/

































































































































































































































































































Текст
А. П. НЕЧАЕВ
ОРГАНИЧЕСКАЯ
ХИМИЯ
Допущено
Министерством пищевой промышленности СССР
в качестве учебника для техникумов
пищевой промышленности
МОСКВА «ВЫСШАЯ ШКОЛА» 1976
547
H59
УДК 547@75)
Рецензенты:
проф. А. Л. Маркман
(Калининский политехнический нн-т) и преподаватель Е. К. Белова
(Московский механико-технологический техникум
пищевой промышленности)
Алексей Петрович Нечаев
ОРГАНИЧЕСКАЯ ХИМИЯ
Редактор М. М. Поплавская. Художник А. В. Исиченко. Художественный
редактор Т. М. Скворцова. Технический редактор 3. В. Нуждина. Корректор
С. К. Марченко
Сдано в набор 15/IX-75 г. Подп. к печати 9/1-76 г. Формат 60х90'/и- Бум. тип.
№ 3. Объем 18 печ. л. Усл. печ. л. 18. Уч.-изд. л. 18,17. Изд. № Хим.—522
Тираж 40 000 экз. Цена 65 коп.
План выпуска литературы издательства
«Высшая школа» (вузы и техникумы) на 1976^ г. Позиция № 239
Москва, К-51, Неглпнная ул., д. 39/14,
Издательство «Высшая школа»
Ордена Трудового Красного Знамени Ленинградское
производственно-техническое объединение «Печатный Двор» имени А. М. Горького Союзполиграф-
прома при Государственном комитете Совета Министров СССР по делам
издательств, полиграфии и книжной торговли. 197136, Ленинград, П-136,
Гатчинская ул., 26. Зак, 153
Нечаев А. П.
Н 59 Органическая химия. Учебник для техникумов. М.,
«Высш. школа», 1976.
288 с. с ил.
В основу построения учебника положен принцип классификации
органических соединений по функциональным группам.
В книге теоретические положения органической химии рассмотрены с
позиций электронной теории строения веществ. В доступной форме автор
раскрывает физический смысл квантовых чисел, на примерах рассматривает
химическую связь в молекулах органических веществ, направленность,
гибридизацию электронных орбитален. строение <J- и л- связей.
Рассматриваются углероды, нефти, спирты, альдегиды, кетоны,
органические кислоты, липиды и другие классы органических веществ.
Особое внимание уделено тем классам органических веществ, которые
составляют основную массу органического вещества пищевого сырья и
готовой продукции (спиртам, углеводам, белкам, витаминам).
20504—098 547
Н 239—76
001@1)—76
© Издательство «Высшая школа», 1976 г,
ПРЕДИСЛОВИЕ
Предлагаемая книга предназначается в качестве учебника по
органической химии для учащихся средних специальных учебных
заведений пищевой промышленности. Современная технология
переработки сельскохозяйственного сырья и получения пищевых
продуктов базируется на глубоком знании химических и биохимических
процессов, протекающих на всех этапах производства, начиная от
заготовок сырья и кончая получением из него готовых продуктов.
Питательная ценность последних определяется количеством и
составом содержащихся в них белков, углеводов, липидов, витаминов
и других соединений. Очевидно, что без глубоких знаний
органической химии немыслимо управление современной технологией
получения продуктов питания, поэтому задача настоящего пособия—
дать эти знания техникам-технологам пищевой промышленности.
Курс органической химии излагается на основе современных
теоретических представлений. Автор стремился уделить особое
внимание общим закономерностям, останавливаясь в то же время
на свойствах наиболее важных органических соединений.
Особенность настоящего учебника состоит в том, что в нем
особое внимание уделено тем классам органических соединений,
которые составляют основную массу органического вещества
пищевого сырья и готовой продукции: белкам, углеводам, липидам,
органическим кислотам и т. д,
Автор отказался от широко распространенной классификации
органических соединений по строению углеродного скелета,
положив в основу настоящего учебника классификацию органических
соединений по функциональным (характеристическим) группам.
Раздел книги, посвященный химии углеводов, написан доц.
Т. В. Еременко.
Данное руководство — первая попытка создать учебник по
органической химии для техников-технологов пищевой
промышленности. Естественно, что он имеет недостатки, автор будет
благодарен за все критические замечания.
Считаю своим долгом выразить особую благодарность
рецензентам докт. хим. наук, проф. А. Л. Маркману и преподавателю Е. К-
Беловой, а также докт. хим. наук, проф. В. Г. Кульневичу, канд.
хим. наук, доц. Н. В. Зотчик, канд. техн. наук, доц. М. П. Попову
и сотрудникам кафедры органической химии Московского
технологического института пищевой промышленности, просмотревшим
рукопись, за ценные замечания и советы.
ВВЕДЕНИЕ
Предмет органической химии и причины выделения ее в
самостоятельную дисциплину. Органическая химия изучает соединения
углерода с другими элементами, которые называются
органическими соединениями, и законы, которым подчиняются превращения
этих веществ.
Чем вызвано, что изучение соединений только одного элемента—
углерода — составляет содержание целой науки? Причины
заключаются в многочисленности и разнообразии органических
соединений, в их специфическом строении и свойствах, особенностях
протекания органических реакций, а также в большом
практическом значении соединений углерода.
Число известных органических веществ составляет несколько
миллионов. Некоторые простые соединения углерода, такие как
окись и двуокись, соли угольной и синильной-кнслот, изучаются
в курсе неорганичной химии. Неорганических веществ известно
значительно меньше (несколько десятков тысяч).
Многочисленность соединений углерода вызвана рядом причин:
способностью его соединяться с атомами большинства других
элементов; образовывать углерод — углеродные цепи различного
строения с практически неограниченным числом атомов;
соединяться в кольца или циклы:
Mill I ! I I I
—С—С—С—С—С— —С—С—С—С—С-
I I I ! I I ! ! I 1
прямая углерод — углеродная —С—
цепь I
разветвленная углерод —углерод- циклическая
ная цепь углеродная цепь
Атомы углерода способны образовывать друг с другом не только
одинарные, но и двойные, и тройные связи (каждая черточка
условно обозначает одну связь):
—С—С— — С=С— —С==С—
! I
одинарная связь двойная связь тройная связь
В органической химии чрезвычайно распространено явление
изомерии, заключающееся в существовании соединений с одннако-
вым элементарным составом и молекулярной массой, но различными
физическими и химическими свойствами. Наличие изомеров
приводит к резкому увеличению числа органических соединений. Так,
углеводород эйкозан С20Н1г существует в виде 336 319
изомеров.
Большинство органических соединений при комнатной
температуре представляет собой легко горючие газы, жидкости или
твердые вещества, температуры плавления которых обычно не
превышают 400° С. Большинство неорганических соединений —
твердые, не горючие, высокоплавкие вещества.
Многие реакции, в которых участвуют органические вещества,
протекают медленно, с незначительным выходом. Реакции в
водных растворах между неорганическими электролитами протекают
мгновенно, с количественным выходом.
Органические соединения сложнее неорганических. При их
изучении мы имеем дело с более высокоорганизованной материей.
Большинство соединений углерода имеют большое практическое
значение: нефть, пластмассы, каучук, синтетические и
искусственные волокна, красители, органические соединения, применяемые
в сельском хозяйстве (инсектициды, фунгициды, гербициды,
ростовые вещества), медицинские препараты, витамины и ферменты и
другие.
Нефть является не только важнейшим источником энергии,
но и основным сырьем для получения большинства органических
соединений. Выделяемые из нефти или получаемые при ее
химической переработке многие из них, в свою очередь, являются сырьем
для разнообразных отраслей химической промышленности.
Созданные в последние десятилетия искусственные
полимерные вещества (пластмассы, каучуки, волокна, пленки,
полимерные покрытия) не только восполнили убыль в традиционных
материалах, известных человеку с давних пор, но благодаря многим
ценным свойствам привели к перевороту в ряде отраслей
промышленности. Применение органических соединений в сельском
хозяйстве способствовало резкому росту урожайности, снижению
себестоимости и увеличению выпуска сельскохозяйственной продукции.
Использование в медицине синтетических лекарственных
препаратов избавляет человека от многих недугов, что является одной
из причин увеличения средней продолжительности жизни.
Все это и было причиной выделения органической химии в
самостоятельную науку.
В то же время было бы неправильным считать, что между
органическими и неорганическими соединениями существует
пропасть. «Подобно тому, как одна форма движения развивается
из другой, — пишет Ф. Энгельс, — так и отражение этих форм,
различные науки, должны с неизбежностью вытекать одна из
другой» *.
* Ф. Энгельс. Диалектика природы. К. Маркс и Ф. Э н г е л ь с.
Соч., т. 20. М., Госполитиздат, 1961, с. 65.
5
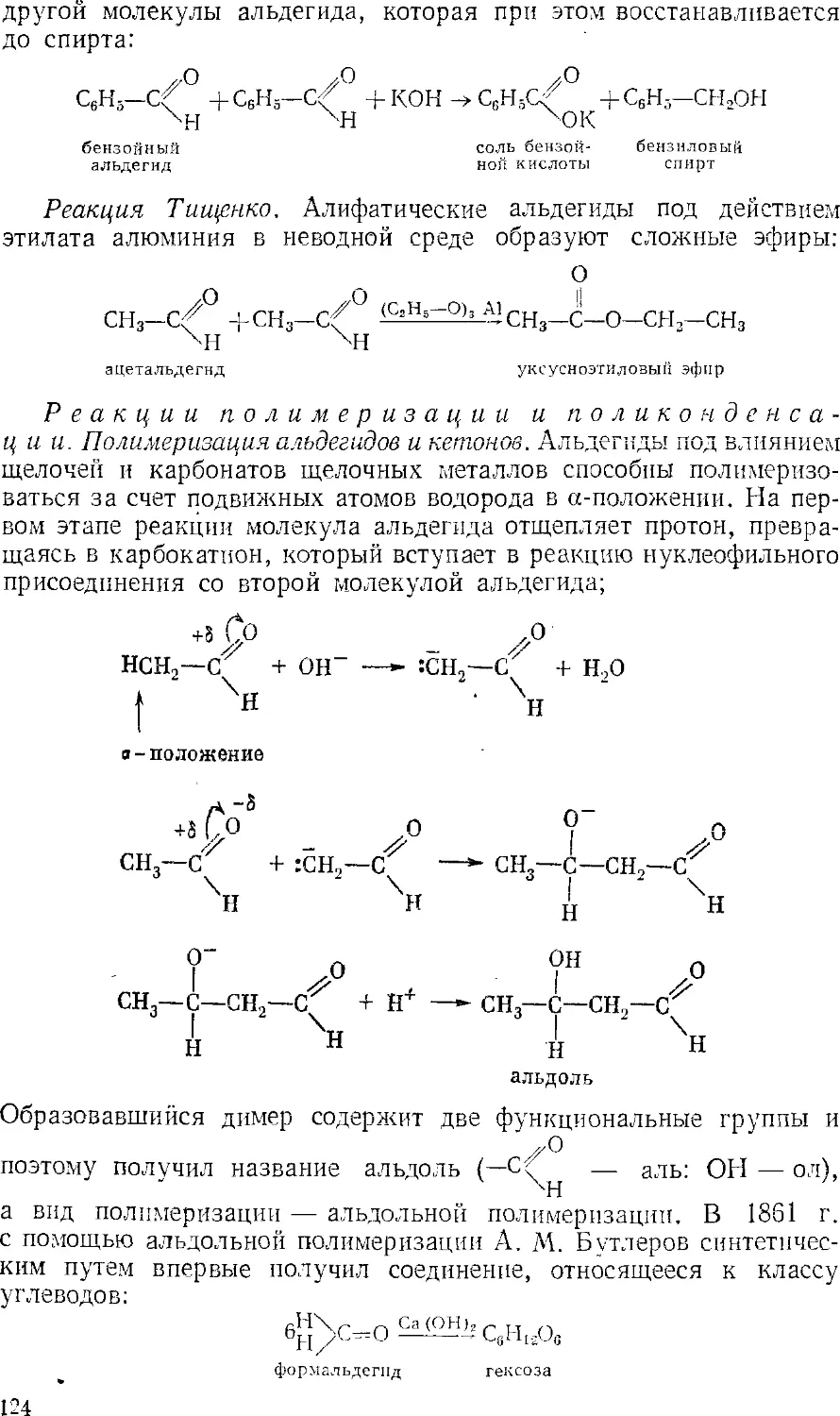
Краткий исторический очерк развития органической химии.
Человек знаком с органическими соединениями очень давно. Его
пища, одежда, топливо и другие предметы первой необходимости
состоят из органических веществ. Постепенно человек
приобретает большой практический опыт по переработке и использованию
природного органического сырья. Он научился делать пищу более
вкусной и сытной, получать пьянящие напитки (вино, пиво, брагу,
напитки из меда), дубить кожу, варить мыло, красить ткани,
приготовлять целебное питье, получать мази. Это были первые
химические производства на Земле, созданные человеком. Этот период
развития химии получил название древнейшего и продолжался
приблизительно до IV в. нашей эры. И хотя в это время был
высказан ряд правильных положений общего характера и накоплен
большой практический материал, химии как науки, по существу, не
было.
Последующий период (приблизительно до XVI в.) получил
название периода алхимии. Основное внимание исследователи в это
время уделяли поискам «философского камня», особого вещества,
с помощью которого можно было бы превратить неблагородные
металлы в благородные, в первую очередь в золото; получению
универсального растворителя; эликсира долголетия. Химические
соединения разделяли на группы по их свойствам или внешнему
виду.
В группу масел (маслообразных жидкостей) включались
серная кислота и оливковое масло. Названия некоторых соединений,
данные им в тот период, сохранились и до наших дней:
купоросное масло (концентрированная серная кислота),
нашатырный спирт — водный раствор аммиака.
Начиная с конца XVI в. темпы развития химии резко
возрастают, накапливается обширный материал о свойствах и
элементарном составе отдельных соединений. Значительные успехи были
достигнуты и в изучении органических соединений. В начале
XIX в. выдающийся шведский ученый И. Берцелиус A779—1848),
отмечая ряд особенностей органических соединений, выделил
органическую химию в самостоятельную науку. Берцелиус^ как и
его современники, считал, что органические вещества могут быть
получены только в живом организме с помощью «жизненной силы».
Поэтому эта ветвь химии и была названа «органическая химия»,
т. е. химия соединений, синтезированных в живом организме или
с его помощью. Это - идеалистическое представление об участии
«души» или «силы» в явлениях жизни, в том числе и в синтезе
органических соединений, получила название витализма, от латинского
vitalis — жизненный. Но уже при жизни Берцелиуса успехи
органической химии наносят сокрушительный удар этим
представлениям. В 1828 г. ученик Берцелиуса — Вёлер синтезировал из
неорганических веществ (цианистого калия и сульфата аммония)
органическое вещество — мочевину. В 1842 г. наш соотечественник
И. Н. Зинин получил анилин, который до этого получался из
природного красителя. Французский ученый Бертло A854) синтези-
ровал вещество, относящееся к классу жиров, а в 1861 г. знаменитый
русский химик А. М. Бутлеров осуществил синтез одного из саха-
ров. Стало ясно, что органические вещества могут быть получены
синтетическим путем.
Современный период развития органической химии начинается
с 60-х годов XIX в., когда была создана А. М. Бутлеровым теория
химического строения органических соединений. С этого времени
начинается ее бурное развитие. За последние 110 лет органическая
химия добилась больших успехов. Дальнейшее развитие на основе
новейших достижений физики получила теория строения
органических соединений, созданы новые методы исследования, достигнуты
большие успехи в синтезе новых органических соединений. Многие
разделы органической химии развиваются настолько интенсивно,
что выросли в самостоятельные разделы химии: биохимия, химия
высокомолекулярных соединений, элементорганических соединений,
красителей, природных соединений, витаминов.
Достижения органической химии открыли большие
возможности для совершенствования технологии и повышения уровня
экономики пищевой промышленности. Понимание химизма
протекающих процессов, введение разнообразных химических добавок
позволяют повысить качество пищевых продуктов, их
питательность, физиологическую ценность, сохранность, снизить их
себестоимость. Успехи органической химии позволили организовать
производства синтетического спирта, моющих средств,
синтетического клея и высвободить большие количества пищевого сырья
и продуктов, которые раньше расходовались на технические нужды.
Создание новых полимерных материалов позволило не только
усовершенствовать ряд машин и технологических процессов, но
и создать принципиально новые виды дешевых упаковочных
материалов, повышающих сохранность пищевых продуктов.
Органическая химия прочно вошла в жизнь человеческого
общества. Сейчас невозможно себе представить его существование
без нефти и продуктов ее переработки, полимерных материалов,
искусственных и синтетических волокон, лекарственных
соединений, моющих средств.
Своими успехами органическая химия во многом обязана
трудам таких выдающихся русских и советских химиков-органиков,
как Н. Н. Зинин, А. М. Бутлеров, Е. Е. Вагнер, В. В. Марковни-
ков, Н. Д. Зелинский, А. Е. Фаворский, С. В. Лебедев, М. Г.
Кучеров, А. Н. Несмеянов и многие другие.
Директивами XXIV съезда КПСС по пятилетнему плану
развития народного хозяйства СССР на 1971—1975 гг.
предусматривается дальнейшее развитие химической и нефтехимической
промышленности. Выпуск продукции должен быть увеличен в 1,7 раза,
в том числе пластических масс и синтетических смол в 2, каучу-
ков — в 1,7 и товаров бытовой химии — в 1,9 раза. Производство
химических волокон в 1975 г. будет доведено до 1050—1100 тыс. т
A0,5-108 — 11,00-108 кг). Должен быть значительно увеличен
выпуск консервантов и антисептиков, биологически активных
веществ для медицинских нужд и сельского хозяйства, реактивов,
различных пленок для расфасовки пищевых продуктов, химических
заменителей жиров и другого пищевого сырья, расходуемого на
технические цели, синтетического глицерина, красителей, лаков,
вспомогательных веществ для улучшения качества тканей,
трикотажных изделий, обуви и искусственной кожи.
Элементарный состав и источники органических соединений.
Находясь в четвертой группе периодической таблицы элементов,
углерод может образовать соединения практически почти со всеми
элементами периодической системы. Однако в состав большинства
органических соединений входят атомы небольшого числа
элементов; углерода, водорода, кислорода, азота, серы, фосфора,
галогенов. Они получили название органогенов. Из перечисленных
элементов, кроме углерода, который, естественно, входит в состав
всех органических соединений, практически в каждом
органическом соединении присутствует водород, широко распространен
кислород, остальные органогены принимают меньшее участие в
построении молекулы органических соединений.
Первые органические вещества были выделены человеком из
растительных и животных организмов. Постепенно все большую
роль в получении органических соединений начинают играть
продукты коксования каменного угля, нефть "и химический синтез.
Перечень основных источников органических веществ,
расположенный-по их значимости, выглядит следующим образом. Природные
источники: нефть, природные и попутные газы, каменный уголь и
сланцы, древесина, продукты сельского хозяйства. Синтетические
методы: химический синтез и микробиологический синтез.
Хотя нефть, природные и попутные газы и являются основным
сырьем для получения большинства органических соединений, но и
другие источники не потеряли своего значения. В последние годы
появился новый, мощный источник органических веществ —•
микробиологический синтез. Способность микроорганизмов синтезировать
сложнейшие органические соединения, многие из которых пока еще
не удается создать химическим путем, используется для
получения ферментов, витаминов, антибиотиков, аминокислот.
Синтетическим путем можно не только воспроизвести сложнейшие соединения,
создаваемые природой, но и получать новые, ранее не известные,
с лучшими, чем природные, свойствами.
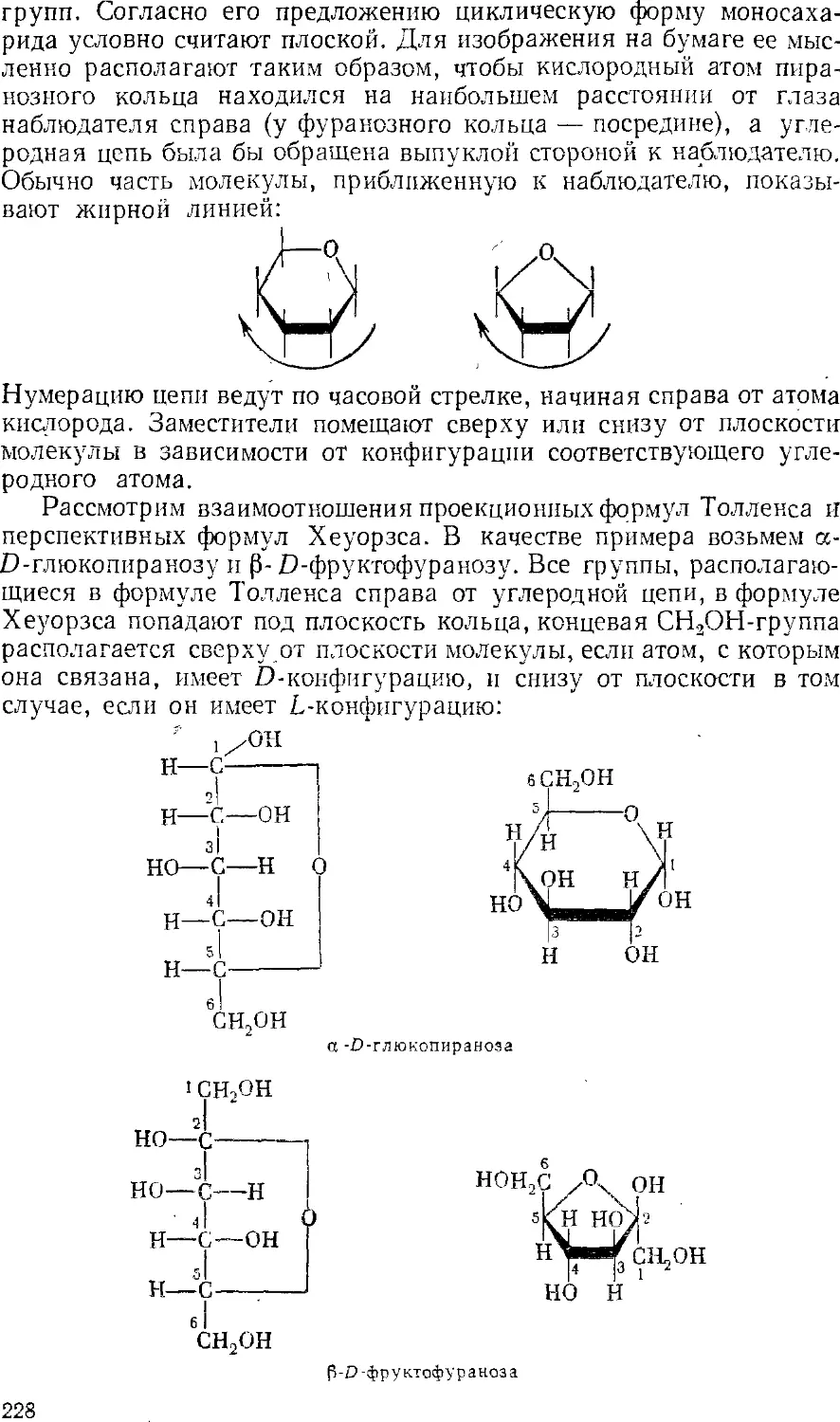
Краткие сведения о развитии теоретических представлений в
органической химии. Первой теорией строения химических соединений
была электрохимическая теория Берцелиуса, примененная к
органическим соединениям в виде теории радикалов (Берцелиус, Ли-
бнх, Вёлер). Авторы ее обратили внимание на то, что во многих
превращениях органических веществ одна из частей молекулы,
названная ими «радикал», не изменяется, а переходит из исходного
соединения в продукт реакции.
Молекула органических соединений, по мнению авторов этой
теории, состоит из двух противоположно заряженных радикалов,
связанных электростатическим взаимодействием. Однако посте-
8
пенно накапливались экспериментальные данные, которые не нашли
объяснения с позиций «теории радикалов», и в 40-х годах XIX в.
на смену ей приходит «теория типов», созданная двумя учеными —
Жераром и Лораном. Ее авторы в противоположность сторонникам
теории радикалов основное внимание уделяли той части молекулы
органического соединения, которая изменяется в процессе
реакции. Они считали, что существует глубокое сходство в поведении
органических и неорганических веществ. По их мнению,
органические вещества можно рассматривать как производные
неорганических, образованные замещением атомов в его молекуле на
радикалы, которые они называли «остатками». Полученные при этом
органические Еещества вступают во все реакции, свойственные
неорганическим веществам, возглавляющим этот «тип» соединений.
Основных типов было четыре: тип водорода, хлористого водорода,
воды, аммиака
Например, к представителям «типа воды» ими были отнесены
О
н ч
н /
веда
О
СН3}°
метиловый
спирт
СНз\о
сн3 j
днметнловый
эфир
с2н3о ¦>
н J
уксусная
кислота
о
СН3О Л
С2Н3О )
уксусный
ангидрид
Расположив органические соединения по типам, авторы не
только установили место для большинства известных в то время
химических соединений, но по аналогии указали и некоторые
способы получения еще неизвестных представителей отдельных групп.
В то же время сторонники этой теории стояли на идеалистических
позициях. Они считали, что строение молекул органических
соединений не может быть познано в ходе химического эксперимента.
Идеалистическое учение, отрицающее познаваемость объективного
мира, получило название агностицизма.
К 60-м годам прошлого века в органической химии накопился
большой экспериментальный материал, ставящий под сомнение
многие положения теории типов. К этому же времени было
сформулировано положение о четырехвалентности атома углерода (Кольбе,
Кекуле), его способности соединяться с другими углеродными
атомами, с образованием углерод — углеродных цепей (Кекуле,
Купер). Шотландский ученый Купер предложил систему
изображения строения химических соединений с помощью черточек.
Возникла острая необходимость в создании научной теории
строения органических соединений. Автором такой теории стал
выдающийся химик А. М. Бутлеров A828—1886).
Теория химического строения органических соединений А. М.
Бутлерова. Теория химического строения органических соединений
была наиболее полно изложена А. М. Бутлеровым в его труде
«Введение к полному изучению органической химии». Ученик
А. М. Бутлерова В. В. Марковников указывал впоследствии,
что эта книга составила тогда «эпоху в развитии теоретических
представлений, положенных в основу современной химии, и
открывала обширный горизонт для совершенно новых исследований».
Ее возникновение в этот период именно в России не было
случайностью. В стране в это время происходит подъем революционно-
демократического движения, бурно развиваются естественные науки.
Россия в этот период по уровню развития ряда отраслей науки
превосходит Запад.
В своей теории А. М, Бутлеров выступает как ученый
материалист, он признает реальность существования атомов и молекул и
возможность познания в ходе эксперимента внутренней структуры
молекул органического соединения.
Сущность теории A.JV1. Бутлерова состоит в следующем:
1. Свойства органических веществ определяются не только
природой и количеством входящих в состав молекулы атомов, но
и порядком их соединения, получившим название химического
строения.
2. В органических веществах существует определенный
порядок соединения атомов в молекуле, заключающийся в
последовательности соединения атомов и характере соединяющих их связей.
Из понятия химического строения вытекает, объяснение явления
изомерии, изученное А. М. Бутлеровым. Оно связано с различным
химическим строением молекул, имеющих одинаковый элементарный
состав и молекулярную массу. Например, углеводород бутан
(С4Н10) существует в виде двух изомеров, различающихся строением,
т. е. порядком соединения атомов в молекуле:
н н н н н н н
III! - 111
Н—С^С—С-С-Н Н—С-С—С—Н
н н н н н
н
нормальный бутан
н-с-н
н
изобутан
3. Химическая структура молекулы может быть представлена
только одной рациональной формулой, которая выражает все
основные химические свойства данного вещества. Это положение имеет
большое практическое значение, око дало возможность на
основании формулы органического соединения предсказать его свойства
и поведение в химических реакциях.
4. Атомы, входящие в молекулу органического соединения,
оказывают взаимное влияние друг на друга и на ее химические
свойства.
Вопросы взаимного влияния атомов в молекуле органического
соединения, во многом определяющие его поведение в химических
10
реакциях, были подробно изучены учеником А. М. Бутлерова
В. В. Марковниковым.
Сформулированные А. М. Бутлеровым теоретические положения
легли в основу современной теоретической органической химии,
которая, по существу, и начинается с его работ. А. М. Бутлеров
создал большую химическую школу, представители которой
развивали дальше теорию химического строения, создавая новые,
оригинальные научные направления.
Краткие сведения об электронных представлениях в
органической химии. Теория химического строения органических
соединений А. М. Бутлерова не касалась вопроса природы сил,
объясняющей возникновение связи между атомами, и о свойствах этой связи.
1. Строение электронных оболочек атома углерода.
Известно, что вещества состоят из молекул, а молекулы из
атомов. Атом — мельчайшая частица элемента, носитель всех его
химических свойств. В химическом отношении он неделим. Атомы
различных элементов характеризуются их атомной массой. В
результате открытия катодных и анодных лучей, явления
радиоактивности было установлено, что атомы не являются неделимыми
частицами. Дальнейшими исследованиями было показано, что они
состоят из ряда частиц, в том числе протонов, электронов, нейтронов.
Атомы всех элементов содержат очень малое по размеру ядро, в
котором сосредоточены все положительные заряды и 0,99% его массы,
и вращающиеся вокруг него отрицательно заряженные частицы —
электроны. Протоны — устойчивые элементарные частицы с
массой, близкой к углеродной единице. Заряд протона равен заряду
электрона и противоположен по знаку. Масса электрона равна
5,49 • 10~4 углеродной единицы. Электроны вращаются вокруг ядра,
как планеты вокруг солнца, однако законы движения электронов
значительно сложнее, чем планет.
В 1913 г. датский физик Нильс Бор теоретически построил
планетарную модель атома самого простого элемента — водорода,
основанную на трех допущениях:
а) электрон может вращаться вокруг ядра не по любым, а только
по определенным, «дозволенным» орбитам;
б) при вращении по этим орбитам электрон не поглощает и
не излучает энергии. Излучение или поглощение энергии происходит
только при переходе электрона с одной орбиты на другую;
в) энергия, выделенная или поглощенная при переходе
электрона с одной орбиты на другую, равна разности энергий,
которыми обладает электрон в начальном и конечном состояниях.
Движение электронов в атомах Бор рассматривал как простое
механическое перемещение.
Достижениями современной физики установлено, что электроны
обладают свойствами частиц и волн, поэтому электрон движется
по всему объему, образуя электронное облако (электронная орби-
таль), которое для разных электронов имеет различную форму.
Основная характеристика, определяющая движение электрона
в поле ядра, — его энергия. Для характеристики каждого электрона
11
в атоме разработана система четырех параметров, которые получили
название квантовых чисел.
Главное квантовое число п характеризует энергию электрона
или энергетический уровень, на котором он находится, и принимает
значение целых чисел от 1 до оо.
Орбитальное или побочное квантовое число / соответствует
энергетическому состоянию электрона данного уровня. Электроны
одного энергетического уровня несколько
различаются по запасу энергии и
группируются в подуровни. Орбитальное
квантовое число определяет форму электронного
облака и принимает значение любых целых
чисел от 0 до (п — 1), где п — главное
квантовое число. Например, при п = 4,
/ = 0; 1; 2; 3. Подуровни электронов имеют
и буквенное обозначение: s; p; d; /.
Число подуровней равно главному
квантовому числу п
Рис. 1. Схематический
вид электронного облака
s-электронов
п=1 1 = 0
п = 2 1 = 0; 1
п = 3 1 = 0; 1; 2
Один подуровень s
Два подуровня s, р
Три подуровня s; р; d
Как уже указывалось, . форма электронного облака зависит
прежде всего от подуровня. Электроны s-подуровня имеют облако
шаровой формы (сферическая орбиталь) (рис. 1).
Облако электронов р-подуровня имеет форму объемной восьмерки
(р-орбиталь) (рис. 2). Магнитное квантовое число т определяет
положение орбитали в пространстве и обозначается в этом случае
индексами при букве Р — Рх; Ру; Pz (рис. 2). Число т может при-
Рис. 2. Схематический вид электронного облака р-элект-
ронов
нимать значение любых целых чисел в пределах от —I до +/,
включая 0, где / — побочное (орбитальное) квантовое число.
Число возможных значений магнитного квантового числа, т. е.
число энергетических состояний, в которых могут находиться
электроны данного подуровня, равно 2/ + 1.
Энергетическое состояние электронов, характеризуемое
одинаковыми значениями 3-х квантовых чисел, схематически принято
12
обозначать энергетическими ячейками, которые рисуют в виде
прямоугольника, а электроны в виде стрелок в этих ячейках:
Спиновое квантовое число s характеризует вращение
электрона вокруг своей оси. Оно может принимать два значения:
+1/2 и — 1/г. т- е- электроны могут вращаться в противоположных
направлениях, но с постоянной скоростью.
Два электрона с одинаковыми значениями квантовых чисел
(п, I, т), но с противоположно направленными спинами (\\)
называются спаренными, или неподеленной парой.
Таблица 1
Распределение электронов по энергетическим уровням и подуровням
Энергетический
уровень
1
2
3
4
Подуровень
обозначение
S
S
Р
S
р
d
s
Р
d
f
побочное
квантовое
число /
0
0
1
0
1
2
0
1
2
3
Магнитное квантовое
число т
0
0
— 1; 0; +1
0
— 1; 0; +1
—2;—1;0;+1; +2
0
—1; 0; +1
—2; —1; 0; +1; 4-2
—3; —2; —1; 6;
+ 1; +2; +3
Максимальное
число
электронов в
подуровне
2 B1+ 1)
2
2
6
2
6
10
2
6
10
14
Максимальное
число
электронов
в уровне 2п2
2
8
18
32
Согласно принципу Паули в атоме не может быть двух
электронов с одинаковыми значениями всех четырех квантовых чисел,
следовательно, число электронов в любой подгруппе равно 2 B1 + 1).
В подуровне s может быть не более 2 электронов, в подуровне р — 6,
в d-подуровне — 10 и в /-подуровне — 14 электронов. В
энергетической ячейке могут быть только два электрона, причем с
противоположными спинами. Распределение электронов по
энергетическим уровням приведено в табл. 1. -Общепринятой является
следующая запись распределения электронов по уровням: буквами
s, р, d, / указывают подгруппу, число электронов в подгруппе
обозначается показателем степени, а номер оболочки — цифрой,
которая ставится перед буквой.
13
При изучении органической химии, естественно, особый интерес
представляет строение электронных оболочек атома углерода:
2s2;
1!
U
t t
Как видно из приведенной записи и схемы, на внешней
электронной оболочке атома углерода находятся только два неспарен-
ных электрона, а так как валентность элемента равна именно числу
неспаренных электронов, а не общему числу электронов на внешней
оболочке, то для углерода она должна быть равной двум. Однако
атом углерода способен относительно легко переходить из этого
состояния (оно иногда называется обычным или основным) в
возбужденное, принимая следующий вид:
t
t
t
В соответствии с этой записью в молекуле простейшего ор'гани-
н
I
ческого соединения метана Н—С—Н в образовании трех связей
н
С — Н должны участвовать три р-электрона, а в образовании
четвертой С — Н связи s-электрон, т.е. связи С — Н должны
быть неравноценными. Это положение не
подтверждается опытными данными.
Американским ученым Полингом было
введено представление о гибридизации
электронных облаков. В молекуле метана атом
углерода находится в первом валентном
состоянии (sp 3-гибридизация), четыре гибриди-
зованные орбитали имеют одинаковую
конфигурацию и энергию (рис. 3) и расположены
друг относительно друга под углом 109°28'.
Кроме sp 3-гибридизации атом углерода
может находиться в состоянии sp2- и sp-гиб-
ридизации. В 5р2-гибридизации участвуют
одна s- и две р-орбитали, оси
образовавшихся гибридизованных орбиталей расположены в одной
плоскости под углом 120°, а негибридизованная р-орбиталь
направлена перпендикулярно к плоскости гибридизованных орбита-
лей (рис. 4).
В sp-гибридизации участвуют одна s- и одна р-орбитали, а
образовавшиеся две гибридизованные орбитали расположены на одной
прямой в разные стороны от ядра атома углерода (рис. 5),
14
Рис. 3. Модель
электронной оболочки
атома углерода в
состоянии зр3-гибридизашш
2. Химическая связь. Взаимодействие атомов,
сопровождающееся перестройкой их электронных оболочек, может привести
к возникновению химической связи, которая обусловливает
образование молекулы.
При написании структурных формул органических соединений
связи между атомами обычно изображаются в виде черточек,
соединяющих их символы. Каждая черточка символически изображает
единицу валентности.
Химические связи делятся на типы по принципу их
электронного строения.
Большинство неорганических соединений построены с помощью
электровалентной, или ионной, связи, которая возникает между
атомами или группами атомов в результате электростатического
у
Рис. 4. Модель электронной
оболочки атома углерода в
состоянии ь/^-гибридизации
sp
Рис. 5. Модель электронной оболочки
атомов углерода в состоянии sp-гиб-
ридизации
взаимодействия. При этом происходит передача электрона от
одного атома к другому, причем первый превращается в положительно
заряженный ион (катион), второй в отрицательно заряженный
нон (анион). Схематично это можно представить следующим
образом:
А ->А++: В~
(точками условно изображены электроны). Две разноименно
заряженные частицы связываются силами электростатического
взаимодействия.
Ионная связь обладает рядом особенностей: ненасыщенностью,
значительной полярностью. Соединения, молекулы которых
построены с помощью ионной связи, имеют высокие температуры
плавления, их растворы проводят электрический ток.
Большинство молекул органических соединений построены с
помощью ковалентной связи. Ковалентная связь возникает за счет
образования одной или нескольких электронных пар, которые
становятся общими для двух соединяющихся атомов:
15
Атомы, образующие ковалентную связь, обладают близкими
свойствами или, как говорят в химии, близким по своему
значению сродством к электрону. Образование связи происходит при
сближении атомов с неспаренными электронами, спины которых
антипараллельны. Условно это можно представить следующим
образом:
/
Возникает единое электронное облако, плотность которого между
ядрами особенно велика, это и обеспечивает устойчивость
молекулы. На рис. 6 показано распределение электронной плотности
Рис. 6. Распределение электронной плотности в
молекуле водорода
Н — С
Рис. 7. Схема
образования простой о-связи
в молекуле водорода, построенной с помощью ковалентной связи.
Кроме водорода, по этому типу построены молекулы хлора,
азота и, как уже указывалось, большинства органических
соединений.
В органической химии существует три вида ковалентной связи:
простая (она условно обозначается одной черточкой), двойная
(изображается двумя черточками) и тройная (изображается тремя
черточками).
Образование простой ковалентной связи происходит путем
обобщения двух электронов, максимальное перекрытие
электронных облаков происходит по прямой, соединяющей два атома.
Такая ковалентная связь называется а-связью (сигма-связь) (рис. 7).
В молекуле одного из простейших органических соединений —
этана С2Н6 — имеется семь сигма-связей, расположенных друг
относительно друга под углом 109с28', атомы углерода находятся
16
в состоянии 5р3-гибридизации (рис. 8), или в первом валентном
состоянии.
Иначе осуществляется связь между атомами углерода,
связанными двойной связью, например, в молекуле этилена
= С\
Н
В этилене атомы углерода находятся во втором валентном
состоянии Eр2-гибридизация). У каждого углеродного атома две
гибридизованные орбитали затрачены на связь с атомами водорода,
одна на связь с другим углеродным
атомом, следовательно в молекуле
этилена имеется пять сигма-связей
(рис. 9). Остаются две негибри-
дизованные р-орбитали, по одной
у каждого углеродного атома.
Они расположены
перпендикулярно к плоскости, в которой
находятся сигма-связи, и перекрываются
боковыми частями гантелей,
образуя дополнительную связь между
атомами углерода. Такая связь
называется я-связью (пи-связью).
я-Связи широко распространены в органических соединениях,
с их помощью могут быть связаны не только атомы углерода, но
и атом углерода с кислородом, азотом, серой и др. В тех случаях,
»когда с помощью п-связи соединяются атомы, различные по своей
электроотрицательности, образующие ее электроны смещены в
сторону более электроотрицательного атома: я-связь поляризована.
103° 2 В'
10Э°28'
109°28'
Рис. 8. Модель молекулы этана
7t
"V'
it
а б
Рис. 9. Схематическое изображение строения этилена
Примером органических соединений, в молекуле которых
атомы углерода связаны тройной связью, является ацетилен
Н — С = С — Н. Атомы углерода в ацетилене находятся в
состоянии sp-гибридизацин, у каждого углеродного атома одна из двух
гибридизованных орбнталей затрачена на связь с атомом водорода,
вторая на связь с другим атомом углерода. В молекуле ацетилена
имеется три а-связи. Два углеродных атома дополнительно свя-
17
заны двумя л-связями, плоскости которых взаимно
перпендикулярны (рис. 10).
Во всех описанных случаях образование ковалентной связи
происходит за счет обобщения электронов, ранее принадлежавших
двум соединяющимся атомам, но возможен и другой механизм
образования ковалентной связи. Ковалентная связь может образоваться
за счет электронной пары только одной из реагирующих частиц.
Такая ковалентная связь получила название координационной
связи, или донорно-акцепторной. Атом или ион, отдающий свою
Tt
6
¦н
Рис. 10. Схематическое изображение строения ацетилена
электронную пару для образования связи, называется донором,
обобщающий чужую электронную пару — акцептором. Рассмотрим
два примера.
При взаимодействии аммиака с соляной кислотой протон
соединяется с аммиаком за счет неподеленной пары электронов атома
азота:
н Г н -|+
H:N:+H+-> H:N:H
Н L Н
ион аммония
Заряд в этом случае находится не на присоединившемся протоне,
а на атоме азота иона аммония. Возникшая новая N — Н-связь
ничем не отличается от остальных ковалентных связей, которые
имелись в молекуле аммиака.
Во втором случае присоединяется не ион, а нейтральный атом;
при этом на атоме, отдающем электронную пару, возникает
положительный заряд, а на присоединяющемся атоме — отрицательный.
В результате два атома оказываются связанными как бы двояко:
ковалентной связью с помощью отданной электронной пары и
электровалентной связью
Этот случай координационной связи получил название семиполяр-
ной связи. Наличие семиполярной связи в молекуле сильно
сказывается на ее свойствах,
18
Ковалентная связь обладает рядом характерных свойств:
межъядерным расстоянием, направленностью в пространстве, энергией
образования, полярностью. Расстояние между центрами атомов,
связанных с ковалентными связями (межъядерное расстояние,
длина связи), — постояннаяо величина. Длина С — С-связи —
1,54 А, С = С-связи — 1,34 А, а С = С-связи — 1,2 А. Одной из
особенностей ковалентной связи является ее определенная
пространственная направленность, которая может быть охарактеризована
с помощью валентного угла. Валентный угол между связями атома
углерода зависит от типа гибридизации и в определенной степени
от вида атома, с которым он связан. Так, в молекуле метана СН4
(sp 3-гибридизация) угол между направляющими связей 109°28',
т. е. атомы водорода располагаются в углах правильного тетраэдра
(рис. 11). Гипотеза о те-
траэдричееком строении
молекулы метана была
впервые высказана более
ста лет назад французским
ученым Ле-Белем и
голландским исследователем
Вант-Гоффом и послужила Н'4
основой для создания
раздела органической
химии, называемого
стереохимией. В молекуле этилена
(зр2-гйбридизация)
валентный угол между о-связя-
ми—20°.
В оценке химической связи большую роль играют два
показателя: энергия связи и ее полярность. Первая характеризуется
энергией, которую нужно затратить на разрыв связи (ккал/моль).
Полярность связи обусловлена несовпадением центра тяжести
отрицательных и положительных зарядов и количественно
характеризуется величиной дипольного момента. Величина диполыюго
момента определяется рядом факторов и зависит в том числе от
взаимного влияния атомов в молекуле органического соединения.
В определенных условиях ковалентные связи проявляют
способность к поляризации (поляризуемость связи), при этом происходит
смещение электронов по отношению к ядрам и самих ядер.
Повышенной способностью к поляризации отличаются электроны я-связи.
Особый вид связи, возникающий между электроотрицательным
атомом, содержащим по крайней мере одну необобщенную
электронную пару (О, N, S) и ковалентно связанным атомом водорода,
обладающим повышенной подвижностью, получил название
водородной связи. По своему характеру она является
электростатической и обозначается пунктиром:
Рис.. 11. Пространственная модель молекулы
метана
СН,
СНз
сн3
СНз
111
Н—О ...Н—О ...Н—О ...Н—
19
Специфическая роль протона в образовании водородных связей
заключается в том, что лишенный электронных оболочек, он
способен сближаться с двумя электроотрицательными атомами на
очень близкое расстояние, располагаясь на соединяющей их
прямой. Энергия водородной связи мала C—10 ккал/моль или 126 X
X 103 — 41,9 -103 Дж/моль). Водородные связи оказывают большое
влияние на свойство многих органических соединений.
Классификация органических соединений. В настоящее время
принято деление органических соединений на несколько больших
групп. В основе этой классификации лежит строение углеродного
скелета — основы органического соединения. Как указывалось,
углеродные цепочки могут быть прямыми или разветвленными,
а также замыкаться в циклы (схема 1). В состав последних, кроме
атомов углерода, могут входить атомы других элементов:
кислорода, азота, серы. Такие соединения получили название
гетероциклических («гетерос» — по-гречески другой).
Схема I
Органические соединения
1
Ациклические соединения,
алифатические соединения
Карбоциклические
соединения
Циклические соединения
1
f
Гетероциклические
соединения
Алициклические
соединения
Ароматические
соединения ~
В молекулах ациклических, или алифатических, соединений
(иногда они называются соединениями жирного ряда) атомы
углерода соединены в прямые или разветвленные цепи, не содержащие
колец или циклов.
Циклические соединения делятся на две группы:
карбоциклические — циклы, молекулы которых состоят из атомов углерода,
и гетероциклические, кольца которых наряду с атомами углерода
содержат атомы других элементов. В карбоциклических
соединениях выделяется группа соединений, содержащих бензольные ядра,
они получили название ароматических. Остальные
карбоциклические соединения входят в состав группы алицпклических
соединений.
Самые простые органические соединения — углеводороды. В
построении их молекулы участвуют атомы только двух элементов —
углерода и водорода. Замещая водороды на группы, содержащие
атомы других элементов (они получили название характеристиче-
20
ских или функциональных групп), можно получить разнообразные
производные углеводородов. Функциональные группы обладают
характерной реакционной способностью и определяют химическое
поведение и принадлежность соединения к определяемому классу.
Основные классы производных углеводородов и функциональные
группы приведены в табл. 2.
Таблица 2
Основные классы производных углеводородов
Класс
Галогенопроиз-
водные
Оксисоединения
[спирты, фенолы)
Оксосоединения
(альдегиды, кетоны)
Карбоновые
кислоты
Нитросоединения
Амины
Сульфокислоты
Функциональная
(харак7-еристическая)_грушта
F, а,
Вг, I
-он
>с=о
О
||
-С-ОН
—NO2
-NHa
-SO3H
Галоген
Гидроксил
(окси-группа)
Карбонил
(оксо-группа)
Карбоксил
Нитрогруппа
Аминогруппа
Сульфогруппа
Примеры представителей
С2Н5С1—хлористый этил
С2Н5ОН — этиловый спирт
С6Н5ОН—фенол
СН3—С<( —уксусный альдегид
О
II
СН3—С—СН3 — ацетон
О
II
СН3—С—ОН—уксусная кислота
СН3—СН2—NO2 — нитроэтан
С6Н5—КО2— нитробензол
СН3—СНЯ—NH2 —этяламин
С6Н5—SO3H — бензолсульфокис-
лота
В молекуле органического соединения может содержаться несколько
одинаковых или различных функциональных групп. Подробно
эти вопросы будут разработаны в соответствующих разделах
учебника.
ЧАСТЬ ПЕРВАЯ
УГЛЕВОДОРОДЫ
Углеводороды — наиболее простые по составу органические
соединения, состоящие только из атомов углерода и водорода.
Общая формула углеводородов С„Нт. Они различаются по
строению углеродного скелета (прямые углерод — углеродные цепи,
разветвленные, замкнутые в циклы) и по характеру связей между
атомами углерода (предельные, или насыщенные, и непредельные,
или ненасыщенные, углеводороды). По первому признаку (стр. 20)
углеводороды принято делить на три большие группы:
алифатические, алициклические и ароматические.
АЛИФАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
АЛКАНЫ
Строение, понятие о гомологическом ряде, изомерия, Алканы —
алифатические углеводороды, в молекуле которых атомы углерода
связаны между собой простой о-связью, а остальные их
валентности максимально (предельно) насыщены атомами водорода.
Отсюда и другое название этих соединений — предельные или
насыщенные углеводороды. Родоначальник всех алканов — метан СН4.
В молекуле метана, как и в молекулах других алканов, атом
углерода находится в состоянии яр3-гибридизации.
Каждый последующий представитель ряда метана отличается
от предыдущего на группу СН2 (метиленовая группа) (табл. 3).
Общая формула соединений этого ряда C,2H2ra+2- Такой ряд
родственных органических соединений с однотипной структурой,
близкими химическими свойствами называется гомологическим рядом;
члены этого ряда — гомологами; группа СН2, на которую
отличаются два соседних гомолога, — гомологической разностью состава.
Три первых представителя гомологического ряда метана не
имеют изомеров, начиная с бутана в ряду алканов наблюдается
явление изомерии, т. е. существование нескольких соединений с
одинаковым качественным и количественным составом, но
различающихся по физическим и химическим свойствам.
Строение бутана может быть представлено с помощью двух
формул:
СН3-СН.,-СНа-СН. СНз-СН-СНз
«•бутан I
Ui3
нзобу ran
22
Алканы (гомологический ряд метана)
Таблица 3
Название
Формула
Т. пл., °С
Т. кип.. °С
Метан
Этан
Пропан
я-Бутан
Изобутан
н-Пентан
я-Гексан
н-Гептан
н-Октаи
н-Нонан
н-Декан
я-Гексадекан
н-Эйкозан
н-Триаконтан
4
СНз—СН3
сн3—сн.—сн,
СН3-(СН3J-СН3
СН3—СН (СН3)—СН3
СН3—(СН2K—СН3
СН3-(СН2),-СН3
СН3-(СН,M-СН3
CHS-(CH2N-CH3
СН3—(СН2O—СН3
СНз—(СН2)8-СН3
СН3—(CH3)ij—СН3
CH3-(CH;)i8-CH3
СН3-(СН,),8-СН3
—182,5
— 183,3
— 187.7
-138,3
— 159,6
—129,7
—95,4
—90,5
—56,8
—53,5
—29,7
+ 18,2
36.8
65,8
—161,5
—88,6
-42,1
-0,5
—11,7
36,1
68,7
98,4
125,7
150,8
174,1
280,8
342,7
446,4
Такой вид изомерии получил название структурной изомерии. На
примере изомеров бутана мы сталкиваемся с одним из видов
структурной изомерии — изомерией углеродного скелета.
Углеводороды с неразветвленной углеродной цепью называют
углеводородами нормального строения (н-бутан); если цепь
углеродных атомов разветвлена, то углеводороды называются
углеводородами изостроения (изобутан).
С увеличением числа углеродных атомов (и) в молекуле алкана
число изомеров (т) быстро возрастает: при п = 5, т = 3; при п = 6
т = 5; при п = 10, т = 75; при п = 20, т = 336 319 изомеров.
Структурные формулы, понятие о первичном, вторичном и
третичном атомах углерода. Приведенные формулы изомеров бутана
получили название структурных. Структурные формулы
показывают не только, какие атомы и в каком количестве находятся в
молекуле данного соединения, но и выражают относительное положение
аттюв и характер соединяющих их связей. Различают полную,
или развернутую, структурную формулу:
Н
Н—С—Н
н
н н
I I
Н—С— С—С—С
н н н н
изопеытан
и краткую, или звеньевую, формулу:
-Н
СН3-СН2—СН—CHj
изопентан
23
В изопентане имеется три вида углеродных атомов: у 1, 4 и 5 из
четырех валентностей только одна затрачена на связь с углеродным
атомом — такие атомы углерода называются первичными; у атома
3 на связь с углеродными атомами затрачены две валентности
(вторичный углеродный атом) и у атома 2 — три валентности (третичный
атом). Атом углерода, у которого все четыре валентности
затрачены на связь с другими углеродными атомами, называется
четвертичным.
Номенклатура. Номенклатуре, т. е. способу наименования
органических соединений, вследствие их многообразия и сложности,
всегда необходимо уделять особое внимание. Название
органического соединения должно не только указывать вид и число атомов,
входящих в его состав, но и давать представление о структуре
молекулы. В научной литературе используются разнообразные
способы наименования органических соединений. Для наименования
алканов применяются: 1) тривиальные или исторические названия;
2) старая рациональная номенклатура и 3) новая (систематическая
номенклатура). Тривиальные названия получили первые четыре
гомолога: метан, этан, пропан, бутан. По существу, историческими
можно считать и названия остальных алканов нормального
строения, хотя они являются производными греческих или латинских
числительных (см. табл. 3). Для наименования изоалканов
необходимо знать название органических радикалов — алкилов, частиц,
условно выделяемых из молекулы углеводорода отнятием одного
атома водорода. Общая формула алкилов СяН2л+1. Их название
получают путем замены окончания «ан» соответствующего алкана
на «ил». Отсюда и их групповое название «алкилы». В формулах
органических соединений алкилы в общем виде обозначают R.
В табл. 4 приведены наиболее распространенные алкилы.
Таблица 4
Наименование наиболее распространенных алкилов
Радикал
Метил
Этил
Пропил
Изопропил
Бутил
Язобутил
Вторичный бутил
Третичный бутил
Формула
СН,—
сн,сн,-т
СНзСН СН3
1
сн,снхн..сн,—
СНз-СН (СН3)"СН2—
СНзСН-СН СН3
СН3
j
сня—с—
j
сня
24
По старой рациональной номенклатуре предельные
углеводороды рассматриваются как производные метана, образованные
замещением атомов водорода в его молекуле на соответствующие
радикалы:
Так, для того чтобы назвать приведенный ундекан, мысленно
выбираем «центральный» атом углерода, соединенный с
максимальным количеством других атомов, таким образом, чтобы связанные
с ним радикалы (R; R^, R2; R3) можно было бы назвать однозначно.
При построении названия радикалы располагают по старшинству,
указывая количество однотипных радикалов (ди-, три-, тетра-
и т. д.). Название завершается наименованием основного
соединения — метана. Приведенный алкан называется метилэтилизопропил-
третбутилметан. Старая рациональная номенклатура используется
только для наименования простых алканов.
Наиболее полной и удобной является современная
(систематическая) номенклатура органических соединений, в основу которой
легли положения, принятые в 1892 г. в Женеве международным
конгрессом химиков. Эти правила дополнялись и уточнялись в 1930 г.
(Льежская номенклатура), в 1957 и 1965 гг. (номенклатура ШРАС).
Мы будем использовать современную номенклатуру в соответствии
с правилами ИЮПАК (правила Комиссии по номенклатуре
органических соединений при Международном Союзе Чистой и
Прикладной химии — International Union of Pure and Applied Chemistry —
сокращенно ШРАС). В соответствии с этими правилами для
наименования алканов нормального строения сохраняются старые
традиционные названия (см. табл. 3), родовое название — алканы,
окончание — «ан».
Для наименования изоалканов они рассматриваются как
производные алканов нормального строения, в молекуле которых вместо
атомов водорода введены соответствующие радикалы. Для
построения названия изоалкана выбирают самую длинную цепь, нумеруя
атомы углерода от одного ее конца до другого цифрами 1, 2, 3, 4
и т. д. Направление нумерации выбирают так, чтобы цифры,
указывающие положение боковых цепей, были наименьшими. При
построении названия первыми указываются наиболее простые
заместители: метил, этил и т. д. Перед названием радикала ставят номер
углеродного атома главной цепи, с которыми он связан, и цифру
от названия отделяют черточкой. Количество одинаковых замести-
25
телей указывается умножающим префиксом — ди-, три-, тетра-
и т. д., а номера углеродных атомов главной цепи, с которыми они
связаны, располагают по возрастающей величине и ставят перед
названием радикалов. Последним называется углеводород главной
цепи. Приведенный выше ундекан в соответствии с этими правилами
должен быть назван: 2, 2, 3, 4-тетраметил-З-этилпентан.
Природные источники. Способы получения. Алканы широко
распространены в природе, это важные компоненты нефти, природных
и попутных газов. Содержание их в нефти отечественного
происхождения колеблется от 30 до 89%. Низшие алканы, в первую очередь
метан (до 98%), — основные компоненты природных газов. Смеси
высокоплавких твердых алканов встречаются в природе в виде
минерала озокерита, который после специальной обработки и очистки
может быть превращен в церезин—воскообразный продукт,
нашедший широкое применение в промышленности. Алканы с числом
атомов углерода от 20 до 30 входят в состав восковых оболочек
семян и листьев.
Основным промышленным источником алканов является нефть,
из которой их выделяют фракционной перегонкой, или получают
разнообразными методами химической переработки.
Важным промышленным способом получения алканов является
химическая переработка каменного угля. Обычно используют
низкосортные бурые угли. Тонко измельченный и смешанный с
тяжелым маслом уголь обрабатывают водородом при температуре
400—600° С и давлении 200—700 атм A9,6 • 10s — 68,6-106 Н/м2)
в присутствии железного катализатора. Образуется сложная смесь
разнообразных продуктов, из которых после фракционирования,
а в отдельных случаях дополнительной обработки получают бензин,
смазочные масла и другие важные для промышленности продукты.
В последние годы, особенно в странах, бедных нефтью и газом,
значительное распространение нашел синтез алкаиов из окиси
углерода и водорода (синтез-газ). Источниками окиси углерода
являются: генераторный газ, водяной газ и синтез-газ, содержащие
окись углерода и различное количество водорода. Синтез-газ
получают из метана и его ближайших гомологов при их взаимодействии
с водяным паром, двуокисью углерода и кислородом (температура
750—900° С) в присутствии металлических катализаторов. Для
синтеза алканов применяют очищенную смесь окиси углерода и
водорода A : 2); катализаторы: кобальт-торий-магниевый, железо-
медный, сплавные железные (температура процесса 170—320° С)
«СО + Bя +1) Н2 -* С„Н2я+а+яН2О
Образовавшаяся смесь алканов называется «синтином».
В лабораторных условиях метан и его гомологи могут быть
получены следующими способами:
1. Реакция Вюрца
йодистый метил
26
открыта в 1855 г. французским ученым Вюрцем A817—1884).
П. П. Шорыгин предложил следующий путь образования этана:
а) образование металлорганического соединения
б) синтез алкана
йодистый этилнатрий
метил
»СН3—
этан
Кроме натрия, можно использовать цинк, магний и др. Если в
реакции участвуют различные галогенопронзводные (например, СН31 и
СН3СН21), образуется смесь алканов, и разделить ее на отдельные
компоненты бывает трудно.
2. Электролиз солей одноосновных органических кислот. Этот
метод был предложен в 1849 г. немецким ученым Кольбе A818—1884):
/Р
2 R—С f -> R—R + 2Ме + 2СО2
;о
R—cf — соль органической кислоты и одновалентного ме-
Р ¦
талла. Например, в случае электролиза ацетата натрия СН3—Of
ХШа
весь процесс может быть представлен следующим образом:
Р , ou n электролизГ СН3-СН3 + 2СО2 (анод)
ЧЖа " l—2NaOH+H2 (катод)
Этим способом могут быть получены алканы с четным числом
атомов углерода в молекуле.
Реакции 1—2 — примеры получения алканов из соединений
с меньшим числом атомов углерода (синтетические методы).
3. Восстановление производных предельных углеводородов (га-
логенопроизводных, спиртов, кислот, металлорганических
соединений). При получении алканов из их производных необходимо
заменить соответствующую группу на атомы водорода. Иногда это
удается легко; например, чтобы заменить атомы цинка в цинкди-
/СНз
метиле Zn< , нужно подействовать водой:
/СН3
Zn( +2HOH->2CH4 + Zn(OH),
ЧСН3
щшкднметил метан
В других случаях это связано со значительными трудностями.
В качестве восстановителя применяются иодистоводородная ки-
27
слота (при нагревании), водород, гидриды металлов, например
литийалюминий гидрид LiAlH4:
йодистый
метил
Использование иодистоводородной кислоты в качестве
восстановителя основано на ее способности диссоциировать:
Водород в момент выделения энергично восстанавливает.
Восстановление молекулярным водородом возможно только в присутствии
катализаторов (платина, палладий, никель).
4. Гидрирование непредельных соединений. Алканы могут быть
получены присоединением водорода к непредельным
углеводородам в присутствии катализатора (платина, палладий, никель):
С2Н4 + Н2 -> С2Н8 С2Н3 + 2Н2 -> С2Н6
этилен этан ацетилен этан
Методы 3 и 4 дают возможность получить алканы из соединений
с тем же числом атомов углерода.
5. Сплавление солей одноосновных предельных кислот с
щелочами. Реакция декарбоксилирования
СН3—Of + HONa -> CH4 + Na,COs
ЧЖа
Физические свойства. Четыре первых гомолога ряда метана —
газообразные вещества (см. табл. 3), начиная от пентана (С5) до
пентадекана (С15) —
жидкости, с С16 и выше —
твердые вещества.
На примере ряда метана
мы знакомимся с
закономерностями в изменении
Рис. 12. Строение цепочки алкана физических свойств в
пределах гомологического
ряда. По мере роста молекулярной массы возрастает температура
кипения, плавления, плотность. Все алканы легче воды. В твердом
состоянии цепочка атомов углерода алканов нормального строения
имеет зигзагообразное строение (рис. 12). Алканы практически
нерастворимы в воде, но хорошо растворимы в органических
растворителях. Метан, этан и высшие гомологи не имеют запаха,
средние обладают запахом бензина.
Химические свойства. Кислоты (серная, азотная), щелочи (едкий
натр, едкое кали), окислители, металлы, в том числе и щелочные,
при обычных условиях на алканы не действуют. Отсюда и возникло
их старое название «парафины» от латинского parum affinis —
лишенные сродства.
Алканы не способны к реакциям присоединения, так как в их
молекулах все связи насыщены, В то же время на солнечном свету
28
они легко вступают во взаимодействие с хлором и другими
галогенами, а при повышенных температурах способны окисляться и
проявляют способность к термическому разложению.
Причины химической инертности алканов станут понятными,
если познакомиться с некоторыми теоретическими представлениями,
существующими в органической химии. Успехи, достигнутые в
последнее время в изучении механизма органических реакций, дали
возможность разделить их по способу нарушения (разрыва) кова-
лентной связи реагирующей молекулы во время реакции.
Если разрыв связи в реагирующей молекуле происходит без
разъединения электронов электронной пары и, следовательно,
образовавшаяся связь возникает за счет уже существующей
электронной пары, то такой вид органической реакции называется гете-
ролитическим. При этом часть молекулы, получившая электронную
пару, превращается в анион, а частицы, ее лишившиеся, — в ка-
ткон; отсюда другое название этого вида реакций — ионные
реакции:
А|:Б-»А++:Б-
Другой вид органических реакций относится к
гемолитическим, в ходе которых происходит разъединение двух электронов
старой пары, а новая электронная пара образуется за счет одного
электрона реагирующей молекулы и одного электрона реагента:
А!~|Б-> А- + Б-
Образовавшиеся частицы А • и Б •, несущие один неспаренный
электрон, получили название свободных радикалов, а реакции
этого типа — радикальные.
В молекулах алканов оба вида связей С — С и С — Н построены
по типу простой а-связи, для которой характерна малая
полярность и поляризуемость. Поэтому для них в первую очередь
типичны реакции, идущие по радикальному типу. Реакции же,
идущие по ионному типу, присущи соединениям с полярными или легко
поляризующимися связями. Для осуществления реакций,
протекающих по свободнорадикальному механизму, необходимы условия
для создания свободных радикалов: высокие температуры, свет и т. д.
Для алканов особенно характерны реакции замещения,
разложения, окисления. Большинство химических превращений алканов
начинается с разрыва С — Н-связи.
Взаимодействие с галогенами. На свету, в темноте при
нагревании или в присутствии катализаторов газообразный хлор
последовательно замещает в молекуле метана все четыре атома водорода
с образованием галогенопроизводных:
СН4+С13 н> СН3С! + НС1 СН3С1 + С13 -> СН2С12-|-НС1
метан хлористый хлористый
метил метилен
29
СНС!3 + С12—* ССЦ + НС1
хлороформ четьгрех-
хлористый
углерод
Бромирование протекает менее энергично, взаимодействие с иодом
осуществить практически не удается, наоборот, фтор
взаимодействует с алканами со взрывом, что делает невозможным, без
принятия специальных предохранительных мер, прямое фторирование
алканов.
Энергия связи С — Н зависит от характера атома углерода
(первичный, вторичный, третичный) и соответственно равна: 99; 94; 90
ккал/моль A14,5-103; 333,5-10-; 376,8 -103 Дж/моль). Поэтому
легче всего замещаются атомы водорода у третичного, труднее
у вторичного и с большим трудом у первичного атомов углерода.
При галогенировании алканов часто образуется сложная смесь
всех возможных продуктов, из которой выделить нужное соединение
бывает очень трудно.
Хлорирование метана под действием света или высоких
температур протекает по цепному радикальному механизму. Молекула
хлора распадается на свободные радикалы:
которые атакуют молекулу СН4,- при этом образуется новый
свободный радикал метил — СН3 •, обладающий высокой
реакционной способностью:
С1-+СН4-»СН8- + НС1
При встрече со второй молекулой хлора метил реагирует с
образованием нового радикала:
СН3. + С1:С1-»СН3С1 + С1.
и т. д., пока не прекратит свое существование свободный радикал,
ведущий приведенную выше цепь превращений. Галогенирование
алканов возможно и при участии катализаторов: хлориды меди,
сурьмы, олова и иод. В этом случае процесс может протекать по
ионному цепному механизму.
Нитрование, При замене атомов водорода в молекуле алканов
на нитрегруппу (—NO2) образуются нитросоединения. Этот
процесс называется нитрованием. Нитруют азотной кислотой и
окислами азота. При обычной температуре азотная кислота не действует
на алкады, при нагревании окисляет. М. И. .Коновалов A856—1906)
разработал метод нитрования алканов в жидкой фазе разбавленной
13%-ной азотной кислотой при температуре 130—140° С:
,.' С5Н13:Н + НО: \О2 -» C6HL,NO2 -{- Н2О \
.^_гексан нитрогексав-- "
Особенно легко ннтрогруппа замещает водород у третичного атома
углерода, труднее у вторичного и с еще большей трудностью у
первичного.
В последнее время особое значение приобрело нитрование
газообразных алканов парами азотной кислоты или окислами азота
30
(парофазное нитрование). Процесс в зависимости от строения ал-
кана, характера нитрующего агента (азотная кислота, окислы азота)
ведется при температурах 200—450° С и давлении до 15 атм A4,7 X
X 10Б Н/м2).
Нитрование протекает по свободно радикальному механизму.
При нитровании алканы частично окисляются. При парофазном
нитровании наряду с нитропроизводиыми исходного углеводорода
образуются нитросоединения с меньшим числом атомов углерода
в молекуле — декструктивное нитрование.
Сульфирование. Серная кислота при обычной температуре не
действует на алканы, однако при слабом нагревании сульфирует,
в результате атом водорода в молекуле алкаиа замещается на
сульфогруппу — SO3H:
ноч
CsH17H-f >S02->C6H17SOaOH + H2O
нск
октан сульфооктан
Сульфооктан — типичный представитель сульфокислот.
Практически получение и применение сульфокислот и их производных
стало возможным после открытия реакций сульфохлорирования и
сульфоокисления.
Соли сульфокислот с 10—18 атомами углерода, широко
применяющиеся в качестве моющих средств, могут быть получены суль-
фохлорированием керосиновых фракций нефти:
СлН2я+2+ЗО2+С1, -> C;!H2,WSO2C1 + HC1
C^HM+1SO2C1 + 2NaOH -> CnH2n+1SO2ONa + NaCl + H2O
Сульфохлорирование проводят при комнатной температуре и
облучении реакционной массы ртутными лампами. Сульфокислоты могут
быть также получены и сульфоокислением алканов:
CnH2n+2 + SO2 + 0,5O2 -> CnH2;!+1SO2OH
Окисление. Предельные углеводороды при комнатной
температуре очень стойки к действию обычных окислителей (КМпО4,
К2Сг207). При высокой температуре алканы сгорают с образованием
двуокиси углерода, воды и выделением большого количества
энергии. Горение продуктов, содержащих алканы, т. е._ нефти,
природных и попутных газов, является важнейшим источником энергии
для человеческого общества.
В последние годы получили промышленное применение
разнообразные методы окисления парафиновых углеводородов при средних
температурах в жидкой и газообразной фазе кислородом воздуха,
обычно в присутствии катализаторов или с помощью окислителей
(МпО2). В результате образуются спирты, альдегиды, кетоны,
кислоты, часто с меньшей молекулярной массой, чем исходный ал-
кан, а также непредельные соединения. Труднее всего окисляется
метан; с повышением молекулярной массы алканов они окисляются
легче. Особое значение приобрело окисление алканов в высоко-
31
молекулярные жирные кислоты, которые затем используются для
производства мыла и моющих средств.
Исходным сырьем для получения синтетических жирных кислот
является смесь парафинов, основная масса которых представлена
углеводородами С16 — С28. Окисление проводят кислородом
воздуха с использованием соединений марганца при температуре
100—110° С. Образуются жирные кислоты с числом атомов углерода
от Q до С20. Наибольшую ценность в промышленности имеют
жирные кислоты С10—С16 и С7—С9. Первая группа жирных кислот
применяется для мыловарения, получения синтетических моющих
средств, пищевых жиров, для производства консистентных смазок,
в производстве синтетического каучука, в промышленности
строительных материалов (линолеум, асбестосмоляные плиты,
заменители битума). Основной потребитель кислот С7—С9 —
промышленность производства синтетических спиртов, используемых в качестве
пластификаторов морозостойких пластмасс. Производство
синтетических жирных кислот непрерывно расширяется. В нашей стране
работает ряд предприятий по получению их из алканов, среди
которых такие крупные химические комбинаты, как Шебекинский
и Волго-Донской.
Термическое разложение. Алканы, как и большинство
органических соединений, устойчивы только при сравнительно невысоких
температурах. При повышении температуры они разлагаются с
разрывом связей С — С (реакции расщепления) и С — Н (реакции
дегидрирования). Химические процессы, происходящие при
термическом разложении углеводородов, носят название крекинга
(разрушения, разламывания) и имеют большое народнохозяйственное
значение.
Крекинг может быть осуществлен без катализатора
(термический крекинг) и с его участием (термокаталитический крекинг).
Термическое разложение при более высоких температурах
G00—800° С) в технике называется пиролизом. Пиролиз приводит
к более глубоким изменениям и частичному образованию углерода.
Характер образующихся при крекинге продуктов зависит от
строения алкана и термического воздействия (температуры,
продолжительности нагрева и т. д.), а также от наличия и вида
катализатора; процесс крекинга часто сопровождается дегидрогенизацией.
При термическом разложении алканов с участием катализаторов
образуются предельные углеводороды с меньшим числом атомов
углерода (по сравнению с исходным соединением), непредельные
(стр. 37) и ароматические (стр. 70) углеводороды, алканы изо-
строения. Так, при крекинге «-бутана могут образовываться:
СН
з
¦CH3—CH3 + CH2=CHa
CHa ^сн2=сн—сн2—а\3-
сн.2
сн3
32
СН2=СН—СН=СН.,
н
Н;С-:
н
н
С:
Н
н
н
->Н:С--
Н
Н
|--С:Н
Й
Наибольшей термической устойчивостью обладает метан (до
800° С); с ростом длимы и увеличением разветвленное™ углеродной
цепочки термостойкость молекулы ал капов снижается.
