









































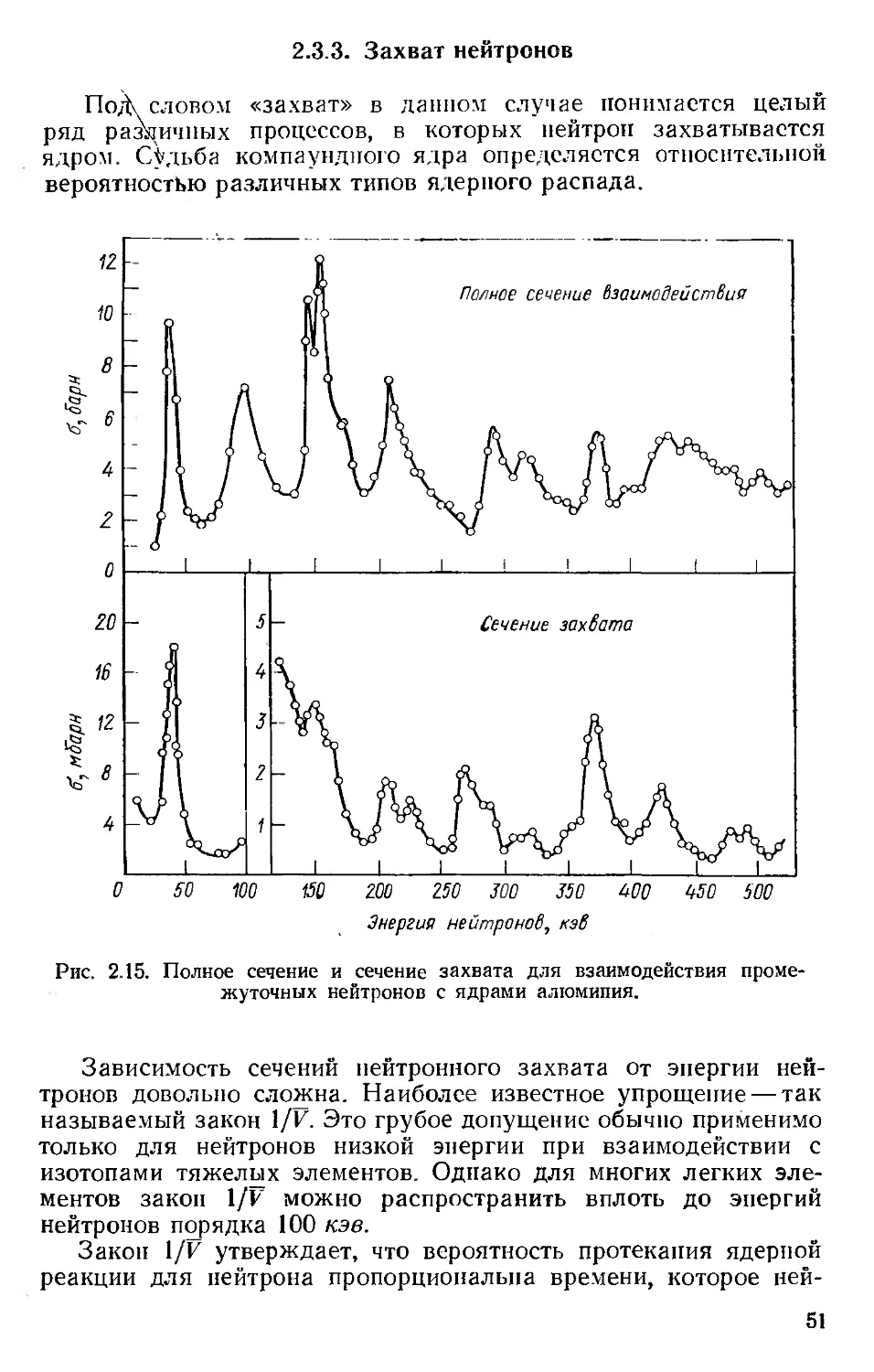













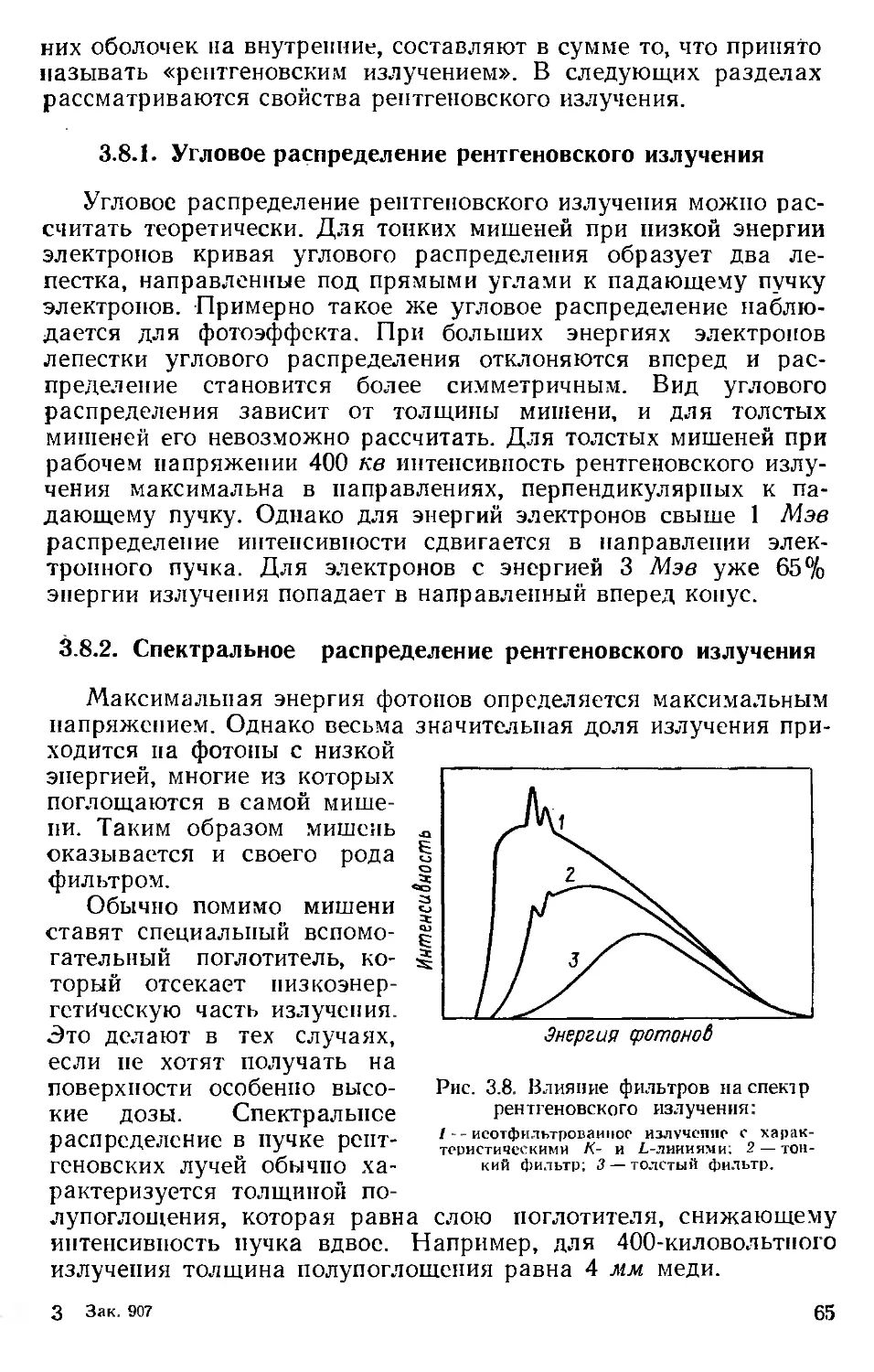


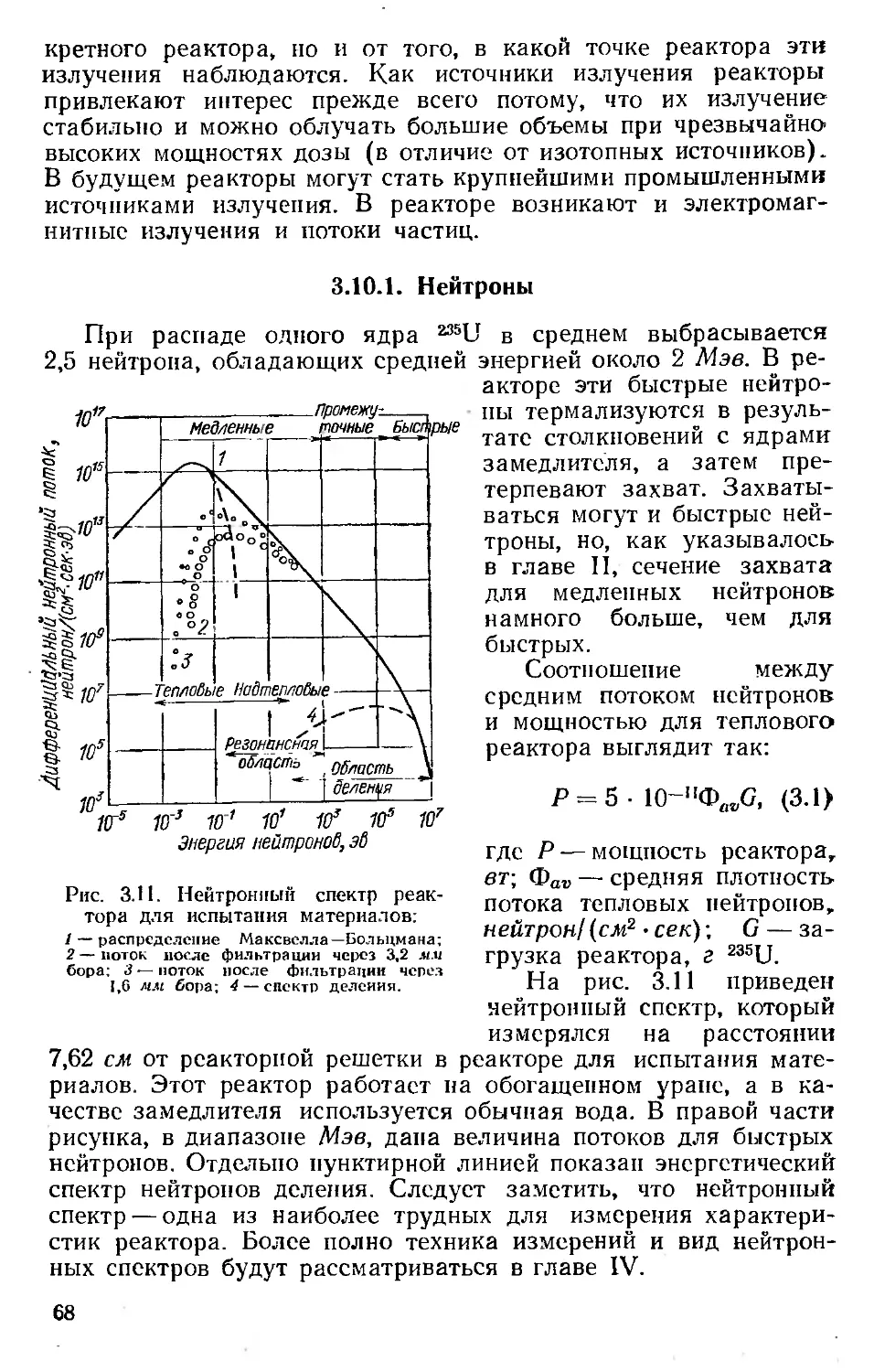

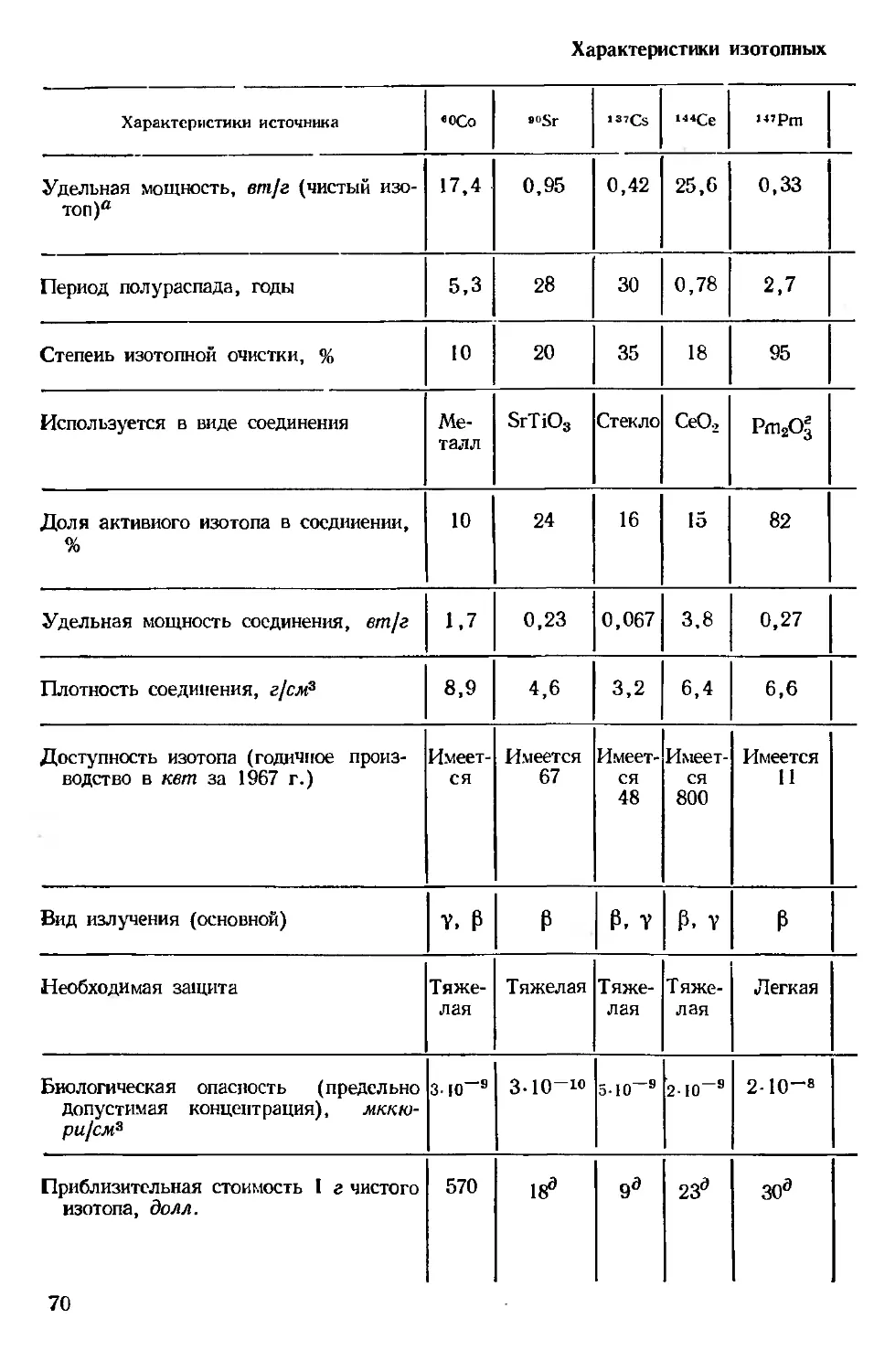






















































































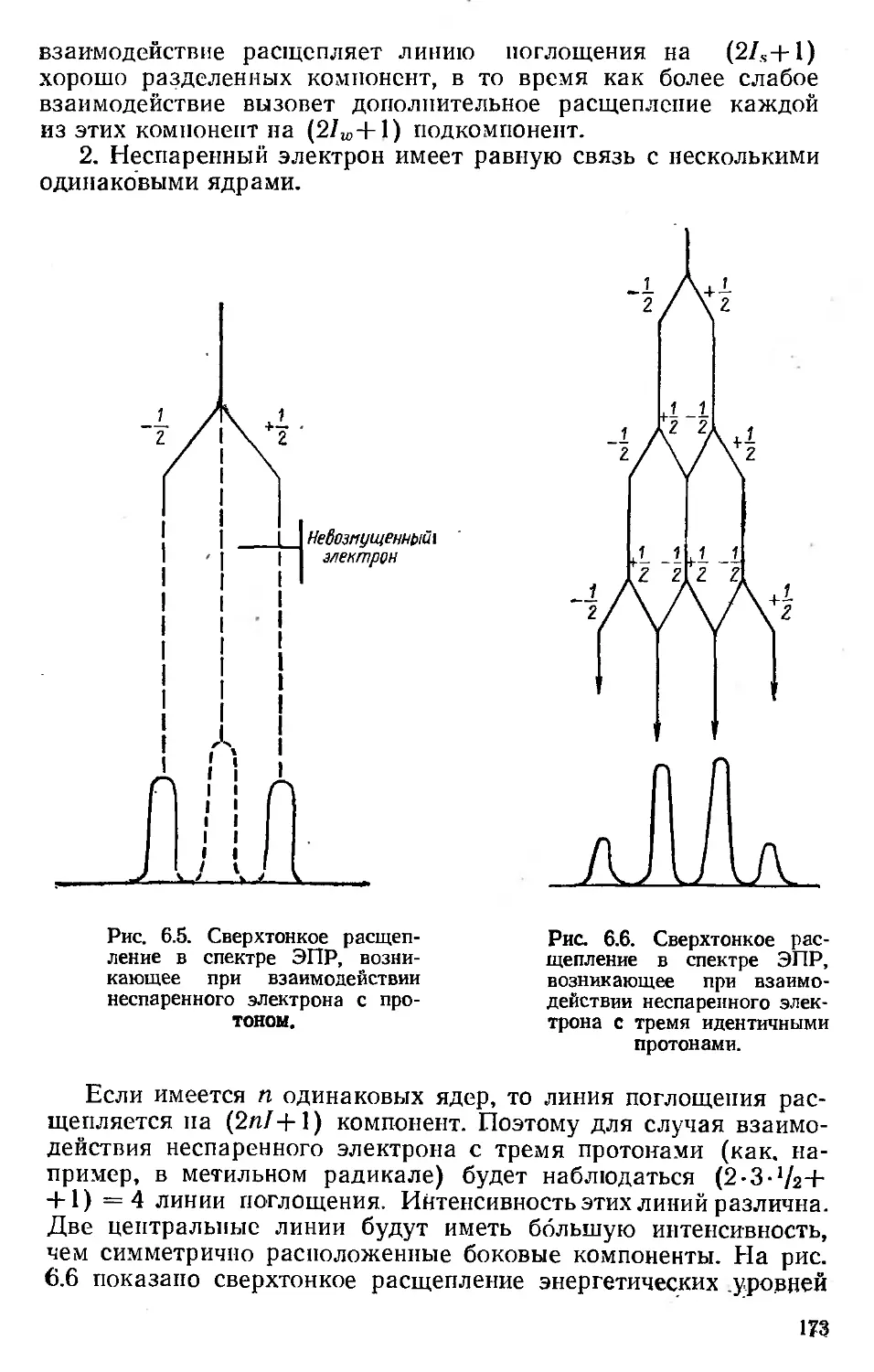







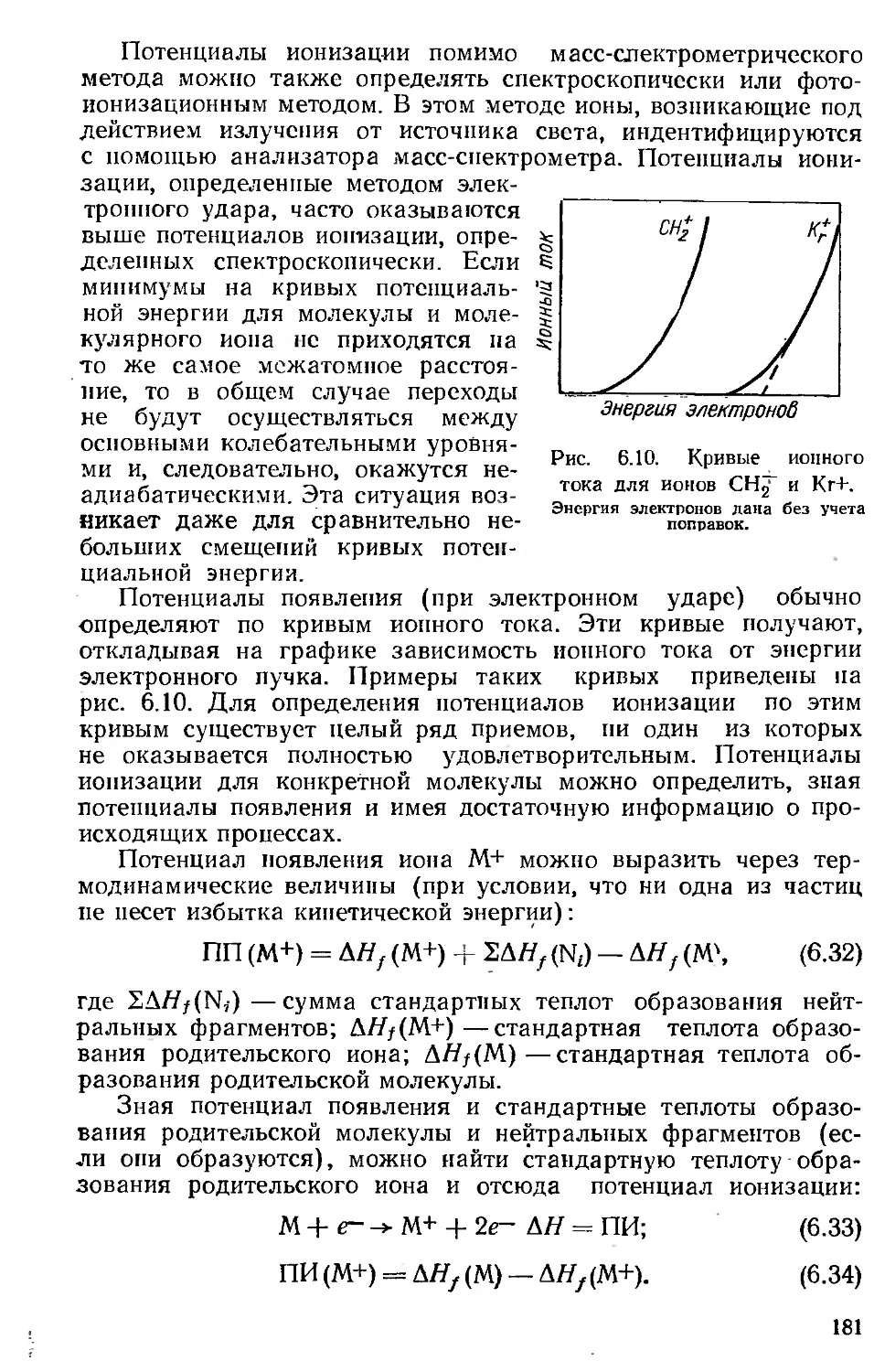
























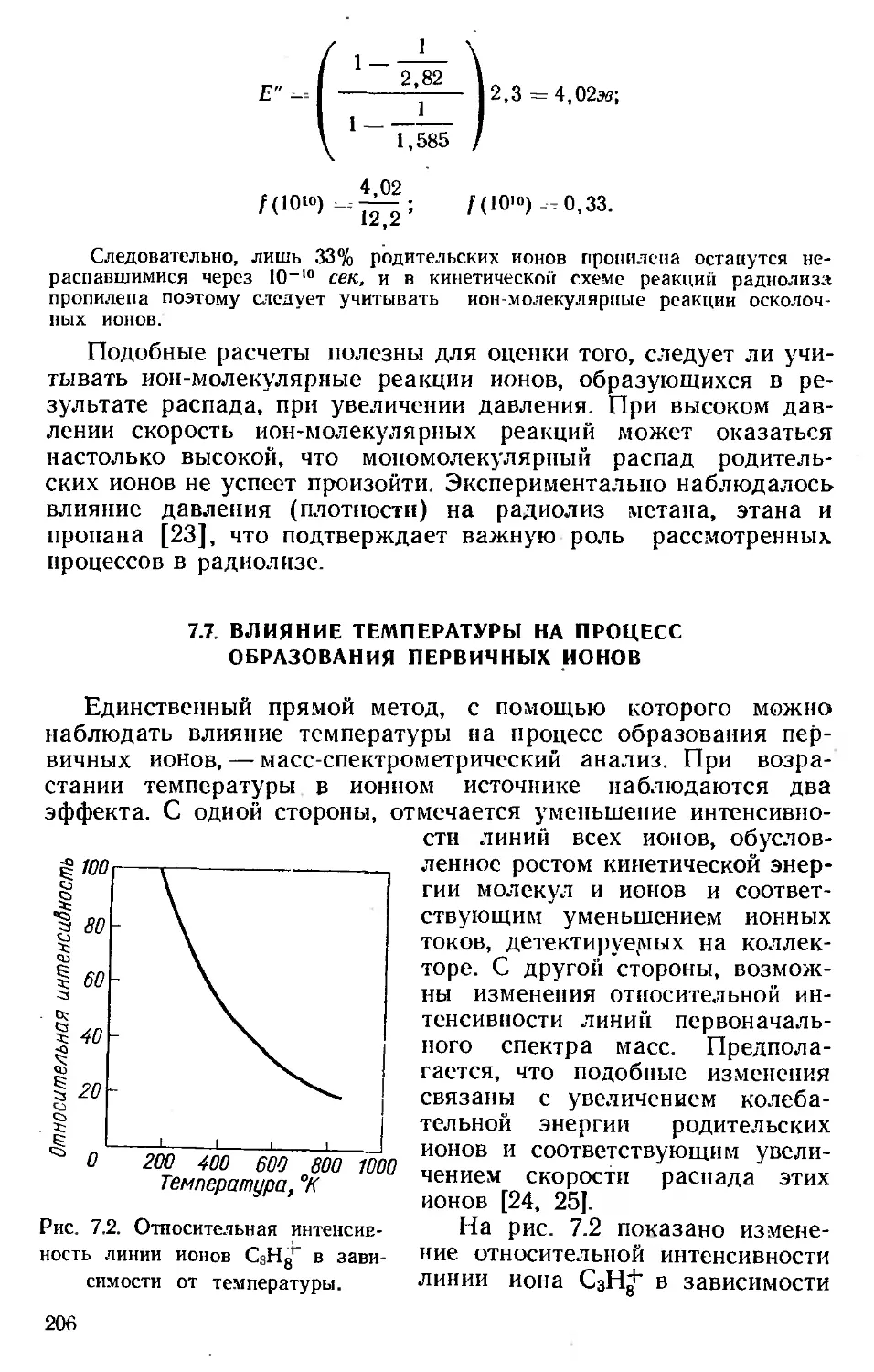

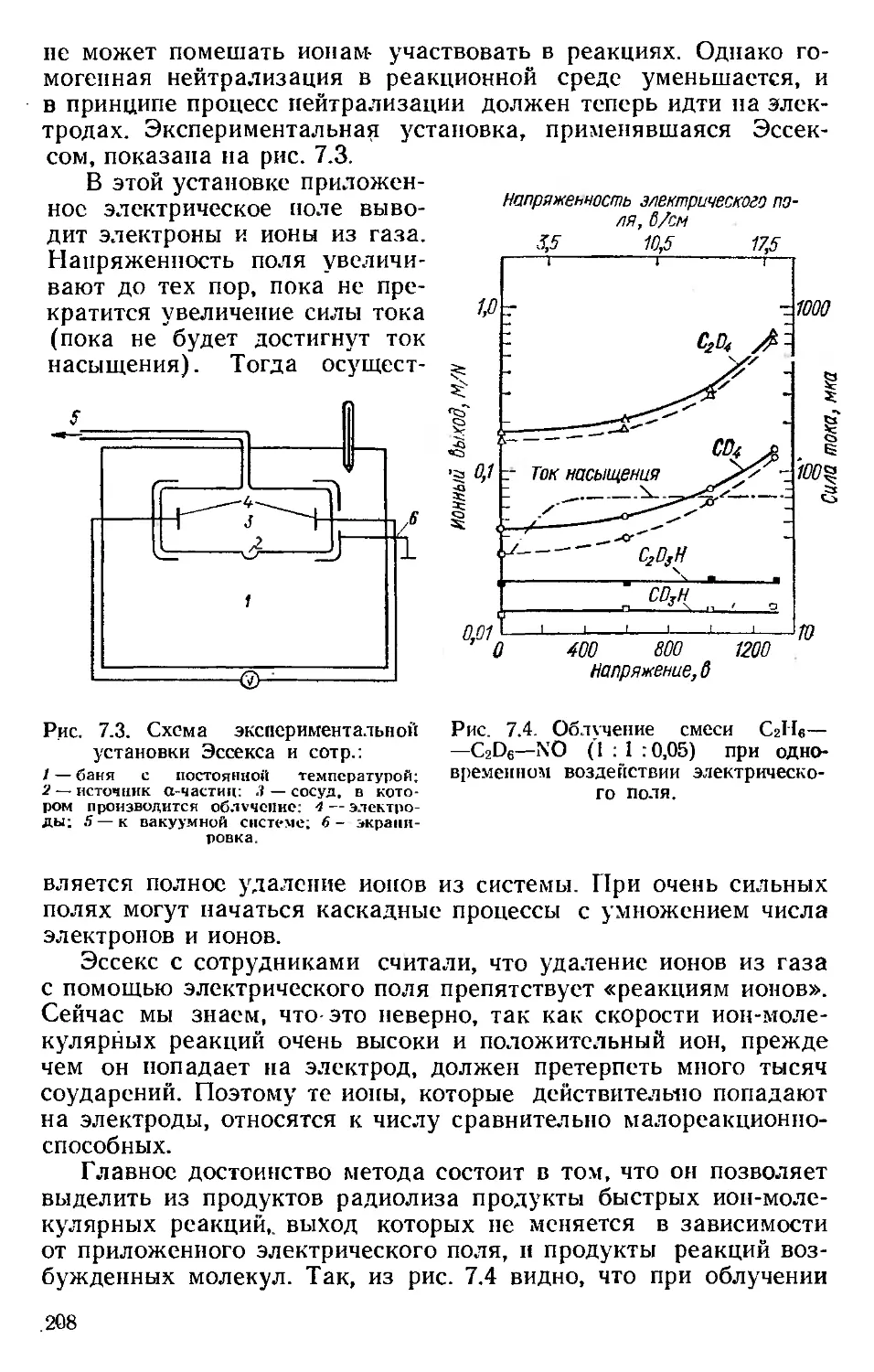




















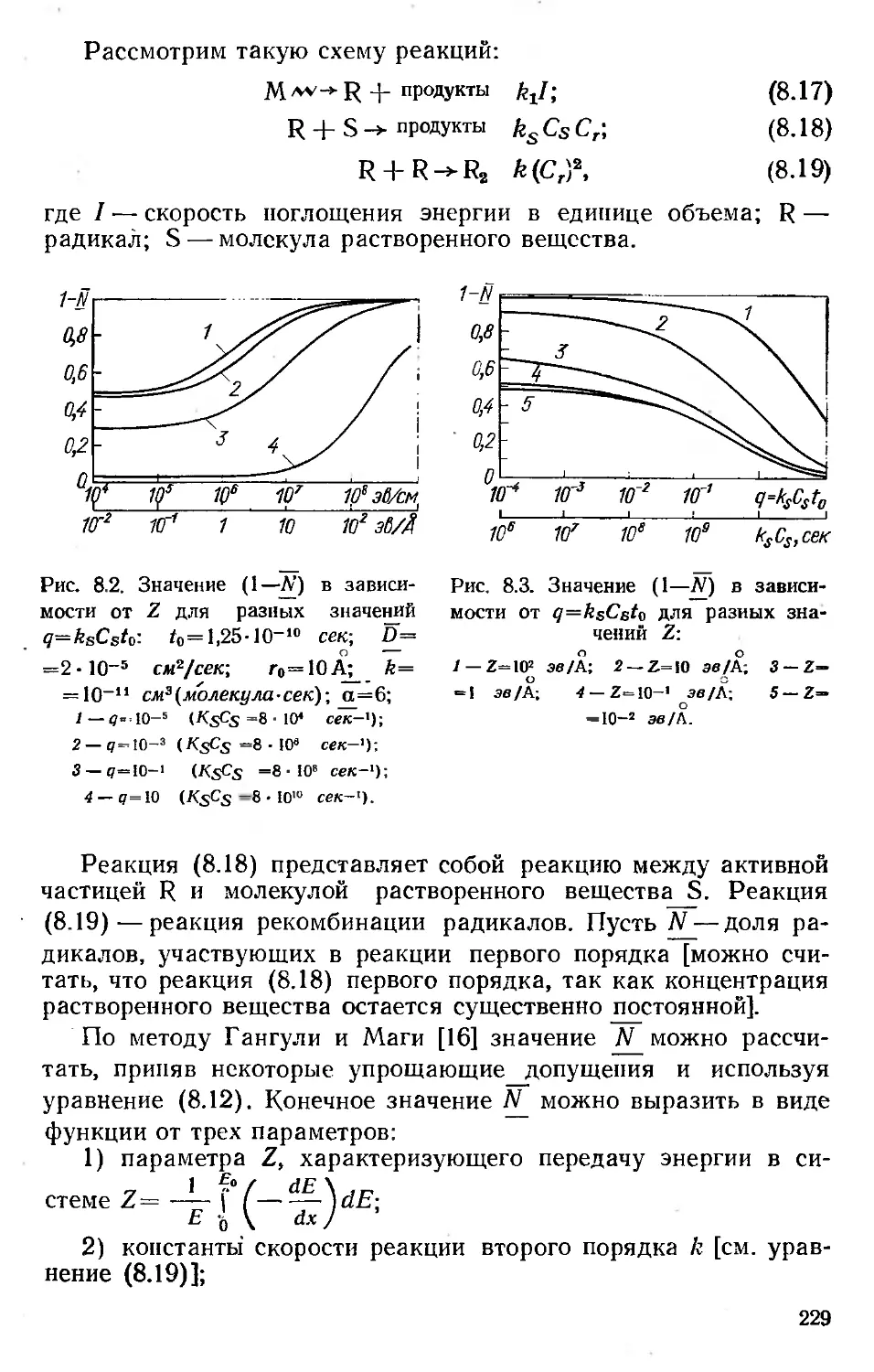





































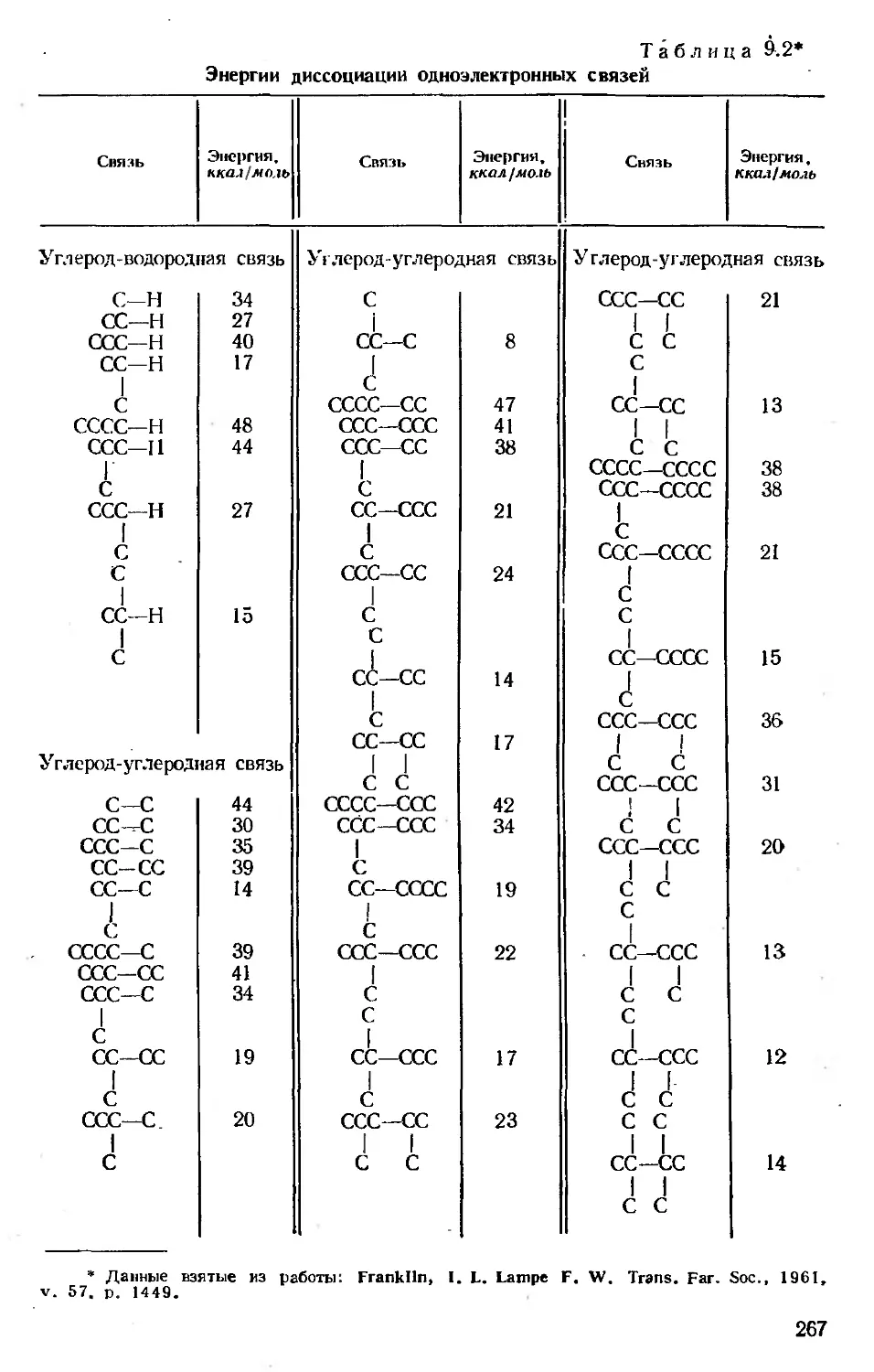











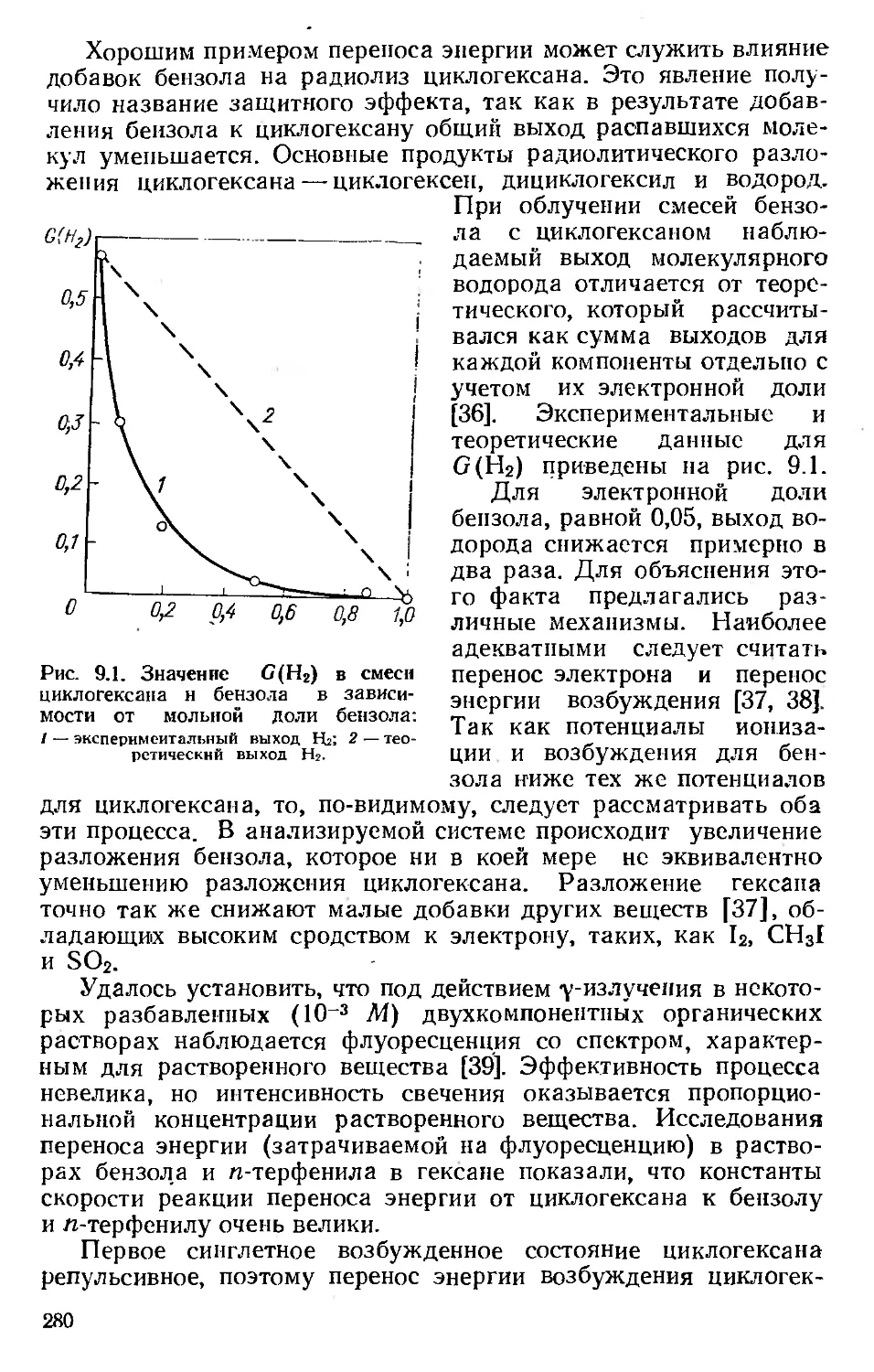






































































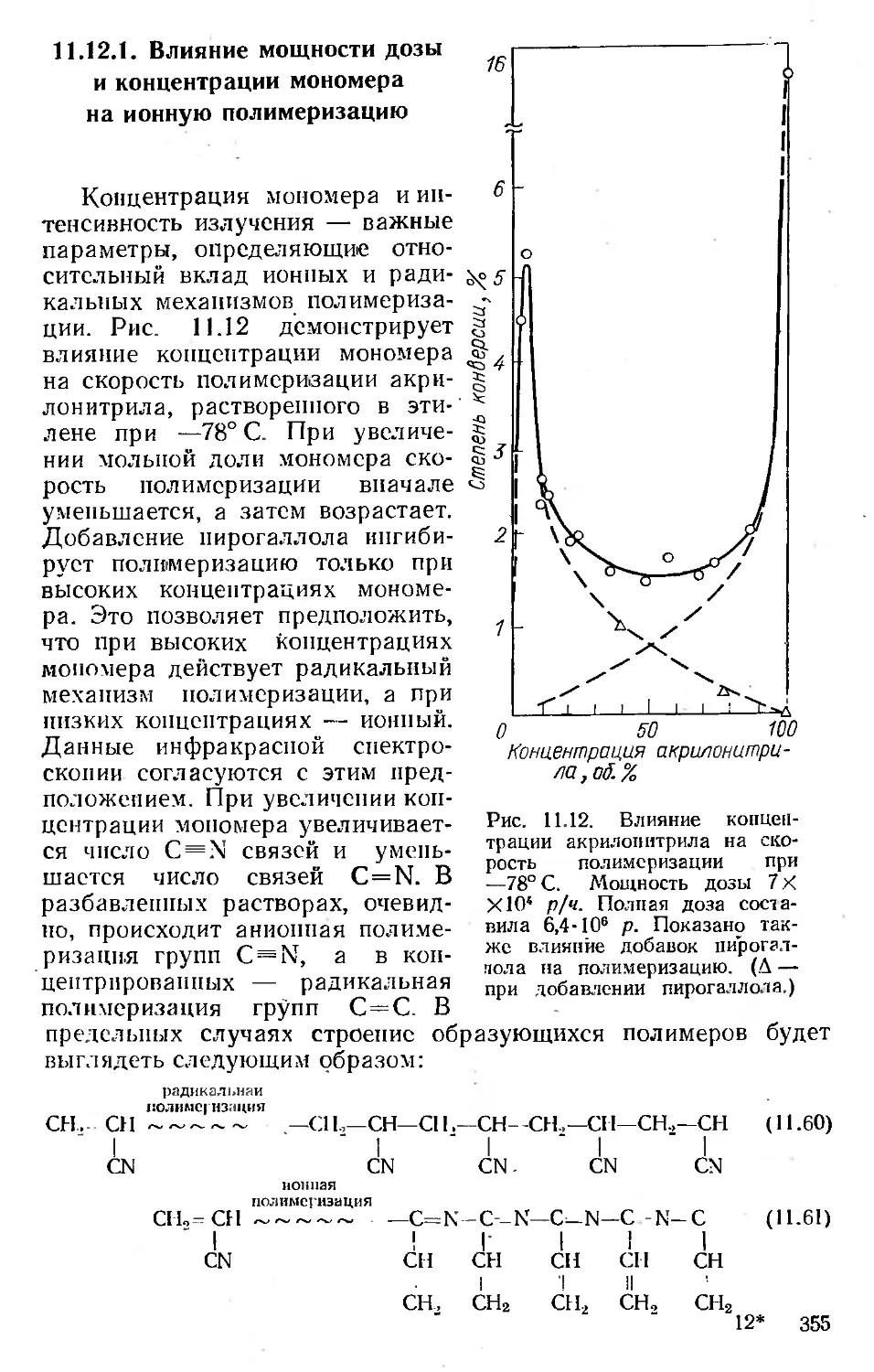















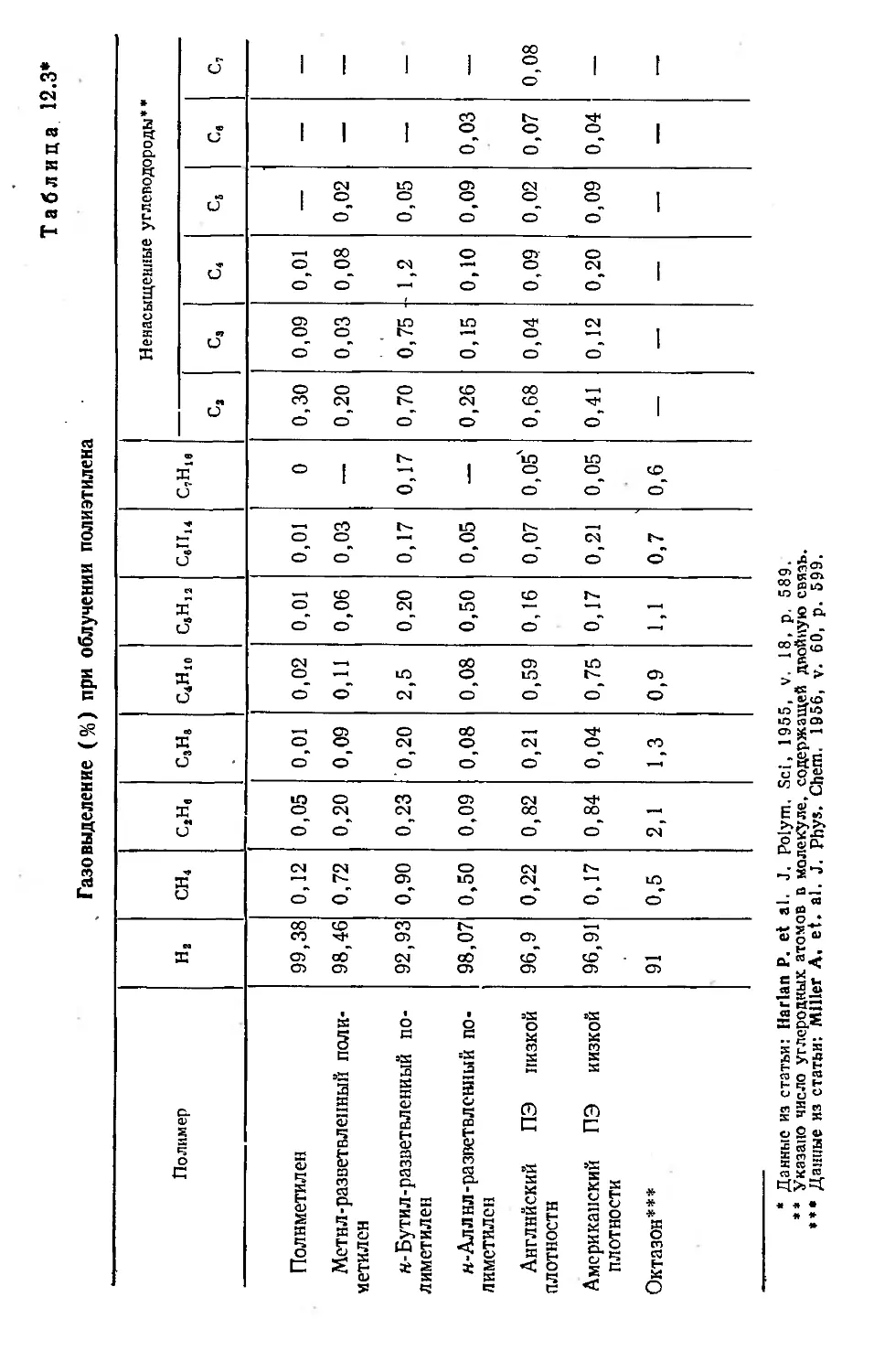





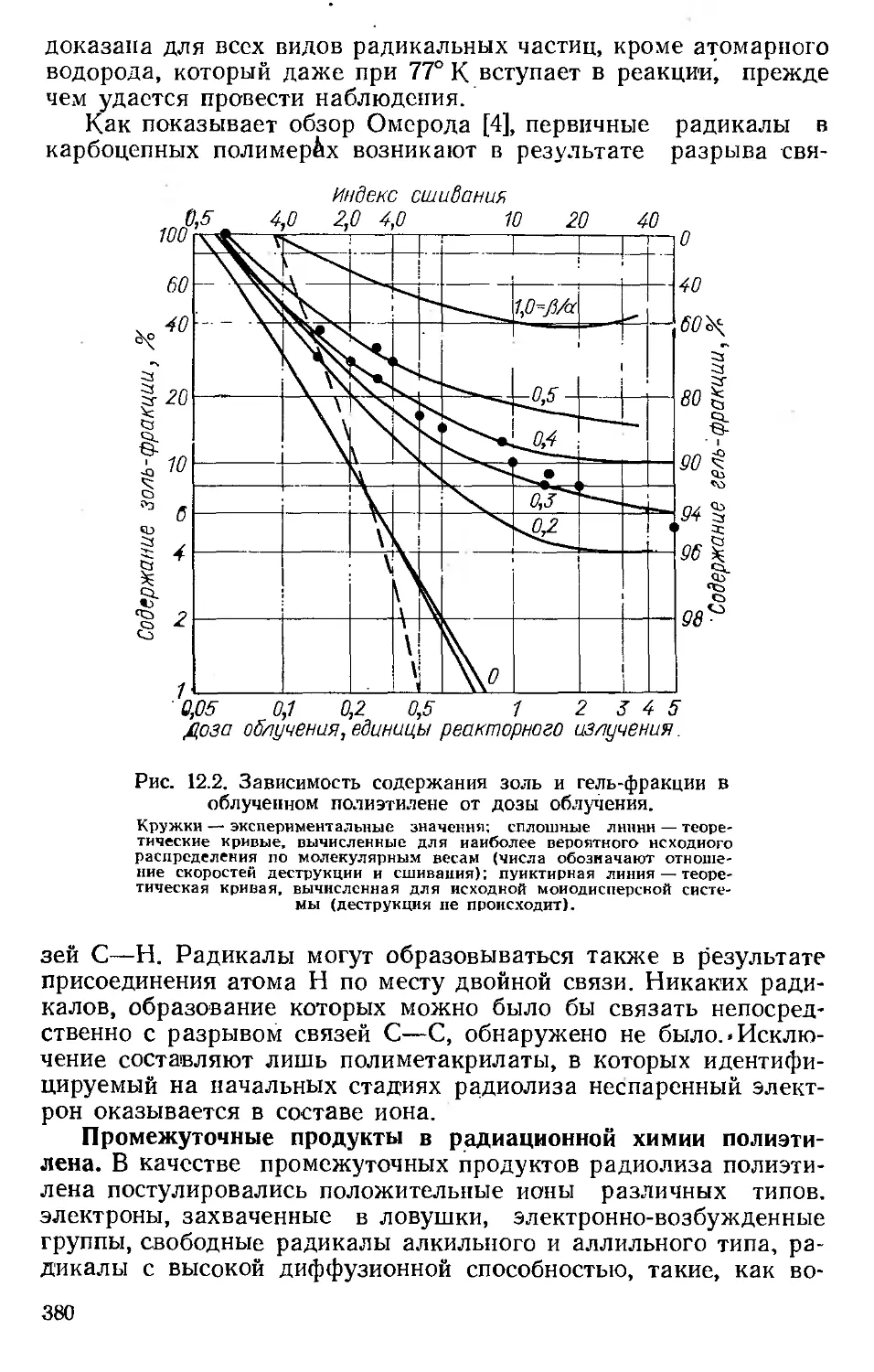









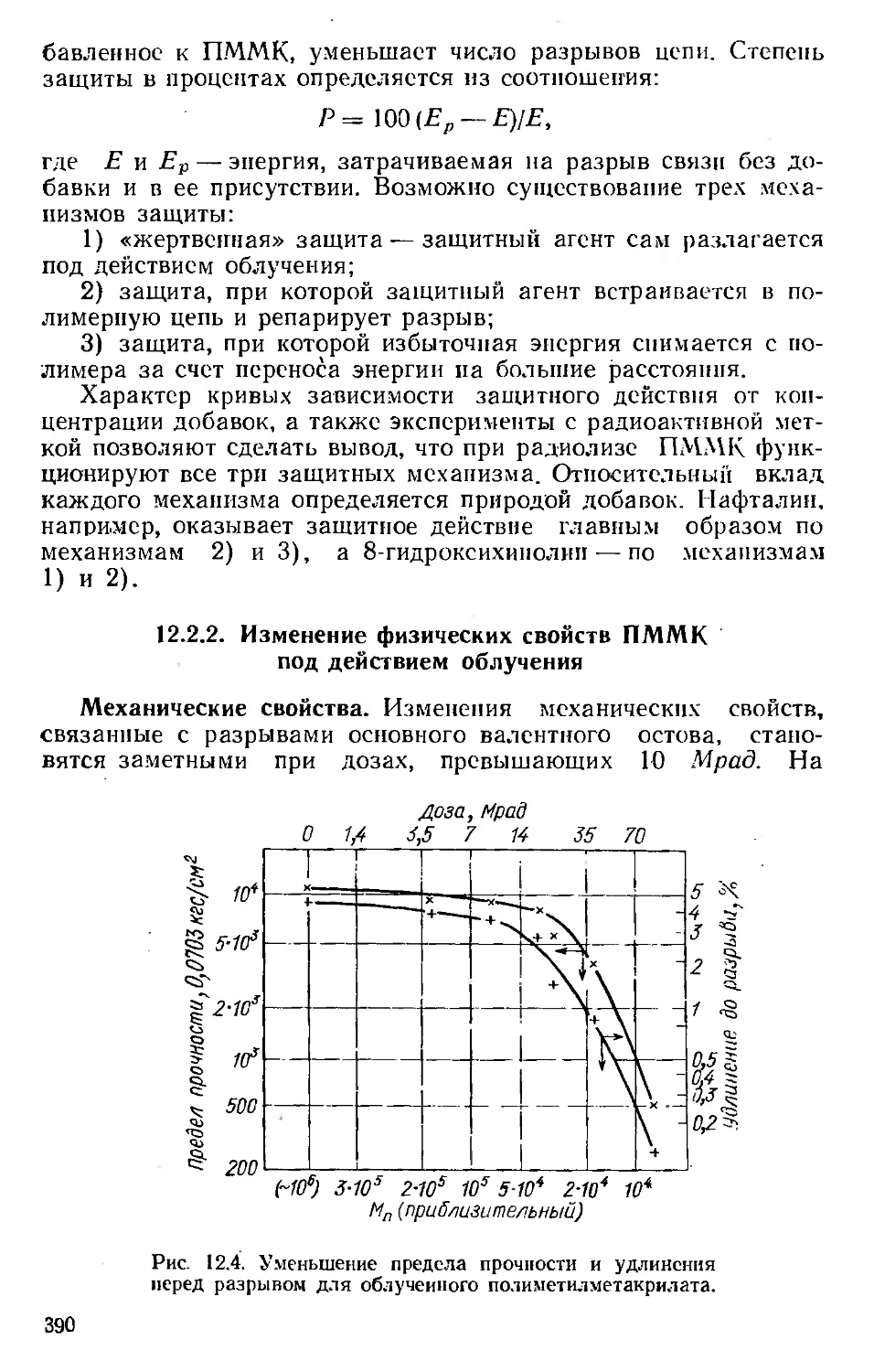








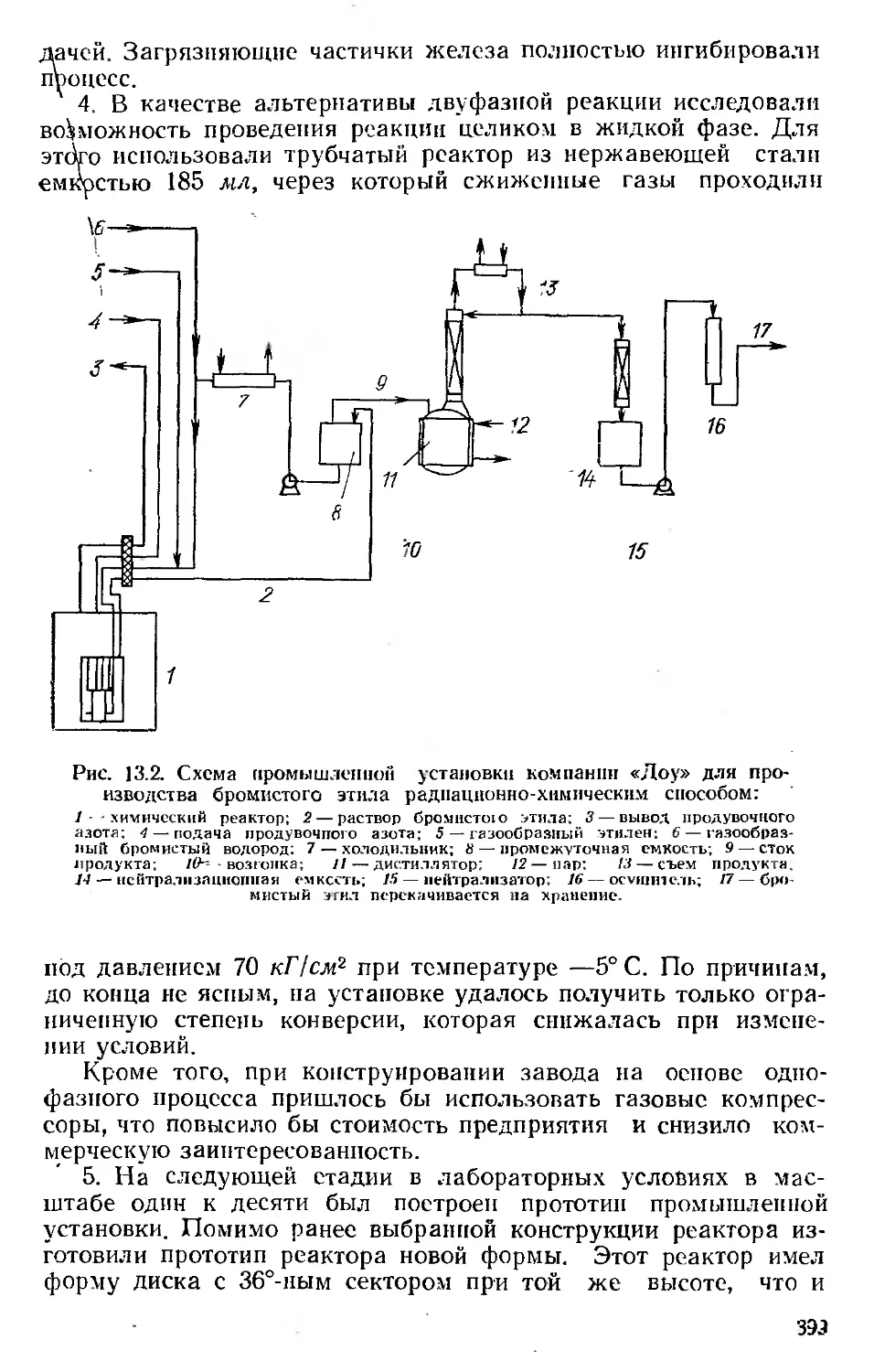
















Автор: Хенли Э. Джонсон Э.
Теги: химия радиация переводная литература твердое тело учебное пособие для студентов атомиздат радиоанализ газовые системы
Год: 1974



