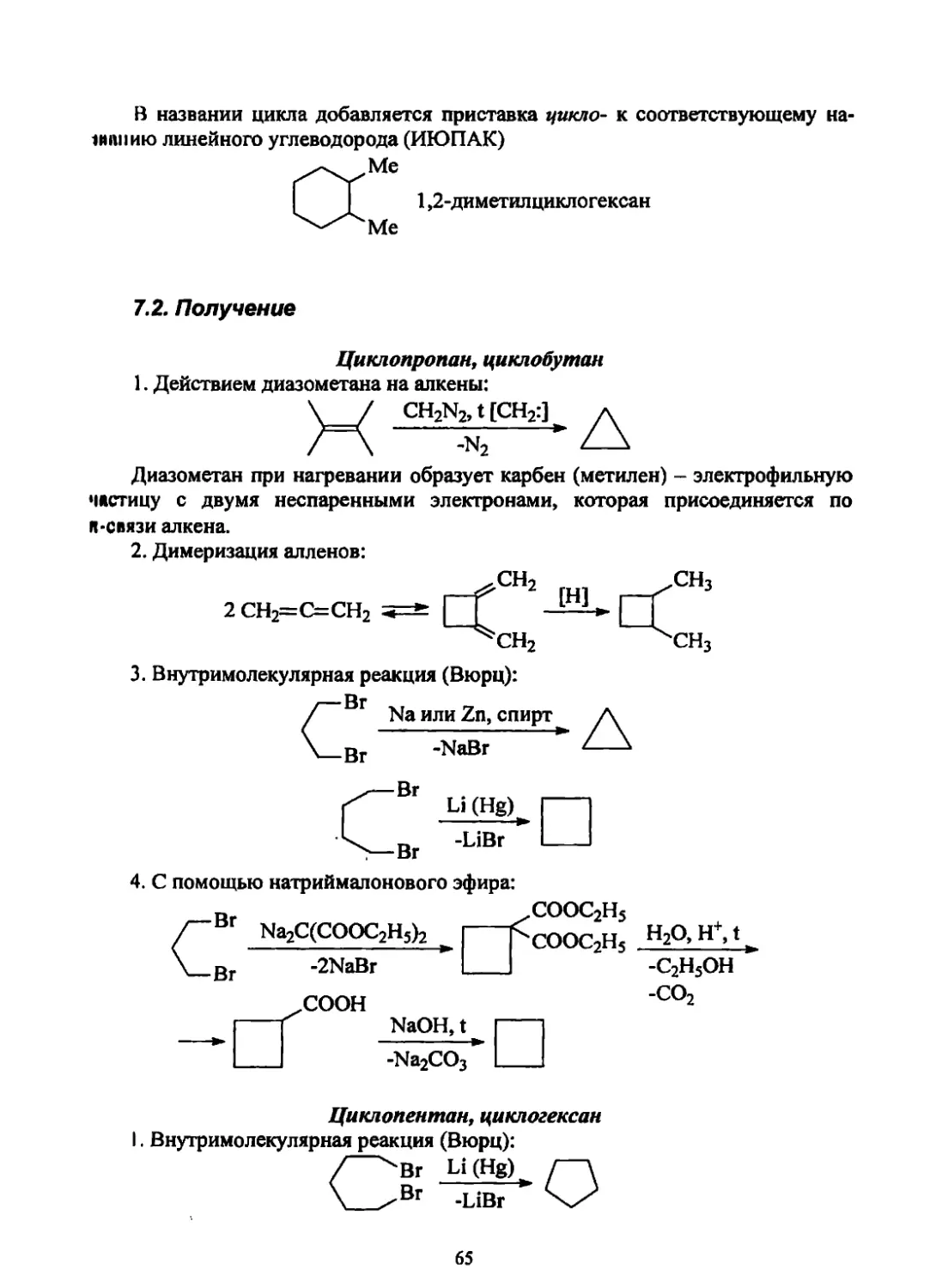
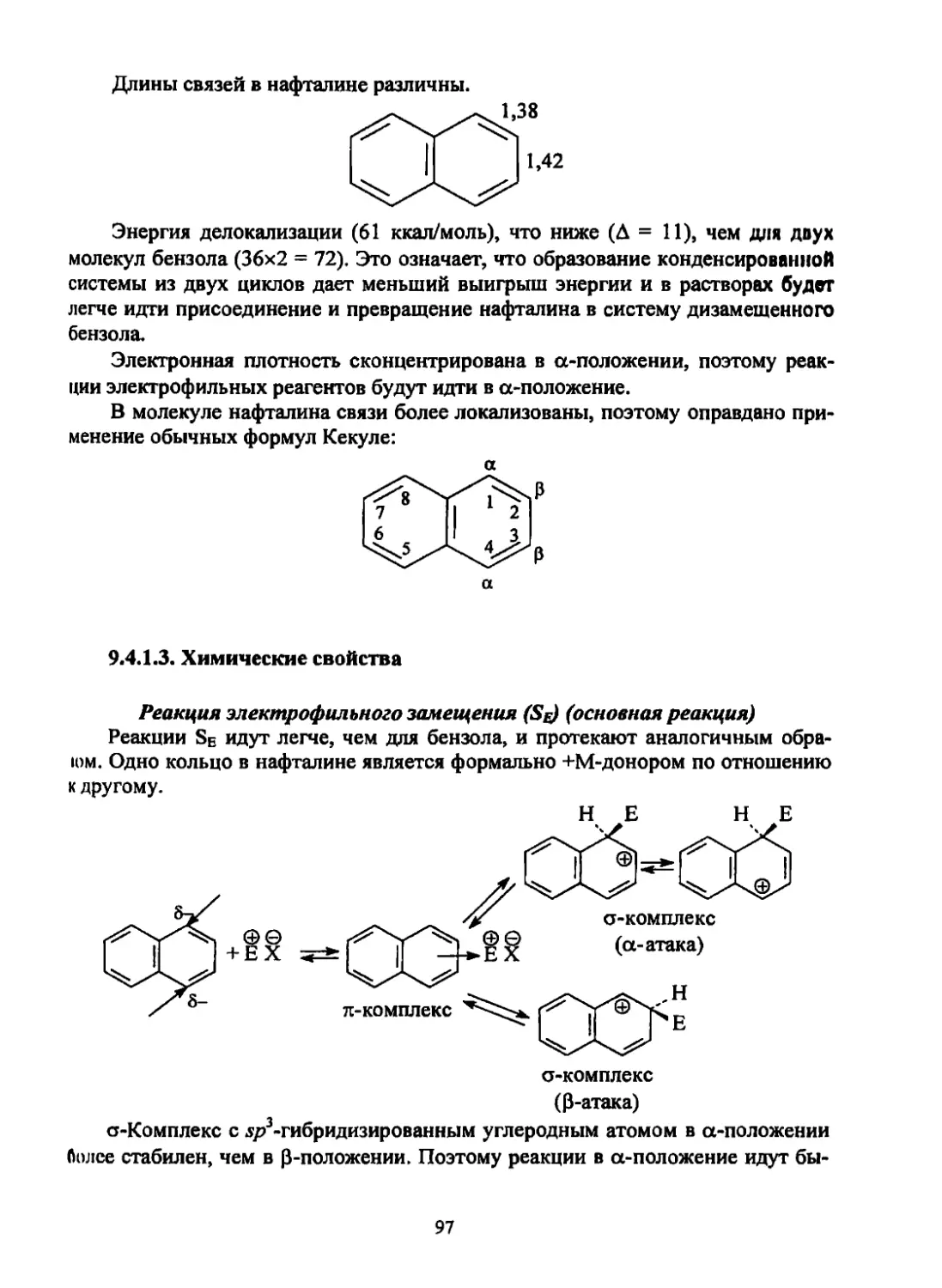
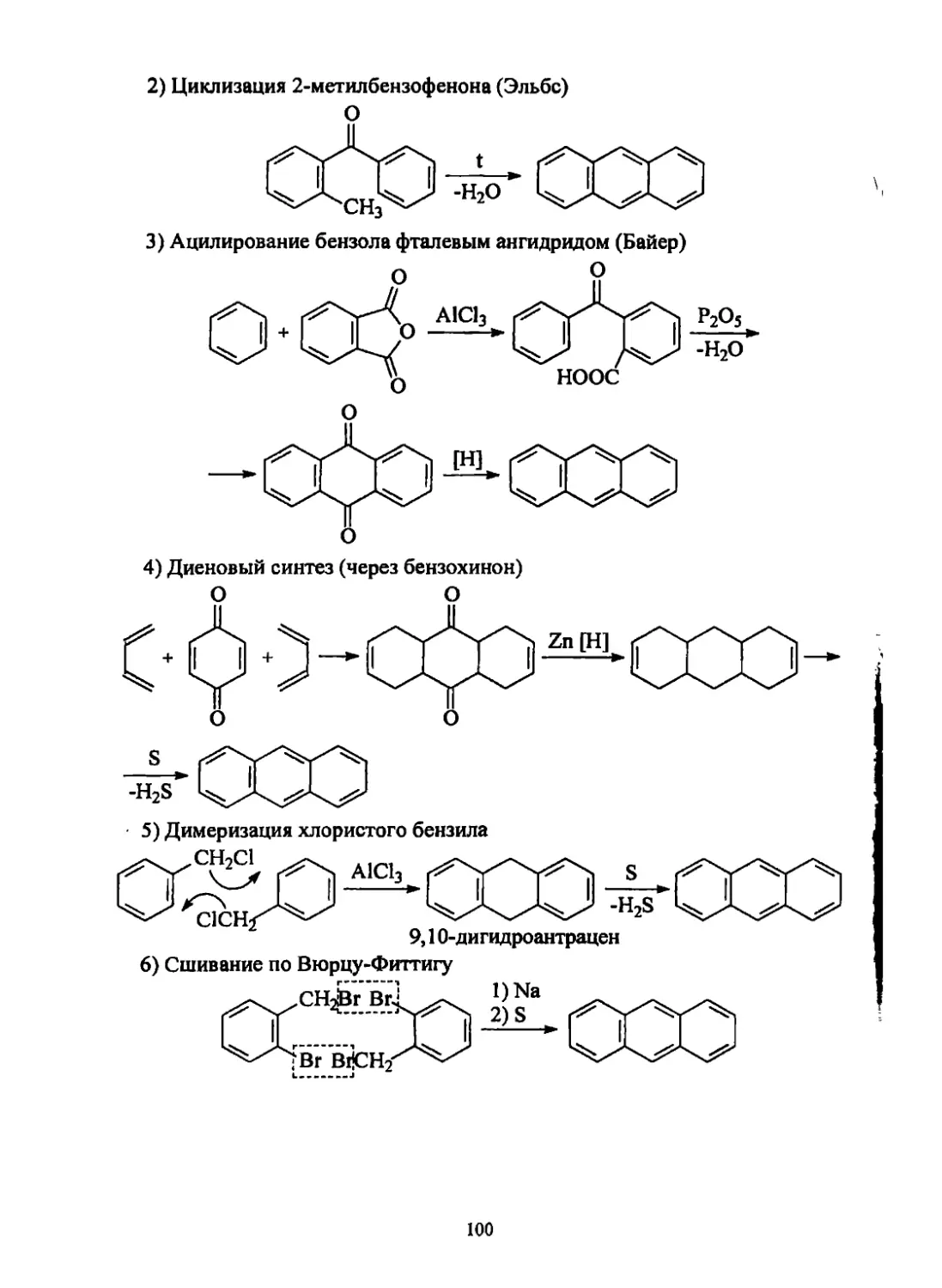
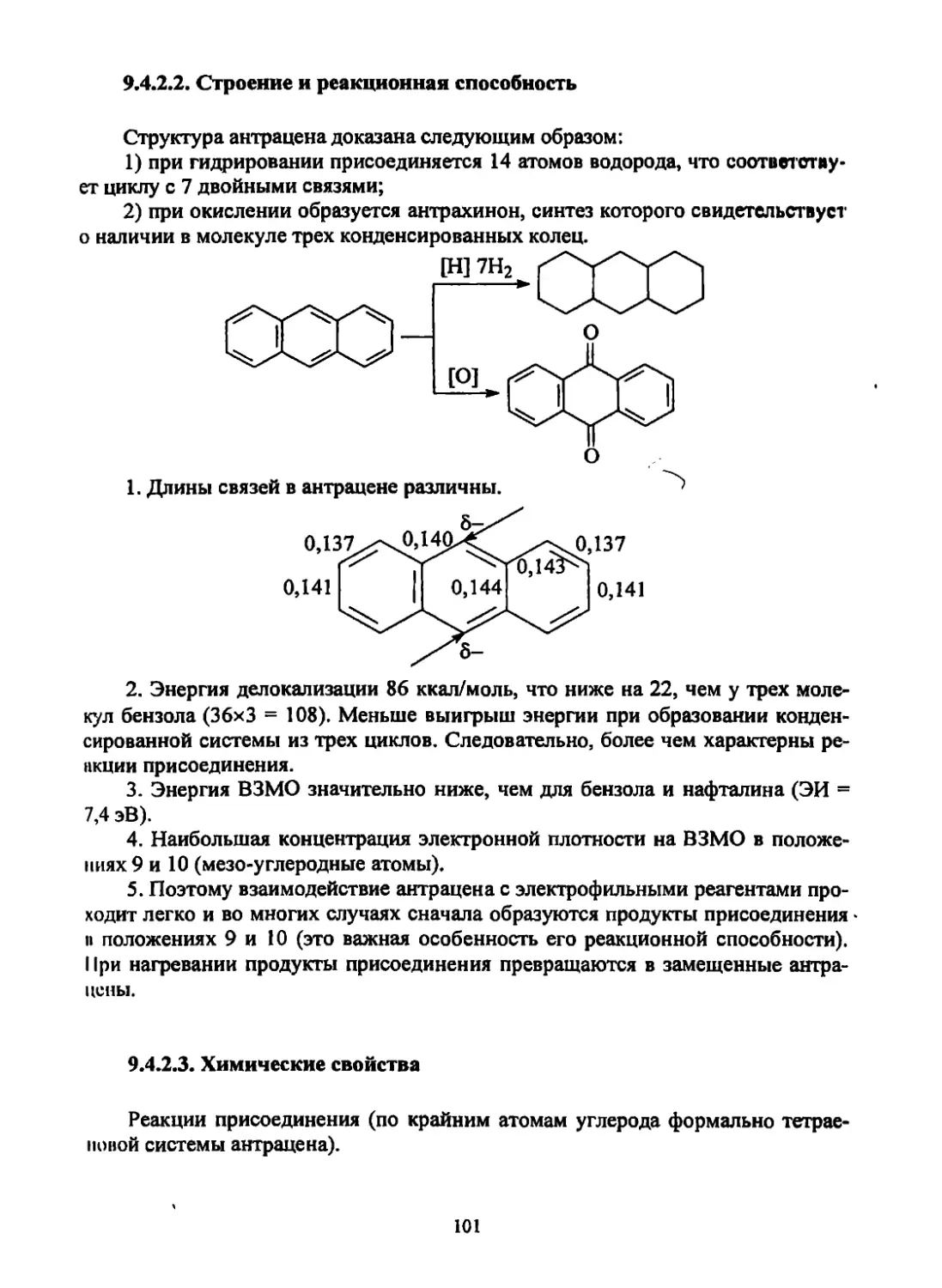
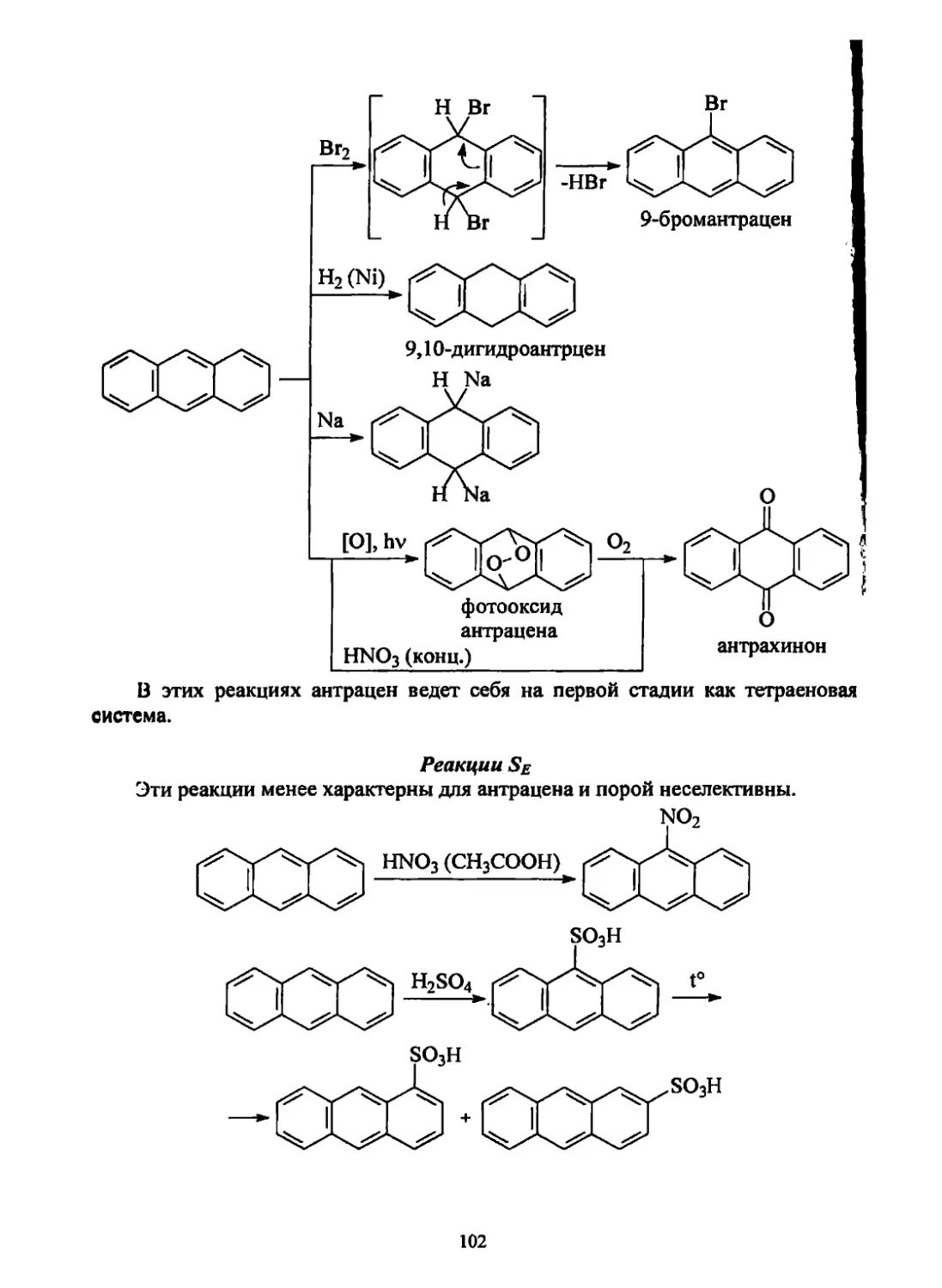
/

















































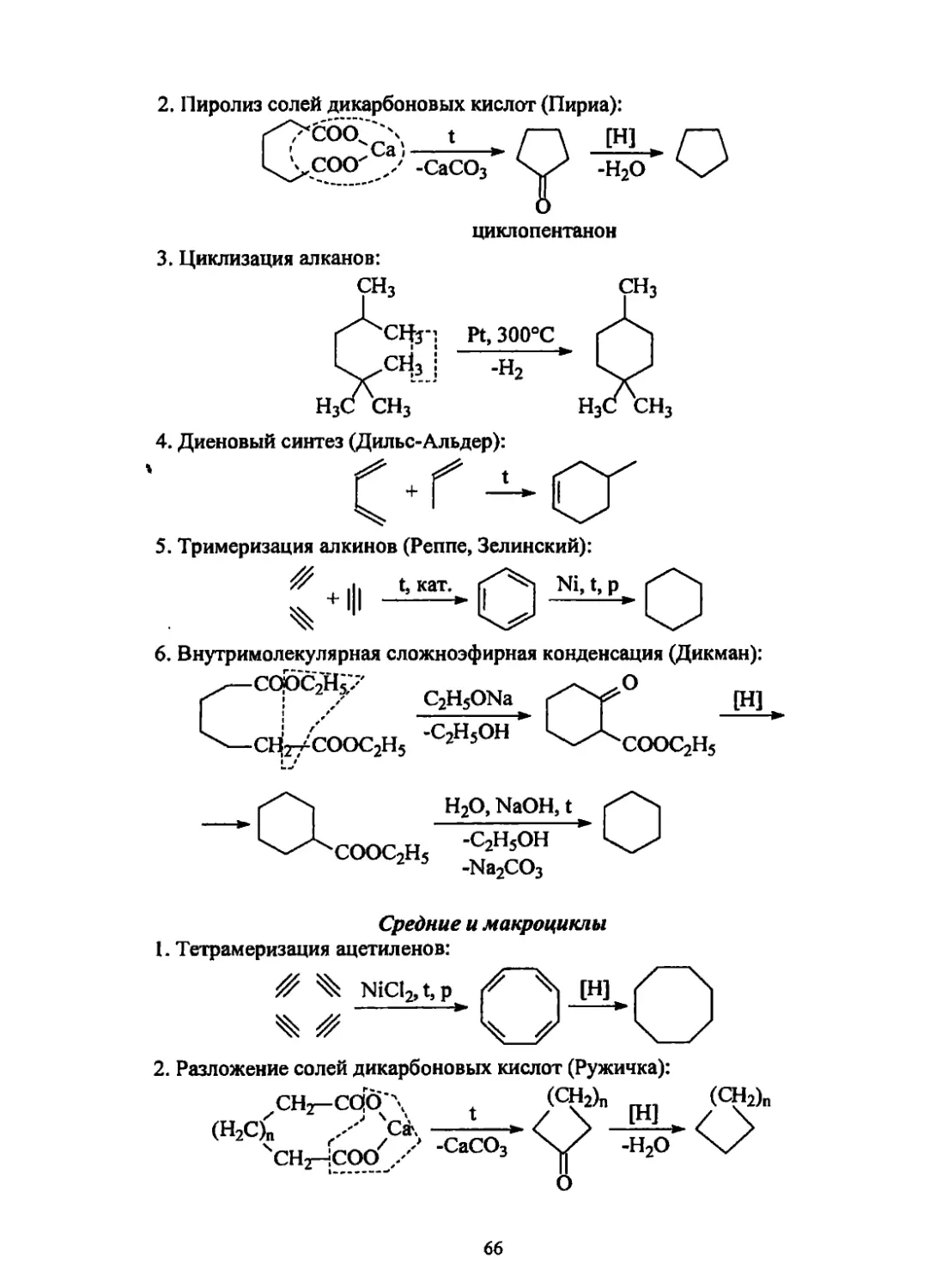


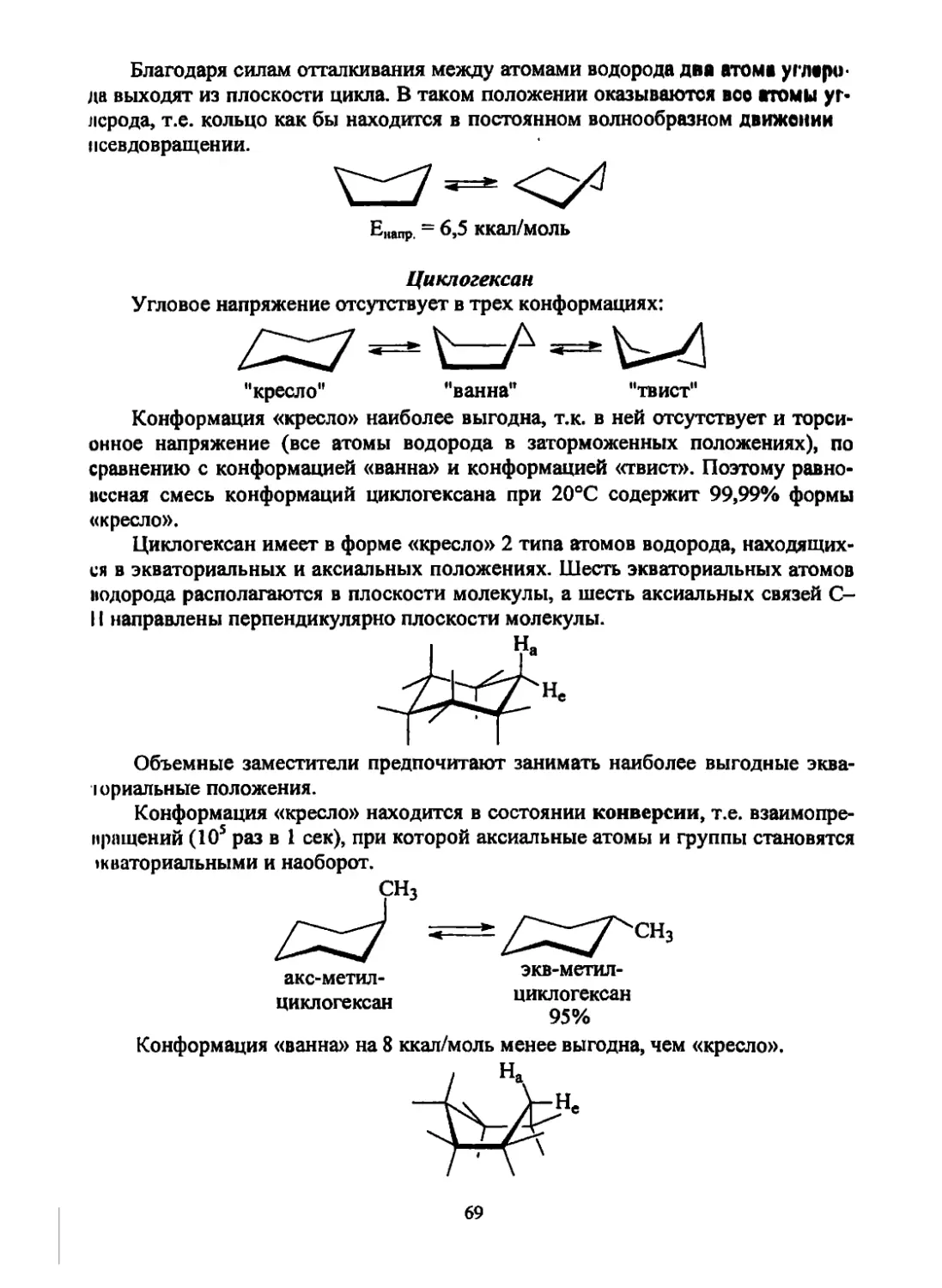

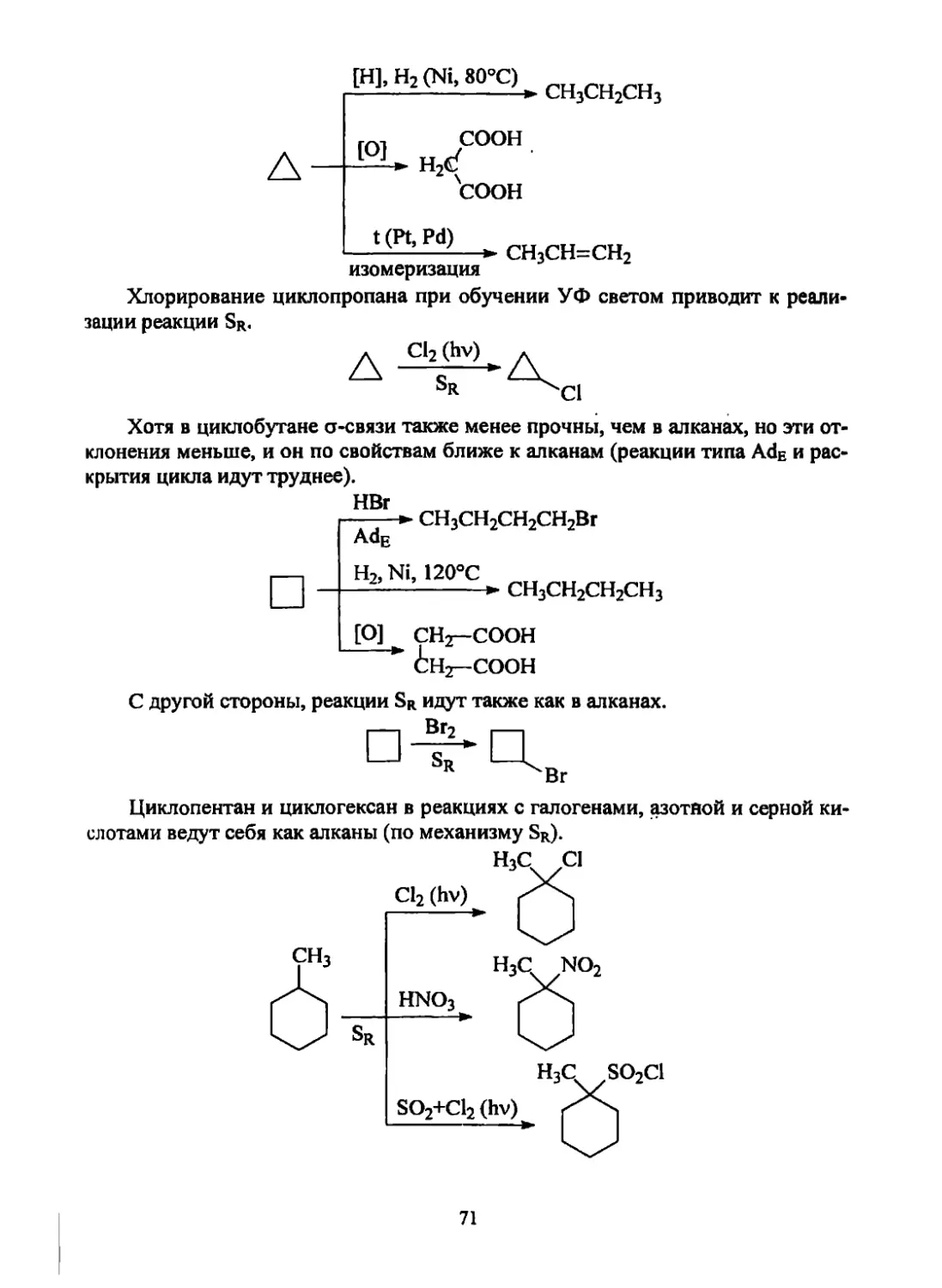
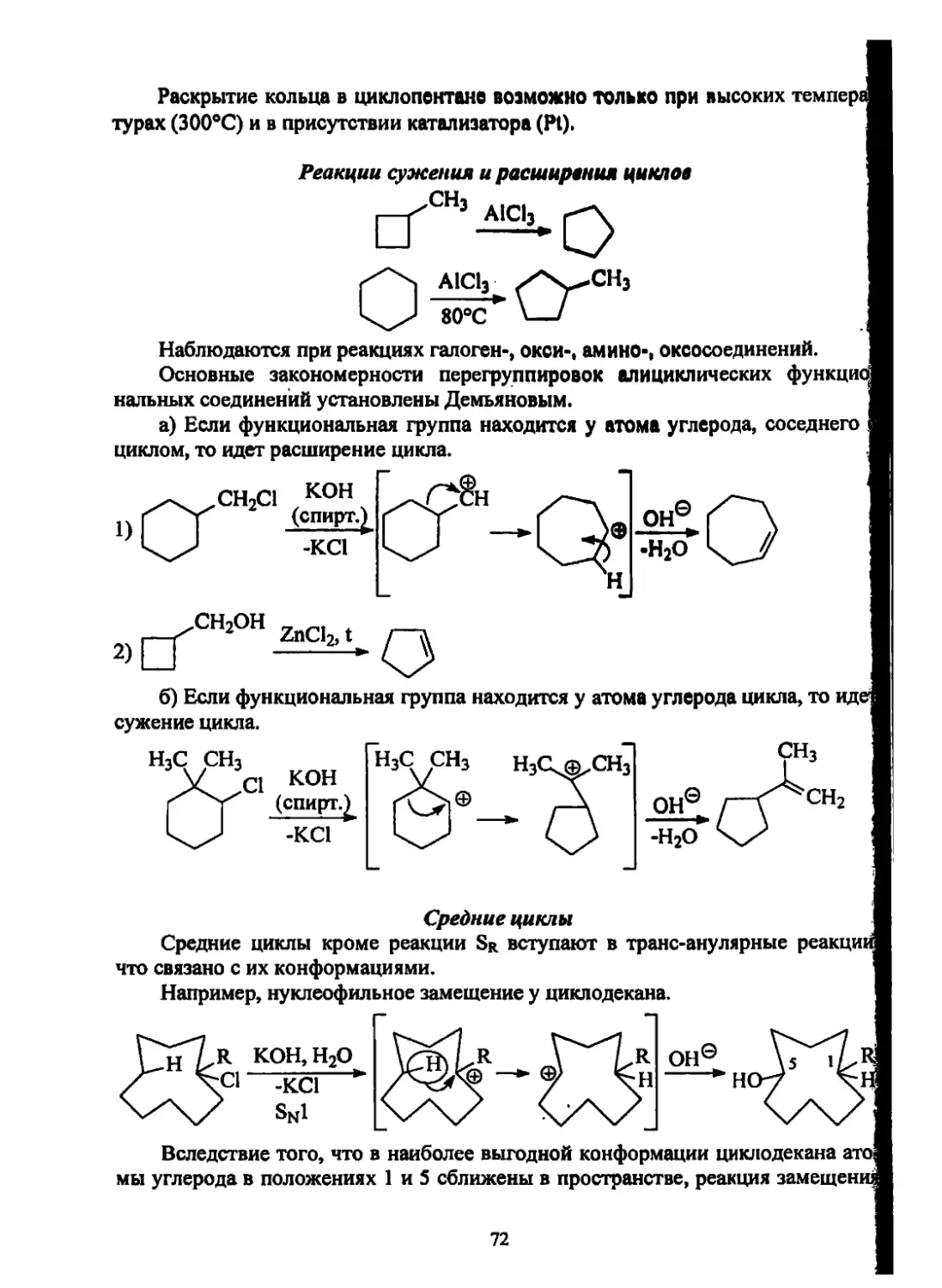



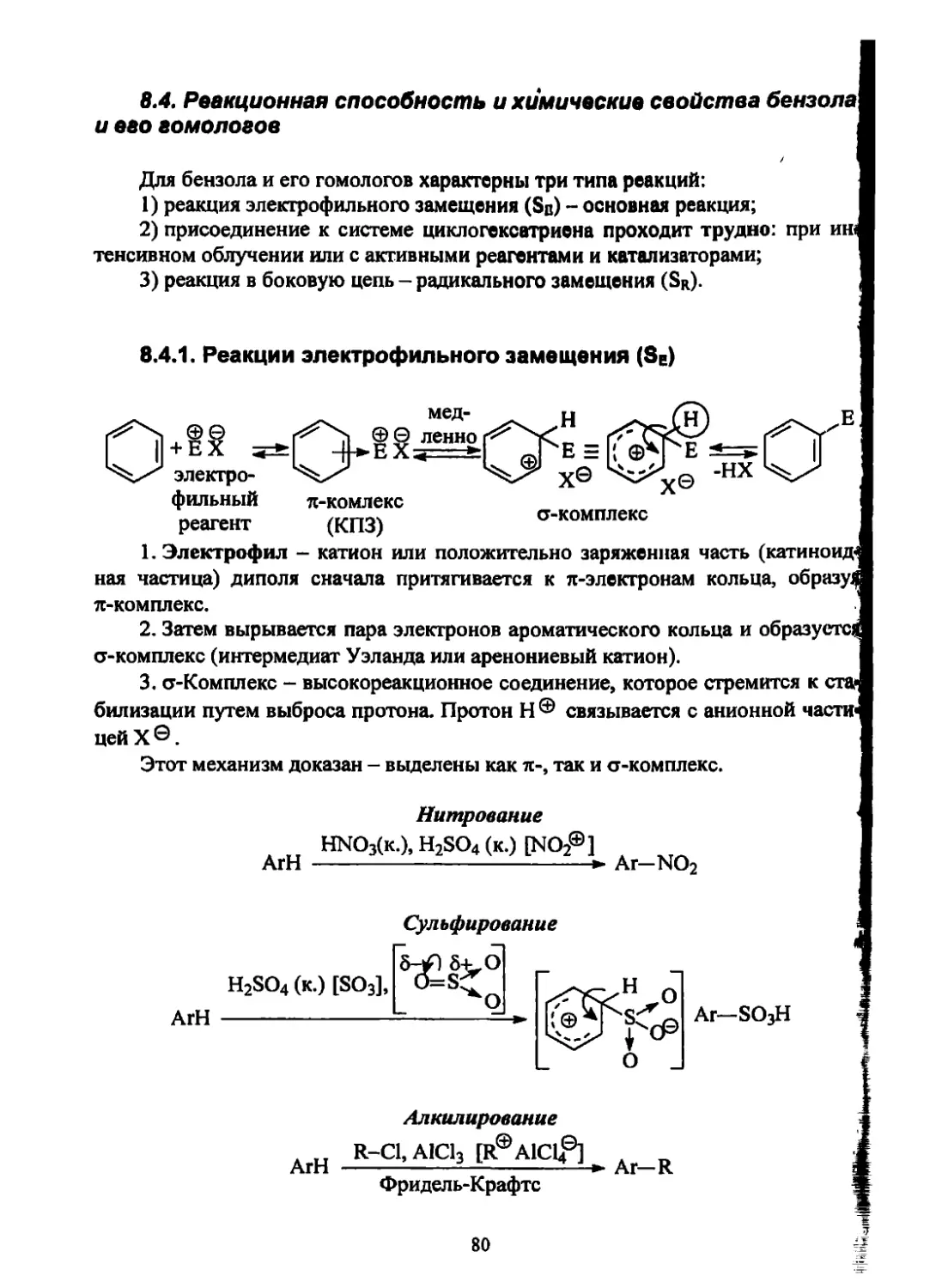

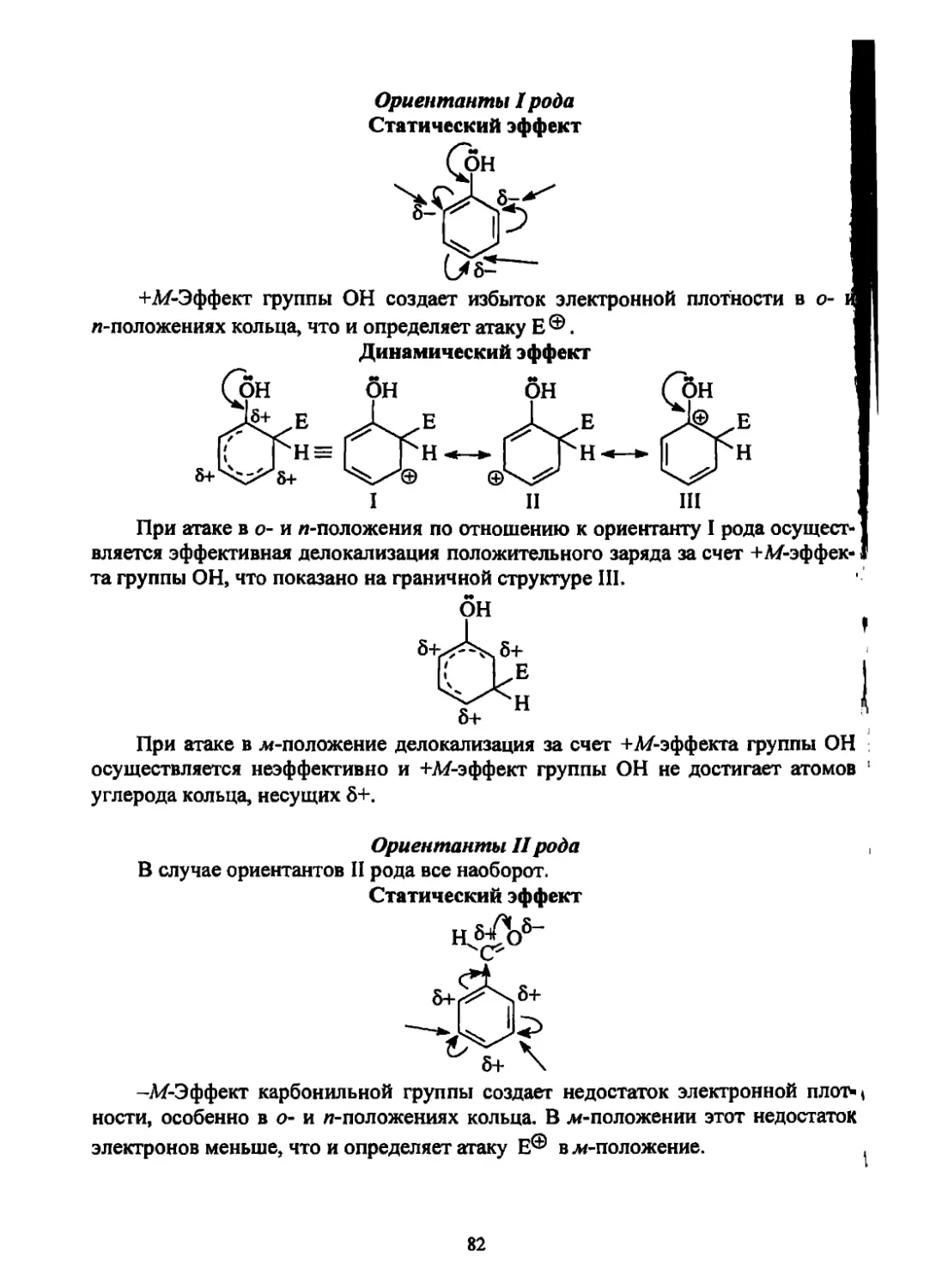
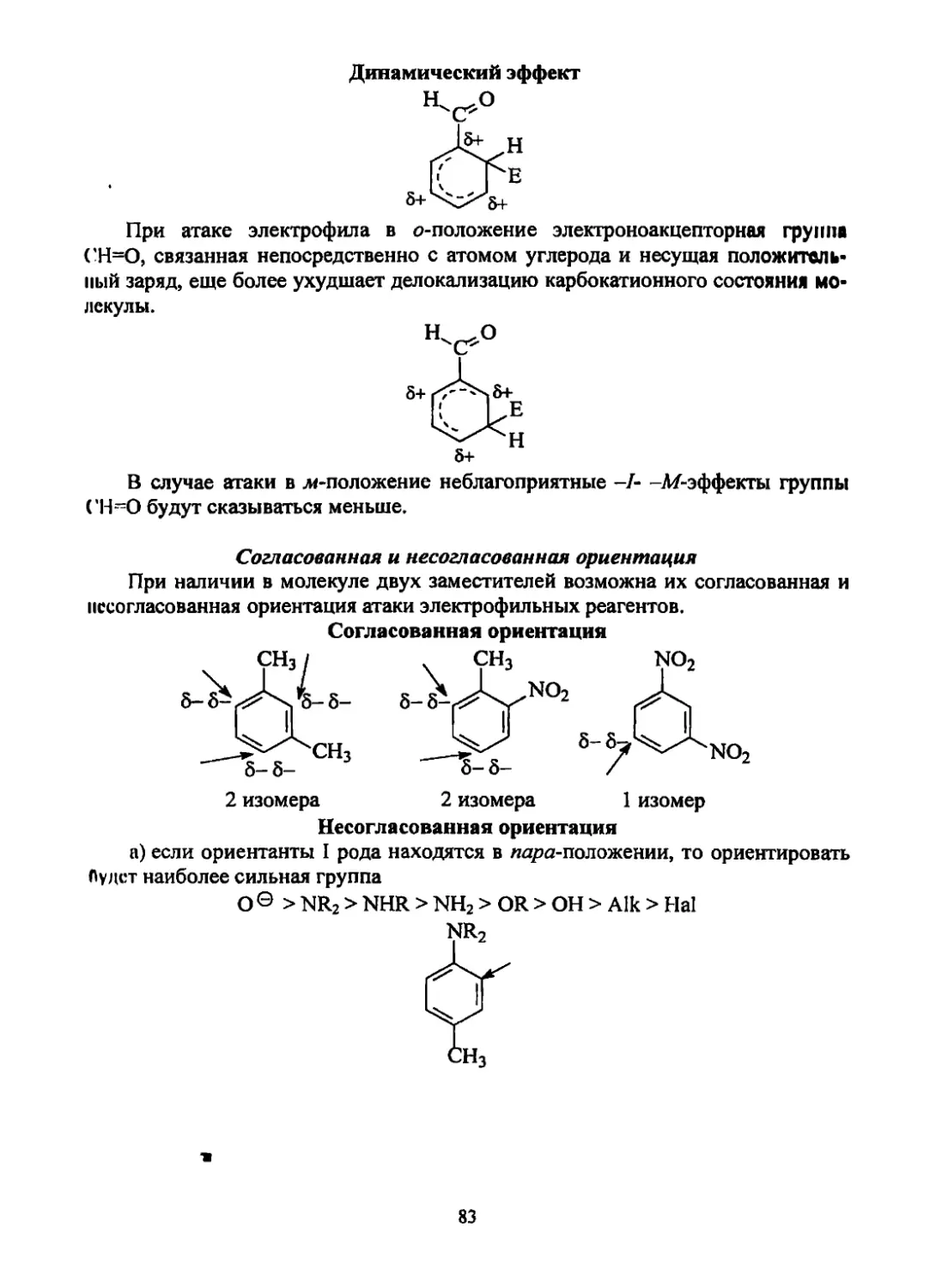
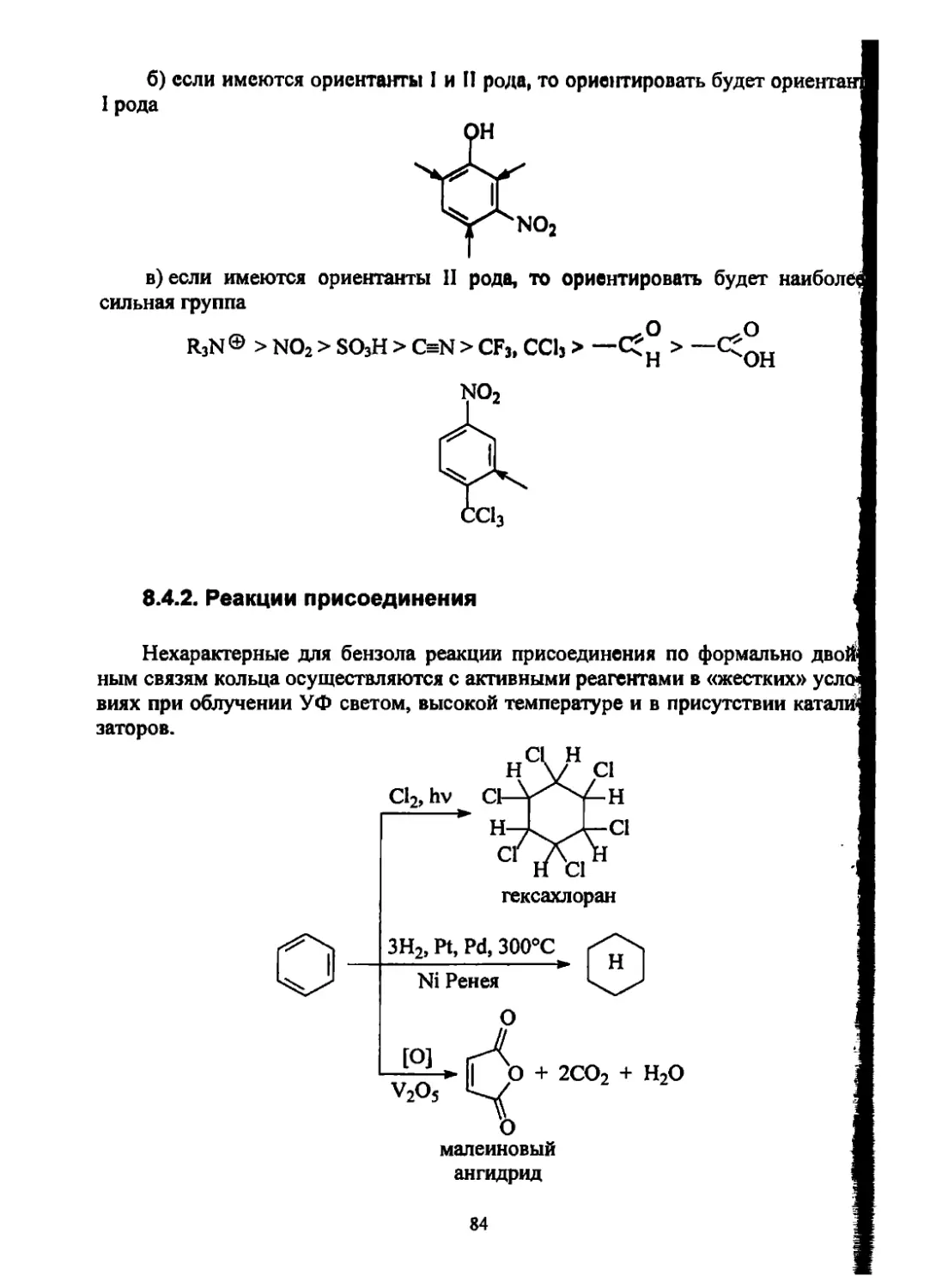
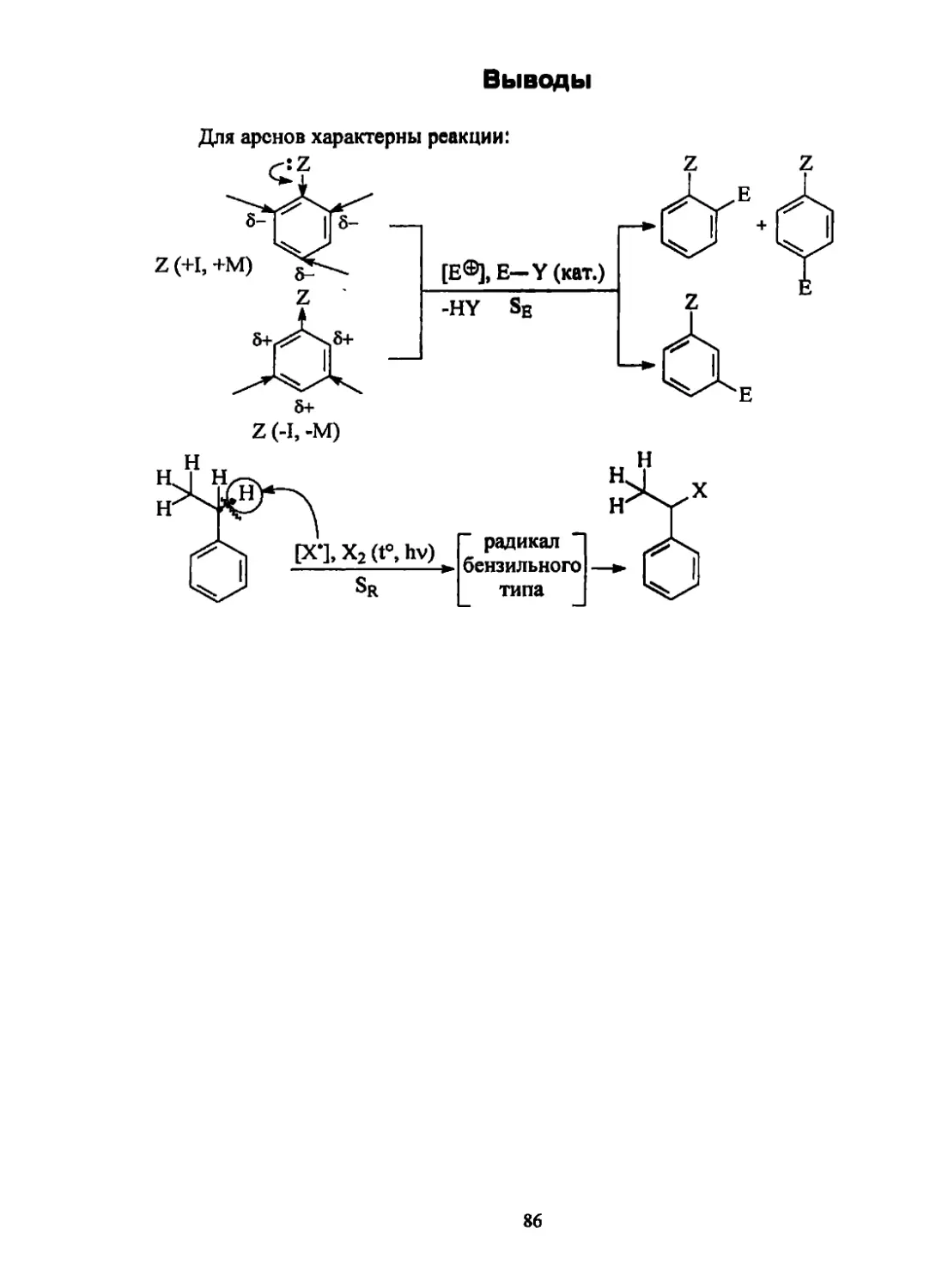
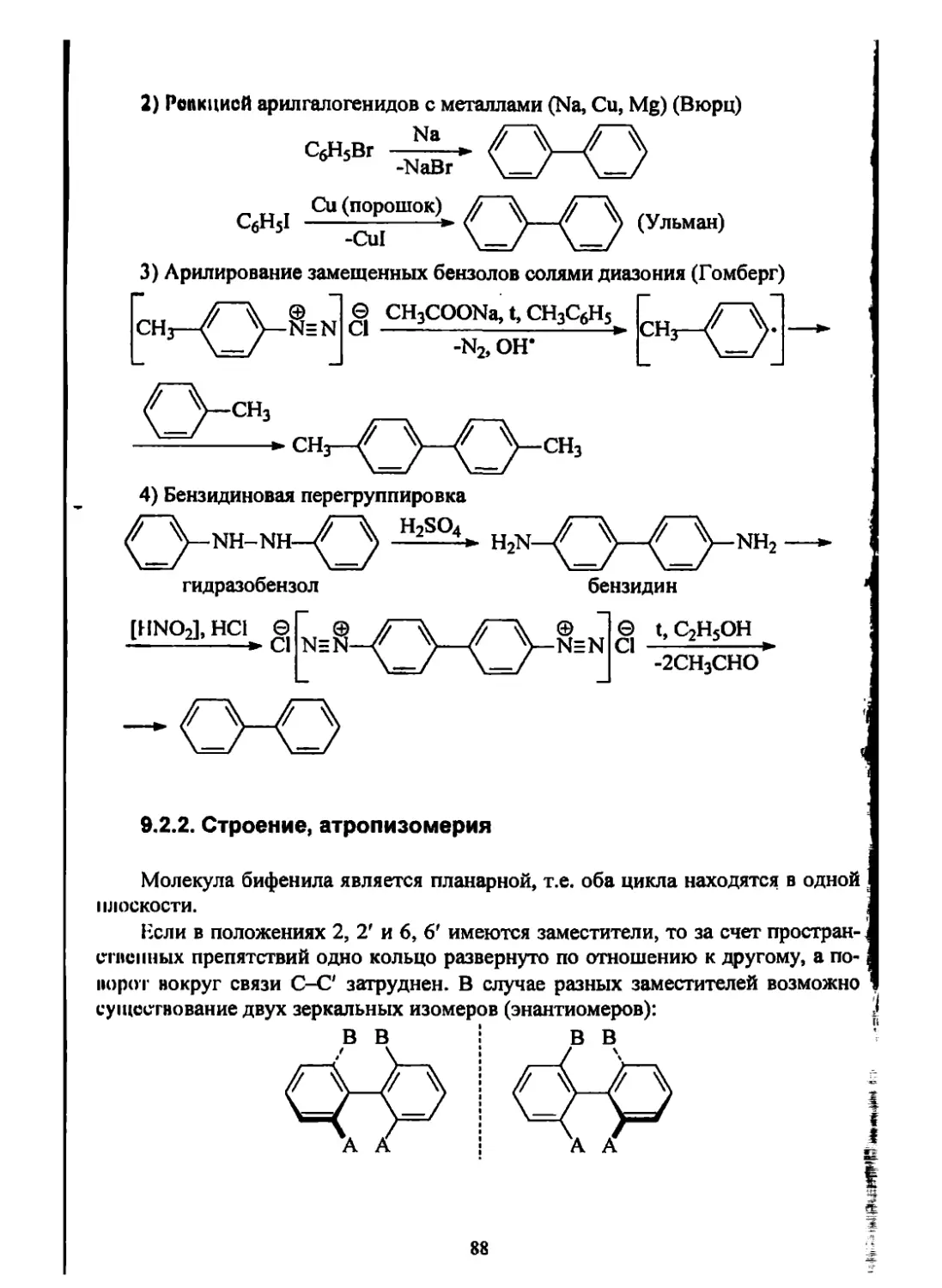
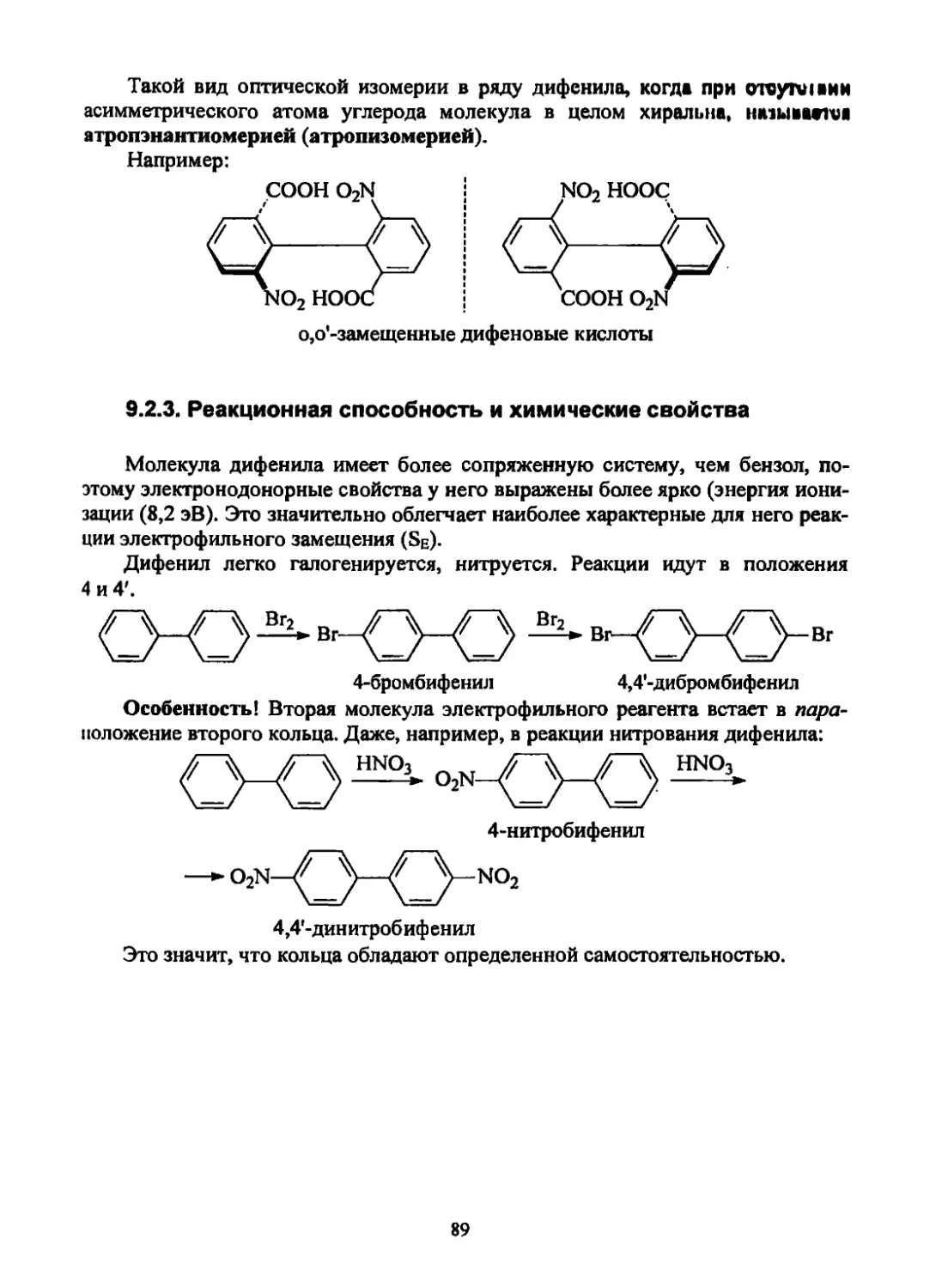

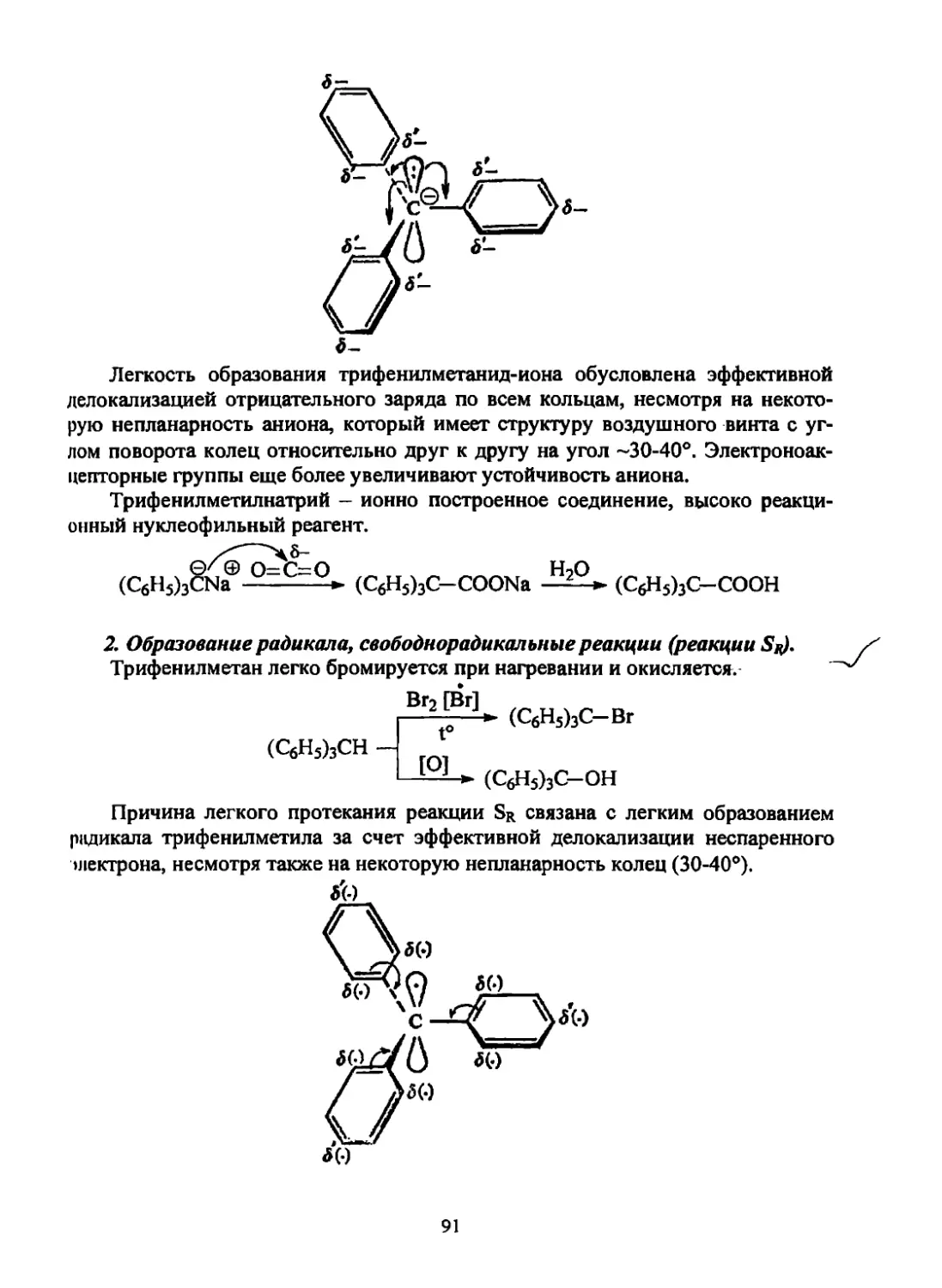


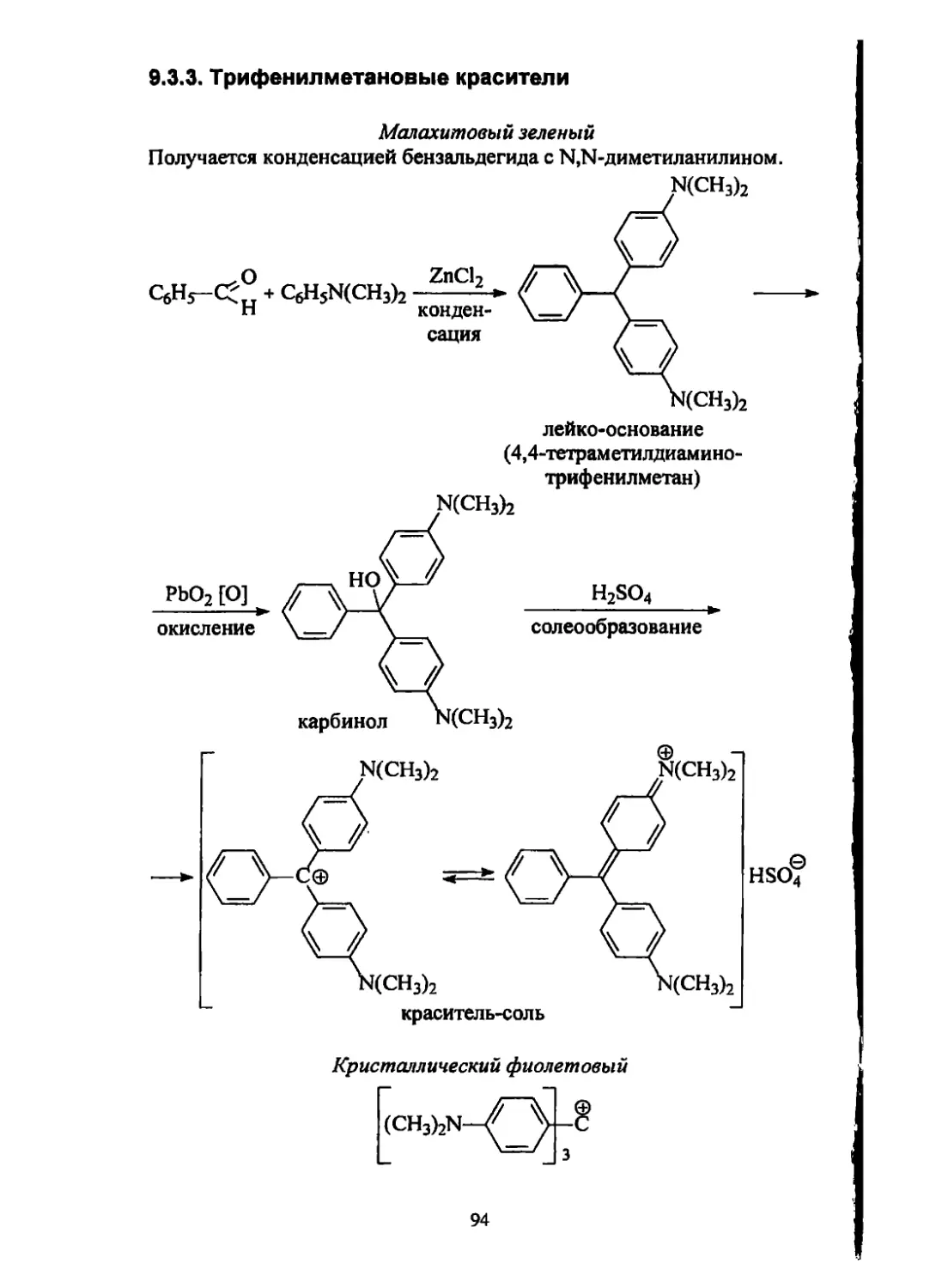

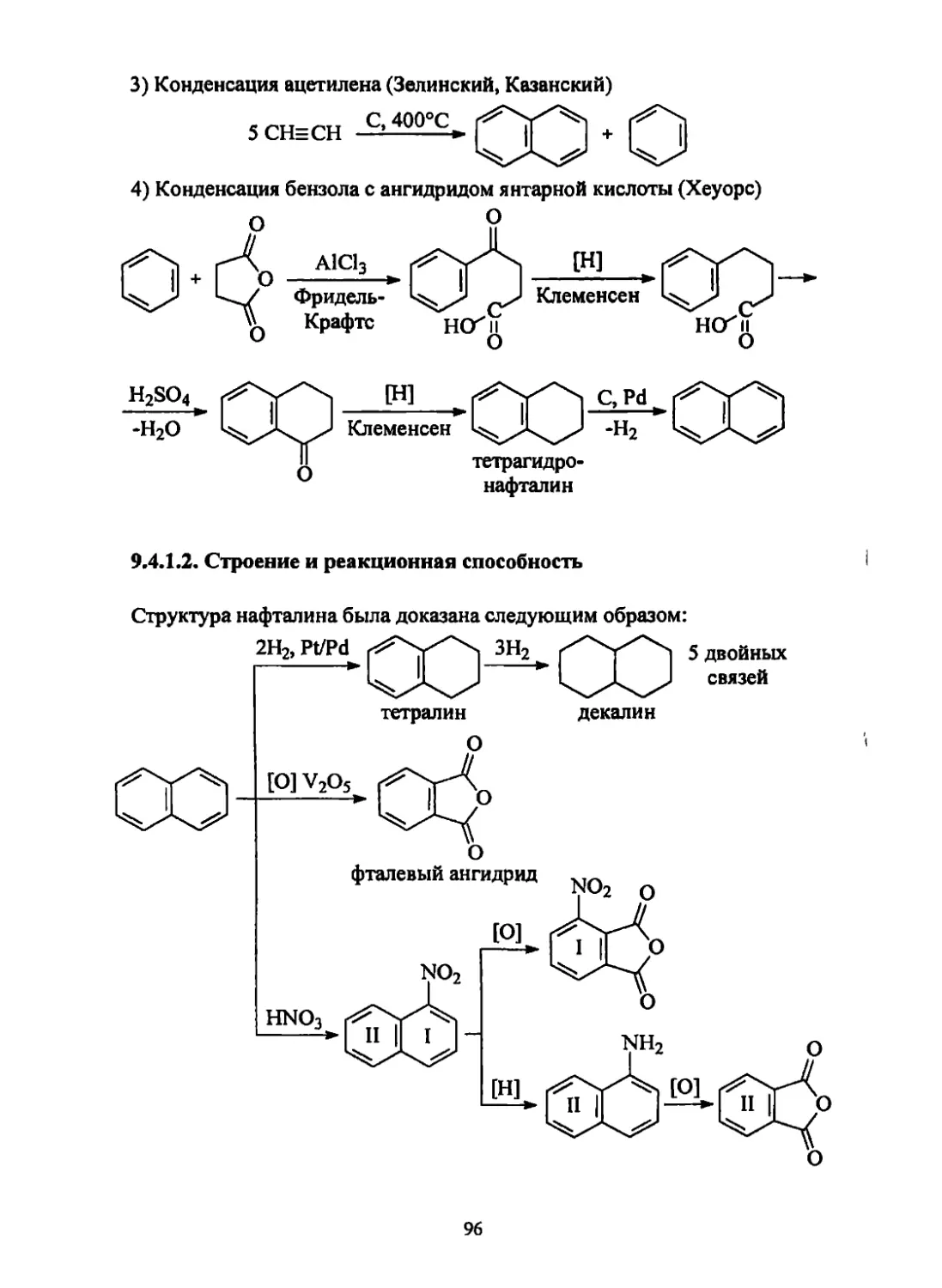


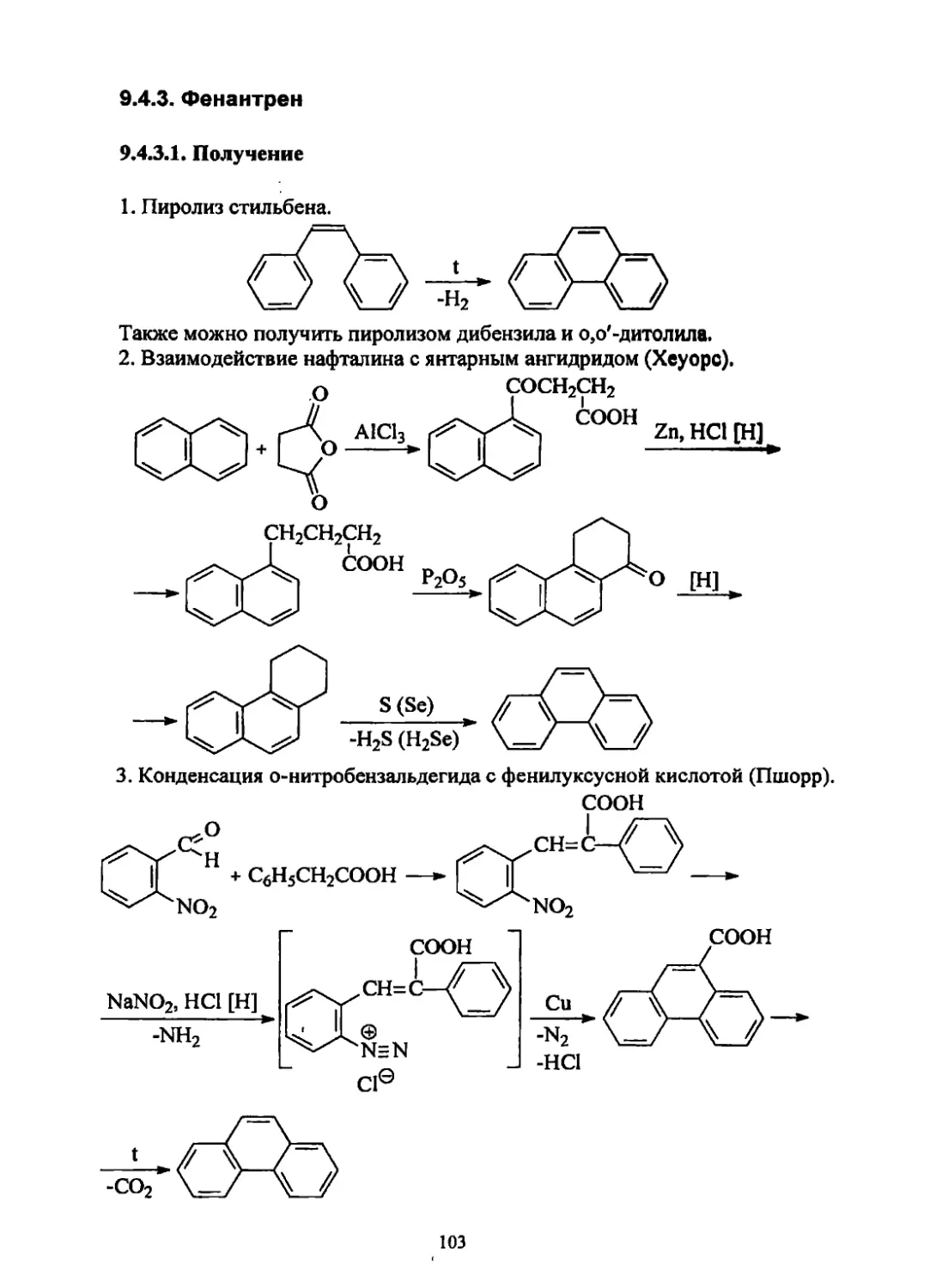
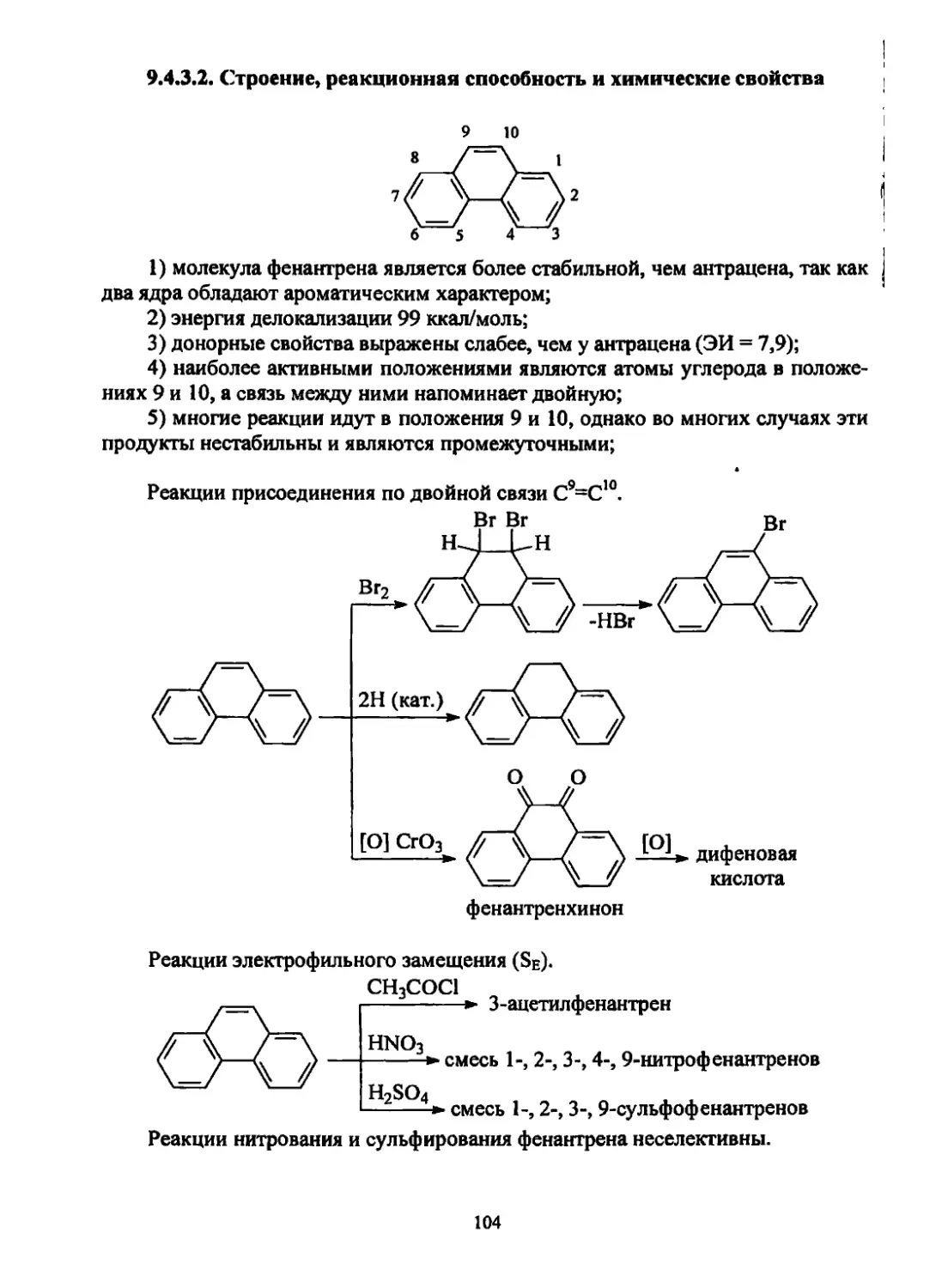
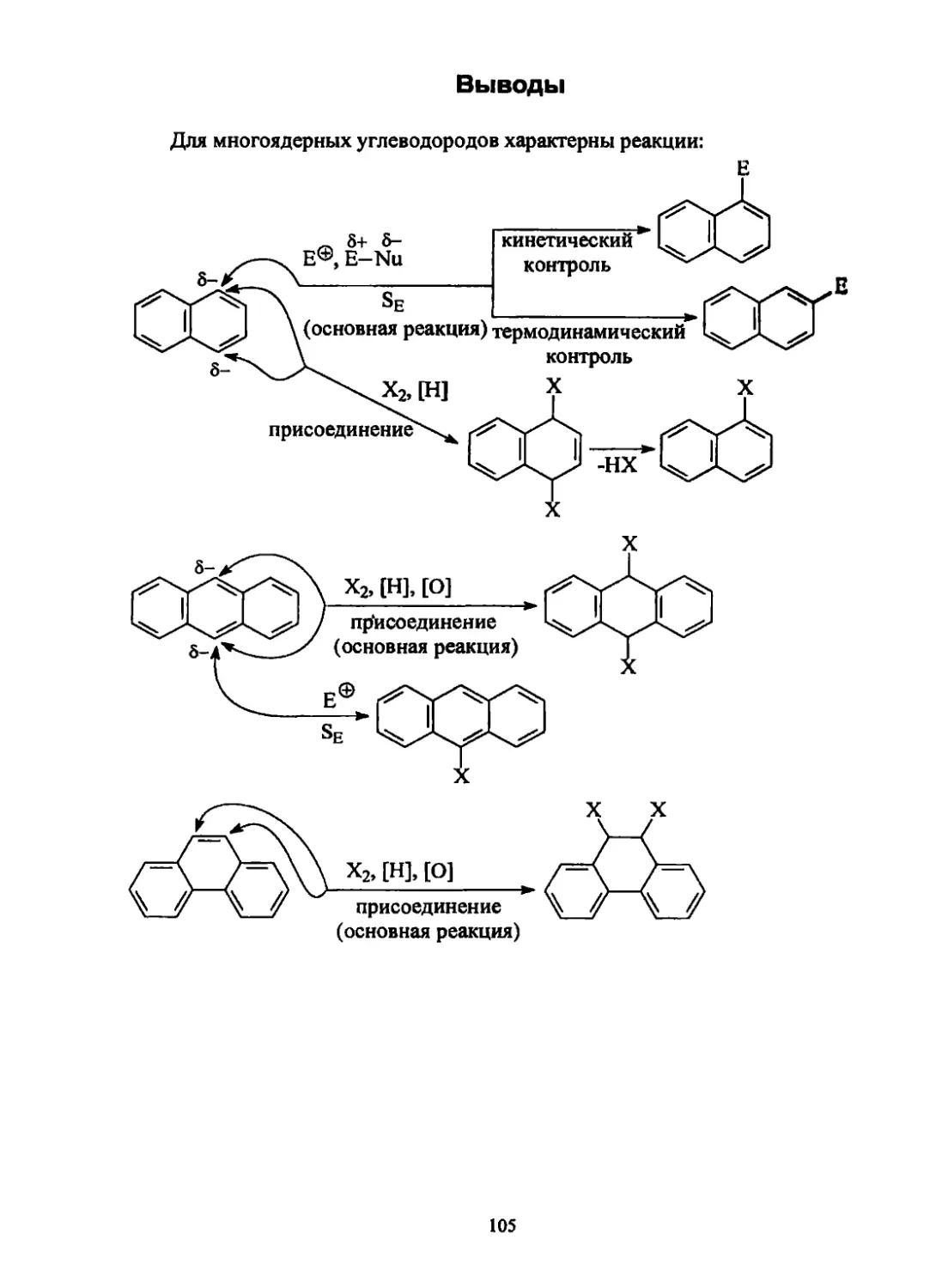







