/



























































































































































































































































































































Текст
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ В ВЕТЕРИНАРИИ
ПРЧВОЧНИК
ХИМИКО-
ТОКСИКОЛОГИЧЕСКИЕ
МЕТОДЫ
Под редакцией Б. И. АНТОНОВА
Москва ВО -Агропромиздат- 1989
ББК 48
Л12
УДК 619: 616—074(031)
Составители: Б. И. Антонов, В. И. Федотова, Н. А. Сухая
Редактор В. Н. Сайтаниди
Лабораторные исследования в ветеринарии: хи-Л12 мико-токсикологические методы: Справочник/Под ред. Б. И. Антонова; Сост. Б. И. Антонов и др.— М.: Агропромиздат, 1989.— 320 с.: ил.
ISBN 5—10—001420—2
Даны апробированные и рекомендованные к применению в ветеринарных лабораториях методы химико-токсикологических исследований кормов, продуктов животного и растительного происхождения, а также воды, почвы, воздуха. Приведены методы лабораторной диагностики при отравлениях животных.
Для ветврачей и фельдшеров, лаборантов ветеринарных лабораторий.
Л
3706000000—095
035(01)—89
99—89
ББК 48
ПРЕДИСЛОВИЕ
Средства химизации широко применяются во всех отраслях । ельскохозяйственного производства и лесного хозяйства. Их ис-иолыуюг для защиты растений и животных от вредителей и бо-|с шей в качестве регуляторов и стимуляторов роста дефолиантов и.। нолях, в лесах и т. д. Выпуск химических средств непрерывно n.ipacracT не только в количественном отношении, но и по ассор-iiiMcury. Предприятия, выпускающие эти сильнодействующие ве-iiieeina, не всегда придерживаются требований закона и допуска-к>| за, рязиепие окружающей среды отходами производства. Правила храпения, использования, транспортировки химических средств ||о|ре('япелями нередко нарушаются. В результате всего этого огромные массы их вовлекаются в природный оборот: почва — растение животов. Отсюда и отравления скота и птицы.
Объем химико-токсикологических исследований в ветеринарных габораториях страны ежегодно возрастает. Ветеринарные лабора- ории осуществляют диагностику отравлений животных, при этом in следуют патологический материал и корма, проводят исследования, направленные на предупреждение отравлений, а также опре-пеляюг остатки пестицидов в кормах с целью предупреждения попадания их в организм животных и загрязнения продуктов жижи иоводства.
I 1срсчень используемых в сельском хозяйстве пестицидов еже-10ДПО изменяется, пополняется новыми ядохимикатами Разрабатываются и утверждаются новые методы их анализа. Многие ранее действовавшие методические указания заменяются новыми, позволяющими проводить лабораторные исследования более качественно и на современном уровне знаний, предлагаются и внедряются и лабораторную практику новые методы диагностики отравлений.
В настоящем справочнике представлены методические указания по исследованию почвы, воды, кормов, продуктов животного происхождения, тканей и органов животных. Из числа наиболее точных и широко применяемых методов для определения ядовитых веществ даны тонкослойная хроматография и колориметрия.
Химические средства представлены в виде групп: хлорорганиче-ские, фосфорорганические, ртутьсодержащие соединения, карбаматные пестициды и пестициды других групп, растительные яды и дру-1 не ядовитые вещества.
Методические указания по проведению исследований даны в единой последовательности: физико-химические свойства пестицидов, основные положения (что и в каком материале определяется), принцип метода, реактивы и растворы, приборы и посуда, подготовка к определению, ход анализа. Затем описываются последовательности выполнения анализа исследований и обработки рез'ль-
татов. Методические указания снабжены соответствующими формулами и таблицами.
Ветеринарные лаборатории в своей работе не могут использовать всего многообразия имеющихся в литературе методов исследований из-за того, что оии недостаточно апробированы, или из-за сложности применяемого оборудования. Иногда методы, предлагаемые различными авторами, при определении одних и тех же показателей дают несовпадающие результаты.
В связи с этим в справочник включены методы лабораторных исследований, апробированные, одобренные и утвержденные Главным управлением ветеринарии Госагропрома СССР и Министерством здравоохранения СССР.
ХЛОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Методические указания по определению хлорорганических пестицидов в воде, продуктах питания, кормах и табачных изделиях методом хроматографии в тонком слое
(Утверждены заместителем Главного государственного санитарного врача СССР от 28 января 1980 г.)
Физико-химические свойства пестицидов (даны в таблицах 1 и 2),
Основные положения. Настоящие методические указания распространяются на определение содержания ДДТ, ДДЭ, ДДД, гексахлорана, альдрина, кельтана, гептахлора, метоксихлора, дактала, тедиона и эфирсульфоната в воде, почве, вине, овощах, фруктах, грибах, зерне, комбикормах, корнеклубнеплодах и зеленых кормах, рыбе, мясе, мясопродуктах, внутренних органах, молоке и молочных продуктах, животном жире, сливочном и растительных маслах, жмыхах, шротах, лузге, меде, сахаре, яйцах и яйцепродуктах, а также в табачных изделиях.
Принцип метода. Метод основан на хроматографии хлорсодержащих пестицидов в топком слое окиси алюминия, силикагеля или пластинок «Силуфол» в различных системах подвижных растворителей после экстракции их из исследуемых образцов и очистке экстрактов. Подвижным растворителем служит гексан или гексан в смеси с ацетоном. Места локализации препаратов обнаруживают после опрыскивания пластинок раствором аммиаката серебра с последующим ультрафиолетовым облучением или после облучения ультрафиолетовым светом пластинок «Силуфол», содержащих о-то-лидип.
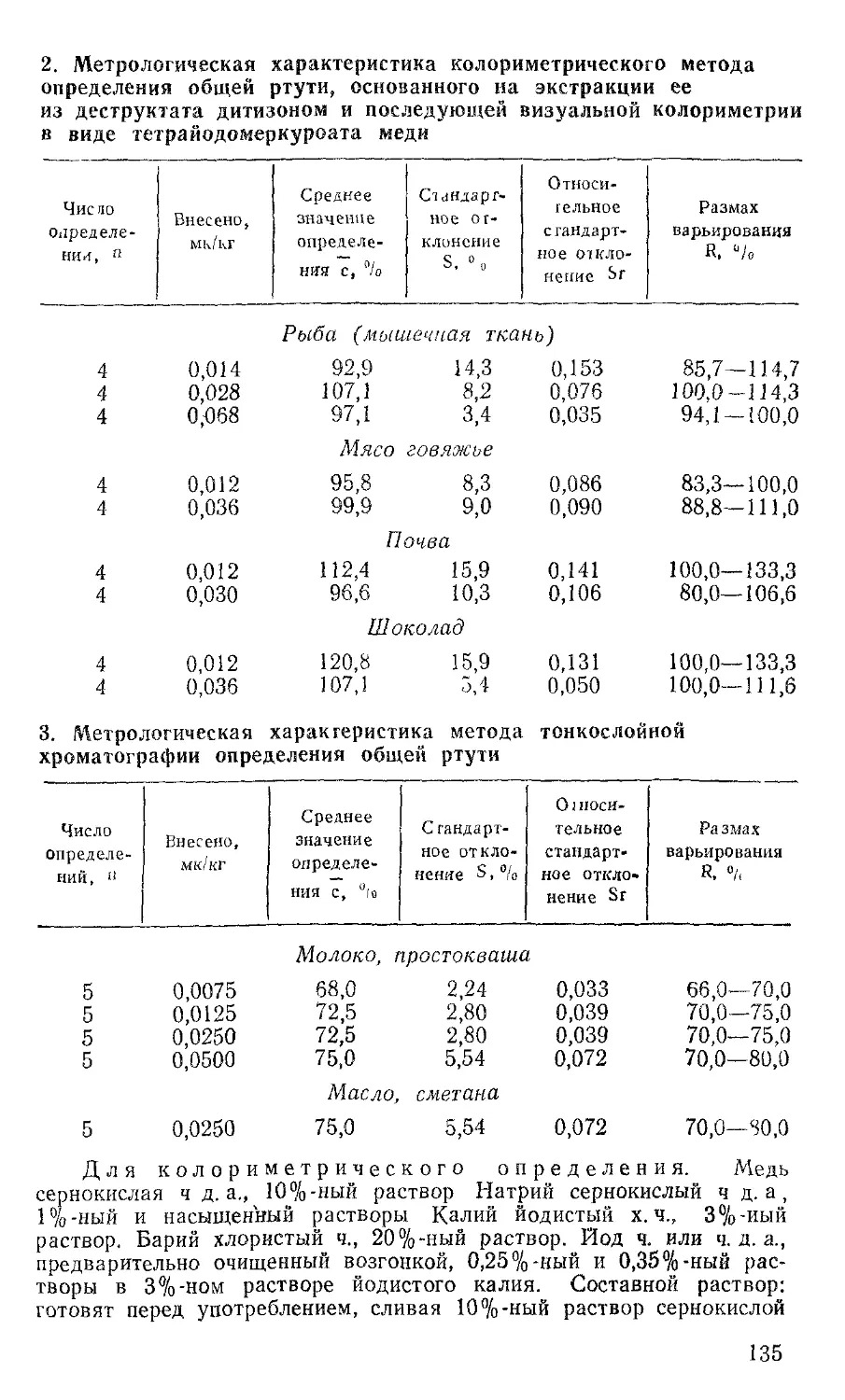
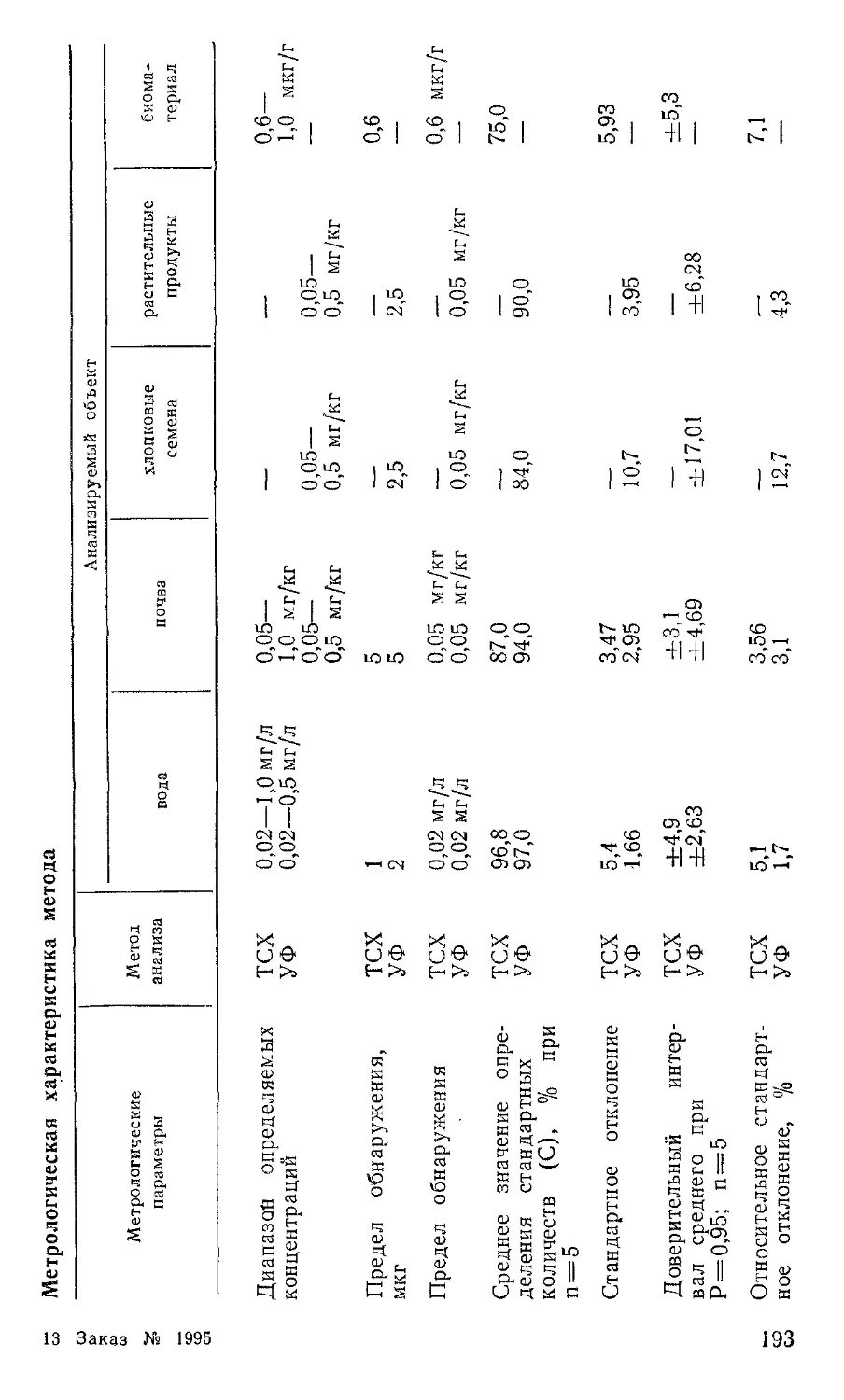
Метрологическая характеристика метода приведена в таблице 3.
Реактивы и растворы. Ацетон х. ч. Аммиак водный х. ч. Алюминия окись II степени активности для хроматографии (А) ч. Просеивают через сито 100 меш. Алюминия окись, пропитанная серной кислотой (Б). Две весовые части окиси алюминия (или окиси кремния) помещают в фарфоровую ступку, заливают одной объемной частью серной кислоты и тщательно перемешивают. Смесь готовят непосредственно перед подготовкой колонок для очистки экстрактов из проб шротов, жмыха, лузги. Бензол х. ч. Гексан ч. Калий щавелевокислый ч. д. а. Кальций сернокислый ч. д. а. Просушивают 6 ч в сушильном шкафу при 160 °C. Просеивают через сито 100 меш. Кремния окись для люминофоров ч. Натрий сернокислый безводный ч. Натрий углекислый кислый х. ч. Натрий хлористый х. ч., насыщенный раствор. Петролейный эфир (т. кип. 40—70°С). Перекись водорода х. ч. (30%-ный водный раствор).
Проявляющий реактив № Г. 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, прибавляют 7 мл аммиака и доводят объем раствора до 100 мл ацетоном, в готовый раствор
Пестицид
Эмпирическая формула
4,4'-дихлор ди фенил-трихлорметилметан-4,4'-ДДТ (п, п'-ДДТ) C14H9C15
2,4'-дихлор дифенилтрихлор метилметан-2, 4-ДДТ (о, п'-ДДТ) 4,4'-дихлордифенил- С14H9CI5
CI4H10CL
дихлорметилметан-4, 4'-ДДТ (п, п'-ДДД)-метаболит ДДТ
1,2,3,4,5,6-гексахлор- циклогексан, а-ГХЦГ С6НеС16
Молекулярная масса
354,5
354,5
320,0
290,6
Температура плавления, °C Растворимость пдк В почве, мг/кг ЛД50 для крыс, мг/кг
в воде, мг/л (25 °C) в органических растворителях
108—109 0,005 Ацетон, гексан, бензол, ксилол, толуол, спирт, хлороформ 1,0 250—400
0,026 То же
112,0 0,020 Ацентон, бензол, гексан, метанол — 3400
1,2,3,4,5,6-гексахуГОр- С6Н6С16
циклогексан, у-ГХЦГ
1,2,3,4,5,6-гексахлорбен- С6С1е
зол-гексахлорбензол
4,4'-дихлордифенил- CuHgCU
дихлорэтилен-
4,4'-ДДЭ (п, п'-ДДЭ)-
метаболит ДДТ
290,86
284,8
318,0
157—158 112,8 2,00 7,80 Бензол, гексан, эфир, четыреххлористый углерод, ацетон, хлоро- форм, ксилол, ацетон, эфир, метанол, дихлорэтан То же 1,0 125
228—231 0,005 0,014 Бензол, спирт, хлороформ, гексан Ацетон, гексан, бензол — 3500
Пестицид Химическое название | Эмпирическая । формула Молекулярная масса Темпера ура, С Растворимое гь в воде, мг/л Органические растворители, в которых растворяется пестицид
плавления кипения (мм рг. ст.)
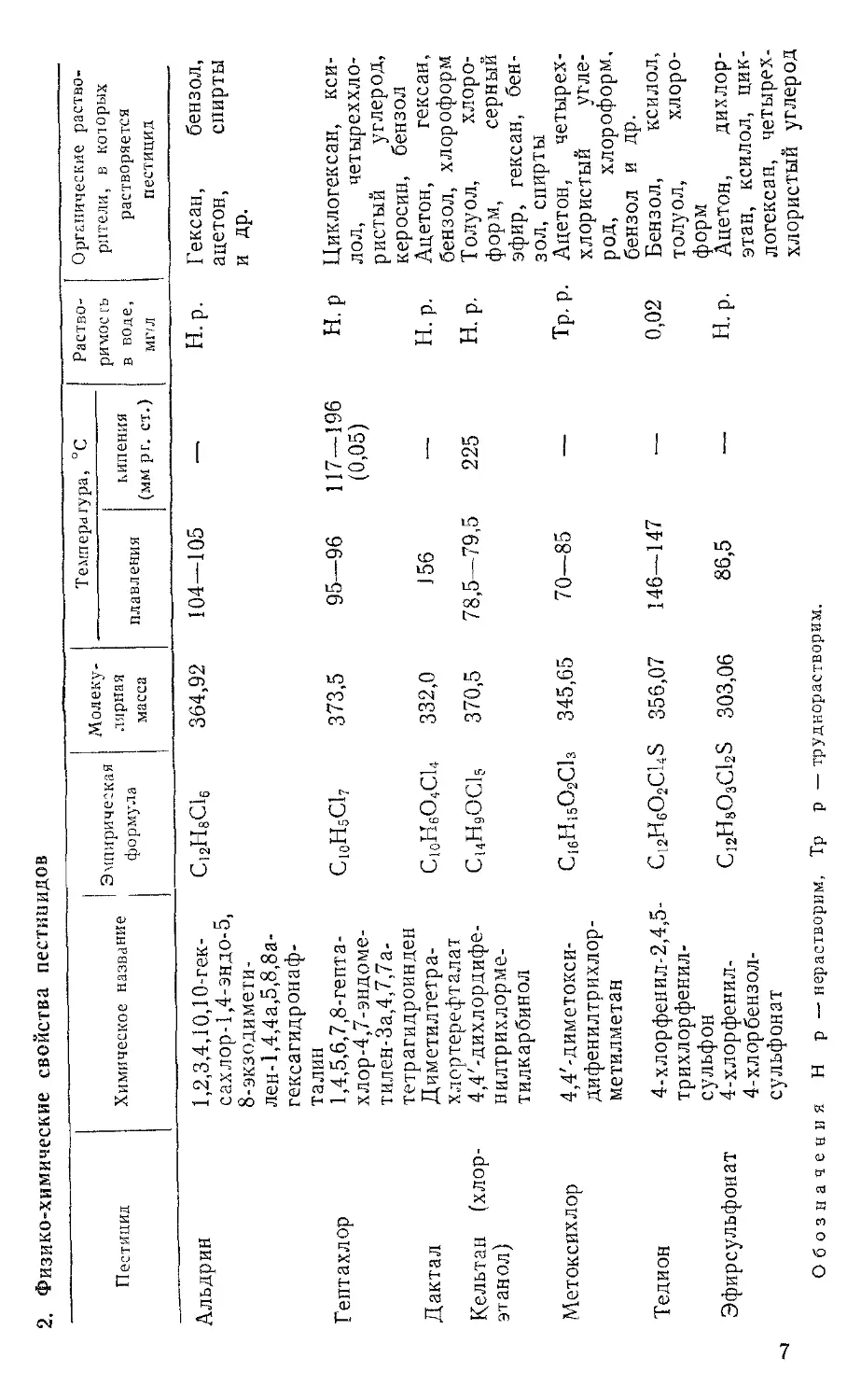
Альдрин 1,2,3,4,10,10-гек-сахлор-1,4-эндо-5, 8-экзодимети-лен-1,4,4а,5,8,8а-гексагидронаф-талин С^НвСЦ 364,92 104—105 — н.р. Гексан, бензол, ацетон, спирты и др.
Гептахлор 1,4,5,6,7,8-гепта- хлор-4,7-эндоме-тилен-3а,4,7,7а-тетрагидроинден С10Н5С17 373,5 95—96 117-196 (0,05) H. р Циклогексан, ксилол, четыреххлористый углерод, керосин, бензол
Дактал Диметилтетра-хлортерефталат С10Н6О4С14 332,0 156 •— Н.р. Ацетон, гексан, бензол, хлороформ
Кельтан (хлор-этанол) 4,4'-дихлордифе-нилтрихлорме-тилкарбинол С14Н9ОС15 370,5 78,5—79,5 225 н. р. Толуол, хлороформ, серный эфир, гексан, бензол, спирты Ацетон, четыреххлористый углерод, хлороформ, бензол и др.
Метоксихлор 4,4'-диметокси-дифенилтрихлор-метилметан C16H15O2C13 345,65 70—85 Тр. р.
Тедион 4-хлорфенил-2,4,5-трихлорфенилсульф он C12H6O2C14S 356,07 146—147 •— 0,02 Бензол, ксилол, толуол, хлоро- форм
Эфирсульфонат 4-хлорфенил-4-хлорбензол-сульфонат C12H8O3C12S 303,06 86,5 Н.р. Ацетон, дихлорэтан, ксилол, циклогексан, четырех-
хлористый углерод
Обозначения Н р — нерастворим, Тр р — труднорастворим.
3. Метрологическая характеристика метода определения хлорорганических пестицидов
Анализируемая проба Предел обнаружения, мг/л или мг, кг Число параллель- : них определе-j НИЙ, П Размах варьирования, R % Среднее значе-I ние степени определения, с °/о Стандартное отклонение, S °/о Относительное стандартное отклонение, Sr °/о Доверительный интервал при п = 5 и а == 0,95, %
Вода 0,005 7 20 93 10 10,7 93,0+7,7
Вино 0,005 7 25 90 12 13,3 90,0+9,0
Овощи 0,050 7 20 83 12 14,4 83,0+9,0
Фрукты 0,050 6 20 81 9 Н,1 81,0+7,4
Зерно 0,050 7 25 75 10 13,3 75,0+7,5
Трава 0,025 10 30 74 12 16,2 74,0+7,6
Рыба 0,050 10 24 83 12 14,4 83,0+7,6
Мясо 0,050 10 30 87 15 17,2 87,0+9,5
Живот- 0,040 6 26 82 12 14,6 82,0+9,9
ный жир Молоко, 0,040 13 30 86 16 18,6 86,0+8,9
сливки, творог Сливоч- 0,050 8 30 83 17 20,0 83,0+12,0
ное масло Сахар 0,020 6 6 97 5 5,1 97,0+4,13
Примечание.
2,0 мг/кг или мг/л.
Диапазон
определяемых концентраций 0,005—
добавляют 0,2 мл перекиси водорода. Раствор следует хранить в колбе с притертой пробкой в темном месте в течение 3 дней. На пластинку 9X12 см расходуется 8—10 мл раствора.
Проявляющий реактив № 2: 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, добавляют 10 мл 2-фе-ноксиэтанола и доводят объем раствора до 200 мл ацетоном, затем добавляют 6 капель 30%-ной перекиси водорода.
Серебро азотнокислое ч. д. а. Серная кислота ч. Силикагель АСК (Воскресенского химкомбината). Силикагель КСК, просеянный через сито 100 меш.
Стандартные образцы: ДДТ, ДДД, ДДЭ, альдрин, изомеры ГХЦГ, гептахлор, метоксихлор, кельтан, эфирсульфонат, дактал, тедиои х. ч. Стандартные растворы: 10 мг соответствующего пестицида растворяют в мерной колбе на 100 мл в гексане и доводят до метки этим растворителем. Стандартные растворы необходимо хранить в стеклянной посуде с притертыми пробками в холодильнике.
Стеклянная вата, очищенная концентрированной серной кисло/ той, промытая дистиллированной водой и высушенная. о-Толидин ч_, 1%-ный раствор в ацетоне. 2-феноксиэтанол. Этиловый спирт-рек-тификат. Хлороформ х. ч. Четыреххлористый углерод х. ч. Диэтиловый эфир (для наркоза). Натрий сернокислый, 2%-ный водный раствор. Натрий сернокислый, насыщенный раствор.
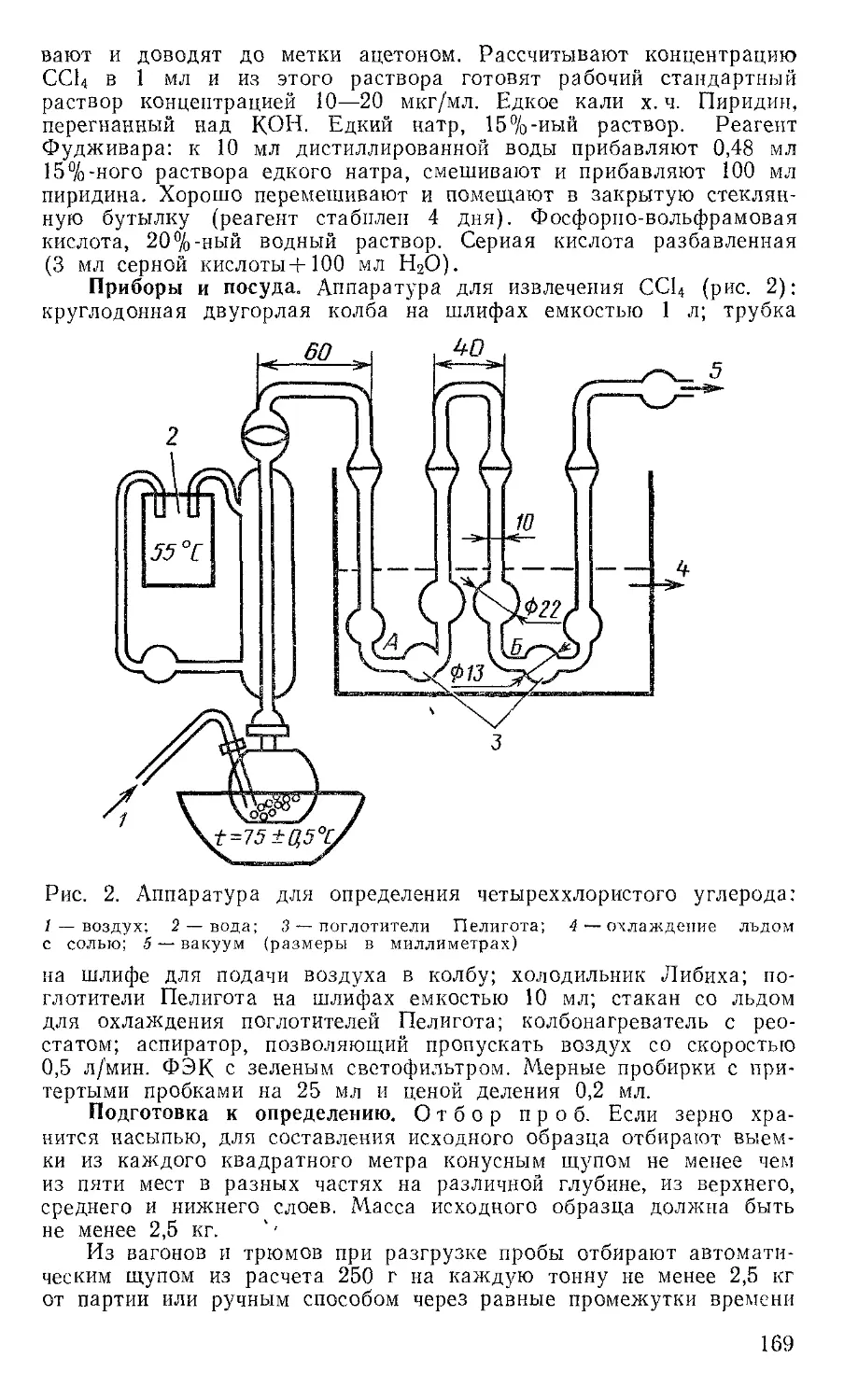
Приборы и посуда. Баня водяная. Вакуумно-ротационный испаритель или прибор для отгонки растворителей. Воронки химические диаметром 6 см. Воронки делительные на 100, 250, 500 мл. Гомогенизатор или измельчитель тканей. Камера для опрыскивания. Камеры для хроматографирования размером 150X200, 105X165 мм. Колбы мерные на 50 и 100 мл. Колбы на шлифах емкостью 100, 250 и 500 мл. Колбы круглодонные на шлифах емкостью 150, 250 и 500 мл. Микропипетки. Пипетки или шприцы для нанесения проб. Пипетки на 1, 5 и 10 м. Прибор для встряхивания. Пластинки стеклянные 9X12, 13X18 см. Пульверизаторы стеклянные для опрыскивания пластинок. Сито на 100 меш. (диаметр отверстий 0,147 мм). Стеклянные хроматографические колонки (диа-метрХвысота, мм) 20X400, 15X150. Ртутно-кварцевая лампа IIPK.-4. Цилиндры мерные иа 25; 50; 100; 250 и 500 мл. Чашки выпарительные № 3 и № 4.
Подготовка к определению. Приготовление пластинок для хроматографирования. Тщательно промытую хромовой смесью, содой, дистиллированной водой и высушенную пластинку протирают этиловым спиртом и покрывают сорбционной массой. Массу готовят следующим образом: а) 50 г просеянной через сито 100 меш. окиси алюминия смешивают в фарфоровой ступке с 5 г сернокислого кальция, прибавляют 75 мл дистиллированной воды и перемешивают в ступке или колбе до образования однородной массы. На пластинку 9X12 см наносят 10 г сорбционной массы (па пластинку 13X18 см — 20 г) и, покачивая, равномерно распределяют по всей пластинке. Пластинки сушат при комнатной температуре 18—20 ч, можно сушить 20 мин при комнатной температуре, а затем 45 мин в сушильном шкафу при температуре НО °C; б) 35 г силикагеля КСК, просеянного через сито 100 меш, смешивают с 2 г серпокислого кальция и 90 мл дистиллированной воды и перемешивают в ступке или колбе до однородной массы. Наносят на пластинки и сушат, как указано выше. Порция рассчитана на 10 пластинок.
Если пластинки с тонким слоем силикагеля темнеют после облучения ультрафиолетовым светом, силикагель перед употреблением следует очистить от примесей. Для этого его заливают на 18— 20 ч разбавленной соляной кислотой (1 : 1), кислоту сливают, промывают силикагель водой и кипятят в круглодопной колбе 2— 3 ч с разбавленной азотной кислотой (1 : 1), промывают проточной водопроводной, затем дистиллированной водой до нейтральной реакции промывных вод, сушат в сушильном шкафу 4—6 ч при температуре 130 °C. Силикагель дробят и просеивают через сито 100 меш
Пластинки для хроматографии «Силуфол UV-254» производства ЧССР перед использованием импрегнируют о-толидином. Для этого каждую пластинку погружают в 0,1%-пый раствор о-толи-дина в ацетоне, налитого в камеру для хроматографирования. После того как фронт растворителя поднимется до верхнего края пластинки, ее вынимают и высушивают на воздухе, избегая прямого солнечного света, пластинка готова к употреблению. Пластинки, импрегнированнЫе о-толидином, хранят в эксикаторе.
Пластинки «Силуфол UV-254» производства ЧССР предварительно промывают дистиллированной водой в хроматографической камере, высушивают на воздухе и непосредственно перед исполь
зованием активируют в сушильном шкафу при температуре 65 °C в течение 4 мин.
Подготовка хроматографических колонок для очистки экстрактов. Хроматографическая колонка для очистки от молочного жира. В нижнюю часть хроматографической колонки (размером 20X400 мм) помещают стекловату или 500 мг обезжиренной ваты. Затем засыпают в колонку силикагель АСК (75 мл для очистки экстрактов из проб свиного жира и 70 мл для всех остальных проб) и уплотняют силикагель постукиванием по колонке. Колонку промывают 50 мл гексана или петролейного эфира, прошедший через нее растворитель отбрасывают. После этого колонка готова для хроматографической очистки экстрактов из проб рыбы, мяса и мясопродуктов, молока и молокопродуктов, меда, яиц и т. п.
Хроматографическая колонка для очистки экстрактов из проб шротов (не обогащенных липидами), жмыхов и лузги. Хроматографическую колонку заполняют на высоту 1 см стеклянной ватой, затем в колонку вносят просеянную окись алюминия (А) слоем 2,5 см или окись кремния слоем 3,5 см, далее засыпают, не утрамбовывая, комочки окиси алюминия (кремния), пропитанные серной кислотой, высота слоя (Б) 2,5 см. Каждый слой последовательно промывают гексаном (всего 30 мл). Для анализа жмыхов и шротов, обогащенных липидами, слои окиси алюминия следует увеличить соответственно до 5 (А) и 3 см (Б), а при использовании окиси кремния — до 6 (А) и 3 см (Б).
Ход анализа.
Экстракция и очистка экстракта из воды и вина Пробу 200 мл помещают в делительную воронку и экстрагируют пестициды, встряхивая в течение 3 мин, гексаном или петролейным эфиром тремя порциями по 30 мл, или диэтиловым эфиром тремя порциями по 50 мл. В объединенные экстракты насыпают 10 г безводного сернокислого натрия или фильтруют через воронку, заполненную на 2/з сернокислым натрием. Экстракты переносят в прибор для отгонки растворителей и отгоняют растворитель до объема 0,2— 0,3 мл. В случае необходимости экстракт чистят серной кислотой.
Экстракция и очистка экстракта из овощей и фруктов. Измельченную пробу 20 г помешают в колбу с притертой пробкой и проводят экстрагирование пестицидов трижды в течение 15 мин на аппарате для встряхивания гексаном или петролейном эфиром порциями по 30 мл. Объединенные экстракты сушат безводным сернокислым натрием, переносят в прибор для отгонки растворителей, отгоняют растворитель до объема 0,2—0,3 мл и наносят на пластинку.
Экстракция и очистка экстракта из зерна и грибов. Из измельченных проб отбирают 20 г зерна, 50 г сырых или 10 г сухих грибов и помещают в колбы с притертыми пробками. Экстракцию пестицидов проводят трижды на приборе для встряхивания гексаном или петролейным эфиром порциями по 30 мл. Объединенные экстракты переносят в делительную воронку, прибавляют 10 мл насыщенного раствора безводного сернокислого натрия в серной кислоте и осторожно встряхивают несколько раз. Отделяют органический слой и повторяют обработку до тех пор, пока кислота не станет бесцветной. Экстракт промывают дистиллированной водой, сушат безводным сернокислым натрием и отгоняют растворитель.
Экстракция и очистка экстракта из яблок, капусты, травы и сена. Пробы 20 г измельченных яблок, 20 г капусты, 40 г травы и 20 г сена заливают 100 мл ацетона в колбах с притертой пробкой. Встряхивают 2—3 мин, прибавляют 20 мл дистиллированной воды и охлаждают па льду 30 мин. Экстракт сливают и фильтруют холодным, экстракцию повторяют. Из объединенных водно-ацетоновых экстрактов отгоняют ацетон, а из водного остатка экстрагируют препараты гексаном тремя порциями по 10 мл в течение 10 мин. Гексановые экстракты очищают серной кислотой, насыщенной безводным серпокислым натрием. Сушат безводным сернокислым натрием Отгоняют растворитель до небольшого объема и наносят на пластинку. Если очистка неполная (после испарения растворителя на колбе остается белый палет), экстракт испаряют досуха, остаток смывают холодным ацетоном 3 раза порциями по 0,2 мл и сразу наносят на пластинку.
Экстракция и очистка экстракта из комбикорма. Для исследования берут навеску 40 г, увлажняют ее в колбе 60 мл дистиллированной воды. Увлажненную навеску оставляют на ночь в колбе с закрытой пробкой. Экстракцию пестицидов проводят дважды 50— 100 мл смеси гексана и ацетона (1:1) при встряхивании в течение 2 ч. Экстракты объединяют в делительной воронке на 500 мл, прибавляют дважды по 50 мл дистиллированной воды и после разделения слоев нижний водный слой сливают в другую делительную воронку и экстрагируют пестициды 40 мл гексана. Водный слой сливают. Гексановые экстракты объединяют, фильтруют через воронку с бумажным фильтром, заполненным на 2/3 безводным сернокислым натрием. Экстракты упаривают на ротационном испарителе до объема 20—30 мл или досуха, растворяя затем сухой остаток в 20—30 мл гексана или петролейного эфира. Экстракт переносят в делительную воронку и производят очистку серной кислотой, как описано выше.
Экстракция и очистка экстракта из шрота, лузги, жмыха. Навески шрота, обогащенного липидами, жмыха (15 г), не обогащенного липидами, и лузги (20 г) делят на равные части и помещают в колбы вместимостью 100—250 мл с притертыми пробками, заливают гексаном (три объема гексана на одну весовую часть шрота), встряхивают на приборе для встряхивания 30 мин. Экстракт фильтруют через воронку Бюхнера, не перенося осадок на воронку. В колбу повторно заливают указанное количество гексана, встряхивают 30 мин, фильтруют, количественно переносят осадок на воронку Бюхнера с помощью 30 мл гексана (3 раза по 10 мл). Полученный экстракт выпаривают до 30 мл на ротационном испарителе или в токе воздуха при температуре не выше 40 °C, остаток делят на две равные части и помещают в морозильную камеру холодильника на 1 ч (не менее). Каждую часть пропускают через отдельную колонку с окисью алюминия или окисью кремния, пропитанных серной кислотой, со скоростью 2 мл/мин промывают колбу и колонку 50 мл охлажденной смеси этилового эфира с гексаном (15:85). Данную операцию необходимо проводить без перерыва, пе оставляя, на следующий день.
Очищенные экстракты объединяют и упаривают до объема 1 мл. Остаток из колбы переносят количественно микропипеткой с помощью резиновой груши в пробирку на 1 мл, колбу и микропипетку 2—3 раза промывают небольшим количеством гексана (всего 0,3— 0,5 мл), сливая его в ту же пробирку. Затем осторожно выпари-
ваюг гексан из пробирки на водяной бане при температуре 50 “С почти досуха (конечный объем приблизительно 2—3 капли). Если общий объем экстракта и промывной жидкости превышает 1 мл, то сначала выпаривают экстракт, постепенно прибавляя к нему промывную жидкость. При наличии в упаренном экстракте белого мазеобразного осадка в пробирку добавляют 5—6 капель гексана и помещают ее на 15—20 мин в морозильную камеру холодильника, затем декантируют дважды таким же количеством гексана и снова упаривают до конечного объема 2—3 капли.
Параллельно с исследуемыми образцами готовят два модельных экстракта. Каждый экстракт получают из 1 г шрота, не содержащего пестицидов (соотношение сухого вещества и пестицида то же, что и в исследуемых образцах). В один из экстрактов перед очисткой на колонке вносят микрошприцем (микропипеткой) определяемые пестициды в количестве 3 мкг, в другой — 0,75 мкг. Упаренные исследуемые и модельные экстракты с помощью микрошприца или микропипетки количественно наносят на пластинку, трижды смывая пробирку небольшим количеством гексана.
Экстракция и очистка экстракта из рыбы, мяса и мясопродуктов. Лйясо и мясопродукты пропускают через мясорубку. Рыбу очищают от чешуи, внутренних органов и тоже пропускают через мясорубку. Пробу 20 г перемешивают с безводным сернокислым натрием и помещают в колбу с притертой пробкой. Пестициды экстрагируют дважды смесью гексана и ацетона или петролейного эфира и ацетона в соотношении 1 : 1 порциями по 50 мл в течение 1,5 ч при встряхивании. Экстракт фильтруют через воронку с бумажным фильтром, заполненным на 2/3 безводным сернокислым натрием, затем растворитель отгоняют, сухой остаток растворяют в 20 мл гексана и вносят его в колонку с силикагелем АСК. После впитывания экстракта в сорбент пестицид элюируют 110 мл смеси бензола с гексаном в соотношении 3: 8 порциями по 25—30 мл. Элюат собирают в круглодонную колбу со шлифом емкостью 250— 300 мл. Через 10 мин после впитывания последней порции растворителя сорбент отжимают с помощью груши. Элюат отгоняют до объема 0,1 мл и наносят на хроматографическую пластинку.
В том случае, если пробы мяса или рыбы содержат большое количество жира, после испарения первого экстрагента (смеси ацетона с гексаном) и растворения сухого остатка в гексане следует провести очистку гексанового экстракта серной кислотой, а затем колоночную очистку, как описано выше.
Экстракция и очистка экстракта животного жира, яйца, яичного порошка. Жир измельчают на мясорубке, яичный порошок тщательно перемешивают, в яйце отделяют желток от белка, взвешивают желток и белок, а для анализа берут только желток. Конечный расчет содержания хлорорганических пестицидов в яйце приводят на все яйцо. Желтки тщательно перемешивают. Пробу 25 г из подготовленного образца заливают 50 мл ацетона, перемешивают и нагревают на горячей водяной бане до закипания растворителя. Колбу охлаждают, добавляют в нее 10 мл охлажденного 2%-кого раствора сернокислого натрия, перемешивают и охлаждают 45 мин на ледяной бане. Затем сливают ацетоновый слой в круглодонную колбу через слой обезжиренной ваты. Экстракцию ацетоном с последующим вымораживанием жира повторяют еще 2 раза.
Из объединенных экстрактов отгоняют ацетон на ротационном испарителе или в приборе для отгонки растворителей (температу-12
ра бани не более 70+2 °C) и трижды экстрагируют петролейным эфиром порциями 20, 10 и 10 мл. Продолжительность первой экстракции 1 ч, последующих 15 мин. Петролейный эфир переносят в делительную воронку с 40 мл 2%-ного водного раствора сернокислого натрия, перемешивают содержимое в течение 2 мин, дают слоям разделиться и водную фазу отбрасывают. Чтобы улучшить разделение слоев, можно добавить несколько миллилитров насыщенного раствора сернокислого натрия.
Операцию промывки экстракта повторяют еще 2 раза, после чего петролейный эфир сливают в стакан с 20 г безводного сернокислого натрия, ополаскивают делительную воронку дважды 5 мл петролсйного эфира. Подсушенный экстракт количественно переносят в мерный цилиндр на 50 мл и доводят объем раствора петролейным эфиром до 30 мл.
Далее наносят 30 мл экстракта в колонку с силикагелем АСК, как указано выше. Для проб свиного жира насыпают 75 мл силикагеля АСК, для всех остальных проб — 70 мл. Очистку экстрактов проводят так же, как описано для проб мяса. Элюат собирают в круглодонную колбу на 150 мл, растворитель упаривают до объема нескольких капель и наносят на хроматографическую пластинку.
Экстракция и очистка экстракта из меда. Пробу меда 30 г смешивают с 3 г безводного сернокислого натрия и трижды экстрагируют пестициды гексаном порциями по 30 мл, каждый раз по 15 мин, тщательно растирая мед стеклянной палочкой в узком химическом стакане. Экстракты объединяют и отгоняют гексан до объема 30 мл или до небольшого объема, далее доводят экстракт до 30 мл гексаном. 30 мл экстракта вносят в хроматографическую колонку с силикагелем АСК и проводят очистку экстракта и испарение растворителя так, как описано выше.
Экстракция и очистка экстракта из сахара. Из павсски 50 г сахара, предварительно растворенного в воде, пестициды экстрагируют в делительной воронке на 250 мл гексаном. Экстракцию пестицидов проводят трижды по 50, 25 и 25 мл растворителя, каждый раз встряхивая по 5 мин. Объединенные гексановые экстракты очищают от коэкстрактивных веществ (красящие, аминокислоты, липиды) сернокислотным способом.
Экстракция и очистка экстракта из молока и цельномолочных продуктов. Для подготовки проб можно использовать один из приведенных способов.
Первый способ. Он применим для работы со сливками, сметаной, молоком и другими цельномолочными продуктами. Для анализа берут 20 г сливок и сметаны, которые разводят равным объемом дистиллированной воды, к 50 мл молока, кефира прибавляют концентрированную серную кислоту (30—40 мл) до полного почернения пробы. Охлажденный до 10—15 °C раствор переносят в делительную воронку и экстрагируют препараты гексаном 2 раза порциями по 25 мл. Для полного извлечения воронку встряхивают 2 мин, затем оставляют ее на 30 мин до полного разделения слоев. Если образуется эмульсия, прибавляют 1—2 мл этилового спирта. К объединенным экстрактам в делительной воронке прибавляют 10 мл концентрирова'ннои серной кислоты, насыщенной сернокислым натрием, и осторожно встряхивают несколько раз. Очистку продолжают до получения бесцветной серной кислоты.
При анализе творога и сыра 50 г творога или 10 г измельченного на терке сыра заливают 40 мл гексана или петролейного
эфира, непрерывно встряхивают 2—3 мин и оставляют па 30 мин. Экстракцию повторяют. Объединенные экстракты в делительной воронке очищают серной кислотой, как указано выше.
Второй способ. Его применяют для анализа молока, кефира, простокваши, кумыса и других цельномолочных продуктов. Пробу продукта (25 мл) помещают в делительную воронку на 300 мл, приливают по 5 мл щавелевокислого калия и насыщенного раствора хлористого натрия, перемешивают, приливают 100 мл ацетона, встряхивают 2 мин. Приливают 100 мл хлороформа и встряхивают 2 мин. Воронку оставляют до полного разделения слоев. Верхнюю фазу отбрасывают, а нижнюю выливают в круглоцониую колбу со шлифом и испаряют растворитель досуха. Остаток смывают 30 мл гсксапа.
Экстракция и очистка экстракта из сгущенного молока, 10 и 20°/а-ных сливок К 10 г продукта прибавляют 10 мл насыщенного раствора хлористого натрия и выливают в делительную воронку вместимостью 150 мл К смеси приливают 40 мл ацетона, встряхивают 2 мин, приливают 60 мл хлороформа, встряхивают 2—3 мин и оставляют до разделения фаз Далее поступают, как при определении пестицидов в молоке
Экстракция и очистка экстракта из сгущенных молочных продуктов Навеску продукта 10 г помещают в стаканчик, заливают 10 мл воды температурой 45—50 °C, перемешивают и переносят в делительную воронку на 150 мл, добавляют 5 мл щавелевокислого калия. Содержимое воронки перемешивают, приливают 80 мл ацетона и встряхивают 2—3 мин. Добавляют 100 мл хлороформа и ьыряхивают 5—7 мин После разделения фаз нижнюю фазу сливают в круглодонную колбу, растворители отгоняют, а сухой остаток растворяют в 30 мл петролейного эфира
Экстракция и очистка экстракта из сухих молочных продуктов. Навеску сухих молочных продуктов 3 г (сливок 2 г) высыпают в стаканчик, приливают 15 мл дистиллированной воды температурой 40—45 °C, размешивают и переносят в делительную воронку вместимостью 300 мл, приливают по 5 мл щавелевокислого калия и насыщенного раствора хлористого натрия. Содержимое воронки перемешивают, добавляют 80 мл ацетона и встряхивают 3—5 мин, приливают 100 мл хлороформа, встряхивают 5 мин и оставляют на 3—5 мин (до разделения фаз). Нижнюю фазу сливают в круглодонную колбу, растворитель отгоняют, а остаток смывают 30 мл гексана.
Экстракция и очистка экстракта из сметаны, 30 и 40%-ных сливок. Навеску продукта 5 г отвешивают в стаканчик, приливают 10 мл насыщенного раствора хлористого натрия и переносят в делительную воронку вместимостью 150 мл Стаканчик обмывают 40 мл ацетона, смывы переносят в делительную воронку, которую встряхивают 2—3 мин, добавляют 70 мл хлороформа и встряхивают 2 мин Воронку оставляют на несколько минут до разделения фаз, нижнюю фазу сливают в колбу для отгонки растворителей, растворители отгоняют, а остаток смывают 30 мл гексана.
Экстракция и очистка экстракта из творога и сыра. Навеску 10 г творога или измельченного на терке сыра растирают с 10 мл насыщенного раствора хлористого натрия и переносят в делительную воронку на 250—300 мл. Прибавляют 80 мл ацетона, встряхивают 2 мин, приливают 100 мл хлороформа и вновь встряхивают.
Нижнюю фазу используют для анализа после отгонки растворителей, растворив остаток в 30 мл гексана.
Далее проводят очистку экстрактов из проб молока и молочных продуктов от молочного жира, подготовленных по второму способу. Для этого 30 мл экстракта вносят в колонку с 70 мл силикагеля АСК. После впитывания экстракта в сорбент пестицид элюируют НО мл смеси бензола с гексаном (3:8) порциями по 25— 30 мл. Элюат собирают в круглодонную колбу на 250—300 мл. Через 10 мин после впитывания последней порции растворителя сорбент отжимают с помощью резиновой груши. После очистки растворители отгоняют под вакуумом.
Экстракция и очистка экстракта из сливочного масла. Сливочное масло (20 г) растапливают на водяной бане в круглодонной колбе, прибавляют 50 мл ацетона, тщательно перемешивают до растворения жира, прибавляют 10 мл ледяной дистиллированной воды и охлаждают на льду до затвердения жира (примерно 30 мин). Сливают ацетоновый экстракт и процедуру повторяют еще 2 раза. Из объединенных экстрактов в круглодонной колбе ацетон отгоняют на водяной бане. Пестициды экстрагируют из оставшегося водного экстракта гексаном тремя порциями по 10 мл в течение 5 мин. Объединенные гексановые экстракты в делительной воронке обрабатывают серной кислотой с сернокислым натрием. Очищенный экстракт сушат безводным сернокислым натрием и упаривают.
Экстракция и очистка экстракта из почвы. К навеске воздушносухой почвы 10 г, помещенной в коническую колбу па 250 мл, приливают 10 мл 1%-ного водного раствора хлористого аммония и оставляют на сутки закрытой. Затем приливают смесь 30 мл ацетона и 30 мл гексана и встряхивают колбу в течение 1 ч на встряхивающем устройстве. Содержимое колбы переносят в центрифужные пробирки. После центрифугирования жидкую часть сливают в конические колбы, почву с помощью 10 мл 1%-ного раствора хлористого аммония и 30 мл ацетона переносят в исходные конические колбы, добавляют 30 мл гексана и проводят экстракцию еще в течение 30 мин. Затем экстракты объединяют. К объединенным экстрактам в делительной воронке приливают 250 мл дистиллированной воды, осторожно встряхивают в течение 5—7 мин, дают жидкостям расслоиться и нижний водный слой сливают в коническую колбу. Гексановый слой пропускают через безводный сульфат натрия (30—40 г сульфата натрия).
Из водно-ацетонового слоя экстракцию пестицидов проводят еще дважды 15 и 10 мл гексана, который затем сушат через тот же сульфат натрия. Гексановые экстракты объединяют. Концентрирование экстрактов проводят либо на ротационно-вакуу.мном испарителе при температуре бани не более 40 °C и времени отгонки 9—11 мин, либо из колбочек с Г-образным отводом при температуре водяной бани 72—75 °C.
Очистку сконцентрированных гексановых экстрактов из проб почв проводят серной кислотой так, как описано выше для других проб, и испаряют растворитель.
Экстракция и очистка экстракта из табака и табачных изделий. Навеску табака 5 г помещают в стеклянный стакан на 500 мл, заливают 50 мл концентрированной серной кислоты и стеклянной палочкой тщательно размешивают до полного равномерного обугливания пробы. Спустя 10—15 мин в колбу добавляют 25 мл гск-
сана, тщательно размешивают содержимое и прибавляют 25 мл четыреххлористого углерода. Экстракцию пестицидов из пробы проводят в течение 15 мин трижды, после чего экстракт последовательно переносят в делительную воронку для однократной или двукратной дополнительной очистки серной кислотой.
Хроматографирование. На хроматографическую пластинку на расстоянии 1,5 см от ее края шприцем или пипеткой наносят исследуемую пробу в одну точку так, чтобы диаметр пятна не превышал 1 см. Остаток экстракта в колбочке смывают тремя порциями (по 0,2 мл) диэтилового эфира, которые наносят в центр первого пятна. Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, содержащие 10, 5 и 1 мкг исследуемых препаратов (или другие количества, близкие к определяемым концентрациям препаратов).
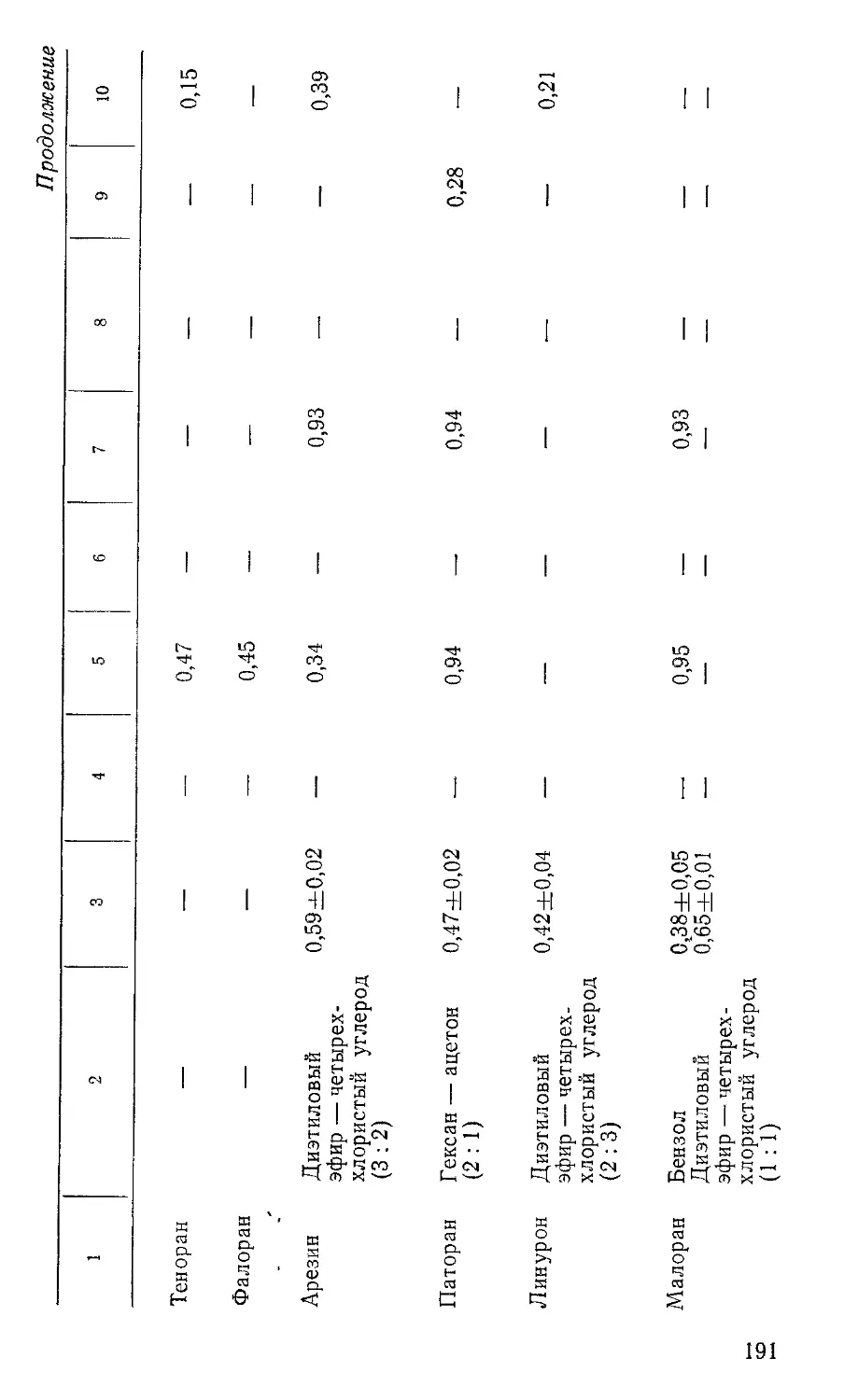
Пластинки с нанесенными растворами помещают в камеру для хроматографирования, на дно которой за 30 мин до начала хроматографирования наливают подвижный растворитель. При использовании пластинок с тонким слоем окиси алюминия или силикагеля в качестве подвижного растворителя применяют гексан или смесь гексана с ацетоном (6 : 1) для препаратов, у которых величина Rf в гексане ниже 0,3. При использовании пластинок «Силуфол» подвижный растворитель — 1°/о-ный раствор ацетона в гексане, а на пластинках «Силуфол», импрегпированных о-толидином,— гексан с диэтиловым эфиром (49:1). Край пластинки с нанесенными растворами может быть погружен в подвижный растворитель не более чем на 0,5 см.
После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Далее пластинку орошают проявляющим реактивом и подвергают действию ультрафиолетового света в течение 10—15 мин (лампа ПРК-4). Пластинки следует располагать на расстоянии 20 см от источника света. При наличии хлороргани-ческих пестицидов на пластинке появляются пятна серо-черного цвета.
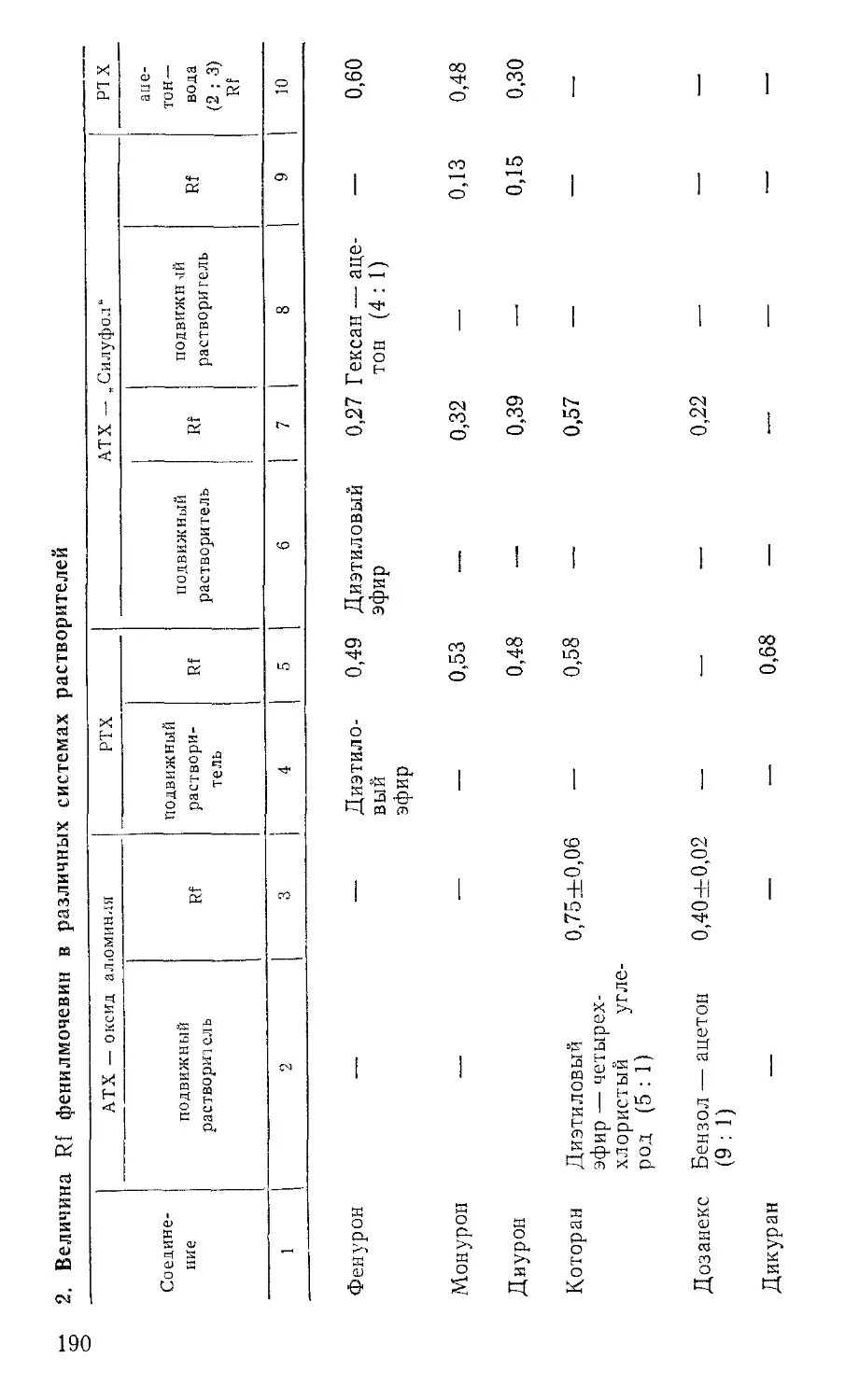
При использовании для анализа пластинок «Силуфол», импрег-нированных о-толидином, их непосредственно после хроматографирования подвергают облучению ультрафиолетовым светом в течение нескольких минут. При наличии хлорорганических пестицидов в этом случае проявляются пятна сине-голубого цвета. Величины Rf пестицидов приведены в таблице 4.
Обработка результатов анализа. Количественное определение осуществляют сравнением площадей пятен пробы и стандартных растворов. Между количеством препарата в пробе, не превышающим 20 мкг, и площадью его пятна на пластинке существует прямая пропорциональная зависимость. При большом содержании препарата следует использовать пропорциональную часть исследуемого экстракта.
Количество препарата в пробе (X, мг/кг или мг/л) вычисляют по формуле
V ^iSg д =-----
PS,
где А—содержание препарата в стандартном растворе, мкг; Si— площадь пятна стандартного раствора, мм2, S2—площадь пятна пробы, мм2; Р — масса или объем исследуемой пробы, г или мл.
4. Величина Rf хлорорганических пестицидов
Пестицид Подвиж ный растворитель Величина Rf
на окиси ал омииия на силикагеле
Г ексахлорбензол Гексан 0,90
Альдрин » 0,83 0,68
ддэ » 0,78 0,66
ДДЭ Гексан + ацетон (6:1) 0,87 —
Гептахлор о,п'-ДДТ Гексан 0,76 0,65
» 0,67 0,54
п,п'-ДДТ » 0,61 0,50
п,п'-ДДТ Гексан+ацетон (6:1) 0,75 —
Линдан Гексан 0,34 0,20
ддд » 0,30 0,40
ддд Гексан+ацетон (6:1) 0,62 —
Метоксихлор Гексан 0,15 —
» Гексан+ацетон (6:1) 0,60 —
Кельтан Гексан 0,05 —
» Гексаи + ацетон (6:1) 0,40 —
» Бензол 0,44 —
Тедион Гексан 0,03 —
» Гексан+ацетон (6:1) 0,55 —
Эфирсульфонат Гексан 0,00 —
» Гексан+ацетон (6: 1) 0,45 —
Дактал То же (2:1) 0,90 —
Определение гамма-изомера гексахлорциклогексана и фенотиазина в продуктах животного происхождения тонкослойной хроматографией *
Химически чистый препарат фснотиазина (тиофениламин, ди-бензоимазин) представляет собой бесцветное или слегка желтоватое кристаллическое вещество. Относительная молекулярная масса 199,26. Температура плавления фенотиазина 180—181 °C. Он хорошо растворяется в ряде органических растворителей (хлороформ, диметилформамид, бензол, ацетон), мало растворим в этаноле, петролейном бфире, практически нерастворим в воде.
Здесь и далее * (звездочкой) помечены методы, утвержденные Минздравом СССР и опубликованные в сборнике «Методы определения микроколичеств пестицидов».— М.: Колос, 1977,
2 Заказ № 1995
17
Фенотиазин легко окисляется, особенно на солнце и во влажных условиях. Такие окислители, как перекись водорода, перманганат калия, гипохлорит калия, хромовый ангидрид, нитрит натрия, окисляют фепотиазин до сульфоксидов и сульфонов. При взаимодействии фенотиазина с дымящей азотной кислотой образуются динитропроизводные (2,7-динитрофенотиазин и др.).
Поступающие в продажу препараты фенотиазина обычно содержат 1% нетоксичного продукта окисления, который имеет зеленый цвет.
Принцип метода. Метод основан на извлечении гамма-изомера ГХЦГ и фенотиазина из пищевого продукта органическими растворителями, удалении мешающих определению примесей путем жидкостного перераспределения,” очистке экстракта на активированной окиси алюминия и хроматографировании в тонком слое силикагеля. Минимально детектируемое количество в хроматографируемом объеме 0,2 мкг гамма-изомера ГХЦГ и 0,1 мкг фенотиазина. Чувствительность метода для гамма-изомера ГХЦГ и фенотиазина 0,003 мг в пробе, или 0,12 мг/кг продукта.
Полнота определения 75—80%. Количественное определение с достаточной точностью можно проводить при содержании 20— 30 мкг препаратов в пробе
Реактивы и растворы. Алюминия окись безводная или II степени активности, активированная в течение 4 ч при 600—800 °C и деактивированная перед употреблением водой (5% воды к массе окиси алюминия) посредством растирания в ступке.
Ацетон х. ч перегнанный. н-Гексан х. ч. перегнанный н-Гек-сан х. ч. перегнанный, насыщенный диметилформамидом Смесь гексана с ацетоном (2:1). Диэтиловый эфир х. ч. 1%-ный раствор дифениламина в ацетоне. Диметилформамид чистый, насыщенный н-гексаном. Натрий сернокислый безводный ч ,д. а и его 2%-ный водный раствор. Пластинки для хроматографии «Силуфол» производства ЧССР. Стандартные растворы гамма-изомера ГХЦГ в гексане (100 мкг/мл и 10 мкг/мл) и фснотиазина в хлороформе (100 мкг/мл и 10 мкг/мл). Подвижный растворитель (гексан-1-диэтиловый эфир + уксусная кислота, 75:25:2). Уксусная кислота ледяная.
Приборы и посуда. Аппарат для встряхивания. Воронки делительные вместимостью 150 и 500 мл. Воронки Бюхнера диаметром 100—120 мм. Ртутно-кварцевая лампа ПРК-4 или ПРК-2 (источник ультрафиолетового света). Колбы Бунзена вместимостью 300 — 500 мл. Колбы для экстракции на 700—1000 мл. Колбы круглотонные на 50, 150 и 250 мл. Колонка хроматографическая стеклянная 470ХЮ мм. В узкий конец колонки помещают ватный тампон и наливают взвесь 10 г деактивированной окиси алюминия в гексане. После осаждения адсорбента и его уплотнения в результате разрежения, создаваемого вакуумным насосом, насыпают слой безводного сульфата натрия высотой 5—6 см и промывают колонку гексаном. Микропипетки. Пипетки Мора Пульверизатор стеклянный. Устройство для удаления растворителей (ротационный испаритель, водоструйный насос и др). Центрифуга (2500 -4000 об/мин).
Подготовка проб.
Мясо (говяжье, куриное). Среднюю пробу массой 0,5 кг измельчают на мясорубке, тщательно перемешивают и берут навеску в 25 г.
Куриные яйца. Средняя проба состоит из 5—10 яиц в зависимости от их величины. Яйца освобождают от скорлупы, помещают в химический стакан и взбалтывают до однородной массы. Для анализа берут часть смеси, соответствующую половине яйца.
Подготовленные навески растирают в фарфоровой ступке с безводным сульфатом натрия до образования сыпучей массы. Пробы количественно переносят в колбы для экстракции, заливают 200 мл смеси н-гексана с ацетоном (2:1) и экстрагируют на аппарате для встряхивания 30 мин Жидкую фазу сливают и повторяют экстракцию со 100 мл смеси. Полученные экстракты объединяют
Молоко. Из тщательно перемешанной средней пробы молока объемом 0,5 л аликвот объемом 25 мл переносят в колбу, туда же добавляют 50 мл ацетона, перемешивают и приливают 50 мл гексана. Пробу экстрагируют 30 мин на аппарате для встряхивания. Затем смесь центрифугируют при 2500—4000 об/мин, в результате чего достигается хорошее разделение фаз: верхняя—гексановая, средняя — твердая (белковая), нижняя—водно-ацетоновая. Гексановую фазу с помощью пипетки Мора и резиновой груши осторожно переносят в колбу для удаления растворителя и отгоняют его до объема 5—10 мл.
Ход анализа. Объединенные экстракты помещают на 1,5—2 ч в морозильную камеру холодильника, а затем фильтруют в колбу Бунзена через воронку Бюхнера с двойным бумажным фильтром, используя при этом водоструйный или вакуумный насос. Колбу дважды ополаскивают порциями охлажденной гексано-ацетоновой смеси (2:1).
Фильтрат количественно переносят в круглодонную колбу и удаляют растворитель до объема 5—10 мл
Очистку экстрактов всех продуктов, включая молоко, проводят посредством жидкостного перераспределения в системе гексан-ди-метилформамид. Для этого объем экстракта доводят гексаном до 25 мл, экстракт переносят в делительную воронку и извлекают из пего гамма-изомер ГХЦГ и фенотиазип посредством трехкратного встряхивания с 10 мл насыщенного гексаном диметилформамида Гексановую фракцию отбрасывают. Для более полного удаления жира объединенный диметилформамидный экстракт промывают 20 мл гексана, насыщенного диметилформамидом. Гексановый слой отбрасывают, а диметилформамидный слой переносят в другую делительную воронку (0,5 л), в которую предварительно наливают 400 мл 2%-ного водного раствора сульфата натрия, добавляют 10 мл гексана, встряхивают и оставляют на 15—20 мин для расслоения. Отделяют слой гексана и экстрагируют еще раз 10 мл гексана.
Объединенные гексановые экстракты отгоняют до объема 10 ми и переносят в хроматографическую колонку с окисью алюминия в момент, когда слой гексана, которым промывают колонку, имеет высоту над сульфатом натрия 10 мм. Хроматографирование проводят на первом этапе самотеком. Элюируют гамма-изомер ГХЦГ и фенотиазин 90 мл гексана, который следует вносить в колонку (предварительно онолоспув им колбу, в которой находился экстракт) после того, как высота экстракта над сульфатом натрия достигнет уровня 5—10 мм.
Для ускорения элюирования можно использовать небольшое разрежение, создаваемое водоструйным или вакуумным насосом 2* 19
Однако разрежение целесообразно лишь после того, как третья порция гексана после экстракции пройдет через колонку.
Из полученного элюата удаляют растворитель досуха. Растворяют сухой остаток в 0,5 мл гексана (следует пользоваться градуированной пробиркой) и наносят аликвоту раствора (20, 40, 60, 100 мкл) микропипеткой на пластинку «Силуфол». Рядом, как обычно, наносят различные количества стандартных растворов гамма-изомера ГХЦГ и фенотиазина.
Пластинку помещают в хроматографическую камеру со смесью гексана с диэтиловым эфиром и уксусной кислотой (75:25:2). После поднятия фронта подвижного растворителя на 10—12 см пластинку вынимают из камеры и оставляют на несколько минут для удаления подвижного растворителя. При этом фенотиазин проявляется в виде четких розовых пятен, которые становятся голубовато-зелеными после опрыскивания 1%-ным раствором дифениламина в ацетоне и ультрафиолетового облучения. Гамма-изомер ГХЦГ проявляется в виде темно-синих или темно-серых пятен. Rf гамма-изомера ГХЦГ равно 0,36, фенотиазина — 0,18 (для ДДТ — 0,53, для альфа-изомера ГХЦГ — 0,43 при данных условиях) .
Количественное определение проводят путем сравнения размеров и интенсивности окраски пятен пробы и стандартных растворов. Для фенотиазина это можно сделать на двух этапах (после извлечения пластинки из камеры и после УФ-облучения), для гамма-изомера ГХЦГ только после облучения.
Расчет проводят по формуле
где X-—содержание препарата в пробе, мг/кг; А — количество препарата, найденное путем визуального сравнения размера и интенсивности пятен пробы и стандартных растворов, мкг; Р — масса исследуемой пробы, г.
Определение полихлорпинена
и полихлоркамфена в воздухе, воде, почве, картофеле и свекле, мясе, молоке, тканях внутренних органов животных, крови, моче тонкослойной хроматографией *
Полихлорпинен (ПХП) — продукт хлорирования хлористого борнила до содержания хлора 64—67%. По внешнему виду ПХП'— вязкое бесцветное масло, по консистенции напоминающее мёд (плотность 1,5—1,6 г/см3). Химически полихлорпинен представляет собой сложную смесь хлорированных пиненов. Он практически нерастворим в воде, хорошо растворяется в органических растворителях.
Выпускаются 20%-ный и 50%-ный масляные растворы, а также 65%-ный концентрат эмульсии.
Полихлоркамфен (ПХК, СюНцСК) представляет собой продукт хлорирования камфена до содержания хлора 67—69%. По внешнему виду ПХК — воскообразное вещество белого цвета с температурой плавления 65—69 °C. Плотность 1,6 г/см3, относительная
молекулярная масса 413,8. Химически полихлоркамфеи представляет собой сложную смесь полихлоркамфенов и камфенов различного строения. Он практически нерастворим в воде, но хорошо растворяется во многих органических растворителях.
Применяют в виде дустов или суспензий, а также в виде эмульсий и растворов.
Принцип метода. Метод основан на извлечении ПХП (ПХК) из пробы н-гексаном, очистке экстракта концентрированной серной кислотой и хроматографировании в тонком слое адсорбента.
При наличии ДДТ и других хлорорганических пестицидов в исследуемой пробе применяется микротонкослойная хроматография. В качестве адсорбента используют силикагель с величиной зерен 3—10 мкм, нанесенный на стеклянные пластинки 6X6 см. Подвижным растворителем служит смесь н-гексана с метиловым спиртом и аммиаком в соотношении 10:4:0,3. При этом достигается разделение ПХП или ПХК и ДДТ.
Если ДДТ и других хлорорганических пестицидов в анализируемой пробе нет, кроме микротоикослойной хроматографии, возможно применение макротопкослойной хроматографии. В качестве адсорбента используют силикагель, просеянный через сито 100 меш. и нанесенный на пластинки 9X12 см.
В качестве подвижного растворителя служит смесь н-гексана с этилацетатом в отношении 5:1. Разделение ПХП или ПХК и ДДТ не достигается.
Хроматограммы проявляют двумя способами.
I способ — раствором азотнокислого серебра и аммиака в ацетоне с последующим ультрафиолетовым облучением. ПХП, ПХК, ДДТ и другие хлорорганическпе пестициды проявляются в виде черных пятен на белом фоне. Минимально детектируемое количество ПХП (ПХК) на микропластинках 0,2—0,3 мкг, на макропластинках 0,5—1,0 мкг.
II с п о с о б — раствором дифениламина и хлористого цинка в ацетоне с последующим выдерживанием пластинки при 140—150 °C. ПХП и ПХК проявляются в виде голубовато-зеленых пятен, ДДТ и ДДЭ — в виде красных, ДДД — голубых, гептахлор — синих пятен. ГХЦГ и альдрин не проявляются. Минимально детектируемое количество ПХП (ПХК) на микропластинках 1,0 мкг, на макропластинках 10 мкг.
Чувствительность определения ПХП и ПХК с помощью дифениламина составляет 2 мкг, с помощью азотнокислого серебра — 0,5—0,6 мкг в пробе. Полнота определения в различных объектах 85—95%, в жировой ткани 65%.
Реактивы и растворы. Азотнокислое серебро. 25%-ный раствор аммиака х. ч. Ацетон х. ч. Вата гигроскопическая, промытая эфиром и высушенная. 0,5 г ваты промывают 5—10 мл диэтилового эфира, упаривают эфир до небольшого объема и наносят на хроматографическую пластинку, которую опускают в камеру с подвижным растворителем. Далее поступают, как при определении препарата.
Если нет пятен, аналогичных по величине Rf-пятнам определяемого препарата, вата пригодна для отбора проб воздуха, н-Гексан х. ч.
Кальций сернокислый (CaSO4-2H2O). В течение 1—2 сут каль
ций сернокислый прокаливают при температуре 160 °C и просеивают через сито 100 меш. Хранят в склянке с притертой пробкой.
Проявляющий раствор № 1: 0,5 г азотнокислого серебра растворяют в 2,5 мл дистиллированной воды, прибавляют 3 мл аммиака и доводят ацетоном до метки в мерной колбе на 50 мл.
Проявляющий раствор № 2: 0,5 г дифениламина и 0,5 г хлористого цинка растворяют в 100 мл ацетона. Реактив готовят перед использованием.
Силикагель КСК очищенный. Силикагель заливают на 18—20 ч соляной кислотой (1 : 1). Кислоту сливают, промывают силикагель водой и кипятят с разведенной азотной кислотой (1:1) в течение 2—3 ч. Промывают сначала горячей водопроводной, а затем дистиллированной водой до нейтральной реакции промывных вод (проба с лакмусовой бумажкой или метилоранжем) и до удаления в промывных водах следов хлоридов (проба с азотнокислым серебром). Далее силикагель сушат 4—6 ч в шкафу с температурой 130 °C при помешивании. Дробят на шаровой мельнице или электромельнице Размолотый и просеянный через сито 100 меш силикагель необходим для приготовления пластинок 9X12 см.
Силикагель с величиной зерен 3—10 мкм, полученный путем фракционирования (см. ниже), необходим для приготовления пластинок 6X6 см. Силикагель с величиной зерен 0,5 мм, полученный просеиванием через сита, необходим для отбора проб воздуха. Спирт метиловый х. ч. Стандартный раствор 0,025 г пестицида растворяют в н-гексане в мерной колбе на 50 мл Концентрация раствора 500 мкг/мл Фильтры беззольные.
Посуда и оборудование. Аспиратор. Аллонжи стеклянные (длина 100—110 мм, диаметр 17 мм), плотно заполненные гигроскопической ватой (1,0—1,2 г). Гофрированные стеклянные трубки (длина 90—100 мм, диаметр 10 мм) с меткой у одного конца, плотно заполненные силикагелем КСК с диаметром зерен 0,5 мм (3—4 г). Трубки с обоих концов должны быть закрыты гигроскопической ватой. Кварцевая лампа ПРК-4 или ПРК-2. Камера для опрыскивания пластинок (стеклянный колпак). Камеры для хроматографирования. Батарейный стакан высотой 160 мм и диаметром 95 мм и батарейный стакан высотой 130 мм и диаметром 70 мм. Капилляры для нанесения проб. Колба для отгонки растворителя грушевидная. Крышка стеклянная (рис 1). Пластинка 60X60 мм с приклеенными с трех сторон стеклянными полосками 5X50 мм. Пластинки стеклянные 6X6 и 9X12 см Прибор для отгонки растворителя. Пульверизатор стеклянный. Стаканы батарейные высотой
Рис. 1. Стеклянная крышка (слева) и пластинка с тонким слоем силикагеля (справа). Штриховкой показан слой силикагеля, к которому прикладывают крышку (размеры в миллиметрах).
200 мм и диаметром 140 мм для фракционирования силикагеля. Шкаф сушильный. Шприц или микропипетка вместимостью 10—-20 мкл. Электрическая мельница для размола силикагеля или шаровая мельница. Патрон для фильтра.
Фракционирование силикагеля. Гранулы силикагеля КСК промывают проточной, затем дистиллированной водой, подсушивают при температуре 120 °C и грубо размалывают ручной «"тлиге! а затем в течение 4—5 ч более мелко размалывают шаровой или электрической мельницей.
Суспензию 250 г измельченного силикагеля в 2,5 л дистиллированной воды помещают в батарейный стакан (/г = 200 мм, d= 140 мм). Высота слоя жидкости в стакане должна быть 19 см. Тщательно перемешивают содержимое стакана и оставляют на 16 мин. После этого надосадочную жидкость переливают г-о второй стакан аналогичного размера и доливают воду до уровня 19 см. В этом стакане суспензию отстаивают 30 мин, надосадочиую жидкость сливают в третий стакан и оставляют на 60 мин. Жидкость переливают в четвертый стакан, отстаивают 120 мин. В четвертом стакане оседают частицы диаметром 3—10 мкм. Ilsдоса 'очную жидкость выливают, а осадок собирают и высушивают при 130— 140 °C в течение суток. Полученною таким образом фракцию силикагеля (3—10 мкм) применяют для приготовления пластинок 6X6 см.
Осадок из первых трех стаканов собирают, высушивают, размалывают и используют для седиментации.
Приготовление пластинок. Пластинки 6X6 см тщательно моют раствором хромовой смеси, проточной, затем дистиллированной водой и сушат.
0,3 г силикагеля (3—10 мкм) и 0,015 г гипса растирают в фарфоровой ступке и прибавляют небольшими порциями дистиллированную воду (2,5 мл). Полученную массу осторожно выливают на стеклянную пластинку и оставляют сохнуть до следующего дня на воздухе. Затем пластинку нагревают в сушильном шкафу при 130—140 °C в течение 20 мин. С трех сторон пластинки соскабливают слой силикагеля шириной 0,7—0,8 см. Хранят пластинки в эксикаторе над хлористым кальцием. Перед использованием пластинку нагревают 20 мин при указанной температуре и охлаждают, сразу наносят анализируемую пробу и стандартные растворы.
Пластинки 9X12 см моют, как описано выше. Размолотый на мельнице силикагель просеивают через сито 100 меш., 14 г силикагеля и 1 г гипса смешивают в колбе, приливают 40 мл дистиллированной воды и взбалтывают 20 мин иа аппарате для встряхивания. Приготовленную сорбционную массу равномерно наносят на пять пластинок. Сушат их при комнатной температуре 17—20 ч. Пластинки выдерживают 20 мин в сушильном шкафу при 130— 140 °C. Хранят в эксикаторе над хлористым кальцием.
Отбор проб воздуха рабочей зоны.
1. Для отбора препарата в капельно-жидком или пылеобразном состоянии исследуемый воздух со скоростью до 10 л/мцн протягивают через вложенный в патрон бумажный фильтр. (Вместо бумажных фильтров ,можно использовать гигроскопическою вату, вложенную в аллонж. Скорость протягивания воздуха 2 л/мин.)
2. Для отбора препарата в парообразном состоянии воздух со скоростью 2 л/мин йротягивают через гофрированную трубку с силикагелем. Необходимо протянуть 120—200 л воздуха.
Ход анализа. Вынимают фильтр из патрона (или вату из аллонжа), переносят в стакан и извлекают препарат трижды н-гек-саиом по 5 мл (или диэтиловым эфиром), отжимая последний стеклянной палочкой в цилиндр.
Адсорбированные силикагелем пары ядохимиката извлекают н-гексаном или диэтиловым эфиром. Для этого конец гофрированной трубки, который был обращен к аспиратору во время отбора пробы, соединяют встык с воронкой. Другой конец опускают в цилиндр, содержащий гексановый раствор ядохимиката, извлеченного из ваты или фильтра. Для промывания трубки с силикагелем через воронку наливают по каплям 50—60 мл н-гексана или диэтилового эфира в течение 15—20 мин.
Высыпают силикагель из гофрированной трубки, приливают К нему и-гексан (10—15 мл) и настаивают 30 мин. Декантируют н-гексан в цилиндр, содержащий гексановые экстракты из ваты и силикагеля. Выпаривают н-гексан досуха. Приливают к сухому остатку 0,05—0,1 мл н-гексана. Отбирают микропипеткой аликвотную часть (10—20 мкл) и наносят на пластинку и хроматографируют.
Вода (речная, озерная, питьевая и др.). 100 мл воды экстрагируют н-гексаном трижды (по 50 мл в течение 3 мин каждый раз). Экстракты объединяют И промывают концентрированной серной кислотой порциями по 25 мл до тех пор, пока свежая порция кислоты не перестанет окрашиваться и мутнеть. Слой кислоты отбрасывают, а н-гексап промывают дистиллированной водой 2—3 раза по 100—150 мл до нейтральной реакции. Фильтруют экстракт через воронку, заполненную безводным сернокислым натрием, и упаривают досуха. К охлажденному остатку приливают 0,05—0,1 мл н-гексана. Микропипеткой отбирают аликвотную часть (10—20 мкл) и наносят па пластинку. Проводят хроматографирование.
Почва. К 50—100 г почвы, просушенной на воздухе и просеянной через сито, добавляют 50 мл дистиллированной воды. К увлажненной почве прибавляют 200 мл смеси н-гексана и ацетона (160 мл н-гексана и 4 мл ацетона). Смесь в колбе энергично перемешивают в течение 1 ч на аппарате для встряхивания. После разделения слоев гексановый слой декантируют. Если гексановый слой отделить трудно, жидкость переносят в узкий цилиндр и пипеткой отбирают верхний гексановый слой. К оставшейся почве добавляют смесь н-гексана с ацетоном в количестве, которое указывалось выше. Экстракцию повторяют. Гексановые экстракты объединяют и проводят очистку серной кислоты описанным выше способом.
Картофель, свекла, мясо, ткани внутренних органов животных (сердце, печень, почки, легкие, селезенка, мозг, мышечная ткань). 25—50 г картофеля, свеклы, их ботвы, мяса, тканей внутренних органов крупных животных или 1—2 г тканей мелких животных после измельчения заливают н-гексаном так, чтобы проба была полностью покрыта им. Встряхивают на аппарате в течение 1 ч. После декантации пробу заливают свежей порцией н-гексана и встряхивают еще 1 ч. Декантируют н-гексан. Вытяжки объединяют и очищают концентрированной серной кислотой, как описано выше. Для уменьшения порций кислоты настаивают экстракт над ней в течение 1 ч и периодически встряхивают. Пробы ботвы свеклы и картофеля надо очищать не только кислотой, но и ацетоном. Для этого к упаренному досуха экстракту приливают холодный ацетон и фильтруют через бумажный фильтр. Если требуется, очистку аце-24
тоиом повторяют. Упаривают досуха, приливают 0,05—0,1 мл н-гексана, отбирают микропипеткой аликвотную часть и наносят на пластинку.
Жировая ткань. 2 г пробы измельчают, растирают в фарфоровой ступке и переносят в химическую колбу. Прибавляют 10—15 мл концентрированной серной кислоты и периодически встряхивают содержимое колбы в течение 1 ч. Прибавляют и-гексан (20—30 мл) и снова периодически встряхивают в течение 1 ч. Экстракт декантируют и экстракцию повторяют. Объединенные экстракты очищают концентрированной серной кислотой, как описано для воды.
Кровь, моча. 5 мл крови или 30—50 мл мочи экстрагируют трижды порциями диэтилового эфира по 10, 30 и 50 мл соответственно. Эфирные вытяжки объединяют и фильтруют через слой безводного сернокислого натрия в колбу для отгонки растворителя. Выпаривают на водяной бане досуха. Затем приливают 0,05—0,1 мл и-гексана, отбирают аликвотную часть и наносят на пластинку.
Молоко. К 50 мл молока в делительной воронке приливают 25—30 мл разбавленной серной кислоты (1:1) и взбалтывают. После створаживания молока проводят экстракцию н-гексаном дважды по 50 мл в течение 5 мин каждый раз. Чтобы ускорить разделение слоев, в воронку добавляют несколько капель этилового спирта. Гексановые экстракты объединяют, приливают к ним 20—25 мл концентрированной серной кислоты, осторожно взбалтывают, оставляют воронку. После разделения слоев нижний слой отбрасывают. К гексану приливают свежую порцию кислоты и в течение 1 ч периодически встряхивают. Нижний слой отбрасывают. Дальнейшая очистка возможна двумя способами:
1) серной кислотой до тех пор, пока свежая порция кислоты не перестанет окрашиваться и мутнеть (3—6 порций кислоты);
2) колоночной хроматографией. Стеклянную колонку высотой 200 мм и диаметром 20 мм заполняют силикагелем марки АСК (объем 70 мл). Предварительно пропускают 50 мл н-гексана и прошедший растворитель отбрасывают. На подготовленную таким образом колонку наносят упаренный до 30 мл экстракт молока. После того как экстракт впитается в сорбент, препараты элюируют 90—100 мл н-гексана. Элюат упаривают досуха, приливают 0,05— 0,1 мл н-гексана, отбирают аликвотную часть и наносят на пластинку.
Хроматографирование. Если в анализируемой пробе содержатся совместно полихлорпинен (полихлоркамфен) и ДДТ, экстракт наносят на пластинки 6X6 см. Расстояние точек нанесения от нижнего и бокового края пластинки 8 мм, расстояние между точками нанесения 10 мм. Стандартные растворы наносят микропипеткой (10—20 мкл), пробы наносят капилляром. Пробы и стандартные растворы наносят так, чтобы диаметр пятна не превышал 3 мм. Стандартный раствор (концентрация 500 мкг/мл) наносят в количестве 5—10 мкг. После нанесения проб и стандартного раствора к пластинке прикладывают крышку (см. рис. 1) так, чтобы она не прикасалась к силикагелю. Пластинку и крышку скрепляют резиновым кольцом и опускают в камеру для хроматографирования.
Подвижным растворителем служит смесь н-гексана с метиловым спиртом и аммиаком (10:4:0,3). После поднятия подвижного растворителя на высоту 4 см пластинку с крышкой вынимают из камеры, крышку снимают, пластинку сушат и опрыскивают 0,5%-ным раствором дифениламина и хлористого цинка в ацетоне.
Далее пластинку выдерживают в сушильном шкафу 10—15 мин при 140—150 °C. При наличии полихлорпинена или полихлоркамфена появляется голубовато-зеленое пятно. Пятна ДДТ и ДДЭ окрашиваются в красный цвет, ДДД — в голубой, пятна альдрина и ГХЦГ не окрашиваются. Значения Rf хлорорганических пестицидов приведены в таблице.
Значения Rf хлорорганических пестицидов и чувствительность определения их с помощью азотнокислого серебра и дифениламина с хлористым цинком
Пестицид Rf Чувстви гельность, мкг
азотнокислое серебро дифениламин + хлористый цинк
Полихлорпинен 0,55+0,05 0,5 1,0
Полихлоркамфен 0,55+0,05 0,5 1,0
ДДТ 0,65+0,4 0,2 1,0—2,0
ДДЭ 0,73+0,02 0,5 80,0
ДДД 0,47+0,04 0,5 0,5
Гептахлор 0,68+0,03 0,3 40,0
Альдрин 0,76+0,02 0,3 Не обнаруживает-
ся
ГХЦГ (у-изомер) 0,30+0,02 0,3 То же
Возможно проявление пятен пестицидов с помощью азотнокислого серебра (проявляющий реактив № 1). Хотя чувствительность определения с помощью азотнокислого серебра высокая, более надежная идентификация достигается при использовании дифениламина с хлористым цинком.
Если в пробах не содержатся ДДТ и другие хлорорганические пестициды, возможно хроматографирование экстрактов на пластинках 9X12 см. Микропипеткой на пластинку наносят исследуемую пробу в одну точку так, чтобы диаметр пятна ие превышал 1 см. На расстоянии 2 см от пробы на пластинку наносят стандартный раствор ПХП (ПХК) и проводят хроматографирование в смеси н-гексана с этилацетатом (5:1). После окончания хроматографирования пластинку сушат на воздухе и опрыскивают раствором азотнокислого серебра и аммиака в ацетоне. Пластинку сушат и подвергают ультрафиолетовому облучению до проявления пятен (10—15 мин). При наличии ПХП (ПХК) и других хлороргаииче-ских пестицидов появляются черные пятна на белом фоне.
Rf для ПХП (ПХК) составляет 0,83+0,05, для ДДТ — 0,85+0,05. Определению мешают также другие хлорорганические пестициды. Количественное определение осуществляется путем сравнения площадей пятен пробы и стандартного раствора.
Расчет. Концентрацию препаратов в воздухе (мг/м3) вычисляют по формуле
BV0 ’
где А — количество препарата, найденное сравнением со стандартом, мкг; В — количество исследуемого раствора, взятое для анали
за, мл; С — общий объем исследуемого раствора пробы, мл; Vo — объем исследуемого воздуха, приведенный к нормальным условиям, л.
Расчет содержания препаратов в пробах воды, почвы, продуктов растительного и животного происхождения, крови, моче производится по формуле
АС
где X— содержание препарата, мг/кг или мг/л; А — количество пестицида, найденное путем сравнения со стандартом, мкг; Р — масса пробы, г; В — количество исследуемого раствора, взятое для анализа, мг; С — общий объем исследуемого раствора пробы, мл.
Если проба содержит менее 10 мкг (ПХП, ПХК), -а пластинку количественно наносят весь остаток экстракта после упаривания. Для расчета используют формулу
Определение дилора в растительных пробах, воде и органах теплокровных животных хроматографией в тонком слое *
Принцип метода. Метод основан на экстракции пестицида н-гексаном (бензолом), очистке экстракта от мешающих определению примесей концентрированной серной кислотой, хроматографировании в слое окиси алюминия. В качестве подвижного растворителя используется н-гексан. Количественное определение препарата проводится путем сравнения площадей пятен пробы и стандарта. Чувствительность метода 0,5 мкг в пробе. Полнота определения 90+10%.
Реактивы и растворы. н-Гексан. Серная кислота концентрированная х. ч. Окись алюминия безводная ч. д. а. Гипс медицинский. 25%-ный раствор аммиака. Серебро азотнокислое. Проявитель (0,85 г азотнокислого серебра, 2,5 мл аммиака и 97 мл дважды дистиллированной воды). Стандартный раствор дилора (100 мкг/мл).
Приборы и посуда. Конические колбы вместимостью 50—700 мл. Воронки. Делительные воронки на 200—500 мл. Ступка с пестиком. Аппарат для встряхивания. Холодильник Либиха со шлифами. Колбы Кляйзена вместимостью 10—30 мл или грушевидная колба для получения экстракта в количестве 0,3—0,5 мл. Пипетки разные (0,1, 1, 5 мл). Хроматографические пластинки, покрытые сорбционной массой, состоящей из 50 г безводной окиси алюминия, 5 г медицинского гипса и 75 мл дважды перегнанной воды. Груши лабораторные для пипеток. Пробирки для центрифугирования конические. Лабораторный штатив. Стеклянные пластинки 9X12 и 12X16 см или «Силуфол» ЧССР. Ртутно-кварцевая лампа.
Ход анализа.
Растительные пробы. 10—50 г растительного образца измельчают, помещают в коническую колбу вместимостью 700 мл и заливают н-гексаном (бензолом) до покрытия. Пестицид экстрагируют
путем взбалтывания в течение часа на аппарате для встряхивания колб. Экстракт фильтруют через складчатый фильтр. Растворитель отгоняют до 0,3—0,5 мл на водяной бане. Остаток переносят количественно в пробирку для центрифугирования и доливают столько же серной концентрированной кислоты. Содержимое пробирки слегка взбалтывают и оставляют до четкого разделения слоев. Верхний слой раствора количественно собирают пипеткой и наносят на хроматографическую пластинку.
Вода. К 0,5 л исследуемой воды в колбе вместимостью 1 л доливают 50—100 мл н-гексаиа и встряхивают на аппарате 30 мин. Гексан тщательно отделяют от воды в делительной воронке (остатки воды мешают определению) и отгоняют на перегонном аппарате до остатка в количестве 0,5 мл.
Остаток, если необходимо, очищают серной кислотой (аналогично растительным пробам).
Органы теплокровных животных. Исследуемые органы взвешиваются полностью. Если органы большие, берут только 100 г, тщательно измельчают ножницами, помещают в ступку и растирают с 5—10 мл н-гексана или бензола. Растертую массу количественно переносят в колбу вместимостью 50—200 мл, заливают растворителем до покрытия и экстрагируют 30 мин на встряхивающем аппарате
Экстракт фильтруют через складчатый фильтр. Далее, как описано для продуктов растительного происхождения.
Хроматографирование. Хроматографическую пластинку с нанесенным экстрактом в вертикальном положении помещают в камеру для хроматографирования (эксикатор) и погружают на 1 см в подвижный растворитель (перегнанный н-гексан). Хроматографирование проводят до поднятия фронта подвижного растворителя на высоту 10 см. Затем пластинки высушивают, опрыскивают проявителем, хроматограммы подвергают ультрафиолетовому облучению до четкого появления стандартных пятен (10—30 мин). Идентификация дилора проводится по величине Rf пятен, равной 0,8.
Количество инсектицида определяют по площади пятна путем сравнения со стандартами.
Расчет. Содержание пестицида вычисляют по формуле
где X — содержание препарата в пробе, мг/кг или мг/л; Л, — содержание препарата в стандартном растворе, мкг; Si—площадь пятна стандартного раствора, мм2; S2 — площадь пятна пробы, мм2; Р — масса или объем исследуемой пробы, г или мл.
Определение тиодана в продуктах растительного происхождения тонкослойной хроматографией и колориметрическим методом *
Тиодан — 1,2,3,4,7,7-гексахлорбицикло-(2,2,1) -гептен-2, илеи-5, 6-диметилсульфит. Светлый порошок с температурой плавления 93— 95 °C. Технический препарат — аморфный порошок коричневого цвета. Тиодан состоит из двух изомеров одинаковой токсичности. Пре
парат нерастворим в воде, растворяется в большинстве органических растворителей.
Принцип метода. Метод основан на извлечении тиодана из растительной пробы петролейным эфиром, очистке экстракта и определении хроматографией в тонком слое или колориметрически. Чувствительность метода при определении в тонком слое 0,1 мг/кг, при колориметрическом определении 0,5 мг/кг яблок и 0,25 мг/кг клубники, земляники, картофеля, томатов и баклажанов. Полнота определения 84—90%.
Реактивы и растворы. 0,025 н. раствор едкого натра в метиловом спирте. Реактив стабилен несколько недель. Пиридиновый реактив. К 10 мл 0,025 н. едкого натра добавляют 50 мл водного пиридина (48 мл пиридина, 2 мл воды). Реактив стабилен 6 ч. Готовят ежедневно. Реактив для обнаружения тиодана: 0,5 г азотнокислого серебра растворяют в 5 мл воды и добавляют 7 мл аммиака плотностью 0,9 г/см3. Раствор доводят до 100 мл ацетоном. Петролейный эфир, очищенный серной кислотой и перегнанный над едким натром; температура кипения 40—70 °C. Пиридин. Перед использованием кипятят в течение 1 ч над едким кали (50 г едкого кали на 700 мл пиридина) с обратным холодильником и перегоняют. Сохраняют в темной хорошо закрытой посуде. Силикагель КСК или кремневая кислота для люминофоров. Гипс медицинский. Уголь активированный марки КАД молотый. Стандартный раствор тиодана в петролейном эфире (100 мкг/мл). Если нет чистого препарата, тиодан извлекают бензолом из дуста в аппарате Сокслета и дважды перекристаллизовывают из этилового спирта.
Приборы и посуда. Фотоколориметр ФЭК-56. Источник ультрафиолетового света. Сосуд для хроматографии. Колбы для отгонки растворителя. Стеклянные пластинки 5X20 или 8X16 см. На одну пластинку 5X20 см со слоем КСК требуется 1,5 г сорбента, 0,075 г гипса и 5 мл воды. На пластинку 8X16 см со слоем кремневой кислоты требуется 1 г сорбента, 0,05 г гипса, 4,5 мл воды. Пластинки выдерживают 30 мип на воздухе и активируют 30 мин при 75 °C.
Ход анализа. Тиодан из яблок, картофеля, томатов, баклажанов, клубники экстрагируют при комнатной температуре 17-—19 ч петролейным эфиром. Для анализа берут аликвотную часть из 50 г яблок и 100 г остальных культур. Пробы не измельчают. Эфир декантируют и фильтруют через вату, предварительно промытую ацетоном. Экстракт концентрируют до объема 5—7 мл. Для колориметрического анализа требуется очистка углем (встряхивание с 0,2 г угля в течение 1 мин). Экстракт фильтруют через маленький бумажный фильтр и трижды ополаскивают фильтр петролейным эфиром, затем отгоняют петролейный эфир досуха. Остаток используют для анализа.
Хроматографическое определение. Сухой остаток после отгона петролейного эфира растворяют в небольшом количестве серного эфира, наносят на пластинку и хроматографируют в петролейном эфире. Как только фронт растворителя поднимется на 10 см, пластинку вынимают, хорошо проветривают и обрабатывают аммиачным раствором азотнокислого серебра. После 2—3-минутного проветривания пластинки подвергают ультрафиолетовому облучению. Тиодан проявляется в виде пятен коричневого цвета с Rf 0,1 и 0,35, соответствующих двум изомерам.
Колориметрическое определение. Для построения калибровочного графика в ряд пробирок вносят 0,15—0,1 мл стандартного раствора, доводят объем петролейным эфиром до 10 мл, отгоняют досуха, добавляют 7 мл пиридинового реагента, закрывают пробирки корковыми пробками и кипятят их содержимое 4 мин на водяной бане. Пробирки охлаждают в ледяной бане, раствор переносят в кювету для колориметрирования. Измерение проводят при синем светофильтре против воды. До начала работы необходимо проверить оптическую плотность пиридина. При розовой окраске его необходимо вторично очистить и перегнать.
Закон Ламберта — Беера соблюдается в интервале 15—100 мкг. Определение тиодана в растительной пробе проводится аналогично построению калибровочного графика. После очистки экстракта, отгона петролейного эфира досуха остаток переносят в пробирку, ополаскивая колбу, из которой проводилась перегонка, 3 раза небольшими порциями (2—3 мл) петролейного эфира. Далее поступают, как описано выше.
Колориметрический метод менее чувствителен, чем хроматографический, поэтому при получении пулевых значений необходимо подтвердить наличие или отсутствие тиодана тонкослойной хроматографией.
Расчет. Для определения содержания тиодана используют уравнение
где X — содержание тиодана, мг/кг; А — количество вещества, найденное по калибровочному графику, мкг; Р— масса исследуемого материала, г.
Временные методические указания по определению тиодана и продуктов его превращения в мясе, органах и тканях животных хроматографическими методами
(Утверждены заместителем Главного государственного санитарного врача СССР 24 августа 1983 г.)
Краткая характеристика препарата. Краткая характеристика тиодана и продуктов его превращения (тиодансульфат, тиодаиди-ол, эфиртиодан, гидроксиэфиртиодаи, лактонтиодан) приведена в Методических указаниях по определению тиодана и продуктов его превращения в растительном материале и почве хроматографическими методами.
Методика определения тиодана в мясе, органах и тканях животных.
Принцип метода. Метод основан на хроматографическом определении тиодана в тонком слое пластинок «Силуфол» или газохроматографическом определении его после экстракции препарата из исследуемых образцов смесью этилового спирта с эфиром и хроматографической очистки экстрактов.
Хроматографическое определение проводится на пластинках «Силуфол UV-254», импрегнированиых о-толидином. Подвижным растворителем служит смесь гексана с эфиром и уксусной кислотой.
Места локализации тиодана в виде двух его изомеров обнаруживают после облучения пластинок ультрафиолетовым светом.
Метрологическая характеристика метода. Нижний предел опре-иелеппя тиодана методом ТСХ составляет 1 мкг, 0,1 мг/кг, методом I ЖХ 0,002 мг/кг. Процент определения составляет 78—92%.
Реактивы и растворы. Ацетон х. ч. н-Гексан ч. Бензол х. ч. 1 ||'гролсйный эфир (температура кипения 40—70 °C). Натрий сернокислый безводный ч. Этиловый спирт-ректификат. Этиловый эфир (иля наркоза). о-Толидин ч. 0,1%-ный раствор в ацетоне. Уголь нтивированиый, КАД молотый. Силикагель КСК. Пластинки «Си-,уфол UV-254», ЧССР. Хроматов N-AW-ДМЦС (0,16—0,20 мм) с >%-пым SE-30 (ЧССР). Азот особой чистоты. Стандартный обра-ец тиодана, стандартный раствор тиодана, содержащий 10 мкг/мл препарата в н-гексане.
Приборы и посуда. Вакуумно-ротацнонный испаритель или прибор для отгонки растворителей. Воронин химические диаметром 6 см. Пробирки градуированные со шлифом, 10—15 мл (тип ПККШ). Камера для хроматографирования размером 150X200 мм. Колбы мерные. Колбы на шлифах емкостью 100 и 250 мл. Колбы круглодопные на шлифах емкостью 150, 250, 500 мд. Микропипет-кп. Пипетки или шприцы для нанесения проб. Пипетки емкостью 1, 5, 10 мл. Микрошприцы 10 мкл. Прибор для встряхивания. Стеклянные хроматографические колонки для очистки экстрактов, 150X1,5 мм. Ртутио-кварисвая лампа для облучения УФ-светом. Цилиндры мерные емкостью 25, 50, 100, 250 мл. Чашки выпарпва-тельныс № 3, 4. Аппарат для встряхивания любого типа. Газовый хроматограф (серии «Цвет» или «Газохром»), снабженный детекторами электронного захвата (ДЭЗ) или постоянной скорости рекомбинации (ДПР). Колонки стеклянные для газовой хроматографии длиной 1 м, вн. диаметр 3 мм. Секундомер. Бумажные фильтры.
Подготовка к определению. Приготовление пластинок для хроматографии. Пластинки для хроматографии «Силуфол UV-254» перед использованием импрегпируют о-толидином. Для этого каждую пластинку опускают на 0,5 см в 0,1%-ный раствор о-толидина и ацетоне, налитый в камеру для хроматографирования. После того как фронт растворителя поднимется до верхнего края пластинки, се вынимают и высушивают на воздухе, избегая прямого солнечного света. Такие пластинки готовы к употреблению, их можно хранить в эксикаторе.
Приготовление хроматографических колонок для очистки экстрактов. В нижнюю часть хроматографической колонки помещают около 500 мг обезжиренной органическим растворителем и высушенной ваты и засыпают безводный сернокислый натрий (высота слоя 1 см), а затем активированный уголь (высота слоя 2 см) и перед употреблением промывают небольшим количеством гексана.
Проведение определения.
Мясо и мясопродукты. Мясо, мясопродукты пропускают через мясорубку. 10 г пробы помещают в колбу с притертой пробкой, пестициды экстрагируют дважды смесью этилового спирта с диэтиловым эфиром в'соотношении 3: 1 порциями по 50 мл в течение 1,5 ч при встряхивании. Экстракт фильтруют через воронку с бумажным фильтром; , заполненным на 2/3 безводным сернокислым натрием. Затем растворитель отгоняют, используя вакуумно-ротационный испаритель или прибор для отгонки растворителей. Сухой
остаток растворяют в небольшом количестве смеси этилового спирта с эфиром в соотношении 2: 1 и наносят его на стартовую линию пластинок «Силуфол» (без обработки их о-толидином) в виде полосы. Еще раз смывают сосуд с сухим остатком 0,2—0,3 мл этого растворителя и количественно наносят его на ту же линию пластинки. Пластинку с нанесенной пробой помещают в камеру для хроматографирования, на дно которой предварительно наливают подвижный растворитель н-гексан — диэтиловый эфир - - уксусная кислота в соотношении 85:15:2. Край пластинки может быть погружен в подвижный растворитель не более чем на 0,5 см.
После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Далее пластинку разрезают на небольшие полоски, опускают их в колбу и проводят экстрагирование пестицида н-гексаном, одновременно смывают тонкий слой с пластинок, дважды по 15—20 мл. Полученный экстракт фильтруют, концентрируют до 5—6 мл и пропускают через насадку хроматографической колонки. Тиодан и продукты его превращения элюируют из колонки 30 мл н-гексана, концентрируют раствор под вакуумом до 5—10 мл и используют его для анализа методом ТСХ или гжх.
Хроматографическое определение тиодана в тонком слое. Для хроматографического определения отбирают аликвотную часть экстракта или весь экстракт, предварительно сконцентрировав его до объема 0,3 мл. На хроматографическую пластинку, импретнированную о-толидином, шприцем или пипеткой наносят исследуемую пробу в одну точку так, чтобы диаметр пятна не превышал 2 см. Остаток экстракта в колбе смывают двумя порциями (по 0,2 мл) диэтилового эфира или гексана и наносят в центр первого пятна. Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, содержащие 1 и 5 мкг тиодана, нли другие, близкие к определяемым концентрации.
Пластннкн с нанесенными растворами помещают в камеру для хроматографирования, на дно которой за 30 мин до хроматографирования наливают подвижный растворитель смесь н-гексана с этиловым спиртом в соотношении 3 : 1. После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и спустя несколько минут подвергают действию УФ-света, располагая пластинку па расстоянии 20 см от источника света.
При наличии тиодана на пластинке появляются пятна сине-голубого цвета. Тиодан обнаруживается в виде двух изомеров. Rf а-тиодана 0,15; Rf р-тиодана 0,40.
Содержание препарата в пробе вычисляют по формуле
где X — содержание препарата в пробе, мг/кг; А—количество препарата, найденное в пробе на пластинке, мкг; Р — количество (масса) исследуемой пробы, г.
Проведение г а з о - хромато графичес к о г о анализа. Условия определения: температура испарителя — 220 °C; термостата детектора — 210 °C; термостата колонок—190 °C Скорость газа-носителя—100 мл/мип. Шкала электрометра — 32
0,25х10~’° а. Время удерживания а-тиодана 3,2 мин, (J-тиодана — 4,3 мин.
Количественное определение проводят, сравнивая высоты или площади пиков исследуемых и стандартных растворов. В колонку хроматографа вводят 5 мкл раствора.
Содержание препаратов в исследуемом объеме вычисляют по формуле
Y.P '
где X — количество препарата, мг/кг; Ci—концентрация стандартного раствора, мкг/мл; Г/1 и Н2 — высота пиков стандартного и анализируемого растворов, мм; У, и Y2 — объемы стандартного и анализируемого растворов, введенные в колонку хроматографа, мкл; Уз — объем экстракта пробы для анализа, мл; Р — навеска пробы, г.
Требования безопасности. Соблюдаются требования безопасн э-сти, обычно рекомендуемые для работы с химическими реактивами и хлорорганичсскими пестицидами.
Извлечения из методических указаний
по диагностике, профилактике, лечению отравлений животных и контролю за предотвращением загрязнения кормов и продуктов животноводства
хпорорганическими пестицидами
(Утверждены Главным управлением ветеринарии
Министерства сельского хозяйства СССР 1 августа 1984 г.)
Характеристика группы, основные препараты
К хлорорганическим пестицидам (ХОП) относятся главным образом хлорсодержащие инсектоакарициды и фунгициды, применяемые для защиты зерновых, зернобобовых, технических культур, Ч1лодовых деревьев, виноградников, овощных и полевых культур, ’лесонасаждений от насекомых и клещей, а также предпосевной обработки семян.
Препараты на основе гексахлорциклогексана (гексалин, гекса-талп, ТАП-85, обогащенный ГХЦГ, технический ГХЦГ, 3%-ный активированный креолин) находят широкое применение в животноводстве в основном для профилактики и лечения псороптоза овец.
В связи с особенностями их токсического действия, способностью долго сохраняться в окружающей среде, накапливаться в организме продуктивных животных ХОП применяются с определенными ограничениями.
В настоящее время в сельскохозяйственном производстве нашей страны используются: гамма-изомер гексахлорцпклогексана (ГХЦГ), гексахлорциклогексан технический, гсксахлорбутадиеп, гептахлор, дилор, дихлорэтан, каптан, кельтан, полихлоркамфен, пентахлорпитробензол, тедион, тиодан, шашки «Гамма», фталан, эупареп и др. Кроме того, хлорорганические соединения входят в качестве действующего начала в состав более десяти сложных препаратов, применяемых в растениеводстве как протравители семян
3 Заказ № 1995
33
(гаммагексан, гексатиурам, гамматиурам, киполят— УЧХ, меркур-бензол, пентатиурам, тигам, фентиурам-молибдат и др.).
Хлорорганические пестициды объединяются большой общностью физико-химических свойств н действия на организм животных. Большинство хлорорганических пестицидов в чистом виде представляют собой кристаллические или аморфные порошки, плохо растворимые в воде и хорошо растворимые в неполярных органических растворителях, жирах и маслах.
Характерной особенностью ХОП является их высокая устойчивость к воздействию факторов внешней среды (персистентность). По принятой гигиенической классификации большинство ХОП относится к разряду стойких и очень стойких пестицидов. Эти свойства пестицидов при определенных условиях могут приводить к накоплению их в значительных количествах в объектах внешней среды, в кормах и продуктах растительного и животного происхождения
Большинство хлорорганических пестицидов — среднетоксичные вещества. Однако степень токсичности находится в большой зависимости от вида животных. Из лабораторных животных наиболее чувствительны кошки, затем крысы, мыши, кролики; из сельскохозяйственных животных на первом месте по чувствительности стоят свиньи, затем овцы, лошади, крупный рогатый скот и куры
Многие хлорорганические пестициды обладают выраженной способностью к накоплению в органах и тканях животных (материальная кумуляция), что создает опасность отравления животных ХОП при поступлении их в организм длительное время в малых дозах.
Длительно задерживаясь (до 3—4 мес) в организме, ХОП медленно выделяются в неизмененном виде или в форме различных метаболитов в основном через желудочно-кишечный тракт п почки ХОП выделяются с молоком у лактирующих животных и с яйцом у птнц, концентрируясь главным образом в жире модока и в желтке яиц.
В организм животных хлорорганические пестициды попадают главным образом через желудочно-кишечный тракт (с кормом, водой). Овцы, свиньи могут получать пестициды с землей, которую они поедают вместе с кормом на пастбище. Реже они поступают респираторным путем (в виде аэрозолей, пыли, дымов) и через неповрежденную кожу в период обработок против эктопаразитов, во время дезинсекции помещений в присутствии животных. Особенно легко проникают через кожные покровы ядохимикаты из растворов, приготовленных на органических растворителях
Наряду с главными источниками поступления ХОП в организм животных, которыми являются растительные и особенно зеленые корма, имеют токсикологическое значение и такие белковые кормовые добавки, как рыбная и костная мука, часто содержащие значительные количества пестицидов.
Основными условиями, приводящими к отравлению животных, являются: неправильное и бесконтрольное применение ХОП, нарушение существующих регламентов по хранению, транспортировке и использованию в растениеводстве и животноводстве, отсутствие контроля за уровнем загрязнения ХОП кормов и воды
Гексахлорциклогексан (ГХЦГ, бензогексахлорид, вер-мсксан, гаммексан, гексахлоран, гексатокс, пультокс, скабикомбин,
iikyiaii и др.) — 1,2,3,4,5,6-гексахлорциклогексан. СвН6С16. М. м.1 290,8. 1сх1шчсскнй препарат — кристаллический порошок светло-серого iiiii'iа с неприятным запахом плесени. В воде практически нерастворим, хорошо растворяется в органических растворителях. Устойчив но внешней среде, долго сохраняется в почве (3—4 г.), при высокой температуре легко возгоняется. Устойчив к кислотам, щелочами ра 1,пагается с образованием трихлорбензола.
Технический гексахлоран является смесью восьми стереоизомеров гексахлорциклогсксапа. Инсектицидную активность препара-ia обусловливает гамма-изомер ГХЦГ, содержащийся в количестве 10 13%.
Остальные изомеры ГХЦГ (альфа-, бета-, дельта-, эпсилон-, составляющие соответственно 55—70, 5—14, 6—8 и 3—4% препарата, п зета-, эта-, тета-изомеры, входящие в препарат в следовых количествах) практически нс токсичны для большинства насекомых п грибов.
Выпускается в форме технического препарата (с содержанием гамма-изомера до 18%), 12%-ного дуста и 25%-пого порошка на фосмуке.
Применяется как инсектицид комплексного действия против вредителей овощных, плодовых, зерновых и технических культур, лесов, против почвообитающих насекомых и для дезинсекции помещений.
Выпас скота запрещается в течение 30 дней после применения 12%-кого дуста ГХЦГ против саранчи, в течение 75 дней запрещается скармливать скоту ботву сахарной свеклы после ее обра-Снцки Запрещается: скармливать скоту траву из междурядий при обработке садов, гузепаю и листья хлопчатника после опыливания, а также выпас скота после опыливания леса и лесополос.
В ветеринарной практике используют технический гексахлоран и обогащенный (содержащий 50—90% гамма-изомера) для борьбы <• псороптозом овец и коз в форме 0,03% водной эмульсии (купание в ваннах). Прн этом убой животных на мясо разрешается производить только через 60 дней после их обработки.
В настоящее время ГХЦГ является основным акарицидным । рецством борьбы с чесоточными заболеваниями овец, регламентированным ветеринарным законодательством. Применение гексахлорана путем опрыскиваний, аэрозолей для лечения животных запрещается. Препарат придает сельскохозяйственной продукции, а (ниже мясу, жнру животных и яйцам птиц специфический неприятным jaiiax и привкус.
Зехнический гексахлоран является пестицидом, среднетоксич-иым для животных ЛД50 для мышей и крыс 400—500 мг/кг. Наиболее чувствительны к нему лошади: минимальной смертельной по loii ГХЦГ для них является 100 мг/кг массы. Аналогичная доза крупного рогатого скота равна 200 мг/кг. Токсическая доза овец 250 мг/кг, а доза 500 мг/кг является для них смертель-Токсическая доза для свиней 500—600 мг/кг, кролики поги-। от дозы 600—700, куры переносят дозу 250, а собаки — мг/кг массы.
Очень чувствителен к 1ексахлорану молодняк всех видов жи- по особенно телята. Препарат обладает выраженным ку-
1 Здесь и далее — молекулярная масса.
мулятивпым действием (КкУМ=1), а также кожно-резорбтивным и местным раздражающим действием (особенно его масляные растворы). В организме гексахлоран длительно (до 3—8 мес) задерживается, особенно в жировой ткани, печени, почках, железах внутренней секреции и центральной нервной системе. Препарат проникает через плацентарный барьер и накапливается в органах и тканях плода. Способен выделяться из организма с молоком животных и с яйцом птиц.
Препарат токсичен для рыб, пчел и полезных насекомых. Запрещается использовать его в санитарной зоне вокруг рыбохозяйственных водоемов, в воде которых наличие ГХЦГ не допускается.
Максимально допустимые уровни (МДУ) содержания ГХЦГ (сумма изомеров) в продуктах животного происхождения: молоко — 0,05 мг/кг, мясо, яйца — 0,1, масло, жир, рыба — 0,2 мг/кг. ДОК в кормах для молочного скота и яйценоской птицы — 0,05 и для откормочных животных и птицы — 0,2 мг/кг корма.
Г амма-изомер ГХЦГ (агрпзерт, билтокс, гаматокс, га-мексан, гортекс, изотокс, линдан, нексол и др.)—1,2,3,4,5,6-гекса-хлорциклогексан гамма-изомер. СеНбС16. М. м. 280,8. Бесцветное кристаллическое вещество без запаха. Плохо растворим в воде. Растворяется в жирах, жирных маслах и в большинстве органических растворителей, слабо — в спирте и керосине. Летуч. Устойчив во внешней среде и при нагревании, кислотоустойчив. При действии щелочей разрушается с образованием трихлорбензола и хлористого водорода. В почве в течение года его содержание снижается на 40—80% от внесенного количества. Активный иисектоакарицид.