Э. ХЕНЛИ, В. ДЖОНСОН
j РАДИАЦИОННАЯ
ХИМИЯ
E. HENLY
Е. JOHNSON
THE CHEMISTRY
AND PHYSICS
OF HIGH ENERGY REACTIONS
UNIVERSITY PRESS 1969
Э. ХЕНЛИ
Э. ДЖОНСОН
РАДИАЦИОННАЯ
ХИМИЯ
Перевод с английского кандидата физико-математических наук
В.Н.ЛЫСЦОВА
МОСКВА АТОМИЗДАТ 1974
УДК 541.15
Хен л и Э., Д ж о н с о п Э. Радиационная химия. Пер. с англ.
Атомиздат, 1974, 416 стр.
В книге изложены современные представления радиационной хи-
мии. Охвачены практически все разделы этой дисциплины: от радио-
лиза газовых систем до радиационной химии твердого тела. Представ-
лен обзор современных методов радиационной химии и уделено вни-
мание использованию их в промышленности.
Книга является учебным пособим для студентов-физиков и хи-
миков; ею могут пользоваться и/научные работники, интересующиеся
современным состоянием радиационной химии.
Рисунков 102, таблиц 75, библиографических ссылок 286.
Spliner
20503—054
X ----------- 54—74
034(01}—74 1
g) Атомиздат, перевод, 1974
Глава I
ИЗЛУЧЕНИЯ И РАДИОАКТИВНОСТЬ.
ОСНОВНЫЕ ЕДИНИЦЫ ИЗМЕРЕНИЯ
1.1. ЕДИНИЦЫ ИЗМЕРЕНИЯ ФИЗИЧЕСКИХ ВЕЛИЧИН
В этой книге мы будем пользоваться абсолютной физической
системой единиц СГС*. Электрические величины будут изме-
ряться в единицах абсолютной электрической (СГСЭ) или аб-
солютной магнитной (СГСМ) системы. Так, например, сила
электростатического взаимодействия между двумя зарядами на
расстоянии I запишется следующим образом:
£ = (1.1)
KZ2
или для двух одинаковых зарядов
£ = —, (1.2)
КР
где величина К зависит от характера среды. По определению
значение К в вакууме равно единице. Размерность силы в си-
стеме СГС дается выражением
F ~ LMT~~. (1.3)
Поэтому для размерности заряда получим
е = Л1,/2Г/27,~1. (1.4)
В радиационной химии часто используется такая единица,
как электронвольт (эв), которая равна энергии, приобретаемой
единичным зарядом (4,803-IO-10 ед. СГСЭ) в электрическом
поле с разностью потенциалов 1 в (1/300 ед. СГСЭ). Поэтому в
4 8-Ю'"10
системе СГС электронвольт равен —, или 1,602х
X 10~12 эрг.
* Международный союз теоретической и прикладной химии (ИЮПАК)
рекомендует пользоваться международной системой единиц СИ, однако эта
система пока применяется недостаточно широко. В текущей литературе боль-
шей частью используется система СГС. Поэтому она будет применяться и
в настоящей книге.
5
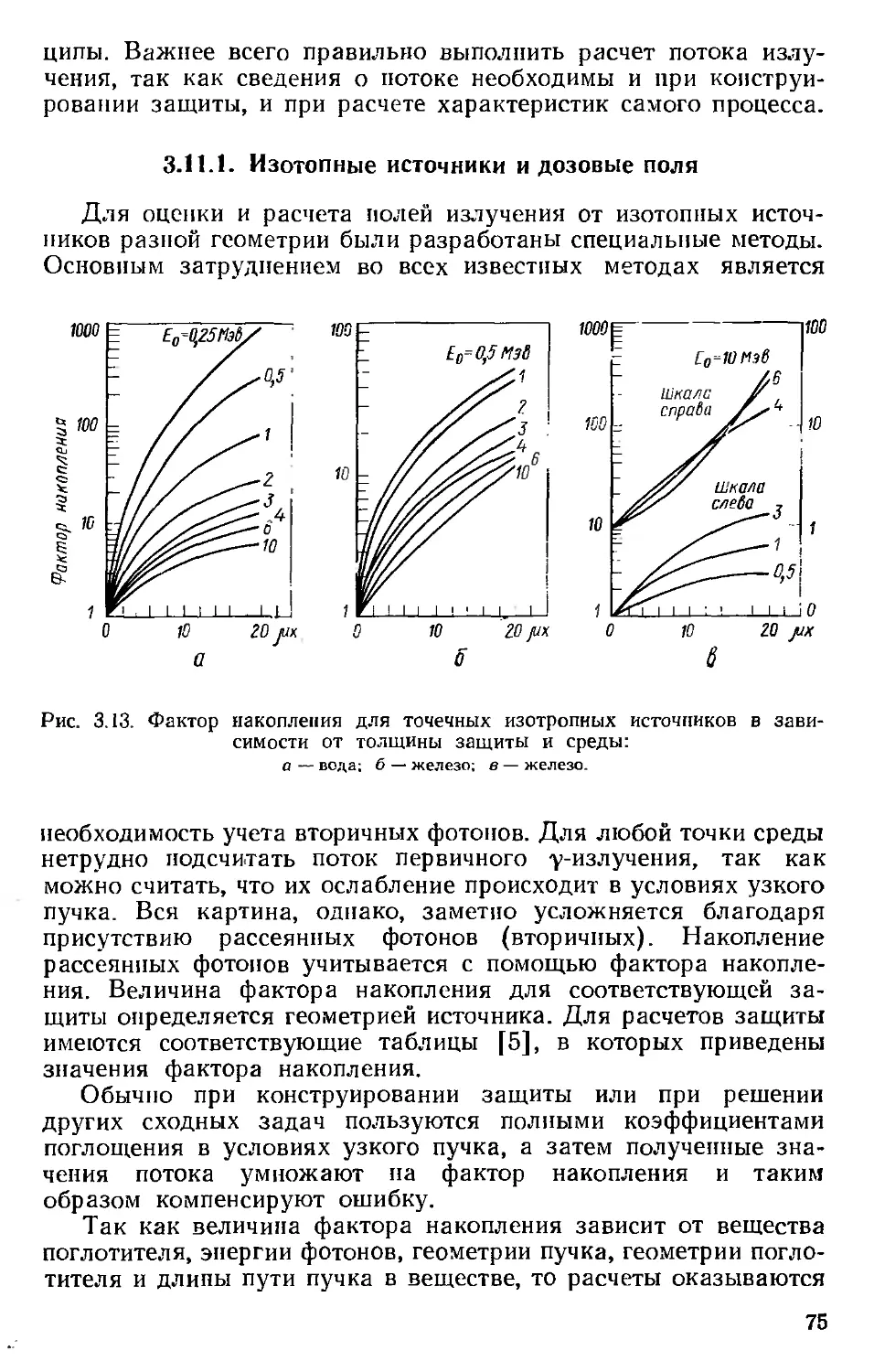
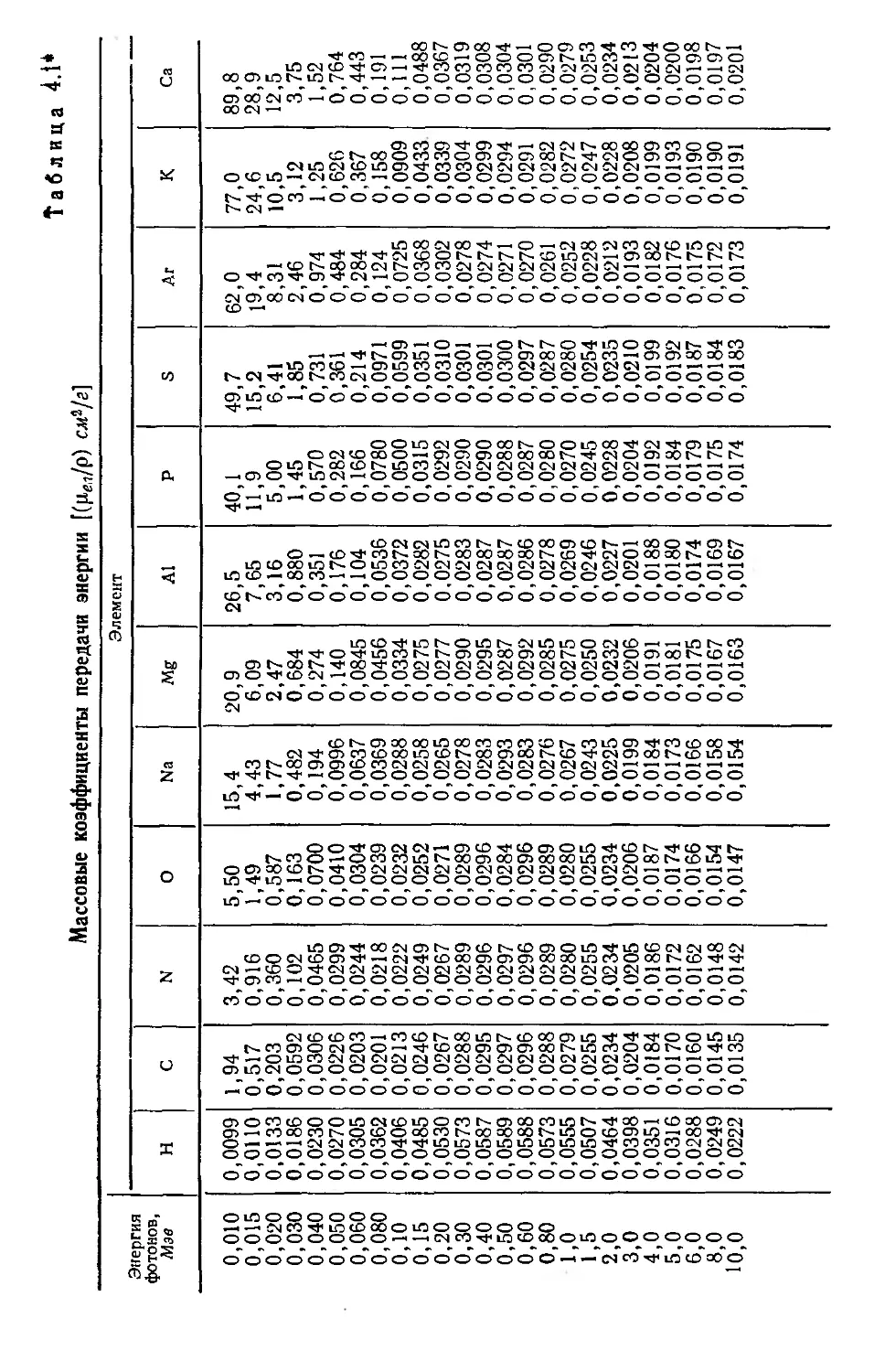
Переводные коэффициенты для некоторых единиц измерения
га
ЕГ
Другая широко используемая
единица энергии — вольт-кулои или
джоуль (дж) равен работе, произ-
водимой при переносе заряда в 1 к
в поле с разностью потенциалов
1 в. 1 дж равен 107 эрг. Умножая
1,602-Ю-12 эрг па 6,02257-1023, по-
лучаем 9,6487-10В * * 11 эрг, или 0,6487Х
Х104 дж, что равно кинетической
энергии грамм-атома электронов
при ускорении в поле с разностью
потенциалов 1 в.
Единицей мощности служит
ватт, который равен 1 дж)сек. В ват-
тах измеряется скорость поглоще-
ния энергии. Для измерения мощ-
ности или количества тепла исполь-
зуют калории, БТЕ *, и киловатты.
Соотношения между этими едини-
цами и их производными приведены
в табл. 1.1.
1.2. СВЯЗЬ МАССЫ И ЭНЕРГИИ.
НЕКОТОРЫЕ КВАНТОВЫЕ ФОРМУЛЫ
В квантовой теории энергия
электромагнитного излучения опре-
деляется его частотой, которую при-
нято обозначать греческой бук-
вой V.
_ Скорость света ]см/сек] с
Длина волны в [tvw] Z
(1-5)
Чаще, однако, используется дру-
гая величина — волновое число, ко-
торое равно числу длин волн, укла-
дывающихся па отрезке в 1 см:
ч . Частота .
Л-‘ — -------------— см
Скорость света
В квантовой теории принимает-
ся, что энергия электромагнитного
излучения прямо пропорциональна
его частоте:
E^hv, (1.6)
* БТЕ — британская тепловая единица,
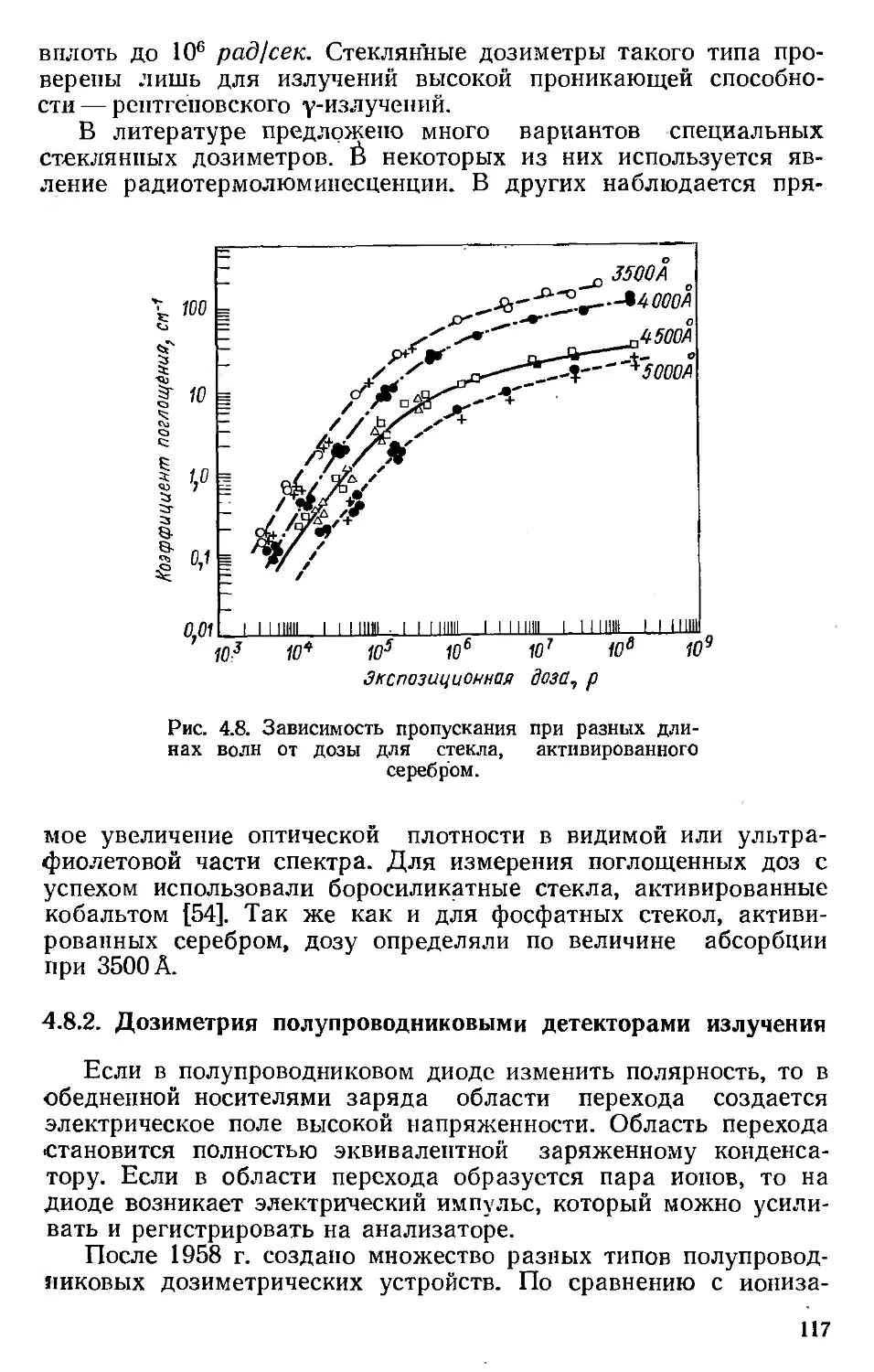
равная 252 кал. — Прим, перев.
6
где h — постоянная Планка, одна из основных физических кон-
стант. Таким образом, если известна длина волны излучения,
то можно рассчитать его энергию и, наоборот, по энергии можно
определить длину волны.
Любому электромагнитному излучению соответствуют кван-
ты или фотоны, энергию которых можно вычислить по уравне-
нию (1.6). Фотоны распространяются как и волны, и поэтому
имеют определенную частоту колебаний, скорость распростране-
ния и длину волны. Фотоны обладают механическим импульсом
и могут передавать его другим частицам. Однако импульс легко
обнаружить лишь у фотонов высокой энергии.
Кинетическая энергия частиц, таких, как протон и электрон,
при сравнительно низких скоростях дается классической фор-
мулой
Е = — mV2.
2
Но если скорость частицы приближается к скорости света, то
ее масса растет в соответствии с законами специальной теории
относител ыюсти
где то — масса покоя частицы, а V — ее скорость. Теория от-
носительности также дает следующее соотношение между мас-
сой и энергией:
Е = гги?. (1.8)
Подставляя уравнение (1.7) в уравнение (1.8), найдем пол-
ную энергию частицы массой т, движущейся со скоростью V:
Е =-------т_°с*- (1.9)
[1 — (У2/с2)]
Для таких частиц, как электрон и протон, часто бывает не-
обходимо знать скорость, так как интенсивность передачи энер-
гии в среде непосредственно зависит от заряда и скорости ча-
стицы.
Пример 1.1. Определите скорость протона с энергией 25 Мэв. Чему будет
равна энергия электрона при той же скорости?
Решение. Если энергия, эквивалентная массе покоя частицы, значительно
превосходит энергию частицы, то применима классическая, нерелятивистская
механика. Для любой частицы масса покоя в электронвольтах дается сле-
дующей формулой:
_ о Атомный вес частицы „ 1
Е men - -----------------• с2 •--------------------.
° Число Авогадро 1,602- 10~12 эрг]эв
7
Для протона
1,008г
г-атом
6,023 • IO2»
атом
г-атом
см \2
2,99 • 10Ю------) х
сек J
1
X 1,602 • 10—I2 эрг!эв
г • см? ____ .эв
Е = 1,008---------- 931 • 106-----.
сек? эрг
Е == 931 • 106 эв.
Так как эта величина значительно превышает заданную энергию частицы,
то можно пользоваться формулами классической механики:
Е = — mV2;
2
25 • 106 эв = — «pV2;
— эрг
V2 = 50 • 10е эв • 1,6 • 10-12—^-
эв
1
1,67 • IO'2* '
Размерность эрга в системе СГС L2MT~2 и, следовательно, скорость про-
тона с энергией 25 Мэв, выраженная в см/сек, равна
— _ / см? см
У = 1/ 50.1013------ к 7 • 10»----.
у сек2 сек
Теперь найдем энергию электрона, имеющего ту же скорость:
1—1 / см \2
Е —---meV2 =------- 9,11 - IO-28 г. /7. ю» -)
2 2 \ сек /
эв
Е = 225. 10-ю эрг . ----;
1,6- 10 I2 эрг
Е » 14 000 эв.
1.3 ЕДИНИЦЫ ИЗМЕРЕНИЯ ИОНИЗИРУЮЩИХ ИЗЛУЧЕНИЙ
Для количественного описания поглощения ионизирующей
радиации в веществе был введен целый ряд единиц измерения.
В последние годы, однако, повсеместно приняты рекомендации
Международной комиссии по радиологическим единицам и из-
мерениям (МКРЕ). МКРЕ предлагает ограничиться следующи-
ми единицами измерений. Основной единицей поглощенной до-
зы служит рад: 1
1 рад =100 эрг/г — дж/кг
8
или
1 рад = _122_э£^ .------™-------= 6,24 • 1013—. (1.10)
г 1,602 • 10~12 эрг г
Мощность дозы выражается в радах, деленных на единицу вре-
мени, например рад/день, рад/сек. Химика или инженера обыч-
но интересует именно поглощенная доза, так как эта доза по-
казывает количество энергии, переданной излучением исследуе-
мому веществу. Величина поглощенной дозы зависит от соста-
ва вещества и спектра излучения.
Специальной единицей экспозиционной дозы служит рентген
(р). При выполнении определенных условий экспозиционную
дозу можно связать с поглощенной. Экспозиционная доза рав-
на 1 р, если в объеме воздуха с массой 0,001293 г под действием
всех электронов, освобожденных фотонами и полностью задер-
жанных в данном объеме, появляется такое количество ионов
одного знака, что их суммарный заряд равен 1 ед. СГСЭ:
1р = 2,58 • 10~4 к/кг. (1.11)
Такая формулировка эквивалентна прежнему определению
рентгена, согласно которому доза в 1 р соответствует образова-
нию 1 единицы заряда СГСЭ в 1 см3 сухого воздуха при нор-
мальных условиях *. В этих условиях 1 см3 сухого воздуха ве-
сит 0,001293 г. По определению рентген применяется только для
электромагнитных излучений (фотонов). В прежних определе-
ниях рентгена эту единицу предлагалось использовать только
для рентгеновского и у-излучепия.
Однако, согласно последним рекомендациям МКРЕ, опреде-
ление рентгена действительно для любых фотонов, исключая
лишь тормозное излучение вторичных электронов.
Если известна энергия U7, необходимая для образования од-
ной пары ионов, а также массовые коэффициенты передачи энер-
гии для воздуха и для рассматриваемого вещества, то по экспо-
зиционной дозе можно найти поглощенную дозу. Это и есть те
условия, в которых подобный пересчет оказывается возможным.
Пример 1.?. Найти поглощенную дозу в радах для воздуха, если экс-
позиционная доза равна 1 р.
Решение.
1 ед. СГСЭ электрон ав
------------• 2,082 • 10»------------. W-----------------
0,001293 г 1 ед. СГСЭ ион (электрон)
эрг рад
X 1,2602 • 10-12 —Г— . .——----------,
эв 100 эрг!г
где W — энергия новообразования для воздуха.
Если использовать величину 117=33,73 эв, то 1
Откуда 1 р=0,0258 W рад.
р=0,869 рад (в воздухе).
Температура 0° С и давление 760 мм рт. ст. — Прим, перев.
9
Доза в биологических эквивалентах рентгена (бэр) равна
дозе в радах, умноженной на некоторые коэффициенты, из ко-
торых наиболее важен коэффициент качества — КК. Коэффи-
циент качества равен отношению поглощенных доз двух раз-
личных видов излучения, которые требуется передать объекту,
чтобы вызвать один и тот же биологический эффект. Вместо
термина «коэффициент качества» используется также термин
«относительная биологическая эффективность», сокращенно
ОБЭ *.
Такая единица дозы, как физический эквивалент рентгена —
фэр, больше не употребляется (1 фэр равен поглощенной дозе
в эргах, полученной тканью нри экспозиционной дозе 1 р).
Существенное различие между единицами поглощенной и эк-
спозиционной доз зачастую не учитывается. Как мы увидим в
главе II, далеко не всегда удается легко перейти от экспози-
ционных доз к поглощенным.
1.4. РАДИОАКТИВНОСТЬ
Явление радиоактивности обусловлено распадом нестабиль-
ных ядер. Единицей радиоактивности служит кюри (1 кюри со-
ответствует 3,7 • 1010 распадам в секунду). Для радиоактивных
источников обычно указывают удельную активность и содержа-
ние радиоактивного вещества.
Любое ядро характеризуется определенным соотношением
числа протонов и нейтронов, при заметном отклонении от ко-
торого ядро оказывается нестабильным. Нестабильные ядра
претерпевают один или несколько видов распада и в конечном
счете превращаются в стабильные ядра.
Лишь немногие из естественных ядер способны к радиоак-
тивному распаду. При распаде ядра испускают либо а-, либо
g-частицы, и оба типа распада могут сопровождаться излуче-
нием у-квантов. Искусственные радиоактивные ядра могут рас-
падаться, испуская позитроны и у-кванты. Ядерпые реакции, та-
кие, как деление ядра, (у, п), (а, п) и (п, р), сопровождаются
вылетом протонов и нейтронов.
Среди продуктов радиоактивного распада ранее других были
исследованы а-частицы. Оказалось, что они представляют собой
дважды ионизованные атомы гелия. а-Частицы, испускаемые
определенным ядром, бывают либо моноэпергетическими, либо
имеют небольшой набор дискретных энергий. Радионуклид,
испускающий а-частицы различных энергий, испускает также
и у-кванты, энергия которых, как правило, коррелирует с энер-
гетическими различиями в спектре а-частиц.
* На самом деле между КК и ОБЭ существуют совершенно определен-
ные различия, связанные с характером рассматриваемой биологической ре-
акции, и если в ряде случаев КК и ОБЭ совпадают, то во многих случаях
эти характеристики заметно различаются. — Прим, перев.
10
у-Излучение — электромагнитное излучение высокой энергии,
испускаемое ядрами. Оно служит тем механизмом, с помощью
которого возбужденные ядра возвращаются в основное состоя-
ние. Энергия у-кваптов при радиоактивном распаде лежит где-
нибудь в диапазоне от 40 кэв до 4 Мэв.
Другим источником электромагнитной радиации является
тормозное излучение, которое возникает при ускорении или тор-
можении заряженных частиц в электрическом поле. Тормозное
излучение имеет обычно непрерывный энергетический спектр.
Примером такого излучения могут служить рентгеновские лучи,
возникающие при торможении быстрых электронов в электри-
ческом ноле атомных ядер.
При бомбардировке высокоэнергетическими электронами ме-
таллической мишени, например золота, возникает рентгеновское
излучение с максимальной энергией, равной максимальной энер-
гии электронов. Его спектр непрерывен и простирается от
энергий, близких к нулю, до энергии электронов. Таким обра-
зом, если говорится о 250-киловольтном рентгеновском излуче-
нии, то это означает, что использовался электронный источник
с напряжением 250 кв. Большая часть фотонов, возникающих в
рентгеновской трубке, имеет энергии гораздо меньшие, чем энер-
гия бомбардирующих электронов. Рентгеновское излучение с
низкой энергией принято называть длинноволновым, с высокой
энергией — коротковолновым.
Не следует считать, что в тормозное излучение превраща-
ется вся энергия быстрого электрона. Часть энергии затрачи-
вается на характеристическое рентгеновское излучение веще-
ства мишени, часть расходуется в других процессах. Эффектив-
ность превращения кинетической энергии в тормозное излуче-
ние зависит от энергии электрона и возрастает при увеличе-
нии электронной энергии и росте атомного номера мишени.
P-Частицы представляют собой электроны. Как правило, их
испускают ядра, в которых нейтрон-протонпое отношение выше,
чем в стабильных ядрах. Энергетический спектр р-излучения
непрерывен и простирается от нуля до максимальной энергии.
P-Излучение сопровождается испусканием второй частицы —
нейтрино, не несущей электрического заряда и обладающей мас-
сой меньше 0,05 массы электрона *. Сечение взаимодействия,
или, другими словами, вероятность реакции нейтрино с части-
цами вещества, настолько мала, что с точки зрения радиацион-
ной химии нейтрино интереса пе представляют.
Позитроны — положительно заряженные частицы с той же
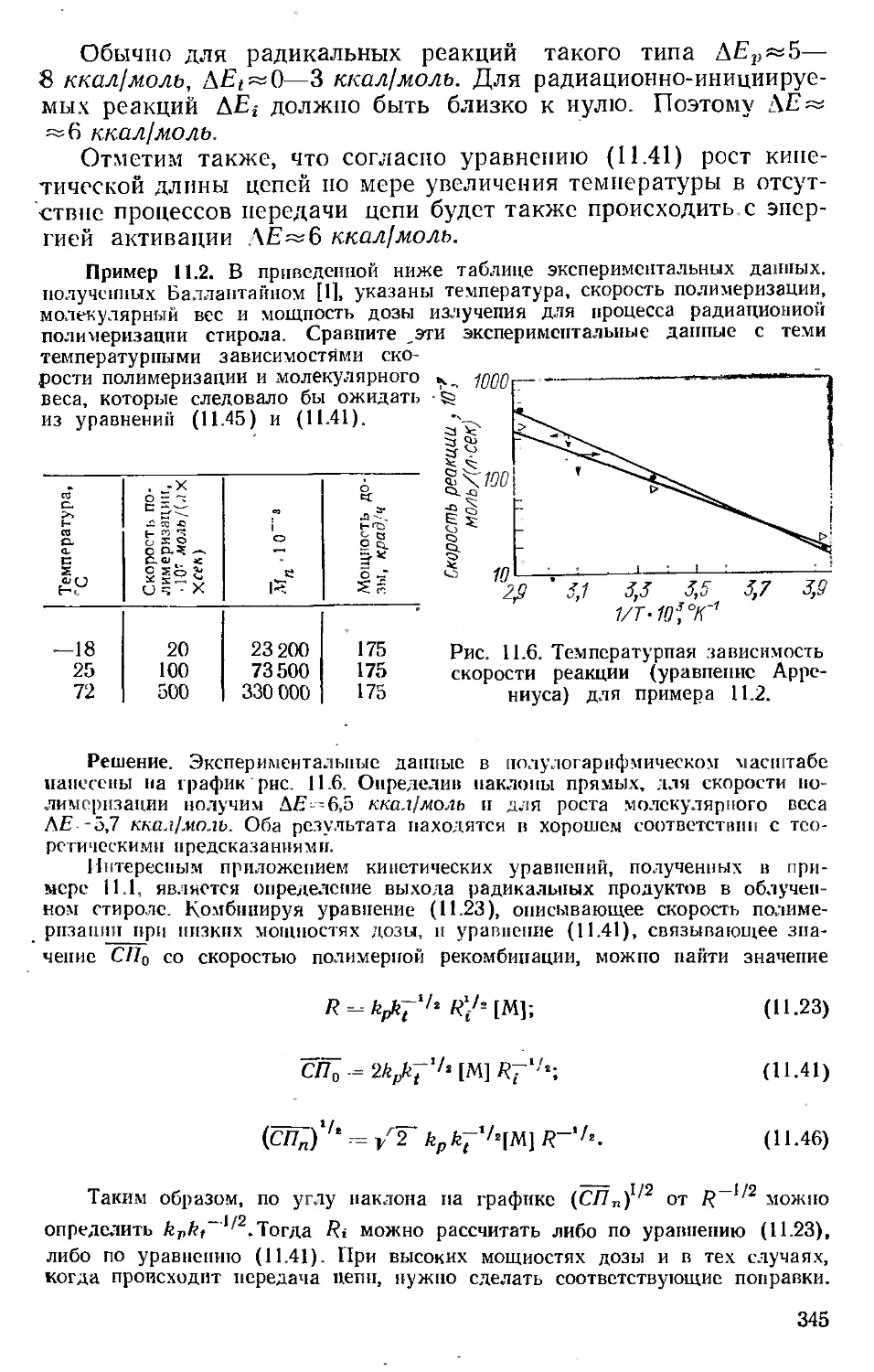
массой, что и электрон. Поэтому их можно рассматривать как
положительные р-частицы. Распад ядер с нейтрон-протонным
отношением, более низким, чем у стабильных ядер, сопровож-
* Сейчас установлено, что для любых видов нейтрино масса покоя
равна пулю. — Прим., перев.
И
дается либо испусканием позитронов, либо /С-захватом. (/(-за-
хватом называют такой физический процесс, при котором ядро
захватывает электрон из /(-оболочки.) Возможен захват элект-
рона также из L- и Л1-оболочек. Но так как /(-электроны в
среднем находятся заметно ближе к ядру, то вероятность захва-
та из /(-оболочки выше, чем из L и М.
Электронный /(-захват сопровождается рентгеновским излу-
чением, соответствующим переходу электронов внешних оболо-
чек на освободившееся место. Наблюдается полный рентгенов-
ский спектр атома, однако /(-линии наиболее интенсивны, /(-за-
хват часто сопровождается испусканием электронов Оже. Обыч-
но эффект Оже наблюдается в тех случаях, когда рентгенов-
ские лучи выбивают электрон одной из оболочек, например
/.-оболочки. Энергия выбитого электрона равна разности энер-
гий рентгеновского фотона и связи электрона в атоме. Электро-
ны Оже образуются также при у-излучепии возбужденных ядер.
Приведем некоторые примеры рассмотренных процессов:
а) испускание а-частиц
2^U ^He + ^Th; (1.12)
б) испускание Р-частиц
23940Th >е--| ^4Ра; (1.13)
в) испускание позитронов
I3N^e++I3C; (1<14)
г) Х-захват
fgCu + е~ > + Т-квант (с энергией 0,571 Мэв). (1-15)
1.5 ЯДЕРНЫЕ РЕАКЦИИ
При превращении ядер в ядерных реакциях, так же как и
в обычных химических реакциях, всегда происходит поглощение
или выделение энергии. Обозначения, используемые для записи
ядерных реакций, выглядят так:
ptHe-^0 4- JH-I-Q, (1.16)
где Q — энергия реакции. Значение Q можно определить, из-
мерив разницу между массой конечных продуктов и массой ис-
ходных ядер. Энергию мы найдем, подставив полученное зна-
чение в уравнение (1.8).
Поглощение или испускание частицы приводит к передаче
ядру какой-то кинетической энергии, величина которой зависит
от вида частицы и природы ядерной реакции. Очень часто этой
энергии вполне достаточно для разрыва химической связи. Воз-
никающие таким образом атомы отдачи принято называть «го-
рячими» атомами. Например, облучение CH3I медленными нейт-
12
ронами приводит к образованию горячих атомов ,281, которые
могут реагировать с другими молекулами CH3I, давая в каче-
стве продуктов реакции СН212 и HI.
Особый случай реакции горячих атомов—реакция Сциллар-
да — Чалмерса. При захвате тепловых нейтронов ядром проис-
ходит превращение (п, у); при этом энергия отдачи атома ока-
зывается достаточной для разрыва химических связей.
1.6. РАДИОАКТИВНЫЙ РАСПАД
Распад радиоактивных ядер часто бывает очень сложным
процессом. Однако к настоящему времени для большей части
радионуклидов выяснен характер распада, определены продук-
(период полураспада 10/к'М)
излучение (99,7%)
__]Ло (период полураспада 5,3года)
'\)3-излучение (0,31Мзд)
ц-излучение (1,17Нэб)
^-излучение (1,ззмз6)
--------------------
^Ls'.период полураспада ЗОлет)
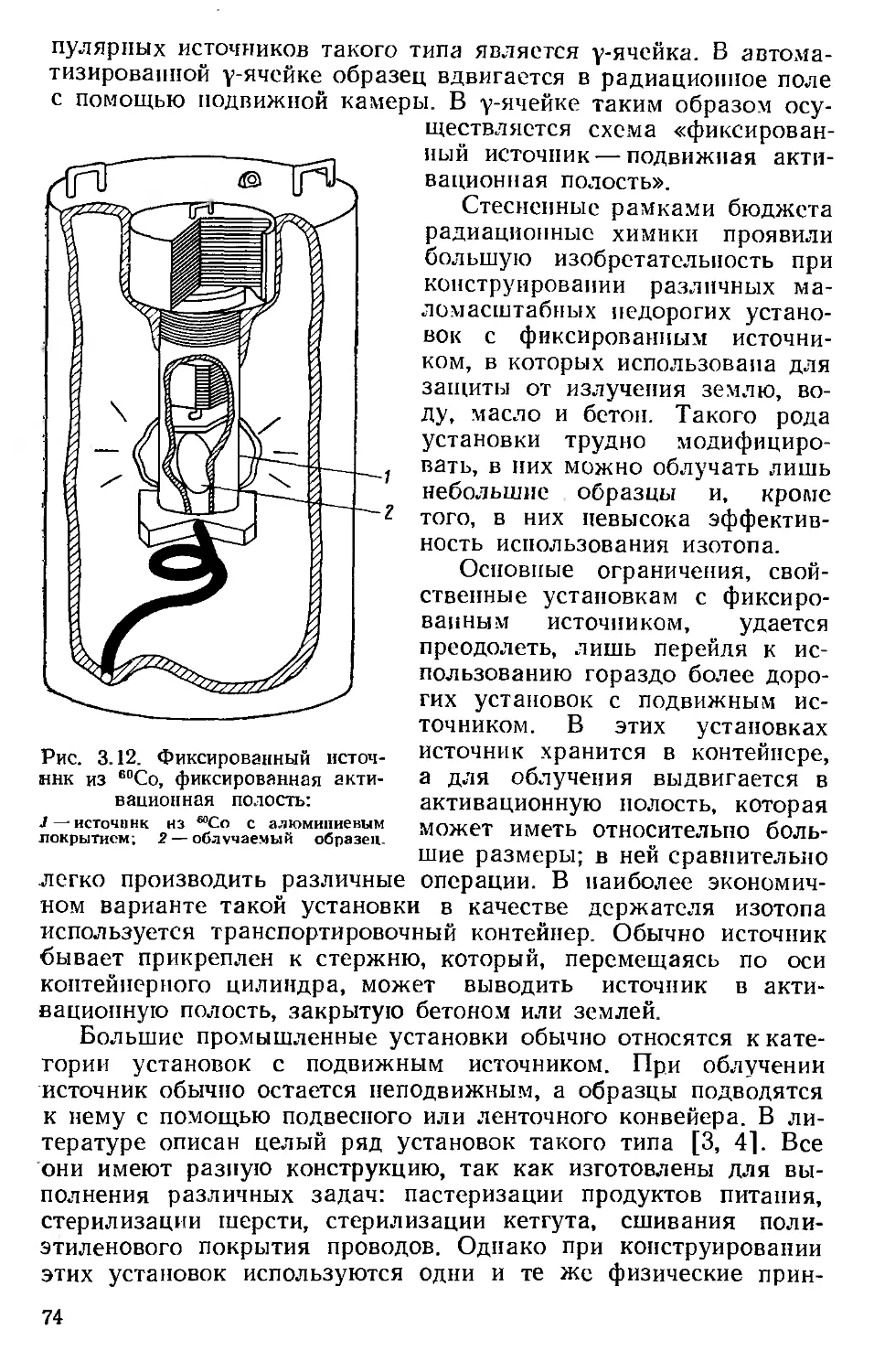
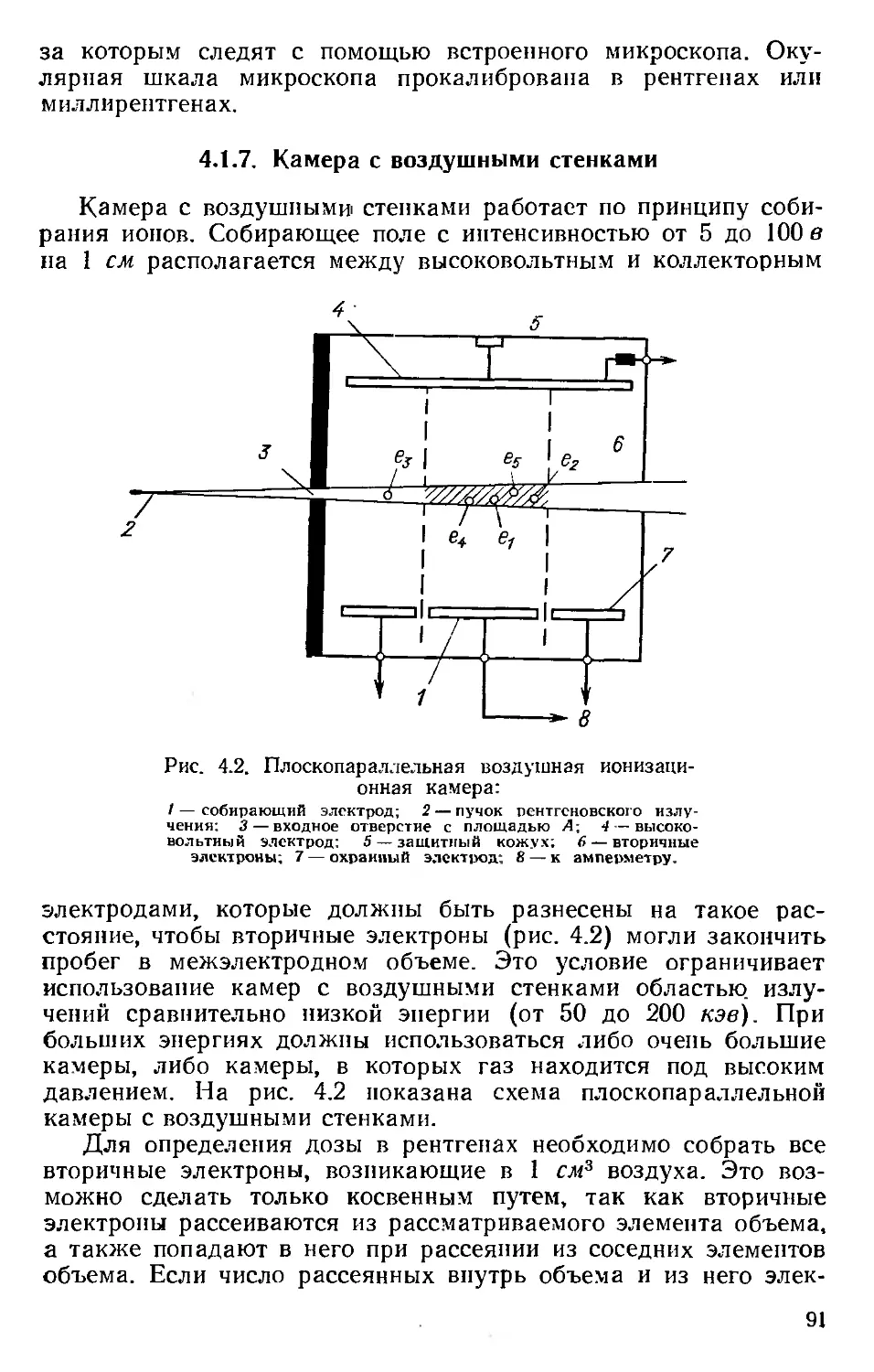
Рис. 1.1. Схемы радиоактивного распада 60Со (а) и ,37Cs (6). Для
0-частиц указана максимальная энергия.
ты радиоактивных превращений и измерены периоды полурас-
пада. Все эти данные можно найти в соответствующих справоч-
никах. Схемы распада для 60Со и 137Cs показаны на рис. 1.1.
Почти все вещества при облучении нейтронами становятся
радиоактивными. Все элементы при взаимодействии с тепловы-
ми нейтронами претерпевают реакцию активации (п, у). Могут
захватываться ядрами также и быстрые нейтроны. Сечения
взаимодействия для этих двух процессов обычно сильно отли-
чаются.
Сечение взаимодействия характеризует вероятность данного
процесса и имеет размерность площади. Для ядерных взаимо-
действий в качестве единицы сечения используют барн, который
равен 10-24 см2. Сечепия взаимодействия для реакций с быстры-
ми нейтронами обычно меньше 1 барн [1], а для реакций с
медленными нейтронами — 105 или 106 барн. Обычно значение
сечений указывается для конкретных процессов и энергий, на-
пример значение полного сечепия взаимодействия для поглоще-
ния нейтронов с энергией 10 Мэв или значение сечения захвата
медленных нейтронов данным ядром. Полное сечение взаимодей-
13
ствия для всех процессов с участием нейтронов характеризует
ослабление нейтронного пучка за счет любых типов реакций,
включая и поглощение и рассеяние. Уменьшение интенсивности
нейтронного пучка при прохождении через вещество мишени
дается выражением
— dn — nNcdx, (1.17)
где dn — ослабление пучка в слое толщиной dx-, п — число па-
дающих частиц; N — число ядер вещества мишени в 1 см3; о —
сечение взаимодействия, которое в данном случае является пол-
ным сечением.
Число падающих частиц задается потоком (плотностью по-
тока), который равен числу частиц или фотонов, входящих в-
данной точке в сферу с единичным сечением за единицу вре-
мени. Интенсивность излучения характеризуется энергией, по-
падающей в данной точке в сферу с единичным сечением за еди-
ницу времени. Единица интенсивности равна 1 эрг!(сек-см2)
или 1 вт[см2.
Статистический анализ радиоактивного распада показывает,
что средняя скорость распада пропорциональна числу имею-
щихся радиоактивных атомов N:
(1.18)
где X—постоянная распада, эквивалентная константе скорости
химической реакции в реакциях первого порядка.
Время жизни радиоактивного изотопа удобно характеризо-
вать периодом полураспада, т. е. временем, за которое распа-
дается половина первоначального количества атомов. Интегри-
рование уравнения (1.18) в пределах от /=0, N=N0 до /=
= td2, IV=’/2^0 Дает
1п2 = М.Л, Л/г = -^. (1.1»)
Л
Часто распадающийся изотоп превращается в другой неста-
бильный изотоп, который при распаде в свою очередь дает но-
вые нестабильные или стабильные изотопы [2]. Пусть изотоп
7V1 распадается, превращаясь в другой нестабильный изотоп
распад которого приводит к появлению стабильного изотопа
7V3. Тогда количество изотопа в любой момент времени мож-
но найти, проинтегрировав уравнение
^ = М\-Л2М2, (1.20)
at
где kjNj — скорость образования N2 из Nf, X2N2— скорость
распада N2-
14
Подставляя вместо Ni выражение Afie~Xi< интегрируя урав-
нение (1.20) в пределах от N2=0 при / = 0 до N2=^N2 iipin t—t,
получаем
Л12 = (е-М-е-'^). (1.21)
л2 —
Относительное содержание двух радионуклидов определяет-
ся соотношением периодов их полураспада.
Для и при t, гораздо меньших периода полураспада
JVb уравнение (1.21) превращается в
-Ь- (1 — е-М, (1.22)
а при t, значительно больших периода полураспада N2, пере-
ходит в
TV2Z2 - Nfa. (1-23)
Это уравнение векового или секулярного равновесия. Если
достигнуто состояние секулярного равновесия, то скорости рас-
пада материнского и дочернего изотопов оказываюстя одина-
ковыми. Нередко это приходится учитывать в радиационнохи-
мических исследованиях. Например, источник в 1 кюри 0-актив-
ного S0Sr, имеющего период полураспада 28 лет и превращаю-
щегося в 90Y (также ^-активный, но с периодом полураспада
всего лишь в 2,5 дня), через четыре периода полураспада 90Y
будет иметь дополнительную активность в 0,94 кюри "Y.
Зная элементарный состав образца и значение сечений взаи-
модействия, можно предсказать, какие радиоактивные изотопы
будут возникать при облучении нейтронным потоком, и рассчи-
тать конечные концентрации радионуклидов для не слишком
сложных систем. Концентрацию данного радиоактивного изото-
па можно определить, зная сечение образования изотопа о,
плотность нейтронного потока ф и концентрацию исходного
изотопа No. Скорость образования изотопа будет равна
-= оФЛ',, — kN R — kN. (1.24)
В случае многоатомных веществ уравнение (1.24) должно
применяться для расчета активности каждого изотопа в отдель-
ности. Если необходимо учесть превращение данного нуклида
в другой радионуклид, то следует использовать уравнение (1.20).
Пример 1.3. Найти полную активность образца кобальта, облученного
потоком медленных нейтронов с плотностью 1012 нейтрон](см2-сек) в тече-
ние 6 месяцев. Сечение образования 60Со из 59Со равно 21,7 барн, период
полураспада юСо составляет 5,26 года *.
* Процесс образования изомера с периодом полураспада 10 мин не учи-
тывается, так как более 99% этого изомера претерпевают переход в изомер
с периодом полураспада 5,26 года.
15
10>2 8,9 г
см2 • сек см3
(1 _e-0.693/5i26);
Решение.
dN
----- = g(IiN0 — kN.
dt
Интегрируя это уравнение в пределах от N=0 при t=0 до N=N при t=t.
Решая относительно N, получаем
А (1 _ e-W) =21,7- 10-м см2
1 г моль 6,02 • 1023 молекул
59 г г моль
N = 8,82 - 1018 атом/см3.
Активность в кюри, соответствующую такой концентрации атомов “Со
в 1 см3, получим следующим образом:
dN
Скорость распада —-----= kN.
dt
Чтобы найти к, используем уравнение (1.19):
0,693 4,2 10-»
к —---------------------------- =--------------;
5,26 (года) • 365 • 24 3600 сек
dN 4,2 10_9 „ атом 1 см3 „ атом
—-------.-=—---------- 8,82 101® ---- •-------= 4,11 • 101°----
dt сек см3 9 г сек-г
Тогда активность в кюри равна
4,11 1010 кюри
-----------«1,10--------- число граммов.
3,7 • 10ю г
Значение 1,10 кюри/г представляет собой удельную активность образца.
Задачи
1.1. Используя величину заряда электрона, показать, что 1 эв=
= 1,6-10-12 эрг.
1.2. Чему равны характеристические длины волн для у-квантов с энер-
гией 1,1'7 и 1,33 Мэв, испускаемых “Со?
1.3. Протон обладает кинетической энергией 50 Мэе. Чему равна его
скорость?
1.4. Электрон движется со скоростью 0,95 сек. Вычислить его кинетиче-
скую энергию с учетом и без учета релятивистской поправки.
1.5. Период полураспада “Со равен 5,26 года. Чему равна постоянная
распада? Если 1 января 1969 г. источник имел активность 10 000 кюри, то
чему она равна сегодня? Какая доля “Со распадается в течение месяца?
1.6. Активность изотопа с периодом полураспада 2 мин определяется с
помощью счета импульсов в течение 5 мин. На какой коэффициент нужно
умножить среднюю скорость счета, чтобы получить начальную активность
изотопа?
1.7. Кусок серебра толщиной 3 см и плотностью 2,7 г/см3, помещенный
в поток нейтронов, уменьшает скорость счета нейтронов в М раз. Чему
равно полное сечение взаимодействия серебра с нейтронами?
1.8. Золотая фольга толщиной 0,02 см и площадью 1 см2 облучается
нейтронами с плотностью потока 1012 нейтрон)(см2-сек). Плотность золота
19,3 г)см3, а сечение взаимодействия с тепловыми нейтронами равно
98,7 барн. Чему равна полная активность в кюри радиоактивного золота 198Аи,
получаемая в течение 1 ч, 1 дня, 1 недели и 1 года? Период полураспада
198Аи равен 2,69 дня.
16
Глава II
взаимодействие излучения с веществом
Три вида излучений наиболее существенны для радиаци-
онной химии. Это заряженные частицы (электроны, протоны,
а-частицы, продукты деления и т. д.), фотоны (рентгеновское и
у-излучение) и нейтроны. Для каждого излучения характерен
свой способ взаимодействия с веществом. Например, заряжен-
ные частицы в воздухе имеют определенный максимальный про-
бег, тогда как у нейтронов и электромагнитного излучения оп-
ределенного пробега не существует. В этой главе рассмотрены
разные способы взаимодействия излучения с веществом.
2.1. ЗАРЯЖЕННЫЕ ЧАСТИЦЫ
Частицы взаимодействуют с веществом либо в процессах
рассеяния, либо в процессах ядерного захвата. Рассеянием на-
зывают такое взаимодействие частицы (или частиц) с вещест-
вом, при котором сама частица не исчезает. Существует два
основных вида рассеяния — упругое и иеупругое. При упругом
рассеянии налетающая частица при любом угле рассеяния не
теряет своей энергии. При неупругом рассеянии * налетающая
частица теряет энергию.
Существуют два основных механизма неупругого рассеяния
в результате столкновения: 1) смещение атомов и 2) возбужде-
ние и ионизация атомов и молекул.
В определенных условиях, если рассеяние происходит под
большим углом, частицы тормозятся; при этом испускаются
кванты электромагнитного излучения, известного как тормозное.
Хотя расходовать энергию на тормозное излучение в неупругих
соударениях могут любые заряженные частицы, относительный
вклад этих механизмов потери энергии различен для электро-
нов и тяжелых заряженных частиц (протонов, а-частиц, дейт-
* Строго говоря, любое рассеяние заряженных частиц является неупру-
гим, исключая лишь столкновение свободных частиц (если не учитывать при
этом тормозного излучения).
Spliner 17
ропов и т. д.). Тормозное излучение — важный механизм поте-
ри энергии электронных пучков как раз того энергетического
диапазона, который наиболее интересен для специалиста по
радиационной химии. Для ос-частиц такой механизм потери
энергии становится существенным лишь для энергий около
миллиарда электронвольт.
Рассмотренные способы взаимодействия справедливы не
только для первичных частиц, но и для высокоэнергетических
электронов, выбиваемых при ионизации. Такие вторичные элект-
роны высокой энергии называют 6-излучением.
2.1.1. Тяжелые заряженные частицы
Потери энергии при неупругих соударениях со связанными
электронами. Движущаяся через вещество заряженная частица
электрон
г
----------------------------------— х
Траектория заряженной настицы
Рис. 2.1. Взаимодействие движущейся
заряженной частицы с массой М и
зарядом Ze и покоящегося электро-
на с массой то. Заряженная частица
пролетает со скоростью V на рассто-
янии г от электрона. Время взаимо-
действия частицы и электрона при-
нимается равным (2г/У).
теряет энергию при электро-
магнитном взаимодействии с
электронами вещества, т. е. со
связанными электронами. В ре-
зультате взаимодействия про-
исходят возбуждение и иони-
зация атомов и молекул, на
что расходуется кинетическая
энергия падающей частицы.
Расстояние, которое частица
успевает пройти до того, как
ее кинетическая энергия станет
равной нулю, называют пробе-
гом частицы. Ниже дается вы-
вод * приближенного выраже-
ния для скорости потери энергии на единицу длины пути
Пусть заряженная частица с массой М, зарядом Ze (Z —
атомный помер, а е — единичный элементарный заряд) проле-
тает со скоростью V на расстоянии г от покоящегося электрона
с массой т0 (рис. 2.1).
Предположим, что электрон не связан и при столкновении
не смещается. Тогда силу, действующую на электрон, можно
рассчитать для точки, в которой он первоначально находится.
Кулоновская сила, действующая на электрон, равна
Ze2
г2 ’
(2.1)
* Этот вывод принадлежит Н. Бору.
18
Импульс р, полученный электроном в направлении, перпенди-
кулярном к линии полета частицы, определяется уравнением:
Р = (кулоновская сила) • (продолжительность столкновиния) =
Энергия, соответствующая этому импульсу и приобретаемая
электроном, равна
р^= 2гУ . (2.3)
2/;г0 /п01/72
Наоборот, частица при каждом столкновении теряет энергию
[см. уравнение (2.3)]. Поэтому, чтобы вычислить потери энер-
гии на единицу длины пути частицы, нужно подсчитать число
соударений на единицу длины пути. Для этого прежде всего
нужно определить вероятность соударения.
Вероятность соударения с электронами, находящимися па
расстоянии от г до r+dr, пропорциональна площади 2nrdr. Эта
площадь составляет некоторую долю от общей площади А:
А = л г2. (2.4)
Вероятность соударения будет равна
2rvdr __ 2rdr
nr'1 г2
(2.5>
Если N — число электронов в 1 см3, то число соударений на еди-
ницу длины пути будет равно числу электронов на единицу дли-
ны пути, умноженному на вероятность соударения:
Число соударений = (nr2Ndx) (= 2nNrdrdx. (2.6)
\ г2 /
Потери энергии па единицу длины пути получим, умножив урав-
нение (2.6) на уравнение (2.3):
dE kiZWN dr 9
dx ~ г ' ’
Если rmax и rmin — максимальное и минимальное расстояния, на
которых происходят соударения, то интегрирование уравнения-
(2.7) дает
dE 4nZ2e4V । гтах
dx ffl0V2 rmin
(2.8)'
Искомое общее выражение для потерь энергии заряженной
частицы дается уравнением (2.8).
Исходя из классических представлений и приняв во внима-
ние лишь квантование энергетических уровней атомов, Н. Бор-
получил выражения для гтах и rmin. Эти выражения, однако,
19
оказались неудовлетворительными, так как при их выводе нс
были учтены волновые свойства вещества.
Минимальное расстояние соударения гтщ должно быть рав-
но половине дебройлевской длины волны X/2=/i/2 rnoF=rmin-
Максимальное расстояние соударения пропорционально скоро-
сти падающей частицы V и обратно пропорционально потенциа-
лу возбуждения I, причем константой пропорциональности яв-
ляется постоянная Планка h; rmax=hV/l. Таким образом:
rmax hVjl 2т0У'г £
rmin /г/2т0У I
Подставляя уравнение (2.9) в уравнение (2.8), получаем
_ dE = 4nZ2e'1N in ^qV'1 (% 10)
торг 1
Потенциал возбуждения I представляет собой геометрическое
среднее всех потенциалов возбуждения и ионизации атомов и
молекул среды.
Точный квантовомеханический подсчет потерь энергии, про-
деланный Бете и Ашкином [ 1], в котором принимаются во вни-
мание и релятивистские эффекты, дает выражение, сходное с
выражением (2.10):
<1Е _ 4.TZ2e4N in 2m0V2 ___„21 (2 11)
dx m0V2 L /(1—₽2) ]
Здесь p=V/c. Множитель 2 в числителе логарифмического
члена относится к тяжелым заряженным частицам (а-частицы).
Для электронов его следует заменить единицей.
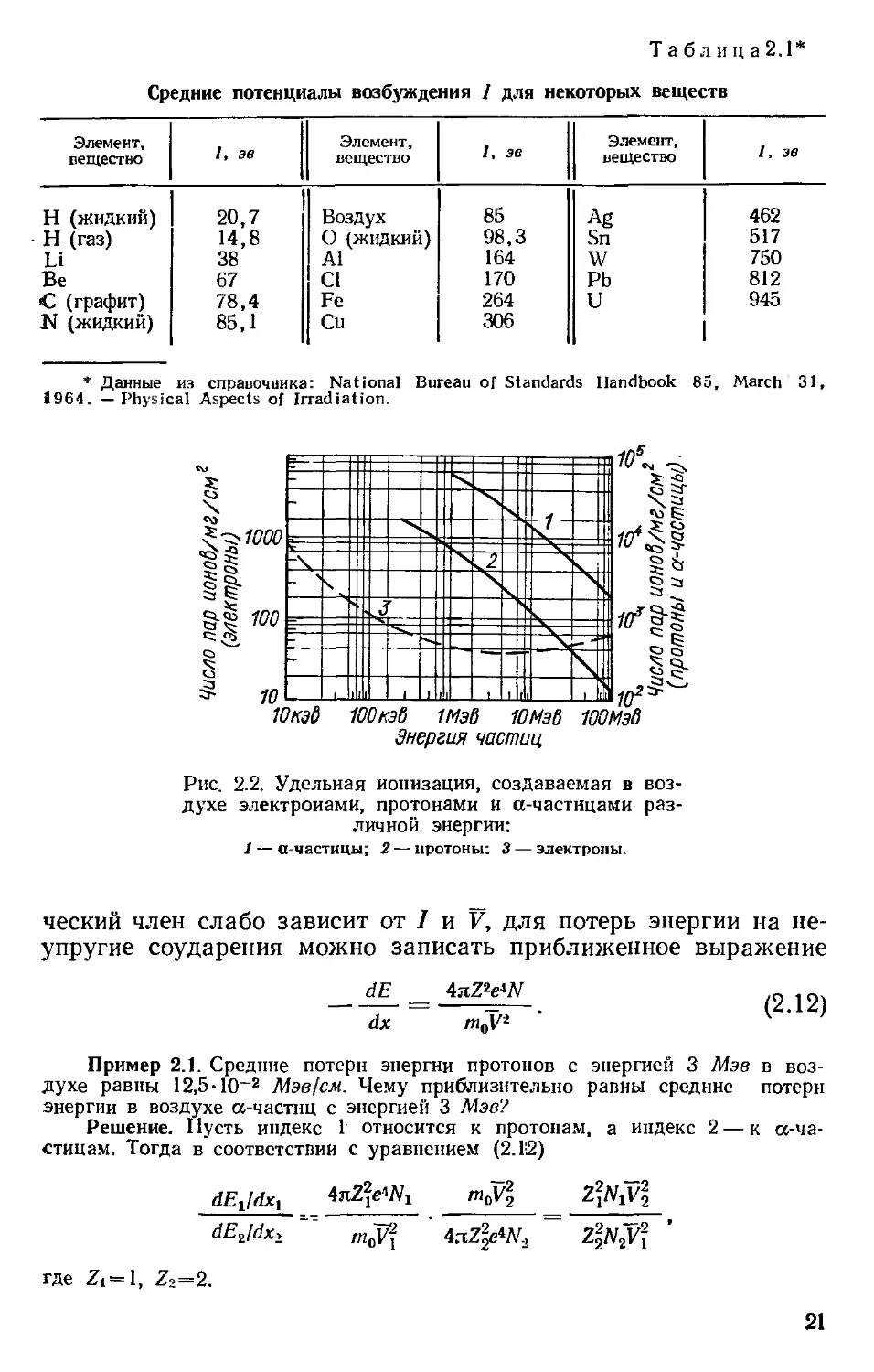
Уравнение (2.10) позволяет объяснить ход кривых на рис. 2.2.
Отметим, что dE/dx (выраженное через^ число образующихся
пар ионов) возрастает при уменьшении V.
В уравнения (2.10) и (2.11) нетрудно подставить значения
всех параметров, кроме I — потенциала возбуждения. В общем
случае величину I определяют экспериментально. Измеряют
энергию, потерянную в данном поглотителе, а затем находят
такое I, чтобы совпали расчетные и экспериментальные значе-
ния потерь. В табл. 2.1 представлены значения / для некоторых
веществ.
Уравнения (2.10) и (2.11) позволяют сформулировать неко-
торые приближенные закономерности. Например, в уравнения
входят только две характеристики падающей частицы — Z и V.
Поэтому можно считать, что две частицы с одинаковыми заря-
дами характеризуются потерями энергии, обратно пропорцио-
нальными квардату их скоростей. А две частицы, движущиеся
с одинаковой скоростью, имеют пробеги, обратно пропорцио-
нальные квадрату их зарядов. Более того, так как логарифми-
20
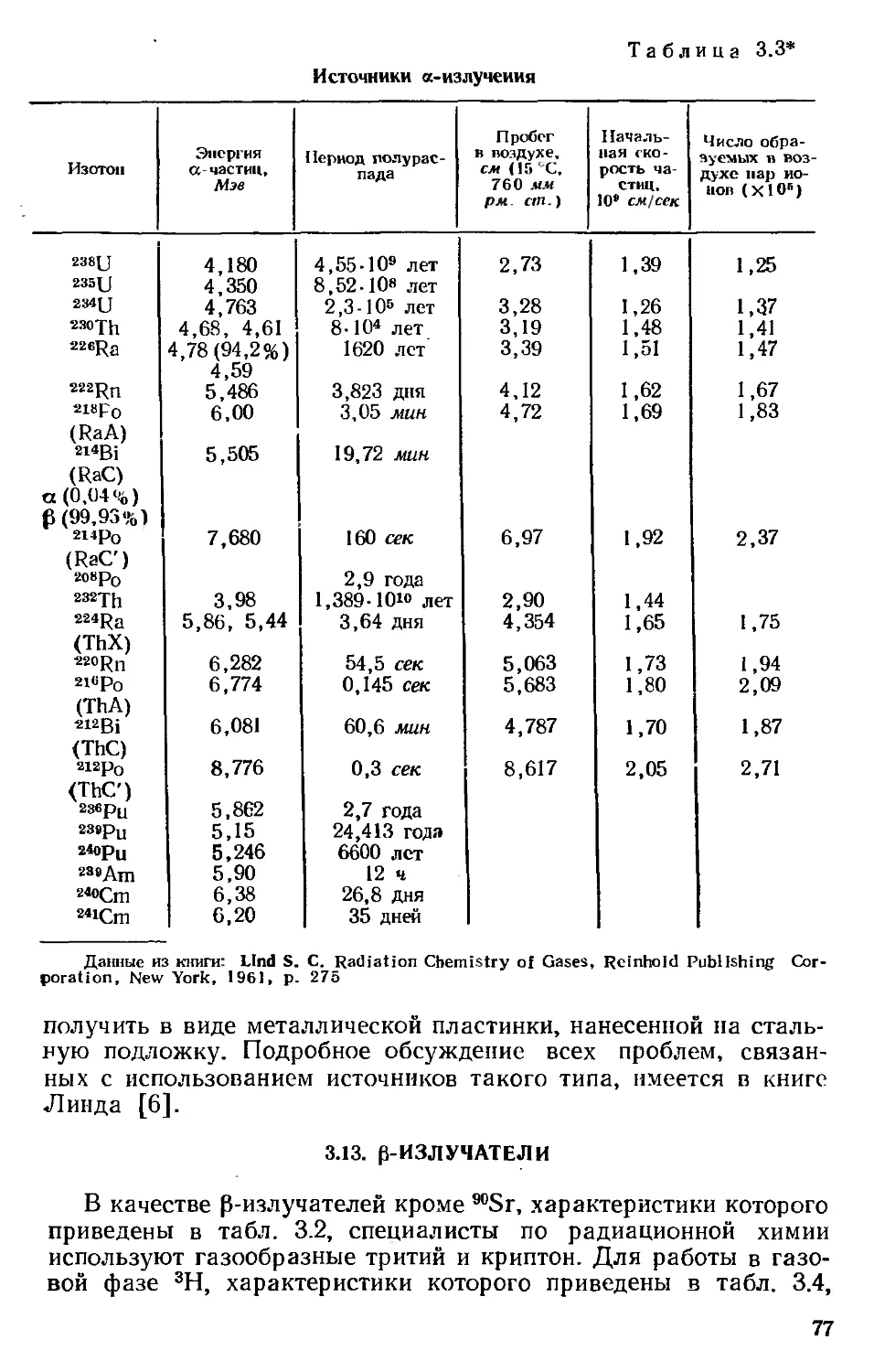
Та бл иц a 2.1
Средние потенциалы возбуждения 1 для некоторых веществ
Элемент, вещество /, эв Элемент, вещество /, эв Элемент, вещество /, эв
Н (жидкий) 20,7 Воздух 85 Ag 462
Н (газ) 14,8 О (жидкий) 98,3 Sn 517
Li 38 А1 164 W 750
Be 67 С1 170 Pb 812
С (графит) 78,4 Fe 264 и 945
N (жидкий) 85,1 Си 306
* Данные из справочника: National
1964. — Physical Aspects of Irradiation.
Bureau of Standards Handbook
85, March
31,
Рис. 2.2. Удельная ионизация, создаваемая в воз-
духе электронами, протонами и а-частнцами раз-
личной энергии:
1 — а-частицы; 2 — протоны: 3 — электроны.
ческий член слабо зависит от I и V, для потерь энергии на не-
упругие соударения можно записать приближенное выражение
dE _ 4aZ2eW
dx ~
(2.12)
Пример 2.1. Средние потерн энергии протонов с энергией 3 Мэв в воз-
духе равны 12,5-10~2 Мэв/см. Чему приблизительно равны средние потерн
энергии в воздухе «-частиц с энергией 3 Мэв?
Решение. Пусть индекс 1 относится к протонам, а индекс 2 — к «-ча-
стицам. Тогда в соответствии с уравнением (2.1!2)
dE1/dxl 4jiZ^e'lN1 ei0V2 Z^N^V?
dE2/dx> ' r/loy2 4aZ^eW2 Z|/V2Vf
где Zi = l, Z3=2.
21
Найдем теперь отношение скоростей Vq/Vj. Так как Е — и Е\ = Е?—
— 3 Мэи, а пц = 1 и ni? — 4, то Vj =6, а =1,5. Отсюда
(4) (6)
— dEjdx, = (12,5 • 10—2) ’ 2 Мэв!см.
(1) (1,5)
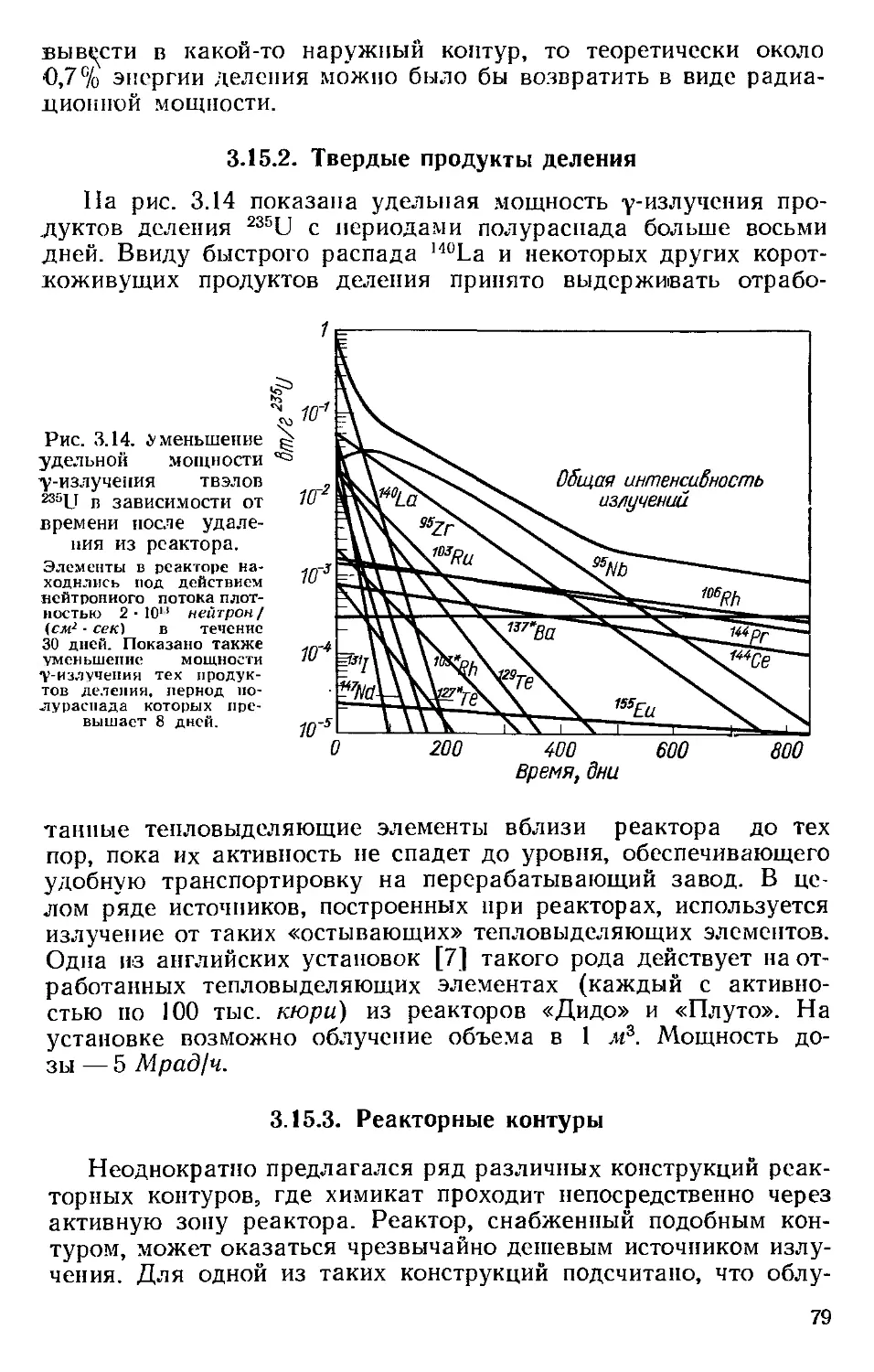
Пробег заряженных частиц. Пробег частицы R можно опре-
делить из следующего соотношения:
Ео
м
о
где Ео — начальная энергия частицы. Интегрирование выполня-
ется вдоль всего пути частицы.
Если вместо dE в уравнение (2.13) подставить результат
дифференцирования Е = тЁ2/2,
—dE/dx подставить уравнение
будет выглядеть так:
dE
— dE/dx
(2.13)
т. е. mVdV, и если вместо
(2.10), то формула для пробега
О
тт0 1
4nZ2e<W J
VsdV
In (2m0V3/7)
(2-14)
трудно использовать. Целесо-
(2-15)
В этом виде уравнение (2.14)
образно сравнивать пробег данной частицы с пробегом другой
частицы, движущейся в данной среде с той же скоростью. На-
пример, обозначив разные частицы индексами 1 и 2 (и заме-
тив, что интегралы в выражении (2.13) для двух частиц с оди-
наковыми скоростями одинаковы), получим
т?! nii ^2
Т?2 т2 /2
откуда легко определить неизвестное значение пробега по из-
вестному. Это выражение применимо для энергии частиц боль-
ше 0,2 Мэв.
Чтобы воспользоваться уравнением (2.15) на практике, не-
обходимо знать пробег стандартной частицы в стандартной сре-
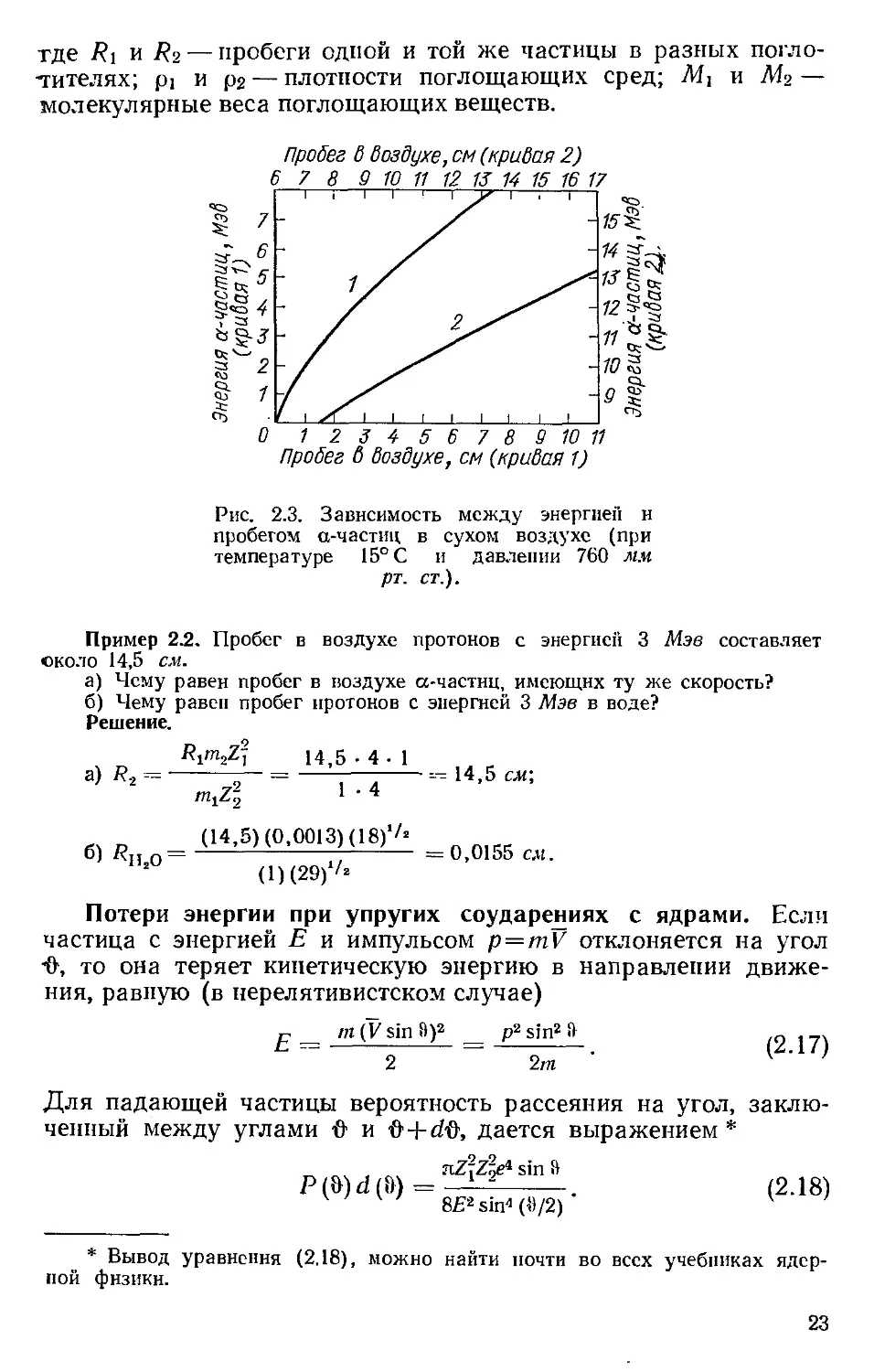
де. В качестве стандартных частиц чаще всего используют
а-частицы, а в качестве стандартной среды — воздух. На
рис. 2.3 показан пробег а-частиц в воздухе в зависимости от
их энергии [2].
Для различных поглощающих сред пробег зависит от их со-
става. Чтобы сравнить пробеги в разных поглощающих средах,
нужно воспользоваться эмпирическим соотношением, основан-
ным на правиле Брегга — Клеемана:
Ri _ Р<№)1/г
^2 Р1 (М2)1/2
(2-16)
22
тде Ri и R2— пробеги одной и той же частицы в разных погло-
тителях; pi и р2 •—плотности поглощающих сред; и М2—
молекулярные веса поглощающих веществ.
Пробег в воздухе, см (кривая 2)
6 7 8 9 Ю 11 12 13 14 15 16 17
Пробег в воздухе, см (кривая 1)
Рис. 2.3. Зависимость между энергией и
пробегом а-частиц в сухом воздухе (при
температуре 15° С и давлении 760 мм
рт. ст.).
Пример 2.2. Пробег в воздухе протонов с энергией 3 Мэв составляет
около 14,5 см.
а) Чему равен пробег в воздухе а-частнц, имеющих ту же скорость?
б) Чему равен пробег протонов с энергией 3 Мэв в воде?
Решение.
Rpn-Z) 14,5 -4-1
a) R-z = ----у- =------:— -----=- 14,5 см;
mjZ; 1 • 4
о о _ <14-5) (0.0013) (18)'^ _
о) г) — — 0,0155 см.
2 (1)(29)*^
Потери энергии при упругих соударениях с ядрами. Если
частица с энергией Е и импульсом p = mV отклоняется на угол
•0, то она теряет кинетическую энергию в направлении движе-
ния, равную (в нерелятивистском случае)
р _ m (V sin S)2 _ р2 sin2 Я „ .
2 2m ’ ' ’ '
Для падающей частицы вероятность рассеяния на угол, заклю-
ченный между углами (1 и О+М, дается выражением *
Р (&)</(&)
sin Я
8£2sin^ (8/2) '
(2-18)
* Вывод уравнения (2.18), можно найти почти во всех учебниках ядер-
ной физики.
23
Потерю энергии АЕ, усредненную по всем направлениям
рассеяния, получим, умножив уравнение (2.17) на уравнение
(2.18) и проинтегрировав по всем возможным углам рассеяния:
— ДЁ = fAE(ll)P(&)d&=- (2.19)
J v v 2m 8£2 J sin* (0/2)
Максимальный угол 0-=n, на который отклоняется падаю-
щая частица, следует использовать в качестве верхнего преде-
ла интегрирования. Нижний предел интегрирования обозначим
fl-min- Интегрирование уравнения (2.19) в этих пределах дает
— А£
р2
~2т ' Е2
2
^min
' min
(2.20)
Минимальный угол рассеяния О-щш определяется наимень-
шим расстоянием, па которое частица, движущаяся со скоро-
стью V, может приблизиться к ядру с зарядом Z. Эксперимен-
тальные и теоретические данные свидетельствуют в пользу то-
го, что это расстояние, с одной стороны, зависит от размеров и
заряда ядра, с другой стороны, от скорости и заряда падающей
частицы. Приближенно это расстояние можно считать равным
размеру ядра (2-10~12 см). В этом случае Отт для легких эле-
ментов равен около 10-6 рад. Поэтому в уравнении (2.20) чле-
нами '&2mm/4 и —1 можно пренебречь по сравнению с
1п (г/О-пип):
пг nZ2Zoe4
\Е = • --!—£_
2т Е2
(2.20а)
Потери энергии на рассеяние на отрезке dx получим, умножив
правую часть уравнения (2.20а) на N\dx, где N\ — число ядер
в 1 см3:
2nZ\Z2^N}
£
dE
dx
ln(2/&mIn).
(2.206)
Здесь p2/2 m мы заменили на Е.
Пример 2.3. Сравните относительную долю потерь энергии на столкно-
вения с ядрами (рассеяние) и на непругие соударения со связанными элект-
ронами для а-частиц с энергией 4 Мэв н для продуктов деления, таких, как
например, 90Sr, если те и другие движутся в углеродной среде.
Решение. Отношение потерь энергии на рассеяние к потерям энергии
на неуиругне соударения с электронами dEe/dEc дается отношением урав-
нений (2.206) и (2.10).
dEs ZnZfZ^N! 2
— ——— ----------In —---
dx Е»'miii
dEc 4nZ2e*N 2mBV2
dx m^V2 1П I
24
Подставляя следующие значения: <f'min=8-10-5, Zi=2, /~80, V=l,40x
X10s см/сек, ni0=9,ll-10-28 г, Z2=6, /V=3,01 • 1Ю23 электрон/г, /V|=0,5x
ХЮ23 ядер!г; Е=4 и 2(2) (9,11 • Ю"28) (1,40-109)2(6,24-105) =22,ЗХ
XНО-4 Мэв и исключая Zi, л и е4, получаем:
dEs = [(2) (6)2 (0,5) (10-*з)/4| In (2/8 10~8) = g i()_g
dEc [(4) (3,01) (10—2S)/(11,15 • 10~4)]/я (2230/80)
Отсюда следует, что потери энергии при рассеянии на ядрах для а-частиц
составляют меньше 1 % потерь энергии при неупругих соударениях с элект-
ронами.
Если же падающей частицей является ион стронция, то в уравнения
нужно подставить следующие значения:
»min = 1.7- 10-3, z2 = 6, / и 80, m0 = 9,ll • 10-23 г,
V = 2,93 • 108 см]сек, Zls = 39, Z1<; = 5
и
2m0V2 = (2) (9,11 • IQ-28) (2,93 . Юв)2 (6,24 - 106) = 9,75.10-в /Изе
dEs [(2)(39)2(6)2(0,5)(10-2з)/4]/я(2/1,7- 10~3) q
dEc [4(10)2(3,01)(10-23)/(4.88-10-5)]/я(97,5/80)
Теперь значительная доля потерь энергии (8,5% от ионизационных потерь)
приходится на упругое рассеяние. Выбор заряда продукта деления Z = 5
(для 90Sr) несколько произволен, так как эффективный заряд 90Sr может
варьироваться от 20 до 0.
2.1.2. Электроны
Взаимодействие электронов с веществом осуществляется те-
ми же способами, что и взаимодействие тяжелых заряженных
частиц. Электроны тратят энергию на неупругие и упругие со-
ударения, а также па тормозное излучение.
Однако относительная роль этих процессов для электронов
и для тяжелых заряженных частиц совершенно различна. Глав-
ным образом это обусловлено небольшой массой электрона.
При высоких энергиях электронов наблюдаются потери па тор-
мозное излучение. При меньших энергиях доминирующими ста-
новятся неупругие соударения с электронами поглотителя.
Потери энергии на неупругих соударениях с электронами.
Для электрон-электронпых взаимодействий можно использовать
уравнение (2.10), полученное для тяжелых частиц, если его мо-
дифицировать, учитывая меньшую массу системы, а также то
обстоятельство, что после столкновения невозможно различить
налетающий и выбитый электроны. Например, если вся энергия
налетающего электрона передается выбитому, то никаких из-
менений в системе не произойдет. В результате логарифмиче-
ский член в уравнении (2.10) следует умножить на (е/2)|/2, где
е — основание натуральных логарифмов:
, /^Х
dx m0V2 \ 1 ) У 2 V ’
25
Для электронов с энергией более 0,5 Мэв следует ввести ре-
лятивистские поправки. В этом случае выражение выглядит так::
dE
dx
moV2£
т0У* 1 2/-(1-Р)2
— [2(1 ——(1 [-₽)] 1П2
о
(2.22)
В этом уравнении не учитываются два процесса, существен-
ные при высоких энергиях: 1) эффект поляризации в конденси-
рованной среде (плотностной эффект Ферми) и 2) тормозное
излучение.
Рис. 2.4. Скорость потери энергии электронами в веществе:
/ — вода; 2 — воздух; 3 — графит; 4 — вода (с поправкой на поляри-
зацию).
Значения —dEfdx, полученные согласно уравнению (2.22),
приведены па рис. 2.4 (кривые потерь на соударения). Срав-
нение уравнений (2.21) и (2.22) показывает, что учет реляти-
вистских эффектов приводит к возрастанию —dEfdx в области
высоких энергий. Вследствие этого в области 1—2 Мэв на кри-
вых появляется минимум.
Потери энергии на тормозное излучение. Если частица с за-
рядом Z] и массой т проходит вблизи ядра с зарядом Z2, то
между ядром и частицей действует кулоновская сила, пропор-
циональная ZiZ2, которая тормозит частицу. В соответствии с
классическими представлениями частица будет излучать элект-
ромагнитную энергию, пропорциональную квадрату ускорения.
26
Точный расчет радиационных потерь энергии выполняется
квантовомехапическими методами и выходит за рамки этой
книги. Однако можно пользоваться уравнением (2.23), которое
дает соотношение между радиационными потерями и потерями
энергии на пеупругие соударения:
(— ^£Мх)рад EZ.y
(2.23)
(—^^/^)нсупр 800
где Е выражается в Мэв и Z> — атомный номер поглотителя.
Энергетические потери на тормозное излучение существенны
только для электронов с высокой
энергией и для больших значений
Z поглотителя (см. рис. 2.4).
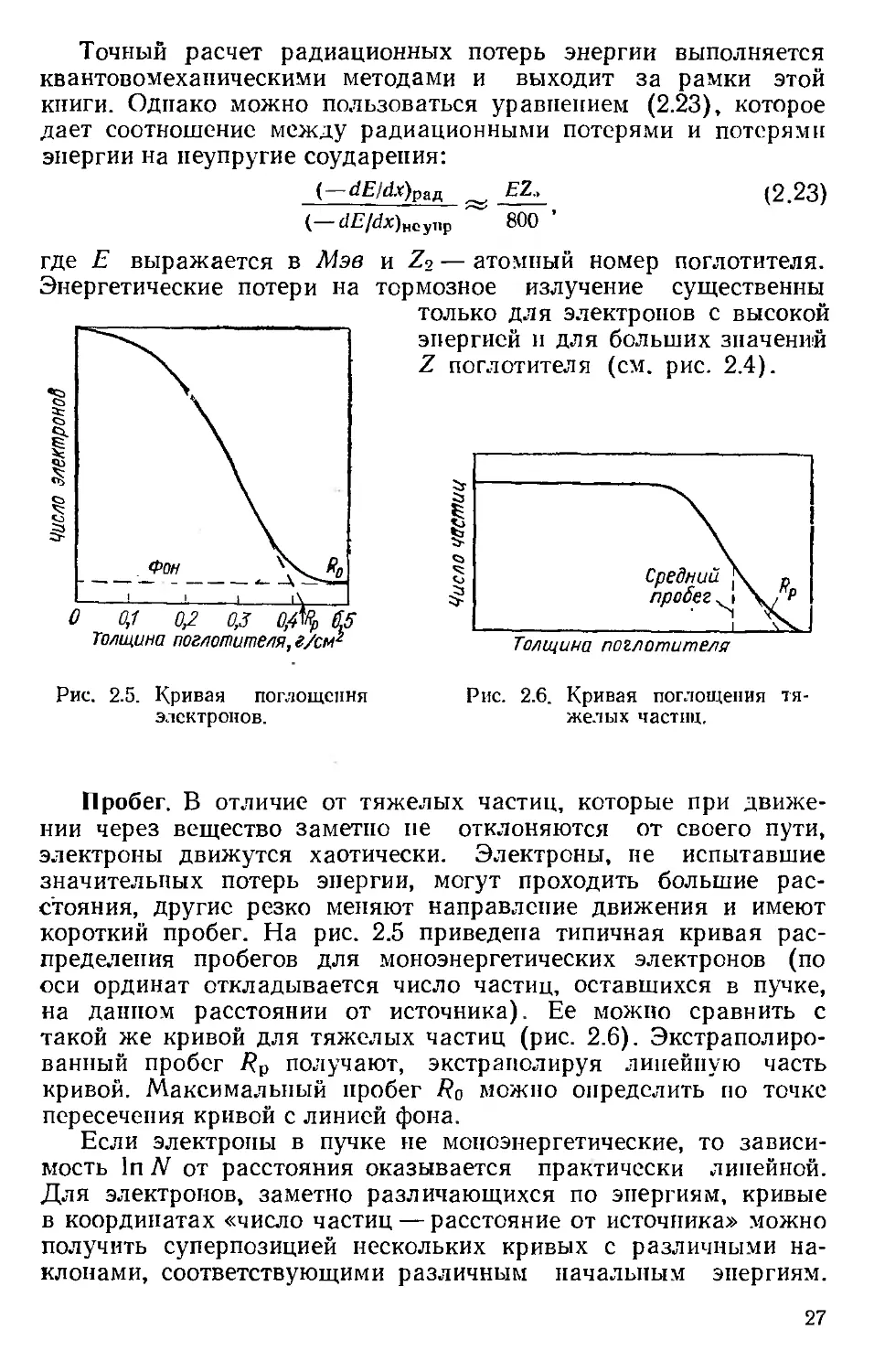
Рис. 2.5. Кривая поглощения
электронов.
Рис. 2.6. Кривая поглощения тя-
желых частиц.
Пробег. В отличие от тяжелых частиц, которые при движе-
нии через вещество заметно не отклоняются от своего пути,
электроны движутся хаотически. Электроны, не испытавшие
значительных потерь энергии, могут проходить большие рас-
стояния, другие резко меняют направление движения и имеют
короткий пробег. На рис. 2.5 приведена типичная кривая рас-
пределения пробегов для моноэнергетических электронов (по
оси ординат откладывается число частиц, оставшихся в пучке,
на данном расстоянии от источника). Ее можно сравнить с
такой же кривой для тяжелых частиц (рис. 2.6). Экстраполиро-
ванный пробег /?р получают, экстраполируя линейную часть
кривой. Максимальный пробег Ro можно определить по точке
пересечения кривой с линией фона.
Если электроны в пучке не моноэнергетические, то зависи-
мость In N от расстояния оказывается практически линейной.
Для электронов, заметно различающихся по энергиям, кривые
в координатах «число частиц — расстояние от источника» можно
получить суперпозицией нескольких кривых с различными на-
клонами, соответствующими различным начальным энергиям.
27
Пробег электронов, выраженный в
мируется эмпирической формулой:
г!см2, хорошо аппрокси-
/? = АЕ — В,
(2.24)
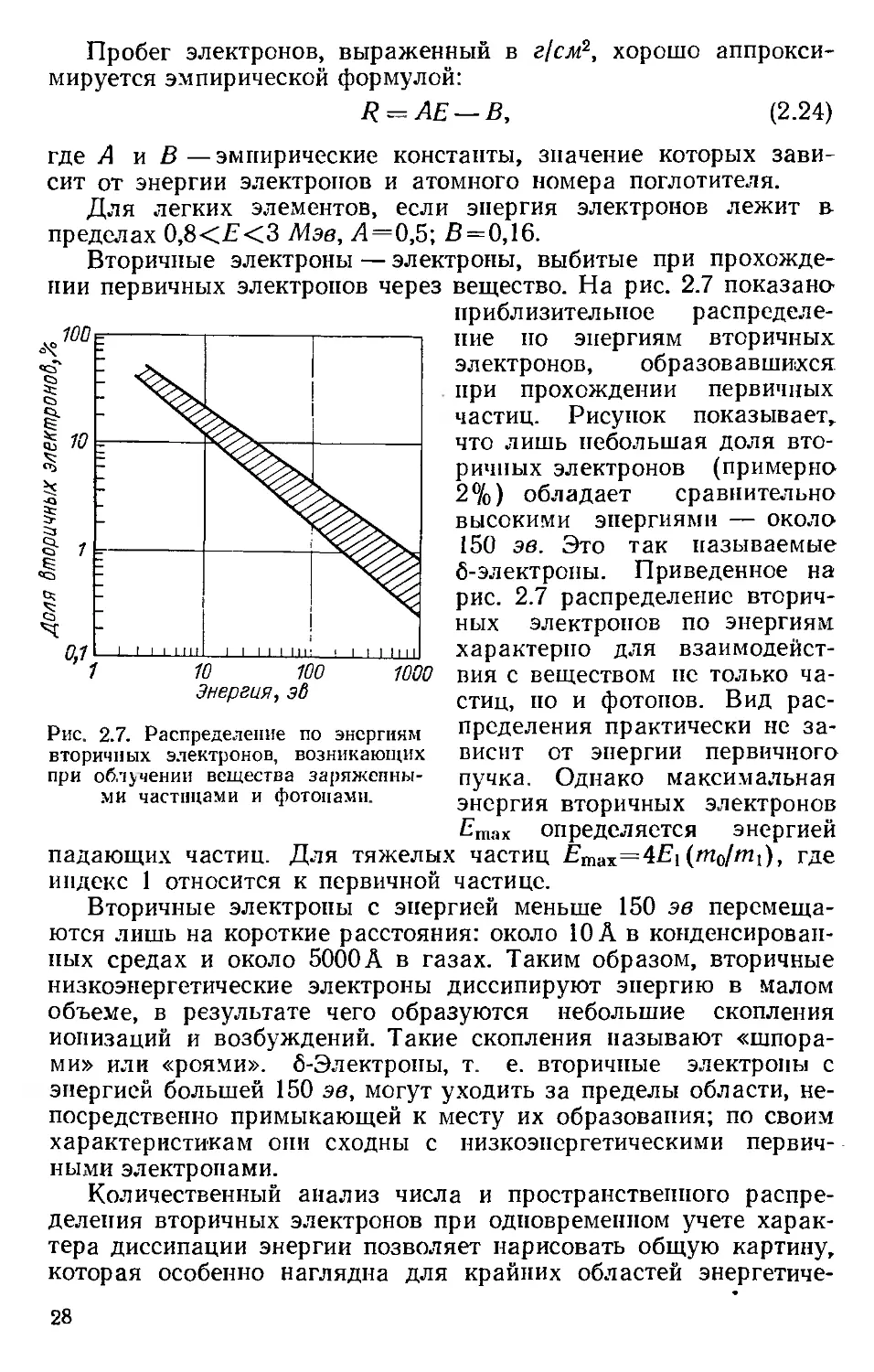
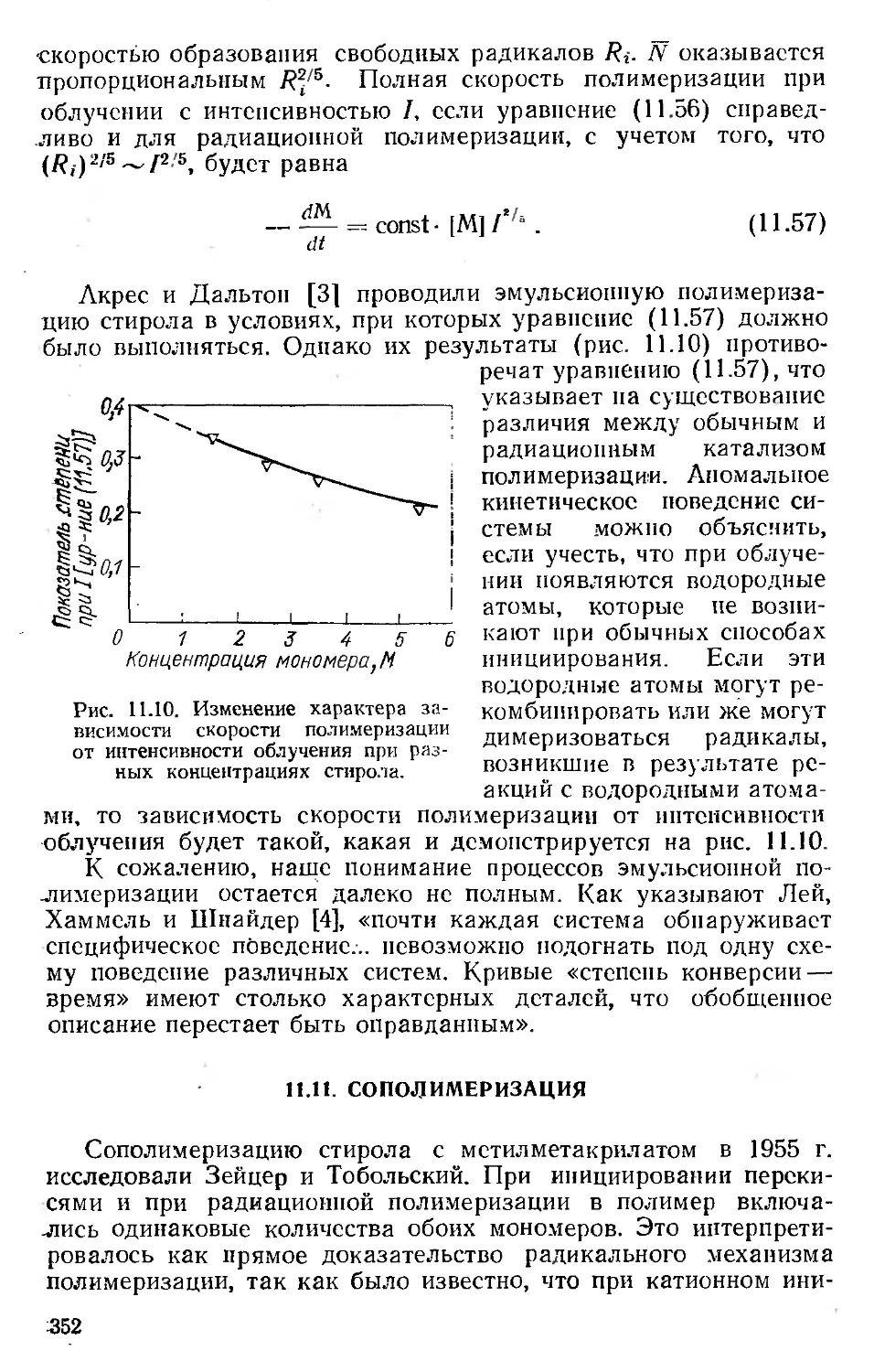
Рис. 2.7. Распределение по энергиям
вторичных электронов, возникающих
при облучении вещества заряженны-
ми частицами и фотонами.
где А и В—эмпирические константы, значение которых зави-
сит от энергии электронов и атомного номера поглотителя.
Для легких элементов, если энергия электронов лежит в
пределах 0,8<Е<3 Мэв, Л=0,5; В = 0,16.
Вторичные электроны — электроны, выбитые при прохожде-
вещество. На рис. 2.7 показано'
приблизительное распределе-
ние по энергиям вторичных
электронов, образовавшихся,
при прохождении первичных
частиц. Рисунок показывает,
что лишь небольшая доля вто-
ричных электронов (примерно
2%) обладает сравнительно
высокими энергиями — около
150 эв. Это так называемые
б-электроны. Приведенное на
рис. 2.7 распределение вторич-
ных электронов по энергиям
характерно для взаимодейст-
вия с веществом нс только ча-
стиц, по и фотонов. Вид рас-
пределения практически не за-
висит от энергии первичного
пучка. Однако максимальная
энергия вторичных электронов
Emax определяется энергией
падающих частиц. Для тяжелых частиц Етах=4Е1(т0/т1), где
индекс 1 относится к первичной частице.
Вторичные электроны с энергией меньше 150 эв перемеща-
ются лишь на короткие расстояния: около 10 А в конденсирован-
ных средах и около 5000А в газах. Таким образом, вторичные
низкоэнергетические электроны диссипируют энергию в малом
объеме, в результате чего образуются небольшие скопления
ионизаций и возбуждений. Такие скопления называют «шпора-
ми» или «роями». б-Электропы, т. е. вторичные электроны с
энергией большей 150 эв, могут уходить за пределы области, не-
посредственно примыкающей к месту их образования; по своим
характеристикам они сходны с низкоэпергетическими первич-
ными электронами.
Количественный анализ числа и пространственного распре-
деления вторичных электронов при одновременном учете харак-
тера диссипации энергии позволяет нарисовать общую картину,
которая особенно наглядна для крайних областей энергетиче-
28
ского спектра первичных частиц, представляющих наибольший
интерес с точки зрения радиационной химии. Низкоэнергетиче-
ские вторичные электроны растрачивают свою энергию в не-
большой области шпоры. Расстояние между отдельными шпо-
рами зависит от скорости потери энергии вдоль пути частицы.
О
ОО
О
О О °
OODOOGDOOOGSDOCW ООООООО
Оо °О о
ОО о °
__ _Ю0Х_ ~
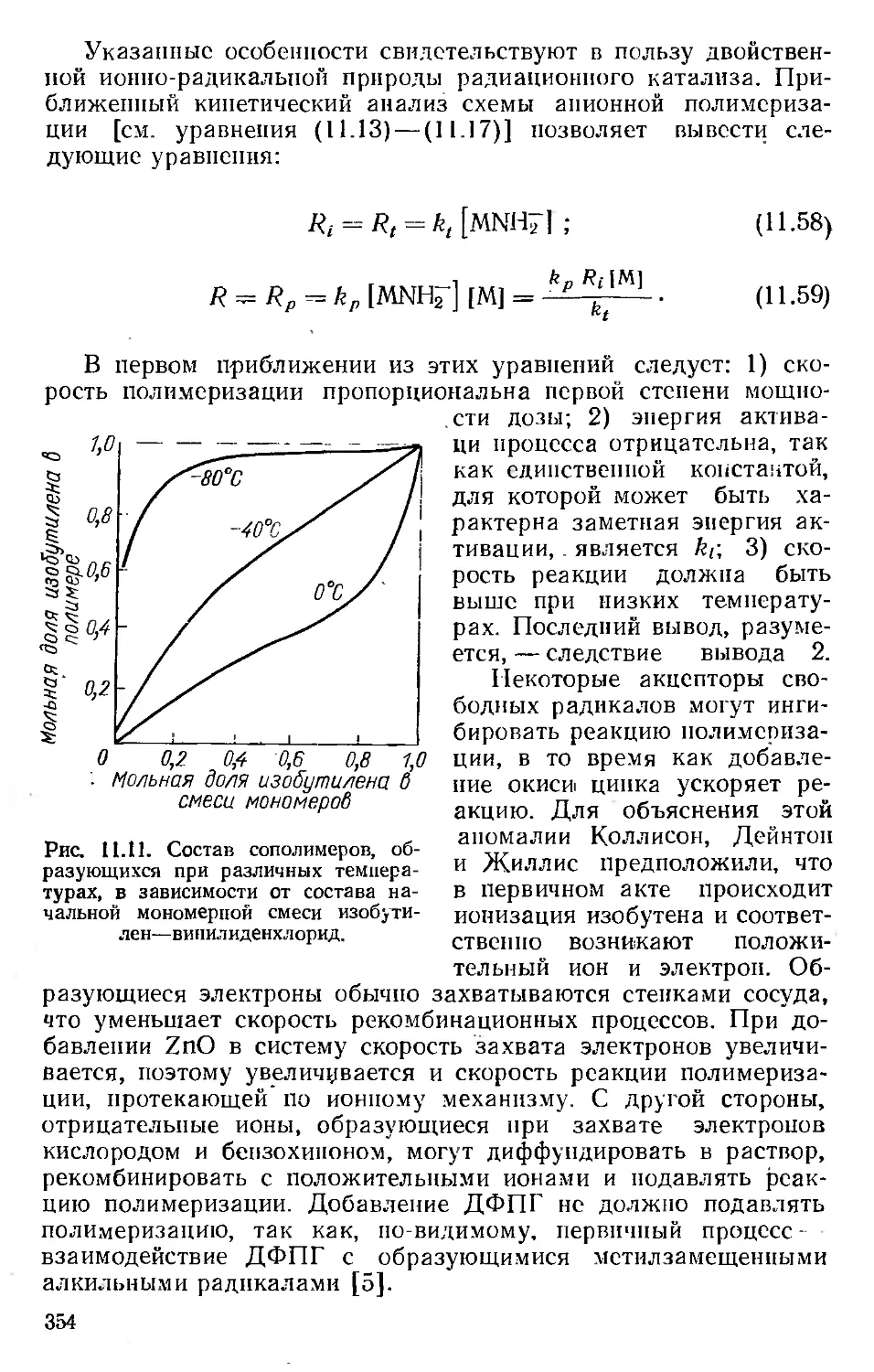
Рис. 2.8. Распределение шпор и первичных иониза-
ции в треках разных частиц:
а — (1-частица: б — быстрый электрон.
Следовательно, при пролете частицы с высокими потерями
энергии должна образоваться цилиндрическая колонка с высо-
кой плотностью ионизации. А при пролете частицы с невысоки-
ми энергетическими потерями возникнут отдельные скопления
ионов (возбужденных молекул и т. п.), распределенные вдоль
пути частицы, словно бусинки на нитке. На рис. 2.8 схематиче-
ски представлено предполагаемое распределение шпор вдоль
пути а-частицы и вдоль пути быстрого электрона. Отметим, что
масштаб для быстрого электрона примерно в 1000 раз меньше,
чем для а-частицы.
В табл. 2.2, которая основывается на оценках, сделанных Ли
[3], дается доля ионных скоплений, содержащих I, 2, 3, 4 или
более пар ионов.
29
Потери энергии на черенковское излучение. Еще один вид
потерь энергии — черенковское излучение *. Черенковским излу-
чением называют видимый свет, испускаемый при движении за-
ряженной частицы в прозрачной среде со скоростью, превышаю-
щей скорость света в этой среде.
Для заряженной частицы, движущейся со скоростью, близ-
кой к скорости света, в каком-либо прозрачном диэлектрике,
Т а б л и ц а 2.2
Относительная частота ионных
скоплений различного размера, %
Число пар ионов в скоплении
таком, например, как вода,
фазовая скорость распростра-
нения электрического и маг-
нитного полей, связанных с
движением данной заряженной
частицы, будет равна с/п (п —
1 2 3 4
43 22 12 10
направлений возникнет
----показатель преломления сре-
>4 ды). Возникающее при этом
электромагнитное излучение
уничтожается гасящей интср-
13 ференцией, если р«<1 (р =
Если рн>1, то в одном из
усиливающая интерференция. Если
скорость частицы превосходит скорость света в данной среде
(с/п), то частица станет двигаться быстрее, чем ее собственное
электромагнитное поле, и при этом будут испускаться любые
частоты, если только pn>L Энергетические потери па черенков-
ское излучение составляют всего около 1000 эв!см в таких ве-
ществах, как вода, и поэтому могут считаться пренебрежимо
малыми по сравнению с потерями на ионизацию и возбуждение.
2.2. ЭЛЕКТРОМАГНИТНОЕ ИЗЛУЧЕНИЕ
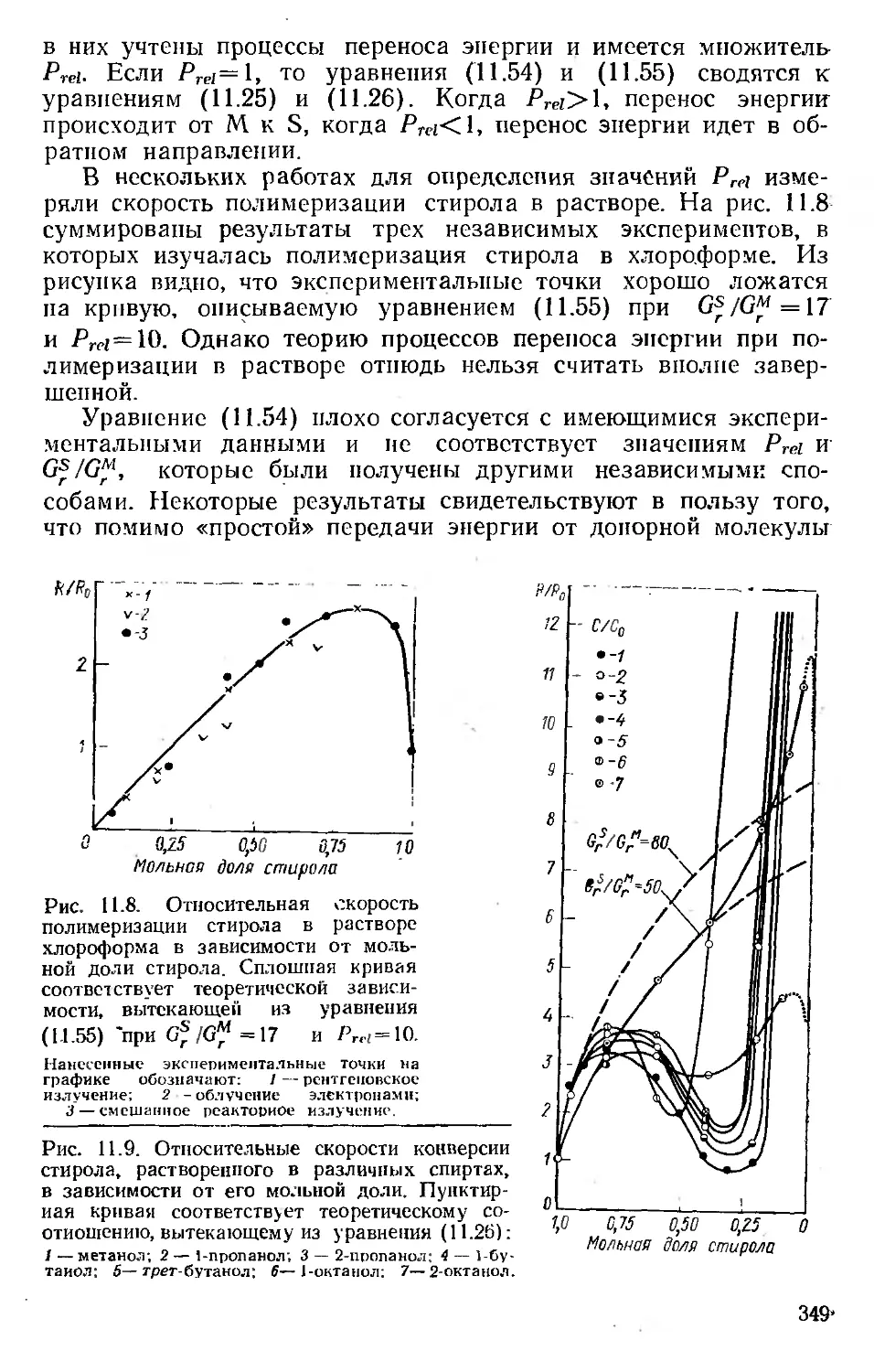
Существует четыре основных вида взаимодействия электро-
магнитного излучения с веществом: 1) фотоэффект, 2) эффект
Комптона, 3) образование пар и 4) фотоялерныс реакции. Каж-
дый из этих видов взаимодействия приобретает существенное
значение для различных энергий фотонов. Отчасти вклад того
или иного вида взаимодействия определяется атомным номером
поглотителя.
При взаимодействии фотонов с веществом относительно
большую роль, чем для частиц, начинает играть тот факт, что
помимо истинного поглощения падающего пучка в поглотителе
происходит и его рассеяние. А так как коэффициенты поглоще-
ния вычисляют по уменьшению интенсивности, то приходится
тщательно учитывать потери па рассеяние.
Открыто советским физиком П. А. Черепковым в 1934 г. — Прим.
перев.
30
2.2.1. Фотоэффект
При фотоэффекте вся энергия фотона передается электро-
ну. Электрон выбрасывается за пределы атома с кинетической
энергией Е, равной энергии фотона hv минус энергия связи
электрона Ф:
E-=hv — Ф. (2.25)
Фотоэффект невозможен на свободном электроне (не связан-
ном с атомом), так как должен выполняться закон сохранения
импульса. Однако фотон поглощается целиком, если электрон
связан с атомом. Сохранение импульса обеспечивается пере-
дачей определенного количества движения атому. У наиболее
прочно связанных электронов, таких, например, как электроны
/С-оболочки, наибольшая вероятность взаимодействия с фото-
ном.
Коэффициент фотоэлектрического поглощения т равен нулю
для фотонов с энергией, меньшей энергии связи электронов.
Коэффициент имеет высокие значения для энергий фотонов,
равных энергии связи электронов. Затем значение коэффициен-
та фотоэлектрического поглощения снова падает по мере роста
энергии фотона.
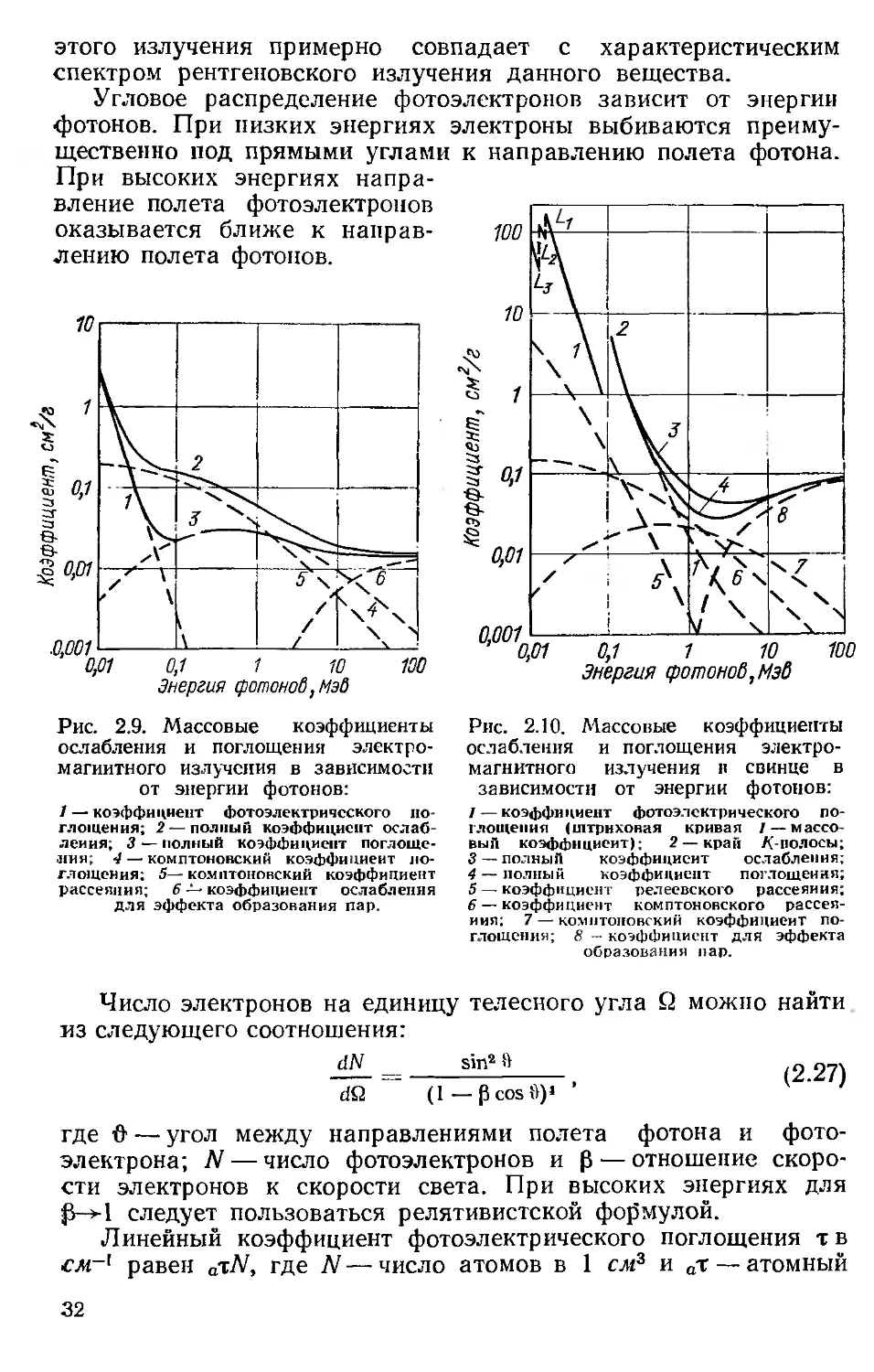
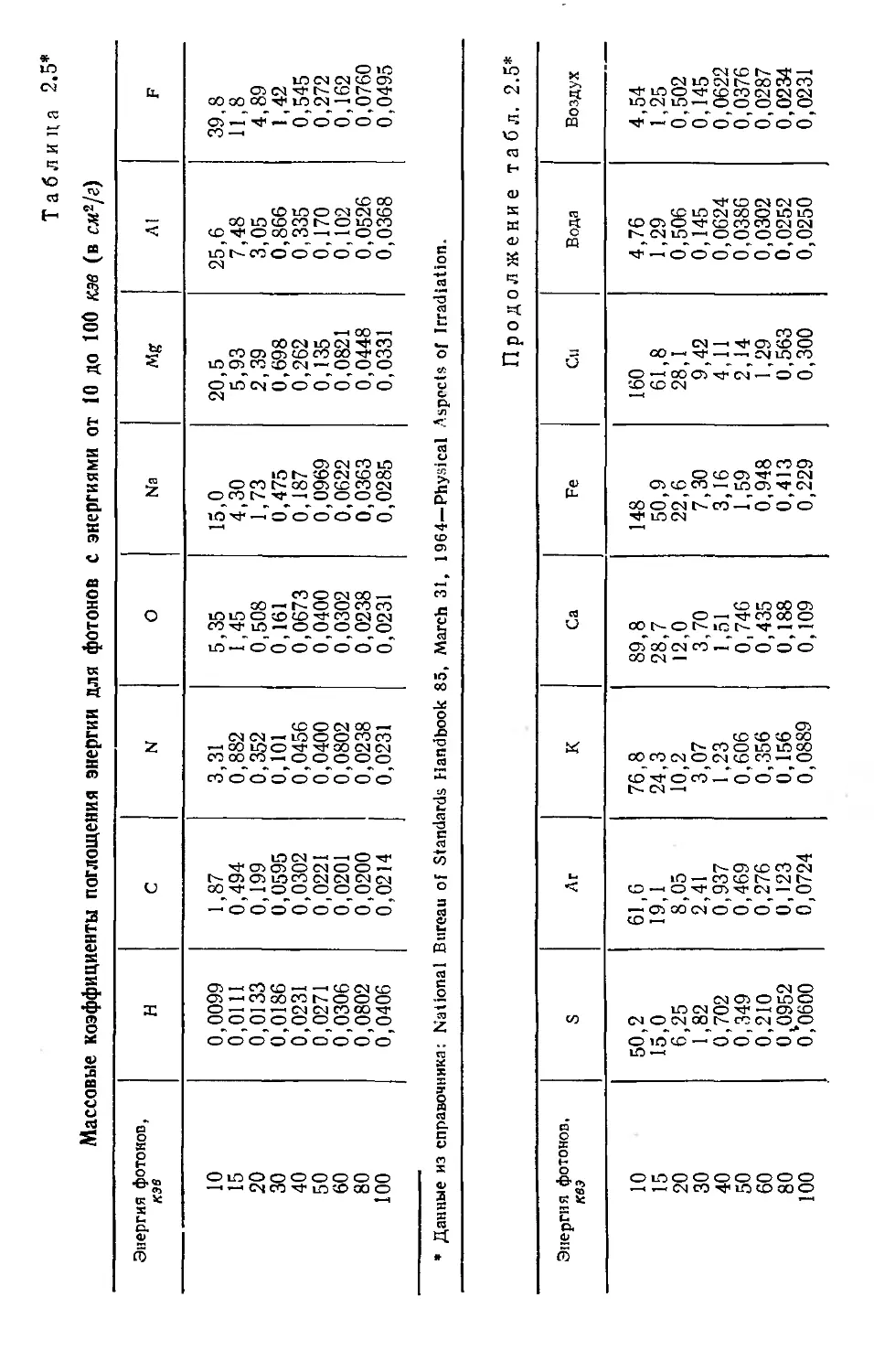
Такая зависимость сечения фотоэффекта от энергии фотонов
иллюстрируется кривыми т/р на рис. 2.9 и 2.10, где приведены
массовые коэффициенты ослабления в зависимости от энергии
для воздуха и свинца. Для свинца при энергиях фотонов ниже
13 кэв возможен фотоэффект на менее прочно связанных L-
электронах. Как только энергия фотонов приближается к 88 кэв
(энергия связи электронов К-оболочки), из К-оболочки начи-
нают выбиваться электроны.
Можно считать общим правилом, что при энергии фотонов
больше энергии связи К-электронов примерно 80% взаимодей-
ствий происходит с К-электронами. Полное сечение фотоэффек-
та на атом определяется приближенным выражением
kZ*
.
(hv)3
где k — константа.
Следовательно, величина сечения взаимодействия
от энергии фотона и атомного номера поглотителя,
наибольшего значения у веществ с высоким Z при низкой энер-
гии фотонов. Резкое возрастание коэффициента фотоэлектриче-
ского поглощения происходит при энергиях, равных энергии
связи электронов. Для тяжелых элементов фотоэлектрический
коэффициент поглощения может увеличиваться в 8 раз.
При переходе электронов наружных оболочек на вакантные
орбитали электронов, выбитых при фотоэффекте, происходит
испускание электромагнитного рентгеновского излучения. Спектр
(2.26)
зависит
достигая
31
этого излучения примерно совпадает с характеристическим
спектром рентгеновского излучения данного вещества.
Угловое распределение фотоэлектронов зависит от энергии
фотонов. При низких энергиях электроны выбиваются преиму-
щественно под прямыми углами к направлению полета фотона.
При высоких энергиях напра-
Рис. 2.9. Массовые коэффициенты
ослабления и поглощения электро-
магнитного излучения в зависимости
от энергии фотонов:
1 — коэффициент фотоэлектрического по-
глощения; 2 —полный коэффициент ослаб-
ления; 3 — полный коэффициент поглоще-
ния; 4— комптоновский коэффициент по-
глощения; 5— комптоновский коэффициент
рассеяния; 6 — коэффициент ослабления
для эффекта образования пар.
Рис. 2.10. Массовые коэффициенты
ослабления и поглощения электро-
магнитного излучения в свинце в
зависимости от энергии фотонов:
I — коэффициент фотоэлектрического по-
I лощения (штриховая кривая / — массо-
вый коэффициент); 2— край К полосы;
3~ полный коэффициент ослабления;
4 — полный коэффициент поглощения;
5 — коэффициент релеевского рассеяния;
6 — коэффициент комптоновского рассея-
ния; 7 — комптоновский коэффициент по-
глощения; 8 — коэффициент для эффекта
образования пар.
Число электронов на единицу телесного угла Q можно найти
из следующего соотношения:
dN_______ sin2 ft
diT (1— P COS ft)» ’
(2-27)
где 0 — угол между направлениями полета фотона и фото-
электрона; N — число фотоэлектронов и р — отношение скоро-
сти электронов к скорости света. При высоких энергиях для
^->1 следует пользоваться релятивистской формулой.
Линейный коэффициент фотоэлектрического поглощения т в
см~1 равен ai;N, где N—число атомов в 1 см3 и ат — атомный
32
фотоэлектрический коэффициент в единицах см* 2/атом. Массо-
вый коэффициент фотоэлектрического поглощения равен т/р.
Для фотоэффекта нет практически никакой разницы между
линейным коэффициентом ослабления и линейным коэффици-
ентом поглощения *, так как можно считать, что возникающее
при фотоэффекте рентгеновское излучение поглощается па рас-
стояниях, сравнимых с пробегом фотоэлектронов.
2.2.2. Эффект Комптона
Комптоновское взаимодействие представляет собой упругое
соударение фотона и слабо связанного или вообще не связан-
ного электрона. В процессе взаимодействия электрон приво-
дится в движение, а энергия фотона уменьшается. Схематиче-
ски эффект Комптона изображен на рис. 2.11.
Рис. 2.11. Эффект Комптона. Взаимодействие фо-
тона и свободного электрона.
При соударении должны выполняться законы сохранения
энергии и импульса. Так как т0с2— энергия, соответствующая
массе нокоя электрона, то уравнение энергетического баланса
будет выглядеть так:
hv — hv' -|- тс2 — т0с2, (2.28)
где т = т0(1—р2)~1/2.
Закон сохранения импульса требует, чтобы
= hv cos8' + rnoc cos 8; (2.29)
С с
hv' sin 8' — moc sin 8. (2.30)
Эти уравнения нетрудно преобразовать алгебраически и
найти изменения длины волны фотона в результате столкнове-
ния ДА.
Д7, = __А_ (1 _ cos 8Л). (2.31)
Av moc
* Вопросы терминологии подробно обсуждаются в разделе 2.2.5.
2 Зак. 907
33
Энергия рассеянного фотона будет равна
hv'
hv
1 -г (/о7т0с2) (1 — cos !>')
(2.32)
Пример 2.4. у-Квант с энергией 0,51 Мэв рассеивается иа угол 180°. Чему
равна энергия рассеянного фотона и энергия комптоновского электрона от-
дачи?
Решение.
/IV
hv’ —----------------------------
! hv \
1 -г I -----i (1 —cos!)')
\ шос- /
_________0,51
1 + 1 • (1 — cos 180°)
0,17 Мэв.
Энергия электрона отдачи равна 0,51—0,17 = 0,3-1 Мэв.
Отметим следующие важные особенности комптоновского
рассеяния у-квантов.
1. Изменение длины волны Лл для данного угла рассеяния
•б не зависит от энергии первичного фотона. Изменение энергии
рассеянного фотона увеличивается с возрастанием энергии па-
дающего фотона.
2. Энергия фотона, рассеянного назад под максимальным уг-
лом (180°), приближается к предельному значению 0,25 Мэв при
росте hv.
Начальное распределение по энергиям
Энергия фотоног, ЛТэя Энергия
0, I 0,5 I ,0 2,0 3,0 4,0 6,0
0—1 1—2 2—3 3—4 4—6 6—8 234 40,8 14,1 7,06 7,13 3,58 43,6 38,8 13,8 7,01 7,03 3,55 39,4 14,4 7,06 7,11 3,55 22,2 8,76 7,66 3,66 15,8 9,95 3,96 17,5 4,70 12,4
1—10 2,13 2,12 2,13 2,16 2,25 2,43 3,44
10—12 1,42 1,41 1,42 1,44 1,47 1,53 1,83
12—14 1,02 1,01 1,02 1,02 1,04 1.07 1,19
14—16 0,760 0,760 0,760 0,768 0,774 0,789 0,846
16—18 0,590 0,590 0,590 0,595 0,599 0,608 0,639
18—20 0,472 0,472 0,472 0,475 0,479 0,484 0,502
20—22 0,386 0,386 0,386 0,388 0,391 0,394 0,406
22—24 0,321 0,321 0,321 0,323 0,324 0,327 0,335
24—26 0,273 0,273 0,273 0,274 0,274 0,276 0,281
26—28 0,234 0,234 0,234 0,234 0,235 0,236 0,240
28—-30 0,203 0,203 0,203 0,203 0,203 0,204 0,207
* Таблица составлена по данным: Cormack D., Johns Н. Е., Brit. J. Radiol, 1952,
воды под действием I фотон!см2. Энергия фотона указана в первом столбце таблицы,
те с другими веществами, кроме воды. Если концентрация электронов в веществе N
34
3. Фотоны, рассеянные под большими углами, теряют боль-
ше энергии, чем фотоны, рассеянные под малыми углами.
4. Фотоны с низкой энергией претерпевают лишь незначи-
тельное изменение энергии, тогда как для фотонов с высокой
энергией изменения энергии при рассеянии весьма значительны.
Высокоэнергетические фотоны передают электронам большую
долю энергии.
Коэффициенты комптоновского взаимодействия. Сечение
комптоновского рассеяния определяется по формуле Клейна —
Нишпны, которая дает следующую величину электронного ко-
эффициента комптоновского взаимодействия:
„ з f 1 — а [2(1 -|- а) 1 ., , „ , I ,
ео - 2лг0-----— --------— In (1 -г 2а) -г
I а2 [ 1 2а 2 J
-•—— In (1 -;-2а)-------1 3” 1 см2/электрон,
2а (1 -|-2а)2 J
(2.33)
где a = hvlm0c2, a го — классический радиус электрона.
Таким образом, полное уменьшение энергии в падающем
пучке измеряется величиной сст, которая определяется по фор-
муле Клейна — Нишины. Доля энергии падающих фотонов на
комптоновских электронов в воде*
Т а б л и ц а 2.3
электронов, ЛЬв
8,0 10,0 12.0 14,0 16.0 18.0 20о0 22,0
9,60 2,73 1,47 0,968 0,702 0,538 0,428 7,86 2,28 1,23 0,820 0,599 0,464 6,66 1,96 1,07 0,713 0,524 5,77 1,72 0,940 0,632 5,10 1,54 0,842 4,56 1.38 4,13
0,350 0,372 0,408 0,466 0,567 0,759 1,27 3,77
0,291 0,306 0,329 0,364 0,420 0,513 0,699 1,16
0,247 0,257 0,272 0,295 0,329 0,381 0,473 0,645
0,212 0,220 0,230 0,245 0,268 0,299 0,352 0,436
V. 25, р. 369. В таблице дается число электронов на интервал (Мэв}, возникающих в I см3
Цифры таблицы следует умножать на ЮЧ Пользоваться таблицей можно и при рабо-
электрон [см\ то табличные величины следует умножать на W/(3,34 Ю23).
2
35
] электрон!см2 поглотителя, которая уносится рассеянными
квантами, выражается коэффициентом
^-f-L 1п(1 |-2а)
mof4 *- а3
2 (1 + «) (2а2 —2а—1)
а2 (1 — 2а)
8а3
3(1 4- 2а)3
см2! электрон.
(2.34)
Пользуясь законом сохранения энергии, можно найти энер-
гию первичных квантов соа, переданную электронам среды, ко-
торая должна быть равна разности общей затраченной энергии
и энергии рассеянных квантов:
е®а е® e^si
(2.35)
где еоа — комптоновский коэффициент поглощения энергии. Ин-
декс е означает, что это коэффициент для электронов. Для вы-
числений, в которых определяется пропускание поглотителя,
удобнее пользоваться полным коэффициентом линейного ослаб-
ления о. Так как в 1 см3 среды имеется N атомов по Z элект-
ронов в каждом, то полный линейный коэффициент ослабления
равен
о = NZea см~'.
(2.36)
Массовый коэффициент ослабления равен о/p (см2/г), где
р — плотность поглотителя.
На рис. 2.9 и 2.10 показана зависимость от энергии фотонов
массовых комптоновских коэффициентов рассеяния, ослабления
и поглощения для воздуха и свинца. Ход этих зависимостей
позволяет сделать следующие выводы:
1. Как массовый коэффициент рассеяния os/p, так и полный
массовый коэффициент ослабления для эффекта Комптона о/р
плавно уменьшаются по мере роста энергии фотонов.
2. Массовый .коэффициент поглощения оп/р возрастает или
остается примерно постоянным вплоть до энергии фотонов
0,5 Мэв, а затем уменьшается.
3. Сечение комптоновского рассеяния на электрон сущест-
венным образом не зависит от Z. Следовательно, массовый ко-
эффициент поглощения практически постоянен и не зависит
от природы поглотителя.
Уравнение Клейна — Нишины можно использовать помимо
расчета полных коэффициентов поглощения и для вычисления
начального распределения комптоновских электронов по энер-
гиям. В табл. 2.3 приведены вероятности возникновения электро-
нов данной энергии при комптоновском рассеянии фотонов с
энергиями 0—30 Мэв.
36
2.2.3. Другие виды рассеяния
Комптоновское рассеяние некогерентно, так как электроны
считаются свободными; поэтому фазовые соотношения между
падающим и рассеянным излучениями оказываются случайными.
Существует, однако, взаимодействие фотонов и со связанными
электронами, которое приводит к когерентному рассеянию, из-
вестному как релеевское рассеяние. При рслеевском рассеянии
фотоны рассеиваются связанными электронами атомов таким
образом, что атом не ионизуется и не возбуждается. Вероят-
ность релеевского рассеяния высока для низких энергий фото-
нов и веществ с большим Z. Эффект релеевского рассеяния
весьма существен для интерпретации измерений коэффициентов
ослабления «узких пучков».
На рис. 2.10 кривая оа/р дает полные комптоновские коэф-
фициенты поглощения, включая и когерентные рассеяния. Ко-
эффициенты когерентного рассеяния наиболее высоки в той об-
ласти, где коэффициенты фотоэлектрического поглощения на
один-два порядка больше коэффициентов комптоновского рас-
сеяния. Таким образом, вклад релеевского рассеяния в полные
коэффициенты поглощения невелик.
Два других процесса рассеяния, в которых атомы участвуют
как целое, — томсоновское рассеяние и ядерпое резонансное рас-
сеяние. Так как оба процесса едва ощутимы в эксперименте, то
здесь они не рассматриваются.
2.2.4. Образование электроно-позитронных пар
Эффект образования нар не имеет аналога в классической
физике. В этом процессе в результате поглощения энергии фо-
тона возникает пара частиц — электрон и позитрон. Полная
энергия поглощенного фотона равна энергетическому эквивален-
ту масс покоя электрона и позитрона Ер и Ес плюс кинетиче-
ские энергии этих частиц:
Ер 4- Ее л- 2m0c2 = hv. (2.37)
Если энергия падающего фотона меньше масс покоя элект-
рона и позитрона в энергетических единицах, то образование
пары не происходит. Для эффекта образования пар необходи-
мо, чтобы hv>2 тос2= 1,02 Мэв. Процесс образования пар помо-
жет происходить в пустоте. Пары возникают лишь в поле заря-
женных частиц, главным образом в поле ядер, но в незначи-
тельной степени и в ноле электронов. Позитрон, потеряв свою
кинетическую энергию, аннигилирует с электроном, в резуль-
тате чего в противоположных направлениях выбрасывается
два у-кванта с энергией 0,51 Мэв.
37
С процессом образования пар связан ряд других второсте-
пенных эффектов, которые не вносят существенного вклада в
процесс передачи энергии. К таким эффектам относятся:
1. Отдача ядер.
2. Аннигиляция позитронов, которые еще обладают опреде-
ленным запасом кинетической энергии.
3. Аннигиляция с испусканием трех фотонов (образование
триплета).
Полный коэффициент ослабления в результате образования
пар обозначают через v.s, он состоит подобно комптоновскому
коэффициенту из коэффициента истинного поглощения ха и ко-
эффициента рассеяния xs. Коэффициент рассеяния всегда очень
мал, и поэтому принято считать х=ха. Массовый коэффициент
ослабления для образования пар равен х/р.
Таблица 2.4
Линейные коэффициенты эффекта образования пар для свинца
Энергия фотонов, Мэв хРЬ, см Энергия фотонов, Мэв хРЬ, см 1 Энергия фотонов, Мэв хРЬ, см 1
1,02 0,0 7,66 0,368 51,0 1,00
1,53 0,011 10,22 0,462 102,0 1,18
2,04 0,042 17,0 0,630 255,0 1,33
3,06 0,116 25,5 0,768 510,0 1,40
5,1 0,255
В табл. 2.4 приведены значения линейного коэффициента
эффекта образования пар х (в см-1) для свинца при различ-
ных энергиях фотонов. Значение х для любого другого элемен-
та можно получить из следующего соотношения:
X
р 207,2 / Z \2
11,35 А \~82~) ’
(2.38)
где А — атомный вес; Z — атомный номер; р — плотность.
2.2.5. Массовые коэффициенты, характеризующие
поглощение энергии в веществе
Терминология, употребляемая для описания взаимодействия
фотонов с веществом, часто приводит к заметной путанице. Та-
кая путаница возникает главным образом из-за того, что термин
«коэффициент поглощения» считают синонимом коэффициента
ослабления. На самом деле коэффициент ослабления характе-
ризует число взаимодействий фотонов с веществом (ничего не
говоря о переданной при этом энергии).
С другой стороны, с помощью коэффициента поглощения
описывается обычно количество энергии, диссипированной вто-
ричными электронами, которые возникли при этих взаимодейст-
38
виях. Поэтому вместо термина «коэффициент поглощения» для
описания поглощения энергии фотонов веществом лучше упот-
реблять термин «массовый коэффициент передачи энергии» *
или «массовый коэффициент поглощения энергии», несколько
отличающийся по смыслу от первого.
Массовый коэффициент поглощения энергии ц.еп/р равен
pfc/p(l—G), где G— доля энергии вторичных заряженных ча-
стиц, теряемая на тормозное излучение, а щ</р — массовый ко-
эффициент передачи энергии. До тех пор пока кинетические
энергии вторичных частиц не окажутся сравнимыми или боль-
шими энергетического эквивалента их массы покоя, массовый
коэффициент передачи энергии щг/р и массовый коэффициент
поглощения энергии реп/р пе будут заметно различаться. По-
этому для практических целей Щт/р=|тд/р-
Массовый коэффициент поглощения энергии для рентгенов-
ского или у-излучения с энергией фотонов hv можно записать
следующим образом:
= Де-+ 4-2^, (2.39)
Р Р Р Р
где та/р — массовый коэффициент поглощения фотоэффекта,
равный т/р (1—6//?v); т/р — массовый коэффициент ослабления
для фотоэффекта; 6 — средняя энергия, испускаемая в виде
рентгеновского излучения; <та/р — комптоновский массовый ко-
эффициент поглощения, равный ст/р (Ee/hv); о/р — комптонов-
ский массовый коэффициент ослабления; Ее — средняя энер-
гия комптоновских электронов на один рассеянный фотон;
ка/р — массовый коэффициент поглощения для эффекта обра-
зования пар, равный х/р(1—2mc2/hv); х/р — массовый коэффи-
циент ослабления для эффекта образования пар; тс2 — энергия,
эквивалентная массе покоя электрона.
Массовый коэффициент ослабления р/р описывает ослабле-
ние интенсивности электромагнитного излучения и определяется
так:
i = — + — + 4- —, (2.40)
Р Р Р Р Р
где o(oh/p •- массовый коэффициент ослабления для когерент-
ного рассеяния. Остальные обозначения разъяснены ранее.
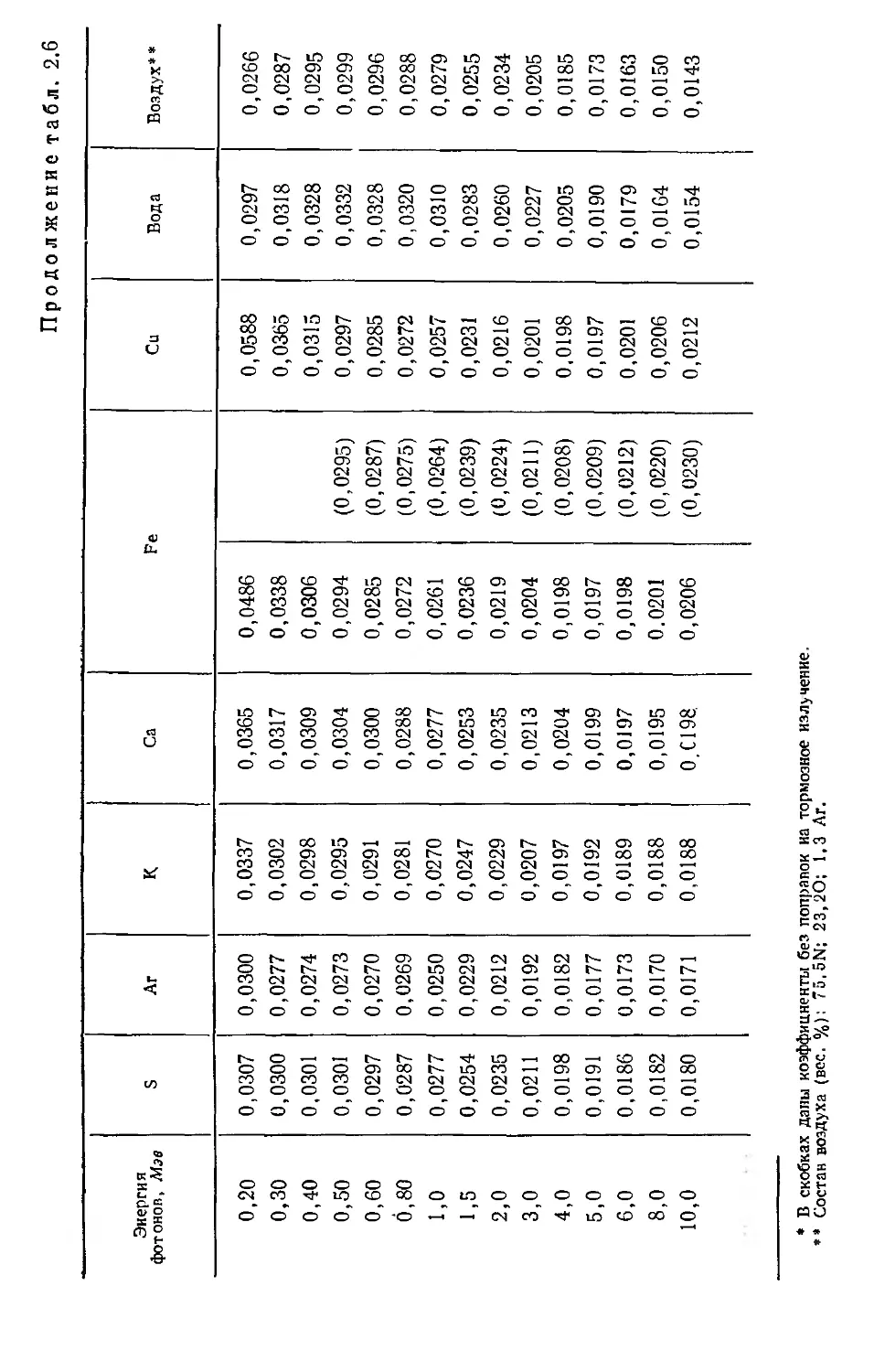
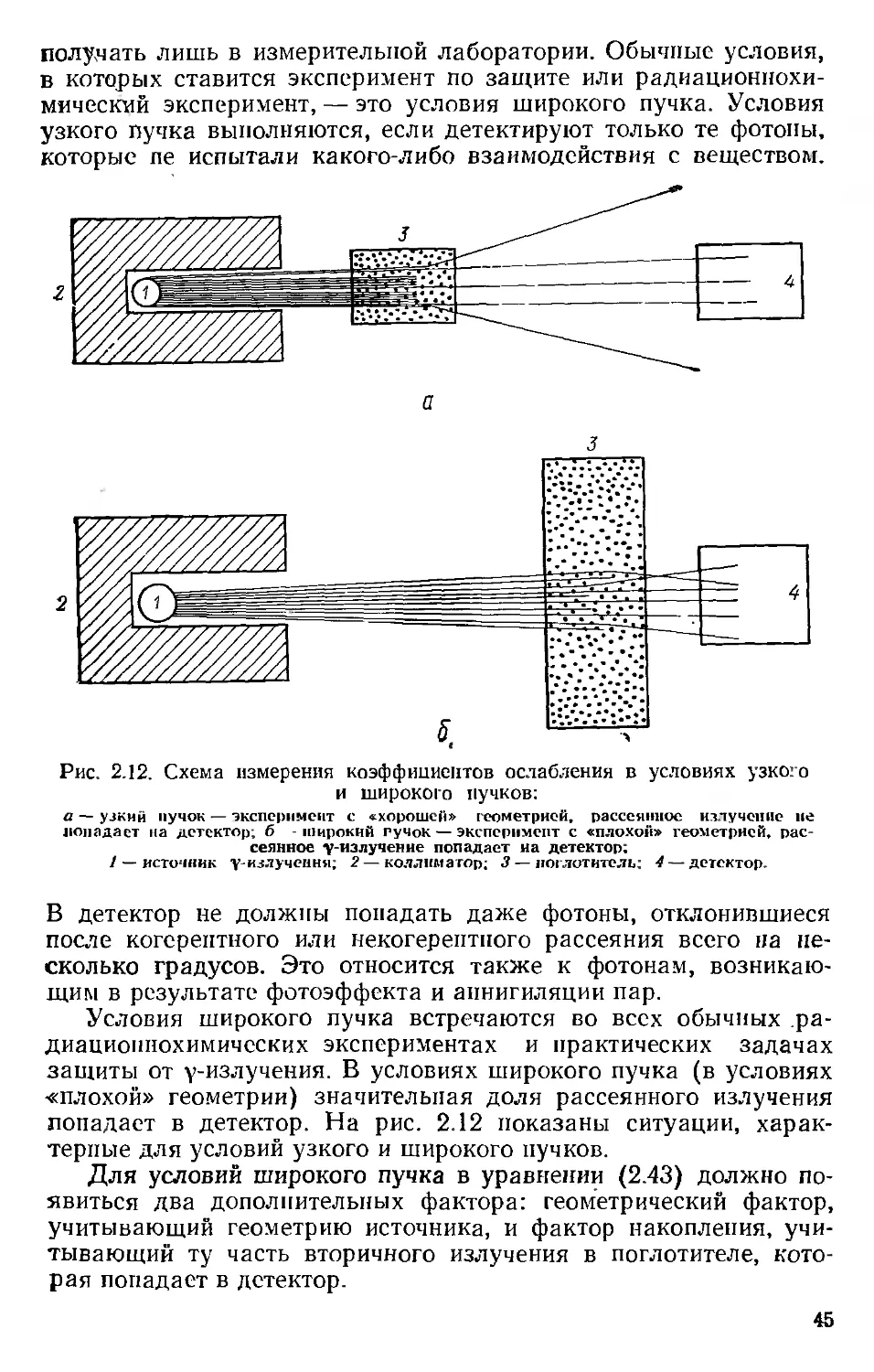
В табл. 2.5 и 2.6 приведены массовые коэффициенты пере-
дачи энергии для ряда распространенных элементов, а также
для воздуха и воды. Непосредственно табличные значения мож-
но употреблять только для тех элементов, которые имеются в
* Этот термин находится в соответствии с рекомендациями МКРЕ и упот-
ребляется в отечественной литературе. См. В. И, Иванов. Курс дозиметрии.
Изд. 2-е, М., Атомиздат, 1970. — Прим, перев.
39
Таблица 2.5*
Массовые коэффициенты поглощения энергии для фотонов с энергиями от 10 до 100 кэв (в cm?/г)
Энергия фотонов, кэв н С N О Na Mg Л1 F
10 0,0099 1,87 3,31 5,35 15,0 20,5 25,6 39,8
15 0,0111 0,494 0,882 1,45 4,30 5,93 7,48 11,8
20 0,0133 0,199 0,352 0,508 1,73 2,39 3,05 4,89
30 0,0186 0,0595 0,101 0,161 0,475 0,698 0,866 1,42
40 0,0231 0,0302 0,0456 0,0673 0,187 0,262 0,335 0,545
50 0,0271 0,0221 0,0400 0,0400 0,0969 0,135 0,170 0,272
60 0,0306 0,0201 0,0802 0,0302 0,0622 0,0821 0,102 0,162
80 0,0802 0,0200 0,0238 0,0238 0,0363 0,0448 0,0526 0,0760
100 0,0406 0,0214 0,0231 0,0231 0,0285 0,0331 0,0368 0,0495
* Данные из справочника: National
Bureau of Standards Handbook 85, March 31,
1964—Physical Aspects of Irradiation.
Продолжение табл. 2.5*
Энергия фотонов, квэ S Лг К Са Fe Си Вода Воздух
10 50,2 61,6 76,8 89,8 148 160 4,76 4,54
15 15,0 19,1 24,3 28,7 50,9 61,8 1,29 1,25
20 6,25 8,05 10,2 12,0 22,6 28,1 0,506 0,502
30 1,82 2,41 3,07 3,70 7,30 9,42 0,145 0,145
40 0,702 0,937 1 ,23 1,51 3,16 4,11 0,0624 0,0622
50 0,349 0,469 0,606 0,746 1,59 2,14 0,0386 0,0376
60 0,210 0,276 0,356 0,435 0,948 1,29 0,0302 0,0287
80 0.0952 0,123 0,156 0,188 0,413 0,563 0,0252 0,0234
100 0,0600 0,0724 0,0889 0,109 0,229 0,300 0,0250 0,0231
Таблица 2.6
Массовые коэффициенты передачи энергии для фотонов с энергиями выше 100 кэв (в см^/г)*
Энергия фотонов, Мэв н с N О Na Mg Al р
0,10 0,0406 0,0213 0,0222 0,0231 0,0285 0,0331 0,0369 0,0494
0,15 0,0481 0,0244 0,0247 0,0250 0,0256 0,0274 0,0281 0,0313
0,20 0,0525 0,0264 0,0265 0,0268 0,0264 0,0276 0,0273 0,0289
0,30 0,0570 0,0287 0,0287 0,0287 ' 0,0277 0,0288 0,0281 0,0288
0,40 0,0586 0,0295 0,0295 0,0296 0,0283 0,0294 0,0287 0,0290
0,50 0,0593 0,0299 0,0299 0,0299 0,0286 0,0296 0,0288 0,0290
0,60 0,0588 0,0296 0,0296 0,0296 0,0283 0,0292 0,0285 0,0286
0,80 0,0573 0,0289 0,0288 0,0288 0,0276 0,0284 0,0278 (0,0279) 0,0278
1,0 0,0555 0,0279 0,0279 0,0279 0,0267 0,0275 0,0268 (0,0270) 0,0269
1,5 0,0508 0,0266 0,0255 0,0255 0,0243 0,0251 0,0245 (0,0247) 0,0246
2,0 0,0464 0,0234 0,0235 0,0234 0,0225 0,0232 0,0227 (0,0229) 0,0228
3,0 0,0398 0,0204 0,0205 0,0206 0,0199 0,0206 0,0202 (0,0205) 0,0204
4,0 0,0351 0,0184 0,0185 0,0187 0,0183 0,0191 0,0188 (0,0192) 0,0191
5,0 0,0316 0,0170 0,0172 0,0174 0,0173 0,0180 0,0178 (0,0184) 0,0183
6,0 0,0288 0,0159 0,0162 0,0165 0,0166 0,0174 0,0172 (0,0179) 0,0178
8,0 0,0249 0,0145 0,0149 0,0153 0,0157 0,0165 0,0165 (0,0173) 0,0173
10,0 0,0222 0,0136 0,0141 0,0146 0,0152 0,0161 0,0162 (0,0172) 0,0172
0,10 0,0599 0,0724 0,0889 0,109 0,219 0,302 0,0250 0,0231
0,15 0,0349 0,0366 0,0431 0,0486 0,0803 0,105 0,0276 0,0249
Энергия фотонов, Мэв S Аг К Са Р
0,20 0,0307 0,0300 0,0337 0,0365 0,0486
0,30 0,0300 0,0277 0,0302 0,0317 0,0338
0,40 0,0301 0,0274 0,0298 0,0309 0,0306
0,50 0,0301 0,0273 0,0295 0,0304 0,0294
0,60 0,0297 0,0270 0,0291 0,0300 0,0285
0,80 0,0287 0,0269 0,0281 0,0288 0,0272
1,0 0,0277 0,0250 0,0270 0,0277 0,0261
1,5 0,0254 0,0229 0,0247 0,0253 0,0236
2,0 0,0235 0,0212 0,0229 0,0235 0,0219
3,0 0,0211 0,0192 0,0207 0,0213 0,0204
4,0 0,0198 0,0182 0,0197 0,0204 0,0198
5,0 0,0191 0,0177 0,0192 0,0199 0,0197
6,0 0,0186 0,0173 0,0189 0,0197 0,0198
8,0 0,0182 0,0170 0,0188 0,0195 0.0201
10,0 0,0180 0,0171 0,0188 0.С198. 0,0206
* В скобках даны коэффициенты без поправок иа тормозное излучение.
♦* Состав воздуха (вес. %): 75,5N; 23,20; 1,3 Аг.
Продолжение табл. 2.6
е Си Вода Воздух* *
0,0588 0,0297 0,0266
0,0365 0,0318 0,0287
0,0315 0,0328 0,0295
(0,0295) 0,0297 0,0332 0,0299
(0,0287) 0,0285 0,0328 0,0296
(0,0275) 0,0272 0,0320 0,0288
(0,0264) 0,0257 0,0310 0,0279
(0,0239) 0,0231 0,0283 0,0255
(0,0224) 0,0216 0,0260 0,0234
(0,0211) 0,0201 0,0227 0,0205
(0,0208) 0,0198 0,0205 0,0185
(0,0209) 0,0197 0,0190 0,0173
(0,0212) 0,0201 0,0179 0,0163
(0,0220) 0,0206 0,0164 0,0150
(0,0230) 0,0212 0,0154 0,0143
таолице. Для сложных соединений с плотностью р массовые ко-
эффициенты передачи энергии можно найти по формуле
Ьть = хх л. х2 + . . .4--Н5-Х (2.41)
Р Pl Р2 Рп
где X], Х2,..Хп —весовые доли различных элементов. На-
пример, в табл. 2.6 массовый коэффициент передачи энергии
для воды при облучении ее фотонами с энергией 1 Мэв находят
по формуле
^-(Н2О) = JL (0,0555) ]- -^-(0,0279; = 0,0310.
2.2.6. Фотоядерные реакции
Фотоны с достаточно высокой энергией могут выбивать из
ядра нейтроны или протоны. Для этого энергия фотона должна
превышать энергию связи * нейтрона или протопа. Для боль-
шей части веществ, за исключением бериллия и дейтерия, энер-
гия, необходимая для фотоядерных реакций, лежит в преде-
лах 6—18 Мэв. Число нейтронов, выбиваемых из материала ми-
шени на один падающий фотон, дается приближенной форму-
лой
п - 0,044 ]/ZW, (2.42)
где п — число нейтронов; 117 — полная энергия фотона в Мэе;
Z — атомный номер.
Сечепия фотоядерных реакций пренебрежимо малы по срав-
нению с сечениями комптоновского рассеяния или эффекта обра-
зования пар. Поэтому учет фотоядерных реакций необходим для
практических целей лишь при расчете защиты электронных ус-
корителей и в некоторых других специальных случаях.
Наиболее интересны две фотоядерные реакции:
4Ве у —> 4Ве -J- п
и
?D у -> }Н + п.
Обычная сокращенная запись этих реакций °Ве(у, н)8Ве и
2D(y, /г)’Н. Эти реакции используются для калибровки элект-
ронных ускорителей, так как с их помощью можно найти пико-
вую энергию тормозного излучения, возникающего при тормо-
жении электронов в мишени, изготовленной из тяжелых метал-
лов. Пороговые энергии этих фотоядерных реакций соответст-
венно 1,666±0,002 и 2,226+0,003 Мэв. В табл. 2.7 приведены
* Энергия связи любого компонента системы равна энергетическому эк-
виваленту разности масс свободных компонентов и массы системы.
43
Таблица 2.7*
Сечения фотоядерных реакций (у, п)
Элемент Пороговая энергия, Мэв Резонансная энергия, Мэв Сечение в резо- од маисе, 10 см*}атом Сечения Комптон- эффекта и эффекта образования пар, 10~24 л2/атам
12С 18,6 22,9 0,013 0,307
16,0 21,5 0,034
14N 16,5 24,2 0,0028 0,379
160 15,6 24,2 0,011 0,461
®3Си 10,9 18,1 0,10 3,56
«Си 9,8 18,6 0,15 3,58
РЬ (природный) 7,9 13,7 0,81 18,8
* Табличные данные цитируются
Phys. Rev., 1953, Bd. 91, S. 659.
но статье; Montalbetti R., Katz L., Goldberg J.
пороговые энергии и сечения некоторых фотоядерных реакций.
В последнем столбце таблицы даны для сравнения суммарные
коэффициенты комптоновского рассеяния и эффекта образо-
вания пар.
2.2.7. Электромагнитное излучение.
Общие замечания
Обычно коэффициенты поглощения определяют, поместив
поглотитель между источником излучения и детектором. Веро-
ятность того, что фотон пройдет сквозь слой поглотителя дан-
ной толщины, не испытав пи одного из возможных видов взаи-
модействий (фотоэффект, комптоновское рассеяние или эффект
образования пар), равна произведению вероятностей отсутствия
взаимодействия для каждого из этих видов взаимодействий в
отдельности. Пучок высокоэнергетических фотонов с начальной
интенсивностью /0, пройдя через слой поглотителя толщиной х,
будет иметь конечную интенсивность Г.
I = /ое-в\ (2.43)
где у. — полный линейный коэффициент ослабления (oa + os +
+т+х), а х — толщина поглотителя. Вид зависимости анало-
гичен закону радиоактивного распада, или закону Ламберта —
Бера.
Полный линейный коэффициент ослабления имеет размер-
ность см~'. Разделив его на плотность р, получим полный мас-
совый коэффициент ослабления с размерностью см2/г. Опреде-
ленный таким образом коэффициент ослабления относится к
условиям опыта, при которых фотонный пучок можно считать
достаточно узким. Практически условия узкого пучка удается
44
получать лишь в измерительной лаборатории. Обычные условия,
в которых ставится эксперимент по защите или радиационнохи-
мический эксперимент, — это условия широкого пучка. Условия
узкого лучка выполняются, если детектируют только те фотоны,
которые пе испытали какого-либо взаимодействия с веществом.
Рис. 2.12. Схема измерения коэффициентов ослабления в условиях узкого
и широкого пучков:
а — узкий пучок — эксперимент с «хорошей» геометрией, рассеянное излучение не
попадает на детектор; б -широкий пучок — эксперимент с «плохом» геометрией, рас-
сеянное Y-излученне попадает на детектор;
/ — источник у излучен ня; 2— коллиматор; 5 — поглотитель: 4 — детектор.
В детектор не должны попадать даже фотоны, отклонившиеся
после когерентного или некогерептпого рассеяния всего на не-
сколько градусов. Это относится также к фотонам, возникаю-
щим в результате фотоэффекта и аннигиляции пар.
Условия широкого пучка встречаются во всех обычных ра-
диациоппохимичсских экспериментах и практических задачах
защиты от у-излучения. В условиях широкого пучка (в условиях
«плохой» геометрии) значительная доля рассеянного излучения
попадает в детектор. На рис. 2.12 показаны ситуации, харак-
терные для условий узкого и широкого пучков.
Для условий широкого пучка в уравнении (2.43) должно по-
явиться два дополнительных фактора: геометрический фактор,
учитывающий геометрию источника, и фактор накопления, учи-
тывающий ту часть вторичного излучения в поглотителе, кото-
рая попадает в детектор.
45
Уравнение (2.43) для условий широкого пучка с учетом фак-
тора накопления запишется так:
Z = /0Be-^v, (2.44)
где В — фактор накопления.
Фактор накопления равен отношению наблюдаемой интен-
сивности прошедшего излучения к интенсивности излучения, ко-
торое попадало бы на детектор в условиях узкого пучка. Фак-
торы накопления для различных сред были рассчитаны лишь
для наиболее характерных геометрий источников [4, 5]. Учи-
тывать величину фактора накопления следует и при расчете по-
глощенной энергии, так как при проникновении электромагнит-
ного излучения в вещество возникают вторичные электроны.
В любом малом объеме будут находиться электроны, возник-
шие в нем, плюс электроны, рассеянные в этот объем из сосед-
них. По мере увеличения расстояния от поверхности раздела
число электронов, рассеянных в малый элемент объема, возра-
стает. На расстоянии, равном среднему пробегу вторичных
электронов, число электронов, рассеянных в малый элемент
объема, окажется максимальным. Если же перейти к погло-
щенной дозе, то максимальная поглощенная доза будет наблю-
даться в точке, расположенной па некотором расстоянии от по-
верхности раздела.
2.2.8. Осколки деления
Осколки деления несут различный заряд и обладают скорее
атомными, нежели ядерными размерами. Поэтому для них рас-
смотренная выше теория ионизационных потерь заряженными
частицами в чистом виде неприменима. Осколки деления за-
трачивают свою энергию па ионизацию, возбуждение и ядерные
столкновения. Относительный вклад этих процессов различен
для различных энергий и зарядов осколков.
Потери на возбуждение и ионизацию в среде с атомным
номером Z будут примерно пропорциональны
(Zavf (Z/m0) V2,
где Zav — средний заряд осколков деления.
Проникая в вещество, осколок деления будет захватывать
электроны и его заряд станет уменьшаться, а следовательно,
начнут уменьшаться и потери па ионизацию и возбуждение
одновременно. По мере замедления осколка деления возрастут
потери энергии на упругие ядерные соударения. Движение
осколка деления заканчивается при ядерном соударении, когда
происходит сравнительно большая передача энергии.
На рис. 2.13 приведена кривая потерь энергии осколками
деления и тут же для сравнения приведена кривая для а-частиц.
Минимум на кривой для осколков деления наблюдается там,
46
где Потери на ионизацию и возбуждение перестают быть основ-
ным механизмом потери энергии и где основным механизмом
становятся потери па упругие ядерные соударения. Масса,
заряд, дйипа пробега в воздухе и начальные энергии, харак-
Рис. 2.13. Кривая энергетических по-
терь осколков деления. Для срав-
нения приведена кривая энергетичес-
ких потерь а-частиц (в целях на-
глядности для а-частиц значения
dE/dx увеличены в 10 раз):
1 — тяжелые осколки деления: 2 —а-ча-
стицы.
терпыс для легких и тяжелых осколков деления ^U, приведены
в табл. 2.8.
Табл н и а 2.8*
Усредненные характеристики осколков деления, возникающих при распаде 23SU
Характеристика Легкие оскслки деления Тяжелые осколки деления
Масса 97 138
Начальный заряд 20 22
Энергия, Мэв 97 65
Средний пробег в воздухе, см 2,5 1,9
Скорость, см)сек 1,4-10» 0,93-10»
Maejed. массы 1,0 0,47
Энергия электронов, имеющих ту же скорость, кэв 0,50 0,26
* Данные таблицы взяты нз статьи: Steinberg М. Advances in Nuclear Science and En-
gineering, E- Henley and H. Konts (Eds), Academic Press Inc., New York, 1962.
2.3. НЕЙТРОНЫ
Основной вид взаимодействия нейтронов с веществом —
взаимодействие с атомными ядрами. Главными механизмами
потери энергии для нейтронов являются: упругое рассеяние (как
при столкновении бильярдных шаров), пеупругое рассеяние
(захватное рассеяние) и ядерные реакции. Быстрые нейтроны,
претерпевая упругие соударения с ядрами, особенно с ядрами
легких атомов (например, водорода), могут порождать заря-
женные частицы. Нейтроны, участвующие в ядерных реакциях,
помимо реакций деления могут также порождать заряженные
47
частицы, например протоны, а- и Р-частицы, а также ,могут
вызывать фотонное излучение. Существует определенная вероят-
ность потери энергии по тому или иному механизму в зависи-
мости от сечения конкретного процесса.
В общем случае сечения взаимодействий ядер с нейтронами
можно приближенно разделить на два компонента — сечение
рассеяния (упругого и пеупругого) и сечепис захвата:
^полн ^Расс “Г Фзахв’
Размер сечения взаимодействия зависит от скорости (кине-
тической энергии) нейтрона и от типа ядра. В соответствии
с энергией нейтроны приближенно делят па следующие группы.
1. Тепловые нейтроны: они находятся в тепловом равновесии
с веществом. Для пих наиболее вероятная скорость в максвел-
ловском распределении по скоростям при температуре 20,44° С
составляет 2200 м;сек. Такая скорость соответствует энергии
нейтронов 0,025 эв.
2. Медленные нейтроны. Это нейтроны с энергией от 0,1 до
300 эв.
3. Промежуточные нейтроны. Их энергия лежит в диапазоне
0,5—10 кэв.
4. Быстрые нейтроны. К ним относят нейтроны с энергией
от 10 кэв до 20 Мэе.
Сечения захвата нейтронов обычно указывают для конкрет-
ных процессов, таких, как активация, деление или ядерная
реакция. Сечения рассеяния подразделяют па сечения упругого
и неупругого рассеяния.
2.3.1. Упругое рассеяние нейтронов
Упругими считают такие соударения, при которых нейтроны
не проникают под поверхность ядра. Эти соударения типа
«столкновений бильярдных шаров» можно исследовать методами
классической механики. Атомы отдачи могут получать энергию
от пулевой до максимальной Ет при лобовом соударении.
Средняя потеря энергии на одпо столкновение, выражаемая в
виде логарифмического декремента, £ определяется выражением
g = In — = In --------i. (2.45)
£0 2Л А — 1 v ’
После п столкновений
In—= п Г И~1)2 1п^Ш------11 (2.46)
Ео 1 2А А — 1 J k ’
где А — атомный вес атома-мишени; Ео — начальная энергия
нейтрона; Е — энергия нейтрона после соударения; Еп — энергия
48
нейтрона после n соударений. Для веществ, у которых Л>10,
уравнение (2.45) можно использовать в приближенном виде
In — =-----------. (2.47)
Еа А -у 2/3 '
Из уравнений видно, что доля начальной энергии нейтрона,
теряемая при столкновениях, не зависит от начальной энергии
нейтрона. «Потерянная» нейтроном энергия превращается в ки-
нетическую энергию движущихся атомов и в конечном счете
растрачивается на ионизацию и возбуждение. Логарифмический
декремент g — важная характеристика вещества, так как позво-
ляет судить о замедляющих свойствах данной среды. Для про-
тонов уравнение (2.46) сводится к Еп=Еое~~п, которое справед-
ливо для водородсодержащих веществ. Нейтроны с энергиями
около 2—3 Мэв замедляются до тепловых энергий примерно
после 20 столкновений.
Пример 2.5. Нейтрон с энергией 10 Мэв «термализуется» в графитовом
замедлителе ядерпого реактора. Сколько столкновений нейтрона с углерод-
ными атомами должно произойти, чтобы энергия нейтрона упала до тепло-
вой (0,025 эв)?
Решение. В соответствии с уравнением (2.47)
2
g = —------- =0.158.
12 4- 2/3
,, Полное уменьшение энергии
Число столкновении ——— ----------------------
Потеря на столкновение
, f 10 - 106 у
In Е0/Еп 5 In 1 " ) \ 0,025 J = -— == 127. 0,158
При низких энергиях, когда длина волны нейтрона велика
по сравнению с радиусом рассеивающего ядра, атом, претер-
Рис. 2.14. Полное сечение вза-
имодействия нейтронов с во-
дородными ядрами в области
низких энергий.
певший соударение, может не получить энергии, достаточной для
разрыва связей с решеткой. Для подобного связанного атома
сечение рассеяния возрастает в (А + 1)2/А2 раз. На рис. 2.14
показано полное сечение (в барнах) водорода для области низ-
ких энергий нейтронов, где можно считать, что Ополн=Орасс-
49
Постоянство сечения для энергии нейтронов выше 1 эв харак-
терно для процесса рассеяния па легких элементах. Возрастание
сечения при энергиях нейтронов ниже 1 эв можно объяснить
переходом от взаимодействия со свободными атомами к ’взаимо-
действию со связанными атомами.
При более высоких энергиях, когда длина волны нейтрона
становится сравнимой с межатомными расстояниями, возникают
дифракционные эффекты. Иногда их называют «теневыми эффек-
тами». Благодаря им возникают осцилляции на гладких кривых.
2.3.2. Неупругое рассеяние
При достаточно высоких энергиях ядра и нейтроны могут
образовывать компаундные ядра, которые затем распадаются.
Если энергия нейтрона недостаточна для того, чтобы перевести
ядро в возбужденное состояние, то такой процесс будет экви-
валентен простому упругому рассеянию. Если же ядро переходит
в возбужденное состояние, то оно помимо нейтрона испускает
также один или несколько у-кваптов.
Для энергий нейтронов ниже нескольких Мэв сечепия пе-
упругого рассеяния малы по сравнению с сечениями упругого
рассеяния даже для тяжелых элементов. Это следует из табл. 2.9,
где даны экспериментальные значения сечений неупругого рас-
сеяния нейтронов различных энергий в разных поглотителях.
Т а б л и и а 2.9*
Сечения неупругого рассеяния моноэнергетических нейтронов, барн
Элемент Энергия нейтронов. Мэв Элемент Энергия нейтронов. Мэв
2,5 3,3 4, I 14 2,5 3,3 4, l 14
с 0,63 Sc 1,88 1,40
Na 0,53 0,57 Mo 1,9
Mg 0,77 0,78 1,08 Ag 2,13
Al 0,96 0,81 0,78 1,13 Cd 2,15 2,35 2,3 1,83
Р 0,7 1,13 Sn 1,65 1,73 2,11 1,81
S 0,54 1,15 Sb 2,00 2,11 2,13 1,90
Cl 0,6 0,90 Те 2,0 1,97
Ca 0,4 1,36 I 1,96 1,88
Cr 1,4 Ba 0,8 1,3 2,2
Fe 1,16 1,51 1,58 1,29 W 2,6 2,35
Co 1,40 Hg 2,6 2,99 3,36 2,65
Ni 0,83 1,35 -—. 1,38 Pb 1,69 1,72 2,42
Cu 1,58 1,67 1,68 1,35 Bi 0,62 0,62 1,14 2,44
Zn 2,18 2,18 1,58 1,37
* Значения, приведенные в таблице, взяты из доклада: Пасечник М. В. «Неупругое
рассеяние быстрых нейтронов атомными ядрами». —Международная конференция по
мирному использованию атомной энергии. Женева. 1955, т. 2. с. 3.
5Й
2.3.3. Захват нейтронов
ПоД словом «захват» в данном случае понимается целый
ряд различных процессов, в которых нейтрон захватывается
ядром. Судьба компаундного ядра определяется относительной
вероятностью различных типов ядерпого распада.
Рис. 2.15. Полное сечение и сечение захвата для взаимодействия проме-
жуточных нейтронов с ядрами алюминия.
Зависимость сечений нейтронного захвата от энергии ней-
тронов довольно сложна. Наиболее известное упрощение — так
называемый закон 1/К Это грубое допущение обычно применимо
только для нейтронов низкой энергии при взаимодействии с
изотопами тяжелых элементов. Однако для многих легких эле-
ментов закон 1/У можно распространить вплоть до энергий
нейтронов порядка 100 кэв.
Закон \/V утверждает, что вероятность протекания ядерпой
реакции для нейтрона пропорциональна времени, которое ней-
51
троп находится в окрестностях ядра. Это_ время, очевидно,
обратно пропорционально скорости нейтрона V:
(2.48)
Основная помеха для количественного описания сечений
нейтронного захвата — резкое увеличение захвата нейтронов в
районе энергий «ядерпого резонанса». На рис. 2.15, например,
приведены полные и захватные сечения для алюминия. Полные
сечения получают нз измерений пропускания нейтронов и, та-
ким образом, учитывают как рассеяние, так и захват. Сечепия
захвата были определены с помощью измерений наведенной
радиоактивности.
2.3.4. Ядерное деление
Ядерпое деление представляет собой уникальный захватный
процесс, в котором захватившее нейтрон ядро распадается и
испускает два или более нейтронов. Наиболее важная реакция
ядерпого деления 235U аппроксимируется уравнением (2.49):
2^П -|- п -> «Мо - 's/La - 2п 7₽ - Q, (2.49)
где ®5Мо и ,39La— характерные легкий и тяжелый осколки
деления, свойства которых приведены в табл. 2.8; Q — энергия
ядерной реакции.
2.3.5. Энергия отдачи
При захвате нейтрона и последующем испускании ядерной
частицы ядро испытывает определенную отдачу. Особый интерес
для радиационной химии представляет появление «горячих»
атомов, которые получили энергию в виде энергии отдачи при
таких ядерных реакциях, как (и, у)- Этот процесс передачи
энергии обсуждается также в главе V.
Испускаемый у-квапт имеет импульс ру=Еу/с. Атом отдачи
будет иметь импульс рг, равный импульсу ру и, следовательно,
энергия атома отдачи будет равна Е, — р2/2М = Е2/2Мс2, гдеЛТ —
масса атома отдачи. Та же формула применима и к испусканию
нейтрино. Учет энергии отдачи существен при анализе радиа-
ционных повреждений органических веществ. Например, для
легкого элемента (Л =20) энергия отдачи может быть порядка
1000 эв.
Пример 2.6. Рассчитать энергию отдачи для атома с массой 10 после
испускания у-кванта с энергией 2 Мэв.
Решение. Энергия, эквивалентная одной единице массы,
£ = Мс2 -= 931 • 10е эв;
52
или, иначе
Е2 4 1012 (Эв)2
Е = —-— -=----------------------------------------———— =
' 2.10^_------------------------. 9-1020^-
моль 6,62 • IO2» молекул сек
4 IO»2 зв2 4 . Ю12 эв
—-------------------------------- — —_-----------= 214 эв.
180 • 1 Q2« эрг эв 186,2 - 108
6 - 1023 молекул 1,6 • 10-12 эрг
Задачи
2.1. Чему равны максимальные пробеги электрона с энергией 4 Мэв
в стали и свинце?
2.2. Число пар ионов, создаваемых в воздухе протоном с энергией 10 Мэв,
равно 1100 на 1 мг/см2. Сколько пар ионов на 1 мг/см2 создает в воздухе
а-частпц, движущаяся с тон же скоростью?
2.3. Потери энергии для протона с энергией 10 Мэв в углероде составляют
4,2007-10'2 Мэв/(мг/см2). Чему будут равны потери энергии в воздухе
в Мэв/(мг/см2) и в Мэв/см? Чему будут равны потери энергии для дейтрона
с той же энергией?
2.4 Пробег а-частицы с энергией 2 Мэв в воздухе равен 1 см. а) Чему
равен се пробег в воде? б) Чему равен пробег в воде дейтрона с той же
скоростью? в) Чему равен пробег в воде дейтрона с той же энергией?
2.5. Вычислить и сравнить потери энергии электронами на излучение
и на соударения с учетом и без учета релятивистских поправок для энергий
электронов 0,01; 0,1; 1,0; 10 и 100 Мэв.
2.6. Вычислить и сравнить относительные потери энергии па соударения
и на рассеяние для электронов с энергией 100 Мэв.
2.7. Используя фотоэлектрический массовый коэффициент поглощения в
воздухе Для фотона с энергией 0.02 Мэв, найти соответствующие коэффи-
циенты в воздухе и свинце для фотона с энергией 0,01 Мэв. Сравните ре-
зультат с литературными данными.
2.8. Чему равны минимальная и максимальная энергии у-кванта, которые
могут возникнуть при комптоновском взаимодействии у-кванта с энергией
2 Мэв со свободным электроном?
2.9. Доказать, что фотон ие может передать всю свою энергию свобод-
ному электрону.
2.10. Сколько упругих соударений должен испытать нейтрон с энергией
5 Мэв в воде, прежде чем он термализуется? Оцепить длину пройденного
нейтроном пути.
2.11. Сечение захвата с энергией 1 эв для Cd равно 20 барн. Какое
сечение захвата_будет иметь Cd для нейтронов с энергией 100 эв, если спра-
ведлив закон 1/V?
Глава III
источники ИЗЛУЧЕНИЙ
Источники излучений, используемые в радиационной химии,
можно разделить на две большие категории. К первой категории
относятся источники, где заряженные частицы разгоняются до
высоких энергий электрическими методами, ко второй — источ-
ники, в которых происходят ядерные превращения. Это радио-
активные изотопы и ядерные реакторы. Среди источников обеих
категорий существуют широкие возможности выбора определен-
ной установки, что обеспечивает разнообразие условий радиа-
ционного воздействия. Необходимый источник излучения можно
подобрать практически для любого эксперимента, представляю-
щего интерес с точки зрения химии.
3.1. УСКОРИТЕЛИ И ЭЛЕКТРИЧЕСКИЕ РАЗРЯДНЫЕ УСТРОЙСТВА
Простейший ускоритель можно построить, скомбинировав
высоковольтный трансформатор с разрядной трубкой. Частицы
набирают энергию, двигаясь от анода к катоду или от катода
к аноду в зависимости от знака заряда. В самом простом случае
электрическая разрядная трубка может питаться от сети с на-
пряжением 110 в через трансформатор на 5000 в. С источниками
этого тина проведены многочисленные эксперименты. Основные
трудности при использовании таких источников обусловлены
отклонениями от заданной энергии ионов, эффектами на стен-
ках и фотохимическими эффектами.
3.1.1. Электрические разрядные трубки
Хотя для радиационнохимических экспериментов чаще ис-
пользуют источники верхнего киловольтного (кэв) или нижнего
мегавольтпого (Мэе) диапазона, многие работы проведены с
киловольтными электрическими разрядными трубками. Как по-
казано на рис. 3.1, в зависимости от плотности тока и отношения
давления газа к напряженности поля в зоне разряда существует
54
Отношение давления газа к на-
пряженности электрического поля
три вида разряда — тихий, тлеющий и искровой. Простейшим
(и, видимо, наиболее дешевым) газоразрядным устройством
является катушка Тесла. В зависимости от условий (давления
газа, величины зазора между электродами и т. п.) катушка
Тесла может работать в режиме тлеющего разряда или в искро-
вом режиме.
Тихий разряд обычно наблюдается при давлениях, близких
к атмосферному, и при низких плотностях тока. Тихий разряд
обусловлен собственной проводи-
мостью газа, возникающей благо-
даря остаточной ионизации. Для
него характерно отсутствие эф- §
фекта пространственного разря- е
да. При увеличении напряжения g
возникает особый вид тихого раз- ё
ряда, так называемый коронный г
разряд
Обычно корона образуется
вблизи электродов в неоднород-
ном электрическом поле, и ее
можно заметить по характерно-
му свечению газового слоя, в ко- Рис. 3.1. Области существования
тором происходит ударная иони- основных типов электрического
зация. Озонаторы, наиболее мае- разряда,
совый пример промышленного
использования электрических разрядных трубок, работают в
режиме тихого разряда. При увеличении плотности тока корон-
ный разряд переходит в искровой. Характерная особенность
искрового разряда — прерывистость. После того как произошел
искровой разряд, газ становится проводящим и разряд гасится.
Затем напряжение восстанавливается, и происходит новый
искровой разряд. Если параллельно с разрядными электродами
включен конденсатор, как это делается в автомобильной системе
зажигания, то разряд называют конденсаторным искровым.
Тлеющий разряд наблюдается обычно при низких давлениях,
хотя в некоторых условиях он может происходить и при атмо-
сферном давлении. Свечение наблюдается в виде топкой пленки
около катода, переходящей в слабо светящийся слой (темное
катодное пространство). В направлении анода лежит другая
темная область (темное астопово пространство). Такое чере-
дование темных и светлых полос возникает благодаря сложному
распределению падения напряжения по трубке и из-за различ-
ных ионных процессов, которые подробно рассматриваются
в книгах, специально посвященных электрическим явлениям
в газах.
Создание напряжений больше нескольких десятков тысяч
вольт с помощью батарей или генераторов оказывается непрак-
тичным. Поэтому были разработаны методы повышения напря-
55
жения первичных источников. С этой целью использовался целый
ряд физических принципов, и сейчас существует много разно-
образных ускорителей заряженных частиц.
3.1.2. Ускоритель Кокрофта—Уолтона
Сконструированный в конце 20-х годов ускоритель Кок-
рофта — Уолтона широко известен. Он сыграл заметную роль
в развитии ускорительной техники. В этом ускорителе выходное
напряжение трансформатора многократно выпрямляется и уси-
ливается. В результате
Рис. 3.2. Схема ускорителя Кокрофта—Уол-
удается получать заря-
женные частицы с энер-
гиями до 1,5 Мэв при то-
ках в несколько милли-
ампер.
В простейшем вариан-
те (рис. 3.2) устройство
состоит из трех эквива-
лентных конденсаторов
Сь С2 и С3, один из кото-
тона- рых подключен к источ-
нику напряжения U. Кро-
ме того, имеются конденсаторы CJ2 и С23. В цикле зарядки, когда
С12 и С23 подключены к С2 и С3, конденсатор С23 заряжается до
напряжения U. Затем происходит переключение (сплошные ли-
нии на рис. 3.2), и заряд распределяется между С23 и С2. Если
емкости конденсаторов одинаковы, то напряжение па С23 и С2
становится равным 1/2 U. После следующего переключения С2з
перезаряжается, а С2 отдаст часть своего заряда па конденса-
тор С12. Процесс подзарядки продолжается до тех пор, пока на
конденсаторе С( не установится напряжение 3 U. Таким обра-
зом, степень усиления напряжения равна числу первичных кон-
денсаторов Сп.
На самом деле вместо механических переключателей исполь-
зуют кенотроны. Применяют также ряд других модификаций
описанной схемы. Ускоритель Кокрофта — Уолтона используется
главным образом для ускорения положительно заряженных
ионов.
3.2. УСКОРИТЕЛЬ ВАН-ДЕ-ГРААФА
В ускорителе Ван-де-Граафа используется следующий основ-
ной принцип. Если заряженное тело привести в соприкосновение
с полым электродом из проводящего материала, то весь заряд
перейдет на полый электрод независимо от фактической раз-
ности потенциалов. Таким образом, если бы не существовало
затруднений с изоляцией высоковольтного электрода и не про-
56
исходила бы утечка из системы переноса зарядов, то при бес-
конечном количестве актов передачи заряда можно было бы
получить бесконечно высокое напряжение.
Как показано на рис. 3.3, электрический заряд переносится
на высоковольтный электрод с помощью ремня из хлопчато-
Рис. 3.3. Схема ускорителя Ван-де-
Граафа:
1 — электрический заряд переносится на ре-
мень из изоляционного материала: 2 — ремень
осуществляет механический перенос заряда на
изолированный высоковольтный электрод, ко-
торый имеет форму полусферы; 3 — здесь за-
ряд автоматически переносится с ремня на
электрод, что позволяет создать высокую раз-
ность потенциалов между терминальным
электродом и нижней частью ускорителя;
4 — атмосфера из сжатого азота изолирует
высоковольтный электрод от остальных час-
тей ускорителя и препятствует возникновению
электрического разряда; 5 — в трубке из
стекла и металла поддерживается очень высо-
кий вакуум; это единственный путь, по кото-
рому электроны могут покинуть высоковольт-
ный электрод; 6 — под действием разности по-
тенциалов между высоковольтным электродом
и нижней частью ускорителя пучок электро-
нов ускоряется до чрезвычайно больших ско-
ростей; 7~с помощью электромагнитных ка-
тушек осуществляется развертка электронною
пучка, в результате которой проходящие вни-
зу образцы подвергаются равномерному облу-
чению; 8— характер лучевой обработки образ-
ца зависит от скорости конвейерной ленты и
энергии электронного пучка.
бумажной ткани, пропитанной неопреном. Электрический заряд
передается на ремень с помощью регулируемого коронного раз-
ряда через систему стальных иголок или экранов, расположен-
ных в непосредственной близости к ремню. Передача заряда на
ремень происходит в области, свободной от электрического поля,
при нулевом потенциале. Постоянное высокое напряжение, ре-
гулируемое в пределах 25- 40 кв, подается между шкивом и
остриями, возле которых происходит ионизация изолирующего
газа в заряде. Образовавшиеся заряды удерживаются на по-
верхности ремня.
Ремень переносит электрический заряд (электроны) к высо-
ковольтному электроду, где через систему специальных щеток,
расположенных так, что потенциал ремня увеличивается, заряд
передается кверху па изолированный высоковольтный электрод.
Таким образом, заряд попадает па терминальный электрод без
непосредственного использования электродвижущей силы источ-
57
инка питания. Так как утечка заряда ограничивает рост на-
пряжения в системе, весь генератор помещают в герметичную
оболочку, заполненную сухим сжатым газом.
Высоковольтный электрод представляет собой полированную
металлическую полусферу, которая поддерживается набором
эквипотенциальных металлических пластин, разделенных изоля-
торами. Пластины последовательно соединены через сопротив-
ления, образующие делитель напряжения, что позволяет созда-
вать регулируемый градиент электрического поля вдоль стопки
пластин.
Разрядная трубка ускорителя состоит из стеклянных торо-
идов, сцементированных с металлическими дисками внутри
опорной колонны. Таким образом вдоль трубки па стопке дисков
(пластин) возникает градиент напряжения. В трубке должно
быть давление 10-5-—10 6 мм рт. ст. Электрическое поле внутри
трубки следует поддерживать достаточно однородным, и при
различных градиентах напряжения должна обеспечиваться фо-
кусировка заряженных частиц. Заряженные частицы, электроны
или положительные ионы, возникают вблизи высоковольтного
электрода со специальным устройством. Для получения поло-
жительно заряженных ионов используется газовый разряд
(обычно в атмосфере Н2).
Генератор Ван-де-Граафа может работать в импульсном
режиме. В этом случае полпая энергия импульса определяется
в большей степени емкостью высоковольтного электрода, чем
общей мощностью генератора. Можно получать электронные
импульсы длительностью в несколько микросекунд при макси-
мальной силе тока в несколько сот миллиампер. Однако это
требует специальных мер предосторожности, которые позво-
ляют свести к минимуму падение напряжения во время им-
пульса.
Генератор Вап-дс-Граафа наиболее важный, по не един-
ственный из электростатических ускорителей. Недавно возро-
дился интерес к так называемому «цилиндрическому» генератору
Феличи, в котором роль ремня выполняет цилиндр из изоли-
рующего материала. Это устройство позволяет получать токи
большей силы, чем генератор Ван-де-Граафа. Конструкция
генератора Феличи быстро совершенствуется.
Генератор Ван-де-Граафа принадлежит к числу существенно
низкотоковых устройств. Мощность генератора ограничивается
эффектами, связанными с накоплением пространственного за-
ряда в точке, где заряд переносится па ремень, невозможностью
передачи через ремень большего тока и низкими скоростями
ремня. Этими принципиальными трудностями отчасти и обуслов-
лено появление большого числа высокотоковых устройств с бо-
лее низкими напряжениями (на 3 млн. в и пиже).
Мишень из тяжелого металла позволяет превратить электрон-
ный пучок ускорителя Ван-де-Граафа в коротковолновое рент-
58
геповское излучение (тормозное излучение). С помощью ускори-
теля можно получать и нейтроны, используя целый ряд ядер-
ных реакций, таких, как реакция (у, п) на бериллии пли реак-
ция взаимодействия нейтронного либо протонного пучка [ (d, п)
и (р, «)] с литием.
3.3. РЕЗОНАНСНЫЕ ПРЕОБРАЗОВАТЕЛИ
Резонансные преобразователи, которые выпускают в настоя-
щее время, способны создавать пучки электронов с энергией
1, 2 и 4 Мэв при средней силе
ный преобразователь был
сконструирован в период
1937—1939 гг. как источник
рентгеновских лучей для лече-
ния рака. В 1948 г. конструк-
цию преобразователя удалось
модифицировать таким обра-
зом, что он стал служить ис-
точником электронов. Надеж-
ность работы и высокие токо-
вые параметры делают резо-
нансный преобразователь осо-
бенно удобным для использо-
вания в промышленности.
На рис. 3.4 показан разрез
резонансного преобразователя.
Низковольтная первичная об-
мотка конической формы окру-
жает нижнюю часть высоко-
вольтной вторичной обмотки,
образованной большим числом
топких плоских пластин. На
каждой пластине возникает
перепад напряжений примерно
в 8,5 кв. Нижняя часть высо-
ковольтной обмотки заземле-
на, верхняя часть заэкраниро-
вана.
Вестендорп [1] так объяс-
няет принцип работы резонанс-
ного преобразователя: «Высо-
ковольтная обмотка обладает
большой индуктивностью, ко-
торая выбрана таким обра-
зом, чтобы при рабочей часто-
те 180 гц наступал резонанс с
нагрузкой. Для функциопиро-
тока вплоть до 6 яа. Резопанс-
Рис. 3.4. Схема резонансного преоб-
разователя:
1— фокусирующая катушка: 2—первичная
обмотка; 3 — стеклянный стержень; 4 —
экраны; 5 —ускоряющие электроды; 6 —
катодное устройство; 7 — пружина; 8 - -
разрезная латунная зашита: 9— устройст-
во для охлаждения изолирующего газа;
10 — стальной кожух; 11 — индуктивный
регулятор; 12— прижимная пластина; 13—
вторичные катушки; 14—слоистая защита;
15 — привод регулятора: 16 — свинцовая
прокладка: 17 — стеклянное покрытие;
/«‘( — двигатель регулятора; 19 -окно уско-
рительной трубки.
59
вания этой большой резонансной системы, амплитуда колебаний
в которой может оказаться порядка миллиона вольт и более,
достаточно сравнительно небольшой входной мощности, расхо-
дуемой на работу электроннолучевой трубки и на компенсацию
потерь в трансформаторе. Поэтому для питания резонансной си-
стемы оказывается достаточно сравнительно маленькой кониче-
ской первичной обмотки. Механической аналогией резонансного
преобразователя может служить качающийся маятник или ме-
ханическая система, в которой колебания .массы происходят в
резонанс с упругой опорой. В таких механических системах при
малых мощностях небольшая сила, воздействующая на систему
с нужной частотой, сможет поддерживать большие амплитуды
колебаний, в которых запасена значительная энергия».
Разрядная трубка проходит через центральную часть высо-
ковольтной обмотки. На верху трубки находится катод, затем
следуют промежуточные электроды, помещенные в стеклянную
оболочку. В нижней выводной части трубки имеется окошко,
через которое проходит пучок. Потери интенсивности пучка при
прохождении через окошко заметно ухудшают характеристики
преобразователя. При высоких значениях токов (6 ма) окошко
должно быть сравнительно толстым. Кроме того, средняя энер-
гия электронов в пучке значительно ниже максимальной. Все
это приводит к тому, например, что для пучка электронов с
энергией 1 Мэв при толщине титанового окошка 0,18 мм теряется
почти 20% энергии.
Важным вспомогательным устройством во всех типах пре-
образователей является отклоняющая система, которая смещает
электронный пучок относительно конвейерной ленты, проходя-
щей под трубкой. В преобразователях, где велики потери энергии
в окошке, отклоняющая система позволяет также поддерживать
его температуру ниже критической. В отклоняющей системе
обычно используется магнитное поле, которое 180 раз в секунду
смещает электронный пучок поперек конвейерной ленты. Одно-
временно, чтобы предотвратить перегревание окошка, другая
система развертки смещает пучок в перпендикулярном направ-
лении (направлении малого размера окошка) с частотой
200 000 гц.
Для таких устройств, как резонансные преобразователи, за-
висимость дозы от глубины проникновения в образец оказы-
вается сложной функцией характеристик окошка и самого
преобразователя, так как выходной электронный пучок не
является моноэнергстическим.
3.4. ТРАНСФОРМАТОР С ИЗОЛИРОВАННЫМ СЕРДЕЧНИКОМ
Новейший из поступающих в продажу ускорителей — транс-
форматор с изолированным сердечником, изобретенный Робертом
Ваи-де-Граафом. Это главным образом низковольтный промыш-
60
леииый источник излучения, способный при сравнительно малых
затратах давать на выходе высокую мощность излучения. Транс-
форматор предназначен для работы на промышленных линиях.
Устройство состоит из ряда вторичных обмоток, переменное
напряжение которых выпрямляется и складывается с напряже-
нием предшествующих и последующих элементов. Обмотки воз-
буждаются участками сердечника, которые заизолированы друг
Рис. 3.5. Схема трансформатора
с изолированным сердечником без
системы ускорения электронов (и)
и эквивалентная электрическая
схема (б):
/ — вторичная обмотка: 2 — первичная
обмотка: <?—магнитный поток; 4—эле-
менты магнитного сердечника; 5 — вы-
соковольтный электрод.
от друга последовательно включенными выпрямителями и кон-
денсаторами. Трансформатор помещается в стальном танке,,
заполненном изолирующим газом.
Магнитный сердечник в системе генерации напряжения раз-
делен на сегменты, изолированные друг от друга, и поэтому
каждый сегмент сердечника может находиться под полным на-
пряжением, создаваемым иа данной стадии. Высоковольтный
электрод от остальной части сердечника отделяет большой зазор.
Благодаря очень небольшим зазорам между сегментами сер-
дечника и большому сечению зазора между сердечником и
высоковольтным электродом магнитное сопротивление цепи
остается низким. В маленькие зазоры вкладываются листы
топкого пластика.
На рис. 3.5 приведена схема трансформатора. Высоковольт-
ный электрод находится внизу под центральной колонной. Верх-
няя часть колонны и наружная часть сердечника служат маг-
питопроводом и находятся при нулевом потенциале. Питание-
первичной обмотки осуществляется с помощью мотор-геператора
или генератора сигналов с частотой 10 кгц. Каждый сегмент
сердечника имеет вид сковороды, на которую надета ленточная
обмотка, представляющая собой вторичную обмотку трансфор-
61
матора. На каждой из вторичных обмоток индуктируется напря-
жение около 25 кв.
Переменное напряжение, возникающее во вторичных об-
мотках, превращается в постоянное с помощью кремниевого
выпрямителя. Сложение напряжений вдоль колонны осущест-
вляется с помощью конденсаторов, последовательно включенных
между сегментами сердечника. В последних вариантах конструк-
ции трансформатора с изолированным сердечником используются
трехфазпый ток и стандартные частоты питания.
3.5. ДИНАМИТРОН
Динамнтрон, так же как и трансформатор с изолированным
сердечником или резонансный преобразователь, относится к
числу сравнительно низковольтных высокотоковых устройств.
В этом ускорителе исполь-
зуется каскадная система вы-
прямления, в которой все вы-
прямители параллельно под-
ключены к высокочастотному
осциллятору, как это показано
па рис. 3.6.
Четыре больших высоко-
частотных электрода, располо-
женные внутри цилиндрическо-
го сосуда со сжатым газом, пе-
редают напряжение от осцил-
лятора к выпрямительной си-
стеме. Через емкостную связь
радиочастотное напряжение
подается па коронные кольца,
которые механически прикреп-
лены к каждому выпрямителю
и находятся в атмосфере ди-
электрического газа высокого
Рис. 3.6. Схема динамитропа:
.1 — высокочастотный генератор: 2 - резо-
нанс; 3--выпрямители: 4 — высокочастот-
ный электрод; 5- выходное напряжение;
6 — высокочастотный дроссель; 7 — корон-
ные кольца; 8— ионный источник; 9 - пярпрния
ускорительная трубка; 10 — вакуумная си- давления.
стема; //—тороидальная катушка. Все ВЫПряМИТСЛИ НЭХОДЯТСЯ
под одним и тем же высоко-
частотным напряжением. Постоянный ток, идущий через цепоч-
ку выпрямителей, создает высокое постоянное напряжение на
высоковольтном электроде. Заряженные частицы образуются в
ионном источнике, связанном с высоковольтным электродом, и
в вакуумной трубке происходит ускорение заряженных частиц.
3.6. ИМПУЛЬСНЫЙ ГЕНЕРАТОР
В импульсном генераторе ряд параллельно включенных кон-
денсаторов заряжается от сравнительно низковольтного выпря-
мительного устройства. Затем с помощью искровых разрядников
:62
происходит последовательный разряд конденсаторов. Комбина-
ция импульсного генератора с вакуумной разрядной трубкой
лежит в основе конструкции ускорителя Брэша, Вэли и Хюбера,
который был назван ими «капаситропом». Вместе с генератором
Ван-де-Граафа это один из первых ускорителей, использован-
ных для радиационпохимических экспериментов. Сейчас такие
ускорители не выпускают.
3.7. ЛИНЕЙНЫЕ УСКОРИТЕЛИ
Если при достаточной силе тока нужно получить пучки
частиц с высокой энергией (>4 Л4эв), то необходимо решить
проблему изоляции, которая существует для всех устройств,
где ускорение осуществляется в постоянном электрическом поле.
Есть два пути решения проблемы. Но и в первом, и во втором
случаях частица набирает энергию небольшими порциями, мно-
гократно пересекая электрическое или магнитное поле. В первом
случае происходит ускорение
частиц на замкнутых орбитах,
как это делается в бетатроне,
циклотроне и синхротроне. Во
втором — ускоряются частицы,
движущиеся по линейной тра-
ектории. При высоких напря-
жениях этот метод позволяет
получать большую силу тока.
Частицы -в линейном ускорите-
ле разгоняются либо движу-
щимся электромагнитным по-
лем, либо радиочастотной элек-
тромагнитной волной, фазовая
скорость которой должна рав-
няться скорости движения ча-
стиц. Основные компоненты
линейного ускорителя показа-
ны па рис. 3.7.
Современный линейный ус-
коритель, потребляющий зна-
чительные мощности в радио-
частотном диапазоне, в конеч-
ном счете есть результат раз-
работок, проведенных в воен-
ное время в Англии и США
при создании радиолокацион-
ной техники. В Англии в каче-
стве источника радиоволн ис-
пользовали эффективные и
компактные магнетроны. Про-
Рис. 3.7. Схема линейного уско-
рителя:
/ — источник питания: 2 — постоянное
напряжение; 3 - модулятор; 4 им-
пульсное напряжение; 5 - СВЧ-генера
тор; 6 — импульсы СВЧ-колебаний; 7 —
СВЧ-волновод; 8—инжектор электронов;
9 — ускорительный волновод.
в®
мышлепность США разработала конструкции мощных клист-
ронов. И тот и другой тип генераторов применяется до сих пор.
При распространении радиочастотных колебаний внутри
полой гладкостепной металлической трубки вдоль оси трубки
создается электрическое поле, которое используется для уско-
рения. С помощью металлических дисков или диафрагм, встроен-
ных в трубку, фазовую скорость распространения радиоволн
делают равной скорости электронов.
Электроны, инжектированные во время импульса в направ-
лении оси трубки, оказываются в ускоряющем поле и движутся
дальше в фазе с волной, забирая от нее энергию и ускоряясь.
Инжектор электронов и источник радиоволн синхронизируются
с помощью триггерных импульсов, посылаемых задающим ге-
нератором. Для того чтобы при разумной длине разгона полу-
чить достаточную энергию частиц, необходимо обеспечить
напряженность ускоряющего поля в десятки киловольт па 1 см.
Это в свою очередь требует, чтобы входная мощность радиоволн
была порядка мегаватт. Поэтому нужны импульсные источники
радиоволн. Отвечающее этим требованиям импульсное оборудо-
вание, основным компонентом которого является импульсный
магнетрон, удалось сконструировать для военных радиолока-
ционных установок.
После 1949 г., когда в Станфорде построили первый линей-
ный ускоритель с клистроппым источником радиоволн, наме-
тился постепенный переход от ускорителей с магнетронными
источниками радиоволн к клистронным липейпым ускорителям.
Вначале использовались генераторы радиоволн, работающие в
S-полосе (2855 Мгц), одпако начиная с 1959 г. стали использо-
вать также и £-полосу (1300 Мгц).
Для линейных ускорителей число ограничений минимально
по сравнению с любым другим типом ускорителя. При прием-
лемых затратах можно построить линейный ускоритель с энер-
гией частиц в миллиарды электронвольт или ускоритель, который
при энергии частиц 100 Мэв будет давать на выходе мощность
40 кет. В настоящее время длительность импульсов линейного
ускорителя можно варьировать от паносекундпого до миллисе-
кундного диапазона. Миллисекундный диапазон стал доступен
лишь после того, как были сконструированы клистроны, рабо-
тающие в L-полосе.
3.8. ГЕНЕРАТОРЫ РЕНТГЕНОВСКОГО ИЗЛУЧЕНИЯ
Любой из описанных здесь ускорителей, так же как и про-
•стую трубку Кулиджа, можно использовать для генерации элек-
тромагнитного излучения, если направить пучок электронов на
мишепь из тяжелого металла. Тормозное излучение, возникаю-
щее при торможении электронов в поле ядер, и характеристи-
ческое излучение, возникающее при переходе электронов с внеш-
64
них оболочек на внутренние, составляют в сумме то, что принято
называть «рентгеновским излучением». В следующих разделах
рассматриваются свойства рентгеновского излучения.
3.8.1. Угловое распределение рентгеновского излучения
Угловое распределение рентгеновского излучения можно рас-
считать теоретически. Для тонких мишеней при низкой энергии
электронов кривая углового распределения образует два ле-
пестка, направленные под прямыми углами к падающему пучку
электронов. Примерно такое же угловое распределение наблю-
дается для фотоэффекта. При больших энергиях электронов
лепестки углового распределения отклоняются вперед и рас-
пределение становится более симметричным. Вид углового
распределения зависит от толщины мишени, и для толстых
мишеней его невозможно рассчитать. Для толстых мишеней при
рабочем напряжении 400 кв интенсивность рентгеновского излу-
чения максимальна в направлениях, перпендикулярных к па-
дающему пучку. Однако для энергий электронов свыше 1 Мэв
распределение интенсивности сдвигается в направлении элек-