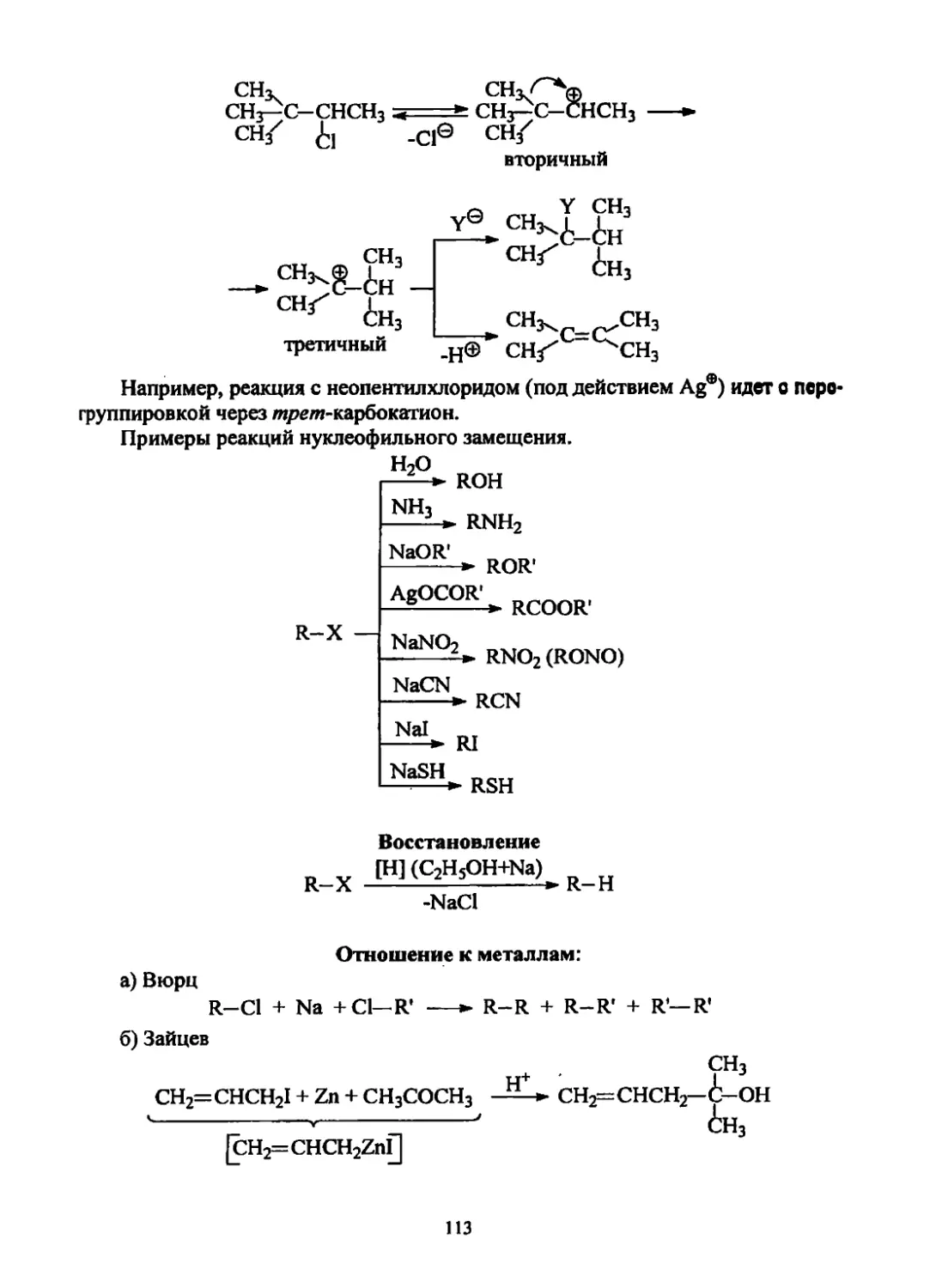











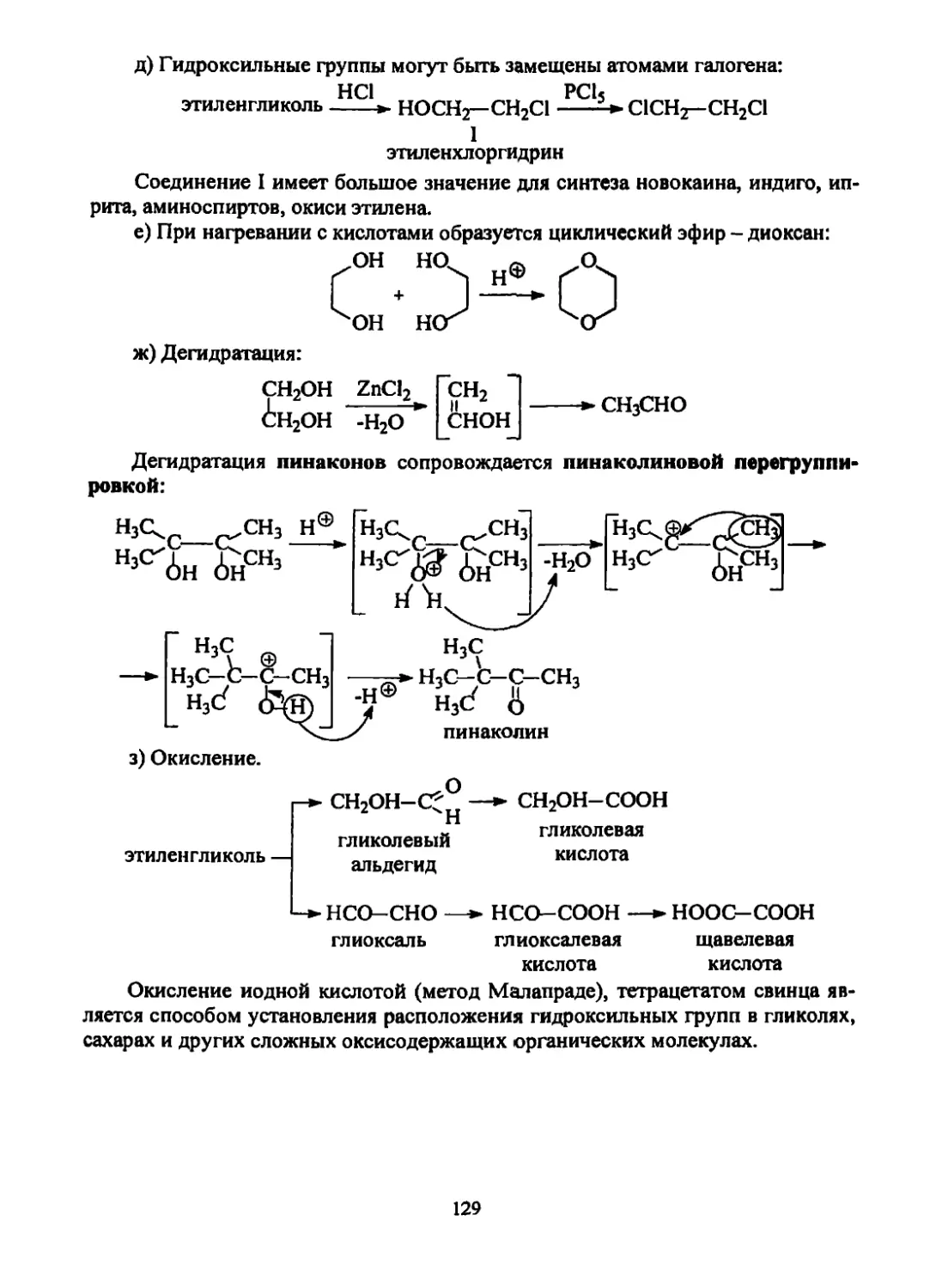


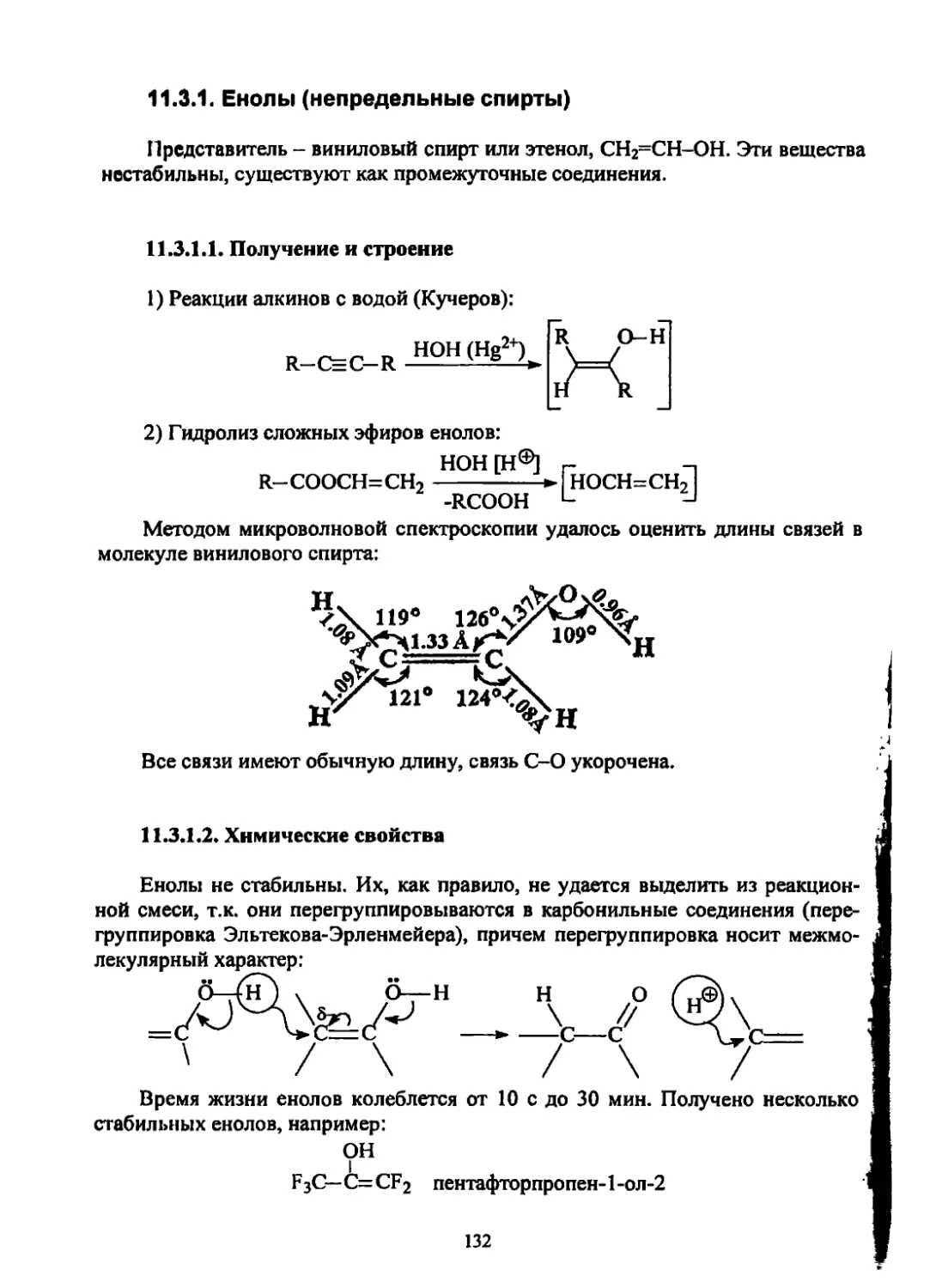

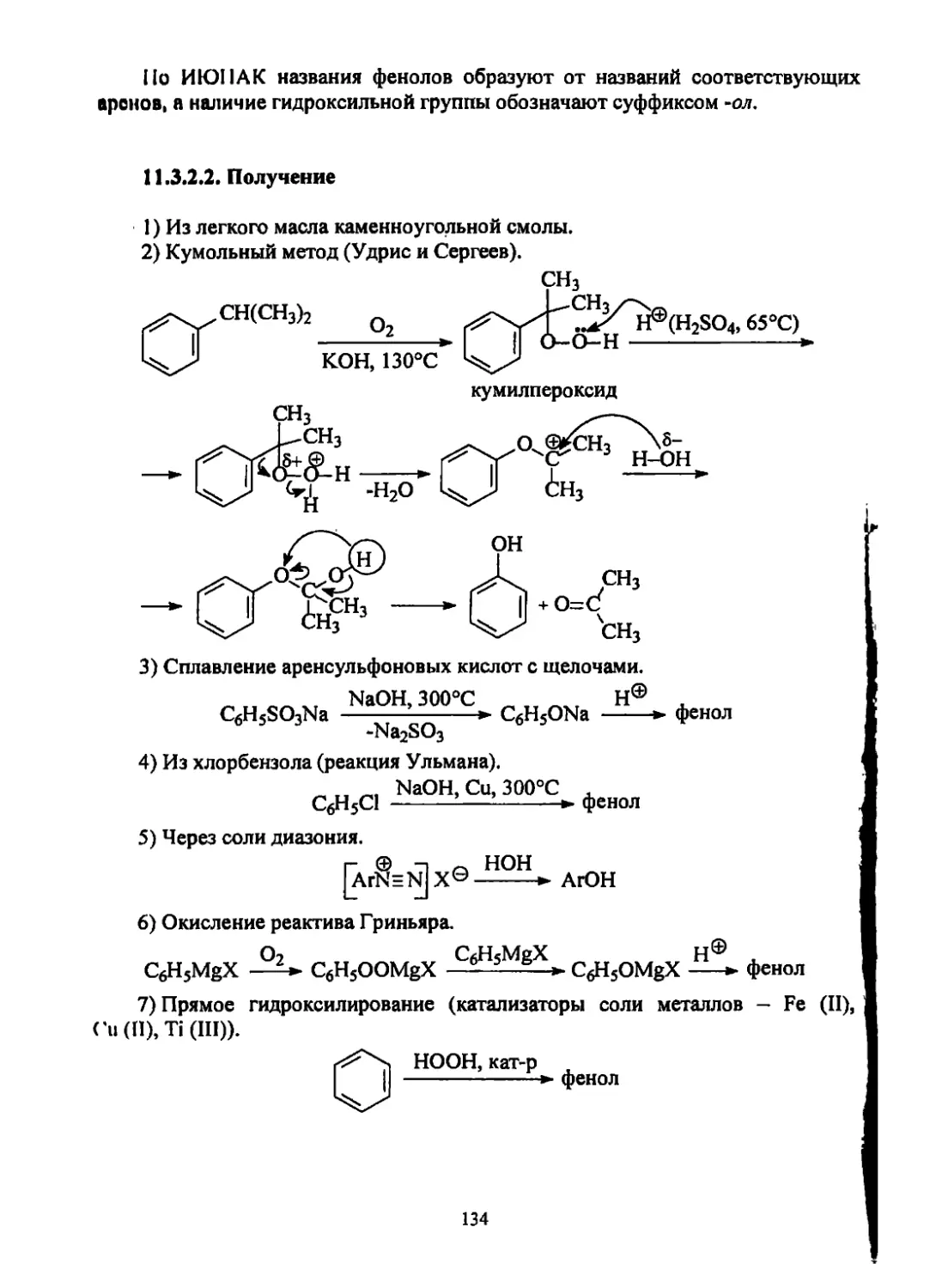
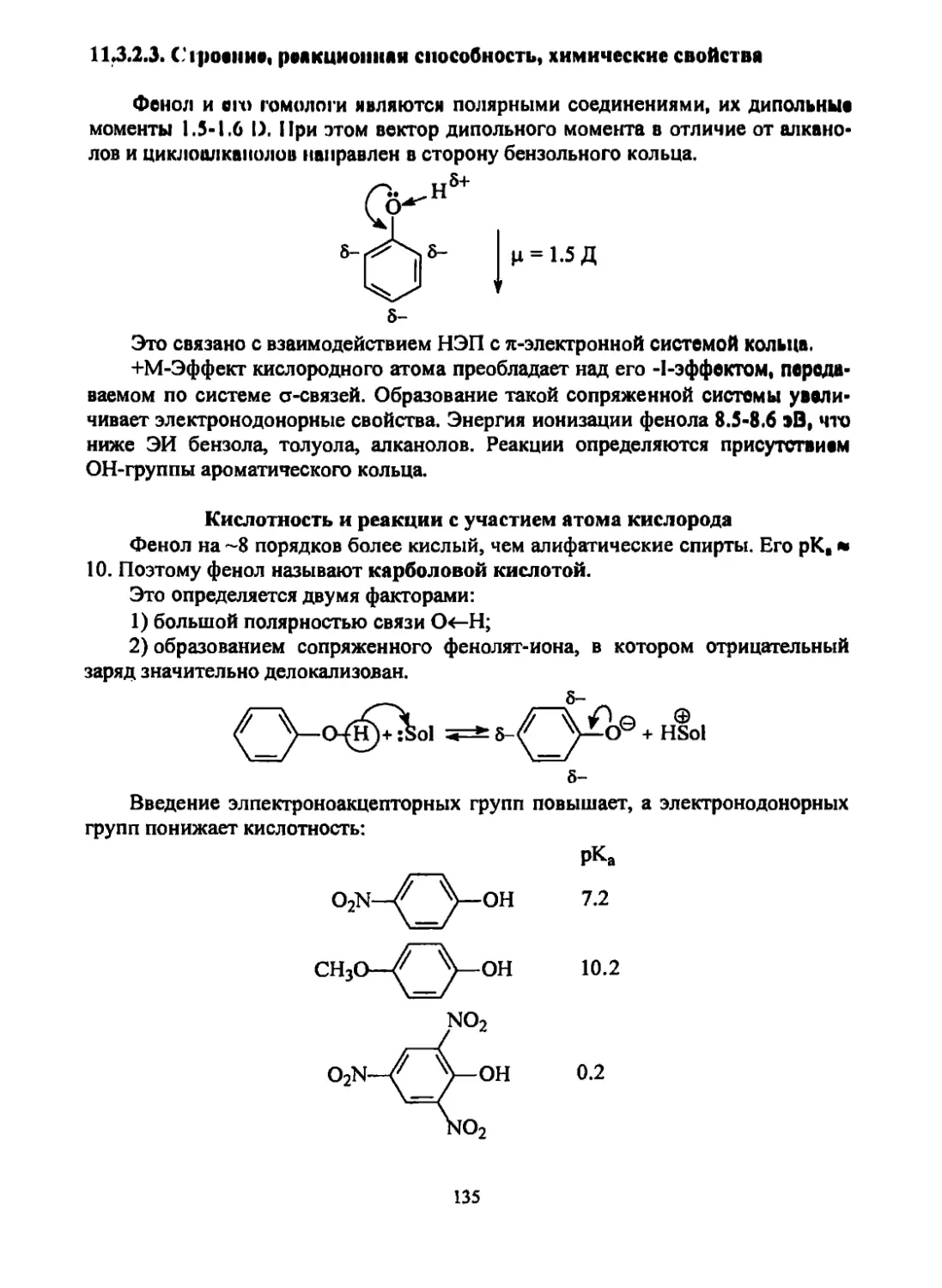
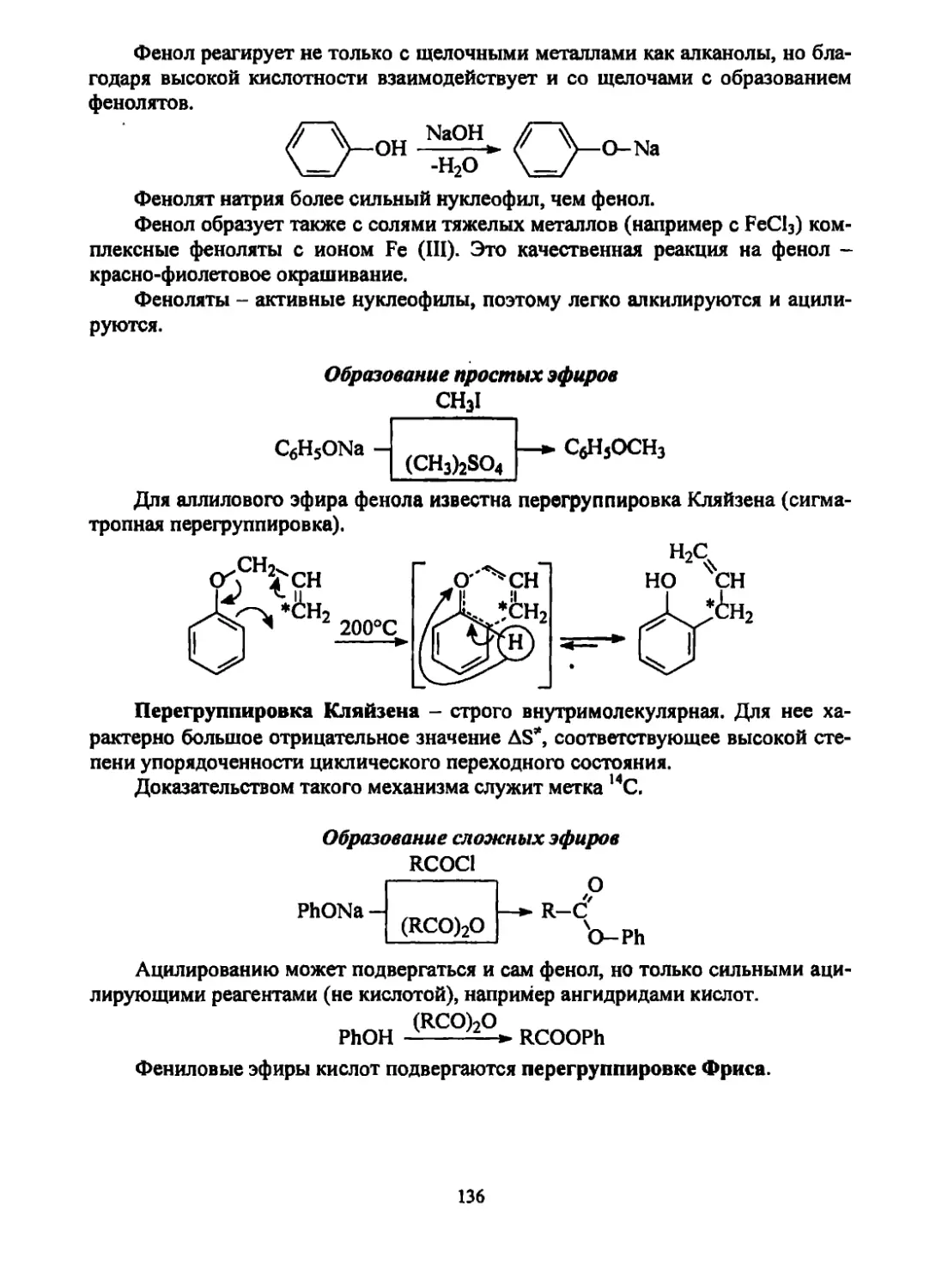
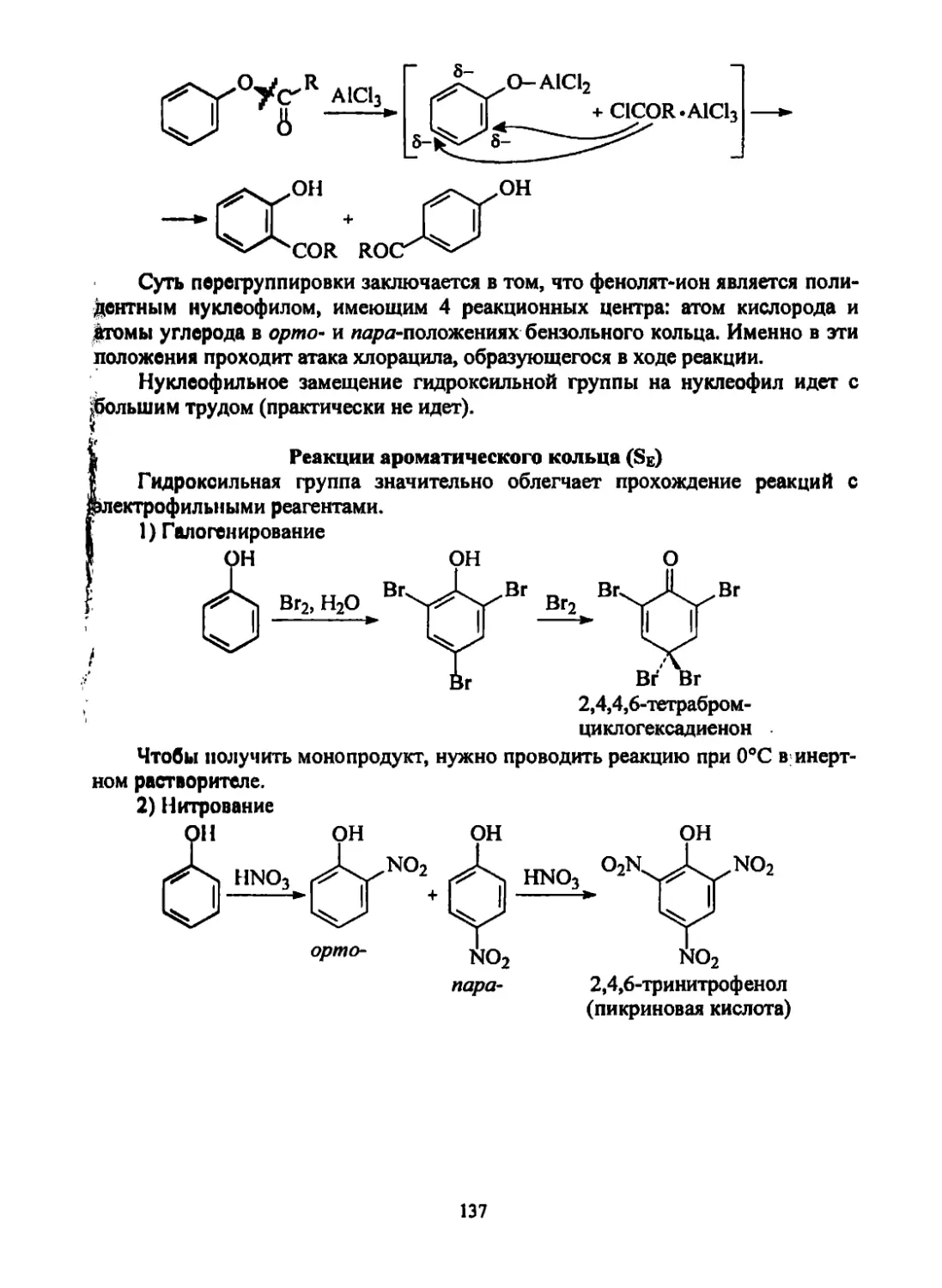
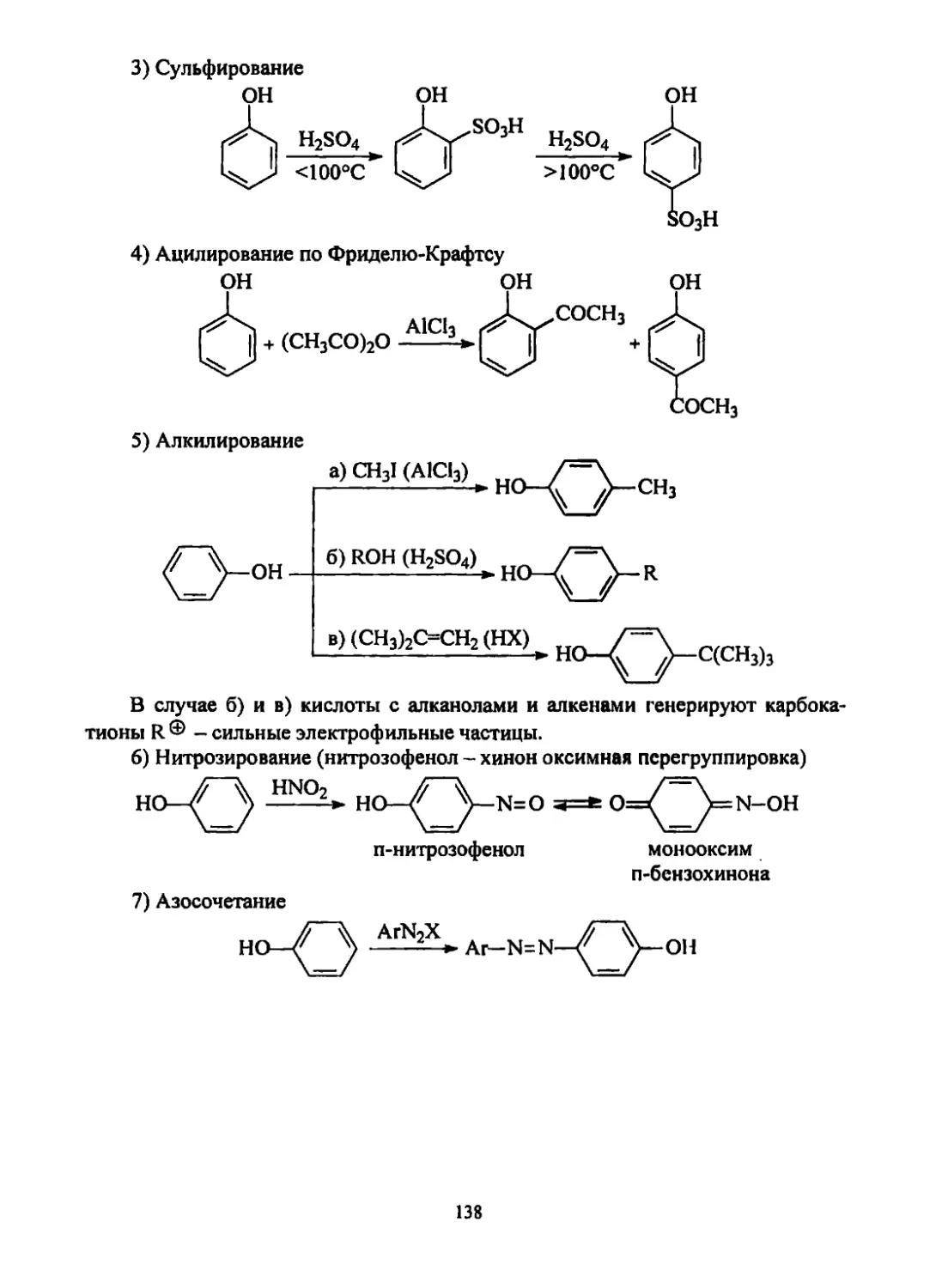
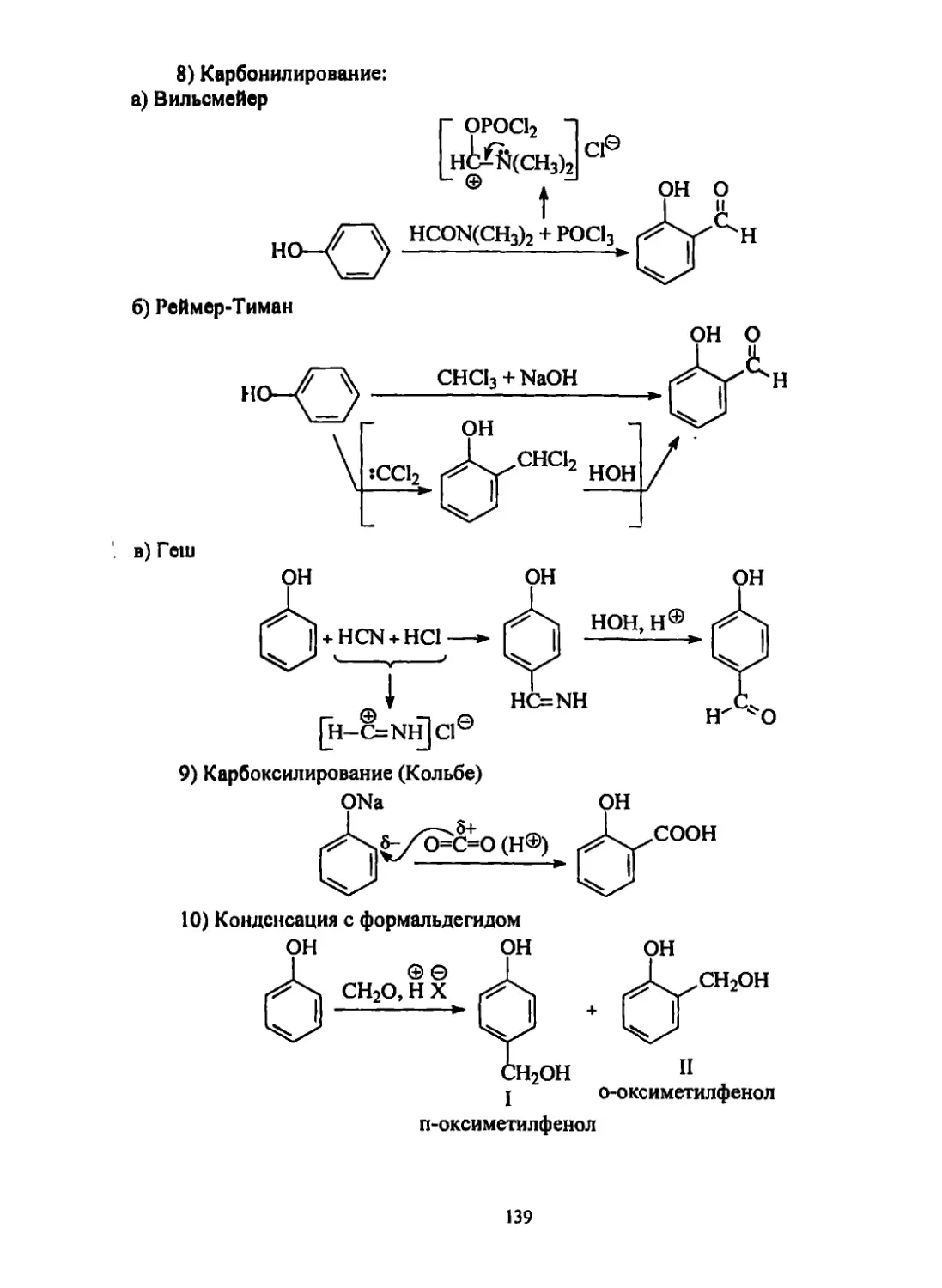
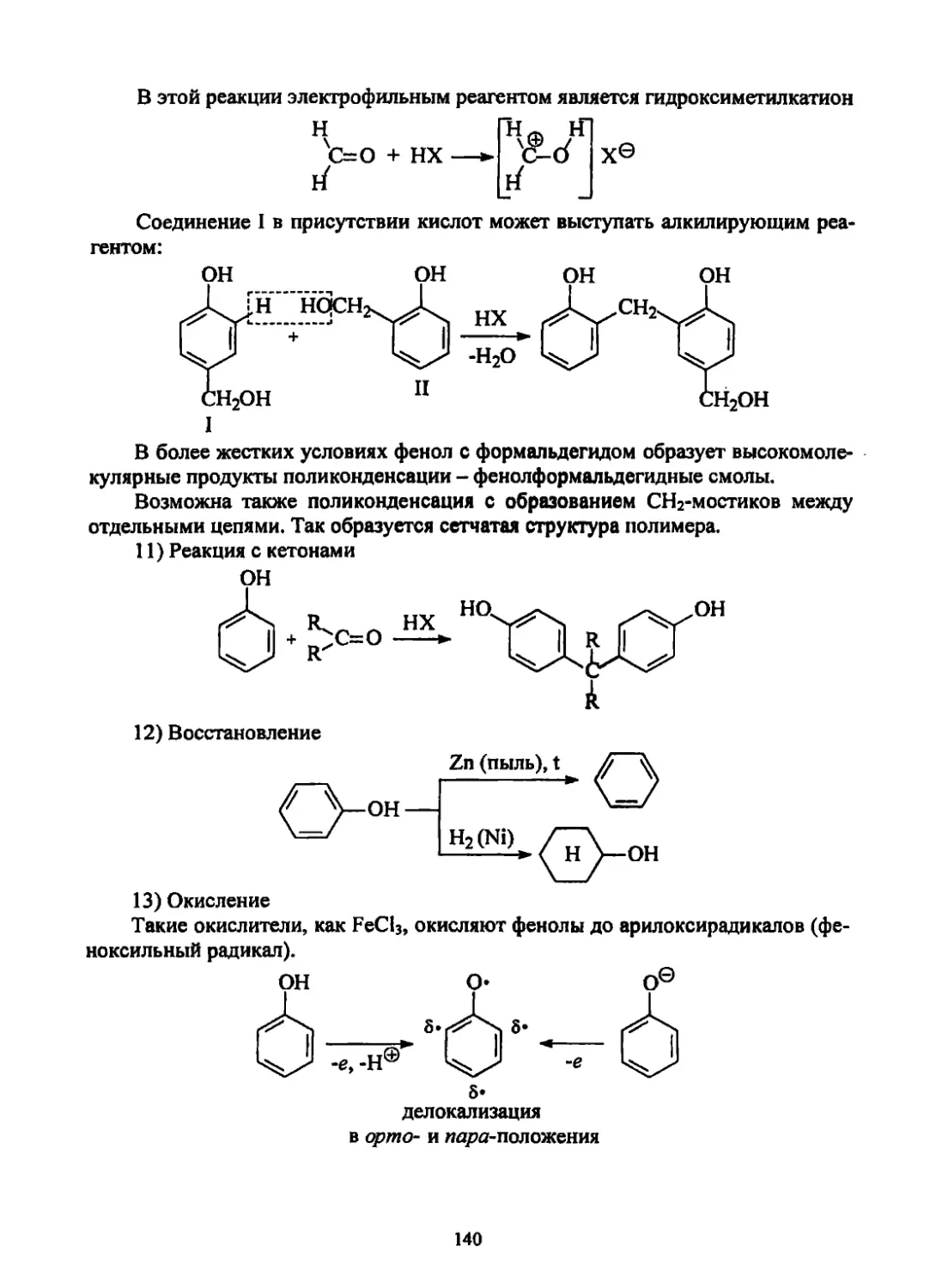
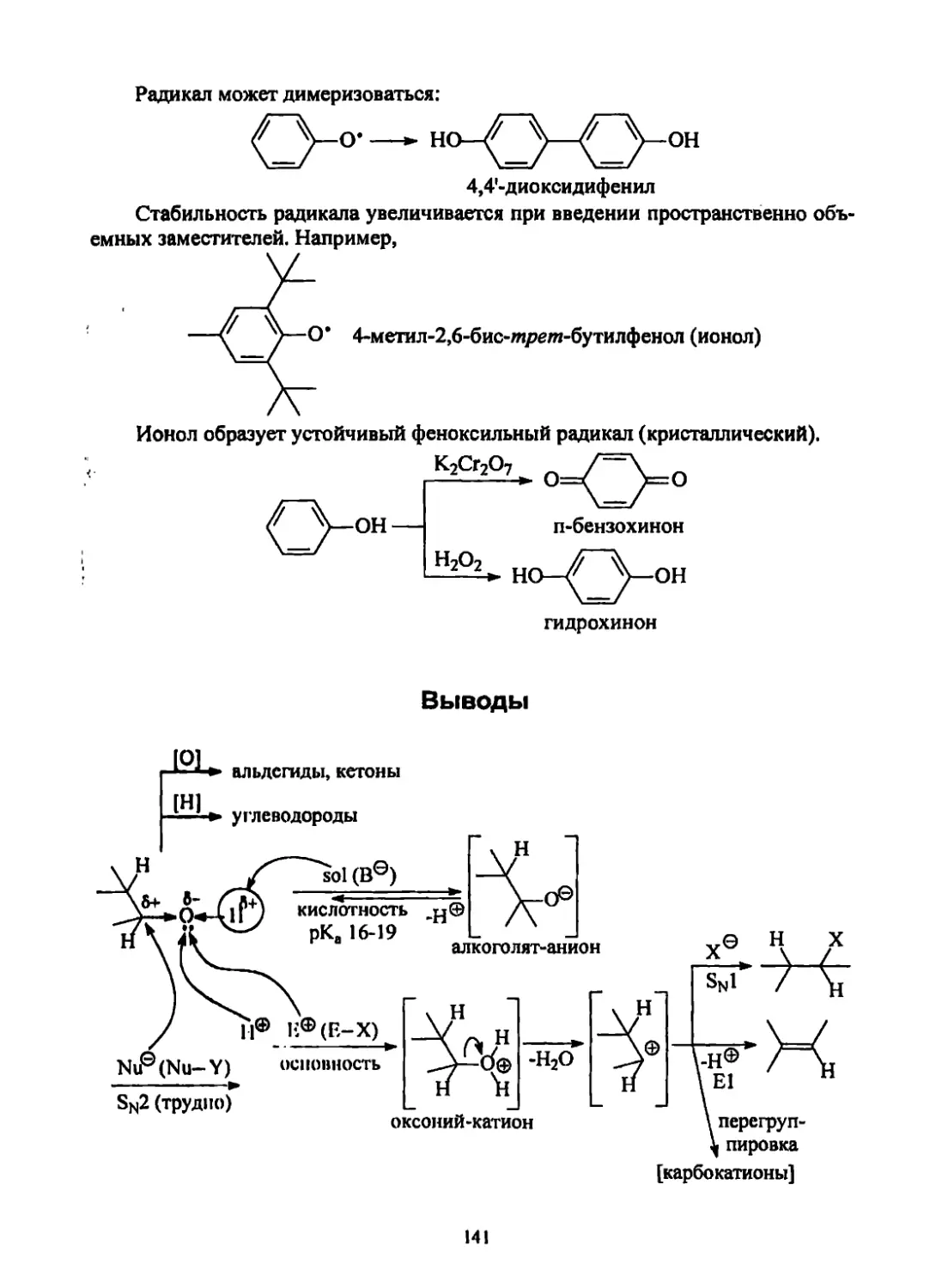
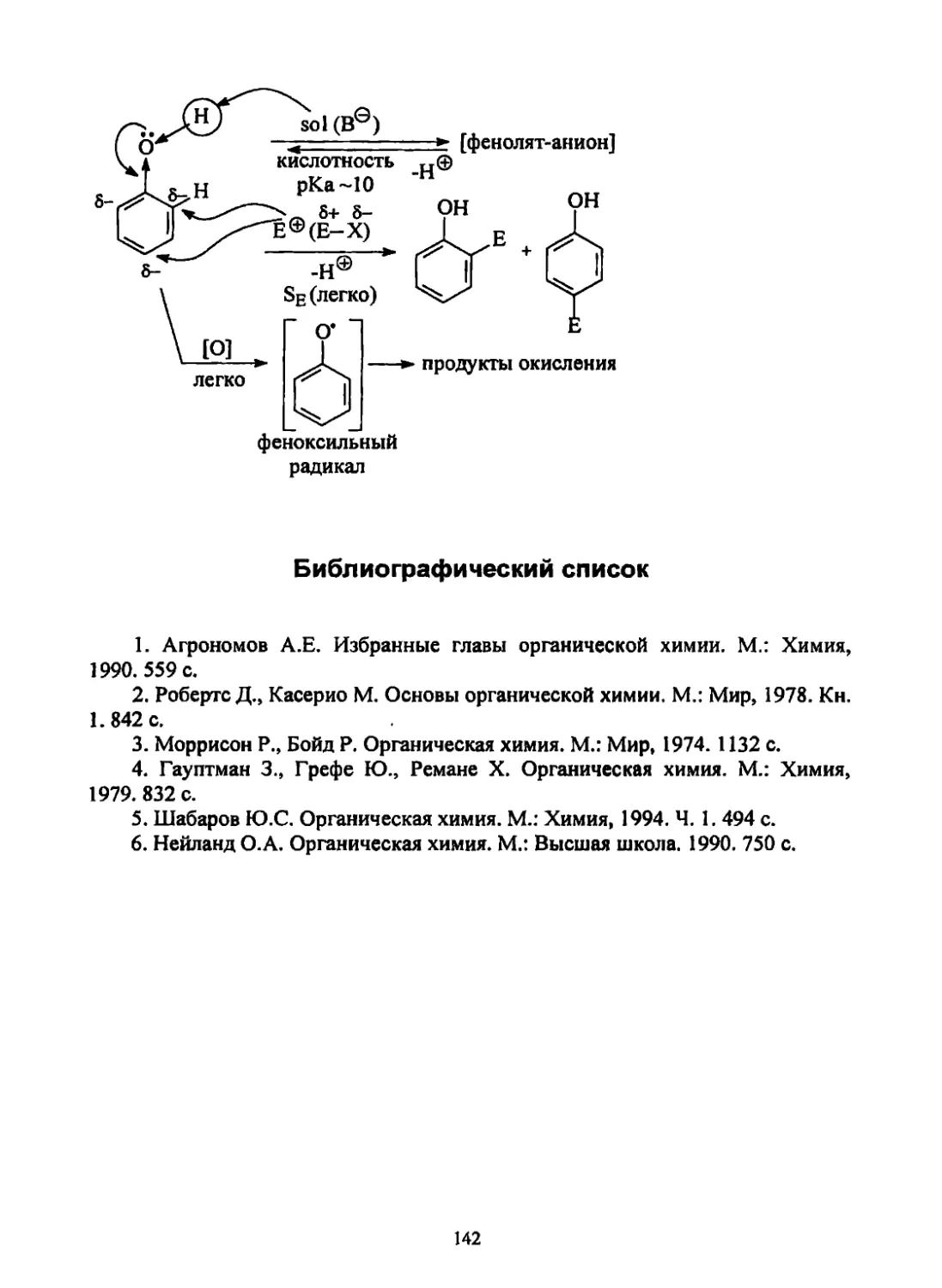









Текст
Щепин В. В.
Органическая химия.
Краткий курс.
Часть 1.
УДК 547(075.8)
ББК 24.2я73
Щ574
Щепин, В.В.
Щ 574 Органическая химия: краткий курс: учеб, пособие для студен-
тов-бакалавров хим. фак-та / Перм.ун-т. — Пермь, 2006.-Ч. 1. - 147 с.
ISBN 5-7944-0651-8
Пособие предназначено для студентов II курса химического факультета
Пермского университета, получающих образование химика-бакалавра. Матери-
ал курса органической химии систематизирован по основным классам органи-