Выпускается в форме 50%-ного смачивающегося порошка, 90%-ного технического препарата, 2 и 4%-ных гранулированных препаратов и 16%-ного концентрата минерально-масляной эмульсии (ГГММЭ), а также в виде дымовых шашек «Гамма».
Широко применяется в сельском хозяйстве против комплекса вредителей на плодовых, овощных, полевых и технических культурах, в лесах, для сухого протравливания семян и обработки зернохранилищ и складов. Выпас скота и кошение трав разрешается не раньше чем через 30 дней после его применения (2—4%-ные грануляты) для обработки почв. При обработке картофеля, хлопчатника и сахарной свеклы 16%-ной минерально-масляной эмульсией этот срок увеличивается до 75 дней, при обработке садов, гороха, люцерны — 60 дней, а после аэрозольной обработки лесополос 90%-ным техническим гамма-ГХЦГ — выпас скота запрещается.
Гамма-изомер ГХЦГ является высокотоксичным соединением для теплокровных животных. ЛДд0 для белых мышей 86 мг/кг, для морских свинок— 100—127, для крыс 125—230 мг/кг массы. Кролики гибнут от 60—200 мг/кг, собаки — от 40—200, свиньи — от 400—500, куры от 250—350 мг/кг. Молодняк животных более чувствителен к препарату: телята погибают от дозы 5—10 мг/кг, подсвинки — от 100—400 мг/кг массы. Отмечается значительная индивидуальная чувствительность к гамма-изомеру ГХЦГ. Обладает выраженной кожно-резорбтивной токсичностью, особенно при попадании на кожу в виде масляных растворов или в органических растворителях. Оказывает местное раздражающее действие на кожу и слизистые оболочки.
Кумулятивное действие у гамма-изомера ГХЦГ выражено слабо (Кн5м более 10). Сравнительно недолго задерживается в жире 36
ti цругнх тканях организма. Быстро выводится через почки и ки-iiii-iiiufi тракт. Способен выделяться с молоком животных и яйцом in пн I амма-изомер ГХЦГ очень токсичен для пчел. СКя> Для рыб 0.03 -0,06 мг/л.
В ветеринарной практике гамма-изомер ГХЦГ широко ис-полыуют как наиболее эффективное средство борьбы с псоропто-нм овец и коз в виде активированного креолина, гексалина, гек-< палка, ТАП-85, гамма-изомера гексахлорана минерально-масляной ииульспи (ГГММЭ) и дымовых шашек Г-17.
3%-«ый активированный креолин — препарат жидкой конси-। 1СПЦШ1, темно-коричневого цвета, содержащий 54—56% легкосред-iieii фракции каменноугольного масла, 25% мыльного эмульгатора и 3% гамма-изомера ГХЦГ.
Применяют в купонных ваннах в форме 0,03 %-ной водной V цьсии (по гамма-изомеру ГХЦГ).
Убой животных разрешается через 60 дней после обработки, прощается обрабатывать дойных животных.
Гексалин (6% э. к. гамма-изомера ГХЦГ)—маслянистая жид-। осн. черно-бурого цвета с запахом креолина. Содержит 6% гамма изомера ГХЦГ, растворенного в смесн каменноугольных обес-фсноленных масел легкосредпей фракции и эмульгатора.
Применяют в купонных ваннах в форме 0,03%-ной (по гамма-|| юмору ГХЦГ) водной эмульсии
Убой животных па мясо, подвергнутых купанию в эмульсии tcKi-алипа, разрешается не ранее чем через 40 дней после обработки Запрещается обрабатывать дойных животных.
Гексаталп (6% э. к. гамма-изомера ГХЦГ на таловом пеке) — маслянистая жидкость темно-коричневого цвета с запахом креолина Содержит 6% гамма-изомера ГХЦГ, растворенного в смеси каменноугольного масла и эмульгатора.
Применяют в купонных ваннах в форме 0,03%-ной водной •мульсии (по гамма-изомеру).
Убой животных на мясо разрешается через 60 дней после обработки. Запрещается обрабатывать дойных животных.
Гамма-изомера гексахлорана минерально-масляная эмульсия (ГГММЭ)— маслянистая жидкость консистенции густых сливок, «еро-желтого цвета, со слабым специфическим запахом Содержит 16% гамма-изомера ГХЦГ, 15% индустриального масла, до 35% полимеров, 4—6% сульфидно-спиртовой барды и около 33% воды. Имеет кислую реакцию.
Применяют в купонных ваннах в форме 0,025—0,03%-ной (гю ।амма-пзомеру) водной эмульсии. Убой животных на мясо разрешайся через 40 дней после их обработки. Молоко от овцематок ра (решается использовать в пищу через 4 сут после купания.
'ГАП-85 (с 3 или 6% гамма-изомера ГХЦГ)—твердый акарицидный препарат консистенции полутвердого мыла, темно-коричне-шио цвета, со специфическим запахом крезола Состоит из мыла, фенольных креолиновых масел и содержит 3 или 6% гамма-изо-м.-рл ГХЦГ.
Применяют в купонных ваннах в форме 0,025—0,03%-ной подпой эмульсии. Убой животных на мясо разрешается через 1.0 диен после обработки. Запрещается обрабатывать дойный скот.
Дымовые шашки Г-17. Применяют для защиты крупного роки ш о скота, лошадей и оленей обработкой загонов и пастбищ про
тив мошек, мокрецов, комаров путем задымления с нормами расхода 1—3 шашки на 1 га. При горении шашек содержащийся в них гамма-изомер ГХЦГ переходит в аэрозольное состояние.
Выпас скота не допускается в течение 30 дней после обработки.
Применение гексахлоран-креолиновых эмульсий для обработки крупного рогатого скота запрещено вследствие их высокой токсичности и выделения ГХЦГ с молоком.
Максимально допустимые уровни (МДУ) содержания гамма-изомера ГХЦГ (линдана) в продуктах животного происхождения: молоко — 0,05 мг/кг, молочные продукты—1,25 (в пересчете на жир), мясо, яйца-—0,1, масло сливочное, жир, рыба — 0,2 мг/кг.
Комбинированные препараты на основе г е к -сахлорциклогексана. Технический 90%-ный препарат гзм-ма-изомера ГХЦГ и линдан часто являются составной частью сложных протравителей семян комплексного действия, к которым относятся гаммагексан, гамматиурам, тигам, фентиурам и фенти-урам-молибдат.
Гаммагексан (гексахлорбензол+гамма-изомер ГХЦГ). Механическая смесь, содержащая 30% гексахлорбензола (ГХБ) и 20% гамма-изомера ГХЦГ. Физико-химические свойства определяются его компонентами.
Выпускается в виде 50%-ного смачивающегося порошка. Применяется как инсектофунгицид для протравливания семян.
Токсичность для теплокровных животных обусловлена его составными частями (см. гамма-изомер ГХЦГ и гексахлорбензол).
Гамматиурам (гамма-изомер ГХЦГ + тирам). Механическая смесь, содержащая гамма-изомер ГХЦГ и ТМТД (тирам) в равных количествах — по 25%. Выпускается в виде 50%-ного смачивающегося порошка. Применяют в качестве инсектофунгицида для протравливания семян.
ТМТД практически нерастворим в воде, умеренно растворим в органических растворителях. Является среднетоксичиым пестЙй,и-дом для теплокровных животных: ЛД50 для белых мышей 1250— 7400 мг/кг, для крыс— 175—1084, для кроликов 210 мг/кг, смертельная доза для овец 225 мг/кг, для свиней — 400 мг/кг массы. Кумулятивные свойства ТМТД выражены значительно. Остаточные количества в пищевых продуктах и в кормах животных не допускаются.
Гамма-изомер ГХЦГ высокотоксичное соединение.
Тигам (тирам + гамма-изомер ГХЦГ). Механическая смесь, содержащая 50% тирама (ТМТД) и 20% гамма-изомера ГХЦГ. Выпускается в виде 70%-ного смачивающегося порошка. Применяют как инсектофунгицид для протравливания семян. Физико-химические свойства, санитарно-гигиенические нормативы и токсичность для теплокровных животных определяются его составными частями. Среднетоксичен для животных, обладает выраженной способностью к накоплению в организме.
Фентиурам (тирам + гамма-нзомер ГХЦГ +трихлорфенолят меди). Механическая смесь, содержащая 40% тирама (ТМТД), 15% гамма-нзомера ГХЦГ и 10% трихлорфенолята меди. Красно-бурый порошок с резким запахом фенола. В воде не растворяется. Выпускается в форме 65%-ного смачивающегося порошка. Применяется как фунгицид, инсектицид и бактерицид для протравливания семян.
Среднетоксичен для теплокровных животных: ЛД50 для мышей
I мг/кг, обладает местнораздражающим действием, способность । накоплению в организме животных выражена умеренно.
Санитарно-гигиенические показатели определяются входящими и препарат составными частями.
Фснтиурам-молибдат. Механическая смесь, содержащая те же питанные части, что и фентиурам, но, кроме того, включает 4% молибдата аммония. Выпускается в форме 65%-ного смачивающеюся порошка. Назначение, санитарно-гигиенические нормы и ток-< ичность для животных аналогичны фентиураму.
Гексахлорбензол (ГХБ, аматин, антикариа, гсксадин, побунт, перхлорбензол, бунткурс)— гексахлорбензол-1,2,3,4,5,6. <',,С16. М. м. 284,8. Кристаллы светло-серого цвета, с неприятным i,шахом. Не растворяется в воде, хорошо растворяется в органических растворителях, устойчив к действию кислот и щелочей.
Выпускается в форме 30%-ного смачивающегося порошка. Применяют для сухого протравливания семян зерновых культур (пшеницы, ржи, гречихи, сои и др.).
Малотоксичен для теплокровных животных: ЛД50 для лабора-юрпых животных 1700—4000 мг/кг. Обладает выраженными кумулятивными свойствами (Ккум=1). Раздражает слизистые оболочки. МДУ в зерне пшеницы 0,01 мг/кг, ПДК в воде 0,05 мг/л. Является составной частью сложных протравителей семян: гамма-гексана (30% гексахлорбензола и 20% гамма-нзомера ГХЦГ), .ексатиурама (30% гексахлорбензола и 50% ТМТД), гексахлор-ьензола+гептахлора (30% гексахлорбепзола и 20% гептахлора), меркурбензола (20% гексахлорбензола и 1% этилмеркурхлорида), юксичность которых для животных определяется входящими в смесь составными частями.
Гексахлорбутадиен (ГХБД, перхлордивипил)—гекса-хлорбутадиен-1,3. С4С16. М. м. 260,7. Тяжелая маслянистая жидкость желтовато-бурого цвета со специфическим скипидарным запахом. В воде почти нерастворим, растворяется в органических растворителях и жирах, летуч. Очень стоек в обычных условиях внешней среды.
Выпускается в форме 94%-ного технического препарата.
Применяют как почвенный фумигант на виноградниках. Остатки препарата сохраняются в почве 2 года. МДУ в винограде 0,01 мг/кг.
Является высокотоксичным пестицидом: ЛД5о для белых мышей 41—90 мг/кг, для крыс 100—165 мг/кг массы. Допустимая концентрация в воде водоемов 0,01 мг/л. Препарат способен проникаю через кожные покровы н оказывать общее токсическое действие, местно на кожу и слизистые оболочки действует раздражающе.
Обладает выраженным кумулятивным действием (Кнум = 2,5) п оказывает эмбриотоксическое действие. Токсичен для пчел и noli mux насекомых.
Гептахлор (велзикол 104, гептазол, гептанал, ГПХ, I 3314) — 1,4,5,6,7,8,8-гептахлор-4,7-эндометилен-бицикло[4.3.0] нопа--iiien-1,5. CioHjCl?- М. м. 373,3. Чистый препарат—белое кристаллины кое вещество. Технический препарат — воскообразное вещество коричневого цвета с камфарным запахом. Не растворяется в воде, хорошо растворяет'ся в маслах и органических растворителях, ус-Ю11ЧПВ в условиях внешней среды. Содержит 65—72% гептахлора и примесь ядовитого летучего гексахлорциклопептадиена. В про-HII се метаболизма в почве и растениях переходит в эпоксид, от-
личающпйся более высокой токсичностью для теплокровных животных.
Выпускается в форме 22%-ного эмульгирующегося концентрата. Применяют его в качестве инсектицида для предпосевной обработки семян всех культур, исключая корнеплоды пищевого и кормового назначения. Длительно (до 3—4 лет) сохраняется в почве.
Высокотоксичнос соединение с выраженным кожпо-резорбтив-ным действием: ЛД50 для белых мышей 82—210 мг/кг, для крыс — 500, для кур 400 мг/кг; минимальная токсическая доза для телят-сосунов 15—25 мг/кг, для овец—25—30, для кроликов 40— 100 мг/кг массы. Оказывает местное раздражающее действие на кожу и слизистые оболочки. Обладает выраженным кумулятивным действием (КнУм = 1). В организме животных накапливается преимущественно в жировой ткани и длительно (более 20 дней) задерживается в ней.
При попадании в водоемы может оказывать токсическое действие на всех гидробионтов. СКзо для рыб 0,008—0,019 мг/л. Высоко-токсичен для полезных насекомых. После опрыскивания растений сохраняет токсичность для пчел в течение 3—4 сут. ЛД50 для пчел 0,53 мкг/особь.
В организме животных гептахлор окисляется и превращается в эпоксид гептахлора, который способен накапливаться в тканях (особенно в жире) и выделяется с молоком животных и яйцами (желтком) птиц.
Содержание гептахлора в пищевых продуктах и в кормах для животных и птицы не допускается.
ДДТ (дихлордифенилтрихлорэтап, пентацид, азотокс, неоцид, дуплексан, пентахлорин и др.)—1,1-ди-(4-хлорфснил)-2,2,2-трихлор-этан. C14H9CI5. М. м. 345,5.
Технический препарат — вещество парафиноподобной консистенции, содержащее 75—76% основного действующего начала. Хорошо растворим в органических растворителях, плохо в воде, термоустойчив, слаболетуч, устойчив во внешней среде (в почве может сохраняться до 12 лет).
Среднетоксичен для животных: ЛД50 масляных растворов для лабораторных животных от 200 до 500 мг/кг, ЛД50 в виде сухого вещества для лошадей более 300 мг/кг, кроликов — 800, собак — 1300, коз 2200 мг/кг. Телята переносят дозу 500 мг/голову, свиньи'—500 мг/кг корма, коровы — 25 г/голову. Обладает выраженными кумулятивными свойствами. Выделяется с молоком животных и яйцами птиц. Токсичен для пчел. СК50 Для рыб от 2,1 до 27 мкг/л.
МДУ ДДТ и его метаболитов в зерне хлебных злаков 0,02 мг/кг, в молоке — 0,05, в мясе и яйцах—-0,1, в рыбе 0,2 мг/кг.
ДОК (сумма изомеров и метаболитов) в кормах для сельскохозяйственных животных и птицы — 0,05 мг/кг.
Применение препаратов ДДТ для обработки пищевых и фуражных сельхозкультур и сельхозживотных запрещено.
Дилор (бета-дигидрогептахлор, БЛ 2487, ГС 9100)—2,4, 5,6,7,8,8-гептахлор-4,7-эндомстилен-бицикло[4.3.0]нонен-5. СюНтС17. М. м. 375,3 Кристаллический порошок кремового цвета, хорошо растворяется в органических растворителях, плохо — в воде (0,014 мг/л).
Выпускается в форме 80%-ного смачивающегося порошка. Применяют в качестве инсектицида для обработки корне- и клуб-40
। ioiiиijx культур, многолетних трав путем опрыскивания. Иногда о ж полыуют в комбинации с фунгицидами (хлорокисью меди, hhihim) и гербицидами (бетанолом, 2М-4Х).
М.ыююксичное соединение: ЛДз0 для белых мышей 1890 мг/кг, hi крыс более 8000 мг/кг, для кур 2 г/кг массы.
(>оладает незначительным кожно-резорбтивным действием и ||ыценной кумуляцией (Ккум = 2,3—3,5). Способен выделяться с oiiiikom животных и яйцами (в желтке) кур. После однократного " /и пня его содержание в желтке снижается на 50% в течение 11 дней. Токсичен для пчел и рыб ПДК в воде рыбохозяпст-HIII.IX водоемов — 0,0005 мг/л.
Лопустимые остаточные количества в кормах для откормочных ПИП..IX и птицы — 0,1 мг/кг, в кормах для продуктивных живот-
14 не допускается.
каптан (акаптан, закаптан, ванцид-89, мелинур, ортоцид, ... 406 и др)—М-трихлорметилтио-1,2,5,6-тетрагидрофталимид. 11(1< I ^OaS М м 300,5 Кристаллическое вещество желтого или I и и и цвета с неприятным специфическим запахом В воде не рас-и1|1пс|ся, хорошо растворяется в органических растворителях, ус-‘iIhiiii к действию кислот, в присутствии щелочей быстро гпдро-11уе । ся.
Выпускают в форме 50%-ного смачивающегося порошка При-И1ИЮ1 в качестве фунгицида для обработки винограда, плодовых I |1ГП1.('В, овощных и ягодных культур.
M.Iлотоксичный пестицид: ЛДзо для крыс 9—12,5 г/кг. Отме-IUII, что препарат слаботоксичен для поросят, свиней и кур. Окании । местное раздражающее действие на слизистые оболочки и оку Не обладает кожно-резорбтивпым действием. Кумулятивные iiiiicnia не выражены Обладает выраженным эмбриотоксическим > йешпем; мутагенного, тератогенного и канцерогенного действия yi ыповлено
Малотоксичен для пчел, которых для профилактики отравления ипшруют на сутки. CKso для рыб 0,25 мг/л ПДК в воде рыбохо-। ih щепных водоемов—0,0006 мг/кг.
Кельтан (ДТМС, дикофол, кетан, хлорэтанол, ФВ-293. / I гше (4-хлорфенил)-2,2,2-трихлорэтанол-1. C14H9CI5O. М м 370,49.
I и 1|побразная вязкая жидкость коричневого цвета, без запаха, , p/н пюряется в воде, растворяется в органических растворителях, । ipушастся щелочами.
Выпускают в форме 20%-ного эмульгирующегося концентрата и м,1>% пого смачивающегося порошка. Применяют в качестве акарп-n’lii для обработки плодовых, овощных, ягодных, цитрусовых и iiui’ieeKHX культур
Является среднетоксичным инсектицидом: ЛД50 для белых мы-|’П 135—430 мг/кг, для крыс 700—900 мг/кг. Обладает выражении кожно-резорбтивным действием и кумулятивными свойствами
1,2) . В организме животных биотрансформируется с обра-
• ипшн'М двух метаболитов. Способен накапливаться в тканях жи- пи,IX (главным образом в жире) и длительно задерживаться
ц|ц‘ 20 дней) в них. Выделяется с молоком лактирующих животин и яйцами (желтком) птиц. Обладает гонадотоксическим и эм-। ни и 11<спческим действием (в дозах, превышающих ДОК в кор-,х) Препарат нетоксичен для пчел и энтомофагов. СК-.» для нН 0,1 -1,5 мг/л. Остаточные количества в воде рыбохозяйствеп-н< пилоемов не допускаются. ДОК кельтана в кормах для жп-
вотных на откорме — 0,05 мг/кг, для лактнрующнх животных и яйценоской птицы — не допускается.
Кельтан в смеси с каратаном (высокотоксическое соединение) входит в состав комбинированного пестицида акартана (18% кельтана, 9% каратана), обладающего средней токсичностью для теплокровных животных и умеренной — для пчел и рыб. CKso для рыб (по препарату) ~ 0,45 мг/л (при экспозиции 24 ч).
Пентахлорнитробензол (ПХНБ, ботрилекс, брассикол, квинтозан, террахлор, тритизон, фользан и др.) — питропентахлор-бензол. C6C1sNO2. М. м. 295,4. Кристаллический порошок от светлосерого до коричневого цвета с неприятным специфическим запахом. Не растворяется в воде, хорошо — в органических растворителях. Устойчив при хранении. Выпускают в форме 96%-ного технического препарата, 25 и 50%-ного смачивающегося порошка. Входит в состав комбинированного препарата пентатиурама в сочетании с ТМТД. Применяют для сухого протравливания семян зерновых, а также как фунгицид и инсектицид для обработки хлопчатника, овощных и декоративных растений.
Является малотоксичным пестицидом: минимальная смертельная доза для белых мышей 800 мг/кг, ЛДюо 2500 мг/кг, ЛДзо для белых крыс 1650 мг/кг. Обладает выраженными кумулятивными свойствами (Ккум около 1). Накапливается в жире животных, выделяется с молоком. Эмбриотоксического действия у пего не установлено.
Максимально допустимый уровень в зерне 1 мг/кг.
Полихлоркамфен (ПХК, килфен, мелипакс, муртокс, октафен, токсафен, фенатокс, хлорфен) — сложная смесь полихлоркам-фенов и полихлоркамфанов различного строения. CloHioCl8-М. м. 413,8. Воскообразное вещество или вязкая жидкость темно-коричневого цвета. Содержит 67—69% связанного хлора. В воде не растворяется, хорошо растворяется в органических растворителях. Под действием щелочей разлагается с выделением хлористого водорода.
Выпускают в форме 50%-ного эмульгирующегося конце!Играта.
Применяют в качестве инсектоакарицида для обработки овош-ных культур и многолетних трав. Препарат стоек во внешней среде.
Является высокотоксичным соединением: ЛД50 для белых мышей 45—190 мг/кг, для крыс — 60—400, для кроликов — 150, для собак—15—60, для овец и коз — 200, для цыплят — 67,3, для кур — 480, для телят 50 мг/кг массы; минимальная токсическая доза для телят 5 мг/кг, для овец — 25, для коз 50 мг/кг массы.
Обладает выраженным кожно-резорбтивным действием и умеренно раздражающим кожу и слизистые оболочки. В зависимости от дозы и вида животных оказывает выраженное или умеренное кумулятивное действие. Выделяется с молоком лактирующих животных. Высокотоксичен для рыб: СК50 для рыб 0,002—0,009 мг/л. ПДК в воде рыбохозяйственных водоемов не допускается. Малотоксичен для пчел и энтомофагов, можно применять на семенных посевах трав во время цветения.
Содержание остаточных количеств полихлоркамфена в продуктах животного происхождения не допускается.
Запрещается использовать на корм животным траву, ботву картофеля и сахарной свеклы ранее чем через 2,5 мес после применения пестицида.
Содержание полихлоркамфена в кормах для лактирующих жи-...... п яйценоской птицы не допускается, для животных и птицы >1 шкорме — допускается 0,25 мг/кг корма.
Полихлорпинен (ПХП, стробан, хлортен) — хлорировании смесь терпенов. CioH)t>Clg. М. м. 413,8. Вязкая маслянистая мас-.1 желто-корнчневого цвета со скипидарным запахом. Содержит 'И 69% хлора. В воде не растворяется, хорошо растворяется в ор-, пнпческих растворителях, обладает средней летучестью, разруша-' leu щелочами. Стоек во внешней среде. Выпускают в форм? >Ti% пого эмульгирующегося концентрата и 20%-ного масляного piirniopa. Применяют в качестве акарицида и инсектицида для об-||.|но'1ки Овощных, зернобобовых культур н семенных трав.
Среднетоксичен для животных: ЛД50 для белых мышей 240— I'.’O мг/кг, для кроликов—417, для кошек и собак — 310—317, для । циней— 384, для овец и крупного рогатого скота—234—267, для к\р 640 мг/кг массы.
Обладает выраженной кожно-резорбтнвной токсичностью и местным раздражающим действием. Способен оказывать умеренное кумулятивное действие. При поступлении в организм животных распределяется по всем органам и тканям, задерживаясь в них in 30 дней. Проникает через плацентарный барьер. Выделяется с молоком лактирующих животных и яйцом птиц. Токсичен для пчел. 1 Кьо для голубого карпа 15 мг/л. Содержание в воде рыбохозяй- i венных водоемов не допускается.
Использование на корм животным ботвы картофеля, гороха, । лхарной свеклы и травы междурядья и силосование разрешается не рапсе чем через 2,5 мес после применения пестицида.
Содержание полихлорпинена в продуктах питания животного происхождения и в кормах для лактирующих животных и яйценоской птицы не допускается, в кормах для откормочных животных м птицы допускается 0,25 мг/кг корма.
СК-9 (SK-9, хлорированный скипидар). Основу препарата (/0%) составляют продукты хлорирования альфа-пиненовой фракции скипидара в смеси с техническим маслом и эмульгаторами. Со-иержит до 54% хлора. Темно-коричневая вязкая жидкость с запасом хвои. Выпускается в форме эмульгирующегося концентрата.
Применяют в ветеринарной практике в качестве активного ин-. смоакарицида в борьбе с иксодовыми, чесоточными, куриными и ичцами и паразитирующими насекомыми путем купания, опрыски-и.тиия, обтирания водной эмульсией препарата в концентрации от 0,!i до 5% (в зависимости от вида животного и паразитирующих насекомых) или в форме мазей (10%-ных) и дуста (5%-ного).
Менее токсичен, чем другие хлорированные терпены. В применяемых концентрациях не оказывает вредного влияния на орга-шим животных.
Тедион (акаритокс, дюфар, онимерт, тетрасульфон, тетради-< >|1>п, тетразул) — 2,4,4,5-тетрахлордифенилсульфон. Ci2H6Cl4O2S. М м. 356,06. Кристаллическое белое вещество. Не растворяется в пипс, хорошо растворим в большинстве органических растворителей, и Hiii'iiiB к действию кислот.
Выпускают в форме 30 и 50%-ного смачивающегося порошка Применяют как акарицид на овощных и цитрусовых культурах, > и in и р и де, люцерне, .хлопчатнике и других растениях
Препарат малотоксичен для теплокровных животных: ЛД50 для ...... мышей и крыс 5000—10000 мг/кг массы. Обладает слабым
местно-раздражающим, кожно-резорбтивным и кумулятивным действием. Безопасен для пчел и других полезных насекомых. СКзо для рыб 0,1 —15 мг/л (при экспозиции 24 ч). Обработку всех культур прекращают за 20 дней до уборки урожая.
В овощах и фруктах допускается содержание остаточных количеств тедиона в количестве 0,7 мг/кг.
Тиодан (мамскс, малике, эндосульфан, эндофен и др.)—1, 2,3,4,7,7,-гексахлорбицикло [2.2.1 ]гептсн-2-плен-5, 6-диметиленсульфит. С9Н6С1бОз8 М. м. 406,9. Мелкокристаллическое вещество коричневого цвета Плохо растворяется в воде, хорошо — в органических растворителях, нелетуч, под воздействием щелочей разрушается. Выпускается в форме 50%-ного смачивающегося порошка и 35%-пого эмульгирующегося концентрата. Применяют в качестве инсектоакарицида для обработки хлопчатника и ягодных культур. Высокотокспчен для теплокровных животных: ЛД50 для белых мышей 10—32 мг/кг, для крыс — 40—100, для кроликов—28,0, для овец — 26,0, для кур 93 мг/кг. Кожпо-резорбтивиое действие выражено сильно. ЛДзо при нанесении на кожу для крыс 35 мг/кг, для кроликов 120 мг/кг. Кумулятивные свойства выражены слабо (КкуМ = 3—6). Препарат довольно быстро выделяется из организма и слабо накапливается в жировой ткани
Токсичен для рыб (СК50 для рыб 0,6—5 мкг/л) и сравнительно малотоксичен для полезных насекомых. Пчелы погибают в случае попадания инсектицида на них во время обработки растений. Время ожидания на хлопчатнике 20 дней.
Остаточные количества тиодана в пищевых продуктах не допускаются.
Фталан (ортофталан, тиофол, фалтан, фолнет)—N-трихлор метилтиофталимид. Cgl-^CbNOgS М. м. 296,6 Белый или кремовый кристаллический порошок, не растворяется в воде, слабо растворяется в органических растворителях, нелетуч.
Выпускают в форме 50%-ного смачивающегося порошка.
Применяют в качестве фунгицида для борьбы с грибковыми заболеваниями плодовых, цитрусовых, овощных, декоративных культур, винограда. Малотоксичен для теплокровных животных (ЛД-,Г для крыс 7000 мг/кг), не проникает через кожные покровы, концентрированные водные растворы обладают местным раздражающим действием Кумулятивные свойства выражены слабо. (Ккум для крыс=13). Не токсичен для пчел (достаточно изоляции на 5—6 ч во время обработки)
При длительном скармливании в малых дозах угнетает росч животных и оказывает эмбриотоксическое действие.
Обработку растений заканчивают за 20 дней до уборки урожая МДУ в растительных продуктах 2 мг/кг.
Эупарен (дихлорфлуанид, байер 47531)—N-(диметиламидосульфо)-М-(фтордихлорметилтио)-анилин. C9H11Q2FN2O2S2. М. м 333,0. Белый кристаллический порошок, практически нерастворим в воде, умеренно растворяется в органических растворителях, раз рушается в щелочной среде. При наличии влаги гидролизуется ( образованием токсического метаболита.
Выпускают в форме 50%-ного смачивающегося порошка. При меняют в качестве фунгицида для обработки винограда и земля ники. Малотоксичен для животных: ЛД50 для белых мышей и кры< 1850—2500 мг/кг, для кроликов около 1000 мг/кг; обладает уме репными кумулятивными свойствами (KKVM более 4), слабым мест
। । im р.ндражающим действием. He оказывает гонадотоксичссксг о, >мГ>|>1101оксичсского и тератогенного действия. ПДК в воде водое-м<>|| бьнового водопользования 0,025 мг/л.
Иля пчел не опасен. Токсичен для рыб. Метаболит эупарена в р.1 ш токсичнее исходного препарата.
Содержание в растительных продуктах не допускается.
Патогенез и токсикокинетика отравления животных хлорорга-пи'Ц’скими пестицидами.
Хлорорганические пестициды (ХОП) являются ядами политроп-н<>1 о действия с преимущественным нарушением функции централь-поп нервной системы и поражением паренхиматозных органов (осо-ikiiho печени). Они проявляют себя в основном как судорожные >< па центрального медуллярного действия, возбуждающие М- н II холинореактивные структуры ЦНС и периферических отделов
Патогенез токсического действия ХОП связан с реакцией де-х ||<>рирования их в тканях организма животных, в результате ко-lopoii образуются свободные радикалы, оказывающие прооксидант-пое влияние Вследствие этого в организме животных значительно ко (растает процесс перекисного окисления липидов В тканях животных происходит интенсивное образование токсических перекисей (Ц111ИДОВ, приводящее к мобилизации и истощению ресурсов естест-кепных антиоксидантов, нарушению проницаемости клеточных мембран, снижению активности тиоловых ферментов. Особенно подвержены воздействию перекисей сульфгидрильные группы белков печени, мозга и надпочечников.
Все это приводит к нарушению раз шчных физиологических и биохимических процессов в организме животных и к возникновению дистрофических процессов в органах п тканях, особенно в печени и мбзге.
Все хлорорганические пестициды являются веществами, спо-(обными к кумуляции. Выраженной кумуляцией обладают: гекса-х чорциклогексан, гексахлорбензол, гексахлорбутадиен, гептахлор, Л.ДТ, пентахлорнитробензол, полихлоркамфен Умеренными кумуля-швными свойствами обладают: днлор, кельтан, полихлорпинен, |унарен. Слабо кумулируют: линдан, каптан, тедион, тиодач, фталан
При поступлении в организм ХОП быстро всасываются п распределяются по всем органам и тканям животных в количествах, швнеящих от дозы поступившего пестицида.
Преимущественна ХОП накапливаются в органах и ткан"х, богатых жирами и зипидами, концентрируясь главным образом во внутреннем жире, в спинном и головном мозге, печени, почках, крови, репродуктивных органах
Содержание ХОП в крови животных отражает уровень их содержания в органах и тканях. Однако при этом следует учитывать, •ио содержание ХОП во внутреннем жире в 1,5—2 раза прсвыша-( I их содержание в крови.
При остром отравлении ХОП обнаруживаются в органах и 1КЯПЯХ животных уже в течение первых 3—6 ч с момента поступ-зипия пестицидов в, организм.
При длительном‘поступлении ХОП в организм животных в малых дозах накопление их в органах и тканях до определенных : о-ппчсств (в зависимости от чувствительности методов) происх^ ч, и 1счепие разного времени, зависящего от количества поступают1 '
пестицида и его кумулятивных свойств. В крови ХОП обнаруживаются с первых дней поступления в организм.
При остром отравлении животных техническим. ГХЦГ он обнаруживается в мышечной ткани крупного рогатого скота до 10— 20 дней, у овец и свиней — до 4—6 лед, сохраняется в жире крупного рогатого скота до 60 дней, овец — до 240, свиней до 180— 240 дней Выделяется с молоком коров и овец в течение 14— 15 дней и более
При длительном поступлении технического ГХЦГ с кормом в организм крупного рогатого скота, с молоком коров выделяется 20—21% пестицида от уровня содержания его в корме; остаточные количества его сохраняются в мышечной ткани до 2 мес, а в жире до 3—6 мес с момента прекращения поступления
При наружной обработке овец и свиней ГХЦГ сохраняется в мышечной ткани, в зависимости от применяемой концентрации, у овец до 1—2 мес, у свиней — до 1,5—2 нед.
При содержании ГХЦГ в околопочечном жире до 2—3 мг/кг кровь животных его не содержит Отсутствие остатков ГХЦГ в жировой ткани гарантирует чистоту печени, почек и мышц в отношении этого пестицида.
При остром отравлении животных гамма-изомером ГХЦГ (линданом) выделение его основного количества из организма животных происходит в первые 10—20 дней, по незначительные его остатки обнаруживаются при этом в органах и тканях кроликов до 30 дней (в жировой ткани до 40—45 дней), свиней до 30 дней (в жировой ткани до 45—60 дней), крупного рогатого скота до 50—60 дней.
При хроническом отравлении линдан полностью выводится из внутренних органов и мышечной ткани кроликов через 30—45 дней (но сохраняется в жире до 2 мес), а с молоком коров его выделение заканчивается к 30-му дню после прекращения поступления в организм животных. В среднем ежедневно с молоком коров выделяется 1,3—5,4% линдана от поступившей дневной дозы
При наружной обработке животных: при опрыскивании ко^бв линдан выделяется с молоком в основном в течение недели, по остатки его обнаруживаются в нем несколько недель, освобождение от линдана органов и тканей полностью заканчивается через 45—50 дней; при опрыскивании свиней эмульсией или суспензией линдан не обнаруживается в жире через 4 и 6 нед соответственно; при купке овец в креолиновой эмульсии содержание линдана в тканях отмечается до 2 мес, в жире он сохраняется до 10—12 нед, при купке овец в ГГММЭ наиболее высокое содержание лнндана отмечается в первые 15 дней и не обнаруживается через 60 дней.
У кур выделяется из организма в течение суток до 7% от поступившего с кормом количества линдана (с яйцом выделяется до 5%) В зависимости от дозы выделение линдана с яйцом продолжается до 8—50 дней
Гексахлорбензол (ГХБ) при длительном поступлении в организм животных быстро накапливается в жировой ткани до уровня, в 10—20 раз превосходящего исходную концентрацию (в зависимости от дозы) Период полувыведения ГХБ из жировой тканн овец составляет 87—98 дней, из жира крупного рогатого скота — 10,5 нед, а из молочного жира — 4—8 нед В организме птиц (куры) ГХБ сохраняется свыше 5 нед после прекращения поступления. С яйцом кур выделяется до 50% поступившего количества ГХБ
Гептахлор и его метаболит эпоксид гептахлора сохраняются и организме (в основном в жировой ткани) при остром отравлении кур и кроликов в течение 3 мес.
При хроническом отравлении гептахлор сохраняется в органах и тканях кур более 3 нед после прекращения его поступления. С, яйцом кур выделяется в течение суток около 15% от поступившего количества гептахлора. У коров при хроническом отравлении в течение суток выделяется с молоком 12—13% гептахлора, который обнаруживается в нем до 60—80 дней после прекращения nori упления пестицида.
Выделение ДДТ с молоком коров прекращается через 2—3 нед после окончания поступления его с кормом и через 3 нед после о инократного опрыскивания 0,5%-ной эмульсией. При обработке коров субтоксическими дозами его выделение с молоком происходит в течение 42—45 дней.
Дилор накапливается преимущественно в жировой ткани животных и сохраняется в ней на протяжении 1—2 мес. Длительно выделяется с молоком лактирующих животных. В яйцах кур его содержание после однократного введения уменьшается на 50% через 8—11 дней. При хроническом отравлении овец выделение дилера продолжается более 30 дней после прекращения поступления.
Кельтан при хроническом отравлении выделяется из ор|аиизма кур и кроликов в течение 21 дня, а из организма овец — более 60 дней после прекращения поступления. Метаболиты пестицида сохраняются в организме еще дольше. Значительные количества кельтана выделяются с молоком.
При остром отравлении полихлоркамфеном (ПХК) он обнаруживается в организме и яйцах кур до 60 дней, в органах и тканях кроликов—до 80—90, подсвинков — до 100—120, овец до 90—120 дней
При длительном поступлении полихлоркамфена в организм животных в малых дозах (1/10 ЛДзо) он обнаруживается в органах и тканях кур до 14—15 дней, кроликов — до 70 дней после прекращения поступления.
При остром отравлении животных полихлорпиненом (ПХП) он обнаруживается в органах и тканях кур до 32 дней, в организме кроликов до 25—30 дней
При хроническом отравлении полихлорпиненом его выделение из организма кроликов в основном заканчивается через 30 дней после прекращения поступления, а из организма свиней через 70 дней. Выделение с молоком свиноматок прекращается через 48—50 дней
При наружной обработке птиц ПХП обнаруживается в их ор-ипах и тканях в течение 30—45 дней. При обработке коров он выделяется с молоком в течение 3—6 дней (в зависимости от концентрации эмульсии).
Тиодан при остром отравлении животных выделяется из орга-шнма кроликов 15, кур 10, овец 30 дней.
Диагностика отравлений животных хлорорганическими пестицидами.
Диагностику отравлений животных хлорорганическими пестицидами проводят комплексно с учетом анамнестических данных, клинической картины, патологоанатомических изменений и резуль-пнов химико-токсикологического анализа кормов, воды и патоло-। цдеского материала.
Дифференциальная диагностика отравлений хлор органическими пестицидами основывается на результатах химиче ского анализа на содержание ХОП в кормах, содержимом желудка и кишечника, в тканях и органах-накопителях (внутренний жир, головной или спинной мозг) павших или выну ж теино убитых и в крови больных животных, а также на результатах проведения бактериологических и вирусологических исследований
При дифференциальной диагностике необходимо учитывать заболевания животных, которые сопровождаются развитием характерного для отравления ХОП симшомокомплекса глубоких функ цпопальных нарушений периферической и центральной нервной системы и судорожно паралитическими явлениями.
Клиническая картина острого отравления животных ХОП сходна с клиникой острого отравления свинцом, барием, мышьяком, фосфидом цинка, мочевиной, ртутноорганическими и фосфорорганическими пестицидами, карбаматами, некоторыми ядовитыми растениями (беленой, дурманом, красавкой, хвойником, вехом ядовитым, омежником, полынью, плевелом, хвощом н др), а также с клиникой некоторых заболеваний обмена веществ (ацидоз, алкалоз, гипомаг-неземия, авитаминоз и ряда инфекционных и паразитарных заболеваний (болезнь Ауески, листериоз, инфекционный энцефаломиелит, бешенство).
Острое отравление животных практически возможно хлорорга-нпческими пестицидами, относящимися к группе высокотоксичных соединений, такими как гептахлор (и его эпоксид), гсксахлорбута диен, дихлорэтан, линдан (гамма-изомер ГХЦГ), полпхлоркамфеи и тиодан, способными оказывать токсическое действие при попадании в организм животных в малых дозах. Однако часто имеет место отравление креолин-гексахлораповой эмульсией при обработке овец и крупного рогатого скота, при выпасе крупного рогатого скота на участках, обработанных против саранчовых техническим ГХЦГ с высокими нормами расхода.
Хроническое отравление животных более вероятно при длительном поступлении в организм ХОП с выраженной или умеренной способностью к материальной кумуляции, это прежде всего гекса-хлорцнклогексан (ГХЦГ), гексахлорбутадиен, гептахлор, кельтан, полихлоркамфен, полпхлорнитробензол, в меньшей степени — дилор, дихлорэтан, полпхлорпинен, эупарен.
Химико-токсикологический анализ кормов, воды, органов и тканей животных на наличие хлорорганических пестицидов проводят официально утвержденными методами.
В органах и тканях животных, павших при отравлении ХОП, уровень минимального содержания пестицидов составляет: поли-хлоркамфена у кур в головном мозге 1,5 мг/кг, в печени 0,5 мг/кг; у овец в головном и спинном мозге 6 мг/кг, печени—11,5, почках— 5,0, внутреннем жире 9 мг/кг; у подсвинков в головном и спинном мозге 2,5 мг/кг, печенн — 2,5, почках—0,9, внутреннем жире 3,5 мг/кг; у телят в печени 2,8 мг/кг, почках — 3,4, мозге 2,6 мг/кг; гексахлорциклогексана у крупного рогатого скота (при тяжелой форме отравления) в головном мозге 10 мг/кг, спинном мозге — 16,0, печени — 6,0, почках 3 мг/кг; полихлорпинена у кроликов (при хронической подострой интоксикации) в печени 10 мг/кг, почках 5 мг/кг; тиодана у кроликов в головном мозге 0,5 мг/кг, печени — 0,9, почках — 0,4, внутреннем жире 3,0 мг/кг; у овец в
> io шипом мозге — 0,3, спинном мозге — 0,6, печени — 0,1, поч-ип 0,2, внутреннем жире 0,9 мг/кг.
Вегеринарно-санитарная оценка мяса животных, содержащего о< । <п очные количества хлорорганических пестицидов.
При остром и хроническом отравлениях хчорорганическими пе-,।пиццами мясо животных при наличии в нем остаточных коли-чк in пестицидов по opi анолептическим и биохимическим показа-Ц'тям почти не отличается oi мяса здоровых животных (кроме i'H'i.icb отравления ГХЦГ и ДДТ).
При поступлении хлорорганических пестицидов в организм животных в малых дозах они могут накапливаться в нем в значи-ii'iii,пы\ количествах, не вызывая клинического проявления инток-< икании Следовательно, отсутствие клинической картины отравления у животных не может служить основанием для использования проектов убоя без предварительного (контрольного) определения остатков хлорорганических пестицидов
Отсутствие остатков хлорорганических пестицидов в жировой 1КЯИП животных (при их убое) гарантирует чистоту печени, почек и мышц в отношении этих пестицидов.
При обнаружении в мышечной ткани и паренхиматозных органах остатков хлорорганических пестицидов, содержание которых и органах и тканях животных не допускается, туши животных вме-i ie с внутренними органами подлежат технической утилизации
Мясо животных, содержащее хлорорганические пестициды в количествах, не превышающих установленный для них максимально допустимый уровень (МДУ) остаточных количеств, и при благоприятных органолептических, биохимических и бактериологических показателях выпускается на общих основаниях. Все внутренние органы, включая желудочно-кишечный тракт, вымя, мозг, направляют на техническую утилизацию.
При содержании в мясе хлорорганических пестицидов в количествах, в 2—4 раза превышающих установленный для них максимально допустимый уровень, оно после проварки может быть допущено только для переработки в мясокостную муку на корм скоту. Все внутренние органы животных подлежат технической утпли lanitu
Партии мяса, содержащие хлорорганические пестициды в количествах, не превышающих две величины установленного для них МДУ, могут быть использованы в качестве подсортировки для из-I отопления колбасных изделий, для приготовления мясных консервов тонкого измельчения (паштетов), стерилизуемых высокими тем-|и ратурамп.
Мясо животных, вынужденно убитых при отравлении хлорор-I иппчсскими пестицидами, в котором не обнаружены остатки пе-11ПЦИДОВ, используется в порядке, предусмотренном действующими Правилами ветеринарного осмотра убойных животных н ветеринар-пн санитарной экспертизы мяса и мясных продуктов.
Санитарную оценку продуктов убоя животных, отравленных х |орорганическими пестицидами (в переносимой дозе), необходимо проводить на основании комплекса результатов органолептических, о la К'рпологических, физико-химических и хпмико-токспкологичсских и, । ледовапий.
Допустимые остаточные количества хлорорганических пестицидов в кормах (ДОК) и в продуктах животноводства (МДУ).
Не спускается содержание остаточных количеств в молоке, 4 in м 1995 49
молочных продуктах, мясе, жире и яйцах гептахлора (эпоксида гептахлора), полихлоркамфена и полихлорпинена.
Максимально допустимый уровень (МДУ) содержания ГХЦГ (сумма изомеров) и гамма-изомера ГХЦГ (линдана) в молоке*— 0,05 мг/кг, в мясе и яйцах он не должен превышать 0,1, в сливочном масле, животном жире и рыбе — 0,2 мг/кг.
Наличие остатков гексахлорбензола в жире животного происхождения, в молоке и молочных продуктах допускается не более 0,5 мг/кг (Рекомендации ВОЗ, 1975).
Максимально допустимый уровень содержания ДДТ и его метаболитов временно установлен для молока 0,05 мг/кг, для мяса и янц — 0,1 и для рыбы 0,2 мг/кг.
Молоко, содержащее хлорорганические пестициды в количествах, превышающих МДУ, может быть переработано на тощий творог, тощий кефир, обезжиренное сухое и сгущенное молоко, а также на кисломолочные продукты смешанного типа брожения (кефир, ацидофильно-дрожжевое молоко, кумыс).
Сливки и сливочное масло, содержащие ХОП выше МДУ, могут быть использованы в кондитерских и других изделиях с таким расчетом, чтобы в готовой продукции остатки их не превышали МДУ, или же на технические цели.
Яйца с наличием хлорорганических пестицидов могут быть использованы в кондитерском производстве с учетом содержания ХОП в продукции в пределах, допустимых МДУ.
Не допускается наличие остаточных количеств в кормах для молочного скота и яйценоской птицы следующих пестицидов: гептахлора, дилора, кельтана, полихлоркамфена, полихлорпинена. Гептахлор не допускается также и в кормах для откормочного скота и птицы.
В кормах для молочного скота н яйценоской птицы допускается содержание остаточных количеств ГХЦГ (суммы изомеров) не более 0,05 мг/кг и ДДТ (сумма изомеров) 0,05 мг/кг.
В кормах для откормочных животных и птицы допускайся содержание остаточных количеств следующих пестицидов: ГХЦГ (сумма изомеров) не более 0,2 мг/кг, ДДТ (сумма изомеров) — 0,05, дилора — 0,1, кельтана — 0,05, полихлоркамфена*—0,25 и полихлорпинена 0,25 мг/кг.
Корма, содержащие допустимые остаточные количества хлорорганических пестицидов, следует исключать из рациона продуктивных животных и птицы каждые 2—3 нед и заменять их кормами, свободными от пестицидов.
Скармливание кормов, содержащих допустимые остаточные количества хлорорганических пестицидов, откормочным животным и птице необходимо прекращать за 30—60 дней до убоя на мясо (в зависимости от пестицида и вида животного).
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Определение амидофоса в молоке и мясе тонкослойной хроматографией *
Амидофос—0-метил-0(2-хлор-4-трет-бутилфенил) -метиламидофос-<]>ат. Синонимы: руэлсн, монтрол, Дауко 152, Дау М-1261.
Химически чистый амидофос — белое кристаллическое вещество <• температурой плавления 62 °C. Относительная молекулярная масса 291,5. Технический продукт—маслянистая, нерастворимая в воле жидкость, хорошо растворяющаяся в большинстве полярных рас-торителей. Температура кипения амидофоса 117—118 °C при 0,01 мм рт. ст.
Принцип метода. Метод основан на экстракции амидофоса из проб, хроматографическом разделении в тонком слое окиси алюминия или силикагеля, гидролизе препарата щелочью и проявлении чиазотированным паранитроанилином. При проявлении хромато-|раммы амидофос обнаруживается в виде розовых пятен с Rf, равным 0,56—0,60. Для количественного определения амидофоса сравнивают интенсивность окраски и размеры пятен со стандартной шкалой. Чувствительность метода 5 мкг в пробе.
Реактивы и растворы. Ацетон х. ч. Петролейный эфир (1кип 40—70 °C) х. ч. Хлороформ х. ч. Бутанол х. ч. Соляная кислота х. ч. Едкий натр х. ч. Паранитроанилин, перекристаллизованный из горячей воды. Амидофос х. ч. Окись алюминия для хроматографии II степени активности. Силикагель КСК- Гипс чистый медицинский.
Реактив I: 1,5 н. раствор едкого натра или едкого кали в смеси метилового и н-бутилового спиртов, взятых в соотношении 1:1.
Реактив И: а) 0,1%-ный раствор паранитроанилина в 0,1 н. растворе соляной кислоты; б) 4%-ный раствор нитрита натрия. Растворы «а» и «б» перед использованием смешивают в соотношении 10:1. Реактив I, а также растворы «а» и «б» готовят заранее и храпят в холодильнике при 4 °C.
Приборы и посуда. Хроматографическая камера (герметично закрывающийся стеклянный сосуд). Микроизмельчитель тканей РТ-2. Центрифуга (8 тыс. об/мин). Стеклянный или металлический валик с ограничительными кольцами для нанесения слоя окиси алюминия с гипсом толщиной 0,2 мм. Станок для нанесения сорбента па пластинки. Фарфоровая ступка и пестик. Сита 100 и 150 меш. Стеклянные пластинки 13X18 см или другого размера или «Силуфол» ЧССР. Бюксы на 100 мл. Колбы иа 150 и 500 мл. Делительные воронки на 100—200 мл. Микропипетки. Бородки конические. I (ульверизаторы.
Приготовление' .пластинок. Для приготовления пластинок 90 г окиси алюминия II степени активности тщательно смешивают с Юг гипса и просеивают через сито 150 меш. На матовую поверхность ил.,станки насыпают сорбент и разравнивают валиком, чтобы по
лучился слой толщиной 0,2 мм. Для закрепления нанесенного слоя пластинки выдерживают 1 мин над водяным паром. Затем пластинки опрыскивают из пульверизатора дистиллированной водой до полного насыщения сорбента, что отчетливо видно по образованию глянца. Пластинки сушат 20 ч в горизонтальном положении при комнатной температуре (20—22 °C).
Для приготовления хроматографических пластинок со слоем силикагеля и гипса 60 г просеянного через сито 100 меш. силикагеля смешивают с 3,5 г гипса В смесь наливают 180 мл дистиллированной воды и тщательно перемешивают. Две чайные ложки гомогенной массы распределяют по поверхности пластинки ровным слоем. Пластинки сушат в горизонтальном положений при комнатной температуре 24 ч.
Ход анализа.
Л1ясо. Пробу массой 20 г заливают 10 мл дистиллированной воды и гомогенизируют в микроизмельчителе 1--2 мин при 1000 об/мин. Гомогенат заливают 20 мл ацетона, оставляют на час при комнатной температуре и центрифугируют 20 мин при 8 тыс об/мин Водно-ацетоновый слой отфильтровывают через смоченный хлороформом бумажный фильтр в делительную воронку, добавляют 3 мл насыщенного раствора хлористого натрия, перемешивают, приливают 50 мл хлороформа и снова перемешивают легким покачиванием воронки После расслоения нижний ацетопо-хлороформный слои сливают и фильтруют через бумажный фильтр в круглотонную колбу или химический стакан вместимостью 150 мл Затем в делительную воронку вносят еще 50 мл хлороформа и снова сливают и фильтруют нижний ацетоно-хлороформный слои. Слитые нижние слои объединяют. В экстракт вносят 10 г безводного сульфата натрия, перемешивают п фильтруют через бумажный фильтр в химический стакан. Экстракт выпаривают досуха на водяной бане или в ротационном испарителе
Молоко. К пробе (20 мл) добавляют 20 мл ацетона. Дальнейший хог экстракции такой же, как и для мяса. После выпарива ния экстракта в сосуд наливают 0,5 мл ацетона, ополаскивают суд и наносят ацетон на хроматографическую пластинку на расстоянии 2 см от нижнего края. Эту процедуру повторяют 3— 4 раза, причем каждый раз ацетон с растворенным в нем остатком препарата наносят в одну точку на хроматографической пластинке
Пластинки помешают для разгонки в хроматографическую камеру с системой ацетон — петролейный эфир или ацетон — н-гексан (1 : 1), предварительно выдержанную в закрытом состоянии с растворителями 60 мин.
После подъема фронта растворителей на высоту 10 см от линии старта пластинку вынимают из камеры и подсушивают 10— 15 мин на воздухе.
Для проявления хроматограммы пластинку опрыскивают из пульверизатора реактивом I до слабого насыщения, подсушивают 10—15 мин на воздухе, подогревают над плиткой или газовой горелкой и через 10—15 мин опрыскивают реактивом II. При наличии амидофоса в пробах на хроматограмме проявляются пятна розового цвета с Rf от 0,56 до 0,60.
Количественное определение амидофоса в исследуемом материале проводят путем сравнения интенсивности окраски и размеров пятен со шкалой стандартов. Для приготовления стандартной шкалы растворы амидофоса в ацетоне 1:20 000, 1:10 000 и 52
I !><)()(), соответствующие 5, 10 и 20 мкг препарата в 0,1 мл рас-inop.i, наносят на хроматографическую пластинку, разгоняют и проявляют. Размеры пятен измеряют или сравнивают визуально.
Расчет. При вычислении количества амидофоса учитывают, 'по и< молока и мяса экстрагируется 50% препарата. Расчет про-ИО/1Я1 ио формуле
2.4-1000 '' р
где X — количество препарата в 1 кг исследуемого продукта, мг/кг; 2 - коэффициент экстракции; А—количество препарата, выделенное из пробы, мкг; Р — масса пробы, г.
Определение амифоса в воде, яблоках и свекле тонкослойной хроматографией *
Химически чистый амифос — О,О-диметил-5-(2-ацетамидоэтил) дигиофосфат. Бесцветные кристаллы с температурой плавления 22—23 °C. Растворяется в большинстве органических растворителей, малорастворим в воде. Технический продукт — светло-коричневая жидкость со слабым неприятным запахом, разрушается сильными щелочами, неустойчив при хранении на свету при температуре выше 40 °C.
Принцип метода. Метод основан на извлечении амифоса из пробы диэтиловым эфиром, очистке экстракта соляной кислотой и хроматографировании в тонком слое силикагеля КСК. Чувствительность метода 5 мкг в пробе.
Реактивы и растворы. Ацетон. Гипс. 0,5 н. раствор гидрата окиси натрия. Диэтиловый эфир. 10%-ный раствор уксусной кислоты. Натрий сернокислый безводный. Силикагель КСК (100 меш.). 0,5 н. раствор соляной кислоты. Стандартный раствор амифоса (100 мкг/мл). В мерной колбе на 100 мл растворяют 0,01 г препарата в ацетоне. Хлороформ. Проявляющие реактивы: 1) 0,5%-ный водно-ацетоновый раствор азотнокислого серебра (одна часть воды п три части ацетона); 2) бромфеноловый реактив (растворяют 0,05 г бромфенолового синего в 10 мл ацетона). Этот раствор доводят водно-ацетоновым раствором азотнокислого серебра до 100 мл.
Приборы и посуда. Воронки делительные вместимостью 100 мл. Камера для хроматографирования. Камера для опрыскивания пластинок. Капилляры для нанесения проб. Колбы для отгонки растворителя. Пипетки на 1—2 мл. Ротационный испаритель ИР-1. Стеклянный пульверизатор. Стеклянные пластинки 9X12 см или «Силуфол» ЧССР. Цилиндры мерные вместимостью 25 и 100 мл.
Приготовление пластинок. 14 г силикагеля и 1 г гипса смешивают в фарфоровой ступке, приливают 40 мл дистиллированной воды. Смесь хорошо перемешивают и наносят на пластинки, которые сушат при комнатной температуре 20—24
Ход анализа.
Вода. 100 мд воды экстрагируют 30 мл хлороформа 10 мин. Экстракт сушат безводным сернокислым натрием и упаривают до небольшого объема.
Яблоки, сахарная свекла. 50 г измельченной пробы яблок или свеклы заливают эфиром до полного покрытия массы, энергично
встряхивают 10 мин. Эфир декантируют, а пробу заливают свежей порцией эфира. Экстракцию повторяют трижды. Экстракты объединяют и фильтруют через слой безводного сернокислого натрия. Осторожно упаривают досуха. К сухому остатку приливают небольшими порциями 0,5 н. раствор соляной кислоты общим объемом 5 мл. Кислотный раствор фильтруют через бумажный фильтр, к фильтрату приливают 0,5 н. раствор гидрата окиси натрия до pH 7 и добавляют 15—20 мл хлороформа. Отделяют водный слой. Хлороформный экстракт сушат над безводным сернокислым натрием, упаривают до небольшого объема и хроматографируют.
Хроматографирование. Пластинку с нанесенными пробами и стандартными растворами помещают в хроматографическую камеру, куда до начала хроматографирования наливают хлороформ. После поднятия фронта растворителя на 10 см пластинку высушивают на воздухе до удаления подвижного растворителя. Затем пластинку помещают в камеру, в которую наливают смесь растворителей— ацетон с хлороформом (1:2). После развития хроматограмм пластинку сушат на воздухе до полного удаления следов растворителей, опрыскивают пластинку проявляющим реактивом, содержащим бромфеноловый синий и азотнокислое серебро, затем опрыскивают ее 10%-ным раствором уксусной кислоты.
Расчет. Содержание пестицида в пробе вычисляют по формуле
где X — содержание амифоса в пробе, мг/л или мг/кг; А — количество препарата, найденное путем сравнения со стандартами, мкг; Р — масса исследуемого продукта (г) или объем пробы (мл).
Методические указания по определению амифоса в растительном материале и мясе методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 19 октября 1979 г.)
Характеристика препарата. Амифос (ДАЕР, № 7232)—О,О-ди-метил-5-2- (ацетиламино)-этилдитиофосфат. Бесцветное кристаллическое вещество без запаха, слабо растворим в воде, хорошо — в ацетоне, хлороформе, н-гексане и других органических растворителях. Мол. масса 243,28.
Амифос выпускается в виде 40%-ного препарата. Технический препарат — светло-коричневая жидкость со слабым запахом. Разрушается щелочами и не устойчив на свету при температуре выше 40 °C.
Амифос применяется как инсекто-акарицид внутрирастительного действия. По токсическим свойствам для насекомых и по эффективности действия этот пестицид близок к рогору и антпо; среднетоксичен для крыс (ЛД5о = 41О—475 мг/кг) и высокотоксичен для сельскохозяйственных животных: для овец ЛД50 = 43 мг/кг и для кур ЛД5о = 29 мг/кг. Кумулятивные свойства слабо выражены.
Амифос рекомендован для борьбы с вредителями плодовых, цитрусовых, ягодников, виноградной лозы, сахарной свеклы, хлоп-54
ч;ппика и других культур. Обработка препаратом прекращается за 20 дней до уборки урожая.
ДОК в пищевых и растительных продуктах — 0,3 мг/кг, ПДК в воздухе рабочей зоны—0,3 мг/м3.
Принцип метода. Метод основан на извлечении амифоса из растительного материала и мяса ацетоном, очистке экстракта путем переведения пестицида в водную фазу, концентрировании экстракта и хроматографировании в тонком слое силикагеля марки КСК в системе растворителей хлороформ — ацетон (1:1). Проявление производят смесью бромфенолового синего и азотнокислого серебра с последующей обработкой хроматограмм 10%-ным раствором уксусной кислоты.
Метрологическая характеристика метода. Нижний предел определения—0,2 мг/кг. Процент определения в пробах, содержащих от 4 до 10 мкг амифоса, в среднем равен 87. Средняя ошибка определения составляет ±5,2. Метод избирателен в присутствии хлорофоса, ДДВФ, антио, фосфамида, бутифоса, фо-залона и фталофоса.
Реактивы и растворы. Ацетон х. ч. н-Гексан х. ч. Хлороформ ч. д. а. Натрий сернокислый безводный ч. д. а. Гипс медицинский. Силикагель марки КСК, просеянный через сито 100 меш. Азотнокислое серебро, х. ч. Бромфеноловый синий (индикатор). Уксусная кислота, 10%-ный водный раствор. Проявляющий реактив: 50 мг бромфенолового синего растворяют в 10 мл ацетона и доводят до 100 мл 1%-ным раствором азотнокислого серебра в водном растворе ацетона (3 ч. ацетона и 1 ч. воды).
Приборы и посуда. Хроматографическая камера (можно использовать эксикатор). Весы аналитические. Делительные воронки на 100 и 150 мл. Пульверизаторы. Шуттель-аппарат. Шприцы медицинские на 1 мл. Фильтры бумажные. Чашки фарфоровые выпарительные на 20 и 100 мл. Баия водяная. Стеклянные пластинки 9X12 или «Силуфол» ЧССР. Конические колбы с притертыми пробками на 150 и 200 мл. Воронки для фильтрования. Градуированные пипетки на 1, 2, 5 и 10 мл. Сушильный шкаф. Фарфоровая ступка. Сито на 100 меш. Мельница для размалывания гранулированного силикагеля. Стеклянные палочки. Камера для обработки хроматограмм. Мерные цилиндры.
Приготовление пластинок с тонким слоем сорбента. Стеклянные пластинки размером 9X12 см моют хромовой смесью, затем водопроводной и дистиллированной водой и сушат в вертикальном положении. Сухие пластинки перед нанесением сорбционной массы протирают спиртом или эфиром. Для приготовления сорбционной массы берут 35 г силикагеля, просеянного через сито 100 меш., 2 г гипса, 90 мл дистиллированной воды и вносят в коническую колбу с притертой пробкой и встряхивают в течение 15 мин. Приготовленную массу в количестве 10 г наносят на пластинку и равномерно распределяют по поверхности.
Пластинки сушат в течение 16—20 ч при комнатной температуре в горизонтальном положении. Готовые пластинки хранят в эксикаторе.
Проведение определения.
Экстракция. Исследуемую пробу кормов (овес, кукуруза, ячмень, картофель, комбикорма и т. д.) и мяса измельчают, отбирают по 20 г, помещают в коническую колбу с притертой пробкой и проводят экстрагирование амифоса ацетоном дважды порциями
по 40 мл, встряхивая по 15 мин. Экстракты отделяют фильтрованием. через бумажный фильтр в фарфоровую выпарительную чашку, затем переливают в делительную воронку и приливают столько же хлороформа. Содержимое энергично встряхивают 1 мин, хлоро-формно-ацетоновую фазу отделяют в выпарительную чашку и концентрируют на водяной бане при 40 °C до 2—3 мл, а затем досуха при комнатной температуре.
Для извлечения амифоса из сухого остатка экстрагирование проводят дистиллированной водой трижды порциями по 5 мл, тщательно растирая остаток стеклянной палочкой.
Экстракты собирают через бумажный фильтр в делительную воронку, объединяют и для извлечения пестицида из водного раствора проводят экстрагирование хлороформом дважды порциями по 15 мл, встряхивая в течение 1 мин. Хлороформные экстракты сливают в фарфоровую чашку через бумажный фильтр с безводным сульфатом натрия, объединяют и выпаривают досуха, как описано выше.
Хроматографирование. Сухой остаток растворяют в 0,2 мл ацетона и с помощью медицинского шприца или микроли-петки наносят на пластинку со слоем сорбента Еще раз смывают чашку 0,2 мл ацетона и наносят в ту же точку.
Для идентификации и количественной оценки пестицида на ту же пластинку наносят стандартный раствор амифоса, содержащий 5—10 мкг действующего вещества. Если в пробе больше 10 мкг, се разводят и берут для анализа соответствующую часть. Затем пластинку помещают в хроматографическую камеру под углом 45° с подвижной системой растворителей хлороформ — ацетон (1 : 1). После подъема фронта подвижной фазы на 10 см от линии старта пластинку вынимают и суша г в вытяжном шкафу до испарения растворителей. Для проявления хроматограммы пластинку обрабатывают из пульверизатора проявляющим реактивом до насыщения слоя сорбента. После этого пластинку помещают на 5—7 мин в сушильный шкаф при температуре 40—50 °C, а затем охлаждают и опрыскивают 10%-ным раствором уксусной кислоты с целью удаяц ления маскирующего темио-синего фона. Амифос проявляется на желтом фоне пластинки в виде темно-синего пятна с Rf = = 0,60+0,02.
Обработка результатов анализа. Количественную оценку пестицида производят путем визуального сравнения интенсивности окраски и величины пятен пробы и стандартного раствора амифоса.
Содержание препарата в пробе (X) в мг/кг рассчитывают по формуле
А
где А — количество амифоса, найденное путем сравнения с пятнами стандартного раствора (мкг); В — масса исследуемой пробы (г).
Определение байтекса в молоке и мясе тонкослойной хроматографией *
Байтекс — О,О-диметил-О- (З-метил-4-метилмеркаптофенил) -тиофосфат. Синонимы: Байер S 1752, Байер 29493, фептпон, тигувон, энтекс, лебайцид. Химически чистый препарат — бесцветное масло 56
.inaxa с температурой кипения 109 °C при 0,01 мм рт. ст. Отдельная молекулярная масса 278. Технический байтекс (95 — ,, цый)—желто-коричневая маслянистая жидкость со слабым-шктерпым запахом, перегоняется при 105 °C и 0,01 мм рт. ст. и гекс хорошо растворяется в метиловом и этиловом спиртах, эфи-ацетоне, многих углеводородах, а также в хлорированных углево-шдах и маслах. В петролейном эфире препарат малорастворим, иоде при комнатной температуре растворимость составляет 54— мг/л.
Принцип метода. Метод основан на экстракции препарата из <) и хроматографировании в тонком слое силикагеля с гипсом, качестве подвижного растворителя используют смесь петролей-о эфира с ацетоном (2 : 1). Препарат проявляют раствором азот-ицелого серебра, которое, взаимодействуя с фенолятами, обра-«ццимися в результате гидролиза байтекса спиртовой щелочью, . г темно-серые пятна на белом фоне.
Количественное определение байтекса проводят путем визуаль-। о сравнения интенсивности окраски и размера пятен со стан-ртной шкалой. Чувствительность метода 3 мкг в пробе, или ' мг/кг.
Реактивы и растворы. Ацетон х. ч. Петролейный эфир х. ч. юроформ х. ч. Уксусная кислота ледяная х. ч. Едкое кали х. ч. иловый спирт 96°-ный (ректификат). Метиловый спирт 96°-ный (ректификат). Бутиловый спирт х. ч. Серебро азотнокислое х. ч. Силикагель КСК (100 меш.). Гипс медицинский. 100%-ный байтекс. 1,5 н. раствор КОН в смеси метилового и бутилового спирта (1:1). 1%-ный водный раствор азотнокислого серебра. Дистиллированная вода.
Приборы и посуда. Хроматографическая камера (стеклянный цилиндрический сосуд с притертой крышкой). Гомогенизатор для и мельчения тканей. Центрифуга (6—8 тыс. об/мин). Термостатиче-। кая водяная баня. Конические колбы на 100 мл. Роторный испаритель. Делительные воронки па 150 мл. Фарфоровые чашки для выпаривания. Пульверизаторы. Мензурки на 50 и 100 мл. Пипетки ра шые. Микропипетки. Фильтры бумажные обеззоленные. Стеклянные пластинки для хроматографии 13X18 см или «Силуфол» ЧССР.
Приготовление пластинок. На обезжиренную пластинку 13X18 см наносят слой сорбента. Для приготовления сорбента бе-IP г 50 г силикагеля и смешивают с 2,5 г гипса. Смесь заливают 150 мл 5,0%-ной уксусной кислоты и тщательно перемешивают. Дне чайных ложки полученной гомогенной массы распределяют ш> поверхности пластинки ровным слоем. Пластинку подсушивают и горизонтальном положении при комнатной температуре 24 ч, пос-'ie чего она готова к употреблению.
Ход анализа.
Мясо. Пробу нежирного мяса массой 10 г заливают 5 мл .шетона и гомогенизируют 2 мин при 1000 об/мин. Гомогенат помешают в коническую колбу. Четыре раза ополаскивают сосуд, в ко-юром измельчали ткань, порциями ацетона по 5 мл, выливают .шетон в колбу с пробой. Смесь гомогената с ацетоном оставляют при комнатной температуре на 30 мин, затем ее центрифугируют 15 мии при 6—8 гыс. об/мин. Жидкую фазу через бумажный фильтр отфильтровывают в делительную воронку, куда затем наминают 10 мл насыщенного раствора поваренной соли и 20 мл
хлороформа. Содержимое воронки хорошо перемешивают. Посте полного разделения слоев нижний слой (смесь ацетона с хлороформом) сливают и фильтруют через бумажный фильтр. В делительную воронку наливают еще 10 мл хлороформа и процедуру повторяют. Для обезвоживания экстракта на поверхность фильтра насыпают 5 г сульфата натрия. Экстракт выпаривают при комнатной температуре под тягой, на водяной бане или в роторном испарителе.
Молоко. К пробе 10 мл добавляют 20 мл ацетона. Дальнейший ход экстракции байтекса такой же, как при экстракции из мяс Сухой остаток растворяют в 0,5 мл ацетона, выпаривают раство) • тель до объема 0,1—0,2 мл и наносят микропипеткой на хрома графическую пластинку. Эту процедуру повторяют 3 раза, нано ацетоновый смыв в одну и ту же точку. Пластинку помещают хроматографическую камеру с системой петролейный эфир — ацетон (2:1) и выдерживают, пока растворитель не поднимется на 10 см. Пластинку вынимают из камеры и высушивают в течение часа па воздухе. Для проявления хроматограммы пластинку опрыскивают из пульверизатора 1,5 н. раствором едкого кали в смеси метилового и бутилового спиртов (1:1), а через 15—20 мин опрыскивают 1%-ным водным раствором азотнокислого серебра. Проявление ускоряется, если после опрыскивания азотнокислым серебром пластинку 2 мин подержать над плиткой или другим источником тепла. Байтекс проявляется в виде темно-серых пятен на белом фоне.
При наличии в пробах байтекса Rf составляет 0,67—0,72. R1 для оксианалога байтекса — 0,24—0,26.
Количественное определение байтекса в исследуемом материале проводят путем сравнения интенсивности окраски и величины пятен со шкалой стандартов. Для приготовления стандартной шкалы готовят растворы байтекса в ацетоне 1: 100 000, 1: 50 000, 1 : 20 000, 1 : 10 000, 1 : 5000. По 0,1 мл каждого разведения наносят на хроматографическую пластинку. После разгонки и проявления получают пятна, соответствующие 1, 2, 5, 10 и 20 мкг байтекса. Пятна измеряют и сравнивают визуально.
Расчет. При вычислении количества байтекса учитывают, что из молока и мяса экстрагируется в среднем 80% препарата. Расчет проводят по формуле
1,25x1-1000
Р
где X — количество препарата в продукте, мг/кг; А — количество препарата, выделенное из пробы, мкг; Р — масса пробы, г; 1,25— коэффициент экстракции.
Энзиматическое агар-диффузное определение фосфорорганических инсектицидов в продуктах животного происхождения *
Принцип метода. В основу метода положен принцип количественного определения антибиотиков, модифицированный Санди для определения остатков фосфорорганических пестицидов в растениях Фосфорорганические пестициды диффундируют в содержащий холинэстеразу слой агара и угнетают фермент. Затем слой с агаром
pnOaiыпают ацетилхолином, при энзиматическом расщеплении корою образуется уксусная кислота. Она изменяет окраску содер-! .ицегося в агаре индикатора — бромтимолового синего. Диаметры и ан площади участков агара с измененной окраской зависят от кониин рации инсектицида в растворе. Сильные ингибиторы холинэсте-l>.i ,и могут быть определены сразу, а слабые (эфиры тио- и дитио-Ф"< форпой кислот)—после активации.
Реактивы и растворы. Хлороформ. Спирт этиловый 96°-ный. Аннон ч. д. а. Агар. Бромтимоловый синий (индикатор). Едкий и 11 р. Нормальная лошадиная сыворотка. Насыщенный водный рас-пюр брома. 0,1 М раствор тиосульфата натрия. Ацетилхолин. Индикаторная бумага (pH 6,6—8,1) или pH-метр. Стандартные рас-ц|цры инсектицидов в этиловом спирте или ацетоне.
Приборы и посуда. Гомогенизатор. Водяная баня. Ножкины, ('к'клянные воронки. Круглодонные колбы на 25, 50, 100 и 1000 мл. Делительные воронки на 100 мл. Бюксы на 25 и 50 мл с пришлифованными крышками. Стеклянные мерные цилиндры разных размеров. Чашки Петри. Пипетки на 0,1; 1, 5 н 10 мл. Пробковые < перла диаметром 8—10 мл.
Ход анализа. Приготовление среды на основе агара. 16 г агара растворяют при постоянном помешивании в 800 мл кипящей дистиллированной воды. Раствор фильтруют через марлевый фильтр, охлаждают до +50°C и определяют pH. Обычно pH колеблется or 5,6 до 7,7 и может постепенно снижаться. Для получения более стабильной реакции в среду добавляют 10%-ный водный раствор едкого натра: при pH 5,6—5,9 добавляют 3 мл, при pH 6,3—6,7 — 0,5 мл, при pH 7,5—7,7 — 0,3 мл. После перемешивания среду ос-’) являют иа 20—24 ч, затем расплавляют путем подогревания и доводят температуру до 50 °C. Добавляют 100 мг бромтимолового синего, растворенного в 2 мл 0,1 н. едкого натра, и 20 мл лошадиной сыворотки, разведенной до 200 мл дистиллированной водой. Измеряют pH среды и доводят до 7,8—8,0. Необходимо все время поддерживать температуру среды на уровне 50 °C. Готовую среду разливают в чашки Петри (примерно 35 мл в чашку), чтобы образовался слой толщиной 5—6 мм.
Экстракция. Пробы тканей массой 20 г гомогенизируют или тщательно измельчают ножницами, заливают 20 мл хлороформа и экстрагируют в течение 2 ч. Экстракт фильтруют через бумажный фильтр, остаток заливают 20 мл хлороформа, перемешивают и фильтруют. Хлороформный экстракт выпаривают в вытяжном шкафу или на ротационном испарителе.
Пробы молока, мочи, крови и других жидких биологических субстратов объемом 20 мл экстрагируют без встряхивания 2 ч в делительной воронке 20 мл хлороформа. Экстракт отделяют, остаток промывают 20 мл хлороформа. Обе порции экстракта объединяют, фильтруют и выпаривают. Остаток растворяют в 1 мл этилового спирта или ацетона.
Активация пестицидов. Производные тио- и дитиофосфорных кислот обладают слабым ингибирующим действием на холинэстеразу in vitro, поэтому путем окисления бромом их переводят в сильно ингибирующие дериваты. Для этого к растворенному в 0,5 мл спирта или ацетона экстракту, содержащему инсектицид, добавляют 0,1 мл насыщеннрго водного раствора брома и оставляют на 30 мин при комнатной температуре. Нейтрализуют излишек брома 0,1 мл 0,1 М раствора тиосульфата натрия, вносят одну каплю
0,05%-ного раствора бромтимол синего, растворенного в 0,1 н. растворе NaOH, и добавляют по каплям 0,1 н. раствор едкого натра до появления синей окраски. Общий объем доводят спиртом (ацетоном) до 1 мл.
Проведение анализа. В затвердевшей среде в центре чашки Петри просверливают корковым сверлом отверстие, извлекают высверленный столбик агарового геля и вносят в лунку 0,1 мл раствора экстракта. Чашки накрывают крышками и оставляют на 18— 20 ч при комнатной температуре или ставят на 6—8 ч в термостат с температурой 38 °C, чтобы инсектицид диффундировал в агар
В аналогичных условиях определяют также ингибирующее действие чистых инсектицидов, которые используются для построения калибровочной кривой
После диффузии в чашки Петри наливают по 15 мл 0,5%-ного водного раствора ацетилхолина. Чашки переносят в гюдяную баню с температурой 40 °C на 30 мин. Зова диффузии инсектицида в агаре проявляется в виде ровных кругов ингенсивно-спнего или сине-зеленого цвета на желтоватом фоне остальной части агара. Диаметры кругов измеряют циркулем и по линейке определяют их размер. Если известно, какой инсектицид находится в исследуемой пробе, то проводят его количественное определение, сопоставляя с калибровочной кривой диаметры пли площади кругов. В остальных случаях arap-диффузным методом выясняют, имеются ли в экс! ракте угнетающие холинэстеразу вещества, и для их идентификации применяют хроматографию или другие методы исследования.
В таблице приведены пределы определения '2б фосфорорганических инсектицидов в продуктах животного происхождения.
Пределы чувствительности определения инсектицидов энзиматическим агар-диффузным методом, мкг в 0,1 мл пробы
Инсектицид । Lcj активации С активацией брой^м
Хлорофос ДДВФ 0,01 — 100 0,001—100 —
Дибром 0,001 — 100 —
Тролен 10—100 0,002—100
Трихлорметафос-3 10—100 0,002—100
Бутонат 0,1 — 100 —
Циодрин 0,001-100 —
Байтекс 10—100 0,01—100
Фосфамид — 10—100
Этоксифос — 0,01—100
Фталофос — 1—100
Корал 1—100 0,002—100
Метили птрофос 10—100 0,01—100
Сумитион — 0,01—100
Дитиоп 10—100 0,002—100
Дурсбан 100 0,002—100
Карбофос — 1—100
Цидеал 100 1—100
Метилапетофос 100 2—100
Амидофос 0,1-100 —
При более высоких, чем указано в таблице, количествах препара-|<>н в экстрактах требуется разведение. Для построения калибровочной кривой диаметры или площади кругов откладывают на графике против логарифмов концентраций или количеств действующих in шести.
При определении пестицидов в продуктах животного происхож-л( пня вносят поправку на полноту извлечения. Хлороформом указанные в таблице пестициды извлекаются из биоматериала в количестве 30—80%, причем из разных тканей экстракция одного и того се препарата также может быть разной. Поскольку метод высо-очувствителен, можно считать, что препараты из биоматериала «сдрагируются на 50%, и соответственно нужно вносить поправку при расчетах.
Количество препарата в органах и тканях животных вычисля-ч г по формуле
20 0004
где X— количество препарата в 1 кг биологического продукта, мг; А — количество препарата в исследуемой пробе, определенное по калибровочному графику, мкг; Р — масса пробы, г; 20 000 — коэффициент.
Определение бутифоса в хлопке-сырце и продуктах его промышленной переработки тонкослойной хроматографией *
Бутифос — 5,8,5-трибутилтритиофосфат. Нерастворим в воде, но растворяется в бензоле, спиртах, хлороформе. Обладает резким неприятным запахом. Относительная молекулярная масса 314,29.
Принцип метода. Метод основан на экстракции органическим растворителем, очистке экстракта и последующем хроматографировании в тонком слое силикагеля КСК. Подвижным растворителем служит смесь н-гексана с ацетоном (4:1). Количественное определение проводят путем сравнения пятен проб и стандартных растворов. Чувствительность метода в хлопке-сырце и волокне хлопчатника 0,1 мг/кг, в шелухе семян и хлопковом шроте 0,5, семенах хлопчатника и хлопковом рафинированном масле 1,0 мг/кг.
Реактивы и растворы. Силикагель КСК. Окись алюминия для хроматографии II степени активности. Окись алюминия, обработанная соляной кислотой. 30—40 г окиси алюминия II степени активности заливают избытком 5 %-ной соляной кислоты и кипятят 3— 4 ч' при непрерывном помешивании. Кислоту сливают, окись алюминия заливают избытком дистиллированной воды и кипятят 10 мин при помешивании. Обработку сорбента водой повторяют несколько раз до получения нейтральной реакции промывной воды. О1деляют декантацией воду, переносят окись алюминия в фарфоровую чашку и прокаливают при помешивании до прекращения выделения «фонтанчиков». Хранят в склянке с притертой пробкой.
Кальций сернокислый безводный ч д. а. Просушивают 18—20 ч и сушильном шкаф% при 160 °C Калий углекислый безводный ч. п-Гексан перегнанный. Ацетон перегнанный. Четыреххлористый углерод. Ацетонитрил ч. Стекловата. Фильтр для тонких осадков (синяя лента).
Проявляющий реактив: 0,05 г бромфенолового синего (индикатор) растворяют в 10 мл ацетона и доводят объем до 100 мл 0,5%-ным раствором азотнокислого серебра в воде и ацетоне (0,5г азотнокислого серебра растворяют в смеси 24 мл ацетона и 72 мл дистиллированной воды).
40%-ный раствор уксусной кислоты. Бутифос, 70%-ный технический препарат (концентрат эмульсии). Для приготовления стандартного раствора бутифоса необходимо очистить технический препарат следующим образом: 200 мл препарата бутифоса пропускают через колонку, заполненную стекловатой на высоту 1 см, 40 г окиси алюминия и обработанную соляной кислотой. Колонку предварительно промывают 40—50 мл четыреххлоркстого углерода. Прошедший через колонку бутифос собирают в колбу на 200 мл с притертой пробкой. При этом получают раствор, содержащий приблизительно 60—68% бутифоса. Концентрацию бутифоса в очищенном растворе определяют стандартным методом (М.РТУ 6-01-11—63) по синему фосфорно-молибдеповому комплексу. Проверочное определение концентрации проводят не реже одного раза в 3 мес. Стандартный раствор с содержанием бутифоса 0,6 мкг/мл готовят каждые 3 дня последовательным разбавлением очищенного раствора бутифоса н-гексаном.
Приборы и посуда. Пластинки для хроматографии 9X12 см или «Силуфол» ЧССР. Шкаф сушильный. Прибор для отгонки растворителя. Камера для хроматографирования. Пульверизаторы стеклянные. Набор пипеток и микропипеток Пробирки вместимостью 1 мл с притертыми пробками. Воронки конические. Воронки делительные вместимостью 250 мл. Колбы конические вместимостью 250 мл с притертыми пробками. Хроматографические колонки. Баня водяная. Сито 100 меш
Приготовление пластинок. 35 г силикагеля смешивают с 2 г сернокислого кальция и тщательно растирают в фарфоровой ступке. Смесь переносят в колбу, приливают 80 мл дистиллированной воды и встряхивают 10—15 мин для образования однородной массы. Полученную массу наносят на пластинку и равномерно распре’ деляют покачиванием пластинки. Сушат пластинки 10—12 ч при комнатной температуре, хранят их в эксикаторе. Перед употреблением пластинки активируют (выдерживают в термостате 10—15 мин при 100 °C).
Ход анализа.
Хлопок-сырец и волокно. Пробу массой 100 г помещают в аппарат Сокслета и экстрагируют четыреххлористым углеродом 7— 8 ч. Полученный экстракт отгоняют до объема 30—40 мл и после охлаждения пропускают через хроматографическую колонку, заполненную на высоту 10 см смесью окиси алюминия с силикагелем (3:1) и промытую 10—15 мл четыреххлористого углерода. После пропуска экстракта колонку промывают тремя порциями растворителя по 10 мл. Элюат собирают в колбу для отгонки растворителя, отгоняют его до объема 1—2 мл и количественно переносят в пробирку вместимостью 10 мл. Упаривают экстракт на водяной бане до объема 1 мл. Наносят по 0,1 мл на пластинку и хроматографируют.
Шелуха семян. 10 г шелухи делят на две равные части и помещают в две колбы вместимостью 250 мл каждая. Экстрагируют каждую часть четыреххлористым углеродом тремя порциями — по 40, 20 и 20 мл в течение 15, 10 и 5 мин соответственно. Полу-
•и'ппыс экстракты от каждой части объединяют и пропускают че-рг I отдельные колонки, заполненные смесью окиси алюминия с < иликагелем (3:1) на высоту 10 см. Предварительно колонку промывают 10—15 мл четыреххлористого углерода. После пропуска ин тракта колонку промывают 30 мл четыреххлористого углерода порциями по 10 мл.
Элюаты от обеих частей пробы собирают в одну перегонную колбу, растворитель отгоняют до 1—2 мл. Содержимое колбы количественно переносят в пробирку и постепенно упаривают до 1 мл. Экстракт наносят на пластинку и хроматографируют вместе со стандартами.
Семена хлопчатника. 10 г семян делят на две равные части, и 1мельчают в фарфоровой ступке и экстрагируют последовательно 15 и 25 мл н-гексана. Полученные экстракты объединяют и пропускают через отдельные колонки (диаметр 12 мм), заполненные окисью алюминия на высоту 15 см и предварительно промытые 10—15 мл н-гексана. После прохождения экстракта колонку промывают тремя порциями по 15 мл ацетонитрила. Элюаты от обеих частей пробы собирают и отгоняют до 1—2 мл. Остаток состоит из растворимой и нерастворимой в ацетонитриле частей Последнюю количественно переносят в мерную пробирку и доводят ее объем до 5 мл н-гексаном. Хроматографируют по 0,1 мл. Параллельно с анализируемым образцом готовят два модельных экстракта, экстракт получают из 2 г исходного образца семян хлопчатника. Перед экстракцией вносят в один образец 10 мкг, а в другой — 5 мкг бутифоса.
Рафинированное хлопковое масло. 30 мл анализируемого масла помещают в делительную воронку и экстрагируют тремя порциями ацетонитрила по 60, 30, 30 мл в течение 20, 10, 10 мин соответственно. Полученные экстракты фильтруют через фильтр для тонких осадков (синяя лента), затем пропускают через колонку, заполненную смесью окиси алюминия с силикагелем (3:1) на высоту 20—25 см и промытую 50 мл ацетонитрила. Экстракт и элюат (30 мл ацетонитрила) собирают в колбу для отгонки растворителя и отгоняют до 1 мл. Содержимое колбы количественно переносят в пробирку на 1 мл.
Параллельно с анализируемым образцом готовят два модельных экстракта. Каждый экстракт получают из 10 мл того же рафинированного масла. Перед экстракцией в один образец масла вносят 30 мкг бутифоса, в другой—15 мкг. Экстракцию проводят тремя порциями ацетонитрила по 20, 10 и 10 мл. Объединенные экстракты фильтруют через фильтр для тонких осадков, затем пропускают через колонку, заполненную на высоту 10 см смесью окиси алюминия с силикагелем (3:1) и промытую 10—15 мл ацетонитрила. Экстракт и элюат (10—15 мл) собирают в колбу для отгонки растворителя и отгоняют до 1 мл, затем количественно переносят в пробирку на 1 мл. Наносят на пластинку и хроматографируют.
Хлопковый шрот. 20 г шрота делят на две равные части и экстрагируют последовательно 30 и 20 мл н-гексапа. Экстракты фильтруют через складчатый фильтр, который затем промывают 10 мл н-гексана. Растворитель отгоняют, пробы собирают в одну пробирку, доводят объем н-гексаном до 1 мл. Параллельно с анализируемым образцом готовят два модельных экстракта. Каждый экстракт получают из 2 г исходного образца хлопкового шрота.
Перед экстракцией в один образец вносят бутифос в количестве 10 мкг, в другой — 5 мкг.
Хроматографирование, На хроматографическую пластинку наносят исследуемую пробу и параллельно стандартные или модельные растворы с известным содержанием бутифоса. После высыхания пятен пластинку помещают в камеру для хроматографирова ния, в которую предварительно наливают смесь н-гексана с ацетоном (4: 1). Смесь готовят ежедневно. После поднятия фронта растворителя на высоту 10—И см пластинку вынимают, выдерживают в вытяжном шкафу 3—5 мин для удаления паров растворителя и опрыскивают раствором проявителя. Через 15—20 мин пластинку опрыскивают раствором уксусной кислоты. На желтом фоне ясно проявляются голубые пятна бутифоса Величина Rf бутифоса 0,45—0,5.
Расчет Содержание бутифоса в анализируемой пробе вычисляют по формуле
АК
Р ’
где X— содержание бутифоса в анализируемой пробе, мг/кг; А — количество бутифоса, найденное путем сравнения со стандартными или модельными растворами бутифоса, мкг; Р — масса анализируй мой пробы, г; К — коэффициент, соответствующий взятой для хроматографирования аликвоте экстракта.
Методика обнаружения бутифоса в молоке, крови и моче животных при диагностике отравлений
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 18 декабря 1975 г.)
Характеристика препарата. Бутифос — трибутилтри гиофосфат. Легко подвижная жидкость коричневого цвета с очень резким неприятным запахом Температура кипения 154 °C Ядохимикат нерастворим в воде, но хорошо растворим во многих органических растворителях. Выпускается промышленностью в виде 70%-ного концентрата эмульсии.
Принцип метода. Метод основан на извлечении бутифоса ацетоном, очистке экстрактов от мешающих анализу коэкстрактивных веществ путем перераспределения в несмешивающихся растворителях и колоночной адсорбции, концентрировании экстрактов и хроматографировании в тонком слое силикагеля в системе растворителей гексан — ацетон (4:1). Для проявления бутифоса применяют бромфеноловый синий в смеси с азотнокислым серебром Чувствительность метода на силикагеле 0,5 мг/кг, а на пластинках «Силуфол»— 0,1 мг/л. Процент определения составляет 854=15.
Реактивы и растворы. Ацетон х. ч. н-Гексан. Окись алюминия для хроматографии II степени активности. Силикагель марки КСК. Гипс. Бутифос — 70%-ный. Бромфеноловый синий (индикатор). Азотнокислое серебро х. ч.
Проявитель: 0,05%-ный раствор бромфеноловою синего в 1% ном водно-ацетоновом растворе азотнокислого серебра (вода — ацетон 1:3).
Приборы и посуда. Хроматографическая камера. Весы анали-тческие Мерные цилиндры с притертой пробкой на 100 мл. Химические воронки Фильтры бумажные. Чашки фарфоровые выпарительные на 20 и 100 мл. Водяная баня Делительные воронки на 150 мл. Шприцы емкостью 1 мл или микропипетки Стеклянные пластинки 9X12 см или «Силуфол» ЧССР. Сушильный шкаф. Фарфоровая ступка. Стеклянные колонки диаметром 15 мм. Стек-т.т«ная вата. Сито на 100 меш. Пульверизаторы Камера для опрыскивания хроматографических пластинок Мельница для размалывания гранулированного силикагеля.
Приготовление пластинок с тонким слоем сорбента. Берут стеклянные пластинки размером 9X12 см, моют хромовой смесью, затем водопроводной и дистиллированной водой и сушат в вертикальном положении при комнатной температуре. Для приготовления сорбционной массы в фарфоровую ступку вносят 35 г силикагеля КСК, просеянною через сито 100 меш, 2 г гипса и тщательно перемешивают, затем переносят в коническую колбу с притертой пробкой. Приливают 90 мл дистиллированной воды и энергично встряхивают в течение 15 мин. Приготовленную массу в количестве 10 г наносят на пластинку и сушат при комнатной температуре в строго горизонтальном положении Готовые пластинки храпят в эксикаторе.
Ход определения. Пробу молока, крови или мочи в объеме 10 мл переносят в мерный цилиндр с притертой пробкой емкостью на 100 мл, приливают 40 мл ацетона, экстрагируют встряхиванием 1—2 мин и скоагулированную массу отделяют фильтрованием через бумажный фильтр, а водно-ацетоновый экстракт собирают в фарфоровую выпарительную чашку и ацетон удаляют выпариванием па водяной бане при температуре 40 °C. Оставшийся водный раствор переливают в делительную воронку, приливают 20 мл н-гек-сана и экстрагируют встряхиванием 1 мин Отделившуюся водную фазу выбрасывают, а гексановый экстракт собирают для очистки на колонке.
Стеклянную колонку диаметром 15 мм заполняют на 15 см окисью алюминия для хроматографии II степени активности и промывают н-гексаном (40 мл), а затем гексановый экстракт вносят в колонку.
Для элюирования бутифоса через колонку пропускают 120 мл н-гексана порциями по 15—20 мл, элюаты собирают, объединяют и выпаривают на водяной бане при температуре 40 °C до 2 мл, а затем при комнатой температуре до 0,1—0,2 мл и наносят шприцем или микропипеткой на пластинку с тонким слоем силикагеля или на пластинку «Силуфол». Фарфоровую чашку, в которой концентрируют элюат, дважды смывают 0,2 мл н-гексана и наносят его в ту же точку на пластинку.
Для идентификации пятен и их количественного определения на ту же пластинку наносят стандартные растворы бутифоса, содержащие предполагаемые количества в пробе Хроматограмму развивают в системе растворителей гексан—-ацетон (4:1) После подъема фронта подвижной фазы на 10 см от линии старта пластинку вынимаюу и сушат при комнатной температуре в вытяжном шкафу до испарения растворителей. Хроматограмму обрабатывают из пульверизатора смесью водно-ацетонового раствора бромфенолового синего и азотнокислого серебра и помещают в сушильный шкаф на 5—7 мин при 50 °C.
5 Заказ № 1995
65
После охлаждения пластинку опрыскивают 10%-ным раствором уксусной или 2%-ным раствором лимонной кислоты для удаления маскирующего фона На желтоватом фоне пластинки с силикагелем бутифос проявляется в виде темно-синего пятна с Rf = = 0,50+0,02, а на пластинке «Силуфол» с Rf = 0,33+0,02.
Количественное определение бутифоса производят путем визуального сравнения интенсивности окрашивания и размера пятен пробы с пятнами стандартных растворов.
Содержание бутифоса в мг/кг рассчитывают по формуле
Где X— количество препарата, мг/кг; А— количество препарата, найденное путем сравнения пятен пробы с пятнами стандартных растворов, мкг; В — объем исследуемой пробы, мл.
Обнаружение бутифоса в биологических субстратах, наличие у больных животных характерной клинической картины интоксикации, снижение уровня ацетилхолинэстеразы крови и выраженные патоморфологические изменения в органах павших и вынужденно убитых животных свидетельствуют об отравлении этим пестицидом
Определение ДДВФ в растительном материале, почве и воде тонкослойной хроматографией *
Принцип метода. Метод заключается в экстракции ДДВФ хлороформом или водно-спиртовым раствором с последующей реэкстракцией хлороформом, очистке экстрактов от сопутствующих веществ при помощи колоночной хроматографии и идентификации препарата методом хроматографии в тонком слое Подвижным растворителем служит смесь бензола с ацетоном (9:1). Проявление пестицида осуществляется последовательной обработкой 2 н. раствором едкого кали в метаноле, 0,05 н. раствором азотнокислого серебра в разбавленной азотной кислоте и ультрафиолетовым о+ лучением пластинок. Минимально детектируемое количество 10 мкг. Чувствительность метода 0,2 мг/кг.
Реактивы и растворы. Этиловый спирт. Хлороформ. Активированный уголь КАД молотый. Натрий сернокислый безводный Силикагель КСК или двуокись кремния для люминофоров Гипс медицинский. 2 н. раствор едкого кали в метаноле 0,05 н. раствор азотнокислого серебра в разбавленной азотной кислоте (0,85 г азотнокислого серебра растворяют в 75 мл воды, добавляют 25 мл концентрированной азотной кислоты плотностью 1,4 г/см3 и 2— 3 капли 30%-ной перекиси водорода).
Приборы и посуда. Аппарат для встряхивания. Баня водяная Воронки химические. Воронки делительные вместимостью 250— 500 мл. Груша резиновая. Мерные цилиндры вместимостью 250 и 100 мл. Микропипетки. Прибор для отгона растворителей под вакуумом. Пипетки разные Пульверизатор. Хроматографические колонки 300X20 мм. Хроматографические пластинки 8X16 см или «Силуфол» ЧССР. Хроматографическая камера. Эксикатор для хранения пластинок. Колбы конические вместимостью 500 мл с пришлифованными пробками.
Приготовление пластинок. 1 г двуокиси кремния или силикагеля КСК и 0,1 г медицинского гипса помещают в коническую колбу, 66
добавляют 3,5—4 мл воды и встряхивают 1 мин. Выливают смесь на пластинку, разравнивая стеклянной палочкой. Сушат пластинку при комнатной температуре 30 мин, затем помещают на 30 мин в сушильный шкаф с температурой 75 °C. Охлаждают и хранят готовые пластинки в эксикаторе над силикагелем.
Ход анализа.
Яблоки, листья, стебли и колосья пшеницы. Среднюю измельченную растительную пробу массой 50—100 г заливают 100—200 мл 20%-ного этилового спирта в колбе вместимостью 500 мл и встряхивают 30 мин. Фильтруют экстракт в делительную воронку. Добавляют 100 мл хлороформа и встряхивают 5 мин. Сливают хлороформный слой в коническую колбу. Водную фазу еще раз экстрагируют 100 мл хлороформа. Сушат объединенный хлороформный экстракт безводным сульфатом натрия.
Почва. 100 г почвы заливают 200 мл хлороформа и периодически помешивают в течение 1 ч. Фильтруют хлороформную вытяжку в коническую колбу. Экстракцию почвы проводят еще 2—3 раза хлороформом. Экстракты объединяют.
Вода. 100 мл воды помещают в делительную воронку, экстрагируют хлороформом 2—3 раза, высушивают над безводным сернокислым натрием. Хлороформ упаривают до 10—15 мл на водяной бане при температуре 50 °C под вакуумом, создаваемым водоструйным насосом. Остаток переносят на хроматографическую колонку, состоящую из слоя безводного сульфата натрия (2 см), 2 г активированного угля КАД молотого и еще одного слоя (2 см) сульфата натрия. Предварительно через колонку пропускают 20—30 мл хлороформа для уменьшения адсорбционной емкости угля. Из колонки ДДВФ элюируют 100 мл хлороформа. Упаривают хлороформ под небольшим вакуумом до 0,3—0,5 мл. Остаток количественно наносят на пластинку с силикагелем КСК или с двуокисью кремния для люминофоров, параллельно наносят стандартные растворы ДДВФ.
Хроматографирование. Пластинку погружают в сосуд (хроматографическую камеру) с подвижным растворителем (смесь бензола с ацетоном 9:1). После того как подвижный растворитель поднимется на высоту 10 см, пластинку вынимают, дают растворителю испариться. Пластинку равномерно опрыскивают из пульверизатора раствором 2 н. едкого кали в метаноле и помещают на 20 мин в сушильный шкаф с температурой ПО—120 °C. Затем пластинку охлаждают и опрыскивают 0,05 н раствором азотнокислого серебра в разбавленной азотной кислоте.
Через 0,5 ч после подсыхания пластинки помещают под ртутно-кварцевую лампу и облучают 10 мин. ДДВФ проявляется в виде коричневато-фиолетового пятна на белом фоне. Не следует допускать избытка раствора азотнокислого серебра. При чрезмерном количестве азотной кислоты поверхность пластинки становится рыхлой, а это затрудняет обнаружение пятен ДДВФ. Величина Rf ДДВФ 0,57—0,66. Количество препарата на хроматограммах определяют путем сравнения интенсивности окраски и размеров пятен проб и стандартных растворов.
Расчет. Содержание ДДВФ в анализируемой пробе вычисляют по формуле
где X— содержание ДДВФ в пробе, мг/кг или мг/л; А — количество ДДВФ, найденное путем сравнения со стандартными растворами, мкг; Р — масса или объем пробы, г или мл.
Методические указания по определению ДДВФ в тканях животных методом хроматографии в тонком слое
(Утверждены заместителем Главного государственного санитарного врача СССР 18 ноября 1977 г.)
Краткая характеристика препарата указана ранее.
Методика определения ДДВФ в тканях животных. Основные положения. Принцип метода. Метод основан на экстракции ДДВФ из тканей животных ацетоном или его 80%-ным водным раствором, очистке с использованием разной растворимости ДДВФ и ко-экстрактивных веществ в водных растворах ацетона и этанола, хроматографии в тонком слое силикагеля.
Метрологическая характеристика метода. Нижний предел определения 0,16 мг/кг.
Избирательность метода. Дибром и хлорофос определению не мешают.
Реактивы и растворы. Ацетон х. ч. и его 8О°/о-пый водный раствор. Гидроокись натрия, 5%-ный раствор. Натрий сернокислый безводный. Резорцин, 5%-ный раствор. Смесь хлороформа и этилацетата в объемном соотношении 9:1. Смесь петролейного (фракция с 1кип 40—60 °C) и диэтплового эфиров в объемном соотношении 3 : 2. Стандартный раствор ДДВФ в смеси эфиров 40 мкг/мл.
Приборы и посуда. Чашки для выпаривания на 100 мл. Конические центрифужные пробирки на 10 мл. Воронки диаметром 5 и 10 см. Воронки делительные па 50 и 150 мл. Колбы конические на 150 мл. Микроразмельчитель тканей РТ-2. Пипетки разные. П[Й1-бор для встряхивания АВУ-1. Сосуд для хроматографического разделения. Стеклянные капилляры (12—15 см). Стеклянные пульверизаторы. Центрифуга типа ЦАС-31 или ЦЛР-1. Хроматографические пластинки «Силуфол».
Подготовка к определению. 25 г тканей животных измельчают ножом или ножницами. Подготовленные таким образом пробы (кроме жировой ткани) переносят в сосуд размельчителя тканей. Посуду и оборудование, использованные для предварительного размельчения, смывают 25 мл 80%-ного ацетона, который затем приливают к пробе. Включают прибор при больших оборотах ротора на 5 мин. Затем приливают еще 50 мл ацетона той же концентрации и включают еще на 5 мин прибор при малых оборотах ротора. Для более полной денатурации белков содержимое сосудов быстро нагревают в водяной бане до начала кипения, а затем охлаждают водой из водопровода до комнатной температуры. Ацетоновый раствор отделяют от частиц пробы центрифугированием (в течение 10 мин при 2000 об/мин) или фильтрованием (через широкопористый складчатый фильтр). При работе с рефрижераторной центрифугой ЦЛР-1 в ней поддерживают температуру 8±1 °C. При использовании прибора ЦАС-31 содержимое центрифужных сосудов предварительно охлаждают до 5—6°C В морозильной камере холодильника.
Пробы жира после предварительного измельчения дополнитель-п : растирают в ступке пестиком, затем переносят в колбу на 1>0 мл. Ступку смывают 75 мл чистого ацетона, который приливают к пробе. Жиры растворяют в ацетоне при встряхивании на аппарате в течение 30 мин. Содержимое колбы охлаждают до 0 “С и приливают 25 мл ледяной воды. Ацетоновый раствор отделяют or частиц пробы фильтрованием.
Ход анализа. Очистка экстракта. Ацетоновый раствор переносят в ротационный испаритель и отгоняют растворитель при ц-мпературе не выше 50 °C. Водную фазу переносят в делительную воронку на 150 мл, приливают 50 мл смеси эфиров и энер-Ы1ЧН0 встряхивают 3 мин. Слоям дают разделиться. Нижний слой сливают и отбрасывают. Эфирный слой сливают через горловину воронки в выпарительную чашку, куда предварительно вносят 5 мл 20%-ного этанола. Эфиры испаряют под тягой вытяжного шкафа. Водно-спиртовой остаток из чашки переносят на плотный складчатый фильтр н фильтруют в делительную воронку на 50 мл. Выпарительную чашку смывают 3 мл 20%-ного этанола, который также переносят на фильтр.
К собранному фильтрату приливают 8 мл дистиллированной воды и извлекают ДДВФ, энергично встряхивая 3 мин с 8 мл смеси эфиров. Нижний слой после разделения сливают и отбрасывают. К эфирной фракции добавляют 0,3—0,5 г безводного сульфата натрия и оставляют на 5 мин. Эфирный раствор переносят в коническую центрифужную пробирку. Сульфат натрия промывают в делительной воронке 2 мл диэтилового эфира, который также приливают в коническую пробирку. В последнюю помещают стеклянный капилляр, запаянный в верхней части, и помещают в водяную баню, предварительно нагретую до 36—40 °C, испаряют растворитель до объема 0,1—0,2 мл. При этом нельзя допускать полного испарения растворителя.
Остаток из пробирки с помощью капилляра, запаянный конец которого отламывают, количественно переносят на хроматографическую пластинку на расстоянии 15 мм от края. Диаметр пятен не должен превышать 6 мм. Пробирку смывают 0,1 мл смеси эфиров, которые наносят в ту же точку. С обеих сторон точки нанесения пробы наносят 0,1—0,2 мл стандартных растворов ДДВФ
Хроматографирование. Пластинку помещают в хроматографическую камеру, на дно которой налита смесь хлороформа и этилацетата высотой 5—7 мм. После того как фронт растворителя поднимется на 10—И см, пластинку вынимают и оставляют на воздухе для испарения растворителей. Затем последовательно опрыскивают растворами гидроокиси натрия и резорцина. Для образования окрашенных пятен пластинку' осторожно нагревают над плиткой или помещают в сушильный шкаф на 5—7 мин при 60 °C. При наличии ДДВФ в пробе на уровне «свидетеля» образуется красное пятно.
Обработка результатов анализа. Количество пестицида (X, мг/кг) рассчитывают по формуле
где А — количество пестицида, найденное на пластинке в анализируемой пробе, мкг; Р — навеска анализируемого образца, г.
Методика обнаружения хлорофоса и ДДВФ в патологическом материале
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 16 февраля 1975 г.)
Характеристика препаратов.
Хлорофос (трихлорфон, диптерекс, Байер Л-13/59). Белое кристаллическое вещество без запаха, плавящееся при 83—84 °C. Действующее начало — О,О-диметил- (1 -окси-2,2,2-трихлорэтил) -фос-фонат. Мол. масса 257,45. Препарат растворим в воде (при 25 °C растворяется 15,4 г в 100 мл). Хорошо растворим в хлороформе, бензоле, спирте, этилапетате; плохо растворим в н-гексапе, п-пен-тане, петролейном эфире
ДДВФ (дихлорфон, нуван). Действующее начало — О,О-диметил-0,2,2-дихлорвинилфосфат. Это бесцветная жидкость с (кип 74 °C. ДДВФ хорошо растворим в большинстве органических растворителей, растворимость в воде около 1%,
Принцип метода. Метод основан на извлечении хлорофоса и ДДВФ из органов и тканей хлороформом, очистке экстрактов от мешающих анализу коэкстрактивных веществ путем перераспределения пестицидов в воду, а затем в хлороформ, концентрировании экстрактов и хроматографировании в тонком слое силикагеля КСК- В качестве подвижной фазы используют систему н-гексан — ацетон в соотношении 1 : 1. Проявление хлорофоса и ДДВФ производят смесью резорцина с карбонатом натрия с последующей обработкой раствором щелочи.
Пестициды проявляются в виде малиновых пятен на белом фоне пластинки. Количественное определение препаратов производят путем визуального сравнения интенсивности окраски и размера пятен проб с пятнами стандартных растворов хлорофоса и ДДВФ.
Чувствительность метода — 0,5 мг/кг, точность определения — 88—95% при содержании пестицидов в пробе от 10 до 20 мкг. х
Реактивы и растворы. Хлороформ х. ч. Ацетон х. ч. н-Гексан. Едкий натр или едкое кали. Силикагель марки КСК. Сульфат натрия безводный. Гипс. Хлорофос 80%-ный. ДДВФ 50%-ный. Резорцин. Карбонат натрия.
Проявители: 1) 2% ный водный раствор резорцина и 10%-ный раствор карбоната натрия. Растворы смешивают перед опрыскиванием соответственно в соотношении 2:3; 2) 20%-ный раствор едкого натра или едкого кали.
Приборы и посуда. Хроматографическая камера. Весы аналитические. Делительные воронки на 150 мл. Пульверизаторы Шуттель-аппарат. Шприцы емкостью 1 мл или микропипетки. Фильтры бумажные. Чашки фарфоровые выпарительные на 20 и 100 мл. Водяная баня. Стеклянные пластинки 9X12 см или «Силуфол» ЧССР. Сушильный шкаф. Фарфоровая ступка. Сито на 100 меш. Конические колбы с притертыми пробками. Мельница для размалывания гранулированного силикагеля. Камера для опрыскивания хроматографических пластинок.
Приготовление пластинок с тонким слоем сорбента. Стеклянные пластинки размером 9X12 см моют хромовой смесью, затем водопроводной и дистиллированной водой и сушат в вертикальном положении. Сухие пластинки перед нанесением сорбционной массы протирают спиртом или эфиром. Для приготовления сорбционной 70
массы в фарфоровую ступку вносят 35 г силикагеля, просеянного через сито 100 меш., 2 г гипса и содержимое тщательно перемешивают, затем переносят в коническую колбу с притертой пробкой, приливают 90 мл дистиллированной воды и встряхивают в течение 15 мни. Приготовленную массу в количестве 10 г (2 чайные ложки) наносят на пластинку и равномерно распределяют по поверхности. Пластинки сушат в течение 16—20 ч при комнатной температуре в строго горизонтальном положении. Готовые пластинки хранят в эксикаторе.
Ход определения. Исследуемую пробу патологического материала (печень, почки, скелетные мышцы, селезенка, головной мозг и т. д.) массой 20 г измельчают ножницами, помещают в коническую колбу, заливают 40 мл хлороформа и экстрагируют встряхиванием на шуттель-аппарате в течение 30 мин. Экстракт отделяют фильтрованием через бумагу в фарфоровую выпарительную чашку и концентрируют выпариванием досуха на водяной бане при температуре не выше 40 °C.
Для извлечения хлорофоса и ДДВФ из сухого остатка экстрагирование проводят дистиллированной водой трижды порциями по 5 мл, тщательно растирая остаток стеклянной палочкой Экстракты собирают через бумажный фильтр в делительную воронку, объединяют и для извлечения ядохимикатов из водного раствора проводят экстрагирование хлороформом дважды порциями по 15 мл, встряхивая в течение 1 мин. Хлороформные экстракты сливают в фарфоровую выпарительную чашку через бумажный фильтр, заполненный безводным сульфатом натрия, предварительно промытым хлороформом. Экстракты объединяют и выпаривают досуха без нагрева в вытяжном шкафу. Сухой остаток растворяют в 0,2 мл хлороформа, который с помощью шприца или микропипетки наносят на пластинку со слоем сорбента Эту процедуру выполняют трижды.
Для идентификации пестицидов и их количественного определения на ту же пластинку на расстоянии 1,5—2 см друг от друга наносят стандартные растворы хлорофоса и ДДВФ, содержащие от 5 до 20 мкг действующего вещества и помещают в хроматографическую камеру под углом 45° с подвижной системой растворителей гексан — ацетон в соотношении 1:1. Камеру предварительно выдерживают в закрытом состоянии с подвижной фазой в течение 1 ч. После подъема фронта подвижной фазы на 10 см от линии старта пластинку вынимают и сушат при комнатной температуре в вытяжном шкафу до испарения растворителей.
Для проявления хроматограммы пластинку опрыскивают из пульверизатора до насыщения слоя сорбента смесью 2%-ного раствора резорцина и 10%-ного раствора гидрата карбоната натрия в соотношении 2 : 3 соответственно и помещают в сушильный шкаф на 5—8 мин при 100 °C.
На белом фоне пластинки хлорофос и ДДВФ проявляются в виде слабо заметных пятен малинового цвета соответственно с Rf 0,42+0,03 и Rf 0,72+0,02. После охлаждения пластинку опрыскивают до насыщения 20%-ным раствором едкого натра или едкого кали и опятв.помещают в сушильный шкаф. Через 2—3 мин интенсивность окраски ДДВФ усиливается, а в дальнейшем малиновая окраска переходит в оранжевую, а затем в желто-зеленую; слабо-малиновая же окраска хлорофоса усиливается и через 5— 8 мин становится ярко-малиновой.
Количественное определение ядохимикатов проводят путем визуального сравнения интенсивности малиновой окраски пятен пробы и их величины с пятнами стандартных растворов.
Содержание пестицидов (в мг/кг) (X) рассчитывают по формуле
где А — количество ядохимиката, найденное в пробе путем сравнения со стандартными пятнами, мкг; В — масса исследуемой пробы, г.
Колориметрическое определение хлорофоса в продуктах растительного происхождения [капуста, картофель, зерно, огурцы, яблоки) и молоке *
Хлорофос — О,О-диметил-2,2,2,-трихлор-1-оксиэтилфосфонат. Синонимы: трихлорфон, диптерекс, тугон, вег} вон, дилекс, Байер 13/59.
Чистый препарат представляет собой кристаллическое вещество без запаха. Температура плавления 73—74 °C. Растворимость в 100 г воды 12 г, бензола 15, эфира 17, хлороформа 75, гексана 0,08 г. В щелочной среде хлорофос превращается в ДДВФ — О, О-диметил-О-2,2-дихлорвинилфосф ат.
Принцип метода. Метод основан на экстракции хлорофоса из пробы растворителем, из остатка (после удаления растворителя) водой, окислении смесью бихромата калия с серной кислотой и определении продуктов разложения хлорофоса (ДДВФ и ДДТ) на основе реакции Фудживара. Чувствительность метода 0,2—0,3 мг/кг. Ошибка определения составляет ±10%. Хлороформ мешает опра^ делению, поэтому не должно быть его паров в воздухе. Продукты разложения хлорофоса данным методом не обнаруживаются.
Реактивы и растворы. Серный эфир х. ч. Кислота серная х. ч. плотностью 1,84 г/см3. Бихромат калия х. ч. Едкий натрий х. ч. 30%-ный и 2% ный водные растворы едкого натра. Кислота орто-фосфорная х. ч. плотностью 1,7 г/см3 или ледяная уксусная кислота х. ч. Сульфат натрия безводный свежепрокаленный. Пиридин х. ч. или ч. Если пиридин дает положительную реакцию по Фуджи-вару, его очищают следующим образом: к 250 мл пиридина добавляют 15 мл 2%-ного раствора едкого натра, крупинку перманганата калия и кипятят 30—40 мин с обратным холодильником. При охлаждении добавляют едкий натр до образования двух фаз (20 г), отделяют пиридиновый слой и перегоняют, используя для нагрева глицериновую баню.
Приборы и посуда. Колбы конические вместимостью 250 и 300 мл с притертыми пробками. Прибор для гидролиза - (кругло-донная колбочка вместимостью 50—100 мл с обратным холодильником на шлифе). Колбы на 500 мл с обратным холодильником. Либиховский холодильник. Делительные воронки на 50 мл. Колбы круглодонные на 50 мл. Чашки Петри. Стаканы химические вместимостью 80—100 мл. Воронки химические диаметром 8 см. Цилиндры мерные на 100 и 250 мл. Колориметрические пробирки па
10—15 мл. Термометр на 100 и 200 °C. Резиновая груша. Баня водяная. Баня глицериновая. Электроплитка. Фотоэлектроколорпметр, Фильтры.
Ход анализа.
Молоко. I метод: к 50 мл молока добавляют 0,3 мл концентрированной ортофосфорной или уксусной кислоты и 50 г свежепро-каленного безводного сульфата натрия. Чтобы не образовалось комков, сульфат натрия добавляют небольшими порциями при тщательном перемешивании смеси. Из полученной смеси хлорофос экстрагируют 100 мл серного эфира в течение 10 мйн1 при энергичном встряхивании. Экстракт фильтруют через бумажный фильтр. После измерения объема удаляют растворитель на водяной бане при 45—50°C (до сухого остатка).
II метод: 50 мл молока выпаривают в чашке Петри на водяной бане (или батарее) при температуре не выше 55 °C до получения творогообразной массы. При помощи стеклянной палочки массу переносят в химический стакан. Экстрагируют хлорофос 30— 40 мл серного эфира, прибавляя его дробно по 6—8 мл. Смесь при экстракции слегка подогревают (не выше 40 °C) на водяной бане и энергично помешивают стеклянной палочкой. Экстракты фильтруют через бумажный фильтр в круглодонную колбу, растворитель удаляют на водяной бане (45—50 °C) до получения сухого остатка.
Растительные продукты. Пробу продукта массой 25—50 г измельчают1 2 и экстрагируют 25—50 мл серного эфира 30—40 мин. Раствор фильтруют через маленький складчатый фильтр в круглодонную колбу. Экстракцию повторяют 3—4 раза, встряхивают каждый раз пробу с 10—20 мл растворителя 5 мин. Растворитель из объединенного экстракта испаряют на водяной бане (50—60 °C) до образования влажного остатка. Следы растворителя выдувают при помощи резиновой груши.
Извлечение хлорофоса из остатка.
Молоко. К маслянистому остатку в колбе добавляют 2 мл дистиллированной воды и взбалтывают 5 мин при подогреве на водяной бане с температурой ие выше 55 °C Смесь охлаждают под струей холодной воды или в холодильнике до затвердения жира. Водный экстракт фильтруют через бумажный фильтр в колбу со шлифом. Операцию проводят 3 раза, добавляя 2 раза по 2 мл дистиллированной воды и один раз 1 мл. Экстракты объединяют.
Растительные продукты. К сухому остатку приливают 2 мл дистиллированной воды. После 15—20-минутного перемешивания водный экстракт сливают в колбу прибора для гидролиза. Остаток снова смывают дистиллированной водой (3 раза по 1 мл). Полученные экстракты присоединяют к основному раствору.
Количественное определение. К объединенному водному экстракту в колбе добавляют 1—2 мл серной кислоты плотностью 1,84 г/см3 и 0,6—1 г бихромата калия Присоединяют колбу к об
1 Если сульфат натрия плохо прокален, то для лучшего отделения жидкой фазы створоженного молока от растворителя необходимо увеличить объем растворителя до 150—200 мл.
2 Зерно не измельчают. При анализе яблок следует использовать только кожуру толщиной 3—4 мм. К картофелю перед экстра-
гированием надо добавить сульфат натрия (20 г).
ратному холодильнику, кипятят содержимое колбы 5—6 мин, считая с момента закипания раствора. После 1—2-минутного отстаивания колбу отсоединяют от холодильника и охлаждают под струей холодной воды. Полученный раствор нейтрализуют 30%-ным раствором едкого награ, внося его по каплям при помешивании до перехода желто-бурой окраски в желто-зеленую.
Добавляют в колбу 3—4 мл 30%-ного раствора щелочи и 8 мл пиридина, смесь тщательно перемешивают и нагревают 15 (±2) мни на водяной бане с температурой 70+5 °C. Смесь охлаждают, переносят в делительную воронку, отделяют щелочной слой. К перенесенному в пробирку (через верхнюю часть делительной воронки) пиридиновому слою добавляют 2 мл дистиллированной воды. Содержимое пробирки перемешивают и измеряют оптическую плотность окрашенного раствора на фотоэлектроколориметре в кювете с толщиной слоя 20 мм при зеленом светофильтре. Раствором сравнения служит дистиллированная вода.
Содержание хлорофоса в пробе определяют по калибровочной кривой, для построения которой берут от 0,1 до 1,2 мд стандартного раствора хлорофоса в воде (100 мкг/мл), доводят объем дистиллированной водой до 5 мл, добавляют 1—2 мл серной кислоты плотностью 1,84 г/см3 и 0,6—1г бихромата калия. Далее проводят все операции, как при анализе пробы.
