/












































































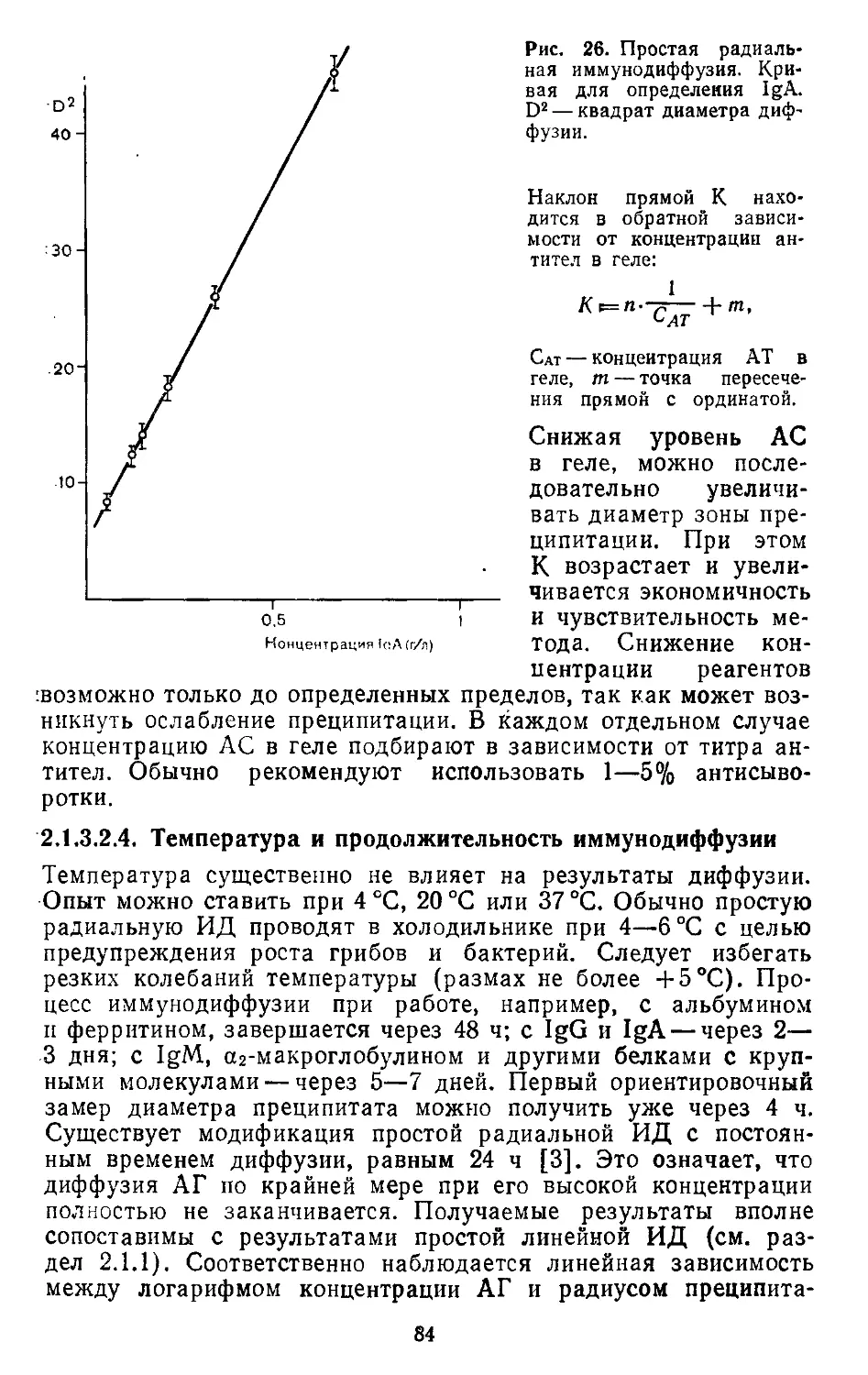















































































































































































































































































































































































Текст
ИММУНОЛОГИЧЕСКИЕ
МЕТОДЫ
Immimologische Arbeitsmethoden
Herausgegeben von
Prof. Dr. sc. med. Helmut Friemel
Bereich Medizin der Wilhelm — Pieck — Universitat Rostock
VEB GUSTAV FISCHER VERLAG JEXA
1984
ИММУНОЛОГИЧЕСКИЕ
МЕТОДЫ
Под редакцией
Г. Фримеля
Перевод с немецкого кандидата биологических наук
А. П. Тарасова
Москва «Медицина»
1987
ББК 53.4
И53
УДК 616-078.73
Издание рекомендовано к переводу академиком Р. В. ПЕТРОВЫМ, ди-
ректором Института иммунологии АМН СССР.
Иммунологические методы/Под ред. Г. Фримеля, Пер.
И53 с нем. А. П. Тарасова. — М.: Медицина, 1987, 472 с.: ил.,
[2] л. ил.
В книге представлены современные методы клинической и эксперименталь-
ной иммунологии. Рассмотрены принципы определения активности антител, реак-
ции антиген — антитело, электрофоретическое исследование белков, культивиро-
вание лимфоцитов и других клеток иммунной системы, изотопные методы и пр.
Материал представлен согласно единому плану: принцип метода, методика, оцен-
ка и практическое использование.
Для иммунологов, иммунохимиков, микробиологов, вирусологов, врачей-ла-
борантов.
4109000000—338
И 039(01)—87 120—87
ББК 53.4
ИММУНОЛОГИЧЕСКИЕ МЕТОДЫ
Монография
Зав. редакцией В. С. Залевский
Редактор В. А. Архипова
Художественный редактор Г. А. Красильщикова
Переплет художника Е. Э. Рыбниковой
Технический редактор Н. М. Гаранкина
Корректор Л. П. Тарарина
ИБ № 4719
Сдано в набор 29.04.87. Подписано к печати 25.08.87. Формат бумаги бОХЭО'/н. Бумага
тип. № 1. Гарнитура литерат. Печать высокая. Усл. печ. л. 29,75. Усл. кр.-отт. 30,5.
Уч.-изд. л. 32,76. Тираж 15 000 экз. Заказ 990. Цена 2 р. 60 к.
Ордена Трудового Красного Знамени издательство «Медицина». 101000, Москва, Петро-
веригский пер., 6/8.
Московская типография № И Союзполнграфпрома при Государственном комитете СССР
по делам издательств, полиграфии и книжной торговли. 113105, Москва, Нагатнискан
УЛ., д. 1.
© VEB Gustav Fischer Verlag Jena, 1984
© Перевод на русский язык. Издатель-
ство «Медицина», Москва, 1987
СОДЕРЖАНИЕ
1. АНТИТЕЛА.................................................... 9
1.1. Получение антисывороток от различных животных. X. Амброзиус
(Н. Ambrosius)................................................. 9
1.2. Получение моноклональных антител путем слияния клеток. X. Фи-
бих (Н. Fiebig)................................................20
1.3. Образование антител культурами клеток. 3. Вихнер (S. Wichner) 34
1.4. Определение аффинности антител. X. Фибих, X. Клейст (Н. Fiebig,
Н. Kleist).....................................................42
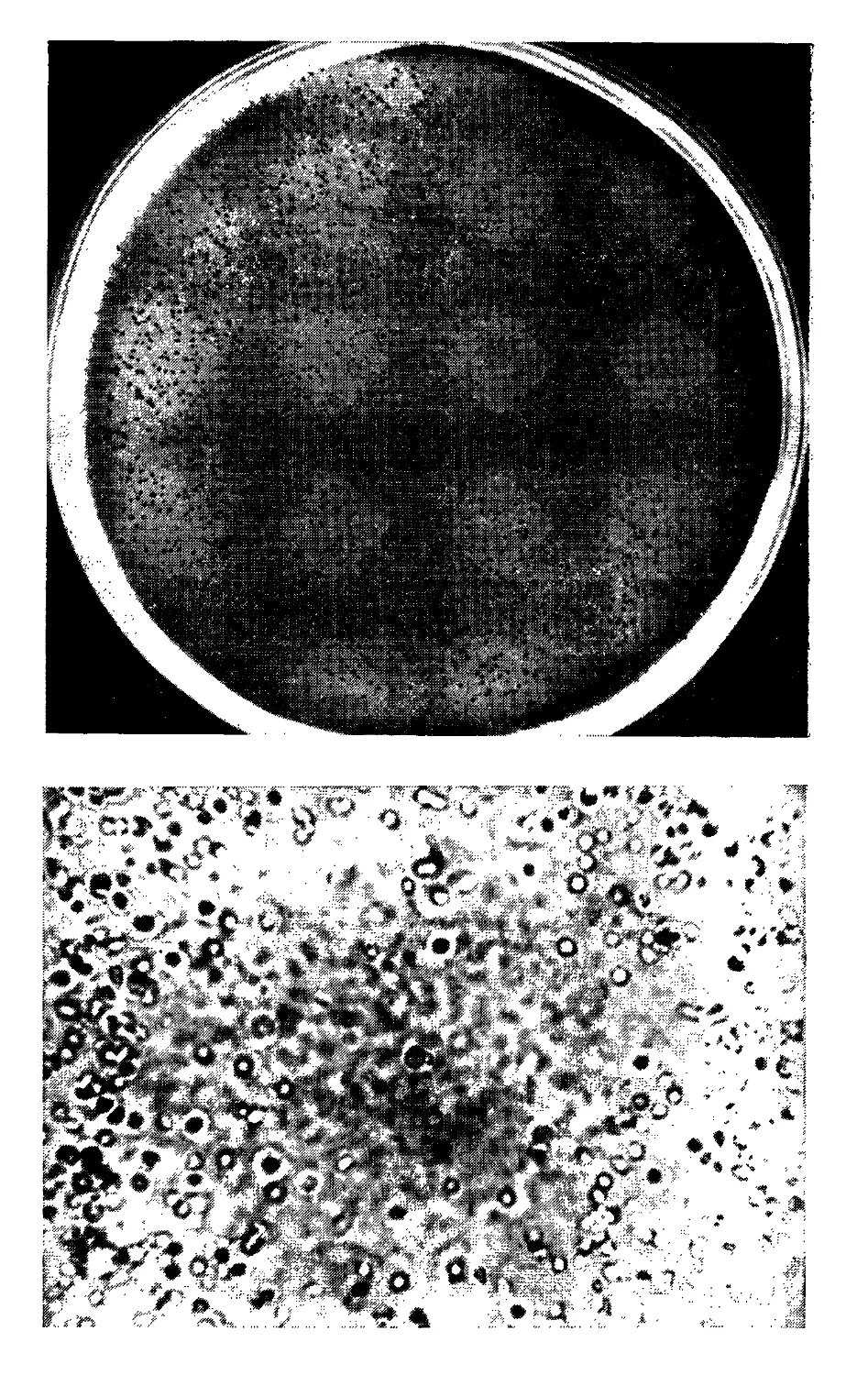
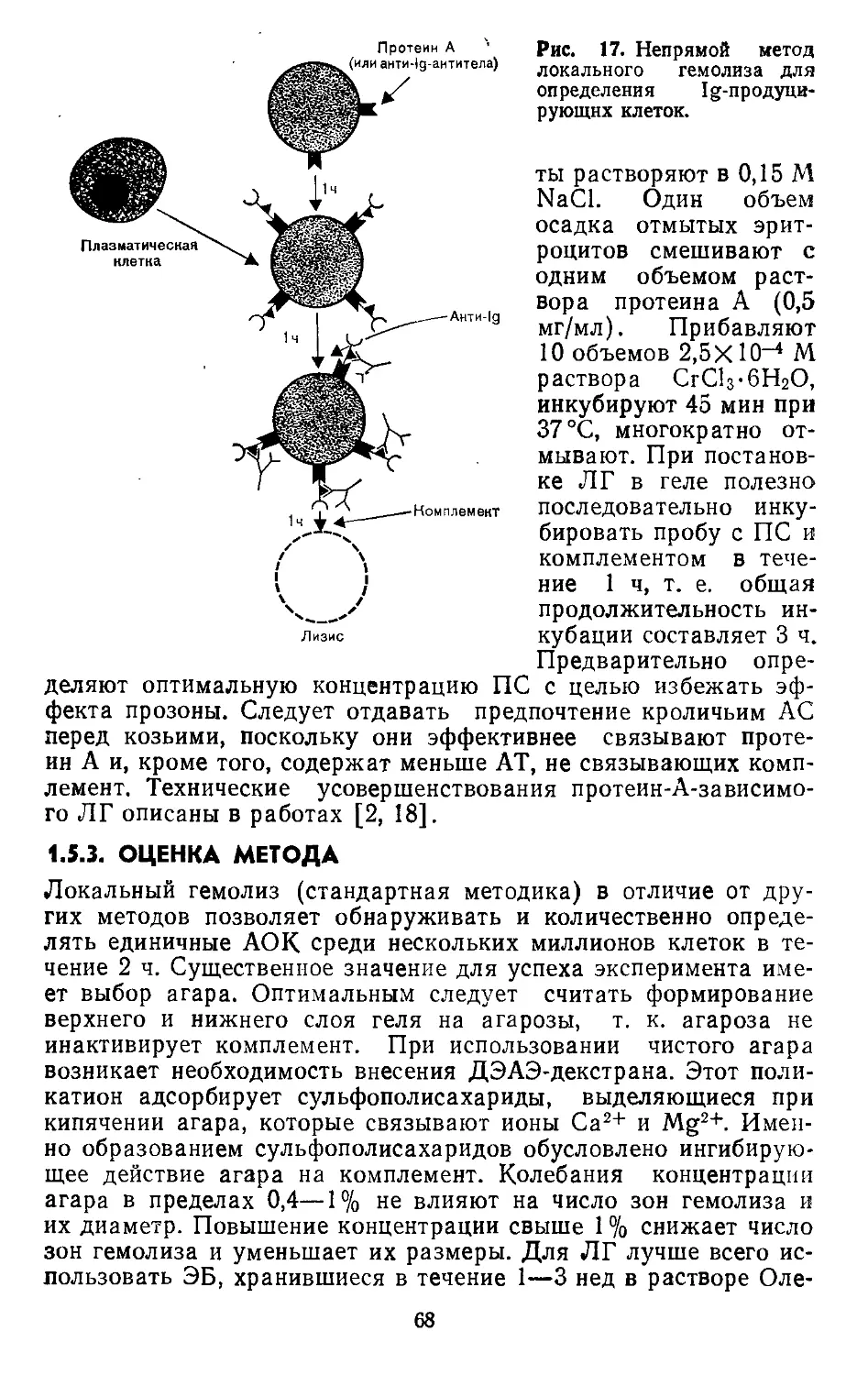
1.5. Метод локального гемолиза. К- Мальберг, Э. Зигль (К. Malberg,
Е. Siegl)......................................................57
2. РЕАКЦИИ АНТИГЕН — АНТИТЕЛО............................................73
2.1. Иммунодиффузия. Э. Бем (Е. Behm)..................................... 73
2.2. Иммуноэлектрофорез (микрометод). X. Фримель (Н. Friemel) 89
2.3. Электроиммунный анализ. Э. Бем (Е. Behm)..............................97
2.4. Встречный электрофорез. К- Вильмс (К. Wilms).........................103
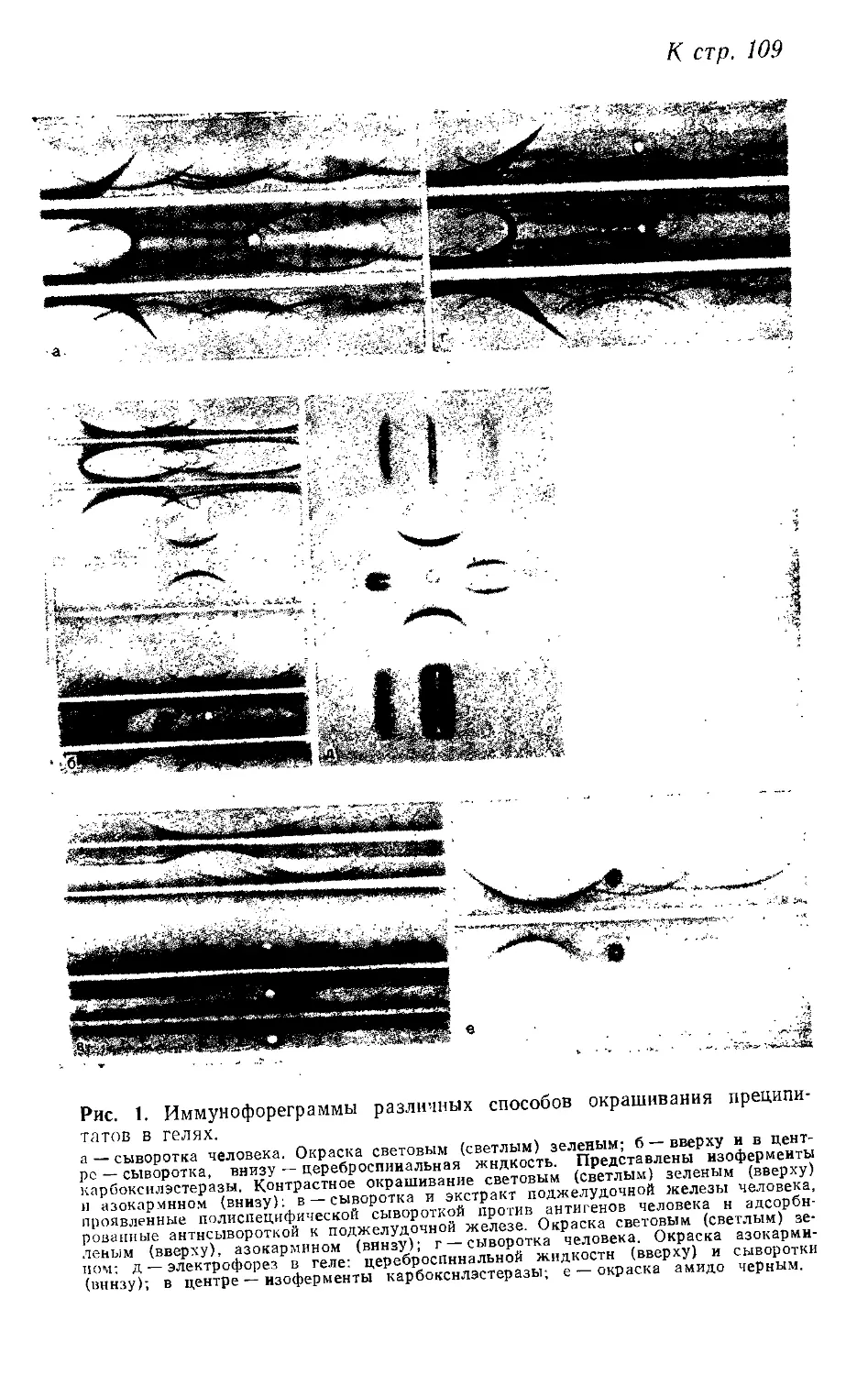
2.5. Анализ иммунных преципитатов в гелях. X. Фримель (И. Friemel) 108
2.6. Фотометрическое определение иммунных агрегатов. X. Тишнер
(Н. Tischner)....................................................115
2.7. Определение растворимых иммунных комплексов. П. Фальк
(Р. Falck)..........................................................120
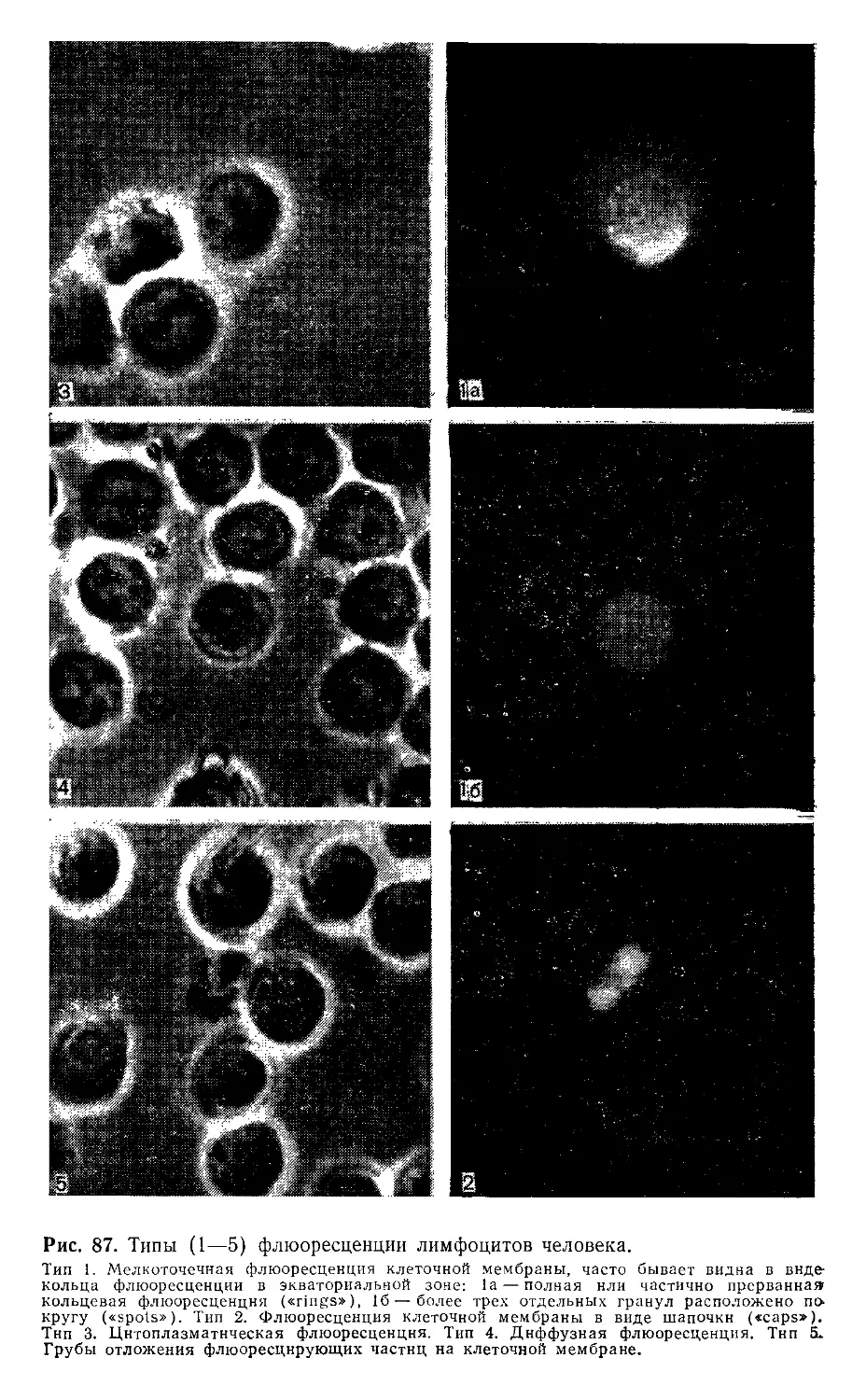
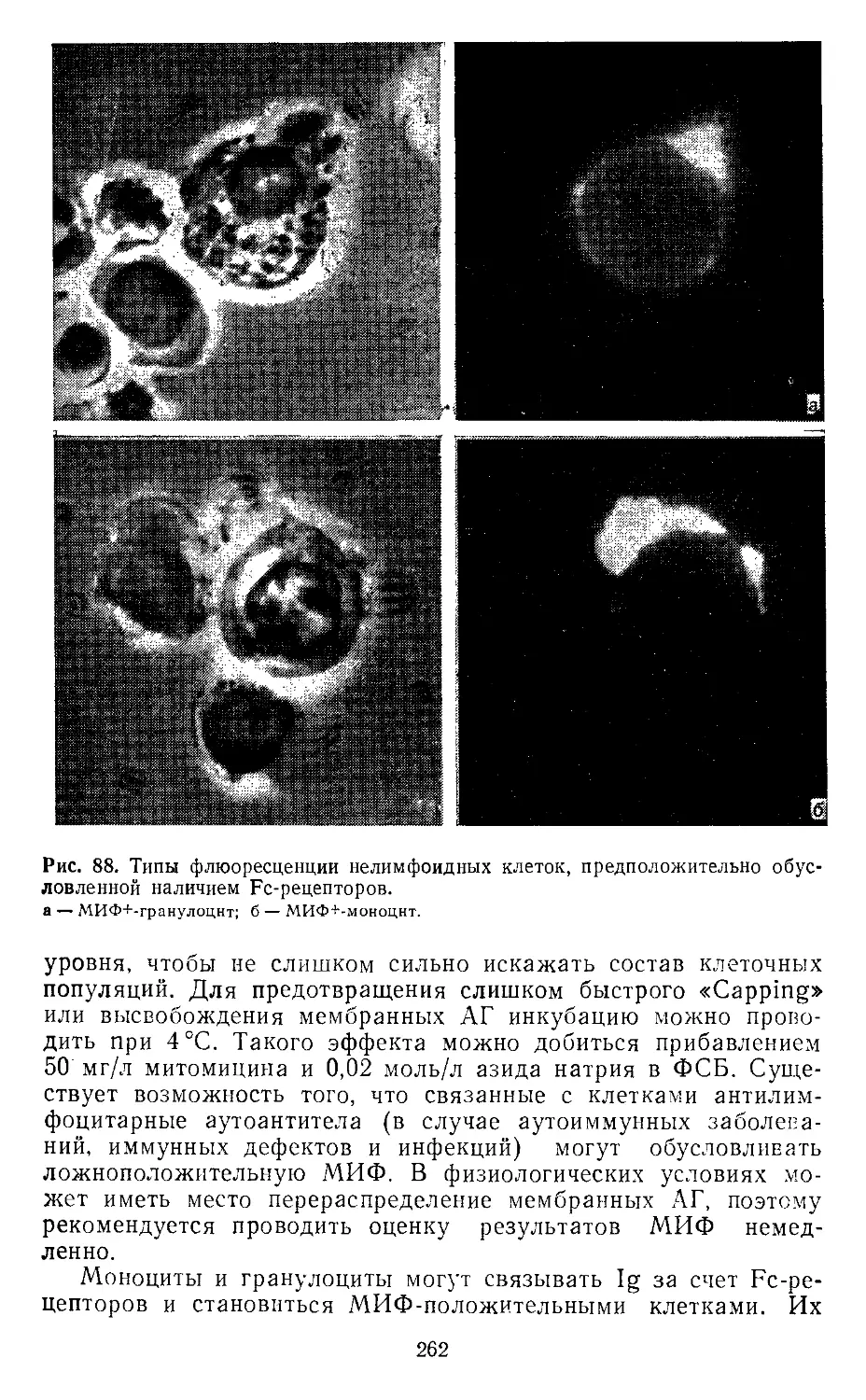
2.8. Иммунофлюоресценция. X. Шторц (И. Storz).............................128
2.9. Радиоиммунологический анализ. П. Фальк (Р. Falck) . . 148
2.10. Иммуиоферментный анализ. X. Фримель, Г. Хольцхайот (Н. Frie-
mel, G. Holzheidt)............................................
2.11. Титрование комплемента. М. Зейфарт (М. Seybarth) .... 171
2.12. Комплементзависимая цитотоксичность. 3. Вегенер (S. Wegener) 176
2.13. Реакция связывания комплемента тромбоцитами. 3. Леееренц
(S. Leverenz)....................................................186
2.14. Реакция связывания комплемента. А. Штельцнер (A. Stelzner) 193
2.15. Антителозависимая клеточная цитотоксичность. Ю. Миллек (J. МИ-
leek).........................................................205
2.16. Реакция гемагглютинации. К- Мальберг (К. Malberg) . . . 211
2.17. Агглютинация латекса. К- Вильмс (К. Wilms)................219
2.18. Реакция агглютинации лейкоцитов. 3. Вегенер (S. Wegener) . . 222
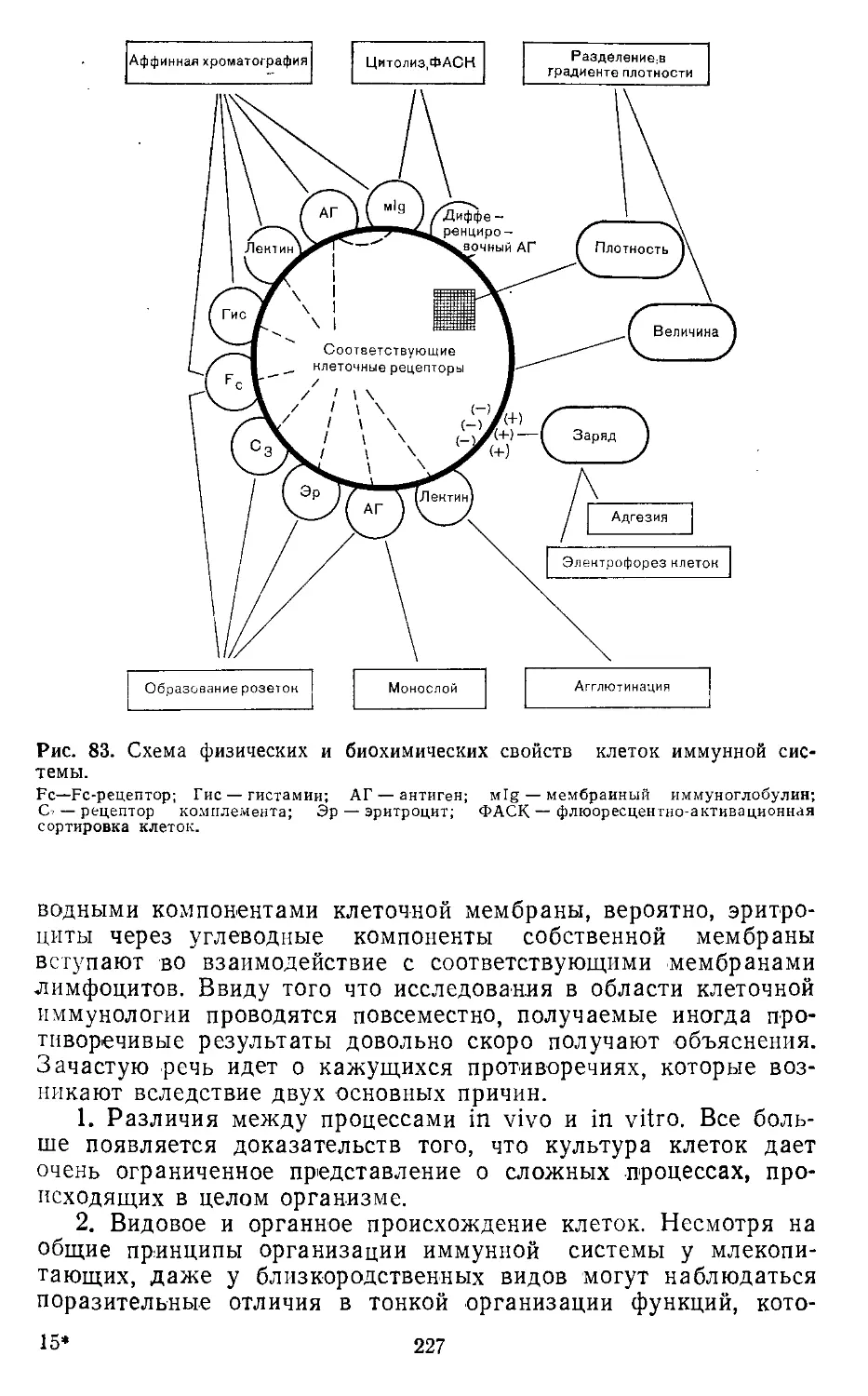
з. клетки иммунной системы.................226
3.1.
3.2.
3.3.
3.4.
3.5.
3.6.
3.7.
3.8.
3.9.
3.10.
Разделение клеток иммунной системы. Р. Эккерт (R. Eckert)
Определение клеточных маркеров методом мембранной иммуно-
флюоресценции. В. Шторх, Й. Эммрих (W. Storch, J. Emmrich)
Метод Е-розеток. Г. Фримель (И. Friemel).....................
Метод ЕАС-розеток. X. Крузе, Г. Эггерс (И. Kruse, G. Eggers)
Метод иммунных розеток. X. Зауэр (Н. Sauer)..................
Цитотоксические лимфоциты. X. А. Шульце (И. A. Schulze)
Реакция бласттрансформации лимфоцитов. X. Шютт (Ch. Schutt)
Смешанная культура лимфоцитов. X. Шютт (Ch. Schutt)
Реакция торможения миграции лейкоцитов (капиллярный метод).
X. Фримель (Н. Friemel)......................................
Реакция миграции лейкоцитов под слоем агарозы. Й. Эммрих,
Ф. Фельбер (J. Emmrich, F. Felber) ........
226
254
269
272
278
282
294
303
308
ЗП
5
3.11. Определение электрофоретической подвижности макрофагов.
X. Келер (Н. Kohler)..............................................317
3.12. Реакция торможения адгезии лейкоцитов. 3. Альбрехт, Г. Пастер-
нак (S. Albrecht, G. Pasternak)...................................321
3.13. Реакция торможения распластывания макрофагов. К. Дрёсслер
(К. Drossier).....................................................327
3.14. Выявление хемотаксических факторов. И. Бланк (I. Blank) . . 333
3.15. Кожнореактивный фактор. Б. Фальбущ, В. Цшише (В. Fahlbusch,
W. Zschiesche)....................................................338
3.16. Кожные реакции замедленного типа. В. Цшише, X. Хайнеке
Н. Heinecke)......................................................342
3.17. Трансплантация кожи. Э. Зигль (Е. Siegl).......................348
3.18. Анафилактические реакции. X. Реннер, Ш. Шницлер (Н. Renner,
S. Schnitzler)....................................................354
3.19. Культивирование макрофагов и моноцитов. П. Дёрфлинг (Р. Dor-
fling) ...........................................................366
3.20. Выделение макрофагов из суспензии сплеиоцитов. П. Дёрфлинг,
3. Вихнер (Р. Dorfling, S. Wischner)............................373
3.21. Фагоцитоз. А. Штельцнер (A. Stelzner)..........................378
4. ИММУНОХИМИЯ.....................................................390
4.1. Получение препаратов иммуноглобулинов. И Брок (J. Brock) 390
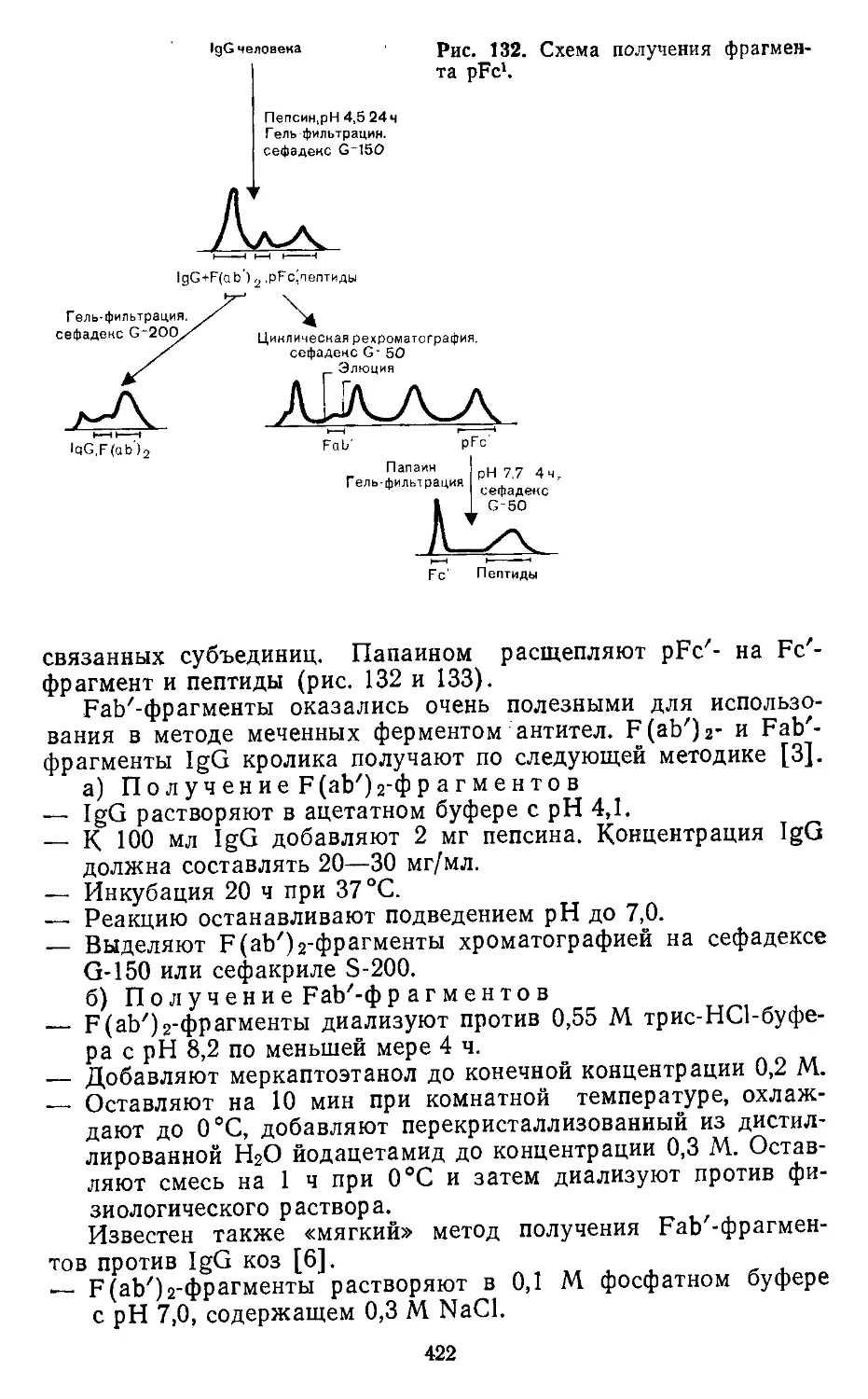
4.2. Фрагментирование иммуноглобулинов. И. Брок (J. Brock) . . 413
4.3. Иммуносорбенты. И. Брок (J. Brock)..............................427
4.4. Приготовление конъюгированных антигенов. И. Брок (J. Brock) 432
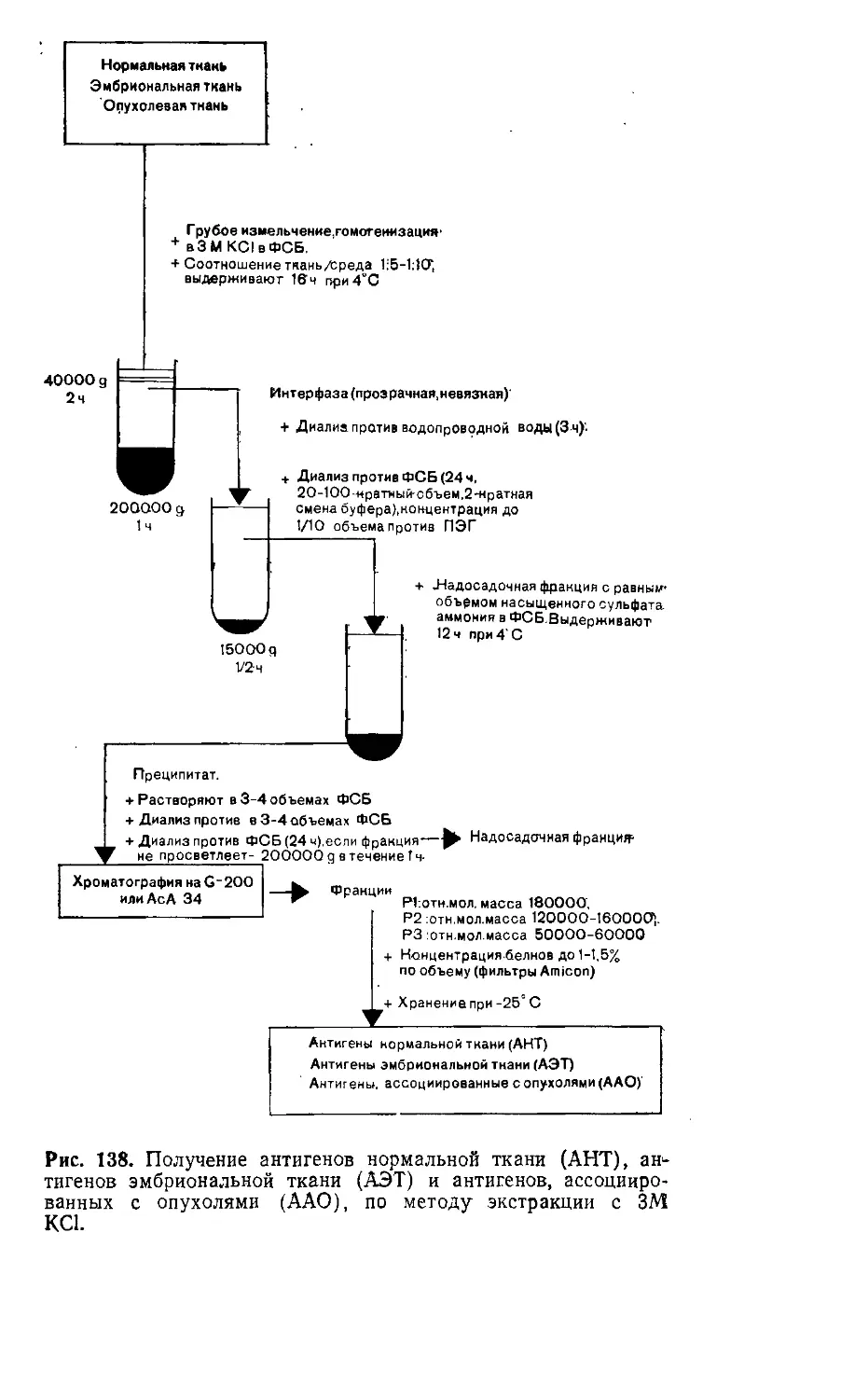
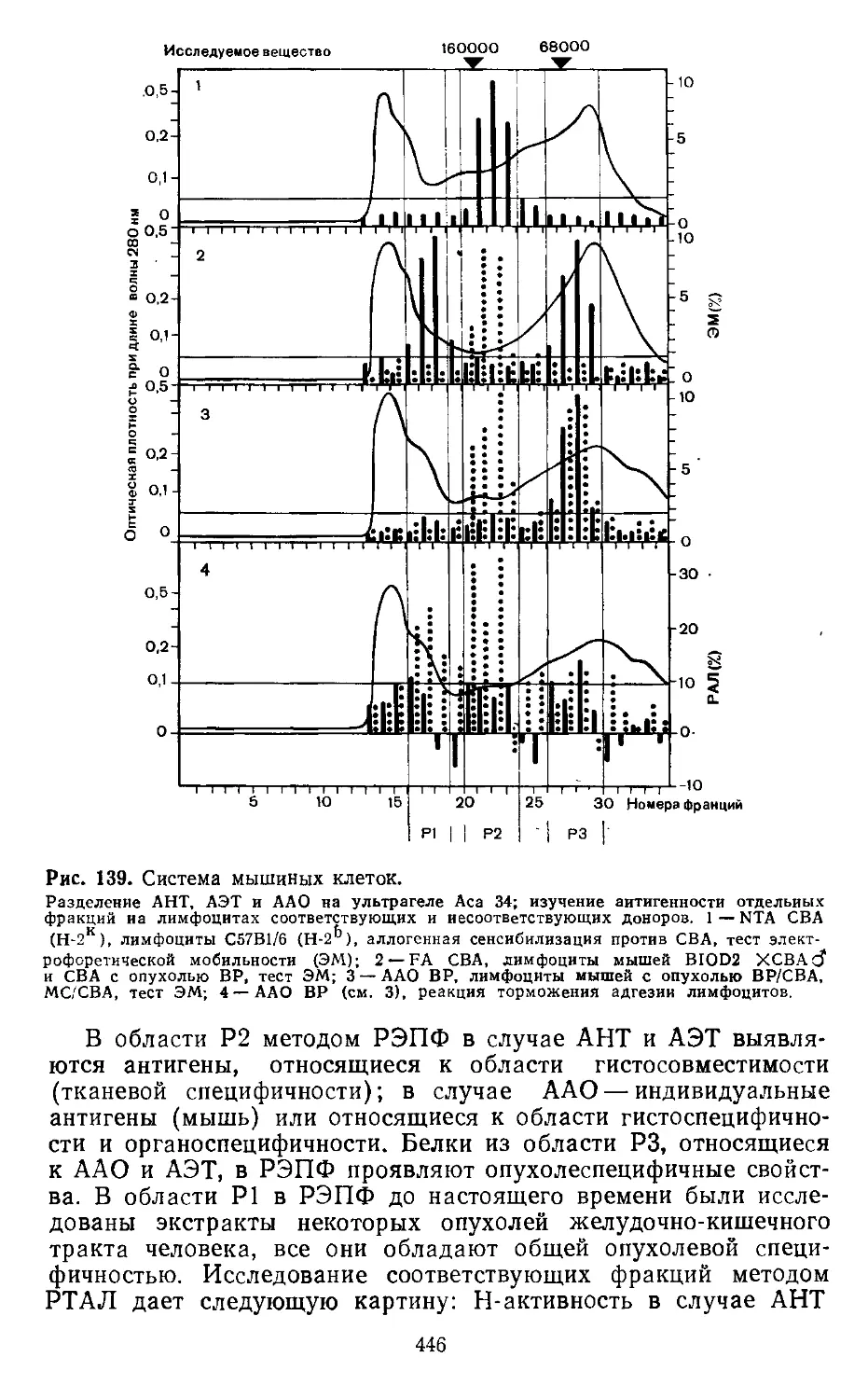
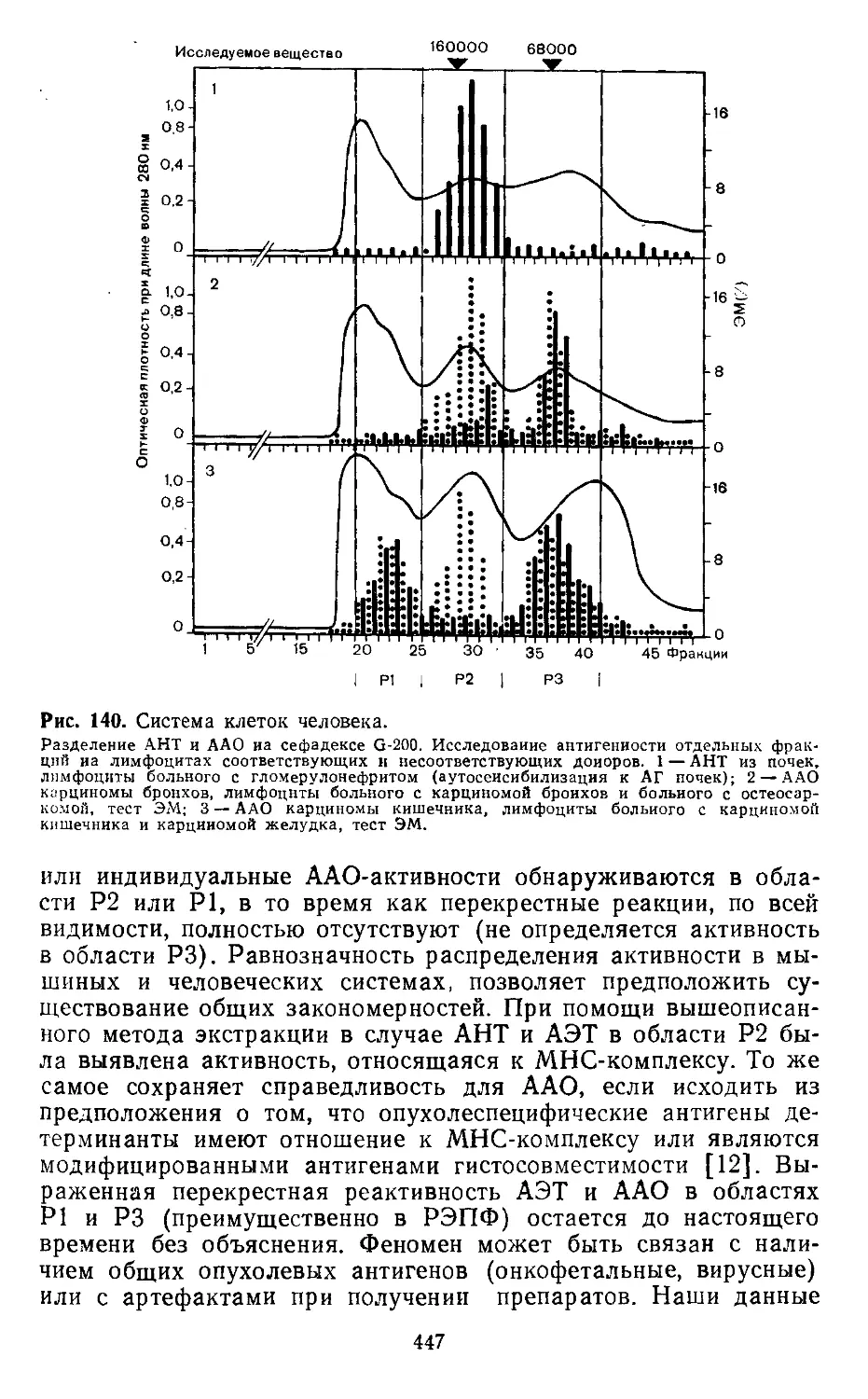
4.5. Получение тканевых экстрактов (3 M-KCI-метод). X. Вернер
(Н. Werner) ....................................................442
4.6. Выделение трансфер-фактора. И. Шрёдер (I. Schroder) . . . 451
4.7. Введение радиоактивной метки в белки. X. Шульфе (Н. A. Schulze) 457
Предметный указатель.................................................467
СПИСОК СОКРАЩЕНИЙ
ААО — аитигеи, ассоциированный
с опухолями
ААТ — аутоантитела
АГ — антиген
АЗКЦ — антителозависимая кле-
точная цитотоксичность
АМА — антимитохоидриальиые
антитела
АМС — аитимакрофагальная сы-
воротка
АНТ — антиген нормальной ткани
АНАЭ — а-нафтилацетатэстераза
АЭТ — аитигеи эмбриональной
ткаии
АС — антисыворотка
АТ — антитело
АФП — а-фетопротеии
АЯФ — антиядерный фактор
Б ДБ — бис-диазотированный бен-
зидин
БНФ — бесклеточная надосадоч-
иая фракция
БПК — бензилпенициллина калие-
вая соль
БСА — бычий сывороточный аль-
бумин
ВИЭФ — встречный иммуноэлект-
рофорез
ГА — гемагглютинация
ГАТ — гипоксантин, амииопте-
рин, тимидин (среда)
ГГФРТ — гипоксаитингуаниифос-
форибозилтраисфераза
ГЗТ — гиперчувствительность за-
медленного типа
ГЭПЭС — N-2-гндроксиэтилпнпе-
разин-Ы-2-этансульфо-
иовая кислота
ДМСО — додецилсульфоксид
ДНФ — 2,4-динитрофеиол
ДРИД — двойная радиальная им-
мунодиффузия
ДСН — додецилсульфат натрия
ДЭЛ — диамид экстракта лейко-
цитов
ИД — иммунодиффузия
ИК — иммунный комплекс
ИЭФ — иммуноэлектрофорез
КИПЭ — клетки индуцированного
перитонеального экссуда-
та
КоиА — коикаиавалии А
ЛДГ — лактатдегндрогеназа
ЛПС — липополисахарид
ЛРК— лимфоциты с рецептора-
ми комплемента
МБГСЭ — т-малеимидобеизоил-N-
гидроксисукцинимидный
эфир
МИР — метод иммунных розеток
МИФ — мембранная иммуиофлюо-
ресцеицня
МКАТ — моноклональные антитела
НАФ — неполный адъювант
Фрейида
ОЛГ — обратный локальный ге-
молиз
ОП — оптическая плотность
ПААГ — полиакриламид
ПАФ — полный адъювант Фрейнда
ПВХ — поливинилхлорид
ПМЯЛ — полиморфно-ядерные лей-
коциты
ПХМБ — парахлормеркурибензоат
ПЭГ — полиэтиленгликоль
РАА — р-ция адгезии лейкоцитов
РИА — радиоиммунологический
анализ
РЛЦ — реакция лимфоцитолиза
РТАЛ — реакция торможения ад-
гезии лейкоцитов
РОК — розеткообразующие кл-ки
РЭФП — реакция электрофоретиче-
ской подвижности
СИ —степень йодирования
СКЛ — смешанная культура лей-
коцитов
СППТ — N-сукцинимидил-З- (2-пи-
ридилтио) -пропионат
СЭБ — суспензия эритроцитов
барана
ТГ — титр гемолизина
ТК — тимидиикииаза
ТНБК — 2,4,6-тринитробензол-
сульфоиовая кислота
ТНС — тетразолий нитросииий
ТНФ — 2,4,6-тринитрофенил
ТРИТЦ — тетраметилродаминизо-
тиоциаиат
ТХУ — трихлоруксусная кислота
ТФ —трансфер-фактор
ФИТЦ — флюоресцеииизотиоциаиат
ФСБ — фосфатио-солевой буфер
ЧГГ — гамма-глобулин человека
ЧСА—сывороточный альбумин
ЭБ — эритроциты барана
ЭКДИ — 1-этил-1,3 (3-диметилами-
нопропил) карбодиимид
Ig — иммуноглобулин
LIF — фактор, тормозящий миг-
рацию лейкоцитов
MIF — фактор, тормозящий миг-
рацию макрофагов
MSF — фактор, тормозящий
электрофоретическую по-
движность макрофагов
Rz — букв, число чистоты фер-
мента
МЕМ — электрофоретическая по-
движность макрофагов
ПРЕДИСЛОВИЕ К 3-МУ ИЗДАНИЮ
Настоящее, третье, издание книги было задумано с учетом
изменений, вызванных развитием иммунологии. Расположение
материала книги по схеме: антитела-^реакции антиген — анти-
тело-?-клетки иммунной системы-5-иммунохимия должно облег-
чить ориентировку. Авторский коллектив в составе 41 ученого,
согласившийся принять участие в работе, предоставил для этой
цели свои знания и методологический опыт в области иммуно-
логии. Редактор, авторы и издательство благодарят всех, кто
прислал свои замечания и предложения по улучшению книги,
которые в значительной мере были учтены.
Благодаря слаженной работе всех участников вышла в свет
эта книга, которая, как мы надеемся, окажется весьма полез-
ной при внедрении современных иммунологических методов в
лабораторную практику.
Росток, июль, 1984 г. Редактор
I. АНТИТЕЛА
1.1. ПОЛУЧЕНИЕ АНТИСЫВОРОТОК
ОТ РАЗЛИЧНЫХ ЖИВОТНЫХ
1.1.1. ПРИНЦИП МЕТОДА
Для большинства иммунологических и серологических методов
исследования необходимо иметь соответствующие антисыворот-
ки. Антисыворотки к так называемым стандартным антигенам,
например человеческому IgG, имеются в продаже, к нестандарт-
ным антигенам антисыворотки приходится получать в лаборато-
рии. Основные требования к таким сывороткам — специфич-
ность и достаточное содержание антител (АТ). Иммунологиче-
ская специфичность не абсолютна, она тесно связана со струк-
турой антигена (АГ). Белки ввиду относительного многообразия
составляющих их аминокислот и практически неограниченной
возможности их комбинирования представляют собой природ-
ные соединения с исключительно разнообразным строением.
С точки зрения иммунологии это означает, что антитела к опре-
деленному белковому антигену могут реагировать только с ним
и родственными по структуре белками. Под последними следует
понимать гомологичные протеины родственных биологических
видов, а также белки, образующие одно «семейство», например
гонадотропины. Все они имеют общую a-цепь, в то время как
р-цепи содержат полипептидные последовательности, индивиду-
альные для каждого гормона. Способность к перекрестным ре-
акциям антител с антигенными детерминантами структурно
близких белков находит практическое применение. Допустим,
требуется получить антитела к какому-либо гормону гипофи-
за человека; получение антисыворотки невозможно ввиду отсут-
ствия достаточного количества человеческих гормонов. В то же
время получение этих гормонов из гипофиза свиней или быков
вполне осуществимо.
При использовании бычьего гормона для получения антител
существует высокая вероятность того, что индуцированные АТ
будут давать выраженную перекрестную реакцию с аналогич-
ным гормоном человека и, следовательно, окажутся пригодными
для постановки специфических проб. Не следует, конечно, ожи-
дать, что антитела к какому-либо определенному белку будут
реагировать со структурно неродственным белком; избиратель-
ность иммунологических реакций обусловлена их высокой спе-
цифичностью. Углеводы, липиды и особенно нуклеиновые кис-
9
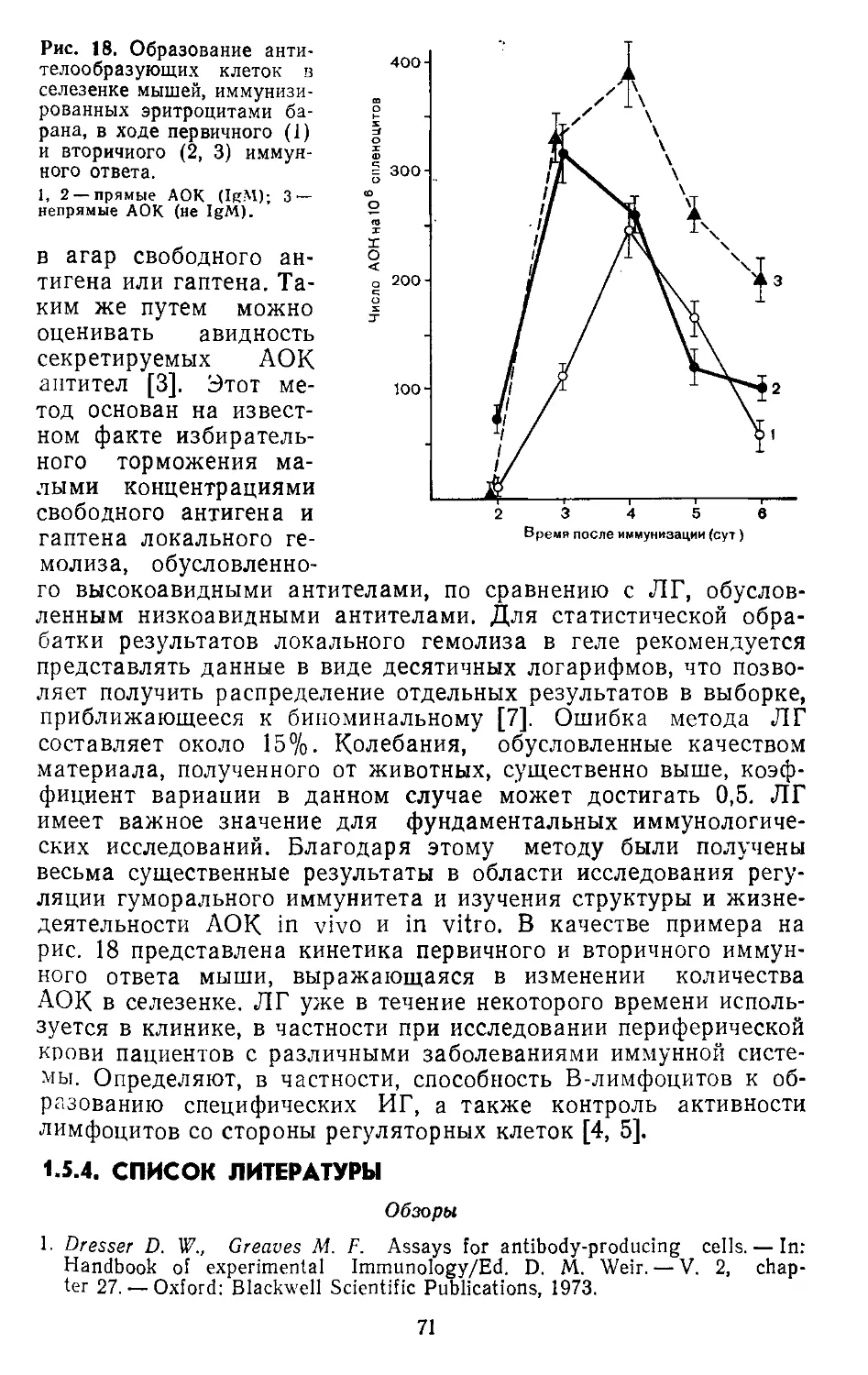
Вторичный ответ
Первичный ответ
1------>
4 Время (нед}
Рис. 1. Схематическое изображение хода различных процессов при первич-
ном и вторичном иммунном ответе кролика иа введение белковых АГ.
лоты в отличие от белков состоят из сравнительно малого числа
структурных элементов, возможность комбинирования которых
ограничена по сравнению с белками. Можно с большой степенью
вероятности предположить, что вещества такого типа, будучи
выделены из разных источников, будут обладать выраженным
структурным сходством. С иммунологической точки зрения это
означает, что антитела к углеводам, липидам и нуклеиновым
кислотам в отличие от АТ к белкам будут активно вступать в
перекрестные реакции с филогенетически неродственными анти-
генами. Подобные взаимодействия отличаются низкой специ-
фичностью, ограничивающей их использование в иммунологиче-
ских исследованиях. Это порождает необходимость разработки
дополнительных тест-систем. Для практической иммунологии
можно сделать следующее заключение: антитела к белкам об-
ладают высокой специфичностью, они удобны для определения
антигенов; антитела к липидам, углеводам и нуклеиновым кис-
лотам малоспецифичны, что приводит к не всегда желательным
перекрестным реакциям (рис. 1). Для получения сывороток с
высоким титром АТ необходимо учитывать и соответствующим
образом использовать механизмы регуляции гуморального им-
мунного ответа. Например, введение больших доз антигена
может индуцировать образование супрессорных клеток, подав-
ляющих выработку АТ, и, наоборот, использование феномена
«иммунологической памяти» может существенно облегчить по-
лучение сыворотки с высоким титром антител. Введение антиге-
на приводит не только к образованию антител, но и к появле-
нию «клеток памяти» — лимфоцитов со специфической реактив-
ностью в отношении вызвавшего иммунную реакцию антигена.
«Клетки памяти» распространяются по всему организму и оседа-
ют в периферических органах лимфатической системы. Повтор-
ное введение антигена приводит к заметному увеличению выра-
10
ботки антител по сравнению с первичным иммунным ответом;
антитела вторичного иммунного ответа циркулируют в крови
более продолжительное время (см. рис. 1). Поскольку образо-
вание «клеток памяти» — процесс довольно длительный, не сле-
дует выбирать слишком маленький промежуток времени между
первичным и повторным введением антигена. Показано, что для
мышей оптимальный промежуток времени после первого введе-
ния АГ составляет 90 дней [14]. Скорость возникновения имму-
нологической памяти зависит от природы антигена, принято
считать что у кроликов и морских свинок она возникает не ра-
нее чем через 6 нед, у крыс и мышей не ранее чем через 4 нед,
у низших позвоночных не ранее чем через 3 мес. Гуморальный
иммунный ответ можно усилить при помощи адъювантов, этот
способ известен давно. Наиболее распространены полный адъ-
ювант Фрейнда и гидроокись алюминия. В последнее время в
качестве адъювантов применяли мурамилпептиды. Полный
адъювант Фрейнда (ПАФ) выпускается в виде коммерческого
препарата, в его состав входит минеральное масло, эмульгатор
и убитые микобактерии туберкулеза. ПАФ смешивают с водным
раствором антигена до получения стабильной водно-масляной
эмульсии. На процесс эмульгирования следует обращать особое
внимание, так как адъювантный эффект наиболее выражен у
стабильных эмульсий. Гидроокись алюминия не обладает столь
выраженными стимулирующими свойствами, как ПАФ, но по
сравнению с последним имеет то преимущество, что при много-
кратном введении не вызывает изменений в периферических
лимфатических органах. Вопрос выбора адъюванта решается в
каждом случае индивидуально. По-видимому, идеальные анти-
сыворотки можно получать, используя гибридомную технологию
(см. раздел 1.2), разработанную в лаборатории Мильстайна
(Кембридж). Получают клеточные гибриды из лимфоцитов им-
мунизированного животного и культивируемых in vitro миелом-
ных клеток; из полученных гибридов отбирают клоны клеток,
синтезирующих определенные АТ. Моноклональные антитела
обладают абсолютно одинаковой специфичностью в отличие от
поликлональных антител, содержащихся в обычных сыворотках,
имеющих различную специфичность и дающих перекрестные ре-
акции.
1.1.2. МЕТОДИКА
1.1.2.1. ЭКСПЕРИМЕНТАЛЬНЫЕ ЖИВОТНЫЕ
Несколько лет назад Р. F. Schollander на заседании физиологов
сказал: «Для специалистов по медицинской энзимологии чело-
век состоит из сердца и почек собаки, мышц лягушки, кишеч-
ника крысы, генов дрозофилы и т. д. Для меня всегда было за-
гадкой такое „антропоидное поведение'1 животных». Для 99%
иммунологов и тех, кто пользуется иммунологической техникой,
И
не существует других животных, пригодных для получения ан-
тисывороток, кроме кроликов, морских свинок, крыс и мышей.
Это тем более необъяснимо, что еще в начале XX века было
известно, что сила иммунного ответа и его специфичность зави-
сят от степени биологической близости животных, от которых
получают антиген, и животных — продуцентов антисыворотки.
Общих рецептов на этот счет не существует, но можно приве-
сти примеры, показывающие, что необходимо тщательно выби-
рать экспериментальных животных, не ограничиваясь приняты-
ми в обычных случаях видами. В качестве иллюстрации можно
привести работу van der Giessen с соавт. [6]. В ней были пред-
приняты попытки получить антисыворотки к 4 классам имму-
ноглобулинов человека. Иммунизации подвергались обезьяны,
кролики, морские свинки, бараны, куры и одна коза; для инъ-
екции использовали продукты биосинтеза соответствующих мие-
лом и 3 различные адъюванта. Антисыворотки кур, козы и ба-
ранов не обладали субклассовой специфичностью. По отношению
к птицам, которые филогенетически далеко отстоят от человека,
этого следовало ожидать. Обратная картина должна наблю-
даться у обезьян, они филогенетически близки человеку, их им-
мунная система должна относительно легко улавливать анти-
генные различия субклассов иммуноглобулинов. И действитель-
но, обезьяны представляют собой удобный источник антисыво-
роток к субклассам IgGa, IgG3 и IgG4, но не IgGj. При получе-
нии антисывороток к какому-либо гормону, помимо иммуно-
логических, имеют значение физиологические факторы. В этом
случае стараются выбирать животное, наименее чувствительное
к данному гормону, поскольку гормональный фон оказывает
влияние на иммунный статус организма. Так, например, птицы
реагируют на введение гормонов, в том числе инсулина, не так
резко как млекопитающие, в то же время они образуют антите-
ла приблизительно с такой же интенсивностью. В многочислен-
ных экспериментах было установлено, что животные различных
инбредных линий по-разному реагируют на один и тот же анти-
ген. В качестве примера, имеющего практическое значение,
можно привести работу Shen с соавт. [17] о получении специ-
фических антисывороток к антигенам мембран лимфоцитов.
Одни линии и гибриды активно продуцировали антитела, в то
время как другие не образовывали их даже в следовых количе-
ствах. Низших позвоночных, например рыб, очень удобно ис-
пользовать для получения антисывороток, хотя они очень удоб-
ны в содержании и продуцируют большие количества антител.
Рыбы особенно пригодны для работы с так называемыми «кон-
сервативными антигенами», т. е. с такими, которые медленно
изменялись в ходе эволюции. Медленно эволюционирующие ан-
тигены млекопитающих, как правило, не вызывают иммунного
ответа у млекопитающих другого вида ввиду минимальных
структурных различий. В таких случаях экспериментальных жи-
вотных нужно выбирать из филогенетически отдаленных клас-
12
сов позвоночных. В частности, карликовый сом продуцирует ан-
тисыворотки с высоким титром АТ против групповых антигенов
крови человека в ответ на введение им слюны секреторов. Рыбы
оказались удобными продуцентами антител, применяемых для
изучения филогенетического родства белков иммунохимически-
ми методами. [7]. Следует отметить, что у низших позвоночных
иммунный ответ зависит от температуры тела. Интенсивное ан-
тителообразование наблюдается только при содержании живот-
ных у верхней границы физиологических значений температуры.
Не последнее место в выборе подопытных животных занимают
экономические соображения. Для получения большого количе-
ства АТ не всегда есть возможность содержать 100 кроликов
или 1 лошадь. С этой точки зрения представляет интерес следу-
ющее наблюдение: беременных коров можно иммунизировать
без вреда для плода, после отела удается получить несколько
литров молозива, в котором содержание антител в несколько
раз превышает их концентрацию в сыворотке крови. Эту техно-
логию использовали для получения больших количеств АТ к
Н-антигену сальмонелл, ферритину, IgG кролика и человека.
1.1.2.2. ПОЛУЧЕНИЕ АНТИСЫВОРОТОК
К БЕЛКОВЫМ АНТИГЕНАМ
Ранее было отмечено, что оптимальный способ иммунизации за-
висит от природы антигена, вида подопытного животного и це-
ли, которой будет служить антисыворотка. В качестве примера
можно привести получение антисывороток путем иммунизации
кроликов и карпов белковыми антигенами в нашей лаборато-
рии. Этот метод применим и к другим моделям. Далее описыва-
ется способ иммунизации, основанный на применении очень ма-
лых количеств антигена. В заключение рассматривается вопрос
о возможности получения моноспецифических сывороток.
1.1.2.2.1. Иммунизация кроликов белковыми антигенами
Материалы и оборудование: для получения одного антигена не-
обходимо 3 кролика любой породы, желательно с большими
ушами для удобства отбора крови из ушных вен; 0,14—14 мг
антигена на кролика; ПАФ; шприц для инъекций (2 мл) с иг-
лой № 16, раствор NaCl1 (9 г/л).
Проведение иммунизации: первую иммунизацию проводят с
ПАФ. Каждому кролику вводят смесь 0,5 мл антигена (20—
2000 мкг) с 0,5 мл ПАФ. Для получения стабильной эмульсии
смесь многократно набирают в шприц и с силой выпускают че-
рез тонкую иглу. О качестве препарата судят по тому, что кап-
ли эмульсии при попадании в воду не распадаются в течение
получаса и более. Смесь антиген-ПАФ в количестве 1 мл вво-
1 В тех случаях, когда концентрация NaCl не указана, речь идет об изо-
тоническом растворе натрия хлорида. — Примеч. пер.
13
дят внутрикожно в подушечки стоп всех четырех конечностей.
Последующую иммунизацию проводят через 8 нед после первой,
таким же количеством антигена в 0,5 мл 0,15 М NaCl. В первый
день последующей иммунизации антиген вводят внутримышеч-
но в верхнюю часть бедра, а на 2-й и 3-й дни внутривенно в
ухо. Через неделю иммунизацию повторяют по той же схеме.
Забор крови производят через 7—10 дней после последней им-
мунизации.
1.1.2.2.2. Иммунизация карпов белковыми антигенами
Материалы и оборудование: для получения одного антигена не-
обходимы 5 карпов с массой тела 500—1500 г. Рыб адаптируют
к температуре воды 24 °C в течение 2 нед и содержат при этой
температуре весь период иммунизации. Как правило, карпы
продуцируют летом больше антител, чем зимой. Необходимо
иметь 1—2 мг антигена на каждую особь, ПАФ, неполный адъ-
ювант Фрейнда (НАФ), шприц (2 мл) с иглой № 16, раствор
NaCl (60 г/л).
Проведение иммунизации: первичную иммунизацию прово-
дят с ПАФ. Раствор антигена в количестве 0,25 мл (100—
1000 мкг) тщательно смешивают с ПАФ и вводят получившуюся
эмульсию внутрибрюшинно. Повторную иммунизацию проводят
через 2 мес. Затем 0,25 мл раствора антигена (1000—2000 мкг)
смешивают с 0,25 мл НАФ и вводят внутрибрюшинно. Через
2 нед забирают кровь из сердца и анализируют сыворотку. Ес-
ли количество антител недостаточно, то через 4 нед проводят
третью иммунизацию с 0,5 мл раствора антигена без адъюван-
та. Забор крови производят через 2 нед после последней имму-
низации.
1.1.2.2.З. Метод эффективной иммунизации кроликов
небольшими количествами антигена
Материалы и оборудование (см. в разделе 1.1.2.2.1). В основу
этого метода положена индукция локальной иммунной реакции
лимфатических узлов малой дозой антигена. Образующиеся
клетки памяти распределяются по всему организму, последую-
щее введение соответствующего антигена стимулирует популя-
цию сенсибилизированных лимфоцитов, начинающих интенсив-
но синтезировать антитела. Готовят эмульсию антигена и ПАФ.
Содержание антигена должно быть не менее 10 мкг/мл. Каждо-
му кролику вводят около 1 мл эмульсии. Большим и указатель-
ным пальцами находят подколенный лимфатический узел зад-
ней ноги. Для этого необходим определенный навык, приобре-
таемый при работе с животным, которому за неделю до этого
вводили ПАФ под кожу верхней части бедра.
Эта инъекция приводит к увеличению лимфатических узлов,,
которые становятся доступными для пальпации. Однако эти жи-
вотные вследствие структурных изменений лимфатических узлов'
по меньшей мере в течение 2 мес непригодны для иммунизации.
14
Животных, ранее бывших в опыте, не следует иммунизировать.
После того как лимфатический узел найден, его фиксируют кон-
чиками пальцев. В лимфатический узел тонкой иглой вводят
0,3 мл эмульсии. При попадании материала в лимфатический
узел последний вздувается под пальцами экспериментатора.
Наш опыт показывает, что даже при инъецировании окружаю-
щих лимфатический узел тканей наблюдается выраженный им-
мунный ответ. Точно также антиген вводят в симметричный
лимфатический узел. Оставшиеся 0,4 мл смеси вводят равными
частями подкожно в подушечки стоп задних конечностей. Не
ранее чем через 6, а лучше через 8 нед проводят повторную им-
мунизацию. Для этого антиген вводят с НАФ или в растворе
NaCl. Каждому кролику вводят не менее 10 мкг материала;
по 0,1—0.2 мл смеси вводят подкожно в каждую конечность.
Подкожные инъекции повторяют с недельным интервалом до
достижения достаточного титра антител.
1.1.2.2.4. Получение моноспецифических сывороток при
отсутствии полностью очищенного антигена
Во многих случаях очень сложно или даже невозможно полу-
чить нужный антиген в чистом виде. Существует несколько спо-
собов получения моноспецифических сывороток без полной очи-
стки антигена.
Иммунизация белками, разделенными в полиакриламидном
геле: при помощи диск-электрофореза, изоэлектрофокусирова-
ния можно получить отдельные фракции антигена, свободные
от примесей. Поскольку полиакриламид обладает свойствами
адъюванта, можно проводить иммунизацию непосредственно ку-
сочками полиакриламида, содержащими соответствующий ком-
понент. Достаточно взять 1 мкг белка. В остальном иммуниза-
цию проводят так же, как в случаях с небольшими количества-
ми антигена.
Иммунизация антигеном из зоны преципитации при иммуно-
электрофорезе: этот метод существенно отличается от обычных
способов очистки антигена. Подопытное животное, например
кролика, иммунизируют неочищенным препаратом антигена.
После получения сыворотки с высоким титром АТ проводят им-
муноэлектрофорез в микромодификации на предметном стекле.
Затем идентифицируют линию преципитации соответствующего
антигена, для чего используют селективное окрашивание, фер-
ментативные реакции или сравнение со стандартным антигеном.
После идентификации иммуноэлектрофорез проводят парал-
лельно на большом количестве стекол. Линию преципитации ан-
тигена тщательно вырезают, избегая мест перекрещивания с
другими линиями преципитации. Вырезанные фрагменты отмы-
вают в течение 2 дней в растворе NaCl с многократной сменой
раствора, затем в течение 1 дня дистиллированной водой с до-
бавлением цитостатиков, например мертиолята. После отмывки
фрагменты геля подвергают механическому измельчению
15
Рис. 2. Иммуноэлектрофореэ
с применением антисыворотки
к бычьему ^-глобулину.
В качестве АГ использовали грубо*
очищенную фракцию глобулинов
(вверху) н сыворотку крупного ро-
гатого скота (внизу).
(скальпелем и ножница-
ми) и суспендируют в
1 мл раствора NaCl. Сус-
пензию эмульгируют с
ПАФ. Эмульсию вводят
кролику в лимфатические
узлы и подушечки стоп. В отличие от других схем иммунизации
повторную иммунизацию проводят через 6 нед с использованием
ПАФ, а не НАФ. Эмульсию преципитат-адъювант в количестве
0,4 мл медленно вводят в вены уха, остальное количество вво-
дят подкожно в конечности. Через 7 дней определяют титр сы-
воротки иммунизированных животных в иммуноэлектрофорезе.
При низких титрах проводят повторную иммунизацию отмытым
преципитатом без адъюванта подкожно. Повторные иммуниза-
ции проводят с недельным интервалом. Если же антисыворотка
не обладает моноспецифичностью, прибегают к иммунизации
других животных преципитатами, полученными при помощи оли-
госпецифической сыворотки. До настоящего времени нам всегда
удавалось получить моноспецифическую сыворотку от одного
или двух животных, задействованных в эксперименте (рис. 2).
Получение моноспецифической сыворотки путем адсорбции:
олигоспецифическую сыворотку можно сделать моноспецифиче-
ской, удалив антитела с нежелательной специфичностью с по-
мощью адекватных сорбентов. Как правило, для этого исполь-
зуют нерастворимые иммуносорбенты, получаемые при соедине-
нии антигена с инертным носителем, например, при помощи
глютарового альдегида.
1.1.2.3. ПОЛУЧЕНИЕ АНТИСЫВОРОТОК
К НИЗКОМОЛЕКУЛЯРНЫМ ВЕЩЕСТВАМ
Антитела к небольшим молекулам представляют значительный
интерес в биохимических и фармакологических исследованиях,
их можно также использовать в клинической диагностике. По-
лучение таких антител осложнено тем, что низкомолекулярные
вещества, как правило, неиммуногенны и сами по себе не инду-
цируют антителообразование. Антигенные детерминанты таких
веществ (участок поверхности антигена, связываемый антите-
лом) состоят из небольшого числа аминокислотных или угле-
водных остатков. Чтобы перевести небольшие молекулы в им-
муногенное состояние, можно агрегировать их в частицы боль-
16
шего размера, или присоединить к соответствующему носителю..
Предпочтительнее пользоваться вторым способом, важное зна-
чение при этом имеет правильный выбор носителя. Ниже при-
водится несколько примеров, они помогают понять ход последу-
ющих рассуждений. Наиболее часто в качестве носителя ис-
пользуют альбумин. Так были получены антитела к витамину А.
путем его присоединения к человеческому сывороточному аль-
бумину (ЧСА) и последующей иммунизацией кроликов [3]. Им-
мунизируя кроликов конъюгатом трийодтиронина с бычьим сы-
вороточным альбумином (БСА), можно индуцировать образо-
вание антител к трийодтиронину, с помощью которых можно
определять до 20 пкг трийодтиронина. Перекрестная реакция с
тироксином наблюдалась примерно в 0,4% случаев [8]. Значе-
ние природы носителя для успеха иммунизации становится оче-
видным из следующего примера: лизин-вазопрессин и парати-
\ реоидный гормон конъюгировали с БСА, яичным альбумином и
- ' некоторыми синтетическими полимерами аминокислот. Провер-
\ ’> ка конъюгатов на 70 кроликах и морских свинках показала.
полное отсутствие иммуногенности. Присоединение этих гормо-
нов к тиреоглобулину позволило получить препараты, индуци-
рующие образование антисывороток с высоким титром АТ у
' всех животных [18]. Антитела, получаемые при иммунизации
конъюгатами на основе белка, в основном принадлежат к клас-
х су IgG. Это не всегда удобно, так как для реакции связывания
'' комплемента предпочтительнее использовать антитела класса
IgM. Для индукции этих антител удобно использовать в качест-
ве носителя бактериальные клетки [5]. Конъюгация АМФ с
клетками Е. coli позволила получить кроличьи антитела к АМФ
IgM и IgG. В качестве перспективных носителей предлагают
использовать пневмококки, полисахаридный антиген которых
относится к тимуснезависимым антигенам и индуцирует интен-
сивный иммунный ответ, вероятно, путем подавления механиз-
мов его регуляции. К формалинизированным клеткам пневмо-
кокка штамма R36A присоединяли неиммуногенные вещества:
полимер у-2-М-морфолинил-этил-Е-глутамина, лизоцим и сополи-
мер глутаминовой кислоты, D-лизина и аланина. Проиммунизи-
рованные этими конъюгатами кролики образовывали антитела в
больших количествах: 16 мг/мл сыворотки, 15 мг/мл и 10 мг/мл-
соответственно [13].
1.1.2.4. ПОЛУЧЕНИЕ АНТИСЫВОРОТОК
К АНТИГЕНАМ КЛЕТОЧНЫХ МЕМБРАН
Одной из сложнейших задач является получение сывороток с
высоким титром АТ к мембранным компонентам различных
клеток. Перекрестные реакции с аналогичными компонентами
Других клеток могут сущ^^юятр’^Иекайсатъ результаты. Веро-
ятно, стандартные антистайрдЯФё? ^Дчества удастся по-
лучить, используя те^^гогию клеточных гйбриДов. Ввиду раз-
2—990
нообразия проблем, связанных с мембранными антигенами, мы
ограничимся всего несколькими примерами для иллюстрации
применяемых методов. Известно получение гетерологической
кроличьей антисыворотки против Т-лимфоцитов мыши путем
иммунизации препаратами мембран тимоцитов. После первых
этапов адсорбции эти антитела реагировали с 95—100% клеток
тимуса и 35—40% клеток селезенки в иммунофлюоресцентной
пробе и в тесте на цитотоксичность. Последующей адсорбцией
удалось повысить специфичность антисыворотки к мембранному
антигену до такой степени, что она реагировала только с 0-по-
зитивными спленоцитами. Иммунизацией кроликов препаратами
эмбрионального мозга человека была получена антисыворотка,
которая после соответствующей адсорбции реагировала с оп-
ределенными субпопуляциями Т-лимфоцитов. Она оказалась
пригодной для дифференциальной диагностики Т-клеточных
лейкозов (хронические лимфоцитарные лейкемии) [2]. Оказав-
шееся необычно полезным разделение Т-лимфоцитов мыши по
Ly-антигену, которое впервые позволило четко разделить Т-лим-
фоциты на субпопуляции по функциональным признакам, так-
же основано на получении специфических антисывороток [17].
Благодаря им стало возможным выводить инбредные линии
мышей с определенными иммунологическими характеристиками.
«Голых» мышей принято считать В-клеточными мышами, кото-
рые представляют собой идеальных доноров для получения ан-
ти-В-лимфоцитарных сывороток.
В частности, введением клеток из лимфатических узлов «го-
лых» мышей кроликам была получена специфическая антисы-
воротка против В-клеток [9]. Она с успехом применяется для
анализа стадий созревания В-лимфоцитов при их превращении
в антителообразующие клетки. Антисыворотка, полученная пу-
тем введения субпопуляции клеток мышиной плазмоцитомы кро-
ликам, после адсорбции не реагировала с клетками тимуса или
с нестнмулированными спленоцитами. Она не реагировала со
спленоцитами, стимулированными ФГА или Кон-А, но взаимо-
действовала со спленоцитами, стимулированными липополиса-
харидом (ЛПС) или со стимулированными В-лимфоцитами
[16]. Все вышеприведенные примеры показывают, какое важное
значение могут иметь антисыворотки при исследовании процес-
сов клеточной дифференцировки и регуляции.
1.1.2.5. ПОЛУЧЕНИЕ АНТИТЕЛ ИЗ ДРУГИХ
БИОЛОГИЧЕСКИХ ЖИДКОСТЕЙ
Выше была описана возможность использования молозива ко-
ров в качестве высокотитражного источника антител. Введени-
ем термоагрегированного у-глобулина человека совместно с
ПАФ за 7, 6 и 2 нед до отела в дозе 300 мг на одну иммуниза-
цию было получено молозиво с высоким титром соответствую-
щих антител [4]. Можно предположить, что иммунизация мень-
18
шими дозами антигена окажется столь же эффективной. Асци-
тическая жидкость мышей в качестве источника АТ использу-
ется давно [ 11]. Наибольшее количество антител образуется
при введении антигена вместе с асцитообразующим материалом
в полость брюшины. С точки зрения выхода антител безразлич-
но, был ли асцит индуцирован введением клеток асцитной кар-
циномы Эрлиха или ПАФ. В среднем у одной мыши можно по-
лучить 5 мл асцитической жидкости, титр антител в которой
составляет 50—100% от титра сыворотки. Это относится и к
антителам к ДНК в асцитической жидкости мышей [10]. Для их
получения рекомендуют использовать линию DDV, поскольку
животные этой линии образуют наибольшие количества асци-
тической жидкости.
Хорошие результаты получены при использовании клеток
саркомы 180/TG для индукции асцита у мышей Charles—River.
На основании имеющихся данных можно сделать следующие
рекомендации: мышам (исследовано много штаммов) с недель-
ным интервалом 7-кратно вводят 0,4 мл раствора антигена
[0,2 мг антигена в ПАФ (1:1) внутрибрюшинно]. Через 7—
10 дней после окончания инъекций отбирают асцитическую жид-
кость через полую иглу после прокалывания брюшной стенки.
По меньшей мере у 80% животных отбирают по 5 мл и более
асцитической жидкости.
1.1.3. ОЦЕНКА КАЧЕСТВА ПОЛУЧАЕМЫХ СЫВОРОТОК
Поскольку сыворотки сильно различаются по своему предна-
значению, например, сыворотки для иммунофлюоресценции, ра-
диоиммунологического анализа (РИА), или цитотоксических
реакций к определенным популяциям клеток, то невозможно
предложить универсальный тест для их оценки. Основным тре-
бованием является контроль специфической сыворотки в соот-
ветствующей тест-системе или в тест-системе с большей чувст-
вительностью. Например, недопустимо проверять специфичность
сывороток для РИА (очень чувствительная проба) посредством
иммуноэлектрофореза. В этом случае следует ставить реакцию
торможения в радиоиммунологическом тесте. Целесообразно
распространить этот принцип на все методы оценки качества
сывороток.
1.1.4. СПИСОК ЛИТЕРАТУРЫ
1. Binaghi R. A., Perrudet-Badoux A., Boussac-Aron У. Rapid and high produc-
tion of precipitating antibodies in rats. — J. Immunol. Methods, 1975, 7,
65—68.
2. Brouet J. C., Toben H. Characterisation of a subpopulation of human T-lim-
phocites reactive with an heteroactiserum to human brain. — J. Immunol.,
1976, 116, 1041—1047.
3. Conrad D. H., Wirth G. H. Characterisation of antibodies to vitamin A.—
Immunochem., 1973, 10, 273—275.
4. Fey H. R., Biltler R., Marti F. The production in the pregnant cow of anti-
2* 19
human immunoglobulin to be used for the antiglobulin test.—Vox Sang.,
1973, 25, 245—253.
.'5. Furuchl K-, Sasaki T., Koyama J. Production of IgM and IgG antibodies
specific for AMP in rabbits by immunisation with AMP-E. coli conjugate.—
J. Biochem, 1973, 74, 451—457.
6. Giessen van der M., de Lange B., van der Lee B. The production of predipi-
tating antiglobulin reagents specific for the subclasses of human IgG.—
Immunology, 1974, 27, 655—663.
7. Hadge D„ Fiebig FL, Ambrosius H. Evolution of low molecular weight immu-
noglobulins I. Relationship of 7S immunoglobulins of various vertebrates
to chicken IgY. — Devel. Compar. Immunol., 1980, 4, 501—514.
8. Heisch R. D., Hujner M. Highly specific antibodies to triiodthyronine. —
Acta biol. med. germ., 1972, 28, 861—864.
9. Kakiuchi T., Nariuchi FL, Tamura N. Preparation and effects of an anti-B-
cell serum. — 1976, 116, 1224—1230.
10. Kitagawa Y., Okuhara E. A method for producing anti-DNA antibodies in
ascetic fluid of mice. — J. Immunol, methods, 1976, 10, 151—159.
11. Krebs M., Schimke R., Ambrosius H. Peritonealflussigkeit als zusatzliche
Quelleder der Anticorpergewinnung.— Allergie und Immunol., 1973, 19,
49—50.
12. Mayer R. G., Walker J. FL Immunochemical methods in the biological scien-
ces: enzymes and proteins. — London, Academic Press, 1980.
.13 . McDonald H. C., Odstrchel G„ Maurer P. H. Hyperimmune response to a
protein and a polypeptide antigen coated on Pneumococcus R 36A.— J. Im-
munol., 1972, 109, 881—883.
.14 . Nakashima L, Ohta F., Kobayashi T. et al. Effect of antigen doses and time
intervals between antigen injections on secondary, tertiary and quarternary
antibody responses. — Immunology, 1974, 26, 443—457.
.15 . Nielsen K. Preparation of antisera to the ц-chain of IgM. — J. Immunol,
methods, 1976, 11, 77—82.
16. Proctor M. L., Herscowitz FL B. Preparation and properties of an anti-plasma
cell serum directed against a defined plasmocytoma cell population. — J. Im-
munol., 1975, 115, 1642—1649.
17. Shen F. W., Boyse E. A., Cantor FL Preparation and use of Ly antisera.—
Immunogenetics, 1975, 2, 591—595.
18. Skowsky W. R., Fisher D. A. The use of thyroglobulin to in duce antigenecity
to small molecules. — J. Lab. Clin. Med., 1972, 80, 134—144.
1.2. ПОЛУЧЕНИЕ МОНОКЛОНАЛЬНЫХ
АНТИТЕЛ ПУТЕМ СЛИЯНИЯ КЛЕТОК
(ГИБРИДОМНАЯ ТЕХНОЛОГИЯ)
1.2.1. ПРИНЦИП МЕТОДА
Количество возможных вариантов антител у млекопитающих
очень велико, оно составляет приблизительно 1ХЮ7- К одной
детерминанте может образовываться до 8000 вариантов моле-
кул антител. В условиях обычного иммунного ответа из всего
видового потенциала антител у данного индивида образуется
небольшое количество типов антител, представляющих собой
случайную выборку из числа всех возможных вариантов. В свя-
зи с этим невозможно получить сыворотки от различных инди-
видов, обладающие одинаковым набором антител. В случае
сложных антигенов, например ксеногенных клеток, иммунный
ответ наблюдается в отношении приблизительно ста поверхно-
20
Синтез
нуклеотидов
de novo
ГГФРТ
8-Азагуанин
Рис. 3. Механизм селекции на среде ГАТ.
стных антигенов, каждый из которых обладает несколькими де-
терминантами. Учитывая случайный выбор вариантов антител
к каждой отдельной детерминанте, становится понятным, что
антисыворотки от двух разных особей могут довольно сильно
отличаться между собой. Невозможность воспроизвести на дру-
гом животном набор антител, получаемых от какого-либо опре-
деленного животного, сильно осложняет процесс стандартиза-
ции сывороток. Для получения сывороток против отдельных де-
терминант ксеногенных клеток приходится использовать весьма
трудоемкие и длительные методы адсорбции, что часто приво-
дит к выделению антисывороток с крайне низким титром анти-
тел. Эти недостатки можно устранить, применяя моноклональ-
ные антитела к определенным клеточным детерминантам; мо-
ноклональные антитела продуцируются постоянно культивируе-
мыми линиями клеток.
В 1975 г. Кёлер и Милыптейн впервые добились слияния
короткоживущих лимфоцитов, продуцирующих антитела, и по-
стоянно растущих клеток плазмоцитомы. Это открытие дало
возможность получать теоретические «бессмертные», непрерывно
культивируемые клоны гибридных клеток — продуцентов анти-
тел (гибридомы). Образуемые гибридомами моноклональные
антитела имеют сходное молекулярное строение и обладают
одинаковой специфичностью. Первоначально слияние проводили
при помощи вируса Сендай. Применение полиэтиленгликоля
(ПЭГ) в качестве агента, стимулирующего этот процесс, позво-
лило существенно увеличить частоту слияний. Неслившиеся
лимфоциты отмирают через несколько дней после гибридизации.
Отделение неслившихся плазмоцитомных клеток от гибридом
возможно только при использовании селективной среды ГАТ
(см. ниже). Предпосылкой для ГАТ-зависимой селекции являет-
ся наличие вариантов плазмоцитомных клеток, дефектных по
гипоксантин-гуанин-фосфорибозилтрансферазе (ГТФРТ) или по
тимидинкиназе (ТК). Эти клетки отмирают в культуральной
среде, содержащей гипоксантин, аминоптерин и тимидин (ГАТ),
поскольку не обладают способностью обойти аминоптериновый
блок основного пути биосинтеза ДНК за счет биосинтеза ги-
поксантина и тимидина (рис. 3). Только гибридные клетки, по-
21
лученные слиянием ГГФРТ-негативных или ТК-негативных
плазмоцитомных клеток и нормальных лимфоцитов (ГГФРТ- и
ТК-позитивных), выживают в среде ГАТ, преодолевая амино-
птериновый блок. Дефектные по ГГФРТ линии плазмоцитом
получают путем селекции клеток, резистентных к 8-азагуанину.
Линии, дефектные по ТК, выделяют по резистентности к 5-бром-
дезоксиуридину. Среди растущих на среде ГАТ гибридом выде-
ляют и селекционируют те клоны, которые продуцируют имму-
ноглобулины (1g) нужного класса и определенной специфич-
ности.
Выявленные антителопродуцирующие клоны гибридных кле-
ток (гибридомы) можно культивировать in vivo и in vitro, в за-
висимости от этого моноклональные антитела получают из ас-
цитической жидкости или из надосадочной фракции культу-
ральной среды.
1.2.2. МЕТОДИКА
1.2.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Среду для культивирования клеток RPMI-1640 (SIFIN, Бер-
лин) разводят бидистиллированной водой и вносят следующие
добавки: бикарбонат натрия — 2 г/л (х. ч.), 20 мМ ГЭПЭС
(Ы-2-гидроксиэтилпиперазин-2-этансульфоновая кислота, х. ч.),
2 мМ L-глутамина (отбирают из замороженного маточного
200 мМ раствора), 5ХЮ”5 М 2-меркаптоэтанола (х. ч., из 0,1 М
маточного раствора), 100 мг/л гентамицина (Фармахим, Со-
фия). Раствор стерилизуют и добавляют по 100 мл стерильной
сыворотки плода коровы (SIFIN, Берлин) на 1 л среды. В та-
ком виде среда может храниться в течение 6 нед при 4 °C. При
работе с надежными клеточными культурами можно заменять
фетальную сыворотку коров, неонатальной сывороткой или в
некоторых случаях нормальной лошадиной сывороткой. Вместо
гентамицина можно использовать смесь пенициллина и стреп-
томицина.
Среда для слияния клеток: к среде RPMI-1640 добавляют
2 г/л бикарбоната натрия, 20 мМ ГЭПЭС и 100 мг/л гентами-
цина; сыворотку не добавляют.
Селективная среда ГАТ состоит из полной среды для куль-
тивирования, к которой добавлено 20 мл/л стерильного маточ-
ного раствора ГАТ. Конечные концентрации компонентов:
1ХЮ-4 М гипоксантина, 4ХЮ-7 М аминоптерина, 1,6ХЮ-5 М
тимидина. Среда ГТ (гипоксантин, тимидин) состоит из полной
культуральной среды, к которой добавлено 10 мл/л стерильного
маточного раствора ГТ.
Приготовление маточных растворов ГТ и ГАТ: в дистиллиро-
ванной воде растворяют хроматографически чистый гипоксантин
(Реанал, Будапешт) до концентрации 1ХЮ-2 М путем нагре-
вания при 45—50 °C в течение 1 ч. К этому раствору добавляют
22
тимидин (Sigma, Мюнхен) до конечной концентрации 1,6Х
Х10~3 М. Этот 10-кратный раствор (ГТ) фильтруют в стериль-
ных условиях и хранят при температуре —20 °C.
Раствор аминоптерина 4ХЮ~5 М получают путем добавления
1,76 мг аминоптерина (Schuchardt, Мюнхен) и 0,5 мл 1 М NaOH
к 90 мл дистиллированной воды. Доводят объем дистиллирован-
ной водой до 100 мл, нейтрализуют раствор 0,5 мл 1 М НС1 и
фильтруют в стерильных условиях.
ГАТ (50-кратный раствор) готовят смешиванием растворов ГТ
и аминоптерина в соотношении 1 : 1, раствор разливают по 2 мл
и хранят при —20 °C. Следует защищать растворы, содержа-
щие аминоптерин, от действия света.
Раствор ПЭГ: 5 г ПЭГ (относительная молекулярная масса
1540, ф. ч. Polyscience Inc., США, или относительная молеку-
лярная масса 4000 чист, для газовой хроматографии, Merck,
Дармштадт) смешивают с 5 мл культуральной среды для слия-
ния, одновременно растворяют и стерилизуют автоклавировани-
ем в течение 20 мин. Непродолжительное время раствор ПЭГ
может храниться при 4 °C. Для длительного хранения раствор
лучше замораживать. Посуда для культивирования:
— микропланшеты для титрования с плоским дном и крышкой
96X200 мкл (Flow Labs, ФРГ);
— планшеты для культуры тканей с плоским дном и крышкой
24X2 мл (Linbro);
— пластиковые культуральные сосуды (5, 25, 50 мл).
Кроме того, необходимы: стеклянные пипетки (1, 2, 5 и 10 мл),
пастеровские пипетки, автоматические пипетки и выдерживаю-
щие автоклавирование сменные наконечники к ним (тип 1 и 2),
центрифужные стаканы (10 и 25 мл).
Вся посуда, применяемая для работы с культурой ткани, долж-
на быть стерильна. Прочие материалы и посуду для стерильных
манипуляций и подсчета клеток см. в разделе 1.3.
Оборудование: ламинарный бокс (VEB, Electromat, Дрезден),
СОг-инкубатор, устройство для стерильной фильтрации (мемб-
ранные фильтры), инвертированный микроскоп Telaval (VEB,
Карл Цейсс, йена), водоструйный насос, центрифуга с охлажде-
нием (К24).
1.2.2.2. ЛИНИИ ПЛАЗМОЦИТОМ
ГАТ-чувствительные, ГГФРТ-дефектные линии плазмоцитом из-
вестны в настоящее время у мышей [2, 5, 8, 9, 10, 14], крыс [4]
и человека [1, 13] (табл. 1). Эти клеточные линии можно селек-
ционировать по резистентности к 8-азагуанину (от 20 до
60 мг/л). До настоящего времени реверсий не наблюдалось. Все
же время от времени желательно культивировать эти клетки в
среде, содержащей 8-азагуанин. Аналогичным образом дефект-
ные по ТК клетки могут быть получены селекцией в среде, со-
держащей 5-бромдезоксиуридин (30 мг/л). Для получения мо-
23
Таблица 1. ГАТ-чувствительиые линии плазмоцитомиых клеток,
пригодные для получения гибридов
Вид (штамм) Линия плазмоцитом Число хромо- сом Иммуно- глобулины Литератур- ный источник
Мышь BAlB/c МРС 11-45.6TG1.7 62 y2b [10]
BAlB/c РЗ X 63-Ag8 65 Vi [8]
BAlB/c NS1-A 4/1 65 X Внут- [9]
BAlB/c Х63-А 8.653 58 рикл. [5]
BAlB/c S Р 2/0-Ag 14 72 — [14]
BAlB/c Крысы — Lou FO 210-RCY 3.Ag 1,2.3. 72 39 и [4]
Человек Миелома GM 1500 GM 1500 6Tg-AL-2 ?2 X [1]
Миелома U-266 SKO-007 — ?2, [13]
Миелома ARH-77 LICR-LON-HMy 2 — Y1, х [13]
номинальных антител оптимальными являются линии плазмо-
цитом, полностью утратившие способность синтезировать цепи
собственных иммуноглобулинов. Получаемые из них гибридные
клетки содержат только активные гены L- и Н-цепей антитело-
образующих лимфоцитов, поэтому образование гибридных мо-
лекул иммуноглобулинов исключено.
1.2.2.З. ПОЛУЧЕНИЕ АНТИТЕЛООБРАЗУЮЩИХ
ЛИМФОЦИТОВ
В дальнейшем речь пойдет только о слиянии клеток плазмоци-
томы мыши с лимфоцитами того же вида. Слияние клеток плаз-
моцитомы и лимфоцитов человека или крысы имеет свои осо-
бенности.
1.2.2.З.1. Иммунизация
Для иммунизации используют инбредные линии мышей (ВА1
В/с), сингенные, по отношению к плазмоцитоме или аллогенным
штаммам. В последнем случае для лучшего роста гибридных
клеток после слияния следует использовать гибриды Fi мышей
ВА1В/с и мышей, иммунизированных с целью выделения у них
нормальных клеток селезенки. Для культивирования гибридом
in vivo также необходимо использование гибридов Fi. Для про-
ведения иммунизации подходят любые схемы, стимулирующие
образование выраженного гуморального иммунного ответа. Ре-
комендуется повторное введение антигена за 4 дня до слияния.
Антиген в этом случае вводится внутрибрюшинно или внутри-
венно без адъюванта. Лучший эффект достигается при исполь-
зовании более высоких доз антигена, чем на предыдущих стади-
ях иммунизации.
24
1.2.2.3.2. Получение клеток
Мышей забивают цервикальной дислокацией, извлекают в усло-
виях стерильности селезенку и готовят суспензию отдельных
клеток. Клетки селезенки трижды отмывают средой для слия-
ния, не содержащей сыворотки. Суспензию, содержащую
2Х107 лимфоцитов, переносят в центрифужный стакан объемом
25 мл. Если число лимфоцитов меньше предусмотренного, то к
суспензии добавляют еще 2ХЮ8 ядерных клеток селезенки.
1.2.2.4. СЛИЯНИЕ КЛЕТОК
Плазмоцитомные клетки в количестве 2ХЮ7 (например, Х63
Ag 8.653), взятые в логарифмической фазе культивирования и
дважды отмытые культуральной средой для слияния, добавля-
ют к отмытым клеткам селезенки мыши (рис. 4). Суспензию
клеток тщательно перемешивают и центрифугируют при 300 g.
После отсасывания надосадочной фракции осадок ресуспенди-
руют в небольшом количестве среды. Затем в течение 2 мин по
каплям добавляют 1 мл раствора полиэтиленгликоля (ПЭГ),
следя за равномерным смешиванием жидкостей. Избыток ПЭГ
разводят добавлением бессывороточной среды для слияния. Для
этого при постоянном покачивании сосуда с интервалом в 2 мин
последовательно прибавляют I, 2, 4 и 8 мл среды для слияния.
Добиваются равномерного распределения клеток во всем объ-
еме жидкости. Клетки собирают центрифугированием при 300 g,
надосадочную фракцию отбрасывают. Все процедуры проводят
в условиях минимальной обсемененности микробами (в лами-
нарном боксе) при комнатной температуре.
1.2.2.5. СЕЛЕКЦИЯ ГИБРИДНЫХ КЛЕТОК
НА СРЕДЕ ГАТ
Слившиеся клетки аккуратно ресуспендируют в 40 мл среды
ГАТ. К этой суспензии для поддержания роста добавляют
1ХЮ7 свежевыделенных ядерных клеток селезенки (см. раздел
I.2.2.3.1). Можно также применять перитонеальные макрофаги,
получаемые смывом раствора глюкозы. Как показывают наши
наблюдения, нет необходимости облучать поддерживающие
клетки дозой в 2 Дж/кг (200 рад), хотя некоторые авторы это
рекомендуют. Из суспензии клеток отбирают 192 пробы по
200 мкл и вносят их в плоскодонные планшеты для микрокуль-
тур альных работ. Культуры клеток инкубируют в водонасыщен-
ной атмосфере, содержащей 5% СОг при 37 °C. Начиная с 4-го
дня после слияния, производят замену половины надосадочной
фракции в лунках с культурой свежей порцией среды ГАТ; за-
мену повторяют каждые 3 или 4 дня. Для этого половину над-
осадочной фракции клеточной культуры аккуратно отсасывают
стерильными пастеровскими пипетками, соединенными с водо-
25
+ Среда ГАТ
192 отдельные культуры (200 мкл)
4 дня замена половины среды
ГАТ свежей
Проверка активности антител
Рис. 4. Гибридомная технология: слияние клеток, ГАТ-селекция, выявление
гибридом.
струйным насосом. Под действием ГАТ дефектные по ГГФРТ
плазмоцитомные клетки отмирают. Остаются неслившиеся клет-
ки селезенки и гибридомы, образовавшиеся из клеток плазмоци-
томы и ГГФРТ-положительных лимфоцитов. Скорость деления
клеток селезенки невелика, в то время как скорость деления
гибридных клеток примерно такая же, как у клеток плазмоци-
26
томы, поэтому быстрорастущие клоны гибридом становятся раз-
личимы в виде плотных колоний клеток. Рекомендуется как
можно раньше заносить в протокол количество растущих гиб-
ридных клонов, поскольку позднее это становится затрудни-
тельным из-за слияния отдельных клонов и образования дочер-
них колоний. Подсчет количества клонов гибридных клеток луч-
ше всего производить при помощи инвертированного микроско-
па Televal на 6-й день, а затем незадолго до проверки антите-
лообразующей способности (с 15-го до 18-й день). В обычных
условиях частота слияний составляет 10~5 (образуется 1 гиб-
ридная клетка на 105 исходных клеток плазмоцитомы), что со-
ответствует образованию 1 гибридного клона в 1 культуральной
лунке. При оптимальных условиях достигается частота слияний,
равная 10“4.
1.2.2.6. ОПРЕДЕЛЕНИЕ АНТИТЕЛОПРОДУЦИРУЮЩЕЙ
СПОСОБНОСТИ ГИБРИДОМ
При обычных условиях культивирования через 2—3 нед количе-
ство клеток в гибридном клоне достигает величины, делающей
возможной определение антител. Из культуральных лунок отби-
рают при помощи автоматической пипетки по 50 мкл надосадоч-
ной фракции. Для обнаружения антител можно использовать
любой высокочувствительный метод. Наиболее оправдал себя
на практике метод, представленный на рис. 6.
1.2.2.6.1. Определение моноклональных антител
к растворимым антигенам
при помощи радиоиммунологического анализа
А. Гибкие планшеты для микротитрования (V-образные) или
штампованные поливинилхлоридные (ПВХ) листы для упа-
ковки таблеток «нагружают антигеном». Для этого углубле-
ния в ПВХ-листах заполняют на 2—12 ч раствором антигена
при комнатной температуре. Концентрация антигена может
составлять от 5 до 200 мг/л. Раствор антигена может исполь-
зоваться для «нагрузки» много раз, при этом следует учиты-
вать, что разведенные растворы истощаются быстрее.
Б. Нагруженные ПВХ-листы трижды отмывают 0,15 М NaCl.
В. Внесением нейтрального белка (10 г/л человеческого или
бычьего сывороточного альбумина, разведенного в соотноше-
нии 1:100 сывороткой плода коров или нормальной лошади-
ной сыворотки) «закрывают» те участки поверхности носите-
ля, с которыми не связался антиген.
Г. Трижды проводят отмывку 0,15 NaCl. После высушивания
на воздухе ПВХ-листы, нагруженные антигеном, могут не-
сколько месяцев храниться при 4 °C без потери связывающей
активности.
27
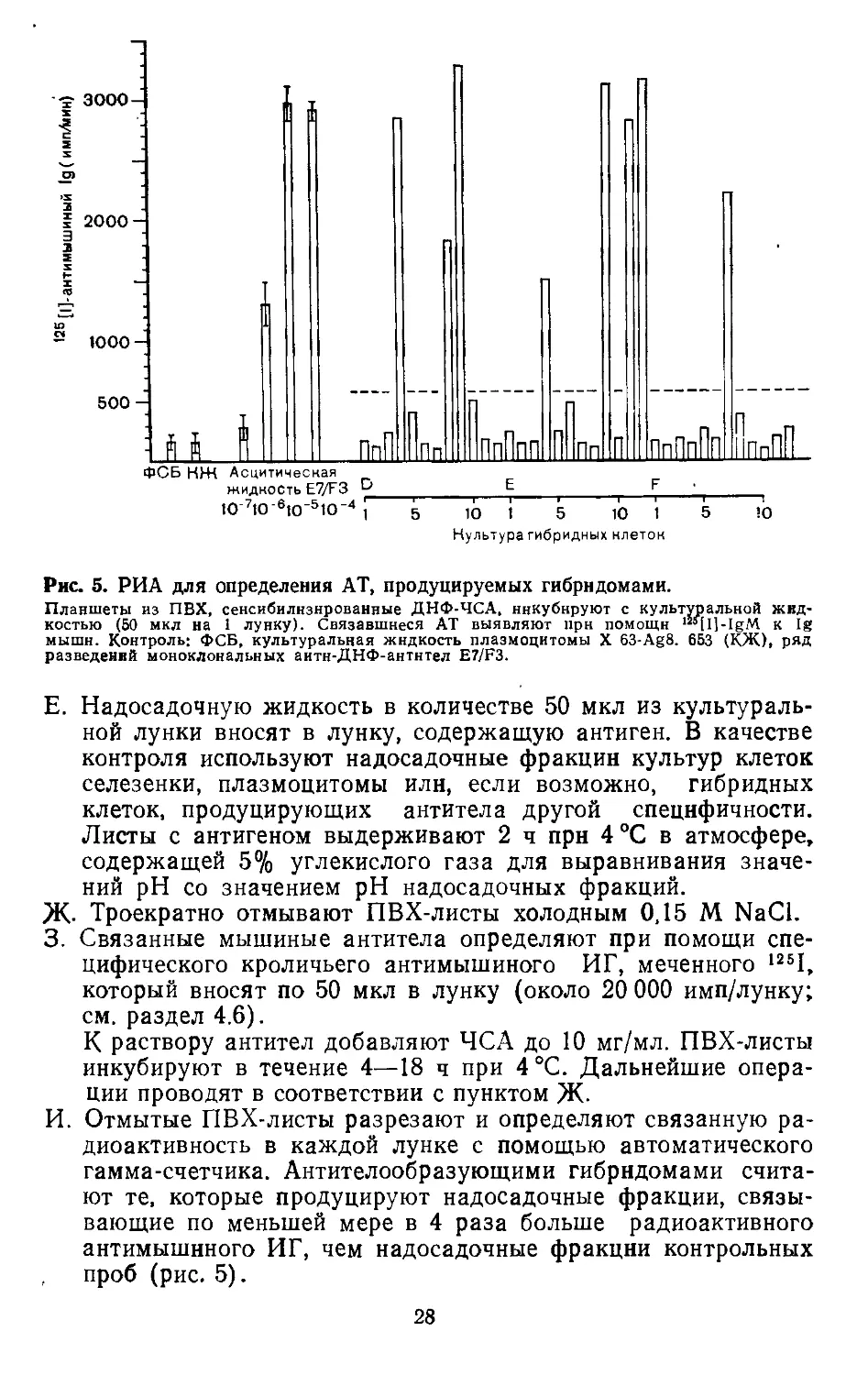
Рис. 5. РИА для определения АТ, продуцируемых гибрндомами.
Планшеты из ПВХ, сенсибилизированные ДНФ-ЧСА, ннкубнруют с культуральной жид-
костью (50 мкл на 1 лунку). Связавшиеся АТ выявляют при помощи ‘"[Il-IgM к 1g
мыши. Контроль; ФСБ, культуральная жидкость плазмоцитомы X 63-Ag8. 653 (КЖ), ряд
разведений моноклональных аитн-ДНФ-антнтел E7/F3.
Е. Надосадочную жидкость в количестве 50 мкл из культураль-
ной лунки вносят в лунку, содержащую антиген. В качестве
контроля используют надосадочные фракции культур клеток
селезенки, плазмоцитомы или, если возможно, гибридных
клеток, продуцирующих антитела другой специфичности.
Листы с антигеном выдерживают 2 ч при 4 °C в атмосфере,
содержащей 5% углекислого газа для выравнивания значе-
ний pH со значением pH надосадочных фракций.
ж. Троекратно отмывают ПВХ-листы холодным 0,15 М NaCl.
3. Связанные мышиные антитела определяют при помощи спе-
цифического кроличьего антимышиного ИГ, меченного 1261,
который вносят по 50 мкл в лунку (около 20 000 имп/лунку;
см. раздел 4.6).
К раствору антител добавляют ЧСА до 10 мг/мл. ПВХ-листы
инкубируют в течение 4—18 ч при 4 °C. Дальнейшие опера-
ции проводят в соответствии с пунктом Ж-
И. Отмытые ПВХ-листы разрезают и определяют связанную ра-
диоактивность в каждой лунке с помощью автоматического
гамма-счетчика. Антителообразующими гибрндомами счита-
ют те, которые продуцируют надосадочные фракции, связы-
вающие по меньшей мере в 4 раза больше радиоактивного
антимышиного ИГ, чем надосадочные фракции контрольных
, проб (рис. 5).
28
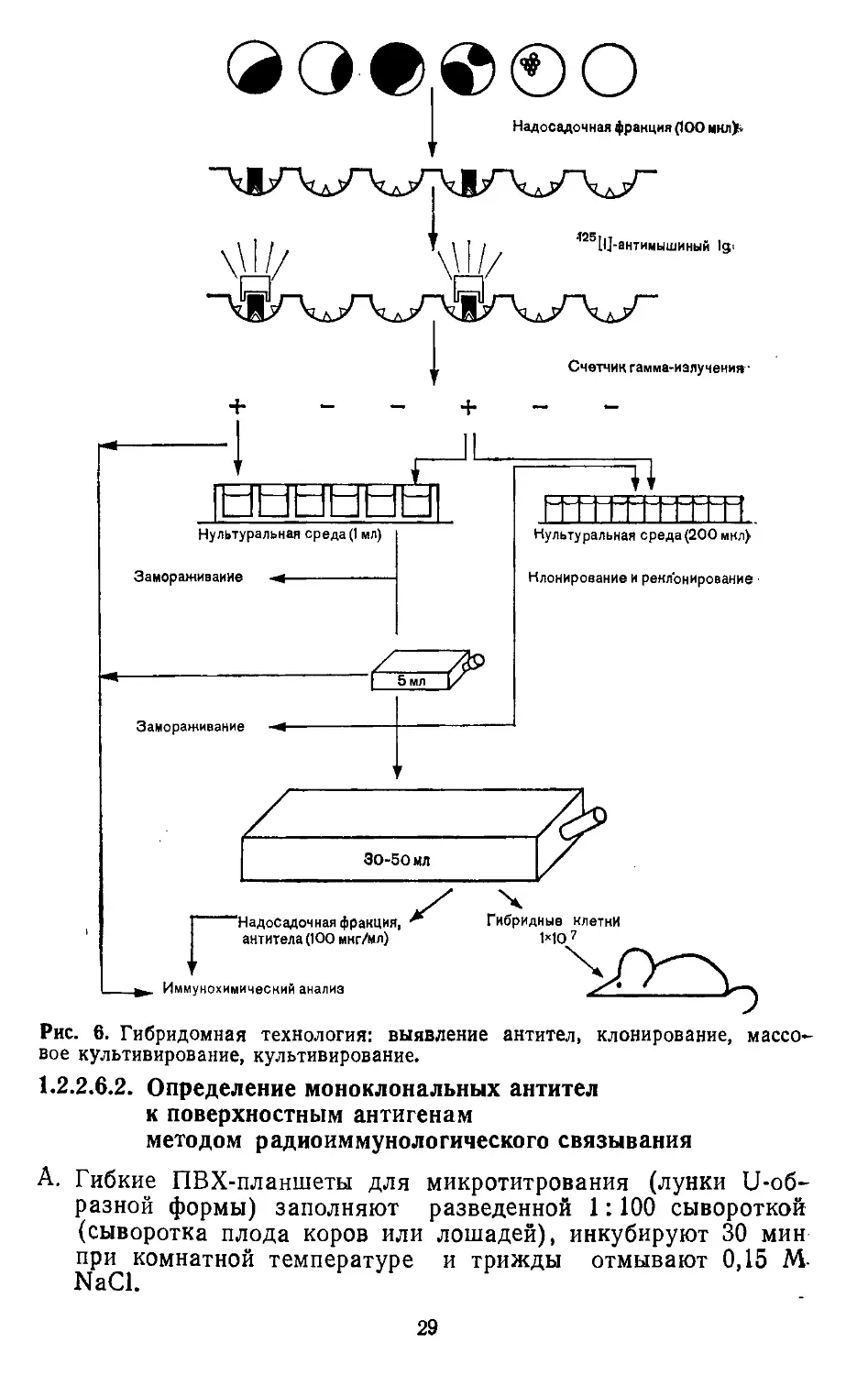
®ое®©о
Надосадочная фракция (100 мнл>«
Счетчик гамма-излучения*
Рис. 6. Гибридомная технология: выявление антител, клонирование, массо-
вое культивирование, культивирование.
1.2.2.6.2. Определение моноклональных антител
к поверхностным антигенам
методом радиоиммунологического связывания
А. Гибкие ПВХ-планшеты для микротитрования (лунки U-об-
разной формы) заполняют разведенной 1:100 сывороткой
(сыворотка плода коров или лошадей), инкубируют 30 мин
при комнатной температуре и трижды отмывают 0,15 М-
29
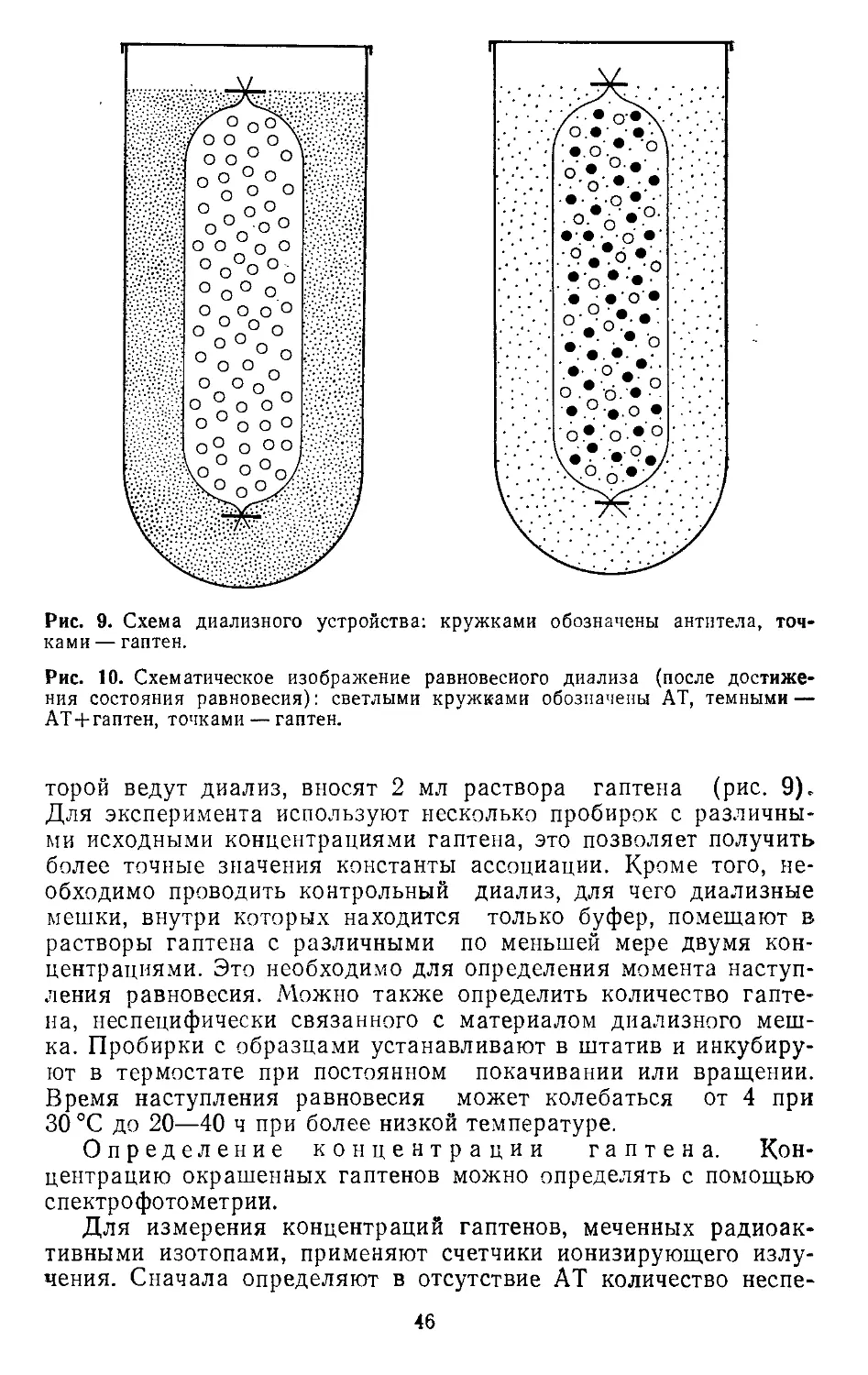
6. В каждую лунку вносят по 5ХЮ5 клеток (например, лимфо-
циты крови) в 50 мкл раствора NaCl; планшеты центрифу-
гируют при 300 g в течение 5 мин. В каждую лунку вносят
по 50 мкл 0,5% глютарового альдегида. В этом растворе
клетки могут сохраняться в течение нескольких недель при
4 °C. Перед употреблением клетки трижды отмывают рас-
твором NaCl, в течение 2 ч инкубируют с ЧСА (10 г/л) и
еще раз троекратно отмывают раствором NaCl.
В. Вносят по 50 мкл надосадочной фракции клеточных культур
или контрольного образца. Остальные этапы см. в разделе
1.2.2.6.1.
Вместо меченных изотопами антител можно использовать конъ-
югированные с ферментами антитела для иммунофлюоресцент-
ного анализа (ИФА) (см. 2.10).
В качестве альтернативы можно применять другие разновидно-
сти РИА (см. 2.9): локальный гемолиз в геле (см. 1.5), нейт-
рализацию фагов и непрямую флюоресценцию (см. 2.8).
1.2.2.7. КЛОНИРОВАНИЕ
Антителопродуцирующие клетки называют гибридомами. В экс-
перименте не всегда образуется только один гибридный клон.
Довольно часто образуется несколько гибридных клонов. Эти
немоноклональные культуры подлежат клонированию. Первич-
но моноклональные культуры также следует время от времени
реклонировать, поскольку возникают клетки, не продуцирующие
антитела, но зачастую растущие быстрее антителопродуцирую-
щих и даже заглушающие рост последних. Для клонирования
чаще всего применяют метод предельного разведения. Гибрид-
ные клетки осторожно суспендируют в 2 мл культуральной сре-
ды, определяют концентрацию жизнеспособных клеток. Суспен-
зию разводят так, чтобы в 1 мл содержалось 150 гибридных
клеток. К 1 мл этой суспензии добавляют 2ХЮ7 сингенных яд-
росодержащих клеток селезенки и доводят объем до 20 мл
культуральной средой. В планшет с плоскодонными лунками
вносят 96 аликвот по 200 мкл. В тех лунках, где образовались
моноклональные культуры (микроскопия), проверяют наличие
антител.
1.2.2.8. КУЛЬТИВИРОВАНИЕ ГИБРИДОМ IN VITRO
Когда клетки становятся достаточно многочисленными, их пере-
носят в лунки объемом 1—2 мл. Для этого за 2 дня до переноса
гибридом в планшеты для культивирования (например, Linbro
с 24 лунками) вносят ядросодержащие сингенные клетки селе-
зенки (по 5Х105 клеток в лунку). Гибридомы аккуратно сус-
пендируют при помощи движений поршня пипетки и переносят
по 100 мкл из каждой исходной лунки в 2 лунки объемом 1 мл.
.Начиная с 5-й нед после слияния, трижды производят замену
30
среды (ГТ, а не ГАТ). В дальнейшем можно вести культивацию
в обычной культуральной среде. В лунках объемом 2 мл гиб-
ридомы так разрастаются, что их переносят в планшеты с че-
тырьмя лунками по 5 мл. Стабильные гибридомы можно пере-
носить в культуральные сосуды по 25—50 мл. На этой стадии
можно заменять сыворотку плода коровы нормальной сыворот-
кой коров или лошадей. В этих сосудах среду следует также
менять каждые 3—4 дня. На этом этапе можно получить до-
статочное количество клеток для культивирования in vivo на
сингенных мышах. Отбираемые при смене среды моноклональ-
ные антитела можно использовать для характеристики специ-
фичности и молекулярных особенностей ИГ. В среднем концент-
рация моноклональных антител в культуральной среде состав-
ляет 10—50 мкг/мл.
1.2.2.9. КУЛЬТИВИРОВАНИЕ ГИБРИДОМ IN VIVO
Сингенным мышам предварительно вводят внутрибрюшинно
0,5 мл полужидкого парафина (DAB 7, VEB Leunawerke, Mer-
seburg) или пристана (Koch-Light Lobs). Через 1—4 нед после
предварительной обработки внутрибрюшинно вводят по 2Х107
гибридных клеток каждому животному. В асцитической жидко-
сти мышей концентрация моноклональных антител может до-
стигать 5—10 мг/мл. Содержащиеся в асцитической жидкости
гибридомы можно перевивать другим мышам после предвари-
тельной обработки (см. выше).
1.2.2.10. КОНСЕРВИРОВАНИЕ ПРИ НИЗКОЙ
ТЕМПЕРАТУРЕ
Необходимо замораживать, гибридомы как можно раньше, на-
чиная со стадии, когда культуры развиваются в лунках объ-
емом 2 мл, не дожидаясь получения массовой культуры. Это
позволит возобновить культуру в случае ее гибели на поздних
стадиях вследствие внесения инфекции. Стабильные культуры
гибридом можно подвергать многократному замораживанию.
В стеклянные ампулы вносят в стерильных условиях по 106 гиб-
ридных клеток, суспендированных в среде RPMI 1640 с добав-
лением 20% сыворотки плода коровы и диметил сульфоксида
(70 г/л). После охлаждения до 4°C клетки переносят в сосуд
Дьюара с сухим льдом, выдерживают их в течение 2 ч и пере-
носят в жидкий азот. При размораживании клетки помещают в
водяную баню при 37 °C, немедленно после оттаивания их пере-
носят в центрифужную пробирку, содержащую 5 мл культу-
ральной среды. Осажденные центрифугированием клетки снова
суспендируют в среде и переносят в заранее подготовленные-
лунки с двухдневной культурой спленоцитов. Проводят иссле-
дование на антителообразующую способность, в зависимости от
результатов которого либо выделяют массовую культуру, либо<
31
Надосадочная фракция культур гибридом
-Y2b
\\1// \\|//
п1 1m
,25(П-аити 1g. -И-
Счетчик гамма-излучения
t
+ + ----
Фис. 7. Определение класса (субкласса) гибридомных АТ при помощи
-меченных 125[I]-антител к иммуноглобулинам.
проводят реклонирование. Если гибридные клетки взяты из сре-
ды, содержащей ГТ или ГАТ, то их следует культивировать на
ГТ среде.
1.2.2.11. ИММУНОХИМИЧЕСКИЙ АНАЛИЗ
ГИБРИДОМНЫХ АНТИТЕЛ
Типирование гибридомных антител должно проводиться на воз-
можно более ранних стадиях (см. раздел 1.2.2.6.1). Изотип свя-
завшихся с ПВХ-листами антител определяют путем проявления
меченной 1251-антисывороткой к тотальному препарату иммуно-
глобулина или к различным тяжелым цепям (ц, yi, уга, угЬ,
-уза (рис. 7). Обычно используют большое количество метки
(до 150 000 имп/50 мкл) в отличие от экспериментов с высоко-
очищенными антителами. Биохимическими методами изотип
можно определять при помощи электрофореза в полиакрил-
амидном геле (ПААГ) с додецилсульфатом натрия (ДСН), раз-
деляя восстановленные моноклональные антитела. Единообра-
.зие антител доказывается при помощи электрофореза и изо-
электрофокусирования ИГ получивших витальную метку 14С.
32
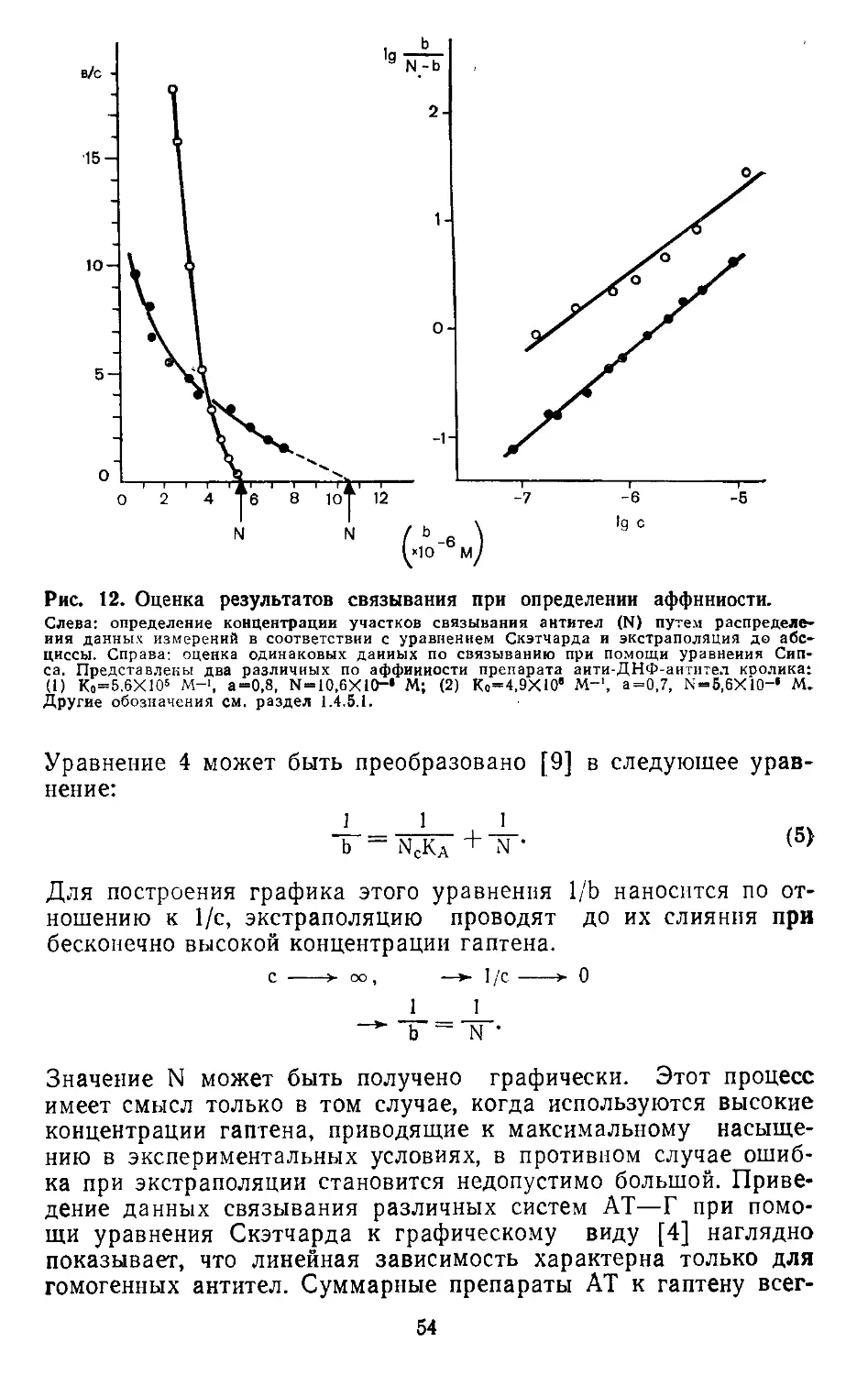
1.2.3. ОЦЕНКА МЕТОДА
Основное преимущество моноклональных антител (МКАТ) —
их специфичность. Правда, сама природа взаимодействия АГ-АТ
может обуславливать иногда возникновение перекрестных реак-
ций и с моноклональными антителами. Еще одной отличитель-
ной чертой МКАТ является их молекулярная однородность и
доступность в относительно больших количествах при сравни-
тельно низкой себестоимости. Поскольку гибридомы теоретиче-
ски бессмертны и легко культивируются после хранения в за-
мороженном состоянии, они могут быть использованы для полу-
чения долгосрочно воспроизводимых результатов. МКАТ от-
крывают новые перспективы в таких областях исследования
как изучение антигенов клеточных мембран (антигены диффе-
ренцировки и антигены, ассоциированные с опухолями). Работ
в этой области много, мы же ограничиваемся цитированием
только 3 литературных источников [6, 11, 12].
1.2.4. СПИСОК ЛИТЕРАТУРЫ
1. Croce С. М., Linnebach A., Hall W. et al. Production of human hybridomas
secreting antibodies to measle virus. — Nature, 1980, 288, 488—489.
2. Fazekas de St. Groth S., Scheidegger D. Production of monoclonal antibodies:
strategy and tactics. — J. Immunol. Meth., 1980, 35, 1—24.
3. Galfre G., Howe S. C., Milstein C. et al. Antibodies to major histocompati-
bility antigens produced by hybrid cell lines. — Nature, 1977, 266, 550—552.
4. Galfre G., Milstein C., Wright B. Rat x rat hybrid myelo mas and a mono-
clonal anti Fd portion of mouse IgG. — Nature, 1977, 131—133.
5. Kearney J. F., Radbruch A., Liesegang B., Rajewsky K. A new mouse myelo-
ma cell line that had lost immunoglobulin ex pression but permits the
construction of antibody secreting hybrid cell lines. — Immunol., 1979, 123,
1548—1550.
6. Kennet R. H., McKearn T. J., Bechtol К- B. Ed Monoclo nal antibodies-hybri-
domas a new dimention in biological analyses. — New York and London;
Plenum Press, 1981.
7. Klinman. N. R., Press J. L. The В-cell specificity repertoire: its relationship
to definable populations. — Transplant. Rev., 1975, 24, 49—83.
8. Kohler G., Milstein C. Continuous cultures of fused cells secreting antibody
of predefined specificity. — Nature, 1975, 256, 495—497.
9. Kohler G., Milstein C. Derivation of specific antibody — producing tissue cul-
ture and tumor lines by cell fusion. — Eur. J. Immunol., 1976, 6, 511—519.
10. Margulies D. H., Kuehl W. M., Scharff M. D. Somatic cell hybridisation of
mouse myeloma cells. — Cell, 1976, 8, 405—415.
11. Melchers F. M„ Potter M., Warner N. (Ed.). Lymphocyte hybridomas. Cur-
rent topics in microbiology and immunology. — Berlin: Springer, 1978, 81.
12. Moller R. H. (Ed.). Hybrid myeloma monoclonal antibodies against MHC
products. — Immunol. Rev., 1979, 47, 1—252.
13. O'Hare M„ Edwards P. New success with human Hybrids. — Immunol today,
1981, i—ii.
14, Schulamn M., Wilde C. D., Kohler G. A better line for making hybridomas
secreting specific antibodies. — Nature, 1978, 276, 269—270.
15. Stahll C„ Staehelin T„ Miggiano V. et al. High frequencies of antigen-speci-
fic hybridomas: dependence on immunisation patterns and prediction by
spleen cell analysis. — J. Immunol. Meth., 1980, 32, 297—307.
3-990 33
1.3. ОБРАЗОВАНИЕ АНТИТЕЛ
КУЛЬТУРАМИ КЛЕТОК
1.3.1. ПРИНЦИП МЕТОДА
Метод, описанный в данном разделе, основан на способности
суспензии клеток лимфоидных органов формировать культуры,
продуцирующие антитела вне исходного организма. Образова-
ния антител in vitro можно добиться различными путями:
1. От животных, подвергшихся стимуляции определенным анти-
геном, получают культуру клеток, которые продолжают in
vitro синтезировать антитела без дополнительного введения
антигена.
2. От иммунизированных животных после угасания иммунного
ответа получают культуру клеток. Повторное введение того
же антигена вызывает вторичный иммунный ответ in vitro.
3. Культуру клеток, полученных от неиммунизированного жи-
вотного, используют для выработки первичного иммунного
ответа путем непосредственного контакта с антигеном.
Индукция первичного иммунного ответа, сопровождающаяся
проявлением in vitro всех этапов полного гуморального иммун-
ного ответа, методически наиболее трудна. Удовлетворительные
результаты впервые были получены в 1967 г. одновременно тре-
мя группами исследователей [2, 10, 9]. Методы, предложенные
Mishell и Dutton (рис. 8, а) [10] и Marbrook (рис. 8,6) [9], по-
лучили широкое распространение и стали стандартными мето-
дами. Позднее к ним добавился метод микрокультивирования
Lefkovitz (рис. 8, в) [8]. При всех трех способах получения
культур клеток используется исходная питательная среда, обо-
гащенная специфическими добавками и сывороткой плода коро-
вы. В питательную среду вносят антиген (обычно эритроциты
барана, ЭБ) и суспензию клеток (чаще спленоциты, или какую-
либо субпопуляцию спленоцитов мыши); инкубацию проводят
в течение 3—7 дней в атмосфере, обогащенной СОг.
При наиболее распространенном способе культивирования,
предложенном Mishell и Dutton, используют пластиковые чаш-
ки Петри небольшого размера. Этот способ очень прост в ис-
полнении, однако для оптимизации роста клеток требуются пи-
тательные добавки. Система выращивания клеток, предложен-
ная Marbrook, основана на использовании стеклянных сосудов,
нижняя часть которых закрыта мембраной, непроницаемой для
клеток, но пропускающей питательные вещества. Клетки растут
на поверхности мембраны. Сосуд с клетками погружают в со-
суд большего размера с питательной средой. Хотя такой метод
требует больших затрат рабочего времени и материалов, он по-
зволяет оптимизировать процесс питания клеток. Его преимуще-
ством является и то, что в одном сосуде можно использовать
несколько мембран, каждая из которых «заселена» клетками
34
Рис. 8. Системы культивирования клеток для выявления антителообразо-
ваиня.
а — по методу Mishell и Datton; б — по методу Marbrook; в —по методу Lefkovitz.
одного типа. Lefkovitz предложил использовать планшеты для
микротитрования, что позволяет обходиться небольшими коли-
чествами среды и клеток. Правда, при этом способе нет воз-
можности получать антителообразующие клетки в значительных
количествах. Преимуществами его являются простота реализа-
ции, малый расход реактивов, возможность выделения отдель-
ных клонов клеток. Мы приводим описание модифицированного
метода Mishell и Dutton, получившего наибольшее распростра-
нение [15].
1.3.2. МЕТОДИКА
1.З.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: однополые мыши инбредных штаммов
СВА, С57Ы или DBA в возрасте 8—12 нед; эритроциты барана
в растворе Олсвера или в дефибринированной крови; чашки
Петри диаметром 50 мм (Anumbra, Венгрия); колбы Эрленмейе-
ра объемом 25, 100, 500 и 1000 мл; градуированные пипетки (1,
10 и 25 мл); подставки для пипеток; пипетки для подсчета
эритроцитов и лейкоцитов; стеклянный стерилизующий фильтр
G5 (Saale — Gias, Иена, ГДР); стеклянные стаканы объемом
на 400 мл; центрифужные пробирки (10 и 20 мл); камеры для
подсчета клеток по Barker и Thoma; силиконовые пробки для
колб Эрленмейера; шприцы (10 мл) с иглами № 16; алюминие-
вая фольга; бунзеновская горелка; ножницы; пинцеты; формы
для льда; чашки с замороженным льдом с плоской поверхно-
стью льда; среды Игла-МЕМ и Хенкса в сухом виде (Госу-
дарственный институт иммунопрепаратов и питательных сред,
Берлин); сыворотка плода коровы (например, «Rehatuin» Rebe-
ls Chemical Company, Phoenix USA); заменимые аминокисло-
ты: Ser, Pro, Gly, Asp кислота (x. ч., или для культуры тканей);
L-глютамин, х. ч.; пируват натрия, х. ч.; пенициллин, стрептоми-
цин 2-меркаптоэтанол (Koch-Light, Англия); NaHCO3; 2,5%
раствор трипанового синего в 0,15 М NaCl; тридистиллирован-
ная вода; сухожаровой шкаф, термостат, настольная центрифу-
з* 35
га, микроскоп, весы (тарирующие); баллоны со сжатым возду-
хом, содержащим 7,5% СОг; эксикатор диаметром 40 см; лабо-
раторное помещение, приспособленное для стерильных работ.
1.3.2.2. ПОДГОТОВКА СТЕКЛЯННОЙ ПОСУДЫ
Работы по культивированию требуют строгого соблюдения сте-
рильности; можно использовать только безупречно чистые по-
суду и растворы. Стеклянную посуду ополаскивают водопро-
водной водой и кипятят в течение часа в растворе моющих
средств. После повторного ополаскивания посуду замачивают в
20% серной кислоте не менее чем на 6 ч. Затем посуду тщатель-
но моют водопроводной водой и дважды ополаскивают дистил-
лированной водой. Лабораторное стекло высушивают в су-
шильном шкафу при 100 °C до исчезновения воды и пятен кон-
денсации пара, после чего посуду заворачивают в алюминиевую
фольгу и стерилизуют 2 ч при 180 °C. Подготовленную таким
образом посуду хранят в помещении, предназначенном для сте-
рильных работ.
1.З.2.З. ПРИГОТОВЛЕНИЕ ПИТАТЕЛЬНЫХ СРЕД
Раствор Хенке а. В соответствии с прописью изготовителя
9,77 г сухого вещества растворяют в 1 л тридистиллированной
воды, с помощью 1 М NaOH доводят pH до 7,4, стерилизуют
фильтрованием и хранят при 4 °C.
Среда Игл а-М Е М. В соответствии с прописью изготови-
теля 10,8 г сухого вещества растворяют в 1 л тридистиллиро-
ванной воды, затем к этому раствору добавляют следующие
компоненты (из расчета на 1 л раствора): 8 мг глицина, 10 мг
L-Ser, 12 мг L-Pro, 14 мг L-Asn, 9 мг L-Ala, 13,3 мг L-Asp, 15 мг
L-Glu, 290 мг L-Geln, ПО мг пирувата натрия. После полного
растворения всех компонентов pH доводят до 7,4 с помощью
1 М NaOH и фильтруют через фильтр G 5. Раствор может хра-
ниться при +4 °C в течение 4—6 нед.
Раствор антибиотиков. Растворяют 1 г сульфата
стрептомицина и 1 000 000 ЕД бензилпенициллина натриевой
соли в 100 мл тридистиллированной воды, стерилизуют фильтро-
ванием, разливают по 2 мл в стерильные сосуды и хранят в за-
мороженном состоянии. Исходный раствор в 100 раз концентри-
рованнее рабочих растворов.
Раствор 2-м е р к а п т о э т а н о л а. Растворяют 0,7 мл
2-меркаптоэтанола в 100 мл стерильной тридистиллированной
воды; 0,25 мл полученного раствора смешивают с 24, 75 мл сте-
рильного раствора Хенкса; хранят при 4°. Концентрация исход-
ного раствора в 20 раз выше концентрации рабочего раствора.
Раствор NaHCO3. По меньшей мере 1 раз в 4 нед гото-
вят свежий раствор бикарбоната натрия в тридистиллированной
воде и стерилизуют фильтрованием.
36
Предпочтительнее готовить полную питательную среду для
клеточных культур ex tempore из отдельных составляющих ее
растворов. Для получения 100 мл питательной среды смешива-
ют 82 мл среды Игла-МЕМ, 10 мл сыворотки плода коровы (ко-
нечная концентрация 10%), 5 мл раствора 2-меркаптоэтанола
(конечная концентрация 0,005 %), 2 мл раствора бикарбоната
натрия (конечная концентрация 0,2%).
1.З.2.4. ПОЛУЧЕНИЕ СУСПЕНЗИИ
КЛЕТОК СЕЛЕЗЕНКИ
Получение суспензии клеток селезенки и последующая работа с
культурой клеток должны проводиться в стерильных условиях с
соблюдением всех правил асептики и антисептики. Подходящим
помещением для этих целей может служить бокс со шлюзом.
Необходимое количество мышей забивают без эфирного нарко-
за путем цервикальной дислокации. Для стерильного удаления
селезенки делают поверхностный надрез и отодвигают шкуру в
стороны от разреза по направлению к конечностям. После
вскрытия брюшной полости селезенку стерильно извлекают и
помещают в охлажденный до 4 °C раствор Хенкса, налитый в
чашку Петри, диаметром 12 см, установленную на горизонталь-
ной поверхности льда. На каждую извлеченную селезенку рас-
ходуется 5 мл раствора Хенкса. Селезенки измельчают стериль-
ными пинцетами без следов ржавчины. Путем осторожного из-
мельчения кусочков органов стараются перевести в суспензию
возможно большее число спленоцитов. Клеточную суспензию
набирают в 10-миллилитровый шприц (игла № 16). При этом
большие скопления клеток остаются, а малые распадаются при
прохождении через узкую иглу. Содержимое шприца переносят
в центрифужные пробирки объемом 20 мл и центрифугируют в
течение 5 мин при 150 g в настольной центрифуге без охлажде-
ния (в стерильном боксе). Надосадочную фракцию отсасыва-
ют, осадок дважды отмывают равным объемом охлажденного
раствора Хенкса путем ресуспендирования. Суспензию клеток
при отмывке несколько раз набирают и спокойно выпускают
пипеткой (10 мл). После отмывки осадок клеток ресуспендиру-
ют в таком объеме раствора Хенкса, чтобы клетки, полученные
из 4 или 5 селезенок, содержались в 1 мл суспензии. После от-
бора небольшой порции для подсчета клеток и определения их
жизнеспособности суспензию хранят в ледяной бане не более
чем 1 ч. Определение количества клеток и их жизнеспособности
проводят в нестерильных условиях, при помощи окрашивания
трипановым синим, в камере Biirker. Приготовленная вышеопи-
санным способом суспензия клеток селезенки содержит 96—98%
жизнеспособных лимфоцитов, выход составляет в среднем 70—
100 млн спленоцитов с каждой селезенки.
37
1.З.2.5. ПОЛУЧЕНИЕ СУСПЕНЗИИ ЭРИТРОЦИТОВ
Применяемые в качестве антигена эритроциты барана отбирают
в условиях стерильности. Эритроциты, независимо от того, хра-
нили ли их в растворе Олсвера или в дефибринированной кро-
ви, трижды отмывают в 10-кратном объеме охлажденного сте-
рильного раствора Хенкса. Из полученного осадка клеток гото-
вят 20% суспензию эритроцитов; точное число клеток подсчиты-
вают в камере Thoma. Из основной суспензии готовят исходный
раствор антигена, содержащий в 1 мл 2ХЮ8 эритроцитов.
1.З.2.6. ПОЛУЧЕНИЕ КУЛЬТУР КЛЕТОК
Рассчитывают количество культуральной среды, необходимое
для опыта, и готовят суспензию из хранящихся в ледяной бане
клеток селезенки так, чтобы в 1 мл среды содержалось 107 жиз-
неспособных клеток. Суспензию разливают по 2 мл в стеклян-
ные чашки Петри диаметром 50 мм. Для индукции антитело-
образования в ряд чашек вносят по 0,1 мл исходной суспензии
ЭБ (конечная концентрация 107 эритроцитов в пробе). Эритро-
циты распределяют равномерно по чашке осторожными враща-
тельными движениями. Посев культуры клеток следует произ-
водить непрерывно, поскольку бикарбонат натрия вызывает
нарастающее ощелачивание среды, а клетки селезенки очень
чувствительны к колебаниям pH в щелочной области. Чашки с
культурой немедленно помещают в эксикатор (он выполняет
роль инкубатора), который заполняют через антибактериальный
фильтр воздухом, содержащим 7,5% СОг. Стационарная газо-
вая фаза достигается пропусканием в течение 30 с через экси-
катор газовой смеси под давлением 5 атм. Описанный выше ме-
тод позволяет получать в стеклянных чашках Петри оптималь-
ное число антител при соблюдении следующих условий: в чашку
вносят 2 мл полной питательной среды; содержание—107 кле-
ток в 1 мл, содержание антигена (эритроциты барана) —5X106
клеток в 1 мл.
1.З.2.7. ОЦЕНКА АНТИТЕЛООБРАЗОВАНИЯ
Культуры инкубируют в стационарной газовой фазе в течение
4 дней при 37 °C без встряхивания и добавления питательных
веществ. После извлечения чашек с культурой из инкубатора
работу проводят в нестерильных условиях. Культивируемые
клетки, образующие отчетливо видимый «газон» на дне чашки,
соскабливают с поверхности стекла. Для этого используют стек-
лянную палочку, на нижний конец которой надет кусок пласт-
массового шланга, край которого выступает примерно на 1 мм.
Суспензию клеток переносят в центрифужную пробирку (10 мл)
и центрифугируют 5 мин при 150 g. После удаления надосадоч-
ной жидкости осадок клеток суспендируют в 1—2 мл охлаж-
38
денного раствора Хенкса и хранят в ледяной бане. Определе-
ние жизнеспособности выросших клеток проводят в камере Вйг-
рег при помощи раствора трипанового синего. Подсчет антите-
лообразующих клеток (АОК) производят при помощи методов
прямого и непрямого локального гемолиза в геле (см. раздел
1.5). С клетками из каждой чашки проводят по меньшей мере
два параллельных определения. Интенсивность антителообразо-
вания определяют как количество АОК на каждые 106 жизне-
способных клеток.
1.3.2.8. МОДИФИКАЦИИ МЕТОДА
Система, о которой шла речь выше, позволяет наблюдать пер-
вичный иммунный ответ in vitro даже в том случае, когда в ка-
честве антигена используются не цельные эритроциты, а соот-
ветствующий лизат. Она также может использоваться для на-
блюдения за индуцированным in vivo иммунным ответом, напри-
мер, с ее помощью можно оценивать in vitro вторичный иммун-
ный ответ на эритроциты барана. Следует помнить, что первич-
ное образование антител к ЭБ возможно при использовании
клеток селезенки кролика, но не крысы. Иногда первичную ин-
дукцию антителообразования удается получать при использо-
вании динитрофенол-фиколла (ДНФ-фиколл) и ДПС Oi27; о по-
явлении антител в этом случае можно судить по реакции ло-
кального гемолиза (см. раздел 1.5). Сыворотку плода коровы
заменяют иногда человеческой плацентарной сывороткой или
подходящей донорской сывороткой. Можно использовать как
пластиковые чашки Петри, так и стеклянные. Покачивание
культур при инкубации практически не влияет на уровень анти-
телообразования. Ежедневное подкармливание культур клеток в
соответствии с оригинальным методом [10], хотя и увеличивает
число антителообразующих клеток, все же не оказывает замет-
ного влияния на качественные результаты опыта. Такая моди-
фикация повышает стоимость исследования и задерживает про-
ведение по времени эксперимента, особенно если не обеспечено
«подкармливание» культур в конце недели. При соблюдении со-
ответствующих условий вместо чашек Петри можно использо-
вать планшеты для микротитрования с плоским дном [13], на-
пример фирмы Nunc, Дания. В каждую лунку вносят ЗХЮ8
с.членоцитов в 0,3 мл культуральной среды, стимулируют их
ДНФ-фиколлом и эритроцитами барана; образование АОК при
этом вполне сопоставимо с тем, которое наблюдается на стек-
лянных чашках Петри, в расчете на 106 жизнеспособных кле-
ток. Имеются предварительные данные об индукции первично-
го иммунного ответа у лимфоцитов из миндалин и крови чело-
века. От 23 здоровых доноров были выделены лимфоциты и
моноциты центрифугированием в градиенте плотности [1].
В 13 случаях наблюдали стимуляцию под влиянием ДНФ-фи-
колла, ЭБ и конъюгированных с тринитрофенильной группой
39
(ТНФ) ЭБ. Показать образования антител в культуре клеток
человека можно, удлиняя время культивирования до 7 дней и
увеличивая продолжительность воздействия конплемента в ре-
акциях прямого и непрямого локального гемолиза до 2 ч [1].
Зоны гемолиза, образуемые АОК человека, имеют меньший диа-
метр, чем зоны, образованные аналогичными клетками мыши.
Выход АОК из клеток донорской крови можно увеличить, до-
бавляя к культуральной среде кон-А (1 мкг/мл) либо добавляя
автологичную и АВ-сыворотку вместо сыворотки плода коровы,
либо снижая содержание моноцитов путем одночасовой адгезии
на стекле полученных центрифугированием в градиенте плот-
ности форменных элементов крови. Лимфоциты из миндалин
выделяют точно так же, как и клетки селезенки мыши. При
культивировании клеток из миндалин необходимо добавлять
нистатин (Sqibb, Англия) в количестве 20 ЕД/мл.
1.3.3. ОЦЕНКА МЕТОДА
Современное состояние знаний о механизме антителообразова-
ния в значительной степени основывается на данных, получен-
ных при исследовании первичной стимуляции культур клеток
[14]. Для оценки современного и будущего значения, а также
преимуществ данной системы следует учитывать следующие мо-
менты:
1. Процесс антителообразования во всей его сложности вычле-
няется из организма животных и делается доступным для
прямых наблюдений.
2. Все стадии процесса происходят в известных постоянных,
воспроизводимых и направленно изменяемых условиях.
3. Становится возможным анализ роли различных типов кле-
ток, принимающих участие в антителообразовании — Т-лим-
фоцитов, В-лимфоцитов, макрофагов путем их совместного
или раздельного культивирования.
4. Вещества, секретируемые в ходе кооперативного взаимодей-
ствия клеток, например, медиаторы регуляции, могут быть
выделены и идентифицированы, а механизм их действия де-
тально изучен.
5. Механизм антителообразования в организме человека до-
ступен изучению почти исключительно методом культивиро-
вания клеток [5, 6].
6. Применение системы культивирования клеток человека дает
возможность выделять клетки, стимулированные определен-
ными антигенами, и затем получать моноклональные анти-
тела человеческого происхождения [4, 7, 11, 17].
7. В будущем, вероятно, смогут быть выявлены причины де-
фектов гуморального иммунитета, сопровождающиеся пато-
логическими проявлениями, станет возможным корригировать
отклонения в образовании антител адекватными терапевти-
ческими мерами.