ческих веществ. Кратко описаны основные способы получения важнейших ти-
пов органических соединений, что является достаточной информацией для вы-
полнения предлагаемых студентам упражнений и задач по органической химии.
Основное внимание уделено реакционной способности и химическим
свойствам органических веществ, а также анализу факторов, влияющих на
электронное строение органических молекул и определяющих их реакционную
способность. Теоретическая база, позволяющая выявить химическое поведение
разных типов органических соединений, основана на представлениях о различ-
ных механизмах, в рамках которых и происходят их трансформации.
Пособие знакомит студентов с методами синтеза и химическими свойст-
вами важнейших классов органических веществ. Их знание дает возможность
анализировать электронную структуру органических соединений и на основе
статического и динамического подходов прогнозировать и объяснять их реак-
ционную способность по отношению к разнообразным реагентам.
Рецензенты: зав. каф. физической и коллоидной химии Пермской государст-
венной фармацевтической академии д-р хим. наук, проф.
ВЛ.Гейн;
ст. науч. сотр. лаборатории активных реагентов ИТХ УрО РАН,
д-р хим. наук Г.Г. Абашев
Печатается по решению редакционно-издательского совета Пермского
университета
ISBN 5-7944-0651-8
© Щепин В.В., 2006
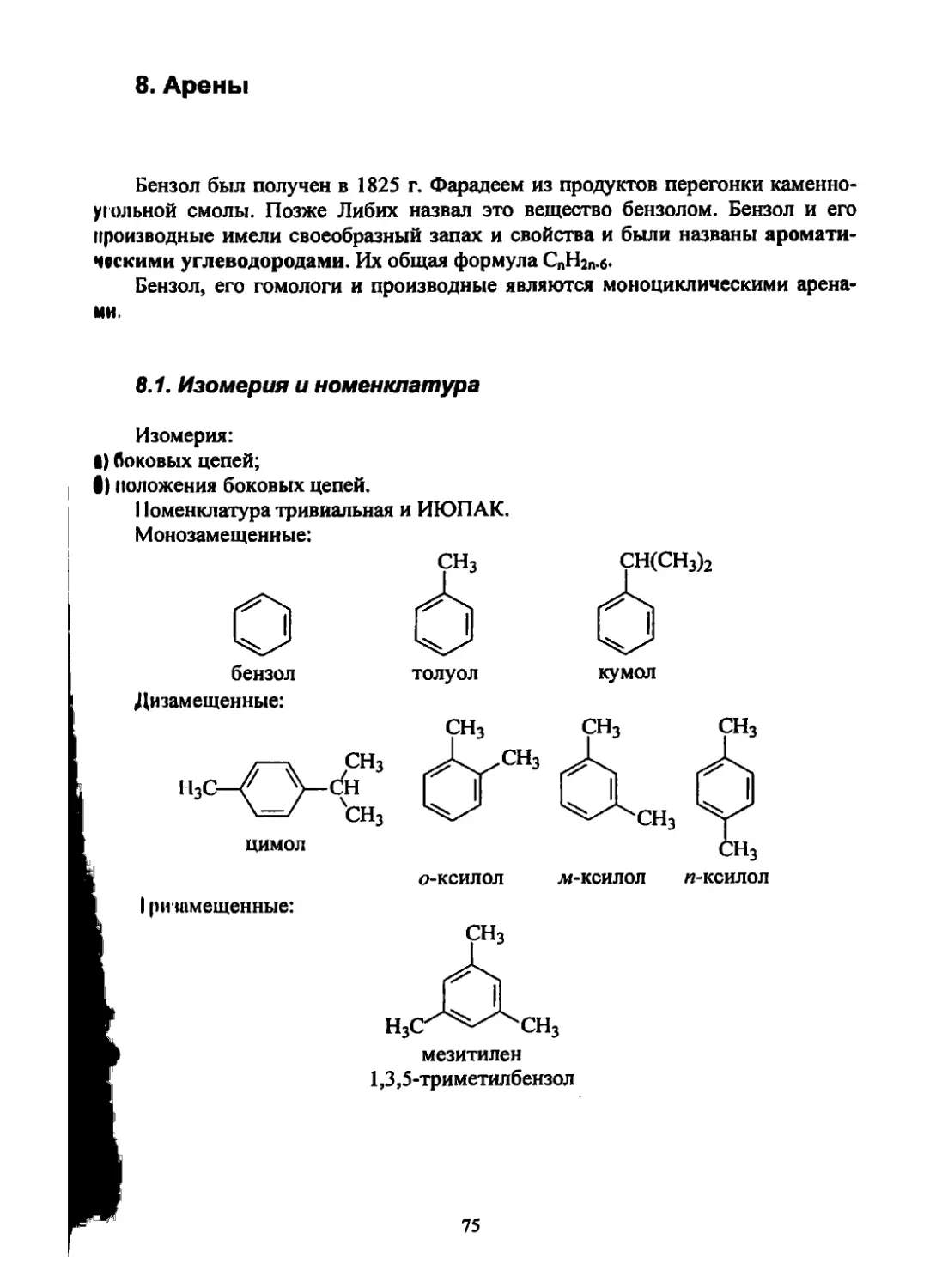
1. Валентное состояние атома углерода, а- и л-связи
1.1. Орбитали
Наиболее распространенными в органической химии являются орбитали
двух типов -я мр.
Эти орбитали и занимающие их электроны отличаются друг от друга
своими энергиями и конфигурацией той части пространства, где вероятность
нахождения электрона максимальна.
Для ^-атомной орбитали (j-AO) харак-
пость нахождения электрона в
p-KO-pXiPy,pz.
терна шаровая симметрия, а распределение
электронного облака зависит только от ра-
диуса (г). Граница поверхности шара нахо-
дится на расстоянии 1.4 А от атомного ядра