тронного пучка. Для электронов с энергией 3 Мэв уже 65%
энергии излучения попадает в направленный вперед конус.
3.8.2. Спектральное распределение рентгеновского излучения
Рис. 3.8. Влияние фильтров на спектр
рентгеновского излучения:
/ - - исотфильтроваиное излучение с харак-
теристическими К- и L-линиями; 2 — тон-
кий фильтр; 3 — толстый фильтр.
Максимальная энергия фотонов определяется максимальным
напряжением. Однако весьма значительная доля излучения при-
ходится на фотоны с низкой
энергией, многие из которых
поглощаются в самой мише-
ни. Таким образом мишень Л
оказывается и своего рода §
фильтром. <5
Обычно помимо мишени 3
ставят специальный вспомо- g
гательный поглотитель, ко-
торый отсекает низкоэнер-
гетическую часть излучения.
Это делают в тех случаях,
если не хотят получать на
поверхности особенно высо-
кие дозы. Спектральное
распределение в пучке рент-
геновских лучей обычно ха-
рактеризуется толщиной по-
лупоглощения, которая равна слою поглотителя, снижающему
интенсивность пучка вдвое. Например, для 400-киловольтного
излучения толщина полупоглощения равна 4 мм меди.
з Зак. 907
65
Толщина полупоглощепия, как и следует ожидать, зависит
от толщины фильтра (рис. 3.8). На рис. 3.8 показаны также
К- и L-пики характеристического излучения, которые на прак-
тике незначительно меняют картину. Они либо отфильтровы-
ваются, либо дают лишь малую долю общей интенсивности.
Поэтому обычно спектральную кривую рентгеновских лучей счи-
тают гладкой и непрерывной.
3.8.3. Интенсивность рентгеновского излучения
Полный выход фотонов равен площади, которую ограничи-
вают кривые на рис. 3.8. Полный выход для телесного угла 4л;
растет практически линейно с увеличением атомного номера
мишени. Увеличение энергии электронов приводит к экспонен-
циальному возрастанию выхода. Эффективность превращения
энергии электронов в полезное излучение в большой степени
зависит от их энергии. Для электронов с энергией 200 кэв эта
величина составляет около 0,2%. с энергией 2 Мэв — 7%, с
энергией 3 Мэв—10%, с энергией 4 Мэв—12% [2]. Столь
низкая эффективность преобразования энергии электронов в из-
лучение заставляет заботиться о том, чтобы мишень была термо-
стойкой и хорошо охлаждалась.
На рис. 3.9 суммированы результаты ряда измерений интен-
сивности рентгеновского излучения в направлении электронного
пучка в зависимости от энергии бомбардирующих электронов
для толстых мишеней из тяжелых металлов.
3.9. ПОЛУЧЕНИЕ НЕЙТРОНОВ НА УСКОРИТЕЛЯХ
Для получения нейтронов на ускорителях можно использо-
вать реакции (р, ti), (d, п) и (у, п). Некоторые из наиболее
часто используемых реакций представлены в табл. 3.1. Энергия
Таблица 3.1
Ядерные реакции, используемые для получения нейтронов
Вид излучения Реакция £ • min Мэв । Вид излучения Реакция Е . mm Мэв
а-Частицы вВс (а, л) 12С 11Ве (а, np4N 7Li (а, п) 10В 5,71 Протоны Т (р, л) 3Не 9Ве (р, п) ВВ 7Li (р, п) 7Вс 1,19 1,88
Дейтроны 2Н (d, л) 3Нс 3H(d, и) 4Не вВе (d, п) 10В 2,45 14,05 Фотоны D(y, п)П вВе (у, п)8Ве 238U (у, п) 23,U 0.1 0,16 6,0
нейтронов зависит от характера ядерпой реакции в мишени, энер-
гии падающей частицы, угла, под которым выбрасываются нейт-
66
рон, и поэтому в каждом конкретном эксперименте может за-
метно меняться.
Выход нейтронов также оказывается сложной функцией вида
ядерпой реакции, энергии бомбардирующих частиц и других
Интенсивность рентгеновского излучения
Рис. 3.9. Зависимость интенсивности
рентгеновского излучения от энергии
бомбардирующих электронов для тол-
стой мишени из металла с высоким
атомным номером:
1 - интенсивность. р1(ма-мин), на расстоянии
I м (в направлении электронного лучка);
Д интенсивность, рКквт • мин), на расстоянии
1 м (в направлении электронного пучка);
.V — интенсивность. рКквт -мин), на расстоянии
I м (перпендикулярно к направлению элек-
тронного пучка).
Рис. 3.10. Выход нейтронов в зависи-
мости от энергии бомбардирующих ча-
стиц для нескольких ядерных реакций:
/ — толстая мишень из льда I)2O; D(d. л)3Пе;
2 мишень Т Т толщиной 2,5 мг/см-'.
T(d, п)4Нс; 3 — толстая мишень; 'Bc(d, л),0В;
4— 236U(y, л)237Ъ’: 5 — рВе(т, л)8Ве; 6 — толстая
мишень; ?1л(р. л)’Вс: 7 - толстая мишень —
vBe(p. л)*В.
характеристик первичного пучка. На рис. 3.10 показаны ней-
тронные выходы в зависимости от энергии бомбардирующих
частиц для большей части широко используемых ядерных ре-
акций.
3.10. ЯДЕРНЫЕ РЕАКТОРЫ КАК ИСТОЧНИКИ ИЗЛУЧЕНИЯ
Ядерные реакторы представляют собой чрезвычайно сложные
источники излучения. Характер, энергетический спектр и ин-
тенсивность излучения зависят не только от параметров кон-
3:
67
кретного реактора, ио и от того, в какой точке реактора эти
излучения наблюдаются. Как источники излучения реакторы
привлекают интерес прежде всего потому, что их излучение
стабильно и можно облучать большие объемы при чрезвычайно-
высоких мощностях дозы (в отличие от изотопных источников).
В будущем реакторы могут стать крупнейшими промышленными
источниками излучения. В реакторе возникают и электромаг-
нитные излучения и потоки частиц.
3.10.1. Нейтроны
Рис. 3.11. Нейтронный спектр реак-
тора для испытания материалов.
1 — распределение Максвелла—Больцмана;
2—поток после фильтрации через 3,2 мм
бора; 3 — поток после фильтрации через
1,6 мм бора; 4— спектр деления.
При распаде одного ядра И5ЪТ в среднем выбрасывается
2,5 нейтрона, обладающих средней энергией около 2 Мэв. В ре-
акторе эти быстрые нейтро-
ны термализуются в резуль-
тате столкновений с ядрами
замедлителя, а затем пре-
терпевают захват. Захваты-
ваться могут и быстрые ней-
троны, но, как указывалось
в главе II, сечение захвата
для медленных нейтронов
намного больше, чем для
быстрых.
Соотношение между
средним потоком нейтронов
и мощностью для теплового
реактора выглядит так:
Р = 5- 10-"Фаг,С, (3.1>
где Р — мощность реактора,
вт\ Фащ — средняя плотность
потока тепловых нейтронов,
нейтрон/ (см2 • сек); G — за-
грузка реактора, г 235U.
На рис. 3.11 приведен
нейтронный спектр, который
измерялся на расстоянии
:акторе для испытания мате-
риалов. Этот реактор работает на обогащенном уране, а в ка-
честве замедлителя используется обычная вода. В правой части
рисунка, в диапазоне Мэв, дана величина потоков для быстрых
нейтронов. Отдельно пунктирной линией показан энергетический
спектр нейтронов деления. Следует заметить, что нейтронный
спектр — одна из наиболее трудных для измерения характери-
стик реактора. Более полно техника измерений и вид нейтрон-
ных спектров будут рассматриваться в главе IV.
7,62 см от реакторной решетки в
68
3.10.2. Электроны
Радиоактивные продукты деления помимо прочих частиц
излучают большое число электронов. Средняя энергия испускае-
мых электронов 0,4 Мэв, а скорость их образования описы-
вается эмпирическим уравнением Л = 3,8- 10~6 tr1>2, где А—ско-
рость, электрон/(сек-деление); t — время после деления, дни.
До тех пор пока продукты деления не будут выделены либо
выведены из тепловыделяющих элементов, как это делается в
хемоядерном реакторе, электроны будут в основном поглощаться
'.и их нельзя использовать для практических целей.
3.10.3. у-ИЗЛУЧЕНИЕ
Кроме у-излучения, сопровождающего деление, электромаг-
нитное излучение большой интенсивности испускают сами про-
дукты деления. В меньших масштабах электромагнитное излу-
чение возникает при реакциях захвата нейтронов и при
торможении заряженных частиц. От 5,5 до 5,7% энергии деления
, расходуется на у-излучение.
3.11. БОЛЬШИЕ ИЗОТОПНЫЕ ИСТОЧНИКИ ИЗЛУЧЕНИЯ
Существует более 1300 изотопов, которые в принципе можно
использовать в качестве источников излучений. Эти изотопы
либо специально изготавливают в ядериых реакторах с по-
мощью реакций (у, п), (п, р) или (п, а), либо выделяют среди
тех сотен миллионов кюри продуктов деления, которые полу-
чают в качестве отходов в процессе переработки ядерного го-
рючего.
Если, однако, ограничиться лишь изотопами с достаточно
длительными периодами полураспада, имеющими при комнат-
ной температуре агрегатное состояние твердого тела, практи-
чески доступными и умеренными по цене, то выбор сведется к
списку изотопов, характеристики которых приведены в
табл. 3.2.
Если же говорить об изотопных источниках, легко доступных
для радиационно-химических исследований, то список следует
вообще уменьшить до двух изотопов: ®°Со и 137Cs. Технология
изготовления источников из 137Cs, однако, разрабатывается
только сейчас. Несмотря на то, что в Брукхейвенской нацио-
нальной лаборатории (США) и в Харуэлле (Англия) теперь
имеются цезиевые источники с активностью 105 кюри и, кроме
того, многие цезиевые источники изготовлены для целей радио-
терапии, пока нельзя считать, что этот изотоп широко исполь-
зуется в радиационной химии.
Наиболее распространенным изотопным источником излу-
чения является ®°Со. Кобальтовые источники изготавливают в
69
Характеристики изотопных
Характеристики источника «оСо 8°Sr 137CS “4Се “’Pm
Удельная мощность, вт/г (чистый изо- топ)" 17,4 0,95 0,42 25,6 0,33
Период полураспада, годы 5,3 28 30 0,78 2,7
Степень изотопной очистки, % 10 20 35 18 95
Используется в виде соединения Ме- талл SrTiO3 Стекло СеО., Pm2O^
Доля активного изотопа в соединении, % 10 24 16 15 82
Удельная мощность соединения, em/a 1,7 0,23 0,067 3,8 0,27
Плотность соединения, г/см3 8,9 4,6 3,2 6,4 6,6
Доступность изотопа (годичное произ- водство в кет за 1967 г.) Имеет- ся Имеется 67 Имеет- ся 48 Имеет- ся 800 Имеется 11
Вид излучения (основной) Y, ₽ ₽ ₽, Т ₽. Y Р
Необходимая защита Тяже- лая Тяжелая Тяже- лая Тяже- лая Легкая
Биологическая опасность (предельно Допустимая концентрация), мккю- ри/см3 3-Ю-9 3-10-ю 5-10—9 2-10“ 9 2-10“8
Приблизительная стоимость I г чистого изотопа, долл. 570 18е 9й 23d 30й
70
Таблица 3.2
источников излучения
17 0ТГП 204Т1 гюро 228Th 232Ц 23Spu г" Ат 242Ст 2“Ст
15,6 0.67 141 170 4,4 0,56 0,11 120 2,8
0,35 4 0,38 1,9 74 89 458 0,45 18
7,5б 20е 95 95 85 80 90 90 90
Тт2О3 Т1.О8 Металл ThO., ио2 PuO., Металл СГП2О3 СГП2ОЗ
6,57 17,7 95 83 75 70 90 82 82
1,03 0,12 134 141 3,3 0,39 0,1 98 2,3
7,7 9,0 9,3 9 10 10 11,7 11,75 11,75
Потен- циально досту- пен Потен- циально досту- пен Имеется Потен- циально Досту- пен Потен- циально досту- пен Доступен в ограни- ченных количе- ствах Доступен в ограни- ченных количе- ствах Потен- циально досту- пен Потен- циально досту- пен
₽ ₽ а а, у а, у а а а, п а, п
Легкая Легкая Легкая Тяже- лая Тяже- лая Легкая Легкая Легкая Легкая
10-е 9-10-9 2-10-9 2-10-12 9-10-12 7-10-13 2-10-12 4-10-11 ЗЮ-12
156 67 2800ж 6600 1540 500э 200“ 2000“ 1000“
71
Характеристики источника в0Со »°Sr 13’Cs 14 «Се “’Pm
Приблизительная стоимость, долл!вт 33 19й 21й I5 91й
Удельная активность чистого изотопа, кюри}г изо 142 87 3180 914
Активность, кюри/вт 65 150 207 124 2770
Период полураспада для спонтанного деления, годы — — — — —
а С учетом вклада дочерних изотопов при равновесии (в тепловых ваттах).
В этой концентрации ,70Тш можно получить облучением ,69Тгп без изотопного
разделения.
6 В этой концентрации 204Те можно получить лишь после разделения изотопов или
при облучении потоками очень большой интенсивности.
г Можно получить в виде металла. Температура плавления металла 1080° С
(Weigei F. Ап new, Chem., 1963. v. 75. p. 451). ожидаемая мощность па единицу объема
2,3 вт!см\
д Оценка стоимости дастся с учетом плана работ на Хапфордском изотопном заводе
<USAEC Report HW-77770).
виде стержней, трубок, полосок, дисков и таблеток. Такие источ-
ники имеют удельные активности от 1 кюри/г до более чем
500 кюри]г. Имеются кобальтовые источники с активностью до
миллионов кюри. Кобальтовые источники обладают также тем
редким преимуществом, что их изготавливает по крайней мере
пять различных предприятий: Брукхейвенская национальная ла-
боратория, Ок-Риджская национальная лаборатория, компания
«Дженерал электрик», компания «Атомная энергия Канады» и
Управление по атомной энергии Великобритании. За прошедшие
10 лет было продано более 10 млн. кюри еоСо. При этом суще-
ствуют практически безграничные возможности увеличения про-
изводства. Так, например, при условии достаточного рыночного
спроса завод Комиссии по атомной энергии США в Савапна-
Ривере мог бы выпускать до 8 млн. кюри 60Со на реактор в
год в качестве побочного продукта.
По конструкции кобальтовые источники, так же как и другие
крупные изотопные источники, можно разделить на три боль-
ших категории:
72
Продолжение табл. 3.2
i’OTm 20<Т] 2 10ро г ««Th 232U еаврп «'Am Z42Cm f44Cm
10 100 20 40 350 894 1820 17 357
- 6000 428 4500 4100к 114к 17 3,25 3310 84
385 640 32 24 26 30 30 28 30
— — — — 8-1013 l,4-10io 1,4-1013 7,2-10“ 1,4-10’
е Легкая защита возможна лишь в том случае, если нет необходимости экранировки
от нейтронов.
Пересмотр в сторону повышения минимальных цен, указанных в докладе:
Hoisted—AiЬаиgh. Hanford Diversification Report, p. IS, Jan. 15, 1964.
3 По данным Комиссии по атомной энергии США [см. также Nucieonics, 1963.
V. 21 (3), р. 63].
и Ожидаемая стоимость при производстве на промышленном плутониевом
реакторе (при стоимости Ри 10 долл/г).
к Активность а-распада с учетом вклада дочерних изотопов при равновесии.
1. Фиксированный источник — фиксированная активационная
полость. В установках этого типа источник обычно находится в
транснортировочпом контейнере.
2. Фиксированный источник — подвижная активационная по-
лость. В этой конструкции источник также неподвижен, но
полость с образцом может перемещаться.
3. Подвижный источник — фиксированная активационная по-
лость. В таких установках источник выдвигается из контейнера
или укрытия и производит облучение образца.
Большая часть сравнительно недорогих лабораторных источ-
ников излучения, которые используются радиационными хими-
ками, относится к категории фиксированных источников. На
'рис. 3.12 приведено схематическое изображение одного из первых
’ кобальтовых источников Брукхейвенской лаборатории, выпол-
ненного в виде полого цилиндра. Источники этого типа, заря-
женные активностью от 50 до 3000 кюри, могут давать мощность
дозы от нескольких тысяч до 106 рад[ч при облучении образцов,
объем которых не превышает 1000 см3. Одним из наиболее по-
73
Рис. 3.12. Фиксированный источ-
ник из 60Со, фиксированная акти-
вационная полость:
J —• источник нз ®°Со с алюминиевым
покрытием; 2— облучаемый образец.
легко производить различные
пулярных источников такого типа является у-ячейка. В автома-
тизированной у-ячейке образец вдвигается в радиационное поле
с помощью подвижной камеры. В у-ячейке таким образом осу-
ществляется схема «фиксирован-
ный источник — подвижная акти-
вационная полость».
Стесненные рамками бюджета
радиационные химики проявили
большую изобретательность при
конструировании различных ма-
ломасштабных недорогих устано-
вок с фиксированным источни-
ком, в которых использована для
защиты от излучения землю, во-
ду, масло и бетон. Такого рода
установки трудно модифициро-
вать, в них можно облучать лишь
небольшие образцы и, кроме
того, в них невысока эффектив-
ность использования изотопа.
Основные ограничения, свой-
ственные установкам с фиксиро-
ванным источником, удается
преодолеть, лишь перейдя к ис-
пользованию гораздо более доро-
гих установок с подвижным ис-
точником. В этих установках
источник хранится в контейнере,
а для облучения выдвигается в
активационную полость, которая
может иметь относительно боль-
шие размеры; в ней сравнительно
операции. В наиболее экономич-
ном варианте такой установки в качестве держателя изотопа
используется транспортировочный контейнер. Обычно источник
бывает прикреплен к стержню, который, перемещаясь по оси
контейнерного цилиндра, может выводить источник в акти-
вационную полость, закрытую бетоном или землей.
Большие промышленные установки обычно относятся к кате-
гории установок с подвижным источником. При облучении
источник обычно остается неподвижным, а образцы подводятся
к нему с помощью подвесного или ленточного конвейера. В ли-
тературе описан целый ряд установок такого типа [3, 4]. Все
они имеют разную конструкцию, так как изготовлены для вы-
полнения различных задач: пастеризации продуктов питания,
стерилизации шерсти, стерилизации кетгута, сшивания поли-
этиленового покрытия проводов. Одпако при конструировании
этих установок используются одни и те же физические прин-
74
ципы. Важнее всего правильно выполнить расчет потока излу-
чения, так как сведения о потоке необходимы и при конструи-
ровании защиты, и при расчете характеристик самого процесса.
3.11.1. Изотопные источники и дозовые поля
Для оценки и расчета нолей излучения от изотопных источ-
ников разной геометрии были разработаны специальные методы.
Основным затруднением во всех известных методах является
Рис. 3.13. Фактор накопления для точечных изотропных источников в зави-
симости от толщины защиты и среды:
а — вода; б — железо; в — железо.
необходимость учета вторичных фотонов. Для любой точки среды
нетрудно подсчитать поток первичного у-излучения, так как
можно считать, что их ослабление происходит в условиях узкого
пучка. Вся картина, однако, заметно усложняется благодаря
присутствию рассеянных фотонов (вторичных). Накопление
рассеянных фотонов учитывается с помощью фактора накопле-
ния. Величина фактора накопления для соответствующей за-
щиты определяется геометрией источника. Для расчетов защиты
имеются соответствующие таблицы [5], в которых приведены
значения фактора накопления.
Обычно при конструировании защиты или при решении
других сходных задач пользуются полными коэффициентами
поглощения в условиях узкого пучка, а затем полученные зна-
чения потока умножают на фактор накопления и таким
образом компенсируют ошибку.
Так как величина фактора накопления зависит от вещества
поглотителя, энергии фотонов, геометрии пучка, геометрии погло-
тителя и длины пути пучка в веществе, то расчеты оказываются
75
достаточно сложными. Используют метод моментов, полино-
миальный Спенсера, Фано, Гольдстейна и Вилкенса, асимптоти-
ческий и метод Монте-Карло. Результаты некоторых расчетов
такого рода отражены на рис. 3.13, где показаны значения
фактора накопления для точечных изотропных источников в
различных средах в зависимости от толщины защиты, энергии
фотонов и линейного коэффициента поглощения.
Хотя дозовые факторы накопления заметно отличаются от
единицы, все же они не столь существенны, как это может пока-
заться при беглом знакомстве с рис. 3.13. Для малых вариаций
в толщине защиты фактор накопления можно аппроксимировать
таким линейным уравнением:
В — 1 + арх, (3.2)
где константа а оценивается но наклону соответствующей кривой
ла рис. 3.13; р. — линейный коэффициент ослабления; х — тол-
щина защиты. Такая аппроксимация вполне допустима, так как
•ослабление в зависимости от толщины поглотителя изменяется
экспоненциально, тогда как фактор накопления оказывается
медленно возрастающей функцией толщины.
3.11.2. Самопоглощение в изотопных источниках
В крупных у-источниках, так же как и в источниках, испу-
скающих заряженные частицы, самопоглощение оказывается
серьезной проблемой. Помимо того, что бесполезно теряется
радиационная мощность источника, самопоглощение может при-
водить к выделению такого количества тепла, что в ряде случаев
его достаточно для сжигания образца. Действительно, при пере-
работке продуктов деления тепло, выделяющеся в результате
самопоглощения, обычно используется для испарения жидки*
•отходов. Чтобы количественно оцепить самопоглощение, необ-
ходимо знать удельную активность источника, геометрическое
распределение изотопа в источнике, коэффициент поглощения
и длину пробега у-квантов в веществе.
3.12. а-ИЗЛУЧАТЕЛИ
В ранний период изучения радиационных эффектов в газах
особенно важными изотопными источниками оказались «-излу-
чатели. В табл. 3.3 приводятся список а-излучающих изотопов и
их характеристики.
Из всех изотопов, приведенных в списке, наиболее удобен
для экспериментов в газовой фазе Rn. Так как продукты его
распада RaA и RaC отлагаются на стенках экспериментальных
сосудов, то обычно Rn помещают в тонкостенные стеклянные
шарики, которые крепятся затем в держателе для образца.
Использование радона в такой форме дает относительно мало
преимуществ по сравнению с использованием Ро, который можно
76
Таблица 3.3*
Источники а-излучеиия
Изотоп Энергия а-частиц, Мэв Период полурас- пада П робег в воздухе, см (15‘ С, 760 мм рм ст.} Началь- ная ско- рость ча- стиц, 10® см/сек Число обра- зуемых в воз- духе пар ио- нов (хЮв)
238JJ 235U 4,180 4,350 4,55-Ю9 лет 8,52-10* 8 лет 2,ТЬ 1,39 1,25
234JJ 4,763 2.3-103 лет 3,28 1,26 1,37
230Th 4,68, 4,61 8-104 лет 3,19 1,48 1,41
226Ra 4,78(94,2%) 4,59 1620 лет 3,39 1,51 1,47
222Rn 5,486 3,823 дня 4,12 1,62 1,67
218Po (RaA) 214J3i (RaC) a (0,04%) f (99,93%) 6,00 5,505 3,05 мин 19,72 мин 4,72 1,69 1,83
214po (RaC') говро 7,680 160 сек 2,9 года 6,97 1,92 2,37
232Th 3,98 1,389-101о лет 2,90 1,44
224Ra (ThX) 220Rn 5,86, 5,44 3,64 дня 4,354 1,65 1,75
6,282 54,5 сек 5,063 1,73 1,94
21<ip0 (ThA) 6,774 0,145 сек 5,683 1,80 2,09
212J3i (ThC) 6,081 60,6 мин 4,787 1,70 1,87
212p0 (ThC') 236 pu 23epu 24opu 238Am 24°Cm 24lCm 8,776 5,862 5,15 5,246 5,90 6,38 6,20 0,3 сек 2,7 года 24,413 года 6600 лет 12 ч 26,8 дня 35 дней 8,617 2,05 2,71
Данные из книги: Lind S. С. Radiation Chemistry of Gases. Reinhold Publishing Cor-
poration, New York, 1961, p. 275
получить в виде металлической пластинки, нанесенной на сталь-
ную подложку. Подробное обсуждение всех проблем, связан-
ных с использованием источников такого типа, имеется в книге
Линда [6].
3.13. р-ИЗЛУЧАТЕЛИ
В качестве fJ-излучателей кроме 90Sr, характеристики которого
приведены в табл. 3.2, специалисты по радиационной химии
используют газообразные тритий и криптон. Для работы в газо-
вой фазе 3Н, характеристики которого приведены в табл. 3.4,
77
Таблица 3.4
Газообразные источники ^-излучения
Изотон Период полураспа- да, годы Максимальная энер- гия (3-частиц, Мэв Метод получения изо- топа
зн 12,46 0,018 6Li (п, а) 3Н
85Кг 10,6 0,67 Деление ядер
обладает целым рядом преимуществ. Так же как и Rn, его
можно смешивать с изучаемым газом, причем дополнительной
защиты не требуется, так как испускается достаточно мягкое
излучение (средняя энергия 5,69 кэв).
3.14. ИЗОТОПНЫЕ ИСТОЧНИКИ НЕЙТРОНОВ
В табл. 3.5 перечислен ряд радионуклидов, которые в сово-
купности с бериллием могут использоваться для получения ней-
тронов. Две первые комбинации, 124Sb—Be и 226Ra—Be, — фото-
нейтронные источники, и поэтому для них характерны интенсив-
ные поля у-излучения. В четырех других комбинациях активация
ядерных реакций, в которых рождаются нейтроны, вызывается
а-частицами. Источник обычно состоит из смеси или сплава
а-излучателя с мишенным элементом — Be.
Таблица 3.5
Характеристики изотопных источников нейтронного излучения
Источник Период полурас- пада Выход нейтронного излучения, • 1 06 нейтрон./{сек-кюри) Средняя энергия нейтронов, Мэв
i2«Sb—Be 60 дней 1,6 0,024
226 Ra—Be 1622 года 1,3 0,2
333Pu—Be 24 600 лет 1,6 3-5
241Am—Be 470 лет 2,2 3-5
2iop0—Be 138 дней 2,5 2—4
3.15. ИСТОЧНИКИ ИЗЛУЧЕНИЯ, СВЯЗАННЫЕ С РЕАКТОРОМ
Целый ряд существующих или потенциально возможных
источников излучения можно изготовить лишь вблизи ядерного
реактора. Такие источники, как правило, хотя и непригодны для
исследовательских целей, могут оказаться чрезвычайно ценными
в тех случаях, когда нужна высокая интенсивность облучения.
3.15.1. Газообразные продукты деления
Значительная часть продуктов деления возникает в виде
радиоактивных газов, ксенона, криптона и паров иода. Если бы
эти газы, по преимуществу являющиеся у-излучателями, удалось
78
вывести в какой-то наружный контур, то теоретически около
С,7% энергии деления можно было бы возвратить в виде радиа-
ционной мощности.
3.15.2. Твердые продукты деления
На рис. 3.14 показана удельная мощность у-излучения про-
дуктов деления 235U с периодами полураспада больше восьми
дней. Ввиду быстрого распада 140La и некоторых других корот-
коживущих продуктов деления принято выдерживать отрабо-
Рис. 3.14. уменьшение
удельной мощности
у-излучения твэлов
235JJ п зависимости от
времени после удале-
ния из реактора.
Элементы в реакторе на-
ходились под действием
нейтронного потока плот-
ностью 2 10й нейтрон/
(см2 сек) в течение
30 дней. Показано также
уменьшение мощности
•у-излучения тех продук-
тов деления, период по-
лураспада которых пре-
вышает 8 дней.
тайные тепловыделяющие элементы вблизи реактора до тех
пор, пока их активность не спадет до уровня, обеспечивающего
удобную транспортировку на перерабатывающий завод. В це-
лом ряде источников, построенных при реакторах, используется
излучение от таких «остывающих» тепловыделяющих элементов.
Одна из английских установок [7] такого рода действует на от-
работанных тепловыделяющих элементах (каждый с активно-
стью по 100 тыс. кюри) из реакторов «Дидо» и «Плуто». На
установке возможно облучение объема в 1 м3. Мощность до-
зы — 5 Мрад/ч.
3.15.3. Реакторные контуры
Неоднократно предлагался ряд различных конструкций реак-
торных контуров, где химикат проходит непосредственно через
активную зону реактора. Реактор, снабженный подобным кон-
туром, может оказаться чрезвычайно дешевым источником излу-
чения. Для одной из таких конструкций подсчитано, что облу-
79
чение 36 300 кг материала до дозы 10 Мрад потребует затрат
всего лишь 100 долларов [8].
3.15.4. Радиационные контуры
Весьма популярна идея получить внутри реактора в резуль-
тате ядерной реакции (гг, у) радиоактивную жидкость, которую
затем можно перекачивать в близлежащую установку, где жид-
кость будет служить источником у-квантов. Сконструированы
источники излучения, в которых используются циркуляционные
контуры, заполненные жидким натрием, индием и индий-гал-
лиевым сплавом. Построен ряд экспериментальных установок.
3.15.5. Импульсные источники излучения
В настоящее время широко используются ускорители и реак-
торы, способные давать высокие дозы излучения за время,
варьирующее от наносекунд до микросекунд. К реакторам такого
типа относятся реакторы «Трига» и «Годива», а также более
новый реактор «Пульстар». Реактор «Трига» работает на
тепловыделяющих элементах С — ZrH. При работе в импульс-
ном режиме этот реактор создает дозы до тысяч Мрад в им-
пульсе. Длительность импульса — несколько микросекунд. Реак-
тор «Годива» в Лос-Аламосе представляет собой 50-килограм-
мовый металлический ансамбль без замедлителя или отражаю-
щих стенок. «Годива» также развивает за время импульса мощ-
ность дозы порядка тысяч мегарад. Длительность импульса —
несколько микросекунд.
Для работы в импульсном режиме можно модифицировать
генератор Ван-де-Граафа и линейный ускоритель. Эти установки
обеспечивают длительность импульсов в диапазоне от 10~9 до
10~8 сек. Работа этих и других импульсных устройств более
подробно рассматривается в главе VI.
Глава IV
ДОЗИМЕТРИЯ ИЗЛУЧЕНИЙ
В полноценном радиационно-химическом эксперименте необ-
ходимо измерить количество энергии, поглощенной образцом, и
обнаружить происшедшие в нем химические изменения. Как ни
странно, но точное измерение поглощенной дозы излучения (до-
зиметрия) часто оказывается не менее сложной задачей, чем вы-
явление характера и объема химических превращений. Труд-
ности могут быть обусловлены: 1) природой и формой источника
излучения; 2) составом и геометрией образца; 3) физическими
условиями, в которых проводится облучение. Трудности первого
типа возникают, например, при использовании в качестве источ-
ников излучения продуктов деления или смеси нескольких изо-
топов. Трудности второго и третьего типов встречаются при
облучении гетерогенных образцов и в случае высоких темпера-
тур и давлений.
Существует целый ряд проверенных методов определения
поглощенной дозы.
Для измерения дозы используют ионизационные камеры,
калориметры, устройства для собирания заряда, а также хими-
ческие системы, радиационно-химический выход * в которых
прокалиброван каким-либо абсолютным методом или сравнива-
ется с известными потоком и энергией пучка. Дозиметрическое
методы, с помощью которых измеряется непосредственно доза
в радах или в рентгенах, называют абсолютными или первич-
ными. Например, к первичным методам относится измерение до-
зы в рентгенах с помощью ионизационных камер. Первичным
методом является также измерение поглощенной дозы в радах
с помощью калориметра. Дозиметры, в которых используются
вторичные методы измерения дозы, должны быть обязательно
прокалиброваны с помощью первичных дозиметров. Любой из
методов при правильном использовании должен давать точную
величину потока излучения в точке, где производятся измерения.
* Радиационно-химическим выходом (G) называют число образовавших-
ся или распавшихся молекул на 100 эв поглощенной энергии.— Прим, перев.
81
Помещая свой образец в калиброванное радиационное поле,
экспериментатор должен быть уверен, что па образец воздей-
ствует определенный поток излучения, измеренный в единицах
с размерностью энергия/(площадь X время).
4.1. ИЗМЕРЕНИЕ ПОГЛОЩЕННОЙ ДОЗЫ
С ПОМОЩЬЮ ИОНИЗАЦИОННЫХ КАМЕР
Принцип измерения поглощенной дозы с помощью иониза-
ционных камер состоит в том, что подсчитывается число всех
ионов, образующихся в определенном объеме газа (обычно воз-
духа). Зная энергию, необходимую для образования одной пары
ионов в газе, можно рассчитать количество энергии, переданной
данному объему газа, и таким образом определить поглощенную
дозу. На практике, чтобы точно измерить поглощенную дозу,
приходится принимать целый ряд мер предосторожности.
Используется несколько типов ионизационных камер: полост-
ные и наперстковые, плоскопараллельные, камеры с воздушными
стенками и экстраполяционные камеры. Наиболее важная раз-
новидность ионизационных камер -— камера с воздушными
стенками. Именно такие камеры используются обычно для
калибровочных измерений. На практике удобнее всего работать
с полостными камерами, и специалисты по радиационной химии
чаще всего ими пользуются. Плоскопараллельная камера — одип
из вариантов камеры с воздушными стенками, а экстраполяцион-
ная представляет собой полостную камеру, предназначенную
для работы с источниками излучения широкого энергетического
спектра.
4.1.1. Полостная камера
Для измерения дозы с помощью полостной камеры непо-
средственно используется формула Брэгга — Грея. Основной
принцип работы камеры состоит в том, что измеряется иониза-
ция в газовой полости, окруженной твердым веществом. Газовая
полость достаточно мала, поэтому находится в том же потоке
излучения, что и окружающий материал. Поглощенная доза
D (в эв) в твердом веществе, внутри которого находится по-
лость, дается выражением
D = 7^Sm, (4.1)
где 7 — число пар ионов, образующихся в 1 г газа; W— энергия,
затрачиваемая на образование одной пары ионов в газе, эв!пара
ионов; Sm — отношение массовой тормозной способности мате-
риала к массовой тормозной способности газа в полости для
рассматриваемого ионизирующего излучения.
Как уже отмечалось, полость должна быть настолько малой,
чтобы спектр энергии вторичных электронов, ее пересекающих,
во всех отношениях совпадал со спектром энергии вторичных
82
электронов в облучаемом твердом веществе в отсутствие по-
лости. Короче, должны осуществляться условия электронного
равновесия. Они соблюдаются, если доля первичного излучения,
взаимодействующего с газом в полости, пренебрежимо мала,
а стенки камеры имеют достаточную толщину, чтобы установи-
лось равновесие между первичными фотонами (или электро-
нами) и возникающими в веществе вторичными электронами.
Толщина стенок должна быть примерно равна пробегу вторич-
ных электронов в твердом веществе.
По практическим соображениям использование полостных
ионизационных камер ограничивается измерениями рентгенов-
ского или у-излучения с энергией квантов выше 20 кэв, а также
при соответствующей коррекции измерениями нейтронного из-
лучения. Эти ограничения обусловлены главным образом тем,
что не всегда удается получить условия электронного равно-
весия, и тем, что невозможно определить в неравновесных
условиях величину Sm для различных источников излучения [1].
Если же можно осуществить условия электронного равновесия
или ввести соответствующие поправки, то полостная иониза-
ционная камера пригодна для измерений почти с любыми источ-
никами излучения.
Электронное равновесие может не соблюдаться в следующих
случаях: 1) при помещении полостной ионизационной камеры
в среду, которая существенно ослабляет пучок; 2) если камера
находится вблизи источника и интенсивность излучения быстро
убывает с расстоянием; 3) если камера находится вблизи гра-
ницы между средами разного состава.
При выполнении условия электронного равновесия погло-
щенную дозу в данной точке среды можно определить с по-
мощью любой ионизационной камеры, проградуированной в
рентгенах, или же с помощью абсолютной ионизационной ка-
меры, если правильно их разместить. Поглощенная доза в
воздухе или воздухоэквивалентном веществе, получившем при
фотонном облучении экспозиционную дозу 1 р, равна
-------ед- С—Э . 2,082 • 109
0,001293 г
X 1,602 Ю-'2-^-.
эв
эв
электрон gg
1 ед. СГСЭ пара ионов
- * 1 р- = 0,86 рад.
100 эрг/г
Если ионизационная камера, проградуированная в рентгенах,
находится в поглощающей среде и сама камера достаточно
мала, чтобы не возмущать условия электронного равновесия
в среде, то поглощенная доза в радах для среды дается выра-
жением
Осоеда = 0,86 1^л/р)среДа
(Р₽п/Р)воздух
где X — доза (в р).
(4.2)
83
4.1.2. Абсолютная полостная камера
Абсолютная полостная камера изготавливается из воздухо-
эквивалентного материала, и ее полость заполнена воздухом.
Воздухоэквивалентпым материалом называется вещество, для
которого коэффициент передачи энергии, рассчитанный на один
электрон среды, тот же, что и у воздуха. Это вещество с эф-
фективным атомным номером, близким к 6. Им может быть
либо соответствующий элемент (углерод), либо сложное веще-
ство (многие пластмассы).
Если камера размещена в среде соответствующим образом,
то она будет измерять поглощенную дозу в тонком слое мате-
риала собственной стенки
^стенка = 0,86ASm, (4.3а)
где А—заряд в ед. СГСЭ, переносимый ионами одного знака,
возникающими в 0,001293 г воздуха полости.
Поглощенная доза в среде будет даваться выражением
Г) _ (Иеп/р)среда г) /Л
^среда — ^стенка- yt.ou;
(Меп/Р/стенка
Значения массового коэффициента передачи энергии для
наиболее часто встречающихся элементов и для некоторых ве-
ществ при фотонном облучении приведены в табл. 4.1. В урав-
нении (4.36) может потребоваться небольшой поправочный
коэффициент, учитывающий возмущения поля рентгеновского
излучения при помещении камеры в среду.
4.1.3. Средняя энергия, затрачиваемая
на образование одной пары иойов
Значение W, равное 33,7 эв, представляет собой взвешенное
среднее нескольких определений энергии ионообразования при
облучении электронами. Как и следовало ожидать, для разных
газов значения W различны. Эти значения собраны в табл. 4.2.
Таблица 4.2
Средняя энергия ионообразования в различных газах при облучении электронами
Газ Взвешенное среднее, эв/пара ионов Газ Взвешенное среднее, aefnapa ионов
н, 36,6±0,3 Хе 21,9±0,3
Не 41,5±0,4 Воздух 33,73±0,15
n2 34,6±0,3 СО3 32,9±0,3
о2 30,8±0,3 СН4 27,3±0,3
Ne 36,2±0,4 С.,Н2 25,7±0,4
Аг 26,2±0,2 С.,Н4 26,3±0,3
Кг 24,3±0,4 с2н6 24.6±0,4
84
Массовые коэффициенты передачи энергии [(ЦеЛ/р) сл4/г]
Таблица 4.1*
Энергия фотонов, Мэв Элемент
Н С N О Na Mg Al р S Ar К Ca
0,010 0,0099 1,94 3,42 5,50 15,4 20,9 26,5 40,1 49,7 62,0 77,0 89,8
0,015 0,0110 0,517 0,916 1,49 4,43 6,09 7,65 11,9 15,2 19,4 24,6 28,9
0,020 0,0133 0,203 0,360 0,587 1,77 2,47 3,16 5,00 6,41 8,31 10,5 12,5
0,030 0,0186 0,0592 0,102 0,163 0,482 0,684 0,880 1,45 1,85 2,46 3,12 3,75
0,040 0,0230 0,0306 0,0465 0,0700 0,194 0,274 0,351 0,570 0,731 0,974 1,25 1,52
0,050 0,0270 0,0226 0,0299 0,0410 0,0996 0,140 0,176 0,282 0,361 0,484 0,626 0,764
0,060 0,0305 0,0203 0,0244 0,0304 0,0637 0,0845 0,104 0,166 0,214 0,284 0,367 0,443
0,080 0,0362 0,0201 0,0218 0,0239 0,0369 0,0456 0,0536 0,0780 0,0971 0,124 0,158 0,191
0,10 0,0406 0,0213 0,0222 0,0232 0,0288 0,0334 0,0372 0,0500 0,0599 0,0725 0,0909 0,111
0,15 0,0485 0,0246 0,0249 0,0252 0,0258 0,0275 0,0282 0,0315 0,0351 0,0368 0,0433 0,0488
0,20 0,0530 0,0267 0,0267 0,0271 0,0265 0,0277 0,0275 0,0292 0,0310 0,0302 0,0339 0,0367
0,30 0,0573 0,0288 0,0289 0,0289 0,0278 0,0290 0,0283 0,0290 0,0301 0,0278 0,0304 0,0319
0,40 0,0587 0,0295 0,0296 0,0296 0,0283 0,0295 0,0287 0,0290 0,0301 0,0274 0,0299 0,0308
0,50 0,0589 0,0297 0,0297 0,0284 0,0293 0,0287 0,0287 0,0288 0,0300 0,0271 0,0294 0,0304
0,60 0,0588 0,0296 0,0296 0,0296 0,0283 0,0292 0,0286 0,0287 0,0297 0,0270 0,0291 0,0301
0,80 0,0573 0,0288 0,0289 0,0289 0,0276 0,0285 0,0278 0,0280 0,0287 0,0261 0,0282 0,0290
1,0 0,0555 0,0279 0,0280 0,0280 0,0267 0,0275 0,0269 0,0270 0,0280 0,0252 0,0272 0,0279
1,5 0,0507 0,0255 0,0255 0,0255 0,0243 0,0250 0,0246 0,0245 0,0254 0,0228 0,0247 0,0253
2,0 0,0464 0,0234 0,0234 0,0234 0,0225 0,0232 0,0227 0,0228 0,0235 0,0212 0,0228 0,0234
3,0 0,0398 0,0204 0,0205 0,0206 0,0199 0,0206 0,0201 0,0204 0,0210 0,0193 0,0208 0,0213
4,0 0,0351 0,0184 0,0186 0,0187 0,0184 0,0191 0,0188 0,0192 0,0199 0,0182 0,0199 0,0204
5,0 0,0316 0,0170 0,0172 0,0174 0,0173 0,0181 0,0180 0,0184 0,0192 0,0176 0,0193 0,0200
6,0 0,0288 0,0160 0,0162 0,0166 0,0166 0,0175 0,0174 0,0179 0,0187 0,0175 0,0190 0,0198
8,0 0,0249 0,0145 0,0148 0,0154 0,0158 0,0167 0,0169 0,0175 0,0184 0,0172 0,0190 0,0197
10,0 0,0222 0,0135 0,0142 0,0147 0,0154 0,0163 0,0167 0,0174 0,0183 0,0173 0,0191 0,0201
Продолжение табл. 4.1
Энергия фотонов, Мэв Вещество S 4 П. *3 Энергия фотонов, Мэв Вещество Я J е. у: а с О' 1 (!лея/р)возд 1
полистрирол (СвНв) плексиглас, люсит , (С5Н8О2)Д 1 полиэтилен (СН2)^ бакелит С43Н38О? вода воздух i 1 X 1 плексиглас, лю.:ит (С5Н8О2 полиэтилен (СН2) ! бакелит С-3Н38О7 вода воздух
о л (^гп/0)во. е? =5. о о э 1 £ 0
с С <
0,010 1,79 2,92 1,66 2,43 4,89 4,66 0,912 0,50 0,0319 0,0320 0,0339 0,0314 0,0330 0,0297 0,966
0,015 0,478 0,788 0,444 0,651 1,32 1,29 0,889 0,60 0,0318 0,0319 0,0338 0,0313 0,0329 0,0296 0,966
0,020 0,188 0,311 0,176 0,257 0,523 0,516 0,881 0,80 0,0310 0,0311 0,0329 0,0305 0,0321 0,0289 0,965
0,030 0,0561 0,0892 0,0534 0,0743 0,147 0,147 0,869 1,0 0,0300 0,0301 0,0319 0,0295 0,0311 0,0280 0,965
0,040 0,0300 0,0426 0,0295 0,0368 0,0647 0,0640 0,878 1,5 0,0274 0,0275 0,0291 0,0270 0,0283 0,0255 0,964
0,050 0,0229 0,0288 0,0232 0,0259 0,0394 0,0384 0,892 2,0 0,0252 0,0252 0,0267 0,0247 0,0260 0,0234 0,966
0,060 0,0211 0,0243 0,0218 0,0226 0,0304 0,0292 0,905 3,0 0,0219 0,0220 0,0232 0,0216 0,0227 0,0205 0,962
0,080 0,0213 0,0226 0,0224 0,0217 0,0253 0,0236 0,932 4,0 0,0197 0,0198 0,0208 0,0194 0,0205 0,0186 0,958
0,10 0,0228 0,0235 0,0241 0,0227 0,0252 0,0231 0,948 5,0 0,0181 0,0183 0,0191 0,0179 0,0190 0,0173 0,954
0,15 0,0264 0,0267 0,0280 0,0261 0,0278 0,0251 0,962 6,0 0,0170 0,0172 0,0178 0,0168 0,0180 0,0163 0,960
0,20 0,0287 0,0289 0,0305 0,0283 0,0300 0,0268 0,973 8,0 0,0153 0,0156 0,0160 0,0153 0,0165 0,0150 0,956
0,30 0,40 0,0310 0,0317 0,0311 0,0319 0,0329 0,0337 0,0305 0,0312 0,0320 0,0329 0,0288 0,0296 0,966 0,966 10,0 0,0144 0,0147 0,0149 0,0144 0,0155 0,0144 0,935
’ Данные из справочника: National Bureau of Standards, Handbook 85, Washington, 1964.
.Для a-излучения получают значения W, немного отличающиеся
от табличных. Однако к инертным газам это не относится (за
исключением гелия).
4.1.4. Значения отношения Sm
Величина отношения Sm меняется в зависимости от вида
материала и энергии излучения. Поэтому, чтобы получить со-
ответствующие значения Sm, необходимо знать оба параметра.
В том случае, если выполняются условия электронного равно-
весия, можно прямо использовать расчетные значения Sm, при-
веденные в табл. 4.3 и 4.3а. Для материала, состоящего из
нескольких элементов, Sm которого неизвестно, вначале рас-
считывается Sm (относительно воздуха) для каждого входящего
в данный материал элемента. Эта величина умножается па
весовую долю элемента в образце, и производится суммиро-
вание по всем элементам, образующим материал. Обнаружено,
что правило суммирования при расчете Sra в том виде, как
оно сформулировано выше, лучше выполняется, если учиты-
ваются различия в значениях Sm для водорода, входящего в
состав насыщенных и ненасыщенных соединений. Эти поправки
имеются в соответствующих таблицах.
4.1.5. Измерение ионных токов
В точной ионизационной камере эффективность собирания
ионов должна быть высокой. Для этого используется электри-
ческое поле, перпендикулярное к стенкам камеры. Прилагаемое
напряжение должно быть достаточным для того, чтобы создать
ток насыщения. Иными словами, заряд должен собираться па
электроды с той же скоростью, с какой он возникает в актив-
ном объеме. Неполное собирание ионов может обусловливаться
либо первичной рекомбинацией, либо рекомбинацией в про-
цессе собирания. Первичная рекомбинация, как правило, не
зависит от мощности дозы. Однако этот эффект может оказаться
существенным для медленных частиц при высоком давлении
газа в камере.
Общая рекомбинация в заметных масштабах происходит
в газах, где образуются отрицательные ионы, при очень высо-
ких интенсивностях излучения, а также в случае дефектной кон-
струкции камеры. Если в камере имеются участки с напряжен-
ностью поля, недостаточной для того, чтобы создать ток насы-
щения, то, естественно, происходит рекомбинация.
На рис. 4.1 показан разрез полостной ионизационной ка-
меры, разработанной для измерения потока излучения от
кобальтового источника высокой интенсивности. Более подроб-
ные сведения о конструкции и работе ионизационных камер
можно найти в книге Прайса [3].
87
Таблица 4. 3
Средние значения отношения массовых тормозных способностей вещества и воздуха с учетом эффекта поляризации для монэ-
энергетического начального пучка электронов в условиях электронного равновесия
Начальная энер- гия, Afae H С N о С (графит) . и и С £
насыщенные соединения ненасыщенные соединения насыщенные соединения ненасыщенные соединения высокохлори- рованные сое- динена я амины, нит- раты гетероцикли- ческие соеди- нения 1 ? II о
0,1 2,52 2,59 1,016 1,021 1,047 0,976 1,018 0,978 0,994 1,014 0,859 0,875 0,852 0,711 0,555 0,468
0,2 2,52 2,59 1,015 1,019 1,043 0,978 1,016 0,979 0,995 1,013 0,870 0,886 0,869 0,734 0,595 0,508
0,3 2,48 2,55 1,014 1,018 1,040 0,979 1,016 0,981 0,995 1,011 0,876 0,891 0,876 0,745 0,614 0,526
0,327 2,48 2,54 1,014 1,018 1,040 0,979 1,015 0,981 0,995 1,010 0,877 0,892 0,878 0,747 0,617 0,530
0,4 2,46 2,53 1,014 1,018 .1,038 0,980 1,015 0,981 0,996 1,009 0,879 0,894 0,882 0,752 0,625 0,539
0,5 2,44 2,51 1,013 1,017 1,037 0,980 1,015 0,982 0,996 1,007 0,881 0,896 0,885 0,757 0,633 0,548
0,6 2,44 2,50 1,012 1,016 1,035 0,980 1,013 0,981 0,995 1,005 0,882 0,898 0,887 0,761 0,639 0,555
0,654 2,43 2,49 1,011 1,014 1,034 0,979 1,012 0,981 0,994 1,004 0,883 0,899 0,889 0,762 0,642 0,557
0,7 2,42 2,48 1,010 1,013 1,033 0,978 1,011 0,980 0,993 1,003 0,883 0,899 0,890 0,764 0,645 0,560
0,8 2,40 2,46 1,009 1,012 1,031 0,978 1,010 0,979 0,992 1,001 0,884 0,901 0,892 0,767 0,649 0,565
1,0 2,39 2,44 1,004 1,008 1,026 0,975 1,005 0,977 0,988 0,998 0,885 0,902 0,895 0,771 0,655 0,572
1,2 2,37 2,42 1,001 1,004 1,022 0,973 1,002 0,974 0,985 0,995 0,885 0,904 0,898 0,775 0,660 0,578
1,308 2,36 2,42 0,999 1,002 1,019 0,971 1,000 0,972 0,983 0,993 0,885 0,905 0,899 0,775 0,663 0,581
1,5 2,35 2,39 0,995 0,998 1,015 0,967 0,996 0,969 0,980 0,907 0,902 0,666 0,584
* Данные нз справочника: National Bureau of Standaids, Handbook 85, p. 8, Washington, 1964.
Таблица 4.3 а
Средние значения отношения массовых тормозных Способностей вещества И воздуха с учетом эффекта поляризации для комп-
тоновских электронов отдачи в условиях электронного равновесия
Энергия фотонов, Мэв II С N о С (графит) О. cd и и е V) Л Л
н асыщениые соедине - НИЯ ненасыщенные соеди- нения насыщенные соедине- ния ненасыщенные соеди- нения высокохлориров энные соединения амины, нитраты гетероциклические соединения 1 о 1 II о
0,15 2,73 2,85 1,020 1,027 1,058 0,970 1,022 0,972 0,992 1,017 0,835 0,823 0,776 0,657 0,458 0,380
0,25 2,62 2,72 1,017 1,022 1,050 0,974 1,019 0,976 0,994 1,015 0,853 0,850 0,816 0,696 0,538 0,443
0,4 2,55 2,63 1,016 1,020 1,045 0,977 1,017 0,978 0,995 1,013 0,866 0,867 0,842 0,723 0,575 0,487
0,6 2,50 2,57 1,014 1,018 1,040 0,979 1,016 0,980 0,995 1,011 0,874 0,879 0,857 0,740 0,605 0,518
1,0 2,44 2,50 1,008 1,012 1,032 0,977 1,009 0,978 0,991 1,005 0,881 0,888 0,873 0,758 0,634 0,548
1,5 2,39 2,45 1,001 2,005 1,023 0,972 1,003 0,973 0,985 0,999 0,883 0,894 0,883 0,768 0,650 0,567
2,0 2,36 2,42 0,994 0,997 1,014 0,966 0,995 0,967 0,978
2,5 2,32 2,37 0,987 0,990 1,007 0,960 0,988 0,962 0,973
Пример 4.1. Полостная ионизационная камера из люсита облучается
у-квантами с энергией 1 Мэв. Ионизационный ток проходит через сопротив-
ление 1,013-106 ом, па котором падение напряжения измеряется с помощью-
калиброванного вибрационного электрометра. Определить интенсивность из-
лучения, если при насыщении электрометр дает отсчет 9.1 мв, а объем воз-
духа в камере равен 3,65 см3 при стандартных температуре и давлении.
Рис. 4.1. Схематический разрез полостной ионизационной камеры:
/ — наполнение из плавленой серы; 2- графит; 3— отверстие для воздуха; 4— внеш-
ний кожух из нержавеющей стали; 5 — керамическая изоляция: 6 — измерительный
прибор; 7 — медный провод: 8— керамическая изоляция; 9 — кабельное покрытие их
нержавеющей стали.
Решение. _
D - lWSm-
— / 0,0091 в I электрон (ион) I 33,7 эв I
\ 1,013- 10е ом | 1,6- IO-18/? I пар ионов | 3,65 см3
см3 0,0301
0,0012932 г 0,0280 )>
D - 4,3 • 1014 эв!(г сек).
4.1.6. Наперстковая камера
Относится к числу наиболее распространенных небольших
ионизационных камер конденсаторного типа. В ней имеется
небольшой воздушный объем, заключенный в тонкостенную
пластиковую оболочку. На внутреннюю стенку наносится со-
ответствующее проводящее покрытие, а в центре находится про-
водящая проволока, изолированная от оболочки. По сути дела,
камера представляет собой небольшой конденсатор, и перед
использованием ее заряжают до некоторого стандартного на-
пряжения. Под действием ионизирующего излучения часть
заряда нейтрализуется. Экспонированную камеру помещают в
отсчетное устройство, которое также используют для ее под-
заряда. Отсчетным устройством служит струпный электрометр.
Оставшийся на камере заряд определяется по отклонению нити,
90
за которым следят с помощью встроенного микроскопа. Оку-
лярная шкала микроскопа прокалибрована в рентгенах или
миллирентгенах.
4.1.7. Камера с воздушными стенками
Камера с воздушными стенками работает по принципу соби-
рания ионов. Собирающее поле с интенсивностью от 5 до 100 в
на 1 см располагается между высоковольтным и коллекторным
Рис. 4.2. Плоскопараллельная воздушная ионизаци-
онная камера:
/ — собирающий электрод; 2 — пучок рентгеновскою излу-
чения: 3 — входное отверстие с площадью А; 4 — высоко-
вольтный электрод: 5 —защитный кожух; 6 — вторичные
электроны; 7—охранный электрод: 8 —к амперметру.
электродами, которые должны быть разнесены на такое рас-
стояние, чтобы вторичные электроны (рис. 4.2) могли закончить
пробег в межэлектродном объеме. Это условие ограничивает
использование камер с воздушными стенками областью излу-
чений сравнительно низкой энергии (от 50 до 200 кэв). При
больших энергиях должны использоваться либо очень большие
камеры, либо камеры, в которых газ находится под высоким
давлением. На рис. 4.2 показана схема плоскопараллельной
камеры с воздушными стенками.
Для определения дозы в рентгенах необходимо собрать все
вторичные электроны, возникающие в 1 сл<3 воздуха. Это воз-
можно сделать только косвенным путем, так как вторичные
электроны рассеиваются из рассматриваемого элемента объема,
а также попадают в него при рассеянии из соседних элементов
объема. Если число рассеянных внутрь объема и из него элек-
91
тронов одинаково, как это имеет место иа рис, 4.2, то говорят,
что соблюдается электронное равновесие.
Пример 4.2. Найдите дозу в рентгенах для участка около входной щели
камеры (см. рис. 4.2), если известны следующие данные: площадь входной,
щели А=3 см2; приращение разности потенциалов на электродах Д1/=1 в;
плотность воздуха р=0,001293 г/см2; длина электрода £=10 см; емкость из-
мерительной системы С=100 пф.
Решение. Полный заряд, собираемый камерой, равен
Q = VC= (1) (10 Ю-n) (3 109) = 30 • IO-2 ед. СГСЭ.
Согласно определению рентгена
Q 0,001293
лГ р
3 10 -1 ед. СГСЭ
3 см2
10 см
0,001293
0,001293
4.1.8. Экстраполяционная камера
Экстраполяционные камеры применяются в тех случаях,
когда источник излучения обладает широким энергетическим
спектром. Теория Брэгга — Грея справедлива для электронов и
нейтронов, так же как и для других видов излучения. Однако,
как указывалось выше, для целого ряда источников или кон-
фигураций источников могут возникать заметные трудности.
Они связаны главным образом с несоблюдением тех условий,
которые рассматривались в разделе 4.1.1. Например, если источ-
ник является р-излучателем, то электроны могут обладать ши-
роким спектром энергий. При этом очень трудно соблюсти
условия электронного равновесия. Чтобы устранить этот недо-
статок, конструируют камеры с переменным уменьшающимся
объемом газовой полости. Одновременно с уменьшением воз-
душной полости измеряют токи насыщения, и в' пределе полу-
чают граничное значение отношения тока насыщения к объему
при объеме, стремящемся к нулю. Для этого граничного зна-
чения справедлива формула Брэгга — Грея.
Основное затруднение при работе с экстраполяционными
камерами — выбор значений Sm. Некоторые методы усреднения
дают вполне приемлемые значения. Подробнее этот вопрос
обсуждается Файлла [4].
4.2. КАЛОРИМЕТРИЯ
Калориметрия относится к числу первичных методов опре-
деления поглощенной дозы. Калориметрические методы позво-
ляют непосредственно определять поглощенную дозу, пе тре-
буют от экспериментатора больших усилий. При правильном
выборе методики можно получать столь же точные результаты,
как и при ионизационных измерениях. Калориметрическая тех-
ника, однако, имеет и специфические недостатки, которые огра-
92
ничивают ее широкое применение. Калориметрические методы
обладают низкой чувствительностью (обычно необходима мощ-
ность дозы, превышающая 1 крад/ч), требуют больших затрат
времени, а аппаратура, необходимая для работы, оказывается
сложной и громоздкой. Затраты поглощенной энергии на другие
процессы, кроме нагревания образца (например, на химические
превращения), приводят к ошибкам. В большей части случаев,
однако, химические превращения и другие процессы можно
учесть, вводя соответствующие поправки.
При калориметрическом определении поглощенной дозы не-
обходимо измерить увеличение температуры небольшой изоли-
рованной массы вещества [5]. Система должна быть сконструи-
рована таким образом, чтобы теплоизолирующий .материал
существенно не искажал спектра излучения. Масса же мешалок,
нагревателей, термопар, термисторов и т. п. должна быть малой
по сравнению с массой вещества, в котором регистрируется
подъем температуры.
В точной калориметрии при определении поглощенной дозы
необходимо также соблюдать условия электронного равновесия.
Кроме того, интенсивность излучения должна быть достаточно
большой по сравнению с тепловыми потерями, а объем погло-
щающего вещества — достаточно малым по сравнению с гра-
диентом поля излучения.
При калориметрических измерениях обычно пользуются по-
нятиями тепловой мощности Р или количества тепла Q. Если
в изолированном от окружающей среды образце (при погло-
щении излучения) создается тепловая мощность Р, то темпера-
тура образца будет меняться по такому закону:
dTc _ Р' k (Тс Те) (4 4)
dt ~ С ’ 1 f
Ш Тс—температура образца, в котором проводятся калори-
метрические измерения; Те— температура окружающей среды;
С — теплоемкость вещества образца; k — коэффициент тепло-
проводности изолирующего материала; Р — тепловая мощность
которая в общем случае равна сумме двух мощностей: Ри — из-
меряемой мощности и Ро — постоянной тепловой мощности, со-
здаваемой в системе, например, благодаря работе мешалок или
при выделении джоулева тепла в термисторах; t — время.
4. 2.1. Типы калориметрических систем
В соответствии со способом измерения Тс, длительностью из-
мерений, относительными значениями С и k, способом калибров-
ки, при котором определяются С или k или оба значения сразу,
калориметрические системы можно разделить на три основных
типа: адиабатические, квазиадиабатические и изотермические.
93
В адиабатических калориметрах Тс поддерживается рав-
ной Те, и скорость изменения температуры дается уравнением
= 4 5)
Калибровка заключается в определении С. Энергию Q, пере-
даваемую калориметру за период времени tf —tf, можно найти
из уравнения
Q = C(T/-Tl)-P9(tf-tl\ (4.6)
где Ti и Tf — начальная и конечная температуры калориметра
соответственно.
Квазиадиабатический метод используется в калориметрии
главным образом в тех случаях, когда необходимо существенно
сократить время измерений. Значение Те поддерживается по-
стоянным. Количество поглощенного тепла дается выражением
Q = С (Tf - Т() 4- k J (Тс - Tb) di, (4.7)
где Ть — балансная, или стационарная, температура, которую
.калориметр приобретает через бесконечный промежуток вре-
мени:
Т„ = Те + -^-. (4.8)
к
-Значение k должно быть малым. Поэтому зависимость темпе-
ратуры от времени до и после нагревания будет приблизи-
тельно линейной. Калибровка заключается в определении С и k.
В изотермическом методе температура Те поддерживается по-
стоянной. Эксперимент заканчивается как только достигается
стационарное значение температуры Тс. Из уравнения (4.4)
следует
^-^ = 4(1-6-^) (4.9)
к
и через бесконечный промежуток времени
P^k(Tc— Те). (4.10)
Таким образом, разность температур Т™ — Те является мерой
теплового потока от образца при постоянной температуре. Ка-
.либровка калориметра состоит в определении одного лишь
значения k. Временная константа калориметра — отношение C/k.
Для уменьшения времени измерения значение С стараются сде-
лать малым.
Как указывалось, помимо перехода в тепло энергия может
диссипировать с помощью других механизмов, таких, как запа-
сание энергии, образование дефектов в кристаллической струк-
34
туре, химический распад и т. п. По-видимому, для большой части
элементов и неорганических твердых веществ, облучаемых при
комнатной температуре, запасание энергии происходит в огра-
ниченных пределах [6]. Однако после продолжительного облу-
чения в графите реакторов вблизи комнатных температур может
сохраняться более 2000 дж'г.
В органических и многих неорганических веществах и водных
растворах весьма значительными могут быть химические эф-
фекты. Майлери и сотрудники [7], например, рассмотрели воз-
можные реакции в полистироле. Обычно принимается, что для
воды и водных растворов в закупоренных сосудах после опре-
деленного промежутка времени непрерывного облучения с по-
стоянной интенсивностью устанавливается равновесное давление
Н2 и О2 н дальнейший радиолиз воды не происходит.
4. 2.2. Измерение поглощенной дозы
калориметрическими методами
Калориметрические методы используют в радиационной хи-
мии главным образом для калибровки химических систем, таких,
как дозиметр Фрике, или для
сравнения с измерениями, про-
веденными с помощью иониза-
ционных камер.
Одно из первых измерений
такого рода — калибровка до-
зиметра Фрике Хоханделем и
Гормли [8]. Применявшийся
калориметр показан па
рис. 4.3. Он состоял из цилин-
дрической медной рубашки,
окружавшей тонкостенную по-
серебренную пирсксовую кол-
бу, в которой находилось от 3
до 6 мл воды, использовав-
шейся в качестве калориметри-
ческого тела. За изменениями
температуры при облучении
следили с помощью медь-коп-
стаптановых термопар, нахо-
Рис. 4.3. Калориметр Хохапделя и
Гормли, предназначенный для изме-
рения энергии у-излучения, поглощен-
ной в воде:
/ — нирексовая колба с толщиной стенки
0.09 мм: 2 — кобальт в стенках латунного
цилиндра; 3 — свинец: 4 — термопара;
5— замазка; 6 - медиан рубашка; 7—к
вакуумной системе: 8— место расположе-
ния источника: 9 — пенополистирол.
дившихся в воде и в медной
рубашке. Температура рубаш-
ки непрерывно регулировалась
с помощью нагревателя, нахо-
дящегося в полости, где поме-
щался источник из 60Со (см.
рис. 4.3). Температура рубаш-
ки поддерживалась равной
95
температуре воды. Используемый кобальтовый источник обес-
лечивал мощность дозы от 25 до 225 р!сек, что приво-
дило к подъему температуры со скоростью от 0,003 до
0,03 град/мин.
Температура прослеживалась в течение 3—5 ч, пока не уста-
навливались равновесные концентрации продуктов радиолиза
воды (Н2, Os, Н2О2) и не исчезали малые эффекты, связанные
с выделением теплот различных реакций.
При расчете скорости поглощения энергии в системе Хохан-
дель и Гормли делали следующие допущения:
1. Температура спая термопары, погруженной в воду, равна
температуре воды и температуре колбы. Между колбой и мед-
ной рубашкой может происходить свободный теплообмен.
2. Полную теплоемкость системы (и полное поглощение
энергии в ней) можно рассчитать, исходя из (известной) тепло-
емкости и весовых долей воды и стекла. Поглощением энергии
в веществе термопары и в топком слое серебра на пирексовой
колбе можно пренебречь.
После того как было измерено значение поглощенной энер-
гии, в тех же геометрических условиях определяли скорость
-окисления ферросульфата и рассчитывали значение G (число
^измененных молекул на 100 эв поглощенной энергии).
Характерный набор экспериментальных данных выглядит так:
Скорость нагревания . .
Масса воды.............
Масса стекла...........
Полная теплоемкость . .
Скорость окисления ионов
0,014сС/лия
5,98 г
0,30 г
6,03 кал/°C
69,0-10“® моль/(л
• мин)
Энергия, поглощенная водой в калориметрической системе,
дэавна
0,014°С
мин
16,03 кал
I °C
— = 0,0100 кал/(г-мин).
Принимая плотность ферросульфатного раствора равной
1,02, найдем поглощенную в нем энергию (0,0100) • (1,02) —
=0,0102 кал/(г-мин). Значение радиационно-химического вы-
хода равно
„ 69-10“®Л4 I г-мин I л I 6-1023 молекул 3,83-10“18 кал I
G —---------—~——------1-----1-----------------------=
л-мин I 0,0102 кал |1000 г| моль 100 эв |
= 15,5 окисленной молекулы ферросульфата на 100 эв погло-
щенной энергии. Серия измерений дает среднее число окислен-
ных ионов Fe2+ на 100 эв поглощенной энергии, равное 15,6+0,3.
Позднее был проделан целый ряд сходных измерений с кало-
риметрами той же системы Г9, 10]. Несколько иную схему изме-
рений применил Брипёльфссон [11], который работал с ледяным
•96
калориметром, состоявшим из сосуда Дьюара объемом 470 мл,
целиком заполненного водой и льдом и окруженного цилиндри-
ческой рубашкой с водой и льдом. Изменение объема смеси
воды и льда в сосуде Дьюара определяли с помощью капил-
лярной трубки находившейся за пределами кобальтового источ-
ника излучения. Преимущество такой системы состоит в том, что
любая световая энергия, возникшая при переходе из промежу-
точных возбужденных состояний в основное, будет удерживаться
в калориметре.
Ксено и Лоу [12], также использовавшие кобальтовый источ-
ник излучения, измерялй тепловую мощность, выделявшуюся
в магниевом калориметрическом теле в квазиадиабатическом ре-
жиме. Затем в полиэтиленовых контейнерах тех же размеров
и формы облучали ферросульфатные растворы и находили зна-
чение G. В калориметрическом теле имелись внутренние поло-
сти, что позволяло получать такую же, как в растворах, сред-
нюю электронную плотность. Так как основным механизмом
поглощения энергии было комптоновское рассеяние, мощность
дозы в растворе находили, умножив тепловую мощность кало-
риметра в ваттах на I г па отношение числа электронов на 1 г
в растворе и в магнии.
Рид й Джонс [13, 14] для определения W (в воздухе) исполь-
зовали сдвоенные квазиадиабатические калориметры с полыми
калориметрическими телами цилиндрической формы. Использо-
валось у-излучение источников 60Со или ,37Cs либо тормозное
излучение с энергией 22 Мэв. Размеры калориметрического тела
были малыми по сравнению с сечением и проникающей способ-
ностью пучка. Энергия, поглощен-
ная в единице массы стенки иониза-
ционной камеры, сравнивалась за-
тем с числом пар ионов, образовав-
шихся в камере.
Для излучений высокой энергии
описан микрокалориметр [15], в ко-
тором определяется локальная по-
глощенная доза в неметаллических
твердых телах. Калориметр изготав-
ливается из угля или угольных час-
тиц в полистироле. Отношение угле-
рода к водороду равно 1:1. Калори-
метр использовался для дозиметрии
излучения 60Со и электронных пуч-
ков с энергиями до 20 Мэв. При
мощности дозы 50 рад/мин или
более этот калориметр обеспечивает
точность около 1%.
В качестве первичного стандарт-
ного устройства для измерений по-
Рис. 4.3а. Принципиальное
устройство адиабатического ка-
лориметра:
/ — термопары: 2 — калориметри-
ческое тело; 3 — внешняя рубашка;
4 — нагреватели.
4 Зак 907
97
глощепной дозы предложен калориметр [16], в котором калори-
метрическим телом служит графитовая или алюминиевая сфера
диаметром 1 см. Сфера закреплена па полистироловых штифтах
внутри адиабатической рубашки с внутренним диаметром 1.4 см
и наружным диаметром 2,2 см. Схема калориметра показана па
рис. 4.3а. Температуры определяются с помощью хромель-
константаповых термопар. Для скрепления двух полусфериче-
ских частей калориметрического тела используется слой про-
водящей краски (с сопротивлением 250 ом), который одновре-
менно служит калибровочным нагревателем.
4.3. ИЗМЕРЕНИЕ ПОГЛОЩЕННОЙ ДОЗЫ
МЕТОДОМ СОБИРАНИЯ ЗАРЯДОВ
В принципе можно точно определить поглощенную дозу,,
направляя в образец с определенной тормозной способностью'
Рис. 4.4. Схема устройства для
собирания заряда, с помощью ко-
торого проводилась калибровка
дозиметра Фрике:
1 — образец; 2 —парафин; 3 *- плати-
новая проволока; 4— направление к
сумматору тока; 5 — алюминиевое
окошко: 6 — электронный пучок.
поток заряженных частиц с из-
вестной энергией. Измерив заряд,,
собранный в образце, можно
определить значение потока и,
умножив ее на энергию частиц,
найти дозу. Метод собирания за-
ряда может использоваться для
измерения доз излучения с точ-
ностью около 0,5%.
На рис. 4.4 показана схема
подобного устройства. Электрон-
ный пучок, напряжение которого
измерено калиброванным вольт-
метром, выходит из генератора
Ван-де-Граафа через ' тонкое
(3 мм) алюминиевое окошко.
Затем пучок попадает в погло-
щающую ячейку с раствором
ферросульфата. Радиус ячейки
подобран так, что обеспечивается
полное поглощение электронного
пучка. В стандартных экспери-
ментах использовали токи от
6-Ю-11 до 7-10~7а. Ток, входя-
щий в раствор, отводился с по-
мощью платиновой проволочки
по экранированному кабелю па сумматор, который измеряет
полный собранный заряд.
Следует, однако, учесть несколько источников возможных,
ошибок и ввести поправки на: 1) потери энергии в пучке при
прохождении через различные окна; 2) перенос некоторого ко-
личества энергии в раствор электронами обратного рассеяния,
98
которые не учитываются в общем заряде; 3) тормозное излу-
чение; 4) утечки тока и 5) рассеяние электронов на диафраг-
мирующем отверстии (см. рис. 4.4). Если учесть соответствую-
щие поправки, то можно провести очень точные измерения по-
глощенной дозы. Метод собирания заряда использовался для
измерений на электронных пучках генератора Ван-де-Граафа
[17, 18], пучках калиброванных р-излучателей [19], на протонных
и электронных пучках, получаемых в циклотроне [20, 21].
4.4. ХИМИЧЕСКИЕ ДОЗИМЕТРЫ
В методах дозиметрии, которые были рассмотрены в преды-
дущих разделах, те или иные физические устройства исполь-
зуются главным образом для измерения интенсивности падаю-
щих пучков излучения. Все эти методы достаточно надежны и
позволяют непосредственно измерять поглощенную дозу. Тем
не менее ни один из них из-за разного рода ограничений не
получил достаточно широкого распространения на практике.
Более целесообразным оказывается использование вторичных
дозиметров, которые калибруются с помощью одного из пер-
вичных методов, рассмотренных в разделах 4.1—4.3.
К настоящему времени предложены буквально сотни раз-
личных химических дозиметров [22-—28]. Большинство из них не
имеет широкого распространения, так как либо недостаточно
хорошо изучены, либо не отвечают требованиям, предъявляемым
к идеальному химическому дозиметру. Эти требования можно
сформулировать следующим образом:
1. Радиационно-химический выход дозиметрической реакции
должен обладать относительной независимостью от концентра-
ции реагента и продукта реакции. В идеальном дозиметре выход
линейно возрастает с увеличением дозы.
2. Применяемые реагенты и техника анализа радиационно-
химической реакции должны быть достаточно простыми. Реа-
генты с достаточной степенью очистки имеются в свободной
продаже.
3. Химическая система сохраняет стабильность при воздей-
ствии света, тепла, при хранении до и после облучения.
4. В нужном диапазоне лоз радиационно-химическая реак-
ция обладает достаточным выходом, который легко удается из-
мерить.
5. Показания дозиметра не зависят от мощности дозы в до-
статочно широком диапазоне.
6. Показания дозиметра не зависят от величины линейной
передачи энергии (ЛПЭ).
7. Общая точность измерений около ±1%.
8. Дозиметр пригоден для работы в смешанных радиаци-
онных полях.
4* 99
Ни один из химических дозиметров, прошедших тщательную
проверку, не удовлетворяет всем этим требованиям одновре-
менно. Однако существует несколько типов дозиметров, для ко-
торых значительная часть этих требований выполняется.
Наиболее широко используемым и тщательно проверенным
лабораторным дозиметром является ферросульфатпый дозиметр
Фрике. Этот дозиметр и ряд других систем подробно рассмат-
риваются в следующих разделах.
4.4.1. Ферросульфатная дозиметрия
При облучении в кислом растворе ферросульфат окисляется
в феррисульфат. Первое сообщение об этой радиационно-хими-
ческой реакции было опубликовано Фрике в 1927 г. Двумя го-
дами позднее Фрике предложил использовать раствор ферро-
сульфата в 0,8 н. H2SO4 в качестве дозиметра рентгеновского
излучения. Поведение этой химической системы при облучении
должно было имитировать работу камеры с воздушными стен-
ками.
Первый шаг в приготовлении дозиметрического раствора
состоит в очистке воды и посуды. Такая предварительная ра-
бота оказывается необходимой во всех радиационно-химических
экспериментах с водными системами. Аллен [29] рекомендует
следующую процедуру очистки: «В Брукхейвепской лаборатории
очищенную воду получают повторной дистилляцией обычной
дистиллированной воды вначале из кислого бихромата, затем из
щелочного перманганата и, наконец, дистилляцию ведут без до-
бавления каких-либо реагентов в сосуд из плавленого кварца.
Во время дистилляции пары воды проходят через колонну, за-
полненную стеклянными спиралями. Небольшой участок в верх-
ней части колонны обогревается снаружи, для того чтобы разор-
вать водяную пленку па внутренней поверхности и таким обра-
зом воспрепятствовать просачиванию примесей под влиянием
капиллярных эффектов. Дистиллированная вода должна про-
ходить только через стеклянные и металлические трубки. Ника-
кие контакты с пластиком или резиной недопустимы. Дистилля-
торы защищают от пыли или паров в окружающем воздухе с
помощью трубок, заполненных активированным углем».
Опыт показывает необходимость всех этих предосторожно-
стей. Сходные меры предосторожности предпринимаются и в
других лабораториях, где ведется работа с водными системами.
В дополнение к описанной процедуре Харт [30] пропускает
пары воды через трубку, нагретую до нескольких сот градусов
с тем, чтобы выжечь органические загрязнения. Но даже после
такой обработки в воде все еще есть загрязнения, которые мо-
гут влиять на такие реакции, как образование перекиси водоро-
да в воде, насыщенной воздухом. По-видимому, наиболее чистую
воду удается получать после у-облучения трижды перегнанной
100
воды, насыщенной воздухом, где возникающая перекись водоро-
да разрушает органические вещества. Затем перекись в воде в
свою очередь разрушают воздействием ультрафиолетовой радиа-
ции ртутной лампы [31].
Наиболее тщательная очистка воды оказывается бесполез-
ной, если с той же тщательностью не будут очищены сосуды, в
которых производится облучение. Стеклянная посуда, которая
соприкасается с воздухом, по-видимому, покрывается пленкой
из жиров и других органических веществ. Большая часть этих
загрязнений остается на поверхности, и их можно удалить мою-
щими растворами. Часть загрязнений, однако, попадает в ульт-
рамикроскопические трещины, где становится недоступной воз-
действию обычных средств. Под действием рентгеновского или
у-излучения эти вещества попадают в воду и реагируют с обра-
зующимися свободными радикалами. Поэтому оказывается
необходимым предварительно облучать заполненные очищенной
водой реакционные сосуды дозами 106 р или более до тех пор,
пока не появится заметное окрашивание стекла. После этого
сосуды споласкивают очищенной водой и держат заполненными
до момента опыта. Хороший способ очистки сосудов перед об-
лучением — пропускание через них потока паров, полученных
при кипячении очищенной воды*.
Ферросульфатпый дозиметр готовят по такому рецепту: при-
мерно 2 г FcSO4 7 НгО или FeSO4 • (NH4)2SO4-6H2O, 0,3 г
NaCl и определенное количество концентрированной (95—
98%-ной) H2SO4, достаточное для получения 0,8 п. кислоты, раз-
водят в дистиллированной воде, после чего доводят общий объ-
ем до 5 л. Полученный раствор должен быть 0,0014 М по фер-
росульфату, 0,001 М по NaCl и 0,8 н. по H2S!O4. Такой
ферросульфатный раствор подвержен медленному окислению,
однако если не выставлять его па солнце, то он остается доста-
точно стабильным в течение 2—3 месяцев.
В первоначальных опытах Фрике количество окисленного
железа определялось титрованием. За исчезновением ионов
двухвалентного железа можно следить также по образованию
комплекса с ортофенантролином, который определяется спектро-
фотометрически. Сейчас общепринят способ измерения концент-
рации ионов трехвалентного железа прямым спектрофотометри-
ческим методом по поглощению, при 305 или 224 нм (см.
главу VIII).
Перейдем теперь к рассмотрению некоторых трудностей, воз-
* Эти предосторожности необходимы при радиационно-химических эк-
спериментах с водными растворами, однако не обязательны для ферросуль-
фатной дозиметрии. Здесь достаточно выполнения обычных правил, т. е.
следует пользоваться чистой посудой и качественной дистиллированной во-
дой. Тем не менее в большей части лабораторий применяются все указан-
ные меры предосторожности.
101
никающих при ферросульфатной дозиметрии. С частью из них
удается справиться, с другими — не удается.
1. Чувствительность к органическим загрязнениям. Чрезвы-
чайно высокие требования, предъявляемые к чистоте посуды и
реагентов, обусловлены высокой чувствительностью системы к
органическим загрязнениям. Дьюхерст обнаружил, что чувстви-
тельность можно заметно понизить добавлением в небольших
количествах ионов хлора. Поэтому при использовании стандарт-
ного рецепта дозиметра достаточно выполнять обычные лабора-
торные правила.
2. Температурная зависимость коэффициента молярной
экстинкции. Поглощенная доза определяется с помощью спект-
рофотометрических измерений количества образовавшегося
Fe3+. В табл. 4.4 приведены коэффициенты молярной экстинк-
Таблица 4.4
Коэффициент молярной экстинкции Fe3+ р 0,8 н. H.,SO4 при 3050 А (по дан-
ным ряда лабораторий)
Лаборатория Автор е
Аргоннская национальная лаборатория Хаут 2225
Кембриджский университет Своллоу 2167
Брукхейвенская национальная лаборатория Аллен 2195
Эдинбургский университет Миллер 2201
Ок-Риджская национальная лаборатория Хохандель 2240
Институт Слоан-Кеттеринга Лафлин 2172
Париж Лефорт 2205
Средняя величина 2201
Стандартное отклонение 0,4%
ции е при 3050А, которые использовали некоторые авторы. Так
как е при этой длине волны заметно зависит от температуры
(в интервале от 20 до 30° С меняется на 0,69% на 1°С) то из-
мерения следует проводить при 25° С или нужно-вводить соот-
ветствующие поправки.
3. Диапазоны измерения поглощенных доз. Значение радиа-
ционно-химического выхода окисленных ионов Fe3+ различна
в насыщенных воздухом и деаэрированных растворах (см. гла-
ву VIII, раздел 10). Если облучать насыщенный воздухом рас-
твор, то весь растворенный кислород расходуется при дозе около
50 крад, после Мего значение G падает с 15,5 до 8. Таким об-
разом «нормальный» верхний предел по дозе для аэрированной
системы Фрике составляет около 50 крад. Если раствор перед
облучением насыщается кислородом вместо воздуха, то диапа-
зон измеряемых доз можно увеличить в пять раз — примерно до
2-105 рад. Если же через раствор непрерывно пропускать кис-
102
лород или использовать деаэрированные растворы, то можно
достаточно точно измерить дозу вплоть до 106 рад [32].
Другой способ повышения верхнего предела дозиметра Фри-
ке состоит в прибавлении к раствору 0,1 М CuSO4. При этом
выход окисленных ионов Fe3+- падает до 0,66 и перестает зави-
сеть от присутствия кислорода
Такая система позволяет изме-
рять очень большие поглощенные
дозы. Использование дозиметра
Фрике для измерения малых по-
глощенных доз (меньше 1000 pad)
ограничивается чувствительно-
стью аналитической техники.
4. Влияние внешних условий.
Выход дозиметра Фрике прак-
тически не зависит от темпера-
туры * в диапазоне от 0 до 65°С.
Выход не зависит также от кон-
центрации ионов Fc2+ в диапа-
зоне от 5-102- М до 10-4 М и от
концентрации H2SO4 в пределах
1,5—0,1 М. Однако при измене-
нии содержания тяжелых ионов
в растворе меняется массовый ко-
эффициент передачи энергии.
Поэтому, добавляя элементы с
высокими атомными номерами,
которые не влияют на радиаци-
онно-химический выход системы,
можно добиться того, чтобы эф-
фективный атомный номер дози-
метра -соответствовал эффектив-
ному атомному номеру любой
исследуемой системы.
5. Влияние линейной передачи
энергии. Имеется целый ряд
данных о зависимости выхода до-
зиметра Фрике от ЛПЭ фотонных излучений и потоков частиц.
Данные, приведенные в табл. 4.5 и на рис. 4.5, так же как и
данные М,индера [33], показывают, что в диапазоне энергий от
100 кэв до 30 Мэв выход дозиметра для фотонов и электронов
не меняется. Как показано на рис. 4.5, где приведены значения
G для аэрированных и деаэрированных растворов, выход начи-
нает падать для ЛПЭ выше 0,1 эв/А и приближается к асимпто-
тическому значению 3,0±0,9 для тяжелых продуктов деления.
Расстояние, А
Рис. 4.5. Средние радиационно-
химические выходы для окисле-
ния Fe2+ (в присутствии кисло-
рода или без него), восстановле-.
ния Се4+ и образования Н2 в за-
висимости от начальной ЛПЭ
падающих частиц. Приведены
данные как для однозарядных,
так и для многозарядных частиц.
Верхняя шкала рисунка показы-
вает среднее расстояние между
первичными ионизациями.
* Температурный коэффициент для окисления ионов железа, вызванного
у-излучением, составляет 0,03—0,04%/°С в диапазоне 0—70° С. Однако для
излучений с высокими ЛПЭ температурный коэффициент оказывается выше.
103
Т аблица 4.5
Значения и для ферросулыратного дозиметра
Излучение G (Fe’+) Метод измерения энергии Авторы
160 Мэв, протоны 30 Me (пик), рентгенов- ское излучение (фильтро- ванное, Е = 7,6 Мэв **) 2 Мв (пик), рентгенов- ское излучение Со60, у-излучение (1,25 Мэв) 2 Мэв электроны Р32, р-частицы (Еср = 0,70 Мэв) 220 кв (пик), рентгенов- ское излучение (фильтро- ванное, 0,65 мм Al, Е — ~ 56 кэв **) Сов0, у-излучение 100 кв (пик), рентгенов" ское излучение (фильтро- ванное, 0,65 мм Al, Е = = 33 кэв **) 50 кв (пик), рентгенов- ское излучение (нефильт- рованное) 8 кв (ник), рентгенов- ское излучение Тритий, р-частицы (£ер = 5,5 кэв, внутрен- ний источник) 12 1,991 3,47 Мэв, протоны 0,63 6Li (п, а)3Н, ядра от- дачи 210Ро, а-частицы (5,3 Мэе, внутренний источник) leB (п, a)7 Li, ядра от- дачи 16,5±1 16,3±0,6 16,0±0,5 15,5±0,5 15,6±0,3 15,7 15,8±0,3 15,45±0,11 15,21 ±0,04 15,39±0,04 15,0±0,5 15,68±О,7 15,42±0,4 14,7±0,5 14,0±0,8 13,4±0,6 12,9±0,2 9,81 8,00 6,90 6,89 5,69±0,12 5,10±0,10 4,22±0,8 Поток протонов от наведенной ра- диоактивности Ионизация Ионизация *** Ионизация Калориметрия Ионизация *** Калориметрия Собирание заряда 1 4Л-СЦИНТИЛЛЯ- J ционный счетчик Ионизация Калориметрия Ионизация Неабсолютная калибровка Ионизация Счет и определе- ние плотности газа • Собирание заря- да . Измерение потоке нейтронов Абсолютный счел Измерение потоке нейтронов Эренберг и Зе- ланд * Хэйбиттл, Саун- дерс и Своллоу * Вейсс, Берн- штейн и Куппер * Хэйбиттл, Саун- дерс, и Своллоу Хоханадель и Горм- ли Лазо, Дьюхерст и Бартон * Шулер и Аллен* Пейт и Стейн * Пейт и Стейн * Хэйбиттл, Саун- дерс, и Своллоу * См. цит. литера- туру [Н] См. цит. литера- туру [12] Хэйбиттл, Саун- дерс и Своллоу Бэк и Миллер * Коттин и Лефорт* Макдоннел и Харт * Харт, Рамлер и Роклин * Шулер и Барр * Тромбор и Харт* Харт, Рамлер и Роклин *
104
П родолжение табл. 4.5
Излучение О (Fe+S) Метод измерения энергии А вторы
2SBU, продукты Деления 3,0±0,9 Измерение потока нейтронов Эренберг и Зе- ланд *
* См. цит. литературу [23], стр. 111.
** Эффективная средняя энергия излучения.
*** Значения G, полученные ионизационными измерениями, пересчитывали на значение
^воздух = 34 эв/пара ионов, если авторы использовали другие значения этой константы.
6. Влияние мощности дозы. Выход дозиметра не меняется
или меняется слабо вплоть до мощности дозы 107 рад!сек, от
107 до 1010 рад/сек выход падает с 15,5 до 8 (табл. 4.6).
Таблица 4.6
Влияние мощности дозы на G (Fes+) при облучении быстрыми электронами*
С г >2, ММОЛЬ Fe^ ммоль Е, Мэв Длительность импульса, мксек Мощность до- зы, рад! сек 10е °(Fes+)
1,0 1,0 1,0 Контроль 0,002 15,6±0,05
1,0 1,0 2,0
3,0 — 4,0 1,7 9,6 14,3
3,0 1,0 1,0—1,2 5,0 0,016 15,0±1,6
3,0 1,0 1,0—1,2 5,0 0,18 13,4+0,.6
3,0 1,0 1,0—1,2 5,0 0,75 10,8±0,9
3,0 1,0 1,0—1,2 5,0 2,4 10,1 ±0,5
1,0 1,0 15 1,4 0,09 15,2±0,4
1,0 1,0 15 1,4 0,69 14,5±0,4
1,0 1,0 15 1,4 2,26 11,3±0,5
1,0 1.0 15 1.4 2,92 12,2±0,5
1,0 1,0 15 1,4 8,0 11,8±0,5
1,0 1,0 15 1,4 8,0 13,0±0,4
1,0 1,0 15 1.4 8,0 8,±0,5
•Данные из справочника: National Bureau of Standards. Handbook 85, p. 17, V. 5,
Government Printing Office, Washington, 1964. Physical Aspects of Irradiation.
Пример 4.3. 10 см3 насыщенного воздухом дозиметрического раствора
Фрике, помещенного в трубку диаметром 1 см, облучались в течение 10 мин
в полости цилиндрического источника у-излучепия из 6сСо. Оптическая плот-
ность (ОП) при 3050 А в сантиметровой кювете при 30е С до начала облуче-
ния была равна 0,003 после облучения 0,260.
1. Чему равна полная доза в радах, полученная раствором?
2. Чему была равна поглощенная доза в радах, если в том же сосуде
в течение 10 мин облучали 10 см3 метанола?
3. Чему была равна поглощенная доза, если бы в ту же трубку поместили
на 10 мин Pb(NO3)2?.
Решение. Коэффициент молярной экстинкции для феррисульфата при
25° С (см. табл. 4.4) равен 2201. При 30° С он равен 2201 [1+0,007(5)]=2278.
105
Концентрацию вещества по заданной оптической плотности находят с помощью
закона Бэра (см. главу VI, пример 6.1). Удельная плотность раствора равна
1,023 г/см?. Значение 0=15,5 молекул/100 эв.
1. Полная поглощенная доза в радах определяется через разность оп-
тических плотностей следующим образом:
D эрг (О77конеч — ОПНЙ„) моль 1 л см3 6,023 • I023 молекул
г /е л 1000 см3 р г моль
100 эв I 1,6 • 10 эрг
15,5 молекул | эв
1 0,260 — 0,003 1 1 6,023 1023 100 X
D,,ad~ 100 (2278) (1) 1000 1,023 15,5
1,6 • го-»2
X-------------= 6,87 • 103 рад.
2. Так как энергия излучения лежит в комптоновском диапазоне, то долю
энергии, поглощенную каждым компонентом, можно считать пропорциональ-
ной числу электронов:
W!A)M
Dm ~ (Z/А)! '
TRe индекс М относится к метанолу, а 1 — к дозиметрическому раствору
Фрике. Для метанола (ZlA)M= (6+ 4+ 8) /32= 18/32=0,562; для раствора
Фрике (Z/4)r=0,533. Следовательно, Дм=6,87-103 (0,562)/(0,533)=7,22Х'
ХЮ3 рад.
3. Для Pb(NO3)2 существенным становится фотоэлектрическое поглоще-
ние, поэтому метод электронных долей и соответствующее уравнение непри-
менимы. Согласно сообщению Хохапделя и Дэвиса [34] массовые коэффици-
енты передачи энергии для РЬ(ЬЮз)2 и для дозиметра Фрике равны соответ-
ственно 0,0319 и 0,023 см2/г. Отсюда найдем поглощенную дозу
OPb(NO8), = 6-87 • 103 (0,0319/0,025) = 8,77 • 103 рад.
При решении этого примера мы сделали целый ряд .упро-
щающих допущений:
1. Замена ферросульфатпого раствора на метанол не при-
вела к изменению спектра вторичных электронов или спектра
первичного пучка. Хотя для излучения 60Со такое допущение в
общем случае достаточно оправдано, в ряде ситуаций следует
учитывать изменение энергетического спектра первичного пучка
при прохождении через вещество и соответствующее изменение
массовых коэффициентов передачи энергии.
Этот эффект проанализирован теоретически и эксперимен-
тально. На рис. 4.6, например, показано изменение массового
коэффициента передачи энергии для 0,8 н. H2SO4 в зависимости
от расстояния (в воде) от источника 60Со. Увеличение коэффи-
циента обусловлено изменением первичного и вторичного спект-
ров излучения.
Метод электронных долей, который применяем только к по-
глощению моноэнергетического излучения в области компто-
новских энергий для обоих веществ, оказывается ненадежным,
106
если применять его к веществам, атомные номера которых за-
метно различаются. Например, метод пригоден для пары
KNO3/KNO2 и мало пригоден для пары KNOs/CsBr.
В методе предполагается, что все взаимодействия (как пер-
вичные, так и вторичные) пропорциональны относительному чис-
лу электронов. .Строго говоря,
это неверно.
2. Интенсивность излучения
в контейнере с любым образ-
цом предполагалась той же,
что и в фсрросульфатном рас-
творе. Для используемого типа
источника это только прибли-
зительно правильно. (В. Берн-
стсйп и Р. Шулер [35] показа-
ли, что около 2% излучения в
полости цилиндрического ис-
точника 60Со является низко-
энергетическим и поэтому про-
исходит фотоэлектрическое
поглощение.)
О 10 20 30 40 50 ВО 70
Расстояние от источника, см
Рис. 4.6. Учет изменения коэффици-
ента передачи энергии 0,8 н. H2SO4
в зависимости от расстояния от ис-
точника 60Со.
4.4.2. Церийсульфатная дозиметрия
Радиационно-химическая реакция восстановления Се4+ до
Се34- используется для дозиметрии в области 105—108 рад. Эта
дозиметрическая система особенно полезна при измерениях в
радиационных нолях с высокой интенсивностью, там где излу-
чение высокой интенсивности не позволяет использовать стан-
дартный ферросульфатный дозиметр.
Чтобы получить с помощью церийсульфатной дозиметриче-
ской системы воспроизводимые результаты, необходимо обра-
тить особое внимание на чистоту реакционных сосудов и реа-
гентов. Посуда должна быть тщательно очищена, а для приго-
товления растворов следует пользоваться только трижды пере-
гнанной водой, приготовленной согласно указаниям в разде-
ле 4.4.1. Дозиметр обеспечивает точность ±1%. Для получения
воспроизводимых результатов необходимо иметь в системе не-
большую начальную концентрацию (>10-5) ионов Се3+.
Рекомендуется применять следующую процедуру:
1. Приготовьте раствор сернокислого окисного церия на
0,8 н. серией кислоте в такой концентрации (от 0,001 до
0,05 Л4), чтобы после облучения восстанавливалось 20—80%
имевшихся ионов. При более высоких концентрациях пропор-
циональность выхода дозы не сохраняется, тац как (7(Се3+)
зависит от концентрации и Се4+ и Се3+.
2. Проводите облучение раствора в достаточно большом со-
суде, чтобы обеспечивались условия электронного равновесия.
107
3. Измерьте оптическую плотность при 3200 А. Коэффициент
молярной экстинкции при этой длине волны зависит от темпе-
ратуры в диапазоне 15—25° С и, по-видимому, чувствителен к
изменениям pH среды. В литературе приводятся различные зна-
чения коэффициента молярной экстинкции. Поэтому следует,
очевидно, провести независимое определение этого значения.
Для цериевого раствора на 0,8 н. H2SO4 в литературе приводи-
лись значения коэффициента молярной экстинкции: 5580 и
5565.
4. Рассчитайте дозу, поглощенную цериевым раствором.
5. Пересчитайте эту дозу па интересующий пас образец.
Значение G(Ce3+) церийсульфатного дозиметра равно 2,5 ±
±0,18. Значение G не зависит от температуры вплоть до 35° С,
от мощности дозы вплоть до 2-Ю6 рад/сек при непрерывном об-
лучении и 2 • 108 рад/сек при импульсном. Зависимость от ЛПЭ
при сравнении действия у-излучения 60Со и электронов с энер-
гиями до 14 Мэв не обнаружена. Для более плотно ионизирую-
щего излучения зависимость G от ЛПЭ показана па рис. 4.5 и
в табл. 4.7.
Таблица 4.7
Значения G для церийсульфатной дозиметрической системы [23]
Излучение G (Се3+) Предполагаемое зна- чение G (Fe3^-) или метод измерения энергии Авторы
8—14 Мэв, электроны 2,5±0,18 Калориметрия Таймути и Таул; Петерсон Таймути и Таул
6оСо у-излучсние 2,5±0,26 15,6
(1,25 Мэв) 2,33±0,03 15,45 Петерсон
2,45±0,08 15,5 Барр и Шулер
2,44±0,03* 15,6 Джонсон и Вейсс
200 кв (пик), рентге- новское излучение 3,15+0,10 15,5 Джонсон и Вейсс
10 Мэв, дейтроны 2,80±0,04 Собирание заряда Барр и Шулер
11 Мэв, ионы гелия 2,90—0,06 То же Барр и Шулер
г10Ро, а-частицы (5,3 Мэв, внутренний ис- точник) 3,20+0,06 Абсолютный счет Лефорт и Таррелло
210Ро, а-частицы (3,4 Мэв, фильтрация че- рез слюду) 2,88±0,02 4,7 Вейсс и Миллер
юв (n, a)7 Li. ядра от- дачи 2,94±0,12 4,22 Барр и Шулер
* Это средняя величина выхода, полученная из шести независимых значений, приведен-
ных в литературе. Величина выхода приведена к стандартным условиям. Предполагается,
что коэффициент молярной экстинкции для Се4+ прн 320 0 А равен 5609 л/моль..
108
4.4.3. Другие химические дозиметры
Как уже отмечалось, к настоящему времени предложено
большое число химических дозиметров: Большая часть этих
дозиметров прокалибрована с помощью ферросульфатного дози-
метра, а некоторые — с помощью ионизационных камер. Одна
из главных причин, побуждающих разрабатывать новые типы
дозиметров, — потребность в повышении их чувствительности.
Так, например, разработаны химические дозиметры, позволяю-
щие измерить поглощенную дозу всего лишь 0,5 рад.
Органические соединения галогенов под действием ионизи-
рующих излучений выделяют кислоты. Дозиметры этого типа
были предложены в 1951 г. Тэплином, Дугласом и Санчесом
[36], а также Хенли и Миллером [37]. Жидкие углеводороды, та-
кие' как хлороформ, четыреххлористый углерод или тетрахлор-
этилен, могут использоваться в виде однофазной, двухфазной
или эмульсионной систем. В двухфазных и эмульсионных вод-
ных системах кислота эстрагируется водным слоем. Количество
кислоты, возникающей при функционировании дозиметра в ра-
бочем диапазоне, обычно линейно изменяется с ростом дозы, и
его можно определить ацидометрическим титрованием или по
изменению цветности pH-чувствительных красителей, таких, на-
пример, как бромкрезол пурпурный.
Распад соединений галогенов происходит с высоким радиа-
ционно-химическим выходом, достигающим 1000. Реакция весь-
ма чувствительна к присутствию кислорода. Так как значения
G велики, то даже излучение с небольшой интенсивностью при-
водит к заметным химическим эффектам, поэтому можно изме-
рять очень маленькие (порядка 1 рад) дозы. Добавляя следо-
вые количества органических ингибиторов цепных реакций, на-
пример спиртов или фенолов, можно снизить G меньше чем до
30, и тогда удается измерить дозы порядка 106 рад.
Как правило, системы, содержащие хлорированные углево-
дороды, весьма чувствительны к температуре, интенсивности из-
лучения и наличию примесей. Присутствие хлора, поглотителя
со сравнительно высоким Z, делает такие системы еще более
чувствительными к изменениям ЛПЭ. Большая часть обширных
исследований дозиметров такого типа проводилась по заказу
армии США в расчете на то, что на основе галоидированных
углеводородов удастся разработать недорогой дозиметр для
личного состава. Эти системы, однако, нашли лишь ограни-
ченное применение.
Водные растворы бензола и бензойной кислоты также пред-
лагались в качестве дозиметров. Основными продуктами радио-
лиза бензола являются фенол и дифепил, содержание которых
в системе нетрудно определить. Бензойная кислота подвергается
радиационному окислению до оксибензоатов, которые легко
определяются флуорометрически.
109
Для диапазона высоких доз (от 10е до 108 рад) в качестве
дозиметра были предложены растворы муравьиной кислоты, на-
сыщенные кислородом [38]. Продуктами радиационно-химических
реакций являются перекись водорода, двуокись углерода и
водород.
Дозиметрия в газовой фазе часто оказывается сложной за-
дачей. Был предложен и пользовался определенным успехом
ацетиленовый дозиметр [39, 40]. При 30° С G(—С2Н2)=71,9.
В качестве газофазного дозиметра использовалась также
закись азота. Предполагаемый механизм радиационного разло-
жения закиси азота выглядит следующим образом:
N2O------- N + NO или N2 + О; (4.11)
N - Ь N2O-► N2-г NO; (4.12)
О -t- N2O -> 2NO или N2 О2; (4.13)
2NO + О22NO2; (4.14)
G(—N2O) равно 12,8 для рентгеновского и у-излучения. Однако,
при высоких поглощенных дозах (>104 рад) начинаются об-
ратные реакции, которые ограничивают область действия дози-
метра. Этот дозиметр применяется в смешанных радиационных
полях, и его использование в этой связи обсуждается в следую-
щем разделе.
4.5. ДОЗИМЕТРИЯ В СМЕШАННЫХ РАДИАЦИОННЫХ ПОЛЯХ
В смешанных радиационных полях, где имеются нейтроны,
помимо трудностей, связанных с наведенной радиоактивностью,
возникает также и ряд других. В дозиметрах на водной основе,
например, нейтроны взаимодействуют с водой, создавая прото-
ны, которые являются источником излучения с высокой ЛПЭ.
Однако сечение захвата быстрых нейтронов в воде невелико.
Следовательно, некоторые типы водных дозиметров можно ис-
пользовать и в нейтронных полях. Например, выход церий-
сульфатного дозиметра для быстрых нейтронов равен 2,85 по
сравнению с выходом для у-квантов, равным 2,40. В результате
эффективный выход G оказывается равным 2,70.
В принципе поглощенную дозу в смешанных радиационных
полях можно определить с помощью ионизационной камеры.
В первую очередь, однако, необходимо определить процентное
содержание компонентов и провести соответствующие вычисле-
ния. Если радиационное поле создается только за счет быстрых
нейтронов, коротковолнового рентгеновского излучения или
у-квантов, то ситуация чрезвычайно упрощается. Этот случай
рассматривается в разделе 4.10, посвященном нейтронной дози-
метрии.
Реакторное излучение включает нейтроны и фотоны широко-
го энергетического спектра, и именно в этой ситуации оказы-
110
вается затруднительным точное определение дозы. В ряде слу-
чаев с успехом удается использовать калориметрический метод.
Однако целый ряд ограничений, таких, как необходимость ра-
боты в малых объемах, удаленность от вспомогательного обо-
рудования, необходимость использовать материалы с низкими
сечениями захвата нейтронов, затрудняет применение метода.
Однако при правильном подходе калориметрический метод по-
зволяет добиваться падежных результатов [41-—44]. В общем,
однако, дозовые ограничения и другие трудности заставляют
радиационных химиков искать иные типы дозиметров для сме-
шанных радиационных полей. Некоторые из таких дозиметров
рассматриваются в следующих разделах.
Дозиметр, основанный на радиолитическом разложении ща-
велевой кислоты, был первоначально предложен Драганичем
145]. Процедура работы с этим дозиметром выглядит следующим
образом:-
1. Приготовляют исходный раствор щавелевой кислоты с
концентрацией 0,05 М.
2. Дозиметрические ампулы с раствором тщательно откачи-
вают.
3. Проводят облучение растворов. Уменьшение концентрации
щавелевой кислоты определяют по оптической плотности при
248 нм после добавления комплексующего агента. Комплек-
сующий агент готовят из двух растворов.
Раствор А. 32,5 мг бензидина растворяют в 2 мл 30%-ной
ускуспой кислоты и водой доводят объем до 250 мл.
Раствор Б. 125,0 мг ускуснокислой меди растворяют в воде
и доводят ую 250 мл.
Затем смешивают равные части растворов А и Б, к ним до-
бавляют 4 части воды; 1 мл полученной смеси добавляют к
5 мл образца.
Значение G для щавелевокислого дозиметра равно 4,9. По-
грешность около 6% в диапазоне доз 1,6—160 Мрад. Зависи-
мость выхода от ЛПЭ и интенсивности излучения подробно не
исследовалась, однако известно, что значение G для протонов
отдачи и продуктов деления равно 0,9.
Для измерения больших доз в смешанных радиационных по-
лях широко используются радиационно-химические реакции об-
разования газообразных продуктов при разложении органиче-
ских и неорганических жидкостей. В диапазоне доз от 104 до
108 рад дозиметрической реакцией с точностью от 3 до 6%
может служить выделение водорода из циклогексана с выходом
G(H2)=5,25. Такой дозиметр был впервые предложен Шуле-
ром и Алленом [46] в 1955 г.
В 1956 г. Хартек и Дондес предложили использовать для
реакторной дозиметрии, и в частности для дозиметрии продук-
тов деления, закись азота. По этому методу, который исполь-
11
зуется в Бруклине. Харуэлле и на другйх реакторах, не так
давно были опубликованы обзоры [47, 48].
Дозиметр обладает тем преимуществом, что в нем обра-
зуются только газообразные продукты, состоящие главным об-
разом из N2, О2 и NO2. Выход, по-видимому, совершенно не за-
висит от ЛПЭ, а следовательно, и от интенсивности излучения.
Дополнительным преимуществом является отсутствие темпера-
турной чувствительности. Дозиметр может использоваться
вплоть до 150° С без тепловых эффектов. Экспериментальная
процедура выглядит следующим образом:
1. Сосуд, изготовленный обычно из кварца, если он рассчи-
тан на давления ниже 5 атм, или из алюминия в случае более
высоких давлений, заполняется N2O.
2. После облучения сосуд, погружают в ловушку с жидким
азотом и подключают к вакуумной системе. После этого изме-
ряют количество пескондепсировавшихся N2 и О2. Затем темпе-
ратуру сосуда поднимают до температуры смеси сухой лед —
ацетон и измеряют давление N2O. Наконец, температуру образ-
ца доводят до комнатной, при которой освобождается NO2.
3. Зная концентрацию NO2, N2+O2 или N2O, полную дозу
можпо рассчитать по следующим формулам:
£(NO2) = 3,80 102lm(%NO2); (4.15)
Е (N2) = 1,59 1021m (% N2,[ 064; (4.16)
£(N2O) = 1,34 • 1021m(%N2O)1-04, (4.17)
где £(NO2), £(N2) и £(N2O)—поглощенная энергия, выра-
женная в эв; m—масса газа в г; (%) —объемная процентная
концентрация данного газа.
Наиболее серьезный недостаток дозиметра — сравнительно
низкая точность (10%) и необходимость учета нейтроннозахват-
ных реакций, в которых участвует N.
Пример 4.4. Стейнберг, Лоффельхальц и Пружанский [47] приводят сле-
дующие экспериментальные результаты исследования разложения N2O,
помещенного в стеклянный сосуд объемом 22 см3, под действием продуктов
деления.
Рассчитайте поглощенную дозу излучения и проверьте, согласуются ли
полученные значения G с табличными.
112
Решение. В соответствии с уравнением для идеального газа массу газа
можно найти по формуле
т = (М)
РУ
RT
0,Н г,
где V — объем, равный 22 см3; М — молекулярный вес, равный 44; R — га-
зовая постоянная, равная 82,06 атм-см3/(°К-г-моль); Т — температура, рав-
ная 298° К; Р — давление, равное 2,55 атм. Используя уравнения (4.15)—
(4.17), получим:
Е (NO2) = 3,80 • 1021 (0,11) (0,16) =--- 0,67 • 102° эв;
Е (Nz) = 1,59 1021 (0,11) (O.32)1’064 -0,54 - 10» эв;
Е (N2O) = 1,34. 1021 (0,11) (О.4)1-04 = 0,58 - 1020 эв.
Причина расхождений в значениях поглощенной энергии не ясна. Погло-
щенйую дозу, однако, можно определить, используя среднюю величину
0,6-1020 38 1 рад • г
Е =----------------------------= 8,7 • 101°
0,11 г | 6,24 - 1013 эв
Пиело молекул, образовашихся или распавшихся в результате реакции,
равно:
NO.
N2 =
- n2o =.
22 | I 2,55 1 0,0016 1 6,02 - 1023
22 400 22 1 1 1 2,55 1 6,02 1023
22 400 1 22 1 1 1 2,55 0,0032 ।
‘22 400 1 1 gno2 | 0,004 2,41 0,6 - | 6,02 - 1023 IO2® — = 4; IO2®
= 8,02
2,41 • 1018;
4,82 10i«;
6,04 - lQi«;
gn2-
4,82
0,6
6,04
G мп —-------- — Ю,5.
—N2O — 0>6
4.6. ДОЗИМЕТРИЯ С ПОМОЩЬЮ ПЛАСТИКОВЫХ ПЛЕНОК
В тех случаях, когда высокая точность измерения не обя-
зательна, наиболее удачными химическими дозиметрами оказы-
ваются разнообразные пластики. Преимущества пластиков — их
низкая стоимость, прозрачность для излучений, гибкость, уни-
версальность и доступность. Для дозиметрии используют такие
пластики, как целлофан, полиэтилен, поливинилхлорид и поли-
эфиры. Ввиду указанных преимуществ дозиметрия с помощью
пластиковых пленок первой получила промышленное распрост-
ранение в процессах стерилизации пищевых и медицинских про-
дуктов, где требуется контролировать уровень доз, полученных
отдельными образцами. Другая важная область приложений для
113
пластиковых дозиметров — измерение распределения доз по
глубине образца и оценка доз в разных участках образца при
сканирующем облучении электронным пучком.
Под действием излучений в пластиках происходят такие хи-
мические реакции, как образование двойных связей, дегалоге-
низации, сшивки, полимеризация, деполимеризация, выделение
газов. Возможны иные химические изменения, которые происхо-
.дят в результате окисления иди восстановления красителей или
некоторых других соединений, включенных в полимерную мат-
рицу. Все эти эффекты, так же как и одновременно.происходя-
щие физические явления (захват электронов в ловушки, обра-
зование или разрушение кристаллических областей), можно за-
регистрировать, измеряя пропускание света, напряжение растя-
жения, теплоту плавления, окраску, спектр свободных радика-
лов, вязкость, растворимость, твердость, относительное удлине-
ние, пластическое течение или рентгеновскую дифракцию. Все
эти методы предлагались в свое время для использования в
дозиметрии. Однако лишь три метода получили действительно'
широкое распространение.
1. При облучении цвет поливинилхлорида меняется от зеле-
ного через желтый и янтарный до красновато-коричневого. Ок-
раска достигает максимума при выдерживании в течение 3—4
дней при компатпой температуре, а при выдерживании при
80—85° С максимальная степень окрашивания достигается через
5—10 мин. Окраска, если характеризовать ее по абсорбции
света при 3960 А, пропорциональна поглощенной дозе в диапа-
зоне 105—107 рад. Стандартный поливинилхлоридный дозиметр
широко используется для промышленного контроля процесса
стерилизации. Применяется процедура, описанная Артанди и
Стоунхиллом [49].
2. Полимеры с включенными в них красителями обесцвечи-
ваются под действием радиации. Существует бесчисленное мно-
жество комбинаций красителей и полимеров. Наиболее полно
изучена и наиболее широко используется комбинация промыш-
ленного целлофана с голубым диметоксидифенил-бис-азо-бис-
8-амино-1-нафтол-5,7-дисульфоновым красителем. .Хенли и Рич-
мэн [50] провели калибровку этой системы для электронов с
энергиями 0,8, 2 и 3 Мэв и для у-излучения 60Со в диапазоне
доз от 200 000 до 10-106 р. Доза определяется с помощью из-
мерений оптической плотности при 6500 А.
3. Метод, основанный на использовании эффекта индукции
дрансненасыщенности в длинных цепях парафинов, таких, как
полиэтилен, хотя и не получил столь широкого распространения,
как два предыдущих, тем не менее также позволяет создать
удобный, часто применяемый дозиметр. Степень ненасыщен-
ности, которая измеряется по поглощению в инфракрасной об-
ласти, не зависит от интенсивности облучения и температуры и
оказывается пропорциональной дозе вплоть до 100 Мрад.
414
4.7. ДОЗИМЕТРИЯ С ПОМОЩЬЮ ФОТОГРАФИЧЕСКИХ ЭМУЛЬСИИ
Проявление скрытых изображений,-создаваемых заряженны-
ми частицами при прохождении через фотографические эмуль-
сии, было одним из наиболее ранних методов детектирования
излучений [52]. В результате проявления вдоль трека частицы
формируются зерна серебра, образующие изображение. Получе-
ние подобных изображений вместе с фотографированием следов
частиц в пузырьковых камерах и камерах Вильсона является
одним из основных методов детектирования и исследования эле-
ментарных частиц. В ядерной физике используют специальные
эмульсии, в которых по сравнению с эмульсиями, применяемыми
в дозиметрии, содержится в 4 раза больше AgBr, а размер зе-
рен значительно меньше (от 0,1 до 0,6 мкм).
Фиксация треков элементарных частиц — лишь один из ас-
пектов ’ многообразных применений фотографических эмульсий
для целей дозиметрии. Эмульсии используют в авторадиографи-
ческих методах, с помощью которых можно проследить за дви-
жением радиоактивных изотопов в
применения эмульсий — индиви-
дуальный фотоконтроль дозы из-
лучения. Подбор соответствую-
щих пленок позволяет следить за
дозами в диапазоне от 0,1 до
1000 р. Почернение пленки, кото-
рая содержит достаточное коли-
чество тяжелых металлических
ионов, сильно зависит от энергии
фотонов и электронов в области
низких энергий. Поэтому стан-
дартные кассеты с пленками,- вы-
даваемые для индивидуального
фотоконтроля, представляют со-
бой довольно сложную комбина-
цию. В кассету помещают две
или более пленок с различной
чувствительностью для измерений
в разных диапазонах доз, а так-
же пакеты фильтров для выделе-
ния вклада различных частиц.
Сравнительно новой областью
является фотографическая дози-
метрия в диапазоне высоких доз
от 104 до 108 р. Эта методика ста-
ла возможной после разработки
специальных фотоэмульсий.
Промышленные рентгеновские
пленки также можно использо-
организмах. Важная область
Рис. 4.7. Почернение промышлен-
ных рентгеновских пленок при
различных дозах и мощностях
дозы. Источник излучения 60Со:
о — пленка «Анско Сьюпрей А», мощ-
ность г дозы (р/ч) 4,0 • Ю2; 4,5 • I04;
2.5 10е; б — пленка «Кодак»' типа К:
1 — три пленки. 2 — одна пленка.
115
вать для дозиметрии высоких уровней облучения. На рис. 4.7
показана зависимость плотности почернения от дозы для рент-
геновских пленок «Анско Сьюпрей А» и «Кодак» типа К. Для
увеличения точности использовали три слоя пленки, что, соглас-
но опубликованным данным, позволяет добиться точности
±5%.
Хотя для пленки «Лиско Сьюпрей А» зависимости плотности
почернения от мощности дозы в диапазоне от 2-Ю2 до 5Х
Х105 р!ч нет, но выше 105 р/мин наблюдается определенный
эффект. Отмечена сильная зависимость от энергии излучения в
диапазоне ниже 0,6 Мэв и от температуры.
4.8. ТВЕРДОТЕЛЬНЫЕ ДОЗИМЕТРЫ
Дозиметры, в которых используются физичесике явления,
происходящие под действием радиации в твердых телах, полу-
чили широкое распространение. В дозиметрах применяют твер-
дые вещества, в которых при облучении происходят фото- и
термолюминесцентные явления, соединения, образующие посто-
янные центры окрашивания (см. раздел 4.8.1) и претерпеваю-
щие химическое разложение, например нитраты, а также орга-
нические люминофоры и полупроводники, в которых при облу-
чении происходит изменение электрической проводимости.
4.8.1. Стеклянные дозиметры
Хотя под действием радиации в стеклах происходят скорее
физические, чем химические изменения, стеклянные дозиметры
обычно относят к классу химических. Первым типом стеклян-
ного дозиметра был дозиметр из фосфатного стекла, активиро-
ванного серебром. Сейчас он используется в американской ар-
мии для индивидуального, контроля. В дозиметре используется
эффект радиофотолюминесценции. Флуоресценция облученного
стекла происходит под воздействием света с длиной волны в
ближней ультрафиолетовой области.
Ионы Ag+ играют роль ловушек для электронов, освобож-
денных под действием радиации. Захватывая электрон, ионы
серебра восстанавливаются до нейтральных атомов, которые
становятся одновременно центрами флуоресценции и окраши-
вания. Появление центров окрашивания также можно исполь-
зовать в целях дозиметрии, однако, как показано на рис. 4.8, в
диапазоне гораздо более высоких доз. Дозу в этом случае оп-
ределяют с помощью измерений оптической плотности на спект-
рофотометре при длине волны 3500 А.
Хотя, как показывает рис. 4.8, выход стеклянного дозиметра
в области доз выше 106 рад становится нелинейным, при тща-
тельных измерениях можно добиться точности ±2% для доз
до 5-106 рад, и результат не будет зависеть от мощности дозы
116
вплоть до 106 рад!сек. Стеклянные дозиметры такого типа про-
верены лишь для излучений высокой проникающей способно-
сти — рентгеновского у-излучений.
В литературе предложено много вариантов специальных
стеклянных дозиметров. Ё некоторых из них используется яв-
ление радиотермолюмипесценции. В других наблюдается пря-
Рис. 4.8. Зависимость пропускания при разных дли-
нах волн от дозы для стекла, активированного
серебром.
мое увеличение оптической плотности в видимой или ультра-
фиолетовой части спектра. Для измерения поглощенных доз с
успехом использовали боросиликатные стекла, активированные
кобальтом [54]. Так же как и для фосфатных стекол, активи-
рованных серебром, дозу определяли по величине абсорбции
при 3500 А.
4.8.2. Дозиметрия полупроводниковыми детекторами излучения
Если в полупроводниковом диоде изменить полярность, то в
обедненной носителями заряда области перехода создается
электрическое поле высокой напряженности. Область перехода
становится полностью эквивалентной заряженному конденса-
тору. Если в области перехода образуется пара ионов, то на
диоде возникает электрический импульс, который можно усили-
вать и регистрировать на анализаторе.
После 1958 г. создано множество разных типов полупровод-
никовых дозиметрических устройств. По сравнению с иониза-
117
ционными камерами и сцинтилляционными устройствами полу-
проводниковые детекторы обладают рядом уникальных
преимуществ. У них очень мало время срабатывания. Чтобы
перебросить электрон из валентной зоны в зону проводимости,
достаточно всего лишь нескольких электронвольт. Эта энергия
составляет примерно 1/10 энергии образования пары ионов в
Рис. 4.9. Схема диффузионного детектора с п+ — р-переходом (а) и по-
верхностно-барьерного детёктора (б):
а) /— лодвод напряжения; 2 — область, обдненная носителями заряда; 3— место кон-
такта о выводом; 4 — облучение происходит с этой стороны: 5 — полупроводниковая
область л+-тида, образовавшаяся в результате диффузии фосфора; 6 — металличе-
ский электрод (не выпрямляющим).
б) 1— подвод напряжения; 2 — область, обедненная носителями заряда; 3—место
контакта с выводом; 4 — облучение происходит с этой стороны; 5 —тонкий золотой
электрод: 6 — протравленная и окисленная поверхность; 7 - металлический электрод
(не выпрямляющий)'.
обычных условиях, что обусловливает заметное улучшение ста-
тистики счета. Для твердотельных устройств характерны хоро-
шая линейность зависимости амплитуды импульса от величины
поглощенной энергии, а также высокое разрешение.
Основные недостатки существующих полупроводниковых
устройств: малое значение выходных сигналов, изменение рабо-
чих характеристик при изменениях во внешней среде, пролет
частиц с большой длиной пробега за пределы детектора (из-за
малых линейных размеров детектора), чувствительность струк-
турных и характеристик детектора к действию радиации.
Два основных типа используемых детекторов показаны на
рис. 4.9. На рис. 4.9, а приведен пример диффузионного детек-
тора. Область п+ — р-перехода создается в результате диффу-
зии донорной примеси, такой, например, как атомы фосфора, в
кремниевый полупроводник p-типа. Наоборот, полупроводнико-
вый диффузионный детектор с р+—n-переходом можно полу-
чить в результате диффузии гелия или бора в кремниевый по-
лупроводник п-типа.
На рис. 4.9, б приведена схема поверхностнобарьерного де-
тектора. Поверхностный слой р+-типа создается в результате
окисления протравленной поверхности. Поверхйостный контакт
получают напылением тонкой золотой пленки. Во время работы
118
детектора пленка из золота, нанесенная на p-слой, становится
отрицательной по отношению к кремниевому кристаллу и-типа.
Рис. 4.10, а демонстрирует использование в качестве детек-
тора излучений области п— р— n-перехода. На рис. 4.10,6
приведена схема включения полупроводникового детектора.
Рис. 4.10. Схемы измерений (а) с двумя типами полу-
проводниковых детекторов излучения (б):
а) I — излучение; 2 — эмиттер; 3 — коллектор; 4 — сигнал.
б) 1 — излучение; 2 — область перехода; 3 — усилитель; 4 — пере-
счетно е устройство.
Учитывая чрезвычайно быстрое развитие полупроводниковых
методов детектирования излучений, можно надеяться, что в те-
чение ближайших лет полупроводниковые детекторы заменят
ионизационные камеры при решении целого ряда задач.
4.9. ОПРЕДЕЛЕНИЕ ПОГЛОЩЕННОЙ ДОЗЫ
С ПОМОЩЬЮ СЧЕТЧИКОВ
Поглощенную дозу можно измерять с помощью таких счетчи-
ков, как, например, счетчик Гейгера— Мюллера, пропорцио-
нальный или сцинтилляционный счетчики. Обычно с помощью
счетчиков удается лишь установить, сколько частиц попадает
на 1 см2 поверхности. Однако при использовании некоторого
дополнительного оборудования можно определить не только
число, по и энергию частиц, и связать эти данные со значением
поглощенной дозы в той среде, которая нас интересует.
4.9.1. Пропорциональные счетчики
Пропорциональные счетчики предназначены для счета ионов
и устроены таким образом, что падение напряжения оказы-
вается пропорциональным как числу образовавшихся ионов, так
и числу актов ионизации. Поэтому пропорциональный счетчик
можно использовать для определения числа и энергии заряжен-
119
ных частиц. Теоретически все это относится также и к фотонам.
Однако на практике дискриминация по энергиям возможна
лишь при низких энергиях фотонов, так как в этом случае счет-
чик фиксирует различия в энергии выбитых электронов.
Дискриминация по энергиям в пропорциональном счетчике
осуществляется путем отбора импульсов, амплитуда которых
выше или ниже определенного произвольно выбранного уровня.
В небольших пределах число отсчетов можно варьировать, по-
вышая или понижая разность потенциалов между анодом и ка-
тодом. Пропорциональные счетчики обычно работают при атмос-
ферном давлении. Их часто используют в качестве проточных
бесстеночпых счетчиков, где газ циркулирует между образцом
и ионизационной камерой.
Хотя по сравнению со счетчиками Гейгера — Мюллера в про-
порциональных счетчиках для импульсов с малой амплитудой
необходимо большее усиление, тем не менее с их помощью
можно измерять большие интенсивности излучения за счет сни-
жения разрешающей способности по времени вплоть до 0,1 сек.
Кроме дополнительных усилителей, пропорциональные счетчики
нуждаются в стабилизированном высоковольтном питании.
В комплект также должны входить дискриминаторы напряже-
ния. Все это делает пропорциональные счетчики гораздо более
дорогими по сравнению со счетчиками Гейгера — Мюллера. Кон-
струкция стандартного пропорционального счетчика обсуж-
дается в статье Вагпера и Херста [55].
Пример 4.5.
1. Какое напряжение создается в пропорциональном счетчике с емко-
стью 10 пф при прохождении а-частицы с энергией 3 Мэв?
2. Если усиление счетчика равно 10 000, то какова энергия частиц неиз-
вестного a-источника, создающего импульсы с амплитудой 20 в?
Решение.
1. Считая, что на образование пары ионов затрачивается 35 эв, получим,
что всего образуется 3-106/35 =8,6-104 пар ионов. Заряд, попадающий на
пластинку, равен произведению числа ионов на заряд электрона:
Q = (8,6- 104)(1,6- 10~») = 13,8- 10-» к;,
Q 13,8 • 10-1»
U = ’— -------=1,38- Ю-з в.
С 10 • Ю-I»
2. Усиление равно 10 000. Следовательно, 17=13,8 в для 3 Мэв. Тогда
энергия частиц неизвестного a-источника равна (20) (3)/( 13,8) =4,34 Мэв
4.9.2. Сцинтилляционные детекторы
Вещества, способные испускать видимый свет под действием
ионизирующей радиации, были известны еще во времена Рент-
гена, использовавшего для качественной дозиметрии люминес-
ценцию ZnS. Для ряда веществ высвечивание после действия
радиации можно объяснить переходом атомов и молекул из
возбужденных состояний, возникших под действием радиации,
120
в основные состояния. Время, в течение которого высвечивается,
например, половина возбужденных атомов и молекул, характе-
ризует механизм перехода. Если константы высвечивания ока-
зываются порядка 10-8—10~9 сек, то сцинтилляционный процесс
называют флуоресценцией. При больших характеристических
временах высвечивания процесс называют фосфоресценцией.
Сцинтилляторы, выпускаемые промышленностью в настоящее
время, изготавливают главным образом либо из органических
кристаллов, либо из кристаллов Nal. Световой выход этих сцин-
тилляторов равен примерно одному фотону на 100 эв погло-
щенной энергии. Преобразование светового импульса в электри-
ческий сигнал осуществляется с помощью фотоэлектронного
умножителя — ФЭУ. Этот прибор состоит из двух основных эле-
ментов: фотокатода, из которого под действием света выби-
ваются электроны, и системы динодов — металлических пласти-
нок, каждая из которых находится под более высоким напря-
жением, чем предыдущая, в результате чего обеспечивается
умножение числа электронов. Диноды, разность потенциалов
между которыми составляет обычно около 100 в, испускают 3
или 4 электрона на один падающий электрон. В стандартном
фотоумножителе бывает 10—11 ступеней усиления.
Число электронов, собираемых на выходе ФЭУ, прямо про-
порционально энергии, теряемой частицей в кристалле сцинтил-
лятора. Так, если электрон с энергией 1 Мэв создает ,на выходе
умножителя напряжение 1 в, то (при отсутствии влияния по-
Таблица 4.8
Характеристики сцинтилляторов *
Сцинтиллятор Плотность, г,/см* Длина волны свечения с максимальной интенсив- ностью, А Импульс, отн. ед i -'° Время высвечивания, сек.
s •• I А а> ст -,о а-е° о . SSg
Кристалл антрацена 1,25 4400 100 9 3-10-8
Кристалл транс-стиль- бена 1,15 4100 60 9 4—8.10-8 (р)
Жидкие фосфоры 0,86 3500—4500 40—60 9 2-8-10-8
Пластиковые фосфоры 1,06 3500—4500 28—48 .— 3-5-10—«
Nal (Т1) 3,67 4200 210 44 3-Ю-7
Lil (Ей} 4,06 4700 70 95 1,2-Ю-6
CsI (Т1) 4,51 4200—5700 55—95 — 1,1-Ю-в (Р) 4-10-10-8 (быстрая ком по-
ZnS (Ag) порошок 4,10 4500 200 100 нента) 4—10-10-е (медленная ком- понента)
* Из книги: price W. J. Nuclear Radiation Detection,'McGrow-Hill. New York 1964,
p. 162.
121
бочных факторов) электрон с энергией 2 Мэв создаст на выходе
напряжение 2 е. Таким образом, при условии точной калибровки
и линейности усиления сигналов значение импульса напряжения
на фотоумножителе будет соответствовать энергии падающей
частицы.
В табл. 4,8 перечислены типичные сцинтилляторы, которые
используются в настоящее время. Примерами органических
сцинтилляторов-монокристаллов могут служить аптрацен и
т/шнс-стильбеи. К жидким фосфорам относятся такие вещества,
как п-терфенил, 2-фенил-5-(4-дифенил) -оксазол и другие соеди-
нения, в состав которых входят бензольные кольца. Пластико-
вые фосфоры изготовляют, включая ароматические соединения
в полимеры типа бутадиена или полистирола. В неорганические
сцинтилляторы, Lil или CsI, добавляют обычно небольшие
количества активатора (Ей или Т1).
4.10. ДОЗИМЕТРИЯ НЕЙТРОНОВ
Дозиметрия нейтронов чрезвычайно осложняется из-за того,
что нейтронный поток почти всегда сопровождается другой ра-
диацией, главным образом у-излучением. Одновременное при-
сутствие излучений с высокой и низкой плотностью ионизации
порождает чрезвычайно сложные проблемы как при измерении
доз, так и при интерпретации экспериментальных данных. Взаи-
модействие нейтронов с веществом зависит от их энергии в го-
раздо большей- степени, чем взаимодействие с веществом элект-
ромагнитного или других видов излучений. Поэтому необходи-
мы как дозовые, так и спектральные измерения.