40
Следует помнить, что изучение первичного иммунного ответа
в культуре клеток позволяет получать данные, которые можно
переносить на живой организм с известными оговорками. Все
существующие системы культивирования используют клеточные
суспензии, содержащие продукты отмирания клеток, которые
могут оказывать искажающее влияние на рост культуры. Выс-
вобождающиеся в процессе жизнедеятельности биологически
активные вещества могут изменять потребности некоторых ти-
пов клеток в питательных веществах. Ограниченность общих
выводов, которые обычно делают при работе с культурой кле-
ток, связана с тем, что удается выращивать in vitro клетки не-
большого числа видов животных, причем далеко не все антиге-
ны в этих условиях индуцируют иммунный ответ. Между индук-
цией и образованием антител в культуре протекает много каче-
ственно различных стадий: захват антигена, «узнавание» анти-
гена, переваривание и презентация антигена, кооперативное
взаимодействие клеток, деление и дифференциация клеток, био-
синтез белка. Каждая из этих стадий требует своих особых, оп-
тимальных условий среды и, кроме того, весьма чувствительна к
различным неблагоприятным воздействиям. Этим объясняется
ют факт, что многочисленным исследовательским группам не
удалось включить метод культивирования АОК в свой арсенал,
н зачастую даже при успешном культивировании не удалось до-
биться эффективной продукции антител. По нашим собствен-
ным наблюдениям, антителообразования не наблюдается в при-
близительно 10% экспериментов. При постановке параллельных
серий экспериментов количество антителообразующих клеток в
разных чашках дает разброс 23%, причем для метода локаль-
ного гемолиза разброс не превышает 15%. Неодновременная
постановка опытов приводит к еще большему разбросу резуль-
татов (от 600 до 4000 АОК в различных чашках). Максимум
антителообразования в культуре обычно наблюдается к 4-му
дню, причем в ряде случаев он к этому времени уже прошел,
в других — еще не наступил. В последнее время все чаще ис-
пользуют 2-меркаптоэтанол в качестве обязательного компо-
нента культуральных сред. Это вещество благоприятно влияет
на выживаемость клеток и заметно повышает количество АОК
[31.
Функциональная активность системы культивирования за-
висит от типа используемой сыворотки. Почти во всех без ис-
ключения случаях используют сыворотку плода коровы. Не все
компоненты этой сыворотки, однако, пригодны в одинаковой
мере. Неблагоприятное воздействие некоторых составляющих
сыворотки может быть нейтрализовано добавлением 2-меркап-
тоэтанола [12]. Несмотря на высокую стоимость, подвержен-
ность искажениям и ограниченность возможных областей при-
менения, методы исследования первичного иммунного ответа in
vitro могут стать незаменимым средством анализа механизмов
гуморального иммунного ответа.
41
1.3.4. СПИСОК ЛИТЕРАТУРЫ
1. Boyutn A. Separation of leukocytes from blood and bone marrow. — Scand.
J. Clin. Lab. Invest., 1968, 21 (suppl. 77), 77.
2. Bussard A. E., Lurie M. Primary antibody response in peritoneal cells. —
J. Exp. Med., 1967, 125, 873.
3. Chen C., Hirsch J. G. The effects of mercaptoethanol and of peritoneal mac-
rophages on the antibody-forming capacity of nonadherent mouse spleen in
vitro.—J. Exp. Med., 1972, 136, 604.
4. Croce C„ Linnebach A., Hall W. et al. Production of human hybridomas secre-
ting antibodies to measle virus. — Nature, 1980, 288, 488.
5. Delfrassy J., Galanaud P„ Dormont J., Wallon C. Primary in vitro antibody
response from human peripheral blood lymphocytes. — J. Immunol., 1977,
118, 630.
6. Hoffman M. K. Antigen-specific induction and regulation of antibody synthe-
sis in cultures of human blood mononuclear cells. — Proc. Nat. Acad. Sci.,
1980, 77, 1139.
7. Kozbor D., Roder I. C. Requirements for the establishment of high-titered
human nomoclonal antibodies against tetanus toxoid using the Epstein Barr
virus technique. — J. Immunol., 1981, 127, 1275.
8. Lefkovits I. Induction of antibody forming cell clones in microcultures.—
Europ. J. Immunol., 1972, 2, 360.
9. Marbrook J. Primary immune response in cultures of spleen cells. — Lancet,
II, 1667, 1279.
10. Mishell R. I., Dutton R. IF. Immunization of dissociated spleen cell cultures
from normal mice. — J. Exp. Med., 1967, 126, 423.
11. Olson L., Kaplan H. S. Human-human hybridomas producing monoclonal
antibodies of predefined autogenic specificity. Proc. Nat. Acad. Sci., 1980,
77, 5429.
12. Opitz H., Opitz U., Lemke H. et al. The role of fetal calf serum in the primary
immune response in vitro. — J. Exp. Med., 1977, 145, 1029.
13. Schimpl A., Wecker E. Replacement of T-cell function by a T-cell product.—
Nature, 1972, 237, 15.
14. Schreter M. H. The antibody response in vitro: Dissection of a complex
system. — Lymphokines.— 1980, 2, 31.
15. Wichner S., Dorfling P„ Hilscher H. Beitrag zur Methode der Antikorperbil-
dtmg in Zellkulturen. — Allergie und Immunol., 1973, 19, 155.
16. B. J. M. Zegers, С. J. Heijnen, IF. Kuis et al. T-cell regulatory function in
patients with various immunodeficiencies as studied by in vitro antibody
response of the blood lymphocytes. — In: Primary immunodeficiencies. In-
serm symposium No 16. Eds/M. Seligmann, W. H. Hitzig. Elsevier. Amster-
dam: North-Holland Biochemical Press, 1980, 129.
17. Zurawski V. R. Jr., Black P. M„ Haber E. Continuously proliferating human
cell lines synthesizing antibody of predetermined specificity. — In: Monoclo-
nal antibodies, Hybridomas. A new Dimension in Biological Analyses/Eds.
R. H. Klennet, T. J. McKearn, К. B. Bechtol. — New York and London: Ple-
num Press, 1980, 19.
1.4. ОПРЕДЕЛЕНИЕ АФФИННОСТИ АНТИТЕЛ
1.4.1. ВВЕДЕНИЕ
Эффективность иммунных сывороток в организме и в реакциях
АГ—АТ зависит не только от содержания антител, но и от силы
связывания антигенов этими антителами. Сила связывания АТ
может быть охарактеризована авидностью и аффинностью.
42
1.4.1.1. АВИДНОСТЬ
Иммунизация позвоночных высокомолекулярными антигенами
вызывает образование антител к различным детерминантам ан-
тигена. Более того, антитела к одной и той же детерминанте ге-
терогенны. Если бы было возможным определить каким-либо
методом взаимную силу притяжения всех содержащихся в сы-
воротке антител с полидетерминантным антигеном, то получен-
ное значение отражало бы авидность системы. Речь шла бы при
s i ом об относительных значениях, которые не были бы сравни-
мы с данными, получаемыми на других моделях. При анализе
такого рода взаимодействий следует учитывать вторичные реак-
ции, приводящие к образованию нерастворимых комплексов;
часто эти вторичные реакции используют для индикации. В та-
ких сложных системах об авидности судят по способности сы-
вороток образовывать нерастворимые комплексы с различными
концентрациями антигена. Оставляя в стороне неточности изме-
рения, можно утверждать, что данные об авидности антисыво-
ротки (АС) имеют ограниченное значение для выяснения рас-
пределения АТ по силе их связывания с отдельными детерми-
нантами.
Изучение авидности, хотя и помогает в решении некоторых
биологических вопросов, все же недостаточно для детального
анализа иммунного ответа.
1.4.1.2. АФФИННОСТЬ
Под аффинностью понимают силу связывания активного центра
молекулы АТ с соответствующей детерминантой АГ [3]. Для
точного измерения аффинности необходимо, во-первых, ограни-
чить гетерогенность исследуемых антител и, во-вторых, не до-
пускать образования нерастворимых комплексов, т. е. исследо-
вать обратимую реакцию. Первое требование может быть до-
стигнуто использованием для иммунизации конъюгатов гаптен-
носитель. В этом случае гаптен выступает в роли доминантной
пммуподетерминанты известного химического строения и анти-
тела образуются преимущественно к гаптену. Пока нет способа
предотвратить образование гетерогенных антител к гаптену.
Для реакции АГ—АТ используют только моновалентные гапте-
ны. что позволяет избегать образования нерастворимых комп-
лексов. Антитела к носителю не влияют на результаты измере-
ний. Обратимая реакция связывания антидетерминанты АТ с
моновалентным гаптеном (Г—) приводит в соответствии с за-
коном действия масс к образованию растворимого комплекса
антитело — гаптен
АТ-ГГч^комплекс АТ—Г.
43
Аффинность в данном случае можно выразить константой ассо-
циации Ка:
или при использовании других символов:
V ь
Ка~ (N — Ь)-с •
где Ка — константа ассоциации, b — концентрация связанного с
АТ гаптена (в молях), с — концентрация свободного гаптена
(в молях), N — концентрация антидетерминант антител (в мо-
лях). Константа ассоциации равна обратной величине значения
концентрации свободного гаптена (с) при условии, что полови-
на антидетерминант связана с гаптеном (Ь), а другая половина
не связана (N—Ь), т. е. N—b = b. Важнейшим условием оценки
аффинности является определение концентрации антидетерми-
нант (N).
В простейшем случае для этого пользуются специфически
очищенными антителами; возможно также определение антиде-
терминант самостоятельными методами, например РИА, и коли-
чественным осаждением (преципитацией). Можно определять
концентрацию антидетерминант при помощи методов оценки аф-
финности в условиях очень высоких концентраций гаптенов.
В таких условиях вероятность насыщения участков связывания
антител довольно высока и, зная Ь, можно вычислить N. Основ-
ная проблема определения аффинности заключается в опреде-
лении первых значений b и с в возможно более обширной обла-
сти значений степени насыщения антидетерминант гаптеном.
Для этой цели было предложено много методов:
равновесный диализ [3],
модифицированный метод осаждения сульфатом аммония
[Ю],
гашение флюоресценции [4],
усиление флюоресценции [8],
торможение преципитации гаптеном [9].
В настоящей главе мы ограничимся описанием первых трех ме-
тодов. Реакции АГ—АТ могут быть изучены и другими метода-
ми, например путем измерения степени поляризации флюорес-
ценции [2, 7]. Последний метод пригоден также для исследова-
ния кинетики реакций и равновесных состояний.
1.4.2. РАВНОВЕСНЫЙ ДИАЛИЗ
1.4.2.1. ПРИНЦИП МЕТОДА
Равновесный диализ представляет собой классический метод
определения параметров связывания низкомолекулярных ли-
гандов с биополимерами, в том числе аффинности антител. Рас-
44
твор АТ известной концентрации приводится в соприкосновение
с раствором гаптена через полупроницаемую мембрану. Оба
компонента растворены в одинаковом буфере для того, чтобы
исключить разницу pH и ионной силы. Молекулы антител не
приходят через мембрану, молекулы гаптена диффундируют че-
рез нее в раствор антител и частично связываются с ними. Со-
стояние равновесия достигается, когда концентрация свободного
гаптена становится одинаковой по обе стороны мембраны. Вре-
мя диффузии зависит от проницаемости мембраны, природы
гаптена и температуры. Измерив концентрацию свободного гап-
тена по обе стороны мембраны, можно определить количество
гаптена, связанного с антителами. В процессе измерения сле-
дует учитывать мембранные эффекты, например, адсорбцию.
Поскольку молекулы антител несут определенный заряд, следу-
ет учитывать эффект Доннана в том случае, если гаптен тоже
заряжен. Обычно для проведения диализа используют целлофа-
новые пакеты или специально сконструированные диализные
ячейки. Данному вопросу посвящен исчерпывающий обзор Ei-
sen [4].
1.4.2.2. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Сосуд для диализа, раствор АТ, раствор гаптена, спектрофото-
метр или радиометр, термостат. В качестве растворителя чаще
всего используют 0,01 М фосфатный буфер pH 7,4 или 0,1 М
трис-НС1-буфер, pH 7,4. Величина pH и ионная сила могут
варьировать в зависимости от целей эксперимента. Для умень-
шения эффекта Доннана целесообразно использовать забуфе-
ренный 0,15 М раствор NaCl. Выбор концентрации гаптена и
антител зависит от ожидаемой величины константы равновесия
(табл. 2).
Таблица 2. Примерная концентрации АТ для
равновесного диализа при различных константах
ассоциации
Константа ассоциации М-' Концентрация антител г/л
1ХЮ5 3,6
1ХЮ6 0,36
1ХЮ7 0,36
1.4.2.З. ПОСТАНОВКА ОПЫТА
В ячейку для диализа (в данном случае диализный мешок) вно-
сят 2 мл раствора антител. Завязанный с обоих концов мешок
помещают в подходящий сосуд, например, закрывающуюся
стеклянную пробирку 10X75 мм. Затем в жидкость, против ко-
45
Рис. 9. Схема диализного устройства: кружками обозначены антитела, точ-
ками — гаптен.
Рис. 10. Схематическое изображение равновесного диализа (после достиже-
ния состояния равновесия): светлыми кружками обозначены АТ, темными —
АТ+гаптен, точками — гаптен.
торой ведут диализ, вносят 2 мл раствора гаптена (рис. 9).
Для эксперимента используют несколько пробирок с различны-
ми исходными концентрациями гаптена, это позволяет получить
более точные значения константы ассоциации. Кроме того, не-
обходимо проводить контрольный диализ, для чего диализные
мешки, внутри которых находится только буфер, помещают в
растворы гаптена с различными по меньшей мере двумя кон-
центрациями. Это необходимо для определения момента наступ-
ления равновесия. Можно также определить количество гапте-
на, неспецифически связанного с материалом диализного меш-
ка. Пробирки с образцами устанавливают в штатив и инкубиру-
ют в термостате при постоянном покачивании или вращении.
Время наступления равновесия может колебаться от 4 при
30 °C до 20—40 ч при более низкой температуре.
Определение концентрации гаптена. Кон-
центрацию окрашенных гаптенов можно определять с помощью
спектрофотометрии.
Для измерения концентраций гаптенов, меченных радиоак-
тивными изотопами, применяют счетчики ионизирующего излу-
чения. Сначала определяют в отсутствие АТ количество неспе-
46
Таблица 3. Определение количества неспецнфически связанного гептена
Исходная концентрация 10-6 М Равновесная кон- центрация при отсутствии неспецифического связывания I0-® М Измеренная концентрация в равновесном состоянии Неспецифическое связывание (% от концент- рации свободного гаптена)
внутри снаружи
9,82 4,91 4,39 4,40 11,6
8,06 4,03 3,51 3,46 15,6
5,20 2,60 2,20 2,24 17,1
2,55 1,27 1,09 1,09 16,5
1,04 0,52 0,455 0,476 11,8
цифически связанного гаптена. Это можно сделать, измерив ко-
личество гаптена в диализном мешке и в жидкости, омывающей
мешок, после наступления равновесия. Количество неспецифиче-
ски связанного гаптена равно разности между первоначально
внесенным количеством гаптена и суммарным количеством гап-
тена по обе стороны диализной мембраны. В табл. 3 приведены
примеры подобных определений для пяти исходных концентра-
ций. Условия опыта следующие: в диализной жидкости — 2 мл
14С-2,4-дннитроанилина, внутри диализного мешка 2 мл буфер-
ного раствора (0,1 М трис-НС1, pH 7,4; 0,1 М КС1). Продолжи-
тельность диализа составляет 5 ч при 30 °C в условиях медлен-
ного вращения; специфическая активность гаптена — 520 имп/
/нМоль. Измерения проводили с помощью жидкостного сцинти-
ляционного счетчика. Данные, приведенные в табл. 3, представ-
ляют собой среднее значение данных, полученных в двух оди-
наковых измерительных стаканах. Учет неспецифически связан-
Таблица 4. Определение количества гаптена, связанного антителами.
Результаты измерений
Концентрация гаптена в диализ- ной жидкости (вне мешка) перед началом опыта 10-® М Концентрация свободного гаптена в диализ- ной жидкости при равновесии 10-е м Равновесная концентрация гаптена после внесения поправки I0-® М Количество гаптена, связан- ного антителами, нМоль Число молей связанного гаптена на 1 Моль АТ
9,82 3,34 4,41 4,28 1,60
8,06 2,52 3,65 4,52 1,69
5,20 1,41 2.39 3,92 1,47
2,55 0,48 1,20 2,88 1,08
1,03 0,083 0,51 1,71 0,64
Условия опыта: диализная жидкость вне мешка—2 мл иС-2,4-диннтроаиилииа
s различных концентрациях. В диализном мешке 2 мл раствора антител (240 мкг/мл,
очищенных антител, специфичных к динитрофенильной детерминанте). Буфер: 0,01 М
трис-НС1, pH 7,5, 0,1 М КС1; продолжительность диализа 5 ч прн медленном вращении
и температуре 30 °C.
Данные в колонке Ш табл. 4 откорригированы на 15% в соответствии с данными
колонки II табл. 3. В колонке IV табл. 4 приводятся данные о количестве связанного
антителами гаптена с учетом исходных концентраций н разностей значений в колонках
И и III (молярное соотношение гаптена, связанного с Ат, приведено в колонке V).
47
ного гаптена облегчает расчет количества гаптена, связанного
антителами (рис. 10). В табл. 4 приведены данные измерений со-
вместно с данными расчета. Поправка за неспецифическое свя-
зывание антигена была принята равной в среднем 15% (см.
табл. 3).
1.4.2.4. ОЦЕНКА МЕТОДА
Методы определения гаптенов могут варьировать в зависимости
от природы гаптена и ожидаемой константы ассоциации. При
использовании окрашенных гаптенов в значениях констант ас-
социации от 103 до 10s М-1 измерить концентрацию гаптена по
оптимальной плотности. При исследовании систем с Ка выше
106 М-1 нужны более чувствительные методы, целесообразно ис-
пользовать гаптены, меченные 14С. Радиоактивные гаптены весь-
ма эффективны для определения Ка в области 106—107 Е-1.
Для определения констант ассоциации в области 107—108 М-!
необходимы гаптены, меченные тритием. При еще более высо-
ких константах ассоциации гаптен находится в основном в свя-
занном состоянии и его прямое определение затруднено. Следу-
ет также учитывать, что при низких Ка необходимые для про-
ведения измерений количества гаптена должны быть достаточ-
но высокими, чтобы определить разницу между свободным и
общим гаптеном внутри диализного мешка. Вследствие этого
точное измерение концентрации АТ затруднено. При проведе-
нии равновесного диализа не обязательно использовать высо-
коочищенные антитела.
1.4.3. ОПРЕДЕЛЕНИЕ АФФИННОСТИ МЕТОДОМ
ОСАЖДЕНИЯ СУЛЬФАТОМ АММОНИЯ
1.4.З.1. ПРИНЦИП МЕТОДА
Определение свободного (с) и связанного (Ь) гаптена необхо-
димо для оценки аффинности, оно требует разделения обоих
компонентов. Иммуноглобулины осаждаются в мягких услови-
ях из раствора при 33—55% насыщения сульфатом аммония бед
потери сродства к гаптену. Поэтому относительно просто изме-
рить количество гаптена, связанного находящимися в осадке
АТ и свободного гаптена в надосадочной жидкости. В дальней-
шем этот метод будет подробно описан для оценки взаимодейст-
вия анти-ДНФ-антител с 8-ДНФ-Ь-лизином [5, 10].
1.4.З.2. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: анти-ДНФ-антисыворотка или специ-
фически очищенные анти-ДНФ-антитела; 3Н-8-ДНФ-Ь-лизин;
нормальная сыворотка, фосфатный буфер (0,01 М фосфат
pH 7,4; 0,15 М NaCl); насыщенный на холоду раствор сульфата
48
аммония; центрифуга с охлаждением; пластиковые пробирки^
автоматические пипетки (50 и 100 мкл); жидкостный сцинтил-
ляционный счетчик.
1.4.З.З. ПОСТАНОВКА ОПЫТА
Для проведения эксперимента можно использовать очищенные-
антитела или просто антисыворотки. Очищенные антитела раст-
воряют в нормальной сыворотке, разведенной в соотношении
1 :5 или 1:10, обладающей низким сродством к соответствую-
щему гаптену. Исследуемые антисыворотки, если они содержат
достаточное количество белка, также разводят в соотношении
1 : 5 или 1:10. При необходимости делают дальнейшие разведе-
ния. К 50 мкл раствора антител приливают пипеткой 50 мкл рас-
твора гаптена. В качестве контроля неспецифического связыва-
ния используют нормальную сыворотку в тех же степенях разве-
дения. В зависимости от необходимой точности для определения
аффинности применяют 5—20 различных концентраций гапте-
на, перекрывающих охватывающую три порядка область кон-
центраций (например, от 10~5 М до 10-7 М). Оба компонента
тщательно перемешивают и выдерживают при 4 °C в течение
1 ч. Затем прибавляют равный объем (100 мкмл) насыщенного
раствора сульфата аммония при температуре 4 °C и немедленно
перемешивают, чтобы избежать денатурации АТ. После 30-ми-
нутной инкубации при 4 °C, в течение которой дважды проводят
кратковременное встряхивание, пробирки центрифугируют в те-
чение 15 мин при 5000 об/мин при той же температуре. Надоса-
дочную фракцию в количестве 100 мкл, в которой определяют
концентрацию свободного гаптена, переносят в 10 мл сцинтил-
лятора (жидкость Брея). Измеряют радиоактивность образца с
помощью жидкостного сцинтилляционного счетчика.
1.4.3.4. ОЦЕНКА РЕЗУЛЬТАТОВ
Количество свободного гаптена (с) определяют непосредствен-
но по радиоактивности материала. Количество связанного с ан-
тителами гаптена (Ь) вычисляют по разности между радиоак-
тивностью надосадочной фракции раствора, содержащего спе-
цифические антитела, и раствора, содержащего нормальную сы-
воротку. Если концентрация антител известна, то может быть
преобразовано в г (молярное соотношение Г/АТ).
1.4.3.5. ОЦЕНКА МЕТОДА
Метод осаждения сульфатом аммония технически проще и тре-
бует меньше времени, чем равновесный диализ. Значения Ка,
получаемые с его помощью, отличаются от получаемых равно-
весным диализом, но обладают хорошей воспроизводимостью
[5, 6]. Обсчет результатов по уравнению Сипса часто приводит
4-990 49
к высоким индексам гетерогенности, что с точки зрения условий
связывания соответствует несколько меньшей гетерогенности по-
пуляции АТ. Антитела с низкой аффинностью так сильно изме-
няются при осаждении, что теряют способность связывания, это
в свою очередь приводит к искажению спектра антител, обус-
ловленному техникой измерения. Это следует особенно учиты-
вать при анализе систем с большой долей низкоаффинных анти-
тел, следует также контролировать результаты, получаемые при
осаждении сульфатом аммония методом равновесного диализа.
1.4.4. ОПРЕДЕЛЕНИЕ АФФИННОСТИ ПУТЕМ
ИЗМЕРЕНИЯ ГАШЕНИЯ ФЛЮОРЕСЦЕНЦИИ
1.4.4.1. ПРИНЦИП МЕТОДА
Еще один метод определения константы ассоциации антител
основан на феномене гашения флюоресценции при взаимодейст-
вии с некоторыми гаптенами [6]. Молекулы антител, облучае-
мые УФ лучами с длиной волны 280—295 нм, испускают флюо-
ресцентное излучение с длиной волны 345 нм. Такое соотноше-
ние длин волн возбуждающего и эмиссионного излучения харак-
терно для триптофана. Другие ароматические аминокислоты
(тирозин и фенилаланин) также флюоресцируют, но их абсорб-
ционные и эмиссионные максимумы находятся в области еще
более коротких длин волн и отличаются малой интенсивностью.
Связывание подходящего лиганда антителами сопровождается
переносом энергии возбужденных остатков триптофана на свя-
занный лиганд, что сопровождается исчезновением флюоресцен-
ции в типичной области. Результатом переноса энергии, таким
образом, является гашение триптофановой флюоресценции мо-
лекул АТ. Энергия возбуждения может передаваться внутри
молекулы антитела на расстояние Fab-фрагмента, т. е. прибли-
зительно на 5 нм. Эффективность переноса энергии зависит, во-
первых, от ориентации возбужденных остатков триптофана по
отношению к связанному лиганду, во-вторых, от степени пере-
крывания спектра флюоресценции антитела и спектра поглоще-
ния лиганда. Особенно хорошо наблюдается тушение с нитро-
ароматическими лигандами, например, ДНФ и ТНФ. Полное
замещение лигандами участков связывания АТ приводит к га-
шению определенной части общей триптофановой флюоресцен-
ции. Эта максимальная степень гашения флюоресценции (Qmax)
должна быть определена в эксперименте, поскольку различные
параметры антител неоднородны в этом отношении. В первом
приближении степень гашения флюоресценции зависит от числа
«занятых» участков связывания антител. При известной кон-
центрации АТ и лиганда по степени гашения Qi/Qmax можно су-
дить о количестве связанного с антителами лиганда (г) и свобод-
ного лиганда (с). В настоящей главе этот метод будет рассмот-
рен применительно к анти-ДНФ-антителам.
50
1.4.4.2. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Спектрофотометр; очищенные специфические анти-ДНФ-антите-
ла, нормальный 1g того же вида, раствор гаптена, например
е-ДНФ-Ь-лизина.
1.4.4.З. ПОСТАНОВКА ОПЫТА
В термостатируемую кювету флюориметра вносят 1 мл раство-
ра антител. После стабилизации температуры образец облуча-
ют УФ светом с длиной волны 280—295 нм и регистрируют ин-
тенсивность флюоресценции на длине волны 345 нм. В получен-
ные данные вводят поправку на флюоресценцию растворителя.
Раствор АТ облучают УФ светом только во время измерения
флюоресценции. Раствор гаптена добавляют вначале порциями
по 10 мкл, затем — по 20 мкл, до тех пор пока объем добавлен-
ного гаптена не достигнет 200 мкл. Концентрация е-ДНФ-Ь-ли-
зпна должна составлять около 10“5 М. После каждого прибав-
ления гаптена смесь необходимо тщательно перемешивать.
Можно наносить раствор гаптена пипеткой на стеклянную па-
лочку с расплющенным концом, который затем переносят в рас-
твор антител и перемешивают. После каждого добавления гап-
тена измеряют интенсивность флюоресценции (Qi). Для оцен-
ки связывания необходимо определять максимальное гашение
флюоресценции (Qmax) для каждого препарата антител. Насы-
щение участков связывания АТ устанавливают титрованием
1X10~3 М раствором гаптена. Получаемое при такой концент-
рации гаптена гашение флюоресценции должно быть откорри-
гировано параллельным титрованием нормального ИГ. Титрова-
ние проводят до молярного избытка гаптена, равного 200:1,
к участкам связывания АТ. При этом практически все участки
связывания оказываются «занятыми». Дальнейшее увеличение
концентрации гаптена не увеличивает специфического гашения
флюоресценции.
1.4.4.4. ОЦЕНКА РЕЗУЛЬТАТОВ
На рис. И представлены кривые титрования для определения
Qmax и аффинности анти-ДНФ-кролика. Количество связанного
гаптена после каждой добавки рассчитывается по следующей
формуле:
Qi
b = n-АК уз->
Ч-тах
где Qi — гашение флюоресценции после внесения соответствую-
щего гаптена; b — количество связанного гаптена; АК — моляр-
ная концентрация антител; п — число участков связывания на
молекулу AT; Qmax — максимальное гашение флюоресценции
при полном связывании антител гаптеном. При помощи (Ь) рас-
4*
51
о
I I I I I I III I I
О 50 100 200 300 12345
Концентрация гаптена (нмоль)
Фис. 11. Гашение флюоресценции при помощи специфических кроличьих
аити-ДНФ-антител при титровании гаптеном — е-ДНФ-Ь-лизином.
Справа: титрование двух различных препаратов АТ с целью определения их аффинно-
сти. Низкоаффинные АТ (1): Ко=6ХЮ5 М-1; высокоаффннные АТ (2): Ко=1,5Х108 М“‘.
Слева: определение максимального гашения флюоресценции (Qmax==70%). Неспецифи-
ческое гашение вследствие высоких концентраций гаптена корригируется параллельным
титрованием с нормальным IgG. Область колебаний Qmax для кроличьих анти-ДНФ-ан-
тител составляет 60—75% (прерывистые линии).
считывают молярное соотношение связанного гаптена с антите-
ламп (г). Количество свободного гаптена (с) равно разности
количеств добавленного гаптена и связанного гаптена (Ь). Оп-
ределение истинной средней константы ассоциации проводят
по уравнению Сипса.
1.4.4.5. ОЦЕНКА МЕТОДА
Изучение гашения флюоресценции при оценке аффинности АТ
имеет по сравнению с другими описанными методами то пре-
имущество, что оно требует очень малого количества антител,
оно в 100—10 раз меньше, чем необходимо при использовании
других методов. Титрование не занимает много времени, при-
близительно 15 мин. Также становится возможным исследова-
ние АТ в экстремальных условиях (температура, pH, ионная
сила, влияние макромолекул), приводящих к денатурации бел-
ков при длительном воздействии. К числу недостатков метода
следует отнести неудобства, связанные с использованием высо-
коочищенных антител и ограниченного круга гаптенов. Наилуч-
шие результаты до настоящего времени получены при работе с
нитрофенильным гаптеном. Данные, полученные методом гаше-
ния флюоресценции, менее точны, чем при равновесном диали-
зе, поскольку определение аффинности основано на вторичном
-феномене связывания гаптена антителами.
52
4.4.5. ОЦЕНКА ДАННЫХ
1.4.5.1. ОБСУЖДЕНИЕ УРАВНЕНИЙ
Вышеперечисленные методы пригодны в равной степени для
определения концентрации участков связывания антител и для
оценки их аффинности. Получаемые в эксперименте данные
измерений касаются концентрации свободного гаптена (с), кон-
центрации связанного гаптена (Ь) и, следовательно, молярного
соотношения связанного с АТ гаптена (г). Взаимодействие ан-
титела с моновалентным гаптеном представляет собой обрати-
мую реакцию, подчиняющуюся закону действующих масс.
АТ 4- Г = комплекс АТ — Г, (1)
Константа ассоциации этой реакции рассчитывается по следую-
щей формуле:
Да“=[АТ](Г)
или с применением символов:
ка = (N —Ь)-с ’
Ка — константа ассоциации (М.-1), b — концентрация связанно-
го с АТ гаптена (М), с — концентрация свободного гаптена
(М), N — концентрация участков связывания АТ (М).
Последнее уравнение может быть преобразовано в координатах
Скэтчарда:
4 = N.KA-b-KA. (4)
Ка может быть рассчитана, только если известна концентрация
участков связывания антител. Эта предпосылка осуществима
при работе с высокоочищенными АТ, но не с АС или гамма-гло-
булиновой фракцией. Поэтому возникает необходимость в до-
полнительных методах (радиоиммуносорбция, количественная
преципитация) или в заранее определенных результатах. Кон-
центрацию участков связывания АТ можно получить экстрапо-
ляцией при графической обработке данных по связыванию в
соответствии с уравнением Скэтчарда [4]. Строят график за-
висимости b/с от Ь. Кривая, полученная соединением экспери-
ментальных точек, экстраполируется для ее пересечения с осью
абсцисс (рис. 12). Точка пересечения кривой и оси абсцисс со-
ответствует N при теоретически бесконечно высокой концентра-
ции гаптена и полном насыщении антител.
с >- оо, —► Ь/с ► О
0=» Кл(N — Ь) -->- N = Ь.
63
Рис. 12. Оценка результатов связывания при определении аффинности.
Слева: определение концентрации участков связывания антител (N) путем распределе-
ния данных измерений в соответствии с уравнением Скэтчарда и экстраполяция до абс-
циссы. Справа: оценка одинаковых данных по связыванию при помощи уравнения Сип-
са. Представлены два различных по аффинности препарата аити-ДНФ-аитител кролика:
(1) Ко=5.6Х1О5 М-\ а-ОД N-10.6X10-® М; (2) Ко-4,9X10® М-1, а=0,7, N-5.6X10-® М.
Другие обозначения см. раздел 1.4.5.1.
Уравнение 4 может быть преобразовано [9] в следующее урав-
нение:
1 _ 1 1
b = NcKa + N •
(5>
Для построения графика этого уравнения 1/Ь наносится по от-
ношению к 1/с, экстраполяцию проводят до их слияния при
бесконечно высокой концентрации гаптена.
с ---> оо , —► 1 /с--> 0
1 1
“* b “ N •
Значение N может быть получено графически. Этот процесс
имеет смысл только в том случае, когда используются высокие
концентрации гаптена, приводящие к максимальному насыще-
нию в экспериментальных условиях, в противном случае ошиб-
ка при экстраполяции становится недопустимо большой. Приве-
дение данных связывания различных систем АТ—Г при помо-
щи уравнения Скэтчарда к графическому виду [4] наглядно
показывает, что линейная зависимость характерна только для
гомогенных антител. Суммарные препараты АТ к гаптену всег-
54
да дают более или менее изогнутую кривую. Такой вид графи-
ка обусловлен гетерогенностью АТ по аффинности. В этих усло-
виях невозможно получить репрезентативную константу ассо-
циации. Наиболее точно можно судить о ней по уравнению Сип-
са [5]. При помощи этого уравнения можно вычислить среднюю
константу ассоциации (Ко), которая больше соответствует рас-
пределению АТ по их аффинности. Уравнение Сипса основано
на предпосылке, что значения аффинности распределяются рав-
номерно и симметрично:
Для практических целей лучше использовать логарифмическую
форму уравнения Сипса:
log(b/N — b) = a-logc 4-a-logKo, (7)
Ко — истинная средняя константа ассоциации (М-1), а — индекс гетероген-
ности (0<а<1).
Значение индекса, равное 1, соответствует гомогенной по аф-
финности популяции антител. Уменьшающиеся значения а ука-
зывают на все большее отклонение Ка определенных популяций
антител от средней константы ассоциации (Ко). Строят график
зависимости log (b/N—b) от log с. По одной точке измерения
можно построить уравнение прямой по методу наименьших
квадратов. Индекс гетерогенности а соответствует при этом на-
клону прямой. Истинная средняя константа ассоциации (так
же, как и Ка) по определению равна обратной величине кон-
центрации свободного гаптена (с) при условии, что половина
связей АТ занята молекулами гаптена. Теоретические сообра-
жения и вариабельность экспериментальных данных часто вы-
зывали критику определения Ко при помощи уравнения Сипса.
Причина этих отклонений — несоответствие реально существую-
щего в природе распределения АТ по аффинности равномер-
ному и симметричному распределению, принятому Сипсом. Бы-
ли описаны сложные математические методы решения, берущие
за основу непредетерминированное и предетерминированное
распределение значений аффинности [I]. К тому же при рабо-
те этими методами требуются такие затраты реактивов (увели-
чение b и с) и времени подсчета данных, что их ценность для
рутинной работы утрачивается.
1.4.5.2. ОПРЕДЕЛЕНИЕ ВАЛЕНТНОСТИ АНТИТЕЛ
При наличии очищенных специфических антител становится
возможным соотнести в молях количество связанного гаптена с
количеством антител [3]. Молярное соотношение Г/АТ=(г)
заменяет b в уравнениях 3—7. Крайние значения г, наблюдае-
мые при полном насыщении участков связывания антител гап-
55
теном соответствуют валентности N-антител. Уравнение Скэт-
чарда применительно к данному случаю выглядит следующим
образом:
— «= пКа — гКд
(8>
(обозначения см. уравнения 3—7).
Для определения средней истинной константы ассоциации (Ко>
можно использовать уравнение Сипса в следующей форме:
log(r/n — г) = a-log с 4-a-log ко. (9)
1.4.5.З. РАСЧЕТ ТЕРМОДИНАМИЧЕСКИХ ВЕЛИЧИН
КОНСТАНТ АССОЦИАЦИИ
Значение константы ассоциации делает возможным расчет сво-
бодной энергии по формуле:
AG° = - RT In Ко,
где R — универсальная газовая постоянная, Т — абсолютная
температура.
Определив Ко для.двух различных температур, можно опреде-
лить энтальпию Н° по формуле, преобразовав ее
b In Ко ДН«
ЬТ •= R-Ta
в уравнение:
, K»j ДН»(Т2 —Т1)
Og К01>- 2.303КТ2-Т1 '
Величину энтропии AS° определяют по формуле:
AGO = ДНО - TAS0.
1.4.6. СПИСОК ЛИТЕРАТУРЫ
1. Bruni С., Germani A., Koch G., Strom R. Derivation of antibody distribution
from experimental binding data. — J. Theor. biol., 1976, 61, 143.
2. Dandliker W. G„ Levison S. A. Investigation of antigen-antibody kinetics by
fluorescence polarization. — Immunochemistry, 1967, 5, 171—173.
3. Eisen H. N. Equilibrium dialysis for measurement of antibody-hapten affini-
ties.— Methods Med. Res., 1964, 10, 106—114.
4. Eisen H. N. Determination of antibody affinity for hapten by means of fluo-
rescence quenching. — ibid. 115,
5. Fiebig H., Ambrosius H. Valenz und affinitat von Anti-Dinitrophenyl-Anti-
korpern. — Acta Biol. Med. Germ., 1975, 34, 1681—1695.
6. Larralde C., Farber S. Confidence intervals for affinity constants, heteroge-
neity indices and concentration of reactive sites estimated by 50% ammonium
sulphate method on rat’s anti-DNP sers. — Immunochemistry, 1972, 9, 699.
7. Levison S. A., Portman A. J., Kierzenbaum F„ Dandliker W. B. Kinetic beha-
vior of anti-hapten antibody of restricted heterogeneity by stopped flow fluo-
rescence polarization kinetics. — Biochem. Biophys. Res. Comm., 1971, 43,
258—266.
56
8. Parker C. W. — In: The handbook of experimental Immunology/Ed. by
D. M. Weir. Oxford, Blackwell Scientific Publication, 1967.
9. Pressman 0., Grossberg A. L. The structural basis of antibody specificity. —
Amsterdam, W. A. Benjamin Inc. N. Y. — 1968.
10. Stupp K, Yoshida T., Paul W. E. Determination of antibody-hapten equilib-
rium constant by an ammonium sulphate precipitation technique. — J. Im-
munol., 1969, 103, 625—627.
11. Velick S. F., Parker C. W„ Eisen H. N. Exitation energy transfer and the
quantitative study of the antibody hapten reaction. — USA, Proc. Nat. Acad,
Sci., 1960, 46, 1470.
1.5. МЕТОД ЛОКАЛЬНОГО ГЕМОЛИЗА
1.5.1. ПРИНЦИП МЕТОДА
Метод локального гемолиза используется для выявления клеток,
образующих антитела. Метод позволяет осуществлять количест-
венные исследования сложного процесса образования В-лимфо-
цитами антител in vivo и in vitro. Лимфоциты лимфоидных ор-
ганов, периферической крови или культур клеток смешивают с
густой суспензией индикаторных эритроцитов, распределяют
суспензию тонким слоем и иммобилизуют ее. При последующей
инкубации плазматические клетки синтезируют антитела, кото-
рые диффундируют в окружающую среду и фиксируются на
эритроцитах. В присутствии комплемента происходит лизис
красных кровяных клеток. Образуются зоны гемолиза — свет-
лые крупные пятна (иногда их называют бляшками), различи-
мые невооруженным глазом (рис. 13). В центре каждой зоны
гемолиза находится АОК—антителообразующая клетка (рис.
14). В целом число АОК соответствует числу зон гемолиза. Ес-
ли известно количество внесенных ядросодержащих клеток
(лимфоциты), то можно определить число АОК на 106 клеток
исследуемого лимфоидного органа. Метод локального гемолиза
(ЛГ) позволяет обнаружить и точно определить количество
АОК в большой популяции лимфоцитов. Этим так называемым
прямым методом определяют клетки, образующие IgM-антите-
ла, высокоэффективные при гемолизе (рис. 15, а). Клетки, не
секретирующие IgM-антитела, могут быть обнаружены так на-
зываемым непрямым методом с антииммуноглобулиновой сыво-
роткой, так называемой «проявляющей» сывороткой (рис. 15, б).
Антииммуноглобулиновые антитела связываются с секретиро-
ванными АТ, фиксированными на эритроцитах. Наличие IgG-ан-
тииммуноглобулиновых комплексов на мембране эритроцитов
способствует гемолизу в присутствии комплемента. Количест-
во «непрямых» АОК можно определить по разности данных, по-
лученных прямым и непрямым методами. Точные методы опре-
деления АОК, синтезирующие 1g, не относящиеся к классу IgM,
будут описаны ниже. При помощи метода локального гемолиза
можно обнаружить не только АОК> синтезирующие 1g против
57
Рис. 15. Прямой (а) н непрямой
(б) методы локального гемолиза
в геле.
ПС — проявляющая сыворотка.
эритроцитарных АГ, но и вы-
являть клетки, продуцирую-
щие антитела к белкам, бак-
териальным полисахаридам
и гаптенам, если использо-
а
1д М -антитела
вать индикаторные эритро-
циты, на мембране которых
сорбированы соответствую-
щие антигены. Эритроциты
подвергаются пассивному
лизису после образования
комплексов АГ—АТ на их
поверхности. Используя эри-
троциты с фиксированным
Комплемент
30 мин
Комплемент
ЗОмин
на их поверхности протеи-
ном А из золотистого ста-
филококка [8] или антитела
к IgG [6] в качестве прояв-
ляющей сыворотки, можно
обнаружить любые Ig-npo-
цуцирующие клетки, незави-
симо от их специфичности
(поликлональная В-лимфо-
цинарная реакция). Поскольку
Лизис
в отличие от оригинального ме-
тода поликлональные антитела обнаруживают при помощи про-
теина А и антител, конъюгированных с эритроцитами, эта моди-
фикация получила название непрямого локального гемолия.
Подстановка ЛГ может сильно варьировать в зависимости от
используемых добавок или носителей (табл. 5). Обе важнейшие
модификации ЛГ представлены на рис. 16.
1.5.2. МЕТОДИКА
1.5.2.1. ПРОВЕДЕНИЕ ИССЛЕДОВАНИЯ
1.5.2.2.1. Материалы и оборудование
Для работы необходимы: шприцы для инъекций; иглы № 20;
ножницы и пинцет; гомогенизатор или дедероновое (или из нер-
жавеющей стали) сито; тонкая марля; чашки Петри диаметром
Рис. 13. Зоны гемолиза, стандартная методика (прн рассматривании нево-
оруженным глазом).
Рис. 14. Антителообразующая клетка в центре зоны гемолиза (X100).
59
Таблица 5. Модификации метода локального гемолиза
Предназначение Обозначение
Определение АОК Определение Ig-образу- ющих клеток Тип антигена Добавки к суспензиям клеток Другое оборудование 1. IgM-АОК 2. не IgM-AOK 1. Изотипы 1g 2. Аллотипы 1g 3. Идиотипы 1g 1. Эритроциты 2. Растворимые антиге- ны, сорбированные на эритроцитах 1. Гели (агар, агароза) 2. Полутвердые добавки (карбоксиметилцеллю- лоза) 3. Без добавок 1. Чашки Петри 2. Планшеты для мик- • рокультур 3. Плоские стеклянные камеры Прямой ЛГ Непрямой ЛГ Обратный Л Г Прямой или непрямой; стандартный метод Пассивный ЛГ, прямой или непрямой Локальный гемолиз в геле (метод Джериа » Нордииа, 1963) Ингрема и Бюссара» 1965 Безгелевый ЛГ по Кан- нингему, 1965 Макро-ЛГ Безгелевый ЛГ
Рис. 16. Стандартный метод (вверху) и метод Gunningham (внизу).
60
5 и 8 см; предметные стекла; пробирки; обоюдоклейкая лента
(Scotch Таре № 410, Minnesota Mining Со); водяная баня на
47—49°С, центрифуга (ТЗО или Т23); автоклав; термостату
влажная камера; лупа; микроскоп; счетная камера; механиче-
ское счетное устройство; лампа для микроскопа или другой ис-
точник света; раствор Тюрка; 0,2% раствор трипанового синего
в 0,15 М NaCl; очищенный агар, очищенная агароза (например,
Ferak, Западный Берлин); диэтиламиноэтил-(ДЭАЭ)-декстран
(Pharmacia, Швеция); 2-меркаптоэтанол (Ferak, Западный Бер-
лин); глютаровый альдегид (Serva, ФРГ); КонА; протеин А
(Pharmacia, Швеция); парафиновая смазка; суспензия эритро-
цитов барана (СЭБ), по меньшей мере однонедельной давно-
сти, 20 или 10% в 0,15 М NaCl. Предварительно определяют
гематокрит при помощи гематокритной центрифуги ТН1 или
ТН2, разводят исходную суспензию сывороткой морской свин-
ки. Адсорбированную СЭБ и разведенную в соотношении 1 : 10
средой Игла сыворотку морской свинки используют в качестве-
источника комплемента. Необходима также анти-IgG сыворот-
ка (проявляющая сыворотка, ПС).
1.5.2.1.2. Прямой метод локального гемолиза
Подготовка чашек Петри (посуды): на дно чашек
петри диаметром 8 см наносят слой агара в среде Игла с до-
бавлением ДЭАЭ-декстрана, концентрация агара — 1,4%; до-
давляют ДЭАЭ-декстран до концентрации 1 мг/мл, подавляю-
щий антикомплементарное действие агара. Агар сравнительно'
труднорастворим. Первоначально готовят двойную концентра-
цию взвеси агара (2,8%) в дистиллированной воде, растворяют
ее автоклавированием в течение 10 мин при 120 °C. Одновремен-
но готовят среду Игла с добавлением ДАЭ-декстрана, оба рас-
твора смешивают, когда агар несколько охладится. Величина
pH золя должна составлять 7,4. В каждую чашку Петри вносят
3—4 мл золя агара. После застывания чашки могут храниться
в холодильнике в течение 1—2 нед. Основной слой можно фор-
мировать без добавления ДЭАЭ-декстрана. Перед использова-
нием чашки инкубируют при 37 °C в течение 1,5 ч для удаления»
воды с поверхности агара.
Получение лимфоидных клеток. Грубую кле-
точную суспензию из лимфоидных органов (селезенка, лимфати-
ческие узлы, тимус) можно получить тремя различными мето-
дами: размельчением органа в гомогенизаторе с притертым теф-
лоновым пестиком в среде Игла, протиранием через тонкое си-
то из дедерона или нержавеющей стали, измельчением пинце-
том и ножницами. Последний метод наиболее щадящий (боль-
шая доля живых клеток), но обычно выход клеток невелик.
При обработке больших количеств материала с целью получе-
ния максимального количества клеток экономнее использовать-
сито или гомогенизатор. Время между забоем животного и по-
61
мещением суспензии клеток на водяную баню не должно превы-
шать 15 мин. Обломки клеток и грубые агрегаты удаляют цент-
рифугированием при 1g в течение нескольких минут или фильт-
рованием через тонкую марлю. Гомогенность суспензии улуч-
шается при ее многократном пропускании через шприц с тон-
кой иглой (№ 20). Суспензию клеток костного мозга получают
путем смыва длинных трубчатых костей мелких животных или
путем пункции у человека. При определении АОК в перифери-
ческой крови необходимо получить суспензию лимфоцитов, сво-
бодную от автогенных эритроцитов, в противном случае можно
пропустить лизис тестерных эритроцитов (см. раздел 3.1). По-
лученную суспензию центрифугируют при 350 g и 4 °C, дважды
отмывают в среде Игла и суспендируют в этой среде. Конечный
объем взвеси зависит от концентрации клеток, которая опреде-
ляется в предварительных опытах так, чтобы получать 200—
300 зон гемолиза на чашку. Клетки должны быть использованы
в опыте не позднее чем через 1 ч после приготовления суспен-
зии. Если обстоятельства не позволяют уложиться в этот срок,
то клетки лучше хранить в виде осадка. Число АОК остается в
этом случае неизменным. Всю работу до момента смешивания
с золем проводят при 0—4 °C. Живые клетки подсчитывают на
основании их резистентности к окрашиванию трипановым си-
ним (мертвые клетки окрашиваются в голубой цвет).
Приготовление верхнего слоя, постановка
опыта (см. рис. 16): Расплавленную агарозу или агар (0,7%
раствор в среде Игла, pH 7,4) с добавлением 1 мг/мл ДЭАЭ-дек-
страна разливают в пробирки по 1 мл и выдерживают при 47—
49 °C в водяной бане. Затем добавляют 0,1 мл 10% суспензии
ЭБ и выдерживают еще 10—15 мин при той же температуре,
наконец вносят 0,1 мл охлажденной клеточной суспензии и тща-
тельно перемешивают, избегая образования воздушных пузы-
рей. Немедленно смесь переносят в заранее прогретую чашку
Петри со сформированным нижним слоем и равномерно распре-
деляют ее по поверхности. Во время застывания агарозы чаш-
ки помещают на горизонтальную подставку, чтобы верхний слой
геля был повсюду одинаковой толщины. Подготовленные чашки
инкубируют во влажной камере в течение 2 ч при 37 °C. Нельзя
допускать высыхание поверхности, так как это может умень-
шить размер зон гемолиза ввиду образующихся при высыхании
гипертонических условий инкубационной среды. В заключение
наслаивают 1,5 мл комплемента морской свинки в разведении
1 : 10, инкубируют ЗОмин при 37 °C и производят подсчет зон ге-
молиза. В нашей лаборатории для этой цели с успехом приме-
няли прибор Dokumator DL II (VEB Carl Zeiss, JENA). Под-
счет можно проводить при помощи стереомикроскопа или спе-
циального устройства для подсчета колоний. Сомнительные зо-
ны исследуют под микроскопом. В центре каждой зоны должна
находиться лимфоидная клетка (см. рис. 14). После смывания
компемента средой Игла пластины геля можно фиксировать
62
0,25% раствором глютарового альдегида в 0,9% NaCl или па-
рами формалина. В этом случае подсчет зон гемолиза можно
проводить и позднее.
1.5.2.1.3. Непрямой метод локального гемолиза
Секвенциальный метод. После подсчета зон гемолиза,
образовавшихся в прямом методе ЛГ, смывают комплемент 2—
3 мл среды Игла. Инкубируют чашки в течение 1 ч при 37 °C
с разведенной антиглобулиновой сывороткой («проявляющей»
ПС). Подбор оптимального разведения ПС имеет важное прак-
тическое значение для успешной постановки непрямого ЛГ. Вы-
сокие концентрации сыворотки маскируют «непрямые зоны»,
слишком низкие неэффективно «проявляют» их. Эффективность
«проявления» следует контролировать для каждой серии анти-
глобулиновой сыворотки. Для работы лучше всего использовать
селезеночные клетки мыши, иммунизированной за 8—12 дней
4Х108 ЭБ. В каждой селезенке образуется приблизительно в
10 раз больше непрямых АОК, чем прямых. После удаления
проявляющей сыворотки проводят повторную инкубацию с
комплементом (30 мин); повторно подсчитывают зоны гемолиза.
Разность числа зон гемолиза, определяемых прямым и непря-
мым методами, дает количество АОК, не продуцирующих IgM.
Торможение локального гемолиза (IgM)
2-м е р к а п т о э т а н о л о м или Ко нА. По окончании первой
инкубации слой агарозы инкубируют еще 1 ч при 37 °C с
0,15 М 2-меркаптоэтанолом или раствором КонА (0,4 мг/мл в
0,9% NaCl). Агарозу троекратно отмывают с интервалом в.
10 мин раствором NaCl при 20 °C. Непрямые зоны гемолиза
проявляют, как было описано выше, при помощи ПрС и комп-
лемента. Наоборот, 2-меркаптоэтанол и КонА блокируют обра-
зование прямых зон гемолиза (IgM) и практически не влияют
на формирование IgG-зависимых зон гемолиза. Торможение
IgM-зависимого локального гемолиза анти-ц-
сывороткой. В ходе приготовления верхнего слоя к ЭБ и
лимфоидным клеткам в зоне агарозы добавляют 0,05 мл анти-ц-
сыворотки в оптимальном разведении (конечное разведение-
1 : 2000 и более). После 2-часовой инкубации при 37°C выявля-
ются непрямые зоны гемолиза. Блокада IgM основана на специ-
фическом взаимодействии с анти-р-антителами, препятствую-
щем образованию IgM-зависимых зон гемолиза.
1.5.2.2. БЕЗГЕЛЕВЫЙ МЕТОД
Гемолиз происходит в безгелевой среде. АОК и эритроциты
смешивают в одном слое. Смесь клеток контактирует с комп-
лементом в закрытой камере (см. рис. 16).
Установка камер. Обезжиренные предметные стекла
складывают бок о бок и соединяют обоюдоклейкой лентой по
краям и по середине. Затем снимают верхний защитный слой'
63
тклейкой ленты и складывают оба стекла (как страницы книги),
с тем чтобы получилось их полное наложение. Образующиеся
при этом двойные камеры могут быть снова легко разделены.
Каждая двойная камера имеет объем около 0,15 мл.
Заполнение камер. Для прямого метода используют
следующую смесь: 0,3 мл 20% суспензии ЭБ+0,1 мл свежего
абсорбированного комплемента+0,5 мл среды Игла; для непря-
мого метода — 0,3 мл 20% суспензии ЭБ-|-0,1 мл свежего аб-
сорбированного комплемент а+ 0,2 мл анти-1§-сыворотки в раз-
ведении 1 :40-4-0,4 мл среды Игла. К 0,4 мл каждой из этих
смесей добавляют по 0,2 мл суспензии лимфоцитов с установ-
ленным содержанием клеточных элементов (1—10ХЮ6/мл)>
тщательно перемешивают и вносят по 0,15 мл в камеру. Если
камера заполнена не до конца, объем доводят суспензией эрит-
роцитов. Камеры герметизируют парафином, инкубируют 1 ч при
37 °C. Подсчет зон гемолиза проводят в течение ближайших 4 ч.
Для предотвращения образования пузырей воздуха в камере
непосредственно перед заполнением суспензию клеток подогре-
вают до комнатной температуры; предметные стекла должны
быть безукоризненно чистыми. В этом смысле хорошо зареко-
мендовало себя многочасовое отмывание в проточной водопро-
водной воде с последующей отмывкой дистиллированной водой
и абсолютным этанолом.
1.5.2.3. ПАССИВНЫЙ МЕТОД ЛОКАЛЬНОГО
ГЕМОЛИЗА
Основную сложность пассивного метода ЛГ представляет со-
бой непосредственный контакт индикаторных эритроцитов с ан-
тигеном в одном слое, причем эритроциты не должны самопро-
извольно разрушаться и в то же время должны быть доступны
для антител и комплемента. Принципиально известны два спо-
соба присоединения антигена к эритроцитам — химический и
иммунологический. Описаны многочисленные варианты этих ме-
тодов. Мы приводим выборочно некоторые из них с учетом при-
меняемых антигенов и способов их конъюгирования с эритро-
цитами.
Получение индикаторных эритроцитов
Белки: известны три метода химической конъюгации. Прежде
всего это метод с применением ЭКДИ [1-этил-1,3 (3-диметил-
аминопропил) карбодимида-HCl]. К 3 мл раствора белка (10—
80 мг/мл в зависимости от используемого блека) добавляют
0,1 мл 50% СЭБ (лучше использовать эритроциты козы), за-
тем 0,5 мл раствора ЭКДИ (100 мг/мл). В качестве растворите-
ля для всех компонентов применяют фосфатно-солевой буфер
(ФСБ). После одночасовой инкубации при 2°C в покое эритро-
циты отмывают ФСБ с 1% сывороткой. При помощи ЭКДИ
получают сохраняющие стабильность в течение нескольких ме-
сяцев эритроциты-мишени, особенно если к нативным или тио-
64
гликолированным эритроцитам присоединяют сукцинил- или
3- (2-пиридилтио)пропионил-производные белков [9, 10].
Самый простой метод конъюгирования белков с эритроцита-
ми основан на использовании хлорида хрома. Процесс
идет при следующих оптимальных условиях: 1 объем 50% сус-
пензии ЭБ смешивают с 1 объемом 0,5 мМ СгС13 и */ю объема
2% БСА, тщательно перемешивают. Исходный 1% водный рас-
твор хлористого хрома хранится в темной посуде при 4 °C в те-
чение нескольких недель. Все компоненты смеси приготовляют
на 0,15 М NaCl. Рекомендуется соблюдать последовательность
операций. Смесь инкубируют в течение 1 ч при 37 °C, перемеши-
вая ее несколько раз. Эритроциты трижды отмывают ФСБ (фос-
фат ингибирует реакцию). Описаны и другие условия постанов-
ки реакции [16]. Для постановки реакции локального гемолиза
возможно использование ЭБ, конъюгированных с сывороточны-
ми белками при помощи бис-д и а з от и р о в а н н о г о бен-
зидина (БДБ) [12]. Белок в количестве 10—15 мг растворя-
ют в 18 мл ФСБ, приливают к 0,5 мл 50% суспензии ЭБ, тща-
тельно перемешивают на водяной бане со льдом. БДБ, который
хранится при —70 °C, размораживают, берут 0,2 мл и немед-
ленно смешивают с 2,8 мл 0,1 М фосфатного буфера (pH 7,4).
Полученную смесь (3 мл) немедленно приливают к охлажден-
ной до 0 °C СЭБ. Конъюгация происходит при 0°С в течение
30 мин при постоянном помешивании. Объем смеси доводят
ФСБ до 50 мл. Отмывают эритроциты ФСБ. Приготовленные
таким образом индикаторные эритроциты сохраняются при
4 °C в течение нескольких часов.
Методы иммунологической конъюгации основаны
на прямом или непрямом связывании белков с антителами к
эритроцитам барана. АОК к гетерологическим иммуноглобули-
нам, бычьему и человеческому (БГГ, ЧГГ) обнаруживаются
сравнительно просто. ЭБ сенсибилизируют субгемолитическими
количествами анти-ЭБ-антител. АОК против БГГ обнаружива-
ют при помощи ЭБ, инкубированных с разведенной сывороткой
против ЭБ крупного рогатого скота. Смешивают равные объемы
2,5% суспензии ЭБ и разведенной в соотношении 1:200 сыво-
роткой против ЭБ (титр гемолизинов неразведенной сыворотки
равен 1: 160, все компоненты растворяют в 0,15 М NaCl), инку-
бируют 1 ч при 37 °C, смесь несколько раз перемешивают. Пос-
ле отмывки эритроциты готовы к употреблению. Вместо вало-
вых иммуноглобулинов можно использовать моновалентные
Fab-фрагменты антител к эритроцитам или IgG кур (IgG птиц
не связывает комплемент млекопитающих) в высоких концент-
рациях, что исключает связывание комплемента с сенсибилизи-
рованными эритроцитами. Оба эти способа можно с успехом
использовать для конъюгирования различных гаптенов. Связы-
вание белков с антителами или их фрагментами глютаровым
альдегидом трудноосуществимо, применительно к ЛГ оно себя
не оправдало. Индикаторные клетки после конъюгации иммуно-
5—990
65
логическим методом можно хранить в холодильнике в течение
многих дней.
Бактериальные полисахариды. Конститутивные
полисахариды, например эндотоксины грамотрицательных бак-
терий или капсульные углеводы пневмококка, могут быть ад-
сорбированы на эритроцитах. В зависимости от метода их по-
лучения они содержат различную долю фосфолипидов и бел-
ков.
Эндотоксин. Для конъюгации эндотоксинов с ЭБ пред-
ложено большое количество сходных способов. Мы рекоменду-
ем следующий метод: ЛПС сальмонелл (коммерческий препарат
или препарат по Вестфалю) перед употреблением разводят да
0,1% концентрации в 0,15 NaCl, затем кипятят в течение 1 ч,
охлаждают и прибавляют к 2—10 мл смеси 1 мл осадка ЭБ.
Инкубируют 30 мин при 30 °C, затем трижды отмывают эрит-
роциты. Особое внимание при выборе ЛПС и эритроцитов сле-
дует обращать на перекрестные реакции антигенов эритроцитов
с ЛПС бактерий.
Капсульные антигены пневмококка. Строение
этих АГ точно установлено, например, АГ пневмококка III ти-
па представляет собой полимер З-р-О-гликозил-4-р-О-глюкуро-
новой кислоты. Собирают надосадочную фракцию 8—12-часовых
культур, pH которых доводили до 7,0 добавлением ИаНСОз.
К Ю мл 1% суспензии ЭБ добавляют 0,9 мл фильтрата надоса-
дочной фракции и инкубируют 45 мин при 37 °C. После инкуба-
ции эритроциты трижды отмывают. Оптимальный объем фильт-
рата для сенсибилизации ЭБ определяют эмпирически. Если
сорбции подлежат нейтральные или безлипидные полисахариды,
например декстран, то для оптимизации сорбции используют
липоиды, например стеариновую кислоту. Процесс сенсибилиза-
ции очень прост: к 1 мл 1% СЭБ добавляют 20 мкг О-стеарин-
декстрана (1% остатков стеариновой кислоты) в объеме 20 или
50 мкл, перемешивают, смесь инкубируют 30 мин при 37 °C.
Трижды отмывают ФСБ.
Гаптены. К эритроцитам можно присоединять значитель-
ное количество гаптенов, применяемых для обнаружения АОК.
Антигенные детерминанты можно фиксировать на мембране
эритроцитов непосредственно или с помощью конъюгатов бе-
лок— гаптен. При последнем способе меньше повреждаются
мембраны эритроцитов, чем при прямом химическом присоеди-
нении гаптена. Мы приводим в качестве примера три различных
способа прямого конъюгирования.
Нитрофенилирование. К эритроцитам могут быть
присоединены самые различные нитрофенильные производные.
Наиболее часто применяемые ДНФ- и ТНФ-гаптены присоеди-
няют при помощи водорастворимых реагентов, таких как бен-
золсульфонат и динитрофторбензол [17].
Сульфоновокислотный метод. 20 мг 2,4,6-тринит-
робензолсульфоновой кислоты (ТНБК) растворяют в 7 мл
66
0,26 М какодилатного буфера с pH 6,9. Добавляют 1 мл осев-
ших ЭБ, суспензию перемешивают 10 мин при 23—25 °C. На-
груженные эритроциты отмывают в холодном ФСБ до тех пор,
пока надосадочная фракция не станет бесцветной. Все процеду-
ры, в том числе и связывание гаптена, проводят в темноте для
предотвращения его фотоиндуцированного отщепления.
Ацилазидный метод. Для присоединения к эритроци-
там 4-гидрокси-3,5-динитрофенилацетазид (NNP)- и З-йод-4-
гидрокси-5-нитрофенилацетазид (NIP)-групп используют ацил-
азидные производные, образующие амидные связи с аминогруп-
пами мембран эритроцитов. Конъюгация NNP-азида с ЭБ про-
ходит в 0,12 М карбонатбикарбонатном буфере с pH 9,25. Сус-
пензию отмытых ЭБ (1%) смешивают с NNP-азидом в концент-
рации 0,001—1 мг/мл. После 16-часовой инкубации при 4 °C
конъюгированные эритроциты отмывают 3 раза.
Диазотирование. Диазотирование относится к про-
стейшим способам конъюгации. Для проведения реакции сме-
шивают равные объемы 0,002—0,005 М диазореактива и 50%
СЭБ, доводят pH до 7,4—7,8, инкубируют 1—2 ч при 20 °C или
оставляют на ночь при 4 °C. Для повышения стабильности
конъюгированных эритроцитов небольшой избыток диазореакти-
ва нейтрализуют прибавлением ароматического амина, избыток
нитрита нейтрализуют 0,1 М мочевиной.
После диазотирования ЭБ отмывают в растворе альбумина или
сыворотки с целью удаления не связанных ковалентно продук-
тов реакции. При помощи этого метода можно обнаруживать
АОК, продуцирующие ИГ против азобензоларсоната, азобензол-
сульфоната и азобензилгалактозида.
Пенициллиновая конъюгация. Бензилпеницилли-
на калиевая соль (БПК) присоединяется к ЭБ в изотоническом
триметиламиновом буфере с pH 10 или в 0,14 М глициновом
буфере с pH 10 [15]. Растворяют 600 мг БПК (1,6 мМоль или
106 ME) в 20 мл буфера и прибавляют 1 мл осадка эритроци-
тов. Смесь инкубируют при 25 °C в течение 50 мин, клетки от-
мывают 4 раза. При описанных условиях бензилпенициллоиль-
иые группы ковалентно связываются через амидные группы с
мембраной эритроцитов.
1.5.2.4. ОБРАТНЫЙ ЛОКАЛЬНЫЙ ГЕМОЛИЗ (ОЛГ)
Основные этапы ОЛГ представлены на рис. 17. В настоящее
время используют преимущественно нагруженные протеином А
индикаторные эритроциты барана, поскольку протеин А пред-
ставляет собой более доступный продукт, чем антиглобулиновые
антитела, очищенные методом аффинной хроматографии. Про-
теин А легче конъюгируется с ЭБ, чем антиглобулиновые анти-
тела.
ОЛГ с протеином А [8]. Протеин А из золотистого ста-
филококка присоединяют к ЭБ при помощи СгС13. Все реаген-
5*
67
Рис. 17. Непрямой метод
локального гемолиза для
определения Ig-продуци-
рующнх клеток.
ты растворяют в 0,15 М
NaCl. Один объем
осадка отмытых эрит-
роцитов смешивают с
одним объемом раст-
вора протеина А (0,5
мг/мл). Прибавляют
10 объемов 2,5хЮ-4 М
раствора СгС1з-6Н2О,
инкубируют 45 мин при
37°C, многократно от-
мывают. При постанов-
ке ЛГ в геле полезно
последовательно инку-
бировать пробу с ПС и
комплементом в тече-
ние 1 ч, т. е. общая
продолжительность ин-
кубации составляет 3 ч.
Предварительно опре-
деляют оптимальную концентрацию ПС с целью избежать эф-
фекта прозоны. Следует отдавать предпочтение кроличьим АС
перед козьими, поскольку они эффективнее связывают проте-
ин А и, кроме того, содержат меньше АТ, не связывающих комп-
лемент. Технические усовершенствования протеин-А-зависимо-
го ЛГ описаны в работах [2, 18].
1.5.3. ОЦЕНКА МЕТОДА
Локальный гемолиз (стандартная методика) в отличие от дру-
гих методов позволяет обнаруживать и количественно опреде-
лять единичные АОК среди нескольких миллионов клеток в те-
чение 2 ч. Существенное значение для успеха эксперимента име-
ет выбор агара. Оптимальным следует считать формирование
верхнего и нижнего слоя геля на агарозы, т. к. агароза не
инактивирует комплемент. При использовании чистого агара
возникает необходимость внесения ДЭАЭ-декстрана. Этот поли-
катион адсорбирует сульфополисахариды, выделяющиеся при
кипячении агара, которые связывают ионы Са2+ и Mg2+. Имен-
но образованием сульфополисахаридов обусловлено ингибирую-
щее действие агара на комплемент. Колебания концентрации
агара в пределах 0,4—1% не влияют на число зон гемолиза и
их диаметр. Повышение концентрации свыше 1 % снижает число
зон гемолиза и уменьшает их размеры. Для ЛГ лучше всего ис-
пользовать ЭБ, хранившиеся в течение 1—3 нед в растворе Оле-
68
вера. Свежие эритроциты малочувствительны к иммуногемоли-
зу; длительно хранившиеся клетки, напротив, чрезмерно хруп-
ки. Следует всегда помнить об генетически обусловленной раз-
личной чувствительности эритроцитов разных особей [1]. По-
этому желательно всегда использовать эритроциты одного и
того же животного. При меньшей концентрации ЭБ образуются
зоны гемолиза большего размера, причем их число не уменьша-
ется. Однако при рекомендованных нами концентрациях до-
стигается большая надежность результатов, особенно если ЭБ
отличаются повышенной хрупкостью. Температура 47—49 °C не
влияет на индикаторные эритроциты, но лишает лейкоциты
жизнеспособности. В связи с этим смешивание лимфоидных
клеток с расплавленной агарозой представляет собой самую
тонкую операцию при постановке ЛГ. Оно должно проводиться
почти одновременно с распределением цитосодержащего золя
по поверхности нижнего слоя стереотипными, отработанными
движениями рук. Удлинение периода инкубации перед внесени-
ем комплемента не оказывает влияния на размер и величину
зон гемолиза. Температура инкубации менее 37 °C приводит к
снижению зон гемолиза. В качестве источника комплемента ча-
ще всего используют сыворотку морской свинки, адсорбирован-
ную ЭБ; этот препарат дает наилучшие результаты с мышины-
ми АТ к ЭБ. Если иммунизируют другие виды животных раз-
личными эритроцитами, то следует также подбирать подходя-
щие сыворотки различных животных в качестве источника
комплемента с целью получения оптимальных результатов. Зо-
ны ЛГ, образующиеся до внесения комплемента и не содержа-
щие в центре клетки с ядром, — ложные и исключаются при
подсчете результатов. Причиной образования ложных зон ЛГ
могут служить трещины или нерастворившиеся кусочки агара,
дегенерировавшие клетки или агрегаты клеток, колонии гемоли-
тических бактери и в редких случаях клетки донора эритроци-
тов, образующие гемолизины. Еще одной причиной образова-
ния ложных зон ЛГ может стать перенос пассивно адсорбиро-
ванных антител, о возможности которого всегда необходимо
помнить [14].
Существуют методы непосредственного определения и подсчета
так называемых непрямых, т. е. He-IgM-продуцирующих АОК.
Эти методы свободны от неопределенностей конвенциональных
методов, в которых подсчет He-IgM-продуцирующих АОК осно-
ван на учете разности количества зон ЛГ наблюдаемых прямым
и непрямым методом. Даже при оптимальном разведении ПС
вследствие анти-ЕаЬ-активности ПС, учитывая неодинаковое
торможение образование IgM-зависимых зон ЛГ, эти методы
приводят к заниженным оценкам непрямых АОК. Элиминация
IgM-зависимых зон ЛГ не только снижает затраты времени
(2-кратный подсчет зон ЛГ), но, что еще более важно, исклю-
чает стадию проявления непрямых IgM-образующих клеток по-
лпспецифической анти-IgM сывороткой. При наличии моноспе-
69
цифических АС можно определять различные алло- и изотипы
иммуноглобулинов в реакции непрямого ЛГ. Наиболее прост,
экономичен и чувствителен безгелевый метод. Он особенно удо-
бен при микроманипуляциях и морфологическом исследовании
отдельных живых АОК- Высокая чувствительность метода
обусловлена ограниченной диффузией антител в монослое ин-
дикаторных ЭБ; лизис даже 7 отдельных эритроцитов приводит
к появлению заметной зоны гемолиза. Этим обусловлен и боль-
ший диаметр зон ЛГ, чем тот, который наблюдается при работе
с гелем. Недостатком безгелевого метода является необходи-
мость подсчета зон гемолиза в течение нескольких часрв и не-
возможность фиксации препарата. Можно исследовать лишь
ограниченное число лимфоидных клеток. При концентрации лим-
фоцитов 5Х107/мл бйльшая часть зон ЛГ маскируется автоло-
гичными эритроцитами и лейкоцитами. Этих недостатков лишен
модифицированный метод [12], при котором монослой индика-
торных эритроцитов связан с поверхностью пластика. Предло-
жено много вариантов постановки реакции локального гемолиза,
важнейшие из иих перечислены в табл. 5. Часто вместо геля ага-
ра или агарозы применяют полутвердые субстанции, например,
карбоксиметиллюлозу (КМ) в закрытых или открытых системах.
Системы с КМ-целлюлозой пригодны не столько для рутинных
работ, сколько для наблюдения за АОК в течение длительных
периодов, так как известно, что КМ-целлюлоза создает опти-
мальные условия для выживания клеток. Многие авторы отка-
зываются от чашек Петри, распределяя смесь золя агарозы с
клетками по поверхности предметного стекла с преформирован-
ным слоем 0,1—0,2% агарозы. Эта модификация особенно удоб-
на при работе с моноспецифическими сыворотками и небольши-
ми количествами конъюгированных с различными веществами
эритроцитов. Она непригодна для работы с большими количе-
ствами лимфоцитов ввиду многочисленности зон ЛГ. Сверхтон-
кий слой агара на предметных стеклах особенно удобно при-
менять для цитологических и авторадиографических исследова-
ний. Микромодификация ЛГ позволяет проводить прямое ис-
следование культур В-лимфоцитов в микропланшетах [11].
Пассивный ЛГ представляет собой довольно тонкий метод, при-
менение которого требует от экспериментатора большого мас-
терства. Условия конъюгирования, при которых получают оп-
тимальную поверхностную плотность детерминант на эритроци-
тах, как правило, подбираются экспериментально. Методы
конъюгирования, описанные для РПГА, не пригодны для реак-
ции ЛГ, поскольку эритроциты, подготовленные для гемагглю-
тинации, содержат очень мало детерминант для того, чтобы
после наслоения АТ и комплемента образовались зоны ЛГ. Раз-
витие ЛГ в геле зависит от изотипа и аффинности выделяемых
АОК антител. Заниженное число АОК в реакции пассивного
ЛГ обусловлено неадекватной конъюгацией эритроцитов. Спе-
цифичность ЛГ можно повысить, тормозя реакцию введением
70
Рис. 18. Образование анти-
телообразующих клеток в
селезенке мышей, иммунизи-
рованных эритроцитами ба-
рана, в ходе первичного (1)
и вторичного (2, 3) иммун-
ного ответа.
1, 2 —прямые АОК (IgM); 3 —
непрямые АОК (не IgM).
в агар свободного ан-
тигена или гаптена. Та-
ким же путем можно
оценивать авидность
секретируемых АОК
антител [3]. Этот ме-
тод основан на извест-
ном факте избиратель-
ного торможения ма-
лыми концентрациями
свободного антигена и
гаптена локального ге-
молиза, обусловленно-
го высокоавидными антителами, по сравнению с ЛГ, обуслов-
ленным низкоавидными антителами. Для статистической обра-
батки результатов локального гемолиза в геле рекомендуется
представлять данные в виде десятичных логарифмов, что позво-
ляет получить распределение отдельных результатов в выборке,
приближающееся к биноминальному [7]. Ошибка метода ЛГ
составляет около 15%. Колебания, обусловленные качеством
материала, полученного от животных, существенно выше, коэф-
фициент вариации в данном случае может достигать 0,5. ЛГ
имеет важное значение для фундаментальных иммунологиче-
ских исследований. Благодаря этому методу были получены
весьма существенные результаты в области исследования регу-
ляции гуморального иммунитета и изучения структуры и жизне-
деятельности АОК in vivo и in vitro. В качестве примера на
рис. 18 представлена кинетика первичного и вторичного иммун-
ного ответа мыши, выражающаяся в изменении количества
АОК в селезенке. ЛГ уже в течение некоторого времени исполь-
зуется в клинике, в частности при исследовании периферической
крови пациентов с различными заболеваниями иммунной систе-
мы. Определяют, в частности, способность В-лимфоцитов к об-
разованию специфических ИГ, а также контроль активности
лимфоцитов со стороны регуляторных клеток [4, 5].
1.5.4. СПИСОК ЛИТЕРАТУРЫ
Обзоры
1. Dresser D. W., Greaves М. F. Assays for antibody-producing cells. — In:
Handbook of experimental Immunology/Ed. D. M. Weir. — V. 2, chap-
ter 27. — Oxford: Blackwell Scientific Publications, 1973.
71
2. Jerne N. К., Henry С., Nordin A. A. et al. Plaque-forming cells: methodology
and theory. — Transplant. Rev., 1974, 18, 130—131.
3. lerne N. K., Henry C., Nordin A. A. et al. Plaque techniques for recognising
individualy antibody-formong cells. — In: Methods in Ummunology and Im-
munochemistry/Eds. C. A. Williams and M. W. Chase. — Vol. V, 335—370.—
New York: Academic Press, 1976.
Цитированные литературные источники
1. Bell D. A., Leblond P. F., Leddy J. R. et al. Immune hemolysis. I. Differing
susceptibility of three genetic types of sheep red cells. — J. Immunol., 1972,
108, 467—479.
2. Burns G. F., Pike B. L. Sontanious reverse haemolitic plaque formation.
Technical aspects of the protein A assay. — J. Immunol., 1981, 41, 269—
277.
3. De Lisi C., Goldstein B. The kinetics of hemolitic plaques formation II and
III. —J. Theor. Biol., 1975, 51, 313—346.
4. Fauci A. S. Human В-cell function in a polyclonally induced PFC system.
Cell triggering and immunoregulation. — Immunol. Rev., 1979, 45, 93—
116.
5. Geha R. S. Regulation of Human В cell activation. — Immunol. Rev., 1979,
45. 275—305.
6. Ginsburg W. W., Finkelman F. D., Lipsky P. E. Circulating and mitogen
induced immunoglobulin-secreting cells in human peripheral blood: evaluting
by a modified reverse hemolitic plaque assay. — J. Immunol., 1978, 120,
33—39.
7. Gottlieb C. F. Application of transformations to normalize the distribution
of plaque forming cells. — J. Immunol., 1974, 113, 51—57.
8. Gronowicz A., Coutinho A., Melchers F. A plaque assay for all cells secreting
Ig of given type or class. — Europ. J. Immunol., 1976, 6, 588—590.
9. Jou I. H., Bankeri R. B. Coupling of protein antigens to erythrocytes through
disulfide bond formation: Preparation of stable and sensitive target cells for
immune hemolysis. — Proc. Natl. Acad. Sci-. USA, 1981, 78, 2493—2496.
10. Jou У. H., Jonsos G., Pressman D. Succinilation of haptenprotein conjugates
facilitates coupling to erithrocytes by water soluble carbodiimide: preparation
of stabel and sensitive target cells for use in hemolitic assays. — J. Immunol.,
1981, 42, 79—92.
11. Kappler J. W. A micro-technique for hemolitic plaque assays. — J. Immunol.,
1974, 112, 1271.
12. Kennedy 1. C., Axelrad M. A. An improved assay for hemolitic plaque for-
ming cells. — Immunology, 1971, 20, 253—257.
13. Lefkovits I., Cosenza H. Assay for plaque forming cells.— In: Immunolo-
gical methods/Eds. I. Lefkovitz, B. Pernis. New York, Academic Press, 1979,
p. 277—285.
14. Muchmore A. V., Koshi I., Dobley N., Blaese R. M. Artifactual plaque forma-
tion in vitro and in vivo due to passive transfer of specific antibodies. —
J. Immunol., 1976, 116, 1016—1019.
15. Naor D., Негу C., Fudenberg H. H, An in vitro immune response to penicil-
lin.— J. Immunol., 1971, 107, 302—305.
16. Sweet G. H., Welborn F. C. Use of chromium chloride as the coupling agent
in a modified plaque assay of cells producing antiprotein antibody. — J. Im-
munol., 1971, 106, 1407—1410.
17. Trump G. N. Preparation of dinitrofluorbenzene-treated sheep erythrocytes
for detection af anti-DNP-plaque forming cells. — J. Immunol., 1972, 109,
754—760.
18. Van Oudenaren A., Hooijkaas H., Benner P. Improvement of the protein. A
aue assay for immunoglobulin secreting cells by using immunoglobulin-
eted guinea pig serum as a source of complement. — J. Immunol.
Methods, 1981, 43, 219—224.
2. РЕАКЦИИ АНТИГЕН-АНТИТЕЛО
2.1. ИММУНОДИФФУЗИЯ
2.1.1. ОБЩИЕ ЗАМЕЧАНИЯ
Развитие техники иммунодиффузии (ИД) стало возможным
благодаря использованию гелевых носителей. В 1946 г. Уден
описал метод простой линейной иммунодиффузии. Он основан
на следующем принципе: стеклянные пробирки заполняют жид-
ким агаром, содержащим антисыворотку. После застывания ге-
ля (агара) раствор АГ (например, сыворотка) наслаивают на
гель (нисходящая ИД), или же нижний, открытый, конец про-
бирки с гелем погружают в сыворотку так, что АГ проникают в
столб геля снизу (восходящая ИД по Удену). В зависимости от
специфичности заключенных в геле АТ, в ходе ИД возникает
одна из нескольких полос преципитации, длину пробега которых
от линии старта можно измерить. Длина пробега полосы преци-
питации при прочих равных условиях пропорциональна кон-
центрации АГ. Простая линейная диффузия по Удену внесла
существенный вклад в исследование теоретических основ ИД.
Впоследствии этот метод был вытеснен более совершенными.
В 1947 г. Ухтерлони в Швеции и в 1948 г. Elek в Англии описа-
ли двойную радиальную иммунодиффузию. В 1963 г. Манчини
впервые описал простую радиальную ЙД (см. 2.1.2 и 2.1.3).
2.1.1.1. ГЕЛИ И ИХ СВОЙСТВА
Основная функция геля — локализация преципитата. Она ста-
новится возможной, если растворенные АГ и АТ легко диффун-
дируют в геле, но образующиеся иммунные комплексы ввиду их
величины остаются внутри ячеек геля. Благодаря локализации
преципитата достигается следующее:
а. В множественных системах АГ-АТ отдельные компоненты ре-
акции диффундируют с различной скоростью, обусловлен-
ной неодинаковой величиной молекул и их концентрацией.
В связи с этим могут образовываться множественные прост-
ранственно разделенные преципитаты, т. е. создается возмож-
ность анализа множественных систем.
б. По форме и расположению преципитатов можно судить о
диффузионных свойствах и, следовательно, об относительной
молекулярной массе АГ или АТ.
в. Порядок расположения линий преципитации по отношению
друг к другу позволяет делать выводы об полном или час-
73
тичном иммунохимическом сходстве, а также об отсутствии
такового.
г. Исходя из величины ареала и удаленности от линии старта
иммунных комплексов, можно проводить их количественное
определение.
Наиболее распространены гели агара и агарозы; гели жела-
тины, пектина, крахмала, полиакриламида или ацетата целлю-
лозы применяются в реакциях иммунодиффузии реже.
2.1.1.2. ДИФФУЗИЯ И ПРЕЦИПИТАЦИЯ В ГЕЛЕ
При концентрации агара 20 г/л средний размер пор геля со-
ставляет Знм. Можно считать достоверным, что обычно приме-
няемые концентрации геля от 3 до 15 г/л существенно не влия-
ют на диффузию большинства компонентов реакции АГ—АТ.
Поэтому для всех компонентов процесса до их соединения спра-
ведливы законы свободной диффузии. В соответствии со вторым
законом Фика концентрация вещества (С) на определенном
расстоянии от линии старта (h) является функцией времени (t)
6С 8С
& t=D Ы* •
D — коэффициент диффузии, зависит от температуры, вязкости среды и ра-
диуса диффундирующей молекулы.
где R — универсальная газовая постоянная, N — число Авогадро, Т — темпе-
ратура в градусах Кельвина, ц — вязкость среды, г — молекулярный радиус.
При прочих равных условиях D представляет собой постоянную
величину. Это означает, что для получения сравнимых результа-
тов при ИД необходимо стандартизовать такие показатели, как
концентрация геля, температура и время диффузии, концент-
рация АГ и АТ. Это особенно важно, когда к моменту оценки
диффузия еще не завершена (простая линейная ИД, простая
радиальная ИД в модификации Фехей и Мак-Келви [8]).
Встреча компонентов реакции АГ—АТ изменяет или прекраща-
ет свободную диффузию. Последнее имеет место в случае обра-
зования стойких преципитатов, например при простой или двой-
ной радиальной ИД. Важно отметить, что наиболее выражен-
ная преципитация наблюдается в зоне эквивалентности. Опти-
мальную пропорцию можно определить при взаимном проник-
новении компонентов реакции. Это становится более понятным
из рис. 19. В данном случае мы полагаем, что два наиболее
важных фактора образования преципитата — коэффициент диф-
фузии и относительные концентрации — примерно равны. В ме-
сте первого контакта молекул АГ и АТ (линия 2) образуются
преципитаты, которые увеличиваются в размерах по мере по-
74
Рис. 19. Схематическое изображе-
ние процессов, протекающих при
двойной иммунодиффузии.
1 — градиент концентрации АГ и АТ вско-
ре после внесения растворов в резервуары;
2 — первый контакт партнеров реакции;
3 — процесс диффузии в основном окончен,
преципитат полностью сформирован.
ступления эквивалентных коли-
честв партнеров реакции. Пре-
ципитация становится полной
при нивелировании градиента
концентрации (линия 3). По
Ухтерлони система называется
сбалансированной, если место
первого образования иммунных агрегатов и место их последую-
щего отложения совпадают. Если один из компонентов реакции
диффундирует медленнее, чем другой, то комплексы АГ—АТ,
возникшие при первом контакте, медленно растворяются вслед-
ствие смещения зоны эквивалентности под влиянием притока
более медленно диффундирующего партнера компонента. Вновь
образующиеся комплексы формируются все ближе и ближе к
резервуару быстро диффундирующего компонента.
В соответствии с вышеизложенным в данном случае можно
говорить о несбалансированной системе. Перемещение линии
преципитации является, таким образом, следствием образова-
ния преципитата, распада комплексов, повторного выпадения
преципитатов и т. д.
2.1.1.3. ФАКТОРЫ, ВЛИЯЮЩИЕ
НА ПРЕЦИПИТАЦИЮ
Образование первичных комплексов после контакта АГ и АТ
происходит в течение нескольких минут. Для полной преципи-
тации необходимо 24—48 ч. Для высокомолекулярных компо-
нентов при наличии «слабых» сывороток время полной преци-
питации увеличивается до 8 дней. Температура не играет осо-
бой роли, она может быть 0—4 °C, 20 °C или 37 °C. Антисыво-
ротки лошади дают лучшую преципитацию при температуре,
ниже комнатной; эта закономерность отсутствует при использо-
вании сыворотки морской свинки. Колебания pH в пределах
6,4—8,5 на процесс преципитации влияния не оказывают. АС
млекопитающих дают оптимальную преципитацию при физио-
логических концентрациях NaCl; АС птиц лучше преципитиру-
ют специфические АГ в средах с высокой ионной силой, напри-
мер, в 1,2 М NaCl. Наличие детергентов, органических раство-
рителей, мочевины и других соединений может препятствовать
образованию иммунных агрегатов. Они могут вызывать дена-
турацию или распад молекул белка на субъединицы. АС лоша-
75
Рис. 20. Схема классифика-
ции методов иммунодиф-
фузии по Ухтерлони.
Темные области — растворы АГ;
заштрихованные области — АТ
или гель с антисывороткой; бе-
лые области — нейтральный
гель.
ди, как правило, дают
преципитацию в зоне
эквивалентности (обыч-
но довольно широкая),
они образуют резкие
тонкие полосы. Преципитаты, образуемые АС морских свинок
или АС «типа морских свинок» со специфическими АГ, образуют
более широкие зоны. При избытке АТ такого типа образуются
так называемые «флаги», «шлейфы» и др. Преимуществом сы-
вороток «типа морской свинки» является возможность исследо-
вать с их помощью обширную область концентраций, так как
даже при избытке антител возникают преципитирующие комп-
лексы.
2.1.1.4. КЛАССИФИКАЦИЯ МЕТОДОВ
ИММУНОДИФФУЗИИ
Различают простую и двойную иммунодиффузию. В первом
случае диффундирует один компонент,-во втором оба, в зависи-
мости от того происходит ли диффузия по одной общей оси или
во все стороны радиально, из резервуара в среду. ИД называет-
ся линейной или радиальной. Даже расположенные под углом
друг к другу резервуары представляют известные удобства для
определенных целей. Общая схема методов диффузии представ-
лена на рис. 20.
2.1.1.5. СИСТЕМЫ АНТИГЕН—АНТИТЕЛО
В зависимости от состава различают простые, сложные и мно-
жественные системы. В простых системах АГ, несущий детерми-
нанту «а», реагирует с АТ, несущим антидетерминанту «анти-
а». В результате взаимодействия АГ с АТ в геле появляется ли-
ния преципитации. Если молекула АГ содержит несколько де-
терминант различной структуры (а, Ь, с), к каждой из которых
в сыворотке имеются соответствующие АТ со специфичностью
А, В, С, то в этом случае образуется только одна линия пре-
ципитации, т. е. система является простой. К сложным систе-
мам относят такие, в которых ввиду структурного сходства
различных молекул антигенов наблюдаются перекрестные реак-
ции. О множественных системах говорят в тех случаях, когда
одновременно происходят реакции нескольких простых или
сложных систем. Эти данные приведены на рис. 21.
76
Рис. 21. Варианты систем АГ—АТ при постановке реакции иммунодиффу-
зии (объяснение в тексте).
2.1.1.6. СПЕЦИФИЧНОСТЬ, ЧУВСТВИТЕЛЬНОСТЬ,
ОШИБКИ МЕТОДА
Высокая специфичность представляет собой важнейшее пре-
имущество методов иммунологического анализа. Для количест-
венного или качественного определения изучаемого АГ в рас-
творе нескольких веществ необходимо иметь соответствующие
моноспецифические сыворотки. Моноспецифичность проверяют в
данном случае методом двойной радиальной ИД или методом
иммуноэлектрофореза, при необходимости нежелательные АТ
устраняются адсорбцией. Что касается чувствительности мето-
дов ИД, то, согласно мнению многих авторов, при концентра-
ции белка 50—5 мкг/мл в соответствующих системах АГ—АТ
еще можно наблюдать образование преципитатов. Если в лун-
ку вносят 2—10 мкл раствора, то удается определить присутст-
вие белка в растворе в количестве 10—500 нг. Разброс данных
при постановке серии количественных определений составляет
около 5% (3—7%).
2.1.2. ДВОЙНАЯ РАДИАЛЬНАЯ ИММУНОДИФФУЗИЯ
2.1.2.1. ПРИНЦИП МЕТОДА
В слое агара равномерной толщины на определенном расстоя-
нии друг от друга делают лунки для АГ или АС. После внесе-
ния растворов в лунку эти компоненты начинают диффундиро-
вать радиально в гель. Встречаясь друг с другом, АГ и АТ на-
77
чинают образовывать иммунные комплексы, которые преципи-
тируют в ячейках геля и становятся видимыми как линии пре-
ципитации. Двойную радиальную иммунодиффузию проводят
на предметных стеклах (микрометод).
2.1.2.2. МЕТОДИКА
2.1.2.2.1. Материалы и оборудование
Для работы необходимо иметь моно- и полиспецифические сы-
воротки, чистый агар (специальный агар Noble или очищенный
агар Difko, США), который можно заменить агарозой. В каче-
стве растворителей для агара используются самые разнообраз-
ные растворы, мы приводим состав трех из них:
— 0,15 М NaCl;
— забуференный раствор NaCl с pH 7,2, состоящий из 9 частей
0,15 М NaCl и 1 части 0,07 М фосфатного буфера по Серен-
сену. Состав фосфатного буфера: КН2РО4— 9,073 г/л;
Na2HPO4-2H2O—11,87 г/л; раствор КН2РО4 в количестве
29,6 мл доводят раствором Na2HPO4 до 100 мл. В результа-
те получается 0,07 М фосфатный буфер с pH 7,2;
— натрий-ацетат — натрий-диэтилбарбитуратный буфер с pH 8,6
и ионной силой 0,1 (веронал-ацетатный буфер по Backhausz)
Na-диэтилбарбитурат — 9,75 г; Na-ацетат — 6,47 г; 0,1 М
НС1 — 60 мл; дистиллированная вода до 1000 мл.
2.1.2.2.2. Приготовление геля и проведение иммунодиффузии
Расплавляют 1,5 г агара в 100 мл буфера (одного из трех пред-
ложенных) на водяной бане. Порциями по 3 мл горячий агаро-
вый золь наносят на строго горизонтально установленные пред-
метные стекла, для того чтобы получить равномерный слой ага-
ра толщиной приблизительно 1,5 мм. После застывания агара
пластинки готовы к употреблению. Рекомендуется сначала на-
носить на стекла по 1 мл горячего золя агарозы (10 г/л), кото-
рый затем высушивают при 70 °C. Образующаяся при высуши-
вании пленка агара усиливает сцепление геля с поверхностью'
стекла. В зависимости от характера иммунохимических иссле-
дований в агаре делают лунки для АГ или АТ при помощи го-
товых штампов или вырезают их стеклянной (можно металли-
ческой) трубочкой. Лунки должны отстоять друг от друга по
меньшей мере на 4 мм, расстояние более 10 мм употребляется
редко. На рис. 22 схематично представлены возможные вариан-
ты расположения лунок в слое геля. После внесения растворов
иммунодиффузию проводят во влажной камере при температу-
ре 4°, 20° или 37 °C. Продолжительность диффузии зависит от
целей исследования, она должна быть не менее 24 ч и в неко-
торых случаях может достигать 6—7 дней. Более продолжи-
тельная инкубация не приводит к изменениям преципитатов.
В случае продолжительной ИД пластины следует ежедневно-
78
Рис. 22. Двойная радиальная иммунодиффузия: возможные варианты рас-
положения резервуаров для растворов АГ (заштрихованы) и сыворотки
(черные).
контролировать. По окончании ИД можно либо непосредствен-
но оценивать результаты по влажному препарату, либо сначала
отмыть непреципитирующие субстанции. Для этого замачивают
пластины 2—3 раза на 12 ч в 0,15 М растворе NaCl. Затем на
поверхность геля кладут фильтровальную бумагу, смоченную
дистиллированной водой, и гель высушивают при 40—50 °C или
при комнатной температуре. Высушенные пластины геля окра-
шивают обычно амидо черным 10 В, после чего оценивают ре-
зультаты иммунодиффузии.
2.1.2.2.3. Оценки результатов
Количество линий преципитации. Во множествен-
ных системах всегда существует вероятность того, что два или
более комплекса АГ—АТ вследствие одинаковой скорости диф-
фузии будут образовываться в одном и том же участке геля,
формируя, следовательно, одну линию преципитации. Кроме
того, концентрация АТ и АГ или соотношение компонентов ре-
акции может быть настолько неблагоприятным, что видимые
преципитаты не будут образовываться. Поэтому число видимых
линий преципитации, как правило, меньше числа систем АГ—АТ
или в лучшем случае равно ему.
Расположение преципитатов между лунками.
При оптимальном соотношении между АГ и АТ после оконча-
ния диффузии линия преципитации располагается примерно на
середине расстояния между лунками. Если один из компонен-
тов реакции присутствует в большем количестве, чем другой, то
линия преципитации сдвигается в сторону лунки с меньшим со-
держанием реагента. Если соотношение концентраций реаген-
тов может измениться в месте первичного выпадения предме-
тов, например, вследствие избыточного поступления одного из
79
Рис. 23. Расположение линий преципитации при сравнительных исследова-
ниях методом двойной радиальной иммунодиффузии.
а —1—4 варианты преципитации; б —анализ органоспецифических антигенов. В цент-
ре— антисыворотка к экстракту почек (АСЭП): ЭП — экстракт почек; СЧ — сыворотка
человека. В сыворотке человека и в экстракте почек присутствует один общий АГ (иден-
тичная реакция). Второй антиген присутствует только в экстракте почек (перекрестная
реакция с первым АГ) (препарат проф. Frlemel).
компонентов, это приводит к смещению линии преципитации.
Часть иммунных агрегатов, особенно в зоне избытка антител,
может оставаться на старом месте в виде вуали и расширения
линии преципитации. Именно возможность возникновения таких
процессов обусловливает необходимость ежедневных проверок
пластин геля при длительно протекающей иммунодиффузии.
Взаимное расположение линий (сравнительный ана-
лиз). Для выявления полной или частичной идентичности обыч-
но располагают лунки в углах треугольника, в две помещают
растворы сравниваемых АГ, в третью — АС. Принципиально
возможны четыре варианта расположения линий преципитации:
1. Обе линии преципитации полностью сливаются. Это говорит
об идентичности обоих антигенов.
2. Линии преципитации пересекаются. Это значит, что реаги-
рующие с АС детерминанты АГ неидентичны и, следователь-
но, сами антигены различны.
3. Одна линия длиннее и продолжается за другую в виде так
называемой „шпоры”. „Шпора” часто бывает тоньше основ-
ной линии преципитации. Линия преципитации от второй
лунки с АГ сливается с первой линией. Это означает, что оба
антигена обладают некоторыми общими детерминантами
и образуют с соответствующими АТ иммунные комплексы,
приводящие к слиянию линий. Кроме того, один из АГ имеет
больше детерминант, чем другой, что при наличии соответ-
ствующих АТ в антисыворотке приводит к образованию,
„шпоры”.
4. Обе линии преципитации перекрещиваются и сливаются од-
новременно. Это означает, что оба антигена содержат как
80
Рис. 24. Двойная радиальная иммуно-
диффузия. Титрование раствора АГ
(титр 1 : 16).
одинаковые, так и различные де-
терминанты, которые вступают в
реакцию с антителами полиспе-
цифической антисыворотки.
Основные типы преципита-
ции представлены на рис. 23.
2.1.2.2.4. Модификация метода
Вышеописанная методика имму-
нодиффузии на предметных
Исходный
1.32 раствор АГ
стеклах представляет собой микровариант метода, предложен-
ного Ухтерлони в 1949 г. В условиях макрометода слой геля,
создаваемый в чашках Петри, составляет 2—3 мм, лунки и со-
ответственно объемы реагентов довольно велики, лунки отодви-
нуты друг от друга на значительные расстояния, линии преци-
питации длинные и хорошо выраженные.
Титрование. Для титрования растворов АГ или АС рас-
полагают лунки так, как показано на рис. 24. Одинаковые объ-
емы растворов антигена, приготовленных двукратным разведе-
нием, вносят в периферические лунки. Такой же объем антисы-
воротки вносят в центральную лунку. При оценке иммунодиф-
фузии учитывают:
1. Расстояние от линии преципитации до центральной и перифе-
рической лунок.
2. Наибольшее разведение антигена, при котором еще заметно
образование преципитата.
3. Интенсивность и ширину полос преципитации.
Наибольшее разведение, при котором еще образуются види-
мые преципитаты, дает значение титра. Этим способом можно
также определять количество преципитирующих антител в раз-
личных АС, которые, например, можно сравнивать в реакции
иммунодиффузии со стандартным раствором АГ.
Полуколичественное определение антигенов.
Титрование часто используют для полуколичественного опреде-
ления АГ [2]. Вокруг центральной лунки с антисывороткой
формируют четыре лунки для антигена. Чем выше концентра-
ция АГ, тем ближе к центральной лунке будут расположены
полосы преципитации. Путем сравнения со стандартным раство-
ром АГ можно определить неизвестную концентрацию антигена
в исследуемой пробе.
2.1.2.3. ОЦЕНКА МЕТОДА
Двойная радиальная иммунодиффузия представляет собой
прежде всего метод количественного анализа. Ее применяют
для определения количества АГ в жидкостях (сыворотка крови,
6 -990 81
цереброспинальная жидкость, экстракты тканей). Ее также при-
меняют для проверки чистоты препаратов, при получении анти-
сывороток животных и оценке эффективности иммунизации.
Титрование и полуколичествепное определение АГ расширяет
возможности применения метода. Определение отдельных анти-
генов в многокомпонентных растворах требует применения мо-
носпецифических антисывороток. Метод двойной радиальной им-
мунодиффузии можно использовать также для анализа трудно-
растворимых субстанций, таких, как кератин и прокератин,
с условием предварительной их солюбилизации ДСН, гуанидин-
хлоридом (до 6 М), мочевиной (до 8 М). После солюбилизации
эти вещества продолжают реагировать с соответствующими АТ
в геле агарозы.
2.1.3. ПРОСТАЯ РАДИАЛЬНАЯ ИММУНОДИФФУЗИЯ
2.1.3.1. ПРИНЦИП МЕТОДА
На плоской поверхности создают равномерный слой геля, со-
держащего антисыворотку. Из лунок, проштампованных в геле
и заполненных раствором антигена, молекулы АГ диффундиру-
ют в гель и образуют кольца преципитации с соответствующи-
ми АТ. Диаметр колец преципитации увеличивается до тех пор,
пока не истощится раствор антигена. Этот метод впервые опи-
сал Манчини (Mancini) в 1963 г. Известны многочисленные мо-
дификации этого метода, мы приводим описание, основанное
на методике, изложенной в монографии Мауег и Walker (см.
список литературы).
2.1.3.2. МЕТОДИКА
2.1.3.2.1. Материалы и оборудование
Для работы необходимы моноспецифические АС, стандартные
препараты, контрольные сыворотки, буферные растворы, агар,
рамки для закрепления предметных стекол, штативы для этих
рамок, измерительное устройство с точностью до 0,1 мм, микро-
шприцы для внесения объемов 1—10 мкл.
2.1.3.2.2. Последовательность этапов исследования
Растворяют 1,5 г агара в 100 мл буфера (см. раздел 2.1.2) на
водяной бане, охлаждают до 48 °C и смешивают с АС, жела-
тельно подогретой до той же температуры, в небольшой про-
порции. Наносят пипеткой 2 мл смеси на предметное стекло,
установленное строго горизонтально на подставке, нагретой до
35—40 °с. Золь агара, содержащий АТ, распределяется по по-
верхности предметного стекла и застывает слоем толщиной
приблизительно 1 мм. Рекомендуют также наносить на пред-
метные стекла по 1 мл горячего раствора агара (10 г/л) и вы-
82
Рис. 25. Простая радиаль-
ная иммунодиффузия. Эта-
пы работы.
суживать их при 70 °C
перед использованием.
Если пластины для
простой радиальной
иммунодиффузии будут
храниться перед упот-
реблением в течение
2—3 нед (во влажной
камере при 4°C), то к
гелю добавляют кон-
серванты (мертиолят,
0,1 г/л, азид натрия,
0,2 г/л). На предмет-
ном стекле в слое ге-
ля вырезают по 8 лу-
нок при помощи от-
шлифованной стеклян-
ной трубки, соединен-
ной с водоструйным на-
сосом (таким образом
создают пониженное давление). В каждую из лунок вносят по-
2 мкл раствора антигена. Реакцию простой радиальной ИД
проводят во влажной камере. Если линии преципитации доста-
точно выражены, оценку результатов можно проводить прямо
на влажной пластинке. В тех случаях, когда оценку проводят
немедленно, пластинки с гелем отмывают 2—3 раза в 0,15 М
NaCl; каждая отмывка длится 12 ч. Этим достигается удаление
непреципитировавших белков. После отмывки препараты высу-
шивают и окрашивают, например, амидо черным 10 В. После-
довательность операций представлена на рис. 25.
2.1.3.2.3. Калибровочная кривая
К моменту завершения диффузии наблюдается прямая линей-
ная зависимость между исходной концентрацией АГ и площадью
преципитата. Это можно видеть на примере графика определе-
ния IgA (рис. 26). Для построения графика берут логарифм
концентрации АГ и как функцию от него площадь преципитата,
квадрат диаметра зоны преципитации или логарифм диаметра
преципитата. Во всех случаях зависимость носит линейный ха-
рактер и описывается уравнением:
5 = К-<Здг +$о,
где S — общая площадь зоны преципитации, включая So, So — площадь стар-
товой лунки, Qa — площадь антигена.
6*
83
ротки.
Рис. 26. Простая радиаль-
ная иммунодиффузия. Кри-
вая для определения IgA.
D2 — квадрат диаметра диф-
фузии.
Наклон прямой К нахо-
дится в обратной зависи-
мости от концентрации ан-
тител в геле:
1
К г=п-~^— + т,
САТ
Cat — концентрация АТ в
геле, т — точка пересече-
ния прямой с ординатой.
Снижая уровень АС
в геле, можно после-
довательно увеличи-
вать диаметр зоны пре-
ципитации. При этом
К возрастает и увели-
чивается экономичность
и чувствительность ме-
тода. Снижение кон-
центрации реагентов
возможно только до определенных пределов, так как может воз-
никнуть ослабление преципитации. В каждом отдельном случае
концентрацию АС в геле подбирают в зависимости от титра ан-
тител. Обычно рекомендуют использовать 1—5% антисыво-
2.1.3.2.4. Температура и продолжительность иммунодиффузии
Температура существенно не влияет на результаты диффузии.
Опыт можно ставить при 4 °C, 20 °C или 37 °C. Обычно простую
радиальную ИД проводят в холодильнике при 4—6 °C с целью
предупреждения роста грибов и бактерий. Следует избегать
резких колебаний температуры (размах не более +5°C). Про-
цесс иммунодиффузии при работе, например, с альбумином
и ферритином, завершается через 48 ч; с IgG и IgA — через 2—
3 дня; с IgM, а2-макроглобулином и другими белками с круп-
ными молекулами — через 5—7 дней. Первый ориентировочный
замер диаметра преципитата можно получить уже через 4 ч.
Существует модификация простой радиальной ИД с постоян-
ным временем диффузии, равным 24 ч [3]. Это означает, что
диффузия АГ по крайней мере при его высокой концентрации
полностью не заканчивается. Получаемые результаты вполне
сопоставимы с результатами простой линейной ИД (см. раз-
дел 2.1.1). Соответственно наблюдается линейная зависимость
между логарифмом концентрации АГ и радиусом преципита-
84
ции. Раньше считалось, что эта модификация уступает по чув-
ствительности методу, предложенному Манчини. Недавние ис-
следования указывают, что оба метода имеют одинаковую чув-
ствительность [5].
2.1.З.2.5. Подготовка пластин для иммунодиффузии
Во избежание погрешностей следует обращать особое внимание
на поддержание температуры расплавленного агара при внесе-
нии антисыворотки (48 °C, максимально 50 °C), на правильный
расчет концентрации АС в геле, на количество агара, наносимое
на каждую пластину, и на строго горизонтальное расположение
пластины. Поскольку толщина слоя геля уменьшается по на-
правлению к периферии пластины, то следует добиваться того,
чтобы кольца преципитации располагались не ближе 5 мм от
края.
2.1.3.2.6. Значение свойств антигена
Как правило, иммунохимические и физико-химические свойства
стандартного и исследуемого антигенов должны быть сходны.
Иногда это требование трудновыполнимо. Например, диффу-
зионные свойства могут изменяться, когда молекулы антигена
обладают склонностью к агрегации или, наоборот, к диссоциа-
ции. Это приводит к изменению условий диффузии, возможно
даже к маскировке одних детерминант антигенов и демаскиров-
ке других. Тенденцию к агрегации можно наблюдать, в частно-
сти, в очищенных препаратах сывороточного альбумина после их
повторного растворения. Если такой раствор предназначен для
использования в качестве стандартного при калибровке, то аг-
регаты удаляют гель-фильтрацией на сефадексе G-200. IgA со-
держится в сыворотке в виде молекул с различной степенью
полимеризации. Обычно соотношение моно-, ди- и тримеров не-
известно. Приходится в данном случае ориентироваться на ус-
редненные результаты анализов большого числа сывороток нор-
мальных доноров или на существующие стандарты ВОЗ. В не-
которых случаях завышенные результаты получаются вследст-
вие преобладания быстродиффундирующих фрагментов (субъ-
единиц) [7]. Это наблюдение справедливо для IgM, при работе
с которым наблюдаются по меньшей мере две линии преципи-
тации. Длительное хранение сывороток при 4 °C может приво-
дить к частичному распаду белков вследствие остаточной про-
теолитической активности.
2.1.З.2.7. Модификации метода
По различным причинам в стандартную методику приходится
вводить ряд изменений. Для повышения чувствительности ме-
тода используют более толстые слои геля и более вместитель-
ные лунки для АГ (до 20 мкл). Слабые преципитаты становятся
лучше различимыми после обработки танином. Танин использу-
ют в концентрациях от 0,2 до 4% при pH 1,5. Усиление действия
85
танина наблюдается при обработке прокрашенных препаратов
раствором дигидроксифениланилина. Использование подобных
приемов позволяет определять даже IgE у верхней границы
нормальной концентрации [10]. Усиление преципитации наблю-
дается при добавлении к смеси агароза — АС карбовакса 20 М
в количестве до 20 г/л [1]. Внесение неионных полимеров (по-
лиэтиленгликоль, карбовакс, оксидвакс) в лунки для АГ или
обработка пластин для ИД антииммуноглобулиновой сыворот-
кой также усиливает преципитацию [10]. Некоторые изменения
при постановке простой радиальной ИД связаны с экономией
антисыворотки.
Определенное небольшое количество АС до или после диф-
фузии АГ наносится на область предполагаемой преципитации
на поверхности геля. АГ, диффундирующие радиально, и АТ,
диффундирующие соответственно вертикально, встречаются
в толще геля и образуют преципитаты. При этом следует избе-
гать подогревания геля. Простая радиальная ИД пригодна
в равной мере для количественного и для качественного анали-
за [6] иммунохимически близких белков. Если количество
в лунках постоянно, можно судить о титре АТ в антисыворотках
различных животных по величине колец преципитации при точ-
но известном содержании АС в геле. Для определения титра АТ
смешивают гель в установленной пропорции с АГ, антитела
диффундируют в гель (так называемая обратная радиальная
ИД). Радиус колец преципитации пропорционален титру. Вме-
сто агара или агарозы в реакции простой радиальной ИД мож-
но использовать пленку ацетата целлюлозы [9]. Разработан ин-
тересный вариант данного метода, позволяющий количественно
оценить степень структурного сходства родственных антиге-
нов [4].
2.1.З.З. ОЦЕНКА МЕТОДА
В пределах своей чувствительности простая радиальная ИД
пригодна для определения любых АГ при условии, что имеются
соответствующая моноспецифическая АС и чистые или стан-
дартные АГ для калибровки системы.
Обычно ПРИД используют для определения белков в био-
логических жидкостях, таких как сыворотка крови, цереброспи-
нальная жидкость, секреты желез или экстракты различных ор-
ганов и т. д. (рис. 27). Этот метод прочно удерживается в ла-
бораториях. Многие белки можно определять, используя ком-
мерческие наборы стандартов или даже готовые иммунодиффу-
зионные пластинки. Специфичность метода описывается в раз-
деле 2.1.1. Ошибка метода при работе с одной серией реагентов
составляет 3—7% (литературные и собственные данные). Конт-
рольные измерения изо дня в день в течение длительного вре-
мени дают коэффициент вариации 8—10% для оценки ИГ. Из-
вестно, что в норме содержание многих сывороточных белков
86
б
Рис. 27. Простая радиальная иммунодиффузия.
а — определение IgG на пластинах 100X70 мм (препарат д-ра Linneke, Внсмар); б —
определение IgM на предметных стеклах. Два противоположных резервуара на узких
сторонах стекла служат для внесения контрольной пробы.
сильно колеблется, например IgG от 8 до 18 г/л. С учетом этого
средняя ошибка определения нижних границ концентрации со-
ставляет для сывороточного IgA— 18,3%, для IgG— 17,4%, для
IgM—15,7%, для гаптоглобина—13,6%, для трансферрина —
9,8%. Соответствующие значения для верхней границы состав-
ляют 8,6%, 6,5%, 7,3%, 7,1% и 5,2% (60—70 измерений после-
довательно в течение 26 дней). Необходимо помнить, что кон-
центрации ИГ, определяемые методом простой радиальной ИД
у здоровых лиц, не укладываются в нормальную кривую рас-
пределения результатов; об этом следует помнить при статисти-
ческой обработке данных[11]. Относительно простая постановка
опыта, несложное оборудование, надежность метода способст-
вуют широкому использованию простой, радиальной ИД. К чис-
87
лу недостатков метода следует отнести большую длительность
реакции по сравнению с другими методами, в основе которых
лежит образование иммунных преципитатов. С точки зрения
клиницистов, например специалистов в области клинической им-
мунологии, этот недостаток не столь существен, поскольку си-
туации, когда необходимо знать концентрацию ИГ в день взя-
тия пробы, встречаются редко.
2.1.4. СПИСОК ЛИТЕРАТУРЫ
Обзоры и монографии
1. Mayer R. J., Walker J. Н. Immunochemical methods in the biological sciences:
enzymes and proteins. — London, 1980. Academic Press, 1980.
2. Stites D. P. Clinical laboratory methods for detection of antigens and anti-
bodies.— In: Basic and clinical immunology. Lange Medical Publications./
Eds, H. H. Fudenberg, D. P. Stites et al. — Los Angeles, 1980.
Цитированная литература
1. Bohout В. A., van Asten Noordijk J. J. L. Divergent results in radial immu-
nodiffusion. III. Isolation and specificity test of class specific antibodies. —
J. Immunol. Methods, 1979, 27, 101—109.
2. Darcy D. A. Antigen and antibody concentration in relative units on diffu-
sion plates. — In: Methods in Immunology and Immunochemistry./Eds.
C. A. Williams and M. W. Chase. — New York: Academic Press, 1971, 200—
209.
3. Fahey J. L., McKelvey E. M. Quantitative determination of serum immuno-
globulins in antibody-agar plates. — J. Immunol., 1965, 94, 84—90.
4. Gaffney P. J., Mahmoud M„ Kirkwood T. B. L. Quantitative immunorecogni-
tion by a radial diffusion technique. — J. Immunol. Meth., 1977, 14, 25—29.
5. Lamerz R., Fateh-Moghadam A., Knedel M. Zur quantitativen iminunologi-
shen Bestimmung von Serumproteinen. — Ztschr. klin. Chem. klin. Biochem.,
1973, 11, 491—500.
6. Mancini G., Nash D. R„ Heremans J. F. Further studies on single radial
immunodiffusion. 111. Quantitative analysis of related and unrelated anti-
gens.— Immunochemistry, 1970, 7, 261—264.
7. Mulder J., Sloots L. С. E., Verhaar M. A. T. Molecular heterogeneity effects
of IgM immunoglobulins in radial immunodiffusion. — J. Immunol. Meth.,
1972, 2, 89—98.
8. Oberdorfer A., Blaufuss H. Zur bestimmung prazipitierender Antikorper mit
der umgekehrten einfachen radialen Immunodiffusion. — Ztschr. klin. Chem.
klin. Biochem., 1972, 10, 385—390.
9. Pizzolato M. C., Pizzolato M. A., Agostini A. Single radial immunodiffusion
of undiluted serum proteins. — Clin. Chem., 1972, 18, 237—238.
10. Schwartze G., Thiman И. H„ Fiedler H. Zur quantitativen Bestimmung des
Immunoglobulins E in Serum mit der einfachen radialen Immunodiffusion.—
Dtsch. Ges.-Wesen, 1977, 32, 299—303.
11. Stein S. F., Ware J. H., Wood R. Serum immunoglobulins. Methods for the
determination of normal values in international units. — J. Immunol. Meth.,
1977, 16, 371—384.
88
2.2. ИММУНОЭЛЕКТРОФОРЕЗ (МИКРОМЕТОД)
2.2.1. ПРИНЦИП МЕТОДА
Иммуноэлектрофоретический анализ представляет собой соче-
тание электрофореза в агаровом геле с иммунодиффузией. Им-
муноэлектрофорез (ИЭФ) впервые описали Грабар и Уильямс
в 1953 г. В 1955 г. Шейдеггер предложил микромодификацию
этого метода, применив обычные предметные стекла. Принцип
ИЭФ состоит в следующем. Вначале проводят электрофоретиче-
ское разделение белков в забуференном геле агара; после раз-
деления в канавку, которая идет в направлении миграции бел-
ков, вносят преципитирующую иммунную сыворотку. АГ и АС
диффундируют в геле навстречу друг другу, и в месте их взаи-
модействия возникают дугообразные линии преципитации, чис-
ло, положение и форма которых дают представление о составе
исходной смеси антигенов (рис. 28).
2.2.2. МЕТОДИКА
2.2.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы нужно иметь предметные стекла, пробирки с проб-
ками, пипетки (10 мл), очищенный агар или агароза, консер-
вант (например, мертиолят), препаровальную иглу или неболь-
шой шприц с иглой; микропипетку со шлангом и мундштуком
или микрошприц (50—100 мкл), фильтровальную бумагу, на-
пример, FN 14; NaCl ч. д. а, ледяную уксусную кислоту, амидо
черный 10 В, веронал-натрий, NaCH3COO-3 Н2О, 0,1 н. НС1;
комплект приборов для электрофореза, в который входят ис-
точник тока, электрофоретическая камера, штампы, водяная
баня, стеклорез, уровень, ванночки для окрашивания предмет-
ных стекол.
Рис. 28. Принцип иммуноэлектрофореза.
89
2.2.2.2. ПРИГОТОВЛЕНИЕ АГАРОВОГО ГЕЛЯ
Суспендируют 1,5 г очищенного агара в 100 мл веронал-ацетат-
ного буфера (pH 8,6, ионная сила 0,1), добавляют мертиолятдо
конечной концентрации 0,1% и нагревают суспензию на водяной
бане до полного расплавления агара (раствор становится про-
зрачным).
Состав буфера: веронал-натрий —29,43 г
ацетат натрия (ЗН2О) — 19,42 г
0,1 М НС1 180,0 мл
дистиллированная вода до 3000,0 мл
Горячий прозрачный раствор агара разливают по пробиркам
(заполнять только до половины), закрывают пробками, дают
остыть при комнатной температуре и после охлаждения хранят
в холодильнике при 4 °C.
2.2.2.3. ПРИГОТОВЛЕНИЕ АГАРОВЫХ ПЛАСТИНОК
Тщательно очищенные и обезжиренные предметные стекла над-
писывают стеклорезом и помещают на горизонтальную плос-
кость (рис. 29). Несколько пробирок с 1,5% гелем агара нагре-
вают на водяной бане. При помощи предварительно нагретой
пипетки (10 мл) на каждое предметное стекло наносят по 2,5 мл
горячего золя агара. Раствор агара должен равномерно распре-
делиться по обезжиренной поверхности стекла и после затверде-
ния образовать однородный слой геля. -
22.2.4. ФОРМИРОВАНИЕ ЛУНОК
И КАНАВОК В ГЕЛЕ
При помощи штампов в слое геля вырезают лунки для антиге-
нов и канавки для антисывороток. Выбор подходящего штампа
определяется задачей исследования. Очень удобны штампы с из-
меняемым профилем (см. рис. 29). Вырезанные фрагменты геля
удаляют иглой или при помощи водоструйного насоса. В обра-
зовавшиеся лунки вносят примерно по 2 мкл исследуемого ма-
териала микрошприцем. Полоски геля, образовавшиеся после
вырезания канавок, также следует удалить до электрофореза,
так как после это будет трудно сделать вследствие нагревания
геля. Вырезанные фрагменты геля удаляются гораздо легче,
если пластинки на 1—2 мин поместить в морозильник.
2.2.2.5. ПРОВЕДЕНИЕ ЭЛЕКТРОФОРЕЗА
Электрофоретическую камеру присоединяют к источнику посто-
янного тока, выдерживающего нагрузку до 100 мА (рис. 30).
Электродные отсеки заполняют веронал-ацетатным буфером
с pH 8,6 и устанавливают камеру строго горизонтально по
уровню.
90
Рис. 29. Юстировочный столик, штатив для пластин и гелевый штамп.
Рис. 30. Камера для проведения иммуноэлектрофореза с источником пи-
тания.
Рис. 31. Сосуды для диффузии и элюции. Справа штатив для пластин.
2.2.2.6. ВНЕСЕНИЕ АНТИСЫВОРОТКИ
После электрофоретического разделения белков в продольную
канавку вносят 0,03—0,05 мл преципитирующей иммунной сы-
воротки. Сыворотка не должна переливаться через край канав-
ки, иначе расположение линий преципитации будет искажено.
2.2.2.7. ИММУНОДИФФУЗИЯ, ЭЛЮИРОВАНИЕ,
ОКРАШИВАНИЕ
Иммунодиффузия продолжается не менее 24 ч во влажной ка-
мере (рис. 31). Затем сразу же начинают элюировать белки,
которые не участвовали в преципитации, с помощью 0,15 М
NaCl. Элюирование проводят 3 сут с ежедневной сменой рас-
твора. После этого пластины геля накрывают фильтровальной
бумагой (не оставляя пузырьков воздуха) и высушивают при
комнатной температуре или в сушильном шкафу при температу-
ре не выше 40 °C. Для окрашивания белков чаще всего приме-
няют амидо черный 10 В, лиссаминовый (светлый) зеленый,
азокармин В или пунцовый 2R. Липопротеиды окрашивают су-
даном черным В. При этом важно окрашивать полностью высу-
шенный препарат, так как судан черный В нерастворим в воде
и не воспринимается влажным гелем или образует неспецифи-
ческие осадки. Об окрашивании ферментов см. раздел 2.5. Им-
мунофореграмма белков сыворотки человека, проявленная по-
лиспецифической антисывороткой кролика, представлена на
рис. 32.
2.2.2.8. МОДИФИКАЦИЯ МЕТОДА
Использование чистых АГ. Иногда одного окрашива-
ния недостаточно для идентификации неизвестного антигена.
В этом случае полезно сравнивать исследуемый АГ с известны-
ми чистыми АГ. Реагенты располагают так, как показано на
рис. 33. Различие в электрофоретической подвижности являет-
ся доказательством неидентичности антигенов.
Рис. 32. Иммуноэлектрофореграмма сыворотки человека, проявленная поли-
специфической кроличьей антисывороткой.
92
Рис. 33. Идентификация
антигенов при помощи чис-
того АГ: сравнение элект-
рофоретической подвиж-
ности.
Рис. 34. Идентификация
антигена при помощи мо-
носпецифической антисы-
воротки.
Рис. 35. Идентификация
антигенов по методу Ос-
сермана.
Рис. 36. Идентификация
антигенов по методу Гере-
манса.
Полиспецифическая иммунная сыворотка
Известный АГ
Применение моноспецифичес к их иммунных
сывороток. Если исследователь заранее предполагает, что
в изучаемом образце присутствует какой-либо определенный
антиген, он может попытаться идентифицировать его при помо-
щи моноспецифической сыворотки (рис. 34). Линия преципита-
ции, образованная на одной стороне иммуноэлектрофореграммы
полиспецифической сывороткой, может слиться с другой лини-
ей, которую на другой стороне образует моноспецифическая
сыворотка. Такое слияние свидетельствует об идентичности дан-
ного компонента антигенной смеси тому АГ, к которому была
получена моноспецифическая сыворотка. Однако этот простой
93
и изящный метод идентификации АГ часто бывает неприменим
из-за отсутствия соответствующих моноспецифических сыво-
роток.
Метод Оссермана. Для идентификации по Оссерману
требуется контрольный раствор, содержащий только известные
антигены в возможно меньшем числе (рис. 35). Еще лучше ис-
пользовать чистые антигены. Контрольный раствор АГ вносят
в одну из канавок, и он, диффундируя в сторону канавки с АС,
образует линию преципитации, параллельную канавкам. В том
месте, где контрольный АГ встречается с идентичным компо-
нентом исследуемой смеси антигенов, линия преципитации ис-
кривляется и переходит в дугу идентичного антигена. Четкая
картина получается лишь тогда, когда контрольный раствор со-
держит минимальное число известных АГ. В противном случае
на иммуноэлектрофореграмме возникает множество беспорядоч-
но расположенных полос и дуг, интерпретировать которые
трудно или даже невозможно.
Метод Гереманса. В основе метода Гереманса лежит
реакция двух различных АГ с антисывороткой, содержащей АТ
к ним обоим. Исследуемый АГ сравнивают с контрольным
по электрофоретической подвижности и одновременно проводят
реакцию на иммунологическую идентичность. С этой целью
в канавке для АС оставляют агаровую перемычку, положение
которой определяется в предварительном опыте. Поэтому ме-
тод Германса называют также «методом разделенной канавки»
(рис. 36). Перемычка должна находиться в том месте, где ожи-
дают слияния полос преципитации исследуемого и контрольно-
го АГ (в случае их идентичности). Если по обе стороны пере-
мычки будут находиться идентичные или перекрестно реагирую-
щие АГ, произойдет полное слияние дуг преципитации или (при
неполной идентичности) образование шпоры.
2.2.3. ОЦЕНКА МЕТОДА
Иммуноэлектрофорез — один из широко распространенных ме-
тодов качественного анализа антигенов. Располагая соответст-
вующими преципитирующими АС, с его помощью можно иссле-
довать любую антигенную смесь. В частности, успешно анализи-
руются белки сыворотки крови, цереброспинальной жидкости,
мочи, молока, экстрактов из органов, а также белки раститель-
ного и бактериального происхождения. В клинике этот метод
чаще всего используется при диагностике парапротеинемий и
иммунодефицитных состояний. В судебной медицине с его по-
мощью анализируют системы гаптоглобина и группоспецифиче-
ского компонента Gc. В научно-исследовательской работе им-
муноэлектрофорез служит основным методом идентификации
белков, содержащихся в сложных смесях. Он незаменим как ме-
тод последовательного наблюдения за процессом очистки бел-
94
Рис. 37. Устройство для проведения электрофореза в геле агара.
ковых препаратов. Его часто используют также для контроля
подлинности и чистоты этих препаратов. Естественно, что ме-
тод, имеющий столь широкое применение, должен был видоиз-
меняться и совершенствоваться в различных направлениях.
Были предложены новые носители: гель агарозы, ПААГ, аце-
тат-целлюлозные пленки. Вместе с тем ИЭФ стали комбиниро-
вать с различными методами специфического окрашивания
и флюорохромироваиия, чтобы выявлять ферменты, углеводы,
липопротеиды, нуклеопротеиды. В результате комбинации с дру-
гими методами анализа возникли диск-иммуноэлектрофорез,
иммуноэлектрофокусирование, радиоиммуноэлектрофорез и др.
Для количественного определения с помощью полиспецифиче-
ских АС недавно был предложен двухмерный ИЭФ. Особая мо-
дификация двухмерного ИЭФ может повысить его чувствитель-
ность до уровня РИА. Это достигается электрофоретической
элюцией антигена из малых стеклянных резервуаров объемом
0,1—10 мл [3]. Для ИЭФ с охлаждением Виме сконструировал
прибор, автоматически поддерживающий постоянную темпера-
туру и силу тока в процессе разделения (рис. 37). Этот прибор
очень удобен для определения электрофоретической подвижно-
сти различных веществ. Использование в этом приборе термо-
электрического охлаждения (батарея Пельтье) следует особен-
но рекомендовать в случае работы с ферментами и другими
термолабильными веществами [2]. Недавно был предложен про-
стои способ разделения белков в двухмерном электрофорезе
[1]. Известен чувствительный способ определения непреципи-
тирующих систем АГ—АТ, основанный на применении ЛАКАТ,
так называемый каскадный ИЭФ, включающий фиксацию АГ
после электрофореза, присоединение к АГ антител, удаление
связанных АТ и их количественная оценка [5]. В случае имму-
нофиксационного электрофореза разделяются АТ, а не антиген.
Это быстрый и экономичный метод, он особенно пригоден для
Массового обследования с целью выявления парапротеинемии,
95
и для получения препаратов белков [4]. При ИЭФ нередко наб-
людаются нарушения нормального течения процесса, и не всег-
да удается сразу обнаружить их причину. Если, например, ис-
пользуется недостаточно очищенный агар, но это чаще всего
приводит к трем нарушениям: плохому образованию геля, силь-
ному электросмосу, плохому обесцвечиванию препаратов. Недо-
статочно чистый агар всегда подлежит очистке. Приступая
к работе с новой порцией агара, нужно сначала испытать его
годность. Часто обнаруживается, что одни и те же АГ имеют
разную электрофоретическую подвижность и по-разному окра-
шиваются при употреблении различных партий агара. Причи-
ной нарушений при ИЭФ может быть гидролиз агара, вследст-
вие многократного или слишком длительного нагревания.
О гидролизе свидетельствует ослабление гелеобразующих
свойств и возникновение неровностей на поверхности геля. Элек-
трофоретическое разделение может быть нарушено при исполь-
зовании неподходящего буферного раствора. Большинство буфе-
ров служит хорошей питательной средой для бактерий и плес-
невых грибков. Поэтому запас буфера следует хранить в холо-
дильнике или добавлять к нему консервант. Буфер, заполняю-
щий электрофоретическую камеру, следует менять не реже од-
ного раза в неделю. При многократном использовании электрод-
ного буфера необходимо после каждого разделения менять по-
люса электродов. Величина напряжения на клеммах камеры
обычно не позволяет судить о физической силе тока, проходя-
щего через гель, поэтому в цепь нужно последовательно вклю-
чить миллиамперметр. В процессе разделения сила тока почти
всегда повышается. Это явление обусловлено нагреванием геля,
которое нарастает с увеличением продолжительности электро-
фореза. Однако после первоначального подъема сила тока
в цепи может упасть. Часто это происходит из-за подсыхания
полосок фильтровальной бумаги, соединяющих гель с буфером,
или даже из-за подсыхания самого геля. В этом случае следует
уменьшить ионную силу буфера или напряжение на клеммах
камеры. Кроме того, высыхание можно предотвратить, если пе-
ред электрофорезом обернуть пластинку геля и полоски бумаги
полимерной пленкой. Удобнее всего работать с источником, ко-
торый обеспечивает постоянную силу тока (стабилизациятока).
•Самой собой разумеется, что результаты ИЭФ зависят от ка-
чества антигенов и антисывороток.
2.2.4. СПИСОК ЛИТЕРАТУРЫ
Обзоры и монографии
1. Arquembourg Р. С. Immunoelectrophoresis. Theory, methods, identification,
interpretation. — Basel, Karger, 1975.
.2. Axelsen N. H. (Ed) Quantitative immunoelectrophoresis: New developments
and applications. — Scand. J. Immunol., Suppl. No. 2, Oslo, 1975.
96
Цитированная литература
1. Crowle A. J. Templates for antiserum application in Immunoelectrophoresis
and two-dimensional electroimmunophoresis. — J. Immunol. Methods, 1977,
14, 197—200.
2. Kleist КЦ Frietnel H., Brock J. et al. Thermoelectishe kuhleinrichtung fur die
Gel- und Immunoelektrophorese. — Ztschr. med. Labortechnik, 1970, 11, 351—
355.
3. Kroll J. Immunoelectrophoretic quantitation of trace proteins. — J. Immunol.
Meth, 1976, 13, 333—339.
4. Otto S. Reversed immunofixation agar gel electrophoresis. Immunol. Letters,
1982, 4, 85—86.
5. Shainhoff J. R., Dardic B. D. Cascade immunoelectrophoresis: Combined
electrophoretic and solid-phase processing of immunoreactive protein by zonal
immobilisation. — J. Immunol. Methods, 1981, 42, 229—241.
2.3. ЭЛЕКТРОИММУННЫЙ АНАЛИЗ
2.3.1. ПРИНЦИП МЕТОДА
Гель агарозы, содержащий АС, распределяют равномерным
слоем по плоской поверхности. Из лунок, вырезанных в геле,
под воздействием постоянного электрического тока АГ мигриру-
ют в слой агарозы, где они взаимодействуют с 1g. В результа-
те этого взаимодействия образуются преципитаты АГ—АТ
в краевой зоне «молекулярного облака». Процесс продолжается
до тех пор, пока все молекулы АГ не окажутся включенными
в состав преципитатов, приобретающих вытянутую, заостренную
форму. Длина зоны преципитации при прочих равных условиях
находится в прямой зависимости от концентрации АГ (под «ус-
ловиями» следует понимать концентрацию геля, концентрацию
АС в геле, толщину его слоя и режим электрофореза). Предпо-
сылкой метода служит неподвижность (довольно условная)
Ig в геле. Она достигается применением агарозы с низкими зна-
чениями электроэндоосмоса буфера с pH 8,6. При таких усло-
виях возникает очень незначительный катодный ток воды, анод-
ное движение АТ практически не проявляется [6]. Метод впер-
вые был предложен Дореллом в 1966 г. В 1967 г. Меррил пред-
ложил способ количественного определения Ig в АС-содержа-
щем геле, названный им «электроиммунной диффузией». Это на-
звание утвердилось, хотя оно не вполне правильно. Явление им-
мунодиффузии следует как раз избегать при определении АГ
этим методом. Термин иммуноэлектрофорез соответствует су-
ществу метода Дорелла не вполне точно, так как речь идет
в действительности об «электрофоретической» иммунодиф-
фузии. Поэтому можно использовать термин «электроиммунный
анализ».
В лабораторной практике часто употребляют термин «ра-
кетный иммуноэлектрофорез».
7—990 97
2.3.2 МЕТОДИКА
2.З.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы моноспецифические, стандартные и
контрольные сыворотки, буферные растворы. Можно использо-
вать самые разнообразные буферы. В качестве примера мы при-
водим состав трех из них: натрий ацетат-натрий-диэтилбарбиту-
ратный буфер pH 8,6 (см. 2.1.2.2.1).
Барбитуратный буфер pH 8,6, 0,075 М [9]:
Барбитурат натрия 77,0 г
Барбитуровая кислота 13,8 г
Лактат кальция 3,85 г
Азид натрия 1,0 г
Дистиллированная вода до 5000 мл
Перед употреблением буфер можно разводить в 2 раза.
Вероналовый буфер pH 8,6, 0,075 М [11]:
Диэтилбарбитурат натрия 131,4 г
Диэтилбарбитуровая кислота 20,7 г
Лактат кальция 4,0 г
Дистиллированная вода до 10 л
Прочие буферные растворы описаны в других работах [И].
Рекомендуется барбитуровую или диэтилбарбитуровую кислоты
растворить в 1—2 л воды при нагревании, внести прочие ком-
поненты, а затем довести смесь до нужного объема дистиллиро-
ванной водой. Можно использовать агарозу производства раз-
ных фирм, например, Feinchemie, Kallis KG (ГДР); Behring-
werke AG (ФРГ); Ferak (Западный Берлин); Lindustrie biolo-
gique Franijaise (Франция) и др. Желательно всегда пользо-
ваться агарозой одной марки и одной серии. Новые образцы
агарозы следует подвергать предварительной оценке на год-
ность. Кроме этого, необходимы электрофоретическая камера
с охлаждением, микрошприцы для отбора проб (1—10 мкл), из-
мерительное устройство для определения расстояний с точно-
стью до 0,1 мм.
2.3.2.2. ПРОВЕДЕНИЕ ОПЫТА
Расплав агарозы (1,5%) в барбитуратном буфере с pH 8,6при
температуре 48 °C смешивают с моноспецифической сыворот-
кой. Концентрация АС в зависимости от титра, свойств АГ и
других факторов может колебаться в широких пределах. Чаще
всего используют концентрации 1—5%. На предметное стекло
размером 120X60X2 мм наносят 10 мл смеси, равномерно рас-
пределяют ее по поверхности. Пластины рекомендуется подго-
тавливать нанесением слоя водной агарозы (см. 2.1.3.2.2). Пла-
стины располагают на подогреваемом (35—40 °C) горизонталь-
ном столике. После застывания геля образуется равномерный
слой толщиной около 1 мм. Рекомендуется применение U-образ-
98
Рис. 38. Электроиммуииый ана-
лиз: этапы работы.
ных рамок из пластика вы-
сотой 1 мм. Два стекла
складывают с помощью та-
кой рамки, образуется пло-
ская камера, которую за-
полняют золем АС-содержа-
щей агарозы, стараясь из-
бегать образования пузы-
рей. После застывания геля
одно стекло удаляют; плас-
тина с гелем готова для ис-
пользования. На расстоянии
8 мм от длинного края пла-
стины в слое геля проделы-
вают лунки для АГ диамет-
ром 4 мм. Лунки должны
отстоять друг от друга по
меньшей мере на 6 мм.
В каждую лунку вносят 10 мкл раствора АГ и проводят элект-
рофорез. Лунки для АГ расположены на катодной части пласти-
ны. Напряжение может составлять 20 В/см; температура должна
поддерживаться на уровне 4—10 °C. Продолжительность элект-
рофореза может составлять 3—18 ч для разных систем АГ — АТ.
Оценка результатов проводится на влажном препарате немед-
ленно по окончании электрофореза. Если прецитаты недостаточ-
но различимы, пластины с гелем отмывают для удаления непре-
ципитирующих белков, высушивают и окрашивают; затем про-
водят оценку результатов. На рис. 38 представлены основные
этапы метода, на рис. 39 — электрофореграмма.
Рис. 39. Электроиммуииый анализ: определение количества альбумина.
7* 99
2.3.2.3. ОБРАБОТКА ПЛАСТИН,
ОЦЕНКА РЕЗУЛЬТАТОВ
Лунки в геле следует вырезать очень тщательно, края должны
быть ровными, гель не должен отставать от стекол, наполнять
лунки следует до краев. Электрофорез начинают немедленно
после внесения образца в лунки, это делается для предотвраще-
ния ИД. Лунки для АГ можно заполнять даже под слабым на-
пряжением, которое затем увеличивают до полного. Концентра-
цию АГ следует подбирать таким образом, чтобы образовыва-
лись преципитаты длиной 10—50 мм. Все разведения АГ готовят
на буфере для электрофореза. При оптимальных условиях элек-
троиммунного анализа (ЭИА) формируются узкие, остроконеч-
ные преципитаты. Точные измерения показывают, что площадь
образующихся преципитатов находится в прямой зависимости
от концентрации белка (рис. 40). При условии, что преципита-
ты будут иметь описаннную выше форму, для практических це-
лей можно ограничиться измерением длины зоны преципи-
тации.
2.3.2.4. ЭЛЕКТРОФОРЕЗ
Преципитаты приобретают характерную вытянутую форму
только при достаточной длительности электрофореза. Необхо-
димое для этого время зависит от исследуемой системы и от
приложенного напряжения. Создать достаточно эффективное
напряжение можно только при надлежащем охлаждении каме-
ры. Обычно напряжение варьирует от 5 до 20 В/см. Ионная
сила буферов составляет 0,02—0,1. В зависимости от имеюще-
гося оборудования можно вносить те или иные методические из-
менения. При большой длительности электрофореза и низком
напряжении можно отказаться от охлаждения и уменьшить со-
держание АС в геле до 0,3%•
2.3.2.5. ОПРЕДЕЛЕНИЕ ИММУНОГЛОБУЛИНОВ
При ЭИА иммуноглобулины, как правило, проявляют незначи-
тельную подвижность как в катодном, так и в анодном направ-
лении. Это наблюдение относится к IgG и IgM. Вокруг старто-
вой лунки образуется широкий двойной «шлейф». Для преду-
преждения этих явлений пользуются следующими приемами:
I. Вместо агарозы используют 1% агар, учитывая его повышен-
ный электроэндоосмос. Образуются довольно широкие, с за-
кругленным концом преципитаты, направленные к катоду.
2. Широкое распространение получила обработка белков, на-
пример сыворотки, 2 М раствором KCNO. Один объем сы-
воротки смешивают с двумя объемами раствора цианата ка-
лия при комнатной температуре и оставляют смесь на ночь
или равные объемы обоих растворов инкубируют 30 мин при
1С0
Рис. 40. Электроиммуиный анализ:
кривая для определения альбумина.
Концентрация альбумина (г/л)
45 °C. Затем делают необ-
ходимое разведение верона-
ловым буфером и проводят
электрофорез. Цианат реа-
гирует со свободными NH2-
и SH-группами [8], повы-
шая общий отрицательный
заряд белковых молекул,
что приводит к более быст-
рому их передвижению в
сторону анода.
3. Похожие результаты полу-
чаются при обработке об-
разцов сыворотки 0-пропиолактоном [9]. Это вещество не ме-
няет антигенные свойства 1g.
4. Для заливки пластин с гелем используют карбамилирован-
ную АС. Электрофорез проводят при pH 5 — изоэлектриче-
ской точке карбамилированных 1g. В этих условиях иммуно-
глобуллины АС остаются неподвижными в геле, a 1g иссле-
дуемого образца мигрируют только в сторону катода [2].
5. Образцы сыворотки обрабатывают глютаровым альдегидом,
что приводит к конъюгированию 1g с электрофоретически бо-
лее подвижными белками, в первую очередь с альбумином.
Антигенные детерминанты 1g остаются «свободными», по-
этому преципитаты образуются в анодной области в условиях
электрофореза при pH 8,6.
2.3.2.6. МОДИФИКАЦИИ МЕТОДА
ЭИА можно проводить на ацетат-целлюлозных пленках [10].
Для осуществления такого анализа требуются меньшие количе-
ства АС. Дальнейшим развитием ЭИА является двумерный ко-
личественный иммуноэлектрофорез. При помощи ЭИА с неболь-
шими модификациями можно идентифицировать различные АГ
и оценивать количество перекрестно реагирующих АТ в антисы-
воротке [4]. Обратный ЭИА позволяет устанавливать титр АТ
в АС. Электрофорез проводят при pH 4,85, т. е. изоэлектриче-
ской точке альбумина. Мигрируют только АТ и только в катод-
ном направлении (см. выше). Для определения продуктов рас-
щепления фактора комплемента 3 (С Зс и С 3d) используют
«двухслойный» ЭИА [3]. АГ сыворотки начинают двигаться
в геле без АС, переходят в гель, содержащий АС к С Зс, снова
переходят в зону геля без АС и, наконец, оказываются в слое
геля с АС к С 3d. Секреторный SIgA и секреторный компонент
(СК) могут определяться одновременно в одной пластине с ге
101
лем, состоящим из смеси агара и агарозы и содержащим каль-
циевую соль этилендиаминтетраацетата (ЭДТА) и карбамили-
рованную сыворотку. В этих условиях SIgA мигрирует в сторо-
ну катода, СК—в сторону анода [5]. В заключение следует
упомянуть о том, что ЭИА может быть использован для опре-
деления нерастворимых белков эпидермиса, ногтей и волос, на-
пример кератина. Электрофорез проводят в присутствии дена-
турирующих реагентов (ДСН, мочевина) [7].
2.3.3. ОЦЕНКА МЕТОДА
ЭИА по Лореллу используют для количественного определения
белка в жидкостях организма. Методу ЭИА можно в принципе
дать ту же оценку, что и простой радиальной ИД, однако он
имеет большее количество ограничений. Дополнительные слож-
ности возникают с АГ, медленно мигрирующими при pH 8,6.
Более сложное оборудование для ЭИА также относится к чис-
лу его недостатков. Существенным преимуществом ЭИА по
сравнению с простой радиальной ИД является быстрота получе-
ния результатов. Коэффициент вариации при ежедневном конт-
роле составляет 3—6%.
2.3.4. СПИСОК ЛИТЕРАТУРЫ
1. Axelsen N. Н., Svendsen Р. J. Reverced rocket Immunoelectrophoresis.—
Scand. J. Immunol., 1973, suppl. 1, 2, 155—157.
2. Bjerrum O. J., Ingild A., Lowenstein H., Weeke B. Carbamylated antibodie
used for quantitation of human IgG. — Scand. J. Immunol., 1973, 2, suppl. 1,
145—148.
3. Brandslund I., Siersted H. C„ Svehag S. E., Teisner B. Double decker rocket
immunoelectrophoresis for direct quantitation of complement C 3 split pro-
ducts with C3d specificities in plasma. — J. Immunol. Methods, 1981, 44,
63—71.
4. Grubb A. Analysis of immunochemical relationship between antigens by
electrophoresis in agarose gels containing antibodies. — Scand. J. Clin.-Lab.
Invest., 1972, 29, suppl. 124, 7—19.
5. Kosaka T., Asahina T„ Kobayashi N. Differential quantification of SIgA and
SC by two-directional rocket method. — Immunology, 1980, 40, 597—604.
6. Laurell С. B. Electroimmunoassay. — Scand. J. Clin. Lab. Invest., 1972, 29,
suppl. 124, 21—37.
7. Lee L. D., Baden H. P„ Cheng С. K. Rocket immunoelectrophoresis in the
presence of denaturating agents.—J. Immunol. Methods, 1978, 34, 155—
162.
8. Raisys V. A., Arvan D. A. Determination of proteins in biological fluids by
electroimmunodiffusion and two-directional immunoelectrophoresis. — Clin.
Chem., 1971, 17, 745—750.
9. Stephan W., Eram U. Quantitative Simultan-Immunoelektrophorese. Ztschr.
klin. Chem. klin. Biochem., 1971, 9, 224—228.
10. Watkins J. B., Atkins B., Holborrow E. J. Quantitative estimation of protein
by electroimmunodiffusion on cellogel acetate membranes. — J. Clin. Pathol.,
1971. 24. 665—667.
11. Weeke B. Rocket Immunoelectrophoresis. — Scand. J. Immunol., 1973, 2,
suppl. No 1, 37—46.
2.4. ВСТРЕЧНЫЙ ЭЛЕКТРОФОРЕЗ
2.4.1. ПРИНЦИП МЕТОДА
Принцип электросинереза или встречного электрофореза при-
менил в 1969 г. Бедарида для обнаружения преципитатов
АГ—АТ. Иммунопреципитация происходит в слое носителя,
причем в отличие от метода Ухтерлони контакт АГ с АТ проис-
ходит не вследствие свободной диффузии, а под влиянием по-
стоянного электрического поля. Белки представляют собой ам-
фолиты; ввиду различий аминокислотного состава они могут
мигрировать в щелочной среде с разной скоростью и в разных
направлениях (анодном, катодном). Если АГ и соответствую-
щая преципитирующая АС мигрируют в разных направлениях,
становится возможным их движение друг навстречу другу в слое
геля или на ацетат-целлюлозной пленке. Преципитация и одно-
временно накопление в одном месте участников реакции наблю-
дается между стартовыми лунками обоих компонентов в течение
30—180 мин в зависимости от приложенного напряжения.
Встречный иммуноэлектрофорез (ВИЭФ) предназначен для об-
наружения и идентификации преципитирующих систем АГ—АТ
при условии, что АГ обладает электрофоретической подвиж-
ностью, отличной от той, которой обладают иммуноглобу-
лины.
Эксперимент проводится с компонентами, мигрирующими в
противоположных направлениях или в одном направлении, но
с различной скоростью. В настоящее время ВИЭФ применяют
в основном для определения поверхностного антигена гепати-
та В (HBsAg), а-фетопротеина (АФП) и антител к ним. Необ-
ходимым условием для ВИЭФ является наличие моноспецифиче-
ской преципитирующей сыворотки. Движение АТ в электриче-
ском поле может искажаться под влиянием электроэндоосмоса.
Электроэндоосмос представляет собой медленное движение
жидкой среды в сторону катода, возникающее вследствие увле-
чения жидкости частицами твердой фазы с прилегающими
к ним положительно заряженными молекулами воды.
Катодный ток жидкости может снизить скорость движения
частиц белка к аноду или даже изменить его на обратное. АФП
и HBsAg перемещаются в аа-области к аноду, в то время как ИГ
в щелочной среде перемещаются к катоду. При контакте обоих
партнеров реакции образуются резкие полосы преципитации,
хорошо заметные при боковом освещении и на темном фоне.
Модификации этого метода получили в работах разных авто-
ров самые различные названия: иммуноэлектродиффузия [1],
контраиммуноэлектрофорез [4], иммуноэлектроосмофорез [10],
встречный (cross — over) электрофорез [8], иммунопреципита-
ционный электрофорез [3]. Предложенный в 1970 г. Коном ме-
тод определения АФП основан на этом же принципе [5].
103
2.4.2. МЕТОДИКА
2.4.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы можно использовать любое электрофоретическое
устройство с регулируемым напряжением (до 300 В), например,
камеру FK66 (Росток) или Цейссовский прибор для электро-
фореза; предметные стекла или стеклянные диапозитивные пла-
стины 5x5 см, агар (например, Difco Agar Noble) или агарозу,
или смесь агар — агароза; ацетат-целлюлозная мембрана (Sar-
torius, Геттинген); трубки для вырезания лунок в геле, водо-
струйный насос для удаления вырезанных гелевых цилиндров;
влажную камеру, фильтровальную бумагу для замыкания кон-
тактов геля с буфером. Антисыворотки (например, лошадиная
анти-HBsAg сыворотка, анти-АФП сыворотка, Institut fiir Imf-
stoffe (Дессау), Behringwerke (Марбург), буферные растворы,
состав которых приводится ниже.
Буферные растворы:
Барбитал-натрий
CH3COONa-3H2O
0,1 М НС1
Дистиллированная вода
или
г
г
мл
А
9,75
6,47
60,0
до 2000 м
pH 8,6; ц=0,05
Б
8,142 г
0,476 г
90,0 мл
до 1000 мл
pH 8,3—8,4; ц = 0,1
2.4.2.2. ПРИГОТОВЛЕНИЕ ГЕЛЯ
Расплавляют 1,5 г агара, агарозы или смеси обоих веществ
в 100 мл барбитал-ацетатного буфера на кипящей водяной бане.
В качестве консерванта можно добавить 2 мл 0,1% раствора тио-
мерсала (мертиолята). Стеклянные пластины обезжиривают,
слегка подогревают и наносят на них равномерным слоем агар
Натод
Рис. 41. Схема расположения лунок при проведении встречного иммуно-
электрофореза.
А — место нанесения АГ, Б — место нанесения антисыворотки, В — место нанесения оха-
рактеризованного АГ для выявления антител.
104
Рис. 42. Схема расположения лунок для реагентов прн постановке ВИЭФ
по Kohn [5].
Рис. 43. Прецнпнтат НВз-антнген/анти-НВв-антитела в геле агарозы (боль-
шое увеличение).
из расчета 0,15 мл на 1 см2. После застывания геля на расстоя-
нии около 7 мм друг от друга вырезают лунки диаметром 3 мм,
гелевые цилиндры удаляют с помощью водоструйного насоса.
Если одновременно хотят обнаружить антитела, то лунки выре-
зают триадами (рис. 41). В зависимости от цели исследования
расположение лунок может быть и таким, как показано на
рис. 42 (в и г); особое внимание следует обращать на точное
соблюдение расстояний между лунками. Если лунки располо-
жены так, как это показано на рис. 42 (в), то, варьируя соотно-
шение АГ—АТ и заполняя исследуемым образцом оба резер-
вуара (без положительного контроля), можно наблюдать (соот-
ветственно избегать) феномен прозоны вследствие избытка АГ
или АТ. При помощи пипетки в лунки с катодной стороны вно-
сят антиген, с анодной — антисыворотку.
2.4.2.3. ПРОВЕДЕНИЕ ЭЛЕКТРОФОРЕЗА
Оба отделения камеры для электрофореза заполняют барбитал-
ацетатным буфером. Пластины с гелем укладывают в соответ-
ствии с расположением полюсов и замыкают электрическую
цепь полосками бумаги, смоченной буферным раствором. При
напряжении 150—200 В (3—4 мА на 1 см ширины агарового
геля) продолжительность электрофореза составляет от 1 до 3 ч.
Увеличивая напряжение до 300 В, можно сократить продолжи-
тельность электрофореза до 30 мин, но для этого необходимо
обеспечить охлаждение камеры. В геле агара преципитаты
HBsAg—анти-НВэ-антитела образуются возле лунки с антисы-
вороткой, в геле агарозы — посередине между лунками с АГ и с
АТ (рис. 43, 44).
105
Рис. 44. АФП/анти-
АФП преципитат в ге-
ле агара.
а — антисыворотка, б — ис-
следуемая сыворотка, в —
позитивная контрольная сы-
воротка.
2.4.2.4. ВСТРЕЧНЫЙ ИММУНОЭЛЕКТРОФОРЕЗ
НА АЦЕТАТ-ЦЕЛЛЮЛОЗНЫХ МЕМБРАНАХ
Смоченные в буферном растворе мембраны из ацетат-целлюло-
зы слегка высушивают фильтровальной бумагой, кладут их на
три слоя фильтровальной бумаги и специальным штампом де-
лают углубления, вмещающие 5 мкл жидкости. Эти углубления
хорошо сохраняются при последующей обработке мембран и
помогают точно определять положение полос преципитации. На-
пряжение 100 В позволяет завершить электрофорез за 20 мин.
Линии преципитации на мембране не видны, для их визуализа-
ции необходимо окрашивание. Мембраны отмывают проточной
водопроводной водой, окрашивают амидо черным 10 В или ни-
грозином и обесцвечивают водой. Весь процесс занимает не бо-
лее 15 мин. ВИЭФ на ацетат-целлюлозных мембранах имеет не-
сомненные преимущества при массовых обследованиях, так как
расход антисыворотки составляет всего лишь 5 мкл на один
анализ.
2.4.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Результаты реакции оценивают визуально, в некоторых случаях
используют лупу; пластины геля рассматривают при косом ос-
вещении или на темном фоне. Реакция считается положитель-
ной, если картина преципитации такая же, как в контрольных
лунках. В случае слабой преципитации или при сомнительных
результатах делают ряд разведений исследуемого образца или
АС для предотвращения эффекта прозопы. Едва заметные пре-
ципитаты можно сделать более отчетливыми, если хорошо от-
мытые пластины геля погрузить на 10 мин в 1% раствор тан-
иина. Окрашивание пластин амидо черным 10 В или нигрозином
обычно не улучшает результаты. Расположение преципитатов
106
зависит как от природы геля, так и от концентрации АГ и АТ.
В геле агарозы преципитаты располагаются посередине между
лунками; в геле агара АТ быстрее продвигаются к катоду,
и преципитаты образуются ближе к лунке с АГ. Для выявления
АГ лучше использовать гель агарозы или смешанный гель ага-
ра-агарозы. АТ удобнее определять в геле агара. Однако при
использовании агара следует учитывать опасность того, что
преципитаты могут образовываться непосредственно у лунки
с АГ и в случае использования мутной АС их невозможно об-
наружить. Еще одним источником ошибок может стать продол-
жительность электрофореза; время электрофореза должно быть
тщательно установлено в системе контрольный АГ — конт-
рольная АС (заниженное время приводит к ложноотри-
цательным результатам, завышенное, при трехлуночной си-
стеме расположения образцов, — к ложноположительиым).
Неспецифические преципитаты могут формироваться в виде сла-
бых диффузных полос, если использовать очень свежую или,
наоборот, очень старую сыворотку. Опытный экспериментатор
должен уметь отличать их от истинных преципитатов. Количест-
венная оценка результатов возможна при использовании ряда
последовательных разведений исследуемого образца и сыворо-
ток с известным титром.
2.4.4. ОЦЕНКА МЕТОДА
Оценка метода обнаружения HBsAg. ВИЭФ пред-
ставляет собой аналитический метод второго поколения и по
сравнению с двойной ИД по Ухтерлони (первое поколение) име-
ет следующие преимущества:
1. Чувствительность ВИЭФ в 10 раз выше, могут быть исполь-
зованы антигены, выявление которых методами диффузии не
оправдывает себя.
2. Результаты можно учитывать через 1 ч, максимум через 3 ч,
тогда как для полноценной диффузии необходимо не менее
24 ч.
Попытки усовершенствования методики привели к разработке
специальных модификаций для обнаружения HBsAg. Упрощение
методики достигается использованием ацетат-целлюлозной мем-
браны [И], длительность процесса сокращается применением
высоковольтного электрофореза [2]. Все же микромодификации
не способны повысить чувствительность метода. ВИЭФ высоко-
специфичен при поиске HBsAg, но он уступает чувствительно-
сти методам третьего поколения, РИА, иммунофлюоресцентный
анализ (ИФА). Методы третьего поколения относительно доро-
ги, поэтому ВИЭФ имеет преимущество перед ними при массо-
вых обследованиях.
Оценка метода обнаружения АПФ. Чувствитель-
ность метода двойной диффузии при определении АФП состав-
ляет около 10 мг вещества на 1 л сыворотки. Применение ВИЭФ
107
позволяет повысить чувствительность в 1,5—3 раза [7], т. е. до
3,6 мг/л. Небольшое повышение чувствительности достигается
окраской нигрозином или применением флюоресцирующих АТ
[6]. Положительная реакция на АФП позволяет предположить
наличие первичного рака печени или злокачественной тератобла-
стомы. Ложноположительные результаты определения АФП
при помощи ВИЭФ наблюдаются примерно в 3 случаях из 100
[7]. Отрицательные результаты ввиду относительно малой чув-
ствительности ВИЭФ не позволяют исключить диагноз рака пе-
чени. Более чувствительные методы, такие как пассивная ге-
магглютинация и радиоиммунологический анализ могут обна-
руживать повышение АФП в крови и при других раковых забо-
леваниях, причем необязательно при метастазировании в печень.
Поэтому только неуклонное повышение уровня АФП в течение
всего периода наблюдения типично для начинающегося рака пе-
чени. Для более точного контроля рекомендуется применять ко-
личественные методы Манчини и Лорелла.