При термическом крекинге происходит гемолитическое
расщепление углерод — углеродной связи с образованием свободных
радикалов
например:
этан свободные метальные радикалы
Свободные радикалы вследствие высокой реакционной способности
существуют очевь непродолжительное время. Продолжительность
жизни метальных и этильных радикалов — 10~3 -f- 10~4 с. В
результате их превращений и образуется все многообразие продуктов
крекинга.
Отдельные представители алканов. Применение предельных
углеводородов. Простейший представитель алканов — кетан (СН4),
Другое название метана — болотный газ; оно связано с его
образованием при гниении растений на дне болот. Метан, встречающийся
в угольных шахтах, где скопления его могут привести к взрывам,
получил название рудничного газа. Это основной компонент
природных газов, содержание его в нем может достигать 98—99 °о,
в значительных количествах метан присутствует в газах
нефтепереработки. Он широко применяется как топливо в технике
и быту и является важным сырьем в химической
промышленности (схема 2).
В технике и быту широко используются и другие алканы. Смеси
пропана н бутана применяются в качестве топлива и сырья для
получения спиртов, альдегидов, кетонов, кислот, нитропарафпнов,
пропилена, бутадиена. Пентаны используются для получения
изопрена (C3HS).
В результате переработки алканов получаются диены,
ацетилены. Жидкие алканы применяются в качестве моторного топлива.
Высшие парафины используются также в качестве сырья для
химической переработки.
АЛКЕНЫ
Алкенами, или этиленовыми углеводородами, называются соедн-
нения, содержащие в молекуле одну двойную связь у С = Ь /
поэтому они относятся к группе непредельных алифатических
углеводородов. Общая формула СЛЫ2Л, т. е. алкены содержат на
два атома водорода меньше, чем алканы с тем же числом атомов
углерода. Старое название соединений этого ряда — олефины.
2 -А. П. Нечаев 33
Схема 2
cs2
сероуглерод
Промышленный и
бытовой
газ (топли-
ЕО)
HCN
+ S
Nil,
со + н,
синтез газ
сн4
CH—GH
ацетилен
пиролиз
хлорирование
нитрование
CH3NO2
нитрометан
CH3OH
метиловый
спчрт
СН2О
муравьиный
альдегид
1
нсоон
муравьиная
кислота
Пластмассы[
Сажа
СН3С1
хлористый
метил
СН2С12
мвтттлен
СНС13
ядороформ
СС14
четырех-
хлористый
углерод
Силиконы
CF2C12
фреон
Голландские химики обнаружили, что этилен при соединении с
хлором образует маслянистое вещество, поэтому он получил название
маслородного газа (oleum — по-латински масло).
Гомологический ряд, строение, номенклатура, изомерия.
Гомологический ряд алкенов начинается с этилена С2Н4. В молекуле
этилена атомы углерода находятся во 2-м валентном состоянии
(sp 2-гибридизация), т. е. из четырех (одна s- и три р-орбиталями)
гибридизованы только три (s и две р). Поэтому в молекуле этилена
пять ст-связей, направленных под углом 120° и лежащих в одной
плоскости, и одна я-связь, образованная не гибридизованными
р-орбиталями, восьмерки которых лежат в плоскости,
перпендикулярной плоскости расположения а-связей, образующих молекулу
этилена (см. рис. 9). Следовательно, двойная связь между атомами
углерода образована и а- и я-связя,ми. Формулы и названия
первых представителей ряда этилена приведены в табл. 5.
По рациональной номенклатуре олефины рассматриваются как
производные этилена, в молекуле которого один или несколько
атомов водорода заменены углеводородными радикалами. Так,
например, СН2 = СН—СН2 — СН3 будет называться этнлэтилен.
Таблица 5
Алкены
(гомологический ряд этилена)
Название
Этилен, этен
Пропилен, пропен
а-Бутилен, 1-бутен
(З-Бутилен, 2-бутен
Изобутилен, 2-ме-
тилпропен
а-Амилен, 1 -пентен
Формула
СН2=СН3
CHg—СН=СН.2
СН*=СН—СН,—СНз
СНэ—СН=СН—СН3
цис-форма
транс-форма
СН,-С=--СН2
1
СНз
сн3—сн2—сн,—сн=сн2
Т. пл., "С
— 169,2
— 185.2
—185,3
—138,3
—105,8
—140,4
—165,2
Т. кип., °С
— 103,7
—47,7
— 6,3
3,7
0,9
—6,9
30,0
В отдельных случаях требуется уточнить положение радикалов, что
достигается нумерацией ненасыщенных углеродных атомов или
указанием на симметричность или несимметричность молекулы
СН3-С=СН-СН.2—СНЭ
I, I-диметил-2-этилэтилен
CH3
I, l-диметнлэтилен или
hecuM.it. диметилзтилен
35
По номенклатуре ИЮПАК родовое название углеводородов
с двойной связью — алкены. Для наименования алкенов
окончание «ан» соответствующего алкана заменяется на «ен», цепь
углеродных атомов нумеруется таким образом, чтобы положение двойной
связи обозначалось наиболее низкими номерами. В названии
указывается номер первого ненасыщенного атома углерода. В
остальном поступают так же, как и при наименовании алканов:
СНз СНЭ
СН3—С=СН—СН—СН2—СН3
2, 4-дийетнл-2-гексен
Структурная изомерия в ряду алкенов связана со строением
углеродного скелета и положением двойной связи:
СН,=6{ CHj—СН3—СН3 СН,=СН—СН— СН3
¦
1-пентеи '
З-четил-1-бутск
СН3—СН=С—СНз
2-.четил-2-бутен
В ряду алкенов встречается еще один вид изомерии —
геометрическая изомерия, которая является частным случаем стерео-, или
пространственной изомерии (стр. 150), связанной с
существованием соединений, различающихся пространственным расположением
входящих в их состав групп.
При геометрической изомерии, иначе называемой цис-транс-
изомерией, пространственные изомеры отличаются расположением
заместителей у ненасыщенных атомов углерода относительно
плоскости двойной связи. Например, в молекуле 2-бутеиа СН3 — СН =
= СН — СН3 метальные группы могут быть расположены по одну
сторону двойной связи (цис-фориа) и по разные стороны от
двойной связи (транс-форма):
н3с н
\/
с
с
/\
и/; н
ц ill -форма
н
Зс
н
¦\;.ч
\/
С
с
н
/
СНз
c-rhcpMa
Появление геометрической изомерии вызвано особенностью
двойной связи, свободное вращение вокруг которой невозможно.
Цис- и транс-изомеры существенно отличаются по своим
физическим и химическим свойствам (см. табл. о).
Получение алкенов. В природе алкепы встречаются в
небольших количествах, В технике их получают главным образом при
36
переработке нефти, природных и попутных газов. Они могут быть
также выделены из продуктов химической переработки каменного
угля (коксовый газ).
Олефины С2 — С5 выделяют из газообразных продуктов,
образующихся при термическом крекинге. В зависимости от вида сырья
и условий проведения крекинга содержание их в образующихся
газовых фракциях может колебаться от 15 до 55%.
Значительное количество олефинов (этилена и его ближайших
гомологов) образуется при пиролизе нефти и газообразных
парафинов. При высокотемпературном пиролизе A000°—1500° С) этилен
получается совместно с ацетиленом. Деструкция молекул при
термическом разложении сопровождается дегидрированием, что также
приводит к образованию олефинов.
Дегидрированием называется реакция, сопровождающаяся
отнятием водорода и образованием ненасыщенных соединений:
СН3—СН3 -jj-> СН2=СН2
Дегидрирование протекает значительно легче и быстрее при
применении специальных, избирательно действующих катализаторов
(селективные катализаторы), ускоряющих только этот процесс.
Широко применяется дегидрирование для получения этилена из этана,
«-бутилена — из «-бутана и изоамилена — из изопентана:
СН,—СН,—СН,—СН3 —гг СН3—СН,—СН=СН,
н-бутан 1-бутен
СН3—СН—СН,—СН3 —^ СНз—СН—СН=СН3
СН, СНз
метилбутан 3-метил-1 ->бутен
Необходимо отметить, что при термическом разложении и
дегидрировании нефтяного сырья, в том числе алканов С3—С5, образуется
смесь разнообразных продуктов. Меняя условия (температура,
давление, катализатор), подбирают режимы, при которых в наибольшем
количестве образуются требуемые продукты.
Высшие олефины (С]о—С20) получают полимеризацией этилена
в присутствии алюминийорганических катализаторов.
Существует несколько лабораторных способов синтеза алкенов.
Отнятие воды от спиртов (дегидратация спиртов). Спирты при
нагревании в присутствии водоотнимающих катализаторов (серная
кислота, фосфорная кислота, окись алюминия, хлористый цинк)
теряют молекулу воды с образованием олефина. Этот метод в силу
своей простоты длительное время применялся для получения
этилена из этилового спирта:
СН,-СН, ^-^СН,=СН2
н он
эггиловый спирт
При дегидратации спиртов более сложного строения водород
отщепляется от наименее гидрогенизированного (связанного с наимеиь-
37
шим количеством водорода) углеродного атома (правило А. М.
Зайцева):
• СН, СН3
СН3—СН2—СН—С—СН3 -5-й CHS—СН,—СН=С—СН3
Получение из галогенопроизводных. Олефины могут быть
получены отнятием галогеноводородов от галогеналкилов с помощью
спиртовых растворов щелочей или твердой щелочи:
СНз СН3
I |
СН2—С—СН3 + NaOH -» СН2=С—СН3 + NaCl + Н2О
ТН СП
Водород в соответствии с упомянутым правилом Зайцева отнимается
от наименее гидрогенизированного атома. Галогеналкилы
различного строения с неодинаковой легкостью превращаются в
непредельные соединения: легче всего отщепляют молекулу галогеново-
дорода третичные галогеналкилы, труднее первичные, вторичные
занимают промежуточное -положение.
Олефины могут быть получены отщеплением двух атомов галогена,
расположенных у соседних углеродных атомов:
СНз—СН2—СН—СН—СНз + Zn -> СН3—СН2—СН=СН—CH3 + ZnBr2
Вг Вг
или двух атомов галогена, расположенных у одного углеродного
атома:
2СН3-СНС12 + 2Hg -> СНз—СН=СН—СНз + 2HgCla
Получение из ацетиленовых соединений
СН^СН Щ СН2=СН3
Физические свойства. Первые три гомолога ряда этилена — газы (см, табл. 5),
начиная с амилена до олефина С17 — жидкости, с С18 — твердые вещества.
Гомологи нормального строения кипят при температуре более высокой, чем их
изомеры разветвленного строения (с тем же числом углеродных атомов). Температура
кипения цыс-изомеров выше, чем транс; при сравнении их температур плавления
установлена обратная закономерность. Олефины мало растворимы в воде,
плотность их меньше единицы и возрастает с увеличением молекулярной массы.
Химические свойства. Для этиленовых углеводородов наиболее
характерны реакции присоединения, они легко окисляются и
полимер изуются. Химическое поведение алкенов связано с наличием
в их молекуле двойной связи. Ненасыщенные атомы углерода в
молекуле алкенов связаны между собой о- и л-связями. п-Связь менее
«прочная», чем о-связь. Для разрыва о-связи требуется 83 ккал/моль
38
C47,5-103 Дж/моль), а двойной связи — 146 ккал/моль F14,0 X
X 103 Дж/моль), т. е. меньше, чем для разрыва двух а-связей —¦
166 ккал/моль F95 • 103 Дж/моль). Поэтому двойная углерод —
углеродная связь обладает способностью разрываться в определенных
условиях как бы наполовину; при этом атомы углерода,
присоединяя новые группы, становятся насыщенными:
я-связь А В
Необходимо также помнить, что электроны, образующие я-связь,
более доступны действию атакующего реагента, чем электроны
а-связи. Естественно, что реагенты, обладающие недостатком
электронов (они называются электрофильными), взаимодействуют с оле-
финами наиболее эффективно:
Вг2
/ Ч
—с—С—
Вг Вг
I i
—С—С-^
| |
н а
H2SO4
к —с—с—
! [
Н OSO,H
н он
Эти реакции, протекающие по гетеролитическому механизму, носят
название реакций электрофильного присоединения. Реакции с нук-
леофильными реагентами (обладающими избытком электронов), а
также реакции, протекающие по гемолитическому механизму
с участием свободных радикалов, для олефинов менее
характерны.
Присоединение галогенов. Алкены очень легко присоединяют
галогены с образованием дигалогенопроизводных: ¦
СН2=СН2 + Вг2 -> СНоВг—СН2Вг
этилен 1,2-дибромэтан
Реакция начинается с электрофильной атаки молекулой брома
двойной связи. Под влиянием электронной плотности олефина молекула
галогена поляризуется и захватывает я-электроны двойной связи
с образованием неустойчивого я-комплекса. Затем я-связь и связь
бром — бром гетеролитически разрывается. Положительно
заряженный ион брома за счет электронов я-связи образует обычную а-связь
39
с одним из ненасыщенных углеродных атомов, другой
углеродный атом, лишившись электрона, заряжается положительно,
в результате образуется карбокатион (он иногда называется
а-комплексом):
t
СН2=СН2 + Вг2-~ СН2=СН2 -» СН2—СН2 + ВГ
Вг
Карбокатион атакуется анионом брома с образованием 1,2-дибром-
этана:
СНа—СН + Вг -> СН.2—СН2 (нуклеофпльная
Вг Вг Вг
карбокатыон 1,2-дибромэтан
j атака
Таким же образом идет присоединение к двойной связи HBr, H2SO4,
Н2О и других неорганических веществ. Присоединение брома
(обесцвечивание бромной воды) — качественная реакция на двойную
связь.
Из галогенов с олефинами наиболее энергично взаимодействует
фтор (со взрывом), труднее всего — иод. Введение заместителей,
поляризующих двойную связь, в молекулу алкена, применение
смешанных галогенных соединений (СИ, С1Вг), молекулы которых
поляризованы, ускоряет присоединение. Галогенирование алкенов
широко применяется в промышленности и в аналитической
химии.
Присоединение галогеноводородов:
СН2=СН, + НВг -> СН3—CH.Br
этилен бромэтсш
В случае несимметрично построенных олефинов водород
предпочтительнее присоединяется к наиболее гндрогенизированному из
ненасыщенных атомов углерода, галоген — к его партнеру:
СН.,—СН=СНгН-НС1 -, СН3—СНС1—СН3
пропилен 2-хлорпропан
Это положение получило название правила Марковникова, в честь
знаменитого русского органика В. В. Марковникова A838—1904),
его открывшего. Правило Марковникова является одним из
положений теории о взаимном влиянии атомов в молекулах органических
соединений. По современным представлениям атомы, входящие в со-
40
став молекулы, могут оказывать влияние не только на соседние
атомы, но и на атомы, удаленные от них. Рассмотрим два примера:
Н
Н— С— С=С-Н B)
I Н Н
н
В первом случае в связи с различием в электроотрицательнссти
хлора и углерода Q электронная плотность связи С1—С будет
сдвинута в сторону более электроотрицательного атома хлора. При этом
электронная плотность у атома углерода Q снизится, что в свою
очередь вызовет смещение электронной плотности связи Q—С,
(условно это показано стрелками), хотя и в меньшей степени.
Постепенно сдвиг электронов в углерод — углеродной цепи будет
затухать. Все это приводит к некоторому перераспределению
электронной плотности: снижению ее в одной части молекулы и увеличению
в другой.
Может быть и обратный случай (пример 2). Углерод является
более электроотрицательным атомом, чем водород, поэтому в связях
С—Н метильной группы (группа СН3) электронная плотность а-связи
будет несколько смещена в его сторону, что, естественно, приведет
к повышению у него электронной плотности и в свою очередь
вызовет ее повышение в связи Q—С3. Электроны, осуществляющие эту
связь, приобретают способность взаимодействовать с электронами
л;-связи. В результате этого происходит смещение электронной
плотности я-связн в сторону углеродного атома С3, я-связь поляризуется.
Поляризацию я-связи принято обозначать изогнутой стрелкой,
показывая направление смещения электронной плотности. Поэтому
у атома углерода С2 будет наблюдаться некоторая нехватка
электронной плотности, а у атома углерода С3 некоторый избыток. Подобные
частичные заряды (они не будут равны полному заряду электрона)
условно принято обозначать греческой буквой дельта (б), знак <;—»
указывает на частичное увеличение электронной плотности, «-J-»
ее нехватку.
Описанный вид взаимного влияния атомов в молекулах
органических соединений получил название индукционного эффекта. В
первом примере имеет место отрицательный индукционный эффект
(электронная плотность вследствие введения заместителя
снижается), во втором — положительный (электронная плотность
возрастает).
При взаимодействии пропилена с галогеноводородом ион
водорода (Н+), образовавшийся при распаде последнего, в первую
очередь будет присоединяться к углеродному атому С, несущему ча-
41
стичный отрицательный заряд, т. е. к наиболее гидрогенизирован-
ному:
+в -в + + Вг_
СН3—СН=СН2 + Н-*СН3—СН—СНз^СН.,—СНВг—СН3
пропилеи карбокатнон 2-бромпропан
Правило Марковникова соблюдается только при гетеролитйче-
ском механизме присоединения галогеноводородов к алканам, если
присоединение протекает по радикальному механизму (например,
в присутствии перекисей), порядок присоединения обратный.
Из галогеноводородов наиболее легко присоединяется HI.
Гидратация (присоединение воды). При присоединении воды
к олефинам образуются спирты:
R—СН=СН2 + НОН -> R— СН—СН3
ОН
Водород направляется к наиболее гидрогенизированному атому
углерода. В технике осуществляется прямая гидратация
(катализаторы — фосфорная кислота, фосфаты, окись алюминия;
повышенная температура):
СН2=СН.2+ НОН катализатг? сНз-СНа-ОН
и сернокислотная гидратация:
СН2=СН2 + H2SO4 -> СН3—СН2—OSO2OH
этилен этнлсерная кислота
СНз—СН2—OSO2OH + Н2О -» СН3—СН2—ОН + H2SO4
этиловый спирт
Способность олефинов к гидратации возрастает с увеличением длины
углеродной цепочки (до известных пределов) и количества алкилов.
Гидрирование. Алкены в присутствии катализаторов (платина,
палладий, никель) легко присоединяют водород, превращаясь в
предельные углеводороды:
R-CH=CH2 + H2 ^f R-CH2-CH3
При отсутствии катализатора даже атомарный водород и водород
в момент выделения на олефины не действует.
На примере реакций гидрирования алкенов мы знакомимся с
явлением гетерогенного катализа. Скорость гидрирования зависит от
вида и активности катализатора, условий гидрирования и строения
алкенов. На платине и палладии гидрирование успешно идет при
комнатной температуре, на никеле — при нагревании.
Гидрирование ненасыщенных связей широко применяется в технике и
аналитической практике. В пищевой промышленности громадное значение
имеет гидрирование жиров (стр. 215).
Много сделали для изучения гидрирования наши отечественные
ученые С. А. Фокин, С. В. Лебедев, Н. Д. Зелинский и зарубежные
исследователи Сабатье, Сендеран, Вильштеттер.
Окисление олефинов. Олефины легко окисляются. Направление
окисления и характер образующихся продуктов зависит от
строения алкенов, вида окислителя и условий окисления,
42
При окислении разбавленным раствором перманганата калия
на холоду в щелочной среде (окисление по Е. Е. Вагнеру) образуются
гликоли (стр. 104):
ЗСНз—СН=СН2-Ь2КМп04 + 4Н20 -> ЗСН3—СН—СН2 + 2МпО2 + 2КОН
пропилен ^ ^
\ ,2-пропиленгликоль
При более энергичном окислении олефинов (азотная кислота,
перманганат калия в кислой среде, хромовая кислота) происходит
разрыв молекулы по двойной связи с образованием кислот и кетонов:
СНз-СН=СН-СНз sp™^» ™лота 2СН3-СООН
2-бутен уксусная кислота
сн3-с=сн-сн3хромова* КИСЛ01! сн3-с=о+сн3соон
СН3 СНз
2-метил-2-бутен ацетон уксусная
кислота
Анализируя продукты окисления, можно сделать заключение
о строении исходного алкена.
При окислении олефинов кислородом воздуха в присутствии
серебряных катализаторов образуются окиси олефинов:
2СН2=СН2 + О2 -> 2СН2—СН2
о
окись этилена
Это важный промышленный способ получения окиси этилена.
Озонирование алкенов. Для установления строения алкенов
используется реакция озонирования:
сн3—сн=с—сн,—сн3+о3 -> СНз—с/ ' \с—сн2—сн3
1ЧО-О/!
СН3 Н СНз
З-метил-2-пентен озонид
Образовавшийся озонид (неустойчивое, легко взрывающееся
вещество) разлагают водой и, анализируя продукты реакции,
устанавливают строение исходного алкена:
/°\
снз-с/ \с-сн2-сн3+н2о ->
н сн3
-» СН3—СНО+О=С—СН2—СН3+Н2О2
уксусный метилэтилкетон
альдегид
Полимеризация алкенов. Полимеризацией называется соединение
одинаковых молекулДмономерсда) .с образованием-нового,, сложного
вещества с большой молекулярной массой (полимер). Если в образо-
43
p4yp процесс_носит
название сополимерязацяи^ Количество молекул мономера (п), соеди-
нившихся при образовании полимера, 1называётся степенью
полимеризации. Практически полимерные материгГльГсостаятПиз~смеси
молекул, составленных из различного числа мономеров, поэтому под
молекулярлоЁ-ЛШссой_гюлнмер_а понимаю! _его__среднюю_молекуляр-
ную массу... Получение ^полимеров всегда ?0?1аат_ш1двхх_этапов:
получёние_мошмераа-(эти вопросы разбираются при рассмотрении
соответствующих классов соединений) и получение, из .них,., щдимер_ов
(полимеризация).
Из олефинов в качестве мономеров наиболее широкое применение
нашли этилен, пропилен, бутилены:
лСН2=СН„ -> [—СН2—СН3—]„
этилен
пСН3~ СН=СН3
пропилен
Свойства полиолефинов во многом зависят от способа
полимеризации. Различают_л_инеиную (цепную) и ступенчатую (миграционную)
полимери!ацшо. В первом случаё"промежуточныё"гГродукты реакции
не удается изолировать, а конечный продукт_отличается_ в_ысокой
молекулярной массой^Во втором"_случае проме2ку^го^шые_г1р^о^д^™ь1_мо-
гут быть,.выделены. Образовавшийся полимер.обычно состоит из
небольшого числа молекул мономераТ'"
В зависимости от механизма полимеризация может
осуществляться по ионному дли радикальному пути. Примером ступенчатой"
ионной полимеризации может служить осуществленное А. М.
Бутлеровым A873) соединение двух молекул изобутилена:
СН3 снз СН3
! | |
сн8-с=снгёо%^;-н^-б7CH3-c+^-_i^-JI? сн3-с-сн2
полиэт
-СН-
СН3
1лен
-сна—
полипропилен
СН3 СНз СН3 СН3
изобутилен
СН3 СН3
—fFCHg—С—СН2—С=СН. + СН3—С—СН=С—СН3 —
| ! | |
СН3 СН3 СНз СНз
. 80% 20%
днизебутилен-
СНз
-> СНз—С—СН2—СН—СНз
СН3 СНз
2,2,4-триыетилпентан (изаоктан)
В присутствии 60°о-ной серной кислоты реакция ограничивается
образованием днмера. Гидрированием смеси «динзобутплена» полу-
44
чают 2,2,4-триметилпентан (изооктан). Реакция имеет важное
практическое значение.
Особо важна для промышленного получения полнолефинов с
высокой молекулярной массой цепная полимеризация,
осуществляемая по свободнорадикальному механизму. Полимериз^щ1^1ачина-
ется с акт1Ш1дюв^дия.^а^р^лпбд__молекз{лы_под влиянием
температуры, света" и т. д. Акт1^ирсщанцая,.ма:1екула
вызьГваёт"активирование и полимеризащ1ю.бодьшрго,числагддугих мЪлекул, с которыми
она сталкивается.- Первая фаза протекает относптельно'медленио,
вторая — практически мгновенно. Для цепной радикальной
полимеризации характерно отсутствие промежуточных продуктов — ди-
меров, тримеров и т. д. Различают три главные стадии:
1. Инициирование (образование свободного радикала)
энергия
Для инициирования часто применяются вещества (инициаторы),
распадающиеся в условиях реакции на свободные радикалы.
2. Рост цепи. Первоначально образовавшийся радикал,
соединяясь с молекулами олефинов, образует новые радикалы с большой
молекулярной массой
М- + М — AU- - М -> М3. "^ Мп-
3. Обрыв цепи. На этой стадии полимерный радикал
превращается в макромолекулу полимера
Полиолефины в настоящее время находят широчайшее
применение. Полиэтилен применяется для изоляции электропроводов,
изготовления пленок, труб, посуды. Полипропилен, не уступая
полиэтилену в химической стойкости, превосходит его по прочности
на разрыв и более стоек к высоким температурам, применяется для
изготовления изоляции, деталей радиоаппаратуры и машин, труб,
канатов, сетей, фильтрующих тканей.
Реакции замещения. При высокотемпературном хлорировании
пропилена происходит замещение атомов водорода, причем хлор
вступает в а-положение к двойной связи:
СН,=СН—СН —^ СН2=СН—СН,С1 + НС1
пропилен хлористый аллил
Отдельные представители. Применение. Высокая реакционная
способность, доступность и низкая стоимость исходного сырья
превратили олефины в один из наиболее важных классов органических
соединений, широко применяемых в разнообразных синтезах. Из
большого числа гомологов особую ценность для промышленности
представляют первые четыре.
Этилен С2Н4. Этилен — газ, без цвета и запаха, мало растворим
в воде. С воздухом образует взрывоопасные смеси. Является
исходным веществом для получения разнообразных продуктов органиче-
45
Схема 3
рнэация
Cl,
(—СН2—СН2—) „
лолыэтнлен
СН2С1—СНгС1
СНС1=СН2
полимери-
[—СНС1—СНг-]п
дихлорэтан
хлористый винил
полихлорвинил
НС1
СН,—СН2С1
хлористый этнл
НОС1
Н?О
этиленхлоргидрин
GH3— CH2—ОН
этиловый спирт
GH2—СН—СН—GH2
дивинил
Каучук
H,SO
СН,—СИ,—OSO,H
зтилсерная кислота
NH,
CH3—CH2—NH2
сн3-
-сн2—он |
этиловый спирт
С2Н
5-о-с2н5
диэтиловый эфир
зтилаыин
C6H5—CH2—CH3
сбн5-сн=сн2
этилбензол
горение
стирол
—СН—СН2—
CO + H2
Сварка, резка
металлов
окись этилена
СН,—СНО
о2
СН,—СООН
уксусный альдегид
уксусная кислота
пропяэиэвын альдегид
, снг
прспионовая кислота
ссь
СН2С1—(СН2)Я—СС13
Синтетические волокна
к полимерные материалы
ского синтеза, имеющих первостепенное народнохозяйственное
значение (схема 3). Большая его часть расходуется на получение
полиэтилена, окиси этилена, этилового спирта. Промышленные пути
получения — выделение из продуктов переработки нефтяного
сырья, коксового газа, дегидрирование этана.
Пропилен СН3—СН=СН.2. Второй по значению гомолог.
Получается из продуктов переработки нефти, пиролизом бутана (совмест-
46
Схема 4
CH2=CH—CH3
пропилен
полимеризация
Г—сн2—сн—"
! с* 1д
1— 3 _
п
полипропилен
Н2О
ОН
сн3—сн—сн3
изспропиловый спирт
сь
ог
со + н-
СО + Н,9
CsHg
НН3 + О2
СН2С1—СНС1—С
1,2-днхлорпропан
СН2—СН СН-С1
хлористый аллнл
Н2С—СН—СН3
0
окись пропилена
о
ацетон
ск2—сн—сне
акролеин
сн3—сн—сон
м„,
изомасляный альдегид
CHg CH^ CH2 ^-s^ rr
масляный альдегид
сн3—сн—соон
СНз
изомаеллная кислота
С6Н5-СН(СН3J
нзопропилбензол
СН2==СН— CN
акрялонтприл
но с этиленом), дегидрированием пропана. Является исходным
сырьем для получения изопропилового спирта, полипропилена,
окиси пропилена и ряда других продуктов (схема 4).
47
Бупшлены и изобупшлен С4Н8. Получают из продуктов
переработки нефтяного сырья (бутан-бутеновая фракция). Изобутилен
является исходным веществом в синтезе изооктана п полиизобути-
лена; н-бутены—сырье для получения дивинила.
АЛКИНЫ
Алкинами, или ацетиленовыми углеводородами, называются
соединения, содержащие в молекуле одну тройную связь. Общая
формула алкинов С„Н2Л-2- Они содержат в молекуле на четыре атома
водорода меньше, чем алканы с тем же числом углеродных атомов, и
изомерны алкадиенам (стр. 54).
Гомологический ряд, строение, номенклатура, изомерия. Первый
представитель класса алкинов — ацетилен Н—С=С—Н, в молекуле
которого, так же, как и у других гомологов, ненасыщенные атомы
углерода связаны между собой тройной связью и находятся в третьем
валентном состоянии (sp-гибридизация), т. е. у них из четырех орби-
талей (одна s и три р) гибридизованы только две (s и р). Из трех
связей между ненасыщенными атомами углерода одна а- и две я-связи.
В молекуле ацетилена все четыре атома расположены на одной
прямой, углы между о-связями —180° (стр. 18). Формулы и названия
основных гомологов приведены в табл. 6.
Таблица б
Алкины
(гомологический ряд ацетилена)
S Название
Ацетилен, этин
Метил ацетилен, про-
пин, аллилен
Дииетилацетилен,
2-бутин, кротонилен
Мети лэти л ацетилен,
2-пентин, валерилен
нс=сн
СНз—Cs=C
СН3—С==еС
СНз—СН,-
Формула
н
—СНз
-С-_^С—СН3
Т. пл., 'С
—81,8
— 102,7
—32,3
— 101,0
Т, кип., °С
-81
—23,2
+27,0
+56,1
При наименовании алкинов тривиальное название практически
сохранилось только для первого гомолога — ацетилена. По
рациональной номенклатуре алкины рассматриваются как производные,
ацетилена, в котором один или оба водорода заменены на алкилы:
СН3—С=С-СН„-СН3 СН3-С=С— СН-СН3
I
метнлэтплацетилен
М^ПТЛНЗСПрОПИЛЙЦОТИЛСН
По номенклатуре ПЮПАК родовое название углеводородов с
тройной связью — алкины. Для наименования алкниов по ИЮПАК
окончание <'ап» в названии соответствующего алкаиа заменяется на «ин»,
перед названием цифрой указывается номер первого ненасышен-
48
ного атома углерода. Цепь углеродных атомов в молекуле алкина
нумеруют таким образом, чтобы ненасыщенные атомы углерода
обозначались наиболее низкими номерами. В остальном поступают
так же, как и при наименовании алканов:
СН3—СН—С=С—СН3 4-метил-2-пентин
СН,
Структурная изомерия в ряду алкинов связана со строением
углеродного скелета и положением тройной связи:
нс=с-сн-
СНз
-СН3
-сн2
СН3—С=С—СН2~
З-метил-1-бутин
—СНз 1-пентнн
-СН2 2-пентин
В связи с линейным строением молекул алкинов геометрическая
изомерия у них отсутствует.
Способы получения. Наибольшее промышленное значение из
алкинов имеет ацетилен, получаемый в технике разложением
карбида кальция водой или термической деструкцией
углеводородов.
Получение ацетилена из карбида кальция (Вёлер, 1862):
Карбид кальция разлагается водой:
-> НС=СН+Са (ОНK
Са
карбид
кальция
По этому способу, хотя он и требует значительного расхода
электроэнергии, до настоящего времени получают основное количество
ацетилена для синтезов и сварочных работ. Он особенно выгоден в
странах с большими запасами дешевого каменного угля.
Получение ацетилена термическим разложением углеводородов.
В последние годы быстро развивается способ получения ацетилена
термической деструкцией углеводородов. Основное сырье — алканы
Q—С4; бензины:
СН6 —S СН^СН +2Н3
Получение из метана требует наибольшей затраты энергии, но это
компенсируется его доступностью и низкой стоимостью. Высокие
49
температуры, необходимые для пиролиза, могут быть достигнуты
частичным сжиганием исходного сырья (окислительный пиролиз):
7CH4-f ЗО2 — 2СН=СН + С + 2СО + 8Н2 + 4Н2О
метан ацетилен
Сейчас разработаны способы получения ацетилена пиролизом при
температуре 2 000~С (получение в «плазме») с выходом до 80%.
Получение из дигалогенопроизводных алканов:
Щ'"вУ\
...... . ....
QHs-C-C—CH3 + 2Na0H — C2H>-teC—CH3 + 2NaBr+2H2O
' ' метилэтилацетилен
;вг н
2, 3-дибромпентан
Отнятие двух молекул галогеноводородов осуществляется
спиртовыми растворами щелочей, порошкообразным едким кали, амидом
натрия.
Получение алкинов алкилированием металлорганических
производных ацетилена (стр. 52).
Физические свойства. Гомологи С2—Q — газы, С5—С15 — жидкости, С15 и
выше — твердые вещества. У алкинов наблюдаются те же закономерности
изменения температур кипения и плавления, что и у алкенов. Положение тройной
связи существенно влияет на температуру кипения.
Химические свойства. Химическое поведение алкинов связано
с наличием в их молекуле тройной связи и особенностями ее
строения. Для алкинов в первую очередь характерны реакции
присоединения, протекающие ступенчато сначала с образованием алкенов
(или их производных), затем алканов. Можно было бы предполагать,
что алкины, содержащие в молекуле две л-связи, присоединяют
другие группы значительно легче, чем алкены. На практике это не
подтверждается. Электроны, образующие связь между ненасыщенными
атомами углерода в молекуле алкина, несколько втянуты внутрь
молекулы (ближе находятся к ядру), поэтому алкины труднее, чем
алкены, присоединяют электрофильные реагенты и легче — нуклео-
фильные. Вторая группа реакций связана с кислотными свойствами
1-алкинов и их способностью образовывать ацетилениды.
Реакции присоединения. 1. Гидрирование алкинов. Водород
в присутствии катализаторов (платина, палладий, никель)
восстанавливает алкины в алканы. Реакция протекает ступенчато, через
промежуточные образования алкенов:
сн=сн + н, ЕЕ=!1°_р сн.2=сн2+н3
ацетилен этилен этан
2. Присоединение галогенов.
СН=СН + Вг2 _ СНВг=СНВг + Вг2 — CHBr,—СНВг2
ацетилен 1, 2-днбоомэген 1, 1, 2, 2-тетрабромэтан
Присоединение галогенов к тройной связи происходит так же, как
и присоединение к алкенам, но с меньшей скоростью,
50
3. Присоединение галогеноводородов. Присоединение галогено-
водородов к алкинам происходит по правилу В. В. Марковннкова,
но менее активно, чем к алкенам. Реакция протекает в две стадии:
CHsCH -— СН2=СНС1 -— СН3—СНС12
ацетилен хлористый 1, 1-дихлорэтан
винил
4. Присоединение воды (реакция гидратации). В присутствии
серной кислоты и сульфата двухвалентной ртути алкины легко
присоединяют воду с образованием уксусного альдегида:
СН=СН + Н2О HaSQ'-f-Hgscu
"О
п
СН,=С(
,0
сн,-с^
¦н
виниловый уксусный
спирт альдегид
Образовавшийся на первом этапе реакции виниловый спирт
неустойчив и перегруппировывается в уксусный альдегид. Эта реакция,
открытая в 1884 г. М. Г. Кучеровым и названная его именем, имеет
большое промышленное значение. Присоединение воды к гомологам
ацетилена происходит в соответствии с правилом В. В. Марковни-
кова:
ОН 1 О
CH3-Cs5CH + Н2О - 4 s - LCH3-C=CH2J -> CH3-C-CH3
метилацетилен ацетон
5. Присоединение спиртов:
сн=сн + нос2н5 -- — сн2=сн—о-сн2—сн3
ацетилен этилопый этилвиниловый эфир
спирт
Реакция протекает в присутствии едких щелочей с образованием ал-
килвиниловых эфиров.
6. Присоединение синильной кислоты:
СН=СН+ HCN — СН2=СН—CN
ацетилен акрилонитрил
Реакция протекает при участии в качестве катализатора раствора,
содержащего хлористую медь, продукт реакции — нитрил
акриловой кислоты имеет важное промышленное значение (акрилонитрил,
стр. 53).
Полимеризация алкинов. Ацетилен легко вступает в реакцию
полимеризации с образованием, в зависимости от условий реакции
и природы катализаторов, разнообразных продуктов, широко
применяющихся в качестве сырья для синтеза каучуков, пластических
масс, химических волокон, пленкообразующих материалов.
1. Образование бензола (Н. Д. Зелинский, Б. А. Казанский):
зсн=сн 4^—> с6нв
ацетилен активированный бензол
уголь
51
2. Образование винилацетилена. При полимеризации ацетилена
в присутствии водного раствора однохлористой меди CuCl и
хлористого аммония NH4C1 получают вииилацетилен и дивннилацетилен:
2СН=СН — СН=С—СН=СН2
ацетилен впнилацетилен
зсн=сн — св>=сн—с=с—сн=сн2
ацетилен дивпнилацетилен
При присоединении к винилацетилену хлористого водорода
образуется хлоропрен, который является мономером в производстве поли-
хлоропрена:
СН.,=СН—С-СН + НС1 ^СН2=СС1-СН=СН2
шшилацетилен хлоропрен
3. Образование циклооктатетраена:
СН=СНЧ
соединения / \
НС СН
НС, . СН
чсн=сн''
цикл оси; гатетраен
4. Образование купрена:
лСК=СН Ц1 (С.Н^
купрен
Купрен — желтый аморфный порошок с малой теплопроводностью,
применяется в качестве изоляционного материала.
Реакции замещения. Атом водорода у ненасыщенного атома в
молекуле алкинов (СНеэСН; R—С=СН) вследствие неравномерного
распределения электронной плотности tj _"с = с <— Н°
приобретает способность отрываться в присутствии сильных
оснований в виде протона.
Ацетилен проявляет как бы кислотные свойства. Так, при
взаимодействии ацетилена с аммиачным раствором окиси серебра
выпадает осадок желто-серого цвета — ацетиленида серебра:
НС=СН + 2 [Ag (NH3J] ОН — AgC=CAg-b 4NH3 + 2H2O
ацстилекид
серебра
Аналогично протекает реакция с аммиачным раствором
однохлористой медп:
СН3— С=СН + [Си (МНзЫ он — СИ3—С=ССи + 2\Н3 + Н2О
Ацетилениды серебра и меди применяются для разнообразных
синтезов, в сухом состоянии они крайне взрывоопасны. Образование
ацетиленидов серебра — качественная реакция на тройную связь.
Ацетилениды магния используются для получения гомологов
ацетилена (стр. 50):
НС=СН -J-CHc-Mgl — HC=GMgl +CH4
HC-CMg] + c,hj - нс=с-с2н5 -;-iMgt,
52
Схема 5
Промышленное использование ацетилена
НС1
i »)СН=5С—С№=СН2
вини.чацегилен-
—»j CH2=CH—C=C—CH=CH2 I
дивинилацетилен
СН,=СН—СС1=СН,
полихлоропрен
циклооктатетраен
С1НС=СС1,
купрен
трихло'
С12
СНС1=СНС1
рэтилен
СНС1,—СНС1,
дихлорэтилен
тетрахлорэтилен
HCI
СН2=СНС1
[СН2—СНС1—]п
хлористый винил
полихлорвинил
Н,0
СН=СИ
ацетилен
СН,-СНО
уксусный альдегид
R-OH
СН,=СН—OR
простые
виниловые эфиры
полимеры эфиров
СН3—СН2—ОН
этиловый спирт
СН3—СООН
уксусная кислота
этилацетат
СН3СНОН—СН2—СНО
альдол
сн,соон
сн,=сн—о—со—сщ
(С,Н6О2)П
вишшацетат
Поливинил-
ацетат
СНпО
сн=с—сн,он
сн3сосн3
пропаргиловыи
спирт
CHjOH—С=С—СН,ОН
оутандиол
сн,он—снон—сн2он
СН2=С-СН=СН2
глицерин
синтетический
каучук
HCN
иаопреи
CH,=GH—CN
сь
акрилонитрил
I сварка и резка
I металлов
синтетическое
волокно
синтетический
каучук
X
I
Отдельные представители. Применение. Из алкинов особое
промышленное значение имеет ацетилен. Простота получения,
относительно низкая стоимость, химическая активность, разнообразие
реакций, в которых он способен участвовать, сделали ацетилен
важнейшим исходным соединением для многочисленных промышленных
синтезов (схема 5). Ацетилен широко используется для резки и
сварки металлов, так как при его сгорании выделяется большое
количество тепла, а температура ацетиленокислородного пламени
превышает 3000° С.
Ацетилен — бесцветный газ, без запаха, хорошо растворимый
во многих органических растворителях. Смеси ацетилена с
кислородом и воздухом, особенно при повышенном давлении, исключительно
взрывоопасны, поэтому при работе с ним нужно проявлять крайнюю
осторожность. Ацетилен хранят и транспортируют в специальных
баллонах, заполненных пористыми материалами (активированный
уголь, алюмосиликаты), пропитанными ацетоном.
АЛКАДИЕНЫ
Диеновые углеводороды (алкадиены) содержат в молекуле две
двойные связи. Общая формула С„Н2,,-2.
Классификация, номенклатура, изомерия. Способы получения
и особенно свойства диенов зависят от взаимного расположения
двойных связей, поэтому диены принято классифицировать по
этому принципу, деля их на три группы.
СН,
СНз
СН,
сн.
сна
сн.
Формула
Диены с
=с=сн,
—СН=С=СН3
Диены
=СН—СН=СН>
=С—СН=СН., "
1
СН3
Диены
Алкадиеновые углеводороды
Название
тривиальное
по ИЮПАК
Таб
Т. пл,, °С
кумулированными связями A,2-диены)
Аллен
Метилаллен
Пропадиен
1,2-Бутадиен
— 146,0
— 13Ь,3
с сопряженными связями A,3-диены)
Дивинил
Изопрен
1,3-Б\ттадиен
2-,Метил-1,3-
бутадиен
— 108,9
— 145,0
с изолированными двойными связями
=СН—СН,—СН=СН,
=сн- сн ,-сн,сн -сн.
Дивиннлме-
тан
Диаллнл
1.4-Пента-
дпен
1,5-Гекса-
диен
— 148,3
— 140,7
лица 7
Т. кип.*
°С
—32,0
10,3
— 3,0
34,0
25.9
59,9
54
I. Диены, у которых двойные связи находятся у одного атома
углерода и содержат группировку >С=С=С< (аллен и его
гомологи). Они называются диенами с кумулированными, или аллено-
выми связями, по названию аллена — первого гомолога,
содержащего эту систему.
П. Диены, у которых двойные связи разделены одной простой
связью. Они содержат группировку >С— С—С=С< и называются
диенами с сопряженными или конъюгированными связями.
III. Диены, в молекуле которых двойные связи разделены двумя
и более простыми связями >С=С—(СН^л—С=С<. Они назы-
!
! I
ваются диенами с изолированными связями. Формула и названия
наиболее важных диенов приведены в табл. 7.
При наименовании диенов широко применяются тривиальные
названия: аллен, изопрен. Некоторыми из них разрешено
пользоваться и по ИЮПАК. В отдельных случаях при построении
названия симметричных диенов их рассматривают как состоящие из двух
радикалов: дивинил, диаллил (табл. 7). По ИЮПАК наименование
диенов строится по тому же принципу, что и наименование алкенов.
Перед окончанием «ен» ставится приставка «ди» (два), указывающая
на количество двойных связей в молекуле:
СН3
i
сн,,-с=сн—сн=сн-сн3
1 2 8 4 5 6
2-метил-2, 4-гексадиен
Изомерия в ряду алкенов связана со строением углеродного
скелета и положением двойных связей. 1,2-Диены (аллен и его
гомологи) мало изучены и пока не имеют большого практического
применения. Диены с изолированными связями ведут себя в
химических реакциях так же, как и алкены, с той разницей, что в
реакциях могут участвовать две двойные связи. Наибольший интерес
в химическом отношении и наибольшее значение в химической
промышленности имеют диены с сопряженными двойными связями.
1,3-Бутадиен, изопрен — мономеры, из которых построены
важнейшие природные и синтетические материалы. Поэтому основное
внимание будет уделено именно этой группе диенов.
Способы получения. 1,3-Бутадиен и изопрен — важнейшие
мономеры для производства синтетического каучука, вырабатываются
в промышленности в очень больших количествах. Известно много
способов получения 1,3-бутадиена. Особое промышленное значение
имеют: каталитическое превращение этилового спирта'в 1,3-бутадиен
(метод С. В. Лебедева) и получение 1,3-бутадиена дегидрированием
н-бутана или н-бутилена.
Получение 1,3-бугпадиена из этилового спирта. В 1910 г. С. В.
Лебедев A874—1934) установил возможность получения 1,3-бутадиена
из этилового спирта в присутствии смешанных катализаторов.
В 1928 г, коллектив химиков, руководимый С. В. Лебедевым, вышел
55
победителем международного конкурса на разработку
промышленного метода получения синтетического каучука, объявленного
Высшим Советом народного хозяйства СССР, а в 1932 г. в нашей стране
уже начали работать заводы по получению синтетического каучука
из 1,3-бутадиена, синтезированного по методу Лебедева. Это было
первое в мире крупное промышленное производство синтетического
каучука. Синтез осуществляется по следующей схеме:
2сн3-сн2-он A'fdotcz- сн2=сн-сн=сн2 + н2+н2о
этиловый спирт 1,3-бутадиен
В настоящее время этот способ целесообразно применять только
при использовании дешевого синтетического этилового спирта.
Получение 1,3-бутадиена из бутана и бутенов. Дегидрирование
н-бутана и н-бутенов с образованием 1,3-бутадиена — наиболее
важный и перспективный способ его получения. В промышленности
осуществляется в двух видах: двухступенчатое дегидрирование н-бутана
и одноступенчатое дегидрирование н-бутана и н-бутенов. Сырье —
бутановая фракция газов нефтепереработки или попутных газов;
н-бутены — из газов деструктивной переработки нефтяного сырья.
1. Двухступенчатое дегидрирование н-бутана:
СНз—CH2—CHa—CH3 ~-
н-бутан
— СН2=СН—СН,—СН3
1-бутеи C4%)
— СН3—СН=СН—СНа
2-бутен F6%)
-Н/
сн=сн.
1,3-бутадиен
Катализатор 1 — окись хрома, нанесенная на окись алюминия;
катализатор 2 — окиси железа и хрома.
2. Одноступенчатое дегидрирование н-бутана:
СН3—СН2—СНа—СН3 -занГ СН,=СН—СН=СН2
к-бутан 1,3-бутадиен
Катализатор — смесь окиси алюминия и окиси хрома. Получение
1,3-бутадиена по одноступенчатой схеме осуществляется по более
простой технологии и требует меньших энергетических
затрат, чем по двухступенчатой схеме, и поэтому экономически
выгоднее.
Получение 1,3-бутадиена из н-бутенов (третий вариант) обычно
осуществляется по технологической схеме второй части
двухступенчатого метода.
Получение изопрен а. В настоящее время отсутствует
единый, общепринятый метод получения изопрена. В каждом
конкретном случае выбор метода связан с наличием сырья,
энергетическими ресурсами и т. д. Наиболее перспективными являются;
получение изопрена дегидрированием изопентана и нзопентенов и
синтез из пропилена.
Дегидрирование изопентана и нзопентенов. Сырье: н-пентан,
изомернзованный в изопентан, пзопентан газового бен-
56
зина, изоамилены, выделенные из продуктов деструкции бензина:
СН3
СН3
|
сн.,-сн
СН2
сн3
2-метплбутан
сн2=с—сн„—сн,—
2-метил-1-бутеп
сн3
_ СНз—С=СН-СН3 -
2-ме-шл-2-бутен F0%)
СНз
изопрен
¦СН2=СН—СН—СН3—
З-ыетил-1 -бутен
Дегидрирование протекает в присутствии следующих катализаторов:
окись хрома, молибдена или вольфрама, нанесенная на окись
алюминия.
Возможно одностадийное дегидрирование изопентана в изопрен:
СНз СН3
си3—сн—сн2—сн3 —-^ сн2=с—сн--=сн2
Синтез изопрена из пропилена. Первая стадия — образование
димера:
2сн2=сн-сн3 15°-25оса сн2=с-сна-сна-сн3
пропилен :
сн3
2-метил-1-пентен (димер)
Димеризацию проводят при давлении 200 атм A9,6-106 Н/м2),
катализатор — трипропилалюминий.
Изомеризация 2-метил-1-пентена в 2-метил-2-пентен протекает
при температуре 150—300э С в присутствии фосфорной кислоты на
алюмосиликате:
СН2=С-СН,-СНг—СН3 — СНа-С=СН—СНо—CHS
СН3
2-метил-1-пентен
2-метнл-2-пентен
Заключительный этап — образование изопрена (температура 650
800' С, катализатор — бромистый водород):
СН3-С^СН—CH.,—CHS —pvr CHn=C—СН=СНа
сн,
2-м?тил-2-пенген
СНз
изопрен
Химические свойства диенов с сопряженными двойными связями.
1,3-Днены отличаются рядом особенностей в своем химическом
поведении, которые обусловлены их электронным строением.
В молекуле 1,3-бутадиена все атомы углерода лежат в одной
плоскости, а четыре параллельные друг другу орбитали я-электронов,
расположенные перпендикулярно к плоскости, в которой лежат ядра
атомов углерода, объединены в одну молекулярную орбиталь
(рис. 13). Образование общей молекулярной орбитали является
следствием взаимодействия между собой двух отряженных двойных
57
связей. В этом случае отдельные пары я-электронов не закреплены
(локализованы) за определенными связями, а распределены (дело-
кализованы) по всей электронной системе, т. е. по всем
находящимся в сопряжении связям, с образованием общего электронного
облака. Максимальная электронная плотность возникает (см.
рис. 13) между Q и С2, С3 и С4 атомами углерода, меньшая — между
С2 и С3. Атомы С2 и С3, кроме 0-связи, как бы дополнительно связаны
между собой. Вследствие взаимодействия сопряженных связей
происходит некоторое выравнивание межъядерных расстояний. Длина
связей между Q и С2, С3 и С4 1,36 Ао(длина обычной двойной связи
1,34 А), длина связи С2—С3 1,46 А (длина обычной связи С—С
1,54 А).
Рис. 13. Модель 1, 3-бутадиена
По существу, в молекуле 1,3-бутадиена нет двойных и одинарных
связей в обычном понимании. Поэтому формула СН2=СН—СН=СН2
не точно отражает ее строение. Предложен следующий способ
написания формул соединений с сопряженными двойными связями
СН2—СН—СН—СН2. В этой формуле с помощью пунктирных
линий пытаются показать распределение электронной плотности
между углеродными атомами. Приведенные особенности строения с
1,3-диенов и определяют их свойства.
Диеиы с сопряженными двойными связями более устойчивы по
сравнению с 1,2-диенами и диенами с изолированными связями.
Отличительной особенностью сопряженных диенов является их
способность присоединять не только по двойным связям (к Q и С,,
С3 и С4), но и к концевым (Q и С4) атомам углерода с образованием
двойной связи между С2 и С3:
СН2С1—СНС1—СН=СН2 (присоединение 1, 2)
3, 4-дихлор-1-бутен
CHoCl—СН=СН—СН2С1 (присоединение 1, 4)
!, 4-дихлор-2-бутен
Выход 1,2- и 1,4-продуктов зависит от характера присоединяющего
вещества и условий взаимодействия.
При присоединении хлора к 1,3-бутадиену образуется 50%
1,4-изомера, а брома 2.'3 от общего количества продукта. Аналогично
идет присоединение галогеноводородов:
I— СН3—СНС1—СН=СН3
З-хлор-1-буген
СН./Л—СН=СН-СН3
1-хлор-2-бутен
СН2=СН—СН=СН2 -^-,
1, 3-бутадиен
I
I
1. 3-бутадиед
58
Присоединение водорода в момент выделения идет в положение
1,4, в присутствии катализатора — в 1,2 и 3,4:
H,/Ni
СН2=Ш—СН=СН2 —
1, 3-бутадиен
¦ СН3-СН3-СН2-СНз
н-бутан
3—сн=сн—сн3
2-бутен
Возможность двоякого 1,2- и 1,4-присоединения к диенам с
сопряженными связями будет понятным, если рассмотреть механизм этой
реакции на примере присоединения хлора. Реакция начинается
с электрофильной атаки хлором двойной связи (стр. 39) с
образованием неустойчивого я-комплекса A), превращающегося в карбока-
тион B). В результате притяжения положительным зарядом на С2
Bа) подвижных я-электронов соседней двойной связи (показано
стрелками) они перемещаются в центр молекулы, а С4, потерявший
электрон, заряжается положительно B6). Но при этом будет
наблюдаться обратный процесс. Наиболее устойчивым состоянием
образовавшегося катиона будет промежуточное состояние, изображенное
соединенными двойной стрелкой формулами 2а и 26 и формулами
3 и 4. На второй стадии анион хлора нуклеофильно атакует карбо-
катион и присоединяется к углеродным атомам С2 и С4, обладающим
частичным положительным зарядом:
СН2=СН—СН=СН2
153- бутадиен
СН2С1—СН—СН=СН2
С12
CH2=CH—CH=CH2
Т+8 5-
п-комплекс
A)
СН,С1-
-сн-,
сг
2а
26
1,2
соединение
Карбокатион (о - комплекс 2)
или
нсГ СН,С1—СН—СН—СН, C)
или
СН,С1—СН=-=СН^=СНо D)
СН2С1—СНС1—СН=СН2
3,4 _ дихлор - 1 - бутен
сГ
1L-присоедине-
ние
СН2С1—СН=СН—СН2С1
1;4 -дихлор - 2 бутен
Диеновые синтезы. Для синтеза разнообразных органических
веществ и количественного определения 1,3-диенов имеет большое
значение присоединение к ним в положение 1,4 соединений, содержа-
59
щих двойную связь, или диенофилов. Эти реакции носят название
диеновых синтезов или синтезов Дильса — Альдера, по имени
ученых, их открывших:
^СН2 о
г» СН-С'/ СН
1! чо;|
I +! |
сн сн-с/ сн—сн,-сн—с.
лсн3
маленноБЬШ
1,-3-бутадиен ангидрид
Полимеризация диенов. В присутствии ряда реагентов 1,3-диены
пол имеризуются:
сн.,=с—сн =сн,+сн„=с-сн =сн,+... —
I ! .
сн, сн3
—... сн.,—с=сн—сн,—сн с=сн—сн—...
) I
СН3 СН3
Реакция имеет важное народнохозяйственное значение.
Отдельные представители. Применение. 1,3- Бутадиен
СН2=СН—СН=СН3 — бесцветный газ с резким запахом. В
промышленности получают из этилового спирта, к-бутана, бутиленов и
ацетилена. Это соединение — один из основных мономеров для
получения каучука и латексов (стр. 278), применяется также для синтеза
других промышленных продуктов, в том числе перхлордивинила —
средства для борьбы с филоксерой на виноградных плантациях.
СН2=С—СН=СН2
Изопрен I — бесцветная жидкость.
Входит в состав большой группы важнейших природных соединений:
природного каучука, терпенов (стр. 273), каротиноидов (стр. 278).
Является одним из основных мономеров при получении
синтетического каучука. Главные способы промышленного получения:
дегидрирование изопеятана н изопентенов, получение из пропилена,
синтез из ацетилена.
АЛИЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДЫ
ЦИКЛОАЛКАНЫ
Циклоалканы, или цикланы, — углеводороды, содержащие в
молекуле циклы (кольца), построенные из атомов углерода (карбоцик-
лнческие соединения), связанных между собой а-связыо. Из
рассмотренной ранее классификации органических соединений (см. стр. 20)
следует, что цикланы входят в состав алицнклнческих соединении.
Общая формула цпклоалканов С„Н,,, следовательно, молекулы
цикланов, не имеющие заместителей, состоят из связанных между
собой и замкнутых в кольца групп СН.2 (метнленовая группа); отсюда
и другое их название — полиметиленовые соединения.
GO
Гомологический ряд, номенклатура, изомерия. Формулы и
названия наиболее важных гомологов приведены в табл. 8, здесь
же приведены и широко применяющиеся упрощенные формулы,
при написании которых подразумевается, что атомы углерода и
связанные с ним атомы водорода находятся в вершинах
многоугольника
\/
циклогексан
По рациональной номенклатуре цикланы рассматриваются как
замкнутые цепочки, построенные из групп СН, A). Номенклатура
ИЮПАК
Щ СН2
СН,
сн2-
„СН—СН,
сн2
Н3С—СН в СН—СН—СН3
"сн
С2Н5
пентаметилен метил цш<лопентан 1-метил-2,5-диэтил-4-изопропилциклопентан
A) B) C)
дает цнкланам те же названия, как и алканам с тем же числом атомов
углерода, но с прибавлением частицы «цикло». Если в цикле
замещено больше одного атома водорода на алкилы, то указывается номер
углеродного атома цикла, у которого расположены заместители.
При этом нумерация должна быть произведена таким образом, чтобы
все атомы цикла, несущие боковые цепи, получили наименьшие
номера C).
Структурная изомерия цикланов связана с различной величиной
циклов, видом и строением заместителей и их расположением:
СН2—СН2—СН3
СН3
-сн3
—СН,—СН,
пропилциклопропан
циклогексан метилциклопентан
сн3
СН—СН3
1-метнл-2-этлл-
циклопропан
Н,С
Н3С
изопропнл-
цнклопропан
I, 2-дмметнлцнкло-
бутаь
1, 3-диметилцикло-
бутаи
Геометрическая изомерия в ряду цикланов возникает при
наличии в цикле двух заместителей, расположенных у различных атомов
углерода.
61
Таблица
Циклоалканы
Название
Циклопропан;
триметилен
Цнклобутан;
тетрабутилен
Циклопентан;
пентаметилен
Циклогексан
Циклооктан
Формула
сн2
Cj Н 2 С Н j
! 1
СН,
сн2
сн2
сн2р
сн2
СН2^ /:ьнг
сн.,
сн2 сн2
СН2
СН2
/ \
\ /
сн2
сн2
СН,
сн2
/\
1
/\
0
Т. пл., °С
—126,6
—50,0
—93,3
6,5
14,3
Т. кип.,
°С
-34,4
13,0
49,5
81,4
148 (при
349 мм
рт. ст.)
Нахождение в природе. Способы получения. Цикланы и их
производные широко представлены в природе. Они входят в состав
нефти, эфирных масел и некоторых других природных соединений.
Нефть является основным природным источником цикланов с пятью
и шестью атомами углерода в цикле, содержание которых превышает
в нефти отдельных месторождений 50%. Это дало основание
В. В. Марковникову назвать этот класс соединений нафтенами (наф-
та — по-гречески нефть). В эфирных маслах (см. стр. 273)
представлены, главным образом, циклические углеводороды с десятью
атомами углерода (терпены) и их разнообразные производные. В
растительных и животных тканях широко распространены производные
гексаокснцпклогексана — инозиты,
Синтетическим путем цикланы получают или из алифатических
соединений (создание цикла), либо из производных цикланов.
Получение из дигалогенопроизводных алканов:
СН2Вг
СН3
/СН,
сн2
ZnBr,
1, 3-дибромпропан ^П2
циклопропан
62
Способ особенно удобен для получения трех- и четырехчленных
циклов.
Прямая циклизации алканов:
СЫ3 СН2-СН2
СН3—С—СН2—СН—СН3-^-^~ СНз—С—СНа—СН—СН3+ Н2
СН3 СН3 СН3
2, 2, 4-триметилпентан 1, 1,3-триметилциклопеитан
Получение из ароматических углеводородов (см. стр. 65):
бензол циклогексан
Каталитическое гидрирование бензола и его гомологов —
наиболее важный способ получения циклогексана. В качестве
катализаторов применяют никель, платину, палладий.
Физические свойства. Характеристика цикланов приведена в табл. 8. Для
них наблюдаются те же закономерности в изменении физических свойств, что
и для других углеводородов. Отличие состоит в более высоких температурах
кипения и плавления, чем у соответствующих алканов.
Химические свойства. Цикланы, содержащие циклы различного
размера, неодинаково ведут себя в химических реакциях.
Соединения с трех- и четырехчленными циклами отличаются повышенной
реакционной способностью, для них характерны реакции
присоединения. Углеводороды, содержащие циклы с пятью и более атомами
углерода, более устойчивы, для них характерны реакции замещения.
Действие галогенов:
СН,—СН,
СН,
циклопропан
сн2—сн2
ч;сн
сн,-сн2
¦циклопентан
Зг2 — Вг—СН3—СН2—СН2—Вг
1, 3-добромпронан
сн2—сн2
2-fBr2- ^CHBr + HBr
бромцикло-
пентан
В первом случае реакция сопровождается разрывом цикла и
присоединением атомов брома. Во втором случае бром замещает атом
водорода в молекуле циклопентана.
Действие галогеноводородов:
СН,-СН2
\ / +НВг-*СН3—СН,—СН,Вг
сн2
С цикланами, содержащими пять и более атомов углерода в
цикле, галогеноводороды не взаимодействуют.
63
Действие водорода (гидрогенолиз). В присутствии катализаторов
(платины, палладия, никеля) водород присоединяется к
циклопропану с образованием пропана:
сн2—сн2
сн.
Сн8-сн8-сн3
С увеличением размеров цикла его устойчивость возрастает и для
осуществления гидрогенолиза требуются все более высокие
температуры. Начиная с циклогексана, цикланы не способны к реакциям
гидрогенолиза.
Окисление цикланов. Цикланы, даже циклопропан и циклобутан,
довольно стойки к действию окислителей. При повышенных
температурах, под действием сильных окислителей происходит разрыв цикла
с образованием двухосновных кислот с тем же
I ^//\чх | числом атомов углерода в молекуле:
СН,
, СН.,-СН2—СООН
СН2—СН2—СООН
аднпиновая кислота
2444'
Рис. 14. Отклонение
валентных углов в
циклопропане
СН2
СН., СН,
\/ "
сн2
циклогексан
Из приведенных реакций следует, что
химическое поведение цикланов тесно
связано с размерами циклов. Исследование в этом
направлении было начато еще в прошлом веке. В 1885 г. А. Байер,
рассматривая циклы как плоские фигуры, высказал предположение,
что угол отклонения валентных связей в цикле от нормального
направления, т. е. от угла 109° 28' (см. стр. 19), является мерой
относительной устойчивости цикла (рис. 14, табл. 9), т. е., например, для
трехчленных циклов угол а равен: а = A09° 28' — 60°)/2 = 24'J 44'.
Таблица 9
Величина отклонения валентных связей циклов от нормальных
Название
Циклопропан
Циклобутан
Циклопентан
i
Угол отклонения
(и) !
+2444' :
--944' '
+044' |
1
Название
Цнклогексан
Циклогепган
Циклооктан
Угол отклонения
(а)
—540'
—9-33'
—15-5Г
Полученные цифры хорошо объясняли химическое поведение
циклопропана, циклобутана, цнклопентана, но противоречили
экспериментальным данным о стойкости средних и больших циклов.
Объяснение этому было дано после.открытия явления конформацпи
64
органических соединений (см. стр. 229). В результате взаимного
отталкивания соседних групп СН2 циклы «изгибаются» (перестают
быть плоскими), что приводит к уменьшению углосого напряжения
и напряжения, вызванного отталкиванием групп СН.,. Для цнклогек-
сана существует две формы (конформации) без углового напряжения
(«ванна» п «кресло», рис. 15). Наиболее устойчива форма «кресло».
Рис. 15. Кочформация циклогексаиа:
с —форма «.-вая.чыз; 0 — форма «кресло»
Отдельные представители. Применение. Циклопропан обладает
наркотическим действием, применяется в хирургии.
Цпклопентан применяется в разнообразных синтезах и в качестве
добавки, улучшающей качество моторного топлива.
Циклогексан применяется для получения аднпиновой кислоты,
цкклогексанола и цпклогексанона (см. стр. 104, 127, 141).
АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
Ароматические углеводороды — соединения, содержащие в
молекуле особую циклическую группировку из шести атомов углерода,
которая носит название бензольной группировки (бензольное ядро):
С
-С' С-
—с с—-
с
(бензольное ядро)
Название углеводородов этой группы (бензола, нафталина,
антрацена и их многочисленных а разнообразных производных)
«ароматические соединения» случайное и сегодня потеряло свой
первоначальный смысл. Действительно, первые открытые соединения или
обладали специфическим, иногда приятным запахом, или были выделены
3 А. П. Нечаев
65
из природных сильно пахнущих продуктов. Но количество
«ароматных» веществ среди многочисленных известных в настоящее время
соединений этой группы невелико. В то же время в строении,
физических свойствах и химическом поведении этих веществ наблюдается
ряд особенностей, связанных с наличием в их молекуле бензольных
группировок.
Различают одноядерные (одна бензольная группировка в
молекуле) и многоядерные ароматические углеводороды.
ОДНОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ УГЛЕВОДОРОДЫ
Гомологический ряд, строение, номенклатура, изомерия.
Первый и наиболее важный представитель гомологического ряда
одноядерных ароматических углеводородов — бензол С6Нв. Отсюда и
общее название гомологического ряда — ряд бензола. Общая
формула С„Н2Л_6. Наиболее важные гомологи и их характеристика
приведены в табл. 10. Химическая формула бензола, которой широко
пользуются и по сей день, была предложена еще в 1865 г. немецким
ученым Кекуле A829—1896), однако споры о строении бензола
продолжались до самого последнего времени. По мнению Кекуле,
молекула бензола представляет собой шестичленныи цикл, состоящий из
атомов углерода, соединенных чередующимися одинарными и
двойными связями. Каждый атом углерода связан с одним атомом
водорода, т. е. бензол является 1,3,5-циклогексантриеном-.
СН
СН СН
] J
СН СН
ч/
СН
(бензол)
(формула Кекуле)
Формула Кекуле правильно отражает элементарный состав,
соотношение углерода и водорода и равноценность всех атомов водорода
в молекуле бензола. Однозамещенные бензола при отсутствии
изомерии радикалов (например: СвН5СН3 — толуол, СаН6Вг — бром-
бензол) не имеют изомеров. В то же время формула Кекуле не
отвечает на вопрос, почему для бензола, содержащего в молекуле
три двойные связи (ненасыщенное соединение), характерны в
первую очередь реакции замещения, протекающие в мягких условиях.
Почему он не обесцвечивает бромную воду (качественная реакция
на двойную связь)?
Не объясняет формула Кекуле и большую устойчивость
бензольного ядра к действию окислителей и высоких температур. Кроме
того, согласно этой формуле должны существовать два изомера
у 1,2-дизамещенных бензола, что противоречит экспериментальным
данным. Ответы на эти вопросы были получены благодаря
применению в последние годы при исследовании бензола наряду с
химическими физических методов.
66
Таблица 10
Гомологический ряд бензола
Название
тривиальное
по ИЮПАК
Формула
Т. кип.,
Бензол
Толуол
Ксилол
ерто-Ксилол
.««/w-Ксилол
нара-Ксилол
Зтилбензол
Пропилбензол
К у мо л
na/w-Цимол
Бензол
Метилбензол
1,2-Диыетилбензол
1,3-Диметилбензол
1,4-Диметилбензол
Этилбензол
Пропилбензол
Изопропилбензол
1-Метил-4-изопропил-
бензол
С6Н3
СаН5СН3
С6Н4 (СН3J
с6н5с2н5
С6Н0С3Н7
С6Н5СН (СН3J
CSH4 (CH3) (С3НГ)
5.5
-95,0
-25.2
-479
3 3,3
-95,0
-99,5
-96,0
-67,2
8ОД
110,6
144,4
139,3
138,4
136,2
159,2
152,4
177,1
По современным представлениям атомы углерода в молекуле
бензола образуют правильный плоский шестиугольник со сторонами
1,4 А (напоминаем, что длина простой —С—С— связи 1,54 А, двой-
/ \
ной — 1,32 А). Каждый атом углерода связан с одним атомом
водорода, длина С—Н-связи 1,09 А от вершины шестиугольника. Углы,
образованные связями Н—С—С и С—С—С, равны 120° (рис. 16, а).
Все шесть атомов углерода и водорода лежат в одной плоскости.
Атомы углерода в молекуле бензола находятся во втором валентном
состоянии Eр'2-пюрндизация), т. е. из четырех (одна и три р-орби-
талн) гнбрнднзованы только три (s и 2р) орбитали, следовательно,
каждый атом углерода связан с соседними углеродными атомами и
атомом водорода освязями, затрачивая на это 3 электрона.
Четвертый электрон не закреплен за определенным атомом углерода. Шесть
незакрепленных (делокализованиых) электронов образуют единую
устойчивую замкнутую я-систему (рис. 16, б), дополнительно
связывая углеродные атомы. Поэтому в молекуле бензола, по существу,
нет ни одинарных, ни двойных связей.
У бензола наблюдается явление кругового сопряжения.
Реальная молекула бензола обладает меньшей на 36 ккал/моль A50,7 X
X 103 Дж'мм) энергией (энергия сопряжения) по сравнению с
энергией молекулы, для которой справедлива формула Кекуле A,2,3-цик-
логексантриен). Поэтому углерод — углеродные связи в молекуле
бензола обладают повышенной прочностью. Отсюда и особенности
в физических свойствах и химическом поведении бензола и его
гомологов. Это в первую.очередь относительная легкость образования и
устойчивость бензольного ядра к повышенным температурам,
стойкость его к окислителям и инертность по сравнению с олефинами
в реакциях присоединения, легкость протекания и своеобразие
реакций электрофнльного замещения (нитрования, сульфирования, га-
67
логенирования и т, д.). Рядом особенностей, как будет показано
в последующих разделах, обладают производные бензола.
Совокупность всех этих свойств получила название «ароматический
характер».
н
Рис. 16. Строение бенлола
Учитывая указанные особенности строения бензола, написание
его структурной формулы представляет сложную задачу. В
последнее время для обозначения бензола используют формулы, в которых
с помощью стрелок, пунктирных линий или окружности,
вписанной в шестиугольник, стремятся показать выразненность
электронной плотности между атомами углерода:
Хотя эти формулы более точно отражают некоторые особенности
строения бензола, формула Кекуле продолжает широко
применяться. Однако, используя ее, нельзя забывать о ее
недостатках.
Дл;
исто г
гклх.
Для наименования ароматических соединений часто применяются
оричеекпе названия: бензол, тотуол. По номенклатуре ИЮПАК
ароматические углеводороды рассматриваются кат
•зводиые
бензола, положение
68
заместителей указывают цифрам:!, следя за
тем, чтобы номера атомов углерода, у которых расположены
заместители, были наименьшими:
CH3
метилбензол
(толуол)
СНз
11
1-метил-З-этнлбензол
В случае двух заместителей вместо цифр можно пользоваться
приставками 1,2-орто (о-), 1,3-мета (м-), l-i-пара (п-):
СН,
1,2-диыетилбензол
(орто-ксплол)
1,3-диметилбензол
I wma-кснлсл)
1.4-диметилбензол
(пара-кстлсл)
Общее название ароматических углеводородов — арены, общее
название радикалов — арилы (Аг). Наиболее распространенные
арилы:
С6Н5— фенил (от старого названия бензола «фен»)
QHi—фенилен (о-, м-, п-)
СН3—СС.Н4— толил (о-, м-, п-)
C6HSCH2— бензил
С6Н5СН< бензилиден
Однозамещенные бензола не имеют изомеров. Изомерия ди- и
полизамещенных бензола связана с различным расположением
заместителей (например, о-, м-, «-) и их строением.
Способы получения. Основные источники получения
ароматических углеводородов — продукты сухой перегонки (коксования)
каменного угля и нефти.
Выделение ароматических углеводородов из продуктов
коксования каменных углей — наиболее старый и до ЕО-х годов основной
способ их получения. При нагревании свыше 1000° С без доступа
воздуха (сухая перегонка) уголь разлагается с образованием твердых
(кокс), жидких (каменноугольная смола, аммиачная вода) и
газообразных (газы коксования) продуктов перегонки.
