воздуха альдегиды—
обязательные промежуточные продукты реакции (см. 17.1.1), оста-
новиться на этой стадии окисления, как уже указано, чрезвычайно
трудно. Объясняется это тем, что альдегиды очень легко образуют
перекиси и гидроперекиси, превращающиеся далее в карбоновые
кислоты.
Прямое окисление метильной группы в альдегидную удается
осуществить обычно только в тех случаях, когда в ароматическом
кольце имеются достаточно сильные электроноакцепторные заме-
стители, затрудняющие дальнейшее окисление. В качестве окисли-
телей при этом используют МпО2 в кислой среде, Мп2(5О4)з или
же SeO2. Так, применяя Mn2(SO4)3, окисляют 2,4-толуолдисульфо-
кислоту (29) до 4-формил-1,3-бензолдисульфокислоты (30), а
4-нитро-2-толуолсульфокислоту (31)—до 5-нитро-2-формилбензол-
сульфокислоты (32) [16].
601
2,4-Динитробензальдегид предпочитают Получать Через азоме-
тин (см. 14.1.3).
Диоксид селена с успехом используется для окисления 4-ме-
тилбензантрона (33) в 4-бензантронкарбальдегид (34) [16], а
также для получения многих гетероциклических альдегидов из
соединений, содержащих метильную группу в а- или у-положениях
к пиридиновому азоту. Так, из лепидина (35) может быть получен
цинхонинальдегид (36), а из хинальдина (37)—хинальдинальде-
гид (38).
уравнению:
АгСНз + SeO2 —> АгСНО + Н2О + Se
ИЗ о- и
Для прямого получения соответствующих альдегидов
п-нитротолуолов используется интересный способ: окисление ведут
хромовым ангидридом в смеси уксусной кислоты, уксусного ан-
гидрида и серной кислоты при низкой температуре. При этом
образуется нитробензилидендиацетаты, которые затем гидролизу-
ют разбавленной соляной или серной кислотой и получают о- и
п-нитробензальдегиды [35]:
+ CrOg
O2NC6H4CH3 + 2(СН3СО)2О.. -• >
—-2Сг1зСииг1
+ НС1; +Н2О
—> O2NC6H4CH(OCOCH3)2 r> O2NC6H4CHO
—tl з UO tl
По-видимому, этот способ может найти применение и для син-
теза других альдегидов.
Практически единственным и очень важным методом получения
п-аминобензальдегида является нагревание спиртового раствора
n-нитротолуола с полисульфидом натрия и щелочью [36]:
п-О2МСбН4СН3 —> n-H2NCsH4CHO
Выход продукта достигает 50% от теоретического. Механизм
реакции неизвестен.
602
Одним из общих методов получения альдегидов служит гидро-
лиз дихлорметильных производных, получающихся при радикаль-
ном хлорировании (см. 14.1.1).
АгСНз —► ArCHCh —> АгСН=О
Альдегиды можно получить и путем окислительных превраще-
ний монохлорметильных производных. Один из старых способов
получения о-нитробензальдегида (42) состоит в том, что о-нитро-
бензилхлорид (39) действием анилина переводят в N- (о-нитробен-
зил)анилин, который далее окисляют хромовой кислотой или
перманганатом калия до соответствующего азометина (41). При
кислотном гидролизе последнего вновь отщепляется анилин и по-
лучается о-нитробензальдегид (42).
Как видно, эта реакция обратна рассмотренной выше (см. 16.5)
реакции восстановительного алкилирования аминов.
Родственный, но значительно более общий характер имеет ре-
акция Соммле [37], с помощью которой альдегиды получают из
хлорметильных производных действием уротропина и последую-
щим гидролизом образующихся четвертичных аммониевых солей
при нагревании. При этом промежуточно образуется А-метилен-
амин, который изомеризуется в азометин; с отщеплением от по-
следнего метиламина получается альдегид.
АгСН2С1 + CeHijNj —► [АгСНгЫ^СеНи] С1 —>
+ Н2О
—> ArCH2N=CH2 —+ ArCH=NCH3 „ц > АгСН=О
—CH3NH2
Ароматические альдегиды получаются в этой реакции с выхо-
дом 50—80%, причем электроноакцепторные заместители в ядре
затрудняют ее течение. Исходя из бисхлорметильных соединений,
можно получать м- и n-диальдегиды, но о-диальдегиды не обра-
зуются, возможно, из-за стерических препятствий.
Галогенметильную группу также можно окислить до альдегид-
ной при действии 2-нитропропана (в присутствии этилата натрия),
превращающегося в ацетоноксим [38]. Этим методом получают с
хорошим выходом о-толуиловый и фталевый альдегиды и другие
продукты.
Бензальдегид производят из бензилхлорида действием
Na2Cr2O7 в присутствии щелочи в водном растворе [16]:
ЗСеН5СН2С1 % Na2Cr2O; -|- NaOH -—> ЗСбН3СНО -р Сг2О3 ~р 2Н2О 3NaCl
§03
Кроме описанных здесь окислительных реакций для получения
ароматических альдегидов используется ряд других методов
(см. 6.2.3 п 6.3.3).
17.1.4. Получение ди- и триарилметановых красителей
Окисление групп СН2 или СН, связывающих два или три
арильных радикала, в которых имеются электронодонорные заме-
стители, считается одним из основных способов получения ди- и
триарилметановых красителей.
Обычно окисление содержащих такие группы ди- и триарилме-
танов проводят на холоду при действии РЬО2 в разбавленной
серной кислоте, а иногда в соляной или уксусной кислоте. Приме-
няется также МпО2, реже FeCl3, бихроматы [39, с. 266].
Раньше предполагали, что окисление, например, триарилмета-
нов (43) идет через промежуточную стадию образования соответ-
ствующих спиртов (карбинольных оснований) (44), с последую-
щим катализируемым кислотой отщеплением от них гидроксиль-
ной группы и образованием красителя (45):
Однако экспериментальные данные опровергают предположе-
ние о промежуточном образовании спиртов (44), так как в одина-
ковых условиях такие спирты при действии кислот превращаются в
катионы (45) медленнее, чем исходные соединения (43) при дей-
ствии на них кислот с окислителями. В связи с изложенным счита-
ют [40], что окисление диарил- и триарилметанов происходит
путем отнятия окислителем двух электронов от iV-атома диалкил-
аминогруппы с последующим или одновременным депротонирова-
нием центрального атома углерода молекулой воды и непосред-
ственным превращением в катион красителя (45).
Другой возможный механизм этой реакции — прямой отрыв от
центрального углеродного атома водорода с парой электронов, т. е.
в виде гидрид-иона. Это предположение подтверждается тем, что
многие акцепторы гидрид-иона гладко превращают триарилмета-
новые производные (43) в катионы соответствующих красителей
504
(45). Например, при действии на (43) трифенилметилхЛорида гид-
рид-ион уходит, образуя трифенилметан.
С хорошим выходом красители образуются и при действии на
(43) трихлоруксусной кислоты; последняя, отрывая ион Н~, вос-
станавливается с распадом на хлороформ и муравьиную кислоту.
Высказано предположение, что окисление с помощью
РЬО2' также идет путем отрыва гидрид-иона [40].
17.1.5. Реакции дегидрирования
Реакции каталитического гидрирования ароматических
соединений (см. 16.2.4) всегда сопровождаются выделением тепла
и уменьшением объема. Их поэтому проводят при сравнительно
невысоких температурах и под давлением. Эти реакции обратимы,
и их можно направить в сторону дегидрирования, если вести
процессы на аналогичных или даже на тех же катализаторах, на
которых происходит гидрирование, но при значительно более вы-
сокой температуре, по возможности без давления.
Например, циклогексанол (46) в присутствии никелевого ката-
лизатора при 360 °C превращается в фенол (47). С медными и
меднохромовыми катализаторами при 280—325 °C дегидрировани-
ем циклогексанола получают технически ценный циклогексанон
(48) [16], используемый в синтезе капролактама.
ОН ОН О
J J II
О •’ о - о
(47) (46) (48)
Реакции дегидрирования, а также окислительного дегидриро-
вания, при котором образующийся водород полностью или частич-
но превращается в воду, используются в промышленности для
ароматизации нефти, при синтезе формальдегида из метанола, при
получении диеновых углеводородов и в ряде других процессов.
Одним из важнейших является получение стирола (50) дегид-
рированием этилбензола (49) при 600—630 °C над окисными ката-
лизаторами, обычно содержащими 87% Fe2O3, 5% Сг20з и8% КгО:
-Н2
С6Н6СН2СНз ---► С6Н5СН=СН2
(49) (50)
Процесс ведут при атмосферном давлении, но в этих условиях
степень конверсии достигает всего 40—50%. Если пары этилбензо-
ла разбавить водяным паром так, чтобы давление паров этилбен-
зола составляло 0,01 МПа, равновесная конверсия возрастает до
80—90% [29, с. 572]. Образующийся стирол после добавления ин-
гибитора полимеризации (обычно вводят гидрохинон) подвергает-
ся перегонке под вакуумом. Отделяющийся этилбензол возвраща-
ется в процесс.
505
Аналогично из изопропилбензола (51) получают а-меТилСтирол
(52), из смеси (53), содержащей 65% м- и 35% и-этилтолуолов,—
смесь изомерных винплтолуолов (54), а из смеси изомерных ди-
этилбензолов (55) — дивинилбензолы (57). В последнем случае
реакция идет через стадию образования винилэтилбензолов (56).
СНз СНз
I I
С6Н5СНСНз —> С6Н5С=СН2 СН3С6Н4СН2СНз —> СН3С6Н4СН=СН2
(51) (52) (53) (54)
С2Н5С6Н4С2Н5 —> С2Н5С6Н4СН=СН2 —> СН2=СН—С6Н4—СН=СН2
(55) (56) (57)
Все эти продукты вырабатываются промышленностью основно-
го органического синтеза в больших количествах и применяются
при получении полимеров.
Что касается производства промежуточных продуктов, то в нем
реакции дегидрирования пока находят лишь ограниченное приме-
нение; в частности, новый процесс получения 1-нафтола (58) за-
ключается в дегидрировании смеси 1-тетралона (26) и 1-тетралола
(27), образующейся при прямом окислении тетралина воздухом
(см. 17.1.3):
Смесь (26) и (27) можно дегидрировать при 500—650 °C в
присутствии водяного пара, используя в качестве катализатора
СаО. Степень превращения составляет 30%, выход целевого про-
дукта— около 80%. В качестве катализаторов могут быть также
использованы никель, медь, железо, кобальт, хром или платина,
нанесенные на оксиды кальция, магния, меди, стронция, цинка, на
кизельгур или силикагель. Выход 1-нафтола при этом находится в
пределах 83—91% [31]. Хорошие результаты достигнуты при ра-
боте на катализаторе, состоящем из металлического никеля и меди,
оксидов хрома и сульфатов щелочных металлов в смеси с нитрата-
ми и нитритами. Особенность этого катализатора заключается в
том, что он вызывает одновременное дегидрирование (27) в (26) и
превращение последнего в (58). Оптимальная температура процес-
са 360—390 °C, выход 1-нафтола 88% [41].
Нашел промышленное применение двухстадийный процесс па-
рофазного дегидрирования указанной смеси. В первой стадии при
200—325 °C на медно-никелевом катализаторе протекает дегидри-
рование (27) в (26). Вторую стадию, образование (58), ведут на
платиновом катализаторе при 350—450 °C. Общий выход 1-нафто-
ла 72% [42].
506
На отечественной установке получения 1-нафтола, созданной в
НИОПиК, дегидрирование также ведется в паровой фазе непре-
рывным способом в системе из двух последовательно соединенных
контактных аппаратов. Образовавшийся 1-нафтол выделяют из
реакционной смеси ректификацией, с последующей кристаллиза-
цией [31].
Новый процесс получения 1-нафтола имеет значительные пре-
имущества перед применяющимися до сих пор процессами, осно-
ванными на кислотном гидролизе 1-нафтиламина и щелочном
плавлении 1-нафталинсульфокислоты (см. 8.1.4 и 8.1.1). Это непре-
рывный и практически безотходный процесс, в котором почти не
используются химические реагенты, а образующийся продукт имеет
высокую степень чистоты и не содержит примесей. Легко увеличить
мощность производства, что при старых методах вызывает серьез-
ные затруднения. На основе 1-нафтола, получаемого новым спосо-
бом, могут быть получены многие промежуточные продукты, в том
числе 1-нафтиламин, не содержащий примеси канцерогенного
2-нафтиламина. Большая потребность в 1-нафтоле связана также
с производством применяемого в сельском хозяйстве инсектици-
да— 1-нафтил-У7-метилкарбамата (севин).
В лабораторной практике дегидрирование часто используют в
качестве последней стадии синтеза полициклических ароматиче-
ских углеводородов, особенно если предшествующее восстановле-
ние приводит к образованию гидрированных продуктов.
Чрезвычайно эффективны в качестве катализаторов дегидриро-
вания платина и палладий, нанесенные на уголь. Реакцию с ними
обычно проводят в паровой фазе при 300—310 °C, сублимируя
углеводород над катализатором в вакууме и токе СО2. В других
случаях ее ведут и в растворах, например в трихлорбензоле, при
200—210 °C. Вместо дегидрирования с катализаторами можно
применять сплавление с серой или лучше с селеном при 300 °C.
Обзор этих способов см. [43, с. 171].