2.4.5. СПИСОК ЛИТЕРАТУРЫ
1. Duquesnoy К. J., Becker G. G. Rapid screening for hepatitis antigen by
immunoelectrodiffusion using paper discs. — Transfusion, 1970, 10, 221.
2. Frank К. H., Reimann W. Schneller, empfindlicher Nachweis des Au-SH-Anti-
gens mittels Mikroubereinwanderungselektrophorese.— DTSCH. Ges.-wesen,
1971, 26, 1042.
3. Geserick G. Das Australia-Antigen (Au/SH/HAA). Serologische untersuchun-
gen zu Nachweismethodik, Vorkommen, Eigenschaften und Bedeutung des
Antigens. — Dissertation B, Berlin, 1972.
4. Goeke D. J., Howe C. Rapid detection of Australia antigen by counterimmu-
noelectrophoresis. — J. Immunol., 1970, 104, 1031.
5. Kohn J. Method for the detection and identification of apfetoprotein in
serum. —J. Clin. Path., 1970, 23, 733.
6. Kohn. J. Alphapfetoprotein: Laborbestimmung und klinische Bedeutung. —
Arztl. Lab., 1972, 18, 307.
7. Lehmann F. Q., Lehmann D. Vergleich verschidener Methoden zum serolo-
gischen Nachweis von ai-fetoprotein im serum. — Ztschr. klin. Chem. klin.
Biochem., 1973, 11, 58.
8. Pesendorfer F„ Krassnitzki O., Wewalka F. Immunoelektrophoretischer Nach-
weis von «Hepatitis associated antigen» (Au-SH-antigen).— Klin. Wschr.,
1970, 48, 58.
9. Porstmann T., Porstmann B., Schmechta H. et al. Entwicklung eines Doppel-
Sandwich Enzymimmunoassays zum quantitativen Nachweis von HBsAg.—
Dtsch. Ges.-wesen, 1980, 35, 598—600.
10. Prince A. M., Burke K. Serum hepatitis antigen (SH): Rapid detection by
high-voltage immunoelectroosmophoresis.— Science, 1970, 169, 593.
11. Vergani C. Crossover electrophoresis for the rapid detection of serum hepati-
tis (Au) antigen and antibody. — J. clin. Pathol., 1971, 24, 86.
2.5. АНАЛИЗ ИММУННЫХ
ПРЕЦИПИТАТОВ В ГЕЛЯХ
Для анализа иммунных преципитатов в гелях (иммунодиффу-
зия, иммуноэлектрофорез) применяют различные варианты
окрашивания, ферментативные реакции, радиоактивную и им-
108
мунофлюоресцентную метки, определение электрофоретической
подвижности. Кроме того, при иммунодиффузии и иммуноэлек-
трофорезе применяют референс-антигены и моноспецифические
сыворотки (см. 2.1, 2.2).
2.5.1. ОКРАШИВАНИЕ КАК СПОСОБ АНАЛИЗА ПРЕЦИПИТАТОВ
Окрашивание преципитатов в гелях повышает контрастность
препарата, а также делает видимыми неразличимые невоору-
женным глазом преципитаты. Перед окрашиванием необходимо
удалить из геля все белки, не имеющие отношения к формиро-
ванию преципитатов (отмывка гелей 0,15 М NaCl). Окрашива-
ние преципитатов позволяет получать более тонкую и дифферен-
цированную характеристику антигенов (1 — белки, 2 — липопро-
теиды, 3 — гликопротеиды, полисахариды, нуклеопротеиды, ме-
таллопротеиды) (см. цв. вклейку, рис. 1).
2.5.1.1. ОКРАШИВАНИЕ БЕЛКОВ
Для выявления белков в составе преципитатов чаще всего ис-
пользуют амидо черный, световой зеленый, азокармин, пунцо-
вый, нигрозин, кумасси синий и бромфеноловый синий. Боль-
шинство красящих растворов перед употреблением фильтруют.
Продолжительность окрашивания зависит от толщины слоя геля,
но оно редко длится более 10—20 мин. Обесцвечивание геля
проводят до тех пор, пока участки геля, не содержащие белка,
становятся совершенно прозрачными. Часто для обесцвечива-
ния достаточно простой водопроводной воды.
Амидо черный. Применяются как водные, так и спирто-
вые растворы.
Хорошо зарекомендовала себя смесь следующего состава:
Амидо черный 10 В 1,0
Ледяная уксусная кислота 100,0
Дистиллированная вода до 1000,0
Продолжительность окрашивания высушенных иммунофоре-
грамм составляет около 10 мин; обесцвечивание проводят 7%
раствором уксусной кислоты или водопроводной водой. Полосы
преципитации имеют темно-синюю окраску, фон — бесцветный.
Световой (светлый) зеленый. Краситель окрашива-
ет у-глобулины почти так же интенсивно, как альбумины. Линии
преципитации имеют ярко-зеленый цвет. Состав смеси сле-
дующий:
Световой зеленый SF 2,0
Ледяная уксусная кислота 100,0
Дистиллированная вода до 1000,0
Продолжительность окрашивания— 1—2 ч. Обесцвечивание про-
водят водопроводной водой в течение нескольких минут.
Азокармин. Этот вид окрашивания позволяет получать
темно-красные полосы преципитации. Окрашивание очень стой-
109
кое, при фотографировании получаются контрастные снимки.
Состав смеси:
Азокармин 2,5
Ледяная уксусная кислота 100,0
Дистиллированная вода до 1000,0
Окрашивание длится 10 мин. Обесцвечивание проводят водопро-
водной водой.
Пунцовый. Этот краситель окрашивает преципитаты
в светло-красный цвет. Краситель особенно удобен для окраши-
вания преципитатов в условиях определения ферментативной ак-
тивности.
Пунцовый 2R 3,0
Ледяная уксусная кислота 100,0
Дистиллированная вода до 1000,0
Продолжительность окрашивания составляет несколько часов.
Отмывают препарат водопроводной водой.
2.5.1.2. ОКРАШИВАНИЕ ЛИПОПРОТЕИДОВ
Для выявления липопротеидов в преципитатах пригодны судан
черный, жировой красный и нильский голубой. Необходимо,
чтобы пластины геля были совершенно сухими; влажный гель
плохо прокрашивается. Недостаточно высушенные препараты
могут обусловливать выпадение неспецифического осадка крас-
ки, поскольку все вышеперечисленные красители водонераство-
римы.
Судан черный. Состав красящего раствора:
Судан черный 1,2
1 М NaOH 20,0
Дистиллированная вода 380,0
Абсолютный этанол до 1000,0
Смесь непродолжительное время кипятят, после остывания пе-
реливают в темную склянку с хорошо притертым горлышком.
Для окрашивания полностью высушенные пластины помещают
на 3 ч в красящий раствор. Обесцвечивают 50% этанолом до
полного просветления фона. Липопротеиды окрашиваются
в темно-синий цвет.
Жировой красный. Окрашивание этим красителем при-
дает препарату незначительную контрастность. Липопротеиды
окрашиваются в бледно-красный цвет. Состав смеси:
Жировой красный 2,0
Этанол 70% 1000,0
После непродолжительного кипячения и охлаждения раствор
фильтруют. В свежеприготовленном растворе пластины геля вы-
держивают несколько часов; обесцвечивают 70% этанолом.
Одновременное окрашивание липопротеидов
и белков. Комбинируя азокармин с Суданом черным, можно
за один цикл окрашивания получить белки в виде красных,
ПО
а липопротеиды в виде темно-синих полос. Состав красящей
смеси:
Азокармин В 0,5
60% этанол, насыщенный
судаком черным до 1000,0
Обесцвечивание фона проводят 50% этанолом, содержащим 1%
уксусную кислоту.
2.5.1.З. ГЛИКО-, НУКЛЕО- И МЕТАЛЛОПРОТЕИДЫ
По аналогии с электрофорезом на бумаге окрашивание углево-
дов можно проводить перйодатным методом Шиффа. Разумеет-
ся, неоправданно применение агара в качестве носителя, по-
скольку он сам, будучи углеводом, дает положительную реак-
цию. Поэтому предпочтительнее использовать ацетат-целлюлоз-
ную мембрану. Нуклеопротеиды можно окрашивать пирони-
ном Y. Продажные препараты пиронина содержат примеси, ко-
торые можно удалить многократной экстракцией хлороформом.
20% раствор пиронина Y
в дистиллированной воде 10,0
0,2 М ацетата натрия 45,0
0,2 М уксусная кислота 45,0
Высушенные пластины с гелем выдерживают в красящем рас-
творе в течение 60 мин, фон обесцвечивают в ацетатном буфере.
Преципитаты, содержащие нуклеопротеиды, окрашиваются
в красный цвет.
Выявление металлопротеидов. Преципитаты, содер-
жащие ионы меди, окрашиваются ализариновым синим в синий
цвет. Реакция специфична в отношении ионов.
Исходный раствор: насыщенный раствор ализаринового си-
него в уксусной кислоте
Рабочий раствор: 1 часть исходного раствора+9 частей
70% уксусной кислоты (использовать немедленно).
Высушенные пластины с гелем помещают на 30 мин в красящий
раствор. Обесцвечивают пластины 70% СН3СООН. Белки, со-
держащие ионы меди, окрашиваются в синий цвет. Железосо-
держащие белки, например сывороточный трансферрин или ге-
моглобинсвязывающие белки, выявляют при помощи реакций,
сопровождающихся образованием берлинской лазури или бен-
зидиновой пробой.
2.5.2. ОПРЕДЕЛЕНИЕ ФЕРМЕНТАТИВНОЙ АКТИВНОСТИ
ИММУННЫХ ПРЕЦИПИТАТОВ
Определение ферментативной активности может служить не
только для характеристики иммунных преципитатов, но и для
повышения чувствительности метода. Блокада активных центров
фермента в результате иммунной реакции наблюдается редко.
111
Как правило, активность фермента сохраняется и после реакции
антиген — антитело. Ферменты и изоферменты можно выявлять
после элюции из геля, окрашивать непосредственно в геле, поль-
зоваться гелем, содержащим субстрат, применять сэндвич-ме-
тод или метод контактной пленки. Ввиду термолабильности
большинства ферментов пластины с гелем в ходе электрофореза
необходимо охлаждать. Обычно пластины с гелем, подлежащие
выявлению ферментативной активности, не высушивают. От-
дельные ферменты, например, карбоксиэстеразы сохраняют ак-
тивность и в высушенных пластинах. Мы приводим только не-
сколько методик выявления ферментативной активности в им-
мунных преципитатах в гелях. Исчерпывающие данные имеются
в публикациях Uriel и Clausen [2,1].
2.5.2.1. ОКСИДОРЕДУКТАЗЫ
Л а кта тд еги д р оген а з а (ЛДГ) КФ 1.1.1.27:
Раствор 1. KCN 50 мг
Хлористый диметилтиазо-
лилтетразолий (или нитро-
синий хлористый тетразо-
лий) 30,0 мг
0,1 М фосфатный буфер pH 7,5
Раствор 2. Феназинметосульфат 10,0 мг
Дистиллированная вода 10,0 мл
Оба раствора можно хранить раздельно в течение 4 нед при
4 °C. Раствор 2 хранят в темной склянке. Среду для инкубации
готовят непосредственно перед использованием.
Среда для инкубации: Раствор 1 7,2 мл
Раствор 2 0,2 мл
НАД 5,0 мг
DL-лактат лития 25,0 мг
Определение изоферментов ДЛГ проводят во влажной пластине
после удаления непреципитировавших белков. Реакция заканчи-
вается через 1—2 ч при 37 °C. Первые видимые полосы прояв-
ляются через 20 мин. Фиксацию проводят при комнатной темпе-
ратуре в течение 30 мин спиртовым раствором уксусной кис-
лоты:
Этанол 150,0 мл
Ледяная уксусная кислота 10,0 мл
Дистиллированная вода 40,0 мл
Препараты, содержащие компоненты с активностью ЛДГ, дают
полосы преципитации сине-фиолетового цвета.
Церулоплазмин (пара-дифенолоксидаза, КФ 1.10.3.2).
21,6 мг пара-фенилендиамина растворяют в 100 мл натрий-аце-
татного буфера (0,1 М, pH 5,7), подогревают до 37 °C и немед-
ленно используют. Инкубация продолжается 2 ч, после инкуба-
ции проводят отмывку смесью спирта и натрий-ацетатного бу-
фера:
112
0,2 М ацетат натрйй 50,0
0,2 М уксусная кислота 50,0
50% этанол 100,0
Церулоплазмин, обладающий оксидазной активностью, окраши-
вается в сине-фиолетовый цвет. Для контроля в параллельном
опыте используют ингибирующее влияние азида натрия на окси-
дазы (добавляют к инкубационной смеси 1 мл 0,1 М NaNs)h
которое предотвращает окрашивание.
2.5.2.2. ГИДРОЛАЗЫ
Карбоксилэстераза (КФ 3.1.1.1)
В качестве субстрата используют а-нафтилацетат, расщепляе-
мый ферментом на нафтол и уксусную кислоту, с солями диазо-
ния а-нафтол реагирует, образуя азокрасители, которые могут
служить индикатором реакции.
Среда для инкубации готовится непосредственно перед употреб-
лением:
а-Нафтилацетат 10,0 мг
Ацетон 0,25 мл
Оба компонента смешивают, затем добавляют:
0,1 М фосфатный буфер
pH 7,4 25,0 мл
Прочный синий В 20,0 мг
Раствор необходимо профильтровать.
Инкубируют при комнатной температуре в течение 30—
60 мин. Преципитаты, обладающие активностью карбоксилэсте-
разы, окрашиваются в коричнево-красный цвет.
Щелочная фосфатаза
Раствор для инкубации:
Нафтилфосфат натрия
Прочный синий В
0,1 М веронал-солянокис-
лый буфер pH 9,0
Раствор фильтруют. Пластины
60 мин при комнатной температуре, фиксируют 5% уксусной
кислотой, отмывают водой в течение 1 ч. Ферментативная ак-
тивность проявляется винно-красным окрашиванием.
(КФ зл.3.1)
20,0
20,0
мг
мг
мл
20,0
инкубируют в течение 30—
2.5.3. РАДИОАВТОГРАФИЯ, ИММУНОФЛЮОРЕСЦЕНЦИЯ
Радиоавтография представляет собой более чувствительный,
чем окрашивание, метод исследования связывающей способно-
сти веществ и их подвижности в геле. Чистые АГ метят 13Ч,
82Br, 69Fe, вносят их в смесь АГ. После преципитации высушен-
ные пластины с гелем накладывают на куски рентгеновской
пленки и оставляют их на несколько дней или недель. Радиоак-
тивные преципитаты вызывают потемнение соответствующих
участков пленки. Определение железосвязывающих трансферри-
8—990
ИЗ
R
Рис. 45. Определение
электрофоретической мо-
бильности при иммуно-
электрофорезе.
А— альбумин, Т — трансферрин,
G — IgG, R — резервуар с анти-
геном, IS — канавка для иммун-
ной сыворотки, АВ — расстояние
между вершинами дуг А и Т,
АХ — расстояние между верши-
нами дуг А и G.
нов, например, оказывается довольно простой процедурой —
к смеси АГ перед анализом добавляют небольшое количество
S9Fe. После высыхания пластины с гелем накладывают на рент-
геновскую пленку; полосы трансферрина становятся хорошо
различимыми на пленке. При помощи радиоактивных изотопов
можно исследовать связывание витаминов, гормонов и некото-
рых лекарственных препаратов с сывороточными белками. Если
антигены или антитела маркировать флюоресцирующими ве-
ществами, например, флюоресцеинизотиоцианатом (ФИТЦ), то
образующиеся иммунные преципитаты также будут флюоресци-
ровать. Это позволяет проводить анализ в УФ лучах. Перед ис-
следованием препаратов в УФ свете необходимо удалить избы-
ток флюоресцирующего красителя из изучаемого препарата
с помощью диализа или гель-фильтрации.
2.5.4. ОПРЕДЕЛЕНИЕ ЭЛЕКТРОФОРЕТИЧЕСКОЙ
ПОДВИЖНОСТИ
Определение относительной электрофоретической подвижности
может служить для характеристики неизвестных белков. Абсо-
лютной электрофоретической подвижностью называют действи-
тельную длину пробега исследуемого вещества, деленную на на-
пряжение и время (размерность 10-5 см2 В-1 сек-1). Для опре-
деления нулевой точки электроэндоосмоса, которая при электро-
форезе в агаровом геле не совпадает с точкой старта, исполь-
зуют вещества, электрически нейтральные и обладающие огра-
ниченной диффузией (леван, декстран, цианкобаламин). В слу-
чае, если иммуноэлектрофорез протекает в геле агара, путь,
пройденный молекулами белка, представляет собой результи-
рующую величину электрокинетического потенциала и электро-
осмотического потока. Поэтому электрофоретическую подвиж-
ность белка получают сложением видимой электрофоретической
подвижности и электроосмотической подвижности. Можно вооб-
ще не определять нулевой точки электроосмоса для изучаемого
белка, если на той же пластине вместе с ним мигрируют по
крайней мере два белка с известной электрофоретической по-
движностью. В этом случае подвижность исследуемого белка оп-
ределяется по следующей формуле (рис. 45):
АХ
их = иа-(иа-иь)-дв-,
114
где Ux — подвижность исследуемого белка; Ua и 15ь— подвижности известных
белков; АХ — расстояние между белками с известной и неизвестной подвиж-
ностью; АВ — расстояние между белками с известной подвижностью.
В качестве референс-белков удобно использовать сывороточ-
ный альбумин и сывороточный трансферрин человека. Абсолют-
ная подвижность сывороточного альбумина при pH 8,6 составля-
ет 6,1-10-5 см2 В-1 сек-1, трансферрина — 3,1-10~5 см2 В-1 сек-1,.
Относительную электрофоретическую подвижность определяют
как подвижность исследуемого белка по сравнению с сыворо-
точным альбумином. Подвижность сывороточного альбумина
принимают равной 1,0.
2.5.5. СПИСОК ЛИТЕРАТУРЫ
1. Clausen J. Immunochemical techniques for the identification and estimation
of macromolecules (2-nd edition). Amsterdam, Elsevier Biomedical press,
1981.
2. Uriel J. Characterisation of precipitates in gels. — Methods Immunol. Immu-
nochem., 1971, 3, 294—317.
2.6. ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
ИММУННЫХ АГРЕГАТОВ
2.6.1, ПРИНЦИП МЕТОДА
Инкубация раствора АГ с моноспецифической сывороткой
приводит к образованию иммунных агрегатов. Эта разновид-
ность иммунных комплексов рассеивает проходящий световой
поток, интенсивность которого можно измерить прямо или кос-
венно. При турбодиметрических измерениях определяют раз-
ность AU между исходной величиной Uo и получаемой величи-
ной Ui светового потока. В случае нефелометрии определяют
количество света, отраженного под определенным углом к на-
правлению падения светового потока. Интенсивность рассеянно-
го света, или разность светового потока, прямо пропорциональ-
на числу и величине иммунных комплексов [1, 2]. Соблюдая
постоянные объемы реагирующих растворов, разведение сыво-
ротки, время и температуру инкубации, можно определять кон-
центрацию антигена в растворе при помощи калибровочной кри-
вой. Калибровочную (стандартную) кривую получают, измеряя
рассеяние света в растворах с известным содержанием антигена.
На рис. 46 представлен ход лучей в лазерном нефелометре. Осо-
бая геометрия устройства позволяет улавливать свет, рассеи-
ваемый иммунными комплексами (ИК), под очень небольшими
углами. В современных лазерных нефелометрах достигается
полное поглощение света, идущего в прямом направлении; этого
эффекта не удается достичь с обыкновенными лампами ввиду
непараллельности их лучей. Высокое постоянство излучения ла-
зера позволяет получать хорошо воспроизводимые результаты^
8*
115
—| 17,77 |
Цифровой
_ l, „ _ вольтметр
Система диафрагм Кювета Диафрагма Система линз Фотодиод
Гелий-неоновый
лазер
Рис. 46. Ход лучей в лазерном нефелометре.
На других факторах, влияющих на воспроизводимость (длина
волны, качество фотодиодов, обусловленное кюветой рассея-
ния), мы не будем останавливаться, так как они влияют на ме-
тодику косвенным образом. Следует упомянуть о том, что при
турбидиметрических измерениях рекомендуется пользоваться по
возможности коротковолновым светом, поскольку он лучше рас-
сеивается по разным направлениям.
Особенно важно избегать при работе длины волн, совпадаю-
щие с областями поглощения гемоглобина (400—420 и 500нм).
Хорошо воспроизводимые результаты были получены нами при
длине волны 450 нм.
2.6.2. МЕТОДИКА
2.6.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Фосфатио-солевой буфер: 11,9 г Na2HPO4-2H2O и 9,1 г NaH2PO4
растворяют по отдельности в 1000 мл 0,15 М NaCl; 85,2 мл рас-
твора Na2HPO4 доводят до 100 мл раствором NaH2PO4; получа-
ют буферный раствор с pH 7,5. Моноспецифическая сыворотка;
стандартный раствор: стандартная сыворотка для лазерной не-
фелометрии с известным содержанием белка (смесь сывороток
здоровых доноров, стабилизированная удалением термолабиль-
ных компонентов); фреон (С2С13Рз) для осветления мутных сы-
вороток; центрифуга на 16 000 g; микрокюветы из синтетических
материалов (кюветы Sarstedt для лазерной нефелометрии); ла-
зерный нефелометр, например He-Ne лазерный нефелометр
(Behring Werke, Марбург).
2.6.2.2. ПОСТРОЕНИЕ СТАНДАРТНОЙ КРИВОЙ
Для каждого белка строят стандартную кривую на основе 5—
6 разведений. Лучше использовать геометрический ряд разведе-
ний стандартных сывороток для лазерной нефелометрии в фос-
фатно-солевом буфере с pH 7,5 от 1: 10 до 1:640. Для IgG
и IgA используют разведения 1:20— 1:640, для IgM и СЗс —
1:10—1: 160. Коммерческие препараты АС для лазерной нефе-
лометрии обычно разводят в соотношении 1:5 (50 мкл АС +
+ 200 мкл буфера) в сосудах, не содержащих даже следов
пыли. Лучше использовать стеклянные сосуды, так как пласт-
массовая посуда притягивает пыль ввиду своих электростатиче-
ских свойств. В измерительные кюветы вносят по 100 мкл раз-
116
веденного стандартного раствора и по 200 мкл различных раз-
ведений сыворотки. В случае IgG берут только 10 мкл стандарт-
ного раствора. Образцы инкубируют в течение 1 ч при комнат-
ной температуре, затем проводят измерения на лазерном нефе-
лометре. Кюветы опускают в шахту нефелометра, данные счи-
тывают с цифрового табло.
2.6.2.3. ОПРЕДЕЛЕНИЕ БЕЛКА
В СЫВОРОТКЕ БОЛЬНЫХ
Исследуемые сыворотки подвергаются осветлению (центрифуги-
рование при 16000 g в течение 10 мин) и обработке фреоном
(1 мл сыворотки смешивают с 1,5 мл фреона, перемешивают
в горизонтальном направлении, центрифугируют 10 мин при
1000 g). Такая обработка приводит к получению относительно
прозрачной сыворотки. Сыворотки разводят в отношении 1 +
+100 (50 мкл сыворотки + 5 мл буфера). К 100 мкл разведенной
сыворотки прибавляют 200 мкл, разведенной 1 + 4 антисыворот-
ки. Смесь инкубируют в кюветах, проводят измерение (табл. 6).
Таблица 6. Схема разведений и используемых объемов
Разведение стандарта Разведение АС Разведение исследуемой сыворотки Объемы реагентов
стандарт или иссле- дуемая сыворотка анти- сыворотка
IgG 1:20, 1,40, 1:80 1:60, :320, 1:640 1+4 1 + 100 10 мкл 200 мкл
1:100, 1:200, 1:400, 1:800, 1:1600 1+4 1+500 100 мкл 200 мкл
IgA 1:20, 1:40, 1:80 1:160, 1:320 1+4 1+100 100 мкл 200 мкл
IgM 1:10, 1:20, 1:40 1:80, 1:160 1+4 1+100 100 мкл 200 мкл
СЗс 1:10, 1:20, 1:40 1:80, 1:160 1+4 1+100 100 мкл 200 мкл
2.62.4. ОЦЕНКА РЕЗУЛЬТАТОВ
Получаемые на цифровом табло значения светорассеяния в
вольтах и соответствующие значения концентраций стандартных
растворов наносят на двойную логарифмическую или простую
миллиметровую бумагу. Данные измерений образуют калибро-
вочную кривую. На рис. 47 и 48 представлены калибровочные
117
Рис. 47. Калибровочная кривая для IgG.
кривые для IgG и IgM.
Одной и той же калибро-
вочной кривой можно
пользоваться, пока рабо-
та осуществляется с од-
ними и теми же реаген-
тами. Рекомендуется все
же контролировать изме-
рения со стандартной сы-
вороткой. Значения све-
торассеяния, полученные
с исследуемыми сыворот-
ками, переводят в кон-
центрацию с учетом ис-
пользованных разведений. Полученные значения должны нахо-
диться в средней части стандартной кривой, в противном случае
приходится пользоваться другими разведениями.
2.6.2.5. ОПРЕДЕЛЕНИЕ ЦИРКУЛИРУЮЩИХ
ИММУННЫХ КОМПЛЕКСОВ
Находящиеся в сыворотке иммунные комплексы могут быть оп-
ределены при помощи преципитации ПЭГ 6000 (см. раз-
дел 2.7). Исследуемые образцы сывороток после обработки
фреоном (см. выше) разводят боратным буфером в соотноше-
нии 1 + 2. Состав буфера: 6,7 г Н3ВО3, 13,4 г Na2B4O7-ЮН2О
растворяют в 1000 мл Н2О; pH 8,4. Смешивают 200 мкл разве-
денной сыворотки с 2 мл боратного буфера, во втором сосуде
смешивают 200 мкл разведенной сыворотки с 2 мл боратного
буфера, содержащего 4% ПЭГ 6000. Инкубируют 45 мин при
комнатной температуре. Проводят измерения на лазерном нефе-
лометре. Значения контроля (исследуемая сыворотка+борат-
ный буфер) не должны превышать 0,3 В. Значения светорассея-
ния в пробах, содержащих ПЭГ, соответствуют концентрации
иммунных комплексов. Результаты можно оценивать косвенно
по калибровочной кривой с искусственно полученными иммун-
ными комплексами с известным содержанием белка. Чаще
всего используют преципитирующие in vitro комплексы БСА —
анти-БСА. Для получения таких комплексов смешивают равные
количества БСА в 0,15 М NaCl и анти-БСА крысы, инкубируют
в течение 30 мин при 37 °C и разводят в соответствии с геомет-
рическим рядом.
2.6.3. ОЦЕНКА МЕТОДА
Область измерений лазерного нефелометра охватывает данные,
характерные для большинства нормальных и патологических
состояний. Правда, можно предположить, что в случае парапро-
118
теинемий количество ан-
тигена будет соответство-
вать зоне избытка АГ
(восходящая часть кри-
вой Гейдельбергера). Из-
быток АГ можно опреде-
лить очень просто. После
считывания результатов
реакции к смеси добавля-
ют 100 мкл разведенной
антисыворотки. Через
15 мин производят по-
вторное измерение. Если
результаты повышаются
более чем на 25%, то
Рис. 48. Калибровочная кривая для IgM.
имеет место избыток антигена. В этом случае измерения прово-
дят с более высокими разведениями. Лазерная нефелометрия
в последние годы получает все более широкое распространение.
Точность метода очень высока. Коэффициент вариации, по дан-
ным литературы, колеблется для одной серии в пределах
1-7%.
Ежедневные повторные измерения указывают на вариацию
в пределах 4—10% [1, 5]. Эти результаты можно еще улучшить,
если автоматизировать анализ. Существует очень высокая кор-
реляция между данными радиальной иммунодиффузии и лазер-
ной нефелометрии [1]. Наибольшие отклонения наблюдаются
при использовании мутных сывороток. Мутность не всегда
можно устранить соответствующими процедурами. С этим всег-
да приходится считаться в лабораторных исследованиях.
2.6.4. СПИСОК ЛИТЕРАТУРЫ
1. Conrad A., Schurman J., Kreuz F. Н., Sieber A. Elaboration of a method for
the quantitative determination in the measurement of serum proteins by lazer
nephelometry in the clinical routine laboratory. — Clin. Chem., 1978, 16, 299—
305.
2. Gressner A. M., Wallraf P. Der Einsatz der Laser-Nephelometrie zur bestim-
mung und rechnerunterstiitzen. Auswertung der Fibronectinkonzentration in
verschidenen Korperfliissigkeiten. — J. Clin. Chem. Clin. Biochem., 1980, 18,
797—805.
3. Gross J. Nephelometry: Principles and Methods. Abstracts of the Behring
Symposium, International Meeting on Nephelometry, Paris, 22—23, 11, 1980.
4. Lakomec H. J., Jacobi E., Husmann K., Krusketnper H. L. Zur Diagnostik des
systemishen Lupus erythematosus. — Dtsch. med. Wschr., 1979, 104, 980—
982.
5. Munster P. J. J., van Hoelen G. E. J. M., Samwel-Manting M., Holtman-van-
Meurs M. A turbidimetric immunoassay (T1A) with automated individual
blank compensation. — Clin. Chem. Acts, 1977, 76, 377—388.
6. Ripoll J. P., Roch A. M., Quash G. A., Grange J. An automatic continuous flow
method of determination of antipelyamine antibodies in human sera. — J. Im-
munol. Methods, 1980, 33, 159—173.
7. Ziegler G. B., Strobel G. J. Laser nephelometrische Plasma-proteinbestimmung:
Immunokomplexe als Fehlerquellen. — Diagnostic Intens intherapie, 1978, 3,
74—78.
2.7. ОПРЕДЕЛЕНИЕ РАСТВОРИМЫХ
ИММУННЫХ КОМПЛЕКСОВ
2.7.1. ПРИНЦИП МЕТОДА
Экзо- или эндогенные АГ могут образовывать в организме им-
мунные комплексы (ИК) с соответствующими АТ. Этот процесс
может стать причиной системной или органной патологии. Судь-
ба циркулирующих ИК зависит от их величины и от индивиду-
альной активности фагоцитирующей системы. АТ в составе ИК
могут включать каскад активации комплемента; кроме того,
IgG- и IgM-антитела могут оказывать влияние на функции
клеток, связываясь с их Fc-рецепторами [11]. Во многих рабо-
тах описано повышение концентрации циркулирующих ИК при
системных, аутоиммунных и инфекционных заболеваниях (леп-
ра, шистосомиаз), а также при злокачественных новообразова-
ниях. Значение анализа растворимых ИК для постановки диаг-
ноза и понимания патогенеза еще раскрыто не до конца [14].
Величина и состав растворимых ИК зависят как от свойств АГ,
так и от свойств АТ и в то же время от их относительной и аб-
солютной концентрации [1]. Высокомолекулярные растворимые
ИК преимущественно образуются из олиговалентных АГ и
IgM-антител. Соотношение АГ/АТ в составе ИК зависит от от-
носительных концентраций обоих компонентов; образующиеся
при избытке АГ иммунные комплексы, как правило, меньше по
размеру, чем формирующиеся в зоне эквивалентности. В отли-
чие от олиговалентных поливалентные АГ легче преципитиру-
ются избытком АТ. Взаимосвязь авидности и величины ИК, об-
разующихся при использовании анти-ДНК-антител, можно про-
следить на гибридах NZB/WF1 [12]. Наши знания о природе АГ,
входящих в состав ИК, еще недостаточны. В некоторых случаях
в составе ИК находили ДНК, инсулин, IgG и антигены вирусов
гепатита; и все же антигенный состав ИК, как правило, не уда-
ется охарактеризовать полностью. Специфические АГ можно
обнаружить только в случае довольно ограниченного числа за-
болеваний. Принято считать, что ИК, возникающие в условиях
небольшого избытка АГ, представляют наибольшую опасность
ввиду длительности их циркуляции и высокой комплементакти--
вирующей способности [10].
2.7.2. МЕТОДИЧЕСКИЕ ВАРИАНТЫ
В 1976—1981 гг. было описано около 40 различных тест-систем.
Это позволило провести в рамках международных исследований
сравнение 18 различных методов определения ИК. Только 6 из
них оказались пригодными в отношении точности, специфично-
сти, чувствительности и воспроизводимости результатов [7].
Вместо широко применявшихся ранее физических методов в на-
стоящее время чаще используют биологические.
120
Таблица 7. Методы определения растворимых НК, не основанные
на антигенной специфичности
Физические
1. Гель-фнльтрация
2. Центрифугирование в градиенте плотности
3. Криопреципитация
4. Осаждение ПЭГ
Биологические
Бесклеточные системы, взаимодействие с:
1. Комплементом
2. Конглютиннном
3. Ревматоидным фактором
Системы с использованием клеток:
1. Взаимодействие с Fc-рецепторами
1.1. Проба на агрегацию тромбоцитов
1.2. Торможение АЗКЦ
1.3. Тест торможения активности макрофагов
2. Взаимодействие с С-рецепторами: тест с клеточной линией Raji
3. Взаимодействие с протеином А
2.7.2.1. ОСАЖДЕНИЕ ПЭГ
Метод [2] основан на различной растворимости мономеров ИГ
в составе ИК при наличии в среде полиэтиленгликоля (ПЭГ)
6000. Исследуемые сыворотки хранят при 4 °C после добавления
0,02% азида натрия. Сыворотки разводят 0,1 М боратным буфе-
ром (pH 8,4) в соотношении 1 :24. К 4 мл разведенной сыворот-
ки добавляют равный объем 7% раствора ПЭГ 6000 в 0,1 М
боратном буфере и выдерживают смесь при 4 СС в течение 18 ч.
После центрифугирования при 20 000 g в течение 20 мин осадок
1 раз отмывают 3,5% раствором ПЭГ, затем растворяют его
в 5 мл 0,1 М NaOH. Содержание белка определяют по Лоури
или по оптической плотности при длине волны 280 нм. В нор-
мальных сыворотках преципитаты не дают значений оптической
плотности более 0,12. Оценка результатов: как и в случае ульт-
рацентрифугирования, гель-фильтрации и криопреципитации,по-
ложительный ответ можно дать только после дополнительного
определения IgG или Clq в сыворотках или преципитатах. Этот
метод, широко применяемый в лабораториях, подвергся в по-
следнее время критике [5], авторы даже объявили метод совер-
шенно непригодным. Они полагают, что ПЭГ следует приме-
нять только для получения ИК, которые в дальнейшем будут
подвергнуты более детальному иммунохимическому анализу.
2.7.2.2. ТЕСТ НА СВЯЗЫВАНИЕ Clq
Антитела (IgG 1, 2, 3 и IgM), входящие в состав ИК, и обла-
дающие свойством связывать Clq, можно легко обнаружить при
помощи Clq, меченного 125[1]. Выделение ИК со связанным
121
Рис. 49. Стандартная кри-
вая, построенная с исполь-
зованием термоагрегиро-
ванного у-глобулина, в
7S-IgG.
Суспензия в нормальной сыво-
ротке в присутствии ЭДТА. 1 —
в нормальной сыворотке; 2 — в
сыворотке, прогретой при 66 °C;
3 —7S-IgG в нормальной сыво-
ротке.
12S[I]-Clq проводят
в присутствии 2,5%
ПЭГ 6000. Clq с со-
храненной реактив-
ностью получают из
сыворотки человека,
используя Н2О2 и лак-
топероксидазу. Приме-
няемый для анализа препарат должен обладать специфической
активностью около 1 мкКи/мкг. Если он разделен на аликвоты
по 10 мкКи и хранится при —70 °C, его можно считать пригод-
ным для работы в течение 6 нед. Перед употреблением 10 мкКи
125[I]-Ciq смешивают с 5 мл веронал-NaCl буфера (pH 7,2,
1% БСА) и центрифугируют в течение 40 мин при 18 000 g.
Из надосадочной фракции отбирают пробы по 50 мкл (около
0,1 мкКи) в каждую пробирку. К 50 мкл исследуемой сыворотки
добавляют 100 мкл раствора ЭДТА (0,2 МЭДТА pH 7,5) и ин-
кубируют 30 мин при 37 °C. После охлаждения смеси до 0°С
к ней прибавляют 50 мкл 125[I]-Clq и 1 мл 3% раствора ПЭГ
(30 г ПЭГ 6000 в 1 л 0,1 М Н3ВОз, 0,025 М тетраборат Na,
0,075 М. NaCl, pH 8,3). Пробиркам со смесью дают постоять
60 мин и центрифугируют их в течение 20 мин при 1500 g, над-
осадочную фракцию отсасывают и отбрасывают, измеряют ра-
диоактивность. Полученное значение радиоактивности относят к
количеству 125[I]-C[I]q, осаждаемому 1 мл 20% ТХУ. Ранее для
уменьшения неспецифической преципитации 125[I]-Clq применяли
прогревание сывороток в течение 30 мин при 56 °C. Использова-
ние ЭДТА позволяет повысить чувствительность метода до
50 мкг, а также предотвратить как переход в иммунные комп-
лексы Clq-связывающих компонентов из сыворотки, так и рас-
пад существующих ИК (рис. 49). Оценка получаемых результа-
тов: использование термоагрегированного человеческого IgG в
качестве стандарта позволяет повысить чувствительность мето-
да до 50 мкг, однако это не всегда бывает достаточно. Особое
внимание следует уделять чистоте препаратов Clq. Присутству-
ющий в них в качестве примеси IgG может соединяться с ревма-
тоидным фактором и симулировать наличие ИК. Потенциально
с Clq могут также связываться ДНК, гепарин, различные поли-
сахариды и фибриноген.
122
2.7.2.3. ТВЕРДОФАЗНЫЙ АНАЛИЗ
С ИСПОЛЬЗОВАНИЕМ Clq
Адсорбированный на твердую фазу Clq сохраняет способность
связываться с Fc-концом IgG 1, 2, 3 и IgM., входящих в состав
ИК. Использование твердой фазы позволяет сравнительно прос-
то удалять компоненты, не связывающие Clq [3]. Адсорбция до-
стигается инкубацией в течение 3 дней при 4 °C пластиковых
пробирок с 1 мл раствора Clq (10 мг человеческого Clq в 1 л
ФСБ pH 7,2; 0,01 М Na2HPO4, 0,15 М NaCl). Пробирки трижды
ополаскивают холодным ФСБ и инкубируют с ФСБ, содержа-
щим 0,1% раствор желатина. Затем проводят троекратное опо-
ласкивание ФСБ, после чего пробирки готовы к использованию
(их можно хранить в холодильнике). К 50 мкл исследуемой сы-
воротки добавляют 100 мкл 0,2 М ЭДТА pH 7,5, инкубируют
смесь в течение 30 мин при 37 °C. После охлаждения до 0 °C
отбирают аликвоты различных разведений сыворотки объемом
50 мкл, смешивают их с 950 мкл 0,05% раствора твин-20 в ФСБ
(ФСБ-твин) и вносят в «сенсибилизированные» Clq пробирки,
которые затем инкубируют 1 ч при 37 °C и еще 30 мин при
4 °C. Несвязавшийся материал удаляют 3—4-кратным ополаски-
ванием 1,5 мл ФСБ-твина, 125[I]-IgG человека в количестве
1 мкг, содержащего специфические антитела (около
0,25 мкКи/мл), растворяют в 1 мл ФСБ-твина, вносят в пробир-
ки и инкубируют их в течение 1 ч при 37 °C, затем еще в течение
30 мин при 4 °C. После удаления 12S[I]-AT измеряют радиоак-
тивность твердой фазы. Неспецифическое связывание 12S[I]-ан-
тител проверяют на пробирках, «нагруженных» только жела-
тином. Если вместо желатина использовать БСА, а концентра-
цию твин-20 повысить до 0,1%, то неспецифическое связывание
уменьшается до такой степени, что им можно пренебречь [3].
Связывание 1g человека с адсорбированным Clq в области кон-
центраций 0,1—10 мкг
носит в отсутствие мо- - ioooo-j
номерного IgG линей- §
ный характер (рис. 50). f
В присутствии 25 мкл °
нормальной человече- s
ской сыворотки этим |
методом удается выя- S.
св
О
О
X
Рис. 50. Связывание агре- g
тированного человеческого “
IgG с Clq, адсорбирован- «
ным на пластиковых про- §
бирках. «
Меченный 125[1] агрегированный х
IgG вносили в различных кон-
центрациях, степень связывания
определяли после отмывки.
123
Рис. 51. Определение агрегирован-
ного IgG в присутствии 25 мкл
нормальной сыворотки человека.
вить 1 мкг Ig человека; пока-
затель связывания выходит на
плато при 25—50 мкг Ig че-
ловека (рис. 51).
Оценка получаемых
результатов. Как упоми-
налось выше, чувствительность
метода на модели Ig человека
составляет 1 мкг/мл. По срав-
нению со связыванием Clq
(меченным 1251) в жидкой фазе
становятся весьма заметными
искажения, вносимые состав-
ными частями сыворотки, не
относящимися к ИК. Приме-
няемый для работы Clq не
должен содержать примесей
иммуноглобулинов. Отсутствие
ИГ следует устанавливать ме-
тодом ИЭФ с моно- и полиспецифическими АС. Было предложе-
но использовать в качестве твердой фазы пластиковые шарики
[9], при этом отмечалось снижение количества адсорбируемого
Clq на 10%, что существенно снижало издержки анализа. Дру-
гим вариантом твердофазного связывания с Clq может стать
применение энзим-меченных антител [13]. Использование
в анализе класс-специфических АТ позволяет отдифференциро-
вать IgG от IgM в составе ИК- Хорошим индикатором может
стать 125[1]-протеин А, заменяющий 125[I]-IgG против АГ чело-
века.
2.7.2.4. СВЯЗЫВАНИЕ ИММУННЫХ КОМПЛЕКСОВ
СО СТАФИЛОКОККОМ А
Клеточные системы для количественной оценки связывания рас-
творимых ИК можно разделить на две группы в соответствии
с задействованными клеточными рецепторами. В тесте с исполь-
зованием линии клеток Raji—это рецепторы типа C3(b, d).
Тромбоциты, макрофаги или К-лимфоциты связывают раство-
римые ИК за счет Fc-рецепторов. При постановке клеточных
проб Fc-рецепторного типа основное значение приобретает из-
бирательное связывание IgG, входящего в состав ИК, но не
мономерного IgG. Описана индикаторная система, в которой
первичным агентом связывания служат инактивированные на-
греванием клетки Staphylococcus aureus, штамм Cowan I [8].
Содержащийся в их наружной мембране протеин А обладает
селективными свойствами по отношению к Fc-рецепторам.
124
Рис. 52. Стандартная кривая опреде-
ления агрегированного глобулина в
присутствии нормальной сыворотки
человека.
Для реакции применяли меченный 125[П
IgG человека (1) или поливалентные спе-
цифические антитела (2) [8].
Подготовка стафи-
лококков. Штамм Cowan I
после культивации инактиви-
руют нагреванием в течение
2,5 мин при 80 °C и хранят при
—70 °C. После оттаивания
микробные клетки остаются
пригодными для работы в те-
чение 4 нед. Для постановки
пробы используют 0,8% (по
объему) суспензию микробов
в БСА — ФСБ (0,5% БСА,
0,01 М Кг НРО4, 0,15 М NaCl,
0,1% NaN3, pH 7,2).
Индикаторная система. Для количественной оценки
растворимых комплексов 50 мкл человеческой сыворотки сме-
шивают с 150 мкл ЭДТА-буфера (0,2 М натриевая соль ЭДТА,
pH 7,4), затем добавляют 1 мл 6% раствора ПЭГ 6000 в 0,1 М
боратном буфере (см. выше). Пробирки с пробами оставляют
на ночь при 4 °C, затем центрифугируют их 20 мин 1000 g. Каж-
дый осадок ресуспендируют в 1 мл 5% раствора ПЭГ и снова
центрифугируют. На этом этапе удаляется мономерный IgG.
Надосадочную фракцию сливают, осадок растворяют в 200 мкл
ФСБ; добавляют при комнатной температуре аликвоту объемом
100 мкл к такому же объему суспензии стафилококков и инку-
бируют 2 ч при 37 °C. Связывающиеся в этих условиях с про-
теином А иммунные комплексы после предварительного добав-
ления к смеси 0,5 мл 0,5% раствора БСА—ФСБ осаждаются
вместе с микробами центрифугированием при 1000 g в течение
10 мин. Перед последующей обработкой пробирки выдерживают
некоторое время при 0—4 °C. Осадки дважды отмывают БСА—
ФСБ и растворяют в 100 мкл ФСБ, содержащего 5 мг/мл IgG
кролика. Определение связавшихся ИК проводят при помощи
100 мкл препарата меченных 12S[I] кроличьих антител к IgG
человека (0,04 мкКи/мкг). Для постановки этой пробы необхо-
дим избыток меченых АТ. Меченые АТ титруют клетками золо-
тистого стафилококка, нагруженными человеческим IgG. Реак-
цию индикации проводят приблизительно в течение 12 ч при
4 °C, непрореагировавшие АТ остаются в надосадочной фракции
после центрифугирования в течение 10 мин при 1000 g. Осадок
трижды отмывают ФСБ, затем измеряют радиоактивность. При
измерениях, проводимых с 1g человека в пределах 0—200 мкгг
125
стандартная кривая имеет линейный характер; обработка осад-
ка поливалентными 125[1]-анти-^О-антителами приводит к вы-
сокому, почти 50% уровню неспецифической сорбции, вследст-
вие чего стандартная кривая приобретает уплощенный вид
(рис. 52).
Оценка результатов. Метод не подвержен искажени-
ям со стороны клеточных процессов и сывороточных антител
в отличие от тест-систем, использующих живые клетки (тест
агглютинации тромбоцитов, тест с линией клеток Raji, тест тор-
можения активности макрофагов). Будучи комплементнезависи-
мым методом, он позволяет получать результаты, которые не за-
висят от содержания комплемента в сыворотке. К недостаткам
метода следует отнести то, что он применим к ИК, содержащим
только IgG. Чувствительность по 1g человека составляет
10 мкг/мл и почти не зависит от степени агрегации белка.
2.7.3. ПРИМЕНЕНИЕ АГРЕГИРОВАННОГО
НАГРЕВАНИЕМ IgG ЧЕЛОВЕКА
В КАЧЕСТВЕ СТАНДАРТА
Оценка чувствительности методов обнаружения ИК, как прави-
ло, связана с применением стандарта по 1g человека. По таким
показателям, как активация комплемента, связывание с макро-
фагами и способность индуцировать феномен Артюса, 1g чело-
века сходен с растворимыми ИК. Применяя растворимые ИК,
можно стандартизовать различные тест-системы на ИК и выра-
жать получаемые значения в эквивалентах 1g человека. Имеют-
ся работы, посвященные изучению его характеристик и стабиль-
ности [4, 6]. Ультрацентрифугированием агрегированного при
63 °C IgG человека было показано, что степень агрегации зави-
сит от продолжительности тепловой обработки (рис. 53). Усиле-
ние процесса агрегации наблюдалось после хранения при
—70 °C. Введение 0,5% БСА заметно тормозило этот процесс.
Агрегаты 1g человека, состоящие из 48—80 молекул IgG, после
хранения в течение 15 мес не изменяли своей способности акти-
вировать комплемент и связываться в двух других тестах при
добавлении 0,5% БСА [4, 6].
Приготовление 1g человека. Растворяют 20—50 мг
мономерного человеческого IgG в 1 мл ФСБ и инкубируют
20 мин при 63 °C. Последующие операции проводят при 0—
4 °C. Легко седиментирующие агрегаты удаляют из опалесци-
рующего раствора центрифугированием в течение 15 мин при
3000 g. Термическая обработка переводит в агрегированную
форму не более 50% исходного материала. Различные способы
определения ИК неодинаково зависят от размера частиц 1g че-
ловека или ИК, поэтому построение стандартной кривой требу-
ет его фракционирования и удаления мономеров IgG. Для уст-
ранения нежелательных компонентов можно применять гель-
фильтрацию через сефакрил S-200, сефарозу 4 В, и особенно
126
Рис. 53. Седиментационный профиль агрегированного глобулина [6].
IgG человека инкубировали при 63 °C в течение 10 мни (1); 20 мии (2); 30 мни (3):
контроль (4). После удаления нерастворимых агрегатов препараты фракционировали
методом зонального центрифугирования при 263 000g в течение 2 ч (ротор SW — 40Ti)
в градиенте плотности сахарозы с добавлением Б С А.
ультрацентрифугирование Ig человека в градиенте плотности са-
харозы 10—30%. В присутствии маркеров можно получать агре-
гаты вполне определенной величины. Градиент приготовляют на
БСА-боратном буфере с pH 7,2 (0,1 М Н3ВО3, 0,14 М NaCl,
0,5% ВС А). К числу недостатков центрифугирования относится
то, что выход Ig человека составляет не более 10 мг, кроме того,
необходимо удалять сахарозу диализом.
2.7.4. ОЦЕНКА МЕТОДА
В основе методов определения растворимых ИК лежит исполь-
зование комплемента или ИГ, входящих в состав ИК, а также
С или Fc-рецепторов клеток. На чувствительность этих методов
оказывают влияние различные факторы (pH, ионная сила, раз-
мер ИК, соотношение АГ/АТ, наличие эндогенного комплемента,
природа АГ). С учетом этого становится понятной малая сопо-
ставимость 18 различных способов определения ИК [7].
Сравнительное изучение показало, что для вывода о нали-
чии растворимых ИК желательно параллельное проведение ис-
следования двумя различными способами, основанными на раз-
ных тест-системах. Для сравнения результатов желательно ис-
пользовать достаточно изученные референс-препараты (Ig чело-
века, ИК)- Достоверность наличия циркулирующих ИК может
быть повышена исследованием системы активности комплемен-
та (С-профиль) и обнаружением отложения компонентов ком-
племента или иммуноглобулинов в стенке сосудов [5].
127
2.7.5. СПИСОК ЛИТЕРАТУРЫ
1. Barnett Е. V. Circulating immune complexes: their immunochemistry, detec-
tion and importance. — Ann. Int. Med., 1979, 91, 430—440.
2. Digeon M., Laver M., Riza J., Bach J. F. Detection of circulating immune
complexes in human sera by simplified assays with polyethylene glycol. —
J. Immunol. Methods, 1977, 16, 165—183.
3. Falck P., Meffert H. et al. Radioimmunologischer Nachweis von loslichen
Immuncomplexen in Serum. — Dermatol. Monatsschr., 1979, 165, 276—281.
4. Kaufmann R. H., van Es L. A., Doha M. R. Aggregated human immunoglobu-
lin G stabilised by albumin: a standard for immune complex detection.—
J. Immunol. Methods, 1979, 31, 11—22.
5. Klein M., Simonovitch K. The significance and limitations of current methods
for detecting immune complexes. — J. Reumatol., 1981, 8, 188—192.
6. Knutson D. V., Kijlstra A., Lentz H., van Es L. A. Isolation of stable aggre-
gates of IgG by zonal centrifugation in sucrose gradients containing albu-
min.— Immunol. Comm., 1979, 8, 337—345.
7. Lambert P. H. et al A WHO collaborative study for the evaluation of methods
for detection of immune complexes in serum. — J. Clin. Lab. Immunol., 1978,
1, 1—15.
8. Me. Dougal J. S., Beredcha P. B. et al. Binding of IgG aggregated and
immune complexes in serum to staphylococci containing protein A. — J. Clin.
Invest., 1979, 62, 627—636.
9. Pohl D. A., Tsai С. C., Roodman S. T. Detection of immune complexes using
a solid phase Clq polystyrene ball assay. — J. Immunol. Methods, 1981, 40,
313—330.
10. Scherzer H„ Ward P. A. Lung injury produced by immune complexes of
varying composition. — J. Immunol., 1978, 121,947—952.
11. Sedlacek H. H. Pathophysiological aspects of immune complex deseases.—
Klin. Wschr., 1980, 58, 543—550, 593—605.
12. Steward M. W., Katz F. E., West M. J. The role of low affinity antibodies in
immune complex desease. — Clin. Exptl. Immunol., 1975, 21, 121—130.
13. Wehler C., Andrews J. M„ Bing D. H. The use of solid phase Clq and hor-
seradish peroxidase conjugated goat and anti-goat anti-IgG for the detection
of immune complexes in human serum. — Molecul. Immunol., 1981, 18, 157—
162.
14. Zubler R. H„ Lange G. et al. Detection of immune complexes in unbeaded
sera by a modified 125I-Clq binding test. — J. Immunol., 1976, 116, 232—
235.
2.8. ИМУНОФЛЮОРЕСЦЕНЦИЯ
2.8.1. ПРИНЦИП МЕТОДА
Иммунофлюоресценция — это метод, основанный на использова-
нии специфичности иммунологической реакции и чувствительно-
сти флюоресцентной микроскопии. В отличие от других методов
маркировки постановка пробы и точная локализация иммунной
реакции на поверхности клетки или ткани происходят довольно
быстро. Один из компонентов иммунной реакции, как правило
АТ, метится флюоресцирующим красителем (маркировка,
конъюгирование). После возбуждения флюоресцирующего
агента положение АГ становится доступным непосредственному
наблюдению вследствие развития флюоресценции (рис. 54).
Предложенный в 1941 г. Кунсом, этот метод после многочислен-
128
Прямой метод
Непрямой метод
Инкубация Инкубация
Объяснение условных знаков
АГ
АТ Анти-АТ
Рис. 54. Принцип иммуиофлюорес-
центного анализа.
Флюорохром Антикомплемент Комплемент
ных усовершенствований прочно вошел в лабораторную прак-
тику. Метод включает следующие важные этапы:
— получение, характеристика и маркировка антисывороток;
— получение антигенного субстрата;
— проведение анализа, т. е. обработка флюоресцирующими АТ;
— микроскопический анализ флюоресценции препаратов.
В настоящее время используют преимущественно коммерче-
ские препараты флюоресцирующих АС. Если необходимых
флюоресцирующих сывороток нет, можно воспользоваться не-
прямым методом флюоресцентного маркирования, присоединив
к исследуемому объекту АГ, к которому имеются меченые АТ.
Очень редко приходится разрабатывать собственный препарат
меченых флюоресцентным красителем АТ. Работа с флюорес-
цирующими АТ, особенно оценка получаемых с их помощью ре-
зультатов, требует определенных навыков. Всегда следует про-
водить анализ с учетом неспецифической или нежелательной
специфической флюоресценции. Необходимо учитывать факто-
ры, которые могут оказывать влияние на флюоресцентный ана-
лиз. К ним относятся:
— желательная специфическая флюоресценция: специфическое
связывание меченых АТ с соответствующим АГ;
— неспецифическая флюоресценция: неспецифическое отложе-
ние флюоресцирующих конъюгатов на различных тканевых
структурах, например, вследствие избытка красителя или
взаимодействия меченых АТ с Fc-рецепторами; включение
метки в другие сывороточные белки, кроме 1g, и неспецифи-
ческое связывание этих белков, например альбумина, фибро-
нектина, аутофлюоресценция;
9-990
129
— нежелательная специфическая флюоресценция: наличие пе-
рекрестных реакций или недостаточная специфичность са-
мого конъюгата. В случае прямого иммунофлюоресцентного
анализа возможно неспецифическое отложение меченых АТ.
Путем соответствующего контроля (см. ниже) удается избе-
жать ложных результатов, обусловленных присутствием посто-
ронней иммунофлюоресценции.
2.8.2. МАРКИРОВКА ИММУНОГЛОБУЛИНОВ
Получение и очистка АС описаны в разделах 1.1 и 1.2. Для по-
лучения у-глобулиновой фракции можно использовать 4—5-
кратное переосаждение сульфатом аммония. Избыток соли ме-
шает маркировке и должен быть предварительно удален диали-
зом. Перед маркировкой необходимо также определить реактив-
ность АТ (см. разделы с 2.1 по 2.4) и содержание белка в пре-
парате. Практически для всех экспериментов можно в качестве
флюоресцирующего красителя использовать ФИТЦ (флюорес-
цеинизотиоцианат). Для двойной маркировки можно использо-
вать добавочное введение ТРИТЦ (тетраметилромадинизотио-
цианат). Спектр флюоресценции этих красителей различен. Ус-
ловия их конъюгирования с Ig одинаковы. Мечение Ig не долж-
но сопровождаться снижением их специфической активности.
Увеличение числа молекул красителя, связанных с каждой мо-
лекулой Ig, одновременно повышает чувствительность и усили-
вает неспецифические реакции с таким конъюгатом. Оптималь-
ное молярное соотношение флюорохрбм/Ig составляет 2—3 для
ФИТЦ и 1—2 для ТРИТЦ [5, 13]. Известны краткосрочное
(2 ч при комнатной температуре) и длительное маркирование
(12—18 ч при 4°C), которому мы отдаем предпочтение. После
введения метки в молекулу Ig избыток красителя удаляют.
Различными видами очистки, например адсорбцией ацетоновым
порошком тканей, можно удалить АТ, содержащие наибольшее
количество красителя. Рекомендуется для каждого конъюгата
определять молярное соотношение краситель/белок.
2.8.2.1. МАРКИРОВАНИЕ БЕЛКОВ ФИТЦ И ТРИТЦ
2.8.2.1.1. Материалы и оборудование
ФИТЦ, изомер I (например, фирмы Baltimore Biological, США)
или ТРИТЦ; 0,1 М. Na2HPO4; диметилсульфоксид (ДМ.СО) (на-
пример, Реахим, СССР); карбонат-бикарбонатный буфер с
pH 9,5 (17,3 г NaHCO3, 8,6 г Na2CO3 в 1000 мл дистиллирован-
ной Н2О); 0,01 М и ФСБ с pH 7,2 (1,72 г Na2HPO4‘2H2O, 0,5 г
КН2РО4, 7,2 г NaCl, дистиллированная Н2О до 1000 мл); 0,01 М
фосфатный буфер с pH 7,5; сефадекс G-25 средний (Pharmacia,
Швеция); ПЭГ 20000 (Ferak, Зап. Берлин). Диализный матери-
ал, магнитная мешалка, хроматографическая колонка [4].
130
2.8.2.1.2. Методика
Перед маркировкой раствор белка (концентрация 15—25 г/л)
в течение ночи диализуют при 4 °C против карбонат-бикарбо-
натного буфера. На каждый грамм белка расходуется 12,5 мг
флюоресцентного красителя (ФИТЦ, ТРИТЦ), этим достигается
оптимальное соотношение краситель/белок. Растворяют 1 мг
ФИТЦ в 2 мл 0,1 М Na2HPO<, 1 мг ТРИТЦ растворяют в 1 мл
ДМСО. Раствор красителя медленно, по каплям, добавляют
к раствору белка при постоянном помешивании. Необходимо
контролировать и поддерживать pH на уровне 9,3. Конъюгиро-
вание продолжается в течение 18 ч при 4 °C в темноте. После
окончания процесса избыток красителя удаляют гель-фильтра-
цией на колонке с сефадексом G-25. Сефадекс кипятят в тече-
ние 4 ч, густую суспензию заливают в колонку, нижнее отвер-
стие которой закрыто нейлоновой сеткой или стеклянной ватой.
После того как сефадекс осядет, колонку заполняют и уравно-
вешивают ФСБ. Раствор, содержащий меченые белки и свобод-
ный краситель, осторожно наносят на колонку, колонку промы-
вают ФСБ. Меченые белки движутся через сефадекс быстрее,
чем свободный краситель, и при прохождении через колонку
видны в виде окрашенной полосы. Для концентрирования по-
лученных фракций мы рекомендуем диализ против раствора
ПЭГ (250 г ПЭГ на 1 л 0,01 М фосфатного буфера с pH 7,5).
Раствор концентрируют до нужной степени и собирают. Колонку
с сефадексом можно многократно использовать для фильтра-
ции. В перерывах между работой к раствору, заполняющему ко-
лонку, добавляют консервант (мертиолат).
2.8.2.2. ФРАКЦИОНИРОВАНИЕ
И ОРИЕНТИРОВОЧНОЕ ОПРЕДЕЛЕНИЕ
СООТНОШЕНИЯ КРАСИТЕЛЬ/БЕЛОК
2.8.2.2.1. Материалы и оборудование
ДЭАЭ-целлюлоза 0,6—0,8 мэкв/г (Ренал, Будапешт), 0,02 М
фосфатный буфер с pH 7,5, растворы NaCl (0,1 М; 0,2 М; 0,4 М;
0,6 М; 0,8 М). Маленькая хроматографическая колонка или
пластиковый шприц. Спектрофотометр UV vis (Zeiss, йена)
[4, 7].
2.8.2.2.2. Методика
ДЭАЭ-целлюлозу уравновешивают 0,01 М фосфатным буфером
с pH 7,5, заполняют сорбентом колонку. На колонку наносят
исследуемую фракцию меченых белков. Вначале колонку про-
мывают буфером без NaCl, затем буферными растворами с воз-
растающей концентрацией соли (0,05 М; 0,1 М; 0,2 М; 0,3 М;
0,4 М). Растворы для элюции получают смешиванием фосфат-
9*
131
Таблица 8. Ориентировочное определение концентрации белка (Кб)
и молярного соотношения краситель/белок (Кр/б) конъюгатов путем
измерения оптической плотности (ОП) на различных длинах волн [4, 12].
Конъюгаты белок— ФИТЦ разводят 0,1 М NaOH, конъюгаты
белок —ТРИТЦ—ФСБ
ФИТЦ ТРИТЦ
v, . , , ОП280—(0,35x017455)
Кб (мг/мл) =------------------—-—
1 >4
Ко/б = 2’87хОПш
р/ ОП280-(0,35ХОП40.)
При оп- ОПл».
тималь- ——>0,5<Ч,5
ном Кр/б ОП280
пгт (O>56xnsso
^280— г-:
1 >4
____4,47хОГТ55о____
ОП288—(0,56x017550
>.я п
ного буфера и растворов NaCl в соотношении 1 + 1. Белки, ме-
ченые ФИТЦ, сходят с колонки с 0,4 М NaCl, белки, меченые
ТРИТЦ, —с 0,05—0,01 М NaCl.
Удаление избыточно меченых АТ можно осуществить осаж-
дением протаминсульфатом [5]. Для точного определения ко-
личества белка мы рекомендуем пользоваться биуретовым ме-
тодом. Ориентировочно количество белка можно определить
спектрофотометрически, используя в качестве стандарта рас-
твор у-глобулина. Содержание ФИТЦ определяют, используя
в качестве стандарта раствор флюоресциндиацетата. Зная коэф-
фициент экстинкции ФИТЦ и ТРИТЦ, можно определить ко-
личество связанного красителя и молярное соотношение краси-
тель/белок (табл. 8).
2.8.2.3. АДСОРБЦИЯ ПОРОШКОМ ТКАНИ
Если конъюгат в прямой реакции с тканью дает неспецифиче-
скую или нежелательную специфическую флюоресценцию, то
адсорбцией конъюгатов порошком соответствущей ткани можно
устранить это явление. Этот метод оказался полезным при об-
работке коммерческих препаратов, дающих перекрестные реак-
ции с тканями различных животных.
2.8.2.З.1. Материалы и оборудование
Необходимо иметь свежую мышиную или крысиную печень,
ФСБ с pH 7,2 ацетон, гомогенизатор, термостат, марлю.
2.8.2.3.2. Методика
Извлекают печень мыши или крысы (промывают ее перед из-
влечением холодным ФСБ через систему нижней полой вены),
измельчают ножницами, затем гомогенизируют.
Гомогенат ткани фильтруют через несколько слоев марли,
марлю аккуратно выжимают. К фильтрату при постоянном по-
132
мешивании добавляют 4-кратное количество ацетона. После
центрифугирования (20 мин, 4000 g) осадок промывают дистил-
лированной водой до полного исчезновения следов гемоглобина.
Осадок высушивают ацетоном ,3—5 раз (суспендируют в ацето-
не и центрифугируют 20 мин при 2000 g). Порошок сушат в те-
чение ночи при 37 °C, разделяют на порции и хранят без досту-
па воздуха при 4 °C. Адсорбцию проводят, смешивая 50 мг су-
хого порошка печени с 1 мл конъюгата и инкубируя их в тече-
ние 1 ч при комнатной температуре. Порошок ткани удаляют
центрифугированием в течение 30 мин при 15000 g. Адсорбиро-
ванный конъюгат диализуют, разделяют на порции и лиофили-
зируют.
2.8.2.4. ХРАНЕНИЕ СЫВОРОТОК И КОНЪЮГАТОВ
Конъюгаты и сыворотки можно хранить без потери ими актив-
ности при —20 °C в течение года и более. Повторное замора-
живание и оттаивание снижает активность в меньшей степени,
чем хранение при комнатной температуре или микробный про-
рост. Порции, на которые разливают препараты при хранении,
не должны быть слишком маленькими, иначе происходит холо-
довое высушивание образцов, которое резко снижает актив-
ность. Идеальным способом хранения конъюгатов является лио-
филизация и последующее хранение при 4 °C. Сильно разве-
денные сыворотки или конъюгаты рекомендуется использовать
в день разведения. При хранении материала при 4 °C рекомен-
дуется добавлять консервант (мертиолат).
2.8.3. ПОЛУЧЕНИЕ АНТИГЕННЫХ СУБСТРАТОВ
В качестве антигенных субстратов, как правило, используют
срезы ткани, клетки, клеточные органеллы. Во время подготов-
ки объекта следует обращать внимание как на морфологиче-
скую сохранность объекта, так и на сохранность антигенного со-
става. В большинстве случаев приходится отказываться от
фиксации тканей и срезов. Если все же фиксация необходима
в силу отсутствия криотома, растворимости АГ или невозмож-
ности удержать объект на предметном стекле, то необходимо за-
ранее определить, какое воздействие оказывает фиксатор на ан-
тигенность. При плохом прилипании объекта к стеклу можно по-
пытаться очистить его нитросерной кислотой, обработать смесью
хромовых квасцов с желатином, формваром (Serva) или мета-
силикатом [6]. Перед работой анализируемые препараты под-
сушивают на воздухе в течение 10—20 мин. Препараты можно
сохранять в отсутствие доступа воздуха при —20 °C, если пред-
варительно дать им подсохнуть при 40—50 °C в течение 30 мин.
Известны способы консервации антигенов на срок более года
[7]. После осторожного подсушивания препарат нагревают
в течение 10 мин при 100 °C и осторожно завертывают в алю-
133
I
---Стеклянная
палочка
Рис. 55. Специальные приспособления для им-
мунофлюоресцентного анализа.
а—устройство для шок-замораживания (медный ци-
линдр, погружаемый в сосуд Дьюара с жидким азо-
том); б — инкубационная камера; в — устройство для
получения отпечатков ткаии; г — штамп для пред-
метных стекол; д — устройство для повторного отыс-
кания микрообъектов.
миниевую фольгу. Препараты сохраняют при —20 °C, перед
употреблением длительно акклиматизируют. Для быстрого ана-
лиза препарата рекомендуют окрашивание акридиновым оран-
жевым (100 мг/л, 10 с, отмывка водопроводной водой); рассмат-
ривают в тех же условиях, что и препараты, окрашенные
ФИТЦ. В зависимости от величины препарата приходится про-
сматривать различное число полей зрения. Маленькие объекты
удобно рассматривать на специальных шаблонах (рис. 55).
2.8.З.1. ПРИГОТОВЛЕНИЕ КРИОСРЕЗОВ
2.8.З.1.1. Материалы и оборудование
Для работы необходимы: изопентан (Fluka), жидкий азот, ОСТ
(Miles, США); предметные стекла нефлюоресцирующие, очи-
щенные нитросерной кислотой (20 г нитрата калия в 1 л кон-
центрированной серной кислоты); криотом.
2.8.3.1.2. Методика
Образцы только что полученной ткани (толщиной не более
5 мм) подвергают шок-замораживанию изопентаном, охлажден-
ным жидким азотом. Для охлаждения можно также использо-
вать смесь ацетона и сухого льда. Небольшие кусочки ткани
(например, биопсийный материал) перед замораживанием за-
ключают в ОСТ, 20% желатин или печень крысы. Высыхание за-
мороженной ткани можно предотвратить, помещая образцы в
небольшие пластиковые пакетики или сосуды. В случаях быст-
рого использования (до 8 нед) ткань можно хранить при
—20 °C. Если возникает необходимость более длительного хра-
нения, рекомендуется использовать жидкий азот. Криосрезы
толщиной 4—6 мкм прикрепляют к предметному стеклу и обра-
батывают, как описано выше.
2.8.3.2. ПРЕПАРАТЫ КУЛЬТУР КЛЕТОК,
МАЗКИ И ОТПЕЧАТКИ
Из суспензии культуры клеток (оптимальное число клеток
в 10 мкл — 5-104Хувеличение объектива) можно приготовлять
мазки для иммунофлюоресцентного анализа. Для предотвраще-
ния смывания клеток при последующих процедурах необходимо
проводить специальную обработку стекла (см. выше) или фик-
сировать препарат. Клеточная суспензия лучше распределяется
по стеклу при добавлении сывороточного альбумина в количест-
ве 5 г/л. После этого необходима фиксация препарата. Культу-
ры клеток на покровных стеклах очень удобны для иммуно-
флюоресцентного анализа. Покровные стекла перед анализом
укрепляют на предметных стеклах пиафлексом или энтелланом
(см. ниже). Отпечатки особенно удобно использовать для изу-
135
чения ядер. Поверхностью свежего среза несколько раз прикаса-
ются к предметному стеклу. Перез каждым новым срезом кусо-
чек ткани несколько выдвигают из держателя (см. рис. 55,в).
2.8.3.3. СРЕЗЫ ПАРАФИНИРОВАННЫХ ТКАНЕЙ
Если необходимо очень высокое качество гистологических пре-
паратов или отсутствует криотом, то образцы тканей можно
в добавление к фиксации спиртом или формалином [8] заклю-
чать в парафин. Влияние этих манипуляций на иммунологиче-
ские свойства АГ выяснены еще недостаточно. Если в резуль-
тате фиксации антигены маскируются, обработка ферментами
может частично или полностью восстановить их доступ-
ность [8].
2.8.4. ИММУНОФЛЮОРЕСЦЕНТНЫЙ АНАЛИЗ
Известны прямой и непрямой методы иммунофлюоресцентного
анализа (см. рис. 54). В прямом методе искомый антигенный
субстрат отыскивают в ткани посредством соответствующей АС.
Сам антиген может быть антителом; в таком случае конъюгат
будет представлять собой меченое анти-антител о. Непрямой ме-
тод применяется при определении антител (антигенов) в жид-
костях или в том случае, когда, как упоминалось выше, нет под-
ходящей меченой антисыворотки. Специальной формой непря-
мого метода является фиксация комплемента антителами. Чув-
ствительность метода, возможность ошибок и количество необ-
ходимых контрольных операций возрастают параллельно ус-
ложнению процедуры иммунофлюоресцентного анализа. В ходе
анализа реакция АГ—АТ протекает непосредственно на суб-
страте, все непрореагировавшие компоненты удаляют смыва-
нием.
2.8.4.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы ФСБ с pH 7,2; лепиналовый буфер с
pH 7,2; 0,575 г лепинала, 0,185 г лепинал-натрия, 0,028 г СаС1,
0,168 г MgC12-6H2O, 8,5 г NaCl, дистиллированная вода до
1000 мл. Среда для заключения ткани: глицерин на лепинало-
вом буфере с pH 8,6 (10,3 г лепинал-натрия, 6,2 г NaCl, дистил-
лированная вода до 1000 мл). Подводят pH до 8,6 HCI и смеши-
вают с глицерином в соотношении 1 + 3. Также нужны пипетки
с поршнем, инкубационная камера (см. рис. 55), кюветы для
окрашивания, устройство для встряхивания.
2.8.4.2. МЕТОДИКА
Растворы при помощи поршневых пипеток осторожно наносят
на препараты, следя за тем, чтобы они были полностью покры-
ты жидкостью. Инкубацию проводят при комнатной температу-
136
ре во влажной камере в течение 30 мин. После инкубации рас-
творы отсасывают очень осторожно, чтобы не сдвинуть с места
соседние препараты. Отмывают препараты фосфатно-солевым
буфером в кюветах, на аппарате для встряхивания. Буфер меня-
ют с различной частотой в зависимости от цели исследования
(см. ниже).
Прямой метод иммунофлюоресцентного анализа
— Препарат отмывают ФСБ в течение 10 мин (однократная
смена буфера), высушивают.
— Инкубируют препарат с различными разведениями конью-
гата.
— Препарат промывают ФСБ в течение 30 мин (троекратная
смена буфера).
— Препарат высушивают и заключают в подходящий мате-
риал.
Непрямой метод иммунофлюоресцентного анализа
— Препарат инкубируют с исследуемым раствором.
— Отмывают ФСБ в течение 20 мин (двухкратная смена бу-
фера).
— Инкубируют с различными разведениями конъюгата.
— Отмывают в ФСБ в течение 30 мин (троекратная смена бу-
фера).
— Препарат высушивают, заключают в подходящий материал.
Определение комплементсвязывающих антител
После первой инкубации с инактивированной сывороткой
(30 мин, 56°C), последующей отмывки, как при прямом иммуно-
флюоресцентном анализе, и высушивания проводят следующие
манипуляции:
— Инкубация со свежей человеческой сывороткой (разведение
1+9) в качестве источника комплемента.
— Отмывка в фера). ФСБ в течение 20 мин (двухкратная смена бу-
— Инкубация с антителами к комплементу человека (СЗ), ме- чененными ФИТЦ.
— Отмывка в ФСБ в течение 30 мин (троекратная смена бу-
фера).
— Высушивание и заключение в подходящую ткань.
Комплемент рекомендуется разливать на небольшие порции и
хранить в жидком азоте. Нужную порцию комплемента размо-
раживают перед употреблением и разводят лепиналовым буфе-
ром с pH 7,2.
Применение двойной флюоресцентной метки
Для одновременного анализа различных АГ в одной и той же
ткани, например в клетках одного типа, применяют антисыво-
ротки, меченные различными флюорохромами (ФИТЦ,ТРИТЦ).
137
Примеры использования этого метода приведены в главе 3.2,
Такая маркировка применяется при диагностике лимфом и точ-
ного анализа антигенов, вызывающих образование аутоантител.
2.8.4.3. ОКРАШИВАНИЕ ДОПОЛНИТЕЛЬНЫМ ЦВЕТОМ
Окрашивание дополнительным цветом применяется для облег-
чения ориентации в полях зрения, облегчения установки нужных
деталей, повышения контрастности. Усиления контрастности
можно достичь за счет подавления фоновой флюоресценции
синим Эванса (0,1 г/л, 30—60 с, последующая отмывка ФСБ
дважды по 5 мин). Для облегчения установки нужных деталей,
особенно в случае объектов с незначительной нежелательной
флюоресценцией, можно воспользоваться дополнительным окра-
шиванием альбумином, меченным родамином или бромистым
этидием (10 мг/л, 2 мин). Ядра нежизнеспособных клеток при-
обретают оранжево-красную окраску.
2.8.4.4. КОНТРОЛЬ КАЧЕСТВА АНАЛИЗА
В иммунофлюоресцентном анализе используют различные виды
контроля для предотвращения ошибок и правильной оценки
данных.
Контроль собственой флюоресценции объекта: проверяют
объект без специальной обработки, например только после про-
мывания.
Контроль конъюгата: объект инкубируют только с конъюга-
том. Этот вид контроля применяется в непрямом методе ана-
лиза. Контроль позволяет установить наличие собственной
флюоресценции препарата, а также наличие нежелательных ре-
акций конъюгата с объектом.
Отрицательный контроль: в прямом методе анализа приме-
няется для гарантии отсутствия соответствующего антигена
(ткань, заведомо не содержащая соответствующего АГ). В не-
прямом методе анализа используют сыворотку, которая не со-
держит АТ к исследуемому объекту, например донорские сыво-
ротки. Обработку материала проводят, как для положительного
контроля. Отрицательный контроль устанавливает неспецифиче-
скую флюоресценцию объекта вследствие неспецифического свя-
зывания ИГ. По отношению к этому контролю оценивают раз-
ведения исследуемых сывороток. При высоких разведениях наб-
людается такая же картина, как при контроле конъюгата.
Положительный контроль: в прямом методе используют пре-
парат, оказавшийся положительным в ранее проведенных ис-
следованиях. В непрямом методе в качестве положительного
контроля используют заведомо положительную сыворотку.
Контроль устанавливает реакционную способность ткани или
конъюгата. В случае многоступенчатых реакций контролю дол-
138
жен подвергаться каждый этап. Определение комплементсвязы-
вающих антител:
Контроль комплемента: ткани или клетки, заведомо не со-
держащие комплементсвязывающие АТ, инкубируют с компле-
ментом или конъюгатом. Контроль показывает, содержит лн сы-
воротка с комплементом антитела к антигенам исследуемого
препарата. Для оценки специфичности АГ или АТ можно вос-
пользоваться адсорбцией сывороток определенным АГ с после-
дующим отделением иммунных комплексов. Можно применять
предварительную инкубацию препарата с антителами соответ-
ствующей специфичности, но принадлежащими другому биоло-
гическому виду (лучше всего к тому, от которого получена сы-
воротка для конъюгата); положительная реакция в этих случа-
ях выражена слабее или вообще не наблюдается.
2.8.4.5. ТИТРОВАНИЕ В ШАХМАТНОМ ПОРЯДКЕ
Каждую новую серию конъюгата с целью определения опти-
мального разведения для непрямого метода проверяют титро-
ванием в шахматном порядке с позитивными и негативными
контрольными сыворотками. Схема перекрестной проверки пред-
ставлена в табл. 9.
Таблица 9. Пример титрования в шахматном порядке для определения
оптимального рабочего разведения конъюгата
Сыворотка Конъюгат, разведение 1:2 1:5 1:10 1:20 1:40 | КП ♦ 1:80 1’160
Позитивная 1:640 0 0 0 0 0 0 0
сыворотка 1:320 ± ± ± ± ± ± 0—ВСП
1:160 + + + + + ± 0
1:80 ++ ++ Ч—h Ч—h —ь + 0
1:40 ч—н ++ Ч—F 4-4- --+ + ±
1:20 Ч—1—Ь Ч—1—Ь +4-4- ++ + ±
Негативная 1:80 0Х 0 0 0 0 0 0
сыворотка 1:20 0Х 0Х 0Х 0Х 0 0 0
Контроль конъюгата 0Х 0Х 0Х 0 0 0 0
Обозначения:
0 — отрицательная реакция, т. е. отсутствие субъективного восприятия
специфической флюоресценции;
X — и нтенсивная неспенифическая (или нежелательная специфическая флюо-
ресценция;
± — слабая специфическая флюоресценция; субъективно ее можно спутать с
отрицательным контролем;
+ , ++, + + Н---специфическая флюоресценция от отчетливой до очень
сильной.
КП — конец плато, здесь при разведении конъюгата 1 : 80;
всп — выход титра сыворотки на плато, здесь 1 : 320;
Рекомендуемое рабочее разведение конъюгата: 2—4-кратная концентрация
конъюгата в КП; рекомендуется разведение 1:30.
139
2.84.6. СРЕДЫ ДЛЯ ЗАКЛЮЧЕНИЯ
И ХРАНЕНИЯ ПРЕПАРАТОВ
Заключение препаратов в водосодержащие среды при pH 8,6
обеспечивает самую высокую степень флюоресценции и наи-
меньшее «выцветание». Препараты, высушенные и заключенные
в неполярные среды, например иммерсионное масло (Piaflex хи-
мический комбинат Пистериц), энтеллан (Merck, Дармштадт),
UV инертную (Serva, Гейдельберг), обладают только 50% ин-
тенсивностью флюоресценции и вдвое более сильным «выцве-
танием» по сравнению с препаратами, заключенными в водные
среды [13]. Среда для заключения расходуется экономно и по
возможности без образования пузырей воздуха. Референс-пре-
параты в высушенном виде хранятся при 4 'С, в среде для за-
ключения — при —20 °C.
2.8.5. ОБОРУДОВАНИЕ ДЛЯ
ИММУНОФЛЮОРЕСЦЕНТНОГО АНАЛИЗА
При возбуждении светом определенных длин волн флюорохро-
мы начинают флюоресцировать. Кванты флюоресцентного излу-
чения обладают меньшей энергией, чем кванты возбуждающего
излучения, это означает, что флюоресцентное излучение имеет
большую длину волны, чем возбуждающее (правило Стокса).
Необходимо добиваться четкого разделения возбуждающего и
флюоресцентного излучения. Этого можно достичь использова-
нием фильтров и подбором длин волн возбуждающего излу-
чения.
Микроскоп. Иммунофлюоресценцию исследуют при помощи
специальных микроскопов, включающих такие детали, как спе-
циальные источники света, фильтры, УФ-оптика и др. Нет смыс-
ла рекомендовать для работы какой-нибудь один тип микроско-
па, подходит любой вариант, обеспечивающий необходимые ус-
ловия. Для работы с ФИТЦ рекомендуется узкополосное голу-
бое освещение, для ТРИТЦ — зеленое. В качестве источников
света, как правило, используют ртутные лампы высокого дав-
ления (линейчатый спектр излучения). Возбуждающее излуче-
ние голубого и зеленого цвета получают пропусканием света та-
ких ламп через светосильный интерференционный фильтр. Мож-
но использовать галогеновые лампы иа 12 В (50 илн 100 Вт),
они более удобны в обращении, быстрее нагреваются, дешевле
прн одинаковой с ртутными лампами интенсивности излучения.
Фильтры необходимы для того, чтобы пропускать из спектра
лампы только тот участок, который необходим для возбужде-
ния флюорохрома (первичный фильтр, или фильтр возбужде-
ния), а также задерживать возбуждающее излучение и пропус-
кать только излучение флюоресценции (вторичный илн запи-
рающий фильтр) (рис. 56). Прочие важные характеристики
140
Рис. 56. Ход лучей в флюоресцентном микроскопе на примере Fluoval 2 VEB
Carl Zeiss.
1 — источник света; 2 — первичный фильтр; 3 — дихроматическое разделяющее зеркало;
4 — объектив; 5 — запирающий фильтр; 6 — окуляр; 7 — проекторное устройство.
Рис. 57. Кривые пропускания света важнейших типов фильтров, применяемых
в флюоресцентной микроскопии.
Прочие характеристики фильтров см. в табл. 10.
Таблица 10. Классификация важнейших типов фильтров, применяемых
для флюоресцентной микроскопии
Обозначение фильтра А КР 490 В КР 560 с BG 12 D BG 23 Е GG 5 р OG 4
Тип фильтра Металлический интерференционный Стеклянный адсорбционный Стеклянный пропускающий
Характеристика фильтра Узкополосный Широкополосный
Функция фильтра на примере голу- бого возбуж- дающего излу- чения Первич- ный фильтр Первичный или вто- ричный вспомога- тельный фильтр, су- живающий нлн отсе- кающий красный свет Первич- ный фильтр Первич- ный вспо- мога- тель- ный фильтр, отсе- кающий крас- ный свет Первич- ный вспо- мога- тель- ный фильтр сужива- ющий Вто- рич- ный фильтр
фильтров можно получить из рис. 57 и табл. 10. Проницаемость
фильтров или их комбинаций для изучения может быть низко-
или широкополосной, в качестве границы между этими града-
циями принята полоса излучения шириной 50 нм.
Сужение полосы спектра возбуждения или флюоресценции оп-
равдано только в случае применения фильтров с четким «ок-
ном» пропускания и высокой прозрачностью, но и в этих случа-
ях наблюдается снижение интенсивности. Более удобным ока-
зывается узкополосное возбуждение, получаемое за счет погло-
щения или блокирования аутофлюоресценции, которая возбуж-
дается излучением близкого УФ или фиолетового цвета. Несмот-
ря на снижение интенсивности флюоресценции, в этих случаях
контрастность изображения и, следовательно, разрешающая спо-
собность увеличиваются. Оптика, как правило, должна быть све-
тосильной, т. е. обладать широкой апертурой, поскольку интен-
сивность изображения пропорциональна квадрату апертуры. Оп-
тимальным можно считать увеличение объекта, равное 20—100,
поскольку в этом случае объектив может служить для освеще-
ния объекта. Сила объектива при этом удваивается для флюо-
ресцентного излучения. Поэтому стараются использовать объек-
тивы с максимальной апертурой, причем не обязательно апохро-
матические. Увеличивающая оптика второй ступени (окуляры,
проекционная оптика) должна, напротив, обладать минималь-
ным увеличением для получения изображения максимальной ос-
142
вещенности. Иногда представляет большой интерес помещение
флюоресцирующих объектов в нефлюоресцирующую ткань
и микроскопия в других условиях освещения или в фазовом
контрасте. Особенно выгодные возможности для этого предо-
ставляет люминесцентная микроскопия. Комбинация флюорес-
ценции с микроскопией в светлых или темных полях зрения
только в редких случаях дает удовлетворительные результаты.
Лучшие результаты дает применение фазово-контрастной мик-
роскопии со специальными объективами, пригодными также
и для иммунофлюоресцентного анализа. Хорошее контрастиро-
вание (слабый эффект Гало) весьма выгодно при рассматрива-
нии мелких объектов (микроорганизмы, отдельные клетки), но
не дает существенных преимуществ при микроскопии срезов
тканей. В работе с такими объектами (большая площадь) лучше
использовать интерференционное контрастирование. В любом
случае контраст зависит от разности коэффициентов преломле-
ния объекта и среды, в которую он заключен. Изменением среды
(например, увеличением пропорции глицерина) можно добить-
ся изменения контрастности. При комбинации флюоресцентного
анализа с оптической поляризацией следует помнить, что ди-
хроматический разделяющий слой зеркала (см. рис. 56) сам по
себе обладает способностью поляризировать свет.
2.8.6. ОЦЕНКА ИММУНОФЛЮОРЕСЦЕНТНЫХ
ПРЕПАРАТОВ
2.8.6.1. СУБЪЕКТИВНАЯ ОЦЕНКА
ИССЛЕДУЕМЫХ ОБЪЕКТОВ
Если флюоресцирующие объекты оцениваются по контрасту,
т. е. по сравнению флюоресценции объекта с флюоресценцией
фона, всегда следует учитывать данные отрицательного контро-
ля. Если при отрицательном контроле на соответствущих струк-
турах не наблюдается нежелательной флюоресценции, вопрос
решается просто: по типу да/нет. Как правило, приходится стал-
киваться с ситуацией, когда необходимо решать, не усиливается
ли специфическая флюоресценция нежелательной флюоресцен-
цией (отрицательный контроль). Это означает, что минимально
воспринимаемая специфическая флюоресценция зависит от сте-
пени неспецифической нежелательной флюоресценции. Вследст-
вие несовершенства органа зрения (адаптаия) при незначитель-
ном контрасте необходимо чаще прибегать к сравнению с из-
вестными препаратами. Если наблюдения ведутся над одним
единственным АГ, интенсивность флюоресценции будет расти
пропорционально увеличению концентрации меченых АТ, пока
не останется свободных детерминант. Если оценивают содержа-
ние нескольких АГ, различных по количеству и по аффинности
к меченым АТ, то интенсивность флюоресценции не зависит от
143
концентрации АТ в применяемом растворе. Специфическую
флюоресценцию можно оценивать по степени интенсивности
(см. табл. 9). Эта система оценки зависит от всех методических
и инструментальных факторов, влияющих на контрастность,
в том числе от положительного и отрицательного контроля. Ста-
новится ясно, что такая система оценки имеет смысл только
в пределах одной лаборатории.
2.8.6.2. ТИТРОВАНИЕ
Одним из вариантов количественной оценки АТ в растворах мо-
жет служить ступенчатое разведение раствора с целью опреде-
ления максимального разведения, при котором еще наблюдает-
ся специфическая флюоресценция. Это разведение принимают за
титр АТ. Для того чтобы сравнивать результаты с результатами
других лабораторий, необходимо время от времени проводить
исследования со стандартными сыворотками. Этим способом
можно установить разницу в чувствительности применяемых ме-
тодов и стандартизовать данные поправками титра.
2.8.6.3. МИКРОФЛЮОРИМЕТРИЯ—
МИКРОФОТОГРАФИЯ
Количественная оценка флюоресценции при помощи флюори-
метрии порождает много проблем [14, 15]. Необходимой пред-
посылкой является стандартизация метода. Следует стремиться
сократить до минимума ошибки и по возможности определить
их абсолютное значение (ошибки, связанные с «выцветанием»,
фокусировкой, толщиной исследуемого объекта). В отличие от
адсорбционной микрофотометрии, в которой сравнение произво-
дят с данными, получаемыми без объекта исследования, микро-
флюориметрия вынуждена использовать флюоресцентный стан-
дарт, чтобы проводить по нему калибровку оборудования и
реагентов. Нити уранового стекла, заключенные в пиафлексили
энтеллан, могут служить легкодоступными стандартами [13].
Поскольку интенсивность флюоресценции зависит от толщины
объекта, то очень трудно проводить сравнение разных срезов
(из-за неодинаковой толщины). В этом случае можно восполь-
зоваться соотношением интенсивности флюоресценции различ-
ных клеточных структур (контраст) или изменением интенсив-
ности флюоресценции во времени (выцветание) [13, 16]. Для
документации исследований необходимо делать микрофотогра-
фии флюоресцирующих объектов. Следует отметить, что микро-
фотография не может служить доказательством положительной
реакции даже в сравнении с микрофотографией заведомо нега-
тивного стандартного препарата вследствие эффекта Шварц-
шильда (Schwarzshild). Слабая интенсивность освещения всег-
да вызывает эффект Шварцшильда при микрофотографирова-
нии, другими словами делает необходимым добавочное время
144
освещения к автоматически регулируемому времени достиже-
ния необходимого усиления окраски. Величина эффекта Шварц-
шильда, т. е. удлинение времени освещения, зависит от интен-
сивности флюоресценции, типа пленки и эмульсии. Вследствие
этой и других проблем (выцветание, эффект темного поля) не-
обходимо соблюдать следующие условия: применять высокочув-
ствительные сорта фотопленки, например ORWO-RS2, NP27, оп-
ределять время экспозиции на образцах исследуемой фото-
пленки, помнить, что установка автоматической регуляции экс-
позиции не приносит ощутимых преимуществ, так как необходи-
мы пробные испытания с каждой новой серией фотоэмульсии.
Ввиду незначительных изменений оптических условий при лю-
минесцентной микроскопии можно производить смену объекти-
вов, рассчитывая изменения времени экспозиции заранее. Для
цветной фотосъемки можно использовать пленки, применяемые
как для съемок при естественном освещении (ORWO chrom
UT18), так и при искусственном освещении (ORWO chrom
UK17). Пленки для искусственного освещения рекомендуются
в тех случаях, когда приходится фотографировать объекты пос-
ле дополнительного окрашивания синим Эванса или бромистым
этидием. Для съемки объектов с монохроматическим флюорес-
центным излучением черно-белые пленки используются наравне
с цветными.
2.8.6.4. ИСТОЧНИКИ ОШИБОК
Приводимое ниже перечисление источников возможных ошибок
не претендует на исчерпывающую полноту, оно скорее представ-
ляет собой выборку из наиболее часто встречающихся вариан-
тов ошибок. Обычно легко путают верхний и нижний, а также
правый и левый края предметных стекол. Для предотвращения
таких ошибок рекомендуется асимметрично располагать образ-
цы или маркировать предметные стекла всегда в одном и том
же углу.
— Долго или неправильно хранившийся конъюгат может вы-
зывать образование флюоресцирующих выпадений по всему
субстрату. Опыт повторяют с заведомо пригодной сывороткой.
— Если во влажной камере во время инкубации недостаточно
жидкости или камера не препятствует испарению жидкости,
может наблюдаться так называемый краевой эффект. Кон-
центрация меченых АТ будет большей на периферии и при-
ведет к ложноположительному результату.
— Если было допущено присыхание сывороток или конъюгата
к препарату, возникает сильная неспецифическая флюорес-
ценция, которая исключает правильную оценку результатов.
— Если в лаборатории проводят эксперименты с другими флюо-
рохромами, например с акридиновым оранжевым, то даже
ничтожные следы этого соединения, попадающие в препарат,.
10—990
145
могут вызывать флюоресценцию ткани, особенно ядер, при-
водя к ложноположительным результатам.
В случае применения высокотитражных сывороток возможно
взаимное влияние двух различных препаратов, подвергаю-
щихся одновременной отмывке.
-— При любом методе окрашивания препаратов с флюорохром-
содержащими АТ следует пользоваться одним и тем же объ-
ективом для получения сравнимых результатов, так как
субъективно воспринимаемая интенсивность флюоресценции
зависит от апертуры объектива.
-— Смеси антител к различным АГ исследуемого препарата мо-
гут давать различного рода искажения ввиду взаимного
влияния АТ. Например, антимитохондриальные АТ исклю-
чают возможность одновременного определения антител
к обкладочным клеткам и мешают определению АТ к глад-
кой мускулатуре.
2.8.6.5. ТЕНДЕНЦИИ РАЗВИТИЯ МЕТОДА
Наибольший интерес представляют собой две тенденции. В пер-
вом случае АГ присоединяют к твердой фазе (сефадекс, плас-
тиковые поверхности), что позволяет улучшить количественную
оценку иммунофлюоресценции. Во втором случае предпринима-
лись попытки повысить чувствительность тест-системы без уси-
ления неспецифических реакций. Этого можно достичь присо-
единением АТ к флюоресцирующим частицам, например липосо-
мам, или присоединением АТ к промежуточно связывающимся
соединениям, например биотину [2. 10].
2.8.7. ПРИМЕНЕНИЕ МЕТОДА
ИММУНОФЛЮОРЕСЦЕНЦИИ В КЛИНИЧЕСКОЙ
ИММУНОЛОГИИ, ОБНАРУЖЕНИЕ АУТОАНТИТЕЛ
Важнейшей областью применения непрямого иммунофлюорес-
центного анализа является обнаружение аутоантител (ААТ)
в сыворотках пациентов с аутоиммунными заболеваниями. Во
многих случаях этот метод занял прочное место в лабораторной
диагностике, например, в определении антиядерного фактора
(АЯФ), антимитохондриальных антител (АМА), АТ к щитовид-
ной железе, АТ при пузырчатке и пемфигоиде. Выбор антиген-
ного субстрата для определения ААТ должен учитывать их спе-
цифичность (тканевую или органную). Поскольку многие разно-
видности ААТ можно обнаруживать или исключать при помо-
щи одного субстрата, часто применяют комбинации тканей раз-
личных органов, таких как печень, желудок, почки крыс или
мышей, иногда с добавлением ткани щитовидной железы чело-
века. Иммунофлюоресцентный анализ применительно к обнару-
146
жению ААТ имеет несомненные преимущества по сравнению
с другими методами, а именно простота методики, малый расход
времени и реагентов, высокая чувствительность и гибкость ана-
лиза, доступность коммерческих препаратов конъюгатов. Опре-
деляющим моментом анализа является дифференциация раз-
личных типов флюоресценции. Требуется большой опыт, чтобы
обнаружить типичную флюоресценцию ААТ в препарате. Гиб-
кости анализа, т. е. возможности определять несколько разно-
видностей ААТ при помощи одного антигенного субстрата, со-
держащего большое количество разных АГ, сопутствует опре-
деленный недостаток; в частности, не всегда можно точно уста-
новить наличие искомого АГ или ААТ в флюоресцирующем
препарате. Картина иммунофлюоресценции, характерная для
различных ААТ, описывается в литературе с учетом относитель-
ной интенсивности флюоресценции, при помощи микрофотогра-
фий или вербально; например, АЯФ — как гомогенную, крапча-
тую, периферическую или нуклеолярную флюоресценцию. Та-
кую информацию нельзя считать достаточной во всех случаях,
поэтому возникает необходимость консультации с другими ла-
бораториями, обмена позитивными сыворотками для получения
сравнимых результатов и т. д. Хотя многие ААТ не обладают
органной и видовой специфичностью, например АЯФ, выбор под-
ходящего субстрата имеет решающее значение, поскольку кон-
центрация или доступность АГ может быть различной у разных
видов [9, 11]. Сравнимость результатов можно обеспечить или
использованием одинаковых субстратов, или сравнением чувст-
вительности применяемых систем. При определении ААТ следует
делать различие между минимальным определяемым титром и
титром, имеющим клиническое значение. Первый зависит от не-
желательной флюоресценции в области расположения соответ-
ствующего АГ и сильно варьирует для различных ААТ; напри-
мер, для АЯФ титр составляет 1: 1, титр АТ к гладкой муску-
латуре— 1:40, а к скелетной мускулатуре 1:80 [1]. Клиниче-
ски значимый титр зависит от распространенности типа ААТ
в группе здоровых или соответственно в группе страдающих оп-
ределенным заболеванием пациентов. Поскольку этот титр за-
висит в значительной степени от вида применяемого антигенно-
го субстрата, невозможно получить общезначимые выводы
о диагностически важных титрах определенных групп больных.
До настоящего времени при использовании тест-систем, ос-
нованных на иммунофлюоресцентном анализе, приходится об-
следовать также большую контрольную группу пациентов. Дан-
ные об обнаружении ААТ могут иметь важное клиническое
значение как в диагностике, так и в решении вопроса о выборе
терапии. Имеет значение только обнаружение присущих для
данной нозологической формы ААТ в диагностических титрах
[17]. Сыворотки рекомендуется с самого начала апробировать
в тех разведениях, которые наиболее подходят для получения
клинически значимых титров. Одновременно испытывают более
10*
147
высокое разведение (4-кратное) с тем, чтобы в случае положи-
тельного результата можно было точнее оценить необходимую
степень разведения.
2.8.8. СПИСОК ЛИТЕРАТУРЫ
.1. Baumann I. Immunfluorescenzoptischer Nachweis von Skelettmuskelantikor-
pern unter besonderer Berucksichtigung der Myastenia gravis. — Promotion-
schrift, Leipzig, 1981.
2. Berman J. IT. Amplification of the biotin-avidin immunofluorescence techni-
que.— J. Immunol. Methods, 1980, 36, 335—339.
.3. Berod A., Hartman В. K.., Pujol J. F. Importance of fixation in immunohisto-
chemistry: use of formaldehyd solution at variable pH for the localisation of
thyrosine hydrolase. — J. Histochem. Cytochem., 1981, 29, 844—850.
-4. Forni L. Reagents for immunofluorescence and their use for studying lym-
phoid cells products. — In: Immunological methods/Eds. J. Lefkovits & B. Per-
nis. New York. Acad. Press, 1979, 151—167.
'5. Frantz H., Mohr J. Reinigung und Spezifitatskontrolle von markierten Anti-
korpern. — Acta histochem. Suppl., 1980, XXII, 41—46.
6. Frietzler M. J. Fluorescent antinuclear antibody test. — In: Manual of clini-
cal immunology, 2nd ed./Eds. N. R. Rose & H. Friedman. — Washington,
Amer. Soc. Microbiol., 1980, 852—857.
7. Gernard K-, Rost D., Hanke E„ Bent H. Eine einfache Fixierung von Antigen-
substraten fur den Autoantikorpernachweis. — Acta histochem. Suppl. 1980,
XXII, 99—100.
8. Hed J., Enestrom S. Detection of immune deposits in glomeruli: The masking
effect of antigenicity of formalin in the presence of proteins. — J. Immunol.,
1981, 41, 57—63.
9. Helmke K.., Scheuermann R. et al. Nachweise und Bedeutung antinucleare
Antikorper in der Diagnostik der Erkrankungen des rheumatischen Formen-
kreises. — Klin. Wschr., 1981, 59, 287—296.
10. Huang A., Kennel S. J., Huang L. Immunoliposome labeling: a sensitive and
specific method for cell surface labeling. — J. Immunol. Methods, 1981, 46,
141—151.
11. Me. Carty G. A., Rice J. R. Characterisation and comparison of commercially
available antinuclear antibody kits using single pattern index sera. — J. Rheu-
matol., 1980, 7, 339—347.
12. Storch W. Ringsversuch zum Nachweiss von Autoantikorpern durch indirekte
Immunfluorescenz. — Allergie u. Immunologie, 1980, 26, 199—205.
13. Storz H. Untersuchungen zum Ausbleiben von Immunfluoreszenzobjekten und
von Moglichkeiten zur Beinflussung dieses Effektes. — Jenaer Rundschau,
1981, 26, 87—88.
14. Storz H. Anmerkungen zur Fluoreszenzmukrofotografie. — Jenaer Rundschau,
1983, 28, 79—81.
15. Storz H. Geratetechnik-bedingte Probleme bei der Immunfluoreszenzmikro-
skopie. — Ztschr. ges. inn. Med., 1983, 38, 101—104.
16. WHO-report. The use and abuse of immunological tests. — Clin. Immunol.
Newsletter, 1981, 2, 178—186.
2.9. РАДИОИММУНОЛОГИЧЕСКИЙ АНАЛИЗ
В 1960 г. Ялоу и Берсон опубликовали статью «Иммунологиче-
ское определение эндогенного инсулина в плазме человека», по-
ложившую начало эпохе радиоиммунологических исследований.
Радиоиммунологические методы имеют особое значение для ко-
личественного определения гормонов [2]. Различия в концен-
трациях этих веществ в биологических жидкостях (сыворотка
148
крови, моча и т. д.) находятся в относительной простой зависи-
мости от различных патологических состояний, что позволяет
-производить in vitro функциональную оценку деятельности щи-
товидной железы, надпочечников и поджелудочной железы. Все
возрастающее значение имеет определение ассоциированных
с опухолями АГ, канцерогенных производных ДНК, фар-
мацевтических препаратов и ИГ радиоиммунологическими ме-
тодами. Эти методы широко применяются при изучении вирус-
ных АГ, противовирусных АТ, в диагнЬстике аутоиммунных за-
болеваний (системная красная волчанка), а также для опреде-
ления циркулирующих иммунных комплексов. Исчерпывающее
изложение вопросов, связанных с проблематикой радиоиммуно-
логического анализа (РИА), можно найти в обзорах [7, 13].
2.9.1. ПРИНЦИП МЕТОДА
Характерной чертой радиоиммунологических методов является
сочетание специфичности, свойственной реакциям АГ—АТ,
с простотой и высокой чувствительностью определения, которые
дает применение радиоактивных изотопов, введенных в состав
АГ или АТ. По сравнению с обычными методами анализа пре-
имущество РИА состоит в том, что отсутствует необходимость
оценивать протекающую реакцию по вторичным проявлениям,
таким как агглютинация, преципитация, лизис эритроцитов.
Важнейшим условием использования РИА является возмож-
ность соединения измеряемой радиоактивной субстанции с ком-
понентами реакции АГ—АТ, как, впрочем, и наличие специфи-
ческих сывороток и высокоочищенных антигенов. Получение
высокотитражных сывороток к большинству белковых гормонов
не представляет особых сложностей. Вещества, являющиеся гап-
тенами, например стероиды, медикаменты и продукты их мета-
болизма, простагландины, циклические нуклеозидмонофосфаты,
можно легко перевести в иммуногенное состояние [3]. АГ и АТ
весьма удобны для прямого и непрямого введения радиоактив-
ной метки [1]. Необходимая для исследований специфическая
радиоактивность метки должна рассчитываться исходя из за-
данной чувствительности. Радиоактивность АГ, применяемых
в РИА, обычно составляет 100—200 мкКи/мкг, радиоактивность
АТ — 20 мкКи/мкг, а для модификации РИА с двойными АТ —
0,1 мкКи/мкг. Возможность проводить конкурентный анализ,
а также анализ в жидкой или твердой фазе приводит к созда-
нию большого числа различных методик. Мы рассмотрим эту
проблему на четырех характерных примерах.
2.9.2 РАДИОИММУНОЛОГИЧЕСКИЙ АНАЛИЗ
Реакция АГ с АТ приводит с течением времени к состоянию
равновесия между свободным и связанным АГ. Характерным
для РИА является то обстоятельство, что меченый и немеченый
149
антигены конкурируют за ограниченное число участков связы-
вания. После инкубации в пределах определенной области кон-
центраций АГ, количество включившегося в состав иммунных
комплексов меченого АГ обратно пропорционально количеству
немеченого АГ в пробе. Основные реакции представлены на сле-
дующей схеме:
АГ АГ —АТ , АГ
„ т^-п^+т
I + АГ ' АГ—АТ АГ
III ~ VI + VII
I — специфическое антитело, II — меченый антиген, III — немеченый антиген
(стандарт или образец), IV — иммунные комплексы, состоящие из АТ и мече-
ного АГ, V — свободный меченый АГ, VI — иммунные комплексы, состоящие
из АТ и немеченого АГ, VIII — свободный немеченый АГ
Для того чтобы происходило конкурентное взаимодействие,
должна существовать определенная степень родства между ме-
ченым и немеченым антигеном. Теоретически РИА можно про-
водить как вариант сатурационного анализа. В этом специаль-
ном случае связывающий компонент Q представлен специфиче-
ским АТ, метка вводится в составе АГ. Детально теоретические
аспекты рассматриваются в работе Walker и Keane [18]. Кроме
того, определенный интерес представляют вопросы, касающиеся
различий связывания меченых и немеченых АГ, влияния темпе-
ратуры, авидности и концентрации компонентов реакции [7,13].
Для оценки качества АС необходимо знание константы авидно-
сти К. Ее можно определить по методу Скэтчарда и Михаэли-
са— Ментен [13]. Отклонения от идеальной формы кривой
можно рассматривать как выражение гетерогенности реакции
связывания, которая обусловлена негомогенностью популяции
антител. Наиболее удачные концентрации АТ оказываются рав-
ными 3/К меченого антигена 4/К. Между чувствительностью ме-
тода (S) и авидностью существует соотношение:
S=0,1/К
Заключение о степени конкуренции между меченым АГ и АГ
исследуемой пробы можно сделать только после отделения об-
разовавшихся ИК от несвязанного меченого антигена. Для от-
деления ИК используют самые разнообразные методы, осно-
ванные на различиях в растворимости, коэффициенте седимен-
тации, заряде, молекулярной массе и др. В некоторых случаях
на способ выделения ИК оказывают влияние специфические
свойства исследуемого АГ. Для того чтобы разделение меченого
несвязавшегося АГ и ИК было удовлетворительным, необходи-
мо соблюдение трех условий:
1) процесс разделения не должен оказывать влияния на состоя-
ние равновесия;
150
2) отделение связанных с АТ молекул от несвязанных должно
носить количественный характер;
3) затрата рабочего времени и стоимость процесса должны быть
незначительными.
Известно много способов достижения вышеперечисленных
условий [13]. Различия в заряде и относительной молекуляр-
ной массе позволяют проводить разделение при помощи элект-
рофореза или гель-хроматографии. В некоторых случаях воз-
можна селективная адсорбция АГ на тонко измельченном таль-
ке, кварце, декстране или целлюлозе, соединенных с активиро-
ванным углем. Связанные АГ можно улавливать при помощи
иммобилизованных АТ, например полимеризованных АТ, или
осаждением ИК специфической АС. Конъюгация АТ с железо-
содержащими частицами энзакрила позволяет проводить разде-
ление в магнитном поле [12]. Различные способы разделения
АГ и ИК требуют неодинаковых затрат времени, их воспроизво-
димость также неодинакова. То же самое относится к возмож-
ности автоматизации отдельных этапов анализа. Наибольшее
внимание привлекают к себе в настоящее время методы, осно-
ванные на использовании двойных АТ. Известным упрощением
метода является использование связанных с целлюлозой вто-
ричных АС, так называемая технология двойных АТ на твердой
фазе. Оптимальное для РИА разведение АС устанавливается
по 50% связыванию меченого АГ. АС в данном разведении
в присутствии меченого АГ смешивается с возрастающим коли-
чеством немеченого АГ. Зависимость связывания меченого АГ
от концентрации стандартного АГ можно представить в виде
различных графиков. Часто используют график зависимости
процента связывания меченого АГ от концентрации немеченого.
Уже упоминавшиеся во введении Берсон и Ялоу предложили ис-
пользовать соотношение связанного меченого АГ к несвязанно-
му в качестве оценки концентрации немеченого АГ. При ис-
пользовании арифметических координат получается экспоненци-
альная кривая, полулогарифмических — кривая с прямолиней-
ным участком, логарифмических — кривая с увеличенным пря-
молинейным участком. Особый интерес представляет возмож-
ность конкурентного взаимодействия с чужеродными вещества-
ми, например медикаментами. Специфичность метода оценивают
также по перекрестной реактивности с применяемыми сыворот-
ками; заключение в этом случае выносится на основании опре-
деления меченого АГ в присутствии сопутствующего АГ. Ошиб-
ки, связанные с вышеперечисленными модификациями, можно
устранить, используя дополнительную очистку реагентов, что,
однако, увеличивает стоимость исследования. Все чаще начина-
ют применять моноклональные антитела для РИА. Уже описа-
ны [16, 17] радиоиммунологические способы определения мор-
фина и человеческого лейкоцитарного интерферона с использо-
ванием моноклональных или полученных аффинной очисткой АТ.
В случае конкурентного анализа белкового связывания вместо
151
антисывороток используют транспортные белки. Следует учи-
тывать, что специфичность их связывания и авидность не всегда,
удовлетворительны.
2.9.2.1. ТВЕРДОФАЗНЫЙ
РАДИОИММУНОЛОГИЧЕСКИЙ АНАЛИЗ
Впервые в качестве водонерастворимого носителя для белков
была применена пара-аминобензилцеллюлоза. В последующее
время было описано большое количество твердых носителей, на-
шедших применение в секвенировании, синтезе и других мани-
пуляциях с белками. Эти носители, соединяясь с АГ или АТ, не
изменяют их иммунологические свойства.
Полимеры, например сефадекс и сефароза, после их обра-
ботки бромцианом приобретают способность связывать белки.
В дальнейшем была установлена способность многих полимеров
(полиэтилен, полипропилен, полистирол), называемых обычно
матрицами, связывать различные биологические макромолекулы
без всякой предварительной химической обработки. Оказалось,
что полимеры обладают способностью связывать АТ, которые
можно использовать для определения различных гормонов (хо-
рионического гонадотропина, лютеотропного гормона, тиреости-
мулирующего гормона, гормона роста и др.) методом РИА.
Связанный с твердой фазой комплекс АГ—АТ легко отделяется
от несвязавшегося биологического материала, кроме того, он
обладает высокой стабильностью. Меченый АГ, необратимо свя-
зываясь с фиксированными на матрице АТ, позволяет проводить
высокочувствительный анализ биологического материала.
АТ можно присоединять к активированной или неактивирован-
ной матрице. Основной проблемой будет в обоих случаях точ-
ное дозирование связавшихся с матрицей АТ.
2.9.2.1.1. Связывание антител с матрицей
Нефракционированные АС кроликов с известным титром ней-
трализации бактериофагов ТЗ и Т4 разводили 0,1 М карбонат-
бикарбонатным буфером с pH 9,6. Сыворотки в различных раз-
ведениях вносили в пластиковые пробирки по 0,5 мл. Пробирки
закрывали и выдерживали в течение 2 ч при комнатной темпе-
ратуре, затем удаляли жидкую фазу. Легко десорбирующиеся
АТ удаляли двукратным ополаскиванием в 0,14 М NaCl (по
1 мл). Затем проводили инкубацию с 0,01 М калий-фосфатным
буфером с pH 7,4, содержащим 0,14 М NaCl и 0,5% ЧСА, для
насыщения свободных участков пластиковой поверхности ней-
тральным белком. Пластиковые пробирки с адсорбированными
АТ можно использовать немедленно или хранить в холодиль-
нике в течение 4 нед.
152
2.9.2.1.2. Мечение антигена
Бактериофаги ТЗ и Т4 после маркирования их 131 [I] или 125[1]
сохраняют полностью иммунологическую реактивность. Йодиро-
вание проводили Т-хлораминовым методом. Специфическая ра-
диоактивность фагов составляла 0,3—7,2 мкКи/мкг. В целом бо-
лее высокая специфическая радиоактивность повышает чувст-
вительность анализа. После удаления радиоактивного йода гель-
фильтрацией через сефадекс и диализа препарат радиоактив-
ных фагов разводят калий-фосфатным буфером с pH 7,4
(см. выше) для предотвращения радиолиза.
2.9.2.1.З. Оптимизация анализа
Вначале определяют оптимальное разведение АТ и меченого
фага для достижения максимального связывания материала
с твердой фазой. Исследовалась также зависимость этих про-
цессов от времени. На рис. 58 представлены данные, относя-
щиеся к фагу ТЗ, меченному 13Ч. Аналогичные данные были по-
лучены для фага Т4. Соотношение связанный/несвязанный (с/н)
меченый фаг отражает воздействие различных факторов. Наи-
лучшие результаты были получены при разведении сыворотки
1:500 или 1: 1000 и при концентрации меченого материала
0,0125 мкКи (около 16000 имп/мин; расчет ведется на 1 пласти-
ковую пробирку). Зависимость от времени исследовали в тече-
ние 48-часовой инкубации при температуре 37 °C. Вначале наб-
людается быстрое увеличение количества связанной метки,
к 12 ч инкубации оно заметно снижается, а затем выходит на
плато.
Время (ч)
Рис. 58. Зависимость связывания мечеиых фагов ТЗ с АТ, адсорбированными
на пластиковой поверхности от времени инкубации, разведения сыворотки,
титра антисыворотки, концентрации меченого свидетеля. Пластиковые про-
бирки обрабатывали двумя антисыворотками к фагу ТЗ с различными титра-
ми нейтрализации. Антисыворотка 7: К=150, антисыворотка 8: К=370.
1 — антисыворотка 8,1 : 100, 0,05 мкКи метки; 2 — антисыворотка 8,1 : 1000, 0,05 мкКн
метки; 3 — антисыворотка 7,1 : 1000, 0,025 мкКи метки; 4 — антисыворотка 8,1 : 1000,
0,025 мкКи метки.
153
Рис. 59. Стандартная кривая ео-
для фага ТЗ.
Пластиковые пробирки обраба-
тывали антисывороткой 1 : 1000.
Количество метки — 0,025 мкКи 50"
на пробирку. Специфическая ак-
тивность 2,3 мкКи/мкг. 1 — вне-
сеиие меченого свидетеля после ~
удаления неактивного АГ; 2— «40-
совместиое введение меченого «•
и немеченого антигенов. ' з
X
<0
” 30-
О)
о
30-
Рис. 60. Стандартная кри-
вая для фага Т4.
4.6
Пластиковые пробирки обраба-
тывали антисывороткой 1 : 1000.
Количество метки — 0,025 мкКи
на пробирку. Специфическая ак-
тивность 7,2 мкКи/мкг. 1 — ме-
4.6*10 4.6 хЮ2 4.6 хЮ3 4,6 хЮ4
Количество фагов (нг).логарифм
ченый свидетель и немеченый
АГ внесены одновременно; 2 —
меченый свидетель внесен по-
сле удаления немеченого АГ.
2.9.2.1.4. Тест-системы РИА на фаги ТЗ и Т4
Пробирки, обработанные специфической АС в разведении
1 : 1000, инкубировали с неизвестным количеством АГ, раство-
ренного в 0,5 мл калий-фосфатного буфера с pH 7,4 в течение
18 ч при 37 °C. После удаления образца с фагом и двукратного
отмывания охлажденным до О °C 0,14 М NaCl в каждую про-
бирку вносят по 0,0125 мкКи меченого АГ, растворенного в
0,5 мл буфера (см. выше). Заключительная инкубация продол-
жается 24 ч; если ее продолжительность сократить до 22 ч, то
это несколько снизит чувствительность анализа. Количество
связавшегося меченого АГ определяют после отмывки проби-
рок раствором NaCl. Измерение радиоактивности проводят на
жидкостном сцинтилляционном счетчике. Эффективность счета
154
125[I] составляла 54%. Получаемая зависимость носит харак-
тер, представленный на рис. 59 и 60. Линейная зависимость
наблюдалась в области концентраций немеченого АГ от 18 до
1800 нг для фага ТЗ и от 46 до 4600 нг для фага Т4.
2.9.2.2. МЕТОД ДВОЙНЫХ АНТИТЕЛ
Определяли IgE в крови нормальных и страдающих аллергией
пациентов при помощи вторичной преципитирующей АС. В ка-
честве первичного связывающего агента использовали моноспе-
цифическую кроличью антисыворотку к Fc-фрагменту IgE. Чис-
тый IgE получали из плазмы пациента, страдающего IgE про-
дуцирующей миеломой. Специфическая активность меченого АГ
составляла 150—250 мкКи/мкг. Все разведения готовили на
0,01 М фосфатном буфере (pH 7,5) с добавлением 1% БСА.
Определение IgE в сыворотке человека. Стан-
дарт IgE или анализируемую сыворотку доводят БСА-фосфат-
ным буфером до объема 0,5 мл. Специфическую кроличью
анти-IgE сыворотку в разведении 1:50 000 вносят аликвотами
по 0,1 мл в измерительные сосуды. В таком же объеме вносят
0,1 нг меченого АГ. После инкубации (24—96 ч при 4 °C) до-
бавляют 0,15 мл козьей АС к IgG кролика и 0,15 мл разведен-
ной в соотношении 1:20 нормальной сыворотки кролика. На-
личие нормальной сыворотки облегчает преципитацию в зоне
эквивалентности на последней стадии инкубации. Этот этап за-
нимает 18—72 ч при 4 °C. Надосадочную фракцию после 30-ми-
нутного центрифугирования при 3500 g отбрасывают. Процент
адсорбированной радиоактивности используется для построения
стандартной кривой. В описанных условиях можно определять
1—14 нг IgE (рис. 61). Добавление ИГ других классов (2 мг
IgA, 0,5 мг IgM, 4 мг IgG) вызывало снижение включения мет-
ки всего лишь на 6%.
2.9.3. МЕТОД ФАРРА
2.9.3.1, ПРИНЦИП МЕТОДА
В 1958 г. Фарр предложил методику определения связывающей
способности антисывороток, в принципе применимую ко всем АГ.
Модифицировав эту методику, можно также определять АТ.
Сходный с ней метод радиоиммунопреципитации с использова-
нием меченых вирусов, например вирусов полиомиелита, приме-
няется для диагностики инфекционных заболеваний. В под-
тверждении диагноза системной красной волчанки (СКВ) ме-
тод Фарра нашел самое широкое применение. Для постановки
эксперимента необходимо наличие радиоактивно меченых АГ.
Его иммунореактивные свойства не должны изменяться после
маркировки, и он должен легко отделяться в связанном с АТ
виде от нес вязавшегося АГ. Для маркировки in vitro и in vivo
155
Рис. 61. Стандартная кривая для оп-
ределения IgE методом двойных ан-
тител.
наиболее подходят 3[Н], 14[С]„
32[Р] и 125[1]. Для облегчения
седиментации связанного АТ
антигена применяют насыще-
ние сульфатом аммония до
50%. В этих условиях преци-
питируют ИГ, а также связан-
ный с ИГ антиген. Для этих
же целей можно использовать
ПЭГ или вторую АС; в любом
случае необходимым условием1
остается полная растворимость
АГ. По своей эффективности
ультрацентрифугирование, при-
менение второй антисыворотки
и хроматография приблизи-
тельно одинаковы. Для отделе-
ния ИК можно использовать
фильтры Миллипор. При СКВ образуются ААТ к ДНК- Эти
ААТ исследуют в отношении специфичности к нативной (н) и
денатурированной (д) ДНК. Ниже будет описан способ, осно-
ванный на применении меченой химическим способом ДНК [6].
2.9.3.2. МАРКИРОВАНИЕ АНТИГЕНА
Растворяют 0,1 мг денатурированной ДНК (содержание белка
1%) или 0,6 мг нативной ДНК (содержание белка 2%) в 0,6 мл
буфера (0,1 М ацетат аммония, 0,04 М уксусная кислота»
pH 5,0). Добавляют к смеси 0,1 мл К1 (0,0025 М), 1 мКи 125[1]
и 0,1 мл Т1С13 (0,0015 М). После нагревания в течение 20 мин
при 60 °C добавляют 0,1 мл 0,05 М Na2SO3 и 0,2 мл буфера
с pH 9,0 (1 М уксусная кислота, 0,05 М аммиак). Нагревают
еще раз в течение 20 мин при 60 °C. Несвязавшийся 125[I] уда-
ляют гель-хроматографией на сефадексе G-50 или диализом
против фосфатного буфера (0,1 М Na2HPO4, 0,14 М NaCl,
pH 8,0, в дальнейшем обозначаемый, как буфер Фарра). Специ-
фическая радиоактивность д-ДНК составляет 2 мкКи/мкг,
а н-ДНК — 0,3 мкКи/м кг.
2.9.3.3. ОПРЕДЕЛЕНИЕ АНТИТЕЛ К ДНК
К 50 мкл разведенной в соотношении 1 + 9 буфером Фарра сы-
воротке пациента добавляют 0,02 мкКи меченой ДНК (100 нг),
растворенной в 50 мкл буфера Фарра. В контрольные пробы
добавляют такое же количество немеченой ДНК- Инкубируют
смесь в течение 1 ч при 37 °C и в течение 16 ч при 4 °C. Добав-
ляют 100 мкл насыщенного раствора сульфата аммония (если
используют д-ДНК) или 100 мкл антисыворотки козы к у-гло-
булину человека, разведенной в соотношении 1 + 2 (если ис-
156
дДНК
Насыщенный (NH4) 2 SO 4
(100 мн л) j---------
125 \ .
[|]-ДНН.О.О2мкНи(5О мил)4-----нДНН---*
Сыворотка.разведение 1; 10
(50 мкл) Антисыворотки,
разведение 1:3 (100 мкл)
4°С
24 ч 4°С
J 2000g
15 мин
J 2000g
30 мин
R
S
надосадочная
франция,100 мл>-
S (надосадочная’
франция,100 мл)
Рис. 62. Определение антител к ДНК.
пользуют н-ДНК). Для определения ДНК, связанной антитела-
ми, измеряют радиоактивность 100 мкл надосадочной фракции
(S) и 100 мкл надосадочной фракции, а также радиоактивность
осадка (R) после центрифугирования (рис. 62). С использова-
нием вышеописанного метода и химически меченой ДНК в ка-
честве антигена можно определять способность сывороток свя-
зывать АГ, а также построить кривые титрования. Кроме того,
увеличивая количество ДНК в пробе при постоянном разведе-
нии сыворотки, можно сравнивать авидность АТ. При наличии
сильного неспецифического связывания рекомендуется использо-
вать детергенты или буферы с высокой ионной силой [5].
2.9.4. МЕТОДЫ, ОСНОВАННЫЕ НА ПРИМЕНЕНИИ
МЕЧЕНЫХ АНТИТЕЛ
Меченые радиоактивными изотопами антитела представляют со-
бой универсальные реагенты для определения различных АГ
(вирусы, гормоны и т. д.). Существенным отличием от обычного
РИА является то, что меченый компонент должен содержаться
в избытке. После окончания реакции в одном из вариантов им-
мунорадиометрического анализа избыток меченых АТ удаляют
АГ, адсорбированным на твердой фазе. В случае использования
так называемого сэндвич-метода необходима длительная отмыв-
ка после первого этапа использования твердой фазы. На рис. 63
и 64 схематически представлены основные этапы анализа. Сте-
пень связывания в иммунорадиометрическом анализе опреде-
ляется путем измерения радиоактивности надосадочной фрак-
ции после центрифугирования. В процессе оптимизации анализа-
определяющими факторами являются неспецифическое связы-
вание, продолжительность инкубации и концентрация мече-
ных АТ. Неспецифическое связывание можно уменьшить, рабо-
тая с высокоочищениыми АТ, кроме того, оно зависит от ион-
ной силы; его уменьшения можно добиться добавлением жела-
тина или сывороточного альбумина. Использование избытка ме-
ченых АТ позволяет сократить время инкубации; правда, это
приводит к снижению чувствительности [10]. Применяемые для
«сэндвич-метода» АТ должны относиться только к классу IgG;
их довольно легко получать, и, кроме того, они пригодны для
157
АГ »гроба)
специфические
АТ
Рис. 63. Принцип им-
мунор адиометрического
анализа.
АГ на твердой фазе
ИннуСаии® •
--------------►
Центриф,! ар. в'чие
Осадок
Аллерген IgE (проба)
на твердой
Фазе
|И[1)анти1дЕ-АТ,
избыток
Рис. 64. Схема «сэндвич-
метода».
Инкубация
Центрифуг ировгние
Надосадочная фракци>
о
о +
работы с самыми различными АГ. Применяя АТ со специфич-
ностью к Н-цепям иммуноглобулинов, можно установить класс
первично связывающих АТ. Использование меченых АТ пред-
ставляется приемлемой альтернативой в тех случаях, когда по-
лучение меченого АГ является неэффективным или дорогостоя-
щим процессом. Меченые 1251 АТ получаются сравнительно лег-
ко, кроме того, они сохраняют специфическую иммунореактив-
ность в течение длительного времени [15]. Методы, основанные
на применении меченых АТ, давно уже вошли в практику мно-
гих лабораторий. Количественное определение АТ затруднено
ввиду их различной авидности, для проведения стандартизации
необходимо применение референс-сывороток [15]. Применяя ан-
титела, модифицированные 2,4-динитрофенилсульфоновой кис-
лотой, можно количественно определять различные АГ, если
имеются меченые 125[1] анти-ДНФ антитела; одновременно
с этим наблюдается повышение чувствительности. Этот вариант
анализа с успехом применяли для определения ферритина,
овальбумина, gs-антигена аденовирусов и НВе-антигена.
Иммунорадиометрическое определение при помощи антител,
специфических к ДНФ-группе. IgG очищают при помощи хро-
матографии на ДЭАЭ-целлюлозе из соответствующих АС.
В концентрации 100 мкг/мл в 0,1 М трис-НС1-буфере с pH 8,8
158
Рис. 66. Стандартная кривая
для определения НВе-антигена.
1 — непрямой <сэндвич-метод>; 2 —
прямой «сэидвич-метод» [10].
IgG используют для обработки пластиковых гранул диаметром
8 мм. Такой препарат IgG используют для получения 2,4-динит-
рофенильного производного. Анти-ДНФ антитела получают им-
мунизацией кроликов ДНФ-БСА. Специфические антитела вы-
деляют, добавляя 10 мг ДНФ-апоферритина к 2 мл антисыво-
ротки, что вызывает иммунопреципитацию. Иммунные комплек-
сы осаждают центрифугированием при 90 000 g в течение 1 ч,
осадок в 1 мл фосфатного буфера с мочевиной (0,01 М фосфат
с pH 8,0; 0,1% нонидет Р40; 8 М мочевина). После хромато-
графии на колонке с 2 мл ДЭАЭ-целлюлозы фракция, содер-
жащая IgG, диализуется против 0,05 М боратного буфера
с pH 8,5 и метится реагентом Bolton-Hunter (специфическая ра-
диоактивность 125[1] 1,2 мкКи/мкг). Адсорбция антигена на
пластиковых гранулах происходит в течение ночи при 20 °C
в 400 мкл нормальной сыворотки. Гранулы отмывают буфером
(0,01 М трис-HCl буфером с pH 8,0, 0,14 М NaCl, 0,1% нони-
дет Р40). В каждую пробиркув носят 0,6—2,5 мкг антигенспе-
цифических ДНФ-АТ и инкубируют 2 ч при 37 °C. Затем вносят
0,1 мкКи 125[1]-АТ, специфических к ДНФ группам, также рас-
159
творенным в 400 мкл нормальной сыворотки. После инкубации
в течение 2 ч при 37 °C и повторной отмывки тем же буфером
измеряют количество специфических 125[I]-антител.
Чувствительность определения овальбумина и ферритина со-
ставляет соответственно 0,8 и 0,2 нг/мл; сравнительно с прямым
твердофазным анализом с применением 125[I]-антител чувстви-
тельность определения увеличивается в 4 или 15 раз соответ-
ственно (рис. 65). Чувствительность за счет эффекта амплифи-
кации при использовании анти-ДНФ-антител увеличивается
также и в отношении НВе-антигена (рис. 66). Известен метод
твердофазного анализа, основанный на применении 125[1]-про-
теина А для определения специфических АТ [9]. Хотя этот ме-
тод основан на избирательном сродстве протеина А к Fc-фраг-
менту IgG, фиксация АГ на матрице должна осуществляться без
участия антител.
2.9.5. ОЦЕНКА МЕТОДА
РИА прочно вошел в арсенал современных лабораторных кли-
нических исследований. Обилие методических вариантов свиде-
тельствует о том, что не всегда можно придерживаться одной
схемы и что следует учитывать качество реагентов. Методы, ос-
нованные на использовании твердой фазы (пластмасса, целлю-
лоза, сефадекс), привели к существенному упрощению процес-
са. За исключением у-счетчика, метод не требует сложного обо-
рудования. Процедуры, занимающие длительное время (отбор
проб, отмывание и измерение радиоактивности), могут быть ав-
томатизированы. Колебания в серии измерений (и = 5) не пре-
вышают, как правило, 10%. Перекрестная реактивность АТ
вследствие фиксации на твердой фазе не претерпевает сущест-
венных изменений. Адсорбция сывороточных белков приводит
к тому, что те из них, которые обладали ингибирующей или
протеолитической активностью, оказываются не в состоянии
влиять на результаты анализа. Метод двойных АТ, при котором
происходит отделение ИК за счет второй сыворотки, обладает
очень высокой воспроизводимостью, но требует много времени
(3—7 дней). К числу преимуществ данной методики относится
небольшой расход сыворотки на одно исследование. Это обстоя-
тельство может иметь важное значение при работе со слабо им-
муногенными веществами, для которых получение АС представ-
ляет известные трудности. Обе основные реакции различаются
по тому, в какой области концентраций они происходят: конку-
рентная реакция протекает при концентрациях порядка нано-
граммов вещества, преципитация — микрограммов вещества. Воз-
можные ошибки во вторичной реакции можно уменьшить, ис-
пользуя очищенные фракции у-глобулина или предварительное
прогревание. Еще одним способом, которым можно избежать не-
специфические реакции, является использование препреципити-
.рующих антител или присоединение вторичной сыворотки
160
к твердой фазе (целлюлоза). Однозначно судить о преимущест-
вах РИА или метода двойных АТ нельзя. На примере опреде-
ления хорионического гонадотропина видно, что присоедине-
ние АТ к твердой фазе бромциановым методом приводит к за-
метному снижению их специфической иммунореактивности, что
означает также снижение чувствительнеости. Сравнение метода
двойных АТ и двух твердофазных методов в отношении опреде-
ления IgE свидетельствует о том, что с нормальными сыворот-
ками первый дает заниженные результаты, а в случае патологии
результаты, получаемые всеми тремя методами, совпадают. Ме-
тод двойных АТ обладает несомненными преимуществами пе-
ред методом преципитации сульфатом аммония [8]. После конъ-
югации активность АТ к гормонам с высокой относительной мо-
лекулярной массой (6000—26000) снижалась на 10—15% с па-
раллельным уменьшением чувствительности. Эти различия, ве-
роятно, обусловлены различной степенью доступности связан-
ных АТ для АГ. Метод Фарра занимает достаточно прочные
позиции в клинической диагностике, по крайней мере в отноше-
нии определения антител к ДНК и РНК. К тому же остаются
нерешенными вопросы стандартизации антигенов и неспецифи-
ческого связывания. Важным преимуществом метода Фарра яв-
ляется то, что используются различные связывающие и преци-
питирующие АТ. РИА с использованием меченых АТ находит
применение для определения веществ, присутствующих в коли-
чествах, измеряемых пикограммами, таких как специфические
IgE-антитела и некоторые вирусные и бактериальные АГ. Осо-
бенно выгодно использовать данную модификацию, когда име-
ются трудности с мечением АГ. При этом преодолеваются слож-
ности с антигенами, которые по своей иммунореактивности отли-
чаются от меченых стандартных АГ. Взаимодействие меченого
АТ с другим АТ (в качестве АГ) приводит к амплификации
данных измерений. Определение содержания тех или иных ве-
ществ на основе реакции АГ—АТ не дает непосредственной кор-
реляции с их биологической активностью. Для подтверждения
заключений на основе РИА требуется дополнительная биопро-
ба. Использование АТ в качестве первичного связывающего
агента позволяет обойти это ограничение. До настоящего вре-
мени в клинике используют анализ пептидов, стероидных гор-
монов, гликопротеидов при помощи радиолигандов и клеточных
рецепторов. Можно получать стандартизованные по сравнению
с антисыворотками препараты АТ, но они не отличаются доста-
точной стойкостью. Особенности радиоиммунологической техни-
ки, такие как необходимость защиты от излучения, ограничен-
ное время жизни реагентов, привели к поиску альтернативных
методов маркировки. Было описано применение бактериофагов,
флюорохромов, ферментов, твердофазных частиц и даже ато-
мов металлов в качестве маркеров. Но все эти методы пока не
могут вытеснить радиоиммунологические методы иследова-
ния [14].
11-990
161
2.9.6. СПИСОК ЛИТЕРАТУРЫ
1. Bolton. А. Е„ Bennie J. G., Hunter W. М. Innovations in labelling technique
for RIA. — In: Protides in the biological fluids/Ed. H. Peters. — Oxford,
Pergamon Press, 1976.
2. Breuer H., Hamel D., Kriiskemper H. L. Methoden der Hormonbestimmung. —
Suttgart, Thieme Verlag, 1975.
3. Broughton A., Strong J. E. RIA of antibiotics and chemotherapeutic agents. —
Clin, chem., 1976, 22, 726—732.
4. Cleiland R., Christenson J. et al. Detection of drug abuse by RIA: a summary
of published data and some new information. — Clin, chem., 1976, 22, 712—
725.
5. Etzrodt H. The use of tween 20 in the double antibody RIA. — Hoppe-Sey-
lers Z. Physiol. Chem., 1976, 357, 1205—1208.
6. Falck P. Radioimmunologisher Nachweis DNA-spezifischer Antikorper. —
Acta biol. med. germ., 1976, 35, 93—101.
7. Hunter W. M. Radioimmunoassay. — In: Handbook of experimental immuno-
logy/Ed. D. M. Weir. — Oxford, Blackwell Scient. Publ., 1978, vol. 1, 1-1.1—
1.40.
8. Kirkpatric A„ Wepsic T. H., Nakamura R. M. Comparision of double-antibody
RIA with Farr-technique RIA and double antibody enzyme assay for a-feto-
protein. — Clin, chem., 1977, 23, 50—59.
9. Marler R„ Jansen M„ Andriole U. T. A new method for measuring antibody
using radiolabeled protein A in solid phase RIA. — J. Immunol, methods.,
1979, 28, 41—49.
10. Miles L. E. M. Immunoradiometric assay, new developments in theory and
practice. — In: Protides of the biological fluids (Ed. H. Peters). Oxford, 1976,
695—704.
11. Neurath A. R., Strick N. 1257-labeled anti-2,4-dinitrophenyl (DNP) antibodies:
a universal reagent for solid phase immunoassays. — Molec. Immunol., 1979,
16, 625—628.
12. Nye L., Forrest G. C. et al. Solid-phase magnetic particle RIA. — Clin. chim.
Acta, 1976, 69, 387—396.
13. Parker C. W. Radioimmunoassay of biologically active compounds. New Jer-
sey, Prentice-Hall Inc. Englewood Cliffs, 1976.
14. Schall R. F., Tenesco H. J. Alternatives to RIA: labels and methods. — Clin.
Chem., 1981, 27, 1157—1164.
15. Schellenberg R. R., Adkinson N. F. Measurement of absolute amounts of anti-
gen-specific human IgE by a RAST elution technique. — J. Immunol., 1975,
115, 1577—1583.
16. Secher D. S. Immunoradiometric assay of human leucocyte interferon using
monoclonal antibodies. — Nature, 290, 501—503.
17. Steiner M„ Spratt J. L. Solid-phase RIA for morphine with use of an affinity-
purified morphine antibody. — Clin, chem., 1978, 24, 339—342.
18. Walker W. H. C., Keane P. M. Theoretical aspects of RIA. — In: Handbook of
radioimmunoassay/Ed. G. M. Abraham. New York — Basel, Marcel Dekker
Inc., 1977, 87—130.
2.10. ИММУНОФЕРМЕНТНЫЙ АНАЛИЗ
2.10.1. ПРИНЦИП МЕТОДА
Иммуноферментный анализ (ИФА) основан на том же принци-
пе, что и РИА, только вместо радиоактивной метки применяется
ферментная. Метод иммуноферментного анализа был предло-
жен в начале 70-х годов тремя независимыми группами иссле-
дователей: Engvall и Perlmann в Швеции, van Weemen и
Schuur в Нидерландах и Rubenstein с сотр. в США. АГ или АТ,
162
вступающее в иммунную реакцию, метится ферментом. По пре-
вращению субстрата ферментом можно судить о количестве
вступившего во взаимодействие компонента реакции АГ—АТ.
Чувствительность ИФА позволяет определять минимальные ко-
личества вещества (нанограммы). Известно много вариантов
постановки ИФА [2]. Различают гомогенный и гетерогенный ва-
риант иммуноферментного анализа. В основе гомогенного ИФА,
применяемого для определения низкомолекулярных субстанций
(гаптены), лежит ингибирование активности фермента при со-
единении его с гаптеном (активность фермента восстанавлива-
ется в результате реакции АГ—АТ) либо, наоборот, потеря ак-
тивности маркерного фермента в результате реакции АГ—АТ
[7]. Поэтому удаление свободного АГ из смеси, содержащей АГ
в связанном виде, в составе иммунных комплексов невозможно.
В противоположность этому при гетерогенном ИФА АГ или АТ
фиксируется „а твердой фазе (как правило, пластик), а непро-
реагировавшие компоненты реакции удаляются многократным
от мыванием.
2.10.2. ВАРИАНТЫ МЕТОДИКИ
В РИА, так же как в ИФА, различают методы конкурентного
и неконкурентного анализа [2].
2.10.2.1. КОНКУРЕНТНЫЙ МЕТОД
Если существует необходимость количественного определения
АГ, который может быть получен в чистом виде, то адекватный
анализ при помощи конъюгата антиген-фермент и фиксирован-
ных на твердой фазе АТ может быть проведен в форме конку-
рентного взаимодействия (рис. 67). Первым этапом в этом слу-
чае будет присоединение АТ к носителю за счет химической ре-
акции или физико-химических взаимодействий. Избыток АТ
удаляют отмыванием и, внося строго определенное количество
меченого АГ и различные количества немеченого АГ, строят ка-
либровочную кривую. По окончании реакции АГ—АТ иммунные
комплексы оказываются присоединенными к твердой фазе за
счет АТ; избыток АГ удаляют отмывкой, добавляют субстрат
для фермента, останавливают реакцию по истечении определен-
ного времени и проводят колориметрическое определение про-
дуктов ферментативной реакции. В другом варианте конкурент-
ного ИФА с твердой фазой первично связывается АГ. После
отмывки добавляют меченые энзимом АТ и стандартный или
исследуемый АГ. Связывание меченых АТ конкурентно тормо-
зится растворенным АГ. Количество продуктов ферментативной
реакции обратно пропорционально концентрации растворенно-
го АГ (рис. 68). Этот метод может включать использование вто-
рого энзим-меченого АТ, специфичного по отношению к IgG,
первично реагирующему с фиксированным на твердой фазе АГ.
Н* 163
АПП
1.
Рис. 67. Схема конкурентного ИФА, антитела сорбированы на твердой фазе,
антиген мечей ферментом [2].
1 - АТ, связывание с твердой фазой; 2 —отмывка; 3 — инкубация с меченным фермен-
том АГ в присутствии (а) и в отсутствие (б) исследуемой пробы; 4 —отмывка; 5 —ин-
кубация с субстратом (светлые кружочки) и измерение количества продуктов реакции
(темные кружочки).
Рис. 68. Схема конкурентного ИФА, антиген сорбирован на твердой фазе,
антитела мечены ферментом [2].
1—связывание АГ с твердой фазой; 2 —отмывка; 3 — инкубация меченных ферментом
АТ в присутствии (а) или в отсутствии (б) исследуемой пробы; 4 — отмывка; 5 — инку-
бация с субстратом (светлые кружочки) в измерение количества продукта реакции (тем-
ные кружочки).
2.10.2.2. НЕКОНКУРЕНТНЫЕ МЕТОДЫ
К числу неконкурентных методов относится метод двойных ан-
тител, или, как его иногда называют, «сэндвич-метод» (рис. 69).
Фиксированные на твердой фазе АТ инкубируют со стандарт-
ным или анализируемым АГ. После отмывания добавляют избы-
ток меченых АТ (специфичных к этому же АГ), несвязавшиеся
АТ отмывают. Продукт ферментативной реакции образуется
в количествах, пропорциональных количеству связанного АГ.
Довольно часто используют немеченые вторые АТ; о реакции
в данном случае судят по связыванию энзим-меченых АТ, спе-
(ифичных по отношению к IgG (вторые антитела). В любом
случае первые и вторые АТ должны принадлежать животным
разных видов. Для иммуноферментного определения антител
часто используется непрямой, неконкурентный ИФА (рис. 70).
Антиген фиксируют на твердой фазе, после отмывания проводят
инкубацию с раствором АТ. Если произошло специфическое
164
1,
Рис. 69. Схема «сэндвич-ИФА> (метод двойных антител для определения ан-
тигена.
I—-АТ, связывание с твердой фазой; 2 —отмывка; 3 —инкубация с пробой, содержа-
щей АГ; 4 — отмывка; 5 —инкубация с конъюгатом АТ-фермент; 6 —отмывка; 7 —ин-
кубация с субстратом (светлые кружки) и измерение количества продукта реакции (тем-
ные кружочки).
Рис. 70. Схема непрямого ИФА для определения антител при помощи фер-
ментмеченого аитиглобулина.
I — связывание АГ с твердой фазой; 2 — отмывкай 3 — инкубация с пробой, содержащей
АТ; 4 — отмывка; 5 —инкубация с конъюгатом аитиглобулнн-фермент; 6 —отмывка;
7 — инкубация с субстратом (светлые кружочки) н измерение количества продукта ре-
акции (темные кружочки).
связывание АТ, их можно количественно определить при помо-
щи меченой ферментом аитиглобулиновой сыворотки, фермента-
тивной реакции. Прочие неконкурентные методы ИФА представ-
ляют собой различные модификации конкурентного метода.
Вместо совместной инкубации меченых и немеченых АГ со свя-
занными антителами можно непосредственно инкубировать
только стандартный или анализируемый АГ, а затем после от-
мывания проводить инкубацию с избытком энзим-меченого АГ,
который взаимодействует с «незанятыми» АТ. Кроме того, стан-
дартный или анализируемый АГ можно инкубировать с избыт-
ком энзим-меченых антител и по окончании иммунной реакции
добавлять эту смесь к антигену, фиксированному на твердой
165
фазе. Определяют количество свободных энзим-меченых АТ, свя-
занных антигеном на твердой фазе. В обоих случаях концентра-
ция продукта ферментативной реакции пропорциональна кон-
центрации анализируемого антигена.
2.10.3. ПРИМЕР ПРОВЕДЕНИЯ ИССЛЕДОВАНИЯ
Последующее описание основывается на гетерогенном ИФА при-
менительно к определению HBs-антигена двойным «сэндвич-ме-
тодом» [10, 11].
2.10.3.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: полистироловые пробирки или план-
шеты (Polypast, Хальберштадт), анти-НВд-гамма-глобулин
морской свинки (первые АТ), анти-НВэ-гамма-глобулин кроли-
ка (вторые АТ), бараньи антитела к IgG кролика, меченые пе-
роксидазой (фракция IgG), HBs-стандарт (Austria II, Abbott
lab., Чикаго), HBs-позитивная сыворотка (вторичный стандарт),
ФСБ (0,1 М фосфатный буфер с pH 7,2, 1 М NaCl), человече-
ский сывороточный альбумин (Институт вакцин, Дессау), орто-
дианизидин (Serva, Гейдельберг), Н2О2 (целлулоидная фабри-
ка, Эйленберг), сорбитовый эфир полиоксиэтилена (Serva, Гей-
дельберг), НС1 5 М, спектрофотометр (например, Бреко! 20,
Карл Цейсс, Йена).
2.10.3.2. МЕТОДИКА
Полистироловые пробирки в течение ночи при 4 °C инкубируют
с 200 мкл анти-НВз-гамма-глобулина морской свинки (10 мг/л
в ФСБ), после инкубации трижды отмывают порциями ФСБ
по 600 мкл. В подготовленные таким способом пробирки добав-
ляют по 200 мкл различных разведений стандарта или анализи-
руемых сывороток. Пробы инкубируют 2 ч при 37 °C или 16 ч
при комнатной температуре, затем трижды отмывают ФСБ. Свя-
завшийся с первыми АТ HBs-антиген на следующем этапе на-
сыщается вторыми АТ. Для этого в пробы добавляют по 200 мкл
анти-НВэ-гамма-глобулина кролика (10 мг/л в ФСБ, содержа-
щем 0,5 мМ ЧСА), снова инкубируют в течение 2 ч при 37 °C,
трижды отмывают ФСБ и вносят в пробы по 200 мкл антител
к IgG кролика, меченых пероксидазой (1,2 мг/л ФСБ, содержа-
щем 0,5 мМ ЧСА). Инкубируют еще 2 ч при 37СС. После трое-
кратного отмывания ФСБ проводят ферментативную реакцию.
В пробы вносят по 500 мкл субстрата (о-дианизин 0,32 мМ,
Н2Ог 0,7 мМ, сорбитовый эфир полиоксиэтилена 1,1 г/л ФСБ).
Инкубируют их 2 ч при комнатной температуре, останавливают
ферментативную реакцию добавлением 500 мкл 5 М НС1 и из-
меряют экстинкцию при длине волны 535 нм в объеме 1 мл.
166
На рис. 71 схематически по-
казан ход работы, на рис.
72 — калибровочная кривая.
2.10.4. ОЦЕНКА МЕТОДА
Область применения и чув-
ствительность ИФА соответ-
ствуют радиоиммунологиче-
ским методам исследования.
Но ИФА по сравнению с
РИА обладает целым рядом
преимуществ: не использу-
ются радиоактивные изото-
пы, стабильность конъюга-
тов позволяет хранить их в
течение длительного време-
ни1, измерение оптической
плотности проводят в опти-
ческом диапазоне, результа-
ты ИФА можно оценивать
полуколичественно без при-
менения аппаратуры (ви-
зуально). ИФА очень легко
поддается автоматизации,
может быть использован,
наконец, в иммуногистоло-
гических исследованиях и
реакциях иммунопреципита-
ции.
Для ИФА используются
более дешевые материалы и
приборы, чем для РИА.
И отчасти в связи с этим об-
ласть применения ИФА Рис. 71. Схема количественного опреде-
очень широка (см.табл. 11). ления HBs-антигена при помощи ИФ.и
Так же как в РИА, специ-
фичность и чувствительность ИФА даже при оптимальных усло-
виях зависят от качества сыворотки. Рекомендуется использо-
вать только высокоавидные и высокоспецифические АТ. Антите-
ла достаточной для ИФА степени чистоты легко могут быть по-
лучены путем иммуноадсорбции (см. раздел 4.3), вс время кото-
рой удаляются неспецифические и могущие быть помехой АТ.
Кроме того, необходимо исключить возможные перекрестные
реакции с другими антигенами. Специфичность реакции может
быть уменьшена неспецифической адсорбцией антител или эн-
1 Конъюгаты фирмы Labsystems сохраняют активность в течение года при
температуре 4°C. — Примеч. пер.
167
зим-меченых компонентов
реакции на твердой фазе.
Самыми сложными и
важными этапами ИФА
являются получение и
очистка ферментных
конъюгатов. Для присо-
единения ферментов к
белкам чаще всего ис-
пользуют глутаральдегид-
ный метод (см. раз-
дел 4.3). Поскольку на-
личие неконъюгирован-
ных белков снижает спе-
цифичность реакции, не-
обходимо проводить
очистку конъюгатов гель-
фильтрацией или любым
другим методом. При по-
лучении конъюгатов пе-
роксидаза — иммуногло-
булин молярное соотно-
шение фермент/ИГ рас-
считывается по соотноше-
Рис. 72. Калибровочная кривая для опреде- НИЮ ЭКСТИНКЦИИ при ДЛИ-
ления HBs-антигена методом ИФА. Н,е ВОЛНЫ 405 И 208 нм.
Иммунологическая харак-
теристика фермент-белковых конъюгатов исследуется при по-
мощи метода иммунодиффузии (см. разделы 2.1, 2.2). Для раз-
работки системы ИФА лучше использовать общедоступные ан-
тигенонезависимые конъюгаты, такие как антитиповые АТ к ИГ
или протеин А, конъюгированные с ферментом [10]. Такие
конъюгаты дают возможность распространять иммунофермент-
ную диагностику на ряд других антигенов. В качестве маркеров
для ИФА чаще всего используют пероксидазу, щелочную фос-
фатазу или глюкозооксидазу (табл. 12). Коммерческие препа-
раты пероксидазы хрена требуют дополнительной очистки aoRZ
более 2,5. RZ (нем. Reinheit-Zahl— буквально число чистоты)
определяется как соотношение экстинкции на длинах волн 405
и 280 нм. Высокоочищенные препараты пероксидазы имеют RZ
3,0 и выше. Воспроизводимость результатов ИФА зависит от
многих факторов, в том числе от условий измерения, оптимиза-
ции фермент-субстратного взаимодействия, толщины слоя и по-
глощающих свойств анализируемых растворов. Негомогенность
материала планшетов (неконтролируемый фактор) отрицатель-
но влияет на воспроизводимость. Отмывки между отдельными
этапами анализа моющими средствами, добавлением БСА и др.
также влияют на нее. Коэффициент вариации ИФА составляет
менее 10%. В качестве твердой фазы для ИФА применяют та-
168
Таблица 11. Применение ИФА в медико-биологических исследованиях
[3, 4, 5, 6, 8, 9, 12, 13, Обзоры 1 и 2]
Объект исследования Примеры
Микроорганизмы (АТ или АГ) Вирусы Коксаки В, гепатита А и В, простего герпеса, инфлюэнцы, краснухи; бруцеллы, холер- ный вибрион, кишечная палочка, сальмонеллы, трепонемы; амебы, плазмодии, токсоплазмы, трипаносомы, столбнячная палочка
Гельминты Белки сыворотки Филярии, шистосомы, трихинеллы а-Фетопротеин, канцероэмбриональный антиген, фактор VIII, ферритин, фибронектин, гаптогло- бин, IgA, S-IgA, IgE, IgG, IgM
Г ормоны Кортизон, человеческий хорионический гонадо- тропин, человеческий плацентарный лактоген, ин- сулин, эстрогены, прогестерон, тестостерон, ре- нин, соматостатин, тироксин, тиреотропин
Медикаменты Хинидин, кодеин, дигоксин, гентамицина сульфат, метотрексат, морфнн, препараты группы пеницил- лина, фенобарбитал, новокаинамид, пропанолол (Propanolol) теофиллин и др.
Разное Аденозин, ДНФ-IgG, С-реактивный белок
Таблица 12. Ферменты-маркеры, наиболее часто используемые в ИФА
Фермент Индикатор реакции Реагент, останавливающий реакцию Длина волны для оценки реакции
Пероксидаза КФ 1.11.1.7 Н2О2 орто-дианизидин (или ортофениленди- НС15 М 535
амии); Na2SO4 2 М
2,2-азино-диэтилбензтиа- золинсульфат (АБТС) NaOH 1 М 405 405
Щелочная фосфатаза КФЗ.1.2.1 Пара-нитрофенилфосфат NaOH 1 М 402
Глюкозооксидаза КФ 1.1.3.4 Н2О2 (пероксидаза) о-ди- анизиднн или АБТС H2SO4 0,5 М 405
D-fJ-галактозидаза КФ 3.2.1.23 Пара-нитрофенил-P-D' галактозид 366
кие материалы, как полистирол, поливинилхлорид, полипропи-
лен, полиакриламид, агарозу, целлюлозу и др. Твердая фаза мо-
жет быть использована в виде пробирок, планшетов для микро-
титрования, бусин, шариков, гранул, пленок и т. д. Необходи-
мым прибором для ИФА является спектрофотометр. При анали-
169
зе больших серий материала желательно использовать автомати-
ческие пипетки и автоматические ридеры (приборы для автома-
тического считывания результатов ИФА, от англ, read — чи-
тать). В будущем ИФА, вероятно, сможет полностью вытеснить
РИА, если удастся преодолеть некоторые сложности: снизить
затраты ручного труда путем автоматизации, исключить кова-
лентное или адсорбционное связывание АГ или АТ с твердой
фазой, а также освоить приготовление конъюгатов ферментов
с небольшими молекулами.
2.10.5. СПИСОК ЛИТЕРАТУРЫ
Обзоры
1. Maggio Е. Т. Emzyme immunoassay. Florida, CRC Press, Boca Raton, 1981.
2. Oellerich M. Enzyme immunoassay in clinical chemistry. Present status and
trends. — J. Clin. chem. biochem., 1980, 18, 197—208.
Цитированная литература
1. Butler 1. E., McGivern P. L., Cantarero L. A. Application of the amplified
enzyme-linked immuno-sorbent assay: comparative quantitation of bovine
serum IgGi, IgG2, IgA and IgM. antibodies. — Amer. J. veterinary research,
1.980, 41, 1479—1491.
2. Clark R., Engvall E. Enzyme-linked immunosorbent assay (ELISA). Theoreti-
cal and practical aspects. Enzyme-Immunoassay (E. T. Maggio ed.). Boca
Raton, Florida, 1981, 167—179.
3. Grenner G., Schmidtberger E. Enzymimmunoassay zum Nachweis von Anti-
HBs (Antikorper gegen Hepatitis B-Oberflachen Antigen). Ztschr. Anal.
Chem., 1980, 301, 125—128.
4. Hannah D., Gotze M. A., Katterman R. Bestiipmung der Gesamtostrogene im
Serum mit einem heterogenen Enzymimmunoassay. — Z. Anal. Chem., 1980,
301, 131—132.
5. Jennings R. T., Smith T., Potter C. IT. Use of the enzyme-linked immunosor-
bent assay (ELISA) for the estimation of serum antibodiesin an influenza
virus vaccine study. — Med. Microbiol. Immunol., 1981, 169, 247—258.
6. Lindenschmidt E. C., Muller F. Demonstration of specific 19S (IgM) anti-
bodies in untreated and treated syphilis. — Br. J. Vener, Dis., 1982, 58,
12—17.
7. Mohr P., Zschische IT. Enzymimmunoassays in der medizinischen Diagnos-
tik. — Wissenschaft und Fortschritt, 1981, 31, 374—379.
8 Oellerich TH. Enzyme immunoassays in clinical chemistry. Present status and
trends.—J. clin. chem. clin. biochem., 1980, 18, 197—208.
9. Poskitt P., Poskitt T. A quantitative enzyme-linked immunoassay for scrum
immune. — J. Clin. Lab. Immunol., 1981, 5, 125—128.
10. Porstmann T. et al. Entwicklung eines Doppel-Sandwich-Enzymimmunoassay
zum quantitativen Nachweis von HbsAg (1. Mitteilung). Dt. Gesund. Wes.,
1980, 35, 598—600.
11. Portsmann T. et al. Entwicklung eines Doppel-Sandwich-Enzymimmunoassays
zum quantitative Nachweis von HBsAg (Mitteilung), 1980, Dt. Gesundh.
Wes. 35, 1587—1593.
12. Salonen E. M. A rapid and sensitive solid-phase enzyme immunoassay for
C-reactive protein. — J. Immunol. Meth., 1982, 48, 45—50.