Кокс — твердая, пористая масса, состоящая в основном из
углерода, применяется в больших количествах в металлургии.
Газы коксования, содержащие водород, метан, окись и двуокись
углерода, азот, этиленовые и ароматические углеводороды (до 40 г
в 1 м3), после отделения последних используются в качестве
топлива,
Из аммиачной воды получают аммиак и соли аммония.
Каменноугольная смола — буро-черная, маслоподобная масса с резким
запахом, содержит большое количество органических соединений
различной природы. Выход смолы около 3,0%. На первом этапе
переработки каменноугольную смолу перегоняют, обычно собирая 4
фракции (табл. 11).
Таблица 11
Основные фракции каменноугольной смолы
Фракции
Легкое масло
Среднее масло
Тяжелое масло
Антраценовое масло
Температура,
До 180
180—240
240—270
270—360
Выход. %
0,5—2,0
12
10
20
Состав
Бензол, толуол, ксилолы,
циклопентадиен, гетероциклы
Фенол, нафталин, крезолы
Нафталин и его производные
Антрацен, фенантрен
Остаток от перегонки — 60% от массы дегтя — называется
пеком. Это твердая, размягчающаяся при нагревании масса темного
цвета.
Из перечисленных фракций разнообразными приемами
выделяются индивидуальные органические соединения. Из 1 т A03 кг)
каменного угля получают 16 кг бензола, 2,5 кг толуола, 50 кг
нафталина, 10—20 кг антрацена.
В последние годы коксохимическая промышленность как
источник ароматических углеводородов уступила первое место нефти.
Содержание ароматических углеводородов в нефти сильно
колеблется, п хотя в отдельных случаях оно достигает 60%, основное
количество ароматических углеводородов получается из нее при
химической переработке (ароматизация нефти), главным образом
пиролизе и каталитическом риформинге, в ходе которого протекают
реакции дегидрирования и дегидроциклизации компонентов нефти
(стр. 80).
Кроме этого, получение ароматических углеводородов может быть
осуществлено дегидрированием циклопарафинов (Н. Д. Зелинский,
1911) с применением в качестве катализатора платины на окиси
алюминия
;.+зна
и дегндраниклизацней парафинов (Б. А. Казанский, А. Ф. Платэ,
В. Л. Молдавский). Катализаторы — окиси хрома, молибдена,
ванадия:
СН3-(СН.:),—СН3->!
\/
С и н т е т и ч е с к и е способ ы полу ч е н и я.
Полимеризация ацетилена (см. стр. 51).
Взаимодействие ароматических и алифатических галогенопроиз-
еодных с металлическим натрием (реакция Вюрца — Фиттига):
QH.Br + C4H9Br + 2Na r^j^r QH—С4Н9
бромоен^ол брот.шстый бугилбеязол
бутил
Алкироеание ароматических углеводородов галогенопроизводными
и олефинами (реакция Фриделя-Крафтса, см. стр. 73).
Физические свойства. Характеристика аренов приведена в табл. 10.
Ароматические углеводороды — жидкости или твердые вещества, легче воды и
практически в ней нерастворимы. Хорошо растворяются в органических
растворителях. Горят ярким коптящим пламенем. Температуры кипения и плавления
аренов сильно зависят от пх строения. При работе с ароматическими веществами
необходимо помнить, что они в большинстве случаев токсичны.
Химические свойства. Как указывалось, вследствие особенностей
строения ароматические соединения отличаются склонностью к
реакциям замещения (а не присоединения) и прочностью бензольного
ядра.
Реакции з а м е щ е н и я. Из реакций замещения для
ароматических соединений особое значение имеют реакции электро-
фильного замещения, с помощью которых получают важные в
промышленном отношении производные бензола.
Нитрование бензола. Под действием концентрированной
азотной кислоты или смеси концентрированных азотной и серной кислот
(«нитрующая смесь») атомы водорода бензольного ядра замещаются
на нитрогруппу:
ССН(. + НОКО2 — CeH5NO2 + Н,0
[бензол нитробензол
Нитрование бензола — типичная реакция электрофилыюго
замещения, поэтому на ее примере мы и разберем механизм этих
превращений. Электрофильное замещение в ароматическом ряду — ионный
процесс, протекающий в две стадии и имеющий на первой стадии
сходство с присоединением к алкенам. Реакции предшествует
поляризация или превращение молекулы реагента с образованием элект-
рофильной частицы (в нашем случае — катион нитрония NO.2) и
аниона:
O4 — NO2-b2HSO4--i- Hp+
нитронпй-
катнон
Катион нитрония притягивается секстетом л-электронов
молекулы бензола с образованием л-комплекса. Затем происходит
разрушение секстета, два из шести электронов, закрепляясь
(локализируясь) у одного из углеродных атомов, образуют освязь с
заместителем. При этом атом углерода переходит из sp2- в sp'J-cocTOHHne.
71
Оставшиеся 4 электрона распределяются между пятью атомами
углерода бензольного ядра. Образуется а-комплекс:
NO,
я-коыплекс сг-комплекс
На второй стадии образовавшийся а-комплекс быстро теряет
протон (он соединяется с HSO4, образуя молекулу серной кислоты),
переходя в ароматическое соединение:
а-комплекс нитробензол
Отщепление протона с восстановлением ароматичности (образование
нитробензола) энергетически выгоднее, чем взаимодействие между
промежуточным катионом (о-комплексом) и анионом (HS07),
которое мы наблюдаем в случае реакции с алкенами.
Галогенироваиие. В обычных условиях в присутствии
катализаторов (А1С13, А1Вг3, FeCl3, FeBr3, ZnCl2) происходит замещение
атомов водорода бензольного ядра на атомы галогена:
+ Вг2 *™И <(^у-Вг + НВг
броыбензол
Роль катализатора заключается в поляризации связи
галоген-галоген в молекуле галогена и создания тем самым необходимых условий
для электрофильного замещения.
В отсутствие катализатора, при нагревании или под действием
света галогены замещают атомы водорода в боковой цепи. Реакция
протекает по радикальному механизму:
С6Н5СН3 + С12 свет' *° С6Н5СН2С1 + НС1
хлористый
толуол бензил
Сульфирование. При действии на бензол и его гомологи
концентрированной серной кислоты или олеума атомы водорода в
бензольном ядре замещаются на сульфогруппу (реакция сульфирования).
<f
;)¦ + hoso,h — f > -so3h -f HoO
бензол i4y ль-
Алкилирование. Действуя на бензол галогеналкилами в
присутствии безводного хлористого алюминия (реакция Фриделя — Крафтса,
1877) или алкенами (катализаторы — фосфорная кислота, трехфто-
72
ристый бор), удается ввести в молекулу бензола алкильную группу
с образованием его гомологов (реакция алкилнровання):
Xx ! Ги CU Г\ ОеЗВ. AiU.j // \ лиги , иг]
этилбензол
сн3
<(""*)+сн2=сн—сн3 - <(' ^—сн—сн3
изопропилбгнзол
(кумол)
Завершая рассмотрение реакций электрофильного замещения,
необходимо остановиться на так называемом правиле ориентации
(или замещения) в бензольном ядре. Реакции замещения после
вхождения в бензольное ядро одного заместителя и образования
монопроизводных бензола на этом не останавливаются, замещение
продолжается с образованием ди-, три- и более производных, кроме
этого, в реакциях могут участвовать и гомологи бензола.
Естественно, возникает вопрос, в какое положение войдет второй
заместитель? При этом нужно помнить, что орто-, мета- и пара-
изомеры существенно отличаются по своим свойствам (см. табл. 10).
Экспериментальным путем было установлено, что положение,
в которое вступает второй заместитель, зависит от природы первого
заместителя, вида действующего реагента и условий проведения
реакции. Первые два условия играют особенно важную роль.
Заместители б зависимости от того, в какое положение они
направляют вхождение второго заместителя, делятся на две группы:
заместители первого и второго рода.
В реакциях электрофнльного замещения заместителя первого
рода направляют вхождение второго заместителя преимущественно
в орто- и /гара-положения. К ним относятся группы: —СН,, —CH2R,
—CRSl —ОН, —OR, —NHa, —NHR, —NR2, галогены.
Заместители второго рода ориентируют (направляют) вхождение
второго заместителя преимущественно в жета-положение. К ним
относятся: —SO3H, —NO,, —СНО, —СООН, —COOR, —CN.
Заместители первого рода — группировки атомов, способные
отдавать электроны бензольному ядру (электронодонорные), при
этом электронная плотность в ядре возрастает, особенно в орто- и
пара-положениях. Естественно, что при атаке молекулы бензола
электрофильными реагентами они и занимают предпочтительно эти
положения. Кроме этого, заместители первого рода облегчают, по
сравнению с бензолом, проведение реакций с электрофильными
реагентами.
Заместители второго рода принимают (оттягивают) электроны
от бензольного ядра (электроноакцепторные). Электронная
плотность в ядре уменьшается, особенно в орто- и пара-положениях.
Электрофильный заместитель предпочтительно вступает в мепш-поло-
жение. Следовательно, заместители второго рода затрудняют по
сравнению с бензолом реакции с электрофильными реагентами. В
73
дальнейшем в соответствующих разделах мы продолжим
рассмотрение этих вопросов.
Реакции п р и с о с д и н е н и я. Ароматические
соединения с трудом вступают в реакции присоединения.
Присоединение галогенов. Под влиянием ультрафиолетового
облучения к молекуле бензола присоединяется три молекулы хлора с
образованием гексахлорциклогексана (гексахлоран):
бензол
гексахлорцпчлогексан
(гексахлоран)
Присоединение водорода (см. стр. 63).
Окисление а р оматичес к их
углеводородов. Бензольное ядро отличается исключительной стойкостью к
действию окислителей: КМпО4 в кислой среде, перекиси водорода,
хромовой смеси и т. д.
При действии окислителей на гомологи бензола окисляются
боковые цепи с образованием ароматических альдегидов и кислот,
причем число образовавшихся карбонильных или карбоксильных групп
равно числу боковых цепей:
-сн,
о
—с—он
—н,о
толуол
бензойная кислота
При очень энергичном окислении происходит разрушение
бензольного ядра. Так, в присутствии катализатора, содержащего
смесь окислов ванадия и молибдена, при температуре 350—4503 С
бензол окисляется кислородом воздуха с образованием малеинового
ангидрида:
I 9О2
СН—С-'
Отдельные представители. Применение. Бензол С6Н6 — жидкость
с характерным запахом, почти нерастворима в воде. Получается при
сухой перегонке каменного угля и химической переработке нефти.
Важнейший представитель ароматических углеводородов.
Толуол (мспшлбснзол) С6КГ)СН:! применяется лля получения
бензола, бензойной кислоты (см. стр. 131), сахарина,
74
Цимол (параметилизопропплбензол) — является основой
многих эфирных масел (см. стр. 274).
Стирол (винилбензол) — понятно пахнущая жидкость,
*КЙП + 144,2= С:
/ V-CH=CH2
Получают дегидрированием этилбензола в присутствии
катализатора:
Н Н
С„Н3—С—СН CjHj—СН=СН2+ Н2
¦¦-¦¦: исходный мономер для
Н Н: получения полистирола
МНОГОЯДЕРНЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Многоядерными ароматическими соединениями называются
вещества, содержащие в молекуле два или более связанных между
собой бензольных ядер. Различают многоядерные соединения с некои-
денсироваиными (изолированными) ядрами — дифенил, трифенил-
метан —и с конденсированными — нафталин:
дифеннл ^
нафталин
трифенилметан
Многоядеркые ароматические углеводороды
с нвкондёнеированнымм (Изолированными* ядрами
В молекулах этих соединений бензольные ядра могут быть
соединены или непосредственно A), или с помощью цепочки, состоящей
из различного количества атомов углерода. Наибольшее значение
имеют дифенил, дифенилметан, трифенилметан и его разнообразные
производные.
Дифенил — кристаллическое вещество, tu:1 = 70° С, получается
при пропускании паров бензола через расплавленный свинец, а так
же выделяется из каменноугольной смолы:
В химическом отношении дифенил напоминает бензол. Применяется
в качестве устойчивого высококнпящего (/кид 254° С) теплоносителя
и для получения красителей.
75
Дифенилметан и трифенилметан получают по реакции Фри-
деля — Крафтса:
К
ЗС8Нв + СНС13 Д|Ь (~\
хлороформ
трифенилметан
Это кристаллические вещества, применяются для получения
разнообразных органических соединений, многие из которых
являются красителями. Красители трифенилметанового ряда: фуксин,
фенолфталеин и др.
Многоядеркые ароматические углеводороды
с конденсированными ядрами
В молекулах этих соединений бензольные ядра имеют общие
углеродные атомы. Важный представитель — нафталин.
НАФТАЛИН
Состав нафталина С1УН8 был установлен А. А. Воскресенским
A858), строение Эрлеимейером A866), последнее лучше всего
отражает формула A):
СП СН 8 г
Р СК СН Р
а а
A) B)
По современным представлениям молекула нафталина имеет
плоское строение, а облако я-электронов распределено в ней менее
равномерно, чем в бензоле B). '
Сравнивая длины связей в молекуле нафталина B) с длиной
одинарных и двойных связей, видно, что они занимают промежуточное
положение. Различают а-положение A,4,5,8-й атомы углерода)
и Р-положение B, 3, 6, 7-й атомы углерода), поэтому для нафталина
существуют а- и Р-монопроиззодные.
Способы получения и свойства нафталина. Нафталин —
бесцветное кристаллическое вещество, tna = 80° С, ?1ШП = 218°С, летуч,
обладает сильным характерным запахом. Хорошо растворим в
органических растворителях. Основной промышленный способ
получения — выделение из каменноугольной смолы. Большая его часть
сосредоточена во фракциях среднего и тяжелого масел (см. табл. 10).
В последнее время практическое значение получило его
выделение при химической переработке нефти.
Химические свойства. Нафталин проявляет ароматический
характер, но легче, чем бензол, вступает в реакции присоединения.
76
Реакции замещения. При этих реакциях предпочтительно
получают а-производные:
CI С1 С1
С1,
/у '•¦¦¦' ' ; 4С12 V* V'' ** — С1
нафталин
- CI CI
а-хлорнаф- перхлориафгалии
талин
SOpH
IbSO4; ,с0°С /'
суль^рирова^ие
«-Нафталин- (а-пзомер при по-
сульфокислота ьытпеиин
температуры перехидиг в
j; 160'С ^ ч-/ ^ —
Р-наф-талн!Гс
К1;слот
N0,
ы-ьигро-
нафталпн
Гидрирование нафталина
нас]|~а;!ин
зна B00'с) ¦' ч/ ч-
тетралин декалин
Нафталин гидрируется легче, чем бензол. Тетралин и декалин
применяются в качестве органических растворителей.
Окисление нафталина. При энергичном окислении нафталина
(/ -= 350—450" С) образуется фталевый ангидрид, катализатор —
пятиокись ванадия (V2O5):
450,
О
II
С
2Н2О + 2СО2
наЛталин
li
О
фталеЕЫЙ
ангидрид
Применение. Нафталин используется для получения фталевого
ангидрида, фталевои кислоты и разнообразных других производных.
77
АНТРАЦЕН
Молекула антрацена С14Н10 имеет плоское строение и может
быть представлена формулой
а -у а
/\\л
5 10 4
а у а
Различают а-положение A, 4, 5, 8); (З-положение B, 3, 6, 7) и
¦у-положение (9, 10), последние иногда называют жзо-положением.
Получают кристаллизацией из каменноугольной смолы.
Антрацен — бесцветное, кристаллическое вещество, /„., = 213° С, он
легко вступает в реакции замещения (заместитель легче всего
вступает в лезо-положеиие) и присоединения. При окислении
антрацена в присутствии солей ванадия и молибденовой кислоты
образуется антрахннон:
О
1!
/>\/\/Х ГП1. Я7,_кпп, г /\/\/\
- [0]; 375 — 500°
I ii I ! "
vw
антрацен
II
антрахинон
Из антрацена, кроме антрахинона, получают разнообразные
красители.
ФЕНАНТРЕН
Фенантрен СЫН1О
8 /"
Выделяется из каменноугольной смолы.
ВЫСШИЕ ПОЛИЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДЫ
Высшие полициклические углеводороды могут быть построены из большого
числа бензольных ядер, связанных между собой различным образом:
1 ексаден хрн^ен I j!
беизгшрен
78
Основной источник их получения — каменноугольная смола. Кроме того,
они образуются в качестве побочных продуктов при пиролизе нефти и газа.
Входят в состав разнообразных лекарственных соединений и могут служить
сырьем для получения красителей. Отдельные представители полициклических
углеводородов обладают канцерогенным действием, т. е. способны вызывать
рак (cancer — рак).
НЕФТЬ И ЕЕ ПЕРЕРАБОТКА
Нефть — добываемое из недр земли жидкое, горючее ископаемое,
состоящее из углеводородов (алканы, циклоалканы, ароматические
углеводороды) с примесью соединений, содержащих азот, серу,
кислород и воду, с растворенными в ней солями. Состав нефти
различных месторождений неодинаков: в одних случаях
преобладают алканы, в других — цикланы или ароматические углеводороды.
Хотя вопрос о происхождении нефти нельзя считать окончательно
решенным, однако, по мнению большинства ученых, она
образовалась в результате сложных химических и биохимических
превращений из остатков микроскопических растений и животных,
накопившихся в течение многих миллионов лет на дне лагун и морей.
Основное количество разведанных запасов нефти находится на
территории СССР, Северной и Южной Америки (США, Канада,
Венесуэла), Ближнего и Среднего Востока (Сирия, Ирак, Иран),
Румынии. По запасам нефти СССР занимает 1-е место, а по добыче
2-е. В 1975 г. добыча нефти в СССР должна достигнуть 480—500
млн. т D80-Ю9 —500-Ю9 кг).
Из нефтяных пластов, расположенных на разной глубине,
нефть поступает на поверхность под давлением либо растворенных
в ней газов (фонтанный способ), либо специально закачиваемой
воды, либо с помощью насосов.
В свежей нефти растворены попутные газы и твердые парафины,
которые отделяются и используются в нефтехимической
промышленности.
Физические свойства. Нефть — жидкость от светло-желтого до черного
цвета, легче воды, с высокой теплотворной способностью 10 500—11 000 ккал./м3
D6 054 800—49 961 400 Дж/м3).
Переработка нефти. После специальной подготовки (удаления
воды, солей) нефть направляется на перегонку (прямая гонка).
Прямая гонка — это основной процесс первоначальной ее
обработки. При прямой гонке нефть нагревают до кипения и отбирают
фракции (погоны), кипящие в определенных температурных
интервалах. Количество отбираемых фракций зависит от состава нефти
и путей их дальнейшего использования (табл. 12).
Продукты, полученные при первичной переработке нефти,
используются в качестве топлива в двигателях и являются сырьем
для дальнейшей переработки. Стремясь увеличить количество
получаемого из нефти бензина (после прямой гонки его образуется
10—20пп), в ходе вторичной переработки высококппящне фракции
и мазут подвергают крекингу для частичного разрушения
(деструкции) «тяжелых молекул». Впервые крекинг нефтяного сырья был
79
осуществлен русским ученым, сотрудником Петербургского
технологического института А. А. Летним, а первый проект
промышленной установки для крекинга разработал в 1891 г. известный русский
инженер В. Г, Шухов.
Таблица 12
Основные фракции перегонки нефти
Наименование
фракции
Бензиновая фракция
Керосиновая
фракция
Соляровые масла
Мазут (нефтяной
остаток)
Т, кнп., "С
До 180
180—300
До 360
Выше 360
Примерный
состав
Г С
С С
С С
Использование
Моторное топливо
Тракторное топливо,
топливо для реактивных
двигателей, бытовое назначение,
сырье для крекинга
Дизельное топливо,
смазочные масла, сырье для
крекинга
Сырье для крекинга,
получения парафина, смазочных
масел, гудрона
В зависимости от исходного сырья и поставленных целей
применяют различные разновидности крекинга, которые объединены
в две большке группы: термический крекинг (проводится без
катализатора) и термокаталптический крекинг. Термический крекинг
осуществляют при температуре 460—560° С и атмосферном или
повышенном давлении (до 100 атм или до 98-1G3 Н/мм). Крекинг
при более высоких температурах G00—800° С) получил название
пиролиза.
Каталитический крекинг проводят в присутствии
алюмосиликатов или платины. В ходе крекинга происходит разложение
сложных углеводородов с образованием соединений с меньшей
молекулярной массой:
CH CH CH
Одновременно протекают реакции изомеризации, циклизации,
уплотнения. В результате образуется смесь разнообразных продуктов,
состав которой сильно зависит от условий крекинга.
При повышении температуры возрастает выход газообразных
продуктов и ароматических углеводородов, Осуществляя вторичную
переработку нефти, важно не только увеличить выход бензина (он
может быть доведет! до 60—70 V6 от массы сырья), но и повысить
его качество. Качество бензина сильно зависит от строения
входящих в его состав углеводородов.
Эталоном хорошего бензина является изооктан B, 2, 4-триметил-
пентан):
СН3 сн3
| |
CHg-С-СНг -СН-СН,
сна
80
Поэтому качество бензина принято оценивать с помощью
«октановых» чисел. Для изооктана оно равно 100. Октановые числа
парафинов нзостроення выше, чем нормального строения; еще выше
они у олефинов, цикланов и особенно у ароматических
углеводородов.
Превращение низкосортных бензинов в высококачественные
(с высоким октановым числом) осуществляется с помощью рифор-
мннга. При этом происходит изомеризация алканов и образование
ароматических соединений. Для повышения качества бензинов
применяют разнообразные добавки, например раствор тетраэтилсвинца
(С,Н5L Pb в бромистом этиле С2Н5Вг.
Отдельные компоненты нефти, полученные в результате ее
первичной и вторичной переработки, и в первую очередь алканы
Сг—С5, этилен, пропилен, ацетилен, цпклогексан, бензол, толуол,
являются важнейшим сырьем для получения многочисленных
органических соединений, в том числе каучука, пластических масс,
синтетических моющих средств и др.
ЧАСТЬ ВТОРАЯ
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
УГЛЕВОДОРОДОВ
При замещении атомов водорода в углеводородах на
функциональные (характеристические) группы (стр. 21) получаются их
разнообразные производные: галогенопроизводные, оксипроизвод-
ные, альдегиды и кетоны, кислоты и т. д. Соединения, содержащие
однотипные функциональные группы, называются соединениями
с однородными функциями (иногда полифункциональными);
разные — соединениями со смешанными функциями (гетерофункцно-
нальными).
ГАЛОГЕНОПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Производные углеводородов, содержащие вместо одного или
нескольких атомов водорода галогены: фтор, хлор, бром, иод,
называются галогенопроизводными. В зависимости от характера
углеводорода, в молекулу которого введен галоген, их делят на
насыщенные (галогеналкилы), ненасыщенные, ароматические (галогенарилы),
а по числу атомов галогена, содержащихся в молекуле, на моно-,
ди-, три- п полнгалогенопронзводные. Наиболее важные
галогенопроизводные приведены в табл. 13,
Таблица 13
Галогенопронзводные углеводородов
Соединение
Хлористый метил
Хлористый этил
Хлористый .метилен
Дихлорэтан
Хлороформ
Четырех хлористый углерод
Хлористый винил
Хлористый аллнл
Хлорбензол
Хлористый бензил
Формула
СН3С1
СН-,С1
сн-ск
CHoCl—СН-С1
CHCij
CCIj '
СН,=СНС1
сн'=сн—сн„сл
СВН:,С1
С6Н=СН.-С1
Т. кип., СС
—24
13
40
88,7
61
76,5
— 14
¦К
132
179
Т. пл., "С
—97
— 130
—96.о
—З.\3
— 63.6
-22.9
- 150,7
— 136
—45
—39
Номенклатура. Изомерия. По рациональной номенклатуре
название галогенонронзводных образуют из названия углеводородного
82
радикала и галогена, указывая в необходимых случаях положение
последнего:
С1
I
с2н;вг сн3--сн~-сн,-сн3 сн,=сна
бромистый вторичный хлористый хлористый
„¦=т;:л Оу гнл винил,
ST ИЛОр ОМИД Б ННИЛУЛОр ИД
СН;С1—СНХ1 СьН5СНоС1
хлористый хлористый
зтплен бензил,
бензнлхлорид
По номенклатуре ИЮПАК положение атома галогена указывают
цифрой, которая вместе с его названием располагается перед
наименованием углеводородного радикала. Нумерацию атомов углерода
главной цепи начинают с того конца, к которому ближе расположен
атом галогена:
СН3 СН3 С1
СН3—СН—.СН—СН.»—СН-СНз
Ч j 4 :j «1
2-хлор-4, 5-днметилгег:сан
В отдельных случаях применяются исторические названия:
СНС13 — хлороформ; СШ3 — йодоформ, Изомерия в ряду галогено-
пронзводных связана с особенностями строения углеродного скелета
и положением атомов галогена:
a
СНз-СН2--СН,-СН.2С1 СН3-С-СН3
1-хлорбуган I
сн3
2-хлор-2-мегил-
пропан
СНз-CH-CH.—Cl
СН3
1-хлор-2-метилпропан
Способы получения. Замещение атомов водорода е углеводородах
на галоген, В результате галогенпрованпя алкаиов (стр. 29),
цикланов (стр. 63), бензола и его гомологов (стр. 72), алкенов
(в определенных условиях) происходит замещение атомов водорода
на галоген.
Однако этот способ имеет практическое значение для получения
только некоторых галогенопроизводных. В промышленности широко
применяется хлорирование метана. Создавая определенные условия,
реакцию направляют преимущественно в сторону образования
одного из компонентов (СН3С1, СН,С12, СНС13, СС14). Этот же способ
нашел применение и для получения хлористого этила (С2Н5С1).
В то же время прямым хлорированием алканов часто не удается
получить индивидуальное соединение, а выделение нужного хлор-
83
производного из образовавшейся смеси веществ обычно является
трудной задачей.
Бензол и его гомологи хлорируются легче, чем алканы. На
холоду в присутствии катализаторов (А1С13, FeCl3) галоген замещает
атомы водорода в бензольном ядре, а при нагревании или на свету
в отсутствие катализатора замещаются атомы водорода в боковой
цепи:
FeCl
СИ,
У
сн,
сн3
I
к)
/7-ХЛОр-
толуол
t°t свет
el
о-хлор-
толуо'л
СН.2С1
А
• ! !
\/
хлористый
бензил
Если катализатор обладает достаточной активностью (А!С13),
все шесть атомов водорода в молекуле бензола могут быть замещены
на хлор:
бензол
-НС1
гексахлор-
бьпзол
Реакция протекает по ионному механизму, образовавшийся при
участии катализатора положительный иен галогена осуществляет
электрофильное замещение в бензольном ядре.
При проведении реакции в газовой среде (пропускание хлора
в пары кипящего толуола) могут быть замещены все три атома
водорода метальной группы (реакция протекает по радикальному
механизму):
С1 С
тглI ' С^
хлористый
бензил
хлористый
бензылидсн
бензитри-
хлорид
Под действием ультрафиолетового излучения в отсутствие
катализатора к молекуле бензола могут присоединяться 3 молекулы
хлора или брома:
СвН6~[-ЗС12 -* СиНеС1й
гексаклор-
циклогексан
(гексахло-
pa.-i)
Это основной промышленный способ получения гексахлорана.
84
При высоких температурах D00—600 С) удается заместить
а то Мы водорода в алкенах, не затрагивая двойной связи:
СН2^-СН2 + С1,~~ СН,=СНС1 + НС1
этилена хлориСтый
is и ни л
СН2=-СН-СН3 + Cl2 d00-5o01? CH,=CH-CH2C1 + HCI
пропилен хлористый аллил
Присоединение галогенов к ненасыщенным углеводородам:
СН,=:СН2 + С12 -* СН,С1—СН2С1
этилен 1, 2-дихлорэтан
Механизм этой реакции ранее уже был подробно разобран (стр. 30).
В промышленности в качестве сырья применяются получаемые при
химической переработке нефти этиленсодержащие фракции.
Присоединение галогеноводородов к ненасыщенным углеводородам
(гидрогалогенирование, стр. 40, 51):
CH^CHa-f HCI -* СН3-СН2С1
ьгилен хлористый
этил
OfeCH-f HCI -t CHa=CHCI
ацетилен хлористый
винил
Гндрохлорированне обычно проводят в жидкой фазе (например,
взаимодействие этилена и хлористого водорода проводится в
хлористом этиле, катализатор — хлористый алюминий).
Гидрохлорирование — важный промышленный способ получения хлористого
этила (С2Н=С1) и хлористого винила (СН2 = СНС1).
Получение еалогенопроизводных из спиртов. В лаборатории для
получения галогенопроизводных алканов широко применяются
спирты (стр. 96). При действии на них пятихлористого фосфора
(РС15), тионилхлорида (SOC12) и галогеноводородов гидроксильная
группа в молекуле спирта может быть замещена на атом галогена:
СоНрн -j- РС13 -> С,Н5С1 + HCI + РОС13
этиловый
спирт
CaHaOH + SOCb
тиокил-
хлорид
хлори-
СТЫ й
этил
^С2Н5С1-
1 " ^ Z
хлор-
окись
фосфора
hS02t + HCIJ
При использовании галогеноводородов для увеличения выхода
необходимо применять водоотнимающие средства (например, CaCU),
которые связывают образующуюся воду и предотвращают гидролиз
полученных галогеналкилов.
85
Физические свойства. Физические свойства галогенопропзводпых зависят
от строения радикала, вида и количества атомов галогена в молекуле. С
увеличением длины углеродной цепочки и с переходом от фтора к иоду температура
кипения моногалогепопроизводпых возрастает, а их плотность снижается.
Плотность подпропзводпых наибольшая, фторпронзводшлх — наименьшая. Днгало-
генопроизиодные — тяжелые масла или твердые вещества. Все галогенопроиз-
водные практически не растворимы в воде, хорошо растворимы в органических
растворителях. Большинство их обладают специфическим, часто резким
запахом, раздражающим слизистую оболочку, некоторые обладают анестезирующим
действием (СНаС12, СНС13), токсичны, являются антисептиками (СН13).
Физические свойства наиболее важных галогенопроизводных приведены в табл. 13.
Химические свойства. Большинство галогенопроизводных
углеводородов — весьма реакционноспособные соединения, широко
применяемые в разнообразных синтезах. Наибольшее значение для
соединений этого класса имеют реакции замещения, они условно
обозначены буквой S, и отщепления — Е. Широко применяется для
разнообразных синтезов взаимодействие галогеналкилов с
металлами. Примером этих превращений могут служить реакция Вюрца
(стр. 26) и Вюрца—Фиттнга (стр. 71), а также реакции с магнием,
которые будут разобраны позднее (стр. 88).
Реакции замещения. Примерами реакций замещения могут
служить следующие превращения:
+ NaI
йодистый
метил
* СН3ОН
СН3С1-
метиловый
¦ спирт
¦NaCN
+ NH,
+ NaNO2
CH3CN
ацетонн-
трил
• CH3NH2
метиламин
CH3NO>
нитронетан
Повышенная способность галогенопроизводных к реакциям
замещения связана с особенностями строения их молекулы. В
молекуле галогеналкила (например, CH3Ci) ковалентная связь между
атомом углерода и галогеном вследствие большей
электроотрицательности атома галогена поляризована. При этом у атома углерода
создается некоторая нехватка электронной плотности, в результате
чего он легко атакуется молекулами или ионами, могущими '
Н
+V6 — а
н -* с — ci
н
86
предоставлять свою электронную пару. Такие реагенты (ОН , I ,
CN , N0-2, NH3), как уже указывалось, называются нуклеофильными
(от греческого слова «нуклеус» — ядро) и условно обозначаются
буквой N, а реакции замещения, протекающие с их участием, —
реакциями нуклеофнльного замещения SA-. Типичным примером
реакций этого типа является взаимодействие галогеноалкнла со
щелочью:
СН3С1 + NaOH -» СН3ОН + NaCl
ХЛОрИ-
CTbTli
мет л л
метиловый
спирт
В ходе этой реакции гндроксильная группа атакует атом углерода
в молекуле хлористого метила:
H:O:->CH3:Ci:
в результате чего по мере приближения между ними возникает
определенное взаимодействие, в то же время связь между метильной
группой и хлором ослабляется. Наступает момент, когда
гндроксильная группа и углеродный атом метильной группы оказываются
связанными между собой, а связь углерода с хлором еще не
окончательно разорвана:
.. Н\/Н ..
Н:0:--- С ••¦:С1:
Н
Это явление получило название промежуточного или переходного
состояния. В дальнейшем между кислородом гидроксильной группы
и углеродом метильной группы возникает ковалентная связь,
осуществляемая электронной парой, предоставленной кислородом,
а связь между углеродом и хлором разрывается
Н:0:СН3+:С1:
В обычных условиях действием воды на галогеналкилы трудно
получить спирты из-за обратимости процесса, однако, применяя
едкий натр или гидроокись серебра, удается довести реакцию до
конца.
Реакционная способность галогенопроизводных в реакциях
нуклеофильного замещения .зависит от строения углеводородного
радикала и природы галогена, большую роль играют также условия
реакции (температура, наличие или отсутствие катализатора) и
характер растворителя, в среде которого взаимодействуют вещества.
В галогенопроизводных алканов в большинстве случаев легче всего
замещается атом галогена, расположенный у первичного атома
углерода, труднее у вторичного и еще труднее у третичного. У
ненасыщенных галогенопроизводных подвижность галогена зависит от
его положения по отношению к двойной связи. Если атом галогена
находится у ненасыщенного углеродного атома или связан с атомом
87
углерода бензольного ядра (например, в молекулах хлористого
винилаСН2=СНС1; хлорбензола С6НГ)С1), то в результате сопряжения
между свободными электронами галогена и л-электрснами двойной
связи пли ароматического секстета его подвижность резко снижается
и он с трудом замещается на другие группы. Если же атом галогена
находится у атома углерода, расположенного рядом с
ненасыщенным углеродным атомом (а-положенне), то он отличается
повышенной реакционной способностью (например, в молекуле хлористого
аллила СН2=СН—СН2С1). Атом галогена, удаленный от двойной
углерод—углеродной связи, по своему поведению не отличается
от галогенопроизводных алканов (например, СН,=СН—(СН2L—
—СН2—С1).
Активность галогенопроизводных возрастает с увеличением
молекулярной массы галогена: атом иода более подвижен, чем
хлора.
Используя реакцию нуклеофильного замещения, можно получить
из галогенопроизводных не только спирты (см. стр. 163), но и
представителей многих других классов органических соединений:
нитрилы, амины, нитросоединения. Во всех этих реакциях в
молекулу нового вещества вводится ал кил (в нашем случае метальную
группу СН3), поэтому галогенопроизводные являются алкилирую-
щими реагентами.
Отщепление галогеноводородов. Галогеналкилы под действием
спиртовых растворов щелочей отщепляют галогеноводороды с
образованием непредельных соединений:
СН, СН,С1
хлористый
этил
спиртов ыи
щелочи
— НС1
Р-Р
>
СНо—СН-2
этилен
Образование магнийорганических соединений. Реакция протекает
в среде безводного диэтилового эфира С2И5ОС2Н3
и приводит к образованию металлорганнческих соединений (стр. 161).
Фторпроиззодные углеводородов. Производные углеводородов,
содержащие в молекуле атомы фтора (фторпроизводные), по способам
получения, свойствам и путям использования резко отличаются
от других галогенопроизводных, поэтому их описание целесообразно
выделить в специальный раздел.
Номенклатура фторпроизводных определяется количеством
атомов фтора, содержащихся в молекуле. Частично замещенные фтором
углеводороды называют по общим для всех галогенопроизводных
правилам. Для наименования полностью фторированных соединений
(перфторуглероды) к названию углеводорода с тем же числом
углеродных атомов прибавляют приставку перфтор. Например,
C.,F6 — перфторэтаи.
Прямое фторирование углеводородов вследствие высокой
химической активности фтора невозможно. В результате выделения.
88
большого количества тепла происходит частичное или полное
расщепление углеродного скелета молекулы. Реакция часто
сопровождается взрывом. Поэтому для получения фтсрпроизводных были
разработаны особые методы.
Действие фторидов металлов на хлор-, бром- и иодпроизводные
углеводородов. Этот способ удобен для получения монофторпроиз-
бодных:
QH.C1 + AgF н> C2H,F -L AgC!
Фторирование углеводородов соединениями, легко отдающими
фтор (CoF3; MnF4; SbF5). Пары углеводорода, разбавленного
азотом, пропускают над CoF3, а образующийся двухфтористый кобальт
обрабатывают фтором; получается исходный продукт:
С7Н13 + 32CcF3 3°°n^°"-? crF]6 + 32CoF3 + 16HF
гептан пер фтор-
гептан
2CoF2-fF2—~2CoF3 "'
Прямое фторирование углеводородов. В настоящее время
осуществлено прямое фторирование углеводородов, сильно разбавленных
азотом:
ыеган четырех-
фтористый
углерод
Физические свойства и химическое поведение фтор производных
зависит от количества атомов фтора, содержащихся в их молекуле.
Температуры кипения перфторуглеродов незначительно отличаются
от температуры кипения алкансв с тем же числом атомов углерода,
но резко отличаются от температуры кипения других галсгено-
производных.
Монофторпроизводные, особенно вторичные и третичные,
нестойки, легко отщепляют фтористый водород CSH17F -> CSH16 + HF,
токсичны.
Дифторпроизводные более устойчивы и мало токсичны.
Полностью фторированные соединения отличаются исключительной
термической и химической стойкостью. Большинство перфторсое-
динений не разлагается при нагревании до 500° С, CF4 — до 300° С;
стойки к действию кислот, щелочей, окислителей, не токсичны.
Перфторсоединения применяются в качестве инертных
растворителей, смазочных масел, для производства пластических масс.
Отдельные представители галогенопроизводных. Хлористый
метил СН3С1., Промышленный способ получения -— хлорирование
метана. Применяется в качестве метилирующего средства,
растворителя, хладоагента в холодильных установках, для получения
кремнийорганических соединений, тетраметилсвинца.
89
Хлористый этил С2Н3С1. Промышленные способы получения:
хлорирование этана, гидрохлорирование этилена. Применяется для
производства тетраэтилсвинца (этнлирующее средство), в качестве
анестезирующего вещества.
Дихлорэтан СН2С1 — СН2С1. Получают хлорированием этилена.
Широко применяется в качестве органического растворителя для
экстракции жиров и восков, обезжиривания меха, шерсти,
металлических изделий перед хромированием и никелированием.
Используется при производстве хлористого винила, этиленгликоля (стр.
46), некоторых видов каучуков.
Хлороформ СНС13. В промышленности получается хлорированием
метана и действием гипохлорита кальция на этиловый спирт:
4Са (OCl),-f 2С,Н5ОН -> 2СНС13 + (НСОО);! Са + 2Са (ОН), + СаС12 + 2НгО
' Применяется для получения фреонов (стр. 90, 91), тефлона (стр.
91), в качестве органического растворителя. При стоянии в
присутствии кислорода воздуха разлагается с образованием фосгена
(СОС1,), являющегося отравляющим веществом (ОВ).
Четыреххлористый углерод СС14. Применяется для получения
фреонов, в качестве растворителя, в том числе и в бытовой химии;
Как растворитель четыреххлористый углерод обладает
существенным преимуществом перед многими другими — он негорюч. Поэтому
применяется для наполнения огнетушителей специального
назначения.
Хлористый винил СН2 =СНС1. Широко применяется в качестве
мономера для получения важного полимерного материала — поли-
винилхлорида:
¦ Г—СН,-СН— "I
L i J.
поливинплхлорид
Из поливиннлхлорида изготовляют электроизоляционные материалы
(пленки, изоляционные трубки), трубы, пленки, упаковочные
материалы, заменители кожи, линолеум. После специальной
обработки поливинилхлорид используется для производства лаков,
лечебного белья.
Хлористый аллил СН.2 = СН — СН2С1. Исходное сырье для
производства глицерина, аллилового спирта.
Хлорбензол СеН5С1. Получают хлорированием бензола. Исходное
сырье для получения разнообразных производных бензола.
Дифтордихлорметан CF2C12. Является представителем особой
группы галогенопроизводных — фреонов (фреон-12). Газ без цвета,
запаха и вкуса (/К11П=— 30° С, /пл=— 155° С). Получается из четырех-
хлорнстого углерода в присутствии пятнхлористой сурьмы:
CClj + 2HF ™С" CC1.F, + 2НС1
фреон-12
90
Широко применяется в качестве хладоагента в холодильных
установках и растворителя инсектицидов, Не токсичен, взрывобезо-
пасен,_не вызывает коррозии металлов.
Тетрафторэпгилен CF2 = CF2. Газ без цвета и запаха, мало
токсичен. Получается пиролизом монохлордифторметана:
2CHFQC1 —— CF2=CF2 + 2HC1
Является исходным сырьем для получения полимера тефлона,
фторопласта-4, отличающегося крайне высокой химической
стойкостью. Практически на него не действуют никакие вещества, за
исключением щелочных металлов.
Понятие об инсектицидах. Некоторые галогенопроизводные,
например, гексахлоран, относятся к так называемым инсектицидам —
химическим веществам, применяемым для борьбы с вредными
насекомыми. Инсектициды в виде порошков или растворов вносятся
в почву вместе с семенами или распыляются с самолетов
сельхозавиации. Они являются эффективным средством борьбы с
вредителями полезных растений, резко сокращая потери урожая. В то же
время их применение требует большой осторожности, так как они
могут уничтожать полезную микрофлору, а, попадая в организм
животных н человека, могут оказывать вредное действие.
КИСЛОРОДСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ
УГЛЕВОДОРОДОВ
Многочисленную и исключительно важную группу
органических соединений составляют кислородсодержащие производные
углеводородов: спирты (фенолы), простые эфиры, органические перекиси,
альдегиды и кетоны, органические кислоты и их многочисленные
производные (сложные эфиры, ангидриды кислот и т. д.).
ГИДРОКСИЛСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ
УГЛЕВОДОРОДОВ (СПИРТЫ, ФЕНОЛЫ И НАФТОЛЫ)
Производные углеводородов, содержащие в молекуле одну или
несколько гидроксильных групп (ОН-групп), называются спиртами
или алкоголями. Группа ОН (гидроксйльная, оксигруппа) является
в молекуле спирта функциональной (характеристической) группой.
В зависимости от строения углеводородного радикала различают:
предельные (насыщенные) спирты A), непредельные (ненасыщенные)
спирты B), ароматические о кси соединен и я C, 4):
СНз—СН;—ОН A) СН3=СН— СН,-ОН B)
этиловый спирт аллиловый спирт
|- OH C)
фенол бензиловыи спирт
91
Последние в зависимости от положения группы ОН принято делить
на фенолы (гидроксильная группа связана с углеродным атомом
бензольного ядра, 3) и собственно ароматические спирты
(гидроксильная группа находится в боковой цепи, 4); в зависимости от
количества гидроксильных групп спирты делятся на одноатомные,
двухатомные, трехатомные и многоатомные.
Одноатомные спирты
Общая формула R — ОН или Аг — ОН, где R — насыщенный
или ненасыщенный радикал алифатического ряда; Аг —
ароматический радикал. Гомологический ряд предельных одноатомных
спиртов и формулы наиболее важных представителей ненасыщенных
спиртов, фенолов и ароматических спиртов приведены в табл. 14.
В зависимости от характера углеродного атома, с которым
связана гидроксильная группа в молекуле спирта, различают:
первичные спирты—гидроксильная группа расположена у первичного
атома углерода A); вторичные — гидроксильная группа находится
у вторичного углеродного атома B) и третичные спирты, оксигруппа
расположена у третичного углеродного атома C):
R—СНг—ОН СН3—СН2—СН2—ОН A)
пропиловый спирт
он он
R-CH-Rj СН3—СН—СН3 B)
изопропиловый спирт
ОН ' ОН
R-C-Rt CH3-C-CH3 C)
I I
R2 CH3
третичный
бутилоЕый спирт
Номенклатура, изомерия. При наименовании отдельных спиртов
широко применяются исторические названия: СН3 — ОН —
древесный спирт, С2Н5ОН — винный спирт. По рациональной
номенклатуре спирты с прямой углеродной цепочкой называют по
наименованию радикала, а с-разветвленной цепочкой рассматривают как
производные простейшего метилового спирта — карбинола. По
номенклатуре ИЮПАК наличие группы ОН в молекуле спирта
обозначается окончанием «ол» с указанием номера углеродного
атома, у которого она расположена; последний обязательно
включается в главную цепь. Нумерацию атомов углерода начинают с того
конца цепи, к которому ближе расположена группа ОН:
°Н з ? '
СН3-СН,-ОН СН,-С—СН3 СН3-СН,—СН,—ОН
\ пропило^ып спирт;
этиловый спирт; 1-пропакол
этанол ^Л;}
трнмотилкарглшол;
2-метп а-2-пропааол
92
Таблица 14
Наиболее важные представители одноатомных спиртов
Название
Формула
Т. ил., "С
Т кип., °С
Метиловый спирт
(метанол)
Этиловый спирт
(этанол)
Пропиловый спирт
(i-пропанол)
Изопропнловый спирт
B-пропанол)
Бутиловый спирт
A-бута пол)
Амиловый спирт A-пен-
танол)
Изоамиловый спирт
C-метил-1 -бутанол)
Аллиловый спирт
Циклсгексанол
Бензиловый спирт
Фенол
о-Крезол
.«-Крезол
п-Крезол
СН3ОН
СН3—СН,-ОН
СН3—СН,—СН2—ОН
СИ.,—СН—СН3
ОН
СН3— (СН.2J—СН.,—ОН
СН3— (СН,K—СНаОН
СН3—СН—СН2—СНаОН
I
СН3
СН-=СН—СНаОН
С6Н„ОН
сен6сн.,он
С6Н5ОН
CeHi (CH3) ОН
С6Н, (СН3) ОН
С6Н4 (СН3) ОН
—97,8
—П0,5
— 127,0
—88,5
—89,6
—78,2
25,4
42,3
30,8
11,9
34,8
64,7
78,3
97,2
82,4
117,5
137,8
132,1
96,9
161
206
182,1
190,9
202,2
202,1
Изомерия предельных одноатомных спиртов связана со
строением радикала и положением гндроксильной группы:
ОН
СН3—СН.—СН-2—СН,—ОН
1-бутанол
СН3—СН—СН2—ОН
сн3
2-мети л-1 -пропанол
СН3-СН2-СН-СН3
2-бутанол
Природные источники и способы получения. Одноатомные спирты
в природе в свободном состоянии встречаются в небольших
количествах, но их производные (в первую очередь простые эфиры,
стр. ИЗ; и особенно сложные эфиры, стр. 145) не только широко
распространены, но в отдельных случаях используются в качестве
промышленных источников получения спиртов. Метиловый спирт
входит в состав лигнина. Высокомолекулярные одноатомные спирты
(С16 — С30) являются структурными компонентами восков (стр. 205).
При их омылении они выделяются в свободном состоянии. Это
важный промышленный способ получения спиртов с числом атомов
углерода в молекуле от 16 до 30.
Синтетические способы получения спиртов. Присоединение воды
к алкеыам. При присоединении воды к этилену образуется
первичный спирт, при гидратации других гомологов в соответствии
93
с правилом Марковникова — вторичные или третичные спирты:
СН2=СН2 + Н,0 -> СНз—СНгОН
этилен этиловый спирт
СН3-СН=СНг+ НоО -> СНз-СН-СНз
пропилен I
ин
БТОРП'ШЫП ПРОПИ-
ЛОБЫЙ СПИрТ
он
СН3~С=СН,-Ь Н2О -> СН3-С—СН3
I 1
СНз СНз
2-метилпропен 2-метил-2-пропанол
В промышленности гидратация олефинов проводится двумя
способами: при участии серной кислоты (сернокислотная
гидратация) и прямая гидратация. В обоих случаях скорость реакции
зависит от длины углеродной цепочки и строения алкена. С
увеличением длины углеродной цепочки (до известных пределов) скорость
реакции возрастает, изоалкены реагируют быстрее, чем олефины
нормального строения. Труднее всего реагирует с водой этилен.
Сернокислотная гидратация алкенов протекает в две стадии:
CHa=CH2 + HOSO./)H -» СН3—СН,—OSCv,OH
этилен этилсервая кислота
СНз-СНг—OSOaOH + НгО -» СНз-СНоОН f H.SO4
этиловый спирт
Прямую гидратацию алкенов проводят в одну стадию, применяя
в качестве катализатора фосфорную кислоту или окисль алюминия:
СН.,=СНг + Н,0 -> СН,—ГН-2ОН
этилен этиловый спирт
Гидратацию этилена проводят при температуре 300—350° С,
давлении 100 атм (98-105 Н/м2), катализатор — смесь фосфорной и
вольфрамовой кислот, нанесенных на силикагель.
Гидратация алкенов — важнейший промышленный способ их
получения, позволяющий использовать дешевое и доступное
сырье — газы крекинга. Прямая гидратация алкенов экономически
более выгодна, чем сернокислотная, проведение которой связано
со значительным расходом труднорегенернруемой серной кислоты.
Восстановление альдегидов и кетонов. При восстановлении
альдегидов образуются первичные спирты, кетонов — вторичные (стр.
92): /0 п
СН3—C<f — СН3-СНоОН
н этиловый спирт
уксусный
альдегид
СНз-С-СНз1Н* СНз-СН-СНз
о он
ац
0-1
ацетон нзопроп иловый
сп ирг
Используя эту реакцию, можно получить этанол из метана:
сн4 ->- ш=сн -> сн3-с/ -> снэ-сн.,он
чн
Гидролиз галогенопроизводных (стр. 87).
Получение спиртов из окиси углерода и водорода. Ранее уже
рассматривалось (стр. 26) получение углеводородов из окиси
углерода и водорода. В ходе этой реакции образуются разнообразные
кислородсодержащие соединения, в первую очередь этиловый
спирт. Меняя условия и катализаторы, реакцию можно направить
в сторону большего образования метилового спирта или высших
спиртов.
Получение спиртов действием металлорганических соединений
на альдегиды, и кетоны (реакции Бутлерова, Тищенко, Вагнера,
Зайцева, стр. 163).
Получение спиртов брожением. Этот метод широко применяется
для получения этилового и некоторых других низкомолекулярных
спиртов (стр. 237).
Гидролиз сложных эфиров. Применяется в промышленности для
получения некоторых высокомолекулярных спиртов из восков:
О О
R—С-О—Rj —— R—С-ОН + Ri—ОН
сложный эфир карбоновая еысокомо-
кислота лекуляр-
иый спирт
Физические свойства. Спирты нормального строения с числом атомов
углерода в цепи до Са — жидкости, содержащие более 11 атомов углерода —
твердые вещества. Легче воды, без цвета, До С3 — с резким «спиртовым»
запахом, С4—Cd с резким, неприятным запахом — «сивушный занах», твердые — без
запаха, Первые гомолога хорошо растворимы в воде, но мере увеличения
молекулярной массы растворимость надает. Низшие спирты легко воспламеняются
и горят бесцветным пламенем. Спирты изостроения кипят при более высокой
температуре, чем спирты нормального строения, причем температура кипения
третичных спиртов выше, чем вторичных. Характеристика спиртов приведена
в табл. 14,
При сравнении температуры кипения спиртов с температурами кипения
представителей других классов органических соединений обнаруживается, что
спирты кипят при более высоких температурах, например:
t- < SC
СН3—СН»—ОН (этиловый спирт) +78,3
СН3—СНо—С1 (хлористый этил) +12,4
СН3—О—СН3 (диметиловый эфир) —23,7
Это связано с тем, что молекулы спирта связаны между собой (ассоциированы)
с помощью водородных связей (стр. 19).
Многие спирты крайне ядовиты: 10—20 мл A0 • 10~6—20 • 10~6м3) метилового
спирта вызывают слепоту, большие количества — смерть.
Химические свойства. Химическое поведение спиртов в первую
очередь связано с наличием в их молекуле группы ОН. В
разнообразных химических превращениях особая роль принадлежит двум
95
типам реакций: разрыву связи между атомами С и О A); и О и Н
(II). Положение гндроксильной группы (расположена она у
первичного, вторичного или третичного атомов углерода) существенно
влияет на ее поведение в этих превращениях:
Н
!
Н (I) (II)
Образование алкоголятов. Спирты, взаимодействуя с
металлическим натрием, образуют алкоголяты — твердые вещества белого
цвета:
2СН3—CH2—OH + 2Na -> 2СН3—СН2—ONa + Н3
этиловый спирт этилат натрия
В присутствии воды алкоголяты гидролизуются с образованием
спирта и щелочи:
СН3—СН2—ONa + НОН -» СН3—СН2—ОН + ХаОН
По строению алкоголяты можно рассматривать как соли щелочей
и очень слабых кислот. Их спиртовые растворы обладают сильными
основными свойствами. Спирты, не меняющие окраски индикаторов,
в определенных условиях проявляют или очень слабые щелочные,
или кислотные свойства, являясь тем самым амфотерными
соединениями. Образование алкоголятов — пример кислотных свойств
спиртов.
Замещение гидроксильной группы в молекуле спирта на атом
галогена. Эти реакции уже были рассмотрены в разделе галогено-
производных (стр. 85). Для введения галогена вместо
гидроксильной группы применяют галогеноводородные кислоты, галогениды
фосфора (РС15, РС13) или тионилхлорпд (SOC1,):
CHsOH + HCi^QH-jCi + H/) (!)
этиловый х лори-
спирт стый
этнл
СаН5ОН + РС15 -» QH5Ci + РОС1» + НС1 B)
хлор-
окись
фосфора
C)
Наиболее удобно для замещения гидроксильной группы
использовать тионнлхлорнд; применение галогенидов фосфора осложняется
образованием побочных продуктов. Образующаяся при
взаимодействии спиртов с галогеноводородамн вода разлагает галогеналкил
на спирт н галогеноводород, поэтому реакция A) обратима. Для
ее успешного проведен!;я необходимо, чтобы исходные продукты
содержали минимальное количество воды: з качестве водоотнимаю-
щих. средств применяют хлористый цинк, хлористый кальций,
серную кислоту.
В реакцию с галогеноводородами труднее всего вступают
первичные спирты, легче третичные, вторичные занимают
промежуточное положение.
Образование сложных эфиров. Реакция этерификации.
Взаимодействие спиртов с кислотами (органическими и неорганическими)
приводит к образованию производных кислот, называемых
сложными афнрамн;
О О
II II
СН3-СН.2ОН + НО-С—СН3 7Z СН3-С—О-СН2—СН3 + Н2О A)
этиловый спирт уксусная уксусноэтпловый эфир
кислота (этилацетат)
СН3-ОН + >S/ —Го' /s€ lib' /К B)
-но/ V>-~H!° он/ ^о ~~ нс-о/ ^о
метиловый '^ wil u ч^з *-> -1
С11ИРТ ыетнлсульфат диметилсульфят
Реакция получила название реакции этерификации от латинского
aether — эфир. Скорость этерификацни зависит от силы кислоты
и природы спирта: с увеличением силы кислоты она возрастает,
первичные спирты реагируют быстрее вторичных, последние быстрее
третичных. Этерификация спиртов органическими (карбоновыми)
кислотами ускоряется добавлением сильных минеральных кислот.
Реакция обратима, обратная реакция получила название гидролиза.
Гидролиз сложных эфиров, проводимый водными растворами
щелочей, называется омылением (стр. 146).
Равновесие прямой и обратной реакции зависит от строения
исходных продуктов. Выделяющаяся при этерификации молекула
воды образовалась из гидроксила кислоты и водорода спирта.
Это было установлено методом «меченых атомов». В молекулу
спирта был введен изотоп кислорода 18О, который был обнаружен
е молекуле сложного эфира:
О О
СНз—ISO—:Н + Н—СУ— С— Я ^ R—С—ГЮ—СН3 -f H2O
«.Меченые соединения» широко применяются в химии, биологии,
физике для разнообразных исследований.
Дегидратация спиртов. Дегидратация или дегидратирование
(отнятие воды) от молекулы спирта приводит к образованию
этиленовых углеводородов A) или простых эфиров B):
СН3—СН2—ОН -> СН.,=СНН-Н2О A)
этиловый спирт этилен
СНз—СНг—О НТ НО -СНо—СНЭ -* СН3-СН3—О—СН3—СН3 + НаО B)
дпэтиловый зфпр
Первичные спирты дегидратируются труднее вторичных, легче
всего отнимается молекула воды от третичных спиртов.
При образовании молекулы воды водород в соответствии с
правилом Зайцева всегда отнимается от наименее гидрогенизирован-
4 А, П. Нечаев 97
ного атома углерода, находящегося по соседству с углеродным
атомом, несущим гидроксильпую группу:
:6н"Н!
сн3—сн—сн—сн3 -. сн3—сн=сн—сн3-ьн2о
2-бутаиол 2-бутен
Гидратацию проводят с помощью водоотнимающпх средств (серная
и ортофосфорная кислоты, окись алюминия, силикагель, хлористый
цинк) при нагревании, Температура дегидратации первичных и
вторичных спиртов 200—350° С. Из двух возможных реакций 1 и
2 (образование алкенов и простых эфиров) первая требует более
высоких температур.
Окисление спиртов. При окислении первичных спиртов
образуются альдегиды, вторичных — кетоны, окисление третичных
спиртов сопровождается разрывом углеродной цепи, обычно около
атома углерода, несущего гидроксильную группу:
СН3-СН2-ОН J— СН3-СНО+ Н2О
этиловый спирт уксусный
альдегид
ОН О
СН3—СН—СН3 — СН3—С—СНз + НгО
2-пропанол ацсюн
Продукты реакции могут подвергаться дальнейшему окислению
с образованием соответствующих карбрновох кислот. Образование
различных продуктов при окислении первичных, вторичных и
третичных спиртов дает возможность использовать эту реакцию
для установления строения спирта.
Окислители: хромовая смесь, перманганат калия, кислород
в присутствии металлической меди. Окисление спиртов ведут при
нагревании, оптимальные температуры зависят от строения спирта
и окисляющей способности примененного окислителя. Первичные
и вторичные спирты окисляются легче, чем третичные.
Дегидрирование спиртов. При пропускании паров спирта над
дегидрирующим катализатором (раскаленная медь, железо, никель)
первичные и вторичные спирты теряют два атома водорода
(дегидрирование), при этом первичные спирты превращаются в альдегиды,
вторичные в кетоны:
На
98
СИз—CH2OH —
этиловый спирт
СН3—СН—СНз "^
[
он
нзопропиловый
ci ирт
СЫз—СНО +
ук су сны й
альдегид
СН3—С—СН3
6
адет он
Отдельные представители спиртов. Метиловый спирт, метанол,
древесный спирт СН3ОН. Прозрачная бесцветная жидкость со
слабым спиртовым запахом. Смешивается с водой, ацетоном, спиртами
во всех отношениях. Крайне ядовит.
Основные способы промышленного получения:
1. Синтез из окиси углерода и водорода СО + 2Нг —СН3ОН
Температура реакции 300—*400° С, повышенное давление,
катализатор — окись цинка и хрома.
2. Неполное окисление метана и его гомологов СН4-Ь О—>СН3ОН
3. Некоторое количество метилового спирта получается методом
сухой перегонки древесины; отсюда и одно из названий метанола —
древесный спирт. Это наиболее старый способ его получения.
Метиловый спирт — один из важных продуктов химической
промышленности, широко применяемый в качестве растворителя и
полупродукта при получении многих органических соединений.
Основные пути промышленного использования метанола приведены
на схеме 6. Особенно большие количества метанола расходуются
для получения формальдегида и в реакциях метилирования. Метанол
один из исходных продуктов в синтезе метионина — важной
аминокислоты (стр. 180), применяемой для стимулирования роста птицы.
Схема 6
сн^он
метп.товый
спирт
неполно? окисление
+ НС1
+so3 >
+СО
+сн— н
+сн=сн + со
+C6U5NH2 ^
HGHO
формалвдегид
СН3С1
хлористый метил
сн3ш2
метиламин
(GH3OJSO2
динети.т-ульфат
СН3СООН
уксусная кислота
СН3—О—СН=СН2
винилкетпловый эфир
СН2=СН—СО О—СН3
иетй-ловий эфир акриловой кислотьт
C6H5-N(CH3J
диметиланилии
GH3SH
метилмеркаптан
Этиловый спирт, этанол, винный спирт СН3 — СН2 — ОН.
Жидкость без цвета, с «винным или спиртовым» запахом, жгучая
99
нэ вкус. Смешивается с водой в любых отношениях. Смесь,
содержащая 95,б°6 спирта и 4,4% воды, перегоняется без разделения
(такие растворы называются азеотропными смесями), отсюда и ее
название «спирт-ректификат». Спирт-ректификат широко
применяется в пищевой и других отраслях промышленности. Удалить
остаток воды и получить чистый «обезвоженный» этанол удается только
в результате специальной обработки или с помощью водоотнимаю-
щих средств, например, прокаленного сульфата меди (лабораторный
метод) или добавлением к спирту-ректификату бензола и
перегонкой смеси спирта, воды и бензола. Сначала отгоняется тройная
смесь, затем смесь спирта и бензола, до полного удаления
последнего, остаток — чистый этанол.
В технике и лабораторной практике концентрацию этилового
спирта обычно выражают не в весовых, а в объемных процентах
пли градусах (G). Концентрацию спирта в градусах определяют
ареометром (спиртометром), градуированным специальным образом.
Так как при смешивании спирта с водой общий объем смеси
уменьшается, то концентрация одного и того же раствора спирта в
градусах н в процентах выражается разными числами.
В промышленности этанол получают гидратацией этилена,
брожением пищевого сырья, гидролизом растительных материалов,
переработкой сульфитного щелока.
1. Гидратация этилена (стр. 42).
2. В основе следующих способов получения этанола лежит
превращение простых углеводов — гексоз (стр. 221), главным
образом, наиболее распространенной и доступной гексозы —
глюкозы (С6Н12О6, стр. 238), под влиянием дрожжей или выделенных
из них биологических катализаторов — ферментов (стр. 268).
Суммарно спиртовое брожение выражается следующим уравнением:
С6Н12О6
гексоза зтиловый
спирт
Кроме этилового спирта и двуокиси углерода также образуются
правда, в значительно меньших количествах, бутиловый и
амиловый спирты, янтарная кислота и другие вещества.
Спиртовое брожение используется не только для получения
спирта, этот процесс лежит в основе многих пищевых производств:
виноделия, пивоварения.
Получение этилового спирта с помощью спиртового брожения
может быть осуществлено различными путями, которые отличаются
друг от друга по характеру сырья, используемого для получения
гексоз, технологии, качеству получаемого этилового спирта и его
стоимости. Основными методами являются: а) брожение пищевого
сырья; б) гидролиз растительных материалов; в) переработка
сульфитного щелока.
а. Брожение пищевого сырья. Сырьем для получения этилового
спирта по этому методу являются крахмалсодержащие продукты:
картофель, зерно кукурузы, ячменя, пшеницы, ржи, проса, овса.
Содержание крахмала в них колеблется от 18 до 60°о. Кроме этих
100
продуктов для получения этанола применяется отход сахарного
производства — меласса, содержащая до 60% сахарозы (стр. 249).
Картофель и зерно после очистки и удаления оболочек (в
необходимых случаях) разрыхляют и разваривают, превращая в
однородную жидкую массу, в которую вводят солод — измельченное н
высушенное проросшее зерно ячменя. Солод содержит
высокоактивные ферменты— амилазы, способные осахаривать крахмалсодержа-
щее сырье:
F (С6Н12О6)„+Н2О -» QoH.^On
крахмал мальтоза
Осахаривание крахмала проводится в течение 10—15 мин при
температуре 60° С.
В последние годы в отдельных случаях вместо солода для оса-
харнвания крахмала стали применять специальным образом
полученные ферментные препараты. Образовавшийся после осахарива-
ния продукт охлаждают и вводят в него дрожжи. Находящийся
в них фермент мальтаза вызывает гидролиз мальтозы с образованием
двух молекул глюкозы:
Ci,H.,Ai + НаО ^ 2С6Н13О6
мальтоза
Раствор, содержащий глюкозу, под влиянием разнообразных
ферментов дрожжей в результате сложных биохимических процессов
превращается в бражку с 10% этилового спирта. Этанол отгоняют
и очищают.
Выход этилового спирта из одной тонны исходного сырья сильно
зависит от его вида, качества и особенностей технологии и
колеблется от 90 до 360 литров.
Остаток после отгонки этанола (кубовый остаток) содержит
сивушное масло — ядовитую маслянистую жидкость крайне
неприятного запаха («сивушный запах»). В состав сивушного масла входят
спирты С3 — Q. Основные компоненты: 1-пропанол G%), 2-метнл-
1-пропанол B4%), З-метил-1-бутанол и 2-метнл-1-бутанол.
б. Экономически выгодно получение этилового спирта из отходов
переработки древесины: опилок и щепы (гидролиз растительных
материалов). Процесс состоит из двух стадий: гидролиза
содержащейся в них целлюлозы с образованием в конечном итоге глюкозы:
(С6Н10О6)„ + пН.2О н> мС6Н12Ов
целлюлоза глюкоза
и сбраживания последней с образованием этилового спирта.
Гидролиз целлюлозы проводят растворами минеральных кислот при
нагревании. Из одной тонны сухой древесины получают 160—200
литров «гидролизного» этилового спирта, применяемого для
технических целей. При комплексной переработке образовавшихся гид-
ролизатов, кроме этилового спирта, получается значительное
количество двуокиси углерода, кормовых дрожжей, фурфурола (стр. 193),
лигнина и других ценных продуктов.
в. Этиловый спирт получают из сульфитных щелоков — отхода,
образующегося при производстве целлюлозы в целлюлозно-бумаж-
101
ной промышленности. Сульфитные щелоки содержат значительное
количество гексоз, которые после специальной обработки могут
сбраживаться.
Из описанных способов получения этанола экономически
наиболее выгоден способ гидратации этилена, особенно прямая
гидратация. Стоимость этилового спирта, выделяемого из пищевого сырья,
наибольшая, гидролизного — ниже. Самый дешевый этанол
получают синтетическим путем, к нему по стоимости приближается
этанол, образуемый из сульфитного щелока. Необходимо помнить,
что при получении 1 т A03 кг) этанола гидратацией этилена удается
сэкономить до 4 т D-Ю3 кг) зерна или 12 т A2-Ю3 кг) картофеля,
которые могут быть использованы для пищевых целей.
Этиловый спирт — один из основных продуктов, выпускаемых
химической и пищевой промышленностью (схема 7):
Схема 7
каталитическое
с2н5он
этиловый
- спярт
разложение
хлорирование
окисление
+ H2SO4
+ СН3СНО
+СН3СООП ^
СН2=СН—СН=СН2
1,3-бутадиен
(С2Н5JО
диэтиловый эфир
Чистый этилен
сн3—сно
ацетальдегид
ацетон
СС13—СНО
хлораль
сн3—сно
ацетальдегид
сн3—соон
уксусная кислота
(C2H5OJSO2
диэтилсульфат
СН2=СН— СН=СН2
1,3 - бутадиен
сн3—соо—с2н5
этилацетат
102
Наибольшее количество этанола используется в производстве
синтетического каучука, этилацетата, диэтилового эфира, хлораля
|СС13—С^ 1 и в качестве растворителя.
Большие количества этанола, образующегося при брожении
пищевого сырья, используют в пищевой промышленности для
получения спиртных напитков (водки, ликеров и т. д.), в парфюмерной
промышленности для изготовления духов, одеколона и в
медицинской промышленности.
Пропиловые спирты. СН3 — СН3 — СН2 — ОН первичный про-
пиловый спирт, 1-пропанол. Бесцветная жидкость с характерным
запахом. Хорошо растворим в воде и многих органических
растворителях. Пропанол выделяют из сивушного масла и используют
для получения пропионового альдегида.
Изопропиловый спирт, 2-пропанол СН3—СН—СН3. Ж.идкость без
ОН
цвета, со спиртовым запахом. Смешивается с водой в малых
отношениях. Получается гидратацией пропилена. Применяется
в качесгве растворителя и для синтезов разнообразных химических
продуктов.
Бутиловые спирты. СН3 — (СН2J — СН., — ОН н-бутиловый
спирт, 1-бутаиол. Получается совместно с ацетоном при ацетонобу-
тнловом брожении углеводов. Применяется в качестве растворителя
лаков и красок, для синтеза сложных эфиров, масляной кислоты.
Бутиловые спирты изостроения получают гидратацией бутиле-
нов, за исключением 2-метил-1-пропанола, который выделяют из
сивушных масел или синтезируют из окиси углерода и водорода;
СН3—СН—СН2—ОН
СН3
2-метил-1-пропанол
Все они применяются для разнообразных синтезов и в качестве
растворителей.
Амиловые спирты С5НПОН. Получают из пентанов С5Н12,
выделяемых из легких погонов нефти, хлорированием и последующим
омылением:
, С5Н12+С12-> С6НиС
CsHuOH
или из амиленов С5Н10, содержащихся в продуктах крекинга и
пиролиза алканов, сернокислотной гидратацией:
СНз—С=СН—СН3 —-^ (СНз)*-С—СН2—СН3
сн3 он
З-метил-1-бутанол и 2-метил- 1-бутанол выделяют из сивушных
масел. Первый применяется для получения уксуснопзоамнлового
103
эфира — вещества с приятным «грушевым» запахом, применяемым
при приготовлении конфет, фруктовых вод, в качестве растворителя
и для разнообразных синтезов.
Высшие с пи р т ы (стр. 206).
Циклогексанол С6НПОН. Бесцветная жидкость со слабым
запахом. Плохо растворим в воде. Получают в промышленности
гидрированием фенола (стр. 111). Применяется для получения адипн-
новой кислоты, в качестве растворителя масел, жиров, синтетических
смол и как добавка при производстве синтетических моющих
средств и хозяйственного мыла.
Виниловый спирт СН2 = СН — ОМ. В свободном состоянии
не существует. Его полимер Г—СН2—СН—1 — поливиниловый
ОН
спирт, белый растворимый в воде порошок, получают омылением
поливпнилацетата pi применяют для получения синтетических
волокон и лекарственных препаратов.
Аллиловый спирт СН2 = СН — СН3 — ОН. Простейший
представитель одноатомных ненасыщенных спиртов. Жидкость с резким
запахом, хорошо растворимая в воде. Получается замещением
водорода метнльнон группы пропилена на хлор и омылением
образовавшегося хлористого аллила:
СН2=СН—СН3 + С1, —:~ СН,=СН—CHoCl +HC1
пропилен хлористый аллил
СН2=СН—СН..С1 + Н,0 -* СН.,=СН—СН.2ОН + НС1
Аллиловый спирт обладает вы:окой реакционной способностью,
применяется для получения глицерина, в производстве смол,
в парфюмерной промышленности.
Бензиловый спирт С6Н3 — СН2ОН. Бесцветная жидкость с
приятным запахом, плохо растворим в воде. Широко распространен
в природе, входит в состав эфирных масел, принимает участие
в формировании запаха цветов, составной компонент ароматобразу-
ющпх веществ пшеничного хлеба. В промышленности получают
омылением хлористого бензила С6Н3СН,С1 + Н2О —нсГ С6Н5СН.2ОН
Химические свойства бензилового спирта связаны с наличием в его
молекуле оксигруппы и бензольного ядра: вступает во все реакции,
характерные для первичных спиртов и производных бензола.
Близкое соседство бензольного ядра и оксигруппы приводит к
повышенной подвижности последней в реакциях замещения.
Бензнловый спирт применяется в парфюмерной промышленности.
Многоатомные спирты
Многоатомные спирты в зависимости от количества гпдрокспль-
ных групп в молекуле делятся на двухатомные (две группы ОН),
или д.чолы, трехатомные (или трполы) и т. д.
У одного атома углерода в молекуле многоатомного спирта
может содержаться не более одной гидроксильной группы. Спирты,
104
содержащие две или три гидроксильные группы у одного С-атома,
выделить в свободном состоянии не удается, в момент образования
они отщепляют воду и превращаются в альдегиды или кетоны:
/ОН
СН3—СНС12 TTTNaCl iCH3—СН
Р
— н2о
хн
ci a
СНз-С-СНз ^щ- LCH3-C-CH3 J ^н7
он он
уксусный
альдсгнд
О
СН3-С-СН3
ацетон
Номенклатура и изомерия. Изомерия многоатомных спиртов
связана со строением углеродного скелета и различием в положении
гидроксильных групп. По ИЮПАК для наименования
многоатомных спиртов к окончанию «ол» прибавляют приставку «ди», «три»
и т. д., соответствующую количеству гидроксильных групп,
положение которых указывается цифрами.