Дегидрирование имеет большое значение также для аромати-
зации непредельно-кольчатых соединений, полученных диеновым
синтезом. При дегидрировании над платиновыми и палладиевыми
катализаторами часто происходят побочные реакции дегидроцик-
лизации. Например, из о-терфенила (59) образуется трифенилен
(60), а из 1,1'-бинафтила (61)—перилен (62) [44, с. 50]:
\59) (6 Л (61) (62)
Реакции дегидроциклизации проходят иногда и при дегидриро-
вании селеном. Например, при нагревании 1-тетралона (26) с
507
селеном до 330 °C наряду с 1-нафтолом образуется 1,2'-бинафти-
лен-2,1/-оксид (63) [45]:
(26) (63)
Обзор реакций окислительного дегидрирования и обсуждение
их механизмов см. [46].
17.2. ОКИСЛЕНИЕ АРОМАТИЧЕСКИХ КОЛЕЦ
Окисление ароматических колец, как правило, идет зна-
чительно труднее, чем рассмотренные выше окислительные реакции
в боковых цепях. Это, однако, в значительной мере зависит от
электронной структуры ароматического соединения. Из ароматиче-
ских углеводородов, бензол, представляющий собой ароматиче-
скую систему с полностью выравненной электронной плотностью,
окисляется труднее всего. Нафталин, в котором эта выравненность
нарушена и имеются связи и атомы с повышенной электронной
плотностью, окисляется значительно легче. По тем же причинам
еще легче идут эти процессы с антраценом, фенантреном и более
сложными соединениями (см. 1.2.3). Во всех случаях электронодо-
норные заместители в кольце облегчают течение реакций окис-
ления.
Возможны по крайней мере три механизма окисления аромати-
ческого кольца [47]: с циклоприсоединением окислителя (напри-
мер озона или тетроксида осмия) к локализованной двойной связи,
с радикальной или с электрофильной атакой окислителем. По-
видимому, наиболее часто встречается радикальный механизм,
который может реализоваться либо, как уже было показано, через
промежуточный радикальный о-комплекс (см.13.1), либо путем
передачи окислителю одного электрона с верхней заполненной
молекулярной орбитали ароматического соединения и образования
катион-радикала, депротонирующегося до (а) или после (б) реак-
ции с радикальной частицей при действии основания:
С другой стороны, принято считать [48], что действие таких
окислителей, как СгО3 или МпО* в растворе осуществляется по
508
третьему механизму — прямой электрофильной атакой ароматиче-
ского соединения с промежуточным образованием положительно
заряженного ст-комплекса (см. 17.2.4 — окисление антрацена).
Окислением ароматических колец могут быть получены фенолы,
хиноны и карбоновые кислоты, весьма важные для синтеза проме-
жуточных продуктов, красителей и химии полимеров.
17.2.1. Получение фенолов прямым окислением
ароматических соединений
Теоретически прямое окисление ароматических соедине-
ний, особенно применительно к получению самого фенола, потреб-
ность в котором продолжает неуклонно возрастать, наиболее пер-
спективно. Этому вопросу посвящена обширная, преимущественно
патентная литература [49].
Разработан промышленный метод синтеза фенола окислением
бензола в водной эмульсии кислородом воздуха с облучением
•у-лучами (60Со) при 200 °C. Недостаток метода — побочное образо-
вание смол [50].
Окисление бензола кислородом в присутствии паров воды
предложено проводить и в псевдоожиженном слое инертного но-
сителя при 670 °C и повышенном давлении [51]. Малые количества
фенола могут быть получены при пропускании воздуха через бен-
зол, содержащий TiCl3 [52]. Аналогично окисляются толуол и
нафталин [53].
Особенно часто в качестве катализаторов окисления упомина-
ются палладий и его соли. Например, сообщается, что фенол обра-
зуется из бензола при комнатной температуре при действии смеси
кислорода и водорода, разбавленной инертным газом в разбав-
ленном растворе НС1 и H2SO4, содержащем небольшие количества
взвеси сульфатов титана и палладия на силикагеле [54]. Окисле-
ние бензола в фенол в присутствии палладия на силикагеле проис-
ходит также в растворе муравьиной кислоты [55].
Изучалось и прямое окисление бензола в уксусной кислоте в
присутствии палладиевых катализаторов для получения фенилаце-
тата. В жидкой фазе наилучший результат получен с палладием и
висмутом на силикагеле. В газовой фазе лучше работает палладий
с золотом с добавкой ацетата кадмия также на силикагеле. Из
хлорбензола и анизола при этом получаются преимущественно
о-производные — о-хлорфенил- и о-метоксифенилацетат. Фенил-
ацетат может быть превращен в фенол путем гидролиза или тер-
молиза, при котором образуется еще и кетен [56]:
С6Н5ОСОСН3 —> С5Н5ОН + СН2=С=О
Особенно интересна реакция окисления бензола в фенилацетат
в уксусной кислоте при катализе хлоридом палладия:
+ V2O2
С6Н6 + СНзСООН ------>• С6Н5ОСОСН3 + Н2О
Эта реакция, получившая название окислительного сочетания
[57], с последующим гидролизом или термолизом фенилацетата
509
могла бы лечь в основу нового метода получения фенола, но, к
сожалению, кислород в таких условиях оказывает ингибирующее
действие [58].
При окислительном сочетании анизола с уксусной кислотой
образуется лг-метоксифенилацетат. Это свидетельствует о том, что
механизм катализа солями палладия иной, чем у катализа метал-
лическим палладием [59].
Соображения о механизме реакции окислительного сочетания и
разнообразные примеры ее использования см. [58].
Не прекращаются попытки прямого синтеза 1-нафтола из на-
фталина. Окисление, как правило, ведут кислородом воздуха в
жидкой фазе в присутствии катализаторов — оксидов серебра, ме-
ди, железа и титана [60]. С выходом 35—45% 1-нафтол может
быть получен парофазным окислением нафталина в присутствии
кислорода и перекиси водорода [61]. Однако практического зна-
чения эти процессы не получили.
В отличие от этого прямое окисление 9,10-антрахинона и осо-
бенно его гидроксильных производных, открытое еще в 1888 г.
(реакция Бона — Шмидта) [16, с. 598; 62, с. 946] продолжают ис-
пользовать в технике. Сам 9,10-антрахинон может быть окислен до
1,4-дигидроксипроизводного — хинизарина (64) нагреванием с
нитрозилсульфатом в присутствии борной кислоты и солей ртути,
взятых в количестве меньшем, чем 1% от массы антрахинона:
О О ОН
(64)
Однако этот способ получения хинизарина [63], как и более
новый, основанный на окислении 9,10-антрахинона кислородом
воздуха в водной среде при 220—300 °C в присутствии бромидов
[64], практического применения не нашел.
1,2,4-Тригидрокси-9,10-антрахинон (пурпурин) (65) получают
окислением ализарина в 93%-ной H2SO4 при постепенном добавле-
нии МпО2 [65]. Этот же продукт может быть получен в аналогич-
ных условиях и из хинизарина (64):
(65)
510
Чаще применяют окисление гидроксиантрйхйнонов высокопро-
центным олеумом, иногда в присутствии борной кислоты. Для
успешного протекания этих реакций необходимо, чтобы в олеуме
содержались хотя бы небольшие количества соединений селена
или ртути, катализирующих окисление. Олеум образует с орто-
расположенными гидроксилами циклические сернокислые эфиры;
благодаря этому электронодонорное влияние гидроксильных групп
ослабляется и окислению подвергается незамещенное кольцо ан-
трахинона. По-видимому, такую же роль играет и борная кислота,
но она может образовывать эфиры и за счет только одного гидр-
оксила, находящегося в a-положении. (Эфиры стабилизируются до-
полнительным взаимодействием бора с карбонильным кислородом
в перн-положении).
Например, окисление ализарина до 1,2,5,8-тетрагидрок-
си-9,10-антрахинона (68) происходит при действии 80%-ного олеу-
ма и комнатной температуре в течение 5 суток. Вначале образуется
сернокислый эфир (66), который окисляется далее до триэфира
(67). По окончании окисления добавляют борную кислоту, реакци-
онную массу разбавляют водой и эфир (67) гидролизуют при
нагревании [66]:
+ н2о
(66) (67) (68)
В аналогичных условиях ведут окисление 1,8-дигидрок-
си-9,10-антрахинона (69) до практически важного 1,4,8-тригидрок-
си-9,10-антрахинона (70) с той лишь особенностью, что борную
кислоту, в количестве 0,5 от массы исходного продукта, добавляют
к олеуму перед окислением [67]:
(69) (70)
Следует отметить, что механизм этих реакций остается неис-
следованным [68].
511
17.2.2. Получение фенолов из карбоновых кислот
В 1948 г. было обнаружено, что при нагревании бензоата
меди(II) образуется смесь продуктов, содержащая фенол, салици-
ловую кислоту и фенилбензоат [69]. Предпринятые позднее де-
тальные исследования механизма и течения этой реакции [11]
позволили разработать новый метод получения фенолов из бен-
зойной кислоты и ее производных; этот метод особенно ценен для
промышленного синтеза фенола, поскольку исходным продуктом
здесь служит толуол, окисляемый в бензойную кислоту. Избыточ-
ные мощности по производству нефтехимического толуола имеются
в ряде стран.
При нагревании бензоата меди(II) (71) в инертных органиче-
ских растворителях (например, в циклогексане) при 250—260 °C
получаются салицилат (72) и бензоат (73) меди(1) и бензойный
ангидрид (74) по суммарному уравнению:
,он
2(C6HsCOO)2Cu —> У—СООСи + С6Н5СООСи + (С6Н5СО)2О
(71) (72) (73) (74)
Очевидно, что происходит окисление (гидроксилирование) аро-
матического кольца за счет превращения Си2+ в Си+. При реакции
идет только орто-гидроксилирование, поскольку образуется сали-
циловая кислота. Поэтому считают, что механизм процесса заклю-
чается в следующем: радикальная атака ароматического кольца
в (71) происходит в циклическом переходном состоянии с одновре-
менной передачей одного электрона связи Си—О атому меди.
Образовавшийся радикал (75) стабилизируется путем отрыва
электрона еще одним атомом меди(II) и связывания протона бен-
зоат-анионом в результате взаимодействия со второй молекулой
бензоата меди. Заключительной стадией превращения является
взаимодействие промежуточно образующихся бензоилсалицилата
меди(1) (76) и бензойной кислоты (77), в результате которого
получаются салицилат меди (72) и бензойный ангидрид (74):
СО
I н ।
н СОС6Н5
+ (C6H5COO)2Cu
-С6Н5СООСи
(71) (75)
(76)
СООСи
+ С6Н5СООН —>
ОСОС6Н5
СООСи
он
+ (С6Н5СО)2О
(77)
(72) (74)
Бензоилсалициловую кислоту и бензойный ангидрид удается в
определенных условиях выделить из продуктов реакции.
512
Степень превращения в салициловую кислоту можно увеличить,
добавляя избыток основного карбоната меди(II)., который связы-
вает бензойную кислоту в соль меди(II), но целесообразнее про-
пускать в реакционную смесь воздух — его кислород окисляет
Си+ в Си2+.
В получаемой в таких условиях смеси кислот содержится до
63% салициловой кислоты, которую можно выделить в виде комп-
лекса с железом (III). Аналогично могут быть получены другие
о-гидроксибензойные кислоты [70].
Если в качестве растворителя при этой реакции использовать
ароматические соединения (толуол, хлорбензол, нитробензол и др.),
то в определенных условиях идет их бензоилоксилирование, причем
группа СбН5СОО вступает избирательно в лгета-положение к за-
местителю, независимо от природы последнего.
Пропускание воздуха, регенерирующего Си2+, позволяет рабо-
тать с каталитическими количествами ее, так что суммарное урав-
нение реакции бензоилоксилирования растворителя приобретает
следующий вид:
Си2+
С6Н5СООН + С6Н5Х +'/2О2 ---> С6Н5СООС6Н4Х-л« + Н2О
Здесь X = СНз, Cl, ЫО2 и др.
Выход бензоилоксипроизводных близок к 50%, побочно полу-
чаются салициловая кислота и продукт ее декарбоксилирования —
фенол [71].
Однако в практическом отношении интереснее оказался вариант
этой реакции, по которому бензойную кислоту в присутствии ката-
литических количеств соли меди (II) нагревают при 220—250 °C с
одновременным пропусканием в реакционную массу водяного пара
и воздуха. Первый продукт окислительного превращения — бензо-
илсалициловая кислота; при гидролизе водой она дает бензойную и
салициловую кислоты, на заключительном этапе происходит де-
карбоксилирование салициловой кислоты с образованием фенола:
СООН
ОСОС6Н5
+ н2о
--------->
—С6Н5С00Н
СООН
ОН
-со2
+ со2
Медь(П) в этом процессе не расходуется, так как регенериру-
ется кислородом из меди(1). Суммарное уравнение реакции полу-
чения фенола из бензойной кислоты имеет вид:
Си2+
С6Н5СООН + '/2О2 —С6Н5ОН + СО2
Метод используется в промышленности, выход фенола дости-
гает 95% [72]. При введении в эту реакцию о- или «-толуиловых
кислот получается .и-крезол, а из .«-толуиловой кислоты — смесь
из 40% о- и 60% л-крезола.