13. Tole K„ Schenck K., Brandzaeg P. Enzyme linked immunosorbent assay for
human IgG, IgA, and IgM antibodies to antigens from anaerobic cultures
of seven oral bacteria. — J. Immunol. Methods, 1981, 45, 27—40.
14. Vejtorp M. Solid phase anti-IgM ELISA for detection of rubella specific
IgM-antibodies. — Acta Path. Microbiol. Scand., 1981, 89, 123—128.
15. Vuento M. et al. Competitive enzyme immunoassay for human plasma fibre-
nectin. — J. Immunol. Meth., 1981, 40, 101—108.
170
2.11. ТИТРОВАНИЕ КОМПЛЕМЕНТА
2.11.1. ПРИНЦИП МЕТОДА
Количественное определение комплемента производится в на-
стоящее время, как правило, методом иммунного гемолиза.
Метод основан на комплементзависимом лизисе нагруженных
АТ эритроцитов барана. Для полного гемолиза необходимо
наличие всех компонентов комплемента. В соответствии с меж-
дународным стандартом за единицу активности комплемента
принимается такое его количество, которое вызывает 50% гемо-
лиз в стандартных условиях. Единица активности комплемента
называется также гемолитической единицей и обозначается
СН50. В рутинных лабораторных исследованиях пользуются
обычно полуколичественным определением гемолиза в агарозе,
оно проще и занимает меньше времени, чем определение СН50.
2.11.2. МЕТОДИКА
2.11.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Растворы: 1) раствор Alsever (глюкоза 21,0 г, цитрат нат-
рия 8,0 г, NaCl 4,0 г, дистиллированная вода до 1000 мл), ав-
токлавируют; 2) калийно-боратный раствор (калия сульфата
10,0 г, борная кислота 4,0 г, дистиллированная вода до 100 мл);
3) барбиталовый буфер (исходный раствор, NaCl 42,5 г, бар-
битал 2,875 г, барбитал-натрий 1,875 г, СаС12-6Н2О 0,164 г,
MgC’i2-6H2O 0,616 г, дистиллированная вода до 1000,0 мл);
4) раствор желатина (желатин 2,0 г, дистиллированная вода
до 100,0 мл); 5) рабочий буфер (барбиталовый буфер, исход-
ный раствор 200 мл, дистиллированная вода 800 мл, раствор
желатина 10 мл, pH должна составлять 7,2, доводят pH 0,1 М.
ХаОН). Все разведения готовят на рабочем буфере.
Оборудование: водяная баня с регуляцией температуры
от 35 до 56 °C, термостат, центрифуга (например, Т 23, Zentri-
fugenbau, Энгельсдорф), гематокритная центрифуга (например,
TH 12, Zentrifugenbau, Энгельсдорф), гемолизины к ЭБ, в ка-
честве амбоцептора (Саксонский сывороточный завод, Дрез-
ден); суспензия ЭБ в растворе Олсвера (1—16-педельной дав-
ности), сыворотка морской свинки, по возможности свежая или
из глубокой заморозки (источник комплемента), сапонин по-
рошкообразный, фотоизмеряющее устройство на 541 нм (на-
пример, Specol Carl Zeiss, Йена), агароза чистейшая, стеклян-
ные пластины подходящего размера (например, 100X200X3 мм)»
разграфленная бумага с абсциссами, имеющими логарифмиче-
ские деления.
171
2.11.2.2. ПОДГОТОВКА СЫВОРОТКИ
ДЛЯ ИССЛЕДОВАНИЯ
Забор крови проводят без антикоагулянтов, пробе дают по-
стоять 1 час при комнатной температуре, центрифугируют
(10 мин при 1000 g) и отделяют сыворотку. Рекомендуется
проводить титрование комплемента сразу после получения сы-
воротки. Если это не удается, то сыворотку ввиду термола-
бильности комплемента рекомендуется хранить в шкафу глу-
бокого охлаждения (—80°C). Применение плазмы в данном
случае не дает никаких преимуществ. После разведения сыво-
ротки калий-боратным буфером в отношении 1 + 2 ее можно
хранить в течение нескольких недель при 4 °C без потери ак-
тивности комплемента [6]. Этот метод консервации применяют
довольно часто. Для определения гемолитической активности
в агарозе пробы сыворотки хранят в течение 24 ч при 4 °C
[6].
2.11.2.3. ПОЛУЧЕНИЕ СУСПЕНЗИИ
ЭРИТРОЦИТОВ БАРАНА
ЭБ трижды отмывают в рабочем буфере (центрифугируют
10 мин при 650 g), из отмытых эритроцитов готовят 4% сус-
пензию. Это соответствует 6.5Х107 клеток в 1 мл. Концентра-
цию суспензии определяют фотометрически и устанавливают
оптическую плотность (ОП) 0,375. Для этого 0,2 мл суспензии
ЭБ лизируют добавлением 4,8 мл дистиллированной воды и
определяют ОП против дистиллированной воды. Конечный
объем суспензии рассчитывают по формуле:
, г 0П
Ve = Va- 0 375 ,
где Ve — конечный объем суспензии, Va — исходный объем суспензии.
2.11.2.4. ТИТРОВАНИЕ ГЕМОЛИЗИНОВ
Титрование гемолизинов проводят в пробирках. В каждую из
8 пробирок вносят по 0,5 мл рабочего буфера. В первую про-
бирку вносят 0,5 мл гемолизина в разведении 1 : 100. Делают
геометрический ряд разведений, перенося по 0,5 мл содержи-
мого первой пробирки в пробирки 2—8. Добавляют 0,5 мл
суспензии ЭБ и инкубируют 30 мин при 37°C. Затем в каждую
пробирку вносят по 5,5 мл рабочего буфера и по 1,0 мл сыво-
ротки морской свинки, разведенной в соотношении 1:200.
После повторной инкубации в течение 60 мин при 37 °C про-
бирки центрифугируют (10 мин, 800 g) и измеряют ОП надо-
садочной фракции. С увеличением концентрации гемолизинов
(уменьшение разведения сыворотки) гемолиз (ОП) возрастает,
достигая плато. Наибольшее разведение гемолитической сыво-
172
ротки, при котором ОП все еще находится на плато, называет-
ся гемолитической единицей. В коммерческих препаратах она
составляет от 1:4000 до 1 :6000. Титрование можно также
проводить в микротитраторе (планшет Takatsy) аналогичным
образом. Каждая новая партия гемолизинов должна тестиро-
ваться с одинаковыми реагентами, контроль следует проводить
один раз в 14 дней.
2.11.2.5. СЕНСИБИЛИЗАЦИЯ ЭРИТРОЦИТОВ
БАРАНА
К 1 объему суспензии ЭБ добавляют равный объем разведен-
ной гемолитической сыворотки, содержащий 4 гемолитические
единицы. Тщательно перемешивают жидкости и инкубируют
в течение 30 мин при 37 °C. Во время инкубации смесь не-
сколько раз встряхивают. Сенсибилизированные эритроциты
должны быть использованы в тот же день, до использования их
хранят при 4 °C. Сенсибилизированные ЭБ называются гемо-
литической системой, которую используют как для титрования
комплемента в пробирке, так и для гемолиза в агарозе.
2.11.2.6. ГЕМОЛИЗ В АГАРОЗЕ
2.11.2.6.1. Постановка опыта
Получают 2% золь агарозы в рабочем буфере и охлаждают
его до 50 °C. Затем на водяной бане (48 °C) смешивают рав-
ные объемы агарозы и сенсибилизированных ЭБ, предваритель-
но нагретых до этой температуры. Суспензию постепенно зали-
вают между двумя стеклами, имеющими зазор 1,5 мм. Мы ис-
пользовали пластины 100X200X3 мм, между которыми вмеща-
лось 23 мл агарозы. После охлаждения одну из пластин уда-
ляют и в геле проделывают специальным штампом по шаблону
отверстия диаметром 3 мм на расстоянии 15 мм друг от друга
(всего до 60 отверстий). В каждое отверстие вносят по 5 мкл
неразведенной сыворотки. Пластину инкубируют 18 часов при
4 °C для того, чтобы прошла диффузия, и еще 2 часа при 37 °C
для того, чтобы произошел гемолиз. После чего зоны гемолиза
становятся хорошо различимыми на свету. Для консервации
гелей их заворачивают в слой фильтровальной бумаги, смочен-
ной физиологическим раствором (без образования под бумагой
пузырей воздуха), насыпают сверху слой целлюлозы толщиной
1 см и кладут груз с таким расчетом, чтобы масса 10 г прихо-
дилась на 1 см2. Через 15 мин груз снимают, убирают целлю-
лозу и бумагу, высушивают гель на воздухе. Пластины для
гемолиза можно сохранять при 4 °C в течение 3—4 дней без
ущерба для качества анализа.
173
2.11.2.6.2. Оценка результатов
Зоны гемолиза оценивают визуально, иногда со слабым увели-
чением. Пластины с гелем можно фотографировать. Величина
зоны гемолиза пропорциональна активности комплемента. Ис-
пользуя стандарты и разведения исследуемой пробы, можно
оценивать процесс количественно [1]. Мы использовали гемо-
лиз в агарозе для полуколичественной оценки в качестве ориен-
тировочного анализа.
2.11.2.7. ТИТРОВАНИЕ КОМПЛЕМЕНТА
2.11.2.7.1. Постановка опыта
Исследуемая сыворотка разводится рабочим буфером в отно-
шении 1 + 2. Если сыворотка уже разведена калий-боратным
буфером, то разведения рабочим буфером уже не требуется.
Разведенную сыворотку в количестве 0,5 мл смешивают с
2,55 мл рабочего буфера, что соответствует разведению 1 : 12,2.
В остальных пробирках готовят ряд разведений в соответствии
с табл. 13. В пробирку № 12, где должен быть 100% гемолиз,
добавляют следовые количества сапонина. Все пробы инкуби-
руют на водяной бане в течение 45 мин при 37°C. Затем цент-
рифугируют 10 мин при 800 g и измеряют оптическую плот-
ность надосадочной фракции в 11-й пробирке (0% гемолиз).
2.11.2.7.2. Обработка результатов
Результаты определения ОП корригируют по значению ОП
контроля (пробирки 11 и 12). Рассчитывают процент гемолиза.
Для построения графической зависимости используют логариф-
мическую бумагу. Значение каждого разведения сыворотки
(12,2, 15,2, 18,5 и т. д.) отмечают на оси абсцисс. На осн ордн-
Таблнца 13. Схема титрования комплемента
Номера п; сбирок
1 2 3 4 5 6 7 8 9 10 111 12
Разведение (1 :) 12,2 15,2 18.5 24,5 30,5 38,0 47,5 59,5 75 93 0% гемолиз 100% гемолиз
Реагенты (мл) Рабочий буфер 0,4 0,4 0,4 0,4 0,4 0,4 0,4 0,4 0,4 0,4
Сыворотка 0,4 1,6 смешивают н переносят IO — —
1 : 12,2 1,6 мл в последующи проопрки
Рабочий буфер 0,8 0.8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 —.
Дистиллнро- 1,2
ванная вода Гемолнтнче- 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8 0,8
ская система
174
нат откладывают соответствующий процент гемолиза. Получае-
мые точки соединяют и по графику тут же определяют разве-
дение сыворотки, при котором наступает 50% гемолиз.
Число гемолитических единиц в 1 мл сыворотки рассчитывают
по формуле:
СН50/мл = .
а — фактор первоначального разведения сыворотки (равен 12,2), х—объем
сыворотки, вызывающий 50% гемолиз.
2.11.3. ОЦЕНКА МЕТОДА
Определение общей активности комплемента при помощи вы-
шеописанного метода позволяет сделать заключение о его ли-
тической способности. Это важная величина, но она не отра-
жает всех биологических свойств комплемента. Приведенная
схема анализа позволяет получить только полуколичественную
оценку, поскольку значение СН50 зависит от используемых
компонентов реакции. Скорость лизиса определяется наиболее
медленной составляющей каскада реакций. Хотя в гемолизе
принимают участие все компоненты от С1 до С9, количествен-
ные изменения их соотношений вышеописанная система не от-
ражает [3]. Для стандартизации исследований можно приме-
нять референс-препарат ВОЗ для определения СН5() [7]. В ре-
зультате обширных исследований было установлено, что коэф-
фициент вариации одноразовых определений СН50 очень не-
значителен. Ошибка метода меньше, чем изменения активности
комплемента, обусловленные циркадными ритмами [3].
Для обычных лабораторных исследований предпочтительнее
проводить гемолиз в геле агарозы. Гемолиз сенсибилизирован-
ных ЭБ применяется для определения активации комплемента
по классическому пути. Для определения активации компле-
мента по альтернативному пути используют несенсибилизиро-
ванные эритроциты морской свинки или кролика [1, 3, 4].
В этом случае следует обращать внимание на катионный со-
став проб. Наши опыты с эритроцитами морской свинки в ра-
створе Олсвера 1—3-недельной давности показали, что опти-
мальной является 1% концентрация. При исследовании альтер-
нативного пути активации комплемента также следует обра-
щать внимание на своевременное охлаждение пластин ввиду
возможного преждевременного гемолиза. Ввиду термолабиль-
ности комплемента следует тщательно соблюдать условия
сбора и хранения исследуемых сывороток. Этот вопрос в общих
чертах приходится решать при постановке гемолиза в агарозе
[6]. Уже упоминавшаяся консервация калий-боратным раство-
ром позволяет осуществлять довольно длительное хранение при
4 °C. Титрование комплемента не дает сведений о состоянии от-
дельных компонентов системы комплемента, например, при
175
нормальном СН50 содержание С4 может быть крайне низким.
Значения СН50 могут быть снижены ввиду снижения уровня
С2. Известны генетические нарушения, встречающиеся весьма
редко, которые приводят к снижению значений СН50 [2].
В настоящее время отдельные компоненты системы комплемен-
та определяют иммунохимически при помощи специфических
антисывороток [3, 4]. Антисыворотки к Cl, СЗ, СЗЬ, активатору
СЗ, С4, С5 имеются в продаже. Определение отдельных компо-
нентов проводят по Манчини, нефелометрически, турбидимет-
рически или при помощи электроиммунодиффузии [4, 5].
Для клинических целей, как правило, достаточно определять
СН50 и содержание компонентов СЗ и С4 {7].
2.11.4. СПИСОК ЛИТЕРАТУРЫ
1. Fong J. F. С., Renaud L. A hemolitic plate method for alter native pathway
complement activity assay. — Amer. J. clin. path., 1978, 69, 156—160.
2. Hobart M. J., Lachmann P. J. Allotypes of complement components in man.—
Transpl. Rev., 1976, 32, 26.
3. Lachmann P. J., Hobart M. J. Complement technology. — In: Handbook of
experimental Immunology. 3-rd ed./Ed. M. D. Weir. Oxford: Blackwell Scien-
tific Publications, 1978.
4. Laurell A. B., Martennsson U., Sjoholm A. G. The development of simple
tests for Clq, Clr, Cis and C2 and the determination of properdin. — In:
Clinical aspects of the complement System/Eds. W. K. Opferkuch et al.—
Stutgart: Thieme-Verlag, 1978.
5. Naish P. F., Barrat J., Collins C. A new haemolitic assay for the second
component of complement (C2) in human serum. — Clin. exp. Immunol.,
1976, 25, 487—492.
6. Truedsson L., Sjoholm G. and Laurell A. B. Screening for deficiencies in the
classical and alternative pathways of complement by hemolysis in gel.—Acta
path, microbiol. scand. Sect. C, 1981, 89, 161—166.
7. WHO-report. Use and abuse of laboratory tests in clinical immunology:
critical considerations of eight widely-used diagnostic procedures. — Report
of IUIS/WHO working group. Geneva, 1981, Allergie Immunol., 1982, 28,
21—35.
2.12. КОМПЛЕМЕНТЗАВИСИМАЯ
ЦИТОТОКСИЧНОСТЬ
2.12.1. ПРИНЦИП МЕТОДА
Известны 4 основные механизма цитотоксических реакций:
1. Комплементзависимая цитотоксичность.
2. Антителозависимый фагоцитоз.
3. Антителозависимая клеточная цитотоксичность.
4. Прямая, опосредованная клетками цитотоксичность.
Реакция АГ и АТ может происходить независимо от нали-
чия комплемента и не сопровождаться видимыми изменениями
клеток. Только после добавления комплемента наблюдается
обусловленная реакцией АГ—АТ активация комплемента, при-
водящая к повреждению клеточной мембраны и цитолизу
176
Антитела.
Рис. 73. Комплементзависимая цитотоксичность, принцип метода.
(рис. 73). Определение комплементзависимой цитотоксичности
удобно проводить на легко получаемых клетках крови. В случае
использования эритроцитов реакция проявляется в виде гемо-
лиза; цитолиз лейкоцитов наблюдают при помощи специальных
методов (фазовый контраст, витальное окрашивание, изотоп-
ные методы).
2.12.2. РЕАКЦИЯ ЛИМФОЦИТОЛИЗА
Классическая форма реакции лимфоцитолиза (РЛЦ) предло-
жена Горер и О’Горман в 1956 г. Применение микроварианта
РЛЦ позволило выявить наиболее часто встречающиеся анти-
гены HLA (human leukocyte locus А) в течение всего несколь-
ких лет. Жизнеспособные лейкоциты при инкубации с цитоток-
сическими АТ в присутствии комплемента подвергаются лизису,,
если они содержат соответствующий АГ. Реакцию оценивают
по исключению красителя. Живые клетки в отличие от погиб-
ших не окрашиваются. Для получения суспензии лимфоцитов
пригоден любой метод, который позволяет получать жизнеспо-
собные лимфоциты с выходом 95—100%. Обычно используют
метод флотации ввиду хорошей воспроизводимости и простоты.
Лимфоциты могут быть отделены от более тяжелых клеточных
элементов крови фракционированием в среде с плотностью,
1,076 г/л.
2.12.2.1. ПОСТАНОВКА ОПЫТА
2.12.2.1.1. Материалы и оборудование
Для работы необходимы: тестерные сыворотки «анти-HLA»;
полиспецифическая сыворотка «анти-HLA» (положительный
контроль); АВ-сыворотка без антител (отрицательный конт-
12—990
177
роль); АВ-сыворотка, инактивированная нагреванием (добавка
к среде); комплемент кролика; фосфатный буфер XVI (1,15 г
двузамещенного фосфата натрия, 0,200 г однозамещенного фос-
фата калия, 0,200 г хлорида калия, 8,00 г хлорида натрия ра-
створяют в 1000,0 мл дистиллированной воды, pH 7,2); раствор
Хенкса (Государственный институт иммунных препаратов и пи-
тательных сред, Берлин); среда для сепарации (5,6 г фиколла)
(Pharmacia, Швеция), 94,4 мл дистиллированной воды, 20,0 мл
Visotrast 370 (Fahlberg List, Магдебург) с плотностью 1,076
или 6,0 г декстрана (сывороточное предприятие, Вернбург),
16,0 мл Visotrast, дистиллированная вода до 100 мл (плот-
ность 1,076 г/л); водный раствор натриевой соли эозина 50 г/л
(Laborchemie, Апольда); азид натрия 0,1%; гепарин (Гедеон
Рихтер, Будапешт); полужидкий парафин; безкислотный фор-
малин с pH 7,2 (содержание формальдегида около 300 г/л);
культуральные среды, RPMI или Игла (например, Институт им-
мунологических препаратов, Берлин); меченная ФИТЦ антисы-
воротка к IgG человека (Behring Werke, Марбург); этидий-бро-
мид (Ferak, Западный Берлин); ЭДТА 60 г/л; найлоновая вата,
найлоновые волокна (Fenwal Lab., США); соломка для питья
(Sweethart Straw, США); глазная вата (Vliestextilien, ГДР);
микрокамеры для постановки реакции (так называемые камеры
Терасаки, Polypast, Хальберштадт или Microtest Tissue Culture
Plats № 3034 США); микрошприцы (Hamilton № 705 с дози-
рующим устройством № РВ-600); фазово-контрастный мик-
роскоп, инвертированный микроскоп для люминесцентной мик-
роскопии, камера для тестирования HLA-DR (Hamax-Kammern,
Норвегия).
2.12.2.1.2. Получение лимфоцитов
Суспензию лимфоцитов получают из свежей гепаринизирован-
ной или дефибринпрованной крови при помощи флотации.
Для этого 2 мл гепаринизированной венозной крови (5 ME ге-
парина) смешивают с 2 мл фосфатного буфера XVI или с со-
левым раствором Хэнкса. Наслаивают эту смесь в центрифуж-
ную пробирку, содержащую 2 мл среды для сепарации. После
центрифугирования в течение 10 мин при 2000 g или 30 мин
при 300 g между плазмой и средой для сепарации образуется
резко очерченный слой лимфоцитов (цветная вкладка II). Сус-
пензию лимфоцитов отсасывают пипеткой и центрифугируют
10 мин при 100 g (дифференциальное центрифугирование для
удаления тромбоцитов), надосадочную фракцию отбрасывают,
осажденные лимфоциты отмывают фосфатным буфером XVI
и снова осаждают центрифугированием в течение 5 мин при
1000 g. Дефибринирование свежеотобранной крови позволяет
удалить примесь тромбоцитов перед флотацией. Дефибриниро-
вание проводят путем встряхивания 10—20 мл венозной крови
с 20 стеклянными бусинами диаметром 4—6 мм в колбе Эр-
178
ленмейера в течение 10—15 мин. Не позднее чем через 2 часа
дефибринировапная кровь должна быть использована для
дальнейшей работы. Готовая суспензия лимфоцитов может хра-
ниться до 24 часов при 4°C в культуральной среде. Длительное
хранение суспензии возможно в жидком азоте (—196 °C) после
добавления криоконсерванта диметилсульфоксида (ДМ.СО).
2.12.2.1.3. Микромодификация реакции лимфоцитолиза
Свежеполученную суспензию лимфоцитов разводят до кон-
центрации 3-109 клеток/л. В ячейки для реакции камеры Те-
расаки осторожно вносят вазелиновое масло (2—3 мл в каме-
ру) с тем, чтобы последующие реакции проходили в отсутствие
зоздуха. При частом употреблении камеры можно отказаться
от использования вазелинового масла, если под крышку поло-
жить полоску фильтровальной бумаги, смоченной водой для
создания водонасыщепноп атмосферы [10].
В каждую лунку добавляют 1 мкл исследуемой сыворотки
и 1 мкл суспензии лимфоцитов. После 30 мин инкубации при
22 °C добавляют 5 мкл комплемента морской свинки и инку-
бируют еще 60 мин при 22 °C.
Для окрашивания мертвых клеток в каждую лунку вносят
no 1—3 мкл раствора эозина (рис. 2, 3 на цв. вклейке). При ис-
пользовании парафиновых камер цветную реакцию фиксируют
через 2 мин 8—10 мкл раствора формальдегида. Для седимен-
тации клеток камеру оставляют на 60 мин при 22 °C. После
фиксации оценку результатов можно проводить через 24 или
48 часов. При использовании камер без вазелинового масла
реакцию останавливают осторожным приливанием фосфатного
буфера XVI (см. рис. 2 на цв. вклейке). После этого смесь
можно немедленно анализировать под микроскопом. После при-
ливания буфера считывание результатов возможно не более
чем через 2—3 часа при выдерживании камеры при 4 °C.
2.12.2.1.4. Оценка результатов
Микроскопию проводят при 100—200-кратном увеличении, по
возможности в условиях фазового контраста (рис. 3, 4, 5, 6 на
цв. вклейке). Количество неживых клеток учитывают по степе-
ни выраженности в крестах или словесно.
Степень выраженности (кресты)
Неживые клетки (%) 0—10 —отрицательная
11—20 — 1+
21—50 — 2+
51—75 — 3+
76—100 —4+
12*
179
Степень выраженности (словесное в ы р а ж е-
н и е)
Неживые клетки (%) 0—10 —отрицательная
11—20 — отрицательная (?)
21—40 —положительная (?)
41—80 —положительная
81—100 —резко положительная
Контроль: ставят отрицательный контроль с АВ-сывороткой
и положительный контроль с высокореактивной полиспецифиче-
ской HLA-сывороткой.
2.12.2.2. КОМПЛЕМЕНТ
В качестве источника комплемента для ЛЦТ используют пудо-
вую сыворотку морских свинок (не менее чем от 6 животных).
Поскольку активность комплемента у различных животных
может сильно вырьировать, желательно, чтобы общий пул сы-
воротки составлял 3—5 л (хранение осуществляют в заморо-
женном состоянии небольшими порциями).
При оценке новых серий комплемента необходимо исполь-
зовать одинаковые сыворотки и планерные лимфоциты для
создания определенных, постоянных условий эксперимента.
Хороший комплемент дает реакцию 4+ в разведении 1+2 [9].
Помимо титра, определяют оптимальное время реакции, для
чего инкубацию проводят в предел ах, 1—3 часов и определяют
минимальное время наиболее выраженной реакции. Комплемент
хранят при температуре —196 °C или —80 °C, транспортируют
в лиофилизированном состоянии при температуре 2°—8 °C. По-
вторное замораживание и оттаивание комплемента недопу-
стимо.
2.12.2.3. ТИПИРУЮЩИЕ СЫВОРОТКИ
Анти-НЬА-сыворотки получают от иммунизированных доноров.
Иммунизация может быть вызвана беременностью (у 10—20%
беременных вырабатываются АТ к HLA-антигенам плода от-
цовского происхождения), переливанием крови или трансплан-
тацией органов. Направленная иммунизация доноров пересад-
кой кожи, переливанием крови или лейкоцитарной массы при-
водит к появлению высокоактивных АТ к HLA-антигенам [12].
Ксено-антисыворотки (кроличьи, шимпанзе) не используются
в рутинной лабораторной практике, поскольку их получение
требует наличия очищенных антигенов или адсорбции получае-
мых сывороток. Ксеногенные моноклональные АТ до настоя-
щего времени используются только в немногих лабораториях
[2, 5], несмотря на их явные преимущества. Моноклональные
АТ следует в первую очередь применять для стандартизации
или исследования антигенных структур [2]. Для определения
180
HLA-антигенов необходимы по меньшей мере 3 различные
тест-сыворотки, из которых хотя бы 2 дают реакцию 3+[9].
Для титрования HLA используют добавку донорской сыворот-
ки (пул составляет приблизительно 120 человек). Эти сыворот-
ки можно заранее поместить в тест-камеры и сохранять в те-
чение нескольких месяцев при —20 °C под слоем вазелина.
Безвазелиновые камеры после внесения в них сыворотки вы-
держивают на воздухе в течение 1 часа при 22 °C для высы-
хания сыворотки. В таком виде тест-камеры можно даже пере-
сылать по почте. Перед использованием в каждую лунку вно-
сят по 1 мкл дистиллированной воды и под крышку подклады-
вают лист влажной фильтровальной бумаги.
2.12.3. ИСТОЧНИКИ ВОЗМОЖНЫХ ОШИБОК
И ИХ ПРЕДОТВРАЩЕНИЕ
2.12.3.1. КЛЕТОЧНЫЕ СУСПЕНЗИИ
Отрицательный контроль (АВ-сыворотка) не должен
давать более 10% .неживых клеток, поскольку при более высо-
ких значениях оценка результатов становится затруднительной.
Большие количества неживых клеток в контроле встречаются
в образцах крови, хранившихся более суток.
Контаминация гранулоцитами. У пациентов с
«левым сдвигом», особенно у потенциальных доноров почки,
суспензия лимфоцитов может содержать значительную примесь
гранулоцитов, которые во всех случаях разрушаются компле-
ментом морской свинки, что весьма затрудняет оценку резуль-
татов [3]. Незрелые гранулоциты имеют такую же плотность,
как лимфоциты, поэтому они не удаляются при отмывке. В та-
ких случаях можно рекомендовать адсорбцию на глазной вате,
к волокнам которой прикрепляются гранулоциты. Для этого
в шприц (20 мл), неплотно заполненный глазной ватой, нано-
сят 10 мл дефибринированной крови или надосадочной фракции
после осаждения декстраном, инкубируют 30 мин при 37 °C в
термостате, затем промывают шприц из пипетки 10 мл раство-
ра Хэнкса, вату осторожно отжимают.
Контаминация тромбоцитами. Тромбоциты уда-
ляют из гепаринизированной крови двукратным отмыванием
при 100 g. Это необходимо, поскольку тромбоциты как носите-
ли HLA-A, HLA-В и HLA-С локусов адсорбируют анти-HLA-
иммуноглобулины и могут вызывать ложноотрицательные реак-
ции.
2.12.3.2. ТИПИРУЮЩИЕ СЫВОРОТКИ
«Проросшие» сыворотки могут вызывать ложноположительные
реакции. В качестве консерванта рекомендуется азид натрия в
концентрации 0,1%. Анти-HLA-сыворотки необходимо прове-
181
рять па наличие слабых перекрестно реагирующих HLA-антител
и антиэритроцитарных антител (особенно анти-А и анти-Le’)
перед их использованием.
2.12.3.3. КОМПЛЕМЕНТ
Способы проверки качества комплемента см. в разделе 2.12.2.
2.12.3.4. ПРОЧИЕ РЕАГЕНТЫ
Вазелиновое масло, используемое для предотвращения высы-
хания жидкости в тест-камерах, не должно обладать токсич-
ностью и способностью ингибировать комплемент. Раствор
формальдегида (для фиксации) доводят карбонатом натрия
до pH 7,2, поскольку при pH ниже 7,0 невозможно проводить
различие между живыми и мертвыми клетками.
2.12.4. ВОЗМОЖНОСТИ ПРИМЕНЕНИЯ МЕТОДА
Определение HLA-A-, -В-, и -С-а н т и г е и о в. Лимфоцито-
токсический тест применяют для определения HLA-антигенов
перед проведением трансплантации и гемотрансфузии [1, 8],
в связи с болезнями, ассоциированными с HLA [4], для уста-
новления отцовства [11]. Поскольку типирование по HLA тре-
бует особого навыка, его проводят только в условиях специаль-
ных лабораторий.
Поиск антител. Лимфоцитотоксический тест может
применяться для определения цитотоксических АТ к HLA-анти-
гепам в сыворотке. Методику проведения теста см. в разделе
2.12.2. Исследуемую сыворотку проверяют по меньшей мере с
20 различными суспензиями лимфоцитов, по возможности со-
держащими HLA-A, -В-, -С-антигены. Проба на АТ к HLA
считается положительной, если одна или две суспензии лимфо-
цитов дают реакцию 2+ в параллельных пробах [9].
Перекрестный тест с лимфоцитами. Этот тест
служит для выявления апти-НЬА-антител у возможных реци-
пиентов органных трансплантатов или клеточных масс (грану-
лоцитарной, тромбоцитарной) [8]. Для проведения анализа
смешивают 1 мкл суспензии лимфоцитов донора с 1 мкл сыво-
ротки предполагаемого реципиента (микрометод см. в разде-
ле 2.12.2).
2.12.5. ТИПИРОВАНИЕ ПО HLA-DR
HLA-DR-антигены (англ. HLA-D-related antigens) тесно ассо-
циированы или идентичны HLA-D-антигенам, они выявлены
только у В-лимфоцитов, макрофагов, клеток эндотелия и спер»
матозоидов [4]. Их определяют модифицированным лимфоци-
тотоксическим методом с В-лимфоцитами. В-лимфоциты выде-
182
ляют на найлоновой вате, -при помощи метода розеток (ЭБ)
или флотации (получаются меченные флюорохромом клетки,
двойная флюорохромная метка).
2.12.5.1. ВЫДЕЛЕНИЕ В-ЛИМФОЦИТОВ
Получают лимфоциты по меньшей мере из 10 мл крови (см.
2.12.2) и суспендируют в 0,5 мл среды (RPMI +5% сыворотки
АВ) [6].
Сепарация В-лимфоцитов на найлоновой
вате (так называемый метод «соломки для
питья»). Приготовление колонки из найлоновой ваты: пласти-
ковую «соломку для питья» (длиной 19,5 и 0,6 см в диаметре)
сваривают по одному концу под углом 45°. В нижнем конце
сварного шва прорезают отверстие диаметром 1 мм, через ко-
торое будет медленно вытекать суспензия клеток. Колонку за-
полняют 0,1 г найлоновой ваты на высоту 6 см (1—2 см от
верхнего конца остаются незаполненными), выдерживают 4 дня
в дистиллированной воде, затем промывают нагретой до 37 °C
средой. Наличие воздушных пузырей недопустимо, поскольку
они вызывают цитолиз. Подготовленная таким способом колон-
ка пригодна для получения 150Х106 клеток. Закупоренные ко-
лонки можно длительное время хранить в замороженном со-
стоянии. Найлоновую вату после употребления можно промы-
вать водопроводной водой и использовать повторно (много-
кратно). После промывания подогретой средой колонку инку-
бируют в горизонтальном положении в течение 30 мин при
37 °C. Суспензию лимфоцитов осторожно наносят на верхний
конец колонки, наклоненной под углом 45°. В заполненную
суспензией колонку вносят еще 0,2 мл среды для предотвраще-
ния высыхания. Колонку кладут в горизонтальном положении
в термостат (37 °C) на 30 мин, а затем ставят вертикально в
сосуд и промывают 5 мл среды, нагретой до 37 °C.
На этом этапе удаляются неадгезировавшие клетки (Т-лим-
фоциты). Еще раз повторяют промывку 5 мл среды. Адгезиро-
вавшие клетки (В-лимфоциты) вымывают средой, охлажденной
до 4 °C, при равномерном надавливании, повторяют процедуру.
Собранные клетки отмывают средой и осаждают центрифуги-
рованием в течение 5 мин при 100 g; доводят концентрацию
клеток до ЗХ Ю9 клеток/л.
Контроль выделения В-клеток. В-лимфоциты
должны составлять не менее 80% выделенной популяции кле-
ток, для того чтобы можно было проводить титрование по
HLA-DR. Позитивный контроль проводят с полиспецифической
HLA-DR-антисывороткой, которая вызывает полный лизис В-
лимфоцитов и не лизирует Т-лимфоциты. Дополнительный
контроль можно проводить маркировкой В-лимфоцитов мечен-
ной ФИТЦ антисывороткой против иммуноглобулинов человека
(см. раздел 2.12.5).
183
Модифицированный микро тест на л и м ф о-
цитотоксичность. Тест проводят в условиях, соответствую-
щих описанным в разделе 2.12.2, но время инкубации увели-
чивают: антисыворотки + В-лимфоциты инкубируют 60 мин,
после добавления комплемента инкубацию проводят еще в те-
чение 120 мин при 22 °C.
2.12.5.2. МЕТОД ДВОЙНОЙ
ФЛЮОРЕСЦЕНТНОЙ МЕТКИ
Выделение лимфоцитов. Лимфоциты из 5 мл гепари-
низированной или дефибринированной крови трижды отмывают
в растворе Хэнкса, ресуспендируют в 1 мл среды и доводят
концентрацию клеток до 5Х109 клеток/л.
Мечение В-клеток. К суспензии лимфоцитов (5Х
X 106 клеток) добавляют 15 мкл меченной ФИТЦ антисыворот-
ки к ИГ человека, инкубируют 10 мин при 37 °C и трижды
отмывают раствором Хэнкса. Ресуспендируют клетки в 0,15 мл
среды и устанавливают концентрацию 8Х109 клеток/л.
Постановка опыта. Антисыворотку (0,5 мкл) смеши-
вают с 0,5 мкл клеточной суспензии под слоем вазелинового
масла и инкубируют 45 мин при 22°С. Добавляют 2 мкл кро-
личьего комплемента и инкубируют 120 мин при 22 °C. Готовят
исходный раствор этидий-бромида Ц0 мг красителя в 50 мл
5% раствора ЭДТА в 0,15 М NaCl). Разводят исходный раствор
этидий-бромида 1:230 (растворитель, как для исходного раст-
вора). Для окрашивания к инкубационной смеси добавляют
0,5 мкл разведенного раствора этидий-бромида. Результаты
учитывают при помощи инвертированного [микроскопа в люми-
несцентном свете (фильтр № 12 для Ploempak 2 Лейц). В-лим-
фоциты могут быть идентифицированы по зеленой флюорес-
ценции (так же как и моноциты); у мертвых клеток ядро окра-
шивается в оранжевый цвет (этидий-бромид). Учитываются
только клетки с зеленой флюоресценцией с неокрашенным
ядром (живые) и клетки с оранжевым ядром (мертвые клетки,
положительная проба).
2.12.5.3. СЫВОРОТКИ ДЛЯ HLA-DR
Антисыворотки к HLA-DR получают от иммунизированных до-
норов. Поскольку они также, как правило, содержат антитела
к HLA-A, -В, -С, последние должны быть адсорбированы тром-
боцитами, содержащими антигены HLA-A, -В -С, и не содер-
жащими антигенов HLA-DR. Для этого сыворотку смешивают
с равным объемом осадка тромбоцитов, инкубируют при 22°C,
постоянно помешивая. Тромбоциты удаляют центрифугирова-
нием (1000—2000 g). Повторяют адсорбцию со свежей порцией
тромбоцитов. Адсорбированные сыворотки не должны реагиро-
вать с Т-лимфоцитами. Тромбоциты получают с донорских пунк-
184
тов. После 25-минутной отмывки 0,14 М NaCl тромбоциты сус-
пендируют в 0,1% азиде натрия и хранят при 4°C до 3 месяцев.
Пул тромбоцитов собирают по меньшей мере из 100 лейкоци-
тарных пленок, что обеспечивает присутствие всех вариантов
HLA-A, -В-, -С-антигенов.
2.12.6. ОЦЕНКА МЕТОДА
При наличии антисывороток достаточно хорошего качества
реакция лимфоцитолиза ввиду ее хорошей воспроизводимости
(свыше 95%) представляется наиболее адекватным методом
типирования по HLA. Наибольшее распространение получил
микрометод. Вместо суправитального окрашивания эозином и
трипановым синим можно метить живые клетки флюоресцеин-
диацетатом или 51[Сг]. Для определения анти-НЬА-иммуногло-
булинов можно применить реакцию лимфоцитолиза в антигло-
булиновом варианте [7].
К числу недостатков метода относится использование в роли
носителя антигенов живых клеток. Это приводит к тому, что
иногда свежевыделенные препараты клеток приходится сохра-
нять в жидком азоте. Типирование HLA-DR в последние годы
приобрело особенное значение в связи с выявлением тесной
ассоциации HLA-DR-антигенов с предрасположенностью к опре-
деленным заболеваниям .[4], а также в связи с ролью этих
антигенов в -приживлении трансплантатов [1]. Воспроизводи-
мость результатов типирования HLA-DR все еще не вполне
удовлетворительна, поскольку aHTH-HLA-DR-сыворотки хороше-
го качества встречаются довольно редко. Пока еще не получе-
ны моноклональные антитела к HLA-DR с полным набором
специфичностей [2]. Определенные преимущества создает ме-
тод двойной флюоресцентной метки, поскольку снижает расход
типирующих сывороток и сокращает потребление крови. Наибо-
лее экономичный метод получения лейкоцитов основан на ис-
пользовании найлоновой ваты.
2.12.7. СПИСОК ЛИТЕРАТУРЫ
1. Albrechtsen D., Bratlie A. et al. Significance of HLA matching in renal Trans-
plantation.— Transplantation, 1979, 28, 280—284.
2. Ax IV. Monoklonale Antikorper durch Lymphozyten-Hibridisierung — Beh-
rings Laboratoriumsblatter, 1980, 30, 89—99.
3. Bender K. Das HLA-System — Biotest—Mitteilungen (1979).
4. Bertrams J., Rittner C. Der HLA-Komplex. —Dtsch. med. Wschr., 1981, 106,
952—958.
5. Ceppelinl R., Carotta G., Trucco M. M. Evaluation of zenogenic monoclonal
antibodies as HLA typing reagents. Histocompatibility testing, 1980, 924,
UCLA, Los Angeles.
6, Danilovs I. A., Ayoub G., Terasaki P. I. В lymphocyte isolation by thrombin-
nylon wool. — Histocompatibility testing, 1980, UCLA tissue Typing Lab.
Los Angeles, p. 287—288.
7. Jager L. Klinische Immunologie und Allergologie. 2. Auflage Fischer Verlag,
Jena, 1983.
8. Johanson A. H. Antiglobulin microcitotoxicity test — NIAD Manual of tissue
185
typing techniques/Ed. Ray J. G. jr, DHEW Publ. No. (NIH) 80—545, 1979,
178—180.
9. Mayr W. R. Die Bedeutung des HLA-Systems fur die Transfusionmedizin.—
Biotest Mitt., 1979, 37, 191—196.
10. Richter K., Brandsadter W., Rackow S. Vorschlag zum 2. Arzneibucli der
DDR, Diagnostische Laboratoriumsmethoden. — Zbl. Pharm., 1979, 118, 949—
955.
11. Thorsby E. The human major histocompatibility complex HLA: some recent
developments. — Transplant. Proc., 1979, XI, 616—623.
12. Wegener S. Gewinnung und Anwendung von HLA-Testseren. — Disserta-
tion B, Wilhelm-Pieck-Universitat Rostock 1983.
13. Wegener R., Haferland W. Probleme der biostatischen Wertung von HLA-
Befunden im Paternitatsgutachten. — Kriminalistik und forensische Wissen-
schaften, 1981, 44, 17—21.
2.13. РЕАКЦИЯ СВЯЗЫВАНИЯ
КОМПЛЕМЕНТА ТРОМБОЦИТАМИ
2.13.1. ПРИНЦИП МЕТОДА
Реакция связывания комплемента (РСК) введена в серологию
тромбоцитов Аккроидом (Ackroyd) в 1951 г. В последующие
годы разные авторы использовали ее с некоторыми методиче-
скими изменениями для определения антитромбоцитарных АГ.
Ее применяют для количественных и качественных исследова-
ний. Так как антигены гистосовместимости обнаружены во
всех клетках организма, в том числе и в тромбоцитах, и при-
обретают все большее значение, возрастает интерес к методам
выявления этих АГ. На IV международной конференции по
гистосовместимости в 1970 г. была предложена одновременно
многими авторами микромодификация РСК [1]. Антитела,
взаимодействуя с соответствующим АГ, связывают комплемент.
Уменьшение количества комплемента определяется при помощи
сенсибилизированных ЭБ (вторая система АГ—АТ). Если ком-
племент не связывается в йервой системе, наблюдается 100%
лизис сенсибилизированных ЭБ.
2.13.2. МЕТОДИКА \
2.13.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: 0,9% раствор NaCl; 5% раствор
ЭДТА в 0,15 М NaCl, 0,1% раствор азида натрия в 0,15 М NaCl,
барбиталовый буфер, гемолизин (гемолитическая сыворотка) к
бараньим эритроцитам, комплемент, тромбоциты, исследуемая
сыворотка, вазелиновое масло, камеры для микрокультивирова-
ния № 3034 (Falcon, Швейцария), гамильтоновские микрошпри-
цы 705N и 710N, микродозатор РВ 600-1 (Hamilton, Нидерлан-
ды), термостат на 37 °C, водяные бани на 37 °C и 56 °C, центри-
фуги, холодильник, вибрационный смеситель, центрифужные
пробирки, пробирки для титрования, мерные пипетки. Для по-
186
стоянкой работы требуется большой запас посуды и материалов.
Вся стеклянная посуда должна быть идеально чистой и свобод-
ной от бактериального загрязнения. Посуду рекомендуется мыть
хромовой смесью.
Барбиталовый буфер, pH 7,4 (исходный раст-
вор):
Барбитал — 5,75 г
Барбитал-натрий — 3,75 г
NaCl — 85,0 г
1 М MgCl2— 5,0 мл
1 М СаС12— 1,5 мл
Бидистиллированная вода до 2000,0 мл
Исходный раствор автоклавируют, перед употреблением разво-
дят в 5 раз дистиллированной водой.
Гемолизин, готовят стабилизированный раствор гемоли-
тической АС 1 : 100. Он сохраняет стабильность в течение не-
скольких недель:
0,9% раствор NaCl — 94,0 мл
5% фенол в 0,9% NaCl — 4,0
Гемолитическая АС— 1,0
Глицерин — 1,0
Перед употреблением гемолизин выдерживают 3—4 дня.
Эритроциты барана: эритроциты барана стерильно
отбирают в гемостабилизатор и перед использованием выдер-
живают около 6 дней при 4 °C.
Комплемент: сыворотки АВ, свежая пудовая сыворот-
ка, проверенная на активность комплемента (минимальный
титр 1:25). АВ-сыворотку здоровых, не получавших гемотранс-
фузий доноров мужского пола, хранят небольшими порциями
в жидком азоте или при —80 СС. В жидком азоте комплемент
сохраняет стабильность в течение нескольких месяцев.
Комплемент морской свинки: смешивают сыворотки живот-
ных с титром комплемента не менее 1:60, немедленно замо-
раживают небольшими порциями или лиофилизируют. Ввиду
высоких титров комплемента сыворотку морских свинок можно
хранить несколько дней при —20 °C. РСК более чувствитель-
на, если в качестве источника комплемента используется сыво-
ротка АВ.
Суспензия тромбоцитов: 10—60 мл крови собирают
в отношении 1+9 в 5% раствор Кта2-ЭДТА, выдерживают смесь
60 мин при комнатной температуре для оседания эритроцитов.
Центрифугируют 10 мин при 200 g и сливают около 4/5 надо-
садочной фракции (плазма, обогащенная эритроцитами). Если
присутствует примесь эритроцитов, плазму центрифугируют еще
раз 10 мин при 200 g. Надосадочную фракцию, содержащую
только тромбоциты, центрифугируют 15 мин при 1600 g. Оса-
док тромбоцитов трижды отмывают 0,9% NaCl с 0,1% азидом
натрия, доводят концентрацию суспензии до 2хЮ6 клеток/мкл,
сохраняют при 4 °C. При соблюдении стерильных условий из-
187
готовления суспензия сохраняет пригодность в течение 12 ме-
сяцев. Для работы используют суспензию 0,5х Ю6 клеток/мкг.
Свежеприготовленная суспензия тромбоцитов связывает меньше
комплемента, чем суспензия, «созревшая» за 24 часа [8];. Сус-
пензию тромбоцитов из крови, взятой в оксалат аммония,
можно использовать тотчас же после приготовления [4].
Сыворотки: для выявления антигенов HLA на тромбо-
цитах и других клетках используют сыворотки беременных,
пациентов, перенесших гемотрансфузию или трансплантацию,
а также сыворотки специально иммунизированных доноров.
Сыворотки получают по возможности стерильно и хранят их
при —20 °C или в жидком азоте. Желательно избегать много-
кратных замораживаний и оттаиваний. Обычно полученные
сыворотки полиспецифичны, но с помощью адсорбции и элю-
ции можно получить сыворотки, приближающиеся к моноспе-
цифическим ;[7]. Все сыворотки инактивируют нагреванием при
56 °C в течение 30 мин. Сыворотки разводят барбиталовым бу-
фером, если разведение более 1: 10, к вероналовому буферу
добавляют 10% инактивированной сыворотки АВ.
2.13.2.2. ПОСТАНОВКА ОПЫТА
2.13.2.2.1. Сенсибилизация эритроцитов барана
ЭБ 3—4 раза отмывают 0,15 М NaCl, последнюю отмывку де-
лают барбиталовым буфером. Из осадка готовят суспензию ЭБ
в том же буфере (4Х106 клеток/мкл). Для сенсибилизации к
суспензии эритроцитов медленно добавляют равный объем ге-
молизина, титр которого подбирают в предварительном опыте.
Смесь инкубируют 30 мин на водяной бане при 37 °C, много-
кратно ее перемешивая, затем охлаждают до 4 °C. Сенсибили-
зированные эритроциты готовят в день опыта.
2.13.2.2.2. Определение титра гемолизина
Готовят разведения гемолитической сыворотки от 1:400 до
1/ 12 800 с двукратным шагом, по 2 мл каждого разведения.
,К 2 мл каждого разведения гемолизина приливают равный
объем суспензии ЭБ (4Х106 клеток/мкл), происходит сенсиби-
лизация. Реакцию ставят в камере для микротитрования под
слоем вазелинового масла. Комплемент добавляют в избытке
(сыворотку АВ в разведении 1 : 10 или сыворотку морской
свинки в разведении 1 : 20).
Наибольшее разведение гемолизина, при котором еще на-
блюдается полный гемолиз, содержит 1 гемолитическую еди-
ницу. Для постановки основного опыта используют 4 гемоли-
тические единицы.
188
Таблица 14. Схема титрования гемолизина
Разведение гемолизина 1:400 1:800 1:1600 1:3200 1:6400 1:12800
Комплемент (мкл) 2 2 2 2 2 2
Барбиталовый буфер (мкл) 0,1% азид натрия в 0,15 М 2 2 2 2 2 2
NaCl (мкл) 2 2 2 2 2 2
Суспензия сенсибилизирован- ных эритроцитов барана 2X106 клеток/мкл 2 2 2 2 2 2
Инкубация 30 мин прн 37 ®С; непродолжительное центрифугирование, оценка.
Пример: если полный гемолиз наблюдается при разведении
гемолизина 1:3200, то для опыта используют разведение
1 :800.
При повседневном проведении РСК с одними и теми же реа-
гентами титр гемолизина определяют 1 раз в неделю. Если
титр комплемента известен, можно провести перекрестное тит-
рование гемолизина. В приведенную выше (табл. 14) схему
титрования вводят разведения комплемента, указанные ниже.
2.13.2.2.3. Определение титра комплемента
Все разведения комплемента готовят на барбиталовом буфере.
Вспомогательное титрование и основной опыт проводят в оди-
наковых условиях. Титр комплемента определяют -каждый раз
в день постановки опыта. Чтобы повысить чувствительность
РСК, приготовляют следующие разведения комплемента: 1:6,
8, 10, 12, 14, 16, 20, 24, 28, 32. Камеру для титрования запол-
няют 6—8 мл вазелинового масла. Все пробы дублируют
(табл. 15).
Таблица 15. Схема титрования комплемента
Разведение комплемента 1:6 1:8 ' 1:10 — 1:28 1:32 Контроль
Барбиталовый буфер 4 4 4 — 4 4 6
(мкл) Комплемент (мкл) 2 2 2 2 2 —
Сме вшиваю т, инку бирукл 60 мин при 37 °C
Сенсибилизированные ЭБ 2 2 2 2 2 2
(мкл) Смешивают, инкубируют 30 мин при 37 С
Результаты реакции легче учитывать после осаждения эритро-
цитов центрифугированием. Степень гемолиза оценивают ви-
зуально по окраске надосадочной фракции и величине осадка
эритроцитов в баллах от 0 до 4 (табл. 16).
189
Таблица 16. Оценка степени гемолиза
Гемолиз (%) 0 25 50 75 100
Балл 4 3 2 1 0
Количество негемолизиро- ванных эритроцитов (%) 100 75 50 25 0
Наибольшее разведение комплемента, которое еще дает
полный гемолиз, принимают за 1 гемолитическую единицу
комплемента. В основном опыте используют 2 единицы ком-
племента. Если, например, комплемент в разведении 1 :24 вы-
зывает полный гемолиз ЭБ, для основного опыта используют
разведение 1 :12. При использовании одной гемолитической
единицы чувствительность РСК возрастает, но возрастает так-
же влияние помех.
2.13.2.2.4. Международная стандартизированная методика
РСК для определения НLA-антигенов на тромбоцитах
Заливают камеры для титрования 6—8 мл вазелинового масла.
В лунки вносят по 2 мкл следующих реагентов:
Антитела (сыворотка в различных разведениях, элюаты).
Антиген (суспензия тромбоцитов 0.5Х106 клеток/мкл).
Комплемент (сыворотка АВ 1 ЕД/мкл).
Смешивают, инкубируют 60 мин при 37 °C.
Сенсибилизированные ЭБ (2Х106 клеток/мкл).
Смешивают, инкубируют 30 мин при 37 °C.
Центрифугируют, учитывают результаты реакции.
Для каждой серии суспензии тромбоцитов и для каждой
сыворотки ставят следующие контроли:
Контроль антигена: барбиталовый буфер 2 мкл
суспензия тромбоцитов 2 мкл
комплемент 2 мкл
суспензия сенсибилизированных ЭБ 2 мкл
Контроль антител: сыворотка 2 мкл
барбиталовый буфер 2 мкл
комплемент 2 мкл
суспензия сенсибилизированных ЭБ 2 мкл
Если нет подходящей центрифуги, микропланшеты оставляют
на 30 мин при комнатной температуре или 4СС для полного
осаждения негемолизированных эритроцитов. Учет реакции
проводят, как и при титровании комплемента, в баллах. В круп-
ных лабораториях проводят количественную РСК на автомати-
ческом микроскопе-денситометре [5]. Причинами ложнополо-
жнтельных или ложноотрицательных результатов могут быть [8]:
— загрязнение центрифужных пробирок, микропланшетов, пи-
петок, шприцев и т. д.;
— нежелательные примеси в сыворотках, суспензиях тромбо-
цитов или растворах;
190
— контаминация сывороток, суспензий тромбоцитов и раство-
ров бактериями или грибами;
— неправильное хранение сыворотки, приводящее и ингибиро-
ванию комплемента;
— подавление цитратом и другими антикоагулянтами актив-
ности комплемента. В связи с этим необходимо тщательно
отмывать клетки от антикоагулянтов. Сыворотки, получен-
ные методом плазмафереза, также ингибируют комплемент;
— потеря некоторыми препаратами тромбоцитов антигенности
при хранении свыше 4 мес при 4 °C;
— изоагглютинины анти-А и анти-В, присутствующие в иссле-
дуемой сыворотке, могут вызывать ложноположительную
реакцию, если суспензия тромбоцитов содержит большую
примесь эритроцитов, которые связывают комплемент;
1— суспензия тромбоцитов может быть использована не ранее,
чем через сутки после ее получения, в противном случае
наблюдается отрицательная реакция.
2.13.3. ОЦЕНКА МЕТОДА
Прежде РСК применяли главным образом для определения
специфических тромбоцитарных АГ и АТ. В новом, более про-
стом, микроварианте ее стали использовать для выявления АГ
или AT HLA-A и -В-локусов тромбоцитов. По воспроизводимо-
сти результатов и затратам времени (2—3 часа) РСК не усту-
пает широко применяемой реакции лимфоцитолиза. С хороши-
ми сыворотками оба метода дают воспроизводимость около
99%. Однако РСК менее чувствительна, чем реакции лейкоаг-
глютинации и лимфоцитолиза, поэтому многие АС оказывают-
ся в РСК неактивными, что ограничивает ее применение. Со-
здать достаточно полный набор типирующих сывороток весьма
сложно. Особую ценность имеют сыворотки реципиентов, полу-
чавших многократные переливания крови. В более чувствитель-
ной реакции лимфоцитолиза эти сыворотки проявляют поли-
специфичность, но в менее чувствительной РСК они нередко
ведут себя как моноспецифические и считаются хорошими реа-
гентами. Самый чувствительный вариант РСК основан на ко-
личественном измерении степени гемолиза при помощи спект-
рофотометра. Эту реакцию ставят в пробирках с относительно
большими объемами АГ и АС, поэтому ее едва ли можно ре-
комендовать как метод для массовых обследований. Реакция
используется главным образом для количественного определе-
ния АГ и АТ. Более простая модификация количественного
определения антигенов HLA-A предложена Коломбани [3].
Как и любой метод исследования, РСК имеет свои преимуще-
ства и недостатки [2, 10]. К преимуществам относится более
легкое получение тромбоцитов из крови, чем лимфоцитов,
а также возможность довольно длительного хранения. Это по-
зволяет использовать клетки из одной и той же взвеси в реак-
191
циях как с известными, так и с плохо изученными или новыми
сыворотками. Реакция учитывается невооруженным глазом,
возможен также автоматический учет ее результатов.
Можно производить количественное определение антигенов.
РСК позволяет определять антигены HLA не только на тром-
боцитах, но и на других клетках, например, на перевиваемых
лимфобластах, фибробластах и опухолевых клетках. При этом
можно не только идентифицировать известные антигены HLA,
но и вести поиск новых специфичностей этой и других антиген-
ных систем [9, 10]. Все варианты выявления антитромбоцитар-
ных АТ в РСК имеют общие недостатки — относительно низ-
кую чувствительность, подверженность влиянию помех и зна-
чительные трудности получения антисывороток, а также то,
что взвесь тромбоцитов нельзя использовать тотчас после при-
готовления. Данные о том, что HLA-A, -В-, -С-антигены выра-
жены на тромбоцитах слабее, чем на лимфоцитах, до настоя-
щего времени не получили подтверждения [6, 11].
Подводя итоги, можно сказать, что микровариант РСК при
тщательной постановке служит весьма полезным методом вы-
явления антигенов HLA и тромбоцитоспецифических антигенов,
а также соответствующих АТ [12]. О взаимодействии АГ
с АТ в этой реакции судят по фиксации комплемента, которую
можно учитывать полуколичественно [1] или количественно
[3]. Именно по фиксации комплемента часто оценивают актив-
ные сыворотки анти-HLA, «моноспецифические» как в РСК, так
и в реакции лимфоцитолиза [8]. В-РСК происходит взаимо-
действие многочисленных реагентов — гемолизина, комплемен-
та, антигена и антител, а также необходим подсчет клеток в
камере; поэтому успешное овладение этим методом требует
большой тщательности и опыта. С помощью РСК можно вы-
являть антигены не только тромбоцитов, но и других клеток,
параллельно или в дополнение к реакции лимфоцитолиза [9].
2.13.4. СПИСОК ЛИТЕРАТУРЫ
~^Г.~Сд1отЬап1 J., D'Amaro J., Gabb В. et al. International Agrement on a Micro-
technique of Platelet Complement Fixation. — Transplant. Proc. Ill, 1971, III,
121—126.
2. Colombani J. Blood Platelet in HLA serology. —- Transplant. Proc., 1971,
III, 1078—1087.
3. Colombani J„ Colombani M. Principles and applications of a quantitative
complement fixation microtechnique. — Symp. Series immunobiol. Standard.,
1973, 18, 89—92.
4. Colombani M., Colombani J., Dehay C., Dausset J. A Microtechnique of Pla-
telet Complement Fixation. Results Obtained with Sera and Eluates as the
source of Antibody. — Histocompatibility Testing 1970. Munksgaard, Copen-
hagen, 1970, 553—559.
5. D’Amaro J., Hensen E„ van Rood J. The Micro-complement Fixation Test.
III. Automatic Quantitative Scoring with a Microscope Densitometer. — Tis-
sue Antigens, 1971, 1, 171—177.
<5. Duquesnoy R. Filip D. J., Tomasulo P. A., Aster R. H. Role of HLA-C
Matching in Histocompatible Platelet Transfusion Therapy of Alloimmunized
Thromobocytopenic Patients. —- Transplant. Proc., 1977, IX, 1827—1828.
192
7. Heinrich D., Mueller-Eckhardt Ch. HL-A-spezifische, C-fixirende antithrobo-
zytare Antikorper. II. Absorptions- und Elutionsuntersuchungen an Schwan-
gerenseren mit HLA-spezifischen Antikorpern. — Blut, 1971, XXIII, 267—
8. Kissmeyer-Nielsen F., Thorsby E. Human Transplantation Antigens. — Trans-
plantation Reviews, 1970, 4, 134—147.
9. Lepage V., Gaudy Y., Terrier E. et al. Screening and use of high titerd Anti-
HLA-DR Sers in PHA-Blast Complement Fixation and B-Lymphozytotoxicity
Techniques. — Tissue Antigens, 1981, 17, 37—42.
10. Mittal K. K., Mickey M. R., Terasaki P. I. Heterogeneity of HLA Specificities
shown by Platelet Complement fixation. — Tissue Antigens, 1971, 1, 279—
11. Mueller-Eckhardt C. Histocompatibilitat und Thrombozytentrans fusion.—
Blut, 1977, 34, 261—270.
12. Mueller-Eckhardt G., Breidenbach M„ Mahn I., Mueller-Eckhardt C. The role
of Alloimmunisation in Platlet Survival Studies. — Blut, 1980, 41, 93—99.
2.14. РЕАКЦИЯ СВЯЗЫВАНИЯ
КОМПЛЕМЕНТА
2.14.1. ПРИНЦИП МЕТОДА
Реакция связывания комплемента (РСК)—это метод серо-
логического анализа, который по своей чувствительности срав-
ним с методами преципитации, агглютинации и нейтрализации,
т. е. позволяет определить до 0,05 мкг±5% азота антител.
РСК представляет собой метод, включающий две независимо
протекающие реакции: первичную и вторичную. Для проведе-
ния РСК необходимы 5 компонентов: диагностируемый АГ и
диагностические АТ, антиген-индикатор (эритроциты барана),
индикаторные АТ (кроличьи амбоцепторы), комплемент. Коли-
чество применяемых в РСК реагентов привело к возникнове-
нию путаницы в различных модификациях этого метода.
2.14.1.1. ВАРИАНТЫ МЕТОДИКИ
Первый вариант: связывание комплемента рассматри-
вается как простая линейная зависимость от количества им-
мунных комплексов. По мере прибавления комплемента линей-
ная зависимость достигает своего максимума. Комплемент по-
степенно добавляют, пока не будет достигнуто 50% связыва-
ние. Титр рассчитывают методом экстраполяции. Его опреде-
ляют как число СН50 (единица активности комплемента, вы-
зывающая 50% гемолиз), которое необходимо прибавить к
0,05 мл антисыворотки, чтобы оставалась свободная 1 CHsq.
В данном методическом варианте определяют количество свя-
зывающегося комплемента, причем возможно одно-, двух- и
трехкомпонентное титрование.
Однокомпонентное: определяют разведение сыворотки, ан-
тигена, комплемента, вызывающее 50% гемолиз.
Двухкомпонентное: определяют необходимое количество
комплемента, вызывающее 50% гемолиз в условиях различных
13—990
193
разведений АС при постоянном количестве АГ или в условиях
различных разведений АГ при постоянном количестве АС.
Трехкомпонентное: определяют количество комплемента,
вызывающее 50% гемолиз для каждого соотношения АГ-АТ.
Второй вариант: используют постоянные количества
комплемента, оптимальную дозу антигена и различные разве-
дения сывороток. Измеряют количество комплемента, связанное
различными формами иммунных комплексов. Количество ком-
племента соотносится графически с количествами сыворотки,
титр определяют как обратную величину количества сыворот-
ки, которое необходимо для связывания от 50 до 100 СН50.
Для выявления АТ можно пользоваться вариантом 1.
При постановке РСК необходимо обратить внимание на
следующие факторы: подбор эритроцитов и их концентраций,
качество амбоцептора, время и температуру инкубации, объ-
емы реагентов, ингибирующее и активирующее комплемент
действие антигенов, сывороток, эффект прозоны и др.
2.14.1.2. ОПРЕДЕЛЕНИЕ ЕДИНИЦЫ КОМПЛЕМЕНТА
Делают несколько рядов разведений комплементсодержащей
сыворотки и смешивают их с сенсибилизированными эритро-
цитами. Середина сигмовидной кривой гемолиза соответствует
наивысшей чувствительности и, следовательно, наиболее при-
годна для титрования.
Процесс гемолиза можно описать следующим уравнением
Y
X = К- р _ Yji/п •
X — количество внесенного комплемента; Y — лизис в %; К — константа, зави-
сящая от концентрации клеток и участвующих в реакции объемов.
Согласно вышеприведенной формуле Krogh, отношение кон-
центрации адсорбированного вещества у к концентрации ад-
сорбирующего вещества х при 1/п пропорционально концент-
рации остающегося всщества. Если в серии опытов
1—у
У '
v = k, то -p = -^-(l_y)i/n.
Преобразование уравнения в логарифмическую форму дает
следующее выражение:
У 11,
log —= log-j- 4- log (1 —у).
Л Л 11
Этому выражению соответствует уравнение прямой u = a-f-b-v.
На графике v откладывают по оси абсцисс, ап — по оси орди-
194
нат, В в этом случае будет касательной к углу наклона:
1 / у \
log X = log К-Ь ~ log (т=у)-
К и 1/п — константы, п определяют исходя из уравнения каса-
тельной. 1/п зависит от количества комплемента, эта величина
растет с уменьшением количества комплемента, log х можно
получить линейной интерполяцией по log . Интенсивность
реакции снижается как в случае избытка антигена, так и ан-
тител. Mayer и сотр. пришли к выводу, что степень связывания
комплемента следует оценивать по остаточной гемолитической
активности с последующим вычитанием этой величины из сред-
ней величины активности в контроле (буфер, антиген, антите-
ла). Было установлено, что чем сильнее сенсибилизация анти-
телами, тем быстрее, при постоянном количестве комплемента
происходит лизис клеток. При одной и той же степени сенси-
билизации снижение количества комплемента вызывает умень-
шение скорости гемолиза. Значение СН50 поэтому зависит от
используемой системы определения активности комплемента.
По Mayer, за 1 СН50 принимают такое количество мл свежей
сыворотки морской свинки, которое вызывает лизис 2,5Х108
оптимально сенсибилизированных эритроцитов из общего коли-
чества 5x10s клеток, суспендированных в 7,5 мл буфера с
ионной силой 0,147 и с оптимальным содержанием Са2+ и
Mg2+ в течение 1 ч при 37°C. Графическое изображение ре-
зультатов гемолиза на логарифмической бумаге позволяет пре-
вратить сигмоидальную кривую в прямую. Калибровочную кри-
вую рассчитывают по выражению
logH% = m-log (Ext X ЮО) -f- b,
где m — угол наклона, вычисляемый графически и аналитически, b — точка
пересечения прямой гемолиза с ординатой Е — 0,01
log Н %Е OiO1 t= log Н % — п • и m/log tg + Ig [lg (E • 100)].
Соответствующую калибровочную кривую получают на фото-
метре в предварительном эксперименте. Графический пример
приведен на рис. 74.
2.14.1.3. ХАРАКТЕРИСТИКА АНТИГЕНОВ
Особое значение для точности РСК имеет вид антигена и спо-
соб его внесения. Оценка АГ проводится в отношении его спе-
цифического антигенного действия и его способности ингиби-
ровать комплемент. Одной из важных характеристик компле-
мента являются кривые изофиксации при различных концент-
рациях комплемента (рис. 75). При этом можно количественно
и качественно сравнивать антигены и иммуноглобулины. Изме-
няя относительные концентрации антигенов, удается наблюдать
расщепления «плеча антител», предпринимались также попыт-
13*
195
Рис. 74. Определение активности комплемента при помощи логарифмической
сетки.
Комплемент I: 50% гемолиза—38 ЕД, 100% гемолиза=26 ЕД. Комплемент II: 50% гемо-
лиза—51 ЕД, 100% гемолиза—27 ЕД.
ки дифференцировать антигены по изменению форм кривых
при минимально определяемых количествах антител. В систе-
ме логарифмических координат можно провести линии макси-
мума антигенов и сывороток, включающих так называемую
упрощенную макси-
мальную линию. Учи-
тывая весьма различ-
ное качество антиге-
нов, на практике быва-
ет очень трудно точно
определить эти линии.
Относительно антисы-
вороток следует ска-
зать, что, очевидно, не-
достаточно установить
Рис. 75. Кривые изофикса-
ции антигена при различных
разведениях комплемента.
SV — разведение сыворотки;
AgV — разведение комплемента;
ML — максимальные линии,
AML — максимальные линии ан-
тигена.
196
оптимальное соотношение АГ и данной сыворотки, т. е. следует
также обращать внимание на тип сыворотки и состав им-
муноглобулинов антисыворотки. Для установления оптимальных
количеств антигена, связывающего комплемент, применяемых
в РСК, необходимо определить следующие параметры: кривые
изофиксации с различными разведениями комплемента, область
прозоны, тип кривых изофиксации и зоны максимума (напри-
мер, максимум антигена), а также дфференцировать ингибирую-
щее комплемент и специфическое антигенное действие, опреде-
лить оптимальное комплементзависимое разведение антигена,
оценить величину ожидаемого рассеяния результатов, расши-
рить имеющиеся сведения путем уточнения типа 1g, установить
группы кривых изофиксации.
2.14.2. ПОСТАНОВКА ОПЫТА
2.14.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Реагенты
Вероналовый буфер:
NaCl 85,0 г
Диэтилбарбитуровая кислота 5,75 г
Диэтилбарбитурат натрия 3,76 г
MgCl2-6H2O 1,68 г
СаС12 (безводный) 0,28 г
Дистиллированная вода до 2000,0 мл
pH
Раствор Витте для консервации компле-
мента:
Сульфат калия 10,0 г
Борная кислота 4,0 г
Дистиллированная вода 100,0 мл
Раствор автоклавируют в течение 1 ч.
Раствор Олсвера:
Глюкоза 24,6 г
Нитрат натрия 9,6 г
Хлорид натрия 5,04 г
Дистиллированная вода 1200 мл
Эритроциты: отбирают в стерильных условиях кровь в
раствор в соотношении l-f-2. Разливают смесь по стерильным
пробиркам. Перед употреблением выдерживают смесь 2 дня.
Сохранность — 6—8 нед.
Гемолизин: используют амбоцепторы государственного
института иммунопрепаратов в Берлине. Амбоцепторы не долж-
ны агглютинировать эритроциты. Разведения свыше 1:1200
не используются.
Комплемент: используют свежие лиофилизированные или
консервированные сыворотки (валовую сыворотку, взятую от
197
возможно большего числа животных). Кровь для сывороток
получают пункцией сердца или вскрытием сонной артерии с
целью предотвращения бактериального загрязнения. Посколь-
ку не всегда имеются коммерческие препараты комплемента,
можно использовать комплемент, консервированный раствором
Витте. Активность таких препаратов сохраняется при 5°C в те-
чение 6 месяцев. В любом случае препарат комплемента не
должен быть загрязнен специфическими антителами, сыворот-
ки, содержащие менее 50 ЕД активности в мл, для работы не
пригодны.
Антигены: для диагностики рекомендуется использовать
официально стандартизованные препараты, но и в этом случае
желательно точно установить, кривые изофиксации.
Оснащение:
Фотометр (например, Carl Ziss, Йена); микропланшеты для
титрования (U-образная форма лунок), микрошприцы на
25 мкл, устройство для отмывки планшет, автоматизирован-
ная микросистема для титрования (для РСК в 0,2 мл), стек-
лянные пробирки и штативы, термостат на 37 °C, водяная баня
для инактивации комплемента (56°C), холодильник, центри-
фугу.
2.14.2.2. ГЕМОЛИТИЧЕСКАЯ СИСТЕМА
Получение суспензии эритроцитов барана
Законсервированные эритроциты барана трижды отмывают
буфером. Получают 3% суспензию. 0,5 мл этой суспензии сме-
шивают с 4,5 мл дистиллированной воды, определяют экстинк-
цию при длине волны 540 нм в односантиметровом кювете.
Получают 1,5% суспензию:
V — наличный объем, Е — измеренная экстинция.
Проверка гемолизинов (амбоцептора)
1 часть = 25 мкл (микропроба) или 200 мкл (макро = РСК)
Определение титра агглютинации:
(исключительно при холодовом связывании):
Состав реакционной смеси:
1 часть амбоцептора (начиная с 1 :30, 1 = 2)
1 часть 1,5% суспензии эритроцитов
Инкубация 18 часов при 5 °C
Визуальный учет
Определение необходимого разведения
гемолизина:
Состав реакционной смеси:
1 часть амбоцептора
198
1 часть 1,5% суспензии эритроцитов
2 части буфера
1 часть комплемента 1 : 30
Инкубация 45 мин при 37 °C
Определяют 4—5-кратный титр 100% гемолиза
Определение авидности:
Состав реакционной смеси:
2 ряда разведений
1 часть комплемента (начиная с 1 :37,5; (=1,25)
2 части буфера
2 части гемолитической системы (1 часть 1% суспензии
эритроцитов + 1 часть рабочего разведения гемолизина)
Инкубация 15 мин и соответственно 45 мин при 37 °C
определяют титр 95% гемолиза (ТГ)
г 95% ТГ за 15 мин
Степень авидности =—----------------
95% ТГ за 45 мин
Степень авидности должна быть по возможности не выше 0,9
и не ниже 0,7. Гемолизин и эритроциты в день эксперимента
смешивают в равных объемах и сохраняют при 5 °C, перед ис-
пользованием прогревают в течение 30 мин при 37°C на водя-
ной бане.
Ряды разведений
f = 2: пример: 1 : 10, 1 : 20, 1 : 40, 1 :80 и т. д.
f=l,25: пример: 1:10,
и т. д.
1:1,25, 1:15,7; 1:19,6, 1:24,5
2.14.2.3. ПРИГОТОВЛЕНИЕ АНТИГЕНА
Часто в практике научных исследований приходится пользо-
ваться нестандартизованными антигенами. Эти АГ следует как
можно тщательнее очистить от сопутствующих примесей, спо-
собных привести к ложноположительным результатам. Поэтому
настоятельно рекомендуется проводить дифференциацию меж-
ду комплементингибирующим и специфическим антигенным
действием. Для этой цели предназначен контроль 2. Для по-
становки контроля 1 необходимы тщательно охарактеризован-
ные реагенты (антигены, сыворотки и т. д.). Учитывая жела-
тельную точность результатов, оценки антигенов проводят мак-
рометодом. Во всех случаях необходимо не только использо-
вать оптимальные дозы АГ, но также контролировать его спе-
цифичность.
Контроль 1 (предварительный опыт)
Состав реакционной смеси:
1 часть АГ (начиная с разведения 1 :8, f = 2)
1 часть АС (начиная с 1 : 10, f = 2)
1 часть комплемента (2 ЕД активности; разведение 1:25
или 1 :20)
199
Рис. 76. Определение комп-
лементингибирующего дей-
ствия АГ.
К — контрольное значение гемо-
лиза.
Инкубация 18 ч при
5 °C, затем 10 мин при
37 °C.
2 части гемолитической
системы
Наблюдение за полно-
той гемолиза в конт-
рольной пробирке
Для этого экспери-
мента используют тит-
рование по схеме так
называемой шахматной
доски или блок-титро-
вание, в котором ана-
лизируют ряд разведе-
ний АГ относительно
ряда разведений анти-
сыворотки. Степень
, торможения гемолиза
оценивается по четырехбалльной системе. В качестве единицы
АГ берут такое его максимальное разведение, которое способно
связывать 3—4 максимальных разведения сыворотки. Исследуе-
мый и контрольный антигены, контрольная антисыворотка не
должны давать торможения гемолиза. В основном опыте ис-
пользуют 2 единицы антигена.
Контроль 2 (предварительный опыт)
Определение комплементингибирующего действия (в пробир-
ках)
Состав реакционной смеси:
0,2 мл комплемента (начиная с разведения 1:37,5; f —1,25)
0,2 мл антигена (начиная с разведения 1:8, f=2)
0,2 мл буфера
Инкубация 18 ч при 5 °C
0,4 мл гемолитической системы
Инкубация 45 мин при 37 °C
Фотометрическое определение 95% ТГ
Расчет фактора потребления комплемента для каждого
разведения АГ
См. пример на рис. 76.
Необходимо проверять наличие неспецифического комплемент-
ингибирующего действия. В целом оно должно проявляться не
200
Рис. 77. Определения факто-
ра потребления комплемен-
та.
v ~ К-95 % НТ R1
КоЬ“ 95 % НТ R2 ’
KoF: для К (2,5) =3,0;
KoF: для К (3,0) =3,6.
более чем в 1—2 раз-
ведениях (при шаге=
= 1,25) по сравнению
с контролем.
Расчет фактора по-
требления комплемен-
та (ФПК). ФПК опре-
деляют для каждого
разведения АГ по фор-
муле:
ФПК =
К-95% ТГ без антигена
= 95% ТГ с антигеном '
для разведения сыво-
ротки 1:5 — 1:20
к=з,о
для разведения сыворотки
начиная с 1:40 К=2,5
Для определения ФПК ставят контроль 2, так как значение
ФПК необходимо знать для многих диагностически важных
АГ в предварительной оценке комплемента. Это можно пояс-
нить примером на рис. 77.
Определение антигенного действия:
Состав реакционной смеси:
1 часть антисыворотки
1 часть антигена (начиная с разведения 1:8, f = 2)
1 часть разведенного комплемента (95% ТГ; количество
комплемента, равное ФПК для соответствующего разведения
АГ)
Инкубация 18 ч, 4 °C
2 части гемолитической системы
Инкубация 45 мин, 37 °C
Визуальная оценка результатов:
1—4-кратный положительный результат
2.14.2.4. ПРИГОТОВЛЕНИЕ КОМПЛЕМЕНТА
Опыт 1.
Подбор ведут в присутствии 2 ЕД антигена
Состав реакционной смеси:
0,13—0,05 мл комплемента (разведение 1:20)
201
0,2 мл антигена
0,27—0,35 мл буфера
Инкубация 30 мин, 37 °C
0,5 мл гемолитической системы
Инкубация 30 мин, 37 °C
Визуальный учет результатов
Опыт 2.
Состав реакционной смеси:
1,0 мл разведенного комплемента (1:37,5; f = l,25)
2,0 мл буфера
2,0 мл гемолитической системы
Инкубация 45 мин 37 °C
Фотометрическое определение 50% или соответственно 95%
ТГ
Оценку результатов см. рис. 74.
2.14.2.5. АНТИСЫВОРОТКИ
Сыворотки пациентов не должны обладать гемолитической ак-
тивностью, их инактивируют в течение 30 мин на водяной бане
(56°C). Последующую инактивацию проводят при той же тем-
пературе в течение 10 мин. Особое значение имеет использо-
вание точно охарактеризованных контрольных иммунных сы-
вороток. Если это человеческие сыворотки, то они должны
иметь максимально высокий титр; смесь сывороток реконвалес-
центов разливают по 1 мл в ампулы и хранят при —20°C или
в лиофилизированном виде. Сыворотки, хранящиеся в шкафу
глубокого охлаждения в открытых сосудах, могут повреж-
даться углекислым газом. Многократное замораживание и от-
таивание также противопоказано. Определяют наличие ком-
плементингибирующей активности, стараются использовать сы-
воротки наиболее подходящего вида животных.
2.14.2.6. ОСНОВНОЙ ОПЫТ
Оценка результатов РСК сильно зависит от того, каким обра-
зом измеряют торможение гемолиза. Фотометрическое опреде-
ление степени 50% гемолиза, несмотря на точность измерения,
практически не всегда удобно. Гораздо чаще используют ви-
зуальную оценку для определения 100% или 50% гемолиза.
Для более точной градации можно использовать четырехбалль-
ную шкалу. За положительный результат принимают 3—4-крат-
ное торможение (0—50% гемолиза).
Опыт 1.
Состав реакционной смеси:
1 часть сыворотки начиная с разведения 1 :10, f=2)
1 часть антигена (2 АЕ)
202
1 часть комплемента (2 ФПК)
Инкубация 18 ч при 5 °C и 10 мин при 37 °C
Внесение 0,5 мл гемолитической системы
Инкубация 15—30 мин при 37 °C
Визуальная оценка торможения гемолиза
Опыт 2.
Состав реакционной смеси:
1 часть сыворотки (разведения начиная с 1:10; f = 2).
1 часть разведенного антигена
0,2 мл разведенного комплемента
Инкубация 18 ч при 5 °C, затем 30 мин при 37 °C
0,4 мл гемолитической системы
Инкубация 45 мин при 37 °C
Визуальная оценка торможения гемолиза
2.14.2.7 . ВАРИАНТЫ КОНТРОЛЯ
Проводят позитивный и негативный контроль антигена и сы-
воротки.
Контроль антигена:
0,2 мл буфера
0,2 мл разведенного антигена
0,2 мл разведенного комплемента
Контроль сыворотки:
0,2 мл буфера
0,2 мл разведенной сыворотки
0,2 мл разведенного комплемента
2.14.2.8 . МИКРОМЕТОД
Любой вариант РСК можно осуществлять в микромодифика-
ции. Необходимым в данном случае является подбор опти-
мальных реагентов. Используемые парциальные объемы могут
составлять 25 или 50 мкл. Особое внимание следует обращать
на чистоту планшетов и тщательное перемешивание компле-
ментов. Водяную баню можно заменить термостатом. Оцени-
вается не столько число лизированных эритроцитов, сколько
количество нелизированного осадка эритроцитов. 100—50% ли-
зис осадков рассматривается как отрицательный результат.
Положительными результатами считаются следующие:
+ + +осадок эритроцитов почти неизмененной величины со
слегка окрашенной надосадочной фазой;
+ Н—Н+осадок эритроцитов максимального размера, гемолиз
отсутствует.
Для предотвращения влияния субъективных оценок резуль-
таты должны оцениваться одним и тем же исследователем.
Определенные проблемы создает соотнесение предварительного
и основного опытов. Трудности возникают, когда титр гемоли-
203
опытах. Многообразия единиц антигена
Рис. 78. Определение титра
50% гемолиза.
По ординате — степень гемоли-
за, по абсциссе: внизу — номера
пробирок, вверху — тнтр сыво-
ротки.
за в обоих опытах раз-
личается, например 50
и 100%. Это связано с
тем, что 100% гемо-
лиз установить слож-
нее, чем 50%. Фото-
метрия, особенно с при-
менением логарифми-
ческой бумаги, по-
зволяет преодолеть эти
сложности. Их легко
избежать, если поль-
зоваться одинаковой
методикой в предвари-
тельном и основном
и комплемента можно не
опасаться, если исходить не из принципа чистоты компонентов,
а из принципа оптимальной, т. е. относящейся к области эквива-
лентности, дозы.
2.14.3 . ОЦЕНКА МЕТОДА
РСК нашла самое широкое распространение в микробиологи-
ческой и серологической диагностике, особый интерес она пред-
ставляет для клинической иммунологии. Результаты исследо-
вания выражаются в титрах. За титр принимают количество
комплемента, связываемое в присутствии оптимальной дозы
антигена и сыворотки, разведенной 1-}-2. Наибольшее распро-
странение получил метод Кольчера, названный также методом
разведения сыворотки. Этим методом в присутствии постоян-
ной дозы антигена определяют обратный титр сыворотки, т. е.
выбирают разведение сыворотки, при котором связывается
такое количество комплемента, что происходит 50% или полное
ингибирование гемолиза. Стремление стандартизовать условия
опыта для определения титра позволяет прийти к следующей
формуле:
&г
Ет = Ед
Ет — активность сыворотки (ЕД/мл); Еа — среднегеометрическое значение ак-
тивности исследуемой сыворотки; St — настоящее среднегеометрическое зна-
чение активности стандартной сыворотки; Sa — активность стандартной сы-
воротки;
204
Титр может быть выражен по 100% торможению гемолиза или,
при более точном титровании, 50% торможению гемолиза.
Пример приведен на рис. 78. Важное значение для оценки
результатов имеет их специфичность. Для этого используют
очистку реагентов,, определяют зону эквивалентности, устанав-
ливают состав иммуноглобулинов антисыворотки, проводят
контроль специфичности при помощи перекрестного титрования
с известными антигенами и антисыворотками. В случае инфек-
ционных заболеваний или иммунизации такие исследования
позволяют наблюдать по меньшей мере 4-кратное повышение
титра. Отдельно взятые значения результатов РСК, так же как
и так называемый базисный титр, следует оценивать весьма
критически.
2.14.4 . СПИСОК ЛИТЕРАТУРЫ
1. Morenz J. Methodik der KBR.— Ztschr. med. Labortechn., 1970, 11, 249—
271.
2. Muller G„ Tielecke К. H., Budek I., Scabinski G. Eine neue automatisierte
Methode fur die KBR. — Ztschr. med. Labortechnik, 1975, 16, 267—271.
3. Palmer D. F. Complement fixation Test. — In: Manual of Clinical Immunology
(2-nd ed.). — Amer. Soc. Microbiol., 1980, 35—47.
4. Stelzner A., Holtz A. Vergleichende Untersuchungen zur Durchfurung der
Komplementbindungsreaktion. 1. Mitteilung: Studied zur Ermittelung der
Antigengebrauchsverdunnung unter Verwendung eines Ornithose—Antigens. —
Ztschr. med. Labortechnik, 1975, 16, 197—210.
5. Stelzner A., Stein G. Moglichkeiten zur hamolytischen Aktivitatsmessung von
Gesamtkomplement — theoretische Grundlagen und methodische Hinweise. —
Wiss. Ztschr. Friedr. — Schiller-Univ. Jena, Math.-Naturwis., 1971, Reihe 20,
933—941.
6. U. S. Dept. Health, Education and Wellfare, Public Health Service Atlanta:
A Guide to the performance of standardized diagnostic CF method and adap-
tation to micro test. — Revised Ed. 1974.
2.15. АНТИТЕЛОЗАВИСИМАЯ
КЛЕТОЧНАЯ ЦИТОТОКСИЧНОСТЬ
Антителозависимая клеточная цитотоксичность (АЗКЦ) может
рассматриваться как вариант клеточной цитотоксичности, при
которой в присутствии специфических АТ к клеткам-мишеням
происходит разрушение последних под воздействием лейкоци-
тов-эффекторов. Специфичность реакции определяется антите-
лами. Эффекторные клетки действуют неспецифически, оии
связываются благодаря Fc-рецепторам с находящимися на по-
верхности клеток-мишеней комплексами АГ—АТ (рис. 79).
Независимый от комплемента цитотоксический эффект впер-
вые наблюдал Moller в 1965 г; более подробные исследования
были проведены группой ученых под руководством MacLennan
[8]. Этот вопрос рассматривается также в обзорах [3, 7, 14].
В реакции АЗКЦ разрушаются все типы клеток, это относится
также к бактериям и паразитам [2]. Для исследовательских
205
Рис. 79. Принцип АЗКЦ.
клетка
(лейкоцит с
Fc-рецепторами)
целей наиболее часто используют эритроциты, лейкоциты,
фибробласты или опухолевые клетки. Для выявления цитолиза
применяют мечение клеток-мишеней 51Сг (в форме хромата
натрия). Хромат поглощается клетками и связывается с бел-
ками в виде трехвалентного хрома [17], в ходе лизиса радио-
активный изотоп быстро высвобождается. В микроварианте
цитотоксического теста (см. 3.6) АЗКЦ можно проводить с не-
мечеными клетками. Для определения единичных эффекторных
клеток был разработан специальный метод [19].
Антитела, участвующие в АЗКЦ, в основном относятся к
классу IgG, в некоторых случаях это могут быть IgM- или
IgE-антитела [4, 7]. АТ без Fc-фрагмента неэффективны. Ком-
плексы АГ—АТ или термоаггрегированный у-глобулин неспе-
цифически тормозят реакцию [14]. На развитие цитотоксиче-
ской реакции оказывают влияние различные факторы. Классы
АТ, возможно, также субклассы, определяют, какой тип эф-
фекторных клеток вступит в реакцию. Клетки-мишени, к кото-
рым присоединился IgG, лизируются эффекторными клетками
с Fc- (IgG)-рецепторами, если клетки-мишени адсорбировали
IgM, то во взаимодействие вступают клетки-эффекторы с
Fc-(IgM)-рецепторами. Непременной предпосылкой АЗКЦ яв-
ляется наличие у лейкоцитов Fc-рецепторов и цитотоксической
активности. Нормальные В-лимфоциты, хотя и имеют Fc-рецеп-
торы, лишены цитотоксичности. Т-киллеры ввиду отсутствия у
них Fc-рецепторов не в состоянии лизировать покрытые анти-
телами клетки-мишени. В случае некоторых генетических на-
рушений, например, у больных синдромом Чедиака — Хигаси
или у бежевых мышей, наблюдается утеря цитотоксической
активности лейкоцитов с Fc-рецепторами [15, 16]. Эффектор-
ными клетками АЗКЦ могут быть лимфоциты, моноциты, гра-
нулоциты, а также эозинофилы [8]. Особенно активны неспо-
собные к адгезии, не фагоцитирующие мононуклеарные лейко-
циты. Вследствие отсутствия классических маркеров Т- и В-кле-
ток эти клетки называются «нуль»-лимфоцитами, или нулевы-
ми клетками. Для эффективных в АЗКЦ нулевых клеток было
предложено название «К-клетки» [13, 14]. С К-клетками в
близком родстве состоят К-клетки (англ, natural killer cells),
206
спонтанно, без помощи АТ, лизирующие некоторые чувстви-
тельные клетки-мишени. К- и К-клетки человека, очевидно, про-
исходят от одной клеточной популяции. Речь идет о крупных
зернистых лимфоидных клетках [18] с особым дифференциро-
вочным антигеном [1]. У человека К- и К-клетки обнаружи-
ваются прежде всего в костном мозге, крови и селезенке и не
обнаруживаются в лимфоузлах, миндалинах, тимусе {5, 10].
Считается, что 5—10% лимфоцитов крови относится к К- или
NK-типу ,[1, 13]. АЗКЦ можно проверять на аллогенных, син-
генных или аутологических компонентах. Физиологическая роль
АЗКЦ, как и других цитотоксических реакций, еще не вполне
ясна. Вероятно, она участвует в механизмах отторжения транс-
плантатов, противоопухолевого иммунитета, а также аутоим-
мунных заболеваний. В практической иммунологии АЗКЦ по-
зволяет обнаруживать АТ в больших разведениях, например
тогда, когда метод определения комплементзависимой цитоток-
сичности недостаточно чувствителен [9]. Выявление антител
в сыворотке больных с трансплантированной почкой также
осуществляется при помощи АЗКЦ. Положительный результат
теста на АЗКЦ также доказывает наличие Fc-рецепторов на
применяемых лейкоцитах. АЗКЦ используют для определения
поверхностных клеточных антигенов, например, Н-2-антигена
мышей. В данном случае специфическая антисыворотка к Н-2-
антигену подавляет активность эффекторных клеток [6].
2.15.2. ПОСТАНОВКА ОПЫТА
Ниже будет описан метод, основанный на цитолизе меченных
51Сг асцитно-лейкемических клеток мыши в присутствии спе-
цифических АТ и эффекторов: сингенных перитонеальных кле-
ток или лейкоцитов крови человека.
2.15.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Подходящей средой является RPMI 1640 с добавлением 10%
инактивированной сыворотки плода коровы. Для поддержания
постоянного pH к сыворотке добавляют 10—20 мМ буфера на
основе ГЭПЭС. Добавляемая сыворотка не должна содержать
у-глобулинов, которые могут образовывать агрегаты и тормо-
зить АЗКЦ. Для постановки эксперимента необходимы чистые,
ополоснутые бидистиллированной водой центрифужные пробир-
ки на 10 и 20 мл, небольшие пробирки (10X40 мм) с круглым
дном и пастеровские пипетки. Необходимы также Ыаг 51СгО4
(Центральный институт ядерных исследований АН ГДГ), лпм-
фопреп (Nyegaard, Норвегия) или смесь фиколл — визотраст
равной плотности, настольная центрифуга с бакет-ротором,
счетчик у-излучения.
207
2.15.2.2. ПОДГОТОВКА КЛЕТОК-МИШЕНЕЙ
Из полости брюшины мышей отбирают асцитическую жидкость,
содержащую лейкемические клетки, индуцированные вирусом
Graffi (перевивку проводят еженедельно на сингенных мышах
штамма XVII/Bln. Осаждают клетки центрифугированием, сус-
пендируют в среде и снова центрифугируют. Содержащиеся в
осадке эритроциты могут быть удалены холодной водой
(1 часть осадка клеток + 2 части дистиллированной воды;
10 сек). После отмывки готовят суспензию, содержащую 1—
5ХЮ7 клеток/мл. В суспензии должно быть не более 5—10%
неживых клеток (окрашивание трипановым синим).
2.15.2.3. МЕЧЕНИЕ КЛЕТОК-МИШЕНЕЙ 51[Сг]
Суспензию клеток асцитической жидкости (0,15 мл) в малень-
кой пробирке (10X40 мм) смешивают со 100 мкКи 51[Сг]
в 0,1 мл 0,15 М NaCl; инкубируют 90 мин при 37 °C, часто
встряхивая. Суспензию переносят пастеровской пипеткой в
центрифужную пробирку, разводят небольшим количеством
среды и центрифугируют 6 мин при 200 g. Надосадочную фрак-
цию осторожно удаляют. Осадок клеток трижды промывают
средой (порциями по 8 мл). Рекомендуется перед последним
центрифугированием дать постоять суспензии клеток 20 мин,
чтобы сорбировавшийся на внешней- мембране Na251[Cr]O4
мог диффундировать в среду. Готовят из осадка суспензию
2,5Х105 клеток/мл. Следует указать, что не все клетки асци-
тической жидкости могут быть клетками-мишенями. Некоторые
типы клеток характеризуются высоким спонтанным уровнем
высвобождения 51 [Сг]. Рекомендуется работать с линиями кле-
ток, адаптированными к культивированию in vitro.
2.15.2.4. ПРИГОТОВЛЕНИЕ АНТИСЫВОРОТКИ
Кролику с интервалом в неделю делают внутривенно 5 инъек-
ций по 108 лейкемических асцитных клеток. Через неделю пос-
ле последней инъекции получают сыворотку, инактивируют ее
30 мин при 56 °C и при —20 °C. Для удаления видоспецифиче-
ских антител сыворотку разводят физиологическим раствором
в соотношении 1: 10 и трижды адсорбируют эритроцитами мы-
ши (3—4 части разведенной сыворотки %1 часть осадка от-
мытых эритроцитов). Затем дважды на холоду адсорбируют
сыворотку спленоцитами мыши (1 часть сыворотки +1 часть
отмытого осадка спленоцитов; инкубация 1 ч). Специфичность
сыворотки можно проверить в реакции комплементзависимой
цитотоксичности (см. раздел 2.12) с лейкемическими, а также
с нормальными клетками тимуса, селезенки, брюшной полости.
208
2.15.2.5. ПРИГОТОВЛЕНИЕ ЭФФЕКТОРНЫХ КЛЕТОК
Перитонеальные клетки мыши получают путем смыва с брю-
шины холодным 0,15 М NaCl или солевым раствором Хенкса;
мононуклеарные лейкоциты человека получают градиентным
центрифугированием гепаринизированной крови по Бейюму
(Boyum) (см. раздел 3.1). Клетки отмывают 2—3 раза, ресус-
пендируют в среде и подсчитывают. Для получения дозозави-
симого эффекта готовят несколько суспензий эффекторных
клеток разной концентрации.
2.15.2.6. ПОСТАНОВКА ПРОБЫ
В маленькие пробирки (10x40 мм), двойное или тройное по-
вторение проб, последовательно вносят по 0,05 мл среды,
0,05 мл антисыворотки, 0,2 мл суспензии клеток-мишеней и
0,2 мл суспензии эффекторных клеток (общий объем 0,5 мл).
Для контроля ставят следующие пробы: клетки-мишени +
+ среда, клетки-мишени + антисыворотка, клетки-мишени+
+ эффекторные клетки. Пробирки центрифугируют (800 g;
2 мин), при этом клетки-мишени и эффекторные клетки входят
в тесное соприкосновение, пробирки закрывают резиновыми
пробками и инкубируют 4 ч при 37°C. Затем добавляют по
0,5 мл охлажденного 0,15 М NaCl, осторожно, но энергично
встряхивают и центрифугируют (5 мин; 1200 g). Из надосадоч-
ной фракции отбирают 0,5 мл для измерения уизлучения. Ра-
циональнее проводить исследование в микропланшетах (Flow —
Labs). В этом случае объем пробы составляет 0,11 мл (0,05 мл
суспензии клеток-мишеней, 0,05 мл суспензии эффекторных
клеток и 0,01 мл антисыворотки). Небольшое число клеток-
мишеней (1Х104) уравновешивается более интенсивным мече-
нием 51{Сг].
2.15.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Для подсчета количества специфически высвободившегося 51Сг
можно пользоваться следующей формулой [13]:
% специфического __ эксп, — споит.
высвобождения макс. — споит. ' *
где: эксп. — радиоактивность, высвобождающаяся при лизисе
«нагруженных» антителами клеток-мишеней эффекторными
клетками; спонт. — радиоактивность, высвобождающаяся в
смеси, содержащей только клетки-мишени и эффекторные
клетки; макс.— радиоактивность, высвобождающаяся при
многократном замораживании и оттаивании клеток или при
их обработке детергентами.
Вышеприведенную формулу используют только в случае,
если спонтанное высвобождение не слишком велико. В случае,
14-990 209
Отношение эффекторных клеток к клеткам -мишеням
(логарифм)
Рис. 80. К-клеточная актив-
ность мононуклеарных лейко-
цитов крови человека, опреде-
ляемая при помощи нагружен-
ных антителами клеток лейко-
за мышей.
Состав пробы: Ю4 клеток-мишеней
(клетки лейкоза мышей Gr/E). ан-
ти-Gr/E — сыворотка (1 : 100); соот-
ношение эффекторных клеток и кле-
ток-мншеней — 2:1, 10:1, 50:1.
Из точки пересечения кривой зави-
симости доза — эффект с линий 50%
лизиса следует, что 1 литическая
единица (количество эффекторных
клеток, вызывающее 50% лизис) со-
ответствует приблизительно 10-крат-
ному избытку эффекторов, т. е. в
условиях данного опыта около
105 клеток.
гели оно составляет свыше 30%, используют формулу:
ЭКСП. — СПОИТ. 1QQ
: общ.
где общ.— общая радиоактивность пробы (осадок и надосадочная фракция).
Измерение высвобождения 51[Сг] при подходящих условиях
опыта позволяет оценить активность киллеров. Высвобожде-
ние изотопа при 15—75% цитолизе прямо пропорционально
логарифму концентрации эффекторных клеток. Если соотнести
специфическое высвобождение 51[Сг] с логарифмом соотноше-
ния эффекторные клетки/клетки-мишени, то получается пря-
мая, при помощи которой можно подсчитать количество эф-
фекторных клеток, необходимое для 50% цитолиза (1 литиче-
ская единица; рис. 80). Метод позволяет сравнивать актив-
ность различных популяций эффекторных клеток. Клеточную
цитотоксичность можно также рассматривать как ферментатив-
ную реакцию. В этом случае можно определить соответствую-
щие кинетические параметры в условиях постоянного числа
эффекторных клеток «и переменного числа клеток-мишеней
[3].
2.15.4. СПИСОК ЛИТЕРАТУРЫ
1. Abo Т„ Balch С. М. A differnetiation antigen of human NK- and K-cells
identified by a monoclonal antibody (HNK-1).— Journ. Immunol., 1981, 127,
1024—1029.
2. Butterworth A. E„ David J. R„ Franke D. et al. Antibody-dependent cell-
mediated damage to 5ICr-labeled Schistosomule of Schistosoma mansoni:
Damage by purified eosinophyles. — Journ. exp. Med., 1977, 145, 136—150.
3. Callewaert D. M., Johnsos D. F„ Kearney J. Spontaneous cytotoxicity of
cultered human cell lines mediated by normal peripheral blood lymphocytes.
III. Kinetic parameters. — Journ. Immunol., 1978, 121, 710—717.
4. Capron A., Dessaint J. P., Joseph M. et al. Interaction between IgG comple-
210
xes and macrophages in the rat. A new mechanism of macrophage activa-
tion.— Eur. Journ. Immunol., 1977, 7, 315—322.
5. Cordier G., Samarut C„ Brochier J., Revillard J. P. Antobodydependent cel-
lular cytotoxicity (ADCC). Characterization of killer cells in human lymphoid
organs. — Scand. Journ. Ummunol., 1976, 5, 233—242.
6. Halloran P„ Schirmacher V., Festenstein H. A new sensitive assay for anti-
body against cell surface antigens based on inhibition of cell-dependent anti-
body-mediated cytotoxicity. — Journ. exp. Med., 1974, 140, 1348—1363.
7. Lamon E. W., Shaw M. W., Goodson S. et al. Antibody-dependent cell-media-
ted cytotoxicity in the Moloney sarcoma virus system В Differential activity
of IgG and IgM with different subpopulations of lymphocytes. — Journ. exp.
Med., 1977, 145, 302—313.
8. MacLennan I. C., Campbell A. C„ Gale D. G. L. Quantitation of K-cells. —
In: In vitro Methods in cell-mediated and tumor immunity/Eds. B. R. Bloom
and J. R. David. New York, Academic press, 1976, 511—522.
9. Moller G., Svehag S. E. Specidicity of Lymphocyte-mediated cytotoxicity
induced by in vitro antibody-coated target cells. — Cell. Immunol., 1972, 4,
1—19.
10. O’Tool C., Saxon A., Bohrer R. Human lymphonode lymphocytes fail to ef-
fect lysis of antibody-coated cells. — Clin. exp. Immunol., 1977, 27, 165—
171.
11. Pearson G. A. In vitro and in vivo investigations on antibody dependent
cellular cytotoxicity. — Current topics in Microbiol. Immunol., 1978, 80,
65—96.
12. Perlmann P„ Holm G., Stejskal V. Cell-mediated cytotoxicity in vitro: Target
cells recognition and effector cell requirement. — Transpl. Proc., 1973, V,
1625—1629.
13. Perlmann P„ Perlmann H., Hellstrom U., Hammarstom S. Purification and
fractionation of human blood lymphocytes. Characterisation of subclasses by
their antibody-dependent cytotoxic potential (К-cells assay). — In: In vitro
Methods in cell mediated and turner immunity/Eds. B. R. Bloom and J. R. Da-
vid. New York: Academic press, 1976, 497—509.
14. Perlmann P., Perlmann H., Larsson A., Wahlin B. Antibody dependent cyto-
litic effector lymphocytes (К-cells) in human blood. — Journ. Reticuloend.
Soc., 1975, 17, 241—250.
15. Roder J., Duwe A. K. The beige mutation in the mouse selectivity impairs
natural killer cell fraction. — Nature, 1979, 451—453.
16. Roder J. C„ Haliotis T., Klein M. et al. A new immunodeficiency desorder in
humans involving NK-cells. — Nature, 1980, 284, 553—555.
17. Sanderson C. J. The uptake and retention of chromium by cells. — Transpl.,
1976, 21, 526—529.
18. Timonen T., Ortaldo J. R., Herberman R. B. Characteristics of human large
granular lymphocytes and relationship to natural killer cells and K-cells. —
J. exp. Med.. 1981, 153, 569—582.
19. Wahlin B., Perlmann P. Detection of К cells by a plaque assay. — In: In vitro
Methods in cell mediated and tumor immunity/Eds. B. R. Bloom, J. R. Da-
vid. — New York: Academic Press, 1976, 523—531.
20. Zighelboim J., Gale R. P. and Kedar E. Polymorphnuclear leucocyte Fc-recep-
tors in antibody-dependent cellular cytotoxicity (ADCC). — Transpl., 1976,
21, 524—526.
2.16. РЕАКЦИЯ ГЕМАГГЛЮТИНАЦИИ
2.16.1. ПРИНЦИП МЕТОДА
Обусловленное реакцией АГ—АТ видимое слипание эритроци-
тов называется гемагглютинацией (ГА).
При помощи ГА можно выявлять антитела к различным
антигенам мембраны эритроцитов. Это реакция прямой гемаг-
14* 211
Рис. 81. Прямая (а) и пассивная (б) гемагглютинация.
Прямоугольники обозначают антиген эритроцитов, кружочки — конъюгированный анти-
ген или гаптен.
глютинации (рис. 81, а). Если эритроциты используются в ка-
честве носителей антигенов, т. е. содержат на своей поверх-
ности чужеродные антигены и гаптены, то и в этом случае
возможна ГА, так называемая пассивная гемагглютинация
Таблица 17. Модификации метода гемагглютинации
Реагенты и процессы Варианты Примечание
Эритроциты Антигены Конъюгированные анти- гены Конъюгированные гаптены Метод конъюгации Добавки среды Метод титрования Свежие Обработанные фермен- тами Фиксированные Эритроцитарные Присоединенные к эрит- роцитам антигены или гаптеиы Белки Полисахариды Нуклеотиды Нитрофильиые группы Медикаменты Абсорбция Химический Иммунологический Стабилизаторы аити-Ig- сывороткп Макрометод Микрометод Автоматический метод Барана, человеческие: О, rh Папани, бромелин Формалин, глутаровый альдегид Прямая ГА Пассивная ГА (ПГА) Сывороточные, оргаио- специфические Бактериальные, ЛПС ДНК, РНК, иуклеотнды ДНФ-, ТНФ-пронзводные Пенициллин ЛПС; таниизированные эритроциты Азо-, амидосвязи анти- тела к ЭБ Фрагменты АТ Сывороточные белки, декстран, проба Кумбса В пробирках Микротитратор Такатси Автоанализатор
212
(ПГА) (рис. 81,6). Как и большая часть иммунологических
методов, ГА является вторичной реакцией АГ—АТ. Первичное
связывание специфических АТ с естественными АГ(ГА) или
искусственно присоединенными антигенными детерминантами
(ПГА) вызывают образование скоплений эритроцитов, дефор-
мацию клеточных мембран и исчезновение дзета-потенциала
[9]. Эритроциты сближаются так, что поли- или бивалентные
АТ образуют перекрестные связи с различными эритроцитами.
Массивное образование поперечных связей между АТ и эри-
троцитами макроскопически выражается как гемагглютинация.
ГА могут вызывать как преципитирующие, так и непреципи-
тирующие АТ. На этом основана высокая чувствительность ме-
тодов ГА для обнаружения АГ. В табл. 17 приведены различ-
ные модификации ГА. Описанные в разделе 1.5 методы при-
соединения белковых и бактериальных антигенов к эритроци-
там находят свое применение в ПГА. Использование ПГА для
выявления АТ к бактериальным полисахаридам детально из-
учено [14].
В качестве примеров будут описаны методы адсорбции бел-
ков на фиксированные, таннизированные эритроциты [6] и ко-
валентное связывание при помощи бифункционального реаген-
та глутарового альдегида. Будет также описано получение бис-
диазобензидина (ВДВ).
2.16.2. МЕТОДИКА
2.16.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы микротитратор Такачи (MIM, Венг-
рия), мешалка, водяная и ледяная бани; смесь ацетон + сухой
лед, бензидин 0,2 М НС1, NaNO2, йодкрахмальная бумага,
глутаровый альдегид (Serva, ФРГ), мертиолят, 0,15 М NaCl,
забуференный 0,15 М NaCl pH 7,2 (ФСБ) (23,9 мл 0,15 М
КН2РО4, 76 мл 0,15 М Na2HPO4, 100 мл 0,15 М NaCl), ФСБ
pH 6,4 (67,7 мл 0,15 М КН2РО4, 32,3 мл 0,15 М Na2HPO4,
100 мл 0,15 М NaCl), формалин (33% водный раствор реди-
стиллированного формальдегида, смешанного с равным объ-
емом ФСБ pH 7,2 двойной концентрации), таннин (Fluka, Швей-
цария) 1:20 000 в ФСБ pH 7,2 (исходный раствор 1:100 в
дистиллированной воде сохраняют при 4°C), 0,15 М фосфатный
буфер pH 7,4 [4,1 мл 0,5 М. Na2HPO4-2H2O (22,25 г/250 мл),
0,9 мл 0,5 М NaH2PO4-H2O (17,25 г/250 мл), 17,7 мл дистил-
лированной воды], эритроциты барана или человека (0, rh)
в растворе Олсвера, ЧСА, среда для разведения (ФСБ
pH 7,2+1% инактивированной адсорбированной эритроцитами
лошадиной сыворотки).
213
2.16.2.2. ПРИГОТОВЛЕНИЕ БИС-ДИАЗОБЕНЗИДИ НА
Бензидин в количестве 0,23 г (канцероген! работать в резино-
вых перчатках) растворяют в 45 мл 0,2 М НС1. Раствор охлаж-
дают на ледяной бане. Растворяют 0,175 г NaNO2 в 5 мл ди-
стиллированной воды, раствор охлаждают и, постоянно поме-
шивая, добавляют по каплям в течение 1 мин к раствору
бензидина. Реакцию диазолирования проводят на ледяной бане
до тех пор, пока йодкрахмальная бумага не перестанет обна-
руживать свободный нитрит (среднее время реакции 30 мин).
Готовый светло-желтый БДБ-реактив на ледяной бане разли-
вают в ампулы по 1 мл (при работе пипетку в рот не брать!!!),
замораживают в смеси ацетон + сухой лед. При —70 °C мате-
риал можно сохранять в течение нескольких месяцев.
2.16.2.3. ФИКСИРОВАНИЕ ЭРИТРОЦИТОВ
Формальдегидный метод: 5 объемов крови смешивают с 5 объ-
емами ФСБ с pH 7,2 и быстро смешивают с 4 объемами фор-
малина (16,5% раствор в ФСБ с pH 7,2). Инкубируют смесь
2 ч на водяной бане при 37 °C, помешивая каждые 15 мин.
По окончании формалинизации эритроциты 6 раз отмывают
ФСБ с pH 7,2 (низкоскоростное центрифугирование или сво-
бодная седиментация в течение 1 дня), готовят 10% суспен-
зию. Консервируют 0,3% формальдегидом, эритроциты сохра-
няются в течение нескольких месяцев, при 4 °C.
Глутаральдегидный метод: трижды отмытый физиологиче-
ским раствором осадок эритроцитов охлаждают до 0°С на во-
дяной бане. 25% глутаровый альдегид разводят до 1% раст-
вором следующего состава: 1 часть 0,15 М Na2HPO4 с pH 8,2+
+9 частей 0,15 М NaCl+ 5 частей дистиллированной воды,
охлаждают смесь на ледяной бане. Разводят осадок эритроци-
тов 1% глутаровым альдегидом до 1—2% суспензии, инкуби-
руют 30 мин при 0 °C при периодическом перемешивании
(фиксация). Фиксированные эритроциты отмывают 5 раз
0,15 М NaCl и 5 раз дистиллированной водой (низкоскоростное
центрифугирование). Сохраняют их в виде 30% суспензии в
дистиллированной воде с добавлением 0,01% мертиолята в.
холодильнике в течение нескольких месяцев.
2.16.2.4. ТАННИЗИРОВАНИЕ
И СЕНСИБИЛИЗАЦИЯ ЭРИТРОЦИТОВ
Отмытые фиксированные эритроциты (10% в ФСБ с pH 7,2)
смешивают с равным объемом раствора таннина 1:20 000 в
ФСБ с pH 7,2. Инкубируют 10 мин при комнатной температу-
ре, трижды отмывают ФСБ с pH 7,2. Для сенсибилизации сме-
шивают 4 мл ФСБ с pH 6,4, 1 мл оптимального разведения
антигена в 0,15 М NaCl и 1 мл 5% суспензии эритроцитов или
214
таннизированных эритроцитов. Инкубируют 20 мин при ком-
натной температуре, трижды отмывают ФСБ с pH 7,2 с 0,2%
ЧСА. Готовят суспензии эритроцитов для титрования в раство-
ре NaCl.
2.16.2.5. КОНЪЮГИРОВАНИЕ
ГЛУТАРОВЫМ АЛЬДЕГИДОМ
К 3,2 мл раствора белка (оптимальную концентрацию опреде-
ляют индивидуально; для бычьего у-глобулина — 2,5 мл/3,2 мл)
в 0,15 М фосфатном буфере с pH 7,4 добавляют 0,1 мл отмы-
того осадка эритроцитов и при постоянном перемешивании
добавляют 0,1 мл 0,25% водного раствора глутарового альде-
гида. Смесь инкубируют 1 ч при комнатной температуре. После
3-кратной отмывки конъюгированные эритроциты можно не-
медленно использовать или хранить в течение нескольких не-
дель в 0,15 М NaCl в холодильнике.
2.16.2.6. ПОСТАНОВКА РЕАКЦИИ
2.16.2.6.1. Реакция прямой гемагглютинации
Для инактивации комплемента с целью предотвращения им-
мунного гемолиза исследуемую сыворотку нагревают в тече-
ние 30 мин при 56°C. Затем готовят геометрический ряд раз-
ведений (1 + 1 или 1+2), смешивают полученные разведения
с суспензией эритроцитов. При постановке реакции в пробир-
ках антисыворотку разводят 0,5 мл среды (0,15 М NaCl или
ФСБ с pH 7,2 с 1% инактивированной лошадиной сывороткой,
адсорбированной тестерными эритроцитами) и добавляют к
каждому разведению по 0,075 мл 2,5% суспензии эритроцитов.
При постановке микротеста сыворотку разводят непосред-
ственно в планшетах при помощи многоканальных пипеток,
перенося по 25 мкл содержимого из одной лунки в другую
(в лунки предварительно внесено по 250 мкл среды). Затем
добавляют по 25 мкл 0,4% суспензии эритроцитов. После пере-
мешивания смесь оставляют на 2—3 ч при комнатной темпе-
ратуре, результат оценивают визуально (можно при помощи
лупы). Реакция считается положительной (есть гемагглютина-
ция), когда на дне сосуда образуется широкий, неправильной
формы «коврик». В случае пробирочного варианта РГА каче-
ственную оценку проводят в баллах от + до + + + + Если
эритроциты седиментировали в виде плотной полугранулы,
реакция считается отрицательной. Количественную оценку реак-
ции выражают в титрах. Обратная величина разведения сыво-
ротки, еще способного вызывать агглютинацию, принимается
за титр. На рис. 82 изображены результаты титрования в мик-
ропланшете.
215
SssSS«»»H®i«
GG@®®©©0©000
GGGGGG000090
©@©@©©000009
©@©©©©000000
©CGGOOOOOOOO
@@© G0OOQOQO0
@©©©©0000009
©©©©©0Q0QQ0O
Рис. 82. Микро-ГА и -ПГА по Takatsy.
Первый ряд: анти-А-сыворотка+эритроциты группы А; второй ряд — аити-А-сыворотка +
4-эритроциты В; третий ряд: аити-В-сыворотка+эритроциты В; четвертый ряд: антн-В-сы-
вороткаЧ-эритроциты А; ряды 5—8-й: сыворотка, содержащая ревматоидный фактор »
ЭБ, сенсибилизированные амбоцептором (тест Ваалера — Розе).
2.16.2.6.2. Реакция пассивной гемагглютинации
Титруемую антисыворотку инактивируют. Адсорбируют ее ин-
дикаторными эритроцитами (1 часть эритроцитов + 4 части
сыворотки; 20 мин при 20 °C) для удаления присутствующих
в большинстве сывороток гетерофильных АТ, особенно АТ к
антигену Форссмана. Титрование см. выше.
2.16.3. ОЦЕНКА МЕТОДА
При помощи РТА определяют антиэритроцитарные антитела.
Особенно часто РТА используется при определении групп
крови. Пассивная гемагглютинация относится к числу наибо-
лее чувствительных полуколичественных методов определения
АТ к растворимым АГ. При помощи РПГА можно определять
до 3—6 нг азота антител. В смысле чувствительности РПГА
равна или даже превосходит РСК. РПГА более чувствительна
в отношении IgM, чем IgG. Из этого становится ясным, что из-
менение титра РПГА связано не только с количеством АТ, но
и с различным их составом [10]. Различия результатов титро-
вания низко- и высокоаффинных АТ могут быть уравнены пу-
216
тем большей антигенной нагрузки эритроцитов [5, 8]. С увели-
чением плотности антигенных детерминат улучшаются резуль-
таты титрования низкоаффинных антител. Склонность сильно
«нагруженных» антигенами эритроцитов к спонтанной агглю-
тинации может быть предотвращена увеличением концентрации
стабилизаторов в среде для суспендирования и разведения.
Из вышеизложенного становится ясно, что самый важный па-
раметр конъюгирования антигенов с эритроцитами — это плот-
ность антигенных детерминант. Оптимальное количество АГ
для «нагрузки» эритроцитов — то, которое дает максимальный
титр РИГА. С эритроцитами связывается менее 2% вносимого
АГ. Вышеупомянутое различие в чувствительности РИГА при
определении IgM и IgG не может быть решено с помощью
пробы Кумбса (антиглобулиновый тест), поскольку необходи-
мые для этой пробы АТ неспецифически адсорбируются на сен-
сибилизированных эритроцитах. Это также означает, что в
РПГА невозможно дифференцировать различные изотипы ан-
тител при помощи антиглобулиновых сывороток. Предложен-
ный для этой цели Кумбсом «red cell linked-antigen antiglo-
bulin test» не получил признания в клинике [15] ввиду его
сложности и меньшей точности, чем РИА и ИФА. При выборе
метода конъюгации белка следует тщательно взвесить преиму-
щества и недостатки каждого метода конъюгации. Например,
с таннизированными эритроцитами в равной мере связываются
все белки, а с эритроцитами, обработанными БДБ,— преиму-
щественно богатые тирозином и гистидином. Глутаровый аль-
дегид присоединяет в основном белки с большим содержанием
свободных аминогрупп. В отличие от связанных ковалентно
адсорбированные АГ сравнительно легко отделяются от мем-
браны. Применение фиксированных эритроцитов делает РПГА
более воспроизводимым методом, к тому же отпадает необхо-
димость получать каждый раз свежие эритроциты. Фиксирова-
ние эритроцитов глутаровым альдегидом предпочтительнее, чем
обработка формалином, так как при первом способе эритро-
циты менее склонны к агрегации при хранении. Кроме того,
появляется возможность одновременно связывать несколько
белковых антигенов. Условия для оптимальной сенсибилизации
0,01—5 мг/мл
0,04—25 мг/мл
2 мг/мл
2 мкг/мл
20 мкг/мл по азоту
РПГА необходимы ежедневные позитив-
эритроцитов различными антигенами, включая концентрацию
таннина, температуру, pH, подбираются опытным путем. По-
дробные данные имеются в соответствующем обзоре [4].
Рекомендуются следующие оптимальные концентрации ан-
тигенов для сенсибилизации эритроцитов [4].
Овальбумин
БСА
Тиреоглобулин
Фибриноген
Тканевые антигены
Для достоверности
ные и негативные контроли, которые исключают бактериальное
217
загрязнение суспензии эритроцитов и антисывороток. Микро-
титрование требует меньшего количества времени и материа-
лов, но оно сопряжено с большей вероятностью ошибок, осо-
бенно при использовании больших разведений антител [2].
РПГА может быть автоматизирована [7, 12]. Ввиду высокой
чувствительности и простоты РПГА стала одним из распро-
страненных методов .полуколичественного определения малых
концентраций непреципитирующих антител. В клинике РПГА
используют в основном для определения аутоантител. В случае
использования больших количеств АГ и специфических АТ
можно проводить количественное определение антигенов (пе-
рекрестные реакции) в реакции торможения пассивной гемаг-
глютинации (РТПГА). Последняя отличается довольно высо-
кой чувствительностью и позволяет определять несколько нано-
граммов белка [1, И), все же РИА чувствительнее и намного
точнее.
2.16.4. СПИСОК ЛИТЕРАТУРЫ
1. Evans J., Steel М„ Arthur Е. A hemagglutination inhibition technique for
detection of immunoglobulins in supernatants of human lymphoid cell lines. —
Cell., 1974, 3, 153—158.
2. Feizl T., Menger E. Microtitration of serum cold agglutinins. — Transfusion,
1970, 10, 33—35.
3. Goding J. IF. The chromic chloride method of coupling antigens to erythro-
cytes: Definition of some important parameters. — J. Immunol. Meth., 1976,
10, 61—66.
4. Herbert IF. I. Passive hemagglutination with special reference to tanned
cell technique. — In: Handbook of experimental immunology/Ed. D. W. Weir,
2-nd ed. Vol. 1, chap. 20, Oxford: Blackwell scientific publications, 1973.
5. Hoyer L. W., Trabold N. C. The significance of erythrocyte antigen site den-
sity.— J. Clin. Invest., 1970, 49, 87—95.
6. Izbicky A. Die Untersuchungen fiber die passive Hamagglutination mit tan-
ninbeladene Erytrhozyten. — Zschr. Immunforsch., 1970, 140, 282—297.
7. Kohler W. H„ Steininger H., Otto R. II. Beschreibung eines voll automati-
schen Pipetier- und Uberpipettiergerates zur quantitativen Antikorperbestim-
mung im Serum. — Zschr. med. L^jortechn., 1974, 15, 285—288.
8. Leikila J., Pasanen V. J. Influence of antigen receptor density on agglutina-
tion of red blood cells. — Int. Arch. Allergy Appl. Immunol., 1970, 39, 352—
360.
9. Mackler M. T., Pesce A. J. Scanning electron microscopy (SEM) shows that
both antigen mobility and membrane deformation occur in the hemagglutina-
tion reaction.—Vox. Sang., 1981, 40, 1—5.
10. Malberg. K- Methoden zum Nachweis und zur Bestimmung humoraler Anti-
korper in der experimentellen Immunologie. — Schriftenreihe der arztlichen
Fortbildung. VEB Verlag Volk und Gesundheit, Berlin, 1970, Band 40,
69—86.
11. Nusbacher J., Berkman E. M„ Wong К. У. et al. Assay of IgG and other
human plasma proteins by quantitotive inhibition of passive hemagglutina-
tion.— J. Immunol., 1972, 108, 893—902.
12. Rees B. IF. G. An automated method for the coated red cell hem/aggluti-
nation technique. — Med. Lab. Techn., 1973, 30, 167—177.
13. Richter A. W„ Kagedal L. Couling of polysaccharides to erythrocytes by
cyanogen bromide. — Scand. J. Immunol., 1974, 3, 365—368.
14. Sachse H., Rackow S. Passive Hamagglutionation zum Nachwes von Anti-
218
korpern gegen bakterielle Polysaccharid-Antigene im Serum. — Vorschlag und
Kommentar. Zbl. Pharm., 1980, 119, 827—834.
15. Wheeler A. W. A method for measuring different classes of human immuno-
globulins specific for the penicilloyl group. — Immunology, 1971, 21, 547—
556.
2.17. АГГЛЮТИНАЦИЯ ЛАТЕКСА
2.17.1. ПРИНЦИП МЕТОДА
Частицы полистерольного латекса нагружают у-глобулиновой
фракцией, содержащей АТ к исследуемому АГ. Частицы ла-
текса служат в качестве носителей и «мостиков» для реакции
АГ—АТ. Нагруженный антителами латекс и антигенсодержа-
щую сыворотку смешивают на прозрачных пластинах и пере-
мешивают покачиванием. В случае положительной реакции уже
через 5—10 мин образуются видимые невооруженным глазом
агглютинаты, которые затем увеличиваются в размере. Чаще
всего этот вариант анализа используется для определения рев-
матоидного фактора и HBs-АГ. Приводимое ниже описание
метода относится к выявлению HBs-антигена. Первые сообще-
ния об этом диагностикуме относятся к 1971 г. [3]. В 1973 г.
появился коммерческий диагностикум .[1], оказавшийся недо-
статочно специфичным. В 1973 г. в нашей лаборатории был
разработан метод, который применяется до настоящего време-
ни [8].
2.17.2. ПОСТАНОВКА ОПЫТА НА ПРИМЕРЕ
ВЫЯВЛЕНИЯ НВ5-АНТИГЕНА
2.17.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: полистироловый латекс (Випа, ГДР),
стеклянные пластины, пипетки, анти-НВэ-сыворотка (например,
анти-НВ8-у-глобулиновая фракция (Serum und Impfstoffwerk,
Дессау, ГДР), насыщенный раствор (NH^SO^, глициновый
буфер, диализная лента.
Состав буфера:
2 М глицин 150,14 г/л — 467 мл
1 М NaOH 40,0 г/л — 100 мл
Смешать буфер в соотношении 1 + 1 с 0,15 М NaCl.
2.17.2.2. ПОЛУЧЕНИЕ
АНТИ-НВ5-у-ГЛОБУЛИНОВОЙ фракции
К 10 мл антисыворотки (независимо от титра) добавляют по
каплям при постоянном помешивании 6 мл насыщенного раст-
вора (NH4)2OH. Осадок собирают центрифугированием и ра-
створяют в 5 мл воды. Раствор диализируют до полного уда-
219
ления сульфата аммония, доводят концентрацию белка в пре-
парате до 20 г/л. Дальнейшая очистка, например хроматогра-
фическая, возможна, но нами не применялась.
2.17.2.3. ПОЛУЧЕНИЕ HBs-АГ-ЛАТЕКСА
К 1 мл 2% анти-НВэ-у-глобулиновой фракции приливают
10 мл 1% суспензии латекса в глицин-NaCl буфере с pH 8,8;
инкубируют 12 ч при 20 °C, а затем еще 12 ч при 4 °C. Обра-
зующийся осадок тщательно суспендируют. Суспензию разво-
дят глицин-NaCl буфером и при помощи латекс-позитивной
контрольной сыворотки устанавливают оптимальные условия
агглютинации. Как правило, наилучшее разведение суспензии
составляет 1+4.
2.17.2.4. ПОСТАНОВКА РЕАКЦИИ
1 каплю латексного реагента смешивают с 1 каплей исследуе-
мой сыворотки на стеклянной пластине и покачивают ее в те-
чение 2 мин. Оценку результатов проводят через 10—15 мин в
косом, падающим снизу свете на темном фоне. Более поздняя
агглютинация рассматривается как неспецифическая. Сыворот-
ка должна быть свежей без примеси клеток. Сыворотки с вы-
соким содержанием липидов и желчных пигментов, а также
ВИЭФ-позитивные сыворотки необходимо перед реакцией раз-
водить.
2.17.3. ОЦЕНКА МЕТОДА
Латекс-тест имеет определенные преимущества при скринирую-
щих обследованиях:
— метод прост и не требует специальной аппаратуры;
— метод относится ко 2-му поколению диагностикумов (чув-
ствительнее ДРИД и ВИЭФ);
— по сравнению с другими способами выявления HBs-АГ ме-
тод требует меньше времени;
Недостатки метода следующие:
— не всякая партия латекса годится для создания диагности-
кума;
— латекс-диагностикум на HBs-антиген выявляет на 20%
больше лиц с этим антигеном, чем ВИЭФ, но только у 30%
больных с положительной реакцией выявляются гистологи-
ческие изменения.
Ниже указаны факторы, обуславливающие большую долю
ложноположительных реакций [2, 6].
1. Ревматоидный фактор {4]: группа ревматоидных факторов
состоит из 195 глобулинов со специфичностью к 78-иммуно-
глобулину. Большая часть ревматоидных факторов реаги-
рует с у-глобулином человека, некоторые также с глобули-
220
нами кролика, лошади или коровы. По нашим данным,
из 89 HBs-позитивных лиц у 26 отмечалась положительная
реакция на ревматоидный фактор.
2. Продукты фибринолиза [7]: добавление тромбина или инак-
тивация при 56 °C в течение 30 мин [5] ускоряет свертыва-
ние фибрина и образование его агрегатов; таким образом,
фибрин больше не оказывает влияния на процесс латекс-
агглютинации. Устранение ложноположительных результа-
тов вышеописанным методом снижает чувствительность тес-
та, поскольку приблизительно 20% положительных реакций
перестают быть таковыми. Ложноположительные результа-
ты вследствие приема медикаментов неизвестны [4], но
нельзя исключить возможность того, что медикаментозное
повреждение печени способно приводить к положительным
результатам в латекс-тесте при отрицательных результатах
анализа на HBs-антиген в РИА. Сам по себе латекс-тест на
ревматоидный фактор дает весьма неспецифические резуль-
таты, что вынуждает использовать в диагностике и другие
параметры. Латекс-тест на гепатит вполне оправдан в эпи-
демиологических исследованиях и при проведении оценки
качества донорской крови. Достоверное выявление HBs-ан-
тигена латекс-тестом не представляется возможным.
2.17. 4. СПИСОК ЛИТЕРАТУРЫ
1. Bonacker L., Stark J. Latex-Agglutination, eine snelle Screeningmethode zum
Nachweis des HAA. — Klin. Wschr., 1972, 50, 166.
2. Desmyter J. Two Latex tests for Australia Antigen in Subjects with and
without Hepatitis. — Vox Sang. Suppl., 1973, 24, 88—94.
3. Leach 1. M„ Ruck B. J. Detection of HAA by the Latex Agglutination Test. —
Brit. Med. Journ., 1971, 4, 597—598.
4 Manthey K. F., Kreutz F. H. Latex-Test zum Nachweis des Hepatitis-assoziir-
te.n Antigens. — DTSCH. med Wschr., 1973, 98, 2070.
5. Miller J. E., Rossman D. P„ Ziegenfuss J. F. et al. Serum hepatitis with
«Masked Australia Antigen». — J.A.M.A., 1972, 221, 8.
6. Ohya T., Ohya F., Tanimura K. Clinical evaluation of antibody sensitized
latex particles method for Australia-antigen in serum. — Nagoya J. med. Sei.,.
1974, 37, 29—32.
7. Perkins H. A., Perkins S. L., Chen E. et al. A latex agglutination test for HAA
(HBsAg). — Hepatitis and blood transfusion. — New York: Grune and Strat-
ton, 1972.
8. Wilms K. Latex-Test zur Bestimmung des «Hepatitis Associated Antigen»
(HAA). —Folia Haematol., 1973, 99, 385—389.
9. Wilms K-, Bader G., Plotz Ch. et al. Zur Wertigkeit des Latex-Testes aid Nach-
weismethode des «Hepatitis В-Antigen» bei Blutspendern. — Dtsch. Ges-Sesen,.
1975, 30, 2141—2143.
2.18. РЕАКЦИЯ
АГГЛЮТИНАЦИИ ЛЕЙКОЦИТОВ
2.18.1. ПРИНЦИП МЕТОДА
Антилейкоцитарные антитела относятся к трем основным ка-
тегориям.
1. Агглютинины (выявляются в реакции агглютинации лейко-
цитов).
2. Комплементсвязывающие антитела (выявляются в реакции
лимфоцитолиза, см. раздел 2.12).
3. Антитела, не связывающие комплемент и не агглютинирую-
щие клеток (выявляются методом иммунофлюоресценции)
[2, 8].
Агглютинирующие антитела вызывают in vitro прямую аг-
глютинацию лейкоцитов, обладающих соответствующим анти-
геном. После окрашивания агглютинаты выявляются под мик-
роскопом. Мы приводим в качестве примера метод микроаг-
глютинации [4] с использованием крови, обработанной ЭДТА.
2.18.2. МЕТОДИКА
2.18.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: 5% ЭДТА, декстран 60 г/л (6,0%
Infucoll), 0,1% толуидиновый синий в 0,15 М NaCl; 6% уксус-
ная кислота, вазелиновое масло, микрошприцы и мпкродоза-
торы, микрокамера (см. раздел 2.12).
2.18.2.2. ВЫДЕЛЕНИЕ ЛЕЙКОЦИТОВ ИЗ КРОВИ,
ОБРАБОТАННОЙ ЭДТА
Смешивают в центрифужной пробирке 9 мл крови и 1 мл 5%
ЭДТА, дают постоять 30 мин, центрифугируют 12 мин при
80 g. Обогащенную тромбоцитами надосадочную фракцию от-
бирают и центрифугируют 15 мин при 500 g. Осадок тромбо-
цитов отбрасывают. Бестромбоцитную плазму смешивают с
осадком эритроцитов. Добавляют 0,5—1 мл 6% инфуколла,
разделяют смесь на порции по 2 мл и ставят на 30 мин в тер-
мостат (37°C). Эритроциты осаждаются декстраном. Обога-
щенную гранулоцитами надосадочную фракцию осторожно со-
бирают и центрифугируют 5 мин при 30 g. Готовят суспензию
с концентрацией 3—5Х109 клеток/л.
2.18.2.3. РЕАКЦИЯ АГГЛЮТИНАЦИИ
ЛЕЙКОЦИТОВ (РАЛ), МИКРОМЕТОД
В микрокамеру вносят при помощи микрошприца 2 мкл инак-
тивированной при 56 °C сыворотки и 1 мкл суспензии лейко-
цитов. Толщина слоя вазелинового масла в камере составляет
222
около 2 мм. Если используется микрокамера Терасаки, в нес
вносят 3 мл вазелинового масла. Инкубируют 60 мин при
37 °C. Затем осторожно встряхивают камеру в течение 3 мин.
Окрашивают смесь добавлением 1 мкл 0,1% толуидинового си-
него в 0,15 М NaCl. Через 5 мин лейкоциты становятся ясно
различимыми. Проводят микроскопию с 60—100-кратным уве-
личением. Если имеется существенная контаминация эритроци-
тами, их гемолизируют добавлением 1 мкл 6% уксусной кис-
лоты.
2.18.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Результаты реакции оценивают в баллах. О наличии агглюти-
нинов говорят, когда наблюдаются группы из 4—5 клеток
(рис. 7, 8 на цветной вклейке).
Разрозненные клетки в поле зрения--
Отдельные мелкие агглютинаты (до 25% клеток)-----Ь
Крупные аггрегаты (до 50% клеток), преобладают разрознен-
ные клетки---Н +
Крупные аггрегаты (до 75% клеток) наряду с разрозненными
клетками---К + +
Агрегированы все клетки (75—100% клеток)--Ь + + +
В каждом опыте ставят отрицательный контроль с сыворот-
кой АВ и положительный контроль с полиспецифической аг-
глютинирующей антилейкоцитарной сывороткой. Особенно ве-
лика опасность ложноположительных результатов вследствие
неспецифической агглютинации.
2.18.4. ИСТОЧНИКИ ВОЗМОЖНЫХ ОШИБОК
И СПОСОБЫ ИХ УСТРАНЕНИЯ
Необходимо обратить внимание на следующие моменты:
— совместимость по AB0. Поиск агглютинирующих лейкоциты
антител рекомендуется проводить с лейкоцитами группы 0;.
— гранулоциты склонны к спонтанной агломерации. Концент-
рация лейкоцитов в суспензии не должна превышать 10X
X109 клеток/л. Если требуется сконцентрировать суспен-
зию центрифугированием, используют ускорение не выше-
200 g;
— в отдельных случаях агглютинация может быть обусловле-
на бактериальным загрязнением.
2.18.5. ОЦЕНКА МЕТОДА
По чувствительности реакция агглютинации превосходит цито-
токсический тест и связывание комплемента тромбоцитами. Вы-
сокой чувствительности противостоит низкая воспроизводи-
мость результатов (около 85%). Поэтому реакция агглютина-
223
ции больше не находит применения при оценке тканей на со-
вместимость (выявления HLA-антигенов). Для обнаружения
цитотоксических антилейкоцитарных антител используется ме-
тод иммунофлюоресценции [8], доступный, правда, для ограни-
ченного числа лабораторий. РАЛ все же должна входить в
число методов, используемых в серодиагностике лабораториями
без иммунофлюоресцентного оснащения. РАЛ применяют в
следующих случаях:
— выявление причин лихорадочных посттрансфузионных реак-
ций [3[. Агглютинирующие анти-НЬА-антитела могут встре-
чаться независимо от цитотоксических анти-НЬА-антител
[1, 9] и вызывать посттрансфузионные реакции (агглюти-
нирующие, антилейкоцитарные антитела у реципиента);
— определение специфических антигранулоцитарных антител,
которые могут быть причиной лихорадочных посттрансфу-
зионных реакций или так называемого эффекта замещения
после переливания гранулоцитов [5, 9];
— выявление антигранулоцитарных аутоантител, обладающих
в большинстве случаев групповой специфичностью [6, 8].
Переливание гранулоцитарной массы под контролем спе-
цифичности антител [2];
— выявление HLA-независимых гранулоцитспецифических ан-
тигенов [5]; в настоящее время вытесняется иммунофлюо-
ресцентным методом [8].
РАЛ с дефибринированной кровью отличается худшей вос-
производимостью, чем при использовании крови, обработанной
ЭДТА, поскольку в первом случае чаще наблюдаются ложно-
положительные результаты. РАЛ в капиллярах гораздо менее
подвержена ошибкам, учитывая высокую чувствительность ме-
тода, его рекомендуют для выявления трудно обнаруживаемых
HLA-антигенов [7]. РАЛ пригодна как метод скрининга аг-
глютинирующих антител. В агглютинационной пробирке сме-
шивают 2 капли суспензии лейкоцитов и 2 капли сыворотки
(около 0,05 мл). Этот вариант РАЛ не требует специального
оснащения. После 90 мин инкубации при 37 °C надосадочную
фракцию отбрасывают. В каждую пробирку вносят 1 каплю
6% уксусной кислоты и слегка покачивают пробирку (гемолиз
остаточных эритроцитов). Капельной пипеткой наносят одну
каплю смеси на обезжиренное этанолом предметное стекло и
делают мазок. Микроскопируют при 60—100-кратном увеличе-
нии.
2.18.6. СПИСОК ЛИТЕРАТУРЫ
1. Bertrams J., Kuwert Е., Linzenmeier G. Leukozyten-Isoantikorper. I. Vorkom-
men agglutinierender und zytotoxischer Antikorper gegen weise Blutzellen im
serum erst- und mehrgebaren der Frauen. — Dtsch. med. Wschr., 1970, 95,
1145—1152.
“2. Engelfriet С. P„ Pegels J. G., von dem Borne A. E. G. K. Immunological
aspects of transfusion therapy. — Bibliotheca haemat., 1980, 46, 120—131.
224
3. Ihle R., Leverenz S., Stobbe H. Nichthamolytische febrile Transfusionreaktio-
nen. — Dtsch, Ges-wesen, 1977, 32, 1231—1234.
4. Kissmeyer-Nielsen J., van. Rood J. J. Micro-agglutination Test. — Manual of
tissue typing techniques. National Institute of Health, Bethesda, 1972, 1—2.
5. Lalezari P„ Thalenfeld B., Weinstein. W. J. The third neutrophil antigen. —
Histocompatibility testing, Munksgaard, Copenhagen 1970, 319—322.
6. Lalezari P„ An-Fu-Yang, Yegen L. et al. Chronic autoimmune neutropenia due
to anti-NA2 antibody. — New Engl. J. Med., 1975, 293, 744—747.
7. Thompson S. J., Severson C. D„ Coppleson L. W. et al. Leucocyte capillary
agglutination: Demonstration of additional leucocyte antibodies in cytotoxi-
cally «monospecific» antisera. — Histocompatibility testing, Munksgaard, Co-
penhagen, 1970, 587—594.
8. Verheugt F. W. A., von dem Borne A. E. G. K„ Decary F. et al. De tection of
granulocytes alloantibodies by an indirect immunofluorescence test. — Brit. J.
Haemat., 1977, 36, 533—544.
9. Wegener R. Vergleichende Untersuchungen zum Vorkommen von Leukozyte-
nantigen auf Granulozyten und Lymphozyten. — Inauguraldissertation, Ros-
tock, 1973.
15—990
3. КЛЕТКИ ИММУННОЙ СИСТЕМЫ
3.1. РАЗДЕЛЕНИЕ
КЛЕТОК ИММУННОЙ СИСТЕМЫ
3.1.1. ВВЕДЕНИЕ
Разделение клеток иммунной системы представляет собой не-
обходимую предпосылку для исследований функциональной
роли отдельных типов клеток, их кооперативного взаимодей-
ствия, дифференцировки и т. д. В конце 60-х годов было об-
наружено, что малые лимфоциты являются гетерогенной попу-
ляцией, состоящей по меньшей мере из двух классов. С этого
момента началось бурное развитие методов разделения иммуно-
компетентных клеток. Результатом этих исследований стало
обнаружение новых субпопуляций лимфоцитов, весьма различ-
ных в функциональном отношении. Исследования в этой об-
ласти продолжаются. Следует подчеркнуть, что далеко не
всегда в специальной литературе правильно используется тер-
мин «популяпия». Довольно часто клетки относят к разным
субпопуляциям только на основании определенных физических
параметров фракционирования, не обременяя себя поиском до-
казательств того, что эти субпопуляции принадлежат к одной
и той же разновидности клеток, находящихся на разных ста-
диях дифференцировки или активации. К числу новых мето-
дов, появившихся в последнее время, следует в первую очередь
отнести те, которые основаны на использовании лектинов. Мы
ограничимся описанием важнейших, относительно простых,
в определенной мере рутинных методов работы и не будем
касаться таких сложных процессов, как флюоресцентно-актива-
ционное разделение клеток, которое можно освоить только, ра-
ботая под непосредственным наблюдением более опытного экс-
периментатора. На рис. 83 схематически изображены способы
разделения клеток, основанные на физических и биохимических
принципах. Здесь речь также идет только о наиболее суще-
ственных комбинациях. Один и тот же признак может быть
выражен в разной степени на клетках одного типа, кроме того,
клетки довольно сильно меняются в процессе дифференцировки
и выделяемая популяция может оказаться загрязненной.
Понятие «рецептор» означает «место связывания», незави-
симо от типа биологического процесса, в котором он участвует.
До настоящего времени остаются неизученными рецепторы эри-
троцитов. Известно, что лектины, например, реагируют с угле-
226
Рис. 83. Схема физических и биохимических свойств клеток иммунной сис-
темы.
Fc—Fc-рецептор; Гис — гистамин; АГ — антиген; Mlg — мембранный иммуноглобулин;
С’ — рецептор комплемента; Эр — эритроцит; ФАСК — флюоресцентно-активационная
сортировка клеток.
водными компонентами клеточной мембраны, вероятно, эритро-
циты через углеводные компоненты собственной мембраны
вступают во взаимодействие с соответствующими мембранами
лимфоцитов. Ввиду того что исследования в области клеточной
иммунологии проводятся повсеместно, получаемые иногда про-
тиворечивые результаты довольно скоро получают объяснения.
Зачастую речь идет о кажущихся противоречиях, которые воз-
никают вследствие двух основных причин.
1. Различия между процессами in vivo и in vitro. Все боль-
ше появляется доказательств того, что культура клеток дает
очень ограниченное представление о сложных процессах, про-
исходящих в целом организме.
2. Видовое и органное происхождение клеток. Несмотря на
общие принципы организации иммунной системы у млекопи-
тающих, даже у близкородственных видов могут наблюдаться
поразительные отличия в тонкой организации функций, кото-
15*
227
рые иногда становятся источником ошибочных интерпретаций.
В качестве примера можно назвать совершенно различное рас-
пределение 6-антигенов у мыши и крысы или различия в строе-
нии Fc-рецепторов Т- и В-лимфоцитов мыши и человека. В ка-
честве причин ошибок при разделении клеточных элементов
часто выступают подлинные артефакты. Причины эти следую-
щие:
а) недостаточные знания об адгезионных свойствах и селек-
тивной чувствительности отдельных субтипов клеток;
б) незначительный выход определенного типа клеток, кото-
рый может быть обусловлен непреднамеренным избирательным
удалением важного в функциональном отношении субтипа, что
приводит в свою очередь к тому, что состав предположительно
чистой клеточной субпопуляции не отражает ее реального со-
става в организме. Например, чистая популяция Т-лимфоцитов,
из которой адгезией удалены Т-супрессоры, будет давать бо-
лее выраженный иммунный ответ, чем существующий in vivo
пул Т-клеток;
в) стимуляция или торможение тех или иных клеточных
функций может иметь место вследствие разделения клеток, что
приводит соответственно к усилению или ослаблению иммун-
ного ответа. Имеются многочисленные примеры артефактов,
связанные с розеткообразованием, очисткой на аффинных сор-
бентах, флюоресцентно-активационным разделением, агглюти-
нацией лектинами и т. д.
Более подробно возможные причины артефактов будут рас-
смотрены в конце описания каждого метода.
3.1.2. РАЗДЕЛЕНИЕ КЛЕТОК ПРИ ПОМОЩИ АДГЕЗИИ
З.1.2.1. ПРИНЦИП МЕТОДА
Фагоцитирующие клетки, в том числе и часть лимфоцитов,
имеют тенденцию к адгезии на твердых поверхностях. Это от-
носится в первую очередь к В-лимфоцитам, особенно к АОК.
Супрессоры, которые в основном относятся к Т-клеткам, встре-
чаются также среди макрофагов и В-лимфоцитов. Механизм
адгезии еще полностью неясен, известно только, что адгезия
тем легче, чем меньше отрицательный потенциал клетки. Раз-
личают две формы адгезии. У фагоцитов это активный про-
цесс, зависящий от интенсивности метаболизма и от темпера-
туры. Эти клетки могут быть отделены от твердой фазы при
помощи сред, не содержащих Са2+ и Mg2+ или содержащих
ЭДТА. Адгезия лимфоцитов не зависит от температуры и на-
личия ЭДТА, но подвержена влиянию концентрации белка в
среде. Для разделения клеток в настоящее время используют
стеклянные и плексигласовые гранулы, сефадекс и найлон.
228
3.1.2.2. РАЗДЕЛЕНИЕ КЛЕТОК
НА СТЕКЛЯННЫХ ГРАНУЛАХ
3.1.2.2.1. Методика
Материалы и оборудование. Для работы необходимы: стеклян-
ная колонка с рубашкой, термостат с циркуляционным насо-
сом, стеклянные бусы диаметром 0,3—0,5 мм (тип R-50,
Trisola, Штейнах), H2SO4 концентрированная, стальное сито
без следов ржавчины, фильтровальная ткань, телячья сыворот-
ка, среда МЕМ с pH 7,2, среда Рабиновича (0,02% ЭДТА в
свободной от Са2+ и Mg2+ ФСБ).
Получение суспензии клеток. Селезенку и лимфатические
узлы (обычно мышиные) погружают в среду МЕМ, содержа-
щую 10% сыворотки телят, разрезают на маленькие кусочки
и осторожно продавливают шпателем через мелкую стальную
сетку. Клетки дважды отмывают в среде МЕМ, ресуспензируя
их и центрифугируя в течение 5 мин при 500 g. Агрегаты уда-
ляют фильтрованием через тонкую ткань. Перитонеальные
клетки получают путем смыва с брюшины средой при помощи
шприца. Выделение лейкоцитов крови проводят по методу
Бёйюма. В зависимости от вида животных градиент плотности
может быть различным.
Подготовка колонки. Размер колонки в зависимости от
количества и видовой принадлежности клеток может изме-
няться от 1,5 до 4,0 см в диаметре и от 10 до 80 см по высоте.
Стеклянные очищают концентрированной H2SO4 и отмывают
дистиллированной водой до исчезновения следов кислоты. Ко-
лонка должна быть оборудована снизу краном, а сверху —
воронкой (рис. 84). Колонку затыкают снизу марлей и запол-
няют средой МЕМ до половины воронки. Затем небольшими
порциями вносят гранулы до нужной высоты. Колонку промы-
вают 5 объемами МЕМ с 10% телячьей сыворотки и охлаж-
дают до 4 °C. Если разделение должно проводиться при 37 =С,
то колонку, среду, гранулы и прочее оборудование перед за-
полнением нагревают при 37 °C для предотвращения образо-
вания пузырей.
Разделение фагоцитирующих клеток и лимфоцитов. Перед
нанесением клеток уровень жидкости в колонке опускают до
уровня носителя. Затем подогретые до 37 °C клетки наносят
на колонку со скоростью 2—3 мл/мин. Для разделения 2хЮ6
клеток в 10 мл среды достаточно колонки размером 2x20 см.
Колонку закрывают и инкубируют. Сливают через 15 мин 1 мл
среды и инкубируют колонку еще в течение 30 мин. Все неад-
гезировавшие клетки удаляют пропусканием через колонку
300 мг МЕМ со скоростью 2—3 мл/ч.
Элюцию адгезированных клеток проводят 200 мл буфера
Рабиновича, причем скорость элюции составляет 2—3 мл/мин
для первых 100 мл, повышаясь далее до 5 мл/мин. Не рекомен-
229
Рис. 84. Схема устройства для фракционирования клеток на стеклянных гра-
нулах.
1 — охлаждающая рубашка; 2 — термостат; 3 —марля; 4 —среда; 5 — кран закрыт; 6 —
кран открыт.
дуется проводить элюцию путем перемешивания гранул в ко-
лонке, поскольку при этом могут высвобождаться лимфоциты,
не элюируемые ЭДТА.
Разделение Т- и В-лимфоцитов. Суспензию лимфоцитов
(2Х108) наносят со скоростью 2—3 мл/мин на колонку раз-
мером 1,5x80 см и выдерживают при 4 °C. Преинкубация стек-
лянных гранул с 10% телячьей сывороткой или добавление
5% сыворотки к среде приводит к снижению адгезивных
свойств Т-лимфоцитов. После того как жидкость, содержащая
лимфоциты, нанесена на колонку (150 мл), носитель выби-
вают.. Для этого нижний кран осторожно вынимают (верхний
закрыт), заливают в воронку среду и, осторожно открывая
верхний кран, собирают гранулы в химический стакан (рис.
84,6 и в). Адгезированные В-лимфоциты отделяют от стеклян-
ных гранул осторожным встряхиванием в течение 30—60 сек
в 150 мл среды МЕМ. Содержащую клетки среду собирают.
Клетки обеих фракций лимфоцитов собирают центрифугиро-
ванием и отмывают 1 раз (500, 5 мин).
Удаление или накопление АОК. Для специфической элими-
нации АОК используют гранулы, нагруженные антигеном (ин-
кубация с АГ в течение 60 мин при 45 °C, концентрация АГ
5 мг/мл); на колонку расходуется около 20 мл раствора АГ.
После добавления телячьей сыворотки (до 10%) гранулы вы-
держивают еще 60 мин при 37 °C или в течение ночи в холо-
230
дильнике. Перед заполнением колонки гранулы отмывают от
АГ (расходуется около 500 мл среды). Нанесение на колонку
и разделение клеток описано выше.
3.1.2.2.2. Оценка результатов
При использовании всех трех вышеописанных методов нужно
учитывать видовое и органное происхождение клеток, отсюда —
возможные модификации. Что касается лейкоцитов крови че-
ловека (Т-лимфоциты или фагоцитирующие клетки), можно
добиться 90% обогащения, в то время как при работе с селе-
зенкой мыши такого результата можно достичь только в отно-
шении лимфоцитов. При разделении Т- и В-лимфоцитов на
адгезионных колонках (стеклянные гранулы, дегалан) обога-
щение Т-лимфоцитов колеблется от 70 до 90% (выход Т-лим-
фоцитов во фракции клеток, не собиравшихся на колонку, со-
ставляет около 50%). Обогащение В-лимфоцитов колеблется
в пределах 70—80% (адгезивная фракция), причем выход со-
ставляет 30—40%. В вышеприведенном примере использовали
спленоциты мыши. Хотя нагруженные АГ колонки и удержи-
вают специфические АОК, этот эффект усилен весьма незна-
чительно по сравнению с неспецифической адгезией АОК на
различных материалах, например на сефадексе. Элиминация
АОК из неадгезированной фракции в зависимости от иммуно-
гена и периода иммунизации может составлять 60—100%. Мак-
симально удается достичь 2—3-кратного концентрирования
АОК, поскольку их доля в адгезивной фракции составляет в
среднем 30—40%.
З.1.2.З. ФРАКЦИОНИРОВАНИЕ КЛЕТОК
НА НАЙЛОНОВЫХ ВОЛОКНАХ
З.1.2.З.1. Методика
Материалы и методы. Для работы необходимы: найлоновая
вата (LP-1 Leuko Рак, Leukozytenfilter, FENWAL Labs. США,
или Semidull Nylon Staple 3 den. 11/2 in 200 E. I. Dupont,
США), шприцы на 10 или 20 мл среды: МЕМ или Dulbecco
ФСБ с добавлением 5% телячьей сыворотки; ФСБ).
Этапы работы. Токсические вещества удаляют из найлоно-
вой ваты отмыванием дистиллированной водой и ФСБ в тер-
мостате. Вату (0,7 или 1,4 г) набивают в шприц до отметки
6 мл или 12 мл соответственно. При необходимости возможно
автоклавирование. Колонку (шприц) промывают 20—30 мл
среды и инкубируют в течение 1 мин при 37 °C. Перед нанесе-
нием клеток через шприц еще раз пропускают 20 мл нагретого
буфера, причем уровень жидкости должен дойти до края ваты.
Клетки, подогретые до 37 °C, наносят на колонку со ско-
ростью 1,0 мл/мин; на колонку объемом 6 мл наносят 100—
150ХЮ6 клеток в 2,0 мл, на колонку объемом 12 мл — 200—
231
300Х106 клеток в объеме 4,0 мл, после чего добавляют еще
0,5—1,0 мл среды. Колонку в вертикальном положении инку-
бируют в течение 45 мин при 37 °C. Элюцию осуществляют
пропусканием 30 или 60 мл разделяющей среды со скоростью
2—3 мл/мин. Затем для удаления слабо связанных клеток
промывают колонку 50—60 мл среды со скоростью 5—6 мл/мин,
эти немногочисленные клетки отбрасывают. Отделение адгези-
рованных клеток осуществляют 5—6-кратным сдавлением ваты
поршнем шприца. В промежутках между сжатиями поршня
найлоновую вату расправляют в новой порции среды. Жид-
кость объединяют, клетки собирают центрифугированием.
З.1.2.З.2. Оценка метода
Фракционирование на- найлоновой вате представляет собой
простой и быстрый метод разделения Т- и В-лимфоцитов. Это
наиболее часто применяемый метод. Данные о степени чистоты
получаемых клеточных популяций варьируют у различных
авторов. На адгезирующие спленоциты мыши после 1-кратного
пропускания через колонку приходится еще 10—20% В-лимфо-
цитов. На колонке преимущественно задерживаются АОК и
фагоцитирующие клетки, однако в виде примеси присутствует
10—20% Т-лимфоцитов. В обеих фракциях выход составляет
не более 25—50% от исходного числа клеток. Элиминация
Ig-положительных клеток из лимфоцитов крови человека со-
ставляет также около 50%. Различия в эффективности разде-
ления могут быть обусловлены также свойствами разделяю-
щих материалов.
Артефакты. В зависимости от состава клеточной суспензии,
который редко бывает постоянным, к носителю могут прикреп-
ляться клетки с различной активностью. Это замечание отно-
сится не только к фагоцитам, но теоретически также к стиму-
лированным лимфоцитам и супрессорным клеткам. В случае
небольшого выхода клеток отмечается также «эффект сита».
Этот эффект может наблюдаться при использовании стекла,
дегалана, найлоновой ваты, материалов для аффинной хрома-
тографии, обработанных лигандами для клеточных рецепторов,
а затем сывороткой. Эффект неспецифического связывания
наблюдается почти всегда. Кроме того, описаны 4 специфиче-
ских источника артефактов для найлона.
1. Адгезия Ти-клеток [1], которая является одной из причин
(помимо утраты К-клеточной активности после обработки
NH4C1, см. раздел 3.1.5) того, что в первые годы после откры-
тия Т-лимфоцитов их вычеркнули из числа кандидатов в К-
клетки.
2. У Т-лимфоцитов мыши из селезенки, лимфатических узлов
или тимуса после контакта с найлоновой ватой может наблю-
даться изменение «Ношш§»-эффекта.
3. Т-лимфоциты человека, разделенные на найлоновой вате,
перестают действовать в смешанной культуре лимфоцитов
232
(СКЛ) как стимуляторы [3]. Причем до настоящего времени
неясно, удаляются при этом клетки-стимуляторы или вспомо-
гательные клетки.
4. После контакта с найлоновой ватой Т-лимфоциты мыши
реагируют на АГ сильнее, чем до сепарации [4]. Объяснением
этого факта скорее может служить адгезия предшественников
супрессорных клеток, чем активация антигеноспецифических
лимфоцитов после сепарации.
Следует указать, что материалы, обладающие выраженным
адгезивным эффектом, нельзя применять для разделения кле-
ток.