Для наименования отдельных спиртов широко применяются
исторические названия:
СН2—СН2 СНз—СН—СН2—СН2 СН2—СН—СН3
он он
\, 2-этандиол
(этиленглпколь)
он он
1, 3-бутанднол
он он он
1, 2, 3-пропантриол
(глицерин)
Способы получения. Получают многоатомные спирты теми же
способами, что и одноатомные, с той лишь разницей, что в молекуле
создается не одна, а несколько гидроксильных групп. Отдельные
представители этой группы соединений получают специфическими
методами.
Физические свойства. По своим физическим свойствам многоатомные спирты —
хорошо растворимые в воде вязкие жидкости или твердые вещества, многие
из которых обладают сладким вкусом.
Химические свойства. Химические свойства многоатомных
спиртов близки к свойствам одноатомных спиртов. В реакциях может
участвовать одна или несколько гидроксильных групп. Особенность
многоатомных спиртов — повышенная подвижность атомов
водорода гидроксильных групп. Многоатомные спирты превращаются
в алкоголяты не только при взаимодействии с натрием и калием,
но и с гидроокисями тяжелых металлов, образуя комплексные
соединения:
Н
н,с—он
Н2С—ОН
этнленгликоль
+ Cu(OHJ +
НО—СН,
.О—СН,
НО—СН2 'н.С—СК'' \э—СНг
I
н
комплексное соединение
105
Отдельные представители. 1,2-Этандиол (этиленгликоль)
ОН
СН2—СН2—ОН. Простейший и наиболее важный представитель
двухатомных спиртов или гликолей, названных так за свой сладкий
вкус (гликос — сладкий). Бесцветная, вязкая жидкость, без
запаха.
Основной промышленный способ получения этиленгликоля —
гидратация окиси этилена (стр. 114), которую проводят избытком
воды в присутствии серной или фосфорной кислот при нагревании
E0—100° С):
СН - СН -f Н,0 -* СН2—СНа
о/ он он
окись
этилена
этиленгликоль
Этиленгликоль — важный продукт химической промышленности.
Широкое применение нашли разнообразные эфиры этиленгликоля.
Диоксан — простой циклический эфир этиленгликоля, получают
при перегонке последнего с серной кислотой:
/он
Н2С
1
Н2С
хон
этт'ле
ноч
СН,
сн3
но/'
•нгликоль
-2Нгб
н
н
/
оС
" I
2С
лиок
ч
СН
1
СН
)/'
Используется в качестве растворителя.
Эфиры этиленгликоля и двухосновных кислот идут на
изготовление пленкообразующих веществ и синтетических волокон.
Этиленгликоль применяют в текстильной, косметической, табачной про-
мышленностях в качестве гигроскопичного материала. Водные
растворы этиленгликоля замерзают при температурах ниже нуля
E0%-ный раствор при —37° С), что дает возможность использовать
их для приготовления антифризов — жидкостей, замерзающих при
низких температурах, используемых для охлаждения
автомобильных двигателей
СН -_СН—СНа
! ! I
он он он
1, 2, З-Пропанпгрнол (глицерин)—единственный практически
важный представитель трехатомных спиртов. Сиропообразная
сладкая жидкость, без цвета, хорошо растворимая в воде. Водные
растворы глицерина замерзают при низких температурах F7%-ный
раствор при —46,5° С) и применяются в качестве антифризов.
Глицерин широко распространен в природе, является основным
спиртом, участвующим в построении молекул разнообразных групп
106
липидов (стр. 198). В промышленности глицерин получают
гидролизом триглицеридов:
О
II
СН.,—О—С—R
О СН2—ОН
'! j
_О_с—Rx + ЗН2О -> СН—ОН + R—СООН + Rx—СООН + R,COOH
r-ur глтл жирные кислоты
Jl LH2—ОН
¦п2 w ^ д2 глицерин
трнглнцерид
Это старый способ, не потерявший своего значения и в настоящее
время. Однако получение глицерина гидролизом триглицеридов
связано с использованием для технических целей больших количеств
ценных пищевых продуктов — жиров и масел, поэтому этот способ
постепенно заменяется синтетическим, основным сырьем в котором
является пропилен:
СН„=СН-СН3 + С12 45°-5001? СН2=СН—СН2С1 + НС1
пропилен хлористый аллил
СН2=СН—СН,С1 + NaOH lo0~'6ul? СН2=СН—СН2ОН + NaCl
аллиловый спирт
-СН2С1—СНОН- -СН2ОН
СН2=СН-СН.,ОН + НС1О-
-СН2ОН—CHCi—CH.OH
монохлоргидрины глицерина
СН2С1— СНОН— СН2ОН 150„с СН2—СН—СНа
+ NaOH | !
СН2ОН— СНС1—СН2ОН ОН ОН ОН
глицерин
В настоящее время отдельные стадии этого синтеза видоизменяются
и модернизируются.
Глицерин широко применяется во многих отраслях
промышленности. Основное его количество расходуется для получения
производных — глифталевых смол, нитроглицерина. Глифталевые смолы
(глифтали) — полиэфиры глицерина и фталевой кислоты —
применяются для изготовления лаков. Глицеринтринитрат или, как
его неправильно называют в технике, нитроглицерин* — сложный
эфир глицерина и азотной кислоты — получают действием азотной
кислоты в присутствии серной на глицерин:
/ СН2-ОН СНа—ONO2
Ц СП
СН—ОН +ЗНОШ2—-^ СН—ONO3 +3H.0
СНе—ОН СН—ONO3
глицерин
глицеринтринитрат '
(нитроглицерин)
* Нигросоединения содержат в молекуле группу NO2> азот которой i enoc-
редственно связан с углеродным атомом (стр. 168).
107
Глицеринтрипитрат — взрывчатое вещество огромной силы,
(взрывается от л(л кого толчка). Для взрывных работ часто
применяется смесь, состоящая из 75% нитроглицерина и 25'!» инфузорной
земли, называемая динамитом, которая безопаснее в работе.
В пищевой промышленности глицерин используется для
приготовления ликеров и безалкогольных напитков, в бумажной и
кожевенной промышленности — для предохранения материала от
высыхания. Он входит в состав многих косметических препаратов
и широко применяется в качестве смягчающего кожу вещества.
Представители спиртов, содержащих в молекуле пять или шесть
гидроксильных групп, — ксилит СН2ОН — (СНОНK — СН2ОН и
сорбит СН2ОН — (СНОН).[ — СН2ОН. Получаются
восстановлением моносахаридов: первый — ксилозы (стр. 243), второй —
глюкозы (стр. 238). Сорбит широко распространен в природе, входит
в состав многих плодов и ягод (в плодах рябины до 7°о сорбита).
Ксилит и сорбит применяются больными диабетом в качестве
заменителя сахара, для приготовления кондитерских изделий,
напитков, зубных паст. Сорбит — первый полупродукт при
производстве аскорбиновой кислоты (витамина С, стр. 271). '
Фенолы, нафтолы
Особую группу составляют оксисоединения, гидроксильная
группа которых связана с атомами углерода бензольного ядра, и
оксипроизводные нафталина. Первые называются фенолами,
вторые — нафтол ами:
ОН СН3 СНз СН3
1 ОН
фенол
(окспбеизол)
он
о-крезол
B-окептолуол)
он
I
V\Oh
л-крезол
C-окситолуол)
ОН
А
!
ОН
л-крезол
D-окситолуол)
ОН
пирокатехин
(о-диоксибензол)
резорцин
(.«-диоксибмзол)
он
гидрохинон
(л-диоксибензол)
—ОН
-он
пирогаллол
(i, 2, З-триоксноепзол)
ОН
!
А-он
I ii
\/
он
оксигидрохпнон
A, 2, 4-трпокснбензол)
108
ОН
ОН
и
флороглюцнн
A, 3, 5-триоксибензол)
-ОН
сс-нафтол
A-нафтол)
р-мафтол
(J-нафгол)
Изомерия фенолов и нафтолов связана с различным положением
гидроксильных групп. Фенолы, кроме этого, могут быть изомерны
ароматическим спиртам:
СНз
f^-OH |^Ч)-СН2ОН
\/ V
о-крезол бензиловый спирт
Способы получения фенолов и нафтолов. Получение фенола
совместно с ацетоном (кумольный способ). Исходное сырье — бензол
и пропилен:
О—О—Н
СН3—СН—СНз НеС—С—СНз
х/ пропилен х.--
бензол изолрошглбен2ол гидроперекись
(кумол) изопропилбензола
ОН
о
р
+ СН3-С-СНэ
ацетон
фенол
Этот метод, разработанный в 1949 г. советскими учеными П. Г.
Сергеевым, Б. Д. Кружаловым, Р. Ю. Удрисом и М. Е. Немцовым,
экономически наиболее выгоден: он дает возможность получать
два ценных продукта — фенол и ацетон с использованием
дешевого сырья — пропанпропиленовой фракции газов крекинга
нефти.
Получение из бензолсульфокислот (стр. 166).
Получение фенола парофазным кшпалитичесшм гидролизом
хлорбензола. На первой стадии из бензола окислительным
хлорированием получают хлорбензол. Катализатор — хлориды меди и
железа, нанесенные на окись алюминия. На второй стадии
хлорбензол гидролизуют с образованием фенола:
! 1 1
\/
зензол
HU-f-
1-Я
- г_ 235—245°
'" * ~Н2°
стадия
CI
|
\/
хлорбензол
400—420
-1-Н2С
2-я
ОН
|
^Л-нс
Ч/ '
фенол
стадия
109
Не потерял своего значения и старый способ получения
фенолов — выделение их из побочных продуктов коксования каменных
углей. При этом каменноугольную смолу обрабатывают растворами
щелочи и выделяют фенолы из полученных продуктов кислотой
с последующей их очисткой:
С6Н5ОН ^п?- CeH5ONa zjfffbo7 C^0H
фенол фенолят фенол
натрия
Двух- и многоатомные фенолы и нафтолы получают из солей
соответствующих сульфокислот и хлорфенолов.
Физические свойства. Фенолы и нафтолы — кристаллические вещества с
характерным запахом, трудно растворимые в воде.
Химические свойства. Химическое поведение фенолов и нафто-
лов связано с наличием в их молекуле оксигруппы и ароматического
ядра. Они вступают практически во все реакции, характерные
для них.
В то же время в химическом поведении фенолов и нафтолов
наблюдается ряд особенностей, связанных с взаимным влиянием
оксигруппы и бензольного ядра. Например, фенолы обладают
большими кислотными свойствами, чем спирты, и способны вступать
в реакцию со щелочами. Это связано с тем, что неподеленная
электронная пара атома кислорода оксигруппы вступает во
взаимодействие с я-электронамн бензольного ядра, электронная плотность
смещается и атом водорода оксигруппы приобретает большую
подвижность:
Соли фенолов получили название фенолятов:
С6Н5ОН + NaOH 7Z C6H5ONd + Н2О
фенол феноляг
натрия
При взаимодействии фенолятов с хлористыми алкплаын и хлоран-
гидридами образуются простые и сложные эфнры. Реакции, сья-
занные с заменой гидроксильной группы в молекуле фенола
(взаимодействие с РС16), протекают значительно труднее, чем у спиртов.
Гьдроксильная группа — заместитель первого рода, поэтому
реакции электрофнльного замещения в бензольном ядре протекают
по
с фенолами значительно легче, чем с оензолом, а заместители
преимущественно направляются в орто- и /тра-положенне:
ОН
нитрование
ОН
л.
Фенол
сульфирование
! VNO»
е-нитрофенол
ОН
NO,
п-нитро-
фнтол
ОН
. f V-so,h
о-фенолсуль-
фокислота
ОН
SO3H
гг-фенолсуль-
фокислота
При восстановлении фенола образуется цнклогексапол. Реакцию
проводят при 150° С и давлении 20 атм A9,6-1Q5 Н/м2), кзталпза-
тор — металлический никель, нанесенный на окнсь алюминия.
Это основной
ОН ОН
I
У'\ н.
фенол
цпкло-
гексааол
промышленный способ получения циклогексанола.
Фенолы (особенно триалкил и многоатомные фенолы и а- и
Р-нафтолы) легко окисляются с образованием разнообразных
продуктов окисления. Легкая окисляемость дает возможность
использовать их в качестве антиокислителей (антиоксидантов).
Внесение небольших количеств антиоксндантов замедляет окисление
молекулярный! кислородом углеводородов, альдегидов и кетонов,
липидов н содержащих их продуктов. Антпокнслителы-гые свойства
фенолов связаны с их способностью реагировать с перекисньшн
111
радикалами R02 •, ведущими цепь окисления с ооразованием в
результате этого малоактивных радикалов, неспособных продолжать
окисление [ НО—<^ Ч—О-::
НО—^~^;-ОН+ RO2- ->ROOH + HO-/~S-O.
гидрохинон пере- малоактивный
кисный радикал
радикал
НО—/ >—O. + ROa--*iwwn-i-^—< >=
Антиокислители фенольнои природы широко применяются для
стабилизации бензинов, замедления старения каучуков. В пищевой
промышленности антиоксиданты фенольнои природы применяются
для сохранения жиров и масел, сухого молока, кондитерских
изделий, рыбных и мясных продуктов, пищеконцентратов. Наибольшее
распространение получили З-третбутил-4-оксианизол A), 3,5-дитрет-
бутил-4-окситолуол B) и нордигидрогваяретовая кислота C):
ОСН3 ОН
^-С(СН3K
он сн3
З-третбутил-4-оксианизол 3, 5-дитретбутил-4-окситолуол
A) ' B)
НО-/ ^—СН,—СН-СН—СН,-/ >-ОН
H°W ! ! * °н
СН3 СН3
кордигидрогваяретовая кислота C)
Отдельные представители. Фенол, оксибензол, карболовая кислота
С6Н6ОН. Бесцветное кристаллическое вещество с характерным
запахом, краснеет на воздухе вследствие окисления, вызывает
ожоги при попадании на кожу. Ядовит. Качественная реакция —
фиолетовое окрашивание с хлорным железом. Получение было
описано ранее (стр. 109). Одни из важнейших продуктов химической
промышленности, используется для получения феноло-форыаль-
дегидных смол, циклогексанола, пикриновой кислоты красителей,
лекарственных соединений, душистых веществ. Водные растворы
фенола применяются в качестве антисептика.
К резолы. Получаются из каменноугольной смолы. Применяются
для изготовления искусственных смол.
Ди- и трифвнолы, нафтолы и их производные. Гидрохинон и
пирогаллол применяются в фотографии. Пирогаллол используется
также при определении содержания кислорода в газовых смесях.
Нафтолы применяются для получения синтетических красителей.
ПРОСТЫЕ ЭФИРЫ
Простыми эфирами называются соединения с общей формулой
R — О — Rx, где R и Ri — углеводородные радикалы. Их можно
рассматривать как продукты замещения атома водорода в молекуле
спирта на радикал.
Номенклатура, изомерия. Для наименования эфиров могут
применяться тривиальные названия (серный эфир; фенетол), но в
большинстве случаев название эфиров складывается из наименования
радикалов с добавлением слова эфир (диметиловый эфир; диэтило-
вый эфир; метилэтиловый эфир). По ИЮПАК, для наименования
простых эфнров к названию углеводорода прибавляют приставку
«алкокси» (RO) или «арнлокси» (АгО). За основу соединения
выбирают радикал с большим числом атомов углерода:
СН,-О-СНз СН3-О-С2Н5
диметиловын эфнр метилэтиловый эфир
метокспмстан метоксиэтан
CjHs-O-QHb CgH5-O-C2H5
серный эфир, диэтнлоеый феиетол
эфир, зтокснзтан
Изомерия эфиров связана с составом и строением радикалов.
Способы получения. Межмолекулярная дегидратация спиртов
(стр. 97).
Взаимодействие алкоголятов с галогеналкилами:
С2Н5—ONa + I—С2Н5 -> С,Н5—О—С2Н5 + Nal
этилат натрия йодистый днэтпловый эфир
этил
Взаимодействие ацетилена со спиртами. Эта реакция была
разобрана ранее (стр. 51), она применяется для получения простых
алкилвиниловых эфиров.
Физические свойства. Первые два представителя: диметиловый эфир
СН3—О—СН3: метнлэтиловый эфир СН3—О—С2Н5 — газы, начиная с диэтило-
еого эфира — жидкости. Температура кипения эфиров ниже температур кипения
соответствующих спиртов, так как их молекулы не ассоциированы за счет
водородных связей. Они обладают приятным (эфирным) запахом, плохо растворимы
в воде, хорошо во многих органических растворителях.
Химические свойства. Простые эфнры устойчивы, обладают
слабой реакционной способностью. Они не гидролизуются, в
обычных условиях не вступают во взаимодействие с
металлическим натрием, на них не действуют щелочи и большинство
кислот.
Взаимодействие с йодистым водородом. При нагревании с
йодистым водородом простые эфиры разлагаются:
113
днэтилоЕЫй эфир этиловый иодистый
спирт этил
Присоединение сильных кислот:
cyv
с2н5.
>0 ¦ НС!
с2н/
Эти соединения можно рассматривать как соли оксония. В
молекуле оксониевых соединений атом кислорода связан с помощью
ковалентных связей с тремя заместителями и ионной связью с
анионом:
Ю: И С1"
хлористый
ДИЭТНЛОКСОИШ"!
Важнейшие представители. Диэтиловый эфир, этоксиэтан,
серный эфир С2Н3 — О — С2Н5. Бесцветная подвижная жидкость
с эфирным запахом, очень горюч. Широко применяется в качестве
органического растворителя, обладает анестезирующим действием.
Простые эфнры фенолов: С6Н5 — О — СН3 — анизол; C6HS —
— ОС2Н5 — фенетсл, высококипящие жидкости с приятным запахом,
применяются в парфюмерной промышленности,
Диоксан — простей внутренний эфир этиленгликоля. Получение
(стр. 106). Жидкость, Применяется в качестве органического раст-
'впрителя,
эпоксиды
Эпоксидамп, или эпокисями, называются простые эфиры
циклического строения с «-окисным кольцом:
>С С.
R/ 4R3
эттоксид (эпокись)
Отличаются высокой реакционной способностью. Наиболее важный
представитель эпоксидов — окись этилена.
Окись этилена СН,—СН3. Летучая жидкость со специфическим
запахом. Хорошо растворима в воде, огнеопасна. Получают прямым
окислением этилена в присутствии серебряного катализатора:
2СН;=СН,-!-О3 -> 2СН2—СН-3
\о/
Вырабатывается в больших количествах и широко используется
в разнообразных синтезах (получение антифризов, моющих
средств, пластических масс, эмульгаторов). Применяется в качестве
пестицида (химические средства для борьбы с вредными организмами).
14
ОРГАНИЧЕСКИЕ ПЕРЕКИСНЫЕ СОЕДИНЕНИЙ
При замещении атомов водорода в молекуле перекиси водорода
Н—О—О—Н на углеводородные радикалы получаются
гидроперекиси (замещен один атом водорода) и перекиси (замещено два
атома водорода):
К_О—О—Н R—О—О—R
гидроперекись перекись
Перекиси получаются при аутоокислении углеводородов, реакция
протекает по сзободнорадикальному маханизму. Гидроперекиси
обладают кислотными свойствами, при взаимодействии с растворами
щелочей образуют соли. Перекиси и гидроперекиси относятся
к сильным окислителям.
ОКСОСОЕДИНЕНИЯ
(АЛЬДЕГИДЫ И НЕТОНЬ!)
Альдегидами и кетонами называются соединения, содержащие
в молекуле оксо-, или карбонильную, группу > С = 0. В молекуле
альдегидов атом углерода карбонильной группы связан с углеводо-
родным радикалом и атомом водорода R—С\ , в молекуле кето-
О
I!
нов с двумя радикалами R—С—R:. Содержащаяся в молекуле
альдегидов группа — C-f получила название альдегидной группы
Н
и часто рассматривается как функциональная группа; группа ^> С =
= О, называемая кето-группой, является функциональной группой
кетонов.
В зависимости от характера углеводородных радикалов,
входящих в молекулу альдегидов и кетоноз, различают предельные,
непредельные, циклические и ароматические оксосоединения.Кето-
ны могут быть, кроме этого, смешанного типа: радикалы, с кото-'
рыми связана кетогруппа, могут принадлежать к разным классам
углеводородов. По количеству карбонильных групп различают
шнокарбонильные, дикарбонильные и поликарбонильные
соединения:
О
СНз—Cf СН3-С-СНз CH2=C-Gf
ацетон, пропанон I п
уксусный альдегид, ^^3
этаналь метакриловый
альдегид
с6н5-с< с6н5-с-с6н3 с6н5-с-сн3
бензофеион мезилфенилке^он,
бензойный альдегид ацетофенон
115
Номенклатура. Изомерия. Альдегиды часто называют по
наименованию кислот, в которые они превращаются при окислении. По
рациональной номенклатуре для наименования кетонов называют
углеводородные радикалы и добавляют слово кетой. По ИЮПАК,
в основе названия альдегидов и кетонов лежит название
углеводорода с тем же числом углеродных атомов, в случае альдегидов к нему
прибавляют окончание «аль», кетонов ¦— «он» с указанием номера
углеродного атома, связанного с кислородом.
Изомерия альдегидов определяется строением углеводородных
радикалов, кетонов — строением радикалов и положением кето-
группы:
СН3-СН2-СН2-с/ СН3—СН2—СН2—С—CHS
\Н 5 4 3 2 1
„ ыетилпропилкетон, 2-пентанон
масляный альдегид, бутаналь
О
р II
СНз—СН—Of СНз—СН2—С—СН2—СНз
I диэтилкетон, 3-пентанон
СН3
изомасляный альдегид,
ди мети л уксусный
альдегид, метилпронаналь
О
СН3—СН—С—СНз
I
СН3
метил изопропилкетон,
З-четнл-2-бутанон
Предельные альдегиды и кетоны
Наиболее важные представители альдегидов и кетонов
приведены в табл. 15.
Способы получения. Окисление спиртов (стр. 98).
Дегидрирование спиртов (стр. 98).
Разложение солей органических кислот. При сухой перегонке
кальциевых и бариевых солей образуются кетоны (если соль
муравьиной кислоты — альдегид);
R—с/ О
}Са-н> R—С—R+CaCO3
О
|! О
R-C-0 ||
>Са -> R—С—Н +СаСО3
н-с-о/ альдегид
о
116
Таблица 15
Наиболее важные представители альдегидов и кетонов
Название
Муравьиный альдегид,
формальдегид
Уксусный альдегид,
ацетальдегид
Пропионовый альдегид,
пропаналь
Масляный альдегид,
бутаналь
Бензальдегид
Ацетон, пропанон
Циклогексанон
Бензофенон
Метилфенилкетон, аце-
тофенсн
Формула
/О
Н-С^
/°
сн3-сн2-с<°
СН3-(СН2J-С^
СНз-С-СНз
li
о
ч /=0
о
QH6C-C6H5
О
II
Т. пл., °С
—92
—122,6
—81
—26
—95,3
—31,2
19,6
Т. кип., °С
—19,2
20,8
49,1
75
178,1
56,1
155,4
307
202,0
Строение образовавшегося кетона зависит от строения кислоты:
если кислотные остатки одинаковы, образуется симметрично
построенный кетон, если соль двухосновной кислоты — циклический,
если смешанная соль — то образуется кетон с двумя различными
радикалами:
О
, СН3—С—СНз + СаСОз
ацетон
О
сн3-с-ох
С"з С О
\\
О
уксуснокислый
кальций
Кетоны и уксусный альдегид могут быть также получены при
пропускании паров кислот (температура 400—500° С) над катализато-
117
рами, содержащими окислы кальция, марганца, цинка:
СНаСООН + НСООН "~§
уксус-пая ыуравьи-
кислота ная
кислота
< I
О
5<Хг С
2СН3СООН
уксусная
кислота
Гидролиз дигалогенопроизводных, При гидролизе дигалогено-
производных, содержащих два атома галогена у крайнего
углеродного атома, образуются альдегиды, у других атомов углерода —¦
кетоны:
Г /0Н1
СН3-СНС1г + 2Н2О -згнсГ Снз -С?- Н
j XOHJ
I, 1-дихлор-
этан
a ci
\/
2, 2-дыхлорпро-
пан
— 2 на
¦ он он
\/
сн.-с—сн<
— Н2О
н
ацетольдегпд
О
йцегои
Получение уксусного альдегида и кетонов из ацетилена и его
гомологов. Реакция Кучерова (стр. 51).
Оксосинтез. Альдегиды могут быть получены в результате
присоединения СО и На к олефинам. Реакция протекает в присутствии
никелевых и кобальтовых катализаторов при 100—200° С и 100—
250 атм (98 ¦ 105 — 245,2 • 105 Н/м2):
R-CH=CH2-fCO + Ha—
-R—СНо—СН,—с/
ЧН
альдегид нормального
строения
-R-CH—СН3
альдегид
изостроения
В реакцию вступают все олефины. Это важный промышленный
способ получения альдегидов, дающий возможность использовать
дешевое сырье — продукты химической переработки нефти.
Для получения ароматических альдегидов и кетонов, кроме
многих из приведенных выше способов, применяются специфические
методы. Мы остановимся на двух наиболее характерных.
118
Прямое введение альдегидной группы (реакция Гаттермана
Коха);
СН, СН3
! ' ' I
/
V N -
тс-луол ' //
п-толуи-
ловын
альдегид
Получение ароматических кетонов по реакции Фриделя — Краф-
тса;
О
|+С1-с-сн3 —С-Ь
бензол хлористый метилфенилкетон
ацетил
Физические свойства. Температуры плавления и кипения наиболее важных
представителей альдегидов и кетонов приведены в табл. 15.
Муравьиный альдегид — газ, остальные альдегиды и кетоны — жидкости
или твердые вещества со специфическим запахом, причем альдегиды обладают
более резким запахом, чем кетоны. Альдегиды и кетоны легче воды, многие
из них хорошо в ней растворимы.
Химические свойства. Альдегиды и кетоны принадлежат к числу
наиболее реакционноспособных органических соединений, причем
альдегиды химически активнее кетонов. Высокая активность оксо-
соединений связана с наличием в их молекуле карбонильной группы
и особенностями ее строения. В карбонильной группе связь между
атомами углерода и кислорода осуществляется двумя парами
электронов и состоит из о- и я- связей. Вследствие большей
электроотрицательности атома кислорода электронная плотность я-связн
смещена в его направлении:
Дипольный момент карбонильной группы 2,7D. В результате этого
у атома кислорода карбонильной группы создается повышенная
электронная плотность, у атома углерода ее недостаток. Атом
углерода карбонильной группы приобретает электрофильные
свойства и активно взаимодействует с нуклеофильными реагентами.
Наоборот, атом кислорода становится нуклеофильным, поэтому
способен взаимодействовать с электрофильными реагентами.
Очевидно, введен-не в углеводородный радикал заместителя,
снижающего электронную плотность у атома углерода карбонильной группы,
увеличивает его способность взаимодействовать с нуклеофильными
119
реагентами и, наооорот, заместители, повышающие электронную
плотность у углеродного атома, уменьшают эту способность. Именно
этим объясняется большая реакционная способность альдегидов и
кетонов алифатического ряда по сравнению с оксосоединениямн
ароматического ряда.
На реакционную способность оксогруппы влияет не только вид
заместителей, но и их объем. С увеличением объема заместителя
реакционная способность альдегидов и кетонов снижается, именно
поэтому альдегиды, карбонильная группа которых связана с одним
радикалом и атомом водорода, обладают большей реакционной
способностью, чем кетоны.
Для альдегидов и кетонов характерны следующие виды реакций:
присоединения, замещения, окисления, конденсации и
полимеризации.
Реакции присоединения. В реакциях присоединения
атом углерода карбонильной группы взаимодействует с нуклеофиль-
ным реагентом, двойная связь разрывается с образованием
аниона, вступающим на втором этапе реакции во
взаимодействие с протоном:
-в О-
+6 „О I
СН3— С( 4-: C=N ^ СНз—С—Н
?ХН ! I
; ; c=n
уксусный
альдегид
о- он
I !
сн3—с—н+н+^сн3-с-н
C=N C=N
нитрил а-оксн-
ПрОПИОНОЕОЙ
кислоты
Примером реакций присоединения могут служить реакции
взаимодействия оксосоединений с водой, водородом, бисульфитом натрия
и др.
Восстановление альдегидов и кетонов. При восстановлении
альдегидов образуются,первичные спирты, кетонов — вторичные
,0
СНз—Cf +Н2^СН3-СН,ОН
\it
п этиловый спирт
о он
II [
СН3—С—СН3 + Н2 -> СНз—СН— СНз
ацетон изопропиловый
спирт
Реакции протекают в присутствии никеля, кобальта, платины,
палладия.
120
Присоединение бисульфита натрия:
-6 ONa ОН ONa
-Н5/О I I I
сн3—с/ + s—он ;=•: сн3—с - s=o
4H I! j !]
о но
Из кетонов вступают в эту реакцию только соединения типа
О
сн3—с—R. Реакция применяется для выделения и
количественного определения оксосоединений. В пищевой промышленности
этим методом устанавливают содержание оксосоединений в
веществах, обусловливающих вкус и аромат пищевых продуктов.
Присоединение металлорганических соединений (стр. 163).
Действие аммиака:
NH3 HNH NH
/Р I _ I II
СН3—Of + :NH3 7Z CH3-C-O ^ CH3—С—OH -» CH3-CH + H2O
ацетальдимин
H
Полученный продукт полимеризуется:
сн3
i
сн
/\
HN NH
I I
3CH3-CH=NH -> HSC—НС СН-СН3
\/
NH
альдеги дамми ак
Присоединение спиртов. При взаимодействии альдегидов со
спиртами образуются ацетали, которые можно рассматривать как
простые эфиры, образованные диолом, содержащим две гидроксиль-
лые группы у одного углеродного атома, и двумя молекулами спирта.
В качестве промежуточного продукта образуется полуацеталь:
ОН О—С,Н5
/^О I СНьОН I -
хн " 1 " i
H H
полуацеталь ацеталь
Реакция протекает легко, при небольшом нагревании, в
присутствии минеральной кислоты. Для получения аналогичных
производных кетонов — кеталей применяются специальные методы.
Ацетали — приятно пахнущие бесцветные жидкости, хорошо
растворимые во многих органических растворителях и плохо растворимые
в воде. Они играют большую роль в процессах биосинтеза сложных
органических соединений, участвуют в формировании запаха ряда
пищевых продуктов. Состав ацеталей вина существенно влияет на
его букет.
121
Реакции замещения. В реакциях замещения
альдегиды и кетоны обменивают атом кислорода на другие атомы.
Взаимодействие с пятихлористым фосфором:
^ 1,1лихлор-
ацетсль^егид этан
О С1 С1
il \/
СН3—С—CH3-j-PCl5 -> СНз—С—СН3 + РОС13
ацегон 2,2-дихлор-
ПрОПаН
Взаимодействие с гидроксиламином:
CH3-C<f + H.2N - ОН -? СН3-С= N-OH 4 Н„О
ги^роксн- I
лаыин Н
альдоксим
О К-ОН
СНз—С—СН34- H2N-OH -> СЫз—С-СН34- Н2О
ке токе им
В реакции применяется солянокислый гидроксиламин (NH3OH) С!
и выделяющаяся соляная кислота может быть оттитрована. Это
дает возможность использовать ее для количественного определения
альдегидов и кетонов.
Взаимодействие с гидразином и его производными.
Взаимодействие одной молекулы гидразина (H2N — NH2) с альдегидами и кето-
нами приводит к образованию гидразонов и далее молекулы азпков:
о
0 сн3-с/
СНз-с/ ¦+ H2N-NH2 ^q CH3-C--= X-NHa ~ТГ^"
Н I
н
гидразон
-> СН3—C=N—N=^C—СН3
k A
альдазин
О о
СГ! 3_с-СН3 +HsN-NHn^j^o CH3-C=N—NH2 —i-i~'-^
СИ3
гндразон
-> СНа—C=N—N=C—CH3
СН.з CPi3
кетазпн
Из производных гидразина для реакции с альдегидами и кетонами
используют фенплгидразин С6Н3 — NH — МН2, 2,4-дннитрофенил-
122
гидразин (OaNJ C6H3 — NH — NH2, семикарбозид HaN—NH-C
3
Образующиеся продукты являются кристаллическими
веществами с характерными температурами плавления, что дает возможность
использовать эту реакцию для выделения, очистки и разделения
альдегидов и кетонов.
Окисление альдегидов и кетонов.
Взаимодействие со слабыми окислителями. Альдегиды легко окисляются даже под
действием слабых окислителей, при этом образуются одноосновные
органические кислоты с тем же числом атомов углерода:
Р
+ 2 [Ag (NH3)J OH -» CH3-C( + 4NH3 + H2O + 2Ag
X)H
ацетальдегид
уксусная
кислота
Образовавшееся при этом серебро тонким слоем покрывает стенки
колбы, поэтому окисление альдегидов аммиачным раствором окиси
серебра получило название «реакции серебряного зеркала». Она
используется для изготовления елочных украшений и зеркал,
является качественной реакцией на альдегиды. Кроме аммиачного
раствора окиси серебра для окисления альдегидов применяется
фелингова жидкость (реактив Фелинга) (стр. 233).
Окисление кетонов протекает с трудом, под действием сильных
окислителей, при нагревании и сопровождается разрывом
углеродной цепи по обеим сторонам от карбонильной группы с
образованием, в общем случае, смеси из четырех кислот (правило Е. Е.
Вагнера и А. И. Попова):
О
II 0
СН3—СН2-С—СН2-СН,-СН3—СН3 —
3-гептанон
ОН
уксусная
кислота
¦ CH3-CH3-C(
XOH
пропионовая кислота
. СНз-СН2-СНа-С<
масляная кислота
чон
валериановая кислота
По образовавшимся продуктам реакции можно судить о строении
кетона.
Реакция Канниццаро. Молекула ароматического альдегида в
присутствии концентрированных щелочей может окисляться за счет
123
другой молекулы альдегида, которая при этом восстанавливается
до спирта:
C6H5-Cf + CeH3—Ccf + КОН-» C6H5Cf +СвН— СН2ОН
\н хн хж
бензойный соль бензой- бензиловый
альдегид ной кислоты спирт
Реакция Тиищнко. Алифатические альдегиды под действием
этилата алюминия в неводной среде образуют сложные эфиры:
О
/ С^ <с=н5-°>з ^СН3—С—О—СИ,—СНз
ацетальдегнд уксусноэтиловый эфир
Реакции полимеризации и
поликонденсации. Полимеризация альдегидов и кетонов. Альдегиды под влиянием
щелочей и карбонатов щелочных металлов способны полимеризо-
ваться за счет подвижных атомов водорода в сс-положении. На
первом этапе реакции молекула альдегида отщепляет протон,
превращаясь в карбокатнон, который вступает в реакцию нуклеофильного
присоединения со второй молекулой альдегида;
НСН2—С + ОН~ —- :СН2—С + Н90
Х ¦ \ '
я-положение
р _ О 9~ О
СН3—С + :СН2—С —*¦ СН,—С— СН2—СУ
\ \ I \
н н ^ н
f о ?н о
сн3—с—сн2—сх + н+ —- сн3—с—сщ—сх
н н А Хн
альдоль
Образовавшийся димер содержит две функциональные группы и
//°
поэтому получил название альдоль (—С^ — аль: ОН — ол),
а вид полимеризации — альдольной полимеризации. В 1861 г.
с помощью альдольной полимеризации А. М. Бутлеров
синтетическим путем впервые получил соединение, относящееся к классу
углеводов:
Н\Г Ca(OH)z г „ п
формальдегид гексоза
124
Низкомолекулярыые кетоны также способны вступать в реакцию
полимеризации:
0 0 ОН О
II II ! 11
СНз-С + НСН2—С—СН3 -> СН3-С-СН2—С—СН3 ^-д
СН3 СН3
ацетон 4-окси-4-метил-2- пентанон
-» СНз—С=СН— С—СН3
I II
СНз О
окись мезитила
Кретоновая конденсация. Взаимодействие альдегидов при
нагревании может идти по типу поликонденсации с потерей молекулы
воды. По названию образующегося при этом простейшего продукта
этот вид конденсации получил название кротоновой конденсации:
СН3—С( +СН3—Cf -> СН3—СН=СН—Of
хн чн чн
формальдегид кротоновый альдегид
Взаимодействие альдегидов с фенолами. Фенол-формальдегидные
смолы. В кислой и щелочной среде альдегиды вступают во
взаимодействие с фенолами с образованием фенолоспиртов:
ОН ОН ОН
\ I I
/\—СН,ОН сн2о /\ +2сн2о /\-СН2ОН
/
4, ЗСН,0
ОН " СН.,ОН
нон2с—f\—снаон
CH2OH
Группа ОН — заместитель 1-го рода, поэтому молекула
формальдегида направляется преимущественно в орто- и пара- положения.
При нагревании фенолоспирты конденсируются с образованием,
в зависимости от условий, различных продуктов:
ОН ОН ОН
/N—СН,ОН , ^\—СН.2ОН , + j^—СНаОН ^
Ч/ '"V "ЛНг0
он он
125
Продукт с молекулярной массой до 1000, называемый ризолом,
используют в качестве лаков и для приготовления пресс-порошков,
в состав которых входят разнообразные наполнители: древесная
мука, стеклянное волокно, графит и другие материалы. После
тщательной обработки (прессования) получают готовый продукт,
обладающий в зависимости от наполнителя и условий обработки
различными свойствами.
Отдельные представители. Применение. Муравьиный альдегид
//°
формальдегид, метаналь Н—С<^ . Бесцветный газ с резким
запахом. Хорошо растворим в воде. 40%-ный водный раствор
называется формалином. Он ядовит. Формальдегид, полимеризуясь
с аммиаком, образует уротропин C6H]2N4. В промышленности
формальдегид получают неполным окислением метана при
температуре 600° С
(окислители — окислы азота) и окислительным дегидрированием
метилового спирта в присутствии металлических (медь, серебро) или
окисных катализаторов:
СНЧОН + 0,50. н> H-Ccf7 + Н2О
\н
Формальдегид — один из важных продуктов органического
синтеза, применяется для получения полимерных материалов,
мономеров каучука, диолов, фармацевтических препаратов.
Формалин используется для протравливания семян перед
посевами, для дубления кожи, хранения анатомических препаратов.
Уротропин применяется в качестве лекарственного вещества и для
консервирования икры.
,р
Уксусный альдегид, ацетальдегид, этаналь СН3—С/ . Лету-
н
чая жидкость с резким запахом. Смешивается с водой и многими
органическими растворителями во всех отношениях. Ядовит.
Получают по реакции Кучерова (стр. 51), дегидрированием этилового
спирта (стр. 98), окислением этилена:
/<°
СН2=СН„ + 0,5О, -н> СН3—С<
хн
Катализатор — раствор хлорида меди и хлорида палладия. Уксус-
ный альдегид полимеризуется в тример 'СН3—G^ ;. Применяется
для получения пластических масс, уксусной кислоты, уксусного
ангидрида, этилового спирта, молочной кислоты и других
продуктов.
126
Бензойный альдегид, бензальдегид С6Н5—С< . Маслянистая
жидкость с запахом горького миндаля. Получают окислением
толуола в присутствии окислов металлов:
/у сна о^. с fy с<н + н*°
толуол бензойный альдегид
или омылением хлористого бензилидена С6Н5СНС12. Применяется
для синтеза красителей.
О
Ацетон, диметилкетон, пропанон CHS—С—СН3. Бесцветная
жидкость со специфическим запахом. С водой и многими
органическими растворителями смешивается во всех отношениях. Горюч.
Получается при сухой перегонке древесины, ацетонобутиловьш
брожением углеводов (стр. 237). Наиболее экономически выгодные
способы: кумольный (стр. 109), дегидрирование изопропилового
спирта (стр. 98). Ацетон —¦ один из важнейших органических
растворителей, широко применяющийся во многих отраслях
промышленности. Большая реакционная способность ацетона дает
возможность широко использовать его в разнообразных синтезах.
Циклогексанон \/~°- Применяется для получения капро-
лактама (полупродукт при производстве капрона) и адипиноаой
кислоты (стр. 141),
О
||
Метилфенилкгтон, ацетофенон СН3—С—С6Н5. Применяется
в пищевой промышленности для ароматизации мыла.
Ванилин НО—-^ V-C/ . Это соединение — производное
6~сн3
фенолов и ароматических альдегидов. Применяется в качестве
ароматизатора при производстве кондитерских изделий,
безалкогольных напитков.
Ненасыщенные карбонильные соединения.
Кетены
В молекуле ненасыщенных оксосоединений присутствуют две
О
реакционноспособные группировки: карбонильная группа —С—
и двойная связь >С=С<. Эти группировки и их взаимное
положение и определяют химическое поведение этих соединений.
Две ненасыщенные связи >С=С< и >С=О могут находиться
127
у одного углеродного атома >С—С—О (кумулированные связи),
могут быть разделены простой связью >С=С—-С=О
(сопряженные связи) и, наконец, могут быть удалены друг от друга.
Представителем первой группы является кетен СН.2=С=О, второй —
акролеин СН2=СН—С <^ . Представители третьей группы обладают
всеми свойствами оксосоедпнений и алканов.
Акролеин СН2=СН—Of , Бесцзетиая жидкость с резким,
ХН
раздражающим запахом, tKhI1 = 56° С, tn:, = —87,7° С. Ограниченно
растворим в воде. Акролеин может быть получен методом альдольнои
конденсации при температуре 300е С в присутствии силиката
натрия, нанесенного на силикагель:
нсно+сн3—c
мальдегид
\
н
ацеталь-
дегид
2—сн2
р-оксипропионовый
альдегид
-- сн.,=сн— ci
акролеин
При каталитическом окислении пропилена также образуется
акролеин:
/°
СНо=СН—СН3 -f Оо -> СН ,= СН— С/ + НоО
\н
Применяется для получения глицерина, акрилонитрила С?1,—
=СН—CN.
/Се/иен СН.,=С=О. Газ, ядовит, t&aa = — 4Г С, ^пл= — 134,6е С.
Получается пиролизом ацетона:
О
il
СНС
-» СН2=С=ОН-СН4
Применяется для получения уксусной кислоты, уксусного
ангидрида, сорбиновой кислоты (стр. 137) и других продуктов:
сн.2=с=о_
СНз-СООН
уксусная
кислота
сн3соон
уксусный
ангидрид
СН3—СИ=СН—С(
/-0
¦ сн,—сн=сн=сн^=сн—соон
сорипновая кислота
ОРГАНИЧЕСКИЕ, ИЛИ КАР5ОНО6ЫЕ, КИСЛОТЫ
Органическими, или карбоновыми, кислотами называются со-
°
единения, содержащие в молекуле карбоксильную группу
Карбоксильная группа определяет основность кислоты, поэтому
в зависимости от количества карбоксильных групп различают
одноосновные (одна карбоксильная группа), двухосновные (две группы)
и многоосновные кислоты. Карбоновые кислоты бывают
алифатические (насыщенные и ненасыщенные), карбоксильная группа в
которых связана с алифатическим радикалом, алициклические,
ароматические и т. д.
ОДНООСНОВНЫЕ НАСЫЩЕННЫЕ И АРОМАТИЧЕСКИЕ
КАРБОНОВЫЕ КИСЛОТЫ
Общая формула одноосновных насыщенных карбоновых кислот
Р °
R—С<Г . ароматических Аг~с\^ - где R и Аг —
углеводородные радикалы.
Старое название алифатических карбоновых кислот — жирные
кислоты или кислоты жирного ряда, так как многие представители
этой группы соединений впервые были выделены из жиров (стр. 200)-
Важнейшие представители приведены в табл. 16.
Таблица 16
Наиболее важные представители одноосновных карбоновых кислот
Название
Муравьиная кислота,
метановая
Уксусная кислота, эта-
новая
Пропионовая кислота,
ГТП1АП!111ЛВЧA
lipuildnUbd л
Масляная кислота, бу-
тановая
Валериановая кислота,
пентановая
Капроновая кислота,
гексановая
Лауриновая клслота,
додекановая
Пальмитиновая
кислота, гексадекановая
Стеариновая кислота,
окгадекановая
Бензойная кислота
Формула
н—соон
сн3—соон
сн3—сн,—соон
СН3-(СН,J-СООН
СН3-(СНоK-СООН
CH3-(CH.,)j-COOH
СН3—(СН.Ьо—СООН
СН3—(СЩц—СООН
СН3—(СН2I6—СООН
QH^COOH
Т. пл.,
°с
8,4
16,6
—22,0
— 8,0
—34,5
—3,9
44,2
64,0
69,4
122,0
Т. кип., °С
100,7
118,1
141,0
163,5
185,0
205,8
298,9
390,0
(с разложением'
360,0
(с разложением'
249,0
Изомерия. Номенклатура. Изомерия карбоновых кислот
определяется строением углеводородного радикала. Для наименования
5 А. П. Нечаев
129
карооновых кислот широко применяются тривиальные названия,
которые часто связаны со старыми способами получения кислот:
муравьиная, уксусная, стеариновая. По ИЮПАК, в основе
наименования кислот лежит название углеводорода с тем же числом
атомов углерода, к которому прибавляется окончание «овая;>,
нумерация начинается с атома углерода карбоксильной группы:
СНо—СН3— СН2—СООН
СН3
3-метплбутановая
кислота
Нахождение в природе. Способы получения. Карбоновые кислоты
широко распространены в природе как в свободном состоянии, так
и в виде структурных компонентов многих природных органических
соединений. Несмотря на это, получение карбоновых кислот из
природных источников большого практического значения не имеет.
Основное их количество получают синтетическими методами.
Окисление органических соединений. Окисление алканов, алкенов,
ароматических углеводородов, спиртов, альдегидов с образованием
кислот было подробно разобрано ранее. Эти методы имеют
большое практическое значение и широко применяются в технике
и лабораторной практике для получения карбоновых кислот.
Оксосинтвз. Карбоновые кислоты могут быть получены при
взаимодействии алкенов с окисью углерода и водой:
R—СН2-СН=СН2 + СО + Н2О—
-> R—СН2—СН2—СН2—СООН
СООН
!
-> R—СН.,—СН—СН3
Реакция протекает при температуре 50—100° С, давлении до 100 атм
(98-Ю5 Н/м2), катализатор — сильные кислоты (H2SO4( H3PO4).
Промышленное получение карбоновых кислот оксосинтезом
может быть осуществлено и в две стадии: синтез альдегидов из
алкенов, окиси углерода и водорода (стр. 118) и последующее окисление
их в кислоты.
Гидролиз нитрилов. При нагревании нитрилов (R—C=N)
с водными растворами щелочей или кислот они гндролизуются,
образуя кислоты и аммиак:
ООО
СНз—C=N -22 СН3—С—NH, -'° СНз-С—ONH4nCH3-C—OH + NH3
ацетонитрил ' алшд уксусной
кислоты
Гидролиз тригалогвнопроизводных ароматических углеводородов,
содержащих три атома галогена у одного углеродного атома:
130
C6H5CCI3-j-2H(
бензотри-
хлорид
Ж -> С6Н5СООН
бензойная
кислота
Гидролиз триглицеридов (стр. 201).
Получение из металлорганических соединений (стр. 163).
Декарбоксилирование двухосновных кислот ^(стр. 140).
Физические свойства. Первые три кислоты (Сх—С3) — подвижные жидкости
с резким запахом, начиная с С4 (масляная кислота) — маслянистые жидкости
с крайне неприятным запахом (запах прогорклого сливочного масла). С С10 —
твердые вещества без цвета и запаха. Нпзкоыолекулярные кислоты хорошо
растворимы в воде, с увеличением молекулярной массы растворимость резко
падает. На физические свойства карбоновых кислот большое влияние
оказывает их способность к ассоциации с образованием линейных или циклических
дилеров за счет водородных связей. Энергия водородной связи 7 ккал/моль
B9,3-103 Дж/моль)
R R
циклический димер ^ ^ " ^ *¦' "
линейный димер
Способностью к ассоциации и прочностью водородных связей и объясняются
относительно высокие температуры кипения кислот по сравнению не только
с углеводородами, имеющими столько же атомов углерода в молекуле, но и
со спиртами.
Химические свойства. Химическое поведение кислот в первую
очередь связано с наличием в их молекуле карбоксильной группы,
а также со строением углеводородного радикала. Рассмотрим
распределение электронной плотности в карбоксильной группе.
Электронная плотность it-связи в группе (> С=О) смещена в сторону
атома кислорода. Вследствие этого у карбонильного углерода
создается недостаток электронной плотности, и он притягивает
к себе неподеленные электронные пары атома кислорода гидроксиль-
ной группы, в результате чего электронная плотность О—Н связи
смещается в сторону атома кислорода, водород становится
подвижным и приобретает способность отщепляться в виде протона:
—С-г-О—Н
К?
Следовательно, кислотные свойства соединений, содержащих
карбоксильную группу, объясняются сопряжением двойной связи
карбонильной группы с неподеленной электронной парой атома
кислорода гидроксильной группы, приводящим к подвижности
атома водорода.
Одноосновные алифатические карбоновые кислоты, за
исключением муравьиной, относятся к слабым кислотам. С увеличением
числа атомов углерода в радикале кислоты константы диссоциации
кислот уменьшаются. Введение в радикал вместо атома водорода
электроноакцепторных группировок, особенно в а-положение,
уменьшает электронную плотность у атома углерода карбонильной группы,
что приводит к увеличению силы кислоты. В свою очередь влияние
5* 131
карбоксильной группы приводит к повышению подвижности атомов
водорода у а-углеродного атома:
Н
//°
| ^ОН
н
Диссоциация карбоновых кислот. В водных растворах карбоновые
кислоты диссоциируют:
О
Л ¦ || _
СНз-Gf +H3O-CH3-C-O+HJO
уксусная
Образование солей:
2СН3СООН + NaOH -> CH3COONa + Н,0
2СН3СООН + Са -> (СН3СОО)8 Са + Н2
Соли карбоновых кислот металлов первой и второй групп легко
гидролизуются, растворы их имеют щелочную реакцию.
Электролиз солей жирных кислот (стр. 27) и образование алканов
при сплавлении солей со щелочами разобраны ранее (стр. 28).
Образование галогенангидридов. При действии на кислоты гало-
генидов фосфора или тионилхлорида образуются галогенангидриды
кислот (стр. 143):
-Р /Р
СН3-С< + SOCL, -> СНз-Cf + НО + SO2
VOH " ХС1
уксусная хлорангидрнд
кислота уксусной
кислоты
Действие галогенов:
СН3—COOH + Cl2 ->CH2C1—COOH+HCI
уксусная монохлоруксус-
кпелота ная кислота
При избытке хлора могут последовательно замещаться все три атома
водорода с образованием трихлоруксусной кислоты (СС13СООН).
Введение хлора (электроноакцепторного заместителя) резко
увеличивает силу кислоты.
Отдельные представители. Муравьиная, метановая кислота
Н—СООН. Содержится в соке крапивы, в выделениях муравьев,
хвое, фруктах. Жидкость с резким запахом, при попадании на кожу
вызывает ожоги (крапивные ожоги). Относится к средним по силе
кислотам. Основной технический способ ее получения —
пропускание окиси углерода через увлажненный едкий натр:
СО + NaOH i201~'a!r°M'S- HC()ONa
2HCOONa + H,SO4 -> 2HCOOH + Na2SOi
Муравьиная кислота вступает во все реакции, характерные
для карбоновых кислот, в то же время обладает рядом особенностей
в своем химическом поведении, связанными с отсутствием в ее
молекуле углеводородного радикала. В отличие от других карбоновых
кислот она легко окисляется с образованием воды и двуокиси
углерода:
Это дает возможность применять ее в качестве восстановителя.
В пищевой промышленности это свойство муравьиной кислоты
используется при получении никелевых катализаторов, в частности
для гидрогенизации жиров (стр. 215):
(НСООJ N i ^~-$ N i + СО2 + Н2
При нагревании с концентрированной серной кислотой
муравьиная кислота разлагается:
Муравьиная кислота применяется в текстильной
промышленности для протравливания тканей при крашении, при дублении
кожи, в пищевой промышленности — для дезинфекции бродильных
чанов и бочек, в медицинской промышленности — для получения
«муравьиного спирта», в химической промышленности — для
синтеза щавелевой кислоты.
Уксусная, этановая кислота СИ3—СООН. Бесцветная жидкость
с резким специфическим запахом. Безводная уксусная кислота
называется «ледяной», так как при замерзании (+ 16,6° С) внешне
она напоминает лед. Уксусная кислота смешивается с водой и
многими органическими растворителями в любых соотношениях. Она
широко распространена в природе, как в свободном состоянии, так
и в виде производных (главным образом сложных эфиров).
Составляет до 85% всех органических кислот в зерне пшеницы и кукурузы.
Старый способ получения уксусной кислоты, известный людям
еще в глубокой древности, — уксуснокислое брожение водных
растворов этилового спирта. Он не потерял своего значения и в
настоящее время для получения «пищевой» уксусной кислоты. При
уксуснокислом «брожении» этиловый спирт окисляется в уксусную
кислоту с помощью особой группы микроорганизмов, называемых
уксуснокислыми бактериями. Суммарное уравнение реакции
«брожения» имеет следующий вид:
CH3-CH,OH-fO, -> СН3СООН + Н3О
Концентрация образовавшегося таким образом уксуса — 5—7%,
из него перегонкой выделяют 70—80%-ную уксусную эссенцию.
Для получения уксуса используют продукты брожения
виноградного и фруктового соков, пиво. Не потерял своего значения и
другой способ — сухая перегонка отходов деревоперерабатывающей
промышленности. Образующийся 'при этом наряду с древесным
133
дегтем водный слой содержит до 10% уксусной кислоты, откуда она
выделяется в виде уксуснокислого кальция. При обработке его
серной кислотой получается древесный уксус. Основное количество
уксусной кислоты вырабатывают, однако, синтетическим путем.
Основные пути:
а) каталитическое окисление ацетальдегида:
2СН3—С<( + Оа-> 2СН8—СООН
'* уксусная
ацетальдегид кислота
б) синтез из метанола и окиси углерода:
СН3ОН + СО -> СН3СООН
Катализатор — фтористый бор, фосфорная кислота, хлориды цинка
и меди. Температура реакции 200—300° С, давление до 1000 атм
(98 -10е Н/м2);
в) в СССР разработан процесс окисления н-бутана при
температуре 145° С и давлении 50 атм D9-10е Н/м2) с выходом уксусной
кислоты до 80 %.
Уксусная кислота — один из важнейших .химических продуктов,
применяется в качестве растворителя, при производстве
искусственных волокон (ацетатный шелк), красителей, сложных зфиров,
лекарственных препаратов. Широкое применение нашла уксусная
кислота в пищевой промышленности в качестве консервирующего
средства при производстве маринадов и как вкусовая добавка.
Масляная, бутановая кислота СН3—(СН2J—СООН. Жидкость
со специфическим запахом. Содержится в виде глицеридов в
сливочном и некоторых других маслах. Получают маслянокислым
брожением углеводсодержащего сырья и окислением н-бутилового
спирта.
Бензойная кислота СвН3—СООН. Типичный и наиболее важный
представитель одноосновных ароматических кислот. Белое
кристаллическое вещество. Распространена в природе в свободном состоянии
и в виде производных. Получают окислением толуола при 140° С
(катализатор — соли кобальта):
Лг+,2Л+Н2О
\/ \/
толуол бензойная
кислота
Степень диссоциации ароматических кислот выше, чем кислот
алифатического ряда; бензойная кислота значительно сильнее,
чем уксусная. Она вступает в те же реакции, что и уксусная, а
кроме того, в реакции, характерные для бензольного ядра.
Бензойная кислота и ее производные используются в синтезе
красителей, фармацевтических препаратов. Сложные эфнры бен-
134
зойной кислоты и одноатомных спиртов (от метилового до амилового)
идут на изготовление духов и одеколонов. Широкое применение
бензойная кислота и ее натриевая соль нашли в пищевой
промышленности для консервации пищевых продуктов. Именно присутствием
бензойной кислоты в мякоти клюквы, брусники, рябины (наряду
с некоторыми другими соединениями) объясняется их способность
к длительному хранению.
Одноосновные ненасыщенные кислоты
В молекуле одноосновного карбоновой кислоты может
содержаться одна или несколько двойных связей *. Общая формула
мононенасыщенных одноосновных карбоновых кислот СЛН2П_1СООН,
диненасыщенных СЛН2П.-3СООН и т. д. Важнейшие представители
этой группы соединений приведены в табл. 17
Таблица 17
Наиболее важные представители одноосновных ненасыщенных
карбоновых кислот
Название
Акриловая
кислота, пропеновая
Кротоновзя
кислота, 2-бутеновая
Метакриловая
кислота, метилпро-
пеновая
Олеиновая
кислота
Сорбиновая
кислота
Линолевая
кислота
Линоленовая
кислота
Формула
СН2=СН—СООН
сн3—сн=сн—соон
СН2=С—СООН
I
СН3
СН3—(СН,O—СН=СН—
—(СН2OСООН
сн3—сн=сн—
—сн=сн—соон
С17Н31СООН
С17НМСООН
Т. пл., °С
13
72
15
18,4
—5
— 11 до —Ц,3
Т. кип., °С
141
189
160
234 (при
100 мм рт. ст.
или 1333,2 Н/ы2)
144
202 (при
1,5 мм рт. ст.
или 19 998 Н/м2)
157 (при
0,001 мм рт. ст.
или 0,133 Н/м-2)
Изомерия. Номенклатура. Структурная изомерия одноосновных
ненасыщенных кислот связана со строением углеродного скелета
и положением двойных связей. У мононенасыщенных кислот
двойная связь может занимать различное положение по отношению
к карбоксильной группе, у ди- и полиненасыщенных одноосновных
кислот еще и различное положение по отношению друг к другу.
* Кислоты, содержащие в радикале тройные связи, в книге не
рассматриваются.
135
Для ненасыщенных кислот существует пространственная
(геометрическая, цис-, транс-) изомерия:
СНз—С-СНз СН3—С-Н
i! II
Н—С—СООН Н—С—СООН
1(яс-кротоновая пгранс-кротоно-
кнслота вая кислота
При наименовании ненасыщенных кислот широко применяются
тривиальные названия (акриловая, сорбиноваякислота).ПоИЮПАК
сохраняются те же принципы, что и при наименовании алкенов и
кислот:
СНз—СН =СН—СООН
2-бутеновая кислота
Способы получения. Для получения двухосновных кислот могут
быть использованы соединения, в молекуле которых уже имеется
карбоксильная группа. В этом случае ранее описанными методами
создается двойная связь, например:
а) отнятием галогеноводородов от галогенозамещенных карбо-
новых кислот
С1
снз-сн-соон спиртово1 сн„=сн-соон
р-р щелочи
сс-хлорпропионовая акриловая кислота
кислота
б) отнятием воды (дегидратация) от оксикислот
НО—СН2—СН2—СООН -> Н,О + СН.2=СН— СООН
р-оксипропионовая кнслога акриловая кислота
Если в молекуле имеется двойная связь, то создается
карбоксильная группа:
СН2=СН—СН3 -—*? СН2=СН—СН2С1 + НС1
пропен хлористый аллил
СН2=СН—СН2С1 + HCN -> СН2=СН—СНг—CN + НС1
нитрил 3-бухеновой кислогы
СН.,=СН—СНг—CN —° СН2=СН—СН2—СООН
3-бутеновая кислота
Физические свойства, Ненасыщенные кислоты — жидкости или твердые
вещества; низшие со специфическим запахом, растворимы в воде; высшие — без
запаха и не растворимы в воде.
Химические свойства. Введение в молекулу двойной связи
увеличивает силу кислоты, поэтому степень диссоциации
одноосновных ненасыщенных кислот выше, чем соответствующих насыщенных.
Влияние двойной связи снижается по мере удаления ее от
карбоксильной группы. Химическое поведение ненасыщенных
одноосновных кислот связано с наличием в их молекуле двух активных
группировок: карбоксильной группы и двойной связи. Они вступают
136
во все реакции, характерные для кислот (образование солеи,
сложных эфиров, хлорангидридов и т. д.), а за счет двойной связи —
в реакции присоединения, окисления, полимеризации.
Кислоты, в молекуле которых двойная связь находится, между
2 и 3 углеродными атомами, отличаются некоторыми особенностями
в своем химическом поведении за счет сопряжения между двойной
связью и карбоксильной группой. Например, при присоединении
галогеноводородов водород направляется к наименее гидрогенизи-
рованному атому углерода:
СН2=СН— С' + НС1 —- СН2С1—СН2—С
он чон
акриловая кислота 3-хлорпропановая кислота
Отдельные представители. Акриловая, пропеновая кислота
СН2=СН—СООН. Бесцветная жидкость с резким запахом.
Смешивается с водой во всех отношениях. Получают гидролизом акрило-
ннтрила
сн2=сн—cn—2сн2=сн—соон+ nh3
акрилонитрил акриловая кислота
и взаимодействием ацетилена с окисью углерода и водой
СН=СН + СО + Н2О -> СН,=СН—СООН
акриловая кислота
Широкое применение нашли эфиры акриловой кислоты,
полимеризацией которых получают ценные полимерные материалы
(органическое стекло).
Метакриловая, метилпропеновая кислота СН.-=С—СООН. Бес-
!
сн„
цветная жидкость. Получают в промышленности из ацетона:
CH3V СНзч .ОН „ .о СНзч -ОН
> >С< ^3 >С<" -й-й СНо=С—СООН
СН/ XCN СН/ ХСООН 2
СН,
ацетон ацетонциан- оксиизомасляная
гидрин кислота летакряловая
кислота
Большое техническое значение имеет метиловый эфир метакри-
ловой кислоты (метилметакрилат), полимеризацией которого
получают «органическое стекло» высокой твердости:
СН3
-СНо—С—
СООСНд
метилметакрилат полиметилметакрилат
Сорбиновая, 2,4-гексадиеновая кислота СН3—СН =СН—СН =
=СН—СООН. Кристаллическое вещество. Будучи совершенно без-
137
вредной, является отличным антисептиком. Применяется для
консервирования мясных и рыбных изделий, сыра, фруктовых
компотов, плодоягодных пюре, соков, овощей.
Олеиновая кислота СН,—(СН2O—СН=СН—(СН2O—СООН.
Маслянистая жидкость без цвета и запаха. Широко распространена
в природе, входит в состав компонентов почти всех жиров. Во
многих она присутствует в десятках процентов, а в некоторых является
основным компонентом. Так, в масле арахиса ее содержание
составляет 50—80%, в оливковом — 70—85%. Получают гидролизом
жиров (стр. 201) с последующим выделением ее из образовавшейся
смеси кислот. Олеиновая кислота является не только важнейшим
компонентом пищи, но и находит широкое применение в технике.
Олеиновая кислота используется в качестве пластификатора при
производстве пластмасс, для производства мыла, в аналитической
химии.
Линолевая кислота СН3—(СН2L—СН =СН—СН2—СН =СН—
—(СН2O—СООН. Светло-желтая маслянистая жидкость,
температура плавления —5,9—5,2°, кипения 202° (при 1,4 мм рт. ст.), и не
растворима в воде, смешивается с метанолом, диэтиловым эфиром во
всех отношениях. Очень широко распространена в природе, входит
в состав триглицеридов многих масел и жиров. Содержание ее в
подсолнечном, кукурузном маслах достигает 60—70%. Масла,
содержащие линолевую кислоту, применяются в производстве олиф,
красок и лаков. Принадлежит к числу незаменимых жирных кислот,
так как человек и большинство животных лишены способности ее
синтезировать. Поэтому для полноценного питания линолевая
кислота должна входить в состав рациона.
Линоленовая кислота СН3—СН2—СН=СН—СН2—СН==СН—
—СН2—СН=СН—(СН2),—СООН. Бесцветная жидкость, входит в
состав глицеридов многих жиров и масел. В льняном масле ее
содержание достигает 30—50%. Принадлежит к числу незаменимых для
человека и животных жирных кислот.
Двухосновные кислоты
В молекуле двухосновных или дикарбоновых кислот содержатся
две карбоксильные группы. В зависимости от строения остальной
части молекулы различают насыщенные, ненасыщенные,
ароматические и другие двухосновные кислоты. Наиболее важные
представители приведены в табл. 18.
Изомерия. Номенклатура. Структурная изомерия насыщенных
алифатических двухосновных карбоновых кислот обусловлена
строением углеродного скелета, ненасыщенных — строением скелета и
положением двойных связей. У ненасыщенных кислот имеет место
геометрическая изомерия:
Н—С—СООН НООС—С—Н
Н—С—СООН Н—С—СООН
уалеиновая кис- фумаровая кислота
лота ( цис-иjOiiep) (mpflrtC-иэомер)
138
Таблица 18
Наиболее важные представители двухосновных карбоновых кислот
Название
Формула
Т. пл., °С
Щавелевая кислота, этан-
диовая
Малоновая кислота, про-
пандиовая
Янтарная кислота, бутан-
диовая
Глутаровая кислота, пен-
тандиовая
Адипиновая кислота, гек-
сандиовая
Малеиновая кислота (цис-)
Фумаровая кислота
(транс-)
Фталевая кислота
Изофталевая кислота
Терефталевая кислота
НООС—СООН
НООС-СН2-СООН
НООС—СН2-СН2—СООН
НООС—(СН2)а—СООН
НООС-(СН2L—СООН
НООС—СН=СН—СООН
НООС—СН=СН—СООН
189,5
136
185
98
152
130
288
Ангидридизуется
348,5
Возгоняется
СООН
У ароматических двухосновных кислот изомерия связана также
со взаимным положением карбоксильных групп в бензольном ядре
СООН
СООН
/—СООН
li1—СООН
фталевая
:оон
изофталевая
СООН
терефта-
левая
Для наименования двухосновных кислот широко
применяются тривиальные названия; по ИЮПАК, родовое окончание
«диовая».
Способы получения. Для получения двухосновных кислот
используют те же методы, что и для одноосновных (окисление диолов,
гидролиз динитрилов, окисление оксикислот с первичной спиртовой
группой).
139
Физические свойства. Двухосновные кислоты — бесцветные
кристаллические вещества, растворимые в воде, растворимость уменьшается с увеличением
молекулярной массы. В водных растворах они диссоциируют на ионы:
НООС—СООН + Н2О Г: (НООС—СООГ + Н-3 О
(НОО—С—СООГ + ШЭ — -ООС—СОС+НЮ
Второй протон отщепляется труднее. Двухосновные кислоты сильнее
одноосновных.
Химические свойства. Двухосновные кислоты вступают во все
химические реакции, характерные для одноосновных кислот:
образование солей, сложных эфиров, ангидридов, галогеноангидридов и
т. д. В реакциях может участвовать одна или обе карбоксильные
группы.
Кислоты, у которых карбоксильные группы находятся близко
друг от друга (щавелевая, малоновая), обладают некоторыми
специфическими свойствами: они легко декарбоксилируются, превращаясь
в монокарбоновые кислоты; водородные атомы метпленовои группы
с малоновой кислоте обладают большой подвижностью:
НООС—СН2—СООН -С СН3СООН + СО2
малоновая кислота уксусная кислота
Дикарбоновые кислоты, у которых карбоксильные группы
находятся в положениях 1,4 и 1,5, при нагревании образуют
внутренние циклические ангидриды:
/,0
СН2—СООН СН2—
сн.,-соон ~НгО сн,—с/
янтарная w
кислота янтарный
ангидрид
Кислоты, содержащие двойные связи и ароматическое ядро,
вступают также в реакции, характерные для алкенов и производных
бензола.
Отдельные представители. Щавелевая, этандиовая кислота
НООС—СООН. Бесцветное, кристаллическое вещество, хорошо
растворимое в воде. Обычно существует в виде кристаллогидрата,
содержащего две молекулы воды. Чрезвычайно широко
распространена в растениях, главным образом, в виде кальциевых солей. В
большом количестве присутствует в щавеле, кислице. Получают ее из
формпата натрия:
2HC00Na ~6-0 'S NaOOC—COONa + Н2
NaOOC—COONa-\-H,,SO4 -> НООС—СООН + .Ma,S04
Щавелевая кислота легко окисляется, это используют в
аналитической химии для установления титра перманганата калия
НООС-СООН ^ 2С0-, + НаО
140
Щавелевая кислота и ее соли широко применяются в
промышленности. При ситцепечатании — как протрава, в деревообделочной
промышленности для обработки орехового и красного дерева.
Используется также для осаждения редких металлов, в качестве
катализатора в реакциях поликонденсацин.
Малоновая, пропандиовая кислота НООС—СН2—СООН.
Бесцветное кристаллическое вещество. Содержится в свекловичном соке.
Получается из монохлоруксусной кислоты:
С1-СН2-СООН ±Щ1 NCCH,COOH ±^° НООС-СН2-СООН
монохлоруксусыая малоновая кислота
кислота
Широко применяется в разнообразных синтезах (витаминов Вь В6,
барбитуровой кислоты) диэтиловый эфир малоновой кислоты.
Адипиновая, гександикарбоновая кислотаНООС—(СН2L—СООН.
Бесцветное кристаллическое вещество. Ограниченно растворима
в воде. Содержится в свекольном соке. Получается окислением
циклогексанола, циклогексанона или их смеси азотной кислотой
при температуре 55—80° С и давлении до 10 атм (98-Ю4 Н/м2):
ОН
"Л НООС-(СН2L—СООН
Адипиновая кислота в техническом отношении — важнейшая
насыщенная дикарбоновая кислота. Применяется в производстве
синтетических смол, для синтеза пластификаторов, найлона. В пищевой
промышленности применяется в качестве заменителя лимонной
кислоты.
Малеиновая и фумаровая кислоты НООС—СН=СН—СООН.
Наиболее простые и важные из двухосновных ненасыщенных кислот.
Являются геометрическими изомерами: малеиновая кислота —
цнс-изомер, фумаровая — транс-изомер. Фумаровая кислота
обнаружена в лишайниках, грибах, мышцах животных; малеиновая
в природе не встречается. Вступают в большинство реакций,
характерных для этиленовых соединений (присоединения, окисления,
полимеризации) и карбоновых кислот (образование солей, эфиров).
Сильно отличаются по физическим свойствам (т. пл. малеиновой
кислоты 130° С, фумаровой 258° С), по растворимости в воде, степени
диссоциации и некоторым химическим свойствам. Так, отнятием
воды от молекулы малеиновой кислоты получают малеиновый
ангидрид, фумаровый ангидрид из-за удаленности карбоксильных
групп в ее молекуле друг от друга не образуется:
<
н-с—с/
о
АОН: Н— С— С/
малепновьш
малеиновая ангидрид
кислота
141
В химическом отношении малеиновая кислота более активна:
под влиянием брома, йода, азотистой кислоты она переходит в фума-
ровую, которая более устойчива.
Малеиновую кислоту в промышленности получают окислением
бензола (стр. 74). Широкое применение для синтеза полимерных
материалов и яблочной кислоты нашел ангидрид малеиновои кислоты.
Фталевые кислоты С6Н4(СООНJ. Ранее указывалось (стр. 139),
что существует три бензолдикарбоновые кислоты, отличающиеся
взаимным положением карбоксильных групп. Фталевые кислоты —
кристаллические вещества, плохо растворимые в воде.
Фталевые кислоты получаются окислением о-, м-, я-ксилолов;
фталевая кислота (о-изомер) окислением нафталина:
f°]f >-(
нафталин
фталевая
кислота
Фталевые кислоты проявляют все свойства кислот (образование
солей, эфиров, амидов и других производных) и вступают во многие
реакции, характерные для дизамещенных бензола. В отличие
от других изомеров фталевая кислота (о-изомер) легко отщепляет
воду с образованием фталевого ангидрида:
f \—СООН
I J1—СООН -н2о
фталевая
кислота
>о
фталевый
ангидрид
который применяется для получения эфиров фталевой кислоты,
используемых в качестве пластификаторов, красителей и глифта-
лей — полиэфирных смол на основе фталевой кислоты и глицерина:
О О ;
С—О—СН2—СН (ОН)—СН2—О—
остаток фталв'
вой кислоты
остаток
глндерина
Терефталевая кислота (n-изомер) применяется в производстве
полиэтилентерефталата, сырья для изготовления волокна лавсан,
пНООС—/ V-СООН + лНОСНо—СН,—
ОН
терефталевая кислота
ьтиленгликоль
-н» (—СО—/' Ч—СООСНл—СНо—О—)
142
ПРОИЗВОДНЫЕ КАРБОЛОВЫХ КИСЛОТ
При замене гидроксила в карбоксильной группе кислоты на
разнообразные атомы и группы получаются производные кислот:
галогеноангидриды, ангидриды, сложные эфнры, амиды кислот
и т. д.