17 Зак. 27i
513
17.2.3. Получение фенолов
из трет-аралкилгидроперекисей
Гидроперекиси образуются из алкилароматических уг-
леводородов при окислении их кислородом воздуха по уже рас-
смотренным цепным радикальным реакциям (17.1.1). Вторичные и
особенно третичные аралкилгидроперекиси в чистом виде доста-
точно стабильны, но бурно разлагаются по цепному механизму в
присутствии солей металлов переменной валентности, металличе-
ской меди и некоторых других соединений. В присутствии катали-
тических количеств сильных кислот (например, 0,1% H2SO4) они
подвергаются изомеризации ионного типа с миграцией арильной
группы к кислороду и образованием нестойкого промежуточного
продукта (а-гидроксиалкиларилового эфира), распадающегося на
фенол и соответствующий кетон (перегруппировка Хока) [73]. Так,
из полученной окислением кумола (изопропилбензол) (78) гид-
роперекиси (а, а-диметилбензилгидропероксид)(79)через проме-
жуточный продукт (80) получаются фенол и ацетон:
(СН3)2СОН
(СН3)2СН (СНз)2СООН А ОН
+ (СН3)2С=О
Таким образом, в результате окисления и этой перегруппировки
алкил исходного алкиларена замещается гидроксильной группой,
т. е. идет окисление ароматического кольца. Перегруппировка Хо-
ка проходит настолько гладко, что оказалось возможным поло-
жить ее в основу не только наиболее распространенного сейчас
технологического процесса получения фенола [2,74], но и ряда
других продуктов (см. ниже). В СССР кумол-гидроперекисный
метод совместного получения фенола и ацетона осуществляется в
промышленном масштабе с 1949 г. [74].
Кумол (78) окисляют кислородом воздуха в тарельчатых реак-
ционных колоннах. С помощью холодильников поддерживается
температура от 120 °C на верхней тарелке до 105 °C в кубе. Подо-
гретый воздух, очищенный от загрязнений, подается противото-
ком в нижнюю часть колонны и барботирует через жидкость на
тарелках. Он увлекает с собой пары кумола и воду; они разделя-
ются в сепараторе, причем кумол после промывки щелочью воз-
вращается на окисление. В нижней части колонны образуется ок-
сидат, содержащий до 30% гидроперекиси. Он охлаждается в
теплообменниках свежим кумолом, идущим на окисление, затем
попадает в вакуум-ректификапионную колонну, где отгоняется ос-
новная часть кумола, возвращаемая в цикл, а в кубе получается
70—75%-ная гидроперекись. Дополнительная вакуум-перегонка
514
при более низком остаточном давлении (обычно 5 мм рт.ст.) по-
зволяет поднять концентрацию гидроперекиси до.90%.
Кислотное разложение гидроперекиси (79) ведут в горизон-
тальном аппарате с перегородками, в котором жидкость перетекает
из одной секции в другую. В аппарат подают гидроперекись,
растворитель — ацетон и серную кислоту, взятую в таком количе-
стве,.чтобы ее концентрация составляла 0,05—0,1%. После нейтра-
лизации образующаяся смесь продуктов попадает на ректифика-
цию, в результате получаются ацетон, фенол, небольшие количе-
ства кумола, возвращаемого в цикл, и кубовый остаток; из
последнего можно выделить побочные продукты реакции: ацето-
фенон, 2-фенил-2-пропанол (диметилфенилкарбинол) и димер «-ме-
тилстирола. Выход фенола и ацетона составляет около 90% от
теоретического.
Важнейшей стадией описанного непрерывного процесса совме-
стного получения фенола и ацетона, от которой более всего зависит
его эффективность, является окисление кумола в гидроперекись.
Эта цепная реакция ингибируется фенолом, алкенами, сернистыми
соединениями и поэтому к исходному кумолу, получаемому по
реакции Фриделя — Крафтса из бензола и пропилена (см. 6.1.3)
предъявляются высокие требования по чистоте. Реакцию нельзя
инициировать с помощью металлов переменной валентности, так
как они вызывают распад гидроперекиси по радикальному меха-
низму. Но и увеличение длительности процесса нежелательно из-за
развития побочных реакций. К ним относится разложение гидро-
перекиси с образованием 2-фенил-2-пропанола, который при де-
гидратации превращается в а-метилстирол, а этот последний явля-
ется ингибитором окисления:
R*
C6HSC(CH3)2 ---> С6Н5С(СН3)2 —> С6Н5С=СН2
I I I
ООН ОН СНз
При разложении пероксирадикалов образуются ацетофенон и
метоксильный радикал:
С6Н5С(СНз)2 —> С6Н5СОСНз + сн3о.
I
оо •
Метоксильный радикал далее превращается в метанол, дающий
продукты кислотного характера.
Поэтому единственный путь увеличения выхода гидропереки-
си — инициирование оеакции окисления кумола. По данным работ
последних лет, при использовании в качестве катализаторов ме-
таллов постоянной валентности, в частности первой и второй групп,
окисление идет на порядок быстрее, чем в отсутствие катализатора.
Эти катализаторы, взятые в концентрации, достаточной для
инициирования реакции, не вызывают распада гидроперекисей
[4,75,76]. Возможный механизм каталитического действия таких
металлов уже обсуждался (см. 17.1.1).
17*
615
Разработан также метод получения фенола из фенилциклогек-
сана (81), окисляемого кислородом воздуха до соответствующей
гидроперекиси (82).
ООН
(81)
+ 02
(82)
Ценность метода в том, что одновременно с фенолом получается
практически важный циклогексанон. Из-за конформационных осо-
бенностей фенилциклогексан вступает в реакцию окисления при-
мерно в 5 раз медленнее, чем кумол, и наряду с основным продук-
том (82) образует 4—5% вторичных гидроперекисей [77].
Аналогичным путем из м- и л-цимолов (м- и л-изопропилтолуо-
лы) получаются м- и л-крезолы, используемые в синтезе полимеров
и инсектицидов.
В результате окисления л- и м-диизопропилбензолов, побочных
продуктов крупнотоннажного производства кумола (см. 6.1.3) об-
разуются бисгидроперекиси (83) и (84). При их разложении полу-
чаются соответственно гидрохинон и резорцин.
>—ОН + 2(СН3)2С=О
-> Hi
Изучение этих процессов было начато в 1949—1950 гг. [2, 74].
Введение двух гидропероксигрупп в одну молекулу происходит в
виде последовательного двухстадийного процесса, и при- этом об-
разуется много побочных продуктов [78]. Но получающиеся бис-
гидроперекиси удается отделить фильтрованием, так как они плохо
растворимы, особенно л-изомер (83). Производство гидрохинона и
резорцина таким способом осуществлено в нескольких странах
|78—81]. Это не удивительно, если принять во внимание очевидные
его преимущества по сравнению с традиционными синтезами
гидрохинона окислением анилина (см. 17.2.4) и резорцина из
.и-бензолдисульфокислоты (см. 8.1.1).
Разработан также новый метод получения 2-нафтола окислени-
ем 2-изопропилнафталина в гидроперекись (85) и кислотным рас-
щеплением ее [2]:
ОН
+ (СН3)2С=О
(85)
СХУаГ ОУ
616
17.2.4. Получение хинонов
Превращение ароматического углеводорода в хинон пред-
ставляет собой стадию более глубокого окисления, чем рассмотрен-
ные выше реакции окислительного гидроксилирования с получе-
нием фенолов. Образование хиноидной системы приводит к потере
соединением ароматического характера и появлению высокой реак-
ционной способности, свойственной непредельно-кольчатым соеди-
нениям.
Важнейшее свойство хинонов — способность обратимо восста-
навливаться в соответствующие двухатомные фенолы (гидрохино-
ны), имеющие ароматический характер, рассмотрено выше
(см. 16.4.2). Оно количественно оценивается по величине нормаль-
ных редокс-потенциалов.
Из двух простейших хинонов 1,2-бензохинон (о-бензохинон),
(86) имеющий очень высокий редокс-потенциал (0,783 В), удается
получить только окислением пирокатехина оксидом серебра в
строго безводном растворителе. Находящий промышленное приме-
нение 1,4-бензохинон (п-бензохинон) (87) значительно устойчивее.
В технике его получают из анилина, проводя окисление
Na2Cr2O7 или МпО2 (пиролюзит) в серной кислоте [82]. Выход
обычно составляет 85—90%. Однако в определенных условиях
1,4-бензохинон может быть получен прямым окислением бензола,
например электрохимическим окислением эмульсии бензола в рас-
творе сульфата и ацетата натрия, причем выход достигает 75%
[83], а также окислением бензола перекисью водорода в уксусной
кислоте [84]. Основное назначение 1,4-бензохинона—восстанов-
ление в гидрохинон (см. 16.4.2).
(86) (87)
О механизме реакции получения 1,4-бензохинона из анилина
известно очень мало. Изучение анодного окисления анилина пока-
зывает [85], что вначале образуется катион-радикал, который ди-
меризуется в /V-фенил-п-фенилендиамин. Повторное окисление
этого продукта приводит к образованию полимера, гидролизующе-
гося в кислой среде до 1,4-бензохинона и n-фенилендиамина. По-
следний при окислении и гидролизе образующегося 1,4-бензохи-
нондиимина также превращается в 1,4-бензохинон.
Гомологи и замещенные производные 1,4-бензохинона (90) ча-
ще всего синтезируют при окислении соответствующих производ-
ных п-аминофенола (88), которые получают через иитрозиро-
вание или азосочетание фенолов и последующее восстановление
517
образующихся нитрозо- и азосоединений. Окисление идет с проме-
жуточным образованием 1,4-хинониминов (89), легко гидролизую-
щихся водой в кислой среде:
Иногда используется и прямое окисление фенолов в хиноны.
Для приготовления небольших количеств хинонов используется
реакция Тойбера — окисление фенолов нитрозодисульфонатом на-
трия «ON(SO3Na)2 (соль Фреми), проходящее в мягких условиях и
дающее хороший выход целевых продуктов [86]. Для получения
окислителя, который необходимо готовить непосредственно перед
синтезом, рекомендуется использовать удобную и простую методи-
ку [87]. Окисление обычно ведут в водно-спиртовом или водно-
ацетоновом фосфатном или ацетатном буферном растворе при
комнатной температуре, используя 2 моль реагента на 1 моль фе-
нола. При занятом лпрп-положении реакция направляется в орто-
положение к гидроксильной группе. Она имеет следующий меха-
низм [88]:
(91)
(87)
Первая молекула окислителя — нитрозодисульфоната, являю-
щегося свободным радикалом, расходуется на образование ради-
кала (91), превращаясь в гидроксиламиндисульфонат. Вторая мо-
лекула окислителя присоединяется к радикалу (91), образуя
нестойкий промежуточный продукт (92), превращающийся в хинон
с отщеплением иминодисульфоната. .
Получить хиноны можно также из метил-, диметил- и триме-
тилфенолов, окисляя их кислородом воздуха при 60 °C под давле-
нием 3 МПа в среде этилцеллозольва, ацетона или диметилформ-
амида в присутствии солей меди(П) [89]. В отсутствие кислорода
воздуха в среде ацетонитрила эти реакции идут при действии
нитрата меди (II), служащего окислителем [90].
Из производных 1,4-бензохинона наибольшее значение имеет
2,3,5,6-тетрахлор-1,4-бензохинон (хлоранил). Его получают при
хлорировании фенола в серной кислоте (см. 5.2.2).
518
Из пяти теоретически возможных нафтохинонов известны толь-
ко три: 1,2-, 1,4- и 2,6-нафтохинон (93), (94) и (95). Их нормальные
редокс-потенциалы см. 16.4.2. Считается, что 1,5-нафтохипон (96)
должен иметь очень высокий редокс-потенциал и поэтому может
существовать только кратковременно, а 2,3-нафтохинон (97) энер-
гетически нестабилен и получить его невозможно:
Мало устойчивый 2,6-нафтохинон может быть получен только из
2,6-дигидроксинафталина (2,6-нафталиндиол) в безводной среде.
Значительно более стабильные 1,2- и особенно 1,4-нафтохинон по-
лучаются окислением соответствующих дигидроксинафталинов в
водной среде при действии FeCl3, СгО3 и других окислителей, а
также окислением аминонафтолов или нафтилендиаминов хромо-
вой кислотой, FeCl3, HNO2 или бромной водой. Имеющие наиболь-
шее техническое применение 1,4-нафтохинон и 2-метил-1,4-нафто-
хинон в промышленности получают прямым окислением нафталина
и 2-метилнафталина бихроматом натрия с серной или уксусной
кислотой; выход продуктов около 50% [19, с. 444].
Известны также 1,4,5,8- и 1,2,3,4-нафтодихиноны (99) и (101).
Их получают окислением соответственно 5,8- и 2,3-дигидроксп-1,4-
нафтохинонов (98) и (100);
(100) (101)
Из девяти возможных хинонов ряда антрацена известны три:
1,2- (102), 1,4- (103) и 9,10-антрахинон (104) [92].
(102) (103) (104)
519
Хиноны (102) и (103) получают окислением соответствующих
аминоантролов, а 9,10-антрахинон (или, просто, антрахинон (104),
обладающий наибольшей стабильностью, — прямым окислением
антрацена, причем выход близок к количественному (нормальные
редокс-потенциала антрахиноновом. 16.4.2).
Из трех названных только 9,10-антрахинон нашел широкое
практическое применение — это одно из главных исходных соеди-
нений в синтезе промежуточных продуктов и красителей.