В полевых условиях оценка полученных результатов с достаточной для практических целей точностью может быть проведена по стандартной (имитационной) шкале, построенной на базе водных растворов хлористого кобальта и кристаллвиолета. Для 100 мкг хлорофоса характерна окраска, которая может быть имитирована раствором, содержащим 8,5 мл 0,02 н СаС12-6Н2О, 0,14 мл 0,0025%-ного кристаллвиолета и 1,36 мл дистиллированной воды.
Поскольку реактивы (особенно кристаллвиолег) различных партий при одних и тех же количествах могут давать неодинаковые оттенки, следует предварительно проверить оптические плотности исходного имитационного раствора и раствора, соответствующего 100 мкг хлорофоса по калибровочной кривой. Разбавлением последнего раствора получают растворы, содержащие меньшие количества хлорофоса. Шкала может сохраняться в темном месте в плотно закрытых пробирках до 6 мес.
Расчет. Содержание хлорофоса в продукте (мг/кг) вычисляют по формулам:
а) при извлечении хлорофоса из молока первым методом
АВ v_______
2) при извлечении хлорофоса из молока вторым методом и при определении пестицида в растительных продуктах
где А — количество хлорофоса, найденное по калибровочному графику или имитационной шкале, мкг; Р — объем или масса пробы молока, мл или г; В — объем экстрагента, мл; С — объем полученного экстракта, мл.
Определение хлорофоса в воде, фруктах, овощах, молоке, мясе и кормах хроматографией в тонком слое *
Принцип метода. Метод основан на извлечении препарата из пробы водой, последующем извлечении из воды органическим рас творителем, хроматографировании в тонком слое силикагеля и до полнительнои очистке на пластинках Подвижным растворителем служит смесь гексана с ацетоном (1 1) Препарат обнаруживаю -после опрыскивания пластинок раствором резорцина с карбонатом натрия и выдерживания при 100 °C или после опрыскивания пла стинок раствором азотнокислого серебра и аммиака в ацетоне и последующего ультрафиолетового облучения Количественно опреде ление проводят путем визуального сравнения интенсивности окрас ки и размеров пятен проб и стандартных растворов Чувствитель ность метода 0,1—0,2 мг/ki Минимально детектируемое количест во 1 мкг
Реактивы и растворы Хлороформ х ч перегнанный Аце тон х ч н Гексан х ч Диэтиловый эфир (для наркоза) Бензол х ч Силикагель КСК или ШСК очищенный Очистку проводят следую щим образом заливают силикагель на 18—20 ч разбавленной соляной кислотой (1 1), кислоту сливают, силикагель промывают водой и кипятят с разбавленной (1 1) азотной кислотой 2—3 ч в колбе с обратным холодильником Промывают силикагель дистил лированной водой до нейтральной реакции промывных вод, сушат 6 ч в сушильном шкафу при 130 °C, периодически перемешивая Дробят и просеивают через сито 100 меш (1600 отверстий на 1 см2) Хранят в склянке с притертой пробкой
Крахмал растворимый Натрий сернокислый безводный х ч Серебро азотнокислое х ч Резорцин х ч Карбонат натрия х ч Соляная кислота, уксусная и ортофосфорная кислоты
Проявляющий реактив Л° 1 2% ный раствор резорцина и 10% ный водный раствор Na2CO3 Перед опрыскиванием растворы смешивают (2 3)
Проявляющий реактив № 2 0,5 г азотнокислого серебра рас творяют в 5 мл дистиллированной воды, прибавляют 10 мл аммиа ка плотностью 0,9 г/см3 и доводят объем до 100 мл ацетоном
Стандартный раствор хлорофоса (10 мг хлорофоса в 100 мл диэтилового эфира)
Приборы и посуда Пластинки для хроматографии 9X12 или 13X18 см или «Силуфол» ЧССР Делительные воронки на 500 и 250 мл Мерные копбы на 100 мл Конические колбы на 250 и 500 мл с притертыми пробками Прибор для отгонки растворителя под вакуумом Водоструйный насос или воздуходувка Пульвери затор стеклянный для опрыскивания пластинок Камера для хро матографирования Стеклянный сосуд с притертой крышкой Можно использовать эксикатор Микропипетки для нанесения стандартных растворов Капиллярные пипетки для нанесения проб Бани водя ные Ртутно кварцевая лампа ПРК 2 или ПРК 4 Сушильнъ й шкаф
Приготовление пластинок. Сорбционную массу гоювяг из силикагеля КСК или ШСК (40 г), крахмала (1 г) и дистиллированной воды (125 мл) Крахмал заваривают в 15 мл воды доливают остальную воду, засыпают силикагель и хорошо перемешивают
10 г сорбционной массы наливают на пластинку и, покачивая ее, равномерно распределяют массу. Пластинки сушат при комнатной температуре, хранят в эксикаторе.
Ход анализа. Для анализа из средней пробы отбирают: овощей, фруктов, зерна 500 г, травы 200, мяса 250 г, молока 500 мл. Мясо измельчают на мясорубке, зерно растирают в ступке, овощи и фрукты измельчают ножом. Отобранное количество материала тщательно перемешивают.
Вода. 200 мл воды в делительной воронке трижды экстрагируют насыщенным водой хлороформом порциями 70, 70 и 50 мл. Каждая экстракция продолжается 5 мин. В объединенный экстракт насыпают около 10 г безводного сернокислого натрия. Оставляют экстракт на 10—15 мин для удаления влаги
Овощи, фрукты, зерно. Из 25 г измельченной пробы (зерно измельчают до частиц в 1 мм) препарат трижды экстрагируют водой порциями по 70, 50 и 50 мл на аппарате для встряхивания; каждый раз экстракция продолжается 15 мин. Водные экстракты объединяют. Чтобы не образовалась эмульсия при дальнейшей экстракции, прибавляют 1 —1,5 г хлористого натрия. Приливают 50 мл насыщенного водой хлороформа и осторожно встряхивают в делительной воронке. Экстракцию хлороформом повторяют трижды. Хлороформные экстракты объединяют, сливая их через слой безводного сернокислого натрия. Если экстракт мутный, то в колбу прибавляют безводный сернокислый натрий и сушат 10—15 мин.
Трава. 20 г мелко нарезанной ножницами травы помещают в колбу и дважды экстрагируют по 20 мин 100 и 70 мл дистиллированной воды, периодически встряхивая. Экстракты объединяют и прибавляют 1 —1,5 г хлористого натрия. Экстрагируют хлорофос трижды в делительной воронке насыщенным водой хлороформом порциями по 50 мл. Хлороформные экстракты объединяют, сливая через слой безводного сернокислого натрия. Если экстракт мутный, его дополнительно сушат безводным сернокислым натрием.
Молоко. К 25 мл молока в конической колбе добавляют по каплям 0,5 мл концентрированной соляной, уксусной или ортофос-форной кислоты, 50 мл серного эфира и небольшими порциями 20 г свежепрокаленного безводного сульфата натрия. Полученную смесь энергично встряхивают 10 мин. Эфирный экстракт сливают по частям в коническую колбу на 50 мл, удаляют эфир на водяной бане при 40—45 °C до объема 1 мл, а затем остаток испаряют при комнатной температуре.
К оставшемуся в колбе жиру добавляют 3 мл дистиллированной воды. Содержимое колбы взбалтывают 1—2 мин при подогревании на водяной бане или под струей теплой воды. Смесь охлаждают холодной водой до затвердевания жира. Водный экстракт хлорофоса через смоченный водой бумажный фильтр в маленькой конической воронке фильтруют в делительную воронку вместимостью 50 мл. Операцию повторяют дважды, внося в колбу с жировым остатком по 2 мл воды.
Водный экстракт хлорофоса в делительной воронке промывают и-гексаном 3 раза порциями по 10 мл. Гексановые экстракты отбрасывают. Очищенный водный экстракт трижды экстрагируют насыщенным водой хлороформом порциями по 15 мл, встряхивая в делительной воронке каждый раз 5 мин. Объединенные хлороформные экстракты сушат 10—15 мин безводным сернокислым натрием (7—10 г) и переносят в прибор для отгонки растворителей.
Мясо. 25 г измельченного па мясорубке мяса дважды экстра!ц-руют по 30 мин дистиллированной водой порциями по 70 мл, периодически перемешивая. Водные экстракты объединяют, прибавляют 1 —1,5 г хлористого натрия, хлорофос экстрагируют насыщенным водой хлороформом порциями по 50, 40 и 40 мл. При встряхивании в делительной воронке в нижнем хлороформном слое образуется стойкая эмульсия. Экстракты объединяют и для разрушения эмульсии постепенно прибавляют безводный сернокислый натрий, помешивание не прекращают до исчезновения эмульсии Колбу с содержимым оставляют на 10—15 мин, затем хлороформ сливают через слой безводного сернокислого натрия в сухую колбу. Колбу с оставшимся сернокислым натрием промывают хлороформом 4 раза по 15 мл и приливают его к экстракту.
Отгонка растворителя. Обезвоженный хлороформный экстракт отгоняют на водяной бане при температуре не выше 40 °C до объема около 0,3 мл при разрежении от водоструйного насоса. Остаток хлороформа упаривают при комнатной температуре досуха, пробу растворяют в диэтиловом эфире, наносят на пластинку и хроматографируют.
Хроматографирование. Пластинку с пробами и стандартными растворами помещают в камеру для хроматографирования, в которую предварительно наливают бензол (для дополнительной очистки пробы на пластинке). Край пластинки с нанесенными растворами должен быть погружен в растворитель не более чем на 0,5 см. После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Пластинку вновь помещают в камеру для хроматографирования с подвижным растворителем (смесь гексана с ацетоном 1:1) и хроматографируют, как указано выше Высушенную пластинку опрыскивают проявляющим реактивом.
Для проб фруктов, зерна, молока и мяса применяют реактив № 1. После опрыскивания проявителем пластинку выдерживают 7—10 мин при 100 °C. Хлорофос проявляется в виде оранжевого пятна с Rf 0,31. Для проб овощей, травы применяют реактив № 2. После опрыскивания реактивом пластинку подвергают ультрафиолетовому облучению в течение 40 мин. Пластинка должна находиться на расстоянии 20 см от источника света. Хлорофос проявляется в
виде серо-черного пятна.
Продукт метаболизма хлорофоса (ДДВФ) определению хлорофоса не мешает, потому что его Rf в этих условиях равно 0,56.
Количественное определение проводят путем сравнения размеров пятен пробы и стандартного раствора. Необходимо отметить, что количественное определение с достаточной точностью можно проводить лишь при содержании пестицида не более 30 мкг в пробе. При большем содержании препарата зоны локализации получаются размытыми, что затрудняет сравнение со стандартом. В этом случае нужно брать пропорциональную часть исследуемого экстракта.
Расчет. Содержание препарата в пробе (мг/кг) вычисляют по формуле
|де А— количество препарата, найденное путем сравнения со стандартом, мкг; Р — масса исследуемой пробы, г.
Определение метафоса в воде, почве и продуктах питания растительного происхождения тонкослойной хроматографией *
Химически чистый метафос— О,О-диметил-О-4-нитрофенилтио-фосфат. Белое кристаллическое вещество с температурой плавления 35—36 °C. Относительная молекулярная масса 263,22. Растворимость в воде при 25 °C около 55 мг/л. Метафос плохо растворяется в парафиновых углеводородах, но хорошо растворяется в ароматических углеводородах и большинстве органических растворителей. Препарат гидролизуется водой, кислотами и щелочами, разрушается солнечным светом.
Принцип метода. Метод основан на извлечении метафоса из проб органическим растворителем, отгонке растворителя, последующем хроматографировании в тонком слое силикагеля КСК или силикагеля КСК с цинковой пылью при использовании в качестве подвижной фазы смеси гексана с ацетоном (4:1).
Зоны локализации препарата определяют после обработки пластинок раствором щелочи или раствором диметиламинобензальдегида (на пластинке силикагель + цинк). Чувствительность метода при обработке первым реактивом 5 мкг метафоса в анализируемой пробе, вторым — 1—3 мкг. Величина Rf метафоса равна 0,38±0,02. Количественное определение метафоса проводят путем сравнения интенсивности окраски и размеров пятен препарата пробы и стандартов раствора. Полнота определения 85%.
Реактивы и растворы. Ацетон х. ч. или ч. д. а. н-Гексан х. ч. 0,25%-ный раствор п-диметиламинобензальдегида в этаноле. Хранят в темной склянке. Раствор перед обработкой пластинок смешивают в соотношении 10:1 с ледяной уксусной кислотой. Кальций сернокислый (CaSO4-2H2O), высушенный при 160 °C в течение 6 ч и просеянный через сито (100 меш.). Хранят без доступа влаги. 4 н. водный раствор натра едкого. Натрий сернокислый безводный* Силикагель КСК. Продажный силикагель измельчают и просеивают через сито 100 меш. Хранят без доступа влаги. Стандартный раствор метафоса в н-гексане (100 мкг/мл). Уксусная кислота ледяная. Цинковая пыль. Хлороформ х. ч. или ч. д. а. Спирт этиловый ректификат. Эфир диэтиловый перегнанный.
Приборы и посуда. Аппарат для встряхивания. Делительные воронки вместимостью 250 мл. Капиллярные пипетки. Колбы конические вместимостью 200 мл. Колбы мерные. Микропипетки. Прибор для отгонки растворителей с набором колб. Цилиндры мерные. Пульверизатор. Хроматографическая камера.
Приготовление пластинок. 14 г силикагеля тщательно смешивают в ступке с 1,5 г сернокислого кальция. Смесь суспендируют в 40 мл дистиллированной воды и равномерно наносят на 7— 8 пластинок 9X12 см (примерно 6 мл суспензии на пластинку). Для приготовления пластинок с цинковой пылью к указанному количеству силикагеля и сернокислого кальция прибавляют 1 г просеянной цинковой пыли.
Пластинки сушат на воздухе при комнатной температуре 17— 18 ч, хранят в эксикаторе над слоем осушителя. Можно использовать пластинки «Силуфол».
Ход анализа.
Вода. 100 мл воды помещают в делительную воронку и триж
ды экстрагируют пестицид эфиром порциями по 20 мл, встряхивая каждый раз 2—3 мин. Объединенные экстракты фильтруют через слой безводного сульфата натрия в колбу для отгонки растворителей. Эфир отгоняют на водяной бане при 40—45 °C до объема 0,2 мл.
Почва. 30 г воздушно-сухой, измельченной в ступке почвы заливают хлороформом и оставляют при комнатной температуре на 4—6 ч. Экстракт фильтруют через слой безводного сульфата натрия в колбу для отгонки растворителей. Почву вновь заливают хлороформом и экстрагируют на аппарате для встряхивания 15 мин. Экстракты объединяют и отгоняют растворитель на водяной бане до небольшого объема.
Сахар. 30 г сахара трижды (по 10 мин) экстрагируют на аппарате для встряхивания диэтиловым эфиром порциями по 30— 40 мл. Экстракты фильтруют через слой безводного сульфата натрия в колбу для отгонки растворителей. Растворитель отгоняют Да водяной бане до небольшого объема.
Овощи, ягоды. Огурцы, свеклу, свежие помидоры, картофель, капусту, черную смородину измельчают, отбирают пробу массой 50 г, заливают 50 мл н-гексана, экстрагируют 20 мин на аппарате для встряхивания. Экстракцию повторяют дважды. Гексановые экстракты объединяют, сушат над безводным сульфатом натрия. Растворитель отгоняют на водяной бане до небольшого объема.
При анализе капусты растворитель отгоняют досуха. Сухой остаток охлаждают до застывания восков, прибавляют 5 мл охлажденного ацетона и охлаждают раствор в течение 1 ч смесью льда с хлористым натрием. Затем пробу быстро фильтруют через слой безводного сульфата натрия (2 г) в небольшой воронке в колбу для отгонки растворителя. Степки колбы трижды ополаскивают охлажденным ацетоном (по 5 мл), фильтруя каждый раз через слой безводного сульфата натрия в одну и ту же колбу. Растворитель отгоняют на водяной бане, колбу с определяемым пестицидом охлаждают.
Хроматографирование. Содержимое колбы после отгонки растворителя при анализе воды, сахара, почвы, растительных проб (кроме капусты) капиллярной пипеткой наносят на пластинку. Стенки колбы еще 3—4 раза ополаскивают небольшими порциями диэтилового эфира (по 0,2 мл), каждый раз наносят раствор в центр первого пятна. При анализе капусты пробу обрабатывают охлажденным ацетоном (0,2 мл) и также количественно наносят на пластинку.
Пластинку с пробами и стандартными растворами помещают в камеру для хроматографирования со смесью гексана с ацетоном (4:1). После подъема фронта растворителя на высоту 10 см хроматографирование прекращают, пластинку помещают в вытяжной шкаф на 5—10 мин для удаления паров растворителей Затем пластинку опрыскивают 4 н. раствором едкого натра и выдерживают в сушильном шкафу 20 мин при температуре 120—130 °C.
Метафос на пластинке проявляется в виде желтых пятен. При использовании плартинок с цинком слой обрабатывают до влажного состояния смесью п-диметиламннобензальдегида с ледяной уксусной кислотой и выдерживают в сушильном шкафу 5—7 мин при температуре 100—120 °C. В этом случае зоны локализации метафоса окрашиваются в ярко-желтый цвет.
Количество метафоса определяют путем сравнения размеров п интенсивности окраски пятен пробы и стандартного раствора.
Расчет. Содержание метафоса в пробе вычисляют по формуле
где X— содержание метафоса в пробе, мг/кг или мг/л; А — количество метафоса, найденное путем сравнения со стандартами, мкг; Р — масса или объем анализируемой пробы, г или мл.
Определение метилнитрофоса и паранитрокрезола в воде, зернофураже, яйцах и патологическом материале тонкослойной хроматографией *
Принцип метода. Метод основан на извлечении метилнитрофоса и паранитрокрезола (продукт гидролиза метилнитрофоса) из зерна, яин и патологического материала н-гексаном, а из воды диэтиловым эфиром и хроматографировании экстрактов в тонком слое окиси алюминия или на силикагелевых пластинках «Силуфол UV-254». Подвижным растворителем служит смесь гексана с ацетоном (2: 1). Пятна препаратов обнаруживают путем опрыскивания пластинок раствором едкого натра и нагревания. Образующиеся нитрокрезоля-ты имеют желтую окраску.
Количественное определение проводят путем сравнения площадей пятен пробы и стандартных растворов с учетом интенсивности окраски пятен. Чувствительность определения метилнитрофоса в тонком слое окиси алюминия 0,1 мг/л воды, 0,25 мг/кг зерна и яиц, 0,5 мг/кг содержимого желудка и помета. Чувствительность определения' паранитрокрезола в 10 раз выше. На пластинках «Силуфол» чувствительность обнаружения метилнитрофоса и паранитрокрезола в 5 раз выше, чем в топком слое окиси алюминия, потому что инертный неорганический люминесцентный индикатор и рефлектирующая алюминиевая фольга повышают интенсивность окраски пятен. Полнота определения в воде 90%, в зернофураже,— 85, в яйцах, помете, содержимом желудка 75%.
Реактивы и растворы. н-Гексан х. ч. Диэтиловый эфир х. ч. 20%-ный раствор едкого натра. Натрий сернокислый безводный х. ч. Окись алюминия для хроматографии. Кальций сернокислый ч. д. а. Сушат 6 ч при 160 °C, хранят в банке с притертой пробкой. Стандартный раствор метилнитрофоса в ацетоне (100 мкг/мл). Стандартный раствор паранитрокрезола в эфире (10 мкг/мл).
Приборы и посуда. Колбы конические вместимостью 100— 150 мл с притертыми пробками Цилиндры мерные на 50—100 мл. Колбы мерные на 100 мл. Воронки делительные на 150 мл. Воронки химические. Чашки фарфоровые. Ступка с пестиком. Пульверизатор стеклянный. Стеклянные пластинки 9X12 см или «Силуфол». Груши резиновые. Шприц медицинский на 1 мл с ценой деления 0,02 мл. Пипетки разные. Хроматографическая камера. Сито (100 меш.). Весы аналитические. Прибор для встряхивания. Сушильный шкаф. Вентилятор. Прибор для вакуумной отгонки растворителя. Хроматографические пластинки «Силуфол UV-254» производства ЧССР (рефлектирующая алюминиевая фольга со слоем силикагеля, содержащего инертный люминесцентный индикатор).
Приготовление пластинок. 50 г окиси алюминия, просеянной через сито 100 меш., и 5 г сернокислого кальция смешивают в фарфоровой ступке, переносят в коническую колбу с притертой пробкой, приливают 75 мл дистиллированной воды и встряхивают 15 мин. 10 г полученной сорбционной массы наносят на пластинку и равномерно распределяют по ее поверхности. Пластинки сушат при комнатной температуре 12 ч и хранят в эксикаторе.
Ход анализа.
Вода. 50 мл воды экстрагируют трижды диэтиловым эфиром порциями по 30, 20, 10 мл в течение 30, 10, 10 мин соответственно. Экстракты переносят в колбу, сушат безводным сульфатом натрия (10 г) в течение 20—30 мин и фильтруют. Колбу с сульфатом натрия промывают дважды порциями по 10 мл эфира, которые фильтруют через тот же фильтр. Затем растворитель испаряют на воздухе или в вакуумном ротационном испарителе при температуре 40 °C. Из колбы испарителя сухой остаток смывают 10 мл эфира, которые переносят в фарфоровую чашку и испаряют па воздухе. Сухой остаток смывают со стенок чашки 2—5 мл растворителя и испаряют досуха.
Зерно. 20 г зерна помещают в колбу с притертой пробкой и экстрагируют трижды н-гексаном порциями по 30, 20, 10 мл, встряхивают в течение 30, 10 п 10 мин соответственно. Экстракты фильтруют, фильтр дважды промывают порциями по 10 мл н-гексана. Растворитель испаряют, как описано выше.
Яйцо. Среднюю пробу массой 20 г растирают в фарфоровой ступке, переносят в делительную воронку' и заливают 40 мл н-гексана. Экстрагируют пестициды из пробы 30 мин при периодическом встряхивании. Гексановый слой декантируют и фильтруют через фильтр с безводным сульфатом натрия (10 г). Экстракцию проводят еще дважды порциями н-гексана по 20 и 10 мл в течение 10 мин каждый раз, пропуская экстракты через тот же фильтр, который затем промывают дважды н-гексаном порциями по 10 мл Растворитель испаряют, как указано выше.
Патологический материал. Средние пробы содержимого желудка и помета массой от 1 до 10 г помещают в колбы с притертыми пробками и экстрагируют пестициды трижды н-гексаном порциями по 20, 10 и 10 мл (для пробы 10 г) в течение 30, 10 и 10 мин соответственно при встряхивании. Экстракты фильтруют через фильтр с безводным сульфатом натрия, который затем дважды промывают порциями по 10 мл н-гексана. Растворитель испаряют досуха одним из указанных выше способов.
Хроматографирование. Сухой остаток в фарфоровой чашке растворяют в двух порциях н-гексана по 0,1 мл и шприцем наносят на хроматографическую пластинку с тонким слоем окиси алюминия или на пластинку «Силуфол» на расстоянии 1,5 см от края в одну точку' так, чтобы диаметр пятна не превышал 1 см.
Пластинку с пробами и стандартными растворами помещают в камеру для хроматографирования, куда за 30 мин до анализа наливают подвижной растворитель — смесь гексана с ацетоном (2:1). Как только фронт растворителя поднимется на 10 см от стартовой линии, пЛастинку помещают на несколько минут в вытяжной шкаф для испарения растворителя. Пластинку' опрыскивают 20%-ным раствором едкого натра. Стеклянные пластинки с тонким слоем окиси алюминия выдерживают в сушильном шкафу 10 мин при 180 °C, а пластинки «Силуфол»—5 мин при 100 °C.
6 Заказ № 1995
81
Если проба содержит метилнитрофос и паранитрокрезол, на пластинке проявляются пятна лимонно-желтого цвета, соответствующие по цвету и значению Rf пятнам стандартных растворов. В тонком слое окиси алюминия Rf паранитрокрезола равно 0,25. Метилнитрофос проявляется в виде двух пятен с Rf 0,55 и 0,42. На пластинках «Силуфол» Rf паранитрокрезола равно 0,53, метил-питрофоса—0,63 и 0,75.
Расчет. Содержание препарата в пробе рассчитывают по формуле
У SXA SstP ’ где X— содержание препарата в пробе, мг/кг; Sx — площадь пятна исследуемой пробы, мм2; S8;— площадь пятна стандарта, мм2; Л — количество препарата в пробе, найденное путем сравнения со стандартными растворами, мкг; Р— масса пробы, г.
Необходимо заметить, что количественное определение метилнитрофоса и паранитрокрезола с достаточной точностью можно проводить только при содержании пестицидов не более 40 мкг в пробе. При большем содержании пятна получаются на пластинке диффузными, что затрудняет сравнение их со стандартами. В этом случае для анализа следует брать аликвотную часть экстракта.
Определение трихлорметафоса-3 в молоке и молочных продуктах тонкослойной хроматографией *
Чистый трихлорметафос-3 — О-метил-О-этил-О-2,4,5,-трихлорфе-пилтиофосфат. Бесцветная маслянистая жидкость с температурой кипения 127 °C при 0,15 мм рт. ст. (летучесть около 8 мг/м3). Трихлорметафос-3 хорошо растворяется в большинстве органических растворителей; растворимость в воде менее 40 мг/л.
Принцип метода. Трпхлорметафос-3 (ТХМ-3) извлекают и^ пробы смесью органических растворителей, экстракт очищают на хроматографической колонке, элюат отгоняют под вакуумом п хроматографируют в тонком слое окиси алюминия. Подвижным растворителем служит н-гексан. Проявляют с помощью растворов едкого натра и феррицианида калия по реакции с 4-аминоаптипи-рином.
Чувствительность метода 0,5 мкг в пробе. Чувствительность метода для молока 0,02 мг/л, сметаны 0,1 мг/кг, творога 0,05, сливочного масла 0,4 и сухого молока 0,1 мг/кг.
Полнота определения в свежем молоке 89,5%, сухом цельном молоке — 79, сухой смеси «Малыш»— 93,8, сухой смеси «Малютка»— 98,2, сливках — 96, сгущенном молоке — 95,0, твороге 90,1%.
Реактивы и растворы. Ацетон х. ч. перегнанный. Бензол х. ч. перегнанный. н-Гексан х. ч. перегнанный или петролейный эфир (1КИ!1 40—70 °C) перегнанный. Окись алюминия для хроматографии, просеянная через сито 150 меш. Гипс (CaSO4-2H2O), выдержанный 6 ч в сушильном шкафу при 150 °C и просеянный через сито 150 меш. 6%-ный раствор едкого натра в смеси метанола и бутанола (1:1). 0,5%-ный водно-ацетоновый раствор феррицианида калия (30 мл дистиллированной воды+ 70 мл ацетона). 4-аминоантипирин. Насыщенный раствор натрия хлористого. 5%-ный раствор оксалата калия или насыщенный раствор оксалата натрия.
Приборы и посуда. Воронки делительные вместимостью 250 мл. Хроматографические колонки 18X390 мм. Прибор для вакуумной отгонки растворителей. Стаканчики химические вместимостью 100 мл Шприц медицинский на 1 или 0,5 мл. Колбы круглодон-ныо на 250—500 мл со шлифом.
Приготовление пластинок. Окись алюминия для хроматографии 1 или II степени активности тщательно растирают в фарфоровой ступке с гипсом в соотношении 10: 1. 60 г смеси переносят в коническую колбу и добавляют раствор 4-аминоантипирина (0,4 г 4-аминоантипирина на 60 мл дистиллированной воды) Смесь тщательно размешивают, наносят на пластинки слоем толщиной 0,2— 0,3 мм. После высыхания пластинки ставят в штатив в вертикальном положении, а затем в сушильный шкаф с температурой 110 °C на 15 мин. Хранят пластинки в темноте не более трех дней. Можно использовать пластинки «Силуфол».
Ход анализа.
Молоко (кефир, простокваша, кумыс и другие цельномолочные продукты). 25 мл продукта помещают в делительную воронку на 300 мл, приливают по 5 мл оксалата калия и насыщенного раствора поваренной соли, перемешивают, приливают 100 мл ацетона, встряхивают 2 мии. Приливают 100 мл хлороформа и встряхивают 2 мин. Воронку оставляют до полного расслоения. Верхнюю фазу отбрасывают, а нижнюю выливают в круглодонную колбу со шлифом, растворитель испаряют досуха. Остаток смывают 30 мл гексана.
Сгущенное стерилизованное молоко, 10—20%-ные сливки К 10 г продукта прибавляют 10 мл насыщенного раствора хлористого натрия и выливают в делительную воронку вместимостью 150 мл. К смеси приливают 40 мл ацетона, встряхивают 2 мин, затем приливают 60 мл хлороформа, встряхивают 2—3 мин и оставляют до разделения фаз. Далее, как при анализе молока.
Сгущенные молочные продукты. 10 г продукта помещают в стаканчик, заливают 10 мл воды с температурой 45—50 "С, перемешивают, переносят в делительную воронку вместимостью 150 мл и добавляют 50 мл раствора оксалата калия. Содержимое воронки перемешивают, приливают 80 мл ацетона и энергично встряхивают 2—3 мин, добавляют 100 мл хлороформа и вновь встряхивают 5—7 мин. После разделения фаз (обычно не более 5 мин) нижнюю фазу сливают в круглодонную колбу, растворители отгоняют, а остаток растворяют в 30 мл петролейного эфира.
Сухие молочные продукты. 3 г сухих продуктов (2 г сухих сливок) отвешивают в стаканчик, приливают 15 мл дистиллированной воды с температурой 40—45 °C, размешивают и переносят в делительную воронку вместимостью 300—350 мл, приливают по 5 мл 5%-ного раствора оксалата калия и насыщенного раствора поваренной соли, предварительно обмывая ими стаканчик. Содержимое воронки тщательно перемешивают, добавляют 80 мл ацетона и энергично встряхивают 3—5 мин, приливают 100 мл хлороформа, воронку вновь встряхивают 5 мии и оставляют на 3— 5 мин (до разделения фаз). Нижнюю фазу сливают в колбу для отгонки растворителёй, а верхнюю отбрасывают. Растворители отгоняют, остаток смывают 30 мл гексана.
Масло, молочный жир. 2 г продукта отвешивают в стаканчик, подогревают до 40 °C и растворяют в 30 мл н-гексана
Сметана, сливки 30—4О°/о-ные. 5 г продукта смешивают с 10 мл 6* 83
насыщенного раствора хлористого натрия. Смесь переносят в делительную воронку, добавляют 50 мл ацетона, встряхивают 2—' 3 мин, приливают 50 мл хлороформа и встряхивают еще 2—3 мин. После разделения фаз нижний слой сливают в круглодонную колбу, растворители оп онягот, а остаток растворяют в 30 мл петро-лейного эфира.
Творог, сыр. 10 г творога или измельченного на терке сыра растирают в ступке с 10 мл насыщенного раствора хлористого натрия и переносят в делительную воронку вместимостью 150 мл. Приливают 80 мл ацетона и встряхивают 2—3 мин, добавляют 100 мл хлороформа и вновь встряхивают 4—5 мин. После разделения фаз нижний слой сливают в круглодонную колбу, растворители отгоняют, а остаток растворяют в 30 мл петролейного эфира.
Очистка экстрактов от примесей. В нижнюю часть хроматографической колонки помещают стекловату, затем насыпают 70 мл силикагеля АСК и уплотняют его постукиванием. Колонку промывают 50 мл петролейного эфира, а затем вводят в нее конечный экстракт. Пестицид элюируют ПО мл смеси бензола с петролей-ным эфиром (3 : 8) порциями по 25—30 мл. Элюат собирают в круглодонную колбу вместимостью 250—300 мл. Последние порции растворителя удаляют с помощью резиновой груши.
Элюат упаривают до 20—30 мл и переносят в круглодонную колбу с оттянутым донышком. Ополаскивают первую колбу 10 мл гексана и смывы помешают во вторую колбу Элюат отгоняют до тех пор, пока он не сконцентрируется в оттянутой части колбы.
Хроматографирование. На расстоянии 1,5 см от нижнего края пластинки на стартовую линию наносят экстракты проб и стандартный раствор трихлорметафоса-З таким образом, чтобы диаметр пятен не превышал 0,5 см. Стенки колбы ополаскивают 0,2—0,3 мл растворителя, который наносят в центр первого пятна. Пластинку ставят в камеру для хроматографирования и выдерживают до тех пор, пока фронт растворителя не поднимется на высоту 10 см Пластинку вынимают, просушивают и ставят для повторной раз' гонки. Как только фронт достигает отмеченной линии, пластинку вынимают из камеры и сушат. Для проявления препарата пластинку слегка опрыскивают 6%-ным раствором едкого натра в смеси метанола с бутанолом и ставят в сушильный шкаф на 5 мии с температурой 80—100 °C. После остывания пластинку 2—3 раза опрыскивают 0,5%-ным раствором феррицианида калия до появления розово-малиновых пятен округлой формы.
Для количественного определения трихлорметафоса-З выводят калибровочную шкалу путем нанесения на пластинку стандартных растворов трихлорметафоса-З, содержащих 0,2; 0,5; 1; 4; 5 и 10 мкг препарата в 0,1 мл растворителя Пластинку хроматографируют дважды в гексане и проявляют по описанной выше методике. Величину Rf полученных пятен сравнивают с величиной Rf пятен пробы. Rf трихлорметафоса-З равно 0,37.
Расчет. Содержимое препарата вычисляют по формуле
где X — содержание препарата в пробе, мг/кг или мг/л; А — количество препарата, найденное в пробе, мкг; Р — масса или объем исследуемой пробы, г или мл.
Определение фозалона в яблоках, листьях яблони, почве и воде тонкослойной хроматографией *
Принцип метода. Метод основан на извлечении фозалона бензолом (из растительных проб) или хлороформом (из воды), очистке экстрактов на хроматографической колонке и качественном обнаружении препарата методом тонкослойной хроматографии. Подвижным растворителем служит смесь бензола с петролейным эфиром (скип 40—60 °C) в соотношении 6:4. Для проявления хромато-I рамм используют ацетоновый раствор бромфенолового синего и азотнокислого серебра. Далее пластинки обрабатывают раствором уксусной или лимонной кислоты.
Чувствительность метода — 3—5 мкг фозалона в пробе. Rf фозалона 0,52—0,55. Метод рекомендуется для идентификации препарата и анализа проб, содержащих менее 0,25 мг действующего вещества в 1 кг продукта. Метод позволяет идентифицировать при совместном присутствии цидеал, метилнитрофос и трихлорметафос-3 В отличие от фозалона цидеал и трихлорметафос-3 проявляются двумя пятнами; Rf цидеала 0,25 и 0,70. Rf трихлорметафоса-3 0,25 и 0,95 Метилнитрофос проявляется одним пятном с Rf = 0,65. Предел обнаружения этих соединений 2—5 мкг в анализируемой пробе
Реактивы и растворы. Бензол. Хлороформ. Хлористый метилен Натрий сернокислый безводный. Уголь активированный. Петролейный эфир (tKlIn 40—60 °C). Проявитель: 10 мл 0,4%-ного раствора бромфенолового синего в ацетоне и 90 мл 1%-ного раствора азотнокислого серебра в 75%-ном водном' ацетоне Двуокись кремния для люминофоров 5%-ный водный раствор уксусной или 2,5%-ный раствор лимонной кислоты.
Приборы и посуда. Аппарат для отгонки растворителей. Колбы конические на 250 мл. Цилиндры мерные на 100 мл. Колонки хроматографические длиной 350—400 мм и диаметром 16—26 мм. Мик-ропипетки на 0,1—0,2 мл. Пульверизатор. Пластинки 8X16 см или «Силуфол».
Приготовление пластинок. Отвешивают 1 г двуокиси кремния и 0,1 г медицинского гипса в коническую колбу на 50 мл, добавляют 3,5—4 мл воды и встряхивают в течение 1 мин. Выливают смесь на пластинку и разравнивают стеклянной палочкой. Оставляют пластинку на 30 мин при комнатной температуре на горизонтальной поверхности. Затем ее помещают на 30 мин в сушильный шкаф с температурой 75 °C Готовые пластинки хранят в эксикаторе над силикагелем.
Ход анализа.
Измельченную пробу яблок (25 г) или яблоневых листьев (5 г) заливают бензолом (2 г бензола на 1 г пробы). Экстрагируют фозалон при встряхивании 1,5—2 ч или оставляют пробу на 16—18 ч. Можно экстрагировать препарат из больших проб и для анализа использовать аликвотную часть экстракта.
Среднюю пробу почвы для анализа предварительно растирают в ступке и просеивают через сито с величиной отверстия 0,25 мм. Для экстракции берут 100 г. Фозалон извлекают хлороформом при встряхивании в течение 2 ч.
Пробу воды предварительно фильтруют через промытую ацетоном вату. Для анализа берут 1000 мл воды. Фозалон извлекают 100 мл хлороформа при трехкратном встряхивании в делительной
вороике. Экстракт фильтруют в круглодонную колбу и растворитель упаривают до 5 мл. Остаток переносят на хроматографическую колонку, заполненную безводным сернокислым натрием (слой 2 см), активированным углем (1г) и безводным сернокислым натрием (слой 2 см). Пропускают через колонку 100 мл бензола. Элюат отбрасывают. Фозалон элюируют 150 мл хлористого метилена, элюат собирают в сухую колбу. Упаривают растворитель на водяной бане до 0,5 мл. Остаток наносят на пластинку
Хроматографирование. Экстракт из воды можно хроматографировать, минуя стадию очистки на хроматографической колонке. Погружают пластинку на глубину 0,5 см в подвижный растворитель (бензол и петролейный эфир в соотношении 6 : 4) и развивают на высоту 10 см. Пластинку сушат на воздухе несколько минут, затем 5 мин в сушильном шкафу при 100—105 °C. Охлажденную пластинку опрыскивают проявляющим реактивом, а после высушивания хроматограмм — раствором уксусной или лимонной кислоты. Фозалон проявляется в виде голубого пятна на лимонно-желтом фоне. Пятна на пластинке сохраняются несколько минут. Rf фоза-лона 0,52—0,55 Чувствительность метода 3—5 мкг в анализируемой пробе. Для количественной оценки метод пригоден лишь в пределах 2—10 мкг препарата.
Методические указания по определению фозалона в молоке, тканях животных и кормах методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 20 декабря 1976 г.)
Краткая характеристика препарата приведена ранее.
Принцип метода. Метод основан на экстрагировании фозалона из исследуемых проб, очистке экстрактов и хроматографическое разделении в тонком слое на пластинках «Силуфол». Для проявления хроматограмм используют раствор азотнокислого серебра и 2-феноксиэтанола в ацетоне.
Метрологическая характеристика метода. Нижний предел определения препарата 0,2 мг/кг. Степень определения 75%.
Реактивы и растворы. Ацетон х. ч. Гексан х. ч. Бензол х. ч. Уксусная кислота ледяная. Стандартный раствор фозалона в гексане. Проявляющий реагент: 0,05 г азотнокислого серебра растворяют в 1 мл воды и доводят объем до 100 мл 10%-ным раствором 2-феноксиэтанола в ацетоне.
Приборы и посуда. Пластинки для хроматографии «Силуфол». Склянки с притертой пробкой на 100 мл. Делительные воронки на 250 мл. Цилиндры на 50 мл. Фарфоровые чашки. Водяная баня. Лампа ПРК-4. Фильтры бумажные. Пробирки с притертой пробкой, градуированные на 1 мл. Микропипетки на 0,1 мл. Пипетки на 5 мл. Пульверизатор стеклянный. Камера для хроматографирования. Фен.
Подготовка к определению. Ткаии животных тщательно измельчают.
Ход определения.
Экстракция и очистка экстрактов из молока и тканей животных. Пробу молока 10 мл или 10 г измельченной ткани животного 86
помещают в склянку с притертой пробкой, заливают 30 мл ацетона, размешивают стеклянной палочкой и помещают в холодильник на 1 ч, через каждые 15 мин склянку встряхивают. Затем сливают через тройной слой марли в делительную воронку. Склянку ополаскивают 10 мл ацетона. Смыв сливаю? в ту же делительную воронку, после чего в делительную воронку с экстрактом наливают 40 мл дистиллированной воды, воронку встряхивают, добавляют в нее 40 мл гексана, еще раз встряхивают в течение 2 мин. После разделения верхний гексановый слой декантируют в фарфоровую чашку и упаривают в токе воздуха досуха.
Экстракция и очистка экстракта из свеклы. Пробу измельченной свеклы 10 г помещают в склянку с притертой пробкой, заливают 30 мл ацетона, размешивают стеклянной палочкой и экстрагируют в течение 1 ч при комнатной температуре, встряхивая через каждые 15 мин. Затем содержимое склянки сливают через двойной слой марли в делительную воронку. Склянку ополаскивают 10 мл ацетона. Смыв сливают в эту же делительную воронку. Далее в делительную воронку с экстрактом наливают 40 мл дистиллированной воды, воронку встряхивают, добавляют в нее 40 мл гексана, встряхивают в течение 2 мин. После разделения верхний гексановый слой декантируют в фарфоровую чашку и упаривают досуха.
Экстракция и очистка экстракта из картофеля. Пробу измельченного картофеля 10 г помещают в склянку с притертой пробкой, заливают 40 мл ацетона, добавляют 0,5 мл ледяной уксусной кислоты, размешивают стеклянной палочкой и экстрагируют в течение 1 ч при комнатной температуре, встряхивая через каждые 15 мин. Затем сливают через тройной слой марли в делительную воронку. Скляику ополаскивают 10 мл ацетона. Смыв сливают в эту же делительную воронку. Далее в делительную воронку с экстрактом наливают 100 мл дистиллированной воды, воронку встряхивают, добавляют в нее 50 мл гексана. Делительную воронку встряхивают в течение 2 мин. После разделения верхний гексановый слой декантируют в фарфоровую чашку и упаривают в токе воздуха досуха.
Экстракция и очистка экстракта из травы. Пробу измельченной травы 10 г помещают в склянку с притертой пробкой, заливают 50 мл теплой воды (40°C), добавляют 0,5 мл ледяной уксусной кислоты, размешивают стеклянной палочкой и оставляют на 18 ч при комнатной температуре, после чего воду сливают. Траву в склянке заливают 5 мл ацетона, встряхивают, добавляют 50 мл гексана, подогретого до 40 °C, экстрагируют в течение 1 ч на водяной бане при 40 °C. Затем гексан сливают в делительную воронку, ополаскивают склянку 10 мл подогретого гексана, который сливают в эту же делительную воронку. Добавляют 60 мл дистиллированной воды, воронку встряхивают в течение 2 мин. Когда жидкости разделятся, декантируют верхний гексановый слой в склянку с притертой пробкой, которую помещают на 1 ч в морозильную камеру холодильника, после чего фильтруют через бумажный фильтр в фарфоровую чашку, а затем упаривают в токе воздуха досуха.
Экстракция и очистка экстракта из комбикорма. Пробу комбикорма 10 г помещают в склянку с притертой пробкой, заливают 40 мл ацетона, добавляют 0,5 мл ледяной уксусной кислоты, размешивают стеклянной палочкой и помещают в холодильник на 1 ч, встряхивая через каждые 15 мин. Затем фильтруют через
тройной слой марли в другую склянку с притертой пробкой. Ополаскивают первую склянку, доливают в нее 25 мл дистиллированной воды и помещают на 2 ч в морозильную камеру холодильника. Далее содержимое склянки сливают через бумажный фильтр в делительную воронку. Склянку и фильтр ополаскивают 5 мл охлажденного ацетона, который сливают в эту же воронку. Доливают 75 мл гексана, встряхивают делительную воронку в течение 1 мин. После разделения жидкостей сливают верхний гексановый слой в фарфоровую чашку, упаривают под током воздуха.
Сухой остаток в чашке растворяют в 0,5 мл гексана, который переносят в градуированную пробирку. Чашку смывают 0,2 мл гексана. Смыв сливают в ту же пробирку. Доводят объем в пробирке гексаном до 1 мл.
Хроматографирование. На хроматографическую пластинку «Силуфол» на расстоянии 15 мм от нижнего края при помощи микропипетки наносят 0,1 мл исследуемого экстракта, подсушивая точку нанесения при помощи фена. Слева и справа от пробы на расстоянии 20 мм наносят 0,2; 0,5 и 1 мл стандартного раствора фозалона. Пластинку с нанесенным раствором помещают в эксикатор, на дно которого установлена чашка Петри с подвижным растворителем бензолом. После того как растворитель поднимется на пластинке на высоту 10 см, пластинку' вынимают из камеры и сушат в течение 1 мии под током воздуха в вытяжном шкафу.
Для проявления препарата пластинку опрыскивают проявляющим реагентом, сушат под током воздуха 2 мин и облучают ультрафиолетовыми лучами.
Фозалон проявляется в виде пятна синего цвета
Величина Rf препарата 0,34—0,36.
Расчет. Содержание фозалона в пробе (X, мг/кг, мг/л) рассчитывают по формуле
где А — количество препарата, найденное на пластинке в анализируемой пробе, мкг; Р — навеска, или объем анализируемой пробы, г, мл.
Методические указания по определению фталофоса и фозалона в воде и рыбе и фозалона в кормах и мясе методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 20 декабря 1976 г.)
Краткая характеристика препаратов приведена ранее.
Принцип метода. Метод основан на извлечении препаратов из пробы opiэпическим растворителем, очистке экстрактов из исследуемых объектов с помощью колоночной хроматографии и перераспределения в несмешивающихся растворителях, концентрировании экстрактов и хроматографическом определении фталофоса и фозалона в тонком слое сорбента. Обнаружение препаратов на хроматограмме проводят после обработки пластинок бромфеноловым проявляющим реактивом с последующим обесцвечиванием фо
на раствором лимонной или уксусной кислоты. Обнаружение фозалона проводят также по хлоримпнфенолу, образующемуся после щелочного гидролиза препарата, путем опрыскивания хроматограммы растворами 4-аминоантипирина и феррицианида калия.
Метрологическая характеристика метода. Нижний предел опре. деления пестицидов 0,5—3,0 мкг в пробе, что составляет на пластинках «Силуфол» для фталофоса и фозалона в рыбе 0,25 мг/кг, в воде — 0,005, фозалона в кормах и мясе 0,3 мг/кг. На пластинках с силикагелем предел обнаружения в 4 раза выше.
Степень определения составляет 85+9%.
Реактивы и растворы. Гексан х. ч. Хлороформ х. ч. Ацетон х. ч. Силикаюль КСК п АСК. Бензол х. ч Крахмал. Гипс. Проявляющие реагенты: бромфеноловый реактив — 0,05 г бромфенолового синего растворяют в 10 мл ацетона и доводят до 100 мл 1% ным раствором азотнокислого серебра в водном ацетоне /ацетон и вода 3:1); лимонная кислота, 1%-ный раствор; уксусная кислота, 10%-ный раствор; 4-аминоантипирин, 2%-ный раствор; феррицианид калия, 10%-ный раствор; едкий натр, 5%-иый раствор. Стандартные растворы фталофоса и фозалона в оксане 100 мкг/мл.
Приборы и посуда. Колбы конические с притертыми пробками на 100—150 мл. Цилиндры мерные на 100 мл. Воронки делительные на 100—250 мл. Воронки химические. Чашки фарфоровые на 20 и 100 мл. Ступка фарфоровая. Пульверизаторы стеклянные. Пластинки стеклянные (9X12 см). Груши резиновые. Шприцы медицинские на 1 мл с ценой деления 0,02 мл. Палочки стеклянные. Пипетки градуированные на 1, 2, 5 и 10 мл. Ножницы. Хроматографическая камера (можно использовать эксикатор). Камера для опрыскивания хроматограмм. Сито 100 меш. Мельница для размалывания гранулированного силикшеля. Весы технические. Весы аналитические. Сушильный шкаф. Прибор для встряхивания. Прибор для вакуумной отгонки растворителя. Баня водяная. Хроматографические пластинки «Силуфол UV-254». Пластинки с тонким слоем силикагеля. Хроматографические колонки высотой 25 см, внутренний диаметр 1,5 см.
Подготовка к определению.
Приготовление пластинок для хроматографирования. Стеклянные пластинки размером 9X12 см моют хромовой смесью, дистиллированной водой, сушат, протирают спиртом или эфиром и покрывают сорбционной массой. Для приготовления сорбционной массы используют две прописи: 1) берут 20,0 г силикагеля КСК, просеянного через сито 100 меш., 0,5 г крахмала, 63 мл дистиллированной воды, крахмал заваривают в 10 мл воды, доливают остальную воду, засыпают силикагель и хорошо перемешивают; 2) смешивают в фарфоровой ступке 35 г силикагеля, просеянного через сито 100 меш., с 2 г гипса; смесь переносят в коническую колбу с притертой пробкой, приливают 90 мл дистиллированной воды и встряхивают в течение 15 мин.
Приготовленную массу в количестве 10 г наносят на каждую пластинку и равномерно распределяют по поверхности. Пластинки сушат в течение 16—20 ,ч при комнатной температуре в горизон-1альпом положении. Готовые пластинки хранят в эксикаторе.
Подготовка хроматографических колонок. В нижнюю узкую часть колонки помещают небольшой тампон ва-|ы, предварительно промытой в гексане и высушенной, затем насы-
пают силикагель АСК на высоту 18 см и уплотняют его, постукивая по колонке. Перед работой колонку промывают 20 мл гексана
Ход анализа.
Экстракция и очистка экстракта при определении фталофоса и фозалона в воде и рыбе. Пробу воды 200 мл наливают в делительную воронку и трижды экстрагируют фталофос и фозалоп хлороформом порциями по 30, 20 и 10 мл, периодически встряхивая воронку в течение соответственно 30, 20 и 10 мин. Экстракты переносят в колбу, сушат безводным сульфатом натрия (10 г) в течение 30 мин и фильтруют в фарфоровую чашку или колбу для отгонки растворителя.
Колбу с сульфатом натрия дважды промывают хлороформом порциями по 10 мл, хлороформ пропускают через тот же фильтр. Затем растворитель испаряют либо из фарфоровой чашки на водяной бане при температуре 60°С, либо при использовании вакуумного ротационного испарителя. Из колбы испарителя сухой остаток смывают 10 мл хлороформа, которые переносят в фарфоровую чашку. Хлороформ испаряют. После удаления растворителя из фарфоровой чашки сухой остаток со стенок смывают 2—5 мл хлороформа, который испаряют на воздухе, а затем проводят хроматографирование, как описано ниже.
Пробу рыбы (мышцы, стенки кишечника, почки, печень, жабры, кожу, кровь) 20 г тщательно измельчают ножницами, помещают в колбу с притертой пробкой и проводят экстрагирование препаратов гексаном дважды порциями по 30 и 10 мл, встряхивая соответственно в течение 60 и 10 мин. Экстракты объединяют и пропускают через колонку, заполненную силикагелем АСК. Затем колонку промывают 100 мл элюирующей смеси петролейного эфира и ацетона в соотношении 9: 1, пропуская ее небольшими порциями (15—20 мл). Экстракт концентрируют либо при использовании вакуумного ротационного испарителя при температуре 50 °C, либо на воздухе без нагревания.
Сухой остаток смывают трижды по 5 мл дистиллированной водой и раствор пропускают в делительную воронку через фильтр, смоченный водой Из водного раствора препараты извлекают хлороформом, экстрагируя двумя порциями растворителя по 10 мл в течение соответственно 30 и 10 мин. Хлороформные экстракты сливают в фарфоровую чашку через фильтр с безводным сульфатом натрия. Фильтр промывают двумя порциями хлороформа по 5 мл. Растворитель испаряют, как указано выше.
Сухой остаток, образовавшийся в фарфоровой чашке после испарения экстрактов из проб, смывают дважды порциями гексана по 0,2 мл, который с помощью шприца наносят на хроматографическую пластинку «Силуфол» или иа пластинку с силикагелем, закрепленным крахмалом, на расстоянии 1,5 см от края в одну точку так, чтобы диаметр пятна не превышал 1 см. Слева и справа от пробы на расстоянии 2 см наносят стандартные растворы фталофоса и фозалона, содержащие предполагаемые в пробе количества (1—30 мкг).
Хроматографирование при определении фталофоса и фозалона в воде и рыбе. Пластинку помещают в камеру для хроматографирования, куда за 30 мин до анализа наливают подвижный растворитель— смесь хлороформа и бензола (2 : 1). После того как фронт растворителя поднимется на 10 см. от стартовой линии, пластинку 90
вынимают и оставляют на несколько минут в вытяжном шкафу для испарения растворителя.
Хроматограмму опрыскивают бромфеноловым проявляющим реактивом, а через 5 мин — 1 % -ным раствором лимонной кислоты. При наличии в пробе фталофоса и фозалона на желтом фоне пластинки проявляются синие пятна, соответствующие по цвету и значению Rf пятнам стандартных растворов. Значения Rf фталофоса на пластинках «Силуфол»—0,63+0,05, фозалона — 0,83+0,05; в тонком слое силикагеля значение Rf соответственно равно 0,4+0,05 и 0,6+0,05. Метаболит фталофоса — фталимид — проявляется на пластинках «Силуфол» в виде пятна зеленоватого цвета с Rf 0,9+0,02.
Для идентификации фозалона на пластинках «Силуфол» рекомендуется также использовать селективный проявляющий реактив 4-аминоантипирин. Пластинку опрыскивают 5%-ным раствором едкого натра и нагревают в термостате при 100 °C в течение 10 мин. Затем пластинку вынимают и опрыскивают последовательно 2%-ным раствором 4-аминоантипирина и 10%-ным раствором феррицианида калня. Фозалон проявляется в виде пятна бордово-ьо-ричневого цвета на слабо-желтом фоне пластинки.
Экстракция и очистка экстракта при определении фозалона в растительном материале и мясе. Исследуемую пробу растительно! о материала (люцерна, кукуруза, овес, корнеплоды, разнотравье, комбикорма) или мяса измельчают, отбирают 10 г, помещают в коническую колбу с притертой пробкой, заливают 30 мл гексана и встряхивают на шуттель-аппарате в течение 15 мин. Экстракт сливают в фарфоровую выпарительную чашку через бумажный фильтр, смоченный гексаном. Для полноты извлечения пестицида экстракцию выполняют дважды.
Экстракты объединяют и концентрируют на водяной бане при 45—50 °C до 1—2 мл, а затем досуха при комнатной температуре. В чашку .приливают 2 мл ацетона, растирают остаток стеклянной палочкой и сливают в делительную воронку через бумажный фильтр, смоченный ацетоном. Для количественного переноса пестицида из чашки в делительную воронку эту процедуру выполняют трижды. К трехкратному ацетоновому раствору (6 мл) приливают 12 мл дистиллированной воды и 15 мл хлороформа, затем энергично встряхивают 1 мин и отделившийся нижний хлороформно-ацетоно-вый слой сливают в фарфоровую выпарительную чашку через бумажный фильтр, заполненный безводным сернокислым натрием, смоченным хлороформом.
Хлороформно-ацетоновый раствор выпаривают на водяной бане при 45—50 °C до 1 мл, а затем при комнатной температуре до 0,1—0,2 мл, который забирают медицинским шприцем или микропипеткой и наносят на пластинку с силикагелем, закрепленным гипсом. Фарфоровую чашку, в которой концентрируют экстракт, смывают еще 2 раза 0,2 мл ацетона и наносят на пластинку в ту же точку.
Хроматографирование при определении фозалона в растительном материале и мясе. Для идентификации пестицида и количественного определения,на ту же пластинку наносят из стандартного раствора 2,5 н 10 мкг фозалона и помещают ее в камеру с подвижной фазой — смесью гексана и ацетона (2:1). Камеру предварительно выдерживают в течение 1 ч в закрытом состоянии для насыщения парами подвижного растворителя. После подъема подвиж-
пой фазы на 10 см 'от линии старта пластинку вынимают и сушат при комнатной температуре в вытяжном шкафу в течение 20 — 30 мин. Затем пластинку опрыскивают проявителем из пульверизатора и помещают на 5—7 мин в сушильный шкаф при температуре 50 °C, после чего ее охлаждают и опрыскивают 10%-ным раствором уксусной кислоты для обесцвечивания темно-сннего фона.
Фозалон проявляется на желтоватом фоне пластинки в виде темно-синих пятен с Rf 0,41—0,49. Частично неотделившиеся ко-экстрактпвные вещества проявляются в виде слабого синего следа ниже фозалона.
Обработка результатов анализа. Количественное определение пестицидов в воде, рыбе, мясе и растительном материале проводят путем сравнения площадей пятен пробы и стандарта при измерении их на миллиметровой бумаге, учитывая визуально интенсивность их окрасок.
Необходимо отметить, что количественное определение фозалона и фталофоса с надежной точностью можно проводить лишь до 20—30 мкг в пробе. При большем содержании препаратов зоны их локализации получаются размытыми, что затрудняет сравнение их со стандартами. В этом случае для исследования следует брать меньшую пробу или аликвотную часть экстракта.
Содержание препарата (X, мг/кг или мг/'л) рассчитывают по формуле
У SjAiC
S2P ’
где Sj — площадь пятка исследуемой пробы, мм2; S2 — площадь пятна стандарта, мм2; А—количество препарата в стандарте, мкг, Р — навеска пробы, г или мл; /< — коэффициент пересчета на объем всего анализируемого экстракта.
Определение фталофоса в молоке и мясе тонкослойной хроматографией *
Фталофос (импдан) — 0,0 диметил- (N-фтальимидометил) -дитио-фосфат. Химически чистый препарат — белое кристаллическое вещество с температурой плавления 72—72,7 °C. В 1 л воды при 25 °C растворяется около 25 мг препарата. Он хорошо растворим в ацетоне, метилэтилкетоне, циклогексаноне и других органических растворителях. Исключение составляют алифатические углеводороды, з которых препарат труднорастворим. Фталофос выпускается в виде 20%-ного эмульгирующегося концентрата, 50%-кого смачивающегося порошка н 10% ного гранулированного препарата.
Принцип метода. Метод основан на извлечении препарата нз пробы ацетоном и последующем хроматографировании в тонком слое окиси алюминия. Подвижным растворителем служит смесь гексана с метанолом (19:7). Проявляют фталофос раствором азотнокислого серебра, которое при взаимодействии с продуктами гидролиза препарата щелочью дает темно-серые пятна на белом фоне.
Количественное определение фталофоса проводят путем сравнения интенсивности окраски и размеров пятен проб со стандартной шкалой. Чувствительность метода 1 мкг в пробе, что составляет 0.1 мг/кг мяса и 0,1 мг/л молока.
Реактивы и растворы. Ацетон х. ч. Хлороформ х. ч. Уксусная кислота ледяная. 20%-ный водный раствор едкого натра. Метиловый спирт. 1%-ный водный раствор азотнокислого серебра. Окись алюминия для хроматографии. Гипс медицинский. Фталофос х. ч.
Приборы и посуда. Хроматографическая камера (стеклянный цилиндрический сосуд с притертой крышкой). Гомогенизатор для измельчения тканей. Центрифуга (8 тыс. об/мин). Делительные воронки на 150 мл Бюксы стеклянные на 50—100 мл. Пульверизаторы. Пипетки разные. Микропипетки. Фильтры бумажные обеззоленные. Пластинки фотографические 13X18 см или «Силуфол». Стеклянный или металлический валик с ограничительными кольцами для нанесения слоя окиси алюминия с гипсом.
Приготовление пластинок. В качестве сорбента используют смесь, состоящую из 98 массовых частей окисн алюминия и 7 массовых частей медицинского гипса. На матовую поверхность стеклянной пластинки насыпают сорбент н разравнивают его валиком с ограничительными кольцами по всей поверхности ровным слоем толщиной 0,2 мм. Для закрепления слоя пластинку выдерживают 30—40 с над водяным паром, затем орошают из пульверизатора 5%-ным водным раствором уксусной кислоты до полного насыщения сорбента, что отчетливо видно по образованию глянца. Пластинки сушат в горизонтальном положении при комнатной температуре 18 ч, после чего они готовы к употреблению.
Ход анализа.
Пробу мяса массой 10 г заливают 2—3 мл ацетона, гомогенизируют 1—2 мин при 1000 об/мин. Гомогенат заливают 20 мл ацетона, оставляют на 1 ч при комнатной температуре, после чего экстракт центрифугируют 15 мин при 8000 об/мин. Водно-ацетоновый слой фильтруют через бумажный фильтр в делительную воронку, добавляют 20 мл хлороформа и перемешивают вращением и легким покачиванием воронки. После расслоения нижний ацего-но-хлороформный слой отделяют и фильтруют через бумажный фильтр в круглодонную колбу вместимостью 30 мл. Экстракт выпаривают досуха на водяной бане или ротационном испарителе.
При определении препарата в молоке к 10 мл пробы добавляют 20 мл ацетона. Дальнейший ход анализа молока такой же, как для мяса. После выпаривания хлороформа сухой остаток растворяют в 0,5 мл ацетона и наносят на пластинку. Пластинку помещают в хроматографическую камеру с системой гексан — метанол (19: 1). Растворители наливают в камеру за 2 ч до хроматографирования.
После того как фронт растворителя поднимется на высоту 10 см, пластинку вынимают нз камеры и подсушивают 10—15 мин на воздухе, чтобы испарились растворители. Затем пластинку опрыскивают 20%-ным раствором едкого натра до насыщения, а через 10 мин—1%-ным раствором азотнокислого серебра. Фталофос проявляется в виде темно-серых пятен на белом фоне с Rf 0,62.
Количественное определение фталофоса проводят путем сравнения интенсивности окраски н размеров пятен проб и стандартных растворов, которые хроматографируют одновременно.
Стандартную шкалу готовят следующим образом. Растворы фталофоса в ацетоне’ (1, 2, 5, 10 и 20 мкг препарата в 0,1 мл) наносят на хроматографическую пластинку, разгоняют и проявляют. Размеры пятен измеряют и сравнивают визуально. При расче
те количества фталофоса учитывают, что из молока и мяса экстрагируется в среднем 50% препарата.
Расчет. Содержание препарата вычисляют по формуле
2С
где X — количество препарата в продукте, мг/'кг; С — количество препарата, найденное в пробе, мкг; Р — масса пробы, г; 2 — коэффициент экстракции.
Определение фозалона, фталофоса, фенкаптона, цидеала и карбофоса в воде, овощах и фруктах, определение байтекса и абата в воде тонкослойной хроматографией *
Принцип метода, Метод основан на извлечении препарата органическим растворителем (н-гексаиом, диэтиловым эфиром или хлороформом) п последующем хроматографировании в тонком слое силикагеля. Подвижными фазами служат чистые органические растворители и их смеси. Препараты обнаруживаются смесью растворов азотнокислого серебра и бромфенолового синего в ацетоне, фон обесцвечивается уксусной или лимонной кислотой. Количественное определение производят путем сравнения интенсивности окраски и размеров пятен проб и стандартных растворов.
Минимально детектируемое количество фенкаптона, фозалона, фталофоса, цидеала и карбофоса 1 мкг, байтекса 2, абата 0,2 мкг. Чувствительность определения в воде фенкаптона, фозалона, фталофоса, цидеала и карбофоса 0,01 мг/л, байтекса 0,005, абата, 0,001 мг/л. Чувствительность определения в овощах и фруктах фенкаптона, фозалона, фталофоса, цидеала, карбофоса 0,1 мг/кг. Полнота определения 92±7,3%.
Реактивы и растворы. н-Гексан х. ч. Хлороформ перегнанный. Диэтиловый эфир (отечественный).
Силикагель КСК, ШСК или КСС-3. Для очистки от примесей силикагель заливают на 18—20 ч разбавленной (1:1) соляной кислотой. Затем силикагель промывают водой и кипятят 2—3 ч с разбавленной азотной кислотой (1:1) в круглодонной колбе с обратным холодильником на песочной бане. Обработанный таким образом силикагель промывают дистиллированной водой до нейтральной реакции промывных вод, сушат в сушильном шкафу 4— 6 ч при 130 °C, периодически помешивая. Силикагель дробят и просеивают через сито 100 меш. (величина отверстия 0,147 мм). Хранят в склянке с притертой пробкой. Можно использовать пластинки «Силуфол».
Уксусная или лимонная кислота х. ч. Крахмал растворимый. Бромфеноловый синий (индикатор). Азотнокислое серебро х. ч. Проявляющий реактив: 0,05 г бромфенолового синего растворяют в 10 мл ацетона и доводят до 100 мл 0,5%-ным азотнокислым серебром в смеси ацетона с водой (3:1). Натрий сернокислый безводный х. ч. прокаленный. Стандартные растворы пестицидов в эфире (100 мкг/мл).
Приборы и посуда. Пластинки для хроматографии. Стеклянную пластинку 9X12 см тщательно промывают содой, хромовой смесью, дистиллированной водой и сушат в вертикальном положении.
Перед нанесением сорбционной массы пластинку протирают спиртом.
Станок для сушки йластинок. Конические колбы вместимостью 250, 500 мл с притертыми пробками. Делительные воронки Яй 250 и 1000 мл. Прибор для отгонки растворителей. Микропипетки на 0,1 и 0,2 мл для нанесения стандартных растворов. Пипетки капиллярные для нанесения проб. Камера для хроматографирования. Можно использовать эксикатор. Колбы мерные на 50 и 100 мл. Пульверизаторы стеклянные. Бани водяные. Прибор для встряхивания. Камера для опрыскивания пластинок.
Приготовление пластинок. Берут 40 г силикагеля КСК или ШСК, 1 г крахмала и 125 мл воды. Крахмал заваривают в 20 мл воды, доливают остальную воду, засыпают силикагель и хорошо перемешивают. 10 г сорбционной массы (две чайных ложки) наливают на пластинку и, покачивая ее, равномерно распределяют по поверхности. Пластинки сушат в течение ночи при комнатной температуре, хранят в эксикаторе.
Ход анализа.
Экстракция из воды. Органический растворитель, применяемый для экстракции пестицидов, предварительно насыщают дистиллированной водой (растворитель и воду встряхивают в делительной воронке 5 мин).
Для определения фозалона, фталофоса, фенкаптона, цидеала, карбофоса и абата берут 200 мл воды. Из этой пробы препарат экстрагируют хлороформом или диэтиловым эфиром (цидеал и фенкаптон можно экстрагировать н-гексаном) трижды порциями по 50, 30 и 30 мл, каждый раз встряхивая пробу в делительной воронке 5 мин.
Для определения байтекса 1000 мл воды подкисляют 1 н. соляной кислотой до pH 3 и дважды экстрагируют хлороформом (100 и 50 мл), встряхивают каждый раз в делительной воронке 5 мин.
Объединенные экстракты каждой пробы сушат 15—20 мин безводным сернокислым натрием (7—10 г) и переносят в прибор дЛя отгонки растворителей. Отгоняют растворитель на водяной бане (хлороформ и н-гексан под вакуумом) при 40—45 °C до небольшого объема (0,1—0,2 мл).
Экстракция из овощей и фруктов. Для анализа фозалона, фталофоса, фенкаптона, карбофоса и цидеала берут 50 г измельченной средней пробы продукта. Из этой пробы препарат экстрагируют хлороформом или диэтиловым эфиром (цидеал и фенкаптон можно экстрагировать н-гексаном) трижды порциями по 50, 50 и 40 мл, каждый раз встряхивая пробу на аппарате для встряхивания 15 мин. Объединенные экстракты сушат 10—15 мин безводным сернокислым натрием (7—10 г), переносят в прибор для отгонки растворителя и .отгоняют растворитель на водяной бане до небольшого объема (около 0,1—0,2 мл). Остаток растворителя испаряют досуха на воздухе.
К сухому остатку прибавляют 10 мл ацетона, счищают не растворившийся в ацетоне воск стеклянной палочкой и прибавляют
2,5 мл воды. Колбы с пробам» помещают в сосуд, заполненный льдом с солью (1 часть соли и 5 частей измельченного льда). Параллельно с пробой охлаждают раствор ацетона (10 мл ацетона+2,5 мл воды), необходимый для дальнейшей работы. Пробу охлаждают 30 мин Выпавший в осадок воск отфильтровывают при легком просасывании через обычный фильтр, вложенный в фильтр Шотта № 2. Промывают осадок на фильтре охлажденным раствором ацетона. К фильтрату прибавляют 50 мл хлороформа. Нижний (ацетоно-хлороформный) слой отделяют, сливая его через воронку с сернокислым натрием (2—3 г), а верхний (водный) дважды промывают хлороформом по 30 мл
Хлороформные экстракты объединяют, сушат 15--20 мин сернокислым натрием (7—10 г), переносят в прибор для отгонки растворителей, отгоняют растворитель на водяной бане ^хлороформ и н-гексан под вакуумом) при 40—45 °C до небольшого объема (0,1—0,2 мл) и хроматографируют.
Хроматографирование. Пластинку с нанесенными растворами помещают в камеру для хроматографирования, в Koronj ю налпгы хлороформ (для определения фозалона, фталофоса, цидеала и карбофоса), четыреххлористый углерод (для определения фенкап-тона), н-гексан в смеси с ацетоном в соотношении 9. 1 (при определении байтекса), хлороформ в смеси с н-гексаном в соотношении 8:2 (при определении абата).
Край пластинки должен быть погружен в растворитель не более чем на 0,5 см.
После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут на воздухе для испарения растворителя. Далее пластинку опрыскивают проявляющим раствором (бромфеноловый синий+AgNOsl и помещают в сушильный шкаф на 10 мин с температурой 35—40 °C. Пластинку обрабатывают 5%-ным раствором уксусной или 2%-ным раствором лимонной кислоты При наличии указанных препаратов появляются синие пятна на желтом фоне. Количественное определение проводят путем визуального сравнения интенсивности окраски и размеров пятен проб и стандартных растворов.
Следует заметить, что количественное определение пестицидов с достаточной точностью возможно при их содержании не более 20—30 мкг в пробе. При большем количестве препаратов зоны локализации получаются размытыми, а это затрудняет сравнение со стандартом. В таких случаях для хроматографирования следует брать пропорциональные части анализируемых экстрактов
Расчет. Содержание препарата вычисляют по формуле
_ _А
Р '
где X— содержание препарата в пробе, мг/кг; А — количество препарата, найденное путем визуального сравнения со стандартным раствором, мкг; Р — масса исследуемой пробы, г.
В таблице приведены значения Rf препаратов в зависимости от подвижного растворителя и марки силикагеля.
Величина Rf семи препаратов
Препарат Подвижный растворитель Силикагель
КСК шск ксс-з
Фталофос Хлороформ 0,47 0,56 0,36
Карбофос » — — 0,59, 0,77
Фозалон » 0,77 0,75 0,72
Цидеал » 0,81 0,81 0,80
Фенкаптон 0,98 0,95 0,85
Байтекс 0,90 —
Абат » 0,70 — —
Фенкаптон Четыреххлористый углерод — 0,50 0,42
Байтекс н-Гексан+ацетон (9:1) 0,46 — —
Абат То же 0,25 — —
» Хлороформ + + н-гексан (8:2) 0,54 — —
Методические указания по определению антио и фосфамида в кормах методом тонкослойной хроматографии
(Утверждены заместителем Главного
государственного санитарного врача СССР 20 декабря 1976 г.)
Краткая характеристика препаратов приведена на с. 99.
Определяют антио и фосфамид в кормах (сене, картофельной и морковной ботве, листьях капусты, свекле, картофеле, овсе, ячмене, комбикормах) хроматографией в тонком слое.
Основные положения. Принцип метода. Метод основан на извлечении антио и фосфамида из кормов хлороформом, очистке экстрактов от коэкстрактивных веществ перераспределением в несме-шивающихся растворителях, концентрировании экстрактов и хроматографировании в тонком слое силикагеля в подвижной фазе — смеси хлороформа и ацетона (9:1) Обнаружение антио и фосфамида производят аммиачно-ацетоновым раствором азотнокислого серебра с предварительной обработкой хроматограмм раствором щелочи.
Метрологическая характеристика метода. Нижний предел определения 0,2 мг/кг. Степень определения 86+13%.
Реактивы и растворы. Ацетон х. ч. Хлороформ х. ч. Едкий натр пли едкое кали х. ч. Серебро азотнокислое х. ч. Силикагель марки КСК. Кальций сернокислый безводный. Сульфат натрия безводный. Лнтио 90%-ный. Фосфамид 96%-ный. Раствор аммиака 25%-ный (удельная масса 0,9). Стандартные растворы антио и фосфамида в ацетоне, содержащие 100 мкг/мл х. ч. действующего вещества.
Проявляющие реагенты: 20%-ный раствор едкого натра или кали, аммиачно-ацетоновый раствор азотнокислого серебра (для получения последнего 0,5 г азотнокислою серебра растворяют в
7 Заказ № 1995
97
5 мл дистиллированной воды, приливают 5 мл 25%-ного раствора аммиака и доводят объем раствора до 100 мл).
Приборы и посуда. Хроматографическая камера (можно использовать эксикатор). Весы аналитические. Делительные воронки на 150 мл. Пульверизаторы. Шуттель-аппарат. Шприцы медицинские на 1 мл. Фильтры бумажные. Чашки фарфоровые выпарительные на 20 и 100 мл. Водяная баня. Стеклянные пластинки 9X12 см. Конические колбы с притертыми пробками на 150 и 200 мл. Воронки для фильтрования. Мерные пипетки на 2, 5 и 10 мл. Микропипетки. Сушильный шкаф. Фарфоровая ступка. Снто на 100 меш. Мельница для измельчения гранулированного силикагеля. Стеклянные палочки. Камера для опрыскивания хроматограмм.
Подготовка к определению. Стеклянные пластинки размером 9X12 см моют хромовой смесью, затем водопроводной и дистиллированной водой и сушат в вертикальном положении. Сухие пластинки перед нанесением сорбционной массы протирают спиртом или эфиром. Для приготовления сорбционной массы берут 35 г силикагеля КСК, просеянного через сито 100 меш., 2 г гипса, 90 мл дистиллированной воды, все переносят в коническую колбу с притертой пробкой и встряхивают в течение 15 мин. Приготовленную массу в количестве 10 г наносят на пластинку и равномерно распределяют по поверхности. Пластинки сушат в течение 16—20 ч при комнатной температуре в горизонтальном положении. Готовые пластинки хранят в эксикаторе. Можно использовать пластинки «Силуфол»
Ход анализа. Экстракция и очистка экстракта. Исследуемую пробу кормов измельчают, отбирают 10 г, помещают в коническую колбу, заливают 40 мл хлороформа и экстрагируют встряхиванием дважды в течение 30 и 3 мин. Экстракты отделяют фильтрованием через бумажный фильтр в фарфоровую выпарительную чашку, объединяют и концентрируют без нагрева в вытяжном шкафу досуха.
Для извлечения антио и фосфамида из сухого остатка проводят экстрагирование дистиллированной водой четырежды порциями по 2 мл, тщательно растирая остаток стеклянной палочкой. Экстракты фильтруют через бумажный фильтр в делительную воронку, объединяют и пестициды экстрагируют из водного раствора хлороформом дважды порциями по 10 мл, встряхивая в течение 1 мин. Хлороформные экстракты сливают в фарфоровую выпарительную чашку через фильтр с безводным сульфатом натрия, объединяют и выпаривают досуха, как описано выше.
Хроматографирование. Сухой остаток растворяют в 0,2 мл хлороформа и с помощью медицинского шприца или микропипетки наносят на пластинку со слоем сорбента. Эту процедуру выполняют трижды.
На ту же пластинку наносят стандартные растворы антио и фосфамида, содержащие 5,0; 2,5; 1,5 мкг пестицида, и пластинку помещают в хроматографическую камеру с подвижной фазой хлороформ-)-ацетон (9:1). После подъема фронта растворителя на 10 см от линии старта пластинку вынимают и сушат в вытяжном шкафу до испарения растворителя. Чтобы обнаружить пестициды, хроматограмму опрыскивают из пульверизатора 20%-ным раствором едкого натра или кали и помещают на 5 мин в сушильный шкаф при температуре 100 °C.
После охлаждения пластинку опрыскивают аммиачно-ацетоно-
вым раствором азотнокислого серебра и опять помещают в сушильный шкаф на 10 мин при той же температуре. На белом фоне пластинки пестициды проявляются в виде темно-серых пятен с Rf 0,72+0,03 для антио и Rf 0,45+0,03 для фосфамида.
Обработка результатов анализа. Количественное определение пестицидов проводят путем сравнения интенсивности окраски пятен пробы и их величины с пятнами стандартных растворов.
Содержание пестицидов (X, мг/кг) рассчитывают по формуле
где А — количество пестицида, найденное путем сравнения со стандартными пятнами, мкг; Р — масса анализируемой пробы, г.
Методика определения антио и фосфамида в органах и тканях, молоке, крови и моче животных при диагностике отравлений
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 18 декабря 1975 г.)
Характеристика препаратов. Антио (формотисн)—О,О-диме-тил-S- (М-метил-Ы-формилкарбамоилметил) -дитиофосфат. Хорошо растворим в спирте, эфире, хлороформе, ацетоне, в растворителях ароматического ряда, плохо растворим в предельных углеводородах и воде, устойчив в неполярных растворителях, неустойчив в щелочной среде. Антио выпускается в виде 25%-ного концентрата эмульсии’ и является высокоэффективным инсектоакарицидом контактного и системного действия.
В животных н растительных организмах под влиянием ферментов, а на обработанных культурах под действием УФ-света, повышенной температуры антио в основном превращается в более токсичный продукт-фосфамид (Н. Н. Мельников, 1968; М. А. Кли-сенко и Л. Г. Адейшвили, 1971; А. Ф. Конюхов, Д. Д. Полоз, Ф. П. Кохтюк, 1974).
Фосфамид (рогор, диметоат, БИ-58)—О,О-диметнл-8-(N-ме-тилкарбамоилметил) -дитиофосфат. Препарат хорошо растворим в большинстве органических растворителей, в воде (39 г/л), но плохо растворим в предельных углеводородах. Он является высокоэффективным инсектоакарицидом контактного и системного действия
Принцип действия. Метод основан на извлечении ядохимикатов из органов и тканей хлороформом, а из молока, крови и мочи ацетоном, очистке экстрактов от мешающих анализу коэкстрактив-пых веществ путем перераспределения в несмешивающихся рас-1ворителях, концентрировании экстрактов и хроматографировании антио и фосфамида в тонком слое сорбента в системе растворителей хлороформ — ацетон в соотношении 9:1. Для проявления ядохимикатов применяют раствор бромфенолового синего в смеси с азотнокислым серебром. Чувствительность метода 0,2 мг/кг (л), процент определения в пробах, содержащих 3—10 мкг пестицидов, и среднем составляет. 83+15,6.
Реактивы и растворы. Хлороформ х. ч. Ацетон х. ч. Едкий натр пли едкое кали. Серебро азотнокислое. Силикагель марки КСК
Гипс. Сульфат натрия безводный. Антио 90%-ный. Фосфамид 96%-ный. Аммиак 25%-ной концентрации (уд. вес 0,9). Проявитель: 0,05%-ный раствор бромфенолового синего в 1%-ном водно-ацетоновом растворе азотнокислого серебра (вода — ацетон 1:3).
Приборы и посуда. Хроматографическая камера. Весы аналитические. Делительные воронки на 150 мл. Пульверизаторы. Шуттель-аппарат. Шприцы емкостью 1 мл или микропипетки. Фильтры бумажные. Чашки фарфоровые выпарительные на 20 и 100 мл. Водяная баня. Стеклянные пластинки 9X12 см. Сушильный шкаф. Фарфоровая ступка. Конические колбы с притертыми пробками на 150 и 200 мл. Воронки для фильтрования. Мерные пипетки на 2 и 5 мл. Сито на 100 меш. Камера для опрыскивания хроматографических пластинок. Мельница для размалывания гранулированного силикагеля.
Приготовление пластинок с тонким слоем сорбента. Стеклянные пластинки размером 9X12 см моют хромовой смесью, затем водопроводной и дистиллированной водой и сушат в вертикальном положении. Сухие пластинки перед нанесением на них сорбционной массы протирают спиртом или эфиром. Для приготовления сорбционной массы в фарфоровую ступку вносят 35 г силикагеля КСК, просеянного через сито 100 меш., 2 г гипса и тщательно перемешивают, затем переносят в коническую колбу с притертой пробкой. Приливают 90 мл дистиллированной воды и энергично встряхивают в течение 15 мин. Приготовленную массу в количестве 10 г (2 чайные ложки) наносят на пластинку и равномерно распределяют по поверхности. Пластинки сушат в течение 16—20 ч при комнатной температуре в строго горизонтальном положении. Готовые пластинки хранят в эксикаторе. Можно использовать пластинки «Силуфол».
Ход определения. Исследуемую пробу патологического материала (печень, головной мозг, почки, мышечную ткань, содержимое желудка и т. д.) измельчают ножницами, берут навеску в 20 г, помещают в коническую колбу с притертой пробкой и проводят экстрагирование ядохимикатов хлороформом дважды порция® по 40 и 20 мл, встряхивая в течение 30 и 3 мин соответственно. Экстракты отделяют фильтрованием через бумажный фильтр в выпарительную чашку, объединяют и концентрируют без нагрева в вытяжном шкафу досуха.
Исследуемую пробу молока, крови и мочи в объеме 10 мл вносят в мерный цилиндр с притертой пробкой емкостью на 100 мл, приливают 40 мл ацетона, экстрагируют встряхиванием в течение 3 мин и скоагулированную массу отделяют фильтрованием через бумажный фильтр, а водно-ацетоновый экстракт собирают в делительную воронку, приливают 40 мл хлороформа и экстрагируют встряхиванием в течение 3 мин. Отделившийся хлороформно-ацето-новый экстракт сливают через бумажный фильтр, заполненный безводным сульфатом натрия, в фарфоровую выпарительную чашку и концентрируют без нагрева в вытяжном шкафу досуха.
Для извлечения антио и фосфамида из сухого остатка экстрагирование проводят дистиллированной водой трижды порциями по 3 мл, тщательно растирая остаток стеклянной палочкой. Экстракты собирают через бумажный фильтр в делительную воронку, объединяют и для извлечения пестицидов из водного раствора экстрагирование проводят хлороформом дважды порциями по 10 мл, встряхивая по 1 мин. Хлороформные экстракты сливают в фар
форовую чашку через фильтр с безводным сульфатом натрия, предварительно промытым хлороформом Экстракты объединяют и выпаривают досуха, как описано выше.
Сухой остаток растворяют в 0,2 мл хлороформа, который с помощью шприца или микропипетки наносят на пластинку со слоем сорбента. Это выполняют трижды.
Для идентификации пятен пестицидов и их количественной оценки на ту же пластинку наносят стандартные растворы антио и фосфамида, содержащие предполагаемые количества в пробе, и пластинку помещают в хроматографическую камеру с подвижной системой растворителей хлороформ — ацетон (9:1). После подъема фронта подвижной фазы на 10 см от линии старта пластинку вынимают и сушат в вытяжном шкафу до испарения растворителей. Хроматограмму обрабатывают из пульверизатора смесью вод-но-ацетонового раствора бромфенолового синего и азотнокислого серебра и помещают в сушильный шкаф на 5—7 мин при 50 °C
После охлаждения пластинку опрыскивают 10°/о-ным раствором уксусной кислоты или 2%-ным раствором лимонной кислоты для удаления маскирующего фона. На желтоватом фоне пластинки пестициды проявляются в виде темно-синих пятен, соответствующие по цвету и величине Rf пятнам стандартных растворов Для аитио Rf 0,74+0,04, для фосфамида — 0,45+0,03. Метод специфичен в присутствии других фосфорорганических пестицидов Так, рицид проявляется с Rf 0,64+0,03, метилнитрофос с Rf 0,27±0,02 и тио-фос с Rf 0,18+0,02.
Количественное определение пестицидов проводят путем визуального сравнения интенсивности окраски пятен пробы и их величины с пятнами стандартных растворов.
Расчет производят по формуле
где X — содержание пестицидов в пробе, мг/кг; А — количество пестицидов в пробе, найденное путем сравнения с пятнами стандартных растворов, мкг; Р — масса (объем) исследуемой пробы, г или мл.
Обнаружение антио и фосфамида в биологических субстратах, наличие у больных животных характерной клинической картины интоксикации, снижение уровня ацетилхолинэстеразы крови и выраженные патологоморфоло!пческие изменения в opianax павших и вынужденно убитых животных свидетельствуют об отравлении этими фосфорорганическими пестицидами.
Методические указания по определению антио и фосфамида в мёде методом хроматографии в тонком слое
(Утверждены заместителем Главного государственного унитарного врача GQCP 22 сентября 1975 г.)
Краткая характеристика препаратов приведена ранее
Принцип метода,'Метод основан на извлечении антио и фосфа-мпда из мёда гексаном, очистке экстрактов от восков и пигментов
и последующем хроматографировании в тонком слое силикагеля, закрепленного гипсом Подвижной фазой служит смесь ацетона и гексана в соотношении 1 : 2. Для обнаружения зон локализации препаратов используют смесь ацетоновых растворов бромфенолового синего и. азотнокислого серебра с последующей обработкой хроматограмм лимонной кислотой. Предел обнаружения 0,1 мг/кг.
Реактивы и растворы. Апетон х. ч. Гексан х. ч. Хлороформ х. ч. Раствор соляной кислоты 0,1 М. Силикагель марки КСК, просеянный через сито 100 меш. Гипс медицинский Бромфеноловый синий водонерастворимый (индикатор). Азотнокислое серебро х. ч 5%-ный раствор лимонной кислоты. Смесь растворов бромфенолового синего и азотнокислого серебра: тщательно перемешивают 10 мл 0,4%-ного раствора бром фенолового синего в ацетоне и 90 мл 1%-ного раствора азотнокислого серебра в 75%-ном водном ацетоне. Стандартные растворы антио и фосфамида в ацетоне, содержащие 100 мкг/мл, для чего 10 мг каждого пестицида (х. ч.) растворяют в 100 мл ацетона в мерной колбе Фильтр бумажный (синяя полоса).
Приборы и посуда. Стеклянные пластинки размером 9X12 ем. Мерные колбы на 100 мл. Делительные воронки на 100 мл Пульверизаторы стеклянные. Камеры для хроматографирования Микропипетки для нанесения стандартных растворов. Шприцы емкостью 1 мл для нанесения проб. Баня водяная. Камера для опрыскивания хроматограмм. Чашки для выпаривания на 50 мл.
Подготовка к определению. Стеклянные пластинки 9X12 см моют водой, хромовой смесью, дистиллированной водой и сушат в вертикальном положении Перед нанесением сорбционной массы пластинку протирают спиртом Для приготовления пластинок с тонким слоем сорбента берут 35 г силикагеля и 2 г гипса, тщательно растирают их в ступке, переносят в колбу, приливают 90 мл дистиллированной воды и встряхивают 10—15 мин, не допуская образования пузырьков воздуха. 10 г сорбционной массы наливают на пластинку и, покачивая, равномерно распределяют по поверхно сти Сушат при комнатной температуре в течение 10—12 ч на ровном столе. Хранят в эксикаторе Перед употреблением нагревают 15—20 мин при 120 °C. Можно использовать пластинки «Силуфол».
Ход анализа. Экстракция и очистка экстракта. Навеску мёда 30 г дважды по 10 мин экстрагируют гексаном порциями по 30 мл, тщательно растирая пробу шпателем. Экстракты объединяют и выпаривают растворитель. Сухой остаток растворяют в 5 мл этилового спирта, тщательно растирая восковой налет стеклянной палочкой. Затем добавляют 30 мл дистиллированной воды, нагретой до 50 °C и подкисленной 0,1 М соляной кислотой до pH 2—3 (pH контролируют), тщательно перемешивают, охлаждают до комнатной температуры и фильтруют через бумажный фильтр, смоченный дистиллированной водой, в делительную воронку Из водно-спиртового раствора пестициды экстрагируют дважды хлороформом порциями по 30 мл, энергично встряхивая Экстракты объединяют, сушат безводным сернокислым натрием (5 г). Сливают хлороформ, промывают сернокислый натрий 20 мл хлороформа и присоединяют к основному экстракту. Хлороформ испаряют до 0,3—0,5 мл и наносят на пластинку.
Хроматографирование. На середину хроматографической пластинки на расстоянии 1,5 см от нижнего края при помощи шприца наносят исследуемую пробу в одну точку так, чтобы диаметр пят-102
на не. превышал I см. Чашку дважды обмывают хлороформом порциями по 0,5 мл, слегка упаривают и также наносят этот хлороформ в центре пятна. Справа и слева от пробы микропипеткой наносят стандартные растворы, содержащие 5,0; 3,0 и 1,5 мкг препарата. Расстояние от центра крайнего пятна до края пластинки должно быть не менее 2 см. В хроматографическую колонку наливают свежеприготовленную смесь ацетона и гексана в соотношении 1 : 2. Через 30 мин помещают пластинку с нанесенными растворами так, чтобы край пластинки был погружен в растворитель не более чем на 0,5 см. После того как фронт растворителя поднимется на высоту 10 см, пластинку вынимают, сушат на воздухе и опрыскивают проявляющим реагентом. Подсушивают иа воздухе 5—10 мин и опрыскивают 5%-ным раствором лимонной кислоты: пестициды проявляются в виде синих пятен на желтом фоне.
Величина Rf антио 0,6, фосфамида 0,3. Необходимо учитывать, что длительно хранившийся антио содержит незначительное количество своего метаболита фосфамида, который иногда может регистрироваться на хроматограммах в виде слабоокрашенного пятна с Rf 0,3.
Обработка результатов анализа. Содержание пестицидов (X, mi /кг) рассчитывают по формуле
где А— количество препарата, найденное путем сравнения со стандартами, мкг; Р — масса анализируемой пробы, г.
Методические указания по определению карбофоса в тканях животных методом хроматографии в тонком слое
(Утверждены заместителем Главного государственного санитарного врана СССР 20 декабря 1976 г.)
Принцип метода. Метод основан на извлечении инсектицида из исследуемой пробы ацетоном, очистке экстракта от липидов 60%-ным водным раствором этилового спирта и идентификации карбофоса в тонком слое «Силуфола» после опрыскивания пластинок 2,6-дибром-М-хлорхинонимином с последующим нагреванием при 105 °C.
Реактивы и растворы. Ацетон ч. д. а. Гексаи. Хлороформ. Ледяная уксусная кислота. Натрий сернокислый безводный. 60%-ный этиловый спирт. Смесь петролейного и серного эфиров в соотношении 3 :2. Соляная кислота 2 н. 2,6-дибром-М-хлорхинонимин, 0,5%-ный раствор в гексане. 80 %-ный водный раствор ацетона. Стандартный раствор карбофоса в этиловом спирте 10 мкг/мл.
Приборы и посуда. Микроизмельчитель тканей. Центрифуга. Магнитная мешалкр. Прибор для вакуум-отгонки. Вакуум-насос. Водяная баня. Камера для хроматографирования. Стеклянный шприц на 1 мл. Пульверизатор стеклянный. Фарфоровые выпарительные чашки. Стеклянные конусовидные выпарительные чашки на 30 мл. Делительные воронки на 50 и 100 мл. Пластинки «Силуфол».
Ход анализа.
Экстракция и очистка экстракта из мяса, органов и тканей животных. Пробу тканей 25 г, предварительно измельченных ножницами, переносят в сосуд для гомогенизации и заливают при анализе мышц 50 мл, а при анализе других тканей 25 мл 80%-ного водного раствора ацетона, подкисленного 2 н. соляной кислотой (10 мл кислоты на 100 мл экстрагента). Ткань измельчают на микроизмельчителе в течение 3—5 мин при 3000 об/мин. В полученный гомогенат добавляют еще 25 мл подкисленного водного раствора ацетона и переносят в охлажденную пробирку. Пробы помещают в холодильник на 30 мин, а затем центрифугируют при 2500 об/мин в течение 20 мин. Полученный верхний слой осторожно переносят в колбу перегонного аппарата (при необходимости фильтруют), а остаток промывают 25 мл подкисленного 80%-ного ацетона, которым предварительно ополаскивают сосуд для гомогенизации. Пробу еще раз центрифугируют в течение 20 мин и верхний слой переносят в ту же колбу перегонного аппарата.
В водно-ацетоновый экстракт добавляют 5 мл насыщенного раствора поваренной соли и отгоняют под вакуумом при температуре 40—48 °C до исчезновения запаха ацетона (10—15 мин). После отгонки водную фазу (остаток) охлаждают под струей воды и переносят в делительную воронку. Затем колбу перегонного аппарата двукратно смывают 50 мл смеси эфиров и переносят в ту же делительную воронку. Воронку встряхивают интенсивно в течение 5 мин. Слоям жидкости дают разделиться, после чего нижний слой сливают через кран и отбрасывают, а эфирный слой переносят через горловину воронки в выпарительную чашку, куда предварительно вносят 5 мл 60%-ного этилового спирта.
Эфиры полностью испаряют под тягой вытяжного шкафа до образования беловатой пленки на поверхности оставшегося водноспиртового раствора. Этот остаток пропускают через плотный бумажный фильтр, смоченный дистиллированной водой, в делительную вороику. Затем выпарительную чашку смывают 3 мл 60%-ного этанола и 8 мл дистиллированной воды, которые фильтруют в ту же воронку, и экстрагируют препарат 15 мл смеси эфиров в течение 2 мии. Слоям дают разделиться, после чего нижний слой отбрасывают, а эфирный сушат 0,3 г сернокислого безводного натрия и переносят через горловину воронки в конусовидную стеклянную выпарительную чашку. Воронку ополаскивают еще раз 3 мл смеси эфиров и переносят в ту же выпарительную чашку.
Экстракт выпаривают до 0,1 мл и наносят на хроматографическую пластинку «Силуфол» на расстоянии 1,5 см от края при помощи медицинского шприца так, чтобы диаметр пятна не превышал 0,5 см. Выпарительную чашку смывают 0,1 мл смеси эфиров, которую также наносят на пластинку в центр первого пятна.
Экстракция и очистка экстракта из молока. Пробу молока объемом 25 мл в плоскодонной колбе встряхивают с 75 мл ацетона в течение 5 мин и помещают в холодильную камеру (при минус 5 °C) на 1 ч. Полученный экстракт фильтруют в колбу перегонного аппарата. Плоскодонную колбу ополаскивают двукратно 20 мл охлажденного 80%-но; о ацетона и промывают остаток на фильтре. Ацетон отгоняют под вакуумом и дальнейший анализ ведут так же, как при определении карбофоса в мясе.
Экстракция и очистка экстракта из жира. Пробу жира 25 г, предварительно измельченного ножницами, заливают в плоскодон-104
ной колбе 75 мл 80%-ного водного раствора ацетона и встряхивают на магнитной мешалке в течение 30 мин на холоде (при температуре от 0 до минус 4 °C). Полученный раствор фильтруют в колбу перегонного аппарата, добавляют в эту же колбу 5 мл насыщенного раствора хлористого натрия и ведут дальнейший анализ так же, как и при определении карбофоса в мясе.
Хроматографирование. Пластинку с нанесенными растворами помещают в камеру с системой растворителей — смеси хлороформа и гексана (10: 1). Хроматографирование проводят до тех пор, пока подвижный растворитель не достигнет уровня 1 см от верхнего края пластинки. После этого пластинку вынимают из камеры, сушат и опрыскивают 0,5%-ным раствором 2,6-дибром-М-хлорхипони-мина в гексане, а затем помещают в сушильный шкаф температурой 105 °C. Через 5—10 мин при наличии в исследуемой пробе карбофоса на пластинке проявляются долго сохраняющиеся пятна розово-красного цвета, величина Rf равна 0,52—0,56.
Обработка результатов анализа. Для определения метаболитов карбофоса дополнительно используют систему растворителей — смесь гексана, уксусной кислоты и диэтилового эфира (75: 15: 10). В этом случае карбофос и его метаболиты проявляются в виде розово-красных дуг.
Количественное определение препарата проводят путем измерения площадей пятен пробы и стандартов, учитывая визуально также интенсивность их окрасок.
Расчет: содержание количества пестицида в анализируемой пробе (X, мг/кг, мг/л) рассчитывают по формуле
где А— количество препарата, найденного на пластинке в анализируемой пробе, мкг; Р — навеска или объем анализируемого образца, г, мл.
Методические указания по определению дифоса (абата) в продуктах животного происхождения методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 28 декабря 1982 г.)
Характеристика пестицида.
Дифос — О,О,О',О'-тетраметил-О,О'-тио-ди-(парафенил) -тиофос-фат. С.бНгоОбРаЗз. М. м. 466.
Синонимы: абат 500 Е, абат 4 Е, ОМ S = 736.
Химически чистое вещество — белые кристаллы. Технический препарат — коричневая жидкость, содержащая 90—95% действующего вещества. Препарат нерастворим в воде, хорошо растворяется в органических растворителях (ацетоне, гексане, хлороформе, эфире, ацетонитриле). Устойчив к воздействию слабых щелочей (при pH 8 и температуре 20 °C не гидролизуется в течение нескольких недель). Выпускается в форме 30% к. э. Зарубежный аналог дифоса — абат выпускается в форме 50% к. э. и 25% с. п. Относится к малотоксичным соединениям. ЛДб0 при введении препарата
в желудок белых крыс составляет 1360—2300 мг/кг, для белых мышей — 460 мг/кг. Проникает через кожу. Для кроликов при накожном наиесеиии ЛД50 970—1930 мг/кг. Раздражающее действие выражено слабо. Коэффициент кумуляции 5,7.
Применяется в качестве инсектицида контактно-кишечного действия. Высокоэффективен в борьбе с личинками комаров, мух, а также с вредными насекомыми домашних животных. Эффективен и для защиты сельскохозяйственных культур от вредных клопов, трипсов и других насекомых. ДОК в сахарной свекле, овощах, цитрусовых и хлопковом масле 0,3 мг/кг, в мясе и яйце 1 мг/кг, в молоке — не допускается.
Принцип метода. Метод основан на извлечении препарата из тканей животного ацетоном, осаждении коэкстрактивных веществ из водио-ацетонового раствора уксуснокислым свинцом на холоду, последующем переэкстрагировании дифоса в гексан, хроматографировании на пластинках «Силуфол», в двух системах растворителей: н-гексан — ацетон и диэтиловый эфир — н-гексан. Для проявления хроматограмм применяют раствор бромфенолового синего и азотнокислого серебра с последующей обработкой лимонной кислотой. Количественное определение проводят путем визуального сравнения интенсивности окраски и размера пятен препарата из анализируемых проб и стандартных растворов.
Метрологическая характеристика метода. Диапазон определения концентраций 0,2—25 мкг дифоса в анализируемой пробе. Предел обнаружения 0,2 мкг или 0,02—0,05 мг/кг, мг/л. Размах варьирования 704-90%. Среднее значение определения стандартных количеств дифоса 79,8%. Стандартное отклонение 4,8. Относительное стандартное отклонение 0,06 Доверительный интервал среднего при р = 0,95 и п=10 79,8+5,7.
Реактивы и материалы. Ацетон ч. д. а. н-Гексан х. ч. Эфир диэтиловый (для наркоза). Натрий сернокислый безводный ч. д. а. Натрий едкий х. ч. Уксусная кислота (ледяная) ч. д. а. Свинец уксуснокислый или свинец уксуснокислый основной ч. д. а. Лимонная кислота ч. д. а. Бромфеноловый синий ч. д. а. Серебро азотнокислое ч. Дифос — стандартный раствор в ацетоне, содержащий 100 мкг/мл препарата.
Приборы, аппаратура и посуда. Пластинки «Силуфол» производства ЧССР. Центрифуга лабораторная стационарная ИЛС-3 или аналогичная (объем стаканов 80—100 мл). Шкаф вытяжной. Шкаф сушильный. Аппарат для встряхивания АВУ-1 или аналогичный. Весы технические. Весы торсионные типа ВТ. Посуда химическая (колбы конические с притертыми пробками 100 мл; воронки делительные 200 мл; воронки конические 5 см; пробирки мерные 10 мл; цилиндры 10, 50, 100 мл; пипетки, микропипетки, стаканы химические). Чашки фарфоровые выпарительные диаметром 5—7 см. Камеры хроматографические. Камера для опрыскивания хроматограмм. Опрыскиватели стеклянные.
Подготовка к определению.
Приготовление 20%-ного водного раствора уксуснокислого свинца. 20 г уксуснокислого свинца растворяют в 80 мл дистиллированной воды. При использовании основного уксуснокислого свинца 20 г его растворяют при легком подогревании в 80 мл 5%-ной уксусной кислоты и pH раствора доводят едким натрием до 5,4—5,6. Хранят в закрытой посуде.
Приготовление 1'1%-ного водного раствора уксусной кислоты. 10 мл ледяной уксусной кислоты смешивают с 90 мл дистиллированной воды.
Приготовление проявляющего реактива. Реактив 1: 25 мг бромфенолового синего растворяют в 5 мл ацетона и объем доводят до 50 мл 0,25% -ным раствором азотнокислого серебра (0,125 г азотнокислого серебра растворяют в 15 мл дистиллированной воды и добавляют 35 мл ацетона). Храпят в закрытой посуде из темного стекла при температуре 4—6 °C в течение 1 мес.
Реактив 2: 2%-ный раствор лимонной кислоты — 2 г лимонной кислоты растворяют в 98 мл дистиллированной воды. Хранят в закрытой посуде при температуре 4—6 °C в течение 5—6 дней.
Отбор проб производят в соответствии с унифицированными правилами отбора проб сельскохозяйственной продукции, пищевых продуктов и объектов окружающей среды для определения микроколичеств пестицидов, утвержденными заместителем Главного Государственного санитарного врача СССР 21 августа 1979 г.
Проведение определения.
Экстракция и очистка экстрактов. Навеску мяса, печени, почек, легких, сердца по 10 г, жира 2—3 г измельчают ножницами и помещают в колбу с притертой пробкой, молока берут 10 мл. Пробы заливают 30 мл ацетона, тщательно перемешивают и экстрагируют на аппарате для встряхивания в течение 60 мин. Ацетоновый экстракт фильтруют в чистую колбу через воронку с ватным фильтром. Пробы промывают 10 мл ацетона и пропускают через тот же фильтр. К объединенному ацетоновому экстракту приливают 20 мл дистиллированной воды, 2 мл раствора уксуснокислого свинца и помещают в испаритель холодильника на 1 ч. (Экстракты жира лучше оставлять в испарителе холодильника на ночь.) После этого содержимое переносят в центрифужные стаканы и центрифугируют на протяжении 3—5 мин при частоте вращения 3000 в мин. Надосадочную жидкость сливают в чистую делительную воронку. (Если на поверхности экстракта имеются взвешенные частицы, его дополнительно фильтруют через воронку с ватным фильтром.) В делительную воронку добавляют 10 мл 10%-иой уксусной кислоты и 60 мл дистиллированной воды. Дифос реэкстрагируют из этой смеси трижды н-гексаном, используя каждый раз по 10 мл экстракта. При плохом разделении слоев, в связи с образованием эмульсии, добавляют 5 мл насыщенного раствора натрия сернокислого. Объединенные гексановые экстракты сушат над натрием сернокислым безводным в течение 10—15 мин. Упа ривание экстракта проводят в вытяжном шкафу прн комнатной температуре до объема 0,2—0,3 мл.
Хроматографирование. Сконцентрированный гексановый экстракт наносят на пластинку «Силуфол» на расстоянии 1,5 см от нижнего края и 3,5 см от правого вертикального края. Диаметр нанесенного пятна не должен превышать 0,5 см.
Для выявления дифоса используют двухмерную хроматографию. Сначала пластинку помещают в хроматографическую камеру горизонтально (на левый край), куда налита смесь ацетона с гексаном (1:5). После того как фронт растворителей пройдет всю пластинку, ее подсушивают на воздухе и данную процедуру повторяют еще дважды.
Слева от нанесейной пробы наносят на пластинку стандартный раствор дифоса. Пластинку помещают в вертикальном положении
в камеру со смесью растворителей эфир — н-гексан (1:2). Общий объем подвижных растворителей в камере подбирают таким образом, чтобы хроматографические пластинки погружались на 0,4— 0,5 см После того как фронт растворителей поднимется на высоту 10 см от старта, пластинку вынимают и сушат на воздухе.
Проявление. После испарения растворителей пластинку опрыскивают бромфеноловым синим с азотнокислым серебром и помещают на 5 мин в сушильный шкаф при температуре 30 °C. Затем пластинку опрыскивают раствором лимонной кислоты. Дифос проявляется в виде синих пятен на лимонно-желтом фоне. На обработку одной пластинки расходуют по 3—5 мл проявляющих реактивов. Величина Rf составляет 0,32.
Обработка результатов. Количество дифоса в анализируемом объеме определяют путем сравнения площади и интенсивности окраски пятна пробы и стандартных растворов препарата с использованием формулы
у Л-1,25
М ’
где X— количество препарата в исследуемом материале, мг/кг, мг/л; А— количество препарата, выявленное на пластинке, мкг; М— масса исследуемой пробы, г, мл; 1,25 — поправка с учетом потерь пестицида в процессе анализа.
Методические указания по определению базудина и оксибазудина в растительном материале, почве и воде тонкослойной и газожидкостной хроматографией
(Утверждены заместителем Главного государственного санитарного врача СССР 27 августа 1978 г.)
1. Краткая характеристика препарата.
1.1. Базудин — О,О-диэтил-О-(2-изопропил-4-метилпиримидил-6) тиофосфат. C12H21O3N2SP. Мол. масса 304,36. Синонимы: алоксон, алфатокс, диазинон, Гейги 24480, дицид, экзодин.
Действующее вещество базудина — бесцветная маслянистая жидкость с температурой кипения 89 °C при 0,1 мм рт. ст. Давление паров при 20 °C составляет 8,4-10-3 мм рт. ст. Летучесть 1,39 мг/м1 * 3 при 20 °C.
Инсектицид нестабилен в кислой и щелочной среде. Растворимость в воде 40 мг/л. Хорошо растворим в большинстве органических растворителей.
Препарат токсичен для теплокровных животных, при оральном введении ЛД50 для мышей составляет 50—135 мг/кг, для крыс — 100—250 мг/кг.
Выпускается в форме 5 и 10%-ных гранул, 60%-ного эмульгирующегося концентрата, 40%-ного смачивающегося порошка и 50%-ного порошка для обработки семян.
В природных условиях базудин превращается в ряд продуктов, среди них окисленный аналог (оксибазудин) более токсичен, чем исходный продукт.
1.2. Оксибазудин — О.О-диэтил-О-^-изопропилА-метилпирими-дил-б)—фосфат (окисленный аналог базудина). Мол. масса 288,36.
Это бесцветная маслянистая жидкость, коэффициент преломления при 20 °C 1,453, хорошо растворим в большинстве органических растворителей. ПДК не установлены.
2. Методика определения базудина и оке и базу д и и а.
2.1. Принцип метода. Метод основан на экстракции базудина и оксибазудина из анализируемой пробы органическим растворителем (из растительных объектов — хлороформом или ацетоном с водой в соотношении 1:1, из почвы — смесью ацетона с 0,05 н. водным раствором хлористого кальция в соотношении 1:1, из воды — хлороформом или ацетоном), перераспределении между двумя не-смешивающимися растворителями с последующим определением тонкослойной или газожидкостной хроматографией. При определении методом тонкослойной хроматографии, если необходимо, пробу дополнительно чистят на колонке.
Метрологическая характеристика метода гжх тех
Минимально детектируемое количество 1 нг 1—3 мкг
Нижний предел определения Р — размах варьирова- ния, %: 0,05 мг/кг 0,2 мг/кг
для базудина 90—94 90—94
для оксибазудина 80—90 80—90
с — среднее значение определения, % 85 85
S — стандартное отклонение, % Доверительный интервал при Р = 0,95 и п = 5, % 6,7
±7,8
Избирательность метода. Актеллик, валексон, метафос, фталофос, фозалон, карбофос, гардона, трихлорметафос-3 определению не мешают.
2.2. Реактивы и растворы. Ацетон ч. и-Гексан х. ч. Хлороформ х. ч. Ацетонитрил ч. Натрий сернокислый безводный ч. д. а. Уголь активированный марки КАД молотый или ОУ-А. Силикагель КСК или пластинки «Силуфол UV-254». Натрий хлористый х. ч. Кальций хлористый х. ч. Окись алюминия для хроматографии. Азот особой чистоты. Водород. 5%-ный SE-30 н 3%-ный ХЕ-60 на хро-матоне N-AW-DMCS (0,16—0,20).
Проявляющие реагенты: 1) 0,5%-ный раствор 2,6-дибром-Н-хлорхиионимина в н-гексане; 2) 1%-ный раствор 4-(п-нитробензил) пиридина в ацетоне; 10%-ный раствор тетраэтиленпентамина в ацетоне; 3) смесь бромфенолового синего и азотнокислого серебра в ацетоне: а) 0,5%-ный водно-ацетоновый раствор азотнокислого серебра (1 ч. воды : 3 ч. ацетона); б) 0,03 г бромфенолового синего растворяют в 10 мл'ацетона, а затем разбавляют раствором а до 100 мл; в) 5%-ный раствор уксусной кислоты.
Стандартные растворы базудина и оксибазудина 5—10 мкг/мл в ацетоне и в н-гексане.
2.3. Приборы и посуда. Колбы конические со шлифами и пробками на 250—500 мл. Колбы круглодонные со шлифами на 100 и 500 мл. Делительные воронки. Воронки. Пробирки с делениями со шлифами на 10—15 мл. Колонки для адсорбционной хроматографии размером 300X15 мм. Аппарат для встряхивания колб. Ротационный вакуумный испаритель или другой прибор для отгонки растворителей под вакуумом. Холодильник. Хроматографическая камера. Микропипетки 0,1—0,2 мл. Хроматоскоп (хемоскоп). Камера для опрыскивания. Опрыскиватель. Газовый хроматограф с детектором термоионным. Для определения только базудина можно использовать детектор постоянной скорости рекомбинации или по захвату электронов. Секундомер.
2.4. Ход анализа.
Отбор и подготовка проб. Растительную пробу (овощи, фрукты, зеленую массу растений) измельчают ножом на кусочки 0,5 см3 и отбирают среднюю пробу 25 г. Почву в естественно-влажном состоянии или воздушно-сухую просеивают через почвенное сито и для анализа отбирают среднюю пробу 10 г. Проба воды — 100 мл.
Проведение определения. Анализируемые навески помещают в конические колбы емкостью 250 мл, пробы воды — в делительные воронки, куда добавляют по 10 г хлористого натрия.
Из растений базудин и оксибазудин экстрагируют 100 мл смеси ацетона с водой (1:1) или хлороформом, из почвы — 50 мл смеси ацетона с 0,05 н. водным раствором хлористого кальция (1 : 1).
Из воды экстракцию проводят хлороформом или ацетоном, для этого к объему пробы добавляют равный объем ацетона.
Из всех проб экстракцию проводят трижды при помощи механического встряхивания в течение 30 мин. Водно-ацетоновые экстракты охлаждают в холодильнике или в смеси соли со льдом. Пробы отфильтровывают в делительные воронки, хлороформные экстракты фильтруют через безводный сернокислый натрий, хлороформ отгоняют под вакуумом досуха.
Водно-ацетоновые вытяжки объединяют и подвергают очистке. Для этого в делительные воронки приливают 20 мл гексана (или петролсйного эфира) и встряхивают. Экстракцию повторяют 3 раза. Гексановые фракции фильтруют через безводный сернокислый натрий в круглодонные колбы и концентрируют под вакуумом при температуре не выше 40 °C до объема 0,5—1,0 мл. Далее проводят количественное определение.
Сконцентрированные хлороформные экстракты чистят ацетонитрилом. Ацетонитрильной очистке подвергают, если необходимо, пробы после переэкстракции в гексан и его отгонки. Для этого к сухим остаткам добавляют по 5 мл ацетонитрила (ацетонитрил можно заменить диметилформамидом, однако с первым растворителем очистка проходит значительно лучше) колбу промывают еще 5 мл ацетонитрила и доводят общий объем до 50 мл водой. Пробы встряхивают. В делительную воронку добавляют 40 мл н-гексана и проводят экстракцию пестицидов из водно-ацетонитрильной смеси в н-гексан, операцию проводят трижды, каждый раз встряхивая 7— 10 мин. Гексановые экстракты собирают и фильтруют через безводный сернокислый натрий. Объединенные гексановые экстракты
концентрируют до 5—7 мл. Далее, если пробы очистились недостаточно, очистку проводят на хроматографической колонке.
Подготовка хроматографической колонки: колонку заполняют 5 г активированного угля и 6 г окиси алюминия, далее колонка промывается хлороформом (30—40 мл) для уменьшения сорбционной емкости.
Проба вносится в колонку и элюируется хлороформом. Собирают первые 70 мл элюата. Хлороформ отгоняют под вакуумом до 0,2—0,3 мл. Очищенные экстракты анализируют ТСХ и ГЖХ.
Пробы почвы после экстракции смесью ацетона с СаСЬ чистят перераспределением в гексан. Как правило, дополнительной очистки не требуется.
Хлороформные экстракты проб воды рекомендуется чистить подобно растительным образцам.
Условия определения тонкослойной хроматографией. Проба с помощью капилляра или микропипетки наносится па пластинку на расстоянии 2 см от нижнего края, параллельно наносят ряд стандартных растворов пестицидов. Пятна должны быть диаметром не более 0,5 см. Хроматографические пластинки ставят в камеру для хроматографирования в систему н-гексан — ацетон (4:1). После подъема растворителя на 10 см, пластинки вынимают и сушат на воздухе.
Для обнаружения определяемых соединений пластинки просматривают под УФ-светом в хемоскопе или проявляют одним из рекомендуемых реагентов. Для этого пластинки опрыскивают реагентом № 1, нагревают в термостате 3—5 мин при 115 °C, на светлом фоне появляются ярко-красное пятно базудина и желтое пятно оксибазудина. Более высокой чувствительностью обладает реагент № 2: сначала опрыскивают 4-(п-нитробензил) пиридином, далее нагревают 3—5 мин при ПО °C в термостате и вновь опрыскивают тетраэтиленпентамииом; на светлом фоне обнаруживаются темномалиновое пятно базудина и синее пятно оксибазудина. После опрыскивания хроматограммы бромфеноловым синим с последующим снятием фона уксусной кислотой обнаруживаются сине-лиловые пятна пестицидов.
Rf базудина в указанной системе растворителей на пластинках «Силуфол» 0,38, а оксибазудина-—0,1. Пятна пестицидов сравнивают с пятнами стандартов.
Содержание препаратов в исследуемой пробе вычисляют по формуле
где X — содержание препарата в пробе, мг/кг (мг/л); А — количество препарата, найденное в пробе, мкг; Р — навеска исследуемой пробы, г.
Условия определения газожидкостной хроматографией. Базудин можно определять на хроматографе с детектором по захвату электронов (постоянной скорости рекомбинации) или иа термоионном. Для этого может быть использована колонка с 5% SE-Зб на хромосорбе W или на хроматоне N-AW-DMCS длиной 100 см и диаметром 3 мм. Температура колонки 180 °C, детектора 210 °C, испарителя 200 °C, скорость газа-носителя 75 мл/мин.
Для определения базудина и оксибазудина при их совместном присутствии следует использовать детектор термоионный, колонку стеклянную, заполненную 3% ХЕ-60 на хромосорбе W. Температура колонки 190 °C, детектора и испарителя 220 °C. Расход газов: азота 20 мл/мин, водорода 10, воздуха 200 мл/мии. Шкала электрометра 2,5 10~10 а, скорость движения ленты самописца 1 см/мин. Абсолютное время удерживания в указанных условиях для базудина 1 мин, для оксибазудина 1,6 мин Количественное определение проводят методом абсолютной калибровки.
В хроматограф вводят 2 мкл рабочего раствора. Линейность детектирования сохраняется в пределах 0,1—2,5 нг.
Содержание препаратов в исследуемом объекте вычисляют по формуле
где X — количество препаратов в пробе, мг/кг; Ci — количество препарата в стандартном растворе, мкг/мл; Н}—-высота пика стандартного раствора, мм; Я2 — высота пика анализируемого раствора, мм; 1'1 — объем стандартного раствора, введенного в хроматограф, мкл; Y2 — объем анализируемого раствора, введенного в хроматограф, мкл; У3 — объем экстракта пробы анализируемой, мл; Р — навеска пробы, г.
Методические указания по определению гардоны и ее продуктов превращения в растительном материале и почве хроматографическими методами
(Утверждены заместителем Главного государственного санитарного врача СССР 5 июня 1978 г.)
1. Краткая характеристика препарата.
Гардона (винилфосфат, тетрахлорвинфос, рабона) — трансизомер 2-хлор-1-(2,4,5-трихлорфенил)-винилдиметилфосфата — инсектицид широкого спектра действия.
Гардона — белое кристаллическое вещество с температурой плавления 97—98 °C. Давление его паров — 4,2-10~8 мм рт. ст. при 20 °C. Растворимость в воде 11 мг/л. Хорошо растворим в ацетоне, хлороформе, ацетонитриле и диметилформамиде.
В природных условиях гардона разлагается с образованием продуктов, данные о которых приведены в таблице 1.
Продукты разложения гардоны мало растворимы в гексане, хорошо растворимы в ацетоне, хлороформе, метаноле и диметилформамиде.
2. Методика определения гардоны и продуктов ее превращения в растительном материале и почве.
Принцип метода. Метод основан на экстракции препаратов из анализируемых проб органическими растворителями — ацетоном или диметилформамидом, очистке экстракта путем перераспределения между двумя несмешивающимися растворителями и на колонках с активированным углем и последующем определении методами тонкослойной и газо-жидкостной хроматографии. Для ГЖХ определения гардоны используется колонка, заполненная 5% SE-30; для одновременного определения гардоны и продуктов ее разложения —
1. Продукты разложения гардоны
Название вещества %л, °C ЛДЮ, мг/кг
Цис-изомер гардоны 67 5000
2,4,5-трихлорацетофенон (ТХАФ) 45 800
2,4,5-трихлорфенилэтанол-1 (ТХФЭ) 87-88 1000
2,4,5-трихлорфенацилхлорид (ТХФХ) 64—65 1000—2000
2,4,5-трихлорфенил-2-хлорэтанол-1 (ТХФХЭ) 72—73 4000
колонка с 5% ХЕ-60 (длиной 1 м) или 2% ПЭГА (54 см) и 5% OF-1 (41 см) на твердом носителе хроматоне N-AW-DMCS (0,16— 0,20 мм). Детекторы электронного захвата (ДЭЗ) или постоянной скорости рекомбинации (ДПР).
Метрологическая характеристика метода. Количественное ТСХ определение гардоны и продуктов ее превращения проводится сравнением интенсивности окраски и величины пятен свидетелей и пробы. Количественное ГЖХ определение проводится сравнением высот пиков анализируемых и стандартных растворов методом абсолютной калибровки.
Предел обнаружения методом ТСХ составляет 0,2 мг/кг, методом ГЖХ — 0,02 мг/кг для изомеров гардоны и 0,007 мг/кг.—для хлорорганических продуктов превращения гардоны.