(граница поверхности шара ограничивает ту
часть пространства, в которой вероятность
нахождения электрона составляет 90%).
Для р-атомной орбитали (p-АО) харак-
терна цилиндрическая симметрия. Она похо-
жа на гантель с минимумом электронной
плотности у ядра. p-АО имеет две части, ко-
торые разделяет узловая плоскость (вероят-
узловой плоскости равна нулю). Имеется три
Все три АО эквивалентны (вырождены) и отличаются направлением в про-
ирппстве.
В положительной части р-орбитали вероятность нахождения электрона в
данный момент времени максимальная, в отрицательной - минимальна.
3
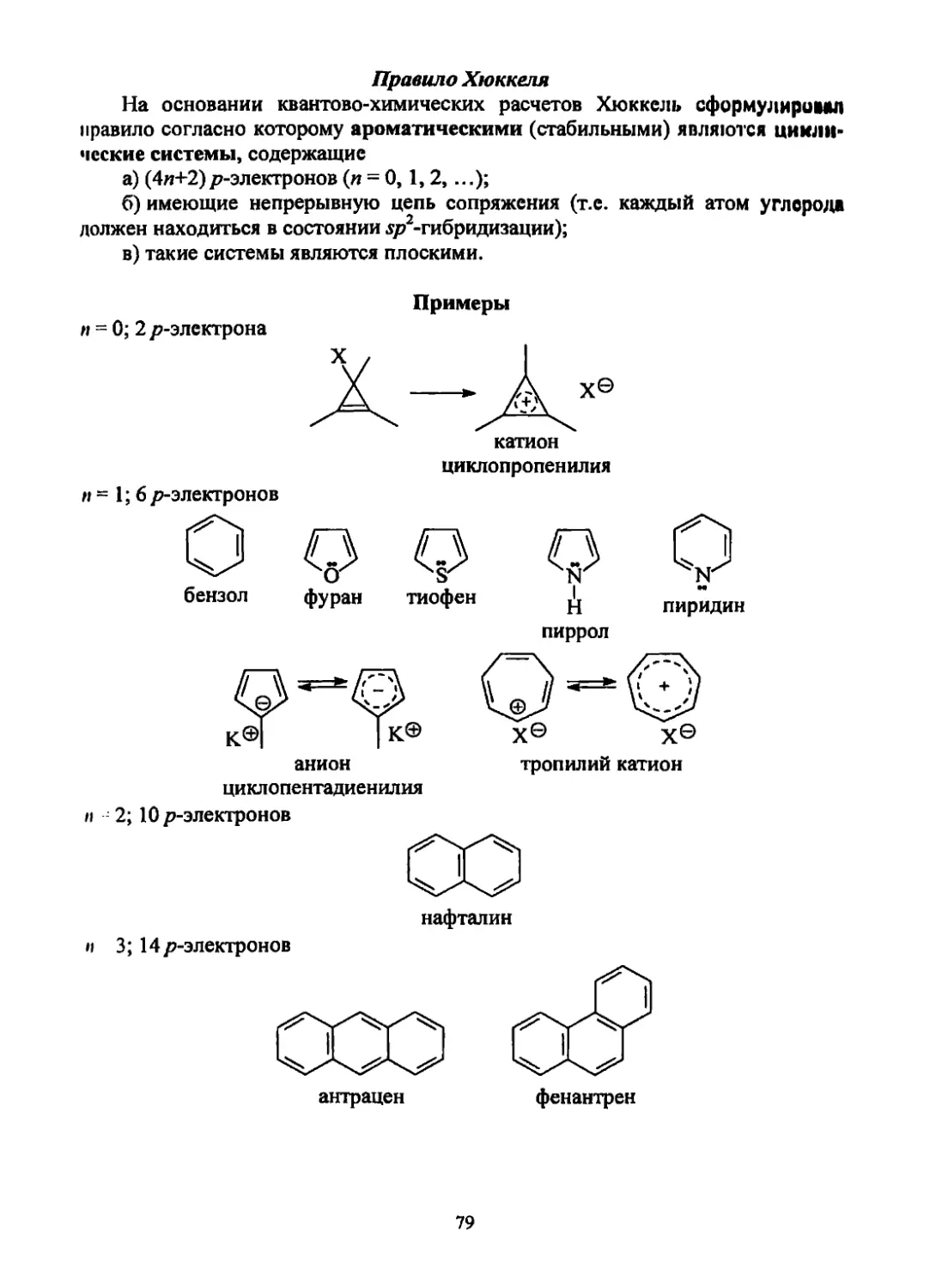
1.2. Основное и возбужденное электронное состояние атома
уалерода
В основном состоянии электронная конфигурация атома углерода (элек-
тронное состояние) записывается так:
С ls22s22plx2ply.
На первом электронном слое, соответствующем к АО, находятся 2 элек-
трона; то же - на 2s АО. По одному неспаренному электрону находится на двух
р-орбиталях, одна р-орбитал ь свободна.