Окисление антрацена в 9,10-антрахинон можно вести кислоро-
дом воздуха в жидкой фазе в присутствии солей металлов перемен-
ной валентности, и в паровой фазе, применяя в качестве катали-
затора V2O5 [2, 16]. Однако традиционным методом является
окисление антрацена Na2Cr2O7 в приблизительно 25%-ной H2SO4.
Считают, что эта реакция начинается прямой электрофильной
атакой .мезо-положения антрацена катионом НСг' О' с образова-
нием и последующим гидролизом а-комплекса (105) [93, с. 135] и
завершается повторным окислением промежуточного продукта
(106) в 9,10-антрахинон (104):
(105)
(106)
(104)
Этот механизм, однако, вызывает определенные сомнения, так
как переход кислорода окислителя в продукт реакции не доказан
[94, с. 133].
Коксохимические заводы обычно поставляют 80—92%-ный ант»
рацен, содержащий примеси фенантрена и карбазола. Для окисле-
ния необходимо использовать 93—96%-ный продукт, поэтому тех-
нический антрацен кристаллизуют из пиридиновых оснований и
тщательно измельчают на шаровых мельницах. Окисление ведут в
гомогенно освинцованных чанах при 80 °C. В заключение реакци-
онную массу нагревают до кипения в течение 2 ч. Отфильтрованный
продукт реакции подвергают тщательной очистке, обрабатывая
концентрированной серной кислотой при 120 °C; при этом идет
сульфирование только примесей. 9,10-Антрахинон выделяют из
раствора разбавлением водой и отфильтровывают. Для оконча-
тельной очистки его перегоняют с перегретым до 240 °C водяным
паром, или еще раз повторяют сернокислотную очистку. Сульфат
хрома (III) или хромовые квасцы, образующиеся в качестве по-
бочного продукта при этом процессе, как уже было указано, нахо-
дят применение в кожевенной промышленности [16, 39].
Очевидно, что получение 9,10-антрахинона из антрацена явля-
ется далеко не совершенным процессом. Поэтому в ряде стран с
520
ним успешно конкурирует синтетическое получение антрахинона из
бензола и фталевого ангидрида (см. 6.3.2).
Из дихинонов ряда антрацена со смежными хиноидными коль-
цами известен 1,4,9,10 антрадихинон (107) и полученный сравни-
тельно недавно 1,2,9,10-антрадихинон (108) [95].
.О О
(107)
(108)
Описаны также 1,2,5,6-антрадихинон (109) и 1,4,5,8-антрадихи-
нон (ПО). Образование 1,4,5,8,9,10-антратрихинона (111) наблю-
дается только в растворах, выделить его не удалось [92].
В ряду фенантрена известны 1,2-, 3,4-, 1,4- и 9,10-фенантренхи-
ноны — формулы (112), (113), (114) и (115).
(112)
U15)
Первые три образуются при окислении соответствующих ами-
нофенантролов, а 9,10-фенантренхинон (или, просто, фенантренхи-
нон) (115), имеющий практическое применение, получают прямым
окислением фенантрена бихроматом в уксусной или серной кислоте.
Реакцию надо вести с осторожностью, так как (115) легко окисля-
ется далее до дифеновой кислоты (см. 17.2.5).Для очистки его
переводят в растворимое в воде бисульфитное соединение. Выход
80% [96].
521
Практически интересны также 3,8- и 3,6-пиренхиноны (116) и
(117), образующиеся при окислении пирена бихроматом в серной
или уксусной кислоте [10].
(ив)
Известны также тетракетон 1,3,6,8(277, 7Н) -пирентетрон (118) и
4,5,9,10-пирендихинон (119).
О
(119)
Тетракетон (118) получают обработкой олеумом 1,3,6,8-тетра-
хлорпирена и применяют далее для окисления в 1,4,5,8-нафталин-
тетракарбоновую кислоту [39] (см. 17.2.5). Дихинон (119) получа-
ется из пирена многостадийным синтезом, начинающимся с озони-
рования по связи С4—С5 [97]; он может быть применен в синтезе
высокотермостойких полимеров.
17.2.5. Получение карбоновых кислот
Карбоновые кислоты, образующиеся в результате окис-
лительной деструкции ароматических колец, получаются, по-види-
мому, через промежуточную стадию хинонов.
На окислительной деструкции ароматических углеводородов
основаны такие крупнейшие производственные процессы, как по-
лучение малеинового ангидрида из бензола и фталевого ангидрида
из нафталина. В обоих процессах осуществляется контактно-ката-
литическое окисление углеводородов в паровой фазе кислородом
воздуха, причем катализаторами для окисления бензола служат
смеси оксидов ванадия и молибдена, а для нафталина — большей
частью чистый V2O5.
Долгое время считалось, что эти процессы заключаются в окис-
лении органического соединения оксидом, который при этом вос-
станавливается, и роль кислорода газовой фазы в том, что он за-
тем вновь окисляет металл [98]. В настоящее время убедительно
показано, что это не так, и что процессы окисления подобно уже
522
рассматривавшимся процессам каталитического гидрирования,
связанным с активацией катализатором молекулярного водорода
(см. 16.2.4) идут через активацию кислорода, хемосорбируемого
поверхностью катализатора с последующим его взаимодействием с
углеводородом [99]. Эта точка зрения подтверждена, в частности,
с помощью изотопной метки кислорода оксида металла, которая не
переходит в продукты окисления.
Тем не менее, для объяснения селективности реакции и до сих
пор продолжают пользоваться представлениями о том, что актив-
ными центрами катализатора являются группы, обладающие
двойной связью М=О, которые подобно химическим окислителям
(см. 17.2.4 — окисление антрацена) подвергают ароматическое со-
единение электрофильной атаке. Регенерация этих центров осуще-
ствляется переносом электронов к молекулярному кислороду и
миграцией кислорода решетки оксида металла к ее восстановлен-
ным участкам [100].
Высказано предположение [101], что окисление бензола в ма-
леиновый ангидрид проходит через стадии: бензол—» фенол-* гид-
рохинон-> 1,4-бензохинон-> 2-гидрокси-1,4-бензохинон (120). По-
следний в результате окислительного декарбоксилирования пре-
вращается в малеиновый ангидрид (121):
(120) (120) (121)
Аналогично окисление нафталина до фталевого ангидрида (12)
может происходить через стадии образования 1-нафтола (58) и
1,4-нафтохинона (94):
(58)
(94)
(12)
Эти схемы, однако, основаны лишь на том, что указанные про-
межуточные продукты были выделены из реакционных смесей.
Не касаясь сложных инженерно-технологических вопросов, да-
дим лишь общее описание производственных процессов. Подроб-
нее см. [12, 101].
Предварительно подогретый воздух поступает в нижнюю часть
испарителя. Навстречу ему по тарелкам стекает бензол или рас-
плавленный нафталин, нагретый до температуры около 100 °C.
523
Образовавшаяся паро-воздушная смесь далее разбавляется необ-
ходимым количеством подогретого воздуха и попадает в контакт-
ный конвертор. Возможны два варианта конструкции конвертора: с
неподвижным слоем катализатора и с катализатором .в псев-
доожиженном слое.
В первом случае катализатор находится в трубках сравнительно
небольшого диаметра—это необходимо для эффективного отвода
тепла, выделяющегося при окислении. Так как температура кон-
тактирования 370—450°C, для съема тепла реакции используется
расплав солей NaNO2 и KNO3 (1 : 1), который циркулирует в меж-
трубном пространстве. Нагретые соли в свою очередь отдают
тепло воде, образуя пар высокого давления. Недостатком конвер-
торов этого типа является неравномерность температурного поля в
зоне реакции, в результате чего часть катализатора фактически не
работает, а другая перегревается. Это вызывает падение выхода
целевых продуктов. Нельзя также работать в области взрывоопас-
ных концентраций паро-воздушных смесей, которая для углеводо-
родов лежит в пределах 1 —10% (об.).
Более совершенными считаются конверторы с гранулированным
катализатором в псевдоожиженном слое. В них эффективно рабо-
тает весь катализатор и создаются условия, позволяющие значи-
тельно легче отводить тепло и точнее поддерживать необходимую
температуру. Это повышает выход продукта на стадии контакти-
рования. Кроме того, взвешенный слой катализатора позволяет
работать с паро-воздушными смесями и в области взрывоопасных
концентраций, так как он обладает взрывогасящим действием
'[101]. Недостатками этой конструкции является неизбежность ис-
тирания гранул и возможность уноса катализатора в виде образу-
ющейся пыли. В связи с этим предъявляются повышенные требо-
вания по прочности гранул катализатора к истиранию, которые не
всегда удается успешно удовлетворить.
Контактные газы, выходящие из конвертора, попадают в теп-
лообменник, где охлаждаются до 160—170 °C водой, образующей
пар, после чего идут на выделение.
Малеиновый ангидрид выделяют в скрубберах, где он ороша-
ется водой и растворяется в ней, образуя малеиновую кислоту.
Затем от раствора отгоняют воду и перегоняют остаток, превраща-
ющийся при этом вновь в малеиновый ангидрид; его подвергают
повторной ректификации.
Для выделения фталевого ангидрида охлажденные контактные
газы поступают в конденсаторы с ребристыми трубами, по которым
циркулирует масло. Фталевый ангидрид конденсируется, и кри-
сталлы его намораживаются на поверхности труб, после чего в
трубы подается горячее масло, продукт плавится и стекает в сбор-
ники, откуда поступает на заключительную ректификацию под ва-
куумом. Побочными продуктами окисления являются 1,4-нафтохи-
нон и малеиновый ангидрид, которые в ряде случаев выделяют.
Для получения малеинового ангидрида кроме бензола окисле-
нию можно подвергать разнообразные углеводороды С4 и Cs, что
524
технически вполне оправдано. Но в настоящее время около 90%
мирового производства малеинового ангидрида, которое возраста-
ет в среднем на 12% в год, основано на окислении бензола [101].
Точно так же быстро растущее производство фталевого ангид-
рида в основном опирается на окисление нафталина, так как рас-
смотренное выше окисление о-ксилола (см. 17.1.1) по полученным
показателям от него еще значительно отстает. Наблюдающийся в
последние годы дефицит нафталина заставляет форсировать ис-
следования по окислению о-ксилола.
Другие карбоновые кислоты, образующиеся при разрушении
ароматических колец, производятся в несравненно меньших коли-
чествах.
Электроноакцепторные заместители стабилизируют ароматиче-
ское кольцо, а электронодонорные облегчают процесс окислитель-
ного разрушения. Эта закономерность была обнаружена еще более
100 лет назад Гребе, показавшим, что при окислении 1-нитронаф-
талина (122) получается 3-нитрофталевая кислота (123), а из
1-нафтиламина (124) — фталевая (11):
NO2
NO2
СООН
СООН
(122) (123)
nh2
oVci
СООН
СООН
(124) (И)
Этот прием позволяет получать некоторые производные фтале-
вой кислоты окислением соответствующих производных нафтали-
на. Например, при окислении 7-гидрокси-1,3-нафталиндисульфо-
кислоты (Г-кислота) (125) перманганатом калия гладко образует-
ся 3,5-дисульфофталевая кислота (126), которую трудно получить
другими методами:
HO3S СООН
(126)
HO3S
СООН
Благодаря электроноакцепторному действию гетероатома при
окислении хинолина (127) бихроматом в серной кислоте или воз-
духом в жидкой фазе разрушается бензольное кольцо и получается
2,3-пиридиндикарбоновая (хинолиновая) кислота (128), а при
525
окислении бензимидазола (129)-4,5 -имидазолдикарбоновая кис-
лота (130) [102]:
N
СООН
^\соон
(129)
ноос
ноос
(130)
(127)
(128)
Оба продукта используются преимущественно в синтезе лекар-
ственных веществ.
Окислением фенантрена, которое можно вести бихроматами в
уксусной или серной кислоте, а также надуксусной кислотой [16]
получается дифеновая (гД'-бифенилдикарбоновая) кислота (131)
через промежуточное образование 9,10-фенантренхинона (115):
НООС СООН
(131»
Дифеновая кислота пока лишь очень ограниченно используется
в технике.
В синтезе красителей, люминофоров, полимеров с высокой тер-
мостойкостью находит применение 1,4,5,8-нафталинтетракарбоно-
вая кислота (133). Ее получают из пирена. Обычно его вначале
переводят прямым хлорированием (см. 5.2.4) в 1,3,6,8-тетрахлор-
пирен (132) при действии олеума образующий 1,3,6,8(27/, 7//)-
пирентетрон (118), а уже его окисляют бихроматом с H2SO4 до
целевой кислоты (133). Другим способом синтеза (133) служит
окисление дикетона — 1,3(2Л)-ацефеналендиона (134). Его полу-
чают по Фриделю — Крафтсу из аценафтена и малононитрила
[38].
(132)
(118) (133) (134)
Деструктивное окисление используется не только для синтети-
ческих целей, но. и как метод установления строения; он особенно
важен для конденсированных ароматических соединений, так как
применение современных спектральных методов структурного ана-
лиза в этой области недостаточно эффективно. Использование де-
структивного окисления для установления строения можно иллю-
стрировать следующим примером [103].
526
В результате несимметричной конденсации двух молекул
1,3-феналендиона был получен 1,7-дигидрокси-3,10-перопиренхинон
(136), который при исчерпывающем восстановлении (см. 16.4.3)
образовал углеводород известного строения—перопирен (137).