х.О
R-Of
галогеро-
акгидриды
'Р
4NH
R-O;
О
амид кислоты <- R—С< —> ангидрид кислоты
сложный эфир
Одновалентный кислотный остаток, образовавшийся после удаления
гидроксильной группы, получил название кислотного радикала
или ацила:
Ацил муравьиной кислоты Н—Cf называется формилом, уксусной
.„0
СН3—С? ацетилом и т. д. К производным кислот относят также
и их соли: эта группа соединений будет рассмотрена позднее
(стр. 132), амиды кислот — в разделе «азотсодержащие
соединения» (стр. 187).
Галогеноангидриды карбоновых кислот
Общая формула галогеноангидрндов карбоновых кислот: R—Cf
V
где Г — фтор, хлор, бром, иод. Название их строится по принципу
галогеналкилов: называется ацил и атом галогена, например
О О
|| хлористый формил; '( бромистый ацетил,
Н—С-С1 - СНз-С—Вг —
143
Получение. Получают галогеноангидриды кислот действием
галогенидов фосфора (РС15, РС13), тионилхлорида на кислоты или
их ангидриды:
/Р //°
CH3-C(f -Ь PC!, -^ СН,-С< + РОС!3 + НС!
уксусная кислота ацетилхлорид
/Р /Р
СН3—СНа—Cf + SOC12-> CHj—CH,—C<f +HC1+SO, ¦
ЧОН - Х-С!
гфогшоновая кислота хлористый пропионмл
Свойства. Галогеноангидриды — жидкости или твердые
вещества с резким запахом, разлагаются водой («дымят» на воздухе):
/Р //°
СНэ— Cf +Н2О->СН3— С( +НС!
ацетилхлорид
Галогеноангидриды отличаются высокой реакционной способностью.
Для них наиболее характерны реакции, связанные с обменом атома
галогена на другие атомы и группы и введение в молекулу
органического вещества ацила, поэтому они и получили название реакций
ацилирования:
hoqHj CH _с/°
о ^Хос2н5
/-и р/7 уксусноэтиловый
1^н3 ^\С1 эфир
ацетилхлорид NH3 ny/
ацетамид
Отдельные представители. Хлористый бензоил QH5—C<f
Жидкость с резким запахом, tmm = 197° С, применяется для
введено
нмя в молекулу группы С6Н5—С<^ .
Р
Хлористый ацетил СН3—-С<^ , Жидкость, tKm =51° С. При-
меняется для введения в молекулу органического соединения
ацетила.
/С!
Фосген О=С<^ . Полный хлорангидрид угольной кислоты,
отравляющее вещество (ОВ). Применяется для разнообразных
синтезов,
144
Ангидриды карбоновых кислот
Получаются при отнятии молекулы воды от двух молекул карбо-
новой кислоты или взаимодействием хлорангидридов с солями этих
кислот:
0 0 ,0
1 I) СН3-С{
СНз— С—ОН f НО—С—СН3 —й-^ /°
уксусная vJ»3
кислота О
О
уксусный
ангидрид
р 1 сн„-с/
v>X 1-Я' ^*\ ] ^-* ач^/""—\_*—™~V_^X Lo хт «f l > \^/
ЧС1 ацетат натрия w CHq—С\
^П
ацетилхлорид w
уксусный
ангидрид
Ангидриды кислот применяются в реакциях ацилирования.
Основные представители: уксусный ангидрид, малеиновый ангидрид.
Сложные эфиры карбоновых кислот
При взаимодействии карбоновых кислот со спиртами образуются
сложные эфиры, в молекуле которых ацил связан со спиртовым
остатком — алкоксильной группой:
R-c/°
алко-
КС!ТЛЬ-
ная
группа
Номенклатура, изомерия. По рациональной номенклатуре
наименование эфира строится из двух частей: названия кислоты и
спиртового радикала. По ИЮПАК, для-наименования сложных
эфиров к названию кислоты прибавляют название спиртового
радикала, изменяя при этом в названии кислоты окончание «овая» на
СН3—с/
«оат». Например, ЧОС2Н5 — этиловый эфир уксусной
кислоты, или этилацетат; этилэтаноат. Изомерия сложных эфиров
определяется изомерией кислот и спиртовых остатков.
Нахождение в природе. Способы получения. Сложные эфиры
широко представлены в природе, но обычно в небольших
количествах: они участвуют в разнообразных процессах, протекающих
в живом организме, и являются ароматобразующими компонентами
ряда растений. В значительных количествах в природе представлены
только сложные эфиры высокомолекулярных спиртов и кислот —
воски (стр. 205).
Синтетические способы получения. Реакция
этерификации (стр. 97).
145
Взаимодействие хлорангидридов кислот со спиртами:
сн3—с/ + нос2н-, . сн3—с/
ХС1 -Hcl NOC2H5
ацетилхлорид , этилацетат
Физические свойства. Сложные эфиры низкомолекулярных и среднемолеку-
лярных кислот и спиртов — жидкости с приятным фруктовым запахом,
высокомолекулярных — твердые вещества без запаха. Плохо или совсем не
растворимы в воде, хорошо растворимы во многих органических растворителях.
Химические свойства. Гидролиз. Это наиболее важная реакция
для сложных эфиров:
< < +Ri
xORx ~H2° XOH
Гидролиз ускоряется в присутствии кислот, щелочей и ферментов.
Гидролиз сложных эфиров с помощью ферментов (эстераз) играет
большую роль в сложных биохимических процессах, протекающих
в живом организме.
Переэтерификоция. Под переэтерификацией понимают замену
спиртового остатка в молекуле сложного эфира, происходящую при
нагревании эфиров со спиртами. Реакция ускоряется в кислой или
щелочной среде:
О
хО !!
R-C< + R2OH -?. R -C-OR2 + RiOH
NORi
Восстановление сложных эфиров. При восстановлении образуются
два спирта:
О
r_C_OR! Щ R—CH2OH+ RiOH
Восстановители: натрий в кипящем спирте, алюмогидрид лития.
Отдельные представители. Этилформиат Н—С<^ . Жид-
хос2н3
кость (tmm = 55° С) обладает запахом рома, применяется в пищевой
промышленности в качестве ромовой эссенции.
/7°
Уксусноэтиловый эфир, этилацетат СН3—С< . Жидкость
хОС,Нб
Dип = 78° С) применяется в качестве растворителя, для синтеза
разнообразных химических препаратов.
/° '
Уксусноизоамиловый эфир СН3—С' . Жидкость DИП =
чос5ни
= 139° С) с запахом груши. Применяется в пищевой
промышленности в качестве грушевой эссенции,
146
Масляноэтиловый эфир СН3—(СН2J—С . Прозрачная
хо-с2н5
жидкость с запахом ананаса (tKm = 121° С.) Применяется в качестве
ананасовой эссенции.
Особую группу сложных эфиров составляют сложные эфиры минеральных
кислот, представителен которых является диметилсульфат (CH3JSO4. Получают
действием олеума на метиловый спирт:
SO3 + 2СН3ОН ->- (CH3JSO4 + Н.2О
Диметилсульфат применяется в качестве метилирующего средства (введения
в молекулу метальной группы).
оксикислоты
Оксикислотами называются производные углеводородов,
содержащие в молекуле два вида функциональных групп: гидроксильные
О
(—ОН) и карбоксильные \—С—ОН/. По количеству карбоксильных
групп различают одно-, двух- и многоосновные оксикислоты, а по
числу гидроксильных групп, содержащихся в молекуле (включая
гидроксильную группу, входящую в состав карбоксильной
группы), — двух, трех- и многоатомные оксикислоты
В зависимости от строения углеводородного радикала, с которым
связаны окси- и карбоксильные группы, оксикислоты делятся на
алифатические (насыщенные и ненасыщенные), ароматические и
т. д. Примеры оксикислот:
ОН
I
СН3—СН—СООН — молочная кислота (одноосновная, двухатомная оксикислога)
НООС— СН—СН2—СООН — яблочная (двухосновная, трехатомная оксикислота)
ОН
НООС—СНОН—СНОН—CQOH — винная (двухосновная, четырехагомная
оксикислога)
СООН
— галловая (одноосновная, чегырехатомная ароматическая окси-
ОН
Структурная изомерия, номенклатура. Структурная изомерия
оксикислот вызывается особенностями строения углеродного
скелета (изомерия скелета), положением функциональных групп и их
взаимным расположением. В зависимости от положения оксигруппы
по отношению к карбоксильной различают а-, |3-, у- и т. д.
оксикислоты:
Г f Г
—С—С—С—СООН
147
При наименовании оксикислот широко применяются тривиальные
названия: молочная, лимонная, винная. По рациональной
номенклатуре в основе наименования лежит название соответствующей
кислоты, положение гидроксильной группы указывается буквами
греческого алфавита а, fi, у:
ОН
сн3—сн—сн2—соон
у Р а
р-окси.масляная кислота
По ИЮПАК, оксикислоты называют, прибавляя приставку «окси»
(диокси, триокси и т. д.) к систематическому названию кислоты.
Номер один присваивается атому углерода карбоксильной группы.
Приведенную выше оксикислоту следует назвать 3-оксибутановая
кислота.
Способы получения. Оксикислоты получают синтетическим путем
из кислот (или их производных) введением в молекулу оксигруппы,
из спиртов — введением карбоксильной группы или из альдегидов
и кетонов. Для получения некоторых кислот (молочной, лимонной)
широко применяются биохимические методы.
Гидролиз галогенозамгщвнных кислот. Этот способ удобен из-за
доступности а-галогенопроизводных кислот:
СНз—СНС1— СООН + НОН ^Ы СНз-СН—СООН+НС1
I
2-хлорпропановая
кислота
он
2-оксипропановая
кислота
Гидратация непредельных кислот:
.» ( о о
/¦—Ч ^ H,,SO4 /W
СН.,=СИ—С— ОН + Н2О —* СН2—СН,—С— ОН
он
акриловая кислота 3-оксипропановая кислота
Присоединение воды к а-, р-непредельным кислотам вследствие
эффекта сопряжения идет не по правилу В. В. Марковникова: гид-
рокснл направляется к более удаленному от карбоксильной группы
ненасыщенному атому углерода. '
Получение из альдегидов и кетонов (циангидринный синтез):
.О
н3-с<-
хн
уксусный
альдегид
ОН
j
—^СН3—С—CN ^
н
ННТриЛ C.-QKCH-
пропноновой
кислоты
он
1
jf~ СН3—СН—СООН
молочная кислота
Этот способ применяется для получения а-оксикнелот.
148
Физические свойства. Оксикислоты — бесцветные вязкие жидкости или
кристаллические вещества. Низшие хорошо растворимы в воде, с ростом
молекулярной массы растворимость уменьшается.
Химические свойства. Химическое поведение оксикислот, как
и других гетерофункциональных соединений, т. е. соединений,
содержащих в молекуле разные функциональные группы, определяется
природой этих групп. Оксикислоты вступают в большинство
химических реакций, характерных для кислот (наличие группы СООН)
и спиртов (присутствие группы ОН). Строение углеводородного
радикала также сказывается на их химическом поведении: ароматические
оксикислоты вступают во многие превращения, характерные для
соответствующих производных бензола. В одних химических
реакциях каждая из указанных функциональных групп может
участвовать независимо друг от друга, в других ход реакции и характер
образующихся продуктов зависит от их взаимного влияния, в
некоторых случаях окси- и карбоксильная группы могут
взаимодействовать между собой.
Кислотные свойства. Введение в молекулу кислоты оксигруппы
повышает силу кислоты: оксикислоты — более сильные, чем соот^
ветствующие им карбоновые кислоты. Они могут образовывать соли
[реакция A)]; сложные эфиры B); амиды C); галогеноангидриды D).
В реакции D) с РС15 может взаимодействовать и спиртовая группа
с образованием СН3—СН—С—С1.
С1 О
Спиртовые свойства. Примером являются следующие
превращения:
он
I
СНз—СН—СООН—
молочная кислота
о
-^-— СН3-С-СООН A)
——> СН3-СН-СООН B)
I
С1
Ы^Р-i, СН3-СН-СООН C)
rI7_H«coci СНз_сн_С00Н
ОСН3
сн_С
ООСС1
Возможно окисление вторичной спиртовой группы с
образованием кетокислоты [реакция A)]; замена спиртовой группы на атом
галоида B); образование простых эфиров C); образование
сложных эфнров D).
Отличие в поведении а-, |3-, у- и б-оксикислот. В этих реакциях,
протекающих с выделением воды, сказывается взаимное влияние
групп СООН и ОН.
149
1. Взаимодействие а-оксикислот. При нагревании а-оксикислот
выделяется вода и образуется циклический сложный эфир — лактид:
CHg—сн—он "но;—с=о сн__СН_О__С:==О
| Э Т
o=c—юн н'.о—нс—снэ 2 о==с—о—нс—сн3
лактид
2. fi-Оксикислопш при нагревании теряют воду с образованием
непредельных кислот:
р а
СН„—СН—СНг—СООН —-д СН3—СН=СН—СООН
' крогоновая кислота
ОН
р-оксимасляная кислота
3. у- и р- Оксикислоты при нагревании образуют внутренние
сложные эфиры — лактоны:
СН3-СН-СН2-СН2-СООН ____, СН3-СН-СН2-СН2-С^°
он ' '-о 1
V-оксивалериановая кислота лактон
Стереохимия углерода. Известно два основных вида изомерии —
структурная и пространственная. Многочисленные виды
структурной изомерии были уже подробно разобраны в предыдущих разделах:
теперь остановимся на пространственной изомерии. К
пространственной изомерии относятся: 1) геометрическая (цис- транс- изомерия);
2) оптическая и 3) поворотная изомерия. Раздел органической химии,
занимающейся исследованием пространственного расположения
атомов в молекулах и его влиянием на свойства органических
соединений, получил название стереохимии. Первый из перечисленных
видов пространственной изомерии — геометрическая изомерия —
был рассмотрен в разделе алкенов. Она характерна и дляфумаровой
и малеиновой кислот.
Оптическая изомерия. В 1815 г. французский химик Бив
обнаружил, что некоторые органические вещества в жидком и
растворенном состояниях способны вращать плоскость поляризованного света.
Ранее эта особенность была отмечена для кристаллов кварца: при
прохождении через них поляризованного луча происходит вращение
плоскости поляризации, причем одни кристаллы отклоняют ее
вправо, другие—-влево. В 1848 г. Л. Пастер обнаружил эту
способность у винных кислот. Это явление, названное оптической
изомерией, получило объяснение в работах Вант-Гоффа и Ле-Беля.
Оно связано с определенной пространственной ориентацией кова-
лентных связей. Все четыре валентности атома углерода
тождественны и направлены к вершинам правильного тетраэдра, в центре
которого находится атом углерода. Если в вершинах тетраэдра
расположить различные заместители, то молекула станет
асимметричной. Пространственные модели этой молекулы будут относиться
150
друг к другу, как предмет к своему зеркальному изображению.
Это явление характерно для многих оксикислот. Так, в молекуле
молочной кислоты содержится атом углерода (он отмечен
звездочкой), связанный с четырьмя различными группами, такой
углеродный атом получил название асимметрического атома углерода:
Н
!
сн3—с*—соон
!
он
молочная
кислота
Наличие асимметрического атома углерода приводит к
образованию двух пространственных изомеров (рис. 17), они отличаются
Рис, 17. Модель зеркальных тетраэдров (на примере
молочной кислоты)
друг от друга, как несимметричный предмет от своего изображения
в зеркале (как левая и правая руки). Отсюда и другое название
оптической изомерии — зеркальная изомерия.
Если тетраэдрические модели молочной кислоты спроектировать
на плоскость, то получатся проекционные формулы, названные
по имени ученого, предложившего их, — формулами Фишера:
СООН
Н-
-ОН
СН3
СООН
но-
-Н
СН,
зеркало
При пользовании формулами Фишера нужно помнить, что верхняя
и нижняя группы (для молочной кислоты СООН и СН3) лежат за
плоскостью чертежа, а боковые (в нашем случае Н и ОН) перед
плоскостью чертежа. Асимметрический атом углерода находится на
пересечении прямых, связывающих эти группы. Такие модели
нельзя совместить при вращении в плоскости чертежа (они
относятся друг к другу, как предмет к своему зеркальному изображению
или как левая и правая руки),
151
Подобные пространственные изомеры называются стереоизоме-
рами, или оптическими антиподами. Число их D = 2", где п —
количество асимметрических атомов углерода.
Молочная кислота существует в виде двух пространственных
изомеров (оптических антиподов), каждый из которых проявляет
оптическую активность — обладает способностью вращать
плоскость поляризованного света. Один из изомеров — правовращающий —
обозначается знаком «-}-», другой — левовращающнй — знаком «—».
Оптически неактивная смесь (или соединение) равных количеств
лево- и правовращающих антиподов получила название рацемата
или рацемической смеси (±).
По химическим и большинству физических свойств оптические
антиподы не отличаются друг от друга. Различие состоит лишь в
3.
Рис. 18. Поляриметр
том, что один из них лево-, другой правовращающий, и в их
способности кристаллизоваться в так называемых энантиоморфных
формах, т. е. асимметрических формах, относящихся друг к другу, как
предмет к своему зеркальному изображению.
Рацематы отличаются по своим физическим свойствам
(температура плавления и т. д.) о т активных стереоизомеров.
Для измерения оптической активности применяется прибор,
названный поляриметром (рис. 18).
Свет от источника 6, проходя через поляризатор 1, в качестве
которого употребляется неподвижно укрепленная призма Николя,
поляризуется. Поляризованный луч, попадая на анализатор
(вторую призму Николя 2), может полностью проходить через нее, когда
оси кристаллов обеих призм строго параллельны, или полностью
задерживаться, если они повернуты на 90°. Для наглядности
прохождение поляризованного света через призму Николя часто
сравнивают с прохождением лезвия ножа через книгу: если они
расположены в одной плоскости, нож проходит, если книгу повернуть на
90°, пет (рис. 19).
152
Для измерения оптической активности исследуемое вещество
в жидком состоянии или в виде раствора помещают в
поляриметрическую трубку 3, которую устанавливают между поляризатором и
анализатором. Если исследуемое вещество оптически неактивно,
то световой луч, проходя через него, не изменяя плоскости
поляризации, попадает в анализатор. Если оно активно, то плоскость
поляризации изменяется, и для прохождения луча через анализатор
его нужно повернуть на угол, соответствующий вращению
находящегося в трубке вещества. Величину этого угла измеряют-с помощью
Рис. 19. Прохождение
поляризованного света через призму
Николя
круговой шкалы 4. Измеренное вращение зависит от природы
вещества, концентрации, длины трубки / и обычно пересчитывается
в удельное вращение [ее]. Удельным вращением называется угол
вращения, наблюдаемый при толщине стоя 1 дм и концентрации
исследуемого вещества, равной 1 г/мл A03 кг/м3).
Разобранный пример стереоизомерии молочной кислоты —
наиболее простой. Большей сложностью отличается стереоизомерия
винной кислоты: НООС — СНОН — СНОН — СООН. В молекуле
винной кислоты имеется два асимметрических атома, и в
соответствии с ранее приведенной формулой нужно ожидать существование
2 пар оптических антиподов:
СООН
*
-он
-он
IIO-
соон
*
н
н—,
*
СООН
НО F—Н
!
СООН
B)
НС
СООН
4^-н
СООН
C)
СООН
но—)-—н
н—|^—он
соон
D)
Каждый асимметрический атом в молекуле винной кислоты может
обеспечивать или левое (—), или правое (--J-) вращение. Возможны
следующие комбинации:
1)
3)
4) -
153
При рассмотрении приведенных формул можно убедиться, что
структуры 3 и 4 являются оптическими антиподами, а 1 и 2 при
повороте на 180° в плоскости чертежа совпадают, т. е. они
соответствуют конфигурации одной молекулы, получившей название мезо-
винной кислоты. Мезовинная кислота не обладает оптической
активностью вследствие внутримолекулярной компенсации.
Следовательно, для винных кислот возможно существование двух оптических
антиподов C и 4), их рацемической смеси и неактивной мезовинной
кислоты. Если сравнивать стереоизомеры 3 и 4 с мезоформой, то
окажется, что они имеют одинаковое расположение групп Н и ОН у
одного асимметрического атома углерода и разное — у другого.
Такие стереоизомеры называются диастереоизомерами. Они
отличаются друг от друга не только по величине угла вращения, но и
по физическим и химическим свойствам.
Вопрос об абсолютной конфигурации оптически активных
соединений, т. е. о действительном расположении групп в
пространстве, в течение длительного времени не нашел своего разрешения.
Условно, по предложению русского ученого Розанова, было решено,
соединения, содержащие гидроксил в проекционных формулах
справа от асимметрического атома углерода, относить к D-ряду,
слева — к L-ряду. С помощью разработанных в последние годы
физических методов исследования была установлена абсолютная
конфигурация молочной кислоты, правовращающей оказалась
L-форма.
Синтетическим путем и в ходе многих биохимических процессов
часто получаются рацемические смеси, разделение которых на
оптические антиподы, учитывая, что их физические и химические
свойства одинаковы, представляет трудную задачу. Наиболее
эффективными являются три метода разделения.
Физический метод. При кристаллизации рацемических
смесей некоторых антиподов образуются кристаллы, отличающиеся
друг от друга, как несимметрический предмет от своего зеркального
изображения. Они могут быть разделены механическим путем.
Этот метод был открыт Пастером и применен им для разделения
неактивной винной кислоты в виде ее натриевоаммонневой соли.
Биохимический метод. Основан на способности отдельных
видов микроорганизмов предпочтительней поглощать из
рацемической смеси один из антиподов.
Химический метод. Используется способность оптических
антиподов превращаться с помощью химических реакций в диастереоизо-
меры, которые существенно отличаются по свойствам:
-*(+)?> А ¦ DB-t
(-f)-DA и (—)LA-fDB-^(f)DA Z?B + (—)LX ¦ DB— \—
( \ГД ПИ_1
рацемат смесь днаотереоизомеров * ¦*
выделение
антиподов
Поворотная изомерия. Поворотная изомерия возникает в
результате ограниченно свободного вращения групп атомов вокруг
154
сигма-связи. Возникающие при этом изомеры получили название
поворотных изомеров или конформаций. Поясним это на примере.
В молекуле этана СН3—СН3 одна метильная группа может
поворачиваться относительно другой, не нарушая при этом порядка
связей, валентных углов и длины связей (рис. 20), образуя различные
поворотные изомеры. Две крайние конфигурации (они получили
название «заслоненной» и «заторможенной») (рис. 20, а, б) несколько
отличаются Друг от друга по запасу энергии.
В простых органических соединениях (алканы) вследствие
малого энергетического барьера переход из одной конформационнои
н
Н" Н
Заслоненная Заторможенная
(эклиптическая) (шахматная)
а 5
Рис. 20. Поворотная изомерия
формы в другую осуществляется легко, но выделить изомеры
в чистом виде не удается. В сложных молекулах, например в цик-
ланах (стр. 65), конформационное состояние молекулы существенно
влияет на ее реакционную способность. В большинстве случаев при
работе с органическими веществами приходится иметь дело со смесью
конформационных модификаций.
Отдельные представители. Молочная, а-оксшгропионовая
кислота СН3—СНОН—СООН. Существует в виде двух оптических
антиподов и рацемата. Мясомолочная кислота (+), правовращающая.
Кристаллическое вещество, ЫН = 4- 3,82° A0%-ный водный
раствор). Содержится в мышечном соке. Левовращающая (—), обладает
теми же свойствами, что и правовращающая, [а]13 = — 3,82°.
Молочная кислота брожения (=р) — рацемат. Гигроскопичная
сиропообразная жидкость. Наиболее важный способ получения —
молочнокислое брожение:
CeHi2Oe -*¦ 2СН3—СНОН—СООН
гексоза молочная кислота
При молочнокислом брожении Сахаров обычно образуется
рацемическая форма молочной кислоты. Молочнокислое брожение
играет очень большую роль в производстве молочнокислых
продуктов (простокваши, ацидофилина, кефира, кумыса), при
изготовлении жидких дрожжей для хлебопечения, квашении капусты,
огурцов, силосовании кормов.
Молочную кислоту получают синтетическим путем 'из уксусного
альдегида:
О
СН3—с/ МРЛ CH3-CH-CN I^tt СН3—СН—СООН
ХН ! !
уксусный *^"
альдегид молочная кислота
или омылением сс-хлорпрошгоновой кислоты (СН3—СНС1—СООН).
Молочная кислота используется в кожевенной промышленности
при обработке кож, текстильной промышленности в качестве
протравы и в медицине. Очень широкое применение она нашла в
пищевой промышленности при изготовлении конфет, безалкогольных
напитков. Эфиры молочной кислоты СН3—СН—СООН употребляют-
оос17н33
ся в качестве улучшающих добавок в хлебопечении, при
производстве кондитерских изделий, мороженого.
Яблочная, оксиянтарная кислота НООС—СН2—СНОН—СООН.
Встречается в виде двух оптических антиподов и рацемата. В
растительном мире широко распространена левовращающая L яблочная
кислота. Является одной из главных органических кислот в
яблоках, рябине, груше, кизиле, махорке (до 6,5%), Бесцветное
кристаллическое вещество, [а]'В = —2,3° A0%-ный водный раствор).
Получают гидролизом бромянтарной кислоты:
НООС—СН2—СНВг—СООН z^ НООС—СН,—СНОН—СООН
или гидратацией фумаровой или малеиновой кислот при 200° С:
НООС—СН=СН—СООН + Н„О -> НООС—СН—СН,—СООН
1
он
Левовращающую яблочную кислоту выделяют из плодов рябины.
Применяется в пищевой промышленности в производстве
кондитерских изделий и безалкогольных напитков.
Винная, диоксиянтарная кислота НООС—СНОН—СНОН—
—СООН. Встречается в виде двух оптических антиподов, рацемата
и мезовинной кислоты (табл. 19),
В природе встречается правовращающая (D-винная) кислота,
получившая название виннокаменной, так как она впервые была
выделена из «винного камня», отхода, получаемого при изготовлении и
выдержке виноградных вин и представляющего собой кислую
калиевую соль НООС—СНОН—СНОН—COOK. Получают винную
кислоту из «винного камня» или синтетическим путем. При нагревании
раствора винной кислоты она переходит из одной стереоформы в другую.
156
Таблица 19
Свойства винных
Наименование
Винная, левовращающая — L-кислота
Виннокаменная (правовращающая — D-
кислота)
Рацемат (виноградная кислота)
Мезовннная
кислот
Т. пл., "С
170
170
206
140
[а]д для 20%-ного
водного раствора
— 12
+ 12
Неактивная
з>
Исследования винных кислот сыграли выдающуюся роль в
изучении явления изомерии. В 1830 г. Берцелнус, установив, что
виннокаменная кислота имеет одинаковый состав с винной
кислотой, ввел понятие изомерии. В пятидесятых годах прошлого века
Л. Пастер, работая с винными кислотами, выяснил природу
рацематов и методы их разделения.
Винная кислота используется в пищевой промышленности при
изготовлении фруктовых вод, химических разрыхлителей теста,
в аналитической химии для определения ионов калия, в медицине.
Широкое применение нашли и соли виннокаменной кислоты. Калне-
во-натриевая соль NaOOC—CHOH—СНОН—СООК-4Н,О,
получившая название сегнетовой соли, является одним из компонентов
фелинговой жидкости, которая используется для качественного и
количественного определения восстанавливающих веществ:
альдегидов, Сахаров (стр. 233). Сегнетовая соль применяется и в
радиотехнике (пьезокристаллы).
СООН
СН,
Лимонная кислота НО—С—СООН трехосновная, четырехатом-
СН2
СООН
наяоксикислота. Основная органическая кислота плодов цитрусовых,
в лимонах ее содержание достигает 9% на сухую массу.
Кристаллическое вещество. Хорошо растворима в воде. Лимонную кислоту
получают из листьев махорки, лимонов и биохимическим путем
с помощью гриба Aspergillus niger.
Применяется в пищевой промышленности при производстве
конфет, кремов, безалкогольных напитков, в текстильной
промышленности при крашенин тканей и в медицине.
Рицинолевая кислота Н3С—(СН2M—СН—СН,—СН=СН—(СН,),—
I
ОН
—СООН. Мононенасыщенная, одноосновная, двухатомная оксикис-
лота. Является основной кислотой касторового масла (80—95%).
Касторовое масло широко используется в технике в качестве
смазывающего вещества и в медицине как слабительное.
157
Оксибензойная кислота QH4(OH)(COOH). Существует в виде
трех изомеров (о, м-, л-кислоты), важнейшим является о-
оксибензойная, или салициловая кислота:
ОН
V-COOH
Бесцветное кристаллическое вещество, трудно растворяется в воде.
Получается действием двуокиси углерода на фенолят натрия
(реакция Кольбе — Шмитта);
ONa ОН ОН
сог /\—COONa
фенолят салицил-ат салициловая
натрия натрия кислота
Применяется в аналитической химии, медицине, анилокрасочной
и пищевой промышленности, при консервировании фруктов.
Производные салициловой кислоты являются лечебными препаратами:
О
II
~ ,соон /х/с-ос6н5
1 U I II
о—с—сн3 I ;—он
[I "^'
6
Спирин салол
соон
Галловая, 3,4,5-триок.сибензойная кисло/па
НО"
ОН
таллическое вещество, хорошо растворимое в кипящей воде. В
природе встречается как в свободном, так и в связанном состоянии,
обычно в растениях, содержащих дубильные вещества (чернильные
орешки, дубовая кора, листья чая). Получают гидролизом танина.
Применяется для получения пирогаллола, образующегося при ее
нагревании, синтеза красителей, в аналитической химии.
ОКСОКИСПОТЫ [АЛЬДЕГИДС И КЕТОКИСЛОТЫ]
Альдегидо- и кетокислотамн называются гетерофункцнональные
соединения, содержащие в молекуле две характеристические
группы: кислотную (карбоксильную) и альдегидную или кетон-
ную. Кроме приведенной классификации (альдокнслоты, кето-
кислоты) оксосоединения в зависимости от положения аль-
158
дегидо- и кетогруппы по отношению к карбоксильной группе делятся
на а-, |3-, у- н т. д. альдегиде- и кетокислоты:
О
СН3-6-СООН
а-кетокислота
о
II
СН з—С—СНа—СООН
Р-кетокислота
Номенклатура, изомерия. Для наименования оксосоединений
часто применяют тривиальные названия:
О
СН3—С—СООН
пировиноградная
кислота
По номенклатуре ИЮПАК, для наименования оксокислот перед
названием кислоты ставится приставка «оксо» с указанием номера
углеродного атома, связанного с кислородом карбонильной группы
(номер 1 имеет атом углерода карбоксильной группы):
>С—СН,—СН,—СООН 4-оксобутановая кислота
W'
О
СН4—С—СН2—СООН 3-оксобутановая кислота
Получение, свойства. Оксокислоты получают из кислот
введением в их молекулу карбонильной группы или, наоборот, из
альдегидов и кетонов созданием в их молекуле карбоксильной
функциональной группы.
Альдегидо- и кетокислоты обладают всеми свойствами кислот
(образуют соли, сложные эфиры, амиды) и оксосоединений.
Основные представители. Глиоксалевая. или глиоксиловая кислота
О.
у-С—СООН единственно возможная а-альдегидокислота.
Существует в виде кристаллогидрата, содержащего одну молекулу воды.
Глиоксалевая кислота распространена в природе, особенно в
большом количестве она содержится в незрелых фруктах. Играет важную
роль в обменных процессах, протекающих в растениях и
микроорганизмах.
О
Пировиноградная кислота СН3—С—СООН. Наиболее важный
представитель кетокислот. Бесцветная жидкость с запахом уксусной
кислоты, tKim = 165° С, /цл = 13,6° С. Смешивается с водой во всех
отношениях. Широко распространена в природе, является
важнейшим промежуточным продуктом при распаде углеводов (стр. 219)
159
в растениях, в спиртовом и молочнокислом брожении. Получается
нагреванием (пиролизом) винной кислоты, отсюда и ее тривиальное
название — пировиноградная кислота:
О
НООС—СНОН—СНОН—СООН г?°512!*° СН3—С—СООН
винная кислота
пировиноградная
кислота
За счет взаимодействия соседних кето- и карбоксильной
группировок активнее, чем обычные кетоны, вступает в характерные для них
реакции:
4?
сн3—с
+5
4
с—он
Пировиноградная кислота применяется в медицине,
О
Ацетоуксусный эфир СН3—С—СН2—СООС2Н5. Исключительный
интерес с теоретических позиций и большое практическое значение
в разнообразных синтезах играет этиловый эфир ацетоуксуснои
кислоты (ацетоуксусный эфир), существующий в двух изомерных
формах, которые легко переходят друг в друга и находятся в состоянии
подвижного равновесия. Это явление получило название подвижной
(динамической) изомерии, или таутомерии. Одна из изомерных форм
ацетоуксусного эфира содержит кетогруппу (кето-форма), другая
гидроксильную группу у ненасыщенного углеродного атома (еноль-
ная форма); отсюда и название этого' вида таутомерии — кето-
енольная таутомерия:
О ОН
Ч I
СНз—С—СН2—COOQsHs Zl СН3—С=СН—СООС2Н5
кетонная форма
енольная форма
Основной способ получения ацетоуксусного эфира — сложно-
эфирная конденсация:
СН3—С—ОС,Н5+СНз—С
Ч :i
0 0
уксусноэтилоЕЫЙ
эфир
СН3—С—СН,—СООС2Н3+СН5ОН
il
О
ацетоуксусный
эфир
Ацетоуксусиый эфир участвует во всех реакциях, характерных
для кетонов (присоединение бисульфита натрия, синильной кислоты,
взаимодействие с фенилгндразином) и енолов (взаимодействие с
пятихлористым фосфором, ацнлирование, образование красно-
фиолетового окрашивания при взаимодействии с хлорным
железом). Ацетоуксусный эфир широко применяется в органическом
синтезе.
ЧАСТЬ ТРЕТЬЯ
ЭЛЕМЕНТОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Молекулы большинства рассмотренных органических
соединений (углеводородов, спиртов, альдегидов и кетонов, кислот) состоят
из атомов небольшого числа элементов (органогенов). Ранее уже
было сказано, что атомы углерода обладают способностью
соединяться практически со всеми элементами периодической системы
Д. И. Менделеева. Органические соединения, в молекуле которых
атом углерода непосредственно связан с атомами этих элементов,
получили названия элементорганических соединений. Первыми
изученными элементорганическими соединениями были соединения
углерода с металлами (этот раздел органической химии получил
название химии металлорганических соединений), по мере
расширения наших знаний возникли разделы, посвященные органическим
соединениям кремния, серы, фосфора и т. д.
МЕТАЛЛОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Из многочисленных и разнообразных соединений этой группы
особое значение имеют соединения магния, цинка, алюминия.
Название металлорганических соединений образуется из двух частей;
названия радикала и названия связанного с ним металла: (CH3JZn —
диметилцинк; C2H5MgCl — хлористый этилмагний.
Способы получения. Взаимодействие металлов с галогеналкилами.
Реакция особенно часто применяется для получения магнийорга-
нических соединений:
йодистый йодистый
метил метил-
магний
Реакцию проводят в среде безводного диэтилового эфира.
Образовавшееся магнийорганическое соединение не выделяется из
раствора. При использовании его в дальнейших синтезах
реагирующее с ним вещество непосредственно вводится в реакционную
массу.
Действие сплавов на галогеналкилы:
4PbNa + 4C2H5Br -> Pb (C.2H5L + 4NaBr + 3Pb
сплав тетраэтил-
свинец
6 А. П. Нечаев 161
Получение алюминийорганических соединений (реакция Циглера):
2А1 + ЗН3 + 6СПН2„ -> 2 (С„Н2П+1K А1
Свойства. Металлорганические соединения обладают большой
химической активностью. Связь С—Me поляризована, причем металл
является положительным концом диполя, углерод —
отрицательным.
Реакции замещения. Образование углеводородов. При
взаимодействии металлорганических соединений с водой A), хпир-
—б +6
том B), кислотой C), амином вследствие поляризации связи С—Me
атом углерода, несущий частичный отрицательный заряд,
соединяется с подвижным водородом, образуя молекулы углеводорода.
Другая часть молекулы металлорганического соединения
взаимодействует с остатком реагирующей молекулы. Примеры этих
превращений приведены ниже:
СНз-MgI -j- Н-О-Н -» СН4 + Mgl (ОН), A)
йодистый метан
метилмагний -
С2Н5ОН + CH3MgI н> QH.OMgl + CH4 B)
QH5MgBr + CH3COOH -» С6Н6 + CH3COOMgBr C)
бромистый
фенилмагшш
В общем виде схема этих реакций будет иметь следующий вид:
R— Mgr + Н—А -* R— Н + Г—MgA D)
В реакции B) объем выделившегося метана легко может быть
замерен, это дает возможность использовать ее для определения числа
гидроксильных групп (метод Чугаева — Церевитинова — Терен-
тьева).
Взаимодействие магнийорганических соединений с ацетиленом
(стр. 52).
Реакции присоединения. Присоединение
металлорганических соединений к соединениям, содержащим оксогруппу
(альдегидам, кетонам, производным кислот), происходит следующим
образом:
ОМеГ
+S -8 -а +5 j
>С = О + R—Mg—Г —— >G—R
т. е. углеводородный радикал магнийорганического соединения
присоединяется к карбонильному углероду, а остальная часть
молекулы — к атому кислорода; при разложении образовавшихся
продуктов водой получаются спирты. Подбирая соответствующие кар-
бонилсодержащпе соединения, синтезируют первичные, вторичные
162
и третичные спирты (реакции Тищенко, Вагнера, Зайцева,
Бутлерова):
H8C=O + CH3MgI -> СН3—CH2-OMgI -2—CH3CH2OH+MgI (ОН)
ЭТИЛОВЫЙ
спирт
OMgl ОН
/,0 i н о !
СН3—С<^ +CH3MgI -> СНз-СН —— CHS—CH—СНз + MgI (ОН)
" I втор, пропи-
Lri3 ловый спирт
О OMgl ОН
СНз—С—CH3+CH3MgI ->СН3—С—СН3 ^2-СН3—С—CH3 + MgI (ОН)
сн3 сн3
трет,
бутиловый спирт
OZnCl ОН
О ' I
СН3—с/ + Zn (СН3J -» СН3—С—СН3 -^ СН3—С—СН3
!
СН3 СНз
С помощью последней реакции А. М. Бутлеровым впервые были
получены третичные спирты. Бутлеров и его ученики применяли для
синтеза спиртов цинкорганические соединения. Позднее в работах
французского химика В. Гриньяра было показано, что
целесообразнее использовать в таких синтезах не цинкорганические
соединения, а магнийорганические.
Присоединение двуокиси углерода:
R—MgCl+COa-» R—COOMgCl —~ R—COOH + MgCla
Органические соединения цинка и магния. Цинкорганические
соединения относятся к числу первых синтезированных металлор-
ганических соединений (Франкланд, 1894). Получаются действием
галогенопроизводных на цинковые стружки или медь-цинковый
сплав. Цинкорганические соединения — низкокипящие жидкости,
без цвета, самовоспламеняющиеся на воздухе, что требует
проведения работы с ними в среде инертного газа.
. Магнийорганические соединения играют наибольшую роль среди
органических соединений металлов II группы. Обычно получаются
с помощью реакции Гриньяра — взаимодействие галогеналкилов
с магнием в среде сухого органического растворителя (диэтиловый
эфир, тетрагидрофуран, третичные амины). Наилучшие результаты
получаются при использовании первичных галогеналкилов.
Широко применяются для разнообразных синтезов и
аналитических исследований.
Алюминийорганические соединения. Их получают по реакции
Циглера (стр. 162). Алюминийорганические соединения —
бесцветные, вязкие жидкости, самовоспламеняются на воздухе. Широкое
применение в разнообразных синтезах получил триэтилалюминий.
Он используется для получения полиолефинов (особенно
полиэтилена низкого давления), каучука, высших спиртов.
6* 163
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Хотя кремний, так же как и углерод, находится в четвертой
группе, его соединения значительно отличаются от соединений
последнего. Основными группами кремнииорганических соединений
являются:
алкилпроизводные силана (SiH4): H3Si—SiH3 — дисилан,
Si (CH3L — тетраметилсилан;
гидроксильные производные силана: RgSiOH — триалкилсила-
нол, RSi (ОНK — алкилсилантриол;
аминопроизводные силана (силазаны): (CH3KSiNH2 — триме-
тилсилазан, (CH3KSi—NH—Si (CH3K — гексаметилдисилазан;
соединения, содержащие группу Si—О—Si, — силоксаны;
(CH3KSi—О—Si (СН3)з — гексаметилдисилоксан.
Для синтеза кремнииорганических соединений используют четы-
реххлористый кремний (получают действием хлора на кремний):
SiCl4 + LiAlH4 -> SiH4 + LiAlCl4
четырех- силан
хлористый
кремний
SiCl4 + RMgr -> RSiCl3 ™§? R2SiCla Ш?. R3SiCl Ш? R4Si
Тетраметилсилан Si (CH3L — жидкость, /кип = 26° С, по
химическим свойствам несколько напоминает алканы. Хлорпроизвод-
ные — алкилхлорсиланы хорошо гидролизуются водой.
Гидроксильные производные (силанолы) легко теряют воду:
2 (С2Н=K Si—ОН -> (С2Н5K Si—О—Si 4C2HSK + H2O
В последнее время разнообразные продукты полимеризации
кремиииорганических соединений применяются в качестве
электроизоляционных, смазочных и каучукоподобных материалов.
СЕРУСОДЕРЖАЩИЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В органических соединениях сера проявляет валентность,
равную двум (восстановленная сера), четырем и шести (окисленная
сера). К соединениям первой группы относятся тиоспирты и тио-
эфиры, ко второй группе — сульфоксиды, сульфиновые кислоты,
сульфоны и сульфокислоты:
О О
R—S—H
тиоспирт
R-S-R
тиоэфир
О
D Ч D .
К—3—1\\
[1
о
сульфоны
R—S—Rj
сульфоксиды
О
О
сульфокиелоты
R—S—ОН
сульфиновые
кислоты
Тиоспирты, тиоэфиры. Тиоспирты (тиолы, меркаптаны) и
тиоэфир ы (дналкилсульфиды, ди ар ил сульфиды) можно рассматривать
164
как производные сероводорода Н—S—Н. В первом случае в
молекулу введен один углеводородный радикал, во втором — два.
Тиоспирты называют следующим образом: к названию радикала
добавляют слово «меркаптан» или аналогично спиртам заменяют «ол»
на «тиол» (ИЮПАК). Для наименования тиоэфиров к названию
радикалов добавляют слово «сульфид»:
CH3SH — метилмеркаптан; метантиол
GjHsSH — этилмеркаптан; этантиол
С2Н5—S—C2HS — диэтилсу льфид
Тиолы и тиоэфиры получают при взаимодействии галогеналкилов
и спиртов с сероводородом и сульфидами:
QjHsI-Ь KSH-* KI+C2H5— SH
йодистый этантиол
метил
с2н5он + h,s ^^-Ш^л c2h5sh + н2е
этиловый
спирт
2C2H5I + K2S -> 2KI + (С2Н5), S
йодистый диэтил-
этял сульфид
Тиолы — ннзкокипящие жидкости с резким, крайне
неприятным запахом, тиоэфиры — нейтральные жидкости, кипят при
более высоких температурах, труднорастворимы в воде.
По химическим свойствам тиолы приближаются к сероводороду
(образование солей меркаптидов R—S—Me), проявляя в то же время
некоторые свойства спиртов (взаимодействие с хлористыми ацилами).
При- окислении тиолов в мягких условиях образуются дисульфиды,
при действии сильных окислителей — сульфокислоты:
т—оН—
С— C2H5-S-S-C2H3
диэтилсульфнд
— С2Н5—SO2H
эта нсу льф окис л ота
Тиолы добавляют к природному газу для обнаружения по запаху
его утечки в жилых и производственных помещениях.
Иприт (р\ $-дихлордиэтилсульфид) С1—СН,—СН2—S—СН2—
—СН2—С1 является отравляющим веществом (ОВ).
К соединениям, содержащим восстановленную серу, могут
быть отнесены два важных в практическом отношении соединения:
сероуглерод и тиомочевина.
Сероуглерод S=C=S. Жидкость с неприятным запахом, tKan =
= 46,3° С. Очень горюч, токсичен. Сероуглерод не растворим в воде,
хорошо растворим в органических растворителях. Получается при
165
взаимодействии серы с углеродом (температура 850—900 С) или из
метана:
Температура реакции 600° С, катализатор — окислы кремния и
алюминия. Применяется в качестве растворителя, в производстве
вискозного шелка, каучука. Является фунгицидом.
S
Тиомочевина, тиокарбамид h2n—С—NH2. Белое
кристаллическое вещество, хорошо растворимое в воде. Применяют в ряде
синтезов и в качестве ростового вещества.
Сульфокислоты. Из органических соединений окисленной серы
наиболее важными являются сульфокислоты — соединения,
содержащие в молекуле сульфогруппу SO3H, атом серы которой связан
с углеродом углеводородного радикала. Получение и свойства
сульфокислот алифатического ряда было разобрано ранее (стр. 31).
В настоящем разделе познакомимся с ароматическими сульфокис-
лотами.
Основным способом получения сульфокислот ароматических
углеводородов является реакция сульфирования:
С6Н6 + H2SO4 = C6H5SO3H + Н2О
Сульфирование проводят олеумом на холоду или
концентрированной серной кислотой при нагревании, для введения второй сульфо-
группы температуры повышают до 240° С, третьей — до 300° С.
Сульфирование — типичная реакция электрофильного замещения,
электрофильным агентом является бисульфониевый ион (SO3H+)
или трехокись серы (SO3). Сульфокислоты отделяют от серной
кислоты, используя различия в растворимости их кальциевых и
бариевых солей.
Ароматические сульфокислоты — бесцветные кристаллические
вещества, хорошо растворимы в воде, гигроскопичны.
Сульфокислоты являются сильными кислотами. Для них
характерны три группы реакций: реакции сульфогруппы A);
реакции замещения сульфогруппы B,3); реакции бензольного
ядра.
Образование солей:
C6H5SO3H + NaOH -> C6HaSO3Na + Н2О
Гидролиз сульфокислот. При действии на сульфокислоты
перегретого пара образуются бензол и серная кислота:
C6H5SO3H + Н2О -> С9Н6 + H2SO4
Сплавление солей сульфокислот с едкими щелочами:
C6H5SOaNa + 2NaOH -> C6H3ONa + Na2SO3 + H2O
166
Обработав фенолят натрия кислотой, получают фенолы. Это один
из промышленных способов получения фенолов:
2C6H5ONa + H2SO4 -> 2С6Н5ОН + Na2SO4
Сульфогруппа относится к заместителям второго рода и
ориентирует вхождение новых заместителей в же/ш-положение.
Сульфокислоты применяются в технике для получения
красителей, лекарственных препаратов. Введение сульфогруппы в
ароматическое ядро часто применяют для повышения растворимости
соединений в воде. Хлорангидриды сульфокислот C6H5SO2C1
используют для получения эфиров, амидов и т. д. Производные амидов
CeH5SO2NH3 применяют при изготовлении лекарственных
препаратов.
ЧАСТЬ ЧЕТВЕРТАЯ
АЗОТСОДЕРЖАЩИЕ
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Органические соединения, содержащие в молекуле атомы азота,
широко представлены в природе и играют большую роль в технике.
К ним относятся белковые вещества, многие 'важнейшие
физиологически активные соединения, полимерные материалы, красители,
лекарственные препараты. В настоящем разделе будут рассмотрены
те азотсодержащие органические соединения, в молекуле которых
атом азота непосредственно связан с атомом углерода: нитросоеди-
нения, амины, диазо- и азосоединения, аминокислоты.
НИТРОСОЕДИНЕНИЯ
Определение, строение, номенклатура, изомерия. Нитросоеди-
нениями называются вещества, содержащие в молекуле нитрогруппу
(—NO2), азот которой непосредственно связан с атомом углерода.
В состав молекулы органического соединения могут входить одна
или несколько нитрогрупп. В зависимости от строения
углеводородного радикала, с которым связана нитрогруппа, различают
алифатические (насыщенные и ненасыщенные), ароматические
и гетероциклические нитросоединения. Нитрогруппа в молекуле
нитросоединения может быть связана с первичным, вторичным или
третичным атомами углерода. Строение ннтрогруппы отличается
рядом особенностей, влияющих на физические и химические
свойства нитросоединений. Атом азота в нитрогруппе связан с одним
из кислородных атомов двойной связью, осуществляемой двумя
парами электронов, а с другим кислородным атомом — семиполярной
связью при помощи донорно-электронной пары, предоставленной
азотом и кулоновским взаимодействием положительно заряженного
атома азота и отрицательно заряженного атома кислорода. Это может
быть изображено формулами (I, II, III, 1У),некоторая выравненное^
электронной плотности может быть передана с помощью формулы V.
R:
N:
I
• •
:0:
• *
V
II
4
0
III
0,
R—N'
IV "" V
168
Для наименования нитросоединений к названию
соответствующего углеводорода прибавляют приставку «нитро-», указывая номер
углеродного атома, у которого расположена нитрогруппа:
СНЭ—NO3
нитрометан
NO2
1
сн3—сн—сн3
2-нитропропан
С6Н5-Ш2
нитробензол
Изомерия нитросоединений связана со строением углеродной
цепочки и положением нитрогруппы. Нитросоединения изомерны
эфирам азотистой кислоты
r_NO, R—О—N=0
нитросордннение эфир азотистой
кислоты
Способы получения. Основным способом получения ннтросоеди-
нений является реакция нитрования. Нитрование может
проводиться азотной кислотой, нитрующей смесью (смесь азотной и
серной кислот), окислами азота. В зависимости от строения исходных
веществ, условий нитрования, характера нитрующего агента в
результате реакции получаются несколько различные продукты.
Общая схема реакции имеет следующий вид:
R—H ) [ R—N0.
I нитрование
или > > I или
f
Аг—Н \ [ Аг—N02
Нитрование алканов и цикланов может быть осуществлено по
реакции Коновалова (стр. 30) или в газовой фазе (парофазное нитрование,
стр. 31). Нитрование ароматических соединений — типичный
пример реакций электрофилыюго замещения (стр. 71). Нитрогруппа
является заместителем второго рода, поэтому вторая и третья
нитрогруппа вступают в жета-положения. Если получение
нитробензола может быть успешно осуществлено при температуре 30—40° С,
то введение второй нитрогруппы требует более высоких температур
(90—100° С), а третья — 130—150° С и более сильного нитрующего
агента.
Физические свойства. Нитросоединения жирного ряда — высококипящие
жидкости, с приятным запахом, мало или совершенно не растворимые в воде.
Плотность первых четырех представителей гомологического ряда (до нитро-
бутана) больше единицы, последующих — меньше единицы. Ароматические
нитросоединения — жидкости или твердые вещества, с запахом горького
миндаля, ядовиты. Нитросоединення обладают большим дипольным моментом.
Повышенная полярность нитросоединений, высокие температуры кипения и
плавления (например, по сравнению с изомерными им эфирами азотистой
кислоты) связаны с наличием в их молекуле семиполярной связи.
Химические свойства. Химическое поведение нитросоединеиий
определяется наличием в молекуле нитрогруппы и ее особенностями,
а также строением углеводородного радикала и их взаимным
влиянием.
Восстановление нитросоединений. При восстановлении
нитросоединений образуются первичные амины. Особенно большое про-
169
мышленное значение имеет восстановление ароматических нитро-
соединений:
CeH5-NO2 J^q C6H5NH3
нитробензол аннлнн
В зависимости от условий восстановления (в кислой, щелочной
или нейтральной средах) и характера восстановителя в ходе
реакции образуются различные промежуточные продукты, многие из
которых нашли широкое применение в технике.
Действие щелочей на нитросоединения. При введении в молекулу
углеводорода нитрогруппа вследствие ее электроноакцепторных
свойств резко повышает подвижность атомов водорода в а-положе-
нии. Первичные и вторичные нитросоединения приобретают
способность растворяться в щелочах с образованием солей. При действии
,.0-, /ONa нг, /ОН
R_CH2-№f ^H?Jr_CH=n/' JHr_ch=n/
\q \q \q
нитросоединение соль аци-формы аци-нитро-форма
кислоты образуется нитросоединение в аци-нитро-форме, которая
затем переходит в нитро-форму. Взаимное превращение двух форм
нитросоединений является типичным примером
R-CH3-Nf —R— СН=С/ .
Ч-0 Х)Н
нитро-форма аци-нитро-форма
динамической изомерии (таутомерии)..
Действие азотистой кислоты на нитросоединения. При
взаимодействии первичных нитросоединений с азотистой кислотой
образуются нитроловые кислоты, вторичных — псевдонитролы:
R-CH2-NOa+HO—N=0——» R-CH—N=0 ^ R-C=N-OH
-Н2О | |
N02 NO,
питроловые
кислоты
N0,
R—CH—NO2 + H-O—N=0——-^ R-C-NO
R R
псевдонитролы
Третичные нитросоединения с азотистой кислотой не реагируют.
Щелочные соли нитроловых кислот и их растворы окрашены
в ярко-красный цвет, растворы псевдонитрилов в эфире и
хлороформе — в синий цвет. Эта реакция позволяет различить
первичные, вторичные н третичные нитросоединения.
Реакции бензольного ядра ароматических нитросоединений.
Нитрогруппа предпочтительно ориентирует вхождение второго
заместителя при электрофильном замещении в жета-положение,
170
при нуклеофильном в орто- и пара-положения. Примером
производных нитросоединений ароматических углеводородов
является 2, 4, 6-тринитрофенол (пикриновая кислота):
он
O2N4 Jv /NOg
N02
пикриновая кислота
Пикриновая кислота и ее соли применяются в качестве взрывчатых
веществ и в аналитической химии.
Применение. Нитропарафины применяются в промышленности
в качестве растворителей, добавок к дизельным топливам,
снижающих температуру их воспламенения, при производстве взрывчатых
веществ, пластмасс, в реактивной технике, в качестве
полупродуктов в синтезе аминов, альдегидов и кетонов, жирных кислот.
Ароматические нитросоединения широко применяются для получения
красителей, пластмасс, душистых и взрывчатых веществ.
Отдельные представители. Нитрометан СН3 — N02. Жидкость,
''кип = 101,2° С. Применяется в качестве растворителя.
Хлорированием нитрометана получают трихлорнитрометан (хлорпикрин)
СС13 — NO2, который применяется для борьбы с грызунами в
хлебохранилищах и складах и в разнообразных синтезах.
Нитроэтан СН3 — СН2 — NO2. Жидкость, tKim = 113Э С.
Применяется для получения гидроксиламина:
- СН3-СН2-\О2 (Н+>; "^ СНз-СООН + NH2OH
нитроэтаи гидроксил-
амин
Ншпроциклогексан C6HnNO2. Жидкость, tKUn = 205° С.
Получают нитрованием циклогексана. Применяется в качестве
полупродукта в синтезе капролактама.
Нитробензол C6H5NO2. Жидкость желтоватого цвета, с запахом
горького миндаля, tmm = 211° С. Плохо растворим в воде и хорошо
во многих органических растворителях. Исходный продукт в
производстве анилина (стр. 176), широко применяется в анилинокрасоч-
ной, парфюмерной,, фармацевтической промышленностях.
Тринитротолуол, тол, тротил
СН3
NO,
Твердое вещество, tnu — 80° С. Широко применяется в качестве
взрывчатого вещества.
171
АМИНЫ
Аминами называются органические соединения, содержащие
в молекуле группу —NH2, называемую аминогруппой. Амины
можно рассматривать и как производные аммиака, в молекуле
которого атомы водорода замещены на углеводородные радикалы. Одно-
замещенные производные аммиака называются первичными, двухза-
мещенные — вторичными, трехзамещенные —• третичными аминами:
R Н Н
! ! j
R—N-H «- H-N-H н> R—N—Н
вторичный аммиак первичный
амин амин
R—N—R
третичный
амин
В зависимости от строения радикала различают алифатические
(насыщенные и ненасыщенные), алициклические, ароматические
(содержащие аминогруппу в ядре или в боковой цепи) и
гетероциклические амины.
Номенклатура. Изомерия. По рациональной номенклатуре амины
рассматриваются как алкилзамещенные аммиака; название их
составляется из двух частей: названия радикала и слова амин (I, II).
По ИЮПАК, к названию углеводорода прибавляют окончание «амин»
с указанием номера углеродного атома, связанного с аминогруппой
(III):
СНЯ NH2
CH3CH2-NH2 CH3—NHCH—СН3 СН3-СН-СН2-СН3
эгиламин (I) ыетилнзопропиламин (II) 2-бутанамин (III)
Изомерия аминов определяется количеством и строением
радикалов, а также положением аминогруппы.
Способы получения. Среди многочисленных и разнообразных
способов получения аминов отметим три.
Восстановление нитросоединений (реакция Зинина). В 1842 г.
Н. Н. Зинин восстановлением нитробензола сернистым аммонием
впервые синтетическим путем получил анилин. Реакция сыграла
выдающуюся роль в создании анилинокрасочной промышленности
и получении разнообразных органических красителей синтетическим
путем. В настоящее время восстановление нитробензола в технике
осуществляется двумя путями:
а) восстановлением нитробензола железными стружками в
присутствии растора хлористого железа
4С„Н5Ш2 + 9Fe + 4Н.2О -> 4C6H5NH.3 + 3Fe3O4
нитробензол анилин
172
б) каталитическим гидрированием нитробензола молекулярным
водородом
C6H5NO2 —\ С6Н5—NH2 + 2H2O
нитробензол анилин
Реакция протекает при температуре 270—300° С, давлении до 1,5 атм
A4,7 • 104 Н/м2) в присутствии металлов или их окислов (например,
меди, нанесенной на двуокись кремния).
Алкилирование аммиака (реакция Гофмана). При действии
аммиака на галогеналкилы атомы водорода в аммиаке постепенно
замещаются на углеводородные радикалы. В результате реакции
образуется смесь солей первичных, вторичных и третичных аминов,
а также четвертичного аммониевого основания:
СН31 + NH3X -» (CH3NH3)+ Г ~н-± CH3NH2 + NH4I
йодистый соль метиламина метиламин
метил
CHgNHj + CHsI -» [(СН3J NH2]+ Г —% (СН3J NH +NH4I
метиламин соль диметилзмина диметилямин
(СН3J NH + CH3T -» [(CHg)g NH]+ Г-&U (CH3)g N + NH4I
диметилаынн соль триметиламина триметил-
амин
(СН3K N + CH3I -» [(CHSL N]+ Г
триыетил- йодистый тетра-
амин метиламмоний
Алкилирование аммиака является примером реакций нуклео-
фильного замещения: атом углерода в йодистом метиле вследствие
поляризации связи С—I имеет частичный положительный заряд и
нуклеофильно атакуется аммиаком, азот которого отдает на
образование связи донорную электронную пару. Недостаток этой
реакции — образование трудноразделимой смеси аминов.
Для выделения индивидуальных соединений реакционную массу
обрабатывают щелочью и перегоняют с водяным паром, остаток
после отгонки — гидроокись тетраметиламмония [(СН3L N]+OH-.
Отогнанную смесь аминов разделяют специальными химическими
методами.
Взаимодействие спирта с аммиаком. Этим способом получаются
r_OH + NH3 350°C| AU^ R—NHa+H2O
главным образом низшие алкиламины (Сх — С4).
Физические свойства. Простейшие ампны алифатического ряда — газы
с запахом аммиака, более сложные —¦ жидкости. Растворы их имеют
неприятный запах испорченной рыбы. Высшие амины — твердые вещества.
Растворимость алифатических аминов в воде уменьшается с увеличением молекулярной
массы. Ароматические амины — жидкости или твердые вещества, без цвета,
не растворимые или плохо растворимые в воде (табл. 20).
173
Таблица 20
Важнейшие
Название
Метиламин
Диметиламин
Триметиламин
Пропил амин
1,6-Гексаметилендиамин
Анилин
Метиланилин
Димети лани лин
представители аминов
Формула
CH3NH2
(CH3JNH
(CH3KN
C3H7NH2
H2N(CHoNNH2
QH5NH2
QH,NHCH3
QH5N (CH3J
T
пл., °C
—
— 117
—
42
—6
—
2,5
Т. кип., СС
—7,6
7
3,5
49
196
184
196
194
Химические свойства. Химическое поведение аминов
определяется наличием в их молекуле аминогруппы. На внешней
электронной оболочке атома азота пять электронов (они условно
обозначены звездочками), в молекуле амина так же, как и в молекуле
аммиака, азот затрачивает на образование трех ковалентных связей три
электрона, а два электрона остаются свободными:
CH3*N*H
Н
метиламин
Наличие свободной электронной пары у атома азота дает ему
возможность присоединять протон, поэтому амины, подобно аммиаку,
проявляют основные свойства: образуют гидроокиси, соли. В этих
реакциях амины, отдавая электронную пару на образование новой
связи, ведут себя как нуклеофильные реагенты.
Образование гидроокисей. Водные растворы аминов имеют
щелочную реакцию, окрашивают красный лакмус в синий цвет вследствие
образования алкилзамещенных гидроокиси аммония:
й Г
¦ .5н10Н"
н
метиламин
н J
гидроокись
метиламмония
На основные свойства аминов влияет строение радикала, с
которым связана аминогруппа. Алифатические амины обладают
большими основными свойствами, чем ароматические, последнее
объясняется сопряжением свободной электронной пары атома азота
с л-электронами бензольного ядра, снижающим ее подвижность.
Образование солей
+
н
c,h5n?h
хлористый
этнлаымоний
ci-
174
Реакции алкилирования и ацилирования. Амины легко
вступают в реакции алкилирования и ацилирования:
СНз—CHsNHa + CH3I -> CH3-CH2—NH—CH3 + HI
этдламнн йодистый метилэтила мин
метил
CH3-CH2NH3+ (СН3СОJ О -» СН3—CH2NH—СО-СНз + СН3—СООН
этиламин уксусный этилацетиламнд нлн
ангидрид втилацетамид
Действие азотистой кислоты на алифатические амины. При
взаимодействии первичных аминов с азотистой кислотой образуется
спирт и выделяется азот, вторичные амины превращаются в нитро-
зоамины (соединения, содержащие группу —N=0), с третичными
аминами азотистая кислота не реагирует:
R—NHa + HNOa-»
R R
>NH + HNO2-> \n—N=O + HaO
r/ r/
Реакция позволяет различать первичные, вторичные и третичные
амины. Реакция ароматических аминов с азотистой кислотой
протекает иначе (стр. 177).
Реакции бензольного ядра в ароматических аминах. Аминогруппа
является заместителем первого рода, она повышает электронную
плотность в ядре бензола, особенно в орто- и пара-положениях,
и облегчает реакцию электрофилыюго замещения.
Применение. Амины применяются в качестве селективных
растворителей, в производстве красителей, поверхностно-активных
веществ, фармацевтических препаратов. Алкиламины используются
при производстве ускорителей вулканизации каучука,
инсектицидов. В промышленности полимерных материалов большую роль
играют диаминопроизводные углеводородов.
Отдельные представители. Моноамины алифатического ряда.
Моно-, ди-, триметиламины. Используются в производстве
красителей, фармацевтических препаратов. Этиламин применяется
при крашении тканей, в медицине, как компонент ракетного
топлива.
1,6-Гексаметилендиамин NH3—(СН2N—NH2. Твердое вещество,
получается из адипиновой кислоты:
НООС-(СНзL-СООН —^H4NOOC-(CH2J-COONH4 ^-3
адипиновая кислота диаммонийадепат
-* HaNCO~(CH2L—CONH2 -» NC-(€H2L-CN ±1% H2N—(CH3N-NH3
адиподиннтрил 1,6 -гексаметилендиамин
Гексаметклендиамин играет большую роль в производстве
полимерных материалов, в молекуле которых мономеры соединены амид-
ными группами HNCO, получивших название полиамидов. Приме-
175
ром полиамидов является продукт поликонденсации диамина
и двухосновной карбоновой кислоты —¦ найлон:
пНООС— (CH2h—COOH + nH.2N—(СН2)8—NH2 =^2"~')Ha°
адипиновая кислота гексаметилендиамии
^...—СО—(CH2)j—CONH—(CH^s—NHCO-(CHaL—COi\H-(CHaN—Ш—...
найлон
и продукт поли конденсации капролактама — капрон:
H2C
СН2 -н> .. .NH—(СН2M—СО—NH—(CH,M—CO—NH-
н2с с=о капрон
\/
NH
капролактам
Полиамиды, выпускаемые во многих странах под различными
названиями, благодаря высокой прочности, упругости и
химической устойчивости нашли широчайшее применение для
изготовления технических и бытовых тканей, лент, сетей и т. д.
Аминобензол, анилиН С6Н5—NH2, Бесцветная жидкость, с
характерным запахом. Получается восстановлением нитробензола
(стр. 170). Легко бронируется, сульфируется, окисляется.
Сульфопроизводное анилина —т я-сульфоанилин (сульфаниловая
кислота) NH2—С6Н4—SOSH применяется для производства
лекарственных препаратов, носящих название сульфамидных
препаратов.
Анилин —¦ один из важнейших продуктов современной
органической химии. Применяется в производстве красителей,
фармацевтических препаратов, вспомогательных веществ для резиновой-
промышленности, получения полимерных материалов.
ПОНЯТИЕ ОБ АЗО- И ДИАЗОСОЕДИНЕНИЯХ
Диазосоединения содержат в молекуле характерную
группировку, состоящую из двух атомов азота, связанную с одним
ароматическим радикалом: ArN2X, где X—-С1~, ОН~ и т. д. В азосоедине-
ниях группировка из двух атомов азота связана с двумя радикалами:
Аг—-N =N—Atj. Азо- и диазосоединения в зависимости от характера
радикалов могут быть алифатического или ароматического ряда,
именно последние играют большую роль в разнообразных
промышленных синтезах.
Диазосоедшения
Строение. Изомерия. Диазосоедннеиия существуют в нескольких изомерных
формах. Наибольшая роль принадлежит треп формам: солям диазония
[Аг—N+=NJX~, образующимся в кислом растворе; диазогидрату Аг—N=N—ОН,
существующему в средах, близких к нейтральным, и дназохалам Аг—M=N—ОМе,
176
существующим в щелочной среде. Эти формы диазосоединений связаны между
собой взаимными превращениями (таутомерия). При действии на растворы солей
диазония слабым основанием они переходят в гидроокись диазония, которая
быстро изомеризуется в диазогидрат, последний при действии щелочей образует
соли — диазотаты. Действуя на них соляной кислотой, снова можно получить
соли диазония:
[c6H5N=n] C1 - щ [c6H5N=i\] ОН" ~ CeH5N=N—ОН 71 C6H5N=N—ONa
хлористый гидроокись диазогидрат диазотат натрия
фенилдназоннй фенил диазония
Способы получения. Основной способ получения ароматических диазосоеди-
нений —действие азотистой кислотой на соль первичного ароматического амина
в кислой среде (реакция диазотирования). Реакция протекает в несколько
стадий: сначала образуется соль амина, затем азотистая кислота, которая и
вступает в реакцию диазотирования:
CeH5NH3 + HCl -> CeH5NHa • НС1
анилин солянокислый анилин
NaNO3 + HCl -> NaCl + HNO2
QH5NH2 • HCl-f- HNO2 -* C6H5N3C1 + 2HaO
хлористый
фенил-
дназоний
Реакцию проводят при охлаждении, применяя избыток соляной кислоты.
Свойства. Из изомерных форм диазосоединений особое значение имеют соли
диазония, обладающие высокой реакционной способностью. Соли диазония
в сухом состоянии легко взрываются, поэтому при работе с ними их обычно
не выделяют из растворов.
Превращения диазосоединений могут быть объединены в две группы:
реакции, протекающие с выделением азота, и реакции, протекающие без выделения
азота.
Примерами первой группы реакций является образование фенолов, гало-
генопроизводных ароматических соединений, нитрилов:
CeH5N2Cl + НОН -> С6Н5ОН + Na + HC1
хлористый фенол
фенил-
диазоний
CeH5NaCl+CuI
иод-
бензол
C6H5N2CI + CuCN -» CeH5CN + N
нитрил
бензойной
кислоты
Примером второй группы реакций, протекающих без выделения азота,
является реакция азосочетания. Реакция протекает при действии солей
диазония на фенолы (в слабощелочной среде) или ароматические амины (в
слабокислой среде). Замещение атомов водорода бензольного ядра в молекуле
фенолов или аминов на группу Аг—N=N— происходит в пара- или орго-положениях
по отношению к окси- или аминогруппе.
177
Реакция азосочетания — основной способ получения азосоединений:
$—OH-1-NaOH-»
Г//\
К/
%/ _ J \=/
хлористый фенол
фенилдиазоннй
диметиланилин
-> /~Ч—n=n—/~Ч—n<^ *3+на
\=/ \=/ Ч;нз
л-диметиламикоазобензол
Азосоединения
Полученные при реакции азосочетания ароматические соединения относятся
к группе азосоединений. Азосоединения играют большую роль в производстве
красителей, многие из которых являются их производными. К азосоединениям
относятся и некоторые индикаторы, меняющие свою окраску в зависимости
от реакции среды, например метиловый оранжевый, конго красный. Так, конго
красный в щелочной среде имеет красную окраску, в кислой — синюю:
NHa NHa
so3h SO3H
коиго красный
По современным представлениям окраска органических соединений связана
с наличием в их молекуле особых группировок, получивших название
хромофорных групп или хромофоров (носителей цветности, от греческих слов
«хромое» — цвет, «форос» — носитель). Характерной структурной особенностью
хромофоров является наличие у них ненасыщенных атомов и двойных связей.
К этим группам относятся:
—N=N— \с=О ^N=0 -n{ N>C=C/
/ / \0 / \
азогруппа оксогруппа нитрозо- нитрогруппа этиленовая
группа связь
Некоторые из них являются сильными хромофорами (азогруппа), другие очень
слабыми (оксогруппа). Сложными хромофорными группами является система
сопряженных двойных связей.
Для превращения окрашенного вещества в краситель в его молекулу вводят
специальные группировки, получившие название — ауксохромы (ауксо —
увеличение). Эти группы не только увеличивают интенсивность окраски и ее
глубину, по и обусловливают сродство окрашенного вещества к материалу.
Ими являются аминогруппы, оксигруппы, сульфогруппы.
Нанесение и закрепление красителя на материале получило название
крашения. Химизм крашения сложен и до конца не изучен. В отдельных случаях
173
удается непосредственно нанести красители па ткань (прямые красители). В
других случаях должны быть внесены специальные вещества, способствующие
закреплению красителя (протравы), этот вид крашения получил название
протравного. Иногда под действием кислорода воздуха нанесенное вещество
непосредственно на ткани превращается в краситель. Этот вид крашения называется
кубовым.
АМИНОКИСЛОТЫ
Аминокислотами называются соединения, содержащие в
молекуле два типа функциональных групп: аминогруппу —NH2 и
карбоксильную группу —СООН. Следовательно, аминокислоты
относятся к гетерофункциональным органическим соединениям.
В зависимости от количества функциональных групп различают:
моноаминомонокарбоновые кислоты, моноаминодикарбоновые
кислоты и т. д.:
сня—сн—соон ноос—сн2—сн—соон
j !
NH2 NH2
моноаминомонокар- моноаминодикарбоновая
боновая кислота кислота
В зависимости от положения аминогруппы по отношению к
карбоксильной группе различают а-, $-, у-аминокислоты. По строению
кислоты, в молекулу которой введена аминогруппа, аминокислоты
делятся на алифатические, ароматические и гетероциклические.
В специальную группу выделяют аминокислоты, содержащие окси-
и тиольные группы.