3.1.3. РАЗДЕЛЕНИЕ КЛЕТОК
НА АФФИННЫХ КОЛОНКАХ
З.1.З.1. ПРИНЦИП МЕТОДА
Аффинная сепарация основана на избирательной сорбции кле-
ток различных субпопуляций носителями, содержащими веще-
ства, к которым клетки данной субпопуляции имеют высокое
сродство. Связывание может быть обратимым или необрати-
мым. Специфически реагирующими компонентами клеточной
мембраны могут служить мембранные Ig, а также рецепторы
комплемента (СЗ), Fc-фрагмент антител, антигены, лектины,
гистамин. В качестве носителя для АГ, антииммуноглобулина
или комплексов АГ—АТ первоначально служит стекло или
дегалан. Поскольку неспецифическое связывание у стекла или
дегалана довольно сильно выражено, разделение клеток лучше
проводить на материалах со слабым неспецифическим связы-
ванием. К этим материалам относятся гранулы из сыворотки
телят, сефадекс, сефароза, биогель (полиакриламид) и сферон
(гидроксиалкилметакрилатный гель). В зависимости от усло-
вий опыта связывающая способность биогеля колебалась от
40% до 0. Привязывание к биогелю азофенил-р-лактозы или
ДНФ позволяло с высокой эффективностью выделять АОК к
этим гаптенам. Присоединение к носителям белков, таких как
антииммуноглобулин, представляет известные трудности и при
этом не делает процесс разделения клеток достаточно эффек-
тивным.
Нагруженный АГ сферон удерживает до 60% АОК, однако
элюцию клеток невозможно проводить свободным АГ, как при
использовании биогеля. Ниже приведены еще несколько мето-
дов:
а) агглютинин Helix pomatia (НРА) связывает обработан-
ные нейраминидазой Т-лимфоциты человека. Через колонки с
НРА-сефарозой поэтому в первую очередь проходят В-лимфо-
циты. Т-лимфоциты элюируют М-ацетил-П-галактозамином;
б) нагруженным антигеном желатином покрывают центри-
фужные пробирки фирмы Falcon. Расплавление желатина, на-
233
ступающее при 37 °C, высвобождает АОК, ранее связанные с
желатиновым слоем (обогащение до 30 раз);
в) протеин A Staphylococcus aureus реагирует с Fc-участ-
ком IgG. Монослой эритроцитов, нагруженных протеином А,
связывает благодаря этому клетки, содержащие антитела.
При помощи гипотонического шока или лизостафина эти клет-
ки можно снова получить в свободном виде;
г) В-лимфоциты мыши связывают через Fc-рецепторы с мо-
нослоем эритроцитов, нагруженных антителами (например,
комплекс АГ—АТ). Свободные В-лимфоциты получают при
помощи гипотенического шока. Чистота разделенных данным
методом Т- и В-лимфоцитов достигает 90%.
З.1.З.2. МЕТОДИКА
3.1.3.2.1. Материалы и оборудование
Получение суспензии клеток см. в разделе 3.2.2. Для работы
необходимы: ФСБ, МЕМ с 5% сывороткой телят (pH 7,2), ко-
лонка с термостатируемой рубашкой, лиофилизированная те-
лячья сыворотка; 25% глютаровый альдегид, 1,0 М фосфатный
буфер Серенсена с pH 7,0, эфир, этанол 96%, сухой лед, 0,1 М
уксусная кислота, 0,3 М бикарбонатный буфер с pH 10,0, мы-
шиный глобулин, антисыворотка против мышиного глобулина,
сефадекс G-200, бромциан, борат-PPS с pH 8,0, антисыворот-
ка к IgG человека, Fab-фрагменты 1g человека, IgG человека,
NaOH, ЭДТА, сефароза 4В для разделения, нормальная сыво-
ротка человека, дегалановые и стеклянные гранулы 100—
500 мкм в диаметре.
З.1.З.2.2. Выделение Т-лимфоцитов путем элиминации
В-лимфоцитов на протеиновых гранулах,
нагруженных комплексами АГ—АТ
Для получения белковых гранул используют лиофилизирован-
ную телячью сыворотку (ТС). Порошок, полученный из 5,0 мл
ТС (15 г/100 мл), смешивают с 0,5 мл 1 М буфера Серенсена
с pH 7,0 и 0,18—0,20 мл 25% глютарового альдегида и тут же
при постоянном перемешивании выливают в охлажденный
эфир (смесь этанол — сухой лед; —60°C). Точное количество
глутарового альдегида должно быть таким, чтобы полимери-
зация белка завершалась в течение 45—60°C. Замороженные
белковые гранулы в плоском сосуде с крышкой в течение не-
скольких часов выдерживают на холоде. В ходе длительного
оттаивания процесс полимеризации белка завершается. По кон-
систенции все еще довольно мягкие гранулы переносят в эта-
нол, где остатки эфира экстрагируются, а гранулы приобретают
необходимую твердость.
На величину получаемых гранул можно воздействовать
снижением концентрации белка (до 10—12%), более быстрым
перемешиванием и более быстрым впрыскиванием материала.
234
Активация,
конъюгация
Перенос в ФСБ
Мешалка —
Рис. 85. Схема получения белковых гранул (носитель для колоночной хрома-
тографии).
Получаемые гранулы просеивают в виде суспензии в этаноле
через пластмассовые бусы (рис. 85). Гранулы более 1 мм в
диаметре отбрасывают. Для присоединения комплексов АГ—
АТ к гранулам их (свежеполученные или уже использованные)
отмывают ФСБ, затем активируют 25% раствором глутарового
альдегида в ФСБ, инкубируют еще 2 ч с глобулином мыши или
крысы (3—4 мг/мл) и 2 ч с соответствующим антиглобулином,
например с 3 мл разведенной 1 :5 АС иа 50 мг гранул. Ис-
пользуемая АС должна давать заметные линии преципитации
в пробе Ухтерлони в разведении 1:8—1:16. После инкубации
гранулы отмывают 5-кратным объемом ФСБ. Для разделения
лимфоцитов используют колонки объемом 1,5x30 см, объемом
50 мл (на 100Х106 клеток) или 2,5X30 см, объемом 150 мл
(на 300—400X106 клеток).
Гранулы, колонки и разделяющую среду (МЕМ с 5% те-
лячьей сыворотки) перед разделением прогревают до 37 °C.
Перед нанесением клеток колонку промывают двумя объема-
ми разделяющей среды и дают уровню среды установиться по
верхнему краю носителя. Со скоростью 3 мл/мин на колонку
наносят суспензию спленоцитов мыши — ЮОхЮ6 в 17 мл или
300—400Х106 в 50 мл; колонку инкубируют 30 мин при 37°С.
Удаление несвязавшихся клеток осуществляют пропусканием
через колонку 2—3 объемов разделяющей среды со скоростью
2—3 мл/мин. Методику элюции с колонки см. в разделе 3.1.2
и рис. 84. Белковые гранулы регенерируют 0,1 М уксусной
235
кислотой, ФСБ, 0,2 М карбонат-бикарбонатным буфером с
pH 10 (по 10 мин каждым раствором) и снова ФСБ. После
нагрузки гранул АГ их можно использовать 3 раза, причем
не рекомендуется одни й те же гранулы нагружать различ-
ными белками. В качестве альтернативы приходится прибегать
к очистке гранул трипсином. Гранулы остаются стабильными
в 50% этаноле при 4 °C в течение 8 ч.
3.1.3.2.3. Разделение Т- и В-лимфоцитов человека
на анти-ГаЬ-сефадексе
Сефадекс с размером частиц не менее 90 мкм в диаметре в
количестве 1 г кипятят в дистиллированной воде и доводят
pH до 10,3—10,5 20% NaOH. После прибавления 100 мг бром-
циана pH снова подводят до этой величины добавлением по
каплям NaOH и выдерживают смесь в течение 10 мин. Акти-
вированный сефадекс отмывают борат-ФСБ буфером с pH 8,3
и немедленно прибавляют 24—30 мг очищенных АТ против
Fab-фрагментов человека и перемешивают в течение 4 ч. После
повторной отмывки 8—10 мл нагруженного сефадекса в раз-
деляющей среде (МЕМ + 5% телячьей сыворотки + 2,5 мМ
ЭДТА) вносят в небольшую колонку или шприц, инкубируют
1 ч при 37°C, промывают 10 мл разделяющей среды и охлаж-
дают до 4 °C. Для разведения клеток, которое происходит при
4°C, на колонку емкостью 8 мл наносят 100—200Х106 лимфо-
цитов крови человека в 10 мл среды для разделения со ско-
ростью 0,5 мл/мин. После удаления всех клеток с колонки
связавшиеся клетки элюируют 30 мл разделяющей среды, со-
держащей 10 мг/мл у-глобулина человека.
3.1.3.2.4. Разделение Т- и В-лимфоцитов на сефарозе-СЗ
Отмытую сефарозу 4В 2 мл перемешивают с сывороткой че-
ловека, разведенной 1 :5 в течение 1 мин при 37°C. При этом
происходит присоединение к сефарозе активированных моле-
кул компонента комплемента СЗ [5]. Свежеотмытой активи-
рованной сефарозой заполняют маленькую колонку (0,7x5см),
нижний конец которой заполнен на высоту 2—3 мм слоем
стеклянных гранул. Разделение клеток ведут при 4 °C. Лим-
фоциты крови человека (20Х106), суспензированные в 2 мл
разделяющей среды, наносят на колонку со скоростью
0,5 мл/мин.
При элюции с первыми 15 мл элюата уходит большая часть
несвязавшихся клеток, всего же колонку промывают прибли-
зительно 30 мл среды МЕМ. Связавшиеся клетки элюируют
сывороткой, содержащей АТ к СЗ человека. На колонку нано-
сят 1 мл этой сыворотки (титр 1 :512 по агглютинации), ждут
15 мин и элюируют клетки 30—40 мл среды.
236
З.1.З.З. ОЦЕНКА ВАРИАНТОВ МЕТОДА
Для всех трех вышеописанных методов общим является то,
что степень чистоты выделенных Т- или В-лимфоцитов дости-
гает почти 100%, т. е. она выше или равна той, которую удает-
ся получить при помощи нагруженных стеклянных гранул или
дегалана [5]. Это существенное преимущество уравновешивает
недостаток, обусловленный сложной процедурой разделения.
Неспецифическое селективное связывание АОК с сефадексом,
белковыми гранулами или сефарозой не может быть полностью
исключено. Кроме этого, уменьшение потерь клеток, достигае-
мое ЭДТА, не селективно в отношении Т- или В-лимфоцитов.
Методы разделения на колонках обладают еще и тем преиму-
ществом, что их легче стандартизировать в отличие от мето-
дов разделения в чашках Петри, пробирках и пр., так как в
последнем случае более или менее сильное встряхивание мо-
жет стать причиной расхождения результатов. Кроме того,
следует отдавать предпочтение методам, которые не вызывают
стимуляции клеток. Вышеописанные методы в этом смысле
пригодны для получения Т-лимфоцитов, так как элюируемые
В-лимфоциты нельзя рассматривать как нестимулированные.
Возможность разделения Т- и В-лимфоцитов при помощи
Fc-рецепторов комплексов АГ—АТ до настоящего времени
полностью не исследована. Сепарированные лимфоциты мыши
или крысы характеризуются вполне определенными чертами,
присущими Т- или В-клеткам в таких тестах, как цитотоксич-
ность, флюоресценция, розеткообразование, антителообразова-
нне после переноса, РТПХ, продукция лимфоцитов и др.
Артефакты. Хотя неспецифическое связывание различных
популяций лимфоцитов (в первую очередь В-лимфоциты) на
вышеперечисленных материалах весьма незначительно, все
же им нельзя совершенно пренебрегать. Так, например, сефа-
декс связывает АОК. Различные эффекты, наблюдаемые после
разделения лимфоцитов на сывороточных гранулах, сефарозе
или сефадексе, позволяют предположить, что при определен-
ных условиях могут быть связаны также супрессорные клетки.
В случае разделения при 37°C нельзя исключить появление
так называемого «Shedding-феномена», если клетки предвари-
тельно были нагружены антителами или лигандами к рецеп-
торам (собственные наблюдения авторов). Наконец, следует
помнить о видовых различиях клеток. Количественное селек-
тивное связывание В-лимфоцитов мыши с комплексами АГ—АТ
за счет Fc-рецепторов, по всей видимости, не применимо к
Т-лимфоцитам человека, у которых имеются в относительно
больших количествах Fc-рецепторы для IgG и IgM [6]. По-
скольку все же образование Fc-розеток Т-лимфоцитами чело-
века требует особо длительного культивирования, без примене-
ния вышеупомянутых методов сепарации нельзя обойтись.
Различия свойств лимфоцитов грызунов и человека указывают
237
на то, что переносить результаты разделения клеток с одного
вида на другой нужно осторожно (см. также раздел 8.1.5).
В настоящее время нельзя определить, насколько эффективны
колонки с СЗ-сефарозой при разделении лимфоцитов других
(кроме человека) биологических видов.
3.1.4. РАЗДЕЛЕНИЕ КЛЕТОК МЕТОДОМ ЦИТОЛИЗА
З.1.4.1. ПРИНЦИП МЕТОДА
Иммунологическими исследованиями установлено, что поверх-
ность клетки состоит из различных АГ. К ним относятся транс-
плантационные, дифферецировочные, тканеспецифические, мем-
бранные АГ, обусловливающие серологические различия раз-
ных типов клеток и тканей. У лимфоцитов мыши, первого хо-
рошо изученного вида, было обнаружено большое количество
дифференцировочных АГ. Наиболее важны для разделения
клеток 9-антигены и антигены лимфоцитов Lyt 1, 2, 3 (Т-клет-
ки), MBL А (В-клетки), а также и мембранные глобулины,
концентрация которых на поверхности Т-лимфоцитов лежит
ниже границы чувствительности в тесте цитотоксичности [7].
Антигены Leu 1, 2а, 2Ь и 3, являющиеся эквивалентами анти-
генов Lyt, описаны также у человека [7]. Кроме того, специ-
фические АГ плазматических, стволовых, цитотоксических и
NK-клеток были обнаружены не только у мыши, но также у
других подопытных животных и у человека. Подробное пере-
числение этих АГ выходит за рамки настоящей работы; боль-
шая часть этих АГ имеет значение только для специальных
исследований. Разделение клеток при помощи цитолиза пред-
ставляет собой препаративную форму цитотоксического теста,
предложенного в 1956 г. Горером и О’Горманом. Клетки, ко-
торые были инкубированы с АТ к мембранным АГ, лизируются
после добавления комплемента. Количество лизированных
клеток определяют путем подсчета, в случае использования
соответствующих приспособлений нелизированные клетки
можно получать в препаративных количествах.
З.1.4.2. МЕТОДИКА
3.1.4.2.1. Материалы и оборудование
Для работы необходимы: МЕМ, сыворотка плода коровы,
центрифужные пробирки, настольная центрифуга, термостат
или водяная баня; микротитраторы Takatsy, гемоцитометр,
трипановый синий, ПАФ, шприцы, антисыворотки, компле-
мент.
3.1.4.2.2. Иммунизация
В литературе описано большое количество различных схем
иммунизации. Часто делают попытки получить сыворотки с
особенно высокими титрами АТ, для чего применяют высокие
238
дозы АГ и частые повторные иммунизации. Следует учитывать
иммуногенность АГ, поскольку большие дозы АГ с выражен-
ной иммуногенностью могут приводить к результатам, проти-
воположным ожидаемым. В гетерологических системах общим
правилом должно стать использование по возможности малых
доз АГ в сочетании с длительными интервалами между их
введением (2—4 нед). Такая схема приводит, как правило,
к получению сывороток хорошего качества.
В сингенных системах с низкой иммуногенностью АГ опти-
мальная доза намного выше. Использование ПАВ позволяет
улучшить результаты иммунизации. Первый раз АТ вводят
по возможности в подколенный лимфатический узел, в после-
дующие разы — подкожно, внутривенно или внутрибрюшинно.
Внутримышечное введение АГ с адъювантами часто вызывает
образование абсцессов и не дает никаких реальных преиму-
ществ (см. раздел 1.1).
3.1.4.2.3. Анти-тэта-сыворотки
Анти-Thy 1.1 (тэта-AKR)-сыворотка: СЗН-анти-АКК-тимус
Анти-Thy 1.2 (тэта-СЗН-анти-АКК-тимус)-сыворотка: AKR-
анти-СЗН-тимус. Суспензию тимоцитов (40Х106/мг) по кап-
лям вносят в равный объем ПАФ и перемешивают до получе-
ния стабильной водно-масляной эмульсии. По 0,02—0,03 мл
этой эмульсии вводят в каждую заднюю лапу, т. е. примерно
по 1 X 106 тимоцитов на мышь. Последующие инъекции следуют
с интервалом 2—3 недели, мышам при этом вводят по 5ХЮ6
клеток в объеме 0,2 мл внутрибрюшинно. Первый отбор крови
проводят через одну неделю после последней иммунизации.
В среднем необходимы 3 иммунизации; с каждой мыши полу-
чают 0,2—0,3 мл сыворотки. Как правило, адсорбцию прово-
дить не приходится. В особых случаях рекомендуется прово-
дить адсорбцию сывороткой или клетками особей того же био-
логического вида. При проведении адсорбции сывороткой по-
лученную АС смешивают с равным объемом нормальной сы-
воротки «донора» АГ и инкубируют в течение 30 мин при
37 °C.
З.1.4.2.4. Крысиная антисыворотка к тимусу мышей
В соответствии с предыдущим разделом тимоциты мышей
(СВА, 50Х106 клеток/мл) смешивают с ПАФ и вводят крысам
Вистар по 0,1 мл в каждую заднюю лапу (т. е. по 5,0Х106 кле-
ток на крысу) подкожно. Однократный (!) забор крови про-
водят через 9 дней. От каждой крысы получают до 3 мл сы-
воротки.
Адсорбция. Поскольку образование АТ против АГ, обуслов-
ливающих различие систем гистосовместимости крысы и мы-
ши, начинается тогда, когда титр АТ к тимусным АГ уже от-
носительно высок, то, как правило, достаточно одно- или дву-
кратной адсорбции сыворотки мышиной печенью.
239
К 5 мл разведенной в соотношении 1 : 5 сыворотки добав-
ляют 0,2 г однократно отмытого ацетонового порошка мыши-
ной печени, что соответствует 1 г свежей печени. Адсорбиро-
ванная сыворотка способна вызывать гибель в цитотоксиче-
ском тесте 99—100% тимусных клеток, 38% клеток селезенки,
50—60% клеток лимфатических узлов, но не приводит к ги-
бели клеток костного мозга у мышей СВА [8].
З.1.4.2.5. Кроличья антисыворотка против
В-лимфоцитов мыши
1 этап: мышей иммунизируют, вводя внутрибрюшинно по
0,2 мл 25% суспензии ЭБ. Кровь берут через 5 дней. Полу-
чают АС к эритроцитам барана (IgM), обязательно инакти-
вируют комплемент (30 мин при 37°C).
30 мин при 37 °C. Клетки осаждают с 3 мл АС (1 этап)
30 мин при 37 °C. Клетки осаждают центрифугированием
и 2 раза отмывают ФСБ. Готовят смесь с ПАФ.
Кролику подкожно ниже подколенного лимфатического
узла вводят по 0,5 мл препарата в каждую лапу. Через 3 нед
проводят 2-ю инъекцию подкожно в верхнюю часть бедра и
еще через 3 нед такую же дозу (без ПАФ) вводят внутрибрю-
шинно. Спустя 14 дней берут первую пробу крови из вены.
Еженедельно без ущерба для здоровья животного можно от-
бирать до 60 мл крови, а при больших интервалах — до 90 мл
(кровь берут из ушной вены). Неадсорбированная сыворотка,
помимо В-лимфоцитов, вызывает гибель приблизительно 50%
Т-лимфоцитов в лимфатических узлах, селезенке и тимусе, что,
вероятно, связано с наличием антигена Форссмана в Т-лимфо-
цитах мыши. После 2—4-кратной адсорбции эритроцитами ба-
рана сыворотка реагирует только с В-лимфоцитами (57% —
лимфоциты селезенки, 30%—лимфатических узлов, 0%—ти-
муса, 9%—костного мозга). Для адсорбции неразведенную
инактивированную сыворотку смешивают с равным объемом
60% суспензии эритроцитов. После 3—4-кратного повторения
достигается необходимая конечная концентрация, вызываю-
щая цитолиз.
3.1.4.2.6. Комплемент
Источником комплемента служит нормальная сыворотка кро-
лика или морской свинки. Поскольку эти сыворотки в основном
цитотоксичны, и, кроме того, активность комплемента может
варьировать, их следует исследовать в сравнении с известной
АС и субтоксическими дозами комплемента в цитотоксическом
тесте. Сыворотки, содержащие активный комплемент, объеди-
няют для получения стандартного препарата. Слаботоксиче-
ские сыворотки можно адсорбировать агаром или агарозой.
Обычно достаточно 20 мг агара для адсорбции 1 мл компле-
мента, разведенного в соотношении 1 :3. Адсорбцию ведут при
0—4 °C, длительность процесса 60 мин. Каждые 10 мин смесь
240
перемешивают, а после окончания инкубации — центрифуги-
руют. Объединенный препарат комплемента разделяют на не-
большие порции, быстро замораживают и хранят при —30 °C
(и ниже). Для проведения пробы на цитотоксичность допу-
стимо только однократное размораживание комплемента.
3.1.4.2.7. Определение цитотоксичности сывороток
Для определения специфичности сыворотки и эффективности-
адсорбции лучше всего использовать тест на цитотоксичность.
Проще всего это сделать следующим образом [8]: в лунки
микротитратора (или маленькие пробирки) вносят по 20 мкл
суспензии клеток (2Х106/мл), 20 мкл сыворотки (ряд разве-
дений) и 20 мкл комплемента, инкубируют 45 мин при 37 °C,
добавляют 20 мкл трипанового синего и подсчитывают количе-
ство живых клеток. При работе с каждой суспензией клеток
ставят двойной контроль антисыворотки. Подсчитывают клетки
на равной площади поверхности камеры (например, ]/3 поверх-
ности камеры Burker), в контроле (только комплемент, только
АС или только сыворотки плода коровы) должно содержаться
150—200 клеток. Этот метод так же точен, как и общепринятая
методика, но он имеет два преимущества:
1) результаты получаются быстрее,
2) количество мертвых клеток в исходной суспензии не
имеет значения в случае избытка антител.
Необходимое разведение комплемента желательно опреде-
лять заранее. Обычно для проведения цитолиза клеток селе-
зенки или лимфатических узлов мыши комплемент (морская
свинка) не разводят, а для цитолиза мышиных тимоцитов
делают разведение в 3 раза.
3.1.4.2.8. Количественный цитолиз
Берут 1 объем клеток (100Х106/мл), 2 объема антисыворотки»
1 объем комплемента.
Смесь инкубируют в течение 60 мин при 37 °C, каждые
10 мин встряхивают, центрифугируют, дважды отмывают сре-
дой МЕМ с 5% телячьей сыворотки, погибшие клетки от-
фильтровывают.
З.1.4.З. ОЦЕНКА МЕТОДА
Разделение клеток путем цитолиза — наиболее простой из всех
методов при условии наличия необходимых сывороток. Метод
позволяет за короткое время разделить большое количество
клеток. Выделенные клетки можно рассматривать как нести-
мулированные. Могут наблюдаться артефакты, хотя и редко,
в тех случаях, когда АС не обладает нужной специфичностью.
Не следует ожидать, что подлежащая элиминации субпопуля-
ция клеток будет лизирована на 100% (эффективность цитоли-
за зависит от качества сыворотки и от концентрации АГ на
16—993
241
поверхности клеток-мишеней), к тому же никакой другой ме-
тод не дает 100% чистоты выделяемой субпопуляции. Еще од-
ним преимуществом является возможность разделения В- или
Т-лимфоцитов на субпопуляции, что невозможно осуществить
большинством других методов.
В настоящее время большая часть моноспецифических АС,
в том числе моноклональных АТ, выпускается в виде коммер-
ческих препаратов. Самостоятельно получить и охарактеризо-
вать сыворотки для цитолиза довольно просто, но необходим
известный опыт, чтобы полученные АС специфически и в до-
статочной степени элиминировали нужную субпопуляцию кле-
ток.
3.1.5. РАЗДЕЛЕНИЕ КЛЕТОК ПРИ ПОМОЩИ
РОЗЕТКООБРАЗОВАНИЯ
3.1.5.1. ПРИНЦИП МЕТОДА
По принятому в иммунологии определению «розетки» обра-
зуются при фиксации эритроцитов на поверхности различных
клеток. Этот феномен обусловлен специфическим взаимодей-
ствием компонентов мембран обоих типов клеток. У розетко-
образующих клеток (лимфоциты, макрофаги и др.) этими ком-
понентами могут быть рецепторы антигенов комплемента, Fc-
фрагментов и др., глобулины или случайно дающие перекрест-
ные реакции с мембранами эритроцитов компоненты (спонтан-
ное розеткообразование).
В случае эритроцитов, помимо мембранных компонентов,
это могут быть адсорбированные или конъюгированные АТ или
АГ (методику розеткообразования см. в разделах 3.3—3.5),
Важнейшие примеры розеткообразования эритроцитами и лим-
фоцитами различных биологических видов представлены в
табл. 18.
Таблица 18. Спонтанное образование розеток
Центральные клетка Эритроцит
Т-лимфоцит (человек) Т-лимфоцит (человек) В-лимфоцит (человек) В-лимфоцит (человек) Т-лимфоцит (морская свинка) Т-лимфоцит (крыса) Т-лимфоцит (собака) Т-лимфоцит (кошка) Барана Человека Мыши Обезьяны Кролика Морской свинки Человека, морской свинки Мыши, крысы, морской свинки
242
З.1.5.2. МЕТОДИКА
3.1.5.2.1. Материалы и оборудование
Разделение клеток см. в разделе 3.1.2.
Розеткообразование в разделе 3.3—3.5.
3.1.5.2.2. Разделение розеток
Разделение розеток и свободных лимфоцитов проводят цент-
рифугированием в градиенте плотности. Условия центрифуги-
рования могут варьировать (400 g в течение 30 мин; 450 g в
течение 45 мин или 860 g в течение 20 мин) в зависимости от
целей эксперимента (см. раздел 3.1.6). При успешном разде-
лении розетки осаждаются, а свободные лимфоциты остаются
в интерфазе. От эритроцитов освобождаются гемолизом.
З.1.5.З. ОЦЕНКА МЕТОДА
Фракционирование при помощи розеток вследствие его отно-
сительной простоты является одним из наиболее часто приме-
няемых методов разделения Т- и В-лимфоцитов наряду с се-
парированием на найлоновой вате. Известно большое количе-
ство методических вариантов, зачастую малооправданных, что
приводит к трудностям интерпретации получаемых результа-
тов. Степень чистоты получаемых клеток в целом удовлетво-
рительная, однако для получения суспензии Т- или В-лимфо-
цитов с чистотой более 90% приходится повторять дважды
процедуру розеткообразования и отделения ввиду нестабиль-
ности некоторых розеток.
Артефакты. При повторном разделении клеток методом ро-
зеткообразования происходит обогащение выделяемых клеток
субпопуляцией с повышенной аффинностью. К числу недостат-
ков этого метода относится то, что нельзя рассматривать вы-
деленные клетки как нестимулированные. Т-лимфоциты, полу-
ченные методом розеткообразования с эритроцитами барана,
усиленно реагируют в смешанной культуре лимфоцитов, в реак-
ции бласттрансформации КонА, при стимуляции фактором
роста Т-клеток, а также проявляют активность, характерную
для К- и NK-клеток [9]. Следует отметить, что активность.
К-клеток заметно снижалась или полностью исчезала, когда
после выделения розеток ЭБ лизировали NH4C1, а не дистил-
лированной водой, как обычно [10]. Что касается выделения
В- и Т-лимфоцитов путем связывания клеток с Fc-рецепторамн
комплексов АГ—АТ (ЕА-розетки), то к этому методу приме-
нимы те же ограничения, что и при разделении на колонках
(см. раздел 3.1.3).
16* 243
3.1.6. РАЗДЕЛЕНИЕ КЛЕТОК
В ГРАДИЕНТЕ ПЛОТНОСТИ
3.1.6.1. ПРИНЦИП МЕТОДА
Между величиной и плотностью клеток существует тесная
взаимосвязь. Обычно меньшие по размеру клетки обладают
относительно меньшим объемом цитоплазмы по сравнению с
ядром и, следовательно, большей плотностью. Разумеется,
из этого правила есть исключения. Клетки, различающиеся по
величине и плотности, могут быть разделены методом изопик-
нического пли изокинетического центрифугирования, а также
осаждением в градиенте плотности. При изопикническом цент-
рифугировании клетки располагаются в слоях градиента с со-
ответствующей плотностью. В случае изокинетического цент-
рифугирования в менее плотном градиенте клетки с большим
диаметром седиментируют быстрее. Тот же принцип лежит в
основе осаждения клеток в градиенте плотности.
З.1.6.2. МЕТОДИКА
3.1.6.2.1. Материалы и оборудование
Для работы необходимы: центрифуга с охлаждением, бакет-
ротор, градиентный смеситель, вещества для формирования
градиента, такие, как альбумин, сыворотка эмбрионов коров,
фиколл, лимфопреп, перколл (Pharmacia, Швеция), визотраст
или другие рентгеноконтрастные вещества.
3.1.6.2.2. Центрифугирование в градиенте плотности
Ввиду разнообразия применяемых методов фракционирования
клеток в градиенте плотности невозможно в рамках настоящей
главы дать их детальный разбор. В зависимости от биологи-
ческого вида, органного происхождения, стадии жизненного
цикла и активации плотность лимфоцитов, макрофагов, грану-
лоцитов варьирует в пределах 1,04—1,12 г/мл. Обычно раз-
брос величин плотности составляет около половины этого
интервала. Условия центрифугирования подбираются индиви-
дуально в зависимости от характера эксперимента, основные
варианты представлены в табл. 19.
Если требуется разделить клетки в соответствии с их плот-
ностью, но нет никаких предварительных указаний на то, как
это сделать, можно попытаться разделить клетки в линейном
градиенте, исследовать, в какой области градиента преобла-
дает определенный тип клеток, а затем провести разделение
в прерывистом градиенте.
244
Таблица 19. Изопикническое центрифугирование клеток и градиенте
плотности фиколла
Градиент Биологический вид Тип клеток g Время, мии
5,5—43% Морская свинка Лимфоциты/макрофаги 800 90
4—48% Мышь Л имс юциты/макрофаги 600 120
4,1-43% Человек Лиме >оциты/гранулоциты 850 90
4,1—43% Мышь Л имфоциты/гранулоци- ты/макрофаги/тучные клетки 2200 39
Изопикническое разделение. В качестве одного из вариантов
градиентного центрифугирования, помимо перечисленных в
табл. 19, можно использовать следующий.
Градиент БСА 15—28%; 3800 g в течение 45 мин или
4000 g в течение 30 мин. Разделению подвергаются лимфоциты
мыши и крысы. При приготовлении градиента надо выбирать
инертные, которые не диссоциируют на ионы и образуют вод-
ные растворы с низкой вязкостью.
Растворы таких веществ должны обладать незначительным
осмотическим давлением во избежание повреждения разделяе-
мых клеток. Лучше всего этим требованиям отвечают фиколл,
перколл и альбумин. Альбумин предварительно должен быть
отдпализирован, деионизирован, его pH должен быть доведен
до 5,1. Это значение pH хорошо переносится клетками и пре-
пятствует образованию агрегатов. Диализ необходим для уда-
ления солей, которые могут присутствовать в альбумине и по-
вышать осмотическое давление. Добавлением гомологичной
нормальной сыворотки разницу в осмотическом давлении мож-
но еще уменьшить. На дно центрифужных пробирок часто на-
слаивают так называемую «подушку», т. е. небольшое количе-
ство раствора высокой плотности (45% фиколл или 40% аль-
бумин). Затем вносят суспензию клеток в растворе фиколла
или альбумина низкой плотности и подслаивают собственно
градиент. Можно также примешивать клетки непосредственно
к линейному градиенту, но при этом следует избегать образо-
вания агрегатов. После центрифугирования клетки отсасывают
при помощи перистальтического насоса, собирают фракции,
содержащие клетки. Жидкость из пробирки можно отсасывать
сверху или снизу, через прокол в стенке пробирки.
Значительно чаще, чем непрерывный, используют ступенча-
тый градиент плотности. По своему составу градиенты могут
значительно варьировать. Мы ограничимся только двумя при-
мерами.
Одновременное разделение эозинофилов, нейтрофилов и мо-
ноцитов человека в градиенте фиколл/гипак,-
245
(A) 15,0 мл 9% фиколла + 10,0 мл 50% гипака; плотность
1,14 г/мл.
(Б) 17,5 мл 9% фиколла + 10,0 мл 50 гипака; плотность
1,13 г/мл.
(В) 20,0 мл 9% фиколла+10,0 мл 50% гипака; плотность
1,12 г/мл.
(Г) 24,0 мл 9% фиколла + 10,0 мл 50% гипака; плотность
1,06 г/мл.
Составляют градиент осторожно, наслаивая друг на друга
по 2,0 мл растворов уменьшающейся плотности. Сверху на-
слаивают 2,0 мл гепаринизированной крови, разведенной в
соотношения 1:2 0,15 М NaCl. Центрифугируют 40 мин при
1000 g и 22°C. Клетки распределяются тогда между различ-
ными слоями следующим образом: между плазмой и слоем
Г — моноциты и лимфоциты с 97—100% чистотой, между Г и
В — нейтрофилы с 94—99% чистотой, между В и Б — ненрофи-
лы (50—98%) и эозинофилы (2—50%), между Б и А — эози-
нофилы с 80—99% чистотой [11].
Разделение гранулоцитов и фракции лимфоциты/моноциты
человека в градиенте фиколл/триомбраст:
(А) 10 объемов 34% триомбраста плотность 1,075 г/мл при 22°C
24 объема 9% фиколла
(Б) 10 объемов 34% триомбраста плотность 1,097 г/мл при 22°C
24 объема 14,6% фиколла
На градиент, составленный из 5 мл фракции А и 5 мл
фракции Б, наслаивают 10 мл разведенной в соотношении 1:2
гепаринизированной крови. Центрифугируют 40 мин при 400 g
и 22°C. Легкая фракция содержит лимфоциты (свыше 60%),
моноциты (свыше 35%) и гранулоциты (около 1%).
Плотная фракция содержит 98% гранулоцитов, их выход
составляет 60% [12].
Изокинетическое разделение. Изокинетическое разделение
используют при необходимости разделять клетки одинаковой
плотности, но различной величины. Если используется градиент
фиколла 2,6—5,5%, то центрифугирование проводят при 18,7
и 97 g в течение 30—14 мин. Результаты можно существенно
улучшить, дополнив изокинетическое центрифугирование изо-
пикническим. Все многообразие методов разделения клеток
можно видеть на примере разделения моноцитов и лимфоцитов
человека в градиенте перколла. Раствор перколла (30 мл) с
плотностью 1,060 г/мл в среде, не содержащей Са2+ и Mg2+,
центрифугируют в течение 1 ч при 26 000 g для формирования
градиента плотности. На этот градиент наслаивают 3,0 мл
суспензии моноциты/лимфоциты (15Х106/мл). Разделение мо-
ноцитов и лимфоцитов происходит в ходе изокинетического
центрифугирования в течение 5 мин при 400 g. Выход высоко-
гомогенной фракции моноцитов составляет около 50% от их
содержания в крови [13].
246
3.1.6.2.3. Седиментация в градиенте плотности
Фракционирование клеток различной величины чаще всего
проводят в седиментационной камере по принципу Miller и
Phillips [14]. Диаметр таких камер обычно варьирует от 11
до 21 см, но известны и камеры меньшего размера [14].
Для стабилизации разделяющей жидкости и предотвращения
конвенции градиент формируют из сыворотки эмбрионов ко-
ров. Готовят суспензию клеток (107/мл) в 3% сыворотке эм-
брионов коров. В камере диаметром 11 см седиментируют
20 мл, диаметром 21 см—100 мл суспензии клеток. Градиент
формируется в процессе центрифугирования, которое происхо-
дит при 4 °C н длится 3,5—4,5 ч. Клетки сорбируют фракциями
по 12—15 мл.
З.1.6.З. ОЦЕНКА МЕТОДА
Если величина и плотность клеток коррелируют между собой,
центрифугирование в градиенте плотности и седиментация в
градиенте дают одинаковые результаты. Однако если клетки
при одинаковой величине обладают различной плотностью или
при одинаковой плотности различной величиной, то один из
двух методов разделения может оказаться более эффективным.
Воспроизводимость методов, основанных на центрифугирова-
нии, очень высока. Например, при разделении клеток одинако-
вого происхождения в линейном градиенте альбумина (15—
30 фракций) положение пика отклоняется в среднем на
0,0003 г/мл, т. е. приблизительно на четверть величины сред-
ней фракции. Также отмечаются заметные вариации профиля
разделения клеток у различных индивидов. Выход составляет
89±9%, погибшие клетки образуют осадок. Еще одной про-
блемой центрифугирования в градиенте плотности является
необходимость создания по всему градиенту условий, одина-
ково хорошо переносимых всеми клетками. В противном слу-
чае возможна утеря клетками биологической активности.
Для разделения Т- и В-лимфоцитов градиентные методы не
подходят. В нестимулированном состоянии оба типа клеток
различаются весьма незначительно по плотности и величине.
Поскольку эти последние свойства сильно варьируют в зави-
симости от степени дифференцировки и активации клеток, по-
лучение профилей их распределения по плотности в величине
представляет собой картину, отражающую внутриклеточные
процессы в Т- и В- субпопуляциях [14]. После стимуляции АГ
или митогенами как В-, так и Т-лимфоциты дают очень боль-
шой разброс по плотности и величине в начале иммунного от-
вета. На поздних этапах популяция становится гомогенной.
Помимо лимфобластов, разделить можно и такие субпопуля-
ции, как Т-хелперы и Т-супрессоры. Степень чистоты макро-
фагов достигает 98%, гранулоцитов — свыше 90% [Н]. Из пе-
247
ритонеальной жидкости эозинофилы выделяются с чистотой
88%, тучные клетки — с чистотой 95% [15]. При помощи четы-
рехступенчатого градиента за один этап работы удается полу-
чить из крови одновременно эозинофилы (чистота 80—99%),
нейтрофилы (чистота 94—99%), моноциты (чистота 97—100%)
[И]. Из опухолей, содержащих приблизительно 2% лимфоци-
тов, получали фракции, содержащие 15% В- и 43% Т-лимфо-
цитов [16]. Из костного мозга, хотя и с ограничениями, удает-
ся выделять стволовые клетки и лимфоциты [17, 18].
Артефакты. Не следует думать, что для успешного решения
задачи разделения достаточно подобрать градиент нетоксиче-
ского материала. Известны случаи избирательной потери Т-лим-
фоцитов вследствие контакта с градиентами фиколл — гипак
или фиколл — метризоат [19, 20]. У лимфоцитов из крови сви-
ней наблюдали повышение числа розеток, обусловленных на-
личием Fc-рецепторов к IgG, с 9 до 33% после контакта с 4%
фиколлом или 14% декстраном [21].
3.1.7. РАЗДЕЛЕНИЕ ПРИ ПОМОЩИ ЛЕКТИНОВ
З.1.7.1. ПРИНЦИП МЕТОДА
До 1980 г. в литературе имелось всего несколько сообщений
об использовании лектинов для разделения лимфоцитов, при-
чем речь идет в большинстве случаев об агглютинине из зем-
ляного ореха. Наиболее широко применяются два метода:
а) агглютинация части лимфоцитов путем инкубации с лекти-
ном, отделение агглютинированных клеток центрифугирова-
нием;
б) аффинная хроматография на колонках с лектинами (см.
раздел 3.1.3). Связанные клетки могут быть элюированы с ко-
лонки с лектинами раствором углевода, по отношению к ко-
торому лектин специфичен.
3.1.7.2. МЕТОДИКА
3.1.7.2.1. Материалы и оборудование
Для работы необходимы: суспензия клеток (см. раздел 3.1.2),.
лектины (например, агглютинин земляного ореха), центрифу-
га, седиментационная камера, среда для клеток, сыворотка
эмбрионов коров, галактоза или другие углеводы, соответ-
ствующие специфичности лектинов.
3.1.7.2.2. Агглютинация тимоцитов мыши агглютинином
земляного ореха
Тимоциты (6—8Х108/мл) смешивают с равным объемом аг-
глютинина земляного ореха (1 мг/мл ФСБ), инкубируют
10 мин при комнатной температуре. Затем 1 мл клеток (3—
248
4Х108/мл) наслаивают на 30 мл клеток 20% сыворотки эмбрио-
нов коров в ФСБ. В качестве седиментационной камеры можно
использовать высокий узкий цилиндр. Через 30 мин после на-
несения пипеткой или насосом отбирают верхние 15 мл. Обе
фракции клеток собирают центрифугированием, затем обраба-
тывают 0,25 М. галактозой в ФСБ в течение 10 мин, периоди-
чески осторожно встряхивая или засасывая раствор пипеткой.
После обработки клетки отмывают и готовят из них суспен-
зию необходимой концентрации.
З.1.7.З. ОЦЕНКА МЕТОДА
PNA, агглютинин земляного ореха (Arachis hypogaea)1, пред-
ставляет собой наиболее хорошо изученный лектин. PNA свя-
зывает незрелые тимоциты мыши [22—25].
SBA2, агглютинин соевых бобов (Glycine max), обладает
специфичностью в отношении В-лимфоцитов мыши [25, 26].
V1L3, лектин из вики (Vicia faba), реагирует с цитотокси-
ческими Т-лимфоцитами человека, но не с К- или NK-клетками
[27].
WGA4, агглютинин зародышей пшеницы (Triticum vulgare)3,
агглютинирует часть лимфоцитов мыши, но до настоящего вре-
мени не нашел применения в разделении лимфоцитов, разли-
чающихся по функциональной активности [28—30]. Данные о
других выделенных к настоящему времени лектинах настолько
неопределенны, что этот вопрос здесь не будет обсуждаться.
Лектины не дают полного разделения клеток; точные данные
в литературе по этому вопросу отсутствуют. Однако, вероятно,
использование лектинов окажется хорошим вспомогательным
средством в фракционировании клеток.
3.1.8. ПРОЧИЕ МЕТОДЫ
Три специальных метода разделения клеток будут критически
рассмотрены и оценены ниже. Электрофорез клеток и флюорес-
центно-активационная сортировка представляют собой слож-
ные методы, требующие специальной аппаратуры, поэтому они
не могут быть подробно рассмотрены в настоящей книге. Тех-
нология монослоя предполагает наличие отработанной системы
культивирования ткани, необходимой для разделения лимфо-
цитов.
1 PNA — аббревиатура от англ, pea nut agglutinin. — П р и м е ч. пер.
2 SB А — аббревиатура от англ, soia bean agglutinin. — Пр имен. пер.
3 VFL — аббревиатура от англ. Vicia-faba-Lektin. — Пр и меч. пер.
4 WGA — аббревиатура от англ, «wheat germ agglutinin». — П р и м е ч.
пер.
249
3.1.8.1. ЭЛЕКТРОФОРЕЗ КЛЕТОК
Находясь в суспензии, под действием постоянного электриче-
ского поля клетки мигрируют к аноду. Миграция становится
возможной вследствие возникновения электрического потен-
циала на поверхности клеточных мембран при контакте со
средой. Потенциал обусловлен особенностями структуры мем-
браны. Распределение ионов на мембране и ионизация функ-
циональных групп белков приводит к образованию суммарного
отрицательного заряда клеток [31].
Различные типы клеток обладают неодинаковой электрофо-
ретической подвижностью. Это особенность убывает в следую-
щем порядке: эритроциты, Т-лимфоциты, В-лимфоциты, макро-
фаги/моноциты. Различия в подвижности Т- и В-лимфоцитов
обусловлены различием концентрации нейраминовой кислоты
в клеточной мембране. Аналитический электрофорез клеток
позволяет получить диаграмму частотного распределения кле-
ток в суспензии по их подвижности. Для каждого органа су-
ществует характерная диаграмма с одним или несколькими
пиками. Обычно при проведении электрофореза без носителя
получают 15—20 фракций, но даже при наличии выраженных
пиков имеется их заметное перекрывание. Если взять суспен-
зию спленоцитов мыши, отбросить фракции клеток со средней
подвижностью и объединить фракции с соответственно высокой
и низкой подвижностью, то можно получить препараты Т- и
В-лимфоцитов высокой степени чистоты [31].
Артефакты. Стимуляцию лимфоцитов в данной системе в
принципе можно исключить. Это, пожалуй, и является един-
ственным преимуществом метода, имеющего многочисленные
недостатки. К их числу относится низкий выход клеток. Если
отбрасывать перекрывающиеся области, возникает вероятность
селективных потерь. Выход клеток может снижаться вслед-
ствие их агрегации, обусловленной высвобождением ДНК из
клеток под действием применяемого буфера. Препаративный
электрофорез существенно отличается от аналитического еще
и тем, что используется нефизиологичный глициновый буфер с.
низкой ионной силой и напряжение электрического поля в
15 раз более высокое. Это означает, что ионное облако вокруг
клеток и, следовательно, их потенциал существенно изменены,
а результаты электрофоретического разделения могут весьма
существенно отличаться от результатов аналитического элект-
рофореза.
З.1.8.2. СОРТИРОВКА КЛЕТОК
Этот метод стал универсальным, особенно с того времени,
как получила развитие гибридомная технология. Возможно
специфическое разделение на основе как АГ, так и АТ, мечен-
ных флюорохромами. В плане достигаемой чистоты метод вы-
250
Таблица 20 (обзорная). Сравнительная оценка методов разделения
лимфоцитов
Метод Издержки Достигаемая чистота Выход Артефакты Общая опенка метода
Разделение в градиенте плотности + 4-4-74-4-4- ++/+ + +
Образование розеток +/++ 4- 4-4- -/++ +
Электрофорез клеток +4—Ь 4- 4- 7> ——
Адгезионные методы 4- 4- 4-4- +++ +
Аффинная хроматогра- фия 4-4- +(+) -/++ +
Цитолиз + 4-4- 4-4-+ — +
Сортировка клеток 4—i—F 4-4-4- + -/++ -4-
Разделение на монослое 4—Н 4- +++ —/4—Ь —
Лектиновый метод +(4-) ? —/+4- ?
сокоэффективен, но количество клеток, которые могут быть
разделены этим методом, крайне незначительно. Цитофлюори-
метрия может быть с успехом применена для цитологического
анализа и клонирования клеток. К числу недостатков метода
относится высокая стоимость оборудования и необходимость
использования абсолютно моноспецифических сывороток или
моноклональных АТ1.
3.1.8.3. РАЗДЕЛЕНИЕ НА МОНОСЛОЕ
Использование монослоя дает возможность определять у Т-
лимфодитов наличие специфических рецепторов в отношении
трансплантационных или опухолевых АГ. В ходе инкубации
суспензии клеток с монослоем клеток—мишеней эффекторные
клетки связываются с соответствующими рецепторами. Элими-
нация эффекторных клеток в этих условиях не может быть
полной, цитотоксические лимфоциты удаляются лучше, чем не-
стимулированные.
Артефакты. Следует принимать во внимание ложное разде-
ление, обусловленное блокадой эффекторных клеток вследствие
отрыва клеток-мишеней или выделения их АГ [32].
1 Существо метода заключается в том, что суспензию клеток, часть из
которых мечена флюорохромом, например при помощи ФИТЦ-иммуноглобули-
нов, пропускают через капиллярную камеру, одновременно освещая их лучом
лазера. Проходя через камеру поштучно, клетки флюоресцируют. Детектор
флюоресценции связан с клапаном, позволяющим направлять в разные капил-
ляры поштучно флюоресцирующие и нефлюоресцирующие клетки. Таким обра-
зом достигается высокая эффективность разделения клеток. — П р и м е ч.
лер.
251
3.1.9. ВЫБОР МЕТОДА, ОБЩАЯ ОЦЕНКА
Выбор метода должен определяться поставленной задачей с
учетом возможных артефактов. В табл. 20 приведены суммар-
ные данные, характеризующие различные аспекты разделения
клеток. Таблица составлена на примере метода образования
розеток, но можно использовать любую другую реакцию. Из-
держки этого метода незначительны в случае простого разде-
ления Т-лимфоцитов, они возрастают, когда в пределах попу-
ляции Т-лимфоцитов разделение проводят по наличию Fc-ре-
цепторов для IgG или IgM. Не образующие розетки лимфоци-
ты не дают артефактов (—). В случае розеткообразующих
клеток приходится считаться с наличием артефактов ( ++)-
Общая оценка метода (+) или (—).
3.1.10. СПИСОК ЛИТЕРАТУРЫ
1. Gordon D. S., Shore S. L. Antibody-dependent cellular cytotoxicity to virus-
infected target cells: Role of nylon wooladherent T cells as effectors. — Cell.
Immunol., 1980, 50, 19—29.
2. Bellavia A., Micklem H. S. T lymphocytes and thymocytes with different
localisation properties separated by nylon-wool filtration. — Transplantation,
1977, 23, 290—292.
3. Potter M. R., Moore M. Human mixed lymphocyte culture using separated
lymphocyte populations. — Immunology, 1977, 32, 359—365.
4. Kagan J., Ben-Sasson S. Z. Antigen induced proliferation of murine T lym-
phocytes in vitro. 1. Characterisation of the lymphocyte culture system. — J'.
Immunol. Meth., 1980, 37, 15—27.
5. Casli P„ Perussia B. Human T and В lymphocytes separation on Sepharose
C-3 «activated» columns. — Bolletino, 1976, 55, 73—74.
6. Moretia A., Mingari M. C., Colombatti M. et al. Fc receptors on human T
lymphocytes: Loss of Fc-mu and expression of Fc-gamma receptors by T
cells stimulated in mixed lymphocyte reaction. — Scand. Journ. Immunol.,
1981, 13, 447—451.
7. Ledbetter J. A., Evans R. L., Lipinski M. et al. Evolutionary conservation of
surface molecules that distinguish T lymphocyte helper/inducer and cytoto-
xic/suppressor subpopulations in mouse and man. — J. exp. Med., 1981, 153,
310—323.
8. Eckert R. An improved method for producing an antiserum against T lympho-
cytes of the mouse.— J. Immunol, meth., 1982, 51, 45—48.
9. Silva A., Lopez Botet M., Alvarez J. et al. Enhancement of the functional acti-
vities of human T cells after their interaction with SRBC. — J. Immunol.,
1981, 126, 393—397.
10. Savary C. A., Phillips J. H., Lolzova E. Inhibition of murine inatural cell-
mediated cytotoxicity by pretreatment with ammonium chloride. — J. Immu-
nol. Meth., 1979, 25, 189—192.
11. Bicalho H. M. S., Contijo С. M., Nogueira-Machado J. A. A simple technique
for simultaneous human leukocyte separation. — Journ. Immunol. Meth., 1981,
40, 115—166.
12. Aguado M. T., Pujol N., Ribiol E. et al. Separation of granulocytes from
peripheral blood in a single step using density gradients of ficoll-urographin.
A comparative study with separation by dextran. — J. Immunol. Meth., 1980,
32, 41—50.
13. Hardin J. A., Downs J. T. Isolation of human monocytes on reorienting gra-
dients of Percoll. — J. Immunol. Meth., 1981, 40, 1—6.
14. Shortman K-, Fedler J. M„ Schlegel R. S. et al. Subpopulations of B-lympho-
cytes. Physical separation of functionally distinct stages of В cell different
252
tiation. — Contemporara Topics in Immunobiol. Ed. W. O. Weigle, Chapt 1,.
p. 1—45, 1976.
15. Dullens H. F. van Basten Ch., de Weger R. A. et al. Isolation of mast cells-
from murine peritoneal cell population by velocity sedimentation at unit gra-
vity.— J. Immunol. Meth., 1981, 40, 367—372.
16. Svennevig J. L„ Lovik M„ Svaar H. Isolation and characterization of lympho-
cytes and macrophages from solid, malignant human tumors. •— Int. J. Can-
cer, 1979, 23, 626—631.
17. Dorshkind K-, Klimpel G. R., Rosse C. Natural regulatory cells in murine-
bone marrow: Inhibition of in vitro proliferative and cytotoxic responses to-
alloantigens. — J. Immunol., 1980, 126, 2584—2588.
18. Ropke C. Characterization of Thy-1,2, positive cells in mouse bone marrow
by sedimentation and mitogen stimulation. — Immunology, 1981, 42, 385—
389.
19. Bast B. J. E. G., Catty D., Manten-Slingerland R. et al. Surface IG om rabbit
lymphocytes. — Eur. J. Immunol., 1980, 9, 997—1003.
20. Hokland P., Heron I. Analysis of the lymphocyte distribution during Isopa-
que-Ficoll isolation of mononuclear cells from human peripheral blood. —
J. Immunol. Meth., 1980, 32, 31—39.
21. Binss R. M., Licence S. T. A major subpopulation of Fc receptors bearing,
lymphocytes revealed by rosette formation in dextran media: Studies with
pig, sheep and rat lymphocytes. — J. Immunol. Meth., 1981, 43, 153—162.
22. Reisner L, Linker-1 sraeli M„ Sharon N. Separation of mouse thymocytes-
into two subpopulations by the use of peanut agglutinin. — Cell. Immunol.,
1981, 25, 129—134.
23. Reisner Y., Ikehara S., Hodes M. Z. et al. Allogeneic hemopoietic stem cell
transplantation using mouse spleen cells fractionated by lectins: In vitro
study of cell fractions. — Proc. Natl. Acad. Sci. USA, 1980, 77, 1164—1168.
24. Hardt C., Pfizenmaier K-, Rollinghoff NY Alloreactive and H 2-restricted'
Lyt-23 cytotoxic T lymphocytes derive from a common pool of antecedent
Lyt-123 precursors. — J. exp. Med., 1980, 152, 1413—1418.
25. Nakano T., Imai Y., Naiki M. et al. Characterization of mouse helper and
suppressor T cell subsets separated by lectins. — J. Immunol., 1980, 125,
1928—1932.
26. Nakano T., Oguchi Y., Imai Y. et al. Induction and separation of mouse hel-
per T cells by lectins. — Immunology, 1980, 40, 217—222.
27. Kimura A., Orn A., Holmquist G. et al. Unique lectin-binding characteristics-
of cytotoxic T lymphocytes allowing their distribution from natural killer
cells and «К» cells. — Eur. J. Immunol., 1979; 9, 575—578.
28. Boldt D. H., Lyons R. D. Fractionation of human lymphocytes with plant
lectins. 3. Identification of cells regulating the in vitro response to L-phyto-
hemagglutinin.— Immunology, 1980, 39, 519;—527.
29. Dilinger-Centerlind M. L„ Hellstrom U., Robertsson E. S. Mitogenic respon-
siveness of human T lymphocyte subpopulations: Regulation by suppressive
Fc-receptor-bearing T cells and influence of fractionation procedures. — Scand.
J. Immunol., 1980, 12, 13—21.
30. Lehtinen T., Perlmann P„ Hellstrom U. et al. Proliferative and cytolitic
reactivities of human lymphocytes fractionated on wheat germ agglutinin —
sepharose columns before or after alloactivation in mized lymphocyte cul-
ture.— Scand. J. Immunol., 1980, 12, 309—320.
31. Jensson H. L., Eckert R. Zellelektrophorese.— In: H. Friemel Immunologische-
Arbeitsmethoden, 2. Aufl. Jena: Gustav Fischer Verlag, 1980, 449—467.
253
3.2. ОПРЕДЕЛЕНИЕ КЛЕТОЧНЫХ МАРКЕРОВ
МЕТОДОМ МЕМБРАННОЙ
ИММУНОФЛЮОРЕСЦЕНЦИИ
3.2.1. ПРИНЦИП МЕТОДА
Мембранная иммунофлюоресценция (МИФ) используется для
локализации АГ на интактной мембране. Для визуализации
АГ применяют специфические АТ, меченные (конъюгирован-
ные) красителем, флюоресцирующим при облучении светом с
адекватной длиной волны. Тот же самый принцип применяется
в гистологических исследованиях, но в МИФ исследуют сус-
пензию живых клеток, что позволяет в определенной мере су-
дить об их функции. На рис. 86 схематически представлена
прямая и непрямая маркировка (метод двойных АТ). МИФ
впервые была предложена Moller в 1961 г. для определения
антигена Н-2 мыши, в дальнейшем этот метод нашел широкое
применение для определения поверхностных мембранных им-
муноглобулинов (Smlg) лимфоцитов при характеристике В-
,клеток. Ниже приводится описание метода применительно к
:SmIg лимфоцитов человека.
Рис. 86. Принцип мембранной иммунофлюоресценции (МИФ).
а — прямая МИФ: флюорохромированные АТ к мембранным антигенам; б — непрямая
МИФ (метод двойных антител): на первом этапе с мембранными АГ связываются спе-
цифические немеченые АТ, на втором этапе с АТ связываются флюорохромированные
аитивидовые АС, например, антитела к v-глобулину кролика, если первые АТ были по-
лучены иммунизацией кроликов; в — непрямая МИФ, оба АТ флюорохромнрованы.
254
3.2.2. НЕОБХОДИМЫЕ УСЛОВИЯ
З.2.2.1. ПРИГОТОВЛЕНИЕ
И МАРКИРОВКА АНТИТЕЛ
Приготовление меченных флюорохромом АТ уже было описано
ранее. Из свойств конъюгатов особое з-начение имеет молярное-
соотношение белка и флюорохрома, поскольку оно использует-
ся для количественной оценки соотношения специфической
флюоресценции и неспецифического окрашивания. Требования,
предъявляемые к конъюгатам, используемым в МИФ, очень
высоки (табл. 21 и 22).
Оптимальные рабочие разведения АС, АТ или конъюгатов
подбираются индивидуально для каждой системы. В ходе тит-
рования желательно работать в области плато и использовать
разведение сыворотки на две ступени выше, чем то, которое
соответствует концу плато. Поскольку при МИФ не всегда
удается достигнуть плато, то для достоверности можно ис-
пользовать неразведепные или слабо разведенные сыворотки,.,
например 1:10. Вторые АТ особенно часто используют без
разведения в непрямом МИФ. Условия конъюгации описаны в
соответствующей статье [14].
3.2.2.2. МИКРОСКОПИЧЕСКОЕ ОСНАЩЕНИЕ
К микроскопам, используемым в МИФ, предъявляются сле-
дующие требования.
1. Возможность попеременно и одновременно проводить наблю-
дения в фазовом (или интерференционном) контрасте, про-
ходящем и отраженном свете.
2. Сильный источник света (например, ртутная лампа высокого
Таблица 21. Критерии оценки конъюгатов
1. Специфичность сыворотки
2. Источник сыворотки (иид животного)
3. Серия и фирма-изготовитель
4. Общее количество белка
5. Количество белка антител
6. Количество и характер примеси других белков
7. Общее содержание флюорохрома
8. Содержание флюорохрома, связанного с белками
9. Содержание не связанного с белками флюорохрома
10. «Флюорохромный профиль» (см. пп. 7, 8, 9)
11. Соотношение флюорохром/общий белок
12. Соотношение белок АТ/общий белок
13. Соотношение белок АТ/флюорохром
14. Вид и количество консервантов и других добавок
15. Данные о контроле специфичности иа модельной системе с указанием на
неспецифические реакции
16. Оптимальное разведение в стандартной тест-системе
255
Таблица 22. Требования к конъюгату для МИФ [14]
1. Содержание антител (не меньше 1 г/л)
2. Молярное соотношение флюорохром : АТ должно составлять 1 : 2
3. Содержание общего белка (около 2 г/л)
4. Соотношение белок АТ/общий белок 0,5
5. Антитела должны представлять собой Fab- или Р(аЬ)2-фрагменты для
предотвращения связывания комплемента
6. АТ не должны давать перекрестных реакций
7. Должен отсутствовать «балластный белок» эозинофилов и базофилов
8. При низком общем содержании белка (ниже 2 г/л) для стабилизации
добавляют альбумин (10—40 г/л)
9. Антитела должны обладать высокой авидностью
10. Конъюгаты должны быть испытаны в модельной системе на специфич-
ность
давления НВО 200), расположенный по возможности вбли-
зи от препарата и снабженный охлаждающим устройством
для предотвращения повреждений интерференционного
фильтра.
3. Полный набор возбуждающих фильтров, ультрафиолетовых,
узкополосных и селективных, для работы с ФИТЦ и ТРИТЦ,
соответствующие интерференционные зеркала и запирающие
фильтры.
4. Возможность работы с одной и с двумя флюорохромными
метками [12].
5. Объективы самого высокого качества [большая апертура,
система обычных и иммерсионных сред (масло, вода или
глицерин)] для микроскопии и фотографии.
6. Окуляры с небольшим увеличением (не болееХб.З).
7. Автоматическая камера для микрофотографирования.
Для решения специальных вопросов используют добавочные
приспособления.
Микрофлюориметрия: в этом методе измеряют интенсив-
ность флюоресценции отдельных клеток и подсчитывают по-
верхностную плотность антигенов. Для этой цели измеряют
интенсивность флюоресценции нескольких клеток с известной
поверхностной плотностью АГ и определяют среднее значение
флюоресценции.
Зависимость интенсивности флюоресценции от количества
антигена может быть продемонстрирована калибровочными
кривыми, для построения которых используют конъюгацию
131 [1]-иммуноглобулинов с эритроцитами в различных количе-
ствах, а затем сравнивают интенсивность радиоактивного излу-
чения и МИФ для одинаковых количеств 1g.
Проточный флюоресцентный денситометр: современные при-
боры этого типа (например, FACS IV, Becton Dickinson, США)
сочетают способность к идентификации и разделению клеток и
клеточных компонентов по их величине или флюоресцентному
.маркеру [3, 6, 12].
256
Телевизионный увеличитель: конвенциональный флюорес-
центный микроскоп соединен с телекамерой, увеличителем
изображений, монитором и видеорекордером. Такая комбина-
ция позволяет повысить чувствительность МИФ при одновре-
менном уменьшении интенсивности возбуждающего света.
Флюорохромы поэтому выцветают не так быстро, а флюорес-
цирующие структуры живых клеток можно наблюдать в тече-
ние 30 и более мин.
3.2.3. МЕТОДИКА
З.2.З.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: визотраст 370 (Fahlberg List,
Магдебург, ГДР) или готовые растворы для разделения, на-
пример: фиколл — гипак (Pharmacia, Швеция); ФСБ (1,15 г
Na2HPO4, 0,2 г КН2РО4, 0,2 г КС1, 8 г NaCl, Н2О до 1000 мл,
pH 7,2—7,3). Для стабилизации ФСБ добавляют ЧСА (10 г/л).
Надо также иметь набор антисывороток, или препаратов АТ,
АС кролика против IgM человека и ФИТЦ-гаммаглобулин ко-
зы с антикроличьей специфичностью («особо чистый», Институт
иммунных препаратов, Берлин), нормальную кроличью сыво-
ротку.
Оснащение для инкубации клеток: пробирки (8X80 мм),
пастеровские пипетки, камеры и пипетки для подсчета клеток,
СН3СООН (30 г/л), качалка (Labortechnik, Ильменау), цент-
рифуга с охлаждением (Т23 или К23, Лейпциг).
Оснащение для фиксации: предметные и покровные стекла,
парафин, вентилятор, глицерин.
Техническое оснащение для ИФ-микроскопии: микроскопы
Laboval 2а.fl и Fluoval II (Carl Zeiss, Йена) для работы с
ФИТЦ и ТРИТЦ-меткой.
Объективы: Achromat 40/0,95 160/0,17; Achromat HI 100/1,40
160/0,17 (масляная иммерсия). Для фазово-контрастной мик-
роскопии в проходящем свете: фазово-контрастный конденсор
Phv Apl, 0,9е; объективы: Achromat 40/0,65 160/0,17, Achromat
HI 100/1,40 160/0,17 Phv.
Оснащение для микрофотографии обычно имеется в стан-
дартном наборе. Для черно-белых съемок применяют рентге-
новскую пленку RS2 и NP27; для цветных съемок —•
ORWOCHROM VI 120 или Кодак Ektachrome High Speed 27
DIN.
3.2.3.2. РАЗДЕЛЕНИЕ ЛИМФОЦИТОВ
Обычно используют разделение по Бейюму. Если в распоря-
жении исследователя нет фиколл — гппака, то можно исполь-
зовать раствор рентгеноконтрастных средств (например, визо-
траст 370) с плотностью 1,077 г/мл.
Г—990
257
1. Гепаринизированную или дефибринированную венозную
кровь разводят ФСБ.
2. В центрифужные пробирки наслаивают по 4 мл разве-
денной крови на 4 мл разделяющего раствора.
3. Центрифугируют 20 мин не более чем при 430g, собирают
интерфазу, содержащую лимфоциты, при помощи пастеровской
пипетки. (В случае недостаточного разделения центрифугиру-
ют еще 5 мин.)
4. Лимфоциты ресуспензируют в тройном количестве ФСБ,
центрифугируют (5 мин при 250 g), трижды повторяют отмывку.
Хотя после разделения остается небольшое количество эритро-
цитов в препарате, оно не мешает проведению МИФ. Избыточ-
ное загрязнение эритроцитами можно устранить гипотониче-
ским током (осадок клеток суспензируют в 1 мл дистиллиро-
ванной воды при 4 °C, через 30 сек добавляют избыток ФСБ
и центрифугируют, осадок ресуспензируют в ФСБ). Сепариро-
ванные клетки на 85—95% состоят из лимфоцитов.
3.2.3.3. ИНКУБАЦИЯ ЛИМФОЦИТОВ
Прямой метод:
1) устанавливают концентрацию лимфоцитов в суспензии
2Х Ю7 клеток в 1 мл. По 0,25 мл суспензии вносят в пробирки
и центрифугируют (5 мин, не больше чем при 180 g);
2) осадок (5Х106 клеток) суспензируют в 0,05 мл ФИТЦ-
aHTH-Ig-сыворотки в оптимальном разведении), инкубируют,
постоянно перемешивая, 30 мин при комнатной температуре;
3) трижды отмывают ФСБ, исследуют осадок микроскопи-
чески.
Непрямой метод:
1) см. прямой метод, п. 1;
2) осадок (5Х106 клеток) суспензируют в 0,1 мл (оптималь-
ное разведение) кроличьей антисыворотки к 1g человека, инку-
бируют 30 мин при постоянном помешивании при комнатной
температуре;
3) трижды отмывают ФСБ, осадок клеток суспензируют в
0,05 мл антивидовых глобулинов меченных ФИТЦ (в данном
случае антикроличьих АТ), инкубируют при постоянном пере-
мешивании в течение 30 мин при комнатной температуре;
4) трижды отмывают ФСБ, осадок клеток исследуют микро-
скопически.
3.2.3.4. ФИКСАЦИЯ, ОЦЕНКА РЕЗУЛЬТАТОВ
Рекомендуется осадок, состоящий из меченых жизнеспособ-
ных лимфоцитов, немедленно ресуспензировать в ФСБ и при-
ступить к оценке результатов (нанести каплю препарата на
предметное стекло, накрыть покровным стеклом и герметизи-
258
ровать парафином, в случае хранения препарата в течение
30 мин и более (при 4°C). При получении фиксированного пре-
парата для длительного пользования 1 каплю суспензии кле-
ток наносят на предметное стекло, высушивают 10 мин при
комнатной температуре, затем в течение 20 мин в токе холод-
ного воздуха от вентилятора, по возможности в темноте. Вы-
сушенный препарат фиксируют 95% этанолом, ополаскивают
холодным ФСБ, снова высушивают в токе холодного воздуха и
покрывают препарат 1 М забуференным глицерином. Еще од-
ним вариантом фиксации может быть приготовление мазков.
Осадок клеток, меченных флюорохромом, суспендируют в
10 мкл ЧСА (200 г/л), делают мазок, высушивают его на воз-
духе и покрывают глицерином. Такие препараты могут сохра-
няться в течение 1 недели. После темновой адаптации препара-
ты сравнивают по флюоресцентному и фазово-контрастному
изображению. Для оценки МИФ проверяют не менее 100 кле-
ток (лучше 200—300). Для подсчета результатов рекомендует-
ся использовать счетно-механические приспособления, особенно
если необходимо учитывать различные варианты МИФ. Типич-
ные картины МИФ представлены на рис. 87. В качестве поло-
жительных примеров можно рассматривать типы МИФ 1 и 2.
На рис. 1, а и б пункты флюоресценции расположены по
сферической поверхности, они проецируются в виде кольца
в экваториальной области. Для удобства оценки результатов
желательно, чтобы клетки были расположены по возможности
отдельно друг от друга, поскольку места контакта клеток от-
личаются особо сильной флюоресценцией, что создает труд-
ности для определения флюоресценции «Сар»-типа. В мазках
по сравнению с препаратами суспензий примерно вдвое боль-
шее число лимфоцитов обнаруживает флюоресценцию «Сар»-
типа. Равномерная флюоресценция мембран с диффузной
флюоресценцией клеток может свидетельствовать о начинаю-
щемся повреждении мембран или равномерном распределении
цитоплазматических и мембранносвязанных 1g при повышенной
проницаемости клеточной мембраны. Типы неспецифической
МИФ представлены на рис. 87 (фото 3, 4 и 5). Тип 3, редко
встречающийся и характеризующийся интенсивной цитоплазма-
тической флюоресценцией, по всей видимости, обусловлен ин-
тенсивным биосинтезом 1g. Диффузная флюоресценция, охва-
тывающая ядра и слабо выраженная в области мембран, ха-
рактерна для сильно разрушенных клеток, сквозь поврежден-
ные мембраны которых проходят меченые АТ. В ходе исследо-
ваний изоантигенов Н-2 на «убитых» клетках из асцитической
жидкости мыши было показано, что диффузная иммунофлюо-
ресценция обусловлена повышенной проницаемостью повреж-
денных мембран. Образование крупных флюоресцирующих час-
тиц (например, флюоресцирующих обломков клеток) является
неспецифическим феноменом, обусловленным недостаточной от-
мывкой препарата.
17*
259
Рис. 87. Типы (1—5) флюоресценции лимфоцитов человека.
Тип 1. Мелкоточечная флюоресценция клеточной мембраны, часто бывает видна в виде-
кольца флюоресценции в экваториальной зоне: 1а—полная нли частично прерванная
кольцевая флюоресценция («rings»), 16 — более трех отдельных гранул расположено по
Кругу («spots»). Тип 2. Флюоресценция клеточной мембраны в виде шапочки («caps»).
Тип 3. Цитоплазматическая флюоресценция. Тип 4. Диффузная флюоресценция. Тнп 5и
Грубы отложения флюоресцирующих частиц на клеточной мембране.
3.2.4. ВОЗМОЖНЫЕ ОШИБКИ И СПОСОБЫ ИХ УСТРАНЕНИЯ
З.2.4.1. АНТИТЕЛА И КОНЪЮГАТЫ
Основная причина ошибок — неспецифическое связывание ме-
ченых АТ с исследуемым объектом.
Неспецифическое флюорохромирование. Конъ-
югаты, содержащие много свободного флюорохрома, могут при
недостаточном разведении и плохой отмывке вызывать неспе-
цифическое окрашивание (контроль Г).
Неспецифическое связывание белков. Конъ-
югаты могут содержать посторонние белки, меченные флюоро-
хромом. Эти белки могут неспецифически откладываться на
клеточных мембранах. Рекомендуется проводить в таких слу-
чаях усиленную отмывку, если это не помогает, стараются по-
добрать оптимальное разведение конъюгата или адсорбировать
конъюгат ацетоновым порошком ткани [14]. Конъюгация белка
с флюорохромами может сама по себе вызывать изменение его
свойств (уменьшение растворимости, сдвиг изоэлектрической
точки в кислую сторону, изменение антигенсвязывающей спо-
собности), поэтому желательно в процессе конъюгации не
использовать чрезмерно высокие дозы флюорохрома. Неконъю-
гированные белки аутологической сыворотки могут отклады-
ваться сами по себе на объекте, если они не удаляются отмы-
ванием, на последующих этапах работы это может приводить
к неспецифическому связыванию конъюгата (см. раздел 3.2.5).
Неспецифическое связывание с Fc-рецепто-
рами. Fc-рецепторы обнаруживаются на В-лимфоцитах, акти-
вированных Т-лимфоцитах, гранулоцитах, моноцитах п даже
тромбоцитах (рис. 88). Они обладают способностью связывать
ИК и агрегированные Ig по Fc-фрагменту. ИК или измененные
Ig, присутствующие в используемых АС и конъюгатах, могут
неспецифически связываться с клетками, стимулируя картину
МИФ. Этого можно избежать, используя Р(аЬ)г-фрагменты.
Комплексы и агрегаты можно удалить ультрацентрифугиро-
ванием. Поскольку реагрегация протекает довольно быстро,
центрифугирование следует проводить перед каждым анализом.
Недостаточная специфичность антисыворо-
ток и антител АТ с посторонней специфичностью, т. е.
не к мембранным АГ (например, р2-микроглобулину), могут
искажать результаты опыта. Полезным может оказаться обога-
щение препарата специфическими АТ. Оно предполагает изго-
товление нерастворимого иммуносорбента, который инкубиру-
ют с АС и затем элюируют специфически связавшиеся АТ.
3.2.4.2. ТЕСТ-СИСТЕМА И ОЦЕНКА РЕЗУЛЬТАТОВ
При аккуратной работе можно ограничиться использовани-
ем ЗХ106 клеток на одну пробу. Для сепарированных лимфо-
цитов должно быть не менее 70% от теоретически ожидаемого
261
Рис. 88. Типы флюоресценции нелимфоидных клеток, предположительно обус-
ловленной наличием Fc-рецепторов.
а — МИФ+-гранулоцнт; б — МИФ+-моноцнт.
уровня, чтобы не слишком сильно искажать состав клеточных
популяций. Для предотвращения слишком быстрого «Capping»
или высвобождения мембранных АГ инкубацию можно прово-
дить при 4 °C. Такого эффекта можно добиться прибавлением
50 мг/л митомицина и 0,02 моль/л азида натрия в ФСБ. Суще-
ствует возможность того, что связанные с клетками антилим-
фоцитарные аутоантитела (в случае аутоиммунных заболева-
ний, иммунных дефектов и инфекций) могут обусловливать
ложноположительную МИФ. В физиологических условиях мо-
жет иметь место перераспределение мембранных АГ, поэтому
рекомендуется проводить оценку результатов МИФ немед-
ленно.
Моноциты и гранулоциты могут связывать 1g за счет Fc-ре-
цепторов и становиться МИФ-положительными клетками. Их
262
следует дифференцировать по морфологическим критериям ( ве-
личина, соотношение ядро — плазма, анализ в фазовом контрас-
те) и исключать при подсчете.
3.2.4.3. ОСНАЩЕНИЕ ОПТИЧЕСКИМИ ПРИБОРАМИ
Как уже отмечалось в разделе 3.2.2, для МИФ необходима
оборудование, используемое в люминесцентной микроскопии,
включая интерференционные фильтры и разделяющие зеркала.
Микроскопия в проходящем свете используется только в осо-
бых случаях, для этого служит интерференционный фильтр.
Самые небольшие изменения, такие как положение конденсора
и недостаточная центровка источника света, могут исказить ре-
зультаты анализа. Апертура конденсатора должна быть всегда
полной. Количество света, попадающего в препарат, можно
увеличить, если на верхнюю линзу конденсора нанести одну
каплю нефлюоресцирующего иммерсионного масла. Для пред-
отвращения ошибочных заключений необходимо сравнение ре-
зультатов флюоресцентного анализа с фазово-контрастным
изображением, это обязательное условие.
3.2.5. КОНТРОЛЬ СПЕЦИФИЧНОСТИ
Контроль специфичности на модельных объек-
тах.
А. Наилучший контроль специфичности проводят с исполь-
зованием стабильных клеточных линий с известными мембран-
ными маркерами, такими как RPMI Molt 4Т (Smlg-, Fc~, Сз~)
или Raji (Smlg-, Fc+, Сз+). Можно также использовать оха-
рактеризованные лейкемические клетки (например, В-лимфоци-
ты при хроническом лимфолейкозе) или клетки плазмацитомы.
Отмытые эритроциты АВ используют для оценки неспецифиче-
ского связывания в прямой и непрямой МИФ. Если имеется
более или менее чистый препарат исследуемого АГ, то, присо-
единив его к инертному носителю, частичкам агарозы или се-
фадекса, можно создать модель для проверки контроля специ-
фичности.
Контроль специфичности исследуемого
объекта (непрямой метод)
Б. Вместо 1g против АГ человека (например, кроличьего)
применяют нормальную сыворотку или у-глобулин кролика. Не-
обходимо с помощью этих реагентов исключить неспецифиче-
ское связывание сывороточных белков кролика, в особенности
у-глобулинов.
В. Реакция торможения I: перед инкубацией с 1g кроликов
против АГ человека проводят инкубацию с аналогичным 1g ко-
зы. Места связывания кроличьего 1g блокируются и происходит
торможение связывания антикроличьих у-глобулинов (2-е анти-
тела). Торможение имеет место в случае специфичности 1g
кролика против АГ человека.
263
Г. Реакция торможения II: перед инкубацией антикроличье-
го у-глобулина, меченного флюорохромом, проводят инкубацию
с немеченым антикроличьим у-глобулином. Этот способ при-
меняется для проверки специфичности вторых антител.
Д. Вместо меченого антикроличьего у-глобулина используют
антикозий у-глобулин. Эта операция применяется для поверки
неспецифического связывания с клетками меченых АТ, а также
для контроля неспецифических «сэндвичей» с немеченым анти-
человеческим Ig.
Е. Ig против АГ человека вообще не используется. Инкуба-
цию проводят исключительно с меченым антикроличьим у-гло-
булином (прямая проба со вторым АГ), исключая неспецифи-
ческое связывание меченых АТ или свободного флюорохрома.
Контроль специфичности исследуемого объ-
екта (прямой метод)
Ж. Вместо меченого Ig против АГ человека применяют ме-
ченую АС другой специфичности или нормальную сыворотку
(можно препарат у-глобулина) для проверки на неспецифиче-
ское связывание с мембранами.
3. Реакция торможения: перед инкубацией с меченым Ig
против АГ человека проводят инкубацию с тем же, но немече-
ным Ig для предотвращения связывания меченых АТ с Smlg
(поверхностный мембранный Ig).
Особо важное значение имеют контроли А, Б, В. Контроль
специфичности на модельных объектах должен по возможности
проводиться при оценке новых партий антисывороток, с тем
чтобы можно было получать сравнимые результаты. Контроль
специфичности на исследуемом объекте должен проводиться
при каждом анализе. Контроль Б проводят с использованием
одинаковых концентраций специфических и неспецифических
сывороток. Реакция торможения при постановке МИФ имеет
некоторые сложности, связанные со специфичностью взаимо-
действия эпитопов с паратопами.
Таблица 23. Количество МИФ-позитивных лимфоцитов в периферической
крови здоровых взрослых людей, %
Среднее значение по нескольким лабораториям Крайние значения
Общий Ig 21 16—28
IgC 7,1 4,0—12,7
IgA 2,2 1,0—4,3
IgM 8,9 6,7—13,0
IgD 6,2 5,2—2,8
X 13,9 10,0—18,6
X 6,8 5,0—3,2
1. IgE — МИФ-позитивные клетки встречаются крайне редко.
2. IgD и IgM часто встречаются на одной клетке.
3. Количество ^0+-лимфоцитов может быть завышено в связи с тем. что не уда-
ется полностью исключить лимфоциты с Fc-рецепторами или малые моноциты.
264
Таблица 24. Определение антител к мембранам островковых клеток
Реагенты:
1. Поджелудочная железа 12-диевных крыс Вистар
2. Коллагеназа (Worthington 170 ЕД/мг, Seromed Gmbh)
3. Раствор Хенкса, ЧСА
Этапы:
— выделение островков ограниченным перевариванием 20 поджелудочных
желез коллагеназой (7,5 г в 5 л раствора Хеикса);
— получение островковых клеток инкубацией островков в течение 30 мин
в безканальцевом растворе Хенкса с последующим механическим измель-
чением (5 сек);
— определение жизнеспособности клеток при помощи трипаиового синего;
— проведение концентрации клеток в суспензии до 1,2ХЮ6/мг (раствор
Хенкса в ЧСА);
Определение поверхностных антител к островко
в ы м клеткам:
— 0,1 мл суспензии клеток смешивают и инкубируют в течение 45 мин с
0,1 мл разведенной в соотношении 1 + 1 активированной комплементом
сывороткой пациента;
— однократная отмывка 5 мл раствора Хенкса с ЧСА (40 г/л);
— осадок в течение 30 мин инкубируют с 0,1 мл конъюгата;
— однократная отмывка 5 мл раствора Хенкса с ЧСА (40 г/л);
— приготовление препарата______________________________________
При постановке непрямого МИФ даже в условиях избытка
специфических тормозящих АТ могут обнаруживаться МИФ-
позитивные лимфоциты. Это позволяет предположить существо-
вание обмена специфических АТ на клеточной мембране в со-
ответствии с равновесным состоянием, при котором высоко-
авидные АТ обмениваются с АТ невысокой авидности.
Контроли Д и М могут быть полезными для выяснения при-
чин грубых неспецифических реакций в методе двойных АТ.
3.2.6. ВОЗМОЖНОЕ ПРИМЕНЕНИЕ МЕТОДА
В клинике МИФ наиболее часто применяется для диагностики
иммунодефицитных состояний при лимфопролиферативных за-
болеваниях. Средние данные, относящиеся к МИФ-позитивным
лимфоцитам, приведены в табл. 23. О различных способах
применения МИФ для исследования структуры, синтеза и ди-
Таблица 25. Определение нагруженных антителами бактерий в моче
Реагенты:
— ФИТЦ-антисыворотка к 1g
— 10 ммоль/л фосфатный буфер с 0,145 М NaCl с pH 7,4 (буфер А)
Проведение опыта:
— осаждают бактерии из 5 мл мочи, взятой из мочеточников центрифугиро-
ванием;
— бактерии дважды отмывают буфером А;
— инкубируют осадок бактерий в течение 30 мин при 37 °C с 0,1 мл мечен-
ной ФИТЦ аитииммуноглобулииовой сывороткой
— осадок дважды отмывают буфером А и растворяют в 0,1 мл глицерина,
забуференного фосфатами;
— готовят мазки из проб объемом по 50 мкл;
— оценивают флюоресценцию с обязательным контролем по фазово-контраст-
ному изображению
Оценка: препарат рассматривается как положительный, если более
20% бактерий обнаруживают краевую флюоресценцию
265
Рис. 89. Выявление антител к
мембранам островковых клеток
поджелудочной железы.
Инкубация сыворотки больного с ме-
ченными ФИТЦ IgG против АГ челове-
ка (препарат любезно предоставлен
д-ром Ziegler, Карлсбург).
намики мембранных АГ
[15] следует сделать не-
сколько коротких замеча-
ний. В принципе при нали-
чии соответствующих АТ
мембранные АГ можно вы-
явить на всех клетках.
Определенный клиниче-
ский интерес представляет
выявление мембранных ан-
тигенов на макрофагах,
плазматических клетках, гранулоцитах и опухолевых клетках,
для чего, помимо АГ, все шире используют лектины [1, 8, 15].
Оценка результатов: линейная или гранулярная
флюоресценция клеточных мембран.
Новые возможности возникают при комбинации МИФ с дру-
гими методами: твердофазной иммунофлюоресценцией [5],
электрофорезом с НДС в ПААГ. Если связать антиген с носи-
телем, то по аналогии с розеткообразованием (см. раздел 3.5)
можно идентифицировать и количественно определять антиген-
связывающие клетки (принцип флюоресцентной иммуноадге-
зии). Применение в клинике нашло также определение иммун-
ных комплексов при помощи клеток Raji благодаря наличию
в них рецепторов к комплементу и Fc-фрагментам, а также оп-
ределение аутоантител к антигенам клеточных мембран (пе-
чень, поджелудочная железа и опухоли) [2, 9] (табл. 24,
рис. 89).
Методы изучения локализации инфекции в мочевыводящих
путях по обнаружению в моче бактерий, «нагруженных» анти-
телами [4], недавно были заметно улучшены [10] (рис. 90,
табл. 25), что расширяет их применение.
3.2.7. ОЦЕНКА МЕТОДА
МИФ обладает не только обычными преимуществами иммуно-
гистологических методов, но также позволяет проводить коли-
чественные и качественные исследования клеточных мембран
в нативном состоянии. Однозначная локализация флюоресцен-
ции возможна благодаря комбинации фазово-контрастной и
флюоресцентной микроскопии. Методически более простая пря-
мая МИФ дает удовлетворительные результаты только в слу-
чае высокой чувствительности системы (препараты АТ с вы-
266
Рис. 90. Выявление нагруженных антителами бактерий в моче (Е. coli) при
помощи меченных ФИТЦ IgG против АГ человека.
сокими титрами, фотолюминесценция). Непрямая МИФ более
чувствительна, хотя и менее специфична, часто наблюдаются
неспецифические реакции, которые можно установить с по-
мощью продуманной системы контроля.
К числу преимуществ непрямой МИФ относится и то, что
меченные флюорохромом вторые антитела пригодны для са-
мых различных диагностических систем, использующих первые
АТ одного вида животного. Мечение флюорохромом обоих АТ
в непрямой системе способно повысить чувствительность до
максимума. Повышение чувствительности должно сопровож-
даться повышением специфичности. Желательно использовать
вместо полных AT F(ab)- или Р(аЬ)2-фрагменты. Если отсут-
ствуют АС к фрагментам Ig, можно использовать протеин А
золотистого стафилококка для блокады Fc-фрагментов [13].
Для определения В-клеток используют смесь АТ к легким це-
пям и и X для определения Т-клеток — моноклональные сыво-
ротки к Т-клеточным АГ [7]. Желательно проводить предвари-
тельный контроль новых серий антисывороток с модельными
системами, содержащими известные мембранные маркеры (ли-
нии клеток). Стандартизация МИФ и создание микровариантов
обусловливают растущую в клинике популярность МИФ при
подсчете В- п Т-клеток в диагностике первичных и вторичных
иммунодефицитных состояний, классификации лимфопролифе-
267
ративных заболеваний [7]. МИФ является основным методом
определения В-клеток, а наряду с Е-розеткообразованием так-
же и Т-клеток (при использовании моноклональных АТ). Осо-
бое внимание нужно обращать на моноциты, которые следует
рассматривать как ложноположительные клетки при выделении
лимфоцитов в градиенте фиколл — изопак. Если отсутствует
устройство для фазово-контрастной микроскопии, моноциты
можно обнаруживать по захвату частиц латекса или по окра-
шиванию на пероксидазу. Неживые клетки можно обнаружить
по прокрашиванию их ядра этидий-бромидом (добавление
0,5 мл раствора этидий-бромида в концентрации 2 мг/л в кон-
це инкубации) [40]. Антитела к АГ клеточных мембран пред-
ставляют все возрастающий интерес для диагностики аутоим-
мунных заболеваний печени (АТ к мембране гепатоцитов) и
некоторых форм сахарного диабета (АТ к мембранам остров-
ковых клеток поджелудочной железы).
3.2.8. СПИСОК ЛИТЕРАТУРЫ
1. Ben-Ze’ev A., Duerr A., Solomon F. et al. The outer boundery of the cytoskele-
ton: a lamina derived from plasma membrane proteins. — Cell 1979, 17,
859—865.
2. Dobersen M. J., Scharff J. E., Ginsberg-Felliner F. Cytotoxic autoantibodies
to beta cells in the serum of patients with insulin dependent diabetes melli-
tus. — N. Engl. J. Med., 1980, 303, 1493—1498.
3. Edwards A. J. Antilymphocyte serum produced by immunization with puri-
fied mouse thymocyte plasma membrane. — J. Immunol., 1979, 27, 225—239.
4. Gleckman R. A critical review of the antibody coated bacteria test. — J. Urol.
(Baltimore), 1979, 122, 770.
5. Gupta S., Schwartz S. A., Good R. A. Subpopulation of human T lymphocy-
tes. VII. Cellular basis of Concanavalin A induced T cell mediated suppres-
sion of immunoglobulin production by В lymphocytes from normal humans. —
Cellular Immunology, 1979, 44, 242—251.
6. Hoffmann R. A., Kung P. C., Hansen IF. P. et al. Simple and rapid measure-
ment of human T lymphocytes and their subclasses in peripheral blood. —
Proc. Natl. Acad. Sci. USA, 1980, 77, 4914—4917.
7. 1U1S/WHO Working Group Report on critical consideration of eight widely
used diagnostic procedures: the use and abuse of immunological test. Clini-
cal Immunology Newsletter, 1981, 2, 177—187.
8. Kieda C„ Roche A. C„ Delmotte F. et al. Lymphocyte membrane lectins.
Direct visualisation by the use of fluoresceinyl-glycosylated cytochemical
markers. — FEBS Letters, 1969, 99, 329—332.
9. Lernmark A., Baekkeskov S. Islet cell antibodies. Theoretical and practical
implications. — Diabetologica, 1981, 21, 334—350.
10. Naumann G„ Nimmich W„ Budde E. et al. Zur methodik der Nachweis anti-
korperbeladener Bakterien im Urin. — Dtsch. med. Wschr., 1981, 106, 1418—
1419.
11. Okudaira K-, Nakai H., Hayakawa T. et al. Detection of antilymphocyte anti-
body with two colour method in systemic lupus erythematosus and its hetero-
geneous specificities ahainst human T cell sibstes. — J. clin. Invest., 1979,
64, 1213—1220.
12. Pandolfi F., Strong D. M., Slease R. B. et al. Serological and functional cha-
racterization of human T cell subsets. — Clin. exp. Immunol., 1981, 43, 319—
328.
13. Ross G. D. Identification of human lymphocyte subpopulations by surface
marker analysis. — Blood, 1979, 53, 799—811.
268
34. Storch HZ. Immunofluorescenz-Fibel.— VEB Gustav Fisher Verlag, Jena,
1979.
,15. Tung J. K. S. K„ van Epps D. E. Identification of human polymorphonuclear
leukocyte specific marker using fluoresceinated fucose binding lectin from
lotus tetragonolobu seeds. — J. Clin. Lab. Immunol., 1979, 2, 171—176.
3.3. МЕТОД E-PO3ETOK
3.3.1. ПРИНЦИП МЕТОДА
Т-лимфоциты человека обладают свойством образовывать спон-
танные розетки с ЭБ. Рецептор ЭБ рассматривается как мар-
кер тимусзависимых лимфоцитов. Определение розеткообразую-
щпх клеток (РОК) стало стандартным методом анализа и пре-
паративного выделения Т-клеток [3]. Человеческие лимфоциты
и ЭБ смешивают в соотношении 1 : 50, иногда в присутствии
РОК-стабилизаторов, после инкубации подсчитывают долю
лимфоцитов, связавших три и более ЭБ (микроскопия).
3.3.2. МЕТОДИКА
-3.3.2.1 . МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: 0,15 М NaCl, среда Игла-МЕМ с pH
7,4, подведенная с помощью 1 М NaOH, гепарин без консер-
вантов (например, Gedeon Richter, Будапешт), раствор фи-
колл — визотраст с плотностью 1,077 г/мл, трипановый синий
0,2% в 0,15 М NaCl.
Также нужно иметь центрифужные пробирки на 10 мл (си-
ликонизированные), пастеровские пипетки, центрифуги, гема-
токритную центрифугу, камеру для подсчета клеток, микроскоп
с фазово-контрастной оптикой, ротор для ресуспендирования
клеток.
3.3.2.2. ПОЛУЧЕНИЕ ЛИМФОЦИТОВ
Для определения РОК необходимы лимфоциты, по возмож-
ности минимально загрязненные аутологическими эритроцитами
[11]. Для получения лимфоцитов подходит метод Бейюма (см.
также раздел 3.1): 2 мл гепаринизированной крови (100 ЕД
гепарина/мл) разводят 6 мл среды Игла-МЕМ и осторожно на-
слаивают в центрифужную пробирку на 2 мл сепарирующего
раствора. Центрифугируют 40 мин при 600 g. Лимфоциты скап-
ливаются в виде полосы белого цвета над сепарирующим ра-
створом. Их отсасывают пастеровской пипеткой и отмывают
средой Игла-МЕМ, центрифугируют 3 раза по 10 мин при 600 g
и еще один раз при 200 g, устанавливают плотность суспензии,
равную ЗХ106 клеток/мл.
269
3.3.2.3. ИНКУБАЦИЯ
Из трижды отмытых в 0,15 М NaCl ЭБ делают 0,5% суспензию
в среде Игла-МЕМ. Равные объемы суспензии лимфоцитов и
ЭБ (например, 0,5 мл) смешивают в центрифужной пробирке,
центрифугируют в течение 5 мин при 200 g и сохраняют при
4 °C в течение ночи.
3.3.2.4. ОЦЕНКА РЕЗУЛЬТАТОВ
Важным этапом является ресуспендирование осадка. Требуется
определенный опыт, чтобы при покачивании и вращении цен-
трифужных пробирок не разрушить розетки. В нашей лабора-
тории для ресуспендирования пользовались ротором (угол ка-
чания 45°, 6 об/мин, 15 мин). При помощи пастедовской пипет-
ки заполняют счетную камеру и в условиях фазового контраста
подсчитывают по меньшей мере 200 лимфоцитов с целью оп-
ределения РОК- При подсчете РОК учитывают лимфоциты
с 3 и более адгезировавшими эритроцитами. Жизнеспособность
клеток определяют окрашиванием 0,2% раствором трипанового
синего. Розетки фиксируют 0,8% раствором глутарового альде-
гида. В течение 1—2 недель розетки можно хранить в 1%
агарозе.
3.3.2.5. МОДИФИКАЦИИ МЕТОДА
Сокращение используемых объемов крови достигается сле-
дующей модификацией: в пробирку вносят 200 мкл среды Иг-
ла-МЕМ, 100 мкл суспензии лимфоцитов (ЗХЮ5 клеток) и
100 мкл 5% суспензии ЭБ, инкубируют 10 мин при 37 °C, ре-
суспендируют (ротор), центрифугируют 15 мин при 200 g и ос-
тавляют на ночь при 4°C. В среднем наблюдается 61 ±8%
РОК. Обработка эритроцитов АЭТ (2-аминоэтилизотиоурония
гидробромид) стабилизирует розеткообразование и дает до
80% розеток. Приготовление ЭБ, обработанных АЭТ: 1 мл
осадка отмытых ЭБ смешивают с 4 мл раствора АЭТ (0,402 г
АЭТ в 10 мл дистиллированной Н2О, pH подводят 4М NaOH
до 9,0). Смесь инкубируют 15 мин при 37 °C, затем отмывают
трижды в 0,15М NaCl. Осадок суспендируют в среде Игла-
МЕМ с 20% сыворотки эмбрионов коров до получения 10%
суспензии ЭБ. Максимально АЭТ — ЭБ могут храниться в те-
чение недели при 4 °C. Для получения розеток смешивают
0,5 мл суспензии лимфоцитов (2ХЮ6/мл), 0,25 мл сыворотки
эмбрионов коров и 6—8 капель АЭТ — ЭБ, центрифугируют
5 мин при 25 g и оставляют на ночь при 4 °C. ЭБ можно также
стабилизировать ЧСА [4]: 50 мкл суспензии лимфоцитов
(2Х106/мл) смешивают с 50 мкл 1% суспензии ЭБ и 20 мкл
ЧСА (20% в среде Игла-МЕМ) в маленьких стеклянных про-
бирках инкубируют 75 мин при 4 °C, затем добавляют 200 мкл
270
среды и ведут подсчет. Добавление 20 мкл смеси этидий-броми-
да с акридиновым оранжевым позволяет дифференцировать
живые (зеленое окрашивание) и неживые клетки (красное
окрашивание). Результаты, полученные этим методом, корре-
лируют на 95% с результатами, полученными при помощи ме-
тода моноклональных АТ (ОКТЗ). Для реакции торможения
розеткообразования используют суспензию лимфоцитов (4Х
Х106/мл) в среде Игла-МЕМ. В пробирку вносят 100 мкл сус-
пензии лимфоцитов, 200 мкл антилимфоцитарных АГ в разве-
дении 1 : 2000—1 : 256000 инкубируют 90 мин при 37 °C, добав-
ляют 100 мкл 0,5% суспензии ЭБ, инкубируют 10 мин при
37 °C, центрифугируют 5 мин при 200 g, выдерживают 90 мин
на ледяной бане. После осторожного ресуспендирования прово-
дят подсчет розеток. Минимальную концентрацию, при которой
отмечается торможение антилимфоцитарных АТ (концентра-
ция, вызывающая 25% снижение розеткообразования по срав-
нению с контролем) определяют графически.
Известен микрометод определения РОК в планшетах Тера-
саки. Под слоем вазелинового масла смешивают 2 мкл суспен-
зии лимфоцитов (8Х103 клеток), 1 мкл суспензии ЭБ, 3 мкл
2% раствора альбумина, центрифугируют и после 1 ч инкуба-
ции при 4 °C производят подсчет, используя в качестве фикса-
тора глутаровый альдегид.
3.3.3. ОБЩАЯ ОЦЕНКА МЕТОДА
Метод Е-розеток сравнительно прост, но ввиду плохой воспро-
изводимости трудно поддается стандартизации. Это создает
трудности при сравнении результатов, полученных разными
группами исследователей. Добавление сыворотки эмбрионов ко-
ров или ЧСА [6, 7] стабилизирует розетки и повышает их вы-
ход. Оптимальное соотношение лимфоцитов и ЭБ колеблется от
1 : 10 и 1 : 50. Автологичные эритроциты тормозят розеткооб-
разование. Подсчет «немедленных» розеток (сразу после цен-
трифугирования) практикуется очень редко. Фактором, опреде-
ляющим успех метода, является щадящее ресуспендирование.
Т-клетки можно определять с высокой корреляций относительно
метода Е-розеток путем подсчета АНАЭ (а-нафтилацетатэсте-
раза-позитивных клеток [5, 10]). Количество АНАЭ-позитив-
ных клеток коррелирует с Тр,-, p-глюкуронидаза-позитивными и
Ту-клетками [2]. При помощи моноклональных АТ можно се-
лективно блокировать рецептор ЭБ [8]. Этот рецептор встреча-
ется только у Т-клеток. У 30% фетальных тимоцитов одновре-
менно обнаруживают рецепторы ЭБ и комплемента. Некоторые
Другие клетки (гепатоциты, клетки опухолей, фибробласты и
Др.) также образуют розетки с ЭБ. Гепатоциты дают до 90—
100% Е-розеток. Вероятно, феномен образования Е-розеток
обусловлен наличием определенных компонентов мембран, от-
сутствующих у В-клеток. В-клетки образуют розетки с эрит-
271
роцитами мыши [6]; ДЭАЭ-декстран усиливает розеткообразо-
вание [11]. Использование КонА (10 мкг/мл) дает максимум
РОК через 48 ч. Можно автоматизировать подсчет РОК [2].
Ввиду простоты и широты применения метод Е-розеток следу-
ет рассматривать как стандартный метод.
3.3.4. СПИСОК ЛИТЕРАТУРЫ
1. Aristizabal L., Garcia-Moreno L. F. The effect of Concanavalin A on the
formation of different types of E-rosettes. — J. Reticuloendothel. Soc., 1981.
29, 117—125.
2. Brown R. A. A new methodfor semi-automated quantitation of E-rosettes
using a particle size analyzer. — J. Immunol. Meth., 1979, 29, 117—131.
3. Elliott В. E. A sensitive method for the separation of rosette forming cells. —
In: Immunological Methods/Eds. Lefkowits I. and B. Pernis. — New York:
Academic Press, 1979, 241—259.
4. Grunow R. Dissertation. Berlin, 1981.
5. Gudat F. Zur diagnostischen Bedeutung der T- und B-Rosetten-Bildung und.
der unspezifischen sauren Esterase in der Differenzialdiagnose der akuten und
chronischen Leukamien. — Schweiz, med. Wschr., 1981, 111, 1325—1330.
6. Hokland P., Ellegard J., Heron I. Human lymphocyte and mouse erythrocyte
rosettes optimization of the assay. — Scand. J. haematol., 1981, 26, 187—
194.
7. Hudson L., Hay F. C. Practical Immunology.-—Oxford: Blackwell Sci. Publ.,
1980, 301—302.
8. Ramoun M. Identification of a human T lymphocyte surface protein associated
with the E-rosette receptor. — J. exp. Med., 1981, 153, 207—212.
9. Menon M., Stefani S. S. DEAE—dextran and T-cell rosette formation. —
Immunol. Commun 1979, 8, 145—154.
10. Muller J. Nonspecific esterase in human Lymphocytes. — Int. Arch. Allergy,
1981, 6, 410—421.
11. Nowaczik M., Skopinska-Rozewska E. The influence of autologous erythrocy-
tes on active T-cell resetting with sheep red blood cells and on DNA synthe-
sis.— Arch. Immunol. Ther. Eexp., 1981, 29, 391—396.
12. Yoshizawa Y. A method for preservation of lymphocyte rosettes in agarose. —
Immunol. Commun., 1979, 8, 185—191.
13. Zielinski A., Oehling A. Cytochemical markers for T-ц and T-gamma. A com-
parative study. — Allergol. Immunopathol., 1982, 10, 115—120.
3.4. МЕТОД EAC РОЗЕТОК
3.4.1. ПРИНЦИП МЕТОДА
Метод EAC-розеток предназначен для выявления рецепторов-
к комплементу на поверхности мононуклеаров (лимфоциты,
моноциты). Эритроциты, сенсибилизированные амбоцептором и
«нагруженные» комплементом, связываются с мембраной мо-
нонуклеаров, содержащих рецепторы комплемента, с образова-
нием розеток. Рецепторы взаимодействуют преимущественно-
с компонентом СЗ (рис. 91). Доказательством того, что в дан-
ном случае розетки образуются при участии рецепторов ком-
племента, может служить изменение числа розеток при инакти-
вации комплемента или внесении АТ к комплементу в систему.
Количество лимфоцитов, образующих EAC-розетки, частич-
но коррелируют с количеством лимфоцитов, на. поверхности-
272
Рис. 91. Схема взаимодействия
различных компонентов прн
образовании ЕАС-розеток.
Лимфоцит С
рецепторами к-
Комплекс ЕАС комплементу
АГ эритроцита Амбоцептор Комплемент
антизрит-
роцитарной
сыворотки
которых обнаруживаются
мембранные иммуногло-
булины [2], т. е. преиму-
щественно с В-лимфоци-
тами. Возможно, что не
все В-лимфоциты облада-
ют рецепторами комплемента [7]. В отдельных случаях рецеп-
торы комплемента обнаруживаются также на Т- или «нуль»-
клетках [5]. Создание окончательной классификации субпопу-
ляций лимфоцитов представляется преждевременным ввиду то-
го, что разные методы исследований дают различные, частично'
перекрещивающиеся результаты [5, 7]. Метод ЕАС-розеток был
впервые предложен в 1968 г. Lay и Nussenzweig, позднее мно-
гие авторы стали применять его для исследования различий’
между популяциями лимфоцитов [3, 4, 8]. Для получения ком-
плекса эритроциты— АТ — компонент СЗ используются, как
правило, эритроциты барана и кроличьи гемолизины, иногда
IgM кролика. Можно использовать эритроциты человека и ан-
тисыворотку кролика. В качестве источника комплемента
берут мышиную или человеческую сыворотку (АВ-сыворотку).
Поиск лимфоцитов с рецепторами комплемента (ЛРК) чаще
всего проводят у мышей и у человека. У человека значительные
отклонения в количестве ЕАС-розеткообразующих лимфоцитов
наблюдается преимущественно при заболеваниях лимфатиче-
ской системы. Обычно доля ЛРК в периферической крови в та-
ких случаях снижена [1, 5]. Наиболее рациональной системой
для метода ЕАС-розеток оказалась система, состоящая из ЭБ,
АС кролика к ЭБ и комплемента мыши [6].
3.4.2. МЕТОДИКА
3.4.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: силиконизированные и центрифуж-
ные пробирки (25 и 10 мл), пастеровские пипетки, гематокрит-
ная центрифуга, центрифуга Т-23, ротор (10—20 об/мин), пик-
нометр и аналитические весы, или весы Вестфаля (Johannes-
Hammer, Лейпциг), камера для подсчета клеток (Вйгкег или
другой тип), седиментационная камера по Sayk (Dorenburg,
Берлин), микроскоп, водяные бани на 37°C и 56°C. Цельную
кровь барана, разведенную раствором Олсвера (1 + 1), сохраня-
ют до 8 дней в холодильном шкафу при 4 °C. Также необходимо
иметь кроличьи гемолизины против эритроцитов барана (Сак-
сонское сывороточное предприятие, Дрезден), свежую мыши-
ную сыворотку, 0,15 М NaCl, среду Игла, изотонический буфер’
18—990
273
для культивирования клеток (Институт иммунных препаратов
и питательных сред, Берлин), гепарин (Gedeon Richter, Буда-
пешт), визотраст 370 (Fahlberg List, Магдебург), декстрансуль-
фат (натриевая соль, отн. мол. масса 500) (Pharmacia, Шве-
ция). pH среды Игла и изотонического раствора для культиви-
рования клеток доводят до 7,3—7,4. Готовят разделяющий ра-
створ следующего состава:
100 мл раствора декстрансульфата в воде (60 г/л)
20 мл визотраста, 30 мл бидистиллированной воды
Устанавливают плотность 1,078—1,081 при помощи пикно-
метра и аналитических весов (или весы Вестфаля). Разделяю-
щий раствор хранят на холоду в темной склянке с притертой
пробкой.
3.4.2.2. ВЫДЕЛЕНИЕ ЛИМФОЦИТОВ
Гепаринизированную кровь в количестве 2—4 мл (100 ЕД ге-
парина на 1 мл) разводят средой Игла в соотношении 1:4 и
1 : 6 и осторожно наслаивают на 3—5 мл разделяющего рас-
твора в центрифужных пробирках на 25 мл, центрифугируют
20—40 мин при 600 g и комнатной температуре. Лимфоциты
скапливаются в виде белого слоя на границе разделяющего
слоя и среды Игла. Их отбирают пастеровской пипеткой и
трижды по 10 мин отмывают средой при комнатной темпера-
туре. Центрифугируют дважды при 600 g и 1 раз при 200 g для
уменьшения примеси тромбоцитов. Концентрацию суспензии
доводят до ЗХ'Ю9 клеток/л (среда Игла). Различные варианты
контроля дают в среднем 93% лимфоцитов, 4% моноцитов, 3%
гранулоцитов. Долю моноцитов можно снизить до 1 % и меньше
по методу Gu [10], однако это для метода ЕАС-розеток не име-
ет особого значения.
3.4.2.3. СЕНСИБИЛИЗАЦИЯ ЭРИТРОЦИТОВ БАРАНА
ЭБ, смешанные с раствором Олсвера, осаждают центрифугиро-
ванием и дважды отмывают ФСБ или 0,15 NaCl. Ресуспенди-
руют осадок в среде Игла и доводят гематокрит до 0,05. Сен-
сибилизацию ЭБ проводят инактивированием гемолизином кро-
лика, разведенным до субагглютинирующей концентрации изо-
тоническим буфером. Для получения оптимальных концентра-
ций амбоцептора проверяют несколько степеней разведения,
выраженных в титрах гемолизина. При титре гемолизина
1:12 000 и титре агглютининов 1:50 исследовали разведения
1 : 100, 1 : 200, 1 : 500 и 1 : 1000. Наилучшие результаты получа-
ются при разведении 1 : 500. При использовании инактивиро-
ванных кроличьих антисывороток к антигену Форссмана или
ЭБ необходимые разведения не совпадают. По данным различ-
ных авторов, степень разведения колеблется от 1 : 500 до 1 :
2000 [9]. Суспензию эритроцитов (гематокрит 0,05) смешива-
274
ют с равным объемом разведенного амбоцептора и инкубируют
в течение 30 мин при 37 °C на водяной бане. Суспензию эри-
троцитов в ходе инкубации многократно перемешивают. После
инкубации сенсибилизированные ЭБ дважды отмывают в изо-
тоническом буферном растворе, ресуспендируют в среде Игла
и устанавливают гематокрит 0,05.
3.4.2.4. ПОЛУЧЕНИЕ КОМПЛЕКСА АНТИТЕЛА —
ЭРИТРОЦИТЫ — КОМПЛЕМЕНТ
В качестве комплемента используют свежую сыворотку мышей,
разведенную средой Игла в соотношении 1 : 10. К 1 мл суспен-
зии сенсибилизированных эритроцитов (гематокрит 0,05) до-
бавляют 1 мл разведенной мышиной сыворотки.
Смесь инкубируют 30 мин при 37 °C, трижды отмывают ФСБ
и доводят средой Игла гематокрит до 0,005. Для контроля ак-
тивности комплемента можно использовать инактивированную
мышиную сыворотку (30 мин, 56°C).
3.4.2.5. ИНКУБАЦИЯ
Суспензию эритроциты — антитела — комплемент (ЕАС) с ге-
матокритом 0,005 (0,5 мл) в центрифужных пробирках объе-
мом 10 мл с 0,5 мл суспензии лимфоцитов инкубируют 15 мин
при 37°C на водяной бане, периодически перемешивая. Затем
центрифугируют 5 мин при 200 g и наконец суспендируют при
комнатной температуре в роторе (10 качаний в минуту) или
вручную (очень осторожно).
3.4.2.6. ОЦЕНКА РЕЗУЛЬТАТОВ
Клеточную суспензию (1—2 капли) наносят на сетку счетной
камеры, используя для этого пипетку с широким носиком. Пос-
ле наложения покровного стекла производят подсчет розеток»
нерозеткообразующих мононуклеаров (микроскопия). За РОК
принимают клетки, несущие три и более эритроцита на своей
поверхности. Часто встречаются розетки в виде морулы.
Однако ее не следует путать с группами агглютинировав-
ших эритроцитов, т. е. в центре морулы должна присутство-
вать ядерная клетка. Подсчитывают 200—300 клеток и вычис-
ляют процент розеток. В норме ЕАС-розетки составляют 10—
20% клеток (рис. 92). Для точной дифференциации РОК от
других клеток рекомендуется седиментация клеточной суспен-
зии в камере Sayk. Обычно достаточно морфологической оцен-
ки при окраске по Паппенгейму. Розеткообразующие моноци-
ты с уверенностью можно отдифференцировать гистохимически
по наличию пероксидазы или а-нафтилацетатэстеразы. Иногда
среди клеток, образующих ЕАС-розетки, моноциты не обнару-
живаются.
18*
275
Рис. 92. Лимфоциты с рецепто-
рами к комплементу.
а — лимфоциты; б — лимфоцит; в —
лимфоидная клетка.
3.4.3. ОЦЕНКА МЕТОДА
Наибольшая сложность
при использовании мето-
да ЕАС-розеток заклю-
чается в достижении эф-
фективного разведения
амбоцептора при сохра-
нении активности комп-
лемента. Вместо мыши-
ного комплемента можно
использовать комплемент
человека (свежая АВ-сы-
воротка). Активность
комплемента сохраняется
исключительно в свежих
или же непродолжитель-
ное время хранившихся
при —20 °C сыворотках.
Связывание комплемента
с комплексом ЭБ — АТ
наблюдается не только
в случаях сенсибилиза-
ции эритроцитов IgG. В
качестве доказательства
можно рассматривать от-
сутствие существенных
различий в числе розеток
(ЕАС и ЕА) при поста-
новке эксперимента с
эритроцитами человека,
анти -D- иммуноглобулина
и комплемента человека.
Метод ЕАС-розеток с
эритроцитами барана осо-
бенно требователен к
соблюдению намеченных температуры и времени инкубации, а
также частоты вращения пробирок с суспензией клеток. При
появлении большого числа розеток нельзя исключить возмож-
ность спонтанного розеткообразования Т-лимфоцитами с ЭБ.
Инкубация при 37°C и встряхивание суспензии тормозят обра-
зование спонтанных розеток и способствуют образованию ЕАС-
розеток (см. раздел 3.3). Образование ЕАС-розеток можно за-
тормозить предварительной обработкой лимфоцитов трипсином
276
или антилимфоцитарной сывороткой [7]. Эритроциты можно от-
делить от РОК с рецепторами комплемента воздействием обра-
ботанной папаином антисывороткой мыши к компоненту СЗ.
Розеткообразование не наступает, если EAC-комплекс обрабо-
тать антикомплементарной сывороткой.
При помощи метода EAC-розеток можно создать детализо-
ванную функциональную классификацию. Он также имеет
большое значение для оценки иммунного статуса при заболе-
ваниях лимфатической системы (изменение числа ЕАС- и спон-
танных розеток). Количество лимфоцитов с рецепторами к СЗ
не вполне соответствуют числу В-лимфоцитов, вероятно, только
часть В-лимфоцитов зависит от специальной функции СЗ-ре-
цепторов [7]. Исследования на мышах с удаленным тимусом
и облученных мышах показали, что одна из субпопуляций В-
клеток может потерять СЗ-рецепторы. На мембранах В-
лимфоцитов новорожденных мышей присутствуют преимущест-
венно рецепторы к иммуноглобулинам, рецепторы к СЗ появ-
ляются через несколько недель. По Parish [7], В-лимфоциты
делятся на 4 субкласса: Fc+CR+, Fc+CR-, Fc_CR+, Fc~CR~
(CR — рецептор комплемента). Хотя РОК представлены пре-
имущественно В-лимфоцитами, все же нельзя исключить, что
Т-лимфоциты или «нуль»-клетки несут рецепторы к компле-
менту [5]. Эти предположения подтверждаются обработкой лим-
фоцитов нейраминидазой.
Поскольку выделяемые различными методами субпопуляции
лимфоцитов частично перекрываются, результаты, полученные
методом EAC-розеток для определения В-лимфоцитов, не явля-
ются полными. Классификация субпопуляций должна прово-
диться с учетом спонтанных розеток, ЕА-розеток (Fc-рецепто-
ры) и мембранных иммуноглобулинов (иммунофлюоресценция).
3.4.4. СПИСОК ЛИТЕРАТУРЫ
1. Eggers G., Kruse Н. Spontanrosettenbildende und Fc-Rezeptortragende
Lymphozyten bei Erkrankungen im Kindesalter. — Folia haematol., 1979, 106,
14—21.
2. Ehlenberger A. G., McWilliams M., Phillips-Quagliata J. M. Igbearing and
complement-receptor lymphocytes constitute the same population in human
peripheral blood. — J. Clin. Invest., 1976, 57, 53—56.
3. Fauci A. S. Mechanism of corticosteroid action on lymphocyte subpopulations.
I. Redistribution of circulating T- and B-lymphocytes to the bone marrow. —
Immunology, 1975, 28, 669—680.
4. Huber H., Michlmayr G., Huber Ch. Immunological characterization of lym-
phoproliferative disorders by membrane markers. — Klin. Wschr., 1976, 54,
699—708.
5. Knapp IF. Charakteriesierung der Lymphozytenpopulation des Menschen. —
Dtsch. Med. Wschr., 1977, 102, 802—806.
6. Nowell P., Jensen J.', Winger L. T cell variant of chronic lymphoid leukaemia
with chromosome abnormality and defective response to mitogens. — Brit.
J. Haematol., 1976, 33, 459—468.
7. Parish C. R. Separation and functional analysis of subpopulations of lympho-
cytes bearing complement and Fc receptors. — Transplant. Rev., 1975, 25,
8. Sapir /., Frensdorff A. Isolation of lymphocyte subpopulations from rabbit
277
peripheral blood. Comparison of methods based on recovery of rosette-for-
ming cells and on recovery of Ig bearing cells from a digestible immunoad-
sorbent— J. Immunol. Meth., 1976, 10, 183—195.
9. Schroit A. J., K.edar E., Gallily R. A rapid and sensitive technique for the
detection of Fc-receptors on macrophages.— J. Immunol. Meth., 1976, 12,
163—170.
10. Yu D. T. Y. Human lymphocyte subpopulations: Giant human red blood cell
rosettes. — J. Immunol. Meth., 1977, 15, 67—76.
3.5. МЕТОД ИММУННЫХ РОЗЕТОК
3.5.1. ПРИНЦИП МЕТОДА
Лимфоциты могут агрегировать in vitro ксеногенные эритроци-
ты с образованием розеток или так называемых кластеров
(рис. 93). Впервые этот феномен был описан в 1964 г. у мы-
Рис. 93. Метод розеток: различные формы розеток.
а —кольцо эритроцитов вокруг центральной клетки не замкнуто; б — две расположенные
рядом лимфоидные клетки, одна из которых — розеткообразующая; в — розетка в форме
«морулы»; г — полярная розетка.
278
Рис. 94. Кинетика появле-
ния РОК в ходе иммунного
ответа швейцарских мышей.
1 — первичная реакция после
внутривенного введения 108 ЭБ;
2 — вторичная реакция после
внутонвенной бустер-инъекцнн
108 ЭБ на 45-й день после пер-
вичной иммунизации.
шей, иммунизирован-
ных ЭБ. Иммуноадге-
зия носит антигенспе-
цифический характер.
Адгезия эритроцитов
на поверхности цент-
ральной клетки проис-
ходит без участия ком-
племента, за счет ан-
тигенных рецепторов.
Эти рецепторы предс-
тавляют собой Ig-no-
добные структуры нг
мембране Т-клеток или
Ig на В-клетках. Прц
связывании АГ-детер*
минант с лимфоцита-
ми происходит перера-
спределение рецепторов на поверхности клетки, допускающее
полярное расположение эритроцитов вокруг центральной клет-
ки (см. рис. 93). У несенсибилизированных животных процент
лимфоцитов, взаимодействующих с определенными АГ-детер-
минантами, низок. В отношении ЭБ, как мультидетерминант-
ных АГ, количество РОК составляет обычно 10—103 на 106
лимфоцитов [2]. После иммунизации происходит 5—100-кратное
увеличение РОК (рис. 94, 95). Антигенспецифические розетки
отражают активность синтеза специфических АТ иммунокомпе-
тентными клетками. В зависимости от иммунологического ста-
туса малые лимфоциты, бластные, а также плазматические
клетки и макрофаги (вследствие пассивного отложения Ig)
могут выполнять роль РОК- Контакт лейкоцитов и эритроцитов
достигается низкоскоростным центрифугированием или просто
инкубацией.
Хотя при инкубации образование розеток протекает медлен-
нее, Т- и В-лимфоциты с низкой авидностью эффективнее во-
влекаются в розеткообразование, чем при центрифугировании.
Насколько АОК являются также и РОК, зависит от продолжи-
тельности и температуры инкубации. У мышей, иммунизиро-
ванных ЭБ, свыше 50% от числа клеток, образующих зоны ге-
молиза, несут рецепторы, обусловливающие образование розе-
ток. Если контакт лейкоцитов и эритроцитов происходит при
279
Рис. 95. Кинетика образо-
вания лимфоцитов с рецеп-
торами к ДНФ у морской
свинки после введения в
подушечки стоп ДНФ34-ЧСА,
суспендированного в пол-
ном адъюванте Фрейнда.
Первичная и вторичная реакции
после бустер-ииъекции одинако-
вой дозы АГ на 14-й день, по-
сле первичной иммунизации.
1 _ РОК в лимфатических уз-
лах, 2 — РОК. в селезенке.
4 °C в течение длитель-
ного времени, то об-
наруживается сущест-
венно меньше РОК и
совсем не обнаружива-
ется АОК, т. е. опреде-
ляются РОК, связыва-
ющие АГ за счет уже
существующих рецеп-
торов клеточной по-
верхности. Согласно исследованиям Charriere [2], у несенсиби-
лизированых мышей количество обнаруживаемых РОК возрас-
тает с ростом соотношения эритроциты/лейкоциты. У иммуни-
зированных животных при увеличении этого соотношения свы-
ше оптимального (8:1) число РОК не увеличивается. Основ-
ное количество исследований по РОК проводится с эритроцита-
ми барана, реже—курицы и голубя. Использование эритроци-
тов в качестве носителей растворимых АГ наряду с пассивной
гемагглютинацией открывает новые возможности для примене-
ния метода иммунных розеток [8]. Из всех вариантов метода
иммунных розеток наилучшим образом зарекомендовал себя
описанный ниже.
3.5.2. МЕТОДИКА
З.5.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: среда Игла или Паркера, ФСБ с pH
7,2, состоящий из 9 частей 0,15 М NaCl и 1 части фосфатного
буфера по Серенсену. Цельная кровь в растворе Олсвера (1 +
+ 1) сохраняет пригодность в течение 8 дней в холодильнике
(4°C). Нужны также центрифужные пробирки (10 мл), гемато-
критная центрифуга, камера для подсчета клеток (камера Bar-
ker).
3.5.2.2. ЭТАПЫ РАБОТЫ
Вся работа, за исключением подсчета розеток, проводится при
0—4 °C. Приготовление суспензии клеток из лимфоидных орга-
нов см. в разделе 1.5. Клетки трижды отмывают средой Игла
280
и доводят концентрацию до 107 живых клеток на 1 мл суспен-
зии. Трижды отмытые в ФСБ эритроциты при помощи гемато-
критной центрифуги переводятся в 5% суспензию (приблизи-
тельно 109 клеток на 1 мл).
Инкубация: 0,9 мл клеточной суспензии исследуемого орга-
на смешивают в стеклянной пробирке с 0,1 мл суспензии эри-
троцитов и инкубируют в течение ночи (16 ч) на ледяной бане.
Затем добавляют 4 мл охлажденной среды (разведение 1:5).
Суспензию осторожно перемешивают и вносят одну каплю при
помощи пипетки с широким концом в счетную камеру, после
наложения покровного стекла подсчитывают розетки. В качест-
ве РОК учитывают клетки не менее чем с 4 присоединившими-
ся эритроцитами. Число РОК относят к 103 или 10® лимфоид-
ных клеток. Число клеток, находящихся в суспензии, можно
уменьшить, если при подсчете будет иметься достаточно репре-
зентативное число лейкоцитов.
3.5.3. МОДИФИКАЦИИ МЕТОДА
т-POK связывают, как правило, меньше эритроцитов, чем В-
клетки, и легче диссоциируют при ресуспендировании. В связи
с этим необходимо фиксировать розетки глутаровым альдеги-
дом. После инкубации при 4 °C и осторожного ресуспендирова-
ния добавляют 2,5% раствор глутарового альдегида до конеч-
ной концентрации 0,6%, перемешивают и инкубируют 25 мин
при 4 °C. Центрифугируют 5 мин при 800g, надосадочную фрак-
цию отбрасывают.
Дифференцирование розеток от неспецифических скоплений
эритроцитов проводят при помощи окраски толуидиновым си-
ним (1 капля 1% раствора). Для выделения функционально
не измененных РОК (например, сепарация) диссоциацию не-
стабильных Т-розеток подавляют добавлением азида натрия до
0,05%.
3.5.4. ОЦЕНКА МЕТОДА
Метод иммунных розеток (МИР) в том виде, в каком он при-
веден, охватывает преимущественно Т- и В-лимфоциты, макро-
фаги в значительной степени исключаются из розеткообразо-
вания [2]. Повышение числа РОК указывает на активную им-
мунную антигенспецифическую реакцию, включающую
механизмы гуморального и клеточного иммунитета. Все же да-
леко не все лимфоциты, участвующие в иммунном ответе, обра-
зуют розетки. Зрелые плазматические клетки, утерявшие по-
верхностные рецепторы, некоторые популяции эффекторных
клеток клеточного иммунитета не образуют розеток. Относи-
тельно розеткообразования Т-хелперов имеются противоречи-
вые данные '[4, 7]. Наличие Т-РОК среди супрессоров, по всей
видимости, твердо установлено [6].
281
Клетки иммунологической памяти и некоторые субпопуля-
ции эффекторных клеток также в значительном проценте слу-
чаев образуют РОК [5]. Хотя иммунологическое значение РОК
не вполне ясно, МИР занимает прочное место среди простых
и чувствительных методов исследования иммунологических ре-
акций. Ошибка метода в пределах одной серии составляет 20%,
коэффициент вариации, обусловленный качеством материала
животных, колеблется в пределах 0,5—1. Издержки при поста-
новке МИР весьма незначительны, единственной длительной
процедурой является микроскопическое исследование. Источ-
ником ошибок может быть образование неспецифических розе-
ток, обусловленное Fc-рецепторами, существенно затрудняющее
определение РОК. Кроме того, следует обращать внимание на
то, что так же, как в методе спонтанных розеток (см. раздел
3.3.), в любой системе может оказаться высокая доля лимфо-
цитов с рецепторами к эритроцитам определенного вида, что
может маскировать действительное число антигенспецифиче-
ских клеток розеток.
3.5.5. СПИСОК ЛИТЕРАТУРЫ
1. Braganza С. М„ Stathopoulos G., Davies A. J. S. et al. Lymphocyte: erythro-
cyte rosettes as indicators of the heterogeneity of lymphocytes in a variety of
mammalian species. — Cell., 1975, 4, 103—106.
2. Charriere J., Dardenne M., Bach J. F. Antigen recognition by T-lymphocy-
tes.— Cell. Immunol., 1973, 9, 32—44.
3. Dlugonska H. The kinetics of rosette forming cells. Production in the primary
and secondary response to 2,4-dinitrophenyl human serum albumin. — Bull,
d. L’academie polonaise des sciences biol., 1980, XXVIII, 25—30.
4. Elliot В. E., Haskill J. S. Rosette forming ability of thymus derived lympho-
cytes in humoral and cell mediated immunity. II. Helper cell activity. — J.
Exp. Med., 1975, 141, 600—607.
5. Elliot В. E„ Haskill J. S., Axelrad M. A. Rosette forming ability of thymus
derived lymphocytes in cell mediated immunity. I. Delayed Hypersensitivity
and in vitro cytotoxicity. — J. Exp. Med., 1975, 141, 584—599.
6. Eardley D. D., Shen F. W„ Cone R. E. Antigen binding T cells: dose response
and kinetic studies on the development of different subsets. — J. Immunol.,
1979, 122, 140—145.
7. Kontiainen S., Andersson L. C. Antigen binding T cells as helper cells/Sepa-
ration of helper cells by immune rosette formation. — J. Exp. Med., 1975, 142,
1035—1039.
8. Lesavre P„ Bach J. F. Properties of bispecific rosette-forming cells. — Ann.
Immunol. (Inst. Pasteur), 1976, 127, 25—30.
3.6. ЦИТОТОКСИЧЕСКИЕ ЛИМФОЦИТЫ
3.6.1. ПРИНЦИП МЕТОДА
Цитотоксический тест используется для определения антигенов
клеточных мембран, поверхностных маркеров, АТ и эффектор-
ных клеток. Проведение теста основано на разрушении клеток-
мишеней под непосредственным воздействием лимфоцитов или
при помощи АТ, например, это имеет место в торможении
282
роста монослоя культур клеток. Эффект наблюдают визуально
или по переходу радиоактивной метки. Как эффекторные клет-
ки функционируют цитотксические Т-клетки (Тс), К-клетки,
цитотоксические макрофаги и индуцированные лектинами кил-
леры. Тс-клетки являются продуктами иммунного ответа, их
рецептор специфичен по отношению к индуцирующему АГ.
К-клетки действуют совместно с антителами (см. раздел 2.15).
Эту функцию могут выполнять различные клетки, но никогда
В-клетки. Соединение лимфоцита с клеткой-мишенью, адсорби-
ровавшей АТ, происходит по Fc-рецептору. К- и NК-клетки
иммунологически неспецифичны и не являются продуктами
иммунной реакции. Эти клетки обладают рецепторами к опухо-
левым клеткам, для своей деятельности они не нуждаются в
наличии АТ. К- и NК-клетки ввиду отсутствия Т- и В-маркеров
также называют «нуль»-клетками. Не исключено, что они явля-
ются предшественниками Т-клеток. Оба вида клеток различают
по их способности разрушать разные клетки-мишени. Индук-
ция эффекторных клеток происходит in vivo и in vitro, значе-
ние клонирования клеток постоянно возрастает [2]. В качестве
клеток-мишеней используют свежеприготовленные клетки
(тимоциты, спленоциты, эритроциты, макрофаги, опухолевые
клетки), а также клетки некоторых линий и стимулированные
митогенами спленоциты. Для радиоактивного мечения исполь-
зуется целый ряд изотопов. Радиоактивное вещество должно
равномерно распределяться по клетке независимо от метаболи-
ческой активности и клеточного цикла, высвобождаться при
необратимом повреждении клетки-мишени, должно быть до-
статочно активным и не реутилизироваться. Интенсивность ли-
зиса зависит также от числа эффекторных клеток, клеток-ми-
шеней, от соотношения этих двух типов и даже от объема смеси
и геометрии реакционного сосуда. Чаще используют высвобож-
дение метки из клеток-мишеней, но находит применение и мар-
кировка оставшихся жизнеспособными клеток-мишеней. Тс-, К-
и NK-клетки обладают сходными механизмами лизирования
клеток-мишеней. Синтезированные Т-клетками антигенспеци-
фические мембранные рецепторы способствуют контакту Тс-
клеток с клетками-мишениями за счет взаимодействия микрофи-
ламентов обоих типов клеток [4]. На процесс связывания ока-
зывают влияние двухвалентные ионы (Mg2+ и Са2+), метабо-
лизм и клеточный цикл. Этот эффект, вероятно, связан с
изменением проницаемости мембраны. Возможно, повреждение
мембраны происходит изнутри клетки-мишени под воздействи-
ем внешнего сигнала эффекторной клетки. Один лимфоцит
может реагировать со многими клетками-мишенями [9]. Су-
ществует целый ряд методических сложностей, связанных с вос-
производимостью результатов, взаимосвязью определенных суб-
популяций лимфоцитов с эффекторными клетками, подсчетом
числа эффекторных клеток на высоте иммунного ответа и др.,
приведенных в соответствующих обзорах [14, 18].
283
З.6.1.1. ОПОСРЕДОВАННАЯ КЛЕТКАМИ
ЦИТОТОКСИЧНОСТЬ
Традиционно различают тест на высвобождение 51 [Сг] и ми-
кроцитотоксический тест. Речь идет не о миниатюризации экс-
перимента, а о продолжительности инкубации. Поэтому пред-
почтительнее использовать термины «быстрый и «медлен-
ный» тест. В дальнейшем и те и другие термины будут исполь-
зоваться в равной степени.
3.6.1.1.1. «Быстрый» тест
Тест основан на маркировании клетки-мишени радиоактивным
Na2Cr2O4. 51 [Сг] представляет собой у-излучающий изотоп с
энергией квантов 323 кэВ и периодом полураспада 27,8 дня.
Большая часть клеток включает его в цитоплазму, где и про-
исходит восстановление до трехвалентного хрома. Высвобож-
дение хрома происходит только вследствие разрушения мем-
браны. Повторного включаения хрома в живые клетки не на-
блюдалось. Лимитирующим фактором для применения 51 [Ст]
является высокий уровень спонтанного высвобождения. Время
инкубации составляет 2—24 ч. Тест предназначен для оценки
цитолитической активности самых различных эффекторных
клеток. Количественная оценка результатов облегчается, если
построена кривая зависимости доза — эффект, по которой оп-
ределяют цитотоксичность при различных соотношениях эф-
фекторные клетки —клетки-мишени.
Известна очень чувствительная модификация реакции тор-
можения,- в которой смешивают меченые и немеченые клетки-
мишени, что приводит к конкурентному торможению. По кри-
вым титрования можно судить о мембранных АГ и об их взаи-
модействии с цитотоксическими лимфоцитами. На практике
применяют большое количество различных вариантов мечения
и высвобождения метки. Для длительной инкубации применя-
ют 3 [Н] -пролин, 125 [I]-дезоксиуридин, 3[Н]-уридин [10, 19].
3.6.1.1.2. «Медленный» тест
Определяют количество клеток-мишеней после инкубации с эф-
фекторными клетками. Этот эксперимент отличается от пробы
с высвобождением 51 [Сг] по количеству клеток-мишеней для
визуальной оценки (200—300), по способу культивирования, по
длительности инкубации (более 24 ч) и по форме представле-
ния результатов. Помимо вышеперечисленных изотопов, для
маркировки используют S6[Rb]Cl, "m[Tc]O4, 7S[Se]-метионин
и 3 [Н] -лейцин [1]. Мечение клеток-мишеней можно проводить
до и после инкубации с учетом свойств меченых соединений
[13].
Комбинация двух способов маркирования позволяет в од-
ном и том же эксперименте учитывать цитолитический и ци-
тотоксический эффекты. Продолжительное время инкубации да-
284
ет возможность исследовать влияние роста и деления клеток на
цитолитическую активность эффекторных клеток. Считается, что
в ’’медленном” тесте определяют зависимую от Т-клеток актив-
ность, которая в первые 24 ч совпадает с результатами, полу-
ченными в ’’быстром” тесте. На более поздних стадиях проис-
ходит реактивация нелитических вторичных Т-клеток, которые
сами по себе не обладают цитотоксичностью. При помощи мик-
роцитотоксического теста можно исследовать реакции клеточ-
ного иммунитета в течение значительно более длительного вре-
мени после аллоиммунизации, чем это позволяет сделать тест
с высвобождением 51[Сг].
3.6.1.1.3. Спонтанная клеточная цитотоксичность
Из довольно старого наблюдения, что лимфоциты нормальных
доноров с различной степенью эффективности реагируют с опу-
холевыми клетками-мишенями, развился метод характеристики
маркеров тимуснезависимых лимфоцитов. Естественные цито-
токсические клетки присутствуют в неодинаковых количествах
у различных биологических видов [8]. Как правило, это моно-
нуклеары, возможно промоноциты [12]. NK-клетки не способ-
ны к адгезии и фагоцитозу, у них нет рецептора СЗ и отсутст-
вуют мембранные иммуноглобулины. Наличие в этих клетках
Fc-рецептора несущественно для осуществления литического
процесса. В отличие от К-клеток контакт с клетками-мишенями
осуществляется не за счет антител. Обсуждался вопрос о раз-
личной авидности Fc-рецепторов К- и NK-клеток [10]. По-
скольку оба типа клеток встречаются в одной и той же субпо-
пуляции лимфоцитов, предполагают, что различия их активно-
сти обусловлены разными пусковыми механизмами. NK-клетки
мыши теряют активность при 37 °C. На процесс индукции in
vitro оказывает влияние характер культивирования (добавки
к питательной среде) [11]. Интерферон и индукторы интерфе-
рона стимулируют активность NK-клеток [7]. Как и в случае
Тс-клеток, культивирование и клонирование NK-клеток может
изменять их свойства [2]. NK-клеткам приписывают важную
роль в осуществлении противоопухолевого контроля [5].
3.6.2. МЕТОДИКА
На простых животных моделях можно поставить реакцию опо-
средованной клетками цитотоксичности, в которой после алло-
генной иммунизации in vivo и in vitro, например, после имплан-
тации химически индуцированной опухоли, определяют цито-
токсические лимфоциты. Клетки-мишени могут быть представ-
лены в виде монослоя или в виде суспензии.
Используют следующие среды:
Культуральная среда I: среда Игла-МЕМ с 0,02 М ГЭПЭС,
100 г телячьей сыворотки на 1 л, 100 000 ЕД пенициллина на
1 л, 0,1 г стрептомицина на 1 л.
285
Культуральная среда II: RPMI 1640 с 0,004 М глутамина на
1 л, 0,01 М ГЭПЭС, 0,01 М ИаНСОз, 0,00002 М меркаптоэтано-
ла, 100 000 ЕД пенициллина и 0,05 г стрептомицина на 1 л,
10% телячья сыворотка.
Сцинтилляционная жидкость: 4 г РРО, 0,2 РОРОР, 60 г нафта-
лина, 100 мл метанола, 20 мл этиленгликоля, диоксан до 100 мл.
3.6.2.1. ЦИТОТОКСИЧЕСКИЕ ЛИМФОЦИТЫ
ПОСЛЕ АЛЛОГЕННОЙ ИММУНИЗАЦИИ
З.6.2.1.1. Иммунизация
Используют самок мышей инбредных линий А (Н-2а) и СВА
(Н-2К) в возрасте 8—12 нед. А-мыши получают по 2Х107 кле-
ток селезенки СВА внутрибрюшинно. Контрольным животным
вводят равное количество сингенных спленоцитов. Для построе-
ния графика изменений числа цитотоксических клеток живот-
ных забивают группами по 4 особи через каждые 2 дня в те-
чение 26 дней [16].
3.6.2.1.2. Клетки лимфатических узлов
как эффекторные клетки
Обработку лимфатических узлов (ингвинальные, мезентериаль-
ные, аксиллярные и цервикальные) проводят при 5 °C, их очи-
щают от жировой и соединительной ткани и помещают в среду
I, измельчают в гомогенизаторе Поттера и переносят в центри-
фужные пробирки, профильтровав через найлоновую сетку.
Суспензию центрифугируют в течение 6 мин при 100 g, осадок
дважды отмывают средой I в тех же условиях. После 90-ми-
нутной инкубации в чашке Петри (удаление адгезирующих
клеток) подсчитывают число клеток при помощи трипанового
синего и доводят концентрацию суспензии до 30XIО6 клеток/мл.
3.6.2.1.3. Фибробласты как клетки-мишени
В качестве клеток-мишеней используют культуру фибробла-
стов, выделенных из новорожденных мышей СВА (или эмбрио-
нов). Обработку материала ведут при 37°С. У 4 животных
отрезают головы, конечности и удаляют внутренности. Тела
разрезают на мелкие кусочки и отмывают изотоническим ФСБ
до обесцвечивания промывных вод. Инкубируют 4 мин в растворе
трипсина (2,5 г трипсина/л). Надосадочная фракция содержит
обломки клеток и эритроциты. Трипсинизацию проводят в Эр-
ленмейеровской колбе на магнитной мешалке при 37°C. Коли-
чество трипсина должно приблизительно в 20 раз превосходить
количество ткани (по объему). Процесс прерывают каждые
10—15 мин и надосадочную фракцию, содержащую клетки,
фильтруют через найлоновый фильтр в центрифужные про-
бирки. Процесс повторяют 6—8 раз. Суспензию клеток центри-
фугируют 10 мин при 200 g и один раз отмывают культуральной
средой I. Выход составляет 35Х106 клеток.
286
Суспензию переносят в культуральные сосуды на 1000 мл
(сосуды Ру), добавляют по 100 мл среды I и инкубируют при
37 °C. Через 24 ч меняют среду. Через 3—5 дней можно сделать
первый пассаж клеток или собирать клетки для опыта. Для
этого среду сливают и ополаскивают клеточный «газон» ФСБ.
Затем наливают по 2 мл раствора трипсина и версена (ЭДТА)
(Chelaplex III, Berlin-Chemi, ГДР). Через 4—6 мин при 37°С
клеточный слой разрыхляется. Надосадочную фракцию отбра-
сывают, клетки смывают предварительно прогретой средой I и
подсчитывают. К 20Х106 клеткам добавляют 1,5 мБк 3 [Н] -ти-
мидина. Устанавливают концентрацию суспензии 2,5Х105/мл
и после интенсивного перемешивания разливают но 2 мл в
культуральные пробирки 160X16 мм. Пробирки кладут под
углом 20°. Через 3 дня образуется монослой клеток. Несвязав-
шуюся радиоактивность удаляют трехкратным отмыванием
ФСБ, затем вносят в пробирки по 1,5 мл среды I.
З.6.2.1.4. Проба на высвобождение 3[Н]-тимидина
В пробирки вносят по 1 мл аллогенных иммунных и синген-
ных обработанных лимфоцитов. Пробы ставятся в 3 паралле-
лях. Инкубируют 20 ч при 37 °C в косом положении. Сливают
надосадочную фракцию, трижды ополаскивают пробирки ФСБ
и суспендируют клетки в протозоле (New Englang Nuclear,
США). После переноса суспензии в стаканчики для сцинтилля-
ции измеряют радиоактивность на жидкостном сцинтилля-
ционном счетчике (например, LKB, Bromma, Швеция). Для
полноты данных необходимо определить нулевой контроль
(клетки-мишени в присутствии среды) и минимальную актив-
ность (прочно связанную с клетками-мишенями радиоактив-
ность, не высвобождаемую под воздействием эффекторных кле-
ток). Для этого культуру клеток-мишеней обабатывают трито-
ном Х-100 (неионный детергент; Ferak, Западный Берлин) или
подвергают многократному замораживанию и оттаиванию.
После центрифугирования в течение 10 мин при 200g проводят
процедуры, описанные выше. Внесение клеток в протозоль вы-
зывает сильную хемилюминесценцию пробы, которая пропадает
примерно через 30 ч. Хранение образцов на холоде этот проме-
жуток времени уменьшает. Образцы могут обладать различной
степенью гашения, т. е. по-разному снижать эффективность
подсчета распадов. В таких случаях рекомендуется построить
калибровочную кривую, параметры которой (коэффициенты
Уравнения 3-й степени) вводят в измерительное устройство.
Счетчик радиоактивности выдает данные, выраженные в числе
распадов в минуту (Desintegration per minute, DPM). Цито-
токсичность определяют по уравнению 7 (см. раздел 3.6.3). Ес-
ли проанализировать рост цитотоксичности (%) в зависимости
от времени, прошедшего после аллогенной иммунизации, то
первый положительный ответ регистрируется через 30 ч, макси-
мум клеточного иммунного ответа наблюдается к 5—6 дню и.
287
достигает 44%, затем кривая затухания антигенной стимуляции
снижается и после 26 дней сенсибилизация вообще не обнару-
живается.
3.6.2.1.5. Оценка метода, модификации
Маркировка фибробластов 51 [Сг] широко используется. Обра-
-ботка образцов в связи с этим сильно упрощается. Изменения
не представляются особенно трудоемкими. Маркировка часто
^оказывается неудачной в связи с потерей клетками способности
прикрепляться к стеклу, они перестают расти или, соответст-
венно, отделяться от стекла.
Попытки метить фибробласты в суспензии 51 [Сг] в течение
короткого периода времени (4 ч) оказались неудовлетворитель-
ными. Здесь следует отметить, что препараты хрома от различ-
ных изготовителей различаются по содержанию в них неактив-
ного хрома, которым и обусловлен вышеприведенный эффект.
Во многих моделях фибробласты являются оптимальными
клетками-мишенями. Для поддержания постоянного соотноше-
ния эффекторы: мишени (Э: Ж) клетки, закладываемые в
одну пробу, можно облучить дозой 30 Гр перед экспериментом.
Маркировка 3 [Н]-тимидином выживших клеток-мишеней после
инкубации с эффекторными клетками может повысить точность
данных о значении цитолитического потенциала, поскольку
включение 3 [Н]-тимидина строго пропорционально количеству
клеток-мишеней.
3.6.2.2. ЦИТОТОКСИЧЕСКИЕ ЛИМФОЦИТЫ
МЫШЕЙ С ОПУХОЛЯМИ
В СИНГЕННОЙ СИСТЕМЕ
Для работы используют солидную, индуцированную бензпире-
ном у мышей СВА, опухоль (ВР-ROS/Ol). Гистологически опу-
холь представляет собой недифференцированную полиморфно-
клеточную саркому.
З.6.2.2.1. Иммунизация
Самкам мышей СВА вводят внутримышечно в мышцы задней
конечности 0,5X10® клеток опухоли ВР. Через 10 дней извле-
кают лимфатические узлы.
3.6.2.2.2. Клетки лимфатических узлов в качестве
эффекторных клеток
Лимфатические узлы обрабатывают как было описано выше,
ингвинальные узлы с той стороны, где была привита опухоль,
отбрасывают вследствие инфильтрации опухолевыми клетками.
.Готовят суспензию ЮХ'10® клеток/мл.
288
3.6.2.2.3. Опухолевые клетки в качестве клеток-мишеней
В данном случае опухолевые клетки подготавливают так же,
как фибробласты (см. выше). Опухолевую ткань от трех жи-
вотных измельчают, трипсинизируют и культивируют. Клетки
20Х106 в 10 мл среды II переносят в культуральный сосуд на
200 мл и обрабатывают 10% СО2 в водонасыщенной атмосфе-
ре. Через 3—4 дня появляются отдельные островки адгезиро-
вавших опухолевых клеток, которые пассируют до получения
компактного монослоя клеточной культуры. Клетки обрабаты-
вают трипсином (1 г/л), отмывают, 5Х'1О6 клеток инкубируют
45 мин при 37°C с 3,7 мБк 51 [Сг]. Клетки трижды отмывают,
готовят суспензию из расчета 5Х'Ю5 клеток/мл.
3.6.2.2.4. Высвобождение 51[Сг]
К 0,1 мл клеток-мишеней добавляют различные количества
эффекторных клеток для получения различных соотношений
э — м. В качестве контроля используют клетки лимфатических
узлов неиммунизированных животных. Пробы инкубируют 6 ч
при 37 °C при постоянном встряхивании. После центрифугиро-
вания отбирают аликвоты по 0,5 мл и определяют радиоактив-
ность в гамма-счетчике. Проводят так называемый ”нуль-кон-
троль” (клетки-мишени) только со средой, для определения
уровня фона в начале анализа, в конце анализа и контроль
максимального высвобождения радиоактивности при лизисе
клеток тритоном Х-100 (см. выше).
Цитотоктичность рассчитывают по уравнению № 6 (см. раз-
дел 3.6.3). В случае вышеописанной модели при соотношении
э: м, равном 100: 1, наблюдается 36% цитолиз. Максимальная
высвобождаемая радиоактивность составляет 84%. Это означа-
ет, что 16% радиоактивности прочно связываются с клетками.
3.6.2.2.5. Микроцитотоксический тест с визуальной
оценкой результатов
Этот тест проводят на микропланшетах (Falcon Ка 3034, США).
Клетки-мишени не метят. В лунки вносят приблизительно по
500 клеток ВР в 20 мкл среды. Через 24 ч надосадочную фрак-
цию отсасывают, около 200 клеток при этом остаются адгези-
рованными к поверхности микропланшета. В 20 мкл вносят в
лунки 1X 10s лимфоцитов, что соответствует соотношению
500:1, закрывают камеру крышкой и инкубируют в атмосфере
с 10% СО2 в течение 48 ч. После двукратной отмывки фиксиру-
ют препарат метиловым спиртом, окрашивают раствором Гим-
за и подсчитывают количество оставшихся в живых клеток-
мишеней.
Результаты оценивают по уравнению 4 (см. раздел 3.6.3) с
той лишь разницей, что вместо импульсов подсчитывают клет-
ки. Значение цитотоксичности составляет около 45%.
19—990
289
3.6.2.2.6. Оценка результатов, модификации
Клетки ВР можно получать из асцитической жидкости после
внутрибрюшинного заражения животных. Если клетки не рас-
тут в полости брюшины, следует поместить кусочек ткани в
культуральный сосуд с сывороткой эмбрионов коров. Через не-
которое время образуется монослой клеток, которой можно
субкультивировать. Микроцитотоксический тест также проводят
с радиоактивной меткой. Измеряют радиоактивность культу-
ральной среды или адгезировавших клеток. Выявление живых
клеток можно упростить при помощи проекционного микроско-
па. В качестве клеток-мишеней можно использовать макрофаги
и тимоциты. По сравнению с фибробластами тимоциты более
устойчивы к цитолизу. Макрофаги можно использовать в каче-
стве клеток-мишеней после их обработки поли-Ь-лизином.
Предынкубация клеток-мишеней в течение 4 ч при 37 °C помо-
гает удалить связанный с мембранами избыток хрома. Наличие
зависимости между числом отмывок эффекторных клеток и сте-
пенью цитолиза указывает на существование сывороточных
факторов торможения цитолиза. Предварительной инкубацией
эффекторных клеток в течение 12 ч при 37°C большую часть
ингибиторов цитолиза можно удалить. Влияние различных ра-
диоактивных соединений на величину цитотоксичности опреде-
ляют при помощи клеток-мишеней, меченных двойной радио-
меткой. Для стандартизации теста с успехом используют хра-
нящиеся на холоде препараты клеток. Монослой клеток-мише-
ней удобнее в работе, чем суспензия клеток [3].
3.6.2.3. ЦИТОТОКСИЧЕСКИЕ КЛЕТКИ ПОСЛЕ
АЛЛОГЕННОЙ СТИМУЛЯЦИИ
З.6.2.З.1. Эффекторные клетки из смешанной
культуры лимфоцитов (СКЛ)
Селезенки респондеров и стимуляторов мягко разрушают
в гомогенизаторе Поттера в ФСБ, дают клеткам осесть в тече-
ние 8 мин, надосадочные фракции отбрасывают, дважды от-
мывают клетки. Клетки стимулятора 100Х106, суспендирован-
ные в 10 мл ФСБ, инкубируют в течение 20 мин при 37 СС
с 250 мкл митомицина С (Ferak, Западный Берлин); трижды
отмывают клетки. В культуральных флаконах на 200 мл сме-
шивают в 15 мл среды II 20Х106 клеток респондера и 10Х
ХЮ6 клеток стимулятора. Культивируют 5 дней при 37°C во
влагонасыщенной атмосфере с 10% СОг. Клетки отделяют от
стенок палочкой с резиновым наконечником, дважды отмывают
и готовят 3 суспензии с концентрацией 10Х106, 5Х Ю6 и 2,5х
X 106 клеток/мл соответственно.
В качестве контроля используют сингенные стимулирующие
клетки, обработанные митомицином С.
290
3.6.2.3.2. Обработанные митогенами клетки
в качестве клеток-мишеней
Селезенку стимулятора обрабатывают как было описано вы-
ше. Стимулирование проводят 100 мкг Кон (Pharmacia, Шве-
ция), затем 20X10® спленоцитов в 10 мл среды II инкубируют
в 100 мл сосудах в присутствии КонА. Клетки отделяют палоч-
кой с резиновым наконечником, суспензию клеток подслаивают
раствором фиколл/визотраст с плотностью 1,077. После 15-ми-
нусного центрифугирования при 200 g отбирают клетки, скопив-
шиеся над разделяющей фазой, и отмывают их. Для маркиро-
вания 10Х106 клеток используют 10 мБк 51 [Сг]. Готовят сус-
пензию клеток 1X 10®/мл.
3.6.2.3.3. Высвобождение 51 Сг
Цитотоксичность оценивают в микропланшетах с лунками V-
образной формы. Конечный объем составляет 100 мкл. Кривую
титрования строят для 3 различных соотношений эффекторы :
: мишени.
Инкубацию ведут 2 ч при 37 °C в вышеуказанных условиях.
Для статистической достоверности достаточно использования
двойных проб. Планшеты центрифугируют, отбирают по 50мкл
надосадочной фракции и измеряют радиоактивность гамма-
счетчиком. Цитотоксичность определяют по уравнению 6 (см.
раздел 3.6.3). На модели, использующей лимфоциты мышей
С57В1 анти-Balb/c, при соотношении э : м, равном 10 : 1, цито-
токсичность составляет 65%. По сравнению с результатами, ко-
торые наблюдаются при использовании эффекторных клеток,
полученных иммунизацией in vivo, индекс цитотоксичности при-
мерно в 3 раза выше. Это явление обусловлено СКЛ и вклю-
чением в процесс Т-клеток, ранее не участвовавших в реакции.
3.6.2.3.4. Оценка метода, модификации
Стимуляция эффекторных клеток in vitro в СКЛ весьма расп-
ространена, часто в дополнение к СКЛ используют также им-
мунизацию in vivo. Миниатюризация метода особенно удобна
при работе с эффекторными клетками человека. Известны мо-
дификации, в которых конечный объем пробы составляет всего
8 мкл [15]. Расчет цитотоксичности ведется не по отношению
к спонтанному высвобождению метки (клетки-мишени+ среда),
а по отношению к лимфоцитам обработанных сингенных жи-
вотных. Это обусловлено тем, что лимфоциты контрольных
животных также вызывают лизис клеток-мишеней. Этот лизис
может обусловливать дополнительные 15% высвобождения мет-
ки по сравнению со спонтанным высвобождением. Соотношения
э : м, равные 10:1, 5:1, 2,5: 1, были отобраны в качестве оп-
тимальных из исследованных соотношений 0,5:1—100:1. Они
позволяют воспроизводить подъем кривой, а также сравнивать
Различные экспериментальные модели у животных.
59*
291
3.6.3. РАСЧЕТ
Расчет индекса цитотоксичности возможен для самых различ-
ных модификаций экспериментов. Измеряют радиоактивность
надосадочной фракции и осадка.
Zim — радиоактивность надосадочной, фракции клеток-мишеней, инкубирован-
ных с сенсибилизированными лимфоцитами; ZKO — то же самое, но с контроль-
ными лимфоцитами; Z]m — радиоактивность нелизированных клеток-мншеней,
инкубированных с сенсибилизированными лимфоцитами; ZK0 — то же самое, но
с контрольными лимфоцитами; Zqes — общая радиоактивность пробы (осадок
и надосадочная фракция); ZMax — максимальная радиоактивность надосадоч-
ной фракции после обработки клеток тритоном Х-100; ZMin — минимальная
остаточная радиоактивность клеток-мишеней после обработки тритоном Х-100;
Zsr — спонтанное высвобождение радиоактивного изотопа (клетки=мншени-|-
+среда).
А: значение цитотоксичности (ZT) можно отнести к общей ра-
диоактивности и выразить в виде % увеличения (надосадоч-
ная фракция) или уменьшения (осадок).
ZT = Z)so х 100%.
ZGES
ZT = Zko.~-Zim - X 100%.
Оба значения идентичны.
Б: если исходить из цитотоксического индекса (ZI)
zi=4^>
ZKO
то в соответствии с преобразованием
(уравнение 1)
(уравнение 2)
(уравнение 3)
ZT = (1 — ZI) х 100%
% снижения или увеличения радиоактивности выражается в
форме
ZT е= Zk0_ х 100 %,
Zko
7Т _ ZIM — ZKo
Zges — ZKO
X 100%.
(уравнение 4)
(уравнение 5)
Уравнения 4 и 5 дают одинаковые результаты. Уравнение 4
пригодно для визуальной оценки микроцитотоксического теста,
когда вместо числа распадов учитывают число клеток.
В: если учесть, что не вся радиоактивность клеток-мишеней
высвобождается под воздействием эффекторных клеток, а толь-
292
ко некоторая величина Zmax (связанной с клетками остается
Zmin), то
ZT = '/мах-^КО Х 100% ’ (уравнение 6)
ZTt=~ ——1ГЛ X 1С0%. (уравнение 7)
гко — ’min
Уравнения 6 и 7 также дают одинаковые результаты.
Данные, получаемые при использовании различных формул,
отличаются друг от друга. Различие зависит в первую очередь
от величины ZKO (Zko) и Zmax (Zmin). При ZGes>ZKo уравне-
ние 5 переходит в уравнение 1, при Zce s=Zmax уравне-
ние 6 переходит в уравнение 5. При высоких значени-
ях Zmax и низких значениях радиоактивности уравне-
ния 1,5 и 6 дают близкие результаты; высокий %
спонтанного высвобождения метки с контрольными лимфоцита-
ми приводит к сходным данным, получаемым по формулам 5
и 6. Эти данные больше тех, которые рассчитываются по урав-
нению 1. Часто для подсчета ZT используют не значение радио-
активности, получаемое под воздействием контрольных клеток,
а значение, получаемое при спонтанном процессе (Zsr). Про-
блемы возникают в случае постановки обоих контролей и если
Zko>Zsr. Это имеет место при культивировании in vitro, когда
ZT контрольных клеток доходит до 15% [17]. В каждом случае
приходится приводить сведения относительно методов расчета,
использованных в цитотоксической реакции.
3.6.4. СПИСОК ЛИТЕРАТУРЫ
1. Brooks С. G. Studies on the microcytotoxicity test. III. Comparison of
75Se-Methionine with 3H Proline, Na2slCrO4 and 125J Deoxyuridine for pre
labelling target cells in long term cytotoxicity tests. — J. Immunol., 1978,
22, 23.
2. Dennert G., Yogeeswaran G., Yamagata S. Cloned cell lines with natural
killer activity. — J. Exp. Med., 1981, 153, 545.
3. Dorfman N. A., Civin С. I., Wunderlich J. R. Susceptibility of adherent ver-
sus suspension target cells derived from adherent tissue culture lines to cell-
mediated cytotoxycity in rapid 6lCr-releas assays. — J. Immunol. Meth., 1980,
32, 127.