Положение кислородных заместителей в (136) было установлено
окислительной деструкцией, в результате которой вначале образо-
валась 2-гидрокси-3,4,9,10-перилентетракарбоновая кислота (135),
а ее строение, в свою очередь, доказано окислительным превраще-
нием в смесь 2,2', 3,3' 4,4/-бифенилгексакарбоновой кислоты (138)
и 9,10-антрахинон-1,2,5,6-тетракарбоновой кислоты (139):
(138)
(139)
17.3. ОКИСЛЕНИЕ АЗОТСОДЕРЖАЩИХ ГРУПП
Окисление азотсодержащих групп имеет значительно
меньшее значение, чем рассмотренные выше реакции. Тем не ме-
нее, в ряде случаев оказывается необходимым от первичного амина
перейти к нитрозо- или нитросоединениям, которые нельзя полу-
чить прямым нитрозированием или нитрованием. Окисление пер-
вичной аминогруппы идет в обратном порядке через те же стадии,
что н реакция восстановления нитрогруппы (ср. 16.6):
ArNH2 —> ArNHOH —> ArNO —> ArNO2
527
Различие в том, что при окислении не удается остановиться на
стадии образования /V-арилгидроксиламинов, которые с большой
легкостью окисляются далее до нитрозосоединений.
В качестве окислителей обычно применяют надкислоты, чаще
всего — мононадсерную кислоту (кислоту Каро) HOSO2OOH.
В тех случаях, когда реакция останавливается на стадии образо-
вания нитрозосоединения, для доокисления обычно применяют
азотную кислоту [16, с. 622] или бихромат. Например, 2,5-динит-
робензойную кислоту (142) можно получить из 4-нитро-о-толуиди-
на (140), окисляя его вначале кислотой Каро до нитрозосоединения
(141), а последнее в свою очередь — бихроматом в кислой среде
[104];
(140)
(142)
Окисление изомерных толуидинов кислотой Каро см. [105].
При окислении 1,4-бензохинондиоксима [106] в аналогичных
условиях образуется с хорошим выходом п-динитробензол:
NOH NO2
NOH NO2
Ароматические амины окисляются до нитрозосоединений
30%-ной перекисью водорода в уксусной кислоте при комнатной
температуре. При 70—80 °C с большим избытком того же окисли-
теля реакция идет дальше с образованием нитропродуктов [107].
Этот метод, по-видимому, удобнее, чем- использование 90%-ной
перекиси водорода в смеси с уксусным ангидридом или трифто-
руксусной кислотой [108], так как при реакции не образуются
продукты гидроксилирования.
В 30%-ном олеуме 30%-ная перекись водорода используется
для окисления 2- и 4-аминопиридинов в соответствующие нитропи-
ридины. Выход продуктов достигает 60—70% [109]. Однако в тех
же условиях из 3-аминопиридина образуется З.З'-азоксипиридин.
Причины такого течения реакции в очень сильнокислой среде не-
ясны.
В обычных случаях азокси- и азосоединения образуются при
окислении первичных аминов в щелочной среде, т. е. в условиях,
когда промежуточные продукты — Al-арилгидроксиламины и нит-
розосоединения — могут взаимодействовать друг с другом, так же
как при восстановлении нитросоединений. Этой реакцией пользу-
528
ются для получения некоторых практически важных азокрасите-
лей. Например, при окислении 2-п-аминофенил-6-метил-5-бензоти-
азолсульфокислоты (143) гипохлоритом натрия при 70—80 °C по-
лучается очень прочный краситель — хлораминовый желтый (144)
[16, с. 623]-.
(144)
Другой путь получения азосоединений при окислении открыт
сравнительно недавно [110]. Он состоит в окислительном сочета-
нии гидразонов и амидразонов с аминами и фенолами. Ниже пред-
ставлены типичные примеры этой реакции для гидразона (145) и
амидразона (146):
(146)
Метод особенно удобен для синтеза гетероциклических азосое-
динений, которые нельзя получить обычным способом диазотиро-
вания и азосочетания. Полагают, что в этой реакции активным
электрофилом, атакующим амин или фенол, является катион, об-
разующийся при окислении амидразона (или гидразона):
\ I 10] \ | +
XN— C=N—NH2 »- ,N—C=N—NH
Особую ценность в этом синтезе имеют 2-сульфониламидразо-
ны
N—C=N—NH—SOjR
529
Они обладают большой устойчивостью при высоких значениях pH
по сравнению с незамещенными амидразонами [111]. '
При окислении надкислотами третичных аминов образуются
соответствующие А-оксиды типа O-*-N(Alk)2Ar. Аналогичные про-
дукты получаются при окислении азотсодержащих гетероаромати-
ческих соединений, например пиридин-А-оксид, хинолин-А-оксид,
хиноксалин-А-оксид и др.
Благодаря тому, что кислород А-оксидной группы является
электронодонорным заместителем, а азот гетерокольца несет поло-
жительный заряд и, следовательно, является акцептором электро-
нов, эти соединения обладают ценным комплексом свойств, широко
используемым в синтезе. Например, в отличие от пиридина, пири-
дин-А-оксид гладко нитруется в положение 4, причем нитрогруппа
в полученном соединении обладает нуклеофильной подвижностью
и может быть замещена при действии нуклеофильных реагентов:
Обзор методов получения и свойств А-оксидов см. в моногра-
фии [112], а также в обзорах [113].
17.4. ОКИСЛЕНИЕ СЕРУСОДЕРЖАЩИХ ГРУПП
Окисление меркаптогрупп в тиолах (147) проходит ста-
дии образования дисульфидов (148), сульфиновых (149) и суль-
фокислот (150):
ArSH —>• ArSSAr —> ArSO2H —> ArSO3H
(147) (148) (149) (150)
Первая стадия идет особенно легко, например, в щелочной
среде под влиянием кислорода воздуха. В качестве окислителя
может применяться также иод. Для более глубокого окисления
применяются гипохлориты, надкислоты, перманганат калия, азот-
ная кислота и др. Ниже приводится несколько практически инте-
ресных примеров использования этих реакций.
Как известно, взаимодействием анилина с сероуглеродом и
серой получают 2-меркаптобензотиазол (каптакс) (151). Окисле-
ние этого тиола обычно ведут, действуя на его водно-щелочные
растворы растворами гипохлорита натрия или гексациа-
но- (III) феррата (железосинеродистого) калия. При теоретическом
количестве окислителя с количественным выходом получается
ди-2-бензотиазолилдисульфид (альтакс) (153). Если реакция про-
водится в присутствии первичных или вторичных аминов, обра-
зуются 2-бензотиазолсульфенамиды (152). Окисление (151) в тех
530
же условиях большими количествами NaCIO, а также Н2О2 или
КМ.пО4 приводит через (153) к 2-бензотиазолсульфиновой кислоте
(154) и далее к 2-бензотиазолсульфокислоте (155) [114, с. 609—
611; 115]:
ют, +nhr2
--------->
(151) (152)
Каптакс, альтакс и сульфенамиды применяются при вулкани-
зации каучуков. Об использовании 2-бензотиазолсульфокислоты в
реакциях нуклеофильного замещения см. 7.1.4 и 7.2.
В отличие от сульфаниловой кислоты, образующейся при запе-
кании сульфата анилина (см. 3.3.2) ее изомер — ортаниловая кис-
лота (157) получается из о-нитрохлорбензола через дисульфид
(156) (см. 9.1.1) по схеме [116]:
(156)
(157)
Окисление (156) ведется хлором в смеси концентрированных
соляной и азотной кислот. Ортаниловая кислота используется в
синтезе азокрасителей.
Находят применение также реакции окисления сульфидов, ко-
торые при помощи приведенных выше окислителей могут быть
превращены в сульфоксиды и сульфоны. Примером, может служить
синтез 4,4'-диаминодифенилсульфона (161) из п-нитрохлорбензола
через сульфид (158), сульфоксид (159) и сульфон (160) по схеме:
Продукт (161) используется в синтезе термостойких полимеров,
а также как лекарственное вещество.
631
ЛИТЕРАТУРА
1. Майрановский В. Г. — Журн. ВХО, 1977, т. 22, № 3, с. 334—347. 2. Сит-
тиг М. Процессы окисления углеводородного сырья. Пер. с англ./Под ред.
С. Ф. Гудкова. М. Химия, 1970. 3. Riley Н. Л. — Chem. and Ind (London), 1966,
№ 24, p. 979—988; Камнева А. И., Лунина M. А. и др. — ЖФХ, 1'976, т. 50,
вып. 11, с. 2826—2829. 4. Цысковский В. К-, Цысковская И. В., Овчинников В. И.
и др —Хим. пром., 1976, № 7, с. 493—496. 5. Семенов Н. Н. Химическая физика
(Физические основы химической кинетики). Черноголовка, ИХФ АН СССР, 1975.
6. Эмануэль Н. Л/. — Успехи химии, 1978, т. 47, вып. 8, с. 1329—1396. 7. Хар-
лампович Г. Д., Чуркин Ю. В.—Журн. ВХО, 1977, 'т. 22, № 1, с. 54—61.
8. Towle Р. Н., Boldwin R. Н.— Hydrocarb. Proc., 1964, v. 43, № 11, р. 149—153.
9. Борщенко В., Махиянов Г. Пиромеллитовый диангидрид, получение и приме-
нение М., ЦНИИТЭнефтехим, 1974. 10. Flavell W., Yarsley V. Е. — Chem. Bri-
tain, 1967, v. 3, № 9, р. 375—381.
11. Ворожцов Н. Н. мл. — В кн.: Химия синтетических красителей. Т. З/Под
ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса Л., Химия, 1974,
с. 1804. 12. Гуревич Д. А. Фталевый ангидрид. М., Химия, 1968. 13. Eur. Chem.
News, 1977, № 22, р. 40. 14. Хчеян X. Е., Самтер Л. И., Мак Н. Е. и др. — Хим.
пром.' 1977, № 1, с. 17—19. 15. Landau R. — Ind. Eng. Chem., 1961, v. 53, № 10,
p. 32A—41A. 16. Ворожцов H. H. Основы синтеза промежуточных продуктов и
красителей. Изд. 4-е. М., Госхимиздат, 1955. 17. Friedman. L., Fishel D. L.,
Schechter H.—J. Org. Chem., 1965, y. 30, № 5, p. 1453—1457. 18. Кийко И. И.—
Хим. пром., 1975, № 1, с. 13—17. 19. Доналдсон Н. Химия н технология соеди-
нений нафталинового ряда. Пер. с англ./Под ред. А. И. Королева. М., Госхим-
издат, 1963. 20. Красовицкий Б. М., Болотин Б. А1. Органические люминофоры.
Л., Химия, 1976.
21. Красовицкий Б. АГ, Шевченко Э. А., Кузнецов А. М. и др. Авт. свид.
СССР № 282309, 1970. 22. Венкатараман К-, Айер В. Н.— В кн.: Химия синте-
тических красителей. Т. 5/Под ред. К- Венкатарамана. Пер. с англ, под ред.
Л. С. Эфроса. Л., Химия, 1977, с. 191. 23. Суворов Б. В. Окислительный аммо-
нолиз органических соединений. Алма-Ата, Наука, 1971. 24. Кагарлицкий А. Д.,
Суворов Б. В., Рафиков С. В. и др. — ЖПХ, 1973, т. 36, вып. 8, с. 1848—1852.
25. Пат. ФРГ 1948714, 1971; Chem. Abstr., 1971, v. 75, 19992. Пат. США 2838558,
1958; Chem. Abstr., 1958, v. 52, 17191A. 26. Ogata У., Sakanishi K-, Sawaki Y.—
J. Chem. Soc. Japan. Ind. Chem. Sec., 1965, v. 68, № 11, p. 2244—2248. 27. Суво-
ров Б. В., Букейханов И. Р. — Журн. ВХО, 1977, т. 22, № 1, с. 62—68; Окисли-
тельные реакции в органическом синтезе. М., Химия, 1978, с. 123—159. 28. Si-
mon G., Germain J.-E. — Bull. Soc. chim. France, 1975, № 11—12, p. 2617—2621.
29. Лебедев И. И. Химия и технология основного органического и нефтехимиче-
ского синтеза. Изд. 2-е. М., Химия, 1975. 30. Найт X., Сверн Д. — В кн.: Син-
тезы органических препаратов. Сб. 6. Пер. с англ. М., ИЛ, 1956, с. 19—21.
31. Трофимов В. И., Левкое Я. Л., Якубсон А. М. — Хим. пром. 1973, № 3,
с. 192—195. 32. Трофимов В. И., Кафаров В. В., Круглик А. Е. и др. — Нефте-
химия, 1976, т. 16, № 1, с. 125—129. 33. Носк Н., Depke W.— Chem. Ber., 1950,
Bd. 83, № 4, S. 317—327. 34. Мельников И. И. — В кн.; Реакции и методы ис-
следования органических соединений. Т. 1/Под ред. С. С. Наметкина, В. М. Ро-
дионова, Н. Н. Мельникова. М., Госхимиздат, 1951, с. 99—177. 35. Либерман С.,
Коппор Р. — В кн.: Синтезы органических препаратов. Сб. 2. Пер. с англ. М.,
ИЛ, 1949, с. 366—368; Тзанг С., Вуд Э., Джонсон Дж. — Там же. Сб. 3. М., ИЛ,
1952, с. 348—351. 36. Кампень Э., Будде В., Шефер Дж. — Там же. Сб. 4. М„
ИЛ, 1953, с. 30—31. 37. Анжиал С. Дж. — В кн.: Органические реакции. Сб. 8.