Аминокислоты играют исключительно важную роль в жизни
животных и растительных организмов, являясь теми кирпичиками,
из которых построены молекулы важнейшего природного
полимера — белка. Поэтому широко распространено деление аминокислот,
предложенное специалистами в области биоорганической химии,
на аминокислоты природные (обнаруженные в растительных и
животных организмах) и синтетические, получаемые в лабораториях
и не имеющие природных аналогов. Природные аминокислоты,
в свою очередь, делятся на протеиногенные (входящие в состав бел-
коз) и непротеиногенные (не входящие в состав белков). Среди
природных аминокислот выделяют группу так называемых незаменимых
аминокислот, которые не могут синтезироваться в животном
организме и организме человека.
В настоящее время в природных объектах обнаружено свыше
150 аминокислот, 22 из них входят в состав белков. Основные
аминокислоты приведены в табл. 21.
Номенклатура. Изомерия. Для наименования аминокислот
широко применяются тривиальные названия. По рациональной
номенклатуре аминокислоты рассматривают как производные
соответствующих органических кислот, положение аминогруппы
указывается буквами: a, f>, у и т. д. (табл. 21). По ИЮПАК, для
наименования аминокислот группу NH2 называют приставкой «амино»,
указывая цифрой номер углеродного атома, с которым она связана,
затем следует название соответствующей кислоты.
179
Таблица 21
Наиболее важные представители аминокислот
Название
Формула
Природные аминокислоты, входящие в состав белков
I. Моноа*миномонокарбоновые кислоты
Глицин, гликокол, амино-
уксусная кислота
а-Аланин, а-аминопропи-
оновая кислота
Валин, а-аминоизовале-
риановая кислота
Лейцин, а-амино-7-метил-
валериановая кислота
Изолейцин, а-амино-р-ме-
тилвалериановая кислота
Фенилаланин, а-аыино-р-
фенилпропионовая кислота
H2N—CHa—COOH
СН3—СН-СООН
NH2
сн3—сн—сн—соон
! I
CHg NH2
сн3—сн—сн2—сн—соон
I !
СН3 NH,
СН3—СН2—СН—СН—СООН
СН3 NH2
С6Н6-СН2-СН-СООН
NH,
II. Моноаминодикарбоновые кислоты
Аспарагиновая кислота,
аминоянтарная кислота
Глутаминовая кислота, а-
амнноглутаровая кислота
НООС—СН—СН2—СООН
|\Нг
НООС—СН—СН2—СН2—СООН
NH2
III. Диаминомонокарбоновые кислоты
Лизин, а-, Е-диаминокап-
роновая кислота
НООС-СН-(СНгK-СН2
NH2
Серии, а-амино-C-оксипро-
пионовая кислота
Треонин, а-амино-C-окси-
масляная кислота
Цистеин, а-амино-р-тио-
пропионовая кислота
Метионин, а-амино-у-ые-
тилтиолмасляная кислота
Тирозин, а-амино-р-окси-
фенилпропионовая кислота
NH2
IV. Окси- и тиолоаминокислоты
НООС—С Н —СНо—ОН
NH,
CH3-CH-CH-COOH
I I
OH NH2
сн.—сн—соон
SH NH2
H3C—S—CH2—CH»—CH—COOH
NlH,
HOOC—CH— CH2—f ^—OH
NHa
180
Продолжение табл. 21
Название
Формула
Т. пл.,
°С
Гетерой иклические аминокислоты
Пролин, а-пирролидин-
карбоновая кислота
Триптофан, а-амино-р-ин-
долилпропионовая кислота
Н2С С-СООН
сн
не
—с—сн2—сн—соон
Jc сн
NH»
382
Аминокислоты, не входящие в состав белков
о-Аминобензойная кислота,
антраниловая кислота
я-Аминобензойная кислота
СООН
/ч
-NH,
СООН
а
145
187
Ч/
NH2 I
Изомерия аминокислот зависит от строения углеродного скелета,
положения аминогруппы по отношению к карбоксильной группе, от
наличия асимметрических атомов углерода. Все природные
аминокислоты, за исключением аминоуксуснои кислоты, содержат один или
несколько асимметрических атомов углерода и, следовательно, могут
существовать в виде нескольких стереоизомеров, количество
которых определяется по форм}'ле, приведенной ранее. Каждой паре
оптических антиподов соответствует один рацемат.
Различают D- и L-ряды аминокислот:
СООН
I
H2N-C-H
СН,
L-конфигурацня
L (+)-аланин
СООН
Н—С— NH2
СН3
D-конфигурация
D (—)-алакии
Все входящие в состав белков аминокислоты (протеиногенные)
представляют собой L-формы. D-формы встречаются в природе
редко.
181
Способы получения. Гидролиз белков под влиянием ферментов,
кислот или щелочей. Этим способом можно получить любую из 22
кислот, входящих в состав белков. Однако разделение образовавшейся
при гидролизе смеси аминокислот представляет собой трудную
задачу.
Практическое значение имеет гидролиз биомассы дрожжей,
выращенных на углеводсодержащем сырье и включающих до 40%
белков. Сырьем для получения биомассы микробиологическим путем
может служить также двуокись углерода, спирт, парафины нефти,
природный газ, отходы деревоперерабатывающеи промышленности.
Полученные при гидролизе аминокислоты выделяют хроматографи-
ческими методами.
Действие аммиака на соли хлорзамещенных органических кислот:
С1—СН2—COONH4 + NH3 -» NH2-CH2-COONH4 + HC1
аммониевая соль аммониевая соль
монохлоруксусной аминоуксусной
кислоты кислоты
Действие цианистого аммония на оксосоединения (реакция
Н. Д. Зелинского). По этому способу получают а-аминокислоты:
О NH2 Ш2
СНз-С-Н ±™Ь9Л CH3-CHr-CN -На°:- [Н^1 СНз-СН-СООН
уксусный аминонитрил а-аминопропионовая
альдегид кислота
Получение ^-аминокислот по методу В. М. Родионова. В основе
реакции лежит способность малоновой кислоты за счет подвижных
атомов водорода вступать во взаимодействие с альдегидами по типу
альдольной конденсации (продукт 1). В образовавшемся соединении
оксигруппа заменяется на аминогруппу (продукт 2), а полученное
вещество декарбоксилируется:
СООН СООН
сн3— с/ +нсн -»сн3—сн—с|н -Ь-
уксусный" СООН ОН СООН
альдегид малоновая A)
кислота
СООН
СНз-СН-СН -^г- СНо-СН—СН2-СООН
NH.2 СООН Шг
B) р-амыномасляная кислота
Присоединение аммиака к непредельным кислотам:
СН2=СН—COOHI-2NH3 -> H2N—CH2—CH2—COONHj
акриловая аммониевая соль |3-амшю-
кнслота пропионово1г кислоты
Микробиологический синтез. В промышленности многие
аминокислоты получают микробиологическим путем, выращивая на
специальной питательной среде микроорганизмы, продуцирующие
182
(производящие) в процессе своей жизнедеятельности определенные
аминокислоты. Таким путем получают, например, лизин и глута-
миновую кислоту.
Кроме приведенных общих методов, для синтеза отдельных
аминокислот существуют свои, экономически выгодные, и нашедшие
не только лабораторное, но и промышленное применение методы.
Физические свойства. Аминокислоты — бесцветные, кристаллические
вещества с высокими температурами плавления (табл. 21). Плавятся с разложением,
нелетучи. Они хорошо растворимы в воде и плохо во многих органических
растворителях.
В большинстве случаев аминокислоты D-ряда сладкие на вкус, L-ряда —
горькие или безвкусные.
Высокие температуры плавления аминокислот, их плохая растворимость
в органических растворителях, нелетучесть и некоторые другие нетипичные
для органических соединений свойства связаны с особенностями строения их
молекул. В молекуле свободной аминокислоты аминогруппа, обладающая основ-
НЫД1И свойствами, вступает во внутримолекулярное взаимодействие с кислотным
карбоксилом, образуя внутреннюю соль. Строение ее может быть описано
следующей формулой:
NH3—(СН2)Л—СОО-
Имея в молекуле одновременно кислотную и основную группы,
аминокислоты в водных растворах ведут себя как типичные амфотерные соединения.
В кислых растворах они проявляют основные свойства, реагируя как
основания, в щелочных — как кислоты, давая соответственно дзе группы солей:
NH3—(СН2L—СОСГ+НС1 н> [NH3— (CH2L—СООН]+ СГ
NH3—(СН2L—СОСГ-f-NaOH н> [HaN— (CH2L—COCT]+Na + H2O
Амфотерные свойства аминокислот дают им возможность играть в живом
организме роль буферных веществ, поддерживающих определенную
концентрацию водородных ионов.
Химические свойства. Две функциональные группы NH2 и
СООН, содержащиеся в молекуле аминокислоты, и определяют их
химическое поведение.
Реакции, связанные с наличием
карбоксильной группы. Образование солей (стр. 132),
Образование сложных эфиров:
NH3—СНа—СОО~ + С2Н6ОН + НС1 -» [h3N—СН2—СООС3Н5]+С1- + Н2О
амивоуксусная соль этилового эфира
кислота амииоуксусной кислоты
2 [н3М—СН2—СООСаН5]+ СГ -f- Ag2O -» 2NHa—CHa—COOC2H5 + 2AgC!.a -f- H2O
этиловый эфир амиио-
уксуеной кислоты
Метиловые и этиловые эфиры аминокислот — жидкости, легко
перегоняющиеся в вакууме, поэтому эта реакция применяется для
разделения смеси аминокислот.
Образование галогеноангидридоз:
H3N—CHa—COO--f-PCl5 -фЧНз—СН3—C2-C1J СГ + РОС1а
183
соль хлорангпдрида
аминоуксусной кислоты
При взаимодействии хлорангидрида аминокислоты с аммиаком
образуется амид кислоты:
О
С1-
H3N-CH2-C// Cl-+HNH2_-rLNH3-CH2-C-NH;!
\Cl I на
соль амида аминоуксус-
ной кислоты
Декарбоксилирование:
СН,—СН-СООН н> СН3—СН,—NH2
' т т зтнламии
NH.J
а-амнвопропионовая
кислота
Реакции, связанные с наличием
аминогрупп ы. Образование солей (стр. 183).
Ацилирование аминокислот:
О О
1! ¦ '|
NH3—СНа-СОО- + (СН3_С=ОJ О -> СН3-С—NH-CH.-C-OH +СН3СООН
уксусный ацетилами ноуксусиая
ангидрид кислота
Алкилирование аминокислот. При алкилироваиии аминокислот
галогеналкилами образуются вторичные аммониевые основания.
Внутренние соли этих оснований называются бетаинами:
+ + Р
H3N—СНа—СОО- + ЗСН31 -* N (СН3K—СН,—С/
бетаин аминоуксусной
кислоты
Действие азотистой кислоты на аминокислоты. При действии
азотистой кислоты на аминокислоты выделяется азот и образуется
оксикислота, т. е. аминокислоты ведут себя в этой реакции, как
первичные амины:
H3N— СНа—COO- + HNOa н> НО—СН2—СООН
Эта реакция лежит в основе количественного метода определения
аминокислот по Д. Ван-Сляйку.
Реакции, связанные с наличием и в з аи м н ы м
влиянием а м и н о - и карбоксильной групп.
Отношение аминокислот к нагреванию:
1. При нагревании а-аминокислот образуются циклические
амиды, называемые дикетошшеразинамн:
СН3-СН-СО—;ОН
¦"Н'—NH + HN—|Н
СН3—СН—С=О
NH NH +2Н3О
!но —со—сн—сн3 о=с—сн—сн3
184
а*аминопропионовая 2, 5-диметнл-З, 6-дике-
к iic лота топиперазнн
2. При нагревании р-аминокислот отщепляется аммиак и
образуется непредельная кислота:
СН2-СН-СООН -> CH.2=CH-COOH + NH3
' акриловая кислота
NH2 п
Р-аминопропионовая
кислота
3. При нагревании у- и 6-аминокислот образуются внутренние
циклические амиды — лактамы:
HaN-CH3—СН2—СН2—СООН -> Н2С-СН.2 + Н2О
V-аминомасляная кислота l I
п2С С=О
\/
NH
лактам ^-ами-
номасляиой
кислоты
Образование комплексных солей меди. При взаимодействии
а-аминокислот с гидратом окиси меди образуется комплексная соль
2СК2-СООН О=С—О. /NH.J—СН2
2~
NH2
Н2С—H2N
Образование ди-, три- и полипептидов. Аминокислоты,
соединяясь амино- и кислотными группами, образуют цепи различной
длины, получившие название пептидов;
— NH-C-
-CONH—C—CONH—
пептидная цепь
В зависимости от количества остатков аминокислот, участвующих
в образовании пептида, различают дипептиды, трипептиды,
полипептиды. Связь —N—С— называется пептидной связью. Линейная
Н О
пептидная цепь всегда имеет на одном конце свободную
аминогруппу, на другом — карбоксильную.
Последовательность соединения остатков аминокислот в
молекуле пептида влияет на его свойства. Так, при взаимодействии
двух различных аминокислот (условно обозначим их через А и В)
можно получить два дипептида: АВ и ВА, различающихся по своим
физическим и химическим свойствам. Например, при
взаимодействии гликоколя и аланина могут образоваться глнцилаланин A)
или аланилглицин B):
NH2—СН3—СО—NH—СН—СООН CH3-CH-COHN—СН3-СООН
СНз NH2
A) B)
185
Для синтеза пептидов определенного строения обычно
применяются не сами аминокислоты, а их производные. Такими
«активированными» соединениями могут быть, например, хлорангидриды
кислот. Функциональные группы, которые не должны участвовать
в реакции, заранее «защищают», а после получения нужного пептида
защиту снимают:
О
li N'aOH
C0H5CH2OCONH—СН2—С—Cl + NH2—СН2—СООН
хлорангидрид карбобензоксиглицина
-»CeH5CH2OCONH—CH2-CO—NH—СН2-СООН
карбобензоксиглицилглицина
В нашем случае в молекуле глицина группа СООН «активирована»
заменой гидроксила на атом хлора, а аминогруппа защищена карбо-
бензоксигруппой С6Н5СН2ОСО. После получения пептида защита
снимается восстановлением полученного производного водородом
на палладиевом катализаторе:
CeH5CH2OCONH—CH2—CONH—СН,—СООН->
-> NH2—CH2—CO—NH—СН2—СООН +СвН5СН3 + СО2
глицилглицин толуол
Применение аминокислот. Отдельные представители.
Исключительная роль аминокислот в жизнедеятельности живого организма
связана в первую очередь с тем, что они входят в состав белков.
При попадании в желудочно-кишечный тракт белки пищи под
действием ферментов распадаются на составляющие их
аминокислоты, которые затем используются организмом для построения
своих собственных белков — тканей, крови, кожи, волос и т. д.
Следует отметить, что организм человека обладает способностью
синтезировать только некоторые из нужных ему аминокислот.
Имеется ряд аминокислот (они получили название незаменимых),
которые организм построить не может и должен получать с пищей.
К ним относятся: лизин, лейцин, изолейцин, метионин, фенилала-
нин, триптофан, треонин, валин.
Среднесуточная потребность взрослого человека в аминокислотах
(в кг) приведена в табл. 22.
При нехватке одной из аминокислот в белке он становится
недостаточно полноценным и не полностью усваивается организмом.
Поэтому аминокислоты, полученные синтетическим или
микробиологическим путем, должны добавляться в пищу для повышения ее
полноценности. Для этой цели широко используется, например,
лизин, которого недостает в растительной пище (белке пшеницы).
Смеси аминокислот применяются в медицине, для питания больных.
Отдельные аминокислоты используют в разнообразных синтезах,
в аналитической химии, некоторые аминокислоты применяются
в пищевой промышленности в качестве вкусовых добавок. Для этой
цели используется, например, мононатриевая соль глутаминовой
кислоты, придающая продукту вкус и запах куриного бульона.
186
Таблица 22
Среднесуточная потребность организма человека в аминокислотах
Аминокислота
Тригпофан
Лейцин
Аргинин
Цистин
Изолейцин
Валин
Треонин
Лизин
Метионин
Потребность, кг
0,001
0,005
0,006
0,003
0,004
0,004
0,003
0,004
0,003
Аминокислота
Фенилаланин
Пролин
Гистидин
Тирозин
Алании
Серин
Глутаминовая кислота
Аспарагиновая кислота
Гликокол
Потребность, кг
0,003
0,005
0,002
0,004
0,003
0,003
0.0016
0,006
0,003
Из аминокислот, не входящих в состав белков, следует отметить
о-аминобензойную (антраннловую) кислоту, которая
—соон
антраниловая кислота
применяется в синтезе красителей и лекарственных препаратов,
в пищевой промышленности.
АМИДЫ КИСЛОТ
Амидами кислот называются продукты замещения гидроксильнои
группы в карбоксиле на группу NH2.
Название амидов слагается из двух частей: слова амид и
названия кислоты или ацила:
СН3
/Р
змтгд уксусной
кислоты; ацетамид
Р
амид бензойной
кислоты; бензамид
Способы получения. Действие аммиака на хлорангидриды
кислот (стр. 144).
Гидролиз нитрилов (стр. 189).
Сухая перегонка аммониевых солей карбоновых кислот:
СН2—CONH4-> СН3—CNH2 + HaO
уксуснокислый ацетамид
аммоний
Свойства. Амиды кислот — твердые вещества, за исключением
формамида, являющегося жидкостью, ограничено растворимы
в воде, растворимость уменьшается с увеличением молекулярной
массы. Растворы имеют нейтральную реакцию. Проявляя очень
187
слабые основные свойства, они образуют соли только с сильными
кислотами:
Г /> 1
|_СН3—CNHj
СНз—CNH2 + HC1 -> |_СН3—CNHj ¦ НС1
ацетамид солянокислый
ацетамид
Соли амидов легко разлагаются водой. При гидролизе амидов
образуются кислоты, при нагревании с водоотнимающими
средствами — нитрилы:
ру
ствами — нитрилы:
// ГН+1' ГОН~1 II
СНз—CNH2 + НаО -—— - СНз-С—ОН + NH3
ацетамид уксусная
кислота
СН3—С—NHa -^ CH3-C=N + Н2О
ацетамид ацетоиитрил
/Р
Отдельные представители. Ацетамид СН3—С—NH2.
Применяется в бумажной и кожевенной промышленности.
О
Карбамид, мочевина, диамид угольной кислоты H2N—С—NH,.
Кристаллическое вещество без цвета и запаха. Хорошо растворимо
в воде. Конечный продукт азотистого обмена млекопитающих.
Один из важных продуктов химической промышленности. Карбамид
получается из двуокиси углерода и аммиака:
СОа-т- NH3 -> H2N—С—NHa
II
О
карбамид
Применяется для получения полимеров, в качестве азотного
удобрения, для синтеза лекарственных соединений, в качестве
кормового азота, для производства гербицидов.
НИТРИЛЫ
Нитрилами называются продукты замещения водорода в
молекуле цианистоводородной кислоты HCN на алкилы. Их можно
рассматривать как производные карбоновых кислот. Общая формула
R—C=N. Углеводородный радикал в молекуле нитрилов может
быть насыщенным, ненасыщенным, ароматическим и т. д. Название
нитрилов строится из названия соответствующей кислоты и слова
нитрил:
СНз-CsN СН3=СН—C=N
нитрил уксусной нитрил акриловой
кислоты, кислоты, акрилонитрил
ацетоиитрил
188
Нитрилы получают из амидов карбоновых кислот при нагревании
с водоотнимающими средствами:
R-CONH2 -* R-C=N + Н2О
и при взаимодействии галогеналкилов с солями синильной кислоты:
R_C1 + AgCN -> R—C=N + AgCl
Низшие нитрилы — растворимые в воде жидкости со слабым
эфирным запахом. Основная реакция нитрилов — гидролиз с
образованием соответствующих кислот:
R-C=N + 2Н2О -> R—COO" + NH+
Реакция может протекать в кислой или щелочной средах.
Ацетонитрил СН3—CN. Применяется в качестве растворителя
и в разнообразных синтезах.
Акрилонитрил СН2=СН—CN. Используется при получении
полимерных материалов:
CN
п СН,=СН—CN -> I—СН,—СН—
ЧАСТЬ ПЯТАЯ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гетероциклическими соединениями называются вещества, в
молекуле которых содержатся циклы, содержащие наряду с атомами
углерода атомы других элементов (гетероатомы). Наибольшее
значение имеют гетероциклические соединения, содержащие атомы
кислорода, азота и серы.
Выделение гетероциклических соединений в особую группу
связано с рядом особенностей в их химическом поведении и все
увеличивающимся значением этой группы органических соединений.
Производные гетероциклических соединений имеют важное
физиологическое значение, к ним относятся нуклеиновые кислоты,
хлорофилл, многие витамины, алкалоиды. Гетероциклические
соединения нашли широкое применение в качестве
лекарственных препаратов. Особое значение приобрели соединения
этой группы, содержащие в молекуле пяти- и шестичленные
циклы.
Гетероциклические соединения делятся на группы по количеству
гетероатомов, содержащихся в цикле (один, два и т. д.), по размерам
цикла. В особую группу выделяют гетероциклы с
конденсированными ядрами.
При наименовании гетероциклов широко применяются
тривиальные названия: например, пиррол, фуран, пиридин.. Для
наименования производных атомы, образующие цикл,
нумеруют, начиная от гетероатома, или обозначают буквами
греческого алфавита (I):
в НС -СНВ
(I) (Ш
Для изображения гетероциклических соединений применяются
упрощенные формулы, которые широко используются для цик-
ланов и ароматических соединений, с указанием вида
гетероатома (II).
190
ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
Пягнчленные гетероциклические соединения
с одним гетероатомом
К этой группе соединений относятся фуран, тиофен, пиррол
и их производные:
НС СН НС СН НС—СН
II II !! II I!
СН НС СН НС СН
НС
II
НС
\/
NH
фуран тиофен пиррол
В молекуле пятичленных гетероциклов в результате сопряжения
п-электронов двойных связей и электронной пары гетероатома
возникает общее электронное облако из шести р-электронов (I),
поэтому они по своим химическим свойствам несколько напоминают
бензол (ароматический характер). Ароматические свойства в
наибольшей степени выражены у тиофена, несколько слабее у фурана
и пиррола:
tl)
Способы получения. Получение из 1,4-дикарбонилсодержащих
соединений. При дегидратации 1,4-дикетонов (I) образуется
производное фурана, при взаимодействии с аммиаком — пиррол, с
фосфорными соединениями серы — производные тиофена:
НС СН Н2С СН2 НС СН
D Г- Г- ТЭ ¦ 2 ТЗ /
производное (I) производное
фурана тиофена
НС—СН
11 11
R-C C-R
NH
производное
пирролз
Получение фурана из фурфурола:
НС С-СНО НС СН
фурфурол фуран
191
Получение тиофена из бутана:
з—СНг—СН2—CH3
700= С
нс-
il
НС
-СН
СН
+ 3H2S
тиофен
Получение пиррола из ацетилена:
НС СН НС—СН
|i| + 1:1 +NH3-> i II
НС СН НС СН
\/
NH
пиррол
Химические свойства. Реакции электрофильного замещения.
Пятичленные гетероциклы с одним гетероатомом, как уже
указывалось, обладают ароматическими свойствами, поэтому для них
характерны реакции электрофильного замещения. Заместитель
предпочтительно направляется в а-положение:
НС-
НС
-СН
СН
V
тиофен
Вг2
с
(
с
HNOS
•
с
СН3СОС1
А12О, "
|
II
1 !!
i-бромтиофен
\S/
с-тио
фок
&-иит
зенсуль-
ислота
ротиофен
4s/ Ч:осн3
а-ацетилтиофен
Ароматические свойства у фурана и пиррола выражены слабее, их
кольца обладают меньшей стойкостью и под действием кислот
разрушаются. Поэтому при сульфировании и нитровании требуется
создание специальных условий, например, сульфирование
комплексом, состоящим из серного ангидрида и диоксана:
фуран
Сн2 сн2
I I
СН2 СН2
so3
комплекс
сн2
С.Н2
i
СН2
1, 4-диоксан
192
Восстановление пятичлеямых гетероциклов. Пятичленные циклы
с различной степенью трудности вступают в реакции присоединения,
примером их может служить присоединение водорода:
НС-
НС
-СН
II -
СН
2Н,
фуран
сн2—сн2
сн2 сн2
/
тетрагкдро-
фуран
Реакция восстановления фурана протекает при температуре 100° С,
давлении 150 атм A4,7 -10° И/м2) в присутствии скелетного никеля.
Взаимные превращения циклов. При нагревании до 400—450° С,
в присутствии А12О3 и участии H.2S, NH3 и КаО происходит взаимное
превращение циклов:
i |
H.O
H,0
фуран
4s/
тиафен
пиррол
Отдельные представители и их производные. Фуран —
бесцветная жидкость, i,iim = 81,4е С, с ззпахом хлороформа, не растворима
в воде. Легко окисляется на воздухе. Наиболее важные
производные — фурфурол и пироелнзевая кислота:
¦сУ \i
фурфурол
\.
Р—соон
ппрослиз^зая
кислота
Фурфурол — бесцветная маслообразная, высококипящая
жидкость A62° С) с запахом печеного хлеба. Образуется пси кислотном
гидролизе пентозанов (стр. 238). Применяется в разнообразных
синтезах, при производстве пластмасс, в качестве растворителя.
Тиофеп — бесцветная жидкость с запахом бензола, iKUT1 — 84s С.
Применяется в разнообразных синтезах.
Пиррол — бесцветная жидкость, /К!Ш = 131° С, с запахом
хлороформа. Обладает слабыми кислотными свойствами, при
взаимодействии со щелочами образует соли, которые при взаимодействии
с галогекопроизводньми образуют алкилпронзводные пиррола:
кон i i ch,i
"" У~1 «-СН
NH
а-метилпиррол
NH
пиррол
NK
пиррол-
калий
сн3
N-метпл-
пиррол
7 А. П. Нечаев
193
Производные пиррола широко распространены в природе, к их
числу относятся такие важные соединения, как хлорофилл,
гемоглобин, гетероциклические аминокислоты, витамин В12. Многие
из этих соединений содержат в молекуле особую группировку из
четырех пиррольных ядер, называемую ядром порфина:
НС
\\
NH N-
.сн
сн
ядро порфина
При замещении атомов водорода в ^-положении в пиррольных
кольцах на радикалы образуются порфирины. Многие важные
природные соединения являются металлическими производными пор-
фиринов. Пигменты крови человека и животных — гемоглобины,
содержат атом железа, являясь хромопротеидами. Их молекула
состоит из двух частей — из белковой части (глобинов) и простети-
ческой группы — гема.
Зеленые пигменты растений — хлорофиллы, являются магний-
производными: в их молекуле атомы азота пиррольных ядер связаны
с магнием:
хлорофилла
Хлорофиллу принадлежит выдающаяся роль в фотосинтезе:
усвоении углекислого газа зелеными растениями за счет световой
энергии с образованием органического вещества. Фотосинтез
является основным процессом образования органического вещества
на Земле. Этот процесс может быть представлен следующим образом:
6СО3+12Н2О
хлорофилл
194
Особую группу производных пиррола составляют его бицикли-
ческие производные. В их молекуле содержится два ядра: пирроль-
ное и бензольное. Представителем этих соединений является индол:
NH
К производным индола относится и уже известная нам
аминокислота — триптофан (табл. 21). Многие производные индола
относятся к стимуляторам роста растений, красителям (индиго).
Пятичленные гетероциклы
с двумя гетероатомбми
Представители этой группы соединений содержат в цикле два
гетероатома, один из них — атом азота. Общее название этих
соединений — азолы:
, N
! Ii
X /
ок^гол
N
изоксазол
{ j
гиазол
j
изотиазол
II
I
N
\/
NH
I,—N
ii I
\/
NH
имидазол
Производными их являются важные в физиологическом отношении
соединения и многие лекарственные препараты.
ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
Шестичленные гетероциклы
с одним гетероатомом
Из шестичленных гетероциклических соединений с одним
гетероатомом познакомимся с двумя: пираном и пиридином, а также
с их производными.
Группа пирана. Производные пирана широко распространены
в природе.
СН,
НС СН
'| '^
НС СН
\/
пиран
К ним можно отнести и подробно разобранные гексозы и продукты
их поликонденсации, имеющие в молекуле пиранозный цикл. К
производным пирана относятся пигменты (красящие вещества)
растений, алкалоиды. Представителями этой группы соединений являются
7* 195
распространенные в природе флавоновые пигменты (I; II). Они
обусловливают окраску деревьев, многих цветов, чая.
ОН О
ОН
НО—'
но—:N
-он <?
-он
-он
хриэин
(I)
кверцетнн
(П)
К производным пирана следует отнести и катехины:
он сн2
с<
НО—
с-
Н
—ОН
-ОН
н
эпикатехин
Катехины содержатся в большом количестве в листьях чая,
бобах какао. Окислительные превращения катехинов играют
важную роль во многих процессах пищевой технологии: производстве
чая, какао, виноделии. Они придают пищевым продуктам
специфический вяжущий вкус и окраску.
Группа пиридина. Пирпдни и его гомологи получают из
каменноугольной смолы или синтетическим путем из ацетилена и синильной
кислоты:
пиридин
Пиридин — жидкость с крайне неприятным запахом, tKn!l = 115° С,
хорошо растворима в воде. Вступает в реакции электрофильного
замещения (галогенируется, нитруется, сульфируется), хотя и со
значительным трудом. Замещающая группа обычно вступает в мета-
положение. Основные свойства пиридина проявляются в его
способности образовывать соли:
+ НС1->
С1-
NH.
пиридин
солянокислый
Пиридин применяется в производстве красителей, лекарственных
соединений. Амид Р-пиридннкарбоновой кислоты является
витамином PP.
190
Понятие об алкалоидах
Алкалоидами называется группа сложных по своему составу
природных органических соединений. Большинство из них относится
к азотсодержащим гетероциклическим соединениям. В больших
количествах и при систематическом употреблении они являются
сильными ядами, в малых количествах широко применяются в
качестве лекарственных препаратов. Представителями алкалоидов
являются кониин; никотин; морфин:
ОН
сн2
кониия
никотин
морфин
Синтез и исследование алкалоидов — один из наиболее сложных
азделов органической химии.
ЧАСТЬ ШЕСТАЯ
БИООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
И НЕКОТОРЫЕ СОЕДИНЕНИЯ
ИЗОПРЕНОИДНОГО ХАРАКТЕРА
Липиды
Липидами называется сложная смесь органических веществ,
выделяемых из растительных и животных объектов. Они обладают
близкими физико-химическими свойствами, в первую очередь
нерастворимостью в воде и хорошей растворимостью в ряде
органических растворителей (диэтиловом эфире, бензоле,
хлороформе).
Основную массу этих веществ составляют сложные эфиры
трехатомного спирта глицерина и высокомолекулярных жирных
кислот— глнцернды (стр. 107, 200). Кроме глицеридов в состав липидов
входятвоски (стр. 205), фосфо- и гликолипиды (стр. 207),
жирорастворимые пигменты (каротиноиды, стр. 278; хлорофиллы, стр. 194),
стерины, жирорастворимые витамины (А, Е, Д, К, стр. 272),
а также продукты их разнообразных превращений.
Липиды широко распространены в природе, являясь
обязательной составной частью каждого живого организма. Они выполняют
роль структурных компонентов протоплазмы клеток и запасных
веществ.
Структурные липиды (главным образом фосфолипиды) образуют
сложные комплексы с белками (липопротеиды, стр. 267) и
углеводами, из которых построены мембраны клетки и клеточных
структур, и участвуют в разнообразных процессах, протекающих в
клетке.
Запасные липиды (в основном триглицериды) являются
энергетическим резервом организма. В растениях они накапливаются
главным образом в плодах и семенах (табл. 23), у животных и рыб —
в подкожных жировых тканях и в тканях окружающих внутренние
органы, а также в печени, в мозговой и нервной тканях. Содержание
их зависит от многих факторов (вида, возраста, режима питания
и т. д.).
Значительное количество липидов находится в молоке
животных: дельфина — 46%, кита — 45%, слона — 20%, козы — 4,8%,
коровы — 3,6%. Активными «создателями» лпппдов являются
некоторые микроорганизмы, содержание липидов в которых может
достигать 20—30% от веса биомассы,
198
Таблица 23
Содержание липидов в семенах и плодах растений
Культура
Подсолнечник
Хлопчатник
Соя
Лен
Арахис
Оливки (мякоть)
Конопля
Тунг (орех)
Рапс
Сурепица
Горчица
Клещевина (ядро)
Содержание
жира, %
51,0
23,0
20,0
44,0
50,0
50,0
33,0
40,0
41,0
40,0
42,0
60,0
Культура
Пшеница
Рожь
Кукуруза
Рис'
Овес
Просо
Гречиха
Арбуз (семена)
Какао (бобы)
Кокосовая пальма (копра)
Кедр (орех)
Содержание
жира, %
2,7
2,5
5,6
2,9
7.2
4,5
3,8
30,0
52,0
70,0
28,0
В живом организме лнпиды выполняют разнообразные функции.
Им принадлежит важная роль в формировании и старении
организма, в деятельности его защитных механизмов. Запасные липиды
являются аккумулятором химической энергии и используются
организмом при недостатке питания и заболеваниях. Подкожные
жировые ткани предохраняют животных от охлаждения, а внутренние
органы — от механических повреждений.
Лппиды — важные компоненты пищи, во многом определяют
ее пищевую ценность и вкусовое достоинство.
Исключительно велика роль липидов в разнообразных
процессах пищевой технологии. Порча зерна и продуктов его переработки
при хранении (прогоркание) в первую очередь связана с изменением
его липидного комплекса. Лппиды, выделенные из ряда растений
и животных, являются основным сырьем для получения
важнейших пищевых и технических продуктов: растительного масла,
животных жиров, в том числе сливочного масла, маргарина,
глицерина, жирных кислот, мыла, моющих средств, витаминов
А, Е, Д.
По химическому составу липиды обычно делят на две группы:
простые и сложные. Простые липиды не содержат азота и фосфора,
к ним относятся в первую очередь сложные эфиры одноатомных
и многоатомных спиртов и высокомолекулярных жирных кислот
(глицериды, эфиры диолов, воски), простые эфиры спиртов,
углеводороды, жирные кислоты, спирты и некоторые другие
соединения.
В состав сложных липидов входят фосфолипнды, соединения, при
гидролизе которых образуются наряду со спиртами (чаще всего
глицерином) и высокомолекулярными жирными кислотами, фосфорная
кислота и аминоспирт.
Иногда в самостоятельные группы липидов выделяют
жирорастворимые пигменты, стерины, жирорастворимые витамины. Липиды,
199
выделенные разнообразными методами из природных ооъектов,
получили название сырого жира или просто жира. Жиры,
добываемые из растительного сырья, называются растительными маслами.
В настоящем разделе будут рассмотрены только основные группы
липидов.
ПРОСТЫЕ ЛИПИДЫ
Глицернды
Определение. Классификация. Глицеридами называются
сложные эфиры глицерина и высокомолекулярных карбоновых (жирных)
кислот. Они составляют основную массу липидов (в отдельных
случаях до 95—97%) и являются, по существу, жирами.
В состав жиров в основном входят триглицериды, но
присутствуют ди- и моноглицериды:
О
ii
СНа—О—С—R
СН—О—С—Rf
1 il
1 11
I 6
СНа—О—С—R2
6
триглиаерид
о
СН2—O-C-R
СН—О—C-Ri
сн2—он
днглицерид
О
СНг—О—С—R
СН—ОН
1
сн2—он
моноглицерид
где R, Rx, R2 — радикалы жирных кислот.
Жирные кислоты глицеридов. В жирах обнаружено свыше
двухсот жирных кислот, однако большинство из них присутствует
в незначительном количестве. В состав многих жиров входят
в небольшом количестве низкомолекулярные кислоты (С2—С10).
Кислоты с числом атомов углерода выше 24 присутствуют в восках.
Наиболее распространенные в жирах кислоты (основные жирные
кислоты жиров) содержат от С12 до С18 (табл. 24).
Большинство природных жиров и масел представляет собой смесь
глицеридов, отличающихся сочетанием относительно небольшого
числа жирных кислот. Учитывая, что одним из структурных
компонентов всегда является глицерин, свойства масел обусловливаются
составом и расположением жирных кислот. Несмотря на
относительно небольшое число основных кислот E—8), участвующих
в образовании глицеридов, количество возможных триглицеридов
может быть значительным:
Количество разных жирных кислот в жире . 5 6 7 8 9 10
Количество возможных трнгл.-щеридов 75 126 196 285 405 550
Поэтому состав жиров достаточно сложный.
200
Физические свойства. Триглицериды — жидкости или твердые вещества
без цвета и запаха, не растворимы в воде, но хорошо растворимы в
органических растворителях.
Таблица 24
Основные жирные кислоты жиров
Кислота
Число
углеродных
атомов
Формула
Насыщенные кислоты
Лаурнновая
Миристиновая
Пальмитиновая
Стеариновая
Арахиновая
Бегеновая
Церотиновая *
Монтановая *
Мелиссиновая *
12
14
16
18
20
22
26
28
30
СНз—(СН.)ю-СООН
СН3—(СНо^-СООН
СНз—(СНг)й—СООН
СНз—(СН?,Iв—СООН
СН3—(СНгI8—СООН
СН3—(СН,),0—СООН
СН3—(СН.,),-4—СООН
СН3—(СН^—СООН
СН8—(CH^g—СООН
Олеиновая
Эруковая
Линолевая
Линоленовая
Арахидоновая
Чаулмугровая
18
Is
18
20
18
Ненасыщенные кислоты
СНз—(СНпO—СН=СН—(СН.,),—СООН
СН,—(СН,O—СН=СН—(СН..")и—СООН
СН3—fCHnL—СН^СН—СН2—СН=СН—(Ь
сн3—сн2—сн=сн—сн2—сн=сн—сн
=СН—(СН2O—СООН
СНзЧСН,L-(СН=СН-СН,K-СН =СН-(СН.,K-СООН
Ь—СООН
СООН
Рнцинолевая
18
Оксикислоты
СНз-(СН2M-СНОН-СН2—СН=СН—(СНаO—СООН
* Встречаются в восках.
Химические свойства. Глицериды вступают во все химические
реакции, характерные для сложных эфиров, однако в их химическом
поведении имеется ряд особенностей, связанных со строением
жирных кислот и глицерина.
Гидролиз триглицеридов. Под влиянием фермента липазы,
кислот, щелочей или специальных смесей (контакт Петрова —
продукт взаимодействия дымящей серной кислоты с высоко-
кипящими фракциями нефти) триглицериды гидролизуются
с образованием сначала ди-, а затем моноглицеридов и в
конечном итоге — жирных кислот и глицерина. Гидролиз сильно
ускоряется, если его проводить при 220—260° С и 25—60 атм
201
B4,5 -105 — 58,8 -105 Н/м2) (безреактивное расщепление жиров):
О
р
CH2—ОН
CH2O—CR
I
CHO—CRH-HOH
6
CH2O—CR2
б
триглицерид
сн.2—он
~,-i CHO—CRj+RCOOH-
Li,
О
диглицернд
сн2—он
—r,-cooh* CH—ОН
СН,—ОН
—R2-COOH
О
CHa-OH
моноглицерид
Результат гидролиза выражается схемой:
О
CH3O-CR
-СРг + ЗН»О l*i
II
О
СН2О—CR3
II
О
CH2—ОН
~ СН-ОН + R-COOH -,- Ri-
I
СН2ОН
+R2—COOH
Гидролиз триглицеридов широко применяется для получения
жирных кислот, глицерина, моно- и диглицеридов. Гидролитический
распад жиров, липидов зерна, муки, крупы и других жирсодер-
жащих пищевых продуктов является одной из причин ухудшения
их качества и в конечном итоге порчи. Особенно ускоряется этот
процесс с повышением влажности хранящихся продуктов.
Гидролиз водными растворами щелочей приводит к образованию
щелочных солей жирных кислот (мыла, стр. 217) и называется
омылением.
Алкоголю. Глицериды при нагревании с метиловым или этиловым
спиртом образуют соответствующие эфиры жирных кислот с
высвобождением глицерина. Такой обмен спиртов в сложных эфир ах
получил название алкоголиза
О
СН;О—CR СН2—ОН О
CHO—CR] +ЗСН3ОН ^ СН—ОН +3R—С—ОСН3
6 СН2—ОН
,Н2О—CR2
j|
6
?02
(катализаторы: едкий натр, алкоголяты щелочных металлов, серная
кислота, хлористый водород). Алкоголиз применяется для
промышленного и лабораторного получения сложных эфиров жирных
кислот.
Ацидолиз. При нагревании триглицеридов со свободными
жирными кислотами до 250—300° С происходит обмен ацилов:
О О
СН2О—С—R СН2О—С—R
CHO-CRi+ 21^-0001-1 ^СНО—CR1 + 2RCOOH
О [ 6
Н20—CR CH2O—CR
II II
О О
Реакция протекает в присутствии следующих катализаторов:
серная кислота, вода, трехфтористый бор. Процесс ускоряется с
увеличением силы свободной жирной кислоты.
Переэтерификация. Глицериды в присутствии катализаторов
(серная кислота, метилат натрия, натрий и калий) способны
обмениваться ацилами, эта реакция получила название переэтерифика-
ции. По-видимому, при этом происходит и частичный обмен ацилов
внутри молекулы триглицерида:
О
СН2О—CR
СНО—CR
6
СН2О—CR
О
О
СН2О—CR
+ CHO-CRi
6
CH2O—CRi
6
(
Э
СНгО—CRi
ПСНО—CR -
! о
СН2О—CR
О
О
CH2O-C-R
¦f СНО—С—R
6
СН2О—CRi
li
6
Переэтерификация приводит к изменению свойств жиров и масел,
поэтому она широко применяется в пищевой промышленности для
получения продуктов с заданными свойствами.
Реакции 1—4 являются примером превращений, связанных с
наличием в молекуле триглицерида сложноэфирных групп. Не меньшее
значение имеют реакции глицеридов, связанные с превращением
углеводородных радикалов.
Гидрогенизация глицеридов. Гидрирование жиров молекулярным
водородом в промышленности проводят при температурах 180—240сС
в присутствии никелевых и медно-никелевых катализаторов, как
правило, при давлении, близком к атмосферному. Подбирая
соответствующие условия реакции, удается осуществить этот процесс
селективно (избирательно), гидрируя сначала в основном радикалы
лнноленовой кислоты до линолевой, затем линолевой до олеиновой,
а уже потом радикалы олеиновой (если это необходимо) до стеари-
203
новой кислоты, и получить продукт с заранее заданными свойствами
(называемый саломасом, стр. 215, 216):
О
I!
СН2—О-С— (СН,O—СН=СН-(СН2O-СН3
СН—О—С—(СН„O—СН=СН-СНо—СН=СН-(СНдL—СНз Cu7k
о
Н3—О—С—(СН,O—СН=СН-СН2—СН=СН-СН,-СН=СН-СН2—СН3
о
о
СН2—О-С—(СН.1)--СН=СН-(СНаO—СН3
Ц*-* СН—О-С—(СН2),-СН=СН-СН2-СН-(СН2L—СН3 с?5П
Cu/N
о
СН2—О—С—(СН2O-СН=СН—СНа—СН=СН—(СН2L—СНз
5
о
о о
!i N
СН2—О-С—(СНаO-СН=СН—(СНгO—СН„ СНа—О—С—С17НЗГ)
_Л?_ I н I
Cu/Ni СН—О—С—(СН2O—СН=СН—(СН2O—СН3 сП7^Т СН—О—С—С1ТН33
о
о
СН,—О—С—(СН,O—СН=СН—(СН2O—СН3 СН2—О—С— С17Н~5
II :|
о о
Окисление жиров. Жиры и масла, особенно содержащие радикалы
ненасыщенных жирных кислот, окисляются кислородом воздуха.
Первыми продуктами окисления являются разнообразные по
строению перекиси и гидроперекиси. Они получили название первичных
продуктов окисления.
В результате сложных превращений перекисей образуются
вторичные продукты окисления: спирты, альдегиды, кетоны,
кислоты и их производные с углеродной цепочкой различной длины.
Образовавшиеся продукты способны к дальнейшим превращениям,
в частности полимеризации.
Направление и глубина окисления зависят в первую очередь
от состава жиров и масел: с увеличением степени непредельности
жирных кислот, входящих в состав глицеридов, скорость их
окисления возрастает. Влияют, кроме этого, способы и режимы их
получения и хранения, а также присутствие влаги, следов металлов,
температура, свет.
В основе современных представлений об окислении жироз и масел
лежит перекисная теория окисления органических соединений
Баха — Энглера и теоретические представления о цепных
реакциях, сформулированные советским ученым акад. Н. Н. Семеновым.
204
В общем виде процесс выглядит следующим образом:
- + O2->ROO-
RH-* ROOH + R-
RO^+RO-; R-+K-; RO2+R-
—CH3—CH=CH—CH2 |-Oa-s>CH2—CH=CH—CH j-
+ HOO-— CH2—CH=CH— CH— +O2->
-> —CH2—CH=CH-CH— CH=CH—CH— + -CH.2—CH2— -->
I
O—0- 0—0-
->- CH=CH—CH— CH,—CH— CH— ¦->—СНСУ [-НО'
1 " I
о—он о—он
Большое влияние на скорость окисления оказывают
антиокислители (ингибиторы) — вещества, добавление которых приводит
к обрыву цепей окисления. Среди них наибольшее значение имеют
окислители фенольной природы (стр. 111, 112): бутилокситолуол,
бутилоксианизол, пропилгаллаты. Из природных антиокислителей
наибольшее применение нашли токоферолы.
Накопление продуктов окисления в жирах и маслах приводит
к изменению их свойств и снижению пищевой ценности, а некоторые
из продуктов окисления оказывают вредное влияние на организм.
Поэтому необходимо принимать меры по предотвращению или
замедлению окисления жиров и масел, используемых для пищевых целей.
В то же время процесс окисления может быть использован в технике
для получения продуктов с новыми положительными свойствами,
например, образование защитных пленок при глубоком окислении
масел («высыхание», стр. 216).
Воски
Определение, классификация. Восками называются входящие
в состав липндов сложные эфиры высокомолекулярных
одноосновных кислот и одноатомных высокомолекулярных спиртов R—СН2—
О
II
—Q—C-—Rb где R—СНа—О— остаток алифатического высокомоле-
О
кулярного спирта; RL—С— остаток карбоновой (жирной) кислоты.
В состав восков входят главным образом кислоты, содержащие
24—32 атома углерода и спирты, углеродная цепочка которых
состоит из 16—30 атомов углерода (табл. 25).
Выделенные из природных источников воски содержат
значительное количество примесей свободных жирных кислот, спиртов,
углеводородов.
205
Т а б л и ц а 2э
Основные спирты восков
Историческое
название
Миристиловый
Цетиловый
Стеариловый
Карнаубиловый
Цериловый
Марициловый
Мелиссиловый
ИЮПАК
1-Тетрадеканол
1-Гексадеканол
1-Октадеканол
1-Тетракозанол
1-Гексакозанол
1-Триаконтанол
1 -Гентриаконтанол
Число
углеродных
атомов
14
16
18
24
26
30
31
Формула
СН3—(СН„Ь—СН2ОН
СН3-(СН2"I4-СН2ОН
СНз—(CH2)i6—СН,ОН
СНз—(СН2)аа—СН,ОН
СНз—(СН»)Ч—СН.2ОН
СНз-(СН:2)м-СН,ОН
СН3-(СН2),9—СН,ОН
В зависимости от происхождения различают растительные,
животные воски, вырабатываемые насекомыми, и ископаемые.
Нахождение в природе, способы получения. Воски широко
распространены в природе. В растениях они покрывают тонким слоем
листья, стебли, стволы и плоды, предохраняя их от смачивания
водой, высыхания, действия микроорганизмов.
Из растительных восков промышленное значение имеют воски,
покрывающие листья пальм (карнаубский воск), воски липидов риса
и подсолнечника.
Из восков животного происхождения наибольшую роль играют
спермацет и спермацетовое масло, шерстяной жир; из восков
насекомых — пчелиный воск. Первые два продукта выделяют из
маслообразной массы, содержащейся в голове кашалота и в длинном
канале, проходящем от его головы до хвоста. Твердый кристаллический
продукт белого цвета, состоящий главным образом из цетилового
эфира пальмитиновой кислоты, называется спермацетом, а жидкий
продукт, оставшийся после его выделения, — спермацетовым
маслом. Из одного кашалота получают 3—5 т C-Ю3 — 5-Ю3 кг)
спермацетового жира.
«Шерстяной жир» — жиропот овечей шерсти, от желтого до
темно-коричневого цвета; с резким неприятным запахом. После
специальной обработки из него получают слабоокрашенный
мазеобразный продукт — ланолин — сложного химического состава,
основной компонент которого — эфиры кислот С10 — С20 и
спиртов С18 — С20. В шерсти овец содержится 5—10% «шерстяного
жира».
Пчелиный воск получают из пчелиных сот вытапливанием или
экстракцией после удаления меда. Он состоит главным образом
из эфиров кислот С26 — С30 и спиртов С14 — С31 и содержит до 15%
углеводородов.
Ископаемые воски (горный воск, воск бурых углей) содержат
до 70% сложных эфиров кислот и спиртов с числом атомов углерода
больше 24.
Свойства. Большая часть восков — твердые, упругие вещества,
не растворимые в воде и при комнатной температуре довольно
плохо растворимые во многих органических растворителях. В хими-
206
ческом отношении воски (особенно содержащие остатки
насыщенных жирных кислот) отличаются значительной стойкостью; с трудом
омыляются, стойки к действию кислорода воздуха.
Применение. Свойства восков определяют и пути их
использования. Они применяются в качестве электроизоляционных покрытий,
являются важным исходным продуктом для получения
разнообразных моющих средств, используются в косметике при изготовлении
кремов и помад. В бытовой химии воски применяются в качестве
компонентов лощильных составов, используемых для полировки
паркетов, мебели и в медицинской промышленности.
СЛОЖНЫЕ ЛИПИДЫ
Фосфолипиды
Определение, распространение в природе. Фосфолипидами
называется наиболее разнообразная и важная группа сложных липидов,
в состав которой входят: фосфорная кислота, насыщенные и
ненасыщенные жирные кислоты, спирты (глицерин, инозит, стр. 106, 208
сфингозин, стр. 210), азотистые основания (холин, зтаноламин,
серии), связанные между собой эфирной или амидной связью.
Фосфолипиды являются обязательной составной частью растений
и животных; их содержание колеблется в широких пределах.
Особенно много их в нервной и мозговой тканях (до 30%). Среднее
содержание фосфолипидов в зерне и семенах некоторых культур
приведено в табл. 26.
Таблица 26
Кулыура
Соя
Хлопчатник
Лен
Подсолнечник
Клещевина
Пшеница
Рожь
Содержание фосфолипидов в зерне и семенах
Содержание
фосфолпппдов, %
1,8
1,7
0,6
0,7
0,3
0,54
0,6
Культура
Кукуруза
Рис
Овес
Просо
Гречиха
4
Содержание
оосфолииидов, %
0,89
0,20
0,37
0,33
0,93
Роль фосфолипидов в жизнедеятельности живого организма
чрезвычайно велика. Вместе с белками и другими соединениями они
участвуют в построении мембран («перегородок») клеток и
субклеточных структур, выполняя роль «несущих конструкций»,
способствуют переносу химических веществ, а также осуществляют другие
функции в биохимических процессах, протекающих в живом
организме.
Основные группы фосфолипидов. Среди фосфолипидов наиболее
распространены и играют особо важную роль соединения, в составе
которых два спирта: глицерин и сфингозин.
207
Фосфоглицериновые э ф и р ы , Общая формула фос-
фоглицериновых эфиров:
О
I!
О СН2—О—С—R
II !
R2—С—О—СН
СН-О—Р—ОХ
j
где R и Rx — остатки жирных (карбоновых) кислот; X — водород
или азотистое основание.
Наиболее простое строение имеют широко распространенные
в природе фосфатидные кислоты (X = Н):
О
CH2--O-C-R
Ri—С—О—СН О
о сн2-о-р-сг
I
О"
мснофосфатидыая кислота
Е растениях, животных тканях и микроорганизмах обнаружены
молекулы, построенные из нескольких остатков фосфатидных
кислот:
О О
S: II
СН2—О—С—R ¦ Н2С—О—С—R
R—С—О—СН О О
II : i] []
6 СНа—О—Р—О— СН2—СН— СН2 СН—О—С—Rj
I
о^
i I у0 I
он о—р-о—сн3
6-
триглицерофосфат, ?1ардиолипин
В фосфатидилинозитах «X» представлен циклическим спиртом —
инозитом:
О
н„с—о—с—в
н он
н он
нонофосфашдилкнозит
208
В состав молекулы фосфатпдилннознта могут входить несколько
остатков инозита, оксигруппы которых эстернфнцированы
кислотами различного строения.
Наиболее распространены среди фосфолипидов холинфосфа-
тиды и этаноламинфосфатиды. В молекуле первых X представлен
азотистым основанием—холнном НО—СН2—СН3—N (СН3)^—(ОН)-:
О
СН2-О-С— R
О
||
Н—О—С—Rx
О
I
СН2—О—Р—О—СН2—СН2—N (СН3)з
О"
фосфатидилхолин (лецитин)
В фосфолипидах масличных семян и животных содержание холнн-
фосфатндов достигает 30—50 %.
В молекуле этаноламннфосфатндов (irx иногда называют
кефалинами) содержится остаток этаноламина НО—СН2—СН2—NH2:
О
СН2—О—С—R
О
I
СН—О—С—Ri
О
СН,—О—Р—О—СН,
-СН,—NH,
О"
фосфатидилэтаноламин
Содержание этаноламинфосфатидов в фосфолипидах масличных
семян и животных тканях достигает 20—25%.
В состав группы фосфолипидов, называемых иногда кефалинами,
входит еще один компонент — серинфосфатиды:
о
сн2-
о
¦Н—О—С—Rx
О NH2
(S ]
СН—О—Р—О—СН2—СН—СООН
I
фосфатидилсерня
209
среди продуктов их гидролиза обнаружена аминокислота серии
НО—СНа—СН—СООН . В тканях живых организмов серинфос-
1|
NH2
фатиды присутствуют в виде солей калия, натрия и магния.
Фосфосфингозиновые эф и р ы. Второй большой
группой фосфолипидов являются производные аминоспирта сфинго-
зина — фосфосфингозиновые эфиры:
СН3—(СН2);2—НС=СН—СН—СН—СН3
I |
ОН NH2 ОН
сфиигозин
Сфингозин входит в состав сфингомиелинов и цереброзидов. При
их гидролизе образуется кроме сфингозина и жирной кислоты
в первом случае холин и фосфорная кислота, а во втором —
галактоза:
СН3
Л
СНЯ
I
(СН3I2 \
СН
СН
I
НО—С—Н
I
н—с—
(СНгI2
!
СН
II
СН
I
но—с—н н
Н—С N—
I
н
сфингоииелин
1
остаток сфингозина
остаток жирной кислоты
; о
¦: Ч .
Т-С—R:
О
!! v +
—Р— Ю-СН2—СНа— N (С
I
А- остаток холина
остаток сфингозина
Н—С-
Н
Н ; О
I : ||
—N Г^
-о-гс<°"
остаток жирной кислоты
! СН2ОН
-—К
н
ННО
I I
он н
ОН
Н
остаток галактизы
церсброзид
210
Молекула цереброзида не содержит фосфора, поэтому эти
соединения относят к группе гликолипндов. Производные сфингозина
вместе с фосфолипидами — основные компоненты липидов мозга.
Свойства, получение, применение. Фосфолишгды — бесцветные
или белые вещества, без запаха, хорошо растворимы в диэти-
ловом эфире, хлороформе, плохо в ацетоне.
В промышленности фосфолипиды получают в качестве
побочного продукта при производстве масел.
Фосфолипиды нашли широкое применение в медицине, в качестве
улучшителей и эмульгаторов в ряде отраслей пищевой
промышленности (хлебопекарной, кондитерской, масложировой).
МЕТОДЫ ИССЛЕДОВАНИЯ ЛИПИДОВ
Анализ липидов и продуктов их превращения, учитывая
особенности их состава, является сложной задачей, требующей
применения наряду с химическими методами современных
физико-химических методов исследования (хроматографии, спектроскопии, рентге-
ноструктурного анализа и т. д.).
Изучение липидов начинается с их выделения; наиболее
эффективно это удается осуществить, экстрагируя липиды органическими
растворителями (диэтнловый эфир, бензин) или их смесями
(хлороформ -г метанол).
В живом организме часть липидов связана с белками и
углеводами, образуя разнообразные по сложности и прочности комплексы
и соединения. Растворители обладают неодинаковой способностью
разрушать эти комплексы и выделять липиды, поэтому состав
липидов до известной степени зависит от выбора растворителя.
В практике пищевой промышленности состав и качество жиров и
масел характеризуют с помощью разнообразных «чисел»,
подразумевая под ними расход определенных реагентов на реакции с жиром.
Наибольшее значение имеют числа: кислотное, омыления, йодное.
Кислотным числом называется показатель, характеризующий
количество свободных жирных кислот, содержащихся в жире.
Он выражается в миллиграммах едкого кали, затраченного на
нейтрализацию свободных жирных кислот, содержащихся в 1 г A0~3 кг)
жира. Учитывая, что хранение пищевых продуктов, содержащих
жиры и масла, всегда сопровождается гидролизом последних,
по величине кислотного числа можно, до известной степени, судить
о их качестве. В заводской практике кислотное число используется
при расчете количества щелочи, необходимого для щелочной
рафинации жиров и масел (стр. 215).
Число омыления равно количеству миллиграммов едкого кали,
необходимого для омыления глицеридов и нейтрализации свободных
жирных кислот в 1 г @,001 кг) жира или масла:
С3Н5 (OCORK + 3KOH -> С3Н3 (OHK + 3RCOOK .
триглицерид
RCOOH + КОН -> RCOOK + Н2О
211
По числу омыления можно судить о средней молекулярной массе
входящих в состав жира жирных кислот и определить при
мыловарении количество щелочи, необходимое для омыления жира.
Йодное число — показатель, характеризующий непредельность
жирных кислот, входящих в состав жира. Он выражается в процентах
иода, эквивалентного галогену, присоединяющемуся к 100 г (КГ1 кг)
жира. Существует несколько методов определения йодного чиста.
Одним из наиболее распространенных является бромометрический
метод. При этом применяется раствор брома в безводном метиловом
спирте, насыщенном бромистым натрием. Бром образует непрочное
комплексное соединение с бромистым натрием:
Отщепляясь, бром реагирует с ненасыщенными глицеридами:
О
СНа—О—С—(СНаO—СН=СН (СН2O СН3
о
СН—О—С—С17Н35 -tNaBr • Вг, ч>
О
!!
СНа-О-С-С17Н35-
О Вг Вг
СН2-О-С (СН2O—СН—СН (СН2), СН3
о
СН—О—С—С17Нзз +NaBr
О
На—О—С—С1?Нзя
Количество непрореагировавшего брома определяют иодометри-
чески:
NaBr- Bra + 2KI->2KBr+NaBr + Ia
I2 + 2NaaS2O3 -> 2NaI + Na2S4Oe
Зная исходное количество брома, можно легко вычислить
йодное число жира.
Йодное число широко применяется для определения вида жира,
способности его к «высыханию», расчета потребного количества
водорода на его гидрогенизацию.
Величины указанных констант для многих жиров, не
подвергшихся разрушению, колеблются в незначительных пределах и
характеризуют вид жира и его качество (табл. 27).
Для детального изучения липидов их разделяют на отдельные
классы, каждый из которых подвергают дальнейшему исследованию.
В зависимости от особенностей изучаемого класса соединений
применяются различные методы.
212
Таблица 27
Основные виды жиров и масел
Жиры и масла
Содержание и состав жирных
кислот, %
Характеристика
температура
застывания
число
омыления
йодное
число
Соевое
Арахисовое
Хлопковое
Подсолнечное
Рапсовое
Оливковое
Кокосовое
Пальмовое
Пальмоядерное
Масло какао
Льняное
Касторовое
Тунговое
Говяжий
Бараний
Свиной
Китовый
Кашалотовьш
Масла
14—25
20—21
22—25
10—12
2-6
9—18
До 90
44—57
79—83
58—60
6—9
До 0,6
3—10
75—86
79-80
75-78
До 90
94-98
82—91
10
43—56
17—21
40—42
91—94
94-97
90—97
Cj's 46—65
C}s 39—70
C;8 45—56
c;a 16—35
Cf5 46—70
qs 25—42
Эруковая
11 56
С»,. 6-44
C\, 70—82
Q2 44—52
C}4 13—18
C50 39—47
C}s 36-43
Co 45—50
q, Ю-1Э
C»e 23-25
С?в 31-34
qs 41—60
Рацинс
левая к-та
80—94
Элеостеари-
новая
кислота
66—82
-18
—2,5 — 3,0
2—4
—16—18
0—10
0—10
0—6,0
16—25
31—41
19—24
21,5—27
-18- (-27)
— 10— (—18)
-2-(-17)
191 — 193
185—186
191«-198
186—194
167—181
167—181
185—200
251—264
196—210
240—257
192—196
191—195
176—186
185—197
120—140
92,2--92,5
101—116
119-136
94—103
94—103
72—89
7—12
52—58
12—20
34—38
175—190
82—90
145—176
45—60
52-62
33—49
10-22
43—52
38-48
48—64
78—90
Животные жиры
C;s 43—44
С»3 24-29
q3 36—42
С»8 25—31
С}8 34—44
С»е 25—32
30-38
32—45
22 32
17—20
190-200
192—198
193—200
181—193
125—150
32—47
31—46
46—66
100—161
62—92
213
ПОЛУЧЕНИЕ И ПЕРЕРАБОТКА ЖИРОВ
И МАСЕЛ
Основные источники жиров и масел. Главным источником
растительных масел являются плоды и семена растений: сои, арахиса,
хлопка, подсолнечника, рапса, оливкового дерева, пальмы
(например, «копра»), льна, какао. Масла извлекают также из маслосодер-
жащих отходов некоторых производств: кукурузные зародыши,
рисовая мучка, семена косточковых плодов, виноградные косточки.
Основное количество масла в мире вырабатывается из сои, арахиса,
хлопка. В СССР растительное масло получается главным образом
из семян подсолнечника, хлопчатника, сои, льна, конопли.
Животные жиры в большом количестве содержатся в тканях
крупного и мелкого рогатого скота (говяжий и бараний жиры),
свиней (смалец), китов и кашалотов и коровьем молоке.
Строгая научная классификация жиров и масел отсутствует.
В зависимости от источников получения жиры делятся на жиры
растительные, животные и жир микроорганизмов. В свою очередь
они могут быть разделены на жиры жидкие при нормальной
температуре B0° С) и твердые.
Твердые жиры растительного происхождения (масло какао,
пальмовые масла) отличаются относительно высоким содержанием
насыщенных жирных кислот (лауриновой, миристиковой,
пальмитиновой, стеариновой), жидкие — ненасыщенных (олеиновой,
линолевой),
- По отношению к окисляющему действию кислорода жидкие
растительные масла условно делятся на высыхающие,
полувысыхающие, невысыхающие. Животные жиры делятся на жиры
наземных животных, молочные жиры и жиры морских млекопитающих
и рыб. Жиры наземных животных (сало говяжье, баранье, свиное)
содержат значительное количество насыщенных жирных кислот,
имеют твердую консистенцию и относительно невысокие йодные
числа. Жиры морских млекопитающих и рыб в зависимости от
источника получения сильно отличаются друг от друга по своим
физико-химическим свойствам, многие из них содержат значительное
количество ненасыщенных жирных кислот с несколькими (до шести)
двойными связями. Жиры микроорганизмов относятся к той области
химии липидов, изучение которой только начинается. Но уже
сейчас молено говорить о некоторых присущих им особенностях я
в первую очередь о высоком содержании жирных кислоте нечетным
числом атомов углерода в молекуле.
В настоящее время производство растительных масел растет
быстрее, чем животных жиров. Это связано с их большей
физиологической ценностью и экономической целесообразностью их
производства: производства растительных масел требует меньших
затрат труда, чем животных жиров.
Извлечение жиров. Основное количество масла извлекается из
специально подготовленного растительного сырья (экстракционным
или форпрессово-экстракционным способами). Содержание его в
214
остатке (шроте) при использовании этих методов не превышает 1%.
Экстракция масла (извлечение его органическими растворителями)
проводится бензином или гексаном при температуре 50—55° С в
специальном аппарате, получившем название экстрактор. При
извлечении масла из высокомасличного сырья часть его может быть
извлечена прессованием (отжим под повышенным давлением), а
оставшееся количество экстракцией (форпрессово-экстракционный
метод). Качество масла, полученного прессовым методом, несколько
выше, чем экстракционного. В технике в зависимости от
особенностей сырья и аппаратурного оформления применяются различные
варианты этих методов. f
Рафинация масел. Полученное после отгонки растворителя
масло содержит значительное количество примесей, мешающих его
дальнейшему использованию или придающих ему нежелательные
вкусовые качества. Очистка масла осуществляется с помощью
целого комплекса разнообразных физико-химических процессов
и называется рафинацией.
Не растворимые в масле примеси удаляют отстаиванием, а затем
фильтрацией. Фосфолипиды, примеси белковых и слизистых веществ
-удаляют гидратацией (обработка водой, слабыми растворами
кислот). Выделенные и очищенные фосфолипиды являются ценным
продуктом, широко применяющимся в хлебопекарном,
кондитерском, макаронном производствах, в комбикормовой промышленности,
животноводстве.
Свободные жирные кислоты, содержащиеся в -сырых маслах,
удаляют обработкой щелочью (щелочная рафинация), образующиеся
при этом соли жирных кислот (мыла) отделяют (соапсток) вместе
с увлеченными примесями. Так как при щелочной нейтрализации
из масла удаляют не только свободные жирные кислоты, но и
разнообразные примеси, оно осветляется. Масло, прошедшее стадии
очистки, может быть сильно окрашенным и обладать неприятным
запахом. В этих случаях для удаления красящих веществ его
подвергают адсорбционной очистке («отбелке») — обрабатывают
порошкообразными веществами (адсорбентами), способными поглощать
(адсорбировать) окрашенные примеси. В качестве адсорбентов
используются природные глины, активированный древесный уголь.
Сильно пахнущие компоненты масла извлекают обработкой
водяным паром под глубоким вакуумом (дезодорация). Очищенное от
примесей «рафинированное» масло применяется в питании, пищевой
промышленности, а также в качестве сырья при гидрогенизации
и получении маргарина.
Гидрогенизация жиров. В отдельных отраслях промышленности
(маргариновая, мыловаренная, кондитерская) в значительных
количествах требуются твердые жиры. Эти потребности не могут быть
удовлетворены за счет ресурсов твердых животных и растительных
жиров. Превращение жидких растительных масел в твердые или
полутвердые жиры достигается гидрогенизацией.
Гидрирование жиров осуществляется в основном
электролитическим водородом при участии никелевых и медно-никелевых
215
катализаторов при температурах 180—220 С в аппаратах
(автоклавах) непрерывного действия при давлении 0,5—1,5 атм D9 ¦ 103—
— 147-Ю3 Н/м2). Меняя условия гидрогенизации, можно получить
продукты (саломасы) разной степени «насыщенности»,
предназначаемые для различных целей. Процесс гидрогенизации осложняется
рядом побочных реакций, в первую очередь изомеризацией
(смещением двойных связей, переходом радикалов жирных кислот
в транс-форму). Эти процессы существенно влияют на свойства
и пищевую ценность готовых продуктов. Полученный при
гидрогенизации саломас после удаления катализатора и рафинации
используется в различных отраслях промышленности.
Переэтерификация и гидропереэтерификация жиров. В последнее
время в промышленности получила развитие переэтерифнкация
жиров, дающая возможность из высокоплавких животных жиров
и их смесей с растительными жирами получить жиры с любым
глпцеридным составом, а следовательно, с любыми свойствами.
Переэтерификацня жиров проводится при температурах 15—50° С,
в вакууме или в токе азота в присутствии в качестве катализаторов
метилата натрия, этилата натрия, сплава натрия и калия.
Полученные продукты не содержат, подобно гидрированным
жирам, нежелательных транс-изомеров и обладают высокой
биологической ценностью. В отдельных случаях гидрогенизацию жиров
совмещают с их переэтерификацией, расширяя тем самым
возможности этих процессов.
Производство маргарина. Маргарином называется искусственно
полученный пищевой продукт, напоминающий по ряду свойств
сливочное масло. В его состав входят растительное масло,
гидрированные и животные жиры, молоко и специальные компоненты,
придающие ему соответствующий цвет и запах. Маргарин нашел
широкое применение в питании населения и в разнообразных
отраслях пищевой промышленности.
Получение и переработка животных жиров. Основной способ
получения животных жиров — вытапливание их из жирсодержа-
щих тканей. Различают мокрую вытопку (сырье в течение всего
процесса находится в непосредственном контакте с водой и паром)
и сухую. В отдельных случаях с целью повышения выхода жира
на измельченную ткань воздействуют слабыми растворами щелочей
и карбонатов (NaOH, Na2CO3).
Высыхание жиров. Растительные масла, содержащие глицериды
полиненасыщенных жирных кислот, обладают способностью
поглощать кислород воздуха, претерпевая при этом сложные химические
изменения. Этот процесс получил название высыхания масел, а такие
маоча — высыхающими. Высыхание масел протекает особенно
быстро, когда оно распределено на поверхности тонким слоем, при
првышенной температуре и в присутствии катализаторов,
называемых сиккативами. На практике это свойство высыхающих масел
используется для приготовления олифы. Олифой называют
растительное масло, содержащее сиккативы (вещества, ускоряющие
высыхание масел), частично окисленное и полимеризованное в ре-
216
зультате нагревания при доступе воздуха. Олифы применяются
в качестве пленкообразователей для защиты металлических и
деревянных конструкций зданий и сооружений. Добавлением
к олифам минеральных пигментов (красок) получают масляные
краски. Наилучшие по качеству олифы получаются из льняного,
конопляного, тунгового масел. В связи с ростом спроса на
пленкообразующие материалы и необходимостью сократить расход пищевых
растительных масел для технических целей в последние годы в
большом количестве в качестве пленкообразсвателей применяют
синтетические смолы. Особое распространение нашли глифталевые и
пентафталевые смолы, продукты полимеризации фталевого
ангидрида с глицерином или пентаэритритом.
ПОВЕРХНОСтаО-АКТИВНЫЕ
И МОЮЩИЕ ВЕЩЕСТВА
Поверхностно-активными веществами называются соединения,
адсорбирующиеся на поверхности раздела фаз и способные снижать
поверхностное натяжение. В состав молекулы этих соединений
входят Две группы: гидрофобная и гидрофильная. У поверхностно-
активных веществ липидной природы гидрофобная группа обычно
представлена углеводородной цепочкой (—R), состоящей из 10—20
атомов углерода, гидрофильная — группами СООН, COONa,
COO", —SO3Na, —NH2. Молекулы этих веществ располагаются
в поверхностном слое таким образом, что гидрофильная группа
оказывается обращенной к воде, а гидрофобная выталкивается
из нее. Все поверхностно-активные вещества могут быть разделены
на два класса: ионогенпые соединения, которые при растворений
в воде диссоциируют на ионы, и неионогенные, не диссоциирующие
на ионы в водных растворах. В зависимости от характера
продуктов, образующихся,при диссоциации ионогенных соединений,
различают анионоактивные и катионоактивные. Большинство моющих
средств относится к анионоактивным.
Анионоактивные диссоциируют на длинноцепочечный анион и
катион (Na+, K+). К ним относятся мыла, алкилсульфаты, алкил-
сульфонагы, алкиларплсульфаты
R—COONa zl R-COO~ + Na+
соль жирной аннон катион
кислоты
Катионоактивные образуют при диссоциации длинноцепочечный
катион и анион:
[C6H-,-CH2N (С2Н5K]"С1-— [CflH5CH2N (С2Н3K]+ + СГ
триэтилбензиламмонийхлорид катион анион
К непоногенным поверхностно-активным веществам относятся
эфиры пропнленглнколя, моно- и диглицериды.
Аниоиоактивные вещества. Мыла. Мылами называются соли
высокомолекулярных карбоновых кислот, растворы которых обла-
217
дают моющим действием. Практическое значение имеют натриевые
и калиевые соли кислот с 10—20 атомами углерода в молекуле.
Для получения мыла исходный жир или смесь жирных кислот,
в том числе полученных синтетическим путем, обрабатывают при
кипячении раствором едкого натра (варка мыла). При этом
происходит гидролиз глицеридов и нейтрализация образовашихся жирных
кислот. Полученный продукт получил название мыльного клея.