Условия эксперимента на ядерных реакторах, основных
источниках нейтронов в радиационной химии, требуют разме-
щения экспериментального оборудования па значительном рас-
стоянии от экспериментатора. Часто это приводит к серьезным
затруднениям. В процессе работы образцы (и оборудование)
могут становиться радиоактивными. Кроме того, при определе-
нии дозы следует учитывать не только первичные нейтронные
и у-потоки, но и вклад наведенной радиоактивности.
Так как у нейтронов нет электрического заряда, то они тор-
мозятся главным образом в результате ядерных взаимодействий.
Поэтому детектирование нейтронов основывается на вторичных
эффектах. Почти все процессы взаимодействия нейтронов с
веществом, рассмотренные в главе II, могут служить основой
измерительной техники. Если удается вызвать реакции (п, а),
(п, р), (п, у) или (п, деление ядра), то задача после этого сво-
дится к тому, чтобы определить выход реакции и продетектиро-
вать вторичное излучение. Другой способ состоит в том, чтобы
получить под действием нейтронов радиоактивные изотопы, ко-
торые можно обнаружить затем по характеристическому излуче-
122
нию. Третий метод сводится к измерениям интенсивности заря-
женных частиц отдачи, например протонов.
4.10.1. Активационный метод дозиметрии
и спектры нейтронных потоков
Детектирование нейтронов с помощью измерений наведенной
радиоактивности в веществе с известным сечепием захвата —
один из очевидных примеров использования процессов, рассмот-
ренных в главе II. Активационные детекторы нейтронов обла-
дают целым рядом преимуществ, которые, делают их особенно
пригодными для целей радиационной химии.
1. Активационный метод позволяет проводить измерения
нейтронных потоков в очень широком диапазоне интенсивностей.
Подбирая вещества с высокими или низкими сечениями захвата
нейтронов, можно измерить практически любые нейтронные
потоки.
2. Метод позволяет снимать показания дозиметра в точке,
удаленной от места измерений. Дозиметр, можно вынуть из нейт-
ронного пучка и определить его активность в другом месте и в
другое время.
3. В активационном методе можно использовать детекторы
любых удобных размеров. Детектор можно изготовить в виде
тонкой фольги, которая прикрепляется к контейнеру с образцом.
Размеры фольги могут быть достаточно малыми, для того чтобы
не вносить возмущений в нейтронный поток.
4. Подбор материала детектора позволяет обеспечивать се-
лективность активационного дозиметра в отношении энергии
нейтронов. Э1о в свою очередь делает возможным измерения
спектра нейтронных энергий в реакторе.
Разные химические элементы для данной энергии нейтронов
обладают сильно различающимися сечениями захвата. Поэтому
для измерений нейтронного спектра можно использовать акти-
вацию нескольких веществ одновременно. Особенно успешным
оказалось применение так называемых «пороговых детекто-
ров» — веществ, которые приобретают наведенную радиоактив-
ность лишь после того, как энергия нейтронов превысит опреде-
ленное минимальное значение. В табл. 4.9 приведены характе-
ристики наиболее часто используемых пороговых реакций.
Если одновременно используется несколько детекторов с раз-
ными порогами, то с помощью последовательных вычитаний
можно найти величину потока нейтронов между любыми двумя
пороговыми энергиями, что Позволяет в конце концов получить
нейтронный спектр в биде гистограммы.
Для очень топкой фольги, помещенной в ядерный реактор,
активность при насыщении Ns будет равна
Ns = N [ иа (Е) Ф (Е) dE, (4.18)
о
123
Таблица 4.9
Характеристики пороговых реакций
Реакция Эффективный порог, Мэв Эффективное течение, мбарн Реакция Эффективный 1 порог, Мэв Эффективное сечение, мбарн.
2s7Np (деление) u»In (п, у) 23SU (деление) sip (п. р)31 88Ni (п, р) Б8Со s2S (п, р) S2P e4Zn, (n, p) e4Cu 0,2 0,45 1,45 2.5 2,9 3,0 4,0 0,223 540 75 420 310 320 84Fc (и, р) Б4М 24Mg (n, р) 24Na 68Fc (n, р) 88Mg 27А1 (n, р) 24Na i2C (n, 2n) «С i27l (n, 2n) i26l 4,2 6,3 7,5 8,2 20 10 610 60 по 130
где N — концентрация атомов мишени; сечение активации оа и
поток Ф зависят от энергии нейтронов.
Детекторные мишеии в реакторе подвергаются воздействию
тепловых (0<Е<0,4 эв), промежуточных, или резонансных,
нейтронов (0,4 эв<£'<10 кэв) и быстрых нейтронов (10 кэв<
<Д<10 Мэв). Вкладом быстрых нейтронов в сечение актива-
ции можно пренебречь, и поэтому
NS = N
j oa(E)(D(E)dE+ J
тепловые промежуточные
Оа(Е)Ф(Е)<1Е]. (4.19)
Так как для тепловых нейтронов сечение активации меняется
в соответствии с законом 1/V, то удобно, вводя относительное
сечепие активации ооа, скорость нейтронов Уо и концентрацию
тепловых нейтронов nt, переписать уравнение (4.19) следующим
образом:
( S^-nt(V)VdV+ (
J у J
тепловые г промежуточные
Ns
ои(Ю«(Ю«^р
(4.20)
Обозначив интегралы в правой части через Nst и Nse, по-
лучим
Ns = Nst + NSe = NntV0o0a + Nse.
(4.21)
Для того чтобы разделить активацию, вызванную действием
тепловых нейтронов Nst и действием промежуточных Nse, ми-
шени заворачивают в кадмиевую фольгу. Кадмий обладает не-
обычно высокими сечениями захвата тепловых нейтронов
(7200 барн), и, следовательно, радиоактивность, наведенная в
мишенях, обернутых кадмием, приблизительно пропорциональ-
на потоку промежуточных нейтронов. Кадмиевое отношение Re
124
определяется как отношение активности открытой фольги при
насыщении к активности фольги, обернутой кадмием:
Rc-~. (4.22}
^se
Подставляя 7?с в уравнение (4.21), получаем:
= (4.23)
^se
Практически значение активности, наведенной в фольге,
обернутой кадмием, будет несколько меньше, чем Nse, так как
кадмий частично поглощает и промежуточные нейтроны. В том
упрощенном варианте, который здесь проводится, этой поправ-
кой, зависящей от относительной толщины слоя кадмия и ми-
шени, можно пренебречь.
Ряд важных соотношений можно получить теперь, комбини-
руя уравнение (4.23) с уравнениями (4..20) и (4.21):
Rс — 1 =... ntV°^a---------. (4.24}
J ос (£) Ф (£) dE
0,4 эв
. В области промежуточных нейтронов сечение захвата боль-
шей части поглотителей меняется_пропорционалыю 1/Е в отли-
чие от зависимости 1/Е1'2 или 1/F, преобладающей при низких
энергиях. Если теперь мы примем, что резонансный поток Ф
пропорционален К/Е, то уравнение (4.24) будет выглядеть так:
Rc-l
nt^o°oa
Ра(£)
Е
(4.25)
dE
Константа К определяется таким выражением:
_________ЕдСГрд__________
и? п 7 яр
(Яс—1). I —— dE
0,4 эв
(4.26)
Интеграл в уравнении (4.25) называется резонансным ин-
тегралом. Значение резонансного интеграла измерено для самых
различных активационных детекторов. Характеристики актива-
ционных детекторов, а также некоторые другие данные приведе-
ны в табл. 4.10. Значение о0а сечения активации для тепловых
нейтронов относится обычно к энергии 0,025 эв, которая соответ-
ствует скорости нейтронов 2200 м!сек.
125
Таблица 4.10
Характеристики активационных детекторов (тепловые и резонансные детекторы)*
Элемент Детектор Продукт активации
сечение актп- 1 нации для тепловых нейт- ронов, барн2* резонансная энергия, зв3* резонансный интеграл, барн образу кяцийся изотоп период полу- распада вид радиоактивности
In- 145 1,45 2640 uein 54,1 мин Р (1 Мэв) У (спектр энергий)
Au 96 4,9 15554* ie«Au 27 дней 1 (0,963 и 0,41) Мэв
I 5,5 20—200 140 . 128} 24,99 мин } (2,0 Мэе)
Dy 2600 54 — 1в5£)у 139,2 мин Р (1,25; 0,88 и 0,42 Мэе) у (0,09; 0,36 и 0,76 Мэе)
Co 34,0 135 49,3 восо5* 5,28 года Р (0,31 Мэв) у 1,33 и 1,17 Мэе)
Mn 13,4 330 11,8 Б6Мп 2,58 ч Р (2,81 и 0,822 Мэв)
Na 0,56 3000 0,24 24Na • 15,0 ч Р (1,39 Мэв) у (1,37 и 2,75 Мэе)
V 4,5 4200, 13000 2,2 52у 3,76 мин р (2,1 и 1,5 Мэе)
Cl 0,56 26 000 — 28C1 37,5 мин Р (5,0; 2,8 и 1,1 Мэё) у (2,2 и 1,6 Мэв)
• Таблица из книги: Price W. J. Nuciear Radiat ion Deiection, New York, McGrow-Hill,
1964, p. 334.
8* Для энергии нейтронов 0,025 эв. Из книги: Hughes J., Harvey J. A. Neutron Cross
Sections.
•• Положение главного пика.
** Из статьи: Keiber С. N. Nucleonics, i952, v. 20, р. 162. В этой статье резонансные
•интегралы приведены с учетом поправок на толщину фольги.
°* в®Со с периодом полураспада i 0,7 мин превращается в <0Со с периодом полураспада
5,28 года.
Пример 4.6. Кадмиевое отношение в урап-графитовом реакторе равно
примерно 33 (см. цит. литературу [56]). Вычислить: 1) соотношение между
тепловой и резонансной активацией для золота, принимая, что золото ведет
себя в области тепловых энергий как поглотитель 1/V, а в области энергий
промежуточных нейтронов как поглотитель 1/£; 2) соотношение потоков
тепловых и промежуточных нейтронов для любого интервала энергий
dEjE In е.
Решение.
ОС
1. Отношение оа теплового к \ <3adEiE для детектора 1/Г(1/£‘/2)
0,4 эв
равно:
_________1/(0,025)1/2 1 (0,4)1/г
^a(dE/E) ~ “ E,/tdE ~ 2 (0,025)^ Л
0,4 за
Тамм образом, для золота вклад поглощения в соответствии с законом
1/V равен 0,5(96) =48, тогда как резонансный интеграл составляет 1555 барн
(см. табл. 4.10).
126
2. Отношение теплового потока к резонансному для любого интер-
вала dE/E—Xne. дается уравнением (4.25)
(/?.-!) = 2-^- = (33-1)
Л
или
ntVo
К
— 16/1п е.
Измерив спектр и величину нейтронных потоков в реакторе, можно
вычислить поглощенную дозу при условии, что значение потока у-квантов
также известно. В расчете следует учесть все возможные способы передачи
энергии, как это демонстрируется в примере 4.7.
Пример 4.7. В работе Фенжера [57] приводятся результаты серии экс-
периментов, в которых потоки у-излучения в тепловой колонне нейтронов
датского реактора Dr2 измеряли с помощью химического дозиметра на ос-
нове щавелевой кислоты. Чтобы найти значение плотности потока у-излуче-
ния, нужно определить какой вклад в общую дозу вносят нейтроны. Для
этого следует учесть передачу энергии в результате: 1) появления атомов,
отдачи при испускании у-квантов; 2) захватного у-излучения; 3) упругого-
рассеяния быстрых нейтронов' и 4) рассеяния нейтронов промежуточных
энергий.
Щавелевая кислота облучалась в кварцевых ампулах длиной 5 см, диа-
метром 8 мм при 25° С в тепловой колонне нейтронов реактора, где имелось
только нейтронное и у-излучение. Плотности потоков нейтронов, измеренные
активационными методами, составляли; тепловых (£„<0,4 эв) 4,5-1012 ней-
трон! (смг-сек), промежуточных (0,4 эв<£„<10 кэа)’5-1010 нейтрон/(см2 У
Xсек), быстрых (со средней энергией 2 Мэв) 8-109 нейтрон!(см2-сек) 30%
щавелевой кислоты разлагалось в течение 2,8 ч. Вычислить дозу, передавае-
мую у-излученисм, и часть дозы, передаваемую нейтронами. Примем, что ак-
тивацией стенок ампулы можно пренебречь.
Решение.
1. Вклад атомов отдачи, возникающих при испускании у-квантов. Для
тепловых нейтронов сечения реакции (п, у) с элементами, входящими в
состав дозиметрической системы, равны:
Он = 0,0332 • 10—см2;
ао = < 2 • IO-2» СЖ2;
ос = 4,5 - 10_27 см2.
Очевидно, оо и ос пренебрежимо малы по сравнению с Он. При реакции
(п, у) с водородом возникают фотоны с энергией 2,23 Мэв. Энергия отдачи,
при этом равна
Ег = Е^/ШМ = 1330 эв,
где £? — энергия у-квапта; М.— масса атома отдачи.
Общее число атомов отдачи за 1 ч будет равно
4,55 1012 нейтрон 3600 сек 2
см2 • сек ч 18
6,02 IO2» 0,332 • 10-2* см2
— 3,64 • 10п атом отдачи/(мл ч).
Доза в радах, передаваемая за 1 ч атомами отдачи, равна:
3,64 10*4 атом отдачи I 1330 эв 1,6-10~14 рад/ч\ мл
-----------------------------------------------------== 7 75 . 103
мл • ч | эв I г
127
2. Захватное у-излучение. В ядерной реакции H(n, y)D возникают
у-кваиты с энергией 2,23 Мэв, для которых коэффициент поглощения равен
0,027 см2/г, или 0,027 см~' в поглощающей среде дозиметра. Средняя длина
пробега будет равен 0,3 см при характеристических размерах дозиметра 5 см.
и 8 мм. Доза, передаваемая у-излучснием дозиметрической системе за 1 ч
будет равна:
3,65 • 1014у-квант 2,23 Мэв 0,027 0,3 см 1,6 • 10~ 8 рад/ч МЛ
мл • ч У СМ Мэв г
= 1,05 • 106 рад/ч.
3. Быстрые нейтроны (£„>1 Мэв), Еср—2 Мэв. Средняя потеря энергии
яа столкновение описывается выражением
1 £о ,
In----= 1
Е
L (Л--1? (Д-1)
‘ 2Д П (Л 4-1) ’
тде Ео=2 Мэв, А = 2 и £=1 Мэв. Сечение рассеяния os равно 0,037 см~1. Счи-
тая, что каждый нейтрон претерпевает однократное рассеяние, найдем ве-
личину передаваемой дозы:
8 - 109 нейтрон 3600 сек 1 Мэв 0,037 1,6 10—8 рад г/см2
см2 • сек ч нейтрон СМ Мэв а ~
= 6,3 • 103 рад/ч.
Полная передача дозы в результате рассеяния быстрых нейтронов, оче-
видно, слишком мала, чтобы стоило проводить подробный расчет числа
соударений.
4. Рассеяние нейтронов промежуточных энергий (0,4 эв<Еп<Л0 кэв).
В области промежуточных энергий как энергетический спектр, так и вели-
чина сечений заметно меняются. Поэтому возникает необходимость в опреде-
лении среднего значения сечения рассеяния:
1 J43B
оср = J Os (Еп)
0.4 эв
Используя результаты измерений энергетического спектра и зависимость
сечения рассеяния от энергии для водорода, Фенжер получил оср =
=7-10_ 18 эв-см2.
Так же как и быстрые, промежуточные нейтроны теряют при каждом
столкновении 0,5 своей энергии. Следовательно, полная доза, передаваемая
нейтронами промежуточной энергии, равна:
0,5
5 • 1010 .нейтрон
см2 сек
3600 сек 7-10 18 эв • см2
ч
I 6,7 • 1022 атом Н
I г
1,6 10—14 рад
X-------------н— = 0,68 • 106 рад/ч.
эв
Суммируя результаты расчетов, запишем:
Вид излучения дв3ы, рад/ч
Атомы отдачи........................... 7,75-103
Захватное у-излучение ................. 1,05-10Б
Быстрые нейтроны....................... 6,30-103
Промежуточные нейтроны................. 0,68-106
128
ЛПЭ для протонов отдачи с энергией 0,5 Мэв, возникающих при рассея-
О
нии нейтронов, приблизительно равно 7 эв/А. Для плотноиопизирующего из-
лучения с таким значением ЛПЭ значение G для щавелевокислого дозиметра
снижается с 4,9 (выхода, характеризующего действие у-излучення) до 0,9
(см. раздел 4.5). Число грамм-молекул щавелевой кислоты, распавшихся
под воздействием излучений с высокой ЛПЭ, будет равно
(6,3 103 4- 6,8 • 1№ + 7,75 • 103) рад
ч
0,9 молекул I эв I
100 эв I 1,6 10~14 рад-г |
моль
6,02 • 1028 молекул
— 6,47 10“7 моль/г.
Общее количество кислоты, распавшейся в 0,05 М растворе, будет равно
(0,05) (0,3)/1000= 1,5-10“5 моль/г. Количество кислоты, распавшейся под
действием захватного у-излучения, будет равно:
1,05 • 10 рад 4,9 молекул Эв моль
ч 100 эв 1,6 10—14 рад-г 6,02 IO®» моль
= 5,34 • 10—7 моль/г.
Общая доза, полученная за счет реакторного у-излучепия, равна:
(150 — 1,5 — 5,34) • 10-’ моль 1 6,02 - 1028 моль 100 эв |
г 2,8 ч 4,9 молекул]
1,6 • 10—4 рад • г „ „
—------------------= 276 • 104 рад/ч.
эв
Задачи
4.1. Какое напряжение возникает на конденсаторе емкостью 10 пф, если
происходит разделение заряда 10”’° к?
4.2. Частица с энергией 2 Мэв регистрируется воздушным пропорцио-
нальным счетчиком с коэффициентом, усиления 1000 и емкостью 10 пф. Чему'
равно импульсное напряжение, возникающее на счетчике?
4.3. При прохождении у-квантов с энергией 3 Мэв в калиброванном
пропорциональном счетчике возникают импульсы напряжения с амплиту-
дой 30 в. Источник а-частиц с неизвестной энергией дает две группы им-
пульсов: одпу с амплитудами 40 в, другую с амплитудами 38 в. Найти
энергетический спектр неизвестного а-источника.
4.4. Средний пробег у-квантов с энергией 1 Мэв в сцинтилляционном
кристалле KI плотность^-3,13 г/см3 равен 7 см. Квантовый выход световых
фотонов равен 1 на 50 эв поглощенной энергии, причем 80% фотонов попа-
дает на фотоумножитель. Эффективность конверсии фотонов в электроны
на фотокатоде составляет 7%; в фотоумножителе имеется 10 динодов с че-
тырехкратным усилением. Чему будет равна амплитуда импульса напряже-
ния, возникающего на конденсаторе емкостью 10 пф? Чему будет равна ам-
плитуда импульса напряжения в тех же условиях для у-кваптов с энергией
1,25 Мэв?
4.5. Вычислить ожидаемый рост давления при поглощении дозы
в 107 рад в циклогексановом, водном и азотпозакисном дозиметрах.
4.6. Коэффициент теплопроводности адиабатического калориметра оце-
нивается в 0,05 ккал/(м2-ч-град). Поглощающий объем представляет собой
углеродную сферу диаметром 2 см. Устройство терморегуляции поддерживает
разность температур в 1° С между поглощающим объемом и адиабатической
стенкой. Постройте график изменения температуры в зависимости от вре-
мени, если дозиметр находится в радиационных полях с мощностью дозы
5 Зак. 90/
129
IO4, 105, 106 и 107 р/ч. Достаточна лч чувствительность данной калориметри-
ческой системы для правильной калибровки источника 90Sr активностью
1 кюри?
4.7. Под действием радиации оптическая плотность ферросульфатного
раствора в сантиметровой кювете при 25° С возрастает на 0,34. Чему равна
полученная доза, если раствор облучался: 1) (i-частицами с энергией 0,01 Мэв;
2) рентгеновским излучением, I Мэв; 3) продуктами деления и 4) (3-части-
цами с энергией 40 Мэв? Подсчитайте дозу в радах и в эв/мл.
4.8. Плотно спрессованный образец Pb(NO3)2 . облучается в пробирке
диаметром 3 см и толщиной стенок 0,3 см. Калибровка, проведенная с по-
мощью ферросульфатного раствора в той же
Рис. 4.11. Коллектор комп-
тоновских электронов:
1— проводящая фольга; 2— изо-
лятор; 3 — металлический погло-
титель ^-излучения; 4 — комп-
тоновские электроны; 5- выход
коллектора.
пробирке, дала значение мощности дозы
103 рад/ч. Источник излучения представляет
собой бесконечную плоскость. Пробирка иахо-
5" дится на расстоянии 5 см от источника. Какую
дозу получит образен Pb(\O;i)2, если источ-
никами являются: “"Со, 0-излучатель с энер-
гией частиц 0,7 Мэв, а-излучатель 210Ро, рент-
геновская трубка 10 кв (пик)?
4,9. Гросс н Мэрфи (Cross and Murphy.
Selected Topics in Radiation Chemistry, Vienna,
IAEA, 1961, p. 549) предложили в качестве
твердотельного гамма-нейтронного дозиметра
устройство, показанное на рис. 4.11. В этом
устройстве выбитые вперед комптоновские
электроны создают ток, сила которого про-
порциональна потоку у-излучения. Изолиро-
ванный металлический блок имеет достаточную
толщину для того, чтобы поглощать все или почти все у-кванты, попадающие
в блок. При этом в металле возникает отрицательный заряд. Найти значение
тока, создаваемого в таком устройстве под действием у-излучепия 60Со с мощ-
ностью дозы 1 р/сек.
4.10. Оцените значение тока, возникающего в цилиндре Фарадея под дей-
ствием электронного пучка с энергией 3 Мэв, создающего в водном растворе
мощностью дозы 5000 рад/мин.
4.11. Найдите, сколько отсчетов в секунду на единицу потока делает
нейтронный счетчик. Счетчик объемом 60 см3 заполнен газом BF3 под дав-
лением 100 мм рт. ст. Средний пробег нейтронов в объеме счетчика состав-
ляет 12,7 см. Для *°В сечепие взаимодействия с нейтронами, обладающими
скоростью 2200 м!сек, составляет 750 барн.
4.12. Сечение взаимодействия ,*°В с нейтронами энергии 0,025 эв состав-
ляет 750 барн. Чему равно сечение взаимодействия с нейтронами энергии
10 кэв, принимая, что справедлив закон 1./V?
4.13. Облучая пороговый детектор из 32S, получили 32Р с активностью
0,1 кюри. Каким должен быть поток быстрых нейтронов?
4.14. Для кобальтовой фольги, помещенной в реактор, кадмиевое отно-
шение равно 81. Если плотность потока тепловых нейтронов составляет
1010 частиц Кем2--сек), то. чему будет равен нейтронный ноток в спектральном
интервале от 2,7 до 2700 эв?
4.15. В своей статье Райт (Wright, Trans. Faraday Soc., 1952, v. 12,
p. 65) сообщает, что золотая фольга под действием тепловых нейтронов
в Харуэлльском реакторе приобретает активность 1000 импКмин-мг), что соот-
ветствует плотности потока 5,42-1012 нейтрон/(см2-сек). Кадмиевое отноше-
ние равно 2,70. Плотность потока быстрых нейтронов равна 2-I09 нейтрон/
I(см2-сек). Подсчитайте, сколько ионов Fe2+ в ферросульфатном растворе
окислится до Fe34 под действием нейтронов в 1 см3 раствора за 1 ч.
Гл а в а V
ХИМИЧЕСКИЕ ПОСЛЕДСТВИЯ ПОГЛОЩЕНИЯ
ИЗЛУЧЕНИЙ ВЫСОКОЙ ЭНЕРГИИ В ВЕЩЕСТВЕ
Заряженная частица, движущаяся через вещество, взаимо-
действуем с внутримолекулярными электронами. В зависимости
от энергии взаимодействия происходит возбуждение, ионизация
или множественная ионизация молекул. Таким образом, в си-
стеме, на которую действует излучение высокой энергии, можно
обнаружить молекулы с одним или более электронами, находя-
щимися в возбужденном состоянии, ионы, возбужденные ионы и
свободные радикалы.
5.1. ВРЕМЕННАЯ ШКАЛА ПЕРВИЧНЫХ ПРОЦЕССОВ
Рис. 2.7 иллюстрирует события, происходящие при движении
заряженной частицы через жидкость. Первичные заряженные
частицы приводят к появлению вторичных электронов различ-
ных энергий, которые в свою очередь также могут создавать
электроны. В системах, подвергающихся воздействию излучения
е0Со, большая часть электронов, возникающих при первичных
взаимодействиях, обладает энергиями не более нескольких ты-
сяч электронвольт. Эти электроны возбуждают и ионизуют
молекулы в небольшом объеме роя или шпоры. Точная форма
шпоры зависит от того, насколько плотно располагаются иони-
зации. Примем пока, что каждая шпора или рой представляет
собой сферу диаметром около 20 А.
Временная шкала первичных процессов определяется ско-
ростью ионизирующей частицы, так как в зависимости от скоро-
сти меняется время, в течение которого находится в окрестно-
стях молекулы. Электрон с энергией 1 Мэв проходит диаметр
молекулы среднего размера за время около 10~18 сек.
Временная шкала дальнейших процессов изучена достаточно
подробно только для -воды. Как показывает табл. 5.1, электрон
с энергией 1 Мэв останавливается в воде примерно через
10-9 сек. В шпорах и треках 6-электронов существуют в это
время ионы, возбужденные молекулы, возбужденные ионы, ра-
5* 131
дикалы, возбужденные радикалы и. электроны со средней энер-
гией около 6 эв. Эти электроны, которые называют слабовоз-
бужденными электронами *, растрачивают свою энергию при со-
ударениях с молекулами (на молекулярные колебания), на ди-
польные взаимодействия и в конечном счете термализуются.
В конденсированной среде слабовозбужденные электроны
захватываются либо положительными ионами, либо нейтраль-
ными молекулами, которые образуют отрицательные ионы.
Электрон, захваченный молекулой воды, называется гидратиро-
ванным электроном, а при захвате молекулами других раство-
рителей (например, метанола или этанола) —сольватированным
электроном.
5.2. ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
РАДИАЦИОННОХИМИЧЕСКИХ РЕАКЦИЙ
В табл. 5.1 перечислен целый ряд промежуточных продуктов
радиационнохимических реакций, с которыми читатель может
быть не знаком. Ниже поэтому приводятся краткие пояс-
нения.
Свободными радикалами называют атомы и молекулы,
имеющие неспаренпый электрон и способные образовывать хи-
мические связи. В зависимости от значения энергии резонанса
свободные радикалы делятся на нестабильные и стабильные.
Примерами нестабильных свободных радикалов могут служить
СН3, Н, С2Н5 и СН2. Сравнительно стабильны и имеют пони-
женную реакционную способность такие радикалы, как NO,
NO2 и С1О2. Примерами радикалов, занимающих по стабиль-
ности промежуточное положение между рассмотренными двумя
типами, могут служить трифенилметил (Ph3C) и дифенилпи-
крилгидразил (ДФПГ).
Ионами называются атомы и молекулы с избытком положи-
тельного или отрицательного заряда. Эти заряды не стабилизи-
рованы ассоциированным зарядом противоположного знака.
Возбужденными ионами называют ионы, в которых один или
более электронов находятся в возбужденном состоянии.
Возбужденными свободными радикалами называют радика-
лы, в которых один или более электронов находятся в возбуж-
денном состоянии.
И он-радикалами называют свободные радикалы с избытком
положительного или отрицательного заряда. В ибн-радикалах,
таким образом, помимо неспаренногр электрона имеется избы-
точный электрический заряд.
* Слабовозбужденные электроны, энергия которых меньше нижнего
уровня возбуждения молекул среды, выделил в особую группу Р. Платцман
в 1955 г. Слабовозбужденные электроны обладают сравнительно большим
временем жизни. — Прим, перев.
132
Таблица 5.1
Временная шкала первичных процессов, происходящих в воде
под действием радиации'
Время, сек Процессы, протекающие за это время в воде
10-18 Электрон с энергией 1 Мэв проходит расстояние, равное диа-
IO-» Ю-le метру молекулы воды Возбуждение или ионизация Электрон с энергией 10 эв проходит расстояние, равное диа- метру молекулы воды
Ю-14 Период молекулярных колебаний. Диссоциация возбужденных
10-13 молекул! Время протекания ион-молекулярных реакций Время между соударениями в жидкостях. Внутренняя конвер- сия электронно-возбужденных состояний. Время термализации вторичных (слабовозбужденных) электро-
10-12 Радикалы проходят мономолекулярный слой, если их коэффи-
10-11 10—1® 10-10—10-8 дисит диффузия ~5-10—5 см21сек Время релаксации дипольных молекул воды Время между соударениями в газах Время протекания диффузионно контролируемых радикал-ради-
10-8—10-8 кальных реакций в шпорах и треках а-частиц Флуоресценция. Время высвечивания возбужденных синглетных
10-’—ю-5 10-’—10-8 состояний Время жизни гидратированного электрона Время протекания реакций радикалов с молекулами растворен- ного вещества (в зависимости от энергии активации)
10-4—10-3 Время диффузии радикалов на расстояния около 5000 А (сред-
10-3 нее расстояние между шпорами для электронов с энергией 1 Мэв) Время высвечивания триплетных возбужденных состояний
5.3. ВОЗБУЖДЕННЫЕ МОЛЕКУЛЫ
Молекулы, обладающие избыточной энергией, электронной,
колебательной
В основном
или вращательной, называются возбуждёнными,
состоянии энергия молекулы минимальна. На
рис.- 5.1 показана кривая по-
тенциальной энергии для ос-
новного электронного состоя-
ния двухатомной молекулы. На
том же рисунке .приведена схе-
ма колебательных и враща-
тельных уровней энергии. Для * 1
Рис. 5.1. Кривая потенциальной энер-
гии для основного электронного со-
стояния двухатомной молекулы:
1 — вращательные уровни: 2 — колебатель-
ные уровни; 3 — энергия диссоциации свя-
зи; 4 — нулевая энергия колебаний.
Расстояние между ядрами
133
возбужденных электронных состояний имеется своя система
колебательных и вращательных уровней, если только само элек-
тронное состояние не относится к числу нестабильных. Для не-
стабильного (репульсивиого) электронного состояния на кривой
потенциальной энергии отсутствует минимум (верхняя кривая
рис. 5.3).
5.3.1. Диссипация энергии возбуждения
Время, в течение которого молекула поглощает или испус-
кает радиацию, мало по сравнению с периодом молекулярных
колебаний. Следовательно, от точек в основном состоянии, соот-
Рис. 5.3, Переход из основного в
репульсивное электронное состоя-
ние:
1 — репульсивное состояние; 2 — основ-
ное состояние.
Рис. 5.2. Кривые потенциальной энер-
гии основного, и возбужденного со-
стояния:
1 — возбужденное состояние”; 2 — флуорес-
ценция; 3 — основное состояние.
линии, пересекающие кривую возбужденного состояния
(рис. 5.2). Если возбужденное состояние оказывается неста-
бильным, как, например, в случае, иллюстрируемом рис. 5.3,
молекула диссоциирует за период молекулярных колебаний
~ 10-13. сек. Если же потенциальная кривая возбужденного со-
стояния имеет минимум, то для диссоциации может потребо-
ваться 10~4 сек или больше.
134
Если потенциальная кривая возбужденного состояния сходна
с кривой, изображенной на рис. 5.2, то, испуская квант излу-
чения, молекула может вернуться в основное состояние. Этот
процесс называется флуоресценцией. Рассматриваемая молеку-
ла при соударениях может растратить часть колебательной
энергии. Тогда длина волны флуоресцентного излучения будет
больше (а энергия меньше), чем длина волны излучения, вызы-
вающего переход в возбужденное состояние. Если же возбуж-
денная молекула полностью растратила колебательную энергию,
то она окажется в низшем колебательном состоянии (состояние
Е на рис. 5.2). В этом состоянии возбужденная молекула может
находиться сравнительно долго (Л/г~ 108 сек), так как энер-
гия электронного возбуждения не растрачивается столь легко,
как энергия колебательного 'возбуждения. Как свидетельствуют
экспериментальные данные, флуоресценция обычно обусловлена
переходами на основной уровень из низших электронно-возбуж-
денных состояний с той же мультиплетностью, хотя первона-
чально могут возбуждаться любые высшие состояния.
В ковалентной химической связи электроны спарены — их
спины противоположны по знаку. При переходе в синглетное
возбужденное состояние электрон, поглотивший энергию, пере-
ходит на высшую орбиту без изменения ориентации спина и та-
ким образом остается спаренным. Возбуждение в триплетное
-состояние связано с переворачиванием спина. В результате спи-
ны двух электронов оказываются параллельными. Триплетным
состояниям, как правило, соответствуют самые низкие энерге-
тические уровни молекулы.
Молекулы в триплетном состоянии играют важную роль в
радиационной химии, так как они обладают свойствами бира-
дикалов. Для любого возбужденного синглетного состояния
имеется соответствующее триплетное состояние. Не следует, од-
нако, считать, что такие состояния характерны только для воз-
бужденных молекул. Основное состояние кислорода, например,
оказывается триплетным, и этим объясняется механизм целого
ряда реакций, в которых он участвует.
Безызлучательный переход с изменением мультиплетности.
Если кривая потенциальной энергии возбужденного состояния
пересекается с потенциальной кривой возбужденного состояния
иной мультиплетности (рис. 5.4), то в точке пересечения кривых
возможен переход в другое состояние, если только колебатель-
ные уровни нового состояния располагаются ниже, чем в ис-
ходном. Схема, приведенная на рис. 5.4, показывает синглетное
возбужденное состояние и соответствующее ему триплетное
состояние. Согласно законам квантовой механики, на триплет-
синглетпые излучательные переходы наложен достаточно стро-
гий запрет, так как правила отбора требуют, чтобы AS = 0.
Поэтому время жизни триплетных возбужденных состояний за-
метно больше, чем синглетных. Обычно оно равно 10-4—iCHce/c,
135
хотя встречаются триплетные состояния с временами жизни по-
рядка секунд. Излучение, возникающее при переходе из возбуж-
денного триплетного состояния в основное, имеет большую
длину волны по сравнению с излучением, возникающим при
флуоресценции. Излучение, связанное с переходом из триплет-
ного состояния, называют фосфоресценцией.
Рис. 5.4. Кривые потенциальной
энергии синглетного и триплетно-
го возбужденных состояний:
/— возбужденное синглетное состояние;
2 — триплетное состояние; 3 — фосфо-
ресценция; 4 — основное состояние.
Время жизни триплетного воз-
бужденного состояния в большей
степени зависит от физических
характеристик среды. На син-
глетное возбужденное состояние
в этом смысле среда не влияет.
Помимо флуоресценции и фос-
форесценции существует много
других способов потери энергии
возбужденными молекулами. Это
внутренняя конверсия, безызлу-
чательная передача энергий, пре-
диссоциация, мономолекулярные
и бимолекулярные (химические)
реакции.
Внутренняя конверсия — про-
цесс, при котором электронная
энергия возбужденного состояния
затрачивается на возбуждение
высших колебательных уровней
основного состояния. Внутренняя
конверсия происходит в том слу-
чае, если один из высших коле-
бательных уровней основного со-
стояния располагается ниже со-
ответствующих колебательных
уровней возбужденного состоя-
ния. В результате конверсии молекула приобретает избыточную
колебательную энергию, которая быстро растрачивается при
соударениях с окружающими молекулами. В конечном счете
происходит безызлучательный переход в состояние с той же
мультиплетностью (рис. 5.5).
Безызлучательный перенос энергии. Перенос энергии элект-
ронного возбуждения от одной молекулы к другой осуществ-
ляется многими путями, однако во всех случаях энергия воз-
буждения донорной молекулы должна быть больше энергии
возбуждения молекулы-акцептора. В простейшем варианте пере-
дача энергии происходит при столкновении возбужденной и не-
возбужденной молекул. Энергия возбуждения, полученная при
этом молекулой-акцептором, может оказаться достаточной. для
ее диссоциации, как это происходит, например, при соударениях
возбужденных атомов ртути с другими молекулами. Такая ре-
136
акция происходит при возбуждении ультрафиолетовым излуче-
нием (с длиной волны 2537 А) ртутных паров в смеси, например,
с водородом или парами ацетона. Возбужденные атомы Hg
передают свою энергию другим молекулам, что приводит к их
распаду.
Перепое энергии может про-
исходить также в результате
флуоресценции донорной молеку-
лы и поглощения излучения
акцепторной молекулой.
В твердых телах энергия воз’
буждения может очень быстро
передаваться от одной молекулы
к другой, если эти молекулы рас-
полагаются в упорядоченной кри-
сталлической решетке. Такой про-
цесс называется миграцией экси-
тона. Если в кристалле имеются
примесные молекулы, уровни воз-
буждения которых располагаются
ниже уровней возбуждения ос-
новных молекул, то такие при-
месные молекулы могут захваты-
вать мигрирующую энергию и
растрачивать ее на флуоресцен-
цию. При этом параметры флуо-
ресцентного излучения будут ха-
рактерны именно для примесных
молекул.
Возможны также процессы внутримолекулярного переноса
энергии. Например, в ацетоне первичное поглощение энергии,
как свидетельствуют спектральные данные, происходит в кар-
бонильной группе, однако диссоциирует при этом С—С-связь.
Энергия электронного возбуждения, как правило, перераспре-
деляется по молекуле чрезвычайно быстро. Поэтому правильнее
считать, что молекула возбуждается как целое, а не рассмат-
ривать возбуждение отдельных химических связей и групп.
Известно, что происходят, например, такие процессы распада:
С2Н6 -> С2Н4 -f- Н2;
Рис. 5.5. Схема процесса внутрен-
ней конверсии:
1 — возбужденное состояние; 2 — основ-
ное состояние.
сн4-сн2+н2.
Процессы такого рода играют важную роль в радиационной
химии.
Предиссоциация имеет место в том случае, если потенци-
альная кривая возбужденного состояния пересекается с потен-
циальной кривой репульсивпого состояния (рис. 5.6). В точке
пересечения молекула может изменять свое состояние на ре-
137
пульсивное. Однако возбуждение может быть растрачено на
флуоресценцию, и молекула вернется в основное состояние.
Процесс, при котором молекула переходит в репульсивное со-
стояние, называется предиссоциацией.
Рис. 5.6. Схема процесса предиссо-
циации:
1— репульсивное состояние: 2— возбужден-
ное состояние; 3 — точка пересечения; 4—
основное состояние.
В принципе существует ряд
других механизмов, с помощью
которых возбужденная моле-
кула может растратить или пе-
редать свою энергию, но все
они по существу представляют
собой комбинацию уже рас-
смотренных механизмов и не-
которых других, которые об-
суждаются ниже.
Мономолекулярные реак-
ции. Возбужденные молекулы
могут возвращаться в основ-
ное состояние путем внутри-
молекулярных перестроек, па-
пример, в результате изомери-
зации (транс- или цис-). Воз-
можно, также, что энергия
возбуждения окажется доста-
точной для разрыва связи.
Если кривая потенциальной
энергии возбужденного состоя-
ния является репульсивной, то
диссоциация происходит за
время, меньшее одного перио-
да колебаний (.10-13 сек).
Бимолекулярные реакции.
Возбужденная молекула мо-
жет вступать в реакции с дру-
гими молекулами, находящимися либо в основном, либо в воз-
бужденном состоянии. Например, недавно высказано предполо-
жение, что выход молекулярного водорода при облучении воды
(см. главу VIII) частично обусловлен взаимодействием между
двумя возбужденными молекулами воды:
2Н2О* -> Н2 -f- 2ОН.
Другие хорошо изученные бимолекулярные реакции возбуж-
денных молекул — это реакции присоединения и реакции пере-
дачи энергии. Последний тип реакций уже рассматривался.
5.3.2. Перечень процессов, в которых растрачивается энергия
возбужденных молекул
Для возбужденной молекулы возможны следующие спосо-
бы диссипации энергии:
1. Флуоресценция — избыток энергии высвечивается.
138
Потенциальная энергий
Радиационный
перенос энергии
Молекула В
Возоуж денном
состоянии
Поглощение
рентгеновских
'U р-кбантоВ.
оссроресценциЯ
Триплетное
состояние
безызлучатель-
ный переход 6
основное
состояние
. Химические
реакции
Продукты
Флуоресценция
Внутренняя
конверсия
9
Воздействие электро-
нов и других
заряженных частиц
Поглощение
сбета
^молекула
В основном
состоянии
Рис. 5.7. Схема процессов, в которых приобретается или растрачивается энергия возбуждения.
2. Внутренняя конверсия-—энергия электронного возбуж-
дения преобразуется в колебательную энергию, которая рас-
трачивается при соударениях. После ряда соударений молекула
возвращается в основное состояние (см. рис. 5.5).
3. Безызлучательный переход в триплетное состояние, после
чего молекула либо вступает в какую-то реакцию, либо воз-
вращается в основное состояние, испуская фосфоресцентное из-
лучение (см. рис. 5.4).
4. Диссоциация — возбуждение в репульсивное состояние
(см. рис. 5.3) или предиссоциация (см. рис. 5.6).
5. Передача энергии — энергия электронного возбуждения,
передается другой молекуле, вызывая диссоциацию или коле-
бательное возбуждение этой молекулы, которая затем возвра-
щается в основное состояние. Процессы передачи энергии та-
кого типа называют тушением.
6. Мономолекулярные и бимолекулярные реакции.
На рис. 5.7 схематически представлены рассмотренные про-
цессы.
5.4. РЕАКЦИИ СВОБОДНЫХ РАДИКАЛОВ
Свободные радикалы возникают при нагревании, в фотохи-
мических реакциях, в окислительно-восстановительных процес-
сах под действием высокочастотного разряда, при прохождении
заряженных частиц через вещество или при вылете ядер .от-
дачи. Многие методы получения свободных радикалов хорошо
изучены и существуют их подробные описания [1, 2]. Реакци-
онная способность свободного радикала зависит от его струк-
туры и метода получения. В этой главе будут рассматриваться
только радикалы с высокой реакционной способностью, такие,
как N, Н, ОН, СН3, СН2, С2Н5, пропиловый, изобутило-
вый и т. д.
При анализе возможных радикальных реакций следует
учитывать их энергетику. Например, если свободный радикал
X реагирует с молекулой АВ
Х +АВ->АХ+В,
то вероятность этой реакции достаточно высока только в том
случае, если реакция экзотермическая. Реакция
1|Н2->НЦ-Н
крайне маловероятна, так как энергия водород-водородной
связи существенно больше, чем энергия связи водород — иод.
До тех пор, пока атом иода не получит достаточной активаци-
онной энергии, вероятность осуществления этой реакции будет
оставаться ничтожной.
При выяснении возможности данной реакции бывает полез-
но определить соотношение между константами скоростей пря-
140
мой (kf) и обратной (kr) реакций. Примем, что уравнение для
константы скорости реакции имеет следующий вид:
Константа скорости реакции = Лехр(—AE/RT), (5.1)
где 4 — константа (предэкспоненциальный множитель): Д£ —
энергия активации.
kj Aj exp (— Д£ j/RT)
kr Ar exp (— AEr/RT)
Считая, что Л/=АГ, получаем:
Пример 5.1. Найти отношение скоростей следующих реакций (для 300° К):
Н -{- Вг2 —- НВг 4- Br, hEt = 0,9 ккал-,
Br -J- НВг —Вг2 + Н, Д£2 = 41,4 ккал.
Решение. В соответствии с уравнением (5.2)
Хотя энергетика реакций играет обычно доминирующую роль, следует
учитывать и другие факторы, в частности стерические.
5.4.1. Реакции свободных радикалов.
Реакции замещения
Одни из распространенных типов свободнорадикальных ре-
акций— реакции замещения. К числу таких реакций, связанных
с отрывом водорода, относятся:
СН3 + С2Нв->СН4 + С2Н5; (5.3)
h-C3F7 + Н2 -> h-C3F7H + Н; (5.4)
Н + С2Н6 -> Н2 + С3Н5; (5.5)
С2Н5 + С6Н14 -> С2Н6 + С6Н13. (5.6)
Часто эти реакции называют реакциями переноса, и если речь
идет о переносе водорода, их обобщенная запись выглядит так:
R + ХН RH + X. (5.7)
В табл. 5.2 приведены энергии активации для ряда реакций
замещения. Энергии активации найдены из температурных за-
висимостей скоростей реакций. Таблица показывает, что, на-
пример, реакция атомов I с молекулами водорода гораздо ме-
нее вероятна по сравнению с реакцией атомарного водорода с
иодом.
141
Т а блица 5.2*
Энергии активации некоторых свободнорадикальных реакций
Реакция ДЕ, ккал {моль | Реакция &е, ккал/моль
СНз-РС2Нв - СН44-С2Нб СН3+СН4 -> сн4+сн3 • 10,4 1+н2 - >Н1+Н 33,4
14,3 Н+С12 - » НС1+С1 2,4
Н+СзНе С2Н5+Н2 6,4 Н4-Вг2 > HBr-1-Br 0,9
С14-Н2 НС1+Н .6,0 Н-И2 - > HI+I 0
Вг-рн2 - НВг+Н (7,6 ОН4-С2Н6 ► Н2О+С2Н5 5,5
Данные из книги: Бенсон С. Основы химической кинетики. Пер. с англ., М., «Мир», t964.
5.4.2. Реакции свободных радикалов.
Рекомбинация
Энергию активации рекомбинации радикалов обычно прини-
мают равной нулю и считают, что эта реакция идет при первом
же соударении (табл. 5.3). Однако рекомбинация атомов про-
Таблица 5.3
Энергия активации для реакций рекомбинации радикалов
Реакция ДЕ, ккал {моль Реакция ДЕ, ккал{ моль
+N-FN2 > 2N2 0 адн-ОЛз . с4н10 0
I-FI-FM - 12+М 0 СНз4-С2Н5 >С3Н8 0
4-Br4-M ->- Вг2-рМ 0 1 з C2F в 0
Нд+СНз - С2Н6 1,7 СгН5-]-С2Н5 > С2Н4-1- 0,8
исходит лишь в том случае, если присутствует третий компо-
нент, который уносит избыток энергии. В противном случае
образовавшаяся молекула вновь диссоциирует на атомы. Для
многоатомных радикалов (исключая метильные радикалы при
низком давлении) присутствие третьего компонента не обяза-
тельно, так как избыточная энергия может перераспределяться
по различным колебательным степеням свободы. При высоких
давлениях (больше 100 мм рт. ст.) необходимость в третьем
компоненте отпадает также и для метильных радикалов.
К типичным реакциям рекомбинации относятся
Н 4~ Н 4- М (третий компонент) -> Н2 4- М; (5.8)
N + N + M->N2+ М; (5.9)
. H-CgH? 4~ Н-С4Н9 CfHjgj
СН3 4-СН3 С2Не;
CFaCl 4- CF2ClCF2 -> CsFeCl2;
С2Н5 4~ С2Н5 -> С4Н10.
(5.10)
(5.П)
(5.12)
(5.13)
142
5.4.3. Реакции свободных радикалов.
Диспропорционирование
При диспропорционировании в результате рекомбинации
двух одинаковых радикалов образуются две стабильные моле-
кулы, причем одна из них оказывается более ненасыщенной,
чем другая. Реакции диспропорционирования конкурируют с
реакциями рекомбинации, хотя энергии активации для дис-
пропорционирования обычно бывают несколько выше. Напри-
мер, отношение констант скоростей kjkc для приведенных ни-
же реакций оказывается меньше 0,14:
k
2С2Н5—UC4H10; (5.14)
ь
2ад_<иад
ад.
(5.15)
Для большей части изученных свободнорадикальных систем
отношение констант скорости реакции диспропорционирования
к константе скорости реакции рекомбинации оказывается мень-
ше единицы, т. е. fea/fec<l [3, 4]. Значения отношения k^k- для
некоторых свободнорадикальных систем приведены в табл. 5.4.
Таблица 5.4
Отношение констант скоростей реакций диспропорционирования и
рекомбинации для алкильных радикалов
Радикалы ‘Л Радикалы kdlkc
2(С2Н5) 0,14 2(цнкло-С6Нв) 0,2
2(н-СзН7) 0,16 2(цикло-С6Нп) 0,5
2(изо-С3Н7) 0,64 ch8+qh5 0,06
2(н-С4Нв) ' 0,7 СН3-|-н-С3Н7 0,05
2(изо-С4Нв) 0,17 0,70
2(mpem-C4HB) 3,0 СН5Ц-втор-С3Н7 0,43
5.4.4. Реакции свободных радикалов.
Реакции присоединения
Присоединение радикала к ненасыщенным молекулам с об-
разованием нового радикала также относится к числу харак-
терных свободнорадикальных реакций:
СНа + ад->С3Н7; (5.16)
н + ад->ад. (5.17)
Реакция (5.17) оказалась весьма существенной при ради-
олизе углеводородов с участием атомов Н тепловых энергий.
Если реакция присоединения радикала идет.с высокой эффек-
тивностью, то ее принято называть реакцией акцептирования
143
(перехвата) радикала, а стабильную молекулу, участвующую
в реакции, — акцептором, радикала. Другим примером реакции
акцептирования радикала может служить следующий процесс:
СН3 Ч~ NO -> CH3NO или CH2NOH. (5.18)
Радикальные реакции полимеризации также дают хороший
пример реакций присоединения радикалов к стабильным моле-
кулам. .
Энергии активации для реакций присоединения алкильных
радикалов к ненасыщенным молекулам составляют около 6—
8 ккал/моль. Это сравнительно малые значения. Поэтому сле-
дует ожидать, что реакции присоединения свободных радикалов
должны идти во многих радиационно-химических системах,
особенно если концентрация олефинов в этих системах доста-
точно высокая.
5.4.5. Реакции свободных радикалов.
Диссоциация
Получив достаточную энергию активации, алкильные ради-
калы могут диссоциировать. Радикалы меньшего размера, чем
пентильный, диссоциируют, давая либо атомы Н, либо метиль-
ный или этильный радикалы:
СН5 -> С2Н4 + Н; (5.19)
СН3СН2СН2 -> СН3 + СН2СН3; (5.20)
СН3СН2СН2СН2 -> С2Н6 + С2Н4; (5.21)
С,Н15^СеН12 = СН3. (5.22)
Энергия активации для реакций типа (5.19) с выделением
водородного атома оказывается около 40—50 ккал)моль. Реак-
ции типа (5.20), (5.22) с распадом на радикалы требуют энер--
гий активации около 20—30 ккал!моль. Для таких радикалов
как СН3СОО и С2Н5СОО. энергия активации диссоциации
составляет всего лишь около 5 ккал/моль. У радикалов с ре-
зонансной стабилизацией структуры, например у бензильного
радикала, энергия активации процессов диссоциации оказы-
вается очень высокой.
5.4.6. Обнаружение свободных радикалов
Один из наиболее широко применяемых косвенных методов
обнаружения свободных радикалов и других промежуточных
продуктов — исследование влияния па выход реакций интенсив-
ности подачи энергии в систему. Интенсивность или мощность
дозы излучения можно менять различными способами. Напри-
мер, между источником и образцом можно помещать различные
поглотители. Прерывистость облучения обеспечивается либо
144
быстрым включением и выключением источника, либо за счет
перемещения образца относительно радиационного поля.
Как правило, подача энергии в систему осуществляется не-
прерывно, т. -е. в течение определенного периода времени обра-
зец находится под воздействием излучения источника .Проме-
жуточные продукты реакций достигают при этом стационарных
концентраций, которые уже пе меняются в течение всей экспо-
зиции. Если же стационарная концентрация радикалов меня-
ется, то должна измениться и скорость реакции. В случае
прямой пропорциональности между скоростью реакции и интен-
сивностью облучения выход реакции должен определяться
средней скоростью поглощения энергии и не зависеть от того,
как распределен но времени сам процесс поглощения энергии.
Если же прямая пропорциональность между интенсивностью
облучения и скоростью реакции отсутствует, прерывистость об-
лучения должна влиять па выход реакции.
Длительность импульсов облучения подбирается обычно в
соответствии с периодом полупревращения промежуточных
продуктов реакции. В фотохимических экспериментах она обе-
спечивается с помощью диска с вырезанными секторами, кото-
рый вращается между источником излучения и препаратом.
Скорость вращения диска определяет скорость подачи энергии
в систему.
Полезные сведения о механизме реакции может давать так-
же плавное (без прерывистости) изменение интенсивности
облучения. Стационарная концентрация радикалов или других
промежуточных продуктов меняется при изменении интенсив-
ности, и это непосредственно влияет на скорость реакции, как
показывает анализ, такой, например, кинетической схемы:
rAr + R; (5.23)
R + A^.B4-C; (5.24)
R+R^R2, (5.25)
где R2, А, Ви С — стабильные молекулы, a R — радикалы.
Если концентрация радикалов высока, то высокой оказы-
вается и вероятность рекомбинации радикалов [см. реакцию
(5.25)]. Если концентрация радикалов низкая, то преобладаю-
щей может оказаться реакция (5.24).
Определение присутствия р'адикалов осуществляется самыми
разнообразными методами — косвенными и прямыми. В газо-
вой фазе с этой целью часто используют такие акцепторы ради-
калов, как NO, пропилен и 12. Сравнительно недавно начали
применять комбинированную диэлектрическую и ЭПР-спектро-
скопию. В твердых телах, газах и жидкостях осуществляется
прямое наблюдение свободных радикалов методами оптической
спектроскопии. В жидкостях одинаково успешно использова-
145
лись как акцепторный метод, так и метод ЭПР. Метод ЭПР
оказался чрезвычайно полезным и при исследовании твердо-
тельных систем.
5.5. РЕАКЦИИ ИОНОВ
Ионы участвуют в разнообразных реакциях. Некоторые из
этих реакций нетрудно предугадать. Положительные ионы мо-
гут захватывать электрон и нейтрализоваться, реагировать с
отрицательным ионом, диссоциировать, давая реакционноспо-
собные продукты (или стабильные молекулы) и ионы, а также
передавать свой заряд в иоп-молекулярпых реакциях. Все эти
процессы следует учитывать .при анализе промежуточных про-
дуктов реакций.
5.5.1. Рекомбинация ионов
Так как в процессе ионизации затрачивается значительная
энергия, ионы обладают большим запасом потенциальной энер-
гии. Поэтому очень часто нейтрализация молекулярных ионов
сопровождается диссоциацией.
Потенциал ионизации воды равен 12,61 эв. Следовательно,
в результате реакции
rl2O"*" -|- С~ —> Н2О
возникают молекулы Н2О в высших возбужденных состояниях.
Поэтому считают, что при нейтрализации иона Н2О+ возникают
Н и ОН радикалы. Энергии нейтрализации более чем доста-
точно для диссоциации молекулы воды. Не удивительно также,
что образовавшиеся радикалы часто обладают избыточной
кинетической энергией (горячие радикалы).
Большая часть наших сведений о ионных реакциях полу-
чена в масс-спектрометрических экспериментах. Часть реакций,
однако, изучена кинетическими методами. Взаимодействие по-
ложительных и отрицательных ионов остается малоизученной
областью. Известно по крайней мере, что такая нейтрализация
ионов протекает чрезвычайно быстро.
5.5.2. Присоединение электрона
Молекулы могут присоединять электроны, образуя отрица-
тельные ионы. Захват электронов — резонансный процесс. При
этом, если присоединяется электрон с энергией, превышающей
тепловую, всегда происходит диссоциация. Однако она может
произойти и в том случае, если захваченный электрон имеет
тепловую энергию. Вот некоторые примеры реакций присоеди-
нения электрона с последующей диссоциацией:
С12 З- е —> Cl -|- С1;
(5.26)
146
НС1 + er- ci- + H;
SO2 + e- -> SO- + O;
C2H5I + e-->C2H5 + I-
H2O 4- e- -> H I- OH" ;
Oj -f- -+• o + О .
(5.27)
(5.28)
(5.29)
(5.30)
(5.31)
Образующиеся отрицательные ионы способны участвовать
в различных реакциях, однако будут преобладать реакции ней-
трализации с положительными ионами. Другие реакции отри-
цательных ионов могут вести к образованию новых продуктов.
О- 4- Н2О -Н ► ОН 4- ОН~; (5.32)
Н- 4- Н2О -н ► Н2 4- он-; (5.33)
О794-Н2-> онй? + Н; (5.34)
Н-4-4О^ ОН- 4- Н2; (5.35)
SO- 4- SO2 -> SOF 4- SO. (5.36)
Для нескольких газофазных реакций отрицательных ионов
определены константы скорости и сечения взаимодействия. Эти
параметры оказались сходными с соответствующими характе-
ристиками для положительных иоиов. В растворах, особенно в
водных растворах, изучены довольно многие реакции отрица-
тельных ионов.
5.5.3. Реакции диссоциации ионов
Образующиеся под действием излучения ионы могут дис-
социировать, распадаясь на радикалы, на один или несколько
новых ионов:
сн3он+ - >СН3О+4-Н; (5.37)
CHt- >СН3 + Н+; (5.38)
cjtf- ->С7НГ + СН3; (5.39)
сМ- > 4НЙ 4- снз; (5.40)
ch3ch2ch2ch2nh^ - >c2h54~C4NH^. (5.41)
Предполагается, что наблюдаемый в результате диссоциации
спектр масс обусловлен главным образом мономолекулярным
распадом родительского иона и в значительно меньшей степени
распадом некоторых дочерних ионов. В определенных пределах
существует зависимость между прочностью порванной связи
и относительным содержанием частиц соответствующей массы.
Этой закономерности нельзя придавать слишком общее значе-
147
яие, так как известен ряд случаев, в которых она не соблю-
дается. Однако, как показано в следующей главе, относитель-
ную вероятность распада различных ионов можно примерно
оценивать по прочности одноэлектронных связей.
5.5.4. Реакции переноса заряда
Реакции переноса заряда оказываются чрезвычайно важны-
ми в радиационной химии газовых и жидких систем. Приведем
некоторые примеры таких реакций:
Не+ Ne-> Ne+4-Не;
Н^ + Н2О->Н2О+Н-Н2;
Аг+4-С2Н4-> Аг 4-С2Н4~.
(5-42)
(5.43)
(5.44)
Перепое заряда — резонансный процесс: он протекает легче
в том случае, если близки потенциалы ионизации обоих компо-
нентов. Если потенциал ионизации нейтрального компонента
меньше, то при переносе заряда выделяется энергия, соответст-
вующая разности потенциалов. Выделенная энергия может
передаваться дочернему иону и вызывать его диссоциацию
Аг+ + СН4 -> Аг 4- СН^ + Н.
(5.45)
5.6. ИОН-МОЛЕКУЛЯРНЫЕ РЕАКЦИИ
Иоп-молекулярпые реакции — один из наиболее важных
типов реакций радиационной химии, широко исследованный в
теоретическом и экспериментальном аспектах. Причем не толь-
ко в связи с задачами радиационной химии, но и в качестве
важного класса химических реакций вообще. К ион-молекуляр-
ным реакциям относятся процессы взаимодействия ионов и
нейтральных молекул. Предполагается, что соударение молеку-
лы и иона приводит к образованию комплекса, последующая
перестройка которого приводит к возникновению наблюдаемых
продуктов реакции. Ион-молекулярные реакции можно разде-
лить па три большие категории: реакции переноса одной части-
цы, реакции конденсации и реакции присоединения.
5.6.1. Реакции переноса одной частицы
Н2О+ + Н,О -> Н3О+ + ОН;
СН4+ + СН4->О# + СН3;
СН3ОН+ + СН3ОН -> CH3OHt + СН3О;
(5.46)
(5.47)
(5.48)
148
+ H2->H3+ + H;
Ar+ + H2->ArH+ + H.
(5.49)
(5.50)
Реакции (5.46) — (5.50) являются реакциями переноса водо-
родного атома. Эта разновидность ион-молекулярпых реакций
относится к числу наиболее интенсивно изучаемых. Константы
скорости для этих реакций оказываются очень высокими (что,
впрочем, справедливо почти для всех ион-молекулярпых реак-
ций). Реакции переноса водорода имеют большое значение,
особенно в процессах радиолиза и газовой фазе.
Другая разновидность реакций переноса одной частицы —
реакции переноса протона. У высших алканов перенос протона
приводит к диссоциации. Например, в системе, заполненной
С3Н8. наблюдаются не ионы С3Н9+, а С2Н5+- и С3Н7+-ионы. Эти
ионы возникают, очевидно, из родительского иона С3Н9+. Из-
вестен целый ряд реакций переноса протона [5J. Среди них:
СН^ + NH3 NHf + СН4;
С2Н^ + NH3 NH4+ + С2Н4;
С2Н5' + Н2О -> Н3О+ + С2Н4;
СНб“ 4-СН4-> СНб" Д-СН4;
Нз~ -j- цикло-С3Н6 -> Н2 + emop-C3H7';
СНб" + цикло-С4Н8 ->СН4 4- «п2ор-С4Н9".
(5.51)
(5.52)
(5.53)
(5.54)
(5.55)
(5.56)
Органические молекулы, обладающие постоянным диполь-
ным моментом, такие, как амипы или спирты, чаще всего участ-
вуют в реакциях переноса протона.
Перенос гидридного иона НГ к положительному молекуляр-
ному иону — очень важная разновидность реакций между ал-
кильными ионами и нейтральными молекулами алканов. В не-
которых случаях это единственная реакция, в которую вступает
алкильный ион' помимо нейтрализации. Перенос гидридного
иона происходит также между алкильными ионами и олефина-
нами, но гораздо реже. Перенос гидридного иона оказывается
единственной реакцией, протекающей между алкильными иона-
ми и циклоалканами (кроме циклопропана). Приведем несколь-
ко примеров реакций переноса гидридного иона:
C4H8D+ + (СН3)2 CDCH2CH3 C4H8D2 + CM (5.57)
C2Ht + С3Н8 -> C2He + QHt; (5.58)
C2D3Ht + CeH12 CsAHg + С6Нп; (5.59)
С3Н5+ + С3Н8 -> С3Н6 + С3Н7+. (5.60)
149
Реакция переноса гидридпого иона часто оказывается очень
эффективной. Например, реакция (5.58) практически единст-
венная реакция, с помощью которой образуются этильные ионы
при радиолизе метана. Реакция идет, если в реакционную смесь
добавлено 0,01% СзНв. Реакции переноса гидридного иона ока-
зываются наиболее существенными радиационно-химическими
реакциями высших алканов. В табл. 5.5 суммированы сведения
об относительных скоростях реакций переноса гидридного иона
для некоторых молекулярных ионов.
Таблица 5.5
Относительные скорости реакций переноса гидридного иона при 300 °К
Вещество С1°9 C3Dy~ С2Г>^ Вещество C4d^ c3d+ CzD5*
Циклопропан .— <0,02 <0,05 н-Гексан 2,37 2,13
изо-Бутан — 0,58 0,87 Метилциклопентап 2,36 1,91 1,29
н-бутан -— 0,78 — Циклогексан 3,51 2,41 1,36
Циклобутан 1,00 1,00 1,00 н-Гептан 4,04 3,88 1,65
Неопентан — 0,049 1,05 Метилциклогексан 3,50 2,69 —
изо-Пентаи 1,26 1,04 1,09 Циклопентан — 0,71 0,34
н-Пентан 1,42 — 1,07 Циклогексан — 0,81 0,49
Циклопентан 1,56 1,44 1,14
К той же категории ион-молекулярных реакций можно от-
нести реакцию переноса иона Н~. Пример такой реакции:
. CmHrm+RH2->CmHw + R+, (5.61)
где СтН^—ион одного из низших олефинов; a RH2 — насы-
щенный углеводород.
Не следует думать, однако, что реакции насыщенных угле-
водородов с олефиновыми ионами всегда имеют подобный ха-
рактер. Точно также возможен перенос и гидридного иона:
CmH2+ + RHa -> СтН2т+1 + RH+. (5.62)
В реакциях (5.63) — (5.67) происходит перенос иона Н^-:
с2н^+с3н8- >С2Не + сЖ; (5.63)
C3I< 4- цикло-С5Н10 - >C3H8 + C6Ht; (5.64)
ОД+ + СА- >C2He + C4Ht; (5.65)
С2Н^ + цикло-С6Н12 - >СХ + С2Н6; (5.66)
QHg' 4- цикло-С6Н12 - (5.67)
Другой вид реакций переноса одной частицы — реакции
переноса Н2. По-видимому, и Н2 и Н? переносятся как единое
150
целое, причем оба атома водорода забираются от одного угле-
родного атома. Вот некоторые нримеры-переноса Иг:
цикло-СвН12 С4НЙ -> С6НЙ + CiH10; (5.68)
цикло-С6НЙ + C3Df -> СвНЙ + CaDgIL;; (5.69)
иикло-СвН12 цикло-С3Нв -> СвНю C3DeH2; (5.70)
цикло-СдНа" + C3De ->C3DeH3 цикло-СйНб". (5.71)
Относительные скорости реакций переноса Нг и НГ меня-
ются примерно одинаковым образом. Однако скорости реакций
могут меняться очень сильно в зависимости от того, какие ком-
поненты в них участвуют.
5.6.2 Реакции конденсации
В отличие от реакций переноса одной частицы в реакциях
конденсации происходит существенная перестройка переходного
комплекса, возникающего при соударении. Можно привести
следующие примеры реакций конденсации:
СН^ + СН4^С2Н^ + Н2; (5.72)
CHf 4-СН4^С2Н^ + Н2; (5.73)
СаН^ + СН4->С3Н^+Н2; (5.74)
С2Н2+ Ч-С2Н2^СЛ^ + Н2; (5.75)
СМ + С4Нв^С7Н^+-СН3; (5.76)
Г4Ч-С2Н81^НЙ’ + С2Н4. (5-77)
5.6.3. Реакции присоединения
В реакции присоединения переходный комплекс оказывается
конечным продуктом:
да+Ч-СзИД^ОД!^; (5.78)
С2Н8Вг+ + С2Н8Вг (С4Н10Вг2)+; (5.79)
С2Н44-С2Н4_С4Н^. (5.80)
Время жизни подобных комплексов согласно масс-спектро-
метрическим наблюдениям составляет по крайней мере I0-6 сек.
Константы скоростей всех ион-молекулярных реакций обыч-
но очень велики [вплоть до 2-10-9 см3/(молекула• сек)]. Столь
высокие константы скоростей реакций означают, что феномено-
логические сечения реакций равны или превосходят геометриче-
ские сечения молекул. Многие ион-молекулярные реакции и их
параметры представлены в табл. 7.3.
151
5.7. ГОРЯЧИЕ РАДИКАЛЫ И АТОМЫ
«Горячими» называют такие атомы и радикалы, которые не
находятся в тепловом равновесии с окружением и обладают
'избыточной кинетической или электронной энергией. Горячие
атомы обычно образуются в результате отдачи в ядерных реак-
циях, таких, например, как
eLi(n,y)3H; (5.81)
3Не(п, р)3Н. (5.82>
Тритиевые атомы отдачи, возникающие в реакции (5.81),
обладают энергией 2,73 Мэв, а в реакции (5.82)—0,19 Мэв.
Горячие атомы и радикалы возникают также при других про-
цессах. Например, горячие метиленовые радикалы образуются
D результате некоторых фотохимических реакций. В этих слу-
чаях возможно появление и горячих водородных атомов. Ра-
зумеется; термин «горячий» весьма условен. Он может характе-
ризовать избыточную энергию от сотых долей электронвольт-
до нескольких мегаэлектронвольт. При фотолизе йодистого во-
дороДа светом с длиной волны 2537 А'образуются горячие во-
дородные атомы с избыточной кинетической энергией
41 ккал!моль. Если при образовании горячих радикалов избы-
точная энергия не перераспределяется по внутренним степеням
свободы, то опа превращается в кинетическую энергию.
5.8. РАДИАЦИОННАЯ ХИМИЯ И ФОТОХИМИЯ
В любой системе, подвергающейся воздействию ионизирую-
щей радиации, при анализе механизмов реакций следует учи-
тывать как реакции возбужденных молекул, так и реакции
ионов. Ни для газообразных, ни для водных или неводных си-
стем до сих пор не удалось установить с достаточной уверен-
ностью, какой относительный вклад в образование промежу-
точных и конечных продуктов реакций вносит энергия, затра-
ченная па прямое электронное возбуждение, и энергия,
затраченная на ионообразовапие. Выяснение роли молекул в
возбужденных состояниях при образовании! промежуточных
продуктов реакций оказывается одной из основных задач ра-
диационной химии. Вот почему очень часто параллельно с ра-
диационно-химическим для той же системы проводят фотохими-
ческий эксперимент.
В фотохимическом эксперименте обычно прежде всего выяс-
няют спектр поглощения рассматриваемой молекулы, чтобы
установить, какая часть молекулы поглощает световую энергию
с данной длиной волны. Затем, облучая систему монохромати-
ческим светом, анализируют происходящие при этом химиче-
ские реакции. Часто одновременно подробно исследуют спектры
152
флуоресценции и фосфоресценции системы. Нередко обнару-
живается, что световая энергия, поглощенная в одном участке
молекулы, мигрирует к другому участку, где может вызывать
диссоциацию валентной связи. Например, энергия, поглощен-
ная карбонильной группой ацетона, мигрирует к С — С связи,
которая и разрывается:
СН3СОСН3 СН3 + СОСН3. (5.83)
Фотохимики, изучавшие реакции подобного рода, выявили
различные механизмы, с помощью которых энергия возбужде-
ния может передаваться внутри молекулы, и тем самым внесли
значительный вклад в понимание процессов миграции энергии.
В этом отношении и фотохимия и радиационная химия стал-
киваются с весьма сходными проблемами. Однако в ряде важ-
ных аспектов радиационная химия и фотохимия существенно
различаются:
1. При воздействии ионизирующей радиации электронное
возбуждение молекулы происходит в результате прямых воз-
мущающих воздействий. При этом молекулы могут возбуж-
даться в «запрещенные состояния», которые остаются незасе-
ленными в фотохимических экспериментах.
2. В радиационно-химическом эксперименте возможно мно-
жественное возбуждение-молекул, т. е. одновременный переход
па высшие уровни нескольких электронов.
3. В радиационной химии важную роль играют реакции Йо-
нов, для появления которых требуются различные энергии.
Однако недавно в связи с развитием фотохимий вакуумного
ультрафиолета у экспериментаторов появилась возможность
работать с монохроматическим светом таких энергий, которые
вполне достаточны для ионизации большей части молекул. Ста-
новится возможным, таким образом, изучение реакции опреде-
ленных ионов в газовой фазе.
Характеризуя эффективность энергетических затрат в фото-
химической реакции, фотохимики используют понятие кванто-
вого выхода. Квантовым выходом называют число образовав-
шихся или распавшихся молекул на -один квант поглощенного
света. Сходство между квантовым и радиационно-химическим
выходом вполне очевидно. Так же как и в радиационной химии,
в фотохимии применяются определенные методы для измерения
количества поглощенной световой энергии. Системы, исполь-
зуемые с этой целью в фотохимии (аналоги дозиметрических),
называют актинометрическими. Одна из лучших актинометри-
ческих систем — раствор урапилоксалата, который разлагается
под действием света согласно реакции
(UOs+) + (СООН)а ПОГ + СО2 + СО + Н2О. (5.84)
Число молекул, реагирующих при поглощении одного кван-
та света, т. е. квантовый выход, равно 0,5 в нормальных усло-
153
виях. Выход меняется в зависимости от длины волны, темпера-
туры раствора,, относительных концентраций ураниловых и
оксалатных ионов, pH, присутствия ионных добавок. Кванто-
вый выход системы не зависит от того, проводится ли освеще-
ние непрерывно или в импульсном режиме.
Энергия светового кванта равна
E = hv = h —. (5.85)
Л
При прохождении через бесконечно топкий слой поглотите-
ля dl интенсивность светового потока уменьшается пропорцио-
нально концентрации поглощающего вещества:
-у- = — kcdl. (5.86)
Здесь / — интенсивность светового потока; с — концентрация
поглощающего вещества; k — константа пропорциональности.
Интегрирование уравнения (5.86) для слоя конечной толщи-
ны дает известный закон Ламберта — Бэра:
In — = — kcl. (5.87)
/о
•Если в уравнении (5.87) концентрация с выражена в моль/л,
то k называют коэффициентом молярной экстинкции. /0 и I —
интенсивности падающего и прошедшего света.
Молекула, поглотившая световой квант, переходит в состоя-
ние электронного возбуждения. Если молекула изолирована,
то энергия возбуждения вновь истратится на излучение в виде
флуоресценции, и молекула вернется в основное состояние
A -j- hv -> А* (скорость реакции — /0); (5.88)
A*—-»-A + hv. (5.89)
Скорость флуоресценции будет равна
-^- = МЛ’). (5.90)
at
Если в системе имеются в достаточной концентрации другие
молекулы, то энергия возбуждения может растрачиваться при
соударениях. Происходит «тушение флуоресценции». В такой
системе начинается конкуренция между процессами тушения и
флуоресценции:
А* + М А + М*. (5.91)
154
Скорость уменьшения числа возбужденных молекул будет
тогда определяться уравнением
- = ke И’) ' <5’92)
at
Выход флуоресценции дается соотношением
=-------МАП-------- (5,93)
/„ ^(A*)(Al) + fce(A*) ’
где 1е — число .испущенных квантов света; 1а — число поглощен-
ных световых квантов.
Измерения выхода флуоресценции позволяют оцепить эф-
фективность процессов передачи энергии.
5.9. ОСНОВНЫЕ КИНЕТИЧЕСКИЕ СООТНОШЕНИЯ.
ОБОЗНАЧЕНИЯ И РАЗМЕРНОСТИ
Константы скорости реакций обычно представляют в виде
уравнения Аррениуса:
k = Ае~^г, (5.94)
где А — предэкспонепциальный множитель й АЕ— энергия ак-
тивации.
Константу скорости реакции k определяют, как правило,
косвенными методами. В тех случаях, когда k определяется
прямым методом, например по появлению продукта или по рас-
ходованию субстрата в ходе реакции, k называют абсолютной
константой скорости реакции. Большая часть измеренных кон-
стант скорости реакций гидратированного электрона с различ-
ными соединениями является абсолютными.
Размерность константы скорости реакции первого порядка
k=—-—, (5.95)
время
константы скорости реакции второго порядка
k— (концентрация)-1 (время)-1, (5.96)
константы скорости реакции третьего порядка
k — (концентрация)-2 (время)-1. (5-97)
Концентрацию можно выражать числом молекул в кубиче-
ском сантиметре или числом молей на литр. В первом случае
из уравнения (5.96) получим:
k = см3 - молекула-1 сек~1. (5.98)
Во-втором случае получим:
k = л-моль~1-сек~1. (5.99)
155
При использовании молярного способа выражения концентра-
ций часто применяется такая запись:
k = М~х-сек~\ (5.100)
где М — молярность.
Далее будут использоваться все три способа выражения кон-
стант скорости реакций второго порядка.
Временем полупревращения (или периодом полупревраще-
ния) называют время, в течение которого концентрация реа-
гирующего вещества убывает вдвое. Средним временем реак-
ции называют время, в течение которого концентрация реаги-
рующего вещества убывает в е раз. Здесь е — основание на-
туральных логарифмов. Для химической реакции, которая опи-
сывается уравнением
2А-+В, (5.101)
скорость реакции можно записать как
—-М- = й(Л)®. (5.102)
dt
Однако при сравнении констант скорости реакций нередко ис-
пользуют такую запись, из которой становится ясно, что в ходе
реакции одновременно исчезают две молекулы:
Скорость реакции = 2k (Л) (Л). (5.103)
Такая запись очень часто используется при составлении
таблиц констант скоростей реакций. Однако обычно для выра-
жения константы скорости реакции используют уравнение
(5.102). Как правило, для обозначения концентраций пользу-
ются пе круглыми, а квадратными скобками.
5.9.1. Кинетика конкурентных реакций
Анализ кинетики конкурентных реакций очень часто оказы-
вается необходимым при рассмотрении радиационно-химических
реакций в водных растворах. Различные свободные радикалы,
возникающие в растворе, могут реагировать с одним и тем же
растворенным веществом. Наоборот, один и тот же радикал,
например Н, может участвовать в конкурентных реакциях с
разными растворенными веществами.
Пусть радикал R реагирует с двумя растворенными вещест-
вами Л и В. Вероятность реакции радикала с веществом А
будет равна kAA, с веществом В — kBB.- Если реакции с Л и В
единственные реакции, в которые вступает радикал /?, то ве-
роятность реакции радикала именно с молекулой А дается вы-
ражением
йдИ]
kA + kB '
(5.104)
156
В общей форме для нескольких растворенных веществ уравне-
ние (5.104) перепишется следующим образом:
,^И]
Er£ 1 i ]
(5.105}
где ki [i] — вероятность реакции радикала с i-м растворенным
веществом.
Если полная концентрация промежуточного продукта R из-
вестна, то в случае конкуренции двух реакций с веществом А
или В доля радикалов, реагирующих с веществом А за опреде-
ленный период времени, будет даваться выражением
МЛ] [Я]
/ = J [/?1~ dL (5.106}
Если радикал может вступать в реакцию только с самим
собой или с растворенным веществом В, при реакции с которым
радикал дает стабильный продукт, то
R+R^RR, 2^[7?][R];
r + b^rb, МЯ1[В1;
=2Л1[Я]2 4-Л2[Я] [В].
at
Концентрацию R в любой момент времени найдем,
уравнение (5.109) и принимая [В] постоянной:
[Я] =-----------------.
С ехр кг [В] t — 2k,
(5.107}
(5.108}
(5.109}
интегрируя
(5.110}
Постоянную интегрирования С найдем, задав граничные
условия, соответствующие условиям данного эксперимента.
Обычно выбирают такой момент времени, для которого из-
вестна полная (или максимальная) концентрация радикала Ro.
Если это момент времени t=0, то
С = (5 111}
[/?о!
где [7?0] — концентрация радикала R при 7=0. Подстановка
уравнения (5.111) в уравнение (5.110) дает:
[Я] =--------------М£ЦЯо]----------------- (5.112}
[k [В] -У 2.^ [7?0]J [exp k2 [В] Л - 2k, [Яо]
Доля радикалов R, реагирующих с веществом В за время от
1=0 до i=t', равна
f = 7 . fe2..[B] [В] di. (5.113}
J [Bol
157
Подстановка выражения для [/?] в уравнение (5.113) дает
/= f -----------------[k'fgl12 f/?nl-----------di. (5.114)
J 1*2[Л14А[/?o)[expk>] [B]<] — 2fei[/?0]
t=o
Задачи
1. Используя соотношения
ln/C =
AG
RT
ЛН
RT
AS°
и In K„ = In A —---,
RT
показать, что для реакций вида
А + В - C-J-D
различия в энергиях активации прямо пропорциональны теплотам реакций,
если считать АСР=О.
2. Наблюдаемая скорость радиационного разложения KNO3 хорошо
•объясняется следующей схемой реакций:
NOif NO7 + О;
О + NOJ" -> О2 + NO~;
О + NO~ -> NCXf.
Покажите, что такая система позволяет описать выход нитрита следующим
уравнением:
Ах2 -|-х --- Ы,
где х — концентрация NOs", Ы — значение, пропорциональное поглощенной
дозе в момент времени t; А —постоянная.
3. Найдите среднее число молекулярных соударений в 1 сек для газа
Н2 при давлении 1 атм и температуре 300° К. Диаметр молекул 2,72 Л.
4. Покажите, что теория соударений предсказывает температурную-зави-
симость для предэкспоненциалыюго множителя А.
5. Что можно сказать об относительных значениях радиационно-химиче-
ского выхода G для реакций:
Н, + С12 -> 2НС1;
Н2 + Вг2 - 2НВг;
Н, 4-1., -2HI.
Глава VI
СОВРЕМЕННЫЕ МЕТОДЫ РАДИАЦИОННОЙ ХИМИИ
Прогресс науки происходит скачкообразно. Иногда делается
крупный шаг вперед, иногда маленький. Как правило, шаги
научного прогресса знаменуйтся новыми успехами теории, но
очень часто это бывает новая техника исследований. После
1950 г. в распоряжении химиков оказались такие новые методы
исследования, как флэш-фотолиз, спектроскопия электронного
парамагнитного и ядерного магнитного резонанса, газовая хро-
матография, импульсный радиолиз и масс-спектрометрия высо-
кого разрешения. Эти экспериментальные методы заметно уско-
рили развитие радиационной химии, поэтому четыре наиболее
важных из них — флэш-фотолиз, импульсный радиолиз,
ЭПР-спектроскопия и масс-спектрометрия — будут рассмотрены
в данной главе.
6.1. ФЛЭШ-ФОТОЛИЗ
Источником энергии в методе флэш-фотолиза служит разряд
батареи конденсаторов через трубку, заполненную инертным
газом, аргоном или криптоном. При этом возникает яркая
вспышка света с энергией вплоть до 105 дж и с длительностью
около 10-4 сек. В результате поглощения столь интенсивного
светового потока в реакционной системе в большом количестве
появляются возбужденные молекулы, свободные радикалы и
другие промежуточные продукты реакций. Затем через систему
пропускают световой импульс от другого источника и записы-
вают спектры всех поглощающих компонент (рис. 6.1).
Флэш-фотолиз представляет собой уникальный метод обна-
ружения промежуточных продуктов реакций .Одновременно он
позволяет изучать время жизни и способы распада возбужден-
ных молекул, тогда как в обычном фотохимическом экспери-
менте концентрации промежуточных продуктов, достаточные
для их непосредственного наблюдения, получить не удается.
159
В этом нетрудно убедиться, если рассмотреть энергетический
аспект экспериментов по флэш-фотолизу.
Пример 6.1.
1. Подсчитайте, какую концентрацию радикалов нужно создать в экспе-
рименте по флэш-фотолизу, чтобы оптическая плотность была равна 0,1 (при
условии, что коэффициент молярной экстинкции радикала равен 5000).
Рис. 6.1. Схема эксперимента по флэш-фотолизу:
1 — вспомогательный источник света (лампа аналитической системы);
2— фильтр; 3— кварцевая трубка с образцом; 4 — спектрограф;
5 — импульсная лампа; 6 — батарея конденсаторов.
2. Если радиус реакционного сосуда равен 1 см, длина равна 100 см,
сколько фотонов должно быть поглощено?
3. Если среднее время жизни наблюдаемого радикала 10~3 сек, то сколько
фотонов в 1 сек должно поглощаться системой?
4. Средняя длина волны световой вспышки 3000 А, па реакцию расходуется
лишь 1% подаваемой энергии. Подсчитайте необходимую энергию вспышки.
Решение. Согласно закону Бэра
1п -у- =^D —г [С] I,
где /о — интенсивность падающего света; / — интенсивность света, прошед-
шего через образец; D — оптическая плотность; [С] — концентрация, моль/л;
е — коэффициент молярной экстинкции; I — длина светового пути.
1. 0,1 = 5000 • [С] • 100 см = 2,0 • 10~7 моль1 л.
2. Емкость реакционного сосуда равна (100 лг2) см3 или л/10 л.
(л/10) • 2 10“7 = 6,28 • 10_8 моль = 3,77 • 1016 молекул (радикалов).
Для обозначения 1 моль фотонов (Nhv) фотохимики используют термин
эйнштейн. Так как У=6.023-1023,
6,28 • 10-е . 6,023 • Ю28 = 3,77 - 101® фотонов.
3. Если среднее время жизни радикала т равно 10-8 сек, то концентра-
ция радикалов в стационарном состоянии будет равна /ат, Где 1а — интен-
сивность света. Чтобы получить 3,77-1016 радикалов, необходима интенсив-
ность света 3.77-1019 фотон/сек.
4. Если средняя длина волны вспышки 3000-10—® см, то необходимую
энергию световой вспышки получим, умножая энергию кванта на число
^реагирующих молекул:
энергия молекул 6,62 . 10~27 эрг сек
= 6,023 . X
эйнштейн-----------------------------моль ? эрг молекула
дж
460
I
CM
X 3 • lOio ---- .
сек
дж
= 390 000-------
3000 • 10~8 СМ эйнштейн
Энергия = 390 000 • 6,28 • 10“8 = 0,024 дж.
Так как используется лишь 1% энергии вспышки, то полная необходи-
мая энергия вспышки равна:
0,024
Полная энергия вспышки = ------= 24 дж.
0,01
В процессе передачи энергии от батареи конденсаторов к поглощающей
свет системе происходит достаточно много потерь. Поэтому в большинстве
экспериментов по флэш-фотолизу требуются энергия порядка 20—100 дж.
Конкретное значение энергии зависит от многих переменных, таких, как
среднее время жизни и коэффициент молярной экстинкции поглощающего
продукта.
Максимальная энергия, которую можно получить при вспышке газораз-
рядной лампы, равна энергии, запасенной в конденсаторной батарее. Эта
энергия равна Г/2 CV2, где С — емкость батареи, ф, V — напряжение, в. На-
пример, при емкости батареи 10 мкф и напряжении 20 кв запасенная энер-
гия равна
(10 . 10~6) (2 IO*)2 = 2000 дж.
6.1.1. Длительность вспышки
Длительность вспышки часто принимают равной временй,
в течение которого световая интенсивность остается не ниже
половины максимальной. Еще более распространенная харак-
теристика продолжительности вспышки — время, в течение ко-
торого интенсивность светового потока убывает в е раз по
сравнению с максимальной. Здесь е —основание натуральных
логарифмов.
В том случае, если энергия вспышки не является существен-
ным фактором, продолжительность вспышки не является су-
щественным фактором, продолжительность вспышки может
быть очень малой. Действительно, существуют импульсные лам-
Т абл иц а 6.1
Импульсные лампы для флэш-фотолиза
Продолжи- тельность вспышки, сек Энергия вспышки, дж Литература
8-10-1° 2-10-’ 310-’ - 1—2-10-® ЗЛО-® 6-I0-® 0,5 1,4 50—300 500 Kerns Q. A., Kersten F., Сох G. С.. Rev. Sci. Instr., 1959, v. 30, р. 31 Fisher Н. J. Opt. Soc. Amer., 1957, v. 47, p. 981 Looms J. S. T., North R. J. Proc. 3 Int. Congr. on High—Speed Photography, London, 1956 Stuhl F., Wefge К. H. Naturforsch., 1963, Bd. 18 (a), S. 900 Porter G., Windsor M. W. Disc. Faraday Soc., 1958, v. 17, p. 178
Зак. 907
161
пы с продолжительностью вспышки 10-9 сек. Однако энергия,
выделяющаяся при вспышке такой лампы, достигает лишь мил-
лионных долей джоуля. Продолжительность вспышки определя-
ется емкостью конденсатора и длиной лампы и в некоторой
степени также напряжением на конденсаторе. В табл.. 6.1 при-
ведены параметры нескольких импульсных ламп.
6.1.2. Ограничения измерений скорости реакций
Время полупревращения для реакций первого порядка об-
ратно пропорционально константе скорости реакции. Поэтому
при продолжительности вспышки от 1 до 10 .мксек можно
проследить лишь за реакциями, константа скорости которых не
превышает 106 сек-1.
Время полупревращения для реакции второго порядка равно
Для превращений промежуточного продукта х реакция
имеет второй порядок и, следовательно,
-^=H(Q]2. (6.2)
Из уравнения (6.1) видно, что время полупревращения бу-
дет меняться в зависимости от концентрации промежуточного
продукта. Подставим теперь в уравнение (6.1) выражение для
Сх, соответствующие закону Бэра:
'•/. = Дг М
В любом эксперименте по флэш-фотолизу предварительной
стадией является определение спектра поглощения промежуточ-
ного продукта [1]. После того как спектр поглощения известен,
выбирают определенную длину волны, для которой характерно
относительно высокое поглощение промежуточного продукта,
и на этой длине волны проводят кинетические и другие измере-
ния [2]. Кривые относительного пропускания получают, прово-
дя денситометрию фотопластинок, либо в результате прямых
фотоэлектрических измерений, при которых сигнал с фотоэле-
мента подается на вход осциллографа.
Как в фотоэлектрическом методе, так и при депситометриро-
вапии пластинок фактически измеряется произведение концент-
рации на коэффициент экстинкции. В фотоэлектрическом ме-
тоде детектирующая система состоит обычно из фотоумножи-
162
теля, электронной схемы и осциллографа, по шкале которого
после соответствующей калибровки можно непосредственно
определять процент пропускания образца в зависимости от вре-
мени. На рис. 6.2 показана типичная осциллограмма такого
рода. Вводя затем поправки на светорассеяние, можно перейти
от процентов пропускания к оптической плотности и, следова-
тельно, получить произведение концентрации на коэффициент
экстинкции.
Раздельное определение
концентрации и коэффициента
экстинкции часто оказывается
чрезвычайно трудной задачей.
Обычно стараются провести
очень тщательный кинетиче-
ский анализ реакции и затем
сопоставляют свойства проме-
жуточного продукта со свой-
ствами Других соединений,
изученных обычными метода-
ми. Другой метод состоит в
измерении концентрации всех
известных продуктов, после
Рис. 6.2. Осциллографическая запись
процесса химического превращения
поглощающего свет вещества.
чего концентрацию неизвест-
ного промежуточного продукта можно найти путем вычитания.
Например, при фотолизе СЮ2 единственными соединениями,
содержащими хлор, являются СЮ и СЮ2. Определяя в любой
момент времени концентрацию СЮг (для которого известны
спектр поглощения и коэффициент экстинкции), можно найти
концентрацию СЮ, возникающего при распаде СЮг. Строя
обычный график зависимости оптической плотности от концен-
трации, легко найдем величину абсолютных коэффициентов
экстинкции СЮ для использованных длин волн. Во многих слу-
чаях, однако, удается получить лишь верхнюю границу коэф-
фициента экстинкции, и тогда концентрацию можно измерить
лишь приблизительно.
Закон Бэра, на котором основаны все спектрофотометри-
ческие измерения, должен быть дополнительно проверен для
исследуемой системы. Для отдельного соединения при фикси-
рованной длине волны этот закон соблюдается очень точно.
На практике отклонения от закона Бэра могут возникать,
во-первых, из-за того, что при данной длине волны свет погло-
щает несколько соединений, во-вторых, из-за немонохроматич-
ности освещения, если в данном интервале длин волн коэффи-
циент экстинкции очень резко меняется. Если спектр погло-
щающего соединения известен, то подобный источник ошибок
легко устранить. Достаточно выбрать-такую длину волны, в
окрестностях которой спектр оказывается сравнительно глад-
ким.
6*
163
6.2. ИМПУЛЬСНЫЙ РАДИОЛИЗ
Метод импульсного радиолиза основывается практически
на тех же принципах, что и флэш-фотолиз: основной задачей
является создание достаточно высоких концентраций промежу-
точных продуктов, для того чтобы можно было наблюдать их
непосредственно. В большей части случаев самый удобный ме-
тод наблюдения — спектрофотометрия. Однако можно исполь-
Рис. 6.3. Схема эксперимента по импульсному радиолизу:
1 — электронный пучок; 2 — зеркало; 3— кювета с образцом; 4 — вогнутое зер-
кало; 5 — линза; 6 — источник света; 7 — свинцовая защита; 8 — осциллограф;
9 — спектрофотометр.
зовать и другие методы, такие, как ЭПР-спектроскопия и изме-
рения проводимости. Метод импульсного радиолиза возник
после того, как появились импульсные электронные ускорите-
ли, создающие микросекупдные импульсы достаточно высокой
энергии. Малое время импульса — необходимое условие, так как
константы скоростей для реакций многих промежуточных про-
дуктов весьма высоки (~1010 М~1 -сек~1); поэтому времена по-
лупревращения для этих реакций лежат в микросекундном
диапазоне. Значения G, как правило, невелики, а коэффициенты
экстинкции для различных промежуточных продуктов могут
различаться очень сильно. Например, коэффициент экстинкции
гидратированного электрона при 7200 А равен 15 800 М~1'СМ~1.
Чтобы оптическая плотность в кювете длиной 15 см оказалась
равной 0,1, достаточно создать концентрацию гидратированных
электронов всего лишь 4,1 ммоль/л. В то же время для наблю-
дения а-этанольных радикалов, коэффициент экстинкции кото-
рых не превышает 240, необходимо создать в тех же условиях
концентрацию ~28 ммоль/л. Типичная схема эксперимента по
импульсному радиолизу показана на рис. 6.3.
164
6.2.1. Энергетические требования
Доза, необходимая для возникновения в растворе наблюдае-
мых концентраций промежуточного продукта, зависит от значе-
ния G, абсолютной константы скорости реакции и коэффициен-
та экстинкции анализируемого продукта. Значение этих пара-
метров лучше всего проиллюстрировать с помощью нескольких
примеров.
Пример 6.2. Чему будет равна концентрация промежуточного продукта,
образующегося в кювете объемом 4 см3 под действием импульсов электрон-
ного ускорителя с энергией 3 Мэв? Сила тока 20 ма, длительность импульса
1 мксек, G=2, размеры кюветы ГХ1.Х4 см.
Решение. Энергия в импульсе равна
3 • 10® • 0,02 а 10-® сек= 0,06 дж'.
2 молекул I моль i-w:- 1 ю? Эрг । 0,06 дж I
100 эв |6,02 • 1023 молекул | 1,6 • 10~12 epaj дж | I
1000 cm3
X-----------------= 31 • 10~6 MOAbjA.
4 см3 л
Пример 6.3. Если коэффициент молярной экстинкции равен 1000 и G—
=2, то какая энергия должна быть поглощена в кювете объемом 4 см3 в те-
чение 1 мксек, чтобы получить оптическую плотность 0,2 (размеры кюветы
1X1Х4 см)? Какой должна быть сила тока в ускорителе с энергией 3 Мэв,
чтобы обеспечивалось поглощение заданной энергии? •
Решение.Оптическая плотность=е[С]/; 0,2= 1000-[С]-4; [С]=5-10-5 моль/л;
5 • 10-6 моль л 4 см2 6,02 • 1023 молекул 100 эв 1
Л 1000 см3 моль 2 молекул 1
1,6- 10“12 эргI
эв
дж
107 эрг
— 0,96 дж;
силу тока 1 в ускорителе найдем из уравнения 3-10® в-I н-10-6 сек=0,96дж;
/=0,32 а=320 ма.
6.2.2. Импульсный радиолиз.
Источники импульсного излучения
В идеале следовало бы иметь ускоритель переменной энер-
гии с продолжительностью импульса от 10-3 до 10“В 9 * * сек. Во
всяком случае продолжительность импульса должна быть мала
по сравнению с временем полупревращения, а форма импульса
должна приближаться к' прямоугольной. В этом отношении
эксперименты по импульсному радиолизу сходны с экспери-
ментами по флэш-фотолизу. Принципиально различны, однако,
источники импульсного излучения и кюветы, в которых проте-
кают реакции.
В настоящее время для импульсного радиолиза используют
линейные ускорители, ускорители Ван-де-Граафа и импульсные
источники рентгеновского излучения. Наиболее широко приме-
165
няют линейные ускорители. С их помощью удается создавать
стабильные пучки заряженных частиц с высокой энергией и
большой силой тока. Чем выше энергия частиц, тем больше
глубина проникновения пучка и, следовательно, тем больше
может быть длина оптического пути в детектирующей системе.
(Оптическая плотность прямо пропорциональна длине кюветы.)
Более высокие энергии частиц и большая сила тока позволяют
также работать-с импульсами очень малой длительности.
Импульсные источники рентгеновского излучения могут
создавать дозы в импульсе от 3000 до 7000 рад; причем нетруд-
но получить импульсы с продолжительностью 17—20 нсек. Ос-
новная трудность при работе с такими источниками заключа-
ется в том, чтобы обеспечить достаточно точную дозиметрию.
Для целого ряда, экспериментов вполне пригоден ускоритель
Ван-де-Граафа с энергией 3 Мэв (в особенности в тех случаях,
когда желательно уменьшить стоимость работ). Основное огра-
ничение при использовании ускорителя Ван-де-Граафа в том,
что нельзя получать очень высокие токи и соответственно ма-
лую длительность импульсов. На терминальном электроде гене-
ратора Ван-де-Граафа запасается огромная энергия, однако
лишь малая ее часть доступна для полезного применения.
Если напряжение терминального электрода падает ниже на-
пряжения ускоряющих электродов, то никакого пучка в трубке
не возникает. Поэтому в импульсном эксперименте исполь-
зуется лишь малая доля запасенной энергии. Для импульсного
радиолиза применяют ускорители Ван-де-Граафа с энергией
3 Мэв, силой тока 1 с, с длительностью импульса от нескольких
десятых микросекунды до 5 мксек. Для импульсных эксперимен-
тов в наносекундном диапазоне, однако, более перспективны-
ми источниками оказываются линейные ускорители и импульс-
ные источники рентгеновского излучения.
6.2.3. Определение промежуточных продуктов
оптическими методами
После того как в изучаемой системе произошло поглощение
энергии, промежуточные продукты исследуют с помощью бе-
зынерционных оптических систем, сходных с теми, которые ис-
пользуются при флэш-фотолизе. Свет от импульсной лампы,
синхронизированной с импульсами ускорителя, проходит через
облучаемую кювету и регистрируется спектрографом. Если
спектр промежуточного продукта известен, можно выбрать
определенную длину волны и следить за изменением поглоще-
ния во времени. В этой схеме эксперимента свет от источника
с постоянной интенсивностью проходит через реакционный со?
суд и попадает в монохроматор. Фиксированная длина волны
регистрируется фотоумножителем, выход которого подключен
166
к осциллографу (рис. 6.4), прокалиброванному в координатах
оптическая плотность — время.
Существенная особенность импульсного радиолиза по срав-
нению с флэш-фотолизом состоит в том, что при импульсном
радиолизе наблюдается ряд побочных радиационных эффектов.
Может произойти радиационное окрашивание р оптической
Рис. 6.4. Осциллографическая запись поглощения света
промежуточным соединением. Образец становится про-
зрачным по мере того, как промежуточное соединение
расходуется в какой-то реакции.
системе, а при высоких энергиях — появиться наведенная радио-
активность. Для большей части систем последний эффект пре-
небрежимо мал. Соответствующие меры предосторожности поз-
воляют обеспечить нормальное функционирование оптической
системы.
Иногда в полученные результаты приходится вносить ис-
правления, связанные с необходимостью учета черенковского
излучения.
Кинетические исследования. Если анализируемая реакция
первого порядка
А—->-В, (6.5)
то выражение для скорости реакции выглядит так:
-^- = МА]. (6.6)
Интегрируя в пределах от t=0 [А] = [Ао] до t=t [А]=[А];
получаем:
<6-7>
167
Если считать, что при данной длине волны свет поглощает
только промежуточное соединение А, то
А> = е[Л0] /;
1п £>0/е/
In О/е/
ИЛИ
InPo/D)^/. (6-9)
где Do — оптическая плотность в момент времени t = 0; D—-.оп-
тическая плотность в момент времени t=t.
Таким образом, чтобы найти константу скорости реакции
kr, не обязательно знать коэффициент экстинкции. Для реакции
псевдопервого порядка
ь
Л + С—'+В(С0»>Д,) (6.10)
/гг=^г[Со] и уравнение (6.9) переходит в
-l-ln(D0/D)==^. (6.11)
iVoj
Для бимолекулярной реакции
Д + В->С (6.12)
при условий, что [Д] = 1В] и свет поглощает только соедине-
ние А,
--^L = kr[Af, (6.13)
at
-------L = M (6,i4)
Dt Do e.1 '
Отсюда следует, что для вычисления константы скорости
реакции второго порядка нужно знать значение коэффициента
экстинкции.
Как правило, коэффициенты экстинкции неизвестны, и для
определения констант скорости реакций приходится использо-
вать косвенные методы. Можно, например, определить (кинети-
ческими методами) значение G для появления конкретного про-
межуточного продукта. Этим способом удалось получить хоро-
шие оценки значения G для гидратированного электрона.
Осциллограмму (см. рис. 6.4) получают обычно в координа-
тах оптическая плотность — время. Непосредственно по осцил-
лограмме можно найти скорость накопления или распада про-
дукта в единицах оптической плотности в единицу, времени.
.Так как оптическая плотность прямо пропорциональна концент-
рации продукта, то, зная поглощенную дозу и длину оптиче-
168
ского пути, вычислим произведение eG. Если значение G извест-
но, то можно найти значение коэффициента экстинкции.
Пример 6.4. Анализ осциллограммы эксперимента по импульсному ра-
диолизу позволяет сделать вывод, что скорость образования продукта в среде
с плотностью I г/см3 при мощности дозы I09 рад/сек составляет 36 000 единиц
оптической плотности в секунду. Если длина оптического пути 1,2 см, чему
равно произведение Ge?
Решение.
36000
сек
= в [С] /;
Концентрацию в молях на литр нужно выразить через число молекул.
Тогда е[С], деленное на дозу, даст величину произведения Ge:
11 сек рад
10® рад эрг
100 —
г
л I 36 000 (моль) см | 1 6,02 • 1023 молекул
1000 г] сек (л) |1,2 см моль
1,6- 10—12 эрг „ „„„ „ молекул
X-----------— = Ge = 28 000----—.
эв 100 эв
Для реакций псевдопервого порядка константы скорости
можно определить по стационарной концентрации промежуточ-
ного продукта во время импульса. Если ведутся наблюдения за
реакцией какого-то поглощающего свет продукта, взаимодейст-
вующего с молекулами растворенного вещества, то при констан-
тах скоростей порядка 5-Ю10—1011 Мт1 сек~1 концентрацию
этого растворенного вещества следует поддерживать достаточ-
но низкой, чтобы оставались измеримыми времена полупревра-
щения. Однако при слишком низких концентрациях всегда
существует опасность локального обеднения системы растворен-
ным веществом за время измерения. При очень высоких кон-
центрациях растворенного вещества (ммоль/л) и для быстрых
реакций время полупревращения может снизиться до сотых
долей микросекунды. Для ускорителей с микросекундной дли-
тельностью импульса этого время оказывается малым по срав-
нению с временем импульса. Концентрация промежуточного
продукта быстро достигает стационарного значения. Скорость
выведения промежуточного продукта из системы в результате
реакции с растворенным веществом становится равной скорости
образования промежуточного продукта.
Скорость образования = скорость выведения = k [А] [В]. (6.15)
Для реакции псевдопервого порядка
А-p В-*С, [В] >»[AJ. (6.16)
169
Если А — промежуточный продукт, за поглощением которого
мы следим, то
= Al И] [В]; (6.17)
[Л] = Rplkx [В]. (6.18)
Но [A]=Dx/el, где Dx— равновесная оптическая плотность (оп-
тическая плотность промежуточного продукта в стационарном
состоянии), и
—(—У — (6.19)
р \ dt /о е/ К '
где (dDldt)o — начальная скорость образования промежуточно-
го продукта А в единицах оптической плотности. Поэтому
Для нескольких импульсов определяется значение Отс, а по
начальным наклонам осциллографических кривых находят
(dD/dt)0 в отсутствие вещества В.
Однако, как уже отмечалось, большая часть реакций имеет
второй порядок и, следовательно, часто возникает необходи-
мость находить коэффициенты экстинкции. Определение коэф-
фициентов экстинкции представляет и самостоятельный инте-
рес, так как без этого невозможна полная интерпретация спект-
ра’поглощения. Как показано выше в экспериментах по им-
пульсному радиолизу, непосредственно измеряют произведение
G на коэффициент экстинкции. При определении констант ско-
ростей бимолекулярных реакций основная погрешность, как
правило, вносится за счет неточности значений коэффициента
экстинций.
6.3. ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС
Все вещества, молекулы которых имеют неспаренные элект-
роны, являются парамагнетиками. Из области магнитного поля
с низкой напряженностью парамагнетики втягиваются в об-
ласть с высокой напряженностью. Как правило, электроны в
молекулах спарены, их спины скомпенсированы, и магнитный
момент у таких молекул отсутствует.
Неспаренные электроны обнаруживают либо с-помощью из-
мерений магнитной восприимчивости, либо методом электрон-
ного парамагнитного резонанса (ЭПР). Для уверенных измере-
ний магнитной восприимчивости необходимы сравнительно вы-'
сокие концентрации свободных радикалов. Поэтому ценность
данного метода при изучении свободных радикалов, образую-
щихся в ходе реакций, невысокая. Более пригоден для этой
цели метод электронного парамагнитного резонанса.
170
Магнитный момент и спин неспаренного электрона, кото-
рый не участвует во взаимодействии с каким-либо ядром мо-
лекулы, ориентируются в пространстве случайным образом.
Если приложить к системе постоянное магнитное поле, то спи-
ны и магнитные моменты неспаренных электронов .будут
ориентироваться, либо параллельно, либо антипараллельно при-
ложенному нолю. Электроны с -параллельной ориентацией спи-
лов обладают энергией па 42g$H меньше, а электроны с анти-
параллельной ориентацией будут иметь энергию на 'IzgfyH
больше энергии электронов в нулевом поле. Разность энергии
двух состояний (ДЕ) определяется формулой
АЕ = §₽// = Av. (6.21)
Здесь g — фактор спектроскопического расщепления или гидро-
магнитное отношение для свободного электрона.
Фактор спектроскопического расщепления является мерой
вклада спинового и орбитального движений электрона в полный
момент количества движения. Для свободного электрона g=
= 2,0023*. Значение Н в уравнении (6.21)—напряженность
приложенного магнитного поля. Магнетон Бора — ^-постоянная,
определяющая отношение момента количества движения к маг-
нитному моменту, которая выражена в единицах СГСЭ.
При заданной напряженности магнитного .поля переменное
электромагнитное поле только с одной частотой v может пере-
вести электрон из низшего состояние в высшее. .Обычно в иссле-
дованиях методом электронного парамагнитного резонанса
используют магнитные поля с напряженностью в тысячи эрстед
и радиоволны сверхвысоких частот.
Поглощение электромагнитного излучения можно наблю-
дать, если электроны с нижнего уровня переходят на верхний.
В то же время электроны будут переходить с верхнего уровня
на нижний, что будет сопровождаться испусканием излучения.
В итоге поглощение энергии наблюдается лишь в том случае,
если электронов в нижнем состоянии больше, чем в верхнем.
Для системы, находящейся в тепловом равновесии, дело об-
стоит именно так. В нижнем состоянии электронов больше,
чем в верхнем. Практически распределение неспаренных элект-
ронов между двумя состояниями при тепловом равновесии
описывается выражением
А. = е-дЕ/fer t (6.22)
^2
где Si —число электронов на верхнем уровне; S2 — число элек-
тронов на нижнем уровне; АЕ — разность энергии между двумя
уровнями; k — постоянная Больцмана.
♦ Небольшое отклонение ^-фактора-от значения 2,0000 обусловлено ре-
лятивистской поправкой. — Прим, перев.
171
Для любых двух заданных уровней отношение SJS2 умень-
шается при понижении температуры. Следовательно, нужно
стараться проводить наблюдения ЭПР при низкой температуре.
Дополнительное увеличение чувствительности метода может
быть достигнуто при увеличении •напряженности магнитного
поля и повышении резонансной частоты.
6.3.1. Спектры ЭПР
Механизмы, с помощью которых электроны, находящиеся
на верхнем уровне, теряют энергию и возвращаются в низшее
состояние, называются процессами релаксации. Передача энер-
гии спинов тепловым колебаниям решетки твердого тела как
целого называется спин-решеточным взаимодействием. Эффек-
тивность этого взаимодействия характеризуется временем спин-
решеточной релаксации, т. е. время, за которое первоначаль-
ный избыток энергии, полученный спинами, уменьшается в
е раз. Поэтому сильное спин-решеточное взаимодействие при-
водит к коротким временам релаксации, а слабое — к длинным.
Если орбита неспаренного электрона охватывает атом,
имеющий ядро с магнитным моментом и спином, то в этом
случае происходит взаимодействие ядра с электроном. Магнит-
ный момент ядра создаст небольшое дополнительное поле, ко-
торое либо усилит внешнее поле, либо ослабит его. В результа-
те уровня энергии электрона расщепляются. Например, при
взаимодействии неспаренного электрона с протоном, который
имеет спин ’/2, возможны две ориентации спина протона отно-
сительно приложенного магнитного поля с проекциями па
направление поля ±'/2. Если приложенное магнитное поле
меняется с определенной частотой, можно наблюдать две линии
поглощения: одну при больших, другую при меньших полях,
чем в случае невозмущенпого электрона (рис. 6.5).
Чем больше значение ядерпого спина, тем более заметным
становится расщепление. Для ядра со спином / должно наблю-
даться (21+1) линий поглощения, расположенных на равных
расстояниях друг от друга. Ядерпое взаимодействие, которое
вызывает расщепление одного электронного уровня на несколь-
ко подуровней, называется сверхтонким взаимодействием, а
разделение линии поглощения на компоненты — сверхтонким
расщеплением. Значение этого расщепления может быть приня-
то за меру взаимодействия между ядром и электроном.
В большинстве свободных радикалов неспаренный электрон
взаимодействует сразу с несколькими ядрами, в результате чего
картина сверхтонкого расщепления сильно усложняется, и
спектр становится трудно разрешимым. Однако имеются два
случая, в которых спектр можно легко проанализировать.
1. Взаимодействие с ядром со спином /« значительно боль-
ше взаимодействия с другим ядром со спином Более сильное
172
взаимодействие расщепляет линию поглощения на (2/,-t-l)
хорошо разделенных компонент, в то время как более слабое
взаимодействие вызовет дополнительное расщепление каждой
из этих компонент на (2/^+1) подкомпонент.
2. Неспаренный электрон имеет равную связь с несколькими
одинаковыми ядрами.
Рис. 6.5. Сверхтонкое расщеп-
ление в спектре ЭПР, возни-
кающее при взаимодействии
неспаренного электрона с про-
тоном.
Рис. 6.6. Сверхтонкое рас-
щепление в спектре ЭПР,
возникающее при взаимо-
действии неспарепного элек-
трона с тремя идентичными
протонами.
Если имеется п одинаковых ядер, то линия поглощения рас-
щепляется па (2п/+1) компонент. Поэтому для случая взаимо-
действия неспаренного электрона с тремя протонами (как. на-
пример, в метильном радикале) будет наблюдаться (2-3-'/2+
+ 1) =4 линии поглощения. Интенсивность этих линий различна.
Две центральные линии будут иметь большую интенсивность,
чем симметрично расположенные боковые компоненты. На рис.
6.6 показано сверхтонкое расщепление энергетических .уровней
173
нсспаренного электрона при взаимодействии с тремя эквива-
лентными протонами. Взаимодействие с первым протоном
расщепляет электронный уровень на два уровня ±’/2- Взаимо-
действие со вторым протоном расщепляет кажый из этих
уровней на два подуровня ±7г- Наконец, взаимодействие с
третьим протоном расщепляет каждый из подуровней еще на
два подуровня ± ’/2-
Поскольку предполагается, что связь и взаимодействие с
тремя протонами одинаковы, то подуровень —7г, возникающий
при1 взаимодействии со вторым протоном из уровня + 7г, совпа-
дает с подуровнем + 7г> возникающим при взаимодействии со
вторым протоном из уровня —72- Следовательно, в конце кон-
цов начальная линия поглощения будет расщеплена на четыре
равноотстоящих компоненты с распределением интенсивностей
1 :3:3:1.
Эксперимент по ЭПР обычно проводится при фиксированной
частоте электромагнитного излучения, но с изменяющейся на-
пряженностью магнитного поля. Поэтому расстояния между
энергетическими уровнями выражают в гауссах. Сверхтонкая
структура изолированных атомов Н обнаруживает расщепление
в 504 гс. В то же время расщепление уровней неспаренного элект-
рона в результате взаимодействия с протонами метильного ра-
дикала составляет всего лишь около 25 гс или примерно 5%
расщепления в свободном Н-атоме. Причина этих различий
заключается в том, что в метильном радикале неспаренный
электрон локализован главным образом на углеродном атоме,
тогда как в водороде неспаренные электроны занимают 1 s-ор-
биталь. Таким образом, энергетические характеристики рас-
щепления позволяют получить существенную информацию о
природе неспаренного электрона, участвующего во взаимодей-
ствии.
Хотя мы ограничились лишь анализом взаимодействия не-
спаренных электронов с протонами, во взаимодействиях такого
рода будет участвовать любое ядро, имеющее магнитный мо-
мент. 13С-, 14N-, 17О-, 19F-, 335-изотопы, ядра которых обладают
магнитным моментом и, следовательно, при взаимодействии с
неспаренным электроном дают сверхтонкое расщепление.
В твердых телах, где свобода перемещений ограничена,
спектры ЭПР тех же самых молекул могут быть иными, чем в
жидкости. В твердых телах спектр ЭПР будет зависеть от по-
ложения образца в магнитном поле, так как химические связи*
будут иметь фиксированные направления. Так, например, неспа-
ренный электрон в этильном радикале имеет наиболее стабиль-
ную конфигурацию, в которой валентные связи углерода
СНг-группы направлены не к вершинам тетраэдра, а лежат в
плоскости (гибридизация sp2) и неспаренный электрон нахо-
дится на л-орбитали, перпендикулярной к плоскости. В твердых
телах при определенных ориентациях взаимодействия неспарен-
174
него электрона с протонами СН2- и СНз-групп оказываются
эквивалентными, тогда как в жидкости расщепление, возникаю-
щее за счет взаимодействия с протонами СН2- и СНз-групп,
оказывается различным.
6.3.2. Применение ЭПР-спектроскопии в радиационной химии
Применение ЭПР-спектроскопии для анализа промежуточ-
ных продуктов становится общепринятым методом. Главная
трудность заключается в том, чтобы получить достаточно высо-
кие концентрации радикалов, обеспечивающие хорошее разре-
шение спектров. Хорошего разрешения в ЭПР-спектрах уда-
ется добиться для жидкофазных реакций различных алкильных
радикалов и для многих полимерных реакций в твердом состоя-
нии.
Помимо трудностей, связанных с чувствительностью прибо-
ров, при изучении методом ЭПР промежуточных продуктов
реакций возникает проблема измерения концентрации свобод-
ных радикалов, создающих данный сигнал. Обычно для этого
в резонатор ЭПР-спектрометра помещают стандартный обра-
зец с известной концентрацией неспаренных электронов.
Образец позволяет провести калибровку спектрометра. В ка-
честве ЭПР-стандартов используют несколько веществ, среди
них 2,6-ди-трет-бутил-(3,5-ди-трет-бутил-4-оксо-2,5-циклогексади-
еп-1-илидеп)-/г-толуокси, обычно именуемый гальвиноксилом.
Вначале концентрацию гальвиноксила определяют по спектру
поглощения (коэффициент молярной экстинкции при 4280 А
равен 180 000 М_|-слН) в растворе циклогексана. После этого
изменяют спектр ЭПР стандартного образца и калибруют
спектрометр. Теперь концентрацию неспаренных электронов,
возникающих под действием излучения в исследуемом образце,
можно определить, сравнивая площади под кривой поглощения
в образце и стандарте.
Для изучения кинетики исчезновения радикалов необходи-
мо (как и в импульсном радиолизе) провести точную дозимет-
рию и определить значения G образования радикалов. Скорость
образования радикалов V равна интенсивности излучения D,
умноженной па G:
V = b-G. (6.23)
В стационарном состоянии (при непрерывном облучении)
скорость образования радикалов будет равна скорости их ис-
чезновения. Для реакции между двумя одинаковыми радика-
лами .(например, этильными)
k.
Ra + Ra-^RaRa (6-24)
.175
скорость исчезновения равна
(6.25)
dt 4
и, следовательно,
g-x - = kr. (6.26)
[Яо]2
Для реакций между различными радикалами уравнение (6.26)
перепишется следующим образом:
P(Gg + gfe). = ъ (6.27)
Ситуации, соответствующие уравнению (6.26) или (6.27),
легко различить в эксперименте. В первом случае концентрация
радикалов должна быть пропорциональна квадратному корню
из мощности дозы, во втором для реакции между разными ра-
дикалами их концентрация будет линейно зависеть от D. Зная
интенсивность излучения, значение G и концентрацию радика-
лов, нетрудно определить абсолютную константу скорости реак-
ции.
Метод ЭПР непригоден для изучения быстрых превращений
радикалов. В лучшем случае время разрешения составляет око-
ло 200 мксек. При высоких концентрациях радикалов можно
добиться меньшего времени разрешения, однако при высокой
интенсивности облучения возникают новые ограничения. Боль-
шие токи в резонаторе спектрометра будут приводить к диэлек-
трическим потерям, которые в свою очередь сильно снизят
чувствительность спектрометра, так как минимальное измеряе-
мое значение магнитной проницаемости образца сильно зависит
от добротности резонатора Q (Q-энергия, накопленная в резо-
наторе/рассеянная энергия). Несмотря на указанные ограниче-
ния, метод ЭПР используется для изучения быстрых реакций
в целом ряде систем.
6.4. МАСС-СПЕКТРОМЕТРИЯ
В любом масс-спектрометре* пучок электронов известной
энергии, взаимодействуя с молекулами в газовой фазе, создает
ионы, которые в соответствии с их массой и зарядом разделя-
ются затем во времени или в пространстве.
* Термины «масс-спектрограф» и «масс-спектрометр» часто употребля-
ют как синонимы. Между ними, одиако, существует различие. В масс-
спектрографе ионы различной массы фокусируются в плоскости фотопла-
стинки, на которой создается скрытое изображение. В масс-спектрометре
пучки ионов фокусируются иа коллекторе и затем детектируются электри-
ческим методом.
176
Сейчас в продаже имеется большое число масс-спектромет-
ров различной конструкции. Этим обусловлено их широкое ис-
пользование в лабораторной практике. Области применения и
типы конструкций масс-спектрометров настолько разнообразны,
что далее мы сможем рассмотреть лишь самые основные ва-
рианты конструкции.
6.4.1. Общие сведения
Та часть прибора, в которой образуются ионы, называется
ионным источником. Схема ионного источника приведена на
рис. 6.7. Источником электронов может служить нагретая нить.
Нить обычно изготовлена из
вольфрамовой полоски толщиной
0,002—0,005 см. Электроны вы-
брасываются из пити и ускоря-
ются под действием напряжения,
приложенного между нитью и
пластинами Pi и Рг.
В электронном пучке сущест-
вует небольшой разброс по энер-
гиям, значение которого отчасти
определяется температурой нити.
Этот разброс различными спосо-
бами стараются свести к мини-
муму. Фокусировка пучка элек-
Рис. 6.7. Ионный источник:
1 — нить; 2 — отталкивающий электрод;
3 —ловушка; 4 — иоиы.
тронов осуществляется обычно с помощью магнитного поля, при-
ложенного вдоль направления пучка. В 60°-ных приборах маг-
нитное поле создается с помощью постоянного магнита, позво-
ляющего получить напряженность в несколько сот гаусс.
Через узкие щели в пластинах Р\ и Р% электронный пучок
попадает в ионизационную камеру. В ионизационной камере
электрическое поле отсутствует, так как пластина Р$ имеет
тот же потенциал, что и пластина Р%. Электронный пучок
собирается в ловушке 3.
Исследуемый газ вдувают в ионизационную камеру (см.
рис. 6.7) перпендикулярно к направлению электронного пучка.
Ионы, созданные электронным пучком, под действием неболь-
шой разности потенциалов между пластиной Fi и отталкиваю-
щим электродом Р направляются через щель в пластине Fi в
ускоряющую камеру. Там под действием разности потенциалов
между пластинами Fs и F& обычно постоянной и равной 1000
или 2000 в, ионы ускоряются. Пучок ускоренных ионов допол-
нительно фокусируется с помощью узких щелей, что позволяет
уменьшить сечение пучка и угловой разброс ионов. В случае
необходимости можно установить дополнительные щели.
Выходящий из источника ионный пучок попадает в попе-
речное магнитное поле (рис. 6.8).
177
Кинетическая энергия ионов будет равна eV, где е — заряд
иона; V — ускоряющий потенциал. Ионы с одинаковым зарядом
‘будут иметь одинаковую энергию, однако их импульсы могут
различаться, так как импульс зависит также от массы.
Рис. 6.8. Схема масс-спектрометра:
J — ионный пучок; 2— ускоряющее напряжение; 3—входная Щель; 4 — газ;
5 —источник; 6— детектор; 7 —магнитная призма (анализатор).
Для энергии ионов справедливо равенство:
eV=l/2mV2. (6.28)
В магнитном поле движущийся ион испытывает воздействие
<силы eVH, перпендикулярной к направлению магнитного поля и
направлению движения. В результате ион начинает двигаться
-по круговой орбите, радиус которой определяется равновесием
.центробежной и отклоняющей сил:
еу// = -^
(6.29)
.Здесь Н — напряженность магнитного поля; г — радиус орбиты.
Подставляя уравнение (6.28) в уравнение (6.29), получаем
2тУ
еН2
(6.30)
Отсюда ясно, что, подбирая напряженность магнитного поля
:и ускоряющий потенциал, можно фокусировать в определенной
точке ионы заданной массы.
Ионы, миновавшие магнитный анализатор, проходят затем
через ряд ограничительных щелей к коллектору, где регистри-
руется ионный ток. Ионные, токи в масс-спектрометрах обычно
•очень малы, поэтому приходится конструировать весьма чувст-
вительные детектирующие устройства. После усиления относи-
тельные значения ионных токов записываются на диаграммной
бумаге, где другой координатой выбрана масса ионов.
178
Каждое химическое соединение обладает собственным харак-
теристическим масс-спектром. Это позволяет использовать масс-
спектрсметр в качестве аналитического прибора. Например,,
масс-.спектры изобутана и н-бутапа очень схожи. Однако рас-'
пределение интенсивностей пиков, соответствующих различным
массам, заметно различается, что позволяет масс-спектрометри-
чески идентифицировать эти соединения.
В масс-спектрографах высокое разрешение обеспечивается с
помощью целого ряда устройств. Наиболее распространены при-
боры с двойной фокусировкой. Комбинация электростатиче-
ской и магнитной фокусировки обеспечивает высокое разреше-
ние.
Обычно в масс-спектрометрах поддерживается низкое давле-
ние, примерно 10-6 мм рт. ст. Однако проводились эксперименты
и при давлениях 1—10 мм рт. ст. При высоких давлениях соу-
дарения между ионами чрезвычайно усложняют картину и
затрудняют идентификацию образующихся ионов. Однако эк-
сперименты специально проводят при высоких давлениях в
тех случаях, когда исследуют иои-молекулярные реакции (ион-
молскулярные реакции рассматриваются в разделу 6.5.1 и в.
главе V).
6.4.2. Особые конструкции масс-спектрометров
Существует несколько специальных конструкций масс-спект-
рометров: квадрупольный; омегатрон; прибор, основанный hq
принципе совпадений, и масс-спектрометр, в котором измеря-
ются времена пролета ионов. Последний тип приборов
особенно ценен при изучении
образования и распада корот-
коживущих продуктов в газо-
фазных реакциях, таких, как
реакции, инициируемые удар-
ными волнами, или некоторые
Рис. 6.9. Схема масс-спектрометра по
времени пролета:
1 — ионизационная камера: 2 — источник
электронов; 3 — сетки; 4 — ионный пучок;
5 — дрейфовое пространство; 6 — детектор.
радиационно-химические реак-
ции. Время отбора ионов в
этих приборах составляет де-
сятые доли микросекунды, а
полный спектр регистрируется
в течение 10—20 мксек.
Характерные детали конструкции масс-спектрометра, осно-
ванного на измерении времени пролета ионов, показаны на
рис. 6.9. Импульсный электронный пучок проходит в иониза-
ционную камеру, создавая сгусток ионов. Эти ионы выбрасы-
ваются из ионизационной камеры с помощью электрического
поля, действующего в течение нескольких микросекунд. Поле
направляет ионы к сеткам, и затем они ускоряются под дейст-
вием постоянного поля. После этого сгусток ионов входит в
179
дрейфовое пространство и движется пр направлению к детек-
тору.
В том случае, если начальная кинетическая энергия ионов
равна пулю, время пролета дается формулой
_ f т \*/г
\ 2е (аУг + bV2) )
где е — заряд, а У3_и Кг— потенциалы, под действием которых
ускоряются ионы; К — константа, зависящая от конструкции
прибора и условий эксперимента.
В реальном эксперименте существует определенный раз-
брос ионов по кинетическим энергиям, что приводит также к
разбросу по временам пролета. Влияние этого эффекта умень-
шают за счет сокращения периода ускорения. Большую часть
времени пролета ионы движутся с постоянной скоростью, кото-
рую они успели набрать за время ускорения.
Для радиационных химиков, исследующих явления элект-
ронного удара или реакции ионов в газовой фазе, масс-спект-
рометр оказывается важнейшим инструментом исследования.
Помимо определения массы ионов, возникающих при соударе-
ниях с электронами, можно изучать разнообразнейшие ионные
реакции, такие, как мопомолекулярпый распад, взаимодействие
с другими ионами и молекулами. Кроме того, анализ явлений,
происходящих при электронном ударе, сам по себе представля-
ет важнейшую область исследований.
6.5. ПРИЛОЖЕНИЯ МАСС-СПЕКТРОМЕТРИИ
6.5.1. Потенциалы ионизации и потенциалы появления
Потенциалом появления (ПП) называют минимальную
энергию, необходимую для образования данного ядра (и со-
путствующих незаряженных молекулярных фрагментов, если
таковые должны возникнуть) из данной молекулы, иона или
радикала. Соединения, вступающие в реакцию, и продукты
реакции должны находиться в основном состоянии и не иметь
избыточной кинетической энергии. Потенциал ионизации —
частный случай потенциала появления.
Потенциалом ионизации (ПИ) называют минимальную
энергию, необходимую для удаления электрона из нейтральной
молекулы, атома или радикала (в основном состоянии) и об-
разования атомного или молекулярного иона (в основном со-
стоянии).
Истинным, или адиабатическим, потенциалом ионизации
(первым ионизационным потенциалом) называют разность
энергий между основным колебательным уровнем наиболее
низкого электронного состояния молекулы и основным колеба-
тельным уровнем молекулярного иона.
ISO
Энергия электронов
Рис. 6.10. Кривые ионного
тока для ионов СН£“ и Кг+.
Энергия электронов дана без учета
поправок.
Потенциалы ионизации помимо масс-слектрометрического
метода можно также определять спектроскопически или фото-
ионизационным методом. В этом методе ионы, возникающие под
действием излучения от источника света, и идентифицируются
с помощью анализатора масс-спектрометра. Потенциалы иони-
зации, определенные методом элек-
тронного удара, часто оказываются
выше потенциалов ионизации, опре-
деленных спектроскопически. Если
минимумы на кривых потенциаль-
ной энергии для молекулы и моле-
кулярного иона не приходятся па
то же самое межатомное расстоя-
ние, то в общем случае переходы
не будут осуществляться между
основными колебательными уровня-
ми и, следовательно, окажутся не-
адиабатическими. Эта ситуация воз-
никает даже для сравнительно не-