Реактивы и растворы. н-Гексан х. ч., перегнанный. Ацетон х. ч. Ацетонитрил ч., перегнанный. Димегилформамид х. ч. Хлороформ ч. д. а., перегнанный. Этиловый эфир уксусной кислоты ч. Натрий сернокислый б/'в, х. ч. Уголь активированный ОУ-А. Силикагель КСК. Кремния окись для люминофоров. Пластинки «Силуфол UV-254». Азот ос. ч. Хроматон N-AW-DMCS (0,16—0,20 мм) с 5% SE-30, 5% ХЕ-60, 2% ПЭГА и 5% QF-1. Хромосорб W (80— 100 меш.) с 5% SE-30, силанизированный. Проявляющие реактивы для ТСХ: 1) 1%-ный раствор 4-(п-нитробепзил)-пиридина ч. в ацетоне; 2) 10 %-ный раствор тетраэтиленпентамина ч. в ацетоне; 3) 0,21 г азотнокислого серебра растворяют в 1,2 мл воды, добавляют 1,2 мл 25%-ного аммиака и разбавляют до 25 мл ацетоном;
4) 0,21 г азотнокислого серебра растворяют в 0,6 мл воды, добавляют 1,2 мл 2-феноксиэтанола и разбавляют до 25 мл ацетоном.
Стандартные растворы: транс- и цис-изомеров гардоны (200 мкг/мл в ацетоне и 1 мкг/мл и 0,1 мкг/мл в гексане); ТХАФ, ТХФЭ, ТХФХ и ТХФХЭ (0,1 мкг/мл и 0,05 мкг/мл в гексане).
Приборы и посуда. Колбы конические со шлифами и пробками (тип К.НКШ). Колбы круглодонные со шлифами (тип ККШ). Делительные воронки (тип VII). Воронки (тип 1а). Пробирки градуированные со шлифом, 10—15 мл (тип ПККШ). Колонки для адсорбционной хроматографии размером 300X15 мм. Холодильник (тип ХПТ-300). Микропипетки 0,1—0,2 мл. Хроматографическая камера. Пульверизатор. Камера для опрыскивания ТСХ пластинок. Аппарат для встряхивания колб любого типа. Вакуумный ротационный испаритель любого типа. Хроматоскоп (хемоскоп). Термостат любого типа. Прибор для облучения УФ-светом с лампой ПРК-4
8 Заказ № 1995 ИЗ
любого типа. Газовый хроматограф серии «Цвет» с детекторами по захвату электронов (ДЗЭ) или постоянной скорости рекомбинации (ДПР). Микрошприцы, 10 мкл. Колонки хроматографические стеклянные длиной 1 м, вн. диам. 3 мм. Секундомер- Бумажные фильтры. Вата, обработанная ацетоном.
Подготовка к определению. В адсорбционную хроматографическую колонку помещают небольшой ватный тампон, затем насыпают 2 см сернокислого натрия б/в, 1 г угля ОУ-А, снова слой 2 см сернокислого натрия б/в. Перед употреблением колонку промывают 50 мл хлороформа.
Проведение определения.
Экстракция ацетоном. Из 50 г измельченного материала, отобранного методом квартования, гардону и продукты ее превращения экстрагируют ацетоном (75 мл 3 раза) при встряхивании в течение 1 ч. Объединенные экстракты фильтруют в круглодонные колбы. Ацетон упаривают до 10 мл на ротационном испарителе. Добавляют в колбу 5 мл воды и помещают в холодильник (0— 4 °C) на 1 ч. Выпавшие воска отфильтровывают, а фильтрат переносят в делительную воронку. Ополаскивают колбу небольшими порциями воды и добавляют ее к основному фильтрату. Далее проводят экстракцию гексаном (10 мл 2 раза), встряхивая делительную воронку 1 мин. После полного разделения слоев отделяют гексановый слой в чистую сухую делительную воронку, добавляют в нее 20 мл ацетонитрила и встряхивают 1 мин. Отделяют нижний ацетонитрильный слой, доводят его объем до 50 мл водой и встряхивают в делительной воронке с гексаном (50 мл 2 раза). Гексановые экстракты сушат натрием сернокислым б/в и упаривают гексан до 1—3 мл на ротационном испарителе.
Экстракция диметилформамидом (яблоки). Из 50 г измельченных яблок, отобранных методом квартования, гардону и продукты ее превращения экстрагируют при встряхивании в течение 2 ч 100 мл диметилформамида (ДМФА). Фильтруют пробы через бумажный фильтр в делительные воронки. Остаток промывают на фильтре дважды по 15 мл ДМФА. К фильтрату добавляют 100 мл воды и встряхивают воронку. Далее проводят экстракцию гексаном, используя последовательно 40 мл, 20 мл 2 раза. Объединенный гексановый экстракт сушат сернокислым натрием б/в. Упаривают гексан на ротационном испарителе при температуре не выше 40 °C до объема 1—3 мл.
Очистка экстрактов от примесей. В подготовленную и промытую хлороформом хроматографическую колонку вводят гексановый экстракт. Гардону и продукты ее превращения элюируют из колонки 40 мл хлороформа, который вытекает из колонки со скоростью 3 мл/мин. Элюат собирают в круглодонную колбу. Хлороформ отгоняют на ротационном испарителе до 0,2—0,3 мл. Очищенные экстракты используют для ТСХ и ГЖХ анализа.
Условия хроматографирования в тонком слое. На расстоянии 2 см от нижнего края пластинки на стартовую линию наносят экстракты проб и стандартный раствор в виде пятен диаметром 0,5 см. Стенки колб смывают несколькими каплями растворителя, который наносят в центр пятна. Хроматографические пластинки ставят в камеру для хроматографирования и ожидают, пока фронт растворителя поднимется на высоту 10 см. Хроматографируют в системах гексан — ацетон (4:1) и гексан — этиловый эфир уксусной кислоты (7:3). Пластинки вынимают и сушат на воздухе. Для
обнаружения определяемых соединений используют один из хромогенных реагентов.
1. Опрыскивают пластинки реагентом № 1, нагревают в термостате 3—5 мин при 115 °C, затем опрыскивают реагентом № 2. Обнаруживаются яркие синие пятна на светлом фоне.
2. Опрыскивают пластинки реагентом № 3 или № 4. Облучают УФ-светом от ртутно-кварцевой лампы до появления серых или коричневых пятен на светлом фоне.
3 Обнаруживают пятна на пластинках «Силуфол UV-254», просматривая их под хроматоскопом, используя тушение флуоресценции.
Rf определяемых соединений представлены в таблице 2. Величины Rf полученных пятен пробы сравнивают с величинами Rf стандартов.
Содержание препаратов в исследуемом объекте вычисляют по формуле
где У — содержание препарата в пробе, мг/кг; А — количество препарата, найденное в пробе, мкг; Р — навеска исследуемой пробы, г.
Условия газо-жидкостного хроматографирования. Для определения гардоны может быть использована колонка с 5% SE-30 на хромосорбе W (80—100 меш.) или хроматоне N-AW-DMCS (0,16— 0,20 мм) длиной 1 м, вн. диам. 3 мм. Температура колонки 180 °C, термостата детектора 210 °C, испарителя 200 °C, скорость газа-носителя (азот ос. ч.) — 75 мл/мин.
Для определения гардоны и ее продуктов превращения следует использовать колонки на основе хроматона N-AW-DMCS (0,16— 0,20 мм): а) с 5% ХЕ-60 (длиной 1 м, вн. диам. 3 мм); б) с 2% ПЭГА (54 см) и 5% QF-1 (41 ем). При применении колонки «а» выдерживается следующий режим: температура колонки при определении изомеров гардоны 175 °C, при определении хлорорганичс-ских продуктов превращения гардоны 145 °C, температура термостата детектора 210 °C, испарителя 200 °C, скорость газа-носителя 75 мл/мин, продувочного газа (азот ос ч.) 150 мл/мин.
В этих условиях линейный динамический диапазон для изомеров гардоны составляет 0,2—5 нг, для хлорорганнческих соединений — 0,02-—0,2 нг.
Хлороформные очищенные экстракты анализируемых проб количественно переносят в пробирки с делениями, отгоняют хлороформ досуха. Окончательно пары хлороформа удаляют током воз духа или азота. Остаток растворяют в точно известном количестве гексана и используют для ГЖХ анализа. При проведении экстракции по способу экстракции диметилформамидом гексановые пробы известного объема непосредственно используют для ввода в колонку. В колонку вводят микрошприцем 2—5 мкл раствора.
Содержание препаратов в исследуемом объекте вычисляют по формуле
v
MV ’
где X— содержание препарата в пробе, мг/кг; Ci—концентрация стандартного раствора, мкг/мл; Hi и //2— высоты пиков стандартного и анализируемого растворов, мм; V] и Vs— объемы стандартного и анализируемого растворов, вводимые в колонку хроматографа, мкл; V3 — объем экстракта пробы для анализа, мл; Р —• навеска пробы, г.
2. Значения Rf гардоны и продуктов ее превращения
Система подвижных растворителей Гардона Цис-изомер гардоны ТХАФ ТХФХ ТХФЭ ТХФХЭ
Гексан — ацетон 0,23 0,23 0,58 0,51 0,42 0,42
(4: 1) 0,35 0,35 0,73 0,69 0,43 0,52
Гексан — этилаце- 0,21 0,17 0,62 0,62 0,57 0,57
тат (7 : 3) 0,34 0,28 0,72 0,75 0,67 0,67
В числителе — значения Rf на пластинках «Силуфол UV-254», в знаме-
нателе— значения Rf на пластинках из силикагеля КСК или кремниевой кислоты для люминофоров.
Методика определения гардоны в органах и тканях животных при диагностике отравления
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 6 января 1978 г.)
1. Характеристика препарата. Гардона (рабонт, тетрахлорвин-фос, S-8447, винилфосфат) — высококонтактный пестицид против ряда вредителей сельскохозяйственных культур и эктопаразитов животных. Препарат плохо растворим в воде (при температуре 20 °C растворяется 0,01 мг/л), большинстве растворителей, устойчив к действию температур до 100 °C. Медленно гидролизуется в кислой и нейтральной средах, быстрее в щелочной.
2. Принцип метода. Определение основано на извлечении ядохимиката из органов смесью хлороформа и гексана (1:2), а из крови — ацетоном, очистке экстрактов от коэкстрактивных веществ путем’ перераспределения в несмешивающихся растворителях, концентрировании экстрактов, хроматографировании гардоны в тонком слое сорбента в системе растворителей хлороформ — ацетон (98:2), проявлении пестицида аммиачным раствором азотнокислого серебра в ацетоне и облучении хроматографической пластинки ультрафиолетовыми лучами.
Чувствительность метода при использовании пластинок «Силуфол» 0,1 мг/кг, при использовании стеклянных пластинок — 1,0 мг/кг. Процент определения составляет 80+10.
3. Реактивы и растворы. Ацетон ч. д. а. Аммиак водный 25%-ный ч. д. а. Гардона 50%-ная с. п. н-Гексан х. ч. Натрий сернокислый безводный х. ч. Серебро азотнокислое х. ч. Хлороформ для наркоза. Силикагель марки КСК, просеянный через сито
100 меш. Гипс медицинский. Проявляющий реактив: 0,1 г азотнокислого серебра растворяют в 1 мл дистиллированной воды, добавляют 1 мл аммиака и доводят до 20 мл ацетоном.
Стандартный раствор гардоны в гексане: 200 мг гардоны (по д. в.) растворяют в 100 мл гексана. Из этого раствора берут 10 мл, переносят в другую мерную колбу на 100 мл и доводят до метки гексаном. 1 мл этого раствора содержит 200 мкг пестицида.
4. Приборы и посуда. Хроматографическая камера. Делительные воронки на 100—200 мл. Пульверизатор. Микрошприц или микропипетка. Чашки фарфоровые выпарительные. Конические колбы с притертыми пробками. Воронки стеклянные. Камера для опрыскивания хроматограмм. Хроматографические пластинки «Силуфол» производства ЧССР или стеклянные пластинки размером 9X12. Источник УФ-света — лампа ПРК-4.
Приготовление пластинок с тонким слоем сорбента. Стеклянные пластинки 9X12 моют водой, хромовой смесью, дистиллированной водой и сушат в вертикальном положении. Перед нанесением сорбционной массы пластинку протирают спиртом. Для приготовления пластинок с тонким слоем сорбента берут 35 г силикагеля, 2 г гипса, тщательно растирают в ступке, переносят в колбу, наливают 80 мл дистиллированной воды и встряхивают 10 мин, не допуская образования пузырьков воздуха. 10 г (примерно 1,5 чайной ложки) сорбционной массы наливают на пластинку и, покачивая, равномерно распределяют по пластинке. Сушат при комнатной температуре в течение 10—12 ч на ровном столе. Хранят в эксикаторе.
5. Ход определения. Исследуемую пробу патологического материала тщательно измельчают, берут навеску 10 г, помещают в колбу и проводят экстрагирование ядохимиката смесью хлороформа и гексана (1 : 2) дважды порциями по 20 мл (по 15 мин каждый раз). Экстракты отделяют через бумажный фильтр с безводным сернокислым натрием (5—6 г) в выпарительную чашку. После окончания фильтрования сернокислый натрий промывают 5 мл смеси растворителей. Экстракты упаривают досуха.
10 мл гепарированной крови помещают в коническую колбу, добавляют 20 мл ацетона и экстрагируют в течение 15 мин. Ско-агулированную массу отделяют через бумажный фильтр, который промывают 5 мл чистого ацетона. Водно-ацетоновый экстракт собирают в делительную воронку, приливают 30 мл хлороформа. Отделившийся хлороформно-ацетоиовый слой сливают в фарфоровую чашку и выпаривают досуха.
Сухой остаток в выпарительной чашке растворяют в 1 мл ацетона, добавляют 10 мл дистиллированной воды, переносят в делительную воронку и экстрагируют дважды в течение 2 мин 10 мл гексана, еще раз чашку ополаскивают 1 мл ацетона, добавляют воду и повторяют экстракцию гексаном. Верхний гексановый слой декантируют через бумажный фильтр, заполненный безводным сернокислым натрием, в выпарительную чашку, фильтр с сернокислым натрием промывают 5 мл чистого гексана в ту же выпарительную чашку. Экстракты упаривают до объема 0,2—0,3 мл и наносят на хроматографическую пластинку. Чашку дважды ополаскивают 0,5 мл гексана, неск'олько упаривают и наносят в ту же точку.
Для идентификации пятен пестицида и их количественной оценки на ту же пластинку наносят стандартный раствор гардоны, содержащий предполагаемое количество препарата в пробе.
Хроматографирование гардоны производится в два этапа: сначала пластинку помещают в хроматографическую камеру с гексаном до подъема фронта растворителя на 10 см, пластинку вынимают, сушат и вновь помещают в хроматографическую камеру с растворителем. Затем помещают в камеру с системой растворителей хлороформ — ацетон в соотношении 98 : 2 до подъема фронта растворителей на высоту 10 см. Пластинку вынимают и сушат в вытяжном шкафу до испарения растворителей.
Хроматограмму обрабатывают из пульверизатора аммиачным раствором азотнокислого серебра в ацетоне и облучают под лампой ПРК-4 светом в течение 5—8 мин. В качестве источника УФ-света можно также использовать прибор для флуоресцентного исследования.
На светло-сером фоне пластинки проявляются пятна темно-серого цвета с Rf 0,33+0,003. Такие распространенные пестициды, как ДДТ, ГХЦГ, хлорофос, не мешают определению гардоны.
Количественное определение гардоны производят путем визуального сравнения интенсивности окрашивания и размера пятен пробы с пятнами стандартного раствора.
Расчет содержания препарата в пробе производится по формуле
АС ----, РВ
где X-—содержание препарата, мг/кг; А— количество пестицида, найденное путем сравнения со стандартом, мкг; Р— масса пробы, г; В — количество исследуемого раствора, взятое для анализа, мл; С — общий объем исследуемого раствора, мл.
Если проба содержит менее 10 мкг гардоны, на пластинку количественно наносят весь остаток экстракта после упаривания. Для расчета используют формулу
А
Х==~Р~'
Предлагаемый метод предназначен для прижизненной и посмертной диагностики отравления сельскохозяйственных животных гардоиой. При постановке диагноза на отравление следует учитывать не только результаты определения гардоны в патматериале, но и наличие пестицида в воде, кормах, а также наличие характерных клинических, биохимических и патологоанатомических изменений с учетом анамнестических данных о применении ядохимиката в хозяйстве.
Методика экспрессной диагностики отравления пчел фосфорорганическими ядохимикатами
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 22 июня 1977 г.)
Принцип метода. Методика основана на свойстве эстераз (ферментов) гидролизовать 1-нафтиловый эфир уксусной кислоты, промежуточный продукт распада которого (1-иафтол) с солью — прочный синий Б — образует вещество, придающее жидкости розовое
окрашивание. Диагноз на отравление устанавливают по степени угнетения активности эстераз в мозгу исследуемых погибших пчел.
Метод является качественным и позволяет определить отравление пчел фосфорорганическими ядохимикатами в течение 9— 30 дней после их гибели в зависимости от условий хранения пат-материала. Продолжительность анализа 15—20 мин.
Реактивы, растворы и их приготовление. 1-нафтиловый эфир уксусной кислоты. Соль — прочный синий Б. Спирт этиловый. Натрий хлористый. Кислота серная, относительная плотность 1,84. Калий двухромовокислый. Вода дистиллированная. Раствор № 1: 5 мг 1-нафтилового эфира уксусной кислоты растворяют в 7,5 мл 96%-ного этилового спирта. Раствор № 2: 5 мг соли прочного синего Б растворяют в 8 мл дистиллированной воды. Оба раствора готовят в день проведения исследований. Физиологический раствор: 0,85 г хлористого натрия -растворяют в дистиллированной воде и общий объем доводят до 100 мл.
Хромовая смесь: 5 г двухромовокислого калия растворяют в 100 мл серной кислоты.
Приборы и посуда. Весы торсионные. Ступки фарфоровые. Пробирки лабораторные. Пипетки на 1—2 мл. Цилиндр мерный на 10 мл. Штатив для пробирок. Порошок из стекла. Посуда должна быть химически чистой, промытой хромовой смесью.
Ход анализа. Из общей пробы отбирают 10 погибших пчел с расставленными крыльями и вытянутым хоботком и с помощью ножниц у них отделяют головы. Изолированные головы растирают в ступке с порошком стекла, добавляют 4 мл физиологического раствора и переносят в пробирку.
Аналогичным образом готовят контрольный гомогенат голов от 10 здоровых пчел и обе пробирки после встряхивания оставляют на 5—10 мин для отстаивания При отсутствии здоровых пчел в качестве контроля используют нормальную лошадиную сыворотку, разведенную физиологическим раствором или дистиллированной водой в соотношении 1 : 40
Берут две чистые пробирки, в каждую вносят по 0,75 мл раствора № 1 и по 1,25 мл раствора № 2. В первую пробирку (контрольную) вносят 0,5 мл гомогената голов от здоровых пчел или разведенной лошадиной сыворотки, во вторую (опытную)—0.5 мл гомогената голов от исследуемых погибших пчел Анализ проводят при комнатной температуре.
В контрольной пробирке после добавления 0,5 мл разведенной лошадиной сыворотки раствор приобретает розовую окраску через несколько секунд, а при внесении гомогената голов от здоровых пчел — через I—2 мин. Цвет этих растворов отличается по оттенку.
В опытной пробирке после внесения гомогената голов пчел, погибших от отравления фосфорорганическими ядохимикатами, цвет раствора остается желтым. Спустя 30 мин и более этот раствор иногда приобретает бледно-розовую окраску. В контрольной пробирке раствор со временем темнеет.
Положительный результат анализа считается установленным в том случае, если ра,створ, содержащий 1-нафтиловый эфир уксусной кислоты и соль про'чного синего Б, в течение 2 мии после внесения гомогената голов исследуемых погибших пчел не приобретает розовую окраску.
Срок, в течение которого правильно диагностируется отравле
ние пчел, зависит от времени сохранения активности эстераз в мозгу здоровых насекомых со дня их гибели.
Эстеразы в мозгу пчел сохраняют свою активность при храпении мертвых насекомых:
а) в сухом месте при доступе воздуха—до 30 дней;
б) в сухой герметично закрытой посуде — до 15—19;
в) в воде при доступе воздуха—до 15—17;
г) в воде в герметично закрытой посуде — до 17;
д) сухими в закрытом целлофановом футляре — до 16;
е) мокрыми в закрытом целлофановом футляре — до 9 дней.
Положительный результат исследования пчел экспрессным методом при наличии на пасеке массовой их гибели, совпадающей с химическими обработками растений, указывает на отравление пчел фосфорорганическими ядохимикатами.
Временные методические указания по определению цианокса в мёде методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 31 июля 1984 г.)
Краткая характеристика препарата. Цианокс (цианофос) — О,О-диметил-О- (4-циапофенил) тиофосфат.
C9H10NO3SP, молекулярная масса 370,5.
Действующее вещество представляет собой жидкость светло-желтого цвета, температура плавления 14—15 °C. Цианокс практически нерастворим в воде, хорошо растворим в спиртах, кетонах, ароматических углеводородах.
ЛДбо для крыс 996 мг/кг.
Применяют в качестве инсектоакарицида на плодовых, цитрусовых и технических культурах.
Принцип метода. Метод основан на извлечении цианокса из водного раствора мёда гексаном, разрушении эмульсии этиловым спиртом и определении хроматографией в тонком слое окиси алюминия при обнаружении по реакции с AgNOs.
Метрологическая характеристика метода. Диапазон определяемых концентраций 0,5—30 мкг пестицида в анализируемой пробе; предел обнаружения 0,5 мкг или 0,05 мг/кг; размах варьирования 80—90%; среднее значение определения стандартных количеств цианокса 85%; стандартное отклонение 8,12%; относительное стандартное отклонение 0,09; доверительный интервал среднего прн Р = 0,05 и п = 9 85+8,29.
Селективность метода. Метод избирательный в присутствии хлорорганических пестицидов, так как под воздействием ультрафиолетовых лучей пятна цианокса исчезают.
Реактивы и материалы. Алюминия окись для хроматографии II степени активности. Аммиак водный 25%-ный ч. д. а. Ацетон ч. д. а. Гексан х. ч. Кальций сернокислый 2-водный ч. д. а., прокаленный при 160 °C в течение 6 ч. Натрий сернокислый безводный ч. д. а. Пергидроль, 30%-ный водный раствор х. ч. Серебро азотнокислое ч. д. а. Спирт этиловый ректификат. Цианокс, х. ч. или технический препарат.
Приборы, аппаратура, посуда. Аппарат для встряхивания АВУ-1 или аналогичный. Весы торсионные типа ВТ. Камеры хроматографические. Камера для опрыскивания хроматограмм. Пластинки стеклянные для хроматографии размером 9Х12 см. Посуда химическая (колбы на 100 мл, воронки делительные, пипетки, стаканы химические, цилиндры мерные). Опрыскиватели стеклянные. Термостат или сушильный шкаф. Чашки для выпаривания. Флакончики с притертыми пробками. Микропипетки на 0,1 и 0,2 мл.
Подготовка к определению. Приготовление стандартного раствора цианокса: 10 мг препарата растворяют в 100 мл ацетона (содержание пестицида составляет 100 мкг в 1 мл). Хранят на холоде в плотно закрытой посуде.
Приготовление проявляющего реактива: 0,1 г азотнокислого серебра растворяют в 1 мл дистиллированной воды, приливают 1,25 мл аммиака и 30 мл ацетона. Затем к раствору добавляют 0,1 мл пергидроля. Раствор готовят в день проведения анализа. На одну пластинку используют 6—8 мл проявляющего реактива.
Приготовление хроматографических пласти-н о к. Стеклянные пластинки моют хромовой смесью, затем водопроводной и дистиллированной водой и высушивают. Перед нанесением сорбента их протирают спиртом. Для приготовления сорбционной массы 50 г окиси алюминия для хроматографии, просеянной через сито 100 меш., смешивают равномерно в фарфоровой ступке с 5 г кальция сернокислого, переносят в колбу и прибавляют 75 мл дистиллированной воды. Смесь встряхивают в течение 20 мин до образования однородной массы, которую в количестве 8—10 г наносят на пластинку. Сушат пластинки с нанесенным сорбентом в строго горизонтальном положении в течение 12—18 ч при комнатной температуре, хранят в экскикаторе.
Проведение определения. Экстракция. Навеску меда в количестве 10 г помещают в химический стакан, растворяют в 20 мл дистиллированной воды и переносят в делительную воронку. Затем приливают 90 мл гексана, встряхивают воронку в течение 2 мин и оставляют на 5 мин для разделения жидкостей. Нижний (водный) слой отбрасывают, а к верхнему с целью разрушения эмульсии добавляют 0,5 мл 96%-hoi о этанола. Через 5 мин делительную воронку 4—5 раз энергично встряхивают и после просветления верхнего слоя нижний отбрасывают, а содержимое воронки переносят в чистый химический стакан и сушат 10—15 г безводного сернокислого натрия в течение 15—20 мин.
Затем растворитель упаривают в вакуумном ротационном испарителе или в токе теплого воздуха от электротепловентилятора (температура воздуха возле чашек не более 40 °C) до небольшого объема или оставляют на ночь под вытяжкой. Досуха остаток гексана упаривают при комнатной температуре, затем содержимое растворяют в 0,5 мл гексана, количественно переносят в мерную центрифужную пробирку и наносят на пластинку.
Хроматография в тонком слое. Гексановый экстракт с помощью микрошприца или микропипетки наносят на хроматографическую пластинку на расстоянии 1,5 см от ее нижнего края. Диаметр пятна не должен превы'шать 1 см. Рядом с опытной пробой наносят известные количества цианокса в ацетоне. Пластинку с пробой н стандартными растворами цианокса ставят левым краем в хроматографическую камеру с гексаном. После прохождения фронта рас
творителя через всю пластинку ее вынимают из камеры и оставляют на несколько минут для испарения гексана. Затем пластинку хроматографируют в системе подвижных растворителей гексан — ацетон (21 : 3), повернув ее на 90° по сравнению с первой разгонкой. После подъема растворителей до линии фронта пластинку вынимают из камеры и оставляют на воздухе на 5—10 мин.
Проявление пластинок. После испарения растворителей пластинку ставят в камеру для опрыскивания, обрабатывают проявляющим реактивом и сразу же помещают на 10 мин в термостат с температурой 36—40 °C.
Остатки цианокса проявляются в виде темно-серых пятен на белом фоне с R1 0,58—0,60.
Обработка результатов. Количественное определение проводят путем сравнения интенсивности окраски и величины пятна пробы и стандартных растворов цианокса, используя формулу расчета
Л32
Х=—-, SjP
где X— количество цианокса в анализируемом образце, мг/кг; А— содержание цианокса в хроматографируемом объеме стандартного раствора, мкг; S1—площадь пятна на хроматограмме стандартного раствора, мм2; S2 — площадь пятна на хроматограмме пробы, мм2; Р — масса анализируемой пробы, г.
Временные методические указания по определению остаточных количеств омайта в мёде методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 31 июля 1984 г.)
Краткая характеристика препарата. Омайт (пропаргит) —• 2- (п-трет-бутилфеиокси) -циклогексил-2-пропалгилсульфит — темно-коричневая жидкость, нерастворим в воде, растворим в большинстве органических растворителей. Молекулярная масса 326,0.
ЛДзо для крыс и мышей 1800—2000 мг/кг. Малотоксичен для пчел.
Используется для борьбы с растительноядными клещами на плодовых культурах и хлопчатнике путем опрыскивания.
Принцип метода. Метод основан на определении омайта методом тонкослойной хроматографии, после предварительного извлечения его из анализируемой пробы хлороформом и очистки полученной хлороформной вытяжки от восковой пленки путем пропускания последнего через колонку с окисью алюминия.
Хроматографирование проводят на пластинках «Силуфол».
Метрологическая характеристика. Диапазон определяемых концентраций 0,3—3 мг/кг. Предел обнаружения 10 мкг в пробе или 0,3 мг/кг. Размах варьирования 70,0—85,0%. Среднее значение определения 77,5%. Доверительный интервал при а = 0,95 п = 5 77,5+11,5%.
Реактивы и растворы. Ацетон ч. д. а. Серебро азотнокислое х. ч. Кислота лимонная х. ч. Окись алюминия для хроматографии. Хлороформ х. ч. н-Гексан х. ч. Подвижная фаза: н-гексан — ацетон
(4:1). Проявляющий реактив: 0,03 г бромфенолового синего растворяют в 10 мл ацетона и доводят до 100 мл 0,5%-ным водно-ацетоновым раствором азотнокислого серебра (1 часть воды и 3 части ацетона); 5°/о-ный раствор лимонной кислоты в смеси вода— ацетон (1:3). Стандартный раствор № 1 омайта в ацетоне с содержанием препарата 500 мкг/мл. Стандартный раствор № 2 омайта в ацетоне с содержанием 100 мкг/мл. Стандартные растворы устойчивы в течение 2 мес в холодильнике.
Приборы и посуда. Хроматографические пластинки «Силуфол UV-254». Делительные воронки емкостью 100 мл. Мерные колбы емкостью 100 мл. Пульверизаторы стеклянные. Камера хроматографическая. Шприцы медицинские емкостью 1 мл. Баня водяная. Чашки фарфоровые для выпаривания диаметром 4—5 см. Колонки стеклянные высотой 20—25 см и внутренним диаметром 1,3 см. Микропипетка емкостью 0,1, 0,2 мл с оттянутыми концами. Цилиндры мерные емкостью 50—100 мл.
Подготовка к определению. Приготовление хроматографической колонки. Колонку заполняют хроматографической окисью алюминия (высота слоя 10 см). Адсорбент перед употреблением промывают 15 мл хлороформа.
Отбор проб для анализа производят в соответствии с «Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов», утвержденными Минздравом СССР 21 августа 1979 г. № 2051—79.
Проведение определения. Экстракция препарата из анализируемой пробы. К 30 г мёда (если мёд законсервирован, его предварительно прогревают на водяной бане при 50 °C до полного разжижения) добавляют 2 мл горячей дистиллированной воды, хорошо размешивают стеклянной палочкой и помещают в делительную воронку, куда добавляют в 2 приема по 30 мл хлороформа. Воронку встряхивают по 10 мин. Хлороформные экстракты объединяют и упаривают на водяной бане до 2 мл при температуре 60 °C.
Хроматография в тонком слое. Полученный экстракт количественно вносят в хроматографическую колонку, откуда омайт элюируют 40 мл хлороформа. Элюат вновь упаривают на водяной бане до 2—3 мл при температуре 60 °C. Последние 2—3 мл элюата удаляют в струе воздуха без подогрева. Полученный сухой остаток растворяют в 0,1 мл хлороформа. На стартовую часть хроматографической пластинки количественно наносят 0,1 мл раствора пробы. Параллельно наносят серию стандартных растворов с содержанием 10; 20; 30; 50; 100 мг действующего вещества омайта. Диаметр пятен не должен превышать 1 см. Пластинку сушат на воздухе, затем помещают в камеру, на дно которой за 20 мин до хроматографирования помещают подвижной растворитель н-гексан — ацетон (4: 1). После подъема подвижной фазы на высоту 10 см пластинку вынимают из камеры и сушат на воздухе до полного испарения растворителя. Далее пластинку опрыскивают проявляющим реагентом. Омайт проявляется в виде пятен синего цвета на желтом фоне пластинки.
Rf омайта в системе н-гексан — ацетон (4:1) составляет 0,45+0,05. Окраска пятен устойчива в течение 1 ч.
Количественное определение омайта производят путем сравнения площади пятен стандартного раствора с площадью пятен раствора
пробы. Площадь пятен Определяют с помощью промасленной миллиметровой бумаги.
Обработка результатов. Содержание омайта в мг/кг рассчитывают по формуле
где X — содержание омайта, мг/кг; А — количество омайта, найденное путем сравнения со стандартом, мкг; Р — навеска анализируемой пробы, г.
Ферментный метод определения фосфорорганических пестицидов в патологическом материале
(Одобрен Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Метод рекомендуется для определения фосфорорганических соединений (хлорофоса, метафоса, карбофоса, трихлорметафоса-3, метилнитрофоса, метилмеркаптофоса, ДДВФ, диброма) в органах и тканях животных.
Принцип. Метод основан на способности фосфорорганических соединений (ФОС) угнетать активность холинэстеразы с учетом времени изменения окраски бромтимолового синего вследствие образования уксусной кислоты при гидролизе ацетилхолина ферментом. О количествах пестицида в пробе судят по степени угнетения активности фермента, выраженной в процентах. Изменение окраски учитывают визуально. Расчет производят по калибровочному графику. Чувствительность метода 0,1 мг/кг анализируемого продукта. Точность метода 80%.
Приборы и посуда. Баня водяная. Термометр химический. Пробирки Колбы мерные емкостью 100 и 500 мл. Пипетки 1, 2, 5 и 10 мл. Склянки широкогорлые с притертыми пробками. Стаканы химические.
Реактивы. Ацетилхолинхлорид. Бромтимоловый синий. Борная кислота. Калий хлористый. 0,1 н. раствор соляной кислоты. 0,1 и. раствор едкого натра. Хлороформ х. ч. Холинэстераза V и VII классов или сыворотка крови лошади.
Приготовление реактивов. 1. Основной индикаторнобуферный раствор (раствор № 1). Для приготовления раствора берут 100 мг бромтимолового синего, растирают в ступке с 2 мл 0,1 н. раствора едкого натра и количественно переносят в мерную колбу емкостью 500 мл. Ступку ополаскивают несколько раз небольшим количеством прокипяченной дистиллированной воды, которую сливают также в мерную колбу. В эту же колбу добавляют 8 мл 0,1 н. раствора едкого натра и 25 мл 0,1 М раствора борной кислоты в 0,1 М растворе хлорида калия (в 25 мл дистиллированной воды растворяют 187,5 мг хлорида калия и 155 мг борной кислоты). Полученный раствор имеет интенсивно-синий цвет (pH 8,65—8,7), стоек при хранении в склянке из нейтрального стекла с притертой пробкой
2. Индикаторно-буферный раствор (раствор № 2) предназначен для приготовления колориметрического стандарта. Для его приготовления берут 9,85 мл основного раствора № 1 и смешивают с
0,15 мл 0,1 н. раствора соляной кислоты. Раствор окрашен в зеленый цвет.
3. 1%-ный раствор ацетилхолинхлорида: 0,2 г ацетилхолинхло-рида растворяют в 20 мл дистиллированной воды, вносят 2— 3 капли раствора № 1 и усредняют 0,1 н. раствором едкого натра по бромтимоловому синему до появления зеленой или сине-зеленой окраски. Раствор пригоден для работы в течение 7 дней при хранении в холодильнике.
4. Индикаторно-буферный раствор ацетилхолина (раствор № 3). В химическом стаканчике смешивают 27 мл раствора № 1 с 3 мл 1%-ного раствора ацетилхолинхлорида (цвет раствора синий). Готовят перед проведением анализа.
5. Холинэстераза разводится дистиллированной водой с таким расчетом, чтобы время изменения цвета в контроле составляло 10—15 мин.
При использовании лошадиной сыворотки, если время протекания реакции в контроле значительно меньше 10 мин, сыворотку разводят дистиллированной водой
Определение. Исследуемый материал тщательно измельчают, после чего навеску в 20—30 г помещают в склянку с притертой пробкой, заливают адекватным количеством хлороформа и экстрагируют не менее 1 ч при периодическом перемешивании. Затем пипеткой, нижний конец которой обернут ватой, набирают 1 мл хлороформенного экстракта и переносят в химический стаканчик. В контрольный стаканчик вносят 1 мл хлороформа. Оба стаканчика помещают в водяную баню (60 °C) или под струю воздуха для полного испарения хлороформа. К сухому остатку прибавляют по 1 мл дистиллированной воды и стаканчики помещают в водяную баню на 3 мин. После этого стаканчики охлаждают до комнатной температуры и вносят в них по 1 мл раствора холинэстеразы или сыворотки крови лошади. Содержимое хорошо перемешивают и ставят стаканчики в водяную баню (38—39 °C) на 30 мин По истечении этого времени в две пробирки берут по 0,5 мл содержимого каждого стаканчика, прибавляют по 4,5 мл раствора № 3 и отмечают время. Для приготовления колориметрического стандарта в отдельную пробирку вносят 0,5 мл содержимого контрольного стаканчика и 4,5 мл раствора № 2. Отмечают время уравнивания цвета в контрольных и опытных пробирках с цветом стандарта.
Процент угнетения активности холинэстеразы (X) высчитывают по ф< рмуле
В-1С0
Х=100 — —-— , о
где X— угнетение активности холинэстеразы, %; В — время уравнивания цвета в контрольных пробирках с цветом стандарта, мин; Ь — время уравнивания цвета в опытных пробирках со стандартом, мин
Если в опытных пробирках отмечено угнетение активности холинэстеразы более 10%, что свидетельствует о наличии ФОС в исследуемом материале, проводят количественное определение ФОС по калибровочному графику.
Построение калибровочного графика проводят по препарату, наличие которого предполагается в исследуемом материале. Если же в материале подозреваются неизвестные фосфорорганические
соединения, то калибровочный график строят по одному из ядохимикатов, перечисленных выше. Для этого необходимо иметь химически чистое соединение или препараты с известным содержанием активно действующего вещества (АДВ). Из заведомо чистого образца исследуемого материала в склянки с притертыми пробками берут пять навесок по 10 г и вносят в них соответственно 1,0; 5,0; 10,0; 20,0; 50,0 мкг препарата по АДВ. Через 30 мин навески заливают 10 мл хлороформа и экстрагируют 1 ч. Хлороформные экстракты исследуют в описанном выше порядке. На оси ординат откладывают величины угнетения активности холинэстеразы, на оси абсцисс — количество препарата в мг/кг (по АДВ). Рабочий участок, построенный таким образом, имеет форму равнобочной гиперболы.
РТУТЬСОДЕРЖАЩИЕ СОЕДИНЕНИЯ
Методические указания по диагностике, профилактике и лечению отравлений сельскохозяйственных животных ртутьорганическими пестицидами *
Извлечение из приложения № 1: Методика дробного определения ртути в трупном материале
Принцип метода. Метод основан на деструкции форменных элементов тканей и последующем осаждении ртути в деструктате йодидом меди. Чувствительность метода 0,05 мк/кг, точность 98%.
Реактивы и растворы. Йодид меди — взвесь: 212 г. йодида калия растворяют в небольшом количестве воды и смешивают с 800 мл 20%-ного раствора сульфата меди. С образовавшегося осадка йодида меди жидкость сливают декантацией. Осадок промывают несколько раз большим количеством воды до светло-желтого цвета жидкости над осадком. Отмытый осадок йодида меди обрабатывают несколькими миллилитрами 2,5 н. раствора сульфита натрия до получения бесцветной взвеси. Добавляют насыщенный раствор сульфата натрия до коагуляции осадка. Жидкость сливают декантацией. Осадок переносят на фильтр и промывают на фильтре водой до слабоположительной реакции промывных вод на сульфат-ион. Фильтр прокалывают п взвесь смывают в мерный цилиндр. Объем доводят до 1 л водой. Взвесь хранят в банке темного стекла.
Этанол. Кислота азотная конц., х. ч. Кислота серная конц., х. ч. Меди сульфат 20 и 10%-ный растворы Натрия сульфат 1%-ный раствор. Натрия бикарбонат 8%-ный раствор. Натрия сульфит 2,5 н. раствор. Раствор йода 0,25%-ный в 3%-ном растворе йодида калия (поглотительный раствор). Раствор йода 0,35%-ный в 3%-ном растворе йодида калия.
Раствор составной: готовят перед употреблением смешением растворов 10%-ного сульфата меди и свежеприготовленного 2,5 н. раствора сульфита натрия, взятых в соотношении 1 :2. Если при этом раствор не становится прозрачным через 1—2 мин, каплями добавляют насыщенный раствор сульфита натрия до получения прозрачного раствора. После этого добавляют 1,5 объема бикарбоната натрия.
Стандартный раствор ртути с содержанием 0,01 мг ртути в 1 мл 0,25%-ного раствора йода в 3%-ном растворе йодида калия. Готовится растворением 135,3 мг дважды перекристаллизованной двухлористой ртути в 1 л 0,25%-ного раствора йода в 3%-ном растворе йодида калия. Для приготовления раствора, содержащего 0,01 мг ртути в 1 мл, полученный раствор разбавляют в 10 раз поглотительным раствором.
Приборы и посуда. Баня водяная. Колбы на 200—500 мл. Пипетки 1, 5, 10 мл. Цилиндры 100—500 мл. Стаканы химические на 500 мл. Воронки стеклянные, пробирки.
Изолирование ртути. Средние пробы печени и почек по 20 г (исследование смеси органов не допускается) помещают в две конические колбы емкостью 200 мл. В каждую пробу приливают 10 мл воды, 1 мл этанола и 10 мл концентрированной азотной
кислоты. К смеси добавляют по каплям 10 мл концентрированной серной кислоты с такой скоростью, чтобы постоянно поддерживалась реакция разложения азотной кислоты, но окислы азота при этом не должны выделяться пз колбы. После окончания добавления серной кислоты колбу оставляют при комнатной температуре на 10—15 мин до прекращения выделения окислов азота. Затем содержимое колбы нагревают на кипящей водяной бане в течение 20 мин (в случае, если кусочки тканей остались неразрушенными из-за обволакивания их жиром, необходимо осторожно растереть их палочкой о стенки колбы).
При бурной реакции в колбу добавляют 30—50 мл воды. Горячий деструктат смешивают с двойным объемом горячей воды и, не охлаждая, фильтруют через двойной, предварительно увлажненный бумажный фильтр. Фильтр и остатки жира промывают не менее 3—5 раз горячей водой. Промывные воды (разведенный мине-рализат) объединяют в мерной колбе и определяют ртуть.
Определение ртути. К половине объема деструктата добавляют 5 мл 2,5 п. раствора сульфита натрия, воды до 250 мл и 10 мл взвеси йодида меди (если взвесь йодида меди приобретает бурую окраску вследствие выделений йода, то прибавляют дополнительно по 1—2 мл сульфита натрия). Если взвесь йодида меди окрасится в розовый или ярко-оранжевый цвет, дополнительно добавляют еще 30 мл взвеси. Через 30 мин взвесь отфильтровывают через плотный бумажный фильтр и промывают 1%-ным раствором сульфата натрия до полного отмывания желтой окраски и pH 5—6 последней фракции промывных вод.
Промытый осадок обрабатывают на фильтре точным объемом 0,35%-ного раствора йода в 3%-ном растворе йодида калия. Объем его зависит от окраски взвеси (ориентировочные данные представлены в таблице 1).
1. Зависимость окраски взвеси от количества ртути и объема раствора йода, необходимого для растворения ртути
Количество ртути, мг Окраска взвеси йодида меди Объем рас гвора йода для растворения [>TyJИ, МЛ
10 мл 40 мл
0,001—0,005 Бесцветная 6
0,01—0,025 Светло-розовая Бесцветная 10
0,05—0,1 Розовая Светло-розовая 20
0,2—0,5 Кирпично-красная Розовая 50
0,5—1,0 » Ярко-розовая 50
2,0 » Кирпично-красная 100
Колориметрическое определение ртути производится параллельно в трех объемах полученного раствора. Исключение составляет случай, когда взвесь йодида меди обрабатывалась только 6 мл раствора йода, которые используются полностью для одноразового колориметрирования.
Объемы раствора йода подбирают для колориметрирования с таким расчетом, чтобы содержание ртути было в пределах 2—4, 3—6, 6—10 мкг стандартной шкалы (оптимальная область колориметрирования) .
Объем колориметрируемого раствора доводят до 6 мл 0,25 %-ным раствором йода в йодиде калия и добавляют по 4 мл составного раствора. Заключение о количестве ртути дают по среднему значению из трех определений: количество найденной ртути выражают в мг в пересчете на 100 г органа.
Формула расчетов
gy-2-ЮО у1«-1000 где X— количество ртути в 100 г органа, мг; а—количество ртути в колориметрируемом объеме, мкг; у — объем раствора йода, взятый для растворения, мл; yt — объем раствора йода, взятый для колориметрирования, мл; п — навеска органа, г.
Стандартную шкалу готовят в интервале от 1 до 10 мкг и хранят в холодильнике не более 3—5 дней (табл. 2).
2. Стандартная шкала раствора ртути
№ пробирки Содержание ртути, мкг Объем стандартного раствора ргути (0,01 МГ 8 1 мл), мл Объем ПОГЛО1И-гельного paciBopa, мл Объем сое равного раствора, мл
1 1,0 0,1 5.9 4
2 2,0 0,2 5,8 4
3 4,0 0,4 5,6 4
4 6,0 0,6 5,4 4
5 8,0 0,8 5,2 4
6 10 1,0 5,0 4
7 — — 6,0 4
Контрольная
Методические указания по определению ртутьорганических пестицидов в овощах, продуктах животноводства, кормах и патматериале хроматографическими методами (Утверждены заместителем Главного государственного санитарного врача СССР 23 января 1975 г.)
Краткая характеристика препаратов. Соли алкил-, метоксиэтил-и фенилртути представляют собой белые кристаллические вещества со специфическим зйпахом. По сравнению с алкилртутью метоксиэтил- и фенилмеркурацетат менее летучие соединения. Т пл ИХ СО~ ответственно 65 и 148 °C. Они растворимы в метаноле, ацетоне, бензоле. Фенилмеркурацетат хорошо растворим в воде и этаноле.
Принцип метода. Идентификация ртутьорганических пестицидов основана на реакции их йодистых солей с дитизоном с образованием окрашенных в оранжевый цвет дитизоиатов, алкил- и арил-ртути, которые определяют в тонком слое окиси алюминия Для извлечения соединений ртути из биологических материалов применяют два способа первый — экстрактивная перегонка алкилртути из солянокислой среды с последующей экстракцией веществ толуо лом, второй способ — применяют жидкость жидкостное распределение и ртутьорганические пестициды извлекают смесью 1 н соляной кислоты и этанола, а из вытяжки вещества экстрагируют бензолом
Метрологическая характеристика метода Степень определения фенилртути равна 55—60%, а метил-, этил и метоксиэтилртути — 80—90% Воспроизводимость метода 90%, относительная ошибка определения при полуколичественной оценке 30—40% Нижний предел определения 0,5 мкг органической ртути в навеске, т е 10 мкг/кг при величине пробы 50 г
Реактивы и растворы Тиосульфат натрия, 0,05 М раствор Готовят из фиксанала и хранят в склянках из темного стекла Рабочий 0,0025 М раствор тиосульфата натрия готовят перед Употреблением, смешивая 5 мл 0,05 М раствора, 45 мл дистиллиро ванной воды 50 мл этилового спирта Дитизон, 0,2% ный раствор (основной) в хлороформе в делительной воронке на 100 мл хлороформа добавляют 50 мг дитизона, 200 мл дистиллированной воды и 5—10 мл концентрированного аммиака Смесь энергично встряхивают 2 мин После разделения слоев хлороформный слой отбрасывают, водный раствор дитизона промывают 20 мл хлороформа Трубку делительной воронки высушивают фильтровальной бума гой, прибавляя в воронку 200 мл хлороформа и разбавленную (I 1) соляную кислоту до отчетливо кислой реакции Смесь встря хивают до тех пор, пока дитизон не перейдет в хлороформ (слой хлороформа при этом окрашивается в темно зеленый цвет, водная фаза обесцвечивается) Хлороформный слой отделяют в другую делительную воронку и промывают водой трижды по 50 мл Полученный раствор дитизона в хлороформе сливают в темную склянку Хранят ее в темноте на холоде Раствор устойчив в течение 1 мес Дитизон, 0,002% ный раствор готовят перед употреблением, разбавляя основной раствор хлороформом в 100 раз
Калий йодистый, 3 М раствор свежеприготовленный (раство ряют 10 г соли в 20 мл воды и добавляют 2 капли 1 н гидроокиси натрия) Соляная кислота, 1 н раствор (смешивают 50 мл концеп трированной соляной кислоты удельной массой 1,9 с 550 мл дистиллированной воды) Смесь 1 н соляной кислоты и этанола (7 3) по объему Хлорная медь, 0,5 М раствор в 2,5 н соляной кислоте Можно использовать хлористую медь, растворяя 5 г соли в 20 мл концентрированной соляной кислоты и добавляя 80 мл дистиллированной воды
Стандартные растворы ртутьорганических пестицидов растворяют по 10 мг метилмеркурйодида, этилмеркурхлорида, метокси-этилмеркурацетата или фенилмеркурацегата в 10 мл бензола Можно использовать препаративные формы пестицидов В этом случае взвешивают количество препарата, эквивалентное 10 мг активно действующего вещества, и вносят в 10 мл бензола Растворы хранят на холоде Они устойчивы в течение 3 мес Рабочие растворы ртутьорганических соединений в концентрации 2 мкг/мл’ 0,1 мл
основного раствора пестицида вносят в колбу на 50 мл и доводят объем до метки ацетоном Растворы устойчивы 2 нед Бензол, то луол, хлороформ х ч или ч д а
Приборы и посуда. Гомогенизатор Механический встряхива-тель Хроматографическая камера объемом 1—2 л Воронки для фильтрования диаметром 5 и 10 см Делительные воронки на 500 250 и 50 мл Мерные цилиндры на 25, 50 и 100 мл Пипетки на 1, 2, 5 и 10 мл Микропипетки на 0,1—0,2 мл Пробирки с притертой пробкой градуированные на 10 мл Чашки фарфоровые для выпа ривания Колбы на 250 и 500 мл
Подготовка к определению.
Синтез метилртути В круглодонную колбу на 50 мл вносят 5 мл йодистого метила и 7 г металлической ртути Колбу при соединяют к обратном^! холодильнику и смесь облучают в течение 6—10 ч ультрафиолетовой лампой низкого давления до тех пор, пока смесь не затвердеет в желтую массу Метилмеркурйодид экстрагируют ацетоном, фильтруют, концентрируют и перекристалли зовывают из метанола или ацетона ТПл вещества 140,5 °C
Получение этилмеркурхлорида из гранозана 100 г розового гранозана переносят в сухую колбу на 1 л, куда добавляют 250 мл юрячего 1 н раствора соляной кислоты Содержимое взбалтывают в течение 1 ч на механическом встряхивателе Суспензию фильтруют через бумажный фильтр в колбу на 750 мл, а осадок промыва ют 150 мл горячей 1 н соляной кислоты К фильтрату добавляют 100 мл бензола и вновь колбу помещают во встряхиватель на 1 ч Затем содержимое переливают в делительную воронку на 500 мл Нижнюю солянокислую фазу сливают в колбу на 1 л, а бензоль ный экстракт пропускают через фильтровальную воронку со стекловатой и хлористым натрием (слой 1 см) в фарфоровую чашку для выпаривания Фильтр промывают 10 мл бензола и присоеди няют к экстракту Растворитель отгоняют в струе воздуха (тяга) Для удаления воды чашку оставляют на ночь На следующий день остаток снимают со стенок чашки, растирают и используют в ка честве стандарта Выход продукта 50% с содержанием основного вещества 97,4%
Приготовление хроматографических пластинок. Окись алюминия II степени активности для хроматографии, просеянная через сито 100 меш, 50 г смешивают в фарфоровой ступке с 3,5 г просеянного и прокаленного гипса, а затем высыпают в колбу емкостью 500 мл Добавляют 60 мл воды и смесь встряхивают 2 мин Свободную от пузырьков воздуха массу распределяют на пластинках размером 9X12 см (по 2 чайные ложки) и высушивают на воздухе Перед употреблением пластинки со слоем адсорбента прокаливают в сушильном шкафу при 105 °C в течение 2 ч и хранят в эксикаторе над безводным хлористым кальцием
Ход анализа.
Экстракция метил- и этилмеркурхлорида из пробы и очистка экстракта Пробу 10 г гомогенизированного или растертого образ ца (зерно, корм, рыба, мясо, патматериал) помещают в круглодон ную колбу на 250 мл, куда добавляют 60 мл 1 н раствора соля ной кислоты, 2 мЛ> 0,5 М раствора хлорной меди, несколько ку сочков пемзы или стеклянных капилляров Сухие образцы увлаж няют 10 мл воды Колбу присоединяют к холодильнику с помощью шлифа и нагревают на электроплитке с прокладкой асбеста При
появлении пены верхнюю часть колбы охлаждают холодной водой. После прекращения вспенивания нагревание усиливают.
Отгон (дистиллят) собирают в делительную воронку, куда предварительно наливают 6 мл толуола. Собирают 60 мл дистиллята. Колбу охлаждают, вносят в нее 20 мл толуола, присоединяют к холодильнику и вновь производят дистилляцию в ту же воронку. Холодильник промывают 5 мл смеси 1 н. соляной кислоты и этанола (7:3) и раствор сливают в приемник. Полученный отгон энергично встряхивают 5 мин. После расслаивания нижнюю фазу отбрасывают, а толуольный экстракт промывают 25 мл воды, встряхивая воронку 3—4 раза. Затем экстракт фильтруют через стекловату или обычную вату, промытую толуолом, в делительную воронку на 50 мл. Фильтр промывают 5 мл растворителя. К экстракту добавляют 5 мл 0,0025 М раствора тиосульфата натрия в разбавленном этаноле и смесь энергично встряхивают 2 мин. После разделения фаз нижнюю сливают в третью делительную воронку, а толуольный экстракт еще раз обрабатывают 5 мл раствора тиосульфата натрия.
К объединенным этанольным растворам тиосульфата натрия добавляют 2,5 мл 3 М раствора йодистого калия, 5 мл бензола и содержимое энергично встряхивают 2 мин. Верхний бензольный экстракт сливают в фарфоровую чашку. Если в конечном экстракте присутствует вода, то бензол осторожно переносят в другую сухую чашку, оставляя водную фазу. Экстракт концентрируют до 0,1—0,2 мл, предварительно добавив к нему 3—4 капли 0,02%-ного раствора дитизона При этом раствор приобретает зеленоватый оттенок. Если экстракт желтеет, то следует еще раз добавить раствор дитизона до устойчивой зеленоватой окраски.
Экстракция метил-, этил-, метоксиэтил- и фенилртути экспресс-методом и очистка экстракта. Пробу 25 г растертого или гомогенизированного образца (органы, ткани животных, рыба, мясо, пат-материал, яйцо или 50 г овощей, целого зерна, почвы, зеленых кормов) перенести в колбу на 250 мл. Внести 100 мл горячей (80 °C) 1 н. соляной кислоты и этанола (7: 3) и 2 мл растворт хлорной меди. При анализе молока в колбу объемом 250 мл влить 50 мл молока, добавить 50 мл спирта, 2 мл раствора хлорной меди, 10 мл концентрированной соляной кислоты и содержимое быстро перемешать. Затем внести 50 мл кипящей воды и вновь перемешать. Комбикорм (25 г) смочить 20 мл воды. Встряхивать колбы в течение 1 ч. После этого внести 10 мл 40%-ного раствора фосфорно вольфрамовой кислоты, перемешать и через 10 мин содержимое отфильтровать через складчатый бумажный фильтр.
Осадок промыть 50 мл смесн соляной кислоты и этанола (7:3). К фильтрату добавить 50 мл бензола и колбы встряхивать 1,5 ч. Содержимое перенести в делительную воронку на 250 мл, используя 10 мл бензола. Отбросить нижнюю водную фазу, а экстракт промыть 50 мл воды, слегка перемешивая содержимое (3— 4 раза). Нижнюю фазу отбросить, а бензольный экстракт перенести в следующую делительную воронку на 250 мл, профильтровав его через вату, промытую бензолом. Реэкстрагировать соединения ртути 0,0025 М раствором тиосульфата натрия (2 раза по 10 мл) в течение 2 мин
К реэкстракту добавить 5 мл 3 М раствора йодистого калия и ртутьорганические пестициды, извлечь 5 мл бензола в течение 2 мин. Бензольный экстракт концентрируют, как и в первом случае, до-132
бавив к нему 3—4 капли раствора дитизона, или используют для газохроматографического анализа. В последнем случае 1 мл экстракта встряхивают с 1 мл 1%-ного раствора йодистого калия в 0,1 н. NaOH в течение 1 мин перед анализом.
Приготовление свидетелей. Пробы 0,5—1 мл стандартных разбавленных растворов этилмеркурхлорида или других соединений (1—4 мкг) вносят в делительную воронку на 50 мл, содержащую 10 мл 0,0025 М тиосульфата натрия. Содержимое перемешивают, добавляют 2,5 мл 3 М раствора йодистого калия, 5 мл бензола и экстрагируют соединения ртути 2 мин. Полученные экстракты упаривают, обработав 0,02%-ным раствором дитизона.
Хроматографирование. Пробы или свидетели наносят на пластинку микропипеткой или пипеткой Пастера в следующей последовательности: свидетель (1 мкг), образец (1-я повторность), образец (2-я повторность), свидетель (2 мкг). Остаток в чашке смывают 0,002%-ным раствором дитизона в хлороформе и наносят его на пластинку. Размер пятен не должен превышать 5 мм. Места нанесения проб располагают на расстоянии 1,5—2 см от края пластинки, и они не должны погружаться в растворитель. В камеру для хроматографирования (эксикатор) наливают 50 мл смеси гексана и ацетона (4: 1), помещают ленту фильтровальной бумаги шириной 5 см так, чтобы края ее достигали верхнего уровня сосуда. Смачивают ленту смесью растворителей и через 20—30 мин помещают пластинку в вертикальном положении. Камеру герметично закрывают, смазывая крышку Апьезоном L или высоковакуумной смазкой, и после того как растворитель поднимется на высоту 9—10 см, шмспгнку выкимагог, огмечаюг фроггг растворителя и высушивают при комнатной температуре.
Органические соединения ртути выявляются в виде желто-оранжевых пятен со следующим значением Rf: фенилртуть 0,35+0,02, метоксиэтилртуть 0,42+0,02, метилртуть 0,48+0,02. Значение Rf ртутьорганических соединений может колебаться в зависимости от условий хроматографирования, поэтому идентификацию органической ртути в пробах следует проводить с учетом Rf свидетелей. Количество органической ртути в пробе определяют сравнением интенсивности окраски и площади пятна свидетелей и образцов не более чем через 1 ч после высушивания хроматограммы, так как в процессе хранения пластинки пятна обесцвечиваются.
Для количественного определения веществ зоны, соответствующие пятнам органической ртути, соскабливают с пластинки. Адсорбент помещают на бумажный фильтр, промытый хлороформом, и элюируют дитизонаты 2 мл того же растворителя. Измеряют оптическую плотность экстракта с синим светофильтром на фотоэлектроколориметре при длине волны 485 нм. Параллельно измеряют оптическую плотность свидетелей (1—5 мкг). Для сравнения используют хлороформ.
Обработка результатов анализа. Содержание метил-, этил-, ме-токсилэтил- или фенилртути в исследуемом образце (А, мг/кг) вычисляют по формуле
где А — содержание'органической ртути в навеске, мкг; Р — масса пробы, г.
Методические указания по определению содержания общей ртути в мясе, мясопродуктах, яйцах, рыбе, молочных продуктах, шоколаде, почве колориметрическим способом или при помощи тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 19 октября 1979 г.)
Принцип метода. Метод основан на деструкции анализируемой пробы смесью азотной и серной кислот в присутствии этилового спирта и дальнейшем определении ртути колориметрическим способом или при помощи тонкослойной хроматографии.
Колориметрическое определение основано на осаждении ртути йодидом меди или на экстракции ее дитизоном и последующем визуальном колориметрическом определении в виде тетрайодомер-куроата меди путем сравнения со стандартной шкалой.
Диапазон определения концентраций 0,25—2,00 мкг в колориметрируемом объеме. Предел обнаружения 0,25 мкг или 0,125 мг/кг.
Метрологическая характеристика метода дана в таблицах 1 и 2.
Метод тонкослойной хроматографии определения, основанный на экстракции ртути из деструктата дитизоном, переэкстракции бромидом калия, последующем хроматографическом определении в виде дитизоната в тонком слое «Силуфола» или окиси алюминия. Подвижный растворитель — смесь гексана и ацетона (4: 1). Предел обнаружения составляет 0,25 мкг, или 0,02 мг/кг. Метрологическая характеристика метода дана в таблице 1.
1. Метрологическая характеристика колориметрического метода определения общей ртути, основанного на осаждении ее из деструктата йодидом меди
Число определений, п Внесено, мг/кг Среднее значение определения С, ’/( Стандартное отклонение S, */0 Относительное стандартное отклонение Sr Размах варьирования R, %
13 0,0125 Мясо и 83,6 мясопродукты 3,52 0,042 81,4—85,8
5 0,025 83,3 0,00 0,000 —
16 0,125 80,3 7,35 0,092 76,4—84,2
10 0,250 81,7 6,07 0,074 77,4—86,0
5 0,050 80,0 Яйца 0,00 0,00 ,
Реактивы и растворы. Для деструкции. Вода дистиллированная. Этиловый спирт 96%-ный Азотная кислота х. ч., концентрированная и разбавленная (1:3). Серная кислота х. ч., концентрированная. Натрий сернокислый ч. д. а., 2,5 н. раствор (свежеприготовленный) .
2. Метрологическая характеристика колориметрического метода определения общей ртути, основанного на экстракции ее
из деструктата дитизоном и последующей визуальной колориметрии в виде тетрайодомеркуроата меди
Число определении, п Внесено, мк/кг Среднее значение определения с, % Стандартное отклонение S, %, Относи- тельное стандартное отклонение Ьг Размах варьирования В. %
Рыба (мышечная ткань)
4 0,014 92,9 14,3 0,153 85,7—114,7
4 0,028 107,1 8,2 0,076 100,0-114,3
4 0,068 97,1 3,4 0,035 94,1 — 100,0
Мясо говяжье
4 0,012 95,8 8,3 0,086 83,3—100,0
4 0,036 99,9 9,0 0,090 88,8-111,0
Почва
4 0,012 112,4 15,9 0,141 100,0—133,3
4 0,030 96,6 10,3 0,106 80,0—106,6
Шоколад
4 0,012 120,8 15,9 0,131 100,0—133,3
4 0,036 107,1 5,4 0,050 100,0—111,6
3. Метрологическая характеристика метода тонкослойной хроматографии определения общей ртути
Число определений, “ Внесено, мк/кг Среднее значение определения с, % Стандартное отклонение $ > % Относительное стандартное отклонение Sr Размах варьирования R, °/i
5 0,0075 Молоко, простокваша 68,0 2,24 0,033 66,0—70,0
5 0,0125 72,5 2,80 0,039 70,0—75,0
5 0,0250 72,5 2,80 0,039 70,0—75,0
5 0,0500 75,0 5,54 0,072 70,0—80,0
5 0,0250 Масло, 75,0 сметана 5,54 0,072 70,0—80,0
Для колориметрического определения. Медь сернокислая ч д. а., 10%-ный раствор Натрий сернокислый ч д. а, 1%-ный и насыщенный растворы Калий йодистый х. ч., 3%-иый раствор. Барий хлористый ч., 20%-ный раствор, йод ч. или ч. д. а., предварительно очищенный возгонкой, 0,25%-ный и 0,35%-ный растворы в 3%-ном растворе йодистого калия. Составной раствор: готовят перед употреблением, сливая 10%-ный раствор сернокислой
меди с 2,5 н раствором сернокислого натрия в соотношении 1 :5, смесь перемешивают до растворения образовавшегося осадка и почти полного обесцвечивания
Взвесь йодида меди 212 г йодистого калия растворяют в 1— 2 л дистиллированной воды, смешивают с раствором сернокислой меди (160 г в 1 л дистиллированной воды) и оставляют на 30 мин С образовавшегося осадка декантируют жидкость, осадок многократно промывают дистиллированном водой (по 2—3 л) до светло-желтой окраски промывных вод Добавляют сначала насыщенный раствор сернокислого натрия (10—12 мл) для коагуляции осадка, а затем 2,5 н раствор сернокислого натрия (10—20 мл) до полного обесцвечивания надосадочной жидкости и осадка Надосадочную жидкость декантируют, осадок переносят на фильтр, вложенный в большую воронку (диаметр 25—30 см), и промывают дистиллированной водой до почти отрицательной реакции на сульфатной (с 20% ным раствором хлористого бария) Фильтр прокалывают, осадок смывают в мерный цилиндр и доводят объем до 1 л, взвесь хранят в темной склянке (не более 1 мес)
Стандартный раствор ртути 0,0135 г перекристаллизованной двухлористой ртути (HgClj) или 0,0226 г двуйодистой ртути (Hgl2) растворяют в 100 мл 0,25% ного раствора йода (100 мкг ртути в 1 мл) Рабочий раствор готовят разведением в 100 раз основного стандартного раствора 0,25% ным раствором йода (1 мкг ртути в 1 мл) непосредственно перед определением
Для определения методом тонкослойной хроматографии Аммиак ч. д а , концентрированный и 5%-ный водный раствор Ацетон ч д а Гексан х ч Буферный раствор 150 г натрия фосфорнокислого двухзамещенного х ч и 38 г углекислого калия х ч растворяют в воде в мерной колбе на 1 л
Основной раствор дитизона в делительную воронку помещают 100 мл хлороформа, растворяют в нем 50 мг дитизона, прибавляют 200 мл дистиллированной воды и 5—10 мл концентрированного раствора аммиака Смесь энергично встряхивают 2 мин Пос<%е разделения фаз хлороформный слой отбрасывают, водный промы’-вают 20 мл хлороформа, последний отбрасывают Трубку делительной воронки высушивают фильтровальной бумагой, прибавляют в воронку 200 мл хлороформа и разбавленную (1 1) соляную кислоту до отчетливо кислой реакции Смесь встряхивают до тех пор, пока дитнзон не перейдет в хлороформ (слой хлороформа при этом окрашивается в темно зеленый цвет, водная фаза обесцвечивается) Раствор дитизона в хлороформе отделяют в другую воронку и промывают водой трижды по 50 мл Трубку воронки высушивают фильтровальной бумагой, раствор дитизона сливают в темную склянку. Хранят его в темноте на холоде Раствор устойчив в течение 1 мес Рабочий раствор А к одному объему основного раствора дитизона прибавляют 4 объема хлороформа (применяют всегда свежеприготовленный раствор) Рабочий раствор Б к одному объему основного раствора дитизона прибавляют 49 объемов хлороформа (применяют всегда свежеприготовленный раствор).
Бромид калия х ч, 40% ный водный раствор Роданид калия х ч или ч д а , 0,1 н раствор (9,72 г растворяют в воде в мерной колбе на 1 л) Серная кислота х ч, концентрированная Комплексон III (трилон Б) ч д а, 0,1 н раствор (растворяют 37,2 г в воде в мерной колбе на 1 л) Стандартные растворы растворяют 0,1668 г нитрата ртути Hg(NOs)2 'AHjO или 0,1350 г перекристал-
лизованнои двухлористой ртути растворяют в воде в мерной колбе на 100 мл с добавлением 0,1 мл концентрированной азотной кислоты Раствор содержит 1 мг ртути в 1 мл. Рабочий раствор готовят разведением водой в 10 раз основного стандартного раствора
Дитизонат ртути в делительную воронку на 50 мл помещают 10 мл раствора А дитизона, вносят 0,5 мл рабочего стандартного раствора соли ртути и тщательно встряхивают содержимое воронки После этого в воронку прибавляют 25 мл 5% ного раствора аммиака и энергично встряхивают до получения прозрачного оран жево красного нижнего слоя Трубку делительной воронки высушивают фильтровальной бумагой Раствор дитизоната ртути в хлоро форме (нижний слой) фильтрованием через небольшой слой обезжиренной ваты в трубке воронки переносят в темную склянку, 1 мл раствора содержит 5 мкг ртути Хранят его в темноте на холоде Раствор устойчив 5 дней Хлороформ перегнанный Окись алюминия II степени активности для хроматографии Кальций сернокислый (CaSO4 2Н2О), высушенный при температуре 160— 180 °C в течение 6 ч
Приборы и посуда. Для деструкции Весы лабораторные технические Гомогенизатор Колбы конические на 500—750 мл Пипетки градуированные Цилиндры градуированные Воронки химические диаметром 3—6 см Баня водяная
Для колориметрического определения Весы аналитические Колбы мерные на 100 мл Пробирки для колориметри рования Стаканы химические
Для ТСХ определения Воронки делительные на 50, 500 и 1000 мл Микропипетки на 0,1 мл Капиллярные пипетки Стек лянные пластинки размером 9X12 см Ступка с пестиком Камера хроматографическая или эксикатор Чашки фарфоровые на 50— 100 мл Пластинки «Силуфол» или приготовленные следующим образом 3 г просеянной окиси алюминия тщательно смешивают с 0,3 г сернокислого кальция, суспендируют в 5 мл воды и равно мерным слоем наносят на стеклянные пластинки размером 9X12 см, сушат пластины при комнатной температуре в течение 17—18 ч, хранят в эксикаторе над слоем осушителя
Подготовка к определению. Пробу 200—250 г образца мяса, мясопродуктов, рыбы, шоколада измельчают на мясорубке и тщательно перемешивают Среднюю пробу яиц из 10 штук смешивают в смесителе или гомогенизаторе при малом числе оборотов, избегая сильного вспенивания Масло рекомендуется расплавить на водяной бане и отобрать среднюю пробу
Ход анализа
Деструкция пробы мяса, мясных продуктов, рыбы, яиц, молочных продуктов От образца отбирают пробу 40 г, помещают в коническую колбу на 750 мл и равномерно распределяют по дну Последовательно прибавляют 1 мл этилового спирта, 15 мл дистиллированной воды, 15 мл концентрированной азотной кислоты и перемешивают
Деструкция пробы шоколадных изделий Образец 25 г помещают в коническую, колбу на 750 мл и приливают 75 мл разбав ленной азотной кислоты (1 3)
Деструкция почвы Образец почвы 50 г помещают в коническую колбу на 750 мл, приливают 10 мл концентрированной азотной кислоты
После добавления кислот пробы перемешивают и оставляют на 10 мин при комнатной температуре. Колбу закрывают воронкой (диаметр 3 см) и по каплям добавляют 20 мл концентрированной серной кислоты, регулируя скорость так, чтобы постоянно поддерживалась реакция разложения азотной кислоты, по не происходило выделения окислов азота из колбы. При бурном течении реакции возможны потери ртути. По окончании внесения серной кислоты колбу оставляют в вытяжном шкафу на 15 мин при комнатной температуре — до прекращения выделения бурых паров окислов азота, после чего колбу нагревают 30—40 мин на кипящей водяной бане (при анализе печени и почек). При исследовании рыбы, мышечной ткани, молока и образцов с большим содержанием жира количество серной кислоты должно быть доведено до 25 мл и соответственно увеличено время нагревания до 45—60 мин. При бурном течении реакции (выделение окислов азота или сильное пенообразование) в колбу приливают 30—50 мл кипящей дистиллированной воды или снимают ее с водяной бани на 1—2 мин.
Деструкцию проводят до полного просветления придонного слоя жидкости в колбе.
К горячему деструктату добавляют 100 мл кипящей дистиллированной воды и фильтруют в коническую колбу (объемом не менее 500 мл), содержащую 10 мл 2,5 н раствора сернистокислого натрия или 20 мл насыщенного раствора мочевины, через предварительно увлажненный двойной обеззоленный бумажный фильтр с красной лентой (диаметр 15—20 см). Колбу из-под деструктата и фильтр несколько раз промывают кипящей дистиллированной водой. Общий объем деструктата и промывных вод около 250—300 мл В случае необходимости полученный фильтрат деструктата может быть оставлен на 10—20 ч до последующих операций.
Метод колориметрического определения. Последующую операцию выделения ртути из деструктата проводят одним из следующих способов.
Первый способ В колбу с охлажденным фильтратом деструктата добавляют 10 мл взвеси йодида меди. Если содержим^ колбы окрашивается в бурый цвет вследствие выделения йода, добавляют 5—10 мл сернистокислого натрия. Содержимое колбы тщательно перемешивают и оставляют до полного осаждения осадка. Если образующийся осадок окрашен в ярко-розовый или кирпично-красный цвет, что свидетельствует о содержании ртути в образце в количестве более 25 мкг, анализ повторяют, уменьшив навеску в 2 раза. При навеске 20 г сокращают количество азотной кислоты до 10 мл и время прогрева на водяной бане до 15 мин. Дальнейший ход анализа остается без изменений.
Когда же содержание ртути в 20 г пробы превышает 25 мкг, анализ может быть проведен по методу А. И. Крыловой.
Через 1 ч максимально возможную часть жидкости осторожно сливают, стараясь не взмутить осадок, и отбрасывают. Оставшуюся жидкость сливают по палочке через однослойный увлажненный обеззоленный бумажный фильтр с красной полосой, плотно уложенный в воронку диаметром ие более 5—6 см так, чтобы края фильтра на 2—3 мм выступали из воронки. Фильтр промывают 1%-ным раствором сернокислого натрия до исчезновения желтой окраски. Осадок количественно переносят на фильтр и промывают его сначала смесью ацетона с 1%-ным раствором сернокислого натрия (1:1) около 50 мл, затем 1%-ным раствором сернокислого
натрия, нанося жидкость по краю фильтра. Отмывание осадка проводят до исчезновения желтой окраски промывных вод и до pH не менее 3 (по универсальной индикаторной бумаге). Промывные воды отбрасывают.
Полоской фильтровальной бумаги удаляют остатки жидкости из узкой части воронки и осадок подсушивают на воздухе в течение 15 мин. Затем его обрабатывают на фильтре определенным объемом 0,35%-ного раствора йода в зависимости от цвета осадка (табл. 4). Для этого необходимое количество раствора йода отмеривают в цилиндр или пробирку и проводят обработку фильтра небольшими порциями из пипетки (по 1—2 мл), нанося жидкость по краю фильтра. Полученный фильтрат доводят до выбранного объема. Фильтрат можно хранить в пробирках с пришлифованными пробками в темном месте в течение суток.
4. Ориентировочная схема проведения анализа осадка при колориметрическом методе определения ртути
Цвет осадка Примерное содержание ртуги в образце, мкг Объем 0,35 "/и-ного раствора йода для обработки осадка, мл Объем раствора ртути для колориметрирования, мл
Белый
С розовым оттенком Бледно-розовый Ярко-розовый
0-5 5—15
15—25 Свыше 25
10 6—3
15 3—1
25 1
Повторить де-
струкцию, умень-
шив навеску
Второй способ извлечения ртути из деструктата. Охлажденный деструктат (или аликвотную часть его) помещают в делительную воронку соответствующего объема, прибавляют 1—2 мл разбавленного (0,001 %-ного) раствора дитизона, встряхивают в течение 1 мин, разделяют фазы, отделяют органический слой в другую делительную воронку емкостью 25—50 мл. Экстракцию раствором дитизона повторяют до появления устойчивой зеленой окраски. К объединенному экстракту в делительной воронке прибавляют 6 мл 0,35%-ного раствора йода и встряхивают в течение 2 мин. Нижнюю фазу отбрасывают, а водную сливают в пробирку, прибавляют по каплям 1 н. раствор йода до уравнивания окраски исследуемого раствора с 0,25%-ным раствором йода. Последующее определение проводят по общей схеме.
Колориметрирование. Количественное определение ртути производят визуально путем сравнения со стандартной шкалой. Объемы опытных растворов для колориметрирования подбирают исходя из оптимума шкалы: от 0,25 до 2,00 мкг. Аликвотную часть раствора анализируемой пробы (1—6 мл) помещают в пробирку, доводят объем до 6 мл 0,25%-ным раствором йода, прибавляют из бюретки 3 мл составного раствора и тщательно перемешивают. Пробирки для колориметрирования подбирают одинаковой формы и размера, они должны быть из бесцветного стекла.
Одновременно готовят стандартную шкалу. Визуальное сравнение окраски пробы со стандартной шкалой производят через 15 мин после добавления составного раствора. Приготовление стандартной шкалы: в пробирки, отобранные для колориметрирования, вносят точное количество рабочего раствора ртути (1 мкг/мл), указанное в таблице 5, доводят объем до 6 мл 0,25%-ным раствором йода, добавляют 3 мл составного раствора и тщательно перемешивают.
5. Схема подготовки стандартной шкалы
Количество рабочего раствора ртути, мл Количество 0,25 %-ного раствора йода, мл Содержание ртути, мкг Количество рабочего раствора ртути, мл Количество 0,25 %-ного раствора йода, мл Содержание ртути, мкг
0 6,00 0 1,25 4,75 1,25
0,25 5,75 0,25 1,50 4,50 1,50
0,50 5,50 0,50 1,75 4,25 1,75
0,75 5,25 0,75 2,00 4,00 2,00
1,00 5,00 1,00 5,00* 1,00 5,00
• Пробирку шкалы с содержанием ртути 5 мкг используют для ори-ентировки при планировании хода повторного анализа образцов с концентрацией более 2 мкг в растворе Йода, взятого для колориметрирования.
Содержание ртути в пробе (X, мг/кг) вычисляют по формулам. Для первого способа
V.P
где А— количество ртути, найденное по стандартной шкале, мкг’; V — объем раствора йода, использованный для обработки осадка на фильтре, мл; —объем анализируемого раствора, взятый для колориметрирования, мл; Р — навеска образца, г.
Для второго способа
х_ ЛУгб
PV.2V3'
где А — количество ртути, найденное по стандартной шкале, мкг; V!—объем деструктата всей пробы, мл; Г2— объем деструктата, взятый для анализа, мл; V3 — объем анализируемого раствора, взятый для колориметрирования, мл; Р — навеска образца, г; 6 — объем раствора йода, взятый для реэкстракции ртути из раствора дитизона, мл.
Метод тонкослойного определения. Охлажденный фильтрат деструктата переносят в делительную воронку на 500—1000 мл, прибавляют по 20 мл растворов комплексона Ш и роданида калия и осторожно экстрагируют ртуть дитизоном (рабочий раствор Б) порциями по 10 мл 4—5 раз. После четкого разделения фаз дитизоновые экстракты объединяют в другой делительной воронке, куда прибавляют 50 мл 0,25 н. раствора серной кислоты и
10 мл 40%-ного раствора бромида калия. Содержимое воронки энергично встряхивают в течение 1 мин. После четкого разделения слоев органическую фазу отбрасывают, водную промывают 10 мл хлороформа, последний удаляют.
Затем водную фазу обрабатывают буферным раствором (Ills мл) до pH 5,5—6,0 (универсальная индикаторная бумага), прибавляют по 10 мл растворов комплексона III и роданида калия и извлекают ртуть дитизоном (рабочий раствор Б) дважды порциями по 5 мл. Каждый раз пробу энергично встряхивают в течение 1 мин. Дитизоновые экстракты переносят в делительную воронку, содержащую 40 мл 5%-ного раствора аммиака, по 10 мл растворов комплексона III и роданида калия, и энергично встряхивают для удаления избытка дитизона (нижний слой приобретает желтоватую окраску). Трубку делительной воронки высушивают фильтровальной бумагой, органическую фазу переносят в фарфоровую чашку, растворитель испаряют на воздухе в затемненном месте.
Остаток из чашки после испарения растворителя наносят количественно на пластинку при помощи хлороформа. Все операции выполняют при затемнении (используют черную бумагу). Справа от пятна пробы наносят стандартный раствор дитизоната ртути, содержащий предполагаемое в пробе количество ртути, слева — «холостую» пробу. Пластинку помещают в затемненную камеру с системой растворителей смеси гексана и ацетона (4:1). После подъема фронта растворителя иа высоту 10 см хроматографирование прекращают. О наличии ртути в пробе свидетельствует появление на хроматограмме оранжево-красного пятна дитизоиата ртути с Rf 0,42—0,45.
Количество ртути определяют сравнением интенсивности окраски и площади пятен проб и стандартных растворов. Содержание ртути в пробе (X, мг/мл или мг/кг) вычисляют по формуле
где А — количество ртути в анализируемой навеске пробы, мкг; В — объем (навеска) пробы, взятый для анализа, мл (г); 1,4— коэффициент, учитывающий воспроизводимость метода.
При проведении анализа необходимо учитывать, что применяемые реактивы и материалы могут быть загрязнены ртутью. Поэтому параллельно с пробами требуется ставить контрольный опыт на чистоту реактивов.
Обработка результатов анализа. Содержание в анализируемых образцах ртути рассчитывают по формулам, приведенным в данном методическом указании ранее.
Методические указания по определению ртути в рыбе и молочных продуктах хроматографическим методом
(.Утверждены заместителем Главного государственного
I иннтарного врача СССР 12 января 1975 г.)
Принцип метода. ‘ Метод основан на минерализации анализируемых проб, экстракции ртути раствором дитизона, реэкстракции
бромидом калия и последующем хроматографическом определении ртути в виде дитизоната в тонком слое сорбента
Метрологическая характеристика метода. Нижний предел определения 0,5 мкг ртути в исследуемой навеске продукта Степень определения 70—75% Минимально детектируемое количество ртути на пластинке составляет 0,1 мы
Реактивы и растворы. Аммиак концентрированный ч д а и 5°/о-ныи водный раствор Ацетон ч д а Бндистилляг получают повторной перегонкой дистиллированной воды в стеклянном приборе Применяется для приготовления всех реактивов Буферный раствор 150 г натрия фосфорнокислой) двузамещенною х ч и 38 г углекислого калия х ч растворяют в бидистилляте в мерной колбе на 1 л Гексан х ч Гидроксиламин солянокислый х ч, 20%-ный водный раствор
Дитизон, основной раствор в делительной воронке растворяют в хлороформе 50 мг дитизона, прибавляют 200 мл дистиллирован пой воды и 5—10 мл концентрированного раствора аммиака Смесь энергично встряхивают 2 мин После разделения слоев хлороформ ный слой отбрасывают, водный раствор дитизона промывают 20 мл хлороформа Трубку делительной воронки высушивают фильтро вальной бумагой, прибавляют в воронку 200 мл хлороформа и раз бавленную (1:1) соляную кислоту до отчетливо кислой реакции Смесь встряхивают до тех пор, пока дигизон не перейдет в хло роформ (слой хлороформа при этом окрашивается в темно зеленый цвет, водная фаза обесцвечивается) Хлороформный слой отделяют в другую воронку и промывают водой трижды по 50 мл Трубку воронки высушивают фильтровальной бумагой, полученный раствор дитизона в хлороформе сливают в темную склянку Хранят его в темноте на холоде Раствор устойчив в течение 1 мес
Рабочий раствор А к одному объему основного раствора дитизона прибавляют 4 объема хлороформа (применяют всегда све жеприготовленный раствор) Рабочий раствор Б к одному объему основного раствора дитизона прибавляют 49 объемов хлороформа (применяют всегда свежеприготовленный раствор) Калия б^О мид х ч, 40%-ный водный раствор Калия перманганат х ч растирают в ступке Калия роданид, 0,1 н раствор 9,72 г роданида калия ч д а растворяют в бидистиллированной воде и доводят объем до 1 л Кислота азотная концентрированная х ч Кислота серная концентрированная х ч, разбавленная (1:3) и 0,25 н вод ный раствор Комплексон III (трилон Б), 0,1 н раствор 37,2 г комплексона III ч д а растворяют в воде и доводят водой до 1 л Фильтры бумажные (красная лента) Хлороформ перегнанный
Приборы и посуда. Весы аналитические Воронки делительные на 50, 500 и 1000 мл Камера хроматографическая Капиллярные пи петки Колбы конические на 250 мл Колбы мерные на 100 и 1000 мл Микропипетки Стеклянные пластинки размером 9X12 см (или пластинки «Силуфол») Ступка с пестиком Цилиндры мерные Чашки фарфоровые на 50—100 мл
Подготовка к определению связана с приготовлением хроматографических пластин с закрепленным слоем сорбента Просеянную окись алюминия для хроматографии 3 г тщательно растирают в ступке с 0,3 г сернокислого кальция, предварительно высушенного при температуре 180 °C в течение 6 ч и просеянного, суспензируют в 5 мл воды и равномерным слоем наносят на стеклянную пла стинку размером 9X12 см Сушат пластины при комнатной темпе
ратуре в течение 17—18 ч, хранят в эксикаторе под слоем осушителя.
Ход анализа.
Экстракция и очистка экстракта из рыбы. Пробу продукта 25 г измельчают, помещают в коническую колбу на 250 мл, куда добавляют 20 мл воды, 1 г азотнокислого натрия и постепенно (избегать сильного разогрева пробы) 25 мл концентрированной серной кислоты. Обработанную таким образом пробу выдерживают на кипящей водяной бане 7—10 мин, прибавляют к ней 50 мл би-дистиллированной воды, охлаждают, фильтруют через складчатый фильтр (красная лента) в коническую колбу емкостью 250 мл. Колбу и фильтр промывают трижды разбавленной серной кислотой (1:3) порциями по 15 мл. В конце фильтрования промывают только фильтр трижды по 5 мл. К фильтрату в колбе постепенно при перемешивании в течение 30—40 мин прибавляют 5 г растертого в ступке перманганата калия. После добавления последней порции перманганата пробу тщательно перемешивают и выдерживают 15 мин при комнатной температуре.
Для обесцвечивания раствора прибавляют при перемешивании 12—15 мл 20%-ного раствора солянокислого гидроксиламина (полученный в результате раствор не должен содержать двуокиси марганца) Объем пробы доводят до 300 мл водой, переносят в делительную воронку, куда добавляют 20 мл растворов комплексона III и роданида калия, и осторожно экстрагируют дитизоном (рабочий раствор Б) порциями по 10 мл 4—5 раз. После отстаивания эмульсии дитизоновый экстракт переносят в другую делительную воронку. К объединенным в делительной воронке дитизоновым экстрактам прибавляют 50 мл 0,25 н. серной кислоты и 10 мл 40%-ного раствора бромида калия. Содержимое воронки энергично встряхивают в течение 1 мин.
После четкого разделения слоев органическую фазу отбрасывают, водную промывают 10 мл хлороформа, последний удаляют. Затем водную фазу обрабатывают буферным раствором (11 —12 мл) до pH 5,5—6,0 (проверяют по универсальной индикаторной бумаге), прибавляют по 10 мл растворов комплексона III и роданида кал: я и извлекают ртуть дитизоном (рабочий раствор Б) дважды порциями по 5 мл. Каждый раз пробу энергично встряхивают в течение 1 мин. Дитизоновые экстракты переносят в делительную воронку, содержащую 40 мл 5%-ного раствора аммиака, 10 мл комплексона III и роданида калия, и энергично встряхивают для удаления избытка дитизона (нижний слой приобретает желтоватую окраску). Трубку делительной воронки высушивают фильтровальной бумагой, органическую фазу переносят в фарфоровую чашку, растворитель испаряют на воздухе, остаток хроматографируют.
Экстракция и очистка экстракта молока и простокваши. Навеску продукта 50 г помещают в коническую колбу на 250 мл, добавляют 1 г азотнокислого натрия и постепенно 25 мл концентрированной серной кислоты. Обработанную таким образом пробу оставляют на ночь. Затем осторожно при перемешивании прибавляют 50 мл воды (при разогреве пробу охлаждают) и фильтруют через складчатый фильтр (красная лента) в коническую колбу на 250 мл. Дальнейший' ход анализа аналогичен анализу проб рыбы.
Экстракция и очистка экстракта из масла, сметаны. Навеску продукта 25 г помещают в коническую колбу на 250 мл. Масло расплавляют на водяной бане. К пробе прибавляют 1 г азотно-
кислого натрия и постепенно, избегая сильного разогрева, 25 мл концентрированной серной кислоты. Выдерживают пробу в течение 15 мин при комнатной температуре, прибавляют при перемешивании 15—20 мл воды и оставляют на ночь. Затем прибавляют еще 60 мл воды, охлаждают на ледяной бане, фильтруют. Дальнейший ход анализа аналогичен анализу проб рыбы, только после фильтрования пробы к ней постепенно прибавляют 4 г перманганата калия, затем 10—12 мл 20%-ного раствора солянокислого гидроксиламина.
В каждом случае параллельно с анализируемой пробой проводят анализ аналогичной навески — «холостой» пробы.
Хроматографирование. Остаток в чашке после испарения растворителя наносят количественно на пластинку при помощи хлороформа. Все операции выполняют при затемнении (используют черную бумагу). Справа от пятен пробы наносят стандартный раствор дитизоната ртути, содержащий предполагаемое в пробе количество ртути, слева — «холостую» пробу. Пластинку помещают в затемненную камеру с системой растворителей — смеси гексана и ацетона (4:1). После подъема фронта растворителя на высоту 10 см хроматографирование прекращают. О наличии ртути в пробе свидетельствует появление на хроматограмме оранжево красного пятна дитизоната ртути, Rf 0,42—0,45.
Количество ртути определяют сравнением интенсивности окраски и площади пятен пробы и стандартных растворов.
Обработка результатов анализа. Количество ртути в пробе (Л, мг/кг) вычисляют по формуле
где Л — количество ртути в анализируемой навеске продукта, мкг; В — навеска продукта, взятая для анализа, г; 1,4 — коэффициент, учитывающий воспроизводимость метода.
Методические указания по определению гранозана и неорганических соединений ртути в воде методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 6 августа 1981 г.)
Настоящие методические указания являются дополнением к Методическим указаниям по определению общего содержания ртути в мясе, мясопродуктах, рыбе, молочных продуктах, шоколаде, почве, утвержденным 19 октября 1979 г.
Методические указания предназначены для использования в полевых условиях при содержании соединений ртути в воде, значительно превышающем ПДК.
Краткая характеристика препарата. Действующим началом гранозана является этилмеркурхлорид — QHsHgCl, мол. масса 265,12. Белое кристаллическое вещество со специфическим запахом, Тпл 192,5 °C. Плохо растворим в воде, хорошо — в горячем спирте, ацетоне и 10%-ном растворе гидроокиси натрия (на холоду до 20%). Обладает значительной летучестью, может посту-144
пать в воздух в виде паров даже при комнатной и более низкой температуре
ПДК гранозана в воде составляет 0,0001 мг/л
Принцип метода. Метод основан на экстракции соединений рту ти из воды хлороформным раствором дитизона, удалении избытка свободного дитизона и хлороформа, хроматографировании пробы в гонком слое силикагеля КСК или пластинок типа «Силуфол» По движной фазой является смесь растворителей н гексан — ацетон (4 1) О наличии ртути свидетельствует появление на хромато грамме желтого пятна дитизоната ртути, образованного этилмер курхлоридом, Rf 0,58+0,04, или оранжево красного пятна дитизо ната Heopi анических соединений ртути, Rf 0,35±0,03
Метрологическая характеристика метода Пре дел обнаружения 0,0006 мг/л ртути, размах варьирования 17%, среднее значение определения 88%, стандартное отклонение 9,5%, относительное стандартное отклонение 11 %
Реактивы и растворы. Этилмеркурхлорид — стандартный раствор этилмеркурхлорида (ЭМХ), содержащий 1 мг/мл ртути, готовят растворением 0,132 г ЭМХ в этиловом спирте в мерной колбе ем костыо 100 мл
Стандартный раствор дитизоната ЭМХ в делительную воронку емкостью 50 мл помещают 10 мл рабочего раствора Б дитизона, вносят туда 0,05 мл стандартного раствора ЭМХ и энергично перемешивают содержимое воронки Затем прибавляют 25 мл 5% ного раствора аммиака и снова энергично встряхивают до получения прозрачного оранжево красного нижнего слоя Трубку делительной воронки высушивают фильтровальной бумагой Хлороформным рас твор дитизоната ЭМХ (нижний слой) фильтрованием через не большой слой обезжиренной ваты в трубке воронки переносят в темную склянку 1 мл этого раствора содержит 5 мкг ртути Хра нят раствор в темноте на холоде Раствор устойчив в течение 5— 7 дней
Азотнокислая ртуть Hg (NO3)2 V2H2O — стандартный раствор азотнокислой ртути 0,1668 г азотнокислой ртути растворяют в воде с добавлением 0,1 мл концентрированной азотной кислоты и дово дят объем до 100 мл Получают раствор с содержанием ртути 1 мг/мл
Стандартный раствор дитизоната ртути готовят из азотнокис лой рт>ти аналогично приготовлению стандартного раствора дити зоната ЭМХ
Дитизон Дитизон, основной раствор В делительной воронке растворяют в хлороформе (50 мл) 50 мг дитизона, прибавляют 200 мл дистилтированной воды и 5—10 мл аммиака Смесь энер гично встряхивают в течение 2 мин После четкого разделения фаз хлороформный слой отбрасывают, водную фазу промывают 20 мл хлороформа, последний удаляют Трубку делительной воронки вы сушивают фильтровальной бумагой, прибавляют в воронку 200 мл хлороформа и разбавленную (1 1) соляную кислоту до отчетливо кислой реакции Смесь встряхивают до тех пор, пока дитцзон не перейдет в хлороформ (слой хлороформа при этом окрашивается в темно зеленый цвет, водная фаза обесцвечивается) Хлороформ ный раствор отделяют в другую делительную воронку и промыва ют водой трижды порциями по 50 мл Затем трубку воронки вы сушивают фильтровальной бумагой, раствор дитизона в хлоро
10 Заказ № 1995
145
форме сливают в темную склянку Хранят его в темноте на холоде Раствор устойчив в течение 1 мес
Рабочий раствор А К одному объему основного раствора дитизона прибавляют 49 объемов хлороформа Используют свежеприготовленный раствор
Рабочий раствор Б К одному объему основного раствора дитизона прибавляют 4 обьема хлороформа Используют свежеприготовленный раствор
Гексан нормальный Ацетон Спирт этиловыи ректификат Ам миак водный 5% ный водный раствор Хлороформ Соляная кислота Натрий уксуснокислый х ч Уксусная кислота ледяная Буферный раствор 82 г уксуснокислого натрия растворяют в дистиллированной воде, прибавляют 57 мл ледяной уксусной кислоты и доводят объем до 1 л водой
Калий роданистый х ч 0,1 н раствор роданистого калия 9,72 г роданистого калия растворяют в дистиллированной воде в мерной колбе на 1 л
Трилон Б 0,1 н раствор трилона Б 37,2 г трилона Б раство ряют в дистиллированной воде в мерной колбе на 1 л
Силикагель КСК товарный силикагель измельчают, просеивают через сито 100 меш Хранят без доступа влаги
Кальций сернокислый двухводный сушат в сушильном шкафу при температуре 160 °C в течение 6 ч, хранят без доступа влаги
Фильтровальная бумага Вата гигроскопическая
Приборы и посуда Весы аналитические Воронки делительные емкостью 50, 250, 1000 мл Колбы мерные емкостью 100, 1000 мл Цилиндры мерные емкостью 10, 50, 250 мл Пипетки емкостью 1, 10 мл, с делениями на 0,01 и 0,1 мл Микропипетки емкостью 0,1 и 0,2 мл Ступка с пестиком емкостью 100 мл Камера хромато графическая (диаметр 9 5 см, высота 16 см) Эксг.катор для хра нения хроматографических пластинок Стеклянные пластинки 9X12 см Мельница для размола силикагеля Сито капроновое 100 меш Пластинки типа «Силуфол» (ЧССР, Хемапол)
Приготовление пластинок для хроматографии' 14 г силикагеля смешивают в ступке с 1 г сернокислого кальция и суспендируют смесь в стопке в 40 мл дистиллированной воды Периодически помешивая, полг ченную суспензию равномерно нано сят на 7—8 тщательно вымытых стеклянных пластинок Сушат пластинки при комнатной температуре в течение 17—18 ч, хранят в эксикаторе над слоем осушителя
Отбор, хранение и доставка проб. Отбор проб производится в соответствии с унифицированными правилами отбора проб сельско хозяйственной продукции, пищевых продуктов и объектов окружаю щей среды для определения микроколичеств пестицидов утвержденными заместителем Г лавного государственно! о санитарного вра ча СССР 21 августа 1979 s
Проведение определения. Экстракция соединений ртути из воды В делительную воронку емкостью 1000 мл по мещают 500 мл воды, прибавляют по 40 мл ацетатного буфера, растворов трилона Б, роданида калия и 10 мл рабочего раствора А дитизона Содержимое воронки энергично встряхивают в течение 3—5 мин После четкого разделения фаз хлороформный экстракт переносят в другую делительную воронку с 15 мл 5% ного раствора аммиака для удаления избытка свободного дитизона Содержимое воронки энергично перемешивают до получения прозрачного
нижнего слоя. Если хлороформный слой при этом имеет зеленоватую окраску, в воронку прибавляют несколько капель 5 %-кого раствора аммиака и снова встряхивают. После этого трубку воронки высушивают фильтровальной бумагой, хлороформный экстракт переносят в фарфоровую чашку емкостью 50—100 мл и в затемненном месте на воздухе удаляют хлороформ до объема 0,3 мл.
Параллельно с анализируемой пробой обрабатывают 500 мл дистиллированной воды («холостая» проба).
Хроматографирование. Остаток в чашке с помощью хлороформа количественно микропипеткой наносят на пластинку (операцию нанесения повторяют 5—6 раз). Рядом с пробой наносят по 0,05; 0,10; 0,20 мл каждого из стандартных растворов ди-тизонатов ртути (равные объемы стандартных растворов наносят в одну точку), что соответствует 0,25; 0,5 и 1,0 мкг ртути. Пластинку помещают в установленную в затемненном месте хроматографическую камеру, куда за 5—10 мнн до хроматографирования наливают смесь растворителей н-гексан — ацетон (4:1). После подъема фронта растворителя на высоту 10 см от линии старта хроматографирование прекращают. О наличии гранозана в пробе свидетельствует появление на хроматограмме желтого пятна днти-зоната ЭМХ, Rf 0,58—0,62. Дитизонат неорганических соединений ртути имеет оранжево-красную окраску, Rf 0,35.
Количество ртути определяют по площади пятна. Для этого вычисляют площадь пятна соединения ртути из пробы и площадь пятна стандарта, близкого по интенсивности окраски к пятну из пробы. Площадь пятен вычисляют аналогично вычислению площади прямоугольника, используя в качестве сомножителей размеры пятен по вертикальной и горизонтальной оси.
Расчет результатов анализа. Содержание соединений ртути в пробе вычисляют по формуле
Ла2Ь2-1,32
РахЬ{
где А — количество ртути в пятне стандарта, мкг; X — количество ртути в пробе, мг/л; Qi — размер пятна стандарта по вертикальной • оси, мм; bt— размер пятна стандарта по горизонтальной оси, мм; а2 — размер пятна пробы по вертикальной оси, мм; в2— размер пятна пробы по горизонтальной оси, мм; Р — объем анализируемой пробы, мл; 1,32 — коэффициент пересчета содержания ртути на гранозан.
КАРБАМАТНЫЕ ПЕСТИЦИДЫ
Экспрессный метод обнаружения тетраметилтиурамдисульфида (ТМТД) в зерне
(Утвержден заместителем Главного государственного санитарного врача СССР 20 декабря 1976 г.)
Тетраметилтиурамдисульфид (ТМДТ, арозан, померзан, помар-зол, терзан, тирам, тиурам, тыоаде, тетратион A). C6H12N2S4. В чистом виде представляет собой белый кристаллический порошок с мол. массой 240,44 и температурой плавления 155—156 °C. Плохо растворим в воде, спирте, эфире, хорошо растворим в разбавленных щелочах, ацетоне, хлороформе, гексане. Выпускается в виде 50 %-ного смачивающегося порошка.
Применяют в сельском хозяйстве в качестве эффективного фунгицида для протравления семян кукурузы, пшеницы, гороха, сахарной свеклы, льна, против патогенных грибов из расчета 2—8 кг/т, а для семян хлопчатника — 30 кг/т.
Принцип метода. Метод основан на разрушении красящих растительных пигментов под воздействием водного раствора щелочи с последующим извлечением препарата гексаном, взаимодействии с реактивным силикагелем, импрегнированным сульфатом меди, с образованием тиурамата меди.
Чувствительность метода 15 мг/кг корма.
Реактивы и растворы. 0,5%-ный раствор едкого натра. Гексан х. ч. Силикагель марки КСК или КСМ. Соляная кислота х. ч. 1%-ный раствор сульфата меди. Фильтровальная бумага. Азотная кислота.
Приготовление реактивного силикагеля. Силикагель марки КСК или КСМ заливают на 18—20 ч соляной кислотой, разбавленной 1 : 1, затем кислоту сливают, силикагель промывают водой и кипятят в течение 2—3 ч с разбавленной 1 : 1 азотной кислотой. Дают силикагелю отстояться, сливают азотную кислоту и промывают дистиллированной водой до нейтральной реакции промывных вод и сушат при температуре 130 °C в течение 4—6 ч (в сушильном шкафу). После этого силикагель измельчают, просеивают через сито с отверстиями 0,2 мм, а затем через сито с отверстиями 0,04 мм для отсеивания мелкого силикагеля.
Просеянный силикагель заливают на 1 ч 1%-ным раствором сульфата меди. Жидкость сливают, силикагель подсушивают вначале на фильтровальной бумаге, а затем в сушильном шкафу при температуре 100 °C. Хранить силикагель следует в плотно закрытых склянках.
Приборы и посуда. Сушильный шкаф. Набор сит. Стаканы химические. Склянки широкогорлые с притертыми пробками для экстрагирования на 100—200 мл. Пробирки центрифужные с притертыми стеклянными или полиэтиленовыми пробками.
Описание определения. 10 г зерна заливают 10—20 мл 0,5%-ного раствора едкого натра, периодически встряхивают в течение 30 мин. Затем добавляют 10 мл н-гексана и встряхивают в
течение 30 мин (при исследовании гороха и кукурузы не следует энергично встряхивать, так как образуется стойкая эмульсия). После разделения слоев экстракт гексана декантируют, фильтруют через бумажный фильтр, к фильтрату прибавляют 0,2 г реактивного силикагеля и встряхивают 30—50 с. В присутствии ТМТД силикагель окрашивается в зеленый цвет.
Колориметрическое определение купроцина-1, купроцина-И, манеба, марцина, полимарцина, поликарбацина, тиазона, ТМТД, цинеба, цирама и эдитона в продуктах питания растительного происхождения и биологических средах *
Купроцин-1. Действующим началом препарата является комплексное соединение этилен-бис-дитиокарбаматов меди и цинка (Cu:Zn=l:9). Купроцин-I представляет собой порошок желтоватого цвета с температурой плавления 135°C (разлагается). Плохо растворяется в воде, хорошо — в разбавленных щелочах, диметил-формамиде.
Купроцин-П. Действующее начало препарата — комплексное соединение К,К'-диметилдитиокарбаматов меди и цинка (Cu:Zn = = 1:9). Порошок темпо-желтого цвета. Хорошо растворяется в разбавленных щелочах, хлороформе, ацетоне, диметилформамиде.
Манеб. Действующее начало — этилен-бис-дитиокарбамат марганца (C4H6N2S<iMn). Порошок светло-желтого цвета с температурой плавления 120 °C (разлагается). Плохо растворяется в воде, но хорошо растворяется в разбавленных щелочах, диметилформамиде, пиридине (разлагается).
Марцин. Действующее начало — комплексное соединение этилен-бис-дитиокарбаматов марганца и цинка (Мп : Zn= 1 : 7). Порошок желтоватого цвета. Плохо растворяется в воде, хорошо растворяется в разбавленных щелочах, диметилформамиде, пиридине (разлагается).
Полимарцин. Полимерный комплекс этилен-бис-тиурамдисуль-фидных мономеров, связанных между собой посредством цинка и марганца. Органические радикалы, составляющие полимарцин, не существуют в свободном состоянии. Препарат растворяется в разбавленных щелочах и диметилформамиде.
Поликарбацин. Действующее начало — комплекс полиэтилентиу-рамдисульфида с этилен-бис-дитиокарбаматом цинка (1:3). Порошок белого или серого цвета. Температура плавления 120 °C (разлагается) Плохо растворяется в воде, хорошо растворяется в разбавленных щелочах, диметилформамиде, пиридине (разлагается).
Тиазон. 3,5-диметилтетрагидро-1,3,5-тиадиазин-2-тион. Относительная молекулярная масса 162,2. Кристаллическое вещество без запаха, цвет от белого до темно-серого. Температура плавления 99,5 °C. Растворяется в ацетоне и диоксане, плохо растворяется в воде, бензоле, метиловом и этиловом спиртах.
ТМТД. Действующее начало — тетраметилтиурамдисульфид (C6H12N2S4). Порошок белого цвета с неприятным запахом. Температура плавления 155—156 °C. Плохо растворяется в воде и спирте, хорошо — в ' разбавленных щелочах, ацетоне, хлороформе, диметилформамиде.
Цинеб. Чистый препарат (цинковая соль этилен-бис-дитиокарба-миновой кислоты)—белый, очень мелкий, легкопылящий порошок. Относительная молекулярная масса 275,73. Технический препарат имеет серовато-белый или желтовато-белый цвет. Температура вспышки 135—145°C. В воде практически нерастворим (10 мг/л), растворим в пиридине и разбавленных щелочах.
Цирам. Технический препарат (цинковая соль М,М-диметилди-тиокарбаминовой кислоты)—белый или серый порошок. Относительная молекулярная масса 305,79. Температура плавления чистого препарата 246 °C, температура воспламенения 149 °C. Очень плохо растворяется в воде (65 мг/л), растворяется в хлороформе, разбавленных щелочах, сероуглероде и ацетоне, не растворяется в спиртах и эфирах.
Эдитон. (5,5'-этилен-бис-4,6-диметилтетрагидро-1,3,5-тиадиазин-тион-2). Относительная молекулярная масса 350,43. Белый порошок с температурой плавления 140—144 °C (разлагается). В воде не растворяется, плохо растворяется в большинстве органических растворителей, растворяется в ацетоне, диметилформамиде, разбавленных щелочах.
Принцип метода. Метод основан на кислотном гидролизе фунгицидов, очистке выделяющегося сероуглерода от сопутствующих соединений, абсорбции сероуглерода спиртовым раствором диэтил-амина, взаимодействии продукта реакции с ацетатом меди, колориметрическом определении образующегося дитиокарбамата меди. Чувствительность метода 15—20 мкг пестицида в пробе, или 0,03—0,1 мг/кг продукта. Полнота определения 90%.
Реактивы и растворы. 1,5%-ный спиртовый раствор диэтилами-на. 7 н. раствор серной кислоты (для определения поликарбацина). 5 н. раствор серной кислоты (для определения цирама, цинеба, тиазона, эдитона, купроцина-I, купроцина-П, полимарцина, манеба, марцина и ТМТД). 0,05%-ный спиртовый раствор уксуснокислой меди. 10 %-ный водный раствор уксуснокислого свинца. Стандартные растворы препаратов: 0,01 г химически чистого препарата растворяют в 100 мл 0,1 н. раствора едкого натра в мерной колбе' 1 мл раствора содержит 100 мкг препарата. Спирт этиловый.
Приборы и посуда. Мерный цилиндр на 100 мл. Отводная трубка, доходящая почти до дна колбы. Пипетки разные. Поглотители. Поглотительная колонка вместимостью 300—500 мл, заполненная аскаритом или натронной известью и активированным углем. Фотоэлектроколориметр. Холодильник Либиха. Широкогорлая колба с боковым отростком. Аспиратор.
Ход анализа.
Экстракция пестицидов. 200—500 г измельченного продукта (яблоки, виноград, груши, огурцы и др.) или 5—10 г биоматериала (печень, кал и др.) помещают в двугорлую колбу (широкогорлую колбу с боковым отростком) и приливают 100 мл 5 н. раствора серной кислоты (при определении поликарбацина приливают 100 мл 7 н. раствора серной кислоты). Колбу соединяют с обратным холодильником. Отводную трубку, доходящую почти до дна колбы, соединяют с поглотительной колонкой. Отводную трубку холодильника последовательно соединяют с двумя поглотителями. Второй поглотитель присоединяют к аспиратору. В первый поглотитель наливают 5 мл 10%-ного раствора ацетата свинца, во второй — 5 мл 1,5%-ного спиртового раствора диэтиламина.
Содержимое колбы доводят до кипения. Включают аспиратор
и с небольшой скоростью (один пузырек в секунду) протягивают воздух в течение 1 ч (при определении цирама и цинеба —1,5 ч). Выделяющийся сероуглерод увлекается током воздуха и абсорбируется во втором поглотителе. Первый поглотитель служит для очистки сероуглерода от H2S и других сульфидов. Содержимое второго поглотителя количественно переносят в пробирку и приливают 0,5 мл 0,05%-ного спиртового раствора ацетата меди (лучше свежеприготовленного). Общий объем раствора составляет 10 мл. При наличии сероуглерода раствор дитиокарбамата меди окрашивается в желто-бурый цвет.
Колориметрирование. Через 2—3 мин после развития окраски раствор колориметрируют на ФЭК-М с синим светофильтром в кюветах при толщине слоя 5 или 10 мм, в зависимости от интенсивности окраски. Количественную оценку полученных результатов проводят по калибровочной кривой, построенной в координатах: оптическая плотность конечного колориметрируемого раствора и концентрация препарата. Для построения калибровочной кривой вносят 10, 25, 50, 75, 100...500 мкг препарата в навеску продукта и проводят определение по описанной выше методике.
Расчет. Количество препарата в пробе вычисляют по формуле
где X— содержание препарата в пробе, мг/кг или мг/м3; А— количество препарата, найденное по калибровочному графику, мкг; Р — масса пробы, г.
Определение дикрезила в биологических средах тонкослойной хроматографией *
Дикрезил — смесь мета- и паракрезиловых эфиров N-метилкар-баминовой кислоты. Белое кристаллическое вещество с розовым оттенком. Относительная молекулярная масса 165,19, температура плавления 43,5—45 °C. Препарат хорошо растворяется в ацетоне, бензоле, хлороформе, но не растворяется в воде.
Принцип метода. Метод основан на экстракции пестицида из проб, хроматографическом разделении в тонком закрепленном слое окиси алюминия, гидролизе щелочью до крезолов и соединении крезолов с диазотированным паранитроанилином. Сочетание диазотированного паранитроанилина с продуктами щелочного гидролиза различных арил-Ы-метилкарбаматов (севии, дикрезил, мезурол) дает различную окраску. При хроматографическом разделении в подобранной системе растворителей значения Rf различаются, чем и обусловливается специфичность метода. Так, севии и 1-нафтол на хроматограмме имеют Rf соответственно 0,5 и 0,29 и при проявлении окрашиваются в синий цвет. Rf мезурола равно 0,71 (мезурол проявляется в виде желтого пятна). Дикрезил и его метаболиты на хроматограммах обнаруживаются в виде оранжевых пятен. Rf дикрезила равно 0,55—0,60, а мета- и паракрезилов (основные метаболиты)— 0,32—0,34.' Чувствительность метода 0,3 мкг в нанесенной пробе.
Реактивы и растворы. Окись алюминия (для хроматографии). Гипс чистый. Паранитроанилин, перекристаллизованный из горячей
воды. Азотистокислый натрий х. ч. Бензол, метилэтилкетон и хлороформ. Метанол перегнанный. Ледяная уксусная кислота. Соляная кислота х. ч. (лучше фиксанал). Гидрат окиси калия х. ч. или ч. д. а. Бутанол.
Реактив № 1: 1,5 н. раствор КОН в смеси метилового и н-бутилового спиртов, взятых в соотношении 1:1.
Реактив № 2: а) 0,1 %-ный раствор паранитроанилина в 0,1 н. растворе соляной кислоты; б) 4%-ный раствор нитрита натрия. Растворы «а» и «б» перед использованием смешивают 10: 1. (Реактив № 1, а также растворы «а» и «б» готовят впрок и хранят в холодильнике.) Система подвижного растворителя (бензол с ме-тилэтилкетоном 19:1).
Стандартный 0,01%-ный бензольный раствор дикрезила (0,01 мл раствора содержит 0,001 мг дикрезила).
Стандартный 0,01%-ный раствор метакрезола (0,01 мл раствора содержит 0,001 мг метакрезола).
Приборы и посуда. Хроматографическая камера. Гомогенизатор для измельчения тканей. Делительные воронки на 100—200 мл. Бюксы на 25—30 мл. Микропипетки на 0,1 мл. Пульверизаторы. Стеклянные пластинки 13x18 см (или другого размера) с матовой поверхностью или пластинки «Силуфол». Сито 150 меш. Стеклянные колбы на 500 мл.
Ход анализа. 5 г ткани (печень, почка, мышца, кровь) гомогенизируют и заливают 10 мл хлороформа. Экстрагируют в плотно закрытых бюксах 2 ч при комнатной температуре. При однократном экстрагировании хлороформом из гомогенатов тканей извлекается 40—44% дикрезила. Из мочи и меланжа препарат лучше экстрагировать в делительных воронках.
Дикрезил из мочи экстрагируют хлороформом. К Ю мл мочи приливают 20 мл хлороформа и встряхивают 2 ч (экстрагируется 56% препарата).
Для обнаружения дикрезила в меланже в качестве экстрагента используют и-гексан (2,5 части н-гексаиа на одну часть меланжа). За 2 ч и-гексан экстрагирует 30% дикрезила.
При экстрагировании препарата из жидких сред (моча, меланж) не следует сильно встряхивать смесь, достаточно легкого покачивания делительной воронки. Экстракт от биологической среды отделяют фильтрованием через беззольный бумажный фильтр или осторожным сливанием в стакан. Экстракты выпаривают досуха под струей воздуха в вытяжном шкафу. Сухой остаток растворяют в 0,2 мл метанола и наносят на хроматографическую пластинку по общепринятым правилам тонкослойной хроматографии. Сухой остаток смывают метанолом 2—3 раза.
Хроматографирование. Для разгонки хроматографические пластинки с нанесенными пробами помещают в хроматографическую камеру с системой растворителей бензол — метилэтилкетон. После подъема фронта растворителя иа высоту 10—12 см от линии старта пластинку вынимают из камеры, подсушивают на воздухе, обрабатывают из пульверизатора реактивом № 1 и еще раз подсушивают на воздухе. После обработки пластинок из пульверизатора реактивом № 2 (диазотированный паранитроанилин) при наличии в исследуемых пробах дикрезила или его метаболитов на хроматограмме проявляются пятна оранжевого цвета. По величине Rf и окраске пятен проводят качественное определение дикрезила и его метаболитов.
Путем сравнения пятен хроматограммы со шкалой стандартов (необходимо учитывать степень экстракции препарата) можно про водить количествеииое определение препарата в исследуемом материале Шкалу стандартов готовят параллельно с исследованием проб, для чего на хроматографическую пластинку наносят чистый стандартный раствор, содержащий 1, 2, 5, 10, 15 и 20 мкг препарата После разгонки и проявления хроматограмм сравнивают пятна исследуемых проб со шкалой стандартов
Колориметрическое определение триаллата в мясе и тканях внутренних органов животных *
Действующим началом триаллата (авадекс ЬВ, препарат СР2 3426, диптал) является S 2,3,3 трихлораллил Ы,Ы-диизопропил-тиокарбамат Относительная молекулярная масса триаллата 304,67. Химически чистый препарат — кристаллическое вещество бледно бурого цвета Температ>ра плавления около 30'С при 4 мм рт ст. 1риаллат хорошо растворяется в ор1анических растворителях (ацетон, спирт, бензол), в маслах, в воде практически нерастворим. Имеет специфический запах Выпускается в виде 46% ною концентрата эмульсии
Принцип метода Метод основан на извлечении триаллата из пробы н гексаном, адсорбционной очистке экстракта, испарении гексана и взаимодействии пестицида с пиридином в щелочной среде с образованием окрашенного в вишнево сиреневый цвет соединения, определяемою колориметрическим путем Микрограммовые количества альдрина, линдана, ДДТ не дают цветной реакции, хлордан и гептахлор образуют соединения желто коричнево! о цве та, полихлорпииен образует комплексы коричнево красною цвета. Чувствительность метода 5 мкг триаллата в пробе или 0,5 мкг/кг, полнота определения 90%
Реактивы и растворы. Ацетон и 1 ексан Диэтиловый эфир. 10% ный раствор едкою кали свежеприготовленный
Пиридин очищенный в бутыль с пиридином насыпают сухое едкое кали и оставляют на сутки, пиридин декантируют в колбу для отгонки и перегоняют его при температуре 115 °C Силикагель АСК Стандартный раствор триаллата Растворяют 10 мг триаллата (по АДВ) в небольшом количестве н гексана в колбе на 100 мл и доводят объем до метки растворителем Этиловый спирт
Приборы и посуда. Воронки Колбы конические с притертыми пробками Колбы мерные на 100 мл Колонки хромато! рафические высотой 250 мм, с внутренним диаметром 10 мм Чашки фарфоровые Цилиндры мерные вместимостью 10 и 25 мл Вакуумный ротационный испаритель Фотоэлектроколориметр ФЭК 56 Колонки хроматографические В нижнюю узкую часть колонки помещают небольшой тампон из промытой в н гексане и высушенной ваты Затем насыпают силикагель АСК слоем 18 см и уплотняют его Перед работой колонку промывают 10—20 мл н гексана
Ход анализа. Пробу мяса, внутренних органов (печень, почки, сердце, легкие, селезенка) или содержимое желудка измельчают и берут навески 5—10 г Триаллат экстрагируют н гексаном двумя порциями по 20—25 мл при встряхивании в течение 1,5—2 ч каждый раз Объединенные экстракты пропускают через колонку, заполненную силикагелем АСК Колонку промывают 100 мл смеси
н гексана с диэтиловым эфиром (6 4) Хроматографирование экстракта и элюирующей смеси через колонку проводят небольшими порциями (около 20 мл) После очистки экстрактов раствори тель испаряют с помощью вакуумного ротационного испарителя при 50 °C или на воздухе без нагревания
Сухой остаток переносят в пробирку двумя порциями по 0,5 мл н гексана, прибавляют 1 мл пиридина и 0,5 мл свежего отфильтрованного 10% ного раствора КОН Содержимое пробирки энергия но встряхивают и нагревают в кипящей водяной бане 2 мин После охлаждения содержимое пробирки растворяют в 2 мл этилового спирта и фильтруют через бумажный фильтр Фильтр промывают спиртом, доводя объем фильтрата до 5 мл Колориметрирование проводят в кюветах с расстоянием между рабочими гранями 1 см при длине волны 490 нм (ФЭК 56, светофильтр № 5) Кон тролем служит этиловый спирт Содержание пестицида определяют по калибровочному графику
Для построения калибровочного графика в пробирки вносят 0,05, 0,1, 0,2, 0,4, 0,6, 0,8 и 1,0 мл стандартного раствора техни ческого препарата, содержащего в 1 мл 100 мкг активно дейст вующего вещества триаллата, прибавляют 1 мл пиридина, 0,5 мл 10% ного раствора КОН и выполняют все операции, как описано выше Для построения кривой на оси абсцисс откладывают количество триаллата в микрограммах, а на оси ординат соответствую щие значения оптической плотности
Расчет Содержание триаллата в пробе вычисляют по формуле
где X — содержание триаллата в анализируемой пробе, мг/кг, А — количество пестицида, найденное по калибровочному графику, мкг, Р — масса пробы, г
Методика определения эптама в биологических объектах тонкослойной хроматографией
(Утверждена Главным управлением ветеринарии Госагропрома СССР 17 марта 1986 г)
Принцип метода. Методика основана на извлечении пестицида из объектов ацетоном, очистке экстракта и идентификации препарата способом тонкослойной хроматографии
Метрологическая характеристика методики. Предел обнаружения 0,5 мкг эптама в пробе (0,05 мг в 1 кг) Размах варьирования 68—91% Среднее значение определения стандартных количеств эптама 77,2% Стандартное отклонение 7,33 Относительное стандартное отклонение 0,09 Доверительный интервал среднего при Р = 0,05 и п = 9 равен ±2,98
Специфичность методики (избирательность метода) Эптам дифференцируют от фосфорорганических пестицидов и симмтриазинов по способу выявления пятен на пластинке и величине Rf
Реактивы и материалы Ацетон х ч Гексан ч Эфир диэтило вый х ч Натрий сернокислый безводный ч д а Натрия гидрокарбонат ч, 5% ный водный раствор Натрия хлорид ч д а Свинец
уксуснокислый Вода дистиллированная Стандартный раствор эпта-ма с концентрацией 0,1 мг/мл в гексане Серебро азотнокислое х ч Бромфеноловый синий (индикатор) Лимонная кислота ч Фильтры бумажные (красная полоса) Алюминия оксид для хроматографии ч (2 степени активности)
Приборы и посуда. Колонки хроматографические стеклянные, диаметр 8 мм Посуда стеклянная химическая (колбы конические с притертой пробкой па 100 мл, воронки делительные, воронки химические, пипетки, пробирки и т д) Аппарат для встряхивания Центрифуга лабораторная, стандартная, стационарная ЦЛС 3 или друюи аналогичной марки Холодильник бытовой Чашки фарфоровые выпарительные на 20 и 100 мл Пластинки хроматографические «Силуфол» Камера хроматографическая Пульверизаторы стек лянныс
Подготовка к определению.