После его охлаждения и разделки получают клеевое мыло. Для
получения ядрового мыла к мыльному клею добавляют
концентрированный раствор поваренной соли (высаливание); образуются
два слоя, верхний — ядро, после отделения, промывки и охлаждения
является высококачественным мылом. Свойства мыл зависят от
состава жирных кислот; соли ненасыщенных кислот обладают
большей растворимостью, чем насыщенных. Жидкими мылами
являются калиевые соли жирных кислот.
Моющее действие мыла (способность удалять находящиеся на
поверхности загрязнения) связано с его поверхностно-активными
свойствами. Водные растворы мыл — коллоидные системы,
содержащие наряду с мылами жирные кислоты (R—СООН), которые
образуют в результате гидролиза едкий натр, а также кислые
мыла (RCOOH-RCOONa) и ионы RCOO и Na+. В водных
растворах молекулы (или ионы) мыла в результате межмолекулярного
взаимодействия образуют шарообразные тела, причем
углеводородные цепи обращены внутрь шара, а гидрофильные группы
расположены снаружи. К. отрицательно заряженной поверхности
таких образований притягиваются молекулы воды и мыла.
Образующиеся частицы называются мицелами и имеют следующий состав:
(R—COO~),j-(R—COOH^^HjO)^. При стирке белья или мытье рук
молекулы мыла окружают частицы «грязи», образуя вокруг нее
оболочку. При определенных условиях частицы грязи отрываются
от поверхности и-переходят в водно-мыльную среду (солюбилизация),
кроме этого, они могут под действием мыла распадаться на более
мелкие частицы и, прилипая к пузырькам воздуха, переходить
в пенную фазу (флотация). Все это приводит к очистке поверхности
от грязи.
Мыла применяются главным образом для мытья.
Использование мыла в быту (стирка), технике (мытье тканей, судов)
снижается; они заменяются синтетическими моющими средствами
(детергентами). Это связано с их большей моющей способностью
по сравнению с мылами (в 10—30 раз), низкой стоимостью,
доступностью сырья для их изготовления и рядом других преимуществ.
Например, в жесткой воде вследствие образования нерастворимых
солей магния, кальция и железа моющие действия мыл снижаются:
2R—COONa + Mg2+ н» (R—COOJMgJ +2Na+
Кроме этого, они оказывают нежелательное действие на ряд тканей.
Нужно также помнить, что для производства мыла используются
высококачественные жиры, что приводит к уменьшению пищевых
ресурсов,
218
Алкилсульфаты — соли эфиров серной кислоты, получаются
по следующей схеме:
парафины -> жирные кислоты -> жирные спирты -> соли алкилсульфатов
Они не чувствительны к жесткой воде и обладают прекрасной моющей
способностью.
Алкилсульфонаты — соли жирных сульфокислот получаются
сульфохлорированием алканов (стр. 31). Не чувствительны к
действию жесткой воды.
Неионогенные поверхностно-активные вещества.
Представителем этой группы являются эфиры полиэтиленгликоля
RCOO (СН2—СН.2О) „ Н R—COO (CHa—CH2O)n OCRt
моноэфир диэфир
моноглицериды НО—СН2—СНОН—СН2ОСО—R и другие продукты.
Они широко применяются в различных отраслях пищевой
промышленности в качестве эмульгаторов и веществ, улучшающих качество
пищевых продуктов.
УГЛЕВОДЫ
Углеводы и их производные уже давно занимают исключительное
место в технике и повседневной жизни человека.
Успехи биоорганической химии позволили оценить подлинную
роль углеводов и в самом процессе жизнедеятельности.
Разнообразные производные углеводов, находясь в составе клеток любого
живого организма, выполняют роль конструкционного материала,
субстратов, регуляторов специфических биохимических реакций, а также
поставщиков энергии. В соединении с липидами, белками и
нуклеиновыми кислотами углеводы образуют сложные
высокомолекулярные комплексы, лежащие в основе сз'бклеточных структур и
представляющих собой основу живой материи.
Являясь первичными продуктами фотосинтеза, углеводы
представляют собой первые органические вещества в кругообороте
углерода в природе и, таким образом, служат как бы мостом между
минеральными и органическими соединениями.
Углеводы широко распространены в природе, особенно в
растительном мире: до 80% сухого вещества растений приходится на долю
углеводов. Содержание их в живых организмах — около 20%
сухого вещества.
Название «углеводы» возникло на основании анализа первых
известных представителей этой группы соединений. Оказалось, что
эти вещества состоят из углерода, водорода и кислорода, причем
соотношение числа атомов водорода и кислорода у них такое же, как
в воде: на два атома водорода — один атом кислорода. Таким
образом, их рассматривали как соединение углерода с водой. В
дальнейшем стало известно много углеводов, не отвечающих этому условию,
однако названия «углеводы» до сих пор остаются общепринятыми.
Классификация углеводов. Большой класс углеводов
подразделяют на две группы: простые углеводы (или простые сахара) и
сложные углеводы (или сложные сахара).
219
Простыми углеводами (моносахаридами, или монозами)
называются такие углеводы, которые не способны гидролизоваться
с образованием более простых углеводов. Большинство этих веществ
имеет состав, соответствующий общей формуле СгаН2лО„, т. е.
число атомов углерода у них равно числу атомов кислорода.
Сложными углеводами (полисахаридами, или полиозамп)
называют такие углеводы, которые способны гндролизоваться с
образованием простых углеводов. Общая формула этих веществ С,гаН2,гОя,
т. е. число атомов углерода у них не равно числу атомов кислорода.
К сложным углеводам относятся довольно разнообразные по
своим свойствам вещества, и поэтому их делят на две подгруппы:
сахароподобные сложные углеводы (олигосахариды) и несахаро-
подобные сложные углеводы (высшие полисахариды).
Олнгосахариды при гидролизе распадаются на 2—8 молекул
моносахаридов («олигос» — по-гречески немногий).
Высшие полисахариды при гидролизе распадаются на очень
большое число моноз, исчисляемых иногда сотнями и тысячами.
МОНОСАХАРИДЫ
Строение моносахаридов. Важнейшими представителями
моносахаридов являются глюкоза (виноградный сахар) и фруктоза
(фруктовый сахар). Оба они являются изомерами и имеют одну
молекулярную формулу СдН^Ое.
При энергичном восстановлении глюкозы йодистым водородом
был получен 2-иодгексан СН3—СН2—СН2—СН2—CHI—СН3 —
вещество с неразветвленной цепью углеродных атомов, а значит, и
сама глюкоза имеет неразветвленную. цепь. Наличие альдегидной
группы доказывается способностью глюкозы присоединять
синильную кислоту (что характерно для альдегидов):
ОН ОН
R—О.{ +HCN->R-C-CN iH
| |
н н
глюкогептоновая
кислота
Кроме того, глюкоза обладает^ сильной восстанавливщощвй.
способностью^ и_лег_ко_окисдяётся7.ТГапример, бромной водой,
образуя кислоту с тем же числом углеродных атомов:
(QHuO5)Gf +HOBr->(C,HuO3)C< +НВг
глюконовая
кислота
Учитывая образование глюкозой сложных эфиров с
органическими кислотами, Вертело (I860) заключил, что глюкоза является
многоатомным спиртом. В 1869 г. А. А. Колли показалл_чтр_^лю-
коза представляет собой -пятргатомнын -сшфх. Из всего сказанного
следует, что глюкоза представляет собой пятиатомный альдегидов
220 " ' "' "
спирт следующего -строения:
СН2ОН—СНОН—СНОН—СНОН—СНОН—СООН—С=О
I
н
Фруктоза при восстановлении йодистым водородом тоже дает
2-иодгексан, т. е. она также имеет неразветвленную цепь
углеродных атомов ir присоединяет синильную кислоту, следовательно,
содержит карбонильную группу. Однако окисление фруктозы
происходит с разрывом углеродной цепи:
С6Н,,О6 ХЛ О.?- С( + >С—СНОН-СНОН-С,^
\он юн он-7 чон
фруктоза щавелевая винная кислота
кислота
а следовательно, фруктоза является кетозой. Как и глюкоза,
фруктоза реагирует с пятью молекулами уксусного ангидрида, образуя
5 сложноэфирных группировок, т.е. она содержит 5 гидроксиль-
ных групп.
Таким образом, фруктоза представляет собой пятиатомный
О
кетоспнрт СН2ОН—С—СНОН—СНОН—СНОН—СН,ОН
Моносахариды, содержащие, подобно глюкозе, альдегидную
группу, получили название альдоз; моносахариды, содержащие
кетонную группу, стали называть кетозами.
По числу углеродных атомов моносахариды делят на:
триозы С3НеО3 (С3)
тетрозы C4HSO4 (Cj)
пснтозы С5Н10О5 (С6)
гексозы СаН12О6 (Св)
И Т. Д.
Чтобы подчеркнуть, что в состав моносахарида входит
альдегидная группа и что этот моносахарид содержит, например, шесть
углеродных атомов, объединяют названия в слово «альдогексоза»
(аналогично кетогексоза, альдопентоза и т. д.).
Итак, по своему химическому строению моносахариды
представляют собой многоатомные альдегидо- или кетоспирхы.
Стереохимия моноз. Пространственные конфигурации
моносахаридов, D и L-p я д ы. Моносахариды содержат асимметрические
углеродные, атомы. Альдотетррзы, например, имёют~два
асимметрических углеродных атома, альдопентозы — три, альдогек-
созы — четыре. В результате этого у моносахаридов имеется
большое число стереоизоМероБ. Дл"я~ альдогексозьт," имеющей
четыре асимметрических атома углерода, число стереоизомеров
в согласии с формулой D = 2я будет равно 24 = 16. У альдогексоз
(так же, как и у кетогексоз) половина оптически деятельных стерео-
изомеров является антиполами другой половины. Таким образом,
16 стереоизомеров альдогексоз образуют 8 пар антиподов. Напри-
221
мер, природному моносахариду D-глюкозе соответствует
синтетически полученный'антйпод L-глюкоза:
if <°
| ЧН | ХН
н—с—он но—с—н
но—с—н н—с—он
.'* !¦•
Н—С—ОН НО-С-Н
Н—С—ОН НО—С—Н
СН2ОН СН3ОН
D-глюкоза L-глюкоза
Нумерацию цепи ведут, начиная от альдегидной группы в альдо-
зах, а в кетозах, начиная от того конца цепи, к которому ближе
кетогруппа. Определение взаимного расположшия._Н1_1иОН-групп
у каждого асимметрического, центра представляет -трудную-задачу.
В 1906 г. А. М. Розанов предложил судить о принадлежности того
или иного моносахарида к D- или L-ряду по расположению в
пространстве атомных групп у последнего асимметрического
углеродного атома (он же — предпоследний атом в цепи): если
конфигурация совпадает с конфигурацией D-глицеринового альдегида, то
моносахарид относят к D-ряду, если конфигурация совпадает с конфн-
гурацией L-глицеринового альдегида, то моносахарид принадлежит
к 1-ряду.
СНО СНО
I I
;Н_С^-ОН| ;НО—C^-HJ
СН2ОН СНоОН
D- глицериновый /.-глицериновый
альдегид альдегид
(СНОН)„ СНО (СНОН)^ СНО
—С*-ОН| :НО—C^H
СН,ОН СН20Н
Д-р яд i-ряд
Таким образом, весь стереохимнческий D-ряд моносахаридов
до гексоз включительно можно изобразить схемой 8. Антиподами
(зеркальными изображениями) каждого из представленных в схеме 8
моносахаридов являются соответствующие моносахариды L-ряда.
В случае соединении со многими асимметрическими
углеродными атомами обозначения «D» или «L» вовсе не служат указанием на
направление вращения плоскости поляризации растворами Сахаров,
так как вращательная способность зависит от расположения
заместителей у всех асимметрических углеродных атомов. Например,
природная D-фруктоза вращает плоскость поляризации влево.
Для указания направления вращения используют знаки «4-»
222
моносахаридов
Схема 8
н
альтроза
глюкоза
гулоза
галактоаа
талоза
(прппое вращение) и «—» (левое вращение), так же, как это принято
ранее в окснкнслотах (стр. 153). Тогда природная фруктоза и
глюкоза будут обозначаться:
D (—)-фруктоза; D (-|- )-глюкоза.
Циклическая структура моносахариде в. Хотя
альдегидные и кетонные формулы моносахаридов хорошо объясняли
многие свойства этих веществ, однако постепенно накапливались
факты, которые этими формулами не могли быть объяснены.
1. Отсутствие некоторых реакций на альдегидную группу.
Оказалось, что моносахариды, давая ряд реакций на альдегидную
группу (окисление до СООН, реакция с синильной кислотой,
восстановление аммиачного раствора окиси серебра, реакция с фелин-
говой жидкостью), в то же время не дают некоторых альдегидных
реакций (не реагируют с бисульфитом натрия, не дают окрашивания
с фуксиксернистой кислотой и т. д.).
2. Мутаротация. Все моносахариды обладают оптической
активностью, однако угол вращения плоскости поляризации
свежеприготовленных растворов моносахаридов по мере стояния изменяется,
пока не достигнет некоторой постоянной величины. Это изменение
угла вращения, или изменение оптической активности, растворов
сахароз при стояния получило название мутаротации. Например,
для jD-глюкозы это изменение происходит от +106 до +52.5°,
что изображают обычно так: +106° —> -(- 52,5°.
3. Количество диастереоизомериых моносахаридов. Если исходить
из формул Фишера, то для альдогексоз должно существовать
16 диастереоизомеров. Действительно, это количество альдоз было
найдено в природе или синтетически получено Фишером. Однако
дальнейшие исследования показали, что каждый из этих
моносахаридов способен существовать в двух совершенно индивидуальных
кристаллических формах с различными константами, в том числе
с резко различными углами вращения. Так, например, были
получены две D-глюкозы с углами вращения +106° и +22,5°.
Характерно, что при растворении обеих форм моносахарида всегда
получался раствор, угол вращения которого менялся вследствие мута-
ротацни и постепенно достигал для обеих форм одной и той же всегда
строго постоянной величины, которая для глюкозы составляла
+52,5°. Таким образом, число реально существующих альдоз
ровно вдвое превышало число предсказанных на основе законов
стереохимии. Удвоение числа днастереонзомерных форм говорило
о наличии в молекуле моносахаридов еще одного дополнительного
асимметрического углеродного атома. Таким образом, количество
асимметрических углеродных атомов в альдопентозах должно
равняться четырем, а в альдогексозах — пяти.
4. Особые свойства одной из ги.дроксильных групп. Последнее
серьезное противоречие оксикарбонильным формулам Фишера состоит
в том, что одна из гндрокспльпых групп моносахаридов резко
отличается по своим свойствам ог других гпдроксилов. Так,
например, при нагревании моносахаридов со спиртами в присутствии
224
небольшого количества сухого хлористого водорода происходит
образование хорошо кристаллизующихся веществ—глйкозидов (стр. 227,
234, 244). Гликозиды в присутствии разбавленных минеральных
кислот очень легко гидролизуются, причем из одной молекулы
глпкозида образуется одна молекула моносахарида и одна молекула
спирта. Если исходить из формулы Фишера, то в глюкозе
присутствует 5 гидроксплов, каждый из которых мог бы дать соединения
типа простого эфира. Почему же только один гидроксил участвует
в образовании гликозида? Далее, если образовалось вещество,
относящееся к простым эфирам, то почему оно так легко
подвергается гидролизу, тогда как основное свойство простых эфиров —
это стойкость к гидролизу?
Все эти факты нашли объяснение, когда было доказано, что
каждый моносахарид может существовать в виде ряда таутомерных
форм. В растворах моносахаридов наряду с альдегидными и кетон-
нымн формами всегда содержатся циклические таутомерные формы.
Еще А. А. Колли в 1870 г. предложил для глюкозы циклическую
полуацетальную формулу с трехчленным окисным кольцом:
/Н
СН2 (ОН)—СН (ОН)—СН (ОН)—СН (ОН)—СН С<
1_о- V°H
Наличием подобного кольца он объяснял отсутствие некоторых
альдегидных реакций у глюкозы.
Позже, в 1883 г., Толленс предложил аналогичную формулу, но
с пятичленным окисным кольцом:
/Н
СНо(ОН)—СН (ОН)—СН—СН (ОН)—СН (ОН)—С.
О '¦
Циклические формулы называют также окисными или полуаце-
тальиыми, Механизм образования циклической формы из
альдегидной можно представить следующим образом. Как известно,
альдегиды легко присоединяют спирты, превращаясь в полуацетали
(стр. 121):
..о*" ; ¦ /он
R—C{ +R-OH:~>R-C- OR
\н \н
альдегид полуацеталь
Альдегидная группа моносахарида, расположенная в пространстве
близко к одной из гидроксильных групп, взаимодействуя с ней,
образует внутренний полуацеталь:
Н
альдегидная форма
8 А. П. Нечаеа
ш
С
циклическая форма
(окисная, полуацетальная)
225
Образование циклических форм объясняет все выше перечисленные
факты, которые нельзя было объйснить альдегидными формулами:
1. То, что моносахариды не дают некоторых альдегидных
реакций, объясняется тем, что в водных растворах моносахаридов
преобладают циклические формы. Альдегидные же формы находятся в
небольших количествах, в отдельных случаях составляют
несколько десятых долей %.
2. Явление мутаротацин: в циклической форме моносахарида
появляется дополнительно еще один асимметрический углеродный
атом, входивший ранее в карбонильную группу. В зависимости
от расположения гидроксила у этого нового асимметрического
центра различают так называемые а- и р-формы (или аномеры):
если гндроксил у вновь образовавшегося асимметрического атома
углерода расположен по одну сторону от углеродной цепи с гидрок-
силом, определяющим принадлежность kD- или L-ряду (или по одну
сторону с окисным кольцом, образованным из этого гидроксила), то
такая форма называется а-формой (а-аномероы); если же полуаце-
тальный гидроксил располагается по другую сторону углеродной
цепи от гидроксила, определяющего принадлежность кй- или L-ряду
(или по другую сторону от окисного кольца, образованного из этого
гидроксила), то такая форма называется Р-формой (или р-аномером):
,он
¦НО
Н—С—ОН
НО—С—Н
Н—С—ОН
н
полуа детальные ,
ИЛИ ГЛПКОЗИДТШе тт р /
гпдроксилы
о
н—с-
но-с-н
I
н—с—он
н-с
о
сн,он
a-D-глюкоза
СН2ОН
В кристаллическом виде моносахариды существуют в
циклической форме и являются устойчивыми соединениями, во многих
случаях они- могут быть выделены в виде двух аномеров. Однако при
растворении индивидуального аномера в воде быстро наступает
таутомерное превращение, приводящее к образованию равновесной
смеси нескольких форм моносахаридов. Внешне это проявляется
в изменении угла вращения сахара.
3. Удвоение числа дпастереоизомеров становится также
объяснимым появлением нового асимметрического углеродного атома.
В циклической форме альдогексозы имеют уже пять
асимметрических атомов углерода, следовательно, образуется 25 или 32 диасте-
реоизомера, а альдопентозы образуют, соответственно, 24 или
16 днастереопзомеров.
4, Образование гликозидов с. участием одного гидроксила сахара
хорошо объясняется существованием циклической формы. Если
у альдегидной формы глюкозы все гидроксилы одинаковы — это
226
спиртовые гндроксилы.то у циклической формы есть один гидроксил,
резко отличающийся от других. Это гидроксил, образовавшийся
из альдегидной или кетонной группы, который называется, полу-
ацетальным или гликозидньш гидрэксилом. Он более реакционно-
способен, чем остальные спиртовые гидроксилы, поэтому легко
вступает в реакцию со спиртом, образуя глнкознд:
.ОН ' /OR
Н-С^- Н-С-—
Н—С—ОН
н-с-он
НО—С—Н О + ROH -гв-г НО—С—Н О + Н2О
н—с—он
Н-С
(Н-)
н—с—он
н-с
СН2ОН СН,ОН
Если гликозид образован из а-формы сахара, то это ос-гликозид,
а если из р1-, то (З-глшгазпд.
Таким образом, циклические формы сахароз объяснили все
существовавшие до их открытия противоречия.
Характер окисных колец. Кольцо циклической формы сахара
может быть шестичленным и пятичленным. Окпсные формы моно-
сахаридоз с полным правом можно рассматривать как производные
соответствующих гетероциклических соединений (стр. 191, 195):
шестичленные как производные пираиа, а пятичленные как
производные фурана (точнее как производные тетрагндропирана и тетра-
гпдрофурана, соответственно). Формы моносахаридов, содержащие
шестичленные циклы, называют пиранозами, а пятичленные — фура-
нозами:
СН3 СН,
СН СН
СН
СН
пиран
СН2ОН
с=о
НО—С—Н
!
н-с-он
н-с-он
I
СН2ОН
D-фруктоза
сн-сн
СН СН
\0/
фуран
сн2 сн2
сн,_
сн.2
тетрагндроппран
СН2ОН
С;
НО—С—Н
Н—С—ОН
Н—С—ОН
!
сн,-
СН2-СН2
СН» СИ,
\0/ "
тс грагпдрофуран
СН2ОН
I
но—с
о
о
но—с-н
н—с—он
н-с
сн»он
Р-0-фр>ктофураноза
Хеуорзс предложил новый способ условного перспективного
изображения структур моносахаридов, очень наглядно
показывающий взаимное пространственное расположение гидроксильиых
227
групп. Согласно его предложению циклическую форму
моносахарида условно считают плоской. Для изображения на бумаге ее
мысленно располагают таким образом, чтобы кислородный атом пира-
нозного кольца находился на наибольшем расстоянии от глаза
наблюдателя справа (у фуранозного кольца — посредине), а
углеродная цепь была бы обращена выпуклой стороной к наблюдателю.
Обычно часть молекулы, приближенную к наблюдателю,
показывают жирной линией:
Нумерацию цепи ведут по часовой стрелке, начиная справа от атома
кислорода. Заместители помещают сверху или снизу от плоскости
молекулы в зависимости от конфигурации соответствующего
углеродного атома.
Рассмотрим взаимоотношения проекционных формул Толленса и
перспективных формул Хеуорзса. В качестве примера возьмем а-
D-глюкопиранозу и f$-D-фруктофуранозу. Все группы,
располагающиеся в формуле Толленса справа от углеродной цепи, в формуле
Хеуорзса попадают под плоскость кольца, концевая СН2ОН-группа
располагается сверху от плоскости молекулы, если атом, с которым
она связана, имеет Ь-конфигурацию, и снизу от плоскости в том
случае, если он имеет L-конфигурацию:
¦ОН
6 СН,ОН
н
а -jD-глюкопираноэа
i CH2OH
но—
но—с—н
¦ 4|
н—с—он
н—с-
CHjOH
но н
СН2ОН
Р-О-фруктофураноза
228
Таким образом, в формулах Хеуорзса полуацетальный гидрок-
сил и концевая СН,ОН-группа располагаются у ос-аномеров по
разные стороны условно плоского кольца молекулы, а у р-аноме-
ров — по одну сторону.
Таутомерия моносахаридов в растворах. Сахара были
исторически одними из первых веществ, для которых наблюдалось
явление та>шмерии. Таутомерия Сахаров — это так называемая коль-
чато-цепная или оксо-окси-таутомерня. Моносахариды, в
зависимости от условий реакции и примененных реагентов, реагируют
в одной из таутомерных форм: пиранозной, фуранозной или
ациклической:
сн,он
сн„он
н он
сн.он
н-
но-
н=
'н ^
но\?н н
-он
-н
н
он
—он
снда
сн,он
н он
он
н он
таутомерные формы
Л-глюкозы
Существование таутомерии для моносахаридов подтверждено
экспериментально путем исследования их оптической активности,
а также с помощью ЯМР- и ИК-спектроскопип. Явление мутаро-
тацин связано со взаимными превращениями таутомерных форм
моносахарида и установлением равновесия между ними. Положение
равновесия зависит от структуры и стереохимии моносахарида,
но не зависит от исходной таутомерной формы данного сахара.
Так, свежеприготовленные водные растворы а- и j^-D-глюкозы
имеют удельное вращение \a\D +106э и +22,5" соответственно.
С течением времени удельное вращение первого падает, а
второго возрастает, в обоих случаях достигая постоянной
величины —52,5°.
Понятие 'О конформационной изомерии. Для Сахаров в циклическом виде
возможен еще один вид изомерии — конформашюнная изомерия, связанная
с расположением в пространстве углеродных атомов шестичленного цикла.. Для
пиранозного кольца из-за его несимметричности, связанной с наличием в нем
кислорода, возможно большее число конформаций, чем для цнклогексана.
Предпочтительность той или иной конформаций определяется соотношением
размеров и числа заместителей данного реального сахара и их пространственным
229
расположением. Данные рентгеноструктурных исследований моносахаридов
показали, что пиранозы существуют в виде кресловидной конформации, причем
такой, в которой наибольшее число объемных заместителей расположено
экваториально. Так, D-глюкоза имеет вид
,-он
Способы_получения. Гидролиз_ди^ и полисахаридов -=-0Дин из
основных способов 1юлучешТя моносахаридов, имеющий большое
практическое значение. Гидролиз происходит под влиянием кислот
или ферментов: ~"
-» 2CeH12Oe
дисахарид моносахарид
(СвН10О5)я + «Н2О -> nCeH12Oe
полисахарид
Альдольная конденсация. Первый синтез сахаристого вещества
был произведен А. Л1. Бутлеровым из формальдегида (стр. 124):
Н2С—СН
I II
НО О
I! г \\
0 _....; о
Н2С—СН -t IH'CH -> Н2С—СН—СН
но о—¦
о
но но о
н2с—сн—сн + ;'н:сн—сн—сн -> н2с-сн—сн-сн—сн-сн
но он о— : он он о
он он он он он о
Полученный густой сироп содержал сахаристое вещество, назЕан-
ное Бутлеровым «метиленитаном». Впоследствии Э. Фишером из
такого сиропа, содержащего смесь гексоз, была выделена гексоза,
названная им «акрозой».
Оксинитрильный синтез. Этот метод, разработанный в 80-х
годах прошлого века, несмотря на его многостадийность, до сих
пор остается важнейшим методом наращивания цепи моносахари-
230
дов. Для примера возьмем схему получения гептоз из глюкозы:
CN
Н—
но-
Н—
н—
чн
—он
—н
—он
HCN
-он
н—
но—
Н—
н-
—он
—Н
с/
СН2ОН
D-глюкоза
(гексоза)
(I)
он
он
2Н2О
-ОН
—он
СН2ОН
оксинитрилы
B)
/.О
н—
¦НО—
н-
н-
—он
—н
—ОН
он
Н—
'¦но-
Н—
Н
СН2ОН
глюкогеп-
тоновые
он
н
-ОН
-н
С(
р
чн
,он
о
[Н]
амальгама
натрия
н—
но—
-он
Н-
СН2ОН
-он
-Н
—он
—он
сн,он
Метод укорачивания цепи. Для получения моносахаридов можно
применять так называемые укорачивания углеродной цепи. Эти
методы используются для получения некоторых малодоступных Сахаров.
Они основаны на окислении моносахаридов, содержащих
большее количество углеродных атомов, чем в нужном моносахариде:
н—
но—
н—
н—
-он
-Н
—ОН'
-он
НО—
Н—
н—
н
—н
—он
—он
СН.-.ОН
СН2ОН
D-глюкоза
?>-арзб[Шозз
Наиболее старый метод — распад по Руффу, в силу своей
простоты применяется и до сих пор. Он основан на окислении альдозы
231
перекисью водорода в присутствии солей (чаще всего ацетата)
трехвалентного железа. В результате окисления углеродный атом
карбонильной группы моносахарида отщепляется в виде
углекислоты, а атом, следующий за ним в цепи, окисляется до
альдегидной группы.
Физические свойства. Моносахариды представляют собой твердые
кристаллические вещества. Все моносахариды гигроскопичны, хорошо растворимы в воде,
образуя сиропы, из которых их выделить в кристаллическом виде часто бывает
трудно. В спирте моносахариды растворяются плохо, в эфире вовсе не
растворяются. Растворы моносахаридов имеют нейтральную па лакмус реакцию и
обычно обладают сладким вкусом. Сладость разных моносахаридов весьма
различна. Например, фруктоза приблизительно в три раза слаще глюкозы.
Растворы моносахаридов обладают оптической активностью, для них характерно
явление мутароташш.
Химические свойства. Каждый представитель ^группы
моносахаридов в растворах существует одновременнов^видё^Тнёсрадлькйх
'таутомерных форм. Рассматривая различные реакции Сахаров, мы
будем даватьГ "формулу лишь той формы, которая непосредственно
вступает в реакцию. В основном будет приводиться ациклическая
форма, несмотря на то, что она в растворах содержится лишь в очень
малых количествах, в то время как подавляющее число молекул
существует в циклических формах. Это можно объяснить явлением
таутомерии; как только некоторое количество одного таутомера
израсходовано, другие немедленно превращаются в него, пока
вновь не установится равновесие между различными таутомерными
формами, постоянное для данных условий (температуры,
растворителя и др.).
Таким _о_бразом моносахариды проявляют свойства спиртов,
карбонильных соединений" и~~полуацеталей.
Окисление. Моносахариды легко окисляются, причем в
зависимости от условий получаются разнообразные продукты. Для
получения альдоновых кислот окисление альдоз обычно осуществляют
в кислой среде хлором, бромом, гипохлоритом, разбавленной азотной
кислотой. При более энергичном окислении, например
концентрированной азотной кислотой, помимо альдегидной группы окисляется
первичная спиртовая группа и образуются двухосновные окси-
кислоты, так называемые сахарные кислоты. Продуктами окисления
альдоз являются также уроновые кислоты. При образовании
уроновых кислот окисляется первичный спиртовой гидроксил
альдозы и в конце цепи образуется карбоксильная группа, тогда
как альдегидная группа остается неизменной:
//°
Н0СН.,-(СНОН)^—С<
•Н
альдоз-J
о о
,О I! хО |! х,0
НОСИ.,—(С!К)НL—(У НОЙ—(CHOHh—Q? НО—С—(СНОНL—(У
" ' NDH ЧЮН ХН
232
альдомовая сахарная уроновая
кислога кислота кислота
При окислении кетоз происходит расщепление их
молекул.
Восстановление. Осторожное восстановление моносахаридов ведет
к образованию соответствующих многоатомных спиртов (тетритов,
центитов, гекситов и пр.).
Реакция «серебряного зеркала». Подобно обычным
альдегидам, моносахариды легко дают реакцию «серебряного зеркала»
при взаимодействии с аммиачным раствором окиси серебра
[Ag (NH3Ji ОН:
/Р /Р
2 I Ag (NH3):] ОН + (C5HnO5) Cf -> (C5HUOF) Cf + 2Agi -Ь 3NH3 + Н2О
"¦Н 4)NH4
Реакция с фелинговой жидкостью. Для моносахаридов ха-
рактернатакже реакция с фелинговой жидкостью, представляю1
щей собой водно-щелочной раствор комплексной соли,
образовавшейся из гидрата окиси меди и натрнйкалневой соли
винной кислоты:
4 ОК Н j ч ОК
н—с—oN \о—с—н л
н-с—о-; хо-с-н чн
I ЧОК
,0 Н—С—ОН
При нагревании моносахаридов с фелинговой жидкостью окисная
медь восстанавливается до закнсной, а альдегидная группа
моносахарида окисляется до карбоксильной. Красная закись меди
выпадает в осадок. Реакцию используют для количественного
определения Сахаров.
Реакция с синильной кислотой была подробно рассмотрена
ранее (оксннитрнльный синтез, стр. 230).
Взаимодействие с фенилгидразином (стр. 122),
Действие щелочей приводит к _изомеризац1Ш моноз в смесь
Сахаров, „отличающихся" "строением~л'ли конфнгурациеп
первого и' второго атомов углерода. Так, из< глюкозы, манноаы
233
или фруктозы получается равновесная смесь этих трех Сахаров:
/О /О
С</ СН—ОН с/
i н
Н—С—ОН
НО-С-Н
Н—С—ОН
Н-С-ОН
СН2ОН
D-глюкоза
С—ОН
НО-С-Н
Н-С-ОН
Н—(
:—он
СН2ОН<—
ендиол
хн
НО-С-Н
НО-С-Н
Н—С—ОН D-манноза
Н—(
:—он
СН2ОН
СН2ОН
с=о
НО—С—Н
—" ]
Н—С—ОН D-фруктоза
Н—С—ОН
снаон
Подобные превращения могут претерпевать все моносахариды.
Происходящее при этих превращениях изменение конфигурации
у второго углеродного атома альдоз носит название эпимеризации,
а альдозы, отличающиеся лишь конфигурацией второго углеродного
атома, называются эпимерами.
Образование простых эфиров. При алкилировании сахара
реагируют в циклической (полуацетальной) таутомерной форме.
СН„ОН
СН,ОН
О—СН,
О—СН,
н о—сн.
Л-глюкоза
234
Гликозидный гидроксил легко взаимодействует даже с
метиловым спиртом в присутствии хлористого водорода. Спиртовые
гидроксилы в этих условиях не затрагиваются; для их алки-
лирования необходимы более сильные алкилирующие агенты,
например галогеналкилы (в присутствии окиси серебра, см. стр. 234).
При действии на полностью алкилироЕанные моносахариды
разбавленных минеральных кислот происходит гидролиз только у первого
углеродного атома, остальные оксиметильные группы не гидро-
лизуются:
сн2—о—сн3
н о—сн3 н . о—сн
Образование сложных эфиров. При действии ацилирующих
агентов (например, ангйдрТ?д6в"или галогенангидридов кислот)
происходит образование сложных эфиров по всем гидроксильиым
группам:
О
СН2ОН
!!
сн,—о—с—сн3
но\?н
5(СН3СО2)О
=5СНТСООН*
н он
D-глюкоза
пентаацетат
D-глюкозы
Определение размера окисного кольца моносахаридов. Существует два
основных метода определения размеров окисного кольца моносахаридов. Важнейшим
является так называемый метод метилирования (метод Хеуорзса). Метод
метилирования основан па различии химических свойств полуацетального н
спиртового гидроксилов. Глюкоза подвергается вначале исчерпывающему
метилированию. Полученная таким образом полностью метилированная глюкоза
содержит пять метокснльных групп: четыре из них, образованные из спиртовых
гидроксилов, являются простыми эфирными группировками, которые устойчивы
к действию как щелочных, так и кислых агентов. Метальная же группа,
находящаяся у первого углеродного атома, легко омыляется при обработке
кислотами, так как она заместила атом водорода полуацетального (гликозидного)
гидроксила. Поэтому после нагревания пентаметилглюкозы с разбавленной
минеральной кислотой получают тетраметилглюкозу, имеющую свободный полу-
ацетальньш гидроксил и, следовательно, способную существовать как в
циклической, так и в открытой таутомерной формах. Если такое соединение
подвергнуть окислению концентрированной азотной кислотой, то окисляются
углеродные атомы, содержащие карбонильную группу и свободный гидроксил, в
результате чего образуется двухосновная кислота. Как следует из анализа
продуктов реакции, в том случае, если бы глюкоза имела б-окисное кольцо (пираноз-
ная форма, левая часть схемы), конечным продуктом окисления была бы триме-
235
токснглутаровая кислота. Напротив, если кольцо является у-окнсным (фура-
позыая форма, правая часть схемы), в результате окисления должна получаться
соответственно дпметоксияитарная кислота. В действительности при проведении
всех этих реакций в конечном счете была получена только триметоксиглутаро-
вая кислота, что совершенно однозначно доказывало наличие б-окисного (пира-
нозного) кольца в глюкозе.
?)-глюкофураноза
Н
-с/
,он
Н—С—ОН
но—с—н о
н—с—он
н—с
D-глюкопираноза
СНпОН
|С!!Л/Д?2О
I /ОСН3
н-с-
н—с—осн3
I
СН3О—С—Н О
н—с—осн3
I
СНоОСН,
i /ОН
н—с-
н—с—осн3
СН3О—С—Н О
I
н—с—осн3
н—с
СН.ОСН,
н-с/
Н—С—ОСН3
СН3О—С—Н
I
; Н-С-ОСН3
н-с-он
СНоОСНз
i[OJ
h-cZ
он
н-с-он
НО-С-Н о
н-с !
н-с-он
сн,он
;CH3I/Ag-.O
I /ОСН3
Н-С—ОСН3
СН3О—С—Н
i
н—с
Н-С—ОСНз
н-с^;°
J
н—с—осн3 н—с
Н3СО—С—Н ^ Н—С—ОСНз
Н—С—ОН СН3О—С—Н О
I | !
Н—С—ОСНз Н-С "-
СН2ОСН3
j[OJ
Н—С—ОСН3 Н—С—ОСН3
!3О—С—Н СНзО-С-Н
Н—С—ОСНз
Н— С—ОСНз
чон
он
трнметокси-
глутаровая
кислота
диметоксиянтар-
ная кислоти
236
Брожение гексоз._ Брожением называется^сложный процесс
р а сщёпленйя~м6нос'аха р и до в под~вЩ},янйёмр азличных^мпкроор га-
низмов, в большинстве случаев сопровождающийся выделением
газообразных продуктов (СО2, Н, и др.) и приводящий в конечном
итоге к образованию таких веществ, как спирт, молочная кислота
и т. д.
Р.плпчают разные видь1 брожения:.
1. Спиртовое брожение:
СвН12Нв —~ 2С2Н5ОН + 2СО2
2. Маслянокислое брожение:
CfiH12Oa -» СН3СНоСН2СООН + 2СО2+ 2Н2
масляная кислота
3. Молочнокислое брожение:
QH12O6 -» 2СН3СН (ОН) СООН
молочная кислота
В этом случае получится смесь стереоизомеров со слабым левым
или правым вращением.
4. Лимоннокислое брожение:
6<
I ЧОН
с'н2
i /ОН
С( +2Н.2О
| СООН
сн2
1 /уо
х-он
лимонная
кислоча
Существуют и другие виды брожения. Не все моносахариды
сбраживаются одинаково; галактоза, например, сбраживается
труднее глюкозы.
Процессы брожения играют большую роль не только в
повседневной жизни, но и широко используются в
промышленности.
В организмах высших животных непрерывно протекают
процессы биохимического расщепления и синтеза моносахаридов.
При мышечном сокращении в результате расщепления углеводов
образуется молочная кислота. Этот процесс называется
гликолизом.
237
Дегидратация с циклизацией. Пентозы при нагревании с
разбавленной НС1 или H2SO4 образуют фурфурол (стр. 193):
С-С
. ОН Н О
пси юза
//О
чн
Гексозы в этих условиях цнклизуются с разложением. Реакция
дегидратации с циклизацией позволяет легко отличить гексозы
от пентоз.
Отдельные представители. Гексозы. Глюкоза:
СНО
ы—I—он-
но—|—н
н—[—он
н—|—он
СН2ОН
?>(-г)-глюкоза
он
а-О(+)-глюкопирапоза
D (-Ь)-глюкоза (виноградный сахар, декстроза) очень широко
распространена в природе, содержится в соке винограда и других
сладких плодов и в небольших количествах в организмах животных
и человека.
Глюкоза входит в состав важнейших полисахаридов, например,
тростниковый сахар, молочный сахар; такие важнейшие
высокомолекулярные полисахариды, как крахмал, гликоген (животный
крахмал) и клетчатка, целиком состоят из остатков глюкозы,
соединенных друг с другом различным способом.
Главным способом технического получения глюкозы является
гидролиз крахмала и клетчатки. Гидролиз крахмала с помощью
разбавленных минеральных кислот ведется в автоклавах при
повышенном давлении и 150JC. В зависимости от продолжительности
процесса гидролиза образуется либо патока (содержит 32—40%
глюкозы), либо техническая глюкоза F5—99% глюкозы).
Глюкозу для медицинских целей получают путем очистки —
перекристаллизации — технической глюкозы из водных или водно-
спиртовых растворов.
Применяется глюкоза в пищевой промышленности как дешевый
заменитель тростникового сахара. Глюкоза используется в
текстильном производстве и в некоторых других производствах в
качестве восстановителя. В медицине чистая глюкоза применяется в виде
растворов для введения в кровь при ряде заболеваний, в производ-
238
стве таблеток и т. д. Из глюкозы получают витамин С. При
восстановлении глюкозы получается шестнатомный спирт сорбит:
Cc;
сн,он
Н—
НО—
н—
н—
-он
—н
—он
-он
[Н]
н-
но—
н—
н—
-он
—н
—он
—он
сн,он
?)-глюкоза
СН2ОН
D -сорбит
При окислении глюкозы бромной или хлорной водой образуется
глюконовая кислота:
Н—
НО—
н—
Н—
хн
-он
—н
ГО]
н—
но—
-он <НОВг> н-
-он
СН2ОН
?)-глюкоза
Н—
4 он
-ОН
—Н
—ОН
—ОН
СН..ОН
глюконолая
кислота
Глюконовая кислота в виде кальциевой соли применяется в
медицине. При более энергичном окислении в кислой среде (например,
концентрированной азотной кислотой) одновременно с
альдегидной группой окисляется первичная спиртовая группа, превращаясь
сначала в альдегидную, а затем во вторую карбоксильную группу,
образуя сахарную кислоту:
Н—
HQ-
Н—
Н-
-ОН
—н
-он
-он
[О]
Н-
но—
(HNO3)
н—
-он
—н
—он
сахарная
кислота
сп.,он
н он
,0
с;
239
При нагревании глюкозы с фенилгидразином образуется плохо
растворимое в воде кристаллическое соединение озазон:
б'Х н2: N—NH—СеН5
С(( CH=N—NH—С„Н5
Н-С-ОН
НО—С—Н
Н—С—ОН
н—с-он
сн,он
Н —С—О Н:
но-с-н
__ 1
— Н„О J_[ Q Q^
1
Н-С-ОН
СН2ОН
_CH = N—NH—С6Н5
С=-;О +
НО—С—Н
Н-С-ОН
Н—С—ОН
СН2ОН
Н, N—NH— С6Н:,
=HsO
+ H,N-!—NH-CeH
— NH,
-CaHs-NH2
CH=N—NH—CsHg
C = N— NH—C6H3
HO-C-H
H—C—OH
H—C—OH
CH.OH
озазон D-глюкозы
Таким образом, реакция моносахаридов с фенилгидразином
протекает несколько иначе, чем у обычных альдегидов: сильно
выраженная способность моносахаридов к окислению,
обусловленная наличием в молекуле наряду с карбонильной группой еще
и гидроксильных групп, приводит к образованию в молекуле второй
карбонильной группы, в результате чего к каждой молекуле
моносахарида присоединяется два остатка феннлгидразина. Реакция
образования озазонов широко используется для идентификации
Сахаров, а также для выделения их из смесей, Выделенные кристаллы
озазонов очищают перекристаллизацией, определяют их
температуру плавления. Так как озазоны различных Сахаров имеют
разные температуры плавления, то можно определить характер'данного
сахара.
Галактоза:
СНО
Н
но
НО
н
СН2ОН
-ОН
-Н
I—н
-OH
I
-CH,GH
D-галактоза вместе с глюкозой входит в состав некоторых глико-
зидов н полисахаридов. Остатки галактозы входяг в состав сложней-
240
ших биополимеров — ганглиозидов или гликосфинголипидов. Они
обнаружены в нервных узлах (ганглиях) человека и животных
и содержатся также в ткани мозга, в селезенке и в эритроцитах.
Получают галактозу главным образом гидролизом молочного
сахара. При восстановлении галактозы образуется недеятельный
спирт дульцит. При окислении галактоза дает сначала галакто-
нсвую, а затем слизевую кислоту.
Манноза:
сно
сн,он
но-
но-
н-
н-
-н
-н
-он
-он
СН2ОН
н
Р-Д(-7-)-маннопираноза
Манноза сравнительно мало распространена в природе. Она
содержится в ячмене, в корке апельсина. Получают ее гидролизом
полисахарида маннана, содержащегося в грибах и некоторых
дрожжах. Как видно из формул, манноза отличается от глюкозы
конфигурацией заместителей только у второго углеродного атома,
т. е. она является эпимером глюкозы.
При восстановлении маннозы образуется спирт маннит
(содержится в грибах, маслинах, жасмине), при окислении образуется
манноновая кислота:
СН.,ОН
с/1
'¦-н
но—
но—
Н—
—н
—н-
—он
-ОН
СН2ОН
D-манноза
HOMO—
н—
н—
—н
—н
—он
-он
сн„он
¦ D-маннит
Л
[О1
но-
но-
чон
—н
-н
—он
н-
н—он
СН2ОН
D-ыанпоновая
кислота
241
Фруктоза:
сн,он
н-
н-
с=о
-н
-он
-он
сн2он
?>( — Ьфруктоза
н,он
нон2с
a-D( — )-фруктопираноза
СН2ОН
но н
B-D(— )-фруктофураноза
D-фруктоза (фруктовый сахар, левулеза) в свободном состоянии
содержится во фруктах, меде. Входит в состав многих сложных
Сахаров, например тростникового сахара, из которого может быть
получена гидролизом. Образует сложно построенный
высокомолекулярный полисахарид инулин, содержащийся в некоторых
растениях. Фруктозу получают также из инулина. При восстановлении
фруктозы образуются сорбит и маннит:
СН2ОН
С=О
НО Н
СНоОН
сн,он
н—
Н—
—он
-он
н—
НО—
1 н—
н—
СН2ОН
D -фруктоза
-он
—н
—он
—он
но—
но—
н—
Н—
СН2ОН
D-сорбит
—н
—н
—ОН
-он
си,он
?}-маннит
При окислении фруктоза дает винную и щавелевую кислоту:
СН2ОН
с=о
НО—С—Н
Н—С—ОН
н—с-он
сн.,он
[О]
/ж
н—с—он
II—С—ОН
Пен т о з ы. Пентозы встречаются в природных условиях
главным образом как составные части молекул более сложно
построенных веществ, например сложных полисахаридов, носящих назва-
242
нне пентозанов, а также растительных камедей. Пентозы в
значительном количестве A0—15%) содержатся в древесине, соломе
и т. д.
Арабиноза:
сно
сно
н—с-он
но—
н-
н-
-н
-ОН
-он
н-
но-
но-
-он
_тт
-н
но-
CHjOH
?>( —Ьарабиноза
СН2ОН
Ц+)-арабиноза
-ОН
-н
-н
НОН2С
сн„он
он
Р-Ц+)-арабофураноза
В природе преимущественно встречается ?-(+)-арабиноза. Она
содержится в вишневом клее, свекле и аравийской камеди
(гуммиарабик), откуда ее и получают. Из свеклы арабиноза получается
вместе с глюкозой, которую удаляют брожением.
При восстановлении арабпнозы получают многоатомный спирт
арабит (встречается в лишайнике), при окислении — арабоновую
кислоту.
Рибоза:
Л
Н—U-OH
Н—[— ОН
н—{— он
сн2он
D-рибоза
НОН2С
но он
a-D-рибопираноза
но он
P-D-рибофураноза
Рибоза играет важную биологическую роль. Она входит в
состав рибонуклеиновых кислот и некоторых других биологически
важных соединений. Получают рибозу эпимеризацией арабинозы.
При восстановлении рибозы образуется недеятельный спирт рибит,
при окислении — рибоновая кислота.
Ксилоза:
н-
но-
н-
-он
-н
-он
СН2ОН
^(^)"ксилоза
В природе распространена 0-(+)-ксилоза. Она образуется
при гидролизе полисахарида ксилозана, содержащегося в соломе,
243
отрубях, древесине, шелухе подсолнечника. Гидролнзаты,
содержащие ксилозу, используются для выращивания некоторых видов
дрожжей, которые в качестве белкового источника применяются
для кормления сельскохозяйственных животных.
При восстановлении ксилозы получают спирт ксилит, при
окислении — нейлоновую кислоту:
СНоОН
[Н]
Н—
НО—
но-
чн
-он
—н
-он
СНоОН
н—
но—
н—
-он
—н
—он
СНоОН
'2'
кенлит
[О]
н—
.НО—
н—
—ОН
—н
-он
СНоОН
кеялонован кислота
Некоторые природные соединения — производные
моносахаридов. Фосфорные эфиры. Из фосфатов Сахаров, играющих важную
роль в фотосинтезе, большое значение имеют: 1,6-днфосфат
фруктозы и 1-фосфат глюкозы:
О
J
сн,он
(ho)J>-o-h2c
сн-о-р(он)
но н
1.6-дпфосфзт D-фруктофуракозы
н он
1-фосфат D-глюкопирако^ы
Гликозиды. Сахара в природе часто встречаются в виде гликозидов
(циклических ацеталей):
сахар
гликозид
гидролиз
агликон
Аг.шконы представляют собой оксисоедннення жирного и
ароматического рядов. Они нередко являются органическими ядамн.
Многие гликозиды применяются в медицине. Замечательно, что
244
подавляющее большинство известных природных гликозидов
является р-гликозидами.
Амигдалин C^H^OuN -3H2O. Содержится в семенах горького
миндаля, в косточках персика, абрикосов и слив, вишен, яблок,
груш, в листьях лавровишни и пр. Под действием кислот
амигдалин распадается на две молекулы глюкозы, молекулу бензойного
альдегида и синильной кислоты:
NCi3lN * глюкоза ^
ампгдалнн бензаль-
дегид
Синигрин C10H1TO9NS2. Это гликозид семян горчицы и корней
хрена. Калиевая соль синигрина под влиянией находящегося в
семенах горчицы энзима распадается на глюкозу, аллилгорчичное
масло и кислый сернистокислый калий:
/S-QHjA
т_т г^ r^vr ст-х с*f ' с* т-г о ' т-т с* >—^т_г f^T-j \т с* ^ i К"Т-Г^Г^
синигрин S1^ -'bUs \ глюкоза аллилгорчичное масло
Витамин С (стр. 271).
ПОЛИСАХАРИДЫ
Олигосахариды (сахароподобные полисахариды)
В зависимости от числа молекул простых Сахаров, образующих
при гидролизе молекулы олигосахарида, различают: дисахариды,
гидролизующиеся на две молекулы монозы, трисахариды, гидроли-
зующиеся на три молекулы монозы, и т. д. Наибольшее значение
имеют дисахариды.
Дисахариды
Строение. Все дисахариды построены по типу гликозидов: они
представляк)Т_собой_ производные циклической формы
моносахарида, у которого атом водорода в полуацетальном гидроксиле
замещен остатком" другой молекулы моносахарида, что хорошо видно
из сравнения общей формулы гликозида и формулы какого-либо
дисахарида:
СН2ОН СН2ОН
о-с6нпо5.
он
глнкозпд дисахарид
Остатки двух молекул моносахарида, соединенные в молекуле
дисахарида, могут быть одинаковыми или разными.
245
Одна молекула моносахарида всегда участвует в, построении
молекулы дисахарида своим полуацетальным гидроксилом. Другая
молекула моносахарида может соединяться либо полуацетальным
гидроксилом, либо одним из спиртовых гндроксилов. В последнем
случае в-молекуле дисахарида будет оставаться свободным один
полуацетальный гидроксил.
Отсутствие или наличие в молекуле дисахарида полуацеталь-
ного гпдроксила сильно отражается на~'свойствах дисахарпдов.
Если при образовании дисахарида обе молекулы участвовали своими
у обоих остатков моносахаридов
циклические формы являются закрепленными, альдегидная_грхпда,
без гидролиза такого дисахарида образоваться не^мо}кет. Такой
дисахарид не обладает восстанавливающими свойствами и
называется невосстанавливающим дисахаридом:
сн,он
-н„о
l°JL>]° -о
невосстанавливающиц
дисахарид
Если же при образовании дисахарида одна_молекула__мо1шса-
харпда участвовала своим полуацетальным _?идроксщшм, а
вторая — спиртовым, то в молекуле дисахарида сохраняется один полу-
ацетнльный гидтхжснл. В этом случае циклическая форма одного
остатка моносахарида не является закрепленной, она может перейти
в альдегидную форму, и тогда дисахарид будет обладать воссхана-
влнвающими свойствами и называться восстанавливающим дисаха1
рндом:
сн,он сн9он
СхЧоОН
о
СН2ОН
/г- очн
он
сн,он
сн.он
осстлнлЕЛНвающин дисахарид
Получение. Хотя способы синтеза днсахаридов известны, прак-
тическн их получают из природных источников. Выделение индн-
246
видуальных олигосахаридов представляет довольно сложную
задачу. Для извлечения олигосахаридов из природного материала
обычно прибегают к водной экстракции. Центральной задачей при
получении олигосахаридов почти из любого источника является
выделение индивидуальных соединений из смеси моно- и
олигосахаридов. Для этой цели разработано несколько методов,
основанных на хроматографии.
Физические свойства. Дисахариды — типичные сахароподобные
полисахариды. Это твердые вещества или некристаллизующиеся сиропы, хорошо
растворимые в воде. Как аморфные, так и кристаллические дисахариды обычно
плавятся в некотором интервале температур и, как правило, с разложением.
Химические свойства. Химическое поведение дисахаридов
складывается из свойств гидроксильных групп восстанавливающего
звена (у восстанавливающих дисахаридов) и свойств гликозидов.
Будучи по своему строению гликозидами, дисахариды легко гидро-
лизуются разбавленными минеральными кислотами\й "сравнительно
стойки к щелочному гидролизу. Кислотный гидролиз приводит к
расщеплению гликозидной связи и образованию моносахаридов.
Восстанавливающие дисахарнды проявляют реакции, характерны,ё~дл~я
карбоннльной~груня-Бг~в такой же степени, как .это свойственно
соответствующим моносахаридам: они мутаротируютв растворах,
восстанавливают фелннгову жидкость, окисляются до альдожшых
кислот, вогстаттавлйваются до многоатшшнх^Ш11Р10Е,-~Рбразуют
озазоны, присоединяют синильную кислоту. Дисахарнды являются
многоатомными спиртами, а поэтому они, подобно моносахаридам,
растворяют гидроокись меди с образованием синего окрашивания.
Отдельные представители: Мальтоза (солодовый сахар).
Название возникло в связи со способом получения мальтозы: она
получается из крахмала при действии солода (maltum — по-лзтыни
солод). В результате гидролиза мальтоза расщепляется на две
молекулы глюкозы:
CHO HO2QHO
глюкоза
Обладает восстанавливающими свойствами, следовательно, один
полуацетальный гидроксил в ее молекуле остается свободным.
В молекуле мальтозы кислородный мостик, связывающий два остатка
глюкозы, соединяет первый атом углерода одного остатка с
четвертым атомом углерода другого остатка глюкозы:
6 С
Н2ОН
EJ
н/н
'к
т
3
н
0
н/1_0_
2
он
оСН.
1/
\?н
3
н
он
0
\
н/
он
н
.1
1
он
Мальтоза кристаллизуется из водных и водно-спиртовых
растворов с одной молекулой воды C12H23OU • Н30. Водные растворы
247
мутаротируют. Удельное вращение после завершения мутаротапии
Ы136Э
Солодовый сахар является промежуточным продуктом при
гидролизе крахмала, он широко распространен в растительных
и животных организмах. Солодовый сахар значительно менее
сладок, чем тростниковый сахар (в 0,6 раза при одинаковых
концентрациях).
Мальтоза восстанавливает фелингову жидкость, дает гидразон
и озазон. При осторожном окислении мальтозы образуется
одноосновная мальтобионовая кислота:
СНпОН
н
он и
мальтоза
СН„ОН
CHLOH
ОН
0 И
соон
но
мальтобионовая кислота
Целлобиоза. Это дисахарид, образующийся при расщеплении
целлюлозы (клетчатки). Целлобиоза, подобно мальтозе, построена
из двух остатков глюкозы, но соединены они неа-1,4-, а [5-1,4-глико-
зидной связью:
СН,,ОН СН-ОН-
но
оы
он
248
Целлобиоза является промежуточным продуктом при гидролизе
клетчатки так же, как мальтоза при гидролизе крахмала.
Кристаллическая целлобиоза представляет собой р-изомер с
/пл = 225° С, растворы ее обладают мутаротацией, причем [a]D
меняется от +14,2 до +34,6°. Целлобиоза обладает всеми
свойствами восстанавливающих Сахаров.
Лакпшзи (молочный сахар). Название этого дисахарида возникло
в связи с получением его из молока (по-латыни lactum). При
гидролизе лактоза расщепляется на глюкозу и галактозу.
2О н> С6Н12О6 + С6Н1гО6
глюкоза галактоза
Лактоза обладает восстанавливающими свойствами,
следовательно, в ее молекуле имеется один свободный полуацетальный
гидроксил, Этот гидроксил принадлежит остатку глюкозы, а
кислородный мостик, связывающий два остатка моносахаридов в
молекуле лактозы, соединяет первый атом углерода остатка галактозы
с четвертым атомом углерода остатка глюкозы:
сн,он
сн,он
н
н
остаток
галактозы
Лактозу получают из молока: в коровьем молоке содержится
4—5,5% лактозы, женское молоко содержит 5,5—8,4% лактозы.
Лактоза отличается от других Сахаров отсутствием
гигроскопичности: она не отсыревает. Молочный сахар применяется
как фармацевтический пр'епарат и как питание для грудных
детей.
Водные растворы лактозы мутаротнруют. После завершения
мутаротации устанавливается удельное вращение [a]D = +52,2°.
Лактоза в 4 или 5 раз менее сладка, чем сахароза.
Сахароза (тростниковый или свекловичный сахар). Названия
возникли в связи с ее получением либо из сахарной свеклы, либо
из сахарного тростника. Тростниковый сахар был известен за много
столетий до нашей эры. Лишь в середине XVIII в. этот дисахарид
был обнаружен в сахарной свекле и только в начале XIX в. он был
получен в производственных условиях. Сахароза очень
распространена в растительном мире. Листья и семена всегда содержат
небольшие количества тростникового сахара. Он находится также
в плодах (абрикосах, персиках, груше, ананасе). Его много в
кленовом соке, пальмовом соке, кукурузе. Это наиболее известный и
249
широко применяемый сахар. При гидролизе из него образуются
глюкоза и фруктоза:
C12H2.,OU + Н3О -> С6Н15О6 + QH12O6
сахароза глюкоза фруктоза
Сахароза — невосстанавливающий сахар, у нее нет свободного
полуацегального гидроксила. Сахароза гидролизуется легче, чем
многие другие дисахариды. Это связано с тем, что один из остатков
моносахаридов, входящих в его молекулу, а именно остаток
фруктозы, находится в фуранозной форме:
б СН2ОН
сн„он
но н
Сахароза кристаллизуется без воды, tan = 184,5° С. Удельное
вращение водного раствора сахарозы [a]D = +66,5°. Растворы
сахарозы не обладают мутаротацией. При гидролизе сахароза
превращается в смесь равных количеств глюкозы и фруктозы. Фруктоза
характеризуется более сильным левым вращением, чем глюкоза
правым Ыд = —92° и [a]D = +52,5\ соответственно, поэтому
раствор гидролизованной сахарозы имеет левое вращение. В связи
с изменением в процессе гидролиза правого вращения раствора на
левое гидролиз тростникового сахара получил название инверсии
(инверсия — обращение). Смесь равных количеств глюкозы и
фруктозы, получающаяся в результате инверсии тростникового сахара,
называется инвертным сахаром. Природным инвертным сахаром
является мед, состоящий в основном из глюкозы и фруктозы.
Сахарозу производят в огромных количествах. В СССР этот
дисахарид получают исключительно из сахарной свеклы, которая
содержит 16—20% сахарозы (сахарный тростник содержит 14—26%
сахарозы). Промытую свеклу измельчают н в аппаратах
(диффузорах) многократно извлекают сахарозу водой, имеющей
температуру около 80~ С. Полученную жидкость, содержащую, кроме
сахарозы, большое количество различных примесей, обрабатывают
известью. Известь осаждает в виде кальциевых солей ряд
органических кислот, а также белки и некоторые другие вещества. Часть
извести при этом образует с тростниковым сахаром растворимые
в холодной воде кальциевые сахараты, которые разрушают
обработкой двуокисью углерода. Осадок углекислого кальция отделяют
фильтрацией, фильтрат после дополнительной очистки упаривают
в вакууме до получения кашицеобразной массы. Выделившиеся
кристаллы сахарозы отделяют при помощи центрифуг. Таким обра-
250
зом, получают сырой сахарный песок, имеющий желтоватый цвет,
маточный раствор бурого цвета, некристаллизующийся сироп,
свекловичная патока или меласса. Сахарный песок очищают
(рафинируют) и получают в готовом виде.
Высокомолекулярные полисахариды
Высшие полиозы являются высокомолекулярными веществами,
содержащими сотни~~й~ тысячи остатков" моносахаридов, поэтому
исследователям долго не удавалось изучить их строение. Лишь за.
последние 30—40 лет химики добились больших успехов в
выяснении строения этих соединений.
Общим для строения полиоз является следующее:
1. Остатки моносахаридов связаны в молекулах высших полиоз
при помощи кислородных мостиков, соединяющих эти остатки
в длинные цепи.
2. При образовании цепимолекула моносахарида своим полуа-
цетальным гидроксилом" взаимодействует с каким-либо спиртовым
гидроксилом второй молекулы моносахарида, эта вторая молекула
взаимодействует своим полуацетальным гидроксидом со спиртовым
гидроксилом третьей молекулы моносахарида и т. д. Например,
образование высшей полиозы из молекул гексозы С6Н12О6 можно
выразить таким образом:
СН2ОН
сн,он
сн,он
Таким образом, каждый остаток моносахарида связан с
соседними остатками гликозидными связями, поэтому высшие полиозы
можно рассматривать как полигликозидьь...
3. Полигликозндные цепи, образующие молекулы
полисахаридов, могут быть неразветвленными и. рдзве/твленными. .
4. Остатки моносахаридов, входящие в состав молекулы
„высшего полисахарида, могут быть одинаковыми, но могут_и
различаться; в первом случае это будут гомопшшсахариды, во втором —
гетерополисахарнды.
Важнейшими гомополисахаридами, состоящими из остатков
глюкозы, являются крахмал, гликоген и клетчатка (или целлюлоза).
251
Крахмал
Крахмал образуется при фотосинтезе в зеленых листьях в виде
зерен. Эти зерна особенно легко обнаружить в микроскоп, используя
известную реакцию с иодом: крахмальные зерна окрашиваются
в синий или сине-черный цвет. По накоплению крахмальных зерен
можно судить об интенсивности фотосинтеза. Крахмал в листьях
расщепляется на моносахариды или олигосахариды и переносится
в другие части растений, например, в клубни картофеля или зерна
злаков. Здесь вновь происходит отложение крахмала в виде зерен.
Наибольшее содержание крахмала в следующих культурах ("о);
Рис (зерно) — 62—82 Пшеница (зерно) —57—75
Кукуруза (зерно) — 65—75 Картофель (клубни) — 12—24
Свойства. Крахмал представляет собой белый аморфный
порошок, напоминающий муку («картофельная мука»). Обычный
крахмал содержит всегда от 10 до 20% воды, которую можно удалить
лишь тщательным высушиванием при 100—110° С, причем
температура должна повышаться медленно во избежание разложения
крахмала. Высушенный крахмал очень гигроскопичен. При
настаивании с водой, даже горячей, крахмальные зерна лишь
разбухают и только небольшая часть полисахаридов крахмала переходит
в раствор. Для, приготовления раствора- (крахмального клейстера)
крахмал размешивают в небольшом количестве воды и выливают
постепенно в кипящую воду при сильном размешивании. Раствор
крахмала имеет правое вращение [a]D = 195°.
Раствор иода с йодистым калием вызывает синее окрашивание
'растворов крахмала, которое исчезает при кипячении и вновь
появляется при охлаждении. Иодкрахмальная реакция применяется для
открытия как крахмала, так п иода. На ней основано широкое
применение крахмала в качестве индикатора в иодометрии — одном
из методов аналитической химии.
Крахмал практически не обладает восстанавливающей
способностью: он не восстанавливает ни гидроокись меди, ни фелингов
раствор. Однако некоторые окисляющие реактивы (динитросалицмло-
вая кислота) дают возможность обнаружить в крахмале ничтожное
количество свободных альдегидных групп: одну на сотни и тысячи
глюкозных остатков. Пользуясь зтими реактивами, определяют
молекулярную массу полисахаридов крахмала.
Открыть в крахмале свободные спиртовые группы при помощи Си(ОН),,
как в случае дпеахэрпдов. не удается, так как сам крахмал дает лишь
коллоидные растворы, поэтому «растворения-) Си(ОНJ произойти не может. Однако
методами метилирования и ацплировашш можно обнаружить в среднем три
свободные гидрокенльные группы на каждый остаток глюкозы.
При быстром нагревании крахмала, содержащего обычно 10—
20% воды, происходит расщепление ее гигантской молекулы на
более мелкие простые молекулы — полисахариды, называемые дек-
стринами. Степень полимеризации (х) в декстринах меньше, чем
степень полимеризации (п) в крахмале. Еще быстрее декстриннзация
идет в присутствии кислот. Декстрины с постепенно убывающей
величиной молекулы окрашиваются иодом в сине-фиолетовый,
фиолетовый, красно-фиолетовый, оранжевый и желтый цвета. Слегка
декстринизнрованный крахмал, все еще окрашивающийся иодом
в синий цвет, но лучше растворяющийся в воде, чем обычный
крахмал, называется растворимым крахмалом.
Ряд декстринов можно получить, если произвести расщепление
крахмала с помощью фермента а-амилазы.
Как при ферментативном, так и при кислотном расщеплении
крахмала гидролиз обычно не заканчивается образованием
декстринов, хотя и существуют ферменты, которые расщепляют крахмал
только до декстринов. Обычно ферменты пищеварительных соков
при температуре около 40° С гидролизуют крахмал до конца, т. е.
до глюкозы: также действуют минеральные кислоты при
длительном кипячении.
Схема постепенно идущего гидролиза крахмала как
ферментативного, так и кислотного в упрощенном виде
выражается так:
(С6Н10О5)„ -> (QH1AO5)A- -* С12Н2?ОП -* С6НПО„
крахмал ряд мальтоза глюкоза
декстринов
При хлебопечении крахмал муки частично превращается в
декстрины, которые более растворимы в воде, а потому легче
перевариваются и усваиваются.
Фракции крахмала и их строение. Крахмал не является
однородным веществом: он представляет смесь полисахаридов,
построенных по двум различным типам, поэтому крахмал можно разделить
на две основные фракции: амилозу и амилопектин. Для разделения
фракций крахмала существует много различных методов. Один из
простейших методов заключается в том, что крахмальные зерна
обрабатывают горячей водой (около 80° С). При этом амилоза
переходит в раствор, тогда как амилопектин лишь разбухает и остается
в зернах. В последние годы разработаны методы получения
кристаллической амилозы.
Соотношение между амилозой и амилопектином колеблется
в крахмалах разных растений. В большинстве случаев содержание
амилозы составляет 10—20°о, амилопектнна 80—90%.
Амилоза, Молекулы амилозы — это очень длинные цепи,
состоящие из остатков глюкозы, совсем неразветвленные или очень мало
разветвленные.
Каждый глюкозный остаток связан атомом кислорода своего
полуацетального гидроксила с четвертым атомом углерода
следующего глюкозного остатка. Все глюкозные остатки находятся в
а-форме и поэтому связь глюкозы в молекуле амилозы называют
а-1,4-глюкозидной связью. Точно такая же связь соединяет два
остатка глюкозы в молекуле мальтозы; отсюда становится понят-
253
ным, почему при гидролизе амилозы образуется мальтоза. Строение
молекулы амилозы можно выразить следующей формулой:
СН2ОН
СН2ОН
н
он
Как видно из формулы, подавляющее большинство остатков
глюкозы содержит три свободные гидроксильные группы: у второго,
третьего и шестого атомов углерода. Свободный полуацетальный
гидроксил в молекуле амилозы находится лишь у одного из глю-
козных остатков в самом «начале» цепи. В связи с этим
восстанавливающая способность амилозы ничтожна.
Амилоза растворима в горячей воде, но эти растворы нестойки:
при стоянии быстро выпадает осадок амилозы. Раствор амилозы дает
с иодом интенсивное синее окрашивание. По данным рентгенострук-
турного анализа, молекула амилозы свернута в спираль. Внутри
спиралевидной молекулы остается канал диаметром около 5 мк,
в котором могут располагаться подходящие по -размеру молекулы,
образуя особого типа комплексы — так называемые соединения
включения. Одним из них является соединение амилозы с иодом,
окрашенное в синий цвет.
Амилопектин. Молекулы амилопектина, подобно молекулам
амилозы, состоят из цепей глюкозных остатков, но в отличие от
амилозы в амилопектине они сильно разветвлены. Подавляющее
большинство глюкозных остатков связано в амылопектине так же,
как и в амилозе сс-1,4-глюкозидной связью. Однако в точках
ветвлений цепи имеются и ос-1,6-глюкозидные связи, т.е. первый атом
углерода одного глюкозного остатка связан с шестым атомом
углерода другого глюкозного остатка при помощи сс-глюкозидной связи.
Ниже при помощи формулы Хеуорзса изображен небольшой
участок молекулы амилопектина:
СН.ОН СН2ОН
О к— О
Н H/L-c-J\OH K/l_o-J\OH
254
Цепи из глюкозных остатков в молекуле амилопектина ветвятся
многократно. «Ветвистая» структура амилопектина подтверждается
данными ферментативного расщепления этого полисахарида.
Физико-химические методы исследования амилопектина
свидетельствуют о сферической форме его молекул (рис. 21).
Общее число глюкозных остатков в молекуле амилопектина
значительно выше, чем в молекулах амилозы. Молекулярная масса
амилопектина порядка 106 — это значит, что степень
полимеризации равна примерно 6000.
Большинство глюкозных остатков в молекуле амилопектина,
так же, как и в молекуле амилозы, имеет по три свободных гидро-
ксила: у второго, третьего и четвертого атомов углерода. Глюкоз-
ный остаток в точке
ветвления имеет лишь два
свободных гидроксила (у второго и
третьего атомов углерода),
в то время как глюкозный
остаток, в конце цепи имеет
четыре свободных гндроксила
(у второго, третьего,
четвертого и шестого атомов
углерода). При большом общем
числе глюкозных остатков
в среднем в каждом остатке
глюкозы будет по три
свободных гндроксила.
Из всех глюкозных
остатков в молекуле амилопектина
лишь один, находящийся в
самом начале цепи, содержит
полуацетальный гидроксил.
Понятно, что амилопектин обладает лишь ничтожной
восстанавливающей способностью. С иодом амилопектии дает фиолетовое
окрашивание.
Применение крахмала. Крахмал является одним из основных
компонентов пищевых продуктов. Мука, хлеб, картофель, крупы
относятся к главным источникам углеводов в нашем питании.
Очищенный крахмал применяют в производстве кондитерских и
кулинарных изделий, колбас. В текстильной промышленности он
используется для производства шлихты, апретуры, загустителей
к краскам. Крахмал применяется в спичечной, бумажной,
полиграфической, косметической промышленности, в переплетном деле.
В медицине и фармакологии крахмал идет на приготовление
присыпок, паст (густых мазей), а также необходим в производстве
таблеток и т. д.
Подвергая крахмал кислотному гидролизу, можно получить
глюкозу в виде чистого кристаллического препарата или в виде
патоки — окрашенного некристаллизующегося сиропа. Патока
широко используется в кондитерской промышленности. В последние
Рис. 21. Строение крахмала
255
годы появилось производство так называемых модифицированных
крахмалов, подвергшихся специальной обработке или содержащих
улучшающие их свойства добавки. Модифицированные крахмалы
широко применяются в различных отраслях промышленности.
Гликоген
Гликоген, или животный крахмал, играет исключительно важную роль
в организмах животных как запасный полисахарид; все процессы
жизнедеятельности, в первую очередь мышечная работа, сопровождаются расщеплением
гликогена, отдающего сосредоточенную в нем энергию. В тканях организма
из гликогена в результате ряда сложных превращений может образовываться
молочная кислота.
Гликоген содержится во всех животных тканях. Особенно его много в
печени (до 2О°о), в мышцах (до 4%). Он содержится также в некоторых низших
растениях, дрожжах и грибах.
Гликоген можно выделить путем обработки животных тканей 5—10%-ной
трихлоруксусной кислотой на холоду с последующим осаждением извлеченного
гликогена спиртом.
Гликоген — белый аморфный порошок, хорошо растворяющийся даже в
холодной воде с образованием опалесцирующих растворов. Растворы гликогена
имеют правое вращение, удельное вращение их очень близко вращению
крахмала: [a]D= -1-196°.