больших смещений кривых потен-
циальной энергии.
Потенциалы появления (при электронном ударе) обычно
определяют по кривым ионного тока. Эти кривые получают,
откладывая на графике зависимость ионного тока от энергии
электронного пучка. Примеры таких кривых приведены па
рис. 6.10. Для определения потенциалов ионизации по этим
кривым существует целый ряд приемов, ни один из которых
не оказывается полностью удовлетворительным. Потенциалы
ионизации для конкретной молекулы можно определить, зная
потенциалы появления и имея достаточную информацию о про-
исходящих процессах.
Потенциал появления иона М+ можно выразить через тер-
модинамические величины (при условии, что ни одна из частиц
не песет избытка кинетической энергии):
ПП (М+) = АДу (М+) + 2ДЯ, (Nz) — ЬНf (М\ (6.32)
где SA/7/(N,-)—сумма стандартных теплот образования нейт-
ральных фрагментов; ДД/(М+)—стандартная теплота образо-
вания родительского иона; Д/УДМ)—стандартная теплота об-
разования родительской молекулы.
Зная потенциал появления и стандартные теплоты образо-
вания родительской молекулы и нейтральных фрагментов (ес-
ли они образуются), можно найти стандартную теплоту обра-
зования родительского иона и отсюда потенциал ионизации:
М + е- -> М+ + 2е- ДЯ = ПИ; (6.33)
ПИ (М+) = ДЯу (М) — ДЯу (М+). (6.34)
181
Интепретация кривых ионного тока облегчается при срав-
нении с кривыми ионного тока для калибровочного газа, тако-
го, например, как криптон, потенциал ионизации которого
измерен с высокой точностью при помощи других методов.
6.5.2. Ион-молекулярные реакции
Ион-молекулярные реакции изучают, работая с масс-спек-
трометром в таком режиме, при котором обеспечиваются бимо-
лекулярные соударения образующихся первичных ионов с нейт-
ральными молекулами. Для этого необходимо поддерживать
в ионном источнике сравнительно высокие давления (выше
10-4 мм рт. ст.).
Первичные ионы, возникающие в ионном источнике, там же
в источнике претерпевают соударения с нейтральными молеку-
лами газа. В результате происходящих реакций образуются
вторичные ионы. Например,
CHt -|- СН4 CHt + СН3 (6.35}
или в более общем случае
Х+ + YH ХН+ + Y. (6.36)
Вторичный ионный ток (ток ионов ХН+) равен концент-
рации первичных ионов (ip), умноженной на концентрацию
нейтральных молекул в источнике (п), на расстояние d, прохо-
димое первичными ионами от места образования до выходной
щели, и на сечение реакции Q:
is ~ ip nQd. (6-37}
Считая, что Q обратно пропорционально корню квадрат-
ному из градиента потенциала отталкивающего электрода Vr
(это допущение будет рассмотрено в главе VII) и не зависит
от температуры газа в источнике, получим:
« = (6.38>
где В — константа.
Кинетическая энергия (КЭ) первичных ионов равна произ-
ведению заряда е на расстояние d, пройденное первичными
ионами, и на Vr:
КЭ = —mV*= deVr; (6.39)
= (6-40>
182
Подставляя выражение для Vr в уравнение (6.38) и ком-
бинируя его с уравнением (6.37), получаем:
is = i ±пВ(-^~V'*. (6.41)
s р V \ 2de J '
Вторичный ионный ток is непосредственно связан со ско-
ростью образования вторичных ионов [по реакции (6.36)]. Если
расстояние d поделить на скорость первичных ионов, то мы
найдем время, в течение которого первичные ионы находятся
в источнике. Это время, умноженное на ток первичных ионов,
равно концентрации первичных ионов в источнике [Х+].
Поэтому
G = = [Х+] [nJ В (—V'' (6.42)
dt \ 2de J
Так как В — константа и другие сомножители не меняются
при фиксированных условиях опыта, уравнение (6.42) можно
переписать в виде
-(^=fef[X+] [пЬ (6.43)
at
где kr — константа скорости бимолекулярной реакции (6.36).
Приведенные рассуждения, хотя и недостаточно строгие,
дают качественное представление о том, как можно найти
константу скорости реакции. Если пользоваться значениями,
непосредственно измеряемыми в масс-спектрометре, то урав-
нение (6.41) можно переписать применительно к реак-
ции (6.36):
= kr [Х+] Rt [ХН+], (6.44)
*х+
где Rt — время пребывания в ионном источнике частиц. Пер-
вичный й вторичный ионные токи, так же как и концентра-
ция молекул газа, измеряются в масс-спектрометре непосред-
ственно. Время пребывания вторичных ионов в источнике можно
вычислить, зная размеры ионного источника, градиент напря-
жения и массу ионов.
Определение констант скоростей ион-молекулярных реак-
ций— одна из важных задач радиационной химии. Высокие
значения констант, от 10-9 до 10~10 см3!(молекула • сек), пока-
зывают, насколько легко протекают все стадии этих реакций.
С помощью масс-спектрометра можно исследовать ион-мо-
лекулярные реакции разного характера. Сложные бимолеку-
лярные реакции, как, например,
СН| + СН4 -> С2Н| + Н2; (6.45)
183
С2нг + С2Н4 -> С3Щ + СН3;
C2Hf + С2Н4 с4нт + н,
и простые реакции мопомолекулярного распада,
CH3CN+->CHt + CN;
сХ->с2н^ + н2,
(6.46)
(6.47>
такие, как
(6.48)
(6.49)
уже изучены весьма подробно. Масс-спектрометр оказался
чрезвычайно полезным при определении энергии диссоциации
свободных радикалов и при изучении кинетики диссоциации
ионов.
Для изучения вторичных и более высоких порядков ионных
реакций, а также процессов образования сгустков ионов скон-
струированы специальные масс-спектрометры высокого давле-
ния, в которых применяются протонные пучки, пучки а-частиц.
и фотоионизационные источники [3, 4].
6.6. ОТРИЦАТЕЛЬНЫЕ ИОНЫ
Масс-спектрометр можно использовать для изучения масс-
спектров и потенциалов появления отрицательных ионов. От-
рицательные ионы могут образовываться:
в реакциях резонансного захвата электрона
М + е-^М-; (6.50)
SFe -г е~ -> SF7; (6.50а)
в реакциях присоединения электрона с последующей диссо-
циацией
АВ + ₽~А + В~; (6.51)
C2N2 Ч- е- -> CN + CN- ; (6.51а)
в реакциях, ведущих к образованию пары ионов,
АВ + е- -> А+ + В- -J- е-; (6.52)
NO + е~ -> N+ + О~ -г е-. (6.52а)
Глава VII
РАДИАЦИОННАЯ ХИМИЯ ГАЗОВ
7.1. ВВЕДЕНИЕ
В развитии радиационной химии газовых систем можно вы-
делить три периода. В течение первого доминирующую роль
играла теория «ионных ассоциаций» С. К. Линда. Постепенно
по мере выявления роли свободных радикалов в радиационно-
химических реакциях теория Линда отходила па задний план.
Третий период наступил, когда выяснилось, что эксперимен-
тально измеренные константы скорости ион-молекулярных
реакций чрезвычайно высоки. Поэтому большее значение стали
придавать реакциям неассоциированных ионов по сравнению
со свободнорадикальными реакциями.
Новейшие исследования показали, что при анализе кине-
тики радиационно-химических превращений в газах следует
учитывать в той или иной степени все три ранее изученных
механизма плюс различные процессы передачи энергии.
Радиационная химия газов — весьма сложная область, что,
впрочем, можно сказать и о большей части других систем, изу-
чаемых радиационной химией. Детальный анализ конкретных
газовых реакций выходит за рамки этой книги. Основное вни-
мание будет уделено характеристикам, общим для всех реак-
ций в газах под действием излучения. Как правило, имеется
лишь качественная информация, и поэтому развиваемые здесь
представления о механизмах целого ряда процессов следует
рассматривать как предварительные.
7.2. ТЕОРИЯ ИОННЫХ АССОЦИАЦИИ ЛИНДА
Первый обзор процессов, происходящих в газах под действи-
ем а-частиц и электронов, опубликовал Линд в 1921 г. Затем
эта книга была переработана в 1928 г.,, а позднее в 1961 г.
На протяжении многих лет книга Линда оставалась основной
сводкой всех сведений о радиационной химии газов.
Теория ионных ассоциаций призвана была объяснить наб-
людавшиеся в ряде случаев ионные выходы, превышавшие 1.
185
Линд предположил, что возникающие при облучении ионы
индуцируют в соседних молекулах электрические дипольные
моменты, взаимодействие которых с родительским ионом при-
водит к образованию ионной ассоциации (рис. 7.1)
При нейтрализации родительского иона выделяется энер-
гия, достаточная для того, чтобы между ионом и нейтраль-
ными молекулами, входящими в ассо-
циацию, прошли химические реакции.
Например, для радиолиза C2N2 под
действием а-частиц был измерен ион-
ный выход M.JN чуть больше 7. Основ-
ной продукт радиолиза — твердый по-
лимер.
Предполагается, что он возникает в
результате следующей реакции:
и (C2N2) av-(C2N2'„твердый (П = 7). (7.1)
В этом случае ионная ассоциация в
Рис. 7.1. Пример ионной среднем должна состоять из 6 моле-
ассоциации. кул, группирующихся вокруг централь-
ного иона. При нейтрализации иона
выделяется достаточная энергия для полимеризации всех участ-
вующих в ассоциации частиц.
В другом случае для радиационной полимеризации ацети-
лена обнаружен ионный выход*, равный примерно 21. Единст-
венным продуктом реакции оказывается твердый полимер
купрен. Согласно теории ионных ассоциаций центральный аце-
тиленовый ион должен быть окружен агрегатом примерно
из 20 нейтральных молекул ацетилена.
Однако было известно несколько реакций, для которых
теория ионных ассоциаций не соответствовала эксперименталь-
ным фактам. Конверсия параводорода в ортоводород происхо-
дит с ионным выходом > 1000 и хорошо объясняется с по-
мощью цепного механизма, в котором роль активных частиц
играли атомы Н. Ионный выход для распада, озона под дей-
ствием а-частиц оказался равным 5—15-Ю3 (в зависимости от
давления в системе). Такие высокие значения М/N также под-
разумевали цепной механизм.
Несмотря на экспериментальные подтверждения важной
роли свободных радикалов в рассмотренных и сходных с ними
системах, теория ионных ассоциаций оставалась в ходу вплоть
до конца 40-х годов. Пересмотр механизма радиолиза ацети-
лена в 1948 г. (1] привел к дальнейшему существенному ослаб-
лению позиций теории ионных ассоциаций. Подробный анализ
соединений, возникающих при радиолизе ацетилена, показал.
* Ионным выходом М/N называют отношение числа измененных молекул
М к числу образовавшихся пар ионов N. — Прим, перев.
186
что купрен составляет лишь 80% образовавшегося продукта,
а остальные 20% составляет бензол. Согласно теории ионных
ассоциаций следовало бы предположить, что образуется два
типа агрегатов. Один — состоящий из трех молекул ацетилена
и второй — из 18 молекул. Наблюдаемый при этом ионный
выход М/N окажется равным 21. Но так каю 20% ацетилена
расходуется на образование бензола, то М/N (по превращению
С2Н2) будет равен 0,2-21=4,2, а не 3, как предсказывает
теория ионных ассоциаций. Наблюдаемый выход продуктов
удалось прекрасно объяснить, используя представления о сво-
боднорадикальном механизме радиолиза [2]:
C2H2av-C2H + H; (7.2)
СгН + 2С2Н2^С6Н5; (7.3)
С6Н5 циклизация (Ф-); (7.4)
ф. + ОД^ед + СгН; (7.5)
СвН5 (или Ф-) + nC2H2-> R'; (7.6)
СвН5 (или Ф-) + R' полимер. (7.7)
Тем не менее известно, что ионные ассоциации в газах
образуются. Подвижность ионов в газах, рассчитанная теорети-
чески, оказалась в 3—5 раз выше наблюдаемых значений, что
можно объяснить образованием ассоциаций ионов с нейтраль-
ными молекулами. Размеры агрегатов зависят от давления
газа, поляризуемости молекул и характера межмолекулярпых
взаимодействий. В водяных парах, например, ионы инертных
газов образуют ассоциации, в которые входит до шести моле-,
кул воды. Недавно удалось идентифицировать [3] такие ионные
ассоциации, как
NHt(NH3)4-20 и Н8О+(Н2О),_18.
Однако конечная судьба ассоциаций после нейтрализации
родительского иона остается невыясненной. Вряд ли вторич-
ные межмолекулярные взаимодействия смогут удержать моле-
кулы в ассоциации при нейтрализации центрального иона.
Образование агрегатов может. препятствовать завершению
реакций, которые протекают в их отсутствие. Маги и Фуна-
баши [4] пересмотрели роль ионных ассоциаций в радиацион-
ной химии газов и указали, что крупные агрегаты могут обра-
зовываться лишь при давлениях, близких к давлению насы-
щения. Кроме того,- они отмечают: «Благодаря образованию
ассоциаций существует интересная возможность индуциро-
вания химических реакций минорных компонент смеси». Под
минорными имеются в виду компоненты, относительная кон-
187
центрация которых в системе мала. Избирательное включение
таких компонент в ассоциации может способствовать-реакциям
с их участием. Большей частью, однако, радиационно-индуци-
ровапные реакции примесных компонент связывают с процес-
сами передачи заряда или энергии возбуждения, либо с обра-
зованием гидридных ионов.
Масс-спектрометрический анализ (5] позволил обнаружить
существование таких ионных комплексов, как (С4Н1о1г) +
и (С6Н1412) + которые образуются в реакциях
(С2Н51)+ + С2Н51-4С4ВД+; (7.8)
(C3H7I)+ + C3H7I->(CeH14I2)+. (7.8а)
Подобные комплексы образуются в результате так назы-
ваемых липких соударений [6] и обладают периодом полурас-
пада, заметно превышающим 10“12 сек. Для того чтобы прои-
зошло липкое соударение, энергия пары сталкивающихся
частиц должна перераспредилиться по всему комплексу. Время
жизни комплекса прямо пропорционально энергии связи комп-
лекса и числу колебательных степеней свободы. Относительная
длительность существования комплекса зависит от конкурен-
ции таких процессов, как нейтрализация заряда и внутренняя
конверсия. Рассмотренные комплексы могут стабилизиро-
ваться при соударениях и вступать в другие реакции.
7.3. ЗАКЛЮЧИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
О РОЛИ ИОННЫХ АССОЦИАЦИЙ
В газах существуют ионные ассоциации. Появление ион-
ных ассоциаций наиболее вероятно при давлениях насыщения
в системах, содержащих молекулы с высокой поляризуемостью.
Однако доказательства того, что нейтрализация центрального
иона в ассоциации вызывает химические реакции так, как это
предполагается в теории Линда, отсутствуют.
В ряде систем удалось непосредственно идентифицировать
ионные комплексы, однако характер реакций этих комплексов
все еще не ясен.
7.4. РЕАКЦИИ ИОНОВ. ИОН-МОЛЕКУЛЯРНЫЕ РЕАКЦИИ
В течение десятилетия, последовавшего за выходом в свет
переработанной монографии Линда, накапливавшиеся экспери-
ментальные и теоретические результаты заставили перенести
основное внимание с ионных ассоциаций на свободные радика-
лы как главную разновидность активных промежуточных
продуктов. Выдающимся теоретическим достижением была
концепция Эйринга, Хиршфельдера и Тейлора (7], согласно
которой свободные радикалы возникают в пезультате ион-мо-
188
лекулярных реакций. Эти радикалы стали считать основными
промежуточными соединениями, ответственными за радиоли-
тическое разложение газов.
7.4.1. Теория Эйринга — Хиршфельдера — Тэйлора
Чтобы раскрыть механизм конверсии параводорода в ортово-
дород при облучении а-частицами, были проведены подробные-
исследования этого процесса. Масс-спектральпые измерения
показали, что в системе присутствуют ионы Н^, Н+ и .
При более высоких давлениях отмечалось преобладание ионов
Н+. Образование атомов водорода может идти несколькими1
путями — в результате прямой диссоциации (из возбужденных
состояний), при нейтрализации ионов и, наконец, в резуль-
тате вторичных реакций
Ht + Н2 = Ht + Н; (7.9)
Ht + Н2 = Н4- + Н + Н2. (7.10)-
Реакция (7.10) требует энергии активации больше 2,5 эв,
поэтому ее вероятность невысока. Реакция (7.9) идет с чрез-
вычайно высокой скоростью и является наиболее вероятным
источником атомов Н. Для расчета скорости реакции (7.9)
Эйринг, Хиршфельдер и Тэйлор использовали теорию активи-
рованного комплекса, в соответствии с которой процесс сле-
дует представить таким образом:
Ht -р Н2 Ht Ht + Н. (7.11)»
Здесь Н+ — активированный комплекс.
Принимая трансмиссионный коэффициент* равным единице,,
получаем:
kr=.^~^, (7.12)
где k — постоянная Больцмана; kr — константа скорости .реак-
ции; h — постоянная Планка; К* — константа равновесия для
обратимой реакции превращения исходных соединений в акти-
вированный комплекс.
Константу равновесия можно переписать следующим
образом:
К* =
НТ
______1__е-л.1:0/нт
112 Hg
(7.13)
* Трансмиссионный коэффициент указывает долю комплексов, превра-
щающихся в конечный продукт. Числовое значение его, очевидно, лежит
между 0 и Г. Обычно трансмиссионный коэффициент принимают равным 1.—
Прим, перев.
189*
где f*£ — полная сумма по состояниям на единицу объема
активированного комплекса; / и + — полные суммы по
состояниям на единицу объема исходных соединений; ДЕ0—
энергия активации реакции при температуре абсолютного нуля.
Энергия активации равна разности энергий реагирующих
веществ и переходного комплекса:
Для реакции (7.11) константа скорости будет равна:
f*
ЪТ '
& = JzL_________4 e-A£f//?r
r h f f ,
'll. 'h+
(7-15)
При оценке полной суммы по состояниям активированного
комплекса примем, что молекула Н2 и ион в комплексе
достаточно разделены, чтобы считать колебательные и вра-
щательные суммы по состояниям теми .же, что и у свободных
молекулы и иона. Тогда в выражении для константы скорости
эти суммы по состояниям сократятся и останутся только
суммы по состояниям поступательного движения:
kr = — .----------------------------[1 + 2 (27 + 1) е-д₽о/^] (7.16)
h (2лтНг (2птн+кТУ'‘
№ №
ИЛИ
(2л6Т)’/«
[1 -|-S(2/+ 1)е-д£о/лг|.
(7.16а)
‘Сомножитель (1+2(27+1) е AF°ft7) представляет собой
вращательную сумму по состояниям самого комплекса. Враще-
ние комплекса приводит к возникновению центробежной силы,
противодействующей поляризационным силам притяжения,
скрепляющим комплекс. Тогда энергия активации определя-
ется разностью вращательной энергии комплекса E*rot и поля-
ризационной энергии E*Poi, создаваемой взаимодействием мо-
лекулы и иона:
ДЕ0
_ р* . р*
— ^-rot *-ро1 ,
ае2
(7.17)
(7-18)
Е* 1
190
где е — заряд электрона; а — поляризуемость; г — расстояние-
между центрами частиц в комплексе;
^rot —’
8л2р.г2
(7.19>
где ц (приведенная масса системы) =-------5
т иг ,
Н2 И2+
I — ротационное квантовое число.
Вычитая уравнение (7.18) из уравнения (7.19), получаемЕ1
выражение для энергии активации. Продифференцировав это-
выражение по г и приравняв его нулю, найдем максимальное
значение г. Пользуясь этим значением г, получим для энергии
активации следующее уравнение:
128 л4 (тн)2 ае2 ‘ (7.20)
Подставляя уравнение (7.20) для энергии активации в уравне-
ние (7.16а) и заменяя суммирование интегрированием, полу-
чаем для последнего члена в уравнении (7.16а): .
f ₽' + . (7.21)*
о
Подставляя уравнение (7.21) в уравнение (7.16а), находим:
. kT
Rr — ---
Т h
т , \’/г ,,
Н4 \ (2ла/*’7’) '« 4л2 (<пн) е
mHi т . J (2лкТ)г/г /г2
2 ' ' ft»
(7.22>
или
( тн+ \/а
kr = I------— I 2птн(а),/‘ е.
\ 2 /
Для водорода а = 0,8-10~24 см3, е — 4,8-10~10 ед. СГСЭ, тн —
= 1,673-10-24 г;
6,68-10~24 13/г 1
11,16-10-« J г1*
1,67-10-24 г\
6,28-----------) .(О.в-Ю-24) см/‘ X.
молекула J
X (4,8-10-w ед. СГСЭ)
10_8 см3
kr =2,66- ------------- = 1,6-101Б см3/(моль-сек),
молекула-сек
19t
Найденное значение константы скорости реакции (7.9)
можно теперь сравнить с известными константами скоростей
для быстрых газофазных реакций:
М -j- Н + Н -> Н2 + М; kr — 3 • 1013 см\моль еек>‘,
СН3 -г СН3 -> C2He; kr = 3,8 1013 см3/{моль • сек\
Константа скорости иоп-молекулярной реакции (7.9)
оказывается почти на два порядка больше, чем константы ско-
ростей наиболее быстрых реакций нейтральных частиц в газо-
вой фазе.
После тщательного учета эффекта взаимодействия со стен-
ками, размеров сосуда, скоростей элементарных реакций и пр.
Эйрипг, Хиршфельдер и Тэйлор [7] сумели рассчитать ионный
выход М/N, прекрасно согласующийся с экспериментальными
данными. Основная цель их работы состояла в доказатель-
стве того факта, что активные свободные радикалы появля-
ются в результате ион-молекулярных реакций, скорость кото-
рых заметно превосходит скорости любых реакций, известных
к тому времени.
Сейчас, принято считать, что реакции обмена протекают,
вероятно, главным образом по ионно-цепному механизму (см.
раздел 7.9.2), а свободнорадикальные механизмы (такие, на-
пример, как Н + Н2->-Н2+Н, анализируемый методом дейтеро-
обмена) вносят, по-видимому, небольшой вклад.
Несмотря на успех расчетов Эйринга, Хиршфельдера
и Тэйлора, объяснивших обменные реакции под действием
a-излучения, прошло почти 20 лет, и должны быть эксперимен-
тальные свидетельства [8] высоких скоростей ион-молекуляр-
ных реакций, подтверждающие расчеты, прежде чем роль
ионов и ионных реакций в процессах газофазного радиолиза
была оценена по достоинству.
7.4.2. Определение скоростей ион-молекулярных реакций
масс-спектрометрическим методом
Уравнение (6.44) описывает ион-молекулярную реакцию
-с помощью величин, непосредственно измеряемых в масс-спек-
трометре:
= kr [X] Rt (7.23)
где [X] — концентрация нейтральных молекул. Для реакции
Х+ + YH = ХН+ + Y (7.23а)
•192
время пребывания иона в ионном источнике дается выраже-
нием
(7.24)
где S — расстояние от электронного пучка до выходной щели;
mi — масса первичного иона; е — заряд электрона; Rv — гради-
ент потенциала отталкивающего электрода в источнике.
Концентрацию нейтральных молекул в источнике можно
определить, найдя полное число ионов, собираемых на оттал-
кивающем электроде, если к нему приложен отрицательный
потенциал, и зная величину сечения ионизации для данного
газа.
Пример 7.1. Рассчитайте константу скорости реакции
сн+ч-сн, >сн++сн8,
если концентрация СН4 в ионном источнике равнр 2,5 -10" молекулам3;
сн+
S = 0,2 см и Ro = 12,5 ejcM, а —-= 0,68 • 10~3.
Решение.
0,68 • 10~3 см3
2,5 • 10“ молекул
(2) (0,2 сл<) (2,67 • 1Q—23 г)___
ед. СГСЭ \ f в \
4,8- 10-ю--------- ) / 12,5--- ] (молекул)
молекул J \ см j
Разделив правую часть на 300,
к ед. СЭ, получим:
чтобы перейти от напряжения в вольтах
0,68 • Ю-з слс3 г 1,068 10—23 г • см? • 300з*/2
_ ь .
2,5- 10“ молекул----------------' см3
а 60-10-ю г-------
сек2
10—8 см3
молекул сек
Таким образом, ион-молекулярные реакции могут наблю-
даться и измеряться в масс-спектрометре, работающем при вы-
соком давлении. Тот факт, что та или иная реакция не наблю-
дается с помощью этого метода, свидетельствует лишь о том,
что верхний предел константы скорости реакции лежит где-то
около 10~п см5/(молекула-сек).
Возможность данной иоп-молекулярной реакции можно
определить термодинамическим методом. Необходимым, хотя и
7 Зак. 907
193
недостаточным условием протекания ион-молекулярной реакции
в замкнутой системе является ее экзотермичность.
Пример 7.2. Найдите ДЯ для реакции
С»Н+ + СН4 - изо-СзН^ Н,.
Решение.
ДЯ/ (продуктов)—ДЯ/ (исходных веществ) =ДЯ (реакции). Для ион-
молекулярпой реакции ДЯ<0. Теплоту образования для иона С2Н5+ находят
из следующего уравнения:
е~~ + С2Нв - С2Н± + Н + 2е~.
Потенциал появления (ПП) этильного иона равен 12,88 эв=297,23 ккал;
[ДЯу(С2Щ) + ДЯу (Н)] — ДДДСаНб) = 297,23 ккал;
&Hf (С,11±) --- 297,23—52,1 —20,1 == 225 ккал.
Точно так же для иона изо-СзН7+ справедливо:
С3Н8 >- изо-С3Н^!” J- Н 4- е~;
ПП -= 11,58 эв=206,9 ккал;
ДЯ/(С3н+)= 190 ккал.
Теперь запишем общее уравнение:
bHf (С3Н+) + ДЯ, (Н2) + ДЯ,(С2н+) - ДЯ, (СН4) = ДЯ;
ДЯ = (190) + (0) — (225) — (—17,88);
ДЯ •— 17,1 ккал.
Теплоты образования ионов можно также найти по вели-
чине сродства к протону. Сродством молекулы к протону на-
зывают взятое с обратным знаком изменение энтальпии в ре-
акции
М + Н+-> МН+— (А//)
-(ДЯ) = сродство к протону = ДЯ^(Н+) + АЯу(М) — (МН+) .
Значения сродства к протону для некоторых молекул приведе-
ны в табл. 7.1.
Таблица 7.1
Сродство к протону
Вещество Сродство к протону, эв Вещество Сродство к протону, эв Вещество Сродство к протону, эв
Н2 3,0 С6нв 6,3 Н2О 8,2 .
сн 5,1 Fe 3,7 СН3ОН 9,1
сн2 5,2 Cl 5,4 NH3 9,15
сн3 , 5,1 Вг 5,8 CH3NH-> 9,35
сн4 5,4 I 6,3 (CH3)2NH 9,3
с2н4 6,8 О 4,8 H2S 7,6
С3Н6 6,6 он 6,2
194
7.4.3. Теория ион-молекулярных реакций
Для констант скоростей ион-молекулярных реакций Жиомуа
и Стивенсон [9] получили* следующее выражение:
k (7.25)
\ 2m. J
где Q — сечение реакции (7.23а), которое описывается уравне-
нием
' 4л2е2а \‘/г Г 2mt Т/г
. р J L e7?aS .1
(7.26)
»] — вероятность того, что соударение вызовет реакцию; а —
поляризуемость; р— приведенная масса; т.\ — масса первич-
ного нона.
Уравнение (7.25) справедливо в том случае, если Q обрат-
но пропорционально корню квадратному из градиента потен-
циала отталкивающего электрода.
В общем случае для достаточно сложных молекул экспери-
ментально измеренные сечения реакций не являются столь
простой функцией Rv. Это связано, по-видимому, с тем слож-
ным влиянием, которое внутренние координаты реагирующих
соединений оказывают на ход реакции. Однако для очень
многих реакций зависимость (/?е)-1/2 хорошо выполняется.
Уравнение (7.26) показывает также, что Q не зависит от тем-
пературы. Этот факт и обратно пропорциональная зависимость
Q от Rv демонстрируются данными табл. 7.2.
Таблица 7.2*
Влияние температуры на сечение ион-молекулярной реакции
Температура, О Q, Л2
Rv~- 2,57 в!см Rv=5. 14 в/см /^==1 о, 3 в/см
333 181 130 97,5
377 179 133 97,0
425 177 132 96,7
537 176 131 98,5
614 185 140 108
* Данные из статьи: Stevenson D. Р., Shlssler D. О. J. Chem. Phys., 1958, у. 29,
р. 2822.
Приведены сечения для реакции Аг+(Кг+ или D+)+H2(D2
или HD) -> АгН(КгН+ или DH+) +Н (или D).
* В лучшем случае эта теория дает лишь порядок константы скорости
реакции. Более полное обсуждение проблемы проведено в работе Хей-
тмана [10].
7* 195
Подставляя уравнение (7.26) в (7.25)
ным единице, получаем
и принимая т] рав-
(7.27)
В примере 7.3 будет показано, что уравнение (7.27) иден-
тично уравнению (7.22). Следовательно, расчет Эйринга, Хир-
шфельдера и Тэйлора не только дает правильный порядок
значения для константы скорости конкретной иоп-молекуляр-
ной реакции, но и предсказывает правильную зависимость от
массы реагирующих компонент и от температуры. Следует
помнить, однако, что ни теория «активированного комплекса»,
ни рассмотренная в этом параграфе теория не дают полного
и адекватного описания всех ион-молекулярных реакций.
Пример 7.3. Доказать, что уравнение (7.27) эквивалентно' уравнению
(7.22), если т) = 1.
Решение. Анализируя выражение для приведенной массы в уравне-
нии (7.22), отмечаем, что
"Чтн+' mH,mH+
2 2
тн, + тн+~ тн+
mimo
mi + m2
что и требовалось доказать.
Характеристические скорости реакций для большей части
иоп-молекулярпых процессов настолько велики, что тепловая
активация либо вообще не нужна, либо оказывает ничтожное
воздействие. Это подтверждается экспериментальными дан-
ными, приведенными в табл. 7.2. Ряд теоретических сообра-
жений [11] также приводит к этому результату.
Уравнение (7.25) описывает константу скорости бимоле-
кулярной реакции между ионами и молекулами при максвел-
196
ловском распределении по скоростям. Подстановка в уравне-
ние (7.25) соответствующих числовых значений позволяет
найти сечение реакции, как это показано bi следующем примере.
Пример 7.4. Используя данные примера 7.1, найдите сечение реакции
СН.+ + СН4 г сн5г + СН3.
Решение. Запишем уравнение (7.25)
Используем численные данные примера 7.1:
си3 / 4,8-10~10-12,5 в-0,2 см
3,8-10—»------------= I-------11-----------------------
молекул сек \ молекул- 2 - 2,67 • 10~23 г-см-332
Отсюда после соответствующих выкладок:
Q-=61,7 ----—--
молекула
7А.4. Роль ион-молекулярных реакций
в радиационной химии газов
Для большей часто ион-молекулярных реакций эксперимен-
тально измеренные константы скорости оказались чрезвычайно
высокими. Расчеты Стивенсона [12] показали достаточно точ-
ное соответствие между эмпирическими сечениями реакций Q
и классическими геометрическими сечениями ad2, что можно
интерпретировать как указание на то, что каждое столкнове-
ние приводит к реакции. Строго говоря, это неверно, так как
существуют сравнительно медленные ион-молекулярные реак-
ции. Однако расчеты Стивенсона выявили тот факт, что кон-
станты скоростей ион-молекулярных реакций приближаются
к максимальным ко.нстантам скоростей, возможным для бимо-
лекулярных реакций. Некоторое представление о значении ион-
молекулярных реакций для радиационной химии газов можно
получить из следующего примера.
Пример 7.5. Подсчитайте, сколько соударений должен претерпеть до ней-
трализации ион Н2+ при давлении газа 1 атм. Мощность дозы 300 pjceK.
а константа скорости рекомбинации ионов с электронами составляет
10~6 см?!(молекул-сек). Концентрацию ионов Со можно считать стацио-
нарной *.
Решение. По определению
1р ='2,08-10в ион/с-и3;.
300 р/сек « 6-Ю11 ион Кем3-сек).
* В расчетах, которые здесь приведены, не учитывается различие между
массовыми коэффициентами поглощения и № для воздуха и водорода, так как
эта поправка, очевидно, окажется малой по сравнению с другими погреш-
ностями.
197
Среднее время жизни иона (т) до нейтрализации с электроном в бимоле-
кулярной реакции будет равно
2,718
(7.28)
Так как ионы (заряды) возникают с постоянной скоростью (6 • 10й
ионКсм3 • сек), то Со — стационарная концентрация. В стационарном состоянии
скорость образования равна скорости исчезновения:
6-1011 ионов
см3-сек
kr(Ca)3
6- 10й ионов
10-6 сл3(С0)2
см3-сек
ионов-сек
1№
ионов
см3
Подставляя это значение в выражение для т, получаем:
2,718
т =-------------------------- = 2,718-10~3 сек.
см3 ионов
10-в-----------10в--------
ионов-сек см3
Число соударений Z, которое претерпевает данная молекула в единицу
времени (сек), дается выражением
Z — nd2nV,
где d — диаметр молекулы; п — число молекулы в 1 см3; V—средняя ско-
рость молекул.
О
Величина сечения nd2, приведенная в табл. 7.3, равна 27,4 А2. Средняя
скорость молекул V при 300° К равна
8-1,38- Ю-®—- ЗОО'К
К
3,14 3,3.10^24 г
см
1,8.105 ----
сек
Число соударений в секунду будет равно
- « , молекул 4 л _ см
Z -=0,27.10’-» ся2-0,27. 1020 -— • 1,8-10* --- ;
см? сек
„ , . оударений
Z = 0,12.10» ——---------.
сек
Среднее время жизни до нейтрализации с электроном
2,718-Ю-3 сек. За это время ион испытывает 0,12-10п-2,7Х
Х10-3~0,3 • 108 соударений. Так как вероятность реакции при
каждом соударении очень высока (см. табл. 7.3), то стано-
вится ясно, насколько существен учет ион-молекулярных реак-
ций при исследовании радиолиза газов.
Родительские ионы в газовой фазе могут участвовать
в других быстрых реакциях, таких, как нейтрализация или
мономолекулярный распад. В ряде случаев эти реакции могут
конкурировать с ион-молекулярными. реакциями, и в разных
условиях эксперимента следует учитывать относительные ско-
•198
Таблица Z.3*
Сечения и константы скоростей для ион-молекулярных реакций
Реакция Q. ' 10“16 СЖ» Л
k, моль'сек
н+ + Н2 > н+ -Ь н 27,4 126-Юм
н+ + О2 - н2о+ + о 126 580
d+ + d2->d+ + d 26,8 86,8-10W
d+ + O2 >do+ + d 88 280
Н2О+ 4- Н2О •» Н3О+ + он 95,0 76-10'0
D2O+ -1- D2O - D3O+ 4- OD 105 61,5-lOW
D2O-i- 4- H2 - HD2O+ 4- H 63 38,7
C2h+ 4- D3O - HD2O+ 4- C2H3 440 445-lOw
CgHg -j~ D2O -> HD2Oi 4“ C3H7 440 540-10Ю
CH+ + CH4 -> CHj- 4-CH3 — 57,4
CD+ 4- CD4 - CD j" 4- CD3 57,2 47,5-lOw
CDt + C2He >GD4H+4-C2H5 8,1 6- Id»
cd+ + C3H8 - CD4H+ 4- C3H7 4 3-10Ю
CD+4-H,S -CD4H+4-SH 12 8,4-lOW
C2H3l 4- C2H4 » С2НГ 4- C2H- 32 18-10Ю
Ch+4-CH4 >C2Hj"-[-H2 — 48-10Ю
сит 4- CH4 - C2h+ 4- H2 — 12,410io
CH i 4- C2H2 - C3H+ 4- H2 39 36-10Ю
CH+ 4- CH4 c2h+ 4- h2 4- h 54 16- IQio
СН^- + С2Н2 -С3Н + 4-Н 19 18-10Ю
CH+ 4- CH4 -► C2h+ 4- H2 4- H 25 —
c2h+ + C2H4 > c3h+ 4- CH3 22 14-10Ю
СгН<^ C2H2 -> 4~ ^2 35 22
C2H j 4- C2H2 - c4h+ 4- H 42 27
c2h+ + c2h4 * c4h+ 4- h2 34 23
C2h+4-C2H4 ^C4h+4-H 14 9,6
СгН+4.С2Н4 >C4h+4-H 4 3
199
Продолжение табл. 7.3*
Реакция Q. 10”16 CM2 k. моль сек
Сзн+ + СзНв -> С4н+ + С2Н4 140 127
W -j- C3FIg > C4plg~ -j- C2H4 74 66
CH+ + D2 - CH4D+ + D — 15-10«
ch+ + h2 >ch+ + h — 4-108
ch+ + d2 >CH2D+ + H2 — 10,2-101°
ch+ + d2 ch3d+ — 10,2-101°
ch+ + H2 - CH+ + H — 13,8-101°
CH+ 4- H2 -> CH+ + H 35-10Ю
C2h+ + C2He C3h+ + CH4 16 - —
C2H+ + C2He > СзН+ + CH4 30 —
C2H+ + C2He -> C4h+ + H2 + H 1 —•
1+ 4- c2hsi > hi+ + c2h4 2,2 0,93-101°
CH3I+ + CH3I C2H6I+ + I 3,4 1,6-101°
♦ Данные из обзора: Lampe F. W., Franklin I. L., Field F. A. Progress in reaction ki-
netics. New York, Pergamon Press, 1961.
рости всех возможных реакций. Эти аспекты радиолитических
превращений в газовой фазе будут рассмотрены далее.
7.5. РЕАКЦИИ РЕКОМБИНАЦИИ ИОНОВ.
РЕКОМБИНАЦИЯ ПОЛОЖИТЕЛЬНЫХ ИОНОВ И ЭЛЕКТРОНОВ
Рекомбинация положительных ионов с электронами проте-
кает во всех радиационно-химических системах, если электроны
не входят в состав каких-то отрицательных ионов. Как показа-
но в главе VI, возможны следующие типы реакций:
рекомбинация с испусканием излучения
М+ + е ->М*;
М* -» М + hv;
(7.28а)
(7.286)
рекомбинация с последующей диссоциацией
М+ + е- А + В;
(7.29а)
200
рекомбинация трех частиц
М 4- е- 4- X -> А -]- Y. (7.296)
Относительный вклад каждой из этих реакций можно оце-
нить, зная соответствующие константы скоростей. К сожале-
нию, определены лишь немногие из этих констант. Тем не ме-
нее известно, что наиболее существенный вклад вносят лишь
реакции (7.29а) и (7.296). Оценки показывают, что константы
скорости для этих реакций оказываются в пределах 10-5—
10-7 см3/(молекула-сек) [13, 14]. Рассмотренные константы, та-
ким образом, на несколько порядков выше, чем соответству-
ющие константы ион-молекулярных реакций. Однако скорости
реакций определяются также и концентрацией реагентов. При-
мер 7.5 показывает, как можно оценить относительный вклад
ион-молекулярных реакций и реакций нейтрализации.
7.5.1. Рекомбинация положительных и отрицательных ионов
В закрытых системах, не содержащих в значительных коли-
чествах соединений с высоким сродством к электрону, концент-
рация отрицательных ионов обычно заметно меньше, чем кон-
центрация положительных ионов. В стандартных условиях при
низком давлении в масс-спектрометре [15] отношение числа
положительных ионов к числу отрицательных оказывается
выше 103. Однако при высоких давлениях молекулы или оскол-
ки молекул с высоким сродством к электрону могут эффектив-
но захватывать вторичные электроны с низкой энергией. Напри-
мер, галогены, алкилгалоиды и галогеноводороды, NO, О2, Н2О
и спирты легко образуют отрицательные ионы. В присутствии
таких веществ основной реакцией нейтрализации оказывается
взаимодействие положительных и отрицательных ионов [16].
При рекомбинации положительных и отрицательных ионов
существенны следующие две реакции: обмен зарядами
А~4-В+->А + В (7.30а)
и рекомбинация трех частиц
А+ -|- В~ + Z -> АВ + Z. (7.306)
Константа скорости реакции (7.30а) равна примерны
10~8 см3/(молекула-сек) при стандартных температуре и дав-
лении [13]. Константа скорости реакции (7.306) оценивается
около 10~4—10-5 см3/ (молекула • сек).
Хотя константы скоростей реакций (7.29) и (7.30а) весьма
высоки, концентрация ионов обычно остается достаточно низ-
кой; и заметной конкуренции между этими реакциями и ион-
молекулярными реакциями не наблюдается.
201
Все ионы, возникающие при радиолизе, в конечном счете
нейтрализуются в том или ином процессе. Положительные
ионы, участвующие в реакции нейтрализации с электронами
или отрицательными ионами, не обязательно принадлежат
к числу ионов, образующихся в первичных актах. Процесс ней-
трализации может идти по разным конкурирующим путям
в зависимости от состава системы. При очень высоких интен-
сивностях излучения (см. пример 7.5) реакции нейтрализации
могут конкурировать с ион-молекулярными реакциями.
7.6. МОНОМОЛЕКУЛЯРНЫЙ РАСПАД ИОНОВ
К другому типу процессов, конкурирующих с ион-молеку-
лярными реакциями первичных ионов, относится мономолеку-
лярный распад.
Обычно эксперименты по радиолизу в газовой фазе прово-
дят в закрытых системах, причем на образующиеся ионы не
действуют градиенты электромагнитного поля, и в системе
отсутствуют какие-либо потоки. В масс-спектрометре, однако,
система оказывается проточной (ведется непрерывная откач-
ка), и образующиеся ионы подвергаются воздействию электри-
ческих и магнитных полей. В этих условиях ионы претерпевают
мономолекулярный распад, что и сказывается па картине
стандартных масс-спектров.
Возникает вопрос, насколько велик при этом относительный
вклад мономолекулярного распада и какую . роль играют дру-
гие реакции первичных ионов, такие, как реакции нейтрализа-
ции и иой-молекулярные реакции. Иными словами, насколько
характерен полученный масс-спектр именно для первичных
ионов, образующихся при радиолизе в газовой фазе.
Частоту соударений иона в данной систем нетрудно рассчи-
тать. А так как ион-молекулярпые реакции происходят прак-
тически при первом же соударении, то следует сравнить ско-
рость ионной диссоциации с частотой соударений. В газе- при
давлении 1 атм происходит 1010 соударений в 1 сек, а при бо-
лее высоких давлениях частота соударений >1010.
Единственной теорией, позволяющей подойти к оценке
скорости распада ионов, является квазиравновесная теория
масс-спектров [17—19]. В этой теории диссоциация ионов ана-
лизируется с помощью теории абсолютных скоростей реакций,
причем принимается, что наблюдаемый спектр масс обусловлен
конкуренцией ряда мономолекулярных реакций распада воз-
бужденного иона.
В принципе следовало бы рассмотреть возбужденный ион
со всеми его колебательными и вращательными степенями сво-
боды и априорно попытаться рассчитать соответствующий
масс-спектр. Возможности теории оказались для этого недоста-
точными, однако удалось установить правильные качественные
202
зависимости масс-спектра от температуры, энергии электрон-
ного пучка и изотопного состава среды.
В упрощенной форме теория сводится к классическому за-
кону мономолекулярного распада:
&г== , (7.31)
где А — предэкспоненциальный множитель, равный приблизи-
тельно 1014 сек-1; Е — полная внутренняя энергия (энергия
возбуждения); ДЕ0— энергия активации данной реакции;
Е=ЗЛ'—6 — число колебательных степеней свободы в молеку-
ле, состоящей из У атомов.
Уравнение (7.31) приближенно описывает эксперименталь-
ные результаты; оно оказывается полезным лишь в тех слу-
чаях, когда при известных энергии возбуждения и энергии
активации нужно оценить относительные скорости реакций.
Некоторые сведения об энергиях возбуждения и активации
можно получить,зная потенциалы появления данной разновид-
ности ионов [20].
7.6.1. Приближенный расчет относительных скоростей
реакций мономолекулярного распада
Доля ионов, оставшихся недиссоциированпыми в течение
времени I, приближенно дается выражением*
f ^Е\ D(E')dE, (7.32)
о
где Р(£)—функция распределения для энергии возбуждения;
Е' — энергия, превосходящая наименьшую энергию, необходи-
мую для диссоциации иона (большая, чем энергия активации).
Функция распределения D(E), которую можно использо-
вать в этом уравнении, выглядит так:
D(E) = J=, (7.33)
где Е — средняя энергия возбуждения и, следовательно,
/= -%- • (7-34)
Время, в течение которого ион попадает на детектор масс-
спектрометра, работающего при низком давлении, составляет
около 10-5 сек. Поэтому ионы, обладающие данной энергией
* Используемый здесь подход квазимономолекулярной теории в лучшем
случае является качественным, и для более строгого рассмотрения вопросам
мы отсылаем читателя к работе [16]. Однако применяемый метод впол-
не позволяет оценить скорости реакций хотя бы по порядку величины.
203
возбуждения, имеют константу скорости распада, по крайней
мере равную 105 сек~1. Из уравнения (7.31) следует
kr (Е') = 105 сек-1 = А^'- ; (7.35)
£'“—<7-36’
1 ~ ( А У ‘
Доля родительских ионов, не распавшихся в течение 10-5 сек,
будет равна
f.- = V- » (7-37)
где Pt — концентрация родительских ионов; £г-— концентрация
всех присутствующих ионов. При этом предполагается, что йсе
ионы, для которых наблюдается спектр масс, образуются в ре-
зультате мономолекулярной диссоциации родительских ионов.
Строго говоря, это неверно, так как возможно и прямое образо-
вание ионов, отличных от родительских. Например,
CH3CH2SSCH2CH3 av-> HSSH+ + 2Н2С = СН2.
Известно, однако, что интенсивность линий родительских
ионов в масс-спектрах остается относительно постоянной при
изменении энергии бомбардирующих электронов более чем
вдвое по сравнению с потенциалом ионизации. В первом при-
ближении fi для конкретного иона можно определить экспери-
ментально, измерив относительные интенсивности линий в масс-
спектре, соответствующие различным ионам.
Как показано в примере 7.5, при обычных давлениях моле-
кула претерпевает около 10*° соударений в 1 сек. Поэтому не-
обходимо выяснить, какая доля ионов с данной энергией воз-
буждения распадается за 10-10 сек. С реакциями диссоциации
могут конкурировать ион-молекулярные реакции первичных
ионов. По аналогии с уравнением (7.36) запишем для новой
энергии возбуждения
Е" =-------. (7.38)
/ lQi° \f=T
1 k A J
Энергия возбуждения Е" снова оказывается решением уравне-
ния (7.35) для
kr(E") = 1010 сек-1. (7.39)
Так как расчеты относятся к одной и той же реакции (на-
пример, реакции диссоциации в родительском ионе), то можно
приравнять Д£о в уравнениях (7.36) и (7.38). Отсюда получим
204
Если Е' определено, то соответственно. можно найти и Е".
Уравнение для доли родительских ионов, не распавшихся в те-
чение 10~10 сек, будет выглядеть так:
F"
f=-5F- (7.41)
Некоторые сведения о средней энергии возбуждения Ё в
уравнении (7.41) можно получить из масс-спектрометрических
данных [13, 21, 22] с помощью соответствующей интерполяции
кривых для потенциала появления. Средние потенциалы возбуж-
дения можно также рассчитать, используя квазиравповесную
теорию.
Стивенсон [20] рассчитал долю родительских ионов, не дис-
социировавших в течение 1О-10 сек, .и показал, что процесс рас-
пада алкановых и алкеновых ионов может конкурировать с
ион-молекулярными реакциями этих ионов. Существенно, что
при этом наряду с реакциями первичных ионов должны прини-
маться во внимание реакции ионов, образующихся при распаде
(так как родительские ионы могут распадаться, не вступая в
иоп-молекулярные реакции). Удалось установить, что в боль-
шей части случаев спектр масс, полученный при низких дав-
лениях, характерен для первичных ионов, возникающих при
газофазном радиолизе, и в первом приближении этот спектр
отражает начальное распределение ионов.
Пример 7.6. Рассчитайте долю родительских ионов пропилена, сохранив-
шихся через 10-10 сек, если вначале доля родительских ионов составляла
19% общего числа ионов (согласно масс-спектрометрическим измерениям
при 10-3 мм рт. ст.) и средняя энергия возбуждения равна 6,1 эв. Предэкс-
поненциальный множитель примите равным ,1014 сек-1.
Решение. Из уравнения (7.40) следует
F = 3N — 6 = 27 — 6 = 21;
Из уравнения (7.34) найдем
f(105)=-^; °>190=^f7’> £'= 2,3 эв;
205
1
I 2,82 I
E" -= I ----------- I 2,3 = 4,02эв;
I 1 —-------I
\ 1,585 /
4,02
f(1°W) ~ 12~2 ’ Hl°,0)-0,33.
Следовательно, лишь 33% родительских ионов пропилена останутся не-
распавшимися через 10-10 сек, и в кинетической схеме реакций радиолиза
пропилена поэтому следует учитывать ион-молекулярпые реакции осколоч-
ных ионов.
Подобные расчеты полезны для оценки того, следует ли учи-
тывать ион-молекуляриые реакции ионов, образующихся в ре-
зультате распада, при увеличении давления. При высоком дав-
лении скорость ион-молекулярных реакций может оказаться
настолько высокой, что мопомолекулярпый распад родитель-
ских ионов не успеет произойти. Экспериментально наблюдалось
влияние давления (плотности) на радиолиз метана, этана и
пропана [23], что подтверждает важную роль рассмотренных
процессов в радиолизе.
7.7. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА ПРОЦЕСС
ОБРАЗОВАНИЯ ПЕРВИЧНЫХ ИОНОВ
Единственный прямой метод, с помощью которого можно
наблюдать влияние температуры на процесс образования пер-
вичных ионов, — масс-спектрометрический анализ. При возра-
стании температуры в ионном источнике наблюдаются два
эффекта. С одной стороны, отмечается уменьшение интенсивпо-
Рис. 7.2. Относительная интенсив-
ность линии ионов СаН8г в зави-
симости от температуры.
сти линий всех ионов, обуслов-
ленное ростом кинетической энер-
гии молекул и ионов и соответ-
ствующим уменьшением ионных
токов, детектируемых на коллек-
торе. С другой стороны, возмож-
ны изменения относительной ин-
тенсивности линий первоначаль-
ного спектра масс. Предпола-
гается, что подобные изменения
связаны с увеличением колеба-
тельной энергии родительских
ионов и соответствующим увели-
чением скорости распада этих
ионов [24, 25].
На рис. 7.2 показано измене-
ние относительной интенсивности
линии иона С3Н+ в зависимости
206
от температуры. Данные, приведенные па рис. 7.2, свидетельст-
вуют в пользу того, что в радиационной химии газов следует
учитывать возможные изменения начального распределения
ионов под влиянием температуры. Естественно, что некоторые
молекулярные ионы оказываются гораздо более чувствительны-
ми к изменениям температуры, чем другие.
7.8. ЗАКЛЮЧИТЕЛЬНЫЕ ЗАМЕЧАНИЯ О РОЛИ
ИОН МОЛЕКУЛЯРНЫХ РЕАКЦИЙ В РАДИОЛИЗЕ ГАЗОВ
Большой экспериментальный материал, накопленный к на-
стоящему времени, показ^ваёт, что ионные реакции относятся
к числу наиболее существенных химических процессов, проис-
ходящих в различных системах под действием ионизирующей
радиации. Установлены следующие факты:
1. Константы скоростей для большей части ион-молекуля'р-
ных реакций весьма высоки, поэтому вероятность этих реакций
также оказывается высокой.
2. Необходимым, но недостаточным условием ион-молеку-
лярной реакции в замкнутой системе является ее экзотермич-
ность.
3. Спектры масс, полученные при низких давлениях, прибли-
женно характеризуют начальное распределение ионов.
4. Мономолекулярный распад родительских ионов весьма
чувствителен к избыточной энергии ионов и может зависеть от
давления.
5. Начальное распределение ионов может оказаться чувст-
вительным к температуре.
Сводка сечений и констант скоростей ион-молекулярных ре-
акций приведена в табл. 7.3.
7.9. СПЕЦИАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ РАДИОЛИЗА ГАЗОВ
Для сравнения вклада ионных и свободнорадикальных реак-
ций в газах радиационные химики используют разнообразные
методы. В систему добавляют инертные газы (чтобы проследить
за процессами передачи заряда или энергии), акцепторы ради-
калов, такие, как I2, HI, N2O, NO и О2, вводят изотопную мет-
ку и, наконец, удаляют ионы из системы с помощью электри-
ческих полей.
7.9.1. Использование электрического поля
в экспериментах по радиолизу газов
Эта методика была разработана Эссексом и сотрудниками
[26], попытавшимися разделить ионные реакции и реакции дру-
гих активных промежуточных продуктов, удаляя ионы из систе-
мы с помощью электрического поля. Конечно, приложенное поле
207
не может помешать ионам- участвовать в реакциях. Однако го-
могенная нейтрализация в реакционной среде уменьшается, и
в принципе процесс нейтрализации должен теперь идти па элек-
тродах. Экспериментальная установка, применявшаяся Эссек-
сом, показана на рис. 7.3.
В этой установке приложен-
ное электрическое поле выво-
дит электроны и ионы из газа.
Напряженность поля увеличи-
вают до тех пор, пока не пре-
кратится увеличение силы тока
(пока не будет достигнут ток
насыщения). Тогда осущест-
Рис. 7.3. Схема экспериментальной
установки Эссекса и сотр.:
1 — баня с постоянной температурой;
2 — источник а-частиц: 3 — сосуд, в кото-
ром производится облучение: 4 — электро-
ды; 5— к вакуумной системе; 6- экрани-
ровка.
Напряженность электрического по-
ля, В/см
Рис. 7.4. Облучение смеси СгИ6—
—C2D6—NO (1:1 :0,05) при одно-
временном воздействии электрическо-
го поля.
вляется полное удаление ионов из системы. При очень сильных
полях могут начаться каскадные процессы с умножением числа
электронов и ионов.
Эссекс с сотрудниками считали, что удаление ионов из газа
с помощью электрического поля препятствует «реакциям ионов».
Сейчас мы знаем, что-это неверно, так как скорости ион-моле-
кулярных реакций очень высоки и положительный ион, прежде
чем он попадает на электрод, должен претерпеть много тысяч
соударений. Поэтому те иопы, которые действительно попадают
на электроды, относятся к числу сравнительно малореакционно-
способных.
Главное достоинство метода состоит в том, что он позволяет
выделить из продуктов радиолиза продукты быстрых ион-моле-
кулярных реакций,, выход которых не меняется в зависимости
от приложенного электрического поля, и продукты реакций воз-
бужденных молекул. Так, из рис. 7.4 видно, что при облучении
.208
смеси С2Н6 и C2D6 выход таких соединений, как C2D3H и CD3H,
не меняется под действием приложенного поля, в то время как
выход C2D4 и CD4 увеличивается в 3—4 раза.
Положительные ионы и электроны, мигрирующие к соответ-
ствующим электродам, отнюдь не ускоряются до энергий, соот-
ветствующих приложенному напряжению, так как претерпевают
в это время многочисленные соударения. Как показывают оцен-
ки, наибольшую энергию приобретают электроны. Однако и для
них эта энергия не превышает 15—25 эв. При соударениях с мо-
лекулами электроны такой энергии вызывают главным образом
возбуждения без ионизации. О том, что дело обстоит именно так,
свидетельствует сохранение тока насыщения постоянным, хотя
выход молекулярных продуктов меняется (см. рис. 7.4). Если бы
ионизация происходила в заметных масштабах, то значение со-
бираемого тока должно было бы зависеть от напряжения. Таким
образом, анализ выхода различных продуктов радиолиза в этих
условиях показывает, какие из них образуются в результате
возбуждения родительских молекул.
При интерпретации результатов, полученных рассмотренным
методом, следует, однако, соблюдать осторожность. Например,
первичный процесс, ведущий к образованию виниловых радика-
лов в радиолизе этилена, в существенных масштабах протекает
только при действии поля, но практически отсутствует, если
поля пет [27].
7.9.2. Добавление инертных газов
Добавление инертных газов при радиолизе может повлиять
на течение самых разнообразных ионных реакций. Это происхо-
дит в результате процессов передачи заряда или энергии, обра-
зования гидридных ионов или благодаря участию инертных га-
зов в других ион-молекулярных реакциях. Яркий пример влия-
ния инертных газов —их воздействие на радиационно-индуциро-
ванный обмен водорода и дейтерия [28, 29].
При облучении смеси Н2 и D2 образуются молекулы HD.
В основе процесса лежит ионная цепная реакция с ионным вы-
ходом М/N около 18 000. Доказательства цепного характера
реакции были получены, когда выяснилось, что присутствие
некоторых инертных газов (Кг и Хе) полностью останавливает
радиационно-индуцированную реакцию, но не влияет на фото-
или термоиндуцировапный обмен водорода и дейтерия.
Как и при а-радиолизе орто-параводородной смеси, первич-
ными ионами оказываются ионы Н+ (или D+), Н+ (или D+).
Так как сечение реакции
велико, то образуются ионы и атомарный Н. В присутствии
Кг или Хе, потенциалы ионизации которых ниже, чем у Н, ско-
209
рость обмена дейтерия и водорода резко снижается. В присутст-
вии же-Не, Ne или Аг, потенциалы ионизации которых выше, чем
у Н2, происходит умеренное ингибирование обменной реакции
(если концентрация инертного газа достаточно высока) и уско-
рение реакции (если концентрация инертного газа низкая). Вна-
чале предполагали, что идущие при этом реакции можно пред-
ставить как простую передачу заряда. Однако более тщательный
анализ энергетики реагирующей системы, а также масс-спек-
трометрические и фотохимические данные позволили найти ме-
ханизмы, более соответствующие наблюдаемым явлениям.
• Экспериментально удалось установить, что длина цепи очень
велика (~ 10000). В связи с этим было выдвинуто предполо-
жение о ионно-цепном механизме реакции и об ингибирующем
воздействии ионов инертных газов на развертывание цепи.
Следующая, схема реакций объясняет ионно-цепной меха-
низм обмена и влияние инертных газов.
Инициация цепи:
Ht + H2-Ht+H. (7.42)
Рост ионной цепи:
Ht-bD2->H2+HDt. (7.43)
Инициация ингибирования:
Ht -|-R->RH+ -i- Н (7.44)
(R — любой инертный газ).
Ингибирование цепи:
Ht -Ь Хе (или Kr) -> ХеН+ -f- Н2. (7.45)
В присутствии Ne, Не или Аг
Ne+ + Н2 NeH+ 4- Н, (7.46)
ускорение реакции
NeH+ + Н2 -> Ne 4- Н|. (7.47)
Обрыв цепи:
Н/ + М (отрицательно заряжена) _> Н2 -j- Н М (7.48)
или
Н8 +М-т-е--»-Н2-г Н 4-М. (7.48а)
Ионная цепь развертывается с участием в качестве актив-
ной частицы иона Н^*, для которого характеристическая кон-
станта скорости реакции составляет 1,4-IO-9см3/(молекула-сек).
Умеренное ингибирование обмена происходит при взаимодей-
ствии атома инертного газа с ионом Несогласно реакции (7.44).
Однако для Не, Ne и Аг эта реакция становится существенной
только в том случае, если концентрация инертного газа сравни-
210
ма с концентрацией Н2. Но достаточно лишь следов Хе или Кг
для того, чтобы практически полностью остановить обмен. Ато-
мы этих газов, очевидно, препятствуют развертыванию цепей.
Энергетику реакций (7.44), (7.45) и (7.46) можно рассмот-
реть с помощью стандартных термодинамических методов. Теп-
лота реакции (7.46) между ионом инертного газа и водорода
дается выражением
АН = — Е (RH+) i- / (Н ^) ~ 2AHf (Н) — / (R+), (7.49)
где £(RH+)—энергия связи гидридного иона инертного газа;
/(Н+) и /(R+)—потенциалы ионизации водородного атома и
атома инертного газа; Д77/(Н) — теплота образования атома Н.
Например, подставляя соответствующие численные значения для
реакции образования нона гидрида гелия, получаем:
АН = — Е (RH+) + 313 -h 103 — 563;
. АЯ-- — £(RH')~ 147.
Такой результат показывает, что реакция (7.46) с высокой
вероятностью будет экзотермической. [Масс-спектрометрическим
методом удалось установить, что реакция (7.46) является экзо-
термической для ионов любых инертных газов, кроме Хе+]
В ряде случаев влияние добавок инертных газов удается
объяснить процессами передачи заряда или энергии. Например,
с помощью процессов передачи заряда или энергии можно объ-
яснить ускорение радиационного разложения бутана в присут-
ствии аргона [30, 31]:
Аг-ь -i- C4HJ0 -► СЯН? 4- СН3 4- Аг; (7.50)
Аг* 4- С4Н10 С3Н7- 4- СН3 4- Аг 4- е~. (7.51)
Здесь Аг* — возбужденный атом аргона, энергия которого по
крайней мере на 10,5 эв выше, чем в основном состоянии. •
7.9.3. Добавление акцепторов свободных радикалов
Добавление к системе таких акцепторов свободных радика-
лов, как 12 или HI, весьма распространенный прием. Однако
интерпретация полученных данных часто осложняется, так как
атом галогена легко образует отрицательный ион. Последую-
щие реакции отрицательного иона типа тех, которые были рас-
смотрены’ в разделе 7.5.1, могут усложнить общую картину.
Акцепторы добавляют в систему для того, чтобы перехва-
тывать свободные радикалы в таких реакциях, как
СН34-12^СН314-1; (7.52)
Н 4-Ia-> HI 4-1. (7.53)
211
Образующиеся алкилиодиды удается обнаружить с помощью
газовой хроматографии.
Часто компоненты системы метят с помощью трития. Одно-
временно тритиевый газ может служить 'источником излучения.
Для этого часть атомов водорода в различных соединениях за-
мещается на тритий. Механизм этого процесса весьма сложен и,
очевидно, включает реакции ионов трития с компонентами си-
стемы, а также реакции ионов и возбужденных молекул с мо-
лекулами Т2.
Другую разновидность акцепторов представляют собой сле-
довые количества дейтерированных молекул или обычных моле-
кул, таких, как пропан или бутан, которые добавляют для вы-
явления реакций гидридного иона с другими ионами или других
типов реакций [32, 33].
Например, если добавить к циклогексану от 3 до 15% дейте-
роциклопропана и смесь облучить, то больше 90% образующе-
гося пропана будет состоять из молекул C3DeH2, что свидетель-
ствует о переносе молекулы Н2.
В качестве акцептора радикалов широко используют окись
азота. При радиолизе этана с небольшими добавками NO выход
продуктов, образование которых считают обусловленными сво-
боднорадикальными реакциями, падает почти до нуля.
Олефины также использовались в качестве акцепторов ради-
калов. Например, атомы Н, возникающие при тепловой диссо-
циации, можно обнаружить посредством реакции
ед + Н-^ОД. (7.54)
Другой распространенный прием — облучение смеси дейтери-
рованной и недейтерированной форм той же самой молекулы.
Этот прием введения изотопной метки оказался одним из наи-
более удачных методов исследования механизмов сложных
реакций. Например, анализ продуктов радиолиза эквимолярной
смеси С2Н6—C2De показал, что выделяющийся водород состоит
главным образом из молекул Н2 и D2. Такой результат можно
рассматривать как подтверждение механизма «молекулярного
отрыва»:
С2Н6^-С2Н4 + Н2. (7.55)
Однако это скорее пример использования изотопной метки, чем
применения акцепторов свободных радикалов.
Для оценки выхода свободных электронов и выяснения меха-
низмов процесса нейтрализации можно применять электронные
акцепторы, такие, как N2O и SF6 [34, 35]. Для обнаружения про-
межуточных ионов с низкой реакционной способностью [36] ис-
пользовались специальные акцепторы зарядов, в качестве кото-
рых пригодны вещества со сравнительно низкими потенциалами
ионизации.
212
7.10. НЕЙТРАЛЬНЫЕ АКТИВНЫЕ ЧАСТИЦЫ.
ОБРАЗУЮЩИЕСЯ ПРИ РАДИОЛИЗЕ ГАЗОВ
Нейтральные активные частицы могут возникать при радио-
лизе в газовой фазе в результате таких ион-молекулярных
реакций
CHt + СН4 СН| -г сн3> (7.56)
а также непосредственно при диссоциации возбужденных мо-
лекул
СН4 СН4 СН2 -Н2; (7.57)
С2Н6^-С2Н^СН3гСН3. (7.58)
Свободные радикалы и ион-радикалы могут образовываться
при передаче заряда, диссоциации ионов, передаче энергии воз-
буждения, а также в некоторых реакциях с участием акцепто-
ров. Ряд реакций наиболее простых радикалов достаточно хо-
рошо изучен. Известны их энергии активации, порядок величи-
ны констант скоростей, характер реакций (перенос Н, диспро-
порционирование и т. д.).Однако сравнительная роль различных
реакций (ион-молекулярных, прямого возбуждения и пр.) в об-
разовании свободных радикалов при радиолизе в газовой фазе
известна недостаточно хорошо. Ясно, что реакции типа (7.57) и
(7.58) могут происходить, однако свободные радикалы этим пу-
тем возникают, очевидно, в значительно меньшей степени, чем
в реакциях с участием ионов.
7.11. ИОННЫЕ МЕХАНИЗМЫ РАДИОЛИЗА
Первичный акт радиолиза — возникновение ионов, радикалов
и возбужденных молекул, разнообразные реакции которых при-
водят в конечном счете к образованию разного рода продуктов.
Для радиолиза метана и пропана было показано, что с помощью
ионных механизмов реакций можно объяснить выход важней-
ших продуктов. Однако сейчас известно, что только ионные ме-
ханизмы не объясняют всех процессов; происходящих в этих
системах. Экспериментально установлено, что некоторые про-
дукты появляются в результате реакций возбужденных ней-
тральных молекул, однако невозможно количественно описать
эти реакции. Оценки показывают, что при радиолизе метана,
пропана и бутана приблизительно от 10 до 20% общей массы
продуктов появляется в результате реакций распада нейтраль-
ных молекул [37].
Каждая индивидуальная система в определенной степени
уникальна, и тем не менее можно с высокой степенью вероятно-
сти утверждать, что преобладающими процессами радиолиза
являются процессы с участием ионов. При отсутствии детальной
213
кинетической информации приемлемую качественную картину
выхода различных продуктов радиолиза часто удается по-
строить, приняв во внимание возможные ионные реакции. Сле-
дует только помнить, что используемые ионные механизмы
описывают реальные процессы, происходящие в системе с огра-
ниченной точностью. Ниже будет приведен пример расчета та-
кого типа, относящийся к радиолизу метана.
Пример 7.7. Дайте качественную оценку выхода возможных продуктов
при радиолизе метана. Масс-спектр метана характеризуется следующими зна-
чениями относительной интенсивности ионов различных типов:
Ион Интенсивность
СН+........................ 0,48
CHjf........................ 0,40
CHj-........................ 0.08
СН+........................ 0,04
С+......................... 0,01
Эти интенсивности примем за меру относительной концентрации каж-
дого типа ноиов. Ион С+ учитывать ие будем.
Решение. Согласно данным табл. 7.3, могут происходить сле-
дующие ион-молекулярные реакции первичных ионов:
CH i -Ь СН4 .СН5+4-СН3; (7.59)
СН+ + СН4 ► C.H+4-Hs; (7.60)
СН+4-СН4-: ► С.Н+ 4-Н; (7.61)
СН+4-СН4^ ► QH+4-H; (7.61а)
СН+ 4- сн4 -! >-СгН+4-Н2 4-Н; (7-62)
СН+ 4-CH.t- ► СН+4-Н24-Н. (7.63)
Кроме них возможны также ион-молекулярные реакции меж-
ду ионами, образующимися в реакциях (7.59) — (7.63), и молеку-
лами метана или молекулами, продуктов. В первом приближе-
нии при отсутствии прямых сведений о вторичных процессах
этими реакциями можно пренебречь. Основными реакциями вто-
ричных ионов будут тогда нейтрализация электронами, гидрид-
ными ионами или при переносе протона. Остальные реакции
вторичных ионов, так же как и другие неионные реакции, можно
рассматривать лишь в качестве поправок.
Реакции нейтрализации:
СНТ + е~ -> СН3 + Н2; (7.64)
CHt + е~ -> СН4 4- Н. (7.65)
214
Из этих двух реакций более выгодна термодинамически *
реакция (7.64)
АН{7.64) = A#z (СН3' + A#z (Н2Ч — А/7/(СН^) = — 253 ккал/моль;
АЯ(7.65) = A#z(СН4) АЯу (Н) — АДу(CHf) s — 186 ккал/моль.
Для этильного иона существуют два пути реакции:
М + C2HjT + е -> С2Н5;
сХ-Ье-^ед + Н.
(7-66)
(7-67)
Обе реакции возможны. Однако реакция (7.67) окажется экви-
валентной (7.66) ввиду того, что идет реакция (7.54). Для вто-
ричных ионов, образующихся по механизмам (7.61) — (7.63),
можно предполагать, что нейтрализация приведет к появлению
молекул С2Н4 [из вторичных ионов реакций (7.61) и (7.62)],
С2Нб [из вторичных ионов реакции (7.61а)], С2Н2 [из вторичных
ионов реакции (7.63)].
Образование первичных ионов можно объяснить с помощью
следующей схемы:
+ (7.68)
СН4 л*- СН2 4- Н + Н; • (7.69)
СН4 СН+ + Н2 + Н. (7.70)
Перенос гибридного иона приводит к нейтрализации целого
ряда положительных ионов. В рассматриваемой системе эта ре-
акция может идти с ионами, возникающими в реакциях (7.60)
и (7.62), и с молекулами продуктов (перенос гидридного иона
от метана происходит с малой вероятностью):
С2Н| + RH -> С2Н6 R- ; (7.71)
С2НГ - Ь RH -> С2Н4 + R+ , (7.72)
где RH — любой алкан, кроме СН4.
Реакции переноса протона легко идут с некоторыми ионами
CH? + RH -> СН4 4- RHt; (7.73)
С2Н| -1- RH С2Н4 - г RHt; (7.74)
С2Н| + RH -> С2Н2 + RHt, (7.75)
где RH — молекула, отличная от СН4.
* Следует, однако, иметь в виду, что только из термодинамических соо-
бражений нельзя получить абсолютных критериев возможности реакции. Вы-
бор пути реакции определяется также целым рядом других факторов. Тем не
менее при отсутствии дополнительных сведений термодинамика может ока-
заться полезной.
215
Возникает вопрос, какие критерии нужно использовать для
выбора между этими возможностями. Имеющейся информации
недостаточно для широких обобщений. Тем не менее нужно от-
метить, что реакции переноса гидридного иона происходят до-
статочно легко, особенно при взаимодействии с ионом С2Н^' .
Поэтому примем, что основные реакции для этого иона — пере-
нос гидридного иона и нейтрализации электронами. Для иона
СНт основными реакциями будем считать перенос протона и
нейтрализацию, причем перепое протона должен происходить
главным образом на молекулы продуктов, таких, как этан и
пропан. Для иона С2Н^ будем рассматривать реакции переноса
гидридного иона, нейтрализации и переноса протона.
Реакции свободных радикалов. Радикал СНз образуется
преимущественно в реакциях (7.59) и (7.64) и может вступать
во взаимодействие с метильными и этильными радикалами и ато-
мами II:
2СН3->С2Н6; (7.76)
СН3 !-Н->СН4; (7.77)
СН3-;-С2Н5-^С3Н8. (7.78)
Энергия активации реакции (7.78) примерно такая же, как
и реакции (7.76), однако концентрация радикалов С2Н5 ока-
жется ниже концентрации СН3. Легко идет реакция (7.77), но
так как концентрация радикалов СН3 примерно вдвое выше кон-
центрации атомов II, преимущественно будет осуществляться
реакция (7.76). Радикалы СН3 могут участвовать также в реак-
циях замещения, по так как для этих реакций требуется замет-
ная энергия активации, они имеют второстепенное значение.
Радикал С2Нд образуется в реакциях (7.66), (7.61а) и (7.54).
Кроме (7.78) для радикала С2Н3 возможны следующие реакции:
СЛЧ-СН^СА + СНз; (7.79)
+ (7.80)
С,Н54-Н^С2Н6. (7.81)
Концентрация атомов Н приблизительно сравнима с концен-
трацией этильных радикалов, однако известно, что реакция
(7.54) отличается высокой эффективностью и, следовательно,
концентрация атомов Н очень мала. Основными реакциями
этильных радикалов поэтому будут (7.78) и (7.80). Реакцию
(7.79) можно не принимать во внимание, так как ее энергия
активации около 15 ккал/моль.
Кроме реакций (7.81) и (7.54) атомы Н могут участвовать
в такой реакции:
Н + Н+М->Н2+М. (7.82)
216
Реакция (7.82) требует присутствия третьей частицы (М).
Известно, что реакция (7.54) происходит с атомами Н тепловых
энергий. Поэтому даже если начальная концентрация молекул
С2Н4 мала, реакция (7.54) преобладает над реакцией (7.82).
Таким образом, учитывая все возникающие ионы, можно
предположить, что радиолиз метана приведет к образованию
этана, этилена, пропана, бутана, ацетилена и водорода.
Происхождение различных продуктов радиолиза:
а) Этан. Основные источники образования этана — реком-
бинация этильных радикалов и перенос гидридного иона на
С2Н^~. Реакция (7.59) будет давать метильные радикалы с вы-
ходом M/7V=O,48, а ионный выход этана по реакции (7.71) со-
ставит 0,40. Максимальный выход Л1/Л' = 0,24+0,40 = 0,64. Ней-
трализация ионов С2Н± по реакции (7.66) будут, разумеется,
снижать этот выход.
б) Этилен. Хотя этилен может образовываться при ней-
трализации ионов, возникающих в реакциях (7.60) и (7.61),
реакция (7.54) сопровождается превращением большей части
этилена в этильные радикалы. Поэтому этилен появится лишь
в очень малых количествах. Максимальный ионный выход для
этилена M/N = 0,08.
в) Пропан. Так как выход метильных радикалов значи-
тельно выше выхода этильных, то следует ожидать, что этильные
радикалы будут в основном расходоваться в реакции (7.78).
Часть этильных радикалов рекомбинирует согласно реакции
(7.80). Образуются этильные радикалы только при нейтрализа-
ции С2Н5 и в реакции (7.54). Выход пропана >0,08.
г) Бутан. Единственный источник бутана — рекомбинация
этильных радикалов согласно реакции (7.80). Учитывая выше-
сказанное, можно сделать вывод, что выход бутана должен быть
меньше выхода пропана.
д) Ацетилен. Нейтрализация ионов С2Н^~ является един-
ственным источником образования ацетилена. Поэтому ионный
выход этого продукта не может быть выше .0,04.
е) Водород. Основными источниками водорода являются
реакции (7.60), (7.61), (7.61а) или (7.62), (7.63), (7.68), (7.69),
(7.70), а также реакция нейтрализации (7.64). Максимальный
ионный выход М/N для водорода будет равен по различным
реакциям:
Реакция:
(7.60) 0.40
(7.64).....................................0.48
(7.61), (7.61а) либо (7.62)..............0,08
(7.63) ...... ......................... 0,04
(7.68) 0,20
(7.69) 0,04
(7.70) 0,03
Общий выход M/N..............1,27
217
ж) Разложение метана. Ионный выход разложения
метана для разных реакций можно рассчитать тем же способом:
0,96
0,80
0,16
0,08
Реакция:
(7.59) .
(7.60) .
(7.61) .
(7.63) .
Сводная таблица ионных выходов
Расчетный Измеренный.
сн4 .... —2,00 —2,50
к .... . 1,27 1,90
СгНв . . . . 0,64 0,70
с.,н4 .... . 0,08 (макс.) 0,01
С2Н3 .... . 0,04 (макс.) 0,01
CsHs-l-C^jo 0,08 0,06
Используя ионные механизмы, можно, таким образом, полу-
чить результаты в грубом качественном соответствии с экспе-
риментальными данными. Однако никоим образом нельзя счи-
тать, что принятые механизмы действительно полностью харак-
теризуют реальные химические процессы в системе. Так, напри-
мер, наверняка известно, что в системе идут реакции (7.57) и
(7.58), вслед за которыми происходит взаимодействие метиль-
ного радикала с метаном и, возможно, с молекулами — про-
дуктами радиолиза. Кроме того, в небольших количествах обра-
зуется полимер, и имеются свидетельства того, что процесс
полимеризации идет за счет развития ионной цепи, инициируе-
мой ионом СН+.
Имеются также обоснованные доказательства существования
нескольких дополнительных ион-молекулярпых реакций (реак-
ций конденсации):
С2НГ
С2нг + СН4 СзН| 4- Н2.
В первом приближении, однако, ионная схема реакций весь-
ма полезна и дает грубое качественное представление о том,
какие продукты должны появиться и каковы их относительные
количества.
Выше указывалось, что принимались во внимание только
реакции первичных ионов и вкладом радикалов, образующихся
при прямом возбуждении молекул, пренебрегали. Однако при
высоких давлениях, папример в масс-спектре, ион С2Н^" исче-
зает и появляется С3Н:1'. При еще более высоких давлениях по-
является ион С3Н+. Фотохимические исследования в области
218
вакуумного ультрафиолета показывают, что радикалы СН3, СН2
и СН являются важной разновидностью активных первичных
частиц.
Задачи
1. Рассчитайте для иона Н число соударений в секунду при давле-
ниях 10-в, 10-3 мм рт. ст. и 10 атм.
2. Для мощности дозы 1000 р!сек оцепите среднее время жизни иона
11+ до его нейтрализации.
3. Рассчитайте константу скорости и сечение для реакции
С2н+ + СН4 * СзНу- + Н2,
если £=8,0 в!см\ d=2,5 мм; а=1,2-10-24 см3.
4. Для иона Н2О+ потенциал появления равен 12,61 эв, а стандартная
теплота образования Н+ составляет 365 ккал)моль; найдите изменение энталь-
лии в реакции
Н.О+ + Н2О Н3О+ + ОН.
Примеча ние. Стандартная теплота образования ОН может быть рассчитана
мз данных об энергии диссоциации связи.
5. Рассчитайте изменение энталышн в реакции
С2Нд" Д-С2Н4 -> СвН+^-СгН»,
если потенциал появления С2Н3+ равен 317 ккал!моль.
6. Средняя энергия возбуждения СН4+ составляет 5,2 эв, найдите долю
родительских ионов метана, сохраняющихся в течение 10 ‘10 сек. (Исполь-
зуйте масс-спектрометрические данные примера 7.7 и примите прелэкспонеп-
циальный множитель равным 1014 сек-1.)
7. Масс-спектр пропана выглядит следующим образом:
Ион . Интенсивность
QH+.................. 29,2
С3н+................. 23,1
Calls'............... 12,9
С3Н+................. 17,5
й о *
Ион Интенсивность
С2Н^............... 100,0
C.Hj-............... 59,5
С2И3Ь............... 40,3
С,Н7................. 8,3
а) Определите относительную концентрацию различных ионов.
б) Используя относительные концентрации ионов и данные табл. 7.3,
дайте качественную оценку возможных продуктов радиолиза пропана.
8. Теплопроводность этана равна приблизительно 0,00027 кал/(сек)(см2) X
X (°С/сл). Оцените среднюю температуру в алюминиевом цилиндре
(02 с.кХЮ см) с этаном под давлением 10 атм при облучении электронным
пучком с энергией электронов 2 Мэв при токе 1 -10-6 а в течение 10 мин. До-
статочно ли высока эта температура, чтобы изменить выход первичных
ионов?
Глава VIII
РАДИАЦИОННАЯ ХИМИЯ ВОДЫ
И ВОДНЫХ СИСТЕМ
Впервые разложение воды под действием ионизирующего
излучения подробно исследовали в 1913 г. Дуан и Шойер [1],
которые обнаружили, что облучение воды а-частицами приво-
дит к образованию водорода, кислорода и небольших количеств
перекиси водорода. Однако механизм радиационного разложе-
ния воды был неясен вплоть до 1927 г., пока Г. Фрике* не
начал свои исследования радиационно-химических реакций.
В паши дни радиационная химия воды и водных растворов —
один из наиболее подробно разработанных разделов радиацион-
ной химии [2].
8.1. ДЕЙСТВИЕ ИОНИЗИРУЮЩЕГО ИЗЛУЧЕНИЯ НА ВОДУ
Фрике и сотрудники обнаружили, что рентгеновское излуче-
ние в отличие от а-частиц, не вызывает заметного разложения
воды, особенно если перед облучением ее тщательно очищали.
В присутствии растворенных веществ заметно значительное раз-
ложение воды. При этом количество разложившейся воды не
зависит от количества растворенного вещества в широком диа-
пазоне концентраций.
Для объяснения механизма химических реакций в разбав-
ленных растворах Фрике постулировал, что под действием из-
лучения вода переходит в некую «активированную» форму, ко-
торая затем инициирует химические реакции. Природа «акти-
вированной воды» оставалась неизвестной. Однако удалось про-
вести целый ряд параллелен между действием ионизирующей
радиации и ультрафиолетового излучения. В частности, Фрике
показал, что облучение водных растворов радиацией с длиной
волны короче 1900 А вызывает многие реакции, сходные с теми,
* Сравнительно полную библиографию ранних работ Фрике можно найти
в статье, посвященной его 70-летпему юбилею в журнале Rad. Res, 1962,
v. 17, р. 253.
220
которые наблюдаются под действием рентгеновского излучения.
Возбужденные молекулы воды при этом могли диссоциировать
Н2О^Н г ОН. (8.1)
Дальнейшие исследования, однако, показали, что разложе-
ние воды и водных растворов гораздо более сложный процесс,
и его нельзя объяснить простым образованием радикалов. Для
выяснения сложных механизмов радиолиза воды потребовалось
разобраться в пространственном распределении активных ча-
стиц, способах их образования, радиационно-химическом выходе
различных процессов, а также реакциях других промежуточных
продуктов, участвующих в наблюдаемых химических явлениях.
8.2. ПЕРВИЧНЫЕ АКТИВНЫЕ ЧАСТИЦЫ
В табл. 8.1 указаны возможные активные частицы и реакции
их образования в жидкой воде, происходящие при воздействии
ионизирующей радиации. Время образования первичных актив-
ных частиц, включенных в табл. 8.1, примерно равно периоду
молекулярных колебаний (IO”14—10“15сек).
Таблица 8.1
Активные частицы при радиолизе Н2О
Активная частица Энергия образования, эв Реакция образования
н2о* 7,4 или 9,2 Н.,О~ >Н2О*
H2Or-|-e- 12,56 Н2О~ Н2О+-|-е-
ОНИ 18,1 Н2О~ .ОН++Н+е~+
+К.9
н- 19,6 Н,О~ >Н++ОН+е-
ОЪ 18,8 Н2О~ ->О++Н„-|-е-
О- е~ (5,6)-гН2О Н..О-|-е- - 0--J-2H
Н- е''(7,5)+Н2О Н,О+е- >Н~+ОН
КЭ — кинетическая энергия.
Данные о присутствии того или иного типа иопов получены
с помощью масс-спектрометрических измерений. Масс-спектр
воды приведен в табл. 8.2.
Таблица 8.2
Ион Относитель- ное содержа- ние Ион Относитель- ное содержание Ион Относитель- ное содержание
н2о+ 100 Hi- 5 О- 1,5
ОН!- 23 О! 2 н- 0,6
Данные из статьи Mann М. Hustrulib A., Tate J. Phys. Rev.. 1940, v. 58, p. 340.
221
Через 1СН4—10~12 сек после возникновения частицы, указан-
ные в табл. 8.1, исчезают. Далее приведены наиболее вероятные
реакции этих частиц:
Н2О* ь Н2О* ->Н2О2 J-Н; (8.2)
Н2О*->ОН Н -J- 2 эв КЭ; (8.3)
Н2О* -> ОН* -- Н; (8.4)
Н2О г Н2О - >Н3ОЬ ;-ОН; (8-5)
ОН+ + 2Н2О Н3О4- г 2ОН; (8-6)
Н3О+ -|- е ~ - >Н3О*; (8.6а)
Н+ 4- Н2О - > Н3О^; (8-7)
о+ + н2о - >1Г;-Н2О; (8-8)
о- + н2о- >ОН4-ОН“; (8-9)
H“ + H2O- > Н2 ОН~ ; (8.10)
£ (вторичный) —» е~~ (термализованный) или е (гидратированный). (8.11)
8.2.1. Доказательства существования первичных активных
частиц
Прямые доказательства существования атомов Н в облучен-
ном льду при 4,2° К или в замороженных облученных растворах
при 77° К можно получить, анализируя парамагнитные свойст-
ва этих систем. В последнем случае удалось охарактеризовать
ряд реакций, захваченных в ловушки атомов II. Радикалы ОН,
возникающие при электрическом разряде в водяных парах, об-
наруживаются по абсорбционным и эмиссионным спектрам око-
ло 3064 Л, а также с помощью масс-спектрометра. В облученном
льду те же радикалы можно идентифицировать благодаря их
парамагнитным свойствам. Существование радикалов Н2О уда-
лось доказать масс-спектрометрически при анализе продуктов
реакции атомов Н, возникающих в газовом разряде, с молеку-
лами О2 в присутствии инертных газов [4]. Спектры поглощения
радикала НО2 можно непосредственно наблюдать в облученных
растворах 0,1 и. H2SO4 [5].
Существование гидратированного электрона Платцмап посту-
лировал еще в 1953 г. [6]. Однако кинетические данные, свиде-
тельствующие о присутствии этой частицы, не были получены
вплоть до 1959 г. [7, 8]. Прямые спектроскопические доказатель-
ства удалось получить в 1963 г. [9]. В этом же году получили
кондуктометрические подтверждения существования гидратиро-
ванного электрона [10].
222
8.2.2. Стационарные концентрации активных частиц
Суммарный результат реакций (8.2) — (8.10) состоит в том,
что появляются частицы Н, ОН, е~ц и небольшие количества Н2
и Н2О2. Реакция (8.5) относится к числу ион-молекулярных ре-
акций и происходит за время около 10-14 сек. Эта реакция мо-
жет успешно конкурировать с прямыми реакциями диссоциации,
и, вероятно, поэтому ионы Н+, О+ и ОН+ почти нс образуются,
а если и образуются, то в очень небольших количествах. В поль-
зу этого говорят и высокие энергии образования ионов Н+, О+
и ОН+ (см. табл. 8.1).
Вторичные электроны, возникающие в процессе ионизации
термализуются и сольватируются в течение 10-11 сек [см. реак-
цию (8.11)]. Сольватированный электрон может реагировать с
молекулами воды (если вода подвергнута особой очистке) при
отсутствии растворенных веществ с образованием H-f-OH- и с
другим сольватированным электроном, давая Н2, или с Н+, об-
разуя Н. Стационарная концентрация любых активных частиц
оказывается весьма низкой.
Пример 8.1. Рассчитайте приблизительную стационарную концентрацию
радикалов в чистой деаэрированной воде при облучении с мощностью дозы
103 рад!сек. Примите, что средняя скорость радикал-радикальных реакций
составляет 10’3 см3/(моль-сек) и G (образования радикалов) = 10.
Решение. В стационарном состоянии скорость образования радикалов
равна скорости их исчезновения. Будем считать, что радикалы исчезают в ре-
зультате радикал-радпкальных взаимодействий
kr
R-LR--M,
где R — радикал; М — молекула; kr — константа скорости реакции.
low смз
Скорость исчезновения — рг 1R]2 -----IR]2;
моль-сек
10 радикал I 103 рад I моль I
Скорость образования = ------ --------- -------------- X
100 эв I сек | 6-1023 радикал |
6,24-1013эв | 1 г моль
X —---------- -----= 1,04- IO—»------ .
рад-г I см3 см3-сек
Скорость исчезновения равна скорости образования:
см3 моль
1013---------[R]2 1,04-10-8--------
моль-сек см3-сек
8.3. ПРОСТРАНСТВЕННОЕ РАСПРЕДЕЛЕНИЕ
ПЕРВИЧНЫХ АКТИВНЫХ ЧАСТИЦ
Схематическое изображение распределения в пространстве
первичных ионизаций показано на рис. 2.8. Как указывалось в-
главе II, среднее расстояние между первичными ионизациями
223-
75 эв. Эта энергия диссипирует
Таблица 8.3*
Среднее расстояние
между первичными ионизациями
Вид ионизирующий Среднее рас-
частицы и ее энергия стояние,А
Электрон, 0,5 Мэв 5000
1 кэв 50
100 эв 5
«-Частица, 5 Мэв 8
1 Мэв 2
* Из книги: HochandelC. J. Comparative
effects of radiation, N.Y., John Wiley,
1960. p. 159.
зависит от скорости ионизирующей частицы и ее заряда.
В табл. 8.3 приводятся средние расстояния между первичными
ионизациями для различных энергий падающих частиц.
Как указывалось в главе V, вторичные электроны, выбивае-
мые при первичных ионизациях, имеют; среднюю энергию около
при образовании первичных
активных частиц, которые ло-
кализуются в небольшом эле-
менте объема (шпоре) с диа-
метром около 20 А. Вторичный
элемент создает в объеме шпо-
ры около пяти диссоциирован-
ных молекул воды. Некоторые
вторичные электроны получают
очень высокую энергию, и то-
гда они образуют 6-электроны.
Существует две теории, в
которых рассматривается судь-
ба вторичного электрона. Со-
гласно первой — теории Саму-
эля — Маги [11] — электрон,
после того как он термализуется, может возвратиться к роди-
тельскому положительному иону и нейтрализовать его. Образую-
щаяся при этом сильно возбужденная молекула воды распа-
дается на радикалы Н и ОН [см. реакцию (8.3)]. Согласно
второй теории — теории Ли — Платцмана [12] — вторичный элек-
трон удаляется от родительского иона на расстояние около 150 А
и образует гидратированный электрон. Родительский ион, всту-
пая в ион-молекулярную реакцию, дает в качестве ее продуктов
НзО++ОН.
По сути дела, в модели Ли — Платцмана образуется та же
пара свободных радикалов. Эти радикалы в отличие от модели
-Самуэля — Маги, где радикалы помещаются в элементе объ-
-ема радиусом около 10 А, разделены расстоянием около 150 А.
Результаты, полученные с помощью диффузионно-кинетической
теории, свидетельствуют в пользу теории Самуэля — Маги.
При движении через жидкость быстрой частицы шпоры хао-
тически распределены в пространстве, и среднее расстояние ме-
жду ними составляет около 5000 А. Шпоры, образуемые медлен-
ными частицами, перекрываются и в результате возникает ци-
.линдрическая колонна с диаметром около 20 А. Частицы, обра-
зовавшиеся в шпорах, Н, ОН, НО2 и e~q, диффундируют наружу
и вступают во взаимодействие с молекулами растворенных ве-
ществ или друг с другом. Заметная доля радикалов одного вида
будет рекомбинировать внутри шпор и создавать молекулярные
продукты радиолиза Н2 и Н2О2. Другая часть радикалов, также
довольно заметная, будет при рекомбинации образовывать мо-
лекулы Н2О. Оставшиеся радикалы диффундируют в объем
224
растворителя. Количество ооразовавшихся в этих процессах
молекул Н2 и Н2О2 характеризуется выходом молекулярных
продуктов, количество образовавшихся частиц е~ , Н и ОН ха-
рактеризуется выходом радик
Процессы, о которых идет
как процессы, постепенного р
теория (см. раздел 8.5) поз-
воляет рассчитать радиус
шпор в зависимости от вре-
мени, а также долю радика-
лов внутри шпор, не проре-
агировавших к моменту вре-
мени t. Типичные результа-
ты, полученные с помощью
диффузионной теории, пока-
заны на рис. 8.1. Здесь при-
ведены . данные для трека,
состоящего из изолирован-
ных сферических шпор ра-
диусом 10 А, каждая из ко-
торых содержит по 12 ради-
калов. Кривая рассчитана
по уравнению (8.12) для ре-
акций одного радикала при
постоянной концентрации
растворенного вещества (10
альных продуктов.
здесь речь, можно рассматривать
сплывапия шпор. Диффузионная
Рис. 8.1. Размер шпор и доля нереком-
бинировавших радикалов в зависимости
от времени.
3 моль!л). Использованы следую-
щие значения параметров:
D — 4,0-10-5 см~!сек\
k — ks ^= 10~п см*Цсек-радикал).
Рис. 8.1 показывает, что радикалы в шпорах исчезают очень
быстро [13].
Если концентрация растворенного вещества достаточно вы-
сока, то вместо обычных реакций рекомбинации Н и ОН — ра-
дикалов в шпорах, приводящих к образованию Н2 и Н2О2, будут
идти реакции с растворенным веществом и поэтому снизится
выход молекулярных продуктов. Как будет позднее показано,
этим можно воспользоваться для выяснения того, идут ли про-
цессы взаимодействия акцепторов со свободными радикалами
шпор. В радиационной химии применяют специальную симво-
лику, которая позволяет отличать выход молекулярных и ради-
кальных продуктов от наблюдаемого выхода, т. е. действитель-
ного выхода тех или иных веществ в условиях данного экспери-
мента.
Символами Gh, Gh£, GH2o£ и т. д. обозначают выход ради-
кальных и молекулярных продуктов, а символами G(H2)<
G(H2O2), G(H) —наблюдаемые выходы. Так, например, в аэри-
рованной воде G(H2O2) = 1,2 в то время как Gh2o2 =0,72.
8.4. ЛИНЕЙНАЯ ПЕРЕДАЧА ЭНЕРГИИ
И ВЫХОД ПЕРВИЧНЫХ ПРОДУКТОВ
Скорость потери энергии на единицу длины пути ионизирую-
щей частицы dEfdx называется линейной передачей энергии
(ЛПЭ) *. В водных системах ЛПЭ для различных видов излу-
чения можно экспериментально охарактеризовать по значению
выхода радикальных и молекулярных продуктов. В табл. 8.4
Таблица 8.4
Выход радикальных и молекулярных продуктов в облученном
деаэрированном растворе 0,8 н. H.,SO4
Вид излучения GH GOH СН2 СН2О2
в0Со 3,65 2,95 0,45 0,80
D+, 18 Мэв ...... 2,39 1,75 0,71 1,03
D4-, 8 Мэв 1,71 1,45 1,05 1,17
Не2+, 32 Мэв . ... . 1,28 1,06 1.14 1,25
10B(n, a)7Li 0,23 0,41 1,66 1,57
приведены Gn, Goh, Gh2 и Gh2o2, измеренные в деаэрированном
растворе 0,8 п. H2SO4 при облучении радиацией различных ис-
точников. Выходы Gh2 и Сц2о, можно измерить непосредствен-
но, по выходы радикальных продуктов Gn и Gon, как это далее
будет показано, должны быть рассчитаны, исходя из представ-
лений об определенном кинетическом механизме.
Так как энергия, необходимая для образования одной пары
ионов, не зависит от энергии ионизирующего излучения **, раз-
личные значения G для выхода, например, П2 при облучении
у-квантами 60Со и дейтронами с энергией 8 Мэв объясняются,
очевидно, иными факторами помимо природы излучения. Счи-
тают, что при высокой ЛПЭ (в случае цилиндрического трека)
плотность радикалов и других активных частиц много выше,
чем плотность тех же частиц при низкой ЛПЭ (в случае сфери-
ческих шпор). Поэтому в первом случае рекомбинации между
активными частицами идет легче, чем во втором. Следователь-
но, выходы молекулярных и радикальных продуктов должны
* Определение ЛПЭ, которое дается здесь авторами в общем случае,
неверно. ЛПЭ характеризует не линейные потери энергии частицы, а значение
энергии, локально переданной веществу. При этом ЛПЭ является усред-
ненной величиной, и конкретное ее значение зависит как от способа усред-
нения, так и от пределов, в которых это усреднение проводится. (См. Ива-
нов В. И. Курс дозиметрии. Изд. 2-е М., Атомиздат, 1970.) — Прим, перев.
** Строго говоря, это верно лишь для частиц, скорость которых равна по
крайней мере скорости протона с энергией 2 Мэв.
226
измениться. Для высокой ЛПЭ выход молекулярных продуктов
будет большим, а выход радикальных продуктов малым.
Данные, представленные в табл. 8.4, показывают, почему под
действием рентгеновского излучения при малой мощности до-
зы вода разлагается в гораздо меньшей степени, чем при дей-
ствии тяжелых частиц. При облучении тяжелыми частицами вы-
ход молекулярных продуктов оказывается выше выхода ради-
кальных, тогда как при облучении у-кваптами! 60Со выход ради-
кальных продуктов выше.
В табл. 8.5 приводится ряд начальных значений ЛПЭ для
разных видов излучения в фсрросульфатном и цсриевосульфат-
пом растворах. Методы, с помощью которых значение ЛПЭ мож-
но найти из кинетических данных, рассматриваются далее.
Таблица 8.5
Начальные значения ЛПЭ для разных видов излучения
Вид излучения
Л11Э,
о
зе/А
Вид излучения
ЛПЭ,
о
м/А
««Со .............
Электроны, 2 Мэв..........
36S, Р-частицы (средняя энергия
46 кэв).................
Рентгеновское излучение, 250 кв
» 10 кв
» 8 кв
(3-Частицы трития (средняя энер-
гия 5,5 кэв) ........
Дейтроны, 20 Мэв.........
» 8,4 Мэв............
» 5,2 Мэв............
0,02 H 12,0 Мэв 1.7
0,02 Ile2+, 38 Мэв - . 2,2
T+, 2,7 Мэе 3,0
0,07 И' , 0,9 Мэе 3,0
0,10 Нс2+, 12 Мэв 5,0
0,20 Не2+, 5,3 Мэв (21°Ро) 8,8
0,28 «Li(n, а 205 Мэе)3Н, 2,73 Мэв — 10,0
Нс2+, 3,4 Мэв 12,0
0,36 Продукты деления, 65 Мэе (мае-
0,45 са 138) • . -700,0
0,55
1,30
8.5. ДИФФУЗИОННО-КИНЕТИЧЕСКАЯ ТЕОРИЯ
В диффузионно-кинетической теории решается задача пред-
сказания выхода радикальных и молекулярных продуктов по
известному начальному пространственному распределению ак-
тивных частиц. Среднее расстояние между шпорами, начальный
радиус шпор и средняя концентрация активных частиц в их
объеме, коэффициенты диффузии частиц и константы скорости
соответствующих реакций считаются известными. По этим дан-
ным нужно рассчитать долю радикалов, избежавших рекомби-
нации в процессе расплывания шпор.
В общей форме диффузионное уравнение записывается сле-
дующим образом:
д-С-^г' = DzV2G - kfii - ZktJCtj + ZkeCe +2km,nCmCn, (8.12)
8* 227
где Di — коэффициент диффузии; V2 — оператор Лапласа; —
константа скорости реакции первого порядка исчезновения ча-
стиц г; ki} — константа скорости реакции второго порядка исчез-
новения частиц I при взаимодействии с частицами /; ke — кон-
станта скорости реакции первого порядка исчезновения частиц i
при взаимодействии с электроном; km, п — константа скорости
реакции второго порядка исчезновения частиц i при взаимодей-
ствии men; C,(r, t) —средняя концентрация частиц i в точке г
в момент времени t.
Для Н-радикалов в воде, принимая что для них имеют ме-
сто реакции
k.
Н+Н-^Н2; (8.13)
Н + ОН^Н2О; (8.14)
^+Н+‘4Н, (8.15)
справедливо:
2Ж = £V2 [Н] - h [Н] [ОН] -t- ka [е-] [Н+] . (8.16)
Начальная концентрация атомов водорода зависит от ЛПЭ
излучения, коэффициента диффузии водорода, констант скоро-
стей различных реакций, концентраций реагентов и значения их
коэффициентов диффузии. Исходя из сравнительно хорошо из-
вестных констант скоростей и коэффициентов диффузии, удалось
объяснить, по крайней мерс качественно, наблюдаемые экспери-
ментальные данные. Первая довольно удачная попытка такого
рода была сделана Самуэлем и Маги [11]. Они считали, что на-
чальное распределение радикалов в шпоре или треке гауссово
и что характер распределения сохраняется и при расплывании
шпоры. Такое предположение принято называть гипотезой за-
данной диффузии.
Еще один постулат требует, чтобы радиус гауссова распре-
деления с течением времени менялся так, как будто идут толь-
ко диффузионные процессы.
Основываясь на этих предположениях, Самуэль и Маги рас-
считали отношение (Gh2 + Gh2o2)/G(—Н2О) для случая одного
радикала в отсутствии растворенных веществ и получили значе-
ние 0,28, которое хорошо согласуется с экспериментальным
значением, равным 0,26. Куперман [14, 15] подробно проанализи-
ровал этот расчет и провел свои собственные вычисления.
Особую ценность диффузионная теория приобретает в тех
‘случаях, когда необходимо учесть влияние различных факторов
на скорость рекомбинации радикалов.
228
Рассмотрим такую схему реакций:
М av-* R + продукты k-J\
R + S -> продукты ks Cs Cr\
R + R->Ra k(Cr}\
(8-17)
(8.18)
(8.19)
где I — скорость поглощения энергии в единице объема; R —
радикал; S — молекула растворенного вещества.
Рис. 8.2. Значение (1—Л’) в зависи-
мости от Z для разных значений
q=ksCstb'. tB= 1,25-IO-10 сек; D=
=2-10-5 см.2/сек; ro=lOA£ k=
= 10_ 11 см3(молекула-сек); a=6;
1 — <7-'lO-5 (KfiCs-8 104 сек—1);
2— q~ 10-3 (KsC,s =8-10е сек—1);
3 — cy=10—1 (KgCs =8 108 сек-1);
4 — ?=10 {KsPs — 8 • ID10 сек—1).
Рис. 8.3. Значение (1—N) в зависи-
мости от q=ksCstB для разных зна-
чений Z:
1-Z=-VP эв/А; 2 —Z=I0 эв/А; 3-2-
= 1 эв /А; 4 — Z=10-' эв/А; 5 — 2-
-Ю-2 эв/А.
Реакция (8.18) представляет собой реакцию между активной
частицей R и молекулой растворенного вещества S. Реакция
(8.19) — реакция рекомбинации радикалов. Пусть N — доля ра-
дикалов, участвующих в реакции первого порядка [можно счи-
тать, что реакция (8.18) первого порядка, так как концентрация
растворенного вещества остается существенно постоянной].
По методу Гангули и Маги [16] значение N можно рассчи-
тать, приняв некоторые упрощающие допущения и используя
уравнение (8.12). Конечное значение N можно выразить в виде
функции от трех параметров:
1) параметра Z, характеризующего передачу энергии в си-
„ 1 dE\,c
стеме Z=---- (------}dE;
Е 0 \ dx J
2) константы' скорости реакции второго порядка k [см. урав-
нение (8.19)];
229
3) параметра q=ksCstQ, где t0 — время диффузии из точеч-
ного источника.
На рис. 8.2 отложено значение (1—N), равное доле радика-
лов, участвующих в реакции второго порядка [см. уравнение
(8.19)], в зависимости от Z при постоянном k для различных
значений параметра q. Использованы следующие значения па-
раметров: f0=l,25-10-10 сек- 75=2-10~5 см21сек; число радика-
лов в шпоре, а = 6.
Рис. 8.3 наглядно показывает, что выход молекулярных про-
дуктов увеличивается с ростом ЛПЭ. Нетрудно видеть, что при
низкой ЛПЭ и высоких значениях q = ksCsta выход молекуляр-
ных продуктов будет зависеть от присутствия растворенного ве-
щества.
8.6. ЗАВИСИМОСТЬ ВЫХОДА МОЛЕКУЛЯРНЫХ ПРОДУКТОВ
ОТ КОНЦЕНТРАЦИИ РАСТВОРЕННОГО ВЕЩЕСТВА
Влияние концентрации растворенного вещества па выход
НгО2 иллюстрируется табл. 8.6 и 8.7 [17, 18], где приведены ре-
зультаты, относящиеся к облучению кислых аэрированных рас-
творов КВг и KCL В обоих растворах радикалы ОН акцепти-
руются растворенными веществами согласно реакциям:
СГ + ОН + Н+ -> С1 4- Н2О; (8.20)
Вг- ОН -> ОН- 4- Вг. (8.20а)
Таблица 8.6 Таблица 8.7
Начальный выход Н,О2 Начальный выход Н2О2
в аэрированных растворах КВг в аэрированных растворах КС1
КВг, моль!л G(H2O2)
0,8 и. H2SO4 рН=2
IO-2 0,675 0,555
10-3 0,675 0,66
ю-« 0,725 0,70
10-5 0,76 0,73
0 0,78 0,75
КС1, моль}л G(H2O2)
0,811. H2S04 pll=2
1 0,18 0,36
10-1 0,67 0,84
Ю-з 0,95 0,97
Ю-з 1,06 1,03
10“4 1,12 —
Таблица 8.8
Значение G (Н2) в различных растворах
G(H„) Раствор Концентрация, моль]л G(H2) Раствор Концентрация, моль! л
0,45 kno2 4-Ю-5 0,38 CuS04 1,25-Ю-з
0,41 kno2 1,2-Ю-з 0,32 CuSO4 1,74-10-a
0,36 KNO, 1,6-10-2 0,21 CuSO4 1,0
0,28 KNO, 0,165 0,45 H2SO4 2,5-10-3
230
Табл. 8.8 показывает, как меняется выход молекулярных про-
дуктов (в частности, молекулярного водорода) в зависимости
от концентрации для различных
демонстрирует влияние акцеп-
торов свободных радикалов на
экспериментально наблюдае-
мый выход молекулярных про-
дуктов и выход, рассчитанный
при некоторых упрощающих
допущениях по уравнению
(8.12) [19]. Экспериментальные
точки откладывались с учетом
различий в абсолютном выходе
112О2 и Н2 и в реакционной
способности акцепторного рас-
твора разной концентрации
при взаимодействии с одним
и тем же радикалом. Учет этих
факторов позволил нанести
все экспериментальные точки
на одну кривую.
растворенных веществ. Рис. 8.4
Рис. 8.4. Отношение G (молекуляр-
ных продуктов) к GM в зависимо-
сти от концентрации растворенного
вещества. GM — выход молекуляр-
ных продуктов в отсутствие акцепто-
ров.
8.7. ВЛИЯНИЕ ИНТЕНСИВНОСТИ ИЗЛУЧЕНИЯ
При очень высоких интенсивностях излучения следует ожи-
дать гомогенного распределения активных частиц в реакционной
среде в отличие от ситуации при низких мощностях дозы, когда
активные частицы концентрируются в шпорах и цилиндрических
треках. Следовательно, при очень интенсивном излучении долж-
ны выполняться кинетические закономерности, характерные для
гомогенных систем. По для того чтобы считать систему гомо-
генной, концентрация активных частиц в ней должна превысить
начальную концентрацию активных частиц в шпорах, которая
равна примерно 1 М.
Эта концентрация активных частиц должна создаваться за
время, сравнимое со временем диффузии на расстояние в не-
сколько молекулярных диаметров (около 10-11 сек). Для соблю-
дения этого условия мощности дозы следует довести до уров-
ня 1019 рад]сек, что, очевидно, превышает любой разумный
предел. Тем пе менее закономерности гомогенной кинетики
применимы к анализу влияния мощности дозы, .так как рассмат-
риваемые процессы разыгрываются в микросекундные времен-
ные интервалы. По сравнению со временем реакций внутри
шпор, составляющим от 10~8 до 10-9 сек, микросекунда—очень
большое время, и при анализе кинетических процессов в систе-
ме следует учитывать только те активные частицы, которые со-
хранились после завершения всех начальных реакций в шпорах.
Но ввиду того, что шпоры распределены гомогенно (при дейст-
231
вии рентгеновского излучения или электронов), всю систему
можно рассматривать как гомогенную. Таким образом выпол-
няются условия стационарного состояния для процессов образо-
вания и расхода активных частиц. Используя схему реакций
(8.17) — (8.19) для стационарного состояния, можно записать:
= = (8.21)
4kki \ 1 /й j
kl cl ) ~ \
О О / )
(8.22)
Рис. 8.5. Значение N в зависимости
от ksCs Для различных значений
1[эв/(см3-сек)]: Ач=2,8-10~11 см3Цмо-
лекула-сек)-, fei=0,05 радикал/эв-,
1 — 1=4 . [О'6; 2 — 1=4 • I018: 3 — 1=4 10м;
4 - 1=4 I022; Б — 1=4 • 1024.
Доля радикалов, прореагировавших с растворенным вещест-
вом, будет равна:
^==l_^s££(/1 , j
2/^ А2С2 )
На рис. 8.5 приводится график зависимости N от ksCs, рас-
считанный в соответствии с уравнением (8.23), где принимали
Л] = 0,05 радикал!эв, fes=2,8-10—11 см?/(сек-радикал) и k=
= 10-11 см2!(сек-радикал).
Пример 8.2, Оцените интенсивность излучения при которой начинается
перекрывание шпор или треков, приняв, что сохраняется только 19% пер-
воначально возникших радикалов и среднее расстояние между шпорами в
любой момент времени равно п~^3. Начальное число радикалов в шпоре
а=12; кроме того, выполняется зависимость, показанная на рис. 8.1.
Решение. Число шпор в единице объема в момент времени I при условии,
что каждая шпора содержит а радикалов, будет равно
INt
п = , (8.24)
где N— число радикалов, образующихся при поглощении 1 эя; 1—интен-
сивность излучения, эвЦсм3-сек), а—число радикалов в шпоре.
Согласно графику рис. 8.1, 10% радикалов сохранится через 2,8-10~7 сек.
Радиус шпор при этом будет равен 550 А. Величина I, при которой шпоры
232
начнут перекрываться, будет соответствовать среднему расстоянию между
шпорами, равному радиусу шпор г:
(a \1/s
-==- ; (8.25)
IM /
а
I . (8.26)
Nir3
Число сохранившихся радикалов в шпоре равно 1,2.
' 1>2 радикал
( радикал \
I Ю-t-------- ) (2,8-10-’ сек) (550-10“8 см)3
\ 100 эв J
/ = 2,7-1013 эв[(см3-сек).
Отсюда следует, что только при чрезвычайно высоких интенсивностях
облучения может возникнуть гомогенное распределение радикалов.
8.8. РЕАКЦИИ АКТИВНЫХ ЧАСТИЦ
Активные частицы вступают в реакции, характер которых
зависит от природы растворенного вещества. Наиболее частыми,
однако, являются окисление при взаимодействии с ОН-радика-
лами, восстановление при взаимодействии с атомами Н и соль-
ватированными электронами, а также нейтрализация ионов
сольватированными электронами.
Реакции в шпорах, завершающиеся примерно через 10~8 сек
после поглощения энергии, приводят к образованию в соответст-
вии с уравнениями (8.2) — (8.11) таких продуктов, как радика-
лы, сольватированные заряженные частицы, молекулярный во-
дород и перекись водорода, образующиеся в последующих
реакциях. Многие реакции этих активных частиц друг с другом
и с молекулами воды хорошо изучены; для них удалось изме-
рить константы скоростей. Уравнения реакций и соответствую-
щие константы скоростей приведены в табл. 8.9.
Если судить по значению констант скоростей, приведенных
в табл. 8.9, при умеренных интенсивностях излучения и ней-
тральных pH в отсутствие растворенных веществ в системе (ис-
ключая Н2 и Н2О2) основными промежуточными продуктами ре-
акций в облученной воде являются , Н, ОН, Н2 и Н2О2. При
низких или высоких pH характер промежуточных продуктов не-
сколько изменяется.
При рН=11—14 ОН-рйдикал находится в равновесии с ионом
ОН-, что ведет к появлению иона О~ благодаря реакции
ОН + ОН~ О~ + Н2О. (8.27)
233
Таблица 8.9
Типы реакций и коистаиты скоростей реакций
промежуточных продуктов в облученной воде
Реакция k, моль 1сек 1
е-+Н3О+->Н+Н2О %+% >Н2+2ОН- е-+н2о2^он+он- е-+Н2О н-гон- н-рн -н2 . Н4-ОН >Н,0 он-рон -н;о2 Н3О+-РОН~^2Н2О Н2О е~-рп—*н2-рон~ он-рн2 >н,ори н-рн2о2 -н2о рон он-рп2о2 - но,-рн9о но2-рно2->н2о2-ро2 2,2.101° 1,2.101°* 1,2-101° 16± 1 2-Юю 1,2-101° 1,1-101° 1,4-1011 2,5-1010 4,5-10’ 9-10’ 4,5-10’ (рН=0,4) 4,7-10° (pH—2,7) 2,5-10е (р1Ь 5,5) 2,9-10’
*3десь для константы скорости реакции использовано выражение 2k.
8.8.1. Реакции восстанавливающих активных частиц
В водных системах существуют две восстанавливающие ак-
тивные частицы * — гидратированный электрон е~ и атомарный
водород Н*. Во многих системах реакции обеих частиц приво-
дят к появлению одинаковых продуктов. Это задержало откры-
тие гидратированного электрона до тех пор, пока кинетическими
методами не удалось доказать существование двух типов вос-
станавливающих частиц.
Гидратированные электроны превращаются в водородные
атомы в следующих реакциях:
е^ + НаОч-Н + ОН- (8.28)
и
е~-рН+->Н. (8.29)
* Еще до открытия гидратированного электрона в радиационной химии
существовало представление о двух типах активных восстанавливающих ча-
стиц. Первую обозначали символом Н1 и считали, что это водородный атом,
вторую обозначали символом II и считали, что это, по-видимому, гидрати-
рованный электрон. Такого рода обозначения можно встретить в литературе,
относящейся к периоду между 1957 и 1962 гг. Сейчас установлено, что ак-
тивная частица, обозначавшаяся как Н, действительно является гидрати-
рованным электроном, а И1 — атомарный водород.
234
Реакция (8.28) — медленная, тогда как реакция (8.29) идет
очень быстро (см. табл. 8.9). В кислых растворах быстро
превращается в атомы Н, и тогда набор активных частиц в си-
стеме сводится лишь к двум радикалам — Ни ОН. Например,
в кислых 0,1 М растворах гидратированный электрон превра-
щается в водородный атом за I0-9—10-10 сек.
В нейтральных и щелочных растворах это не так, поэтому
могут встречаться различные промежуточные продукты. Сход-
ство между реакциями гидратированного электрона и атомар-
ного. водорода можно проиллюстрировать следующими приме-
рами:
Cu2++H->Cu+ + Н+;
Cu2+ + e7q -> Си+ ;
Н + О2->НО2;
eaq + С>2 С>2 ’
Н+ + ОГ-*НО2;
О он
II I
СН3С—СН3 + Н -> СН3С—СН3;
О О-
II I
СН3С-СН3 + е^ -> СН3С—СН3;
- 0“ ОН
I I
СН3С—сн3 + Н+ -> СН3С—сн,.
(8.30)
(8.30а)
(8.31)
(8.31а)
(8.316)
(8.32)
(8.32а)
(8.326)
Реакции, в которых сольватированный электрон и атомарный
водород дают различные продукты, идут с перекисью водорода
и хлоруксуспой кислотой. В первом случае
Н -Ь Н2О2 Н2О + ОН; (8.33)
+ ПА -> ОН" + ОН. (8.34)
Отношение констант скоростей реакций #(8.з4)/£(8.зз) равно при-
мерно 500.
В разбавленных растворах хлоруксусной кислоты б(Н2) воз-
растает благодаря реакции (8.29) и реакции
Н + С1СН2СООН Н2 + С1СНСООН. (8.35)
При увеличении концентрации хлоруксусной кислоты (она
относится к числу сравнительно слабых кислот) с реакцией
(8.29) начинает конкурировать реакция недиссоциированных
молекул кислоты:
е- + С1СН2СООН Cl" + СН2СООН. (8.36)
235
Появление в растворе НС1 позволяет отличить реакцию
(8.36) от реакции (8.35).
Так как у гидратированного электрона имеется свой спектр
поглощения [10], то с помощью метода импульсного радиолиза
удается непосредственно измерить константы скоростей многих
реакций, в которых участвует е~ . В методе импульсного радио-
лиза реакции гидратированного электрона можно сравнительно
просто отличить от реакций атома Н, так как при столь высокой
интенсивности излучения появляется возможность провести
спектральные измерения.
Ниже даны некоторые характеристики гидратированного
электрона:
Длина волны максимума поглощения........................ 7200 А
О
Коэффициент экстинкции при 7200 А ......................15 800Л1—1 см~1
* °
Коэффициент экстинкции при 5780 А.......................10 бООЛ!”1 см~1
Время полупревращения (е“+Н2О).......................... 780 мксек
о
Радиус распределения заряда ........................ 2,5—3,0 А
Энергия гидратации (расчетная) ........................ 1,82 эв
Коэффициент диффузии....................................4,7-10-5 см2/сек
£°(е-+Н3О+^i/aHa+H2O) .................................. -2,56 в
Гидратированный электрон представляет собой гораздо бо-
лее мощный восстанавливающий агент, чем атом Н. Окисли-
тельно-восстановительный потенциал Е° атомарного водорода
равен —2,1 в для электрода Н, Н+|Нг (окисление атома Н),
тогда как для гидратированного электрона Е°=—2,56 в. Это
значение рассчитано Баксендейлом [20]. Примененный им ме-
тод может служить примером определения окислительно-восста-
новительного потенциала.
Пример 8.3. Найдите Е° для реакции
«79+h+ = VH4:
[Ha]V«
к ~ ~.
KJ [Н+]
Решение. Для этой реакции необходимо определить Д6’°, так как AG°=
=—nfE°, где f — число Фарадея; п—заряд; Е° — окислительно-восстанови-
тельный потенциал. Запишем прямую и обратную реакции, константы ско-
рости которых известны:
kf
е~ + Н2О — - Н + ОН~ , kf st 16 M~i сек-i;
kr
JI + ОН------> е- + НгО, kr = 1,8- 10’Л1-1 сек-1.
236
Константа равновесия Ki будет равна
kf [Н] [ОН-] 16
К‘-^” К|ГнД--7Л17Г"8'910’ ’
Дб° = — RT In К я 9120 кал/моль.
Искомую константу равновесия К можно получить, умножив Ki на обрат-
ную величину константы равновесия Kw для реакции
[ОН-] [Н+1
1--= 10-м (доо = о зоо кал/моль)
[Н8О]
и на обратную величину константы равновесия Кн, для реакции
ТН2^Н:
[Н]
= '[и j1// = 500 кал/моль),
в результате получим
1 1
К1' Kw ’ *Н,
к_ IH21V-
[%] И
ДО0 — 9120 — 19 300 — 48 500 = — 58 680 кал/моль;
п/Е° = — 58 680; £° = — 2,56 в.
Следовательно, гидратированный электрон на 0,46 в более
сильный восстановитель, чем атом Н*.
В табл. 8.10 приведены окислительно-восстановительные по-
тенциалы Е° для ряда важных окислительно-восстановитёльных
агентов в водных системах.
Таблица 8.10
Окислительно-восстановительные потенциалы
Электрод
£°, в (Il+«I.O М)
ОН I он-
h. Н+ I н2
Н2О22Н I Н2О
НО2, Н+ I Н2О2
Н+, НО2 I Н2О, он
О2, 2Н I Н2О2
Н+ | у2н2
Н+, О2 | но2
Н+ I н
—2,8
—2,1 (окисление атома Н)
1,8
1,7
—1,35
—0,68 (восстановление Н2О2)
—0,00 (восстановление НО2)
+0,30 (восстановление НО2)
+2,1 (восстановление атома Н)
* Полученное значение Е° несколько выше, чем найденное Баксендей-
лом, благодаря тому, что использованы другие значения констант скоростей.
237
8.8.2. Реакции окислительных активных частиц
Основными окислительными агентами в облученной воде яв-
ляются ОН-радикалы. В ряде случаев, однако, следует учиты-
вать вклад вторичных активных частиц — радикалов НО2 и
Н2О3, ионов О<у и О^-- Эти активные частицы наблюдаются при
облучении насыщенной кислородом воды. Некоторые оптические
Таблица 8.11
Оптические свойства некоторых
промежуточных продуктов
в насыщенной кислородом
облученной воде
Активная частица Коэффициент молярной экстинкции, см^1 Длина волны, о А
но.. Н50 2300
0? 1900 4300
0Г 1060 2400
он 370 2600
Предполагаётся, что Н2Оз возникает в реакции
свойства окислительных актив-
ных частиц приведены в
табл. 8.11. Радикалы НО2 об-
разуются в результате реакций
Н-|-О2->НО2; (8.31)
ОН 4- Н2О2 -> Н2О Ь НО2; (8.37)
е- + О2->°-, (8.31а)
вслед за которой идет ре-
акция
О2~ -г Н+ НО2.
Частицы Н20з обнаружива-
ются только при облучении с
высокой мощностью дозы.
ОН + НО2->Н2О3.
(8.38)
Выход Н2О3 для рН>2, G(H2O3) ~ 1,6.
При обычных интенсивностях облучения концентрация ОН-
и НО'2-радикалов недостаточно высока, чтобы привести к обра-
зованию Н2О3. Чаще эти радикалы участвуют в других реак-
циях, в которых возникают другие типы продуктов.
Гидроксильный радикал обнаруживает сходство с очень сла-
бой кислотой и при достаточно высоких pH диссоциирует
ОН + ОН О~ 4- Н2О. (8.38а)
Скорости реакции с различными растворенными веществами
для двух форм гидроксильного радикала — ОН и О- --различ-
ны [21]. Для реакции (8.38а) рК=П,9±0,2.
8.9. ВЛИЯНИЕ pH НА ВЫХОД РАДИКАЛЬНЫХ
И МОЛЕКУЛЯРНЫХ ПРОДУКТОВ
В водных системах действительный состав активных частиц
будет зависеть от pH, так как возможны реакции диссоциации,
такие, как НО2ч^Н++О^ (рН=4,5) или (8.38а). Кроме того,
быстрые реакции типа е~„+Н+ могут преобладать над осталь-
238
ними настолько, что иные реакции гидратированного электрона
не будут идти. Поэтому значение радиационно-химического вы-
хода активных частиц зависит от pH. В табл. 8.12 приведены
значения G для разных активных частиц в зависимости от pH.
Данные табл. 8.12 свиде-
тельствуют, что G(—Н2О), рав-
ный Goh + 2Gh2o2, при кислых
pH выше, чем при нейтральных
и щелочных. Одно из предло-
женных объяснений этого фак-
та состоит в том, что в кислых
растворах оксониевые ионы ре-
агируют с электронами внутри
шпор (существенно увеличивая
их размеры). Возникающие
при этом атомы Н обладают
гораздо большей скоростью
диффузии и выходят в объем
растворителя, что приводит к
увеличению выхода радикаль-
ных продуктов.
Таблица 8.12
Выход радикальных и молекулярных
продуктов в облученной воде
при разных значениях pH
Радиа- ционно- химический выход H2O 0,8 H. H2SO4 pII=iO,S
G 2,58 — 2,6
eaq
GII 0,55 3,65 0,52
CO1I 2,59 2,95 2,6
CII2 0,45 0,45 0,45
CH2O2 0,72 0,80 0,65
8.9.1. Определение выхода молекулярных продуктов
Облучение деаэрированных разбавленных водных растворов
различных неорганических солей (Се4+, Fe2+, Br~, I-, Н2О2,
As3+, Fe(CN)|+ и др.) ведет к выделению молекул Н2 с одина-
ковым выходом, который нс зависит от величины pH. В значи-
тельной степени теории радиолиза водных растворов основы-
ваются именно па этом факте. Накопление молекул Н2 и Н2О2
определяет выход молекулярных продуктов. Как видно из
табл. 8.12, Gh2 составляет 0,45.
Выход Н2О2 по удается определить столь же прямым и про-
стым способом, как Gh2. Известно, одпако, что реакция
Н2О2 г 2Fe2+ ^2Fe3 H + 2ОН"
идет довольно медленно, и ее константа скорости равна при-
мерно 61,9 М-’-секг1. Это позволяет прокалибровать количест-
во перекиси, возникшей во время облучения в разбавленном
растворе Fe2+ (10~6 М), по выходу реакции постиррадиацион-
ного окисления ионов Fe2+. Эксперименты такого рода позволи-
ли довольно точно измерить радиационно-химический выход
Н2О2.
Значения Gh2o2, приведенные в табл. 8.12, получены в опы-
тах с более сложными системами, такими, как насыщенные
кислородом водные растворы кислот и цериевосульфатные рас-
творы. Gh2o2 в отличие от выхода молекулярного водорода за-
висит от pH среды. Значение Gh2o2 при очень высоких pH
239
(~14) падает до 0,45. Отчасти это объясняется тем, что начи-
нает идти реакция (8.38а), а реакционная способность 0“ и
ОН в отношении образования Н2О2 заметно различается.
8.9.2. Определение выхода радикальных продуктов
Выход радикальных продуктов удается определить после
тщательного изучения механизмов реакций, протекающих в при-
сутствии растворенных веществ, таких, как сульфат железа,
сульфат церия, кислород, водород, СО, разбавленная муравьи-
ная или бромистоводородпая кислота (с добавками или без до-
бавок перекиси). Возможность экспериментального определения
выхода радикальных продуктов зависит от правильности при-
нятой кинетической схемы. Если схема правильная и экспери-
мент тщательно выполнен, то выход радикальных продуктов
удается измерить с достаточной точностью.
Далее в качестве примера будет рассмотрено определение
выхода радикальных продуктов в водной системе, содержащей
Вг, Н2О2 и О2*. В том же примере читатель познакомится с
принятой в радиационной химии системой обозначений.
Пример 8.4. Аллен и Чапский исследовали радиационно-химические реак-
ции в нейтральных и кислых растворах, содержащих ионы Вг- [22]. В не-
которых опытах в те же растворы вводили О2 и Н2О2. Пользуясь приведен-
ной ниже схемой реакции, определите выходы G(H) и G(e~ ).
Решение. В разбавлеппых кислых или нейтральных растворах КВг, на-
сыщенных • кислородом с добавками или без добавок перекиси водорода,
имеют место следующие реакции:
Н2О ~ -> Н, ОН, е~ , Н2, Н2О;
(1) Вг- 4- ОН -> ОН- 4- Вг; (8.39)
(2) Вг 4- Н.2О2 НО2 4- Н+ 4- Вг~; (8.40)
(3) е“4-Н2О2 ОН 4-ОН—, (8.34)
Н2О
(4) а-+О2 —НО.4-ОН-; (8.31а, б)
(5) Н + О2 - НО2; (8.31)
(6) 2НО2 - Н2О24-О2; (8.41)
(7) %+Н+ - Н2О 4- Н; (8.29)
(8) Н 4- Н2О > н2о + ОН. (8.33)
Ион Вг- акцептирует радикалы ОН и препятствует реакции ОН+Н2->-
->Н2О+Н. Реакция (8.39) идет очень быстро, так же как и реакции (8.40),
(8.34), (8.31а) и (8.29) (см. табл. 8.9). Реакции (8.31) и (8.33) сравнительно
медленные, как и реакция (8.41).
* Как было недавно выяснено, химические механизмы, рассмотренные
в примере 8.4, представлялись слишком упрощенно. Реакция ОН-радикала с
Вг- зависит от pH, и образующийся в этой реакции атом немедленно взаи-
модействует с Вг-, давая Вг2“. Именно Вг2- реагирует затем с Н2О2.
240
Будет рассмотрено четыре варианта систем, содержащих ионы Вт-.
I. Кислый раствор бромида, насыщенный О2.
II. Кислый раствор бромида, . насыщенный О2 и содержащий добавку
НгО2 в такой концентрации, что [Н+]2»[НгО2].
IIJ. Нейтральный раствор бромида, насыщенный О2.
IV. Кислый раствор бромида, насыщенный О2 и содержащий добавку
Н2О2 в такой концентрации, что [Н+] « [Н2О2].
Для упрощения кинетической схемы будем пользоваться порядковыми
номерами. реакций, записанными слева от соответствующих уравнений.
Вариант I. В кислом растворе [Н'-]3>[О2] или [Н2О2] и поэтому реак-
цию (7) можно считать единственной реакцией е~. Тогда для начального
выхода [Н2О2] мы можем записать:
[Н2О2]нач - Go = Сц2ож — (2) + (6). (а)
В реакциях, выход которых должен учитываться в уравнении (а), участ-
вуют радикалы Вт, НО2, ОН н Н.
Вг образуется в реакции (1) и расходуется в реакции (2). Поэтому
можно записать
(1)-(2). (б)
1Ю2 образуется в реакциях (2) и (5) и расходуется в реакции (6).
Скорость исчезновения радикала в результате рекомбинации с таким же
радикалом записывается согласно принятым правилам как 2fe6[HO]22, тогда
как скорость появления Н2О2 в той, же реакции запишется как /г6[НО2]2.
Поэтому
(2) + (5) = 2.(6). (в)
ОН-радикал образуется с выходом Goh и исчезает в результате реакции
(1). Поэтому
°он = (1). (г)
Н-радикал образуется в результате реакции (7) настолько быстро, что
выход Н-радикалов можно считать равным Gh, принимая, что [Н] = [е~ ]-[-
+[Н]. Атомы Н исчезают в результате реакции (5). Следовательно,
GH = (5). (д)
Подставляя уравнения (б); (в), (г) и (д) в уравнение (а), получаем
[Н2О2]нач = Go = СИ1Ог + -у (GH - GOH). (8.42)
Измерив начальную концентрацию Н2О2 в исследуемом растворе и зная
Gji2o2> найдем значение '/2(бн — Gon).
Вариант II. В кислый раствор бромида, насыщенный О2, добавлена
Н2О2 в такой концентрации, что [Н]»[Н2О2]. Снова е~ участвует только в
реакции (7).
Так как в систему добавлена Н2О2, следует учитывать реакцию (8), и
выход Н2О2 теперь будет равен G(H2O2):
G (Н2О2) = GHiO£ - (2) - (8) 4- (6). (а)
Активными радикалами являются Вг, ОН, Н и НО2.
Как и в варианте I, выход ’атомов Н определяется величиной Gh- Рас-
ходуются атомы Н снова в реакции (5), но так как добавлена Н2О2, следует
учесть конкурирующую реакцию (8):
«Н = (5) + (8)- (б)
241
Вг образуется в реакции (1) и расходуется в реакции (2). Поэтому, как
•и в варианте 1,
(1) = (2). (в)
ПО2. Те же реакции, что и в варианте I,
(2)-(5) = 2.(6). (г)
ОН-радикал образуется при радиолизе с выходом Gon и в реакции (8).
Расходуется в реакции (2), которая заметно преобладает над реакцией (1):
GOH + (8) = (2). (д)
Подставляя в уравнение (а) выражение для (2), получим
0(Н2Ог) = 0Н2о2—GOI1 2-(8) ; 6. (е)
В реакциях (8) и (6) участвуют радикалы НО2 и П, которые можно
исключить из конечного уравнения. Отметим прежде всего, что в выражения
для скоростей реакций (5) и (8) входит концентрация [И]
(8) _ fe8 [Н2О2] (Н|
(5) ' fe6[O2](H] (Ж)
или
Подставляя полученное выражение в уравнения (б) для радикала Н, найдем
/г6 [О-.]
Он-(5)-г(8) = (8) + (8)-^-
ИЛИ
(8)=rZZKWZ- (3)
+ МН2О2]
Теперь (8) выражено через измеряемые величины концентрации О2 и
Н2О2. Складывая стехиометрические уравнения для радикалов Н, ОН и НО2,
т. е. уравнения (б), (д) и (г), можно выразить (6) через измеряемые ве-
личины
GH + (5) + (2) + GOH + (8) = (5) + (8) 4- 2-(6) + (2);
Подставляя уравнения (з) и (и) в (е), получим
G (Н2О2) = GHzq2 Goh 2GH GH GOH
feslQzl 1 2 + 2 + ks [H2O2]
ИЛИ
2GH . (GH GOH)
G (Н2О2) — ^н2О2 MO2] 1 2 + ЫН2О2]
242
Подставляя выражение для начального выхода перекиси Go, полученное в
варианте I Гем. уравнение (8.42)], найдем
fes[O2] GH
!+ Л8[Н2О2] G(H2O2) — Go
(8.43>
Наклон графика 1/G(H2O2)—Go в зависимости от [О2]/[П2О2] и величина
отрезка, отсекаемого на оси ординат, дадут величину выхода Gn (или точ-
нее G восстанавливающих активных частиц).
Вариант III. В нейтральном растворе бромида, насыщенном О2, ос-
новными процессами являются реакции (4), (5), (1), (2) и (6). Так как
концентрация Н+ невелика, реакция (4) преобладает над реакциями (3) и
(8). Стехиометрические уравнения радикальных реакций будут выглядеть-
следующим образом:
Н: GH = (5). (а>
ОН: GOII=(1). (б>
НО2: (4) + (5)4-(2) = 2.(6). (в)
е~ : G = Ю- (г>
°’ eaq
Вг: (1) = (2). (Д)
G0-GHsOi-(2) + (6); (е)
(6) = GH + GOH 4- G^_ ;
aq
Go - gh2o + V (GH -I- G - GOIIy (8.44)
Измерив начальный выход перекиси и зная GH о , найдем величину Оц +
+ G g— —Gon-
aq
Вариант IV. Разбавленный кислый раствор бромида насыщен О2 и
содержит добавленную И2О2. В присутствии добавки Н2О2 начинается замет-
ная конкуренция между промежуточными продуктами и растворенными ве-
ществами. Одиако ввиду того, что реакцию (8) можно считать медленной по
сравнению с реакцией (5) и при условии [О2]>[Н2О2], конкуренцию между
этими реакциями можно не учитывать:
G(H2O2) = GH!O2-(2)-(3)4-(6). (а)'
Стехиометрические уравнения для промежуточных продуктов деть следующим образом: будут выгля-
Н: О„+(7)=-(5). (б)
eaq : Gg_+(3) + (4) + (7). (В)
а?
ОН: GOH + (3)^(1). (г>
НО2: (2) +(4)4- (5) = 2.(6). (д>
Вг: (1) = (2). (е)
Складывая стехиометрические уравнения, мы сможем выразить (6), через
измеряемые величины:
Gn + G£_ 4- GOH
g<?
243
Уравнения (2) и (3) найдем следующим образом:
(7) Л7 [Н+] (4) МО>1
(3) МН2О2] ’ (3) ЫН2О2] ’
Подставляя выражения для (7) и (4) в уравнение (в), получим
G
е—
feJOJ g fe7|H+] ~ = (3)- <3)