Приготовление стандартного раствора Отвешивают 10 мг стандартного пестицида и растворяют его в мерной колбе на 100 мл в гексане (концентрация 0,1 мг в 1 мл)
Приготовление 20%-ного раствора свинца уксуснокислого 20 г уксуснокислого свинца растворяют в 80 мл дистиллированной воды При использовании 20% иого раствора основного уксуснокислого свинца к 20 г его добавляют 80 мл 5% ного раствора уксусной кислоты и смесь подогревают на водяной бане до 50—60 °C, После растворения устанавливают pH раствора 10%-ным едким натром в пределах 5,4—5,6 (по pH метру)
Приготовление насыщенного раствора натрия хлорида 36— 40 г натрия хлорида растворяют в 100 мл дистиллированной воды при подогревании до 60—70 °C Затем раствор охлаждают
Приготовление проявляющего реактива. Реактив № 1 а) 25 мг бромфенолового синего растворяют в 5 мл ацетона, б) 250 мг азот нокислого серебра растворяют в 10 мл дистиллированной воды и добавляют 35 мл ацетона Оба реактива смешивают и хранят в темной посуде в холодильнике
Реактив № 2 — 2%-ный раствор лимонной кислоты 2 г лимонном кислоты растворяют в 98 мл дистиллированной воды
Проведение определения. Экстракция препарата и очистка экстракта
Молоко К 20 мл молока добавляют 50 мл ацетона и экстрагируют иа аппарате для встряхивания 1 ч или 12 ч при периодическом перемешивании Отделяют осадок центрифугированием К надосадочнои жидкости добавляют 20 мл воды и 0,5 мл 20% ного раствора уксуснокислого свинца Выдерживают 10 мин и прилива ют 0,5 мл 5% ного раствора гидрокарбоиата натрия и 1 мл насыщенного раствора натрия хлорида Смесь оставляют в морозильной камере холодильника на 1—2 ч После этого жидкость сливают в охлажденные центрифужные стаканы и центрифугируют 3 мин при 3 тыс об/мин Надосадочиую жидкость отфильтровывают через бумажный фильтр, добавляют к ней 40 мл дистиллированной воды и реэкстрагируют пестицид 2 раза смесью гексана с эфиром в соотношении 2.1, используя каждый раз по 25 мл смеси экстр-агентов.
Ткани животных Для исследования отбирают пробу мышц, печени, почек 10 г, жира 2—3 г. Пробу мелко измельчают и экстрагируют 30 мл ацетона 1 ч на аппарате для встряхивания или 12 ч при периодическом перемешивании Экстракт отфильтровывают че-
рез бумажный фильтр, остаток промывают 15 мл ацетона. К объединенному экстракту добавляют 30 мл дистиллированной воды, 0,5 мл 20%-ного раствора уксуснокислого свинца. Выдерживают 10 мин и приливают 0,5 мл 5%-ного раствора гидрокарбоната натрия п 1 мл насыщенного раствора хлорида натрия. Смесь охлаждают в морозильной камере бытового холодильника 1—2 ч. (Экстракты жира оставляют в морозильной камере па ночь.) После этого центрифугируют в охлажденном стакане 3 мин при 3 тыс. об/мин. Надосадочную часть отфильтровывают в чистую посуду. После этого к ней добавляют 40 мл дистиллированной воды и реэкстрагируют пестицид 2 раза смесью гексана с эфиром в соотношении 2:1, используя каждый раз по 25 мл экстрагента.
Зеленые растения и содержимое желудочно-кишечного тракта. 10 г измельченной зеленой массы или содержимого желудочно-кишечного тракта заливают 50—60 мл ацетона, экстрагируют на аппарате для встряхивания 1—2 ч или 12 ч при периодическом перемешивании. Затем экстракт отфильтровывают через бумажный фильтр или ватный тампон. Остаток промывают 40 мл ацетона. Объединенный экстракт переносят в выпарительную чашку и упаривают до объема 35—40 мл. Затем в экстракт добавляют 30 мл дистиллированной воды, 1 мл 20%-ного раствора уксуснокислого свинца. Выдерживают 10 мин и приливают 1 мл 5%-ного раствора гидрокарбоната натрия, 1 мл насыщенного раствора хлорида натрия и помещают смесь в морозильную камеру холодильника на 1 — 2 ч. Охлажденную смесь центрифугируют 3 мин при 3 тыс. об/мин. Надосадочную жидкость отфильтровывают, добавляют к ней 40 мл дистиллированной воды и реэкстрагируют пестицид 2 раза смесью гексана с эфиром в соотношении 2:1, используя каждый раз по 25 мл экстрагента.
Дополнительная очистка экстракта на колонке с оксидом алюминия. Объединенные экстракты анализируемых проб промывают 10—20 мл дистиллированной воды, сушат безводным сернокислым натрием, переносят в выпарительную чашку и концентрируют до объема 2—3 мл.
Для очистки экстракта используют колонку диаметром 8 мм. В нижнюю узкую часть колонки помещают небольшой тампон из белой ваты, затем насыпают слои: безводного сернокислого натрия— 2 см, оксида алюминия — 1 см и снова безводного сернокислого натрия — 0,5 см. Перед внесением пробы колонку промывают 5 мл гексана.
Концентрированные пробы переносят в подготовленную и промытую колонку. Прошедший раствор собирают. Окончательно пестицид экстрагируют из колонки 5—6 мл смеси бензола с гексаном в соотношении 1 : 1. Концентрирование экстрактов проводят при комнатной температуре под тягой вытяжного шкафа, не допуская подогревания или выпаривания экстракта досуха.
Хроматографирование. Полученный экстракт (сконцентрированный до объема 0,2—0,3 мл) количественно наносят при помощи микропипетки на хроматографическую пластинку «Силуфол» на расстоянии 1,5 см от ее нижнего края так, чтобы диаметр пятна не превышал 0,5 см. Справа и слева от пробы наносят на расстоянии 2 см стандартные растворы, содержащие 1, 5, 10 или 20 мкг эп-тама. Затем пластинку помещают в хроматографическую камеру, заполненную смесью гексана с ацетоном в соотношении 2:1. После того, как фронт растворителя поднимется на высоту 10 см от ли-156
нии старта, пластинку вынимают, просушивают на воздухе и помещают в сушильный шкаф при температуре ПО °C иа 10 мин. Затем пластинку вынимают из сушильного шкафа, обрабатывают из пульверизатора раствором азотнокислого серебра с бромфеноловым синим (реактив № 1) и опять выдерживают в сушильном шкафу при температуре 110 °C 10 мин. Пластинку вынимают из сушильного шкафа и через 1 мин осторожно опрыскивают 2%-ным водным раствором лимонной кислоты (реактив № 2).
Эптам проявляется в виде сине-голубого пятна на светло-желтом фоне. Величина Rf около 0,85.
Количество эптама в анализируемой пробе определяют путем сравнения площади и интенсивности окраски пятен пробы и стандартных растворов препарата по формуле
100
МК ’
где X — содержание препарата в пробе, мг на 1 кг; А — количество препарата, обнаруженное на пластинке в опытном образце, в мкг; М — масса исследуемой пробы, г; 100/К — коэффициент для внесения поправки на потери пестицида при экстракции и подготовке пробы к исследованиям, где К — определяемость препарата, %.
Колориметрическое определение эптама и тиллама в растительном материале, почве, воде и биологических средах *
Действующим началом эптама (ЭПТК) является З-этил-М.Ы-ди-н-пропилтиокарбамат. Чистый эптам — прозрачная жидкость с резким специфическим запахом и температурой кипения 232 °C при 760 мм рт. ст. Плотность 0,9546 при 30 °C, относительная молекулярная масса 189,32. Эптам хорошо растворяется в большинстве органических растворителей, в воде его растворимость составляет 375 мг/л при 20 °C. Выпускается в виде 76,8%-ного концентрата эмульсии (бурая жидкость) и 5%-ного гранулированного препарата.
Действующее начало тиллама (ПЭБК) — 5-и-пропил-М-этил-М-н-бутилтиокарбамат. Чистый тиллам — светлая жидкость с температурой кипения 142,5 °C при 20 мм рт. ст. Плотность 0,9458 г/см3 при 30 °C, относительная молекулярная масса 203,35. Тиллам растворяется в обычных органических растворителях, в воде при 20 °C растворимость составляет 92 мг/л. Препаративные формы —• 76,4%-ный концентрат эмульсии (бурая жидкость с выраженным специфическим запахом) и 5%-ный гранулированный препарат.
Принцип метода. Метод основан иа извлечении эптама и тиллама из растительного материала и почвы путем перегонки с паром, из биологических сред н-гексаном, кислотном гидролизе препаратов до вторичного амина и колориметрическом определении последнего по образованию окрашенного в желтый цвет комплекса дитиокарбамината меди. Чувствительность метода 0,05 мг/кг растительного материала, почвы и воды и 0,005 мг в анализируемом объеме при определении в воздухе и биологических средах. Полнота определения 90—95%.
Реактивы, растворы и материалы. Стандартные растворы эптама и тиллама в гексане (100 мкг/мл). Медно-аммиачиый реак
тив 1 г сернокислой меди растворяют в 5 мл воды и объем дово дят до 250 мл концентрированным раствором аммиака 50%-ный раствор едкого натра 5% ный раствор лимоннокислого натрия Натрий сернокислый безводный х ч 0,9% ный раствор хлористого натрия Раствор сероуглерода в бензоле 1 объем сероуглерода смешивают с 20 объемами бензола
Растворители изооктаи, эфир диэтиловый, бензол и н гек сан х ч 1 % ный раствор фенолфталеина
Приборы и посуда. Баночки вместимостью 50 и 200 мл с при тертыми пробками Баня водяная с термометром Водоструйный насос Воронки делительные вместимостью 300 мл Колба Бунзена на 250 мл Пипетки Пастера Пипетки химические разные Пробир ки круглодонные широкие из толстостенного стекла с притертыми пробками и храдуировкой (вместимость 20 мл, высота 150 мм, диа метр 30 мм) Цробирки узкие с притертой пробкой (высота 150 мм, диаметр 15 мм) Цилиндры с притертыми пробками (диаметр 20 мм высота 200 мм, вместимость 25 мл) Фильтр Шотта № 2 Прибор для перегонки с паром Штатив для пробирок
Ход анализа.
Растительный материал, вода почва 200 i растительного мате риала (свеклы, помидоров) или 200 г почвы заливают 0,5 л дне тиллированной воды в двухлитровой широкогорлой колбе, прили вают 12 мл ледяной уксусной кислоты и перегоняют с паром Со бпрают 200 мл дистиллята (желательно, чтобы время отгона не превышало 1 ч) Полученный дистиллят при 200 мл исследуемой пробы воды переносят в делительную воронку на 300 мл, прибав ляют 2 капли концентрированной соляной кислоты и экстрагируют эптам или тиллам из водной фазы двумя порциями изооктана по 15 мл (вместо изооктана можно применять н гексан) Для очистки изооктановый экстракт (после удаления водной фазы) из дели тельной воронки количественно пропускают через фильтр Шот та № 2, заполненный безводным сернокислым натрием (высота слоя 1 см) и окисью алюминия для хроматографии II степени активности (высота слоя 2—3 см) С помощью водоструйного на coca отсасывают экстракт в колбу Бунзена или круглодонную ши рокогорлую пробирку с отводом Затем количественно переносят экстракт в широкую пробирку с притертой пробкой и приливают 1 мл концентрированной серной кислоты
Кровь Пробу крови вносят в центрифужную пробирку, смо ченную 5% иым раствором лимоннокислого натрия
Внутренние органы (печень почки, селезенка, сердце легкие мозг) Пробу ткани отмывают от крови охлажденным 0,9% ным раствором хлористою натрия и измельчают до получения однород ной кашицы 0,5 мл крови или 0,5 г кашицы помещают в пробирку с притертой пробкой и приливают 3 мл н гексана Эптам (или тиллам) извлекают при постоянном энергичном встряхивании в те чение 20—30 мин Гексан количественно переносят в другую сухую пробирку с притертой пробкой Экстракты проб крови количест венно пропускают через фильтр Шотта, заполненный безводным сернокислым натрием (высота слоя 1 см) Элюат собирают в про бирку с притертой пробкой К полученным гексановым растворам приливают по 1 мл концентрированной серной кислоты
Гидролиз и колориметрирование. Пробирки с серной кислотой закрывают пробками и энергично взбалтывают 2 мин Открывают пробирки и помещают их в водяную баню с температурой 85 °C
на 30 мии для гидролиза эптама или с температурой 90 °C иа 20 мин для гидролиза тиллама; содержимое пробирок периодически встряхивают. Пробирки охлаждают в ледяной воде. Слой растворителя при помощи резинового баллончика и пипетки (при малых объемах применяют пипетку Пастера) удаляют, оставляя сернокислый слой в пробирке. Небольшое остаточное количество растворителя определению не мешает. Во время охлаждения кислоту в пробирках разбавляют водой до объема 20 мл, добавляют 2 капли 1 %-кого раствора фенолфталеина и осторожно нейтрализуют раствор 50 %-ным едким натром до появления устойчивой розовой окраски.
Жидкость из пробирки переливают в цилиндр вместимостью 25 мл с притертой пробкой (пробирку ополаскивают 2—3 мл воды). Температура жидкости в цилиндре должна быть в пределах 20—25 °C. В цилиндр прибавляют 1 мл медно-аммиачиого реактива и 7 мл раствора сероуглерода в бензоле. Цилиндр закрывают пробкой и встряхивают 4 мии для развития окраски бензольного слоя. После 6-минутного отстаивания отбирают пипеткой окрашенный в желтый цвет верхний слой. Фильтруют его для обезвоживания через воронку, заполненную 2 г безводного сернокислого натрия, в узкую пробирку с притертой пробкой.
Оптическую плотность окрашенной жидкости измеряют на фотоэлектроколориметре при толщине слоя 10 мм и синем светофильтре. Раствором сравнения служит бензол.
Для построения калибровочного графика (10—200 мкг) в пробирки с притертыми пробками вносят 0,1—2 мл стандартного раствора препарата с интервалом в первых двух пробирках в 0,1 мл, в последующих в 0,2 мл. Объем жидкости во всех пробирках доводят гексаном до 2 мл. В контрольную пробирку вносят 2 мл гексана. Далее анализ ведут, как описано выше. На оси ординат откладывают оптическую плотность, а на оси абсцисс — количество препарата.
Расчет. Содержание эптама и тиллама в растительном материале находят по формуле
где X — содержание эптама или тиллама в пробе, мг/кг; А— количество эптама или тиллама, найденное по калибровочному графику, мкг; Р — масса пробы, г.
Содержание пестицида в биологических средах выражают в миллиграммах на 100 г.
Примечание. При определении пестицидов в растительном материале в ряде случаев требуется удаление побочных продуктов гидролиза. Для этого кислый водный раствор (после гидролиза и доведения объема в пробирке до 20 мл) очищают двумя порциями бензола по 10 мл в делительной воронке вместимостью 100 мл. Отделяют и отбрасывают обе бензольные вытяжки. Бензол после последней очистки удаляют как можно тщательнее. Очищенный водный раствор снова переносят в пробирку, добавляют 50%-ный раствор едкого натра по фенолфталеину и т. д.
Методические указания по определению севина в биологических субстратах и воде методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР от 20 декабря 1976 г.)
Краткая характеристика препарата. Севин (карбарил)—1-наф-тил^-метил-карбамат. C12HnO2N. Молекулярная масса 201,23. Высокоэффективный инсектоакарицид с широким спектром действия. Химически чистый препарат представляет собой бесцветные кристаллы без запаха. Тпл 142 °C. Давление паров менее 0,005 мм рт. ст. при 26 °C. В 1 л воды комнатной температуры растворяется 0,099 г севина. В большинстве органических растворителей (хлороформ, ацетон, гексан и др.) препарат растворяется хорошо. Устойчив в нейтральной и кислой среде. В щелочных растворах (pH 10) быстро гидролизуется до 1-нафтола. Устойчив на свету, при повышенных температурах и хранении. При сильном нагревании разрушается до 1-нафтола и метилизоционата. Выпускается в виде 85%-ного смачивающегося порошка. Используется для защиты растений, в ветеринарной практике — для борьбы с различными эктопаразитами сельскохозяйственных животных.
Принцип метода. Метод основан иа экстракции препарата гексаном или хлороформом, хроматографировании на пластинках «Силуфол», гидролизе препарата спиртовым раствором щелочи до 1-нафтола и образовании соединения с солью диазония — прочный синий Б или прочный красный ГГ. Использование для проявления хроматограмм готовых стабилизированных солей диазония (диазоле) прочный синий Б и прочный красный ГГ позволило в несколько раз понизить предел определения и значительно упростить выполнение исследования.
Метрологическая характеристика метода. Нижний предел определения 0,01—0,001 мг/кг. Степень определения 75%.
Реактивы и растворы. Спирт этиловый. Гексан х. ч. Хлороформ х. ч. Ацетон х. ч. Прочный синий Б (или прочный красный ГГ). Кали едкое ч., х. ч. (15%-ный раствор на 60%-ном спирте). Натрий сернокислый безводный. Петролейный эфир ч., х. ч. Натрий хлористый. Уксусная кислота, 0,25 н. раствор.
Приборы и посуда. Хроматографические пластинки «Силуфол». Камера хроматографическая. Чашки выпарительные фарфоровые. Вакуумный ротационный испаритель. Пульверизаторы стеклянные (химические). Химическая посуда (пипетки, воронки, воронки делительные, стаканы химические, колбы).
Подготовка к определению. Ткани внутренних органов тщательно измельчают ножницами или в гомогенизаторе.
Ход анализа.
Экстракция и очистка экстракта из тканей животных (мышцы, печень, почки, легкие, сердце). К 2—3 г измельченной ткани приливают 5 мл гексаиа и прибавляют 2—3 г безводного сернокислого натрия. После этого ткани растирают пестиком и вместе с экстрагентом переносят в колбу для экстракции. Общий объем растворителя при этом должен составлять 30—50 мл.
Экстрагирование проводят в течение 2—3 ч при периодическом перемешивании пробы. Если необходимо, то экстракт можно ос-160
тавлять в таком состоянии до следующего дня. После этого экстракт отфильтровывают и упаривают в вакуумном ротационном испарителе или в выпарительных чашках при температуре не выше 60—70 °C. Особенно осторожно нужно удалять последние порции растворителя.
При исследовании более крупных проб экстракт следует очищать от жиров и других мешающих определению севипа веществ. Навеску ткани 5—20 г (или даже более) экстрагируют аналогичным способом, но с применением больших количеств растворителя. Сухой остаток после выпаривания смывают 20 мл метанола в делительную воронку, туда же добавляют 30 мл дистиллированной воды и 2—3 г хлористого натрия. Водно-спиртовой раствор 2—3 раза промывают петролейным эфиром. Затем севин дважды экстрагируют из водно-спиртового раствора 30—40 мл хлороформа. Далее хлороформ осторожно выпаривают в вакуумном ротационном испарителе или водяной бане с температурой не выше 60 °C. Последние порции хлороформа нужно удалять особенно осторожно н только при легком подогревании.
Экстракция и очистка экстракта из молока и крови. Пробу молока 10—12 мл вносят в колбу и затем туда же осторожно приливают 50 мл хлороформа и добавляют последовательно небольшими порциями безводный сернокислый натрий (из расчета 2—2,5 г на 2 мл молока). Пробу при этом осторожно перемешивают. Затем колбы с пробами встряхивают в течение 1 ч. Содержимое колб фильтруют и фильтрат обрабатывают так, как описано выше.
Экстракция и очистка экстракта из мочи. Мочу разводят 0,25 н. уксусной кислотой (1:1 или 1:2) и экстрагируют. Небольшие порции мочи (до 5 мл) лучше экстрагировать хлороформом. Хлороформ выпаривают, сухой остаток смывают гексаном или метанолом и наносят на пластинки.
Экстракция и очистка экстракта из воды. Пробу воды 10— 50 мл экстрагируют гексаном. Растворитель осторожно выпаривают до небольшого объема (0,5—1 мл) и наносят на хроматографические пластинки.
Хроматографирование. Сухой остаток после выпаривания растворителя смывают 1—0,5 мл гексана в пробирку и концентрируют слабым током теплого воздуха до необходимого объема. Затем весь раствор или его часть собирают микрошприцем или микропипеткой и наносят на хроматографические пластинки.
Хроматографирование проводят на стандартных пластинках «Силуфол». Размер наносимого пятна не должен превышать 5 мм в диаметре. Точка старта расположена в 1,5—2 см от нижнего края пластинки. В качестве подвижного растворителя используют систему, состоящую из смеси гексана и ацетона (3: 1). При такой системе растворителей Rf севина и 1-нафтола колеблются в зависимости от серии пластинок и качества реактивов соответственно в пределах от 0,33 до 0,37 и от 0,51 до 0,58. После того как подвижный растворитель поднимется на 8—10 см, пластинки вынимают из камеры, высушивают на воздухе и опрыскивают 15%-ным раствором едкого кали в 60%-ном спирте для гидролиза севина. Пластинки вновь просушивают на воздухе и опрыскивают для проявления препарата и продукта его метаболизма 1-нафтола 0,2%-ным раствором соли прочный синий Б (или прочный красный ГГ). При этом на белом фоне проявляются четкие компактные
11 Заказ № 1995
161
пятна определяемых веществ: в случае обработки пластинок солью прочный синий Б — красного цвета, а прочный красный ГГ — синего цвета. Окраска препарата стабильная и не теряет интенсивности в течение нескольких часов.
Обработка результатов анализа. Содержание севи-на в анализируемой пробе (X, мг/кг или мг/л) рассчитывают по формуле
А
где А — количество препарата в анализируемой пробе, найденное на пластине, мкг; Р — навеска или объем анализируемого образца, г или мл.
ПЕСТИЦИДЫ ДРУГИХ ГРУПП
Определение динитроортокрезола (ДНОК) в воде, картофеле, винограде и яблоках тонкослойной хроматографией *
Динитроортокрезол — кристаллическое вещество светло-желтого цвета с относительной молекулярной массой 198,1 Растворим в щелочах, воде (до 0,03% при 25 °C), спирте, ацетоне, бензоле, эфире, дихлорэтане. Температура плавления 86,4 °C В сельском хозяйстве используется натриевая соль динитроортокрезола
Принцип метода. Метод основан на экстракции пестицида из пробы, отгонке растворителя, хроматографировании остатка в тонком слое силикагеля КСК с цинковой пылью Подвижной фазой служит сначала хлороформ, затем смесь хлороформа с метанолом (9:1). Пятна препарата на хроматограмме обнаруживают путем восстановления его до аминосоединения, которое проявляют раствором п диметиламииобепзальдегида с уксусной кислотой Чувствительность метода 2 мкг пестицида в пробе воды (0,01 мг/л), 3 мкг в пробе картофеля, яблок и винограда (0,06 мг/кг) Полнота определения 85—90%
Реактивы и растворы. Вата медицинская обезжиренная н-Гек-сан х ч. 0,25%-ный раствор п-диметиламинобензальдегида в этаноле (Перед употреблением 10 мл раствора смешивают с 1 мл ледя ной уксусной кислоты )
Диэтиловый эфир х ч Кальций сернокислый безводный х ч Кислота соляная разбавленная (1:1) Метиловый спирт х ч 0,05%-ный раствор едкого натра Натрий сернокислый безводный х ч Силикагель КСК Товарный силикагель измельчают, просеивают через сито 100 меш и хранят без доступа влаги
Стандартный раствор: 2—3 г технического препарата ДНОК растворяют в 50 мл воды, раствор фильтруют, переносят в делительную воронку и подкисляют до pH 3 разбавленной соляной кислотой ДНОК экстрагируют трижды эфиром порциями по 30 мл Объединенные в другой делительной воронке экстракты промывают водой и высушивают над безводным сульфатом натрия Эфир отгоняют Полученные светло-желтые кристаллы 2,4 динитро-6 метилфенола используют для приготовления стандартного раствора 10 мг препарата растворяют в смеси гексана с эфиром (20: 5) в мерной колбе на 100 мл и доводят объем до метки В 1 мл раствора содержится 100 мкг ДНОК Уксусная кислота ледяная Хлороформ х ч. Цинковая пыль х ч Этиловый спирт 96%-ный
Приборы и посуда. Баня водяная Воронки делительные вместимостью 250 мл Воронки химические Камера хроматографическая Капиллярные пипетки Колбы конические на 250 мл с пришлифованными пробками Колбы мерные на 100 мл Мельница для размола силикагеля Микропипетки Прибор для отгонки раствори телей с набором колб вместимостью 50—100 мл Сито капроновое 100 меш Стеклянные пластинки 9X12 см или «Силуфол» Ступки с пестиком Сушильный шкаф Цилиндры мерные Эксикатор 11* 163
Ход анализа.
Вода. 200 мл воды помещают в делительную воронку и подкисляют разбавленной соляной кислотой до pH 3. ДНОК экстрагируют диэтиловым эфиром трижды, порциями по 20—25 мл. Объединенные экстракты высушивают над безводным сульфатом натрия и количественно переносят в колбу для отгонки растворителя. Эфир отгоняют на водяной бане.
Виноград, яблоки, картофель. Виноград разминают, картофель и яблоки измельчают. Пробу массой 50 г помещают в коническую колбу, заливают 50—60 мл 0,05%-ного раствора едкого натра и экстрагируют 20—30 мин при периодическом встряхивании. Щелочной экстракт фильтруют через вату и переносят в делительную воронку. Пробу 2—3 раза промывают раствором щелочи, который объединяют с экстрактом в воронке. Содержимое воронки подкисляют до pH 3 разбавленной соляной кислотой. Пестицид экстрагируют эфиром трижды порциями по 20—25 мл. Чтобы не образовалась эмульсия (особенно при анализе картофеля), экстракцию выполняют при осторожном встряхивании делительной воронки. Экстракты объединяют. Далее поступают, как при анализе воды.
Хроматографирование. Содержимое колбы после отгонки растворителя растворяют в 0,2 мл диэтилового эфира и капиллярной пипеткой наносят на пластинку. Стенки колбы 3—4 раза ополаскивают небольшими порциями растворителя. Каждый раз наносят раствор в центр первого пятна так, чтобы диаметр его не превышал 1 см. Пластинку с нанесенными пробами и стандартными растворами помещают в хроматографическую камеру с хлороформом. После подъема фронта растворителя на высоту 10 см пластинку вынимают, удаляют пары хлороформа и хроматографируют повторно в системе хлороформ — метанол (9:1). После подъема растворителей на высоту 10 см пластинку помещают в вытяжной шкаф на 5—10 мин, затем обрызгивают до влажного состояния раствором п-диметиламинобензальдегида с уксусной кислотой и помещают в сушильный шкаф с температурой ПО °C на 7—10 мин. При наличии ДНОК на хроматограммах пробы и стандартов проявляется красно-оранжевое пятно с Rf 0,22—0,25.
Количественное определение пестицида проводят сравнением размеров и интенсивности окраски пятен ядохимиката пробы и стандартного раствора.
Расчет. Количество ДНОК в пробе рассчитывают по формуле Х = А:Р, где X— количество пестицида в материале, мг/кг; А— количество пестицида в анализируемой пробе, мкг; Р — масса пробы, г.
Определение каратана, акрекса в воздухе, воде, огурцах, яблоках, биологическом материале и диносеба в биологическом материале тонкослойной хроматографией *
Действующим началом каратана (аратан, динокап, милдекс) является 2,4-динитро-6-втор-октилфенилкротонат. Относительная молекулярная масса каратана 364,40. Химически чистый препарат— вязкая темная жидкость с температурой кипения 138—
140 °C при 0,05 мм рт. ст. Каратан хорошо растворяется в бензоле, пентане, хлороформе и в других органических растворителях. Выпускается в виде 22—25%-ного смачивающегося порошка.
Действующее начало акрекса (динобутон, дессин, динофен, МС-1053) — 2-втор-бутил-4,6-динитрофенилизопропилкарбонат. Относительная молекулярная масса 326,00. Акрекс — кристаллическое вещество желтого цвета с температурой плавления 60—61 °C. Препарат хорошо растворяется в ацетоне, ксилоле, спирте; растворимость в гексане 1,9%. Выпускается в виде 50%-ного смачивающегося порошка. В организме теплокровных животных акрекс превращается в диносеб.
Действующее начало диносеба (ДНБФ, килосеб)— 4,6-динитро-2-втор-бутилфенол. Относительная молекулярная масса 240,12. Диносеб — темно-желтое кристаллическое вещество с температурой плавления 42 °C. Растворимость в воде 0,07% при 25 °C, в спирте 23,4% Хорошо растворяется в органических растворителях.
Принцип метода. Метод основан на экстракции пестицида органическим растворителем, отгонке растворителя, хроматографировании пробы в тонком слое силикагеля КСК с цинковой пылью. Зоны локализации препаратов на пластинке обнаруживают путем восстановления их до аминосоединений и проявления нингидриновым реактивом (акрекс и каратан) или раствором п-диметиламино-бензальдегида (диносеб).
Чувствительность метода при определении в воздухе 1 мкг акрекса (0,04 мг/м3), 2 мкг каратана (0,08 мг/м3); в воде и огурцах 3 мкг акрекса (0,015 мг/л, кг), 5 мкг каратана (0,025 мг/л, кг); в яблоках 5 мкг акрекса (0,1 мг/кг), 10 мкг каратана (0,2 мг/кг), в животных тканях 1 мкг акрекса, 1 мкг диносеба (0,001 мг/кг), 2 мкг каратана (0,002 мг/кг). Полнота определения 85—90%.
Реактивы и растворы. Ацетон х. ч. Вата медицинская обезжиренная. Гексан нормальный х ч 0,25%-ный раствор п-диметилами-нобензальдегида в этаноле. Диэтиловый эфир х. ч. Кальций сернокислый безводный. 2 н. раствор соляной кислоты. Уксусная кислота концентрированная х. ч. 5%-ный раствор лимоннокислого натрия. Натрий сернокислый безводный. 0,85 %-ный раствор хлористого натрия (физиологический раствор). 0,25%-ный раствор нингидрина в спирте. Для приготовления раствора нингидрин перекристаллизовывают. Для этого 3 г нингидрина растворяют в 25 мл соляной кислоты и добавляют 1 г активированного древесного угля. Раствор с углем нагревают до кипения и фильтруют в горячем состоянии (стеклянный фильтр № 3). Фильтрат оставляют на 4 ч при комнатной температуре, затем на ночь в холодильнике. Кристаллы собирают на стеклянный фильтр, промывают холодной водой, высушивают на воздухе. Хранят в темной склянке.
Раствор для проявления хроматограмм: перед употреблением смешивают 10 мл 0,25%-ного раствора нингидрина с 1 мл ледяной уксусной кислоты. Спирт этиловый 96%-ный. Силикагель КСК или КСК-2,5. Товарный силикагель измельчают, просеивают через сито 100 меш. Хранят без доступа влаги. Уголь активированный БАУ. Цинковая пыль.
Стандартные растворы препаратов: раствор каратана готовят из 25%-ного порошка — 40 мг препарата растворяют в н-гексане или ацетоне в мерной колбе на 100 мл; стандартный раствор акрекса готовят аналогичным способом из химически чистого препарата (10 мг) или 50%-ного порошка (20 мг). Диносеб выделяют
из технического препарата (25—36%-пая аминная или натриевая соль). Для этого 5 г препарата разбавляют в делительной воронке 10 мл дистиллированной воды и экстрагируют 10 мл этилового эфира. После разделения слоев нижний водный слой, содержащий препарат, подкисляют 2 н. соляной кислотой до pH 3. Диносеб экстрагируют трижды эфиром порциями по 20 мл. Эфирные экстракты объединяют фильтрованием через слой безводного сульфата натрия. Эфир отгоняют, остаток растворяют в 4—5 мл этилового спирта. Спиртовый раствор охлаждают смесью льда с хлоридом натрия в холодильнике. Выпавшие кристаллы отфильтровывают и высушивают фильтровальной бумагой. 10 мг полученного диносеба растворяют в н-гексане в мерной колбе на 100 мл (100 мкг/мл). Хранят стандартные растворы в затемненном месте, на холоде.
Приборы и посуда. Баня водяная. Камера хроматографическая. Капиллярные пипетки. Колбы конические вместимостью 25—50 мл с пришлифованной пробкой. Мельница 1ля размола силикагеля (кофемолка). Микропипетки. Прибор для опонки растворителей с набором грушевидных колб вместимостью 50—100 мл. Пульверизаторы стеклянные. Сито капроновое 100 меш. Сушильный шкаф. Стеклянные пластинки 9X12 см или «Силуфол». Ступка с пестиком. Цилиндры мерные. Эксикатор.
Ход анализа.
Вода. 200 мл воды помещают в делительную воронку. Пестицид экстрагируют 10—15 мл хлороформа, встряхивая воронку 2— 3 мин. Экстракт фильтруют через слой безводного сульфата натрия в конической воронке (5—6 г) в колбу для отгонки растворителя. Экстракцию повторяют дважды, экстракты объединяют. Сульфат натрия в воронке промывают 2 раза 5 мл хлороформа, промывную жидкость объединяют с экстрактом. Растворитель отгоняют на водяной бане до 0,5 мл. Остаток хлороформа удаляют осторожным продуванием воздуха.
Огурцы. 50 г средней пробы огурцов при определении акрекса или кожуру, снятую со 100 г огурцов при определении каратана. мелко нарезают, помещают в коническую колбу, заливают 50— 60 мл в-:ексана или хлороформа и экстрагируют 30 мин при периодическом встряхивании. Экстракт фильтруют через безводный сульфат натрия, переносят в колбу для отгонки растворителя. Экстракцию повторяют. Вновь полученный экстракт переносят в ту же колбу, пробу промывают 2 раза 10—15 мл растворителя. Промывную жидкость объединяют с экстрактом и отгоняют растворитель на водяной бане до объема 0,5 мл. Остаток растворителя удаляют продуванием воздуха.
Яблоки. 50 г средней пробы яблок измельчают, помещают в коническую колбу и заливают 50—60 мл охлажденного гексана. Экстрагируют пестицид 30 мин на холоде (смесь льда с хлористым натрием) при периодическом встряхивании. Экстракт фильтруют через слой безводного сульфата натрия (5—6 г) в колбу для отгонки растворителя. Гексан отгоняют на водяной бане, экстракцию повторяют. Вновь полученный экстракт фильтруют в ту же колбу, что и предыдущий. Пробу дважды промывают охлажденным гексаном по 10—15 мл, промывную жидкость путем фильтрования через сульфат натрия объединяют с экстрактом. Гексан отгоняют на водяной бане до 0,5 мл, остаток гексана удаляют продуванием воздуха.
Сухой остаток в колбе охлаждают, растворяют в 1,5—2 мл
охлажденного ацетона Осадок со стенок снимают стеклянной палочкой Все операции проводят при охлаждении пробы смесью льда с хлористым натрием. Смесь быстро фильтруют через слои безводного сульфата натрия (2 г) в небольшой воронке в колбу для отгонки растворителя Стенки еще трижды споласкивают небольшими порциями (1,5—2 мл) охлажденного ацетона, фильтруя каждый раз растворитель через сульфат натрия в колбу для от-юнки Ацетон отгоняют на водяной бане до 0,5 мл, остаток растворителя удаляют продуванием воздуха Колбу охлаждают.
Кровь, моча Стенки пробирки для отбора пробы крови смачивают 5%-ным раствором лимоннокислого натрия 1 мл крови переносят в делительную воронку, прибавляют 1 мл физиологического раствора и экстрагируют ядохимикат диэтиловым эфиром по 7—10 мл трижды, встряхивая колбу каждый раз 2—3 мин Экстракт переносят в колбу для отгонки растворителя путем фильтрования через безводный сернокислый натрий (3—4 г), находящийся в конической воронке Слой Na2SO4 в воронке промывают 2 раза 3—4 мл эфира Растворитель отгоняют на водяной бане 1—2 мл мочи помещают в делительную воронку и экстрагируют пестициды эфиром, как при определении их в крови
Внутренние органы животных Органы ополаскивают физиологическим раствором, высушивают фильтровальной бумагой Для анализа отбирают пробу массой 1—2 г Исследуемую ткань тщательно измельчают, помещают в коническую колбу и заливают 10 мл смеси н гексана с ацетоном (9. 1) при определении акрекса и диносеба или 10 мл н гексана при анализе на каратан В течение 30 мин колбу периодически встряхивают Экстракт фильтруют через безводный сернокислый натрии в колбу для отгонки растворителя на водяной бане Экстракцию повторяют Второй экстракт количественно переносят в колбу, в которой находится первый экстракт Растворитель отгоняют на водяной бане до 0,5 мл, остаток его удаляют осторожным продуванием воздуха
Кал 0,5 г кала помещают в колбу' и растирают стеклянной палочкой Далее пробу обрабатывают так же, как при определении пестицидов в тканях внутренних органов
Хроматографирование. Содержимое колбы после отгонки растворителя при определении пестицидов в воде, О1урцах и животных тканях растворяют в 0,2 мл диэтилового эфира или н гексана При определении пестицидов в яблоках содержимое колбы растворяют в 0,2 мл охлажденного ацетона Раствор капиллярной пипеткой наносят на пластинку Стенки колбы 3—4 раза ополаскивают небольшими порциями растворителя Каждый раз раствор наносят в центре пятна так, чтобы диаметр его не превышал 1 см
При определении пестицидов в животных тканях стандартные растворы акрекса и диносеба наносят в одну точку
Пластинки с нанесенными пробами помещают в хроматографическую камеру с системой растворителей (смесь гексана с ацетоном 4 1) при определении каратана и акрекса или с хлороформом при определении акрекса и диносеба в животных тканях После подъема фронта растворителя на высоту 10 см хроматографирование прекращают, пластинку переносят в вытяжной шкаф на 5— 10 мин для удаления паров растворителя Затем пластинку обрыз-inaarcn нингидриновым реактивом до влажного состояния и помещают в сушильный шкаф с температурой 100 °C на 5 мин при определении акрекса и на 5—7 мин при определении каратана На
оранжевом фоне пластинки проявляется яркое малиновое пятно акрекса с Rf 0,48+0,02 или сиреневое пятно каратана с Rf 0,5.
При определении акрекса и диносеба (приветствуют совместно) верхнюю часть пластинки покрывают стеклом, а открытую нижнюю часть (до Rf 0,5) обрабатывают до влажного состояния свежеприготовленной смесью раствора п-диметиламинобензальдеги-да с уксусной кислотой. Убирают стекло и обрабатывают верхнюю часть пластинки нингидриновым реактивом. Пластинку помещают в сушильный шкаф с температурой 100—105 °C на 5—7 мин. На белом фоне слоя проявляется оранжево-красное пятно диносеба (Rf от 0,28 до 0,30), на оранжевом фоне — ярко-малиновое пятно акрекса (Rf 0,78). Количественное определение пестицидов проводят путем сравнения сразу после проявления размеров и интенсивности окраски пятен пробы и стандартного раствора.
Расчет. Количество акрекса, каратана и диносеба в твердом материале вычисляют по формуле Х = А:Р, где X— количество пестицида в пробе, мг/кг или мкг/г; А — количество пестицида в пробе, найденное сравнением со стандартом, мкг; Р— масса продукта, взятая для анализа, г.
Методические указания по определению четыреххлористого углерода в зерне фотоколориметрическим методом
(Утверждены заместителем Главного государственного санитарного врача СССР 15 октября 1976 г.)
Четыреххлористый углерод — бесцветная легколетучая жидкость с неприятным запахом. СС14-ТКИп. 76,8 °C, плотность 1,595, молекулярная масса 158,8, упругость пара 91 мм рт. ст. при 20 °C. В 100 мл воды растворяется 0,08 г четыреххлористого углерода. Легко растворим в спирте, ацетоне и других органических растворителях. Невоспламеним, при соприкосновении с пламенем раздач гается, образуя фосген. Четыреххлористый углерод применяют в качестве фумиганта зерна. Допустимые остаточные количества СС14 в зерне 50 мг/кг, муке, крупе — 10 мг/кг.
Принцип метода. Метод основан на выдувании в ацетон СС14 током воздуха из зерна при кислотной обработке последнего и дальнейшем определении фотоколориметрическим методом по реакции Фудживара (взаимодействие СС14 с пиридином в щелочной среде). Фотометрируют пробы при длине волны 530 нм.
Метрологическая характеристика метода. Диапазон определяемых концентраций 10—200 мкг в пробе. Предел обнаружения 1 мкг/мл или 0,1 мг/кг. Варьирование степени определения 70— 90%. Среднее значение степени определения стандартных количеств пестицидов 80%, число параллельных определений 5. Стандартное отклонение 7,9%. Относительное стандартное отклонение 9,8%. Доверительный интервал среднего при а = 0,95 и п = 5 равен 80+9,3%.
Реактивы и растворы. Ацетон (перегнанный) не должен давать окраску контроля. Основной стандартный раствор: в мерную колбу емкостью 25 мл вносят приблизительно 10 мл ацетона, закрывают пробкой и точно взвешивают на аналитических весах. Затем быстро прибавляют около 0,15 мл СС14, закрывают, взвеши
вают и доводят до метки ацетоном. Рассчитывают концентрацию ССЦ в 1 мл и из этого раствора готовят рабочий стандартный раствор концентрацией 10—20 мкг/мл. Едкое кали х. ч. Пиридин, перегнанный над КОН. Едкий натр, 15%-иый раствор. Реагент Фудживара: к 10 мл дистиллированной воды прибавляют 0,48 мл 15%-ного раствора едкого натра, смешивают и прибавляют 100 мл пиридина. Хорошо перемешивают и помещают в закрытую стеклянную бутылку (реагент стабилен 4 дня). Фосфорпо-вольфрамовая кислота, 20%-ный водный раствор. Сериал кислота разбавленная (3 мл серной кислоты-Ь 100 мл Н2О).
Приборы и посуда. Аппаратура для извлечения СС14 (рис. 2): круглодонная двугорлая колба на шлифах емкостью 1 л; трубка
Рис. 2. Аппаратура для определения четыреххлористого углерода:
/ — воздух; 2 —вода; 3 — поглотители Пелигота; 4 — охлаждение льдом с солью; 5 — вакуум (размеры в миллиметрах)
на шлифе для подачи воздуха в колбу; холодильник Либиха; поглотители Пелигота на шлифах емкостью 10 мл; стакан со льдом для охлаждения поглотителей Пелигота; колбонагреватель с реостатом; аспиратор, позволяющий пропускать воздух со скоростью 0,5 л/мин. ФЭК с зеленым светофильтром. Мерные пробирки с притертыми пробками на 25 мл и ценой деления 0,2 мл.
Подготовка к определению. Отбор проб. Если зерно хранится насыпью, для составления исходного образца отбирают выемки из каждого квадратного метра конусным щупом не менее чем из пяти мест в разных частях на различной глубине, из верхнего, среднего и нижнего слоев. Масса исходного образца должна быть не менее 2,5 кг.
Из вагонов и трюмов при разгрузке пробы отбирают автоматическим щупом из расчета 250 г на каждую тонну не менее 2,5 кг от партии или ручным способом через равные промежутки времени
путем пересечения не менее 10 лотков по всей их ширине в месте свободного падения сыпучих.
Выемку зерна, хранящегося в мешках, проводят конусным щупом из 5%-ных единиц (но не менее 5 мешков) в количестве 0,5 кг. Из первого места упаковки пробы берут сверху, из второго — из середины, из третьего — снизу.
Получают исходный образец. После осмотра и установления однородности всех выемок их смешивают и методом квартования отбирают среднюю пробу необходимой величины. Средний образец, отбираемый в хранилищах, должен быть равен 10 кг.
Навеску для анализа зерна берут методом квартования (эту операцию необходимо проводить быстро из-за большой летучести фунгицида). Средний образец рассыпают на столе в форме квадрата, который диагоналями делят на четыре части, две из них (противоположные) отбрасывают, а две вновь раскладывают в форме квадрата и так до тех пор, пока не останется количество продукта, по массе приблизительно в 5 раз превышающее величину навески для анализа.
В лаборатории на анализ берется не менее чем по три навески каждого образца.
Подготовка стандартной шкалы. Вносят пипеткой 0,5; 1; 2; 3; 4; 5 мл рабочего стандартного раствора CCL в мерные пробирки на 25 мл и доводят ацетоном до 5 мл. Параллельно ставят две контрольные пробирки с ацетоном по 5 мл. В каждую пробирку приливают по 10 мл раствора Фудживара, закрывают и смешивают. Снимают пробки и помещают пробирки в баню при постоянной температуре 85—87 °C точно на 15 мин. Уровень воды в бане должен быть выше уровня раствора в пробирках. Через 15 мин вынимают пробирки из бани и охлаждают помещением их в водяную баню комнатной температуры примерно на 5 мин. В каждую пробирк}' прибавляют дистиллированную воду до объема точно 16 мл, закрывают и смешивают.
Фотометрируют в 10-миллиметровых кюветах при 530 нм (зе-ij леный светофильтр), принимая за 0 контроль реактивов. Интервал времени после удаления из бани 10—15 мин, постоянно принимают один и тот же интервал. Строят градуировочный график зависимости оптической плотности от концентрации в мкг.
Ход анализа. Подсоединяют аппаратуру для извлечения CCU в соответствии с рис. 2. Смазывают все шлифы небольшим количеством глицерина. Вносят по 4 мл ацетона в каждую из двух последовательно соединенных трубок Пелигота и помещают их в стакан с хорошо раскрошенным льдом. Быстро взвешивают 100 г образца пшеницы и переносят ее немедленно в круглодонную колбу. Приливают в эту же колбу смесь из 300 мл воды, 60 мл разбавленной, как описано выше, серной кислоты, 10 мл 20%-ной фосфорно-вольфрамовой кислоты. Подсоединяют трубку подачи воздуха к колбе и закрывают ее винтовым зажимом на резиновой части. Подают в кожух холодильника теплую воду (40 °C).
Нагревают колбу на колбонагревателе до энергичного закипания смеси, уменьшают нагрев и присоединяют трубки Пелигота к аспиратору, который предварительно настраивают на просасывание воздуха со скоростью до 0,5 л/мин. Винтовым зажимом на трубке регулируют равномерно подачу через поглотители Реостат устанавливают так, чтобы был нагрев с энергичным кипением, но не настолько, чтобы капельки воды конденсировались в горячем холо
дильнике. Пропускают воздух в течение 1 ч. После этого установку отключают, переносят ацетон из поглотителей Пелигота в мерную пробирку, промывая поглотитель А ацетоном из поглотителя Б. Ополаскивают поглотители ацетоном в той же последовательности и доводят объем в пробирке до 10 мл.
Для того чтобы определить подходящий объем раствора для фотоколориметрического анализа, оптическая плотность которого в 10-миллиметровой кювете должна составлять 0,1—0,5, приливают предварительно 1 мл аликвоты из раствора в пробирку, добавляют 4 мл ацетона, 10 мл реактива Фудживара и продолжают, как при подготовке градуировочного графика. Если поглощение меньше, чем 0,05, берут две дополнительные аликвоты — 3 или 5 мл для каждого определения. При поглощении больше 0,05 берут дополнительную аликвоту такого размера, чтобы адсорбция была 0,1 или более. Однако, если поглощение предварительной аликвоты настолько велико, что не укладывается в пределы стандартной кривой (при содержании СС14 более 10 мг/кг) отбирают меньшую аликвоту. Из двух различных по величине аликвот учитывают среднее'значение оптической плотности. Когда в зерне содержится СС14 10 мг/кг и более, необходимо повторить анализ с меньшей навеской — 25 г, так как в этом случае при рекомендуемых условиях препарат не полностью извлекается из зерна.
Обработка результатов анализа. Содержание препарата в анализируемой пробе (X, мг/кг) рассчитывают по формуле
где А — количество СС14, найденное по градуировочному графику, мкг; И — объем аликвоты, взятой для проведения цветной реакции из общего объема пробы 10 мл, мл; Р—-масса пробы, взятой на анализ, г.
Определение бромидов в зерне и растительном материале хроматографией в тонком слое *
Метил бромистый. СН3Вг. Относительная молекулярная масса 94,9. При комнатной температуре представляет собой бесцветный газ со слабым запахом, при температуре 4 °C сгущается в подвижную жидкость. Хорошо растворяется в дихлорэтане, маслах, спирте. В 100 г воды растворяется 1,83 г бромистого метила. Летучесть при 20 °C составляет 7900 мг/л. Пары бромистого метила в 3,3 раза тяжелее воздуха.
Принцип метода. Метод основан на озолении пробы с последующим определением ионов брома хроматографией в тонком слое. Минимально определяемое количество 2 мкг. Чувствительность метода 0,5 мг/кг. Полнота определения 80+5%. Метод предназначен для определения бромидов в зерне и пищевых продуктах растительного происхождения после фумигации их бромистым метилом.
Реактивы и растворы. Спирт этиловый. Едкое кали х. ч. Окись алюминия для хроматографии. Кальций сернокислый (CaSO4-2H2O) ч. д. а. просушивают в сушильном шкафу 6 ч при 160 °C. Калий бромистый ч. д. а. Стандартный раствор бромистого калия (100 мкг брома в 1 мл). 15 мг бромистого калия растворяют в 100 мл спир
та Ацетон х. ч. н-Бутанол х ч Аммиак плотностью 0,9 г/см3. Проявляющий реактив А: 1 % ный раствор азотнокислого серебра в 0,5 н растворе аммиака (3,8 мл аммиака доводят до 100 мл дистиллированной водой). Проявляющий реактив Б: 0,1 %-ный раствор флуоресцеина в этиловом спирте
Приборы и посуда. Муфельная печь. Тигли Микропипетки. Пластинки для хроматографии, покрытые окисью алюминия, скрепленной гипсом или пластинки «Силуфол». Камера для хроматографирования. Пульверизатор. Пипетки для нанесения проб
Ход анализа. 5 г растительного вещества заливают в тигле 10—20 мл 1 %-кого спиртового раствора едкого кали и выдерживают 24 ч. Оставшийся спирт удаляют на водяной бане при температуре 80 °C Затем пробу сжигают на плитке. Обугленный остаток помещают в муфельную печь и озоляют 10 ч при интенсивном повышении температуры до 600 °C Озоленная проба должна быть бело-серого цвета После охлаждения озоленную пробу экстрагируют 3—4 раза этиловым спиртом Общий объем экстракта должен составлять 30 мл Экстракт фильтруют и упаривают до 0,2 мл. Если содержание брома больше, берут аликвотную часть
Хроматографирование. Упаренную пробу наносят на хроматографическую пластинку Колбу тщательно ополаскивают 3 раза этиловым спиртом, который также наносят на пластинку в центр пятна и хроматографируют Пластинку помещают в камеру для хроматографирования с подвижным растворителем, состоящим из ацетона, н-бутанола, аммиака и воды в соотношении 13:4:2:1. После того как растворитель поднимется на 10 см по пластинке, ее вынимают из камеры и оставляют на воздухе для испарения растворителя
Для обнаружения галоидов пластинку опрыскивают 1%-ным раствором азотнокислого серебра в 0,5 н растворе аммиака, сушат на воздухе и опрыскивают 0,1 %-ным спиртовым раствором флуоресцеина После высушивания на воздухе пластинку подвергают ультрафиолетовому облучению 10 мин Галоиды проявляются в виде темных пятен.
Количество препарата в пробе определяют путем сравнения интенсивности и размеров пятен пробы и стандартного раствора.
Расчет. Количество препарата в пробе вычисляют по формуле
где X — содержание препарата в пробе, мг/кг; А — количество препарата, найденное путем сравнения со стандартным раствором, мкг; Р— масса пробы, г. Величина Rf бромида 0,40+0,05.
Извлечения из методических указаний по диагностике, профилактике и лечению отравлений сельскохозяйственных животных гербицидами
(Утверждены Главным управлением ветеринарии Госагропрома СССР 30 октября 1987 г.)
Производные феноксиуксусных кислот.
2,4-дихлорфеноксиуксусная (2,4-Д), 2-метил 4-хлорфеноксиуксус-ная кислота (2М-4Х), их соли и эфиры применяют в качестве гер
бицидов для борьбы с сорняками в посевах различных культур, особенно зерновых.
Соединения 2,4-Д и 2М-4Х могут поступать в организм сельскохозяйственных животных через кожу, органы дыхания и желудочно-кишечный тракт.
Аминная соль 2,4-Д (диметиламмониевая соль 2,4-Д). Кристаллическое вещество белого цвета, хорошо растворимое в воде. Промышленный гербицид — темно-бурая, водорастворимая жидкость, содержащая 40 и 50% 2,4-дихлорфеноксиуксусной кислоты.
Гербицид среднетоксичен для теплокровных животных: ЛД50 для крыс 960 мг/кг, для мышей 337, для 4-месячных кроликов 400 мг/'кг. Обладает высокими кумулятивными и эмбриотропными свойствами. Коэффициент кумуляции для белых мышей составляет 1,03, для белых крыс—1,79. Коэффициент острого отравления (S) лежит в пределах 2,03—2,33.
Коэффициент видовой чувствительности к препарату равен 2,84. ПДК в кормах не более 0,05 мг/кг.
2,4-Д бутиловый эфир (БЭ-2,4, бутапон, 2,4-ДБ). Технический 2,4-ДБ — темно-бурая вязкая жидкость с запахом фенола, плохо растворимая в воде, хорошо — в органических растворителях. Обладает летучестью.
Эфиры 2,4-Д средне- и малотоксичны для теплокровных животных: ЛД50 бутилового эфира для крыс 480—1500 мг/кг, октилового эфира 1200—1300 мг/кг. Остаточные количества всех производных 2,4-Д в продуктах питания не допускаются. ПДК 2,4-Д в кормах составляет для лактирующих животных 0,1, для откормочных 0,6 мг/кг корма.
2М-4Х (агроксон, метаксон, дикотекс, УТ-10). В чистом виде белое кристаллическое вещество, плохо растворимое в воде, хорошо — в органических растворителях.
По гербицидным свойствам и характеру действия на растения, а также по токсичности для теплокровных животных 2М-4Х близка к 2,4-Д, но отличается большей избирательностью. МДУ в зерне хлебных злаков, картофеле и растительном масле 0,05 мг/кг. ПДК в кормах для лактирующих животных и яйценоской птицы 0,05, для животных и птицы на откорме — 0,5 мг/кг.
Производные феноксипропионовых и фенокси-у-м асляных кислот.
2М-4ХП (ранкотекс, мекопроп, Сис 67 МПРОП). В чистом виде кристаллическое вещество, без запаха; технический продукт имеет запах хлоркрезола, растворимость в воде при 20 °C 620 мг/л. Соли лучше растворимы в воде: натриевая —25 г в 100 мл воды, аммонийная— 32, диэтиламппная — 52, калиевая—48 г в 100 мл воды.
Среднетоксичен для человека и теплокровных животных: ЛД50 для крыс и мышей 700—650 мг/кг, для пчел и других полезных насекомых малотоксичен.
2М-4ХМ (тропотокс, легумекс, Сис 67 МБ). В чистом виде — бесцветное кристаллическое вещество, слаборастворимое в воде (44 мг/л), хорошо — в органических растворителях. Щелочные соли 2М-4ХМ растворимы в воде.
Среднетоксичеп для теплокровных животных: ЛД50 для крыс 600 мг/кг (по кислоте), для мышей — 700 мг/кг.
2,4-ДМ (бутирак,' бутоксон). Белое кристаллическое вещество, плохо растворимое в воде и хорошо в органических растворителях.
Во внешней среде 2,4-ДМ разлагается до 2,4-Д и 2,4-дихлорфенола. Остатки в виде 2,4-Д могут сохраняться в почве до 4 мес.
Среднетоксичен для теплокровных животных: ЛД50 для крыс 700 мг/кг и малотоксичен для пчел.
Производные симметричных триазинов.
Отравления сельскохозяйственных животных и птицы препаратами из этой группы могут быть вследствие выпаса скота на обработанных площадях ранее «сроков ожидания», а также при скармливании загрязненных кормов, содержащих препараты выше допустимых уровней.
Атразин. В чистом виде белое кристаллическое вещество без запаха, плохо растворим в воде, лучше — в этиловом эфире, метаноле, хлороформе.
Малотоксичен для теплокровных животных: ЛД50 для крыс 1410—3330, для мышей 850—1570 мг/кг. МДУ в овощах, фруктах, винограде, зерне хлебных злаков и кукурузе 0,1 мг/кг, в мясе и яйцах 0,02 мг/кг. В смородине, крыжовнике, малине, молоке остатки атразина не допускаются.
Симазин (хунгазин, бладекс). Белое кристаллическое вещество, плохо растворимое в воде и мало — в органических растворителях.
Для теплокровных животных симазин малотоксичен; ЛД50 для крыс при введении в желудок 1390 мг/кг, для мышей 4100 мг/кг. Однократная токсическая доза для овец и крупного рогатого скота 250 мг/кг. Летальная концентрация симазина для рыб 45 — 350 мг/л. МДУ в косточковых, семечковых и цитрусовых 0,2 мг/кг, в зерне хлебных злаков и кукурузы 1 мг/кг, в винограде и чае — 0,05 мг/кг, в смородине, крыжовнике, малине и землянике остатки симазина не допускаются.
Прометрин (мерказин, гезагард-50, селектин). Белое кристаллическое вещество, плохо растворимое в воде, хорошо — в органических растворителях.
Для теплокровных животных малотоксичен: ЛД5() для мышей и крыс 1,8—3,12 г/кг, для кур 2,95 г/кг. МДУ в картофеле, чесноке, фасоли, горохе, чине, растительном масле 0,1 мг/кг. Остаточные количества прометрина в моркови, сельдерее, петрушке и укропе не допускаются.
Семерон (десметрин). Белый кристаллический порошок с желтоватым оттенком, в воде растворяется в количестве 500 мг/л, растворяется в этиловом спирте и хлороформе. Выпускают в форме 25%-ного смачивающегося порошка.
Для человека и теплокровных животных семерон малотоксичен: ЛД50 для крыс 2 г/кг. Кумулятивные свойства выражены слабо. МДУ в капусте 0,05 мг/кг.
Котофор (дипропетрин). Белый порошок, слаборастворимый в воде, хорошо — в органических растворителях. Выпускают в виде 80%-ного смачивающегося порошка.
Для теплокровных животных малотоксичен: ЛД50 для крыс 4050 мг/кг. МДУ в арбузах 0,1 мг/кг, в хлопковом масле остатки котофора не допускаются.
Мезоранил (азипротрин, бразоран, мезурон). Бесцветный кристаллический порошок без запаха, слабо растворим в воде, лучше в органических растворителях Выпускают в форме 50%-ного смачивающегося порошка.
Для теплокровных животных малотоксичен: ЛД50 Для мышей 632, для крыс 3696 мг/кг.
Производные мочевины.
Дихлоральмочевина (ДХМ). Белое кристаллическое вещество, без запаха. Не растворяется в воде, но растворяется в органических растворителях.
Препарат малотоксичен для теплокровных животных: ЛДз0 для крыс 6800 мг/кг; его пыль может вызывать раздражение слизистой оболочки носоглотки. Остаточные количества дихлоральмоче-вины во всех пищевых продуктах и кормах не допускаются.
Диурон (дихлорфенидим, гербатокс, кермекс). Белое кристаллическое вещество плохо растворимое в воде, хорошо — в органических растворителях.
Выпускают в виде 80%-ного смачивающегося порошка.
Для человека и теплокровных животных диурон малотоксичен: ЛД6о для крыс 3500 мг/кг.
МДУ в хлопковом масле, плодовых, цитрусовых, винограде 0,05 мг/кг.
Арезин (монолинурон). В чистом виде белое кристаллическое вещество хорошо растворимое в спирте, бензоле и других органических растворителях, кроме предельных углеводородов. Растворимость в воде 580 мг/л.
Для теплокровных животных арезин малотоксичен: ЛДИ для крыс 3600 мг/кг. МДУ в картофеле 0,1 мг/кг.
Которая (флуометурон). Белое кристаллическое вещество, хорошо растворимое в органических растворителях. Выпускают в форме 80%-ного смачивающегося порошка.
Для теплокровных животных которая малотоксичен: ЛД50 для мышей 2750, для крыс 4200, для кроликов 3100, для кур яйценоской породы 3668, мясной породы 1850, для уток 920, для овец 606 мг/кг. Слабовыражены раздражающие и кожно-резорбтивные свойства.
МДУ в хлопковом масле 0,1 мг/кг,
Теноран (хлороксурон). Белое кристаллическое вещество, плохо растворимое в воде (37 мг/л). Выпускают в форме 50%-ного смачивающегося порошка.
Для теплокровных животных малотоксичен: ЛД50 для крыс 2000 мг/кг. Переносимая доза для кур 1500, среднесмертельная 2500 мг/кг. Умеренно токсичен для рыб. ЛКзо Для карповой рыбы 15—30 мг/л.
МДУ в хлопковом масле 0,1 мг/кг.
Дозанекс (метаксурон). Светлый кристаллический порошок, слаборастворимый в воде (678 мг/л), растворим в ацетоне, горячем спирте. Гербицид малотоксичен для теплокровных животных: ЛД50 для крыс 1600—2000 мг/кг.
МДУ в овощах и зерне хлебных злаков 0,1 мг/кг.
Паторан (метобромурон). Белое кристаллическое вещество, плохо растворимое в воде, хорошо — в ацетоне, спирте, хлороформе.
Для теплокровных животных малотоксичен: ЛД50 для крыс 2000—3000 мг/кг.
Малоран (хлорбромурон). Чистый малоран — бесцветный или желтоватый порошок без запаха, слаборастворимый в воде (50 мг/л), хорошо —. в ацетоне, диметилсульфоксиде, хлороформе.
Для теплокровных животных малотоксичен: ЛД50 для крыс 1670, для мышей 3150 мг/кг.
Производные карбаминовой и т и о карб амино-вой кислот.
Хлор-ИФК. Белое кристаллическое вещество, плохо растворимое в воде и хорошо — в органических растворителях.
Для теплокровных животных и человека малотоксичен: ЛД50 для крыс 1500—3000 мг/кг. МДУ в корнеплодах моркови, цикория и в луке не должен превышать 0,05 мг/кг.
Бетанал (фенмедифам). Бесцветные кристаллы, слаборастворимые в воде (10 мг/л) и значительно лучше — в органических растворителях (до 200 г/л).
Для теплокровных животных среднетоксичен: ЛД50 для крыс 750 мг/кг.
МДУ в свекле сахарной и столовой, цикории 0,2 мг/кг.
Карбин (барбан, хлоринат). Белое кристаллическое вещество, плохо растворимое в воде, хорошо в органических растворителях. Выпускают в виде 12%-ного концентрата эмульсии темно-янтарного цвета. Для теплокровных животных среднетоксичен: ЛД50 для крыс 600—820, для кроликов около 600, для кур более 700 мг/кг, для пчел 7,9 мкг на пчелу. МДУ в зерне хлебных злаков 1 мг/кг, в овощах и фруктах — 0,1 мг/кг.
Триаллат (авадекс БВ). Кристаллическое вещество, растворимость в воде 9 мг/л, хорошо растворимое в органических растворителях.
Для теплокровных животных малотоксичен: ЛД50 для крыс 1300 мг/кг. Препарат может вызвать раздражение слизистых оболочек.
Эптам. В чистом виде светлая жидкость, нерастворимая в воде, хорошо — в органических растворителях. Выпускают в форме 75%-ного и 84%-ного концентратов эмульсии.
Для теплокровных животных эптам малотоксичен: ЛД50 для крыс 1630, для кроликов 2640 мг/кг.
МДУ в корнеплодах свеклы и растительном масле 0,05 мг/кг. ПДК в кормах для лактирующих животных и яйценоской птиць*|. 0,01, для животных и птицы на откорме 0,02 мг/кг.
Тиллам (пебулат). В чистом виде прозрачная жидкость, плохо растворимая в воде и хорошо — в органических растворителях. Выпускают 76,4%-ный концентрат эмульсии и 10%-ные гранулы.
Для теплокровных животных малотоксичен: ЛД50 для крыс 1120 мг/кг.
МДУ в овощах, томатах, сахарной свекле 0,05 мг/кг. ПДК в почве 0,6 мг/кг, в воде 0,01 мг/л.
Ялан (ордрам, молинат). Химически чистый продукт — жидкость, плохо растворимая в воде (меньше 0,1%). Препарат средие-токсичен для теплокровных животных (ЛД50 для крыс 657, для мышей 545 мг/кг) и рыб.
МДУ для риса 0,2 мг/кг. ПДК в воде 0,025 мг/л.
Ронит (циклоат). Жидкость, плохо растворимая в воде (100 мг/л), хорошо — в органических растворителях. Выпускают в форме 72%-ного концентрата эмульсии. Для теплокровных животных малотоксичен: ЛД50 для крыс 3600 мг/кг.
МДУ в корнеплодах свеклы 0,3 мг/кг.
Сатурн (тиобенкарб). Жидкость светло-желтого цвета, плохо растворимая в воде, хорошо — в спирте, ацетоне и ксилоле. Выпускают в форме 50%-ного концентрата эмульсии. Препарат малотоксичен для животных: ЛД60 для крыс 1300 мг/кг.
Вернам (вернолат). Светлая маслянистая жидкость, в воде при 20 °C растворяется 90 мг/л, во всех соотношениях смешивается с ксилолом, циклогексаном, гидролизуется в щелочной среде. Малотоксичен для животных (ЛДзо для крыс 1750 мг/кг), пчел и других полезных насекомых. В почве разрушается в течение 1,5— 2 мес.
Производные хлорбензойной кислоты.
Из производных бензойной кислоты в качестве гербицидов широко применяют банвел-Д, полидим и суффикс.
Банвел-Д (дикамба, дианат). В чистом виде белое кристаллическое вещество, плохо растворимое в воде, хорошо — в органических растворителях. Для теплокровных животных малотоксичен: ЛД50 для крыс 1200—3000 мг/кг.
МДУ в зерновых 0,05 мг/кг.
Полидим (2 КФ, трисбен 200). В чистом виде твердые кристаллы коричневого цвета, плохо растворимые в воде. Соли 2,3,6-трихлорбензойной кислоты хорошо растворимы в воде.
Для теплокровных животных среднетоксичен: ЛД50 для крыс более 4000, для мышей 800 мг/кг; малотоксичен для рыб и пчел.
Суффикс (карахол). Белый кристаллический порошок, плохо растворимый в воде, растворяется в метаноле, ацетоне, четыреххлористом углероде. Выпускают в форме концентрата эмульсии, содержащего 20% ДВ.
Применяют против овсюга в посевах яровой пшеницы. Среднетоксичен: ЛД5о для мышей 495, для крыс — 907 мг/кг.
Амиды и нитрилы алифатических карбоновых кислот.
Пропанид (пропанил, стам Ф-34, суркопур). Белое кристаллическое вещество с температурой плавления 91—92 °C. В воде растворяется около 225 мг/л, хорошо — в кетонах, ароматических углеводородах.
Для теплокровных животных пропанид малотоксичен: ЛД50 для крыс 1380 мг/кг; токсичен для рыб, обладает кумулятивными и кожно-резорбтивными свойствами.
МДУ в рисе 0,3 мг/кг.
Солан (пентахлор). Белый кристаллический порошок, плохо растворимый в воде (8 мг/л). Растворимость в ксилоле 20, в диизо-бутилкетоне — 46%.
Для теплокровных животных солан малотоксичен: ЛД50 для крыс 10 000 мг/кг.
МДУ в томатах 1,5 мг/кг. ПДК. в воздухе рабочей зоны 1 мг/м\ в воде 0,1 мг/л.
Рамрод (пропахлор, ацилид). Твердое кристаллическое вещество с температурой плавления 67—76 °C, слаборастворимое в воде (700 мг/л), лучше — в органических растворителях.
Для теплокровных животных среднетоксичен: ЛД50 для крыс 1056 мг/кг, для мышей и кроликов 300—500, для кур 120 мг/кг.
МДУ в капусте, луке, чесноке, брюкве, турнепсе и других овощах не должен превышать 0,2 мг/кг, в зерне бобовых и хлебных злаков 0,3 мг/кг.
Лассо (алахлор). Белое кристаллическое вещество, слаборастворимое в воде (148 мг/л), хорошо — в органических растворителях.
Лассо/атразин — 48%-ная текучая суспензия, содержащая 33,6% алахлора и 14,4% атразина. Для теплокровных животных
среднетоксичен: ЛДМ для крыс 462—1100 мг/кг; для пчел и других полезных насекомых малотоксичен.
Толуин. Жидкость, слаборастворимая в воде (50 мг/л), хорошо— в органических растворителях. Технический толуин — темно-фиолетовая нелетучая жидкость, без запаха.
Малотоксичен для теплокровных животных: ЛД50 для крыс 1260 мг/кг. В объектах внешней среды разрушайся через 5 мес.
Дуал (метолахлор). Бесцветная жидкость, без запаха, в воде растворяется 530 мг/л, хорошо — в органических растворителях.
Малотоксичен для теплокровных животных (ЛД50 для крыс 2700 мг/кг), пчел.
Гетероциклические соединения.
Тордон 22К (пиклорам, хлорамп). Бесцветное кристаллическое вещество, в воде растворяется плохо. Аминная и калиевая соли хорошо растворимы в воде.
Для теплокровных животных малотоксичен: ЛД50 для крыс 3750, для мышей 1500 мг/кг.
ПДК тордона 22К Для животных и птицы на откорме составляет 0,1 мг/кг корма.
Сангор. Комбинированный гербицид. Состоит из оксиэтилгидра-зиновой соли 2,4-Д и оксиэтилгидразиновой соли тордона 22К в соотношении 3:1.
Для теплокровных животных малотоксичен: ЛД50 для крыс 4320, для мышей 2048 мг/кг.
Лонтрел. Действующее вещество 3,6-дихлорпиколиновая кислота, белое кристаллическое вещество, плохо растворимое: в 100 г воды растворяется 0,1 г, в 100 г ацетона 25, ксилола 25, метанола 25 г.
Малотоксичен для теплокровных животных (ЛД3о для крыс и мышей 5000 мг/кг), пчел и других полезных насекомых. Период полураспада в почве 49 дней.
Вензар (ленацил, гексилур). Белое кристаллическое вещество,> без запаха. В воде растворяется плохо, хорошо в пиридине.
Для теплокровных животных малотоксичен: ЛД50 для крыс 10 000 мг/кг.
МДУ в сахарной и столовой свекле 0,5 мг/кг, в землянике не допускаются.
Пирамин (феназон, хлоридазон, пиразон). Белое кристаллическое вещество, плохо растворимое в воде (0,04%), растворимое в ацетоне, метаноле, диметилсульфоксиде.
Длй теплокровных животных малотоксичен: ЛД50 Для крыс 3500 мг/кг.
Зенкор (метрибуцин). Бесцветные кристаллы, плохо растворимые в воде, хорошо — в органических растворителях.
Для теплокровных животных малотоксичен: ЛД50 для крыс 2200 мг/кг.
МДУ в картофеле и томатах 0,25 мг/кг.
Синбар (тербацил). Белое кристаллическое вещество, плохо растворимое в воде, хорошо — в органических растворителях.
Малотоксичен для теплокровных животных, в том числе пчел; ЛД50 для крыс 1487 мг/кг.
МДУ в яблоках, грушах, цитрусовых, персиках, абрикосах, сливах, вишне и винограде 0,05 мг/кг.
МГ-натрия. Действующее вещество — натриевая соль гидразида
малеиновой кислоты: З-гидроксипиридазинон-6. Для теплокровных животных малотоксичен: ЛД50 для крыс 7000 мг/кг.
МДУ в картофеле, свекле, луке, чесноке, моркови, томатах, арбузе (кожуре), табаке 8 мг/кг.
Базагран (бентазон). Белое кристаллическое вещество, плохо растворимое в воде, хорошо — в ацетоне, этаноле, диэтиловом эфире.
Малотоксичен для теплокровных животных: ЛД50 для крыс 1100 мг/кг; не токсичен для пчел и других полезных насекомых. ПДК в кормах для сельскохозяйственных животных составляет 0,5 мг/кг
Нортрон (этофумесат). Белое кристаллическое вещество, плохо растворимое в воде (ПО мг/л), в этаноле — 10%, в ацетоне, хлороформе, диоксане и бензоле — 40%. Малотоксичен для теплокровных животных (ЛД50 для крыс более 6000 мг/кг), пчел и других полезных насекомых.
Производные пиридина.
Реглон (дикват). Белое кристаллическое вещество, хорошо растворяется в воде (при 20 °C 700 мг/л). Относят к среднетоксичным препаратам: ЛД50 реглона для крыс 400 мг/кг, для крупного рогатого скота, овец и кур — 300—450, для поросят (30—40 кг) летальная доза равна 20—50 мг/кг. Препарат обладает выраженными раздражающими свойствами.
Производные алифатических карбоновых кислот.
Далапон (даупон, радапон, пропинат). Жидкость, хорошо растворимая в воде и спирте. Образует соли, из которых натриевая применяется как гербицид. Технический препарат содержит 85% ДВ.
Малотоксичен для теплокровных животных: ЛД50 для крыс 6600—8100, для кроликов 3860, для кур 5660 мг/кг.
МДУ во фруктах, винограде, картофеле и свекле 1 мг/кг, в чае — 0,2, хлопковом масле — 0,1 мг/кг, в смородине, крыжовнике и малине не допускается.
Трихлорацетат натрия (ТХА). Действующее вещество трихлоруксусная кислота. Белое кристаллическое вещество, хорошо растворимое в воде, спирте, эфире. Из солей ТХА в качестве гербицида наиболее широко применяют трихлорацетат натрия — кристаллическое гигроскопическое вещество от белого до светло-коричневого цвета, хорошо растворимое в воде. Содержит 90% ДВ.
Относят к малотоксичным препаратам: ЛД50 для крыс 3300, кроликов — 4000, для цыплят — 4280 мг/кг.
МДУ в овощах, фруктах, зерне, картофеле, огурцах, сахарной и столовой свекле, луке, моркови, плодовых не более 0,01 мг/кг.
Комбинированные гербициды.
Банлен. Смесь 2М-4Х и банвела-Д в форме диметиламинной соли, содержащая в водном растворе 27% ДВ. Для теплокровных животных среднетоксичен.
МДУ в зерновых 0,05 мг/кг.
Диамет-Д. Препарат системного действия, состоит из диметиламинной соли 2-метокси-3,6-дихлорбензойной кислоты (2,7%) и диметиламинной соли 2-метил-4-хлорфеноксиуксусной кислоты (41,9%). Для теплокровных ж'ивотных малотоксичен.
МДУ в зерне 0,05 мг/кг.
Диален. Гербицид системного действия. Действующее вещество содержит 3,6% водного раствора диметиламинной соли 2-метокси-3,6-дихлорбензойной кислоты и 36,1% диметиламинной соли 2,4-ди-хлорфеноксиуксусной кислоты.
Среднетоксичен для теплокровных животных. Остаточные количества в зерне хлебных злаков не допускаются.
Агелон. Смесь двух гербицидов: атразина и прометрина. Для теплокровных животных малотоксичен.
МДУ в кукурузе 0,2 мг/кг.
Кортекс-М. Смесь гербицидов, состоящая из рамрода (40%), арезина (13%) и прометрина (7%).
Карагард 3587. Смесь тербутилазина (25%) и тербуметона (25%). Для теплокровных животных малотоксичен: ЛД50 для крыс 1090, для мышей 1340 мг/кг.
МДУ в плодах семечковых культур и винограде 0 1 мг/кг.
Ветеринарно-санитарная экспертиза.
Мясо и субпродукты животных, вынужденно убитых в период острого отравления гербицидами с выраженными клиническими признаками, являются непригодными к использованию на пищевые цели и направляются на техническую утилизацию.
При обнаружении в мышечной ткани и паренхиматозных органах остатков гербицидов, содержание которых в органах и тканях не допускается, туши животных вместе с внутренними органами подлежат технической утилизации.
Мясо животных, содержащее гербициды в количествах, не превышающих установленные для них максимально допустимые уровни (МДУ) остаточных количеств и при благоприятных органолептических, биохимических и бактериологических показателях, выпускают на общих основаниях. Все внутренние органы, включая желудочно-кишечный тракт, вымя, мозг, направляются на техническую утилизацию.
При содержании в мясе гербицидов в пределах, не превышающих 4 величины предельно допустимых количеств или 4 пределаЧ чувствительности официальных методов определения остатков гербицидов, мясо может быть допущено для переработки на сухие животные корма.
Мясо животных, вынужденно убитых при отравлении гербицидами, в котором не обнаружены их остатки, используют в порядке, как указано в разделах 3, 4, 5 и 9 Правил ветеринарного осмотра убойных животных и ветеринарно-саиитарной экспертизы мяса и мясных продуктов, утвержденных 27 декабря 1983 г.
При убое на мясо животных, перенесших отравление гербицидами, необходимо соблюдать допустимые сроки убоя со времени отравления, установленные нормативными документами.
В случае убоя животных ранее установленных сроков со времени перенесенного отравления гербицидами решение о возможности использования в пищу мяса от таких животных принимают в каждом отдельном случае с учетом степени и клинических признаков отравления животных, токсичности и остаточного количества яда, вызвавшего отравление.
Яйца, молоко и молочные продукты, в которых обнаружены остаточные количества гербицидов, использовать в пищу людям запрещается, их направляют на корм животным в соответствии с пдк.
Определение прометрина в почве, воде и растительном материале тонкослойной хроматографией *
Прометрин — 2 метил тио-4,6 бис изопропиламино-симм триазин Химически чистый препарат представляет собой белый порошок с температурой плавления 120—121 °C и относительной молекулярной массой 241,21 Хорошо растворяется в органических растворителях и воде (48 мг/л)
Принцип метода. Метод основан на экстрагировании прометрина opi аническим растворителем и последующем хроматографиро вании в тонком слое окиси алюминия Подвижным растворителем служит смесь гексана с ацетоном или четыреххлористого углерода с эфиром в соотношении 4 1 Пятна прометрина обнаруживаются на пластинке при опрыскивании проявляющим раствором бромфенолового синего и азотнокислого серебра Количественное определение проводится путем визуального сравнения интенсивности ок раски и размеров пятен пробы и стандартных растворов
Чувствительность метода 0,05 мг/л воды и 0,1 мг/кг почвы или растительного материала Полнота определения 80% Метод специфичен при отсутствии других содержащих серу органических веществ, которые окрашиваются бромфеноловым синим и имеют одинаковое с прометрином значение Rf
Реактивы и растворы Ацетон н Гексан Кальций сернокислый ч д а Просушивают в сушильном шкафу 6 ч при 150 °C, хранят в банке с притер гои пробкой Натрий серпокислый безводный све жепрокаленный Хранят в банке с притертой пробкой Окись алю миния для хроматографии II степени активности с размером ча стиц не более 100 меш
Проявляющий реактив № I смесь равных объемов бромфеноло вого синего (0,4% ный раствор в ацетоне) и азотнокислого серебра (2% ный водный раствор)
Проявляющий реактив № 2 4% ный раствор лимонной кисло ты Проявляющий реактив № 3 0,25% ный раствор перманганата калия Стандартный раствор прометрина в гексане (100 или 200 мкг/мл) Хлороформ Четыреххлористый углерод Эфир (для наркоза)
Приборы и посуда Аппарат для встряхивания Баня водяная Воронки делительные вместимостью 250—300 мл Воронки кониче ские Камера для опрыскивания (стеклянный сосуд диаметром 17—18 см и высотой 15—16 см) Камера для хроматографирования (стеклянный сосуд с притертой крышкой) Можно использовать эк сикатор Колбы конические на 250—300 и 100—150 мл Колбы мерные на 50—100 мл Пипетки с оттянутым концом Прибор для отюнки растворителей с набором колб Пульверизаторы стеклян ные Сито металлическое с диаметром отверстий 2—3 мм Фильтры бумажные Шкаф сушильный Шприц туберкулиновый Пластинки «Силуфол»
Ход анализа.
Вода 200 мл воды помещают в делительную воронку, трижды экстрагируют хлороформом порциями по 20 мл Объединенные экс тракты фильтруют через воронку с безводным сернокислым натри ем (5 г) в колбу прибора для отгонки растворителей Отгоняют хлороформ почти досуха (0,1 мл)
Картофель. 100 г чисто вымытого и измельченного картофеля помещают в колбу с притертой пробкой, заливают 100 мл эфира, оставляют на ночь. Растворитель сливают, пробу трижды промывают эфиром порциями по 20 мл. Объединенный экстракт фильтруют через слой безводного сернокислого натрия (5—10 г) в колбу прибора для отгонки растворителей. Эфир отгоняют почти досуха.
Морковь, конопля, горох, клубника, зеленая масса. 100 г измельченного материала помещают в колбу с притертой пробкой, заливают 100 мл н-гексана и оставляют на ночь. Растворитель сливают, пробу трижды промывают н-гексаном порциями по 20 мл. Экстракты собирают в делительную воронку вместимостью 250— 300 мл. Прометрин извлекают тремя порциями 0,1 н. соляной кислоты по 50 мл. Объединенный солянокислый экстракт промывают 80 мл н-гексана и подщелачивают 1 мл 50%-ного раствора едкого натра. Экстрагируют прометрин хлороформом тремя порциями по 20 мл (продолжительность каждой экстракции 3—5 мин). Хлороформный экстракт дважды промывают дистиллированной водой порциями по 20 мл, фильтруют через слой безводного сернокислого натрия в колбу прибора для отгонки растворителей. Хлороформ отгоняют почти досуха.
Хроматографирование. Наносят пробу и стандартные растворы на пластинку так, чтобы диаметр пятна не превышал 10—12 мм. Колбу с пробой трижды тщательно ополаскивают небольшими порциями эфира (0,5 мл), который наносят в центр пятна Пластинку помещают в камеру для хроматографирования, содержащую смесь четыреххлористого углерода с эфиром при анализе почвы и растений или смесь гексана с ацетоном при анализе воды.
После подъема фронта растворителя на высоту 10 см хроматографирование прекращают, пластинку переносят в вытяжной шкаф для удаления паров растворителя. В камере обрызгивают хроматограмму проявляющим реактивом № 1, пластинку помещают в сушильный шкаф с температурой 50 °C на 20 мин. Охлажденную пластинку обрабатывают проявляющим реактивом № 2. При налЙ*» чии прометрина в пробе на пластинке появляется синее пятно, аналогичное пятнам стандартного раствора с Rf 0,5+0,04. Для закрепления окраски пластинку обрабатывают проявляющим реактивом № 3. Количество прометрина в пробе определяют сравнением размеров и интенсивности окраски пятен пестицида на хроматограммах пробы и стандартного раствора.
Расчет. Содержание прометрина в пробе вычисляют по формуле
где X— содержание прометрина в анализируемой пробе, мг/л или мг/кг; А — количество прометрина, найденное путем сравнения со стандартами, мкг; Р — масса или объем пробы, г или мл.
Количественное определение с достаточной точностью возможно лишь при содержании прометрина до 50—60 мкг в пробе. Если проба содержит больше препарата, следует брать аликвотную часть экстракта.
Методические указания по определению рамрода, лассо и дуала в воде, почве и растительных пробах хроматографией в тонком слое
(Утверждены заместителем Главного государственного санитарного врача СССР 27 апреля 1984 г.)
Краткая характеристика препаратов. Р а м р о д — N-изопропил-N-2-хлорацетанилид. Молекулярная масса 211,69 Синонимы: про-пахлор, нитицид, ацилид.
Белое кристаллическое вещество, труднорастворимое в воде, хорошо — в бензоле, ацетоне, хлороформе, спирте, эфире, гексане. Температура плавления 70—72 °C.
Рамрод малотоксичен для теплокровных: ЛД50 при оральном введении для крыс 1200 мг/кг, для кроликов 710 мг/кг. Разрешен к применению в виде 65%-ного смачивающегося порошка на капусте, луке, чесноке, брюкве, турнепсе. МДУ в капусте и других овощах (кроме картофеля) 0,2 мг/кг, растительных пищевых продуктах 0,05 мг/кг. ПДК в воде 0,01 мг/л.
Лассо — 2-хлор-2'6'-диэтил-М-метоксиметилацетанилид. Молекулярная масса 269,77. Синонимы: алахлор, нудор.
Белый кристаллический порошок с температурой плавления 40—41 °C. Труднорастворим в воде, хорошо — в ацетоне, бензоле, спирте этиловом, хлороформе, этилацетате. Разрушается в сильных кислотах и щелочах.
Относится к малотоксичным соединениям: ЛД50 для крыс 1200—1500 мг/кг при оральном введении.
Выпускается в виде концентрата эмульсии и гранулятов.
ПДК и МДУ не установлены.
Д у а л — 2-этил-6-метил-Ы(Ы-метил-2'-метоксиэтил)-хлорацета-нилид-метахлор. Молекулярная масса 283,80. Синонимы: ЦГА-24705.
Бесцветная жидкость с температурой кипения 100 °C. Растворимость в воде 5 мг/л (20 °C). Хорошо растворим в большинстве органических растворителей. Разрушается в сильных кислотах и щелочах.
Малотоксичен для теплокровных животных: ЛД50 при оральном введении для крыс 3170 мг/кг. Разрешен для опытно-производственного применения в виде 50%-ного концентрата эмульсии на посевах свеклы, сои, клещевины
ПДК и МДУ не установлены.
Принцип метода. Метод основан на извлечении гербицидов из растительных проб, почвы и воды органическим растворителем, очистке экстрактов осаждением коэкстрактивных веществ и перераспределением в системе двух несмешивающихся фаз. Количественное определение производят методом хроматографии в тонком слое. Минимально детектируемое количество — 2—4 мкг препарата в анализируемой пробе. Нижний предел определения 0,02—0,04 мг/кг почвы и растений, 0,02 мг/л воды.
Метрологическая характеристика метода. Метод специфичен в присутствии ДДТ и линдана по Rf.
Рамрод Лассо Дуал
Р — размах варьирования, °/о
Растения Почва Вода 84,6—92,3 93,7—97,3 94,5—98,7 89,5—93,7 90,1—96,7 92,6—97,8 86,0—91,5 92,5—97,5 95,5—99,7
С — среднее значение определения, %
Растения 88,4 91,6 85,5
Почва 94,6 92,3 95,0
Вода 96,7 94,8 97,7
п — стандартное отклонение, %
Растения Почва Вода 3,7 3,4 2,7 3,2 2,6 2,8 2,5 2,5 2,4
Доверительный интервал при Р = 0,95 и п = 5
Растения 88,4+3,7 91,6+3,2 85,5+2,5
Почва 94,6+3,4 92,3±2,6 95,0+2,5
Вода 96,7±2,7 94,8+2,8 97,7+2,4
Реактивы и растворы. Хлороформ х. ч. н-Гексан х. ч. Ацетон ч.д. а. Бензол ч. д. а. Натрии сернокислый безводный ч. Аммоний хлористый х. ч. Кислота ортофосфорная ч. Пластинки «Силуфол», «Хемапол» ЧССР. Аммиак водный 25%-ный. Кальций сернокислый ч. д. а. Серебро азотнокислое х. ч. Четыреххлористый углерод. Стандартные растворы гербицидов в хлороформе с содержанием по ДВ 100 мкг/мл.
Коагулирующий раствор: 5 г хлористого аммония растворяю» в 10 мл дистиллированной воды, приливают 10 мл 85%-ной орто-фосфорной кислоты и доводят до 1 л дистиллированной водой.
Проявляющий реактив: 0,5 г азотнокислого серебра, 5 мл дистиллированной воды и 2 мл 25%-ного водного аммиака доводят в мерной колбе до 100 мл.
Приборы и посуда. Колбы конические на шлифах КШ-500 29/32 ТС. Колбы грушевидные ОКТ-14/23 ТС. Делительные воронки ВД-3-600. Воронки конические диаметром 5 см. Колбы мерные на 100 мл. Аппарат для встряхивания АВУ-1. Ротационный испаритель ИР-1М. Хроматографические камеры. Микропипетки. Лампа кварцевая ПРК-4. Камера для опрыскивания. Пульверизаторы.
Подготовка реактивов. Органические растворители необходимо перегнать, сульфат натрия прокалить при температуре 300—400 °C.
Oi6op проб. Отбор проб для анализа проводят в соответствии с Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов, утвержденными заместителем Главного государственного санитарного врача СССР 21 августа 1979 г.
На анализ отбирают 25—30 г растительной пробы, 100 г почвы и 200 мл воды.
Ход определения.
Вода 200 мл воды в делительной воронке экстрагируют трижды по 80, 60 и 50 мл хлороформом Объединенный хлороформенный экстракт осушают сульфатом натрия б/в и отгоняют хлороформ на ротационном испарителе при температуре не выше 50 “С
Почва 100 г воздушно сухой почвы растирают в ступке и просеивают через сито Пробу помещают в коническую колбу на 500 мл с притертой пробкой и экстрагируют гербицид на аппарате встряхивания в течение 2 ч 100 мл хлороформа Растворитель от фильтровывают через складчатый фильтр со слоем безводного сульфата натрия Колбу и фильтр трижды промывают хлоро формой порциями по 10—15 мл Объединенный экстракт упаривают на ротационном испарителе ...осу.ха при температуре не вы ше 50 °C
Растительный материал 25—50 г измельченной растительной пробы экстрагируют хлороформом в течение 2 ч на аппарате для встряхивания Экстракт отфильтровывают через слой безводного сульфата натрия Колбу и фильтр трижды промывают хлорофор мом порциями по 10—15 мл Объединенный экстракт отгоняют досуха на ротационном испарителе при температуре 50 °C Сухой остаток растворяют в 3 мл охлажденного ацетона и прибавляют 25—30 мл охлажденной коагулирующей смеси Колбу помещают на 30—40 мин в холодильник, после че1 о содержимое отфильтровыва ют х-ерез плотный бумажный фильтр в делительную воронку Пре параты экстра! ируют трижды н гексаном порциями по 30 мл Объединенный гексановый экстракт сушат безводным сернокислым натрием и упаривают досуха при температуре не выше 50 °C
Хроматографирование К сухому остатку прибавляют 0,1 — 0,2 мл хлороформа и наносят образцы на хроматографическую пластинку «Силуфол» Стенки колбы ополаскивают несколькими каплями растворителя и наносят в центр пятна
Хроматографирование проводят в системе четыреххлористый углерод—эфир (3 2) После этого пластинку высушивают от растворителей и опрыскивают проявляющим реактивом После облучения под УФ лампой в течение 10—15 мин препараты проявляются в виде темно серых ,ятен Rf рамрода 0,42±0,03, дуала 0,50+0,03, лассо 0,61+0,04
Для дополнительной очистки можно использовать двумерную хроматографию В качестве первой системы подвижных растворителей используют четыреххлористый углерод, а второй — смесь четыреххлористого углерода — эфира (3 2)
Обработка результатов анализа Количественное определение производится путем сравнения интенсивности окраски и площади пя1ен проб и стандартныv растворов
Содержание препаратов (мг/кг, мг/л) вычисляют по формуле v Л = --------------------------- ,
где m — масса препарата, нанесенного на пластинку из стандартного раствора, мкг, S1 — площадь пятна стандартного раствора, мм2, S2 - площадь пятна пробы, мм2, А — масса или объем пробы, взятой на анализ, г или мл
Методические указания по определению фенилмочевинных гербицидов (фенурона, которана, монурона, диурона, дикурана, дозанекса, тенорана, фалорана, арезина, линурона, паторана, майорана] в воде, почве, растительной массе, овощах методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 24 августа 1983 г)
Характеристика препаратов. Краткая характеристика препаратов и их токсиколоп ческая оценка даны в сборнике «Методиче ские указания по определению микроколичеств пестицидов в про дуктах питания, кормах и внешней среде», ч XII, М, 1982, с 162
Методика определения фенил мочевинных гербицидов.
Применимость методов. Метод адсорбционной тонкослойной хроматографии (АТХ) распространяется на определение которана, монурона, диурона, дозанекса, тенорана, фалорана, арезина, лину рона, паторана, малорана в воде, которана, монурона, диурона, тенорана, фалорана, арезина, линурона, патораиа в почве, арезина, линурона, паторана, малорана в картофеле, дозанекса, линурона, па тораиа, малорана в моркови, которана, дикурана, паторана, мало рана в лекарственных растениях, дозанекса в пшенице, теиораиа в ягодах земляники Метод распределительной (обращенной) тонко слойнои хроматографии (РТХ) рекомендуется для определения и повышения надежности идентификации фенурона, монурона, диу рона, тенорана, арезина и линурона в воде при совместном при сутствии этих гербицидов
Принцип метода Определение фечилмочевин методами тонко слойной хроматографии заключается в извлечении соединений из проб органическим растворителем, очистке полученного экстракта, (для растительных проб) концентрированной серной кислотой и хроматографировании на пластинках «Силуфол» или в тонком слое окиси алюминия (АТХ) или силикагеля, импрегнированного вазе линовым маслом (РТХ) Пятна на пластинках обнаруживаются после термического разложения препаратов до ароматических ами нов, диазотирования и азосочетания с а) а нафтолом, либо б) N (1 нафтил) этилендиамином В последнем случае чувствительность обнаружения в 5—10 раз выше
Метрологическая характеристика метода. Минимально откры ваемое количество анализируемых соединений на пластинках «Си луфол» 0,2 мкг, на пластинках с оксидом алюминия 1,0 мкг, на импрепгированных пластинках 0,5 мкг Пределы обнаружения пре парагов в анализируемых средах, средние значения определения, стандартные отклонения и доверительные интервалы приведены в таблице 1
Реактивы и растворы. Ацетон ч д а Гидроокись натрия х ч 50% ный водный раствор Гидроокись калия ч Кальций сернокис лый ч д а Кислота соляная ч Кислота серная ч Натрий азотисто кислый ч д а Натрий сернокислый, безводный ч 1 Нафтол ч д а N (1 нафтил) этилендиамии дигидрохлорид ч Ацетонитрил х ч н Гексан ч Спирт этиловый, ректификат Оксид алюминия для хро матографии II степени активности ч Пластинки для ТСХ «Силу
1. Метрологическая характеристика метода (п = 5, Р = 0,95)
Анализир} емый об ьем Соединение Предел обнаружения, мг л (мг/кг) Среднее значение определения, % Стандартное о г»ло-нение, % Доверительный интервал, %
Вода Все фенил-мочевииы 0,01—0,02 85—95 3—6 4-8
Почва Картофель Дозанекс Арезии 0,1—0,2 60—80 3—8 5—14
Морковь Липурон Паторан Малоран 0,05—0,1 75—78 3—6 4—8
Валериана лекарственная (корни) Паторан 5,0 75 — —
Ромашка аптечная Малоран 0,2 80 — —
Подорожник большой Мак масличный: Которая 0,2 72 — —
семена Дикуран 0,5 75 — —
коробочки 0,25 70 — —
Зерно пшеницы Дозанекс 0,1 80 — —
Земляника (ягоды) Теноран 0,5 83—90 — —
фол UV-254». Хлороформ фарм., перегнанный. Этиловый эфир для наркоза, перегнанный. Проявляющие реактивы:
по способу а. Проявляющий реактив № 1: к 46 мл дистиллированной воды прибавляют 4 мл соляной кислоты и 1 г азотистокислого натрия. Проявляющий реактив № 2: к 50 мл дистиллированной воды прибавляют 2,8 г гидроксида калия и 0,1 г 1-нафтола. Использовать необходимо свежеприготовленные реактивы;
по способу б. Проявляющий реактив № 1: 20 мл соляной кислоты смешивают с 80 мл этилового спирта. Проявляющий реактив № 2: 1 г азотистокислого натрия растворяют в 2 мл дистиллированной воды в мерной колбе на 10 мл и доводят до метки этиловым спиртом. Проявляющий реактив № 3: 1 г дигидрохлорида
Ы-(1-иафтил) этилендиамина растворяют при нагревании в 2 мл дистиллированной воды в мерной колбе на 100 мл и доводят до метки этиловым спиртом.
Стандартные растворы фенилмочевии в этиловом спирте с содержанием 200 мкг/мл химически чистого вещества.
Вазелиновое масло. Силикагель ЛС 5/40 М для тонкослойной хроматографии с 13% гипса («Хемапол», ЧССР). Силикагель КСК № 2.
Приборы и посуда. Механический встряхиватель любого типа. Ротационный испаритель любого типа. Воронки делительные емкостью 100 и 500 мл„ тип VIII. Колбы круглодоииые емкостью 100 и 500 мл, тип КК III. Колбы плоскодонные конические емкостью 25, 100 и 200 мл, тип КН. Колбы мерные емкостью 10 и 50 мл,
тип la Пипетки емкостью 1, 5 и 10 мл. Фильтры бумажные Сито капроновое, 100/120 меш Пластинки стеклянные 9X12 см и 15X15 см Пульверизатор стеклянный для обработки пластинок Камера для обработки пластинок. Камера хроматографическая Шкаф сушильный
Отбор проб. Отбор проб производится в соответствии с Унифицированными правилами отбора проб сельскохозяйственной про дукции, пищевых продуктов и объектов окружающей среды для определения микроколичеств пестицидов, утвержденными заместителем Главного государственного санитарного врача СССР 21 августа 1979 г
Определение феиилмочевин методом тонкослойной хроматографии. При определении феиилмочевин методами АТХ и РТХ подготовка проб проводится следующим образом
Вода Пробу воды объемом 250—500 мл экстрагируют в делительной воронке трижды хлороформом (в случае линуропа и мало-рана можно использовать гексан) при соотношении объемов водной и органической фаз 10:1 Экстракты объединяют, сушат, пропуская через слой безводного сернокислого натрия (5—10 г), и отгоняют растворитель на ротационном испарителе до 0,1—0,2 мл при температуре водяной бани 50 °C (или в приборе для перегонки растворителей). Остаток количественно переносят в градуированную пробирку, ополаскивая колбу, в которой происходила отгонка растворителя, 2—3 раза по 0,5—1,0 мл гексана и затем удаляют растворитель в токе воздуха или на водяной бане до 0,1 мл
Почва. Навеску воздушно-сухой почвы (50 г) растирают в фарфоровой ступке и затем экстрагируют фенилмочевины в плоско донной колбе на 500 мл тремя порциями ацетона (хлороформа) по 75 мл Продолжительность одного экстрагирования 30 мин За тем растворитель из колбы сливают через слой безводного сульфата натрия (5—7 г) и отгоняют на ротационном испарителе при температуре водяной бани 50 °C до объема 0,1—0,2 мл В случае получения загрязненных экстрактов проводят очистку так, как это описано ниже
Овощи (клубни картофеля, корнеплоды моркови, ботва) Из мельченную пробу (50—100 г) экстрагируют на аппарате для встряхивания тремя порциями хлороформа по 100 мл Продолжи тельность одного экстрагирования 20 мин При определении в кар тофеле для экстракции применяют эфир. Экстракты сушат безвод ным сульфатом натрия и растворитель удаляют на ротационном испарителе К сухому остатку приливают 5 мл концентрированной серной кислоты и выдерживают 30—40 мин Затем при охлаждении к пробе приливают 50 мл дистиллированной воды и доводят pH до 8—9 также при охлаждении 50%-ным водным раствором гидро ксида натрия Извлекают фенилмочевины гексаном (3 по 30 мл) Экстракт сушат безводным сульфатом натрия и удаляют растворитель на ротационном испарителе
Лекарственные растения Корни валерианы лекарственной (определение которана) Измельченную пробу корней 5 г заливают на ночь 20 мл ацетона Затем отфильтровывают растворитель, остаток на фильтре дважды промывают ацетоном по 10 мл, растворитель удаляют на ротационном испарителе и далее поступают так, как это описано выше
Ромашка аптечная (определение малорана), подорожник большой (определение которана). Пробу измельченного материала 50 г
заливают 100 мл хлороформа, встряхивают в течение часа и оставляют на ночь. Затем растворитель отфильтровывают через слой безводного сульфата натрия (5—7 г), остаток дважды промывают хлороформом по 75 мл, растворитель объединяют и отгоняют на ротационном испарителе. Далее поступают так, как это описано выше.
Семена мака масличного (определение дикурана). Измельченную пробу 10 г заливают на ночь 50 мл ацетонитрила. Затем растворитель отфильтровывают, а остаток дважды промывают ацетонитрилом по 20 мл. Объединенный экстракт пропускают через колонку, заполненную на высоту 20 см смесью силикагеля и оксида алюминия в соотношении 3:1 и предварительно промытую 30 мл ацетонитрила. Затем дикуран элюируют 45 мл ацетонитрила и удаляют растворитель из элюата на ротационном испарителе. Остаток растворяют в 1 мл ацетона.
Коробочки мака масличного (определение дикурана). Измельченную воздушно-сухую пробу 10—20 г заливают на ночь 50 мл хлороформа. Затем растворитель отфильтровывают, удаляют на ротационном испарителе и далее поступают так, как это описано при определении фенилмочевин в овощах.
Зерно пшеницы и кукурузы (определение дозанекса). Измельченную пробу 10 г трижды экстрагируют хлороформом порциями по 70 мл на аппарате для встряхивания. Продолжительность одного экстрагирования 15 мин. Объединенный экстракт пропускают через слой безводного сульфата натрия (5—7 г) и удаляют растворитель на ротационном испарителе. Далее поступают так, как это описано при определении фенилмочевин в овощах.
Ягоды земляники (определение тенорана). Измельченную пробу 50 г заливают 75 мл хлороформа и помещают на аппарат для встряхивания на 30 мин. Экстракт сливают через фильтр, а гомогенат еще дважды экстрагируют хлороформом по 75 мл. Объединенный экстракт упаривают на ротационном испарителе досуха. К сухому остатку приливают 2 мл 0,5 М водного раствора соляной кислоты и переносят в делительную воронку, ополаскивая колбу двумя порциями соляной кислоты по 2 мл. Затем нейтрализуют раствор 0,5 М водным раствором гидроксида натрия, контролируя с помощью индикаторной бумаги, и экстрагируют теноран хлороформом (3 по 30 мл). Объединенный экстракт сушат безводным сульфатом натрия (10—15 г), упаривают растворитель на ротационном испарителе и растворяют сухой остаток в 1 мл ацетона.
Хроматографирование. АТХ. На хроматографическую пластинку количественно наносят конечный экстракт, полученный после удаления растворителя на ротационном испарителе (при определении фенилмочевин в лекарственных растениях и зерне пшеницы и кукурузы на пластинку наносят */ю часть конечного экстракта), и стандартный раствор соответствующего определяемого вещества. Пластинку помещают в камеру для хроматографирования, в которую за 30 мин до проведения хроматографирования наливают подвижный растворитель. После того, как фронт растворителя поднимется на 10 см, пластинку извлекают из камеры, отмечают границу фронта растворителя и сушат на воздухе в вытяжном шкафу. Затем пластинку помещают в сушильный шкаф при 160—170 °C на 30—40 мин. После охлаждения проводят обнаружение определяемых соединений. При проявлении по способу «а» пластинку обрабатывают проявляющим реактивом № 1 и сразу же проявляющим
2. Величина Rf феиилмочевин в различных системах растворителей
Соединение АТХ — оксид алюминия РТХ АТХ — „Силуфол" РТХ
подвижный раствори!ель Rf подвижный растворитель Rf подвижный растворитель Rf ПОДВИЖН чй растворитель Rf аце- тон— вода <2 : 3) Rf
1 2 3 4 5 6 7 8 9 10
Фенурон __ Диэтиловый эфир 0,49 Диэтиловый эфир 0,27 Гексан — ацетон (4 : 1) — 0,60
Монурон — — — 0,53 —- 0,32 — 0,13 0,48
Диурон 0,48 — 0,39 — 0,15 0,30
Которая Диэтиловый эфир — четыреххлористый углерод (5:1) 0,75+0,06 — 0,58 — 0,57 — — —
Дозанекс Бензол — ацетон (9:1) 0,40+0,02 — — — 0,22 — — —
Дикуран — — — 0,68 — — — — —
Теноран
Фалоран
Арезин Диэтиловый 0,59+0,02
эфир — четыреххлористый углерод (3:2)
Паторан Гексан — ацетон 0,47±0,02 (2:1)
Линурон Диэтиловый 0,42+0,04 эфир — четыреххлористый углерод (2:3)
Малоран Бензол 0,38+0,05 Диэтиловый 0,65+0,01 эфир — четырех-
СО хлористый углерод (1:1)
Продолжение
5 6 7 8 9 10
0,47 — — — 0,15
0,45 — — — —
0,34 __ 0,93 — — 0,39
0,94 — 0,94 — 0,28 —
__ — — — 0,21
0,95 . 0,93
реактивом № 2. При наличии в пробе фенилмочевин на пластинке появляются красно-малиновые пятна. При проявлении по способу «б» пластинку последовательно обрабатывают проявляющими реактивами № 1, № 2 и № 3. При наличии в пробе фенилмочевин на пластинке появляются ярко-синие пятна.
Р Т X. В этом случае развитие хроматограммы проводят в системе подвижных растворителей ацетон — вода (2:3), насыщенной вазелиновым маслом. Для насыщения смеси подвижных растворителей вазелиновым маслом смешивают 100 мл смеси и 5 мл вазелинового масла на аппарате для встряхивания в течение 1 ч. После отстаивания разделяют слои, н в качестве подвижного растворителя используют смесь растворителей.
В таблице 2 приведены системы растворителей для хроматографирования и величины Rf фенилмочевин в этих системах растворителей.
Обработка результатов анализа. Оценку содержания фенилмочевин в пробе проводят путем визуального сравнения размера пятен и интенсивности их окраски со стандартами. Количественное определение может быть проведено с использованием денситометра БИАН-170.
Содержание фенилмочевин рассчитывают по формуле
где X— содержание соединения в анализируемой пробе, мг/л (мг/кг); А — количество соединения, найденного путем сравнения со стандартом или посредством денситометрирования, мкг; Р — количество анализируемой пробы, мл (г).
Методические указания по определению котофора в воде, почве, хлопковых семенах, продуктах питания растительного происхождения и биологическом материале методом тонкослойной хроматографии и УФ-спектроскопии
(Утверждены заместителем Главного государственного санитарного врача СССР 31 июля 1984 г.)
Краткая характеристика пестицида. Котофор — 2-этил-тио-4,6-бис-(изопропиламино)-симм-триазин. C11H21N5S. Молекулярная масса 255,39.
Химически чистый препарат — белый порошок с температурой плавления 104—106°C. Трудно растворяется в воде (16 мг/л), хорошо — в органических растворителях — хлороформе, ацетоне, метаноле.
Технический препарат котофора — смачивающийся порошок, содержащий 80% действующего вещества.
Препарат котофор малотоксичен для теплокровных животных: ЛД50 для крыс составляет 4760 мг/кг, для мышей 2800 мг/кг. МДУ котофора в хлопковом масле 4,5 мг/кг, в жмыхе 0,38 мг/кг, ПДК в воде 1,0 мг/л.
Котофор рекомендован для применения в качестве селективного гербицида для довсходового опрыскивания на посевах овощных, бахчевых культур и хлопчатника.
13 Заказ № 1995
Метрологическая характеристика метода
Метрологические параметры Метод анализа вода
Диапазон определяемых ТСХ
концентраций УФ
0,02—1,0 мг/л
0,02—0,5 мг/л
Предел обнаружения, мкг ТСХ УФ 1 2
Предел обнаружения ТСХ УФ 0,02 мг/л 0,02 мг/л
Среднее значение определения стандартных количеств (С), °/о при п = 5 ТСХ УФ 96,8 97,0
Стандартное отклонение ТСХ УФ 5,4 1,66
Доверительный интервал среднего при Р = 0,95; п = 5 ТСХ УФ ±4,9 ±2,63
Относительное стандартное отклонение, % ТСХ УФ 5,1 1,7
Анализируемый объект
почва хлопковые растительные биома-
семена продукты териал
0,05— 1,0 мг/кг 0,05— 0,5 мг/кг 0,05— 0,5 мг/кг 0,05— 0,5 мг/кг 0,6—
1,0 мкг/г
5 — 0,6
5 2,5 2,5 —
0,05 мг/кг — ,— 0,6 мкг/г
0,05 мг/кг 0,05 мг/кг 0,05 мг/кг —
87,0 — 75,0
94,0 84,0 90,0 —
3,47 — — 5,93
2,95 10,7 3,95 —
±3,1 — ±5,3
±4,69 ±17,01 ±6,28 —
3,56 . 7,1
3,1 12,7 4,3 —
Принцип метода. Метод основан на извлечении котофора из исследуемой пробы органическим растворителем, очистке экстракта и последующем определении методами тонкослойной хроматографии и УФ спектрофотометрии
Избирательность метода. Определению остаточных количеств котофора в воде, почве, хлопковых семенах, растительных продуктах и биологическом материале не мешают близкие по строению и области применения пестициды — мезоранил, прометрин
Реактивы и материалы. Этиловый спирт х ч Ацетон ч д а н Гексан х ч Хлороформ ч д а Этиловый эфир медицинский Углерод 4-хлористый х ч Натрий сернокислый безводный х ч Силикагель КСК Кальций сернокислый ч д а Алюминия окись для хроматографии Бромфеноловый синии, индикатор ч д а, 0,4% ный раствор в ацетоне Бромтимоловый синий, индикатор ч д а Серебро азотнокислое х ч 2% ный водный раствор Лимонная кислота х ч 4%-ный водный раствор Соляная кислота х ч 1 н водный раствор Натр едкий х ч 1 н. водный раствор Уголь активированный марки «БАУ» Фильтры бумажные Пластинки для хроматографии «Силуфол» (ЧССР) Вата обезжиренная (гигроскопическая) Стандартные растворы котофора в хлороформе с содержанием 500 и 100 мкг/мл
Приборы, аппаратура и посуда. Спектрофотометр СФ-4а или другой аналогичный прибор Шкаф вытяжной химический Шкаф сушильный Испаритель ротационный ИР-1М Аппарат для встряхивания жидкости в лабораторной посуде АВУ-1 Баня водяная Камера для опрыскивания Камера для хроматографирования Пульверизатор стеклянный Воронки делительные на 150, 250, 1000 мл Воронки простые конусообразные с коротким стеблем № 5 Колбы конические на 250 мл Колбы круглодонные на 100 мл Колбы мер ные с коническим шлифом на 100, 1000 мл Капилляры стеклянные Пипетки на 0,1 и 10 мл Пластинки хроматографические стеклянные размером 9X12 см Сито лабораторное № 5 Сито лабораторное № 40
Подготовка к определению. Приготовление растворов
Приготовление 1 н соляной кислоты 86 мл 36% ной соляной кислоты (d= 1,1789 г/см3) доводят в мерной колбе до 1 л дистил лированной водой Раствор можно приготовить из фиксанала Хранить в прохладном месте в плотно закрытой склянке Срок хранения 3 мес
Приготовление 1 н раствора едкого натра 40 г едкого натра растворяют в минимальном количестве дистиллированной воды и доводят ею объем до 1 л Рекомендуется использовать свежеприготовленные растворы
Проявляющий реагент № 1 Смесь равных объемов 0,4%-ного раствора бромфенолового синего в ацетоне и 2% ного водного раствора азотнокислого серебра Готовят перед употреблением
Проявляющий реагент № 2 4% ный водный раствор лимонной кислоты Хранить в прохладном месте не более 10 дней
Проявляющий реагент № 3 В 250 мл мерной колбе 1,0 г азотнокислого серебра, 0,1 г бромтимолового синего растворяют в 90 мл дистиллированной воды и доводят объем до метки ацетоном Готовят перед употреблением
Стандартные растворы Стандартные растворы котофора с содержанием 100 и 500 мкг действующего начала препарата в 1 мл хлороформа, готовят из х ч или технического препарата с учетом
процента содержания действующего вещества. Растворы хранить в прохладном месте не более 1 мес.
Приготовление хроматографических пластинок.
Пластинки с тонким слоем КСК.. Тщательно промытую и высушенную пластинку размером 9X12 см протирают ватным тампоном, смоченным в этиловом спирте или диэтиловом эфире, и покрывают сорбционной массой, приготовленной из 35 г силикагеля КСК, 2 г сернокислого кальция и 90 мл дистиллированной воды Сорбционную массу готовят, тщательно растирая в фарфоровой ступке сначала сухую смесь силикагеля с гипсом, а затем и с водой до однородной массы. 10 г сорбционной массы равномерно распределяют по всей поверхности пластинки путем ее покачивания. Сушат пластинки при комнатной температуре 18—20 ч. Хранят в эксикаторе.
Силикагель предварительно очищают от примесей, для чего заливают его на 18—20 ч соляной кислотой (1 : 1), кислоту сливают, промывают водой и кипятят 2—3 ч с разбавленной азотной кислотой (1 : 1), промывают обычной водой, а затем и дистиллированной до нейтральной реакции промывных вод, сушат в сушильном шкафу 4—6 ч при температуре 130 °C. Обработанный и просушенный силикагель дробят и просеивают через сито № 40. Хранят в склянке с притертой пробкой.
Пластинки с тонким слоем окиси алюминия. Для приготовления сорбционной массы на 10 пластинок берут 50 г окиси алюминия, просеянной через сито № 40, 5 г сернокислого кальция. Окись алюминия с сернокислым кальцием тщательно растирают в фарфоровой ступке, переносят в коническую колбу, прибавляют 150 мл дистиллированной воды и встряхивают до образования однородной массы. Полученную массу наносят тонким слоем на заранее приготовленные стеклянные пластинки. Пластинки сушат в течение 12— 15 ч при комнатной температуре. Готовые пластинки хранят в эксикаторе.
Построение калибровочного графика для спектрофотометрического определения. На пластинку наносят 0,02, 0,05, 0,1, 0,2, 0,3, 0,5, 0,7 мл стандартного раствора с концентрацией котофора 100 мкг/мл, что соответствует 2, 5, 10, 20, 30, 50, 70 мкг, и хроматографируют в смеси хлороформ — гексан (4:1). С целью спектро-фотометрирования пластинка, снятая с камеры, проявляется не полностью, а частично. При этом покрывается листком чистой бумаги участок опытной площади и опрыскивается проявляющим реактивом № 3 только область одного из пятен (лучше одна из концевых областей, например с 70 мкг вещества). Затем предполагаемые площади опытных пятен на уровне стандарта количественно вместе с окисью алюминия переносят в центрифужные пробирки. В каждую пробирку заливают по 3 мл хлороформа, закрывают корковой пробкой и оставляют иа 2 ч. Время от времени смесь взбалтывают, затем экстракт центрифугируют при 1500 об/мин в течение 15 мин. После этого хлороформный слой центрифугата осторожно, не взмучивая осадок, выливают в другую центрифужную пробирку и объем доводят хлороформом до 3 мл. Затем раствор переливают в 3-миллиметровую кювету и спектрофотометрируют при длине волны 247 нм. Строят, график зависимости оптической плотности от концентрации вещества. Линейность показаний наблюдается в пределах 2—50 мкг. При содержании котофора в пробе более 50 мкг
необходимо развести ее, и коэффициент разведения учитывать при расчетах.
Отбор проб. Отбор проб производят в соответствии с Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов. Пробы воды, почвы, хлопковых семян, продуктов питания растительного происхождения и биологического материала должны транспортироваться и храниться в стеклянной или полиэтиленовой таре, причем пробы биоматериала следует хранить при температуре минус 5—8 °C.
Проведение определения. Метод ТСХ. Экстракция.
Вода. 1 л анализируемой воды помещают в делительную воронку и трижды экстрагируют хлороформом порциями по 50, 30, 30 мл. Объединенный экстракт сливают через воронку с 5—7 г безводного сульфата натрия в колбу ротационного испарителя и отгоняют хлороформ до объема 0,1—0,2 мл при температуре бани 80 °C.
Почва. 100 г воздушно-сухой почвы растирают в ступке, просеивают через сито № 5, заливают в конической колбе ацетоном до покрытия пробы и оставляют на ночь. Отфильтрованный объединенный экстракт кипятят на водяной бане при температуре бани 80 °C с активированным углем марки «БАУ» (5—7 г) до исчезновения окраски. Растворитель фильтруют через бумажный фильтр «синяя лента» и безводный сульфат натрия в колбу для отгонки растворителя. Уголь на фильтре промывают несколькими порциями ацетона. Ацетон отгоняют с помощью ротационного испарителя до объема 0,1—0,2 мл.
Биологический материал (кровь, печень, почки, легкие, селезенка, сердце, мышечная ткань): 5 г биоматериала тщательно измельчают ножницами, помещают в колбу с притертой пробкой, заливают 30 мл смеси гексан — ацетон (1:1) и периодически встряхивают в течение 1 ч. Сливают смесь гексан — ацетон, пробу повторно заливают смесью и опять периодически встряхивают 1 ч; * экстракцию повторяют трижды. Объединенные экстракты сушат безводным сернокислым натрием (3—5 г) и упаривают при температуре бани 80 °C. К сухому остатку прибавляют 2 мл 1 н. соляной кислоты, тщательно ополаскивают стенки колбы. Смывы сливают в делительную воронку, фильтруя через небольшой слой ваты. Колбу трижды ополаскивают 1 мл 1 н. соляной кислоты, смывы сливают в ту же делительную воронку. В делительную воронку добавляют 4 мл 1 н. раствора едкого натра и перемешивают, а затем по каплям приливают 1 н. раствор едкого натра до нейтральной реакции (по индикаторной бумажке). После этого в воронку прибавляют 10 мл предварительно насыщенного водой хлороформа и встряхивают 1—2 мин. Хлороформный экстракт сливают из воронки в колбу через слой безводного сернокислого натрия. Экстракцию хлороформом повторяют трижды. Объединенный хлороформный экстракт отгоняют на ротационном испарителе при температуре бани 80 °C до объема 0,1—0,2 мл.