Пер. с англ. М., ИЛ, 1956, с. 263—287. 38. Уильямс Дж., Виттен Ч., КриниЦ-
кий Дж. — В кн.: Синтезы органических препаратов. Сб. 4. Пер. с англ. М., ИЛ,
1953, с. 484—486. 39. Фирц-Давид Г. Э., Бланже Л. Основные процессы синтеза
красителей. Пер. с англ./Под ред. С. В. Богданова и И. В. Фодимана. М., ИЛ,
1957. 40. Айянгар Н. Р., Тилак Б. Д. — В кн.: Химия синтетических красителей.
Т. 4/Под ред. К. Венкатарамана. Пер. с англ, под ред. Л. С. Эфроса. Л.. Химия,
1975, т. 4, с. 139.
532
41. Пат. США, 3340311, 1967; Chem. Abstr., 1968, v. 68, 87062. 42. Пат. США,
3356743, 1967; Chem. Abstr., 1968, v. 68, 49352. 43. Клар- Э. Полициклические
углеводороды. Т. 1. Пер. с англ. В. В. Ершова. М, Химия, 1971. 44. Физер Л.,
Физер М. Реагенты для органического синтеза. Т. 3. Пер. с англ./Под ред.
И. Л. Кнунянца и Р. Г. Костяновского. М, Мир, 1970. 45. Ворожцов И. И. мл.
Коптюг В. А. — ЖОХ, 1958, т. 28, вып. 11, с. 2981—2987. 46. Скарченко В. К.—
Успехи химии, 1977, т. 46, вып. 8, с. 1411—1444. 47. Эфрос Л. С. — Там же,
1960, т. 29, вып. 2, с. 162—186. 48. Waters W. A. Mechanism of Oxidations of
Organic Compounds. London, Methuen, 1964. 49. Денисов E. T., Метелица Д. И —
Успехи химии, 1968, т. 37, вып. 9, с. 1547—1566. 50. Chem. Age (London), 1962,
v. 88, № 2249, р. 233.
51. Англ. пат. 903868, 1965; Chem. Abstr., 1963, v. 58, 10223Е. 52. Пат. ФРГ
1184772, 1965; Chem. Abstr., 1965, v. 62, 11737V. 53. Пат. США 3033903, 1962;
Chem. Abstr., 1962, v. 57, 11108E. 54. Нидерл. пат. 6608118, 1966; Chem. Abstr.,
1967, v. 67, 21638. 55. Нидерл. пат. 6611558, 1967; Chem. Abstr., 1968, v. 68,
49288. 56. Агре H. J., Hornig L. — Erdol, Kohle, Erdgas, Petrochemie, 1970, Bd. 23,
№ 2, S. 79—84. 57. Stern E.—Amer. Chem. Soc., Preprint 169-th Session. N. Y„
May, 1970. 58. Моисеев И. И. — Журн. ВХО, 1977, т. 22, № 1, с. 30—45.
59. Eberson L., Gomes-Gonzales L.-—Chem. Commun, 1971, № 6, p. 263—264.
60. Иоффе И. И., Сучков В. В. и др. Авт. свид. СССР 176915, 1965.
61. Пат. США 2792430, 1958; Chem. Abstr., 1958, v. 52, 1231Е. 62. Венкатара-
ман К- Химия синтетических красителей. Т. 2/Пер. с англ, под ред. Н. С. Вульф-
сона. Л., Госхимиздат, 1957, с. 946. 63. Аигл. пат. 346355, 1930; Chem. Abstr.,
1932, v. 26, 1948. 64. Пат. ФРГ 1165180, 1964, Chem. Abstr, 1964, v. 60, 15804Н;
1160124, 1963; Chem. Abstr, 1964, v. 61, 627A. 65. FIAT, v. 1313, p. 210. 66. BIOS,
v. 1484, p. 27. 67. BIOS, v. 1484, p. 22. 68. Winkler J., Jenny W. — Helv. chim.
acta, 1965, v. 48, № 1, p.119—126. 69. Бамдас E. M., Шемякин M. M. — ЖОХ,
1948, t. 18, вып. 2, c. 324—332. 70. Kaeding W. W., Collins G. R. — J. Org. Chem,
1965, v. 30, № 11, p. 3750—3754.
71. Kaeding W. W., Kerlinger И. V., Collins G. R. — Ibid, p. 3754—3759.
72. Kaeding W. W., Lindblom R. O., Temple R. G. e.a.— Ind. Eng. Chem, Process
Design a. Develop, 1965, v. 4, № 1, p. 97—101; Kaeding W. W. — Hydrocarb.
Proc, 1964, v. 43, № 11, p. 173—176. 73. Hock H., Lang S. — Chem. Ber, 1944,
Bd. 77, № 3—4, S. 257—264. 74. Кружалов Б. Д., Голованенко В. И. Совместное
получение фенола и ацетона. М, Госхимиздат, 1963. 75. Цысковский В. К.,
Москович Ю. А. — Кинетика и катализ, 1974, т. 15, вып. 6, с. 1466—1469.
76. Цысковский В. К., Прокофьев Е. К-, Пыльников В. И. и др. — ЖПХ, 1974,
т. 47, № 5, с. 1112—1117. 77. Кошель Г. Н., Глазырина И. И., Фарберов М. И.
и др. — ЖОрХ, 1978, т. 14, вып. 3, с. 534—538. 78. Graham А. — In: Proc. 7th
World Petroleum Congress, Mexico, 1970. 79. Japan Chem. Week, 1974, v. 15,
№ 758, p. 6. 80. Chem. Ind, 1975, v. 27, № 2, p. 78.
81. Olzinger A. N. — Chem. Eng, 1975, v. 82, № 12, p. 50—51. 82. Shea-
ron W. H., Davy L. G, von Bramer H. — Ind. Eng. Chem, 1952, v. 44, p. 1730—
1740. 83. Isomura Y.— J. Chem. Soc. Japan, 1941, v. 62, p. 1167—1177. 84. Пат.
США 2373003, 1945; Chem. Abstr., 1945, v. 39, 4335. 85. Mohilner D. M„
Adams R. N., Argersinger W. J.—J. Amer. Chem. Soc, 1962, v. 84, p. 3618—3624.
86. Zimmer H., Lankin D. C., Horgan S. W. — Chem. Rev, 1971, v. 71, p. 229—
240. 87. Moser W., Howie R. A.—J. Chem. Soc. (A), 1968, p. 3039. 88. Teuber H. J.,
Dietz К. H. — Angew. Chemie, 1965, Bd. 4, S. 871. 89. Пат. ФРГ 2221624, 1972;
Chem. Abstr, 1973, v. 78, 58057. 90. Афанасьев И. Б., Гурьянова Л. Ф., Бара-
нова Н. Г. и др. — ЖОрХ, 1978, т. 15, вып. 3, с. 569—574.
92. Горелик М. В. — В кн.: Химия антрахинона/Под ред. А. И. Королева.
М„ Химия, 1969, с. 5—56. 93. Wiberg К. В. Oxidation in Organic Chemistry.
Part A. New York, Academic Press, 1965. 94. Thomson R. N. — In: The Chemistry
of the Quinonoid Compounds. Part 1/Ed. S. Patai. London, Wiley, 1975. 95. Горе-
лик M. B. — ЖОрХ, 1965, t. 1, вып. 12, c. 2246. 96. Уэндлснд P., Лаланд Дж.—
533
В кн.: Синтезы органических препаратов. Сб. 6. Пер. с англ. М, ИЛ, 1956,
с. 73. 97. Цесси Р. Е., Ньюмен М. С. — Там же. Сб. 10. М, ИЛ, 1960, с. 70. 98.
Mars Р., Van Krevelen D. W.— Chem. Eng. Sci., Spec. Suppl., 1954, v. 3, p. 41—
59. 99. Дзисяк А. П., Боресков Г. К., Касаткина Л. А. и др. — Кинетика и ка-
тализ, 19.61 т. 2, вып. 3, с. 386—393; Боресков Г. К., Грувер В. Щ. и др. —
ДАН СССР, 1974, т. 215, № 2, с. 359—362. 100, Вейсс Ф., Марион Ж..,
Коньон Ж- М. — Кинетика и катализ, 1973, т. 14, № 1, с. 45—72.
101. Молдавский Б. Л., Кернос Ю. Д. Малеиновый ангидрид и малеиновая
кислота. Л., Химия, 1976. 102. Эфрос Л. С., Хромов Н. В.. Давиденков Л. Р. —
ЖОХ, 1956, т. 27, вып. 2, с. 455—458. 103. Солодарь С. Л. — Журн. ВХО, 1976,
т. 21, № 3, с. 306—315. 104. Лангли В. — В кн.: Синтезы органических препа-
ратов. Сб. 3. Пер. с англ. М., ИЛ, 1952, с. 211—214. 105. Fernandez J. Е.,
Schwarz М. L. — J. Chem. Eng. Data, 1970, v. 15, № 3, p. 462. 106. Гареев Г. A.t
Черкашина H. А., Хищенко Ю. С. Авт. свид. СССР 395360, 1973. 107. Hol-
mes R. R., Bayer R. Р. — J. Amer. Chem. Soc., 1960, v. 82, № 13, p. 3454—3456.
108. Emmons W. D. — Ibid., 1954, v. 76, № 13, p. 3470—3472; 1957, v. 79 № 20,
p. 5528—5530. 109. Wiley R. N„ Harman J. L. — Ibid, 1951, v. 73, № 1. p. 494.
110. Hunig S., Balli H. e.a. — Angew. Chem, 1958, Bd. 70, № 8, S. 215—222; 1962,
Bd. 74, № 21, S. 818—824.
111. Hunig S, Brenniger W. e.a. — Angew. Chem, Bd. 80, № 9, S. 343—352.
112. Katritzky A. R., Lagowski J. M. Chemistry of the Heterocyclis IV-oxides. Lon-
don— N. Y, Academic Press, 1971. 113. Иоффе Д. В., Эфрос Л. С. — Успехи хи-
мии, 1961, т. 30, вып. 11, с. 1325—1351; Хамана Масомото.— Химия гетероцик-
лических соединений, 1973, № 9, с. 1155—1171. 114. Вейганд-Хильгетаг. Методы
эксперимента в органической химии. Пер. с нем./Под ред. Н. Н. Суворова. М,
Химия, 1968, с. 609—611. 115. BIOS, v. 1018, р. 36. 116. Вертхейм Е. — В кн.;
Синтезы органических препаратов. Сб. 2. Пер. с англ. М, ИЛ, 1949, с. 403—406.