+ МИД] + feslibO.,]
Из стехиометрических уравнений (2) = (3)4-Gon
2G
1 е
G (Н2О2) = GHiOi + — (GH -I- Gg_ — GOH j — - fe4[Q,| w fe7 [H4-] •
+ k3 [H.Q.] + k3 [H,O2] -
подставив выражение для Go [см. уравнение (8.44)], найдем
2G
е
1 + J 4- -------—
k3 [Н2О2] мн.о.,]
или
2G _
% t МО2] к, [11+]
G0-G(H2O2) ~ *' fe3 [Н2О,] + МН2О2]
Зная Go и измерив С(Н2О2) при различных концентрациях растворенного
вещества, можем определить величину Ge— .
' aq
Пример 8.5. Недавно для определения выхода радикальных продуктов
стали использовать радикал-акценторные реакции взаимодействия СО и ОН и
взаимодействия О2 с атомами Н и гидратированными электронами. Реакция
взаимодействия СО и ОН подробно изучена в ряде фотохимических и ра-
диационно-химических исследований. В газовой фазе гидроксильные радикалы
реагируют с молекулами СО, давая СО2Ч-Н, а в растворе основным про-
дуктом являются радикалы НСОО. Хорошо изучены также реакции е~ и
Н с молекулами О2.
Рассмотрите механизмы химических превращений в водной системе, со-
держащей СО и О2, и покажите, как можно определить выход радикальных
продуктов.
Решение. Система уравнений будет выглядеть следующим образом:
Н2О ~ ► е~ , Н, ОН, Н2, Н2О2;
<1) eaq + °2 - - О- (+Н4- Н2О2); (8.31а,б)
(2) н + о2 - Н2О; (8.31)
(3) ОН-}-СО - НСОО; (8.46)
(4) НСОО + О2 - СО2 + НО2; (8.47)
(5) 2НО2 -» Н2О2 + (8.41)
244
Запишем теперь систему стехиометрических уравнений для промежуточных
продуктов: G(H2O2) = GHjOi+(5). (a)
еад : Ge_ = (1). (6)
Н: ад GH = (2). (в)
ОН: GOH = (3). (r)
НО: (1) + (2) + (4) = 2-(5). (Д)
НСОО: (3) = (4). (e)
Производя замены и суммирование, найдем
G (НА1 — GHaOs + 2 [G + GH + GOH] (8.48)
или 4 % J G (HA) = GHjOt [Gred + GOXJ. (8.49)
Измерения G(H2O2) дали значение 3,58 [23, 24]:
3,58 = GhjOj ~2~ №red 4~ GOXJ.
Значения 6геа и Gnx можно получить отдельно, комбинируя этот резуль-
тат с результатами примера 8.4. Экспериментальное значение Go для началь-
ного выделения 112О2 в насыщенных кислородом разбавленных растворах
КВг равно 0,99.
Подставляя значение Со в уравнение (8.44)
Go = 0,99 = GHtOt + [Gred-Gox] (8.50)
и вычитая уравнение (8.50) из уравнения (8.49), получим
3,58—0,99 = 2,59 + GOH (или Gox). (8.50а)
Для всех водных систем справедливо следующее уравнение материаль-
ного баланса:
2GH, + Gred = 2GHjOii + Gox. (8.51)
Вместе с уравнением (8.49) это дает
GHj Gred = 3,58.
Пользуясь известным значением Gh* =0,45, получим
Gred ~ 3*13.
Как указывалось ранее, если концентрация растворенного ве-
щества высока, то произойдет акцептирование радикалов внут-
ри шпор. Полученные значения G будут отличаться от обычных
значений, характерных для радиолиза воды. Применяется не-
сколько методов, чтобы установить, идут ли радикал-акцептор-
ные реакции в шпорах. Обычно измеряется выход либо молекул
245
Н2, либо молекул Н2О2 в зависимости от концентрации раство-
ренного вещества. Зная, как меняется выход молекулярных
продуктов, можно учесть, какая часть радикалов исчезает из
системы в результате радикал-акцепторных реакций в шпорах,
и, таким образом, оцепить истинные значения радиационно-хи-
мического выхода радикалов. Например, для того чтобы ском-
пенсировать акцептирование радикалов в шпорах в водной си-
стеме СО + О2, нужно ввести 1%-ную поправку значения
G(H2O2). Такой метод не общепринят, однако его используют
достаточно часто.
Как показано в предыдущих примерах, в водных системах
при образовании окислительных и восстанавливающих частиц
выполняется уравнение материального баланса
Ge— + Gh 2Gh2 = Goh + 2Gh2os - (8.52)
aq
Во всех случаях на один грамм-эквивалент образующихся
окислительных частиц возникает один грамм-эквивалент восста-
навливающих частиц. Поэтому, прежде чем приступать к ана-
лизу какого-либо кинетического механизма, следует проверить,
выполняется ли соотношение (8.52) *.
8.10. ДОЗИМЕТР ФРИКЕ
Радиационная химия дозиметра Фрике исследовалась весь-
ма интенсивно. Этому способствовало, во-первых, широкое при-
менение дозиметра и, во-вторых, общее значение подобных ре-
акций для химии.
Основной реакцией в ферросульфатной системе является ре-
акция окисления двухвалентного железа
Fe2+ -}- Н2О2 -> Fe3+ + ОН -'г ОН" . (8.53)
Эта реакция изучена достаточно полно несколькими груп-
пами исследователей. Гидроксильный радикал, образующийся
в этой реакции, можно идентифицировать по его взаимодейст-
вию с различными добавками. Рассматриваемая реакция позво-
ляет, не проводя радиолиза, получить представление о скорости
реакции ОН-радикалов с ионами Fc2+. Поэтому реакция Фен-
тона (8.53) служит отправным пунктом при изучении химиче-
ских механизмов дозиметра Фрике.
Если дозиметрический раствор FcSO4 в 0,8 п. H2SO4 содер-
жит воздух, то принято считать, что в системе идут следующие
реакции:
Н2О----Н2 4- Н2О2 -г Н + ОН ’- е - ;
(г-ьН+->Н (8.29)
* Аллен (Allen А. О. Rad. Res. Suppl., 1964, v. 4, p. 69) предположил, что
в число окислительных частиц должны входить также атомы О.
246
Fe2+ + H2O2 -> Fe3+ 4- ОН + OH~ (8.53)
(реакция Фентона);
Fe2+Н-ОН-> Fe31-+ ОН-; (8.54)
Н -J- О2 (из растворенного воздуха) _> НО2; (8.31)
Fe2+ НО2 -> Fe3+ НО? ; (8.55)
НОГ-*-Н+^Н2О2.
Согласно данным табл. 8.12, для активных промежуточных
продуктов в 0,8 н. H2SO4 значения выходов равны:
Он, ~ 0,45; Goh = 2,95;
Gh2o2 — 0,80; Gn — 3,65.
Суммируя выражения для выхода реакций (8.29), (8.53), (8.54),
(8.31) и (8.55), получим:
G(Fe3+) = 2Gh2o2 Ч- Gon г 3GH = 1,6 + 2,95 -j- 10,95;
G(Fe3 ' ) = 15,5.
Это общепринятое значение G дозиметра Фрике при воздей-
ствии у-излучения 60Со или электронов высоких энергий (0,5—
2 Мэв).
Окисление насыщенных воздухом растворов Fe2+ происходит
согласно реакциям (8.53), (8.54) и (8.55). Константы скоростей
для этих реакций измерялись методом импульсного радио-
лиза [25]:
(1) Fe2+ + ОН -> Fe3+ 4- ОН”,
(2) Fe2+ 4~ НО2 Fe3 + 4- НО? ,
k > 108 М~1 сек~1;
/г 7,3-105 М~' сек.~х ;
(3) 2Fe2+ Ч- Н2О2 -> 2Fe3+ Ь 2ОН“ , k = 61,9М~1 сек .
Самая быстрая реакция происходит между ионами Fe2+ и
ОН-радикалами, самая медленная — между Fe2+ и Н2О2. В рас-
творах, насыщенных воздухом, реакция НЧ-О2 идет настолько
быстро, что се обычно исключают из общей схемы процессов и
сразу включают радикалы НО2 в число первичных продуктов.
Продукт окисления — ионы Fe3+. Их появление сопровож-
дается восстановлением эквивалентного количества О2 до Н2О
и небольшого количества Н2О до Н2. В отсутствие кислорода
конечными продуктами являются ионы Fe3+ и молекулы Н2,
образующиеся в отношении 2:1. Снова происходит окисление
ионов Fe3+, но восстанавливаются теперь молекулы Н2О. Посту-
лируется следующий механизм процесса:
Н2о.—Н, ОН, Н,, Н2О2;
%+н+->н+н2°; (8-29)
247
Fe2+ + H2O2 -> Fe3+ +- ОН + OH“; (8.53)
Fe2+ 4- ОН Fe3+ + ОН~ ; (8.54)
(H + H+)->Ht; (8.56)
Fe2++ (Н + Н+)Fe3++ Н2 (8.56а)
или
Fe2+ (Н2О) + Н Fe (ОН)2+ 4- Н2; (8.57>
Fe(OH)2+-> Fe3+4-ОН~ (8.57а)
Суммируя выходы для этих реакций, найдем
С (Fe3+) = 2Сн2ог 4- Goh 4- GH = 1,6 4- 2,95 4- 3,65 = 8,2; (8.58)
6(Н2) = 0,45 4-3,65 = 4,10. (8.59)
Обычно считают, что за образование молекулярного водо-
рода ответственна реакция (8.57). Реакция (8.56) и (8.56а), так-
же ведущие к образованию молекулярного водорода, были
предложены Риггом, Штейном и Вейсом [26], а реакция (8.57) —
Дэвисом, Гордоном и Хартом [27]. В пользу рассмотренного ме-
ханизма образования молекулярного водорода свидетельствует
тот факт, что при пропускании через фсрросульфатный раствор
атомарного водорода, полученного в электрическом разряде,
образуются ионы Fe3+ [23].
В насыщенных воздухом дозиметрических растворах в тот
момент, когда расходуется весь растворенный кислород, G(Fe3+)
резко падает с 15,5 до 8,2. Может происходить либо общее вы-
горание кислорода в растворе, либо (при высоких мощностях
Интенсивность^ эв^сн1- сек)
Рис. 8.6. Влияние интенсивности об-
лучения на величину G(Fe3+):
О, А — аэрированные растворы:
X — 1,2-Ю-з М [О2] и IO-2 М [Fe2+].
дозы) локальное кислородное
обеднение среды, которое мо-
жет повести к ошибочным ре-
зультатам дозиметрических из-
мерений.
8.10.1 . Выход дозиметра Фрике
при импульсном радиолизе
При интенсивности излуче-
ния ниже 1025 эв/(л-сек)
G(Fe3+) остается постоянным
и равным 15,5. Однако при вы-
соких интенсивностях излуче-
ния ~ 1028 эв/(л • сек) G(Fe3+)
падает до 8 [28]. Влияние
мощности дозы иллюстрирует
рис. 8.6. В наблюдаемом эф-
248
фекте, однако, играет роль не истинная интенсивность излучения,
а полная поглощенная доза за время импульса. Дело в том,
что продолжительность импульсов мала по сравнению с време-
нем жизни радикалов, и эффект проявляется при облучении
раствора с мощностью дозы 106—107 рад/сек в течение более
длительного времени. В' ферросульфатных растворах отсутствует
истинный эффект, связанный с интенсивностью излучения, так
как реакции идут настолько быстро, что ионы Fe24- полностью
расходуются, прежде чем установится стационарное состояние.
8.10.2 . Влияние ЛПЭ на выход дозиметра Фрике
Выход окисленных ионов Fe3+ в зависимости от ЛПЭ иссле-
дован практически для всех возможных ситуаций. Результаты
суммированы в табл. 8.13 и на рис. 4.5.
Таблица 8.13
Значение G (Fe3+) в зависимости от ЛПЭ
Вид излучения ЛПЭ, эв/А 6 (Ре3+)
Электроны, 10 Mse 0,02 15,7±0,3
2 Мэв 0,02 15,45±0,3
у-Кванты в0Со — 0,10 15,68 + 0,07
Рентгеновское излучение, 100 кв -0,15 14,1 + 0,5
60 кв 0,28 13,1 +0,5
8 кв -0,36 13,4+0,6
fi-Частицы, тритий 0,45 12,9 + 0,2
D+, 21,16 Мае -1,2 11,3 + 0,5
ГН , 6,2 Мэв 1,7 8,5-1 0,4
Н+, 1,99 Мэв -2,1 8,О+О,4
а-Частицы, 40 Мэв -— 8,7 + 0,4
20 Мэв .— 7,05±0,4
р02Ю 8,8 5,1 ±0,1
10B(n, a)’Li — 4,38 + 0,08
Механизмы окисления при разных ЛПЭ не изменяются, и
имеют место реакции (8.27), (8.31), (8.53) и (8.55). Однако зна-
чения G(H2) и G(O2H2) и, следовательно, G(H) и G(OH) зави-
сят от ЛПЭ. При высоких ЛПЭ в число первичных активных
частиц часто включают радикал НО2. Этот радикал образуется
в результате реакции в шпорах между гидроксилами и пере-
кисью водорода.
Используя значения G для различных радикальных и моле-
кулярных продуктов, приведенные в табл. 8.4, можно найти
G(Fe3+) и, следовательно, ЛПЭ для данного излучения.
249
Рис. 8.7. Влияние
pH на величину
O(Fe3+).
так и
8.10.3 . Влияние pH на выход дозиметра Фрике
С ростом pH выход ионов Fc3+ уменьшается. Эта зависи-
мость представлена на рис. 8.7. Приемлемое объяснение эффек-
та можно дать, учитывая изменение выходов Gn и бон в кислых
растворах (см. табл. 8.13), а также изменение природы ком-
плексов, включающих ион Fe3+. Реакцию
окисления нельзя проводить выше рН=3, так
как при таких pH начинается выпадение трех-
валентного железа в осадок.
8.10.4 . Роль ионов СП в дозиметре Фрике
В дозиметр Фрике .(см. раздел 4.4.1) вхо-
дит NaCl в концентрации 10-3 М. Ионы С1_
вводятся в дозиметр для того, чтобы снизить
влияние органических загрязнений, которые
могут изменить выход ионов Fc3+ как в аэри-
в деаэрированных растворах. До того, как
установили вредное влияние органических примесей, был про-
веден ряд измерений отношения G(Fe3+) аэрированный/О(Ре3+)
деаэрированный. Значение отношения менялось от 2 до 2,8, как
теперь известно, благодаря влиянию примесей. В насыщенных
воздухом растворах ОН-радикалы взаимодействуют с молеку-
лами органических веществ, давая новые радикалы
ОН RH->H2O + R'. (8.60)
Эти радикалы реагируют с растворенным кислородом, обра-
зуя органические перекиси (RO2), которые ведут себя подобно
радикалу ПО2 и в конечном счете окисляют три иона Fe2+. В де-
аэрированных растворах радикал, образующийся в результате
реакции (8.60), может восстанавливать Fe^ до Fe2+. Поэтому
соотношение G(Fc3+) в аэрированных и деаэрированных рас-
творах -меняется.
Ионы С1_ в кислых растворах, по-видимому, реагируют с
ОН-радикалами и образуют атомы С1, которые успевают окис-
лить ион Fe2+ прежде, чем тот прореагирует с молекулой орга-
нической перекиси. Однако в более концентрированных раство-
рах NaCl образуются ионы С1~.
Химия разыгрывающихся процессов теперь уже не может
быть столь простой, так как следует ожидать, что и атомы CI
и ионы С1~ будут легко реагировать с молекулами воды. Тем не
менее G(Fe3+) в 1 М НС1 не отличается от выхода в растворах
0,8 н. H2SO4 (тс же 15,5).
8.10.5 . Восстановление иона Fe3+
Вплоть до весьма высоких доз излучения в деаэрированных
растворах отмечается пропорциональность между окислением
Fe2+ и поглощенной дозой. Однако при дальнейшем увеличении
250
дозы начинается восстановление ионов Fe3+ атомами Н, и ли-
нейность показаний дозиметра нарушается.
Механизмы процессов окисления и восстановления, очевидно,
существенным образом зависят от относительной концентрации
различных ферро- и феррикомплексов в сульфатном растворе.
Среди идентифицированных комплексов встречаются FcSO4,
Fe(OH)2'1', Fe(H2O)2+ и Fe(OH)2Fe4+.
8.10.6 . Конкурентные реакции
в железосодержащих растворах
В большей части водных систем, рассматриваемых в ради-
ационной химии, происходит конкуренция между молекулами
и радикалами за возможность участия в реакциях с активными
частицами. Конкуренцию можно исключить или снизить, добав-
ляя избирательные акцепторы радикалов или подбирая концен-
трации растворенных веществ таким образом, чтобы ингибиро-
вались определенные реакции. Прекрасный пример анализа
системы с конкурентными реакциями дается Свитом и Тома-
сом [29].
Пример 8.6. Разбавленный деаэрированный ферросульфатпый раствор,
насыщенный Н2 и находящийся под давлением Н2, облучается импульсами
(длительность импульса 1 мксек) электронов с энергией 13 Мэв. Сразу же
после импульса раствор анализируется. Получите соответствующие кинетиче-
ские уравнения и найдите, какая доля атомов II реагирует с ионами трех-
валентного железа.
Решение.
Н,О —> Н, ОН, Н2, Н,О2;
kt
Н + Н Н,; (1)
Fe3+ + Н [Fe3 HI] > Fe2+ + Н+; (2)
ОН + Н2 -> Н2О + Н. (3)
В рассматриваемой системе реакциями
Н2О.2 + Fe2+ - Fe3-!- + ОН + ОН~ (4)
и
ОН -ф- Fe2+ -> Fe3+ + ОН- (5)
можно пренебречь, так как анализ продуктов проводится сразу же после
импульса и, следовательно, медленная реакция (4) не оказывает влияния.
А реакция (5) имеет низкую вероятность по сравнению с реакцией (3), так
как в любой момент времени концентрация Fe2b значительно ниже, чем кон-
центрация Н2. Поэтому следует учитывать конкуренцию лишь между реак-
циями (1) и (2)
= - 2k, [Н]2 - k2 [Н] [Fe3+],
at
(6)
251
Интегрируя уравнение (6) по частям, найдем
fe2 [Fe3+]
IHJ= [С exp fc2 [Fe3+]/] — ’ (7)
где C — постоянная интегрирования; ее можно найти из граничного условия,
согласно которому при /=0 (конец импульса) концентрация атомов Н имеет
конечное значение Г.
„ k, [Fe3+] + 2V
С = —-------. (8)
Подставив значение постоянной в уравнение (7), получим
(*2 [Fe3+] + 2ki I) (exp k.2 [Fes+] t — 2kt I) ‘ '
Если концентрация атомов H к концу импульса равна /, то доля водо-
родных атомов f, реагирующих с ионами Fe3+ начиная с момента 1=0 в те-
чение времени, которое велико по сравнению с периодом полупревращения
(для практических целей его можно считать равным бесконечности), будет
равна:
СО
/ = J W+] [Н]^
о
Подставляя для [Н] выражение (9), найдем:
/ = 7____________k, [Fe3+]3 /_________
J (k2 [Fe3+] + 2kt I) J* IFeS+J/ _ 2ktI
Пусть
(k2 [Fe3+] + 2kiP) IFe®+I' _ 2k± 1 = S;
‘ dS
~=k2 [Fe*+](S + 2M);
dt
Г (fe, [Fe3+])2 dS_____________
J Sk2 [Fe3+] (S + ’
[Fe®+ ]
t = 0 S = k, [Fe3+];
t — oo S = oo.
Интегрируя по частям, получим окончательный результат в следующем виде:
k2 [Fe3+] Г S j°°
2ktl L S + 2^11 [fc3+]
fe3[Fe3+] , /, , 2k,I \
2fe,/ \ k2 [Fe3+] J
252
8.10.7 . Дозиметрическая система
ферросульфат — сульфат меди
Добавление солей меди в дозиметр Фрике приводит к томуг
что показания дозиметра перестают зависеть от присутствия
кислорода. Радикалы Н и НОг восстанавливают ионы Си2+
до Си+. Эти ионы, в свою очередь, восстанавливают Fe3+ до-
Fe2+:
Cu2+ + Н Н+ + Си+;
Н + О2 НО2;
НО2 + Си2+ -> Си+ + Н+ + О2;
Cu+ + Fe3+ Cu2+ + Fe2+.
Добавки Cu2+ особенно полезны при высокой интенсивности-
излучения.
8.11. ЦЕРИЕВОСУЛЬФАТНАЯ СИСТЕМА
При облучении кислых растворов сульфата четырехвалент-
ного церия образуются ионы трехвалентного церия и кислород..
Выход Се3+ не зависит от присутствия кислорода, поэтому
остается одним и тем же как в аэрированных, так и в деаэри-
рованных растворах.
Считается, что в системе идут следующие реакции:
Н2О-----Н, ОН, Н,, Н2О2;
Н+Н; (8.29)
2Се4+ 4- Н2О2 2Се3+ О2 + 2Н+ ; (8.61)
Н + О2->НО2; (8.31)
Се4+ -(- НО2 Се3+ + Н+ •- О.,; (8.62}
Се3++ ОН-> Се4++ ОН-. (8.63)
В растворе сульфата церия молекулярный водород обра-
зуется с выходом G = 0,45, который совпадает с Gh2 и, следова-
тельно, реакция ОН-радикалов с Н2 отсутствует. Существование
реакции (8.63) подтверждается исследованиями [30, 31] реакций
перехода Се4+^Се3+. Реакция (8.61) хорошо изучена обычны-
ми кинетическими методами. Исходя из рассмотренного меха-
низма радиолиза, можем записать:
G (Се3+) = 2Gh2o2 т Gh — Goh •
Подставляя числовые значения G для молекулярных и ради-
кальных продуктов из табл. 8.4, находим:
G (Се3+) = 2 (0,80) 4- 3,65 — 2,95 = 2,30.
253-
Таким образом анализ всех рассмотренных систем показы-
вает, что судьба неорганических ионов в облученных водных
растворах зависит от реакций промежуточных продуктов, воз-
никающих при облучении. Можно сформулировать следующие
общие характеристики этих реакций:
1. Растворенные вещества реагируют с промежуточными
продуктами, если их окислительно-восстановительные потен-
циалы находятся в нужном соотношении.
2. Если ход процесса определяется взаимодействием с ради-
калами Н и ОН, то конечный выход продуктов должен зависеть
от pH раствора.
3. Ионные образования, участвующие в реакциях, должны
находиться в равновесии с различными аквакомплексами или
другими видами комплексов.
4. Для любых механизмов должны соблюдаться условия ма-
териального баланса. Число окисленных эквивалентов равно
числу восстановленных эквивалентов:
2Gh2 -|- Gred 2Gh2o2 т- Gox.
5. Реакции в разбавленных растворах не должны зависеть
ют интенсивности излучения, но могут зависеть от полной погло-
щенной дозы.
6. При высоких концентрациях растворенных веществ будут
происходить радикал-акцепторные реакции в шпорах.
7. Выход продуктов должен зависеть от ЛПЭ.
8.12. РЕАКЦИИ В ВОДНЫХ РАСТВОРАХ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. МУРАВЬИНАЯ КИСЛОТА
Водные растворы муравьиной кислоты исследовались доста-
точно подробно [32]. Предполагается следующий механизм ра-
диолиза разбавленных (10-3 Л4) деаэрированных растворов му-
равьиной кислоты при pH ниже 3:
н2о - - > е- , н, ОН, Н,, Н2О2;
НСООН % + н+ > Н+ + НСОО-; Н; (8.29)
НД-НСООН > Н, + СООН; (8.64)
Н + НСОО- > Н2 + СОО-; (8.65)
он + НСООН > Н,О + НСОО (или СООН); (8.66)
ОН + НСОО- - ОН- + НСОО (или СОО- + Н,О); (8.67)
НСОО + СООН * НСООН + СО,; (8.68)
соо- 4- н,о, - СО2 + ОН + ОН-; (8.69)
СООН + Н2О2 -» Н2О + СО2 +ОН; (8.70)
2СОО- (СОО-)2. (8.7Г
254
Суммарная реакция
НСООН -> СО2 + Н2
описывает цепной процесс окисления муравьиной кислоты:
G (- НСООН) = G (СО2) = G (Н2) = GH2o2 + V2 [Gred + G0H] =
= 0,80 H 72 (3,65 -|-2,95?;
G (— НСООН) = 4,10.
При увеличении концентрации муравьиной кислоты увеличи-
вается выход G(CO2). Рост выхода углекислого газа объяс-
няется протеканием следующих цепных реакций:
НСОО |- НСООН -> СО2 + НСО -|- Н2О; (8.72)
НСО + НСООН -> НСНО + НСОО. (8.73)
Эти реакции чувствительны к изменениям pH. Как было-
показано экспериментально, ионы НСОО- легче реагируют с Н-
и ОН-радикалами, чем недиссоциированпая кислота.
В приведенном расчете значения G(CO2) не принималась во
внимание реакция (8.71) (реакция образования щавелевой кис-
лоты). Эта реакция начинает играть существенную роль только
при pH выше 6—7. По мере увеличения pH G(CO2) падает,
пока при pH = 7 основными продуктами радиолиза не стано-
вятся молекулярный водород и щавелевая кислота.
В системе также образуется окись углерода, но со сравни-
тельно низким выходом. По-видимому, СО возникает при взаимо-
действии небольшой части радикалов COOII с молекулами Н2О2:
СООН + II2O2 -> СО •- Н2О + НО2. (8.74)
При возрастании pH начинает идти реакция гидратирован-
ного электрона с молекулами муравьиной кислоты. Однако про-
дукты этой реакции пока не идентифицированы.
8.12.1. Азотсодержащие соединения
При радиолизе деаэрированных водных растворов глицина
(pH=6,5) происходит образование следующих продуктов:
Н2 NH3, СН3СООН, НСОСООН, СН2О и СО2. Предлагается
[33—35] следующая схема радиолиза:
Н2О---, Н, ОН, Н2, Н2О2;
NH2CH2COOH -> NH3b СН2СОО~; (8.75)
ео- + NH;- СН2СОО- -> МН3 4- СН2СОО“ ; (8.76)
Н + NHt СН2СОО- -> NHt СНСОО- + Н2; (8.77)
ОН 4- NHt СН2СОО- -> NHt СНСОО- + Н2О. (8.78)
255-
Так выглядят основные идущие в системе реакции. Радика-
лы ОН предпочтительно реагируют с а-углеродными атомами,
отрывая атом Н. Гидратированный электрон взаимодействует
с цвиттер-ионом по реакции (8.76). Хорошим подтверждением
существования радикалов СН2СООН и NH2CHCOOH, возникаю-
щих в результате отрыва водорода, может служить образование
небольших количеств янтарной и аспарагиновой кислот.
Вопрос о том, какие из радикальных реакций разыгрываются
в системе, остается пока открытым. Тем не менее можно вы-
делить реакции, которые дадут качественное согласие с наблю-
даемым выходом продуктов:
СН2СООН 4- NHt СН2СОО~ СН3СОО~ + NHtСНСОО- + Н+ ;
(8.79)
2NHt СНСОО- ->NHt = СНСОО” -J- NHt СН2СОО~ ; (8.80)
Н2О + NHt = СНСОО- -> NH3 + НСОСООН; (8.81)
-> ЫНз + СН2О + СО2; (8.82)
Н2О2 -г NHt СНСОО- NHt = СНСОО” + Н2О + ОН. (8.83)
Суммируя, можно отметить следующие общие черты рассмот-
ренных реакций:
1. Реакция отрыва водорода радикалами Н или ОН проис-
ходит при взаимодействии с лабильными атомами Н. Экспе-
рименты по исследованию Н-радикалов в твердых матрицах
различных спиртов [36] подтверждают такой механизм.
2. Реакции диссоциации при взаимодействии с гидратирован-
ным электроном происходят в том случае, если в результате
образуются стабильные ионы или молекулы [37]. Реакции типа
+ RXR 4~ X”
идут очень быстро, если Х = С1, Вг или I. Скорость реакции
.зависит также от природы R. По порядку значения константа
скорости реакций такого типа составляет 108—1010 М~1сек~1.
3. Обычно наблюдаются реакции взаимодействия Н2О2 с
образующимися радикалами или исходным веществом.
4. Реакции радикалов, возникающих из растворенного веще-
ства, при взаимодействии с Н- или ОН-радикалами ведут к
образованию молекулярных продуктов.
5. Отрыв водорода от групп NH3, NHt или NHt не происхо-
дит, т. е. реакции
NH3 + H-5 * NH2 4~ Н2; (8.84)
NHt 4- Н - + NHt 4- Н2; (8.85)
NHt 4- Н - NHt -F Н2; (8.86)
-256
по-видимому, эндотермические. Экспериментально установлено
[38J, что реакция (8.85) действительно является эндотермиче-
ской, и изменение энтальпии составляет около 27 ккал) моль.
Однако реакция NH4? +е~ ->NH3 + H идет. Константа скорости
этой реакции равна приблизительно 103 ЛН • сек~’.
Пример 8.7. Исходя из сформулированных выше правил, попробуйте
предсказать продукты радиолиза водных растворов первичных аминов.
Решение.
Н.,О —> Н, ОН, е~ , н,, Н.,О,;
NH+cH.,tf + е~ NH3 + СНЯ;
II + NH^CIIgT? -> NH^CHT? л- Н.,;
ОН 4- NH+CH.,7? NITfCH/? + Н2О;
2(СН.,Я) ЯСН»СН2Я;
11,0., л- NH+сНЯ NH+ + ЯСНО -р ОН.
(правило 2)
(правило 1)
(правило 1)
(правило 4)
(правило 3)
При малых уровнях разложения амина основными начальными продук-
тами радиолиза будут .\'Н3, ЯСН2СН2Я и ЯСНО.
8.12.2. Водные растворы спиртов
Продуктами радиолиза деаэрированных растворов метанола
являются этиленгликоль, молекулярный водород и формальде-
гид. Приемлемым механизмом, объясняющим выход этих про-
дуктов, может быть такая совокупность реакций:
Н2О е- , Н, ОН, Н2, Н2О2;
СН3ОН + Н СН2ОН Н2;
СН3ОН + ОН -> СН2ОН + Н2О;
2 (СН2ОН) СН2ОНСН2ОН;
СН2ОН + Н2О2 -> НСНО 4- Н2О 4- ОН;
е- + Н+ -> Н.
aq 1
(8.87)
(8.88)
(8.89)
(8.90)
(8.29)
Гидратированный электрон слабо реагирует с молекулами
метанола и поэтому его основной реакцией будет реакция обра-
зования атомов водорода. Начальный выход продуктов должен
быть равен
G(H2) = Оц2-с Gred;
G(CH2O) = Gh2o2;
G (этиленгликоль) = i/2 (Gred -|- G01I) — 72Gh£o
Фактически процесс сложнее, чем это представлено здесь,
так как появление даже небольших количеств продуктов суще-
9 Зак. 907
257
ствеппо меняет конечный результат. Например, такие реак-
ции, как
е~ + НСНО [НСНСГ] (8.91)
будут изменять выход атомов Н. Тем не менее рассмотренный
механизм пригоден для объяснения процесса в общих чертах.
8.12.3. Растворы бензола
Радиолиз водных растворов бензола всегда привлекал вни-
мание,, так как эта система представляет интерес с точки зрения
дозиметрии. Кроме того, существует перспектива ее использо-
вания для промышленного производства фенола. Недавние ис-
следования методом импульсного радиолиза показали, что пред-
лагавшиеся ранее схемы реакций с участием Н- и ОН-радикалов
были ошибочными. В насыщенных воздухом растворах, по-ви-
димому, имеют место следующие реакции:
Н2О--->е~, Н, ОН, Hg, Н2О2;
е-4-Н+->Н;
ОН -ь С6Нв С6Н6ОН;
е~ |-°2->ОГ(Ог+Н+->№,);
Н + О2 НО2;
ОНС6Н6 + О2 ОНСвН6О2;
ОНС6Н6О2 С6Н5ОН 4- НО2;
2НО2 -> Н2О2 4- О2;
НО, 4- ОНСКН„О, -> органические гидроперекиси.
Z О О Z
(8.29)
(8.92)
(8.31а, б)
(8.93)
(8.94)
(8.95)
(8.96)
(8.97)
Уравнение материального баланса для Н2О2 запишем так:
G(H2O2) = Gh2o. + + V2G (CeH.OH) — V2G (гидроперекиси) =
= 0,72 4- V2 (3,13) 4- V2 (0, Ю) — V2G (гидроперекиси) ;
G (H,o2) = 2,38 — 42 G (гидроперекиси).
Экспериментально измеренный выход 0(Н2Ог) равен 2,2±0,4.
Учитывая, что выход гидроперекиси неизвестен, такой результат
следует считать неплохим совпадением.
Любопытно, что ОН-радикал не отрывает водород от арома-
тического кольца, а, наоборот, прикрепляется к нему. Как в
аэрированных, так и в деаэрированных растворах ОН-радикалы
акцептируются молекулами бензола. Имеющиеся данные ука-
зывают па то, что атомы И также присоединяются к бензолу,
образуя циклогексадиениловый радикал СеН?. Конечный резуль-
258
тат облучения деаэрированных растворов бензола — образова-
ние молекулярного водорода, фенола, дифенила и других орга-
нических продуктов, возникающих в результате реакций радика-
лов СвНбОН и СбН7.
В растворах бензола отмечены постирадиационные эффекты.
В результате пострадиационных процессов образуются дифе-
нил и ряд других димерных продуктов. Предполагается, что
фенол образуется в результате реакции диспропорционирования
2 (C6HgOH) С6Н5ОН д- С6Н7ОН.
(8.98)
8.12.4. Окисление органических соединений
Органические молекулы, растворенные в воде, при достаточно
долгом облучении окисляются и конечными продуктами процесса
оказываются углекислый газ и вода. Для насыщенных воздухом
растворов многих органических соединений при умеренной ин-
тенсивности излучения следующий общий механизм радиолиза
может оказаться достаточно хорошим приближением:
Н2О~^е~, Н, ОН, Н2,'Н2О2;
RH + ОН R Н2О;
H2R ф- О2 RH 4- НО2;
(нейтральные pH) Д- О2 О2 ;
(кислые pH) е~ ~ Нт Н;
аа 1 ’
О? + Н+ -> НО2;
Н + О2->НО2;
2НО2Н2О2 О2;
G(—RH) — G(R) — Сон .
Из предыдущего следует
(8.99)
(8.100)
(8.31а)
(8.29)
(8.316)
(8.93)
(8.96)
G (Н2О2) = Сн2оа 4- VzGh 4- 1/2Goh •
Например, облучение насыщенных воздухом разбавленных рас-
творов этанола приводит к таким реакциям:
Н2О~-£>- Н, ОН, Н2, Н2О„;
ад 1 1 с 4.^
+ °2 - °2-(°Г + н !НО2);
Н-|-О2^НО2;
ОН i- C2HSOH С2Н4ОН 4- Н2О;
С2Н4ОН 4- О2 СН3СНО 4- НО2;
2НО2 Н2О2 4- О2.
(8.31а)
(8.93)
(8.99)
(8.100)
(8.96)
9* 259
Откуда
G (Н2О2) — GII;o3 + -f- */2^011 — 3,58;
G (H2O2) (экспериментальный) = 4,0.
Во многих растворах органических соединений образуются
органические перекиси, которые могут инициировать цепные
реакции
RО2RO2; (8.101)’
RO2 + RH RO2H + R (8.102)
и увеличивать, таким образом, выход.
Задачи
1. Рассчитайте скорость исчезновения е~ в растворе 0,1 М IICI, если
производится облучение раствора с мощностью дозы 300 рад/сек.
2. Приведите десять примеров реакций, в.которых взаимодействие с ато-
мами Н н приводит к образованию одинаковых продуктов.
3. Найдите значение Е° для реакции Fe2+-i-H+->Fe3+-!-,/2II2.
4. Рассчитайте теплоту реакции е_-гН2О->е~ .
5. Используя данные табл. 8.4, рассчитайте ЛПЭ для разных видов ра-
диации.
6. Основными продуктами радиолиза иодных растворов этанола явля-
ются водород, бутан-2,3-днол и ацетальдегид. Считая, что ацетальдегид об-
разуется в результате днспропорцнопнрованпя а-этапольпых радикалов, сфор-
мулируйте механизм, объясняющий появление указанных продуктов.
7. Рассчитайте концентрацию радикалов ОН в 4 см3 Н2О, если этот
объем облучен импульсом (0,4 мксек) электронов с энергией 3 Мэв при силе
тока 10 ма. Принимается, что во время импульса радикальные реакции
не плут.
8. Сформулируйте механизм радиолиза разбавленных насыщенных воз-
духом растворов пропанола. Какие основные продукты появятся в резуль-
тате радиолиза?
9. Чему будет равна температура водного раствора, облучаемого в адиа-
батических условиях при интенсивности излучения 10:0 и 1б24 эв/(см3-сек) в
течение 5 сек, 20 сек, 1 мин?
10. Являются ли эквивалентными приведенные ниже реакции
eaq + О: > »
И 4-О. - НО..?
Мотивируйте ваш ответ.
Глава IX
ДЕЙСТВИЕ ИЗЛУЧЕНИЙ НА ЖИДКИЕ
УГЛЕВОДОРОДЫ
9.1. ВВЕДЕНИЕ
Литература, посвященная воздействию ионизирующих излу-
чений па углеводороды, столь же обширна, как и литература
по радиационной химии водных систем. В ранних исследованиях
[1, 2] были получены главным образом качественные резуль-
таты. Например, Кларк и Пикетт изучали воздействие рентге-
новского излучения на самые разнообразные растворы, включая
растворы антрацена, малеиновой кислоты и иода с бензолом.
Опп проследили также за реакциями конденсации альдегидов
и кетонов, для которых обнаружили заметное каталитическое
действие рентгеновского излучения. В иод-бензольной системе
отмечалось поглощение 6,5% иода. Последующая эксперимен-
тальная работа в этой области стала активно развиваться лишь
в конце 40 — начале 50-х годов.
Радиолиз углеводородов и сходных с ним молекул в значи-
тельной мере можно рассматривать с тех же позиций, что и
радиолиз водных систем. Однако углеводородные системы обла-
дают целым рядом уникальных характеристик, которые застав-
ляют выделить изучение радиолиза этих систем в особую
область исследований.
9.2. ПЕРВИЧНЫЕ ПРОДУКТЫ РАДИОЛИЗА УГЛЕВОДОРОДОВ
Ионизирующая частица, движущаяся в углеводородной среде,
вызывает ионизацию, возбуждение и диссоциацию молекул
точно так же, как и в воде (см. главу VIII). Среднее расстояние
между шпорами при у-облучепии алканов оказывается порядка
5000 А. Однако последующие реакции активных частиц, обра-
зовавшихся в шпорах, гораздо сложнее, чем в воде. При облу-
чении углеводородов возникает четыре основных типа активных
частиц: I) ионы положительные и отрицательные (могут нахо-
диться и в основном, и в возбужденном состоянии); 2) воз-
бужденные молекулы; 3) радикалы (возбужденные и в основном
состоянии); 4) электроны (свободные, связанные в ловушках и
сольватированные).
261
Ионы, возбужденные молекулы и радикалы вступают в реак-
ции внутри шпор. Часть активных частиц покидает объем шпор.
Этими активными частицами определяется выход радикальных
продуктов (более строго — выход активных продуктов с высокой
реакционной способностью). Те же частицы, которые прореаги-
ровали внутри шпор и образовали идентифицируемые соедине-
ния, определяют выход молекулярных продуктов. Таким обра-
зом, в углеводородах следует различать выход радикальных и
молекулярных продуктов. Однако природа соответствующих
реакций в углеводородах намного сложнее, чем в воде.
9.3. ВРЕМЕННАЯ ШКАЛА ПЕРВИЧЫХ ПРОЦЕССОВ
Временная шкала первичных процессов приведена в табл. 9.1
и сходна с аналогичной шкалой для воды. Тем не менее следует
подчеркнуть некоторые различия. Например, в результате ион-
ных реакций, которые включают нейтрализацию, диссоциацию
Таблица 9.1
Временная шкала первичных процессов при радиолизе углеводородов
Процесс
Время, сек
Ионизация и возбуждение
Период молекулярных колебаний, время диссоциации
Термализация электрона
Время соударения в жидкости (для ион-молекулярных
реакций)
Нейтрализация ионов (в сферических шпорах, модель
Самуэля—Маги)
Время диффузии радикала на расстояние, равное толщи-
не мономолекуляриого слоя (D- -2—8-10~5 сл<2/сек)
Рекомбинация радикалов в трековой колонне
Рекомбинация радикалов сферических шпор
Время жизни триплетного возбужденного состояния
10-и
Ю-K
От 10-14 до Ю-12,
10-13
От 10-13 до 10 -12
10-12
10-»
10-8
>ю—4
или ион-молекулярные взаимодействия, образуется гораздо
больше, чем в воде, разнообразных продуктов с высокой реак-
ционной способностью. Именно эта особенность делает угле-
водородные системы уникальными. Выбиваемый при ионизации
электрон может термализоваться, образовывать отрицательный
ион, сольватироваться или реагировать с положительным ионом
и нейтрализоваться. Эксперимент показал [3—5], что в жидком
гексаие практически все термализованные электроны захваты-
ваются ионами в шпорах и лишь небольшая часть уходит в
объем раствора.
262
Значение G для разделенных пар ионов составляет око-
ло 0,1 *. Это резко контрастирует с поведением воды при радио-
лизе, где с помощью измерений проводимости и прямыми
спектроскопическими методами удается установить присутствие
сольватированного (гидратированного) электрона с периодом
полупревращения около 0,6 мксек.
Как показывает табл. 9.1, вторичные электроны термали-
зуются за 10~14—10“12 сек. Однако в углеводородных системах
сродство к электрону алканов или алкенов, по-видимому, имеет
такую величину, что, как правило, длительное существование
сольватированного электрона в этих системах невозможно.
Процесс нейтрализации завершается примерно за 10-10—10-9 сек.
Сольватацию электрона в результате поляризующего дей-
ствия его заряда можно ожидать только для полярных жидко-
стей. Возможность наблюдения сольватированного электрона с
помощью быстрых методов спектрографического анализа опре-
деляется скоростью исчезновения электрона в химических реак-
циях, конкурирующих с процессом нейтрализации. В ряде орга-
нических жидкостей, таких, как алифатические спирты [6] (ме-
танол, этанол, н-пропанол и изопропанол) и этилендиамип [7],
сольватированные электроны наблюдались. В растворах дифе-
нила в этаноле образуется дифенильный ион (С^Ны)-. Другие
случаи наблюдений связанного электрона относятся к раство-
рам антрацена и терфенила в этаноле [8].
Таким образом, существует много данных, показывающих,
что в полярных органических средах происходит сольватация
электрона. В ряде случаев, например, в растворе, содержащем
иод, или в системах, рассмотренных выше, формируются пары
удаленных друг от друга ионов. Однако в алканах и алкенах
этого не наблюдается. Очевидно, ионные пары в таких средах
имеют слишком малое время жизни. Теоретические обоснования
сольватации электрона выдвигались и для неполярных жидко-
стей. Сила связи, однако, должна бы быть очень малой и спектр,
если бы он наблюдался, попал бы в инфракрасную область.
Кроме того, время жизни сольватированного электрона скорее
всего должно быть столь малым, что с помощью современных
методов измерить его не удалось бы.
Согласно табл. 9.1 первичные частицы в течение 10-12 сек
должны прореагировать и образовать активные радикальные
продукты или, возможно, некоторые стабильные продукты. Эти
первичные частицы (ионы, возбужденные молекулы) являются
предшественниками наблюдаемых промежуточных продуктов ре-
акций. Однако конкретная роль тех или иных первичных частиц
* Однако при добавлении C2H2OD в качестве акцептора протонов при
радиолизе циклогексана- удается получить выход положительных ионов
вплоть до 2.6 нон/100 эв. См. Buchanan I. W., Williams F. I. Chem. Phys.,
1966. v. 44. p. 4577.
263
в образовании -активных промежуточных продуктов все еще
остается неясной.
Обычно характер продуктов, образующихся при радиолизе
углеводородов, удается в основном объяснить, -зная, какие сво-
бодные радикалы образуются при облучении. Свободные ради-
калы возникают в результате реакций самых различных частиц
(ионов, возбужденых молекул и np.)t появляющихся в первич-
ных актах.
9.4. ВОЗБУЖДЕННЫЕ МОЛЕКУЛЫ
Как предположили Воеводский и Молин [9], возбужденные
молекулы насыщенных углеводородов в результате внутренней
конверсии быстро переходят из высших состояний в низшее,
оптически разрешенное возбужденное состояние. Молекула
имеет избыток энергии около 7 эв, чего более чем достаточно
для диссоциации углерод-углеродных или углерод-водородных
связей. Такая диссоциация может быть одним из первичных
механизмов образования алкильных радикалов, наблюдаемых
в рассматриваемых системах.' Данные фотохимических [10] и
кинетических [11] исследований, а также эффекты, связанные
с ЛПЭ [12] поддерживают представление о важной роли воз-
бужденных состояний в образовании радикалов.
При изучении методом импульсного радиолиза антрацена,
фенантрена и нафталина в этих ароматических системах уда-
лось обнаружить триплетные состояния. Радиолиз чистого цикло-
гексанона и чистого циклопентаиона приводит к образованию
среди прочих продуктов изомерных производных 5-гексенала и
4-пентенала. Около 75% выхода этих олефиновых альдегидов
с разомкнутой цепью образуется через низшие триплетные со-
стояния кетонов; 25% выхода возникает через другие возбуж-
денные состояния, включая, возможно, и низшее синглетное
возбужденное состояние. При облучении в бензоле эти молекулы
претерпевают такую же реакцию изомеризации, но в данном
случае процесс идет за счет передачи энергии от возбужденных
молекул бензола [13].
Возбужденные молекулы, образующиеся в результате реком-
бинации ионов,’ могут служить источником образования свобод-
ных радикалов [14]. С помощью этого механизма удается
объяснить радиолитическое разложение н-гексапа:
СМ + е~ -> С6Н;4;
С6Н;4 ->СвН13 + Н;
-> С-Нп + СНз;
С4Н9-фС2Н5;
-* С3Н, -ф С3Н7.
264
9.5. ИОННЫЕ РЕАКЦИИ. МОНОМОЛ ЕКУЛЯРНАЯ ДИССОЦИАЦИЯ
ИОНОВ
При движении заряженных частиц в среде образуются ионы.
В углеводородных средах они помимо прочих реакций могут
претерпевать мономолекулярпый распад, как это происходит,
например, с нзобутаном:
изо-С4Н]'о -> СН3 4- C3Ht. (9.1}
Возникающий в этом процессе радикал называют осколоч-
ным радикалом, а ион — осколочным ионом. Для того чтобы
процесс такого рода мог произойти, молекулярный ион должен
обладать сравнительно большой избыточной энергией, так как
подобные реакции существенно эндотермичны и в ходе их дол-
жен происходить разрыв одиоэлектропной углерод-углеродной
связи. Для оценки прочности одноэлектронных связей С—С+
можно воспользоваться расчетом, в котором рассматривается
определенный термодинамический цикл:
D (RC-C R') = D (RC:CR) + I (RC-) — I (RC:CR'\ (9.2)
где D(RC-C+R') —энергия диссоциации одноэлектронной связи;
/(RC-) — потенциал ионизации радикала RC-; /(RC:CR') —по-
тенциал ионизации родительской молекулы (с двухэлектронной
связью); £)(RC:CR)—энергия диссоциации связи (RC:CR).
Пример 9.1. Обоснуйте справедливость уравнения (9.2) и рассчитайте
энергию диссоциации углерод-углеродпой связи в иопе этана С2Пб4’
Решение. Искомое значение будет равно энергии диссоциации связи в
реакции
(С.Н+) - СН^+СНз; (1)
D (НзС-СНз’’) = ДЯДСН+) -J- ДЯу (СН3) — A//z(C,h+). (2)
Как найти ДЯ/(С2Н^), показано в примере (7.2):
С,Н6 > С,Н^ -f- е—, ДЯ = ПИ = 11,65 эв;
/(С2Н6) = ДЯ/(С,Н+) -ДЯ^С.Н,.) . (3)
Подставляя (3) в (2), получим
D (Н3С-СН+) = ДЯДСН-,) 4 ДЯ/(СН+) - I (C,He) - ДЯДС^). (4)
Нетрудно найти и ДЯ/(СНз+):
СН3 СН+ 4 е—, ДЯ — ПИ = 9,84 эв,
Ml f (СН+) = I (СН3) + Mlf (СН3), (5)
и подставляя (5) и (4), получим:
D (Н8С СН+) = 2ДЯ, (СН3) 4 / (СН8) — I (С,Нв) — ДЯ, (С,Нв). (6)
265
Но
D (СН3СН3) = 2AWz (СН3) — A//z (С,Н6) (7)
и поэтому, включая (7) в (6), получим
D (НЛС СН+) - D (СН3СН3) Н- I (СН3) - 1 (С2Н6);
D (СН3СН3) = 3,67 эв;
D (Н3С-СН+) = 3,67 4- 9,84 — 11,65 = 1,86 эв.
Представление о мономолекулярном распаде ионов в каче-
стве источника алкильных радикалов подтверждается и тем
фактом, 4jo в разветвленных молекулах увеличивается выход
осколочных радикалов; кроме того, выход осколочных алкильных
радикалов оказывается неэквивалентным. Например, при ра-
диолизе н-пентана 33% первичных радикалов оказываются оско-
лочными. В то же время при радиолизе 2,4-диметилпентаиа
выход осколочных алкильных радикалов составляет 48%, при
радиолизе 2,2,4-триметилпентана — 64%, а при радиолизе нео-
пентана— 79%. Выход осколочных алкильных радикалов будет
высоким, если образующийся в реакции диссоциации осколоч-
ный ион будет в свою очередь претерпевать дальнейший распад.
Примером может служить реакция:
H3O-C4Ht0 -> CH3 -h С3Н^; (9.1)
С3Н7+4-е~-> Н-НОД. (9.3)
Вероятность осуществления этих реакций определяется со-
ответствующими энергиями связей. В табл. 9.2 приведены энер-
гии диссоциации одноэлектронных связей для целого ряда моле-
кул, рассчитанные по уравнению (9.2). Сравнение энергий связи
С—СН3 для неопентана (8 ккал/маль) и н-пентана (39 ккал!
]моль) позволяет, по крайней мере качественно, объяснить раз-
личия в наблюдаемом радиолитическом выходе осколочных ал-
кильных радикалов (79 и 33% соответственно).
9.5.1. Ион-молекулярные реакции
Так как ионы образуются в конденсированной фазе, то суще-
ствуют условия, благоприятные для развития таких ион-молеку-
лярных реакций, как, например, реакция (9.4):
RH+ 4- RH -> RH2h -j- R. (9.4)
Образующиеся в реакции молекулярные ионы в результате
нейтрализации могут, как показано в главе VII, либо диссо-
циировать далее, либо нейтрализоваться без диссоциации.
Обычно сечения ион-молекулярных реакций велики. Однако
следует сопоставить скорости ион-молекулярных реакций со
скоростью процессов нейтрализации и мономолекулярного рас-
266
Таблица 9.2*
Энергии диссоциации одноэлектронных связей
Связь Энергия. ккал/моль Связь Энергия, ккал (моль I Связь Энергия, ккал {моль
Углерод-водородная связь Углерод-углеродная связь Углерод-углеродная связь
C—H 34 С ССС—СС 21
СС—Н 27 1 1 1
ССС—н 40 СС—С 8 с с
СС—н 17 1 с
1 с 1
с сссс—СС 47 СС—СС 13
сссс—н 48 ССС—ССС 41 1 1
ССС—11 44 ССС—СС 38 с с
г 1 СССС—сссс 38
с с ССС—сссс 38
ССС—н 27 СС—ССС 21 1
1 1 с
с с ссс—сссс 21
с ССС—СС 24 1
1 1 с
СС—н 15 с с
1 с 1
с СС—сссс 15
СС—СС 14 1
[ с
с ссс—ссс 36
сс-сс 17 1 1
Углерод-углеродная связь 1 1 с с
с с ССС—ссс 31
С—с 44 сссс—ССС 42 ! 1
CC--C 30 ССС—ССС 34 с с
ссс-с 35 1 ссс—ссс 20
сс-сс 39 С 1 1
сс—с 14 СС—сссс 19 с с
1 1 с
с с 1
СССС—с 39 ССС—ССС 22 СС—ссс 13
ССС—СС 41 1 1 1
ССС—с 34 с с с
1 с с
с 1
СС—СС 19 СС—ССС 17 СС—ссс 12
с с с с
ССС—с. 20 ССС—СС 23 с с
с 1 1 с с 1 1 СС—СС 14
с с
* Данные взятые из работы: Franklin, I. L. Lampe F. W. Trans. Far. Soc., 1961,
v. 57. p. 1449.
267
пада ионов. В ряде случаев, особенно в конденсированной фазе,
характерные константы ион-молекулярных реакций могут ока-
заться недостаточно высокими для того, чтобы конкурировать
с упомянутыми выше двумя процессами.
Не вызывает сомнений, что иоп-молекулярные реакции про-
исходят. Однако их роль в образовании свободных радикалов
трудно оценить. Представления о иоп молекулярных реакциях
использовались для объяснения наблюдаемого распределения
додеканов в продуктах радиолиза «-гексана [15] и циклопропан-
гексановых растворов [16].
Предполагается, что вслед за ион-молекулярной реакцией
происходит реакция нейтрализации [17, 18]
RH^ ~ R Н2. (9.5)
Эта реакция ведет к образованию свободных радикалов и
молекулярного водорода Н2 без промежуточного атомарного
водорода. Например:
цикло-CgHib + е~ цикло-СвНп -}- Н2. (9.6)
9.5.2. Захват электронов с последующей диссоциацией
Целый ряд данных [19—21] показывает, что процесс захвата
электронов с последующей диссоциацией идет в галогензаме-
щенных алканах, таких, как CHCI3, CH3I, C2F2CI2 и др. Уравне-
ния реакций выглядят следующим образом:
ССГ4 + -> С1~ ф- СС13; (9.7)
СН31 + + СН3. (9.8)
Реакция протекает легче, если галоген обладает большим
сродством к электрону и энергия диссоциирующей связи мала.
При облучении систем, содержащих галогензамещенные угле-
водороды, реакции такого рода можно считать одним из досто-
верно установленных источников свободных радикалов.
В табл. 9.3 приведены потенциалы появления (ПП) отри-
цательных ионов в некоторых галогензамещепных алканах.
Таблица 9.3
Потенциалы появления (ПП) для некоторых отрицательных ионов
Вещество Отрицатель- ный ион ПП, эв Вещество Отрицатель- ный ион ПП, эв
cf3sf5 CIO3F СС14 CF2C1CF2C1 CC13F CFF CIO7 Cl- ci- Cl- 0,20 0,20 0,20 0,20 0,20 SF6 CC1..F2 C.F3CI CHC12F SFr Cl- Cl- Cl- 0,05 0,50 0,90 0,90
268
Особенно яркий пример реакции захвата электрона с после-
дующей диссоциацией можно встретить при облучении цикло-
гексана, содержащего всего 1 мол.% СН31 [19, 22]. Выяснилось,
что при радиолизе этой системы СН31 разлагается в значитель-
ных количествах. Этот факт удается объяснить, лишь привлекая
реакцию (9.8). Та же реакция захвата электрона с последую-
щей диссоциацией позволяет объяснить высокий выход радика-
лов СН3 при радиолизе циклогексана, содержащего миллимо-
лярные количества СНз1.
9.5.3. Другие ионные реакции
Кроме реакций, рассмотренных выше, при радиолизе угле-
водородов идут и многие другие реакции. Например:
СН4----> СНТ Н- Н2 -г е-;
С4Н10 —> СН4 -|- С3Н6+ -|- е~-
цикло-С6Н12 Cgl Ijg -i- Н2.
При этом возможны вторичные реакции образующихся ионов.
Однако, если учесть низкий выход разделенных пар ионов в
жидких углеводородах, представляется вполне вероятной успеш-
ная конкуренция быстрых реакций нейтрализации со вторич-
ными ион-молекулярными реакциями.
9.6. ВЫХОД РАДИКАЛЬНЫХ ПРОДУКТОВ
Единственный тип активных частиц, выход которых при
радиолизе углеводородов удается измерить количественно,—
свободные радикалы. Хотя известно, что ионы и возбужденные
молекулы непосредственно участвуют в наблюдаемых реакциях,
количественно учесть их роль при радиолизе углеводородов не
удается.
Выход радикальных продуктов при радиолизе углеводородов
определяется самыми разнообразными методами. Разные методы
в подавляющем большинстве случаев дают хорошее качественное
совпадение выхода радикалов, а во многих случаях отмечается
и прекрасное количественное соответствие. В большей части
методов определения выхода радикальных продуктов использу-
ются акцепторы свободных радикалов. С помощью акцепторов
удается определять не только наблюдаемый выход радикалов, но
иногда и выход первичных радикалов. Те радикалы, которые
реагируют в шпорах и образуют разного рода продукты, есте-
ственно, не вносят какого-либо вклада в наблюдаемый выход
радикалов. Наблюдаемый выход радикалов определяется только
теми радикалами, которые успели продиффундировать за пре-
делы шпор.
269
9.6.1. Обнаружение свободных радикалов
методом электронного парамагнитного резонанса (ЭПР)
Методом ЭПР удалось идентифицировать целый ряд радика-
лов в различных углеводородных системах п- измерить их
относительный выход [23]. Полученные данные суммированы
в табл. 9.4. Если вспомнить, что временное разрешение метода
ЭПР составляет около 0,2 мсек, то, очевидно, регистрировать
удается лишь те радикалы, время жизни которых достаточно
велико.
В системах, где доступно хорошее временное разрешение,
удается провести точные количественные измерения. В этих
условиях часто удается измерить абсолютные константы скоро-
стей реакций исчезновения свободных радикалов точно так же,
как это делается при импульсном радиолизе. Сходство между
двумя методами заключается в том, что с их помощью удается
непосредственно измерять концентрацию частиц с высокой реак-
ционной способностью. Определение абсолютных констант ско-
ростей реакции методом ЭПР иллюстрирует пример 9.2.
Пример 9.2. Отношение числа неспаренных электронов в облученном
образце этапа к числу неспаренных электронов в 1 см3 стандартного образна
(гальвиноксила) равно 2,35-10~4. Стандартный образец содержит в 1 см3
3,31-10” песпаренных электронов. Объем полости с этаном составляет
0,524 ел3, а скорость поглощения энергии в образце равна 2,62-10” эв/сек.
Принимая для образования этильных радикалов значение G=4,4, рас-
считайте константу скорости бимолекулярной реакции исчезновения этильных
радикалов.
Решение. 3,31 • 10”-2,35-10 4 = 7,78-1013 неспарепных электронов на весь
объем образца (0,524 си3) или
7,78-1013 радикал I моль I 1000 см3 „ . , ,
—---------е------ -----------——------------- 2,47-10-’ моль л.
0,524 см3 I 6,02-1023 радикал | л
Константа скорости реакции kr выражается через интенсивность излучения н
другие параметры согласно уравнению (6.26)
k
r IRI2 ’
Интенсивность излучения D=2,62-10” эв/сек-, G=4,4;
2,62-101’эв 1 4,4 радикал I моль I 1000 си3
0,524 см3 сек | 100 эв | 6,02-1023 радикал | л
DG = 3,61 -10—в моль/(л-сек)-,
kr = 3,61 • 10 мольКлсек)---------------------•
г ' ’ (2,47-10-’лоль/л)2 4
fer==5,85-108 лДлоль-сек).
270
Таблиц! 9,4*
Свободные радикалы, наблюдаемые методом ЭПР при радиолизе углеводородов
Вещество Наблюдаемые свободные радикалы Относительная интенсивность
сн4 С.2нв с.,н4 с3н8 Н-С4Н10 изо-С4Н10 с6н12 нео-С5Н12 изо-С5Н12 нео-СпН14 2,' 2, 4-Три- метилпентан 3, З-Диэтил- пентан Циклобутан •Циклопентан Циклогексан Циклогептан Циклопропан 1, 4-Цикло- гексадиен СН3, Н С2Н5, сн3, с.,н3 с2нб, с2н3> сн2снсн2сн2 нзо-С3Н7, С3Н7 СН3, С2Н5, emop-C4H9, С3Н7, С4Н9 mpem-C4H9, СН3, изо-С4Н9, изо-С3Н7 СН3, С2Н5, С5Н9, 1-метилбутил, 1-этилпропил mpe/n-C4H9, неопентил mpem-CjHn, СН3, С2Н6, ИЗО-С3Н7 СН3, С2НЙ, mpem-CfHg, трет-С5Нц mpem-CjHg, изо-С4Н9 (или радикал Cs), изо-СзН,, нео-С6Нц (может быть СН3) 1, 1-Диэтилпропил, 1-метил, 2, 2-диэтилбутил, С2Н5 С2Н6, 3-бутенил, циклобутил Циклопентил’ (единственный наблюдаемый радикал) Циклогексил (единственный наблюдаемый радикал) Циклогептил (единственный наблюдаемый радикал) Циклопропилаллил (единственный наблюдаемый радикал) Циклогексадиенил (единственный наблюдаемый радикал) С2Н5>СН3>С.,Н3 С2Н3>С4Н7>С,Н6 СНз>С,Н5>С3Н7>С4Н9> emop-С4Н9 трет.-С4Н9>изо:С4Н9>СНз>изо-С3Н7 > 1-Метилбутил > 1-этилпропил >С6Н9>СН3>С2Н6 трет-С6Нц>других радикалов трет-С4Н9>других радикалов С,Н5>любого из других радикалов С.,Н6>других радикалов
Данные из статьи Фессендена и Шулера [23].
9.6.2. Обнаружение радикалов химическими методами
Для акцепторной реакции с радикалами и определения их
выхода используется целый ряд веществ. Среди них HI, 3Н1, 12
(обычный и радиоактивный), FeCl3, антрацен, NO, гальвиноксил,
,4СНз1 и другие вещества. В роли акцепторов радикалов эти
вещества обладают различной эффективностью, причем одним
из наиболее эффективных акцепторов оказывается иод. Чрезвы-
чайно важно, разумеется, чтобы добавление акцептора не меняло
природу первичных химических процессов. На отсутствие такого
эффекта можно надеяться с достаточными основаниями, если
поддерживать концентрацию акцептора очень низкой.
Как указывалось в разделе 5.4, для радикала R существует
несколько возможных реакций:
реакция с акцептором
R + А -> RA; (9.9)
реакция с нейтральной молекулой
R4-R'H-> RH + R'; (9.10)
рекомбинация с другим радикалом
R + R(wih R') -> RR(kth RR'). (9.11)
Легче всего реакция (9.9) идет в том случае, если раство-
ренное вещество действует в качестве акцептора. При проте-
кании реакции (9.9) стационарная концентрация радикалов
слишком низка (за исключением объема шпор) для того, чтобы
рекомбинация (9.11) имела какое-либо значение [24]. Однако
обмен радикалов (9.10) может идти, и его следует учитывать
при анализе кинетики акцепторных реакций, так как при обмене
меняется характер первичных радикалов. Если, например, реак-
ция (9.10) представляет собой отрыв водорода, то в соответ-
ствии с данными табл. 5.2 для нее будет характерна энергия
активации 10,4 ккал!моль для радикала СНз и 6,4 ккал!моль
для радикала Н. Поэтому, очевидно, должно наступать равно-
весие, положение которого определяется концентрацией акцеп-
тора и различиями в энергиях активации реакций (9.9) и (9.10).
Пример 9.3. Каким должно быть различие в энергиях активации для
того, чтобы реакция (9.9) преобладала над реакцией (9.10), если при ком-
натной температуре концентрация 1г в пентане составляет 10~4 Л4?
Решение. Чтобы реакция (9.9) доминировала над реакцией (9.10), долж-
но выполняться следующее соотношение:
feB[Rj[A]
Л10 [R] [R'H] * *
Так как Лг=Де—АБ^Г,
A9e~AE^7’[R] [А]
Aloe-AE‘«/R7’[R1[R'H1
272
Принимая Ло—Дщ, найдем
ЬМе^/*7-
—---------->;• 1
[R'H]
620
где A£'r=A£'io — А7Ч; |R'H]— концентрация пентана, равная =8,6 М.
Следовательно,
|А1 10“^
1 1 =------— 1,2- IO-6.
|R'H| 8,6
Для того, чтобы е 'т -1,2-10~6 было много больше единицы, \ЕТ ~
~7 ккал/мом.
Поэтому реакция (9.9) должна иметь энергию активации на 7 ккал/моль
.меньшую, чем энергия активации реакции (9.10).
Иод (обычный или радиоактивный 1311) широко используется
в качестве акцептора свободных радикалов. Реакция
R + I.2->RI + I (9.12)
имеет константу скорости порядка 109 М~1-сек~1. Реакция идет
очень быстро, и выход акцептируемых радикалов можно опре-
делить, измерив количество израсходованного 1г и относительное
количество образовавшихся иодистых алкилов. Йодистые алкилы
обычно определяют с помощью газовой хроматографии. Вариан-
том метода может быть измерение' концентрации иодистых алки-
лов с помощью полупроводниковых детекторов в совокупности
с газовым хроматографом [25]. Этот очень чувствительный ме-
тод основан на сравнительно высокой скорости захвата электро-
нов иодистыми алкилами.
Результаты, полученные при использовании иода в качестве
акцептора, не всегда однозначны. Так, молекула иода может
захватывать электрон и затем диссоциировать
12 + е~ -> Г +1. (9.13)
Образующийся ион I" может прямо вступать в реакции нейтра-
лизации или обмена электрона. Такие реакции могут изменить
характер продуктов радиолиза. Например, в результате реакций
нейтрализации заметно увеличится концентрация атомов I
RH++ I" -> RHI. (9.14)
Нод также может конкурировать с углеводородами,, вступая
в реакцию с атомарным водородом:
Н + I2-> HI1. (9.15)
Эта реакция может конкурировать с реакцией
H + RH->R-)-H2. (9.16)
В результате общий выход радикальных продуктов будет
снижаться, так как подавляются реакции взаимодействия ато-
мов Н с нейтральными молекулами, ведущие к образованию-
273
алкильных радикалов, и реакции присоединения атомов Н
к олефинам (например, CsHj+H-^C^K), которые также ведут
к возникновению свободных радикалов. При понижении кон-
центрации атомов Н чрезвычайно трудно получить достаточно
верную картину выхода радикальных продуктов. В тех случаях,
когда среди продуктов радиолиза имеются олефины, легко идут
реакции присоединения к олефинам HI, 12 и атомов I. Кроме
этих эффектов при высоких концентрациях йодистых алкилов,
возникающих при высоких концентрациях иода, может происхо-
дить реакция захвата электрона йодистыми алкилами с после-
дующей диссоциацией
СН31 е- -> СН34 Г. (9.8)
Поэтому при использовании 12 в качестве акцептора следует,
очевидно, проявлять большую осторожность. Концентрацию иода
следует поддерживать на достаточно низком уровне, чтобы вос-
препятствовать возникновению заметных количеств алкильных
радикалов и HI. Рекомендуется концентрация 1^ ниже 10~:! М.
При таких количествах иода концентрация образующихся йоди-
стых алкилов будет порядка 10-6 М. Таким образом, при низких
концентрациях 12 присоединение Н] к олефинам будет мини-
мальным.
Йодистый водород. Реакцию HI с углеводородными радика-
лами в общем виде можно записать так:
R + HIRH + J- (9.17)
В результате реакции возникают атомы I, которые, реком-
бинируя, образуют 12. Количество образовавшегося иода может
служить мерой выхода радикальных продуктов. В широко рас-
пространенной методике подобных экспериментов принято ис-
пользовать иодистый тритий. Количество образовавшихся мече-
ных углеводородов можно теперь определять, измеряя радио-
активность. Методика позволяет не только производить двойную
проверку числа образовавшихся радикалов, но и определять,
какие именно радикалы возникают.
Как и для молекулярного иода в случае применения HI,
в качестве акцептора также может происходить захват электро-
на с последующей диссоциацией
Hl-t- е~ -> Н -'г I". (9.18)
Этот процесс способен повлиять на измеряемый выход радикаль-
ных продуктов. Кроме того, HI вступает в конкуренцию за
взаимодействие с атомами II
Н HI -> Н2-j- I. (9.19)
В результате реакции должен меняться выход радикальных
продуктов, так как при этом происходит замена атомов Н на
274
атомы 1, Которые не способны отрывать водород и образовы-
вать свободные радикалы.
Поэтому, как и в случае использования 12, концентрация HI
или 3Н1 должна быть меньше 10~3 М. Исследование концентра-
ционных эффектов 3Н1 при радиолизе н-гексана [26] показало,,
что достаточно 0,3-10 3 М 3Н1 для эффективного связывания
всех алкильных радикалов в системе.
Величину отношения констант скоростей реакций (9.12) в
(9.17) можно получить, используя кинетические уравнения для
гомогенных радикальных систем. Показано [27], что в темпера-
турном диапазоне от —78 до +25° С значение отношения
дается выражением (1/0,13)ехр(—800/RT). Это до-
статочно хорошо согласуется со значением того же отношения
(1,4) при комнатной температуре, полученным при упрощен-
ном кинетическом анализе [28].
Гальвиноксил относится к числу стабильных свободных ра-
дикалов, концентрацию которых нетрудно измерить. В качестве
акцептора свободных радикалов при радиолизе гальвиноксил
используется не слишком часто, так как его эффективность ока-
залась невысокой. Сообщалось [29], что реакционная способность
иода в отношении метильных радикалов в 16 раз выше, чем
у гальвиноксила.
Хлорное железо широко используется в качестве акцептора
в различных органических системах. Концентрацию FeCl3 в си-
стеме нетрудно измерить спектрофотометрически по поглощению
при 350 н.м. Отмечалось [30], что в нескольких системах, где
в’ качестве акцепторов радикалов использовали FeCl3, ДФПГ
и 12, соблюдалось соотношение
G (— FeCI3) - G (— ДФПГ) G (— 12\
Однако системы, содержащие FeCl3, не изучались столь
тщательно, как системы, содержащие I2, HI или ДФПГ. До
концентрации 10-3 М FeCl3 растворяется почти во всех органи-
ческих жидкостях.
а, а'-Дифенил-пикрил-гидразил (ДФПГ). Одно время ДФПГ
широко применяли в качестве детектора радикалов при радио-
лизе органических соединений. Затем выяснился ряд недостатков
ДФПГ, затрудняющих его использование как акцептора.
ДФПГ — очень светочувствителен. На молекулу ДФПГ легко
идет перенос энергии от молекул растворителя. Продукты реак-
ций ДФПГ с радикалами трудно выделить с помощью обычных
хроматографических методов. Кроме того, присутствие ДФПГ
вносит ряд трудностей в кинетический анализ системы. Тем не
менее в нескольких работах'при использовании ДФПГ был полу-
чен тот же выход радикальных продуктов, что и при исполь-
зовании 12 [31—34].
Отбор радикалов. Под отбором радикалов подразумевают
методику, при которой меченые соединения — этилен (14С2Н4)
275
или иодистый метил (14СН31)—добавляют в низких концентра-
циях в систему. Благодаря реакциям
Н + 14С2Н4 + ОД; (9.20)
Н 14СН31 -> »СН3 -г HI; (9.21)
“СН31 + -> ,4СН3 + е~ + I (9.22)
в растворе появляются меченые радикалы. Методика основы-
вается на высокой эффективности С2Н4 и CHJ в качестве акцеп-
торов атомов И.
Образовавшиеся меченые радикалы реагируют затем с ради-
калами, возникающими при радиолизе, и дают меченые про-
дукты. После выделения нетрудно определить количество дан-
ного продукта. Метод, отбора радикалов несколько сложнее
других методов с использованием акцепторов. В этом методе
должны быть созданы условия облучения, благоприятствующие
радикал-радикальным реакциям. Облучение, следовательно,
нужно вести с высокой интенсивностью [~ 1022 эв/(л-сек)]_ При
оценке результатов приходится вводить поправки, учитывающие
реакции диспропорционирования. Метод отбора радикалов поз-
воляет включить в состав меченых продуктов лишь небольшую
долю присутствующих радикалов. Однако и этого бывает доста-
точно, чтобы выяснить, какие радикалы имеются и каковы их
относительные концентрации.
Другие акцепторы радикалов. При изучении радиолиза угле-
водородов используют и такие акцепторы радикалов, как кисло-
род, трифенилметан, окись азота, закись азота, олефины и не-
которые мономеры, например, стирол. Олефины в целом обла-
дают меньшей реакционной способностью по отношению
к радикалам (включая атомы Н), чем многие другие акцепторы
(ДФПГ, FeCls, HI, 12). Кислород в ряде случаев оказывается
столь же эффективным акцептором, как и иод. К числу хорошо
изученных акцепторов относится окись азота; особенно подробно
проанализированы реакции окиси азота в газовой фазе.
Широко используется в качестве акцептора трифенилметан,
один из водородных атомов которого отличается высокой реак-
ционной способностью. Закись азота представляет собой эффек-
тивный акцептор электронов. В целом, однако, использование
перечисленных акцепторов ограничивается только 'углеводород-
ными системами.
9.6.3. Выход радикальных продуктов
Выход радикальных продуктов для ряда обычных и раз-
ветвленных алканов и циклоалканов, измеренный рассмотрен-
ными выше методами, приведен в табл. 9.5 и 9.6.
Выход метильных радикалов для обычных циклических и
разветвленных алканов можно выразить в виде простой функции
276
Таблица 9.5*
Выход радикальных продуктов при радиолизе нормальных и разветвленных
углеводородов
Радикал н -Б} тан н-Пентан н-Гексан н Гептан мзо-Пептан
сн3 0,4 0,10 0,08 0,04 0,71
С2Н5 0,94 0,34 0,27 0,21 0,91
H-C3H7 0,09 0,34 0,25 — 0,12
ИЗО-С.ЗН7 0,04 0,20 0,04 0,24 0,66
н-С4Н9 1,04 0,05 0,23 — 0,07
изо-С4Н9 — — —. •—• 0
<?«юр-С4Н9 2,14 0,05 0,05 0,23 0,18
тпре/тг-СтНд — — — — 0
н’С5Нц — 0,78 0,03} 0,22 —
впгср-С511п — 2,84 —
н-С6Н13 — — 0,5 ( 0 02 -—•
«тор-С61 йз -— .—_ 2,3 J —.
Радикалы, образовавшиеся из
родительских молекул:
первичные — -— —
вторичные — — —. ^4,04 |1,44
третичные —
Продолжение табл. 9.5*
Радикал Нео-пентан 2. 4- Диметил- пентаи 2, 2, 4- Триметил- пентан 2,2- Диметил- бутаи н-Тридекан
сн3 2,3 0,28 0,69 0,99 0,012
С2н6 0 0 0 0,6 0,046
Z/-C3H7 0 0 0 <0,1] 0,05
изо-С3Н7 0 0,53 0,12 —
н-С4Н9 0 0 — -—. .
изо-С4Н9 0,16 0,54 0,74 -—. 0.05
влюр-С4Н9 0 0 0 —
mpem-C4H9 0,55 0 0,51 —-
Я-С5Н11 emop-QUn •—• •—• ~ } 0.2 0,06
я-С6Н13 втор-С6Н13 — — 0,07
Радикалы, образовавшиеся из родительских молекул:
первичные 0,78 0,64 0,60 ]
вторичные 0 0,46 0,50 1,2 4,4
третичные 0 0,30 0,05 J
* Данные из доклада Жада и Шулера [16].
от числа углеродных атомов и положения метильных групп в
молекуле [35]. Различают три типа метильных групп:
1) тип А, где к углеродному атому прикреплена одна и только
одна метильная группа (к этой категории относятся метильные
группы всех нормальных парафинов);
277
Таблица 9.6*
Выход радикальных продуктов при радиолизе некоторых циклических
угле водородов
Радикал Цикло- пеитаи Цикло- гексан Радикал Цикло- пентан Цикло- гексан
сн3 0,21 0,22 emop-CjHn 0,10 0,02
с,н5 . 0,06 0,06 Циклопентил 2,86 —-
С3Н7 0,09 0,08 -—. 0,92
С4н9 0,04 0,07 emop-CeHl3 —- 0,05
к-СьНц 0,22 0,03 Циклогексил — 2,93
• Данные из доклада Жада п Шулера [16].
2) тил Б, где к одному углеродному атому прикреплены две
.метильные группы;
3) тип В, где к одному углеродному атому прикреплены три
метильные группы (как например, в изобутане).
Эмпирическая формула, выражающая соотношение между
выходом метильных радикалов и структурой родительской моле-
кулы, выглядит следующим образом:
G (СН3) = ----—_ 1,02 (а) И- 2,79 (б) ф- 7,89 (в). (9.23)
(п 1 )-
Здесь п — число атомов углерода в молекуле; (а), (б), (в) —
число метильных групп типа А, Б и В соответственно.
С помощью этой формулы удается за немногими исключе-
ниями предсказать выход метильных радикалов с погрешно-
стью, не превышающей 15%. Основным исключением из прави-
ла оказывается 3,3-диметилпентан, где расхождение между пред-
сказанным и наблюдаемым выходом составляет 62%. Важная
особенность уравнения (9.23) состоит в том. что оно предсказы-
вает снижение выхода метильных радикалов при радиолизе с
ростом молекулярного веса. Наибольших выходов согласно фор-
муле следует ожидать для сильно разветвленных углеводородов
с низким молекулярным весом.
Пример 9.4. Рассчитайте выход метильных радикалов при радиолизе
2,2-диметилбутаиа.
Решение. п=6;
G ™ 1 I1 -°2 3 + (7.89) (3)];
(6— I)2
G (СН3) = 0,987.
9.7. ВЫХОД МОЛЕКУЛЯРНЫХ ПРОДУКТОВ
Выход молекулярных продуктов, возникающих в результате
рекомбинации радикалов, измерить очень трудно. Обычно, ис-
ходя из количества образовавшихся продуктов, рассчитывают
278
общий выход радикалов, а затем из этого общего выхода вычи-
тают выход- радикальных продуктов, измеренный с применением
акцепторов. Например, при радиолизе циклогексана единствен-
ный наблюдаемый радикал — циклогексильный радикал, а ос-
новные продукты — дициклогексил, циклогексен и водород.
Появление этих продуктов можно легко объяснить, предполо-
жив, что циклогексильные радикалы могут вступать в реакции
рекомбинации и диспропорционирования
2 (цикло-С6Ни) -» дициклогексил цикло-С6П10 -*г цикло-СвН13.
Имея определенные сведения об относительных скоростях
этих реакций, можно оценить общий выход циклогексильных
радикалов: он примерно в два раза больше выхода радикаль-
ных продуктов, измеренного с помощью акцепторов.
Как указывалось в разделе 9.2, выход молекулярных продук-
тов повышается за счет реакций радикалов внутри шпор. Такие
реакции позволяют, например, оценить выход метильных
радикалов, включающихся в молекулярные продукты при радио-
лизе неопентана. Известно, что облучение неопентана ведет к
образованию метильных, изобутильных, неопентильных и трет-
бутильных радикалов. Метильные радикалы внутри шпор могут
реагировать со всеми этими радикалами и образовывать этан,
изопентан, 2,2-диметилбутан и неопентан. Если подобные про-
дукты действительно образуются внутри шпор, то добавление
акцепторов в низких концентрациях не должно повлиять на их
выход. Тогда измерение выхода таких продуктов плюс измере-
ние выхода продуктов, зависящего от добавления акцепторов,
позволит оценить долю образующихся метильных радикалов,
•связанных внутри молекулярных продуктов. (Разумеется, выход
неопентана должен быть определен по концентрации трет-
бутильных радикалов.) Исследования такого рода показали, что
при радиолизе углеводородов менее 70% радикалов диффунди-
руют из объема шпор в среду [20]. Поэтому, согласно приведен-
ным оценкам, выход молекулярных продуктов должен превы-
шать 30% общего выхода радикальных продуктов.
9.8. ПЕРЕНОС ЭНЕРГИИ
Вопросы переноса энергии при радиолизе углеводородов при-
влекли к себе широкое внимание исследователей, и представле-
ние о переносе энергии неоднократно использовалось для интер-
претации результатов. Передача энергии между молекулами
растворителя М и молекулами растворенного вещества S может
идти в форме переноса электрона (как это наблюдается в га-
зах), переноса энергии возбуждения M* + S->-S*+M, образова-
ния пары ионов с последующей нейтрализацией M++S_->-
-►M+S* или в форме процесса внутренней конверсии.
279
Хорошим примером переноса энергии может служить влияние
добавок бензола на радиолиз циклогексана. Это явление полу-
чило название защитного эффекта, так как в результате добав-
ления бензола к циклогексану общий выход распавшихся моле-
кул уменьшается. Основные продукты радиолитического разло-
жения циклогексана— циклогексен, дициклогексил и водород.
Рис. 9.1. Значение О(Нг) в смеси
циклогексана н бензола в зависи-
мости от мольной доли бензола:
I — экспериментальный выход Fb; 2 — тео-
ретический выход Нг.
При облучении смесей бензо-
ла с циклогексаном наблю-
даемый выход молекулярного
водорода отличается от теоре-
тического, который рассчиты-
вался как сумма выходов для
каждой компоненты отдельно с
учетом их электронной доли
[36]. Экспериментальные и
теоретические данные для
G(H2) приведены на рис. 9.1.
Для электронной доли
бензола, равной 0,05, выход во-
дорода снижается примерно в
два раза. Для объяснения это-
го факта предлагались раз-
личные механизмы. Наиболее
адекватными следует считать
перенос электрона и перенос
энергии возбуждения [37, 38].
Так как потенциалы иониза-
ции и возбуждения для бен-
зола ниже тех же потенциалов
для циклогексана, то, по-видимому, следует рассматривать оба
эти процесса. В анализируемой системе происходит увеличение
разложения бензола, которое ни в коей мере не эквивалентно
уменьшению разложения циклогексана. Разложение гексапа
точно так же снижают малые добавки других веществ [37], об-
ладающих высоким сродством к электрону, таких, как 12, СН31
и SO2.
Удалось установить, что под действием у-излучепия в некото-
рых разбавленных (10~3 М) двухкомпонентных органических
растворах наблюдается флуоресценция со спектром, характер-
ным для растворенного вещества [39]. Эффективность процесса
невелика, но интенсивность свечения оказывается пропорцио-
нальной концентрации растворенного вещества. Исследования
переноса энергии (затрачиваемой на флуоресценцию) в раство-
рах бензола и n-терфенила в гексане показали, что константы
скорости реакции переноса энергии от циклогексана к бензолу
и л-терфенилу очень велики.
Первое синглетное возбужденное состояние циклогексана
репульсивное, поэтому перенос энергии возбуждения циклогек-
280
сана к растворенным молекулам бензола нельзя объяснить
с помощью простых соударений между отдельными молекулами.
Перенос энергии в данной системе можно объяснить, предполо-
жив, что существуют упорядоченные молекулярные домены,
в которых возможна миграция энергии. Скорость процессов миг-
рации энергии в доменах очень велика; эти процессы могут
конкурировать с диссоциацией возбужденных молекул. Теорети-
ческий анализ поддерживает представление о том, что в спектре
потерь энергии в конденсированных системах должен существо-
вать максимум, соответствующий коллективному возбуждению
электронов в элементе объема с диаметром около 100А [41].
С миграционной гипотезой согласуется целый ряд эксперимен-
тальных данных [42—44], относящихся не только к влиянию
добавок, но и к влиянию ЛПЭ на радиолиз конденсированных
систем [45].
Выход водорода при радиолизе циклогексана не зависит от
ЛПЭ. Это можно объяснить, если считать, что начальная об-
ласть. в которой образуются атомы Н, достаточно велика.
Важным обстоятельством является также тот факт, что умень-
шение G(H2) при добавлении бензола не компенсируется уве-
личением выхода других продуктов радиолиза циклогексана или
бензола. Следовательно, бензол проявляет истинный «защит-
ный» эффект.
С помощью механизма переноса энергии удалось объяснить
влияние антрацена, кислорода и нафталина на радиационно-
индуцированную изомеризацию н-бутана [46]. Влияние аргона
на радиолиз н-бутана также удалось частично объяснить с по-
мощью переноса энергии [47]. Исследование радиационно-инду-
цированной изомеризации в растворах цис- и транс-стильбена
в бензоле показало, что реакция идет через возбужденные три-
плетные состояния стильбена, возникающие в результате пере-
носа энергии от бензольных молекул в триплетном состоянии.
Согласно оценкам [48—50], G (молекул бензола в триплетном
состоянии) может достигать 10.
9.9. ВЛИЯНИЕ ЛПЭ
Так как обобщающих концепций по этому вопросу не сущест-
вует, для каждой органической системы влияние ЛПЭ должно
рассматриваться отдельно. Одна из главных трудностей при ис-
следовании влияния ЛПЭ состоит в том. что наблюдаемые эф-
фекты следует отделить от эффектов мощности дозы, обуслов-
ленных реакциями одних и тех же активных частиц.
При радиолизе циклогексана выход водорода, по-видимому,
не зависит от ЛПЭ. Изменения в соотношении различных про-
дуктов в н-гексане, циклогексане или н-пептане при облучении
.с разными ЛПЭ объясняли скорее влиянием мощности дозы,
нежели влиянием ЛПЭ. Однако Фальконер и Бартон [51] паблю-
281
дали влияние ЛПЭ при радиолизе циклогексана. Если размеры
шпор велики и в них происходит рекомбинация радикалов, то-
эти реакции будут продолжаться и в условиях перекрывания
шпор. Поэтому никаких заметных отклонений в выходе продук-
тов, характерных для данной ЛПЭ, не должно наблюдаться.
Если в системе образуются водородные атомы и алкильные
радикалы, то в результате возможных реакций рекомбинации
и взаимодействия с другими радикалами конечное распределе-
ние продуктов при действии излучений с высокой ЛПЭ не будет
отличаться от распределения продуктов при высокой интенсив-
ности излучения.
Таким образом, на алифатических углеводородах влияние
ЛПЭ не должно сказываться. Однако эффекты, обусловленные
ЛПЭ, отмечались в бензоле [52], дифениле, ортотерфениле [53J
и в некоторых других органических системах. Сильное влияние
ЛПЭ на радиолиз в водных системах в отличие от углеводород-
ных объясняется существенной конкуренцией в треках частиц,
между реакциями псевдопервого и второго порядка.
9.10. ВЛИЯНИЕ ИНТЕНСИВНОСТИ ИЗЛУЧЕНИЯ
Высокая интенсивность излучения обычно благоприятствует
радикал-радикальпым реакциям в большей степени, чем реак-
циям отрыва и присоединения. В углеводородных системах сле-
дует ожидать, что будут идти следующие реакции:
RH > Н, R, Rj, R2; (9.24)
Н RH Н2 R; (9.25)
Н-; Н Д- Н2; (9.26)
R, -j- RH R,H 4- R; (9.27)
Hi 4- R2 RiR2- (9.28)
Если учитывать только реакции с участием атомов Н, то,
очевидно, влияние интенсивности излучения будет проявляться
в тех случаях, когда скорость реакции (9.25) сравнима со ско-
ростью реакции (9.26). Если обозначить концентрацию атомов Н
через С, а концентрацию молекул RH через Cs, то скорость
реакции (9.25) будет равна ksCsC, а реакции (9.26)—kC2.
Тогда условие влияния интенсивности излучения на радиолиз
будет выглядеть так: kc2^ksCsC. Наоборот, влияние интенсив-
ности излучения на выход Н2 будет незаметно, если во всем
диапазоне интенсивностей облучения скорость одной из реакций
будет заметно превосходить другую.
282
Можно считать (для у-излучения и быстрых электронов), что
шпоры в объеме жидкости распределены равномерно. Следова-
тельно, равномерное распределение можно предполагать и для
активных частиц, особенно, если учесть расплывание шпор со
временем. При выполнении такого условия гомогенности можно,
анализируя конкретные радикальные реакции, выяснить, в ка-
ких ситуациях будут преобладать радикал-радикальные реак-
ции рекомбинации, такие, как (9.26) п (9.28).
При условии равномерного начального распределения радика-
лов уравнение (8.23) дает долю радикалов, вступающих в реак-
цию (9.25) с образованием Н2. Если считать, что влияние интен-
сивности излучения начинает проявляться при Аг=0,5, то не-
трудно найти такое значение интенсивности /, при котором
удовлетворяется это требование. Нужно, конечно, сделать пред-
положение, что справедливо уравнение (8.23).
Пример 9.5. Рассчитайте, при какой интенсивности облучения углеводоро-
да начинает сказываться ее влияние, если в системе имеют место реакции
(9.25) и (9.26). Известно,'что G(/7)=6; /г.«=10-14 см3!(молекул-сек)-, k-=
=0,06 молекул)эв; &=10-н см3!(молекул-сек) и (Д=6-1021 молекул/см3.
Решение. Используем уравнение (8.23) и примем, что влияние интенсив-
ности излучения начинает сказываться при А’=0,5. Отношение
kktI
------ обозначим через Д-
ks cs
А = -0,5 = -^-[(1^4Д)‘А -1].
k^s 2А
Откуда .1 ~2.
Теперь можем найти искомую интенсивность облучения:
2-10~г8-36-1042 эв
/ =--------------------= 1,2-1028
0,06-10-11 см3-сек
ж/см3-сек.
Подобные расчеты можно проделать для любого из обра-
зующихся радикалов, если только известны константы скоростей
соответствующих реакций. Результаты такого расчета в лучшем
случае нужно считать приблизительным, так как приходится
предполагать, во-первых, что радикалы распределены равно-
мерно и, во-вторых, что идут только реакции отрыва и рекомби-
нации. В общем случае, однако, радикалы рекомбинируют хаоти-
чески (т. е. метильный радикал, например, может рекомбиниро-
вать с метильным, этильным или любым другим радикалом,
присутствующим в системе). Расчет, проделанный в примере 9.5,
показывает, что в углеводородах, когда молекулярный водород
образуется только в реакциях (9.25) и (9.26), для проявления
влияния интенсивности!, излучения необходимы очень высокие
ее значения. Влияние ЛПЭ на радиолиз углеводородов, воз-
можно, лучше бы удалось оценить, если бы учитывались кон-
курентные радикальные реакции, такие, как реакции (9.27)
283
и (9.28). В радиолизе углеводородов все еще не выяснена роль-
термализованных атомов Н; кроме того, следовало бы учесть,,
что атомарный водород легко вступает в реакции H + R->-npo-
дукты.
Если расчеты, приведенные в примере 9.5, провести в об-
ратном порядке, то можно оценить долю радикалов, рекомбини-
рующих при различной интенсивности излучения. Основными,
реакциями радикалов (особенно радикалов с низким .молекуляр-
ным весом) являются реакции отрыва и рекомбинации. Учет
различий в энергии активации для этих двух процессов может
оказаться достаточным для объяснения изменений выхода про-
дуктов при низких и высоких интенсивностях излучения.
Резюмируя, можно отметить следующие эффекты, связанные
с влиянием интенсивности излучения при облучении углеводо-
родных систем:
1. При низких интенсивностях излучения основной реакцией,
в которой участвуют водородные атомы и алкильные радикалы,,
будет реакция отрыва водорода от родительской молекулы.
2. При высоких интенсивностях излучения основной реакцией
водородных атомов будет реакция отрыва, а основной реакцией
алкильных радикалов — рекомбинация.
3. При очень высоких интенсивностях излучения как для
водородных атомов, так и для алкильных радикалов основной
реакцией будет рекомбинация.
Эти выводы не обязательны для радикалов высокого молеку-
лярного веса, так как вероятность реакций отрыва снижается
при увеличении числа углеродных атомов в радикале.
9.11. КИНЕТИКА И МЕХАНИЗМЫ РАДИОЛИЗА УГЛЕВОДОРОДОВ
Принято считать, что кинетику радиолитического разложе-
ния углеводородов можно объяснить, пользуясь только свобод-
норадикальными механизмами. Однако при изучении некоторых
систем получены доказательства того, что водород (и следова-
тельно, другие продукты) может образовываться прямо в ре-
зультате реакций замещения. Но этот механизм можно рас-
сматривать скорее как исключение и считать, что основная
масса реакций имеет радикальную природу. Рассматривая слу-
чайное статистическое взаимодействие образовавшихся радика-
лов, можно определить природу продуктов радиолитического
разложения [54]. При таком подходе предполагается, что эффек-
тивность столкновений не зависит от структуры радикала. Так
как энергия активации радикал-радикальных реакций очень
близка к нулю, то это эквивалентно предположению, что значе-
ние предэкспонепц'иалыюго множителя не зависит от струк-
туры. Ряд фактов свидетельствует в пользу такого предполо-
жения.
284
При радиолизе жидких углеводородов выход радикалов
(включая Н-атомы) и непосредственно образующихся продуктов
не превышает 10 частиц на 100 эв, что указывает на отсутствие
цепных реакций. Это резко контрастирует с газофазным термо-
разложением углеводородов, которое часто идет по цепному
механизму. Так как и в тех и в других системах часто присутст-
вуют и одни и тс же радикалы, следует предположить, что основ-
ными реакциями при радиолизе жидких углеводородов являются
реакций рекомбинации.
Прямое измерение констант скоростей для некоторых
радикал-радикальных реакций в углеводородных системах пока-
зало, что скорость этих реакций определяется скоростью про-
цессов диффузии {20, 23, 55]. Поэтому константы скорости реак-
ций одинаковы по порядку значения.
Таким образом, константы скорости для реакций взаимо-
действия радикалов можно найти по уравнению Смолуховского,
зная коэффициенты диффузии и параметры соударения:
k --- 4nNpD/1 000 М ~1 • cek'~1, (9.29}
где N — число Авогадро; р — параметр соударения; Л — коэффи-
циент диффузии.
В качестве примера с помощью уравнения (9.29) будет опре-
делена константа скорости рекомбинации этильных радикалов.
Результат можно сравнить с тем, который был получен в при-
мере 9.2.
Пример 9.6. Рассчитайте константу скорости реакции рекомбинации этиль-
ных радикалов по уравнению Смолуховского, принимая 0=0,75-10-5 см2/сек-,
р=2,0 А.
Решение. _
k = 4n,VpD/1000;
молекул/1000
k = 4 3,14 2 10-8 см 0,75 10-з см2/сек -6,03-102з -^-1-.
моль
k—1,1-I09 л/(моль-сек).
Найденная константа скорости реакции существенно не отличается от
константы, полученной в примере 9.2 [0,6-109 л/(моль-сек)].
Для того чтобы получить хотя бы качественные сведения
о продуктах радиолиза углеводородной системы, нужно иметь
определенную информацию о типах и относительном выходе
образующихся радикалов. Если такой информации нет, то ана-
лиз можно провести, основываясь на некоторых обобщениях,
справедливых для линейных углеводородов. Будем считать, что
основные радикалы образуются в результате следующих реак-
ций: 1) отрыва атома Н от метиленовых групп' родительской
молекулы; 2) отрыва атома II от метильной группы; 3) диссо-
циации углерод-углеродной связи, приводящей к образованию
радикалов меньшего размера, чем родительская молекула. На-
сколько обоснованы эти предположения, позволяют судить дан-
285
лые табл. 9.2, 9.5 и 9.6. Свободные радикалы образуются также
в результате вторичных реакций, таких, как отрыв атома II или
•его присоединение к олефинам.
При умеренных интенсивностях излучения радиолиз угле-
водородной системы можно представить следующим образом:
RH —Н, Rb R2, R3, R„ . (9.30)
Продукты радиолиза будут образовываться в результате слу-
чайного (статистического) взаимодействия радикалов друг
•с другом. Для радикалов R(, R2, Rs, R4 будут иметь место сле-
дующие реакции:
(1) Ri-L Ri -> 2R,Ri;
(2) Ri-tR2 -> RiR2;
(3) Ri -I- R3 R1R3J
(4) Ri *r Ri -> R1R.6
(5) R2 -r R, 2RoR2;
(6) R2 -r R3 — R2R3;
(7) R-2 -r R4 -> R2R4;
(8) R3 _L R3 2R3R3;
(9) R3 ~r Ri R3R4;
(10) Ri-l- R4 -> 2R4R4.
Если Ri — метил, Rs—этил, R3—пропил и так далее, то про-
дуктами радиолиза окажутся этап, пропан, бутан и пр. Этап
будет образовываться только в результате реакции (1), бутан
в реакциях (3) и (5), гексан в реакциях (7) и (8). Если общая
концентрация радикалов [Л] и выход радикалов равен [RJ, [R2]
и т. д., то, например, для концентрации бутана получим следую-
щее соотношение:
IRiJ 1R3] , [RJ2
Mi ' 2[Л]
Таким методом удается найти распределение продуктов ра-
диолиза в хорошем качественном согласии с эксперименталь-
ными результатами. Данный способ расчета, очевидно, подразу-
мевает, что диспропорционирование при рекомбинации радика-
лов либо незначительно, либо вообще отсутствует.
Пример 9.7 иллюстрирует использование данного метода рас-
чета. Количественные, данные взяты из работы де Фриза и Алле-
на [56].
Пример 9.7. Для радиолиза н-пентана известен тип возникающих ради-
калов и их радиационно-химический выход:
(I) G(CH3) = 0,l; (2) G(C.,HS)= 1,1;
<3) G(C3H7)=l,0; (4) G(C4I1„) = O,3;
<5) G(ewop-C5HU) = 5,0; (6) G(H-CsHn) = 2,2.
286
Какими будут значения G для продуктов радиолиза н-пептана при умерен-
ных интенсивностях излучения? *
Решение. При умеренных интенсивностях излучения отрыв водорода бу-
дет происходить в минимальных масштабах и следует рассматривать только-
радикальные реакции. Пусть [СИэ.]=о; [С2Н5|=Ь; [С3Н7]=с; [СД19]=й;
[вгор-С5Пц]==с, [н-С5Иц]=/; общин выход радикальных продуктов пусть,
равен R.
Соответствующие значения G будут равны:
11родукт G (расчет.) G (набл.)
СИ °2 ~0 0,27
СгНб “ 2R
ab СзН8= — -0,01 0,33
Ь2 ас ~ 2R т R -0,06 0,09
be ad -0,11 —
ае изо-С6П14 - — IX -0,05 0,03
af bd с2 . 0,11 0,14
Ci t- Л г 1 <£> С
be изо-С7Н16 — —— R 0,57 0,41
cd bf я-СЛ, = -+- 0,28 0,45
ce H3O-C8Hi8 ' к 0,52 0,59
d2 ef h-C8H18 — •-)- —- 2R R 0,23 0,20
de H3o-C»H20 — К 0,15 0,21'
«-QH.e = К 0,07 0
e2 ef изо-С10Н.,., — ——J- — 2R R 2,42 2,40
f2 H-C|0H.>2 — . 0,28 0,28
* Для сравнения в примере 9.7 приводится также экспериментально на-
блюдаемый выход продуктов. Использованные значения выхода радикальных
продуктов отличаются от данных табл. 9.5. Оценка выхода радикалов была
проведена авторами на основании измеренных значений выхода продуктов.
287
Метод позволяет достаточно хорошо описать выход продук-
тов с высоким молекулярным весом, ио оказывается неточным
для продуктов с низким молекулярным весом. Многие из этих
низкомолекулярных соединений возникают в результате слож-
ных вторичных реакций, таких, как образование С2Н4 с после-
дующим захватом атома Н и возникновением радикала С2Н5.
При радиолизе н-гексана происходят две следующие основ-
ные первичные реакции:
СвН14--->С6Н13-|-Н;
С6Н14---> С6Н12 Н2.
Происходит также диссоциация углерод-углеродных связей,
ведущая к образованию радикалов с низким молекулярным ве-
сом, таких, как метильные, этильные, пропильные и бутильные
радикалы и продукты их последующих превращений. Разрыв
углерод-водородных связей идет преимущественно в метилено-
вых группах. Поэтому боковые цепи в разветвленных продуктах
присоединяются при рекомбинации к углеродным атомам мети-
леновых групп.
9.12. ЭФФЕКТЫ, ВОЗНИКАЮЩИЕ ПРИ ПОГЛОЩЕНИИ
БОЛЬШИХ ДОЗ
При поглощении больших доз конечные продукты сущест-
венно отличаются от начальных. Возникающие продукты сами
могут вступать в реакции с активными частицами по мере того,
•как их концентрация будет становиться заметной.
Так, например, данные ЭПР-спсктроскопии показывают, что
атомы Н обнаруживаются только при радиолизе жидкого ме-
тана, хотя из экспериментов с акцепторами (такими, как оле-
фины) хорошо известно, что водородные атомы образуются при
радиолизе большинства углеводородных систем. В частности,
исследование методом ЭПР-радиолиза жидкого этапа показы-
вает присутствие этильных радикалов, по не позволяет обнару-
жить атомы Н. При добавлении к этой системе 5% C2D4 возни-
кают радикалы C2D4H в концентрации, почти равной концент-
рации радикалов С2Н5. Отсюда можно сделать вывод, что при
введении в достаточном количестве олефина происходит акцеп-
тирование атомов П и образование радикалов. При поглощении
больших доз этот эффект должен усиливаться.
9.13. МАТЕРИАЛЬНЫЙ БАЛАНС
Самый лучший метод проверки материального баланса —
учет всех возникших продуктов. Часто, однако, этот метод ока-
зывается неудобным, так как при радиолизе большей части
углеводородных систем образуется некоторое количество поли-
288
мера. Обычно бывает полезен другой метод, в котором опреде-
ляют отношение числа атомов Н к числу атомов С в продуктах
и в исходном соединении. В н-пентапе, например, отношение
Н/С в исходной.системе равно 2,40. Это значение следует затем
сравнить с отношением общего числа Н-атомов, возникших на
100 эв, к числу атомов С, возникших на 100 эв, во всей массе
продуктов.
Еще один метод проверки материального баланса состоит
в сравнении количества выделившегося водорода с общей не-
насыщенностью конечной системы. При этом количество выде-
лившегося водорода должно быть больше общей ненасыщен-
ности системы, так как источником водорода служат также
реакции:
RI I R - J- II; R -h R -> RR; Н -< Н -> Н2.
Ненасыщенность образующегося полимера можно обычно опре-
делить йодометрическим титрованием. Йодометрия дает лишь
приблизительный результат, однако позволяет провести быструю
проверку материального баланса.
9.14. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА РАДИОЛИЗ
УГЛЕВОДОРОДОВ
Повышение температуры в общем случае благоприятствует,
очевидно, тем реакциям, которые нуждаются в тепловой актива-
ции. Это реакции отрыва водорода, диссоциации связей и реак-
ции мономолекулярного распада родительских ионов (см. раз-
дел 9.5 и пример 9.1). Повышение температуры благоприятствует
также реакциям диспропорционирования и разложения.радика-
лов. Как правило, химические реакции имеют положительные
температурные коэффициенты; поэтому следует ожидать, что
облучение при повышенных температурах приведет к ускорению
большей части реакций.
В общем случае термодеструкцию углеводородов объясняют
с помощью механизма, в котором после начальной диссоциации
углерод-углеродной связи развиваются радикальные цепные
реакции с переменной длиной цепи. Энергия активации для
диссоциации углерод-углеродной связи составляет около
80 ккал/моль. Следовательно, для того чтобы термодеструкция
шла I- измеримой скоростью, необходима температура порядка
500—600° С. Энергия активации реакции распада радикалов или
родительских ионов гораздо меньше, поэтому ускорение радио-
литического разложения будет наблюдаться при значительно
более низких температурах.
В радиолитических процессах при комнатной температуре,
например, если происходит радиолиз н-гексана, образуются
крупные радикалы (в данном случае гексильные радикалы).
Из-за етерических и энергетических факторов эти радикалы
10 Зак. 907
289
обычно не инициируют реакций отрыва водорода. Их основные
реакции— реакции рекомбинации. Реакции отрыва водорода
инициируют радикалы меньших размеров, такие, как метил,
этил или пропил, при условии, что обеспечена необходимая теп-
ловая активация. Повышение температуры благоприятствует
распаду крупных радикалов (и родительских ионов) на мень-
шие радикалы, которые способны отрывать водород от роди-
тельских молекул и таким образом .создавать новые крупные
радикалы, которые в свою очередь *гакже могут диссоциировать.
Схематически этот цепной процесс можно записать следую-
щим образом:
1) RH -> R-J-П;
Д/.
(2) R------->R' стабильная мо 1екула;
(3) R' Ч- RH R'H 4- R.
Одновременно будут идти реакции рекомбинации и диспро-
порционирования радикалов, однако при достаточной тепловой
активации реакции отрыва водорода начнут конкурировать с ре-
комбинационными процессами.
Повышение температуры ведет не только к увеличению об-
щего выхода на единицу поглощенной энергии (единицу погло-
щенной дозы), ио может также существенно изменить относи-
тельные значения выхода для различных продуктов. По мере
повышения температуры примерно до 600° С влияние термоинду-
цированных реакций становится заметным, а при дальнейшем
росте температуры эти реакции начинают преобладать. Если
пренебречь тепловой диссоциацией радикалов или ионов, то от-
ношение скорости термоинициировапия процесса разложения к
скорости радиационного инициирования найдем, зная лишь
число радикалов, возникающих на 100 эв поглощенной энергии,
и приблизительную энергию активации для инициирования тер-
модеструкции. Хотя расчет довольно грубый, он вполне годится
для оценки влияния излучения па термоактивированные реак-
ции. Расчет такого типа выполнен в примере 9.8.
Пример 9.8. Рассчитайте отношение скорости термоинициирования раз-
ложения н-гептана при 600° С к скорости радиационного инициирования при
интенсивности излучения 1016 эв/(см3-сек). G (радикалов )=6. Энергию акти-
вации радикального термоинициирования примите равной 80 ккал/моль.
Решение. Скорость термоинициирования равна /г[№]. Считая, что Л=
— 1013 е~сек~' и [RII]=5 1021 молекул/см3, найдем скорость термо-
И11ицпирования=5- 1034-е~45 молекул/(см3-сек):
1016 эв
Скорость радиационного инициирования =
ССК-СМ3
6 радикал „
X ‘——------- = 6-Ю14 молекул/(см3-сек);
Скорость термоинициирования 5-1034-е~45
Скорость радиационного инициирования 6-Ю14
290
Значение отношения будет расти при повышении температуры, пока, на-
конец, при 800° С радиационный вклад п разложение н-гептапа не станет
пренебрежимо малым.
Расчет того же типа можно использовать для определения температуры,
при которой следует ожидать увеличения скорости радиациоппо-иидунирован-
ных реакций. В качестве энергии активации выбирают наименьшую из энер-
гий активации реакций отрыва водорода, распада радикалов и распада ро-
дительских ионов.
ЗАДАЧИ
9.1. Объясните появление радикала СИ2=СНСИ2СН2, наблюдаемого ме-
тодом ЭПР при радиолизе этилена.
9.2. Используя численные значения дли констант скоростей и концентра-
ций, приведенные в примере 9.5, найдите величину ktkl/k^C^ для N—0,2 с
помощью уравнения (8.23).
9.3. Считая, что энергия активации реакции отрыва водорода порядка
8 ккал!моль, оцените относительную вероятность этой реакции по сравнению
с реакцией рекомбинации метильных радикалов (G=4) при мощностях дозы
10'3, 107 и 105 рад/мин.
9.4. Рассчитайте, когда начнет сказываться влияние мощности дозы, если
метильные радикалы могут участвовать в конкурентных реакциях отрыва
водорода и рекомбинации и известно, что G(CH3)=0,l; ft.s=6,8-104 л/(лтольХ
X сек); k = 3 • 1()8 л/(моль сек).
9.5. Рассчитайте константу скорости рекомбинации метильных радикалов,
если Z?=2-I0-5 см?/сек и параметр соударения равен 2 А.
9.6. Оцените, при какой температуре увеличение скорости разложения
пентана станет критическим, если энергия активации реакции распада роди-
тельского иона равна 30 ккал!моль.
9.7. Как повлияет увеличение температуры па скорость диффузионно-конт-
ролируемых реакций? Будет ли существенным изменение скорости таких реак-
ций в жидкой фазе при увеличении температуры на 20°?
9.8. Используя правило электронных долей, рассчитайте выход водорода
в 5%-но,м растворе бензола в н-гексапе, если G(H2, бензол) =0,04; G(H2,
гексан) =5,8.
9.9. Считая, что предэкспоненниальпые множители равны, найдите, при
каких температурах реакция отрыва водорода сможет конкурировать с реак-
цией рекомбинации. Энергия активации реакции отрыва равна 8 ккал/моль,
реакции рекомбинации — 1 ккал/мо th.
9.10. Используя для выхода радикалов значения G, приведенные в
табл. 9.5, найдите, какие продукты образуются при радиолизе н-гексана и
чему будет равен их выход.
9.11. Используя формулу (9.23) для выхода метильных радикалов, най-
дите выход этих радикалов для всех соединений, указанных в табл. 9.5; ре-
зультаты сравните с экспериментально наблюдаемыми значениями.
10*
ГлаваХ
РАДИАЦИОННАЯ ХИМИЯ ТВЕРДОГО ТЕЛА
10.1. ВВЕДЕНИЕ
Воздействие радиации па твердые тела в значительной мере
сводится к образованию тех или иных дефектов. Среди них мож-
но выделить дефекты, связанные со смещением атомов, и дефек-
ты, связанные с изменением электронных конфигураций. Обе
категории повреждений тесно связаны друг с другом, так как
изменение электронной конфигурации в одном из атомов, конеч-
но, влияет на положение соседних атомов и наоборот. Однако
в некоторых веществах, например в металлах, радиационные
повреждения представляют собой главным образом смещения
атомов, так как возмущения электронных конфигураций быстро
сглаживаются. Поэтому прежде чем выяснять, какие свойства
твердых веществ меняются под действием ионизирующих излу-
чений, нужно разобраться в природе дефектов кристаллической
решетки. Далее мы кратко рассмотрим основные виды дефек-
тов в твердых телах, способы их образования, а также харак-
теристики вещества, которые изменяются благодаря появлению
этих дефектов (более полную информацию читатель может най-
ти в книге Ван-Бюрена [1]). В этой главе в основном будут
рассмотрены такие вещества, которые претерпевают химические
изменения и поэтому интересны для химиков. Радиационные
эффекты в металлах, большей части полупроводников и кера-
мике рассматриваться не будут.
10.2. ПРИРОДА ДЕФЕКТОВ КРИСТАЛЛИЧЕСКОЙ РЕШЕТКИ
В кристаллических телах хорошо изучены разнообразные
нарушения структуры или дефекты. Нарушения структуры де-
лятся па две основные категории — точечные дефекты и дисло-
кации. В свою очередь точечные дефекты подразделяются на
вакансии и атомы внедрения. Дислокации представляют собой
объемные или линейные дефекты, при которых нарушения иде-
альной решетки распространяются на несколько межатомных
292
расстояний. Иногда точечные дефекты называют дефектами
Френкеля и дефектами Шоттки, имея в виду соответственно
атомы внедрения и вакансии.
10.2.1 . Вакансии
В идеальной иопной решетке содержится равное число анио-
нов и катионов. Если ион, положительный или отрицательный,
будет вырван из узла решетки и попадет на поверхность, то
возникнет вакансия. Число ка-
тионных и анионных вакансий
всегда одинаково (при отсутствии
примесей). Если на поверхность
мигрирует избыточное количество
катионов, то создается положи-
тельный заряд, задерживающий
дальнейшую миграцию катионов
н- способствующий миграции ани-
онов. Таким образом, в отсутст-
вие внешних сил число вакансий
противоположного знака стре-
мится выровняться (рис. 10.1).
Дефекты Шоттки в ионных кри-
Рис. 10.1. Точечные дефекты кри-
сталлической решетки. Пунктир-
ные кружки указывают положе-
ние катионных и анионных ва-
кансий.
сталлах можно рассматривать по-
этому как пары вакансий.
Вакансии имеются во всех
кристаллических твердых телах.
В кристалле обычно существует
равновесное число вакансий, за-
висящее от температуры, при которой формировался кристалл.
Если ионный кристалл содержит N атомов, то п дефектов Шот-
тки возникают при удалении п катионов и анионов из объема
кристалла. Число способов, какими можно удалить п ионов одно-
го знака, дается формулой
У(У- 1)(ЛГ—2). . . (TV— 1) = ЛИ ,10 р
п! (N —п)! п! '
Так как число катионных и анионных вакансий равно, то
число способов образования п дефектов Шоттки равно квадра-
ту выражения (10.1). Образование п дефектов Шоттки увели-
чивает энтропию кристалла на
S = k In Г---Т + nS„. (10.2)
I (N—n)\nl I р
Возрастание энтропии приводит в свою очередь к
свободной энергии Гиббса
G = H — TS-,
G — пН„ — nSpT — kT In [-----—------] ,
р р |_ (TV — И)! и! J ’
изменению
(10.3)
(10-4)
293
где пНр — полное изменение энтальпии; nSp—добавочное из-
менение энтропии, связанное с образованием дефектов Урав-
нение (10.4) можно упростить, пользуясь приближением Стир-
линга (In х\~х 1п х—х):
In Г--—----] = 2 [A In А— (N— n)\n(N— п) — nlnnj (10.5)
L (А —и)! п\ J
или
G = пНр — nTSp —2kT[N\nN — (N — ti) In (A — n) — n Inn]. (10.6)
В условиях равновесия при температуре Т свободная энергия
постоянна, поэтому первая производная G по п равна нулю.
Дифференцируя уравнение (10.6) по п при постоянной Т, по-
лучаем:
(= 0 = (Hp — TSp) — 2kT[ln(N— п)—1пп]; (10.7)
\ дп. Jt=o
Gp = 2kTln ; (10.7a)
n
J^=eGP/k\ (10.8)
где Gp — свободная энергия, необходимая для образования па-
ры ваканский.
Так как N п, то уравнение (10.8) можно упростить:
п х Ne^p/kT.
(Ю.9)
Уравнение (10.9) дает число дефектов Шоттки в условиях рав-
новесия при температуре Т.
Пример 10.1. Рассчитайте число дефектов Шоттки в кристалле КС1 при
температуре плавления 1054° К, если свободная энергия, необходимая для
образования пары вакансий, равна 2 эв. Плотность КС1 равна 1,984 г)см3.
Решение. Подставляя численные значения в уравнение (10.9), находим:
1,984 г 1 моль 6-1023 молекул е 2 3e'kT
п ~ см3 .74,6 г МОЛЬ
молекул { Г 2 эв-1,6-10—12 эрг
п = 1,62-1022 ------ехр — ------------------——
СМ3 ( L эв
°к
2-1,38-10-1в эрг
1 1)
—------— J --= 15-101® вакансий1см3.
1054 К U
10.2.2 . Атомы внедрения
Атомы внедрения — это атомы, вырванные со своих мест,
но не занявшие узлов решетки. Атом внедрения вместе с ва-
кансией образует пару, как это схематически показано па
* Образование дефектов приводит к появлению добавочного энтропий-
ного члена, так как меняется число колебательных степеней свободы.
294
рис. 10.2. Как и дефекты Шоттки, дефекты Френкеля существу-
ют во всех кристаллах. Их число зависит от температуры кри-
сталлизации. Выражение для числа дефектов Френкеля полу-
чают способом, аналогичным тому, с помощью которого было
выведено уравнение (10.9). Выражение выглядит следующим
образом:
п (Ж)'/2 (10.10)
где N — число атомов в кри-
сталле; Ni — число межузловых
промежутков, в которых могут
разместиться атомы внедрения;
Gf — свободная энергия образо-
вания пары вакансия — атом
внедрения.
Очевидно, что и в отсутствие
облучения всегда существует ко-
нечное число вакансий и атомов
внедрения, поэтому получить кри-
сталлы без дефектов невозможно. „ т .
тт Рис. 10.2. Точечные дефекты кри-
Не следует думать, однако, сталлической решетки. Сплошные
что все Дефекты, возникшие в кружки указывают положение
кристалле при высокой темпера- атомов внедрения,
туре, сохраняются при комнатной.
Во время охлаждения диффузионные процессы приводят к бы-
строму уменьшению концентрации дефектов. Но кристалл,
сформировавшийся при высокой температуре и затем резко
охлажденный, сохранит заметную долю первоначальных дефек-
тов. Такая «закалка» представляет собой обычный метод увели-
чения числа дефектов в кристалле.
10.2.3 . Дислокации
Вначале представление о дислокациях сформулировали, что-
бы объяснить пластическую деформацию твердых тел. Позднее
было показано, что дислокации нужны при описании процес-
сов роста кристаллов, а также для объяснения других свойств
твердых тел, таких, например, как электропроводность.
Существует два главных типа дислокаций — краевые и вин-
товые. Краевая дислокация (в моноатомных кристаллах) пред-
ставляет собой линию атомов (или ионов), каждый из которых
образует на единицу меньше координационных связей с со-
седними атомами, чем должно быть в идеальном кристалле
(рис. 10.3). Иначе краевую дислокацию можно представить се-
бе как искажение кристаллической структуры, вызванное лиш-
ней атомной плоскостью, вдвинутой в часть объема кристалла.
В ионных кристаллах нужно вдвинуть две полуплоскости: одну
для анионной и другую для катионной решетки. Под нагрузкой
295
краевые дислокации смещаются. Плоскость, в которой лежат
линии дислокации и линии направления движения, называется
плоскостью скольжения. Краевая дислокация под воздействием
нагрузки свободно смещается только в плоскости скольжения.
При добавлении или удалении атомов из лишней полуплоско-
сти происходит переползание дислокации.
ооооооооооооо
OOOOOOQOOOOOO
ооооос/оооооо
оооооо оооооо
оооооооооооо
Рис. 10.3. Краевая дислокация. Символ указывает направле-
ние дислокационной линии, которая перпендикулярна к пло-
скости рисуика.
Винтовая дислокация образована линией атомов с правиль-
ным числом координационных связей, по с нарушенными угла-
ми координации. Если атомы (или ионы) решетки соединить
прямыми, то образуются многогранники, такие, например, как
Рис. 10.4. Винтовая дислокация.
Стрелка показывает ось винтовой
дислокации.
куб или тетраэдр. В результате
винтовой дислокации форма мно-
гогранников искажается (рис.
10.4). Структура винтовой дис-
локации напоминает строение
винтовой лестницы. Винтовые
дислокации иногда называют дис-
локациями Бюргерса.
Существует большое разнооб-
разие возможных дислокаций,
однако все они сводятся к двум
рассмотренным выше типам. На-
пример, поверхностные дефекты
изгиба образованы линейной ком-
бинацией краевых дислокаций.
Особые комбинации дислокаций
образуют границы между зер-
нами материала.
Линии дислокаций не могут заканчиваться внутри кристал-
ла и либо заканчиваются на поверхности, либо образуют замк-
нутый контур. Дислокации в кристалле при пересечении спо-
собны скомпенсировать друг друга. Очень часто при пересече-
нии винтовых дислокаций образуются краевые дислокации.
296
10.2.4 . Центры окраски
Еще один тип дефектов в твердых телах представляют цент-
ры окраски. Под действием ионизирующей радиации большая
часть органических и неорганических соединений приобретает
ту или иную окраску. В некоторых случаях окраска связана с
образованием поглощающих свет новых стабильных соединений
или сравнительно стабильных радикалов [2]. В других случаях
окрашивание связано с образованием ионов, таких, например,
как ион нефталинида, возникающий при облучении. растворов
нафталина в 2-метилтетрагидрофуране в стеклообразном со-
стоянии. Окраску также может обусловливать появление соль-
ватированных электронов [3].
Однако термин «центр окраски» применяется обычно в тех
случаях, если вакансия или точечный дефект захватывает элект-
роны или дырки и приобретает способность поглощать свет.
Центры окраски могут возникать в ионных кристаллах, изоля-
торах и полупроводниках. В литературе этот термин часто ис-
пользуется для описания радиациоппо-йндуцированного окра-
шивания любых веществ. Мы, однако, будем употреблять его в
более ограниченном смысле, называя центрами окраски слабо-
связанные электроны, захваченные вакансиями.
Любой кристалл, выращенный в- равновесных условиях, со-
держит дефекты и дислокации. Наличие дефектов меняет рас-
пределение заряда, поэтому в районе дефектов следует ожидать
сдвига электронных уровней.
Еще до использования ионизирующей радиации центры
окраски получали в щелочно-галоидных кристаллах, создавая
избыток металла или галогена. Если на поверхность наносили
слой щелочного металла, то в объеме кристалла появлялось
окрашивание. Предполагается, что анион мигрирует сквозь ре-
шетку, взаимодействует с атомом металла, ионизуется и отдает
электрон, который захватывается анионной вакансией
(рис. 10.5). Такие кристаллы имеют полосы поглощения в види-
мой области (кристалл КС1—темно-голубой, LiF — розовый,
NaCI — коричневый). Правильность этой модели подтвержда-
ется тем фактом, что характер полосы поглощения не зависит
от природы добавляемого металла. Добавление натрия к кри-
сталлу KCI вызывает точно такое же окрашивание, как и до-
бавление калия. Модель образования центров окраски под-
тверждается также методом ЭПР. Расщепление в спектрах со-
ответствует ^-фактору 1,995 (сравните с g=2,0023 для свобод-
ного электрона).
Электроны, захваченные анионными вакансиями в щелочно-
галоидном кристалле, принято называть F-центрами (от немец-
кого Farben Zentrum — центр окраски). При освещении F-цент-
ров белым светом захваченные электроны могут возбуждаться
в высшие квантовые состояния. Полоса поглощения, которую
297
принято называть F-полосой,- занимает определенную спект-
ральную область с характерным положением максимума. При
освещении кристалла светом с длиной волны, лежащей в F-по-
лосе, и одновременном воздействии электрического поля на-
блюдается фотопроводимость. Если под действием света F-
центры обесцвечиваются, может возникнуть новая полоса по-
Рис. 10.5. Схематическое изображение некоторых типов центров окрас-
ки. Квадраты символизируют ионные вакансии, сплошные кружки —
электроны; пустые кружки — дырки.
глощепия F'. Ее появление, очевидно, обусловлено тем, что
электроны F-центров попадают вначале в полосу проводимости
и затем захватываются другими F-центрами. Так как F-центры
уже имели по одному электрону, возникают новые центры (F')
с двумя электронами на одной вакансии.
При добавлении к щелочно-галоидпому кристаллу (NaCl)
избытка галогена появляются другие полосы поглощения. Их
принято называть V-полосами. Считается, что возникновение
V-центра связано с потерей электрона одним из отрицательных
ионов хлора, окружающих катионную вакансию. Физическая
природа V-центров довольно сложна. Как показано методом
ЭПР, некоторые из V-центров не обладают сферической сим-
метрией. Их возникновение связывают с присутствием молеку-
лярного иона С1£". При этом два соседних иона галогена взаи-
модействуют с положительной дыркой [4]. V-центры сходного
типа были обнаружены в кристаллах LiF [5, 6]. Согласно
298
классификации Зейтца [7], это центры типа Уь Vi-центры пред-
ставляют собой дырки, захваченные вблизи катионной вакан-
сии.
В табл. 10.1 приведены приблизительные положения макси-
мумов /•'-полосы в рядег щелочно-галоидных кристаллов. При
облучении щелочно-галоидных кристаллов ионизирующей ра-
диацией возникают F- и У-центры.
Т аблица 10.1
Характеристики f-полос в щелочно-галоидных кристаллах
Кристалл Положение максимума, о 2 Полуширина полосы погло- щения при 20- С, эв Сила осцил- лятора Кристалл Положение максимума, о А Полуширина полосы погло- щения при 20° С, эв Сила осцил- лятора
NaF 3400 0,62 КС1 5630 0,36 0,90
NaCl 4650 0,47 0,81 КВг 6300 0,36 0,80
NaBr 5400 .— — KI 6850 0,35 —
Nal 5880 .— — RbCl 6240 0,31
KF 4550 0,41 —~ RbBr 7200 0,28 —
Rbl 7750 — —
Значительно труднее установить природу центров окраски в
более сложных ионных кристаллах, таких, как нитраты и пер-
хлораты. Природу этих центров окраски удается выяснить толь-
ко после изучения оптических характеристик в разных условиях
облучения (в частности, температурных) и для разных режи-
мов температурного отжига.
Полосы поглощения различаются по стабильности, что мо-
жет служить непосредственной мерой энергии центров окраски.
Эксперименты с отжигом при различных температурах позво-
ляют пайти температурный коэффициент (энергию активации)
процесса уничтожения центров окраски.
Число центров окраски можно определить с помощью урав-
нения, вначале полученного Шмакулой и затем упрощенного
Пшибрамом [8]. Уравнение справедливо только в том случае,
если полоса поглощения гауссовой или лоренцевой формы:
fd =1,31- 10”-2”2-)2-^ах Н. (10.11)
Здесь f — сила осциллятора; d — концентрация центров окра-
ски; п — показатель преломления; аШах — коэффициент погло-
щения в максимуме, елг-1; Н — полуширина полосы поглоще-
ния, эв.
Чтобы найти концентрацию центров окраски, прежде всего
необходимо построить график зависимости оптической плотно-
299
ста от длины волны и определить Zmax и форму полосы погло-
щения. Коэффициент поглощения а равен произведению мо-
лярного коэффициента экстинкции па концентрацию а=еС.
Следовательно, «max можно найти, разделив оптическую плот-
ность при Zmax на длину оптического пути. Следует также зпать
и силу осциллятора: обычно опа варьирует от 0,6 до 1. Для оди-
ночного электрона действует правило сумм, которое дает мак-
симальное значение силы осциллятора, равное 1. Таким обра-
зом, используя приближенное значение [, можно оценить кон-
центрацию центров окраски в кристалле.
10.2.5 . Другие типы дефектов
Включение примесей также следует считать дефектами ре-
шетки. В химически лабильных системах примеси образуются
под действием радиации. Например, облучение кристаллов
ККОз приводит к появлению NO2 и Ог, которые следует рас-
сматривать как примеси, способные повлиять на миграцию
энергии в решетке. Примеси могут возникать также в резуль-
тате ядерных превращений.
10.3. РАДИАЦИОННЫЕ ПОВРЕЖДЕНИЯ В ТВЕРДЫХ ТЕЛАХ
В твердом теле поглощенная энергия может затрачиваться
на следующие процессы: 1) перемещение электронов (возбуж-
дение, ионизация); 2) перемещение атомов; 3) образование
примесных атомов; 4) захват электронов. Процессы поглоще-
ния энергии заканчиваются примерно' за 10-17 сек. После этого
развертываются молекулярные процессы (включая миграцию
экситонов) — флуоресценция, диссоциация и тому подобные,
которые занимают около 10-13—10~9 сек. Фосфоресценция, диф-
фузия, химические реакции, распад центров окраски, рекомби-
нация электронов и дырок и другие процессы, ведущие к вос-
становлению равновесия, занимают на временной шкале про-
межутки от 10-8 сек вплоть до интервалов, которые пришлось
бы измерять в годах.
Радиационные эффекты в твердых телах разделяют на два
больших класса: преимущественно кристаллографические струк-
турные дефекты и повреждения химической природы. Преобла-
дание того или иного класса повреждений в значительной мере
зависит от материала (металл, ионный кристалл, полупровод-
ник и т. д.) и от характера излучения. Облучение тяжелыми
частицами (быстрыми нейтронами, продуктами деления, бы-
стрыми протонами и т. д.) может вызывать атомные смещения
и химические изменения, тогда как облучение электронами и
у-квантами вызывает главным образом химические изменения.
300
10.3.1. Облучение твердых тел нейтронами
Взаимодействие быстрых нейтронов с веществом осуществ-
ляется главным образом через прямые соударения с ядрами.
В результате соударений выбиваются ионизированные атомы,
которые ведут себя далее как тяжелые заряженные частицы и
могут в свою очередь вызывать ионизации и атомные смеще-
ния. Медленные нейтроны взаимодействуют с веществом почти
целиком через процессы захвата; в результате образуются ато-
мы примесей (дефекты), которые могут обладать достаточной
энергией отдачи для того, чтобы вызывать ионизации, возбуж-
дение и атомные смещения.
В качестве источника быстрых нейтронов обычно использу-
ется реактор, так как нейтронные источники на базе ускори-
телей, как правило, не дают достаточно высоких потоков, необ-
ходимых для исследования радиационных повреждений в твер-
дых телах. Нейтронное излучение реактора всегда сопровожда-
ется у-излучепием, поэтому во всех случаях образец подверга-
ется воздействию смешанного излучения. Радиационно-химиче-
ское разложение в системе под действием быстрых нейтронов
(через посредство выбитых атомов) оказывается обычно пезпа*
чителытым по сравнению с разложением, вызванным у-излуче-
нием, сопровождающим возникновение нейтронов деления.
Среднее число смещений N, вызываемых быстрым нейтро-
ном, можно приближенно определить с помощью уравнения
(10.12):
[о,Ю8 —0,56-lln-^Kl + 1, (Ю.12)
*** ml- £ J L J
где Е — энергия нейтронов; Ет — энергия, затрачиваемая на
атомное смещение; Тт — максимальная энергия первичного сме-
щенного атома.
Оценки показывают, что для большей части кристаллических
твердых тел значение Ет равно примерно 10 эв. Эта энергия
велика по сравнению с энергией образования атомов внедре-
ния. Такое несоответствие возникает потому, что отдача про-
исходит мгновенно и соседние атомы не успевают отрелаксиро-
вать. Тт — максимальная энергия выбитого первичного атома —
дается выражением
у, _ 4ЕМт
т ~~ ~(М + т)2
(10.13)
где т — масса нейтрона; М — масса выбитого атома.
В качестве примера использования этих уравнений ниже
приводится расчет числа смещений, создаваемых нейтронами с
энергией 1,5 Мэв в кристалле NaNOa.
301
Пример 10.2. Рассчитайте число атомных смещений в образце NaNCh при
облучении его потоком быстрых нейтронов (со средней энергией 1,5 Л4эд) с
плотностью 1013 нейтрон!(см2• сек) в течение двух дней.
Решение. Для числа смещений справедливо уравнение (10.12):
£ I £г1Г £rV
iV;S 4T^L0’,08-°-5611n'£-|L1-|-~rl
Чтобы рассчитать Tm, примем, что средняя масса атомов в кристалле
NaXO.3 равна 17:
4(1,5-Ю6) 17 .
(18)2
Тт = 31 • 10« эе;
1 5-106
V ’--------{0,108 — 0,561 1п [6,6-10—6]} [1 + 6.6-10-6]-1-
124-104 г '
N к 4.
Общее число смещений ~ 4-1013-3600-24.2, т. °-
гг0,7-1018 смещений/см3 или я0,3-1018 смещений1г.
10.3.2. Облучение твердых тел тяжелыми заряженными
частицами («-частицами, протонами и др.)
Для высокоэнергетических заряженных частиц основной вид
взаимодействий — кулоновские взаимодействия. Энергия частиц
растрачивается на ионизации, возбуждение, а также атомиые
смещения. Максимальная энергия, передаваемая выбитому ато-
му, дается уравнением (10.13), которое справедливо для лю-
бых столкновений твердых сфер. Средняя энергия, передавае-
мая первичному смещенному атому, равна:
£ = Дг1п(^Л для Tm»£r. (Ю.14)
Смысл обозначений тот Же, что и в уравнении (10.12). Урав-
нение (10.14) дает среднюю энергию первичных атомов отдачи.
Среднее число вторичных атомов, выбитых первичным атомом
из узлов решетки, будет равно:
N = Го,885 + 0,'561 In (Х'” + 1)-1 . (Ю. 15)
I. 4 J Xm
Здесь Xm+1 = ———------f- J М— масса выбитого атома; т —
(М+т)2 Ет
масса частицы. Например, при облучении меди дейтронами с
энергией 500 кэв Е= 195 эв и ТУ—4,5.
10.3.3. Облучение твердых тел электронами и у-квантами
Действие высокоэнергетических электронов (~1—2 Мэв) и
и у-излучения (>1 Мэв) на твердое тело может приводить к
атомным смещениям. Ярким примером может служить обра-
302
зование дефектов в кристаллической решетке сплава меди и зо-
лота под действием у-излучения 60Со. Однако по сравнению с
химическими изменениями (если они возможны в данной си-
стеме) число атомных смещений при действии электронов и
у-излучения настолько мало, что этим эффектом можно прене-
бречь. Первичные атомы, выбитые высокоэпсргетическими
электронами, обладают такой малой энергией, что выход
вторичных смещенных атомов практически равен пулю. Облу-
чение электронами и квантами вызывает главным образом
ионизации, возбуждение и химическое разложение в тех систе-
мах, где могут идти радиационно-химические реакции.
Если принять, что на образование пары ионов затрачива-
ется 35 эв, то электрон с энергией 1 Мэв создаст около 104 пар
ионов и около 2—3 - 104 возбужденных частиц. Эти ионизован-
ные и возбужденные частицы будут распределены вдоль трека