Гликоген легко гидролизуется кислотами и ферментами, образуя в качестве
промежуточных веществ декстрины и мальтозу и при полном гидролизе
превращаясь в глюкозу.
Молекулы гликогена построены по тому же принципу, что и молекулы амило-
пектина (наличие а-1,4- и а-1,6-гликозидных связей). От амилопектина гликоген
отличается лишь тем, что у него больше боковых ветвей, вследствие чего
молекула гликогена более плотная; общее число остатков глюкозы в молекуле
гликогена выше, чем в молекулах амилопектнна. Молекулярная масса гликогена
исчисляется миллионами, степень полимеризации 2 400—24 000. С иодом
растворы гликогена дают окрашивание от випно-красного до красно-бурого в
зависимости от происхождения гликогена, вида животного и других условий.
Окрашивание иодом исчезает при кипячении и вновь появляется при охлаждении.
Клетчатка, или целлюлоза
(С6Н10О5)л является основным веществом растительных клеток,
откуда и произошло это название. Таково же происхождение и
другого названия — целлюлоза (от латинского cellula — клеточка).
Клетчатка составляет от 50 до 70°о древесины. Хлопок представляет
собой почти чистую клетчатку. Волокна льна и конопли состоят
преимущественно из клетчатки. Наиболее чистыми образцами
клетчатки, с которыми приходится встречаться в обыденной жизни,
является очищенная вата, получаемая из хлопка, и
фильтровальная бумага.
В древесине клетчатка содержится наряду с так называемыми
гемицеллюлозами и лигнином. Примером гемнцеллюлоз могут
служить пентозаны — полисахариды пентоз (С,Н8О4)„.
Лигнин является сложным веществом ароматического характера.
Строение. Молекулы клетчатки представляют собой очень
длинные цепи глюкозных остатков, причем кислородные мостики,
соединяющие эти остатки, связывают первый атом углерода одного остатка
256
с четвертым атомом углерода следующего остатка. Таким образом,
по общему типу строения клетчатка сходна с амилозой, но одна
особенность сильно отличает ее от амилозы: все глюкозидные остатки
в молекуле целлюлозы находятся в Р-форме, т. е. если в молекуле
амилозы имеются а- 1,4-глюкозидные связи, то в целлюлозе C-1,4-глю-
козидные связи. Это подтверждается, в частности, образованием,
при частичном гидролизе клетчатки дисахарида целлобиозы,
содержащей р-1,4-глюкозидную связь. Строение целлюлозы можно
представить в виде формулы:
СН2ОН
сн,он
о
\он
он
Калсдый глюкозный остаток клетчатки содержит три свободных
спиртовых гидроксила: у второго, третьего и шестого углеродных атомов.
Общее число глюкозиых остатков в молекуле целлюлозы равно
в среднем 6000—12 000, что соответствует молекулярной массе
10 000 000—20 000 000. Обработка клетчатки при ее очистке обычно
сопровождается некоторым расщеплением ее молекулы: технические
ее образцы имеют молекулярную массу 50 000—150 000.
Сравнивая строение клетчатки со строением амилозы, видно,
что различие сводится только к другому пространственному
расположению кислородных мостиков, соединяющих глюкозные остатки,
по отношению к плоскости циклов этих остатков. Однако эта,
казалось бы, небольшая деталь строения приводит к значительным
различиям в свойствах: в то время как амилоза относится к
питательным веществам и очень легко расщепляется, целлюлоза
является веществом, крайне стойким как к механическим, так и к
химическим воздействиям.
Получение. Для получения чистой клетчатки из хлопка его обрабатывают,
чтобы удалить жир органическими растворителями; другие примеси удаляют
при помощи щелочей.
При получении клетчатки из древесины пользуются ее химической
прочностью: при жесткой обработке древесины удаляются примеси — лигннн и геми-
целлюлозы, в результате остается чистая целлюлоза. При такой обработке
древесины большое значение имеет «сульфитный процесс»: измельченную древесину
нагревают в котлах под большим давлением с бисульфитом кальция Ca(HSO3J>
который растворяет лигнин: таким образом получается сульфитная целлюлоза.
Свойства. Клетчатка характеризуется большой механической и химической
прочностью. Она не растворима в воде и растворима в небольшом числе
растворителей. К ним относятся: 1) реактив Швейцера, представляющий собой раствор
гидрата окиси меди в концентрированном аммиаке; 2) солянокислые растворы
хлористого цинка и некоторых других солей; 3) концентрированная соляная
кислота.
Целлюлоза обладает очень слабой оптической активностью [a]D = —3,21°
(при растворении в реактиве Швейцера).
9 А. П. Нечаев
257
Для проведения полного гидролиза клетчатки ее растворяют в
концентрированной серной кислоте, зате.м разбавляют водой и кипятят долгое время:
(СвН1вО,)л + яН2О н> пС3Н12О3
целлюлоза глюкоза
Целлюлоза почти не проявляет восстанавливающих свойств и не дает
других реакций карбонильной группы, характерных для моносахаридов.
При действии окислителей (озон, перекись водорода, хлорноеатистая
кислота и ее соли, разбавленная азотная кислота, пермангаиат и др.) получаются
сложные смеси продуктов окисления, носящие название оксицеллюлоз.
Химические свойства целлюлозы определяются прежде всего присутствием
гидроксильных .ррупп. Главньши реакциями целлюлозы как многоатомного
спирта являются образование алкоголятов и эфирсв целлюлозы. Действуя
металлическим натрием, можно получить тринатрийалкоголят целлюлозы
[CeHA(ONaK]n.
В отличие от низших спиртов целлюлоза при обработке
концентрированными растворами едких щелочей образует прочные соединения, имеющие
строение алкоголятов:
[С6Н,О2 (OHK]ra-|-nNaOH -* [С6П7О2 (ОН), ONa]rt + nH2O
алкалпцеллюлоза
Этот процесс получил название мерсеризации. При этом происходит набухание
волокна и повышается его восприимчивость к красителям. Щелочная целлюлоза,
или алкалицеллюлоза, при действии воды распадается обратно на целлюлозу
и едкую щелочь:
[C,H7O2(OHJONa];l + «H2O -* [C6H7O3 (OHK]/t~h«NaOH
Однако регенерированная целлюлоза имеет другую физическую структуру.
Мерсеризация хлопчатобумажных тканей широко применяется для
придания им лучшей окрашиваемости и более красивого внешнего вида. Щелочная
целлюлоза имеет также большое значение в технике как промежуточный
продукт в производстве вискозного волокна и эфиров целлюлозы.
Целлюлоза, как и любой другой спирт, образует простые п сложные эфиры.
В зависимости от условий этерификацин замещается большее пли меньшее число
спиртовых гидроксилов целлюлозы. Максимальная степень этерификации
отвечает триэфнру целлюлозы. Общим способом получения простых эфиров
целлюлозы является действие на щелочную целлюлозу галогеналкилов или алкил-
сульфатов в присутствии избытка щелочи:
[С6Н7О2 (ОНK ОМа],г -f- 3nC6H3CH2Cl -f 2nNaOH ->
н> [CeH;Oa (ОСИ2СбН5)з]я + ЗШаС1
ИЛИ
[СбН7О3 (ОНJ ОКа]я + Зи (СН3J SO4+2nNaOH ->
-> [С6Н7О2 (OCH,)]., + 3/iCH3NaSO4
Среди простых эфиров целлюлозы наибольшее применение в технике имеют
метил-, этил- и бензклцеллюлозы. При метилировании целлюлоза приобретает
некоторую растворимость в воде. Мстплцеллюлоза применяется главным
образом как загуститель (взамен крахмала) в текстильной, косметической и пищевой
промышленности. Этилцеллюлоза обладает высокой механической и химической
прочностью, термо- и морозостойкостью. Она используется в производстве
пластмасс, пленок п лаков.
При действии на целлюлозу смесью азотной и серной кислот получают
нитраты целлюлозы, неправильно называемые иногда нитроцеллюлозой или
нитроклетчаткой. Все нитраты целлюлозы горючи, а некоторые взрывчаты.
Максимальное число остатков азотной кислоты, которое можно ввести в клетчатку, равно
трем на каждое звено глюкозы:
[QH?O2 (ОНЫ., + ЗлНМОз -> [С6Н7О2 (О.\О2K]Л
258
Продукт полной этерификации — тринитрат целлюлозы — должен содержать
в соответствии с формулой 14,1% азота. На практике получают продукт с
несколько меньшим содержанием азота A2,5—13,5%), известный в технике под
названием пироксилин. Пироксилин — детонирующее взрывчатое вещество
большой силы, это — дробящее взрывчатое вещество. Применяется в подрывных
работах. Пироксилин не растворим ни в воде, ни в спирте, ни в эфире; такие
растворители, как ацетон и уксусноамиловый эфир, желатинируют пироксилин,
т. е. пироксилин в этих растворителях набухает и образует густую студенистую
массу. Из этой массы прессуют ленты различной толщины и размеров, которые
при высыхании могут быть применены в качестве бездымного пороха. Он
сгорает медленнее пироксилина и, сгорая, не дает дыма. Пироксилин желатинируется
также при помощи нитроглицерина. Образующийся «гремучий студень»
представляет собой особый вид динамита и также применяется в подрывном деле.
Продукт нитрования клетчатки, содержащий 10% азота, отвечает по составу
динмграту целлюлозы: в технике такой продукт известен под названием
«коллоксилин». При действии на него смеси спирта и эфира образуется вязкий
раствор, так называемый коллодий. Коллодий — густая, клейкая жидкость,
оставляющая по высыхании блестящую пленку коллоксилина. Коллодий
применяется в медицине вместо пластыря, чтобы закрыть небольшие порезы. Если
к коллодию добавить камфору @,4 в. ч. па ! в. ч. коллоксилина) и испарить
растворитель, то остается гибкая пленка — целлулоид. Исторически — это
первый известный тип пластмасс. Еще с прошлого века целлулоид получил
широкое применение как удобный термопластический материал для
производства многих изделий. В особенности важно использование целлулоида в
производстве кинопленки и нитролаков. Серьезным недостатком этого материала
является его сильная горючесть, поэтому целлулоид все чаще стали заменять
другими материалами, в частности ацетатами целлюлозы.
При действии на целлюлозу смеси уксусного ангидрида, уксусной кислоты
и серной кислоты или хлористого цинка (последние играют роль катализаторов)
образуется триацетат целлюлозы:
[С6Н7О2 (ОНK]„ + Зп (СН3СОJ О (сн.соон; h8so4>
-> [С6НА (ОСОСН3K]ге + ЗпСН3СООН
Триацетилцеллюлозу называют первичным ацетатом. Неполное ацетилирование
целлюлозы или частичный гидролиз первичного ацетата приводят к вторичному
ацетату:
[С,Н,О, (ОСОСН3)з],+ пН2О -» [С6Н,О2 (ОСОСН3K ОН],, + яСН3СООН
Ацетилцеллюлоза идет на изготовление ацетатного шелка. Из полученных двух
типов ацетнлцеллюлозы готовят растворы, причем первичный ацетат растворяют
обычно в смеси Си2С12 и спирта, а вторичный ацетат — в апетоне. Вязкие
прядильные растворы продавливают через колпачки с тонкими отверстиями
(фильеры; сухое прядение), далее растворители в токе нагретого воздуха испаряются,
а струйки раствора превращаются в тончайшие нити из ацетилиеллюлозы —
ацетатный шелк. Ацетилцеллюлоза используется в производстве тканей,
трикотажа, негорючей кинопленки, пластмасс, электроизсляций.
Общий принцип, лежащий в основе получения ацетатного волокна
(растворение, затем формование нитей), можно осуществлять по-иному, т. е. получить
растворы целлюлозы другим путем. Большое техническое значение имеет
вискозный способ. Сущность этого процесса — образование раствооимого в воде
ксантогената целлюлозы при действии на нее сероуглерода и щелочи:
Г ' S
[С6Н;О.; (OHK]anCS'- "Na0-S CSH7O2; О-С^
целлюлоза L \ SNa 3Jn
ксантогенат целлюлозы
Образовавшееся соединение представляет собой натриевую соль сложного эфира
ксантогеновой кислоты. Ксантогенаты целлюлозы растворяются в воде или
в разбавленной щелочи, образуя так называемые вискозные растворы. Вискоз-
9* 259
ньш раствор подают на прядильную машину, с помощью дозирующего насоса
продавливают через фильеры в так называемую прядильную ванну, содержащую
разбавленную серную кислоту и некоторые сульфаты (мокрое прядение). Под
действием кислоты ксантогенатные группы отщепляются и целлюлоза
регенерируется, образуя гладкую нить вискозного шелка:
СвН7О»|О—с/ I
|_ \ NSNa/3Jn
Вискозный шелк — самый дешевый, так как его можно производить из
сравнительно мало очищенной клетчатки и с применением не очень дорогих реактивов.
Вискозная нить, но более толстая и «нарубленная» на мелкие куски
представляет собой штапельное волокно, из которого получают ткани, заменяющие
хлопчатобумажные. Если вискозу, вместо фильер, продавливать через узкие
щели, то получается прозрачная пленка — хорошо известный всем целлофан.
Первые промышленные способы химической переработки
целлюлозы возникли в связи с развитием бумажной промышленности.
Бумага — это тонкий слой волокон клетчатки, спрессованных и
проклеенных для создания механической прочности, а также
гладкой поверхности для предотвращения растекания чернил.
Первоначально для изготовления бумаги употребляли растительное
сырье, из которого чисто механически можно было получить
необходимые волокна: стебли риса, хлопка, использовались также
собираемые у населения изношенные ткани (тряпки). Однако по
мере развития книгопечатания перечисленных источников сырья
стало не хватать для удовлетворения растущей потребности в
бумаге. Особенно много бумаги расходуется для печатания газет,
причем вопрос о качестве (белизне, прочности, долговечности)
для газетной бумаги значения не имеет. Зная, что древесина
примерно на 50% состоит из клетчатки, к бумажной массе стали
добавлять размолотую древесину. Такая бумага не прочна и быстро
желтеет, особенно на свету. Для того чтобы улучшить качество
древесных добавок к бумажной массе, были предложены различные
способы химической очистки древесины, позволяющие получить из
нее более или менее чистую целлюлозу, освобожденную от
сопутствующих веществ — лигнина, смол и т. д. Для выделения целлюлозы
было предложено несколько способов. По сульфитному способу
измельченную древесину «варят» под давлением с бисульфитом
кальция. При этом сопутствующие вещества растворяются и
освобожденную от примесей целлюлозу отделяют фильтрованием.
Образующиеся сульфитные щелока являются в бумажном производстве
отходами. Однако вследствие того, что они содержат наряду с
другими веществами способные к брожению моносахариды, их
используют как сырье для получения этилового спирта (стр. 101).
Пектиновые вещества, растительные камеди и слизи
Пектиновые вещества — соединения, содержащиеся в
растительных соках, плодах, ягодах, корнеплодах, осаждающиеся
спиртом из водных растворов. С сахарамп и кислотами они образуют
студии. Это свойство используется в пищевой промышленности
260
для приготовления желе и мармеладов. Пектиновые вещества
известны более 100 лет, однако основные черты их строения выяснены
сравнительно недавно. Долгое время считали, что пектиновые
вещества состоят в основном из линейного полимера полигалакту-
роновой кислоты, остатки которой соединены сх-1, 4-связыо, причем
карбоксильные группы частично этерифицированы метиловым
спиртом, а часть атомов водорода карбоксильных групп замещена
катионами. Однако это мнение, по-видимому, является слишком
упрощенным. Доказано, что, как.правило, пектиновые кислоты
представляют собой гегерополисахарпды, состоящие не только из уроно-
вых кислот, но содержащие еще и нейтральные моносахариды:
галактозу, рамнозу и арабинозу. Это представление подтверждается
частичным кислотным гидролизом.
Камеди — вещества, выделяющиеся в виде прозрачных густеющих масс
при повреждении растений: механическом ранении или патологических
процессах, вызываемых бактериями или грибками. Из выделенной растениями
аморфной массы можно извлечь камеди щелочью с последующим осаждением
кислотой. Это гидрофильные вещества, в большинстве случаев хорошо
растворимые в воде с образованием клейких растворов. Таковы аравийская камедь,
или, как ее называют, гуммиарабик, получаемая из сенегальской акации, камедь
тракаганта, вишневый клей и др. Камеди являются ге.терополисахарндами,
состоящими из нескольких моносахаридов, среди которых может быть одна или
несколько уроновых кислот.
Слизнми называют полисахариды, родственные камедям, но присутствующие
в неповрежденных растениях. Их источником служат кора, корпи, листья,
семена. Слизи являются продуктами нормального метаболизма растений н служат
либо пищевым резервуаром, либо веществами, удерживающими волу. Среди
елнзеи встречается много нейтральных гетеро- и гомополлсахаридов.
БЕЛКОВЫЕ ВЕЩЕСТВА
Белками, или белковыми веществами (протеинами), называются
высокомолекулярные органические соединения, молекулы которых
построены из остатков а-аминоклелот. Количество последних
может колебаться очень сильно и достигать иногда нескольких
тысяч.
Белки играют исключительную роль в жизни живого организма.
Функции их весьма разнообразны. Из них состоит основная, масса
протоплазмы клеток, они выполняют каталитические,
строительные, энергетические, обменные, защитные и многие другие функции.
«Повсюду, где мы встречаем жизнь, мы находим, что она связана
с каким-либо белковым телом, и повсюду, где мы встречаем какое-
либо белковое тело, не находящееся в процессе разложения, мы
без исключения встречаем и явления жизни», — писал Ф. Энгельс. *
По существу, вся деятельность организма (развитие, движение,
заболевание и многое другое) связана" "с белковыми
веществами.
* Ф. Энгельс. Антн-Дюрикг. К. М арке и Ф. Энгельс. Соч.,
т. 20. М., Госполятиздат. 1961, с. 83.
261
Все это дало основание Ф. Энгельсу еще в прошлом веке
сделать свое знаменитое заключение: «Жизнь есть способ
существования белковых тел...» *
Белками являются и многие важнейшие физиологически
активные соединения: ферменты, некоторые гормоны и антибиотики и
другие вещества.
Белки являются необходимой составной частью пищевого
рациона человека и животных. Пищевая ценность белков в первую
очередь определяется_ихаминокислотным составом. Несмотря на
разнообразие в строении и функциях, элементарный состав
белковых веществ колеблется незначительно (в % на сухой вес): 50—55С;
6,5—7,ЗН; 21,5—23,50; 15—17,5N; 0,3—2,55. Некоторые белки
содержат в небольших количествах фосфор, селен, металлы
(железо, цинк, медь).
Количество белковых веществ в животных и растительных
организмах неодинаково. Обычно в животных организмах белков больше,
чем в растениях. Неодинаковое количество их присутствует в
отдельных организмах и тканях. Так, содержание белков в мышцах
18—23%, в мозгу 8—9%, сердце 16—18%, крови 7—8,5%. В
растениях находится от 5 до 20% белковых веществ. Особенно богаты
белком семена бобовых и масличных культур (до 20—35%).
Различно содержание белковых веществ и в пищевых продуктах.
Главным источником белков для человека являются мясо животных
(до 20%), рыба (до 18%), молоко (до 3%), сыр (до 22,5%),
пшеничный хлеб (до 8%), изделия из круп A0%).
Строение белков. Согласно общепринятой теории молекула белка
состоит из остатков а-аминокислот, связанных между собой
пептидными связями:
Н О Н Н О
i Н || I I Н ||
н/ \с/ \N/j\c/ \с/ N4/-
! i Ri II ! f
R HO R2 H
Впервые__мысльи-О- неитидных .связях__была_ высказана .выдающимся
русским биохимиком А. Я- Данилевским A838—1923). Состав и
последовательность расположения аминокислотных остатков в
молекуле белка могут быть различными, вследствие чего
разнообразие белков поистине безгранично. Например, для белков,
содержащих всего 100 аминокислотных остатков, число изомеров равно
20100.
Пептидные связи являются не единственными видами связей
в белках. Отдельные пептидные цепи или их участки могут быть
связаны между собой дисульфидными (—S—S—), солевыми и
водородными связями. Солевые связи образуются между свободными
аминогруппами (например, N-концевая аминогруппа,
расположенная на одном конце полипептндной цепи или е-амнногруппы ли-
* Ф. Энгельс. Анти-Дюринг. К. М арке и Ф. Энгельс. Соч.,
т. 20. М., Госпол!гги.;даг, 1901, ^ 82.
262
зина) и свободными кислотными
группами (С-концевая карбоксильная
группа полипептидной цепи,
свободные карбоксильные группы
двухосновных аминокислот). Водородные
связи в белках могут возникать между
атомами кислорода кето- или окси-
группы и атомом водорода окси- или
аминогруппы:
Н
/с=°
r/V:=o...h—n
Ъс/
с—н
н
н
...Н—О
,н
В молекуле белка существуют
также и некоторые другие виды
взаимодействия.
Если определение
аминокислотного состава белка может быть в
настоящее время проведено
относительно быстро, то выяснение
последовательности соединения
аминокислотных остатков — задача
исключительно сложная. Выдающимся
достижением в этой области было
установление структуры и синтез гормона
инсулина (Сенглер, 1963), состоящего
из остатков 51 аминокислоты и
имеющего молекулярную массу 5733
(рис. 22). Сочетанием из двух и трех
букв на рисунке условно
обозначаются остатки аминокислот. Черным
показаны дисульфндные связи.
Большое значение имело
установление структуры фермента рибонук-
леазы, молекула которого состоит
из 124 аминокислотных остатков и
содержит 4 дисульфидных мостика.
В настоящее время известны состав
и последовательность соединения
аминокислотных остатков пептидных
цепей 400 белков.
z м
Различают первичную, вторичную, третичную _и__четвертичную
структуры белковых молекул._Всё белки, независимо от того, к
какой группе они относятся и какие функции выполняют, построены
из относительно небольшого набора (обычно 20) аминокислот. Эти
структурные компоненты, или, как их иногда называют,
«строительные блоки», расположены в различной, но всегда строго
определенной для данного вида белка последовательности. Под первичной
структурой белка понимают вид, число и последовательность
^аминокислотных остатков в полипептидной цепи.
Рис. 23. Вторичная структура молекулы белка (схема
а-структуры полипептидиой цепочки)
Полнпептидные цепи вследствие взаимодействия отдельных ее
участков с помощью водородных связей имеют спиралевидную или
зигзагообразную конфигурацию, которая называется вторичной
структурой белковой молекулы (рис. 23). Полипептидная
спиралевидная цепочка может быть по-разному расположена в
пространстве. Эго пространственное расположение, или, как иногда говорят,
«упаковка», получило название третичной структуры (рис. 24).
Следует отметить, что «свертывание» полипептидной цепи с
образованием третичной структуры происходит не хаотично, а строго
определенно и любые изменения в третичной структуре влияют на
биологическую активность белков.
В последние годы сформулировано представление о так
называемой четвертичной структуре белковой молекулы. В отдельных
случаях молекулы состоят из нескольких полипептидных цепочек,
264
связанных между сооои водородными, ионными и некоторыми
другими нековалентными видами связей. В определенных условиях они
способны диссоциировать на более мелкие «субмолекулы», которые
могут опять соединяться в первоначальную молекулу. Такое
объединение нескольких частиц с третичной структурой получило
название четвертичной структуры. Следует отметить, что далеко не
у всех белков мы встречаем все четыре структуры.
Выделение и свойства белков. Белки экстрагируют из белок-
содержащего материала водой, растворами солей, щелочей, кислот,
водоспиртовыми растворами. Полученньш_таким образом продукт
обычно содержит значительное количество примесей. Для
дальнейшего выделения и очистки белка раствор обрабатывают солями
(высаливание), насыщают спиртом или ацетоном, нейтрализуют.
При этом выделяется
соответствующая фракция белка. Выделить белок
в неизмененном состоянии очень
трудно, для этого необходимо соблюдать
целый ряд условий: низкие
температуры, определенную реакцию среды
и т. д.
Выделенные и очищенные белки
имеют в большинстве случаев вид
белых порошков или сохраняют
природную форму (например, белки
шерсти и шелка). Установлено, что белкн
по форме молекул могут быть
разделены на две группы: фибриллярные
или нитевидные и глобулярные или
шаровидные. Представителем перзой
группы язляется фиброин шелка
(они обычно и сохраняют после выделения природную форму) ко
второй группе относится большинство белков живых организмов.
Многие белки растворимы в воде, щелочах, кислотах, но не
растворимы в органических растворителях. В растворах белки пред:
ставляют собой высокомолекулярные коллоиды и обладают многими
свойствами коллоидных растворов.
Так как белки содержат аминные и карбоксильные группы, то
они являются амфотерными электролитами: в щелочном растворе
проявляют кислотные свойства, в кислом — основные. Поэтому при
пропускании электрического тока через щелочной раствор белка
его молекулы будут двигаться к аноду, а при пропускании через
кислый раствор к катоду. Однако при определенной реакции среды
(так называемая нзоэлектрическая точка), количество
положительных н отрицательных зарядов в молекуле белка будет одинаковым.
Это одна из основных констант белка.
При действии на белок сильных кислот и щелочен, высоких
температур и некоторых других воздействиях они денатурируют.
Денатурация белка хорошо знакомый процесс, который можно,
например, наблюдать при варке куриного яйца. При денатурации
265
Рис. 24. Молекулярная модель
белка миоглобина
нарушается структура белка, изменяются его физико-химические
свойства. ~-— ¦-
Под действием" кислот, щелочей или протеолитических
ферментов белковые вещества распадаются с образованием в конечном
итоге смеси а-аминокнслот. Гидролиз протекает ступенчато, с
образованием все более и более простых продуктов:
белок -> альбумозы (пептоны) -» полипептиды —> дипептиды -> а-аминокислоты
Цветные реакции белков. Для белков характерны некоторые
цветные реакции, связанные с наличием в их молекуле
определенных группировок и аминокислотных остатков.
Бищктовая, реакция. -Появление фиолетового окрашивания при
обработке белка концентрированным раствором щелочи и
насыщенным раствором CuSO4. Связана с наличием в молекуле пептидных
связей.
Ксантопротеиновая реакция — появление желтой окраски в
результате действия на белки концентрированной азотной кислоты.
Реакция связана с наличием в белке ароматических колец.
Миллонова__ реакция— появление вишнево-красного
окрашивания при действии на белок Миллонова реактива (раствор нитрата
ртути в азотистой кислоте). Реакция объясняется присутствием
в белке фенольных группировок.
Сульфгидрильная реакция — выпадение черного осадка
сернистого свинца при нагревании белка с раствором плюмбита (связана
с наличием в белке сульфгндрпльных групп).
Реакция Адамкевича — появление фиолетового окрашивания
при добавлении к белку глиоксалевой и концентрированной серной
кислот. Связана с наличием индольных группировок.
Классификация белков. Белки подразделяются на протеины
(простые белки), состоящие только из остатков аминокислот, и
сложные белки (прртеидыO которые при гидролизе дают
аминокислоты и вещества небелковой природы (фосфорную кислоту,
углеводы, гетероциклические соединения, нуклеиновые кислоты).
Протеины составляют основу запасных веществ семян, а
протеиды — основу ядер протоплазмы клеток. Протеины и протеиды
разделяются на ряд подгрупп.
Up о теины. Альбумины — белки, имеющие сравнительно
небольшую молекулярную массу, хорошо растворяются.в воде. Из
водных растворов высаливаются насыщенным раствором сернокислого
аммония, при нагревании свертываются (денатурируют). Белок яйца —
типичный представитель альбуминов. Многие из них получены в
кристаллическом состоянии.
Глобулины — белки, не растворимые в чистой воде, но
растворимые в теплом 100о-ноы растворе NaCl. Чистый глобулин извлекают,
разбавляя солевой раствор большим количеством воды.
Глобулины — самые распространенные белки, входят в состав
мышечных волокон, крови, молока, яйца, растительных семян.
Проламины незначительно растворимы в воде. Растворяются
в 60—80 "о-ном водном этиловом спирте. При гидролизе пролампнов
266
образуется в большом количестве аминокислота — пролин.
Характерны для семян злаков.
Глкппелины — растворимы только в 0,2 % -ной щелочи. Найдены
в семенах пшеницы, риса, кукурузы.
Протамины — обнаружены только в молоках рыб. На 80%
состоят из щелочных аминокислот, поэтому являются сильными
основаниями. Совершенно не содержат серы.
Протеиноиды — нерастворимые белки, входящие в состав шелка,
волос, рогов, ногтей, копыт и сухожилий. Имеют нитевидную
(фибриллярную) форму молекул. Содержат серу.
Фосфопрогпеины — содержат фосфорную кислоту. Играют
большую роль в питании молодого организма. Примером их является
казеин — белок молока.
П р в т-вид-ы. Сложные белки делятся на группы в
зависимости от состава их небелковой части, которая называется просте-
тической группой.
Липопротеиды — гидролизуются на простой белок и липиды.
Липопротеиды содержатся в большом количестве в составе зерен
хлорофилла и протоплазмы клеток.
Глюкопротеиды — гидролизуются на простые белки и
высокомолекулярные углеводы. Не растворяются в воде, но растворяются
в разбавленных щелочах. Содержатся в различных слизистых
выделениях животных.
Хрояопротеиды — гидролизуются на простые белки и красящие
вещества. Например, гемоглобин крови распадается на белок
глобин и сложное азотистое основание, содержащее железо.
Нукмопротеиды — гидролизуются на простые белки, обычно
протамины, или гистоны, и нуклеиновые кислоты.
Использование белков. Исключительная роль белковых веществ
в живых организмах, ряд их ценных технических свойств
превратили белок в незаменимый продукт питания и важный вид сырья
для многих отраслей промышленности. Все возрастающую роль
играет производство биологически активных белковых препаратов:
ферментов, гормонов, антисывороток, кровезаменителей.
Белок является самой важной и в то же время наиболее
дефицитной частью пищи. Суточная потребность в белке взрослого
человека 100—150 г A0—1,5-Ю кг). Основное качество пищевого
белка — его способность снабжать организм необходимым
количеством аминокислот. "По аминокислотному' составу пищевые белки
делятся на две группы:полноценные (содержащие все необходимые
для организма животных и человека аминокислоты) и
неполноценные, в последних часть незаменимых аминокислот или полностью
отсутствует, или присутствует в незначительном количестве. Отсюда
следует, что при организации правильного питания нужно
учитывать не только количество, но и качество белка. Более того,
необходимое для человека количество белка во многом зависит от его
качества. Главными и наиболее ценными источниками белка
животного происхождения являются молоко и молочные продукты
(творог, сыр), яйца, рыба, мясо и получаемые из них продукты.
267
Источники белков растительного происхождения — зерновые
продукты (мука, крупа, макароны, бобовые). Овощи относительно
бедны белком, но их следует учитывать как дополнительный
источник белков; кроме того, они способствуют лучшей усвояемости
белков.
Несмотря иа обилие и разнообразие источников пищевого белка,
население ряда стран испытывает острый белковый голод. Дефицит
полноценного по аминокислотному составу белка в питании
населения земного шара равен примерно 15 мл. т A5-Ю9 кг). Одним
из путей устранения белкового дефицита являются поиски новых
их источников. В настоящее время проводятся исследования по
более эффективному использованию дешевого растительного белка,
применяемого для кормления скота; получению белков из
микробиологического сырья. Последний путь является особенно
перспективным.
Неограниченные сырьевые источники для производства
кормовых дрожжей (например, использования для их выращивания
парафинов нефти), их высокая продуктивность — за одни сутки 1 т
A03 кг) дрожжей может дать до 100 т A05 кг) потомства, высокое
содержание и качество белка в биомассе привлекают к этому методу
самое пристальное внимание исследователей.
Получение пищевого белка является одной иа основдых проблем
в создании пстсусственной~1Г;-еидае?ичесКой пищи. «Одна из
извечных п коренныХпроблём человечества — обеспечение пищей. Я
полагаю, — заявляет академик А. Н. Несмеянов, — что теперь химия
созрела до такой степени, что должна включиться в помощь
сельскому хозяйству в коренном вопросе — производстве самой дорогой
и дефицитной пищи — белковой, включиться непосредственно, а не
только на вспомогательных фронтах производства удобрений,
инсектицидов, фунгицидов, гербицидов, консервантов и т. д.» *.
Многое в этом направлении уже делается. В Директивах
XXIV съезда КПСС по пятилетнему плану развития народного
хозяйства СССР на 1971—1975 годы записано: «Увеличить выпуск
кормовых дрожжей в 3,5—3,7 раза. Развивать промышленное
производство белково-витамннных концентратов, значительно
расширить производство аминокислот...».
ПОНЯТИЕ О ФЕРМЕНТАХ И ВИТАМИНАХ
ФЕРМЕНТЫ
Ферментами или энзимами называются сложные биологические
катализаторы органическом природы, ускоряющие химические
реакции, протекающие в живом организме.
Слово «энзим» произошло от древнегреческого «эн зюме», что
значит в «дрожжах», а слово «фермент» является производным от
латинского слова iermentum — закваска.
* Вестник АН СССР, 1969, № 1, с. 27.
268
По образному выражению В. А. Кретовича, «ферменты
представляют собой движущую силу всего того бесконечного
разнообразия химических превращений, которые в своей совокупности
составляют лежащий в основе жизни биологический обмен веществ» *.
В то же время ферменты играют очень важную роль в различных
отраслях промышленности, и в первую очередь в пищевой, в
отдельных случаях осуществляя, а в других помогая осуществлять
многие технологические процессы. Достаточно напомнить, что
технологические процессы в таких отраслях пищевой промышленности,
как виноделие и пивоварение, хлебопечение, сыроделие,
осуществляются с непосредственным участием ферментов.
По своей химической природе ферменты — это белковые
вещества (протеиды). Молекула их может состоять или только из белка,
или из двух частей: белковой и небелковой. Последняя получила
название кофермента. Следует отметить, что одни и те же кофер-
менты могут входить в состав молекул разных ферментов.
По химическому строению они делятся на коферменты
алифатического, ароматического и гетероциклического рядов.
Известно около тысячи различных ферментов, часть их подробно
изучена. По современной классификации все ферменты делятся
на шесть классов.
Оксиредуктазы или окислительно-восстановительные ферменты.
Эта большая группа, состоящая из 180—190 ферментов.
Оксиредуктазы ускоряют окисление или восстановление различных
химических веществ. Таи, относящийся к этому классу фермент алкоголь-
дегидрогеназа катализирует окисление этилового спирта в
уксусный альдегид и играет большую роль в процессе спиртового
брожения.
Фермент липоксигеназа окисляет кислородом воздуха
ненасыщенные жирные кислоты. Действие этого фермента является одной
из причин прогоркания муки и крупы.
Трансферазы. Представители этой группы ферментов
катализируют перенос различных групп с одной молекулы на другую.
Например, фермент тирозин — аминогрансфераза-— катализирует
перенос аминогрупп. Ферменты этой группы играют большую роль
в медицине.
Гидролазы. Ферменты этой группы катализируют реакции
гидролиза. Представители этой группы ферментов имеют большое
значение в процессах пищеварения, в пищевой и других отраслях
промышленности. Так, фермент липаза катализирует гидролиз глице-
ридов с образованием свободных жирных кислот и глицерина.
Гидролиз пектиновых веществ протекает с участием пектолитнче-
ских ферментов. Их применение дает возможность повысить выход
и осветлить плодово-ягодные соки.
Представителем группы гидролаз являются амилазы,
катализирующие гидролиз крахмала. Они нашли широкое применение
в спиртовой, хлебопекарной, крахмало-паточной промышленностях.
* В. А. К р е т о в и ч. Введение в энзинологию. М., «Наука», 1974, 5.
269
К гидролазам относится большая группа протеолнтических
ферментов, катализирующих гидролиз белков и пептидов. Они
применяются в легкой и пищевой промышленности. С их помощью
проводят «мягченпе» мяса, кожи, получение сыров.
Лиазы. Катализируют реакции расщепления между атомами
углерода, углерода и кислорода, углерода и азота, углерода и
галогена. К ферментам этой группы относятся декарбоксилазы,
отщепляющие молекулу двуокиси углерода от органических кислот.
Изомеразы. Ферменты этой группы катализируют
разнообразные перегруппировки внутри молекулы органического соединения.
Лигазы. Катализируют образование связей С—О; С—S; С—N;
С—С. Характерной особенностью ферментов является их высокая
эффективность (они способны катализировать сложнейшие реакции
в очень мягких условиях) и строгая направленность. Действие
ферментов чрезвычайно сильно зависит от ряда факторов:
температуры (оптимальная темпеоатура 30—50° С), кислотности среды,
присутствия специфических веществ, носящих название
активаторов и ингибиторов. Первые повышают активность ферментов,
вторые снижают (угнетают ферменты), Сейчас налажено
промышленное производство ряда ферментных препаратов. Применение их
в различных отраслях промышленности дает большой
экономический эффект.
ВИТАМИНЫ
Витаминами называется разнообразная по своей химической
природе группа органических соединений, яеляющихся
необходимым составным компонентом пищи, нехватка их в организме
вызывает болезни недостаточности. Они выполняют в организме
каталитические функции. Основной источник витаминов — растения.
Отсутствие или недостаток в организме витаминов приводит к
глубокому расстройству и в конечном итоге даже к его гибели.
Таблица 28
Суточная потребность в витаминах
Витамин
Аскорбиновая кислота; С
Паптотеновая кислота; В3
Эргокальциферол; D2
Ретинол; А
К
а-Токоферол; Е
Никотинамид; РР
Потребность,
• 10~"d кг/сут
70-100
5—10
0,01 -0,04
1,5—2,0
2,0
2—6
15—25
Витамин
Пнридокспн; Вв
Тиамин; Bj
Биотин; Н
Фолиевая кислота
Рибофлавин; В2
Цианокобаламин; Вь,
Потребность,
• 10-« кг/сут
2.0—2.5
1,5—2,0
0,1—0,3
0,1—0,5
2,0—2,5
0,005—0,05
Витамины были открыты в 1880 г. русским ученым И. И.
Луниным. Свое название они получили по предложению польского
исследователя К. Функа, от слова Vita — по-латыни жизнь. Сейчас
известно свыше двадцати соединений, относящихся к витаминам.
270
По растворимости в воде они могут быть разделены на две группы:
водорастворимые (В2; В,; В6, РР, С и др.) и жирорастворимые
(А, Е, D, К). Потребность человека в витаминах зависит от его
возраста, состояния здоровья, характера деятельности, условий
жизни. Сведения о потребности взрослого человека в основных
витаминах приведены в табл. 28.
Кроме вшамннов существует группа соединений, представители
которой способны переходить в витамины. Они получили название
провитамины. К ним относятся некоторые каротиноиды (стр. 278),
переходящие в витамин А.
Водорастворимые витамины
Витамин С (аскорбиновая кислота). Имеет большое значение
для человека, являясь протнвоцынготным фактором.
Витамин С широко распространен в природе. Он содержится
в лимонах, апельсинах, черной смородине, свежей капусте, в
плодах шиповника, луке, картофеле, красном перце и пр. В СССР
производят витамин С в промышленном масштабе: синтетическим
путем аскорбиновую кислоту получают из глюкозы. Глюкозу
сначала восстанавливают до сорбита, которая путем ферментативного
окисления переходит в кегогексозу — L-сорбозу, затем окисляют
пгаохлоритом натрия в 2-кето-Ь-гулоновую кислоту, образующую
после лактонизацпи и енолизации аскорбиновую кислоту:
сн.он
НО-С-Н 1ШНО-С-Н
н—с—он
н—с—он
I
сн.рн
271
Витамин Bt (аневрин, тиамин)
NH3
-CH2-+N СН3
) ''\ II
\ Л
^S/ \СН2СН2ОН
Недостаток вызывает нарушение в работе нервной системы,
полиневрит («бери — бери»). Полиневрит распространен в странах Юго-
Восточной Азии. Природные источники тиамина — пшеничные и
рисовые отруби, дрожжи. Получается синтетическим путем.
Витамин Ва (рибофлавин)
Н СНа—(СНОНK—СН2ОН
СН
СН
з-С
з-С
С
!
н
\
/
с
N4
/у
с
!
[
с
N\
с=о
NH
;;
!(
0
Недостаток вызывает нарушение аппетита, падение веса, слабость.
В наибольшем количестве присутствует в дрожжах, печени, почках.
Получается синтетическим путем.
Витамин РР (никотинамид)
СН
НС C-CONH2
; j|
НС СН
\N/
Отсутствие витамина РР в пище вызывает пелагру. Наиболее богаты
этим витамином дрожжи, отруби, печень.
Жирорастворимые витамины
Витамины группы А
СНЗХ /СП3 СН3 СН,
ш:/^N:—сн=сн—с=с.н—сн=сн—с=сн—сн2он
Н2С С-СНз
\ /
сн2
Отсутствие витамина А вызывает нарушение роста, снижение
стойкости к заболеваниям, куриную слепоту. Образуется только в
тканях животного происхождения. Наибольшее количество содержится
в рыбьем жире, печени рыб п морских животных.
272
Витамины группы D
I
сня \/Ч/\_
сн,'
витамин D
Недостаток этих витаминов приводит к заболеванию рахитом.
Встречается только в животном организме.
ИЗОПРЕНОИДЫ
Изопренондами называются широко распространенные в
природе органические соединения, молекулы которых содержат
различное число изопреновых группировок (C5HS) и имеют полиизопрено-
бый скелет. Общая формула углеводородов этой группы (C5H8),.t.
К ним относятся такие важные соединения, как терпены, каротн-
нонды, природный каучук.
В группу изопренондов наряду с углеводородами (С5Н8)ге входят
их разнообразные кислородсодержащие производные: оксн-,
альдегид-, кето- и т. д.
Изопреноиды делят на группы по числу изопреновых остатков
(п --=-- 2, 3, 4 и т. д.) н строению углеродного скелета (ациклические,
моно-, ди- и полициклические):
Терпены
Сесквитерпены
Дитер пены
Каротпноиды
Политерпены
С10Н]е
Ci 5H14
с4ан64
(СвНв)а
(С5Н8K
(С5Н8L
(С5Н8)8
(С5Н8)П
Первые, три группы углеводородов и их многочисленные
кислородсодержащие производные объединяются под общим названием —¦
терпенонды.
ТЕРПЕНОИДЫ
Терпеноиды широко распространены в природе. Они входят
в состав эфирных масел, живицы, липидов животных тканей (сква-
лен).
Монотерпены и их кислородсодержащие
производные
Из многочисленных терпенов особое значение имеют
алифатические терпены (молекула их состоит из открытой
углерод-углеродной цепи и содержит три двойные связи, I), моноциклические
терпены (в их молекуле содержится кольцо из атомов углерода
и две двойные связи, II и III) и бициклические терпены (со-
273
держат в молекуле два кольца и двойную связь, IV). При
написании терпеноидов широко применяются условные формулы (III, IV):
СН3—С=СН--СН2— СН — С— СН—СН2
СН3 j СН2
СН,
I
Hn n—пхт
II IV
Эфирные масла. Алифатические и моноциклические терпены
в значительном количестве присутствуют в эфирных маслах
(анисовом, розовом, гвоздичном, тминном и др.). Эфирными маслами
называют жидкие, ароматические, легко летучие смеси
органических веществ, вырабатываемые растениями. Они включают до
1000 компонентов. Эфирные масла хорошо растворимы в
органических растворителях и не растворимы в воде. На воздухе под
действием света и кислорода они осмоляются, изменяют свой цвет и
запах. В составе эфирных масел хвойных и цитрусовых растений
в большом количестве присутствуют тер леновые углеводороды,
в других — спирты и оксосоедннения. Выделяют эфирные масла
из зфнромасличных растений отгонкой с водяным паром или
экстракцией органическими растворителями. Они широко применяются
в парфюмерной, косметической, медицинской и пищевой промыш-
ленностях, а также являются сырьем для получения отдельных
органических соединений.
В СССР возделывается около 20 эфироносов, особое значение
имеют кориандр, анис, тмин, перечная мята, лаванда, герань, роза.
Лимонное масло. Получают прессованием из корок лимона.
Из 1000 штук лимонов получают до 500 г (до 5-10~х кг) масла. Его
главный компонент «,-лимонен.
Мятное масло. Получают из мяты. Основной компонент ментол,
Анисовое масло. Получают из растений семейства зонтиковых.
Основной компонент — эфир анетол:
ОСНЯ
СН=СН
аи ею л
274
Кориандровое масло содержится в семенах кориандра.
Основной компонент — линалоол.
Пихтовое масло. Получают из хвои и веток сибирской пихты.
Основные компоненты: борнеолацетат и камфен.
Алифатические терпены и их кислородсодержащие производные.
По своим свойствам это жидкости легче воды, не растворимые в ней
и хорошо растворимые во многих органических растворителях.
Обладают приятным запахом. Их химическое поведение
определяется наличием в молекуле соответствующих функциональных
групп. Из них наибольшее значение имеют следующие
соединения.
Мирцен СН3—С=СН—СН.2—СН2—С—СН=СН2. Содержится в масле
СН3 СН.2
хмеля.
Гераниол СН3—С=СН—СН.2—СН2—С=СН—СН2ОН . Является одним из
СН3 СН3
основных компонентов розового масла.
¦О
Цитраль СН3—С=СН—СН,—СН,—С=СН—Gf . Одно из важней-
I \Н
сн3 сн3
ших соединений в группе алифатических терпенов. Распространен
в эфирных маслах.
ОН
Линалоол СН3—С=СН—СН2—СНа—С—СН=СН2. Содержится в ко-
СН3 СН3
риандовом и померанцевом маслах. Обладает запахом
ландыша.
Моноциклические терпены и их кислородсодержащие
производные. Большинство моноциклических терпенов можно рассматривать
как производные ментана, содержащие в молекуле две двойные
связи — ментадиены:
С= 4 3G
XT
с—с—с
10 8 9
мешан
A-метил-4-изопропилциклогексан)
275
Особое значение имеют четыре представителя этой группы
соединений: лимонен, ос-терпинеол, ментол и ментан:
СНз СН3 СНз
А /\ А
\J ч/ \/v
он
СНз—С—СНз
он
а-тергшнеол
сн3—с—а
н
ментол
Лимонен содержит один асимметрический атом углерода и
может существовать в двух оптически деятельных формах (D и L)
и оптически недеятельной рацемической форме. Находится в масле
цитрусовых, укропа, тмина.
Ментол принадлежит к числу важнейших кислородсодержащих
производных моноциклических терпенов. Кристаллическое
вещество с приятным запахом. Основной компонент перечной мяты.
Синтетическим путем получается гидрированием тимола:
СИ» СН3
ЗНг / N
'\ \А
ОН i ОН
- СНз-СН-СНз СН3-СН-СНз
тимол ментол
Применяется в медицине как дезинфицирующее средство и в
качестве компонента зубной пасты.
а-Терпинеол — жидкость с запахом сирени. Используется в
парфюмерии.
Бициклические терпены и их кислородсодержащие производные.
Среди бициклических производных особое значение имеют
производные трех групп углеводородов: карана, пинана, камфана:
каран
камфан
Наиболее распространенным из этой группы соединений является
а-пинен, главная составная часть скипидара:
276
а-Пинен — жидкость с /К1Ш = 156° С и запахом хвои.
Получается из скипидара. Применяется для получения камфоры,
инсектицидов, в качестве растворителя.
Скипидар извлекают пз живицы, которая вытекает из надрезов
на стволах "сосны. Живица — раствор канифоли (смесь смоляных
кислот с общей формулой С19Н2вСООН) в скипидаре (терпентиновое
масло), который состоит из смеси различных терпенов. Скипидар
получают из живицы отгонкой с водяным паром. Это бесцветная
жидкость, легче воды, смешивается с большинством органических
растворителей. Скипидар применяется в текстильной
промышленности, лакокрасочной для приготовления лаков, в парфюмерии,
для синтеза камфоры.
=0
Камфора—прозрачная,бесцветная, вязкая кристаллическая
масса, /пл = 175°С. Получается из камфорного дерева (Япония, Тайвань),
отгонкой из измельченной древесины с водяным паром или
синтетическим путем. Синтез камфоры протекает следующим образом:
сн,
TiO,
-CHjCQOH
бьрвеол
камфора
Камфора применяется в качестве пластификатора при получении
целлулоида, в медицине как стимулятор сердечной деятельности.
Дитерпены и их производные
Из представителей этой группы изопреноидов важным является
фитол — непредельный спирт, составной компонент хлорофилла и
витамина К
CHs-CH-(CH2K-CH-<CH2)s-CH-(CH2)a-C=CH-CH2OH
СН3 СН3 СН,
СН3
фит ел
277
КАРОТИНОИДЫ
К этой группе изопреноидов относятся углеводороды (С5Н8)8
и их кислородсодержащие производные. Каротиноиды — красящие
вещества от желтого до ярко-красного цвета. Они широко
распространены в природе; вместе с хлорофиллом содержатся не только
в зеленых частях растений, но и в семенах. В организм животных
они попадают с пищей, Природные каротиноиды можно
рассматривать как производные ликопина, содержащегося в плодах томата,
шиповника, ягодах, фруктах:
При замыкании одного или двух колец на концах молекулы
ликопина образуются ее-, Р- и у-каротины, отличающиеся
количеством и строением боковых колец:
Н3С СН3
\/
н,с сн
g-каротин
Известно 100 каротиноидов. Их г^ожно разделить на
углеводороды, называемые каротинами, и их кислородсодержащие
производные (спирты, альдегиды, эпоксиды, кислоты), часть которых
объединяется под общим названием «ксантофиллы».
Каротиноиды хорошо растворяются в большинстве
органических растворителей и не растворяются в воде. Некоторые
каротиноиды являются провитаминами А (стр. 272). Из одной молекулы
р-каротнна в животном организме образуется две молекулы
витамина А.
КАУЧУК
К высокомолекулярным изопреноидам относится натуральный
каучук, являющийся одним из наиболее важных химических
продуктов. Каучук представляет собой высокоэластичную массу,
добываемую из сока растений (каучуконосов). В промышленном
масштабе каучук получают из сока дерева гевеи, произрастающего
в тропических странах: Южная Америка, Индонезия, Малайя,
Цейлон. Для извлечения каучука млечный сок (латекс), полученный
подсечкой деревьев, коагулируют, добавляя органические кислоты,
прокатывают в листы и сушат. Состав полученного продукта (С5Н8)Я.
278
После обработки сырого каучука серой получают эластичную
резину. Каучук представляет собой полимер со среднЕ'йГ"'Кголе1{уляр-
но'й массой 300.000—400 000, содержащий остаткГЪзопреНа',""сое-
диненные в цис-форме:
Н3Сч /Н Н3СЧ /Н Н3Сч Н
\с=с( \с=сг >с=с<
В связи с бурным ростом потребности промышленности в
каучуке большая часть его получается синтетическим путем.
Впервые производство синтетического каучука было
осуществлено в СССР по методу С. В. Лебедева. Для получения
синтетического каучука в качестве мономеров используют 1,3-бутадиен,
изопрен, хлоропрен, стирол, кремнийорганические соединения.
Используя разные мономеры, меняя условия полимеризации, получают
синтетический каучук, не уступающий, а по некоторым свойствам
превосходящий природный.
ЛИТЕРАТУРА
Потапов В. М., Татаринчик С. Н. Органическая химия. М.,
«Химия», 1973.
Степа н енко Б. Н. Курс органической химии. М., «Высшая школа»,
1974.
Писарей ко Л. П., Хавин 3. Я. Курс органической химии. М.,
«Высшая школа», 1968.
Павлов Б. А., Т е р е и т ь е в А. П. Курс органической химии. М.,
«Химия», 1972.
предметный указатель
Агликоны 244
Адамкевича реакция 266
Адипиновая кислота 139, 141
Азокраснтели 178
Азолы 195
Азосоединения 178
Азосочетания реакция 178
Акриловая кислота 137
Акрилонитрил 51, 189
Акролеин 128
к-Аланин 180
Алкалицеллюлоза 258
Алкалоиды 195
Алкилсульфиды 165
Алкины 48
Алкоголи — см. Спирты
Алкоголиз 202 ч
Алкоголяты 96
Алкоксигруппа 113
Аллиловый спирт 104
Альдегидокислоты 158
Альдегиды 115
— номенклатура 116
— получение 116
— представители 126
— свойства 119
Альдогексозы 221
Альдозы 221
Альдоксим 122
Альдольная конденсация 124, 230
Амигдалин 245
Амиды 187
— получение 187
— представители 188
— свойства 187
Амиловые спирты 103
Амилоза 257
Амплопектин 250
о-Аминобензойная кислота 181
л-Аминобензойная кислота 181
Аминогруппа 172
— С-концевая 263
— Л'-концевая 262
Аминокислоты 179
— изомерия 179
— получение 182
— представители 180
— свойства 183
Амины 172
— номенклатура 172
— получение 172
— представители 115
— свойства 174
Ангидриды кислот 145
Анетол 274
Анизол 114
Анилин 176
Аномеры 226
Антиподы 152
Антрахинон 78
Антраниловая кислота 181
Антрацен 70, 78
L-Арабиноза 243
Арабнт 243
Арахидоновая кислота 201
Арахиновая кислота 201
Ар ил 69
Асимметрический углеродный атом 221
Аспарагнновая кислота 180
Аспирин 158
Ауксохромы 178
Ацетали 121
Аиетальдегид — см. Уксусный
альдегид
Ацетамид 187
Ацетил 143
Ацетилен 53, 54
— электронное строение связи 48
Ацетиленид серебра 52
Ацетиленистая медь 52
Ацетилцеллюлоза 259
Ацетон 127
Ацетоннтрил 188
Ацетоуксусный эфир. 160
Ацетофенон 127
Ацидолиз 203
Ацил 143
Бегеновая кислота 201
Белки 261
— классификация 266
— строение 262
Бензальдегид 127, 245
Бензамид 187
Бензил 69
Бензпловый спирт 104
Бензин 79
Бензойная кислота 134
Бензол 66
— свойства 71
— строение. 67
Бензохнноп 112
2S0
Бетаины 184
Бнуретовая реакция 266
Болотный газ 33
Бора модель водородного атома 11
Борнеол 277
— ацетат 277
Брожение 237
— лимоннокислое 237
— маслянокпслое 237
— молочнокислое 237
— спиртовое 237
Бутадиен — 1,3 54, 58, 60, 279
Бутан 23
Вутандиовая кислота — см. Янтарная
кислота
Бутил 24
Бутилен 48
Бутиловые спитры 103
Бутин 48
Бутлерова теория строения 9
Вагнера реакция 43
Валериановая кислота 129
Валин 127, 180
Ванилнн 127
Вёлера реакция 49
Винилацетилен 52
Виниловый - спирт 104
Винная кислота 153, 156
Винный спирт — см. Этиловый спирт
Вискоза 259
Витамины 270
— водорастворимые 271
— жирорастворимые 272
— В:2 112
— РР 196
Водородная связь 19
Водородный атом, модель 14
Воски 198, 205
Высыхание жиров 216
Вюрца реакция 26
Вюрца — Фитпшга реакция 71
Газ природный 26
Галактоза 240
Галактоновая кислота 241
Галловая кислота 158
Галогеиаигидриды 143
Галогеиопроизводные 85
— получение 83
— представители 89
— свойства 86
Галактопнраноза 240
Гаттермана — Коха реакция 119
Ганглиозид 241
Гексаметилендиамин 175
Гексан 23
Гексахлоран 74
Гексахлорциклогексан 74
Гексозы 238
Геыицеллюяоза 257
Гемиглобин 194
Гептан 23
Гептоза 231
Гераниол 275
Гетероатомы 190
Гетероциклические соединения 190
Гетероцпклы 190
— пятнчленные 191
— — с двумя гетер оатомамн 195
с одним гетероатомоы 191
¦— шестнчленные 195
— — с двумя гетероатомамм 195
с одним гетероатомом 195
Гидразон 248
Гидрогенолиз 64
Гидропереэтерификация 216
Гидрохинон 108
Гликоген 256
Гликозидный гидроксил 227
Гликозиды 225, 244
Гликокол 180
Гликолипиды 198
Гликолн 104
Глиоксалевая кислота 159
Глицерин 106
— трннитрат 107
Глицериновые альдегиды 222
Глицериды 198, 200
Глутаровая кислота 139
Глутаминовая кислота 180
Глюкоза 220, 238
Глюконовая кислота. 239
Глюкопираноза 238
Глюкопиранознды 238
Глюкофураноза 229
Глюкофуранозиды 229
Гомологи 22
Топологическая разность 22
Гомологический ряд 22
Гофмана реакция 173
Гриньяра реакция 163
Двойная связь 17
Декстрины 253
Диазогндрат 177
Диазоний хлористый 177
Диазония соли 177
Диазосоединеиия 176
Дназотировання реакция 177
Диастреоизомеры 154, 224
Диацетилцеллюлоза 259
Дивинил—см. Бутаднен-1,3
Диены 54
Дикетопнпёразин 184
Дильса — Альдера синтез 60
Диметилаыиы 174
п-Диметиламнноазобензол 187
Диметиловын эфир 113
Диметилсульфат 147
Диоксан 114,' 192
Диолы 104
Дпсахарпды 250, 257
Днфеаил 75
Дифторднхл op метан 90
Дихлорэтан 90
Дпэтнловый эфир 113
Дпэтнлсульфид 165
Дульцит 241
Еидиол 234
Енольная форма 160
Женевская номенклатура — см.
Номенклатура ИГОПАК
Жирные кислоты высокомолекулярные
199
Жиры 214
— гидрогенизация 215
Заместители второго рода 73
— первого рода 73
Зелинского, Казанского реакция 51
Зимина реакция 172
Изобутилены 48
Изолейцин 180
Изомерия 10
— геометрическая 36
— динамическая 170
— конформационная 229
— оптическая 150
— поворотная 150, 154
— пространственная 150
Изомеры 10
Изопрен 54, 279
Изопреноиды 273
Имидазол 195
Инверсия 250
Инвертный сахар 250
Ингибиторы 205
Индиго 195 .
Индол 195
Индукционный эффект 41
Инозит 62, 207
Инулин 242
Инсулин 263
Йодистый метил 86
Ицрит 165
Камеди 260
Каменноугольная смола 60
— фракции 70
Каыфан 27G
Камфара 277
Кзмфен 275
Канниццаро реакция 123
е-Капролактам 176
Капрон 176
Капроновая кислота 129
Карбамид 188
Каран 170
Карбид кальция 49
Карбонил, электронное строение 131
Карбоповые кислоты — см. Кислоты
Карбоцнклические соединения 60
Каротиноиды 198, 273, 278
Катсхин 1159
Каучук 278
Квантовые числа 12
Керосин 80
Кетены 127
Кото-инольная таутомерия 160
Кетозы 221
Кетокислоты 158
Кетоксимы 122
Кетоны 115
— получение 116
— свойства 119
Кефалины 209
Кислоты
— алифатические двухосновные 138
— — одноосновные непредельные 135
— предельные 129
— фосфатпдиые 208
Клетчатка 256
Коксование 69
Коллодий 259
Коллоксилин 123
Кольбе, метод получения алканов 27
Коновалова реакция 30
Конформации 64, 115
Крахмал 250
Крезолы 108, 112
Крекинг-процесс 32, 79
Кротоновая кислота 135
Ксантогенаты клетчатки 259
Кеантопротеиновая реакция 266
Ксантофнлы 278
Ксилит* 108, 244.
D-Ксилоза 243
Ксилолы 67
Кумол 73
Купрен 52
Кучерова реакция 51
Лавсан 142
Лактамы 185
Лактиды 150
Лактоза 241, 248
Лактоны 150
Ланолин 206
Лаурнновая кислота 129, 201
Лебедева реакция 55
Лейцин 180
Лецитины 209
Лигнин 256
Лизин 180
Лнкопин 278
Лимонен 176
Лимонная кяслота 157
Лкпалоол 275
Лннолеоая кислота 138, 201
Линолеповая кислота 201
282
Липазы 201
Липиды 198
¦— методы исследования 211
— простые 200
¦— сложные 207
Мазут 80
Малеиновая кислота 139, 141
Малоновая кислота 139, 141
Мальтоза 247
Манноза 241
Мапнит 241
Маргарин 216
Марковникова правило 40, 51
Масляноэтиловый эфир 147
Мезовинная кислота 153
Мелиссиновая кислота 201
Ментан 275
.кета-Изомеры 69
Метакриловая кислота 137
Металлорганические соединения 161
Метан 33
Метаналь — см. Формальдегид
Метанол — см. Метиловый спирт
Метил 24
Метиламин 174
Метилбензол — см. Толуол
2-Метилбутадиен-1,3 — см. Изопрен
Метиловый спирт 99
Метилоранж 178
Метилфенилкетон 119
Метилэтилаиетилен 48
Метилэгнловый спирт 113
Метпонин 80
Меркаптаны 165
Меркаптиды 165
Мерсеризация 258
Миллонова реакция 266
Мирициловая кислота 201
Мирцен 275
Молочная кислота 155
Молочный сахар — см. Лактоза
Моноамины 175
Моносахариды 220
Монтаповая кислота 201
Морфин 197
Мочевина 188
Муравьиная кислота 132
Мутаротация 224
Мыла 217
— моющее действие 217
Мясомолочная кислота 155
Найлон 176
Нафталин 76
— изомерия производных
Нафтены 62
Нафтол ы 108
Нефть 26, 79
— ароматизация 70
— прямая гонка 79
Никотин 197
Нитрилы 188
— целлюлозы 258
Нитробензол 171
Нитроглицерин 107
Ннтрозоамнны 175
Нптросоединения 168
Нитрофенол 11 i
Номенклатура ИЮПАК 25
— рациональная 24
Нуклеиновые кислоты 219
Озазопы 248
— глюкозы 240
Озокерит 26
Окись этилена 114
Оксигидрохинон 108
Оксикислоты 147
— изомерия 147
— получение 148
— представители 155
Р-Оксимасляная кислота 148
Окснмы 122
2-Оксипропановая кислота 148
Оксокислоты 158
2-Оксопропановая кислота — см. Пи-
ровиноградная кислота
Оксосинтез 118
Октановое число 81
Олеиновая кислота 138, 201
Олефины 33
— изомерия 35
— номенклатура 35
— получение 36
— представители 45
— свойства 38
— строение 35
Олигосахариды 247
Олифа 216
Оптическая изомерия 150
Оптические антиподы 152
Органическая химия
— — значение 7
исторический очерк 6
определение 4
причины выделения 4
Органические вещества, источники 8
— — классификация 20
ог'Л'ш-Изомеры 69
Пальмитиновая кислота 129, 201
«ара-Изомеры 69
Парафины — см. Углеводороды
предельные
Пектиновые вещества 260
— кислоты 201
Пентан 23
Пектозаны 242
Пентозы 242
Пептидные связи 185, 262
Пептиды 185
283
Перекиси органические 115
Переэтерификация 146
Пинан 275
а-Пи не н 276
Пиразол 195
Пиран 195
Пиранозы 227
Пиридин 190, 196
Ппровиноградная кислота 159
Пирогаллол 108
Пирокатехин 108
Пироксилин 259
Пиролиз нефти 80
Пирослизевая кислота 193
Пиррол 190
Поверхностно-активные вещества 217
анионоактивные 217
иеионогенные 219
Полиамиды 175
Поливинилацетат 104
Поливиниловый спирт 104
Поливинилхлорид 90
Полимеризация
— ионная 44
— радикальная 45
— цепная 43
Полиметилметакрилат 137
Полиметпленовые углеводороды — см.
Циклоалкаиы
Полистирол 75
Полисахариды 241
Полиэтилен 44
Полуацетали 121
Поляриметр 152
Порфин 194
Принцип Паули 13
Пролин 181
Пропан 23
Пропен 47
Пропил 24
Пропиловые спирты 103
Пропин 48
Пропионовая кислота 129
Радикалы свободные 45
Рацематы 152
— разделение на антиподы 154
Резорцин 108
?>-Рибоза 243
Рибонуклеиновые кислоты 213
Рибофураноза 243
Рпформинг нефти 8]
Рицпнолевая кислота 157, 201
Родионова метод 182
Рудничный газ 33
Спнтсз-газ 26
Слизи 260
Соляровые смела 80
Сорбиновая кислота 137
Сорбит 108, 242
Спирт этиловый 99
284
Спирты
— многоатомные 104
— одноатомные 92
— вторичные 92
— изомерия 92
— номенклатура 92
— первичные 92
— получение 93
— третичные 92
— химические свойства 95
— строение 92
Стеариновая кислота 129, 201
Стекло органическое 137
Стереоизомерия 150
Стерины 198
Стеролы 279
Сульфгидрильная реакция 266
Сульфогруппа 166
Сульфокислоты 166
Сфингозин 207
Танин 158
Таутомерия — см. Изомерия
динамическая
Теория строения Бутлерова 9
Терефталевая кислота 142
ТерилеР! — си. Лавсан
Терпены 273
—"алифатические 274
Терпинеол 276
Тетрафторэтилен 91
Тимол 276
Тполы — см. ТиоспиртЫ
Тиоспирты 164
Тиофен 191
Тиоэфиры 164
Тирозин 180
Тищенко реакция 124
Толуол 67, 74
Треонин 180
Триглицериды 200
Триозы 221
Триолы 104
Триптофан 181
Трисахариды 245
Трихлоруксусная кислота 132
Тройная связь 18
Тростниковый сахар — см. Сахароза
Тротил 171
Углеводороды 22, 219
— ароматические 65
общая формула 66
— — получение 69
— — свойства 71
— ацетиленовые 48
— — номенклатура 48
— — получение 49
— — свойства 50
— длсиовые 54
— непредельные 33
— полиметиленовые 60
— предельные 22
— — изомерия 23
— — номенклатура 24
— — общая формула 22
— — отдельные представители 33
— — природные источники 26
— — свойства 28
— — синтезы 26
— этиленовые — см. Олефины
Уксусная кислота 133
Уксусноизоамиловый эфир 146
Уксусноэтиловый эфир — см. Этил-
ацетат
Уксусный альдегид 126, 145
Уксусный ангидрид 115
Уроновые кислоты 232
Фелингов раствор 123, 233
Фенантрен 70, 78
Фенетол 113
Фенил 69
Феннлаланнн 180
Феннлгидразин 122
Фенол 108
Фенолы 108
— получение 109
— представители 112
— свойства 110
Феноло-форяальдешдные смолы 125
Феноляты 110
Ферменты 268
Фитол 277
Фишера формулы 151
Флороглюцин 108
Формальдегид 126
Формил 143
Фостен 144
Фосфолипиды 198, 207
Фотосинтез !94
Фреон-12 90
Фриделя — Крафтса реакция 71
Фруктоза 221, 242
Фталевая кислота 139, 142
Фталевый ангидрид !42
Фунаровая кислота 139, 141
Функциональные группы 82
Фуран 190
Фуранозы 227 .
Фуранозы 227, 191, 193
Хеуорзса формулы 227
Химическая связь
— — двойная 17
— — диеновые 54
— — конъюгированные 54
— — кумулированные 54
— — сопряженные 54
— — ковалентная 15
координационная 18
л- 17
полярность 19
— — семиполярная 18
а- 16
— — тройная 17
электровалентная 15
— — энергия 19
Хлораль 103
Хлорбензол 90
Хлорвинил 82
Хлористый аллил 90
Хлористый ацетил 144
Хлористый бензнл 72
Хлористый бензоил 144
Хлоропрен 279
Хлорофилл 194
Хлороформ 82, 90, 193
Холин 207
Хромофоры 178
Целлобиоза 248
Целлюлоза — см. Клетчатка
Целлулоид 259, 277
Цереброзид 210
Церезин 26
Цпклоалканы 60
Цнклобутан 62
Циклогексан 61
Цикло-оксо-таугомерия 229
Циклопентан 62
Циклопропан 62
Цимол 75
Цис-транс-Изоыерпя — см.
Геометрическая изомерия
Цистеин 180
Цитр ал ь 275
Чалмугровая кислота 201
Четыреххлористын углерод 90
Число йодное 212
— кислотное 211
— омыление 211
Электроны
— л- 12
— s- 12
— гибридизация 14
Электронное облако 12
Электронное строение связи 17
Элементорганические соединения 161
кремнийорганические 164
серусодержащие 164
Энергия образования связи 19
Эпоксиды 144
Эруковая кислота 201
Этаналь — см. Уксусный альдегид
Этандиовая кислота,— сы. Щавелевая
кислота
Этандиол — см. Этиленгликоль
Этанол — сы, Этиловый спирт
Этанол амин 207
Этен — см. Этилен
285
Этерификация 77
Этил 23
Этилацетат 146
Этилен 4G
Этиленгликоль 106
Этиловый спирт 99
Этилформиат 146
Этин — сы. Ацетилен
Этоксиэтан — см. Диэтиловый
Эфиры простые 113
представители 113
— — свойства 113
— — способы получения 113
— — сложные 145
номенклатура 145
— фосфоглицериновые 208
— фосфосфингозиновые 210
эфир Яблочная кислота 155
Янтарная кислота 139
СОДЕРЖАНИЕ
Стр.
Предисловие 3
Введение 4
Часть первая
Углеводороды
Алифатические углеводороды 22
Алканы 22
Алкены 33
Алкины 48
Алкаднены 54
Алициклические углеводороды 60
Циклоалканы 60
Ароматические углеводороды 6о
Одноядерные ароматические углеводороды 66
Многоядерные ароматические соединения 75
Многоядерные ароматические углеводороды с некопденсированньши
(изолированными) ядрами 75
Многоядерные ароматические углеводороды с конденсированными ядрами 76
Высшие полициклические углеводороды 78
Нефть и ее переработка 79
Частьвторая
Функциональные производные углеводородов
Галогенопроизводные углеводородов 82
Кислородсодержащие производные углеводородов 91
Гидрокснлсодержащие производные углеводородов (спирты, фенолы
и нафтолы) 91
Одноатомные спирты 92
Многоатомные спирты 104
Фенолы, нафтолы 108
Простые эфиры 113
Эпоксиды 114
Органические перекисные соединения 115
Оксосоединения (альдегиды н кетоны) 115
Предельные альдегиды и кетоны 116
Ненасыщенные карбонильные соединения. Кетоны 127
Органические или карбоиовые кислоты ." 129
Одноосновные насыщенные и ароматические карбоновые кислоты 129
Одноосновные насыщенные кислоты 135
Двухосновные кислоты ., . . - 138
Производные карбоновых кислот 143
Галогеноангидриды карбоновых кислот 143
Ангидриды карбоновых кислот 145
Сложные эфиры карбоновых кислот 145
Окснкислоты 147
Оксокислоты (альдегндо- и кетокислоты) . 158
Часть третья
Элементорганические соединения
Металлсрганические соединения 161
Кремпийсфганические соединения 164
Серусодержащие органические соединения 164
287
Часть четвертая
Азотсодержащие органические соединения
Стр.
Нитросоедннения ,...,, 168
Амины 172
Понятие об азо- и дназосоединениях 176
Диаэосоединения 176
Азосоединения . , 178
Аминокислоты 179
Амиды кислот 187
Нитрилы 188
Часть пятая
Гетероциклические соединения
Пятичленные гетероциклы 191
Пятичленные гетероциклические соединения с одним гетероатомоы 191
Пятичленные гетероциклы с двумя гетероатомамн 195
Шестичленные гетероциклы 195
Шестичленные гетероциклы с одним гетероатомоы , , , , 195
Понятие об алкалоидах ¦ 197
Часть шестая
Биоорганические соединения и некоторые соединения изопренондного
характера
Липиды 196
Простые липиды 200
Глицериды 200
Воски 205
Сложные липиды 207
Фосфолипиды 207
Методы исследования липидов 211
Получение и переработка жиров и масел 214
Поверхностно-активные и моющие вещества 217
Углеводы 219
Моносахариды 220
Полисахариды 245
Олигосахариды (сахароподобные полисахариды) 245
Дисахариды 245
Высокомолекулярные полисахариды 251
Крахмал 252
Гликоген 256
Клетчатка или целлюлоза 256
Пектиновые вещества, растительные камеди и слизи ......... 260
Белковые вещества 261
Понятие о ферментах и витаминах . 268
Ферменты 268
Витамины , 270
Водорастворимые витамины 271
Жирорастворимые витамины 272
Изопреноиды . 273
Терпеноиды 273
Монотерпены и их кислородсодержащие производные 273
Дитерпены и их производные 277
Каротиноиды 278
Каучук 278
Литература 279