Это схематически записывают так:
2s АО
1s АО
2рАО
Обозначают каждый электрон стрелочкой.
Здесь наглядно видно, что энергия 25-орбитали больше 1 s-орбитали, а
энергия 2р-орбитали самая большая.
Согласно такому электронному состоянию атом углерода должен быть
двухвалентным. Это противоречит тому факту, что в большинстве органиче-
ских соединений атом углерода - четырехвалентный.
1.3. Гибридизация
Для объяснения такого противоречия было введено и математически обое-
новано понятие о гибридизации орбиталей.
Считается, что при образовании ковалентных связей один из 2s электронов
(или электрон, находящийся на 2$-орбитали) невозбужденного атома углерода
возбуждается (с затратой 100-150 ккал/моль) и переходит на свободную р-орби-
тал ь.
В возбужденном состоянии у атома углерода - уже 4 неспаренных элек-
трона на 252рх2р^2р2-орбиталях.
Взаимодействие орбиталей возбужденного атома углерода между собой
приводит к образованию новых гибридных орбиталей. Энергия же образования
четырех связей за счет четырех новых гибридных орбиталей с избытком пере-
крывает энергию, требуемую для перевода С -> С*. При этом разное число
2р-орбиталей может вступать во взаимодействие с 25-орбиталью.
4
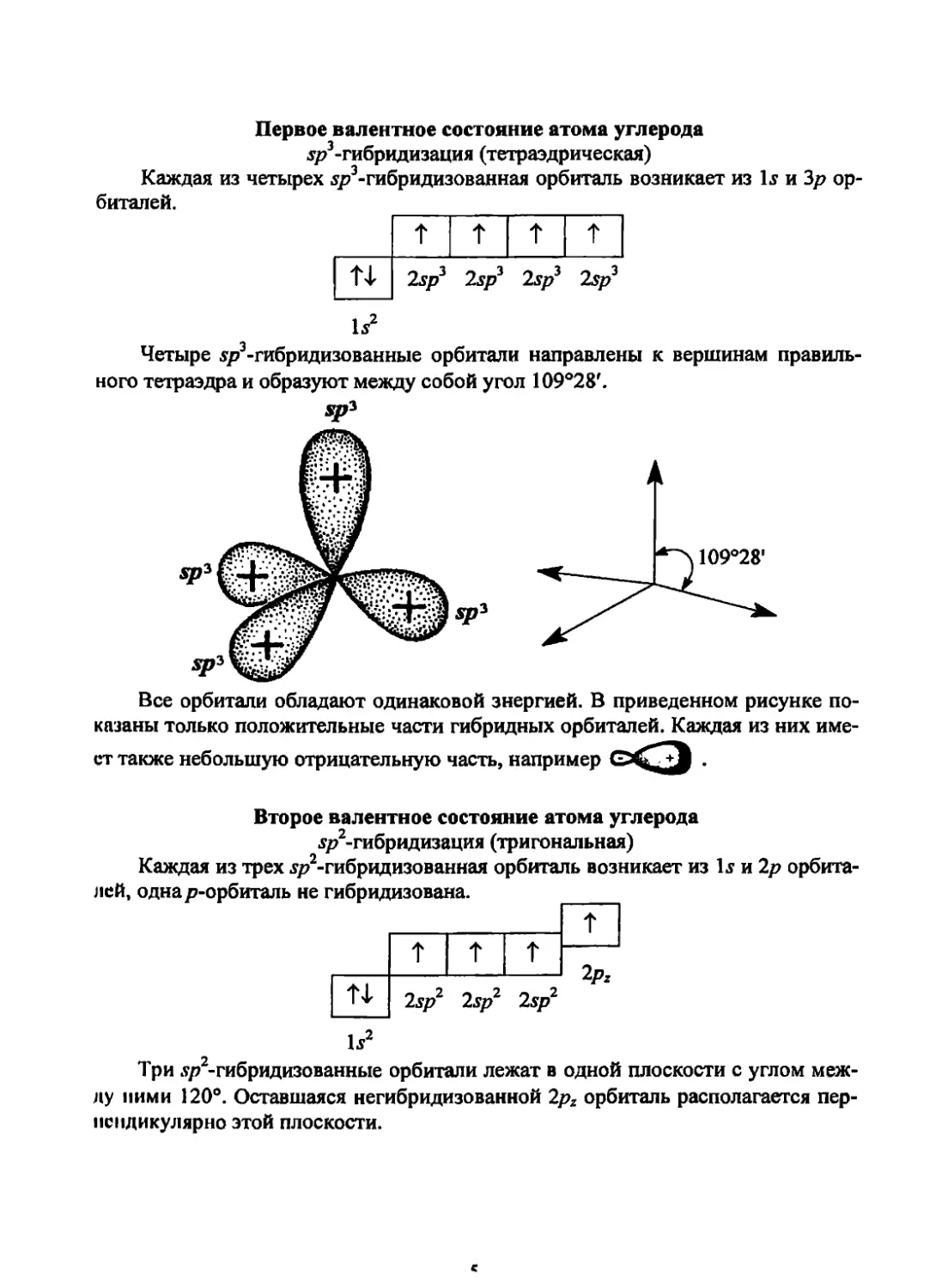
Первое валентное состояние атома углерода
.у/Дгибридизация (тетраэдрическая)
Каждая из четырех $р3-гибридизованная орбиталь возникает из 1j и Зр ор-
биталей.
т
т
т
Т 4- 2sp3 2sp3 2sp3 2sp3
1?
Четыре .$/73-гибридизованные орбитали направлены к вершинам правиль-
ного тетраэдра и образуют между собой угол 109°28'.
Все орбитали обладают одинаковой энергией. В приведенном рисунке по-
казаны только положительные части гибридных орбиталей. Каждая из них име-
ет также небольшую отрицательную часть, например .
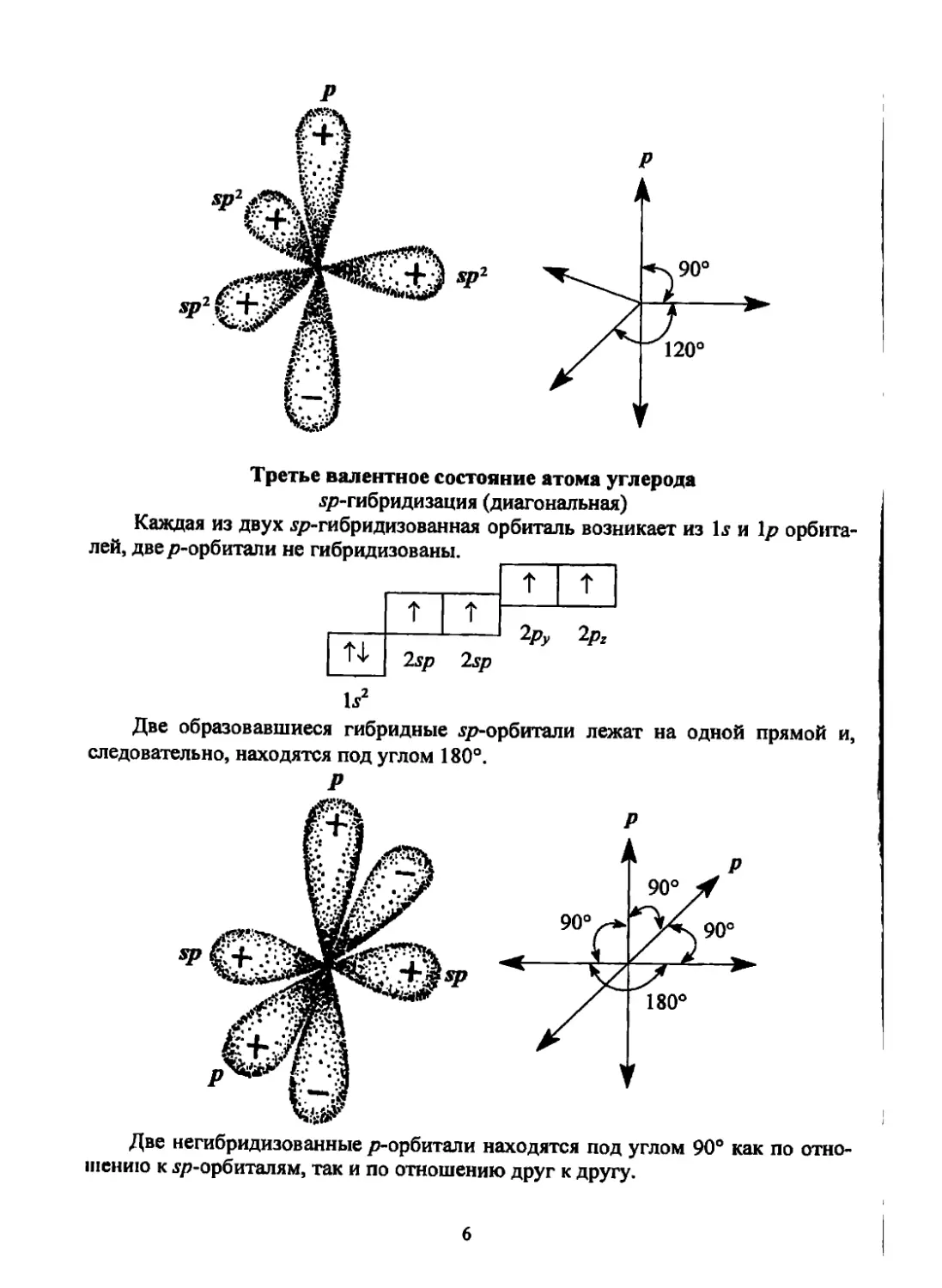
Второе валентное состояние атома углерода
^-гибридизация (тригональная)
Каждая из трех ^-гибридизованная орбиталь возникает из и 2р орбита-
лей, одна р-орбиталь не гибридизована.
T
T
Ti 2sp2 2sp2 2sp2
2p2
1?
Три .ур2-гибридизованные орбитали лежат в одной плоскости с углом меж-
ду ними 120°. Оставшаяся негибридизованной 2pz орбиталь располагается пер-
пендикулярно этой плоскости.
Третье валентное состояние атома углерода
ур-гибридизация (диагональная)
Каждая из двух sp-гибридизованная орбиталь возникает из 1s и 1р орбита-
лей, двер-орбитали не гибридизованы.
т
^"Ру
Две негибридизованные р-орбитали находятся под углом 90° как по отно-
шению к sp-орбиталям, так и по отношению друг к другу.
6
1.4. ar-Сеязи и я-сеязи
п-Связь
При образовании ковалентных связей атомные орбитали связанных атомов
могут взаимодействовать двояким образом.
Например, в молекуле водорода 1л орбитали образуют молекулярную ор-
биталь так, что область перекрывания атомных орбиталей или область макси-
мальной электронной плотности лежит на линии, соединяющей центры атомов.
(Н-Н)
(Н:Н)
Такой тип связи называется с-связь.
о-Связи образуют и гибридные орбитали.
о-Связь характеризуется большой прочностью, так как основная часть
электронной плотности сосредоточена в пространстве между ядрами атомов,
что и приводит к их прочному удерживанию в молекуле. Энергия разрыва
о-связи составляет -90-100 ккал/моль.
л-Связь
При взаимодействии двух р-орбиталей за счет их менее эффективного бо-
кового перекрывания образуется л-связь.
В этом случае создаются две области максимальной электронной плотно-
11 и цыпic и ниже оси связи. Поэтому л-связь менее прочна и более подвижна
(иолмр1пуема), чем a-связь. Энергия разрыва л-связи составляет-65 ккал/моль.
7
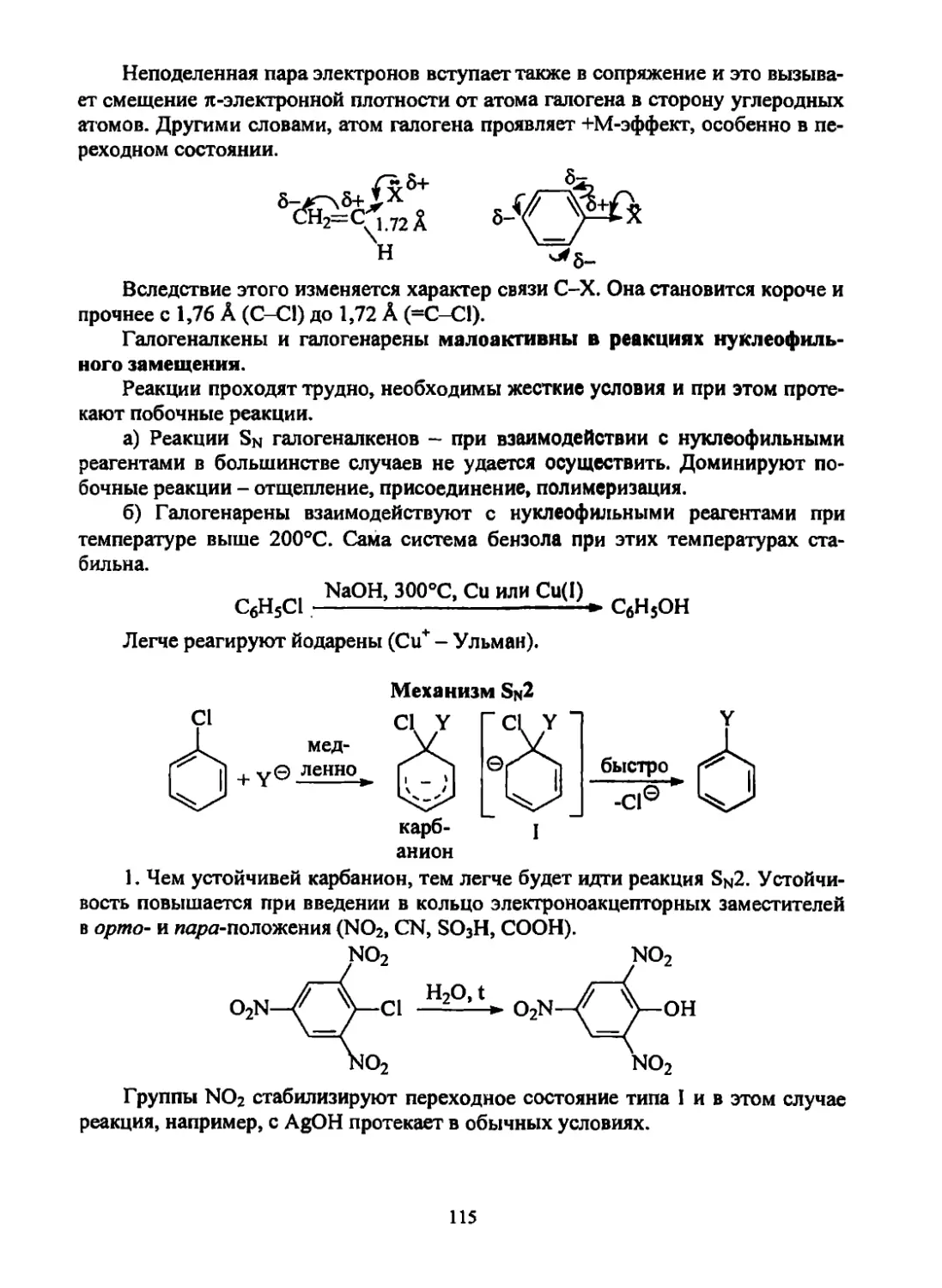
2. Электронные эффекты (механизмы) влияния атома в
молекулах, классификация реакций
Химическое поведение молекулы органического вещества обусловлено ха-
рактером распределения в ней электронной плотности и перераспределения ее в
ходе реакции. Это, в свою очередь, определяется взаимным влиянием атомов и
групп атомов в молекулах. Поэтому знание электронных механизмов влияния
атомов в молекулах позволяет с общих позиций оценить их химические свойст-
ва и реакционную способность.
Совершенно очевидно, что только в молекулах, состоящих из симметрич-
ных частей, электронная плотность равномерно распределена между двумя
атомами центральной связи. Когда же атомы разнородны или молекулы несим-
метричны, то электроны, образующие центральную связь, смещены в сторону
одного из атомов. С целью характеристики сродства атомов к электрону была
создана шкала значений электроотрицательности атомов (Л. Полинг). Электро-
отрицательность - мера способности атома или группы атомов притягивать
электроны из других частей той же молекулы. Чем больше электроотрицатель-
ность атома, тем сильнее его сродство к электрону.
Значения электроотрицательности некоторых атомов:
Н 2,3
Li 1,0 В 2,0 । Cj 2,5 N 3,0 О 3,5 F 4,0
Na 1,0 Mg 1,2 Al 1,5 Si 1,7 P 2,1 S 2,4 Cl 3,0
Са 1,5 Br 2,8
I 2,5
Таким образом, если химическая связь образуется из элементов различной
электроотрицательности, то отрицательный заряд концентрируется на более I
электроотрицательном атоме, химическая связь становится более полярной, f
Перераспределение электронной плотности в молекулах описывается в органи- 1
ческой химии двумя основными механизмами, которые называются индуктив-
ным и мезомерным эффектами. ।
2.1. Электронные смещения в насыщенных молекулах (индук-
тивный эффект, I)
Индуктивный эффект - это эффект смещения электронной пары вдоль
о-связи. Индуктивный эффект обозначают прямой стрелочкой:
------► ◄------
1-эффект
8
Положительным индуктивным эффектом (+1) обладают электронодо-
норные (отталкивающие электронную плотность) атомы или группы атомов. В
соответствии с электроотрицательностью атомы элементов, находящиеся в пе-
риодической системе левее атома углерода, проявляют +1. Поэтому металлоор-
ганические соединения поляризованы так:
5+ 8- 6+ 8- 8+ 8—
Na-кС Li-кС BrMg->:C
Слабый +1 проявляют алкильные радикалы и отрицательно заряженные
атомы, причем его сила возрастает в рядах:
-СН3 < -СН2СНэ < -СН(СНэ)2 < -С(СН3)3
-о0<Л-
Отрицательным индуктивным эффектом (-1) обладают электроноак-
цепторные (притягивающие электронную плотность) атомы или группы ато-
мов.
Заместители обладают тем большим -I эффектом, чем правее и выше в пе-
риодической системе находится соответствующий элемент:
-NR2<-OR<-F
-I <-Br<-Cl<-F
=NR < =О
-NR2<=NR<sN
—^R3 < —8r2
Сила индуктивного эффекта быстро убывает с ростом расстояния между
атомами, что объясняется малой подвижностью электронов о-связи.
6+ 54- 54- 8-
СН3-» СН2-* СН2-» С1
Возмущение, вызываемое, например, атомом хлора, через три связи равно
пулю. Наиболее же сильный индуктивный эффект проявляется на атоме угле-
рода, непосредственно связанного с заместителем.
Индуктивный эффект вызывает статическую поляризацию молекул и по-
«илспис дипольного момента.
Наличие диполя в молекуле с полярной ковалентной связью является при-
чиной многих гетеролитических реакций.
2.2. Электронные смещения в ненасыщенных молекулах (ме-
еомерный эффект или эффект сопряжения, М)
Мпомерный эффект - это эффект смещения /7-электронов л-связи. Мезо-
марный эффект обозначают кривой (изогнутой) стрелочкой:
9
или
М-эффект
Для появления мезомерного эффекта необходимо взаимодействие p-элек-
тронов л-связей или р-электронов с п электронами гетероатомов (X), содержа-
щих на р-орбитали неподеленную электронную пару (НЭП), т.е. в системе
должно возникать общее (единое) поле р-электронов (р-орбиталей). Например:
Положительным мезомерным эффектом (+М) обладают группы, имею-
щие гетероатом с НЭП:
X = NR2 > OR > F
---------► У
SR CI
V
Br
V
T
Убывание +М
Анионы более сильные доноры электронов, чем соответствующие гетеро-
атомы с НЭП:
О
-О > OR.
Эти группы повышают электронную плотность на л-связи.
Способность гетероатома отдавать электронную плотность обратно про-
порционально его электроотрицательности и уменьшается в периоде в перио-
дической системе слева направо, а в группе - сверху вниз.
В группе решающая роль принадлежит размерности (величине) р-орбита-
ли, отчего и зависит его способность перекрываться с р-орбиталями л-связей.
У атома фтора 2р-орбитали одного уровня перекрываются с 2р-орбиталями
атома углерода. Их перекрывание наиболее эффективно, поэтому атом фтора
проявляет максимальный +М-эффект в отличие от других атомов галогенов.
Например, у атома йода 5р-орбитали и их перекрывание с 2р-орбиталями атома
углерода неэффективно.
10
Отрицательным мезомерным эффектом (-М) обладают группы, имею-
щие в своем составе двойную или тройную связь с электроотрицательным ато-
мом.
Эти группировки имеют сильнополярные л-связи, например, карбонильная
. 8/|5-
группа ^С=О . Если карбонильная группа сопряжена с двойной связью, то
сдвиг электронов происходит по всей системе сопряжение, приводя к поляри-
зации всей молекулы:
8+ /is- бч/is- С/ s+/is-
сн2=сн-с=о a+Z V-c=o
I x \==/ I
5“Ъ 5+
Отрицательный мезомерный эффект увеличивается в ряду:
=CR < =NR < =0
=CR < sN
©
=nr<=nr2
0 0 0 0 о
—<— c"^ < — <— c' <—c" <
ж он or vch3 'Vi
о
ЧС1
Для передачи электронных влияний атомов по механизму сопряжение ха-
рактерно, что перераспределение электронной плотности практически не осла-
Поааст и распределяется на всю цепочку сопряженных связей.
Эффект сверхсопряжения (гиперконъюгации)
Эффект сверхсопряжения заключается в том, что алкильные группы,
и моющие а-СН связи и присоединенные к углероду в sp2- или (sp-) состоянии,
прояпляют кроме +1 дополнительный электронодонорный эффект (условно обо-
шячнм его! \), аналогичный +М-эффекту. Сила его возрастает, в отличие от
1I-эффекта алкильных групп, с увеличением числа а-СН связей, т.е. в ряду:
/i8- /05-
(CI13)3C-*CH=CH2 < (СН3)2СН-^СН=СН2 <
05- /05-
< СН3СН2-^СН=СН2 < сн3-*-сн=сн2
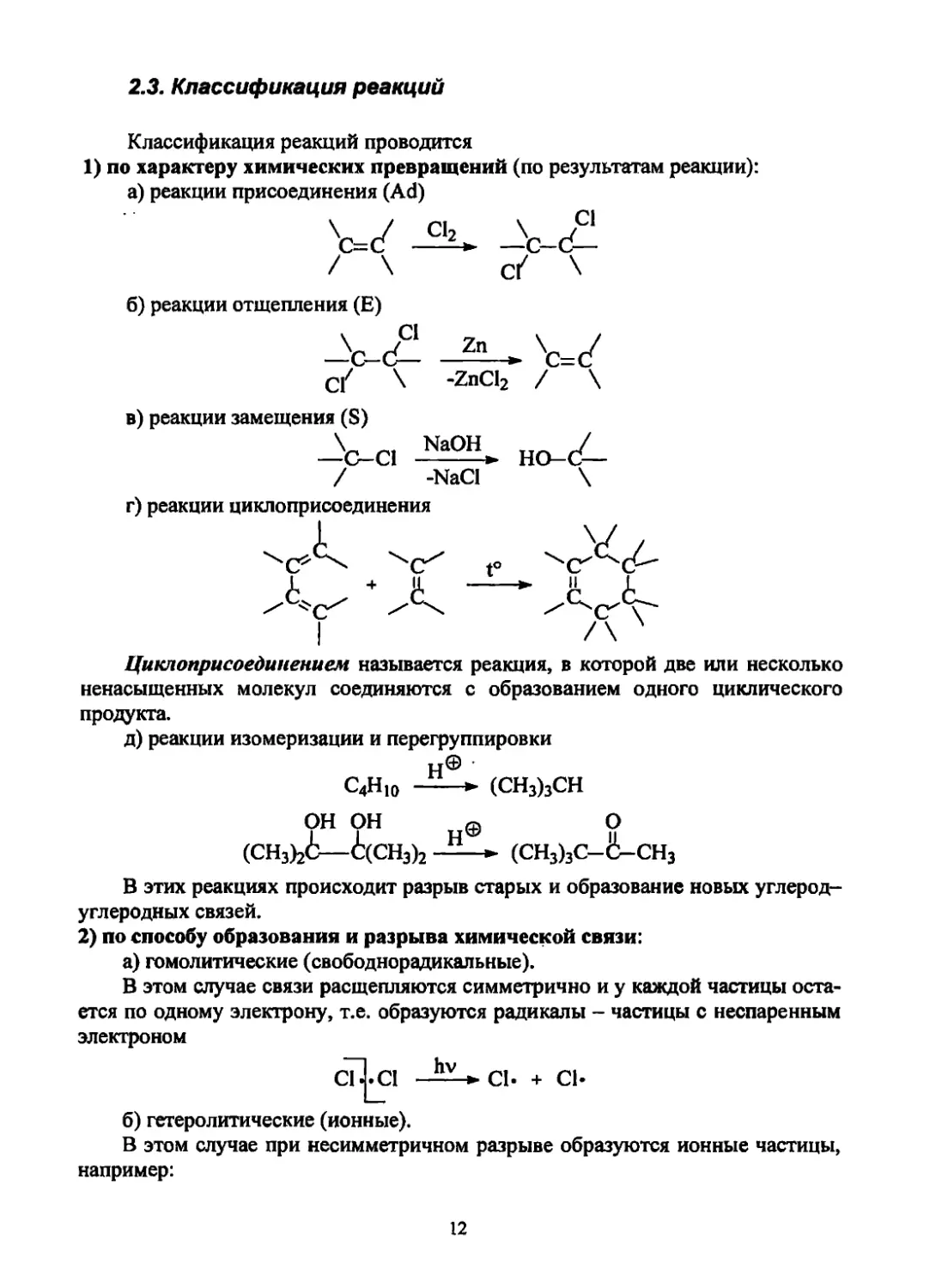
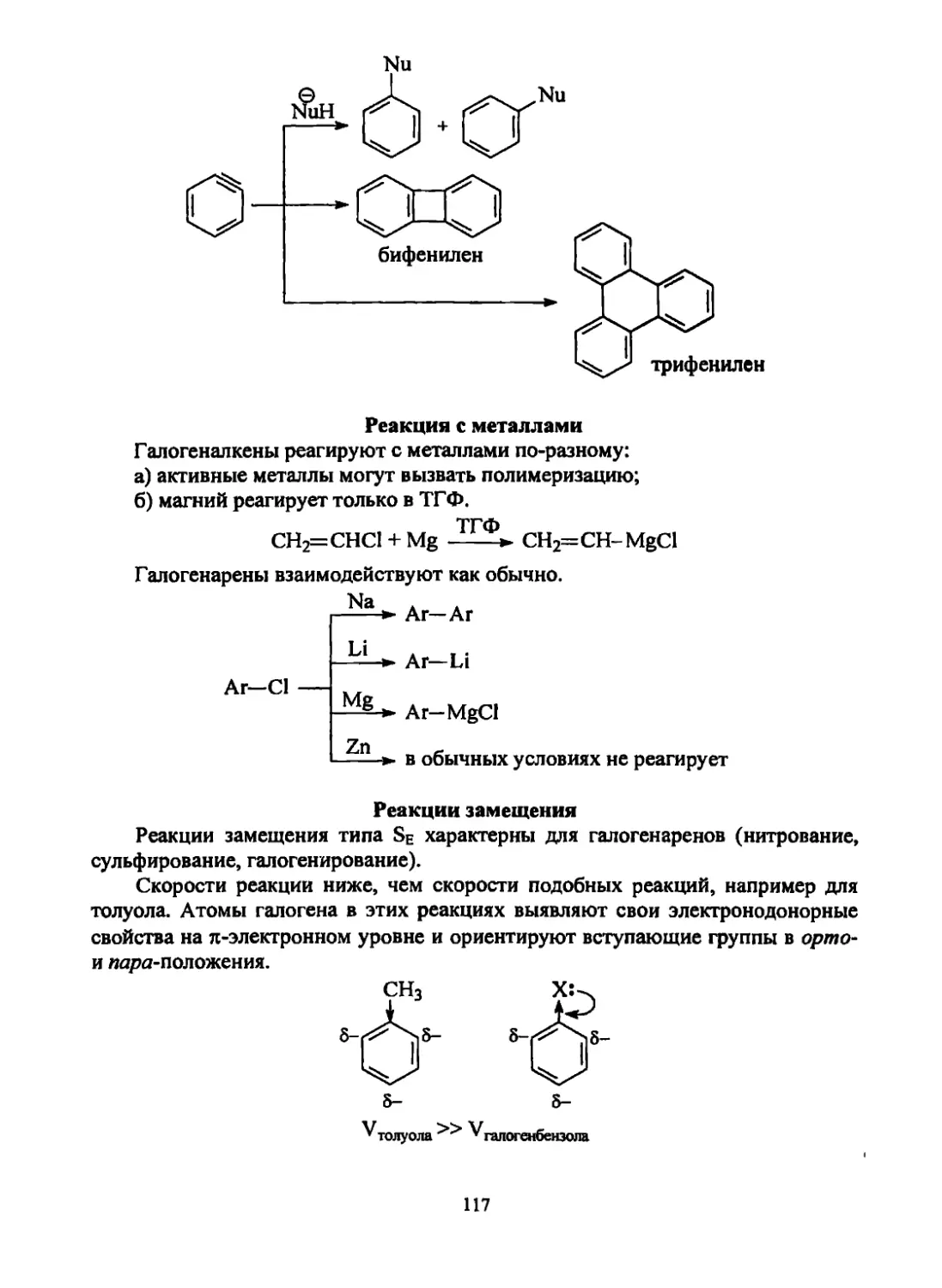
2.3. Классификация реакций
Классификация реакций проводится
1) по характеру химических превращений (по результатам реакции):
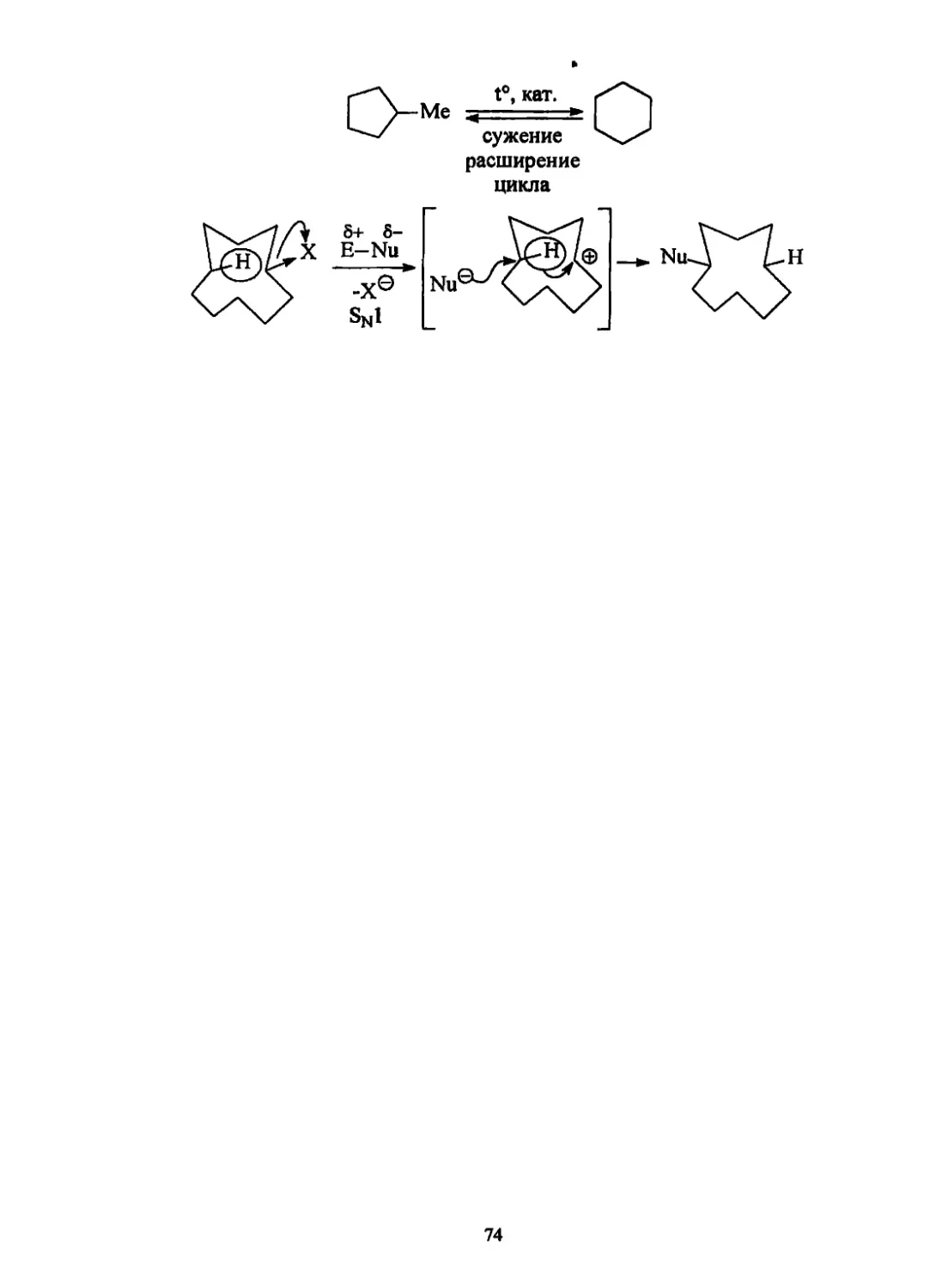
а) реакции присоединения (Ad)
W Л-<£
/ \ cf \
б) реакции отщепления (Е)
с/ \ -ZnCl2 / \
в) реакции замещения (S)
Циклоприсоединением называется реакция, в которой две или несколько
ненасыщенных молекул соединяются с образованием одного циклического
продукта.