УКАЗАТЕЛЬ ВАЖНЕЙШИХ СОЕДИНЕНИЙ
Азобензол 184, 476
Азометины 397
Азулен 18, 27, 29
Акридин 351
Алая кислота 403
Ализарин 279, 296
нитрование 160
окисление 510
сульфирование 129
Ализарин-сафирол 471
Алкилбензолы 198
изомеризация 196
нитрование 138
сульфирование 114
Алкилнафталины, изомеризация 197
1-Алкилтио-9,10-антрахиноны 318
Алкилфенолы 200
Альтакс 531
1-Амино-9,10-антрахинон 246
2-Амино-9,10-антрахинон 235, 246
и-Аминобензальдегид 502
fi-Аминобензойная кислота 497
4-Амино-3-гидрокси-1-нафталинсуль-
фокислота 322
6-Амино-5-гидрокси-1 -нафталинсуль-
фокислота 430, 483
Амино-е-кислота 242
1 -Амино-2-нафталинсульфокислота
105
Аминонафталинсульфокислоты 252
ариламинирование 242
Аминонафтолы 418, 433
2-Аминопиридин 262
о-Аминотиофенолы 309
1-Аминофеназин, диазотирование 425
2-Амино-4-фенилпиримидин 79
о-Аминофенол, диазотирование 418
лг-Аминофенол 115
карбоксилирование 224
и-Аминофенол 482
диазотирование 418
Амины 397
азосочетание 432
алкилирование 200
ацилирование 398
диазотирование 416
нитрозирование 162
Амины
окисление 527
о-Анизидин, щелочное плавление 293
м-Анизидин 474
Анизол 298
ацетоксилирование 374
окисление 509
Анилин 249, 468
азосочетание 431
гидрирование 455
гидролиз 291
хлорирование 181
Антрапиррол 443
Антраруфин 159
9,10-Антрахинон 519
аминирование 379
восстановление 460
нитрование 140, 158
окисление 510
сульфирование 126
хлорирование 186
9,10-Антрахинон-1 -сульфокислота 128
9,10-Антрахинон-2-сульфокислота, ще-
лочное плавление 279
Антрацен 5, 7, 14, 23, 27, 29, 48
метилирование 351
окисление 520
сульфирование 129
1-Антраценсульфокислота 131
2-Антрол, карбоксилирование 223
Антрон 49, 461
Арендиазония соли, восстановление
451
Арендиазосульфонаты натрия 427
Арил (арилдиазо) сульфоны 427, 441
Арилгидразины 451
А-Арилгидроксиламины 473
А-Арилнафтиламины 250
Арилсульфаты 412
Арин(ы) 81, 82
Аценафтен 214, 497
Аценафтенхинон 498
Аценафтилен 5
А-Ацетил-Аш-кислота 400
А-Ацетил-Гамма-Кислота 400
А-Ацетил-И-кислота 400
А-Ацетилсульфапиловая кислота 400
535
Ацетонитрил 57
Ацетоуксусный эфир, азосочетание 434
Ацетофенон 211
аминометилирование 207
Аш-кислота 122, 157, 278, 291,420,433
Бензальдегид 384, 503
нитрование 150
Бенз [а] антрацен 49
Бензантрон, бромирование 190
Бензгидрол 457
Бензидин 476, 479
Бензилидендихлорид 384
Бензимидазол, окисление 526
Бензилиодиды 385
Бензилхлорид 204, 384
окисление 503
А-Бензоил-Аш-кислота 405
А-Бензоил-Гамма-кислота 405
А-Бензоил-И-кислота 405
Бензойная кислота, гидрирование 456
Бензол 5, 7, 9, 18, 27, 29, 38, 47, 57,
372
алкилирование 195, 197
аминометилирование 209
ацетилирование 211
ацетоксилирование 374
бромирование 172, 189
гидрирование 453
нитрование 141, 142
однозамещенный, фенилирование
88
окисление 509, 517, 523
сульфирование 109
формилирование 216
хлорирование 174
Бензолгексасульфокислоты натриевая
соль 320
Беизолсульфокислота 109
Бензонитрил 499
Бензотрихлорид 384
Бензофенон 212
восстановление 447, 457
Бензохиноны 315, 517
восстановление 459
Бензоциклобутадиен 18
1,1'-Биантрахинонилы 354
Биарилы 354
8,8'-Би-1-нафтойная кислота 440
Бисфенол А 203
Бифенил 372
нитрование 160
Бромаминовая кислота 190
4-Бромбифенил 360
5-Бромизатин 189
о-Бромиодбензол 334
2-Бромпиридин 338
2-Бром-4-фенилпиримидин 79
п-Бутилапилин 200
А-Бутиланилпн 393
Ванилин 205
536
Г алогенантрахиноны, аминирование
237
Гамма-кислота 125, 243, 252, 253,
278, 434
Гваякол 293
алкилирование 410
Гексаметилендиамин 465
Гексан 57
Гексахлорбензол 308, 330
Гексацианобензол 341
Гетарины 82
Гидразобензол 476, 480
n-Гидроксибензанилид, нитрование
150
Гидроксибензолсульфокислоты 105
Гидрокснгидрохинон 297
о-Гидроксибензонитрилы 343
2-Гидроксибензофенон 373
4-Гидроксиметил-2-метилхинолин 369
З-Гидрокси-2-нафтойная кислота 221
гидрирование 456
Гндрол Михлера 457
Гидрохинон 286, 297, 431, 459, 517
Г-кислота 125, 252, 525
Глицин (проявитель) 394
Декалин 454
Диазоли 426
1,4-Диамино-2,3-дициано-9,10-антрахи-
нон 341
2,6-Диаминопиридин 262
Диан 203
о-Дианизидин 480
Дибензила производные 385
2,6-Ди-г/7ет-бутилфенол 200
бромирование 171
Дигидробифенил 87
Диметиламин 293
Диметилформамид 57
1,4-Диметоксибензол 89
2,4-Динитробензальдегид 389
о-Динитробензол 289
.и-Дииитробензол 142
аминирование 260
восстановление 469
п-Динитробензол 289, 528
1,3-Динитро-4,6-дихлорбензол 332
Динитронафталины 154, 289
Динитротолуолы 145
2,4-Динитрофенилгидразин 266
2,4-Динитрофенилизотиоцианат 310
1,3-Динитро-4-хлорбензол 148, 332
4-Диоксанил-2-метилхинолин 369
3,5-Дисульфофталевая кислота 525
Дифениленоксид 5
Дифенилолпропан 203
Дифеновая кислота 526
нитрование 160
1,5-Дифенокси-9,10-антрахинон 300
Дихлорбензолы 174, 176
алкоксилирование 298
3,4-Дихлорбифенил 363
2,4-Дихлоризофталевая кислота, ще-
лочное плавление 286
2,3-Дихлор-1,4-нафтохинон 185
2,5-Дихлортерефталевая кислота, ще-
лочное плавление 286
Дихлортолуолы 178
Дихлорфенолы 180
4,5-Дихлорфталевая кислота 183
Дицианонафталины 342
1,4-Ди (этилтио) бензол 318
Дурол, окисление 494
Изоникотиновой кислоты нитрил 345
Изопропилбензол 197
алкилирование 195
дегидрирование 506
окисление 514
Изофталевая кислота 493
Изохинолин, щелочное плавление 295
1-Изохинолон 295
И-кислота 252, 278, 400, 434
1,3-Индандион, азосочетание 434
Индантрон, восстановление 462
Инден 5
1-Инденкарбоновая кислота 443
Индиго, бромирование 190
Индол 18
Иидолизин 16
Иодбензол 334
Каптакс 318, 530
Карбазол 5, 7, 18
Карбоновых кислот соли, диспропор-
ционирование 224
е-Кислота 291
К-кислота 122, 157
Клеве-кислоты 121, 156
Крезидин 408
Крезолы 293, 513, 516
аминометилирование 207
Ксилидины 469
Ксилолы 6, 7
бромирование 189, 385
изомеризация 196
нитрование 145
окисление 493
сульфирование 114
хлорирование 173, 179, 385
Кумол см. Изопропилбензол
Лейкотроп 394
Лейкохинизарин 461
Малеиновый ангидрид 523
Мезитилен 6
бромирование 189
хлорирование 173
2-Меркаптогидрохиноны 315
Метаниловая кислота 115, 118
2-Метилнафталин 5
сульфирование 123
5-Метилрезорцин 283
а-Метилстирол 506
2-Метил-4-этилхинолин 369
2-Метокси-9,10-антрахиноны 303
Метоксибензол 298
1-Метокси-2,5-дихлорбензол 298
1-Метоксинафталин 298
2-Метокси-6-нитробензонитрпл 343
Метоксифенол 293, 408
Метол 395
М-кислота 434
Нафтазарин 475
Нафталевая кислота 497
Нафталин 5, 6, 8, 18, 27, 29, 47, 499
ацилирование 213
гидрирование 454
гидроксилирование 372
метилирование 351
нитрование 139, 153
окисление 510, 523
сульфирование 118
формилирование 216
хлорирование 184
хлорметилирование 205
Нафталиндисульфокислоты 121, 125,
426
нитрование 140, 156
1-Нафталинкарбонитрил 341
Нафталинсульфокислоты 119, 120,440
нитрование 139, 156
щелочное плавление 278
1,4,5,8-Нафталинтетракарбоновая кис-
лота 526
Нафталинтрисульфокислоты 122, 123
Нафтиламины 291, 432
Нафтионовая кислота, щелочное
плавление 292
1-Нафтол 291, 506, 510
азосочетание 432
аминирование 249, 254
гидрирование 455
карбоксилирование 220
нитрование 158
хлорирование 185
2-Нафтол 278, 280, 516
азосочетание 432
аминирование 249, 252
ацилирование 314
гидрирование 455
карбоксилирование 220
нитрозирование 482
сульфирование 123
сульфометилирование 209
хлорирование 185
Нафтохиноны 519
ацетилирование 297
Никотиновая кислота 342
537
о-Нитроанизол 299
Нитроанилины 232, 293
о-Нитробензальдегид 503
Нитробензол 57, 142
восстановление 468, 473
сульфирование 115
хлорирование 182
.и-Нитробензолсульфокислота 115
п-Нитрозофенол 250, 482
Нитроксилолы, восстановление 469
а-Нитрокумолы 349
Нитромезитилен 146
Нитрон 348
Нитроиафталины 153
2-Нитро-1-нафтол 289
лг-Нитротолуол 440
«-Нитротолуол 144, 390
нитрование 144
сульфирование 115
хлорметилирование 204
4-Нитро-2-толуолсульфокислота 115
Нитрофеиолы 285, 289, 293
аминирование 249
п-Нитрофторбензол 331
О-Нитрохлорбензол
алкоксилирование 299
аммонолиз 232
щелочное плавление 285
л-Нитрохлорбензол 181
аммонолиз 232
п-Нитрохлорбензол 147
сульфирование 115
аммонолиз 232
5-Нитро-2-хлорбензолсульфокислота
115
Октадециламин 465
Октафтор-9,10-антрахинон 330
Орсин 283
Ортаниловая кислота 531
Перикислота 156, 242
Пентацен 18
Пентален 18
Перилен 15, 18, 23, 507
нитрование 161
Перхлорнафталин 185
Пиколиновой кислоты нитрил 345
Пикрамииовая кислота 472
Пикриновая кислота, восстановление
472
Пирен 5, 15, 18
нитрование 161
окисление 526
хлорирование 188
Пиренхиноны 522
Пиридин 18, 27, 29, 38, 57
Пиридинпентасульфокислота 320
Пиридинсульфотриоксид 103
Пирокатехин 410
Пиромеллитовая кислота 494
Пиррол 16, 18, 27, 29, 38
Полибромбифенил 189
Полинитробензотригалогениды 74
Полихлорнафталины 185
Псевдокумол, окисление 494
Пурпурин 510
Резорциловая кислота 223
Резорцин 279, 280, 286, 291
азосочетание 431
алкилирование 408
аминирование 251
ацилирование 215
нитрозирование 166
Р-кислота 124, 125
Салициловая кислота
азосочетание 431
нитрование 149
С-кислота 433
2С-кислота 157
Стильбена производные 385
Стильбенхинои 386
Стирол 505
Сульфаниловая кислота 117
Сульфенамиды 531
Сульфонолы 114
Сульфохлориды 116
Терефталевая кислота 226, 228, 493
бромирование 189
Терефталевый альдегид 385
Тетрагидро-п-кватерфеиил 87
Тетрагидрофуран 57
Тетралин 454
окисление 500
1-Тетралол 500, 506
1-Тетралои 500, 506
2,3,5,6-Тетраметиланизол 69
Тетрацен 18, 29
Т-кислота 157, 278, 293
Тиогликолевая кислота 410
л-Тиокрезол 311
2-Тионафтол 314
Тиофен 6, 16, 18, 27, 29
Тиофенол (ы) 308, 312, 318, 485
алкилирование 410
Тобиаса кислота 124, 125, 126, 252
Тобиаса оксикислота 124, 126, 252
о-Толидин 480
2,4-Толилендиамин 145
Толуидины 145, 469
гидрирование 456
нитрование 151
окисление 497
щелочное плавление 293
Толуол 6, 7
алкилирование 195
ацилирование 212
бромирование 172
нитрование 141, 144
538
Толуол
сульфирование 113
хлорирование 178, 383
1,3,5-Триаминобензол, гидролиз 291
Триарнлметаны, окисление 504
1,2,4-Трнацетоксинафталин 297
Трибромбензолы 83, 440
Гримеллитовая кислота 494
Триметилфлуорен 365
2,4,6-Тринитроанизол 74
2,4,6-Тринитротолуол 145
1,3,5-Тринитро-2-хлорбензол 148
Трифенилен 507
Трифтор-1,3,5-триазин 330
Трихлорбензолы 174, 176, 440
алкоксилирование 298
Трихлорфенолы 180, 283
1,3,5-Триэтилбензол, бромирование
189
Тропилийбромид 38
Тротил 145
Фенален 14
Фенантрен 5, 7, 14, 18, 23, 29, 49
метилирование 351
окисление 526
9-Фенантренкарбонитрил 340
Фенантренхиноны 521
л-Фенетидин 474
Фенилацетат, окисление 509
Фенил-Гамма-кислота 243, 253
Фенилгидразин 484
Фенилглицин 394
.и-Фепилепдиамип 469
гидролиз 291
4-Фенил-1-тетралон 71
Фенол (ы) 248, 280, 281, 291, 293,
372, 505, 509
азосочетание 431
алкилирование 198
ацилирование 215, 313, 411
бромирование 189
гидрирование 454, 463
карбоксилирование 219
нитрование 149
нитрозирование 165
окисление 518
формилирование 206, 216
хлорирование 173, 179
Флороглюцин 283, 291
Флуорен 5
нитрование 161
Фрейнда кислота 291
Фрейнда оксикнслота 291
Фталевые кислоты
гидрирование 456
хлорирование 183
Фталевый альдегид 503
Фталевый ангидрид 495, 523
иодирование 189
хлорирование 183
Фтор-9,10-антрахиноны 331, 361
Фторбензол 333
л-Фторбензойна'я кислота 337
Фульфен 18
Фуран 16, 18, 27, 29
Хинальдиновой кислоты нитрил 345
Хиннзарин 510
восстановление 461
хлорирование 188
2-Хиноксалинкарбоксамид 370
Хинолин 18
Хинолиновая кислота 525
2-Хинолон 295
Хиноны 517
арилирование 366
Хлоранизол 60, 298
аминирование 236
Хлоранил 518
ариламинирование 234
Хлоранилины 236, 432
Хлорантрахиноны 160, 237
Хлорбензол 57, 174
алкилирование 195
алкоксилирование 298
аминирование 236
ацилирование 212
нитрование 141, 146
окисление 509
сульфирование 114
хлорирование 176
щелочное плавление 281
8-Хлор-1-нафталинсульфокислота 336
Хлорнафталины 184
алкоксилирование 298
Р-Хлорпропилбензол 383
Хлортолуолы 178
Хлорфенолы 180
Хризен 5
Хромотроповая кислота 291
8-Цианонафта лин-1-сульфокислота
340
о-Цианофенилтиогликолевые кислоты
340
Циклобутадиен 18
1,3-Циклогексадиен 29
Циклогексанкарбоновая кислота 456
Циклогексанол, дегидрирование 505
Циклогексанон 505, 516
N-Циклогексиланилин 464
л-Цимол 68
Чикаго-С-кислота 433
Чикаго-СС-кислота 157
Шеффера кислота 124, 125
Эпсилон-кислота см. е-Кислота
Этилбензол 6, 197
алкилирование 195
дегидрирование 505
окисление 499
ОГЛАВЛЕНИЕ
Предисловие •..........................................................3
Часть 1
Строение и свойства ароматических соединений
Глава 1. Ароматические соединения......................................5
1.1. Источники получения...........................................5
1.2. Строение ароматических соединений.............................8
1.2.1. Ранние представления....................................8
1.2.2. Квантовомеханическая трактовка..........................9
1.2.3. Полициклические соединения.............................13
1.2.4. Гетероциклические соединения...........................15
1.2.5. Определение понятия ароматичности......................17
1.3. Критерии ароматичности.......................................17
1.3.1. Энергетические критерии................................18
1.3.2. Структурные критерии...................................25
1.3.3. Магнитные критерии.....................................28
1.3.4. Заключение.............................................32
Литература........................................................33
Глава 2. Реакционная способность ароматических соединений.............35
2.1. Энергетические характеристики реакционной способности .... 35
2.2. Реакционная способность и ароматичность................37
2.3. Классификация реакций..................................39
2.4. Влияние строения ароматических соединений на реакционную спо-
собность ........................................................42
2.4.1. Электронные эффекты заместителей.................42
2.4.2. Влияние аннелирования............................47
2.5. Влияние строения реагента..............................50
2.6. Влияние растворителя........................................55
2.7. Элементарные стадии реакций ароматического замещения .... 62
2.7.1. Электрофильное замещение.........................63
2.7.2. Нуклеофильное замещение..........................72
2.7.3. Свободнорадикальное замещение....................86
2.8. Квантовомеханическое описание реакционной способности .... 90
Литература........................................................98
Часть 2
Реакции ароматического электрофильного замещения
Г лава 3. Сульфирование..............................................102
3.1. Общие представления о реакции сульфирования и сульфирующие
агенты.......................................................102
3.2. Механизм реакции сульфирования.............................106
540
3.3. Практика сульфирования
Сульфирование
Сульфирование
Сульфирование
Сульфирование
Сульфирование
3.3.1.