Хроматографирование. Подготовленные экстракты количественно наносят при помощи капиллярной пипетки на хроматографическую пластинку «Силуфол» так, чтобы диаметр пятна не превышал 1 см. Центр пятна должен быть на расстоянии 1,5 см от нижнего края пластинки. Колбочку с экстрактом 2—3 раза смывают небольшими 196
порциями хлороформа, который также наносят в центр первого пятна. Справа и слева от пробы па расстоянии 2 см от нее наносят стандартные растворы (шприцем или микропипеткой), содержащие 5, 10 или 20 мкг препарата
Пластинку с нанесенными растворами помещают в хроматографическую камеру, в которую предварительно налита смесь растворителей гексан-ацетон в соотношении 10:3 или четыреххлористый углерод-диэтиловый эфир в соотношении 4:1. Край пластинки не должен быть погружен в раствор более чем на 0,5 см. После того как фронт подвижного растворителя поднимется на 10 см, пластинку вынимают и оставляют на несколько минут на воздухе для испарения подвижного растворителя. Пластинку обрабатывают с помощью пульверизатора проявляющим реактивом № 1. После высушивания пластинки на воздухе ее обрабатывают проявляющим реактивом № 2. Зоны локализации препарата обнаруживаются в виде синих пятен на желтом фоне со следующими величинами Rf:
Система подвижных растворителей Величина Ш
силикагель КСК „Силуфол*1
Гексан — ацетон (10:3) 0,52+0,01 0,50+0,01
Четыреххлористый углерод — диэтиловый эфир (4:1) 0,21±0,01 0,29+0,01
Количество котофора определяют путем сравнения размеров и интенсивности окраски пятен пестицида на хроматограммах пробы и стандартного раствора.
Метод УФ-спектрофотометрии, Экстракция.
Вода, почва. Экстракция проводится, как при методе ТСХ.
Хлопковые семена. Размельченные в ступке хлопковые семена (50 г) трехкратно экстрагируют хлороформом по 50 мл, хлороформные экстракты объединяют, фильтруют через бумажный фильтр в выпарительную колбочку и выпаривают до полного удаления хлороформа. Содержимое выпарительной колбочки переносят в коническую колбу на 250 мл путем двукратного смыва по 25 мл ацетоном.
Затем в эту же колбу добавляют 100 мл ледяной дистиллированной воды. Колбу с содержимым ставят в морозильную камеру холодильника на 1 ч, после чего колбу вынимают из холодильника и содержимое быстро фильтруют через бумажный фильтр в другую коническую колбу и вновь ставят в морозильную камеру. Через 1 ч колбу снимают, содержимое фильтруют в выпарительную колбочку и имеющийся в фильтрате ацетон выпаривают на ротационном испарителе или водяной бане. Оставшуюся водную фракцию экстракта переносят в делительную воронку и трижды экстрагируют хлороформом по 25 мл. Экстракты объединяют, сушат над безводным сульфатом, натрия и выпаривают досуха.
Растительные продукты (картофель, лук, томаты). Из средней пробы продуктов, размельченных миксером или мясорубкой, отбирают навеску в 50 г. Экстракцию проводят трехкратно хлороформом по 100 мл в течение 30—40 мин. Полученные экстракты объ-
едпняют и выпаривают досуха на ротационном испарителе или водяной бане.
Хроматографирование и спектрофотометрическое определение. Колбочку из-под экстракта трехкратно ополаскивают по 0,1 мл хлороформа и полученный смыв наносят на хроматографическую пластинку, отступая на 1 см от ее нижнего края. Рядом с пробой на пластинку наносят стандарт с известной концентрацией котофора. Затем развивают хроматограмму в смеси хлороформ — гексан (4:1). После завершения разгонки (10 см) пластинку вынимают из камеры и высушивают при комнатной температуре в течение 10 мин. Покрыв листком чистой бумаги участок опытной площади пластинки, обрабатывают проявляющим реактивом № 3 лишь зону локализации свидетеля (Rf 0,50—0,60). Далее поступают, как при построении калибровочного графика, спектрофотометрируя конечные растворы при длине волны 247 нм.
Количество котофора определяют по калибровочному графику.
Обработка результатов
Содержание котофора в анализируемой пробе рассчитывают по формуле
где X — содержание котофора в анализируемой пробе, мг/л, мг/кг; мкг /г (в случае биоматериала); А — в варианте ТСХ, количество котофора, найденное путем сравнения со стандартом, мкг; в варианте спектрофотометрического определения, количество котофора, найденное по калибровочному графику, мкг; Р— количество пробы, взятой для анализа, мл, г.
Временные методические указания по определению витавакса в зерне и воде методом тонкослойной хроматографии
(Утверждены заместителем. Главного государственного санитарного врача СССР 31 июля 1984 г.)
Краткая характеристика пестицида. Витавакс (карбоксин) — 3-метил-2-фенилкарбамоил-5,6-Дигидро-1,4-оксатиин представляет собой белое кристаллическое вещество с {Пл 91,5—92,5 °C. Ci2H13NO2S. Растворимость в воде — 0,17 г в 100 г. Хорошо растворяется в хлороформе, ацетоне, бензоле, метаноле и этаноле. Молекулярная масса 235,3.
ЛДйо для мышей и крыс 3200 мг/кг.
ПДК в воздухе рабочей зоны 1 мг/м3, в продуктах питания — не допускается.
Используется для протравливания семян зерновых культур в борьбе с пыльной головней. Эффективен также против ржавчинных грибов на различных культурах. Выпускается в форме 75%-ного с. п.
При окислении его перекисью водорода получается плантвакс.
В растениях в течение 6 нед витавакс подвергается окислению, образуя в основном сульфоксид (5,6-дигидро-2-метил-1,4-оксатиин-З-карбоксанилид-4-оксид) и в небольших количествах сульфон (5,6-дигидро-2-метил-1,4-оксатиин-3-карбоксанилид-4,4-диоксид). В воде при pH 2 и 4 через несколько недель обнаруживают витавакс,
сульфоксид и в незначительных количествах сульфон. В почве окисление витавакса до сульфоксида полностью завершается за 2 нед.
Окисление витавакса до сульфоксида наблюдается в растворах некоторых органических растворителей.
Принцип метода. Вариант 1. Суммарное определение витавакса и его метаболитов Метод определения витавакса и его метаболитов основан на их экстракции органическим растворителем, окислении витавакса перманганатом калия до сульфоксида, очистке экстракта, переэкстракции в органический растворитель и дальнейшем определении в тонком слое адсорбента («Силуфол», силикагель). В качестве подвижной фазы используют смесь хлороформа и эфира в соотношении 5 :2.
Обнаружение зон локализации сульфоксида витавакса в тонком слое сорбента основано на его УФ-разложении до аминосоединения, диазотировании нитритом натрия в солянокислой среде и азосочетании полученных солей фенилдиазония с М-(1-нафтилэтилендиами-ном), либо а-нафтолом, а-нафтиламином
Вариант 2. Определение витавакса. Метод определения витавакса основан на экстракции его из анализируемой пробы орианическим растворителем, очистке экстракта перераспределением пестицида в системе органический растворитель — вода — органический растворитель с последующим определением в тонком слое силикагеля КСК-2,5 или силуфола.
Подвижная фаза — смесь н-гексана и ацетона в соотношении 3:2 или н-гексана и эфира в соотношении 1 : 1.
Обнаружение зон локализации пестицида производят смесью реактивов: 0,05%-ного раствора бромфенолового синего в 50%-ном этаноле и 1%-ного раствора азотнокислого серебра в воде с последующей обработкой пластинок 2%-ным раствором лимонной кислоты. Продукты окисления витавакса данным реактивом не обнаруживаются
Возможно обнаружение витавакса по реакции, описанной выше (вариант 1, УФ-разложение, диазотирование и азосочетание).
Метрологическая характеристика метода. Нижний предел обнаружения сульфоксида витавакса с помощью реакции диазотирования и азосочетания — азосоставляющая Ы-(1-нафтилэтилендиамин) 0,5—1,0 мкг и азосоставляющие а-нафтиламин, а-нафтол 1,0— 1,5 мкг. Диапазон определяемых концентраций 1—30 мкг.
Метрологическая характеристика метода представлена в таблице
Метрологическая характеристика метода
Анализируемый объект Нижнии предел обнаружения, мл/л, мг/кг Среднее значение определения, % Стандартное отклонение. °/о Доверительный интервал среднего, °/о
Вода (варианты 1,, 2) 0,02 88,0 2,6 88,0±3,2 Зерно пшеницы (вари- 0,1 85,0 8,0 85,0±8,7 ант 1) Зерно пшеницы (вари- 0,1 83,0 4,0 83,0±6,1 анг 2)
Реактивы и растворы. Ацетон х. ч. н-Гексан ч. Гипс (кальций сернокислый CaSO4-H2O) в течение 2 сут прокаливают при 180 °C и просеивают через сито 100 меш. Калий едкий ч. д. а. Калия перманганат ч. д. а.
Крахмал водорастворимый ч. Кислота соляная, отн. пл. 1,18. Натрий сернокислый безводный х. ч. Натрий азотистокислый х. ч. Спирт этиловый ректификат 96 %-ный. Хлороформ х. ч. Эфир для наркоза. Пластинки «Силуфол» (ЧССР). Силикагель для хроматографии, КСК-2,5. Измельчают, просеивают через сито 100 меш.
Приготовление проявляющих реактивов. Реактив № 1: а) к смеси, состоящей из 46 мл дистиллированной воды и 4 мл концентрированной соляной кислоты, прибавляют 1 г азотистокислого натрия; б) N-(1-нафтилэтилендиамин)-дигидрохлорид ч. 1%-ный раствор в этиловом спирте; в) а-пафтиламин ч. д. а. 1 % ный раствор в этиловом спирте; г) а-нафтол ч. д. а., 3 г едкого кали растворяют в 50 мл дистиллированной воды, затем прибавляют 0,5 г а-нафтола.
Реактивы готовят непосредственно перед проявлением. Расход реактивов 4—5 мл на одну пластинку.
Реактив № 2: а) 0,05 %-ный раствор бромфенолового синего ч д. а. в 50%-ном этаноле; б) 1%-ный водный раствор азотнокислого серебра ч. д. а. Растворы «а» и «б» смешивают в соотношении 1:1; в) 2 %-ный водный раствор лимонной кислоты х. ч.
Растворы хранят в темной посуде в течение месяца.
Стандартный раствор витавахса готовят растворением 10 мг препарата в ацетоне (50 мл). Раствор содержит 200 мкг/мл. Устойчив при хранении в холодильнике в течение 3 мес.
Приборы и посуда. Аппарат для встряхивания. Баня водяная. Воронки химические, диаметр 6 см. Воронки делительные емкостью 250, 500 мл. Камера для опрыскивания. Камера для хроматографирования размером 150X200 мм. Колбы мерные емкостью 50, 100 мл Колбы круглодонные со шлифами емкостью 150, 200 мл. Лампа кварцевая ПРК-4 или ПРК-7. Микропипетка (для нанесения стандартного раствора). Пластинки стеклянные размером 90Х120 мм.* Пипетки емкостью 1, 5, 10 мл. Прибор для отгонки растворителя. Пульверизатор стеклянный. Цилиндры мерные емкостью 25, 50, 100, 250 мл.
Подготовка к определению. Приготовление пластинок. 40 г силикагеля марки Л либо КСК-2,5 растирают в фарфоровой ступке с 2 г гипса. Прибавляют воду (90 мл) небольшими порциями и размешивают до образования однородной массы На пластинку размером 9X12 см наносят 5 г массы, равномерно распределяют ее по поверхности. Сушат пластинки на воздухе в течение суток при комнатной температуре.
Приготовление «свидетеля». В колбочку вносят 0,025—0,05 мл стандартного раствора витавакса (200 мкг/мл), что соответствует 5—10 мкг препарата, упаривают в токе теплого воздуха досуха, прибавляют 10 мл 0,1%-ного раствора перманганата калия. Оставляют раствор на 15 мин. Затем прибавляют 10 мл 0,3%-ного раствора соляной кислоты и 5 мл 1 н. щавелевой кислоты и ждут до тех пор, пока раствор обесцветится. Экстрагируют хлороформом трижды по 50 мл в течение 3—5 мин каждый раз. Экстракты объединяют, сушат безводным сернокислым натрием и упаривают до небольшого объема. Сконцентрированную пробу количественно наносят на пластинку.
Отбор проб. Отбор проб должен проводиться в соответствии с Унифицированными правилами отбора проб сельскохозяйственной продукции, продуктов питания и объектов окружающей среды для определения микроколичеств пестицидов.
Проведение определения. Экстракция.
Вода. 200 мл анализируемой пробы воды помещают в делительную воронку и экстрагируют хлороформом трижды по 50 мл в течение 5 мин каждый раз. Экстракты объединяют и фильтруют через слой безводного сернокислого натрия и упаривают досуха на ротационном испарителе.
При необходимости определения в воде суммы витавакса и его метаболитов (вариант 1) к сухому остатку прибавляют 10 мл 0,1%-ного раствора перманганата калия и делают дальше так, как описано в приготовлении «свидетеля».
При определении только метаболитов растворы перманганата калия и щавелевой кислоты не прибавляют (остальное делают так же).
Для определения только витавакса в воде по варианту 2 сконцентрированный хлороформенный экстракт (описано выше) наносят на пластинку, покрытую тонким слоем адсорбента, и хроматографируют.
Зерно. 50—100 г зерна пшеницы помещают в коническую колбу, прибавляют 60—120 мл хлороформа и экстрагируют 2 ч на аппарате для встряхивания. Экстракцию повторяют еще раз в течение 30 мин. Экстракты объединяют, сушат безводным сернокислым натрием, фильтруют через бумажный фильтр (синяя лента) и упаривают досуха на ротационном испарителе.
При определении в зерне суммы витавакса и его метаболитов (вариант 1) к сухому остатку добавляют 10 мл 0,1%-ного раствора перманганата калия. Оставляют раствор на 15 мин. Затем прибавляют 10 мл 0,3 %-ного раствора соляной кислоты и 5 мл 1 н. раствора щавелевой кислоты и ждут, пока раствор обесцветится. Прибавляют 20 мл ацетона и ставят в морозильную камеру на 60 мин. Фильтруют раствор через плотный бумажный фильтр (синяя лента). Экстрагируют хлороформом трижды по 50 мл в течение 3—5 мин каждый раз. Экстракты объединяют, сушат безводным сернокислым натрием и упаривают до небольшого объема. Сконцентрированную пробу количественно наносят на пластинку
В случае определения в пробе одних метаболитов в ходе анализа перманганата калия и щавелевую кислоту не прибавляют.
Для определения в зерне витавакса по варианту 2 к сухому остатку, полученному после упаривания сконцентрированного хлороформенного экстракта, добавляют дистиллированную воду (30— 35 мл), тщательно перемешивают стеклянной палочкой и оставляют на 2 ч, периодически перемешивая. Водный экстракт переносят в делительную воронку иа 250 мл через бумажный фильтр (синяя лента). Экстракцию витавакса повторяют еще 2 раза в течение 30 мин и переносят экстракт в ту же воронку. В воронку прибавляют 0,1 г хлорида натрия, 15 мл хлороформа и встряхивают содержимое в течение. 5 мин. Хлороформенный слой отделяют. Экстракцию витавакса из водного раствора повторяют еще 3 раза. Экстракты объединяют, сушат безводным сернокислым натрием и упаривают на ротационном испарителе до небольшого объема (0,1 мл). Остаток наносят на хроматографическую пластинку.
Хроматографирование. При определении в анализируемых пробах суммы витавакса и его метаболитов (вариант 1) используют подвижную фазу хлороформ—эфир (5:2). В качестве «свидетеля» наносят стандартные растворы витавакса, обработанные перманганатом калия (как описано при приготовлении «свидетеля»). После хроматографирования пластинку сушат и облучают УФ-светом в течение 20 мин. Обнаружение зон локализации сульфоксида витавакса производят путем последовательной обработки пластинки раствором нитрита натрия в солянокислой среде и раствором М-(1-нафтилэтилендиамина) либо а-нафтиламина (а-нафтола). При наличии витавакса (метаболитов) появляются пятна фиолетового либо розового цвета соответственно. Величина Rf сульфоксида 0,38 («Силуфол») и 0,65 (силикагель).
При определении витавакса по варианту 2 в качестве подвижной фазы используют смесь н-гексана и ацетона в соотношении 3:2 или н-гексана и эфира в соотношении 1 : 1.
В качестве «свидетеля» наносят стандартный раствор витавакса (200 мкг/мл) в количестве 0,005—0,05 мл, что соответствует 1—10 мкг пестицида. После хроматографирования пластинку сушат и обрабатывают смесью растворов бромфенолового синего в ацетоне и азотнокислого серебра в воде в соотношении 1 : 1. Для устранения фона пластинку обрабатывают раствором лимонной кислоты. Расход проявляющих реактивов на одну пластинку составляет 4—5 мл. Витавакс проявляется в виде синих пятен на (.вегло-желтом фоне. Величина Rf витавакса 0,50 (н-гексан + аце-тон в соотношении 3:2) на силикагеле, 0,38 и 0,65 (н-гексан+эфир в соотношении 1:1) на «Силуфоле» и силикагеле соответственно.
Возможно также обнаружение зон локализации витавакса по реакции диазотирования и азосочетания (вариант 1).
Обработка результатов анализа. Количественное определение производится путем сравнения интенсивности окраски и размера пятен пробы и стандартных растворов.
Содержание препарата в анализируемой пробе рассчитывают по формуле
где X—содержание препарата в анализируемой пробе, мг/л (мг/кг); А — содержание препарата в стандартном растворе, мкг; Si — площадь пятна стандартного раствора, мм2; S2 — площадь пятна пробы, мм2; Р — объем пробы (или масса), взятой на анализ, мл (г).
Временные методические указания по определению голтикса в воде, почве и растениях методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врана СССР 27 апреля 1984 г.)
Характеристика пестицида. Голтикс (метамитрон) — гербицид, рекомендуемый фирмой Байер (ФРГ) для прополки посевов сахарной, кормовой и столовой свеклы. Эффективен в до- и после-всходовый период против двудольных и некоторых злаковых сорняков.
Химическое название 3 метил 4 амино 6 фенил 1,2,4 триазин 5-(4Н) ОН
Молекулярная масса 202, температура плавления 166,6 °C Растворимость, % вода 0,18, метиленхлорид, циклогексан 1—5, изопропанол, толуол до 1 Давление паров 1 10~4 мм рт ст (при 20—70 °C)
Малотоксичен для теплокровных ЛД50 для крыс 3343 мг/кг
Описание методики определения голтикса в воде, почве и растениях
Принцип метода. Методика основана на тонкослойно-хроматографическом определении голтикса после экстракции его из пробы органическим растворителем и очистки экстракта путем перерас пределения между двумя несмешивающимися фазами
Метрологическая характеристика метода. Нижний предел де тектирования — 0,2 мкг Чувствительность определения вода 0,05— 0,1 мг/л, почва 0,1—0,2 мг/кг, корпи свеклы 0,1 мг/кг Среднее значение определения стандартных растворов гербицида вода 82,5%, почва — 80, корни свеклы 70,2% Стандартное отклонение 5,0% (вода), 8,5 (почва), 6,4% (корни свеклы) Относительное стандартное отклонение 0,06 (вода), 0,11 (почва), 0,09 (корни свеклы)
Определению голтикса не мешают бетанал, ленацил, пирамин, ТХА, метафос
Реактивы и растворы
Ацетон ч д а I ексан х ч Этилацетат х ч Хлороформ меди цинский Бензол ч д а Этанол ректификат Кислота серная ч д а Натрий сернокислый, безводный х ч Стандартные растворы гол тикса в гексане, содержащие 10, 50, 100 мкг/мл препарата Стан дартные растворы хранятся в темном прохладном месте в течеш е двух месяцев без заметного разложения
Проявляющий реактив 2% иый водный раствор азотнокисл01 о серебра
Посуда и приборы Аппарат для встряхивания АВУ 1 Микроизмельчитель тканей РТ 2 Испаритель ротационный ИР 1М Источник УФ света (лампа типа ПРК-4) Хроматоскоп Сито металличс ское с диаметром отверстий 1 мм Водоструйный насос КМ 1230 Воронки делительные ВД 3 500 Колбы Кн КШ 500 29/32 ТС Кол бы остродонные ОКШ 50 14/23 ТС Колбы мерные 100 мл Микропипетки на 0,1 мл Пластины для тонкослойной хромаго! рафии «Силуфол»
Подготовка к определению Отбор и подготовка проб Отбор проб на анализ проводят в соответствии с Унифицированны ми правилами отбора проб сельскохозяйственной продукции, про дуктов питания и объектов окружающей среды для определения микроколичеств пестицидов
Для анализа воды отбирают средний образец не менее 1—2 т, при необходимости фильтруют Средний образец почвы (1—2 ki ) сушат до воздушно сухого состояния, растирают, просеивают через сито Из среднего образца корней свеклы вырезают полоски в про дольном направлении и измельчают ножом
Проведение определения. Экстракция
Вода 20—50 мЛ воды трижды экстрагируют этилацетатом порциями по 20—50 мл Органический растворитель пропускают через фильтр со слоем безводного сернокислого натрия и упаривают досуха на ротационном испарителе при температуре не выше 40 °C
Почва. 20 г почвы встряхивают в течение 60 мин со 100 мл смеси ацетона и воды (1:3 по объему). Экстракт фильтруют через бумажный фильтр, остаток дважды встряхивают с водным ацетоном в течение 10 мин. Экстракты объединяют и реэкстрагируют этилацетатом 3X50 мл, встряхивая каждый раз в течение 2 мин. Этилацетат осушают безводным сернокислым натрием и упаривают досуха.
Корни свеклы. Гомогенизируют 20 г пробы с 50 мл смеси ацетон— вода (1:3) в микроизмельчителе тканей в течение 2 мин. Растворитель декантируют и фильтруют через слой ваты, твердый остаток повторно гомогенизируют 2 мин с водным ацетоном и фильтруют через слой ваты. Дальше поступают, как при анализе почвы Чтобы предотвратить образование стойкой эмульсии, встряхивание с этилацетатом (особенно первые 2 раза) необходимо проводить осторожно
Хроматографирование. Сухие остатки растворяют в 2 мл гексана, растворитель упаривают до 0,5—1 мл под струей холодного воздуха (точный объем экстракта замеряют пипеткой) и аликвоту (0,05—0,1 мл) переносят в одно пятно на пластинку. Рядом наносят стандартные растворы, содержащие 1, 5 и 10 мкг голтикса. Пластинку хроматографируют в любой из трех систем подвижных растворителей (в скобках указан Rf голтикса): бензол — ацетон 2:1 (0,5); хлороформ — ацетон 2:1 (0,5); хлороформ — этанол 9:1 (0,45).
После хроматографирования пластинку подсушивают до полного улетучивания органических растворителей, затем просматривают под хроматоскопом. Голтикс обнаруживается в виде темно-синих пятен на желтом флуоресцирующем фоне.
В качестве проявляющего реактива используют также 2 %-ный водный раствор азотнокислого серебра После хроматографирования пластину опрыскивают проявляющим раствором и помещают на 10—15 мин под источник УФ-света. При наличии в пробе голтикса на сером фоне проявляются коричневые пятна, Rf которых совпадает с Rf пятен стандартных растворов.
Обработка результатов анализа. Количество голтикса в пягне определяют путем сравнения размера пятен проб и стандартов.
Содержание голтикса в пробе рассчитывают по формуле
где X— содержание голтикса в пробе, мг/кг (мг/л); А— количество гербицида, найденное путем сравнения со стандартами, мкг; Р— масса (объем) пробы, г (мл).
Методические указания по диагностике, лечению и профилактике отравления сельскохозяйственных животных туром
(Утверждены Главным управлением ветеринарии
Министерства сельского хозяйства СССР 29 февраля 1984 г.)
Общие положения. Тур (синонимы: хлорхолинхлорид, зур, хлор-мекват, шгкоцель, стабилан, сайкосел, эмсурон, лиоцин, кукукаль). Действующее начало (2-хлорэтилтриметиламмоний хлорид) являет
ся эффективным регулятором роста растений (ретардантом) и применяется в сельскохозяйственном производстве для предпосевной обработки зерна злаковых культур с одновременным протравливанием гранозаном перед посевом, а также с мочевиной для подкормки и предотвращения полегания пшеницы, ржи, ячменя, овса в дозе 3,3—6,6 кг/га, для ограничения роста лозы на винограднике и ботвы на картофеле, на свекле, томатах и моркови в дозе 1,5—3 кг/га с целью предотвращения преждевременного прорастания при хранении, для повышения морозоустойчивости и засухоустойчивости злаковых культур в дозе 4—6 кг/га в виде водных растворов 0,5—0,8 %-ной концентрации.
Химически чистый препарат — белое кристаллическое вещество без запаха, мол. масса 158,07, температура плавления 300 °C, гигроскопичен, хорошо растворим в воде, ацетоне, бензоле, толуоле, имеет горький вкус. Технический продукт — 60%-ный водный раствор с коричневым оттенком, pH 8,0, при 20 °C d= 1,12.
Тур сравнительно стоек во внешней среде, на поверхности почвы и в воде разлагается в течение 2—3 нед до холина, аммиака, углекислоты и воды, в емкостях и на стенах тары активность пре парата сохраняется до одного года. Тур не взаимодействует с гербицидами, мочевиной, гранозаном и минеральными химическими удобрениями, поэтому допускается применение его в смеси с указанными соединениями. Препарат тормозит рост стебля злаковых растений, вызывая усиление междоузлий и корневой системы, повышая, таким образом, устойчивость соломины к полеганию без отрицательного влияния на колос и урожайность зерна.
Токсикологическая характеристика тура. Тур относится к сред-нетоксичпым веществам, параметры его токсичности составляют (мг/кг): ЛД50 для крыс самцов 470 (303—728), для самок 410 (282—595), для мышей 400 (286—560), для морских свинок 430 (380—486), для кроликов 150, для собак 75, для овец 200; смертельная доза (ЛДюо) для коров находится в пределах 400—600, ЛД50 200—300, максимально-переносимая 100—166. Препарат выделяется из организма животных в течение 2—8 ч преимущественно с мочой. Кожно-резорбтивными, кумулятивными, тератогенными и канцерогенными свойствами тур не обладает. Остаточные количества препарата в растениях, обработанных в рекомендуемых дозах и убранных в установленные сроки после его внесения, не обнаруживаются, допустимые остаточные количества в зерне — 0,5 мг/кг.
В механизме токсического влияния тура на организм животных преобладат курареподобное действие, проявляющееся в блокировании нервно-мышечных синапсов, кратковременным возбуждением и судорогами. Метаболизм тура в организме животных обусловлен отщеплением атома хлора и образованием холинхлорида, который является естественным метаболитом теплокровных животных.
Отравление сельскохозяйственных животных (коров, телят, овец и лошадей) туром возникает в хозяйствах при попадании препарата в корма и водоисточники, в результате нарушения требований инструкции по хранению и внесению препарата, при несоблюдении сроков уборки зернорых после обработки. Источником загрязнения кормов туром также может быть металлическая и деревянная тара, в которой хранился, препарат, при использовании ее в качестве автопоилок и для перевозки жидких кормовых средств (патоки, мелассы, спиртовой барды), при несоблюдении сроков ожидания и
скармливания свежеобработанной туром зеленой массы растений, при пастьбе животных по свежеобработанному травостою.
Диагностика отравлений животных туром. Клинические симптомы интоксикации туром проявляются в тяжелой, средней и легкой степени. Тяжелая степень отравления характеризуется нарушениями функции центральной нервной системы, которые возникают через 1—3 ч после поступления препарата в организм животных, при этом отмечаются беспокойство, нарушение координации движений, слабость конечностей, в последующем развивается общее угнетение, одышка, тахикардия, тремор скелетных мышц, судороги, сужение зрачков, слюнотечение, затрудненное дыхание вследствие бронхоспазма и отека легких, затем развивается коматозное состояние, и животное гибнет от асфиксии. Смертность высокая, особенно при скармливании жидких кормов и воды, загрязненных туром.
При средней и легкой степени отравления течение патологического процесса более растянуто, у коров, телят и овец наблюдаются диарея, цианоз видимых слизистых оболочек, хрипы в легких вследствие отека. Клинические симптомы отравления у животных продолжаются в течение 8—10 ч, затем общее состояние улучшается, прекращается слюнотечение, через 12—16 ч восстанавливается координация движений.
Прием корма и воды восстанавливается через 2—3 сут, однако стойкая диарея, атония преджелудков и общее исхудание наблюдаются в течение 7—!0 дней. Полное выздоровление животных наступает через 12—15 сут.
Патологоанатомические изменения. При патологоанатомическом вскрытии павших и вынужденно убитых животных после отравления туром наблюдаются выраженное полнокровие паренхиматозных органов, отек легких, множественные точечные кровоизлияния под эндо- и перикардом и под капсулой почек. Печень увеличена, капсула напряжена, с поверхности стекает темная венозная кровь. Слизистая оболочка желудочно-кишечного тракта в состоянии катарально-геморрагического воспаления.
Диагностика отравлений животных туром должна быть комплексной с учетом анамнестических данных, сведений об использовании тура в хозяйствах, контакте с ним животных, результатов клинических и патоморфологнческих исследований, химико-аналитического определения тура в кормах, воде, за исключением инфекционных заболеваний (бешенство, столбняк, ботулизм), путем бактериологических, вирусологических и биологических исследований. Необходимо также выяснить возможность комбинированного отравления животных туром, гранозаном и карбамидом, поскольку они зачастую применяются совместно для обработки посевного зерна. Определение остаточных количеств тура в кормах и воде проводится с помощью хроматографического ионообменного метода, утвержденного Минздравом СССР 27.09.78 г. № 1909 и опубликованного в Методических указаниях по определению микроколичеств пестицидов в продуктах питания, кормах и внешней среде, часть 10 (М., 1980). Биологический метод заключается в нанесении на слизистую глаз кроликов водных растворов из кормов и выявлении эффекта сужения зрачков.
Лечение животных при отравлении туром. Для лечения животных используются следующие лекарственные средства: атропина сульфат, кальция хлорид, кофеин и глюкоза, применение которых должно носить характер экстренной помощи животным.
Атропина сульфат рекомендуется применять подкожно или внутримышечно в водном растворе для лошадей в дозе 1 мг/кг, коровам и взрослым телятам по 0,5, овцам н свиньям 2—3 мг/кг.
Кальция хлорид рекомендуется применять в виде 10 %-ного водного раствора совместно с 40%-ным раствором глюкозы путем внутривенного введения в дозах 1 мл/кг массы тела.
Кофеин-натрий бензойпокислый применяют подкожно лошадям и крупному рогатому скоту 2—5 г, телятам и свиньям 0,5 —1,5 г на голову.
В необходимых случаях при тяжелой степени отравления животных туром дополнительно следует применять кислород в форме ингаляции в виде 50%-ной смеси с обычным воздухом с помощью противогазовых масок и шлангов, присоединенных к кислородному баллону. Газообразный кислород можно вводить также подкожно крупным животным в количестве по 5—10 л и мелким животным по 2—3 л в область подгрудка.
В тяжелых случаях отравления для возбуждения дыхания рекомендуется применять лобелии солянокислый подкожно в дозах крупному рогатому скоту и лошадям 50—150 мг, мелким животным 10—30 мг на голову.
Профилактика отравления животных туром. Общие профилактические мероприятия основываются на строгом выполнении требований «Инструкции по применению препарата тур для предупреждения полегания зерновых культур», утвержденной 8.08.1975 г. МСХ СССР. Нельзя допускать использование животным растений, посевного зерна, зеленой массы злаковых культур, обработанных туром.
Специальные меры предупреждения отравлений животных туром должны включать систему мероприятий по плановому контролю санитарного качества кормов на наличие остаточных количеств препарата.
Запрещается использование емкостей из-под тура для хранения воды, транспортировки и хранения жидких кормов, а также автопоилок на пастбищах после контакта их с препаратом тур.
Не допускается приготовление рабочих растворов тура вблизи водоемов, предназначенных для водопоя животных, разведения водоплавающей птицы и рыбоводства.
Руководители и специалисты хозяйств в зонах применения тура должны располагать планами проведения предпосевной обработки семян, опрыскивания посевов и следить за тем, чтобы сбор урожая кормовых культур, пастьба животных осуществлялись не ранее 12 дней после обработки препаратом.
Ветеринарные врачи хозяйств проводят техническую учебу с работниками животноводства по вопросам профилактики и лечения отравлений животных туром, а специалисты агрохимслужбы — инструктаж механизаторов, рабочих и авиационных специалистов по мерам безопасного применения тура на посевах и при предпосевной обработке семян злаковых культур.
Ветсанэкспертиза мяса и мясопродуктов при вынужденном убое животных после отравления туром проводится в соответствии с Правилами ветеринарного осмотра убойных животных и ветеринарно-санитарной экспертизы мяса и мясных продуктов, утвержденных Главным управлением ветеринарии МСХ СССР 27 декабря 1983 г.
Методика определения хлорхолинхлорида в почве, воде, растительных объектах, органах и тканях животных методом тонкослойной хроматографии
(Утверждена Главным управлением ветеринарии Госагропрома СССР 14 мая 1986 г)
Краткая характеристика препарата. Хлорхолинхлорид — 2 хлор-этилтриметилхлорид аммония Синонимы тур, ЗАР, XXX, ССС, антивылегач, сайкосел, хлормекват, эмсурон
Химически чистый хлорхолинхлорид — белое кристаллическое весьма гигроскопичное вещество, практически не имеет запаха Температура плавления 240 °C (плавится с разложением), растворимость в воде — 74,2, этиловом спирте —35,1, эфире — 0,59, дихлорэтане— 0,48, бензоле — 0,18 г/100 мл при 20 °C
Хлорхолинхлорид относится к среднетоксичным веществам (ЛДзо для крыс 470—720 мг/кг, для белых мышей 409 мг/кг, для овец ЛД1оо 200 мг/кг)
Используется 60% ный водный раствор
Принцип метода. Метод основан на извлечении хлорхолинхлорида из почвы, растительных объектов 0,5% ным раствором соляной кислоты, а из органов и тканей 75% ным водным раствором этилового спирта, очистке экстракта на колонке с силикагелем Л Количественное определение проводят в тонком слое пластинок «Силу фол»
Избирательность метода. Метод специфичен, фосфорорганиче ские и ХОС в данных условиях хроматографирования определению не мешают
Реактивы и растворы. Ацетон х ч Соляная кислота ч Дипи-криламин Хлороформ х ч Силикагель Л 100/250 (Чехословакия) Пластинки «Силуфол UV-254» (ЧССР) Стандартный раствор хлорхолинхлорида
100 мг хлорхолинхлорида растворяют в 100 мл 1% ного раствора соляной кислоты, в 1 мл содержится 1000 мкт Раствор хранят в холодильнике в колбе с притертой пробкой Срок хранения 6 мес
Проявляющий реактив 0,2 г дипикриламина растворяют в 100 мл 50% ного раствора ацетона Проявляющий реактив готовят непосредственно перед опрыскиванием
Посуда и приборы. Аппарат для встряхивания АВУ 1 Колонка хроматографическая высотой 1,5 см, 20 см Фарфоровые чашки Хроматографическая камера Камера для опрыскивания Колбы мерные на 100 мл Воронки химические Микропипетки на 0,1 мл Бумажные фильтры Микрошприц на 10 мкл МШ-10 Стеклянная колонка высотой 25 см, диаметром 1,2 см
Подготовка к определению. Получение кристалличе ского хлорхолинхлорида Для получения хлорхолинхлори да с содержанием 99% основного вещества берут технический препарат, помещаю) в кристаллизатор, нагревают до кипения и быстро охлаждают, помещая кристаллизатор в холодную воду, снег или лед Выпавшее при перекристаллизации вещество отделяют от маточного раствора путем фильтрования Оставшиеся на фильтре кристаллы сушат на воздухе и перекладывают в банку с притертой пробкой
Подготовка хроматографической колонки. Стеклянную колонку высотой 25 см с внутренним диаметром 1,3—1,5 см (можно сделать из бюретки) заполняют силикагелем на высоту 10 см. Носик колонки забивают промытой в ацетоне стеклянной ватой. Колонку закрепляют на штативе. Промывают силикагель 100 мл 5%-ным раствором соляной кислоты, затем дистиллированной водой до нейтральной pH. Колонку используют до 20 раз.
Отбор проб. Отбор проб производится в соответствии с ГОСТ 14189—81.
Проведение определения. Экстракция хлорхолинхло-рида из проб.
Почва. Пробу почвы (50 г) сушат на воздухе и просеивают через сито с диаметром отверстий 1—2 мм, заливают 100 мл 0,5 %-ным раствором соляной кислоты, экстрагируют в течение 1 ч на аппарате для встряхивания. Экстракт фильтруют через бумажный фильтр в фарфоровую чашку. Чашку и фильтр промывают дважды по 20 мл 0,5%-ным раствором соляной кислоты, который сливают в ту же чашку.
Вода. 500 мл воды упаривают на кипящей водяной бане до 20—30 мл.
Растения, зерно, органы и ткани животных.. Пробу 50 г гомогенизируют, заливают 100 мл 0,5 %-ным раствором соляной кислоты, а органы и ткани 25 г заливают 100 мл 75%-ным раствором этилового спирта и экстрагируют в течение 1 ч путем встряхивания на аппарате для встряхивания. Затем экстракт фильтруют в выпарительную фарфоровую чашку через бумажный фильтр.
Очистка экстракта Экстракт выпаривают на кипящей водяной бане до 10—20 мл. Затем его наносят на хроматографическую колонку с силикагелем, водный элюат отбрасывают; пропускают 50 мл 70%-ного раствора ацетона, ацетоновый раствор отбрасывают; пропускают 20 мл дистиллированной воды и отбрасывают; адсорбированный на силикагеле хлорхолинхлорид вымывают 300 мл 0,5%-ного раствора соляной кислоты. Элюат собирают в фарфоровую чашку и выпаривают досуха.
Определение тонкослойной хроматографией. Сухой остаток растворяют в 1 мл дистиллированной воды и 0,05 мл наносят на хроматографическую пластинку в одну точку. Рядом наносят стандартные растворы хлорхолинхлорида с содержанием от 1 до 10 мкг. Пластинку с нанесенными растворами хроматографируют в системе хлороформ — ацетон — дистиллированная вода — 25 %-ный раствор соляной кислоты в соотношении 20 : 20 : 20 ; 10. После поднятия фронта растворителя на 10 см пластинку вынимают из камеры, подсушивают на воздухе и подогревают в течение 5—10 мин над плиткой при t 150—200 °C и опрыскивают 0,2%-ным раствором дипикриламина. При наличии хлорхолинхлорида в пробах проявляются красно-оранжевые пятна на желтом фоне. Rf 0,4—0,6.
Количество препарата вычисляют по формуле
„ AY
где X — количество препарата, мг/кг, мг/л; А — количество препарата, найденное в аликвотной части экстракта, мю; Р — навеска, г, мл; У — конечный объем экстракта; Yt — объем аликвотной части экстракта, нанесенный на хроматографическую пластинку, мл.
14 Заказ № 1995 2 09
Методические указания по определению 2,4-Дихлорфеноксиуксусной кислоты (2,4-Д) в воде, почве, фураже, продуктах питания растительного и животного происхождения хроматографическими методами
(Утверждены заместителем Главного государственного санитарного врача СССР 20 декабря 1976 г.)
Краткая характеристика препарата. Эмпирическая формула 2,4-дихлорфеноксиуксусная кислота (2,4-Д) — белое кристаллическое вещество, стабильное при хранении. С8Н6О3С12. Молекулярная масса 221,04. Тпл 141 °C, 1кип 160 °C при 0,4 мм рт. ст. Константа диссоциации 2,3-10-3. Стабильна при хранении в растворах органических растворителей и в кристаллическом состоянии. При 20 “С 540 мг кислоты растворяется в 1 л воды, 243 г кислоты — в 100 мл эфира, 130 г кислоты — в 100 мл этилового спирта, хорошо растворяется в ацетоне, четыреххлористом углероде и бензоле. Применяется для борьбы с двудольными сорняками в посевах зерновых культур.
Принцип метода. Метод основан на экстракции 2,4-Д из гидролизованной пробы органическим растворителем с последующим определением в виде метилового эфира 2,4-Д с помощью газожидкостной хроматографии или в виде кислоты методом тонкослойной хроматографии. С помощью предлагаемого метода можно определить 2,4-Д, находящуюся в анализируемой пробе как в свободном, так и в связанном состоянии в виде конъюгатов с эндогенными веществами растений. Количественное определение 2,4-Д проводится методом внутреннего стандарта, который добавляется к пробе перед проведением гидролиза. В качестве внутреннего стандарта используется 2,4,5-трихлорфеноксиуксусная кислота (2,4,5-Т).
Метрологическая характеристика метода. Нижний предел определения газожидкостной хроматографией составляет: в воде 0,002 мг/л, в почве 0,01 мг/кг, в траве 0,02, в сене 0,1, в зерне 0,02, в молоке 0,04, в сливочном масле 0,1, в мясе (говядина) 0,08 мг/кг. Нижний предел определения методом тонкослойной хроматографии составляет: в виде 2,4-Д — в воде 0,04, в молоке 0,4 мг/л, в почве 0,2 мг/кг, в траве 0,06, в сене 0,4, в зерне 0,3, в сливочном масле 0,8, в мясе (говядина) 0,6 мг/кг; в виде метилового эфира 2,4-Д — в воде 0,01, в молоке 0,1 мг/л, в почве 0,05 мг/кг, в траве 0,08, в сене 0,1, в зерне 0,08, в сливочном масле 0,2, в мясе (говядина) 0,15 мг/кг. Степень определения методом тонкослойной хроматографии составляет, %: в воде 90—95, в почве 70—80, в траве. 60, в сене 60, в молоке 80, в сливочном масле 70, в мясе 75, в зерне 60.
Минимально детектируемое количество метилового эфира 2,4-Д с помощью детектора постоянной скорости рекомбинации 2 нг, линейный динамический диапазон детектирования не менее 70 нг. Минимально открываемое количество 2,4-Д на тонкослойных хроматограммах 1 мкг.
Реактивы и растворы. Азот особой чистоты. Азотнокислое серебро х. ч. Аммиак, 25%-ный водный раствор. Ацетон х. ч. Бикарбонат натрия, 3%-ный водный раствор. Гексан х. ч. (перегнанный). Диметилсульфат, 5%-ный (в объемных процентах) раствор в абсолютном метиловом спирте. Диэтиловый эфир (перегнанный).
Кальций сернокислый х ч, прокаленный при 160 °C в течение 6 ч Метиловый спирт абсолютный Муравьиная кислота х ч Насыщенный водный раствор хлористого натрия Петролейный эфир (1Кип 40—60 °C) Проявляющий реактив в мерную колбу на 100 мл помещают 0,5 г азотнокислого серебра, 5 мл дистиллированной во ды 7 мл аммиака (25% ный водный раствор) и доводят до метки ацетоном Силикагель КСК Соляная кислота х ч, концентрирован ная Соляная кислота, 6 н водный раствор Соляная кислота, 2 н водный раствор Соляная кислота, водный раствор (1 1) Стан дартный раствор 2,4 Д в метаноле с содержанием 100 мкг/мл Стандартный раствор 2,4,5 Т в метаноле с содержанием 2,5 мкг/мл Стандартный раствор метиловых эфиров 2,4 Д (10 мкг/мл) и 2,4,5 Т (2,5 мкг/мл) в метаноле
Стандартный раствор метилового эфира 2,4 Д в гексане с кон центрацией 100 мкг/мл Сульфат натрия безводный х ч Фосфорно вольфрамовая кислота х ч, 40% иый водный раствор Хлористый натрий х ч Эфирный раствор диазометана
Приборы и посуда Газовый хроматограф с детектором по за хвату электронов (Цвет 5, Цвет 106 и т п) Гомогенизатор (из мельчитель тканей) Колбы двугорлые, круглодоиные на 500 и 250 мл Делительная воронка на 500 мл Колба Бунзена на 500 мл Воронка Бюхнера диаметром 15 см Грушевидные колбы на 100мл Ротационный испаритель ИР 1 Источник ультрафиолетового света Водяная баня Центрифужные пробирки Контактный термометр на 100 °C Мерные колбы на 5, 10 и 100 мл Вакуумный водоструйный насос Холодильник Либиха Камера хроматографическая Микро пипетки Медицинский шприц на 1 мл Стеклянные пластинки раз мерой 9X12 см Сито капроновое 100 меш Пульверизатор стек лянный
Подготовка к определению связана с получением диазометана из нитрозометилмочевины Для этого во взвешенную литровую круглодонную колбу помещают 200 г 24% ного водного раствора метиламина (метиламин используется либо в виде водного раство ра, либо в виде хлоргидрата) и добавляют при охлаждении 155 мл концентрированной соляной кислоты до кислой реакции (индикатор метилрот) Затем приливают такое количество воды, чтобы масса содержимого колбы достигла 500 г, и прибавляют 300 г мочевины После этого содержимое колбы осторожно кипятят с обратным хо лодильником 2 ч 45 мин и энергично 15 мин Раствор в колбе охлаждают до комнатной температуры, растворяют в нем ПО г 95% ною азотнокислого натрия и охлаждают до 0 °C В 3 литровом стакане готовят смесь 600 г льда и 100 г коицеитрированной серной кислоты, охлаждают содержимое стакана смесью льда и со ли В этот стакан при перемешивании приливают содержимое кол бы (холодный раствор метилмочевииы и нитрата натрия) с такой скоростью, чтобы температура не поднималась выше 0 °C Получе ние нитрозометилмочевины проходит по следующим реакциям
CH3NH2 НС1 + H2N—СО—NH^CHs—NH—СО—NH2 + NH4C1, СН3—NH—СО—NH2+HNO2->CH3—N (NO) —CO—nh2+ H2O
Нитрозометилмочевииа всплывает иа поверхность в виде мелких кристаллов, которые немедленно отфильтровывают иа воронке Бюхнера и хорошо отсасывают под вакуумом Затем кристаллы на
фильтре размешивают до образования пасты с 50 мл холодной дистиллированной воды, отсасывают и сушат в вакуум-эксикаторе. Выход нитрозометилмочевины 66—72% (105—115 г) от теоретического. Полученную таким образом нитрозометилмочевину можно хранить в холодильнике длительное время. При температуре выше 20 °C ее не следует хранить более 1 ч. При температуре 30 °C нитрозометилмочевина может разложиться с выделением газообразных продуктов.
Получение диазометана из нитрозометилмочевины проходит по следующей реакции: CH3N(NO)—СО—NH2+KOH->CH2N2+KCNO + + 2Н2О.
В круглодонную колбу на 100 мл помещают 3 мл 50 %-ного водного раствора едкого кали и 10 мл диэтилового эфира. Смесь охлаждают до 5 °C, после чего при взбалтывании прибавляют 1 г нитрозометилмочевины, колбу присоединяют к холодильнику, нижний конец которого снабжен алонжем с отводом, проходящим через резиновую пробку и погруженным в слой эфира на дне приемника. Приемник охлаждают смесью льда и соли.
Реакционную колбу помещают на водяную баню, нагретую до 50 °C. Эфирный раствор в колбе доводят до кипения. Время от времени содержимое колбы перемешивают. Отгонку прекращают после получения первых капель бесцветного дистиллята.
Ход анализа при газожидкостной хроматографии. Экстракция и очистка экстракта из воды. Пробу воды 250— 1000 мл помещают в делительную вороику, подкисляют соляной кислотой до pH 3, прибавляют 1 мл стандартного раствора 2,4,5-Т с концентрацией 2,5 мкг/мл и экстрагируют тремя порциями диэтилового эфира (100, 50 и 50 мл). Объединенный эфирный экстракт переносят в делительную воронку и экстрагируют 3%-ным водным раствором бикарбоната натрия (или 5 %-ным водным раствором гидроортофосфата натрия) (трижды по 50 мл). Объединенный водный раствор промывают двумя порциями петролейного эфира (или гексана) по 50 мл, отбрасывают этот эфир (гексан), а водный раствор подкисляют соляной кислотой до pH 3 и трижды экстрагируют диэтиловым эфиром по 50 мл. Объединенный эфирный экстракт сушат над безводным сульфатом натрия (5—10 г) в течение 15—30 мин при периодическом встряхивании и удаляют растворитель па ротационном испарителе в грушевидной колбе на 100 мл, причем последние порции растворителя удаляют током сухого воздуха. Сухой остаток в колбе метилируют, используя диметилсульфат или диазометан.
Метилирование диметилсульфатом. К сухому остатку в колбе приливают 3 мл 5%-ного раствора диметилсульфата в абсолютном метиловом спирте, тщательно ополаскивают стенки колбы, прибавляют 1 г безводного сульфата натрия, присоединяют обратный холодильник и помещают на водяную баню с температурой 55 °C на 10 мин. После окончания реакции метилирования содержимое колбы охлаждают под водопроводной водой, приливают 3 мл насыщенного раствора хлористого натрия, 1 мл гексана, энергично встряхивают в течение 2 мин и после расслоения фаз вводят в хроматограф 3 мкл гексанового слоя.
Метилирование диазометаиом. К сухому остатку в колбе приливают эфирный раствор диазометана до появления желтоватой окраски (при этом встряхивают содержимое колбы) и оставляют на 10 мин. Затем диэтиловый эфир удаляют током су
хого воздуха, а остаток растворяют в 0,5—1,0 мл гексана и вводят в хроматограф 1—5 мкл раствора
Хроматографирование. Условия хроматографирования следующие Хроматограф Цвет 106 Стеклянная спиральная колонка (длина 2 м, внутренний диаметр 3 мм), заполненная хроматоном-N (0,16—0,20 мм), силаиизированным DMCS, с 5% SE-30 Скорость газа носителя (азот особой чистоты) через колонку 50 мл/мин, скорость продувочного газа (азот особой чистоты) через детектор 150 мл/мин Температура термостата колонок 170 °C, испарителя 220 °C, термостата детектора 220 °C Шкала электрометра 20 10";2 А Скорость диаграммной ленты потенциометра 240 мм/ч Относительное время удерживания метилового эфира 2,4-Д в этих условиях 0,57 Абсолютное время удерживания 4 мин 10 с
Хроматографирование одной пробы проводят дважды Измеряют иа хроматограммах высоты пиков метиловых эфиров 2,4-Д и 2,4,5-Т, вычисляют средние значения отношений этих высот из параллельных определений и по уравнению градуировочного гра фика находят содержание 2,4-Д в воде Для построения градуиро вочного графика к пробам воды приливают 5, 10, 15 и 20 мкг 2,4-Д (в виде раствора в метиловом спирте), 1 мл стандартного раствора 2,4,5 Т с содержанием 2,5 мкг/мл и далее поступают так, как это описано выше Измеряют на хроматограммах высоты пиков метиловых эфиров 2,4-Д и 2,4,5-Т, вычисляют среднее значе ние отношений этих высот из параллельных определений, строят график зависимости отношения высот хроматографических пиков метиловых эфиров 2,4 Д и 2,4,5 Т от содержания 2,4 Д в воде (мг/л) Полученный градуировочный график обрабатывают по методу наименьших квадратов и получают уравнение градуировочной прямой в виде V— А + ВХ, где V — отношение высот хроматографических пиков метиловых эфиров 2,4-Д и 2,4,5 Т, X — содержание 2,4-Д в воде, мг/л, А и В—коэффициенты, которые получают при обработке градуировочного графика по методу наименьших квадратов
Чтобы повысить надежность идентификации 2,4-Д, для анализа проб целесообразно использовать колонки с неподвижными фазами различной полярности (например, SE-30 и ХЕ-60, SE-30 и OV-17) Условия хроматографирования при использовании фенилметилсили-кона OV-17 следующие стеклянная спиральная колонка (длина 2 м, внутренний диаметр 3 мм), заполненная хорматоном-N (0,16— 0,20 мм), силаиизированным DMCS, с 5% OV-17. Скорость тока газа носителя (азот особой чистоты) через колонку 50 мл/мин, скорость тока продувочного газа (азот особой чистоты) через детектор 150 мл/мин Температура испарителя 220 °C, термостата колонок 200 °C, термостата детектора 220 °C Шкала электрометра 20-10-12 А Скорость движения диаграммной ленты потенциометра 240 мм/ч Относительное время удерживания метилового эфира 2,4-Д в этих условиях 0,63 Абсолютное время удерживания 6 мии 48 с
Экстракция и очистка экстракта из травы, сена, зерна. Навеску травы 50 г гомогенизируют в 150 мл дистиллированной воды, гомогенат количественно переносят в круглодоиную колбу на 1 л и приливают 38 мл концентрированной соляной кислоты Навеску измельченного зерна (50 г) соломы или сена (по 10 г) помещают в круглодонную колбу на 1 л и приливают 150 мл 2 н водного раствора соляной кислоты. К подготовленным образцам добавляют
по 1 мл стандартного раствора 2,4,5-Т с концентрацией 2,5 мкг/мл, перемешивают, к колбе присоединяют обратный холодильник и помещают на кипящую водяную баню на 1 ч.
После охлаждения гидролизат фильтруют через бумажный фильтр на воронке Бюхнера, промывают осадок на фильтре без перемешивания тремя порциями по 30 мл 2 н. водного раствора соляной кислоты. К фильтрату добавляют 40%-ный водный раствор фосфорно-вольфрамовой кислоты до 2 %-ной концентрации и фильтруют через бумажный фильтр на воронке Бюхнера. Осадок на фильтре трижды промывают 2 н. водным раствором соляной кислоты порциями по 30 мл. Объединенный фильтрат переносят в делительную воронку и экстрагируют тремя порциями диэтилового эфира (общий объем эфира равен объему фильтрата). Эфирный экстракт промывают небольшими количествами (15—20 мл) дистиллированной воды до нейтральной реакции промывных вод и затем экстрагируют тремя порциями (по 50 мл) 5%-ного водного раствора гидроортофосфата натрия (или раствора бикарбоната натрия).
Водные экстракты объединяют, подкисляют концентрированной соляной кислотой до pH 1 и экстрагируют тремя порциями диэтилового эфира (75 + 50 + 50 мл). Объединенный эфирный экстракт после промывания небольшими (15—20 мл) порциями дистиллированной воды до нейтральной реакции промывных вод сушат над безводным сульфатом натрия (5—10 г) в течение 15—30 мин при периодическом встряхивании и растворитель удаляют на роторном испарителе в грушевидной колбе на 100 мл, последние порции растворителя удаляют током сухого воздуха.
Сухой остаток растворяют в 0,5 мл ацетона и раствор наносят в виде полосы на стеклянную пластинку (5X20 см) с тонким слоем (0,4 мм) силикагеля КСК и дважды хроматографируют в системе растворителей петролейный эфир (или гексан) — диэтиловый эфир — муравьиная кислота (50:50:2), Затем снимают силикагель с пластинки площадью 1 см2 с Rf 2,4-Д 0,5—0,6 и количественно переносят в коническую колбу с притертой пробкой иа 50 мл. Смачивают силикагель в колбе 1 мл дистиллированной воды, приливают 4—5 мл ацетона, встряхивают 1—2 мин и фильтруют через бумажный фильтр. Обработку силикагеля ацетоном проводят еще трижды. Объединенный ацетоновый экстракт сушат над безводным сульфатом натрия, удаляют растворитель на роторном испарителе, затем проводят метилирование образца и газохроматографическое определение 2,4-Д так, как описано выше.
Для построения калибровочного графика в пробы травы, зерна, сена или соломы вносят 10, 15, 20 и 25 мкг 2,4-Д в виде раствора в метиловом спирте, 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл и далее поступают так, как описано выше.
Экстракция и очистка экстракта из сухой почвы. Пробу сухой почвы 100 г, растертой и просеянной через сито с отверстиями размером 1 мм, помещают в коническую колбу на 500 мл с пришлифованной пробкой, приливают 25—35 мл дистиллированной воды, 5 мл концентрированной соляной кислоты, 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл, тщательно перемешивают, прибавляют 150 мл ацетона и помещают на аппарат для встряхивания на 1 ч. Затем растворитель отфильтровывают под вакуумом на воронке Бюхнера через бумажный фильтр, почву на фильтре трижды промывают ацетоном по 20 мл. После этого ацетон удаля
ют на роторном испарителе, остаток из колбы переносят в 100 мл дистиллированной воды в делительную воронку, экстрагируют тремя порциями диэтилового эфира (75, 50 и 50 мл) и далее поступают так, как это описано при определении 2,4-Д в воде.
Для построения градуировочного графика в пробы почвы вносят 10, 15, 20 и 25 мкг 2,4-Д в виде раствора в метиловом спирте, 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл и далее поступают, как описано выше.
Экстракция и очистка экстракта из влажной почвы. Пробу влажной почвы 100—200 г помещают в коническую колбу на 500 мл с притертой пробкой, приливают 5—10 мл концентрированной соляной кислоты, 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл, 150—300 мл ацетона, тщательно перемешивают, помещают на аппарат для встряхивания иа 1 ч и далее поступают так, как описано выше.
Экстракция и очистка экстракта из молока. В пробу молока 25—50 мл вносят 1 мл стандартного раствора 2,4,5-Т с концентрацией 2,5 мкг/мл, прибавляют концентрированную соляную кислоту 4,4—8,8 мл, помещают в круглодоииую колбу на 250 мл, присоединяют обратный холодильник и помещают иа кипящую водяную баню на 1 ч для гидролиза. После охлаждения в колбу приливают 15 мл 40%-ного водного раствора фосфорио-вольфрамовой кислоты, тщательно встряхивают, затем добавляют 100 мл дистиллированной воды и содержимое фильтруют под вакуумом на воронке Бюхнера. Остаток на фильтре промывают 2 и. водным раствором соляной кислоты, фильтрат экстрагируют диэтиловым эфиром трижды по 50 мл и далее поступают так, как это описано выше
Для построения градуировочного графика в пробы молока (25—50 мл) вносят 2, 5, 10 и 20 мкг 2,4-Д (в виде раствора в метиловом спирте), 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл, концентрированную соляную кислоту 4,4—8,8 мл и далее поступают так, как описано выше.
Экстракция и очистка экстракта из сливочного масла. Пробу масла 25 г растворяют в 100 мл гексана, вносят 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл, помещают в двугорлую круглодонную колбу на 500 мл, приливают 100 мл 6 н. соляной кислоты, присоединяют обратный холодильник и помещают на кипящую водяную баню на 1 ч, перемешивая содержимое колбы с помощью электромешалки. После охлаждения содержимого колбы слои разделяют в делительной воронке и водный слой экстрагируют диэтиловым эфиром 2 раза по 50 мл. Гексановый слой экстрагируют 100 мл 6 н. соляной кислоты. Затем водный слой экстрагируют диэтиловым эфиром дважды по 50 мл и данный экстракт объединяют с первоначальным эфирным экстрактом. После этого из объединенного эфирного экстракта 2,4-Д извлекают 0,4 н водным раствором бикарбоната натрия и далее поступают так, как описано при определении гербицида в молоке.
Для построения градуировочного графика в пробы масла 25 г, растворенные в 100 мл гексана, вносят 1 мл стандартного раствора 2,4,5-Т с концентрацией 2,5 мкг/мл, 10, 15, 20 и 25 мкг 2,4-Д в виде раствора в метиловом спирте. Далее поступают так, как это описано выше.
Экстракция и очистка экстракта из мяса. Пробу мяса 25—50 г измельчают с помощью мясорубки, помещают в двугорлую кругло-дониую колбу на 500 мл, вносят 1 мл стандартного раствора
2,4,5-Т с концентрацией 2,5 мкг/мл, приливают 100—200 мл 6 и. соляной кислоты, присоединяют обратный холодильник и помещают иа водяную баню (с температурой 100 °C) на 1 ч, перемешивая содержимое колбы с помощью электромешалки. После охлаждения в колбу приливают 15 мл 40 %-ного водного раствора фос-форно-вольфрамовой кислоты, 100 мл дистиллированной воды и содержимое колбы фильтруют под вакуумом на воронке Бюхнера. Остаток на фильтре промывают 2 н. водным раствором соляной кислоты, фильтрат экстрагируют диэтиловым эфиром трижды по 50 мл и далее поступают так, как это описано выше.
Для построения градуировочного графика в пробы мяса вносят 1 мл стандартного раствора 2,4,5-Т с содержанием 2,5 мкг/мл, 10, 15, 20 и 25 мкг 2,4-Д в виде раствора в метиловом спирте и далее поступают так, как это описано выше.
Ход анализа при тонкослойной хроматографии. Экстракция и очистка экстрактов. При количественном определении содержания 2,4-Д методом тонкослойной хроматографии подготовку образца и очистку экстракта проводят таким же образом как при анализе гербицида газохроматографическим методом, за исключением того, что 2,4,5-Т в пробы не вносят.
Полученный после очистки объединенный эфирный экстракт сушат над безводным сульфатом натрия и упаривают растворитель на ротационном испарителе в грушевидной колбе на 100 мл до небольшого объема (1—2 мл).
Хроматографирование Упаренный экстракт с помощью капилляра или медицинского шприца на 1 мл количественно наносят на хроматографическую пластинку (9X12 см) с тонким слоем силикагеля КСК, закрепленного гипсом. Затем на хроматографическую пластинку наносят 2,5 и 10 мкг 2,4-Д в виде раствора ацетона и проводят хроматографирование в системе растворителей пет-ролейиый эфир (или гексан)—диэтиловый эфир — муравьиная кислота (50:50:2). После окончания процесса хроматографирования пластинку извлекают из хроматографической камеры и сушат на воздухе в вытяжном шкафу. Для обнаружения зоны локализации 2,4-Д пластинку обрабатывают раствором азотнокислого серебра в смеси с дистиллированной водой, аммиаком и ацетоном, сушат и облучают ультрафиолетовым светом в течение 10—15 мин. Если в пробе есть 2,4-Д, то на пластинке появляется серо-черное пятно на белом фоне с величиной Rf 0,5. Количественное определение 2,4-Д проводят путем визуального сравнения размера и интенсивности окраски пятен стандарта с пятном пробы. Минимально детектируемое количество 2,4-Д на хроматографической пластинке 1 мкг.
Перед проведением серийных анализов необходимо определить открываемость метода, т. е. процент возврата 2,4-Д из контрольных проб, к которым предварительно были добавлены различные количества гербицида.
Для повышения надежности идентификации 2,4-Д, особенно при анализе проб с неизвестными обстоятельствами загрязнения, целесообразно после проведения газохроматографического определения содержания гербицида оставшуюся часть пробы анализировать с помощью тонкослойной хроматографии. Для этого гексановый экстракт наносят на хроматографическую пластинку (9X12 см) с тонким слоем силикагеля КСК, рядом наносят 2—10 мкг метилового раствора 2,4-Д в виде раствора в гексаие и хроматографи-216
руют в системе растворителей гептан — ацетон (9:3). Далее поступают так, как это описано выше. Величина Rf метилового эфира 2,4-Д 0,46—0,47.
Сочетание величины времени удерживания и величины Rf метилового эфира 2,4-Д в сравнении с этими же показателями, полученными для стандартного соединения, позволяет более надежно проводить определение остаточных количеств 2,4-Д в анализируемых образцах.
Обработка результатов анализа. Газожидкостная хроматография. Для определения содержания 2,4-Д в пробах (X, мг/л или мг/кг) методом газожидкостной хроматографии используют следующую формулу:
где V — отношение высот хроматографических пиков метиловых эфиров 2,4-Д и 2,4,5-Т; А и В — коэффициенты, которые получают при обработке градуировочных графиков по методу наименьших квадратов.
Тонкослойная хроматография. Для определения содержания 2,4-Д в пробе (X, мг/л или мг/'кг) методом тонкослойной хроматографии применяют следующую формулу:
где А — количество 2,4-Д в пробе, найденное сравнением со стандартом, мкг; Р — объем или масса пробы, мл или г.
Для определения содержания в пробе натриевой, диметилди-этил- или триэтаиоламиниых солей 2,4-Д полученный результат необходимо соответственно умножить на 1,1; 1,46 и 1,67.
Калориметрическое определение хлората магния в хлопковом масле, шроте, жмыхе, семенах, ядрах семян и волокне хлопчатника *
Принцип метода. Метод основан на окислении бензидина хлоратом магния в кислой среде с последующим колориметрированием желтой окраски. Пестицид из волокна, семян, ядер семян, шрота, жмыха экстрагируют ацетоном. После отгонки растворителя остаток обрабатывают водой. Из хлопкового масла препарат извлекают 0,05 н. соляной кислотой, экстракт пропускают через колонку с анионитом АВ-17 в ОН-форме. Хлорат-ион элюируют раствором 2 н. серной кислоты. Чувствительность метода: 0,4 мг/кг шрота, 0,5 мг/кг жмыха или ядер семян, 1 мг/кг семян, 3,0 мг/кг волокна и 0,5 мг/л хлопкового масла.
Реактивы и растворы. Хлорат калия х. ч. Хлорат магния. 0,5%-ный раствор солянокислого бензидина в 0,06 н. соляной кислоте. Кислота серная плотностью 1,84 и ее 18 и 2 н. растворы. Кислота соляная плотностью 1,17 п ее 0,05, 0,06 и 2 и. растворы. Ацетон х.ч. или ч. д. а'. Петролейиый эфир (1Кип 40—60°C). 5%-ные растворы едко! о натра или калия х.ч. Аммовий роданистый я. д. а. 1%-ный раствор азотнокислого серебра ч. д, а. 1%-ный раствор фенолфталеина ч. д. а. Спирт этиловый ректификат'.
Стандартные растворы хлората калия, содержащие 1 и 10 мкг/мл хлоратиона. Точную навеску свежеперекристаллизован-ного хлората калия растворяют в дистиллированной воде.
Аиионит АВ-17: ионообменную смолу АВ-17 растирают в агатовой ступке. Смолу помещают в цилиндр вместимостью 1 л, заливают водой и взмучивают. Для работы отбирают фракцию, оседающую за 60 с. Анионит переносят в колонки и промывают раствором 2 и. соляной кислоты для удаления ионов железа (до отрицательной реакции с роданидом аммония). Избыток соляной кислоты из смолы вымывают дистиллированной водой до отрицательной реакции на ион хлора (реакция с нитратом серебра). Для переведения в ОН-форму аиионит обрабатывают 5%-ным раствором щелочи до равенства концентраций щелочи до и после пропускания через слой анионита. Избыток щелочи вымывают дистиллированной водой до отрицательной реакции на ион гидроксила (реакция с фенолфталеином). Высушивают анионит на воздухе, хранят в закрытом сосуде. В работе используют колонку, на дно которой помещают стеклянную вату и 1 г сухой смолы.
Приборы и посуда. Фотоэлектроколориметр ФЭК-56 или ФЭКН-57 Ионообменная колонка-бюретка диаметром 12 мм. Набор пипеток иа 1, 2 и 5 мл. Колбы мерные на 25, 50 и 100 мл. Воронки делительные на 50, 100 и 200 мл. Фильтры бумажные. Вата стеклянная. Баня водяная. Весы аналитические и технические.
Ход анализа. Построение калибровочного графика. В мерные колбы на 25 мл вводят 0,5, 1,0 ... 5,0 мл стандартного раствора хлората калия, содержащего 1 мкг/мл хлорат-иоиа, и 0,5 1,0, 1,5 и 2,0 мл стандартного раствора хлората калия, содержащего 10 мкг/мл хлорат-иона, приливают 0,75 мл 0,5 %-ного раствора солянокислого бензидина и 15 мл 18 н. серной кислоты.
Колбы нагревают на кипящей водяной бане 1,5 мин. Затем растворы охлаждают под проточной водой до комнатной температуры, объем доводят до метки 18 н. серной кислотой. Растворы перемешивают и фотометрируют при ?v = 430 нм в кюветах с толщиной слоя 5,0 см для стандартных растворов, содержащих 1 мкг/мл хлорат-иона и 1,0 см для растворов, содержащих 10 мкг/мл хлорат-иона. В качестве растворов сравнения используют дистиллированную воду.
Ход анализа.
Определение хлората магния в нерафинированном хлопковом масле. 10 мл хлопкового масла наливают в делительную воронку на 50 мл, прибавляют 10 мл петролейного эфира и 10 мл 0,5 н. соляной кислоты. Содержимое делительной вороики умеренно встряхивают и оставляют, чтобы произошло разделение слоев. Водный слой сливают через фильтр (синяя лента) для удаления следов масла. Экстракцию проводят 3 раза. Объединенные экстракты пропускают через колонку с АВ-17 в ОН-форме со скоростью 2 мл/мин и отбрасывают. Элюируют хлорат-ион раствором 2 и. серной кислоты до отрицательной реакции с 0,5 %-ным раствором бензидина. Элюат упаривают на водяной бане до объема 2—3 мл, количественно переносят в мерную колбу на 25 мл, прибавляют 15 мл серной кислоты и 0,75 мл 0,5%-ного раствора бензидина. Дальнейшее определение проводят, как при построении калибровочного графика.
Определение хлората магния в волокне, семенах, хлопковом необогащенном шроте, ядрах семян и жмыхе (обезжиренных пет-ролейиым эфиром). 3 г волокна, 10 г семян, 5 г шрота, 3 г ядер семян или 5 г жмыха заливают ацетоном (волокно и семена в делительной воронке; шрот, ядра семян, жмых в конической колбе). Толщина слоя ацетона над волокном должна составлять 1 —1,5 см. Для остальных проб используют следующее количество ацетона: семена — 30, 20 и 20 мл, ядра семян— 10, 10 и 10, шрот и жмых — 15, 15 и 10 мл (экстракцию проводят трижды). Пробы с ацетоном встряхивают 1—2 мин, экстракт сливают через фильтр в стакан вместимостью 100 мл. Экстракты объединяют, растворитель упаривают досуха при температуре 70—80 °C. Стакан обмывают дистиллированной водой, остаток количественно переносят через фильтр (синяя лента) в мерную колбу на 25 мл (общий объем воды 3— 4 мл). Дальнейшее определение проводят, как при построении калибровочного графика.
Расчет. Содержание хлората магния (X, мг/кг) вычисляют по формуле
_ 1,145 Л
~ Р '
где А— количество хлорат-иона, найденное по калибровочному графику, мкг; Р— масса или объем пробы, г или мл; 1,145 — коэффициент пересчета на Mg(ClO3)2.
Методические указания по определению зоокумарина в тканях и крови животных, в приманках и препарате [пенокумарин] хроматографическими и спектрофотометрическими методами
(Утверждены заместителем Главного государственного санитарного врача СССР 20 декабря 1976 г.)
Краткая характеристика препарата. Зоокумарин — dl-З-(а-аце-тонилбензил) -4-оксикумарин. Ci9H16O4. Молекулярная масса 308,3. Препарат применяют для борьбы с грызунами в животноводческих помещениях в форме пищевых приманок, содержащих 0,025% препарата. Это сильнодействующее вещество (1-я группа государственной классификации). ЛДюо для серых крыс 4—14 мг/кг, кошек и собак 30—60 мг/кг. Коэффициент кумуляции 0,4. Рацемическая dl-форма пестицида представляет собой бесцветные кристаллы с 1пл 159—161 °C, без запаха. Препарат малолетуч, сохраняет токсические свойства в течение нескольких лет, не растворяется в воде, бензоле, хорошо растворим в ацетоне, хлороформе и диоксане.
Зоокумарин определяют тонкослойной или газожидкостной хроматографией, а также спектрофотометрическим методом.
Принцип методов. Хроматографические методы основаны иа извлечении зоокумарина из биологических объектов хлороформом, очистке экстракта на колонке с окисью алюминия и последующем хроматографическом определении пестицида в закрепленном тонком слое силикагеля по реакции диазосочетания или в виде метилового эфира на газовом хроматографе с электронно-захватным детектором. Спектрофотометрический метод основан на извлечении
зооцида из пенокумарина, кормов, мышечной ткани и крови жи вотных хлороформом, последующей очистке экстракта на колонке с окисью алюминия и определении натриевой соли зоокумарина по характерному поглощению света в ультрафиолетовой области спект ра при 308 нм
Метрологическая характеристика методов. Нижний предел опре деления тонкослойной хроматографией 2 мкг зоокумарина в пробе, что составляет 0,2 мг/кг Минимально детектируемое количество зоокумарина на газовом хроматографе 1 нг, или 0,02 мг/кг Сте пень определения составляет 80+5% Нижний предел определения спектрографическим методом 10 мкг вещества в 0,4 мл пенокума рина, в 0,5 I корма, в 25 г мышечной ткани и крови животных Степень определения зооцида 90+5%
Реактивы и растворы Кислота серная, 1 н раствор Хлоро форм Кислота уксусная Натрий сернокислый безводный Ацетон Аммиак водный Натрий азотистокислый (нитрит натрия) 1% ный водный раствор Гексан Диэтиловый эфир Кислота соляная, 0,33 и раствор (готовят из фиксанала) Натрия гидрат окиси (натр едкий), 2% ный раствор Вода дистиллированная Насыщен ный раствор 2,4 дихлоранилина в 0,33 н соляной кислоте 1 г ве щества смешивают со 100 мл 0,33 и соляной кислоты и встряхи вают 1 ч, хранят раствор в темной склянке в холодильнике, ре актив устойчив в течение трех месяцев Реактив для выявления зоокумарина (хромогенный реагент) смешивают 25 мл 1% ного водного раствора нигрита натрия с 25 мл насыщенного раствора 2,4 дихлоранилина в 0,33 и соляной кислоте, готовят не реже раза в неделю Смесь растворителей, состоящая из гексана и ацетона в соотношении (2 1)
Основной раствор зоокумарина 1 мг/мл растворяют 100 мг вещества в 100 мл ацетона Рабочий раствор зоокумарина 10 мк!(мл разбавляют 1 мл основного раствора в 100 мл ацетона Стандартный раствор зоокумарина и натриевой соли зоокумарина для спектрофотометрических измерений 0,1 мг/мл растворяют 10 мг зооцида в 2% ном растворе гидроокиси натрия в мерной колбе на 100 мт Силикагель КСК Гипс медицинский Окись алюминия для хроматографии Кальций хлористый Калий углекислый (калия карбонат), растертый порошок, прокаленный в течение 6 ч при температуре 160 °C, хранят в эксикаторе над безводным хлористым кальцием Метилирующий реагент в колбу емкостью 100 мл вносят 5 г безводного карбоната калия, 47,5 мл абсолютного ацетона и 2,5 мл диметилсульфата, после перемешивания реактив хранят без доступа воздуха
Ацетон абсолютный 0,5 л ацетона встряхивают 30 мии с 50 г безводного хлористого кальция, обработанный ацетон перегоняют на кипящей водяной бане, первые и последние 50 мл дистиллята отбрасывают, а остальное количество собирают в склянку с притертой пробкой Хранят без доступа воздуха Натрий хлористый, насыщенный водный раствор Метилсилоксан SE-30 (5%), нанесенный на силанизированный хроматон 80—100 меш Все реактивы должны быть квалификации х ч или ч д а
Приборы и посуда. Аналитические весы Шуттель аппарат Хроматографические камеры Эксикатор Спектрометр СФ 4а или другой для измерения поглощения в ультрафиолетовой области спектра Колбы Эрлеимейера на 50, 100 и 250 мл Чашки для выпаривания № 5 и № 10 Мерные цилиндры на 50 и 100 мл Мерные
колбы на 100 мл. Пипетки на 1, 2, 5 и 10 мл. Микропипетки на 0,1 мл. Воронки для фильтрования № 3 и № 5. Пластинки стеклянные размером 9X12 см. Пробирки длиной 15 см, диаметром 1,5 см, градуированные, емкостью 15 мл. Колонки стеклянные для хроматографии длиной 35 см, с внутренним диаметром 1,5 см. В качестве колонки можно использовать нижнюю часть бюретки на 100 мл, разрезав ее пополам.
Газовый хроматограф «Цвет-5-68», ЛХМ-8МДП или другой с электронно-захватным детектором Колонка для газовой хроматографии стеклянная, длиной 0,75 м, диаметром 3 мм. Микрошприцы на 10 мкл. Баня водяная. Мельница шаровая. Ступка фарфоровая. Пластинки «Силуфол».
Ход анализа.
Экстракция зоокумарина из мышечной ткани и крови животных и очистка экстракта. Пробу 25 г гомогенизированного образца (мышечная ткань, кровь и другой материал, не подвергшийся гниению) помещают в колбу Эрленмейера на 250 мл, куда вносят 10 мл 1 н. серной кислоты. Содержимое перемешивают, добавляют 75 мл хлороформа и встряхивают на шуттель-аппарате 40 мин. Затем в колбу вносят через фильтровальную воронку небольшими порциями (чтобы избежать образования комков) 65 г сульфата натрия. Отделившийся хлороформный экстракт фильтруют в колбочку на 100 мл через тампон стекловаты (или обычной ваты). Извлечение препарата повторяют еще раз с 50 мл хлороформа и образец встряхивают 15 мин. Профильтрованный через ватный тампон экстракт соединяют с первой фракцией.
Объединенные порции экстракта пропускают через колонку, которую готовят следующим образом. В ее нижнюю часть помещают слой ваты (1 см), вносят 10 г окиси алюминия и промывают 20 мл хлороформа, затем вносят экстракт. После того как весь экстракт опустится до адсорбента, колонку промывают 100 мл чистого хлороформа, а затем 20 мл ацетона (для удаления коэкстрактив-ных веществ). Зоокумарип элюируют 50 мл смеси ацетона и 2 %-ного раствора гидрата окиси натрия (47:3). Элюат собирают в фарфоровую чашку № 10, добавляют 0,2 мл ледяной уксусной кислоты и выпаривают досуха на кипящей водяной бане.
Для определения зоокумарина на спектрофотометре уксусную кислоту не добавляют.
Экстракция зоокумарина из препарата пенокумарин и очистка экстракта. После двухминутного энергичного встряхивания баллона с препаратом отбирают около 1 г препарата в колбу Эрленмейера на 100 мл. Для хроматографического определения на каждые 20 мг пенокумарина добавляют 1 мл ацетона. Содержимое колбы перемешивают и выдерживают 30 мин при комнатной температуре. Затем 0,4 мл раствора переносят в фарфоровую чашку № 5, добавляют 0,2 мл ледяной уксусной кислоты и выпаривают досуха на кипящей водяной бане.
Для определения зоокумарина на спектрофотометре отбирают около 1 г препарата в колбу Эрленмейера на 100 мл. Пенокумарин растворяют в 2 %-ном растворе гидроокиси натрия из расчета 1 мл растворителя на 20 мг, образца. Содержимое колбы перемешивают и выдерживают на водяной бане при 60 °C до исчезновения пены. Затем раствор фильтруют через бумажный фильтр.
Экстракция зоокумарина из кормов (приманок) и очистка экстракта. Пробу 0,5 г корма (приманки в целых или размолотых
зернах) помещают в колбу Эрленменера на 100 мл, куда вносят 3 мл 1 н. серной кислоты. Содержимое перемешивают, добавляют 25 мл хлороформа и встряхивают на шуттель-аппарате 40 мин. Затем в колбу вносят через фильтровальную воронку небольшими порциями 8—10 г сульфата натрия. Отделившийся хлороформный экстракт фильтруют через ватный тампон в колбочку на 50 мл. Извлечение повторяют еще раз, добавляя 15 мл хлороформа, и встряхивают образец в течение 15 мин.
Экстракт фильтруют через ватный тампон и полученную порцию фильтрата соединяют с первой фракцией, а затем пропускают через колонку, которую готовят так же, как и при исследовании мышечной ткаии или крови. Колонку промывают 20 мл хлороформа. Когда весь экстракт опустится до адсорбента, колонку промывают последовательно 50 мл хлороформа и 20 мл ацетона. Зоокумарин элюируют 50 мл смеси ацетона и 2 %-ного раствора гидроокиси натрия (47:3). Элюат собирают в фарфоровую чашку № 10, добавляют 0,2 мл ледяной уксусной кислоты и выпаривают досуха на кипящей водяной бане При спектрофотометрическом определении зоокумарина уксусную кислоту ие добавляют
Определение тонкослойной хроматографией. Полученный после выпаривания остаток растворяют в 0,5 мл ацетона и полностью наносят на пластинку микропипеткой или пипеткой Пастера. Одновременно готовят стандарты, внося в фарфоровую чашку 5—10 мкг вещества, что соответствует 0,5—1,0 мл рабочего раствора зоокумарина. Образцы наносят на пластинку в следующей последовательности: стандарт (5 мкг), образец (I повторность), образец (II повторность), стандарт (10 мкг). Размер пятна не должен превышать 0,5 см. Места нанесения проб располагают на расстоянии 1,5—2,0 см друг от друга и от нижнего и боковых краев пластинки; пятна не должны погружаться в растворитель.
В камеру для хроматографирования (эксикатор) наливают 60 мл смеси гексана и ацетона (2:1) и через 20 мин помещают пластинку в вертикальном положении. Камеру герметично закры-' вают. Хроматографирование проводят при температуре не ниже 20 °C. После того как растворитель поднимется на высоту 8—9 см, пластинку вынимают, отмечают фронт растворителя и помещают в сушильный шкаф при 105 °C на 3 мин. Затем пластинку охлаждают и опрыскивают из пульверизатора хромогенным реагентом, вновь высушивают при тех же условиях и помещают в эксикатор над парами аммиака на 1 мин. Зоокумарин проявляется в виде коричневых пятен на желтом фоне с Rf 0,4—0,5 в тонком слое силикагеля и Rf 0,29—0.35 на «Силуфоле». Количество пестицида в пробе определяют сравнением интенсивности окраски и площади пятна стандарта и образца. В случае если содержание зооцида в пробе превышает 10 мкг, то следует получить хроматограмму стандартов большей концентрации (20—100 мкг).
Расчет результатов анализа при тонкослойной хроматографии. Содержание зоокумарина в исследуемой пробе (X, мг/кг) вычисляют по формуле
где А — содержание зоокумарина в исследуемой навеске, мкг; Р — масса пробы, г.
Определение газожидкостной хроматографией. Полученный после выпаривания остаток растворяют 3 мл метилирующего реагента и переносят в пробирку длиной 15 см, диаметром 1,5 см, содержащую 500 мг безводного карбоната калия. Пробирку закрывают ватным тампоном и выдерживают в водяной бане при 65 °C в течение 15 мин. Затем содержимое переносят в делительную воронку емкостью 100 мл посредством 30 мл дистиллированной воды и 5 мл гексана. Метиловый эфир зоокумарина экстрагируют путем встряхивания делительной воронки в течение 2 мин, водную фазу отбрасывают, экстракт промывают 15 мл насыщенного водного раствора хлористого натрия и сливают в сухую пробирку.
В испаритель газового хроматографа с электронно-захватным детектором вводят 5—10 мкл экстракта при следующих условиях: расход газа-носителя 80 мл/мин, поддув 120 мл/мин. Температура колонки, детектора и испарителя соответственно 225, 250 и 250 °C. Чувствительность электрометра 1 10-11 А. Скорость протяжки ленты самописца 1 см/мин. Время удерживания метилового эфира зоокумарина 2,5 мин. Высота пика при введении в испаритель 10 нг вещества 80 мм. Линейный диапазон от 5 до 25 нг.
Количественное определение проводят методом абсолютной калибровки, по градуировочному графику, высоте пика. График строят в интервале концентраций от 5 до 20 иг зоокумарина. С этой целью в фарфоровые чашки вносят 5, 10 и 20 мкг зоокумарина в ацетоне. Ацетон выпаривают, а остаток метилируют, как и пробы. Введение в испаритель хроматографа 5 мкл гексанового экстракта соответствует детектированию 5, 10 и 20 нг зооцида.
Расчет результатов анализа при газовой хроматографии. Содержание зоокумарина в анализируемой пробе (л, мг/кг) находят по формуле
где Ск — количество вещества, найденное по градуировочному графику, нг; Vi—объем экстракта, мл; Рг— объем вводимого в испаритель хроматографа экстракта, мкл; Р — масса анализируемой пробы, г.
Концентрация зоокумарина в мышечной ткани и крови животных при отравлении колеблется от 1,0 до 10,0 мг/кг.
Спектрофотометрическое определение зоокумарина в мышечной ткани и крови животных. Полученный после выпаривания остаток в фарфоровой чашке растворяют в 4 мл 2%-ного раствора гидроокиси натрия, фильтруют через бумажный фильтр в пробирку и измеряют оптическую плотность при 308 нм. Для сравнения используют пробу мышечной ткани или крови от того же вида здорового животного.
Спектрофотометрическое определение препарата пенокумарина. Фильтра'г 0,4 мл переносят в колбочку емкостью 50 мл и добавляют 9,6 мл 2%-ного раствора гидроокиси натрия. Раствор перемешивают и измеряют его оптическую плотность при длине волн 308 нм в кюветах с, длиной светового пути 1 см. В кювету для сравнения наливают 2 %-ный раствор гидроокиси иатрия.
Спектрофотометрическое определение зоокумарина в приманках (корме). Полученный' после выпаривания остаток растворяют в 10 мл 2%-ного раствора гидроокиси натрия и фильтруют через
бумажный фильтр в пробирку. Фильтрат 0,2 мл смешивают с 9,8 мл 2 %-ного раствора гидроокиси натрия и измеряют оптическую плотность при 308 нм против «холостой» пробы.
Количество вещества измеряют по градуировочному графику, для построения которого в ряд пробирок емкостью 15 мл вносят 0,25; 0,5; 1,0 и 2,0 мл стандартного раствора натриевой соли зоокумарина (0,1 мг/мл), что соответствует 25; 50; 100 и 200 мкг зооци-да. Затем добавляют 2 %-ный раствор гидроокиси натрия до объема 10 мл и после перемешивания измеряют оптическую плотность растворов при 308 нм.
Расчет результатов анализа при спектрофотометрии. Концентрацию зоокумарина в образце (X, мг/кг) вычисляют по формуле
где С — содержание зоокумарина, найденное по градуировочному графику, мкг; V — общий объем раствора препарата, мл; V;—объем раствора, взятого для анализа, мл; Р— масса анализируемого препарата, г.
Обработка результатов анализа. Формулы для расчета результатов анализа приведены в данном методическом указании ранее.
Временные методические указания по фотоэлектроколориметрическому определению глифтора в органах и тканях животных
(Утверждены заместителем Главного государственного санитарного врача СССР 12 мая 1983 г.)
Краткая характеристика препарата. Глифтор — 1,3-дифторпропа-нол-2. Смесь 70-%ного дпфторгидрина глицерина и 30%-нота а-хлор-у-фторгидрина глицерина. Химически чистое соединение -У бесцветная прозрачная жидкость с температурой кипения 120— 132 °C и относительной плотностью при 20 “С 1,255. С водой смешивается в любых отношениях. Умеренно растворим в хлороформе и диэтиловом эфире. Устойчив в нейтральной и слабокислой среде.
Глифтор — сильнодействующее фторорганическое соединение, ЛДб0 для кроликов и кошек 6—7 мг/кг, для крыс при внутреннем введении — 45 мг/кг. Препарат высокотоксичен для овец и ие оказывает отрицательного действия на организм птиц. Глифтор обладает выраженной кожнорезорбтивной токсичностью.
Выпускается в виде жидкости, окрашенной в синий или черный цвет. Применяется в качестве зооцида для борьбы с сусликами и мышевидными грызунами в зерновых приманках, содержащих 0,3—0,6% глифтора. Норма расхода 3,6—7,2 г/га по действующему веществу, ПДК глифтора в воздухе рабочей зоны 0,05 мг/м3.
Принцип метода. Метод основан на дистилляции глифтора из нейтральной среды с парами воды, гидролизе слабым раствором гидроксида натрия, очистке на колонке катионита и фотоэлектро-колориметрическом определении по реакции фторид-иона с лантан-ализаринкомплексоном в водно-ацетоновой среде. Количественное определение проводят на фотоэлектроколориметре с красным светофильтром или на спектрофотометре при длине волны 615 нм.
Избирательность метода. Определению глифтора не мешают неорганические соединения фтора и пестициды, не содержащие фтора. Фторорганические соединения, перегоняющиеся с парами воды и разлагающиеся в щелочной среде с образованием фторид-иона, реагируют с лантан-ализаринкомплексоном.
Реактивы и растворы, материалы. Ализаринкомплексон ч. д. а. Ацетон ос. ч., х. ч. или перегнанный в присутствии перманганата калия (на 1 л растворителя добавляют 1 г соли). Бумага индикаторная универсальная. Вата гигроскопическая медицинская. Вода бидистиллированная. Катионит КРС 8п (Н+-форма), 0,25— 0,50 мм, ч. Кислота азотная концентрированная х. ч. плотностью 1,40 г/см3. Кислота соляная концентрированная х. ч плотностью 1,19 г/см3. Кислота уксусная ледяная х. ч. Лантан азотнокислый ч. Натрия гидроксид х. ч. Натрий уксуснокислый кристаллический х. ч. Натрий фтористый х. ч. Ализаринкомплексон, 0,0005 М раствор: 0,2405 г реагента растирают в 100 мл бидистиллированной воды. К полученной суспензии добавляют 0,1 н. раствор гидроксида натрия до растворения осадка. Раствор переносят в мерную колбу вместимостью 1 л, добавляют 0,25 г ацетата натрия и после его растворения прикапывают 0,5 н. соляную кислоту до установления pH 4,5—5,0 (по универсальной индикаторной бумаге). После этого доводят объем раствора бидистиллированной водой до метки. Хранят в холодильнике не более 30 дней.
Кислота азотная, 0,01 н. раствор. Кислота соляная, 0,5 н. раствор. Лантан азотнокислый, 0,0005 М раствор: 0,2165 г La(NO3)3-6H2O растворяют в 1 л бидистиллированной воды, добавляя 5—6 капель 0,01 н. азотной кислоты. Натрия гидроксид, 0,1 н. раствор. Натрия гидроксид, 2О°/о-ный раствор. Раствор ацетатный буферный (pH 4,3): растворяют 105 г CH3COONa-ЗН2О и 100 мл ледяной уксусной кислоты в бидистиллированной воде в мерной колбе вместимостью 1 л и объем доводят водой до метки.
Раствор лантан-ализаринкомплсксона: смешивают 2 части (по объему) ацетатного буфера, 10 частей 0,0005 М лантана азотнокислого, 10 частей 0,0005 М раствора ализаринкомплексона и 25 частей ацетона. Полученный раствор хранят в холодильнике не более 7 дней.
Раствор фтора стандартный основной, 100 мкг/мл: растворяют 0,2211 г иатрия фтористого в мерной колбе емкостью 1 л и доводят объем бидистиллированной водой до метки. Полученный раствор хранят в полиэтиленовой бутыли в холодильнике не более 30 дней.
Раствор фтора стандартный рабочий, 5 мкг/мл: 5 мл основного раствора вносят в мерную колбу на 100 мл и объем доводят бидистиллированной водой до ме^ки. Готовят непосредственно перед анализом и полученный раствор переносят в полиэтиленовый флакон.
Посуда и приборы. Аппарат для встряхивания АВУ-1 или подобный. Баня водяная электрическая. Баня песчаная электрическая или электроплитка с закрытой спиралью. Бюретки на 25 мл, используемые в качестве колонок. Весы аналитические лабораторные. Весы технические, разновесы. Гомогенизатор, или измельчитель тканей. Колбы Эрленмейера н/ш вместимостью 200—250 мл. Колбы Эрленмейера или стаканы на 100 мл. Колбы мерные вместимостью 100 и 1000 мл. Пипетки мерные на 1, 5 и 10 мл. Прибор для перегонки с парами воды на шлифах, состоящий из холодильника с
длиной трубки 20—25 см, реакционной колбы Эрленмейера вместимостью 200—250 мл и приемника (пробирки градуированной с притертой пробкой на 25 мл). Пробирки градуированные н/ш на 25 мл Фотоэлектроколориметр ФЭК-М или другой, кюветы с толщиной слоя 30 мм, красный светофильтр. Часы сигнальные.
Подготовка к определению. Подготовка посуды. В результате адсорбции глифтора на стекле посуду очищают следующим образом: в колбы вносят 10 мл концентрированной азотной кислоты и 1 мл этилового спирта. После бурного выделения окне лов азота колбы многократно промывают дистиллированной водой, ополаскивают бидистиллированной водой и сушат в сушильном шкафу при 120 “С. Остальную посуду обрабатывают хромовой смесью, полностью погружая ее в окислитель на сутки. Затем многократно промывают ее водопроводной, дистиллированной и бидистиллированной водой.
Подготовка колонки для ионо-обменной хроматографии. В бюретку на 25 мл помещают тампон гигроскопической ваты. Открыв кран, вносят 10 мл бидистиллированной воды и после начала вытекания воды кран перекрывают. Затем вновь открывают кран и быстро вносят 3 г катионита, суспензированного в 15 мл бидистиллированной воды. После оседания частиц катионита колонку промывают 20 мл бидистиллированной воды и столбик катионита хранят под слоем воды (5—10 см) до использования. Катионит готовят не менее чем за 2 ч до работы.
Отбор и подготовка проб. Отбор проб производится в соответствии с «Унифицированными правилами отбора проб для определения микроколичеств пестицидов в сельскохозяйственной продукции, продуктах питания и объектах окружающей среды», утвержденными заместителем Главного государственного санитарного врача СССР 21 августа 1978 г , учитывая летучесть глифтора. Рекомендуется пробы отбирать в полиэтиленовые банки и до анализа хранить в холодильнике
Проведение определения.
Извлечение глифтора из проб. Измельченный или гомогенизированный образец массой 20 г переносят в колбу Эрленмейера на 250 мл, добавляют 50 мл бидистиллированной воды и помещают на встряхиватель на 30 мин. После встряхивания колбу присоединяют к холодильнику прибора для дистилляции и нагревают на песчаной бане, собирая 25 мл дистиллята в калиброванную пробирку на 25 мл, содержащую 0,6 мл 20 %-ного раствора гидроксида натрия. В конпе дистилляции содержимое пробирки перемешивают.
Щелочной гидролиз глифтора и очистка гидролизата. Пробирку с отгоном выдерживают 20 мин в кипящей водяной бане. Затем содержимое охлаждают под струей холодной водопроводной воды до комнатной температуры. Охлажденный гидролизат пропускают через колонку с катионитом со скоростью вытекания элюата 1 капля в 1 с (при большой скорости обменная эффективность катионита уменьшается, что может привести к сдвигу pH раствора). Колонку промывают бидистиллированной водой до тех пор, пока объем элюата станет равным 25 мл.
Проведение реакции и измерение оптической плотности окрашенного комплекса. Очищенный гидролизат переносят в колбу на 100 мл, добавляют 25 мл раствора лан-тан-ализаринкомплексона и перемешивают. Через 20 мин измеряют 226
оптическую плотность раствора на ФЭК-М при длине волны 615 им (красный светофильтр) в кювете с толщиной слоя 30 мм относительно раствора сравнения (аналогичная проба массой 20 г, подвергнутая дистилляции без гидролиза гидроксидом натрия, но пропущенная через катионит).
Построение градуировочного графика. Для построения градуировочного графика в колбы вместимостью 100 мл вносят стандартный раствор фтора и реактивы по нижеприведенной схеме:
Компоненты градуировочных растворов и их объемы Номер градуировочных растворов
1 । 2 з 4 5 6 7
Рабочий стандартный раствор фтора, мл Вода бидистилли- 0 0,2 0,5 1,0 2,0 4,0 6,0
25 24,8 24,5 24,0 23,0 21,0 19,0
рованная, мл Раствор лантан-ализаринком-плексона, мл 25,0 25,0 25,0 25,0 25,0 25,0 25,0
Количество фтора в 50 мл раствора, мкг 0 1,0 2,5 5,0 10,0 20,0 30,0
Через 20 мин измеряют оптическую плотность растворов относительно раствора сравнения, куда не вносили фтора.
Градуировочный график строят в координатах оптическая плотность — количество фтора в мкг. Объединенный закон Бугера — Ламберта — Бера соблюдается в пределах 1—20 мкг фтора.
Обработка результатов анализа. Содержание глифтора (мг/кг) в анализируемой пробе вычисляют по формуле
СД-2,76
М
где С — количество фтора в пробе, найденное по градуировочному графику, мкг; М — масса исследуемой пробы, г; К — коэффициент, учитывающий полноту извлечения и определения глифтора. Для большинства материалов он равен 1,11; 2,76 — коэффициент пересчета на глифтор.
Качественное обнаружение фосфида цинка по фосфору
(Одобрено Главным управлением ветеринарии
Госагропрома СССР 11 ноября 1987 г.)
Часть дистиллята, полученного при перегонке с водяным паром, смешивают с дымящейся азотной кислотой, выпаривают на водяной бане досуха, растворяют в небольшом количестве воды и производят реакции.
Реактивы. 10%-ный раствор серной кислоты. Бромная вода — насыщенный водный раствор брома, готовят, внося 3 мл брома в 100 мл дистиллированной воды, и хорошо встряхивая; после отстаивания прозрачный раствор сливают и хранят в темной склянке. Раствор молибдата аммония: 15 г молибдата аммония растворяют в 30 мл дистиллированной воды, а затем добавляют немного 10%-ного раствора аммиака и доливают водой до 100 мл. Этот раствор медленно вливают при помешивании в 100 мл азотной кислоты (уд. вес 1,185). Рекомендуется оба раствора сохранять отдельно и сливать при необходимости. Магнезиальная смесь: 50 г кристаллического хлорида магния и 70 г хлорида аммония растворяют в 350 мл 10%-ного раствора аммиака и затем добавляют до 750 мл дистиллированной воды, через несколько часов фильтруют. Реактив Дениже: к 25 мл 10%-ного раствора молибдата аммония добавляют 25 мл серной кислоты (уд. вес 1,83—1,84). После того как смесь охладится, добавляют 0,35 г медных опилок и дистиллированную воду до 250 мл, в течение 1 ч взбалтывают, затем сливают в другую склянку. Реактив стоек.
Ход определения. 1—2 капли раствора молибдата аммония нагревают (не до кипения) и по каплям добавляют исследуемый раствор. При наличии фосфорной кислоты образуется желтый осадок. Исследуемый раствор нейтрализуют аммиаком без избытка, прибавляют магнезиальную смесь, перемешивают и прибавляют ’/з объема 10%-ного раствора аммиака. В присутствии фосфорной кислоты выпадает кристаллический осадок.
Наиболее чувствительной реакцией на фосфорную кислоту является реакция с молибденовой синью. К испытуемому раствору добавляют несколько капель реактива Дениже и нагревают. При наличии фосфорной кислоты появляется синее окрашивание.
Перегонку с водяным паром производят в темной комнате. Конец холодильника опускают в колбочку с 3—5 мл воды. В присутствии фосфора в холодильнике, колбе и на поверхности жидкости заметно свечение. Свечению препятствуют сероводород, фенолу эфир, спирт.
В прибор для определения мышьяка по Зангер-Блеку наливают дистиллят, содержащий фосфор, прибавляют серной кислоты без мышьяка такое количество, чтобы получилась концентрация 1 : 20, прибавляют 2 г цинка и вставляют верхнюю насадку, содержащую вату, пропитанную ацетатом свинца, и кружок реактивной бумажки. Прибор оставляют иа 2 ч. В присутствии желтого фосфора или кислородных соединений трехвалентного фосфора реактивная бумажка окрашивается в канареечный цвет. При обработке реактивной бумажки 10%-ным раствором йодида калия желтая окраска переходит в оранжевую и делается более заметной. Чувствительность 0,001 мг в пробе. Мышьяк и сероводород окрашивают реактивную бумажку, но она отличается от окраски, даваемой фосфором. Сероводород улавливают ватой, пропитанной ацетатом свинца. Мышьяк образует желто-коричневую окраску, которая после обработки раствором йодида калия переходит в коричневую и темно-коричневую.
Количественное определение. Перегонку ведут до тех пор, пока перегоняемые пары не перестанут давать реакцию с азотно-серебря-ной бумажкой. Перегон окисляют бромной водой, выпаривают досуха, растворяют в 10—20 мл воды и осаждают фосфорную кислоту магнезиальной смесью. Осадок прокаливают и взвешивают
пирофосфорнокислый магний М^зРзО?. Найденное количество пирофосфорнокислого магния умножают на 0,2783 и находят количество фосфора в навеске При сомнительных определениях фосфора проводят исследования на наличие цинка.
Количественное определение фосфида цинка по фосфористому водороду
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Основано на кислотном гидролизе фосфида цинка до фосфористого водорода и солей цинка. Обнаружение фосфористого водорода производят путем титрования: Zn3P2+3H2SO4 = 3ZnSO4+2PH3.
Реактивы. 0,1 н. раствор йода: в мерную литровую колбу помещают 30 г йодида калия и 13 г перекристаллизованного йода, растворяют в 50 мл дистиллированной воды, после чего объем доводят водой до метки. Титр устанавливают по натрию тиосульфата. Для этого к 25 мл раствора йода добавляют 0,1 н. раствор тиосульфата натрия до слабо-желтой окраски. Затем добавляют 1 мл 1 %-ного раствора крахмала и окончательно титруют тиосульфатом натрия до обесцвечивания При хранении растворы йода меняют свой титр, поэтому каждый раз их титр проверяют по тиосульфату натрия.
0,1 н. раствор тиосульфата натрия (гипосульфит): 25 г кристаллической соли тиосульфата натрия вносят в мерную литровую колбу и растворяют дистиллированной водой, из которой кипячением удалей углекислый газ, затем доводят водой до метки, оставляют на 12—15 дней, устанавливают титр.
1%-ный раствор крахмала: 0,5 г картофельного или рисового крахмала растирают в небольшом количестве воды, отдельно кипятят 50 мл дистиллированной воды и при помешивании вливают в нее приготовленную крахмальную массу, продолжая кипячение в течение 3—5 мин, и затем охлаждают. Хранят в прохладном месте не более 3 дней.
Раствор соляной кислоты 1 : 3.
Ход анализа. В сухую колбочку помещают 0,1—0,3 г исследуемой соли или 5—10 г отравленной приманки и патматериала, тщательно закрывают ее резиновой пробкой с тремя отверстиями. Через одно отверстие проходит небольшая капельная воронка для кислоты, через два других — изогнутые под прямым углом стеклянные трубочки: длинная — до дна колбочки для ввода воздуха и короткая — для вывода воздуха и образовавшегося фосфористого водорода.
Колбочку помещают в водяную баню при 48—50 °C, нагревают в течение всего опыта.
Короткую трубочку для отвода воздуха и фосфористого водорода соединяют с тремя последовательно соединенными Дрекселями, в два из них вливают по 50 мл 0,1 н. раствора йода, в третий — крахмальный раствор, для определения возможного проскока йода. Всю установку присоединяют к водоструйному насосу или аспиратору для протягивания воздуха со скоростью 4—5 пузырьков в 1 с. В то же время по каплям из капельной воронки вводят 75 мл разведенной соляной кислоты.
Реакция протекает в течение 2—2,5 ч. По окончании реакции из реакционной колбы вытесняют воздух водой, после вытеснения воздуха раствор йода из обоих дрскселей соединяют вместе и титруют 0,1 н. раствором тиосульфата натрия.
При изменении окраски крахмала (посинение) его также титруют. 1 мл 0,1 н. раствора йода соответствует 0,6 мг фосфористого водорода (РН3) или 2,2764 мг фосфида цинка. Опыт ставят с контрольной пробой.
Определение крысида в биологическом материале (по Вантропу]
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Основано на экстракции крысида этиловым спиртом в колбе с обратным холодильником, последующей очистке экстракта и установлении наличия этого яда по изменению окраски при взаимодействии его с бромной водой. Чувствительность метода — 10 мкг в исследуемой навеске.
Реактивы. Ацетон. Петролейный эфир (фракция отгона при 65—70 °C). Хлороформ. 0,5 н. раствор едкого кали. Насыщенная бромная вода (1 капля брома на 1 мл воды). 5 н. раствор едкого натра. 60%-ный раствор серной кислоты.
Ход анализа. 100—150 г измельченного исследуемого материала (рвотные массы, содержимое желудочно-кишечного тракта, печень или другие органы) помещают в колбу с обратным холодильником, добавляют ацетон в жирный материал или спирт в материал без жира до консистенции жидкой кашицы и в течение 15 мин экстрагируют в кипящей водяной бане, периодически перемешивая содержимое колбы встряхиванием. Полученный экстракт фильтруют. Остаток вновь экстрагируют. Эту операцию делают 3 раза.
Собранные фильтраты помещают в фарфоровую чашечку, добавляют 100 мл дистиллированной воды и упаривают на водяной бане до объема 100 мл, остаток переносят в делительную воронку, а фарфоровую чашечку тщательно ополаскивают теплой дистиллированной водой. Смывную воду вводят в ту же делительную воронку. После того как высохнет фарфоровая чашечка, ее трижды промывают петролейным эфиром (по 25 мл каждый раз). Эфир после смывания также помещают в делительную воронку. Эфир извлекает пигменты и липиды. Обработку экстракта в делительной воронке продолжают новыми порциями эфира до обесцвечивания водного слоя. Фарфоровую чашечку, в которой производилось упаривание, смывают 50 мл хлороформа, который сливают в делительную воронку. При плавном перевертывании воронки 40—50 раз производят извлечение крысида.
По окончании смешивания жидкости дают расслоиться, хлороформ сливают, экстрагирование хлороформом по 50 мл делают 3 раза. Хлороформные вытяжки соединяют, фильтруют и частично выпаривают на водяной бане. В полученный остаток добавляют 25 мл раствора едкого калия и нагревают на водяной бане, размешивая стеклянной палочкой, пока раствор станет прозрачным.
Прозрачный раствор переносят в делительную воронку, а сосуд промывают теплой дистиллированной водой (75 мл). Промыв-230
ную воду вводят в ту же делительную воронку. После того как высохнет промытый сосуд, его ополаскивают 50 мл хлороформа, последний также вводят в делительную воронку. В делительной воронке экстракцию производят 5 раз, расходуя 150 мл хлороформа. Хлороформные вытяжки соединяют вместе и промывают трижды дистиллированной водой (по 25 мл).
Хлороформный остаток помещают в фарфоровую чашечку и выпаривают до объема 10 мл.
В маленькую делительную воронку помещают 2 мл полученного экстракта, добавляют 1 мл бромной воды и смешивают. Для обесцвечивания избытка брома добавляют 2 мл раствора едкого натра и вновь взбалтывают.
При наличии крысида слой хлороформа окрашивается в синий или фиолетовый цвет. При неясной окраске реакцию повторяют с остатком экстракта, предварительно выпаренного до объема 2 мл. Часть окрашенного раствора помещают в пробирку, добавляют немного серной кислоты и взбалтывают. При наличии крысида окраска не изменяется.
Колориметрическое определение фостоксина в зерне *
Действующим началом фостоксина является фосфористый водород, который выделяется из фосфида алюминия под влиянием влаги: А1Р + ЗН2О->РН3+А1 (ОН)3.
Допустимое остаточное количество фостоксина в зерне 0,01 мг/кг.
Принцип метода. Метод основан на разложении фосфида алюминия разбавленной серной кислотой, улавливании выделяющегося РН3 азотной кислотой и последующим колориметрическом определении по общему фосфору (по синему фосфорно-молибденовому гетерополикомплексу). Чувствительность метода 0,5 мкг фосфора, полнота определения 96±4%.
Реактивы и растворы. 0,02 М раствор молибденовокислого аммония в 10 н. серной кислоте. 0,13%-ный раствор сернокислого гидразина. 10 н. и 2%-ный растворы серной кислоты, концентрированная серная кислота плотностью 1,84 г/см3. Кислота азотная плотностью 1,34—1,37 г/см3. Перманганат калия перекристаллизованный. Калий фосфорнокислый однозамещенный перекристаллизованный. Окислительная смесь: растворяют 0,5 г растертого в порошок марганцовокислого калия в 10 мл серной кислоты плотностью 1,84 г/см3. Реактив следует хранить в склянке с притертой пробкой не более пяти дней.
Стандартный раствор № 1 (250 мкг фосфора в 1 мл): 0,2742 г однозамещенного фосфорнокислого калия растворяют в 250 мл дистиллированной воды, подкисленной несколькими каплями серной кислоты. Раствор сохраняется 2—3 нед.
Стандартный раствор № 2 (1 мкг фосфора в 1 мл): разбавляют 1 мл раствора № 1 до 250 мл подкисленной дистиллированной водой.
Раствор готовят непосредственно перед употреблением.
Приборы и посуда. Аспиратор или воздуходувка. Реометр на скорость до 2 л/мин. Пробирки колориметрические мерные вместимостью 15 мл. Пипетки вместимостью 1, 5 и 10 мл с минимальными
делениями 0,01 и 0,1 мл. Колбы мерные на 250 мл. Колбы из тугоплавкого стекла вместимостью 100 мл. Термометр на 200 °C. Баня водяная. Электроплитка. Поглотительные склянки вместимостью 150 и 1000 мл.
Ход анализа. 200 г зерна помещают в поглотительную склянку вместимостью 1000 мл, увлажняют 30 мл 2%-ной серной кислоты и закрывают пробкой с двумя отводными трубками. Длинная трубка должна быть погружена в зерно почти до дна поглотительной склянки. Короткую отводную трубку соединяют встык с поглотительной склянкой вместимостью 150 мл, содержащей 50 мл концентрированной азотной кислоты. Продувают через зерно воздух со скоростью 1 л/мин в течение 2 ч. Отсоединяют поглотительную склянку с кислотой, переносят кисло! у в термостойкую колбу на 100 мл и упаривают на плитке досуха. Параллельно для контроля упаривают 50 мл чистой азотной кислоты.
К сухому остатку прибавляют 2,5 мл азотной кислоты плотностью 1,34—1,37 г/см3 и 0,1 мл окислительной смеси. Колбу осторожно встряхивают и нагревают на плитке с асбестовой прокладкой. Нагревание исследуемого раствора продолжают до полного удаления паров азотной кислоты. После охлаждения остаток в колбе растворяют в 7 мл дистиллированной воды и переносят в колориметрическую пробирку. Одновременно или предварительно готовят шкалу стандартных растворов (табл.).
Схема приготовления шкалы стандартных растворов
№ стандарта 1 2 3 J 4 5 G 7 | 8 1 9 10 И
Стандартный раствор № 2, мл — 0,5 1 1,5 2 2,5 3 3,5 4 4,5 5
Дистиллированная вода, мл 7 6,5 6 5,5 5 4,5 4 3,5 3 2,5
Содержание фосфора, мкг — 0,5 1 1,5 2 2,5 3 3,5 4 4,5 5
В пробирки с пробами и стандартными растворами прибавляют по 1 мл раствора молибденовокислого аммония. Пробирки встряхивают и помещают в кипящую водяную баню на 10 мин. После охлаждения растворов прибавляют по 1,6 мл раствора сернокислого гидразина, встряхивают и вновь помещают на 15 мин в кипящую водяную баню. После вторичного охлаждения растворов доводят объем в колориметрической пробирке дистиллированной водой до 10 мл и хорошо встряхивают содержимое пробирки. Измеряют оптическую плотность стандартных растворов на фотоколориметре при красном светофильтре (кюветы 10 мм) и строят график зависимости оптической плотности от концентрации фосфора.
Измеряют оптическую плотность исследуемой пробы, используя в качестве раствора сравнения пробу, полученную при выпаривании чистой азотной кислоты. Определяют концентрацию фосфора в пробе зерна, пользуясь построенным калибровочным графиком.
Расчет. Содержание фостоксина в пробе (X, мг/кг) определяют по формуле
v 1,06 л A = p '
где A — количество фосфора, найденное по графику, мкг; Р — масса пробы зерна, г; 1,06 — коэффициент пересчета на фостоксин.
Определение мышьяка по Зангер-Блеку
(Одобрено Главным управлением ветеринарии Госагропрома СССР И ноября 1987 г)
Оно основано на восстановлении соединений мышьяка до мышьяковистого водорода Последний в зависимости от количества мышьяка окрашивает бумажку, обработанную бромидом ртути или дихлоридом ртути (сулемой), от желтого до темно коричневого цвета
В результате реакции мышьяковисто! о водорода с дихлоридом ртути образуется комплексное соединение As(HgCl)3-Hg2Cl2, дающее окрашивание
Реактивы. Стандартный раствор очищенного мышьяковистого ангидрида Водный раствор серной кислоты (1:7) Хлорид закиси олова (SnCl2 2Н2О) Цинк без мышьяка Вата, смоченная 5% ным раствором ацетата свинца и высушенная Бромно ртутная бумажка Насыщенный раствор йодида кадмия Насыщенный раствор йодида калия
Ход анализа. Для улавливания выделившегося мышьяковистого водорода (гидрида мышьяка) на его пути помещают кружочки из фильтровальной бумаги, пропитанные хлорной или бромной ртутью В присутствии мышьяковистого водорода бумажки окрашиваются в желтый или коричневый цвет Реакция неспецифнчна, так как ее дает и фосфористый водород Сероводород окрашивает бумажку в грязно желтый цвет Таким способом улавливается до 0,001 мг мышьяка, а при помощи бумажки, пропитанной бромной ртутью,— до 0,0001 мг мышьяка Чувствительность реакции можно повысить, если пользоваться газовыводящей трубкой меньшего диаметра
Для открытия мышьяка по этому способу пользуются прибором, состоящим из редукционной колбочки емкостью 100 мл, трубки со шлифом с шарообразным расширением внизу и газовыделяющей трубочкой меньшего диаметра вверху, на которую помещают кружок реактивно,: бумажки В шарообразное расширение трубки кладут вату, пропитанную 5% ным раствором ацетата свинца и высушенную, для задержки сероводорода
Обнаружение мышьяка проводят следующим образом В редукционную колбу вносят 2 г гранулированного цинка, не содержащего мышьяка, 5—10 мг хлорида олова (на кончике ножа), исследуемую жидкость, 40—50 мл разведенной 1 : 7 серной кислоты (без мышьяка) и быстро вставляют заранее подготовленные трубки В шарообразное расширение трубки закладывают пропитанную 5% ным раствором уксуснокислого свинца и высушенную вату. В верхнюю часть трубки помещает кружок фильтровальной бумаги, пропитанной 5% ным раствором бромида ртути (кружок фильтровальной бумаги кладут на внутреннюю трубочку и сверху прижимают трубочкой такого же диаметра, пропущенной через пробку). Вата, пропитанная ацетатом свинца, задерживает сероводород, который
может образоваться одновременно с мышьяковистым водородом. Тампон ваты служит контролем на случай улетучивания сероводорода: если вата окрашивается в серый цвет и окраска появляется по бокам и особенно в верхней части ватного тампона, это служит признаком прохождения сероводорода и опыт считается неудавшимся.
Прибор оставляют на 1 ч. После окончания реакции кружок реактивной бумажки проявляют погружением его вначале в насыщенный раствор йодида кадмия, а затем в насыщенный раствор йодида калия, после чего промывают водой. Окрашивание кружочка реактивной бумаги в коричневый или темно-коричневый цвет указывает на присутствие мышьяка. При пользовании реактивными бумажками, пропитанными бромидом ртути, можно их не проявлять йодидом калия. Особое внимание следует обращать на чистоту применяемых реактивов. Цинк и серная кислота не должны содержать мышьяка. Прежде чем производить определения, следует проверить реактивы на чистоту. Для этого ставят контрольные опыты без исследуемой жидкости. Чтобы результаты исследования были удовлетворительными, требуется соблюдать ряд условий
Для активации выделения водорода обычно рекомендуется прибавлять сульфат меди, что может, однако, привести к понижению чувствительности реакции. Такие же результаты дают соли железа, особенно вредно сказывается присутствие солей ртути и платины, сильно задерживающих восстановление мышьяка до мышьяковистого водорода. Хлорид магния и особенно хлорид олова, наоборот, оказывают благоприятное действие на реакцию восстановления мышьяка и могут даже устранять вредное влияние катионов железа, меди, ртути.
Бромную бумагу готовят пропитыванием кусочков фильтровальной бумаги 5%-ным спиртовым раствором бромида ртути (HgBr2). Бумагу сушат и сохраняют в темном и сухом месте.
Количественное определение. При определении малых количеств мышьяка можно применять описанный выше способ Зангер-Блека. Нужно только, исходя из определенных количеств мышьяковистого ангидрида, приготовить шкалу, с которой сравнивают интенсивность окрашивания кружка реактивной бумаги и определяют количество мышьяка в исследуемой пробе. Для приготовления шкалы берут 0,1321 г очищенного мышьяковистого ангидрида, растворяют в небольшом количестве (4—5 мл) концентрированного едкого натра, переводят в мерную колбу емкостью 100 мл, нейтрализуют серной кислотой в присутствии фенолфталеина и доводят дистиллированной водой до 100 мл. Такой раствор I можно хранить довольно долго. Берут 1 мл раствора и разводят водой в мерной колбе до 100 мл, в 0,1 мл полученного раствора II будет содержаться 0,001 мг мышьяка, как раз то наименьшее количество, которое еще имеется в ряду стандартных окрасок Затем берут 0,3 мл раствора II, соответствующие 0,003 мг мышьяка, 0,5 мл, соответствующие 0,005 мг, и так до 2 мл, а затем поступают, как описано выше при определении мышьяка. Шкалу (Ряд окрашенных бумажек, соответствующих определенным количествам мышьяка) завертывают в черную бумагу и хранят в темном и сухом месте — в эксикаторе с хлористым кальцием. (Пользоваться эксикатором с H2SO4 нельзя, так как пары серной кислоты действуют на шкалу и она выцветает.) В таких условиях она сохраняется около месяца и более. Чтобы устранить влияние влаги,
рекомендуется покрыть шкалу раствором органического стекла в дихлорэтане, тогда она может сохраняться до 15 мес Для малых количеств мышьяка (до 0,02 мг) отверстие внутренней трубочки, по которой выходит газ и на которую накладывают кружок бу маги, должен быть диаметром 8 мм, для больших количеств беру г трубку диаметром 1,6 см, для меньших количеств — 2—4 мм
Удовлетворительные результаты анализа получаются при со блюдении ряда условий Например, следует пользоваться редук ционнои колбой одного и того же размера и трубками для бром нои бумажки одного и того же диаметра, для опытов необходимо брать одно и то же количество цинка и серной кислоты, цинк всегда должен быть одной н той же степени дробления, температу ра постоянной, чтобы реакция шла с одинаковой скоростью Толь ко соблюдая условия опыта, можно получить достаточно точные и воспроизводимые результаты
Очистка мышьяковистого ангидрида Продажный фарфоровид ный мышьяковистый ангидрид растворяют в горячей соляной кис лоте, разбавленной 1 :2, отфильтровывают от нерастворившегося сульфида мышьяка и охлаждают, при этом образуется осадо<> мышьяковистого ангидрида Раствор сливают, осадок несколько раз промывают водой, сушат на водяной бане, возгоняют и по лучают чистый мышьяковистый ангидрид После 12 ч высушивания в эксикаторе над хлористым кальцием препарат употребляют для приготовления шкалы
Методические указания по определению фенотиазина в мёде методом тонкослойной хроматографии
(Утверждены заместителем Главного государственного санитарного врача СССР 5 июня 1978 г)
Краткая характеристика препарата. Фенотиазин (тиодифенил амин, зоотиазин, антиверм) Молекулярная масса 199,28 Химически чистый продукт — светло-желтое кристаллическое вещество Тп > 185 °C, 1кип 371 °C Практически нерастворим в воде Плохо рас творим в большинстве органических растворителей ЛД50 для крыс 5000 мг/кг
Принцип метода. Метод основан на извлечении фенотиазина бензолом, очистке экстракта от восков и пигментов, хроматографи ровании па пластинках «Силуфол» Зоны локализации препарата определяются после обработки пластинок ацетоновым раствором хлорного железа Нижний предел определения 0,05 мг/кг
Реактивы и растворы. Апетон х ч Бензол х ч Окись алюминия для хроматографии II степени активности Хлороформ х ч Пластинки «Силуфол» Силикагель АСК Хлорное железо Проявляющий реактив 0,5% ный раствор хлорного железа в 50 %-ном водном ацетоне Стандартный раствор фенотиазина в бензоле 10 мг фенотиазина, перекристаллизованного в горячем бензоле, раство ряют в 100 мл бензола в мерной колбе Раствор содержит 100 мкг фенотиазина в мл ,
Приборы и посуда. Делительные воронки емкостью 50 мл Мерные колбы на 100 мл Пульверизаторы стеклянные Камера для хроматографирования Шприцы 1 мл для нанесения стандартных растворов и проб Баня водяная Настольный вентилятор Камера
для опрыскивания хроматограмм. Чашки для выпаривания на 50 мл. Стеклянные хроматографические колонки (высота 20—25 см, внутренний диаметр 1,3 см).