4. Grimm E. A., Thoma J. A., Bonavida B. Mechanism of cell mediated cytoto-
xicity at the single cell level. II. Evidence for the first order kinetics of T cell
mediated cytolysis and for heterogeneity of lytic rate. — J. Immunol., 1979,
123, 2870.
5. Herberman R. B., Djeu J. D., Ray H. D. et al. Natural killer cells: Characte-
ristics and regulation of activity. — Immunol. Rev., 1979, 44, 43.
6. Jondal M„ Targan S. Spontaneous and lectin-dependent cellular cytotoxicity
by lymphocyte subpopulations against cell lines susceptible or resistent to
spontaneous cytotoxicity. — Clin. Exp. Immunol., 1978, 33, 122.
7. Lust J. A., Rumar V., Burton R. C. et al. Heterigeneity of natural killer cells
in mouse. — J. exp. Med., 1981, 154, 306.
8. McDermott R. P., Nash G. S., Merkle N. S. et al. Further evidence that anti-
body-dependent and cell-mediated cytotoxicity are rhediated by different pro-
cesses or cell types. — Immunology, 1980, 41, 439.
293
9. Matter A. Microkinematographic and electron microscopic analysis of target
cell lysis induced by cytotoxic T lymphocytes. — Immunology, 1979, 36, 179.
10. Neville M. E. Human killer cells and natural killer cells: distinct subpopu-
lations of Fc receptor bearing lymphocytes. — J. Immunol., 1980, 125, 2604.
11. Oshimi K, Kano S., SumiyaM. et al. Cytotoxicity of cultured mouse spleen
cells against natural killersensitive target cells. — Arch. Allerg. Appl. Immu-
nol., 1981, 64, 395.
12. Peter H. H„ Heidenreich IF. Spontaneous cell mediated cytotoxicity.—Can-
cer Immunol. Immunother., 1980, 8, 79.
13. Porzsolt F., Goldman S. E., Heit IF. Measurement of cell mediated cytotoxi-
city of post-labeling surviving target cells. — Tissue Antigens ly, 398, 1979.
14. Sanderson C. J. The mechanism of lymphocyte-mediated cytotoxicity. — Biol.
Rev, 1981, 56, 975.
15. Schnagl H. У, Boyle IF. A micro-version of the 61Cr release assay for cyto-
toxic lymphocytes. — J. Immunol. Meth, 1978, 24, 79.
16. Schulze H. A., Brock J., Siegl E. et al. Zytotoxische Lymphozyten nach Allo-
imunisierung in vivo: Kinetik, Reproduzierbarkeit und Spezifitat nach primar
Stiemulirung. — Allergie. Immunol, 1980, 26, 310.
17. Schulze H. A., Brock J., Siegl E. Zur Spezifitat der Zellvermittelten Zytotoxi-
zitat: I. Zytotoxische Reactionen im H-2 System nach allogener Immuniesie-
rung in vivo. — Allergie Immunol, 1982, 28, 109.
18. Sela M. The antigens. — New York: Academic Press, 1979, vol. V.
19. Smith G., Nilklin S. 3H-uridine uptake by monolayers as a terminal label in
an in vitro cell-mediated cytotoxicity assay. — J. Immunol. Meth, 1979.
25, 265.
20. Weir D. M. Handbook of experimental immunology (3-rd Ed.). — Oxford:
Blackwell Scientific Publications, 1978.
3.7. РЕАКЦИЯ БЛАСТТРАНСФОРМАЦИИ
ЛИМФОЦИТОВ
3.7.1. ПРИНЦИП МЕТОДА
Активация лимфоцитов под воздействием ФГА послужила в
1960 г. толчком для разработки метода бласттрансформации,
при помощи которого многочисленные исследователи оценива-
ют in vitro клеточный иммунитет, в том числе реакции В-кле-
ток. Методическая сложность РБТЛ породила самые разнооб-
разные модификации, которые в свою очередь затрудняют
интерпретацию результатов, полученных разными исследовате-
лями. Контакт лимфоцитов, несущих соответствующий рецеп-
тор с антигеном или митогеном, вызывает так называемую ре-
акцию бласттрансформации. В соответствии с различными
биологическими процессами, протекающими в трансформиро-
ванных клетках, известно много физико-химических и биологи-
ческих методов. Рекомендуется использовать вариант РБТЛ,
основанный на включении меченного тимидина [7]. Измерение
основано на вторичном вовлечении первично-неактивированных
лимфоцитов в реакцию вследствие продукции митогенных фак-
торов (интерлейкины). Этим объясняется причина продолжи-
тельного времени культивирования (ожидание эффекта усиле-
ния). Тест позволяет исследовать 6 основных моментов:
294
— определение специфической сенсибилизации пациентов
к чужеродным АГ или аутоантигенам, соответствующий анти-
ген исследуют in vitro;
— вследствие внесения митогенов in vitro клетки подверга-
ются неспецифической стимуляции; известно довольно много
селективно действующих Т- или В-митогенов; митогены доказы-
вают способность лимфоцитов к стимуляции; до настоящего
времени не ясно, какие субпопуляции стимулируются теми или
иными митогенами;
— обычная реактивность лимфоцитов может изменяться под
воздействием внешних факторов (условия культивирования или
добавки);
— если к моменту отбора пробы крови иммунная система
испытывает нагрузки (инфекция, отторжение трансплантата),
то большое число лимфоцитов оказываются реактивными, т. е.,
помимо прочего, способными к бласттрансформации in vivo.
Если провести немедленное мечение клеток, отмечается повы-
шенное включение метки в лимфоциты. Этот феномен не толь-
ко указывает на острый процесс, но также может существенно
искажать результаты некоторых тестов;
— пролиферативный ответ на активаторы лимфоцитов мож-
но использовать в качестве индикаторной системы для изуче-
ния регуляции иммунитета. Для этого можно осуществлять
совместное культивирование в аутологичных и аллогенных си-
стемах. В последнем случае исследуемая популяция клеток
рассматривается как индикаторная (респондеры) или как мо-
дулирующая (реактанты);
— определение первичной реакции на аллогенные клетки в
форме СКД (см. раздел 3.8).
3.7.2. МЕТОДИКА
З.7.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы нужно иметь стерильное рабочее место, оснащен-
ное УФ-лампой, стерильные силиконизированные закрываю-
щиеся сосуды. Возможны 2 варианта культивирования (табл.
26).
Необходимы также ФГА (Wellcome, Великобритания), причем
следует избегать замораживания и оттаивания, КонА (Phar-
macia, Швеция), митоген лаконоса (PWM) (Grand Island Biol,
corp, США), специфические антигены (в зависимости от целей
исследования), 3 [Н]-тимидин-Сб (1 ТБк/мМ) (Чешский инсти-
тут по исследованию, получению и применению изотопов);
0.15М NaCl, ТХУ 5%, метанол; РРО, POPOP (New Engl.
Nuclear, США), раствор Тюрка, тридистиллированная вода,
раствор сцинтилятора (0,1—0,2 г РОРОР, 5,0 г РРО, толуол
до 1000 мл). pH среды (табл. 27) перед точной установкой
295
Таблица 26. Оснащение для двух различных вариантов РБТЛ
Вариант I
Вариант II
Центрифуга (см. раздел 3.1)
Камера
Микроскоп
Шуттель-аппарат
Пипетки
Автоклав
Сухожаровой шкаф
Микрошприцы
СОа-инкубатор
Стерильные микропланшеты с
U-образными лунками (напри-
мер, Flow Lab., ФРГ)
Устройство для сбора клеток
(например, Skatron, Норвегия)
Термостат
Полипропиленовые пробирки
(автоклавируемые) с крышкой
(VEB Polyplast, ГДР)
Штатив для установки проби-
рок с культурой, расположен-
ный под углом 15°
Доска с гвоздями
Кружки фильтровальной бума-
ги FN 7 диаметром 2,2 см
(VEB Spezialpapierfabrik, ГДР)
Жидкостный сцинтилляционный счет-
чик (например, LKB Wallac 81 000,
Швеция)
объема подводят 1 М NaOH до 7,4 (37°C). Рекомендуется
иметь смесь антибиотиков, расфасованную на небольшие пор-
ции и хранящуюся в замороженном состоянии. Если использу-
ется СОг-инкубатор, к среде ие добавляют ГЭПЭС. Нужно
иметь также гепарин без консервантов (Gedeon Richter, Вен-
грия), среду Игла-МЕМ (SIFIN, ГДР); L( + )-глутамин
(Ferak, Зап. Берлин), ГЭПЭС (№2-гидроксиэтилпиперазин-№2-
этансульфокислота) (Ferak, Зап. Берлин), стрептомицин, пени-
циллин G; инактивированную сыворотку АВ или сыворотку
эмбрионов коров (Difco, США).
Таблица 27. Среда для РБТЛ
540 мг среды Игла-МЕМ
15 мг глутамина
238 мг ГЭПЭС
Тридистиллированная вода до 50 мл
Сохраняется при 4 °C в течение од-
ной нед
Готовая среда
+ 50 мкг стрептомицина
100 ME пенициллина/мл
+ 20% сыворотки
296
3.7.2.2. МЕТОДИКА
Перед проведением РБТЛ выбирают подходящие антигены и
митогены, а также адекватную модификацию теста. Мы ис-
пользовали в основном 3 модификации теста (рис. 96). Следу-
ет учитывать, что клетки различных животных не всегда удоб-
ны в обращении в такой же степени, как лимфоциты перифери-
ческой крови человека. Необходимое количество крови очень
невелико. После разведения образца раствором Трюка подсчи-
тывают абсолютное число лимфоцитов в счетной камере. В од-
ном флаконе находится 5ХЮ4 лимфоцитов. Для получения кле-
ток в градиенте плотности (см. раздел 3.1) с целью проведения
РБТЛ требуется примерно в 5 раз больше крови.
Известно, что для оптимальной активации необходимо на-
личие небольшого числа макрофагов (моноциты), поэтому не
рекомендуется использовать высокоочищенные препараты лим-
фоцитов.
Культивирование и обработка культур. При всех вариантах
(см. рис. 96) культивирование проводят в объеме 200 мкл.
В идеале следует лимфоциты цельной крови и препараты лим-
фоцитом подвергать аналогичной обработке для РБТЛ. Опти-
мальное время культивирования при использовании митогенов
составляет 3 дня (для лимфоцитов цельной крови 4 дня), при
использовании антигенов — 7 дней. При культивировании в про-
бирках (см. табл. 26, вариант II) используют шуттель-аппарат
для ресуспендирования после внесения метки в культуру и пе-
ред сбором клеток. Процесс мечения прерывают нанесением
100 мкл суспензии клеток на кружки фильтровальной бумаги.
После высыхания отмывают фильтры (табл. 28), по 20—
80 штук в одном стакане. Стакан многократно слегка покачи-
вают, после чего декантируют жидкость.
При использовании микропланшетов и устройств для сбора
клеток стадии отмывки ТХУ и метанолом опускают и ограничи-
ваются только отмывкой водой.
Тест с суппрессорными клетками. До настоящего времени
не разработано общепризнанной тест-системы для работы
с супрессорными клетками. На рис. 97 и 98 представлены два
возможных варианта постановки опыта, основанные на воздей-
ствии на интенсивность синтеза ДНК-
Таблица 28. Отмывка бумажных фильтров с меченым осадком клеток
(вариант II)
Смесь для отмывки Продолжительность отмывки (мин) Число отмывок
1. 0,15 М NaCl 5 2
2. 5% ТХУ 5 2
3. Метаиол 5 1
297
Г епаринизиро-
ванная кровь
(50-100 ЕДА»л)
Нативная кровь,
дефибринированная на
стеклянных гранулах
Спленоциты или
клетки лимфатических
узлов мыши
FV
Разведение
1:3 (среда Игла),
лодслаивание
лимфопрепом (пл.1,077),
центрифугирование
(30 мин,250 д),
сбор лимфоцитов
Размельчение
пинцетом
Перлоновое сито
Двухкратная отмывка
средой Игла
(10 мин.250g)
Двухкратная отмывка средой Игла
(Юмин.ЗООд)
Подсчет
абсолютного
числа
лимфоцитов
Среды с
20% сыворотки
АВ или
автологической
сыворотки
Среда с
добавлением
20% сыворотки
АВ
МО 6/м л
Культуры (200 мкл)
2,5*10 5
По 100 мнл
на бумажные
полоски
| Началка
+ ФГА,антигены,
контрольное
культивирование
(3-7 дней)
5xio 6
Среда с
добавлением
20% сыворотки
плода коровы
16 ч,
37 кБк(25 мкл),
^Н-тимидин
'шивание|
Камера для сбора клеток
Измерение
на LSC, 1 мин
Распечатка данных о
радиоактивности
Рис. 96. Этапы работы при различных модификациях РБТЛ.
V — цельная кровь; FV — препарат лимфоцитов, полученный из градиента фи-
колл — визотраст; LSC — жидкостный сцинтилляционный счетчик; одной звездоч-
кой обозначен метод с использоеванием пробирок (вариант II); двумя звездочка-
ми— метод с использованием устройства для сбора клеток (вариант I); кружоч-
ком — отмывка (см. табл. 28).
97
I 3 I
3 дня 37 кБк Н-тимидин 'Здня
I 16 ч I
Расп/мин РаспЛиин
А В
98
Рис. 97. Вызываемая КонА супрессия поддающихся стимуляции ФГА авто-
логических лимфоцитов из цельной крови (V) или выделенных в системе
фиколл-визотраст (FV); концентрации клеток показаны на рис. 96.
расп/мин В
Торможение (%)=1— д XI00 (другие авторы применяют более
раСП/МИН Л
высокие дозы КонА).
Рис. 98. Индекс супрессорных клеток (ИСК) для выявления короткоживущих
супрессорных клеток [,1].
расп/мин В
ИСК“ расп/мин А ’
3.7.2.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Высушенные фильтры помещают в стаканчики с 10 мл толуо-
лового сцинтиллятора и проводят измерение радиоактивности
в жидкостном сцинтилляционном счетчике в диапазоне энер-
гии квантов 1,26—20 кэВ с двумя поддиапазонами (1,26—
6,35 кэВ и 4—20 кэВ). Эффективность счета составляет только
2%, ошибка измерения— 16%. При любой модификации можно
ограничиться подсчетом величины имп/мин, не проводя пере-
счет на расп/мин. Исходная активность для нестимулирован-
ных лимфоцитов человека не превышает 200 имп/мин, а для
лимфоцитов, выделенных в системе фиколл/визотраст, —
1000 имп/мин (см. рис. 96). У мышиных клеток (при использо-
вании сыворотки эмбрионов коров) эта величина не превыша-
ет 2000 имп/мин. После стимуляции ФГА нормальные лимфоци-
ты цельной крови человека включают метку до величины свы-
ше 104 имп/мин, а лимфоциты, полученные в системе фиколл/
визотраст, — 5Х104 имп/мин. Для клеток лимфоузлов мыши
после стимуляции ФГА характерны величины порядка 104имп/
/мин. В случае острых заболеваний, для которых характерны
299
повышенные значения включения метки, после 3-дневного куль-
тивирования использование индекса стимуляции (ИС) не ре-
комендуется.
имп/мин стимул.___
~ имп/мин контроль
Поскольку не удается уловить неизменно высокий ответ на
антигенный стимул, бывает достаточно данных, выраженных
просто в имп/мин. Для некоторых болезней характерно измене-
ние уровня выживаемости клеток (например, при лейкемиях до
400% по сравнению с 70%, характерными для 3-дневного куль-
тивирования у нормальных людей). В этих особых случаях ре-
комендуется соотносить реактивность клеток с их числом.
Реактивность _____ИМП/мИНпробы ИМП/мИНКонтроля
клеток ~ % выживших клеток *
Толуоловый сцинтиллятор можно использовать многократно
с условием, что его радиоактивность не будет превышать
50 имп/мин на 1 стакан (10 мл). Тщательно отмытые стаканы
заполняют свежим сцинтиллятором и перед использованием оп-
ределяют уровень фона. Данные о суппрессорах см. рис. 97 и
98. В цельной крови индуцированная КонА супрессия достига-
ет 50%, при использовании препарата лимфоцитов, полученных
в системе фиколл/визотраст, — 24%. У нормальных людей эти
данные сильно колеблются, их интерпретация возможна только
в динамике. Индекс супрессорных клеток у здоровых людей со-
ставляет около 2,45.
3.7.3. ОЦЕНКА МЕТОДА
Для выявления специфической сенсибилизации желательно
иметь возможно более чистый АГ с большим содержанием бел-
ка. Необходимо исключить воздействие примесей. Спектр воз-
можных применений простирается от выявления аллергии до
выявления сенсибилизации к простейшим, грибкам, бактериям,
вирусам или определения блокирующих сывороточных факто-
ров, изучения действия фармакологических препаратов in vitro
и определения органоспецифических антигенов. Последний ме-
тод оказался не столь успешным, поэтому для диагностики
опухолевых заболеваний следует отказаться от РБТЛ в пользу
определения лимфокинов. Причина этого состоит в том, что
невозможно исследовать все процессы, протекающие в культуре
клеток в течение 7 дней. При использовании бактериальных,
вирусных или грибковых антигенов необходимо помнить, что
многие из них обладают митогенным действием [6]. Особый
областью применения РБТЛ является оценка митогенного дей-
ствия. Наиболее часто используемые митогены Т-клеток — ФГА
и КонА, В-клеток и Т-клеток — PWM и протеин А '[2]. В кли-
нике исследование митогензависимой реактивности применяется
3 00
Таблица 29. Условия постановки РБТЛ
Постоянно встречающиеся ограничения:
— исходный материал;
— неестественные условия содержания лимфоцитов;
— биологические ритмы;
— величина отбираемой пробы у пациента;
— установление «нормальных значений» у доноров;
— прочие оптимальные условия, как при других пробах в реакциях кле-
точного иммунитета;
Иногда встречающиеся ограничения:
— функция митогенреакгивных клеток (?);
— намеренное или случайное воздействие терапевтических мер;
— многофакторные причины снижения митогенной реактивности;
— влияние иммунорегуляторных лимфоцитов или моноцитов (?);
— причины различной выживаемости клеток;
— артефакты, связанные с приготовлением препаратов клеток и выбором
оптимальной концентрации;
— применимость данных модельного эксперимента;
— определение минимального числа клеток для достижения специфического
эффекта;
— временные эффекты (период рефрактерности);
— планирование времени для проведения наблюдений.
при оценке первичных иммунодефицитных состояний. В ряде
случаев этот анализ проводят при вторичных иммунодефицит-
ных состояниях [8]. Оценка результатов митогенной стимуля-
ции представляется весьма непростым делом. Так, обусловлен-
ная терапией ’’нормализация митогензависимой реактивности”
совсем не обязательно развивается параллельно с улучшением
клинической картины {10]. Наиболее выраженная корреляция
с клинической картиной отмечается в отношении реактивности
к аллогенным клеткам (см. раздел 3.8). Возможности теста для
клинической диагностики представлены в табл. 29.
В последние годы все чаще используют для РБТЛ лимфо-
циты цельной крови. Из 1—2 мл крови можно получить свыше
30 культур. Результаты РБТЛ, как правило, зависят от числа
клеток в культуре, поэтому рекомендуется использовать посто-
янное число лимфоцитов при засеве культуры (см. рис. 97) ли-
бо проводить пересчет на постоянное число лимфоцитов [4].
Использование градиентов фиколл/визотраст? создает до-
полнительные нефизиологические условия. Оно приводит к на-
рушению соотношения различных форм лейкоцитов, в частно-
сти к обогащению суспензии моноцитами, что может вызывать
супрессорный эффект. Отмечается потеря лимфоцитов с боль-
шой плотностью и аутологических розеткообразующих клеток.
Было также показано, что удаление эритроцитов ведет к из-
менению реактивности лимфоцитов [9]. Это больше всего ме-
шает попыткам функциональной оценки РБТЛ. Для тест-систе-
мы на супрессорные клетки характерна обратная закономер-
ность [10]. При помощи индекса супрессорных клеток (см.
рис. 98) выявляют неспецифические, стероидорезистентные, мо-
301
ноцитозависимые эффекты. Индуцируемые КонА супрессорные
функции неспецифичны, активность моноцитов маскируется
эритроцитами [12]. Индекс супрессорных клеток невозможно
определять в цельной крови, в то же время обусловленная
КонА супрессия лучше выявляется в препаратах лейкоцитов из
цельной крови, чем в препаратах, полученных с применением
фиколл/визотраста. Имеются указания на то, что индуцирован-
ная КонА супрессия протекает с участием моноцитов [5, 10,
11]. Возможность перенесения этих данных in vivo в настоя-
щее время обсуждается. Следует отметить, что, помимо изме-
рения скорости синтеза ДНК, необходимо измерять и другие
параметры в культуре лимфоцитов (например, синтез иммуно-
глобулинов или интерферона, спектр лимфокинов, селективное
влияние моноцитов), чтобы получить информативную картину
состояния иммунной системы при всех существующих ограниче-
ниях (табл. 29). Особое внимание следует обращать на дина-
мику процессов.
3.7.4. СПИСОК ЛИТЕРАТУРЫ
1. Bresnihan В., Jasin Н. Е. Suppressor function of peripheral blood mononuc-
lear cells in normal individuals and in patients with SLE.— J. Clin. Invest.,
1977, 59, 106—116.
2. Brochier J. Le Thi Bichthuy. Human В cell differentiation induced by various
mitogens and its T cell control.— In: Human lymphocyte differentiation. Its
application to cancer. INSERM Symp. No 8/Eds. Serrou B. and Rosenfeld C.
Elsevier, North-Holland: Biomedical Press, 1978.
3. Farrant J., Knight S. C. Help and suppression by lymphoid cells as a func-
tion of cellular concentration. — Proc. Natl. Acad. Sci. USA, 1979, 76,
3507—3510.
4. Jensen P. T„ Christensen K- In vitro evaluation of porcine lymphocytes res-
ponse to PHA using a modified «whole blood» technique. — Vet. Immunol.
Immunopatol., 1981, 2, 121—132.
5. Kallenberg С. С. M., de Gast C. The suppression of DNA synthesis by Con
А-activated human lymphocytes. Role oi monocytes in Con А-induced sup-
pression.— Clin. Exp. Immunol., 1980, 41, 583—590.
6. Rasanen L., Arvilommi H. Cell walls, peptidoglycans and teichoic acids of
Gram positive bacteria as polyclonal activators and immunomodulators of
proliferative and lymphokine responses of human T and В lymphocytes. —
Infect. Immunity, 1982, 35, 523—527.
7. Report of the Commetee on Clinical Immunology of the IUIS. — Eur. J.
Immunol., 1976, 6, 231—234.
8. Report of a IUIS/WHO working group: Use and abuse of laboratory tests
in Clinical Immunology. Critical consideration of eight widely used diagnos-
tic procedures. Geneva, 1981, 5, 18—20.
9. Schutt Ch., Glawe D., Westphal H. J. Die 3H-Thymidin-Plattchen Modifika-
tion des LTT. II. Eine Vollblut-Micromethode mit Einsatz spezifischer Anti-
gene.— Allergie. Immunol., 1979, 25, 54—70.
10. Schutt Ch. Zur Aussagekraft des LTT in der klinischen Diagnostik. — Pro-
motion B, Wilhelm-Pieck-Universitat, Rostock, 1982.
11. Smith W. G„ Ursinger W. R., Splitter G. A. Bovine Con A induced suppres-
sor cells. Generation, macrophage requirements and possible mechanisms of
regulatory action. — Immunology, 1981, 43, 91—100.
12. Webb P. J., Brooks C. G. Macrophage like suppressor cells in rats. I. Inhibi-
tion of natural macrophage like suppressor cells by red blood cells. — Celt.
Immunol., 1980, 52, 370—380.
302
3.8. СМЕШАННАЯ КУЛЬТУРА ЛИМФОЦИТОВ
3.8.1. ПРИНЦИП МЕТОДА
Первые сообщения о смешанной культуре лимфоцитов (СКЛ)
появились в 1964 г. в связи с феноменом бластрансформации.
Изучение СКЛ основано на том же принципе, что и РБТЛ (см.
раздел 3.7.1). Сила реакции зависит от степени различия меж-
ду антигенами гистосовместимости [11]. HLA-A, -В-В- и -С-ан-
тигены на поверхности лимфоцитов соответствуют HLA-D-ан-
тигенам, которые могут быть выявлены только при помощи
СКЛ. В то же время DR-антигены (D-related) могут быть об-
наружены при помощи антисывороток (см. раздел 2.12.5). Ги-
потеза о том, что анти-ВК-сыворотки позволяют осуществлять
тонкое типирование всего локуса D [5], кажется в настоящее
время менее вероятной, чем гипотеза о существовании двух
различных локусов (рис. 99). Даже для 1а-антигенов человека
в настоящее время обсуждается трехлокусная модель ’[12], так
что термин «DR» не следует более применять в качестве сино-
нима понятия «человеческие молекулы II класса».
Для оценки реактивности (индивидуума) в отношении ал-
логенного раздражителя готовят так называемую односторон-
нюю СКЛ, при которой для стимуляции используют инактиви-
рованные клетки. Следует заметить, что даже инактивирован-
ные стимулирующие клетки (см. ниже) обладают способностью
выделять митогенные факторы, а также до некоторой степени
усиливать синтез ДНК и биосинтез иммуноглобулинов [9].
Это может стать скрытым источником ошибок. На мышиной
модели хорошо изучены взаимодействия, возникающие в ходе
аллогенной реакции (рис. 100). Правда, до настоящего времени
остается неясным, какие типы клеток участвуют в пролифера-
тивном ответе. Исследование первичной реакции лимфоцитов на
аллогенные клетки используется в следующих случаях:
Рис. 99. Хромосома 6 человека с HLA-комплексом и близко расположенными
локусами (GLO; PGM3) [2]. В настоящее время точно установлено сущест-
вование 12 антигенов DW и 10 антигенов DR. LD—2=минорный D-локус, су-
ществование которого можно предположить на основании рекомбинационно-
го анализа.
303
Рис. 100. Механизм индук-
ции аллогенной реакции
для получения эффекта за-
висимого от клеток лимфо-
цитолиза (модификация
описана в соответствующей
работе [4]).
MF — макрофаг; TlNDH—индук-
тор хелперных клеток; Тн —
Т-хелпер; рТс — предшественник
цитотоксической клетки; Тс —
цитотоксическая клетка; тре-
угольник— DR-антиген; прямо-
угольник — HLA-A/B-антигены.
— проверка СКЛ-ре-
активности в каче-
стве критерия оцен-
ки клеточного им-
мунитета;
— типирование HLA-
-D;
— выяснение взаимо-
зависимости меж-
ду HLA-D-антиге-
нами и предраспо-
ложенностью к оп-
ределенным заболе-
ваниям;
— выбор пары до-
нор — реципиент.
3.8.2. МЕТОДИКА
З.8.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
См. разделы 3.1 и 3.7.2. Кроме того, необходимы низкотемпера-
турный шкаф, ДМСО, биокамера ВО 100, жидкий азот, сухой
лед, полиэтиленовые пробирки, для инактивации клеток мито-
мицин С или кобальтовая пушка.
3.8.2.2. ПРОВЕДЕНИЕ АНАЛИЗА
Если СКЛ применяют для общей оценки функции Т-клеток, то
для стимуляции используют смесь аллогенных клеток. Если
подбирают пару донор — реципиент, то оценивают силу реак-
ции прежде всего донора. Подробнее см. рис. 101.
Инактивация клеток-стимуляторов: 30 Гр дают при помощи
кобальтовой пушки с нагрузкой 2,5—12.5Х10-2 Ки/кг. Если
отсутствует источник радиации, клетки можно инактивировать
добавлением 50 мкг радиоактивного материала на каждый мл
суспензии клеток (30 мин, 37°C). После этого клетки трижды
отмывают.
304
Рис. 101. Этапы работы при получении смешанной культуры лимфоцитов дл»
оценки иммунореактивностн (I, III), в частности для выбора пары донор —
реципиент (I, II, III).
Ли —лимфоцит; ШГО — шкаф глубокого охлаждения; ф — фон; ЛД — лимфоциты до-
нора; ПКС — пул клеток стимуляторов; обработка и измерение описаны в разделе 3.7.2.
Стандартная смесь клеток: перед постановкой диагностиче-
ского теста для исследования больного с предполагаемым
иммунодефицитным состоянием необходимо установить грани-
цы ’’нормального ответа”. Для этого необходимо учитывать це-
лый ряд переменных величин. Желательно иметь стандартный
препарат для получения максимальной аллогенной стимуляции.
Для этого используют пул стимулирующих клеток, полученных
не менее чем от 10 различных доноров. Клетки могут сохра-
няться в замороженном состоянии. В табл. 30 представлены
этапы получения пула. Выход клеток составляют около 70%.
Хотя данный метод замораживания и не является оптималь-
ным, он вполне пригоден для сохранения клетками стимулирую-
щей активности [9]; для сохранения клеток респондера этот
метод не применим.
Культивирование и обработка проб:
Из рис. 101 видно, что в пробе смешивают 105 лимфоцитов
респондера и 105 стимулирующих клеток. Продолжительность
20-990
305
Таблица 30. Низкотемпературная консервация без криостата
А. Замораживание
1. Приготовление суспензии облученных клеток из расчета 5X106 кле-
ток/мл
2. Приготовление 20% раствора ДМСО в сыворотке АВ
3. Заполнение полиэтиленовых пробирок клеточной суспензией (по 0,5 мл)
с последующим добавлением по каплям 0,5 мл раствора ДМСО (при
4 °C)
4. Закупоривание крышек и инкубация при —20 °C в течение 30 мин
5. Хранение в течение 3—15 ч при —70 °C (сухой лед)
6. Перенесение в сосуды для хранения биоматериала
7. Хранение при —196 °C (жидкий азот) в течение произвольно длитель-
ного времени
Б. Размораживание
1. Быстрое разогревание пластиковых пробирок до 0—4 °C при постоян
ном встряхивании в теплой воде (37 °C)
2. Суспензию клеток смешивают с 2 мл холодной среды (медленно,
с постоянным встряхиванием)
3. Отмывают 10 мл среды, центрифугируют 10 мин при 300 g.
4. Ресуспендирование, подсчет клеток при помощи трипанового синего.
5. Приготовление суспензии 10е клеток/мл.
культивирования составляет 7 дней. После внесения метки в
культуру (см. рис. 101) проводят обработку и измерение ра-
диоактивности так же, как для РБТЛ (см. рис. 96, табл. 28).
3.8.2.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Оценку результатов проводят аналогично тому, как это приня-
то для РБТЛ (см. раздел 3.7.2) либо по абсолютным показа-
телям (имп/мин), либо по индексу стимуляции (ИС):
радиоактивность после стимуляции
радиоактивность контроля
_ реакция на тонирование
~ максимальная реакция *
радиоактивность SL — радиоактивность контроля
'° ~ радиоактивность SZP — радиоактивность контроля ® ’
(Сокращения см. объяснение к рис. 101.)
3.8.3. ОЦЕНКА МЕТОДА
СКЛ прочно утвердилась в арсенале методов трансплантацион-
ной и клинической иммунологии, хотя она не дает возможности
провести однозначный отбор оптимальных пар донор-реципи-
ент [3,6]. Для сокращения длительности анализа были пред-
ложены различные модификации, например первичное типиро-
вание лимфоцитов (ПТЛ) [1,8,10].
306
При постановке СКЛ отрицательный результат (ОР = 0—
—20%) указывает на идентичность LD-детерминант (первич-
ная СКЛ); тот же вывод при ПТЛ делают в случае позитивно-
го результата (ОР>70%) (вторичная СКЛ), Использование
’’гомозиготного типирования клеток” не является практическим
решением вопроса, если нет возможности вести перманентные
культуры [1,7]. Типирование по HLA-DR можно существенно
улучшить применением моноклональных АТ. Диагностика реак-
ций отторжения трансплантата при помощи СКЛ невозможна
ввиду длительности анализа.
Ретроспективное исследование выявляет высокую корреля-
цию данных, полученных при помощи СКЛ, с данными клини-
ческих наблюдений [7].
Значение степени несовместимости видно из следующего
примера: лица с двумя различиями в гаплотипе реагируют
вдвое сильнее (ОР=100%), чем лица с одним различием в га-
плотипе (ОР = 40%). Первичный ответ на аллогенные клетки
часто используют для общей оценки функции Т-клеток. Вос-
производимый стандарт пула стимулирующих клеток получают
смешиванием стимулирующих клеток от 10 доноров, отобран-
ных на основании различий по HLA-B [9]. Можно получать
СКЛ на основе цельной крови по аналогии с РБТЛ (на такой
же крови) [9]. Введением сывороточных добавок выявляют ал-
лоантитела и некоторые сывороточные факторы [8]. Точное
соотношение между реакцией отторжения трансплантата, нали-
чием антител к HLA-A- и -В-антигенам или D- и DR-антиге-
нам, СКЛ-реактивностью и претрансплантационными гемотран-
сфузиями до настоящего времени окончательно не выяснено
[3]. Включение автологичного контроля СКЛ, помимо обыч-
ного контроля (см. рис. 101), не рекомендуется, поскольку
аутологичная СКЛ имеет самостоятельное значение в оценке
состояния иммунной системы [13].
3.8.4. СПИСОК ЛИТЕРАТУРЫ
1. Bach F. Н., Inouye Н., Hank J. A. et al. Human Т lymphocyte clones reactive
in primed lymphocyte typing and cytotoxicity. — Nature, 1979, 281, 307—
311.
2. Bertrams J., Rittner C. Der HLA-K.omplex. — Dtsch. med. Wschr., 1981, 106,
352____958.
3. Goeken N. E., Cross D. E., Rodey G. E. et al. Effect of HLA-A and B,
HLA-DR and blood transfusion on cadaveric renal allograft survival. — In:
Report of the 8th International Histocompatibility workshop. 4—10.2.1980.
Los Angeles UCLA, California, 1980, 896—897.
4. Hardt C„ Rollinghoff M., Pfizenmayer U. et al. Lyt 23+ cyclophosphamide-
sensitive T cells regulate the activity of an IL 2 inhibitor in vivo. — J. exp.
Med., 1981, 154, 262—274.
5. De Marchi M., Varetto O., Savina C. et al. Relationship between HLA D and
DR. — In: Report of the 8th International histocompatibility workshop
4—10.2.1980. Los Angeles, UCLA, California, 1980, 893—896.
6. Niethammer D. Treatment of severe combined immunodeficiency. — Blut,
1981, 42, 137—148.
7. Reinmoen N., Yunts E. J., Bach M. L. Lymphoblastoid cell lines of homozy-
20’ 307
gous typing cells used for sensitization in PLT, — Tissue Antigens, 1977, 9,
11—16.
S. Robbins F. M., Hartzmann R. J. Rapid matching for transplantation PHA
primed lymphocytes. — Human Immunol., 1981, 3, 121—131.
'9 . Schutt Ch., Wegener S. Die 3H-Thymidin-Plattchen-Modifikation des LTT.
Die gemieschte Lymphozytenkultur im Vollblut unter Einsatz eines Stimula-
torzellpooles. — Acta biol. med. germ., 1980, 39, 1045—1050.
10. Shaw S., Pollack M. S., Payne S. M. et al. HLA linked В cell alloantigens
of a new segregant series. Population and family studies of the SB anti-
gens.— Human Immunol., 1980, 1, 177—185.
11. Svejgaard A., Hauge M„ Jersild C. The HLA System (2nd ed.). Karger,
12. Tanigiki N., Tosi T. The genetic control of human la allo antigens. A three-
loci model derived from the immunochemical analysis of «supertypic» speci-
ficities.— Immunol. Rev., 1982, 66, 5—38.
13. Damle N. K„ Gupta S. Autologous mixed lymphocyte reaction in man.
V. Functionally and phenotipically distinct human T cell subpopulations
respond to non T- and activated T cells in AMLR. — Scand. J. Immunol.,
1982, 16, 59—68.
3.9. РЕАКЦИЯ ТОРМОЖЕНИЯ МИГРАЦИИ
ЛЕЙКОЦИТОВ
(КАПИЛЛЯРНЫЙ МЕТОД)
3.9.1. ПРИНЦИП МЕТОДА
Реакция торможения миграции (РТМ) используется для выяв-
ления факторов торможения, которые секретируются сенсиби-
лизированными лимфоцитами наряду с другими лимфокинами
после повторного контакта с антигеном или митогенной стиму-
ляции [3]. При помощи РТМ выявляют фактор торможения
миграции макрофагов (MIF, Svejcar, 1963) либо фактор тормо-
жения миграции лейкоцитов (LIF, Rocklin, 1974). MIF тормо-
зит миграцию макрофагов и моноцитов, его видовая специфич-
ность невысока, отн. мол. масса человеческого MIF составляет
20 000—30 000. LIF селективно тормозит миграцию гранулоци-
тов, относительно нестабилен, отн. мол. масса составляет
50000—70 000, он также, вероятно, обладает эстеразной актив-
ностью. Капиллярный метод РТМ предложен Soborg [4]. Для
работы чаще всего используют макрофаги морской свинки или
лейкоциты крови человека. В непрямой РТМ используется бес-
клеточная надосадочная фракция лимфоцитов после их контак-
та с антигеном. Если существует сенсибилизация к соответст-
вующему антигену, наблюдается торможение миграции индика-
торных клеток в капилляре (рис. 102).
3.9.2. МЕТОДИКА
З.9.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы среда для культивирования (Игла-
МЕМ с pH 7,4) со 100 ME пенициллина/мл и 100 мкг стрепто-
мицина/мл, инактивированная сыворотка (морской свинки, че-
308
Рис. 102. Миграция индикаторных
клеток из капилляра.
а — нормальная миграция (контроль);
б — торможение миграции.
человеческая АВ, телячья),
инфуколл 6%, вазиленовое
масло (стерильное), гепа-
рин без консервантов (Ge-
deon Richter, Венгрия), си-
ликоновая смазка NP 12
(хим. завод Niinchrittz, ГДР), пластилин, стерильное рабочее
место (например, ламинарный бокс), камера для миграции
(например, из пиакрила 90X90X 15 мм с 4X4 лунками диамет-
ром 17 мм или планшеты Costar с 24 лунками, США), гемато-
критные капилляры (1X50 мм) центрифуга, термостат, микро-
скоп, камера для счета клеток, аналитические весы.
3.9.2.2. ПОЛУЧЕНИЕ НАДОСАДОЧНОЙ ФРАКЦИИ
ЛИМФОЦИТОВ
Препарат лимфоцитов получают из гепаринизированной крови
в градиенте плотности (см. раздел 3.1). Дважды отмытые
клетки в концентрации 1Х106/мл в среде Игла-МЕМ с анти-
биотиками инкубируют с соответствующим антигеном 20 ч при
37 °C. Бесклеточную надосадочную фракцию получают центри-
фугированием (10 мин, 1500g).
3.9.2.3. ИНДИКАТОРНЫЕ КЛЕТКИ
З.9.2.З.1. Подготовка макрофагов
За три дня до выделения макрофагов морским свинкам вво-
дят внутрибрюшинно по 20 мл вазелинового масла. Получен-
ный экссудат центрифугируют 20 мин при 150g, слой масла
отбрасывают, осадок клеток ресуспендируют в чистых центри-
фужных пробирках и дважды отмывают средой Игла-МЕМ
(10 мин, 150g). Готовят суспензию макрофагов из расчета 1—
2Х'Ю8/мл в среде Игла-МЕМ с 10% сыворотки.
3.9.2.3.2. Подготовка лейкоцитов
Гепаринизированную кровь (30 ЕД гепарина на 1 мл) в соот-
ношении 4+1 смешивают с 6% инфуколлом и оставляют на
1 ч при 37 °C. Содержащую лейкоциты надосадочную фрак-
цию трижды отмывают в среде Игла (10 мин, 150g), готовят
суспензию лейкоцитов 1—2Х108 клеток/мл в среде Игла с до-
бавкой 10% сыворотки.
3.9.2.4. ПОСТАНОВКА РЕАКЦИИ
Капилляры погружают в суспензию индикаторных клеток, за-
полнение происходит под действием капиллярных сил. Верх-
309
Миграция
Рис. 103. Выборочные результаты по реакции торможения миграции.
А — контрольные клетки; Б — 5 мкг/мл КоиА; В — 20 мкг/мл надосадочиой фракции с
КонА-сефарозой; Г — надосадочная фракция (В), обработанная 100 мкл антн-КонА/мл;
Д — 200 мкл кандндина; Е — иадосадочная фракция лимфоцитов человека, стимулиро-
ванных 5 мкг/мл КонА; Ж — как Е, но концентрация в 4 раза выше; 3 — как Ж, но
концентрация в 4 раза выше; И — как 3, но концентрация в 6 раз выше.
ний конец заполненного капилляра затыкают пальцем, вынима-
ют капилляр из клеточной суспензии и втыкают его нижним
концом в пластилин для закупоривания. Подготовленные таким
образом 20—30 капилляров ставят вертикально в центрифуж-
ный стакан и центрифугируют 10 мин при 30—40g. Затем на
уровне границы раздела клетки/надосадочная фракция капил-
ляры надпиливают и обламывают. Закрытый конец капилляра
обмакивают в силиконовую смазку и фиксируют капилляр на
дне лунки (см. рис. 102). Вся работа должна проводиться
в стерильных условиях. Заполнение камер соответствующей
надосадочной фракцией культуры лимфоцитов производится
немедленно после фиксации капилляра. При использовании
планшетов Costar в каждую лунку вносят 1 мл исследуемой
надосадочной фракции. Концентрацию антигенов подбирают в
предварительном опыте, обычно ставят 4 параллельные пробы.
Контроль: индикаторные клетки со средой (положительный
контроль); индикаторные клетки с надосадочной фракцией, со-
держащей MIF (непрямой метод) или, соответственно 5 мкг/мл
КонА в РТМ лейкоцитов (прямой метод). Камеры для мигра-
ции инкубируют 20 ч при 37°C, затем определяют величину
площадки миграции, вырезают ее и взвешивают. Индекс мигра-
ции получают из соотношения средних значений миграции
в опыте и контроле (пример, рис. 103, цв. вклейка рис. 9).
3.9.3. ОЦЕНКА МЕТОДА
РТМ представляет собой классический метод количественной
оценки клеточной сенсибилизации. Разброс результатов состав-
ляет ±10%, поэтому индексы миграции, лежащие за предела-
310
ми 0,80—1,20, обычно считают положительными. Модификации
приведены в разделе 3.10 и обзоре [1]. Механизм торможения
миграции лимфокинами также описан [2].
3.9.4. СПИСОК ЛИТЕРАТУРЫ
1. Morlex J., WOlsten.croft R. A., Dumonde D. C. Measurement of lymphokines.—
In: Handbook of Experimental Immunology (3rd ed)/Ed. D. M. Weir.— Ox-
ford: Blackwell Sci. Publ., 1978, 27.1—27.28.
2. Pick F. Mechanism of action of migration inhibitory lymphokines. — In: Biolo-
gy of the Lymphokines/Eds. S. Cohen, E. Pick, J. J. Oppenheim. — New York:
Academic Press, 1979, 59—119.
3. Pick E., Cohen S., Oppenheim J. J. The lymphokine concept T. Ibid., 1—12.
4. Soborg M„ Bendixen G. Human lymphocyte migration as a parameter of
hypersensitivity. — Acta med. Scand., 1967, 181, 247—256.
3.10. РЕАКЦИЯ МИГРАЦИИ ЛЕЙКОЦИТОВ
ПОД СЛОЕМ АГАРОЗЫ (РМЛА)
3.10.1. ПРИНЦИП МЕТОДА
В основе данного метода также лежит тот факт, что лимфоци-
ты после повторного контакта с антигеном, к которому они
были сенсибилизированы, продуцируют различные лимфокины,
в том числе влияющие на миграцию индикаторных клеток (гра-
нулоциты, моноциты, макрофаги). Как правило, наблюдается
торможение миграции, в некоторых случаях возможно специфи-
ческое усиление миграции. В 1967 г. Soborg и Bendixen пред-
ложили капиллярный вариант РТМ лейкоцитов для оценки кле-
точной иммунореактивности у человека. В 1971 г. Clausen
предложил метод, в котором лейкоциты мигрируют под слоем
агарозы, образуя циркулярные зоны [4]. В случае прямой
РМЛА лимфоциты и индикаторные клетки принадлежат одно-
му индивиду [2], в двухфазной реакции (непрямая РМЛА)
используют культуральную жидкость лимфоцитов обследуемого
индивида и индикаторные клетки нейтральных доноров,
рис. 104 [1,5].
3.10.2. МЕТОДИКА
3.10.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы нужно иметь: раствор Хенкса или ФСБ с pH
7,3; среду Игла-МЕМ с 5% инактивированной сыворотки (ло-
шадиная, телячья, человеческая АВ сыворотка, лошадиная и
телячья сыворотка, адсорбированная на человеческих эритроци-
тах АВ, с добавлением пенициллина (60 ЕД/мл), стрептомици-
на (60 мг/л), NaHCO3 (150 мг/л), pH 7,3 (лучшего буферного
эффекта можно достигнуть добавлением 10 ммоль/л ГЭПЭС,
311
Непрямой
Сепарированные
лимфоциты больного
Прямой
Г ранулоциты больного
♦I +Буфер
1 г + Буфер
45 мин., 37*С
(2,2*10®
клеток/Мл)
24ч,37°С,
2хЮбнлеток/мл
Тест Контроль Тест- Контрольная
Надосадочная суспензия суспензия
культуральная фракция
Миграция под |зч 37°с
слоем агарозы
Рис. 104. Принципы прямого и непрямого методов анализа миграции лейко-
цитов.
Сигма, США, с подведением pH до 7,3, NaOH в концентрации
5 моль/л), агарозу, гепарин (желательно без консервантов,
например, Gedeon Richter, Будапешт), декстран (например,
Infukoll 6%, сывороточный з-д Bernburg, ГДР), метанол, 35%
формалин. Также необходимы центрифужные пробирки с кони-
ческим дном на 10 мл и круглым дном на 20 мл, пастеровские
пипетки, штампы для вырезания отверстий в агарозе (2,0—
2,5 мм), микрошприцы (на 50 и 100 мкл), эппендорфовские
пипетки (5 и 10 мкл, Hamburg-Eppendorf), чашки Петри стек-
лянные или пластиковые, предметные стекла или стеклянные
пластины, алюминиевая фольга, счетные пипетки и счетные ка-
меры, центрифуги, термостат, стерилизатор, микроскоп, рН-
метр, водяной насос, водяная баня, проекционный аппарат (с
увеличением 15). Среды стерилизуют фильтрацией, стеклянные
предметы — в сухожаровом шкафу, пластмассовое оборудование
автоклавируют. Для непрямой РМЛА (культивирование лим-
фоцитов) дополнительно используют визотраст 370 (Fahlberg
List, ГДР), фиколл 400 (Pharmacia, Швеция).
3.10.2.2. ПОДГОТОВКА ПЛАСТИН С АГАРОЗОЙ
Размешивают 1 г агарозы в 45 мл дистиллированной воды,
расплавляют на кипящей водяной бане и охлаждают до 48 °C.
Затем добавляют 45 мл среды Игла (двойная концентрация)
и 10 мл сыворотки, предварительно нагретых до 48 °C. Теплый
312
4
Рис. 105. Оборудование для анализа миграции лейкоцитов в слое агарозы.
1 — игла для вырезания цилиндров агарозы (плоский срез, диаметр 2,5 мм); 2 —шаблон
для лунок (30 отверстий, ограничительный край); 3 — стеклянная пластина после удале-
ния слоя агарозы, высушивания на воздухе и фиксации зон миграции; 4 — сосуд для
стеклянных пластин при валивке агарозы.
раствор агарозы быстро разливают в чашки Петри (по 25—
30 мл в чашки диаметром 10 см), на обезжиренные стеклянные
пластины размером 76X97 мм (по 16 мл) или по 8 мл в счет-
ные камеры Neubauer.
Пластины с агарозой можно сохранять во влажной камере
при 4 °C. Перед внесением суспензии в агарозе проделывают
отверстия при помощи штампа (рис. 105). Вырезанные цилинд-
ры агарозы удаляют при помощи пипетки, соединенной с водя-
ным насосом.
3.10.2.3. ПРЯМАЯ РМЛА
К гепаринизированной крови (30 МЕ/мл) добавляют 6% инфу-
колл (4 мл на 16 мл крови), разливают в узкие (по возмож-
ности) пробирки и оставляют стоять в течение 1 ч при 37 °C
для спонтанной седиментации. Надосадочную жидкость, содер-
жащую лейкоциты, отсасывают, осаждают клетки центрифуги-
рованием в течение 10 мин при 150g. Клетки дважды отмывают
в ФСБ и один раз в среде Игла (8 мин, 100g). Готовят суспен-
зию, состоящую из 2,0—2.5Х108 лейкоцитов/мл в среде Игла
с 10% сыворотки. Для предотвращения снижения pH, обуслов-
313
ленного метаболизмом клеток, к каждому мл суспензии добав-
ляют по 5 мкл 1,8 М ЫаНСОз. На дно центрифужных пробирок
объемом 10 мл вносят по 10 мкл среды Игла (контроль) или
раствора антигенов в среде Игла (проба), к ним добавляют
по 40—70 мкл суспензии лейкоцитов. Пробирки закрывают ре-
зиновыми пробками и инкубируют при 37 °C при постоянном
встряхивании не менее 45 мин. По окончании инкубации клетки
ресуспендируют. Содержимое каждой пробы делят на аликвоты
по 10 мкл и заполняют ими 3—5 отверстий в агарозе. Пластины
с агарозой инкубируют 18 часов при 37°C. Из 16 мл крови па-
циента получается количество лимфоцитов, достаточное для
постановки 4 проб. Рекомендуется использовать модификацию,
в которой вместо 10 мкл используют 100 мкл разведения анти-
гена. После первого этапа инкубации клетки осаждают центри-
фугированием (5 мин, 30g) и готовят суспензию необходимой
плотности.
3.10.2.4. НЕПРЯМАЯ РМЛА
Получение надосадочной фракции культуры лимфоцитов
(1 фаза): 10 мл гепаринизированной крови испытуемого субъ-
екта смешивают со средой Игла (1 + 1), наслаивают в центри-
фужные пробирки на слой смеси фиколла и визотраста с плот-
ностью 1,077 (85,5 мл 6% фиколла+14,5 мл визотраста 370)
и центрифугируют 20 мин при 350 g. Отсасывают интерфазу,
содержащую лимфоциты, клетки дважды отмывают ФСБ и
один раз средой Игла (центрифугирование по 10 мин при
150g), готовят суспензию в среде Игла 2Х Ю6 лимфоцитов/мл.
Аликвоты по 0,45 мл этой суспензии инкубируют 24 ч при 37 °C
со средой Игла (контроль) или с 0,05 мл антигена (проба).
Культивирование проводят в центрифужных пробирках, закры-
тых силиконовыми пробками, по окончании культивирования их
центрифугируют 10 мин при 200g. Бесклеточную надосадочную
фракцию (БНФ) осторожно отбирают и непосредственно ис-
пользуют во 2-й фазе или хранят при —20 °C.
Исследование надосадочной фракции культуры лимфоцитов
(2 фаза): в соответствии с вышеописанным способом из гепа-
ринизированной донорской крови получают суспензию лейкоци-
тов с концентрацией 2,0—2,5хЮ8 клеток/мл. В конических цен-
трифужных пробирках смешивают по 100 мкл исследуемой
БНФ с 10 мкл среды Игла (проба) и, соответственно, 100 мкл
контрольной БНФ с 10 мкл раствора антигена (контроль).
В контрольную пробирку вносят такое же количество АГ, ка-
кое уже содержится в пробирке с исследуемой БНФ, для выяв-
ления возможных реакций со стороны лимфоцитов донора.
С каждой исследуемой пробой параллельно ставят соответст-
вующую контрольную. Количество контрольной БНФ также
должно составлять 400 мкл. К БНФ добавляют по 70 мкл сус-
пензии клеток. Работу с пластинами агарозы проводят как
314
описано выше. Для оценки величины миграции лейкоцитов до-
нора ставят отрицательный контроль с индикаторными лейко-
цитами, с средой Игла, с сывороткой вместо БНФ. Если по ре-
зультатам прямой РМЛА и всех видов контроля показано, что
использованный антиген в различных дозировках не вызывает
реакции лейкоцитов различных доноров, то можно отказаться
от постановки параллельных контрольных проб и ограничиться
одной пробой на каждой пластине с агарозой. Это существенно
снижает издержки анализа.
3.10.2.5. ОЦЕНКА РЕЗУЛЬТАТОВ
Диаметр большей части циркулярных зон можно определять
при помощи линейки в рассеянном свете. Для сохранения ре-
зультатов исследования пластины агарозы инкубируют 30 мин
с метанолом и 30 мин с 35% формалином, ополаскивают водой.
Слой агарозы осторожно отделяют от пластины скальпелем.
Пластины или чашки Петри с агарозой можно высушивать на
воздухе, в таком виде они хранятся практически неограничен-
ное время. Оценку зон миграции можно в этом случае прово-
дить при помощи проекционного аппарата. Исходя из среднего
диаметра, рассчитывают среднюю площадь зоны миграции в
пробе (возможна также планиметрия или вырезание и взве-
шивание зон миграции). Индекс миграции (ИМ) рассчитыва-
ют по следующей формуле:
М _ средняя площадь зоны миграции в присутствии АГ
~ средняя площадь зоны миграции без АГ
Для фотографирования пластин рекомендуется контрастное
окрашивание, например 5% эозином.
3.10.3. ОЦЕНКА МЕТОДА
Преимущества метода Clausen по сравнению с миграцией в ка-
пиллярах заключаются в большей простоте и скорости анали-
за, в лучшей воспроизводимости результатов и более удобной
документации (фиксирование пластин). При меньшем количе-
стве клеток и АГ возможно поставить большее число проб, ко-
торые дают меньший разброс результатов. По нашим наблюде-
ниям, краевая зона области миграции более резко очерчена,
чем при использовании капиллярного метода. Выбор, между
прямой и непрямой РМЛА зависит от поставленных задач
(см. табл. 31).
Контрольная зона миграции должна иметь площадь по
меньшей мере 20 мм2. Разброс данных пораллельных проб не
должен превышать ±0,10. При правильной постановке экспери-
мента разброс составляет около ±0,05. В этих условиях ин-
дексы миграции <0,80 и >1,20 становятся клинически значи-
мыми. Степень торможения миграции позволяет дать только
315
Таблица 31. Сравнение прямой и непрямой РМЛА
Преимущества непрямой РМЛА
1. Количество крови, необходимое для постановки одинакового числа проб,
сокращается втрое
2. Существенно более высокая чувствительность вследствие продолжительно-
сти культивирования; как правило, достаточно около 0,1 количества АГ
для того, чтобы выявить одинаковую реактивность
3. Лучшая стандартизация условий эксперимента ввиду точно известного
количества лимфоцитов
4. Отсутствие неудачных проб, обусловленных недостатком лейкоцитов у па-
циента
5. Возможность использовать БНФ большого числа пациентов в реакции е
одними и теми же индикаторными клетками
Недостатки непрямой РМЛА
1. Увеличение почти вдвое материально-технических затрат
2. Результаты анализа становятся известными на день позже
ориентировочную оценку уровня сенсибилизации, более точные
результаты можно получить при постановке параллельных проб
с различными концентрациями антигенов и лимфоцитов. Целе-
сообразно для каждого нового антигена определить минималь-
ную неспецифическую реактивную дозу (например, токсический
эффект) на несенсибилизированных индивидах, а на сенсибили-
рованных индивидах — минимальную дозу, вызывающую специ-
фическое торможение. При помощи РМЛА можно устанавли-
вать факт сенсибилизации к различным антигенам (бактерии,,
вирусы, грибки, опухоли, клетки трансплантата, растворимые-
белки, углеводы, синтетические полипептиды и др.) в той мере,
в какой они вызывают реакции клеточного иммунитета [1, 2,
3, 6]. Кроме того, БНФ лимфоцитов, подвергшихся митогенной
стимуляции, может быть при помощи РМЛА исследована на-
наличие тормозящих миграцию лимфокинов [1, 3, 10]. Быстрый
анализ особенно важен для диагностики аллергии, поскольку
не требуется отмывки клеток в связи с тем, что нет необхо-
димости учитывать воздействие блокирующих АТ, а условия'
in vivo (связывание аллергена, образование ИК) в достаточной
мере представлены аутологичной плазмой. Все же для исклю-
чения гуморальных факторов (блокирующие, цитофильные
В последнее время РМЛА используют для оценки хемотаксиса
АТ, ИК) рекомендуется работать с отмытыми клетками.
[8]. Лейкоциты и хемотаксический агент вносят в расположен-
ные рядом отверстия. Результаты оценивают по параболиче-
скому изменению зоны миграции в направлении отверстия с хе-
мотаксическим фактором. Для оценки можно также использо-
вать плотность расположения клеток в зоне миграции и ориен-
тацию отдельных клеток [7]. Известна микромодификация
РМЛА, предложенная в 1973 г. Harrington, находящая в насто-
ящее время все более широкое применение [9]. В данной мо-
дификации клетки, содержащиеся в капле агарозы, мигрируют
в окружающую среду, содержащую АГ или другой иссле-
316
дуемый материал. Преимущество этого метода для клинических
целей определяется очень небольшим количеством клеток, со-
держащихся в одной капле (2Х105 клеток в 1 мкл).
3.10.4. СПИСОК ЛИТЕРАТУРЫ
1. Ansfield М. Shalaby М. R., Kaltreider Н. В. An indirect assay for canine-
leukocyte migration inhibitory factor (LIF): Demonstration of its species-
specificity. — J- Immunol. Meth., 1980, 34, 43—48.
2. Bell R. B., Aurelian L., Cohen G. H. Proteins of Herpes Virus type 2.
IV. Leukocyte inhibition responses to type common antigens in cervix cancer
and recurrent infections. — Cell. Immunol., 1978, 41, 86—102.
3. Chapman S. W., Kirkpatrik С. H. The two-step leukocyte migration inhibition
factor (LIF) assay. Its use in evaluation of cellular immune function in
patients with immunodeficiency deseases. — Cell. Immunol., 1978, 37, 209—
220.
4. Clausen J. E. The agarose migration inhibition technique for in vitro
demonstration of cell mediated immunity in man. — Danish Med. Bull., 1975,
22, 181—194.
5. Cochrum К. C., Haynes D. M„ Solvatierra O. et al. Leukocyte migration inhi-
bitory factor (LIF) as an indicator of presensitization and allograft survi-
val.— Transpl. Proc., 1978, 10, 455—-458.
6. Emmrich F. J., Irmscher J., Kotzsch M. Leukocyte migration inhibition under
agarose versus MEM for detection of tumor associated antigens. — Brit. J.
Cancer, 1978, 37, 461—465.
7. Palmblad J., Uden A. M„ Venizelos N. The quantification of neutrophil orien-
tation and migration under agarose — a new method for detecting directed
and random movements. — J. Immunol. Meth., 1981, 44, 37—53.
8. Radermecker M„ Maidague M. P. Depression of neutrophil chemotaxis in
atopic individuals. — Int. Arch. Allergy appl. Immun, 1981, 65, 144—152.
9. Suslov I., McCoy J. L., Herberman R. B. Indirect leukocyte migration inhi-
bition reactions to a 3 M KCL extract of lung addenocarcinoma by lung
cancer patients. — J. Natl. Cancer. Inst., 1981, 66, 233—237.
10. Weisbart R. H„ Golde D. W., Spoiler L. et al. Neutrophil migration inhibition
factor from T lymphocytes (NIF-T): A new lymphokine. — Clin. Immunol.
Immunopathol., 1979, 14, 441—448.
3.11. ОПРЕДЕЛЕНИЕ ЭЛЕКТРОФОРЕТИЧЕСКОЙ
ПОДВИЖНОСТИ МАКРОФАГОВ
3.11.1. ПРИНЦИП МЕТОДА
Если сенсибилизированные лимфоциты инкубировать с»
специфическим антигеном, они приобретают способность выра-
батывать медиаторы, снижающие поверхностный заряд макрофа-
гов морской свинки, что приводит к уменьшению скорости дви-
жения макрофагов по сравнению с контрольными клетками
[5]. Образующийся через 90 мин инкубации медиатор, вызыва-
ющий снижение поверхностного заряда, называется «фактором,
тормозящим электрофоретическую подвижность» (англ, macro-
phage slowing factor, MSF). MSF определяют по электрофоре-
тической подвижности макрофагов после инкубации с исследуе-
мыми лимфоцитами и соответствующим антигеном (прямой
тест) или после взаимодействия с бесклеточной надосадочной
317
фракцией (непрямой тест). Снижение скорости перемещения
•обработанных клеток по сравнению с контрольными макрофа-
гами указывает на сенсибилизацию лимфоцитов [3]. Хотя не-
прямой тест требует больших затрат времени, его проще при-
способить к условиям клинической лаборатории.
3.11.2. МЕТОДИКА
3.11.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы среда для культивирования клеток
Игла-МЕМ (институт иммунных препаратов и питательных
сред ГДР), вазелиновое масло стерильное, морские свинки
(минимальный вес 300 г), цитоферометр (Opton, Oberkochen,
ГДР), регистрирующее устройство с отсчетом времени (напри-
мер, фотометр 20026 и рекордер 23144, Messelektronik, Дрез-
ден), 0,15 М NaCl.
3.11.2.2. ПРОВЕДЕНИЕ ЭКСПЕРИМЕНТА
Для постановки непрямой МЕМ (рис. 106) 106 лимфоцитов, по-
лученных из гепаринизированной крови в градиенте плотности
(см. раздел 3.1), смешивают с исследуемым антигеном в общем
объеме 3 мл среды Игла-МЕМ и инкубируют 90 мин при ком-
натной температуре. В качестве контроля берут просто среду
Игла. Оптимальную концентрацию антигена определяют в пред-
варительном опыте (например, для РРД 20 мкг/мл или для
энцефалолитогенного белка человека 33 мкг/мл). Через 90мин
смесь центрифугируют 10 мин при 300 g, бесклеточную надоса-
дочную фракцию (БНФ) немедленно используют или сохраня-
ют при —20°С. Инкубацию 8Х106 макрофагов морской свинки
с БНФ проводят при комнатной температуре. Препарат макро-
фагов готовят как было описано ранее (см. раздел 3,9) из
перитонеального экссудата морских свинок на 7—14-й день
после введения им 20 мл стерильного вазелинового масла.
Опыт Контроль I Контроль II Контроль III
Рис. 106. Непрямой МЕМ.
318
После инкубации определяют среднее время электрофоретиче-
ской миграции макрофагов в микрофоретической камере (ци-
тоферометр) при 23 °C. Определяют время миграции приблизи-
тельно 30 макрофагов через постоянный отрезок измерительно-
го поля окуляра (16 мкм). Проводят двойные измерения
(в прямом и обратном направлении при смене полюсов). Для
анализа используют только те пары измерений, в которых раз-
личия длительности прямого и обратного движения не превы-
шают 10%. Для анализа отбирают макрофаги с диаметром
около 16 мкм, имеющих 2—3 включения вазелинового масла,
поскольку на таких клетках отмечается наибольший тормозя-
щий эффект. Изменение времени электрофоретической мигра-
ции макрофагов в сравнении с контрольной пробой выражают
как процент торможения (Т%).
Г = /пр-/контр х100(
‘контр
где /Пр — средняя электрофоретическая подвижность макрофагов в пробе,
(контр—средняя электрофоретическая подвижность макрофагов в контрольной
пробе.
Описание контроля см. рис. 106, кроме того, ставят контроль-
ные пробы I и II (макрофаги в присутствии антигена и без не-
го) для исключения неспецифического воздействия АГ на ма-
крофаги. В качестве позитивного контроля можно поставить
пробу с РРД, поскольку практически все индивиды имеют ту
или иную степень сенсибилизации к туберкулину.
3.11.3. ОЦЕНКА РЕЗУЛЬТАТОВ
Определение электрофоретической подвижности макрофагов
может рассматриваться как метод выявления клеточной гипер-
чувствительности, поскольку его данные коррелируют с данны-
ми реакции торможения миграции макрофагов у кроликов при
экспериментальном аллергическом энцефаломиелите и у мор-
ских свинок, вакцинированных БЦЖ [4]. Относительно значе-
ния МЕМ в качестве средства диагностики in vitro злокачест-
венных опухолей имеются различные суждения [1, 7]. Получен-
ное в МЕМ торможение скорости миграции макрофагов >6%
рассматривается как положительный результат. Тщательной
стандартизацией условий постановки опыта можно снизить
разброс результатов до 3%. Для большей надежности резуль-
татов лучше ориентироваться на значения 6% (2 сек). Иногда
0,5хЮ6 лимфоцитов, антиген и 1Х107 облученных (1 Дж)
макрофагов морской свинки инкубируют в объеме 3 мл в те-
чение 90 мин при комнатной температуре, после чего опреде-
ляют электрофоретическую мобильность макрофагов. Облуче-
ние макрофагов должно предотвращать иммунологическое
319
взаимодействие содержащихся в перитонеальном экссудате
лимфоцитов морской свинки с исследуемыми лимфоцитами.
Хотя такое взаимодействие полностью устранить не удается,
облучение повышает чувствительность макрофагов к MSF. Со-
гласно исследованиям Shenton [8], большое количество анти-
генов, например РРД или экстракты тканей, содержащие 3 М
KCI, могут быть использованы в непрямом тесте с применением
таннизированных эритроцитов барана при условии, что коли-
чество индикаторных клеток будет увеличено до ЗхЮ7. При
помощи МЕМ можно определять блокирующие сывороточные
факторы. Для этого различное количество лимфоцитов инку-
бируют с испытуемым антигеном или проводят преинкубацию
лимфоцитов с различными количествами сыворотки. В качестве
еще одной модификации МЕМ можно рассматривать (МЕМ)-
СКЛ-тест, при помощи которого определяют степень гистосов-
местимости пары донор — реципиент [6] (быстрая диагности-
ка). При постановке этой пробы по О,5Х1О6 лимфоцитов доно-
ра и реципиента инкубируют 90 мин при комнатной темпера-
туре. Получаемые центрифугированием БНФ используются так
же, как в непрямом тесте. Eckstein и соавт. [2] подтвердили
эти данные на коммерческой системе для безгелевого электро-
фореза. Через 60 мин инкубации БНФ от специальной культуры
лимфоцитов (по 106 лимфоцитов, 3 ч, 37°C) с ЗхЮ6 индика-
торных клеток можно наблюдать изменения электрофоретиче-
ской мобильности индикаторных клеток по сравнению с нуле-
вым стандартом. Данные МЕМ показывают высокое статисти-
ческое соответствие с результатами СКЛ.
3.11.4. СПИСОК ЛИТЕРАТУРЫ
1 . Arvilommi Н., Dale М. М„ Desai И. N. et al. Failure to obtain positive MEM
tests in either cell-mediated immune conditions in the guinea pig or in human
cancer. — Brit. J. Cancer, 1977, 36, 545—549.
2 .‘ Eckstein R„ Mempel W., Hannig К et al. Recognition of a positive MLC
reaction after four hours. — Blut, 1980, 41, 459—464.
3. Field E. J., Shenton В. K, Meyer-Rienecker H. et al. Die Makrophagen-Elektro-
phorese-Mobilitats (MEM) Methode: Ihre Anwendung in der klinischen Immu-
nologie. — Dtsch. Ges-wesen, 1975, 30, 377—383.
4. Jenssen H. L., Koehler H„ Werner H. Die Anwendung der Zellelektrophorese
zur untersuchung verschidener Immunphanomene. — Dissertation (B), Rostock
1978.
5. Jenssen H. L., Werner H„ Koehler H. Lymphokines wich influence electropho-
retic mobility of macrophagees. — Acta biol. med. germ., 1976, 35, 1499—
1503.
6. Lippert H., Lorenz D., Panzig E. Ergebnisse der Inseltransplantation bei
diabetischen Hunden. — Ztschr. Exper. Chirurg., 1978, 11, 342—347.
7. Pritchard J. A., Sutherland W. H., Teasdale C. The macrophage electrophore-
tic mobility (MEM) test — an investigation of its value as a routine laboratory
test in the detection of malignant desease. — Ann. Clin. Res., 1978, 10, 71—74.
3. Shenton B. K, Jenssen H. L„ Werner H. et al. A comparison of the kinetics
of the macrophage electrophoretic mobility (MEM) and the tanned sheep
erythrocyte electrophoretic mobility (TEEM) test. — J. Immunol. Meth., 1977,
14, 123—139.
320
3.12. РЕАКЦИЯ ТОРМОЖЕНИЯ
АДГЕЗИИ ЛЕЙКОЦИТОВ (РТАЛ)
3.12.1. ПРИНЦИП МЕТОДА
РТАЛ основана на прекращении адгезии лейкоцитов сенсиби-
лизированых доноров к стеклянным или пластиковым поверх-
ностям в присутствии специфического АГ. Метод был перво-
начально предложен для выявления сенсибилизации к опухоле-
вым АГ экспериментально полученных опухолей животных
[1, 2] и неоплазм человека [3, 4, 5]. РТАЛ можно использо-
вать для выявления клеточной сенсибилизации и к другим
АГ [7]. Известны различные модификации метода, основанные
на различных механизмах реакции. Метод «счетной камеры»,
в котором подсчитывают адгезировавшие клетки, основан на
участии Т-клеток и продукции лимфокинов [8], «пробирочный»
метод, в котором подсчитывают число неадгезировавших кле-
ток, основан на участии моноцитов [9], — цитофильные АТ,
продуцируемые сенсибилизированными В-клетками, связыва-
ются с моноцитами. После инкубации с соответствующим АГ
адгезия моноцитов ослабляется. При помощи антисывороток
даже чужеродные моноциты можно сделать реактивными. На
этом явлении основана гуморальная РТАЛ, в которой опухо-
левые АГ и сыворотка больных с новообразованиями инкуби-
руются с трипсинизированными лейкоцитами здоровых доноров
1’10].
3.12.2. МЕТОДИКА
3.12.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Растворы для сепарирования клеток: среда Иг-
ла, раствор Хенкса, изотонический буфер с pH 7,3 (Институт
иммунных препаратов и питательных сред, Берлин), гепарин,
лимфопреп (Nyegaard, Норвегия) или фиколл — гипак (Phar-
macia, Швеция).
Оснащение для приготовления антигенов;
гомогенизатор, ультрацентрифуга, диализные мешки.
Посуд а: сосуды с частично плоской поверхностью (30X
Х13 мм), пластиковые сосуды с плоским дном, микропланше-
ты (например, Falcon N 3034).
Подсчет клеток: камеры для подсчета клеток (Вйгкег,
Thoma и др.); микроскоп, прибор для счета клеток (например,
Picoscale, Medicor, Венгрия); кроме того, необходимы термо-
стат, центрифуги и различная стеклянная посуда.
2!—990 321
3.12.2.2. ПРИГОТОВЛЕНИЕ КЛЕТОК
В РТАЛ можно использовать как лейкоциты, так и лимфоциты.
Мы предлагаем следующую простую модификацию: отслоив-
шуются после спонтанной седиментации гепаринизированной
крови (30—40 мл) плазму наслаивают на лимфопреп. После
центрифугирования (20 мин, 150 g) слой лимфоцитов, содержа-
щийся между плазмой и лимфопрепом, отсасывают. Клетки
трижды отмывают в изотоническом буфере и суспендируют в
среде Игла. При работе с животными можно использовать клет-
ки лимфатических узлов или перитонеального экссудата. Можно-
также использовать спленоциты. Перед подсчетом клеток необ-
ходимо лизировать эритроциты. Суспензию клеток лимфатиче-
ских узлов и селезенки получают путем продавливания этих ор-
ганов через тонкое металлическое сито или осторожного расти-
рания в стеклянном гомогенизаторе. Фильтрованием через
слой марли или капроновой сетки удаляют агрегаты клеток.
Клетки человека выделяют при комнатной температуре, по-
скольку на холоду они склонны к агрегации. Препараты клеток
мыши получают при 4 °C.
3.12.2.3. АНТИГЕНЫ
Антигенсодержащие экстракты из опухолевых, эмбриональных
и других тканей выделяют различными методами. Широкое
распространение получило экстрагирование ЗМ KCI [11], так-
же используется выделение препаратов мембран [9, 12] и экст-
ракция изотоническим раствором хлорида натрия. Для экстрак-
ции изотоническим раствором NaCl ткань измельчают ножни-
цами, смешивают в соотношении 1 + 4 с буфером (ФСБ) и
гомогенизируют (гомогенизатор Уорринга, Поттера или Ultra-
Turrax). Центрифугируют 10 мин при 1000—3000 g, собирают
надосадочную фракцию и центрифугируют 30 мин при 20 000 g
(можно больше). Полученную прозрачную надосадочную фрак-
цию замораживат небольшими аликвотами при —20 °C. Если
после размораживания образуется нерастворимый осадок, его
удалят центрифугированием. В качестве рабочей концентрации
в РТАЛ используют разведение, которое дает значимые разли-
чия между пробами от сенсибилизированных и несенсибилизи-
рованных доноров. Для экстрактов тканей эта величина состав-:
ляет в среднем 50—500 мкг белка на пробу.
3.12.2.4. ПРОВЕДЕНИЕ РТАЛ
3.12.2.4.1. Метод «счетной камеры»
Смешивают 106 лейкоцитов в 0,05 мл среды с 0,05 мл препара-
та АГ (в контроле просто среда) и с 0,1 мл нормальной сыво-
ротки. Пробирки закрывают и инкубируют на водяной бане
30 мин при 37°C. Через каждые 5 мин содержимое пробирок)
322
**• *
Л i'Jf '•&£<$ *, ”'.8 «
*»х***|< *'
<••: .♦ » . 4Л^.9. ' '
’л •
.«в <$ «х\ _;*4 ч9> ф. ;
.4 <>’ Ъ>!4Х> • '
л '-- ^S *'
. • ••» < .•/• ф-
; ' V ’- - 9
-
-’i *«. #-.У
1 *: Х > * ^..®.в.,;.-; :* «5^
"Л’ « . * • . ’.$•.
^'„ •Л,,-/ «S^rf8 °
.. .у -,у.^
*«» SV:; Оо
а?-:
в? а
вло
в6 * **?!. '
9 а
««>
а
е
• ' МЙ
.
л V, >’* ,"
»»<‘* ' V-e ./b'jt">'♦.*? '$'">
• ’ »•• .• фХ8!*
*» *’.<•"» *.*<• *• •'» « *
б
~ *• ,‘й
О; *•
*&. >‘ «*
9 & -
й»
» & ' •' !
. л>-. а:
-. «
q • а
- <&
о а
<&
&
*'3
г!
о
£
и
л
. о
а
. ^ХЭ*в ^?
SJ
а ®
%
9
!^:
<Г
«0»
«•
г
9 Оj&.
а £
# ф
« .
«
а
о.
&
о ”
5> ‘
°°о
© О.
*0 ‘
Og
С
•о
§*\
Л
«5
О
О л
sec °
'S b
4
в.оо’
’е
'“' <J <с*!
о
'а
Рис. 107. Адгезия лимфоцитов опухолеиосителя в контроле без антигена (а)
и в пробе с эмбриональным антигеном (б).
21*
а.Выделение лимфоцитов
Плазма
Лимфопреп
20 мин
х 2000 об/мин
3»
Лимфоциты
Трехкратная отмывка
изотоническим
буфером
Суспензия клеток-
в изотоническом
растворе хлорида натрии
б.Схема постановки РАЛ
Лимфоциты, контроль
Лимфоциты,опыт
1011 ою
Счетная камера
Рис. 108. Схема постановки РТАЛ (подсчет неадгезировавших клеток).
встяхивают. После инкубации суспензию вносят в счетную
камеру пастеровской пипеткой. Камеру выдерживают 1 мии
при 37 °C в водонасыщенной атмосфере (чашка Петри со смо-
ченным куском фильтровальной бумаги). Подсчитывают клет-
ки. В квадрате 0,2X0,2 мм должно содержаться 15—25 клеток.
Подсчитывают клетки в 5 квадратах. Затем отмывают неадгези-
рованные клетки (процедура требует особого навыка). Счет-
ную камеру помещают в горизонтальном положении в сосуд
с физиологическим раствором и осторожно снимают покровное
стекло. Камеру бережно извлекают и на короткое время поме-
щают в вертикальном положении в сосуд с изотоническим ра-
створом. Затем в камеру добавляют 1 каплю среды и покрыва-
ют чистым покровным стеклом. Через 1—2 ч подсчитывают ад-
гезированные клетки в тех же 5 квадратах, определяют процент
адгезированных клеток, (рис. 107). Добавлением исследуемой
сыворотки (0,1 мл) вместо нормальной можно выявлять бло-
кирующие или деблокирующие адгезию сывороточные факторы.
3.12.2.4.2. «Пробирочный» вариант РТАЛ
Суспендируют 106 клеток совместно с АГ в конечном объеме
0,5 мл в специальном сосуде, имеющем частично плоскую по-
верхность, на которой он стоит. Клеток должно быть немного,
чтобы не мог образоваться монослой лимфоцитов на плоской
поверхности. После инкубации в горизонтальном положении
(2 ч, 37 °C) сосуды ставят вертикально, осторожно ополаски-
вают плоскую поверхность суспензией клеток. После перемеши-
вания суспензию переносят пастеровской пипеткой в счетную
камеру и подсчитывают неадгезировавшие клетки (рис. 108).
324
3.12.2.4.3. Прочие модификации
Модификации, как правило, преследуют цель ускорить проведе-
ние анализа. Для этого индикаторные капли метят радиоак-
тивными изотопами и применяют автоматизированный подсчет
адгезированных и неадгезированных клеток [12, 13]. Кроме
того, РТАЛ проводят в микропланшетах или капиллярах с це-
лью уменьшения расхода клеток или антигена [12]. Микро-
планшеты можно анализировать даже при помощи соответст-
вующей программы на ЭВМ.
3.12.2.4.4. Подсчет результатов
В контрольных пробах смешивают лимфоциты несенсибилизи-
рованного донора со специфическим АГ, исследуемые лимфоци-
ты со средой или с неспецифическим антигеном. Торможение
адгезии (ТА) оценивают по следующей формуле:
_ % адгезии в присутствии АГ — % адгезии без АГ
Т (>«) — % адгезии без АГ
Для оценки результатов «пробирочного» метода часто исполь-
зуют такой показатель, как индекс неадгезировавших клеток
(ИНА).
% неадгезировавших клеток со специфическим АГ — % не-
_________адгезировавших клеток с иеспецифическим АГ_
" — % неадгезировавших клеток с неспецифическим АГ
Границу между специфическим и неспецифическим торможени-
ем адгезии устанавливают статистически или эмпирически по
сравнению с реакцией лимфоцитов сенсибилизированных и не-
сенсибилизированных доноров.
3.12.3. ОЦЕНКА МЕТОДА
РТАЛ не требует больших затрат материалов и времени. Кри-
тический момент метода — отделение адгезированных клеток
от неадгезированных. Метод «счетной камеры» может быть
использован для выявления лимфокинов и, соответственно, про-
водиться с несенсибилизированными и индикаторными клетками.
«Пробирочный» метод представляет собой преимущественно
одноступенчатый процесс, как это было показано в параллель-
ных пробах с лимфокинсодержащими надосадочными фракци-
ями [6]. Сообщалось, что после 20 ч инкубации [2] или обра-
ботки индикаторных клеток индометацином [5] непрямую
РТАЛ можно проводить также «пробирочным» методом. Меж-
ду обоими методами РТАЛ не всегда существует соответствие,
например, в послеоперационном периоде у больных с опухоля-
ми «пробирочный» метод дает отрицательные результаты, а ме-
тод «счетной камеры» — положительные результаты. «Проби-
рочный» метод проще в исполнении, неспецифическое торможе-
325
Рис. 109. Торможение адге-
зии лимфоцитов человека в
присутствии различных кон-
центраций эмбриональных
антигенов.
1 — лимфоциты опухоленосите-
лей (13 наблюдений); 2 —лим-
фоциты здоровых доноров
(8 наблюдений).
пие адгезии представ-
ляет собой проблему
для обоих основных
модификаций. В ли-
тературе имеются раз-
личные сведения о доле адгезированных клеток. По нашим
собственным данным («пробирочный метод»), лимфоциты че-
ловека, выделенные с применением лимфопрепа, показывают
адгезию, равную 73±11% (п=20). Клетки лимфатических уз-
лов мыши адегзируют в 78±5% случаев (п=10). Если к куль-
туральной среде добавить 2—10% сыворотки эмбрионов коров,
наблюдается неспецифическое торможение адгезии на 50—60%.
Подобный эффект наблюдается и при добавлении многих дру-
гих белков. На рис. 109 показано, что для работы пригодны
только те АГ, которые вызывают большее торможение адгезии
у сенсибилизированных, чем у несенсибилизированных доно-
ров. В качестве примера приведена реакция лимфоцитов у
больных с новообразованием (45± 14% торможения адгезии)
и лимфоцитов здоровых доноров (25± 13% торможения адге-
зии) с АГ из эмбриональных тканей. Известно, что значитель-
ное относительное число больных с опухолями сенсибилизиро-
ваны к эмбриональным АГ. Постановка РТАЛ затрудняется
тем, что только около 20% АГ позволяют выявлять различия
в адгезии сенсибилизированных и несенсибилизированных па-
циентов. Причина этого явления не установлена. Удалось до-
биться снижения неспецифической адгезии преинкубацией кле-
ток со специфическим АГ [14]. Экстракты тканей имеют огра-
ниченный срок хранения, его лучше осуществлять в жидком
азоте. РТАЛ, как и прочие методы исследования клеточного
иммунитета, пригодна прежде всего для исследования сенсиби-
лизированных и несенсибилизированных коллективов, а не от-
дельных индивидов.
3.12.4. СПИСОК ЛИТЕРАТУРЫ
1. Mortensen R. F. Detection of cellular immunity to murine sarcoma virus-indu-
ced tumors by leukocyte adherence inhibition. — J. Natl. Cancer. Inst., 1979,
62, 157—163.
2. Kalafut F., Hung P. M„ Novotna L. Application of the indirect tube LAI
assay in the study of cell-mediated immunity in rats immunized with В 77
tumor cells. — Neoplasma, 1980, 27, 525—531.
326
3. Flores M„ Marti J. H., Grosser N. et al. An overview: antitumor immunity in
breast cancer assayed by tube leukocyte adherence inhibition. — Cancer, 1977,
39 494____505.
4. Tanaka F., Sawada K., Yonemoto R. H. Analysis of sera from patients with
breast cancer for blocking and induction of leukocyte adherence inhibition. —
Cann. 1980, 71, 589—595.
5. Fritze D., Grunze M„ Kaufmann M. The cellular mechanisms involved in
leukocyte adherence inhibition (LAI) tests of patients with breast cancer. —
Immunol. Letters, 1981, 2, 225—230.
6. Holan V., Schimke R., Ambrosius H. et al. Comparison of the haemocytome-
ter and tube modifications of the leukocyte adherence inhibition assay. —
Folia biol., 1981, 27, 66—75.
7. Powell A., Sloss A. M., Smith R. N. et al. Antigenic specificity and cellular
mechanisms in leukocyte adherence inhibition analysis if immunity to simple
proteins and hapten protein conjugates.—Cancer Res., 1979, 39, 570—575.
8. Powell A., Sloss A. M„ Smith R. N. Leukocyte adherence inhibition: Aspecific
assay of cell-mediated immunity dependent on lymphokine-mediated collabo-
ration between T-lymphocytes. — J. Immunol., 1978, 120, 1957—1963.
9. Goldrosen M. H., Russo A. J., Howell J. H. et al. Cellular and humoral fac-
tors involved in the mechanism of the microleukocyte adherence inhibition
reaction. — Cancer Res., 1979, 39, 587—592.
10. Kotlar H. K, Sanner T. Humoral antitumor response in patients with breast
cancer measured with the leukocyte adherence inhibition technique. — J. Natl.
Cancer Inst., 1981, 66, 265—271.
11. Fujisawa T., Waldman S. R., Yonemoto R. H. Effect of serum factors and
cell washings on leukocyte adherence inhibition in breast cancer patients. —
Cann., 1980, 71, 165—172.
12. Russo A. J., Nordin A. A., Goldrosen M. H. A radio (5ICr) microtube leuko-
cyte adherence inhibition assay: Specific tumor associated immunity in 3 mu-
rine tumor systems. — J. Immunol. Meh., 1979, 31, 259—269.
13. Tsang P. H., Tangnavarad K, Lesnick G. Radioisotopic 5ICr leukocyte adhe-
rence inhibition (LAI) assay. I. Demonstration of antitumor immunity in
patients with breast carcinoma. — J. Immunol. Meth., 1980, 36, 119—135.
14. Morizane T., Kumagi N., Tsuchimoto K. et al. Specific immunodiagnosis of
hepatoma by tube leukocyte adherence inhibition assay and modified method
of repeated tube leukocyte andherence assay. — Cancer Res., 1980, 40, 2928—
2934.
3.13. РЕАКЦИЯ ТОРМОЖЕНИЯ
РАСПЛАСТЫВАНИЯ МАКРОФАГОВ
3.13.1. ПРИНЦИП МЕТОДА
Реакция торможения распластывания макрофагов (РТРМ) ис-
пользуется для выявления клеточного иммунитета и основана
на обусловленных лимфокинами изменениях мононуклеарных
фагоцитирующих клеток. Моноциты и макрофаги обладают
способностью в условиях соответствующей среды распласты-
ваться по стеклянной или пластиковой поверхности, образуя
похожие на псевдоподии плазматические выступы. Способность
к распластыванию может быть существенно ограничена, если
клетки перитонеального экссудата (КПЭ) сенсибилизирован-
ных животных инкубируют со специфическим АГ (прямой ме-
тод) или КПЭ интактных животных взаимодействуют с лим-
фокинсодержащей БНФ (непрямой метод). Метод РТРМ был
327
впервые предложен в 1968 г. Позднее было показано, что тор-
можение распластывания обусловлено растворимыми медиато-
рами [1]. Методика подробно описана в ряде работ [3, 5].
Торможение распластывания может наблюдаться в течение не-
продолжительного периода времени, от 2 до 5 ч. При более
длительном культивировании [18 ч] возможна даже стимуля-
ция распластывания. В клинике используют как прямой (ана-
лизируется суспензия лимфоцитов больного, в которой моноци-
ты служат индикаторными клетками), так и непрямой методы,
в котором БНФ лимфоцитов человека вступает во взаимо-
действие с макрофагами морской свинки.
3.13.2. МЕТОДИКА
3.13.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы вазелиновое масло для инъекций
(Leuna-Werke, ГДР); солевой раствор Хенкса с pH 7,4 (вари-
анты с добавлением и без добавления 5 ME безфенольного
препарата натриевой соли гепарина на 1 мл); среда Игла с pH
7,4 с добавлением 100 ME пенициллина/мл и 100 мкг стрепто-
мицина/мл; 0,01 мл инактивированной гомологичной нормаль-
ной сыворотки и 0,02 мг L-глутамина; ФСБ с pH 7,4; легко-
плавкий парафин для монтажа камер для культивирования;
0,2% ратвор трипанового синего в 0,15 М NaCl; красящий ра-
створ по Паппенгейму (Medizin-chemie, Лейпциг, ГДР). Фикси-
рующая смесь по Bouin (30 частей водонасыщенной пикрино-
вой кислоты, 10 частей формола, 2 части ледяной уксусной кис-
лоты, специфический АГ, в данном случае бычий у-глобулин
(Bekringwerke); полный адъювант Фрейнда (ПАФ) (Difco,
США); шприцы для инъекций на 1 мл, 10 мл, 20 мл, центри-
фужные пробирки, градуированные пипетки (0,2—5,0 мл), пи-
петки для подсчета лейкоцитов, счетная камера Neubauer, пред-
метные стекла, покровные стекла 20X45 мм, плоские шлифо-
ванные стеклянные кольца, клейкая лента постоянной толщины
(Atlas Plastic Таре, 0,2 мм толщиной, 19 мм шириной), сухо-
жаровой шкаф, автоклав, стеклянный стерилизующий фильтр
G5 (Saale-Glas, Йена), устройство для мембранного фильтро-
вания, шприц для мембранных фильтров на 5 мл (Sartorius,
ФРГ), мембранные фильтры с диаметром пор 0,40 мкм (Sar-
torius, ФРГ), термостат, фазово-контрастный микроскоп (Carl
Zeiss, Иена), денситрон (Центральный институт изотопов и
ионизирующего излучения, Лейпциг).
Растворы стерилизуют фильтрацией и хранят при 4 °C,
центрифужные пробирки силиконизируют (помещают на 24 ч
в смесь 1 части эмульсии силиконового масла NE 4 (Химиче-
ский завод Niinchritz, ГДР) и 4 частей дистиллированной во-
ды, отмывают пробирки водопроводной водой, высушивают на
328
Рис. ПО. Камера Cunningham для культивирования КПЭ в реакции тормо-
жения распластывания макрофагов (объяснение в тексте).
1 — пипетка с супензией клеток; 2 — предметное стекло; 3 — пластиковая лента «Atlas
Plastic Таре»; 4 — клейка лента; 5 — покровное стекло (45X22 мм); 6 — герметизация
расплавленным парафином.
воздухе, а затем 1 ч при 200°C). Все прочие стеклянные части
автоклавируют или обрабатывают в сухожаровом шкафу при
180 °C.
3.13.2.2. МОНТАЖ КАМЕРЫ ДЛЯ КУЛЬТИВИРОВАНИЯ
Для инкубации КПЭ особенно удобны камеры, изображенные
на рис. ПО. На обезжиренное предметное стекло на расстоянии
20 мм наклеивают 2 полоски клейкой ленты. Сверху кладут по-
кровное стекло и также укрепляют клейкой лентой. Между
предметным и покровным стеклом образуется полость с посто-
янным объемом, которую по краям герметизируют горячим па-
рафином. Эту полость заполняют сусупензией клеток. Для
формирования камер также удобно использовать двусторон-
нюю клейкую ленту (Scotch № 410). В некоторых модифика-
циях метода используют «открытые» камеры. Плоские стеклян-
ные кольца прикрепляют при помощи горячего парафина
к предметным стеклам, при этом образуется полость вмести-
мостью около 0,5 мл. После внесения суспензии клеток кольца
прикрывают сверху покровным стеклом без герметизации.
3.13.2.3. ПОЛУЧЕНИЕ КЛЕТОК
ПЕРИТОНЕАЛЬНОГО ЭКССУДАТА
Через 5 дней после внутрибрюшного введения 10—20 мл вазе-
линового масла морских свинок забивают и полость брюшины
промывают 50 мл холодного раствора Хенкса, содержащего
5 ME гепарина/мл. Суспензию клеток собирают в силиконизи-
рованные центрифужные пробирки и трижды отмывают раство-
ром Хенкса без гепарина (центрифугирование 10 мин, 150 g).
После подсчета количества неживых клеток готовят суспензию
клеток в культуральной среде (5хЮ6/мл).
329
3.13.2.4. ПРОВЕДЕНИЕ ИССЛЕДОВАНИЯ В КАМЕРЕ
Суспензию клеток делят на две части по 0,5 мл. Одна из проб
изготовлена на чистой культуральной среде, другая — на среде
с добавкой 5 мг бычьего у-глобулина. Обе пробирки инкубиру-
ют 30 мин при 37°C в термостате, желательно при постоянном
встряхивании. После инкубации берут две камеры для пробы,
две для контроля. Герметизацию камер проводят горячим па-
рафином при помощи кисточки. Герметизированные камеры ин-
кубируют еще 2 ч при 37 °C в горизонтальном положении. Ре-
зультаты оценивают при помощи фазово-контрастной микро-
скопии. Все клетки, имеющие псевдоподиеподобные выступы и
темную окраску, рассматриваются как мигрирующие (рис. 111).
В каждой камере считают по 200 клеток. Расчет индекса тор-
можения (ИТ), или, соответственно, процент торможения про-
водят по следующей формуле:
мигрирующие клетки (из 400) в опыте
““ мигрирующие клетки (из 400) в контроле
Процент торможеиия= (1 — ИТ) X100.
3.13.2.5. ПРОВЕДЕНИЕ ИССЛЕДОВАНИЯ
В ОТКРЫТОЙ камере
И ДЕНСИТОМЕТРИЧЕСКИЙ АНАЛИЗ
Различия касаются только формы камер и зависящей от нее
меньшей концентрации суспензии клеток. После инкубации
пробирок и камер (см. выше) кольца удаляют, предметные
стекла споласкивают ФСБ и фиксируют по Bouin (10 мин при
Рис. 111. Фазово-контрастная микроскопия КПЭ в камере Cunningham.
Время инкубации 2 ч без добавления АГ. L—лимфоцит; ri — округлившийся макрофаг;
Тз — макрофаг, состояние которого нельзя оценить однозначно; остальные клетки можно
отнести к категории распластанных.
330
Рис. 112. Комплекс приборов, составляющих деиситрон: телекамера (в дан-
ном случае соединена с микроскопом), дисплей и печатающее устройство,
комнатной температуре). Затем окрашивают препарат по Пап-
пенгейму и микроскопируют. Подсчет и оценку результатов см.
выше. Более объективные и точные результаты получаются при
оценке величины макрофагов на энкстроне, электронно-оптиче-
ском приспособлении, разработанном в ГДР (рис. 112). Сего
помощью можно быстро провести эквиденситометрическое опре-
деление площади отдельных макрофагов и оценить относитель-
ную частоту встречаемости макрофагов разной величины
(мкм2) в пробе и в контроле. На рис. 113 показано влияние
лимфокинсодержащей БНФ на распластывание макрофагов по
стеклянной поверхности. Сдвиг пика в кривой распределения со-
вершенно очевиден, полученное различие в высокой степени
достоверно.
3.13.3. МОДИФИКАЦИИ МЕТОДА
Если в прямой РТРМ антигенореактивные, высвобождающие
лимфокины, Т-лимфоциты и макрофаги (индикаторные клетки)
культивируют совместно, то в непрямой РТРМ этапы реакции
разделены. Сначала лимфоциты, выделенные из перифериче-
ской крови больного или экспериментального животного, инку-
бируют 18—24 ч со специфическим АГ в полной среде для
культивирования. Клетки удаляют центрифугированием, над-
331
Рис. 113. Относительная
частота встречаемости раз-
личных размеров макрофа-
гов.
1 — в контроле; 2 — в исследуе-
мой группе. При помощи ден-
ситометра было измерено 231 и
325 клеток соответственно.
осадочную фракцию
стерилизуют фильтра-
цией и хранят ее при
—20 °C. Поскольку при
культивировании воз-
можно высвобождение
АТ, рекомендуется надосадочную фракцию высолить 40%
сульфатом аммония, отбросить возможный осадок и отдиали-
зировать не выпавшие в осадок белки от соли. Это довольно
важный момент, поскольку цитофильные антитела, так же как
и комплексы АГ — АТ, могут оказывать ингибирующее влияние
«а макрофаги и приводить к ложноположительным результа-
там. На втором этапе КПЭ морской свинки смешивают в соот-
ветствующей концентрации с лимфокинсодержащей надосадоч-
ной фракцией и инкубируют 2 ч при 37°C. Преинкубация не
требуется. Прочие модификации касаются вида камер, типа
культуральных сред и условий культивирования. Так, например,
было показано, что инкубация в атмосфере, содержащей 5%
СО2, улучшает адгезию и распластывание клеток [4].
3.13.4. ОЦЕНКА МЕТОДА
РТРМ основана на влиянии лимфокинов на форму макрофагов
(округлая, распластанная). Это различие можно выражать ко-
личественно в виде индекса торможения (ИТ). ИТ меньше 0,8
соответствует положительному результату и означает наличие
клеточного иммунитета к исследуемому АГ. К незначительным
отличиям следует относиться весьма критически, поскольку
даже в контрольной пробе КПЭ оказываются весьма вариа-
бельными в отношении распластывания, оценка которого, кро-
ме всего прочего, зависит от опыта экспериментатора. Непря-
мая РТРМ, несмотря на большую трудоемкость, позволяет
получать результаты, легче поддающиеся стандартизации, по-
скольку различные БНФ можно исследовать с одной и той же
серией индикаторных клеток. Этим самым существенно снижа-
ется обусловленный биологической вариабельностью разброс
результатов. Поскольку иммунные комплексы уже в малых кон-
центрациях оказывают тормозящее влияние на распластывание
макрофагов и иногда нельзя исключить наличие в БНФ различ-
ных ИК, рекомендуется удаление этих примесей сульфатом ам-
332
мония. Преимуществом описанного в данной главе второго ва-
рианта прямой РТРМ следует считать получение препаратов
с длительными сроками хранения. Денситометрическое опреде-
ление величины макрофагов способствует увеличению объек-
тивности данных. К тому же денситрон отмечает небольшие
отклонения в площади поверхности макрофагов и суммирует их
в значимые разности. К числу недостатков метода можно от-
нести удаление сильно сморщенных макрофагов при отмывке,
что, разумеется, вносит искажение в результаты. Несмотря на
все оговорки, РТРМ следует считать удобным способом быстро-
го количественного определения in vitro состояния клеточного
иммунитета.
3.13.5. СПИСОК ЛИТЕРАТУРЫ
1. Dekaris D„ Smerdel S., Veselic В. Inhibition of macrophage spreading by
supernatants of antigen-stimulated sensitized lymphocytes. — Eur. J. Immu-
nol., 1971, 1, 402.
2. Hess J., Drossier K., Ambrosius H. Grossenmessungen von Makrophagen mit
dem elektronischen Bildauswertgerat Densitron. — Ztschr. med. Labortechnik,
1976, 17, 349—351.
3 Salvin S. B. .Observation on migration inhibition of sensitized peritoneal exu-
date cells as a function of time. — Cell. Immunol., 1974, 10, 310—318.
4. Sandor P. L„ Hinsdill R. D„ Albrecht R. M. Isolation of substances responsible
for lymphokine activity from sensitized mouse spleen cells. — Infection and
immunity, 1974, 9, 1045—1050.
5. Veselic B., Sabioncello A., Rabatic S. Lymphokines mediated macrophage
spreading inhibition in guinea pigs: Characteristics of the reaction and compa-
rison with migration inhibition and lymphocytic transformation tests. — Pro-
ceedings of the Yugoslav Immunological Society, 1976, 4, 24.
3.14. ВЫЯВЛЕНИЕ
ХЕМОТАКСИЧЕСКИХ ФАКТОРОВ
3.14.1. ПРИНЦИП МЕТОДА
Лейкоциты обладают способностью направленно перемещаться
под действием хемотаксического раздражения in vitro. В от-
сутствие такого раздражения лейкоциты движутся беспорядоч-
но, они начинают перемещаться в одном направлении под вли-
янием веществ, вызывающих хемотаксис. Хемотаксическое ори-
ентирование происходит под воздействием градиента концен-
трации. От хемотаксиса следует отличать хемокинез — усилен-
ное. но ненаправленное движение клеток под влиянием
некоторых веществ, содержащихся в окружающей среде [1].
Хемотаксические факторы возникают in vitro при реакциях гу-
морального и клеточного иммунитета. Так, например, комплек-
сы АГ — АТ опосредованно, через активацию комплемента,
оказывают хемотаксическое воздействие на нейтрофилы, макро-
фаги, эозинофилы, базофилы и лимфоциты [2, 3]. В реакциях
клеточного иммунитета, протекающих in vitro, также могут об-
333
Рис. 114. Составные части камеры для хемотаксиса.
разовываться хемотаксические факторы, стимулирующие все вы-
шеперечисленные типы клеток. В 1962 г. Boyden предложил
использовать микропористые фильтры для изучения хемотакси-
са. Метод основан на способности лейкоцитов проходить сквозь
пористую мембрану в направлении градиента концентрации
веществ, вызывающих хемотаксис. Мембрана разделяет камеру
на две части, по одну сторону помещают лейкоциты, по дру-
гую— исследуемое вещество. Величина пор мембраны должна
быть такой, чтобы лейкоциты проходили через нее только в ре-
зультате активной миграции. Метод Boyden можно усовершен-
ствовать, поставив второй фильтр. Поскольку часть мигрирую-
щих клеток отделяется от мембраны, они не полностью выяв-
ляются. Второй, непроницаемый для лейкоцитов, фильтр улав-
ливает эти клетки. Проблему отделения клеток можно решить
при помощи так называемого метода «Leading-front», в кото-
ром миграция клеток заканчивается до того, как лейкоциты
достигнут нижней- поверхности мембраны1.
3.14.2. МЕТОДИКА
3.14.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходима камера для хемотаксиса из пиакрила
(полиметилметакрилат) с внешним диаметром 15 мм и внутрен-
ним 14 мм. На рис. 114 показаны составные части камеры: кор-
пус с внутренней резьбой и держателем для мембраны, 2 мем-
браны, уплотнительное кольцо, верхняя камера с резьбой. Нуж-
но иметь также нитроцеллюлозные мембраны диаметром 12 мм
(Sartorius, ФРГ). Для исследования мононуклеаров диаметр
пор мембраны должен составлять 5—8 мкм, нейтрофилов —
3 мкм. Размер пор непроницаемой для лейкоцитов мембраны —
0,3—0,45 мкл.
334
3.14.2.2. ПОЛУЧЕНИЕ КЛЕТОК
Наиболее часто индикаторными клетками служат КПЭ кроли-
ка или морской свинки, а также лейкоциты крови человека.
Обогащенный гранулоцитами экссудат (95—99% нейтрофилов)
получают внутрибрюшинным введением 100 мл стерильного
•бульона (25 г питательного бульона на 1 л дистиллированной
воды, Институт иммунных препаратов и питательных сред, Бер-
лин) с последующей через 24 ч пункцией брюшной стенки и
промыванием брюшной полости. Обогащенный макрофагами
экссудат (50—80% макрофагов) получают на 5—7-й день пос-
ле внутрибрюшинного введения вазелинового масла кролику
(100 мл) или морской свинке (20 мл). Клетки трижды отмыва-
ют раствором Хенкса или средой Игла при 5—10 °C. Лейкоци-
ты крови человека в зависимости от целей исследования ис-
пользуют в виде тотального препарата или выделяют отдель-
ные типы клеток. Например, нейтрофилы и моноиуклеары по-
лучают разделением в градиенте плотности по Бейюму (см.
раздел 3.2). Нейтрофилы используют в виде суспензии 2—ЗХ
ХЮ6 клеток/мл, макрофаги — 4—5Х106 клеток/мл. К суспен-
зии клеток добавляют либо сыворотку (100—150 мл/л), либо
10—20 г/л сывороточного альбумина.
3.14.2.3. ХЕМОТАКСИС
В нижнее пространство камеры вносят 1 мл исследуемого ма-
териала или контрольной среды. В качестве примера мы рас-
смотрим определение хемотаксических свойств бесклеточной
•надосадочной жидкости, полученной из стимулированной куль-
туры лимфоцитов (рис. 115). После заполнения нижнюю каме-
ру закрывают непроницаемой для клеток мембраной, стараясь
избежать образования пузырей воздуха. Сверху кладут вторую
мембрану, проницаемую для лейкоцитов, и фиксируют винто-
вым запором. В верхнее пространство вносят 1 мл клеточной
суспензии, закрывают камеру покровным стеклом для предот-
вращения испарения. Инкубируют камеры при 37—39 °C. Про-
должительность инкубации нейтрофилов составляет 3 ч, моно-
«уклеаров — 5 ч. По окончании инкубации жидкость из верх-
ней камеры отсасывают, убирают мембраны и, пометив верх-
нюю и нижнюю стороны, аккуратно ополаскивают физиологиче-
ским раствором, через 3 мин фиксируют 96% этанолом. Окра-
шивание можно производить 1% метиленовым синим (10 мин).
Количественную оценку проводят путем подсчета мигриро-
вавших клеток на обеих мембранах. Просматривают по 10 по-
лей зрения на нижней поверхности верхней мембраны и на
верхней поверхности нижней мембраны. Учитывают только те
клетки, которые полностью прошли через верхнюю мембрану.
Подсчитывают среднее количество клеток в поле зрения в
двойных пробах. Для достоверности результатов следует учи-
335
Рис. 115. Мембрана с промигрировавшими гранулоцитами крови человека.
Хемотаксический агент — надосадочная фракция культуры активированных лимфоцитоз,
а — нижняя сторона верхней мембраны; б — верхняя сторона нижней мембраны.
тывать спонтанную миграцию (контроль с чистой культураль-
ной средой). Рассматривают либо разность значений стимули-
рованной и спонтанной миграции, либо их отношение. Для
сценки хемокинетического эффекта исследуемую субстанцию
вносят в верхнюю и нижнюю камеры. О наличии хемотаксиче-
ского эффекта говорят в тех случаях, когда миграция клеток,
обусловленная градиентом концентрации, выше, чем в отсутст-
вие градиента.
3.14.3. ОЦЕНКА МЕТОДА
Метод, предложенный Boyden, и его различные модификации
в настоящее время представляются методом выбора для изуче-
ния хемотаксиса лейкоцитов. Этот метод позволяет получить
ценные данные относительно механизмов хемотаксиса и спе-
цифического и неспецифического воспаления. В клинике его ис-
пользуют для оценки функциональной активности лейкоцитов и
для выявления клеточного иммунитета. Решающее значение для
воспроизводимости результатов имеет выбор фильтров. Поэтому
перед использованием в эксперименте мембраны подлежат ис-
пытанию со стандартным хемотаксическим агентом (например,
фильтрат культуры бактерий) для выяснения, возможна ли
миграция через данные фильтры, не слишком ли она велика
в контроле. Для уменьшения разброса данных следует пользо-
ваться одной и той же партией проверенных фильтров. Кроме
того, для получения сравнимых данных следует пользоваться
суспензиями клеток одинаковой концентрации. Разброс данных
может составлять 10—20%.
К числу недостатков метода можно отнести сложности в оп-
ределении отдельных типов клеток. Хотя известны сообщения
об успешной идентификации мигрирующих клеток, большинст-
во авторов скептически относятся к такой возможности. Этот
недостаток особенно заметен, когда речь идет об исследовании
макрофагов. Обогащенные макрофагами экссудаты содержат’
также нейтрофилы, которые особенно сильно реагируют на хе-
мотаксические стимулы. Поскольку хемотаксическая чувстви-
тельность нейтрофилов намного выше, чем у макрофагов, даже
небольшая примесь нейтрофилов может существенно искажать
результаты исследования. При исследовании хемотаксиса ней-
трофилов проблему определения типов клеток можно обойти
использованием мембран с таким размером пор (0,65 мкм),
который не допускает миграции крупных клеток. Ио в этом
случае приходится подсчитывать клетки во всех слоях фильт-
ра. Поликарбонатные фильтры (нуклеопор) отличаются от
миллипоровских фильтров однотипной цилиндрической формой
пор и меньшей толщиной мембраны (12 мкм по сравнению
с 150 мкм). Их преимущество состоит в том, что клетки, про-
ходя через мембрану, менее деформируются и легче подда-
ются морфологической идентификации. Время инкубации тоже
можно сократить. Чувствительность метода можно повысить,
если определять расстояние клеточного фронта от поверхности
мембраны при помощи микрометра микроскопа. Время инку-
бации при этом сокращается до 1 ч для нейтрофилов и до 3 ч
для мононуклеотидов. Фронт миграции определяют по двум
ведущим клеткам, которые отчетливо выделяются па плоскости.
Фильтр делают прозрачным при помощи ксилола. Этот метод
превосходит другие известные методы с использованием филь-
тров ввиду его большей чувствительности; относительно не-
большие изменения миграции, порядка 10 мкм, являются стати-
стически значимыми. Разброс результатов и воспроизводимость
также лучше. Варьируя концентрации исследуемых веществ,
можно уточнить данные о миграции в зависимости от градиента
и тем самым точнее дифференцировать направленную мигра-
цию от ненаправленной. Этот метод часто подвергают критике
за то, что он применим только к наиболее быстро мигрирую-
щим клеткам. Использованием меченных радиоактивным хро-
мом лейкоцитов и автоматическим подсчетом клеток можно су-
щественно снизить субъективные ошибки [4,5]. Еще один уп-
рощенный метод изучения хемотаксиса основан на использова-
нии бумажных фильтров, смоченных раствором хемотаксическо-
го агента. Мембрану накладывают на такой фильтр, оценивают
22—990
337
толщину слоя клеток, проникших в мембрану [7]. Лейкотаксис
•можно определять, используя в качестве среды агарозу [8].
Но сравнению с мембранными методами возникает дополни-
тельная возможность одновременного определения хемотакси-
ческой и спонтанной миграции. Снижаются необходимое для
проведения анализа количество клеток и затраты времени.
Остается только ждать, вытеснит ли агарозный метод мембран-
ные.
3.14.4. СПИСОК ЛИТЕРАТУРЫ
1. Keller Н. U., Wilkinson Р. С., Abercrombie М. et al. A proposal for the defi-
nition of terms related to locomotion of leukocytes and other cells. — Clin.
Exp. Immunol., 1977, 27, 377—380.
2. Lett-Brown M. A., Boetcher P. A., Leonard E. J. Chemotactic responses of nor-
mal human basophils to C5a and to lymphocyte-derived chemotactic factor. —
J. Immunol., 1976, 117, 246—252.
3. El-Naggar A. K„ van Epps D. E„ Williams R. C. Human B- and T-lymphocyte
locomotion in response to Casein, C5a, and f-Met-Leu-Phe. — Cell. Immunol.,
i980, 56, 365—373.
• 4. Wine A. C., Kellermeyer R. A computer-based method for the quantification
of leukocyte chemotaxis. — J. Lab. Clin. Med., 1976, 88, 487—490.
• 5. Turner S. R. ACDAS: an automated chemotaxis data aquisition system.—
J. Immunol. Meth., 1979, 28, 355—360.
• 6. Hill H. R., Hogan N. A., Mitchell Th. G. et al. Evaluation of a cytocentrifuge
method for measuring neutrophil granulocyte chemotaxis. — J. Lab. Clin.
Med., 1975, 86, 703—710.
7. Addison I. E„ Babbage J. IF. A raft technique for chemotaxis: a versatile
method for clisical studies. — J. Immunol. Meth., 1976, 10, 385—388.
'8. Nelson R. D., Quie P. G., Simmons R. L. Chemotaxis under agarose: a new
and simple method for measuring chemotaxis and spontaneous migration of
human polymorphonuclear leukocytes and monocytes. — J. Immunol., 1975,
.115, 1650—1656.
3.15. КОЖНОРЕАКТИВНЫЙ ФАКТОР
3.15.1. ПРИНЦИП МЕТОДА
Кожнореактивный фактор (КРФ) впервые описан Bennet и
Bloom в 1968 г. По своему происхождению и свойствам он от-
носится к лимфокинам и обнаруживается в бесклеточной над-
осадочной фракции культур лимфоцитов после специфической
и неспецифической стимуляции, в культурах постоянно под-
держиваемых лимфоидных клеток, в органах животных с раз-
вивающейся иммунной реакцией и у животных носителей опу-
холей, вызывающих асцит [9, 10]. КРФ до настоящего времени
определяли в БНФ культур лимфоцитов всех видов лаборатор-
ных грызунов (кролик, морская свинка, крыса, мышь) и чело-
века [2,3]. Его действие видонеспецифично [9]. Специфическая
и неспецифическая индукция образования КРФ требует срав-
нительно непродолжительного контакта индуктора и клеток.
Так, для системы ФГА/периферические лимфоциты человека
338
достаточно всего 4—5 ч. Максимальное высвобождение КРФ*
в зависимости от использованной in vitro системы колеблется
между 6 и 24 ч [9]. КРФ выявляют при помощи внутрикожной
пробы, которая вызывает появление реакции туберкулинового-
типа на месте введения. Оптимальное лабораторное животное-
для этих целей—морская свинка, можно использовать также-
крыс и кроликов.
3.15.2. МЕТОДИКА
3.15.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы нужны морские свинки обоих полов весом 200—
300 г, бесклеточный материал, содержащий КРФ, в качестве
контроля БНФ культуры нестимулированных лимфоцитов с до-
бавлением антигена, можно лектина, или инактивированная на-
греванием (30 мин, 100°C), электрическая машинка для стриж-
ки шерсти (использование химических депиляторов ввиду их
раздражающего действия на кожу не рекомендуется), туберку-
линовые шприцы с ценой делений 0,02 мл.
3.15.2.2. ВЫПОЛНЕНИЕ ЭКСПЕРИМЕНТА
За 24 ч до начала опыта волосы на спине морской свинки'
тщательно выстригают. Внутрикожно вводят 0,1—0,2 мл КРФ-
содержащего материала. На одном животном можно поставить
до 8 проб. Через 16—24 ч после инъекции проводят визуальную
и (или) гистологическую оценку результатов.
3.15.2.3. ВИЗУАЛЬНАЯ ОЦЕНКА
Через 20—30 мин после инъекции возникает участок наблюде-
ния, через 6 ч начинает развиваться эритема с прогредиентным'
уплотнением, которые через 24 ч начинают претерпевать обрат-
ное развитие. Оценку проводят по следующим критериям.
1. Степень эритемы: субъективная оценка от 0 до 3,0 (отсутст-
вие покраснения — темно-красная окраска кожных покровов).
2. Диаметр зоны эритемы (продольный, поперечный).
3. Степень уплотнения (субъективная оценка или измерение:
толщины кожной складки).
3.15.2.4. ГИСТОЛОГИЧЕСКАЯ ОЦЕНКА
Гистологически определяют протекающий в две фазы воспали-
тельный процесс соединительной и жировой ткани. Он начина-
ется приблизительно через 4 ч после инъекции с миграции
лейкоцитов в область посткапиллярных венул, сопровождается
рыхлой периваскулярной и интерстициальной инфильтрацией
преимущественно нейтрофильными гранулоцитами. Через 6—
22* 339
Рис. 116. Периваскулярный инфильтрат, состоящий из макрофагов и лимфо-
цитов, через 24 ч после внутрикожной инъекции КРФ человека (индуциро-
ванного ФГА). Окраска по Гимзе. Х?30.
8 ч наблюдается усиленное накопление моноцитов и макрофа-
гов (рис. 116). Оценка изменений производится качественно
после фиксирования и заключения объектов с последующей
окраской по Гимзе. Полу количественную оценку можно про-
извести, вырезав место реакции, обработав ткань трипсином и
подсчитав количество высвободившихся ядросодержащих кле-
ток.
3.15.2.5. ИСТОЧНИКИ ОШИБОК
Существуют две основные причины ошибок.
1. Контаминация исследуемого материала посторонним раз-
дражающим кожу материалом. Исключение путем точной био-
химической характеристики инъецируемой пробы.
2. Ошибочное восприятие восстановительных процессов во-
круг канала от иглы, за КРФ-обусловленное воспаление.
Ошибку устраняют путем анализа нескольких срезов.
Наиболее точный критерий оценки — гистологический, по-
скольку эритема и уплотнения как сосудистые явления не
обязательно могут быть связаны причинно-следственным отно-
шением с КРФ- Еще один способ устранения ошибок заключа-
ется в одновременном введении под кожу или внутривенно
0,5 М раствора L-фукозы. Поскольку КРФ-индуцированная ре-
акция будет тормозиться за счет конкуренции за рецепторы,
содержащие L-фукозу, станет возможной дифференциальная
диагностика неспецифических раздражений кожи, например,
реакции Артюса [1].
340
3.15.3. ОЦЕНКА МЕТОДА
Результаты получения КРФ, так же как и других методов
диагностики, основанных на определении лимфокинов, в экспе-
риментальных и клинических наблюдениях достаточно полно
коррелируют с другими проявлениями клеточного иммунитета
(бласттрансформация, кожные реакции замедленного типа), но
более чувствительны к иммуносупрессивной терапии. В диаг-
ностических целях имеет смысл исследовать сыворотки на на-
личие КРФ у больных с опухолями со спонтанным образовани-
ем лимфокинов (например, синдром Сезари) [8]. Относительно
постановки КРФ-теста больным с целью оценки их иммуноре-
активности не имеется пока однозначных данных. Химическая
природа КРФ требует дальнейшего изучения. Исследования
с антилимфокиновыми сыворотками показали, что КРФ не
идентичен MIF [4, 5, 7]. Поскольку соответствующие исследова-
ния еще не проводились, нельзя исключить, что КРФ обладает
свойствами хемотаксического фактора в отношении макрофагов.
3.15.4. СПИСОК ЛИТЕРАТУРЫ
1. Baba Т., Yoshida Т., Cohen S. Suppression of cell-mediated immune reactions
by monosaccharides. — J. Immunol., 1979, 122, 838—841.
2. Bakker W. W., van den Berg W., Hoedemaeker P. J. Lymphokines in sensiti-
zed rats. II. Skin reacting factors and lymphonode activating substances ori-
ginating from thymocytes. — Ztschr. Immun. Forsch., 1975, 150, 31—44.
3. Bray M. A., Dumonde D. C., Hanso J. M. et al. Heterogeneity of guinea-pig
lymphokines revealed by parallel bioassay. — Clin. Exp. Immunol., 1976, 23,
333—346.
4. Geczy С. I., Friedrich W., De Week A. L. Production and in vivo effect of
antibodies against guinea-pig lymphokines. — Cell. Immunol., 1975, 19.
65—77.
5. Onozaki K., Haga S„ Miura K. et al. Production of antibody against guinea-
pig MIF. — Cell. Immunol., 1979, 48, 258—266.
6. Warrington R. J., Buehler S. K-, Roberts К. B. Inflammatory factors produ-
ced by sensitized guinea-pig peripheral blood lymphocytes. — Experientia,
1976, 32, 110—112.
7. Yoshida T., Bigazzi P. E., Cohen S. The production of antiguinea-pig lympho-
kine antibody. — J. Immunol., 1975, 114, 688—691.
8. Yoshida T., Edelson R., Cohen S. Migration inhibitory activity in serum and
cell supernatants in patients with Sezary syndrome. — J. Immunol., 1975,
114, 915—918.
9. Zschiesche IF. Biologische Eigenschaften und Identitat des hautreaktiven
Factors. — Nova Acta Leopoldina N. F., 1975, 41, No 217, 377—385.
10. Zschiesche W„ Fahibush B„ Gutsche W. Presence and characterization of
lymphokines in mouse ascites tumor fluids.— Int. Arch. Allergy, 1976, 51,
742.
341
3.16. КОЖНЫЕ РЕАКЦИИ
ЗАМЕДЛЕННОГО ТИПА
3.16.1. ПРИНЦИП МЕТОДА
Кожные реакции замедленного типа наблюдаются у сенсибили-
зированных животных и человека после повторного внутрикож-
ного введения специфического АГ. Такая измененная реактив-
ность организма, описанная Пирке в 1906 г., называется аллер-
гией. Различие между острыми состояниями (анафилактиче-
ский шок, феномен Артюса) и истинными замедленными реак-
циями впервые проведено Sinsser в 1925 г. Классическая кож-
ная реакция замедленного типа (туберкулиновая) в ответ на,
подкожное введение АГ была впервые описана Р. Кохом в,
1890 г. В модификации Манту (внутрикожная проба, 1919 г.)
эта реакция прочно вошла в арсенал экспериментальных и;
клинических методов. Еще одна разновидность замедленной!
кожной реакции, контактная сенсибилизация, впервые описана
Cash в 1911 г. В отличие от анафилактических форм кожные-
реакции замедленного типа переносятся только с иммунными-
лимфоцитами. Любой АГ может вызывать такие реакции..
Контактная сенсибилизация развивается даже к простым орга-
ническим соединениям, которые после внедрения в кожу и со-
единения с белками эпидермиса становятся иммуногенными.
Для развития кожной реакции замедленного типа необхо-
димо соблюдение ряда условий [4].
— Химические и физико-химические свойства АГ. Сенсиби-
лизация может быть осуществлена аппликацией растворимых и
взвешенных АГ в форме водно-масляной эмульсии. Фрейндом
был разработан для этого адъювант следующего состава: ми-
неральное масло—10 мл, эмульгатор А—1 мл, убитые мико-
бактерии туберкулеза — 50 мг, мертиолят — 4 мг. Введение АГ
в полном адъюванте Фрейнда создает оптимальные условия
для развития кожных реакций замедленного типа. Определен-
ную роль играет химическое строение АГ, например, метилиро-
ванные или ацетилированные сывороточные белки индуцируют
почти исключительно реакции замедленного типа. Для развития
контактной сенсибилизации чаще всего используют нанесение
АГ на кожу кисточкой.
— Доза АГ и способ введения. Хотя не существует никаких
общих правил, известно, что малые дозы АГ при их внутри-
кожном введении закономерно приводят к развитию кожных
реакций замедленного типа.
— Напряженность иммунитета. Напряженность иммунитета
зависит от иммуногенности АГ. Оптимальным считается повтор-
ное введение АГ через 2—3 нед после первого. В целом латент-
ный период обратно пропорционален введенной дозе АГ. Со-
стояние напряженного иммунитета может длиться годами и да-
342
же сохраняться пожизненно. По мере старения происходит ос-
лабление иммунитета.
— Вид животного. Кожные реакции удобнее всего вызывать
у кроликов и морских свинок. Можно также использовать
крыс. Для развития кожных реакций у мышей необходимы
•особые условия постановки опыта [3].
— Возраст и упитанность животных. Морские свинки де-
монстрируют оптимальную реактивность в возрасте 2—3 мес,
по мере взросления она уменьшается. В корм животных необ-
ходимо добавлять витамин С (в отсутствие зелени), в условиях
авитаминоза полноценная иммунизация невозможна.
Классическая кожная реакция замедленного типа начинает
развиваться через 6 ч после внутрикожного введения АГ. Раз-
витие начинается с эритемы, постепенно нарастает уплотнение
тканей; все явления достигают максимального развития через
24—48 ч после введения АГ. Гистологически обнаруживают
периваскулярные и интерстициальные инфильтраты, состоящие
из лимфоцитов, моноцитов и макрофагов (у морской свинки
инфильтрация отмечается в жировом слое кожи, у человека —
в коже и подкожной клетчатке). Гранулоциты обнаруживаются
только в инициальной фазе. Проницаемость сосудов в области
инфильтрата повышена. Помимо классической формы реакции,
известна еще так называемая реакция Jones — Mote, которую
выявляют по двум критериям: раннему развитию (уже через
1 нед после иммунизации) и краткой длительности реактивно-
сти.
Эта форма развивается прежде всего после иммунизации
растворимыми белками или комплексами АГ — АТ в неполном
адъюванте Фрейнда (без микобактерий). Используемый пре-
парат должен обладать сильной иммуногенностью. Реакция
Jones — Mote не является самостоятельной формой реактивно-
сти, она скорее представляет собой особую форму течения клас-
сической кожной реакции замедленного типа при одновремен-
ном сильно выраженном образовании регуляторных, тормозя-
щих АТ.
Изредка выделяемая по гистологическим критериям ограни-
ченная базофильная кожная гиперчувствительность представля-
ет собой, очевидно, морфологический эквивалент реакции
Jones — Mote и переносится только с клетками.
По сравнению с туберкулиновой реакцией контактная сен-
сибилизация отличается большим участием гранулоцитов и
меньшим — макрофагов. По мере ее развития возникает скоп-
ление жидкости под эпидермисом и базальный акантоз. Извест-
ны также случаи контактной сенсибилизации с выраженным
участием базофильных лейкоцитов.
343
3.16.2. МЕТОДИКА
3.16.2.1. КОНТАКТНАЯ СЕНСИБИЛИЗАЦИЯ МЫШЕЙ
3.16.2.1.1. Материалы и оборудование
Для работы нужно иметь пикрилхлорид или оксазолон (2-фе-
нил-4-этоксиметиленоксазолон), оливковое масло очищенное
(можно арахисовое, подсолнечное), полный адъювант Фрейн-
да, щипцы для маркировки ушей, мышей (самки).
3.16.2.1.2. Ход исследования
Схема опыта: через 6—8 дней после контакта с антигеном (им-
мунизация) повторный контакт с тем же АГ приводит к раз-
витию местной воспалительной реакции. В качестве места кон-
такта выбирают ухо, реакция приводит к его утолщению. Ре-
комендуется использовать для постановки реакции одно и то
же ухо (например, правое), а для нумерации соответственно —
левое. Схема (см. табл. 32) зависит от силы используемого
АГ и от того уровня реактивности, который намечается до-
стичь.
Схема I применяется при работе со слабыми антигенами
или при необходимости получать длительно сохраняющуюся
реактивность. Схема II или III применяется при работе с силь-
ными АГ, например в терапевтических целях или при скри-
нинге.
Группы животных: выраженность реакции в значительной
мере зависит от штамма (величина набухания, разброс дан-
ных), поэтому в предварительных опытах подбирают оптималь-
ный штамм. Желательно использовать самок, поскольку самцы
часто имеют следы укусов. Ввиду необходимости тщательного
контроля перекрестной реактивности животных метят не
красителем как обычно, а прн помощи ушной бирки. Условия
подбора представлены в табл. 33. Необходимо взвешивать
животных всех групп до начала и по окончании опыта, это по-
может выявить общетоксический эффект исследуемых препара-
тов.
Иммунизация и вызывание реакции: готовят 2—3% раствор
пикрилхлорида в очищенном оливковом масле; 3% раствор
Таблица 32. Схема индукции контактной сенсибилизации
Иммунизация Адъювант +АГ АГ АГ Определение (дни)
1-е 2-е
Схема I Схема II Схема III 0. +7 0. + 14 +7 0. +21 + 14 +7 +22 + 15 +8
344
Таблица 33. Распределение животных по группам для контактной
сенсибилизации
Группа животных Иммунизация Обработка Вызывание реакции
Негативный контроль Масло Нейтральное вещество Антиген
Позитивный контроль Антиген Растворитель Антиген
Испытуемая группа Антиген Нейтральное вещество Антиген
оксазолона — на чистом спирте, иногда вместо спирта исполь-
зуют масло.
При иммунизации по схеме I 0,2 мкмоля вещества суспен-
дируют в 0,15 мл адъюванта (доза на 1 мышь). Инъекцию
делают в основание хвоста. Для накожной аппликации 0,1 мл
смеси наносят на спину животного и втирают стеклянной па-
лочкой на площади 2—3 см2. Депиляция требуется не всегда,
но если в ней возникает необходимость, то волосы выщипыва-
ют за один день до опыта (химическую депиляцию не произво-
дят). Реакцию вызывают, смазывая намеченное ухо с двух сто-
рон тем раствором, который использовали для иммунизации.
При работе с контактными аллергенами следует всегда на-
девать резиновые перчатки.
Измерение: толщину уха измеряют при помощи любого под-
ходящего устройства типа штангенциркуля, избегая сдавлива-
ния тканей уха. Измерение проводят, как правило, через 24 ч,
в некоторых специальных случаях через 48 ч.
Некоторые авторы рекомендуют электромеханическое опре-
деление толщины [2]. Описаны способы определения гиперчув-
ствительности замедленного типа при помощи альбумина, ме-
ченного 131[1] [7]. Оценка результатов: если при определении
толщины уха используют измерительный циркуль для покров-
Таблица 34. Влияние депиляции иа контактную сенсибилизацию мышей
Предварительная обработка Иммунизация Вызывание реакции Толщина уха через 24 ч
Депиляция Масло ПХ/масло 3,7±0,86
Депиляция ПХ/масло ПХ/масло 6,5±0,80
Без депиляции ПХ/масло ПХ/масло 7,6±0,61
Депиляция Этанол ПХ/этанол 5,0±0,71
Депиляция ПХ/этанол ПХ/этанол 9,6±0,61
Без депиляции ПХ/этанол ПХ/этанол 8,3+1,56
ПХ — 2,5% раствор пикрилхлорида в масле или этаноле.
В каждой группе по 10 самок в возрасте 8 нед (10 AB/Jena или SPF).
345
ных стекол, то толщину можно выражать в относительных ве-
личинах или в абсолютных (в мм после соответствующей ка-
либровки). Сравнение средних значений опыта с данными по-
зитивного контроля проводят методами вариационного анализа.
3.16.2.2. КОЖНЫЕ РЕАКЦИИ ЗАМЕДЛЕННОГО ТИПА
У МЫШЕЙ ИЛИ КРЫС
3.16.2.2.1. Материалы и оборудование
Для работы нужно иметь очищенный антиген, полный адъювант
Фрейда, измерительный циркуль, мышей или крыс.
3.16.2.2.2. Этапы исследования
Иммунизация и вызывание реакции. Иммунизацию проводят
очищенным АГ (например, эритроциты барана, белковые препа-
раты, аллогенная суспензия клеток), вводя его в подошву зад-
ней лапы. Обычно вводят 0,01—0,04 мл очень тонкой иглой,
продвигая ее как можно дальше. При необходимости сенсиби-
лизацию повторяют через 7 дней. Реакцию вызывают, вводя
тот же самый АГ через 9—10 дней под кожу задней лапы, ко-
торую не использовали в эксперименте.
Измерение и оценка результатов: в результате реакции про-
исходит опухание той задней лапы, на которой вызывали реак-
цию. Степень опухания оценивают по изменению толщины ла-
пы или ее объема (рис. 117). В обоих случаях требуется выдер-
живать одинаковые условия измерения. Ошибку можно умень-
шить, измеряя средние величины отпечатков лапы, окрашенной
тушью. Измерение проводят через 24 ч и по возможности вто-
рой раз через 48 ч после вызывания реакции. Сравнивают тол-
щину (объем) обработанной и необработанной задней лапы
одного и того же животного. Данные соотносят с результатами
позитивного контроля с адъювантом Фрейнда (ПАФ).
3.16.2.3. КОЖНАЯ РЕАКЦИЯ ЗАМЕДЛЕННОГО ТИПА
У МОРСКИХ СВИНОК
3.16.2.3.1. Материалы и оборудование
Используют самок морских свинок весом 300—400 г. Нужно
иметь антигены, ПАФ, туберкулиновые шприцы, электрическую-
машинку для стрижки шерсти, измерительный циркуль.
3.16.2.3.2. Этапы исследования
Обычно иммунизируют животных эмульсией соответствующего
АГ в ПАФ. АГ вводят подкожно в количестве 0,1—0,2 мл во
все 4 подушечки конечностей или внутримышечно. Количество<
346
АГ может варьировать в зависи-
мости от его природы, например,
БЦЖ смешивают в количестве
1 мг/мл адъюванта. Интервал
между иммунизациями также
может меняться, для БЦЖ опти-
мальный интервал составляет
10 дней. Реакцию вызывают вну-
трикожным введением АГ в вод-
ном растворе в область спины.
Непосредственно перед инъекци-
ей на соответствующем участке
кожи тщательно выстригают во-
лосы. Не следует испытывать
различные концентрации антиге-
нов на одном и том же живот-
Рис. 117. Измерительное устройст-
во для определения объема орга-
нов.
ном, так как ма<лые дозы анти-
генов способны вызвать десенсибилизацию и искажение резуль-
татов других экспериментов.
3.16.2.3.3. Оценка результатов исследования
Кожную реакцию замедленного типа оценивают через 24 и
(или) 48 ч после разрешающей инъекции. Ниже приводятся
критерии оценки результатов реакции.
— Эритема. Определяют величину эритематозной зоны. Если ее
диаметр больше 6 мм, говорят о положительном результате
реакции. Интенсивность эритемы также оценивают в баллах
(крестах) от 0 (отрицательная) до + + + + (темно-красная).
Геморрагический компонент нетипичен, его наличие должно
рассматриваться как признак сопутствующей реакции типа фе-
номена Артюса.
— Уплотнение. Измеряют толщину складки кожи, образуемой
одним и тем же участком поверхности до и после разрешающей
инъекции, или путем сравнения с симметричным участком ко-
жи на противоположной стороне тела.
При сравнении результатов, полученных на различных жи-
вотных, необходимо следить, чтобы разрешающая инъекция
всегда делалась в одно и то же место.
3.16.3. ОЦЕНКА МЕТОДА
Для некоторых специальных целей, например для получения
экспериментальных моделей заболеваний, были разработаны
особые модификации вышеописанного метода. Вызывание ре-
акции на роговице морской свинки может быть достигнуто
интракорнеальным введением 3—5 мкл антигена. Помутнение
роговицы вследствие инфильтрации может быть оценено коли-
чественно на длине волны 530 нм. Описаны также реакции
слизистых защечных мешков хомяка [9] и толстого кишечника
морской свинки [1]. Длительное время реакции замедленного
347
типа изучались на легких после ингаляции антигена. Развиваю-
щиеся явления представляют собой картину интерстициальной
пневмонии, количественную оценку реакции можно проводить
гравиметрически. Результаты кожной реакции в целом корре-
лируют с данными других тестов, полученными in vitro, кото-
рые обычно рассматривают как эквивалент реакций клеточного
иммунитета (реакция бласттрансформации, хемотаксис, тормо-
жение миграции макрофагов или гранулоцитов). Последние
основаны на действии тех или иных лимфокинов, кожные реак-
ции замедленного типа, по всей видимости, обусловлены взаи-
модействием многих лимфокинов (цитокинов).
Антигенная специфичность кожных реакций замедленного
типа, как правило, отличается от специфичности реакций гумо-
рального иммунитета. В экспериментах на животных было по-
казано, что в реакциях замедленного типа принимают участие
другие антигенные детерминанты. Это особенно ярко выражено
при иммунизации конъюгатами гаптен — носитель; специфич-
ность кожных реакций замедленного типа может определяться
как самим носителем, так и областью связывания носителя с
гаптеном [5].
3.16.4. СПИСОК ЛИТЕРАТУРЫ У
1. Askenase Р. W., Boone W. Т„ Binder Н. J. Colonic basophil hypersensiti-
vity.— J. Immunol., 1978, 120, 198—201.
2. Christie G. H. An electro-mechanical method for measuring hypersensitivity
in mice by increase of ear thickness. — J. Immun. Meth., 1975, 8, 257—262.
3. Crowle A. J. Delayed hypersensitivity in the mouse. — Adv. Immunol., 1975,
20, 197—264.
4. Drossier K. Grundlagen der zellvermittelten Immunitat. — In: Grundriss der
Immunbiologie (Hrsg. Ambrosius H., Rudolph W.) Fischer Verlag, Jena,
1978, S. 269—295.
5. Fleischman J. B., Eisen H. N. Antigen recognition in delayed type hypersen-
sitivity.— Cell. Immunol., 1975, 15, 312—320.
6. Heinecke H., Zschiesche IF. The use of contact sensitivity in mice for drug
testing. — Zwierzeta Laboratory]ne, 1975, 12, 11—12.
7. Kostiala A. A. I. Radiometric ear test index as a measure of delayed-type
hypersensitivity in the rat. — Immunology, 1977, 33, 561—571.
8. Lagrange P. H., Mackaness G. B. A stable form of delayed-type hypersen-
sitivity.— J. exp. Med., 1975, 141, 82—96.
9. Marshck M., Toto P., Kerman P. Delayed hypersensitivity in the Hamster
cheek pouch. — J. Immunol. Meth., 1977, 15, 325—330.
10. Phanuphak P„ Moorhead J. W., Claman H. Tolerance and contact sensitivity
to DNFB in mice. IV. Desensitization as a manifestation of increased proli-
feration of sensitized cells. — J. Immunol., 1975, 114, 1147—1151.
3.17. ТРАНСПЛАНТАЦИЯ КОЖИ
3.17.1. ПРИНЦИП МЕТОДА
Пересадка кожи у мышей, крыс и кроликов представляет собой
широко распространенный метод исследования in vivo некото-
рых специфических феноменов реакций клеточного иммунитета.
348
Различия антигенов гистосовместимости донора и реципиента
приводят вследствие сенсибилизации реципиента к более или
менее быстрому отторжению трансплантата. Отторжение
трансплантата рассматривается как результат цитотоксических
реакций, например иммунных реакций замедленного типа.
В отличие от других тканей кожа удобна в обращении и может
сохраняться некоторое время без утраты своих функций. Соб-
ственно трансплантация не является серьезным оперативным
вмешательством и мало влияет на общее состояние животных.
Кроме того, кожа — это единственная ткань, которая имплан-
тируется ортотопически, т. е. попадает в анатомически свойст-
венное ей окружение. За состоянием трансплантата возможен
простой ежедневный надзор. Биопсия и гистологические иссле-
дования тоже, как правило, не представляют особых сложно-
стей.
Мы описываем три вида трансплантации: со спины, с хвоста
и с боков; трансплантат перевивается также соответственно на
спину, хвост или бок.
3.17.2. МЕТОДИКА
3.17.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для пересадки кожи схвоста на хвост нужны гексобар-
битал-Na (1 г в 70 мл 0,15 М NaCl), аэрозоль «Ankerlast»
(Jena-pharm, ГДР), клейкий пластырь, бритвенные лезвия, пин-
цеты, этанол, специальные стеклянные пробирки (рис. 118).
Для пересадки схвоста на спину необходимы гексо-
барбитал-натрий (см. выше), аэрозоль «Ankerplast», смесь ва-
зелинового масла с ланолином 2+1; гипсовые повязки 1,5Х
X15 см, марля, ножницы, пинцеты, стерильный изотонический
раствор хлорида натрия.
3.17.2.2. ПЕРЕСАДКА КОЖИ С ХВОСТА
НА ХВОСТ
Мышам, донорам и реципиентам вводят 0,2 мл раствора гексо-
барбитала-натрия (на 20 г веса) внутрибрюшинно. Сначала
подготавливают операционное поле реципиента. Хвост тщатель-
но протирают спиртом. Очень острым бритвенным лезвием,
отступя 5 мм от основания хвоста, срезают с его верхней по-
верхности кусочек кожи размером 0,2X0,6—0,7 мм. Лучше все-
го держать хвост на левом указательном пальце и не делать,
слишком глубокие надрезы, вызывающие сильное кровотече-
ние. Первый кусочек кожи, срезанный у животного, снова при-
кладывают к ране, но в другом направлении (контроль ауто-
трансплантации). Ниже первого операционного поля срезают
еще два кусочка кожи и отбрасывают их. На эти ранки накла-
дывают кусочки кожи, взятые с хвоста донора. От одного до-
349
4Рис. 118. Пересадка кожи хвоста иа хвост.
'Справа — свободный трансплантат, слева — трансплантат, прикрытый стеклянной труб-
кой.
нора можно получить 10—12 трансплантатов. В заключение
раны обрабатывают анкерпластом и надевают на хвост стек-
лянную трубку с отверстиями для воздуха, защищающую
трансплантаты от механических повреждений. Нижний конец
трубки прикрепляется к хвосту клейким пластырем (рис. 118).
Через два дня трубку удаляют. В течение этих двух дней в ка-
честве подстилки желательно использовать фильтровальную
бумагу.
350
3.17.2.3. ПЕРЕСАДКА КОЖИ С ХВОСТА
НА СПИНУ
3.17.2.3.1. Подготовка трансплантата
Мышь донора убивают и отрезают хвост. Хвост протирают эта-
нолом и разрезают кожу ножницами вдоль хвоста. Пинцетом
полностью отделяют кожу. Ее переносят в стерильную чашку-
Петри и разрезают скальпелем на кусочки размером 5X10 мм.
Верхний и нижний концы не используют. В стерильном 0,15 М
NaCl или в другой подходящей среде кусочки кожи могут хра-
ниться в течение нескольких часов.
3.17.2.3.2. Трансплантация
Донорам вводят внутрибрюшинно 0,2 мл раствора гексобарби-
тала-натрия (на 20 г веса). Наркоз должен быть относительно
глубоким. Кожу спины подтягивают пинцетом и в стерильных
условиях осторожно вырезают кусочек глазными ножницами,
стараясь не повредить сосуды Panniculus carnosus. Поверхность
среза должна быть несколько больше поверхности транспланта-
та, поскольку при заживлении края раны стягиваются. Транс-
плантат переносят на рану и фиксируют анкерпластом. Сверху
кладут кусочек марли размером 15X15 мм, смазанный ланоли-
ном. Влажную гипсовую повязку накладывают на тело доволь-
но плотно, чтобы не было зазора. Гипсовую повязку можно
удалить через неделю (рис. 119).
3.17.3. ОЦЕНКА МЕТОДА
После удаления стеклянных трубок или гипса трансплантаты
должны ежедневно контролироваться для точного определения
времени отторжения. Состояние трансплантатов на хвосте оце-
нивается не всегда легко. Различают острую и хроническую
реакции отторжения. Даже при остром отторжении наблюда-
ется первичное приживление трансплантата (исключение состав-
ляют повторные трансплантаты). Самое раннее через 5 дней
начинается воспалительная реакция, сопровождающаяся по-
краснением и припухлостью. На спине отмечается сухое шелу-
шение и выпадение волос. Края трансплантата некротизируют-
ся, он сморщивается и приподнимается. В конце концов оста-
ется грибовидное образование на фиброзном стебле, которое
легко отпадает. Отторжение происходит под воздействием сен-
сибилизированных лимфоцитов и сопровождается массивной
миграцией полиморфно-ядерных лейкоцитов и моноцитов в эпи-
дермис [3].
Хроническая реакция отторжения отличается от острой бо-
лее сглаженным течением. Эпителий хозяина медленно подсла-
ивается под трансплантат, причем коллагеновые волокна тран-
сплантата вначале сохраняются. О протекании хронической»
351
Рис. 119. Пересадка кожи хвоста на спину.
Слева — подготовленное операционное поле; в середине — операционное поле с транс-
плантатом; справа — гипсовая повязка, покрывающая трансплантат.
реакции отторжения, при небольших генетических различиях
или при частичной толерантности, судить довольно сложно.
Интактный трансплантат приживается без воспаления и без
изменения волосяного покрова. Донор и реципиент должны
быть одного пола. Связанный с Y-хромосомой локус Н может
вызывать отторжение трансплантата даже у сингенных мышей.
Пересадка от самки к самцу возможна, но лучше использовать
«однополые» трансплантаты, поскольку существуют обуслов-
ленные полом различия в скорости приживления трансплантата
даже в пределах одной инбредной. линии. Пересадка с хвоста
на хвост болеее удобна ввиду меньших издержек и относитель-
ной легкости наблюдения. К числу недостатков метода отно-
сится то, что за состоянием трансплантатов трудно следить
из-за их малой величины. Рекомендуется исследование с лупой
и препаровальной иглой. Можно отказаться от использования
стеклянных трубок, если покрыть трансплантаты несколькими
слоями анкерпласта. Трансплантаты с хвоста на спину оцени-
вать гораздо проще. Интактные трансплантаты всегда отчет-
ливо видны. Отделение трансплантата может произойти вслед-
352
ствие его сползания с раны при наложении гипса Или вследст-
вие расчесов после удаления гипса. Рекомендуется после уда-
ления гипса еще раз использовать анкерпласт. В протокол со-
стояния трансплантата наблюдаемые изменения заносят по
возможности в простых и емких символах, например:
+ + + интактный трансплантат;
( + + +) интактный трансплантат, начинающееся воспаление;
+ + трансплантат воспален, отечно изменен, частично
приподнят;
(++) трансплантат начинает некротизироваться;
+ трансплантат полностью некротизирован, частично
приподнят;
— трансплантат отторгнут;
(—) трансплантат оторван (техническая ошибка).
Существуют, разумеется, переходные состояния между от-
дельными стадиями, но их оценка грешит субъективизмом, по-
этому должна проводиться всегда одним и тем же лицом. Из-
вестны местные различия в скорости отторжения, т. е., на-
пример, в зависимости от того, ближе или дальше (от основа-
ния хвоста) был приживлен трансплантат [2]. Фиксировать
трансплантат можно пришиванием, но это занимает больше
времени, чем вышеописанный метод. Можно трансплантиро-
вать также кожу ушных раковин [4]. Описан способ трансплан-
тации сердца новорожденных мышей в ушной карман [1]. Тех-
нически этот метод очень прост. Об отторжении можно судить
по ЭКГ. Недостатком метода является то, что даже у синген-
ных толерантных животных трансплантат прорастает соедини-
тельной тканью, которая не позволяет более улавливать ка-
кие-либо электрические импульсы. Но даже трансплантаты
сердца в некоторых случаях оказываются более долгоживущи-
ми, чем трансплантаты кожи, в пределах одинаковой комбина-
ции штаммов. Снятие ЭКГ требует определенного навыка, жи-
вотное при этом должно находиться в состоянии наркоза.
3.17.4. СПИСОК ЛИТЕРАТУРЫ
1. Davis W. С., McKenzie I. F. С., Melvold R. W. М. A comparison of skin and
heart rejection patterns in H-2 mutant mice. — Transplantation, 1980, 29, 189—
2. Kubai L„ Auerbach R. Regional differences in the growth of skin Trans-
plants. — Transplantation, 1980, 30, 128—131.
3. Silvers W. K, Wachtel S. S., Poole T. W. The behaviour of skin grafts incom-
patible with respect to skin alloantigens in mice rendered tolerant at birth
with lymphoid cells. —J. exp. Med., 1976, 143, 1317—1326.
4. Sugarbaker P. H., Chang A. E. Uncovered skin grafts in mice. — J. Immunol.
Meth., 1979, 31, 167—175.
5. Wachtel S. S., Silvers W. K. The role of passenger leukocytes in the anoma-
lous survival of neonatal skin grafts in mice.— J. exp. Med., 1972, 135, 388—
404.
23-990
353
3.18. АНАФИЛАКТИЧЕСКИЕ РЕАКЦИИ
Анафилактические реакции (реакции I типа) опосредованы мо-
лекулами IgE, которые взаимодействуют с рецепторами тучных:
клеток и базофилов. Появление аллергена приводит к выбросу
этими клетками медиаторов (гистамин, медленно реагирующая
анафилактическая субстанция, хемотаксический фактор эозино-
филов, тромбоцитактивирующий фактор, простагландины, в-
некоторых случаях 5-гидрокситриптамин). Медиаторы вызыва-
ют различные явления, в основном повышение проницаемости
сосудов и сокращение гладкой мускулатуры. Разработка экс-
периментальных моделей анафилаксии на животных стала воз-
можной только после достаточно глубоких исследований меха-
низмов анафилаксии. Экспериментальная анафилаксия исполь-
зуется при разработке эффективных антианафилактических
средств. Далее будут описаны важнейшие методы оценки вы-
свобождения медиаторов in vivo и in vitro.
3.18.1. МЕТОДЫ IN VITRO
3.18.1.1. ИССЛЕДОВАНИЯ НА ТУЧНЫХ
КЛЕТКАХ КРЫС
3.18.1.1.1. Материалы и оборудование
Для работы необходимы однополые* крысы весом 200—250 г,
пипетки, шприцы для инъекций, центрифужные пробирки (си-
ликонизированные), камера Тома, настольная центрифуга, ми-
кроскоп, флюориметр [в качестве замены можно использовать
Specol с приставкой для измерения флюоресценции FK и фото-
увеличителем (PhoM), ртутную лампу на 40 Вт (Osram),
фильтр GG-13, возбуждение на длине волны 365 нм, эмиссия
на 450 нм]; устройство для регистрации сокращений изолиро-
ванного отрезка толстого кишечника морской свинки; буфер
для тучных клеток [8,417 г NaCl, 0,201 г КС1, 0,360 г глюкозы,
довести до 1000,0 мл фосфатным буфером Серенсена с pH 7,2,
если необходимы ионы Са2+, к 1000 мл буфера для тучных
клеток добавляют 0,147 г СаС12Х2Н2О]; раствор Тироде [9 г
NaCl, 1 г глюкозы, 0,2 г КС1, 0,3 г СаС12, 0,1 г MgCl2, 5 мл
0,2 М раствора трис-аминометана; дистиллированная вода до
1,0 л], этиловый эфир; спиртовой раствор нейтрального крас-
ного (0,2%) или метиленового синего (0,025%); 2,5% раствор
толуидинового синего в 25% уксусной кислоте; 1 М NaOH;
3 М НС1; 0,1 М НС1 для калибровочной кривой, ЧСА; орто-фта-
левый альдегид (ОФТ)—0,05% раствор в метаноле; препарат
48/80 (1 мкг/мл приблизительно для 50% высвобождения
гистамина); протамиисульфат (Protamin forte) (37,5 мг/мл
приблизительно для 50% высвобождения гистамина); крысиная
354
антиовальбуминовая сыворотка; гистамин, дигидрохлорид; ат-
ропин; блокатор Нррецепторов, например, мепирамин (Меру-
ramin).
3.18.1.1.2. Выделение тучных клеток крысы
Крыс обескровливают под эфирным наркозом, затем вводят
внутрибрюшинно по 10 мл безкальциевого буфера для тучных
клеток. Энергично массируют живот в течение 90 сек и вскры-
вают брюшную полость. Пипеткой или шприцем без иглы от-
сасывают жидкость из полости брюшины. Центрифугируют ее
5 мин при 350 g, отбрасывают надосадочную фракцию и сус-
пендируют осадок клеток в 0,5 мл буфера для тучных клеток.
Можно использовать буферы другого состава [8]. Для поста-
новки опыта необходимо иметь клетки по меньшей мере от
4 крыс.
Определение количества клеток: на несущее
стекло камеры Тома наносят 1—2 капли спиртового раствора
нейтрального красного или метиленового синего, дают спирту
испариться, затем проводят подсчет клеток обычным способом.
Рекомендуется провести подсчет по всей камере, поскольку
тучные клетки (ТКл) составляют обычно около 3—6% от об-
щего числа клеток. После подсчета суспензию разводят Са2+-
содержащим буфером для ТКл, обычно рекомендуют исполь-
зовать суспензию 5x10s ТКл/мл. Большего обогащения суспен-
зии ТКл обычно не требуется, но если возникает в этом необ-
ходимость, то на 4 мл 38% раствора ЧСА в силиконизирован-
ной пробирке наносят 2 мл суспензии ТКл в безкальциевом
буфере (1.5Х106 клеток/мл), центрифугируют 45 мин при 450g.
Тучные клетки скапливаются в слое раствора альбумина.
Фазу над слоем раствора альбумина отбрасывают. Доводят
объем альбуминового слоя до 10 мл буфером для ТКл и центри-
фугируют 10 мин при 150 g. Тучные клетки осаждаются, их ре-
суспендируют в подходящем буфере и отмывают еще раз.
Доля ТКл в осадке обычно составляет свыше 90%. Известны
и другие модификации метода выделения ТКл [3, 5, 13J.
3.18.1.1.3. Высвобождение медиаторов из тучных
клеток крысы
Высвобождение медиаторов можно вызвать различными спо-
собами, например, добавлением препарата 48/80, протамин-
сульфата, сыворотки против IgG крыс, IgE-содержащей анти-
овальбуминовой сыворотки с последующим добавлением оваль-
бумина, обработкой ТКл реагинсодержащей сывороткой чело-
века с последующим добавлением аллергенов и пр. После
5-минутной инкубации 0,1 мл суспензии ТКл с 0,1 мл исследуе-
мого вещества (в контроле 0,1 мл буфера для ТКл) добавляют
0,1 мл вещества, высвобождающего гистамин, в подходящей
концентрации. После 10—30-минутной инкубации при 37 °C
пробы с ТКл разводят буфером до 2 мл. С каждым контролем
23*
355
и с каждой концентрацией исследуемого вещества ставят по
меньшей мере 3 параллельные пробы. После подведения объема
проб их центрифугируют 10 мин при 350 g и определяют медиа-
торы в надосадочной фракции.
3.18.1.1.4. Определение дегрануляции тучных клеток
Степень дегрануляции определяют в осадке клеток морфологи-
чески (рис. 120). Осадок ТКл суспендируют в 0,1 мл 2,5%>
раствора толуидинового синего в 25% уксусной кислоте. В этом
растворе клетки одновременно фиксируются и окрашиваются.
Микроскопируют 100 тучных клеток. Процент дегранулирован-
ных клеток подсчитывают по следующей формуле:
100[1 — (х— у) (у — г)],
х — совместное действие испытуемого материала и гистаминвысвобождающего
фактора; у — только действие гистаминвысвобождающего фактора; z — кон
троль (спонтанная дегрануляция).
3.18.1.1.5. Выявление медиаторов
Флюориметрическое определение гистамина. К 2 мл надосадоч-
ной фракции добавляют 0,4 мл 1 М NaOH, смесь встряхивают
на качалке. Добавляют 0,25 мл 0,05% раствора ОФА (см. вы-
ше), смесь встряхивают и отставляют на 4 мин при комнатной
температуре. Затем прибавляют 0,2 мл 3 М НС1. Если раствор
мутнеет (выпадение в осадок альбумина), его центрифугируют
5 мин при 2000 g. Проводят фотометрическое определение
гистамина. Гистамин образует с ортофталевым альдегидом про-
дукт конденсации, который при освещении УФ-лучами (365 нм)
флюоресцирует с максимумом около 450 нм. Концентрацию
гистамина определяют по калибровочной кривой (гистамин-
дигидрохлорид, растворенный в 0,1 М НС1, 1—20 мкг/мл).
В 1 мг гистамина дигидрохлорида содержится 0,602 мг свобод-
ного гистамина. Все исследуемые вещества проверяют на нали-
чие неспецифической флюоресценции, кроме проб с исследуе-
мым материалом и гистаминвысвобождающим препаратом, про-
водят следующие измерения:
— определение общего содержания гистамина (пробы с со-
ответствующим количеством тучных клеток 5 мин выдерживают
на кипящей водяной бане, центрифугируют, определяют гис-
тамин) ;
— определение спонтанного высвобождения (обработка проб
описана выше; ТКл суспендированы в буфере);
— определение степени высвобождения под воздействием толь-
ко гистаминвысвобождающего препарата;
— определение степени высвобождения под воздействием толь-
ко исследуемого вещества.
Точность анализа может быть еще повышена определением
гистамина, остающегося в осадке (кипячение проб, см. выше).
Биологический метод определения гистамина. Метод основан
на сокращении изолированного кусочка толстого кишечника
356
Рис. 120. Тучные клетки
крысы,
а — интактные; б — с начинаю-
щейся дегрануляцией; в —де-
гранулированные. Окраска ней-
тральным красным. Объектив
100Х, проектив 8Х» масляная
иммерсия.
морской свинки при
добавлении в среду
гистамина. Посколь-
ку антианафилактн-
ческие средства пода-
вляют высвобождение
гистамина, результа-
ты относят либо к ко-
личеству гистамина,
высвобождаемому под
воздействием гиста-
минвысвобождающих
препаратов, либо к
общему гистамину.
Общего гистамина из
5000—7000 ТКл доста-
точно для субмакси-
мального сокращения
изолированных фраг-
ментов кишечника
морской свинки. Чув-
ствительность кусоч-
ков кишечника контро-
лируется по степени
сокращения со стан-
дартными фармацев-
тическими препарата-
ми (гистамин ЗХ10-7
моль/л; ацетилхолин
1,6х Ю-7 моль/л).
Прочие методы оп-
ределения гистамина.
Обычно достаточно
чувствительности ме-
тодов определения ги-
стамина, описанных
выше. В ряде случаев
возникает необходимость выявлять гистамин в более низких
концентрациях. Имеются многочисленные публикации на эту
тему [1, 4, 10, 11, 16].
Определение медленно реагирующей анафилактической суб-
станции (МРС-А). МРС-А (лейкотриен) также вызывает со-
357
кращение гладкой мускулатуры, поэтому для ее определения
можно воспользоваться такой же биопробой, как и для выявле-
ния гистамина. Одновременное воздействие гистамина и МРС-А
может быть блокировано антигистаминными препаратами (бло-
каторы Hi-рецепторов, например, мепирамин в концентрации
Ю“6—10~7 моль/л); спонтанные сокращения подавляют атро-
пином (10~6—10~7 моль/л). Поскольку мепирамин легко ад-
сорбируется на стекле, рекомендуется использовать для опре-
деления гистамина и МРС-А различные сосуды. Промежутки
между отдельными измерениями должны быть не менее 5 мин.