3.3.2.
3.3.3.
3.3.4.
3.3.5.
Литература .
бензола и его гомологов ....................
производных углеводородов ряда бензола . .
нафталина и его производных.................
антрахинона и его производных ..............
антрацена ...............
108
109
114
118
126
129
131
Глава 4. Нитрование и нитрозирование..................................132
4.1. Общие представления о реакции нитрования и нитрующие агенты 132
4.2. Механизм реакции нитрования..................................134
4.3. Особенности ориентации при нитровании........................138
4.4. Практика нитрования..........................................141
4.4.1. Нитрование бензола и его гомологов .....................142
4.4.2. Нитрование производных углеводородов ряда бензола . . . 146
4.4.3. Нитрование нафталина и его производных..................153
4.4.4. Нитрование антрахинона и его производных................158
4.4.5. Нитрование других полициклических соединений............160
4.5. Нитрозирование...............................................161
4.5.1. Нитрозирование аминов...................................162
4.5.2. Нитрозирование фенолов и енолов.........................165
Литература........................................................167
Г лава 5. Галогенирование в ароматическое кольцо......................168
5.1. Общие представления о реакции, реагентах и механизмах .... 169
5.2. Практика галогенирования.....................................174
5.2.1. Хлорирование бензола и его гомологов....................174
5.2.2. Хлорирование производных углеводородов ряда бензола . .179
5.2.3. Хлорирование нафталина и его производных................184
5.2.4. Хлорирование антрахинона, его производных и других по-
лициклических соединений.......................................186
5.2.5. Бромирование и иодирование..............................188
Литература........................................................191
Глава 6. Введение в ароматическое кольцо углеродсодержащих групп . . . 193
6.1. Реакции с галогеналкилами, непредельными соединениями и спир-
тами ............................................................194
6.1.1. Общие представления об алкилировании...................194
6.1.2. Изомеризация алкильных производных.....................196
6.1.3. Алкилирование углеводородов............................197
6.1.4. C-Алкилирование фенолов................................198
6.1.5. С-Алкплированне аминов.................................200
6.2. Реакции с альдегидами и кетонами............................202
6.2.1. Получение ди- и триарилметановых производных...........202
6.2.2. Реакция хлорметилирования..............................203
6.2.3. Получение альдегидов реакцией с формальдегидом .... 205
6.2.4. Аминометилирование и родственные реакции...............206
6.3. Реакции с производными карбоновых кислот...................209
6.3.1. Общие представления о реакции ацилирования.............209
6.3.2. Получение кетонов......................................211
6.3.3. Реакция формилирования . ..............................215
6.3.4. Реакция карбоксилирования фенолов......................219
6.3.5. Диспропорционирование и изомеризация солей карбоновых
кислот.......................................................224
Литература.......................................................228
Часть 3
Реакции ароматического нуклеофильного замещения
Глава 7. Реакции с азотсодержащими нуклеофильными реагентами . . . 230
7.1. Введение аминогрупп .....................................230
7.1.1. Замещение атомов галогенов.........................231
7.1.2. Замещение нитрогруппы..............................239
641
7.1.3. Замещение амино- и аммониевых групп.............241
7.1.4. Замещение сульфогруппы................................245
7.1.5. Замещение гидроксильной группы........................247
7.1.6. Замещение алкокси-, арилокси- и аиилоксигрупп.........256
7.1.7. Замещение атома водорода..............................260
7.2. Введение гидразиногрупп................................ . . 266
7.3. Введение нитро- и азидогруппы...............................269
Литература.......................................................273
Г лава 8. Реакции с кислородсодержащими нуклеофильными реагентами . . 276
8.1. Введение гидроксильной группы...............................276
8.1.1. Замещение сульфогруппы................................276
8.1.2. Замещение атомов галогенов............................281
8.1.3. Замещение нитрогруппы.................................289
8.1.4. Замещение амино- и диазониевой групп..................290
8.1.5. Замещение атома водорода..............................294
8.2. Введение алкокси- и арилоксигрупп...........................298
Литература.......................................................305
Г лава 9. Реакции с серусодержащими нуклеофильными реагентами .... 307
9.1. Введение меркаптогруппы ....................................308
9.1.1. Замещение атомов галогенов............................308
9.1.2. Замещение диазониевой группы .........................310
9.1.3. Замещение гидроксильной группы........................312
9.1.4. Замещение атома водорода .............................315
9.2. Введение алкилтио- и арилтиогрупп . ................316
9.3. Введение сульфогруппы, алкил- и арилсульфонильных групп . . 319
Литература.......................................................326
Г лава 10. Реакции с аннонами галогенов.............................327
10.1. Замещение атомов галогенов и нитрогруппы...................327
10.2. Замещение гидроксильной группы ............................331
10.3. Замещение диазониевой группы...............................334
Литература.......................................................338
Глава 11. Реакции с углеродсодержащими нуклеофильными реагентами . . 340
11.1. Введение цианогруппы.......................................340
11.1.1. Замещение диазониевой группы........................340
11.1.2. Замещение атомов галогенов и сульфогруппы..........340
11.1.3. Замещение атома водорода............................343
11.2. Введение алкильных групп...................................346
11.2.1. Замещение атомов галогенов и других групп..........346
11.2.2. Замещение атома водорода............................350
11.3. Введение арильных групп....................................354
Литература.......................................................357
Часть 4
Реакции ароматического свободнорадикального замещения
Г лава 12. Реакции с углеродными радикалами.........................360
12.1. Введение арильных групп....................................360
12.2. Введение алкильных и других групп..........................366
Литература.......................................................371
Глава 13. Реакции с гетероатомными радикалами.......................371
13.1. Введение гидроксильной и ацилоксильных групп...............371
13.2. Введение аминогрупп........................................375
Литература................................................... 379
542
Часть 5
Реакции в замещающих группах ароматических соединений
Глава 14. Замещение водорода в боковых цепях и в функциональных группах 380
14.1. Замещение в алкильных группах............................ 380
14.1.1. Свободнорадикальное галогенирование в алкильных группах 381
14.1.2. Получение производных дибензила и стильбена........гс85
14.1.3. Электрофильное замещение в алкильных группах .... 387
14.2. Замещение в аминогруппах..................................391
14.2.1. А-Алкилирование.....................................392
14.2.2. Реакции первичных аминов с альдегидами..............397
14.2.3. А-Ацилирование......................................398
14.3. Замещение в гидрокси- и меркаптогруппах...................407
14.3.1. О- и S-Алкилирование................................408
14.3.2. О- и S-Ацилирование.................................411
Литература......................................................414
Глава 15. Диазотирование. Строение и реакции диазосоединений........415
15.1. Общие представления о реакции диазотирования..............416
15.2. Механизм образования и строение диазосоединений...........420
15.3. Стойкие формы диазосоединений.............................426
15.4. Реакции диазосоединений...................................428
15.4.1. Азосочетание........................................428
15.4.2. Особенности механизма нуклеофильного замещения диазо-
ниевой группы...............................................435
15.4.3. Свободнорадикальные реакции диазосоединений.........438
15.4.4. Фотохимические реакции диазосоединений..............441
Литература......................................................444
Часть 6
Реакции восстановления и окисления ароматических
соединений
Глава 16. Восстановление ароматических колец и функциональных групп 446
16.1. Общие представления об окислительно-восстановительных реак-
циях ............................................................446
16.2. Механизмы реакций восстановления.............................446
16.2.1. Восстановление с передачей электронов..................447
16.2.2. Восстановление с передачей гидрид-иона................449
16.2.3. Восстановление с передачей пары электронов от нуклео-
фильного агента...............................................451
16.2.4. Восстановление водородом..............................452
16.3. Восстановление ароматических колец.......................... 453
16.4. Восстановление ароматических соединений, содержащих углерод-
кислородные связи..................................................457
16.4.1. Восстановление кетонов и альдегидов..............457
16.4.2. Восстановление хинонов и кубовых красителей..........459
16.4.3. Восстановление фенолов................................463
16.5. Восстановление азометинов, амидов и нитрилов.................463
16.6. Восстановление ароматических нитросоединений................466
16.6.1. Получение аминов......................................468
16.6.2. Получение А-арилгидроксиламинов и их перегруппировка в
аминофенолы ... 473
16.6.3. Получение азо- и гидразосоединений. Бензидиновая пере-
группировка ..................................................475
16.7. Восстановление нитрозо- и азосоединенип......................481
16.8. Восстановление диазосоединений...............................484
16.9. Восстановление групп, содержащих серу.....................485
Литература.........................................................487
543
Глава 17. Окисление боковых цепей, ароматических колец и функцио-
нальных групп.................................................489
17.1. Окисление боковых цепей .... ... 490
17.1.1. Получение ароматических карбоновых кислот 490
17.1.2. Получение ароматических нитрилов (окислительный ам-
монолиз) ........................ ... .........498
17.1.3. Получение ароматических альдегидов и кетонов . . . 499
17.1.4. Получение ди- и триарилметановых красителей .... 504
17.1.5. Реакции дегидрирования.......................505
17.2. Окисление ароматических колец......................508
17.2.1. Получение фенолов прямым окислением ароматических
соединений...........................................509
17.2.2. Получение фенолов из карбоновых кислот.......512
17.2.3. Получение фенолов из трет-аралкилгидроперекисей . . 514
17.2.4. Получение хинонов............................517
17.2.5. Получение карбоновых кислот..................522
17.3. Окисление азотсодержащих групп . . .527
17.4. Окисление серусодержащих групп.....................530
Литература...............................................532
Указатель важнейших соединений................................535
Сканировал: Neptunyi
(Магнитогорск)
Все замечания и пожелания присылать по адресам:
rhenium@list.ru
nept2006@yandex.ru
Буду рад также высылаемым книгам от вас!
Лев Соломонович. Эфрос
Михаил Викторович Горелик
химия и ТЕХНОЛОГИЯ
ПРОМЕЖУТОЧНЫХ продуктов
Научный редактор 3. Я. Хавин
Редактор издательства Н. Р. Либерман
Техн, редактор 3. Е. Маркова
Художник И. М. Сенский
Корректор Г. А. Лебедева
ИБ № 419
Сдано в наб. 10.07.79. Подп. к печ. 29.02.80. М-27606. Формат бумаги
60Х90’/1б- Бум. тип. № 2. Литературная гарнитура. Высокая печать.
Усл. печ. л. 34,0. Уч-.изд. л. 38,49. Тираж 3700 экз. Заказ 271.
Цена 2 р. 20 к. Изд. № 1163
Ордена «Знак Почета» издательство «Химия». Ленинградское отделение,
191186, г. Ленинград, Д-186, Невский пр.. 28
Ордена Трудового Красного Знамени Ленинградская типография № 2
имени Евгении Соколовой «Союзполиграфпрома» прн Государственном
комитете СССР по делам издательств, полиграфии н книжной торговли.
198052, Ленинград, Измайловский пр., 29.