Подготовка к определению. Приготовление хроматографической колонки. Заполняют хроматографическую колонку силикагелем АСК (высота слоя 5 см) и окисью алюминия (высота слоя 5 см). Промывают перед употреблением 15 мл бензола.
Проведение определения.
Экстракция и очистка. К 30 г меда (закристаллизовавшийся мед прогревается на водяной бане с температурой 50 °C до полного разжижения) добавляют 2 мл горячей дистиллированной воды, хорошо размешивают, помещают в делительную воронку и дважды экстрагируют равным объемом бензола, встряхивая в течение 10 мин, не допуская образования эмульсии. Экстракты объединяют. Упаривают на водяной бане с температурой 60 °C в струе воздуха от вентилятора до 2 мл, количественно вносят на хроматографическую колонку. Фенотиазин элюируют 40 мл бензола. Для ускорения элюации ее проводят под небольшим давлением, создаваемым резиновой грушей. Упаривают бензол, как было описано выше. Последние 2—3 мл удаляют без подогрева в струе воздуха от вентилятора. Сухой остаток растворяют в 0,2—0,3 мл хлороформа, дают хлороформу испариться до 0,1 мл и наносят на пластинку дважды, обмывая чашку таким же количеством хлороформа. Параллельно наносят стандартный раствор фенотиазина 1 и 3 мкг.
Условия хроматографирования. Пластинку помещают в камеру для хроматографирования со смесью гексана и ацетона (4:1). После того как растворитель поднимется на высоту 10 см от места нанесения пробы, пластинку вынимают, сушат и опрыскивают проявляющим реактивом. Фенотиазин проявляется в виде оранжевого постепенно зеленеющего пятна, Rf 0,5.
Расчет результатов анализа. Количество фенотиазина в пробе определяют путем сравнивания размера и интенсивности окраски пятен пробы и стандарта. Содержание фенотиазина в меде (в мг/кг) рассчитывают по формуле
где X— содержание фенотиазина, мг/кг; А — количество фенотиазина, найденное путем сравнения со стандартом, мкг; Р — навеска анализируемой пробы, г.
Методические указания по определению остаточных количеств формальдегида в почве, воде, продуктах и отходах сахарного производства
(Утверждены заместителем Главного государственного санитарного врача СССР 27 сентября 1978 г.)
Краткая характеристика препарата. Формальдегид (формол, формалин 40%-ный раствор). Молекулярная масса 30,03. Формальдегид— бесцветный газ. Тплавл 92 °C, tKBn 21 °C. Хорошо растворяется в воде, этиловом спирте н эфире. По химическим свойствам 236
формальдегид — сильный восстановитель ПДК в воде водоемов — 0,05 мг/л Формальдегид применяется в качестве антисептика в сахарном производстве
Принцип метода. Метод основан на реакции взаимодействия формальдегида с димедоном (5,5 диметилгидрорезорцин), экстрак ции его производного (формальдимедона) органическим раствори телем и последующем определении методом хроматографии в тон ком слое
Реактивы и растворы. Силикагель марки КСК, просеянный через сито 100 меш Хлороформ х ч Натрий сернокислый безводный х ч Спирт этиловый, ректификат Формалин, 40 %-ный водный раствор Стандартный раствор димедона в спирте 200 мкг/мл Три хлоруксусная кислота, 5%-ный водный раствор Стандартный раствор формальдимедона в хлороформе 100 мкг/мл.
Формальдимедон готовят следующим образом. к 10 мл 40%-ного водного раствора формальдегида при перемешивании прибавляют 100 мл спиртового раствора димедона Полученную смесь нагревают на кипящей водяной бане в течение 30—45 мин Смесь охлаждают на воздухе при температуре 20—25 °C Выпавший осадок формальдимедона отфильтровывают на воронке Бюхнера или через пористый фильтр, дважды промывают горячей водой и перекристаллизовывают из диметилформамида Для этого в коническую колбу помещают 10—15 мл диметилформамида, нагревают до 100 °C и постепенно прибавляют формальдимедон Полученную смесь нагревают до кипения При наличии нерастворивше-гося осадка прибавляют несколько миллилитров диметилформамида до полного его растворения Полученный раствор охлаждают на воздухе до 20—25 °C Выпавший осадок формальдимедона отфильтровывают на воронке Бюхнера и промывают этиловым спиртом дважды по 5 мл каждый раз Осадок сушат до постоянного веса. Формальдимедон имеет 1пл 184—186 °C При отсутствии диметилформамида формальдимедон можно перекристаллизовывать из этилового спирта
Проявляющий реактив 0,5%-ный раствор йода в хлороформе.
Посуда и приборы. Колбы мерные на 100 мл. Воронки химические d = 60 мм Пипетки на 0,1 мл с ценой деления 0,01 мл. Цилиндры мерные на 25 и 100 мл Холодильник Либиха Делительные воронки на 250 мл Баня водяная Колбы грушевидные на 25 и 50 мл Воронки Бюхнера Прибор для встряхивания АВУ. Пульверизаторы стеклянные Камера для хроматографирования. Камера для опрыскивания пластинок Прибор для отгонки с водяным паром (круглодонная или коническая колба на 1000 мл, насадка Вюрца, холодильник Либиха 1 = 200—400 мм, аллонж, приемник колба на 100 мл Вся посуда на шлифах). Весы аналитические ВЛА 200М Пластинки стеклянные 9X12 см или «Силуфол».
Проведение определения. 30—50 г пробы сахара, мелассы, диффузионного сока, жома, воды или 100 мл почвы помещают в коническую колбу на 250 мл, добавляют 80—100 мл дистиллированной воды, 5—10 мл 5%-ной трихлоруксусной кислоты взбалтывают в течение 15 мин и с помощью водяного пара отгоняют формальдегид К 80 м'л полученного дистиллята прибавляют 3 мл стандартного спиртового раствора димедона и нагревают в течение 10 мин на кипящей водяной бане с обратным холодильником или оставляют при комнатной температуре на 24 ч Раствор охлаждают до комнатной температуры и формальдимедон трижды
экстрагируют в делительной воронке хлороформом по 15 мл в течение 5—10 мин каждый раз. Хлороформные экстракты объединяют, пропускают через слой безводного сернокислого натрия в грушевидную колбочку к упаривают на водяной бане при t 40 °C до объема 0,1—0,2 мл.
Очистка экстракта, В случае анализа мелассы водные растворы формальдимедона до проведения экстракции очищают от красящих и других коэкстрактивных веществ колоночным способом с помощью активированного угля — карболена на колонке высотой 100 мм, диаметром 15 мм с последующей десорбцией формальдегида из колонки.
Хроматографирование. Остаток в колбе после отгонки растворителя наносят на тонкий слой сорбента с помощью микрокапилляра в одну точку на расстоянии 1,5 см от нижнего края и боковой стороны хроматографической пластинки. Нанесение проводится так, чтобы диаметр полученного пятна не превышал 8—10 мм. Справа и слева от пробы на расстоянии 1,5—2 см наносят стандартные растворы формальдимедона 1, 5, 10 мкг. Пластинку с нанесенными растворами пробы и стандартов помещают в камеру для хроматографирования, в которую за 15—20 мин налита на высоту 0,5 см подвижная фаза—хлороформ. После того как растворитель поднимется на высоту 10 см, пластинку вынимают и сушат на воздухе в течение 10—15 мин.
Для обнаружения препарата пластинку опрыскивают проявляющим раствором. Формальдимедон на хроматограмме обнаруживается в виде серо-коричневых пятен на белом фоне.
Качественная идентификация препарата осуществляется по величине Rf 0,48.
Обработка результатов анализа. Количественное определение препарата проводят либо инструментальным декситометрическим методом на приборе «Оптон», либо путем визуального сравнения интенсивности и площади пятен пробы с интенсивностью и наиболее близкой по величине площади пятна стандарта. Содержание препарата в пробе (мг/кг) рассчитывают по формуле
А
, В-9,7
где А — количество формальдимедона, найденное путем визуального сравнения со стандартом, мкг; В — навеска пробы, г; 9,7 — фактор пересчета формальдимедона на формальдегид.
Методика определения фтора в биологическом материале и минеральных веществах
(Утверждена Главным управлением ветеринарии Министерства сельского хозяйства СССР 13 марта 1985 г.)
Определение фтора с предварительной отгонкой.
Принцип метода. Фтор отгоняется в виде кремнефтористой кислоты с водяным паром и определяется в отгоне колориметрическим цирконализариновым методом.
Приготовление реактивов.
1. Серная кислота. Для освобождения кислоты от фтора ее кипятят в течение 1 ч с момента появления густых белых паров.
В анализе используют разбавленную кислоту (1:1 или 1 : 1,5) в дистиллированной воде.
2. Ферросицилий — растертый и прокаленный в муфеле при 800 °C в течение 2 ч порошок. Можно применять чистый кварцевый песок или измельченное стекло. Последние смачивают в платиновой чашке концентрированной серной кислотой и осторожно нагревают до появления белых паров, а затем прокаливают при температуре 700 °C.
3. Сернокислое серебро. При отсутствии сернокислого серебра его готовят из азотнокислого серебра согласно протекающей реакции 2AgNOs+ (NH4)2SO4 = Ag2SO44-2NH2NO3 следующим образом: 34 г азотнокислого серебра растворяют в 200 мл дистиллированной воды, 13,2 г сернокислого аммония отдельно также растворяют в 200 мл дистиллированной воды. После полного растворения оба реактива смешивают. Образовавшемуся сернокислому серебру дают осесть, надосадочную жидкость и промывную воду осторожно сливают по стеклянной палочке так, чтобы осадок оставался в стакане. Промывание повторяют несколько раз до исчезновения реакции на нитратион в промывных водах. Осадок фильтруют и высушивают при комнатной температуре в темном месте.
4. Ализариновый красный «С» (мопосульфат натрия). Ci4H2SNa-H2O. Навеску 0,750 г растворяют в 1 л бидистиллированной воды.
5. Азотнокислый (или оксихлорид) цирконий. 0,295 г азотнокислого циркония пли 0,354 г оксихлорида циркония растворяют в 800 мл бидистиллированной воды. Затем, медленно, помешивая, добавляют 33,0 мл концентрированной серной кислоты и 100 мл концентрированной соляной кислоты. После охлаждения раствор доводят до 1 л бидистиллированной водой.
6. Стандартный раствор: 50,0 г фтористого натрия растворяют в 100 мл бидистиллированной воды, фильтруют и упаривают на водяной бане до появления на поверхности твердой корки, которую снимают на стекло и сушат на воздухе. 0,221 г перекристаллизованного фтористого натрия растворяют в 1 л бидистиллированной воды, 1 мл этого раствора содержит 0,1 мг фтора. Стандартный раствор хранят в запарафинированной склянке с притертой пробкой.
Все приготовленные растворы хранят в темном прохладном месте.
Подготовка проб к исследованию. Из измельченных проб мяса или печени берут навеску 25—50 г, молока или мочи 50—200 мл, из высушенных проб кормов, фекалий берут навеску 10,0—30,0 г. Кости подвергают автоклавированию (при 1,5—2,0 атм) до полного обезжиривания и берут навеску 0,5—1,0 г. Пробы минеральных добавок, содержимого желудка и др. берут в зависимости от содержания фтора (1,0—25,0). К пробам биологического материала (кроме костей) и кормов добавляют 0,5—1,0 г окиси кальция (СаО). К жидким пробам добавляют порошок, к сухим — взвесь гидроокиси кальция, настаивают в течение суток и затем высушивают в сушильном% шкафу, постепенно повышая температуру до 170—180 °C, а далее' сжигают в муфельной печи, повышая температуру до 400—550 °C, до полного озоления (до светло-серой золы). Золу в чашках осторожно смачивают водой и переносят в дистилляционную колбу. Чашки ополаскивают 100 мл серной кислоты (1:1) двумя порциями по 40 и 60 мл, которые переносят в ту же
колбу, прибавляют несколько зерен кварцевого песка и 60—70 мг сернокислого серебра для осаждения хлора и проводят отгон.
Отгон фтора производят при температуре 135—145 °C, регулируя ее при помощи трансформатора «ЛАТР». Можно использовать также контактный термометр или подкладывание под дистилляционную колбу асбестовой сетки. Отгоняют 400 мл дистиллята (если материал беден фтором можно 100—200 мл) в слабощелочной среде, а для контроля добавляют 1—2 капли фенолфталеина и несколько капель раствора NaOH. Отгон проводят в 2—3 параллелях.
Обязательно проводить холостой опыт для определения содержания фтора в реактивах. Вместо навески пробы берут 10 мл бидистиллированной воды. При необходимости ставят контрольный опыт для определения процента потери фтора (к образцу добавляют известное количество NaF).
Стандартная шкала. Калибровочный график. В мерные колбочки на 100 мл наливают следующие количества стандартного раствора фтористого натрия и доводят до метки дистиллированной водой:
Стандартный раствор, мл 0 0,5 1,0 1,5 2,0 2,5 3,0
Содержание фтора, мг/100 мл 0 0,05 0,1 0,15 0,2 0,25 0,3
Р-Ра
Содержание фтора, мг/л 0 0,5 1,0 1,5 2,0 2,5 3,0
Добавляют по 5 мл раствора ализарина и оксихлорида (или азотнокислого циркония) в каждую из колбочек и ставят в темное
место. Ровно через 1 ч колориметрируют.
Измерения проводят на правом барабане в кювете с длиной граней 50’ мм при зеленом светофильтре. Данные отмечают на миллиметровой бумаге, где по оси абсцисс откладывают известные концентрации, а на оси ординат — показания фотоэлектрического колориметра (ФЭК).
Колориметрирование. Берут 100 мл отгона и пипеткой добавляют по 5 мл раствора ализарина моносульфата и азотнокислого циркония. Через 60 мин колориметрируют, данные откладывают на оси ординат и по графику высчитывают количественное содер-
жание фтора.
Примечание. Если в пробе концентрация фтора большая (более 10 мг/л), то берут по 50, 20, 10 и 5 мл отгона и дистиллированной водой доводят до 100 мл; при малой концентрации фтора отгон (200, 300 мл) упаривают до 100 мл.
Методика определения гексахлорпараксилола (гексихола) в кормолекарственной смеси, молоке, органах и тканях животных
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 16 июня 1980 г.)
Краткая характеристика препарата. Гексахлорпараксилол (ге-тол, хлоксил, гексихол)— 1,4-бис-(трихлорметил)-бензол — белый кристаллический порошок, нерастворимый в воде, хорошо растворимый в большинстве органических растворителей (ацетон, гексан, 240
этиловый эфир, спирт), молекулярный вес 313,0, удельный вес 1,5 (1 20 °C). Ткип 157 °C, 1пл 110-111 °C, устойчив к действию кислот, разлагается в щелочной среде.
Гексихол — новая препаративная форма препарата с содержанием гидрофобизирующей жидкости (поверхностно-активное вещество), предотвращающей его слеживаемость. Препарат широко применяется в ветеринарной и медицинской практике в качестве ант-гельминтика при трематодозах животных и человека (фасциолез, дикроцелиоз, описторхоз). Терапевтическая доза гексихола для крупного рогатого скота 0,3 г/кг, мелкого рогатого скота — 0,2 г/кг. ЛД50 для белых мышей составляет 18 г/кг при парентеральном и 22 г/кг при энтеральном введении, коэффициент токсичности равен НО.
Кормолекарственные смеси предназначены для дегельминтизации крупного и мелкого рогатого скота. Гексихол входит в состав кормолекарствениых смесей в количестве, зависящем от дозы и вида животного, которому они предназначены.
Принцип метода. Метод основан на извлечении гексихола из кормолекарственной смеси, органов и тканей животных ацетоном с последующим перераспределением из водно-ацетоновой смеси в гексан, из молока, предварительно сожженного кислотой, гексаном и дальнейшим определением ТСХ. Экстракты из молока, органов и тканей животных очищают концентрированной серной кислотой и в колонке с силикагелем АСК.
Избирательность метода. Прочие хлорорганические соединения не мешают определению и не проявляются в количестве меньше 5 мкг в пробе. При наличии в пробе хлорорганических соединений Rf их не совпадает с гексихолом.
Реактивы и растворы. Растворители: ацетон х. ч., гексан х. ч. Натрий хлористый, 0,25%-ный водный раствор. Натрий сернокислый безводный х.ч. Серебро азотнокислое, 1%-ный водный раствор. Серная кислота концентрированная х. ч.
Стандартный раствор гексихола в гексане. Приготовление: 10 мг гексихола растворяют в 10 мл гексана (1 : 1000, в 1 мл— 100 мкг препарата), 1 мл раствора переносят в мерную пробирку па 10 мл и добавляют до 10 мл гексан (1 : 10 000, в 1 мл— 10 мкг), из этого раствора 1 мл переносят в другую пробирку и доливают до 10 мл гексан (1:100 000, в 1 мл—1 мкг).
Пластинки «Силуфол» 150X150.
Приборы и посуда. Хроматографическая колонка высотой 50 см, внутренним диаметром 1,5 см. Камера для хроматографирования. Пульверизатор стеклянный. УФ-лампа. Весы аналитические. Колбы плоскодонные со шлифом, емкостью 250 мл. Делительные воронки емкостью 250 и 500 мл. Конические воронки. Мерные цилиндры емкостью 50, 100 и 250 мл. Чашки фарфоровые для упаривания. Шприц на 1 мл. Пипетки на 0,1, 1, 5, 10 мл. Бумажные фильтры.
Ход анализа.
Кормолекарственные смеси. Отбор образцов проводят согласно ГОСТ 13496.0—70. Правила отбора среднего образца. В случае, если смесь гранулирована, гранулы измельчают. Из средней пробы берут 1 г (для овеГг) или 0,5 г (для крупного рогатого скота) и экстрагируют в колбе на 250 мл дважды по 15 мл ацетона в течение 3 ч на шуттель-аппарате. Экстракты фильтруют через бумажный фильтр в колбу на 500 мл, добавляют 200 мл 0,25 %-ного раствора NaCl и 30 мл гексана, встряхивают 5 мин и переносят в де-
16 Заказ № 1995
241
лительную воронку на 500 мл. После разделения слоев нижний водно-ацетоновый слой сливают в колбу и экстрагируют гексихол еще раз 30 мл гексана. Из объединенного гексанового экстракта (60 мл) отбирают пипеткой 1 мл раствора при содержании в 1 г кормолекарственной смеси менее 100 мг гексихола пли 0,1 мл при содержании более 100 мг, помещают в мерную пробирку на 10 мл и доливают до 10 мл гексаном, из расчета в 0,1 мл содержится 1—3 мкг гексихола.
На хроматографическую пластинку, которую предварительно в течение 5—10 мин облучают УФ-светом, на расстоянии 1,5 см от края наносят 0,1 мл (для овец) или 0,3 мл (для крупного рогатого скота) экстракта. Параллельно наносят серию стандартов гексихола с содержанием 0,5; 1,0; 2,0; 3,0 мкг. Хроматограмму помещают в предварительно насыщенную камеру и дважды разгоняют в гексане, длина пробега растворителя 10 см. После разгонки хроматограмму подсушивают, опрыскивают 1%-ным водным раствором AgNO3, снова сушат ее в токе воздуха, а затем облучают УФ-светом до появления четких темных пятен гексихола па светлом фоне пластинки. Величина Rf препарата 0,57±0,01.
Органы и ткани животных. 10 г соответствующего биосубстрата тщательно измельчают и перетирают в ступке с 25—30 г безводного Na2SO4 до сыпучей консистенции, помещают в плоскодонную колбу емкостью 250 мл, добавляют 60 мл ацетона и экстрагируют гексихол в течение 1,5 ч при комнатной температуре при периодическом встряхивании содержимого колбы, а затем продолжают экстракцию в течение 1,5 ч в холодильнике При t 2—4 °C без «ерем.ецив.аиищ После окоичавия экетрак’в.ии декаели'руют раствор через бумажный фильтр в делительную воронку емкостью 500 мл. Биосубстрат в колбе ополаскивают 30 мл ацетона, который объединяют с первой частью растворителя. К объединенному экстракту добавляют 250 мл 0,25%-ного раствора NaCl и 30 мл гексана, тщательно встряхивают ее содержимое в течение 5 мин. Дают слоям разделиться, нижний водно-ацетоновый слой переносят в другую делительную воронку, гексихол экстрагируют еще дважды таким же количеством гексана. Объединенные гексановые экстракты в делительной воронке очищают серной кислотой до тех пор, пока кислота будет бесцветной Затем экстракт сливают в фарфоровую чашку, упаривают в токе воздуха до 2 мл и дополнительно очищают на колонке с силикагелем АСК.
Предварительно в колонку помещают тампон из стекловаты или ваты, засыпают сорбент на высоту 15 см, уплотняют его постукиванием по колонке деревянной палочкой и промывают 50 мл гексана.
В подготовленную таким образом колонку из фарфоровой чашки переносят экстракт гексихола. После того как экстракт полностью впитается в сорбент, гексихол элюируют 100 мл гексана порциями по 25—30 мл. Последнюю порцию элюата отсасывают резиновой грушей. Элюат собирают в фарфоровую чашку и упаривают в токе воздуха до объема 0,3—0,5 мл и с помощью микропипстки наносят на хроматографическую пластинку. Параллельно наносят серию стандартов гексихола с содержанием 0,3; 0,5; 1,0 и 2.0 мкг. Пластинку обрабатывают, как описано выше.
Пропорциональная зависимость между размером пятна и интенсивностью окраски сохраняется до 10 мкг гексихола в пробе.
Молоко. 30 мл молока помещают в колбу емкостью 250 мл
и добавляют 20 мл H2SO4 для сжигания субстрата. После остывания добавляют 30 мл гексана, встряхивают содержимое в течение 5 мин. Затем переносят в делительную воронку па 250 мл и дают возможность слоям полностью разделиться. Верхний гексановый слой оставляют, а нижнюю часть сливают в колбу и экстракцию гексихола повторяют тем же объемом гексана. Объединенный экстракт в делительной воронке очищают серной кислотой до тех пор, пока кислота будет бесцветной. Далее экстракт очищают на колонке с силикагелем АСК, упаривают до 0,3—0,5 мл и хроматографируют. Стандартные растворы гексихола наносят на хроматографическую пластинку такие же, как при определении в органах и тканях животных.
Обработка результатов анализа.
1. Для определения гексихола визуально сравнивают пятна, полученные при анализе исследуемых образцов кормолекарственной смеси, с пятнами стандартных растворов гексихола.
Кормолекарственная смесь считается пригодной для дегельминтизации животных в том случае, если полученные пятна такой же интенсивности окраски, как модельные пятна, содержащие 1 или 2 мкг гексихола (для овец) или 2 мкг (для крупного рогатого скота).
2. Содержание 1ексихола в анализируемой пробе органов, тканей и молоке животных рассчитывают по формуле
_ _А_ ~ В ’
где X — содержание гексихола в пробе (мг/кг или мг/л); А — количество гексихола, найденного путем визуального сравнения с пятнами стандартных растворов препарата по их интенсивности окраски и площади (мкг); В — навеска исследуемой пробы (г).
Нижний предел определения гексихола 0,05 мг/кг в кормолекарственной смеси, органах и тканях животных и 0,03 мг/л в молоке
Обнаружение гексихола составляет 70+10%.
РАСТИТЕЛЬНЫЕ ЯДЫ
Методика хроматографического определения алкалоидов в патологическом материале и растениях
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 25 марта 1976 г )
Принцип метода. Метод основан на извлечении алкалоидов из испытуемых проб, определении их в остатках путем постановки цветной реакции на индикаторной бумаге с последующей идентифи кацией методом хроматографии в тонком слое окиси алюминия
Чувствительность в зависимости от вида алкалоидов 20—50 мг в пробе для тонкого слоя
Воспроизводимость при разгонке на одной пластинке 100%, относительная ошибка 7%
Реактивы Серная кислота х ч или ч д а Винная кислота ч д а Щавелевая кислота х ч или ч д а Хлороформ х ч или ч д а Аммиак водный ч д а 1,2 дихлорэтан ч д а Калий йоди стый х ч или ч д а Висмут азотнокислый основной ч д а Уксус ная кислота х ч Динониловый эфир фталевой кислоты для хрома тографии ч д а Ацетон х ч или ч д а Спирт этиловый 96% ный Окись алюминия для хроматографии ч
Растворы 5 и 10% ные водные растворы серной кистоты 10 % ный водный раствор аммиака
Подвижный растворитель хлороформ, ацетон, динониловы i эфир Фталиевой кислоты, взятые в соотношении 5 4 1 по объему
Реактив 1 16,5 г основного азотнокислого висмута растворяют в 500 мл 25% ной уксусной кислоты, отдельно растворяют 80 г йодистого калия в 200 мл дистиллированной воды Оба раствора смешивают и выдерживают в темной склянке в течение 1 мес Для проявления берут 20 мл основного раствора, 20 мл ледяной уксусной кислоты и 100 мл воды Чувствительность реакции 3— 5 мкг для бхмажной хроматографии
Реактив 2 200 г винной кислоты и 17 г основного азотно кислого висмута растворяют в 800 мл воды, отдельно растворяют 160 v йодистого калия в 400 мл воды Оба раствора смешивают и хранят в темной склянке Для проявления берут 10 мл основного раствора, 10 г винной кислоты и 50 мл воды Чувствительность реакциг 1 —3 мкг для бумажной хроматографии
Приборы и оборудование. Колбы конические широкогорлые на 0,5, 0,75, 1 л Камера для хроматографирования Пульверизатор стеклянный Стаканы химические емкостью 50, 100, 75, 150 мл Бюксы Аппарат для встряхивания проб Весы аналитические Весы технические Бумага для хроматографии марки М. Центрифуга с емкостью стаканов 100 мл и более Делительные воронки емкостью 0,5, 0,75, 1 л Универсальная индикаторная бумага Реагент, pH 1 —10 Баня водяная Ножницы Электрическая мельница МРГ 1 Пластинки стеклянные размером 9X12, 13X18, 20X20 см Сито 150 меш Камера для люминесцентного анализа
Ход анализа.
Экстракция алкалоидов из алкалоидоносных растений и кормов. Алкалоиды из растительной ткани извлекают двумя способами: водной экстракцией в виде солей алкалоидов и органическими растворителями в виде оснований.
Извлечение алкалоидов в виде солей. Измельченный растительный материал в количестве 100 г нативной массы заливают водой в соотношении для сочных кормов 1 : 4, сухих — 1:10. В склянки с материалом при постоянном помешивании добавляют 10%-ный раствор серной кислоты до pH 3. Затем все тщательно взбалтывают и ставят на водяную баню при температуре 40 °C па 2 ч при периодическом помешивании. Дальнейшее исследование сочных кормов проводят сразу же после окончания прогревания, сухих объектов — через 24 ч после настаивания.
Водную вытяжку из растений фильтруют через бумажный фильтр в делительную воронку и обрабатывают 30 мл хлороформа 2 раза по 5 мин путем легкого взбалтывания с целью очистки от белков и посторонних примесей. Хлороформ отделяют и выбрасывают, к очищенной кислой вытяжке, желательно па холоду, добавляют 10%-пый раствор аммиака до pH 9.
Подщелоченную водную вытяжку переносят в делительную воронку и обрабатывают хлороформом по 50, 30, 20 мл путем легкого взбалтывания в течение 5—7 мин. Хлороформ каждый раз отделяют, все порции соединяют и фильтруют через бумажный фильтр, предварительно смоченный обезвоженным хлороформом, в химический стакан и упаривают досуха.
Извлечение алкалоидов в виде оснований применяется только в отношении сухих кормов и высушенных ядовитых растений. Для выполнения этого анализа берут навеску 100 г измельченного объекта, смачивают 10%-ным раствором аммиака до состояния слабого увлажнения, переносят в склянку, плотно закрывают и оставляют на 30 мин. При проведении данной операции следует избегать сильного увлажнения, так как излишек влаги затрудняет контакт частиц растения с органическим растворителем. По истечении 30 мин исследуемую массу заливают хлороформом или дихлорэтаном в отношении соответственно 1:8, 1:10 и оставляют для настаивания на 24 ч. По истечении указанного срока органический растворитель сливают через бумажный фильтр в делительную воронк}' и 3 раза обрабатывают 5%-ным водным раствором серной кислоты порциями по 50, 30, 20 мл с целью перевода алкалоидов из хлороформа в сернокислую водную вытяжку. Сернокислые водные вытяжки собирают вместе, фильтруют через ватный фильтр и подщелачивают 10%-ным аммиаком до pH 9 и вновь обрабатывают хлороформом 3 раза, добавляя его по 30 мл, алкалоиды в этом случае переходят в хлороформ. Хлороформные извлечения собирают, фильтруют в химический стаканчик, который оставляют при комнатной температуре для испарения хлороформа. В случае обработки больших партий растений в конечной стадии хлороформ отгоняют на водяной бане до малого объема.
Часть сухого остатка, содержащегося в химических стаканчиках, испытывают на алкалоиды и в случае положительных результатов полученные алкалоиды сохраняют в лаборатории в закрытых склянках, в темном сухом месте, так как они могут в последующем понадобиться в качестве свидетеля при хроматографической
идентификации алкалоидов, выделенных из органов и тканей погибшего животного.
Экстракция алкалоидов из патологического материала. Берут навеску измельченных неконсервировапных паренхиматозных органов (мышц, содержимого рубца или желудка) в количестве 100 г, мочу можно брать до 500 мл, но не менее чем 20 мл. Внутренние органы и головной мозг хорошо измельчают, переносят каждый отдельно в широкогорлые колбы емкостью 750 мл, заливают водой в соотношении 1 :4 и подкисляют 5%-ным водным раствором серной кислоты до pH 2 по универсальной индикаторной бумаге. Содержимое колб несколько раз тщательно взбалтывают и ставят в водяную баню на 1 ч при температуре 40 °C. Затем снимают с водяной бани, оставляют на 2 ч для экстракции и застывания жира. Экстракцию можно проводить и без подогревания, оставляя пробы на 16—20 ч при температуре не выше 15 °C. После настаивания жидкость с проб сливают и фильтруют через ватный фильтр в делительную воронку, где 3 раза обрабатывают хлороформом порциями по 25 мл в течение 10 мин путем осторожного взбалтывания. Хлороформный слой после расслаивания сливают и выбрасывают. Если отстаивание хлороформного слоя не происходит ввиду образования стойкой эмульсии, то для отделения хлороформа следует эмульсию подвергнуть центрифугированию при 3000—-4000 об/мин в течение 5 мин.
Водяную кислую вытяжку после очистки ее хлороформом осторожно (желательно на холоду) подщелачивают 10%-ным водным раствором аммиака до pH 9 и содержащиеся в ней алкалоиды извлекают хлороформом 3 раза порциями 25—30 мл. Хлороформ сливают, фильтруют. Хлороформная вытяжка не должна содержать следов водного фильтрата и быть прозрачной, для чего ее вторично фильтруют через бумажный фильтр, предварительно смоченный обезвоженным хлороформом. Полученную хлороформную вытяжку оставляют в химическом стакане для испарения хлороформа и получения сухого остатка.
Операцию выделения алкалоидов из растительной ткани и патологического материала следует отнести к одной из самых ответственных, так как в конечном итоге от хода анализа на данном этапе работы зависит получение правильного результата в будущем.
Обнаружение алкалоидов в изолированном остатке. После изоляции алкалоидов из биологического материала следует этап определения их в полученном остатке. Сухой остаток, изолированный из объекта исследования, растворяют в 1 мл хлороформа. Часть полученного раствора наносят на хроматографическую бумагу (размером 3x7 см) стеклянными капиллярами, очередную каплю раствора наносят после испарения предыдущей. Затем реактивом 1 или 2 проявляют бумагу с нанесенными на нее пробами. При наличии алкалоидов в хлороформных экстрактах появляются красные или малиновые пятна .В случае появления нечетких пятен проявленная бумага отмывается водой до обесцвечивания фона, при этом на белом фоне бумаги четко выявляются пятна алкалоидов красного цвета.
Этот способ обнаружения алкалоидов также применим для их идентификации и в водных растворах, для чего эти растворы наносят на хроматографическую бумагу и после полного высыхания проявляют этими же реактивами.
В случае получения положительной качественной реакции на алкалоиды проводят их определение методом тонкослойной хроматографии.
Хроматографическая идентификация.
Хроматографирование. С краев хроматографической пластинки снимают слой сорбента 3 мм, затем, отступая 1,5 см от нижнего края и боковых сторон, наносят изолированные из проб остатки так, чтобы диаметр пятна не превышал 0,5 см. С этой целью сухие остатки растворяют 1 мл хлороформа и полученный раствор наносят на стартовую линию с помощью микропипетки или пипетки от гемометра ГС в количествах 0,02 мл и более. Для контроля рядом наносят пробы алкалоидов, выделенных из ядовитых растений или образца корма. На одну пластинку размером 9\12 см можно нанести 4 пробы, 13X18 см — 6, а на 20X20 см еще больше.
Пробы на слой сорбента наносят осторожно, не нарушая его целостности, так как нарушение ее приводит к изменению величины и форм пятен в процессе хроматографирования.
Пластинку с нанесенными пробами после полного испарения растворителя помещают в камеру для хроматографирования, па дно которой mi 1 ч до внесения пластинки наливают подвижный растворитель, состоящий из хлороформа, ацетона, динонилового эфира фталевой кислоты, взятых в соотношении 5:4: 1, приготов ленного за 24 ч до хроматографирования. Пластинку погружают в раствор не более чем на 5 мм, а пятна от нанесенных веществ должны быть на 5 мм выше места соприкосновения подвижного растворителя с тонким слоем.
После того как фронт растворителя поднимется до верхнего края пластинки, ее извлекают из камеры и оставляют для испарения растворителя.
Высохшая пластинка просматривается в люминесцентной камере в ультрафиолетовых лучах (лампа ПРК-2, ПКР-4, светофильтр УФС-3). Флуоресцирующие участки хроматограммы обводятся карандашом по стеклу со стороны, обратной нанесенному слою, с описанием цвета свечения, затем пластинка опрыскивается одним из проявляющих (1, 2) реактивов до полной пропитки тонкого слоя, что узнается отсутствием белого слоя при просмотре обратной стороны стеклянной пластинки.
Флуоресцентное проявление позволяет легко отдифференцировать птомаины в остатках, изолированных из патологического материала, которые на хроматограмме светятся в виде беловато-голубоватых пятен, не имеющих четких контуров и располагающихся вблизи фронта растворителя.
После опрыскивания пластинки реактивами 1 или 2 алкалоиды проявляются в виде пятен красного или малинового цвета на общем желтом фоне.
Идентификация алкалоидов в патологическом материале производится на основании совпадения пятен от анализируемой пробы с пятнами алкалоидов, выделенных из растений. Совпадение пятен позволяет сделать заключение о наличии алкалоидов растений в анализируемом объекте, а исходя из их токсикологических свойств — поставить диагноз.
При несовпадении пятен алкалоидов испытуемой пробы со свидетелями осуществляется повторная разгонка, а за свидетелей берутся алкалоиды, значение Rf которых близко к испытуемой,
для чего используется набор алкалоидов, выделенных ранее из ал-калоидоносиых растений зоны деятельности ветеринарной лаборатории (табл.).
При необходимости документирования проведенных исследований делают фотокопии или пересъемку пластинки па кальку, которые затем подклеивают к карточке химико-токсикологического исследования. Для приготовления стандартных растворов используют природные растительные алкалоиды (аконитин, зонгорип, анабазин, ореолин, кондельфин и др.) и сумму алкалоидов, выделенных из растений, произрастающих на территории зоны деятельности ветеринарной лаборатории. Для получения стандарта отвешивают 5 мг сухого порошка алкалоида или остатка, выделенного из растения, переносят в стеклянный бюкс с притертой пробкой и разводят 1 мл хлороформа, этот раствор и является свидетелем по отношению к испытуемой пробе. После использования части раствора бюкс ставят в вытяжной шкаф для испарения хлороформа. Неиспользованная часть сухого остатка сохраняется в закрытом бюксе в холодильнике от 3 до 5 лет. В случае необходимости он может быть многократно использован.
Сухие неиспользованные порошки алкалоидов сохраняются от 10 до 25 лет.
Величина Rf некоторых алкалоидов, полученных в стандартных условиях
Соеципение Rf Иве г флуоресценций
Аконитин 0,61 Нет
Зонгорин 0,51 Све гло-желтая
Лаппаконитин 0,52 Голубая
Кондельфин 0,60 Сине-фиолетовая
Талатизин 0,20 Нет
Лупинин 0,13 Нет
Секурипин 0,72 Желтая
Анабазин 0,22 Нет
Методика определения свободного и связанного госсипола в семенах хлопчатника, хлопковом жмыхе, шроте и комбикормах
(Одобрена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 24 декабря 1982 г.)
Краткая характеристика. Госсипол — растительный полифенол (2,2-ди-1,6,7-триокси-5-изопропил-8-альдегидонафтил). Содержится в семенах, листьях и корне хлопчатника в количестве от 0,4 до 1,7%, в хлопковых шроте и жмыхе — от 0,01 до 0,4%.
Наличие в корме свободного госсипола в количестве 0,008% вызывает снижение прироста массы цыплят, а его концентрация 0,04% приводит к характерной клинике токсикоза у свиней.
Связанный госсипол также обладает токсическими свойствами, так как под воздействием ферментов и микрофлоры кишечника до 50% его переходит в свободный.
Принцип метода. Метод основан на извлечении свободного гос-сипола смесью ацетона и воды (7:3), очистке экстракта петролейным эфиром и соляной кислотой. Связанный госсипол после гидролиза с анилином извлекают гексаном, очистку экстракта проводят соляной кислотой.
Метрологическая характеристика метода
Свободный госсипол Связи, и. госсипол
Минимально определяемое количество госсипола (стандарт) 0,03—0,01 мкг 0,1 мкг
Нижняя граница определения 1,5 мг/кг 2,5 мг/кг
Верхняя граница определения 250 мг/кг 500 мг/кг
Посуда и приборы. Камеры хроматографические. Опрыскиватель. Пластинки «Силуфол». Микропипетки 0,1 мл. Колбы конические, нормального шлифа. Пробирки градуированные 10 мл. Воронки химические. Воронки делительные ВД-3-500. Чашки для выпаривания. Аппарат для встряхивания АВУ-1. Водяная баня. Стеклянные бусинки. Бумажные фильтры. Мельница электрическая.
Реактивы и растворы. Стандартный раствор свободного госси-пола в ацетоне. Стандартный раствор дианилингоссипола в хлороформе. Ацетон х. ч. н-Гексан х. ч. Хлороформ х. ч. Толуол х. ч. Эфир петролейный (1кип 30—60 °C). Спирт этиловый ректификованный. Анилин ч. д. а. Вода дистиллированная. Кислота соляная х. ч. Кислота уксусная ледяная. Олово хлорное х. ч. Железо хлорное х. ч.
Проявляющие реактивы: на свободный госсипол — 2%-ный раствор хлорного олова в этаноле и 5%-ный раствор хлорного железа в 50%-ном этаноле; на связанный госсипол — 5%-ный раствор хлорного железа в 50%-ном этаноле.
Приготовление стандартных растворов.
Свободный госсипол: 3—5 мг госсипола растворяют в 30—50 мл ацетона (0,1 мг/мл); из полученного разведения берут 1 мл в градуированное пробирку на 10 мл и объем доводят ацетоном до 10 мл (0,01 мг/мл).
Связанный госсипол (дианилингоссипол) готовят аналогично свободному, используя хлороформ в качестве растворителя. Стандартные растворы хранят в посуде из темного стекла не более 2 мес при температуре 4 °C.
Ход анализа.
Определение свободного госсипола. Массу исследуемою образца измельчают на мельнице до порошкообразного состояния, тщательно перемешивают и отбирают среднюю пробу (семян, шрота, жмыха 3 г, комбикорма 5 г) с точностью до 0,01 г, помещают в колбу на 250 мл, вносят 50 мл смеси ацетона п воды (7 : 3) и 5—6 стеклянных бусинок. Колбу ставят на аппарат для встряхивания на 30 мин. По истечении времени экстракт отфильтровывают через бумажный фильтр в делительную воронку, колбу обмывают трижды по 10 мл смесью, которую так же пропускают через фильтр в воронку Фильтр с остатками корма помещают в колбу, в которой проводили экстракцию. В делительную воронку вносят 10 мл петролейного эфира и содержимое воронки переме-
шивают. После разделения слоев нижний сливают в другую делительную воронку. Слой эфира в делительной воронке отбрасывают. Очистку петролейным эфиром повторяют. В воронку вносят 100 мл дистиллированной воды и 1 мл концентрированной соляной кислоты, перемешивают, после чего госсипол переэкстрагируют в хлороформ 5—6 раз порциями по 5 мл, интенсивно встряхивая воронку. Объединяют растворы хлороформа в выпарительной чашке, выпаривают досуха в токе воздуха. Сухой остаток из проб семян, жмыхов, шротов переносят количественно ацетоном порциями по 2 мл 4—5 раз, соскребая стеклянной палочкой, сливают растворы в градуированную пробирку на 10 мл, доводят объем ацетоном до 10 мл Сухой остаток из проб комбикорма переносят количественно ацетоном, порциями по 0,5 мл в градуированную пробирку, доводя объем в пробирке до метки 2 мл. Ацетоновый экстракт наносят на пластинку «Силуфол» в количестве 10 и 20 мкл из проб семян, жмыхов, шротов и 100 мкл из проб комбикорма, в 1,5 см от проб наносят стандартный раствор госсипола в количестве 0,5, 1, 2 и 3 мкг.
Пластину помещают в камеру с системой: ледяная уксусная кислота — толуол — гексан (10:8:8), фронт растворителей должен пройти 10 см, после чего пластинку вынимают, подсушивают и опрыскивают одним из проявляющих реактивов. Rf 0,8—0,85 При проявлении хлорным оловом пятна малиновой окраски на белом фоне, узкие, при проявлении хлорным железом — оливково-серые на желтом фоне.
Определение связанного госсипола. В колбу с остатками корма и фильтром добавляют 10 мл ацетона в пробы семян, жмыхов, шротов и 10 мл 72%-ного этанола в пробы комбикорма и по 1 мл свежеперегнанного анилина, закрывают и ставят на водяную баню на 30 мин (температура 60 °C), колбу во избежание прилипания корма ко дну помешивают. Если ацетон выпарится, то вносят еще 10 мл его. Колбу охлаждают в течение 2 мин и вносят 50 мл гексана, ставят иа аппарат для встряхивания на 30 мин Затем содержимое колбы осторожно сливают в делительную воронку, если в воронку попал не смешивающийся с гексаном слой (из проб комбикорма), то его возвращают в колбу. Обмывают стенки колбы несколько раз гексаном до обесцвечивания его, сливая все в воронку. Объединенные экстракты промывают 5— 6 раз по 5 мл 0,2 н. раствором соляной кислоты, энергично встряхивая воронку. Нижний слой (кислотный) отбрасывают. Гексановый слой из делительной воронки сливают в выпарительную чашку, выпаривают в токе воздуха досуха.
Экстракты можно оставить на ночь под тягой или в выпаренном виде убрать в холодильник. Сухой остаток с чашки переносят количественно ацетоном, соскребая стеклянной палочкой, порциями по 2 мл в градуированную пробирку, доводя объем до метки 10 мл (для проб из семян, жмыхов, шротов) и порциями по 0,5 мл, доводя объем в пробирке до метки 2 мл (для проб из комбикорма). На старт пластины «Силуфол» наносят микропипеткой 20 мкл экстракта из проб семян, жмыхов, шротов и 50 мкл из проб комбикорма, в 1,5 см от проб наносят стандартный раствор дианилин-госсипола в количестве 1, 2, 4 мкг. Пластину помещают в камеру с системой: петролейный эфир — ацетон (15:10), растворители должны пройти 10 см, после чего пластину вынимают, подсушивают и опрыскивают 5%-ным раствором хлорного железа в 50%-ном 250
этаноле. Rf 0,6—0,55, пятна узкие, коричневого цвета на желтом фоне.
Обработка результатов. Полуколичественное определение проводят визуально, сравнивая размер пятна и интенсивность его окраски со стандартными растворами. Содержание вещества в исследуемой пробе вычисляют по формуле
££•1,33 А=----- -— ,
АГ
где X'—количество вещества в органе, мкг/г или мг/кг; А — объем экстракта, нанесенного на пластинку, мкл; Б — конечный объем экстракта, мкл; В — количество вещества, обнаруженное на пластине, мкг; Г — навеска, г; 1,33 — коэффициент пересчета на свободный госсипол для комбикормов.
Методика определения свободного и связанного госсипола в органах и тканях сельскохозяйственных животных и птиц
(Одобрена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 24 декабря 1982 г.)
Краткая характеристика. Госсипол — растительный полифенол (2,2-ди-1,6,7-триокси-5-изопропил-8-альдегидонафтил), содержится в семенах, листьях и корне хлопчатника в количестве от 0,4 до 1,7%, в хлопковом шроте и жмыхе— от 0,01 до 0,4%.
Наличие в корме свободного госсипола в количестве 0,02% вызывает снижение живой массы и яйценоскости у кур-несушек. Госсипол обладает кумулятивными свойствами и откладывается в печени, почках, легких, селезенке, сердце, мышцах и яйце в свободной и связанной форме.
Связанный госсипол также обладает токсическими свойствами, так как под воздействием ферментов и микрофлоры кишечника до 50% его переходит в свободный.
Принцип метода. Метод основан на извлечении свободного госсипола из органов ацетоном, очистке экстрактов петролейным эфиром и в системе подвижных растворителей. Связанный госсипол после гидролиза с анилином экстрагируют гексаном, очистку экстрактов проводят соляной кислотой.
Краткая характеристика метода
Свободный госсипол Связанный госсипол
Минимально определяе- 0,03—0,01 мкг 0,1 мкг
мое количество госсипола (стандарт)
Нижняя граница опре- 1,5 мг/кг 10 мг/кг
деления
Посуда и приборы. Камеры хроматографические. Опрыскиватель. Пластины «Сидуфол». Микропипетки 0,1 мл. Колбы конические нормального шлифа. Пробирки градуированные 5 мл. Воронки химические. Воронки делительные ВД-3-250. Чашки выпарительные. Аппарат для встряхивания АВУ 1. Водяная баня. Стеклянные бусинки. Бумажные фильтры. Ножницы.
Реактивы и растворы. Стандартный раствор свободного госсипола в ацетоне. Стандартный раствор дианилингоссипола в хлороформе. Ацетон х. ч. н-ГеКсан х. ч. Хлороформ х. ч. Толуол х. ч. Эфир петролейный (1Кип. 30—60 °C). Спирт этиловый ректификованный. Анилин ч. д. а. Вода дистиллированная. Кислота соляная х ч. Кислота уксусная ледяная. Бура. Железо хлорное х. ч. Проявляющий реактив: 5%-ный раствор хлорного железа в 50%-ном растворе этанола.
Приготовление стандартных растворов.
Свободный госсипол. Госсипол в количестве 3—5 мг растворяют в 30—50 мл ацетона (0,1 мг/мл), из полученного разведения берут 1 мл в градуированную пробирку на 10 мл и объем доводят ацетоном до 10 мл (0,01 мг/мл).
Связанный госсипол (дианилингоссипол) готовят аналогично свободному госсиполу, используя хлороформ в качестве растворителя.
Стандартные растворы хранят в посуде из темного стекла не более 2 мес при температуре 4 °C.
Ход анализа.
Определение свободного госсипола. Для анализа отбирают от туши вынужденно убитого животного или трупа кусочки печени, почек, сердца, легких, селезенки, мышц, тщательно измельчают ножницами из нержавеющей стали, отбирают 5 г с точностью до 0,01 г, помещают в колбу на 250 мл, вносят 50 мл ацетона и 5—6 стеклянных бусинок. Колбу закрывают и ставят на аппарат для встряхивания на 30 мин. После встряхивания ацетон фильтруют через бумажный фильтр в делительную воронку, колбу промывают трижды по 10 мл ацетоном, который сливают в воронку. Фильтр с остатками ткани помещают в колбу, в которой проводили экстракцию. В делительную воронку вносят примерно равный объем петролейного эфира и 10 мл 1 %-ного раствора буры, содержимое воронки осторожно перемешивают, 4—5 раз переворачивая воронку. После разделения нижний слой сливают в другую делительную воронку. Слой эфира, оставшийся в первой делительной воронке, промывают 10 мл 1 %-hoi о раствора буры, 4—5 раз переворачивая воронку, после разделения нижний слой объединяют с тем, что слит в другую делительную воронку, верхний слой отбрасывают. В делительную воронку вносят 100 мл дистиллированной воды и 1 мл концентрированной соляной кислоты, перемешивают. Госсипол в делительной воронке переэкстрагируют в хлороформ трижды, порциями по 5 мл, интенсивно встряхивают воронку, растворы хлороформа объединяют в выпарительной чашке. При образовании эмульсии в делительной воронке вносят 5—10 капель этанола. Чашку выпаривают досуха в токе воздуха. Сухой остаток переносят количественно ацетоном, порциями по 0,2 мл, соскребая стеклянной палочкой, в 1радуированную пробирку, доводя объем до метки 1 мл. Ацетоновый экстракт наносят на пластину «Силуфол» микропипеткой в количестве 100 мкл, в 1,5 см от пробы наносят стандартный раствор госсипола в количестве 0,05; 0,1; 0,2; 0,4 мкг.
Очистка пластин проводится в подвижной системе: петролейный эфир — толуол — ацетон — хлороформ (10:2:4:4), фронт растворителей должен пройти до противоположного края пластины. Затем пластину вынимают, подсушивают и разгоняют в системе: ледяная уксусная кислота—толуол — гексан (10:8:8). Фронт рас
творителей должен пройти 10 см, после чего пластину вынимают, подсушивают и опрыскивают проявляющим реактивом. Rf 0,8—0,85, пятна узкие, оливково-серого цвета на желтом фоне.
Определение связанного госсипола. В колбу с остатками тканей и фильтром добавляют 10 мл 72%-ного этанола и 1 мл свежеперегнанного анилина, закрывают и ставят на водяную баню на 30 мин (температура 60 °C). Колбу охлаждают в течение 2 мин, вносят 50 мл гексана и встряхивают в течение 30 мин на аппарате для встряхивания. Содержимое колбы осторожно сливают в чистую делительную воронку. Если в воронку попал несме-шивающийся с гексаном слой (нижний), его возвращают в колбу. Обмывают колбу гексаном до тех пор, пока гексан не будет окрашиваться, сливают все в делительную воронку. Объединенные экстракты промывают 5—6 раз по 5 мл 0,2 н. раствором соляной кислоты, энергично встряхивая воронку. Нижний слой (кислотный) отбрасывают. Гексановый слой из делительной воронки сливают в выпарительную чашку и выпаривают в токе воздуха досуха. Для выпаривания чашки можно оставить на ночь под тягой или уже выпаренные чашки убрать на ночь в холодильник. Сухой остаток с чашки смывают ацетоном, соскребая стеклянной палочкой, порциями по 0,5 мл в пробирку, довод,ч объем до метки 2 мл. Fla старт пластины «Силуфол» наносят микропипегкой 50 мкл экстракта, в 1,5 см от пробы наносят стандартный раствор дпанплингосси-пола в количестве 0,5; 1; 2; 4 мкг. Пластину помещают в камеру с системой растворителей' петролейный эфир — ацетон (15:10), растворители должны пройти 10 см, после чего пластину вынимают, подсушивают и опрыскивают проявляющим реактивом. Пятна узкие, коричневого цвета на желтом фоне.
Обработка результатов. Определение проводят визуально, сравнивая размер пятна и интенсивность его окраски со стандартным раствором.
Содержание вещества в исследуемой пробе вычисляют по формуле
ДД-1,33 А—
где X — количество вещества в органе, мкг/г или мг/кг; А — объем экстракта, нанесенного на пластинку, мкл; Б — конечный объем экстракта, мкл; В — количество вещества, обнаруженное на пластине, мкг; Г — навеска, г; 1,33 — коэффициент пересчета для свободного госсипола.
Методика определения госсипола в кормах, содержащих продукты переработки хлопчатника
(Утверждена Главным управлением ветеринарии Госагропрома СССР 30 сентября 1986 г.)
Принцип метода. Метод основан на извлечении госсипола из растительного материала в виде госсиполята натрия, очистке экстракта от коэкстрактивных веществ, переводе госсиполята натрия в свободный госсипол, извлечении его хлороформом, отгонке хлороформа с последующим анализом сухого остатка цветной качественной реакцией на госсипол и при наличии последнего определение
способом распределительной хроматографии в тонком слое с полу-количественным расчетом содержания яда путем сравнения со стандартом.
Метрологическая характеристика метода. Минимально детектируемое количество — 0,5 мкг. Нижний предел определения в хлопчатнике 5 мг/кг, в комбикормах — 10 мг/кг. Размах варьирования— 90—95%. Среднее значение определения — 95%. Доверительные интервалы среднего определения при pH 95, п = 5 — 95±3,0.
Избирательность метода. Метод специфичен, другие природные яды (алкалоиды, гликозиды, эфирные масла, лактоны и пестициды — хлорорганические, фосфорорганические, тяжелые металлы) определению не мешают.
Пригодность метода. Метод пригоден для определения госсипола в хлопчатниковых семенах, шроте, жмыхе, гузопае и кормах, содержащих продукты переработки хлопчатника.
Реактивы, Госсипол фармакопейный, 99,8 %-ной чистоты, желтого цвета, с температурой плавления 183±1 °C. Ацетон х. ч. н-Гексан х. ч. Хлороформ х. ч. Эфир петролейный. Спирт этиловый ректификат. Кислота соляная х. ч. Кислота серная х. ч. Нонан. Флороглюцин. Натрий едкий х. ч. или ч. д. а. Вода дистиллированная. Изопропиловый спирт.
Приборы и посуда. Весы лабораторные класса точности 2 с наибольшим пределом взвешивания 200 г. Камера для хроматографии. Опрыскиватель стеклянный. Пластинки «Силуфол». Микропипетки на 0,1 мл. Пипетки на 1 мл. Пипетки на 10 мл. Бюксы стеклянные низкие с крышкой. Колбы конические на 250 мл. Мельница электрическая лабораторная. Цилиндры мерные на 25 мл. Цилиндры мерные на 500 мл. Аппарат для встряхивания АВУ-1. Стеклянные бусы. Воронки химические 7 см. Воронки делительные на 250 и 500 мл. Фильтры бумажные (синяя лента). Сито лабораторное с диаметром отверстий 1 мм.
Растворы. Стандартный раствор свободного госсипола готовят путем растворения 5 мг госсипола в 50 мл хлороформа (0,1 мг/мл). Срок хранения 3 мес в темном прохладном месте.
Система подвижного растворителя: гексан 10 мл, нонан 10 мл, изопропиловый спирт 5 мл.
Проявитель для качественной пробы и хроматограмм: 0,1 г флороглюцина растворяют в 100 мл этилового спирта и в полученный раствор вносят 10 мл соляной кислоты концентрированной. Хранят в темной склянке 2 мес
Экстрагирующая смесь: к 70 мл ацетона добавляют 30 мл 0,2%-ного водного раствора едкого натрия.
Ход анализа. Пробу измельчают на электромельнице, просеивают через сито, навеску 5 г переносят в колбу на 250 мл, заливают 80 мл экстрагирующей смеси, вносят бусинки, закрывают. Колбу оставляют на 24 ч в вытяжном шкафу для настаивания при частом взбалтывании. Экстракт фильтруют через бумажный фильтр в делительную воронку. Колбу с пробой дважды по 15—20 мл промывают экстрагирующей смесью и пропускают через этот же фильтр.
В делительную воронку с экстрагирующей жидкостью вносят 20 мл петролейного эфира или гексана, осторожно 5 мин взбалтывают при частом открывании крана для спуска паров петролейного эфира и после отстаивания нижний слой сливают в химический стакан, а оставшийся в воронке верхний слой отбрасывают. Очистку петролейным эфиром или гексаном повторяют 3—5 раз, а при 254
наличии в комбикорме травяной муки — до бесцветного состояния очистителя.
Очищенную от коэкстрактивных веществ экстракционную жидкость переносят в делительную воронку, туда добавляют 50 мл 2%-ного водного раствора серной кислоты, смесь взбалтывают, выдерживают 30 мин и госсипол трижды переэкстрагируют хлороформом порциями 15, 10 и 10 мл. Хлороформный экстракт фильтруют через бумажный фильтр, предварительно смоченный хлороформом, в стеклянный низкий бюкс и ставят для испарения в вытяжной шкаф. После испарения хлороформа сухой остаток сохраняется в закрытом бюксе в темном сухом месте 3 мес.
Для исследования сухой остаток растворяют в 1 мл хлороформа, с которым проводят качественную пробу на госсипол и при положительной реакции исследование на пластинках «Силуфол».
Качественная реакция обнаружения госсипола: 0,1 мл хлороформного раствора изолированного остатка наносят иа полоску, вырезанную на пластинки «Силуфол», 4X15 см, с левой стороны отступая от края 3 см, на расстоянии 3 см от пробы наносят 0,1 мл стандартного раствора госсипола. После высыхания проб их проявляют путем опрыскивания проявителем дважды через 30 мин. При появлении в пробе цветного пятна (красного пли малинового) дают заключение о наличии госсипола в пробе, что служит основанием для проведения хроматографического анализа. При отсутствии красного пятна в испытуемой пробе на этом этапе прекращают анализ и дают отрицательное заключение.
Хроматографическое определение госсипола. На пластинку «Силуфол», отступая от краев 1,5 и 2 см снизу, наносят микропипеткой хлороформный раствор сухого остатка пробы (семян хлопчатника, хлопкового жмыха, шрота, гузопаи и др.) 0,005, 0,01, 0,03 мл, а из пробы комбикорма 0,03—0,05 мл. В 1,5 см от проб наносят стандартный раствор госсипола 0,01—0,02 мл. После испарения хлороформа пластинку помещают в камеру с подвижным растворителем для разгонки при температуре окружающей среды не ниже 18 °C. Фронт растворителя должен пройти 10 см. После извлечения пластинки из камеры отмечают границу фронта растворителя. Пластинку помещают в горизонтальное положение для испарения растворителей, на что уходит 15—20 мин в зависимости от температуры окружающей среды. Высохшую пластинку опрыскивают проявителем дважды с интервалом 30 мин.
В случае наличия большого количества госсипола в изолированном остатке и невозможности разделения в нанесенных количествах бюкс оставляют открытым для испарения хлороформа и полученный сухой остаток вновь растворяют в 2 мл и более хлороформа и повторяют хроматографический анализ до полученного четкого разделения пятна. Проявление красно-малинового пятна спустя 10—25 мин на одних уровнях значения стандарта и пробы позволяет провести полуколичествеп.чое определение госсипола путем сравнения размера и интенсивности окраски пятна на испытуемой пробе со стандартом. Rf госсипола 0,40
Содержание вещества в анализируемой пробе вычисляют по формуле
0,1 -АВ А =-------.
Бт
где X — массовая доля свободного госсипола, %; Л —содержание госсипола, найденное на хроматограмме пробы путем сравнения со стандартом, мг; Б — объем экстракта, взятый для хроматографирования, мл; В — общий объем растворенного экстракта, мл; т — навеска анализируемой пробы, г; 0,1—коэффициент перевода в проценты.
Определение синильной кислоты (качественная реакция]
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Извлечение синильной кислоты. Выделение синильной кислоты из исследуемого материала проводят перегонкой с водяным паром. 100—150 г измельченного патологического материала (взятого в течение 2—3 ч после смерти животного) смешивают с дистиллированной водой до консистенции жидкой кашицы, подкисляют 10%-ным раствором винной кислоты (С6Н6О6) и переносят в круглодонную колбу. Затем колбу присоединяют к холодильнику. Отдельно нагревают до кипения парообразователь и, когда весь прибор собран, присоединяют парообразователь и начинают подогревать баню под колбой. Перегонку следует вести медленно. Полученный дистиллят исследуют.
Реактивы. 10%-ный раствор соляной кислоты. 5%-ный раствор сульфата закисного железа (FeSO4). 5%-ный раствор хлорида окисного железа (FeCl3). 10%-ный раствор винной кислоты. 10%-ный раствор едкого натра.
Качественная проба. Реакция образования берлинской лазури: из приемника берут 2—4 мл дистиллята, к нему добавляют 10%-ный раствор едкого натра до получения щелочной реакции (1—2 капли), затем прибавляют по 3 капли свежеприготовленных насыщенных растворов сульфата закисного железа и хлорида окисного железа, тщательно взбалтывают, подогревают и осторожно подкисляют 10%-ным раствором соляной кислоты (4—6 капель). В зависимости от количества синильной кислоты появляется синий осадок или окрашивание от синего до сине-зеленого. Чувствительность реакции 20 мкг HCN в 1 мл раствора. Реакция специфична.
При очень малых количествах синильной кислоты окрашивание может появиться спустя 24—48 ч.
Качественное обнаружение синильной кислоты в жмыхах и шротах из плодовых косточек проводят по ГОСТ 13979.8—69.
Количественный анализ: берут определенную навеску измельченного материала и подвергают медленной перегонке с водяным паром. Дистиллят отбирают в один приемник, точно измеряют его объем и определяют количественное содержание вещества.
Определение глюкозидов в кормах (проба с фелинговой жидкостью)
(Одобрено Главным управлением ветеринарии Госагропрома СССР И ноября 1987 г.)
Основано на восстановлении меди, входящей в фелингову жидкость, при взаимодействии с глюкозидами до красной закиси меди.
Реактивы. Фелингова жидкость 4% ный раствор медного купороса смешивают с равным объемом раствора, состоящего из 20 г сегнетовой соли и 15 г едкого натра в 100 мл воды Спирт этиловый 96° ный Спиртовой раствор виннокаменной кислоты
100—200 г измельченного материала помещают в колбу, заливают 96 ° ным спиртом и подкисляют спиртовым раствором виннокаменной кислоты Содержимое колбы взбалтывают и через 1 ч проверяют ее реакцию лакмусовой бумажкой При щелочной реак ции в жидкость добавляют еще раствор виннокаменной кислоты и через 1 ч снова проверяют реакцию Если реакция кислая, то колбу закрывают ватной пробкой и оставляют на сутки при тем пературе 25—30 °C время от времени взбалтывая содержимое Проверяют вытяжку на наличие кислой реакции и осторожно сливают, а оставшийся в колбе материал снова заливают спиртом и оставляют на вторые сутки Спиртовые вытяжки объединяют и фильтруют Остаток пробы переносят из колбы на фильтр и дваж ды промывают спиртом, который присоединяют к вытяжкам Полученную вытяжку выпаривают в фарфоровой чашке на водяной бане при температуре 40 °C до состояния жидкого сиропа
К упаренной вытяжке для осаждения белков приливают по каплям при помешивании этиловый (96°) спирт до прекращения образования осадка После отстаивания жидкость фильтруют и выпаривают до густоты жидкого сиропа Осаждение белков проводят до тех пор пока прибавление спирта не будет давать мути После этого жидкость упаривают до полного удаления спирта, а остаток растворяют в 50 мл воды Если раствор получается мутным, его фи льтрг ют
Испытуемый раствор кипятят с разведенной серной или соляной кислотой, для гидролиза глюкозидов и фильтруют К фильтрату добавляют жидкость Фелинга и кипятят 2—3 мин При наличии глюкозидов (точнее, сахаров, получающихся при гидролизе глюкозидов) образуется желтый осадок гщрата закиси меди, который при нагревании становится красным
Определение рицина в клещевинном, жмыхе методом агглютинации эритроцитов
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г )
Реактивы Физиологический раствор NaCl (0,85% ный), толуол Взвесь эритроцитов в физиологическом растворе NaCl кровь дефибринируют, взбалтывая в пробирке со стеклянными шариками, после дефибринирования 2 мл крови помещают в центрифужную пробирку, отмечая уровень налитой крови на стенке пробирки, при ливают 10—12 мл физиологического раствора NaCl, осторожно смешивают с кровью и центрифугируют при 3000 об/мин 5—8 мин Кровяные тельца оседают на дно Пипеткой отсасывают верхний слой жидкости, не касаясь осадка, вновь добавляют физиологический раствор и еще раз центрифугируют Если после второго центрифугирования верхний слой жидкости окрашен в бледно розовый цвет, то промывание и центрифугирование делают третий раз Промытый осадок эритроцитов разбавляют физиологическим
раствором до отметки на стенке пробирки, осторожно перемешивают, переносят в мерную колбу на 100 мл и доливают физиологический раствор до метки.
Ход анализа. 5—10 г измельченного клещевинного жмыха, взятого из средней пробы, обливают 25—50 мл физиологического раствора NaCl. В колбу приливают 0,5 мл толуола и, перемешав смесь, оставляют ее на 24 ч. После этого водную вытяжку отфильтровывают через бумажный фильтр. Полученный фильтрат будет основной вытяжкой, из которой готовят на физиологическом растворе следующие разведения: 1 : 100; 1 : 300: 1 : 500; 1 : 1000; 1 :2000; 1 : 10 000 и 1 : 15 000.
Для приготовления разведения 1 : 100 берут 0,1 мл основной вытяжки и 9,9 мл физиологического раствора. Последующие разведения готовят из разведения 1 : 100 по формуле
где а — показатель нужного разведения (300, 500, 1000 и т. д.); Ь—количество физиологического раствора, которое нужно добавить к 1 мл разведения 1 : 100 для получения нужной концентрации, мл.
Для приготовления разведения 1 : 10 000 и 1 : 15 000 можно брать из разведения 1 : 100 не 1 мл, а 0,1 мл, в связи с чем и физиологического раствора нужно будет в 10—15 раз меньше.
Для постановки реакции берут по 2 мл каждого разведения приготовленной вытяжки. В две пробирки наливают для контроля по 2 мл физиологического раствора Разведение отмечают на пробирках. Кровяную взвесь перед использованием осторожно перемешивают для получения равномерной эмульсии кровяных шариков. Во все пробирки добавляют по 2 мл эмульсии кровяных телец. Содержимое осторожно смешивают и оставляют на 24 ч в комнате’-при 16—18 °C. Через сутки пробирки слегка встряхивают и просматривают. При полной агглютинации кровяные тельца склеиваются и оседают на дно пробирки в виде зонтика, а жидкость над осадком полностью просветляется. При встряхивании пробирки в ней появляются крупные хлопья. Такая реакция должна быть в контрольных пробирках с вытяжкой из семян клещевины.
При частичной агглютинации на дне пробирки могут быть крупные хлопья и довольно интенсивное окрашивание жидкости. В случае неполной агглютинации жидкость бывает окрашена в красноватый цвет, а на дне имеются мелкие скопления эритроцитов.
При отрицательной реакции эритроциты оседают «пуговкой», а раствор просветляется.
Рицина в клещевинном жмыхе содержится значительное количество, если полная агглютинация произошла в пробирках с разведением 1 : 15 000 и более. Наличие полной агглютинации в пробирках с разведением в пределах 1 : 2000 и 1 : 10 000 соответствует среднему количеству рицина, наличие агглютинации только в пробирках с разведением в пределах 1:500 — 1:1500 — небольшому количеству, и, наконец, отсутствие агглютинации во всех пробирках характеризует полное отсутствие рицина.
Жмыхи, дающие агглютинацию в пределах 1:500—1:1500, следует давать животным в ограниченном количестве. Жмыхи, содержащие средние и повышенные количества рицина, должны подвергаться нагреванию при 150 °C или варке.
Определение соланина в картофеле
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Качественная проба (по В. Н. Ниловой). С клубня картофеля делают несколько срезов толщиной 1 мм: 1) от верхушки до основания по оси, делящей клубень на равноценные половинки; 2) поперечные — у основания и у верхушки клубня; 3) с боков; 4) в участках около глазков. Срезы помещают в фарфоровую чашку или на часозое стекло. На них наносят по каплям вначале крепкую уксусную кислоту (80—90%), затем концентрированную серную кислоту (уд. вес 1,84) и несколько капель 5%-ной перекиси водорода. Почти немедленно в местах среза, содержащих соланин, появляется интенсивное темно-малиновое или красное окрашивание.
Количественное определение.
Реактивы. Этиловый спирт 96°-ный. 1%-ный раствор уксусной кислоты. 5%-ный раствор аммиака. 1%-ный раствор аммиака.
Навеску в 30—50 г сухого, тонко размолотого клубня или ботвы картофеля экстрагируют несколько раз 100—150 мл спирта на водяной бане в колбе на 300—500 мл, соединенной с обратным холодильником; кипящим 96°-ным спиртом экстрагируют полчаса при частом взбалтывании. Через полчаса колбу охлаждают и содержимое фильтруют через воронку Бюхнера в бунзеновскую колбу. Осадок вновь помещают в колбу и экстрагируют 100—150 мл спирта. Эту операцию выполняют 3—4 раза. Все спиртовые вытяжки сливают вместе.
Колбу со спиртовой вытяжкой помещают на водяную баню и спирт отгоняют почти досуха. Остаток растворяют в 150 мл воды, подкисленный уксусной кислотой. Получившийся раствор центрифугируют, центрифугат сливают, остаток снова заливают 1%-ным раствором уксусной кислоты, взбалтывают и опять центрифугируют, промывание делают еще раз, центрифугаты соединяют.
К кислому центрифугату постепенно приливают 5%-ный раствор аммиака до щелочной реакции на лакмус и нагревают 30 мин на кипящей водяной бане. Выпадает хлопьевидный осадок соланина. Если во время нагревания аммиак улетучился, то необходимо его добавить.
Выпавший соланин центрифугируют, осадок растворяют в спирте, отфильтровывают, затем отгоняют спирт, осадок растворяют в подкисленной воде и после центрифугирования осаждают аммиаком. Такую чистку делают 2—3 раза. Последний раз соланин фильтруют через маленькие, предварительно высушенные до постоянного веса фильтры. Осадок на фильтре промывают 1%-ным раствором аммиака. Затем фильтры с осадком соланина высушивают в весовых стаканчиках при 100—105 °C, взвешивают и по весу рассчитывают процентное содержание соланина.
Определение сапонинов в растениях и кормах (гемолитическая проба]
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Реактивы. Физиологический раствор хлористого натрия (9 г хлористого натрия растворяют в 1000 мл дистиллированной воды).
5%-ная взвесь эритроцитов кролика (кровь помещают в стеклянный стаканчик, дефибринируют ее помешиванием в течение 15 мин деревянной палочкой). Фибрин удаляют, а плазму с эритроцитами фильтруют через 2 слоя марли. Фильтрат смешивают с 2—3 объемами физиологического раствора и центрифугируют в течение 8—10 мин. Прозрачную жидкость над осадком эритроцитов отсасывают и до прежнего уровня доливают физиологический раствор. Снова центрифугируют. Эту операцию повторяют до тех пор, пока жидкость над эритроцитами станет бесцветной. Затем берут 0,5 мл осадка отмытых эритроцитов и к ним добавляют 9,5 мл физиологического раствора.
В колбочку помещают 1 г измельченного сена, муки или отрубей, добавляют 10 мл физиологического раствора. Пробу сена ставят на 10 мин в кипящую водяную баню (помешивая), а пробы муки или отрубей экстрагируют 15 мин при комнатной температуре, периодически встряхивая. После этого фильтруют через бумажный фильтр. 2 мл фильтрата помещают в пробирку. В другую пробирку наливают 2 мл физиологического раствора. В обе пробирки добавляют по 0,5 мл 5°/о-ной взвеси эритроцитов. Встряхивают и оставляют на 5—10 мин. При наличии сапонинов в пробирке с фильтратом наступает гемолиз, в контрольной пробирке изменений не наблюдается.
ДРУГИЕ ЯДОВИТЫЕ ВЕЩЕСТВА
Методические указания по обнаружению металлических ядов в патологическом материале, кормах и воде с помощью экспрессных методов исследования (Извлечения)
(Утверждены Главным управлением, ветеринарии
Министерства сельского хозяйства СССР 26 августа 1964 г )
Реактивы и посуда Аммиак концентрированный (20—25% ный) Аммоний молибденовокислый Бензидин основной Бензол Днтизон Калий йодистый (калин йодид) Калий хлорат (бертолетовая соль) Кислота азотная (уд вес 1,22) Кислота лимонная Кислота серная концентрированная х ч Кислота соляная концентрированная х ч Кислота рубеановодородная Кислота уксусная концентрированная (80% пая) Медь сернокислая х ч Натрий кремнекислый Натрий виннокислый Натрии родизоновокислый (родизонат натрия) Нат рий фосфорнокислый (Na2HPO4 12Н2О) Пергидроль (30% ная перекись водорода) Ртуть бромная Свинец уксуснокислый Спирт ректификованный Тиомочевнна Цинк гранулированный (без мышьяка) Колбы Кьельдаля объемом 250—300 мл Пульверизато ры Пипетки, воронки химические Маленькие фарфоровые чашки Прибор Зангер Блека Вата гигроскопическая Фильтровальная бу мага Беззольные фильтры
Прибор Зангер Блека состоит из конической редукционной кол бочки объемом 50 мл и пришлифованной трубки, в нижней части которой имеется расширение для помещения свинцово ацетатной ваты, а вверху впаяна узкая газоотводная трубочка, на которую помещают кружочки бромно ртутной бумаги
Приготовление реактивов Раствор дитизона в бензоле 5 мг дитизона растворяют в 10 мл бензола Раствор приго ден для работы в течение одного дня
Фильтровальная бумага, пропитанная раствором тиомочевины Полоски фильтровальной бумаги размером 5X30 см пропитывают 4% ным раствором тиомочевины и сушат на воздухе Такие полоски бумаги можно хранить в плотно закрытой банке 3 мес
Раствор рубеановодородной кислоты 0,1 г рубеановодороднои кислоты растворяют в 10 мл ректификованного спирта Раствор сохраняется б дней
Фильтровальная бумага, пропитанная кремнекислым натрием Полоски фильтровальной бумаги размером 5X30 см пропитывают 4% ным раствором кремнекислого натрия и высушивают на воз духе Хранят в плотно закрытой банке Срок хранения 1 год
Йодистая медь 5,3 г йодистого калия растворяют в 10—15 мл дистиллированной воды, к полученному раствору' прибавляют 40 мл 10% ного раствора сернокислой меди, образующийся осадок отфильтровывают и пррмывают дистиллированной водой до полного обесцвечивания промывных вод Фильтр с осадком прокалывают и” лой, смывают осадок дистиллированной водой в колбу и доводят до объема 50 мл Взвесь йодистой меди пригодна для работы в течение 6 мес
Раствор родизоновокислого натрия. 0,05 г родизоновокислого натрия растворяют в 10 мл дистиллированной воды. Раствор пригоден для работы в течение 3 дней.
Фильтровальная бумага, пропитанная виннокислым натрием: полоски фильтровальной бумаги размером 5X30 см пропитывают 4%-ным раствором виннокислого натрия и сушат на воздухе. Хранят в плотно закрытой банке до 1 г.
Буферный раствор с pH 2,8. Раствор А: 35,628 г двуметаллического фосфорнокислого натрия (ХаоНРО,- 12Н2О) растворяют в воде и доводят до 1 л Раствор Б: 21,008 г лимонной кислоты растворяют в воде и доводят до 1 л. Для получения буферного раствора с pH 2,8 смешивают 1,58 мл раствора А с 8,42 мл раствора Б Исходные растворы сохраняют в течение 1 мес.
Бромно-ртутные бумажки: из беззольной фильтровальной бумаги вырезают кружочки по размерам трубки насадки на колбочку прибора Зангера-Блека Бумажные кружочки пропитывают 5%-пым спиртовым раствором бромной ртути, сушат на воздухе и хранят в плотно закрытой склянке из темного стекла до 6 мес.
Свинцово-ацетатная вата: гигроскопическую вату пропитывают в 5 %-ном растворе уксуснокислого свинца и сушат. Хранят в плотно закрытой склянке до 1 г.
Раствор молибденовокислого аммония: 5 г молибденовокислого аммония растворяют в 100 мл воды, добавив 35 мл азотной кислоты с удельным весом 1,22 (43%-ный раствор). Срок хранения 3 мес.
Раствор уксуснокислого бензидина: 0,05 г основного бензидина растворяют в 10 мл концентрированной уксусной кислоты (80%) и доводят водой до 100 мл Срок хранения 10 дней
В целях ускорения токсикологических исследований при проведении лабораторной диагностики отравлений животных металлическими ядами (медью, ртутью, цинком, барием и свинцом) разработаны экспрессные (ускоренные) методы обнаружения этих ядов в патологическом материале, кормах, воде и других объектах.!
Вышеуказанные металлы входят в состав широко применяемых в сельском хозяйстве ядохимикатов: парижской зелени, препарата АБ, хлорокиси меди, трихлорфенолята меди, бордоской жидкости, медного купороса, фосфида цинка, хлористого цинка, углекислого бария, хлористого бария, гранозана, меркурана, сулемы, этилмеркурфосфата, углекислого свинца, тетраэтилсвинца и др.
Для обнаружения с помощью экспрессных химических методов бария, свинца, цинка, меди и ртути (каждого в отдельности) требуется около 1 ч, включая разрушение патологического материала, кормов и других исследуемых объектов, а для исследования на фосфид цинка по фосфору и цинку — 2 ч.
Обнаружение металлических ядов — цинка, меди, бария и свинца — основано на применении метода осадочной хроматографии на бумаге, разработанного Вяхиревым и Кулаевым. Цинк определяют по реакции с дитизоном, медь — рубеановодородной кислотой, барий н свинец — родизоновокислым натрием. Обнаружение ртути проводят с помощью реакции с йодистой медью капельным методом на бумаге.
Метод осадочной хроматографии на бумаге позволяет обнаруживать искомый катион, находящийся в смесн с другими катионами, без выделения его в чистом виде. Присутствие других катионов в количествах, превышающих в 100 раз количество иско
мого катиона, не мешает его обнаружению методом осадочной хроматографии на бумаге (ртути — капельной реакцией с йодистой медью на бумаге).
Для определения цннка, меди и ртути применяют экспрессный способ неполного разрушения патологического материала концентрированной серной кислотой и пергидролем; для выделения бария и свинца применяют способ разрушения патологического материала концентрированной соляной кислотой и хлоратом калия. В обоих случаях разрушение навески (20—25 г) патологического материала для получения минерализата проводят за 30—40 мин.
Подготовка материала для исследования. Из патологического материала (содержимое желудка, зоба, кишечника со стенками, кусочки паренхиматозных органов, рвотные массы) и кормов отделяют 20—25 г для исследования на медь, ртуть и цинк, часть материала сохраняют для исследования на барий и свинец, на другие яды и для контроля. Если материал был консервирован спиртом или содержит много воды (рвотные массы, силос), то пробу помещают в фарфоровую чашку н подсушивают на кипящей водяной бане (для удаления избытка воды).
Для исследования на цинк, ртуть и медь разрушение материала проводят концентрированной серной кислотой и пергидролем. 20— 25 г патологического материала или корма помещают в колбу Кьельдаля объемом 250—300 мл, заливают 10—12,5 мл пергидроля, 1—2 мин перемешивают и прибавляют 6—7 мл концентрированной серной кислоты. Содержимое колбы разогревается и наступает бурная реакция. Когда реакция затихнет, колбу осторожно нагревают на электроплитке и прибавляют по 1—2 мл пергидроля до тех пор, пока содержимое колбы сделается прозрачным (полного разрушения органического материала при этом не происходит — остаются неразрушенными жир и продукты распада белков). Для разрушения 20—25 г материала требуется примерно 25—40 мин.
Для исследования на барнй и свинец разрушение патологического материала и кормов проводят концентрированной соляной кислотой и хлоратом калия (бертолетовой солью), чтобы избежать образования труднорастворимых сернокислых солей бария и свинца, выпадающих в осадок. Навеску материала в 20—25 г помещают в колбу Кьельдаля, заливают концентрированной соляной кислотой, нагревают и прибавляют небольшими порциями хлорат калия, пока жидкость не станет прозрачной. Разрушение 20—25 г материала происходит в течение 30—40 мин.
Обнаружение цинка. Часть минерализата (1—2 мл) разбавляют 1 : 1 дистиллированной водой, берут 2—3 капли и нейтрализуют концентрированным аммиаком (по лакмусу), одну каплю нейтрализованного раствора наносят на полоску фильтровальной бумаги, пропитанной тиомочевиной и высушенной, и держат 1 мин над горлом склянки с концентрированным аммиаком, высушивают на воздухе и опрыскивают из пульверизатора раствором дитизона в бензоле. При наличии цинка на бумаге появляется пятно розоватокрасного или красно-малинового цвета.
В контрольном 'опыте вместо минерализата берут дистиллированную воду и проделывают все операции основного опыта. Пятно на бумаге не должно окрашиваться в розовый или красно-малиновый цвет. Нормально присутствующий в органах цинк при этих условиях не обнаруживается.
Обнаружение меди. 2—3 капли минерализата нейтрализуют концентрированным раствором аммиака, каплю нейтрализованного раствора наносят на полоску фильтровальной бумаги, пропитанной раствором кремнекислого натрия и высушенной, держат над горлом склянки с концентрированным аммиаком, подсушивают и опрыскивают из пульверизатора раствором рубеановодородной кислоты. В присутствии меди пятно окрашивается в темно-зеленый цвет.
В случае сомнительной реакции на медь с целью повышения чувствительности метода следует взять 3—5 мл минерализата и выпарить в фарфоровой или кварцевой чашке на электроплитке с добавлением нескольких капель пергидроля для обесцвечивания темнеющей жидкости и проделать реакцию на медь, как указано выше.
Обнаружение ртути. На беззольную фильтровальную бумагу наносят каплю взвеси йодистой меди, выжидают 2—3 мин и наносят на это место каплю минерализата. В присутствии ртути появляется красное или красно-оранжевое окрашивание. Реакция высокочувствительная, ею можно обнаруживать 0,25 мкг ртути в одной капле.
Обнаружение бария. 3—5 капель минерализата нейтрализуют концентрированным аммиаком, одну каплю жидкости наносят на полоску фильтровальной бумаги, пропитанную 4°/о-ным раствором виннокислого натрия и высушенной, подсушивают на воздухе и опрыскивают из пульверизатора раствором родизоната натрия, а затем буферным раствором с pH 2,8.
В присутствии бария пятно окрашивается в оранжево-красный цвет. Присутствие в минерализате цинка, меди, ртути и железа в 100-кратном количестве по отношению к барию не мешает его обнаружению этим методом.
Для дифференциации бария от свинца на оранжево-красное пятно от родизоновокислого бария наносят каплю 10%-ного раствора сернокислого натрия, оранжево-красная окраска исчезает; если же присутствует свинец, окраска пятна не меняется.
Обнаружение свинца. Каплю горячего минерализата (при охлаждении минерализата хлористый свинец выпадает в осадок) наносят на полоску бумаги, пропитанной 4%-ным раствором виннокислого натрия и высушенной, держат над горлом склянки с раствором крепкого аммиака до полной нейтрализации соляной кислоты, подсушивают и опрыскивают из пульверизатора раствором родизоната натрия, а затем буферным раствором с pH 2,8.
В присутствии свинца пятно окрашивается в фиолетовый цвет, а при незначительном количестве свинца — в красно-оранжевый.
Для дифференциации бария от свинца на оранжево-красное пятно, образовавшееся от родизоновокислого бария, наносят каплю 10%-ного раствора сульфата натрия. Оранжево-красная окраска исчезает. Если же присутствует свинец, цвет пятна не меняется.
Определение фенола
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Исследуемый объект (вода, измельченное содержимое желудка и др.) в количестве не менее 500 мл или 500 г помещают в колбу и подкисляют уксусной кислотой до кислой реакции по лакмусу и 264
затем производят перегонку с водяным паром. Для этого колбу устанавливают наклонно (для предотвращения выбрасывания жидкости из колбы в холодильник) и закрывают пробкой с двумя пропущенными через нее стеклянными трубками, причем одну трубку, доходящую до дна колбы, присоединяют к парообразователю, а другую, отводную трубку — к холодильнику Либиха. Образующийся дистиллят исследуют.
Реактивы. Бромная вода. 5%-ный раствор хлорного железа. Эфир. Винный спирт. Салициловая кислота. 10%-ный раствор йодистого калия. 0,1 н. раствор гипосульфита.
Качественные реакции. 1. К раствору прибавляют бромной воды; появляется белый осадок или муть трибромфенола (кристаллы— при исследовании под микроскопом). Реакция чувствительная, но неспецифична. Салициловая кислота, анилин, крезолы дают такую же реакцию. Имеет значение ее отрицательный результат (отсутствие фенола).
2. К пробе прибавляют 1—2 капли 5%-ного раствора хлорного железа; при наличии фенола образуется синее или сине-фиолетовое окрашивание. Окрашивание исчезает от прибавления кислот, избытка воды и винного спирта. Салициловая кислота дает такую же реакцию, но возникающая синяя окраска не исчезает от прибавления спирта. Реакция малочувствительна (1 : 1000).
Количественное определение.
Весовое определение. При достаточном количестве фенола к дистилляту или его определенной части прибавляют бромной воды до исчезающего желтого окрашивания (избыток брома). Осадок отфильтровывают во взвешенном тигле Гуча, промывают водой и сушат до постоянной массы в вакууме или при 90 °C в сушильном шкафу. Умножая найденное количество трибромфенола на 0,2839, находят количество фенола во взятом объеме дистиллята.
Объемное определение (для малых количеств фенола). Фенол извлекают эфиром после предварительного подщелачивания содой. Эфир испаряют, образовавшийся остаток растворяют в воде и к жидкости прибавляют разведенную бромную воду с определенным содержанием брома до появления желтой окраски. Спустя 15 мин прибавляют 10%-ный раствор йодистого калия и титруют выделившийся йод 0,1 н. или 0,01 н. раствором тиосульфата натрия (гипосульфита), т. е. определяют бром, не вступивший в реакцию с фенолом. Вычитая количество оставшегося брома из количества прибавленного брома, находят бром, израсходованный на реакцию с фенолом. 1 мл 0,1 н. раствора брома соответствует 0,003156 г фенола.
Определение поваренной соли в патологическом материале [по Мору)
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Аргентометрический метод (Мора). Метод основан на титровании иона хлора раствором нитрата серебра в присутствии хромата калия как индикатора. В нейтральных или слабощелочных растворах нитрат серебра образует с ионом хлора белый осадок хлорис
того серебра, и только после того, как весь нон хлора окажется связанным, избыток нитрата серебра реагирует с хроматом калия с образованием хромата серебра, осадка красно-кирпичного цвета: NaCl+AgNO3=AgC14-NaNO3; K2CrO44-2AgN03 = Ag2Cr04+ + 2KNO3.
Реактивы. 0,1 н. раствор нитрата серебра. 10%-ный раствор хромата калия.
Ход определения. Измельченный патологический материал (Юг) переносят в мерную колбу на 100 мл и доливают колбу до 3/4 объема дистиллированной водой, хорошо встряхивают и нагревают на водяной бане до 80 °C, после чего колбу снимают и ставят на 30 мин, периодически встряхивая. Затем колбу охлаждают под краном до комнатной температуры, доливают водой до метки, закрыв плотно пробкой, хорошо перемешивают. Фильтруют жидкость через сухой складчатый фильтр в сухой стакан или колбу. Отбирают пипеткой 25 мл фильтрата в коническую колбу емкостью 100 мл, приливают 1 мл 10%-ного раствора хромата калия и титруют 0,1 н. раствором нитрата серебра до появления неисчезающего кирпично-красного окрашивания.
Если вытяжка интенсивно окрашена и дальнейшее титрование затруднительно, рекомендуется взятую навеску материала подсушить на водяной бане, а затем обуглить до такого состояния, при котором содержимое тигля легко распадается прн надавливании стеклянной палочкой. Затем содержимое тигля переносят количественно (дистиллированной водой) в стакан и нагревают при помешивании до кипения. После остывания жидкость фильтруют в мерную колбу, доводят до метки и поступают далее, как описано выше.
Содержание хлорида натрия вычисляют ио формуле
а-0,005844./7, -100
Л'--- —-—-— --------,
be
где X — содержание хлорида натрия, %; а — количество 0,1 н. раствора нитрата серебра, израсходованное на титрование, мл; b — объем вытяжки, взятый для титрования, мл; %—общий объем вытяжки, мл; с — навеска объекта, г; 0,005844 — количество NaCl (г), соответствующее 1 мл 0,1 н. раствора нитрата серебра.
Определение хлорида натрия обратным титрованием по методу Фольгарда
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Оно основано на осаждении хлоридов избыточным количеством азотнокислого серебра и последующим оттитровыванием избытка AgNO3 роданидом аммония в присутствии железоаммонийных квасцов, взятых в качестве индикатора. По разности результатов двух титрований определяют объем раствора нитрата серебра, израсходованного на осаждение ионов хлора.
Последовательно протекают три реакции: AgNO3+NaCl = = AgCl+NaNO3; AgNO3+NH4SCN = AgSCN + NH4NO3; 6NH4SCN + + Fe2(SO4)3=2Fe(SCN)3+3(NH4)2SO4.
Титрование хлоридов по методу Фольгарда проводят в кислой среде и получают более точные результаты, чем при титровании хлорида натрия по методу Мора в нейтральной или слабощелочной среде.
Реактивы. 0,1 н. раствор нитрата серебра: 17 г нитрата сереб-ра+150—200 мл азотной кислоты+дистиллированная вода до объема 1000 мл. 0,1 н. раствор роданида аммония (NH4SCN): 7,6 г роданида аммония (препарат гигроскопичен) растворяют в 1000 мл дистиллированной воды. Насыщенный (40%-ный) раствор железоаммонийных квасцов FeNH4(SC>4)2. Азотная кислота, разбавленная 1 : 1.
Ход определения. Об извлечении хлорида натрия из объекта см. предыдущий метод. К 25 мл водного извлечения прибавляют 20—25 мл 0,1 н. раствора нитрата серебра, 1,5—2 мл раствора железоаммонийных квасцов, 5 мл азотной кислоты, разбавленной 1 : 1, и титруют раствором роданида аммония, приливая его медленно небольшими порциями при постоянном взбалтывании. Титрование прекращают после появления не исчезающего при энергичном взбалтывании коричнево-розового окрашивания.
Рассчитывают по формуле
v (А — В)-0,005844-^-100
Ьс
где X— содержание хлорида натрия, %; А — количество прибавленного 0,1 н. раствора нитрата серебра, мл; В — количество 0,1 н. раствора роданида аммония, пошедшего на титрование, мл; Ь — объем водной вытяжки, взятой для анализа, мл; bt — общий объем водной вытяжки, мл; с — навеска объекта, г; 0,005844 — количество хлорида натрия (г), соответствующее 1 мл 0,1 н. раствора нитрата серебра.
Комбикорма на содержание поваренной соли исследуют в соответствии с ГОСТ 13496.1—74 «Комбикорма, сырье. Методы определения содержания поваренной соли».
Обнаружение свободных минеральных кислот и щелочей
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Исследуемый материал (содержимое желудка, рвотные массы и т. д.) смешивают с небольшим количеством дистиллированной воды. Через 1—2 ч фильтруют. Для отделения белковых веществ фильтрат подвергают диализу. Диализ проводят следующим образом; у стакана с отрезанным дном затягивают дно животным пузырем, пергаментной бумагой или целлофаном, наливают в стакан исследуемый фильтрат и помещают в более широкий сосуд, наполненный дистиллированной водой, уровень жидкости в обоих сосудах должен быть одинаковым. Диализ проводят 5—6 ч, при этом воду в наружном стакане меняют 2—3 раза. Диализаты соединяют вместе, выпаривают на водяной бане до объема 5— 10 мл, исследуют на присутствие кислот и щелочей.
Обнаружение серной кислоты. Жидкость помещают в колбу, прибавляют медных опилок, соединяют с холодильником, на конец которого надет аллонж, опущенный в раствор йода в концентрированном йодистом калии. Колбу нагревают в масляной бане до температуры, при которой начинается реакция меди с серной кислотой, что будет заметно по просветлению окраски раствора йода в приемнике. Жидкость в приемнике подкисляют разбавленной соляной кислотой, нагревают до удаления йода и прибавляют 5%-ный раствор хлористого бария. В присутствии серной кислоты образуется белый осадок сернокислого бария.
Обнаружение соляной кислоты. К 1 мл диализата прибавляют 1 мл 2%-ного раствора азотнокислого серебра и 1—2 мл азотной кислоты. В присутствии иона хлора выпадает творожистый белый осадок хлористого серебра, растворимый в аммиаке.
Обнаружение азотной кислоты. Часть диализата выпаривают с кусочками белых шерстяных ниток. При этом шерсть окрашивается в желтый цвет, переходящий от аммиака в оранжевый. Кроме азотной кислоты шерсть окрашивается в желтый цвет и пикриновой кислотой. При этом будет окрашена в тот же цвет и жидкость.
Каплю исследуемой жидкости смешивают с 2—3 каплями раствора дифениламина в концентрированной серной кислоте. В присутствии азотной кислоты появляется синее окрашивание. Реакцию дают азотная и азотистая кислоты, а также другие окислители.
Количественное определение. В испытуемую пробу прибавляют в избытке цинковую пыль. Колбу с пробой соединяют с холодильником и приемником, в который налито 25—30 мл 0,1 н. раствора серной кислоты. Колбу с жидкостью кипятят 30—40 мин. Азотная и азотистая кислоты восстанавливаются в аммиак, поглощаемый титрованным раствором серной кислоты, к которой прибавлен индикатор метиловый красный. Избыток кислоты оттитровывают 0,1 н. раствором едкого натра и по разности находят количество серной кислоты, вступившей в реакцию с аммиаком.
1 мл 0,1 н. раствора серной кислоты соответствует 0,0017 г аммиака или 0,006302 г азотной кислоты.
Обнаружение едкого натра. Часть исследуемого фильтрата выпаривают почти досуха. Затем на платиновую петлю помещают капельку исследуемого сконцентрированного фильтрата, слегка подсушивают в верхней части пламени спиртовки, после этого вводят в пламя спиртовой горелки. При наличии едкого натра пламя окрашивается в желтый цвет.
Обнаружение едкого кали. К 1 мл исследуемого фильтрата добавляют 1 мл 10%-ного раствора винной кислоты. В присутствии едкого кали образуется белый кристаллический осадок, растворяющийся в разведенных кислотах и растворах щелочей.
Обнаружение аммиака. Часть фильтрата помещают в колбу с пробкой, к нижней поверхности которой прикреплены три бумажки: первая — красная лакмусовая, вторая — смоченная раствором сульфата меди, третья — смоченная щелочным раствором ацетата свинца. Посинение двух первых бумажек указывает на наличие аммиака. Почернение третьей, «свинцовой», бумажки указывает на наличие сероводорода. Этой реакцией открыть аммиак в гниющем материале невозможно.
Извлечения. ТУ 8-22-4—77. Концентрат карбамидный, технические условия
(Утверждено заместителем министра заготовок СССР 16 декабря 1977 г )
Определение содержания карбамида.
Колориметрический метод.
Принцип метода основан на экстракции карбамида водой, образовании окрашенных растворов прн взаимодействии с днметил-аминобензальдегидом и последующем колориметрированием
Аппаратура и реактивы Аппарат для встряхивания жидкости Весы аналитические н технические Воронки стеклянные Воронки Бюхнера Колбы мерные вместимостью 25, 50 и 100 мл. Пипетки стеклянные вместимостью 1, 5 и 10 мл Пробирки стеклянные вместимостью 10—15 мл Стаканы химические вместимостью 50 мл Фотоэлектроколориметр Фильтры бумажные Вода дистиллированная Калий железисто-синеродистый (желтая кровяная соль), 10,6%-ныи раствор Кислота соляная, концентрированная и раствор ее, приготовленный в соотношении 1 5 (1 часть кислоты и 5 частей дистиллированной воды) Кислота уксусная концентрированная Натрий едкии, 20%-ный раствор Спирт этиловый Уголь активированный Цинк уксуснокислый берут 21,9 г вещества в мерною колбу на 100 мл, добавляют 50 мл воды и 3 мл ледяной уксусной кислоты и доводят объем в колбе водой до метки Карбамид, рабочий раствор 100 мг карбамида растворяют в воде в мерной колбе на 100 мл (концентрация 1 мг/мл) Диметиламинобензальдегид
500 мг вещества помещают в мерную колбу на 25 мл, приливают 10 мл спирта этилового, 2,5 мл концентрированной соляной кислоты, доводят спиртом до метки и перемешивают Раствор готовят в день анализа
Очистка диметиламинобензальдегида 5 г вещества растворяют в 50 мл соляной кислоты, приготовленной в соотношении 1 5, приливают по каплям 20% ный раствор едкого натрия до появления белого густого осадка Затем содержимое переносят на воронку Бюхнера с бумажным фильтром, фильтруют прн вакууме и промывают водой до нейтральной pH Осадок высушивают в эксикаторе до постоянного веса
Построение калибровочной кривой Берут в пробирки 1, 2, 3, 4, 5, 6, 7 мл основного рабочего раствора карбамида с концентрацией 1 мг/мл и добавляют соответственно 9, 8, 7, 6, 5, 4, 3 мл воды (концентрация карбамида соответственно равна 0,1; 0,2, 0,3, 0,4, 0,5, 0,6, 0,7 мг/мл) Затем приливают по 1 мл раство ра диметиламинобензальдегида, перемешивают и через 2 мин измеряют оптическую плотность окрашенных растворов на фотоэлект роколориметре с сипим светофильтром (длина волны 400 нм) в кюветах с шириной граней 20 мм против контроля, приготовленного без карбамида (10 мл воды и 1 мл раствора диметиламинобензальдегида) На основании йзвестных концентраций карбамида и соответствующих им величин оптической плотности строят калибровочный график
Проведение испытания Берут 25 г измельченного карбамидного концентрата, помещают в мерную колбу вместимостью
500 мл, приливают 300 мл воды и встряхивают на качалке 5 мин Затем доводят объем водой до метки и перемешивают
2,5 мл экстракта переносят в мерную колбу вместимостью 50 мл, приливают по 0,5 мл раствора желтой кровяной соли и уксуснокислого цинка, доводят объем в колбе водой до метки и перемешивают После этого содержимое фильтруют через бумажный складчатый фильтр, на который предварительно помещают 1 г активированного угля
К. 10 мл фильтрата прибавляют 1 мл раствора димегиламино бензальдегида, перемешивают и через 2 мин колориметрируют, как указано выше
Обработка результатов Расчет содержания карбамида (X) проводят по формуле
X- 160
/7%. 1000 ’
Х%=С-40, X 1/кг=С-400,
где С — количество карбамида, найденное по калибровочной кри вой, мг, Y-—объем экстракта, мл, У1—объем экстракта, взятого Ht анализ, мл, — объем взятого на анализ экстракта при разведении, мл; II— масса навески, г, 100 — коэффициент пересчета в проценты; 1000 — коэффициент пересчета мг в г
Арбитражный метод.
Для проведения арбитражных испытаний используют уреазный метод с применением наиболее длительных приемов экстракции гидролиза карбамида, исключающих вероятность искажения результатов исследований
Метод основан на экстракции карбамида водой, гидролизе его ферментом уреазой, содержащейся в семенах сои, и последующем титровании соляной кислотой
Аппаратура и реактивы Баня водяная Бюретки вместимостью 2 и 5 мл Колбы мерные вместимостью 50, 100 и 500 мл Колбы конические вместимостью 100 мл Пипетки вместимостью 10 мл Весы технические Мельница лабораторная марки ЛЗМ Капельница. Цилиндры вместимостью 100 мл Вода дистиллированная Спирт этиловыи Соляная кислота, 0,1 и 0,5 н растворы Соя (измельчается до величины частиц диаметром не более 1 мм). Индикатор смесь равных количеств 0,2% ного спиртового раствора метилового красного и 0,1% ного спиртового раствора метиленового голубого
Проведение испытания Берут 25 г измельченного на лабораторной мельнице карбамидного концентрата в мерную колбу на 500 мл, приливают 300 мл воды и встряхивают на качалке 5 мин Затем доводят объем колбы водой до метки и перемешивают Из колбы берут 10 мл экстракта (над осадком) и переносят в мерную колбу вместимостью 50 мл, добавляют 1 г свеженз-мельченных семян сои и доводят до метки дистиллированной водой Полученную смесь встряхивают н в течение 2 ч выдерживают при комнатной температуре (содержимое колбы перемешивают через каждые 30 мин).
По истечении 2 ч отбирают пипеткой 10 мл экстракта (над осадком), переносят его в коническую колбу вместимостью 100 мл, прибавляют 5 капель индикатора и титруют 0,5 н. раствором соля
ной кислоты до перехода зеленой окраски экстракта в розово-фиолетовую.
Обработка результатов. Расчет содержания карбамида проводят по формуле
(К— 0,1)- KtKj.0,У15-100
Х% =(У —о,1). 15; Л' г/кг=(У-0,1).150,
где 0,1—количество 0,5 н. соляной кислоты, пошедшее на титрование экстракта пробы без карбамида, мл; 0,015 — количество карбамида, соответствующее 1 мл 0,5 н. соляной кислоты; Y — количество 0,1 н. соляной кислоты, пошедшее на титрование экстракта испытуемой пробы, мл; У]—объем экстракта первоначальный, мл; У" — объем экстракта, взятого на анализ, мл; У3— объем взятого на анализ экстракта при вторичном разведении, мл; У4 — объем экстракта, взятого на титрование, мл; Н — масса навески, г.
За окончательный результат испытания принимают среднее арифметическое показателей двух параллельных определений.
Допускаемые расхождения между результатами параллельных определений не должны превышать ±0,5%.
Методика определения меди в кормах, продуктах животноводства и патматериале
(Одобрена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 17 августа 1982 г.)
Принцип метода. Метод основан на минерализации биоматериалов минеральными кислотами с последующим фотоэлектроколо-риметрическим определением меди по реакции с тетраэтилтиурам-дисульфидом при 435 нм. Нижняя граница определения меди 0,5 мкг или при массе образца 2 г 0,25 мг/кг. В интервале концентраций элемента 0,8—1,6 мг/кг относительная ошибка определения не превышает 9,5 %
Аппаратура, материалы, реактивы и растворы. Фотоэлектроколориметр, имеющий синий светофильтр с максимумом пропускания при 435 нм или спектрофотометр. Кюветы с толщиной слоя 10 и 20 мм. Баня песчаная электрическая или электроплитка с закрытой спиралью. Весы лабораторные. Гомогенизатор или смеситель тканей типа РТ-2. Мельница лабораторная электрическая. Воронки делительные 100 и 250 мл. Воронки для фильтрования № 3. Колбы круглые плоскодонные термостойкие вместимостью 100 мл. Посуда мерная стеклянная лабораторная вместимостью: колбы мерные 25, 50, 100 мл; пипетки 0,1; 1, 2, 5 и 10 мл с делениями 0,002; 0,01 и 0,1 мл; цилиндры измерительные 25, 50 мл с притертыми пробками. Пробирки мерные с притертой пробкой на 5 или 10 мл.
Аммония гидробкись х. ч. Вода дистиллированная дважды перегнанная. Вата гигроскопическая медицинская свободная от металлов: 10 г ваты погружают в 300 мл теплой 0,2 н. соляной кислоты и выдерживают 24 ч; затем фильтруют или отжимают, промывают 500 мл бидистиллята 3 раза и высушивают.
Калия ферроцианид ч д а и 5% ный водный раствор Кислота азотная х ч плотностью 1,40 г/см3 Кислота соляная 0,2 н раствор Кислота соляная х ч плотностью 1,19 г/см3 Кислота хлорная х ч или о с ч плотностью 1,51 г/см3 (57%) Комплексон III (три лон Б) Комплексон III, 0,1 М раствор 16,81 г реагента растворяют в мерной колбе вместимостью 500 мл, используя бидистиллирован-ную воду и объем доводят до метки той же водой
Меди хлорид или сульфат х ч Меди стандартный раствор ос новной, 1 мг/мл взвешивают 67,31 мг СиС12 2Н2О или 98,91 мг CuSO4 5Н2О и количественно переносят в мерную колбу вместимо стью 25 мл, куда предварительно вносят 1 мл концентрированной хлорной кислоты Объем доводят до метки водой, перемешивают и хранят в холодильнике
Меди стандартный раствор рабочий, 5 мкг/мл, готовят разбав лением основного раствора бидистиллированной водой в 200 раз Раствор устойчив одну неделю Спирт этиловый ректификованный Тетраэтилтиурамдисульфид (тетурам, антабус) х ч или таблетиро ванный по фармакопее Р 71 945 19
Тетурама насыщенный раствор в этаноле в мерную колбу на 100 мл вносят 1 таблетку (0,25 г) препарата, добавляют 90 мл спирта и нагревают до кипения После охлаждения раствора объем доводят до метки спиртом и декантируют с осадка
Хлороформ х ч
Подготовка к испытанию
Подготовка посуды В колбы или другие сосуды вносят 1 мл этанола и 10 мл концентрированной азотной кислоты После бурного выделения окислов азота посуду выдерживают 30 мин при комнатной температуре Затем промывают дистиллиройанной водой и сушат при ПО °C Перед использованием посуду ополаскивают бидистиллированной водой
Подготовка измерительного устройства Фото этектроколориметр ФЭК М, ФЭК 56М или подобный с лампой на каливания включают за 30 мнн до измерений, устанавливая синий светофильтр с максимумом пропускания 435 им Возможно испопь зование спектрофотометра СФ 4а СФ 16 или подобного прибора
Отбор пробы Отбор проб производится в соответствии с Унифицированными правилами отбора проб для определения микро количеств пестицидов в сельскохозяйственной продукции, продук тах питания и объекта окружающей среды, утвержденными заме ститслем Главного государственного санитарного врача СССР 21 августа 1978 г
Качественные реакции При диагностике острых отравлений химико токсикологический анализ начинают с исследования содер жимого желудочно кишечного тракта С этой целью исследуемый материал смешивают с бидиститлированной водой, этиловым спир том и хлорной кислотой в соотношении 10 5 10 2 и через 10 мин фильтруют через бумажный фильтр К 5 мл фильтрата добавляют 1 мл насыщенного раствора тетурама в спирте Появление темно зеленого окрашивания свидетельствует о высоком содержании меди В качестве дополнительного теста используют реакцию с желтой кровяной солью Для этого в пробирку вносят 1 мл фильтрата, 5 капель концентрированной уксусной кислоты и 5 капель 5% ного раствора ферроцианида калия В присутствии меди появляется крас но коричневое окрашивание или осадок в результате образования взвеси коллоидального ферроцианида меди
Подготовка пробы. Исследуемый сухой образец массой 50—100 г растирают в мельнице. Органы и ткани животных, патма-териал измельчают ножницами, а затем гомогенизируют, используя пластмассовые стаканы и стеклянные ножи. Жидкие образцы перемешивают встряхиванием в течение 1 мин.
Средний сухой образец корма массой 1 ±0,001 г, сырой печени массой 0,54=0,001 г или крови (сыворотки) объемом 2,0+0,05 мл переносят в круглую колбу вместимостью 100 мл. Добавляют 10 мл концентрированной азотной кислоты и 0,2 мл этанола. После прекращения бурного выделения окислов азота колбу выдерживают на песчаной бане до просветления гидролизата и растворения основной массы материала. Затем вносят 5 мл 57 %-ной хлорной кислоты и нагревание усиливают. Разложение материала продолжают до появления белых паров хлорного ангидрида. Если в момент кипения хлорной кислоты раствор приобретает коричневый оттенок (обугливается), то быстро вносят 5—10 капель азотной кислоты и вновь доводят до стадии выделения хлорного ангидрида На конечной стадии гидролиза нагревание уменьшают, а минерали-заты выдерживают 5 мин на песчаной бане. После охлаждения содержимого в колбы вносят бидистиллированную воду до объема 14 мл Через весь ход анализа проводят контрольный опыт на чистоту реактивов
Построение градуировочного графика. Для построения градуировочного графика в ряд колб вместимостью 100 мл вносят 0; 0,5; 1; 2 и 3 мл рабочего стандартного раствора меди (5 мкг/мл) и по 5 мл 57 %-ной хлорной кислоты. Объем раствора доводят до 14 мл бидистиллированной водой. Затем вносят 1 мл 0,1 М раствора комплексона III, 13 мл этилового спирта и 4 мл насыщенного раствора тетурама в спирте. После перемешивания раствор выдерживают при комнатной температуре 30 мин. Затем раствор переносят в кювету с толщиной слоя 20 мм и измеряют оптическую плотность при 435 нм (синий светофильтр).
При анализе малых количеств меди (менее 10 мкг) применяют переэкстракцию комплекса в хлороформ. Для этого водно-спиртовой раствор переносят в делительную воронку вместимостью 250 мл, добавляют 5 мл хлороформа, 32 мл бидистиллированной воды и экстрагируют комплекс меди с тетурамом 1 мин, энергично встряхивая воронку. После разделения фаз нижний (органический) слой сливают в сухую мерную пробирку на 5—10 мл, фильтруя экстракт через небольшой тампон гигроскопической ваты. Объем экстракта доводят до 5 мл посредством промывания фильтра чистым хлороформом. Затем экстракт вносят в кювету с толщиной слоя 10 мм и измеряют оптическую плотность при 435 нм. Раствором сравнения служит контрольный опыт (экстракт без внесения стандартного раствора меди).
При построении градуировочного графика по оси абсцисс откладывают количество меди в мкг, а по оси ординат — соответствующие значения оптической плотности.
Проведение испытания. К гидролизату объемом 14 мл добавляют 1 мл 0,1 М. раствора комплексона III и 13 мл этилового спирта. Спустя 5 мйн при появлении осадка солей раствор фильтруют через бумажный фильтр в мерную колбу или цилиндр вместимостью 50 мл. Осадок на фильтре промывают 1%-ным раствором хлорной кислоты до первоначального объема раствора (28 мл). Затем вносят 4 мл насыщенного раствора тетурама в спирте, пе-
18 Заказ № 1995
273
ремешивая раствор. Колбы выдерживают при комнатной температуре 30 мин, а последующие стадии анализа выполняют сстлаено построению градуировочного графика.
Полученный окрашенный водно-спиртовой раствор переносят в кювету с толщиной слоя 20 мм и измеряют оптическую плотность раствора при 435 нм. Раствором сравнения служит контрольный опыт на содержание меди в реактивах, включая стадию минерализации. Если оптическая плотность опытного раствора больше оптической плотности максимального стандарта (15 мкг), то разбавляют окрашенный водно-спиртовой раствор 10%-ным раствором хлорной кислоты в 48 %-ном этаноле. Если гидролизат имеет желтый цвет, то фотометрирование раствора производят дважды (до и после внесения раствора тетурама) с последующим учетом неспецифического окрашивания исследуемою раствора. При малых количествах меди в растворе (менее 2.5 мкг) применяют переэкст-ракцию комплекса в хлороформ и повторное измерение оптической плотности органического экстракта при 435 нм в кювете с толщиной слоя 10 мм.
Обработка результатов. Для определения содержания меди в исследуемом материале применяют следующую формулу расчета:
где С — содержание меди в исследуемом материале, мг/кг или мг/л; А— количество элемента в растворе, найденное по градуировочному графику, мкг; К — коэффициент, учитывающий разбавление раствора в ходе анализа; А1 — масса образца, взятого для испытания, г или мл.
За результат анализа принимают среднее значение двух параллельных определений, выраженных с точностью до 0,01 мг/кг. Расхождение результатов параллельных определений не должно превышать 10%.
Методика флуориметрического определения селена в почве, кормах, органах (тканях] животных и продуктах животноводства
(Утверждена Гласным управлением ветеринарии
Министерства сельского хозяйства СССР 7 октября 1976 г.)
Селен халькофильный, редко встречающийся в природе элемент с атомной массой 78,96 В нормальных почвах и большинстве организмов содержание селена колеблется от 0,01 до 1 мг/кг. Более обогащены им пойменные почвы, черноземы, сероземы (0,3— 1 мг/кг;, в дерново-подзолистых и песчаных почвах 0,01—0,2 мг/кг.
Нормальное содержание селена в крови животных составляет 4—10 мкг%, а в печени—10—20 мкг%. Потребность животных в селене не превышает 50—100 мкг/кг рациона.
Высокие концентрации элемента в рационе животных (более 2 мг/кг) вызывают острые и хронические формы токсикоза. При этом ингибируются окислительно-восстановительные ферменты, нарушается синтез метионина и рост опорно-покровных тканей, наблюдаются анемия и цирроз печени.
Недостаток селена в кормах (менее 30 мкг/кг) приводит к появлению беломышечной болезни, некротической дегенерации печени и экссудативного диатеза, для предупреждения которых используют селенит натрия. При явлениях недостаточности концентрация элемента в органах и тканях падает до 1—3 мкг% на сырую массу, а в случае отравлений повышается до 40—100 мкг%. Наиболее высокое содержание селена встречается в почках. Для диагностики токсикозов и признаков селеновой недостаточности у животных проводят анализ кормов, печени, молока и крови.
Принцип метода. Метод основан на разложении биологических материалов смесью хлорной и азотной кислот и переведении селена до селенит-иона, который определяют по реакции с 2,3-диамино-нафтжшном флуориметрически. Чувствительность метода—10 мкг/кг объекта.
Реактивы, растворы и их приготовление. Аммиак водный х. ч. или ч. д. а. Вода дистиллированная, дважды перегнанная. Гексан, перегнанный на водяной бане. Гидроксиламин гидрохлорид. 2,3-диаминонафталин (ДАН). Калия йодид. Кислота азотная х. ч. или ч. д. а. Кислота соляная х. ч. или ч. д. а. Кислота хлорная 57 или 30%-ная х. ч. или ч. д. а. Крахмал растворимый. Натрий селенистокислый ч. д. а. Натрий сернокислый ч. д. а. Натрия тиосульфат фиксанал. Трилон Б х. ч. 12,5%-ный водный раствор аммиака. 2%-ный раствор крахмала свежеприготовленный. 6 н. раствор соляной кислоты. 0,1 н. раствор тиосульфата натрия. 0,2 М. раствор трилона Б, который готовят растворением 9,3 г реагента в 250 мл дважды перегнанной дистиллированной воды. 1%-ный водный раствор гидроксиламина гидрохлорида. Основной стандартный раствор селена (1 мг/мл). Для его приготовления берут 0,219 г селенистокислого натрия, растворяют в 100 мл бидистиллированной воды, подкисленной 1 мл концентрированной соляной кислоты.
Титр раствора устанавливают следующим образом: к 20 мл раствора селенистокислого натрия добавляют 80 мл воды, 10 мл 2%-ного раствора крахмала, 5 г йодистого калия и 10 мл концентрированной соляной кислоты. Колбу закрывают пробкой, ставят в темное место на 5 мин и выделившийся йод титруют 0,1 н. раствором тиосульфата натрия. Обязателен контрольный опыт на содержание селена в реактивах. Количество селенита натрия определяют с учетом, что 1 мл 0,1 н. раствора тиосульфата натрия, пошедшего на титрование, соответствует 1,974 мг селена.
Стандартные растворы селена в концентрации 1 мкг/мл и 0,1 мкг/мл готовят перед употреблением, разбавляя основной стандартный раствор (1 мг/мл) в 1000 и 10 000 раз бидистиллированной водой.
Раствор ДАН готовят следующим образом: в делительную воронку емкостью 250 мл вносят 50 мг реактива, добавляют 5 мл концентрированной соляной кислоты и 45 мл бидистиллированной воды. Содержимое перемешивают до полного растворения ДАН, затем вносят 50 мл гексана и встряхивают 2 мин. После разделения фаз нижний (водный) слой фильтруют через бумажный фильтр во флакон нз темного стекла Полученный раствор ДАН хранят в холодильнйке, а перед анализом необходимое количество этого реактива встряхивают с равным объемом гексана, используя для реакции водную фазу.
Приборы и посуда. Флуориметры, снабженные ртутнокварцевой лампой и светофильтрами: первичный — с максимумом пропускания 18* 275
366 нм, вторичный — с узким максимумом пропускания при 530 нм или с более широким (510—600 нм). Для прибора ЭФ-ЗМ применяют следующие светофильтры: первичный ФК-1, вторичный В2-2.
Колбы мерные емкостью 50 и 100 мл. Колбы круглые плоскодонные из стекла «пирекс» емкостью 50 мл. Пипетки на 1, 2, 5 и 10 мл. Делительные воронки емкостью 250 и 100 мл. Воронки для фильтрования № 3 и № 5.
Подготовка лабораторной посуды для проведения анализа. Посуду кипятят 30 мин в мыльном растворе, содержащем 20 г грилона Б, 20 г каустической соды и 20 г мыльной стружки или порошка на 5 л воды. Затем посуду промывают теплой водой, 3%-ным раствором уксусной кислоты, дистиллированной водой и сушат при 120 СС.
Примечание: при проведении анализа на содержание селена нельзя пользоваться хромовой смесью для обработки лабораторной посуды.