Изучение структуры и синтез лейкотриенов могут открыть ши-
рокие перспективы для биохимического анализа.
3.18.1.2. ИССЛЕДОВАНИЯ НА БАЗОФИЛЬНЫХ
ГРАНУЛОЦИТАХ ЧЕЛОВЕКА [15]
Этот метод служит для определения сенсибилизации базофиль-
ных лейкоцитов у больных с атопией. Вещества, обладающие
способностью подавлять высвобождение медиаторов, инкубиру-
ют с препаратами лейкоцитов. Следует упомянуть о методе,
в котором базофилы нормального донора «нагревают» сыво-
роткой больных с атопией. Добавление соответствующего ал-
лергена приводит к высвобождению медиаторов. Метод требу-
ет специальной экстракции гистамина ввиду относительной
малочисленности базофилов в периферической крови человека
и низкого содержания в них гистамина.
3,18.1.2.1. Материалы и оборудование
Для работы необходимы шприцы на 20 мл, изогнутые иглы;
пипетки, микропипетки (10—100 мкл и 100—1000 мкл); пласти-
ковые центрифужные стаканы (10 мл); сосуды для реакции со
шлифами (10 мл); центрифуга с бакет-ротором (например,
Janetzki Т23); качалки; флюориметр; гепарин (5000 ME).
Растворы
Раствор декстранглюкозы: 3 г декстрана растворяют в 100 мл
0,15 М NaCl; в 25 мл этого раствора растворяют 750 мг D-глю-
козы.
Концентрат NaCl: 3,75 г трис-аминометана, 6,95 г NaCl,
0,370 г КС1, дистиллированная вода до 100 мл, pH 7,6 (имеет
критическое значение); для употребления концентрат разводят
(10 мл доводят дистиллированной водой до 100 мл).
Концентрат кальция: 900 мг СаС12Х2Н2О растворяют в дис-
тиллированной воде (до 100 мл).
Концентрат ЧСА: 33 мг высушенного ЧСА растворяют в
10 мл 0,15 М NaCl (хранение не более недели).
Трис-альбуминовый буфер: 10 мл концентрата NaCl доводят
дистиллированной водой приблизительно до 98 мл, добавляют
1 мл концентрата ЧСА, доводят объем до 100 мл (свежий ра-
створ готовят каждую неделю).
358
Трис-АМС-буфер: к 10 мл концентрата NaCl добавляют 1 мл
кальциевого н 1 мг магниевого концентрата, доводят дистил-
лированной водой до 98 мл и прибавляют 1 мл концентрата
ЧСА; доводят объем до 100 мл (каждую неделю готовят но-
вый раствор). Кроме того, готовят 7,5 М NaOH, ЗМ NaOH,
0,75 М NaOH, 0,12 М НС1, 1,25 М Н3РО4 (8,25 мл концентри-
рованной Н3РО4 доводят до 10 мл), 0,05% ортофталевый аль-
дегид (ОФТ) (5 мл ОФТ непосредственно перед употреблени-
ем разводят в 10 мл метанола).
Гистаминовый стандарт для построения кривой: 1,66 мл ги-
стамина дигидрохлорида (например, Jenapharm, 1 мг/мл) до-
водят до 100 мл 0,01 М НО (соответствует 10 мкг/мл) до-
мина). Гистаминовый стандарт 1 мл разводят до 100 мл 0,12 М
HCI (0,1 мкл/мл) и готовят из этого раствора ряд разведений,
например, 0,1; 0,08; 0,06 и т. д. до 0,005 мкг/мл.
3.18.1.2.2. Выделение лейкоцитов человека
Засасывают в шприц объемом 20 мл 0,04 мл раствора гепари-
на и 2 мл раствора декстранглюкозы. Отбирают 18 мл венозной
крови. После полного перемешивания шприц укрепляют в вер-
тикальном положении в штативе и оставляют на 60 мин для
седиментации эритроцитов при комнатной температуре. После
чего осторожно слой плазмы с лейкоцитами выпускают через
изогнутую иглу в пластиковый центрифужный стакан. Лейко-
циты осаждают 2 мин при 170 g. Сливают плазму. Клетки
дважды отмывают 1—2 мл охлажденного трис-альбуминового
буфера. Центрифугируют каждый раз по 8 мин при 170 g,
следует обращать внимание на полноценное ресуспендирова-
ние, например, путем повторных засасываний и выпусканий
силиконизированной пастеровской пипеткой. Для дальнейших
экспериментов клетки осторожно суспендируют в 12,5 мл
трис-АМС.
3.18.1.2.3. Взаимодействие лейкоцитов с антигеном
К 2 мл суспензии лейкоцитов добавляют 0,4 мл раствора анти-
гена (оптимальную концентрацию АГ необходимо определить
предварительно), инкубируют пробы при 37°C. Каждые 20 мин
все пробы перемешивают. Клетки осаждают центрифугировани-
ем при 600 g в течение 10 мин, в насадочной фракции опреде-
ляют гистамин.
3.18.1.2.4. Экстракция гистамина
В 10-миллилитровые стеклянные сосуды развешивают по
600 мг NaCl и вносят по 2,5 мл н-бутанола, затем добавляют
1,6 мл бесклеточной надосадочной фракции. Прививают 0,2 мл
ЗМ NaOH и ровно 3 мин энергично встряхивают на качалке.
При этом гистамин переходит в фазу бутанола. Сосуды центри-
359
фугируют 3 мин при 900 g. Энергично перемешивают для раство-
рения осадка и снова центрифугируют (8 мин 900 g). По 2 мл
бутаноловой фазы переносят в пробирки объемом 10 мл с 1,2 мл
0,12 М НС1 и 3,8 мл н-гептана. Закрывают сосуды притертой
пробкой и встряхивают в течение ровно 1 мин.
Сосуды оставляют в темноте на холоду до полного расслое-
ния жидкости. В качестве альтернативы можно использовать
центрифугирование 5 мин при 600 g. Слой гептана отсасывают
водяным насосом, отбирают по 1 мл гистаминсодержащего
кислотно-солевого слоя и охлаждают их на ледяной бане.
Быстро добавляют 0,4 мл 0,75 М NaOH и 0,12 мл раствора
ОФА, перемешивают и инкубируют 40 мин на льду или 4 мин
при комнатной температуре. Добавляют 0,2 мл 1,25 М Н3РО4
и инкубируют еще 20 мин на льду. Проводят фотометрическое
определение гистамина, как было описано выше. Нижняя гра-
ница чувствительности составляет около 0,001 мкг гистамина
в мл. В среднем в 106 лейкоцитов содержится около 1 нг гиста-
мина. Гистамин, высвобождающийся в присутствии АГ, соотно-
сят с общим содержанием гистамина. Учитывают также спон-
танное высвобождение гистамина (инкубация суспензии кле-
ток без АГ).
3.18.1.2.5. Реакция дегрануляции базофилов [2, 6]
Метод представляет собой альтернативу аллергениндуци-
рованному высвобождению гистамина из базофилов, к тому же
он существенно проще. К числу его недостатков относится пло-
хая воспроизводимость. При использовании проточной цитоме-
трии [12] или автоматизированного анализа изображений [9]
метод становится более точным, но и более трудоемким. Не
надо забывать, что высвобождение гистамина и дегрануляция
не обязательно протекает параллельно. Мы опишем далее очень
простую модификацию метода [6].
Материал и оборудование: шприцы объемом 20 мл, часовые
стекла, центрифуга, капилляры, счетная камера Neubauer,
микроскоп, трис-NaCl- и трис-АСМ-буферы, 0,025% раствор
толуидинового синего в 30% этаноле (pH подведен до 3,4—
3,6 уксусной кислотой).
Проведение эксперимента: в шприц набирают 0,04 мл гепари-
на (5000 МЕ/мл) и 2 мл 3% раствора декстрана глюкозы в
0,15 М NaCl, 18 мл крови. Эритроциты седиментируют 60 мин
при вертикальном положении шприца. Лейкоциты дважды от-
мывают охлажденным трис-NaCl (по 2 мл), затем суспендиру-
ют осадок в 2 мл трис-ACM. По 0,1 мл этой суспензии перено-
сят в часовые стекла. В контрольные пробы добавляют по
10 мкл трис-АСМ-буфера, в остальные — по 10 мкл раствора
аллергена. Пробы инкубируют 20 мин при 37 °C. Окрашивают
их, добавляя по 270 мкл раствора толуидинового синего (20
мин при комнатной температуре, при легком покачивании).
Заполняют смесью обе стороны счетной камеры и подсчитыва-
360
ют интактные базофилы при увеличении 10X40. Оценку ре-
зультатов проводят по формуле:
число базофилов в контроле —
число базофилов в опыте ...
% дегрануляцин= числ0 базофиЛоВ в контроле’ Х 10°-
Известна модификация с использованием микропланшетов
[2]. Аллергены высушивают на стенках планшета в присутствии
ионов Са2+ и Mg2+, в контрольных лунках этих ионов нет..
Коэффициент дегрануляции базофилов и выделения гистами-
на составляет 0,74.
3.18.1.3. ДРУГИЕ МЕТОДЫ IN VITRO
Принцип всех этих тестов один и тот же: нормальные ткани
(легкие, кожа, кишечник и т. д.) инкубируют с сывороткой, со-
держащей реагенты. После внесения антигена и протекания ре-
акции АГ — АТ измеряют количество высвободившегося гиста-
мина. Опыты можно ставить среди прочего на ткани легких че-
ловека и морской свинки [14].
Пассивно сенсибилизированные легкие морской свинки [14]
Материалы и оборудование: однополые морские
свинки весом около 300 г, 20-миллиметровые шприцы, иглы,
пипетки, микропипетки, центрифужные пробирки, эрленмейе-
ровские колбы (50 мл), титраторы Takatsy, стеклянные фильт-
ры, мельницы для тканей, ледяная и водяная бани, качалка,
центрифуга, овальбумин, коклюшная вакцина.
Растворы: ФСБ с pH 7,0 (6,775 г NaCl, 1,1419 т
NazHPO4 (безв.), 0,408 г КН2РО4, дистиллированная вода до
1000 мл); среда для сенсибилизации (pH 7,8; 8,01 г NaCl,
0,176 г СаС12Х2Н2О, 0,201 г КС1, 0,238 мл 10% раствора
MgCl2, 1,0 г глюкозы, трис-буфер с pH 8,2 до 1000 мл); трис-
буфер (24,53 г трис, дистиллированная вода до 1000 мл, для
получения pH 7,8 смешивают 250 мл раствора трис и 337 мл
0,1 М НС1, доводят дистиллированной водой до 1000 мл; для
получения pH 8,2 250 мл раствора трис с 229 мл 0,1 М НС1
Доводят дистиллированной водой до 1000 мл).
Получение антисывороток
Морских свинок иммунизируют кристаллическим овальбуми-
ном и коклюшной вакциной в качестве адъюванта. Растворя-
ют 10 мг овальбумина в 0,5 мл ФСБ и добавляют 0,5 мл ко-
клюшной вакцины (1,5ХЮ10 клеток). Эту дозу вводят каждо-
му животному внутрибрюшинно. Параллельно 10 мл овальбу-
мина растворяют в 0,5 мл ФСБ, смешивают с 0,5 мл полного
адъюванта Фрейнда и вводят каждому животному внутрикож-
но в 6—10 точек на животе. Через 28 дней получают сыворотки
и хранят их в замороженном состоянии.
361
Пассивная сенсибилизация легких морской свинки
Забивают животных декапитацией, обескровливают, извлека-
ют легкие через разрез грудной клетки. Для постановки опыта
необходимо 3—4 животных. Легкие осторожно отмывают ох-
лажденной средой для сенсибилизации и на мельнице измель-
чают до кусочков 3x0,25x0,25 мм. Кусочки ткани помещают
на 2 слоя фильтровальной бумаги на воронку со стеклянным
фильтром и отмывают охлажденным раствором для сенсиби-
лизации, затем переносят их в 50 мл эрленмейеровские колбы,
добавляют 15 мл разведенной антиовальбуминовой сыворотки
н инкубируют 2 часа при 37 °C при постоянном покачивании и
аэрации. Оптимальное разведение сыворотки определяют для
каждой новой партии (0,125% раствор овальбумина должен
вызывать по меньшей мере 50% высвобождение общего гиста-
мина). Обычно разведение составляет 1 : 2—1 : 4. Антисыворот-
ку разводят раствором для сенсибилизации, кусочки легких
4—5 раз отмывают средой для сенсибилизации и один раз сре-
дой для высвобождения гистамина.
Реакция сенсибилизированных легких с антигеном
Для получения по возможности одинаковых количеств ткани
в качестве стандарта пользуются объемом лунки титратора
Takatsy. Отдельные пробы переносят в центрифужные пробирки
с 1 мл среды для высвобождения. При медленном покачива-
нии инкубируют 5 мин при 37 °C. Затем вносят 2,5 мг оваль-
бумина в 1 мл среды для высвобождения. Инкубируют еще
15 мин. При фармакологических исследованиях препарат ра-
створяют до желаемой концентрации в среде для высвобожде-
ния и вносят его в пробирку с тканью за 5 мин до внесения
антигена. Реакцию останавливают, охлаждая пробу на ледя-
ной бане в течение 10 мин. Центрифугируют пробы 10 мин при
1000 g. Надосадочную фракцию проверяют на высвободивший-
ся гистамин любым известным методом. Для определения об-
щего гистамина пробы инкубируют 5 мин на кипящей бане,
центрифугируют, исследуют надосадочную фракцию.
3.18.2. МЕТОД IN VIVO
Начиная с экспериментов Прауснитца и Кюстнера в 1921 г.
методы исследования анафилаксии in vivo находят очень широ-
кое применение. При поиске антианафилактических фармпрепа-
ратов в качестве модели особенно часто служит пассивная
кожная анафилаксия (ПКА) крыс (рис. 121), когда через
некоторое время после введения IgE-содержащей сыворотки
вводится соответствующий АГ. В реакции активной кожной
анафилаксии (АКА) антиген вводят сенсибилизированным жи-
вотным внутрикожно. Появление внутривенно введенного кра-
362
Рис. 121. Пассивная кож-
ная анафилаксия у кры-
сы.
Справа вверху — положи-
тельный контроль; добавле-
ние кромогликата натрия
ОД мг (справа внизу), 0,2 мг
(слева внизу), 0,4 мг (сле-
ва вверху).
сителя в области реакции АГ — АТ указывает на повышенную
проницаемость капилляров вследствие иммунологически инду-
цированного высвобождения медиаторов.
3.18.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для опыта необходимы однополые крысы весом 200—250 г,
пипетки, шприцы для инъекций, центрифужные пробирки, зон-
ды, эфир, ФСБ, ацетон, 0,5% раствор сульфата натрия, ко-
клюшная вакцина, овальбумин, синий Эванса, центрифуга, ка-
чалка, фотометр.
3.18.2.2. ПАССИВНАЯ И АКТИВНАЯ
КОЖНАЯ АНАФИЛАКСИЯ
3.18.2.2.1. Иммунизация крыс овальбумином
Растворяют 1 мг овальбумина в 0,2 мл ФСБ и вводят внутри-
мышечно; параллельно внутрибрюшинно вводят 2Х1010 ми-
кробных клеток Bordetella pertussis. Сыворотки для ПКА по-
лучают на 14-й день (IgE-содержащие сыворотки против оваль-
бумина; их можно хранить в замороженном состоянии). Опре-
деляют титр ПКА в геометрическом ряду разведений. За титр
принимают разведение, которое вызывает образование окра-
шенного пятна 15—20 мм в диаметре. Для постановки АКА
также используют крыс на 14-й день после иммунизации.
363
3.18.2.2.2. Постановка ПКА
Иод легким эфирным наркозом в 4 точки на животе вводят по
6,1 мл подходящей концентрации антиовальбуминовой сыво-
ротки внутрикожно. Через 48 ч вводят овальбумин с синим
Эванса внутривенно или точно в те же участки, куда вводили
сыворотку. Испытуемый фармпрепарат вводят перорально,
внутривенно или местно. Для выявления антигистаминного дей-
ствия и для сравнения добавочно внутрикожно вводят гиста-
мин и 5-гидрокситриптамин. Через 30 мин животных забивают,
осторожно отсепаровывают кожу живота и определяют размер
и интенсивность окраски пятен. LDso определяют по графику
доза — эффект (% торможения по отношению к дозе в лога-
рифмическом масштабе).
3.18.2.2.3. Постановка АКА
Внутривенно вводят синий Эванса (0,25 мл 1,6% раствора на
100 г веса). Сразу же делают внутрикожную инъекцию0,5 мкг
овальбумина (АГ) в три точки на животе. Через 20 мин обра-
зование контрольных пятен заканчивается и испытуемый
фармпрепарат вводят животному в соответствующей дозе и
форме. После достижения желаемого эффекта антиген вводят
в три точки с другой стороны живота. Еще через 20 мин жи-
вотных забивают и оценивают результаты реакции. Опреде-
ляют площадь пятен и сравнивают интенсивность их окраски.
3.18.2.2.4. Количественная оценка анафилактических реакций
Методы количественной оценки применимы к ПКА и АКА. Ко-
жу экспериментальных животных отсепаровывают, вырезают
пятна ножницами, измельчают их. Кусочки ткани в течение
24 ч при комнатной температуре экстрагируют смесью 14 мл
ацетона с 6 мл 0,5% раствора сульфата натрия. Ацетон и ра-
створ соли последовательно добавляют к кусочкам ткани.
После центрифугирования измеряют оптическую плотность
надосадочной фракции при длине волны 620 нм.
3.18.3. ОЦЕНКА МЕТОДА
Описанные методы применимы как для отбора антианафилак-
тических препаратов, так и для диагностических целей. Любая
модель на экспериментальных животных не может быть меха-
нически применима к людям. Хотя патофизиологические меха-
низмы анафилаксии в принципе одинаковы у млекопитающих
и человека, существуют все же значительные видовые отличия
в клинических проявлениях, составе медиаторов и пр. Поэтому
клинические наблюдения более информативны. Исследование
большого числа различных фармпрепаратов, разумеется, удоб-
нее проводить на животных. Подобный сбор анамнеза у аллер-
виков и тестирование in vivo позволяют получить больше дан-
364
ных, чем методы анализа in vitro. Индуцированное аллергеном
высвобождение гистамина может существенно облегчить диаг-
ностику аллергии, даже при невозможности поставить кожную
пробу, поскольку оно свидетельствует о степени индивидуаль-
ной реактивности базофилов.
3.18.4. СПИСОК ЛИТЕРАТУРЫ
1. Beaven М. A. Histamine: Its role in physiological and pathological proces-
ses.— S. Karger, Basel, 1978.
2. Benvebiste J. The human basophil degranulation test as an in vitro method
for the diagnosis of allergies. — Clinical Allergy, 1981, 11, 1—11.
3. Enerback L., Svensson I. Isolation of rat peritoneal mast cells by centrifu-
gation on density gradients of Percoll. — J. Immunol. Methods, 1980, 39,
135—145.
4. Gleich G. J., Hull W. M. Measurement of histamine: a quality control stu-
dy.— J. Allergy Clin. Immunol., 1980, 64, 295—298.
5. Hammer D. K. Der Einfluss des Reiningungsverffahrens auf die Funktion
von Mastzellen. — Der Krankenhausarzt, 1976, 49, 29—32.
6. Hien T. L. Ausgewahlte Untersuchungen zur Degranulation und Histamin-
freisetzung menschlocher basophiler Granulozyten. — Dissertation. Berlin,
1981.
7. Khandwala A., Damiani P., Weinryb I. Antigen-induced release of histamine
from passively sensitized pig lung slices. — Int. Archs. Allergy appl. Immun.,
1979, 59, 34—44.
8. Kruger P. G. The histamine release process and concomitant structural chan-
ges in rat peritoneal mast cells. — Int. Archs. Allergy appl. Immunol., 1976,
51, 608—626.
9. Leynadier F., Luce H., Abrego A., Dry J. Automated measurement of human
basophil degranulation. — Allergy, 1981, 36, 239—244.
10. Lorents W., Doenicke A. Histamine release on clinical conditions. — Mount
Sinai J. Med., 1978, 45, 357—386.
SI. Lorenz W., Doenicke A., Schonling B. et al. The role of histamine in adverse
reactions to intravenous agents. — In: Adverse reactions of anaesthetic
drugs/Ed. J. Thornton. — North-Holland Biomedical Press: Elsevier, 1981,
169—238.
4 2. Nakagawa T., Stadler B. M„ de Week A. L. Determination of rat mast cells
by flow cytometry. — J. Immunol. Meth., 1980, 33, 87—92.
13. Nemeth A., Rohlich P. Rapid separation of rat peritoneal mast cells with
Percoll. — Europ. J. Cell. Biol., 1980, 20, 272—275.
14. Schmidt M. J., Truex L. L., Hooker C. S. et al. Ethanol inhibition of antigen-
induced histamine release from guinea pig: correlation with intracellular
levels of cycle nucleotides. — Life Sciences, 1979, 24, 2321—2332.
15. Sehrt J., Bergman K. Ch., Dehnert I. et al. Allergeninduzierte Histaminfrei-
setzung zur in vitro Diagnostik des atopischen extrinsic-Asthmas. — Dtsch.
Ges.-wesen, 1977, 32, 303—307.
>6. Taylor К. M., Krilis S., Baldo B. A. A. An enzymatic-isotopic microassay for
measuring allergic release of histamine from blood and mast cells in vitro. —
Int. Archs. Allergy appl. Immunol., 1980, 61, 19—27.
’7. Wilhelms О. H. An improved automated fluorimetric method for determine*
tion of histamine.— J. Immunol. Meth., 1980, 36, 221—226.
365
3.19. КУЛЬТИВИРОВАНИЕ
МАКРОФАГОВ И МОНОЦИТОВ
3.19.1. ПРИНЦИП МЕТОДА
Чаще всего культивируют перитонеальные макрофаги мыши в
моноциты из крови человека. Эти клетки суспендируют в син-
тетической питательной среде с добавкой сыворотки эмбрионов
коров. Макрофаги и моноциты уже после непродолжительной
инкубации фиксируются на стенках культуральных сосудов.
Часть лимфоцитов также проявляет адгезирующие свойства, но
через 12—24 ч культивирования они теряют эту способность-
и легко вымываются, оставляя монослой моноцитов или макро-
фагов. Их культивируют далее или готовят из них суспензию.
Различают кратко- и долговременные культуры. Кратковремен-
ная культура не должна быть «старше» нескольких дней. Куль-
туры служат прежде всего для исследования таких функций
макрофагов, как фагоцитоз или выделение биологически актив-
ных веществ. К их числу относятся факторы, влияющие на
иммунный ответ, например интерлейкины [1, 2, 4]. Долговре-
менные культуры растут много месяцев и служат для иссле-
дования функциональных и морфологических изменений ма-
крофагбв и моноцитов in vitro. Длительная культивация мыши-
ных макрофагов не представляет сложностей.
3.19.2. МЕТОДИКА
3.19.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
а) Для получения перитонеальных клеток необходимы инбред-
ные мышн различных штаммов в возрасте 3—4 мес, изотониче-
ский фосфатный буфер (например, Институт иммунных препа-
ратов и питательных сред, Берлин), после получения сухого
препарата его растворяют в тридистиллированной воде и про-
водят стерилизацию фильтрованием при 4 °C 4—6 нед. Нужно
иметь также раствор гепарина без консервантов (5000 МЕ/мл),
сыворотку эмбрионов коров, инактивированную нагреванием в
течение 30 мин при 56 °C, 70% этанол, вату, кубики льда, ана-
томические пинцеты, ножницы, стеклянные шприцы на 10 и
20 мл, стальные иглы № 16 и 18, стакан на 400 мл, силико-
низированные стеклянные центрифужные пробирки на 25 мл,
настольную центрифугу, б) Для подсчета и дифференциально-
го определения макрофагов и моноцитов используют смесь ци-
трат/NaCl (8 объемов IM NaCl+ 2 объема 1М лимонной кис-
лоты; подводят pH 20% NaOH до 3,0—3,5 при 4 °C после до-
бавления небольшого количества тимола), 0,1% раствор
кристаллического фиолетового в 1М лимонной кислоте, 0,1%
раствор нейтрального красного в дистиллированной воде (кра-
366
ситель хранят при комнатной температуре несколько месяцев),
раствор туши (коммерческую черную тушь разводят в соотно-
шении 1 : 20 0,15 М NaCl, автоклавируют), мерные пипеткидля
лейкоцитов и эритроцитов, камеру Biirker, обогреваемый сто-
лик, световой микроскоп, в) Для получения культур макрофа-
гов и моноцитов нужно иметь раствор Хенкса (например,
Институт иммунных препаратов и питательных сред, Берлин),
концентрат растворяют в тридистиллированной воде, стерили-
зуют фильтрацией, хранят при 4 °C в течение 4—6 нед; среду
Игла-МЕМ (тот же поставщик) и PPMI 1640; к средам добав-
ляют по 2 г гидрокарбоната натрия и по 290 мг глутамина,
растворяют в 1 л тридистиллированной воды, стерилизуют
фильтрацией, хранят 4—6 нед при 4°C. Также необходимы ра-
створы антибиотиков: 1 000 000 ЕД пенициллина +1 г стрепто-
мицина растворяют в 100 мл тридистиллированной воды, стери-
лизуют фильтрацией, делят на небольшие порции и сохраняют
при —20°C, инактивированная сыворотка эмбрионов коров,
нормальная лошадиная сыворотка (инактивированная), челове-
ческая сыворотка (инактивированная), газовая смесь: 92,5 ча-
стей воздуха и 7,5 частей СОг, стеклянные чашки Петри диа-
метром 5 см, пипетки на 1, 5 и 10 мл, мерные цилиндры на 50
и 100 мл, эрленмейеровские колбы на 50 и 100 мл, эксикатор,
термостат, качалка.
Для силиконирования стеклянной посуды ее смачивают рас-
твором силикона (Serva, ФРГ) и высушивают 1 ч при 100 °C.
Способы очистки посуды см. раздел 1.3. Посуду упаковывают
в алюминиевую фольгу и стерилизуют 2 ч при 180 °C. Так же
стерилизуют ножницы и пинцеты. Растворы и среды стерили-
зуют пропусканием через антибактериальный фильтр G5.
3.19.2.2. ПОЛУЧЕНИЕ МАКРОФАГ-
И МОНОЦИТСОДЕРЖАЩИХ
СУСПЕНЗИЙ КЛЕТОК
а) Получение перитонеальных клеток [5]: мышей забивают
цервикальной дислокацией. Шкуру животного смачивают 70%
этанолом. Захватывают пинцетом шкуру на животе и делают
надрез ножницами с таким расчетом, чтобы разрез раздвигался
в краниально-каудальном направлении. В брюшную полость
вводят 3—4 мл жидкости (изотонический буфер с добавлени-
ем 5000 ЕД гепарина и 20 мл сыворотки эмбрионов коров из
расчета на 1л). Жидкость вводят осторожно, стараясь не по-
вредить кишечник, слегка приподнимая брюшную стенку пин-
цетом. После инъекции живот массируют большим и указатель-
ным пальцами 20—30 сек. Перитонеальную жидкость медленно
отсасывают шприцем. Для этого иглу вводят в латеральную
часть живота параллельно брюшной стенке. Рекомендуется от-
сасывать перитонеальную жидкость с обеих сторон живота,
367
Таблица 35. Состав клеток перитонеальной жидкости у различных
штаммов мышей
Штамм) Вес ЖИВОТНОГО (г) Количество» перитонеаль- ных клеток (ХЮв) Из них
лимфоцитов (%) макрофагов (%) гранулоцитов (%)
АВ 36 14,7 65 34 1
СВА (30—39) (12,9—16,5) (61—67) (31—39) (0-2)
20 6,1 74 25 1
C57/BL (18—22) (4,6—8,2) (69—78) (23—28) (0-2)
22 15,1 65 35 1
DBA (21—23) (12,8-17,5) (60—70) (30—39) (0-1) 1
22 7,1 64 35
(20—25) (5,1—9,4) (58—66) (33—41) (1-2)
Примечание. Представлены данные от 5 животных 3-месячного возраста,
в скобках — размах вариации.
тогда из 4 мл введенной жидкости получают 3,5 мл перитоне-
альной жидкости, которую собирают в охлажденные до О °C
силиконизированные центрифужные пробирки. Перитонеальную
жидкость, загрязненную кровью, не используют. Клетки осаж-
дают центрифугированием в течение 10 мин при 150 g, ресус-
пендируют осадок в растворе Хенкса. До засева культуры
клетки сохраняют на льду. Ниже приводится состав клеток пе-
ритонеальной жидкости у различных штаммов мышей (табл.
35).
Внутрибрюшинное введение различных раздражающих
средств за 3—4 дня до получения перитонеальной жидкости су-
щественно увеличивает число клеток и содержание в них мак-
рофагов (табл. 36). Следует указать, что индуцированные пе-
Таблица 36. Влияние различных раздражающих веществ на число
и состав клеток перитонеального экссудата (данные получены
на мышах СВА)
Раздражитель Получено клеток пери- тонеального экссудата 1 (ХЮ °C) Из них
лимфоциты % макрофаги % гранулоциты %
1 мл 0,9% 1 мл 10% гликогена 0,5 мл минерального масла F 1 мл протеозопептона 1 мл тиогликолата М 5,7 (3,5—7,0) 8,2 (6,0—9,3) Q (7,4—12,1) 9,2 (6,8-11,2) 18 (12,7—21,8) 66 (63-66) 54 (46-62) 54 (38—60) 46 (42-49) 27 (22-31) 32 (20-37) 43 (38—51) 38 (34—46) 53 (48-58) 71 (67—74) 2 (0-4) 3 (0-7) 8 (4-9) 1 (0-4) 2 (1-4)
Примечание. См. табл. 36.
368
ритонеальные макрофаги отличаются по целому ряду свойств
от неиндуцированных. К числу таких свойств относятся усиле-
ние пиноцитоза и фагоцитоза, а также усиление секреции био-
логически активных веществ in vitro.
б) Получение мононуклеаров из крови: мононуклеары полу-
чают из гепаринизированной крови (10 ME гепарина/мл), см.
раздел 3.1.
3.19.2.3. ИДЕНТИФИКАЦИЯ И ПОДСЧЕТ
МАКРОФАГОВ И МОНОЦИТОВ
Клетки окрашивают кристаллическим фиолетовым или ней-
тральным красным, идентифицируют и подсчитывают. В зависи-
мости от густоты суспензии ее разводят в пипетке для лейко-
цитов или эритроцитов раствором одного из красителей и пере-
носят в камеру Biirker (смешивают 1 мл раствора кристалличе-
ского фиолетового с 10 мл цитрат/NaCl и 90 мл дистиллиро-
ванной воды или 0,5 мл раствора нейтрального красного с 9 мл
раствора Хенкса и 1 мл сыворотки эмбрионов коров, можно
лошадиной). При окрашивании кристаллическим фиолетовым
клетки подсчитывают через 5 мин после заполнения камеры,
при окраске нейтральным красным подсчет ведут через 5—
10 мин после выдерживания камеры на подогретом до 37 °C
столике. Кристаллический фиолетовый окрашивает ядра кле-
ток. Это дает возможность отдифференцировать макрофаги и
моноциты от лимфоцитов и гранулоцитов. Они выглядят как
крупные клетки с круглым или овальным ядром. В их цито-
плазме присутствуют светопреломляющие гранулы. Эритроци-
ты при окраске лизируются, поскольку раствор кристалличе-
ского фиолетового сильно гипотоничен. Только макрофаги и мо-
ноциты прокрашиваются нейтральным красным, в вакуолях ко-
торых он откладывается. При подсчете клеток рекомендуется
просчитывать 3 группы квадратов камеры Btirker по диагонали
из левого верхнего угла в правый нижний.
Расчет ведут по следующей формуле:
Подсчитанное количество
клеток
----------2---------X фактор1 * разведения X 104 =
= число клеток в 1 мл.
Идентификацию макрофагов и моноцитов в культуре проводят
по захвату ими нейтрального красного или коллоидного угля.
Для этой цели к культуре клеток добавляют 0,2 мл разведенно-
го нейтрального красного или 0,1 мл разведенной туши. Инку-
бируют смесь 10 мин при 37 °C и подсчитывают клетки, окра-
шенные нейтральным красным. Клетки, захватывающие тушь,
1 Фактор разведения — величина, обратная степени разведения.
24—990 369
подсчитывают через 30—60 мин инкубации после отмывания
избытка туши. Подсчет клеток ведут под микроскопом, пере-
вернув чашку Петри вверх дном.
3.19.2.4. ПОЛУЧЕНИЕ КУЛЬТУР МАКРОФАГОВ
И МОНОЦИТОВ
Готовят среду (Игла-МЕМ или RPMI 1640), добавляя 10 мл
сыворотки эмбрионов коров или сыворотки человека группы
АВ и 2 мл растворов антибиотиков к 88 мл среды. Сыворотку
эмбрионов коров лучше использовать для улучшения культуры
мышиных перитонеальных макрофагов, сыворотку АВ — для
культивирования моноцитов из крови человека. Количество
клеток, которое вносят в культуральную среду, определяется
стоящей перед исследователем задачей.
После тщательного перемешивания с культуральной средой
суспензию клеток разливают по чашкам Петри (2 мл на чаш-
ку), которые затем помещают в эксикатор. Клетки культивиру-
ют при 37 °C в водонасыщенной атмосфере, содержащей 7,5 °/о
СОг. После 30—60-минутной инкубации неадгезировавшие клет-
ки удаляют из чашек Петри. Для этого чашки помещают на
5 мин на качалку (60—80 качаний/мин). Надосадочную фрак-
цию отсасывают шприцем, остающийся монослой клеток смы-
вают раствором Хенкса. Для этого в чашки вносят по 2 мл ра-
створа Хенкса и в течение 10 сек резко качают их в горизон-
тальном направлении. Раствор Хенкса отсасывают, повторяют
процесс отмывки 4 раза, затем инкубируют чашки 24 ч при
37 °C, предварительно добавив 2 мл свежей культуральной сре-
ды. В случае работы с перитонеальными клетками получают мо-
нослой, состоящий по меньшей мере на 98% из макрофагов. При
постоянном культивировании макрофагов или моноцитов каж-
дые 3—4 дня необходимо менять среду. При замене среды мо-
нослой клеток несколько раз отмывают раствором Хенкса для
удаления погибших макрофагов.
3.19.2.5. МОДИФИКАЦИЯ МЕТОДА
Если жидкая фаза культуры макрофагов (моноцитов) должна
анализироваться при помощи культуры антителообразующих
спленоцитов, макрофаги желательно культивировать в той же
среде, что и спленоциты (см. раздел 1.3). Иногда возникает не-
обходимость получать большие количества макрофагов, напри-
мер для изготовления антимакрофагальной сыворотки. В этих
случаях суспензию макрофагов (1—2хЮ6 клеток/мл) разлива-
ют по 10 мл в чашки Петри диаметром 12 см. В культуральной
среде можно при этом заменить добавку 10% сыворотки эмбри-
онов коров и 20% инактивированной лошадиной сыворотки.
Обработку культур проводят как описано выше. Культивиро-
вание осуществляют в течение 3—4 дней с ежедневной сменой
370
среды. Образующиеся монослои более чем на 99% состоят на
макрофагов. Макрофаги осторожно отделяют от дна чашки
стеклянной палочкой, на которую надет кусок мягкого пласти-
кового шланга, выступающий за конец палочки на 10 мм.
Для гистологических или гистохимических исследований ре-
комендуется культивировать клетки на стеклянных пластинах.
Для этой цели одну каплю суспензии клеток наносят на сте-
клянные пластины, которые инкубируют 1 ч во влажной каме-
ре. Неадгезировавшие клетки смывают раствором Хенкса и
пластинки помещают в чашки Петри с культуральной средой.
Поверхность пластин должна быть покрыта культуральной сре-
дой. По окончании культивирования стеклянные пластины от-
мывают изотоническим буфером, фиксируют клетки любым под-
ходящим способом и окрашивают соответствующим красителем.
3.19.3. ОЦЕНКА МЕТОДА
Культуру макрофагов и моноцитов оценивают по следующим
критериям:
а) внешний вид и жизнеспособность клеток;
б) клеточная однородность монослоя;
в) биохимическая активность клеток.
Обычно уже через 24 ч культивирования макрофаги выгля-
дят как распластанные удлиненные клетки с большим количе-
ством отростков. В культуре моноцитов это явление наблюда-
ется только через несколько дней. Наличие токсических ве-
ществ в культуральной среде, сдвиг pH или недостаточная
чистота стекол могут препятствовать распластыванию макро-
фагов и моноцитов, а также способствовать их отрыву от стен-
ки сосуда. Довольно часто гетерологические сыворотки (чаще
всего лошадиные), добавляемые в среду, оказываются токсич-
ными для клеток. Поэтому применяемые гетерологические сы-
воротки следует предварительно проверять на цитотоксичность
в отношении макрофагов и моноцитов. Из микробных загрязне-
ний чаще всего наблюдаются дрожжевые и грибковые (недо-
статочно тщательная стерилизация). Сдвиг pH среды может
быть обусловлен микробным загрязнением или улетучиванием
СОг. Сдвиг pH обнаруживается по изменению оранжево-крас-
ной окраски среды в желтый или фиолетовый цвет. Хотя после
30-минутной инкубации суспензии перитонеальных клеток адге-
зирует также довольно большое количество лимфоцитов, уже
через 24 ч инкубации монослой состоит на 98% из макрофагов.
Это относится как к обычным перитонеальным клеткам (ПК),
так и к клеткам индуцированного (протеозопептон) перитоне-
ального экссудата (КИПЭ). В обоих случаях приблизительно
60% от внесенного в культуру числа макрофагов оказываются
в составе монослоя (табл. 37).
При длительном культивировании макрофаги могут в неко-
торых случаях быть вытеснены фибробластами. Оказавшиеся
24* 371
Таблица 37. Влияние продолжительности культивирования иа долю
макрофагов в монослое
Продолжительность культивирования, ч Тип клеток Доля макрофагов среди адгезиро- вавших клеток, % Адгезировавшие макрофаги (% от исходного количества)
0,5 ПК 42 (38-47) 88 (85—90)
кипэ 66 (55—80) 83 (74-90)
24 ПК 98 (96—100) 58 (47—66)
кипэ 98 (96—100) 59 (50—71)
Примечание. Указаны средние результаты 10 определений. Цифры в скобках
обозначают разброс данных.
в культуре фибробласты могут быть удалены обработкой трип-
сином. Создавая оптимальные условия, культуру макрофагов
можно поддерживать в течение более 30 нед. Обычно макрофа-
ги не делятся в культуре. Пролиферацию можно индуцировать,
добавив бесклеточную жидкость культуры фибробластов или
вирус SV-40. При помощи SV-40 удается даже поддерживать
постоянные линии макрофагов, очень похожие по своим свойст-
вам на клетки первичной культуры. К числу биохимических
свойств макрофагов и моноцитов относится, как уже упомина-
лось, образование интерлейкина 1. Было показано, что в усло-
виях культивирования, близких к оптимальным, перитонеаль-
ные макрофаги мыши синтезируют относительно большие ко-
личества интерлейкина 1, который, однако, высвобождается в
культуральную среду в очень незначительных количествах.
ЛПС стимулирует образование интерлейкина 1, но не влияет на
его высвобождение. Частицы латекса усиливают как биосинтез
интерлейкина, так и его высвобождение [2].
3.19.4. СПИСОК ЛИТЕРАТУРЫ
1. Aarden L. A. Revised nomenclatur of antigen-nonspecific T cell proliferation
and helper factors. — J. Immunol , 1979, 123, 2928—2929.
2. Gery J., Davies P„ Derr J. et al. Relationship between production and release
of lymphocyte-activating factor (Interleukin 1) by murine macrophages. I. Ef-
fect of various agents. — Cell. Immunol., 1981, 64, 293—303.
3. Mocarelli P„ Palmer J. Defendi V. A permanent line of macrophages with
normal activity in a primary antibody response in vitro. — Immunol. Comm.,
1973, 2, 441—447.
4. Oppenheim J. J., Gery I. Iterleukin I is more than an interleukin. — Immuno-
logy Today, 1982, 3, 113—119.
5. Stuart A. E„ Habeshaw A., Davidson E. Plagocytes in vitro.-—In: Handbook
of experimental Immunology. Chapter 31/Ed D. M. Weir. — Oxford: Blackwell
Scientific Publications, 1978.
372
3.20. ВЫДЕЛЕНИЕ МАКРОФАГОВ
ИЗ СУСПЕНЗИИ СПЛЕНОЦИТОВ
3.20.1. ПРИНЦИП МЕТОДА
Для изучения роли макрофагов в выработке гуморального и
клеточного иммунного ответа in vitro необходимо удалить мак-
рофаги из суспензии спленоцитов. Обычно применяемые спосо-
бы удаления макрофагов основаны на следующих принципах:
а) способность макрофагов к адгезии на гладких поверх-
ностях; разделение на колонках, заполненных стеклянными бу-
сами, нейлоновой ватой, сефадексом G-10; способность макро-
фагов адсорбировать железосодержащие частицы [1];
б) меньшая плотность макрофагов по сравнению с лимфо-
цитами и гранулоцитами (разделение в градиенте плотности);
в) избирательная токсичность различных агентов в отноше-
нии макрофагов, например антимакрофагальной сыворотки,
экстракта ирландского мха или соединений кремния [32].
Наиболее часто используют адгезию на чашках Петри, об-
работку карбонилом железа и разделение на сефадексе G-10.
3.20.2. МЕТОДИКА
3.20.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
а) Для получения спленоцитов и идентифика-
ции макрофагов: инбредные мыши различных штаммов,
раствор Хенкса и среда Игла-МЕМ, сыворотка эмбрионов ко-
ров, раствор нейтрального красного и раствор туши (см. раз-
дел 3.19.2), ножницы, анатомические пинцеты, стеклянные
чашки Петри (диаметром 5 и 12 см), силиконизированные про-
бирки, стеклянный антибактериальный фильтр G-5, алюминие-
вая фольга, стеклянные шприцы на 10 мл, стальные иглы № 18.
центрифужные пробирки, силиконизированные на. 25 мл, пипет-
ки для лейкоцитов и эритроцитов, камера Burker, сухожаро-
вой шкаф, термостат, настольная центрифуга, микроскоп.
Используемую стеклянную посуду очищают соответствую-
щим образом (см. раздел 1.3). Стерилизацию инструментов и
стеклянной посуды, силиконнрование стеклянной посуды см.
в разделе 3.19.2.
б) Для отделения макрофагов методом ад-
гезии на чашках Петри: спленоциты, раствор Хенкса,
среда Игла-МЕМ без гидрокарбоната, сыворотка эмбрионов
коров, стеклянные шприцы на 10 мл, стальные иглы № 12,
центрифужные пробирки на 25 мл, стеклянные чашки Петри
диаметром 6 см, столик с подогревом, настольная центрифуга.
в) Для отделения макрофагов при помощи кар-
бонила железа: спленоциты, раствор Хенкса, сыворотка
373
эмбрионов коров, карбонил железа порошкообразный, чистей-
ший (Fluka, Швейцария), эрленмейеровские колбы (50 мл), си-
ликонизированные, силиконовые пробки, градуированные пипет-
ки, водяная баня, качалка, настольная центрифуга, магнит.
г) Для отделения макрофагов на сефадекс
G-10: спленоциты; сефадекс G-10 (Pharmacia, Швеция), 10 г
сефадекса замачивают на ночь в 40 мл 0,15 М NaCl, трижды
отмывают порциями по 40 мл 0,15 М NaCl, суспендируют в
25 мл 0,15М NaCl и автоклавируют 40 мин при 110°С; изото-
нический буфер (см. раздел 3.19.2), перед употреблением к 1 л
буфера добавляют 50 мл сыворотки эмбрионов коров и 20 мл
раствора антибиотиков (см. раздел 3.19.2); нейлоновая вата;
кусочки льда; стеклянный шприц на 30 мл силиконизирован-
ный; стальные иглы № 14; лабораторный штатив; градуирован-
ные пипетки; 100-миллиметровые эрленмейеровские колбы;
стакан на 400 мл; центрифужные стаканы на 25 мл; настольная
центрифуга.
д) Для отделения макрофагов при помощи а н-
тимакрофагальной сыворотки: спленоциты, раствор
Хенкса, антимакрофагальная сыворотка (АМС), сыворотка
морской свинки (комплемент), градуированные пипетки, эрлен-
мейеровские колбы на 50 мл силиконизированные, термостат,
качалка, настольная центрифуга.
3.20.2.2. ПОЛУЧЕНИЕ СУСПЕНЗИИ СПЛЕНОЦИТОВ
Извлекают в стерильных условиях мышиные селезенки, очища-
ют их от соединительной ткани, помещают в чашку Петри диа-
метром 12 см с охлажденным до 0°С раствором Хенкса (5 мл
на селезенку) и измельчают на мельчайшие кусочки при помо-
щи двух пинцетов. Образовавшуюся в результате измельчения
суспензию клеток осторожно отбирают шприцем (игла № 18).
Через 5—10 мин крупные скопления клеток оседают, находя-
щиеся в надосадочной фракции спленоциты центрифугируют
5 мин при 150 g, дважды отмывают охлажденным до 0°С ра-
створом Хенкса. Из каждой селезенки получают 5—10Х107
клеток. Жизнеспособность клеток определяют при помощи ок-
раски трипановым синим (см. раздел 1.3), она составляет 90—•
95%.
3.20.2.3. ИДЕНТИФИКАЦИЯ МАКРОФАГОВ
В СУСПЕНЗИИ КЛЕТОК
Макрофаги идентифицируют по способности воспринимать ней-
тральный красный или коллоидный уголь (тушь). Окрашива-
ние нейтральным красным см. в разделе 3.19.2. Для определе-
ния поглощения коллоидного угля 107 спленоцитов культивиру-
ют в силиконизированной чашке Петри в присутствии 0,1 мл
раствора туши; условия культивирования аналогичны услови-
ям макрофагов (см. раздел 3.19.2). После 15—20-часового
374
культивирования клетки осторожно отделяют от дна чашки
Петри и микроскопируют, выявляя фагоцитирующие клетки.
Число фагоцитирующих клеток должно составлять 1000—2000.
3.20.2.4. ОТДЕЛЕНИЕ МАКРОФАГОВ ПУТЕМ
АДГЕЗИИ В ЧАШКАХ ПЕТРИ
Полученные спленоциты суспендируют в среде Игла-МЕМ с до-
бавлением 10% сыворотки эмбрионов коров. Концентрация
клеток должна составлять около 107 клеток/мл. Суспензию раз-
ливают порциями по 3,5 мл в стеклянные чашки Петри диа-
метром 6 см и инкубируют их в течение 1 ч на обогреваемом
столике (37°C). Затем чашки резко качают в горизонтальном
направлении, чтобы взболтать неадгезировавшие спленоциты,
которые отсасывают шприцем и переносят в другую чашку
Петри. Проводят еще одну инкубацию в течение 30 мин, неад-
гезировавшие клетки переносят в новую чашку Петри и еще
раз дают им возможность прикрепиться к стеклу. Клетки, ос-
тавшиеся в жидкой фазе, осаждают центрифугированием, от-
мывают 1 раз раствором Хенкса и используют для дальнейших
исследований.
3.20.2.5. ОТДЕЛЕНИЕ МАКРОФАГОВ
ПРИ ПОМОЩИ ПОРОШКА карбонила
ЖЕЛЕЗА
Спленоциты суспендируют в растворе Хенкса с добавлением
5% сыворотки эмбрионов коров. Концентрация клеток должна
составлять около 2Х107 кл/мл. Эту суспензию разливают пор-
циями по 10 мл в 50-миллилитровые эрленмейеровские колбы
и добавляют по 400 мг порошка карбонила железа, закрывают
колбы силиконовыми пробками. Вносимый карбонил железа
предварительно стерилизуют УФ-излучением. Колбы помещают
на качалку на 30 мин при 37°C (водяная баня или термостат),
число качаний составляет 80—100 в 1 мин. По окончании инку-
бации удаляют из суспензии железосодержащий порошок, а так-
же макрофаги, захватившие частицы порошка. Для этого ко
дну колбы прикладывают сильный магнит, надосадочную фрак-
цию декантируют. Процесс повторяют до тех пор, пока не оста-
нется следов железосодержащего порошка в суспензии клеток.
После однократной отмывки раствором Хенкса клетки можно
использовать для дальнейших исследований.
3.20.2.6. ОТДЕЛЕНИЕ МАКРОФАГОВ
НА СЕФАДЕКСЕ G-10
В качестве колонки для G-10 используют 30-миллилитровый
силиконизированный стеклянный шприц с иглой № 14. В колон-
ку вносят около 3 мл нейлоновой ваты, сверху помещают на-
бухший автоклавированный сефадекс G-10. После вытекания
остатков жидкости колонку промывают 100 мл изотонического
375
буфера. Наносят на колонку 2—4 мл суспензии спленоцитов и
дают ей войти в слой геля. Не собирающиеся на сефадексе
спленоциты вымывают 25—30 мл изотонического буфера. Вы-
текающие фракции собирают в центрифужную пробирку, стоя-
щую во льду. Спленоциты отмывают однократно раствором
Хенкса, после чего они готовы для дальнейшей работы.
3.20.2.7. ОТДЕЛЕНИЕ МАКРОФАГОВ
ПРИ ПОМОЩИ АНТИМАКРОФАГАЛЬНОЙ
СЫВОРОТКИ
Готовят суспензию спленоцитов в растворе Хенкса с концентра-
цией 2Х107 клеток/мл. Разливают ее по 10 мл в 50-миллилит-
ровые эрленмейеровские силиконизированные колбы. Вносят
в суспензию 1 мл АМС и 1 мл комплемента, инкубируют 2 ч
при 37 °C, постоянно встряхивая. Частота качаний 20—30 в
1 мин. Осаждают клетки центрифугированием и трижды отмы-
вают раствором Хенкса.
3.20.3. СРАВНИТЕЛЬНАЯ ОЦЕНКА РАЗЛИЧНЫХ МЕТОДОВ
Все способы отделения макрофагов из суспензии спленоцитов
оцениваются по следующим критериям:
а) содержание макрофагов в суспензии после процесса отделе-
ния;
б) потери клеток при разделении;
в) функциональная активность лимфоцитов после разделения.
Количество макрофагов в суспензии клеток зависит от
способа их выявления. Например, у мышей СВА 2,5—3% кле-
ток быстро пиноцитируют нейтральный красный, и только 1,5—
Таблица 38. Влияние различных методов разделения клеток на их
свойства
Метод разделения Клетки, воспринимаю- щие нейтраль- ный красный %! Фагоцити- рующие клетки, % Т-клетки Жизнеспо- собные спленоциты, % Потери клеток, о'о
Интактная суспензия 2,5-3,0 1,5—2,0 34—39 90—95 —
После адгезии 0,6—0,9 0,2—0,3 35—40 90 40—60
После обра- ботки карбонилом железа 0,6—0,8 0,2—0,3 35—40 90 40—50
После обра- ботки АМС 0,7 0,2 33—38 90 10—15
.376
Таблица 39. Влияние способов удаления макрофагов на выработку
сплеиоцитами первичного и вторичного иммунного ответа иа ЭБ
и жизнеспособность спленоцитов в 4-дневиой культуре
Иммунный ответ Спленоциты Макрофаги удаляли
адгезией карбонилом железа АМС
АОК, % Жизнеспо- собность, % АОК, % Жизнеспо- собность, % АОК, % Жизнеспо- собность, %
Первнч- Неразделенные 100±11 24±4 100±14 24±3 100±3 22±4
ный Разделенные 6±2 13±3 9±7 23±5 2±1 11±2
Вторнч- Неразделенные 100±8 .°4±6 100±6 39±5 100±16 36±6
НЫЙ Разделенные 35±5 18±4 40±6 36±3 16±3 20±3
2% клеток фагоцитируют коллоидный углеводород (табл. 38).
Это различие, вероятно, можно объяснить наличием в селезенке
незрелых макрофагов с недостаточно развитой способностью к
пиноцитозу. Все применяемые методы не полностью удаляют
макрофаги из суспензии спленоцитов. Это относится и к разде-
лению на сефадексе G-10 [5], хотя эти данные не приведены
в таблице. Методами адгезии на чашках Петри, обработкой
спленоцитов карбонилом железа или АМС можно снизить со-
держание клеток, фагоцитирующих частицы туши, до ’/ю, пи-
ноцитирующих нейтральный красный — до у4. Все вышеописан-
ные методы удаления макрофагов из суспензии обладают при-
близительно одинаковой эффективностью, но получаемые с их
помощью препараты спленоцитов весьма варьируют в отноше-
нии эффективности гуморального иммунного ответа. Культиви-
рование по Mishell-Dutton (табл. 39) показывает, что макрофа-
ги наиболее эффективно удаляются при помощи АМС. Эффек-
тивность удаления макрофагов можно повысить, комбинируя
различные методы разделения. Например, сочетанием с адгези-
ей можно уменьшить содержание фагоцитирующих клеток в
суспензии спленоцитов до '/го исходной величины. Такой же
результат получается при комбинации разделения на G-10 с по-
следующей обработкой карбонилом железа [2]. Однако комби-
нация двух различных методов разделения приводит к очень
высоким потерям клеток, например, комбинация разделения
G-10 с железокарбонильным методом дает 80% потерю сплено-
цитов [2]. При сочетании адгезии с железокарбонильным мето-
дом процент потери достигает 50 (табл. 38). Наименьшие по-
тери спленоцитов дает использование АМС (около 15%). Уда-
ление макрофагов не оказывает существенного влияния на
долю Т-клеток или на соотношение Т- и В-клеток (табл. 38).
Спленоциты после обработки карбонилом железа отличаются
сниженной способностью к антителообразованию, но большей
жизнеспособностью, чем спленоциты, полученные после адгезии
или обработки АМС (табл. 39).
377
Таблица 40. Восстановление гуморального иммунного ответа на ЭБ
спленоцитами после добавления ПМ
Клетки Антиген Макрофаги* удалены
адгезией карбонилам железа АМС
Чис .ло АОК в кул >туре
Неразделенные сплено- 65+5 110±14 56±5
циты + 896±119 932±68 577±17
Спленоциты после раз- 32±9 58±11 3+1
деления + 120±21 110±11 16±2
Спленоциты после раз- 84+22 140+23 46+6
деления + 1 % ПМ + 837±81 837±66 440+16
Удаление макрофагов различными методами не влияет на
жизнеспособность спленоцитов. Точно так же при этом не за-
трагивается способность клеток к антителообразованию
(табл. 40). Так, эти клетки демонстрируют слабый иммунный
ответ или полное его отсутствие в отношении ЭБ, что указывает
на эффективность удаления макрофагов. Если же эти клетки
культивировать с добавлением 1 % перитонеальных макрофа-
гов (ПМ) в форме монослоя, наблюдается почти полное вос-
становление иммунного ответа.
3.20.4. СПИСОК ЛИТЕРАТУРЫ
1. Feldmann М., Kontiainen S. Suppressor cell induction in vitro: II. Cellular
requirements of suppressor cell induction. — Eur. J. Immunol., 1976, 6, 302—
305.
2. Glaser M. Macrophage requirement for in vitro generation of specific, secon-
dary cell-mediated cytotoxicity against SV 40-induced tumor-associated anti-
gen in mice. — Eur. J. Immunol., 1980, 10, 342—346.
3. Gorczynski M. R. Control of the immune response: Role of macrophages in
regulation of antibody- and cell-mediated immune responses. — Scand. J. Im-
munol., 1976, 5, 1031—1047.
4. Lemke H., Opltz H. G. Function of 2-mercaptoethanol as a macrophage substi-
tute in the primary immune response in vitro. — J. Immunol., 1976, 117, 380—
395.
5. Nakano M. Polyclonal antibody production in murine spleen cells induced by
Staphylococcus. — Microbiol. Immunol., 1980, 24, 981—994.
3.21. ФАГОЦИТОЗ
3.21.1. ПРИНЦИП МЕТОДА
3.21.1.1. ПРЕДВАРИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Явление фагоцитоза было открыто И. И. Мечниковым в 1883—
84 гг. Оно представляет собой захват чужеродных частиц оп-
ределенными клетками организма с последующим их фермен-
тативным разрушением [2, 3, 4, 10, 11]. У человека способ-
378
ностью к фагоцитозу обладают дифференцированные клетки
мононуклеарно-фагоцитарной системы (МФС, старое назва-
ние — ретикуло-гистоцитарная система, РГС) и гранулоциты.
Способность клеток к фагоцитозу у разных биологических видов
существенно различается. Так, например, для полиморфно-ядер-
ных лейкоцитов (ПМЯЛ) крупного рогатого скота характерна
очень высокая активность фагоцитоза, для ПМЯЛ человека и
лошади — средняя, а ПМЯЛ барана, морской свинки и кролика
вообще лишены ее.
Процесс фагоцитоза можно разделить на 5 стадий.
1. Миграция фагоцитов к очагу инфекции (пассивная по
кровотоку и активная за счет хемотаксиса).
2. Адгезия фагоцита с чужеродной частицей.
3. Поглощение чужеродной частицы в виде фагосомы.
4. Слияние фагосомы с лизосомами с образованием перева-
ривающей вакуоли (фаголизосома).
5. Переваривание захваченного материала.
Предпосылкой фагоцитоза бактериальных клеток является
их способность к адгезии. Подлежащий фагоцитозу материал
вначале адсорбируется на поверхности фагоцита. В месте кон-
такта с бактерией мембраны фагоцита формируют углубление,
затем начинает образовываться псевдоподия, которая в конце
концов полностью охватывает микроорганизм. Охватывающая
микроорганизм часть мембраны отпочковывается в виде от-
дельной вакуоли (фагосома). Довольно часто можно наблю-
дать объединение нескольких фагосом в одну. Амебоидное дви-
жение фагоцита и захват им частиц объясняется частично
электростатическими эффектами, частично структурными изме-
нениями внутриклеточных коллоидов. Захваченные частицы,
как правило, внутри фагосомы полностью разрушаются. Крайне
редко наблюдается выталкивание микроба за пределы мембра-
ны или его персистенция внутри вакуоли. Уже через несколько
минут после захвата частицы лизосомы выбрасывают свое
содержимое внутрь фагосомы, которая превращается таким
образом в фаголизосому. Внутри ПМЯЛ наблюдаются 2 типа
гранул, специфические и азурофильные. Азурофильные гранулы
образуются на стадии програнулоцита; они берут начало от
вогнутой поверхности пластинчатого комплекса. Они больше и
плотней специфических гранул, в них содержится 90% актив-
ности миелопероксидазы и, кроме того, кислая фосфатаза,
арилсульфатаза, p-глюкуронидаза, эстераза и б'-нуклеотидаза.
Специфические гранулы, как правило, не содержат миелопе-
роксидазы, но в них находится почти весь лактоферрин и около
50% лизоцима клетки. Они образуются на выпуклой поверх-
ности пластинчатого комплекса на стадии миелоцита. Иногда
они сливаются с фагосомами раньше, чем азурофильные гра-
нулы. Защитные механизмы фагоцита в настоящее время яв-
ляются объектом многочисленных исследований, предваритель-
ные данные представлены в виде схемы.
379
1. Кислородозависимые механизмы
Пероксидазозависимые
Пероксидазонезависимые:
— образование супероксиданиона;
— перекись водорода;
— гидроксильные радикалы;
— атомарный кислород;
2. Кислородонезависимые механизмы
— кислоты;
— лизоцим;
— лактоферрин;
— кислые и нейтральные гидролазы;
— кислые белки.
В интактных ПМЯЛ существует много антимикробных си-
стем. Некоторые микроорганизмы особенно чувствительны к
кислоте, другие — к лизоциму. В целом антимикробная актив-
ность определяется совокупным действием различных механиз-
мов защиты.
3.21.1.2. ПРИНЦИПИАЛЬНЫЕ ПОДХОДЫ
К ОЦЕНКЕ ФАГОЦИТОЗА
3.21.1.2.1. Проверка захвата частиц и бактерицидности
В экспериментах по захвату частиц и их накоплению в клетке
используют микроорганизмы, частицы коллоидного угля или
полистирола и др.
Для этого достаточно непродолжительной инкубации (20—
30 мин). Необходимо точно устанавливать соотношение части-
цы: клетки, например равное 10:1. Параллельно с захватом
частиц исследуют, как влияет ингибирование обмена веществ,
например охлаждение до 0 °C, на этот процесс. При определе-
нии бактерицидности исследуют не только захват эксперимен-
тальных микроорганизмов, но и их гибель внутри клетки. Про-
цесс фагоцитоза можно исследовать в суспензии клеток, в мо-
нослое или in vivo (измерение клиренса). Во многих тест-сис-
темах одновременно определяют захват и опсонизацию. Это
требует совместного и раздельного тестирования клеток, опсо-
нинов и частиц. Исследование комбинации этих трех факторов
представляет практические трудности, его результаты трудно
интерпретировать.
3.21.1.2.2. Исследование дегрануляции
Эксперимент включает в себя определение ассоциированных с
гранулами ферментов внутри вакуоли или во внеклеточной
среде. Полуколичественные методы основаны на гистохимиче-
ских реакциях клетки, количественные — на выделении вакуо-
лей. Внеклеточную дегрануляцию определяют по уровню фер-
380
ментативной активности в среде, что предполагает наличие кор-
реляции между секрецией содержимого гранул во внешнюю
среду и внутрь вакуолей.
3.21.1.2.3. Определение кислородного обмена фагоцитов
Метод основан на способности ассоциированных с плазматиче-
ской мембраной оксидаз восстанавливать кислород с образова-
нием супероксид-анионов или перекиси водорода. Есть основа-
ния предполагать, что активация кислородного обмена может
служить индикатором процесса захвата. Существуют разнооб-
разные методы прямого определения перекиси водорода и су-
пероксид-аниона.
3.21.2. МЕТОДИКА
В литературе описано большое число методов количествен-
ной оценки фагоцитоза [1, 5, 7, 8, 9, 10, 12, 13, 14]. Объем на-
стоящей книги не позволяет подробно изложить все из них, по-
этому мы ограничимся описанием лишь некоторых.
3.21. 2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимо иметь:
— цитратдекстрозный антикоагулянт: 8 г лимонной кислоты,.
22 г трехзамещенного цитрата натрия (двухводного), 24,5 г
глюкозы растворяют в 1 л воды;
— раствор декстрозодекстрана: 4,5 г NaCl, 25 г глюкозы, 30 г
декстрана (отн. мол. масса 500 000) в 1 л;
— раствор хлористого аммония: 9 частей 0,83% хлористого ам-
мония, 1 часть трис-НС1-буфера с pH 7,2 (20,6 г/л);
— смесь фиколл — визотраст: 9 г фиколла, 20 мл визотраста,
100 мл бидистиллированной Н2О, плотность 1,077;
— субстрат для Р-глюкуронидазы: 31,5 мг паранитрофенил-р-
глюкуронида и 100 мкм тритон X 100 растворяют в 100 мл
0,05 М натрий-ацетатного буфера с pH 5;
— реагенты для определения дефицита миелопероксидазы:
фиксатор (10 мл 37% формальдегида с 90 мл абсолютного
этанола), субстратный раствор (100 мл 30% ЭДТА, 0,3 г
хлористого бензидина, 0,038 г ZnSO4X7H2O, 1 мл дистил-
лированной воды, 1,0 г СНзСООНаХЗН2О, 0,7 мл 3% Н2О2);
подводят pH 1,0 М. NaOH до 6,0.
Коммерческие реагенты:
— ФСБ, раствор Хенкса и среда Игла-МЕМ (Гос. институт
иммунных препаратов и питательных сред, Берлин-Вайсен-
зее, ГДР);
— гепарин (5000 ЕД/мг) (Gedeon Richter, Венгрия);
— сыворотка эмбрионов коров (Flow Laboratories, США,,
можно другой фирмы);
— визотраст (VEB Fahlberg List, Magdeburg, ГДР);
381
— инфуколл (VEB Serumwerk Bernburg, ГДР);
— декстран, фиколл (Pharmacia, Швеция);
— коллоидный уголь CII/143 a (Wagner, Pelikanwerke, ФРГ);
— диизодецилфталат, парадиоксан (Coleman, Matheson and
Bell, США);
— тритон X 100 (Serva, ФРГ, можно другой фирмы);
— масляный красный О (Allied chemical corp., Morristown,
NY, США);
— паранитрофенил-р-глюкуронид (Sigma, США);
— сафранин О (Fischer Scientific Lab., Chicago, США);
— полистироловые бусы, трубочки (Nunc, Дания);
— сетка F 905 (VEB Orvo Wolfen, ГДР).
3.21.2.2 . ПОЛУЧЕНИЕ ФАГОЦИТОВ
Необходимые сведения о выделении гранулоцитов человека
можно получить в главе 3.1; получение перитонеальных макро-
фагов см. в разделах 3.19 и 3.20. Детально этот вопрос рас-
сматривается в ряде работ [1, 5, 8, 12].
Кроме того следует еще упомянуть о следующих методах:
— 8 мл крови смешивают с раствором декстранглюкозы. За-
тем добавляют 6% раствор декстрана 75 в 0,15 М NaCl (5 мл).
Смесь оставляют на 45—50 мин при комнатной температуре для
седиментации эритроцитов. Отсасывают плазму. Остаточные
эритроциты лизируют добавлением 0,83% хлористого аммония
(35 мл к 15 мл плазмы). Центрифугируют 10 мин при 80g, сус-
пендируют осадок в охлажденном до 0°С 0,15 М NaCl. Объ-
единяют несколько осадков и центрифугируют 10 мин при
800 g. Клетки лучше сохранять на льду в 0,15 М NaCl (эта сре-
да больше подходит, чем забуференные среды с бивалентными
катионами, которые вызывают слипание клеток);
— если объектом фагоцитоза являются дрожжи, можно опу-
стить стадию обработки хлористым аммонием, так как эритро-
циты не мешают процессу. Мононуклеары можно получать сле-
дующим образом: гепаринизированную кровь смешивают с ’/з
объема среды Игла, содержащей 15% глюкозы, наслаивают на
слой смеси фиколл — визотраст и центрифугируют 20 мин при
400 g. Фракцию мононуклеаров отсасывают пастеровской пи-
петкой, дважды отмывают ФСБ и готовят суспензию на среде
Игла (IXЮ7 клеток/мл).
3.21.2.3 . ПРИГОТОВЛЕНИЕ ЧАСТИЦ
ДЛЯ ФАГОЦИТОЗА
Чаще используют живые культуры Staphylococcus aureus (SG
511 или 502 A), Staphylococcus epididermidis SG 475, E. coli и
другие энтеробактерии, листерии, коринебактерии, Candida al-
bicans, Saccharomyces cerevisiae. Микроорганизмы выращивают
24 ч (при необходимости 48 ч) на твердых и жидких питатель-
382
Рис. 122. Калибровочная
кривая для оценки фа-
гоцитоза Е. coli 04/К31.
них средах. Собранную биомассу трижды отмывают 0,15М
NaCl. Слишком густую взвесь измеряют при 640 нм, опреде-
ляют концентрацию по калибровочной кривой (рис. 122). Кон-
центрацию микроорганизмов определяют также методами рас-
сева на плотные питательные среды; в некоторых случаях под-
счет микроорганизмов можно проводить в счетных камерах.
При работе с живыми бактериальными культурами следует
обращать внимание на то, чтобы использовались всегда культу-
ры на одной и той же стадии. Добавление 0,01% БСА способ-
ствует выживаемости микроорганизмов. Изготовленная суспен-
зия остается стабильной в течение 1—2 ч.
Убитые культуры микроорганизмов обычно получают на-
греванием в течение 30 мин при 80 °C или обработкой текучим
паром. Убитые микробы трижды отмывают 0,15 М NaCl, ресус-
пендируют и определяют концентрацию суспензии.
Приготовление пекарских дрожжей: 0,5 г пекарских дрож-
жей суспендируют в 0,15 М NaCl и помещают на 30 мин в ки-
пящую водяную баню. Фильтруют через ватно-марлевый
фильтр. При использовании живых дрожжей свежие клетки
(4—5-дневная культура) трижды отмывают в среде Игла с до-
бавлением 1,0% глюкозы. Обычно используют суспензию кле-
ток 108 и 109 клеток/мл. Дрожжи используют в качестве тест-
частиц при выявлении дефектов компонента С5.
Использование взвеси частиц полистирола: готовят 10%
водную суспензию частиц полистирола диаметром 1,091 мкм.
Разводят ее в соотношении 1 + 1 0,2% раствором БСА в 0,15 М
NaCl, центрифугируют. При длине волны 253 нм суспензия
полистирола 1 мкг/мл дает поглощение 1.17Х10-3.
Применение суспензии ЛПС — масляный красный О в ми-
неральном масле [10]: 2 г масляного красного растирают в
50 мл диизодецилфталата (или вазелиновое масло) в фарфо-
ровой ступке. Центрифугируют 20 мин при 500 g. Добавляют
10 мкг надосадочной фракции к 10 мл диоксина, измеряют оп-
тическую плотность на длине волны 525 им. Фактор пересчета
равен 0,92. На втором этапе 40 мг ЛПС (Е. coli 0,26 В6 и др.)
растворяют в 3 мл 0,15 М NaCl. Затем добавляют к этой смеси
383
1 мл раствора масляного красного О в диизодецилсульфате,
суспендируют смесь в течение 90 сек. Суспензию использу-
ют немедленно, ее можно замораживать.
Для постановки реакции фагоцитоза в качестве тест-частиц
можно использовать формалинизированные эритроциты.
3.21.2.4 . ФАГОЦИТОЗ БАКТЕРИЙ,
БАКТЕРИЦИДНОСТЬ
Эксперимент с суспензией живых бактерий: обычно используют
соотношение 3—10 микробов/фагоцит. В общем объеме 2 мл
смешивают 1Х106 фагацитов с 3X106 — 1X107 микробов.
На практике к 1 мл суспензии фагоцитов, к которому
было добавлено 8 ME гепарина, приливают 1 мл сус-
пензии бактерий. Время взаимодействия составляет обыч-
но 30 мин, в некоторых случаях оно больше. После
инкубации отбирают 0,5 мл смеси, добавляют 1,5 мл
0,1% раствора желатина в растворе Хенкса, охлажденного до
Ю°С, и центрифугируют 3—4 мин при 300 g. Из осадка готовят
мазки, которые окрашивают по Паппенгейму. Просматривают
200 клеток (по возможности трижды). Вычисляют процент фа-
гоцитоза. По числу содержащихся в клетках бактерий рассчи-
тывают индекс активности фагоцитоза: число фагоцитирован-
ных бактерий умножают на процент фагоцитирующих клеток;
интенсивность фагоцитоза выражают числами от 1 до 4.
Степень 1: фагоцитировано 1—4 бактерий
Степень 2: фагоцитировано 5—7 бактерий
Степень 3: фагоцитировано 8—10 бактерий
Степень 4: фагоцитировано более 10 бактерий на клетку
Пример проведения расчета при слабом фагоцитозе
Число просчитанных клеток 500
Число иефагоцитировавших клеток 350
Число клеток, фагоцитировавших 1—2 микроба 104
» » » 3—4 микроба 40
» » » 5 и более микробов 7
Индекс фагоцитоза 104X1,5=156
40X3,5=140
7X5=35
331
331 : 500=0,7
При определении уровня фагоцитоза живых микробов от-
дельно проверяют осадок клеток и надосадочную фракцию.
Количество живых микробов определяют по рассеву на плот-
ные питательные среды 0,1 мл исследуемых фракций. Инкуби-
руют 24 (48) ч. При расчете бактерицидности исходят из того,
384
что одна образовавшаяся колония соответствует одному живо-
му микробу.
Для направленного изучения бактерицидности исследуют
кровь пациентов и здоровых доноров с добавлением раствора
антибиотиков (по 5000 ЕД пенициллина и стрептомицина/мл) и
без него, а также проводят бактериальный контроль. Состав
пробы: 0,3 мл раствора Хенкса+ 0,1 мл нормальной сыворотки,
полученной из крови, взятой у 5 доноров, +0,5 мл суспензии
лейкоцитов (107 клеток/мл) +0,1 мл суспензии микробов
(106 микробов/мл). Раствор антибиотиков вносят по 0,02 мл
в пробу. Инкубируют пробы при 37 °C и определяют число
микробов через 20 мин, 1,5 ч и 3 ч. Для этого отбирают но
0,1 мл из каждой пробы, разводят отобранные аликвоты рас-
твором Хенкса в 10, 100 и 1000 раз и наносят на подогретый
агар. Иногда, особенно если были добавлены антибиотики,
для точного подсчета микробов готовят промежуточные разве-
дения (например, 0,2 мл пробы смешивают с 5 мл раствора
Хенкса, центрифугируют 5 мин при 450 g, осадок растворяют
в 1,9 мл ФСБ, наносят на агар). Если хотят сократить дли-
тельность бактериологического исследования, можно определять
находящиеся внутри клетки микробы по флюоресценции при
помощи окраски акридиновым желтым.
Фагоцитоз пекарских дрожжей: готовят суспензию 109 кле-
ток/мл в 0,15 М NaCl. Смешивают 0,1 мл суспензии с 0,1 мл
плазмы больного, инкубируют 30 мин при 37 °C, добавляют
0,2 мл (106) ПМЯЛ, инкубируют 30 мин. Отбирают аликвоты
через интервалы от 5 до 30 мин. Просчитывают 100 ПМЯЛ и
определяют число захваченных дрожжевых частиц на клетку.
Известна следующая модификация: к 50 мкл сыворотки мор-
ской свинки добавляют 50 мкл исследуемой сыворотки (пред-
варительно ее разводят в соотношении 1 + 1 средой Игла с глю-
козой), добавляют 50—200 мкл суспензии лейкоцитов (107 кле-
ток/мл) доводят объем до 450 мкл средой Игла с глюкозой и
инкубируют 30 мин при 37 °C. Затем добавляют 50 мкл суспен-
зии дрожжей (108 клеток/мл) перемешивают, инкубируют
40 мин при 37 °C. Вносят 50 мкл L- 755е-метионина (общая ак-
тивность 100 кБк) перемешивают и инкубируют 1 ч при 37 °C.
Осаждают клетки центрифугированием в течение 5 мин при
1000 g, отмывают их дважды ФСБ, измеряют радиоактивность
на гамме-счетчике. Процент фагоцитированных дрожжей вычи-
сляют по формуле:
имп/мин (лейкоциты+дрожжи)—имп/мин (лейкоциты) '
имп/мин (дрожжи)
Фагоцитоз с использованием раствора масляного красного в
минеральном масле: 0,2 мл суспензии частиц смешивают с 0,8 мл
клеточной суспензии, предварительно нагретой до 37 °C. Через
5 мин инкубации добавляют 6 мл охлажденного до 0°С 0,15 М
раствора NaCl, содержащего 126 мкг/кл N-этилмалеимида (для
25—990 385
100 —
прекращения захвата частиц). Центрифугируют 10 мин при
250 g. Надосадочную фракцию отбрасывают, осадок ресуспен-
дируют в растворе NaCl и N-этилмалеимида (см. выше), дваж-
ды отмывают клетки. Лизируют клетки ультразвуком, происхо-
дит высвобождение масляного красного. Добавляют 1 мл диок-
сана. Центрифугируют 15 мин при 500 g, измеряют оптическую
плотность на волне 525 нм против чистого диоксана. Степень
фагоцитоза (ИФ) определяют как количество минерального
масла (мг), поглощаемого в минуту 107 клетками. Для
подсчета можно использовать следующую формулу:
14ф _ О*~ls25Xфактор пересчета (см. 3.21.2,3)
время инкубации Хчисло фагоцитов *
Исследование фагоцитоза в монослое макрофагов:
1. Фаза: в стерильные полистирольные пробирки вносят по
2 мл суспензии клеток (200 000 клеток/мл). Инокулируют 5 ч
при 37°C, затем отмывают средой Игла. Вносят в пробирки
2 мл культуральной среды с 10% инактивированной (30 мин,
56 °C) сыворотки эмбрионов коров, инкубируют при 37 °C.
2. Фаза А: вносят суспензию микробов (3—10/макрофаг),
инкубируют 30—60 мин при 37 °C, 6 раз ополаскивают пробир-
ки порциями среды по 3 мл для удаления нефагоцитированных
микробов. Немедленно фиксируют препарат смесью 1 части ле-
дяной уксусной кислоты с 3 частями метанола. Окрашивают
препарат по Мау — Grunwald, подсчитывают клетки.
Фаза Б: определение внутриклеточных живых бактерий.
Последовательность операций та же, что и в фазе А до эта-
па фиксации. После отмывки тщательно удаляют все следы
среды. Лизируют клетки, для чего вносят 2 мл 0,01% стериль-
ного раствора БСА (4°C), многократно встряхивают. Большая
часть клеток лизируется через 20 мин. Высвободившиеся бакте-
рии определяют путем рассева на плотные питательные среды.
Фагоцитоз эритроцитов: смешивают 4Х107 фагоцитов с 5Х
ХЮ7 тест-эритроцитов в 5 мл ФСБ. Переносят 2 мл смеси в
охлажденный до 0°C 5 мМ фосфатный буфер и центрифугиру-
ют. Измеряют оптическую плотность надосадочной фракции
при длине волны 420 нм. Степень фагоцитоза определяют по
снижению содержания гемоглобина в бесклеточной фазе при
помощи калибровочных кривых.
3.21.2.5 . УПРОЩЕННЫЙ МЕТОД
ОПРЕДЕЛЕНИЯ КЛИРЕНСА
Пример определения клиренса на крысах: животным вводят
внутрибрюшинно по 5Х107 микробов на 100 г веса (0,1мл/
/100 г). Оптимальная доза может колебаться от 106 до 108 на
100 г веса. С интервалом 1—2 ч из каждой группы эксперимен-
тальных животных забивают по 3 особи; общая продолжитель-
ность эксперимента 16 ч. Стерильно берут 0,5 мл крови из серд-
386
ца, 1 мл перитонеальной жидкости, выделяют легкие, печень,
селезенку и почки. Из ткани органов вырезают цилиндры па-
стеровской пипеткой. Определяют число микроорганизмов пу-
тем культивирования на жидких и твердых питательных средах.
Пример определения клиренса на мышах [5J: используют
коллоидный уголь или меченные 51[Сг] эритроциты барана. Мы-
шам (по меньшей мере 5 особей на опыт) внутривенно вводят
по 0,01 мл суспензии угля из расчета 16 мг на 100 г веса.
В течение 15 мин с интервалом в 2 мин отбирают по 0,025 мл
крови из ретроорбитального пространства. Отобранные 0,025 мл
крови вносят в 2,0 мл 0,1% раствора Na2COs. После гемолиза
концентрацию угля определяют колориметрически на длине вол-
ны 675 нм при помощи калибровочных кривых.
log Ci — log С,
К (клиренс) = —-------
»2 —*1
t—время в мин, С — концентрация углерода в пробе. Корригированное зна-
чение фагоцитоза:
Зл— масса тела
а = уК X ---------------------------.
1 масса печени и селезенки
3.21.2.6 . ФУНКЦИОНАЛЬНЫЙ СКРИНИНГ
ФАГОЦИТОВ
Определение дегрануляции (измерение активности р-глюкуро-
пидазы) [10]: 107 лейкоцитов суспендируют в 0,8 мл ФСБ в
пластиковых пробирках, встряхивают 5 мин при 37 °C. Добав-
ляют 0,2 мл сенсибилизированных ЛПС, флюорохромнрованных
частиц (FC 80), охлаждают 30 мин на льду, центрифугируют
10 мин при 250 g. Проверяют активность фермента в надосадоч-
пой фракции. Инкубируют 0,9 мл субстратной смеси в течение
18 ч с 0,1 мл исследуемой фракции. Добавляют 2 мл 0,1М
NaOH, измеряют оптическую плотность на волне 410 нм.
Расчет:
(ОП410Х20)/(1,84Х 18) =число нмолей вещества, высвобож-
даемых за 1 час 107 лейкоцитами, т. е. степень дегрануляции
выражают в наномолях паранитрофенил-|3-глюкуронида.
Метод определения дефектов миелопероксидазы: наилучшие
результаты дает биохимическое определение Н2О2, указываю-
щее на изменения метаболизма в процессе фагоцитоза. Для
практических целей весьма важно, что активация гексозо-моно-
фосфатного шунта, восстановление тетразолиевого нитроеннего
и связывание экзогенного йода с белками ПМЯЛ коррелируют
с образованием Н2О2. Обычный мазок крови фиксируют 30 сек
смесью алкоголь—формалин. Отмывают дистиллированной во-
дой, и в течение 30 сек проводят окраску на пероксидазу. В со-
держащих пероксидазу клетках выявляются включения, окра-
шенные в темно-голубой цвет.
25*
387
Проба на восстановление тетразолиевого нитросинего (ТНС)
[5, 10]. ТНС восстанавливается нормальными ПМЯЛ до форма-
зана. Смешивают с 0,1 мл крови 0,1 мл 0,1% раствора ТНС
в 0,15 М NaCl, инкубируют 20 мин при 37 °C, еще раз тщатель-
но перемешивают. Включения формазана в клетки определяют
микроскопией. Результат выражают в процентах формазан-по-
зитивных клеток.
Несколько сложнее следующий метод: 1 каплю крови па-
циента наносят на покровное стекло. Инкубируют 20 мин при
37°C во влажной камере, затем осторожно отмывают стекло
стерильным 0,15 М NaCl. Кладут покровное стекло на пред-
метное, куда была нанесена 1 капля ТНС-среды (0,5 мл сыво-
ротки + 0,.3 мл стерильного 0,15 М NaCl + 0,6 мл ТНС, см. ра-
нее). Инкубируют 30 мин во влажной камере при 37 °C, удаля-
ют покровное стекло и высушивают на воздухе. В течение 60сек
фиксируют абсолютным метанолом и отмывают дистиллирован-
ной водой. Окрашивают в течение 5 мин сафранином (1 г са-
франина + 100 мл дистиллированной воды + 40 мл глицерина),
отмывают. Формазан-позитивные клетки отличаются большими
размерами, они напоминают бластные клетки и содержат го-
лубые гранулы. Обычно в препарате определяется около 30%
формазан-позитивных клеток.
Вышеприведенные методы оценки фагоцитоза представляют
собой обобщение очень большого числа публикаций. Более де-
тальную информацию по этим вопросам можно найти в соот-
ветствующих работах [1, 2, 4, 5, 6, 7, 8, 9, 10, 12, 13, 14].
3.21.3. ОБЩАЯ ОЦЕНКА МЕТОДОВ
Оценка способности клеток к фагоцитозу представляет собой
незаменимый метод исследований в современных клинико-им-
мунологических лабораториях. Этот метод используется в ди-
агностике таких заболеваний, как хронический гранулематоз,
дефицит миелопероксидазы, синдром Чедиака — Хигаси, дефи-
цит глюкозо-6-фосфат дегидрогеназы, синдром Иова, синдром
Дауна, синдром Лейнера и др. Определение фагоцитирующей
способности все шире используется в научных исследованиях
по иммуномодуляции, повышению резистентности, регуляции
иммунного ответа и др.
Несмотря на самые оптимистические оценки, очевидна боль-
шая сложность и трудоемкость методов исследования фагоци-
тоза. Особые сложности представляет работа с живыми микро-
организмами. Интерпретация результатов существенно затруд-
няется, когда приходится иметь дело более чем с двумя пере-
менными (живые микроорганизмы, опсонизация и др.). Очень
сложно или даже невозможно переносить данные литературы
на результаты собственных исследований.
В литературе нет исчерпывающих данных о сравнении эф-
фективности различных методов оценки фагоцитоза. Каждый
388
потенциальный исследователь должен сам решить, какой метод
наиболее подходит для его целей, или, используя свой опыт,
определить наилучшее направление исследований фагоцитоза.
3.21.4. СПИСОК ЛИТЕРАТУРЫ
1. De Boer М„ Reijneke R., van de Griend R. et al. Large scale purification and
criopreservation of human monocytes. — J. Immunol. Meth., 1981, 43, 225—
239.
2. Escobar M. R. (Ed). Macrophages and lymphocytes: nature, functions and
interactions. — Adv. Exper. Med. Biol. 121 A and B. — New York and Lon-
don: Plenum Press, 1980.
3. Van Furth R. (Ed.). Mononuclear Phagocytes: functional aspects; Nijhoff,
Hague, 1980.
4 Gabig T. G., Babior В. M. The killing of pathgens by phagocytes. — Ann. Rev.
Med., 1981, 32, 313—325.
5. Herscowitz H. B., Holden H. T., Bellanti I. A. et al. Manual of macrophage
methodology. Collection, characterization and function. Immunol. Ser. 13,
Marcel Dekker, New York and Basel, 1981.
6. Klebanoff S. J. Cytodial mechanisms of phagocytic cells. — Ser. Rev., 1980,
721—736.
7. Lane T. A., Windle В. E. A model for the study of granulocyte concentrate
preservation. — Transfus/on, 1980, 20, 559—562.
8. Mishell B., Shigi S. (Eds.) Selected methods in cellular immunology. — San
Francisco: Freeman Co., 1980.
9. Raynor R. H., Doran I. E„ Reese A. C. A polymorphonuclear leukocyte mono-
layer system for studies of phagocytosis. — J. Reticuloendothel. Soc., 1981,
29, 441—449.
10. Rose N. R., Friedman H. Manual of clinical immunology. Amer. Soc. Micro-
biol. Washington, 1980.
11. Unanue E. R„ Rosenthal A. S. (Ed) Macrophage regulation of immunity.—
London, New York: Academic Press, 1980.
12. Undeutsch Ch., Brunner H. Einfluss von Antikorpern auf die Phagozytose
von Klebsiella pneumoniae durch Alveolomakrophagen. — Zbl. Bakt. Abt. I
Orig. A 249, 43—52 (1981).
13. Vray B., Hoebeke I., Saint-Guillain M. et al. A new quantitative fluorimetric
assay for phagocytosis of bacteria. — Scand. J. Immunol., 1980, 11, 147—
153.
14. White A. G., Walker Sh. M. A new assay for the assessment of staphylococ-
cal killing by human leukocytes. — J. Immunol. Meth., 1981, 42, 203—212.
26 -990
4. ИММУНОХИМИЯ
4.1. ПОЛУЧЕНИЕ ПРЕПАРАТОВ
ИММУНОГЛОБУЛИНОВ
4.1.1. ВЫДЕЛЕНИЕ ИММУНОГЛОБУЛИНА G (IgG)
4.1.1.1. ПРИНЦИП МЕТОДА
В отличие от иммуноглобулинов других классов IgG сравнитель-
но просто выделить в чистом виде. Выделение проводят путем
осаждения сульфатом аммония и хроматографии на анионооб-
менниках, например ДЭАЭ-целлюлозе или ДЭАЭ-сефадексе. Ис-
ходным материалом служит сыворотка. IgG представляет собой
гетерогенную смесь иммуноглобулинов с похожими характери-
стиками, эти 1g можно разделить на анионообменниках, изменяя
ионную силу элюента. Часто перед проведением хроматографии
проводят осаждение сульфатом аммония или риванолом (2-эток-
си-6, 9-диаминоакридинлактат).
4.1.1.2. ПОЛУЧЕНИЕ ПРЕПАРАТОВ
С ПОВЫШЕННЫМ СОДЕРЖАНИЕМ IgG
Осаждение сульфатом аммония. К 100 мл сыворотки добавляют
50 мл насыщенного раствора сульфата аммония. Осадок уда-
ляют центрифугированием, растворяют в 50 мл дистиллирован-
ной воды и снова осаждают 25 мл насыщенного раствора суль-
фата аммония. Осаждение повторяют 4—5 раз. Затем препарат
диализуют против соответствующего буфера. Иногда осаждение
проводят при 40% насыщении соли, что увеличивает выход,
но снижает чистоту препарата. Иногда насыщение сульфата
аммония выражают в молях; насыщенный раствор при 0°С со-
ответствует 3,9 М (514,72 г в 1л), при 20 °C — 4,06 М (536,34 г
в 1 л).
Осаждение сульфатом натрия. К 100 мл сыворотки при ком-
натной температуре добавляют 18 г сульфата натрия (при 4 °C
растворимость сульфата натрия уменьшается). Осадок раство-
ряют в 40 мл фосфатного буфера с pH 8,0 и ионной силой 0,1.
Переосаждают белок, прибавив 4,8 г сульфата натрия. Осадок
после центрифугирования растворяют в 20 мл фосфатного буфе-
ра (см. выше) и переосаждают 2,4 г сульфата натрия. Выпав-
ший осадок отделяют, растворяют в небольшом количестве буфе-
ра, диализуют.
390
Осаждение риванолом. Риванол образует с сывороточными
белками комплексы, которые выпадают в осадок, но растворя-
ются при pH 5. Ввиду высокой изоэлектрической точки IgG по
сравнению с остальными сывороточными белками в слабощелоч-
ной среде риванол не образует комплексов с IgG. На этом осно-
вана очистка IgG. К сыворотке добавляют раствор риванола
(7,5 г/л) при постоянном перемешивании. Осадок удаляют цент-
рифугированием. IgG остается в надосадочной фракции. Рива-
нол можно удалить активированным углем (10 мг/мл) или NaCl
(до 50 г/л). Соотношение сыворотки и риванола может быть
различным. Наилучшее соотношение сыворотка/риванол при
выделении человеческого IgG равна 1+2. На одни объем сыво-
ротки животных берут 1—1,5 объема риванола. При обработке
сыворотки человека используют даже соотношение 1+3—3,5.
Фракционирование органическими растворителями. У этого
вида фракционирования есть недостаток: необходим строгий
контроль температуры и pH. Поскольку органические раствори-
тели легко вызывают денатурацию, необходимо проводить рабо-
ту при температуре ниже 0°С. Чаще используют этанол при
фракционировании белков плазмы человека. В лаборатории к
фракционированию этанолом прибегают редко, однако в про-
мышленности этот метод нашел широкое применение. Так назы-
ваемая II фракция по Кону состоит в основном из IgG.
4.1.1.З. ПОЛУЧЕНИЕ IgG
ИЗ СЫВОРОТКИ ЧЕЛОВЕКА
4.1.1.3.1. Материалы и оборудование
Для работы необходимы сыворотка человека, ДЭАЭ-сефадекс
(Pharmacia, Швеция), 0,1 М н 0,01 М фосфатный буфер с pH 6,5,
0,1 М Н3РО4, 0,5 М NaOH, 1 М К2НРО4, 1 М NaCl.
4.1.1.3.2. Методика
Дают ДЭАЭ-сефадексу А-50 набухнуть, мелкие частицы декан-
тируют. Отмывают 0,5 М NaOH до тех пор, пока промывная
жидкость не освободится от ионов хлора. Отмывают сорбент во-
дой до достижения нейтрального pH. При помощи 0,1 М фос-
форной кислоты переводят обменник в фосфатную форму. До-
бавляют такое количество кислоты, чтобы надосадочная фрак-
ция имела выраженную кислую реакцию. Отмывают избыток
кислоты дистиллированной водой, уравновешивают сорбент 0,1 М
фосфатным буфером с pH 6,5, затем 0,01 М фосфатным буфером
с pH 6,5. К 100 мл сыворотки человека добавляют 20 г набухше-
го уравновешенного ДЭАЭ-сефадекса А-50. Сыворотку можно
предварительно продиализовать против 0,01 М фосфатного бу-
фера с pH 6,5. Смесь периодически суспендируют в течение часа
(4°C). Затем фильтруют и 4 раза отмывают порциями 0,01 М
буфера по 50 мл. Фильтрат и промывную жидкость объединяют
н снова инкубируют с 20 г ДЭАЭ-сефадекса в тех же условиях.
26* 391
Снова фильтруют и отмывают 0,01 М фосфатным буфером. До-
водят pH с помощью 1 М К2НРО4 до 7,5 для предотвращения
образования нерастворимого продукта. Раствор IgG диализуют
против дистиллированной воды, после чего его лиофилизируют.
После лиофилизации IgG приобретает склонность к агрегации.
Использованный сефадекс регенерируют 1 М NaCl, 0,5 М NaOH
и этанолом. Этанол удаляет адсорбированные липиды. Избыток
щелочи отмывают дистиллированной водой, после чего ионооб-
менник переводят в фосфатную форму как было описано выше.
4.1.1,3.3. Обсуждение
Метод приготовления IgG из сыворотки человека состоит по су-
ществу из одного этапа. Хроматография на колонке является
более эффективным методом, чем метод спонтанной седимента-
ции (англ. Batch method). Однако ввиду того, что гель изменяет
объем в зависимости от ионной силы, регенерация геля непо-
средственно на колонке невозможна, гель приходится выбивать
из колонки. При получении больших количеств IgG желательно
до хроматографии провести еще одну стадию очистки. Возмож-
но также использовать для хроматографии QAE-сефадекс, объем
гранул которого не зависит от pH. В качестве элюента исполь-
зуют смесь этилендиамида и уксусной кислоты с pH 7,0, ионной
силой 0,1; регенерируют сорбент натрий-ацетатным буфером с
pH 4,0 с ионной силой 0,1. Липопротеиды удаляют 5% раство-
ром твина-80 в ацетатном буфере. Этилендиамин-ацетатный бу-
фер улетучивается при лиофилизации, что является преимуще-
ством. Перед лиофилизацией концентрацию IgG доводят до
30 г/л, так как в случае меньших концентраций образуются не-
растворимые продукты. Протеин А из клеточной стенки Staphy-
lococcus aureus осаждает IgG 1,2 и 4, но не IgG3. Формалинизи-
рованную культуру золотистого стафилококка можно использо-
вать для специфической очистки IgG3. IgG 1,2 и 4 можно десор-
бировать с иммобилизованного протеина А при помощи глицин-
солянокислого буфера с pH 3,0, а также электрофоретически
[9]. Градиентом pH удается десорбировать IgG 1 и 2 с протеин-
А-сефарозы с 90% и 95% чистотой соответственно [2]. Под-
классы IgG можно также выделить при помощи антисывороток
со специфичностью к отдельным подклассам, конъюгированных
с бромциансефарозой [3].
4.1.1.4. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ КРОЛИКА
Очистку IgG кролика можно осуществлять в следующих усло-
виях:
а) хроматографию проводят на ДЭАЭ-целлюлозе в 0,01 М
фосфатном буфере с pH 8,0 или в 0,0175 М фосфатном буфере
с pH 6,3. Перед хроматографией сыворотку диализуют против
соответствующего буфера. Элюируется чистый IgG, остальные
белки задерживаются на колонке;
392
б) после хроматографии на ДЭАЭ-сефадексе А-50 в
0,0175 М фосфатном буфере с pH 6,3, на ДЭАЭ-целлюлозе в
0,0175 М фосфатном буфере с pH 6,9 или в 0,025 М фосфатном
буфере с pH 6,9 IgG осаждают сульфатом натрия;
в) трехкратно осаждают IgG сульфатом аммония (конечная
концентрация 1,35 М) при pH 7,0, проводят диализ против дис-
тиллированной НгО. Хроматографируют белок на ДЭАЭ-цел-
люлозе в 0,02 М фосфатном буфере с pH 7,2;
г) осаждают IgG сульфатом аммония в конечной концентра-
ции 37, 40 или 43% насыщения с последующей хроматографией
на ДЭАЭ-целлюлозе в 0,0175 М фосфатном буфере с pH 6,3;
д) предварительно осаждают IgG сульфатом аммония или
риванолом, затем хроматографируют на ДЭАЭ-целлюлозе. Элю-
ирование проводят фосфатными буферами:
1) 0,0175 М с pH 6,9; 2) 0,035 М с pH 7,5; 3) 0,05 М с pH 7,5.
Фракции 1 и 2 представляют собой чистый Ig, фракцию 3 очи-
щают гель-фильтрацией на сефадексе G-200 (0,2 М. трис, 0,5 М
глицин, pH 8,2).
При хроматографии на ДЭАЭ-целлюлозе или ДЭАЭ-сефа-
дексе А-50 IgG кролика удается разделить на 2 фракции, «мед-
ленную» и «быструю» (по электрофоретической подвижности).
Сыворотку диализуют против стартового буфера (0,035 М трис,
0,005 М фосфорная кислота pH 8,4), хроматографируют на
ДЭАЭ-целлюлозе в этом же буфере. При этом элюируется «мед-
ленный» IgG. Если вести дальше элюцию смесью 0,5 М трис—
0,59 М фосфорная кислота с pH 4,0, то вначале появляется пик,
отражающий содержание чистого «быстрого» IgG, затем элюи-
руется «быстрый» IgG в смеси с другими сывороточными белка-
ми. При хроматографии сыворотки на ДЭАЭ-сефадексе А-50
также можно выделить две фракции IgG, идентичные иммуно-
электрофоретически, но различающиеся по подвижности при
электрофорезе. Первую фракцию выводит стартовый буфер, вто-
рая элюируется градиентом 0,2—0,3 М. фосфатного буфера с
pH 8,0. Соотношение величин обеих фракций зависит от моляр-
ности стартового буфера.
4.1.1.5. ВЫДЕЛЕНИЕ IgG ИЗ КУРИНОЙ
СЫВОРОТКИ
IgG из куриной сыворотки получают трехкратной преципита-
цией сульфатом натрия (180, 140 и 125 г/л) с последующей
хроматографией на ДЭАЭ-целлюлозе в 0,05 М. фосфатном бу-
фере с pH 8,0.
4.1.1.6. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ
ЛОШАДИ
Лошадиный IgG готовят методом ионообменной хроматографии
из нормальной сыворотки на ДЭАЭ-целлюлозе в 0,01 М фосфат-
ном буфере с pH 8,0. Дальнейшую очистку можно проводить
393
осаждением сульфатом аммония при 37% насыщения с после-
дующей доочисткой на ТЭАЭ-целлюлозе в 0,00525 М фосфатном
буфере. IgG можно выделить из нормальной сыворотки лошади
на ДЭАЭ-сефадексе в 0,0175 М фосфатном буфере с pH 6,3.
Если в идентичных условиях выделять IgG из иммунной сыво-
ротки, получается препарат, загрязненный IgG (Т).
В ходе рехроматографии на ДЭАЭ-сефадексе А-50 в 0,015 М
фосфатным буфере с pH 8,4 IgG не адсорбируется, a IgG (Т)
задерживается на колонке, откуда он может быть элюирован
градиентом фосфатный буфер 0,015 М с pH 8,4 — фосфатный
буфер 0,175 М с pH 6,3. Выделенный таким образом IgG (Т)
содержит примесь IgG, которую удаляют методом зонального
электрофореза. IgG (Т) ранее называли IgA (Т), Т-глобулином
или А-глобулином. Тяжелая цепь IgG (Т) имеет общие детерми-
нанты с IgG лошади других подклассов (IgG а, Ь, с) лошади и
при ионообменной хроматографии элюируется вслед за IgGa,b,
с. IgG а, b элюируется с 0,005—0,01 М фосфатным буфером с
pH 8,0, IgG с — 0,02 М фосфатным буфером с pH 8,0.
4.1.1.7. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ
МОРСКОЙ СВИНКИ
Фракция 7S иммуноглобулинов морской свинки разделяется
на 7S у1 («быстрая») и 7S у2 («медленная»). Их антигенные
детерминанты несколько отличаются друг от друга, так же как
и их биологические свойства. Так, например, 7S у 1 в отличие от
7Sy2 вызывают реакцию пассивной кожной анафилаксии у
морских свинок. Обе фракции осаждают из сыворотки при 50%
насыщении сульфатом аммония. Их разделяют на ДЭАЭ-целлю-
лозе, проводя ступенчатое элюирование фосфатными буферами:
0,005 М с pH 8,0, 0,01 М с pH 6,3, 0,04 М с pH 6,0, 0,1 М с
pH 5,8, 0,3 М с pH 5,4. При этом фракция у 2 элюируется
0,005 М буфером, фракция у 1—0,1 М. Обе эти фракции могут
быть разделены на подфракции у 2а и у2Ь, соответственно у 1а
и у 1Ь.
4.1.1.8. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ КРУПНОГО
РОГАТОГО СКОТА
Из сыворотки крупного рогатого скота при помощи ионообмен-
ной хроматографии на ДЭАЭ-сефадексе А-50 был выделен IgG 2
(известно два подкласса IgG: IgG 1 и 2). Элюцию проводят гра-
диентом 0,01 М трис-HCl с pH 8,6—0,7 М трис-HCl с pH 8,6.
Первый пик представляет собой чистый IgG 2. При помощи бу-
фера, содержащего 0,001 М ЭДТА, IgG 1 выделяют из моло-
зива. После обезжиривания и удаления казеина проводят хро-
матографию на ДЭАЭ-сефадексе А-50; собирают последнюю
порцию элюата, не содержащую IgG 2, концентрируют ее и очи-
щают на сефедексе G-200. Первая часть пика 7S содержит чис-
тый IgG 1.
394
4.1.1.9. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ
КРЫС И МЫШЕЙ
Различают 4 подкласса IgG крыс: IgG 1, IgG 2а, 2b, IgG 2с, ко-
торые можно выделить из сыворотки или асцитической жидко-
сти крысы с соответствующей миеломой [13]. У мыши также
описаны 4 подкласса IgG: IgG 1, IgG 2а, IgG 2b и IgG3. Их вы-
деляют из сыворотки мышей с миеломой. Для этого можно ис-
пользовать зональный электрофорез с последующей электрофо-
кусировкой. Из нормальной мышиной сыворотки IgG получают
преципитацией сульфатом аммония (20—30% насыщения) с
последующей ионообменной хроматографией на ДЭАЭ-целлюло-
зе в 0,01 М фосфатном буфере с pH 8,4. IgG можно также полу-
чать фракционированием этанолом по Кону, П-фракцию осаж-
дают сульфатом аммония (50% насыщения), затем проводят
хроматографию на ДЭАЭ-целлюлозе в 0,01 М фосфатном буфе-
ре с pH 7,5 с 0,015 М NaCl. Довольно часто используют очистку
на сефадексе G-200. Хроматография на ДЭАЭ-целлюлозе не так
эффективна для очистки мышиного IgG как для очистки IgG
других животных. Мышиный IgG довольно прочие связывается
с ДЭАЭ-целлюлозой и элюируется вместе с другими сывороточ-
ными белками (0,04 М фосфатный буфер с pH 8,0). Первый пик
при низкой ионной силе, который при фракционировании сыво-
ротки человека содержит чистый IgG, здесь сильно загрязнен
трансферрином. Лучшие результаты дает зональный электрофо-
рез. IgG 1 и IgG 2 могут быть разделены фракционированием
этанолом [8]. Элегантным методом разделения IgG на подклас-
сы может стать иммуноадсорбция фракции 7S из асцитической
жидкости, полученной после аллоиммунизации.
В клеточной мембране Staphylococcus aureus содержится
протеин А, который связывает IgG 2а, IgG2b и IgG3 мыши,
но не IgG 1, IgA или IgM. После адсорбции на протеине А,
IgG 1 можно отделить от IgA и IgM хроматографией на ДЭАЭ-
целлюлозе [6].
4.1.1.10. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ
СВИНЬИ
Подклассы IgG 1 и IgG 2 выделяют в чистом виде из молозива
[12]. Удаляют жиры, казеин и липопротеиды (см. разделы 4.1.2),
затем проводят хроматографию на ДЭАЭ-целлюлозе, затем
гель-фильтрацию на сефарозе 6В. Элюцию с ДЭАЭ-целлюлозы
проводят градиентом трис- HCl-буфера с pH 7,4, 0,002 М—0,3 М.
IgG 2 элюируется исходным буфером; сначала выходит IgG 1,
загрязненный IgG 2, затем элюируются IgG 1, S-IgA и IgM. Эти
последние можно получить в чистом виде гель-фильтрацией на
сефарозе 6В [12].
395
4.1.1.11. ВЫДЕЛЕНИЕ IgG ИЗ СЫВОРОТКИ ОВЦЫ
Вначале проводят осаждение сульфатом аммония, затем хрома-
тографию на ДЭАЭ-целлюлозе. В 0,025 М трис-НС1-буфере с
pH 7,8 IgG 2 не адсорбируется. При элюции линейным градиен-
том 0,025 М трис-НС1 pH 7,8—0,15 М трис-НС1-буфер с pH 7,8
остатки IgG 2 элюируются вместе с IgG 1, затем выходит IgG 1
4.1.2. ВЫДЕЛЕНИЕ ИММУНОГЛОБУЛИНА A (IgA)
4.1.2.1. ПРИНЦИП МЕТОДА
Выделение IgA представляет известные трудности, поскольку
методы, основанные на использовании молекулярного сита или
на использовании различного заряда белков, не позволяют в
достаточной мере отделить IgA от IgG. Для выделения IgA при-
ходится комбинировать несколько методов, например гель-
фильтрацию, осаждение балластных белков сульфатом цинка и
зональный электрофорез. Довольно часто применяют ионообмен-
ную хроматографию. Небольшие количества IgA можно полу-
чать из сыворотки методом иммуноадсорбции. В молозиве че-
ловека секреторный IgA является основным иммуноглобулином,
но это не характерно для других биологических видов.
4.1.2.2. ВЫДЕЛЕНИЕ IgA ИЗ СЫВОРОТКИ
ЧЕЛОВЕКА
4.1.2.2.1. Материалы и оборудование
Для работы необходимы: сыворотка человека, сефадекс
С-200, колонки для гель-фильтрации (длина 100 см), певикон,
прибор для зонального электрофореза, сульфат декстрана и
хлорид кальция для удаления липопротеидов малой плотности.
4.1.2.2.2. Методика
Исходным материалом служит плазма, из которой были удале-
ны фибриноген и p-липопротеиды. Этот материал фильтруют че-
рез сефадекс G-200 (рис. 123). IgA элюируется несколько рань-
Рис. 123. Хроматография IgA на сефадексе G-200.
После хроматографии фракция 2 была рехроматографирована иа сефадексе G-200. Пер*
вая фракция этого разделения подвергалась рехроматографии на сефадексе G-200 и т. д.
396
ше IgG. Вторую фракцию (см. рис. 123) концентрируют и снова?
хроматографируют на G-200. Собирают первую фракцию (от
второго разделения), рехроматографируют ее и т. д. до 5 раз.
Вторую фракцию 2, 3, 4 и 5 разделения отбрасывают (см.
рис. 123). Первую фракцию после 5 (последнего) разделения
собирают и при помощи зонального электрофореза с певиконом-
в качестве носителя очищают от аг-глобулина и значительна
уменьшают содержание IgG.
4.1.2.2.3. Оценка метода и другие варианты
Выход IgA при использовании этого метода составляет 20%.
Иммуноэлектрофорез не выявляет примеси IgG в препарате
IgA; но при анализе по методу Ухтерлони обнаруживаются
следы этого белка. В некоторых случаях в препарате содержит-
ся, помимо мономера, небольшое количество полимера IgA. Не-
достатком этого метода является его крайняя длительность.
Кроме того, методом препаративного электрофореза получают
IgA с ограниченной электрофоретической подвижностью.
Препарат, отражающий электрофоретическую гетероген-
ность сывороточного IgA, может быть получен методом, схема
которого приводится ниже. Выход составляет 9%, остаточные
количества IgG удаляют при помощи иммуносорбции.
Рекальцифицированная плазма
2 М (NH4)2SO4
4’С
Надосадочная
жидкость I -------------------
Преципитат I
Диализ против 0,025 М вероналового буфера
С pH 6,9; 4 °C, 48 ч
Преципитат II ----------------
Надосадочная жидкость II
Концентрирование, очистка иа ДЭАЭ-сефадексе А-50 седимеитационно-декаи-
тационным методом. Элюция при помощи ступенчатого градиента: по 2 л
каждого элюента на 1 л разбухшего сефадекса. В 0,01 М фосфатном буфе-
ре с pH 8,0 с ионообменииком связывается 3 г белка.
Элюент I Элюент ТТ 0,01 М фосфатный буфер с pH 8,0
Элюент III - 0,04 М фосфатный буфер с pH 8,0
Элюент IV 0,04 М фосфатный буфер с pH 7,5
0,2 М фосфатный буфер с pH 7,5 |
397
Элюат IV
(0,2 М фосфатный буфер с pH 7,5)
I
Диализ против дистиллированной воды, лиофилизация. Растворяют 3,5 г
лиофилизата в 100 мл 0,02 М фосфатного буфера с pH 7,0. Доводят кон-
центрацию белка до 3,33 г/л, затем белок осаждают этанолом в конечной
концентрации 25% при —5 °C 30 мин.
Преципитат III-----------------1
Надосадочная жидкость III
Диализ, 2 М (NH4)2SO4, глицин (20 г/л), температура 20°C
Надосадочная ------------------
жидкость IV
Преципитат IV
Хроматография на ТЭАЭ-целлюлозе
Ступенчатое элюирование
0,02 М фосфатный буфер, pH 6,7
0,035 М фосфатный буфер, pH 6,7
0,05 М фосфатный буфер, pH 6,7
0,065 М фосфатный буфер, pH 6,7
0,08 М фосфатный буфер, pH 6,7
(рис. 124)
I
I
Фракция 3
Многократная иммуноадсорбция на бромацетилцеллюлозе, конъюгированной
с анти-IgG. Из антисыворотки получают у-глобулин осаждением Na2SO4
(двукратное осаждение, 140 г/л), конъюгируют его с бромацетилцеллюлозой
I
Неадсорбированная фракция IgA
Довольно часто IgA получают следующим методом:
1) удаление эу-, псевдоглобулинов: разводят в 10 раз исход-
ную сыворотку 0,02 М буфером Сёренсена с pH 6,0, диализуют
против этого буфера;
2) осаждение сопутствующих белков Z11SO4;
3) проведение препаративного электрофореза.
Выход составляет 5—10%. Отдельные партии препарата мо-
гут быть загрязнены IgG и IgM. Известной оптимизации мето-
да можно добиться, удалив вначале большую часть IgG при
помощи хроматографии на ДЭАЭ-целлюлозе в 0,01 М фосфат-
ном буфере с pH 8,0, а затем выделив IgA из фракции, элюируе-
мой 0,08 М фосфатным буфером. Выделенный данным способом
IgA содержит небольшую примесь IgM. Полученный препарат
преимущественно содержит IgA в мономерной форме (7S),
а также некоторое количество полимера. В небольших количе-
ствах IgA можно получать непосредственно из сыворотки мето-
дом иммуноадсорбции или из иммунных преципитатов, образо-
398
ванных Р(аЬ')г-фрагмента-
ми антител к IgA. Для по-
лучения анти-IgA-сывороток
допустимо использование
препаратов IgA с большим
содержанием IgG. После-
дующей адсорбцией IgG
антисыворотку можно сде-
лать специфической только
к IgA. Препарат IgA с 20%
IgG получают из сыворотки
по следующей методике:
1) трехкратное осажде-
ние сульфатом аммония
(50% насыщения);
2) ионообменная хрома-
тография на ДЭАЭ-целлю-
лозе. Вначале IgG получа-
ют в 0,005 М фосфатном
буфере с pH 8,0, затем про-
водят элюцию градиентом
фосфатного буфера (pH
8,0) 0,005 М —0,1 М. Фрак-
Рис. 124. Хроматография IgA на ТЭАЭ-
целлюлозе (объяснение в тексте).
цию, элюируемую в пределах концентрации 0,0175 М —
0,045 М, собирают, концентрируют и разделяют на сефадексе;
3) гель-фильтрация на сефадексе G-200 в 0,15 М растворе
NaCl, содержащем 0,01 М боратный буфер с pH 8,0.
Чаще всего мономерные и полимерные формы IgA получают
из сыворотки больных с множественной миеломой. Вначале про-
водят зональный электрофорез в вероналовом буфере с pH 8,6
и ионной силой 0,05 (4°C), затем хроматографию последова-
тельно на трех колонках с сефадексом G-200 или био-гелем
А-1,5т. В ходе фильтрации IgG отделяется от мономерного
IgA. Последнюю часть пика IgA, содержащую IgG, отбрасы-
вают. Очистку и выделение IgA из сыворотки больных с миело-
мой можно осуществлять осаждением сульфатом аммония (50%
насыщения) с последующей многократной гель-фильтрацией на
сефадексе G-200 или сефарозе 6В. Можно попытаться провести
очистку комбинаций последовательных осаждений сульфатом
аммония, каприловой кислотой и адсорбцией на ДЭАЭ-целлюло-
зе (Batch-метод).
4.1.2.З. ВЫДЕЛЕНИЕ IgA ИЗ СЫВОРОТКИ
СВИНЬИ
Процесс получения IgA из сыворотки свиньи аналогичен про-
цессу получения IgA из сыворотки человека. Вначале удаляют
липопротеиды осаждением сульфатом декстрана 500. К 1 мл
399
сыворотки добавляют 0,02
мл 10% раствора декстра-
на и 0,1 мл IM CaCU.
Центрифугируют 10 мин
при 10 000g (4°C), осадок
отбрасывают, надосадоч-
ную фракцию диализируют
сначала против дистиллиро-
ванной воды, а затем про-
тив 0,1 М трис-HCl буфера
с pH 8,0, содержащего 1 М
NaCl, и хроматографируют
на колонке (100 см) с сефа-
дексом G-200 (рис. 125).
Объединяют фракции 69—
78, диализируют их против
Фракции
Рис. 125. Хроматография освобожден-
ной от липопротеидов сыворотки свиньи
на сефадексе G-200.
дистиллированной воды и концентрируют ПЭГ 20 000. Диали-
зируют против 0,02 М трис-НС1-буфера с pH 7,4 и по оконча-
нии диализа хроматографируют на ДЭАЭ-целлюлозе. Элюи-
рование проводят ступенчатым градиентом трис-НС1-буфера
с pH 7,4: 0,02, 0,05, 0,075, 0,10, 0,125, 0,15 М. IgA элюируется
0,075 и 0,1 М буфером. Его концентрируют, диализируют про-
тив 0,1 М трис-НС1-буфера с pH 8,0, содержащего 1 М NaCl,
и хроматографируют на сефадексе G-200. Выделяют два пика,
второй собирают и рехроматографируют на сефадексе G-200.
Собирают фракции, соответствующие вершине пика, концент-
рируют их, диализируют против 0,05 М вероналового буфера
с pH 8,6 и разделяют электрофоретически на пластинах 1 % ге-
ля агара (10X20 см). Вносят в центральную канавку 0,5мл
раствора белка (5 мг) и в течение 2 ч ведут электрофорез при
4 °C и напряжении 3Q В/см. После этого IgA элюируют из по-
лоски агара шириной 1,5 см с катодной стороны. Полоску ага-
ра заливают 0,15 М NaCl, замораживают при —20°С, оттаива-
ют, центрифугируют 10 мин при 10000. Надосадочную фрак-
цию диализируют и концентрируют.
4.1.2.4. ВЫДЕЛЕНИЕ IgA ИЗ СЫВОРОТКИ
ЛОШАДИ
Выявление IgA в сыворотке лошади длительное время пред-
ставляло серьезные трудности. Исходным материалом для выде-
ления IgA служит псевдоглобулиновая фракция после осажде-
ния сульфатом аммония (40% насыщения), которую диализиру-
ют против 0,005 М. фосфатного буфера с pH 6,0. В ходе ионооб-
менной хроматографии на ДЭАЭ-целлюлозе мономерный IgA
элюируется 0,06 М. фосфатным буфером с pH 7,0, полимер-
ный — 0,15 М фосфатным буфером с pH 6,0.
400
4.1.2.5. ВЫДЕЛЕНИЕ IgA ИЗ БЫЧЬЕЙ,
БАРАНЬЕЙ И КОЗЬЕЙ СЫВОРОТОК
Фракционирование IgA овец и коз проводят по следующей
схеме:
1) осаждение сульфатом аммония (50% насыщения);
2) ступенчатое фракционирование на ДЭАЭ-целлюлозе фос-
фатными буферами: 0,01 М, pH 7,4; 0,02 М, pH 7,4; 0,05 М
pH 6,5; 0,13 М, pH 6,5 и 0,2 М, pH 6,0;
3) гель-фильтрация на сефадексе G-200.
Иммуноглобулин IgA элюируется 0,13 М фосфатным буфе-
ром с pH 6,5. Молекулы бычьего IgA имеют меньший электроот-
рицательный заряд, IgA из сыворотки и молока коров элюиру-
ется градиентом фосфатного буфера 0,03 М, pH 7,0—0,06 М,
pH 6,5. IgA овец и коз элюируется градиентом фосфатного бу-
фера 0,05 М, pH 6,6—0,13 М, pH 6,3.
4.1.2.6. ВЫДЕЛЕНИЕ IgA КРЫС
Моноклональный полимерный IgA выделяли из асцитической
жидкости крыс, больных плазмоцитомой [10], то же касается
и мономера IgA. Мономерный IgA выделяли методом гель-филь-
трации на ультрогеле АсА22 и АсА34, а также хроматографией
на ДЭАЭ-целлюлозе. Разделение на АсА34 обеспечивает осво-
бождение от следов полимерного IgA и альбумина. Фракциони-
рованием на ДЭАЭ-целлюлозе выделяют IgG. IgA элюируют
0,05 М фосфатным буфером с pH 6,0, содержащим 0,3 М NaCl.
4.1.2.7. ВЫДЕЛЕНИЕ СЕКРЕТОРНОГО IgA (S-IgA)
ИЗ МОЛОЗИВА ЧЕЛОВЕКА
4.1.2.7.1. Материалы и оборудование
Для работы необходимы: молозиво, 1 М уксусная кислота,
ДЭАЭ-целлюлоза, КМ-целлюлоза, 0,01 М фосфатный буфер с
pH 7,6, 0,1 М фосфатный буфер с pH 6,4, 0,005 М ацетатный бу-
фер с pH 5,0, 0,5 М ацетатный буфер с pH 5,0, сефадекс G-200,
хроматографические колонки (2X25 см, 3X30 см, 2,5X100 см).
4.1.2.7.2. Методика
Собирают молозиво через 2—3 дня после родов. Центрифуги-
руют 2 ч при 20 000 об/мин с целью удаления жира. Доводят
pH 1 М уксусной кислотой до 4,6, чем вызывают выпадение ка-
зеина в осадок. После центрифугирования надосадочную фрак-
цию (3 г белка) диализируют против 0,01 М фосфатного буфера
с pH 7,6 и хроматографируют на ДЭАЭ-целлюлозе (колонка
3X30 см). Ступенчатое элюирование проводят градиентом:
1) 0,01 М фосфатным буфером с pH 7,6
2) 0,1 М фосфатным буфером с pH 6,4
Собирают фракцию, элюированную вторым буфером (0,5 г
401
Объем элюции.
Рис. 126. Получение секреторного IgA из мо-
лозива человека.
Белки, элюируемые 0,1 М NaC с ДЭАЭ-целлюлозы,
разделяют иа сефадексе G-200.
белка), диализируют
ее против 0,005 М аце-
татного буфера и хро-
матографируют на
КМ-целлюлозе (ко-
лонка 2X25 см). Элю-
цию ведут линейным
градиентам ацетатнога
буфера с pH 5,0,
0,005 М —0,5 М (по
300 мл каждого буфе-
ра). Фракцию, содер-
жащую S-IgA (подан-
ным иммунодиффу-
зии), концентрируют
и фракционируют на
колонке с сефадексом G-200 (2,5X100 см) в 0,05 М трис-
НС1-буфере (pH 8,0) с 1 М NaCl. Фракцию первого пика, со-
держащую S-IgA, снова разделяют на сефадексе G-200, диали-
зируют против дистиллированной воды и лиофилизируют.
4.1.2.7.3. Прочие методы выделения S-IgA
из молозива человека
Известны менее трудоемкие варианты получения S-IgA из моло-
зива человека.
1. После обеззараживания и осаждения казенна материал
диализируют против 0,01 М фосфатного буфера с pH 7,0 и фрак-
ционируют на ДЭАЭ-целлюлозе. Элюцию проводят линейным
градиентом 0,01 М фосфатного буфера с pH 7,0 и того же буфе-
ра, содержащего 0,1 М NaCl. Вторую фракцию хроматографи-
руют на сефадексе G-200. Вначале элюируется S-IgA. Получен-
ный препарат S-IgA электрофоретически чист. Выход состав-
ляет 5—10 мг на 1 мл молозива.
2. Молозиво смешивают с равным объемом 0,15 М NaCl и
центрифугируют 1 ч при 20 000 об/мин. Средний слой диализи-
руют против 0,01 М трис-НС1-буфера с pH 8,0 и фракционируют
на ДЭАЭ-целлюлозе. Для ступенчатого элюирования применяют
0,01 М трис-НС1-буферы с pH 8,0, 0,03, 0,05, 0,1 и 2,0 М NaCl.
Фракцию, элюируемую буфером с 0,1 М NaCl, концент-
рируют и подвергают восходящей гель-фильтрации на сефадексе
G-200 (2X120 см) в 0,01 М боратном буфере с pH 8,0, содержа-
щем 0,15 М NaCl. Нисходящую часть первого пика собирают и
повторно фракционируют на сефадексе G-200. Разделение пов-
торяют 2—3 раза, причем каждый раз собирают только нисхо-
дящую часть первого пика (рис. 126).
3. После обезжиривания и удаления казеина проводят осаж-
дение сульфатом аммония (0,5 насыщения), осадок растворяют
и диализуют против 0,1 М трис-НС1-буфера с pH 7,4, содержа-
402
Рис. 127. Фракциониро-
вание молозива кролика
иа сефадексе G-200 пос-
ле обезжиривания и уда-
ления казеина.
щего 0,15 М NaCl.
Проводят восходя-
щую гель-фильтра-
цию на сефадексе
G-200. Первый пик
концентрируют и
подвергают восхо-
дящей гель-фильт-
рации на сефарозе
6 В. Второй пик со-
держит S-IgA. Загрязнения удаляют хроматографией на
ДЭАЭ-сефадексе. S-IgA элюируют градиентом 0,01 М фосфат-
ного буфера с pH 7,6 и 0,1 М фосфатного буфера с pH 6,4.
4.1.2.8. ВЫДЕЛЕНИЕ S-IgA ИЗ МОЛОЗИВА
кролика
Выделение секреторного IgA животных проводят так же, как
и S-IgA человека. Сначала молозиво обезжиривают, удаляют
казеин подкислением pH, затем проводят гель-фильтрацию на
сефадексе G-200 в 0,1 М фосфатном буфере с pH 6,8. Далее
проводят ионообменную хроматографию на ДЭАЭ-целлюлозе,,
уравновешенной 0,01 М фосфатным буфером с pH 7,5. Прово-
дят элюцию ступенчатым градиентом того же буфера, содержа-
щего 0,1, 0,2, 0,3, 0,4 и 1,0 М NaCl (рис. 127). S-IgA элюируется
при концентрации NaCl 0,2 и 0,3 М. В заключение проводят
гельфильтрацию на сефарозе 4 В в 0,1 М фосфатном буфере с
pH 6,8 для удаления высокомолекулярных мукополисахаридов.
4.1.2.9. ВЫДЕЛЕНИЕ БЫЧЬЕГО S-IgA
У коров S-IgA выделяют из молозива, слюны и носовой слизи.
Из молозива его выделяют после обезжиривания и удаления
казеина путем хроматографии на ДЭАЭ-целлюлозе и гель-филь-
трации на сефадексе G-200. С ионообменника S-IgA элюируется
0,05 М. фосфатным буфером с pH 7,0. При гель-фильтрации ои
опережает IgG. После рехроматографии на сефадексе G-200
препарат S-IgA еще содержит примесь агрегированного IgG.
S-IgA из слюны также выделяют хроматографией на ДЭАЭ-
целлюлозе и гель-фильтрацией на сефадексе G-200. С ионооб-
менника S-IgA элюируют ступенчатым градиентом фосфатных
буферов: 0,01 М с pH 7,4; 0,02 М с pH 7,2; 0,06 М с pH 7,0;
0,1 М с pH 6,0; 0,2 М с pH 6,0. S-IgA элюируется с 0,06 М
403
'фосфатным буфером с pH 7,0. Первую половину пика после
гель-фильтрации рехроматографируют на сефадексе G-200, по-
лучают иммунохимически чистый S-IgA. Из носового секрета
'S-IgA получают при помощи блок-электрофореза в певиконе и
гель-хроматографии на сефадексе G-200. При гель-фильтрации
'получают два пика: первый наряду с S-IgA содержит IgM, вто-
рой— только S-IgA.
4.1.2.10. ВЫДЕЛЕНИЕ S-IgA ЛОШАДИ
Секреторный IgA лошади ведет себя при ионообменной хрома-
тографии и гель-фильтрации так же, как S-IgA крупного рога-
того скота. Поэтому для его получения можно использовать
аналогичную схему очистки. Как обычно, удаляют вначале жир
и казеин, осаждают белок сульфатом аммония (0,4 насыще-
ния), растворяют осадок, диализируют его против 0,01 М фос-
фатного буфера с pH 7,4 и хроматографируют иа ДЭАЭ-цел-
люлозе. «Медленную» фракцию IgG элюируют 0,01 М фосфат-
ным буфером с pH 7,4, секреторный IgA — 0,06 М фосфатным
буфером с pH 7,0. При гель-фильтрации на сефадексе G-200
S-IgA появляется между первым пиком и 75-фракцией выше
области исключения. Рехроматография на сефадексе G-200 дает
чистый секреторный IgA.
4.1.2.11. ВЫДЕЛЕНИЕ S-IgA
ИЗ СВИНОГО МОЛОКА
Секреторный IgA получают из свиного молока при помощи
хроматографии на ДЭАЭ-сефадексе А-50 (седиментационно-
декантациониый метод) и гель-фильтрации. S-IgA адсорбиру-
ется на ДЭАЭ-сефадексе в 0,1 М трис-буфере с pH 7,4; десорб-
ция происходит в 0,4 М трис-НС1-буфере с pH 7,4. Фракции
концентрируют, диализируют против 0,01 М трис-НС1-буфера
с pH 7,4, центрифугируют, надосадочную фракцию разделяют
на биогеле А-5т в 0,15 М NaCl. В третьем (первый большой)
пике обнаруживается иммуноэлектрофоретически чистый S-IgA.
Молоко с относительно высокой концентрацией IgA получают
после первой недели лактации, когда содержание IgG резко па-
дает. Помимо биогеля А-5т, для гель-фильтрации применяют
также сефадекс G-200 и сефарозу [12]. При использовании
градиента трис-НС1-буфера с pH 7,4 (0,002 М — 0,3 М) вначале
выходит IgG 2 со стартовым буфером, затем—IgG 1, содержа-
щий примесь IgG 2. В средней части градиента элюат содержит
IgG 1, S-IgA и IgM. Эти компоненты могут быть разделены на
сефарозе 6 В [12]. . • _i
404
4.1.2.12. ВЫДЕЛЕНИЕ S-IgA СОБАКИ
Чистый секреторный IgA из молозива собаки получают после
предварительного обезжиривания и удаления казеина методом
гель-фильтрации. Разделение проводят методом последователь-
ной восходящей элюции на сефадексе G-200 и сефарозе 4 В.
Фракцию, содержащую IgA, концентрируют, затем вновь нано-
сят на колонку сефадекса G-200. Первый пик содержит IgM и
IgA, следующий — IgA. Повторная хроматография дает чистый
S-IgA.
4.1.2.13. ВЫДЕЛЕНИЕ S-IgA МОРСКОЙ
свинки
Секреторный IgA выделяют из молока морской свинки после
обезжиривания и удаления казеина методом гель-фильтрации
на сефадексе G-200 или методом препаративного электрофоре-
за в певиконе. В ходе гель-фильтрации S-IgA выходит во вто-
ром пике после IgM и перед IgG 1 и IgG 2.
4.1.2.14. ВЫДЕЛЕНИЕ S-IgA КУР
Секреторный IgA получают из желчи после диализа против
0,1 М ацетатного буфера с pH 4,6, содержащего 2% NaCl. Сли-
зистый преципитат отбрасывают, желчь диализируют последо-
вательно против 0,15 М NaCl и против 0,01 М трис-НС1, pH 8,0.
Затем проводят хроматографию на ДЭАЭ-целлюлозе. Элюцию
ведут 0,01 М трис-НС1-буфером с pH 8,0 с возрастающей кон-
центрацией NaCl (0,06 М, 0,12 М, 0,30 М, 0,50 М). Большая
часть IgA элюируется с 0,30 М NaCl. Далее очистку ведут с
помощью зонального электрофореза в певиконе (0,03 М трис-
HCl-буфера с pH 8,0) и гель-фильтрации на сефарозе 4В. Пре-
парат оценивают по Оухтерлони с использованием анти-а-сыво-
ротки и антисыворотки к желчи кур [7].
4.1.3. ВЫДЕЛЕНИЕ ИММУНОГЛОБУЛИНА М (IgM)
4.1.3.1. ПРИНЦИП МЕТОДА
Иммуноглобулин М присутствует в сыворотке в меньших коли-
чествах, чем IgA и IgG. Поэтому часто в качестве исходного
материала используют сыворотку больных макроглобулине-
мией Вальденстрема для получения IgM. Основные методы
выделения IgM — гель-фильтрация и зональный электрофорез.
Часто используют осаждение фракции эуглобулинов. Липо-
протеиды удаляют сульфатом декстрана и СаС1г.
405
4.1.З.2. ПОЛУЧЕНИЕ IgM ЧЕЛОВЕКА
4.1.З.2.1. Материалы и оборудование
Для работы нужно иметь: СаС12, декстрансульфат, ПЭГ 6000,
сефадекс G-200 или сефарозу 4 В, сефарозу 6 В, биогель А-15,
0,05 М фосфатный буфер с pH 7,2, содержащий 0,5 М NaCl и
2% бутанола, 0,1 М вероналовый буфер с pH 8,6, поливинилхло-
рид, прибор для зонального электрофореза, колонки для гель-
фильтрации длиной 100 см.
4.1.3.2.2. Методика
Выделение IgM проводят в четыре этапа.
1. Удаляют липопротеиды низкой плотности осаждением
декстрансульфатом (относительная молекулярная масса 560 000,
5 г/л) и 0,09 М СаС12.
2. Осаждение макроглобулинов ПЭГ (относительная моле-
кулярная масса 6000), 75 г/л.
3. Гель-фильтрация (после растворения) макроглобулинов
на сефадексе G-200, сефарозе 4 В или биогеле А-15 в фосфат-
ном буфере 0,05 М с pH 7,2 (см. выше), содержащем 0,5 М
NaCl и 2% бутанола.
4. Разделение IgM и а-макроглобулина при помощи зональ-
ного электрофореза на поливинилхлориде в 0,1 М вероналовом
буфере с pH 8,6.
4.1.3.2.3. Обсуждение
Для разделения белков ПЭГ (6000) оказался особенно удоб-
ным, поскольку он не приводит к их денатурации (табл. 41).
Если гель-фильтрация на биогеле А-15 протекает на очень
длинных колонках (3 м) или проводится циклическая рефиль-
трация, то можно наблюдать частичное разделение IgM и
а2-макроглобулина. К зональному электрофорезу в этих усло-
виях можно не прибегать, поскольку первая часть пика сво-
бодна от а-2-макроглобулина.
Иммуноглобулин М из нормальной сыворотки также полу-
чают в 2 этапа при помощи гель-фильтрации на сефадексе
G-200 н электрофореза в колонках. Препарат все же содержит
следы а2-липопротеина. Точно так же IgM можно выделять
методом гель-фильтрации на сефакриле S-300 н аффинной хро-
матографией на протеин-А-сефарозе 4 В. На этапе гель-хрома-
тографии удаляются IgA и IgG. Элюцию с протеин-А-сефарозы
проводят 0,1 М цитратным буфером с pH 2,7, собранные фрак-
ции немедленно нейтрализуют 2,5 М трис-НС1-буфером с pH
8,5 [16]. Часто для выделения IgM из нормальной сыворотки
больных микроглобулинемией Вальденстрема сначала осажда-
ют фракцию эуглобулинов. Для этого сыворотку либо диали-
зуют против дистиллированной воды, 0,0067 М фосфатного бу-
фера с pH 5,8 или 0,002 М фосфатного буфера с pH 6,0 либо
проводят 10-кратное разведение сыворотки дистиллированной
406
Таблица 41. Фракционирование сыворотки Человека НоЛиэТйленгликолем
Белки Концентрация ПЭГ (г/л), при которой данный белок:
определяется преципитате обнаруживает снижение концентрации в иадосадоч- ной фракции ПОЛНОСТЬЮ исчезает из надосадочной фракции
«2- и [3-липопротеиды 30 40 50
IgM 20 40 60
а2-макроглобулин 40 100 120
IgG 30 90 120
IgA 30 120 140
Основной глобулин 40 120 Обнаружива-
Трансферрин «2-липопротеид 30 30 150 150 ются в надоса- дочной фрак- ции при кон- центрации 20—210 г/л
водой. Из сыворотки больных макроглобулинемией достаточно
чистый IgM можно получить одной лишь гель-фильтрацией на
сефадексе G-200. IgM-парапротеины можно выделить относи-
тельно простым путем независимо от электрофоретической мо-
бильности следующим образом [5]: 100 мл плазмы дефибрини-
руют добавлением 100 ЕД тромбина в 1 М СаОг (1 мл). После
30-минутной инкубации при 37 °C при постоянном помешива-
нии фибрин осаждают центрифугированием в течение 30 мин
при 10 000 (4°C). Затем проводят фильтрацию иа ультрагеле
ЛсА 34 в 0,1 М трис-НСЬбуфере с pH 8,0, содержащем 0,15 М
NaCl и 0,02% азида натрия (длина колонки 1 м). Первый пик
содержит IgM. Фракции, содержащие IgM, диализируют про-
тив стартового буфера (0,05 М НагНРС^/лимонная кислота с
pH 6,8). Элюцию проводят линейным градиентом стартового
буфера (300 мл) и 0,1 М буфера ИагНРОл/лимонная кислота
с pH 5,0 (300 мл). Первый пик, появляющийся при градиент-
ной элюции, — IgM. Его диализуют против 0,1 М трис-НС1-бу-
фера с pH 8,0 и концентрируют ультрафильтрацией до 10 мг/мл.
4.1.3.3. ВЫДЕЛЕНИЕ IgM ИЗ СЫВОРОТКИ
КРОЛИКА
Кроличий IgM выделяют тем же самым методом, что и IgM
человека:
Сыворотка (580 мл)
I Осаждение (NH4)2SO4 (0,35 насыщения) 3 раза
Преципитат
| Растворение в 144 мл 0,15 М NaCl, замораживание (—30°C), разморажи-
| ванне, фильтрация, центрифугирование 6,5 г при 50 000 об/мин
407
Осадок
I Растворение в 72 мл 0,15 М NaCl, наслаивание на равный объем 2 М
( CaCU, центрифугирование 11 г при 50 000 об/мин
Осадок
j Растворение в 15 мл 0,15 М NaCl, добавление 0,6 мл 10% раствора
| декстрансульфата (относительная молекулярная масса 2 000 000) н 1,5 мл
| 1 М СаС12, центрифугирование 15 мин. Диализ падосадочной фракции в
| течение 12 ч против 0,15 М NaCl, центрифугирование
Надосадочная фракция
| Гель-фильтрация на сефадексе G-200
Пик IgM
| Концентрирование до 5 мл, проведение электрофореза на колонках с се-
[ фадексом G-100 (буфер: 3 г трис и 14,4 г глицина на 1 л)
30 мг IgM
Благодаря ультрацентрифугированию уменьшается содержание
IgM и частично удаляются липопротеиды. Замораживание и
оттаивание с последующей фильтрацией также приводит к час-
тичному удалению липопротеидов. Полного их удаления дости-
гают с помощью декстрансульфата и СаСБ. Предварительное
осаждение эуглобулинов не имеет особого смысла, поскольку
IgM кролика почти не содержится в осадке эуглобулинов.
Иммуноглобулин М из сыворотки кролика можно получить
комбинацией методов, указанных ниже.
1. Двукратное высаливание сульфатом натрия (180 г/л)
2. Ионообменная хроматография
а) Ступенчатое фракционирование на ДЭАЭ-целлюлозе в
0,0175 М фосфатном буфере с pH 6,9. Элюирование проводят
тем же буфером, содержащим 0,1325 М NaCl и 0,1525 М NaCl.
б) Фракцию, элюированную 0,1525 М NaCl, наносят на
ДЭАЭ-целлюлозу (4 мг белка на 1 мл ионообменника) в 0,05 М
трис-НСЬбуфере с pH 8,6. Собирают фракции, полученные с
использованием стартового буфера и того же буфера, но со-
держащего 0,1 М NaCl. Их диализуют против 0,001 М борат-
ного буфера с pH 8,0, содержащего 0,32 М NaCl, и наносят на
колонку биогеля Р-200 (2,5X100 см).
в) Двукратная хроматография на биогеле Р-200. Восходя-
щая часть первого пика содержит иммуноэлектрофоретически
чистый IgM.
IgM кролика можно также получить методом иммуноадсорб-
ции с использованием антисыворотки к ц-цепи человека [14].
Инфекции, вызванные трипаносомами, повышают уровень IgM
в сыворотке у кроликов, IgM может быть выделен из такой
сыворотки гель-фильтрацией на сефадексе G-200 (1-й пик).
Дальнейшую очистку проводят центрифугированием после хра-
нения при 4 °C в течение ночи и зональным электрофорезом
надосадочной фракции в блоке агарозы в вероналовом буфере
с pH 8,6 [15].
408
4.1.З.4. ВЫДЕЛЕНИЕ IgM ИЗ СЫВОРОТКИ
ДРУГИХ животных
Иммуноглобулин М крупного рогатого скота выделяют из фрак-
ции эуглобулинов сыворотки или молозива, которые диализу-
ют против 0,005 М фосфатного буфера с pH 6,0. Дальнейшую
очистку проводят при помощи ионообменной хроматографии на
ДЭАЭ-целлюлозе и последующей гель-фильтрации. Фракцию
эуглобулинов диализуют против 0,06 М фосфатного буфера с
pH 7,0. Элюцию IgM с ДЭАЭ-целлюлозы проводят ступенчатым
градиентом: 0,06 М фосфатный буфер с pH 7,0, 0,10 М фосфат-
ный буфер с pH 6,0 и 0,20 М фосфатный буфер с pH 6,0. IgM
десорбируется с 0,20 М буфером. Доочистку проводят двукрат-
ной гель-фильтрацией на сефадексе G-200. Аналогичный способ
очистки применяют для получения IgM из сыворотки лошади.
IgM из сыворотки иммунизированных мышей получают хрома-
тографией на сефадексе G-25, G-200 и ультрацентрифугирова-
нием. Сыворотку в количестве 1—3 мл наносят на колонку
(2,5X100 см) с сефадексом G-25, уравновешенным дистиллиро-
ванной водой или 0,0001 М фосфатным буфером с pH 7,0.
Элюируют 0,01 М фосфатным буфером с pH 7,4, содержащим
0,15 М NaCl. При фильтрации выявляются два пика, первый
из которых содержит большую часть сывороточных белков.
Распределение иммуноглобулинов указано в табл. 42.
Таблица 42. Распределение иммуноглобулинов при гель-фильтрации на
сефадексе G-25
1 пик 2 пик
Доля белка
IgA 0,65—0,85 0,15—0,35
IgG 1 0,65 0,35
IgG 2 0,50—0,80 0,30—0,35
IgM Следы 1,00
Для дальнейшей очистки собирают материал 2-го пика и про-
водят гель-фильтрацию на сефадексе G-200 в 0,1 М трис-НС1-
буфере с pH 8,2, содержащем 1 М NaCl. Первая часть 1-го пи-
ка состоит из почти чистого IgM, лишь иногда загрязненного
небольшой примесью а2-липопротеидов. Дополнительное цент-
рифугирование в градиенте плотности сахарозы позволяет по-
лучить чистый IgM. Выход составляет 15—20%. IgM из сыво-
ротки крыс можно выделить методом иммуноадсорбции при по-
мощи антисыворотки к ц-цепям человека [14]. IgM из молози-
ва свиней, после удаления жира, казеина и липопротеидов,
получают ионообменной хроматографией на ДЭАЭ-целлюлозе
при помощи градиента трис-НС1-буфера с pH 7,4 0,002—0,3 М.
IgM элюируется совместно с S-IgA и IgG 1 в средней части
градиента. Чистый IgM получают после гель-фильтрации на
27—990 409
сефарозе 6 В [12]. Из сыворотки свиньи IgM можно выделить
осаждением борной кислотой (диализ против раствора борной
кислоты, 7,5 г/л) с последующей гель-фильтрацией через сефа-
декс G-200 (длина колонки 150 см).
Иммуноглобулин М из сыворотки кур осаждают сульфатом
аммония (насыщение 0,3), проводят ионообменную хроматогра-
фию на ДЭАЭ-целлюлозе. Элюирование проводят ступенчатым
градиентом: 0,06 М NaCl в 0,01 М трис-НС1-буфере с pH 8,О’
и 0,12 М NaCl в том же буфере. Собирают фракцию, элюируе-
мую вторым буфером, и проводят повторную гель-фильтрацик>
на сефадексе G-200. Первая половина 1-го пика содержит IgM
[7].
4.1.4. ВЫДЕЛЕНИЕ ИММУНОГЛОБУЛИНА D [IgD]
4.1.4.1. ПРИНЦИП МЕТОДА
Иммуноглобулин D присутствует в сыворотке в очень низких
концентрациях (30 мг/л), по своим физико-химическим свойст-
вам мало отличается от других 1g, что создает определенные
сложности при его выделении. В качестве исходного материала
используют сыворотки больных IgD-миеломой. Очистка прово-
дится на ДЭАЭ-целлюлозе (градиент фосфатного буфера с
pH 8,0 0,01 М — 0,3 М), а затем гель-хроматографией на сефа-
дексе G-200 в 0,05 М трис-НС1-буфере с pH 8,0, содержащем
0,2 М NaCl. Вместо сефадекса G-200 можно использовать уль-
тра-гель АсА 34 [4]. Фракционирование на ультра-геле позво-
ляет получить очищенный IgD, не содержащий примеси IgG и
других белков, имеющих сходную относительную молекулярную
массу. Дополнительная хроматография на ДЭАЭ-целлюлозе в
0,035 М фосфатном буфере с pH 8,0 позволяет получить миелом-
ный IgD в чистом виде с выходом свыше 90%.
4.1.4.2. ВЫДЕЛЕНИЕ IgD ИЗ СЫВОРОТОК
БОЛЬНЫХ С ДИСГАММАГЛОБУЛИНЕМИЕЙ
В качестве исходного материала служила сыворотка больного
с дисгаммаглобулинемией, характеризующейся повышенным со-
держанием IgD (1,04 г/л). Концентрация IgM также была по- I
вышена (3,1 г/л), содержание IgG составляло 8,9 г/л, IgA не-
определялся. IgD выделяли по следующей схеме:
Сыворотка
| 0,2 насыщения (NH4)2SO4______________Преципитат I
I I
Надосадочная жидкость I
| 0,5 насыщения (NH4);SO4
Преципитат II Налосадочная
I жидкость II
| Суспендируют в 0,2 М NaCl, разделяют на сефадексе G-200 в 0,01 М
| фосфатном буфере с pH 8,0 (длина колонки 100 см)
410
Объедииеиие фракций, содержащих IgD и IgM
Адсорбция суспензией ДЭАЭ-сефадекса
А-50 в 0,01 М фосфатном буфере с pH 8,0
Адсорбированный белок Фильтрат I (IgG)
| Элюция 0,075 М фосфатным буфером с pH 8,0
i J
Фильтрат II (IgD) Адсорбирован-
ный белок
0,2 М NaCl, pH 5,5 (за счет НС1)
Медленное добавление 1 М ацетата цинка до 0,05 М
I
Надосадочиая жидкость III Преципитат III
11 М NH4OH, pH 7,2
Преципитат IV Надосадочная
жидкость IV
Растворяют в 0,2 М NaCl с На2-ЭДТА (20 г/л), диализируют против
0,01 М фосфатного буфера с pH 8,0, хроматографируют на целлюлозе
IDE-52, элюируют градиентом фосфатного буфера с pH 8,0 0,01 М — 0,3 М
Объедииеиие фракций,
содержащих IgD
0,5 насыщения (NH^jSQ*
i Г
Преципитат V Надосадочная
| жидкость V
j Растворяют в 0,2 М NaCl, фракционируют на сефадексе G-200 (длина
Фколонки 100 см)
IgD
| Адсорбция на парааминобензилцеллюлозе, конъюгированной с антитела-
| ми к IgG и IgM
Фильтрат III (IgD)
4.1.4.3. ОБСУЖДЕНИЕ
В ходе гель-фильтрации на сефадексе G-200 IgM элюируется
в свободном объеме, за ним следуют IgD, IgE, IgA и IgG. Про-
цесс выделения IgD контролируют методом Оухтерлони или
иммуноэлектрофореза с применением моноспецифических анти-
сывороток. Моноспецифические сыворотки получают иммуниза-
цией экспериментальных животных миеломным IgD, с после-
дующей адсорбцией иммуноглобулином и сывороткой больного
с агаммаглобулинемией или небольшим объемом нормальной
сыворотки. Довольно часто при выделении IgD из сыворотки
больных миеломой наблюдается фрагментация его молекул,
обычно при хроматографии на ДЭАЭ-целлюлозе. Протеолиз
можно предотвратить добавлением е-аминокапроновой кислоты
[4]. Весь процесс выделения IgD должен проводиться при 4 °C.
4.1.5. ВЫДЕЛЕНИЕ ИММУНОГЛОБУЛИНА Е (IgE)
4.1.5.1. ПРИНЦИП МЕТОДА
Иммуноглобулин Е можно выделить из сыворотки больных
аллергическими заболеваниями с помощью ионообменной хро-
матографии на ДЭАЭ-целлюлозе и гель-фильтрации на сефа-
дексе G-200. Сыворотки больных IgE-продуцирующей миело-
27* 411
мой встречаются крайне редко и практически не используются
в качестве исходного материала. Ограниченное применение на-
ходит получение IgE из сыворотки аллергиков методом имму-
ноадсорбции.
4.1.5.2. ПОЛУЧЕНИЕ IgE ИЗ СЫВОРОТКИ
БОЛЬНЫХ С АЛЛЕРГИЕЙ
Проводят осаждение сульфатом аммония при насыщении 0,5 и
pH 7,0. Осадок диализируют против 0,005 М фосфатного буфе-
ра с pH 8,0 и хроматографируют на ДЭАЭ-целлюлозе. Элюиро-
вание проводят ступенчатым градиентом фосфатного буфера
0,005 М, 0,01 Ми 0,025 М с pH 8,0. Последний буфер элюирует
обогащенную IgE-фракцию, которая может быть подвергнута
дополнительной очистке на сефадексе G-200. IgE обнаружива-
ется в восходящей части пика. Путем повторной хроматографии
на сефадексе G-200 удается освободиться от большей части
IgG.
4.1.5.3. ВЫДЕЛЕНИЕ IgE ИЗ СЫВОРОТКИ
БОЛЬНЫХ С МИЕЛОМОЙ
Для выделения IgE из сыворотки больных миеломой исполь-
зуют метод препаративного электрофореза, фракционирование
сульфатом натрия (180 г/л) и гель-фильтрацию на сефадексе
G-150. Можно также использовать осаждение сульфатом ам-
мония (0,5 насыщения), хроматографию на ДЭАЭ-сефадексе
А-50 и гель-фильтрацию на сефадексе G-200.
4.1.6. СПИСОК ЛИТЕРАТУРЫ
1. Davies М. Е„ Barret A. I., Hembry R. М. Preparation of antibody fragments:
Conditions for Proteolysis compared by SDS-slab-gel electrophoresis and
Quantitation of antibody yield. — J. Immunol. Meth., 1978, 21, 305—315.
2. Duhamel R. C„ Schur P. H„ Brendel K. et al. pH gradient elution of human i
IgGl, IgG2 and IgG4 from protein A sepharose. — J. Immunol. Meth., 1979, ,
31,211—217.
3. Goosen P. Isolation of subclasses of human IgG with affinity chromatogra- .
phy. — J. Immunol. Meth., 1980, 37, 89—93.
4. Jefferis R. A rapid technique or the isolation of human IgD myeloma proteins
Employing ultrogel AcA 34. — J. Immunol. Meth., 1976, 9, 231—234.
5. Jehanly A., Bough D. A rapid procedure for the Isolation of human IgM
Myeloma proteins. — J. Immunol. Meth., 1981, 44, 199—204.
6. Johnson R. J„ Pasternack G. R. Antibody-mediated Suppression of Tumor
Growth. I. Suppression by murine IgG isolated from Alloantiserum. — J. Im-
munology, 1977, 118, 489—493.
7. Lebacq A. M. Purification and Testing of class specific antichiken immuno-
globulin Antibodies. — J. Immunol. Meth., 1979, 25, 101—117.
8. Measel J. VP., Cozard G. C. The ethanol fractionation of mouse serum. —
J. Immunol. Meth., 1976, 13, 289—298.
9. Morgan M., Johnson P„ Dean P. Electrophoretic desorption of immunoglobu-
lins from immobilized protein A and other ligands. — J. Immunol. Meth.,
1978, 23, 381—387.
10. Orlans E„ Peppard J., Reynolds J. et al. Rapid transport of Immunoglobu-
lin A from blood to bile. —J. exp. Med., 1978, 147, 588—592.
412
II. Peppard J. Quantitative Estimation of IgA in rats: A comparison of two
methods. — J. Immunol. Meth., 1979, 31, 129—139.
12. Reyero C., Siockl IF. Isolation of porcine colostral immunoglobulins and
preparation of monospecific anti-a, anti-у and anti-M chain antibodies using
agarose linked immunosorbents.—J. Immunol. Meth., 1977, 15, 211—221.
13. Rousseaux J., Biserte G., Bazin H. The differential enzyme sensitivity of rat
immunoglobulin G subclasses to papain and pepsin. —Molecular Immunology,
1980, 17, 469—482.
14. Sapin C., Druett P. Isolation of Rat and rabbit IgM from Normal serum
using anti-human M antibody polyacrilamide beads immunosorbents.—J. Im-
munol. Meth., 1976, 12, 355—363.
15. Van Tol M„ Veenhoff E„ Seijen H. Rabbit IgM: Its isolation in high yield by
a convenient procedure using serum from trypanosome-infected animals. —
J. Immunol. Methods, 1978, 21, 125—131.
16. Vidal M„ Conde E. P. Human immunoglobulin purification by affinity chro-
matographie on protein A-sepharose. — J. Biochem. Biophys. Methods, 1981»,
4, 155—161.
4.2. ФРАГМЕНТИРОВАНИЕ ИММУНОГЛОБУЛИНОВ
4.2.1. ПРИНЦИП МЕТОДА
Молекулы иммуноглобулинов состоят из двух легких и двух
тяжелых полипептидных цепей, соединенных дисульфидными
мостиками. Путем разрыва дисульфидных связей можно выде-
лить оба типа. Расщепление дисульфидных связей проводят при
помощи меркаптосоединений или сульфитом натрия в присут-
ствии ионов двухвалентной меди. Протеолиз пепсином позво-
ляет получать F(ab')2-фрагменты, папаином — Fab-фрагменты
(англ, fragment antibody) и Fc-фрагмент (англ, fragment
crystalline). Fab состоит из Fd-фрагмента (англ, fragment
difficult, N-концевая часть тяжелой цепи) и легкой цепи.
F(ab')2-фрагмент имеет константу седиментации 5S, относи-
тельную молекулярную массу 100 000 и обладает способностью
вызывать преципитацию антигенов. Восстановлением Р(аЬ')г-
Рис. 128. Фрагментация IgG папаином и пепсином.
СНО — углеводная часть; PC — пирролид«2*он-3-карбоновая кислота.
413
фрагментов получают моновалентные Fab'-фрагменты (3,5S),
которые подобно lFab-фрагментам обладают способностью ин-
гибировать преципитацию в гомологической системе, но сами
не обладают способностью преципитировать соответствующий
АГ (рис. 128).
4.2.2. ПОЛУЧЕНИЕ ЛЕГКИХ И ТЯЖЕЛЫХ ЦЕПЕЙ IgG
4.2.2.1. ВОССТАНОВИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ
МЕРКАПТОЭТАНОЛОМ
4.2.2.1.1. Материалы и оборудование
Для работы необходимы IgG, 0,55 М трис-НС1-буфер с pH 8,2,
меркаптоэтанол, йодацетамид (перекристаллизованный из спир-
тового раствора), триметиламин, 0,15 М NaCl, 1 М пропионовая
кислота, сефадекс G-75, хроматографическая колонка длиной
75 см.
4.2.2.1.2. Расщепление
Готовят раствор IgG (20 г/л) в 0,55 М трис-НС1-буфере с pH
8,2, деаэрируют его с помощью водоструйного насоса и насыща-
ют азотом. Добавляют меркаптоэтанол до конечной концентра-
ции 0,75 М. Оставляют на 1 ч при комнатной температуре, за-
тем охлаждают на ледяной бане и добавляют йодацетамид до
концентрации 0,75 М. Доводят pH до 8,0 триметиламином. Вы-
держивают 1 ч и диализуют против 0,15 М NaCl при 4 °C.
4.2.2.1.З. Разделение легких и тяжелых цепей IgG
Восстановленный IgG диализуют против 1 М пропионовой кис-
лоты при 4 °C, после чего разделяют легкие цепи (L) и тяже-
лые цепи (Н) на колонке с сефадексом G-25. Н-цепи выходят
в свободном объеме, второй пик соответствует легким цепям.
Фракции концентрируют и проводят повторную хроматографию
на сефадексе G-75 в 1 М пропионовой кислоте, после чего вновь
концентрируют и проводят диализ против физиологического
раствора.
4.2.22. ВОССТАНОВИТЕЛЬНЫЙ СУЛЬФИТОЛИЗ
4.2.2.2.1. Материалы и оборудование
Для работы нужно иметь IgG, Na2SO3, CuSO4-5H2O, NH4CI,
(NH4)2CO3, концентрированный раствор аммиака, сефадекс
G-25, сефадекс G-100, муравьиную кислоту, мочевину, хрома-
тографическую колонку длиной 1 м.
4.2.2.2.2. Расщепление
Растворяют 0,5 г белка в 70 мл буфера с pH 8,6 (0,5 М NH4CI,
pH подводят концентрированным NH3). Прибавляют 5 мл 0,1 М
CuSO4 и 0,1 мл концентрированного раствора аммиака. Доводят
414
pH до 8,6, затем прибавляют буфер до объема 80 мл. Еще до-
ливают 20 мл 1,25 М раствора Na2SO3 в буфере с pH 8,6.
Оставляют в закрытом сосуде на 24 ч при постоянном переме-
шивании. Проводят гель-фильтрацию на сефадексе G-25 в
0,25 М карбонате аммония (смена буфера) и лиофилизируют.
Фракционирование легких и тяжелых цепей проводят на сефа-
дексе G-100 в 0,05 М муравьиной кислоте с 6 М мочевиной. По-
лучают 3 пика: IgG (агрегаты Н-цепей), тяжелые и легкие
цепи.