д) реакции изомеризации и перегруппировки
it©
СдНю ► (СН3)3СН
ОН ОН @ о
(CH3)2i—i(CH3)2 —* (сн3)3с-ё-сн3
В этих реакциях происходит разрыв старых и образование новых углерод-
углеродных связей.
2) по способу образования и разрыва химической связи:
а) гомолитические (свободнорадикальные).
В этом случае связи расщепляются симметрично и у каждой частицы оста-
ется по одному электрону, т.е. образуются радикалы - частицы с неспаренным
электроном
I hv
C1J.CI —+ сь
б) гетеролитические (ионные).
В этом случае при несимметричном разрыве образуются ионные частицы,
например:
12
\ Sol \ m
—С-С1 -------► —С® + С1и
Гетеролитические реакции проходят в растворителях (sol). Образующиеся
из полярных молекул (X&+Y6-) ионы могут взаимодействовать как с противоио-
нами, так и с молекулами растворителя и находиться в виде контактной (тес-
ной) ионной пары (А), сольватно-разделенной ионной пары (Б), свободных ио-
нов (В):
sol sol Sol Sol
:y" « —» X®Y®<=- X®|Sol| Y® <- —X® + Y®
иони- А Б ДИСС°’ Sol Sol Sol Sol
зация циация
В
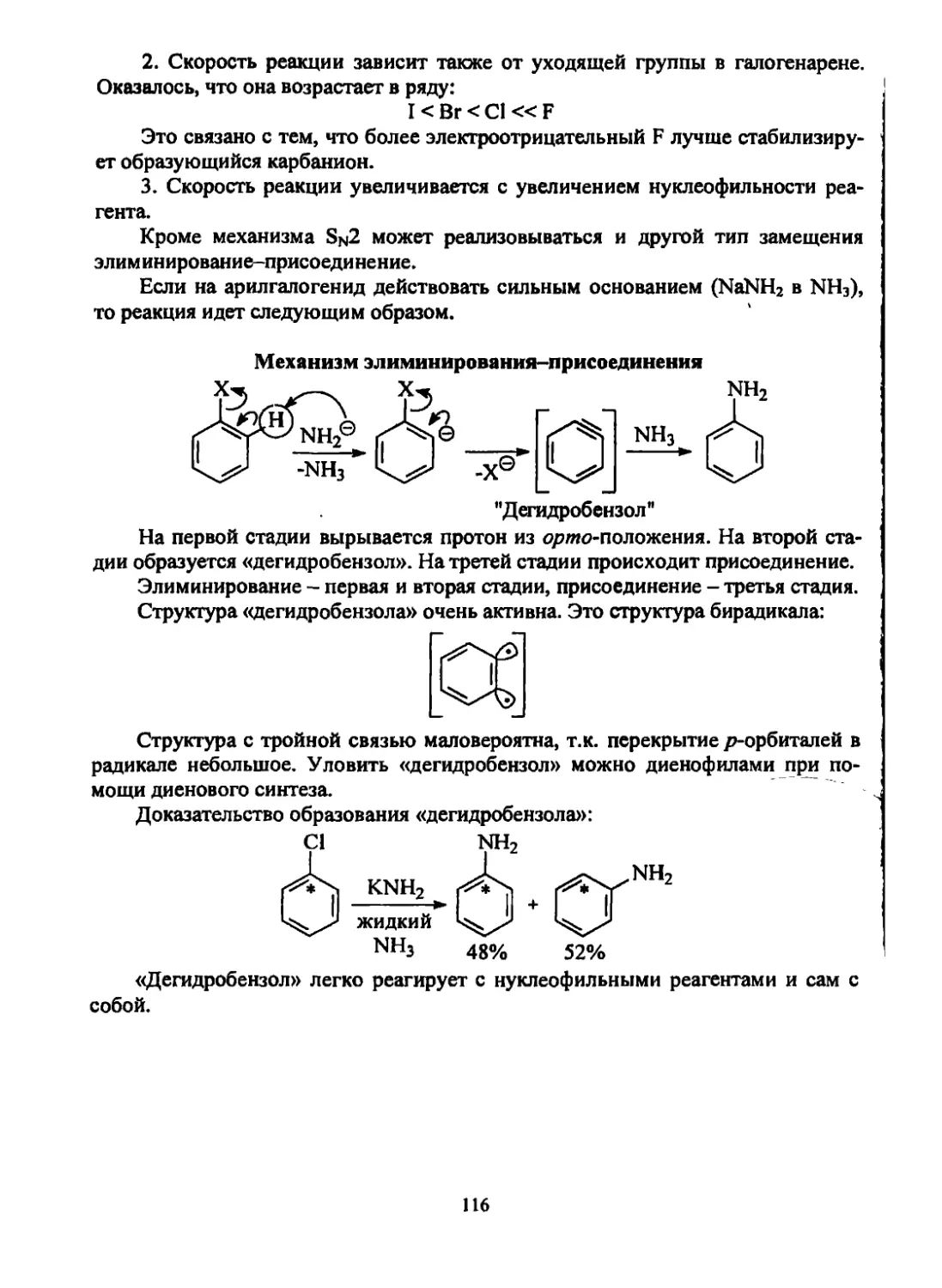
Реагенты, вступающие в гетеролитические реакции, разделяются на нук-
леофильные (Nu) и электрофильные (Е).
Нуклеофильные реагенты - это молекулы или частицы, имеющие избы-
ток электронной плотности и обладающие электронодонорными свойствами.
Их можно классифицировать в зависимости от типа нуклеофильного ато-
ма:
Й-нуклеофилы: Н® (гидрид-ион), BH®Na® (натрий боргидрид), AlH®Li®
(литий алюмогидрид);
С-нуклеофилы: R3C:® (карбанионы), R3C-Me (металлоорганические соедине-
ния), алкены, алкадиены, арены;
>.0 ©
N-нуклеофилы: R2N: Me (амиды металлов), R3N (амины и их производ-
ные);
О-нуклеофилы: R—бРме® (гидроксиды, алкоксиды металлов), НОН, ROH
(вода, спирты);
S-нуклеофилы: R—S^Me® (меркаптиды, сульфиды металлов), HSH, RSH (се-
роводород, меркаптаны);
Hal-нуклеофилы: SOCh (тионилхлорид), РС15, РВг5, РС13, РОС13 (галогениды
фосфора).
Нуклеофильными являются анионы, металлоорганические соединения с
сильнополярными связями, электроноизбыточные молекулы и молекулы, со-
держащие атомы с НЭП.
Нуклеофильность реагента тем выше, чем больше концентрация электрон-
ной плотности на нуклеофильном центре и чем больше способность (поляри-
зуемость) к передаче этой плотности электронодефицитному центру другой
молекулы.
Электрофильные реагенты — это молекулы или частицы, имеющие не-
достаток электронной плотности и обладающие электроноакцепторными свой-
ствами.
13
Их можно классифицировать в зависимости от типа электрофильного ато-
ма:
Н-электрофилы: Н®Х® (сильные кислоты);
С-электрофилы: R3C® (карбкатионы), соединения с сильной полярной свя-
§4" 8— 1 8т 8
зью: R3C—Hal (галогенопроизводные), ЯгС=О (карбонильные соедине-
8т 8-
ния), RC—N (нитрилы);
N-электрофилы: (соли нитрозония), jo=l^ojx® (соли нитро-
ния), [r-tSn]x0 (солидиазония);
Hal-электрофилы: Hah (галогены);
кислоты Льюиса: BF3, А1С13, ZnCl2.
Исходя из того, каков характер реагента, играющего основную роль в осу-
ществлении взаимодействия реагирующих молекул реакции, реакции подразде-
ляются на нуклеофильные и электрофильные.
в) Имеются также перициклические реакции, в которых реорганизация
связей происходит согласованно, т.е. разрыв и образование связей проходят од-
новременно в полностью сопряженном циклическом переходном состоянии.
14
3. Алканы
Углеводороды с открытой цепью, содержащие только простые ковалент-
ные связи, называются насыщенными углеводородами или парафинами, по но-
менклатуре ИЮПАК - алканами.
Общая формула алканов СпН2п+2.
3.1. Гомологический ряд, изомерия и номенклатура
Ряд сходных по строению соединений, в котором каждый последующий
член отличается от предыдущего на группу СН2, называется гомологическим
рядом. Группа СН2 называется гомологической разницей.
Первые члены ряда (С1-С4) имеют тривиальные названия. Названия ос-
тальных гомологов образованы от греческих и латинских числительных:
СН4 - метан
С2Нб - этан
С]0Н22 - декан
СцН24 -ундекан
С12Нгб - додекан
СгоНи - эйкозан
C30H62 - триконтан
CiooH2O2 - гектан
Все члены гомологического ряда обладают близкими химическими свой-
ствами. Это дает возможность систематизировать реакции по гомологическим
рядам или по классам органических соединений.
Изомерами называются соединения, которые имеют одинаковую молеку-
лярную формулу, но отличаются порядком связей атомов или расположением
их в пространстве, т.е. строением.
Структурная изомерия
Изомеры, различающиеся последовательностью соединения атомов в мо-
лекуле, называют структурными изомерами. Например:
СН3
СН3СН2СН2СНз или СН3— (Ь-СН3 или I
бутан изобутан (2-метилпропан)
Число структурных изомеров алканов быстро растет с увеличением числа
углеродных атомов. Так, пентан С5Н12 имеет три изомера, гептан С7Н]6 - 9, ок-
тан С8Н]8 - 18, декан СюН22 - 75.
15
Стереоизомерия
Число изомеров еще увеличивается за счет возможных стереоизомеров
(изомеров, различающихся только расположением атомов в пространстве). На-
чиная с С7Н16 возможно существование хиральных молекул, которые образуют
два энантиомера (зеркальных изомера). Так, из девяти изомеров гептана два яв-
ляются хиральными:
СН3
С2Н5-С-Н
с3н/
СН3
Н-С-С2Н5 ’
чс3н7
Номенклатура
Тривиальная номенклатура использует названия углеводородов гомоло-
гического ряда.
Рациональная номенклатура рассматривает углеводороды как замещен-
ные метана, например,
СН3
диметилэтилизопропилметан
Номенклатура разветвленных алканов основана на следующих правилах
(ИЮПАК):
а) выбирают наиболее длинную неразветвленную цепь, название которой
составляет основу (корень);
б) разветвления называют в качестве заместителей (групп, радикалов).
Алкильные радикалы - это остатки молекулы алкана, образующиеся при
формальном отрыве атома водорода (или двух атомов водорода) от молекулы
алкана.
Названия одновалентных алкильных радикалов (остатков) образуются из
названий соответствующих алканов замещением суффикса -ан на-ил:
СНз-
СН3СН2-
СН3СН2СН2-
- метил
-этил
- пропил
снГСН
CH3CH2CH2CH2-
енг—
CH3CH2CH—
(Ьн3
- изопропил (1-метилэтил)
-бутил
- изобутил (2-метилпропил)
- втор.-бутил (1-метилпропил)
16
СНз^
СН3— С— - трет.-бутил (1,1 -диметилэтил)
СН37
Названия двухвалентных алкильных радикалов образуются при добавле-
нии к названиям соответствующих одновалентных радикалов суффикса -ен или
-идеи (если обе свободные связи находятся у одного атома углерода):
- метилен
СН3СН^ - этилиден
—CHj— СН2— - этилен
СН3СН2СН^ - пропилиден
СНз^ /
“ изопропилиден
СН3СНСН2— - 1,2-пропилен
—СН2СН2СН2----1,3-пропилен
в) нумерацию цепи осуществляют по принципу наименьших локантов (за-
местителей), т.е. заместители должны иметь наименьшие номера:
1 2 3 4 5 6
СНз—СН-СН-СН2-СН-СН3
6н3 <Ьн3 <Ьн3
2,3,5-треметилгексан
(но не 2,4,5-триметилгексан)
г) если в молекуле алкана имеются различные заместители, то они пере-
числяются в алфавитном порядке:
СН3
1 2 3 4 51 6 7 8 9
СНз-СН-СН2-СН—С-СН2-СН2-СН2-СН3
<Ьн3 62н5 <Ьн
СНз7 СН3
5-изопропил-2,5-диметил-4-этилнонан
В зависимости от числа других углеродных атомов, с которыми связан не-
посредственно рассматриваемый углеродный атом, молекулы различают пер-
вичные, вторичные, третичные, четвертичные атомы углерода:
вторичный
первичный / третичный четвертичный
7 / I
СНз—СН-СН2- СН—С^СНг—СН2—СНг—СН3
<5н3 62н5 <Ьн
ен/ СН3
17
3.2. Получение
Реакции с увеличением углеродной цепи
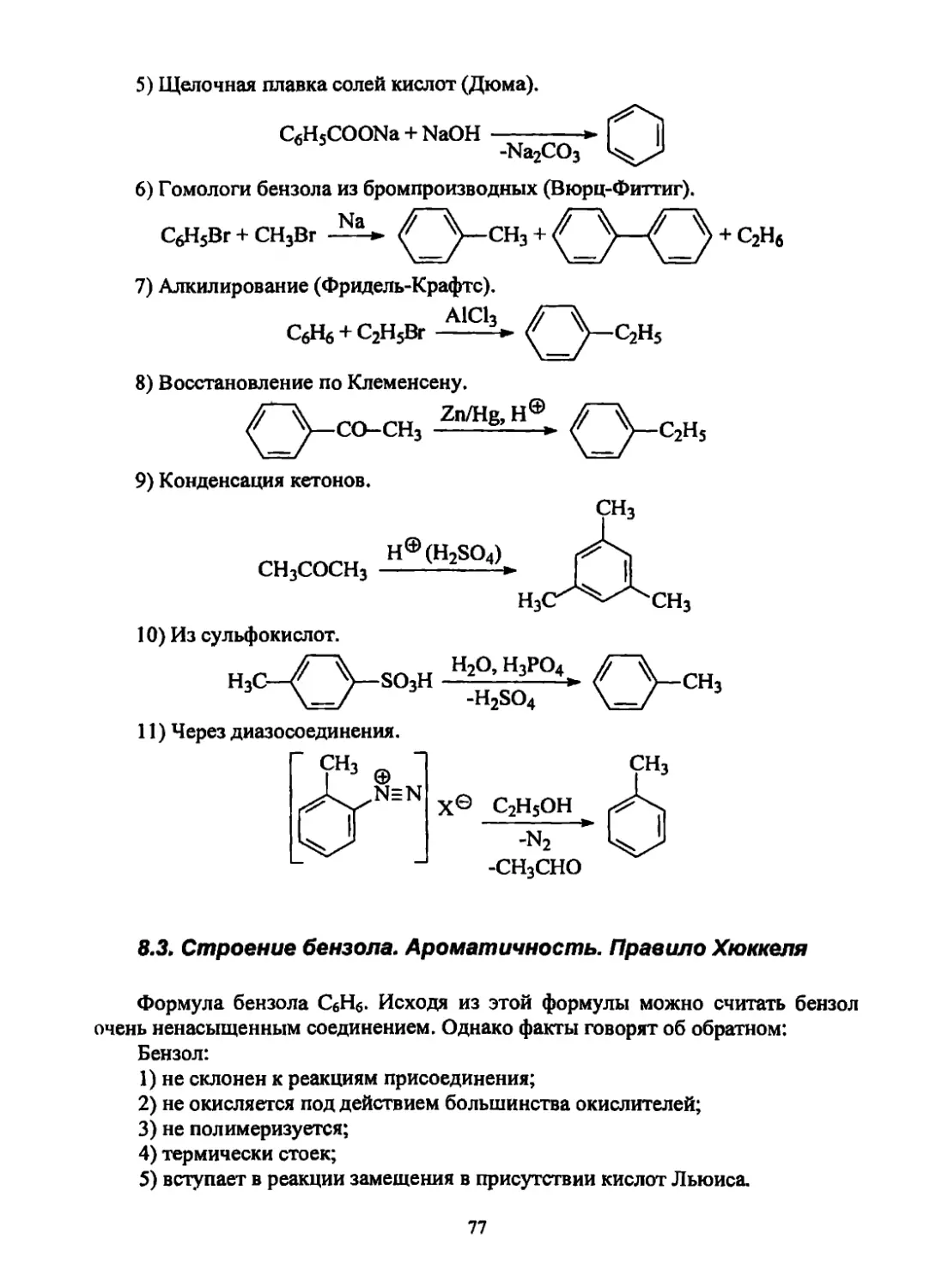
1. Реакция галогеналканов с натрием (Вюрц):
2 R—Вг + 2 Na --------► R-R
-2NaBr
3 R^-Br + 3 R2—Br + 6 Na ------► R1—R1 + R2—R2 + R1—R2
-6NaBr
2. Электролиз солей карбоновых кислот (Кольбе):
2 RCOO“Na+ электРолиз». 2 RCOO- --------------►
-е~ на аноде
—► 2 RCOO’ ------► 2 R’ —► R-R
-СО2
Реакции с укорочением углеродной цепи
3. Декарбоксилирование солей карбоновых кислот (Дюма):
RCOONa + NaQH 2I222Z22*. R—Н
ь j -Na2CO3
Реакции с сохранением углеродной цепи
4. Гидрирование алкенов, алкинов:
Ni Pt Pd
RCH=CH2 + H2 ’ » RCH2CH3
5. Восстановление галогеналканов:
R-I + HI ----► R-H
-I2
R-Br+Mg Э-Ф-Ир> R-MgBr ------—-------► R-H (Гриньяр)
-Mg(OH)Br
6. Восстановление кетонов:
О
R1—C-R Znн+ » r1—CHz-R2 (Клеменсен)
О „ !N-NH2
Rl-fc-R WH2> rU£r 2NaOH,t(200QC)>RLcH^R (Кижнер,
-H2O гидразон "^2 Вольф)
Промышленные способы получения
1. Нефть и природный газ. Природный газ состоит из метана с неболь-
шой примесью этана, пропана и бутана. Из продуктов переработки нефти полу-
чают различные смеси алканов. Фракционной перегонкой бензиновой фракции
нефти можно получить индивидуальные алканы.
18
2. Гидрирование угля. Гидрирование (обогащение водородом) каменного
или бурого угля проводят в автоклавах (реакторах) при высоких температурах
(до 500°С) и давлении (300 атм.). В результате получается смесь различных ал-
канов и циклоалканов, которые используются в качестве моторного топлива.
Процесс идет только в присутствии катализаторов - оксидов и сульфидов нике-
ля, молибдена, вольфрама.
3. Метод Фишера-Тропша (каталитическое гидрирование СО2 и СО). В
качестве катализатора используют никель (Сабатье):
СО2+ 4Н2 Ni^°°C- СН4
со+ зн2 Ni-300°c. сн4
В присутствии катализатора, содержащего кобальт и железо, в основном
образуются алканы:
Со, 290°С
СО + 2 Н2 —--------Алканы
3.3. Физические свойства, пространственное и электронное
строение, реакционная способность
Алканы С1-С4 - газы, Cs-Cj6 - жидкости, Сп и выше - твердые.
С увеличением молекулярной массы растет температура кипения. У раз-
ветвленных алканов температура кипения ниже, чем у неразветвленных, а тем-
пература плавления - наоборот.
Атом углерода в алканах находится в первом валентном состоянии
(.^-гибридизация).
О пространственном строении алканов можно судить по данным рентгено-
структурного анализа и электронографии.
Молекула метана представляет собой тетраэдр, в вершинах которого нахо-
дятся атомы водорода.
н
1 1.096 А
н
Молекулы w-алканов с большим числом углеродных атомов имеют зигза-
гообразное строение, при этом углеродные атомы находятся, с небольшими от-
клонениями, в одной плоскости.
19
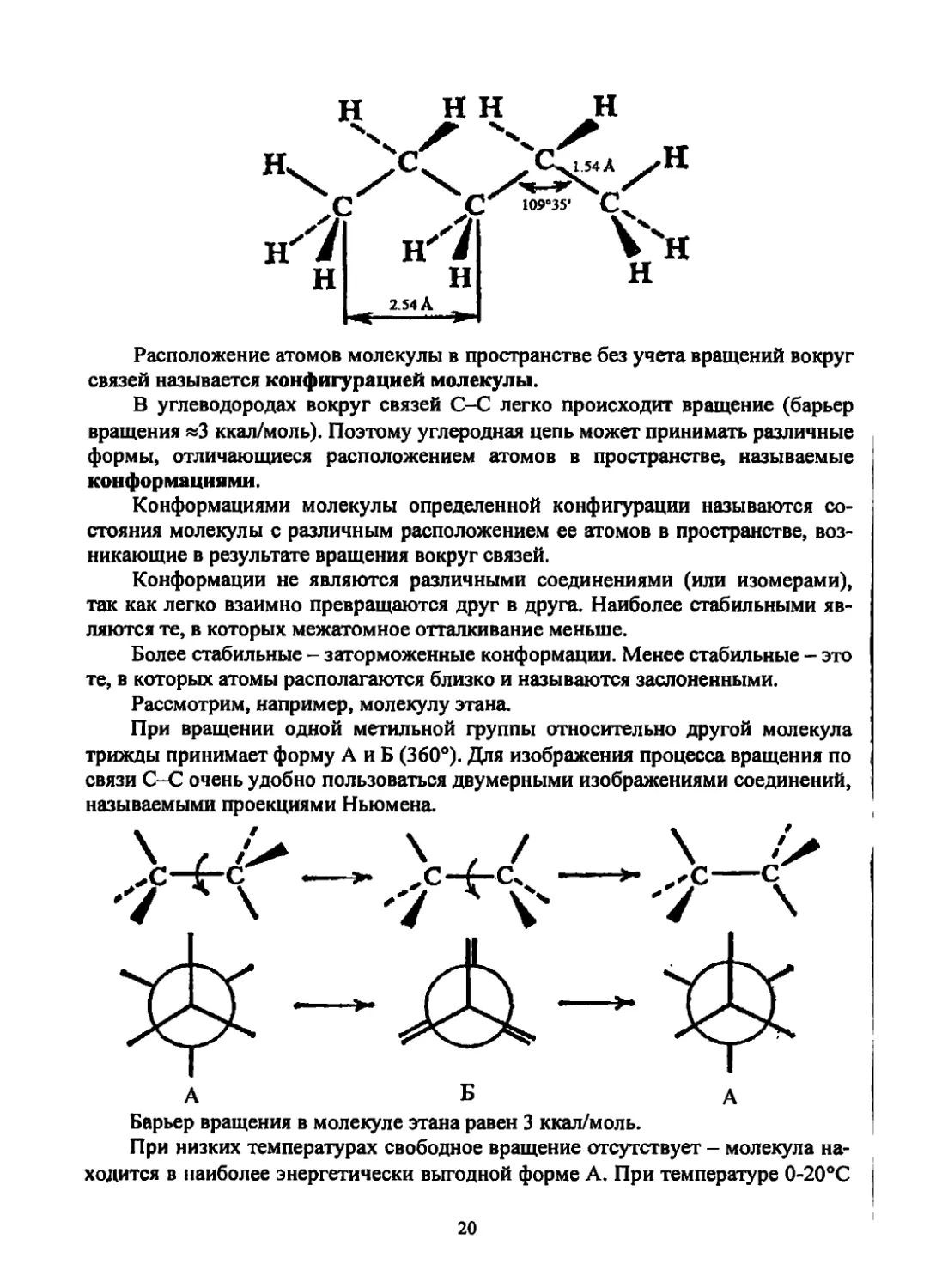
Расположение атомов молекулы в пространстве без учета вращений вокруг
связей называется конфигурацией молекулы.