электрона, который несколько превышает стандартный пробег
электронов в этой среде. С другой стороны, а-частица с той же
энергией образует такое же число ионных пар, но они будут
сосредоточены на гораздо более прямом и коротком отрезке,
что приведет к высокой плотности ионизации.
10.3;4. Действие продуктов деления и ядер отдачи
Если в кристаллическую решетку внедрены делящиеся ве-
щества, а затем, материал подвергается нейтронному облуче-
нию, то возникающие внутри кристалла продукты деления,
представляющие собой массивные заряженные частицы высо-
кой энергии, могут вызвать значительное число ионизаций и
атомных смещений.
Ядра отдачи в кристалле появляются в результате различ-
ных ядерных превращений. В химии горячих атомов часто ис-
пользуют такие источники внутреннего облучения. Для транс-
мутаций- обычно применяют Li (превращается в 3Н) и N (пре-
вращается в 14С).
10.3.5. Другие механизмы образования
радиационных повреждений в твердых телах
Как показано в разделах 10.3.1, 10.3.2 и 10.3.3, воздействие
быстрых нейтронов, тяжелых заряженных частиц, продуктов де-
ления или атомов отдачи, возникающих в результате захвата
медленных нейтронов, приводит к появлению дефектов в кри-
сталлах. Эти дефекты возникают благодаря атомным смеще-
ниям, химическому распаду и образованию центров окраски.
С другой стороны, облучение фотонами и> электронами
сопровождается главным образом изменением электронных кон-
фигураций (что ведет к химическим превращениям, образова-
нию центров окраски, возникновению радикалов и пр.) при
303
практически полном отсутствии атомных смещений. Считается,
что атомные смещения иод действием нейтронов и заряженных
тяжелых частиц возникают в результате прямых соударений.
Помимо этого, предполагается существование нескольких дру-
гих механизмов возникновения атомных смещений в кристал-
лах.
Локальное выделение тепла («тепловой спайк»). Сущест-
вуют два основных типа процессов, приводящих к резкому ло-
кальному повышению температуры. Процессы первого типа вы-
зывают выбитые атомы (первичные атомы) с энергией Е, ра-
страчивающие эту энергию в малом объеме. Такое внезапное
выделение энергии может привести на короткое время к весьма
значительному локальному повышению температуры [9]. На-
пример, передача энергии в 105 эв в объеме, содержащем
около 104 атомов, при коэффициенте диффузии порядка
10-3 см21сек приведет к повышению температуры объема до
1000° К в течение примерно 10-11 сек. Процессы подобного ти-
па существенны только при облучении тяжелыми заряженными
частицами или Нейтронами.
Электронным спайком [8] иногда называют другой тип
процессов, приводящих к локальному повышению температуры.
В этом случае при прохождении заряженной частицы (элект-
рона) практически не происходит непосредственной передачи
энергии решетке, но почти вся энергия затрачивается на элект-
ронное возбуждение, энергия которого затем конвертирует в
тепло. От скорости конверсии энергии электронного возбужде-
ния в энергию решетки зависит, будет ли решетка нагрета до
таких высоких температур, что произойдет образование дефек-
тов. Значение передаваемой энергии будет зависеть от силы
взаимодействия электронов с решеткой. Для металлов, где взаи-
модействие очень слабое, из расчетов Зейтца и Колера [10]
следует, что при возбуждении всех атомов вдоль трека ионизи-
рующей частицы до 10 эв температура решетки поднимается
приблизительно до 500° К. Это значение составляет лишь
1/100 электронной температуры и нс может привести к образо-
ванию заметных дефектов в решетке. Однако при сильном
взаимодействии электронов с решеткой электронный спайк, по
мнению Зейтца и Колера, может приводить к возникновению
дефектов.
Конверсия энергии электронного возбуждения в колебания
решетки может быть также весьма существенной для протека-
ния различных химических процессов. Хотя в настоящее время
нет определенных теоретических или экспериментальных дока-
зательств того, что подобные процессы играют важную роль в
радиационном разложении неорганических или органических
систем, нельзя отрицать, что такая возможность существует.
Механизм Варли. Еще один способ возникновения дефектов
в твердых телах, который был постулирован Варли [11], отно-
304
сится к облучению ионных кристаллов. Суть механизма заклю-
чается в том, что многократная ионизация аниона может при-
водить к возникновению суммарного положительного заряда в
соответствующем узле решетки. Анион, занимающий этот узел,,
становится нестабильным благодаря взаимодействию с кристал-
лическим нолем (ионизованный анион оказывается в окруже-
нии других положительных зарядов). Многократная ионизация
может происходить в результате каскада Оже, который возни-
кает при перестройке электронных оболочек после выбивания
электрона с одной из внутренних оболочек. Например, у неона
многократная ионизация произойдет у 16% атомов, потеряв-
ших электрон /(-оболочки. Нестабильный ионизованный анион
легко выбивается из узлового положения и превращается в атом
внедрения. Возникновение дефектов по механизму Варли, одна-
ко, наиболее вероятно только для простых анионов, таких, на-
пример, как галогены. Многоатомные анионы в результате
многократной ионизации, очевидно, должны претерпевать
распад.
Дюруп и Платцмап [13] разработали метод расчета абсо-
лютного выхода ионизаций внутренних оболочек. Полученные
результаты показывают, что значение G для внутренних иони-
заций быстро уменьшается, если энергия падающих электро-
нов не превосходит по крайней мере в сто раз энергию, необ-
ходимую для соответствующей ионизации. Для электронов с
энергией 1 Мэв значения GK для ионизации /(-оболочек Li, F,
К и С1 равны 0,155; 0,00681; 0,00027 и 0,000373 соответственно.
У фтора значение GL для выбивания электрона с L-оболочки
равно 0,3. У калия и хлора значение G для выбивания элект-
ронов с L- и М-оболочек заметно превосходит GK (в 20—
100 раз).
Хотя существование механизма Варли до сих пор не оспари-
валось, недавно был предложен другой механизм [14], опи-
сывающий те же явления. Согласно этому механизму происхо-
дит образование возбужденных молекулярных ионов (в щелоч-
но-галоидных кристаллах Х2~*), последующий распад которых
ведет к возникновению вакансий и ионов внедрения. Этот ме-
ханизм в настоящее время приобретает широкое признание.
10.3.6. Краткая сводка рассмотренных механизмов
радиационных повреждений твердого тела
Общая картина различных процессов, протекающих при об-
лучении в твердых телах, отражена на рис. 10.6 и в табл. 10.2.
Электроны, выбитые в процессе ионизации, независимо от вида
излучения будут мигрировать на определенное расстояние от
родительского иона (в зависимости от характера среды) и в
конечном счете будут захватываться положительными ионами
или ловушками, образуя центры окраски.
305
Таблица 10.2
Повреждения в твердых телах, вызываемые различными видами излучений
Излучение Энергия, эв Электронные дефекты Атомные смещения
УФ 10—103 Центры окраски Отсутствуют
Рентгеновское Юз-Юз » »
у-Кванты 105—108 » 11 ^значительное количество
Нейтроны 0,01—0,1 Отсутствуют Отсутствуют *
Нейтроны >10 кэв Умеренное коли юствэ Умеренное коли- чество
Заряженные час- тицы (тяжелые) 104—107 Возбужденные состоя- ния, центры окраски То же
Электроны >103 То же Н на 1! стельное количество
Продукты деле- ния ~108 Возбужденные состоя- ния О ;ень много
* Если не учитывать захвата нейтронов и появления атомов отдачи.
.Атомные
смещения
Прямое Отдача
выбивание
Ионизация
е ,
Захват ловушкой
Примесные
атомы
А
1 -Перенос заряда
А++М^/Г+А
. а) Разрыв связи с образа-
Образование от- ----------------.—
рицательного ионе. - ваниен радикала и иона
е~+А-А~ A*~Ri-M+
-Сольватация
-Нейтрализация
e~+A+-A+hv
A^-ReM*
б) Молекулы
и ион-радикала
А^-М+М1-
0) Горячего атома
и иона
А^М++А¥
Ион-молекулярная реак-
ция
А\АРАНЧА"
Возбуждение
Переходы между состояниями
а) безызлучательные
А*~А’+кТ
А*—А +кТ
б) с испусканием излучения
A*-A+hv
A'—A +hp
- Перенос энергии
а) экситонный перенос
б) А^М-М^А
-Нейтрализация
А++е~—А*
А*+е~—А+кТ
- Разрыв связи с образованием
а) радикалов
A*—R'+R"
б) молекул
A*—M+N
6) горячих атомов
А^М+А*
Дефекты,
центры
окраски
Реакции радикалов
R+A--RB+C
R+R-RR
Центры
окраски
Молекулярные продукты
Рис. 10.6. Сводная схема процессов, происходящих под действием радиации
в твердых телах. Ось времени на схеме направлена сверху вниз.
306
10.4. ХАРАКТЕРИСТИКИ ТВЕРДЫХ ТЕЛ, ЧУВСТВИТЕЛЬНЫЕ
К ПОВРЕЖДАЮЩЕМУ ДЕЙСТВИЮ РАДИАЦИИ
Для веществ, представляющих интерес с точки зрения ра-
диационной химии, наиболее важными характеристиками счи-
таются их физические, тепловые, электромагнитные и электри-
ческие свойства. Разумеется, при облучении твердых тел в оп-
ределенной степени меняются и их механические свойства, од-
нако эти изменения в неорганических и органических кристал-
лах имеют второстепенное значение. Существенным механиче-
ским эффектом в сложных неорганических кристаллах явля-
ется общее нарушение сил кристаллического поля, приводящее
к изменениям плотности образца. В случае системы, подверга-
ющейся заметным химическим изменениям под действием ра-
диации, существенным механическим эффектом может оказаться
измельчение образца. В некоторых простых солях (таких, напри-
мер, как LiF) может происходить заметное увеличение твердо-
сти. Одновременно может увеличиваться предел текучее™
(иногда вдвое).
10.4.1. Тепловые свойства
Высокие концентрации точечных дефектов будут приводить к
снижению теплопроводности. Уменьшение теплопроводности
связано с рассеянием фононов. Фопонами называют кванты, со-
ответствующие колебаниям атомов (или групп атомов), кото-
рые можно представить в виде совокупности волн, распростра-
няющихся по кристаллической решетке. Тепловая энергия рас-
пространяется по кристаллу в виде фононов. Если в кристалле
много дефектов, фононы будут рассеиваться и теплопровод-
ность снизится.
Энергия, запасенная в виде дефектов кристаллической ре-
шетки, также важная тепловая характеристика. Образование
одного атома внедрения требует затраты от 2 до 5 эв. Следо-
вательно, в виде дефектов в кристалле может запасаться за-
метная энергия. Важным конкретным примером такого процес-
са служит возникновение дефектов в графитовых замедлителях
ядерпых реакторов. Внезапное выделение этой энергии может
привести к серьезным последствиям *.
Количество энергии, запасенной в кристалле (за счет самых
разнообразных дефектов) и представляющей интерес с точки
зрения химика, можно определить, измеряя теплоту растворе-
ния. Современные методы позволяют обнаруживать малые ко-
личества запасенной энергии вплоть до 0,01 кал/г.
* Такое выделение энергии оказалось главной причиной крупной аварии
реактора в Уиндскейле (Англия), в результате которой реактор пришлось
остановить.
307
10.4.2. Электромагнитные свойства
В окрестности точечных дефектов происходит изменение
электрического потенциала, поэтому точечные дефекты ведут
себя как центры рассеяния электронов, увеличивая электриче-
ское сопротивление.
Часто изменение электрического потенциала, обусловленное
дефектами, приводит к возникновению локализованных элект-
ронных уровней и соответствующих им полос поглощения (об-
разование F-, F'- и V-цснтров и т. п.). Кристалл при этом при-
обретает окраску, в нем обнаруживаются фотопроводимость и
люминесцентные свойства.
Определение природы физических изменений, приводящих
к образованию центров окраски, для сложных молекул часто
оказывается очень трудной задачей и требует применения це-
лой комбинации таких методов, как ЭПР (или ЯМР), оптиче-
ская спектроскопия, идентификация с помощью акцепторов ра-
дикалов (или изотопной метки), тепловой отжиг, оптическое
обесцвечивание и т. д.
Исследование облученных метанола и этанола (в стеклооб-
разном состоянии) дает превосходный пример трудностей, с ко-
торыми приходится сталкиваться при выяснении природы цент-
ров окраски. Методом импульсного радиолиза в жидком эта-
ноле при 298° К удается выявить спектр сольватированного
электрона с широким максимумом при 7000 А и плечом при
5200 Л. Облучение этанола в стеклообразном состоянии (при
77° К) приводит к возникновению оптического спектра с дву-
мя полосами поглощения: с максимумом около 5200 А и мак-
симумом, расположенным в ультрафиолете пиже 3000 А. Если
полоса при 5200 А обесцвечивается при освещении белым све-
том или в результате тепловой обработки, то одновременно
слегка изменяется центральная область и увеличивается интен-
сивность боковых полос спектра ЭПР. Возникающий спектр
совпадает со спектром радикала СН2СНОН. Неспарепный
электрон, ассоциированный с центром окраски, является пред-
шественником этого свободного радикала и, по всей вероятно-
сти, представляет собой сольватированный электрон в стекло-
видной матрице.
10.4. Электрические свойства
Существует ряд электрических свойств твердых тел, измере-
ние которых полезно с точки зрения радиационной химии. Од-
нако большую часть информации удается извлечь, пользуясь
лишь двумя видами измерений — измерениями проводимости
(сопротивления) и фотопроводимости. С их помощью получают
достаточно надежные сведения о концентрации носителей заря-
да (которая определяется количеством электронов, оторванных
308
от родительских положительных ионов), о концентрации и рас-
пределении по энергиям различных электронных ловушек, их
глубине, а также о способности носителей заряда передавать
энергию окружающим молекулам.
В облученной системе число свободных электронов па 1 ел3
равно числу электронов, возникших в единицу времени в 1 см3,
умноженному на их среднее время жизни:
= —. (10.16)
ей
Здесь Gc — число электронов, возникших при поглощении
100 эв энергии; R— мощность поглощенной дозы, 100 эв!сек\
с — проводимость, (ом • си)-1; е — заряд электрона-,' ц— по-
движность электрона, см2/(в • сек); т — среднее время жизни
электрона.
Фотопроводимостью называют способность образца прово-
дить ток при освещении его светом (строго говоря, это опре-
деление должно быть распространено и на облучение образца
рентгеновским или у-излучепием).
Изучая зависимость фотопроводимости от температуры и
длины волны излучения, можно получить цепные сведения о
природе тех центров, которые поставляют электроны прово-
димости. Очень часто эти методы используют одновременно с
методом ЭПР. Например, в комплексных соединениях иода с
пиреном и псрилепом была обнаружена одинаковая зависимость
от температуры как проводимости, так и числа парамагнитных
частиц 115]. Измерение проводимости позволило выяснить влия-
ние примесей на захват в ловушки электронов и дырок в ант-
рацене [16].
10.4.4. Другие характеристики
Почти во всех случаях при облучении неорганических ве-
ществ, претерпевающих химическое разложение (включая и
щелочпо-галоидпые кристаллы), наблюдаются изменения плот-
ности образцов, часто сопровождающиеся изменением картин
рентгеновской дифракции и спектров инфракрасного поглоще-
ния. При рентгеноструктурном анализе облученных неорганиче-
ских кристаллов в общем случае наблюдается уширение линий
дифракции и, если кристалл близок к идеальному, отмечается
изменение интенсивностей отдельных рефлексов. В инфракрас-
ных спектрах облученных кристаллов обнаруживается ушире-
ние основных полос и появление многочисленных боковых мак-
симумов, что, очевидно, связано с общими искажениями взаим-
ных ориентаций плоскостей решетки.
Отмечаются также изменения поверхностных свойств, раст-
воримости, сдвиги точки плавления. Любой физический пара-
309
метр или свойство, зависящее от скорости внутренней диффу-
зии, будут меняться, так как процесс диффузии существенно за- '
висит от концентрации дефектов в объеме [17].
10.5. РОЛЬ КРИСТАЛЛОГРАФИЧЕСКИХ ПАРАМЕТРОВ РЕШЕТКИ
Многие данные указывают па то, что характер (распреде-
ление) продуктов и глубина радиационно-химического разложе-
ния неорганических и органических твердых веществ часто за-
висят от кристаллографических параметров решетки. Облучение
кристаллического хлористого холина при комнатной темпера-
туре приводит к его разложению с величиной G, достигающей
55,0. Уравнение реакции распада можно записать следующим
образом:
[(CH3)2NCH2CH2OH]+Cr ~ (CH3)2NH+C1~+СН3СНО. (10.17)
При температуре около 73—-78° С кристаллы хлористого хо-
лина претерпевают фазовый переход от орторомбической к гра-
нецептрированпой кубической решетке. Если вести облучение
при этих температурах, то происходит весьма заметное умень-
шение радиационно-химического выхода [19].
При облучении алкилгалоидов в стеклообразном состоянии
выход продуктов существенно отличается от наблюдаемого при
облучении в поликристаллической форме [20].
Радиационная полимеризация н-алкил-н-винилсульфонами-
дов в твердой фазе идет с гораздо большим выходом, чем в
жидкой. Еще более нагляден тот факт, что облучение при
—75° С индуцирует полимеризацию в кристаллическом, но не в
стеклообразном состоянии [21].
Бромат бария при облучении образует такие продукты, как
BrOj-, ВгО- и Вг- (возможно также большое количество
ВгО2). При 180° С отмечается резкое изменение радиационно-
химического выхода продуктов. Его непосредственно связывают
с фазовым переходом, который Ва(ВгО3)2 претерпевает при
этой температуре [22].
Можно привести множество других примеров, свидетельст-
вующих о том, что фазовые переходы оказывают глубокое воз-
действие на поведение вещества, подвергающегося облучению
ионизирующей радиацией [23]. Поэтому в радиационной химии
твердого тела чрезвычайно важно знать строение кристалла и
ориентацию молекул в решетке. В качестве примера можно от-
метить, что сведения о пространственной ориентации молекул
были использованы для объяснения образования димеров при
радиолизе твердого гексана [24].
10.6. ПЕРЕНОС ЭНЕРГИИ
В кристаллических телах в отсутствие примесей существует
сильное взаимодействие между электронными состояниями. По-
310
этому нельзя считать, что электронное возбуждение в кристалле
локализовано на отдельной молекуле *. Ситуация в кристалле
резко отличается от ситуации в газовой фазе, растворе или
стеклах. Электронные уровни в этих системах остаются суще-
ственно такими же, как и в изолированных молекулах. В стек-
лах, таких, как борная кислота, и в замороженных растворах,
например, ароматических углеводородов времена жизни воз-
бужденных состояний, энергия которых растрачивается па
флуоресценцию, ничем не отличается от соответствующих вре-
мен жизни в газовой фазе или в растворах.
Передача энергии осуществляется двумя основными спосо-
бами — электронным и тепловым. Перенос энергии электронно-
го возбуждения может принимать самые разнообразные формы,
зависящие от специфики системы Целый ряд механизмов пере-
носа энергии возбуждения рассмотрен в главе V (главным об-
разом те из механизмов, которые встречаются в органических
системах). Однако некоторые механизмы, такие, как миграция
экситона или передача энергии электронного возбуждения ко-
лебаниям решетки, характерны именно для твердого состояния.
Тепловые эффекты в твердых телах имеют иную природу,
так как элементы решетки обладают ограниченной подвижно-
стью.
10.6.1. Миграция экситонов
Перенос энергии посредством миграции экситонов может
происходить только в изолирующих или полупроводниковых
кристаллических телах. К таким телам относятся .молекулярные
и ионные кристаллы, керамика, большая часть полимеров, по-
лупроводники и пр. Экситонами называют возбужденные элект-
ронные состояния в кристаллах. В принципе экситон представ-
ляет собой пару электрон — дырка. Эта связанная пара —
электрон в полосе проводимости и дырка в валентной полосе —
как одно целое может двигаться через кристалл. Движение эк-
ситона через кристалл можно представить себе как ряд реком-
бинаций электронов и дырок с последующим поглощением осво-
божденной энергии. Возбужденная молекула окружена другими
молекулами, которые способны воспринимать и переизлучать
энергию возбуждения. Экситон мигрирует сквозь кристалл до
тех пор, пока не произойдет взаимодействие с фононами (рас-
сеяние на фононах) или атомами примесей, с дислокациями
или точечными дефектами, которое приведет к потере энергии
возбуждения. После этого экситон может быть локализован и
захвачен ловушкой. Энергия экситона растрачивается на флуо-
ресценцию или иногда на химические превращения молекул
ловушки. Роль экситонов в процессах, происходящих в пеорга-
* Это не относится к глубоко лежащим уровням, таким, например, как
уровни /-электронов в редкоземельных элементах.
311
нических (ионных) твердых телах, исключая щелочно-галоид-
пые кристаллы и некоторые окислы, пока не очень ясна. Од-
нако во многих органических системах экситоны играют важ-
ную роль.
Основное значение экситонного переноса энергии заключа-
ется в том, что энергия может передаваться далеко от места
первоначального поглощения. Процессы миграции экситонов в
кристаллах можно изучать, добавляя в кристалл примесные
атомы с хорошо известными характеристиками или увеличивая
концентрацию дефектов. Например, в системе, содержащей
1 долю тетрацена на 10 000 долей антрацена, экситон захваты-
вается молекулами тетрацена, так как электронный уровень
возбужденного состояния тетрацена ниже соответствующего
уровня антрацена. Энергия экситона растрачивается в виде
флуоресценции тетраценовой молекулы.
Энергия экситонов определяется природой кристаллов. В по-
лупроводниках основное состояние кристалла соответствует це-
ликом заполненной валентной зоне, отделенной энергетической
щелью от пустой зоны проводимости [25, 26]. Электроны в воз--
буждеппых состояниях перебрасываются через энергетическую
щель (запрещенную зону) и попадают в зону проводимости.
Энергию экситонов можно определить из спектров поглоще-
ния твердого тела. В ионных кристаллах переходы между зо-
нами обычно требуют энергии, на 1 эв большей энергии первой
полосы поглощения. При рекомбинации экситона может выде-
ляться значительное количество тепла. В кристаллах ДС1, на-
пример, энергия, выделяющаяся при рекомбинации, составляет
около 6 эв. Яркие примеры процессов миграции энергии мож-
но встретить при облучении таких веществ, как KNO3, который
подвергается радиационному разложению в матрице КВг, или
гетерогенных систем (см. раздел 10.16). Для KNO3, ДВгО2 или
KNtO2 (концентрация 0,03 мол. %) в матрице КВг было обна-
ружено, что глубина их радиолиза, рассчитанная с учетом
электронной доли этих веществ, ничтожна по сравнению с ре-
ально наблюдаемым выходом. Объяснить это можно только
тем, что энергия, поглощаемая матрицей КВг, мигрирует к рас-
сеянным в матрице солевым включениям [27].
Приблизительное представление о диапазоне миграции энер-
гии дает тот факт, что добавление в кристалл одной миллион-
ной доли примеси может привести к существенному гашению
люминесценции основного вещества кристалла.
10.7. РАДИАЦИОННЫЕ ЭФФЕКТЫ
В ТВЕРДЫХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Облучение органических систем сопровождается появлением
радикалов, новых стабильных молекул, электронов, захвачен-
ных в ловушки, дырок, различных возбужденных состояний,
312
атомов внедрения и т. д. Для изучения этих явлений обычно
используют методы ЭПР, ЯМР, оптической и инфракрасной
спектроскопии, рентгеновской дифракции, а также измерения
проводимости и фотопроводимости. Следует помнить, однако,
что возможность выявления первичных частиц в большей части
случаев будет зависеть от температуры, при которой ведется
облучение.
10.7.1. Выход радикальных продуктов
Одна из наиболее важных разновидностей промежуточных
продуктов, идентифицируемых при радиолизе твердых органи-
ческих систем, — свободные радикалы. Воеводский и Молин
[28] облучили ряд органических соединений и получили ли-
нейную зависимость концентрации радикалов (определенной
методом ЭПР) от дозы. Облучение велось при низких темпера-
турах. Выяснилось, что выход радикалов в соединениях, не со-
держащих кратных сопряженных связей, гораздо выше, чем в
ароматических соединениях. Например, G(R) в циклогексане
равно »4,2, в бензоле — 0,2, в дифениле — 0,045 и в анили-
не— 0,05 (табл. 10.3). Чем выше сопряжение в системе, тем
Таблица 10.3
Значение G для радикалов, возникающих при радиолизе в некоторых органи-
ческих веществах в твердом состоянии
Вещество Радикал 6 (R)
Малоновая кислота СН(СООН)., .
Циклогексан Сб! 1|1 4,2—4,6
Бензол с«н5 0,2
Циклопентаи С5НЯ 4,2
СН3ОН сн .он 12
СН3СН2ОН СН3СНОН 8,3
СН3СН2СН2ОН СН3СН2СНОН 8,4
RCONHCH.,CH3 RCONHCHCH3 —
СН..СООН СН2СООН -—
СН4 СН.;Н 4,2
С,Н51 с.,н5 —.
СС14 СС13 —
cf4 CF, .—-
сен5сн3 С6Н5СН2 —
С6НПОН а-С6Н10ОН —
ниже значение G(R). Для объяснения столь больших колеба-
ний выхода радикальных продуктов Воеводский и Молин вы-
двинули предположение, что выход радикалов зависит от по-
ложения низшего возбужденного состояния. При этом счита-
ется, что ароматическая молекула, возбужденная в высшие со-
313
стояния (близкие к границе ионизации), быстро за (КН3 сек)
совершает безызлучательный переход в низшее возбужденное
состояние. Эта точка зрения согласуется с тем фактом, что
спектр люминесценции ароматических молекул не зависит от
длины волны возбуждающего света. Воеводский и Молин на-
блюдали в своих экспериментах, что выход свободных радика-
лов существенно растет, когда (энергия первого возбужден-
ного синглетного состояния) становится примерно равной энер-
гии диссоциации связи.
Маги [29] показал, что при слабом взаимодействии между
линейными молекулами (расчет проводился для HJ ) первона-
чально возбужденная молекула должна диссоциировать. Но
при сильном взаимодействии электронное возбуждение распро-
страняется столь быстро, что.диссоциация становится невоз-
можной. Будущее покажет, можно ли распространять эти пред-
ставления па другие типы твердых тел. Однако следует отме-
тить, что значения G для разложения ионных кристаллов обыч-
но значительно ниже, чем для радиолиза органических твердых
веществ.
В органических твердых телах обнаруживается большое чис-
ло радикалов. Малые радикалы, такие, как атомарный водо-
род, нелегко обнаружить из-за их высокой подвижности даже
при низких температурах. Атомы водорода удается идентифи-
цировать, если метан облучают при 4е К. Однако малейшее на-
гревание ведет к их быстрому исчезновению. Существует при-
ближенное правило, согласно которому температура стабилиза-
ции радикала составляет 40—60% температуры плавления чи-
стого материала матрицы [30].
Неспаренные электроны обнаруживают при облучении прак-
тически всех органических веществ. К сожалению, разрешение
в спектрах ЭПР очень часто бывает плохим из-за сложной при-
роды взаимодействий.
Концентрация радикалов как в органических, так и неорга-
нических твердых телах обладает способностью к насыщению.
Иными словами, после того как достигнута некоторая концент-
рация радикалов, дальнейшее облучение может привести лишь
к незначительному ее росту. Концентрация насыщения зависит
от температуры. Однако обычно эта концентрация достигается
в том случае, если скорость исчезновения радикалов становится
примерно равной скорости их образования. Насыщение дости-
гается при концентрациях радикалов всего лишь в несколько
десятых процента. Отчасти эффект насыщения можно также
объяснить нарушениями решетки, возникающими при образова-
нии радикалов. Нарушения решетки способствуют диффузии
радикалов и облегчают радикальные реакции. Теоретические
подходы к этому вопросу рассматриваются в работе Нок-
са [25].
314
10.7.2. Стеклообразные тела
Исследование радиационных эффектов в органических стек--
лах имеет особую ценность, так как позволяет выявить для
разных соединений преобладание тех или иных типов реакций.
Полученная информация в общем случае больше относится к
процессам, протекающим в жидкостях, чем в кристаллах. На-
пример, ароматические углеводороды в метаполовых стеклах
при облучении образуют анион-радикалы [31], которые могут
затем взаимодействовать .со спиртовой матрицей путем перено-
са протона от спирта па анион-радикал:
Аг~ 4- ROH -> АгН 4- RO”.
В метилтетрагидрофурановых стеклах ароматические соеди-
нения легко образуют отрицательные ионы Аг-. Указанная ре-
акция образования ароматического аниона наблюдалась также
для жидкого состояния в разбавленных растворах нафталина
в нескольких видах спиртов [32]. Однако в бутилхлоридпых
стеклах ароматические углеводороды оказываются эффективны-
ми ловушками для положительных дырок, и поэтому наблюда-
ется образование лишь ароматических катионов.
10.8 РАДИАЦИОННЫЕ ЭФФЕКТЫ
В ТВЕРДЫХ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
За исключением щелочпо-галоидиых кристаллов радиацион-
ные эффекты в твердых неорганических соединениях широко не
исследовались. В большей части случаев эксперименты проводи-
ли лишь при комнатной температуре, определяя характер и от-
носительный выход продуктов. Было сделано несколько попы-
ток сравнить наблюдаемый выход продуктов в гомологическом
ряду неорганических солей и провести сопоставление с такими
характеристиками, как свободный объем, свободная энергия об-
разования продуктов, температура термодеструкции, плотность,
поляризуемость катионов, точка плавления, энергия решетки.
Все эти попытки, однако, оказались неудачными.
В настоящее время принято считать, что факторы, обуслов-
ливающие относительные значения G в ряду гомологичных со-
единений, таких, как нитраты, броматы или перхлораты, чрез-
вычайно сложны и, вероятно, определяются структурой кри-
сталла, величиной электронно-фононного взаимодействия, плот-
ностью кристалла, средней энергией колебательных состояний,
эффектом «клетки» и т. д. Эти параметры слишком мало изу-
чены, чтобы можно было проводить какие-то оценки их относи-
тельной роли в процессах радиолиза неорганических твердых тел.
При изучении радиолиза твердых тел, претерпевающих хи-
мическое разложение, всегда следует помнить, что одновремен-
но с химическими превращениями происходит интенсивное раз-
рушение кристаллической решетки, в результате чего изменя-
315
ются условия для диффузионных процессов и процессов пере-
дачи энергии. Эта сторона радиолиза особенно очевидна для
тех соединений, где возникают газообразные продукты, такие,
как кислород или азот.
В некоторых случаях значения G могут меняться из-за пор-
чи исходных солей следовыми количествами других элементов.
Метод следовых добавок оказался весьма полезным при изу-
чении захвата экситонов ловушками и в целом ряде фотохими-
ческих экспериментов в органических кристаллах.
В исследованиях радиолиза неорганических кристаллов ме-
тод следовых добавок достаточно широко не использовался, хо-
тя вполне заслуживает этого. Так, например, было найдено, что
при добавлении небольших количеств серебра к кристаллам
KNO3 меняется радиационно-химический выход нитритов.
При изучении радиолиза неорганических соединений облу-
ченную соль обычно растворяют в воде и затем проводят хими-
ческий анализ. Недостаток этого метода заключается в том, что
анализируемый продукт может возникнуть в результате реак-
ции растворителя с облученной солью и не иметь никакого от-
ношения к истинным продуктам радиолиза. Однако изучение
рентгеновских, инфракрасных и ультрафиолетовых спектров
твердого тела, а также теплоты растворения позволяет в боль-
шей части случаев рассеять сомнения относительно природы
продуктов радиолиза, проводившегося при комнатной темпера-
туре. Если же радиолиз проводится при пониженной темпера-
туре, иногда могут возникнуть 'некоторые трудности. Имеются
указания, что при облучении KNO3 при —196° С с последующим
растворением в цериевосульфатном растворе образовавшиеся
продукты отличаются от продуктов, полученных в аналогичном
эксперименте при комнатной температуре [33].
Для тех немногих солей, радиолиз которых исследован, об-
наруживается непосредственная зависимость между выходом и
температурой облучения, что указывает на важную роль тепло-
вой активации в некоторых реакциях.
Так как увеличение температуры, приводящее к расшире-
нию кристаллической решетки, облегчает радиолитическое
разложение, то, очевидно, повышение давления должно вести к
противоположному результату. Действительно, для некоторых
солей это предположение оказалось верным. Однако для дру-
гих солей (в изученном диапазоне давлений) пе было обнару-
жено никакого эффекта. Далее в соответствующих параграфах
рассматривается влияние температуры и давления на радиолиз
некоторых солей.
10.8.1. Эффект клетки
В твердом теле из-за ограничений, связанных с существо-
ванием решетки, продукты распада молекул не могут столь лег-
ко покинуть место возникновения, как это происходит, напри-
316
мер, в жидкости. Продукты распада оказываются «внутри клет-
ки», и «клетка» благоприятствует рекомбинационным процес-
сам. Продукты небольшого размера, такие как атомарный во-
дород, имеют больше шансов выйти из клетки по сравнению с
крупными атомами кислорода или азота. Не меняя распределе-
ния продуктов, эти процессы, очевидно, будут менять значе-
ние G.
Эффект клетки использовался для объяснения влияния изо-
топного состава на радиолиз нитратов [34], а также для объ-
яснения высоких значений температурных коэффициентов при
радиолизе ионных солей.
10.8.2. Влияние ЛПЭ
Влияние ЛПЭ на радиолиз в твердых телах изучалось срав-
нительно мало. При облучении нитратов тяжелыми заряжен-
ными частицами значение G увеличивается и коррелирует при-
мерно с ходом температурной зависимости радиолиза [35]. Де-
ло в том, что в твердых телах ЛПЭ оказывает влияние на ра-
диолиз не за счет перекрывания шпор, как в жидкостях, а ско-
рее благодаря общему нагреванию и значительным локальным
нарушениям структуры. После воздействия па твердую соль
KNO3 у-излучения при 60°С G=l,46, а при 190° С G=2,84. Об-
лучение KNO3 а-частицами при 30° С характеризуется значе-
нием G=2,2 (табл. 10.4). Разложение KNO3 под действием
продуктов деления [36] происходит с выходом, примерно рав-
ным-6. ЛПЭ для продуктов деления в KNO3 составляет около
600эв/Л, тогда как-для а-частиц ЛПЭ=34 эв/Л и для у-излу-
чения кобальта 0,06 эв/Л.
Таблица 10.4
Начальные значения G для радиационного разложения твердых нитратов
Соль V-Излучение 6OCo а-Излуче- ние
- I 10" 25' 60'' 190" 330° 30’
LiNO3 0,02 3,73 0,7
NaNO3 0,341 0,15 0,308 1,04 5,1 1,3
K.NO3 1,472 1,38 1,46 2,84 5,8( жидкость 2,2
RbNO3 0,79 0,79 0,78 2,54 4,5 —
CsNO3 1,364 1,65 1,66 3,13 7,O' 1,4
Ba (NO3)2 1,462 1,90 1,78 1,81 0,8 твердое тело 1,6
Sr(NO3)2 — 0,5 — —• -—• —
AgNO3 — 0,18 — — — —
Pb(NO3)2 •— - 0,45 — —• — —
Ввиду ограниченной информации невозможно сделать какие-
либо общие предсказания относительно влияния ЛПЭ па ра-
317
диациопные эффекты в неорганических или органических кри-
сталлах. Здесь ситуация в корне отличается от существующей
в радиационной химии жидкостей (особенно воды). Однако не-
которые экспериментальные факты известны.
В броматах, хлоратах и нитратах под действием смешанного
реакторного излучения возникают продукты с тем же распре-
делением, что и под действием у-излучения. Это же наблюда-
ется при радиолизе органических соединений в тех случаях,
когда ядерные превращения происходят в незначительном мас-
штабе. Под действием смешанного реакторного излучения мо-
жет усиливаться разложение азидов и нитратов в результате
реакции ,4N(n, р)14С. Предполагается, что этот эффект обуслов-
лен: 1) конкуренцией реакций первого и второго порядков ти-
па Х+М->продукты и X + Х->продукты, где X — радикальные
или возбужденные частицы, а М — стабильные молекулы;
2) «перезарядкой ионов», заключающейся в обмене электрона-
ми между средой и медленно движущимся попом [37]. Эти
представления расходятся со взглядами Хеннига, Лиса и Мэтью-
сона [38], которые сумели объяснить увеличение разложения
NaNO.3 под действием реакторного излучения без использова-
ния эффектов, требующих высокой ЛПЭ.
10,9. НИТРАТЫ
При радиационном разложении щелочных и щелочноземель-
ных нитратов в качестве единственных продуктов разложения
образуются нитритные ионы и кислород. Однако для солей не-
которых переходных металлов это неверно. Нитраты серебра
и свинца, по-видимому, разлагаются с образованием некоторого
количества окислов. Исследования, проведенные методом ЭПР
при низких температурах, показали, что в этих солях образуются
разнообразные первичные частицы. Однако выяснить, какую
- роль играют эти частицы в общем кинетическом механизме, не
удалось. При комнатной температуре радиолиз целого ряда
нитратов соответствует следующей кинетической схеме [39]:
NO?-------> NO7 4- О, /ггФ; (10.18)
О NO2 —> NO3 , /?2;
(10.19)
О -] ЙОГ NOT 4- О2, k3. (10.20)
Здесь Ф — интенсивность облучения. Скорость образования
иона нитрита дается выражением
- [N°21 = № [NO3-] - k2 [NCV] [О] 4- Ъ [NOT] [О]. (10.21)
318
Пользуясь обычными условиями стационарности, получим
d №1 _ 2МзФ [W12 10 22>
dt fe2[NO~]+MN°r] '
При малых степенях превращения концентрация [NO/ ]
остается существенно постоянной, и, выполняя интегрирование,,
найдем
— —1— [NO/]2 -i- [NOrl - 2/гхФ [NOd t (10.23)
2 fc3 [NO/]
или сокращенно
<zX2 X ЬТ,
(10.24>
,, число молекул NO2
где Л =
доза, эв
г-10-21
aJO-i9
Константы а и b можно найти из экспериментальной зави-
симости выхода от дозы для нитритного иона. Кинетический
закон, согласно которому выход нитрита нелинейно зависит от
дозы, выполняется при радиолизе NaNO3, KNO3, CsNO3,
Ba(NO3) и Pb(NO3)2, но не выполняется при радиолизе AgNO3.
При разложении этой соли помимо NO/ образуется также и
окисел серебра. Радиационное разложение Pb(NO3)2 также
представляет собой сложный процесс, и хотя выход NO/ сле-
дует кинетической зависимости уравнения (10.24), истинный:
механизм разложения гораздо сложнее, чем это показывают
уравнения (10.18) — (10.20). В диапазоне мощностей дозы, раз-
личающихся на пять порядков, не отмечалось никаких измене-
ний выхода продуктов радиолиза твердых нитратов. Но этого
и следует ожидать, так как из-за ограниченной диффузионной
подвижности в твердых телах бимолекулярные реакции свобод-
ных радикалов друг с другом не могут играть существенной
роли.
Значения G для радиационного разложения различных нит-
ратов приводятся в табл. 10.4.
10.9.1. Центры окраски в нитратах
Припсгейм [40] обнаружил в облученном NaNO3 сильную-
полосу поглощения при 3500 А, приписываемую переходу в ион
NO/, электронные уровни которого возмущаются близким со-
седством другого электрона. Возможно, наблюдаемой частицей
является ион NO2-. Позднее в KNO3 было идентифицировано
еще несколько оптических полос поглощения, которые соответст-
вуют определенным ЭПР-спектрам. Возникновение этих полос
приписывалось частицам (NO^), NO3, NO2 и NO|~, однако
окончательных подтверждений получено не было [41]. Полоса,
приписываемая (NO|~), имеет максимумы при 2900 и 5100А.
10.9.2. Спектры ЭПР
Относительно спектров ЭПР облученных нитратов в литера-
туре существуют некоторые противоречия [42—44]. Спектры ЭПР
указывают па существование NO3, NO2, NO^_ и О2. При радио-
лизе Pb(NO3)2 и Sr(NO3)2 при комнатной температуре наблю-
дается характерный спектр NO2. Но тот же спектр в нитратах
щелочных металлов наблюдается лишь в том случае, если облу-
чение ведется при низкой температуре. Существуют определен-
ные доказательства того, -что ион NO~ является предшественни-
ком радикала NO2 [44]. В качестве продукта разложения нитра-
тов постулировалось -также образование NO, однако прямых
химических доказательств появления этого окисла не имеется.
NOg- с высокой эффективностью образуется в результате облу-
чения NaNO3 рентгеновскими лучами при 77° К [45].
10.9.3. Влияние давления
Наблюдалось влияние давления на радиолиз ряда нитратов
[46—47]. Степень проявления этого влияния зависит от природы
соли, температуры и поглощенной дозы (т. е. от степени разло-
жения). Относительно влияния давления на радиолиз KNO3 в
литературе существуют определенные расхождения. Данные
Каннингхэма [46] показывают, что влияние давления в этих си-
стемах проявляется весьма сложным образом.
10.9.4. Влияние температуры
Влияние температуры на радиационно-химический выход нит-
ратов суммировано в табл. 10.4. Как можно видеть, с ростом
температуры происходит общее увеличение G. Единственное ис-
ключение— соль Ba(NO3)2, для которой отмечается резкое сни-
жение выхода. Возможно, что снижение выхода связано с фа-
зовым переходом. Как и следовало ожидать, отмечается боль-
шой рост значений G при облучении солей в жидком состоянии.
10.10. ПЕРХЛОРАТЫ
Перхлораты щелочных и щелочноземельных металлов разла-
гаются при комнатной температуре, образуя CIO^, CIO^, С’-,
С1О2 и О2, а также, возможно, следовые количества окислов ме-
таллов и С1О3 [48—51]. Имеются данные, согласно которым
320
СЮ- является вторичным продуктом, возникающим в резуль-
тате ряда сложных взаимодействий первичных продуктов.
Вее перхлораты окрашиваются после облучения, и окраска,
по-видимому, связана с накоплением С1О2. Основные продукты
радиолиза СЮ3 и С1_ удалось идентифицировать в сухих по-
рошках с помощью инфракрасных и рентгеновских спектров.
СЮ2~, СЮ- и СЮ2 были идентифицированы с помощью обычных
методов химического анализа в растворе. Значения G для ра-
диолиза перхлоратов приведены в табл. 10.5.
Т а б л и ц а 10.5
Значения G для продуктов радиолиза перхлоратов щелочных и щелоч-
ноземельных металлов
Соль C1O4 CIO,- C1O2 C1O2 CIO Cl- O3
LiC104 3,76 2,80 0,15 0,59 0,10 0,12 2,15
NaC104 4,36 3,57 0,17 0,11 0,09 0,42 2,96
ксю4 3,83 2,99 0,18 0,12 0,09 0,45 2,68
RbC104 5,27 4,06 0,20 0,12 0,14 0,75 3,84
CsC104 6,84 5,28 0,22 0,10 0,17 1,07 5,28
Mgcio4 4,67 4,29 0,14 0,07 0,03 0,15 2,62
СаС1О„ 4,15 3,44 0,00 0,51 0,08 0,12 1,99
SrC104 4,53 3,90 0,19 0,14 0,11 0,19 2,61
BaClO4 3,20 1,76 0,84 0,42 0,12 0,06 2,18
10.10.1. Влияние температуры
Основной результат повышения температуры при радиолизе
перхлоратов — распад С1О~ и СЮ- с образованием С1_. Облу-
чение при повышенных температурах (25—300° С) также ведет
к заметному понижению концентрации С1О2, однако сопутствую-
щего увеличения концентрации С1~ нс наблюдается. Предпола-
гают, что с повышением температуры С1О2 вступает в реакцию
Таблица 10.6
Значения <? для продуктов радиолиза КС1О4 при различных температу-
рах облучения
Температу- ра, °c CIO, C1O3 C1O2 C1O2 CIO- Cl- O2
—196 4,0 2,7 0,58 0,18 0,00 0,5 3,0
—80 4,0 2,8 0,49 0,17 0,00 0,6 2,7
—16 3,7 2,8 0,26 0,06 0,06 0,5 2,4
—8 3,5 2,6 0,26 0,09 0,06 0,5 2,6
0 3,4 2,6 0,20 0,07 0,06 0,5 2,2
72 4,1 3,3 0,19 0,09 0,00 0,5 2,7
295 3,8 2,9 0,00 0,00 0,00 0,7 1,0
11 Зак. 907
321
с окислом металла, возникающим при радиолизе; в результате
образуется С1О4 [49].
В различных кристаллических решетках С1О2', СЮ ц СЮ2
проявляют разную степень тсрмостабильности. Например, иоп
СЮ-, как правило, более стабилен в решетках щелочноземель-
ных металлов по сравнению с решетками щелочных металлов.
Изменение G для разных продуктов радиолиза КСЮ4 при раз-
ных температурах облучения показано в табл. 10.6.
10.10.2. Спектры ЭПР
Спектры ЭПР облученных перхлоратов чрезвычайно слож-
ны; в них не удается добиться хорошего разрешения. Анализ
спектров ЭПР свидетельствует в пользу существования в этих
кристаллах радикалов СЮ2 и СЮ3, что подтверждается также
химическими методами.
10.10.3. Кинетика
Кинетика распада перхлоратов очень сложна, и ее не удается
аппроксимировать с помощью реакции первого порядка. Основ-
ные продукты радиолиза — С10^ и С1-. Их выход при разных
температурах существенно не меняется. Следует отмстить, что
выход СЮ^" не обнаруживает линейной зависимости от дозы.
Эти факты, а также результаты исследований терморазложения
облученных солей свидетельствуют в пользу следующего меха-
низма:
СЮ?------ СЮ? -4- О;
О + С1ОГ -> СЮГ + О2;
О 4- С1ОГ -> СЮГ;
СЮ? '•--> Cl” + 2Ог;
СЮ? —> (СЮГ)’.
(10.25)
(10.26)
(10.27)
(10.28)
(10.29)
Здесь (СЮ-) * — возбужденное состояние СЮ^. Может сущест-
вовать также (С1О|~) *. Разложение (С1О^) * происходит сле-
дующим образом:
(СЮ?)’ _> сюг + о2, (10.30)
или -> СЮ2 -ь 02, (10.31)
или -> сю2 о + о-, (10.32)
(СЮ4 ) -j- тепло -> СЮ -J- О2 -{- О. (10.33)
322
10.10.4. Влияние давления
При радиолизе перхлоратов щелочных и щелочноземельных
металлов не удалось обнаружить влияния давления на значение
G (вплоть до давлений аргона ~ 140 атм [52]). Единственный
регистрируемый эффект — уменьшение массы стронциевых, ру-
бидиевых и цезиевых солей (благодаря выходу молекул О2 из
кристаллической решетки).
10.11. БРОМАТЫ
Под действием излучения броматы щелочных и щелочнозе-
мельных металлов разлагаются, образуя Вг-, О2, ВгО^Г, ВгО- и,
возможно, также по аналогии с перхлоратами некоторое коли-
чество ВгО2 [53—56]. Для облучения использовали 60Со, реак-
торное излучение, источники ультрафиолетового излучения [57].
Облучение соли LiBrO3 проводили а-частицами и ядрами три-
тия, образующимися при делении ядер 6Li.
При высоких дозах концентрация окислительных продуктов
достигает стационарного уровня, который зависит от темпера-
туры образца при облучении. Стационарное состояние-—резуль-
тат равновесия между реакцией образования окислительных
продуктов и обратной реакцией образования броматного иона.
При десятикратном изменении интенсивности у-излучения эф-
фектов, связанных с интенсивностью излучения, не наблюдалось.
Все облученные соли приобретают желто-коричневую окрас-
ку и дают слабощелочную реакцию при растворении в Н2О. Ще-
лочные свойства растворов обусловлены, очевидно, гидролизом
иона ВгО-. При отжиге кристаллов ниже точки плавления
окраска и щелочная реакция растворов исчезают. В облученных
сухих порошках с помощью рентгепоструктурного анализа и
анализа инфракрасных спектров удалось, по-видимому, иденти-
фицировать ионы Вг-, ВгО- и ВгО-. Начальные значения G для
радиолиза различных броматов представлены в табл. 10.7.
Таблица 10.7
Начальные значения G продуктов радиолиза кристаллических броматов щелочных
и щелочноземельных металлов при 35° С
I.iBrOs NaBrO3 KBrO3 о U. ё CsBrOj Ca(BrOs)2 Sr(BrO8)j
0 (—ВгО3—) 0,31 1,4 1,3 2,3 5,6 2,4 2,9
G (Вг-) 0,10 (0,59) 0,63 (0,96) 0,48 0,33 0,51
G (окисл. *) 0,21 0,83 0,69 1,4 5,2 2,0 2,4
<? (О.,) — — — — 2,4 1,5 2,0
* G (окисл.)—выход окислительных продуктов.
11* 323
10.11.1. Влияние температуры
Зависимость радиационно-химического выхода от температу-
ры исследовалась для соли CsBrO3. При повышении температу-
ры от —86 до +300°С выход окислительных продуктов плавно
снижался, пока не упал до нуля при 300° С.
10.11.2. Спектры ЭПР
В кристаллической соли КВгО3, облученной при 77° К, уда-
лось обнаружить несколько парамагнитных центров, включаю-
щих бром [58]. Спектры были получены с низким разрешением.
10.11.3. Кинетика и механизм радиолиза броматов
Бойд с соавторами [53—56] | приводят целый ряд доказа-
тельств в рованного пользу следующего распада броматов: механизма радиационно'-иидуци-
ВгОГ > ВгОз -| (10.34)
ВгОГ — ВгОГ*; (10.35)
ВгОГ* -> ВгОГ -г О; (10.36)
ВгО?‘ -> ВгО- 1 О + О; (10.37)
вго; -> ВгО2 4- О; (10.38)
ВгО2 4- е~ -> ВгОГ; (10.39)
ВгО2 -|- е~~ -► ВгОГ’; (10.40)
ВгОГ’ -> ВгО- 4- О; (10.41)
ВгОГ + О -> ВгОГ. (10.42)
Реакция ВгОз -4- О —» ВгО2 4- О2 (10.43)
исключена из рассматриваемого механизма, так как известно,
что концентрация ВгОг достигает стационарных значений и не
меняется при увеличении интенсивности излучения. Этого не на-
блюдалось бы, если бы шла реакция (10.43).
Образование кислорода происходит по реакции
ВгОз + О -> Вг + 2О2.
(10.44)
324
10.12. СУЛЬФАТЫ
Ионы SQ2~, но-видимому, весьма устойчивы относительно
радиационного разложения. Облучение твердых гидратов
FeSO4 и NiSO4 при комнатной температуре приводит к окисле-
нию катиона и восстановлению иона SOJ— до SO'j'. Гидратная
вода с низким радиационно-химическим выходом разлагается
на молекулы Н2 и ионы ОН~. Однако в гидратах CoSO4,
Cr2(SO4)3, CuSO4 и ZnSO4 не наблюдается ни окисления катио-
нов, ни восстановления аниона SC§~. Отмечается лишь незна-
чительное разложение гидратной воды. В гидрате MnSO4 наб-
людается окисление иона марганца и восстановления Н2О, но
нс наблюдается восстановления сульфатной группы.
Сульфаты щелочных и щелочноземельных металлов очень
устойчивы к воздействию ионизирующей радиации. Облучение
Na2SO4 в дозе, превышающей 1022 эв/г, не приводит к сколько-
нибудь заметному разложению соли. Разложение соли не наб-
людается после дозы 3-Ю21 эв/г, даже если вести облучение при
300° С. Стабильность сульфатной группы в гидрате CoSO4 мож-
но, по-видимому, объяснить соотношением окислительных потен-
циалов иона кобальта и воды. Однако объяснить таким обра-
зом устойчивость сульфатного иона в солях никеля и железа
не удается.
Предположительно механизм радиолиза гидратов выглядит
следующим образом:
(MSO4) (Н2О) ~ -> Н2О+ + е—, (10.45)
е + Н2О -> Н2О-; (10.46)
Н2О+ 4- М"+ -> М<"+,)+ 4- Н2О; (10.47)
нго- + н2о+ -> 2Н2О*; (10.48)
Н2О~ + SO4- -> SOf~ + ОН- 4- ОН; (10.49)
2Н2О~ -> Н, 4- 2ОН~; (10.50)
2Н2Оь -> О2 4- 2Н+ 4- Н2. (10.51)
Здесь М"+— катион. Реакции (10.47) и (10.48) можно рассмат-
ривать как конкурирующие. В условиях, благоприятствующих
реакции (10.47), начинает также идти реакция (10.50) или, как
наблюдалось в нескольких случаях, одновременно идут реакции
(10.49) и (10.50). В отсутствие реакций катионов и анионов не-
значительное радиолитическое разложение может происходить
в результате реакций (10.50) и (10.51) [59].
325
10.13. ХЛОРАТЫ
Для нескольких хлоратов исследовалось радиационно-инду-
цнровапное разложение под действием рентгеновского излуче-
ния [50], у-излучения 60Со [58, 60] и смешанного' реакторного
излучения [61, 62]. Среди продуктов радиолиза удалось иденти-
фицировать С1“, СЮ-, СЮ-, О2, СЮ2, С12О6 и СЮ^. С12О6 уда-
лось обнаружить по слабой полосе поглощения при 4500 А в
кристаллах, облученных при комнатной температуре. После рас-
творения облученных кристаллов в разбавленном растворе
Na2CO3 удалось идентифицировать радикалы С1О2 по полосе
поглощения при 3300—4000 А. Хлоридные, хлоритные и гипохло-
ритные ионы идентифицированы обычными химическими мето-
дами в растворах. Известные значения G приведены в табл. 10.8.
Таблица 10.8
Значения G продуктов радиолиза хлоратов некоторых щелочных металлов
Вещество С!— С1О— СЮ,- о2 сю2 С12О6 С104— Литература
ксю3 1,9 0,7 2,6 2,9 0,2 1,4 |50]
КС1О3 — -— 1,2 — •—. — — 158]
КС1О3 — — — 1,57 —. — — |62]
NaClOs 0,23 0,21 0,21 1,95 0,95 — 0,22 160]
10.13.1. Влияние температуры
Облучение КС1О3 при низкой температуре (77° К) приводит
к заметному снижению радиационно-химического выхода О2,
СЮ-, СЮ^ и С1-. Отжиг облученных хлоратов (при 100—210° С)
увеличивает выход С1-, который, по-видимому, образуется в ре-
зультате терморазложения СЮ- и СЮу.
10.13.2. Механизм радиолиза хлоратов
Слишком скудные экспериментальные данные не позволяют
остановиться на каком-либо определенном кинетическом меха-
низме радиолиза хлоратов. В диапазоне больших поглощенных
доз скорость образования хлоритпых и гипохлорнтпых ионов
уменьшается, что указывает па возможность обратных реакций
и других сложных превращений. Хил [50] сообщает о влиянии-
интенсивности облучения на радиолиз КСЮ3. При уменьшении
интенсивности облучения в 3 раза Хил наблюдал заметное по-
нижение выхода основных продуктов. Этот результат заметно
расходится с экспериментальными данными, полученными при
радиолизе других солей.
326
10.14. ЩЕЛОЧНО-ГАЛОИДНЫЕ СОЛИ
К числу простейших неорганических систем относятся щелоч-
но-галоидные соли. Единственными химическими продуктами,
которые могут возникать в них при облучении, являются щелоч-
ные металлы и галогены. Образование центров окраски в щелоч-
но-галоидных кристаллах рассматривалось в разделе 10.2.4. Бы-
ло показано, что центры окраски возникают в результате
захвата электрона или дырки дефектами кристаллической ре-
шетки. Предполагается, что при облучении образуются такие
молекулярные частицы, как Na2, Вг2 или С12 и ионы С1^~, С1^~,
и ВгГ-
Существование равного числа центров с избытком и с недо-
статком электронов в облученном NaCl было продемонстриро-
вано химическими методами [63]. Облученную соль растворяли
в воде и определяли количество выделившегося Н2 и концентра-
цию ионов гипохлорита. Происходящие при этом реакции можно
представить следующим образом:
Na 4- Н2О -> NaOH + у Н2 (10.52)
или
е~ + Н2О -> ОН- 4- -уН2; (10.53)
С1 -|- Н2О СЮ- + Н+-£- На’ (10.54)
Хотя предельная концентрация центров окраски, измеренная
для образцов солей, облученных при комнатной температуре,
составляла 1017 на 1 см3, облучение при комнатной температуре
соли'КС! приводит к образованию такого количества С1О-, кото-
рое эквивалентно разложению 8% соли [48].
Наиболее важные результаты, полученные при изучений
радиолиза щелочно-галоидных кристаллов, касаются идентифи-
кации центров окраски, способов их образования и других свя-
занных с этим проблем. Хороший обзор имеющейся литературы
можно найти в целом ряде работ [64—66].
10.15. АЗИДЫ
Облучение азида натрия рентгеновскими лучами [67] приво-
дит к тому, что, после растворения облученной соли в воде в
растворе можно обнаружить NH3, ОН- и N2 в стехиометриче-
ском соотношении 1:3:4. Возможно, что NH3 и ОН- образуются
в результате гидролиза нитридного иона:
3N?------> №~ 4- 4N2;
№“ 4- ЗН2О -> NH3 4- ЗОН”.
(10.55)
(10.56)
327
Среди продуктов, появившихся после растворения соли, на-
ходившейся под действием реакторного излучения, можно обна-
ружить также и водород'[68]. По-видимому, водород образуется
в результате реакции атомов натрия с водой. Растворение облу-
ченной соли в жидком аммиаке ведет к появлению такой же
окраски, какая обнаруживается при растворении металличе-
ского натрия в жидком аммиаке. Кроме того, в водных раство-
рах облученной соли удается обнаружить следовые количества
гидразина.
• Фотолиз NaN3 под действием ультрафиолетового излучения
с длиной волны 2537 А приводит к выделению азота. Это позво-
лило предположить, что Е-цснтры и положительные подвижные
дырки образуются в равных количествах благодаря взаимодей-
ствию . экситонов с анионными вакансиями. Взаимодействие
V-центров с экситонами должно приводить к образованию
F-центров и двойной вакансии [69]. Освещение NaN3 при 573° К
сопровождается выделением азота и образованием коллоидного
натрия.
Несмотря па то что световое воздействие приводит к появ-
лению сильных F-, V- и /?-полос в кристаллах NaNg, облучение
рентгеновским или у-излучением приводит лишь к появлению
слабых F-полос. Таким образом, между фотохимическим разло-
жением NaN3 и разложением под действием ионизирующей
радиации существует заметное различие.
10.16. ГЕТЕРОГЕННЫЕ СИСТЕМЫ
Если вещество, подверженное радиолизу, адсорбировать на
поверхности! инертной матрицы, такой, как силикагель, окись
магния или другое твердое тело, и провести облучение системы,
то, как показали Аллен [70] и Хенц [71], часть энергии, погло-
щенной твердым телом, будет передаваться к поверхности и вы-
зывать разложение адсорбата. Подобный вывод основывается
на том, что степень разложения адсорбата непропорционально
велика и не соответствует количеству энергии, поглощенной
чистым адсорбатом, которое можно рассчитать, зпая электрон-
ную долю адсорбата. Таким образом, заметная часть энергии,
поглощенной инертной матрицей, передается к поверхности и
вызывает радиолитическое разложение адсорбированных моле-
кул.
Если силикагель или микропористый SiO2—Л120з облучать в
отсутствие адсорбата, то возникает голубое окрашивание. Но
стоит покрыть облученный порошок адсорбатом и эта окраска
исчезнет. Исчезновение окраски сопровождается химическим
разложением адсорбата.
В подобных гетерогенных системах эффективность передачи
энергии на поверхность зависит от поглощенной дозы. Поэтому
зависимость дозы степени радиационному разложению оказы-
328
вается нелинейной. Это можно объяснить тем, что дефекты,
возникающие в твердом теле при облучении, влияют на процесс
переноса энергии. В предварительно облученном твердом теле
перенос энергии осуществляется с меньшей эффективностью, чем
в нсоблученном. Доводом в пользу этого служит тот факт, что
при использовании MgO в качестве матрицы перенос энергии
происходит с гораздо большей эффективностью в хороших кри-
сталлических образцах по сравнению с образцами плохой кри-
сталличности.
Важным аспектом рассматриваемого процесса является соот-
ветствие между энергетическими уровнями поверхности твердого
тела и энергетическими уровнями молекул адсорбата. Доля по-
верхности, которую занимает адсорбат, по крайней мере в неко-
торых случаях, также оказывает влияние на распределение
продуктов радиолиза.
Суммируя имеющуюся информацию о процессах в гетеро-
генных системах, можно сделать следующие предварительные
выводы:
1. Наблюдается перенос энергии сквозь кристаллическую ре-
шетку к поверхности.
2. Эффективность процесса переноса энергии зависит от ха-
рактера вещества, числа имеющихся дефектов, ширины запре-
щенной зоны, степени кристалличности и доли поверхности,
занятой адсорбатом.
ЗАДАЧИ
10.1. Получите уравнение (10.23) исходя из уравнения (10.21).
10.2. Рассчитайте концентрацию атомов внедрения в образце KNOs, на-
ходившемся при температуре плавления, после закалки в жидком азоте.
10.3. Чему равна концентрация центров окраски в образце азида иатрия,
если коэффициент поглощения при 3400 А равен 0,4, а полуширина полосы
при ?.тях равна 740 А.
10.4. Что можно сказать о вероятном механизме радиационного разло-
жения, если для данного атома мала энергия активации провеса диффузии.
10.5. Рассмотрите возможный механизм радиационного разложения хло-
ратов щелочных металлов.
Глава XI
РАДИАЦИОННАЯ ПОЛИМЕРИЗАЦИЯ
Прежде чем приступить к обсуждению процессов радиа-
ционной полимеризации, рассмотрим вкратце природу и основ-
ные свойства полимеров, которые обусловлены их высоким мо-
лекулярным весом, химическим строением и упаковкой молекул
в образце.
1'1.1. СВОЙСТВА ПОЛИМЕРОВ В ТВЕРДОМ СОСТОЯНИИ
Все полимеры за немногими исключениями можно разделить
на две большие группы. Полимеры первой группы — это длин-
ные цепные молекулы с молекулярным весом 104—107, скреп-
ленные друг с другом слабыми вторичными взаимодействиями.
Основу полимерных материалов второй группы образуют трех-
мерные сетки, где связи между цепными молекулами имеют
ковалентный характер.
К первой группе относятся пластмассы— тефлон и полиэти-
лен, волокна — целлюлоза, каучуки — бутадиспстирол (невулка-
низированный). К сетчатым материалам с трехмерной структу-
рой относятся фенолформальдегидные и мочевипо-формальде-
гидные смолы, вулканиеированный каучук. Существуют поли-
мерные материалы с малым числом ковалентных сшивок меж-
ду цепями, например частично вулканизированный каучук.
Для полимерных материалов, как и для других твердых тел,
существует характерная зависимость между приложенной на-
грузкой и деформацией. Типичная кривая зависимости относи-
тельной деформации от напряжения для полимера приведена на
рис. 11.1. Длина отрезка CD от точки текучести до точки раз-
рыва является мерой пластической необратимой деформации.
Линейный начальный участок кривой характеризует упругое по-
ведение материала, которое подчиняется закону Гука:
F/А = У(Д///).
Здесь F— сила, действующая на поперечное сечение образца /1;
Y — модуль Юнга (модуль упругости); I — длина образца. При
увеличении числа сшивок в полимерном образце модуль Юнга,
330
как правило, увеличивается, а предельная деформация умень-
шается.
Эксперименты, где изучается связь между нагрузкой и де-
формацией, выполняют в различных вариантах. Рассмотрим два
из наиболее часто встречаю-
щихся.
Термохимические кривые.
Нагруженный полимер под-
вергают медленному ' нагре-
ванию. Температура, при ко-
торой длина полимера начи-
нает резко расти, называется
температурой текучести. При
прочих равных условиях тем-
пература текучести тем выше,
чем выше степень сшивания
образца.
Измерения ударной проч-
ности. Если время приложе-
ния нагрузки мало по срав-
нению с характеристическим
временем вязкоупругой ре-
лаксации, то в образце появ-
ляются трещины. Существуют
Относительное удлинение M/L
Рис. 11.1. Кривая напряжение — де-
формация, характерная для полиме-
ров:
АВ — область упругого поведения полиме-
ра; В С — вязкоупругого; CD — область
пластической деформации.
различные способы измерений
ударной прочности. Обычно опыт проводят па образце с особы-
ми надрезами, которые позволяют сконцентрировать напряжения
и соответственно локализовать места появления трещин.
11.1.1. Электрические свойства полимеров
Укажем на наиболее важные из электрических свойств по-
лимеров.
Диэлектрическая проницаемость характеризует способность
образца накапливать энергию в электрическом поле. Величиной
диэлектрической проницаемости определяется также уменьше-
ние силы взаимодействия между двумя зарядами в данной
среде. Для большей части твердых полимеров диэлектрическая
проницаемость равна 2—4.
Электрическая прочность. Напряженность электрического
поля, при которой происходит пробой, является мерой электри-
ческой прочности диэлектрика. Напряжение пробоя для поли-
меров оказывается обычно в пределах 106—Ю7 в[см.
Диэлектрическими потерями называют ту часть энергии элек-
трического поля, которая необратимо рассеивается в виде тепла
в результате движения диполей.
Удельная электропроводность полимеров очень мала. Сопро-
тивление полимерных образцов составляет обычно около
1010 ом-см.
331
Следует отметить, что все указанные свойства сложным об-
разом зависят от частоты приложенного электрического поля,
от температуры и других параметров окружения.
11.1.2. Молекулярный вес
В состав полимерного образца, как правило, входят молеку-
лы разной длины. Поэтому при определении молекулярного веса
образца приходится пользоваться тем или иным способом усред-
нения.
1. Средпечисленный молекулярный вес и средпечислснпая
степень полимеризации вводятся следующим образом:
Мп = 1
2/ц
07,
2л/
Здесь tii — число молекул, имеющих определенную длину;' М,—
молекулярный вес; СП/— степень полимеризации 7-й молекулы.
Степень полимеризации равна числу мономерных звеньев в
составе молекулярной цепочки. Среднечисленный молекулярный
вес можно найти, проводя измерения осмотического давления в
растворе или определяя химическими методами число концевых
групп. Оба способа позволяют подсчитать среднее число цепей
в образце. __
2. Средпевесовой молекулярный вес Mw и средневесовая сте-
пень полимеризации СПп, вводятся таким образом:
Mw=
^ntMi ’
.— Уп,СП2,
cnw = 1 1
Квадраты в формулах появляются из-за того, что усредне-
ние проводится с помощью весовых долей молекул данной дли-
ны (а не по числу цепей). Средневесовой молекулярный вес
можно найти из измерений светорассеяния, вязкости, диффузии
или седиментации в растворах. Последние три метода, однако,
могут давать и более сложные виды средних молекулярных
весов.
11.1.3 . Свойства полимеров в растворах
При растворении линейных полимеров молекулы отделяются
друг от друга, и можно изучать их индивидуальные характери-
стики. Такие свойства полимерных растворов, как осмотическое
давление, понижение точки замерзания, повышение точки кипе-
332
«ия, светорассеяние, вязкость или диффузионные характеристи-
ки, можно использовать как для определения молекулярных
весов, так и для определения формы отдельных макромолекул.
Хотя сшитые полимеры и не могут растворяться, они часто
обладают способностью к набуханию в растворителе. Степень
набухания сетки можно связать со степенью сшивания.
11.1.4 . Изменения агрегатного состояния полимеров
Так как полимеры состоят из смеси молекул различного
молекулярного веса, они не имеют определенной точки плавле-
ния (в отличие от льда, например, который плавится при 0°С).
При нагревании аморфные полимеры ведут себя как переохлаж-
денные жидкости, вроде стекол, и, перед тем как превратиться
в вязкую жидкость, проходят через высокоэластичпое состояние.
Истинное плавление может происходить лишь в кристалличе-
ских областях полимерной системы.
11.1.5 . Кристалличность и ориентация молекул
в полимерных образцах
В общем случае полимеры не могут образовывать идеальных
кристаллов как из-за различий в длине молекулярных цепей,
так и благодаря низкой подвижности полимерных молекул при
обычных температурах кристаллизации.
Рис. 11.2. Упаковка полимерных молекул в образце:
а — аморфный полимер; б — частично закристаллизованный
полимер; в — полимер, закристаллизованный при растя-
жении.
Степень кристалличности полимерного образца меняется в
зависимости от химического строения полимера, а также зави-
сит от скорости охлаждения расплава. Некоторые полимеры,
такие, как нейлон, кристаллизуются довольно легко. Улучшить
кристалличность можно с помощью ориентации (растяжения)
333
цепей вдоль одной из осей кристалла, как это показано на
рис. 11.2. Частичная ориентация возможна и в некристаллнзую-
щихся полимерах, таких, как каучуки. При этом образец дает
картину рентгеновской дифракции, сходную с рентгенограмма-
ми кристаллических образцов.
Особую пространственную морфологию полимерных цепей
можно получить посредством стереоспецифической полимериза-
ции. Рассмотрим виниловый полимер —СН2—CHR—СН2—
—CHR—, в котором R может быть фенильной группой (поли-
стирол) или метильной группой (полипропилен). Если в цепи
существует хаотическое пространственное распределение боко-
вых групп, полимер называют атактическим. Возможны также
упорядоченные пространственные структуры, показанные на ри-
сунке. Это изотактические и синдиотактические полимеры.
Атактический
полимер
н- н н н н
„ I 1 I I I
V У У Н С Н С цзотактический
'’/г'/й4 С RX CZRX lZRX тЛиМеР
I I I I I
H H . H H H
H R H R H
V/f\| /f\? /f\?/f\?/Г\ Сшд.иотакт^с-
CZRXCZHXk RXCZHXCZRX
I I I I I
H -H H H H
11.2. ПОЛИМЕРИЗАЦИЯ
Полимеризация — это реакция соединения мономеров, проте-
кающая без изменения элементарного состава и не сопровож-
дающаяся выделением побочных продуктов. В ходе реакции не
образуется стабильных соединений, и все промежуточные про-
дукты представляют собой короткоживущие радикалы или
ионы.
Образование полимерной цепи происходит обычно за доли
секунды, если только этому не препятствуют повышенная вяз-
кость системы или стерические факторы. Хотя, как правило, в
334
реакциях полимеризации участвуют лишь одинаковые виниловые
мономеры, возможна полимеризация в смеси различных моно-
меров. Полимеризация двух или большего числа мономеров
разного строения называется сополимеризацией. Ниже для сме-
си мономеров А и В показано, какие типы полимерных цепей
могут образовываться в результате их сополимеризации:
ААВАААВВВВАВАА — случайный сополимер;
АВАВАВАВЛВАВАВ — периодический сополимер;
ЛААЛВВВВАААА — блок-сополимер;
ВВВВВ ВВВ
АЛААААЛААЛАЛАА—привйтой сополимер (или графт-сополимер).
ВВВВВВ
Обычно, однако, блок- и графт-сополимеры получают не из
двух сортов мономеров, а простой полимеризацией одного моно-
мера в присутствии заготовок полимера — «хозяина».
Процесс цепной полимеризации состоит из четырех основных
стадий:
1. Инициирование (скорость реакции /?г)
Я/ с — с- (11.1)
2. Рост цепи (скорость реакции Rp)
k' с-₽-М„.; (11.2)
k'' +М—р-*М-п+ь (И-З)
3. Обрыв цепи (скорость реакции Rt)
k't 2М„ М2,; (рекомбинация); (П.4)
/г Мй 4-М* > МЛ-|-М/Н (Диспропорционирование). (П.5)
4. Передача цепи (скорость реакции Rs)
k AV 4- М —М„ п- М- (на мономер); (И.6)
AV; + Мт —S-* М„ 4- AV (ча полимер); (П.7)
k М- 4- S —S- 4- М„ (на другие молекулы). п 1 • п (П-8)
335
Инициирование. Многие вещества, которые разлагаются под
влиянием внешних воздействий или самопроизвольно, могут
инициировать полимеризацию. Если в качестве внешнего воз-
действия используется облучение, частица С может быть или
мономером, или любым другим радикалом. В процессе химиче-
ски инициируемой полимеризации катализатор С все время
прикреплен к полимерной цепи.
Рост цепи. Во время роста цепи происходит взаимодействие
радикалов с мономерами. Возможность протекания и скорость
соответствующих реакций определяются стабильностью мономе-
ра и радикала, а также стерическими эффектами, связанными
с наличием тех или иных химических групп в реагирующих ча-
стицах.
Обрыв цепи. Скорость обрыва цепей нередко оказывается
пропорциональной числу радикалов. При высоких концентра-
циях радикалов обрыв цепей происходит достаточно часто, что
обусловливает сравнительно низкие молекулярные веса. В об-
щем случае п=^=т в уравнении (11.5) и два п в уравнении (11.4)
также неодинаковы.
Передача цепи. При передаче цепи число свободных радика-
лов в системе не уменьшается. При этом происходит снижение
молекулярного веса, если только передача не происходит на
сформировавшиеся полимерные цепи.
Ингибирование. Вещества, добавление которых в систему за-
медляет или останавливает реакцию полимеризации, называ-
ются ингибиторами. Обычно механизм их действия сводится
к тому, что они образуют стабильные соединения при взаимо-
действии с промежуточными продуктами высокой реакционной
способности.
Автокаталитический эффект (эффект Троммсдорфа). Авто-
каталитический эффект наблюдается главным образом для ре-
акции полимеризации мономеров типа СН2=СНХ. С повыше-
нием вязкости системы скорость полимеризации увеличивается.
Предполагается, что подобное самоускорение реакции связано
с тем, что при повышении вязкости среды уменьшается скорость
диффузии и скорость обрыва цепей, так как для обрыва цепи
необходимо соударение двух радикалов.
11.3. ИОННАЯ ПОЛИМЕРИЗАЦИЯ
Ионная полимеризация отличается от радикальной тем, что
нестабильные промежуточные продукты несут либо положитель-
ный, либо отрицательный заряд. Соответственно различают ка-
тионную и анионную полимеризацию. Примером катионной
может служить полимеризация изобутилена, инициируемая трех-
фтористым бором в присутствии воды в качестве сокатали-
затора.
336
Инициирование:
BF3 + Н2О -> F3BOH~ + Н'; (11. 9>
Н+ -г СН2 -- С (СН3)2 -> (СН^С1-. (11.10)
Рост цепи:
СН3 СН3
(СН3) С++ СН2 = С (СН3). - СН3С—СН,—с+ (Н.П)
I " I
сн3 сн3
Обрыв цепи:
СН3 R~ С (СН3)2 + 11+
R—С СН3 (11-12)
СН3 ^R—+11+
'Ч
СН2
Катализаторами анионной полимеризации могут служить
металлический натрий, этилат натрия, водные растворы щелочи
и ряд других веществ. Полимеризацию мономера М в присутст-
вии оснований можно, видимо, представить следующим образом.
Инициирование:
К + NII3 -> К+ + NHF + Н2; (11.13)
М + NHi* RNH7- (11.14)
Рост цепи:
RNIIf + М R'NHF. (11.15)
Обрыв цепи:
R'NHr Р + NHF; (11.16)
R'NH? + NH3 -► PNH3 + NHF. (11.17)
Кинетика ионной полимеризации зависит от характера среды.
В средах с высокой диэлектрической проницаемостью разделе-
ние зарядов облегчено. Скорость инициирования возрастает и
скорость реакций обрыва цепи уменьшается, так как сохране-
ние электронейтральности в различных участках системы может
обеспечиваться противоионами.
11.4. СОПОЛИМЕРИЗАЦИЯ
Одновременная полимеризация двух или более мономеров,
ведущая к образованию полимерной цепи, включающей разные
мономеры, называется сополимеризацией. Если в сополимери-
337
зации участвуют два мономера All и M2, то возможны четыре
типа радикал-мономерных реакций:
М; + Mj —*> MjM; ; (11.18)
Mj ф- Мг —MiM- ; (И.19)
М • + М2 —М2М • ; (11.20)
Mj + Мх —- М2М- . (И.21)
Строение образующегося сополимера зависит от соотноше-
ния скоростей конкурирующих реакций. Если, например, ^12—^21,
a kn и k22— малы, то образуется цепь с периодической последо-
вательностью мономеров
—Mr—М2—Mj—М2—Мг—М2—.
11.5. СПОСОБЫ ПРОВЕДЕНИЯ ПОЛИМЕРИЗАЦИИ
Блочная полимеризация или полимеризация в массе. При
этом способе ведут полимеризацию чистого мономера в газооб-
разном, жидком или твердом состоянии. Во время блочной поли-
меризации трудной задачей зачастую оказывается регулирова-
ние температуры системы. Местные перегревы ведут к широкому
распределению по молекулярным весам, что ухудшает механи-
ческие характеристики полимера.
При полимеризации в растворе, когда мономер диспергиро-
ван в растворителе, удается лучше регулировать температуру.
Характерные недостатки этого способа полимеризации: 1) вклю-
чение следовых количеств растворителей в полимер; 2) пере-
дача цепи, снижающая молекулярный вес.
Эмульсионную полимеризацию проводят в суспензиях моно-
мера в водной среде, добавляя соли высших органических кис-
лот (мыла). В начальной стадии полимеризации мономер при-
сутствует в виде мелких капелек, покрытых мыльной оболочкой,
или же он солюбилизирован в мыльных мицеллах. Согласно
существующим теориям эмульсионной полимеризации, иниции-
рование происходит в мицеллах под действием радикалов, об-
разующихся в водной фазе. Мономер диффундирует в мицеллы,
где развертываются реакции роста и обрыва цепи. Процесс про-
должается до тех пор, пока не израсходуется весь мономер
эмульсионной капельки.
Помимо полимеризации в растворе, блочной и эмульсионной,
используют также другие способы полимеризации. Однако все
они по сравнению с перечисленными имеют гораздо меньшее
значение, поэтому для них не проводилось широких радиацион-
но-химических исследований.
338
11.6. КИНЕТИКА полимеризации
В стационарном состоянии промежуточные продукты обра-
зуются и исчезают с равными скоростями. При этом, если спра-
ведлива кинетическая схема, представленная уравнениями
(11.1) — (11.8), из уравнений (11.1) и (11.4) мы получим
Я/ = R, = kf [М-]2. ' (11.22)
Уравнение (11.22) можно использовать для того, чтобы ис-
ключить концентрацию [М^] из кинетического уравнения для
реакции (11.3):
- = R = kP [M„J IM] = kpk^!^ [М]. (11.23)
При выводе уравнения (11.23) предполагалось, что мономер
расходуется только на реакцию роста цепи; реакционная спо-
собность полимерного радикала не зависит от размера цепи и,
наконец, обрыв цепи происходит в результате реакции (11.4).
В случае радиационпо-инициируемой реакции, если G™ — вы-
ход радикалов М-, Ф —поток радиации, р.еэт/р — массовый ко-
эффициент поглощения энергии, получим
7?;- = СгмФ(рг,;/рИМ]. (11.24)
Отсюда для разных вариантов радиационной полимеризации
нетрудно получить соответствующие кинетические уравнения.
Некоторые ситуации рассмотрены в примере 11.1.
Пример 11.1. Выведите кинетические уравнения расходования мономера
для следующих случаев:
1) полимеризация происходит в присутствии вещества S;
2) в системе происходит рекомбинация первичных радикалов;
3) идет сополимеризация двух мономеров;
4) для полимеризации в растворе и для блочной полимеризации при
низких мощностях дозы найдите выражения для СПп.
Решение.
1. Вещество S также может участвовать в инициировании. Если перенос
энергии от S к М не происходит, то справедливо выражение:
R( =-- Ф |G;v [М] + G$ ('S-- [S]) . (И.25)
I p Р )
Вещество S способно принять участие и в процессах роста цепи при до-
статочно высокой реакционной способности радикалов S. Участие вещества S
не изменит скорость полимеризации, хотя может повлиять на величину мо-
лекулярного веса.
Подставляя уравнение (11.25) в уравнение (11.23), найдем:
R =-. kpk~'^ [M]s/1 <DG;M ( A
\ P /
(pfn(S)/p)Gf[S] l‘z*
(МгЛ(М)/р)ОгЛ4|М]
(11.26)
Отметим, что уравнение (11.26) позволяет найти G^ при проведении по-
лимеризации в растворе.
339
2. Если концентрация первичных радикалов достаточно высока, то будут
происходить реакции обрыва цепи (11.4а) и (11.46):
ktc'
С-+С----------СС; (11.4а
ktc
С- + М„. - > СМ„. (11.46)
Уравнения для стационарных концентраций радикалов С- и М- будут
выглядеть следующим образом:
dC-
= IC-F — [С-ЦМ] — [С] ЬМ-„]; (11.27)
dM.
— = 0 = kpc [C • | [M] - kt IM„- j* - klc [C| |M„ ]. (11.28)
Здесь индекс «с» указывает, что в реакции принимают участие радикалы
С-, реакционную способность кторых мы считаем отличной от реакционной
способности радикалом 7ИП. Уравнения (Г1.27) и (11.28) ие позволяют полу-
чить простое общее выражение для 7? = ЛР[7И] [Л4„ •] через концентрации ста-
бильных веществ. Однако для ряда граничных условий такое выражение
удается получить. Если, например, скорость ипциировапия настолько велика,
что Л(с[С-] [Л4„ •] ^>kt [Л4„-]2 (т. е. обрыв цепей происходит за счет реком-
бинации с первичными радикалами), то уравнение (11.28) приобретает сле-
дующий вид:
[М„-Ь-/грс/^с[М]. (11.29)
Подставляя это выражение для [Л4„-] в уравнение (11.23), находим,что
скорость исчезновения мономера не зависит от скорости инициирования:
kpfipc
Ktc
(11.30)
Если мощность дозы возрастает еще больше, то значительная часть мо-
номера будет расходоваться в реакции
С- +М — - м..
Из уравнения (11.23) для полной скорости расходования мономера получим
я = МмНмп1+*рс1С][М]. (11.31)
В уравнении (11.27) при высоких мощностях дозы последними двумя
членами можно пренебречь по сравнению с остальными, тогда
= (11.32)
Подставляя уравнения (11.32) и (11.29) в (11.31), найдем
R = №pciktc + kpcR^ [M]/fc$. (11.33)
На рис. 11.3 изменение скорости полимеризации при увеличении мощно-
сти дозы представлено графически в соответствии с уравнениями (11.23),
(11.30) и (11.33).
3. При стационарной сополимеризации скорость, с которой радикал Mj
трансформируется в М2- [см. уравнение (111.19)], должна быть равна скорости,
с которой М2- трансформируется в Мц [см уравнение (11.20)]. Тогда в со-
340
ответствии с уравнениями (11.19) и (11.20) отношение [MjJ/tAlJ должно
•быть равно:
= й12 [M2]/ft21 [MJ. (11.34)
Скорость расходования мономеров Mi и М2 должна быть равна скорости
их включения в полимер:
dMi
— = kn [Mi-J (МП + k21 [MJ;
o’M,
- = kit [Mi- ] [M2] + k„ [M, • ] [M,].
(11.35)
(11.36)
Разделив уравнение (11.35) на' уравнение (11.36) и используя уравнение
(11.34), получим:
__ [MJ ri [MJ 4- |М2]
dM, [MJ [MJ 4- г, |М2] ’ 1 J
igR.
где Г1=&н/й12 и Г2=й22/^21. Значе-
ния г, большие единицы, будут ука-
зывать на то, что радикалы легче
реагируют с мономерами той же при-
роды, нежели с другими.
4. Степень полимеризации равна
полному числу мономеров, включен-
ных в полимер, деленному на число
актов обрыва цепи (или актов иници-
ирования). Если же возможна пере-
дача цепи на мономер М или ра-
створитель S, то
Уравнение ‘ (11.23) Уравнение (11.30) R не зависит от R; Уравнение 01.33) 1 К=а+ЬВ&
Ri
Рис. 11.3. Зависимость скорости по-
лимеризации от мощности дозы.
(скорость роста цени)/[(скорость обрыва цепи)-{-(скорость передачи цепи на мономстр)-}-
-{-(скорость передачи цепи на растворитель)]. (11.38)
Если считать, что процесс передачи цепи на мономер, описываемый урав-
нением (11.6), сравнительно малосуществен, то
__________________________ Ммл-1|М]
СП ---------------
^[M„.]4-^[M„.][S]
Уравнение (11.39) удобно переписать, используя величину
пень полимеризации в отсутствие процессов передачи цепи:
j_________________________________i_. ks [S)
СП„ СПВ т kp [М] ’
где
______________________ йР[Мл.][М] fep[M]
СП Q =------------------------------=-----------------.
MW
Если обрыв цепей происходит в результате рекомбинации
радикалов согласно уравнению (11.5), то величину СП0 следует удвоить.
(11.39)
СП0 — сте-
(11.40)
(11-41)
полимерных
11.6.1. Кинетика полимеризации.
Нестационарное состояние системы
Если при нестационарной полимеризации обрыв цепей про-
исходит в результате реакции второго порядка, то для концен-
341
трации радикалов в системе можно получить следующее урав-
нение:
at
(11.42)
где [R-]— концентрация радикалов в системе; Ri(t)—скорость
инициирования, которая зависит от времени. Если скорость Ri
постоянна или равна нулю, уравнение (11.42) имеет аналитиче-
Рис. 11.4. Зависимость скорости по-
лимеризации от характеристик пре-
рывистого облучения:
Скорость реакции при импульсном
п, облучении
Скорость стационарной реакции поли-
меризации
1 — синусоидальное облучение; 2 — облуче-
ние импульсами прямоугольной формы.
4[R'-J
df
ское решение. В тех случаях,
когда Ri является периодиче-
ской функцией времени, как,
например, при облучении си-
стемы через вращающиеся сек-
торные прорези, удобно заме-
нить переменные в уравнении
(11.42) на безразмерные вели-
чины:
R' = R-/Rr;
Ri = RiWIRis', (11-43)
t' = t/x.
Здесь индекс s указывает
на то, что соответствующая ве-
личина относится к стационар-
ным условиям, а т — среднее
время жизни радикала. Урав-
нение (11.42) можно пере-
писать в следующей форме:
(11.44)
RdO-MR'-]2.
Теперь в уравнение входят лишь безразмерные величины,
кроме того, его можно нормировать, если выбрать скорость ини-
циирования Ris равной максимальному значению периодически
меняющейся скорости инициирования Ri(t).
Решение уравнения (11.44), полученное с помощью вычисли-
тельной машины для периодических функций Ri(t) синусоидаль-
ной и прямоугольной форм, приведено иа рис. 11.4. По оси орди-
нат отложено значение средней концентрации радикалов (JF)»
деленной на концентрацию радикалов в стационарном состоянии
при интенсивности излучения, равной максимальному значению
излучения прерывистого облучения. При высоких частотах обе
кривые стремятся к одним и тем же асимптотическим значениям,
так как одинаково среднее значение Ri'. При низких частотах
отмечаются заметные различия в скорости реакции для облуче-
ния прямоугольными импульсами и синусоидально меняющимся
излучением.
342
11.7. ПРОСТРАНСТВЕННОЕ РАСПРЕДЕЛЕНИЕ СОБЫТИЙ
ИНИЦИИРОВАНИЯ
Весь предыдущий анализ основывается на предположении о
гомогенном пространственном распределении частиц с высокой
реакционной способностью. Подобная гомогенность при радиа-
ционной полимеризации может обеспечиваться лишь за счет
диффузионного перемешивания радикалов. Если же достаточ-
ного перемешивания не происходит, приходится учитывать про-
странственное распределение радикалов вдоль треков. Когда
шпоры нс перекрываются и скорость реакции велика, то во вре-
мя процесса полимеризации свободные радикалы останутся изо-
лированными. В этих условиях увеличение интенсивности излу-
чения и R{ приведет к появлению новых радикалов. Значение
СПп при этом не изменится. Скорость полимеризации будет
пропорциональна а ne R\/2, как это следует из уравнения
(11.23), полученного для гомогенных условий.
118 ПОЛИМЕРИЗАЦИЯ В ЖИДКОЙ ФАЗЕ
В литературе сообщалось о радиационной полимеризации в
жидкой фазе, когда удавалось получать цепочки более чем из
100 мономерных звеньев. Хотя различные жидкие системы обла-
дают специфическими особенностями, общее представление
о происходящих в них процессах можно получить на примере
полимеризации стирола.
Первые эксперименты по радиационной блочной полимери-
зации стирола проводили при комнатной температуре без ка-
ких-либо добавок. До степеней конверсии мономера в полимер,
равных примерно 60%, скорость реакции оставалась постоянной,
ио затем происходило внезапное увеличение как скорости ре-
акции, так и молекулярного веса.
Выяснилось, что полимеризацию стирола ингибируют следо-
вые количества воздуха, бензохинона и других ингибиторов сво-
боднорадикальных реакций. Поэтому, даже несмотря на резкое
ускорение радиационной полимеризации в конце процесса, ко-
торое не наблюдается при использовании обычных свободнора-
дикальных катализаторов, можно предположить, что кинетиче-
ски механизм сводится к классической свободнорадикальной
схеме. Ускорение полимеризации при высоких степенях конвер-
сии связано, по-видимому, с особенностями радиационной по-
лимеризации, а также с проявлением гсль-эффекта (эффекта
Троммсдорфа).
Качественное объяснение рассматриваемого явления осно
вывается на том, что стирол, подобно бензолу, обладает высокой
устойчивостью к действию радиации. Присутствие виниловой
связи, сопряженной с фенильным кольцом, способствует стаби-
лизации возбужденных состояний и в результате Gr (стиро-
343
ла) =0,69, что даже ниже значения Gr (бензола). С другой сто-
роны, полистирол обладает структурой, более сходной с алкил-
замещеппыми производными бензола, для которых значения Gr
лежат между 1,5 и 4. Поэтому с ростом степени конверсии
можно ожидать увеличения выхода радикалов, скорости поли-
меризации, молекулярного веса и степени ветвления полимера
за счет инициирования в самом полимере.
Дополнительные доказательства существования полимерных
радикалов можно получить при изучении механических свойств
готового материала. Полистирол, полученный методом радиа-
ционной полимеризации, обладает большей прочностью на раз-
рыв по сравнению с образцами полистирола, полученными в ре-
зультате термополимеризации. Очевидно, объяснение заклю-
чается в том, что первый образец имеет больше ветвящихся
молекул и внутренних сшивок.
11.8.1. Влияние мощности дозы
Весьма подробный анализ экспериментов по изучению влия-.
ния мощности дозы па полимеризацию стирола был проделан
Шапиро. На рис. 11.5 суммированы экспериментальные резуль-
Мощность дозы, рад/сек
Рис. 11.5. Влияние мощности
дозы на скорость полимериза-
ции стирола и молекулярный
вес образующегося полимера.
Результаты приведены к ком-
натной температуре (20° С).
Пунктирными линиями пока-
заны экстраполированные за-
висимости.
таты восьми различных исследований. Как показывают данные
рис. 11.5, в диапазоне мощностей доз от 2-Ю-3 до 10 рад]сек
скорость полимеризации стирола и среднечисленный молекуляр-
ный вес образующегося полимера непосредственно задаются
величиной корня квадратного из скорости инициирования [см.
уравнения (11.23) и (11.41)].
11.8.2. Влияние температуры
Общая энергия активации ДЕ для «простой» полимеризации
виниловых мономеров [см. уравнение (11.23)] будет равна:
ДЕ = ЛЕр — -±- &Et 4- — ДЕР (11.45)
2 2
344
Обычно для радикальных реакций такого типа \Ev^b—
8 ккал)моль, —3 ккал!моль. Для радиационно-инициируе-
мых реакций ДЁг должно быть близко к нулю. Поэтому ДЕ~
~6 ккал/моль.
Отметим также, что согласно уравнению (11.41) рост кине-
тической длины цепей по мере увеличения температуры в отсут-
ствие процессов передачи цепи будет также происходить с энер-
гией активации ЛЕ~6 ккал)моль.
Пример 11.2. В приведенной ниже таблице экспериментальных данных,
полученных Баллантайном [1], указаны температура, скорость полимеризации,
молекулярный вес и мощность дозы излучения для процесса радиационной
полимеризации стирола. Сравните ,эти экспериментальные данные с теми
температурными зависимостями ско-
рости полимеризации и молекулярного
веса, которые следовало бы ожидать
из уравнении (11.45) и (11.41).
Температура, Скорость по- лимеризации, 10т мольЦлх Хсеи} е 1< Мощность до- зы, крад-ч
—18 20 23 200 175
25 100 73500 175
72 500 330 000 175
Рис. 11.6. Температурная зависимость
скорости реакции (уравнение Арре-
ниуса) для примера 11.2.
Решение. Экспериментальные данные в полулогарифмическом масштабе
нанесены на график рис. 11.6. Определив наклоны прямых, для скорости по-
лимеризации получим ДЕ =6,5 ккал/моль и для роста молекулярного веса
ЛЕ -5,7 ккал]молъ. Оба результата находятся в хорошем соответствии с тео-
ретическими предсказаниями.
Интересным приложением кинетических уравнений, полученных в при-
мере 11.1, является определение выхода радикальных продуктов в облучен-
ном стироле. Комбинируя уравнение (11.23), описывающее скорость полиме-
ризации при низких мощностях дозы, н уравнение (11.41), связывающее зна-
чение СП0 со скоростью полимерной рекомбинации, можно найти значение
R kpkY4‘ [MJ; (Н .23)
СП~0^ 2Apfe7'/![M] (11.41)
(cTQl,t = yT kpkYth[^]R~'/!- (11.46)
Таким образом, по углу наклона па графике (С/7„)^2 от R~^2 можно
определить kpkt~'1^2.Тогда Ri можно рассчитать либо по уравнению (11.23),
либо по уравнению (11.41). При высоких мощностях дозы и в тех случаях,
когда происходит передача цепи, нужно сделать соответствующие поправки.
345
11.9. ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ
Присутствие растворителя в полимеризационной системе
приводит к тому, что становятся возможными множественное
инициирование, передача цепи, перенос энергии и химические
Рис. 11.7. Относительные скорости
конверсии стирола, растворенного
в бензоле (А=1), толуоле (А--
= 1,5), ксилоле (71=4,5) и этил-
бензоле (А =6,5). A = G$ Hs/G^n.v.
реакции, отличные от полимери-
зации. Хотя благодаря веем этим
вторичным реакциям кинетика
полимеризации усложняется, тем
не менее изучение полимериза-
ции в растворе оказывается по-
лезным методом исследования
радиолиза самих растворителей.
Примером снова может служить
полимеризация стирола.
При отсутствии усложняю-
щих обстоятельств скорость
полимеризации для низких мощ-
ностей дозы дается уравнением
(11.26):
Г Т'*
/? = kpkT',x [М]’/гФС? (1 л- —-----------------------------
\ р J г
(11.26)
На рис. 11.7 показана относительная скорость конверсии сти-
рола, растворенного в бензоле, толуоле, ксилоле и этилбензоле
в зависимости от мольной доли стирола. Видно, что при умень-
шении концентрации стирола вплоть до 20 мол. % эксперимен-
тальные точки хорошо ложатся на теоретические кривые, кото-
рые получены посредством деления уравнения (11.26) на урав-
нение (11.23). По оси ординат отложено значение R/Ro:
r/r0 = [М]'/г Фб;л’ ( у'2
^n(S).Gs[S]
Р
( **ст(М) ЛсМ[М]
к р /
(11.26а)
Эксперименты того же типа, проделанные с растворами сти-
рола в хлористом этиле, хлористом бутиле, циклогексилхлориде,
монохлорбензоле, о-дихлорбензоле, привели к тем же результа-
там. С другой стороны, растворы стирола в CCI4, CHCI3, СНзВг,
СВг4 и CH3CH2I обнаружили аномальное поведение, поэтому
346
при анализе процесса полимеризации в этих системах необхо-
димо принять во внимание возможность переноса энергии воз-
буждения. .
Кинетические уравнения для подобных систем получены Ни-
китиной и Багдасарьяпом, постулировавшими ряд процессов с
участием мономерных молекул М и молекул растворителя S.
Далее звездочкой обозначены промежуточные возбужденные
состояния.
1. Если S обладает высокой радиационной устойчивостью, то
М может передавать энергию S, и это приведет к защитному
эффекту
М М* S* -> S + кэ
(Н-47)
(КЭ — кинетическая энергия).
2. Если S передает энергию М, то это ведет к сенсибилиза-
ции системы, и в результате появляются свободные радикалы
S S* -> М* -► М-.
(11.48)
Кинетическую схему реакций
образом:
можно записать следующим
к5М
(11.49)
*i'
гУШ\ММ/Мг^~ <.*
kS
Если считать, что перенос энергии между М* и S* происхо-
дит очень быстро и процесс идет с равной эффективностью в
обоих направлениях, то можно принять, что
ksM [S*] [М]
(11.50)
В стационарном состоянии для возбужденных молекул S*
и М* можно записать балансные уравнения:
Rt IM] 4- kSM [S*J [Ml = kM [M*J + kM. [M*] + kMS [M*] [S]; (11.51)
Ri [S] + kMS [M*J [S] = ks [S*] -r ks. [S*] + kSM [S*J [М]. (11.52)
347
Теперь уравнения (11.51) и (11.52) можно решить совместно
с уравнением (11.50) и получить выражения для [S*] и [М*].
Умножив эти выражения на kM. и kc., соответственно получим
уравнения для скоростей образования радикалов:
R[m-j ~ км- [M*J =
1 + { ?s + ks ) kMS А/Лч + Ч-) ks.u 1^1}
(11.52a)
Отношение kM-/ (kM+kM.) представляет собой вероятность обра«
зования свободных радикалов М- из М в системе, не содержа-
щей S, и, следовательно, это отношение равно <DpMG;w. Точно
так же ks/(ks.+ks) =OptsG^.
Обозначим Prei следующее отношение:
&MS
, _ kM_ -i-^Вероятность переноса энергии от М* к S
kSM Вероятность переноса энергии от S* к М
^S- +
Из уравнения (11.52а) и соответствующего уравнения для
Rs- получим выражения для полной скорости образования ра-
дикалов и для полной скорости полимеризации:
Я[«-] = Яри + Я[5.] = Ф6\м ( ^ML\ х
\ р J
1
X (1М] + [S])-
(11.54)
Уравнения. (11.54) и (11.55) представляют собой аналоги
уравнений (11.25) и (11.26) с тем единственным отличием, что
348
в них учтены процессы переноса энергии и имеется множитель
Prei. Если Рге(=1, то уравнения (11.54) и (11.55) сводятся к
уравнениям (11.25) и (11.26). Когда Prei>l, перенос энергии
происходит от М к S, когда Рге1<1, перенос энергии идет в об-
ратном направлении.
В нескольких работах для определения значений Prei изме-
ряли скорость полимеризации стирола в растворе. На рис. 11.8
суммированы результаты трех независимых экспериментов, в
которых изучалась полимеризация стирола в хлоро.форме. Из
рисунка видно, что экспериментальные точки хорошо ложатся
па кривую, описываемую уравнением (11.55) при G^/G^* 1 =17
и РГ(>г=1О. Однако теорию процессов переноса энергии при по-
лимеризации в растворе отнюдь нельзя считать вполне завер-
шенной.
Уравнение (11.54) плохо согласуется с имеющимися экспери-
ментальными данными и не соответствует значениям Prei и
Gs/G™, которые были получены другими независимыми спо-
собами. Некоторые результаты свидетельствуют в пользу того,
что помимо «простой» передачи энергии от донорной молекулы
Рис. 11.8. Относительная скорость
полимеризации стирола в растворе
хлороформа в зависимости от моль-
ной доли стирола. Сплошная кривая
соответствует теоретической зависи-
мости, вытекающей из уравнения
(1.1.55) при Gsr /G™ =\7 и /У,., = 10.
Нанесенные экспериментальные точки на
графике обозначают: / — рентгеновское
излучение: 2 -облучение электронами;
3— смешанное реакторное излучение.
Рис. 11.9. Относительные скорости конверсии
стирола, растворенного в различных спиртах,
в зависимости от его мольной доли. Пунктир-
ная кривая соответствует теоретическому со-
отношению, вытекающему из уравнения (11.26):
1 — метанол; 2 — 1-пропанол; 3 — 2-пропанол: 4 — 1-бу-
танол: 5— трет-бутанол; 6— 1-октанол; 7—2-октанол.
349’
к молекуле-акцептору с более пизко расположенным возбужден-
ным состоянием существуют другие более сложные процессы
обмена энергией.
11.10. ПОЛИМЕРИЗАЦИЯ В ГЕТЕРОГЕННЫХ СИСТЕМАХ
Двухфазные полимеризациопные системы возникают: 1) если
мономер или растворитель является осадителем для полимера;
2) если мономер адсорбирован на твердой поверхности и 3) при
эмульсионной полимеризации.
На рис. 11.9 показаны относительные скорости конверсии
стирола, растворенного в различных спиртах, в зависимости от
мольной доли мономера в смеси. Для полистирола любые спир-
ты являются осадителями. На графике приведены также теоре-
тические кривые для полимеризации в растворе, соответствую-
щие уравнению (11.26), в котором Gsr/G™ принималось равным
80 и 50.
График рис. 11.9 можно разбить на три области, каждая из
которых соответствует определенному механизму реакции:
1. 0—30% спирта. Хотя в целом форма экспериментальной
кривой согласуется с предсказаниями уравнения (11.26), необ-
ходимы гораздо большие значения G^, чем известные для спир-
тов, чтобы наблюдалось совпадение экспериментальных и тео-
ретических зависимостей при G^/G1^ =504-80. Повышение ско-
рости полимеризации связано, no-видимому, с эффектом
сепсибилазиции; кроме того, в преципитирующей среде локаль-
ная концентрация мономеров вблизи растущей полимерной цепи
может быть выше средней.
2. 30—70% спирта. В этой области полимер выделяется в
виде прозрачного жидкого слоя, что существенно изменяет ки-
нетику процесса. Чтобы объяснить уменьшение скорости поли-
меризации и уменьшение среднего молекулярного веса в этой
системе, приходится предполагать либо уменьшение константы
/гр, либо увеличение константы kt.
3. 70—95% спирта. В этой области и молекулярный вес по-
лимера, и скорость полимеризации резко возрастают. Полимер
Выделяется в виде порошка, топких пленок или волокнистых
структур. Для реакции полимеризации отмечается значительное
последействие, т. с. реакция продолжается и после удаления об-
разца из радиационного поля. Все эти факты указывают па то,
что происходит заметное подавление процессов обрыва цепей.
Для растворителей неспиртовой природы концентрационные
зависимости проявляются менее ярко. Соответствующие кривые
обычно представляют собой нечто среднее между кривыми обыч-
ной полимеризации в растворе согласно уравнению (11.26) и
кривыми для спиртов, показанными па рис. 11.9. Дополнитель-
но следует учитывать характер образующегося преципитата и
адсорбцию мономеров на полимере.
350
11.10.1. Эмульсионная полимеризация
При эмульсионной полимеризации мономер находится в виде
водной суспензии. Инициирование осуществляется обычно за
счет радикалов, которые возникли нс в мономерной фазе, а в
воде. Значительная часть ранних работ по эмульсионной поли-
меризации была посвящена выяснению первичных процессов в
водном компоненте системы. Например, Дэйптоп, изучая эмуль-
сионную полимеризацию акрилонитрила, показал, что в воде
образуются Н- и ОН-радикалы, которые можно обнаружить за-
тем прикрепленными на концах полимерных цепей.
Радиациопно-инициирусмая эмульсионная полимеризация
стирола происходит примерно в 100 раз быстрее, чем блочная
полимеризация. Образующийся при этом полимер имеет в 10—
30 раз большие молекулярные веса (табл. 11.1). Измерения по-
казали, что общая энергия активации для эмульсионной поли-
меризации равна 3,7 ккал/моль.
Таблица 11.1
Блочная и эмульсионная радиационная полимеризация стирола
Способ полимеризации Интенсивность облучения, р/мин Температура, °C Скорость кон- версии, % в 1 ч Молекулярный вес полимера, -I03
Блочная 4400 —18 0,1 17—26
» 3200 30 0,42 38—110
» 4100 72 2,53 165—348
Эмульсионная 1000 25 36 800—1500
» 1000 35 45 885—1726
1000 45 54 1200—2060
Два фактора обусловливают высокие молекулярные веса и
скорость процесса при эмульсионной полимеризации: 1) высо-
кий выход радикальных продуктов в воде; 2) низкая скорость
процессов обрыва цепей, которые, как предполагают, происходят
только в изолированных полимерных частицах.
Подобные представления хорошо согласуются с классической
теорией каталитической эмульсионной полимеризации. В других
случаях, однако, такого согласия не наблюдается. Так, напри-
мер, уравнение для скорости эмульсионной полимеризации, ка-
тализируемой неорганической перекисью, когда радикал, возни-
кающий в водной фазе, входит в мицеллярную частицу и
остается там до тех пор, пока другой радикал нс приведет к
обрыву цепи, будет выглядеть следующим образом:
= JV/2. (11.56)
N — число частиц на 1 л — можно связать с площадью, покры-
той эмульгатором, скоростью набухания отдельных частиц и
351
•скоростью образования свободных радикалов N оказывается
пропорциональным R2/5. Полная скорость полимеризации при
облучении с интенсивностью /, если уравнение (11.56) справед-
ливо и для радиационной полимеризации, с учетом того, что
(/?()2/5 ~/2'5, будет равна
= const- [М]/г/а .
dt
(11.57)
Лкрес и Дальтон [3] проводили эмульсионную полимериза-
цию стирола в условиях, при которых уравнение (11.57) должно
было выполняться. Однако их результаты (рис. 11.10) противо-
речат уравнению (11.57), что
Концентрация моно мера, М
Рис. 11.10. Изменение характера за-
висимости скорости полимеризации
от интенсивности облучения при раз-
ных концентрациях стирола.
указывает па существование
различия между обычным и
радиационным катализом
полимеризации. Аномальное
кинетическое поведение си-
стемы можно объяснить,
если учесть, что при облуче-
нии появляются водородные
атомы, которые пе возни-
кают при обычных способах
инициирования. Если эти
водородные атомы могут ре-
комбинировать или же могут
димеризоваться радикалы,
возникшие в результате ре-
акций с водородными атома-
ми, то зависимость скорости полимеризации от интенсивности
облучения будет такой, какая и демонстрируется на рнс. 11.10.
К сожалению, наше понимание процессов эмульсионной по-
лимеризации остается далеко нс полным. Как указывают Лей,
Хаммель и Шнайдер [4], «почти каждая система обнаруживает
специфическое поведение... невозможно подогнать под одну схе-
му поведение различных систем. Кривые «степень конверсии —
время» имеют столько характерных деталей, что обобщенное
описание перестает быть оправданным».
11.11. СОПОЛИМЕРИЗАЦИЯ
Сополимеризацию стирола с метилметакрилатом в 1955 г.
исследовали Зейцер и Тобольский. При инициировании переки-
сями и при радиационной полимеризации в полимер включа-
лись одинаковые количества обоих мономеров. Это интерпрети-
ровалось как прямое доказательство радикального механизма
полимеризации, так как было известно, что при катионном ини-
352
циировапии, например, с помощью хлорида олова, в сополи-
мер включалось более 99% стирола, тогда как при анионном
инициировании в полимер входило более 99% метилметакри-
лата.
Однако в 1957 г. наметился отход от свободнорадикального
механизма, после того как Дэвисон. Пиппер и Уоррол обнару-
жили, что под действием излучения при —78° С можно запели-
меризовать изобутсп. Реакция отличалась многими особенностя-
ми, характерными для ионных механизмов. После этого' реакции
сополимеризации стали использовать для выявления вклада
иопных механизмов в полимсризацпопный процесс.
11.12. ИОННАЯ И ЧАСТИЧНО ИОННАЯ
ПОЛИМЕРИЗАЦИЯ В ЖИДКОЙ ФАЗЕ
Радиационная полимеризация изобутилена, которую мы рас-
смотрим в качестве примера, обладает следующими особенно-
стями:
1. При 0° и выше процесс идет в незначительных масштабах.
2. При —80° С реакция протекает сравнительно быстро.
3. При низких температурах ингибируется диизобутсном, ко-
торый известен как ингибитор иопных реакций.
4. Скорость полимеризации прямо пропорциональна интен-
сивности облучения при низких температурах и пропорциональ-
на (/)^2 при 20° С.
5. Кислород и бензохинон при низких температурах ингиби-
руют полимеризацию; окись цинка ускоряет процесс, а дифе-
нилпикрилгидразил никак па него по влияет.
6. Идентифицированы положительные ионы изобутена.
7. Фотополимеризация при 150° С происходит при освеще-
нии ультрафиолетовым излучением с длиной волны 1470 А
(8,44 эв). Если же излучение имеет длину волны 1600 А, реакция
не идет. Этот эксперимент не только свидетельствует в пользу
справедливости ионного механизма, но также показывает, что
потенциал ионизации мономера в жидкой фазе лежит по край-
ней мере на 0,8 эв выше, чем в газовой фазе, где достаточно
излучения с длиной волны 1600 А, чтобы инициировать газо-
фазную полимеризацию.
8. Состав сополимеров, образующихся из смеси изобутилена
с винилиденхлоридом, а также из других мономерных смесей,
сильно зависит от температуры (рис. 11.11). При низких темпе-
ратурах, когда преобладают ионные механизмы полимеризации,
в составе сополимера обнаруживается больше изобутилена.
9. Кажущаяся энергия активации равна 14 ккал!моль при
комнатной температуре и нулю при —78° С.
10. При низких температурах полярные растворители, такие,
как хлористый этил, меняют состав сополимера, в результате
чего он обогащается ионными компонентами.
12 Зак. 907
353
Указанные особенности свидетельствуют в пользу двойствен-
ной ионно-радикалыюй природы радиационного катализа. При-
ближенный кинетический анализ схемы анионной полимериза-
ции [см. уравнения (11.13) — (11.17)] позволяет вывести сле-
дующие уравнения:
/<= Rt = kt [МИН?] ; (11.58)
г 1 Ri [М]
R Rp = kp [MW] [М] = Р-~- • (11.59)
В первом приближении из этих уравнений следует: 1) ско-
рость полимеризации пропорциональна первой степени мощно-
,сти дозы; 2) энергия актива-
Рис. 11.11. Состав сополимеров, об-
разующихся при различных темпера-
турах, в зависимости от состава на-
чальной мономерной смеси изобути-
лен—винилиденхлорид.
разующиеся электроны обычно
ци процесса отрицательна, так
как единственной константой,
для которой может быть ха-
рактерна заметная энергия ак-
тивации, . является kr, 3) ско-
рость реакции должна быть
выше при низких температу-
рах. Последний вывод, разуме-
ется, — следствие вывода 2.
Некоторые акцепторы сво-
бодных радикалов могут инги-
бировать реакцию полимериза-
ции, в то время как добавле-
ние окиси цинка ускоряет ре-
акцию. Для объяснения этой
аномалии Коллисон, Дейнтон
и Жиллис предположили, что
в первичном акте происходит
ионизация изобутена и соответ-
ственно возникают положи-
тельный ион и электрон. Об-
захватываются стенками сосуда,
что уменьшает скорость рекомбинационных процессов. При до-
бавлении ZnO в систему скорость захвата электронов увеличи-
вается, поэтому увеличивается и скорость реакции полимериза-
ции, протекающей по ионному механизму. С другой стороны,
отрицательные ионы, образующиеся при захвате электронов
кислородом и бензохиноном, могут диффундировать в раствор,
рекомбинировать с положительными ионами и подавлять реак-
цию полимеризации. Добавление ДФПГ не должно подавлять
полимеризацию, так как, но-видимому, первичный процесс-
взаимодействие ДФПГ с образующимися мстилзамещенпыми
алкильными радикалами [5].
354
11.12.1. Влияние мощности дозы
и концентрации мономера
на ионную полимеризацию
Концентрация мономера и ин-
тенсивность излучения — важные
параметры, определяющие отно-
сительный вклад ионных и ради-
кальных механизмов полимериза-
ции. Рис. 11.12 демонстрирует
влияние концентрации мономера
на скорость полимеризации акри-
лонитрила, растворенного в эти-
лене при —78° С. При увеличе-
нии мольной доли мономера ско-
рость полимеризации вначале
уменьшается, а затем возрастает.
Добавление пирогаллола ингиби-
рует полимеризацию только при
высоких концентрациях мономе-
ра. Это позволяет предположить,
что при высоких концентрациях
мономера действует радикальный
механизм полимеризации, а при
низких концентрациях — ионный.
Данные инфракрасной спектро-
скопии согласуются с этим пред-
положением. При увеличении кон-
центрации мономера увеличивает-
ся число C = N связей и умень-
шается число связей C=N. В
разбавленных растворах, очевид-
но, происходит анионная полиме-
ризация групп C = N, а в кон-
центрированных — радикальная
полимеризация групп С=С. В
Концентрация акрилонитри-
ла ,og %
Рис. 11.12. Влияние концен-
трации акрилонитрила на ско-
рость полимеризации при
—78° С. Мощность дозы 7 X
Х104 р/ч. Полная доза соста-
вила 6,4-106 р. Показано так-
же влияние добавок пирогал-
лола на полимеризацию. (Л —
при добавлении пирогаллола.)
предельных случаях строение образующихся полимеров будет
выглядеть следующим образом:
сн. радикальная ПОЛИМС1 нзация
СН — СП.,—СН—СП,—СН -СН,—СП—СН.,—сн 1 11*11 (11.60)
1 1111 CN CN CN - CN CN ионная полимеризация СН2=СН — C=N-C--Nr—C-N-C N-C (11.61)
। । р । j । CN СН СН СН СИ СН 1 1 II СН, СН2 СН2 СН, СН2 12* 355
Влияние интенсивности излучения сказывается постольку, по-
скольку скорость радикальной реакции пропорциональна корню
квадратному из мощности дозы, тогда как скорость ионной ре-
акции пропорциональна первой степени дозы. Следовательно,
’низкие интенсивности излучения будут благоприятствовать ра-
дикальной полимеризации, а высокие — ионной. Этот вывод экс-
периментально подтверждается для полимеризации стирола,
акрилонитрила и изобутилена при низких температурах.
11.12.2. Полимеризация в твердой фазе
Ошима и Табата выделяют три механизма полимеризации
в твердой фазе:
1) радикальный механизм, где полимеризация мономеров
происходит при сравнительно высоких температурах;
2) полимеризация при низких температурах, отличающаяся
от ионной или радикальной полимеризации и требующая уча-
стия в процессе возбужденных молекул;
3) полимеризация с размыканием кольца мономера.
Примеры всех трех возможных механизмов дает табл. 11.2.
Таблица 11.2
Полимеризация в твердой фазе
Мономер Температу- ра плавле- ния, °C Температура облучения, °C Энергия акти- вации, ккал/моль Механизм
Акриламид 83 о, 80 4 1
Акриловая кислота 12 24 1
Винилкарбазол 64 20 60 12 1
Цетилметил акрилат 15 —21 —78 0,7 1
Акрилонитрил —83 —83, —196 От 3 до 1 2
Стирол —51, —78 9,2 2
Бутадиен —30 —100, —196 0,1 2
2-Метил-5-вииилпиридин —13 —13, —196 0,3 2
Винилацетат —93 _ 196 0 2-
Изопрен —120 —120, —196 0,2 2
Формальдегид —118 —118, —160 2,2 3
0-Пропиолактон —33,4 —33, —70 —12,6 3
Г ексаметилциклотрисилоксан 64 64 0 9,6 3
Дикетен —6,5 —6,5 —130 —2,4 3
—130, —196 0,11
Оксистирол — —80, —120 0,1 3
Наши сведения о процессах полимеризации в твердой фазе
остаются пока крайне неполными. До сих пор не существует
признанной теории, которая смогла бы объединить разрознен-
ные экспериментальные факты. К числу существенных парамет-
ров, определяющих скорость полимеризации в твердой фазе и
356
характеристики образующегося полимера, следует отнести струк-
туру и размеры кристаллов, присутствие добавок и степень кон-
версии мономера в полимере.
В ряде случаев в заметных количествах образуются стерео-
специфические полимеры. Это может служить указанием на то,
что процесс роста цепи определяется ориентацией мономеров
в кристаллической решетке. Высокий выход стереоспецифичного
полимера наблюдался в случае полимеризации бутадиена и
акрилонитрила, но не акриламида или близких ему мономеров.
По-видимому, при более высоких температурах [см. механизм 1]
рост цепи определяется геометрической структурой кристал-
ла мономера только па начальных стадиях полимеризации. Да-
лее процесс полимеризации происходит па поверхности раздела
между кристаллическим мономером и аморфным полимерным
материалом.
Если полимеризация происходит по механизму (2), удается
обнаружить совершенно неожиданное влияние добавок. Часто
добавки изменяют структуру полимера. Акцепторы радикалов,
ингибирующие полимеризацию в жидкой фазе, почти не оказы-
вают влияния на твердофазную полимеризацию. С другой сто-
роны, целый ряд соединений, полностью подавляющих твердо-
фазную полимеризацию, не оказывает никакого влияния в
жидкой фазе. Например, для изопрена можно обнаружить три
различных кинетических механизма полимеризации в зависимо-
сти от температуры и агрегатного состояния мономера. В жид-
ком состоянии при высоких температурах наблюдается «класси-
ческая» свободнорадикальная полимеризация. При низких
температурах полимеризация приобретает ионный характер.
В твердом состоянии при температуре выше температуры фазо-
вого перехода (—158° С) эксперименты с акцепторами, а также
эксперименты по влиянию мощности дозы указывают на то, что
реакция может иметь ионный характер. Но при температуре
ниже —158° С сразу появляется множество неожиданных эффек-
тов, включая изменение структуры самого полимера. Согласно
одному из возможных объяснений, происходит коллективное
возбуждение мономерного кристалла как целого. В N мономе-
рах возникает коллективная волна возбуждения, и все эти N
возбужденных мономеров мгновенно полимеризуются, образуя
макромолекулу со степенью полимеризации N. Табата и со-
трудники называют такой процесс «электронной полимериза-
цией».
Время возникновения волны возбуждения должно быть по-
рядка 10~14 сек, и весь процесс полимеризации завершится все-
го лишь за 1СН1 сек.
Согласно менее изящному, но отнюдь не менее правдоподоб-
ному механизму, перенос энергии при полимеризации в кри-
сталле происходит в виде ступенчатого процесса от одного ато-
ма к другому.
857
11.13. ПОЛУЧЕНИЕ ПРИВИТЫХ СОПОЛИМЕРОВ
Привитые или блок-сополимеры обычно образуются в резуль-
тате полимеризации мономера А в присутствии полимера Вт.
Если возникающий полимер А„ оказывается присоединенным к
полимеру Вт, то говорят, что образовался привитой (графт)
сополимер AnBm. Как показывает табл. 11.3, в которой перечис-
лены радиационно-привитые полимерные волокна, полученные
вплоть до 1967 г., уже исследовано очень много систем.
Для получения блок- или графт-сополимера на полимерном
остове должен быть инициирован процесс полимеризации. С этой
целью используют два способа:
1) прямое облучение системы, состоящей из мономера и по-
лимера с более высокой радиационной чувствительностью;
2) добавление мономера к предварительно облученному по-
лимеру, в котором имеются либо захваченные свободные ради-
калы, либо перекисные группы.
Схема получения привитых и блок-сополимеров по способу
2) совпадает со схемой 1) с тем лишь единственным различием,
что мономер А добавляется в систему после облучения полимера
(в отсутствие воздуха). При способе 1), когда облучается смесь
мономера и полимера, кроме привитого сополимера может об-
разовываться также гомополимер. В первом приближении отно-
шение гомополимера к сополимеру определяется отношением
G, к G&. К числу прочих факторов, определяющих значение
отношения гомополимера и сополимеру, относятся скорость диф-
фузии, растворимость различных компонент, положение рав-
новесия реакции полимеризации, присутствие ингибиторов
или растворителей, мощность дозы и параметры окружающей
среды.
Как будет показано далее, кинетика привитой сополимериза-
ции гораздо сложнее, чем кинетика полимеризации в гомоген-
358
Таблица 11-3*
Радиационно-привитые полимерные волокна
Тип полимерного волокна Прививаемый мономер Источник излучения
Нейлон-6 Полиамидные волокна. (Агрегатное состояние мономера—газ) Ацетилен, акрилонитрил, акриловая кислота, бутадиен, метил метакриловая кислота, фенил- ацетнлен, . пропаргиловый спирт, пропилен, сти- рол, винилацетат Акрилонитрил, этилен, метилметакрилат, про- «°Со
Нейлон-6 (капрон) Рентге-
Нейлон-6 пилен Випилпирролидон новская трубка (80 кв, 200 ма) »°Со
Нейлон-6 (капрон) Стирол »
Нейлон-6 Стирол »
Нейлон-6 (капрон) Акрилонитрил »
Нейлон-6 Акрилонитрил »
Нейлон-6 Метакриловая кислота »
То же Акриловая кислота, малеиновая кислота »
» Акрилонитрил, бутадиен, этилакрилат, метил- »
Нейлон-66 акрилат, винилацетат Акриловая кислота, акрилонитрил, аллнлакри- в0Со
Полиэтилеитере- лат, бутадиен, 1,3-бутилен, диметакрилат, диви- нилсульфон, этилен, этилакрилат, этилендимета- крилат, метилметакрилат, пропилен, стирол, винилацетат, винилхлорид, н-винилпирролидон, этилендиметакрилат-метилакрилат, этилепдимета- крилат-акриловая кислота, 1,3-бутилен-димета- криловая кислота Полиэфирные волокна 4-Винилпирролидон, метакриловая кислота »
фталат (терилен) Полиэтилен тере- Стирол »
фталат (лавсан) Полиэфир Акриловая кислота, акрилонитрил, бутадиен,
Целлюлоза дивинил сульфон, этилакрилат, этилен, метила- крилат, метилметакрилат, пропилен, стирол, ви- нилацетат, винилхлорид, н-винилпирролидон Целлюлозные волокна Метилакрилат, Метилметакрилат, винилацетат »
То же Стирол в0Со
» Поливиниловый спирт (винилацетат) »
« Акрилонитрил, метакриламид »
» Метилакрилат »
Акрилонитрил, метилакрилат, метилметакри- £
лат, стирол, винилацетат
* В таблице указаны системы, изученные в 115 работах, которые цитируется в обзоре:
D. Gilbert R., Stannett V.Isotopes Rad. Tech., 1967, v. 4, No 4
359
Продолжение табл. 11.3
Тип полимерного волокна Прививаемый мономер Источник излучения
Целлюлоза Акрилонитрил-стирол, метил метакрилат-стирол метил метакрилат-акрилонитрил, метил метакри- лат-а-метил стирол, акрилопитрил-а-мстнлстирол, винилацетат-стирол воСо
» Метакриловая кислота »
Целлюлоза (хлопок) Акрилонитрил, бутадиен, этилакрилат, мети- лакрилат, винилацетат »
То же Акриловая ки.:,-.ота, акри.-.ожпри;, алли.та- крилат, бутадиен, дивинилсульфон,этилен, этила- крилат, метилакрилат, метилметакрилат, пропи- лен, метакриламид, стирол, винилацетат, ви- нилхлорид, н-винилпирролидон »
» Акрилонитрил, стирол (Мономер находится в растворе)
Стирол »
» Акрилонитрил, метилметакрилат, винилацетат »
» Стирол воСо
» Акрилонитрил, стирол, мсатилметакрилат, стирол-акриловая кислота, стиролметилмстакри- лат, акрилонитрил-метил-метакрилат »
» Метакриловая кислота »
» Метакриламид, метилакрилат »
» Бутадиен-стирол, бутадиеп-акрилонитрил, сти- рол-акрилонитрил, бутадиен—стирол—акрило- нитрил £
» Бутадиен, бутадиен-стирол, бутадиен—акри- лонитрил 4 в
» Акрилонитрил, а-мети летирол, винилиденхло- рид, винилтолуен »
» Акрилонитрил, метакриламид, метакриловая кислота, метилметакрилат, стирол »
> Акрилонитрил, бутадиен, этилакрилат, метила- крилат »
Целлюлоза (виско- Стирол Ядериый
за) реактор
То же Стирол в°Со
» Стирол, акрилонитрил В
» Стирол 60Со
» Винилп ирролидон В
У> Бутадиен, бутадиен-стирол, бутадиеи-акрило- иитрил »
> Акриловая кислота, акриламид, аллилакрилат, будатиеп, 1,3-бутилен, диметакрилат, дивинил- сульфоп, этилен, этилакрилат, этилендиметакри- лат, 2-этилгексилакрилат, метилакрилат, метил- метакрилат, пропилеи, стирол, винилацетат, ви- нилхлорид, н-винилнирролидоп, аллилакрилат— акриловая кислота, 1,3-бутилсндимстакрилат-ак- риловая кислота, этилендимстакрилат-акрило- вая кислота, этилендиметакрилат-метилакрилат Акрилонитрил, бутадиен »
Ацетилцеллюлоза »
360
Продолжение табл. 11. 3
Тип полимерного волокна П рививасмын мономер Источник излучения
Ацетилцеллюлоза Стирол иСо
То же Акриловые эфиры »
> Акрилонитрил—стирол, метилметакрилат—ак- рилонитрил, метилметакрилат—стирол, винил- ацетат—стирол, а-метилстирол—акрилонитрил, а-метил стирол—метилметакрилат