4.2.2.3. ОБСУЖДЕНИЕ
Дисульфидные мостики между цепями молекул иммуноглобу-
линов расщепляются меркаптоэтанолом. Для предотвращения
окисления вводят йодацетамид. Восстановление дисульфидных
связей происходит при конечных концентрациях меркаптоэтано-
ла 0,2 М. или даже 0,1 М. IgM восстанавливается 0,2 М мер-
каптоэтанолом при 4°C за 24 ч, при 30°C — за 1 ч. Эффектив-
ным восстанавливающим агентом является дитиотреитол в
концентрации 0,1 М. Йодацетамид добавляют к тиоловым сое-
динениям в молярных соотношениях 1:1, 1,1:1 или 2:1. Вос-
становление меркаптоэтанолом можно проводить в присутствии
денатурирующих агентов (8 М мочевина, 7 М гуанидин-гидро-
хлорид). При этом образуются продукты, нерастворимые в
буферных растворах, но растворимые в 6 М мочевине, 3 М гуа-
нидин-гидрохлориде, 50% уксусной кислоте или 0,5—1°/о раст-
воре додецилсульфата натрия. Нековалентные связи между
легкими и тяжелыми цепями разрывают 1 М пропионовой или
уксусной кислотой, после чего становится возможным разделе-
ние на сефадексе. В пропионовой кислоте легкие цепи присут-
ствуют в виде мономеров, тяжелые цепи частично агрегированы.
Даже в смеси 8 М мочевина—1 М пропионовая кислота на-
блюдается незначительная агрегация Н-цепей. Наблюдаемая в
нейтральных буферных растворах тенденция к агрегации тяже-
лых цепей обусловлена нековалентными связями (тиоловые
группы блокированы йодацетамидом). В 0,004 М ацетатном
буфере с pH 5,4 легкие и соответственно тяжелые цепи присут-
ствуют в виде димеров. Если в этом буфере смешивать димеры
L- и Н-цепей, образуются молекулы IgG, не отличимые ни ме-
тодом Оухтерлони, ни ультрацентрифугированием от нативного
IgG. Рекомбинация алкилированных L- и Н-цепей IgG в раство-
ре пропионовой кислоты может быть осуществлена диализом
против нейтрального буфера и последующей очисткой на сефа-
дексе G-200. Возможна рекомбинация легких и тяжелых цепей
различных биологических видов. Рекомбинация IgG за счет ди-
сульфидных связей возможна только в отсутствие алкилирова-
ния после восстановления меркаптоэтанолом. Нековалентные
связи между цепями не образуются, если проводить ацилиро-
вание е-аминогруппы лизина. Поэтому после расщепления мер-
каптоэтанолом и алкилирования диссоциация проходит легко и
415
цепи хорошо отделяются друг от друга. В качестве ацилирую-
щего агента используют малеиновый или янтарный ангидриды.
Разделение молекулы IgM человека на легкие и тяжелые цепи
после малеинирования, восстановления и алкилирования про-
водят на сефадексе G-200 в 0,01 М трис-НС1-буфере с pH 8,6,
содержанием 0,1 М NaCl. L- и Н-цепи IgG кролика разделяют
на сефадексе G-100 после сукцинилирования в 0,01 М трис-НС1-
буфере с pH 8,2, но не при высоких концентрациях солей ввиду
опасности агрегации. Расщепление дисульфидных мостиков
под действием NajSOs и Си2+ протекает по следующему урав-
нению: RSSR+2Cu2++2SO32-^=^2RSSO3-+2Cu+.
Антитела IgM теряют активность после обработки меркапто-
этанолом, IgG резистентен к меркаптоэтанолу в этом отноше-
нии. Для дифференцировки IgM и IgG антитела инкубируют
1 ч при 37°C с 0,1 М меркаптоэтанолом. Затем определяют
активность антител. Аналитическим ультрацентрифугированием
было показано, что после восстановления IgM человека в тече-
ние 1 ч при комнатной температуре в трис-НС1-буфере с pH 8,6
и последующего алкилирования йодацетамидом образуются
субъединицы IgM с константой седиментации 6,7S, состоящие
из двух ц-цепей и двух легких цепей, связанных нековалентно.
При низкой концентрации белка субъединицы IgM распадают-
ся на 55-компоненты, т. е. на «половинки» четырех цепочечных
субъединиц с относительной молекулярной массой около 100 000.
В одинаковых экспериментальных условиях восстановленный и
алкилированный IgG не распадается на «половинки». Это мо-
жет быть причиной того, что восстановленный и алкилирован-
ный IgM теряет агглютинирующую способность после восста-
новления. В то же время субъединицы IgM с ковалентно свя-
занными ц- и L-цепями сохраняют агглютинирующие свойства.
Следует отметить, что лимфоцитотоксические IgG мыши при
низких концентрациях белка (0,3—0,7 мг/мл) оказываются до
некоторой степени чувствительными к меркаптоэтанолу, однако
это свойство пропадает при более высоких концентрациях IgG
(20 мг/мл [1]).
При помощи 0,1 М меркаптоэтиламина молекулу IgG кро-
лика можно разделить на 2 субъединицы, состоящие из одной
тяжелой и одной легкой цепи. Реакцию проводят в 0,1 М аце-
татном буфере с pH 5,0 при 37 °C в течение 75 мин в атмосфере
азота. Меркаптоэтиламин удаляют ионообменной хроматогра-
фией при pH 5,0 на холоду. Свободные SH-группы инактиви-
руют парахлормеркурибензоатом, который затем удаляют диа-
лизом против 0,025 М NaCl. Подкисление раствора IgM НС1
до pH 2,4 вызывает диссоциацию молекулы на две половинки.
Меркаптоэтиламин уже в концентрации 0,015 М вызывает час-
тичное расщепление молекул IgG. При нейтральном pH наблю-
дается рекомбинация половинок в полные молекулы IgG за
счет нековалентных связей. Заблокированные SH-группы мож-
но восстановить 0,1 М меркаптоэтиламином (3 ч, 37 °C) в
416
0,1 М ацетатном буфере с pH 4,5. Белок очищают на сефадек-
се G-25, в том же буфере при pH 8,0 происходит окисление
сульфгидрильных групп в дисульфидные с полным восстанов-
лением исходной структуры. Способность к диссоциации при
pH 2,4 сохраняет только 32% молекул IgG после трехчасовой
инкубации при 37°C и pH 8,0. За 30 мин инкубации при 30°C
в 0,1 М трис-НС1-буфере, содержащем 0,06 М NaCl, при
pH 8,0 0,02 М меркаптоэтиламин вызывает расщепление дисуль-
фидных связей между субъединицами IgM. Окисление неалки-
лированных субъединиц путем диализа против 0,001 М натрий-
боратного буфера, содержащего 0,16 М NaCl, с pH 8,0 при ин-
тенсивном перемешивании буфера ведет к восстановлению
структуры IgM.
4.2.3. РАСЩЕПЛЕНИЕ ПАПАИНОМ
4.2.3.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы IgG кролика, меркурипапаин, 0,01 М
цистеин и 0,002 М ЭДТА-натриевая соль в 0,1 М натрий-фос-
фатном буфере с pH 7,0, 0,1 М фосфатный буфер с pH 6,5,
0,01 М ацетатный буфер с pH 5,5, 0,9 М ацетатный буфер с
pH 5,5, КМ-целлюлоза, хроматографическая колонка 2,4X35 см.
4.2.3.2. МЕТОДИКА
Растворяют 150 мг IgG и 1,5 мг меркурипапаина в 10 мл
0,1 М фосфатного буфера с pH 7,0, содержащего ЭДТА и цис-
теин. Инкубируют смесь 16 ч при 37 °C и против дистиллиро-
ванной воды, диализуют при энергичном перемешивании в
течение 48 ч при 4 °C. Продукты расщепления подвергают диа-
лизу против 0,01 М ацетатного буфера с pH 5,5 и хроматогра-
фируют на КМ-целлюлозе. Элюирование проводят градиентом
фосфатного буфера 0,01—0,9 М с pH 5,5 (рис. 129). Хромато-
графию выполняют при комнатной температуре для предотвра-
щения кристаллизации Fc-фрагмента. Фракции I и II представ-
ляют собой Fab-фрагменты различных молекул иммуноглобули-
нов, фракция III содержит Fc-фрагменты.
Fc-фрагменты характеризуются выраженной склонностью к
кристаллизации при pH, близком к нейтральному (5,0—7,5), их
можно перекристаллизовать, диализуя растворенную в 0,02 М.
уксусной кислоте фракцию III против 0,1 М фосфатного буфе-
ра с pH 6,5 при 4 °C.
4.2.3.3. ОБСУЖДЕНИЕ
Для расщепления IgG на Fab- и Fc-фрагменты используют
ртутные производные папаина или его кристаллическую фор-
му. Реакция прекращается либо вследствие окисления кисло-
417
Рис. 129. Хроматография фраг-
ментов IgG кролика, полученных
прн обработке папаином, на КМ-
целлюлозе.
Градиент ацетатного буфера: 0,01 М,
pH 5,5—0,9 М. pH 5,5. По ординате —
оптическая плотность при длине вол-
ны 280 им.
родом воздуха, либо при
добавлении N-этилмалеими-
да, йодацетамида или
йодуксусной кислоты. IgG
различных биологических
видов неодинаково расщеп-
ляются папаином. Вышепри-
веденные условия разделе-
ния Fab- и Fc-фрагментов на КМ-целлюлозе не позволяют про-
вести фракционирование человека, хотя в принципе этот ионо-
обменник использовать можно. Выход Fc-фрагмента после
1,5-часового гидролиза в присутствии 0,01 М цистеина и 0,002 М
ЭДТА при pH 7,0 достаточно выражен. После гидролиза про-
водят диализ против 0,01 М фосфатного буфера с pH 7,6, за-
тем разделяют фрагменты на КМ-целлюлозе. В первом пике
выходят Fc-фрагменты, во втором — Fab. Оба пика содержат
также некоторое количество IgG. После концентрирования из
первого пика выкристаллизовывается Fc-фрагмент в 0,2 М
фосфатном буфере с pH 7,6 против 6,002 М фосфатного буфе-
ра при 4 °C. Увеличение длительности протеолиза до 16 ч при-
водит к увеличению выхода низкомолекулярных продуктов.
Папаиновые фрагменты IgG человека могут быть разделены
также на ДЭАЭ-целлюлозе в градиенте фосфатного буфера с
pH 7,7 0,007—0,7 М. При ионной силе 0,01 в трис-НС1-буфере
с pH 8,0 Fab-фрагменты не адсорбируется на ДЭАЭ-целлюлозе,
а Fc-фрагменты связываются. Последние затем элюируют
0,3 М. NaCl в том же буфере.
Цистеин не только активирует папаин, но также вызывает
конформационные изменения субстрата вследствие восстано-
вительного расщепления дисульфитных связей между у-цепями.
При протеолизе IgG кролика папаином было обнаружено, что
восстановление дисульфидных связей между у-цепями Fc-фраг-
мента зависит от концентрации цистеина. Концентрация 0,001 М.
оказывается неэффективной, полное восстановление наблюда-
ется при концентрации 0,02 или 0,05 М. При длительном (свы-
ше 10 ч) гидролизе папаин выщепляет тот участок Fc-фрагмен-
та, где расположена дисульфидная связь. При pH 4,5—5,0
наблюдается особенно интенсивное разрушение Fc-фрагмента.
Из 4 субклассов IgG человека наиболее чувствителен к папаи-
лу IgG3, за ним следуют IgG 1 и IgG 2, наименее чувствителен
JgG 4. Папаин разрушает Fc-фрагменты чувствительных субклас-
418
сов. Гетерогенность Fc-фрагмента выявляется при электрофорезе
в крахмальном геле, например, Fc-фрагменты папаинчувствитель-
ных субклассов IgG отличаются от Fc-фрагментов миеломных
IgG. В присутствии цистеина IgG человека расщепляется, об-
разуя в небольших количествах Fc-фрагмент. Fc-фрагмент сос-
тоит из нековалентно связанных димеров и при электрофорезе
мигрирует в сторону анода быстрее, чем Fc-фрагмент. Расщеп-
ление IgG папаином в присутствии восстанавливающих аген-
тов приводит к отщеплению небольших пептидов от Fc-фраг-
мента. Fab-фрагмент подвергается восстановительному расщеп-
лению 0,2 М меркаптоэтанолом. При этом образуются легкая
цепь и Fd-фрагмент, который выделяется гель-фильтрацией на
сефадексе G-75 в присутствии 1 М пропионовой кислоты. Fd-
фрагмент IgG кролика в этих условиях элюируется в форме ди-
мера. Однако в 8 М мочевине с pH 3,5 его относительная моле-
кулярная масса составляет 21 600, а Fab-фрагмента в тех же
условиях —40 700. Fd-фрагмент IgG человека в 1 М пропионо-
вой кислоте с 6 М мочевиной отделяется на сефадексе G-100>
от легких цепей. Но и в этих условиях обнаруживаются диме-
ры Fd-фрагмента.
Трипсиновые фрагменты IgG человека разделяют при ком-
натной температуре методами ионообменной хроматографии
(см. разделение папаиновых фрагментов). Дальнейшую очист-
ку проводят на сефадексе G-75. IgG инкубируют в 0,1 М трис-
HCl-буфере с pH 8,0, содержащем 0,02 М СаС12; соотношение
фермент/субстрат составляет 1 :50. Помимо tFab- и tFc-, обра-
зуются Fab- и Fc-фрагменты. Протеолиз останавливают через
16 ч добавлением ингибитора трипсина. tFab- и tFc- соответст-
вуют Fab- и Fc-фрагментам, но отличаются от них по электро-
форетической подвижности. Относительная молекулярная масса
(отн. мол. масса) фрагментов составляет 52 000. tFab'- и tFc'-
представляют собой продукты расщепления tFab- и tFc-фраг-
ментов, их отн. мол. масса составляет 28 000. IgG разных суб-
классов неодинаково чувствительны к гидролизу трипсином,,
известны также видовые различия, например, IgG кролика ус-
тойчив к действию трипсина. Протеолиз IgM папаином
(рис. 130) приводит к образованию Fabp. После восстановления
0,1 М меркаптоэтанолом с последующим алкилированием этот
фрагмент при помощи гель-фильтрации в 1 М пропионовой кис-
лоте можно разделить на Fdp и легкие цепи. Fcp быстро рас-
падается на мелкие фрагменты, его можно выделить только
после непродолжительного (45 мин) гидролиза. В отсутствие-
цистеина можно получить (Fc)sp, папаиновым протеолизом. Об-
работкой IgM человека трипсином при 37 °C можно получить
Fabp- и .Р(аЬ)2ц-фрагменты (рис. 131). Обработка трипсином,
при 56°C приводит к расщеплению IgM до Fabp- и (Fc)sp-
фрагментов. Максимальный выход (Fc)sp наблюдается через
30 мин. Реакцию останавливают добавлением ингибитора трип-
сина; фрагменты разделяют на сефадексе G-200. Первый пик
419
Рис. 130. Протеолитическое и восстановительное расщепление IgM.
рапа*"’
Рис. 131. Фрагменты IgM [Fabp.; Ffab'JaH; (Fc)5p]. oo — углеводная часть
содержит (Fc)six> второй большой пик —Fabp,. (Fc)5p и Fabp,
можно доочистить на ДЭАЭ-целлюлозе [5]. Мономерный Fcp
получают мягким восстановлением (Fc)5|j, 0,004 М дитиотреито-
лом с последующим алкилированием 0,01 М йодацетамидом
[5]. Fcp отделяют от соответствующего пентамера гель-филь-
трацией на сефадексе G-200.
4.2.4. РАСЩЕПЛЕНИЕ ПЕПСИНОМ
4.2.4.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: кроличий IgG, кристаллический пеп-
син, 0,1 М ацетатный буфер с pH 4,5, 1 М NaOH, Na2SO4.
4.2.4.2. МЕТОДИКА
Обрабатывают 2 г IgG кролика 20 мг кристаллического пепси-
на в 76 мл ацетатного буфера с pH 4,5, проводят гидролиз в
течение 20 ч при 37°C.
Смесь центрифугируют, pH надосадочной фракции доводят
до 8,0 1 М NaOH. Медленно при комнатной температуре и при
постоянном помешивании добавляют раствор сульфата натрия
(25 г/100 мл) до конечной концентрации 18 г/100 мл. Осадок
собирают центрифугированием, растворяют в воде и диализуют
против 0,1 М ацетата натрия.
4.2.4.3. ОБСУЖДЕНИЕ
При фрагментации IgG пепсином образуются Р(аЬ')2-фрагмен-
ты с константой седиментации 5S, остальная часть молекулы
распадается на низкомолекулярные пептиды. Очистку фрагмен-
тов проводят методом гель-фильтрации {8]. Первое разделе-
ние проводят на сефадексе G-100; нефрагмеитированный IgG
удаляют на сефадексе G-150, дальнейшую очистку ведут на
КМ-сефадексе G-25 в 0,02 М ацетатном буфере с pH 5,5. После
восстановления 0,01 М меркаптоэтиламином в течение 75 мин
в 0,1 М ацетатном буфере с pH 5,0 из F(ab')2-фрагментов обра-
зуются моновалентные Fab'-фрагменты; этот процесс обратим
(окисление), если не проводилось алкилирование. При восста-
новлении 0,1—0,2 М меркаптоэтанолом образуются легкие цепи
и Fd'-фрагменты. Относительная молекулярная масса Fd'-фраг-
мента IgG человека составляет 31 500. Его можно отделить от
легких цепей хроматографией на сефадексе G-100 в 6 М моче-
вине с 0,1 М уксусной или 1 М пропионовой кислотами.
Под воздействием пепсина образуется также pFc'-фрагмент
с отн. мол. массой 26 000. Он выходит с сефадекса G-150 во
втором пике и может быть очищен повторной хроматографией
на сефадексе G-50. pFc'-фрагмент состоит из двух ковалентно
421
IgG человека
Рис. 132. Схема получения фрагмен-
та pFc1.
Пепсин.рН 4,5 24ч
Г ель -фильтрации.
сефадекс G~15O
lgG+F(ab’) 2 .pFc,'пептиды
Циклическая рехроматография,
сефадекс G- 50
связанных субъединиц. Папаином расщепляют pFc'- на Fc'-
фрагмент и пептиды (рис. 132 и 133).
Fab'-фрагменты оказались очень полезными для использо-
вания в методе меченных ферментом антител. F(al/)2- и Fab'-
фрагменты IgG кролика получают по следующей методике [3].
а) П о л у ч ен и е р(аЬ')г-ф Р а г м е н т о в
— IgG растворяют в ацетатном буфере с pH 4,1.
— К 100 мл IgG добавляют 2 мг пепсина. Концентрация IgG
должна составлять 20—30 мг/мл.
— Инкубация 20 ч при 37 °C.
— Реакцию останавливают подведением pH до 7,0.
— Выделяют F(ab')2-фрагменты хроматографией на сефадексе
G-150 или сефакриле S-200.
б) П о л у ч е н и е Fab'-ф р а г м ен то в
— F(ab')2-фрагменты диализуют против 0,55 М трис-НС1-буфе-
ра с pH 8,2 по меньшей мере 4 ч.
— Добавляют меркаптоэтанол до конечной концентрации 0,2 М.
— Оставляют на 10 мин при комнатной температуре, охлаж-
дают до 0 °C, добавляют перекристаллизованный из дистил-
лированной НгО йодацетамид до концентрации 0,3 М. Остав-
ляют смесь на 1 ч при 0°С и затем диализуют против фи-
зиологического раствора.
Известен также «мягкий» метод получения Fab'-фрагмен-
тов против IgG коз [6].
— F(ab')2-фрагменты растворяют в 0,1 М фосфатном буфере
с pH 7,0, содержащем 0,3 М NaCl.
422
— Раствор F(ab')2-фраг-
ментов пропускают
через колонку с сефа-
розой 4 В (10x30
мм), конъюгирован-
ной с IgG козы.
— Элюируют фосфатным
буфером с pH 7,0, со-
держащим 0,2 М NaCl.
— Fab'-фрагменты элю-
ируют тем же самым
буфером с добавлени-
ем 0,01—0,12 меркап-
тоэтаноламина и 0,001
ЭДТА.
F(ab') 2-фрагменты из
IgG крупного рогатого
скота получают по сле-
дующей методике [4].
— 2 г IgG растворяют в
50 мл 0,15 М NaCl.
Рис. 133. pFc1- и Fc'-фрагменты IgG.
— Добавляют 50 мл 0,4 М ацетатного буфера с pH 4,0, затем
80 мг трижды перекристаллизованного пепсина.
— Инкубируют 24 г при 37 °C на водяной бане.
— Доводят pH до 8,02 М NaOH.
— Диализуют надосадочную фракцию против физиологическо-
го раствора в течение 5 дней.
— Добавляют ZnSO* до конечной концентрации 0,025 М для
преципитации IgG и Fc.
- — Перемешивают 2 ч при комнатной температуре, центрифу-
гируют.
— Добавляют ЭДТА-натриевую соль (10 г/л).
— Диализ против ФСБ, продукт реакции состоит из Р(аЬ')г- и
Fab'-фрагментов.
Для получения F(ab')2-фрагментов из небольших количеств
сыворотки пепсиновому протеолизу подвергают глобулиновую
фракцию [9]. Затем проводят гель-фильтрацию на сефадексе
G-150. Во втором пике присутствует F(ab')2- Оптимальные ус-
ловия расщепления IgG различных биологических видов пеп-
сином определяют под контролем диск-электрофореза с ДСН.
Известны различия в чувствительности к пепсину в пределах
субкласса IgG, например, у крыс они выражены следующим
образом: [10] IgG 2c>IgG 2b>IgG 2a>IgG 1.
Протеолиз IgM человека пепсином приводит через 8 ч к его
полному распаду на пептиды. Более короткий по времени про-
теолиз приводит к образованию F(ab")2|i> распадающиеся по-
том на Fab'p,. Фрагменты выделяют методом гель-фильтрации
на сефадексе G-200, их обозначают F(ab')2 (IgM) или Fab pep
(IgM).
423
4.2.5. ФРАГМЕНТАЦИЯ СЕКРЕТОРНОГО IgA
Секреторный IgA с константой седиментации 11S представляет
собой димер; он состоит из четырех a-цепей и четырех легких
цепей. Кроме того, он содержит секреторный компонент (СК)
и J-цепь (англ. Joining peptide — соединяющий пептид). J-цепь
найдена в полимерных иммуноглобулинах, содержащихся как
в секретах, так и в сыворотке.
4.2.5.1. ВЫДЕЛЕНИЕ J-ЦЕПИ
4.2.5.1.1. Материалы и оборудование
Для работы необходимо иметь человеческий S-IgA, меркапто-
этанол, йодацетамид, трис-HCI, триметиламин или Йа250з,
CUSO4, NH4CI/NH3, гуанидин-гидрохлорид, мочевину, NaCl,
0,01 М. фосфатный буфер с pH 9, сефадекс G-200, ДЭАЭ-сефа-
декс А-50, хроматографические колонки 2,5х 100 см и 1,5x30 см.
4.2.5.1.2. Методика
Секреторный IgA человека расщепляют 0,2 М меркаптоэтано-
лом с последующим алкилированием. При гель-фильтрации на
сефадексе G-200 в 5 М гуанидин-гидрохлориде или на сефадек-
се G-100 в 1 М уксусной кислоте образуются два основных пи-
ка. Первый пик содержит два компонента с отн. мол. массами
75 000 и 64 000 (в соотношении 1:4). Компонент с отн. мол.
массой 75 000 является секреторным, с отн. мол. массой 64 000—
a-цепью. Второй пик содержит легкие цепи и J-цепи. J- и
L-цепи разделяют ионообменной хроматографией на ДЭАЭ-се-
фадексе А-50, уравновешенном 0,01 М фосфатным буфером с
pH 9,0, содержащим 8 М мочевину. Элюирование ведут линей-
ным градиентом: тот же буфер, но содержащий 0,7 М NaCl.
4.2.5.1.З. Обсуждение
Для выделения J-цепей из полимерного сывороточного IgA
или IgM достаточно обработать их 0,1 М меркаптоэтанолом;
для выделения S-IgA достаточна 0,2 М концентрация меркапто-
этанола. J- и L-цепи полностью отделяются друг от друга мето-
дом окислительного сульфитолиза. После восстановления мер-
каптоэтанолом и алкилирования J-цепь содержит примесь
L-цепей. Первые могут быть выделены после сульфитолиза, ми-
нуя стадию разделения на сефадексе G-200 в 5 М гуанидин-
гидрохлориде, методом хроматографии на ДЭАЭ-сефадексе.
Элюцию при этом проводят градиентом молярности NaCl от 0
до 0,7 М в 0,01 М фосфатном буфере с pH 9,0, содержащем
8 М мочевину. Секреторный компонент, легкие и тяжелые цепи
элюируются в одной и той же фракции (рис. 134). После рас-
щепления дисульфидных мостиков J-цепь более не связывается
ковалентно с 1g и очень легко, без применения денатурирующих
агентов (гуанидин-гидрохлорид, мочевина),отделяется при гель-
424
Рис. 134. Выделение J-цепи
по методу ионообменной
хроматографии на ДЭАЭ-се-
фадексе А-50 S-сульфони-
рованного секреторного IgA.
фильтрации на сефа-
дексе G-200 и хрома-
тографии на ДЭАЭ-
целлюлозе. J-цепь мо-
жно выделить методом
диск-электрофореза в
10 М мочевине, она
обладает большей эле-
ктрофоретической по-
движностью, особенно после окислительного сульфитолиза,.
чем L-цепи, J-цепь секреторного IgA кролика после вос-
становления и алкилирования также может быть выделена
электрофоретически в 10 М мочевине. Разделение J- и L-цепей
при помощи диск-электрофореза возможно и без мочевины
[11], а также методом иммуноадсорбции [11].
Простой способ разделения J- и L-цепей основан на диализе
против дистиллированной воды. J-цепи выпадают в осадок, в-
растворе остаются J-цепи и небольшое количество L-цепей.
Именно такой процедурой были получены J-цепи IgA, S-IgA
и IgM человека после восстановления и гель-фильтрации в 4 М
гуанидин-гидрохлориде [1Г]. J-цепи секреторных IgA и IgM не-
различаются в иммуноэлектрофорезе и имеют одинаковый ами-
нокислотный состав.
4.25.2. ВЫДЕЛЕНИЕ СЕКРЕТОРНОГО КОМПОНЕНТА
4.2.5.2.1. Материалы и оборудование
Для работы необходимы фракция, содержащая тяжелые цепи и<
секреторный компонент, получаемая гель-фильтрацией продук-
тов окислительного сульфитолиза S-IgA человека, гуанидин-
гидрохлорид; хроматографическая колонка длиной 100 см.
4.2.5.2.2. Методика
Исходный материал содержит секреторный компонент и тяже-
лые цепи. Секреторный компонент отделяется от Н-цепей при*
циклической рехроматографии на сефадексе G-200 в 5 М гуани-
дин-гидрохлориде. Вначале элюируются агрегаты тяжелых це-
пей, следом секреторный компонент, а затем тяжелые цепи..
Степень чистоты препарата оценивают с помощью электрофо-
реза.
28—990
425
Молозиво
Обезжиривание,удаление казеина
Рис. 135. Очистка свободного сек-
реторного компонента.
ДЭАЭ
4.2.5.2.3. Обсуждение
^0,01 М фосфат натрия,pH 7,6
(NH4l2SO4 (27 г/100 мл)
I
Надосадочная фракция
Секреторный компонент мож-
но выделить комбинацией ме-
тодов гель-фильтрации и им-
муноадсорбции после восста-
новления мекраптоэтанолом и
Градиент ацетата натрия
0,005-0.5 М. pH 5.0
*
последующего алкилирования.
Разделение проводят на сефа-
дексе G-200 (колонка 2,4 X
Х'150 см) в трис-НС1-буфере
(pH 8,0) с 1 М NaCl. Третий
пик содержит большую часть
секреторного компонента. Для
дальнейшей очистки 3-й пик
концентрируют и хроматогра-
фируют еще раз на сефадексе
G-200 для удаления остатков
а- и L-цепей, затем проводят
иммуноадсорбцию седимента-
ционным методом с иммоби-
лизованной антисывороткой
кролика к миеломному IgA.
Поскольку секреторный компонент не содержит метионина, он
не подвергается расщеплению бромцианом, на этом свойстве
основана его очистка. В секретах содержится также не связан-
ный с IgA секреторный компонент, который может быть выде-
лен, например, из молозива (рис. 135). Относительная молеку-
Сефадекс С-200,трис-НС1
pH 8.0 (2.4*100 см)
40 50 60 70
Фракции (5 мл)
лярная масса свободного и связанного секреторного компо-
нента одинакова, по данным разных авторов она колеблется
от 58 000 до 97 000. Секреторный компонент (СК) соединен с
S-IgA в основном дисульфидными связями, однако часть моле-
кул СК связана нековалентно. При гель-фильтрации S-IgA на
сефадексе G-200 в 5 М гуанидин-гидрохлориде наблюдается
диссоциация нековалентных связей, что позволяет выделить
СК, хотя и не в совершенно чистом виде. Секреторный компо-
нент кролика в отличие от человеческого СК соединяется с
S-IgA только нековалентными связями. Его можно выделить
гель-фильтрацией на сефадексе G-200 в присутствии 5 М гу-
анидин-гидрохлорида, однако получаемый препарат содержит
примесь L-цепей.
4.2.6. СПИСОК ЛИТЕРАТУРЫ
1. Capel Р., Gerlag Р„ Hageman J. et al. The effect of 2-mercaptoethanol on IgM
and IgG antibody. — J. Immun. Meth., 1980, 36, 77—80.
2. Davies M. E., Barret A. J., Hembry R. M. Preparation of antibody fragments:
426
Conditions for proteolysis compared by SDS slab-gel electrophoresis and’
quantitation of antibody yield. — J. Immunol. Meth., 1978, 21, 305—315.
3. Far A. G., Nakane P. K. Immunohistochemistry with enzyme labeled anti-
bodies: A brief review. — J. Immunol. Meth., 1981, 47, 129—144.
4 Fey H Light chains, Fab and F (ab)2 from Bovine IgG. — Immunochemistry,.
1977, 14, 99—106.
5. Chose G. A. Acomparative conformational studies on the tryptic digestion
fragments of human immunoglobulins M and G. — Biochem. Biophys. Res.
Commun., 1971, 45, 1144—1150.
6. Ishikawa E., Yoshitake S. A general and mild procedure for the purifica-
tion of rabbit Fab’ antibodies. — J. Immunol. Meth., 1980, 38, 117—123.
7. Johnson R. L., Deutsch H. E. Preparation and studies of myeloma Fab sub-
fractions. — Immunochemistry, 1970, 7, 207—215.
8. Kuenzle С. C., Pelloni R. R„ Weibel M. H. The purification of peptic antibody"
fragments from rabbit immunoglobulin G. — Experientia, 1972, 28, 1522—
1523.
9. Madsen L., Rodkey S. A method for preparing IgG F(ab') fragment using'
small amounts of serum. — J. Immunol. Meth., 1976, 9, 355—361.
10. Rousseau J., Biserte G., Bazin H. The differential enzyme sensitivity of rat
immunoglobulin G subclasses to papain and pepsin. — Molecular Immuno-
logy, 1980, 17, 469—482.
11. Se Kang Y„ Galvanio N. Y., Tomasi T. B. Human J-chain: isolation and mole-
cular weight studies. — J. Immunol., 1974, 112, 162—167.
4.3. ИММУНОСОРБЕНТЫ
4.3.1. ПРИНЦИП МЕТОДА
Иммуносорбенты представляют собой нерастворимые полимеры,
с которыми антигены или антитела специфически связываются
в реакции АГ —АТ. Принцип изготовления иммуносорбентов
состоит в том, что один из компонентов реакции АГ — АТ пере-
водят в нерастворимую форму (присоединение к полимеру ко-
валентными связями, например), а второй компонент при кон-
такте с иммобилизованным первым компонентом также пере-
ходит в нерастворимую форму. Растворимые примеси удаляют
отмывкой. Затем изменением pH или ионной силы связь АГ —
АТ разорвать и растворимый компонент элюировать. Теорети-
чески этим методом можно очистить любые АГ или АТ. Пре-
имущества метода заключаются в его простоте, возможности.
регенерировать иммуносорбент, а также в повышении стабиль-
ности иммобилизованного белка.
4.3.1.1. ГЛУТАРАЛЬДЕГИДНЫЙ МЕТОД
ПОЛУЧЕНИЯ ИММУНОСОРБЕНТОВ
Обе альдегидные группы глутарового альдегида взаимодейст-
вуют с белками, в результате чего отдельные белковые моле-
кулы оказываются соединенными мостиками, образуя в целом
сетевую структуру, нерастворимую в воде. Химизм реакции
состоит в образовании оснований Шиффа за счет взаимодей-
ствия альдегидных групп глутаральдегида со свободными ами-
ногруппами белка [8].
28*
427
Конъюгация
NH2—Белон
•O-C-NH-Белок
NH
;ОН
✓ CsN-Белок
•О
-O-C-NH-Белок
>)
о
-он
Нарбамат (неактивный)
Имидонарбамат (активный)
Производное мочевины
N-замещенный имидонарбамат
N-замещеннай нарбамат
Рис. 136. Активация сефарозы бромцианом и присоединение белка к активи-
рованной сефарозе.
4.3.1.2. ИММУНОСОРБЕНТЫ НА ОСНОВЕ СЕФАРОЗЫ
Полисахариды, например сефадекс, сефароза, целлюлоза, акти-
вируются бромцианом. С активированным носителем белки
образуют ковалентные связи [8, 10]. Механизм ковалентного
связывания белка представлен на рис. 136.
4.3.2. МЕТОДИКИ
4.З.2.1. БЕЛКОВО-ГЛУТАРАЛЬДЕГИДНЫЙ
ИММУНОСОРБЕНТ
4.З.2.1.1. Материалы и оборудование
Для работы необходимы водный 2,5% раствор глутарового
альдегида, 0,9% раствор NaCl, 1 М фосфатный буфер с pH 7,0,
0,2 М фосфатный буфер с pH 7,3, 0,01 М ФСБ с pH 7,0, 0,1 М
глицин-НСЬбуфер с pH 2,8, антиген, антисыворотка, гомогени-
затор Уорринга, центрифуга, УФ-фотометр (например, VSU 2,
Carl Zeiss, йена), кварцевые кюветы, мешалка, мембранные
фильтры (Sartorius 13 200).
4.3.2.1.2. Проведение опыта
Растворяют 750 мг белка в 10 мл 0,1 М фосфатного буфера с
pH 7,0. При необходимости значение pH подводят 0,1 М НС1
или 0,1 М NaOH. К этому раствору добавляют 3 мл 2,5% раст-
вора глутарового альдегида, по каплям, при постоянном пере-
мешивании. Через 3 ч при комнатной температуре образуется
гель. Гель измельчают в гомогенизаторе Уорринга в 200 мл
0,2 М фосфатного буфера с pH 7,3. Полученную суспензию
центрифугируют 15 мин при 1000g (4°C). Частицы белка отмы-
вают 0,2 М фосфатным буфером с pH 7,3 до тех пор, пока
оптическая плотность смыва при длине волны 280 нм не станет
428
меньше 0,01. Затем гель суспендируют в 200 мл 0,1 М глицин-
HCl-буфера с pH 2,8 и снова центрифугируют. Повторяют про-
цедуру три раза. Частицы белка суспендируют в 200 мл ФСБ
и осаждают центрифугированием. Этот процесс повторяют до
тех пор, пока оптическая плотность смыва при 280 нм не ста-
нет равной нулю. Отмытый гель переносят в центрифужные
пробирки и добавляют соответствующее количество антисыво-
ротки (антигена). Перемешивают в течение 30 мин при комнат-
ной температуре и оставляют на 1,5 ч. Центрифугируют 15 мин
при 1000 g (4°C), надосадочную фракцию отбрасывают. Отмы-
вают гель ФСБ до тех пор, пока оптическая плотность смыва
при 280 нм не станет менее 0,01. Затем суспендируют гель в
5 мл 0,1 М глицин-НСЬбуфера с pH 2,8 в течение 15 мин, цент-
рифугируют 15 мин при 6000g (4°C), трижды повторяя эту
процедуру. Надосадочные фракции, содержащие очищенный
белок, объединяют и пропускают через микропористый стеклян-
ный фильтр G5. Фильтрат диализуют против изотонического
раствора хлорида натрия, концентрируют методом вакуумной
фильтрации через мембранные фильтры, проводят количествен-
ный анализ по Оухтерлони. Гель можно использовать повтор-
но, для этого его отмывают 0,2 М фосфатным буфером с pH 7,3
и хранят при 4 °C с добавлением мертиолята.
4.3.2.2. ИММУНОСОРБЕНТЫ НА ОСНОВЕ СЕФАРОЗЫ
4.З.2.2.1. Метод А
Материалы и оборудование: сефароза 4 В, бромциан, 1 М NaOH,
NaHCOa, NaCl, Н3ВО3, 1 М уксусная кислота, этаноламин, 1 М
НС1, pH-метр. Бромциан ядовит! Работа с ним должна прово-
диться под тягой.
Проведение опыта. Сефарозу 4 В отмывают дистиллирован-
ной водой, суспендируют в равном объеме дистиллированной
воды. Под тягой к сефарозе добавляют бромциан (0,3 г на 1 мл
сефарозы). Подводят pH до 11,2 с помощью 0,1 М NaOH и
оставляют на 12 мин при комнатной температуре. По окончании
реакции смесь выливают в 1 г охлажденной до 0 °C 0,1 М NaHCO3
с 0,5 М NaCl. После отмывки сефарозу суспендируют в том же
растворе и инкубируют с белком при 4 °C в течение 16 ч (5—
10 мг белка на 1 мл геля). Реакцию ведут при осторожном пе-
ремешивании. Проводят отмывку тем же раствором, суспенди-
руют в 1 М этаноламине с pH 8,0 для насыщения реакционно-
способных групп и мешают 2 ч при комнатной температуре.
Отмывают 0,1 М NaHCO3 с 0,5 М NaCl. Три раза проводят от-
мывку нековалентно связанного белка. Каждый цикл состоит
из двух этапов: 1) отмывки 0,1 М ацетатным буфером с pH 4,0,
содержащим 1 М NaCl; 2) отмывки 0,1 М боратным буфером с
pH 8,0, содержащим 1 М NaCl. Иммуноадсорбцию проводят в
0,1 М боратном буфере с pH 8,0, содержащем 0,5 М NaCl [10].
429
4.3.2.2.2. Метод Б
Материалы и оборудование: сефароза 4 В, Na2CO3, бромциан,
ацетонитрил, NaHCOs, NaCl, Н3ВО3, 1 М уксусная кислота,
1 М NaOH, мочевина.
Проведение эксперимента: 5 мл сефарозы 4 В отмывают
300 мл дистиллированной воды, затем суспендируют в 15 мл
2 М NaHCO3 при 4 °C. Растворяют при осторожном помешива-
нии 2 г бромциана в 5 мл ацетонитрила. На ледяной бане при
постоянном помешивании к сефарозе по каплям добавляют
раствор бромциана в течение 2 мин. Оставляют смесь на 15 мин,
затем быстро отмывают 250 мл 0,1 М NaHCO3 с pH 9,7, содер-
жащим 0,5 М NaCl, 250 мл дистиллированной воды и снова
250 мл 0,1 М NaHCOs с pH 9,7 с 0,5 М NaCl. Отмывку ведут
при 4°C. Конъюгацию проводят с 10 мл раствора белка (3—
4 мл/мл) в 0,1 М NaHCO3 (pH 9,7) с 0,5 М NaCl в течение 2 ч
при 25 °C при легком перемешивании. Следует избегать интен-
сивного перемешивания. Сефарозу выдерживают еще в течение
20 ч при 4 °C для инактивации реакционноспособных имидо-
карбонатных групп, после чего гель отмывают при 4°C:
1) 250 мл 0,00625 М боратного буфера (pH 8,8) с 0,15 М NaCl
2) 50 мл 0,1 М ацетата натрия (pH 4,5) с 0,5 М NaCl
3) 50 мл 2 М мочевины с 0,5 М NaCl
4) 50 мл 0,1 М NaHCO3 (pH 9,7) с 0,5 М NaCl
Повторяют весь цикл отмывки. Иммуноадсорбцию проводят на
колонке, уравновешенной 0,1 М боратным буфером (pH 8,0)
с 0,5 М NaCl [8].
4.3.2.2.3. Десорбция
Колонку тщательно промывают буфером для иммуносорбции
(см. выше). Для десорбции чаще всего используют 0,1 М гли-
цин-НС1-буфер с pH 2,8, а также 5 М гуанидин-НС1-буфер или
3,5 М KSCN в 0,15 М NaCl. Элюированные фракции нейтрали-
зуют, если десорбцию проводили первым способом, или диали-
зуют против ФСБ с pH 7,5. Количественную оценку проводят
по Оухтерлони или методом иммуноэлектрофореза. Для повтор-
ных использований колонку промывают 0,1 М боратным буфе-
ром с pH 8,0, содержащим 0,5 М NaCl. В качестве консерванта
добавляют азид натрия.
4.3.3. ОЦЕНКА МЕТОДА
У глутаральдегидно-белкового иммуносорбента специфическая
биологическая активность белка проявляется только на по-
верхности частиц геля. По сравнению с белковым гелем актив-
ная поверхность сефарозных иммуносорбентов существенно вы-
ше. Для повышения степени конъюгации количество бромциана
вместо обычных 100—150 мг увеличивают до 300 мг на 1 мл
430
сефарозы [8, 10]. Бромциан перед употреблением можно раство-
рять не только в воде, но, например, в диоксане. Контроль pH
не проводят, когда бромциан растворен в ацетонитриле (ме-
тод Б), причем бромциановый раствор добавляют очень мед-
ленно, чтобы избежать преципитации бромциана. Конъюгация
белка с активированными бромцианом сорбентами лучше всего
протекает при pH 8—10. Если лиганды теряют активность
вследствие слишком прочной конъюгации, то процесс присоеди-
нения лиганда проводят при pH 7. Буфер для конъюгации не
должен содержать аминогрупп (как, например, трисаминомета-
новые буферы). АН-сефароза 4 В и СН-сефароза 4 В представ-
ляют собой коммерческие продукты, полученные присоедине-
нием 1,6-диаминогексана или соответственно 6-аминокапроно-
вой кислоты к сефарозе 4 В, активированной бромцианом [10].
При помощи реакции с карбо диимидами, например с 1-этил-З-
диметиламинопропилкарбодиимид-HCl, к AH-сефарозе можно
присоединять лиганды с карбоксильными группами, к СН-сефа-
розе — лиганды со свободными аминогруппами. Эпоксиактиви-
рованная сефароза 6 В способна связывать лиганды с одной
свободной аминогруппой. Эпоксисефарозу получают взаимодей-
ствием сефарозы 6 В с 1,4-бис (2,3-эпокси-пропокси) бутаном
[10]. Эпоксисефароза пригодна для присоединения лигандов че-
рез гидроксильные группы сахара или сульфгидрильные группы.
Белок присоединяется к эпоксисефарозе прочнее, чем к бром-
циан-сефарозе [9]. Неспецифическую адсорбцию белков на
протеин-сефарозный конъюгат, обусловленную наличием основ-
ных групп на белке, конъюгированном с сефарозой, можно по-
давить анионным красителем голубым декстраном [7]. Для по-
лучения иммуносорбентов пригодны также активированный
бромцианом сефадекс G-200 или сефароза 6 МВ с крупными
гранулами [4]. При помощи аффинной хроматографии можно
разделять клетки с различными мембранными рецепторами, на-
пример лимфоциты, причем не обязательно использовать имму-
носорбенты на основе бромциан-сефадексов или сефарозы [1,
3]. Клетки можно присоединять к планшетам для микротитро-
вания через поли-Ь-лизин и глутаровый альдегид, после чего
поверхностные антигены этих клеток легко выявляются при по-
мощи ИФА [2]. Антитела можно также выявлять при помощи
антигена, конъюгированного с бромцианцеллюлозой {5]. Опи-
саны иммуносорбенты, получаемые за счет формирования се-
тевых структур, подобных белковым полимерам, образующим-
ся при обработке глутаровым альдегидом. Сетевые структуры
образуются под воздействием на белки этилхлорформиата или
N-ацетилмоноцистеинтиолактона. Кроме того, в качестве носи-
телей для белковых конъюгатов можно использовать параами-
нобензилцеллюлозу, карбоксиметилцеллюлозу, бромацетилцел-
люлозу, полиаминостирол, гидразидные производные полиакрила-
мида, активированный глутаровым альдегидом гель полиакрил-
амида и агарозы или полиакриламид [6].
431
4.3.4. СПИСОК ЛИТЕРАТУРЫ
1. Broecker Е., Sorg С. Specific separation of cytotoxic T lymphocytes on im-
munoadsorbtive film.—J. Immunol. Meth., 1977, 14, 333—342.
2. Cobbold S., Waldmann H. A rapid solid phase enzyme-linked binding assay
for screening monoclonal antibodies to cells surface antigens. — J. Immunol.
Meth., 1981, 44, 125—133.
3. Fong S., Troucas C. D., Pasquali J. L. et al. Fractionation human lymphocyte
subpopulations on immunoglobulin coated Petri dishes. — J. Immunol. Meth.,
1981, 44, 171—182.
4. Ghetie V., Mota G„ Sjoequist J. Separation of cells by affinity chromato-
graphy on Sp A-sepharose 6MB. — J. Immunol. Meth., 1978, 21, 133—141.
5. Gheuens J., McFarlin D. A multivalent antibody radioimmunoassay (MARIA}
for screening antibody secretion by lymphocyte hybridoma cultures. — J. Im-
munol. Meth., 1981, 47, 183—189.
6. Guesdon J. L„ Avrameas S. Polyacrilamide-agarose beads for the preparation
of effective immunosorbents. — J. Immunol. Meth., 1976, 11, 129—133.
7. Heinzel W., Rahimi-Laridjani I., Grimmlnger H. Immunoadsorbents. Non
specific binding of proteins to albumin-sepharose. — J. Immunol. Meth., 1976,
9, 337—344.
8. Johnson G., Garvey J. S. Improved methods for separation and purification
by affinity chromatography. — J. Immun. Methods., 1977, 15, 29—37.
9. Zola H. Immunoadsorbents prepared from cell membrane antigens: Efficiency
of Incorporation and Shedding of Protein during use.— J. Immunol. Meth.,
1978, 21, 51—58.
10. Firmenschrift. Affinity chromatography principles and methods. — Uppsala,
Sweden, 1976.
4.4. ПРИГОТОВЛЕНИЕ
КОНЪЮГИРОВАННЫХ АНТИГЕНОВ
4.4.1. ПРИНЦИП МЕТОДА
Конъюгаты гаптен — белок используются для получения специ-
фических АТ к гаптену. Существует большое количество спо-
собов присоединения гаптена к белку-носителю. Часто для этой
цели применяют диазопроизводные ароматических соединений.
Можно вводить 2,4-динитрофенильные или 2,4,6-динитрофениль-
ные группы в состав белков при помощи соответствующих суль-
фонатов. Пенициллин обладает способностью реагировать in
vivo с белками, что может быть причиной развития повышенной
чувствительности к пенициллину, которая обусловлена пеницил-
лоил-лизиновой группой. Пенициллоил-конъюгаты получают в
препаративных количествах с небольшим выходом побочных
продуктов (рис. 137). Для получения конъюгатов используют
бифункциональные реагенты, например, карбодиимиды или ди-
изоцианаты— для получения конъюгатов пептидов и нуклеоти-
дов с белками. Диизоцианаты реагируют со свободными амино-
группами. Часто используемым реагентом является толуол-2,4-
диизоцианат, изоцианатные группы которого обладают различ-
ной реакционной способностью. Реакцию с толуол-2,4-диизоциа-
натом проводят в два этапа: вначале диизоцианат присоединя-
ют к пептиду, а затем, удалив избыток лиганда, присоединяют
432
R-C-NH-CH-CH 'c(CH3)2
О yC— N-------CH-COOH
О
R-NH2
1 pH 12
R-C-NH-CH-CH XC(CH3)2
О O = C NH---CH-COOH
I
NH
iV
S HgCI
N--- C =CH C(CH3)9
2 H III
---► R -C C=O NH-CH-COOH
HgCI2 ''O/
H2S
SH
I
N - - C = CH (CH3)2
II III'"'
R-Г C = O NH-CH-COOH.
0Z
pH 7,5
NH2
(CH2)4
Лизиновые
группы белка
S
R-C-NH-CH--CH C(CH3)2
0 = C NH-CH-COOH
I
NH
I
(CH2)4
О CH
\c NH
/ \
Рис. 137. Пенициллоильные конъюгаты. Диастероизомеры
пептид к белку. Меченные ферментом АТ используются в цито-
химии для выявления антигенов (1); меченные ферментом АТ
и АГ находят применение в ИФА, где они позволяют выявлять
антигены, антитела и гаптены в количестве нескольких нано-
граммов (6). Меченный таким образом протеин А представляет
собой универсальный реагент для ИФА.
4.4.2. КОНЪЮГАТЫ ДНФ — БЕЛОК
4.4.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы бычий у-глобулин, динитробензол-суль-
фонат натрия, К2СО3, сефадекс G-25, хроматографические ко-
лонки.
4.4.2.2. МЕТОДИКА
В 50 мл воды растворяют 1,0 г бычьего у-глобулина и 1,0 г
К2СО3, затем добавляют 1,0 г 2,4-динитробензолсульфоната
натрия или калия (в 10-кратном молярном избытке по отноше-
433
нию к содержанию лизина в белке). Инкубацию проводят при
37 °C при постоянном перемешивании. За 24 ч инкубации при
25 °C происходит замещение 50—60 групп на одну молекулу
у-глобулина (максимальное замещение 70 групп). Реакцию про-
водят в темноте, поскольку ДНФ-группы светочувствительны.
По окончании реакции ДНФ-белок очищают гель-фильтрацией
на сефадексе G-25.
4.4.2.3. ДОПОЛНИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Определение ДНФ-групп в белке проводят спектрофотометри-
чески на волне 360 нм. В качестве коэффициента экстинкции
используют молярный коэффициент экстинкции е-ДНФ-лизина,
который при pH 7,4 составляет 17 530. Число ДНФ-групп соот-
носят со средней относительной молекулярной массой белка.
Количество белка определяют микромодификацией метода
Кьельдаля или по сухому весу. Для определения сухого веса
препарат диализуют против дистиллированной воды или раство-
ра ацетата аммония. Препарат высушивают в вакууме при
105 °C, в этих условиях ацетат аммония улетучивается за 1—
3 дня. Например, при содержании белка 16,8 мг/мл и экстинк-
ции 87,5 при 360 нм на молекулу бычьего у-глобулина (отн.
мол. масса 160 000) приходится в среднем 50 ДНФ-групп. Ди-
нитробензолсульфонат натрия очищают путем перекристалли-
зации из спиртового раствора и адсорбции активированным уг-
лем. Его точка плавления превышает ЗЛО °C. Молярный коэффи-
циент экстинкции при 360 нм составляет 280, после нагревания
в 0,02 М NaOH при 90 °C в течение 1 ч коэффициент экстинк-
ции увеличивается до 14 000 (превращение в динитрофенол).
ДНФ-белки хранят в водном растворе при —20°С или 4°С.
Белки стабильны, но их следует сохранять в темноте. Белки
с высокой степенью замещения выпадают в осадок в 0,15 М
NaCl. В концентрациях ниже 1 мг/мл препараты замещенных
белков растворимы в 0,1 М трис с 0,1 М NaCl (pH 7,6) или в
ФСН (pH 7,6). Очистку ДНФ-белков после реакции с динитро-
бензолсульфонатом от непрореагировавших ароматических ди-
нитросоединений проводят не только путем гель-фильтрации на
сефадексе G-25, но также диализом против воды при pH 7,8 и
выше, при помощи ионообменников или осаждением при pH
ниже 4. Если замещены все NH2-rpynnbi, ДНФ-белок нераство-
рим в среде с pH ниже 4. Если степень замещения невелика,
то растворимость белка может быть при pH 1—2 даже боль-
шей, чем при pH 3—4. ДНФ-производные белков предпочти-
тельнее получать с использованием динитробензолсульфоновой
кислоты, хотя, например, динитрофторбензол. реагирует энер-
гичнее. В реакцию с динитробензолсульфонатом вступают поч-
ти исключительно е-аминогруппы лизина. N-концевыми амино-
группами на практике можно пренебречь. Фенольные ОН-груп-
пы тирозина в вышеописанных условиях не вступают в реак-
434
ции, сульфгидрильные группы также не оказывают влияния на
степень замещения. К числу преимуществ данного метода отно-
сится то, что динитробензол сульфонат, в противоположность
динитрофторбензолу, хорошо растворим в воде и для его уда-
ления по окончании реакции не требуются органические раство-
рители. Аналогично ДНФ-белкам могут быть получены ТНФ-
производные путем взаимодействия тринитробензолсульфоновой
кислоты и белка в 0,2 М боратном буфере с pH 9,2. Эффектив-
ность замещения ТНФ-группами определяется по измерению
оптической плотности при 348 нм; молярный коэффициент экс-
тинкции е-ТЕФ-лизина равен 15 400.
4.4.3. ПЕНИЦИЛЛОИЛ-ПОЛИЛИЗИН
4.4.З.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы 100 мг полилизина-НС1 (20 амино-
кислотных остатков на молекулу), 2,4 г бензилпенициллина ка-
лиевой соли, 1 М раствор карбоната натрия с pH 10, 4, 5 М
NaOH, 0,002 М фосфатный буфер с pH 8,0, диализный мешок.
4.4.3.2. МЕТОДИКА
Растворяют 100 мг полилизина-НС1 в 0,4 мл воды и нейтрали-
зуют 5 М NaOH. Затем добавляют 1 мл 1 М карбоната
(pH 10,4) и 1,2 г бензилпенициллина. Доводят pH до 9,6 5 М
NaOH и, если необходимо, добавляют воду для полного раство-
рения пенициллина. По 600 мг бензилпенициллина добавляют
через 16 и 24 ч. После каждого добавления pH подводят до 11,0.
Через 6 ч после последнего внесения пенициллина проводят
диализ против 0,002 М фосфатного буфера с pH 8,0.
4.4.3.3. ОБСУЖДЕНИЕ
Хотя реакция при pH 9,6 протекает медленно, повышать pH не
имеет смысла, так как растворимость полилизина снижается.
При хотя бы частичном замещении полилизин не выладает в
осадок и при более высоких значениях pH. Степень замещения
определяют, оценивая содержание белка по Кьельдалю (необ-
ходима поправка, поскольку гаптен также содержит азот) и
определяя количество пенициллоильных групп в реакции с па-
рахлормеркурибензоатом (ПХМБ). Продукт реакции характе-
ризуется сильным поглощением на длине волны 285 нм. Моляр-
ный коэффициент экстинкции составляет 23 800. Реакцию про-
водят в 0,05 М карбонате с pH 9,2 при 4—50-кратном избытке
ПХМБ. Следует учитывать разведение, вызываемое добавле-
нием ПХМБ, а также экстинкцию, обусловленную непрореаги-
ровавшим ПХМБ.
435
4.4.4. КОНЪЮГАТ АНГИОТЕНЗИНА С АЛЬБУМИНОМ
4.4.4.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы 1-этил-3-(3-диметиламинопропил) -кар-
бодиимид, (1-аспарагин, 5-валин)-ангиотензин, кроличий сыво-
роточный альбумин, диализная леита.
4.4.4.2. МЕТОДИКА
Смешивают 10 мг ангиотензина (10 мкмоль), 5 мг альбумина
(0,1 мкмоль) и 150 мг 1-этил-3(3-диметиламинопропил)-карбо-
диимида (1 мкмоль) в 0,2 мл воды и оставляют на 1—12 ч
при комнатной температуре. Как только в результате реакции
образуется коллоидный раствор, его диализируют против воды
в течение 48 ч. В некоторых случаях коллоид не образуется,
тогда диализ начинают проводить через 12 ч.
4.4.5. АНТИТЕЛА, МЕЧЕННЫЕ ФЕРМЕНТОМ
4.4.5.1. ПРИНЦИП МЕТОДА
Конъюгаты ферментов с антителами применяются в иммуно-
ферментном анализе или в иммуногистохимии. В иммуногисто-
химии находят применение пероксидаза хрена, глюкозооксида-
за, кислая и щелочная фосфатазы, щитохром С; в ИФА — ще-
лочная фосфатаза, пероксидаза хрена, глюкозооксидаза и р-га-
лактозидаза. Используют следующие методы получения конъю-
гатов.
1. «Сшивание» глутаровым альдегидом.
а) одноступенчатый метод (щелочная фосфатаза);
б) двухступенчатый метод (пероксидаза хрена).
2. Конъюгирование после перйодатного окисления углеводов
в составе гликопротеидов (пероксидаза хрена).
3. «Сшивание» Ы.Ы'-о-фенилендималеимидом (р-галакто-
зидаза).
4. «Сшивание» т-малеиимидобензоил-Ы-гидроксисукциними-
дом (р-галактозидаза).
Интактным молекулам IgG следует предпочесть Fab'-фраг-
менты, поскольку их специфическая адсорбция меньше. Для
снижения уровня фона используют специфические Fab'-фраг-
менты. Fab'-фрагменты с незаблокированными SH-группами
присоединяют к р-галактозидазе через Г4,М'-о-фенилендимале-
имид, который избирательно реагирует с сульфгидрильными
группами [4]. После блокады SH-rpynn N-этилмалеимидом или
преобразования Fab'- в Р(аЬ')2-фрагменты конъюгирование с
ферментом можно проводить глутаральдегидным или перйодат-
ным методом. Количество N-этилмалеимида, необходимое для
блокады SH-групп, определяют по интенсивности поглощения
436
на волне 310 нм. После взаимодействия с SH-группами погло-
щение N-этилмалеимида на волне 310 нм исчезает [3]. ш-мале-
имидобензоил-М-гидроксисукцинимидный эфир реагирует как
с аминогруппами (гидроксисукцинимидный эфир), так и с SH-
группами (малеимидная часть) [4]. В первую очередь реаги-
руют аминогруппы антител, во вторую — SH-группы ферментов.
Поскольку у антител нет свободных SH-групп, полимеризация
не происходит.
4.4.5.2. ОДНОСТУПЕНЧАТЫЙ ГЛУТАРАЛЬДЕГИДНЫЙ
МЕТОД ПРИГОТОВЛЕНИЯ КОНЪЮГАТОВ
АНТИТЕЛ С ЩЕЛОЧНОЙ ФОСФАТАЗОЙ
4.4.5.2.1. Материалы и оборудование
Для работы необходимы щелочная фосфатаза
(15 мкмоль субстрата ,хттт ч ,
---------~к мг-----при 37 С) в (NH4)2SO4, IgG (антитела),
глутаровый альдегид, ФСБ; трис-НС1-буфер с pH 8,0, БСА,
азид натрия.
4.4.5.2.2. Методика
К 2 мг иммуноглобулина в 1,0 мл ФСБ добавляют 5 мл щелоч-
ной фосфатазы и диализуют смесь 18 ч при 4 °C против ФСБ..
Добавляют к смеси глутаровый альдегид до конечной концент-
рации 2 г/л и оставляют на 1—2 ч при комнатной температуре.
Ставят на диализ в течение ночи при 4 °C против ФСБ, затем
против 0,05 М трис-НС1-буфера с pH 8,0. После диализа смесь
доводят до объема 4 мл трис-НС1-буфером с pH 8,0, содержа-
щим БСА (10 г/л) и NaN3 (0,2 г/л) [6].
4.4.5.3. ДВУХСТУПЕНЧАТЫЙ МЕТОД
ГЛУТАРАЛЬДЕГИДНОГО «СШИВАНИЯ»
ПЕРОКСИДАЗЫ ХРЕНА С АНТИТЕЛАМИ
4.4.5.З.1. Материалы и оборудование
„ „ /экстинкция 403 нм) .
Пероксидаза хрена, Rz = 3,0 I--------, глютаровый
к к ’ V экстинкция 280 нм/ г
альдегид, сефадекс G-25, 0,1 М фосфатный буфер с pH 6,8,
0,15 М NaCl; антитела (IgG или Fab'), 0,1 М карбонат-бикар-
бонатный буфер с pH 9,5, лизин, сульфат аммония, центрифуга,
хроматографическая колонка размером 0,9X60 см.
4.4.5.3.2. Методика
Растворяют 10 мг пероксидазы в 0,2 мл 0,1 М фосфатного бу-
фера с pH 6,8, содержащего 12,5 г/л глутарового альдегида.
Через 18 ч инкубации при комнатной температуре смесь нано-
сят на колонку (см. выше) с сефадексом G-25, уравновешен-
ным 0,15 М NaCl. Фракции, содержащие активированную пер-
437
оксидазу (экстинкция при 403 нм), объединяют и при необхо-
димости концентрируют (мембрана Diaflo-PM-Ю). К этому
раствору добавляют 5 мг антител (IgG) или 2,5 мг Fab' в сме-
си, состоящей из 1 мл 0,15 М NaCl и 0,1 мл 0,1 М карбонат-
бикарбонатного буфера. Через 24 ч инкубации при 4 °C добав-
ляют 0,1 мл 0,2 М раствора лизина. Еще раз 2 ч ставят на диа-
лиз против ФСБ. Меченные ферментом антитела осаждают
сульфатом аммония (50% насыщения), отмывают тем же рас-
твором и диализируют против ФСБ. Стадию осаждения сульфа-
том аммония опускают при работе с меченными ферментом
РаЬ-фрагментами. Их центрифугируют 20 мин при 20 000 g и
хранят до 3 мес при 4 °C в неразведенном состоянии.
4.4.5.4. ПЕРЙОДАТНЫЙ МЕТОД КОНЪЮГИРОВАНИЯ
ПЕРОКСИДАЗЫ ХРЕНА С АНТИТЕЛАМИ
4.4.5.4.1. Принцип метода
В результате перйодатного окисления углеводов в углеводсо-
держащих ферментах образуются альдегидные группы, кото-
рые взаимодействуют с аминогруппами антител. Продукт реак-
ции стабилизируют восстановлением боргидридом натрия.
-4.4.5.4.2. Материалы и оборудование
Для работы необходимы пероксидаза, хрена (Rz=3), перйодат
.натрия, сефадекс G-10 или р-25, ацетатный буфер с pH 4,0, ан-
титела (IgG соотв. Fab'), карбонат-бикарбонатный буфер с
pH 9,5, боргидрид натрия, ФСБ; сефакрил S-200; колонки раз-
мером 2,5X30 см и 1X30 см.
•4.4.5.4.3. Методика
Растворяют 4 мг пероксидазы хрена в 1 мл дистиллированной
:воды. Прибавляют 5 мкл раствора метаперйодата натрия
(38,5 мг/мл). Выдерживают 20 мин при комнатной температу-
ре и фильтруют через небольшую колонку с сефадексом G-10
или G-25, уравновешенную 0,001 М ацетатным буфером с pH 4,0.
•Собирают окрашенные фракции, содержащие пероксидазу,
.и доводят pH до 9,5 0,2 М NaOH. Сразу после этого добавляют
10 мг IgG или 5 мг Fab' в 1 мл 0,01 М натрий-карбонатного бу-
фера с pH 9,5. Через 2 ч постоянного перемешивания при ком-
натной температуре добавляют 100 мкл раствора боргидрида
натрия (4 мг/мл в дистиллированной воде). Инкубируют 2 ч
при 4 °C и ставят на диализ против ФСБ в течение ночи. Про-
водят гель-фильтрацию на сефакриле S-200 (колонка 2,5X30 см)
в ФСБ. При использовании Fab' собирают пик с отн. мол. мас-
сой 90 000, при использовании IgG — пик с отн. мол. массой
.200000. Эти пики обладают выраженной абсорбцией как на
438
волне 280 нм, так и на 403 нм. К конъюгату пероксидазы до-
бавляют БСА до конечной концентрации 10 мг/мл. Аликвоты
конъюгата сохраняют в стеклянных сосудах при —20°C.
4.4.5.5. КОНЪЮГИРОВАНИЕ p-D-ГАЛАКТОЗИДАЗЫ
ПРИ ПОМОЩИ ФЕНИЛЕНДИМАЛЕИМИДА
4.4.5.5.1. Принцип метода
N.N'-o-фенилендималеимид реагирует с SH-группами в мягких
условиях. Вначале восстанавливают дисульфидные мостики в
молекулах антител. Затем проводят реакцию с малеимидом.
Удаляют избыток малеимнда. Проводят реакцию SH-содержа-
щего фермента с р-галактозидазой.
4.4.5.5.2. Материалы и оборудование
Для работы необходимы IgG, меркаптоэтиламин, фосфатные
буферы (pH 6,0, 7,0, 7,5), ацетатный буфер с pH 7,5 и 5,0, сефа-
декс G-25, сефароза 6 В; М,М'-о-фенилендималеимид, N-этилма-
леимид, БСА; NaCl, MgCl2, p-D-галактозидаза (5 мг/мл; 2,2 М
(NH4)2SO4; 5—10 мкмолей о-нитрофенил-р-Э-галактозида/секХ
Хмг при 37°С pH 7,3), азид натрия, колонки (1,5X40 и 1,0Х
Х40 см), азот.
4.4.5.5.3. Методика
Диализуют 4—8 мг IgG против 0,1 М фосфатного буфера с
pH 6,0. IgG, отдиализованный в объеме 0,9 мл, смешивают с
0,1 мл 0,1 М 2-меркаптоэтиламина и инкубируют 90 мин при
37°C. Проводят гель-фильтрацию на колонке с сефадексом
G-25 (1,0X40 см) в 0,1 М фосфатном буфере с pH 6,0. Колон-
ку предварительно дегазируют и хранят в атмосфере азота.
Элюат с восстановлением IgG немедленно при непрерыв-
ном помешивании переносят в насыщенный раствор Ы,№-о-фе-
нилендималеимида (около 0,75 мМ в 0,1 М ацетатном буфере с
pH 5,0). Реакцию проводят в атмосфере азота при 30°C. Соот-
ношение Н,№-о-фенилендималеимида с антителами составляет
30:1. Через 20 мин смесь разделяют на сефадексе G-25
(1,0X40 см) в 0,02 М ацетатном буфере с pH 5,0. Фракцию
малеимид-IgG концентрируют на мембранах до объема 0,15—
0,5 мл н 0,25 М фосфатным буфером с pH 7,5 доводят pH до 6,5.
Затем добавляют 20 мкл раствора БСА (25 г/л), обработанного
N-этилмалеимидом, 1 мкл 1 М MgCl2 и 0,1 мл p-D-галактознда-
зы (5 мг/мл). Конечный объем пробы составляет 0,25—0,6 мл.
Смесь выдерживают 24 ч при 4 °C и доводят ее объем до 1,0 мл
0,01 М фосфатным буфером с pH 7,0, содержащим 0,1 М NaClr
1 мМ MgCl2, БСА (1 г/л) и азид натрия (1 г/л). В этом буфере
проводят фильтрацию на сефарозе 6 В (колонка размером
1.5X40) [4].
439
•4.4.5.6. КОНЪЮГИРОВАНИЕ p-D-ГАЛАКТОЗИДАЗЫ
ПРИ ПОМОЩИ ш-МАЛЕИМИДОБЕНЗОИОЛ-
N-ГИДРОКСИСУКЦИНИМИДНОГО ЭФИРА
(МБГСЭ)
4.4.5.6.1. Принцип метода
Гидроксисукцинимидный эфир селективно реагирует с амино-
труппами, малеимидный остаток — с SH-группами (p-D-галак-
тозидазы).
4.4.5.6.2. Материалы и оборудование
Для работы необходимы IgG (антитела), p-D-галактозидаза,
МБГСЭ, диоксан, 0,1 М фосфатный буфер с pH 7,0, NaCl,
MgCl2, 2-меркаптоэтанол, 0,01 М трис-НС1-буфер с pH 7,0, БСА,
азид натрия, сефадексы G-25 и G-200.
4.4.5.6.3. Методика
Вносят 0,32 мг МБГСЭ в 15 мкл диоксана в 1,5 мл 0,1 М фос-
фатного буфера с pH 7,0 с 50 мМ NaCl, содержащего 1,5 мг
•специфического IgG. Смесь инкубируют при 30 °C в течение
1 ч, затем наносят на сефадексе G-25 (0,9X30 см) и проводят
разделение в 0,1 М фосфатном буфере, содержащем 50 мМ
NaCl и 10 мМ MgCl2. Элюат (3 мг), в котором содержится бе-
лок, немедленно смешивают с 1,5 мг p-D-галактозидазы в 0,1 М
фосфатном буфере с pH 7,0, содержащем 10 мМ MgCl2 и 50 мМ
NaCl. Инкубируют 1 ч при 30°C и останавливают реакцию
2-меркаптоэтанолом в конечной концентрации 10 мМ. Не-
конъюгированные АТ удаляют гель-фильтрацией на сефадексе
G-200 (колонка 0,9X90 см). Хроматографию проводят в буфе-
ре следующего состава: 10 мМ трис-HCl с pH 7,0, 10 мМ MgCl2,
10 мМ 2-меркаптоэтанола, 450 мМ NaCl. Конъюгированные ан-
титела выходят вслед за свободным объемом. Их хранят в при-
сутствии БСА (1 г/л) и азида натрия (0,2 г/л) [4].
4.4.5.7. ДОПОЛНИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Меченные пероксидазой антитела можно очищать от неконъюги-
рованного иммуноглобулина на ультрагеле АсА-44, на КонА-се-
фарозе (связывание пероксидазы или ее конъюгатов) и на про-
теин-А-сефарозе [1]. Элюцию с протеин-А-сефарозы проводят
0,1 М глицин-HCl с pH 2,8. Элюцию с КонА-сефарозы прово-
дят 0,1 М а-метилманнозидом в 0,1 М ацетатном буфере с
pH 6,0 с 1 М NaCl, 0,001 М MgCl2 и 0,001 М СаС12.
1. Соотношение пероксидаза : IgG определяют следующим об-
разом:
________Е<оз нм конъюгата__
Е<озви пероксидазы 1 мг/мл [мг/мл]
440
2. Концентрацию IgG в конъюгате:
Е 280 IgG г , ,
Е 280 IgG 1 мг/мл 1мг/мл]
Е 280 IgG = E 280 конъюгата — Е 280 пероксидазы, где Е — оп-
тическая плотность.
Молярное соотношение:
концентрация пероксидазыX4
концентрация IgG
Отн. мол. масса пероксидазы хрена 40000. Активность галак-
тозидазы проверяют по гидролизу 4-метилумбиллиферил-р-О-
галактопиранозида или о-нитрофенил-р-й-галактозида. Субст-
ратом для щелочной фосфатазы служит 4-нитрофенилфосфат.
Эффективность конъюгирования глутаральдегидом в одно-
и в двухфазном методе невысока. Конъюгат, полученный двух-
фазным методом, более гомогенен, соотношение в нем перокси-
дазы и иммуноглобулина не равно 1. Эффективность конъюги-
рования перйодатом выше, чем у глутарового альдегида.
4.4.6. КОНЪЮГАТ ПРОТЕИНА А С ПЕРОКСИДАЗОЙ
4.4.6.1. ПРИНЦИП МЕТОДА
М-сукцинимидил-3-(2-пиридилтио) -пропионат (СППТ) является
гетеробифункциональным реагентом. Сукцинимидная часть ре-
агирует с первичными аминогруппами как пероксидазы, так и
протеина А. Реакцию проводят раздельно, она сопровождается
переносом 2-пиридилдисульфидных остатков. Эти остатки в мо-
лекуле пероксидазы восстанавливают. Образующиеся SH-груп-
пы реагируют с 2-пиридилдисульфидными остатками протеи-
на А с образованием алифатического дисульфида и пиридин-2-
тиона.
4.4.6.2. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы пероксидаза хрена, СППТ, протеин А,
дитиотреитол, фосфатный буфер с pH 7,5, NaCl, этанол, сефа-
декс G-25, G-100, колонка (0,6X40 см).
4.4.6.3. МЕТОДИКА
а) Растворяют 1,5 мг пероксидазы хрена в 0,2 мл 0,1 М
фосфатного буфера с pH 7,5, содержащего 0,1 М NaCl. К это-
му раствору быстро добавляют 0,2 мл СППТ в этаноле
(700 мкг/мл) при непрерывном перемешивании, которое прово-
дят 30 мин при 25 °C, а затем выполняют гель-фильтрацию на
сефадексе G-25 в 0,1 М фосфатном буфере с pH 7,5, содержа-
29—990
441
щем 0,1 М NaCl. Фракции, содержащие пероксидазу, концентри-
руют до 0,3 мл.
б) Смешивают 1,42 мг протеина с 0,06 мл СППТ в условиях,
аналогичных указанным выше. После гель-фильтрации на сефа-
дексе G-25 фракции, содержащие протеин А, концентрируют до-
0,3 мл.
в) К пероксидазе, обработанной СППТ, добавляют дитио-
треитол до конечной концентрации 0,05 М. Избыток дитиотреи-
тола и образующийся пиридин-2-тион удаляют гель-фильтра-
цией на сефадексе G-25, смешивают пероксидазу, содержащую
свободные SH-группы, и протеин А, содержащий 2-пиридилди-
сульфидные группы. Объем пробы составляет 1,3 мл. Пробу
инкубируют 20 ч при 25 °C, концентрируют до 0,5 мл. Прово-
дят гель-фильтрацию на сефадексе G-100 (колонка размером
0,6X40 см) в 0,1 М. фосфатном буфере с pH 7,5 с 0,1 М. NaCl
4. 46.4. ДОПОЛНИТЕЛЬНЫЕ ЗАМЕЧАНИЯ
Конъюгат протеина А с пероксидазой представляет собой уни-
версальный реагент для ИФА в варианте выявления антител.
Протеин А и пероксидаза содержатся в конъюгате в соотноше-
нии 1 : 1, выход конечного продукта составляет 62%.
4.4.7. СПИСОК ЛИТЕРАТУРЫ
1. Boorsma D. М., Strefkerk J. G. Peroxidase-conjugate chromatography. Isola-
tion of conjugates prepared with glutaraldehyd or periodate using polyacril-
amide-agarose gel. — J. Histochem. Cytochem., 1976, 24, 481—486.
2. Farr A. G., Nankane P. K. Immunochemistry with enzyme labeled antibodies:
A brief Review. — J. Immun. Meth., 1981, 47, 129—144.
3. Ishikawa E., Yoshitake S. A general and Mild procedure for the purification
of rabbit Fab' antibodies. — J. Immunol. Meth., 1980, 38, 117—123.
4. O’Sullivan H. J., Marks V. Methods for the preparation of enzyme-antibody
conjugates for the use in enzyme immunoassay. — In: Methods in Enzymo-
logy 73, part B, 147—166.
5. Pain D., Surolia A. Preparation of protein A-peroxidase monoconjugate usin
a heterobifunctional reagent, and its use in enzyme immunoassays. — J. Immu-
nol. Meth., 1981, 40, 219—230.
6. Voller A., Bidwell D. E„ Bartlett A. Enzyme Immunoassay in diagnostic Medi-
cine.—Bull. WHO, 1976, 53, 55—65.
4.5. ПОЛУЧЕНИЕ ТКАНЕВЫХ
ЭКСТРАКТОВ p МКСЬМЕТОД)
4.5.1. ПРИНЦИП МЕТОДА
Для изучения различных явлений, относящихся к реакциям им-
мунитета, необходимо выделять антигены из экстрактов тканей.
Большая часть этих АГ (антигены основного комплекса гисто-
совместимости, тканевые АГ, АГ групп крови, рецепторы мито-
442
генов, опухолевых и трансплантационных АГ) находятся на
внешней мембране клеток. Иногда поверхностные АГ обнару-
живаются также и внутри клеток (некоторые вирусные, опухо-
левые и фетальные АГ). Согласно новейшим представлениям
о строении клеточных мембран как о жидкой мозаичной струк-
туре, большая часть иммунологически значимых АГ относится
к «интегральным» белкам цитоплазматической мембраны. Они
относительно прочно связаны с мембраной и не могут быть от-
делены без ее разрушения. При получении растворимых мемб-
ранных АГ выделяют эти белки в молекулярно-дисперсной фор-
ме с сохранением их биологической активности. Любой метод
выделения означает потерю или изменение биологических
свойств по сравнению с нативным состоянием, причем следует
также учитывать возможность артефактов. Существует боль-
шое число способов разрушения плазматических мембран: об-
работка ультразвуком, пониженным или повышенным осмоти-
ческим давлением, кислотами, ферментами, детергентами, меха-
ническое воздействие. Как правило, используют комбинацию
нескольких методов. Из довольно большого числа комбинаций
чаще всего применяют следующие:
1) получение фракции мембран путем выделения отдельных
компонентов клетки, перевод мембранной фракции в раствори-
мое состояние (двухэтапный метод);
2) получение фрагментов мембран путем гомогенизации тка-
ни с одновременным экстрагированием необходимого компонен-
та (одноэтапный метод).
В мембранных исследованиях почти исключительно исполь-
зуется двухэтапный метод, который позволяет получать высо-
коочищенные фрагменты мембран в малоизмененном виде.
Предпосылкой для успешной работы служит наличие суспен-
зии, состоящей из отдельных клеток (асцитическая жидкость,
кровь), продолжительно культивировавшихся тканей или отно-
сительно мягких по консистенции тканей, таких как селезенка
или печень. Часто такие суспензии получить невозможно (мы-
шечная ткань, кишечник, сосуды, солидные опухоли). Из одно-
этапных методов особенно широко используется экстракция
3 М КС1, которая позволяет извлекать АГ даже из плотных
тканей. Ниже будет описан модифицированный метод [2],
которым в условиях нашей лаборатории были получены анти-
гены комплекса гистосовместимости мыши и человека, АГ, ас-
социированные с опухолями, и антигены нормальных тканей.
4.5.2. МЕТОДИКА
Исходным материалом для получения АГ, ассоциированных с
опухолями (ААО), служила опухолевая ткань с гистологиче-
ским подтверждением; участки некроза и нормальную ткань
предварительно удаляли. Антигены нормальной ткани (АНТ)
получали из секционного материала, взятого через 8 ч после
29*
443
Нормальная ткань
Эмбриональная ткань
'Опухолевая ткань
Грубое измельчение,гомогенизация1
+ аЗМ КС1вФСБ,
+ Соотношение тнань/Среда 1:5-1:КУ,
выдерживают 18ч при4°С
200000 g
1ч
Интерфаза (прозрачная,невязная)
+ Диализ против водопроводной воды(Зч)'.
Диализ против ФСБ (24 ч,
20-100-яратныйсбъем.2-нратная
смена буфера),концентрация до
1/10 объема против ПЭГ
15000 g
У2ч
+ Ладосадочная фракция с равным-
объемом насыщенного сульфата
аммония в ФСБ.Выдерживают
12 ч при 4'С
Преципитат.
•►Растворяют в3-4объемах ФСБ
+ Диализ против в 3-4 объемах ФСБ
+ Диализ против ФСБ (24 ч),если фракция—
не просветлеет- 200000двтечениеТч-
Надосадочная франции
Хроматография на G~2OO
илиАсА 34
Франции
Р1:отн.мол, масса 180000,
Р2 :отн,мол.масса 12OOOO-16OOOO).
РЗ :отн.мол.масса 50000-60000
+ Концентрация белнов до 1-1,5%
по объему (фильтры Am icon)
+ Хранение при-25° С
Антигены нормальной ткани (АНТ)
Антигены эмбриональной ткани (АЭТ)
Антигены, ассоциированные с опухолями (ААО)'
Рис. 138. Получение антигенов нормальной ткани (АНТ), ан-
тигенов эмбриональной ткани (АЭТ) и антигенов, ассоцииро-
ванных с опухолями (ААО), по методу экстракции с ЗМ
КС1.
смерти. Антигены эмбриональной ткани (АЭТ) получали из
абортного материала или из эмбрионов животных, например
мышей. Все образцы тканей сохраняли без потери активности
при —25 °C в течение многих месяцев. Принципиальная схема
получения ААО и АНТ представлена на рис. 138. Грубое из-
мельчение ткани, желательно в полузамороженном состоянии,
проводят при помощи ножниц (оптимальный размер кусочков
ткани составляет менее 5 мм в диаметре). Затем кусочки тка-
ни гомогенизируют, например, в гомогенизаторе с ножами
(Biihler, Tubingen, 13000 об/мин). Разрушение ведут неболь-
шими порциями, следя, чтобы температура гомогената не под-
нималась выше 10 °C, длительность каждого разрушения не
должна превышать 1—2 мин. После центрифугирования прово-
дят диализ надосадочной фракции и концентрируют ее против
ПЭГ 20000 до у5—Ую исходного объема. Выпадающие в осадок
белки удаляют центрифугированием. Проводят осаждение, до-
бавляя равный объем насыщенного раствора сульфата аммо-
ния в 0,15 М фосфатном буфере с pH 7,2. После перемешивания
растворов смесь оставляют при 4 °C на 12 часов для достиже-
ния полноты осаждения. Осадок собирают центрифугированием,
растворяют в ФСБ с pH 7,2 и диализуют против ФСБ. Как пра-
вило, раствор не является достаточно прозрачным и перед раз-
делением на колонках его еще раз центрифугируют. Гель-
фильтрацию проводят на сефадексе G-200 или на ультрагеле
АсА 34. Размер колонки зависит от количества разделяемого
материала (например, 32X1200 мм). Элюцию проводят ФСБ
или 0,01 М трис-НС1-буфером с pH 7,3, содержащим 0,85%
NaCl. Регистрируют оптическую плотность элюируемых фрак-
ций на волне 280 нм (Увикорд II, Швеция), разделение прово-
дят при 4°C. На рис. 139 и 140 изображены профили элюции
различных ААО, АНТ и АЭТ мыши и человека, полученных в
одинаковых условиях фракционирования. Изменения оптиче-
ской плотности элюируемого материала в УФ области дают
мало ценной информации, поскольку они в основном обуслов-
лены сопутствующими белками. Объединенные фракции Pl, Р2
и РЗ концентрировали методом диафильтрации (установка фир-
мы Amicon, Нидерланды) до концентрации белка 1—1,5% и
сохраняли их при —25 °C в течение нескольких месяцев без по-
тери активности. Выход антигенов зависит от типа ткани.
В среднем, например, фракция Р2 содержала 0,2—0,8% белка
от веса влажной ткани. При работе более чем с 50 г ткани,
выход снижался. Данные об антигенном составе различных
фракций, приводимые в литературе, существенно различаются
между собой, что, вероятно, связано с использованием различ-
ных методов оценки.
Наши собственные исследования сенсибилизированных лимфо-
цитов в реакции электрофоретической подвижности (РЭФП) и
в реакции торможения адгезии (РТАЛ) представлены на
рис. 139 и 140, а также в табл. 43.
445
Рис. 139. Система мышиных клеток.
Разделение АНТ, АЭТ и ААО на ультрагеле Аса 34; изучение антигенности отдельных
фракций иа лимфоцитах соответствующих и несоответствующих доноров. 1 — NTA СВА
(Н-2К), лимфоциты С57В1/6 (Н-2^), аллогенная сенсибилизация против СВА, тест элект-
рофоретической мобильности (ЭМ); 2 — FA СВА, лимфоциты мышей BIOD2 XCBAd*
и СВА с опухолью ВР, тест ЭМ; 3 — ААО ВР, лимфоциты мышей с опухолью ВР/СВА,
МС/СВА, тест ЭМ; 4 — ААО ВР (см. 3), реакция торможения адгезии лимфоцитов.
В области Р2 методом РЭПФ в случае АНТ и АЭТ выявля-
ются антигены, относящиеся к области гистосовместимости
(тканевой специфичности); в случае ААО — индивидуальные
антигены (мышь) или относящиеся к области гистоспецифично-
сти и органоспецифичности. Белки из области РЗ, относящиеся
к ААО и АЭТ, в РЭПФ проявляют опухолеспецифичные свойст-
ва. В области Р1 в РЭПФ до настоящего времени были иссле-
дованы экстракты некоторых опухолей желудочно-кишечного
тракта человека, все они обладают общей опухолевой специ-
фичностью. Исследование соответствующих фракций методом
РТАЛ дает следующую картину: Н-активность в случае АНТ
446
Исследуемое вещество
160000
68000
Рис. 140. Система клеток человека.
Разделение АНТ и ААО иа сефадексе G-200. Исследование антигенности отдельных фрак-
ций на лимфоцитах соответствующих и несоответствующих доноров. 1—АНТ из почек,
лимфоциты больного с гломерулонефритом (аутосеисибилизация к АГ почек); 2 — ААО
карциномы бронхов, лимфоциты больного с карциномой бронхов и больного с остеосар-
комой, тест ЭМ; 3 — ААО карциномы кишечника, лимфоциты больного с карциномой
кишечника и карциномой желудка, тест ЭМ.
или индивидуальные ААО-активности обнаруживаются в обла-
сти Р2 или Р1, в то время как перекрестные реакции, по всей
видимости, полностью отсутствуют (не определяется активность
в области РЗ). Равнозначность распределения активности в мы-
шиных и человеческих системах, позволяет предположить су-
ществование общих закономерностей. При помощи вышеописан-
ного метода экстракции в случае АНТ и АЭТ в области Р2 бы-
ла выявлена активность, относящаяся к МНС-комплексу. То же
самое сохраняет справедливость для ААО, если исходить из
предположения о том, что опухолеспецифические антигены де-
терминанты имеют отношение к МНС-комплексу или являются
модифицированными антигенами гистосовместимости [12]. Вы-
раженная перекрестная реактивность АЭТ и ААО в областях
Р1 и РЗ (преимущественно в РЭПФ) остается до настоящего
времени без объяснения. Феномен может быть связан с нали-
чием общих опухолевых антигенов (онкофетальные, вирусные)
или с артефактами при получении препаратов. Наши данные
447
Таблица 43. Антигенность и специфичность полученных при помощи
3 М KCI экстрактов ААО, АНТ и АЭТ в РЭФП и РТАЛ [2, 5, 7]
Вид Антиген Тест- система Специфичность Фракции1
Р1 | Р2 РЗ
Мышь РЭПФ Н-2 или не Н-2 специфический 4- —
АНТ2 Перекрестно реагирующий (ал- логен) — — —
РТАЛ Н-2 или не Н-2 специфический + 4- —
Перекрестно реагирующий (ал- логен) — — —
Мышь АЭТ РЭПФ Общий 4- — 4-
Опухолеспецифический Специфический для беременно- +
сти
Мышь ААО РЭФП Индивидуальный3 — + —.
Общий Опухолеспецифический — 4-
РТАЛ Индивидуальный 4- 4- —
Общий —
Человек РЭФП Тканеспецифический4 4- —
АНТ Специальный или общий опу- холеспецифический — —.
Человек РЭФП Общий 4- — 4-
АЭТ Опухолеспецифический Специфический для беременно- 4-
СТИ
Человек РЭФП Гисто- или органоспецифиче- — 4- —
ААО ский Общий Опухолеспецифическнй (4-) — 4-
1 отн. мол. масса: Р1>180 000, Р2 120000—160000/РЗ 50 000—60 000;
2 получены из клеток селезенки иа аллогенной модели сенсибилизации;
5 индивидуальный, обладающий активностью, сравнимой с активностью ассоцииро-
ванных трансплантационных антигенов;
4 при аутоиммунных заболеваниях.
совпадают с результатами, согласно которым опухолеспецифи-
ческие трансплантационные антигены растворимой мембранной
фракции после гель-хроматографии обнаруживаются в обла-
сти с отн. мол. массами 70000—150000 [8, 24]. Методом РТАЛ
опухолеспефицические антигены также определяются в области
отн. мол, масс 70000—150000 [9, 10].
4.5.3. ОБСУЖДЕНИЕ
О механизмах извлечения антигенных экстрактов из тканей
при помощи 3 М KCI мало что известно. Вероятно, этот про-
цесс протекает с участием аутоферментативных реакций. Ме-
тод получения растворимых мембранных компонентов при по-
мощи гипертонических солевых растворов имеет определенные
преимущества перед методами, основанными на использовании
ферментов (р-глюкозидаза) или детергентов (Тритон Х-100).
448
Ферменты способны вызывать разрушение антигенных струк-
тур; при использовании детергентов возникают сложности с
их последующим удалением из готового препарата. К числу
недостатков метода с применением 3 М KCI относятся:
— изменение растворимости отдельных активных компонен-
тов при уменьшении концентрации соли в ходе получения пре-
парата;
— относительно небольшой выход АГ;
— обусловленная одноэтапным методом выделения контами-
нация активных компонентов мембраны компонентами других
составных частей клетки.
Обработка 3 М KCI вызывает селективное высвобождение
некоторых важных гликопротеидов из состава мембраны [11].
До недавнего времени большая часть экстрактов, полученных
с помощью 3 М KCI, использовалась в нефракционированном
виде (или добавлялась стадия осаждения 50% сульфатом ам-
мония) в реакциях клеточного иммунитета: кожной пробе, ре-
акции торможения миграции лейкоцитов, реакции ингибирова-
ния цитотоксичности, РЭПФ [13, 14, 15, 16], а также все
чаще в РТАЛ [17, 18, 19, 20]. После того как стала очевидной
гетерогенность экстрактов, были предприняты многочисленные
попытки их фракционирования биохимическими или иммунохи-
мическим методами, а также при помощи электрофореза. Обна-
ружилось, что одна и та же фракция может проявлять различ-
ные активности в зависимости от метода определения. Общим
является то, что в области отн. мол. масс 80 000—160 000 (Р2) в
основном обнаруживаются гистоспецифические и отдельные ин-
дивидуальные антигены, а в области отн. мол. масс 50 000—
70000 (РЗ) и в области с отн. мол. массами более 200000 (Р1)
выявляются в основном перекрестно реагирующие антигенные
компоненты. Следует подчеркнуть, что объединение антигенов
в отдельные фракции с определенными свойствами (Р1—РЗ)
имеет смысл только в отношении некоторых из них, охаракте-
ризованных в РЭФП, АНТ и ААО. При исследовании таких
антигенов в других иммунологических тест-системах вышепри-
веденное разделение антигенов на группы в соответствии с их
активностью будет лучшей гарантией правильного выбора ан-
тигена для соответствующего теста. К этому всегда рекоменду-
ется прибегать, когда нет точной биохимической характеристи-
ки антигена. Гетерогенность экстрактов, получаемых с приме-
нением 3 М KCI, указывает на причину противоречивых резуль-
татов, наблюдаемых при работе с грубыми экстрактами тканей,
л на необходимость частичной очистки компонентов с извест-
ными иммунологическими свойствами. Новые данные об очист-
ке ААО и АНТ при помощи лектинов, электрофокусирования
[21], иммунных сорбентов [22] или моноклональных антител
[23] позволяют надеяться, что в скором времени будут получе-
ны новые субстраты для исследования реакций клеточного им-
мунитета.
449
4.5.4. СПИСОК ЛИТЕРАТУРЫ
1. Hellstrom К. Е., Brown. J. Р. Tumor antigens. — In: The Antigens Vol. 5/Ed,
M. Sela. — New York: Academic Press, 1979,
2. Muller M., Irmscher J., Fischer R. Immunologisches Tumorprofil. Ein neuar-
tiges Prinzip in der Anwendung des MEM-tests zur differezierten Karzinom-
diagnose. — Dtsch. Ges-wesen, 1975, 30, 1836—1842.
3. Jenssen H. L„ Werner H. Ceti electrophoretic mobility test (EMT) for detec-
tion of tumor associated antigens (TAA). — In: Protides of Biological
Fluids/Ed. H. Peters. — Oxford: Pergamon Press, 1979, 233—238.
4. Lampson L. A., Levy R. A role for clonal antigens in cancer diagnosis and
therapy. — J. Natl. Cancer Inst., 1979, 62, 217—219.
5. Seyfarth M„ Schmitt E., Werner H. et al. Beitrag zur zellvermittelten Immu-
nitat bei Nierenerkrankungen. — Dtsch. Geswesen, 1981, 36, 293—296.
6. Meyer-Rienecker H. J., Jenssen H. L., Werner H. Aspect of cellular immunity
in multiple sclerosis. Antigen reactivity of lymphocytes and lymphokine acti-
vity.—J. Neurol. Sci., 1979, 42, 173—186.
7. Meyer-Rieneecker H. J., Jenssen H. L., Werner H. Aspekte der immunologi-
schen Diagnostik von Hirntumoren. — Arch. Psychiat. Nervenkr., 1979, 227,
121—133.
8. Rogers M. J., Law L. W., Pierotti M. A. et al. Separation of the tumor asso-
ciated transplantational antigens (TSTA) from the alien H-2k antigens expres-
sed on a methylcholantrene induced tumor. — Int. J. Cancer, 1980, 25, 105—
109.
9. Thompson D. M. P., Tataryn D. N„ O’Connor R. et al. Evidence for the expres-
sion of human tumor specific antigens associated with p2-microglobulin in
human cancer and in some colon adenomas and benign breast lesions. — Can-
cer Res., 1979, 39, 604—611.
10. Howell J. H., Russo A. J., Goldrosen M. H. Characterization of murine tumor-
associated antigens by the microleucocyte adherence inhibition assay. — Can-
cer. Res., 1979, 39, 612—618.
11. Ristau E., Schon R., Schoon R., Schlott B. et al. Zum Nachweis und zur
Charakteriesierung tumorassozierter antigener Komponenten aus Zellmembra-
nen eines UV-Licht induzierten Sarkoms der Maus mit Hilfe der MEM Tech-
nik. — Acta. Biol. Med. Germ., 1980, 39, 315—325.
12. Parmiani G. Histocompatibility antigens and tumor antigens. — Cancer Immu-
nol. Immunother., 1980, 8, 215—217.
13. Pasternak G., v. Broen B., Schlott B. et al. Lymphocyte reactivity in animal
and human tumors systems to fetal antigens as detected by MEM, LAI and
LMItechniques. — In: Cell Electrophoresis: Clinical Application and Methodo-
logy, INSERM sympos. No 11 (A. W. Preece and D. Sabolovic/Eds.) Else-
vier/North Holland, Biomedical Press, 1979, 225—234.
14. Jenssen H. L., Werner H., Mix E. et al. Effects of tumor antigen preparations
on lymphocytes of different origin. — Ibid., 211—216.
15. Hartus H. P., Ax W. Electrophoretic mobility test (EMT): studies on lympho-
cytee response and mechanism of the test using a rat tumor model. — Immu-
nobiol., 1981, 158, 151—172.
16. Mross К. B. Zellelektrophorese in der Tumordiagnostik: EM-Test, Grundlagen-
Material-Methoden-Ergebnisse-Thieme Verlag, Stuttgart, 1980.
17. Holt P. G., Fimmel P. J., Finley-Jones L. M. Evaluation of the microplate
leukocyte adherence inhibition test and its reproducibility, sensitivity and
relationship to other tests of cellular immunity. — Cancer Res., 1979, 39,
564—569.
18. Mortensen R. F. Detection of cellular immunity to murine sarcoma virus indu-
ced tumors by leukocyte adherence inhibition. — J. Natl. Cancer Inst., 1979,
62, 157—163.
19. Goldrosen M. H. Summary and future prospects of leukocyte adherence inhi-
bition.— Cancer Res., 1979, 39, 660—662.
20. Varga A., Stosiek P„ Kasper M. Modifikation des Leukozyten-Adharenz-
Inhibitions-Tests (LAI) mittels 5lCr-Markierung der Leukozyten. — Allergie
u. Immunol., 1980, 26, 171—178.
450
21. Pells N. R., Yamagishi H., Shulan. D. J. et al. .Use of preparative electric
focusing, in a sephadex gel slab to separate immunizing and growth facilita-
ting moieties in crude 3 M KCI extract oi murine fibrosarcoma. — Cancer
Immunol. Immunother., 1981, 11, 53—58.
22. De Leo A., Jay G., Appelta A. et al. Cell surface antigens of chemically indu-
ced sarcomas of inbred mice. — Transpl. Proc., 1980, 12, 65—69.
23. Steplewsky Z. Monoclonal antibodies to human tumor antigens. — Transpl.
Proc., 1980, 12, 384—387.
24. Law L. W. Studies of the purification of tumor antigens: progress and prob-
lems.— Arch. Geschwulstforsch., 1981, 51, 294—301.
4.6. ВЫДЕЛЕНИЕ ТРАНСФЕР-ФАКТОРА
4.6.1. ПРИНЦИП МЕТОДА
В 1942 г. Ландштейнеру и Чейзу впервые удалось осуществить
пассивный перенос гиперчувствительности замедленного типа
(ГЗТ) при помощи живых клеток у морской свинки. С тех пор
многими авторами было подтверждено, что жизнеспособные
лимфоциты из лимфатических узлов, селезенки, грудного лим-
фатического протока, перитонеального экссудата, а также лей-
коциты периферической крови сенсибилизированных доноров
обладают способностью переносить ГЗТ несенсибилизирован-
ным реципиентам. Эти данные справедливы как в отношении
экспериментальных животных, так и человека. Продолжитель-
ность пассивно индуцированной ГЗТ в сенсибилизированном
организме зависит от длительности сохранения в нем жизнеспо-
собных лейкоцитов донора (при аллогенном переносе она со-
ставляет несколько дней). Дальнейшими исследованиями было
показано, что пассивный перенос клеточных реакций гиперчув-
ствительности возможен при помощи диализата экстракта лей-
коцитов (ДЭЛ). Методом гель-фильтрации на сефадексе ДЭЛ
был разделен на две биологически активные фракции [1, 2, 3],
которые получили название «трансфер-фактор». Если несенси-
билизированному реципиенту ввести ДЭЛ или трансфер-фак-
тор сенсибилизированного донора, то у реципиента через 24 ч
в отдаленном от места введения участке кожи может развиться
та же самая реакция повышенной чувствительности, что и у до-
нора (рис. 141). Кожные реакции повышенной чувствительности
у реципиента сохраняются в течение многих месяцев, в некото-
рых случаях до двух лет. Это свидетельствует о том, что меха-
низм переноса гиперчувствительности не является чисто пас-
сивным; многие детали этого механизма остаются непонят-
ными.
Во все возрастающих масштабах ДЭЛ и трансфер-фактор
применяются в лечебных целях при хронических инфекцион-
ных заболеваниях и у пациентов с иммунодефицитными со-
стояниями [4]. Эксперименты на животных с применением ДЭЛ
или трансфер-фактора до настоящего времени редко оказыва-
лись удачными [5, 6]. Результаты экспериментов воспроизводи-
мы, если препараты трансфер-фактора вводятся с небольшим
451
Рис. 141. Пассивный перенос повышенной чувствительности к туберкулину
при помощи трансфер-фактора.
1 — туберкулинположительный донор; 2 — сенсибилизированные лимфоциты; 3 — замо-
раживание и оттаивание; 4 — диализ; 5 — лиофилизация; 6 — туберкулинотрицательный
реципиент после введения трансфер-фактора дает положительную кожн>ю реакцию на
туберкулин.
количеством специфического АГ [7, 8, 9, 10]. Этот вопрос под-
робно разбирается в соответствующем обзоре [11]. Трансфер-
фактор, так же как и другие медиаторы клеточного иммуните-
та, высвобождается при инкубации сенсибилизированных лим-
фоцитов с антигеном, его можно отнести к лимфокинам. Отно-
сительно природы и иммунологических свойств трансфер-фак-
тора имеются подробные данные [12]. Выделенный по методу
Лоренса трансфер-фактор [12] имеет активность, эквивалент-
ную активности того количества лейкоцитов, из которых он был
получен.
Таблица 44. Свойства трансфер-фактора
Биохимические Иммунологические
Растворим в воде Проходит через диализную мембрану Отн. мол. масса менее 10 000 Не является белком В состав входят полипептиды н оли- гонуклеотиды Термолабилен (инактивируется при 56 °C за 30 мин) Резистентен к ДНКазе, РНКазе, трипсину Не является антителом Не обладает иммуногенностью Не является трансплантационным АГ Переносит способность к реакциям клеточного иммунитета (торможение миграции клеток, трансформация лимфоцитов, кожные реакции)
452
4.6.2. МЕТОДИКА
4.6.2.1. МАТЕРИАЛЫ И ОБОРУДОВАНИЕ
Для работы необходимы: гепарин без консервантов, декст-
ран 250, инфуколл, апирогенная дистиллированная вода,
0,15М NaCl, сухой лед, этиловый спирт (денатурат), ДНКаза
(панкреатическая), сульфат магния, диализные целлофановые
мешки (например, Visking, США), шприцы, иглы, стеклянные
стаканы на 25 мл, термостат, водяная баня, конические центри-
фужные стаканы на 20 мл, центрифуга, лабораторная посуда
со шлифами, ампулы для лиофилизации, устройство для лиофи-
лизации, миллипоровские фильтры (0,22 мкм) с держателем,
сефадекс G-25, ФСБ с pH 7,4, азид натрия, хроматографические
колонки, перистальтический насос, увикорд (фильтр 254 нм),
автоматический коллектор фракций, рекордер.
4.6.22. ПОЛУЧЕНИЕ ДИАЛИЗАТА
ЭКСТРАКТА ЛЕЙКОЦИТОВ
Доноров с такими заболеваниями, как сифилис, малярия, гепа-
тит и др., необходимо исключить. У доноров должна наблю-
даться выраженная кожная реакция замедленного типа на со-
ответствующий антиген. Препараты из лейкоцитов доноров с
отрицательной кожной реакцией не переносят сенсибилизацию.
Кроме того, для успешного переноса необходимо иметь доста-
точное количество лейкоцитов, которое зависит от используе-
мого метода сенсибилизации. Для «локального» переноса дос-
таточно 0,01 мл взвеси (8,5Х106 лейкоцитов), для «системного»
переноса — 0,1 мл (85Х106 клеток). Кожную реакцию оцени-
вают через 24, 48 и 72 ч. Важнейшим методом является «сис-
темный» перенос, при котором трансфер-фактор вводят внутри-
кожно, подкожно или внутримышечно в область плеча. Кож-
ную пробу с антигеном проводят на ладонной поверхности
предплечья с противоположной стороны. Кровь отбирают в сте-
рильных условиях и разделяют на порции по 20 мл (на каж-
дые 100 мл крови добавляют 500 ME гепарина).
Нет необходимости силиконизировать шприцы и посуду.
Кровь разливают в стерильные пробирки (25X155 мм), добав-
ляют 2 мл раствора декстрана 250 (60 г/л) в 0,15 М NaCl или
раствор инфуколла. Пробирки закрывают, несколько раз встря-
хивают и инкубируют при 37 °C. Эритроциты осаждаются в те-
чение 30—45 мии. Продолжительность седиментации не должна
превышать 1 ч. Желтоватый или слегка красноватый слой
надосадочной жидкости переносят при помощи пастеровской
пипетки в конические центрифужные пробирки. Закрытые про-
бирки центрифугируют в течение 10 мин при 1500 g. Плазму
«осторожно сливают. Осевшие лейкоциты образуют слой над
осадком эритроцитов. 0,1 мл осевших лейкоцитов соответствуют
453
Рис. 142. Этапы получения трансфер-фактора, модифицированный метод.
1 — кровь и декстран; 2 — седиментация эритроцитов; 3 — центрифугирование надоса-
дочиой фракции; 4 — иаслаиваиие иа осадок 0,15 М NaCl; 5— 10-кратиое замораживание
и оттаивание с прибавлением ДНКазы; 6 — диализ; 7 — лиофилизация; 8 — стерилизую-
щая фильтрация; 9 — разлив по ампулам; 10 — инъекция.
85Х106 клеток, этого количества достаточно для получения
одной дозы препарата. Осадок состоит из 70% из лимфоцитов.
Самая нижняя часть конуса пробирки заполнена эритроцита-
ми. На слой лейкоцитов наслаивают 0,1 мл стерильного апиро-
генного 0,15 М раствора NaCl. Кончик пастеровской пипетки
осторожно проводят сквозь слой раствора соли и осторожно
отсасывают лейкоциты в виде суспензии в изотоническом рас-
творе NaCl. Можно объединять полученные суспензии лейкоци-
тов. Для получения ДЭЛ необходимо'полное разрушение лей-
коцитов. Оно достигается 10-кратным замораживанием и оттаи-
ванием суспензии. Пробирки с суспензией клеток заморажива-
ют смесью сухого льда с этанолом, а размораживают на водя-
ной бане при 37 °C. Процесс повторяют до тех пор, пока жид-
кость не станет вязкой. При полном разрушении лейкоцитов
высвобождается ДНК, делающая раствор чрезвычайно вязким
и затрудняющая его дальнейшую обработку. Для деполимери-
зации ДНК в раствор вносят несколько кристаллов ДНКазы и
небольшое количество ионов Mg2+. Гидролиз протекает в тече-
ние 30 мин при 37 °C.
Если ДНКазу, которая отличается чрезвычайной стабильно-
стью, добавлять прямо к суспензии лейкоцитов, то деполимери-
зация ДНК происходит в процессе замораживания и оттаива-
ния и не требует дополнительной инкубации. Для удаления вы-
сокомолекулярных компонентов проводят диализ. Обработанный
ДНКазой экстракт лейкоцитов осторожно переносят в завя-
занный с одного конца диализный мешок, стараясь не испач-
кать экстрактом наружный слой мешка, после чего его завя-
зывают, ополаскивают стерильным апирогенным изотоническим
раствором NaCl и переносят мешок в стерильный сосуд с дис-
тиллированной водой. На 0,1 мл экстракта используют 5 мл
дистиллированной воды. Диализ проходит в течение 24 ч при.
454
4 °C. По окончании диализа диализат переносят в стерильный
сосуд, замораживают смесью сухого льда со спиртом и лиофи-
лизируют. В таком виде продукт сохраняет активность в тече-
ние года при 4 °C. Перед употреблением лиофилизированный
препарат растворяют в стерильной апирогенной воде по 20 мл
на ампулу (соответствует 0,1 мл экстракта). Раствор фильтру-
ют через миллипоровский фильтр (диаметр пор 0,22 мкм) в
стерильных условиях. Этап стерилизирующей фильтрации яв-
ляется безусловно необходимым для очистки препарата от воз-
можного бактериального заражения в ходе диализа. Отдельные
«стадии процесса представлены на рис. 142.
4.6.2.3. ВЫДЕЛЕНИЕ ТРАНСФЕР-ФАКТОРА (ТФ)
ДЭЛ, получение которого описано выше, растворяют в ФСБ
с pH 7,4 в соотношении 1:20, при необходимости центрифуги-
руют 20 мин при 10 000 и проводят стерилизацию фильтра-
цией. Выделение ТФ осуществляют на колонках размером
20X530 или 50X1000 мм. Колонки с сефадексом G-25, закон-
сервированные 0,2% азидом натрия, перед разделением про-
мывают тремя объемами разведенного в соотношении 1 :20
РО4-ЫаС1-буфера (8,5 г/л NaCl, 0,01 М фосфатный буфер,
pH 7,4). Раствор ДЭЛ в стерильных условиях наносят на ко-
лонку. Проводят восходящую хроматографию в вышеприведен-
ном стерильном буфере. Скорость фильтрации может достигать
90 мл/час. Фракции собирают, лиофилизируют и при необхо-
димости .разводят апирогенной дистиллированной водой до изо-
тонического состояния, затем проводят стерилизующую филь-
трацию. На рис. 143 представлен характерный профиль гель-
фильтрации ДЭЛ. Биологическую активность фракций оцени-
вают по так называемому «Recovery assay»1 [3]. Активность
ТФ представлена в виде столбиков черного цвета. Многие ав-
торы подтверждают наличие биологической активности обеих
фракций. Для терапевтических целей объединяют обе активные
фракции, отбрасывая неактивные, и обозначают смесь актив-
ных фракций как ТФ. Вопрос об идентичности ТФ в обоих
фракциях остается открытым.
4.6.3. ОЦЕНКА МЕТОДА
Широкое распространение терапии ТФ, а . также использование
повышенных доз ТФ вызвало увеличение спроса на этот пре-
парат. Часто препарат получают из больших количеств крови
(до 400 мл на дозу). Такие препараты ТФ содержат большие
1 Лимфоциты человека обладают способностью образовывать розетки с
ЭБ. С трипсинизированными ЭБ розетки не образуются. Обработка лимфоци-
тов трансфер-фактором «восстанавливает» их способность к розектообразова-
•нию с трипсинизированными ЭБ. Это явление называется Recovery assay. —
<1 р и м е ч. ле р.
455
0.8
Фракции
Рис. 143. Профиль элюции ДЭЛ при разделении на сефадексе G-25 (fine).
Условия эксперимента: 150 мг ДЭЛ; колонка 50X600 мм; прочие детали описаны в
тексте.
количества биологически неактивного материала и ингибито-
ры ТФ [3]. Поэтому рекомендуется после лиофилизации удалять
неактивный материал путем гель-фильтрации на сефадексе G-25.
Ранее препараты ТЧ получали из органов лимфатической сис-
темы, например миндалин, содержащих до 40% Т-лимфоцитов.
Миндалины можно использовать через несколько часов после
операции или хранить несколько месяцев при —20 °C. После
удаления пирогенов, балластных веществ, эндотоксинов и виру-
сов получают препарат для лечения хронических инфекций и
иммунодефицитных состояний. Препарат содержит олигопеп-
тиды, олигонуклеотиды и другие низкомолекулярные компо-
ненты. Трансфер-фактор не иммуногенен для человека, его
можно применять в течение нескольких месяцев в больших до-
зах. Это позволяет рекомендовать его для лечения иммуноде-
фицитных состояний.
4.6.4. СПИСОК ЛИТЕРАТУРЫ
1. Uotila A. Studies on the chemical nature of dializable transfer factor. Com-
parison of human leukocyte dialysate and dialysates derived from human
serum and from mammalian lymphoid and non-lymphoid organs. — Immuno-
biol., 1979, 156, 353.
2. Wilson G. B., Fudenberg H. H., Horsmanheimo M. Effects of dislyzable leu-
kocyte extracts with transfer factor activity on leukocyte migration in vitro.
I. Antigen-dependent inhibition and antigen-independent inhibition and enhan-
cement of migration. II. Separation and partial characterization of the compo-
nents in DLE producing antigen-dependent and antigen-independent ef-
fects.— J. Lab. Clin. Medicine, 1979, 93, 800.
3. Schroder I., Rovensky J. In vitro determination of effects of dialyzable leuko-
cyte extracts containing transfer factor activity. — Allergol. et Immunopathol.,
1982, 10, 171.
4. Schroder I. Zur Therapie mit Transfer-Factor bei Erkrankungen mit Storung
der zellelaren Immunreaktion. — Ztschr. ges. inn. Med., 1980, 35, 86.
5. Bhardwaj N., Brummer E., Foster L. G. et al. Transfer of DTN to SK-SD and
tetanus toxoid in BALB/c mice by TFd prepared from purified subpopulations
456
of murine lymphocytes. — In: Immune regulators in transfer factor/Eds.
A. Khan et al.— New York: Academic Press, 1979, 285.
6. Brummer E., Foster L. G., Bhardwaj N. et al. A murine model for studying"
the transfer of DTH with dialyzable human transfer factor. — ibid., p. 27.
7. Krohn K, Uotila A., Ashorn R. et al. Augmentation of skin reactivities in-
antigen primed guinea-pigs by transfer factor and other cellular dialysates.—
Ibid., p. 655.
8. Arrenbrecht S., Aker O., Dubs R. et al. Human transfer factor in rats. — In:
Immune regulators in transfer factor/Eds. A. Khan et al. — New York, San
Francisco. London: Academic Press, 1979, 643.
9. Thilsted J. P., Shifrine M. Studies on the transfer of tuberculin specific СМГ
in the dog. — Ibid., 673.
10. (Schroder l.) Шрёдер И., Перепечкина H. П., Тац А. Н., Медуницин Н. В.
Адъювантные свойства препаратов трансфер-фактора из лимфоцитов мин-
далин человека.—Журнал микробиологии, эпидемиологии и иммунологии-
(ЖМЭИ), 1979, № 2, 103.
11. Salaman М. R. The state of transfer factor. — Immunology today, 1982, 3, 4.
12. Khan, Kirkpatric С. H., Hill N. O./Eds. Immune regulators in transfer fac-
tor.— New York, San Francisco, London: Academic Press, 1979.
4.7. ВВЕДЕНИЕ РАДИОАКТИВНОЙ МЕТКИ В БЕЛКИ
4.7.1. ПРИНЦИП МЕТОДА
Меченные радиоактивными изотопами белки, гормоны, антите-
ла, нуклеиновые кислоты и гаптены находят широкое примене-
ние в изучении реакций АГ — АТ и в самых разнообразных им-
мунобиологических исследованиях. Вид метки и ее активность
зависят от цели исследования. Мечение метаболитов 3[Н] (бета-
излучатель, энергия частиц 18 кэВ, период полураспада
12,3 года) И[С]-ацетильными группами (бета-излучатель, энер-
гия частиц 156 кэВ, период полураспада 5730 лет) или 35[S]-
метионином (бета-излучатель, 167 кэВ, 87 дней) довольно быст-
ро приводит к образованию in vivo или in vitro соединений с
небольшой специфической активностью. Можно получить мече-
ные соединения, например антигены, иммунологические свойст-
ва которых идентичны немеченым антигенам. Высокую специ-
фическую активность получают in vitro восстановительным ме-
тилированием (превращение аминогрупп в моно- или диметил-
аминогруппы). Этот метод позволяет без затруднений вводить
в соединения двойную метку — а[Н] и 14[С]. Широкое распро-
странение получило мечение белков радиоактивным йодом. Для
этой цели используют изотопы [1251] и [1311], которые позво-
ляют получать препараты с высокой специфической актив-
ностью. В обращении 125{1] удобнее ввиду его более длитель-
ного периода полураспада (60 дней по сравнению с 8 днями)
и меньшей энергии излучения (35 кэВ по сравнению с 700 кэВ),
чем 131 [I]. Оба изотопа дают у-излучение, кроме того, 131 [I]
обладает также p-излучением. Мечение белка основано на его
взаимодействии с высвобождающимся при окислении свобод-
ным радиоактивным йодом. Включение йода в ароматическое
кольцо тирозина происходит при значении pH, близком к нейт-
ральному. Повышение pH приводит к взаимодействию йода с.
30-990
457
сульфгидрильными группами, а также с гистидином и трипто-
фаном.
Помимо таких окислителей, как персульфаты, йодаты, нит-
риты, гипохлориты, перекись водорода, широкое применение
нашли хлорид йода, хлорамин Т, электрохимический метод,
ферментативное йодирование при помощи лактопероксидазы.
С использованием последних методов можно вводить метку
211 [At] в белки и гормоны. Этот изотоп представляет собой
«-излучатель с энергией частиц 5,9 мэВ, малой длиной свобод-
ного пробега в воде (60 мкм) и коротким периодом полураспа-
да (7,2 ч), что позволяет использовать 2,1 [At] для специфиче-
ской инактивации сенсибилизированных лимфоцитов или для се-
лективной иммуносупрессии in vivo. После маркирования белок
отделяют от несвязавшегося йода и продуктов окисления при
помощи гель-фильтрации, диализа, осаждения, ионообменной
хроматографии и других методов. Меченые продукты прове-
ряют в отношении электрофоретических и иммунологических
свойств, а также стабильности. Для сохранения исходных
свойств белков степень йодирования не должна превышать 1.
Данному вопросу посвящены специальные обзоры [5, 6].
4.7.2. МАРКИРОВАНИЕ С ПОМОЩЬЮ 125[1]
Будут описаны методы химического маркирования БСА при по-
мощи хлорамина Т и электрохимический способ маркирования
ферритина. Используют изотоп 125[1] в виде изотонического
раствора йодида натрия, не содержащего носителя и восста-
навливающих примесей.
4.7.2.1. МАРКИРОВАНИЕ С ПОМОЩЬЮ ХЛОРАМИНА Т
Хлорамин Т представляет собой натриевую соль N-монохлор-
паратолуолсульфонамида. В водных растворах от него отцеп-
ляется хлорноватистая кислота. Для маркирования белков не-
обходима слегка щелочная среда (pH около 7,5), в таких усло-
виях включение йода протекает количественно. Состав реакци-
онной смеси: 10 мг БСА, 0,5 мл 0,5 М КН2РО4, 2,5 мл 0,5 М
NasHPO4, 0,5 мл 0,0005 М KI, 40 МБк Na 12&[1] (5 ГБк/мл),
0,8 мл хлорамина Т (1 мг/мл).
Смесь медленно перемешивают в колбе Эрленмейера в те-
чение 30 мин. Процесс останавливают добавлением 0,1 мл
0,2% раствора Na2S2O5. Степень йодирования зависит от кон-
центрации хлорамина Т, от продолжительности инкубации и от
специфической активности метки (см. раздел 1, 3).
4.7.2.2. ЭЛЕКТРОХИМИЧЕСКОЕ ЙОДИРОВАНИЕ
Белок взаимодействует со свободным радиоактивным йодом,
выделяющимся в ходе анодного окисления йодидов (необходи-
мый потенциал равен примерно 388 мВ). Схема прибора пред-
458
Рис. 144. Электрическая схема
аппарата для электрохимиче-
ского маркирования белка.
Р — платиновый тигель; М — пла-
тиновый катод; R — сопротивление;
К — компенсатор; Н — каломелевый
электрод; EI — источник питания;
Е2 — нормальный элемент; ЕЗ —
вспомогательный источник питания;
G1 — микроамперметр; G2 — вольт-
метр; G3 — гальванометр.
ставлена на рис. 144. В качестве анода используют платино-
вый тигель, в котором находится вещество, подлежащее марки-
рованию. Перед использованием тигель тщательно прокали-
вают во избежание поляризационных эффектов. Катод сделан
в виде платинового цилиндра. Он покрыт слоем коллодия или
диализной мембраной. В качестве электролита используют
0,1 М NaCl. Нужное напряжение устанавливается при помощи
потенциометра. Фактический потенциал определяют при помо-
щи каломельного электрода компенсационным методом при
сравнении с нормальным элементом. Перемешивание компонен-
тов смеси осуществляют магнитной мешалкой. В случае работы
с термолабильными веществами применяют охлаждение. В сос-
тав смеси входят 5 мг ферритина лошади, 1,5 мл 0,1 М NaCl,
0,1 мл 0,0005 М KI, 40 МБк Na I25[I] (4 ГБк/мл). Степень
йодирования зависит от продолжительности электролиза и си-
лы тока, помимо прочих вышеперечисленных условий. При анод-
ном напряжении 700 мВ сила тока в начале процесса состав-
ляет около 100 мкА, спустя приблизительно 100 мин она сни-
жается на 10 мкВ в зависимости от эффективной поверхности
электрода. Увеличение анодного напряжения и продолжитель-
ности йодирования приводит к накоплению продуктов окисле-
ния.
4.7.2.3. ВЫДЕЛЕНИЕ МЕЧЕНЫХ БЕЛКОВ
ИЗ РЕАКЦИОННОЙ СМЕСИ
Отделение меченых белков от несвязавшейся с белками радио-
активной метки, электролита и промежуточных продуктов про-
водят на сефадексе G-25. Реакционную смесь, содержащую
радиоактивный йод, наносят на колонку размером 25x240 мм
и элюируют 0,005 М трис-НС1-буфером с pH 8. Радиоактивность
измеряют при помощи проточной кюветы на жидкостном сцин-
тилляторе, концентрацию белка — при помощи проточной кю-
веты (Увикорд) на волне 280 нм. Качество разделения опреде-
ляют методом бумажной хроматографии (см. ниже). При гель-
фильтрации меченый белок практически полностью отделяется
от непрореагировавшего йода.
го*
459
4.7.2.4. АНАЛИЗ ПРЕПАРАТА
Меченые [1251] препараты оцениваются по специфической ра-
диоактивности и по степени йодирования. Кроме того, прово-
дят проверку электрофоретических и иммунологических
свойств.
4.7.2.4.1. Определение эффективности маркирования
Эффективность маркирования, т. е. долю связавшейся с белка-
ми метки, определяют с помощью осаждения белков ТХУ или
методом восходящей бумажной хроматографии.
Осаждение ТХУ. Из пробы, содержащей радиоактивный
йод, отбирают аликвоту, смешивают ее с 1% БСА. После осаж-
дения 10% ТХУ осадок собирают на мембранные фильтры
(Хемапол, ЧССР, диаметр пор 0,45 мкм), фильтры промывают
и измеряют радиоактивность осадка сцинтилляционным счет-
чиком.
Хроматография на бумаге. Небольшое количество материа-
ла наносят на бумажную полоску 40x250 мм (FN14, Erzge-
birge, ГДР) и проводят хроматографию в 70% водном растворе
метанола. Распределение радиоактивной метки на хроматограм-
ме определяют при помощи сцинтилляционного счетчика. Эф-
фективность маркирования БСА химическим способом состав-
ляет 60—80%, электрохимическим — 30—50%.
Оба метода дают несколько отличающиеся результаты, обус-
ловленные различием в количестве метки, переходящей в ТХУ-
растворимый материал с увеличением концентрации восстано-
вителей. В среднем получаются белки со специфической радио-
активностью 0,7—2,2 МБк/мг белка.
4.7.2.4.2. Определение степени йодирования
Степень йодирования, выраженную числом грамм-атомов йодд,
присоединенных к 1 молю белка, рассчитывают после опреде-
ления концентрации белка в пробе по методу Лоури, а радио-
активности пробы — с помощью сцинтиляционного счетчика.
Степень йодирования (СИ) определяют по формуле:
АТ-ММ
СИ= А^Б-Ш ’
где А — радиоактивность белка (мин-1), А’-радиоактивпость
пробы (мин"1), I — количество йода в пробе (мкг), Б — коли-
чество белка в пробе (мкг), ММ — отн. мол. масса белка.
Количество метки, попавшей в ТХУ-растворимую фракцию
реакционной смеси, не учитывается. В случае БСА степень
йодирования близка к 1, в случае ферритина она значительно
«иже.
469
4.7.2.4.3. Электрофоретические свойства
Одной из характеристик меченого белка являются его электро-
форетические свойства. Для сравнения используют немеченый
белок. Оценку результатов проводят путем окрашивания амидо
черным и радиоавтографии. Для макрорадиоавтографии приме-
няют пленку ORWO-film AF4, которую разрезают на кусочки
соответствующей величины и накладывают на пластины с ага-
ром. Мягкое у-излучение 125[1] особенно удобно для получения
радиоавтографических изображений. Через 3—5 дней экспози-
ции пленку обрабатывают рентгеновским проявителем АЗО или
МН-28.
После проявления пленки проводят окрашивание пластины
с гелем амидо черным для выявления полос белка. Сопоставляя
окрашенные полосы белка в геле (йодированный и нативный
белки) с полосами почернения на радиоавтограмме, можно сде-
лать заключение о качестве меченого белка. Если белок час-
тично денатурирован, часть метки остается у стартового же-
лобка. Оценку фореграммы можно проводить непрерывным
фото- и радиометрическим измерением пластинок и пленок.
На рис. 145 представлена фореграмма частично денатурирован-
ного БСА, окрашенная амидо черным.
4.7.2.4.4. Иммунологические свойства
Изучение иммунологических свойств меченого белка в сравне-
нии с исходными проводят при помощи иммуноэлектрофореза.
В вырезанные лунки вносят меченые и, соответственно, немече-
ные белки. После электрофоретического разделения в боковые
желобки вносят антисыворотку и оставляют пластины до за-
Рис. 145. Электрофорез в геле бычьего сывороточного альбумина, меченного
*25[.1].
Продолжительность фореза 2’/а ч в EL-KF66; вероналовый буфер с pH 8,6; окрашивание
лмидо черным.
461
Рис. 146. Иммуноэлектрофорез интактного ферритина.
Продолжительность фореза З’/з ч; вероналовый буфер с pH 8,6; продолжительность диф-
фузии 24 ч; окрашивание амидо черным.
вершения диффузии. Пластины высушивают, фиксируют и ок-
рашивают амидо черным. Окрашенную пластину можно исполь-
зовать для радиоавтографии. По положению дуг преципитации,
образованию шлейфов и затемнений в месте нанесения белков
судят о возможной денатурации меченых белков. На рис. 146
представлена иммунофореграмма интактного ферритина, мечен-
ного 125[1].
4.7.2.4.5. Стабильность препаратов
Длительное хранение меченых белков приводит к потере ими
йода. Дейодирование происходит как вследствие радиоактив-
ного распада метки, так и вследствие взаимодействия с про-
дуктами радиолиза воды. Кроме того, возможно ферментатив-
ное разрушение меченых компонентов. При хранении меченого
БСА при 4 °C в течение 30 дней наблюдается незначительное
увеличение радиоактивности, не связанной с белком. Если хра-
нение происходит при 37 °C, то уже через 8 дней не связанная
с белками радиоактивность возрастает до 15% от общей радио-
активности.
Меченый 125[1] овальбумин дейодируется быстрее, чем БСА;
ферритин подвергается дейодированию сравнительно медленнее.
Потеря белками йода представляет собой процесс, на который
оказывают воздействие различные физико-химические факторы,
в том числе свет. В ходе хроматографии на бумаге часто наблю-
дается спонтанное дейодирование, зависящее от величины pH и
от изменений концентрации реагентов. Этот эффект может
быть уменьшен добавлением белка-носителя [3].
462
4.7.2.5. ОЦЕНКА МЕТОДА
Иодирование может изменять молекулярные и иммунобиоло-
гические свойства белков. Изменения связаны с заменой ато-
мов водорода на сравнительно более крупные атомы йода, в не-
которых случаях они обусловлены воздействием окислительных
агентов [7]. Как было отмечено выше, маркирование белков
хлораминовым методом приводит к образованию частично де-
натурированных продуктов. При изменении условий может быть
получен интактный в электрофоретическом отношении БСА.
Электрохимическое йодирование представляет собой более ща-
дящий способ маркирования, однако оно используется сравни-
тельно редко ввиду сложности аппаратуры. К числу его пре-
имуществ относится хорошая воспроизводимость степени йоди-
рования. Эффективность йодирования электрохимическим
методом невысока.
4.7.2.6. ВАРИАНТЫ
Известно большое количество окислителей, пригодных для про-
цесса йодирования. Далее будут перечислены важнейшие из
них, описаны наиболее часто используемые модификации в той
мере, в какой они отклоняются от оригинальных методик.
4.7.2.6.1. Модифицированный хлораминовый метод
Известен альтернативный метод йодирования без прямого ис-
пользования хлорамина Т. При помощи хлорамина Т йодируют
гидроксисукцинимидный эфир 3,4-гидроксифенилпропионовой
кислоты. Продукт реакции экстрагируют бензолом и высуши-
вают. Белок модифицируется в результате переноса ацильной
группы на е-аминогруппы лизина или на N-концевые остатки
белка. Преимущество данного метода состоит в том, что белок
не вступает в непосредственный контакт с окислителями и вос-
становителями, йодирование становится возможным даже в от-
сутствие тирозиновых остатков у белка и, таким образом, не
происходит изменения антигенных детерминант белка.
4.7.2.6.2. Иодирование монохлоридом йода
Это старейший метод маркирования, который применялся в пе-
риод, когда не было препаратов с высокой специфической ра-
диоактивностью [2]. Степень йодирования зависит в основном
от количества IC1. Метод отличается хорошей воспроизводи-
мостью. Наряду с лактопероксидазой хлористый йод представ-
ляется средством выбора при маркировании белков, к тому же
•он является очень мягким окислителем, не изменяющим иммуно-
логическую активность меченых белков. Метод основан на изо-
топном обмене. В условиях окислительного варианта монохлор-
йодидного метода радиоактивный йодид непосредственно пре-
вращается в радиоактивный монохлорид йода, который затем
463
взаимодействует с белком. Этот вариант позволяет получать
наиболее высокую специфическую активность. Помимо альбу-
мина и инсулина, этим методом можно также маркировать ан-
титела.
4.7.2.6.3. Ферментативное йодирование
Принцип ферментативного йодирования известен давно [7]. Он
позволяет получать препараты с высокой специфической актив-
ностью. Физико-химические и иммунологические свойства бел-
ка остаются при этом неизмененными. Основные сложности
связаны с выбором фермента, так как можно использовать да-
леко не всякую пероксидазу. Для радиойодирования наиболее
подходящим ферментом является лактопероксидаза.
Чаще всего для работы используют коммерческие препара-
ты пероксидаз, в том числе пероксидазу хрена. Ниже приво-
дятся условия, в которых йодирование полипептидов проводит-
ся с применением лактопероксидазы. Состав смеси: в 0,05 М
фосфатном буфере с pH 7,5 растворяют 50 МБк Na 125{1],
5 мкг полипептида или белка, 4 мкг лактопероксидазы и 1 мкл
0,88 М перекиси водорода; общий объем смеси составляет
50 мкл. После тщательного перемешивания при комнатной тем-
пературе процесс через 1—2 с останавливают добавлением
500 мкл фосфатного буфера. Не связавшуюся с белками радио-
активность удаляют гель-фильтрацией через колонку с сефадек-
сом G-50, уравновешенную натрий-барбиталовым буфером. Для
стабилизации меченого белка к каждому мл раствора добавля-
ют 0,5 мл 1% раствора БСА. Перекись водорода можно вно-
сить непосредственно или получать ее путем реакции глюкозы
с глюкозооксидазой. Вышеописанным методом были получены
меченые препараты пролактина, фрагментов антител и компо-
нента комплемента Clq.
4.7.2.6.4. Другие окислители
Описан метод получения меченых нуклеиновых кислот. Йодиро-
вание происходит в ацетатном буфере с pH 5 при температуре
60 °C. В качестве окислителя используют Т1С13. Метка включа-
ется в состав ДНК в виде 5-йодцитозина [5, 7].
4.7.2.7. ОЦЕНКА МЕТОДА
Йодирование белка представляет собой простой, легко осущест-
вимый процесс. Для этой цели можно использовать самые раз-
нообразные окислители. Например, Clq, несмотря на его ла-
бильность, можно метить и при помощи лактопероксидазы, при
помощи и хлорамина Т [1]. Получаемый меченый препарат
комплемента позволяет во много раз повысить чувствитель-
ность определения иммунных комплексов. Маркирование бел-
ков зависит от очень большого числа параметров (pH, ионная
сила, температура, концентрация реагентов и их химическая
464
природа и т. д.), поэтому у каждого экспериментатора остают-
ся большие возможности для поиска оптимальных вариантов
маркирования соответствующих веществ.
4.7.3. МЕЧЕНИЕ 3[Н] И 14[С]
К числу недостатков йодной метки относится сравнительно ко-
роткая продолжительность жизни изотопов йодов. Тритий и ра-
диоактивный углерод выгодно отличаются в этом отношении.
Сравнительно высокая энергия распада этих радиоактивных
элементов делает их использование в иммунобиологических ис-
следованиях весьма перспективным.
4.7.З.1. МЕЧЕНИЕ МЕТОДОМ ВОССТАНОВИТЕЛЬНОГО
МЕТИЛИРОВАНИЯ
Аминогруппы белка могут быть восстановлены до моно- или
диметиламиногрупп. Метку в метильную группу можно вводить
посредством меченого I4i[C] формальдегида или меченого три-
тием боргидрида натрия. Эффективность реакции существенно
зависит от pH.
Состав смеси: к 0,1 мг белка в 0,1 мл 0,2 М натрий-борат-
ного буфера с pH 9 добавляют 10 мкл 14[С]-формальдегида
(0,04 моль/л). Через 30 с в смесь вносят четырехкратное коли-
чество боргидрида натрия в объеме 2 мкл (0,15 моль/л). Еще
через 60 с к смеси добавляют 10 мкл раствора боргидрида нат-
рия. Непрореагировавшие низкомолекулярные компоненты уда-
ляют диализом. Обычно получаются препараты с невысокой
специфической активностью. Специфическую активность можно
повысить, используя меченный тритием боргидрид натрия [8].
К 0,5 мл белка (5 мг/мл) в 0,2 М боратном буфере с pH 9 до-
бавляют 10—60 мкмоль формальдегида (водный раствор, 3,7%).
Меченный тритием боргидрид натрия растворяют в воде или
в смеси вода/изопропанол в соотношении 1 + 1, или в 0,1 М
NaOH. Радиоактивность реагента должна составлять приблизи-
тельно 70 МБк/мкл.
После 30-минутной инкубации несвязавшуюся радиоактив-
ную метку отделяют на колонке с сефадексом G-25. Меченый
продукт стабилизируют добавлением 10% сыворотки плода ко-
ровы.
4.7.3.2. ПРОЧИЕ РЕАГЕНТЫ
Антитела и фрагменты антител можно маркировать мечеными
алкилирующими агентами: йодуксусной кислотой, этилметан-
сульфонатом, 1-фтор-2,4-динитробензолом. Обычно используют
алкилирующие агенты, меченные тритием [5]. Белки можно
метить тритированным Ы-сукцинимидил-2,3-пропионатом (аци-
лирование). Этот метод пригоден для маркирования монокло-
465
нальных антител [4]. Алкилирующие агенты взаимодействуют
с белками при щелочных значениях pH, связываясь с остатка-
ми лизина, гистидина или сульфгидрильными группами.
4.7.3.3. МЕЧЕНИЕ ПУТЕМ БИОСИНТЕЗА
Этот метод основан на включении в состав всех клеточных
структур аминокислот, меченных 3[Н] или 14[С], за счет мета-
болизма клеток in vivo или in vitro. Для этой цели могут при-
меняться самые разнообразные аминокислоты, но чаще исполь-
зуют лейцин и лизин, которые встречаются практически во
всех белках. Культуру клеток метят в СО2-инкубаторе в тече-
ние 12 ч. Если планируются радиоавтографические исследова-
ния, то предпочтение отдается метионину, меченному 35[S] [5].
4.7.4. СПИСОК ЛИТЕРАТУРЫ
1. Danis V., Trent R., Clancy R. A modified Clq deviation radioimmunoassay for
the use in a routine clinical laboratory. — J. Immunol. Meth., 1980, 33, 175.
2. Doran D. M„ Spar I. L. Oxidative iodine monochloride iodination technique.—
J. Immunol. Meth., 1980, 39, 155.
3. Jackson D. C. Some effects of Chloramine T induced radioiodination on the
physico-chemical properties of oligomeric proteins. — J. Immunol. Meth., 1980,
34, 253.
4. Kummer U., Thiel E., Doxiasis I. et al. Tritium radiolabelling of antibodies to
high specific activity with N-succinimidyl-2,3-3H-peopionate: use in detecting
monoclonal antibodies. — J. Immunol. Meth., 1981, 42, 367.
5. Lefkovits I., Pernis B. Immunological Methods. — New York, San Francisko,
London: Academic Press, 1979.
6. Mayer R. J., Walker J. H. Immunochemical Methods in the biological sciences:
enzymes and proteins. — London — New York—San-Francisko, Toronto, Syd-
ney: Academic Press, 1980.
7. Weir D. M. Handbook of experimental Immunology. Third Edition. Oxford,
London Edinburgh, Melbourne: Blackwell Scientific Publ., 1978.
8. Wilder R. L., Yuen С. C., Subbarao B. et al. Tritium (3H) radiolabelling of
protein A and antibody to high specific activity: Application to cell surface
antigen radioimmunoassays. — J. Immunol. Meth., 1979, 28, 255.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авидность, изучение 43
Агглютинация латекса 219
— — методика 219
---- оценка 220
— тимоцитов мыши агглютинином земля-
ного ореха 248
Агрегаты иммунные, определение фотомет-
рическое 115
---------материалы 116
---------методика 116
-----------белок в сыворотке больных,
определение 117
-----------комплексы иммунные цирку-
лирующие, определение 118
----------- кривая стандартная, построе-
ние 116
--------- оборудование 116
--------- оценка результатов 117
Анализ иммуноферментный 162
----материалы 163
----метод(ы) конкурентный 163
------ неконкурентные 164
---- оборудование 166
---- оценка метода 167
— нммунофлюоресцентиый 136
---- контроль качества анализа 138
---- метод непрямой 137
------прямой 137
---- оборудование 140
----окрашивание дополнительным цве-
том 138
---- определение антител комплементсвя-
зывающих 137
----применение метки флюоресцентной
двойной 137
---- среды для хранения препаратов 140
----титрование в шахматном порядке
139
— преципитатов иммунных в гелях 108
----------- выявление гликопротеидов
111
--------------металлопротеидов 111
-------------- нуклеопротеидов 111
-----------иммунофлуоресценция 113
-----------окрашивание белков 109
----липопротеидов ПО
----преципитаты иммунные, ак-
тивность ферментативная, определение
111
----радиоавтография ИЗ
— радионммунологнческнй 148
----метод(ы) двойных антител 155
------основанные на применении мече-
ных антител 157
------. Фарра 155
---------маркирование антигена 156
---------определение антител к ДНК
156
— •— оценка метода 160
------ твердофазный 152
------ мечение антигена 153
------ оптимизация анализа 153
-------- связывание антител с матрицей
152
------тест-системы РИА на фаги ТЗ и Т4
154
— электроиммунный 97
---материалы 98
--- методика 98
------иммуноглобулины, определение 100
------ обработка пластин 100
------ оценка результатов 100
------электрофорез 100
---модификации 101
--- оценка метода 102
Анафилаксия кожная активная 363
---пассивная 363
Антигены белковые, иммунизация карпов
14
------ кроликов 13, 14
---------материалы 13
---------методика 13
--------- оборудование 13
— конъюгированные 432
Антисыворотка (и) к антигенам белковым,
получение 13
------клеточных мембран, получение 17
---веществам низкомолекулярным, по-
лучение 16
—• крысиная к тнмусу мышей, получение
239
— кроличья против В-лимфоцитов мышей
240
— получение 9
---от животных экспериментальных 11
Анти-тэта-сыворотки 239
Антитела, аффинность, определение 42
— гибридомные, анализ нммунохимнческий
32
— меченные ферментом 436
— моноклональные к антигенам поверх-
ностным, определение 29
— —----растворимым, определение 27
— — получение слиянием клеток 20
467
----— — материалы 22
----—------методика 22
----------- оборудование 22
-----------— технология гибридомная 20
— образование культурами клеток 34
— материалы 35
— методика 35
модификация 39
оценка 40
—--------оборудование 35
---------подготовка посуды 36
---------— сред питательных 36
получение культур клеток 38
-------------------------------- — суспензии клеток селезенки
37
-----------— эритроцитов 38
Аффинность 43
— определение, метод измерения гашения
флюоресценции 50
—-------------материалы 51
-------------- оборудование 51
-------------- оценка результатов 51
— — — — — — постановка опыта 51
— — — осаждения аммония сульфатом 48
---полученные данные, оценка 53
Белки, коиъюгироваиие с эритроцитами 64
— окрашивание 109
--- азокармии 109
---амидо черный 109
---пунцовый 110
---светлый зеленый 109
Витте раствор, приготовление 197
p-D-Галактозидаза, коиъюгироваиие 439,
440
Гели, свойства 73
— функции 73
Гемолиз локальный 57
---материалы 59
---метод безгелевый 63
--------- камеры, заполнение 64
-----------установка 63
— — — непрямой 63
---— — секвенциальный 63
— —------торможение локального гемоли-
за 63
--- прямой 60
— —-----подготовка чашек Петри 60
— получение лимфоидных клеток
60
— • — — — постановка опыта 62
--- оборудование 59
— — обратный 67
оценка метода 68
Гибридомы, клонирование in vitro 30
---in vivo 31
— консервирование при низкой температу-
ре 31
— способность антителопродуцируюшая^
определение 27
Диализат экстрактов лейкоцитов, получе-
ние 453
Диффузия в геле 74
Иммунизация антигенами белковыми кар-
пов 14
— — — кроликов 13, 14
— схемы 238
Иммуноглобулин А, выделение из сыво-
ротки бараньей 401
-----------бычьей 401
----—------козьей 401
-----------лошади 400
----------- свиньи 399 '
------— — человека 397
----крыс, выделение 401
----секреторный бычий 403
------ кролика 403
------ кур 405
------лошади 404
------ морской свинки, выделение 405
------ свиньи 404
—-----собаки 405
----— фрагментация 424
------ человека 401
— D, выделение из сывороток больных с
дисгаммаглобулинемией 410
— Е,, выделение из сыворотки вольных с
аллергией 412
----— — — миеломой 412
— G, выделение 390
—-----из сыворотки кролика 392
----------- крупного рогатого скота 394
-----------крыс 395
-----------курицы 393
-----------лошади 393
----------- морской свинки 394
-----------мышей 395
-----------овцы 396
----------- свиньи 395
----------- человека 391
----расщепление меркаптоэтаиолом 414
------ папаином 417
----сульфитолиз восстановительный 414
---- фрагментация пепсином 421
----цепи легкие, получение 414
------тяжелые, получение 414
— М, выделение 405
------ из сыворотки кролика 407
—----------крупного рогатого скота 409г
----------- Кур 41Q
----—------свиньи 410
------— — человека 406
Иммунодиффузия 73
— методы, классификация 76
— ошибки 77
— радиальная двойная 77
'> 8
-------материалы 78
-------методика 78
— — — — модификация 81
------- оборудование 78
------- оценка метода 81
------- результатов 79
---- простая 82
-------материалы 82
-------методика 82
------- антиген, свойства, значение 85
------- кривая калибровочная построе-
ние 83
-------модификация 85
------- подготовка пластин 85
------- оборудование 82
-------продолжительность 84
------- этапы исследования 82
— специфичность 77
— чувствительность 77
Иммуиосорбент(ы) 427
— белково-глутаральдегидный 428
— на основе сефарозы 428
— получение, метод глутаральдегидный 427
Иммуиофлюоресценция 128
— адсорбция порошком ткани 132
— анализ иммунофлюоресцентный 136
— источники ошибок 145
— криосрезы, приготовление 135
— мазки 135
— маркировка белков ТРИТЦ 130
-------ФИТЦ 130
----иммуноглобулинов 130
— отпечатки 135
— препараты культур клеток 135
---- оценка 143
— применение в иммунологии 146
— срезы тканей парафинированных 136
— субстраты антигенные, получение 133
— фракционирование 131
— хранение конъюгатов 133
— — сывороток 133
Лммуиоэлектрофорез 89
— материалы 89
— методика 90
----антисыворотка, внесение 92
----белки, окрашивание 92
элюирование 92
иммунодиффузия 92
— —модификация 92
— — — метод Гереманса 94
— —--Оссермаиа 94
---- оценка 94
— — приготовление геля агарового 93
------- пластинок агаровых 93
----формирование в геле канавок 90
----------- лунок 90
— — электрофорез, проведение 90
Индекс цитотоксичности, расчет 292
Клетки-мишени, мечеиие 208
— подготовка 208
Клетки макрофагсодержащие, суспензия1,,
получение 367
— моноцитсодержащие, суспензия, получе-
ние 367
— системы иммунной 226
------- разделение 226
---------адгезия 228
---------в градиенте плотности 244
-------— методом розеткообразования 242
----------- цитолиза 238
----------иа гранулах стеклянных 229
—----------колонках аффинных 233
----------при помощи лектинов 248
------- свойства 227
------- фракционирование иа волокнах
нейлоновых 231
— —. — электрофорез 250
---------метод, выбор 252
--------- сортировка клеток 250
--------- разделение на монослое 251
— тучные крыс, выделение 355
-------высвобождение медиаторов 355
-------деграиуляция, определение 356
— цитотоксические после аллогеииой сти-
муляции 290
-----------высвобождение 5,Сг 295
—----------клетки, обработанные митоге-
нами, в качестве клеток-мишеней 291
--------------эффекторные из смешан-
ной культуры лимфоцитов 290
----------- оценка метода 291
---- приготовление 209
Кожа, трансплантация 348
----с хвоста на спину 351
----------- хвост 349
Комплексы иммунные растворимые, опре-
деление 120
—--------методики 121
—----------анализ твердофазный 123
-----------осаждение полнэтилеиглико-
лем 121
-----------применение агрегированного
нагреванием IgG человека в качестве
стандарта 126
—----------связывание со стафилокок-
ком 124
-----------тест на связывание C1G 121
----—. оценка метода 127
----циркулирующие, определение 118
Комплемент, реакция связывания тромбо-
цитами 186
Конъюгаты ангнотензииа, приготовление,,
метод глутаральдегидный двухступенча-
тый 437
— —--------одноступенчатый 437
— — с альбумином 436
— ДНФ-белок 433
— протеина А с пероксидазой 441
Латекс, агглютинация 219
Лейкоциты-, адгезия, реакция торможения
321.
469'
— базофильные, сенсибилизация, определе-
ние у больных с атопией 358
— миграция, реакция под слоем агарозы
311
— — — торможения 308
Лимфоциты антителообразующие, получе-
ние 24
— инкубация, метод непрямой 258
------ прямой 258
— культура смешанная 303
------ анализ, проведение 304
------материалы 304
----------- клетки, смесь стандартная
305
----------- — клетки-стимуляторы, инакти-
вация 304
-----------пробы, культивирование 305
—-------------обработка 305
------— — оборудование 304
----------- оценка метода 306
-------------- результатов 306
— реакция бласт-трансформации 294
— цитотоксические 282
---мышей с опухолями в системе син-
генной 288
-----------— высвобождение 51Сг 289
--------------иммунизация 288
------—-------клетки опухолевые в ка-
честве клеток-мишеней 289
--------------оценка результатов 290
------—-------тест мнкроцнтотоксическнй
с визуальной оценкой результатов 289
---после аллогенной иммунизации 286
---— — клетки лимфатических узлов
как эффекторные клетки 286
-----------проба на высвобождение
3Н-тимидина 287
------— фибробласты как клетки-мншеии
287
— человека, флюоресценция, типы 260
B-Лимфоциты, выделение 183
Т-Лимфоциты, выделение путем элимина-
ции В-лимфоцитов иа протеиновых гра-
нулах 234
Т, B-Лимфоциты, разделение 236
Липопротеиды, окрашивание 110
---жировой красный НО
--- комбинированное ПО
— — Судан черный 110
Макрофаги, выделение из суспензии спле-
ноцнтов 373
— идентификация 369
--- в суспензии спленоцитов 374
— культивирование 366
---модификация 370
— подвижность электрофоретическая, оп-
ределение 417
— подсчет 369
— распластывание, реакция торможения
327
470
Маркеры клеточные, определение мембран-
ной иммунофлюоресцеицией 261, 254
----------- антитела, маркировка 255
-------------- приготовление 255
-----------возможности применение 265
-----------контроль специфичности 263
—----------конъюгаты 261
-----------лимфоциты, инкубация 258
-------------- разделение 257
-----------материалы 257
---------— методика 257
----------- оценка 258, 266
---------— ошибки 261
-----------приборы оптические 263
----— — тест-система 261
Метка радиоактивная, в белки 457
---- анализ препарата 460
---- маркирование йодное 458
—----— йодирование электрохимическое
458
---------с помощью хлорамина Т 458
----мечеиие путем биосинтеза 466
----тритием 465
----углеродом радиоактивным 465
Метод Е-розеток 269
---- инкубация 270
----лимфоциты, получение 269
----материалы 269
----методика 269
— — модификации 270
---- оборудование 269
— — оценка 271
— ЕАС-розеток 272
— — инкубация 275
----комплекс антитела — эритроциты —•
комплемент, получение 275
— — лимфоциты, выделение 274
— — материалы 273
— — методика 273
---- оборудование 273
----оценка 276
---- эритроциты барана, сенсибилизация
274 4
— розеток иммунных 278
— — — материалы 280
— — — методика 280
----модификации 281
— — — оборудование 280
---- оценка 281
----— этапы работы 280
Моноциты, идентификация 369
— культивирование 366 .
— — модификация 370
— подсчет 369
Олсвера раствор 197
Препараты иммуноглобулинов, получение
390
Преципитаты иммунные, активность фер-
ментативная, определение 111
-----------гидролазы ИЗ
.-----—• —• оксиредуктазы 112
Преципитация в геле 74
— влияние некоторых факторов 75
раствор Витте 197
— Олсвера 197
— Хенкса 36
Реакция(н) агглютинации лейкоцитоз 222
______материалы 222
------методика 222
------мнкрометод 222
------ оценка метода 223
--------- результатов 223
— — — ошибки, источники 223
— анафилактическая 354
---исследования на базофильных грану-
лоцитах человека 358
---------тучных клетках крыс 354
---методы in vitro 354, 361
------in vivo 363
— антиген—антитело 73
— бласттрансформацин лимфоцитов 294
---— материалы 295
---— методика 297
--------- культуры, культивирование 297
------—- —- обработка 297
------ оценка метода 300
--------- результатов 299
— гемагглютинации 211
---материалы 213
---методика 213
------ бисдназобензнднн, приготовление
214
---— коиъюгироваиие альдегидом глута-
ровым 215
---— эритроциты, сенсибилизация 214
— — — — танизнроваиие 214
--------- фиксирование 214
--- оценка метода 216
--- пассивной 216
--- прямой 215
— кожные типа замедленного 342
---------сенсибилизация мышей контакт-
ная 344
---------у крыс 346
----------- морских свинок 346
---—- — — мышей 346
— лимфоцитолиза 177
— комплемент 180
— материалы 177
— методика 177
------лимфоциты, получение 178
— микромодификации 179
— оборудование 177
— оценка результатов 179
— миграции лейкоцитов под слоем агаро-
зы 311
----------- оценка метода 315
---— — — — результатов 315
------------ пластины с агарозой, подго-
товка 312
— — ------ реакция Непрямая 314
— —---------прямая 313
— связывания комплемента 193
------- антиген, контроль 203
--------- приготовление 199
--------- характеристика 195
-------антисыворотки 202
-------единица комплемента, определе*
ине 194-
-------комплемент, приготовление 201
-------материалы 197
------- методика, варианты 193
—------микрометод 203
------- оборудование 197
-------оценка метода 191, 204
--------- результатов 202
-------система гемолитическая 198
------------ получение суспензии эритро-
цитов барана 198
------------ проверка гемолизинов 198
-------тромбоцитами 186
---------материалы 186
—--------методика 186
------------международная стандартизи-
рованная 190
--------- оборудование 186
--------- определение титра гемолизина
188
--------------комплемента 189
--------- сенсибилизация эритроцитов ба-
рана 188
— торможения адгезии лейкоцитов 321
---------материалы 321
---------методика 321
—-----------антигены 322
— —---------клетки, приготовление 322
------------метод «четной камеры» 322
--------------«пробирочный» 324
— — оборудование 321
— оценка метода 325
— — миграции лейкоцитов 308
— — материалы 308
— методика 308
— лейкоциты, подготовка 309
------------лимфоциты, фракция надоса-
дочная, получение 309
— макрофаги, подготовка 309
— оборудование 308
оценка метода 310
— — распластывания макрофагов 327
---------материалы 327
—--------камера для культивирования,,
монтаж 329
--------- клетки экссудата перитонеаль-
ного, получение 329
---------модификации 331
— — — — оборудование 327
--------- оценка метода 332
Розетки, разделение 243
Розеткообразоваиие 242
471’
•Система(ы) антиген — антитело множест-
венные 76
----простые 76
----сложные 76
— иммунная, клетки 226
•Спленоциты, суспензия, получение 374
•Среда Игла-МЕМ, приготовление 37
Суспензии клеток макрофагсодержащнх,
получение 367
----моноцитсодержащих, получение 367
----селезенки, получение 37
— спленоцитов, получение 374
— эритроцитов, получение 38
^Сыворотки моноспецифические, получение
прн отсутствии полностью очищенного
антигена 15
— получение, оценка качества 19
— цитотоксичность, определение 241
Тест цитотоксический 282
Титрование комплемента 171
----материалы 171
----методика 171
-------гемолиз в агарозе 173
----— гемолизины, титрование 172
-------подготовка сыворотки 172
----— эритроциты барана, сенсибилиза-
ция 173
------------суспензия, получение 172
---- оценка метода 175
Трансфер-фактор, выделение 451
----методика 455
Фагоцитоз 378
— бактерий, бактерицидиасть 384
— материалы 381
— метод упрощенный, определение кли-
ренса 386
— оборудование 381
— оценка 380, 388
----бактерицидность, проверка 380
----дегрануляция, исследование 380
---- захват частиц, проверка 380
----обмен фагоцитов кислородный, опре-
деление 381
— фагоциты, получение 382
---- скрининг функциональный 387
— частицы для фагоцитоза, приготовление
382
Фактор (ы) кожнореактнвный 338
----материалы 339
----методика 339
---- оборудование 339
---- оценка визуальная 339
------- гистологическая 339
— — — метода 341
-------ошибки, источники 340
— хемотаксические, выявление 333
------материалы 334
--------- методика 334
—--------клетки, получение 335
---------хемотаксис 335
------ оборудование 334
------ оценка метода 336
Фарра метод анализа радиологического
155
Хенкса раствор, приготовление 37
Цнтолнз 238
Цитотоксичность клеточная антителозави-
симая 205
------антисыворотка, приготовление 208
------клетки-мишени, мечение 208
---------- подготовка 208
------клетки эффекторные, приготовле-
ние 209
------материалы 207
------методика 207
------ оценка результатов 209
---- спонтанная 285
— комплементзависимая 176
----возможности применения 182
---- оценка метода 185
----ошибки, источники, комплемент 182
--------- суспензии клеточные 181
----------- сыворотки типирующие 181
----реакция лимфоцитолиза 177
---------материалы 177
------— мнкромодифнкация 179
----:----оборудование 178
--------- — получение лимфоцитов 178
----типирование по HLA-DR 182
—-----------выделение В-лимфоцитов 183
------------метод двойной флюоресцент'
ной метки 184
------------сыворотки для HLA-DR 184
— опосредованная клетками 284
•-----лимфоциты цитотоксичекие пос0
аллогенной иммунизации 286
------методика 285
------тест быстрый 284
---------медленный 284
Экстракты тканевые, получение 442
Электрофорез встречный 103
----материалы 104
— — методика 104
--------- иммуноэлектрофорез встречный на
ацетат-целлюлоидных мембранах 106
--------- приготовление геля 104
----— проведение электрофореза 105
---- оборудование 104
---- оценка метода 107
----• — результатов 106
Эритроциты индикаторные, получение 64
472
К стр. 109
Рис. I. Иммунофореграммы различных способов окрашивания преципи-
татов в гелях.
а — сыворотка человека. Окраска световым (светлым) зеленым; б — вверху и в цент-
ре — сыворотка, внизу — цереброспинальная жидкость. Представлены изоферменты
карбоксилэстеразы. Контрастное окрашивание световым (светлым) зеленым (вверху)
и азокармнном (внизу); в — сыворотка и экстракт поджелудочной железы человека,
проявленные полиспецифической сывороткой против антигенов человека н адсорби-
рованные антисывороткой к поджелудочной железе. Окраска световым (светлым) зе-
леным (вверху), азокармином (внизу); г — сыворотка человека. Окраска азокарми-
иом: д — электрофорез в геле: цереброспинальной жидкости (вверху) и сыворотки
(внизу); в центре ~ изоферменты карбоксилэстеразы; е — окраска амидо черным.
179
Рис. 2. Выделение лимфоцитов методом флотации: сле-
ва — после наслаивания, справа — после центрифугиро-
вания.
Рис. 3. Микровариант реакции лимфоцитолиза: спра-
ва — внесение краски, слева — после заполнения каме-
ры с целью остановки реакции.
Рис. 4. Отрицательная реакция лимфоцитолиза.
Рис. 5. Двукратноположительная реакция лимфо-
цитолиза.
Рис. 6. Четырехкратноположительная реакция
лимфоцитолиза.
К стр. 223
Рис. 7. Отрицательный тест микроагглютинации лейкоцитов (отдельные окра-
шенные клетки в поле зрения — лейкоциты, неокрашенные — эритроциты).
Рис. 8. Положительный тест микроагглютинации лейкоцитов (агглютинаты
лейкоцитов между эритроцитами).
Рис. 9. Реакция торможения миграции лейкоцитов (капиллярный метод).