Ход анализа. 1 г тонко растертого или гомогенизированного образца (печень, комбикорм, сено, почва) или 2 мл крови, молока помещают в колбу емкостью 50 мл. Сухие образцы смачивают 5 мл воды. Затем в колбу добавляют 10 мл концентрированной азотной кислоты и через 12 ч вносят 5 мл 57%-ной или 10 мл 30%-ной хлорной кислоты. Смесь нагревают на слабом огне (газовая горелка или электроплитка с прокладкой асбеста). На данном этапе раствор нагревают до появления паров окислов азота, после чего нагревательный прибор отключают, а через 10—15 мин нагревание повторяют на сильном огне и разложение материала продолжают до появления белых паров хлорной кислоты. Если при этом раствор имеет коричневый оттенок, в него добавляют 2—3 мл концентрированной азотной кислоты и разложение материала повторяют. Затем колбу охлаждают, вносят 5 мл воды и вновь нагревают до появления белых паров хлорной кислоты. В этот момент колбу закрывают воронкой для фильтрования (№ 3) и нагревают еще в течение 10 мин. После охлаждения содержимого колбы в нее' вносят 20 мл воды, обмывая воронку. Полученный раствор фильтруют в колбу емкостью 50 мл через бумажный фильтр, промывая его 10 мл бидистиллированной воды (минерализаты можно хранить закрытыми в холодильнике до 7 дней). Разбавленный бидистиллированной водой минерализат переносят в делительную воронку емкостью 100 мл и извлекают мешающие флуоресцентному анализу примеси 5 мл гексана в течение 2 мин, сливая нижнюю (водную) фазу в ту же колбу (объемом 50 мл). К очищенному раствору добавляют 12,5%-ный раствор аммиака или разбавленную соляную кислоту (1 : 1) до pH 2 (по универсальной индикаторной бумаге) и вносят 2 мл 0,2 М раствора трилона Б и 2 мл 1 %-ного раствора гидроксиламина. Содержимое колбы каждый раз перемешивают и спустя точно 5 мин вносят 2 мл очищенного раствора ДАН (работы с применением ДАН следует проводить при рассеянном свете, в тени, а гексановые экстракты необходимо хранить в темном месте до измерения флуоресценции). Колбу с раствором закрывают воронкой для фильтрования, нагревают 20 мин на водяной бане при 80 °C, после чего охлаждают 30 мин при комнатной температуре. Затем раствор переливают в делительную воронку емкостью 100 мл, добавляют 6 мл гексана и экстрагируют комплекс селена с ДАН в течение 2 мин. После разделения фаз нижний слой отбрасывают, а органическую фазу
сливают в пробирку с притертой пробкой, обезвоживают добавлением сульфата натрия (0,5 г), переливают в кювету или пробирку флуориметра и измеряют интенсивность флуоресценции. Обязательно проводят анализ реактивов Величину флуоресценции контрольного опыта вычитают из значения, полученного для образца.
Для построения калибровочной кривой в ряд колб объемом 50 мл вносят по 5 мл 57 %-ной хлорной кислоты, стандартный раствор селена (0,05; 0,10; 0,15; 0,20 мкг) и 30 мл воды. Полученные растворы обрабатывают 5 мл гексана. Затем добавляют разбавленный водный раствор аммиака или соляную кислоту до pH 2 и далее поступают так же, как и при анализе проб.
Калибровочный график строят в координатах; концентрация селена в мкг — интенсивность флуоресценции в делениях шкалы прибора. Чувствительность прибора устанавливают таким образом, чтобы гексановый экстракт, содержащий 0,2 мкг комплекса селена с ДАН, давал отклонение стрелки гальванометра на всю шкалу Если интенсивность флуоресценции образца превышает интенсивность флуоресценции наибольшего стандарта (0,2 мкг), то органический экстракт разбавляют гексаном, а кратность разбавления учитывают при оценке содержания селена в 1 кг объекта
Содержание селена в образце рассчитывают по формуле
С-1000
где X — концентрация селена в образце, мкг/кг; С — результат, полученный по калибровочной кривой, мкг; Р — навеска пробы, г.
Извлечения из методических указаний по диагностике, профилактике и лечению отравлений сельскохозяйственных животных нитратами и нитритами
(Утверждены Главным управлением ветеринарии Госагропрома СССР 18 июня 1986 г.)
Общие положения. Применение азотных минеральных удобрений в растениеводстве по интенсивной технологии позволяет обеспечить более рациональное использование земли и при относительно меньших затратах средств производства и труда получить высокие урожаи зерновых, овощных, технических, кормовых и других культур, а также улучшить сенокосные угодья и культурные пастбища для продуктивных животных, что в совокупности должно способствовать обеспечению возрастающих потребностей населения в продуктах питания и промышленности сырьем.
В нашей стране применяются следующие азотные минеральные удобрения; нитратные удобрения (натриевая селитра, калийная селитра, кальциевая селитра); аммиачные удобрения (аммиак жидкий — безводный, аммиак водный, аммоний хлористый, аммоний сернокислый); аммиачно-нитратные удобрения (аммиачная селитра, аммиачно-известковая селитра, аммония нитрат сернокислый); амидные удобрения (кальция цианамид, мочевина — карбамид — синтетическая) ; комбинированные или полные удобрения включают азот, фосфор, калий (аммофос, аммофоска, нитроаммофоска и др).
В СССР установлено оптимальное внесение в почву азотных удобрений по азоту в количестве 100—120 кг/га под картофель
и овощи, 140 кг/га под посевы ржи, ячменя и пшеницы, 120 кг/га под культурные пастбища.
Нитратные удобрения хорошо растворимы в воде, поэтому при внесении в почву они быстро всасываются корневой системой растений и под воздействием ферментов нитратредуктазы, нитритре-дуктазы, гидроксиламинредуктазы и редуктазы окиси азота восстанавливаются до аммиака, который, взаимодействуя с а-кетоглюта-ровой кислотой, образует амиды аспарагиновой и глютаминовой кислот, способствующих при участии трансаминаз синтезу всех аминокислот и растительного белка (протеина), необходимого для роста растений и формирования их продуктивности.
Аммиачные и амидные удобрения первоначально подвергаются нитрофикации под воздействием почвенных нитрифицирующих бактерий с превращением в доступную для растений форму нитратов с последующим всасыванием через корневую систему и восстановлению до аммиака, используемого для синтеза аминокислот и протеина.
При скармливании растительных кормов жвачным животным под влиянием гидролитических ферментов рубцовой микрофлоры происходит окислительное дезаминирование растительного белка с образованием кетокислот и аммиака, участвующих в синтезе глютаминовой и других аминокислот, необходимых для синтеза животного белка, роста животных, образования мясной и молочной продукции. В рубце жвачных животных под влиянием фермента уреазы происходит расщепление мочевины на аммиак и углекислоту.
При скармливании жвачным животным азотфиксирующих растений (подсолнечник, кукуруза) в многокамерном желудке происходит превращение нитратов в нитриты и далее в гидроксиламин, окислы азота и аммиак не только за счет редуцирующих ферментов рубца, но и за счет растительных редуцирующих ферментов, поступивших вместе с кормами.
При нарушениях азотистого обмена в организме животных происходит образование весьма больших количеств аммиака, а также карбаминовой и карбамилфосфорной кислот, обладающих весьма высокой токсичностью.
Возрастающий объем применения азотных удобрений, нерациональное их использование, отрицательное влияние неблагоприятных погодных условий на процессы нитрификации в почве и денитрификации в расгениях. а также высокий уровень содержания нитратов и нитритов в воде водоемов для полива растений обусловливают накопление их в кормах сверхдопустимых количеств и возникновение случаев острых и хронических токсикозов животных, снижение санитарного качества и биологической полноценности продуктов животноводства.
В условиях широкой химизации сельскохозяйственного производства необходим комплексный общегосударственный контроль за предотвращением загрязнения кормов, продуктов животноводства и окружающей природной среды азотными удобрениями п их опасными метаболитами.
Условия накопления нитратов, нитритов и нитрозаминов в кормах и оценка их опасности для животных. Накопление нитратов и нитритов в почве и кормовых культурах обусловлено в основном трехкратным и двухкратным внесением повышенных количеств азотных удобрений, особенно в начальный период вегетации моло-278
дых растений и незадолго до сбора урожая, когда растения не способны метаболизировать их для синтеза протеина в оставшийся период вегетации.
Высоким накоплением нитратов отличаются азотфиксирующие растения: кормовая свекла, подсолнечник, кукуруза, картофель, капуста, люцерна, клевер, люпин, овес, ячмень особенно при внесении под их посевы натриевой и аммиачной селитры более 150 кг/га. Большое количество нитратов накапливается в растениях семейства маревых, зонтичных и крестоцветных.
Повышенное содержание нитратов в кормовых культурах отмечается в период засухи, на засушливых участках, при недостаточной инсоляции, при понижении температуры почвы и воздушной среды, при недостатке в почве молибдена, кобальта, серы и калия, при повышенной кислотности, засоленности почвы и внесении в почву больших количеств органических удобрений (жидкий навоз, куриный помет), поскольку все эти факторы резко понижают активность ферментов азотистого обмена — нитратредуктазы и нитритредуктазы.
Вредное влияние больших доз азотных удобрений обусловливает понижение всхожести и энергии роста семян; снижение сосущей силы корневой системы, всасывания воды и питательных веществ, синтеза аминокислот, каротина; накопление небелкового азота в побочной части растений — соломе, ботве.
Опасность азотных удобрений при их внесении в почву в повышенных количествах обусловлена накоплением не только нитратов в кормах, но и накоплением в почве и растениях нитрозаминов, обладающих высокой токсичностью и выраженным гонадотоксиче-ским, эмбриотоксическим, тератогенным и канцерогенным действием.
Нитрозамины (N-дифенилнитрозамин, нитрозодиметиламин, нит-розодиэтиламин и др.) поступают в азотные удобрения в процессе их изготовления при использовании нитрозных газов (окислов азота) при внесении в почву карбаматных пестицидов и гербицидов группы 2,4-Д, при распаде которых образуются амины, вступающие во взаимодействие с нитритами с образованием нитрозаминов.
Нитрозамины образуются также в силосе под влиянием окислов азота при силосовании кукурузы с повышенным содержанием нитратов, из которых под влиянием гидроксиламинредуктазы и редуктазы окиси азота образуются гидроксиламин и окислы азота, способствующие синтезу соответствующих нитрозаминов.
Эндогенные нитрозамины образуются в толстом отделе кишечника жвачных животных при взаимодействии нитритов и окислов азота с биогенными аминами, образующимися из аминокислот (лейцина, валина, орнитина и др.) под влиянием ферментов декарбоксилаз эшерихий коли, сальмонелл и других патогенных бактерий.
Возрастание токсичности кормов за счет накопления нитратов, нитритов, гидроксиламина, окислов азота и аммиака, обусловливающих отравление животных, нередко возникает в следующих случаях:
при скармливании корнеплодов свеклы и свекольной ботвы, подвергавшихся плесневению и гнилостной порче во время неправильного хранения или в период уборки, отравление животных возникает вследствие накопления нитритов под влиянием ферментов денитрифицирующих гнилостных бактерий;
при бесконтрольном скармливании недоброкачественного кукурузного силоса с высоким содержанием свободных окислов азота, гидроксиламина и аммиака, образовавшихся из нитратов под влиянием денитрифицирующих бактерий вследствие большого дефицита в силосной массе углеводов и недостаточного образования уксусной и молочной кислот, необходимых для подавления активности вышеуказанных денитрифицирующих бактерий;
при скармливании клубней свеклы и теплого отвара после ее длительной варки, обусловившей превращение нитратов в нитриты под влиянием редуцирующего влияния сахаров;
при скармлвваниг’ комбикормов и сенажа с повышенным содержанием нитратов, после добавления молочнокислых продуктов (заквасок) и содержания этих кормов в течение 18—24 ч при оптимальных температурных условиях, что ведет к интенсивному превращению нитратов в нитриты под влиянием оксидоредуктазных ферментов молочнокислых бактерий, что ведет к отравлению чаще всего телят и поросят;
при бесконтрольном использовании воды из глубоких колодцев с высоким содержанием нитратов и нитритов или из открытых загрязненных водоемов для водопоя или приготовления кормов нередко возникает отравление животных, особенно телят, жеребят, ягнят, поросят.
Токсическое действие. Азотные удобрения относятся к категории химических соединений повышенной опасности вследствие большого объема их применения (в 100 раз превышающего объем применения многих пестицидов), постоянного поступления их в организм животных с кормами, питьевой водой и образования в кормах и организме продуктивных животных высокотоксичных нитритов и весьма опасных нитрозаминов. Окислы азота и их предшественники (нитраты и нитриты) являются основными химическими мутагенами в окружающей природной среде.
Степень токсичности нитратов и нитритов характеризуется не только величинами ЛД50, установленными для чистых препаратов в эксперименте, но в основном суммарной токсичностью всех метаболитов, образующихся при ферментативном превращении (вместе с зелеными кормами) в многокамерном желудке жвачных животных до гидроксиламипа, окислов азота и аммиака. Нитриты более чем в 10 раз токсичнее нитратов. Жвачные животные более чувствительны к нитратам, а животные с однокамерным желудком — к нитритам. Молодняк более чувствителен, чем взрослые животные.
Голодные животные, а также больные колибактериозом н сальмонеллезом более чувствительны к нитратам, чем здоровые животные. Чувствительность телят к нитратам значительно возрастает при одновременном применении лечебных нитрофурановых препаратов (фуразолидона, фурагина) вследствие суммации токсического эффекта.
Отравление животных нитратами и нитритами наблюдается в случае поступления этих веществ в организм в концентрациях, выше предельно допустимых (табл. 1).
Механизм токсического действия нитратов заключается в превращении их в рубце жвачных животных в нитриты, гидроксиламин, окислы азота и аммиак по естественному метаболическому пути под влиянием окислительно-восстановительных ферментов: нитрит редуктазы, гидроксиламинредуктазы, редуктазы окислов азота.
1. Нормы содержания нитратов и нитритов в кормах для сельскохозяйственных животных (утверждены Главным управлением ветеринарии МСХ СССР 25.10.1982 и 10.06.1983 гг.)
Корма Нитраты по NOi, мг/кг Нитриты по ^о2) 1 МГ/I г Корма Нитраты по NOj, МГ/КГ Нитриты по NO3, мг/кг
Комбикорм для 500 10 Зеленые корма 200 10
крупного ро- Картофель 300 10
гатого скота Свекла 800 10
Комбикорм для 200 5 Силос (сенаж) 300 10
свиней и Зернофураж 300 10
птицы Жом сухой 800 10
Грубые корма 500 10 Травяная мука 800 10
(сено, солома) Жмых и шроты 200 10
Основным процессом токсического действия нитратов, нитритов, гидроксиламииа, окиси азота и нитрозаминов является блокада всех геминовых железосодержащих дыхательных ферментов за счет химического взаимодействия с двухвалентным железом (ферроформы) гемоглобина крови, миоглобина сердца и скелетных мышц и цитохромоксидазы нервной ткани с превращением железа в трехвалентную форму (ферриформу). В результате этого все вышеуказанные ферменты теряют способность воспринимать, транспортировать кислород и обеспечивать тканевое дыхание. В крови образуется высокий уровень метгемоглобина, вернее, нитрозогемоглобина, возникает острая гипоксия, истощение кислородной емкости крови, асфиксия. Блокада цитохромоксидазы обусловливает торможение транспорта электронов в дыхательной цепи цитрохромов, понижение образования молекулярного кислорода и острое нарушение функции центральной нервной системы (ЦНС).
Нитриты угнетают сосудодвигательный центр, что ведет к падению кровяного давления и коллапсу. Накопление аммиака вызывает возбуждение и паралич ЦНС. Все эти явления в совокупности ведут к быстрому летальному исходу.
Хроническая интоксикация животных нитратами обусловлена кислородным голоданием жизненно важных органов и тканей и сопровождается понижением обмена веществ, понижением воспроизводительной функции, снижением жизнеспособности молодняка, возникновением дистрофических явлений, нарушением обмена РНК и ДНК, понижением иммунологического состояния.
Диагностика отравлений. Диагностика отравлений животных нитратами и нитритами должна быть комплексной и основываться на анамнестических данных об уровне внесения в почву азотных удобрений под посевы кормовых культур, на характерных клинических симптомах интоксикации животных, результатах химико-аналитических исследований кормов, воды, содержимого желудочно-кишечного тракта и крови, а также на результатах патологоанатомического вскрытия павших и вынужденно убитых животных.
Предварительно необходимо исключить острые инфекционные заболевания, сходные по клиническому проявлению е отравлением нитратами.
Основными признаками для прижизненной дифференциальной диагностики нитратно-нитритных отравлений являются: наличие более 30% метгемоглобина в крови, шоколадный цвет крови, а также высокое содержание аммиака, окислов азота и нитритов в крови, носовой слизи, слюне и моче животных. Решающее значение имеет обнаружение нитратов в сверхдопустимых количествах в кормах и воде, использованных для кормления и водопоя животных.
Посмертная диагностика отравлений должна предусматривать обнаружение в пробах содержимого рубца или сычуга нитратов, нитритов, окислов азота и аммиака при одновременном определении метгемоглобина в свежих пробах крови.
Следует учесть, что при быстром течении отравления у телят после скармливания зеленой массы подсолнечника, кукурузы, клевера, люцерны и других легкобродящих кормов в рубце этих животных происходит быстрое и полное превращение нитратов и нитритов до аммиака вследствие активности редуцирующих ферментов до момента остывания трупа, поэтому пробы содержимого рубца должны быть взяты сразу после падежа или вынужденного убоя животных и исследованы на содержание нитратов и нитритов в течение до 2 ч или быстро помещены в холодильник до момента исследований.
Ветеринарно-санитарная экспертиза. Убой животных на мясо после перенесенного отравления следует производить пе ранее чем через 72 ч после клинического выздоровления. Мясо вынужденно убитых животных подлежит органолептическому, бактериологическому и биохимическому исследованиям с определением остаточных количеств нитратов и нитритов.
Доброкачественное мясо по всем показателям, но прн содержании в нем нитрат-иона до 100 мг/кг и нитрит-иона до 10 мг/кг можно использовать для пищевых целей как условно годное, требующее более длительного проваривания. При более высоком содержании нитратов и нитритов мясо может быть использовано для приготовления вареных сортов колбас при условии пятикратного разбавления мясом от здоровых животных.
Внутренние органы, кровь, желудочно-кишечный тракт и голова подлежат технической утилизации.
Молоко от коров и яйца кур, перенесших отравление нитратами, можно использовать для пищевых целей при получении их через 72 ч после клинического выздоровления животных.
Профилактика отравлений. Для надежной профилактики отравлений животных нитратами необходимо строго выполнять следующие мероприятия:
обеспечивать строгое храпение минеральных азотных удобрений в условиях, гарантирующих от контакта с животными, кормами, источниками водопоя, а также от загрязнения транспортных средств для перевозки животных и кормов;
норма внесения органических и минеральных азотных удобрений под кормовые культуры не должна превышать 150 кг (по азоту) на гектар с учетом природного запаса азота в почве;
перед массовым скармливанием зеленой массы растений и корнеплодов с новых посевных площадей, а также перед выгоном животных па пастбиша необходимо провести химико-аналитическое исследование проб кормов для определения содержания нитратов и нитритов. В необходимых случаях следует провести биопробу
па нескольких менее ценных животных, давая им корма вволю после 12—16-часового голодания с последующим наблюдением в течение 24 ч;
при обнаружении нитратов и нитритов в концентрациях, превышающих утвержденные предельно допустимые нормы, такие корма можно скармливать животным вместе с доброкачественными кормами, при условии если содержание этих веществ в рационе не будет превышать уровня ПДК с учетом их содержания также в питьевой воде. При расчете объема скармливаемых кормов следует учитывать, чтобы суточная доза нитратов в рационе и воде для водопоя не превышала для крупного рогатого скота 0,2 г; для лошадей и овец 0,4; для свиней 0,6; кроликов 1,0 и для кур 1,0 г нитрат-иона на 1 кг массы тела животных;
при содержании в кормовых растениях нитратов и нитритов свыше 0,2% зеленую массу следует засилосовать с добавлением 40% углеводсодержащих растений, п? закрывая бурты и ямы в течение 2--3 дней во избежание накопления высокотоксичных окислов азота, либо скосить траву на сено, переработать на травяную муку, оставить для получения семян или скосить после снижения концентрации нитратов в растениях до уровня ПДК;
необходимо постоянно приучать жвачных животных к выпаса-нию на пастбищах с повышенным внесением азотных удобрений и не допускать более чем 2-суточного перерыва при использовании таких кормов;
не выгонять голодных животных на пастбища и не допускать перекармливания жвачных животных зеленой массой азотфикси-рующих растений при стойловом содержании. Необходимо предварительно скармливать животным сухие корма с добавлением углеводов;
не допускать использования воды для приготовления кормов и водопоя животных из источников, содержащих более 1 мг/л нитритов и более 45 мг/л нитратов;
нельзя скармливать животным отвар после варки свеклы, а также недопустимо сдабривать молочнокислыми продуктами корма с повышенным содержанием нитратов.
Методика определения нитратов и нитритов в кормах, овощах, бахчевых культурах, крови, патологическом материале, молоке и молочных продуктах.
Принцип метода. Метод определения нитритов основан на фотометрическом измерении интенсивности окраски азосоединения розово-малинового цвета, образующегося при реакции нитритов с альфа-нафтиламином и сульфаниловой кислотой (реактив Грисса) в кислой среде после водного извлечения их из исследуемых проб.
Реакция специфична для нитритов.
Метод определения нитратов основан на водном извлечении их из исследуемых проб, количественном восстановлении нитратов в нитриты и последующем определении нитритов с реактивом Грисса.
Метрологическая характеристика метода. Предел определения нитритов составляет 0,05 мг/кг или мг/л, степень определения нитритов 90+10 ко-
предел определения нитратов составляет 0,5 мг/кг или мг/л, степень определения нитратов 83+17%.
Реактивы и растворы. Сульфаниловая кислота Ч д а Альфа-нафтиламин ч д а Барий сернокислый ч д а Цинковая пыль, порошок цинка Лимонная кислота х ч или ч д а Марганец серно кислый ч д а Натрий азотистокислый NaNO2 х ч Калий азотнокислый KNO3 х ч Сульфаминовая кислота х ч Уксусная кислота, концентрированная (ледяная) 12% ный водный раствор уксусной кислоты 1°/о ный водный раствор сульфаминовой кислоты Уголь активированный марки «Б» или уголь активированный в таблетках (аптечный) Натрия гидроокись х ч Цинк сернокислый ч д а
Приготовление реактива Грисса
а) 0,5 г сульфаниловой кислоты растворяют в 150 мл 12% ного раствора уксусной кислоты,
б) 0,1 г альфа нафтиламина растворяют (при нагревании) в 20 мл дистиллированной воды, фильтруют и смешивают со 150 мл 12% ного раствора уксусной кислоты
Перед } потреблением одну часть раствора «а» смешивают с равной по объему частью раствора «б»
Растворы «а» и «б» хранят отдельно в холодильнике, при не обходимости — в течение полугода
Приготовление стандартного раствора нитрита натрия 0,15 г высушенного до постоянной массы при температуре 80 °C NaNO2 растворяют в 1 л дистиллированной воды, свободной от нитритов
1 мл этого раствора содержит 0,100 мг (100 мкг) NO2 Раствор хранят в холодильнике, он устойчив в течение месяца
Приготовление рабочего раствора нитрита натрия
25 мл полученного станлартного раствора переносят в мерную колбу на 500 мл и доводят объем до метки дистиллированной во дой Раствор должен быть свежеприготовленным 1 мл его содержит 0 0050 мг (5 мкг) NO2 Этот раствор используют для по строения калибровочной кривой
Прш отовление стандартного раствора нитрата калия раство ряют 1 630 г нитрата калия, высушенного до постоянной массы при 105 °C, в дистиллированной воде в мерной колбе на 1 л и до водят объем раствора до метки дистиллированной водой
1 мл этого раствора содержит 1 мг нитрат ионов (NO3) Раствор можно хранить в холодильнике в течение 3 мес
Приготовление рабочего раствора нитрата калия
В мерную колбу на 100 мл переносят пипеткой 1 мл стандартного раствора нитрата калия и доводят объем раствора в колбе до метки 12% ным раствором уксусной кислоты
1 мл этого раствора содержит 0,010 мг (10 мкг) NO3 Раствор должен быть свежеприготовленным Этот раствор используют для построения калибровочной кривой
Пр (’овление реактива для восстановления нитратов в на ш тьг а) барий сернокислый (высушенный при 100 °C до постоянной массы) 100 г, б) марганец сернокислый (прокаленный при 200 °C 1 ч) 10,0 г, в цинковая пыль 2 г, г лимонная кислота 75,0 г, д) сульфаниловая кислота 4,0 г, е) альфа нафтиламин 2,0 г
Каждый реактив тщательно растирают в ступке, затем к реактиву «а» в ступке последовательно добавляют реактивы «б», «в» «д», «е», «г» и тщательно перемешивают до получения однородной массы
Полученный реактив хранят в склянке из темного стекла, он пригоден для использования в те' ение 2 лет
Приготовление 12%-ного раствора уксусной кислоты: 120 мл ледяной уксусной кислоты смешивают с 880 мл дистиллированной воды.
Приготовление 1%-ного раствора сульфаминовой кислоты: 1 г сульфаминовой кислоты растворяют в 99 мл дистиллированной воды.
Приготовление 0,5 М раствора гидроокиси натрия: 20г NaOH растворяют в небольшом количестве дистиллированной воды в мерной колбе на 1 л и доводят объем в колбе до метки дистиллированной водой.
Приготовление 5%-ного раствора сульфата цинка: 5 г сульфата цинка растворяют в 95 мл дистиллированной воды.
Приборы и посуда. Химические пробирки. Пробирки с притертыми пробками. Центрифужные пробирки. Пипетки на 1, 5, 10, 20 мл. Мерные колбы на 100 и 1000 мл. Конические колбы на 100, 250 мл. Химические стаканы на 100, 200 мл. Мешочки для диализа илн пленка для диализа № 3 (выпускается картонажными фабриками). Цилиндры на 10, 50 и 100 мл. Химические воронки. Песочные часы на 2 мин. Фотоэлектроколориметр. Центрифуга на 5000—6000 об/мин. Фильтры обеззоленные среднепористые.
Пленку или мешочки для диализа перед использованием для анализа помещают на 30—40 мин в дистиллированную воду. Во всех случаях при проведении диализа пленку следует брать с запасом по отношению к пробе так, чтобы узел мешочка при проведении диализа не был опущен в воду.
Построение калибровочных кривых.
Калибровочная кривая для определения нитритов. В 11 химических пробирок наливают рабочий раствор нитрита натрия в количествах, указанных в таблице 2. Затем в пробирки наливают дистиллированную воду до объема 10 мл, приливают по 1 мл реактива Грисса и энергично встряхивают.
2. Таблица для приготовления шкалы стандартов на нитриты
№ преби-Р'к Кол-во рабочего раствора, мл Содержание № пробирок Кол-во рабочего рас г-вора, мл Содержание NO2
мг МКГ мг МКГ
1 о,1 0,0005 0,5 7 1,2 0,006 6,0
2 0,2 0,0010 1,0 8 1,4 0,007 7,0
3 0,4 0,0020 2,0 9 1,6 0,008 8,0
4 0,6 0,0030 3,0 10 1,8 0,009 9,0
5 0,8 0,0040 4,0 11 2,0 0,010 10,0
6 1,0 0,0050 5,0
Для построения калибровочной кривой через 15 мин после начала окрашивания определяют оптическую плотность растворов на фотоэлектроколориметре при зеленом светофильтре (длина волны 540 нм, кюветы 10 мм1)., раствор для сравнения — дистиллированная вода.
Затем на оси абсцисс , откладывают количество нитрит-ионов (мкг), а на оси ординат — найденные значения оптической плотности растворов и проводят кривую через точки пересечения.
Эта же шкала стандартов может быть использована для визуального сравнения с опытными пробами.
Калибровочная кривая для определения нитратов. Для приготовления шкалы стандартов в 7 химических пробирок с притертыми пробками (или в 7 центрифужных пробирок с притертыми пробками) последовательно прибавляют по 0,3 г реактива для восстановления нитратов, затем приливают рабочий раствор и 12%-ную уксусную кислоту в количествах, указанных в таблице 3 (общий объем составляет 10 мл).
3. Таблица для приготовления шкалы стандартов на нитраты
№ пробирок Кол-во рабочего раствора, мл Содержание Кол-во 12 %-ной уксусной кислоты, мл
мг М' г
1 0,0 0,0 0,0 10,0
2 1,0 0,010 10 9,0
3 2,0 0,020 20 8,0
4 4,0 0,040 40 6,0
5 6,0 0,060 60 4,0
6 8,0 0,080 80 2,0
7 10,0 0,100 100 0,0
Затем пробирки или центрифужные пробирки) закрывают
пробками и энергично встряхивают в течение ровно 2 мин. Через 10 мин пробы центрифугируют в центрифужных пробирках при 5—6 тыс. об/мин в течение 5 мин. Далее проводят колориметрическое определение интенсивности окраски раствора при длине волны 540 нм, кюветы 10 мм, раствор для сравнения — 12%-ный раствор уксусной кислоты, обработанный, как пробы, сухим восстановителем и прошедший центрифугирование (пробирка № 1).
Затем на оси абсцисс откладывают количество нитрат-ионов (мкг), а па оси ординат — найденные значения оптической плотности растворов и проводят кривую через точки пересечения.
Эта же шкала стандартов может быть использована для визуальных определений.
6. Ход определения.
Корма. Для исследования измельчают пробу корма и берут навеску 2—10 г. Извлечение нитритов и нитратов из проб кормов проводят методом диализа. Для этого точно отмеривают 50— 100 мл дистиллированной воды и используют этот объем для всех дальнейших операций с пробой. Пробу помещают в мешочек для диализа, приливают туда небольшое количество дистиллированной воды из первоначального объема (50 или 100 мл). Воды приливают столько, чтобы она хорошо смочила пробу. Далее пленку завязывают в виде мешочка, весь оставшийся объем дистиллированной воды (от 50 или 100 мл) наливают в химический стаканчик, опускают мешочек в стакан таким образом, чтобы полностью погрузить пробу в воду и оставляют пробу для проведения диализа в течение 1,5—2 ч.
Затем, не вынимая мешочков из стаканов, пипеткой отбирают 10 мл диализата для анализа на нитриты и 5 мл — для анализа на нитраты и проводят анализ, как описано ниже.
Кровь, органы и ткани животных. Для анализа берут 20— 50 мл крови, 2—10 г органов или тканей животных, помещают пробу в диализный мешочек и опускают в химический стаканчик, в который наливают соответственно 50—100 мл прокипяченной горячей дистиллированной воды. Через 1,5—2 ч пипеткой отбирают из стакана 10 мл диализата для анализа на нитриты и 5 мл — для анализа на нитраты и проводят анализ, как описано ниже.
Если раствор в стакане после диализа имеет окраску или муть, такой раствор следует анализировать после удаления пигментов или мути.
Пигменты, окрашивающие диализат, удаляют путем фильтрования небольшого объема диализата через воронку с бумажным фильтром, заполненным на 2/з активированным углем. Уголь на фильтре предварительно промывают 50 мл прокипяченной дистиллированной воды, которую отбрасывают. Затем через фильтр отфильтровывают объем диализата, необходимый для анализа на нитриты и нитраты. В случае необходимости проводят повторное фильтрование этого раствора через свежую порцию активированною угля, промытого водой, до полного исчезновения окраски диализата.
При наличии в диализате мути проводят осаждение белков смесью 0,5 М раствора NaOH и 5%-ного раствора сульфата цинка в объемном отношении 2 : 5. Для этого отбирают из стакана 30 мл диализата, переносят его в другой стаканчик или колбу, добавляют 10—20 мл вышеуказанной смеси. После выпадения осадка диализат фильтруют, измеряют объем фильтрата и отбирают 5 мл для анализа на нитраты и 10 мл для анализа на нитриты.
В этом случае при расчете по формуле содержания нитратов и нитритов за общий объем фильтрата следует принимать сумму объема диализата, оставшегося в первом стаканчике (20 мл) и объема фильтрата, измеренного после осаждения белков и фильтрования.
Молоко, молозиво. Отбирают 20—50 мл молока или молозива, переносят в пленку для диализа п приливают туда 2—4 мл 12 %-ной уксусной кислоты, перемешивают содержимое стеклянной палочкой, завязывают пленку и опускают ее в стакан, содержащий 50—100 мл горячей прокипяченной дистиллированной воды. Затем через 1,5—2 ч отбирают 5 мл диализата для анализа на нитраты и 10 мл для анализа на нитриты и проводят анализ, как описано ниже. В том случае, если в растворе после диализа есть мугь, необходимо провести осаждение белков. Для этого отбирают 20—30 мл диализата, добавляют 10—20 мл смеси 0,5 М раствора NaOH и 5%-ного раствора сульфата цинка в объемном отношении 2 : 5. После выпадения осадка диализат фильтруют, измеряют объем фильтрата и отбирают пипеткой 5 мл для анализа на нитраты и 10 мл для анализа на нитриты.
Общий объем диализата при этом будет составлять сумму первоначального объема диализата, оставшегося в первом стакане и измеренного после фильтрования объема.
Сухое молоко, ЗЦМ. Для анализа берут 2—5 г пробы, помещают в пленку для диализа, хорошо смачивают пробу небольшим объемом дистиллированной воды, приливают туда 2—4 мл 12 %-ной уксусной кислоты, перемешивают стеклянной палочкой, завязывают пленку и опускают ее в стакан, содержащий 50—100 мл горячей дистиллированной воды, диализ проводят в течение 1,5—2 ч. При
расчете общего объема воды необходимо учесть объем воды, взятый для смачивания пробы.
Анализ проводят, как описано для проб молока.
Обрат. Для анализа берут 20—50 мл обрата, приливают к нему 20—50 мл дистиллированной воды, хорошо перемешивают в течение 10 мин и отбирают 5 мл для анализа на нитраты и 10 мл для анализа на нитриты. Если в растворе есть муть или опалесценция, необходимо проводить осаждение белков, как указано выше.
Картофель, сахарная свекла, огурцы, капуста, лук, редька и др. овощи и бахчевые культуры, дающие с водой неокрашенные растворы. После измельчения проб берут навеску 2—10 г, помещают в колб\' и заливают 50—100 мл дистиллированной воды. Перемешивают в течение 10—15 мин и отбирают аликвотные количества для анализа на нитраты и нитриты. В том случае, если раствор имеет окраску, проводят фильтрование небольшого объема экстракта через активированный уголь, как описано выше.
Проведение определения. После того, как в пробирку отобрано 10 мл диализата для анализа на нитриты, туда добавляют 1 мл реактива Грисса (раствор «а» смешивают с раствором «б» в равных объемных частях), встряхивают содержимое пробирки и через 15 мин после появления окраски проводят измерение оптической плотности раствора на фотоэлектроколориметре при зеленом светофильтре (длина волны 540 им) против дистиллированной воды в кюветах с рабочей длиной 10 мм.
Для анализа на нитраты отбирают 5 мл диализата в пробирку с притертой пробкой, в которую предварительно насыпано 0,3 г сухого восстановителя, приливают туда же 5 мл 12 %-ной уксусной кислоты, закрывают пробирку пробкой и энергично встряхивают ее в течение точно 2 мин.
В параллельную пробирку насыпают 0,3 г сухого восстановителя и наливают 10 мл 12%-ной уксусной кислоты, закрывают ее пробкой и встряхивают в течение 2 мин — пробирка для сравнения при измерении оптической плотности пробы.
Через 10 мин переливают содержимое пробирок в центрифужные пробирки и центрифугируют при 5—6 тыс. об/мин в течение 5 мин.
После этого проводят колориметрирование проб на ФЭКе при зеленом светофильтре (длина волны 540 нм, кюветы с рабочей длиной граней 10 мм). В качестве раствора для сравнения используют «холостой» раствор — раствор уксусной кислоты, обработанный сухим восстановителем, как описано выше.
В том случае, если в пробах устанавливается наличие большого содержания нитритов — свыше 20 мкг (интенсивная окраска в пробирке), их следует удалять, так как они несколько завышают результаты определения нитратов.
Для удаления нитритов к диализату в стакане, соответственно 50 или 100 мл, прибавляют 2—4 мл 1 %-ного раствора сульфаминовой кислоты, перемешивают раствор стеклянной палочкой и оставляют на 5 мин. После этого отбирают 5 мл диализата для анализа на нитраты и проводят анализ, как описано выше. При расчетах учитывают увеличение общего объема пробы.
После колориметрирования проб по соответствующим калибровочным кривым находят количество нитритов и нитратов, подстав-288
ляют их в формулу для расчета (в мкг) и определяют содержа-ние нитратов и нитритов.
Если при определении нитратов образуется очень интенсивная окраска, не укладывающаяся при определении оптической плотности в пределы калибровочной кривой, необходимо повторить определение, взяв меньшее аликвотное количество диализата для анализа, например 0,1 мл, и довести анализируемый объем до 10 мл 12 %-ной уксусной кислотой. Далее провести анализ, как указано выше, учитывая при расчетах разведение этого раствора.
Однако, если определяется очень высокое содержание нитратов (г/кг), необходимо повторить анализ данной пробы, соответственно уменьшая навеску и увеличивая объем дистиллированной воды для диализа. Можно также после проведения диализа в первый раз и установления высокого содержания нитратов прибавить в стакан к прежнему диализату, в котором находится мешочек с пробой, еще 100 мл дистиллированной воды и провести дополнительный диализ в течение 1 ч. После этого взять аликвотное количество для анализа и провести анализ на нитраты. При расчетах необходимо учесть это разведение диализата, в данном случае в 2 раза, т. е. общий объем диализата при этом составил не 100, а 200 мл.
Расчет результатов анализа проводят по формуле
где X — содержание нитритов (нитрит-ионов) или нитратов (нитрат-ионов), мг/кг; В — содержание нитрит-ионов или нитрат-ионов, найденное по соответствующим калибровочным кривым, мкг; А — навеска анализируемого образца, г; Yi—общий объем фильтрата, мл (соответственно 50, 100 мл или другие объемы с учетом раз-ведений); У2 — объем фильтрата, взятый для анализа, мл (5 или 10 мл соответственно).
Поскольку при диализе однородная равновесная концентрация минеральных солей устанавливается во всем объеме раствора (в стакане и в растворе в диализном мешочке), при расчете принимается весь взятый для анализа объем раствора или дистиллированной воды для диализа.
Фотоколориметрический гемиглобинцианид-ный метод определения метгемоглобина крови животных, в т. ч. птиц. Метод предназначен для химико-токсикологических, клинических и других исследований свернувшейся (сгустков) и несвернувшейся (цельной) крови на долю метгемоглобина в процентах по отношению к общему гемоглобину. Методы основаны на уменьшении величины оптической плотности содержащегося в исследуемом растворе крови метгемоглобина (гемоглобина) вследствие перевода его в цианметгемоглобин (гемиглобин-цианид) и вычисления процентного соотношения этой величины к такому же показателю 100%-ного метгемоглобина того же раствора крови.
Принцип определе'ния — фотоколориметрический по прямому определению (установлению) величины уменьшения оптической 610
плотности (DA ) раствора крови в одной кювете. Погрешность метода —не более 0,075% при нормальном и 0,227% при 71,1%-ном
19 Заказ № 1995
289
содержании метгемоглобина от общего гемоглобина. Выявляемое™ метода прн оптической плотности 0,001 составляет не более 0,3%.
Оборудование. Колориметр-нефелометр фотоэлектрический типа ФЭК-56 М или другие приборы подобного типа с наличием светофильтра с длиной волны 625—670 нм. Центрифуга лабораторная от 4 до 8 тыс. об/мин типа ЦЛН-2 или ЦУМ-1 и др. Колбы мерные вместимостью 25 см3. Пипетки на 1, 5 и 10 см3. Стаканчики стеклянные на 50 см3. Пипетки глазные. Палочки стеклянные или пластмассовые.
Реактивы и растворы. Ацетонциангидрин, 10%-ный раствор (объем/объем), годен до 1 года. Вода дистиллированная Калий железосинеродистый (соль красная кровяная) х ч. 10%-ный раствор (годен 30 дней при хранении в темном месте).
Подготовка к анализу.
Приготовление раствора крови. Из сгустка крови. От свежего трупа животного (птицы) или из патматериала (из сердца, крупных сосудов) берут в стаканчик сгусток крови массой около 1 г, добавляют 9 см3 воды и сгусток тщательно разминают чистым никелированным пинцетом до обесцвечивания комков фибрина. Затем комки фибрина удаляют, а в стаканчик с гемолизатом эритроцитов приливают 15 см3 воды.
Из цельной крови. В 25 см3 мерную колбу наливают около 20 см3 воды, 1 см3 (точно) гепаринизированной (декальцинированной трилоном Б или оксалатной) крови, промывают внутренний канал пипетки несколько раз содержимым колбы (избегая образования пены), объем гемолизата эритроцитов в колбе доводят водой до метки и тщательно перемешивают — разведение крови в 25 раз.
Гемолизаты из стабилизированной крови или сгустка крови переносят в капроновые центрифужные патроны, центрифугируют 15—20 мин при 8 тыс. об/мин и исследуют не позднее 1 ч после центрифугирования.
Проведение анализа. Устанавливают в рабочее положение светофильтр с длиной волны 630 нм. В левый кюветодержатель помещают 10-миллиметровую кювету с водой, а в правый — такую же кювету с 4 см3 центрифугата раствора крови и устанавливают правый измерительный барабан прибора точно на ноль (по красной шкале). Открывают световое окно с помощью рукоятки шторки и вращением левого барабана добиваются установки стрелки микроамперметра на ноль.
В кювету с раствором крови вносят 1 каплю раствора ацетонциангидрина (АЦГ) и перемешивают содержимое кюветы стеклянной палочкой. При этом взаимодействие метгемоглобина с цианидом (АЦГ) с образованием цианметгемоглобина заканчивается через 2—3 мин.
Через 2—3 мин после прибавления раствора АЦГ открывают световое окно прибора и правым измерительным барабаном устанавливают стрелку микроамперметра на ноль. По красной шкале правого барабана отсчитывают показатель уменьшения оптической плотности (Ц[Д) раствора крови.
Правую кювету освобождают от содержимого и 4—5 раз тщательно промывают водой. Затем ополаскивают кювету 0,5 см3 раствора крови, наливают 4 см3 того же раствора крови и устанав-290
ливают кювету в правый кюветодержатель. К раствору крови в кювете добавляют 1 каплю 10%-ного раствора красной кровяной соли (ККС), перемешивают другой чистой стеклянной палочкой и оставляют стоять 3 мин. За это время весь кровяной пигмент превращается в метгемоглобин.
Устанавливают правый измерительный барабан точно на ноль, а левый (не открывая светового окна), устанавливают на отметку 0,4—0,6 по красной шкале. Открывают шторку светового окна и левым барабаном точно устанавливают стрелку микроамперметра на ноль.
К раствору крови в кювете прибавляют 1 каплю раствора АЦГ, содержимое кюветы перемешивают стеклянной палочкой, устанавливают первый барабан на отметку 0,4 (грубая наводка, без открытия светового окна)
Через 2—3 мин после прибавления раствора АЦГ открывают световое окно и устанавливают правым измерительным барабаном стрелку микроамперметра на ноль. По красной шкале правого барабана снимают показатель уменьшения оптической плотности 630
(D2A) раствора крови, в котором весь гемоглобин был переведен в метгемоглобин.
Обработка результатов. Долю метгемоглобина (в %) к общему гемоглобину (общему пигменту) крови вычисляют по формуле
где DiA — показатель уменьшения оптической плотности раствора 630
крови после прибавления АЦГ; D2 А — показатель уменьшения оптической плотности 100%-ного метгемоглобинового раствора крови после прибавления АЦГ.
Оценка результатов. Для постановки диагноза при отравлении животных и птицы метгемоглобинобразующими ядами определение доли метгемоглобина к общему гемоглобину является решающим диагностическим показателем. Обнаружение в крови более 30% метгемоглобина указывает на возможность отравления, а более 60% — на возможную причину падежа животного от отравления метгемоглобинобразующими веществами (ядами). Обнаружение в крови животного нормального содержания (до 5%) метгемоглобина исключает потребность проведения химико-токсикологических исследований патматериала на метгемоглобинобразующие вещества (нитриты, бертолетовую соль, красную кровяную соль, анилин и др.). Однако для исключения отравления животных нитрат-ионами (без их редукции в нитриты) необходимо патматериал исследовать на нитраты.
Определение аммиака объемным методом
(Одобрено Главным управлением ветеринарии Госагропрома СССР 11 ноября 1987 г.)
Принцип. Аммиак отгоняют, поглощают 0,1 н. раствором серной кислоты, избыток которой оттитровывают 0,1 н. раствором ед-19* 291
кого натра. Метод используется для диагностики отравления животных карбамидом, аммиачной водой и др.
Ход определения. Для анализа берут 50 г содержимого рубца и помещают в колбу Кьельдаля объемом 1 л, прибавляют 200 мл воды, 0,3—0,5 мл изоамилового спирта и соединяют колбу с насадкой для прибора Кьельдаля и холодильником, конец которого опускают в коническую колбочку со 100 мл 0,1 н раствора серной кислоты и 3—4 каплями индикатора метилового красного
Колбу Кьельдаля нагревают и отгоняют аммиак Перегонку ведут до тех пор, пока капли дистиллята не перестанут окрашивать розовую лакмусовую бумажку в синий цвет. Избыток 0,1 н. серной кислоты титруют 0,1 н. раствором едкого натра и по разнице узнают количество миллилитров 0,1 н. серной кислоты, пошедшее на реакцию с аммиаком. Содержание аммиака в содержимом рубца рассчитывают по формуле
(А — 5).1,7-100
где X— содержание аммиака в содержимом рубца, мг%; А— количество 0,1 н раствора серной кислоты, взятое для поглощения аммиака, мл; В—количество 0,1 н раствора едкого натра, пошедшее на обратное титрование серной кислоты, мл; 1,7 — количество аммиака, соответствующее 1 мл 0,1 н. раствора серной кислоты, мг; 50 — навеска содержимого рубца, г.
Методические указания по диагностике отравлений рыб и сельскохозяйственных животных элементарным фосфором
(Утверждены Главным управлением ветеринарии
Министерства сельского хозяйства СССР 24 июня 1983 г.)
Введение. При выбросе промышленными предприятиями в окружающую среду отходов, содержащих элементарный фосфор, возможны отравления рыб и сельскохозяйственных животных Элементарный фосфор под действием воздуха быстро окисляется, превращаясь в фосфины, а затем в фосфаты, которые не представляют большой токсикологической опасности Однако элементарный фосфор, попадая в воду, может сохраняться в ней длительное время в пределах своей растворимости, вызывая отравление гидробионтов. При употреблении такой воды сельскохозяйственными животными возможны отравления. В организме рыб и сельскохозяйственных животных элементарный фосфор также быстро окисляется. Поэтому обнаружить его можно только в виде фосфинов в содержимом желудочно-кишечного тракта и печени. Для диагностики отравления необходимо прежде всего исследовать на наличие элементарного фосфора воду, затем рыб и других гидробионтов, у сельскохозяйственных животных — содержимое желудочно-кишечного тракта, печень, легкие и другие паренхиматозные органы. Обнаружение элементарного фосфора или продуктов его окисления в органах и содержимом желудочно-кишечного тракта погибших животных, рыб и в воде служит основанием для постановки диагноза иа отравление.
Определение элементарного фосфора в воде, рыбе, содержимом желудочно-кишечного тракта и паренхиматозных органах сельскохозяйственных животных.
Принцип метода. Рекомендуются два метода определения элементарного фосфора: экспрессный — по изменению окраски реактивной бумажки, пропитанной раствором азотно-кислого серебра, в приборе Зангер-Блека и фотоколориметрический — по интенсивности окрашивания цветного комплекса с молибдатом аммония.
Приборы, реактивы и посуда. ФЭК-56 М. Прибор Зангер-Блека. Колбы мерные 1-литровые. Холодильник шаровой. Плитки электрические закрытые. Пипетки разные. Колбы плоскодонные на 0,25, 1, 2 л. Колбы мерные на 50 мл. Мензурки. Стаканы химические. Воронки делительные. Пинцет анатомический. Прибор для приготовления отверстий в пробках (дырокол). Ножницы прямые. Фильтровальная бумага. Аммиак 1 : 10.
Раствор серной кислоты: к 850 мл дистиллированной воды прибавляют 150 мл серной кислоты с уд. весом 1,84 х. ч.
Сульфит натрия, 5 %-ный раствор, или 0,5%-ный раствор солянокислого гидроксиламина. Окислительная смесь: смешивают 0,2 и. раствор перманганата калия и раствор серной кислоты в соотношении 1 : 10 (смесь готовят перед употреблением). Молибденовый реактив: а) 10%-ный раствор молибдата аммония х. ч.; б) 50%-ный раствор по объему серной кислоты х. ч. Растворы смешивают в соотношении 1 : 3 по объему соответственно. Раствор олова 2-валепт-ного: к 250 мг хлористого олова (SnCl2) прибавляют 2,4 мл концентрированной соляной кислоты и доводят до 10 мл дистиллированной водой.
Раствор альфадинитрофенола — насыщенный водный раствор, приготовленный без нагревания. Бензол х. ч.
Азотнокислое серебро, 5%-ный водный раствор. Реактивная бумажка с азотнокислым серебром (кружок фильтровальной бумаги, вырезанный по размер}^ приемника прибора Зангер-Блека, погружают на 1—2 с в 5%-ный водный раствор азотнокислого серебра и тщательно подсушивают между слоями фильтровальной бумаги). Готовят перед использованием.
Виннокаменная кислота, 10%-ный раствор. Свинец уксуснокислый х. ч. Вата с уксуснокислым свинцом (готовят 5%-ный водный раствор уксуснокислого свинца, тщательно пропитывают им гигроскопическую вату, после чего ее высушивают в сушильном шкафу). Цинк без мышьяка. Концентрированная серная кислота х. ч. Соляная кислота х. ч., 2 н. раствор.
Ход определения.
Вода. Первый способ. Пробу воды объемом 100 мл заливают в приемную колбу прибора Зангер-Блека. Туда же помещают 3 гранулы цинка без мышьяка. В прибор Зангер-Блека вкладывают реактивную бумажку, пропитанную раствором азотнокислого серебра. Затем в колбу заливают 10 мл концентрированной серной кислоты. Прибор ставят на 1 ч.
После того, как таблетки цинка полностью перейдут в растворимую соль (1—2 ч), извлекают реактивную бумажку. При наличии элементарного фосфора или продуктов его неполного окисления в количестве 0,2 мкг бумажка приобретает коричневую окраску, 0,5 мкг — темно-коричневую, 1 мкг — черную. При наличии в
пробе 2 мкг окраска бумажки становится черной с металлическим блеском. _ ,п
Второй способ. 10 мл исследуемой воды помещают в делительную воронку, подкисляют 2 н. раствором соляной кислоты до з______4 заливают в воронку 40 мл бензола и проводят экстрак-
цию путём встряхивания воронки в течение 20—30 мин. Затем бензольный слой отделяют, промывают несколько раз дистиллированной водой до тех пор, пока промывные воды не будут содержать фосфат-иона (Р<№)- Для определения к 50 мл промывной воды добавляют 2 мл молибденового реактива и 2 капли раствора хлористого олова. Если интенсивность голубого окрашивания не отличается от холостой пробы, промывание заканчивают. 10 мл бензольного раствора помещают в мерную колбу емкостью 50 мл, добавляют в нее 10 мл окислительной смеси, закрывают колбу пробкой и встряхивают в течение 15 мин. Затем колбу помещают на водяную баню и отгоняют бензол. Избыток перманганата калия и выпавшую в осадок двуокись марганца восстанавливают добавлением Ш) каплям раствора безводного сульфита натрия или солянокислого гидроксиламина. Раствор кипятят на плитке в течение 2__з мин а затем охлаждают, добавляют 3 капли индикатора
альфадинитрофенола и по каплям аммиак, разведенный 1 : 10, до желтого окрашивания. Объем раствора доводят дистиллированной водой приблизительно до 45 мл, прибавляют 2 мл молибденового реактива Раствору дают постоять 10 мин, доводят его объем дистиллированной водой до метки и добавляют 2 капли раствора хлористого олова.
Через 15 мин измеряют оптическую плотность раствора по отношению к холостой пробе, содержащей все реактивы и проведенной через все этапы анализа. Оптическую плотность раствора измеряют на ФЭК-56 М с красным светофильтром. При низком уровне содержания фосфора используют кюветы длиной 50 мм, при повышенном___20 мм. Количество фосфора в аликвотной части опре-
деляют по калибровочной кривой.
Рыба внутренние органы животных, содержимое желудочно-кишечного тракта. Пробы рыбы, внутренних органов крупных животных массой 200 г тщательно измельчают, смешивают с дистиллированной водой до густоты кашицы. Содержимое желудочно-кишечного тракта отбирают в том же количестве без добавления воды Пробы помещают в круглодонную колбу емкостью 750— 1000 мл закрывают пробкой с двумя стеклянными трубками, одна из которых доходит до дна колбы и соединяется с парообразователем другая, короткая,—с холодильником. Форштос холодильника опускают в приемную колбочку. По окончании сборки прибора пробу подкисляют 15—20 мл 10%-ного раствора виннокаменной кислоты Отдельно нагревают парообразователь до кипения. Колбу с пробой ставят на водяную баню, соединяют с парообразователем и холодильником; содержимое колбы отгоняют до тех пор, пока объем дистиллята в колбочке не достигнет 50 мл Отгонку проводят медленно, регулируя скорость нагревания парообразователя Затем исследование 50 мл дистиллята продолжают так же, как и воды.
Построение калибровочной кривой. Основной эталонный раствор «А» в пересчете на элементарный фосфор
Навеску очищенной соли КН2РО4 (0,8775 г) растворяют в не
большом количестве дистиллированной воды в мерной колбе емкостью 1 л, после полного растворения соли доводят объем раствора до метки дистиллированной водой. Такой раствор содержит в 1 мл 0,2 мг фосфора. Его хранят в темном месте в плотно закрытой колбе или склянке. В таком виде раствор можно сохранять длительное время. Для приготовления серии рабочих эталонных растворов основной раствор разбавляют дистиллированной водой в 10 или 100 раз. Эталонные растворы готовят в день построения калибровочного графика.
Рабочий эталонный раствор: 50 мл основного эталонного раствора «А» помещают в мерную колбу на 500 мл и доводят до метки дистиллированной водой. Из этого раствора отбирают 10 мл, переносят в колбу на 100 мл и доводят объем колбы до метки. Получают раствор, содержащий 0,002 мг/мл фосфора. После этого готовят серию разведений для построения калибровочного графика. Для этого берут 10 мерных колбочек по 50 мл и в каждую из них вносят рабочий эталонный раствор в соответствии со следующей таблицей.
Номера проб
2 3 4 5 6 7 8 9 10
Кол-во рабочего 0,2 0,5 0,7 1,0 1,5 2,0 2,5 3,0 5,0 6,0 эталонного р-ра, мл
Кол-во внесенного 0,4 1,0 1,4 2,0 3,0 4,0 5,0 6,0 10,0 12,0 в пробу фосфора, мкг
Строят 2 калибровочных графика. Один для эталонных растворов с концентрацией ог 0,008 до 0,06 мкг/мл (колбы № 1—Ns 5). Для колориметрирования используют кюветы длиной 50 мм. Второй график для концентраций растворов от 0,028 до 0,24 мгк/мл (колбы № 3 — Ns 10) с использованием кюветы длиной 20 мм. На оси абсцисс откладывают количество фосфора в мкг, на оси ординат — показатель экстинции.
Расчет определения содержания фосфора в пробе. При анализе фосфора первым способом расчет производят по следующей формуле:
в '
где Р — количество фосфора, обнаруженное в исследованном объекте, мкг/кг или мкг/л; А — количество фосфора, обнаруженное в пробе, мкг; В — масса пробы, г.
При анализе пробы вторым способом расчет производят по следующей формуле:
aS,
где Р— количество фосфора, обнаруженное в исследованном объекте, мкг/кг или мг/л; А-—количество фосфора, найденное по калибровочному графику, мкг; В — объем бензола, взятый для экст
ракции, мл; Bi — объем бензола, взятый для колориметрирования, мл; а — масса воды или навески патматериала, г.
Чувствительность методики при определении фосфора в воде первым способом—0,002 мг/л, вторым — 0,017 мг/л, при определении в патологическом материале и содержимом желудочно-кишечного тракта — 0,001 и 0,008 мг/кг соответственно.
Методические указания по количественному определению муравьиной кислоты в мёде
(Утверждены Главным управлением ветеринарии
Министерства сельского хозяйства СССР 28 августа 1985 г., согласованы с Минздравом СССР 25 июля 1985 г.)
Общие положения. Методика предназначена для определения муравьиной кислоты в меде. Основана на извлечении муравьиной кислоты из раствора меда перегонкой с водяным паром. Количественное определение производится посредством взвешивания осадка, образующегося при восстановлении муравьиной кислотой двухлористой ртути до однохлористой; нижний предел определения 5 мг/кг.
Проведение исследований. Подготовка приемной колбы: в чистую колбу (500 мл) вливают 130 мл дистиллированной воды, по уровню жидкости делают отметку на стенке сосуда. Затем воду выливают, колбу высушивают, наливают в нее 50 мл 5%-ного раствора двухлористой ртути.
В перегонную колбу помещают 50 г меда, добавляют 100 мл теплой (45—50 °C) дистиллированной воды и тщательно перемешивают. Посредством обычной перегонной системы отгоняют муравьиную кислоту с паром из раствора меда в приемную колбу. Во время перегонки выходное отверстие холодильника должно быть погруженным в раствор двухлористой ртути в приемной колбе. Перегонку прекращают к моменту достижения в приемной колбе отметки жидкости (130 мл).
Приемную колбу закрывают пробкой с вмонтированным обратным холодильником и кипятят в течение 1,5 ч на слабом огне. По истечении этого времени содержимое колбы взбалтывают и выливают в воронку с предварительно взвешенным сухим фильтром. Для ускорения процесса фильтрации используют водоструйный насос. Колбу ополаскивают 100 мл горячей (80 °C) дистиллированной воды и выливают содержимое на фильтр. Осадок на фильтре последовательно промывают 30 мл этилового спирта, затем 30 мл серного эфира. Фильтр вынимают из воронки, высушивают в сушильном шкафу при температуре 90—100 °C до постоянной массы.
Учет результатов исследования. Содержание муравьиной кислоты в исследуемом меде вычисляют по формуле
97,5-Х-1000 “ М
где С — содержание муравьиной кислоты в исследуемом меде, мг/кг; X — количество осадка, образовавшегося при анализе навески меда, г; М — навеска исследуемого меда, г; 97,5 — постоянная величины, мг; 1000 — пересчет на 1 кг продукта (г).
Санитарная оценка меда. При обнаружении муравьиной кислоты в количестве до 500 мг/кг мед используют без ограничений, при обнаружении свыше этого количества и ощущении кислого привкуса мед направляют на кондитерские изделия.
Меры предосторожности. При проведении исследований соблюдают правила предосторожности, необходимые при работе с ядовитыми, взрывоопасными и легковоспламеняющимися веществами.
Методические указания по количественному определению щавелевой кислоты в мёде
(Утверждены Главным управлением ветеринарии
Министерства сельского хозяйства СССР 10 декабря 1984 г., согласовано с Минздравом СССР 28 ноября 1984 г.)
Общие положения. Метод предназначен для определения щавелевой кислоты в меде в ветеринарных лабораториях. Методика основана на выделении щавелевой кислоты из меда раствором хлорида кальция с последующим растворением осадка соли в серной кислоте и титрованием его раствором перманганата калия. Чувствительность метода 10 мг/кг.
Проведение исследований. В мерный цилиндр помещают 40 г меда, доливая теплую дистиллированную воду (40 °C) до объема 50 мл, добиваются получения гомогенной смеси, затем все переливают в большую центрифужную стеклянную пробирку, добавляют туда 5 мл 10%-ного раствора хлорида кальция и помещают в водяную баню (60 °C) на 10 мин.
После прогревания пробирку со смесью центрифугируют при 3000 об/мин в течение 10 мин, осторожно сливают надосадочную жидкость, приливают 30 мл дистиллированной воды для промывания осадка и повторно центрифугируют. Слив надосадочную жидкость, осадок растворяют в 10 мл 10%-ной серной кислоты.
Пробирку с растворенным осадком нагревают в водяной бане до 80 °C, испытуемый раствор переливают в коническую колбу и титруют 0,01 н. раствором перманганата калия до розового окрашивания, сохраняющегося в течение 20—30 с. Во время титрации температура реагирующей смеси должна быть 80 °C. Одновременно при тех же условиях титруют 10 мл 10%-ного раствора серной кислоты (контроль).
Учет результатов исследования. Содержание щавелевой кислоты в меде определяют, учитывая объем 0,01 н. раствора перманганата калия, пошедшего на титрование испытуемой пробы и контроля (1 мл 0,01 н. раствора перманганата калия соответствует 0,45 мг щавелевой кислоты).
Оценку содержания щавелевой кислоты в меде проводят по формуле
0,45-(77и — /7к )• 1000 '°=' м ’
где С — содержание кисл'оты в исследуемом меде, мг/кг; Пи — объем раствора перманганата калия, пошедшего на титрование испытуемой пробы, мл; Пк —юбъем раствора перманганата калия, пошедшего на титрование контроля, мл; М — навеска исследуемого меда, г.
Пример: навеска меда 40 г; на титрование испытуемого раствора пошло 6 мл 0,01 н. раствора перманганата калия; на титрование контроля пошло 0,3 мл 0,01 н. раствора перманганата калия. Тогда содержание щавелевой кислоты в меде составит (мг/кг)
0,45-(6— 0,3)-1000 С= —-----------—-------=64,1.
40
Санитарная оценка меда. При обнаружении щавелевой кислоты в количестве до 100 мг/кг мед используют без ограничений; при обнаружении щавелевой кислоты в количестве свыше 100 мг/кг партия меда с целью нейтрализации кислоты подвергается прогреванию при 45—50 °C при постоянном перемешивании в течение 1 ч с добавлением в начале процесса 10% -ного раствора СаОа. Необходимый объем раствора СаСЬ (мл) рассчитывают по формуле У = 0,013-СЛ4, где 0,013—постоянный коэффициент; С — содержание щавелевой кислоты в 1 кг меда, мг/кг; М — величина партии меда, кг.
Методика количественного определения перекисного числа в кормах животного и растительного происхождения
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 23 января 1984 г.)
Аппаратура, посуда, реактивы. Весы лабораторные аналитические Гомогенизатор лабораторный с емкостью стакана не менее 200 см3. Делительная воронка на 250 см3. Колбы стеклянные с притертой пробкой вместимостью 200—250 см3. Баня водяная. Ступка фарфоровая. Цилиндры лабораторные мерные вместимостью 10— 25 см3. Палочки стеклянные. Бумага фильтровальная Вата медицинская гигроскопическая. Чашки выпарительные фарфоровые. Эфир медицинский. Кислота уксусная ледяная. Калий йодистый. Хлороформ. Песок речной чистый, обезжиренный. Натр сернокислый обезвоженный. 0,01 н. титрованный раствор гипосульфита натрия. 1%-ный раствор растворимого крахмала.
Проведение испытания.
Исследование на содержание перекисей в мясокостной и рыбной муке. Навеску корма не менее 5 г, взвешенную с точностью 0,01, помещают в стакан гомогенизатора, куда добавляют 10 г сернокислого натра, 20 см3 хлороформа и 20 см3 ледяной уксусной кислоты. Взвесь гомогенизируют в течение 1—2 мин, затем надосадочную жидкость сливают в химический стакан и дают отстояться 5 мин. После отстаивания в колбу на 200 см3 отбирают 20 см3 верхнего слоя гомогенизата, добавляют туда 0,5 см3 свежеприготовленного насыщенного раствора йодистого калия и ставят в темное место. По истечении 3 мин в колбу приливают 50 см3 дистиллированной воды, в которую предварительно был внесен 1 см3 1 %-ного крахмала, и немедленно титруют 0,01 н. раствором гипосульфита натрия до исчезновения синей окраски.
Для проверки чистоты реактивов проводят контрольное определение, используя те же реактивы, но без навески.
Перекисное число, выраженное в % йода, вычисляют по формуле Х= (У—У])-0,00127-100, где Y—объем раствора гипосуль
фита натрия, пошедший иа титрование в рабочем опыте, см3; ¥>— объем раствора гипосульфита натрия, пошедший на титрование в контрольном опыте, см3; 0,00127 —количество йода, эквивалентное 1 см3 0,01 н. раствора гипосульфита натрия.
Исследование на содержание перекисей в комбикорме, жмыхе, шроте, зерне и других кормах. При определении перекисей в комбикорме, жмыхе, шроте, зерне и других кормах предварительно проводят экстракцию сырого жира из них, а затем определяют перекисное число в последнем. При этом навеску корма берут ориентировочно с расчетом экстрагирования не менее 1 г жира.
В делительную воронку вместимостью 250 см3 последовательно вкладывают фильтр, состоящий из плотного тампона гигроскопической ваты, двух кружков фильтровальной бумаги, диаметр которых несколько превышает диаметр делительной воронки, и вновь ватный тампон. Общая высота фильтра должна быть около 50 мм.
Навеску корма, взвешенную с точностью до 0,01, измельчают на лабораторной мельнице до мучнистого состояния, затем высыпают в химический стакан на 1000 см3 и туда же добавляют 100 г отмытого и обезжиренного речного песка. Смесь тщательно перемешивают стеклянной палочкой и затем высыпают в делительную воронку, укрепленную на штативе. Под воронкой устанавливают выпарительную чашку, предварительно высушенную до постоянной массы.
В делительную воронку постепенно, по мере фильтрации, приливают двойное количество эфира от взятой навески корма. Завершение экстракции жира контролируют фильтровальной бумагой путем смачивания вытекающей капли. В случае отсутствия на бумаге жирового пятна экстракция считается законченной. После этого выпарительную чашку ставят на водяную баню и выпаривают эфир до полного удаления его запаха.
Полученный жир в количестве 1 г, взвешенный с точностью до 0,01, переносят в коническую колбу с притертой пробкой емкостью 200—250 мл, туда же приливают 10 см3 хлороформа и 10 см3 ледяной уксусной кислоты. Затем в колбу быстро приливают 0,5 см3 свежеприготовленного насыщенного раствора йодистого калия и закрывают пробкой. Вращательным движением перемешивают содержимое колбы и ставят в темное место на 3 мин. По окончании выдержки в колбу приливают 100 см3 дистиллированной воды, в которую заранее был добавлен 1 см3 1 % -ного раствора крахмала, и немедленно титруют раствором 0,01 н. гипосульфита натрия до исчезновения синей окраски.
Для проверки чистоты реактивов проводят контрольное определение, используя те же реактивы, но без навески жира. Реактивы считаются пригодными для использования, если на контрольный анализ расход гипосульфита натрия не превышает 0,07 см3.
Перекисное число, выраженное в % йода, вычисляют по формуле
(У--0,00127.100 m
где ¥ — объем раствора гипосульфита натрия, пошедший на титрование в рабочем опыте, см3; У[—объем раствора гипосульфита натрия, пошедший на титрование в контрольном опыте, см3;
0,00127 — количество йода, эквивалентном 1 см3 0,01 н. раствора гипосульфита натрия; т — навеска исследуемого образца жира, г.
За окончательный результат принимают среднее арифметическое двух параллельных определений. Расхождение между параллельными определениями не должно превышать 0,005%. Вычисление проводят до 0,001.
Методика количественного определения кислотного числа в кормах животного
и растительного происхождения
(Утверждена Главным управлением ветеринарии
Министерства сельского хозяйства СССР 23 января 1984 г.)
Аппаратура, посуда, реактивы. Весы лабораторные аналитические. Делительная воронка емкостью на 250 см3. Колбы стеклянные с притертой пробкой вместимостью 200—250 см3. Баня водяная. Ступка фарфоровая. Цилиндры лабораторные мерные вместимостью 25—100 см3. Палочки стеклянные. Бумага фильтровальная. Вата медицинская гигроскопическая. Чашки выпарительные фарфоровые. Эфир медицинский. Спирт этиловый ректификованный. 1%-ный спиртовой раствор фенолфталеина. 1%-ный спиртовой раствор тимолфталеина. 0,1 н. титрованный раствор едкого кали или едкого натра. Песок речной чистый, обезжиренный.
Сущность метода заключается в экстракции сырого жира из кормов животного и растительного происхождения эфиром в стеклянной колонке или делительной воронке с фильтром и дальнейшего определения кислотного числа в полученном жире.
Подготовка к испытанию. В конусообразную стеклянную трубку (колонку), высотой 200—250 мм и диаметром 14—16 мм (для кормов животного происхождения) или делительную воронку вместимостью 250 см3 (для кормов растительного происхождения) последовательно вкладывают ватный тампон из гигроскопической ваты, затем два кружка вырезанной фильтровальной бумаги, диаметр которых несколько превышает диаметр трубки или воронки, и вновь ватный тампон. Общая высота фильтра должна быть около 20 мм для трубки и 50 мм для воронки. Налитый в трубку (воронку) эфир должен вытекать из нее со скоростью 40— 80 капель в 1 мин.
Ход определения. Корма животного происхождения в количестве 10 г или растительного в количестве 100 г, взвешенные с точностью до 0,01 г, двойное количество отмытого и обезжиренного речного песка вносят в фарфоровую ступку. Смесь растирают пестиком до однородной массы и высыпают в стеклянную трубку или делительную воронку, установленные на штативе. Под трубкой или воронкой устанавливают выпарительную чашку, предварительно высушенную до постоянной массы
В стеклянную трубку или воронку постепенно по мере фильтрации приливают двойное количество эфира от взятой навески ! корма. Завершение экстракции жира контролируют фильтровальной J бумагой путем смачивания вытекающей капли. В случае отсутствия < на бумаге жирового пятна экстракция считается законченной.
После этого выпарительную чашку ставят на водяную баню и выпаривают эфир до полного удаления его запаха.
Полученный жир в количестве не менее 1,5 г, взвешенный с точностью до 0,01, переносят в коническую колбу с притертой пробкой емкостью 200—250 мл, туда же приливают 50 мл нейтрализованной смеси, состоящей из серного эфира и 96°-ного этилового спирта (в соотношении 2:1) и взбалтывают. Затем добавляют несколько капель соответствующего индикатора (раствор фенолфталеина при испытании светлых жиров; раствор тимолфталеина при испытании жиров, имеющих темную окраску) и быстро титруют 0,1 н. водным раствором едкого кали или едкого натра до отчетливого изменения окраски индикатора (фенолфталеина — розовый, тимолфталеина — синий).
Для проверки чистоты реактивов проводят контрольное определение, используя те же реактивы, но без навески жира.
Кислотное число, выраженное в мг КОН или NaOH, вычисляют по формуле
a k 5,61 b
где X— кислотное число жира, мг КОН или NaOH; а — количество 0,1 н. раствора едкого кали или едкого натра, пошедшее на титрование, мл; k-—поправка к раствору щелочи для пересчета на точный 0,1 н. раствор; 5,61—количество мг едкого кали или едкого натра, содержащееся в 1 мл 0,1 н. раствора едкого кали или едкого натра; b — навеска испытуемого жира, г.
За окончательный результат принимают среднее арифметическое двух параллельных определений. Расхождение между параллельными определениями не должно превышать 0,4 мг. Вычисление проводят до 0,01.
Временный максимально допустимый
уровень (МДУ] содержания некоторых химических элементов и госсипола в кормах для сельскохозяйственных животных и кормовых добавках
(Утверждено Главным управлением ветеринарии Госагропрома СССР 7 августа 1987 г.)
Рекомендуемый временный максимально допустимый уровень (МДУ) некоторых токсичных элементов и госсипола в кормах для сельскохозяйственных животных установлен на основании результатов экспериментальных исследований по изучению их токсичности для различных видов сельскохозяйственных животных, степени материальной кумуляции в органах и тканях, выделения с молоком и яйцом в количествах, не превышающих величин, установленных
Предельно допустимыми концентрациями тяжелых металлов и мышьяка, в продовольственном сырье н пищевых продуктах, утвержденными Минздравом СССР 31 марта 1986 г.
Кроме того, учитывался естественный фон содержания химических элементов в кормовых растениях, мясе, молоке и яйцах, максимально допустимая суточная доза элементов для различных видов животных, норма ввода кормовых ингредиентов в суточный рацион
Настоящие санитарные нормативы предназначены для использования органами ветеринарного надзора при осуществлении профилактического и текущего контроля за содержанием основных токсичных химических элементов в кормах с целью обеспечения их безвредности для сельскохозяйственных животных и профилактики загрязнения ими продуктов питания животного происхождения, а также для ветеринарно-санитарной оценки кормов и сырья для их изготовления, импортируемых из-за рубежа
Для определения токсичных элементов в кормах используются методы анализа, установленные ГОСТами на продукты питания, а также методы, рекомендованные Минздравом СССР и Госагро-промом СССР.
Контроль за содержанием токсичных элементов в кормах должен быть в первую очередь организован в зоне рудных разработок, выбросов металлургической и химической промышленности, в биогеохимических провинциях с повышенным уровнем содержания элементов в почве, воде, растениях, кормах и тканях животных. Исследования также должны проводиться при хронических интоксикациях сельскохозяйственных животных с признаками поражения печени, желудочно-кишечного тракта, анемии, эритропении.
Специалисты ветеринарной службы республики, области, района должны знать, какие промышленные предприятия и рудные разработки находятся в зоне их обслуживания, а также химический состав промышленных выбросов, возможную их концентрацию и дальность распространения, поддерживать постоянную связь с агрохимической, медицинской и гидрометеослужбой и получать от них информацию по содержанию токсичных элементов в воздухе, почве, воде, растениях и продуктах питания.
Из токсичных элементов наибольшее ветеринарно-санитарное и токсикологическое значение имеют ртуть, кадмий, свинец, мышьяк и селен, поэтому контроль кормов в первую очередь организуется за содержанием этих элементов.
При обнаружении в кормах токсичных элементов выше МДУ производится их подсортировка другими кормами из такого расчета, чтобы общий уровень содержания токсичных элементов в рационе не превышал показатели МДУ. Корма, содержащие повышенные уровни ртути и кадмия, а также в случае обнаружения в них других элементов в количествах, превышающих МДУ в 10 и более раз, подсортировке не подлежат. Вопрос о их реализации должен решаться в каждом конкретном случае отдельно по согласованию с органами здравоохранения.
В зонах промышленных выбросов и рудных разработок, где отмечается повышенный уровень содержания ртути и кадмия в кормах, рекомендуется вводить в рационы животных серу или натрия тиосульфат из расчета 100—120 мг/кг корма.
Временный максимально допустимый уровень (МДУ) некоторых химических элементов и госсипола в кормах для сельскохозяйственных животных, мг/кг корма
Химический элемент Комбикорма Г' : Зерно и зернофураж j Грубые и сочные корма 1 ! Корнеклубнеплоды Корма микробного сиьиеза Минеральные добавки, * s т. ч. неолиты 1 Корма для произвол- 1 ства продуктов детско- го питания
S S со птица крупный и мелкий рогатый скот
1 | откорм 1 яйценоская откорм 1 । молочный 1
Ртуть 0,1 0,1 0,05 0,1 0,05 о,1 0,05 0,05 0,1 о,1 0,05
Кадмий 0,4 0,4 0,3 0,4 0,3 0,3 0,3 0,3 0,3 0,4 0,2
Свинец 5,0 5,0 3,0 5,0 3,0 5,0 5,0 5,0 5,0 50,0 2,0
Мышьяк 1,0 1,0 0,5 1,0 0,5 0,5 0,5 0,5 2,0 50,0 0,5
Медь 80,0 80,0 80,0 30,0 30,0 30,0 30,0 30,0 80,0 500,0 30,0
Цинк 100,0 100,0 50,0 100,0 50,0 50,0 50,0 100,0 100,0 1000,0 50,0
Железо 200,0 200,0 100,0 200,0 100,0 100,0 100,0 100,0 200,0 3000,0 100,0
Сурьма 1,0 1,0 0,5 1,0 0,5 0,5 0,5 0,5 2,0 5,0 0,5
Никель 3,0 3,0 1,0 3,0 1,0 1,0 3,0 3,0 3,0 20,0 1,0
Селен 1,0 1,0 0,5 1,0 0,5 0,5 1,0 1,0 2,0 5,0 0,5
Хром 1,0 1,0 0,5 1,0 0,5 0,5 0,5 0,5 2,0 5,0 0,5
Фтор 50,0 50,0 20,0 20,0 10,0 10,0 20,0 20,0 100,0 2000,0 10,0
йод 6,0 5,0 2,0 5,0 2,0 2,0 2,0 5,0 5,0 50,0 2,0
Молибден 3,0 3,0 2,0 3,0 2,0 2,0 2,0 2,0 3,0 10,0 2,0
Кобальт 2,0 3,0 2,0 3,0 2,0 1,0 1,0 2,0 3,0 20,0 1,0
Г оссипол 10,0 10,0 5,0 10,0
свободный
Извлечение из методических указаний по определению нитратов в продукции растениеводства
(Утверждены заместителем Главного государственного санитарного врача СССР 24 ноября 1986 г.)
1.2 Подготовка проб для анализа. Из общих проб продукции отбирают пробы для подготовки к анализу следующим образом:
1.2 1. Картофель. Клубни моют водой, обсушивают фильтровальной бумагой или чистой тканью. От каждого клубня берут четвертую часть. Отобранный материал перемешивают и выделяют пробу для анализа массой не менее 0,25 кг.
1.2.2. Свекла и другие корнеплоды. Корнеплоды моют водой, вытирают досуха, срезают шейку к тонкий конец корня. Крупные корнеплоды разрезают крестообразно вдоль вертикальной оси и используют для анализа половину или четверть. Из полученного материала выделяют пробу для анализа массой 0,5—0,25 кг.
1.2.3 Капуста. Каждый кочан разрезают на 4 части по вертикальной оси и берут по одной четверти в пробу для анализа. При этом срезают и отбрасывают поверхность предыдущего среза, отбрасывают верхние несъедобные листья и остаток кочерыжки. Из полученного материала выделяют пробу для анализа массой 0,5 кг.
1.2.4. Лиственные овощи очищают от земли, освобождают от несъедобных частей и включений и выделяют пробу для анализа массой 0,25 кг.
1.2 5. Луковичные растения. Отбрасывают несъедобные части. С луковиц удаляют верхние чешуи, срезают и отбрасывают основание корня и сухую шейку. Луковицы делят на две части по вертикали и берут в пробу для анализа только одну половинку. Из полученного материала отбирают пробу для анализа массой 0,25 кг.
1.2 6. Томаты, огурцы. Плоды моют водой, просушивают фильтровальной бумагой или чистой тканью, плодоножки удаляют. Крупные плоды разрезают на 2—4 части вдоль оси, для анализа берут половину или четвертую часть. Из полученного материала выделяют для анализа пробу массой 0,5 кг.
1.2.7. Бахчевые культуры. С плодов снимают верхний слой, не употребляемый в пищу, удаляют также семена и исследуют только съедобную часть. Плоды разрезают на две части по линии от места прикрепления стебля до следа цветка таким образом, чтобы в каждую половину попали затемненные и освещенные солнцем части. Если плоды очень крупные, их разрезают на сегменты в 6— 8 см по окружности плода и берут 2—4 сегмента с противоположных сторон каждого плода. Из полученного материала выделяют пробу для анализа массой 0,5 кг.
2. Качественная оценка нитратов в продукции растениеводства с помощью индикаторной бумаги «Индам». Сущность метода состоит в визуальной оценке окрашенных соединений, образующихся при взаимодействии нитратов с реагентом, нанесенным на бумагу. Нижний предел обнаружения нитратов (в пересчете на нитрат-ион) в анализируемой пробе 50 млн-1 (мг/кг).
Метод не может быть использован при анализе образцов красной свеклы и моркови.
Результаты, полученные этим методом, следует рассматривать как ориентировочные, они не могут служить основанием для отбраковки продуктов, а требуют подтверждения ионометрическим методом.
2.1. Материалы и реактивы. Индикаторная бумага «Индам» с нанесенным чувствительным по отношению к нитратам реагентом, выпускаемая НПО «Селекция» Молдавской ССР. Стеклянные палочки или пипетки. Колбы мерные вместимостью 100 см3. Цилиндры вместимостью 50, 100 см3. Весы аналитические. Пипетки вместимостью 1, 5, 10 см3. Калий азотнокислый. Натрий азотнокислый Дистиллированная вода.
2.2. Подготовка к испытанию.
2.2 1. Подготовка проб к испытанию. Пробы для анализа, отобранные в соответствии с разделом 1.2, измельчают с помощью терки или мезгообразователя. Зеленные культуры измельчают ножницами или ножом до размеров частиц 0,5—1 см. Измельченную пробу тщательно перемешивают, отделяя предметным стеклом или с помощью соковыжималки сок, и используют его для анализа.
2.2.2. Приготовление раствора азотнокислого калия (или натрия) концентрации (по нитрат-иону) KN03 = 3000 мг/'дм3. 4,89 г азотнокислого калия (или 4,11 г азотнокислого натрия), высушенного при температуре 100—105 °C до постоянной массы, взвешивают с точностью до второго десятичного знака, помещают в мерную колбу вме’стимостыо 1000 см3, растворяют в дистиллированной воде и доводят объем до метки водой. Хранят раствор в течение года.
2 2.3. Приготовление растворов сравнения азотнокислого калия (или натрия). Растворы сравнения азотнокислого калия (или натрия) готовят из раствора азотнокислого калия (или натрия), приготовленного по пункту 2.2.2.
2 2.3.1. Раствор сравнения концентрации 1500 мг/дм3. Раствор, приготовленный по п. 2.2.2, разбавляют водой в 2 раза.
2.2.3.2. Раствор сравнения концентрации 750 мг/дм3. Раствор, приготовленный по п. 2.2.3.1, разбавляют водой в 2 раза.
2.2.3.3. Раствор сравнения концентрации 300 мг/дм3. Раствор, приготовленный по п. 2.2.2, разбавляют водой в 10 раз.
2.2.3.4. Раствор сравнения концентрации 150 мг/дм3. Раствор, приготовленный по п. 2.2.2, разбавляют водой в 20 раз.
2.2.3.5. Раствор сравнения концентрации 100 мг/дм3. Раствор, приготовленный по п. 2.2.3.3, разбавляют водой в 3 раза.
2.2.3.6. Раствор сравнения концентрации 50 мг/дм3. Раствор, приготовленный по п. 2.2.3.4, разбавляют водой в 3 раза. Растворы, приготовленные по п.п. 2.2.3.1—2.2.3.6, используют в качестве сравнительных при оценке содержания нитратов в образцах.
2.3. Проведение испытаний.
2.3.1. Капли сока анализируемых продуктов, приготовленных по п. 2.2.1, наносят на активную часть индикаторной бумаги «Индам». На другой лист^ индикаторной бумаги наносят последовательно капли растворов' сравнения, приготовленные по п. 2.2.3.
2.3.2. Измерение концентрации иона нитрата. Оценку концентрации нитратов в пробе проводят путем визуального сравнения интенсивности окраски индикаторной бумаги под действием растворов сравнения и анализируемых образцов.
3. Качественная оценка нитратов в продукции растениеводства с использованием дифениламина. Сущность метода состоит в визуальной оценке окрашенных соединений, образующихся при взаимодействии нитратов с дифениламином. Нижний предел обнаружения нитратов в анализируемой пробе 100 млн~> (мг/кг).
Метод может быть использован при определении нитратов во всех продуктах растениеводства. Результаты, полученные этим методом, следует рассматривать как ориентировочные, и они не могут служить основанием для отбраковки продуктов, а требуют подтверждения ионометрическим методом, описанным далее.
3.1. Материалы и реактивы. Предметное стекло 180x90 мм. Стеклянные палочки или пипетки. Колбы мерные вместимостью 100 см3. Пипетки вместимостью 1, 5, 10 см3. Цилиндры вместимостью 50, 100 см3. Весы аналитические. Серная кислота. Дифениламин. Натрий азотнокислый х. ч. Калий азотнокислый х. ч. Дистиллированная вода.
3.2. Подготовка к испытанию.
3.2.1. Отбор проб для анализа проводят по п. 1.2.
3.2.2. Подготовку проб к испытанию проводят по п. 2.2.1.
3.2.3. Приготовление раствора азотнокислого калия (натрия) концентрации (по нитрат-иону) KNO3 = 3000 мг/дм3 проводят по п. 2.2.2.
3.2.4. Приготовление растворов сравнения азотнокислого калия (натрия) проводят по п. 2.2.3.
3.3. Проведение испытаний.
3.3.1. На предметное стекло, помещенное на лист белой бумаги, на расстоянии 15 мм наносят последовательно капли растворов сравнения, приготовленные по п. 2.2.3. На это же предметное стекло наносят сок исследуемых образцов продуктов, приготовленных к анализу по п. 2.2.1. Одновременно добавляют по одной капле раствора дифениламина в серной кислоте к растворам сравнения и соку анализируемых образцов. Окраска развивается в течение 20—30 с. В зависимости от концентрации нитратов интенсивность окраски меняется от бледно-голубой до интенсивно-синей.
3.3.2. Измерение концентрации иона нитрата. Оценку концентрации нитратов в пробе проводят путем визуального сравнения интенсивности окраски растворов сравнения и сока анализируемых образцов.
4. Количественный ионометрический метод определения нитратов. Сущность метода — извлечение нитратов из анализируемого материала раствором алюмокалиевых квасцов и последующее измерение концентрации нитратов в полученной вытяжке с помощью ионоселективного электрода.
Метод не пригоден в случае, если содержание хлоридов в исследуемом материале превышает содержание иигратов более чем в 50 раз, поэтому метод применим только для анализа сырой продукции, к которой не добавляют хлориды. При анализе лука-репки, капусты и других растений семейства крестоцветных обязательным является фильтрование и разбавление анализируемых вытяжек из испытуемого материала, как описано в приведенной ниже методике. Это устраняет влияние сопутствующих веществ, присутствующих в анализируемых материалах, на величину потенциала электрода.
Чувствительность метода — 6 мг/дм3 анализируемого раствора, нижний предел определения нитратов в анализируемой пробе 306
30 млн-1 (мг/кг). Суммарная погрешность метода оценивается по коэффициенту вариации и составляет ±12%.
4.1. Аппаратура, материалы и реактивы. Ионометр типа ЭВ-74, pH-метр, милливольтметр pH-340, pH-121 или аналогичный прибор с погрешностью измерений не более 5 мВ (0,05 рСмо3). Ионоселективный нитратный электрод 9M-NO3-01, ЭИМ-I, ЭИМ-П или другой электрод, имеющий такие же метрологические характеристики. Электрод сравнения хлорсеребряный насыщенный образцовый 2-го разряда. Весы лабораторные ВЛКТР-500 или другие лабораторные весы общего назначения 4-го класса точности с наибольшим пределом взвешивания 500 г. Весы АДВ-200 или другие лабораторные весы общего назначения 2-го класса точности с наибольшим пределом взвешивания 200 г. Колбы мерные 2-го класса точности, исполнение 1,2, вместимость 50, 100 см3. Дозатор вместимостью 50 см3 с погрешностью дозирования не более 1% или цилиндр вместимостью 50 см3. Мешалка лабораторная электромеханическая. Встряхиватель ВВ-1 или блок экстрагирования БЭ-1. Ступка фарфоровая. Мезгопастообразователь, пластмассовая терка, ножницы, скальпель. Гомогенизатор с частотой вращения не менее 6000 мин"1. Квасцы алюмокалиевые ч. д. а. Калий хлористый х. ч. Калий азотнокислый х. ч. Вода дистиллированная. Бумага масштабно-координатная марки D2.
4.2. Подготовка к испытанию.
4.2.1. Отбор проб для анализа проводят по п. 1.2.
4.2.2. Подготовка проб к испытанию. Пробы для анализа, отобранные в соответствии с разделом 1,2, измельчают с помощью терки, мезгопастообразователя до получения однородной массы. Зеленые культуры предварительно измельчают ножницами или ножом до размеров частиц 0,5—1 см. Измельченную пробу тщательно перемешивают и используют для анализа.
4.2.3. Приготовление раствора алюмокалиевых квасцов с массовой долей 1% (экстрагирующий раствор). 10 г алюмокалиевых квасцов взвешивают с точностью до первого десятичного знака, переносят в мерную колбу вместимостью 1000 см3, растворяют в дистиллированной воде и доводят объем раствора водой до метки.
Раствор хранят в склянке с притертой пробкой не более 1 года. При появлении мути или осадка раствор заменяют свежеприготовленным.
4.2.4. Приготовление раствора азотнокислого калия концентрации с (KNO3)=0,l моль/дм3 (pCno3) =—log Cno3= 1. Ю, 11 г азотнокислого калия, высушенного при температуре 100—105 °C до постоянной массы, взвешивают с точностью до второго десятичного знака, помещают в мерную колбу вместимостью 1000 см3, растворяют в экстрагирующем растворе и доводят объем до метки экстрагирующим раствором.
Раствор хранят в склянке с притертой пробкой не более 1 г. При появлении мути или осадка раствор заменяют свежеприготовленным.
4.2.5. Приготовление''растворов сравнения азотнокислого калия. Растворы сравнения азотнокислого калия готовят из раствора азотнокислого калия, приготовленного по п. 4.2.4 в день проведения анализа, используя для разбавления тот же раствор алюмокалиевых квасцов, который применяется для анализа.
4.2.5.1. Раствор сравнения с концентрацией с (NO3~) = = 0,01 моль/дм3 (рСко3 = 2).
Раствор азотнокислого калия, приготовленный по п. 4.2.4. разбавляют в 10 раз раствором алюмокалиевых квасцов. Для этого отбирают пипеткой 10 см3 раствора с рСмо3=1 в мерную колбу вместимостью 100 см3, добавляют до метки раствором алюмокалиевых квасцов и перемешивают.
4.2.5.2. Раствор сравнения с концентрацией с (N03~) = = 0,001 моль/дм3 (рСко3 = 3).
Раствор, приготовленный по п. 4.2.5.1, разбавляют в 10 раз раствором алюмокалиевых квасцов.
4.2.5.3. Раствор сравнения с концентрацией с (NO3-) = = 0,0001 моль/дм3 (рСмо3 = 4).
Раствор, приготовленный по п. 4 2.5.2, разбавляют в 10 раз раствором алюмокалиевых квасцов.
Растворы сравнения, приготовленные по пп. 4.2.4, 4 2.5.1, 4 2.5.2, 4.2.5.5.3, используют для градуировки прибора, проверки электродов и построения градуировочного графика.
В промежутках между проведением испытаний мембранный ионоселективный электрод погружают в дистиллированную воду, при перерывах в работе порядка суток или более хранят в растворе азотнокислого калия с концентрацией с (NO3~)=0,l моль/дм3. При длительных перерывах между испытаниями (более 5 сут) электрод хранят в воздухе. В обоих случаях перед началом измерений электрод выдерживают в дистиллированной воде не менее 10 мин.
Хлорсеребряный электрод сравнения в перерывах между испытаниями хранят в дистиллированной воде.
4.3. Проведение испытаний.
4.3.1. 10 г испытуемого материала, подготовленного по п. 2.2.1, взвешивают с точностью до первого десятичного знака, помещают в стакан гомогенизатора, приливают 50 см3 раствора алюмокалиевых квасцов с массовой долей 1 % (отношение массы пробы к объему раствора 1:5) и гомогенизируют в течение 1 мин при частоте вращения 6000 мин-1.
При отсутствии гомогенизатора 10 г мезги или пасты корнеклубнеплодов помещают в технологическую емкость, приливают 50 см3 раствора алюмокалиевых квасцов с массой долей 1 % и перемешивают на встряхивателе, ротаторе или с помощью мешалки в течение 3 мин. При отсутствии гомогенизатора при анализе материала, содержащего твердые ткани, пробу массой 10 г растирают в ступке с прокаленным песком или битым стеклом до однородной массы, переносят ее с помощью 50 см3 раствора алюмокалиевых квасцов с массовой долей 1% в технологическую емкость вместимостью не менее 100 см3, а затем перемешивают в течение 3 мин на встряхивателе, ротаторе или с помощью мешалки. В полученной суспензии измеряют концентрацию иона нитрата.
При анализе культур семейства крестоцветных требуется проведение 10-кратного разбавления при величине рСцо3 исследуемых суспензий от 2,5 до 3,0 и 20-кратное разбавление при величине рСно, менее 2,5. Для этого суспензию фильтруют через бумажный фильтр и отбирают пробу 2 см3 и приливают к ней 38 см3 раствора алюмокалиевых квасцов для 20-кратного разбавления или
отбирают пробу 4 см3 и прибавляют к ней 36 см3 раствора алюмокалиевых квасцов для 10-кратного разбавления. В разбавленном фильтрате определяют концентрацию иона нитрата.
4.3.2. Измерение концентрации иона нитрата.
4.3.2.1. Измерение концентрации иона нитрата в единицах рСко3 по шкале прибора.
Прн непосредственном измерении рСмо3 прибор ежедневно настраивают в режиме «рХ» в соответствии с инструкцией завода-изготовителя по растворам сравнения с рСмо., равным 4 и 2. Раствор сравнения с рСмо3 = 3 используют для контроля настройки. Отклонения значений рСцо3 от номинальных значений рСко3 растворов сравнения не должны превышать 0,02 единицы рСмо3-
При работе с приборами, имеющими соответствующие преобразователи величины рСмо3, т. е. отрицательного логарифма концентрации, в значения концентрации, настройку прибора проводят непосредственно в единицах массовой доли нитрата — миллионных долях (мг/кг) по растворам сравнения. Массовую долю иона нитрата в растворах сравнения находят по значениям величины pCwo3 в соответствующей таблице. При настройке прибора отклонения значений массовой доли иона нитрата от номинального значения, найденного по таблице, не должны превышать 4%.
После градуировки прибора электроды тщательно ополаскивают дистиллированной водой, промокают фильтровальной бумагой и погружают в испытуемые пробы. Показания прибора считывают не ранее чем через 1 мин после прекращения заметного дрейфа показаний прибора. При переходе от одной пробы к другой электроды ополаскивают дистиллированной водой и промокают фильтровальной бумагой. Температура испытуемых проб и растворов сравнения должна быть одинаковой. Настройку прибора проверяют по растворам сравнения не менее 3 раз в течение рабочего дня, используя каждый раз свежие порции растворов сравнения. Перед каждой проверкой настройки прибора нитратный ионоселективный электрод выдерживают в растворе сравнения концентрации с (NO3~) =0,0001 моль/дм3 в течение 3—4 мин.
4.3.2.2. Измерение концентрации иона нитрата по градуировочному графику. Градуировочный график строят по результатам измерения ЭДС электродной пары в милливольтах.
При измерении ЭДС в милливольтах тумблер «Род работ» ставят в положение «мВ». Измерения ЭДС электродной пары в растворах сравнения начинают с наиболее низкой концентрации (рСмо3 = 4). Электрод имеет линейную функцию в диапазоне от 1 до 4 ед. рСмо3 с наклоном (56+3) мВ на единицу рСмо3. Если характеристика электрода отличается от заданной, электрод не пригоден для работы.
После градуировки прибора электроды тщательно ополаскивают дистиллированной водой, промокают фильтровальной бумагой и измеряют ЭДС в испытуемых пробах. Перед погружением электродов испытуемые пробы 'перемешивают. После каждого измерения электроды ополаскивают дистиллированной водой и промокают фильтровальной бумагой.1 Показания прибора считывают не ранее чем через 1 мин, т. е. после прекращения дрейфа показаний прибора. Температура испытуемых проб и растворов сравнения долж
на быть одинаковой. Настройку прибора проверяют не менее 3 раз в течение рабочего дня, используя каждый раз свежие порции растворов сравнения.
Величину рСмо3 в испытуемых пробах находят по градуировочному графику, построенному по результатам измерения ЭДС электродной пары в растворах сравнения с рСмоз, равными 1, 2, 3 и 4 единицам.
4.4. Обработка результатов.
Массовую долю нитратов в испытуемом материале (X) в миллионных долях (млн-1, мг/кг) можно рассчитать по формуле
аН -
V+----- -10 K0;i -62-103
__ _100П___________________
х= 1000Н
где 62 — молярная масса иоиа нитрата, г; Н — масса пробы, взятой для анализа, г; V — объем экстрагирующего раствора, см3; 10 " rSo, —концентрация нитратов в вытяжке, моль/дм3; 1000-коэффициент перевода дм3 в см3; о> — массовая доля воды в пробе, %; 100 — коэффициент перевода % в доли единиц; 1 — плотность воды, г/см3; 106—коэффициент перевода долей единицы в миллионные доли (млн-1, мг/кг).
Принимая V = 50 см3, Н = 10 г и проведя соответствующие сокращения и преобразования, получаем следующую формулу для расчета:
/ 0) \
Х= 50+----------- -10 N°3-6200.
\ 10 /
В случае разбавления вытяжки результат анализа увеличивают во столько раз, во сколько раз была разбавлена вытяжка при анализе.
Определение содержания воды в пробе, а также выполнение расчетов можно исключить, используя таблицы 1 и 2 для перевода величин рСко3 в массовую долю нитрата в анализируемой пробе.
1. Перевод величины pCno3 в массовую долю нитрата при анализе арбузов, дынь, огурцов, томатов, лука-пера, столовой капусты
О я о Сотые доли pC^Q
00 01 г 02 1 03 1 С4 05 06 07 08 09
1.6 9188 Массовая доля 8979 8775 8575 нитрата, млн~{ (мг/кг) 8380 8189 8003 7821 7643 7469
1.7 7299 7133 6970 6812 6656 6505 6357 6212 6071 5933
1.8 5798 5666 5537 5411 5287 5167 5049 4935 4822 4712
1.9 4605 4500 4398 4298 4200 4104 4011 3920 3830 3743
2.0 3658 3575 3493 3414 3336 3260 3186 3113 3043 2973
2.1 2906 2840 2775 2712 2650 2590 2531 2478 2417 2362
2.2 2308 2256 2204 2154 2105 2057 2010 1964 1920 1876
2.3 1833 1792 1751 1711 1672 1634 1597 1560 1525 1490
2.4 1456 1423 1391 1359 1328 1298 1268 1239 1211 1184
2.5 1157 ИЗО 1105 1080 1055 1031 1007 985 962 940
2.6 919 898 877 858 838 319 800 782 764 747
2.7 730 713 697 681 666 650 636 621 607 593
2.8 580 567 554 541 529 517 505 493 482 471
2.9 461 450 440 430 420 410 401 392 383 374
3.0 366 357 349 341 334 326 319 311 304 297
3.1 291 284 277 271 265 259 253 247 242 336
3.2 231 226 220 215 , 210 206 201 196 192 188
3.3 183 179 175 171 167 163 160 156 152 149
3.4 146 142 139 136 133 130 127 124 121 118
3.5 116 113 НО 108 105 103 101 98 96 94
3.6 91,9 89,8 87,7 85,8 83,8 81,9 80,0 78,2 76,4 74,7
3.7 73,0 71,3 69,7 ,68,1 66,6 65,0 63,6 62,1 60,7 59,3
3.8 58,0 56,7 55,4 54,1 52,9 51,7 50,5 49,3 48,2 47,1
3.9 46,1 45,0 44,0 48,0 42,0 41,0 40,1 39,2 38,3 37,4
4.0 36,6 35,7 34,9 34,1 33,4 32,6 31,9 31,1 30,4 29,7
2. Перевод величины рСко3 в массовую долю нитрата при анализе картофеля, моркови, столовой свеклы, лука-репки
С о Сот.>>е доли pCNQ
00 01 02 03 С4 05 06 07 08 09
Массовая доля нитрата, млн~г (мг!кг)
1.6 9033 8827 8626 8430 8238 8050 7867 7688 7513 7342
1.7 7175 7012 6852 6696 6544 6395 6249 6107 5968 5832
1.8 5699 5570 5443 5319 5198 5079 4964 4851 4740 4633
1.9 4527 4424 4323 4225 4129 4035 3943 3853 3765 3680
2.0 3596 3514 3434 3356 3280 3205 3132 3061 2991 2923
2.1 2856 2791 2728 2666 2605 2546 2488 2431 2376 2322
2.2 2269 2217 2167 2117 2069 2022 1976 1931 1887 1844
2.3 1802 1761 1721 1682 1644 1606 1570 1534 1499 1465
2.4 1432 1399 1367 1336 1306 1276 1247 1218 1191 1164
2.5 1137 1111 1086 1061 1037 1013 990 968 946 924
2.6 903 883 863 843 824 805 787 769 751 734
2.7 717 701 685 670 654 639 625 611 597 583
2.8 570 557 544 532 520 508 496 485 474 463
2.9 453 442 432 422 413 403 394 385 377 368
3.0 360 351 343 336 323 320 313 306 299 292
3.1 286 279 273 267 261 255 249 243 238 232
3.2 227 222 217 212 207 202 198 193 189 184
3.3 180 176 172 168 164 161 157 153 150 146
3.4 143 140 137 134 131 128 125 122 119 116
3.5 114 111 109 106 104 101 99 97 95 92
3.6 90,3 88,3 86,3 84,3 82,4 80,5 78,7 76,9 75,1 73,4
3.7 71,7 70,1 68,5 67,0 65,4 63,9 62,5 61,1 59,7 58,3
3.8 57,0 55,7 54,4 53,2 52,0 50,8 49,6 48,5 47,4 46,3
3.9 45,3 44,2 43,2 42,2 41,3 40,3 39,4 38,5 37,7 36,8
4.0 36,0 35,1 34,3 33,6 32,8 32,0 31,3 30,6 29,9 29,2
312
Абат 94, 105
Агелон 180
Акрекс 165
Алкалоиды 244
Амидофос 51, 53, 54
Аминиая соль 24-Д 173
Аммиак 268, 291
Амифос 53, 54
Антио 97, 99, 101
Атразин 174
Базагран 179
Базудин 108
Байтекс 56, 94
Банвел-Д 177
Банлсв 179
Барий 264
Бетанал 176
Бромиды 171
Бутифос 61, 64
Вензар 178
Вернам 177
Витавакс 198
Гаммагсксан 38
Гамматиурам 38
Гардопа 112, 116
ГГММЭ (гамма-изомера гексахлорана минерально-масляная эмульсия) 37
Гексалин 37
Гексаталп 37
Гексахлорбензол (ГХБ) 39, 46
Гексахлорбутадиен (ГХБД) 39
Гексахлорпараксилол 240
Гептахлор 39, 47
Гербициды 172
Глифтор 224
Глюкозиды 256
Голтикс 202
Госсипол 248, 251, 253
Гранозан 144
ГХЦГ (гексахлорциклогексан) 34
— гамма-изомер 17, 36
Далапон 179
2,4-Д бутиловый эфир 173
ДДВФ 66, 68, 70
Диален 180
Диамет-Д 179
Дикрезил 151
Дикуран 186
Дилор 27, 40, 47
2,4-ДМ 173
Динитрооргокрезол (ДНОК) 163
Диносеб 165
Диурон 175, 186
Дифос 105
Дихлоральмочевина (ДХМ) 175
2,4-Ди хлор феноксиуксусная кисло-
та (2,4-Д) 210
Дозанекс 175, 186
Дуал 178, 183
Дымовые шашки Г-17 37
Зенкор 178
Зоокумарин 219
Кали едкое 268
Каптаи 41
Карагарт 3587 180
Каратан 165
Карбаматные пестициды 148
Карбин 176
Карбофос 94, 103
Кельтан 41, 47
Кислота азотная 268
— муравьиная 296
— серная 268
— соляная 268
— щавелевая 297
Кислотное число 301
Клещевинный жмых 257
Концентрат карбамидный 269
Кортекс-М 180
Которая 186
Котофор 174, 192
Крысид 230
Купроцин-I, II 149
Лассо 177, 183
Лассо/атразин 177
Лин^рон 186
Лонзрел 178
Максимально допустимые уровни
303
Малоран 175, 186
Манеб 149
Марцин 149
МГ-натрия 178
Медь 264, 271
Мезоранил 174
Метафос 78
Метилнитрофос 80
Минеральные кислоты 267
Монурон 186
2М-4Х 173
2М-4ХМ 173
2М-4ХП 173
Мышьяк 233
Натр едкий 268
Нитраты 277, 306
Нитриты 277
Нортрон 179
Оксибазудин 108
Омайт 122
Параиитрокрезол 80
Паторан 175, 186
Пенокумарин 219
Перекисное числе 299
Пирамин 178 *
Полидим 177
Поликарбацин 149
Полимарцин 149
Полихлоркамфен 20
Полихлорпинсн 20
Прометрин 174, 181
Пропанид 177
Рамрод 177, 183
Реглон 179
Рицин 257
Ронит 176
Ртути неорганические соединения 144
Ргуть 141
— общая 134
Ртутьорганические пестициды 127, 129
Ртутьсодержащне соединения 127
Сангор 178
Сапонины 260
Сатурн 176
Свинец 264
Севин 160
Селен 274
Семерон 174
Серная кислота 268
Симазин 174
Синбар 178
Синильная кислота 256
СК-9 43
Солан 177
Соланин 259
Соляная кислота 268
Соль поваренная 265
Суффикс 177
ТАП-85 37
Тедион 43
Теноран 186
Тетраметнлтиурамдисульфид
(ТМТД) 148
Тиазон 149
Тигам 38
Тиллам 157, 176
Тиодан 30, 44, 47
ТМТД 149
Толунн 178
Тордон 22 К 178
Триаллат 153, 176
Трихлорацетаг натрия (ТХА) 179
Трихлорметафос-3 82
Тур 204
Углерод четыреххлористый 168
Фалоран 186
Фенил мочевинные гербициды 186
Фенкаптон 94
Фенол 264
Фснотиазин 17, 235
Фентиурам 38
Фентнурам-молибдат 39
Фенурон 186
Фозалон 85, 86, 88, 94
Формальдегид 236
Фостоксин 231
Фосфамид 97, 99, 101
Фосфид цинка 227, 229
Фосфор 292
Фосфорорганические инсектициды 58 — пестициды 124
—соединения 51
— ядохимикаты 118
Фталан 44
Фталофос 88, 92, 94
Фтор 238
Хлорат магния 217
Хлорид натрия 266
Хлор-ИФК 176
Хлорорганические пестициды 5
----характеристика группы 33
Хлорофос 72, 75
Хлорхолинхлорид 208
Цианокс 120
Цидеал 94
Цинеб 150
Цинк 263
Цирам 150
Щелочи 267
Эдитой 150
Эптам 154, 157, 176
Эупарен 44
Яды металлические 261
Ялан 176
СОДЕРЖАНИЕ
Предисловие , , ...............................3
ХЛОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ............................5
Методические указания по определению хлорорганических пестицидов в воде, продуктах питания, кормах и табачных изделиях методом хроматографии в тонком слое ... 5
Определение гамма-изомера гексахлорциклогексана и фенотиазина в продуктах животного происхождения тонкослойной хроматографией...................................17
Определение полихлорпинена и полихлоркамфена в воздухе, воде, почве, картофеле и свекле, мясе, молоке, тканях внутренних органов животных, крови, моче тонкослойной хроматографией .........................................20
Определение дилора в растительных пробах, воде и органах теплокровных животных хроматографией в тонком слое . 27
Определение тиодана в продуктах растительного происхождения тонкослойной хроматографией и колориметрическим методом............................................ 28
Временные методические указания по определению тиодана и продуктов его превращения в мясе, органах и тканях животных хроматографическими методами . ... 30
Извлечения из методических указаний по диагностике, профилактике, лечению отравлений животных и контролю за предотвращением загрязнения кормов и продуктов животноводства хлорорганическими пестицидами . ... 33
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ . ... 51
Определение амидофоса в молоке и мясе тонкослойной хроматографией .........................................51
Определение амифоса в воде, яблоках и свекле тонкослойной хроматографией...................................53
Методические указания по определению амифоса в растительном материале и мясе методом тонкослойной хроматографии ..............................................54
Определение байтекса в молоке и мясе тонкослойной хроматографией . .......................... 56
Энзиматическое агар-диффузное определение фосфорорганических инсектицидов в • продуктах животного происхождения ............................................. 58
Определение бутифоса в хлопке-сырце и продуктах его промышленной переработки тонкослойной хроматографией , 61
Методика обнаружения бутифоса в молоке, крови и моче животных при диагностике отравлений......................64
Определение ДДВФ в растительном материале, почве и воде тонкослойной хроматографией..............................66
Методические указания по определению ДДВФ в тканях животных методом хроматографии в тонком слое ... 68
Методика обнаружения хлорофоса и ДДВФ в патологическом материале................................................70
Колориметрическое определение хлорофоса в продуктах растительного происхождения (капуста, картофель, зерно, огурцы, яблоки) и молоке.................................72
Определение хлорофоса в воде, фруктах, овощах, молоке, мясе и кормах хроматографией в тонком слое ... 75
Определение метафоса в воде, почве и продуктах питания растительного происхождения тонкослойной хроматографией 78
Определение метилнитрофоса и паранитрокрезола в воде, зернофураже, яйцах и патологическом материале тонкослойной хроматографией...................................80
Определение трихлорметафоса-3 в молоке и молочных продуктах тонкослойной хроматографией.......................82
Определение фозалона в яблоках, листьях яблони, почве и воде тонкослойной хроматографией.........................85
Методические указания по определению фозалона в молоке, тканях животных и кормах методом тонкослойной хроматографии ................................................86
Методические указания по определению фталофоса и фозалона в воде и рыбе и фозалона в кормах и мясе методом тонкослойной хроматографии...............................88
Определение фталофоса в молоке и мясе тонкослойной хроматографией .............................................92
Определение фозалона, фталофоса, фенкаптона, цидеала и карбофоса в воде, овощах и фруктах, определение байтекса и абата в воде тонкослойной хроматографией .... 94
Методические указания по определению антио и фосфамида в кормах методом тонкослойной хроматографии ... 97
Методика определения антио и фосфамида в органах и тканях, молоке, крови и моче животных при диагностике отравлений ................................................99
Методические указания по определению антио и фосфамида в мёде методом хроматографии в тонком слое . . .191
Методические указания по определению карбофоса в тканях животных методом хроматографии в тонком слое . . ЮЗ
Методические указания по определению дифоса (абата) в продуктах животного происхождения методом тонкослойной хроматографии...........................................105
Методические указания по определению базудина и оксибазудина в растительном материале, почве и воде тонкослойной и газожидкостной хроматографией .... .108
Методические указания по определению гардоны и ее продуктов превращения в растительном материале и почве хроматографическими методами............................112
Методика определения гардоны в органах и тканях животных при диагностике отравления ............................ 116
Методика экспрессной диагностики отравления пчел фосфорорганическими ядохимикатами.............................118
Временные методические указания по определению цианокса в мёде методом тонкослойной хроматографии .... 120
Временные методические указания по определению остаточных количеств омайта в мёде методом тонкослойной хроматографии ..............................................122
Ферментный метод определения фосфорорганических пестицидов в патологическом материале.........................124
РТУТЬСОДЕРЖАЩИЕ СОЕДИНЕНИЯ............................127
Методические указания по диагностике, профилактике и лечению отравлений сельскохозяйственных животных ртуть-органическими пестицидами..............................127
Извлечение из приложения № 1: Методика дробного определения ртути в трупном материале ............ 127
Методические указания по определению ртутьорганических пестицидов в овощах, продуктах животноводства, кормах и патматериале хроматографическими методами . . .129
Методические указания по определению содержания общей ртути в мясе, мясопродуктах, яйцах, рыбе, молочных продуктах, шоколаде, почве колориметрическим способом или при помощи тонкослойной хроматографии.................134
Методические указания по определению ртути в рыбе и молочных продуктах хроматографическим методом . . . 141
Методические указания по определению гранозана и неорганических соединений ртути в воде методом тонкослойной хроматографии..........................................144
КАРБАМАТНЫЕ ПЕСТИЦИДЫ.................................148
Экспрессный метод обнаружения тетраметилтиурамдисуль-фида (ТМТД) в зерне....................................148
Колориметрическое определение купроцина-I, купроцина-Н, манеба, марцина, полимарцина, поликарбацина, тиазона, ТМТД, цинеба, цирама и эдитона в воздухе, продуктах питания растительного происхождения и биологических средах................................................149
Определение дикрезила в биологических средах тонкослойной хроматографией.........................................151
Колориметрическое определение триаллата в мясе и тканях внутренних органов животных............................153
Методика определения эптама в биологических объектах тонкослойной хроматографией...............................154
Колориметрическое определение эптама и тиллама в растительном материале, почве, воде и биологических средах . 157
Методические указания по определению севина в биологических субстратах и воде методом тонкослойной хроматографии ................................................160
ПЕСТИЦИДЫ ДРУГИХ ГРУПП................................163
Определение динитроорток(>езола (ДНОК) в воде, картофеле, винограде и яблоках тонкослойной хроматографией . 163
Определение каратана, айрекса в воздухе, воде, огурцах, яблоках, биологическом материале и диносеба в биологическом материале тонкослойной хроматографией . . . .165
Методические указания по определению четыреххлористого углерода в зерне фотоколориметрическим методом 168
Определение бромидов в зерне и растительном материале хроматографией в тонком слое 171
Извлечения из методических указаний по диагностике, про филактчке и лечению отравлений сельскохозяйственных жи вотных гербицидами 172
Определение прометрина в почве, воде и растительном мате риале тонкослойной хроматографией
Методические указания по определению рамрода, лассо и дуала в воде, почве и растительных пробах хроматографи ей в тонком слое 183
Методические указания по определению фенилмочевинных гербицидов (фенурона которана, монурона, диурона, ди курана, дозанекса, тенорана фалорана, арезина линурона паторана, малорана) в воде почве, расг ыельной массе овощах методом тонкослойной хроматографии 186
Методические указания по опреде тению котофора в воде, почве, хлопковых семенах, продуктах питания растительно го происхождения и биологическом материа ле методом тонкослойной хроматографии и УФ спектроскопии < 2
Временные методические указания по определению витавак са в зерне и воде методом тонкослойной хроматографии 198
Временные методические указания по определению голтикса в воде, почве и растениях методом тонкослойной хрома тографии 202
Методические указания по диагностике, лечению и профилак тике отравления сельскохозяйственных животных туром 204
Методика определения хлорхолинхлорида в почве воде, рас тигельных объектах, органах и гканях животных методом тонкослойной хроматот рафии 208
Методические указания по определению 2,4 дихлорфенокси уксусной кислоты (2,4 Д) в воде, почве, фураже, продук тах питания растительного и животного происхождения хроматографическими методами 210
Колориметрическое определение хлората магния в хлопковом масле, шроте, жмыхе, семенах, ядрах семян и волокне хлопчатника 217
Методические указания по определению зоокумарина в тка нях и крови животных, в приманках и препарате (пеноку марин) хроматографическими и спектрофотометрическими методами 219
Временные методические указания по фотоэлектроколоримет рическому определению глифтора в органах и тканях жи вотных 224
Количественное определение фосфида цинка по фосфористому водороду 229
Определение крысида в биологическом материале (по Ван тропу) 23)
Колориметрическое определение фостоксина в зерне 231
Определение мышьяка по Зангер Влеку 233
Меюдические указания по определению фенотиазина в меде методом тонкослойной хроматографии 235
Методические указания по определению остаточных количеств формальдегида в почве, воде, продуктах и отходах сахарного производства ... ...................236
Методика определения фтора в биологическом материале и минеральных веществах ... ...............238
Методика определения гексахлорпараксилола (гексихола) в кормолекарственной смеси, молоке, органах и тканях животных ................................................240
РАСТИТЕЛЬНЫЕ ЯДЫ........................................244
Методика хроматографического определения алкалоидов в патологическом материале и растениях . . . . 244
Методика определения свободного и связанного госсипола в семенах хлопчатника, хлопковом жмыхе, шроте и комбикормах . . .... 248
Методика определения свободного и связанного госсипола в
органах и тканях сельскохозяйственных животных и птиц 251
Методика определения госсипола в кормах, содержащих продукты переработки хлопчатника ........................ 253
Определение синильной кислоты (качественная реакция) . 256
Определение глюкозидов в кормах (проба с фелинговой жидкостью) ...............................................256
Определение рицина в клещевинном жмыхе методом агглютинации эритроцитов......................................257
Определение соланина в картофеле......................259
Определение сапонинов в растениях и кормах (гемолитическая проба)............................................260
ДРУГИЕ ЯДОВИТЫЕ ВЕЩЕСТВА.........................261
Методические указания по обнаружению металлических ядов в патологическом материале, кормах и воде с помощью экспрессных методов исследования ........................ 261
Определение фенола......................................264
Определение поваренной соли в патологическом материале (по Мору)........................................... 265
Определение хлорида натрия обратным титрованием по методу Фольгарда.........................................266
Обнаружение свободных минеральных кислот и щелочей 267
Извлечения ТУ 8-22-4—77. Концентрат карбамидный, технические условия.........................................269
Методика определения меди в кормах, продуктах животноводства и патматериале ............................... 271
Мею гика флуорнметрического определения селена в почве, кормах, органах (тканях) животных и продуктах живот-но,;и,ц1вт . ................. 274
Извлечения из методических указаний по диагностике, профилактике и лечению отравлений сельскохозяйственных животных нитратами и нитритами...........................277
Определение аммиака объемным методом....................291
Методические указания по диагностике отравлений рыб и сельскохозяйственных животных элементарным фосфором 292
Методические указания по количественному определению муравьиной кислоты в мёде.............................. 296
Методические указания по количественному определению щавелевой кислоты в мёде...................................297
Методика количественного определения перекисного числа в
кормах животного и растительного происхождения . . 298
Методика количественного определения кислотного числа в
кормах животного и растительного происхождения . . 300
Временный максимально допустимый уровень (МДУ) содержания некоторых химических элементов и госсипола в кормах для сельскохозяйственных животных и кормовых добавках ....................................................3)1
Извлечение из методических указаний по определению нитратов в продуктах растениеводства............................304
Предметный указатель......................................313
Антонов Борис Иванович, Федотова Вера Ивановна, Сухая Наталья Александровна
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ В ВЕТЕРИНАРИИ:
ХИМИКО-ТОКСИКОЛОГИЧЕСКИЕ МЕТОДЫ
СПРАВОЧНИК
Зав. редакцией В. Г. Федотов
Художественный редактор М. Д. Северина
Технический редактор Н. Н. Зиновьева
Корректор Н. В. Карпова
ИБ № 6486
Сдано в набор 08.09.88. Подписано к печати 23 11.88. Т-18360. Формат 84X108V32. Бум. кн.-жури. отсч. Гарнитура Литературная. Печать высокая. Усл. печ. л. 16,8. Усл. кр.-отт. 16,8. Уч.-изд. л. 25,6. Изд. № 091. Тираж 29 000 экз. Заказ № 1995. Цена 1 р. 30 к.
Ордена Трудового Красного Знамени ВО «Агропромиздат», 107807, ГСП-6, Москва, Б-78, ул. Садовая-Спасская, 18.
Областная ордена «Знак Почета» типография им. Смирнова Смоленского облуправления издательств, полиграфии и книжной торговли. 214000, г. Смоленск, проспект им. Ю. Гагарина, 2.