» Стирол »
» Метилметакрилат »
» Акриловая кислота, акрилонитрил, аллилакри- лат, бутадиен, 1,3-бутилепдиметакрилат, диви- нилсульфон, этилен, этилакрилат, этнлендиме- такрилат, метилакрилат, метилметакрилат, про- пилеи, стирол, винилацетат, винилхлорид, н-ви- пнлнирролидон, аллилакрнлат—акриловая кисло- та, этилендиметакрилат—акриловая кислота, 1,3-бутилендиметакрнлат— акриловая кислота »
Полиолефиновые волокна
Полиэтилен Стирол »
То же Бутадиен »
» Винилхлорид »
» Метилметакрилат, стирол, винилиденхлорид, дихлорэтилен, трихлорэтилен, тетрахлорэтилен »
Акрилонитрил, этилен, метилметакрилат, про- »
пилен
» Метакриловая кислота »
» Акриловая кислота, акрилонитрил, ацетилен, бутадиен, этилен, метилметакрилат, фенилацети- леп, пропилен, пропаргиловый спирт, стирол, »
винилацетат
» Полипропилен Акриловая кислота, акрилонитрил, 2-метил- 5-ВИНИЛПИрИДНН »
Акрилонитрил, бутадиен, этилакрилат »
То же Бутадиен »
» Акриловая кислота, метилметакрилат »
» Г л ицидилметак рилат »
> Стирол »
» Акриловая кислота »
» Винилпирролидон, винилацетат »
Акрилонитрил, этилен, метилметакрилат, про- пилен^ йя »
Стирол—малеиновая кислота, стирол—фумаро- вая кислота, стирол—диэтилфумарат, стирал— Ядерньг" реактор
итаконовый ангидрид, стирол—малеиновый ан- (для об
гидрид, винилацетат—малеиновый ангидрид, ак- лучения
рилоиитрил—малеиновый ангидрид, 2-метил-5-ви- а-части-
нилпиридинмалеиновый ангидрид цами ис пользова- ли реа -
ЦП ю (л, а)1
361
Продолжение табл. 11.3
Тип полимерного волокна Прививаемый мономер Источник излучении
Полипропилен Ацетилен, акриловая кислота, акрилонитрил, бутадиен, этилен, метилметакрилат, феиилацети- лен, пропаргиловый спирт, пропилен, стирол, винилацетат 6ССо
» Акриловая кислота, акрилонитрил, 2-мстил- 5-винилпиридип »
» Акриловая кислота, аллилакрилат, бутадиен, 1,3-бутилендимстакрилат, дивинилсульфон, эти- лен, этилакрилат, этилендиметакрилат, метилак- рилат, метилметакрилат, пропилен, -стирол, ви- нилацетат, винилхлорид, н-випилпирролидоп Шерстяные волокна »
Шерсть Акрилонитрил, бутадиен »
То же Акрилонитрил, метилметакрилат, стирол »
> Акрилонитрил, аллилацетат. аллилбснзоат, аллилстеарат, стирол
» Акриловая кислота, акрилонитрил, бутадиен, дивинилсульфон, этилакрилат, этилен, метилак- рилат, метилметакрилат, пропилен, винилацетат, винилхлорид, к-вииилпирролидои Смешанные волокна »
Поливинилхлорид Глицидилметак рилат »
» 4-Винилпиридин »
Джут Стирол
Цианоэтилирован- ный хлопок Акрилонитрил »
Акриловое волокно Метакриловая кислота »
То же Акриловая кислота, акрилонитрил, бутадиен, дивинилсульфон, этилакрилат, этилен, метилак- рилат, метилметакрилат, пропилен, винилацетат, винилхлорид, н-винилпирролидон »
ных системах. Система, в которой протекает привитая сополи-
меризация, обычно обладает повышенной вязкостью, и скорость
реакции может определяться скоростью диффузии. Более того,
в зависимости от степени растворимости полимера, интенсивно-
сти излучения и значения коэффициентов диффузии прививка
может происходить не в объеме, а только на поверхности. Го-
могенная привитая сополимеризация происходит в объеме.
Полимеры, несущие перекисные группы, образуются в ре-
зультате предварительного облучения полимера в присутствии
кислорода. Могут возникать либо диперекисныс соединения,
либо гидроперекиси. В любом случае перекиси затем разла-
гаются под действием тепла, и в присутствии мономера обра-
зуется привитой сополимер.
Экспериментов по привитой сополимеризации с предвари-
тельным инициированием (за счет захваченных свободных ра-
.362
дикалов или перекисей) было выполнено сравнительно мало.
Способ, при котором используются захваченные свободные ра-
дикалы, труден и сложен, так как из системы нужно удалить
любые акцепторы радикалов (в частности, кислород), а моно-
мер необходимо ввести немедленно после облучения. С другой
стороны, инициирование с помощью гидроперекисей приводит
к образованию в заметных количествах гомополимера, так как
при нагревании низкомолекулярные свободные радикалы диф-
фундируют в объем и взаимодействуют с мономерами.
В В
Константы скорости для отдельных стадий привитой поли-
меризации существенно отличаются от тех, которые характерны
для радиационной полимеризации в растворах. Наибольшие
различия отмечаются для константы скорости обрыва цепи kt.
Наиболее ярко это продемонстрировал Стейнберг, измерявший
скорость привитой полимеризации стирола на полиэтилене при
непрерывном и прерывистом воздействии у-излучением 60Со.
На рис. 11.13 показана скорость привитой полимеризации в
зависимости от мощности дозы. «Правильная» кинетика, соот-
ветствующая закону квадратного корня [см. уравнение (11.23)],
сохраняется вплоть до мощности дозы 200 крад/ч. Далее неза-
висимо от мощности дозы скорость реакции остается примерно
постоянной. В этой области, как будет показано ниже, скорость
привитой полимеризации определяется скоростью диффузии.
363
Для определения констант скорости kv и kt использовали
метод облучения через вращающийся сектор. На рис. 11.14 при-
веден график, полученный в результате численного решения
уравнения (11.44) при подстановке периодической функции вре-
мени соответствующей
(экспериментальным условиям
Стейнберга. Отсюда, как показа-
но в примере 11.3, можно найти
среднее время жизни радикалов
и константы скорости kp и kt.
Скорость привитой полимеризации, %/ч
Рис. 11.13. Скорость привитой поли-
меризации в зависимости от мощ-
ности дозы.
Рис. 11J4. Зависимость отношения
скоростей реакций Ri(t)/Ris при пре-
рывистом и стационарном иницииро-
вании от периода облучения и вре-
мени жизни радикалов.
Пример 11.3. На рис. 11.15 приведен график зависимости скорости при-
витой полимеризации от периода прерывистого облучения для системы сти-
рол— полиэтилен. Скорость привитой полимеризации в стационарных усло-
виях дается графиком рис. 11.13. Решение уравнения (11.44) для данного
вила функции Ri(t) приведено па рис. 11.14. Найдите среднее время жизни
7 7(/ 1UU 1UUU
Период обяучения/вреня жизни
радикалов, г
Рис. 11.1b. Скорость привитой поли-
меризации в зависимости от периода
нестационарного облучения.
радикалов и константы скорости
kp и kt, принимая, что справедли-
во уравнение (11.42), а макси-
мальная мощность дозы прерыви-
стого облучения составляет
49,8 крад)ч.
Решение. По графику рис. 11.13
можно определить, что для макси-
мальной интенсивности излучения
экстраполированная стационарная
скорость привитой полимеризации
будет равна 8,7% конверсии при-
виваемого мономера за час. Со
гласно графику рис. 11.14 скорость
реакции при высоких частотах об-
лучения должна составлять 44%
от скорости реакции в стационар-
ных условиях. Это дает 3,8% кон-
334
версии в 1 ч. Реально измеренное значение составляет ~4% конверсии в 1 ч,
чем демонстрируется хорошее соответствие между теорий и экспериментом.
Среднее время жизни радикалов можно определить, подсчитав среднюю
величину горизонтального сдвига между кривыми рис. 11.14 и 11.15 после
нормировки ординат обеих кривых. На теоретической кривой можно отметить
точку, для которой период облучения и время жизни радикалов совпадают.
Если теперь мы перенесем эту точку иа экспериментальную кривую, то соот-
ветствующий период облучения будет равен среднему времени жизни ради-
калов (34 сек).
Рис. 11.16. Графическое решение
примера 11.3.
После этого можно определить значения констант скоростей в соответ-
ствии с уравнениями (11.23) и (11.25)
/?( = ФС^(н(М)/Р)[М]. (2)
Если в уравнения (1) и (2) подставить соответствующие значения кон-
центраций и скоростей, то мы получим, что Apfe^_,^2=[2700 моль[(л-ч)]~
Среднее время жизни радикалов равно числу присутствующих в системе ра-
дикалов, деленному на скорость их образования (или исчезновения)':
' = (kt Ri)~l,t- (3)
Из уравнения (3) найдем, что £*=1,1-10* (моль/л)~,сек-1. Ниже для
сравнения приведены константы скорости для гомополимеризации и привитой
полимеризации
(моль/л) 1сек 1 (моль/л) 1сек
Гомополимеризация........................... 55 2,5-107
Привитая полимеризация ..................... 91 1,1-10*
Константы скорости роста цепи оказываются примерно одинаковыми.
Однако константа скорости обрыва цепи для привитой полимеризации со-
ставляет около одной тысячной значения коистаиты скорости обрыва цепи
для гомополимеризации. Этот результат может служить серьезным свиде-
тельством в пользу того, что в привитой полимеризации участвуют мало-
подвижные радикалы, рекомбинирующие в медленных бимолекулярных реак-
циях.
365:
11.13.1. Привитая полимеризация.
Влияние диффузии мономеров
Как видно из рис. 11.13, при высоких интенсивностях излу-
чения скорость привитой полимеризации перестает быть про-
порциональной корню квадратному из интенсивности облучения.
Этот факт, а также некоторые эксперименты, в которых приви-
тую полимеризацию проводили при различной толщине поли-
мерной пленки, заставляют предположить, что в этих случаях
скорость реакции определяется диффузионными процессами.
Тогда, очевидно, можно написать уравнение, связывающее друг
с другом концентрацию мономера внутрй полимера С, коэффи-
циент диффузии мономера D и константы, характеризующие
скорость привитой полимеризации [см. уравнение (11.23)]:
= D - kp (Rjk^ С. (11.62)
dt — дх2
В уравнении (11.62) первый член справа характеризует ско-
рость диффузии мономера в полимер, а второй-—скорость при-
витой полимеризации. Решение этого уравнения, полученное
Чэндлером, Хенли и Трахтенбергом [6], выглядит следующим
образом:
= 2 zW[exp{_(а+^)0-1]. (11.63)
л=1,3,5,...
Здесь Q — количество прививаемого мономера на единицу объ-
ема; a=n2n2D/l; I-—толщина полимерной пленки; К =
=kP(Ri/kt)'/2- ~
Уравнение (11.63) содержит как стационарный, так и неста-
ционарный член и предсказывает зависимость скорости приви-
той полимеризации от следующих параметров:
1) толщины полимерного образца;
2) значения коэффициента диффузии мономера в полимере;
3) уровня насыщающей концентрации мономера в полимере;
4) интенсивности излучения;
5) значения констант скоростей различных стадий реакции
привитой полимеризации;
6) температуры, поскольку она влияет на величину констант
скоростей и коэффициента диффузии.
Хотя в ряде случаев уравнение (11.63) удается использовать
достаточно успешно, для полного описания радиационной при-
витой полимеризации нужно учесть такие факторы, как физи-
ческое и химическое строение мономера и полимера.
366
11.14. ВЛИЯНИЕ ПРИМЕСЕЙ
Непропорционально большое влияние малых концентраций
примесей представляет собой одну из основных трудностей при
изучении цепных реакций вообще и реакций полимеризации в
частности. Нередко даже после очень высокой очистки системы
экспериментальные результаты плохо воспроизводятся. Стироль-
ные системы в этом смысле тоже не составляют исключения.
Работы по выяснению причин такой невоспроизводимое™ рас-
смотрены в обзрре Меца [7]. Отмечено, что удаление следовых
количеств воды может резко ускорить полимеризациопный про-
цесс. Органические реакции обладают разной чувствитель-
ностью к присутствию воды. Наиболее чувствительны реакции,
в которых участвуют карбанионы, менее чувствительны реакции
с участием карбониевых ионов, еще менее чувствительны сво-
боднорадикальные реакции. Поэтому ускорение реакции при
удалении воды из системы, содержащей виниловые мономеры,
можно объяснить либо усилением радикальных реакций, либо
их заменой на реакции с участием карбониевого иона. Дальней-
шее ускорение полимеризации при низком уровне содержания
воды можно объяснить переходом на карбанионный механизм.
Следует подчеркнуть, что все эти предположения имеют пока
предварительный характер.
ЗАДАЧИ
11.1. Если в смеси мономеров М, и М2 отношения г, и г2, характеризую-
щие предпочтительность взаимодействия радикала со сходным по строению
мономером, равны 2 и 0,5 соответственно, найдите зависимость первоначаль-
ного состава сополимера от относительной концентрации мономеров в смеси.
11.2. Выведите уравнение для степени полимеризации при высоких мощ-
ностях дозы.
11.3. Пользуясь приведенными ниже в табл. 1 и 2 данными для радиа-
ционной блочной полимеризации метилметакрилата, определите вид кинети-
ческого уравнения полимеризации. Рассчитайте выход радикальных продук-
тов общую энергию активации. При расчете выхода радикальных
продуктов примите, что происходит как рекомбинация, так и диспропорцио-
нирование радикалов.
Таблица! Таблиц а 2
Скорость полимери- зации [моль, (лсек)]Х XI 0е Мп ,0~5 Мощность .дозы, рад/сек Мощность дозы, рад/сек Темпе- ратура, °C Скорость полимери- зации R, [моль/(я X Хсек)]- 10* Мп-10~8
3 20 5-Ю-з 68,5 —18 72,5 70
8 12 5-10-2 64,5 25 211 162
20 8 5-10-1 63,4 70 499 260
70 4 5 0,77 30 44,5 340.
150 2 50 0,77 50 64 510
250 1,5 300 0,77 70 104 670
367
11.4. Получите уравнение (11.43) в форме, удобной для расчета выхода
радикальных продуктов при: 1) полимеризации в растворе, 2) рекомбинации
и диспропорционировании радикалов, 3) высоких мощностях дозы, 4) пере-
даче цепи на мономер, 5) передаче цепи па полимер.
11.5. По данным рис. 11.11 подтвердите справедливость приведенных ниже
в таблице численных значений отношений и и г2, если Mi — это изобутилен,
а М2 — винилхлорид.
Г1 г2
—78’ . ..............25 О
—40°................. 1,27 0,21
0°...................0,03 1,3
11.6. Рассчитайте выход радикальных продуктов при радиационной поли-
меризации метилметакрилата, используя приведенные в таблице данные.
Мощность дозы, рад/сек-10—5 Скорость полимериза- ции Я, моль/(л-сек) X хю« При условии 100%-ной рекомбинации радикалов При условии - 5 0% - ной рекомбинации и 50%-пой диспропорционирования радикалов
0,2 2.9 kt/k2p - 180 {klc + ktd)lk2 -- 120
1,1 5,9 М^-180 + 120
4,0 6,6 kt/k2 - 180 120
11.7. Как будет выглядеть механизм ионной и свободнорадикальной по-
лимеризации акрилонитрила, растворенного в этилене при —78° С?
Глава XII
ДЕЙСТВИЕ ИЗЛУЧЕНИЙ НА ПОЛИМЕРЫ
Благодаря высокому молекулярному весу полимеров уже"
сравнительно низкие дозы радиации могут приводить к их су-
щественным физическим изменениям. Допустим, что значение G
для какой-то радиациопно-ипдуцированной реакции будет рав-
но 5. Тогда число химических изменений в образце с единичной
плотностью при дозе в 1 Мрад составит примерно 3,2-1018.
Если молекулярный вес вещества образца равен 100, то хи-
мические изменения будут наблюдаться в 0,05% молекул. По
если молекулярный вес равен 106, то в каждой молекуле образ-
ца в среднем произойдет по пять химических изменений. В тех
случаях, когда подобные химические изменения представляют
собой разрывы валентного остова, будет наблюдаться шести-
кратное снижение молекулярного веса полимера. Если же обра-
зуются межмолекулярные сшивки, то молекулярный вес должен
возрасти в 6 раз. При этом образец начинает приобретать трех-
мерную сетчатую структуру.
Приведенный пример, кроме того, показывает, что полимеры
можно разделить на две группы: у одной молекулярный вес
возрастает при облучении, у другой — уменьшается. Первые
можно назвать сшивающимися, а вторые — дсструктирующими
полимерами. В табл. 12.1 перечислены полимеры обеих
групп.
Согласно эмпирическому правилу, которое достаточно хоро-
шо себя оправдывает, полимеры типа (—СИ2—CIIR—)л преи-
мущественно сшиваются, тогда как для полимеров типа
(—СП2—CCH3R—)п преобладают дсструкниоппыс процессы.
Поливиниловый спирт, однако, может служить примером несоб-
людения этого правила. Скорее всего в основе правила лежит
тот факт, что в дсструктирующих. полимерах тетразамещепный
атом С испытывает большие стерическис напряжения. В общем
случае деструктирующие полимеры отличаются высоким- выхо-
дом мономеров при пиролизе в вакууме и обладают низкими
стандартными теплотами образования.
369-
Таблица 12.1*
Действие излучений иа полимеры
.Преимущественно сшивающиеся Преимущественно дсструктмрующмс
полимеры полимеры
Полиэтилен
Полипропилен
Поливинилхлорид
Хлорированный полиэтилен
Хлорсульфонированный полиэтилен
Полиакрилонитрил
Полиакриловая кислота
Полиакрилаты
Полиакриламид
Поливинилпирролидон
Поливинилалкильные эфиры
Поливинилметилкетопы
Полистирол
Сульфонированный полистирол
Природный каучук
Синтетический каучук (кроме полиизо-
бутилепа)
Полисилоксаны
Полиамиды
Полиоксиэтилен
Полиэфиры
Полиизобутилен
Пол ивинилиденхлорид
Полимонохлор трифторэтилен
Политетрафторэтилен
Поли-а-метакрилопитрил
Полиметакриловая кислота
Полиметакрилаты
Поли метакрил амид
Поли-а-метилстирол
Полиэтилентерефталат
Целлюлозные пластмассы
* Из книги: Swallow. Radiation Chemistry of Organic Compounds, New York, Pergamon
Press, 1960.
Химические изменения в облученных полимерах и в их низ-
комолекулярных аналогах весьма сходны по своей природе.
'Сшиванию и деструкции на уровне мономеров соответствуют
конденсация и разрыв связей. Зная поведение мономеров, с до-
статочной надежностью можно предсказать и другие эффекты
при облучении полимеров, такие, как газовыделение, образова-
ние двойных связей, окисление, возникновение центров окраски
и т. д. Например, зная поведение циклогексана при облучении,
можно предсказать, какие продукты будут образовываться при
радиолизе полиэтилена. Если же в выходе и характере продук-
тов радиолиза высоко- и низкомолекулярной систем наблю-
даются различия, то их обычно можно объяснить эффектом
клетки и другими эффектами, характерными для твердых тел.
Следует также отметить, что полимеры вообще (и особенно
промышленные образцы) следует считать по меньшей мерс за-
грязненными различными примесями. В худшем случае образец
представляет собой смесь молекул с различной длиной цепи,
с добавками пластификаторов, антиоксидантов, формовочных
агентов, остатков катализатора и прочего. По этой причине
большая часть известных данных по действию излучений на
промышленные полимеры имеет незначительную научную цен-
ность.
370
В этой главе мы рассмотрим действие излучений па поли-
этилен и полимстйл метакрил ат, сшивающийся и деструктирую-
щий полимеры. Поведение этих полимеров при облучении ти-
пично во многих отношениях, и сделанные выводы могут быть
распространены на большое число сходных систем.
12.1. ДЕЙСТВИЕ ИЗЛУЧЕНИЙ НА ПОЛИЭТИЛЕН
Полиэтилен (ПЭ), несмотря на видимую простоту химиче-
ского строения (лишь обычные СН2-группы), обладает доста-
точно сложной структурой. ПЭ может быть преимущественно
кристаллическим или преимущественно аморфным, разветвлен-
ным или неразветвленным. Размеры и характер кристалличе-
ских областей будут определяться степенью ветвления. Образны
с более высокой степенью кристалличности имеют большую
плотность и меньшую степень ветвления. Такие образцы полу-
чают при низких давлениях каталитическим способом с помо-
щью процесса Циглера. С другой стороны, полимеризация в га-
зовой фазе при высоких давлениях позволяет получать образцы
ПЭ с низкой платностью. Степень ветвления в таких образцах
заметно выше, они менее кристалличны и имеют точку плавле-
ния около 115° С, что заметно отличается от температуры плав-
ления образцов ПЭ с высокой плотностью, которая равна 135° С.
А. Чарлсби, впервые охарактеризовавший процессы радиа-
ционного сшивания, указывает на следующие основные явления,
происходящие при облучении полиэтилена: 1) выделение водо-
рода и низкомолекулярных углеводородов; 2) образование С—С
связей между молекулами (димеризация или сшивание); 3) уве-
личение ненасыщенности; 4) уменьшение степени кристаллич-
ности; 5) изменение окраски (как и многие другие полимеры,
полиэтилен при облучении приобретает желтоватый оттенок);
6) окислительные реакции (особенно вблизи поверхности), ко-
гда облучение проводят в присутствии кислорода.
Все эти эффекты приводят к такому преобразованию физи-
ческих и химических свойств полиэтилена, что он зарегистриро-
ван в патентном бюро как неизвестный ранее новый мате-
риал — облученный полиэтилен.
Преобразование полиэтиленового образца происходит посте-
пенно по мере увеличения числа сшивок, приходящихся на одну
молекулу. Чарлсби отмечает несколько стадий трансформации.
В соответствии с его механическими характеристиками образец
вначале становится каучукоподобным, затем сыроподобным и
наконец стекловидным.
12.1.1. Химические изменения в полиэтилене при облучении
Газовыделение. Радиолиз полиэтилена сходен с радиолизом
низкомолекулярных алканов. Водород является основным газо-
образным продуктом радиолиза. Количество и состав других
371
выделяющихся газов зависят от степени разветвления полиэти-
лена и поглощенной дозы излучения. Хотя некоторые экспери-
ментальные данные противоречат друг другу, можно сделать
несколько обобщающих выводов.
1. Выход водорода при радиолизе полиэтилена не зависит
от степени кристалличности образца и длины цепей полимера.
Значение G для выхода Н2, равное 4, не меняется при увеличе-
нии кристалличности от 50 до 85% (табл. 12.2). Примерно та-
кое же значение G(H2) характерно для радиолиза нормальных
алканов.
Таблица 12.2*
Значения радиационно-химического выхода для газовыделения и образования
ненасыщенных евнзей при радиолизе полиэтилена
Тип полимера Вид излучения и температура облучаемого образца С(П2) G(RII) 6 —(СН=СН)—
ПЭ низкой плот- Реакторное излучение (70'- С) 4,0 0,08 —
ности То же Электроны с энергией 800 кэв 5,0** 0,9** 1,9
Полиметилен То же 4 7** 0 1,2
ПЭ низкой Реакторное излучение 5,0*** — —
ПЛОТНОСТИ То же у-Излучение 60Со 3,75 — —
ПЭ низкой и вы- Электроны с энергией 2 Мэв 3,1 -—. 1,3
сокой плотности То же (от —190 до -j-80“ С) у-Излучепие 60Со 4,0 1,7—2,4
» Реакторное излучение (80' С) 7,0 — — .
» Электроны с энергией 800 кэв — — .—_
Марлекс-50 То же От —170 до -|-34с С 136" С 3,75** 5,5** 0,07** 0,13** ^2,0
» 240° С 5,8** 0,17**
ПЭ низкой плот- Рентгеновское излучение, 2,5 0,15 —
ности То же 50 кв (13е С) Рентгеновское излучение, 3,0 0,36 1,15
ПЭ высокой плот- 50 кв (80 е С) 50 кв (10° С) 2,8 0,03 — 1,35
ности То же 50 кв (80" С) 3,0 0,09 —
* Из книги: Chaplro. Radiation Chemistry of Polymeric Systems. Interscience Pub-
lishers, New York, 1962.
** Исправленное значение G с учетом того, что на 1 p в полиэтилене выделяется энер-
гия 6«IO1S эв/г.
*** Полный выход для всех газов.
2. Газы, иные, чем водород, составляют от 0,4 до 7% общего
выхода газов. Относительно состава и выхода этих газов суще-
ствуют заметные разногласия. Разные исследователи характе-
ризовали эти газы как содержащие: только следы С2Нб и С2Н4,
только насыщенные молекулы, только ненасыщенные молекулы,
главным образом метан, не содержащие метана.
372
Одно из наиболее обстоятельных исследований выполнил
к настоящему времени Харлан [1], данные которого приведены
в табл. 12.3. Характер выделяющихся газообразных продуктов
сильно зависит от состава боковых цепей. Так, например, бу-
тан оказывается основным углеводородом, выделяющимся при
облучении бутил-разветвленного полиметилена. Поэтому Хар-
лан предлагает использовать радиацию в качестве аналитиче-
ского инструмента для определения наличия и состава боковых
ветвей в образце. Появление ненасыщенных газов, очевидно,
свидетельствует о том, что могут идти реакции переноса водо-
рода в главной цепи.
Различия в количестве выделившегося газа при облучении
полимстилена и ПЭ высокого давления также указывают на от-
носительную легкость отрыва боковых цепей. Следует отме-
тить, что в случае октазона, данные по которому приведены
для сравнения, высокое отношение количества выделившихся
углеводородов к водороду обусловлено низким значением отно-
шения числа концов молекулярных цепей к числу углеродных
атомов основного валентного остова.
3. Отношение водород / углеводородные газы непостоянно и
уменьшается при увеличении дозы либо за счет увеличения
числа сшивок, либо за счет возрастания ненасыщенности.
4. В температурном диапазоне от 200° С и до комнатной тем-
пературы выделение Н2 существенно не изменяется. При более
высоких температурах выход увеличивается. Энергия активации
составляет около 0,8 ккал!моль. Выход углеводородных газов
также увеличивается с ростом температуры в этом диапазоне.
В температурном интервале от 10 до 80° С для выделения угле-
водородных газов получены энергии активации вплоть до
2,5 ккал!моль.
Образование ненасыщенных связей. Ненасыщенность в поли-
этилене можно определить с помощью титрования бромом или
иодом, а также по инфракрасным спектрам. Первый метод по-
зволяет определять лишь общую ненасыщенность; анализ ин-
фракрасных спектров позволяет выявить различные формы не-
насыщенности и идентифицировать виниловые (—СН = СН2),
винилиденовые (—CR = CH2) и транс-випиленовые (—СН =
= СН—) группы. Зигзагообразная природа угл'еводородной
цепи не благоприятствует образованию цис-виниленовых групп.
Теоретически всегда нетрудно провести инфракрасные спект-
ральные измерения. Однако практически приходится затратить
немало усилий, чтобы добиться недвусмысленной интерпрета-
ции результатов.
Промышленные полиэтилены содержат в среднем одну двой-
ную связь на молекулу. Встречаются любые формы ненасыщен-
ности. Обычно приблизительно одна виниловая или транс-вини-
леновая группа приходится на три винилиденовых. При облу-
чении эти «висячие» группы с двойными связями начинают
373
Таблица 12.3*
Газовыделение (%) при облучении полиэтилена
Полимер н2 СН4 с2н, СзНд С.Нхо С6н13 с.п14 с,н„ Ненасыщенные углеводороды* *
са С3 С. С, С. С,
Полнметилен 99,38 0,12 0,05 0,01 0,02 0,01 0,01 0 0,30 0,09 0,01 — — —
Метнл-разветвленный поли- метилен 98,46 0,72 0,20 0,09 0,11 0,06 0,03 — 0,20 0,03 0,08 0,02 — —
я-Бутил-разветвлениый по- лиметилен 92,93 0,90 0,23 0,20 2,5 0,20 0,17 0,17 0,70 0,75 ' 1,2 0,05 — —
я-Ал лил-разветвленный по- лиметилен 98,07 0,50 0,09 0,08 0,08 0,50 0,05 — 0,26 0,15 0,10 0,09 0,03 —
Английский ПЭ низкой плотности 96,9 0,22 0,82 0,21 0,59 0,16 0,07 0,05' 0,68 0,04 0,09 0,02 0,07 0,08
Американский ПЭ иизкой плотности 96,91 0,17 0,84 0,04 0,75 0,17 0,21 0,05 0,41 0,12 0,20 0,09 0,04 —-
Октазон*** 91 0,5 2,1 1,3 0,9 1,1 0,7 0,6 — — — — — —
* Данные из статьи: Harlan Р. et al. J. Polym. Sci, 1955, v. 18, p. 589.
** Указано число углеродных, атомов в молекуле, содержащей двойную связь.
Данные из статьи: Miller A. et. al. J. Phys. Chem. 1956, v. 60, p. 599.
исчезать с очень высоким радиациойно-химическим выходом —
G около (—10). В связи с этим можно предложить интересный,
хотя и не слишком обоснованный механизм переноса энергии,
так как исчезновение ненасыщенных групп связано либо с гидро-
генизацией, либо со случайными взаимодействиями.
Рис. 12.1. Изменение ненасыщенности в облучен-
ном полиэтилене в зависимости от дозы:
/ — виниловые группы; 2 — виниленовые группы; 3 — ви-
нилиденовые группы.
Как видно из рис. 12.1, количество виниленовых групп с ро-
стом дозы увеличивается, и зависимость сохраняет линейный
характер вплоть до 10 Мрад. При дальнейшем увеличении дозы
выход виниленовых групп уменьшается. Равновесие между
числом образующихся и разорванных двойных связей, по-види-
мому, наступает тогда, когда одна двойная связь приходится
на 18 углеродных атомов. Сходная картина наблюдается и для
низкомолекулярных алканов.
Механизм разрушения двойных связей до сих пор не ясен.
Уменьшение ненасыщенности не сопровождается уменьшением
выхода Н, как это следовало бы ожидать для реакции
-СН=СН—j-H2 —СН2—СН2—.
(12.1)
375
Точно так же не удастся отметить увеличения числа сшивок,
которое должно было бы происходить при радикальных реак-
циях типа
R
I .
—СН=СН—I-R -СН—СН—. (12.2)
Выход rpawc-виниленовых групп практически нс зависит от
температуры в диапазоне от —200 до +150° С. Это обстоятель-
ство, а также тот факт, что ни акцепторы свободных радикалов,
ни строение образца ПЭ не влияют па образование транс-випи-
леновых групп поддерживают представление о том, что эти
группы возникают в результате отрыва Н от главной валентной
цепи или, возможно, благодаря ионным механизмам. В связи
с этим интересно отмстить, что облучение ультрафиолетом ПЭ,
сенсибилизированного бензохиноном, не ведет к возникновению
ненасыщенности транс-тина. В то же время ультрафиолетовое и
ионизирующее излучение приводят к возникновению сопряжен-
ной ненасыщенности.
Разрывы главного валентного остова и сшивание. Хорошее
соответствие между степенью разветвленности и количеством
углеводородных газов, выделяющихся при облучении
(табл. 12.3), заставляет предполагать, что разрывы главной
цепи происходят в минимальных масштабах. Это предположе-
ние трудно проверить, так как единственный эксперименталь-
ный метод состоит в растворении облученного полимера и от-
делении низкомолекулярной растворимой фракции (золя) от
высокомолекулярной фракции со сшивками (геля). Сшивки при
этом могут маскировать разрывы.
Чтобы количественно оценить соотношение между числом
разрывов, сшивок и значением молекулярного веса, нужно по-
строить соответствующую статистическую теорию.
Для количественного анализа мы должны задать исходное
молекулярно-весовое распределение полимера. Рассмотрим
вначале, какое молекулярно-весовое распределение должно воз-
никнуть, если в бесконечной полимерной цени происходят слу-
чайные разрывы.
Пусть N(x)— число фрагментов с длиной х, где х—коли-
чество мономеров; N — общее число фрагментов; Ро — вероят-
ность разрыва; (1— Ро) —вероятность отсутствия разрыва;
(х—1) — число связей между мономерами в фрагменте длиной х.
Вероятность того, что ни одна из связей, соединяющих х мо-
номеров, не разорвана, будет равна (1—Ро)ж~’. Вероятность
того, что от бесконечно длинной цепи будет оторвано ровно
х мономеров, равна Ро(1—Ро)*-1- Следовательно,
2^ = Р0(1-Р0>/-\ (12.3)
376
Для .Po<J и х»1 можно аппроксимировать это выражение
следующим образом:
N (х) = NPoe~p°x. (12.4)
Такое распределение принято называть «наиболее вероят-
ным», «экспоненциальным» или «случайным» распределением
по молекулярным весам. Среднее значение этой функции даст
нам среднечисленную степень полимеризации СПп:
N [ хРае~р«х dx
СП = _®-------------- -L . (12.5)
" N Ро V
Чтобы получить средневесовую степень полимеризации, мы
должны воспользоваться более общим выражением, в котором
п равно 2:
N | хлРое—dx
= (12.6)
N f xn-i Poe~p«x dx 0
о
Откуда Cnw=2 СПП.
Для молекулярно-весовых распределений, отличающихся от
наиболее вероятного 2МП=^М!,.., соотношение между средними
величинами зависит от формы распределения.
Рассуждения, с помощью которых были пол/чсны уравне-
ния (12.5) и (12.6), нетрудно провести в обратном порядке.
Тогда, начав с наиболее вероятного распределения по молеку-
лярным весам, можно проследить за процессом сшивания моле-
кул. Ро имеет теперь смысл вероятности образования сшивки.
В конечном счете должна образоваться одна большая моле-
кула, послужившая нам отправным пунктом при выводе урав-
нений (12.5) и (12.6). Для описания процесса сшивания удобно
ввести два новых параметра.
Коэффициентом сшивания б называют число сшитых звеньев
на одну средневесовую исходную молекулу. При образовании
геля
б = бс = 1.
Индексом сшивания у называют число сшитых звеньев на
одну среднечисленную исходную молекулу.
Для наиболее вероятного распределения будут выполняться
следующие соотношения:
6 = POMW; У = РОМ„; б = 2у. (12.7)
377
В точке гелеобразования
6C = /’OCMW = 1; ТС = РОСМ„; yf=M,/Mo- (12-8)
• Для наиболее вероятного распределения ус=0,5. Таким об-
разом, на две исходные среднечисленные молекулы приходится
одно сшитое звено и б=2у. При более широком молекулярно-
весовом распределении, если М.1С=пМ.п, то
б = пу. (12-9)
Пример 12.1. Бовей [2] показал, что если в монодисперсную исходную
систему вносить случайные разрывы, то для средиечислеппого и средневесо-
вого молекулярных весов будут справедливы следующие выражения:
Еа-гп Г™" -I- 2 I1 ~ » hl - + fcno - 11 9 Ю)
м0 -“ рспо ’
Мл _ оц_______________
Мо " 1 +{(СЦ0-1)
(12.11)
1. Покажите, что после нескольких разрывов на исходную молекулу s
отношение М1С/МП приближается к значению, характерному для наиболее
вероятного распределения.
2. Для данной системы найдите соотношение между индексом сшивания
у и коэффициентом сшивания 6 и сравните результат с уравнением (12.9).
Решение. Для полимера с высоким молекулярным весом СП0^СП0—1 и
{=$/(СПо—l)^s/Cn0. Отсюда
(1 — ff"° s; (1 — s/Cn0)c"° х e~s. (12.12)
Подставляя уравнение (12.12) в (12.10) и считая, что s/Cn0<gA, получим:
___ ___ 2 ,
Cnw/Cn0 = — (e~s + s - 1). (12.13)
S2
Точно так же найдем
С77„/С77о — 1/(1-f-s). (12.14)
Следовательно,
Ма,/М„ - [2 (1 -J- s)/sz] (е-* + s - 1).
Для ряда значений s получим
s 1 2 3 6 7
Мю/М„ 1,5 1,7 1,8 1,9 1,95
Сравнивая результаты с уравнением (12.6), мы видим, что уже после
трех разрывов па исходную молекулу отношение М„:/Мп составляет 90% от
величины, характерной для наиболее вероятного распределения.
При монодисперспом распределении согласно уравнению (12.9) 6=у.
В точке гелеобразования 6с=Ус=1; на одну исходную молекулу приходится
одно сшитое звено.
Если сшивание и деструкция происходят одновременно, то
уже нельзя пользоваться одним и тем же символом (Ро) и для
378
вероятности с-шиванйя и для вероятности разрыва. Введем два
новых параметра аир следующим- образом:
вероятность сшивания = а/)-
вероятность разрыва = [3D.
где D — доза излучения.
При условии, что эти вероятности не меняются в зависимо-
сти от дозы, отношение p/а должно характеризовать отношение
растворимой фракции и гсль-фракции. Если для гелеобразования
необходимо появление одного сшитого звена на средневесовую
молекулу (0,5 на среднечисленную), то гелеобразование не бу-
дет происходить при
р/а > 2.
Пользуясь параметрами р и а, можно найти выражение для
доли растворимой фракции. Такое выражение было получено
Чарлсби [3]. Найденные им зависимости графически представ-
лены на рис. 12.2. Сплошные кривые относятся к случаю наибо-
лее вероятного исходного распределения, а пунктирная кри-
вая — к монодисперспому распределению, рассмотренному в
примере 12.1.
Таким образом, измеряя содержание геля в образце в за-
висимости от дозы, можно определить значение отношения р/а.
Эксперименты, проделанные в начале и середине 50-х годов,
показали, что в полиэтилене одновременно происходят сшивание
и деструкция. Согласно экспериментальным данным Чарлсби,
приведенным в виде точек на рис. 12.2, отношение p/а. для поли-
этилена лежит где-то около 0,35. Сходные результаты были -по-
лучены Басксттом и Миллером, которые экстрагировали гель-
фракцию из облученного ПЭ и вновь облучали ее. Сейчас, одна-
ко, результаты этих экспериментов находятся под сомнением.
Высказывается предположение, что в этих опытах могла проис-
ходить дополнительная деструкция, либо индуцированная раст-
ворителем, либо окислительного типа.
Согласно существующим взглядам, в вакууме нс происходит
разрывов основной валентной цепи, за исключением точек вет-
вления и концевых связей (у второго или третьего углерода).
Образование свободных радикалов. Мощным инструментом
обнаружения и идентификации свободных радикалов в облу-
ченных полимерах является метод ЭПР. Успех метода ЭПР в
таких системах обусловлен главным образом тем фактом, что
относительно малоподвижные свободные радикалы, связанные
с твердой матрицей, имеют сравнительно большие времена
жизни. Тем не менее, если хотят обеспечить сохранение всех
образующихся первичных радикалов, измерения проводят при
температуре жидкого азота, считая, что в этих условиях ради-
кальные реакции нс идут. Справедливость этого предположения
379
доказана для всех видов радикальных частиц, кроме атомарного
водорода, который даже при 77° К вступает в реакции, прежде
чем удается про-вести наблюдения.
Как показывает обзор Омерода [4], первичные радикалы в
карбоцепных полимер&х возникают в результате разрыва свя-
Рис. 12.2. Зависимость содержания золь и гель-фракции в
облученном полиэтилене от дозы облучения.
Кружки — экспериментальные значения; сплошные линии—теоре-
тические кривые, вычисленные для наиболее вероятного исходного
распределения по молекулярным весам (числа обозначают отноше-
ние скоростей деструкции и сшивания); пунктирная линия — теоре-
тическая кривая, вычисленная для исходной моиодисперсной систе-
мы (деструкция не происходит).
зей С—Н. Радикалы могут образовываться также в результате
присоединения атома Н по месту двойной связи. Никаких ради-
калов, образование которых можно было бы связать непосред-
ственно с разрывом связей С—С, обнаружено не было.«Исклю-
чение составляют лишь полиметакрилаты, в которых идентифи-
цируемый на начальных стадиях радиолиза неспаренный элект-
рон оказывается в составе иона.
Промежуточные продукты в радиационной химии полиэти-
лена. В качестве промежуточных продуктов радиолиза полиэти-
лена постулировались положительные ионы различных типов,
электроны, захваченные в ловушки, электронно-возбужденные
группы, свободные радикалы алкильного и аллильного типа, ра-
дикалы с высокой диффузионной способностью, такие, как во-
380
дород или метильный радикал, отрицательные ионы. В обзоре
Доула [5] суммированы доказательства существования всех этих
частиц.
1. Ионные промежуточные продукты. Доказательства проте-
кания ион-молекулярных реакций в полиэтилене практически
отсутствуют. Доул, однако, предполагает, что образование
транс-винилена может происходить следующим образом:
I , Г 4
СНг"—СН2---> Н2 4 СН4 =СН— (транс).
I I
2. Отрицательные ионы и захваченные электроны. Прямых
доказательств участия карбанионов R- или захваченных элект-
ронов в каких-либо химических реакциях пока не имеется (пред-
полагают, что карбанионы образуются при захвате электронов
свободными радикалами). Однако наблюдалась термолюми-
несценция электронов, захваченных в ловушки.
3. Промежуточные продукты с высокой диффузионной спо-
собностью. Хотя имеется много косвенных доказательств образо-
вания атомов Н, их никогда не удавалось наблюдать непосред-
ственно даже при —196° С. То же можно сказать и об алкиль-
ных свободных радикалах, метиленовых и метильных, хотя в по-
липропилене метильные радикалы идентифицированы.
Однако аллильные свободные радикалы и полиэтильные ра-
дикалы уверенно регистрируются с помощью метода ЭПР и
ультрафиолетовой спектроскопии.
Механизмы радиационно-индуцированных процессов в по-
лиэтилене. Данные ЭПР и результаты анализа газовыделения
из облученного полиэтилена указывают на то, что первичной
радиационно-индуцированной реакцией является разрыв С—И-
связей:
—СН8—СН2—СН2— —> СН2—CH—СН2—+ Н. (12.15)
Другая первичная реакция — образование ненасыщенной
виниленовой группы:
—СН2—СН2—СН2—СН2------>- —СН2—СН=СН -СН2— 4- Н2. (12.16)
Объяснить образование аллильного радикала, который, как
показано, возникает при облучении полимера при 77° К и сохра-
няется вплоть до комнатных температур, удается, если принять,,
что возможна следующая реакция:
—СН2—СН—СН2—+—СН2—СН==СН—
—СН2—СН2—СН2—Ь -СН—СН=СН—. (12.17)
38Г
Выяснить роль водородного атома, образующегося в реакции
(12.15), в количественных терминах не удается. Однако соглас-
но общепринятой точке зрения могут идти такие реакции:
—II-Н—СН2—СН2 СН2------> —СН2—СН—СН24-Н2; (12.18)
Н’ ненасыщенная связь - радикал; (12.19)
2Н- Н2; (12.20)
к-Н-4—CIJ2—СН—СН2-----> — С112—СН2—СН2—. (12.21)
Сшивание, по-видимому, происходит в основном следующими
двумя способами:
2(—СН2—СН—СН2' -> сшивка (12.22)
и
—СН2—СН—СН2—4—СН—СН==СН -> сшивка. (12.23)
Многочисленные второстепенные реакции, такие, как раз-
рушение виниловых и винилиденовых групп или разрывы ос-
новного валентного остова, здесь во внимание не принимались
Уравнение материального баланса, как показывает табл. 12.2,
более или менее выполняется:
= Q (сшивок) 4- Q (ненасыщенных групп). .
Однако для реакции (12 18), предшествующей реакции
(12.22), необходима высокая энергия активации около
8 ккал!моль, и это противоречит всей рассмотренной схеме
Легкость, с которой происходит сшивание при комнатной тем-
пературе, заставляет предполагать, что в реакции (12.15) рож-
даются «горячие» водородные атомы, т. е. атомы, несущие из-
быточную кинетическую энергию. Б таком случае реакция
(12.18) происходит при каждом соударении. Таким же обра-
зом можно объяснить механизм реакции (12.16):
;н; Н
I I
—СН2—СН—СН—СН2-------> -СН,—СН=СН-СН2 Н2. (12.16)
Здесь квадратик вокруг II символизирует горячий атом.
Для объяснения радиационных эффектов в полиэтилене
Вейсс, Дорфман, Шапиро [6] и другие авторы привлекли также
ионные механизмы. Хотя по сравнению с радикальными ионные
механизмы представляются менее убедительными, в их пользу
в определенной мере свидетельствует тот факт, что проводи-
мость полиэтилена при облучении возрастает.
382
Предполагается, что сшивание по ионному механизму ана-
логично конденсации метана, наблюдаемой в масс-спектрометре:'
CHt СН4 -+ C2Ht + Н2, (12.24)
т. е.
II II
СН+ + СН2 -> +С—СП + Н2. (12.24а)
Возможен также механизм сшивания, сходный с хорошо изучен-
ным процессом переноса протона:
CHt + СН4 -+ CHt -I- СН3 . (12.25)
В полиэтилене перенос протона выглядит следующим образом:
CHt+ СН2 -> СН • 4-CHt (12.26)
Затем происходит нейтрализация
II II
СН- + СН+ -L е- -+ СН- + СН- + Н2. (12.27)
Полимерные радикалы оказываются рядом друг с другом
и могут рекомбинировать, образуя сшивку.
Окислительные реакции. Кислород легко реагирует с поли-
мерными радикалами, образуя перекисные радикалы. В резуль-
тате цепного процесса с одним полимерным радикалом реагиру-
ет около пяти молекул кислорода. Уравнение исходной реакции
выглядит так:
О2
—СН2—СН—СН2— -I- О2 > —СН2—с—СН2—. (12.28)
За ней, возможно, следуют:
RO2 + RH R + ROOH; (12.29)
R- 4-О2RO2- . (12.30)
или же скорее внутримолекулярные, чем межмолекулярпыс ре-
акции:
—СН2—СН—СН=СН—СН,—
[о.
—СН2—CI I (О2 )—СН СН-СНг—
(12.31)
(12.31а)
383
—CH2—CH (02Н)—С-=СН—CH-2— — СН2—НС—О2—СН=СН—сн2
I | (12.316)
4
—СН2—CI I (ОН)—С (О)—СН—CI12— —СН-2—нс=о
„ -1-0-СН—СН-СН2 (12.31b)
о, !
I
—СН2—СН (ОН)—С (О)—СН (О,-)—СН,— О- СН—СН (О, )—СН2— (12.31г)
Как для межмолекулярных, так и для внутримолекулярных
превращений обрыв цепей; по-видимому, происходит по такому
механизму:
R- + R-^R—R; (12.32)
RO2- + R- -> ROOR. (12.33)
12.1.2. Изменение физических свойств полиэтилена
под действием облучения
Степень кристалличности и удельный объем. Благодаря
существованию кристаллических областей в полиэтилене на кар-
тине рентгеновской дифракции видны резкие кольца на фоне
диффузного гало. Отдельные небольшие кристаллиты дисперги-
рованы в аморфном полимере. При увеличении дозы облучения
на дифракционных картинах полиэтилена отмечается постепен-
ное уменьшение интенсивности колец. Неизменность расстояний
между кольцами и отсутствие их уширения указывают на то,
что при облучении меняется степень кристалличности, но раз-
меры или структура кристаллитов не затрагиваются. Для пол-
ного исчезновения кристалличности необходимы дозы около
500 Мрад.
Удельный объем полиэтилена зависит от температуры, сте-
пени кристалличности и числа сшивок. Облучение меняет удель-
ный объем ПЭ постольку, поскольку меняются два последних
параметра. Отмечаются следующие эффекты:
1. При высоких температурах (>115° С), когда кристаллич-
ность уничтожена, облученный сшитый ПЭ проявляет при повы-
шении температуры меньшее относительное увеличение удель-
ного объема.
2. При низких температурах и дозах облучения, от малых
до умеренных, плотность полиэтилена под воздействием радиа-
ции уменьшается, так как разрушается более плотная кристал-
лическая фаза.
3. При низких температурах и дозах облучения, достаточно
высоких для уничтожения большей части кристаллической фазы
и образования многих сшивок на молекулу, плотность ПЭ под
воздействием радиации возрастает, так как расстояние между
384
сшитыми углеродными атомами составляет около 1,5 А по срав-
нению с 4 А для несшитых атомов.
Изменение механических свойств полиэтилена. На рис. 12.3
показаны кривые напряжение — деформация для облученного
полиэтилена низкой плотности. Кроме того, для дозы 15 Мр
приведены три кривые, снятые при 150, 25 и —81° С, которые
Рис. 12.3. Прочность па разрыв образцов полиэтилена
низкой плотности. Температура испытаний, если не ука-
зана особо, равна 25° С.
демонстрируют температурную зависимость модуля упругости,
определяемого по наклону кривой напряжение — деформация.
При повышении температуры модуль упругости резко падает
главным образом за счет нарушения кристалличности при высо-
ких температурах.
Необлученпый образец проявляет характерные для всех пла-
стиков механические свойства. Начальная область, где справед-
лив закон Гука, сменяется областью холодного течения и ориен-
тации. В точке С происходит разрыв образца.
Вначале нод действием облучения отмечается как рост
прочности на разрыв, так и увеличение предельной деформации.
При продолжении облучения и та и другая величины достигают
максимума, а затем начинают снижаться. Возможность пласти-
ческого течения уменьшается и улучшаются упругие свойства.
Происходит сдвиг от пластического поведения к упругому. Для
высоких температур (кривая 15 Мр при 150° С) и высоких доз
(150 Мр), когда кристалличность образца в основном уничто-
жена, форма кривых напряжение — деформация становится та-
кой же, как и для каучукоподобных материалов.
Наиболее резко механическое поведение облученных н пеоб-
лученных образцов различается при температурах выше 110° С.
В этой температурной области полиэтилен, облученный в дозе
10—20 Мр, ведет себя как упругий материал: может растяги-
ваться без приложения дополнительной нагрузки до 270—450%
13 Зак. 907
385
и обладает прочностью па разрыв, превышающей 7 кГ[см2. Не-
облученпый полиэтилен в тех же условиях ведет себя как вяз-
кая жидкость, и его прочность па разрыв невозможно измерить.
Эффект памяти. Радиацию можно использовать для того,
чтобы придать полиэтилену и другим сшивающимся полимерам
поразительное и полезное свойство — память. Это явление ос-
новано па способности трехмерной сетки сшивок, образующейся
при облучении, возвращаться к равновесному состоянию. При
циклическом процессе с изменением агрегатного состояния поли-
мерный образец всегда будет восстанавливать равновесные
размеры, которые он имел во время сшивания.
Допустим, что полиэтилен отформован в виде трубки диамет-
ром 1 см и подвергнут сшивающему воздействию радиации, в
результате чего образец приобретает «память». После этого об-
разец можно нагреть выше точки плавления кристаллитов и ра-
стянуть так, что он приобретает новую форму, например форму
трубки с диаметром 2 см. В растянутом состоянии образец
можно охладить, и он будет представлять собой трубку с диа-
метром 2 см. Однако эту новую форму образец будет сохранять
лишь до тех пор, пока он не нагрет выше точки плавления кри-
сталлитов. Как только кристаллиты будут уничтожены, трубка
«вспомнит» свой равновесный диаметр в 1 см и сожмется до
первоначальных размеров. Эффект памяти в полимерах имеет
важные промышленные приложения (см. главу ХШ).
Электрические свойства облученного полиэтилена. При об-
лучении электропроводность полиэтилена а заметно увеличи-
вается. Например, в полиэтиленовых пленках отмечается тысяче-
кратное увеличение проводимости при дозах от нескольких сот
до нескольких тысяч рад. Проводимость резко растет, достигает
величины насыщения и остается постоянной. Когда облучение
заканчивается, проводимость снижается до своего первоначаль-
ного значения (если доза была не слишком высокой).
Ток, индуцируемый при облучении, сильно зависит от интен-
сивности радиации Ф:
о = /гФ". (12.34)
Показатель степени п в уравнении (12.34) лежит между 0,5 и
1,0. По величине п можно судить о том, что происходит с пер-
вичными и комптоновскими электронами в полимерном образце:
если п=1, то электроны свободно движутся к аноду, или же
непрерывно попадают в ловушки после коротких пробегов;
если н=0,5, то электроны, очевидно, быстро рекомбинируют
с положительными ионами, образующимися в процессе иони-
зации;
если « = 0,5—1, то электроны, видимо, попадают в неглубо-
кие ловушки и вероятность выхода оттуда зависит от глубины
ловушки и условий окружающей среды.
-386
Для полиэтилена значение п колеблется между 0,6—0,8. Это
дало пищу для многочисленных теоретических рассмотрений,
в которых решалась задача определения среднего времени жиз-
ни ионов, возникающих при облучении.
12.2. ДЕЙСТВИЕ ИЗЛУЧЕНИЙ НА ПОЛИМЕТИЛМЕТАКРИЛАТ
Полнметилметакрилат (ПММК), образованный повторением
групп
—СН2—С (СН3) (СОЭСН3)—СН,—,
представляет собой бесцветный аморфный полимер, который раз-
мягчается при 80—120° С. Основные изменения, происходящие
под действием радиации, — разрывы главной валентной цепи и
разложение боковых групп. Эти изменения характерны для дс-
структирующих полимеров.
12.2.1. Химические изменения в полиметилметакрилате
при облучении
Газовыделение. При облучении полнметилметакрилат выде-
ляет большие количества газов. Для дозы 75—100 Мрад количе-
ство выделившихся газов столь велико, что его достаточно для
образования пузырьков или «вспенивания» полимера при нагре-
вании. Газы, возникающие главным образом за счет разложения
эфирных боковых групп, состоят из водорода, окиси углерода,
двуокиси углерода и метана. Хотя качественная сторона газо-
выделения сомнений не вызывает, количественные результаты
заметно колеблются. В табл. 12.4 приведены данные Уолла и
Брауна [7], которые дают представление о разбросе данных,
имеющихся в литературе. Отчасти этот разброс связан с разли-
чиями в экспериментальных условиях. Уолл и Браун воздейст-
вовали па свои образцы у-нзлучением в вакууме до дозы
6,5 Мрад. Другие авторы проводили облучение на воздухе, ис-
пользуя смешанное реакторное излучение при более высоких
температурах и дозах.
Т а б л и ц а 12.4
Состав газообразных продуктов радиолиза полиметилметакрилата
Авторы Газовый состав, % G (общий)
112 сн4 СО со2 мономер
Уолл и Браун Другие авторы 13 13—44 34 6-34 28 23—36 20 19—25 0 0 1,6 1,1—1.5
Хотя для отношения водород/метап наблюдаются заметные
различия, все авторы сходятся на том, что (Н2+СН4) состав-
13*
387
ляют примерно половину выделившихся газов. Другая поло-
вина приходится на СО плюс СО2, выделяющиеся в отношении
3/2. Учитывая лишь разложение группы СООСН3, невозможно
записать уравнение материального баланса.
Ненасыщенность. Имеются серьезные доказательства в поль-
зу того, что возникающие при облучении ПММК полосы по-
глощения в ультрафиолете обусловлены сопряженными двой-
ными связями.
Разрывы основного валентного остова и сшивание. Так как
ПММК деградирует при облучении, гсль-фракции нс возникает
и образец остается растворимым. В растворе с помощью клас-
сических методов, таких, как светорассеяние или вискозиметрия,
можно измерить молекулярный вес после облучения. Чарлсби
[3] суммировал значения G для разрывов, полученные четырьмя
различными группами исследователей (табл. 12.5). Все резуль-
Таблица 12.5
Значения G для разрывов основного валентного остова
полиметилметакрилата при температуре около 25' С
Образец Вакуумная обработка перед облучением Значения G
темпера- тура. ‘ С время, ч вакуум воздух
Пленка FB-1 FB-I FB-2 FB-2 Промышленный образец (ориентирован- ный Промышленный образец (неориентиро- ванный) Стружка 0,5-дюймового стержня Тонкий лист Пленка 100 100 120 100 120 1 100 1 120 { 100 ( 120 20 20 20 20 20 20 20 20 20 1,23 1,70 2,00 1,99 2,48 2,28 2,31 2,23 2,23 2,16 2,16 '1,6 0,645 0,894 1,25 1,37 1,52 1,6 1,5 1,7
тэты в табл. 12.5, кроме трех последних, получены Уоллом и
Брауном, использовавшими в качестве источника облучения
60Со. Три последних результата получены при гораздо более
высоких интенсивностях облучения, когда для облучения ис-
пользовали реакторное излучение или электронный поток. Ни-
каких эффектов, связанных с мощностью дозы или ориента-
цией полимера, не отмечалось. Однако кислород снижал сте-
пень деструкции примерно па одну треть.
Хотя общее деструктирующее действие радиации на ПММК
не вызывало сомнений, следовало еще убедиться, насколько
388
оправдано допущение о том, что параллельно не происходит
сшивания и ветвления полимера. Такую проверку удалось про-
извести, выполняя измерения молекулярного веса различными
методами.
Образование свободных радикалов. При комнатной темпера-
туре все метакрилатные полимеры после электронного, гамма-
или ультрафиолетового облучения дают одни и те же спектры
ЭПР. Общепринято, что помимо других радикалов в системе
присутствует радикал
—СН2—С (СН3) (COOR).
Механизм радиолиза ПММК. Весьма вероятным представ-
ляется бирадикальный механизм радиолитического разложения:
0 0 О
’ h
С—0СН3 С—осн3 с-осн3 о
1 .1 —CH»—C—CH—c ~ - CH2C—CH»— (3 |-НСОСН3-> (12.35)
1 1 CH3 CH3 1 | газы CH3 CH2 0 СН3 |i I СОСНз
- —СН2С=С :н2 + сн2=с—
Этот механизм согласуется с большей частью известных
данных. Он объясняет присутствие радикалов, обнаруживаемых
в системе, и предсказывает, что разрыв валентного остова дол-
жен происходить возле четвертичного углерода.
Интересным, хотя и не до конца понятным эффектом являет-
ся зависимость скорости деструкции от присутствия различных
добавок. Как показывает табл. 12.6, почти любое вещество, до-
Таблица 12.6*
Добавка Концент- рация добавки. % но массе Сте- пень защи- ты, % Добавка Концент- рация до- бавки, % но массе Сте- пень защи- ТЫ, %
Бензойная кислота 1,0 76 8-Гидроксихинолии 2 52
Нафталин 2,0 32 Дифенилтиокочевина 3,6 69
Антрацен 2,1 37 а-Нафтол 5,5 82
Фенантрен 2,2 51 Р-Нафтол 5,3 24
Дифенил 2,5 56 Этил мочевина 10,0 12
Фенол 2,7 52 Парафин 10 3
а-Нафтилампн 2,5 78
* Данные из статьи: Alexander Р., Toms D. Rad. Res., 1958, v. 9, p. 5C9.
389
бавленное к ПММК, уменьшает число разрывов цепи. Степень
защиты в процентах определяется из соотношения:
Р = \00(Ер — Е)/Е,
где Е и Ер—-энергия, затрачиваемая на разрыв связи без до-
бавки и в ее присутствии. Возможно существование трех меха-
низмов защиты:
1) «жертвенная» защита — защитный агент сам разлагается
под действием облучения;
2) защита, при которой защитный агент встраивается в по-
лимерную цепь и репарирует разрыв;
3) защита, при которой избыточная энергия снимается с по-
лимера за счет переноса энергии па большие расстояния.
Характер кривых зависимости защитного действия от кон-
центрации добавок, а также эксперименты с радиоактивной мет-
кой позволяют сделать вывод, что при радиолизе ПММК функ-
ционируют все три защитных механизма. Относительный вклад
каждого механизма определяется природой добавок. Нафталин,
например, оказывает защитное действие главным образом по
механизмам 2) и 3), а 8-гидроксихиполип —по механизмам
1) и 2).
12.2.2. Изменение физических свойств ПММК
под действием облучения
Механические свойства. Изменения механических свойств,
связанные с разрывами основного валентного остова, стано-
вятся заметными при дозах, превышающих 10 Мрад. На
Доза, Мрад
Рис. 12.4. Уменьшение предела прочности и удлинения
перед разрывом для облученного полиметилметакрилата.
390
рис. 12.4 показаны изменения предела прочности и предельной
деформации при воздействии на ПММК реакторного излуче-
ния. Уменьшение молекулярного веса в зависимости от дозы
можно рассчитать, зная соотношение между молекулярным ве-
сом и пределом прочности.
Электростатический разряд. При облучении блочного поли-
мера в массе образца накапливаются электроны и создают
пространственный отрицательный заряд. Во многих полимерах
этот заряд можно спять, если ввести в образец заземленный
точечный электрод. При этом в ПММК и других полимерах
появляются прекрасные узоры Лихтенберга.
ЗАДАЧИ
12.1. Сколько сшивок должно приходиться на одну средиевесовую и
одну среднечислеииую молекулы, чтобы могло начаться гелеобразование?
12.2. Принимай значение G для образования водорода равное 4, 1, рас-
считайте значения G для других газов, пользуясь данными табл. 12.1.
12.3. Пусть молекулярно-весовое распределение в исходной системе ха-
рактеризуется отношением М„/М„ =0,25. В полимерных молекулах’ начинают
появляться случайные разрывы. Получите уравнение для нового молекуляр-
но-весового распределения в системе. Как может сказаться исходное рас-
пределение па результатах, приведенных на рис. 12.2?
12.4. Рассмотрите возможность использования приведенных ниже ионных
реакций для объяснения образования сшивок и возникновения ненасыщенно-
сти в полиэтилене:
2—СН;-СН2—CIL— -> —СН:,—СН—СН.,— -|-Н2’-!-—СН,—СН—СН2—
-сн.,—сн.,—сн2—сн.,-~ - —сн.,—сн-сн—сн„—;-н+.
Доведите схему реакций до конца (Weiss. J. Poiym. Sci., 1958, v. 29, p. 425).
Глава XIII
ПРОМЫШЛЕННОЕ ИСПОЛЬЗОВАНИЕ
РАДИАЦИОННО-ХИМИЧЕСКИХ ПРОЦЕССОВ *
Экспериментальная работа по промышленному внедрению
радиационно-химических процессов не могла начаться до конца
40-х годов, поскольку не существовало достаточно мощных
источников излучения. В период 60-х годов основные усилия
были сосредоточены на попытках добиться радиационной сти-
муляции химических процессов без катализаторов при низких
температурах. Первые результаты породили широкий энтузиазм
относительно перспектив промышленного использования излу-
чений. К сожалению, однако, среди сотен проектов, предлагав-
шихся для промышленного внедрения в 40-х и 50-х годах, реаль-
ным оказался только одни — использование радиационного
сшивания полимеров для увеличения их термостойкости и при-
дания усадочных свойств при нагреве. После многих лет прак-
тически бесплодной экспериментальной работы новые надежды
на будущее появились в 60-х годах, когда для промышленного
использования был предложен ряд новых .радиационно-химиче-
ских процессов. Среди них — метод производства бромистого
этила компании «Доу Кемикл», радиационный синтез детерген-
тов, доступных биопереработке (разложению микроорганиз-
мами), компании «Эссо», изготовление радиационнопривитых
мембран и аккумуляторных разделителей компании «РАИ», ра-
диационная вулканизация красок компании «Форд», процесс
изготовления древесно-полимерных пластин и полимерной про-
питки деревянных изделий. Современному состоянию промыш-
ленных приложений радиационной химии посвящен хороший
обзор Джефферсона с соавторами [1].
13-1. СШИТЫЕ ПОЛИМЕРНЫЕ МАТЕРИАЛЫ
Кабельная и проводная изоляция. Облученный полиэтилен
под коммерческим названием «ирратап» ввела в употребление
в 1954 г. компания «Дженерал Электрик». Ирратан выпускали
Глава переведена с незначительными сокращениями. — Прим, перев.
392
в виде пленки или ленты и использовали как электроизолирую-
щий материал. В настоящее время он в продажу не посту-
пает.
Позднее для аэрокосмической промышленности был разра-
ботан другой продукт—высоконаполненная изоляция на поли-
этиленовой основе для кабелей и проводов (ИПО). Разра-
ботку продукта закончили в 1960 г., и сейчас он выпускается
фирмами «Райхем -корпорейшн», «Анаконда» и «Супренант».
Изоляция из облученного полиэтилена не только обладает
всеми высокими показателями, присущими полиэтилену, но и
сохраняет их при повышенной температуре, когда необлучеи-
ные материалы уже не годятся. Коммерческий продукт не об-
наруживает признаков плавления или течения вплоть до темпе-
ратуры тер.моразложения (325° С), в то время как лучший по-
лиэтилен высокой плотности плавится при 135° С. Кроме того,
облученный полиэтилен не обнаруживает тенденций к растрес-
киванию при изменении условий внешней среды.
Помимо ПЭ основными ингредиентами изоляции ИПО яв-
ляются смола, антиоксидант и противовоспламенительное сое-
динение. В некоторых случаях добавляют небольшое количество
мостикообразующего агента — серу, серусодержащие соедине-
ния, селен, селенсодержащие соединения, теллур и теллурсо-
держащие соединения (австралийский патент № 217912, сен-
тябрь 1956 г., П. М. Кук). При этом после радиационной обра-
ботки следует проводить дополнительную тепловую. В конеч-
ном счете точный химический состав варьируют в зависимости
от условий работы изделия и условий прессования.
Изоляция ИПО, однако, сравнительно дорого стоит. Это от-
части обусловлено сложным составом и специальной обработ-
кой изделия, но основной вклад в увеличение стоимости вносят
затраты на облучение. Главным потребителем изоляции ИПО
является аэрокосмическая промышленность, где высокая стои-
мость компенсируется малой массой и химической устойчиво-
стью изоляции.
Стягивающиеся пленки и трубки. При облучении в дозах
свыше 20 Мрад полимеры приобретают «память». Полимер,
растянутый после облучения, если его нагреть (обычно до тем-
пературы выше 113° С), вновь стягивается до первоначальных
размеров и формы. Материалы такого типа широко использу-
ются для изготовления покрытий, кожухов, упаковок, изолято-
ров заданной формы и других изделий. Сейчас стягивающихся
трубок продается за год более чем на 20 млн. долларов.
В табл. 13.1 приведены характеристики стягивающихся при
нагревании трубок, которые выпускает фирма «Райхем». В ка-
честве материала для трубок кроме полиэтилена используют
фторуглеводород, поливинилхлорид и неопрен. В растянутом
состоянии размеры изделий обычно превышают размеры после
усадки на 50—100%.
393
Таблица 13.1
Стягивающиеся трубки «Термофит»
Описание Обозначе- ние
Гибкие, огнестойкие, стягивающиеся при нагревании облученные
трубки из модифицированного полиолефина. Выпускаются с конеч-
ными диаметрами от ~0,5 до 50 мм. Размеры п растянутом состо-
янии примерно на 50—100% больше конечных
Гибкие, прозрачные, стягивающиеся при нагревании облученные
трубки из модифицированного полиолефина. Выпускаются с конеч-
ными Диаметрами от ~0,5 до 25 мм. Размеры и растянутом состо-
янии примерно на 50—100% больше конечных
Прочные, умеренной жесткости, огнестойкие, стягивающиеся при
нагревании облученные трубки из модифицированного полиолефина.
Выпускаются в растянутом состоянии с диаметрами от ~1 до 25 мм.
Конечные размеры — около 50% размера в растянутом состоянии
Прочные, умеренной жесткости, прозрачные, стягивающиеся при
нагревании облученные трубки из модифицированного полиолефина.
Выпускаются в растянутом состоянии с диаметрами от ~1 до 25 мм.
Конечные размеры — около 50 % размера в растянутом состоянии
Прочные, негорючие, прозрачные, стягивающиеся при нагревании
трубки из модифицированного фторуглеводорода. Выпускаются с ко-
нечными диаметрами от 0,25 мм и до 0. Размеры в растянутом со-
стоянии на 50—60% больше конечных
Гибкие, с радиационными сшивками, стягивающиеся при нагрева-
нии трубки из модифицированного поливинилхлорида. Выпускаются
в растянутом состоянии с диаметрами от ~1 до 100 мм. Конечные
размеры — около 50% размера в растянутом состоянии
Гибкие, термостабильные, сшитые, стягивающиеся при нагревании
трубки из модифицированного неопренового каучука. Выпускаются
с конечными диаметрами от ~3 до 25 мм. Размеры в растянутом
состоянии примерно вдвое превышают конечные
RNF
RF
CRN
CR
TFE
PVC
NT
Стягивающиеся пакеты из облученного полиэтилена, пред-
назначенные для упаковки пищевых продуктов, выпускаются
компанией «Грейс». Облученные пленки оказываются примерно
в пять раз прочнее необлучепных. Изделия из такой пленки
можно растянуть примерно в два раза по сравнению с первона-
чальными размерами, а затем, погружая в горячую воду, про-
вести контролируемую усадку изделия. Для облучения пленок
используют дозы от 15 до 20 Мрад. По имеющимся сведениям
в год производится около 1500 т такой пленки.
13.2. МАТЕРИАЛЫ, ПОЛУЧЕННЫЕ С ПОМОЩЬЮ
РАДИАЦИОННОЙ ПРИВИТОЙ ПОЛИМЕРИЗАЦИИ
Древесно-полимерные материалы. Идея прививки мономера
А к полимеру В для придания материалу необходимых свойств
весьма привлекательна, особенно в тех случаях, когда В — не-
дорогой материал, а дорогой мономер А расходуется в иеболь-
394
Дшх количествах. Практически во всех процессах, предложен-
ных для промышленности, компонент В представляет собой не-
дорогие природные высокомолекулярные соединения типа дре-
весины или целлюлозы, синтетические углеводороды вроде по-
лиэтилена или полипропилена.
Прививаемый гидрофобный или гидрофильный мономер А
придает необходимые свойства поверхности материала. Поверх-
ность можно сделать восприимчивой к красителю, проницаемой
для ионов или же диссипирующей электрический заряд (анти-
статической) .
Единственным исключением, когда в результате прививки из-
меняются не свойства поверхности, а свойства всей массы ма-
териала, является пропитка древесины. В этом процессе, кото-
рый сейчас используется в промышленности, прививаемый к
древесине мономер пропитывает весь объем материала. Вначале
древесина пропитывается жидким мономером, таким, как метил-
метакрилат, а затем под действием облучения инициируется
полимеризация. Испытано множество комбинаций древесина —
полимер. Комбинированный материал можно сделать:
1) огнестойким;
2) примерно на 900% более твердым, чем природная дре-
весина, и поэтому более устойчивым по отношению к ударам,
надрезам и царапинам;
3) обладающим повышенной прочностью на сжатие:
4) обладающим повышенным сопротивлением изгибу;
5) адсорбирующим жидкость с меньшей скоростью и по-
этому лучше сохраняющим первоначальные размеры (устойчи-
вость к перекосам и набуханию);
6) можно выявить и усилить естественные окраску и узор
древесины;
7) материал можно пилить, сверлить, обрабатывать на то-
карном станке и шлифовать до появления высококачественной
гладкой поверхности.
Комбинированные материалы такого рода можно использо-
вать для изготовления фурнитуры, полов, оконных рам, подо-
конников, дверей, дверных ручек, декоративной отделки, несу-
щих балок и перекладин, спортивных товаров, яхтенных палуб
и обувных колодок.
При разработке промышленного процесса удалось добиться
снижения дозы излучения, инициирующей полимеризацию, на
75% (до 0,5 Мрад). Для инициирования используют обычно
у-излученпе 60Со, так как для него характерна большая глубина
проникновения в образец и независимость радиационного поля
от формы объекта. Однако радиационную обработку панелей
или фанерных листов удобнее проводить с помощью ускори-
тельных устройств. (Изготовление фанеры и древесно-полимер-
ных плиточных полов представляется в коммерческом отноше-
нии наиболее перспективной областью.)
395 .
Схема производственного процесса получения древесио-ио-
лимерных пластин показана па рис. 13.1. На первой стадии про-
цесса древесину подвергают воздействию вакуума для удаления
кислорода, так как кислород ингибирует полимеризацию. Затфм
в атмосфере инертного газа древесину пропитывают мрномер'ом,
после чего избыток мономера удаляют. Хотя для пропитки
можно применять мономер и в жидкой, и в газообразной фор-
S
Рис. 13.1. Схема производства дре-
вссно-полимерных пластин:
/ - - деревянные заготовки; 2 — мономер;
«?—пронитка: 4 — {!} поры древесины
освобождаются посредством вакууми-
рования; 3 (2) древесина погружает-
ся в раствор мономера; 6 — мономер
пропитывает древесину; 7- изделия
облучаются у-кнаптами е,’Со или друго-
го источника; 8— древесно-полнмерпые
изделия; 9—(3} слив избыточного моно-
мера.
ме, обычно предпочитают вести пропитку жидким способом
под давлением 14 кГ)см2. Как правило, требуется высокая сте-
пень наполнения древесины мономером.
Ионообменные мембраны. Полученные с помощью радиа-
ционной привитой полимеризации мембраны для диализа, элек-
тролиза, аккумуляторных батарей и топливных элементов вы-
пускает компания «РАИ». Мембраны «Псрмиоп» изготавливают
из привитого сополимера и используют в качестве ионообмен-
ных мембран с размерами пор в апгетремном диапазоне. Не-
сколько типов выпускаемых мембран «Псрмиоп» перечислены
в табл. 13.2.
Таблица 13.2
Ионообменные мембраны «Пермион»
Номер мембраны «Пермион» Основной полимер Тип функциональных групп
300 Полиэтилен Карбоновые кислоты
1000 Политетрафторэтилен То же
3000 Фторированный сополимер, этиленпро- »
пилен
1010 Политетрафторэтилен Сульфоновые кислоты
200 Полиэтилен Третичные амины
1200 Политетрафторэтилен То же
210 Полиэтилен Четвертичные амины
1210 Политетрафторэтилен То же
Мембраны «Пермион» отличаются высокой прочностью л
большой устойчивостью к действию химических агентов. Мем-
браны успешно используют в качестве аккумуляторных разде-
лителей. «Пермион 300» широко применяется с этой целью в
Ni—Cd-элементах.
396
Текстильные материалы. К числу успешных промышленных
Йримепепий радиационной привитой полимеризации, по-внди-
Аому, следует отнести обработку электронным пучком хлопко-
в^-пол и эфир ной смеси. Конечный ’ материал хорошо прессуется
и Ьбладает грязеотталкиваюшими свойствами. Процесс разра-
ботан компанией «Диринг Милликен» в 1966 г.
13.3. РАДИАЦИОННЫЙ СИНТЕЗ ДЕТЕРГЕНТОВ,
ДОСТУПНЫХ БИОПЕРЕРАБОТКЕ
Компании «Эссо» в США и «Хёхст» в ФРГ затратили много
усилий на разработку инициируемого у-излучепием одновремен-
ного сульфирования и окисления углеводородов. В процессе
«Эссо» предусматривается прохождение SO2 и О2 через линей-
ные углеводороды в поле у-излучения. SO2 присоединяется к
алкильному радикалу и образуется алкилсульфонильпый ради-
кал
R-4-SO2 -> RSO,-. (13.1)
Присоединяя кислород, этот радикал, превращается в надсуль-
фонильный радикал
RSO2 • + О2 -> RSO2O2•. (13.2)
Затем происходит реакция отрыва водорода и возникает новый
алкильный радикал
RSO2O2 • + RM -> RSO2O2H 4- R •. (13.3)
Надсульфоновая кислота разлагается на надсульфонильный
и гидроксильный радикалы, которые в свою очередь отрывают
водород от углеводородов и снова образуют алкильные ради-
калы. В результате разветвленной цепной реакции процесс мо-
жет продолжаться, даже если удалить источник излучения.
В конце концов, однако, молекулы воды, возникающие в ходе
реакции, ограничивают ветвление.
Компания «Эссо» построила опытный завод. Однако в круп-
ных масштабах процесс пока не используется.
13.4. ПРОЦЕСС ПРОИЗВОДСТВА
БРОМИСТОГО ЭТИЛА КОМПАНИИ «ДОУ»
В марте 1963 г. компания «Доу кемпкл» запустила первый
химический завод, где используется принцип радиационного
катализа. На заводе с производительностью около 500 т бро-
мистого этила в год был применен процесс, запатентованный
Пампелли и Вилкинсоном (канадский патент № 649718, октябрь
1962 г.). Серьезный вклад был сделан также в результате иссле-
дований Армстронга и Спинкса [5] и технических разработок
Хармера и Било [6].
297
Бромистый этил образуется в результате следующей реаку
ции:
СН2 = СН2 -i- HBr -> CI 13СН2Вг. (13.^)
Реакция идет ио радикально-цепному механизму, и рост 1£е-
пи происходит в результате следующих процессов:
Вг • п- СН2 СН2 -> -С; В — СН2Вг; (13.5)
НВг -|- • CI L — СН2Вг -> Вт • + СН3СН2Вг. (13.6)
Реакция (13.5) инициируется радикалами брома, которые об-
разуются при диссоциации бромистого водорода. Альтернатив-
ная схема с участием водородных радикалов оказывается менее
энергетически выгодной.
Промышленный процесс удалось разработать за пять лет.
Трудности, которые пришлось преодолеть при лабораторных
разработках и доводке процесса на опытном заводе, дают пред-
ставление о тех усилиях, которые нужно затратить, чтобы внед-
рить в промышленность радиационный процесс.
13.4.1. Разработка процесса производства
бромистого этила в лаборатории и на опытном заводе
1. На первой стадии, чтобы выяснить оптимальные условия
реакции (характер растворителя, дозировку), бромистый водо-
род и этилен в стехиометрическом соотношении вводили в со-
суд емкостью 500 мл, содержащий 175 мл растворителя. В ка-
честве растворителей использовали двубромистый этилен, че-
тыреххлористый углерод, перхлорэтилеп и бромистый этил. Ока-
залось, что для бромистого этила наблюдается наибольший вы-
ход и наибольшие значения G.
2. Затем был сконструирован сосуд для проведения реакции
в потоке. В ппрексовый шар емкостью 60 мл подавали этилен
и бромистый водород с различной скоростью и в разных весо-
вых отношениях. Реакцию проводили до тех пор, пока не уста-
навливалось равновесие, после этого определяли выход. Таким
образом были получены данные для предварительного конструи-
рования установки.
3. На следующей стадии экспериментов использовался- сосуд
с большей емкостью (550 мл} и с водяным охлаждением. (Экс-
периментальные условия были сходными с условиями в преды-
дущей серии опытов.) Оборудование для подачи газа, однако,
оказалось непригодным, и пришлось вести изменения в кон-
струкцию, чтобы обеспечить весь диапазон скоростей подачи
газа (30—140 г этилена в 1 ч).
В опытах с реактором емкостью 550 мл удалось также вы-
яснить другие факторы, влияющие на процесс. Так, оказалось
выгоднее смешивать бромистый водород с этиленом перед по-
398
дачей. Загрязняющие частички железа полностью ингибировали
процесс.
' 4. В качестве альтернативы двуфазпой реакции исследовали
возможность проведения реакции целиком в жидкой фазе. Для
этЗро использовали трубчатый реактор из нержавеющей стали
ем^стью 185 мл, через который сжиженные газы проходили
Рис. 13.2. Схема промышленной установки компании «Доу» для про-
изводетва бромистого этила радиационно-химическим способом:
/ - химический реактор; 2 — раствор бромистою этила; 3 — вывод продувочного
азота; 4 — подача продувочного азота; 5 — газообразный этилен: 6 — газообраз-
ный бромистый водород; 7 — холодильник; « — промежуточная емкость; 9— сток
продукта; ЛЬ - возгонка; // — дистиллятор; 12— пар: 13— съем продукта.
14— нейтрализационная емкость; 15— нейтрализатор; 16—ocvnmie.ib; 17 — бро-
мистый этил перекачивается на хранение.
под давлением 70 кГ1см2 при температуре —5° С. По причинам,
до конца не ясным, на установке удалось получить только огра-
ниченную степень конверсии, которая снижалась при измене-
нии условий.
Кроме того, при конструировании завода на основе одно-
фазного процесса пришлось бы использовать газовые компрес-
соры, что повысило бы стоимость предприятия и снизило ком-
мерческую заинтересованность.
5. На следующей стадии в лабораторных условиях в мас-
штабе один к десяти был построен прототип промышленной
установки. Помимо ранее выбранной конструкции реактора из-
готовили прототип реактора новой формы. Этот реактор имел
форму диска с 36°-ным сектором при той же высоте, что и
399
раньше. Выбранная геометрия облучения была сохранена в про-
мышленной установке. /
На установке удалось добиться конверсии от 98 до 100%
при скорости подачи реагентов ~ 4 кг/ч при 19° С. Снова было
отмечено ингибирующее влияние продуктов коррозии и загряз-
нений в растворителе или газе.
6. Схема промышленной установки показана на рис. Д3.2.
Реактор изготовлен из никеля и имеет емкость 151 л. Реактор
помещается в бетонной выемке глубиной 1,20 м и покрыт'сна-
ружи стальной обшивкой. Источник из 6пСо с активностью
1800 кюри в виде покрытых алюминием стержней вводится в
специальный колодец в центре реакционного сосуда.
Во время работы установки насос обеспечивает непрерывную
циркуляцию бромистого этила со скоростью около 106 л/мин.
Рециркуляционное отношение равно 179/1, т. е. за каждый цикл
0,5% объема изымается в виде продукта.
13.5. РАДИАЦИОННАЯ ВУЛКАНИЗАЦИЯ КРАСОК
Этот процесс разработан на основе ранних результатов Бал-
лантайна, Чарлсби и Месробяна, которые обнаружили, что по-
лиэфиры, облученные в присутствии мономеров, легко сшива-
ются и образуют жесткие гели. Промышленный процесс был
разработан группой Бурланта в лабораториях компании «Форд».
Работая с полиэфирными окрашенными пленками, Содержа-
щими до 35% мономера, Бурлапт и сотрудники обнаружили,
что при облучении электронами с энергией 300 кэв идет поли-
меризация, скорость которой не зависит от интенсивности облу-
чения ‘вплоть до мощностей дозы порядка 100 рад/мин.
Радиационный метод вулканизации красок имеет хорошие
коммерческие перспективы и может оказаться одним из важ-
нейших промышленных процессов радиационной химии.
13.6. РАДИАЦИОННАЯ ПОЛИМЕРИЗАЦИЯ ЭТИЛЕНА
Полимеризация этилена обычно проводится либо при высо-
ких давлениях и температурах (1000 атм и ~150°С), либо ка-
талитически при низких температурах. В первом случае обра-
зуется разветвленный полимер с низкой плотностью и низкой
температурой размягчения, во втором — более термостойкий по-
лимер с высокой плотностью.
С целью заменить оба эти процесса радиационной полимери-
зацией Стейнберг с сотрудниками из Брукхейвенской нацио-
нальной лаборатории провели большие экспериментальные опыт-
ные и проектно-конструкторские работы. В настоящее время
процесс отрабатывается на опытном заводе. Одно из основных
препятствий имеет механическую природу и связано со спосо-
бом удаления образовавшегося продукта из реакционной зоны.
400
Ёрли этого не делать, то полиэтилен будет загрязнен большими
количествами сшитого материала. При условии, что с этой про-
блемой удастся справиться, техническая и финансовая инициа-
тива быстро обеспечит замену старых методов радиационной по-
лимеризацией этилена.
ЗАДАЧИ
13.1. Ниже в виде таблицы приведены данные Хармера и Биле во непре-
рывному двухфазному гидробромированию этилена в сосуде емкостью 60 мл
при - 10* С. Исходя из этих данных, оцепите размеры источника 60Со и
реакционного сосуда для производства 500 т этилбромида в год. Чему равно
значение G для реакции образования бромистого этила?
Мощность дозы, крад/ч Скорость подачи ВгН, г/ч Скорость подачи этилена, С/Ч Выход продукта. । Мощность ДОЗЫ. крад{ч Скорость подачи ВгН, г,ч Скорость подачи этилена. Выход продукта, %
254 72 24 86 100 84 28 68
254 116 38 61 100 138 44 14
254 180 60 /о 100 180 62 3,5
100 64 22 94
СПИСОК ЛИТЕРАТУРЫ
Глава I
1. Hughes D. J., Schwartz R. В. Neutron Cross Sections, USAEC Report,
BNL-325 (2nd cd.), Brookhaven National Laboratory, July 1, 1958.
2. Goldman D. T„ Roesser J. Chart of the Nuclides, 9th edition, General Elect-
ric Co., Schenektady, N.Y.
Глава 11
1. Bethe H. A., Ashkin J. Experimental Nuclear Physics, John Wiley and Sons,
Inc., New York, 1953, p. 166.
2. Bethe H. A. Properties of Atomic Nuclei. 11. Range-Energy Curves: Alpha
Particles, Protons, and Mesons, USAEC Report BNL-T7, Brookhaven Natio-
nal Laboratory, June 1949.
3. Lea D. E. «Actions of Radiations on Living Cells». The Macmillan Company,
New York, 1947.
4. Berger R. T. Radiation Res., 1961, v. 15, p. 1.
5. Blizard E. P. (cd.). Shielding, v. Ill, Part B, «Reactor Handbook», Inter-
science Publishers, Inc., New York, 1962.
Глава III
1. Westendorp W. F. Resonant-transformer Electron-beam Generators, in
A. Charlesby (ed.), «Radiation Sources». Pergamon Press, Ltd, Oxford, 1964.
2. Traegeser D. A. High Voltage Engineering Co., personal communication.
3. Kuhl O. A., Ballantine D. S. Isotopic Sources of Radiation Power, in
A. Charlesby (cd.), «Radiation Sources», Pergamon Press, Ltd, Oxford, 1964.
4. «Industrial Uses of Large Radiation Sources», v. 11, Symposium Procee-
dings, Salzburg, 1963, Vienna, IAEA, 1963.
5. Blizard E. P. (ed.). «Reactor Handbook», v. Hl, Part B, Shielding, 2d ed.,
Interscience Publishers, New York, 1962.
'6 . Lind S. C. «Radiation Chemistry of Gases»,. Reinhold Publishing Corpora-
tion, New York, 1961.
7. Jefferson S. Experience in Operating Large Gamma-radiation Installations,
in «Industrial Uses of Large Radiation Sources», v. II. Symposium Procee-
dings, Salzburg, 1963, IAEA, Vienna, 1963 (STI/PUB/75).
8. Mins S. In: «Large Radiation Sources in Industry», Symposium Procee-
dings, Warsaw, 1959: Vienna, IAEA, 1960 (STI/PUB/12).
Г л а ва IV
1. «Physical Aspects of Irradiation», National Bureau of Standards, Hand-
book 85, U. S. Government Printing Office, Washington, 1964.
402
‘Д Taimuty S. I. «Obtaining a System of Dosimetry», USAEC File number
\ NP-7613. Stanford Research Institute, March 1959.
3. 'Price W. J. «Nuclear Radiation Detection», McGraw-Hill Book Company,
'{tic., New York, 1964.
4. Failla G. Dosimetry of Ionizing Radiations, Proc. Intern. Conf. Peaceful
Uses At. Energy, Geneva, 1956, v. 14, p. 239.
5. Coops J., Jessup R. S., Van Ness K. «Experimental Thermochemistry»,
E. D. Rossini (ed), Inlerscienee Publishers, New York, 1956.
6. Johnson E. R., Forten J, Radiation-induced Decomposition of Inorganic
Nitrates, Discuss. Faradav Soc., 1961, v. 31, p. 238.
7. Milery P„ Barr N., Geisselsoder J., Laughlin J. S. In: B. Rajewsky (ed.),
«International Congress of Radiology», v. 2. George Thieme Verlag, Stutt-
gart, 1961, p. 1348.
8. Hochanadel C. J., Ghormley J. Calorimetric Calibration of Gamma-ray
Actinometers. ,1. Chem. Ph\s„ 1953, v. 21, p. 880.
9. Lazo R. M., Dewhurst H. A., Burton M. The Ferrous Sulfate Radiation Do-
simetry: A Calorimetric Calibration with Gamma Rays. J. Chem. Phvs.,
1954, v. 22, p. 1370.
10. Zimmer L. T. Dosimetry of Kilocurie 6,)Co Sources. USAEC Report
AECU-3446, Argonne Cancer Research Hospital, October 1956.
11. Brynjolfsson A. In: «Papers Presented at the First Nordic Meeting on
Food Preservation by Ionizing Radiations Held at Riso, April 25—26,
1960», p. 17. Danish Report R150- 16, July 1960.
12. Keene J. P„ Law J. Determination of the G Value of Ferrous Sulfate for
•’"Co Radiation Using Calorimetric Dosimetry. Phys. Med. Biol., 1963,
v. 8 (1), p. 83.
13. Reid W. B., Johns H. E. Measurement of Absorbed .Dose with Calorimeter
and Determination of W. Rad. Res.,1961, v. 14, p. 1.
14. Reid W. B„ Johns H. E. Rad. Res., 1957, v. 7, p. 217.
15. Geisselsoder J., Keopke K., Laughlin J. S. Rad. Res., 1963, v. 20 (2),
p. 423.
16. Petree B., Ward G. The Construction of Calorimetric for the. Measurement
of Absorbed Dose. National Bureau of Standards, Technica‘1 Note 1963,
U. S. Government Printing Office, Washington. November 1962.
17. Saldick J., Allen A. O. Yield of Oxidation of Ferrous Sulfate in Acid So-
lution b\ liigh-energ\ Cathode Rays. J. Chem. Phys., 1954, v. 22, p. 438.
18. Schuler R. H., Allen A. O. Yield of the Ferrous Sulfate Radiation Dosime-
ter: An Improved Cathode-ray Determination. J. Chem. Phys., 1956,
v. 24, p. 56.
19. Donaldson D. M., Miller N. Quantitative Studies of Radiation induced
Reactions. 111. Oxidation of Ferrous Sulfate by Beta Particles. J. Chem.
Phys., 1955, v. 52, p. 578.
20. Schuler R. H., Allen A. O. Radiation Chemistry Studies with Cyclotron.
Beams of Variable Energy - Yields in Aerated Ferrous. Sulfate Solution.
,1. Amer. Chem. Soc., 1957. v. 79, p. 1565.
21. Hart E. J., Ramler W. J., Rocklin S. R. Chemical Yields of Ionizing Par-
ticles in Aqueous Solutions: Effect of Energy of Protons and Deuterons.
Rad. Res., 1956, v. 4, p. 378.
22. Price В. T., Horton С. C., Spinney К. T. Radiation Shielding, Pergamon
Press, 1957.
23. Спинкс Дж., Вудс P. Введение в радиационную химию. Пер. с англ. М.,
Лтомйздат, 1967.
. 24. Kircher J. F. Effects of Radiation on Materials and Components. Kirch-
man and Bowman (eds), Reinhold, 1964.
25 Attix F. H. Nucleonics, 1959. v. 17. p. 10.
26. Artandi C. Nucleonics, 1959, v. 17, p. 61; Chandler V. Nucleonics, 1959,
v. 17, p. 63.
27. Shall P. Nucleonics, 1959, v. 7, p. 68.
28. Harmer D. E. Nucleonics, 1959, v. 17, p. 72.
403
29. Allen A. О. Radiation Chemistry of Water and Aqueous Solutions, D. Van
Nistrand Company, Inc., Princeton, N. J., 1961, p. 19. 1
30. Hart E. J. Mehanism of the Gamma-ray Induced Oxidation of Formic ^cid
in Aqueous Solution. J. Amer. Chem. Soc., 1951, v. 73, p. 68. '
31. Allen A. O., Holroyd R. A. Peroxide Yields in the Gamma Irradiation of
Air-saturated Water. J. Amer. Chem. Soc., 1955, v. 77, p. 5852
32. Haasbrock F. J. Extending the Range of the «Fricke» Dosimeter up to
106 Rads. USAEC Report BNL-763, Brookhawen National Laboratory,
April 1962.
33. Minder W. Selected Topics in Radiation Dosimetry. Symposium Procee-
dings, IAEA, Vienna, 1961, STI/PUB/25, p. 315.
34. Hochanadel C. J., Davis T. W, Radiolysis of Solid Nitrates. J. Chem. Phys.,
1957, v. 27, p. 333.
35. Bernstein W., Schuler R. H. Low-energy Scattered Radiation Inside а Cy-
lindrical Cobalt-60 Source. Nucleonics, 1955, v. 13, No. 11, p. 110.
36. Taplin G. V., Douglas С. H., Sanchez B. Nucleonics, 1951, v. 9, No. 2,
p. 73.
37. Henley E. J,, Miller A. Gamma-ray Dosimetry with Polyvinyl Chloride
Films. Nucleonics, 1951, v. 9, No. 6, p. 62.
38. Hart E. J., Walsh P. D. Dosimetry of Gamma Ray and Neutron Fluxes in
CP-5, Proc. Second U. N. Int. Conf. Peaceful Uses At. Energy. Geneva,
1958, v. 29, p. 38.
39. Dorfman L. M., Shipko F. J, Beta-particle Radiolysis of Acetylene. J. Amer.
Chem. Soc., 1955, v. 77, p. 4723.
40. Field F. H. Benzene Production in the Radiation Chemistry of Acetylene.
J. Phys. Chem., 1964, v/68 (5), p. 1039.
41. Lewis W. B. Trans. Amer. Nucl. Soc., 1963, v. 6, p. 73.
42. Bopp C. D., Parkinson W. W., Sisman O., Towns R. L., Kirkland W. K.
In: Solid State Division Annual Progress Report for Period Ending
August 31, 1961. USAEC Report ORNL-3213, Oak Ridge National Labo-
ratory, December 1961, p. 95.
43. Anderson A. R,, Linacre J. K. Calorimetric Dosimetry of Reactor Radia-
tion. In: Selected Topics in Radiation Dosimetry, Symposium Proceedings.
Vienna, 1960. IAEA, Vienna, 1961, STI/PUB/25, p. 609.
44. «Measurement of Absorbed Dose of Neutrons and Mixtures of Neutrons
and Gamma Rays», National Bureau of Standards Handbook 75.
45. Draganic I. Oxalic Acid: The Only Aqueous Dosimeter for In-pile Use.
Nucleonics, 1963, v. 21, No. 2, p. 33.
46. Schuler R. H., Allen A. O. Radiation-chemical Studies With Cyclotron
Beams. J. Amer. Chem. Soc., 1955, v. 77, p. 507.
47. Steinberg M., Loffelholz M. M., Pruzansky J. In: Industrial Uses of Large
Radiation Sources, Conference Proceedings, Salzberg, Austria, 1963, v. II.
IAEA, Vienna, 1963, STI/PUB/75, p. 89.
48. Dainton F. S. Chemical Dosimetry, Radiation Research, in G. Silini (ed.).
Proceedings of the Third International Congress of Radiation Research.
North-Holland Publishing Co., Amsterdam, 1967.
49. Artandi C„ Stonehill A. A. Poly (vinyl chloride) — New High-level Dosi-
meter. Nucleonics, 1958, v. 16, No. 5, p. 118.
50. Henley E. J., Richman D. Cellophane-dye Dosimeter fpr the 105 to 107 Ront-
gen Range. Anal. Chem., 1956, v. 28, p. 1580.
51. Charlesby A., Davison W Temperature Effects in the Irradiation of Poly-
mers. Chem. Ind. (London) (Rev.), 1957, p. 232.
52. Nitka H. F. Photographic Methods. Nucleonics, 1959, v. 17, No. 10, p. 59.
53. Shulman J. H., Klick С. C., Rabin H. Measuring High Doses by Absorp-
tion Changers in Glass. Nucleonics, 1955, v, 13, No. 2, p. 30.
54. Kreidl N, J., Blair G. E. Recent Developments in Glass Dosimetry. Nu-
cleonics, 1956, v. 14, No. 3, p. 82.
55. Wagner E. B„ Hurst G. S. Advances in the Standard Proportional Coun-
ter Method of Fast-neutron Dosimetry. Rev. Sci. Instrum., 1958, v. 29,
p. 153.
404
Hughes D. J. Pile Neutron Research. Addison-Wesley Publishing Company,
Inc., Reading Mass., 1953, p, 63.
57: Fenger J. Measurement of Gamma Flux in a Thermal Column with an
Oxalic-acid Dosimeter. Danish Report RISO-67, September 1963.
Глава V
1. Steacie E, W. R. Atomic and Free Radical Reactions. A.C.S. monograph
no. 125, Reinhold Publishing Co., New York, 1954.
.2. Trotman-Dlckenson A. F, Free Radicals. Methuen, London, 1959.
3. Kerr J. A., Trotman-Dickenson A. F., Porter G. (ed.). Progress in Reaction
Kinetics. Pergamon Press, Inc., New York, 1961.
4. McDonald C. C„ Goll R. J. J. Phys. Chem, 1965, v. 69, p. 293.
5. Ausloss P. Ann. Rev. Phys. Chem., 1965.
Глава VI
1. Porter G. Techniques of Organic Chemistry, 2nd ed., v. VIII, part 2. Inter-
science Publishers, 1965, p. 1055.
.2. Dorfman L. M., Matheson M. S. Progress in Reaction Kinetics, v. 3. G. Por-
ter (ed.), Pergamon Press, New York, 1965, p. 329.
3. Kebarle P., Haynes R. M., Searles S. Adv. Chem. Series, 1966, v. 58, p. 210.
4. Wexler S., Lifshitz A., Quattrochi A. Adv. Chem. Series, 1966, v. 58, p. 193.
Глава VII
I. Rosenbaum C. J. Phys. Chem., 1948, v. 41, p. 469.
2. Futrell J. H., Siek L. W. J. Phys. Chem., 1965, v. 69, p. 892.
3. Kebarle P., Hogg A. M. J. Chem. Phys., 1965, v. 43, p. 449.
4. Magee J., Funabashi K. Rad. Res., 1959, v. 10, p. 622.
5. Burton M., Magee J. J. Phys. Chem., 1952, v. 56, p. 842.
6. Boelrijk N., Hamill W. Н: J.A.C.S, 1962, v. 84, p. 730.
7. Eyring H., Hirshfelder J., Taylor H. S. J. Chem. Phys., 1936, v. 4, p. 479.
8. Stevenson D. P., Schissler D. O. J. Chem. Phys., 1955, v. 23, p. 1353.
9. Gioumousis G., Stevenson D. P. J. Chem. Phys., 1958, v. 29, p. 294.
10. Henchman M. J. Annual Reports of the Chemical Society, 1966, v. 63.
11. Stevenson D. Mass Spectrometry. C. A. McDowell (ed.), McGraw-Hill
Book Commpany, New York, 1963, p. 607.
12. Stevenson D. P. Rad. Res., 1959, v. 10, p. 618.
13. Wassey H. S. W., Burhop E. H. S. Electronic andMonic Impact Phenome-
na. Clarendon Press, Oxford, 1952, p. 618.
14. Shoen R. I. J. Chem. Phys., 1964, v. 40, p. 1830.
15. Stevenson D. P., Schissler D. O. The Chemical and Biological Action of
Radiations. M. Haissinsky (ed.), Academic Press, London, 1961.
16. Magee J. L., Burton M J.A.C.S., 1952, v. 73, p. 523.
17. Rosenstock H. M., Wallenstein M. B., Wahrhafting A. L,, Eyring H. Proc.
Nat. Acad. Sci., LJ. S., 1952, v. 38, p. 667.
18. Rosenstock H. M,, Kraus M. Current Status of the Statistical Theory of
Mass Spectra, Advances in Mass Spectrometry, v. 2, Pergamon Press,
New York, 1963.
19. Vestal M. L. J. Chem. Phys., 1965, v. 43, p. 1356.
20. Stevenson D. P. Rad. Res., 1959, v. 10, p. 610.
21. Mariner T., Bleakney W. Phys. Sci., 1947, v. 72, p. 807.
22. Friedman L., Long F. A.. Wolfsberg M. J. Chem. Phys., 1957, v. 27, p. 613.
23. Ausloos P., Gorden R., Lias S. G. J. Chem. Phys., 1964, v. 40, p. 1854.
24. Field F. H., Franklin J. L. Electron Impact Phenomenon. Academic Press,
New York, 1957.
25. Erhardt von H., Osberghaus G. Z. Naturforschug, 1960, Bd. 15, S. 575.
26. Essex H. J. Phys. Chem., 1954, v. 58, p. 42.
27. Meisels G. G-, Sworski T. J. J. Phys. Chem., 1965, v. 69, p. 2867.
405
28. Thompson S. О., Schaeffer О. A. J.A.C.S., 1958, v. 80, p. 553.
29. Schaeffer O. A., Thompson S. O. Rad. Res., 1959, v. 10, p. 671.
30. Borkowski R. P., Ausloos P. J. J. Chem. Phys., 1962, v. 37, p. 877.
31. Cermak V„ Herman Z. Cool. Czeck. Chem. Commun., 1965, v. 30, p. 169-
32. Ausloos P., Lias G. J. Chem. Phys., 1965, v. 43, p. 127.
33. Yang K., Gant P. L. J. Phys. Chem., 1961, v. 65, p. 1861.
34. Johnson G. R. A., Warman J. M. Trans. Far. Soc., 1962, v. 61, p. 512.
35. Warman J. M. Nature, 1967, v. 213, p. 381.
36. Doepker R. D., Ausloos P. J. Chem. Phys., 1965, v. 43, p. 3841.
37. Ausloos P., Lias S. G., Sandoval I. B. Disc. Far. Soc., 1963, v. 36, p. 66.
Глава VIII
1. Duane W., Scheuer O. Le Radium, 1913, v. 10, p. 33.
2. Allen A. O. Radiation Chemistry of Water and Aqueous Systems. N. Y,
1961.
3. Kovan L., Moorthy P. W„ Weiss J. J.A.C.S., 1964, v. 86, p. 771.
4. Foner S. N., Hudson R. L. J. Chem. Phys., 1953, v. 21, p. 1608.
5. Gordon S., Hart E. J.. Thomas J. K. J. Phys. Chem., 1964, v. 68, p. 1262.
6. Platzman R. L. Basic Mechanisms in Radiobiology 11, Physical and Che-
mical Aspects. Nat. Acad. Sci., Publ. No. 305, 1953, p. 35.
7. Allen A. O., Barr N. J. Phys. Chem., 1959, v. 63, p. 920.
8. Czapski G., Schwartz H. A. J. Phys. Chem.. 1962, v. 66, p. 471.
9. Boag J. W., Hart G. Nature, 1963, v. 197, p. 45.
10. Boag J. W. Amer. J. Roent., Radium Therapv. Nucl. Med., 1963, v. 40,
p. 896.
11. Samuel A. H., Magee J. L. J. Phys. Chem., 1953, v. 21, p. 1080.
12. Platzman R. L. U.S. Nat. Res. Council, Publ. No. 305, 1953.
13. Kupperman A., Belford G. G. J. Chem. Phys., 1962, v. 36, p. 1412.
14. Kupperman A. Diffusion Kinetiks in Radiation Chemistry, in V. M. Ilais-
sinski (ed.). The Chemical and Biological Action of Radiations, v.
Academic Press, London, 1961, p. 85.
15. Kupperman A, Diffusion Model in Radiation Chemistry, in G. Silini (ed.),
«Radiation Research», North-Holland Publishing Co., Amsterdam, 1967.
16. Ganguly A. B., Magee J. L. J. Chem. Phys., 1956, v. 25, p. 129.
17. Sworski T. J. J.A.C.S., 1954, v. 76, p. 4687.
18. Sworski T. J. Rad. Res., 1955, v. 2, p. 26.
19. Schwartz H. A. J.A.C.S., 1955, v. 77, p. 4960.
20. Baxendale J. H. Rad. Res., Suppl. A, 1964, p. 139.
21. Rabani J., Matheson M. S. J. Phys. Chem., 1966, v. 70, p. 761.
22. Czapski G., Allen A. O. J. Phys. Chem., 1962, v. 66, p. 262.
23. Hochanadel C. J. ORNL Report No. 3815, May 6, 1965, p. 32.
24. Hochanadel C. J. Personal communication,
25. Keene J. P. Rad. Res., 1964, v. 22, p. 14.
26. Rigg T., Stein G., Weiss J. J. Proc. Roy. Soc., 1952, v. A211, p. 375.
27. Davis T. W., Gordon S., Hart E. J. J.A.C.S., 1958, v. 80, p. 4487.
28. Thomas J. K-> Hart E. J. Rad. Res., 1962, v. 17, p. 408.
29. Sweet J. P., Thomas J. K. J. Phys. Chem., 1964, v. 68, p. 1363.
30. Challenger G. E., Masters B. J. J.A.C.S., 1965, v. 77, p. 1063.
31. Challenger G. E., Masters B. J. J. Phys. Chem., 1955, v. 59, p. 1093.
32. Hart E. J., Thomas J. K., Gordon S. Rad. Res., Suppl., 1964, v. 4, p. 79.
33. Garrison W. M. Rad. Res., Suppl. 4, 1964.
34. Weeks В. M., Cole S. A., Garrison W. M. J. Phys. Chem., -1965, v. 69,
p. 4131.
35. Brams R. Rad. Res., 1967, v. 30.
36. Fumimoto M., Ingram D. J. E. Trans. Far. Soc., 1958, v. 54, p. 1304.
37. Anbar M., Hart E. J. J. Phys. Chem., 1967, v. 71, p. 3700.
38. Тальрозе В., Франкевич M. Всесоюзная конференция по радиационной
химии. М., 1957.
,406
Глава IX
J. Clark G. L„ Pickett L. W. J.A.C.S., 1930, v. 52, p. 465.
2. Baumeister P. W., Glocker G. Z. Physik. Chem., 1921, Bd. 97, S. 368.
3. Allen A. O., Hummel A. Disc. Far. Soc., 1963, No. 36, p. 147.
4. Hummel A., Allen A. O. J. Chem. Phys., 1966, v. 44, p. 3426.
5. Freeman G. J. Chem. Phys., 1963, v. 34, p. 988.
6. Sauer M. C., Arai S., Dorfman L. M. J. Chem. Phys., 1965, v. 42, p. 708.
7. Ambar M., Hart E. J. J. Phys. Chem., 196o, v. 69, p. 1244.
8. Arai S., Dorfman L. M. J. Chem Phys., 1964, v. 41, p. 2754.
9. Воеводский В., Молин Ю. Rad. Res., 1962, v. 17, p. 366.
.10. Okabe H., McNesby J. R. J. Chem. Phys., 1962, v. 37, p. 1340.
11. Dyne P. J., Denhartog J., Smith D. R. Disc. Far. Soc., 1963, No. 36, p. 315.
12. Burns W. G., Winter J. A. Disc. Far. Soc., 1963, No. 36, p. 124.
13. Dugie D. L., Freeman G. R. Trans. Far. Soc., 1965, v. 61, p. 1174.
14. Hardwick T. J. J. Phys. Chem., 1960, v. 64, p. 1263.
15. Kevan L„ Libby W. F. J. Chem. Phys., 1963, v. 19, p. 1283.
16. Rzad S. J., Shuler R. H. Paper presented at the 154th meeting of the
ACS, Sept. 1967. Abstract. No. 74, Division of Physical Chemistry (To be
published).
17. Holroyd R. A. Radical Yields in Hydrocarbon Radiolysis. (In press).
18. Ncwilt T. D., Remsberg L. P. J. Phys. Chem., 1960, v. 64, p. 969.
19. Roberts J., Hamill W. .1. Phys. Chem., 1966, v. 70, p. 910.
20. Halroyd R. W. e. a. J. Phys. Chem., 1966, v. 70, p. 910.
21. Fallgatter M. B., Hanrahan R. J. Paper presented at the 154th meeting of
the ACS, Sept. 1967. Abstract No. 75, Division of Physical Chemistry.
22. Williams R. R., Hamill W. H. Rad. Res., 1954, v. 7, p. 158.
23. Fessenden R. W., Shuler R. H. J. Chem. Phys., 1963, v. 39, p. 2147.
24. Shuler R. H. J. Phys. Chem., 1958, v. 62, p. 37.
25. Adloff J. P., Guequeniat P. J. Chromatog., 1963, v. 12, p. 96.
26. Perner D,, Gnarra D. Mellon Inst. Report RRL-147, 1964.
27. Perner D., Shuler R. M. J. Phys. Chem., 1966, v. 70, p. 2224.
28 Mani I., Hanrahan J. J. Phys. Chem., 1966, v. 70, p. 2233.
29. Shuler R. H. J. Phys. Chem., 1964, v. 68, p. 3873.
30. Collinson E., Convay J. J., Dainton F. S. Disc. Far. Soc., 1963, v. 36, p. 153.
31. Holroyd R. A., Klein G. W. J.A.C.S., 1962, v. 84, p. 400.
32. Holroyd R. A„ Klein G. W. J. Appl. Rad. Isotopes, 1963, v. 13, p. 493.
33. Holroyd R. A., Klein G. W. J. Appl. Rad. Isotopes, 1964, v. 15, p. 633.
34. Holroyd R. A., Klein G. W. J. Phys. Chem., 1965, v. 69, p. 194.
35. Shuler R. H., Kuntz R. R. J. Phys. Chem., 1963, v. 67, p. 1004.
36. Mannion J. P., Burton M. J. Phys. Chem., 1952, v. 56, p. 560.
37. Shuler R. H. J. Phys. Chem., 1957, v. 61, p. 1472.
38. Hardwick T. J. J. Phys. Chem., 1962, v. 66, p. 767.
39. Kailman H., Furst M. Phys. Rev., 1950, v. 78, p. 857.
40. Lipsky S., Burton E. M. J. Chem. Phys., 1959, v. 31, p. 1221.
41. Fano LI. Comparative Effects of Radiation. M. Burton e. a. eds, John Wi-
ley and Sons, New York, 1960.
42. Dyne P. J., Denhartog J., Smith D. P. Disc. Far. Soc., 1963, v. 36, p. 135.
43. Collinson E., Conway J. J., Dainton F. S. Disc. Far. Soc., 1963, v. 36, p. 153.
44. Burton M., Chang J., Lipsky S., Reddy M. P. J. Chem. Phys., 1957,
v. 29, p. 1337.
45. Burns J. J. Phys. Chem., 1961, v. 65, p. 2261.
46. Cundall R. B., Griffiths P. A. Disc. Far. Soc., 1963, v. 36, p. 111.
47. Collinson E., Todd J. F. J., Wilkinson F. Disc. Far. Soc., 1963, v. 36, p. 83.
48. Caldwell R. A., Whitten D. G„ Hammond G. S. J.A.C.S., 1966, v. 88, p. 2659.
49. Hentz R. R., Peterson D. B., Srivastava S. B., Barzynski S. B., Burton M.
J. Phys. Chem., 1966, v. 70, p, 2362.
50. Golub M. A., Stephens C. L., Brash J. L. J. Chem. Phys., 1966, v. 45, p. 1503.
51. Falconer J. W., Burton M. J. Phys. Chem., 1963, v. 67, p. 1743.
52. Burns W. G. Trans. Far. Soc., 1962, v. 58, n. 961.
407
53. Burns W. G. Trans. Far. Soc., 1963, v. 59, p. 101.’
54. Wagner C. D. J. Phys. Chern., 1960. v. 64, p. 231.
55. Fessenden R. W., Shuler R. H. 'Disc. Far. Soc., 1963, v. 36, p. 149.
56. De Vries A. E., Allen A. O. J. Phys. Chem., 1959, v. 53, p. 879.
.Глава X
I. Van-Bueren H. G. Imperfections in Crystals. North-Holland Publishing Co.,
Amsterdam, 1961.
2. Ronayne M. R., Hamill W. H., Guarino J. P. J.A.C.S., 1962, v. 84, p. 4230.
3. Hamill W. H., Guarino J. P., Ronayne M. R., Ward J. A. Disc. Far. Soc.,
1963, v. 36, p. 169.
4. Kanzig W. Phvs. Rev., 1955, v. 99, p. 1890.
5. Kanzig W., Woodruff T. O. Phys. Rev., 1958, v. 109, p. 220.
6. Kanzig W. Phys. Rev. Letters, 1960, v. 4, p. 117.
7. Seitz F. Rev. Mod. Phjs., 1954, v. 26, p. 7.
8. Przibram K. Irradiation Colours and Luminescence. Pergamon Press,
London, 1956.
9. Querre Y. In: M. Haissinsky fed.), «Action Chimique et Biologiques State
Physics», Masson, Paris, 1964.
10. Seitz F., Koehler J. S. In: F. Seitz. D. Turnbull (eds), «Solid State Phy-
sics», v. 2. Academic Press, Inc.. New York, 1956.
If. Varley J. H. O. Nature. 1954. v. 174. p. 886.
12. Carlson T. A., Krause M. O. Phys. Rev., 1965, v. 140A, p. 1057.
13. Durup J., Platzman R. L. Disc. Far. Soc., 1961, v. 31, p. 156.
14. Pooley D. Color Center Conferences, U. of 111., 1965.
15. Singer L. S., Kommandeur J. J. Chem. Phys., 1961, v. 32, p. 1013.
16. Hoosterey D. C., Leston G. M. J. Phys. Chem. Solids, 1963, v. 24, p. 1609.
17. Dines G. J., Damas A. C. In: G. M. Schwab (ed.), 5th International Sym-
posium of the Reactivity of’ Solids, Elsevier Publishing Co., New
York, 1965.
18. Rose A. Concepts in Photoconductivity, Chap. 4, Interscience Publishers,
New York, 1963.
19. Serlin I. Science, 1957, v. 126, p. 261.
20. Fenrick H. W., Filseth S. LI., Hanson A. L., Willard J. E. J.A.C.S., 1963,
v. 85, p. 3731.
21. Stacey F. W., Sauer J. C., McKusick В. C. J.A.C.S., 1959, v. 81, p. 987.
22. Boyd G. Private communication.
23. Okamura S., Hayashi K., Kitanishi Y. J. Folym. Sci., 1962, v. 58, p. 927.
24. Kevan L., Libby W. F. Recent Advances in Photochemistry, 1963, p. 183.
25. Knox R. S. Theory of Excitons. Solid State Physics Supplement 3, F. Seitz,
D. Turnbul (eds), Academic Press, New York, 1963.
26. Давыдов А. С. Теория молекулярных экситонов. M., «Наука», 1968.
27. Jones A. R. J. Chem. Phys., 1961, v. 35, p. 751.
28. Воеводский В., Молин Ю. Rad. Res., 1962, v. 17, p. 366.
29. Magee J. L. Rad. Res., 1963. v. 20, p. 71.
30. Bass A. M., Brolda H. P. Stabilization of Free Radicals at Low Tempe-
ratures. NBS Monograph 12, 1960.
31. Shida T.. Hamill W. H. J.A.C.S.,' 1966, v. 88, pp. 2369, 2375, 3683, 3689,
4372. 5371. 5376.
32. Arai S., Dorfman L. M. J. Chem. Phys., 1964, v. 41, p. 2190.
33. Cunningham J. J. Phys. Chem., 1963, v. 67, p. 1772.
34. Cunningham J. J. Phys. Chem., 1961, v. 65, p. 628.
35. Hochanadel C. J. Rad. Res., 1962, v. 16, p. 3, 286.
36. Hall D„ Walton G. N. J. Inorg. Nuclear Chem., 1959, v. 10, p. 215.
37. Cunningham J. Trans. Far. Soc., No. 525, 62 (part 9), 1966, p. 2423.
38. Henning G., Lees R., Matheson M. J. Chem. Phys.. 1955, v. 21, p. 664.
39. Chen T., Johnson E. R. J. Phys. Chem., 1962, v. 66, p. 2249.
40. Pringsheim P. J Chem. Phys., 1953, v. 23, p. 369.
41. Cunningham J. J. Phys. Chem. Solids, 1962, v. 23, p. 843.
408
42. Jacard C. Phys. Rev., 1961, v. 124, p. 60.
43. Golding R, M., Henchman M. J. Chem. Phys., 1964, v. 40, p. 1554;
44. Zeldes H., Livingston R. J. Chem. Phys, 1961, v. 35, p. 563.
45. Cunningham J. J. Phys. Chem., 1967, v. 71, p. 1967.
46. Cunningham J. J. Phys. Chem., 1966. v. 70. p. 30.
47. Chen T. H., Johnson E. R. J. Phys. Chem., 1962. v. 66, p. 2068.
48. Prince L. A. Dissertation, Stevens Institute of Technology, Hoboken, N. J.
49. Prince L. A., Johnson E. R. J. Phys., 1965, v. 69, p. 359, 369, 377.
50. Heal H. G. Can. J. Chem., 1959, v. 37, p. 979.
51. Баберкин А. С. Материалы сессии отделения химических наук АН
СССР. М., 1958, с. 107.
52. Cole Т. Proc. Natl. Acad. Set. U.S., 1960, v. 46, p. 506.
53. Boyd G. E. e. a. J. Phys. Chem., 1962. v. 66, p. 3O0.
54. Boyd G. E. e. a. J. Phys. Chem., 1965, v. 69, p. 1413.
55. Boyd G. E. e. a. J. Phys. Chem., 1966, v. 70, p. 1031.
56. Chase J. M., Boyd G. E. Symposium on the Chemical and Physical
. Effects of High Energy Radiation. ASTM STP? 1966, v. 400, p. 17.
57. Herley P. J., Levy P. J. Chem. Phys., 1967, v. 46, p. 627,-
58. Баберкин А. С. Материалы сессии отделения химических паук АН СССР.
М., 1958, с. 187.
59. Huang S., Johnson Е. R. Radiation Effects Symposium, ASTM Committee
E-10, Seattle, Washington, N. 2—5, 1965.
60. Burchill С. E. Nature, 1962, v. 194. Reported by P. F. Patrick,
K. J. McCallum.
61. Sherman L. J., McCallum K. J. J. Chem. Phys., 1955, v. 23, p. 597.
62. Henning G., Lees R. Matheson M. S. J. Chem. Phys., 1953, v. 21, p. 664.
63. Burns W. G., Williams T. F. Nature (London), 1955, v. 175, p. 1043.
€4. Wiegand D. A., Smoluchowski R. The Production of Defacts in Alkali Ha-
lides, Actions Chemiques and Biologiques des Radiation, M. Haissinsky
(ed.), 7, Masson, Paris, 1964.
65. Compton W. D., Rabin H. Aggregate Centers in Alkali Halide Crystals, in
F. Seitz, D. Turnbul (eds). Solid State Physics, v. 16, Academic Press,
New York, 1964.
66. Gibbon C. F., Kuczynski G. C. Materials Science Research, v. 3, Plenum
Press, New York, 1966, p. 131.
67. Heal H. G. Can. J. Chem., 1953, v. 31, p. 1153.
68. Cunningham J. Trans. Far. Soc., 1966, 62 (Part 9), p. 2423.
69. Cunningham J. C-, Tompkins F. C. Proc. Roy. Soc., 1959, v. Л251, p. 27.
70. Allen A. O. e. a. J.A.C.S., 1964, v. 86, p. 3887.
71. Hentz R. R. J. Phys. Chem., 1962, v. 66, p. 1625.
Глава XI
1. Ballantine D. Chem. Eng. Prog., 1954. v. 50, p. 267.
2. Smith W. V., Ewart R. P. J. Chem. Phys., 1948, v. 16, p. 592.
3. Acres G. J., Dalton F. L. Polymer Sci., Part A, 1963, v. 1, No. 10,
p. 3013.
4. Ley G., Hummel D., Schneider C. Gamma-Radiation-Induced Polimerization
of Some Vinyl Monomers in Emulsion Systems. Advances in Chemistry
Series 66, ACS, 1967, p. 184.
5. Lampe F. J. Chem. Phys., 1959, v. 63, p. 1986.
6. Chandler H., Henley E., Trachtenberg E. Int. J. Apl. Rad. Isotopes, 1962,
v. 13, p. 239.
7. Metz D. Radiation-Induced Polymerization of Pure Liquid-Methylstyrcne.
Advances in Chemistry Series 66, ACS, 1967, p. 170.
Монографии
1. Бовей Ф. Л. Действие ионизирующих излучений на природные и синте-
тические полимеры. Пер. с англ. М„ Изд-во иностр, лит., 1959.
409
2. Chapiro A. Atomic Radiation and Polymers. Pergamon Press, London, I960.
3. Gould R. F. (ed.). Irradiation of Polymers. Advances in Chemistry Series
66. ACS, 1967.
Глава XII
1. Harlan P. e. a. J. Polym. Sci., 1955, v. 18, p. 589.
2. Бовей Ф. Л. Действие ионизирующих излучений на природные п синтети-
ческие полимеры. Пер. с англ. М„ Изд-во иностр, лит., 19э9.
3. Чарлсбн А. Ядерные излучения и полимеры. Пер. с англ. М., Изд-во
иностр, лит., 1962.
4 Omerod М. In: Е. Henley, Н. Kouts (eds). Advances in Nuclear Science
and Engineering, v. 2. Academic Press, New York, 1964.
5. Dole M. Reactive Intermediates in the Radiation Chemistry of Polyethyle-
ne. Advances in Chemistry Series 66, v. 31, ACS, 1967.
6. Chapiro A. Chemical Nature of the Reactive Species Produced in Polymers
by Ionizing Radiation. Advances in Chemistry Scries 66, v. 25, ACS, 1967.
7. Wall L., Brown D. W. J. Res. Nat. Bur. Stds., 1956, v. 57, p. 131.
Глава XIII
1. Jefferson S., Roberts R., Ley F. J., Rogers F. Advances in Nuclear Science,
and Technology. Greebler, E. Henley (ed.). Academic Press, Inc., New
York, 1968.
2. Timmerman R. Plastics World, August 1963, p. 468.
3. Isotopes and Radiation Tecrnology, U.S. Dept, of Commerce, Spring,
1965, p. 263.
4. Kuckacka L. E., Manowitz B. Nucleonics, Jan. 1965, v. 74, p. 85.
5. Artnstrong W. A., Spinks J. W. T. Can. J. Chem., 1959, v. 37, p. 1002.
6. Harmer D. E., Beale J. S. Chemical Eng. Prog., 1964, v. 60, No. 4, p. 33-
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азиды твердые 327
Азотные соединения 318—320; ПО
Акрилонитрил 359—362
полимеризация в растворе 355
Активация нейтронами 61
Активность удельная 70—73
Акцептирование радикалов 143—145,
211, 294
«-Частицы 23
источники 70—73
пробег 23-—25
Аминокислоты 255—257
Антраценовый дозиметр (21
Атактические полимеры 334
Атомные смещения 292—296
Атомы внедрения (дефекты Френ-
келя) 294
Ацетилцеллюлоза, привитая 359—362
Ацетилен 187, 217
Бензол 258
fi-Частины (см. также электроны (3-
излучение) 25—30
Бимолекулярные реакции 138
Бора, магнетон 171
Бромид калия 312
Броматы, твердые 323—324
Брэгга — Грея, условие 82
Вакансии (дефекты Шоттки) 293
Варли, механизм 305
Вековое (секулярное) равновесие 15
Внутренняя конверсия 136
Вода 220—260
выход молекулярных продуктов 239
выход первичных частиц 221
выход радикальных продуктов
240—246
Водные системы 220—260
Водорода перенос, реакция 149
Возбуждения, потенциал 20
Воздушная камера 91—92
Волновое число 6
Вращающийся сектор 342
Выход квантовый 153
Выход .молекулярных продуктов 239
Выход радиационно-химический 81
Газовыделенпе из облученного поли-
этилена 372
Гальвипокисл 275
Гамма-ячейка 74
Гейгера — Мюллера, счетчик 119—120
Гелеобразование 378—379
Гексан 289
Гиббса, свободная энергия 293
Гидратированный электрон 132,
Горячие атомы 152
Горячие радикалы 152
Гука, закон 330
Деструкция полимеров 370
Дефекты в твердых телах 292—296
Джоуль 6
Диметнлбутап 278
Дииа.митрон 62
Дислокации 295—296
винтовые 295
краевые 296
Диспрозиевая фольга 126
Диспропорционирование 143
Дифенплпнкрилгидразил (ДФПГ) 275
Диффузионная кинетика 366
Диффузионная теория 227—230
Дициклогекснл 279
Диэлектрическая проницаемость 331
Дозиметрия 81 —130
активационная 123—126
калориметрическая 92—98
ионизационная 82—92
фотографическая 115
химическая 99—ПО
Дозиметры 81—130
сцинтилляционные 120—122
411
твердотельные 116—122
химические 99—НО
бензойная кислота 109
бензол 109
пленочный из пластика 113—114
поливинилхлоридный 114
полиэтиленовый 114
полиэфирный 114
стеклянный 116
ферросульфатиый (Фрике) 100—
107
фотопленочиый 115
хлороформ 109
целлофан 113
четыреххлористын углерод 109
Единицы 5
систем СГС и СГСЭ 5
экспозиционной дозы 9
поглощенной дозы 8
Жидких углеводородов, радиолиз
261—291
Заряженные частицы 17—30
взаимодействие с веществом 18—
30
Захват электронов с последующей
диссоциацией 268
Защитный эффект 390
Излучение Вавилова — Черенкова 30
Изотактические полимеры 334
Изотопная метка 276
Изотопные источники излучения 69—
75
Ингибирование 336
Инертные газы, влияние на радио-
лиз 209—211
Ион-молекулярные реакции 148—151,
188—200, 266
константы скоростей 199—200
перепое одной частицы 148—151
сечения в кондесированных угле-
водородах 266
Ионизации, потенциал 180
Ионизационные камеры 82—92
Калориметрия в дозиметрии 92—98
Калория 6
Камера полостная 82—83
Качества коэффициент 10
/(-Захват 12
Кинетика 155—158
Клейна —Нишины, формула 35
Кобальт-60 69—74
Кокрофта — Уолтона, ускоритель 56
Комптоновское рассеяние 33—36
Константы скорости реакций 155
Коронный разряд '55
Коэффициенты поглощения 38—43
линейный 32
массовый 39
общий 39
передачи энергии 39
рассеяния 37
Кулиджа, трубка 64
Кулона, закон 5
Кюри, определение 10
Ламберта — Бэра, закон 154, 160
Ли—Грея—Платцмана, модель ра-
диолиза воды 224
Линейная передача энергии (ЛПЭ)
226
Линейный ускоритель электронов 63—
64
Максвелловское распределение 48
Марганец, активация 126
Массовый
коэффициент ослабления 39
коэффициент передачи энергии 39*
коэффициент поглощения 39
Масс-спектрометр, применения 176—
184
Метана, радиолиз 214—218
Метилподид 276
Метильные радикалы 141 —143, 278
МКРЕ, рекомендации 8—9
Моиомолекулярные реакции 138, 155
Мономолекулярный распад 138. 20?
Монте-Карло, метод 76
Муравьиная кислота, водный раствор
254
Наперстковая ионизационная камера
90—91
Натрий, активация 126
Ненасыщенность в облученном поли-
этилене 373
Нейлон, привитая полимеризация на
359—362
Нейтроны 47—52
быстрые 48
взаимодействие с твердым телом
301—302
дозиметрия 122—128
источники 66—68
плотность потока 68
промежуточные 48
резонансное взаимодействие 5[
сечения взаимодействия 51
спектр деления 68
тепловые 48
Нитраты, твердые 318—320
Относительная биологическая эффек-
тивность (ОБЭ) 10
Озон 186
Окислительная деструкция полимеров
383
412
Окислительно-восстановительный по-
тенциал 2.36—237 .
Органические соединения
водные растворы 254—256
Органические твердые тела 312—315
Осколки деления 46—47
Пар, образование 37—38
Пентан 286-287
Первичные процессы 133
Передача цепи 336
Перенос заряда 148
Перенос энергии 137
Перхлораты, твердые 320—323
Планка, постоянная 7
Поглощенная доза 8
измерения ионизационные 82—92
калориметрические 92—98
методом собирания заряда 98
Полимеризация 334—368
блочная 338
в растворе 346—349
кинетика 339—342
привитая 366
радиационная 341—349
эмульсионная 350 —352
Полимеры 330—396
блочные 335
действие излучений на 369—391
кристалличность 333
.молекулярный вес 332
образование свободных радика-
лов 335
орнитированпые 333
привитые 335
электрические свойства 331
Полнметилметакрилат 387—391
газовыделенпе при облучении 387
действие излучений на
механические свойства 390
разрывы основного валентного
остова 388
узоры Лихтенберга 391
Полиэтилен 371—387
действие излучений на
механические свойства' 385
окислительные реакции 383
«памяти» эффект 386
привитая полимеризация 361
степень кристалличности 384
сшивание 382
удельный объем 384
электрические свойства 386
Полиэфиры, привитая полимериза-
ция на 359—362
Появления, потенциал 180
Пре.писсоцнация 138
Примесные атомы в твердых телах
316
Пробег 22, 27
Промышленные приложения 392—401
Пропорциональный счетчик 119, J20
Протона, реакция переноса 149
Рад, определение 8
Радикалы, определение 132
активность 140
акцептирование (захват) 143
выход в облученных мономерах
339
выход в углеводородах 269
образование 140
первичное распределение
в облученной воде 225
реакции 140—146
диспропорционирования 143
диссоциации 144
замещения 141
присоединения 143
переноса электрона 146—147
рекомбинации 142
термодинамика 141
Радиоактивного распада, постоянная
14
Радиоактивность 10—16
Разрывы основного валентного осто-
ва 388
Разряд электрический 55
Рассеяние 17
иеупругое 17, 50
упругое 17, 48
Рекомбинация
ионон 201
радикалов 142
Релеевское рассеяние 37
Рентген, определение 9
соотношение с радом 9
Рентгеновское излучение 64—66
возникновение 64—65
прохождение через вещество 30—
45
спектр энергетический 65—66
характеристическое 66
Репульсивпос состояние 134
Самопоглощение 76
Самуэля — Маги, модель радиолиза
воды 224
Сверхтонкое взаимодействие 172
расщепление 172
Синдиотактические полимеры 334
Скольжения, плоскость 296
Слой половинного ослабления 65
Сополимеризация 337
Спайк тепловой 304
электронный 304
Спектр поглощения 163
нейтронный 68
Спектр деления 68
413
Стирол 343—352
полимеризация в растворе 346 —
350
полимеризация с осаждением 350
сополимеризация 352
эмульсионная полимеризация 351
Стронций-90 69—75
Сульфаты, твердые 325
.Суммы по состояниям 190
•Сциларда— Чалмерса, реакции 13
Сцинтилляционный детектор 121
Сшивание, радиационное
полимеров 377—384
полиэтилена 382
Твердое тело 292—329
действие излучений па 300—307
гетерогенное 328
перенос энергии 311—312
радиационные повреждения 300—
307
электрические свойства 308—309
Терфенил 280
Тлеющий разряд 55
Томсоновское рассеяние 37
Трансформатор с изолированным
сердечником 60—62
Треки, распределение первичных ча-
стиц 29
Триплетное состояние 135
Трифснилметил 276
Троммсдорфа, эффект 336
Углеводороды, жидкие 261—291
влияние мощности дозы 282—284
возбужденные молекулы 262, 264
высокие дозы 288
ион-молекулярный распад 265
кинетика 284—288
первичные процессы 262
перенос энергии 279—281
термодеструкция 290
Угловое распределение излучения 65
Урап-235 52
•Фактор накопления 46
Фентона, реактив 246
Ферросульфатиый дозиметр 100—107,
246—253
величина G 104
величина G при импульсном ра-
диолизе 248
влияние ЛПЭ 103, 249
влияние мощности дозы 105
влияние pH 250
влияние температуры 103
Ферросульфат — сульфат меди
дозиметрический раствор 253
Фильтры 65
•Флуоресценция 135
•Флэш-фотолиз 159
Фольга 125—126
золотая 126
индиевая 126
Фононы 307
Фосфоресценция 136
Фотохимия 152—155
Фотоэффект 31—33
Фотоядерные реакции 43—44
Френкеля, дефекты (атомы внедре-
ния) 293
Фрике, дозиметр (см. ферросульфат-
ный дозиметр) 100—107
Хлор, активация 126
Хлораты, твердые 326
Хлоруксусная кислота, водная 235
Цвиттер-ион 255
Цезий-137 69—75
Центры окраски в твердых телах
297—300
Цепные реакции 335—337
Цериевосульфатпый дозиметр 107—
108
Циклогексан, радиолиз 278—280
Шоттки, дефекты (вакансии) 293
Экситон 137, 311
Экстраполяционная камера 92
Электромагнитное излучение 30—45
Электрическая прочность 331
Электронвольт 5
Электронное равновесие 83
Электронный парамагнитный резо-
нанс (ЭПР) 170—176
Электроны 25 -30
взаимодействие с веществом 25—
30
гидратированные 132
захваченные в ловушки 297—300
Оже 12
сольватированные 132
Энергетические потери 138
Энергия
активации 155
возбуждения 133
кинетическая 7
массы покоя 7
Энергия диссоциации связен 133
Энергия ценообразования 82, 84
Эрстед 171
Этил бромистый 397—401
Эйнштейн 160
Эйринга —Хнршфельдера — Тейло-
ра, теория 189
Эффект клетки 316
Ядерные реакторы как источники
излучения 67—69
Ядериые реакции 12
ОГЛАВЛЕНИЕ
Глава 1. Излучения и радиоактивность Основные единицы измерения 5
Глава II. Взаимодействие излучения с веществом...........17
Глава Ill. Источники излучении............................54
Глава IV. Дозиметрия излучений...........................81
Глава V. Химические последствия поглощения излучений высокой
энергии в веществе...............................131
Глава VI. Современные методы радиационной химии . . 159
Глава VII. Радиационная химия газов......................185
Глава VIII. Радиационная химия воды и водных систем...... 220-
Глава IX. Действие излучений на жидкие углеводороды . . . 261
Глава X. Радиационная химия твердого тела......................292
Глава XI. Радиационная полимеризация . 330
Глава XII. Действие излучений на полимеры.........................369
Глава XIII. Промышленное использование радиационно-химических про-
цессов............................................................ 392
Список литературы................................................ 402:
Предметный указатель........................................... 411
Э. Хенли, Э. Джонсон
РАДИАЦИОННАЯ ХИМИЯ
Редактор В. 11. Вяземцева Художественный редактор А. Т. Кирьянов
Обложка художника С. II. Голубева Тохи, редактор И. Н. Подшебякин
Корректоры О. Р. Харламова, О. М. Герасимова
Сдано в набор 26/III 1974 г. Подписано к печати G/VI1I 1974 г. Формат бОхОО’/тв
Бумага типографская Хе 2 • Усл. печ. л. 26 Уч.-изд. л. 28,58 Тираж 3000 экз.
Зак. изд. 71322 Зак. тип. 907 Цена 3 р. 10 к.
Атомиздат. 103031, Москва, К-31, ул. Жданова, 5/7.
Московская типография № 6 Союзполиграфпрома
при Государственном комитете Совета Министров СССР
по делам издательств, полиграфии и книжной торговли
109088, Москва, Ж-88, Южноясртсвая ул., 24.