В углеводородах вокруг связей С-С легко происходит вращение (барьер
вращения »3 ккал/моль). Поэтому углеродная цепь может принимать различные
формы, отличающиеся расположением атомов в пространстве, называемые
конформациями.
Конформациями молекулы определенной конфигурации называются со-
стояния молекулы с различным расположением ее атомов в пространстве, воз-
никающие в результате вращения вокруг связей.
Конформации не являются различными соединениями (или изомерами),
так как легко взаимно превращаются друг в друга. Наиболее стабильными яв-
ляются те, в которых межатомное отталкивание меньше.
Более стабильные - заторможенные конформации. Менее стабильные - это
те, в которых атомы располагаются близко и называются заслоненными.
Рассмотрим, например, молекулу этана.
При вращении одной метильной группы относительно другой молекула
трижды принимает форму А и Б (360°). Для изображения процесса вращения по
связи С-С очень удобно пользоваться двумерными изображениями соединений,
называемыми проекциями Ньюмена.
Барьер вращения в молекуле этана равен 3 ккал/моль.
При низких температурах свободное вращение отсутствует - молекула на-
ходится в наиболее энергетически выгодной форме А. При температуре 0-20°С
20
соотношение А : Б = 1000 : 1. При высоких температурах легко преодолевается
энергетический барьер (конформация Б) и метильные группы с большой скоро-
стью вращаются одна относительно другой.
В молекулах с более объемными радикалами разница в стабильности меж-
ду заторможенными и заслоненными конформациями больше.
Реакционную способность соединения определяют функциональные груп-
пы. В алканах функциональных групп нет. Поэтому они инертны - за что и на-
зываются парафинами (т.е. не обладающие сродством ко многим реагентам).
К реакциям присоединения они не способны - нет л-связей.
К реакциям полярного замещения - мало способны, так как С-Н связи ма-
лополярны.
Они также весьма прочны и к гомолитическому разрыву, так как Ес-н, с-с ~
90-100 ккал/моль. Это обусловлено типом химических связей в алканах. а-Свя-
зи прочны и локализованы внутри молекул.
Алканы являются очень слабыми электронодонорами. Отрыв электронов
чрезвычайно затруднен. Об этом свидетельствуют высокие энергии ионизации
алканов, однако с увеличением числа a-связей увеличиваются электронодонор-
ные свойства молекул.________________' _____________________
Алкан Метан Этан Бутан Гексан
ЭИ, эВ 12,5 11,6 10,5 10,3
Поэтому с большинством химических реагентов алканы не реагируют при
нормальных условиях.
Они способны реагировать только с теми реагентами, которые могут в оп-
ределенных условиях образовывать чрезвычайно реакционноспособные ко-
роткоживущие частицы с неспаренным электроном - радикалы. Кроме то-
го, могут осуществляться превращения алканов при высоких температурах или
в присутствии катализаторов, в том числе сверхсильных кислот.
Таким образом, для алканов наиболее характерен гомолитический тип
разрыва связей!
3.4. Химические свойства
3.4.1. Реакция радикального замещения (SR)
Механизм
1-я стадия - инициирование.
Она заключается в том, что под влиянием энергии извне в реакционном
сосуде возникают свободные радикалы.
Инициировать реакцию можно:
а) фотолизом (энергия кванта света с X = 300 мм —95 ккал/моль);
б) термолизом (нагреванием);
в) радиолизом (у-облучение).
21
2-я стадия - рост цепи.
Идет преимущественно в том направлении, которое дает наибольший теп-
ловой эффект.
3-я стадия - обрыв цепи.
Происходит за счет рекомбинации радикалов.
1. Галогенирование
Рассмотрим эту реакцию на примере хлорирования метана.
1-я стадия:
гомолитический разрыв связи С1-С1, образование свободного радикала.
Энтальпия диссоциации ДН = 58 ккал/моль.
2-я стадия:
Mi сь
Н3С^Н -------*► Н3С- + HCI
ch3ci + сь
рост цепи (длина цепи до 104)
Гомолитический разрыв связи С-Н. Радикал хлора отрывает атом водорода
и образует метильный радикал, который, в свою очередь, отрывает атом хлора
и образует молекулу хлористого метила. В реакционной среде опять возникает
радикал хлора.
Процессы такого типа называются цепными, так как один радикал может
вызвать хлорирование огромного количества молекул метана.
Расчет показывает, что реакция экзотермична и происходит с выделением
энергии (-24 ккал/моль). __________________________
Связь ДН
С1-С1 58 ккал/моль (затраты)
С-Н 102 ккал/моль (затраты)
Н-С1 -103 ккал/моль (выделение)
С-С1 -81 ккал/моль (выделение)
Д = -24 ккал/моль
3-я стадия:
2С1----------------------------------► С12
2 Н3С-----► С2Н6
Н3С- + С1-----► СН3С1
обрыв цепи
Обрыв цепи происходит за счет рекомбинации (димеризации) радикалов, а
также за счет их «гибели» на стенках реакционных сосудов.
Обрыв цепи можно осуществить, вводя в реакцию ингибиторы Ог, Ь, кото-
рые образуют малоактивные радикалы.
22
В принципе, этот процесс может идти и дальше, в результате чего образу-
ются хлорированные метаны с бблыпим числом атомов хлора: хлористый ме-
тилен, хлороформ, четыреххлористый углерод:
С12 С12 С12 С12
СН4 ----СН3С1--------СН2С12---------СНС13---------СС14
-НС1 -НС1 -НС1 -НС1
В реакцию галогенирования могут вступать и другие алканы. В этом слу-
чае замещение водорода идет преимущественно у третичного, затем у вторич-
ного и первичного атомов углерода. Это объясняется тем, что связь С-Н у тре-
тичного атома углерода разрывается легче, а с другой стороны, третичный ра-
дикал наиболее стабилен.
Энергии диссоциации связи С-Н
Реакция АН (ккал/моль)
СН3СН3 * сн3Сн2 + н« 97
СН3СН2СН3 - —► СН3СНСН3 + н. 94
СН3СНСН3 - <Ьн3 —► СН3ССН3 + н. (Ьн3 90
Несмотря на вышесказанное, хлорирование, например, изопентана приво-
дит к смеси хлоризопентанов:
сн3
I С12
СН3—СН2Г-СН—СН3-------► первичный + вторичный + третичный
45% 33% 22%
Этот результат связан с высокой реакционной способностью радикала
хлора и с тем, что в молекуле изопентана соотношение первичных, вторичных
и третичных С-Н связей равно 9:2:1 соответственно.
Бромирование протекает через менее активные радикалы брома, которые
отрывают только те атомы водорода, которые наименее прочно связаны с ато-
мом углерода. Поэтому в этом случае реакция более селективна.
Таким образом, региоселективность (избирательность) реакции зави-
сит от активности реагента и скорости реакции. Чем активнее реагент и
выше скорость реакции, тем меньше региоселективность.
Так как относительная активность радикала брома меньше активности ра-
дикала хлора в 105 раза, то при бромировании изопентана образуется практиче-
ски чистый трет.-бромбутан, а реакция является региоспецифичной.
С другой стороны, фторирование протекает с огромной скоростью и идет с
разрывом связей С-С и С-Н и иногда носит характер взрыва.
Йод с алканами не реагирует (в этом случае реакция эндотермична).
2. Сульфохлорирование (Рид)
Алканы RH реагируют с SO2 и С12 под действием УФ света, при этом обра-
зуются хлорангидриды алкансульфоновых кислот RSO2CI. Под действием УФ
23
лучей образуются атомы хлора и свободные алкильные радикалы. Последние
взаимодействуют с SO2:
hv
С12 » С1- + СЬ
R-H + С1---► R* + НС1
О
R* + SO2 —** R—S«
11
О
О о
R-S- + С12 —► R-S-C1 + С1-
и и
О о
В результате образуются алкилсульфохлориды, или сульфонилхлориды,
или хлорангидриды алкансульфоновых кислот.
Эта реакция имеет большое значение в производстве моющих средств.
Омылением щелочами сульфонилхлоридов получают соли сульфокислот, кото-
рые являются при R = С12-С20 поверхностно-активными веществами (ПАВ).
3. Сульфирование
Сульфируются соединения с третичным атомом углерода:
, H2SO4+SO3 (олеум) L^3
(СНз)зСН —-— ----——H3C-C-SO3H
(Ьн3
4. Нитрование
Азотная кислота при обычной температуре не действует на алканы, при
высокой температуре окисляет алканы.
а) В жидкой фазе.
Коновалов разработал метод нитрования алканов в жидкой фазе:
НЬЮ3(разб., 12,5%)
К—Н —-------------— ► К—
в ампулах при 140°С
J г нитроалканы
Механизм реакции нитрования вероятно следующий:
НО-ЫО2 НО’ + no2.
ho-no2
----#--» RNO2 + НО’
R-H + ОН’ ----► R.
-Н2О 4
Обрыв цепи:
ROH
R.+ ОН.
б) В газовой фазе (Гесс).
Кратковременное нагревание в специальных реакторах смеси до 400-
500®С.
24
При нитровании алканов разрываются также и С-С связи и образуется
смесь нитроалканов, которые разделяются перегонкой:
СНзДсН-ДсН2 —N —» CH3NO2+C2H5NO2+C3H7NO2+?-C3H7NO2
Y Y 420°C 9% 26% 32% 33%
5. Окисление
1) При температуры выше 300°С алканы воспламеняются и сгорают с вы-
делением большого количества тепла:
5СН4 + 8О2---► 5СО2 + 6Н2О
2) Окисление алканов воздухом происходит в присутствии катализаторов в
газообразной или жидкой фазах и протекает через стадию образования переки-
сей и гидроперекисей до получения кислородсодержащих продуктов: спиртов,
альдегидов, кетонов, кислот и т.д.
Механизм Sr (Семенов, Бах, Эмануль)
t° (19 ккал/моль)
о2 ---------------► —О*
легко бирадикал
r£h’+76^o.1----► R-+ноо.
Рост цепи:
r.+ .O-O- --------------------► R-O-O-
R-O-O. + R-H ----► R-OOH + R-
гидропероксид
гидроперекись
R-O-O. + R-----► R—О—О—R
перекись
Например,
(СН3)2СНОН
спирт
(СН3)2С=О
кетон
* СН3ОН + СН3СНО —СН3СООН
альдегид кислота
RH
-ROH
Сп3 1
-Н2О
3.4.2. Термическое расщепление алканов
При температуре выше 500°С алканы становятся нестабильными и распа-
даются с выделением водорода и образованием углеводородов с более низкой
25
молекулярной массой. В этих реакциях происходит гомолитический разрыв
связей С-Н и С-С. Термические превращения алканов называются крекингом.
Метан наиболее устойчив (Ес-н =102 ккал/моль, £с~с = 88 ккал/моль):
1400°С
2СН4 > НС=СН + Н2С=СН2 + Н2
Этан дегидрируется при более низких температурах:
CHj-CHa —°?°С-*- HteCH + Н2С=СН2 + Н2
Алканы с более длинной цепью образуют ненасыщенные углеводороды
или распадаются на углеводороды с меньшей массой (т.е. идет деструкция С-С
связей):
СН3СН2СН2СН3
СН3СН=СНСН3 + Н2
СН3СН=СН2 + СН4
СН2=СН2 + С2Нб
Приведем, например, механизм направления б):
СН3СН2СН2^СН3
гомолитический
разрыв
СН3СН=СН2 + СН4
н-Алканы с шестью и более углеродными атомами в присутствии катали-
затора способны к циклизации и образованию бензола и его производных:
t°
С6Н14 —
+ 4Н2
Каталитический крекинг (на алюмосиликатах). В этом случае дополни-
тельно проходит изомеризация углеродного скелета и образуются разветвлен-
ные углеводороды, что имеет важное значение в промышленности.
Крекинг тяжелых фракций нефти. При простой перегонке нефти получа-
ется 20% бензина, при каталитическом крекинге - 80% бензина.
Пиролиз проходит при ~700°С. Состав продуктов иной: этилен, ацетилен,
бензол, ароматические углеводороды.
3.4.3. Изомеризация и перегруппировки алканов
Под действием сильных электрофильных реагентов (кислот Льюиса: А1С13,
А1Вг3; сверхкислот: BF3+HF, SbFj+HF, SbFj+FSOsH) алканы превращаются в
изомерные углеводороды с разветвленной цепью. Кислоты Льюиса изомеризу-
ют алканы при повышенных температурах, сверхкислоты - при обычных тем-
пературах.
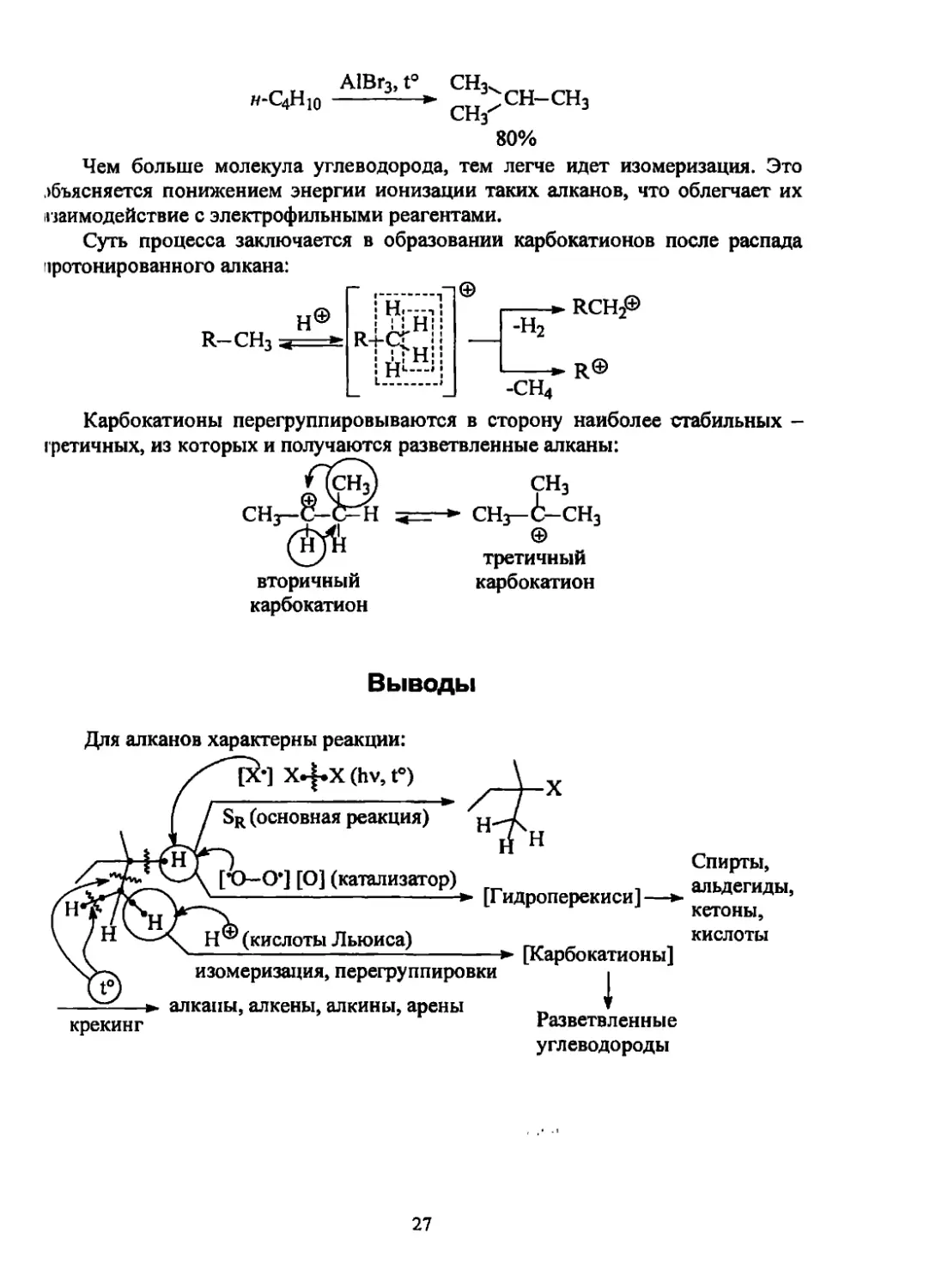
26
А1Вг3, t° СН3.
Н-С4Ню -----'/СН-СН3
Сг1з
80%
Чем больше молекула углеводорода, тем легче идет изомеризация. Это
объясняется понижением энергии ионизации таких алканов, что облегчает их
взаимодействие с электрофильными реагентами.
Суть процесса заключается в образовании карбокатионов после распада
зротонированного алкана:
©
н©
R—СН3
! Н. -
---► RCH,®
-н2
----- R©
-СН4
в сторону наиболее стабильных -
вторичный
карбокатион
Карбокатионы перегруппировываются
гретичных, из которых и получаются разветвленные алканы:
СН3
СНз- i- СН3
©
третичный
карбокатион
Выводы
Для алканов характерны реакции:
[Х-] Хфх (hv, t°)
SR (основная реакция)
(кислоты Льюиса)
X
[О-O’] [О] (катализатор)
[Гидроперекиси]
изомеризация, перегруппировки
алканы, алкены, алкины, арены
крекинг
[Карбокатионы]
Спирты,
альдегиды,
кетоны,
кислоты
Разветвленные
углеводороды
27
4. Алкены
Углеводороды, в молекулах которых кроме и-связей есть л-связи, называ-
ются непредельными.
Углеводороды с одной л-связью называются алкенами (этиленовыми угле-
водородами, олефинами).
Общая формула алкенов СПН2П.
4.1. Изомерия и номенклатура
I) Структурная - углеродного скелета.
2) Изомерия положения двойной связи - положения функции.
3) Геометрическая изомерия.
Напоминание: вращение вокруг С-С связи требует затраты энергии
~3 ккал/моль. Для этого достаточно теплового движения молекул.
Вращение фрагмента СНг= в этилене требует разрыва л-связи и затраты
65 ккал/моль.
"Ш"
Поэтому соединения, содержащие у атомов углерода, связанных двойной
связью, хотя бы по одному разному заместителю, могут существовать в изо-
мерных формах. Эти соединения отличаются расположением заместителей от-
носительно плоскости л-связи и называются в простейшем случае: z/uc-изомер -
одинаковые заместители находятся по одну сторону плоскости двойной связи,
транс-изомер - по разные стороны. Для обозначения геометрических изомеров
предложены также буквенные обозначения: Z для цис- (от нем. zusammen - вме-
сте) и Е для транс-изомера (от нем. entgegen - напротив).
НН Н СН3
НзС^^сНз h3(?=Si
цис (Z) транс (Е)
Если у двойной связи имеются три или четыре различных заместителя, то
обозначения Z и Е выбираются по старшинству, т.е. необходимо найти два
старших заместителя и определить: по одну (Z) или по разные стороны (Е) они
находятся относительно плоскости двойной связи.
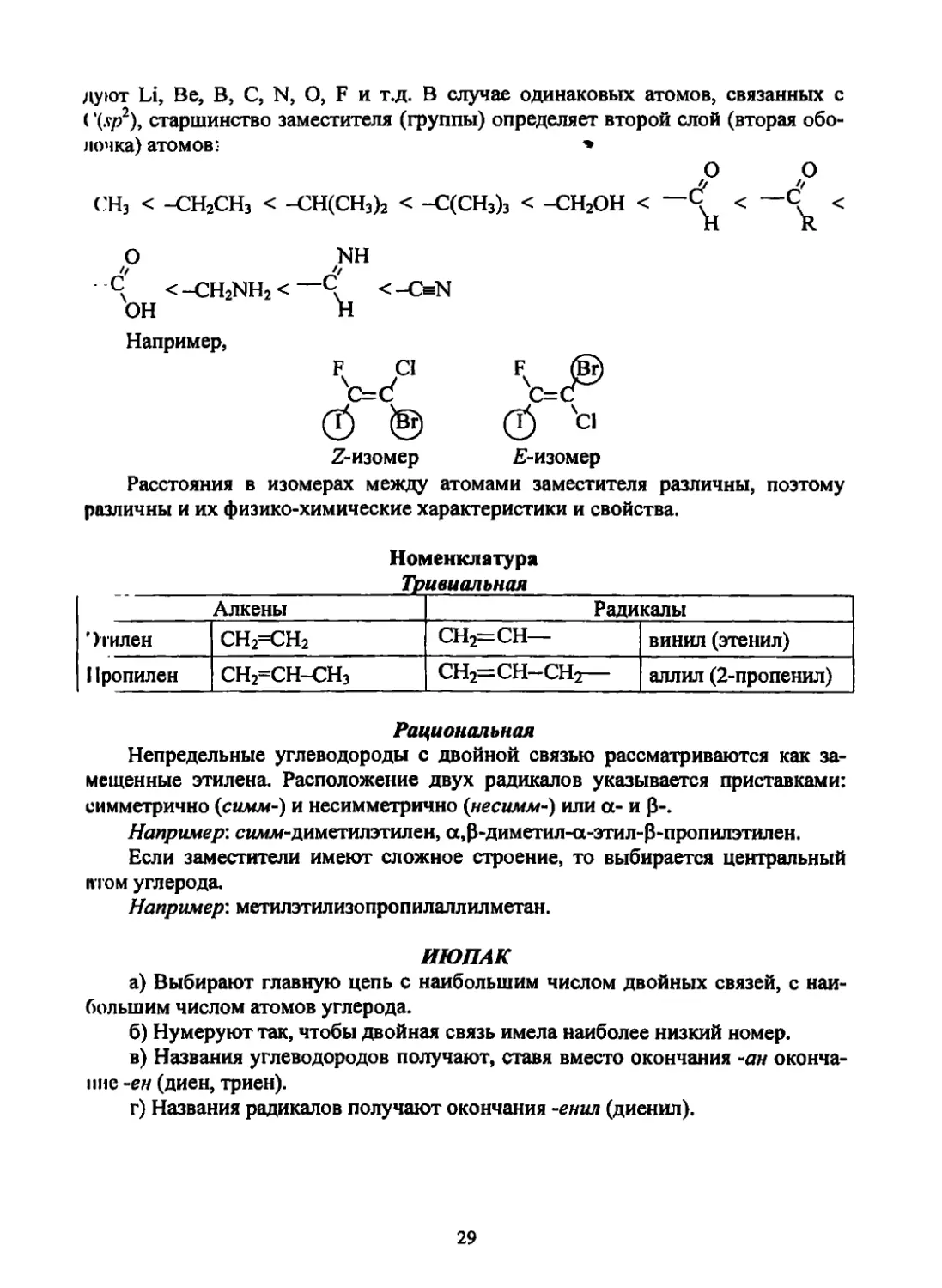
В основе определения старшинства находится атомный номер элемента.
Отсюда самым младшим атомом и заместителем является водород, затем сле-
28
дуют Li, Be, В, C, N, О, F и т.д. В случае одинаковых атомов, связанных с
(\.чр2), старшинство заместителя (группы) определяет второй слой (вторая обо-
лочка) атомов: *
О О
СН3 < -СН2СН3 < -СН(СН3)2 < -С(СН3)з < -СН2ОН < ~~
О
с/ < -CH2NH2 < —С/ < -ON
ОН Н
Например,
Z-изомер Е-изомер
Расстояния в изомерах между атомами заместителя различны, поэтому
различны и их физико-химические характеристики и свойства.
Номенклатура
Тривиальная
Алкены Радикалы
'Усилен СН2=СН2 СН2=СН— винил (этенил)
Пропилен СН2=СН-СН3 СН2=СН-СН2— аллил (2-пропенил)
Рациональная
Непредельные углеводороды с двойной связью рассматриваются как за-
мещенные этилена. Расположение двух радикалов указывается приставками:
симметрично {симм-} и несимметрично {несимм-} или а- и р-.
Например: сшш-диметилэтилен, а,Р-диметил-а-этил-р-пропилэтилен.
Если заместители имеют сложное строение, то выбирается центральный
атом углерода.
Например: метилэтилизопропилаллилметан.
ИЮПАК
а) Выбирают главную цепь с наибольшим числом двойных связей, с наи-
большим числом атомов углерода.
б) Нумеруют так, чтобы двойная связь имела наиболее низкий номер.
в) Названия углеводородов получают, ставя вместо окончания -ан оконча-
ние -ей (диен, триен).
г) Названия радикалов получают окончания -енил (диенил).
29
сн2=сн— этенил
СНз—сн=сн-сн7- 2-бутенил
сн2=&-сн=сн- 1,3-бутадиенил
сн2=с^ винилиден
д) Заместители (радикалы) перечисляют в названии в алфавитном порядке.
Например,
С2Н5 СН3
СН2= сн- 6н—СН- СН3
I 2 3 4 5 6
4-метил-З -этил гексадиен-1,4
4.2. Получение
Реакции получения алкенов основываются на отщеплении атомов или
атомных группировок от алканов и их производных. Меньше применяются ре-
акции, в которых алкены образуются из соединений с тройной связью или не-
сколькими двойными связями, и реакции конденсации.
Реакции получения алкенов относятся к реакциям отщепления или элими-
нирования (Е).
1. Дегидрирование и крекинг алканов (промышленный метод). Алке-
ны получают отщеплением двух атомов водорода от молекулы алкана. Эти ре-
акции идут при повышенной температуре и в присутствии катализаторов.
2. Отщепление воды от спиртов (Е2, Зайцев). Отщепление воды достига-
ется различными способами. Отщепление воды проходит по правилу Зайцева,
которое гласит: протон отщепляется от наименее гидрогенизированного атома
углерода, соседнего (вицинального) с атомом углерода, содержащим гидро-
ксильную группу,
а) при нагревании в присутствии сильных кислот:
OSO3H“
сн3сн2<Ьнсн3
ОН
H2SO4,t
-Н2О
СН3СН=СНСН3
б) при повышенной температуре на катализаторе:
ОН
СН3-СН2-(Ьн-СН3--------2 3 > СН3СН=СНСН3
300-350°С
3. Отщепление галогеноводорода или галогена от галогеналканов.
а) Действием спиртовых растворов щелочей:
RCHr-СН2Вг ^ОН^ннрт-).» RCH=CH2
-NaBr
-Н2О
30
Cl
(СН3)2СН-6н-СН3 —► (CH3)2C=CHCH3
Отщепление галогеноводорода идет по правилу Зайцева.
б) Обработка дигалогеналканов цинком:
ch2ci-ch2ci » сн2=сн2
4. Разложение аммониевых оснований при нагревании (Гофман, Е2).
Отщепление воды проходит по правилу Гофмана, т.е протон отщепляется от
ипиболее гидрогенизированного атома углерода, соседнего с атомом углерода,
содержащим функциональную группу.
N(CH3)3~|_
СНз— СНг- СН-СНз]0Н —► СН3—CHj—СН=СН2
-N(CH3)3
5. Гидрирование диенов и алкинов.
При использовании селективных катализаторов гидрирования удается гид-
рировать диены и алкины до алкенов:
Н2
СН2=СН-СН=СН2 —=-► СНз—сн=сн-сн3
Ni
Pd/РЬСОз
R—СН=СН—R
6. Через илиды фосфора (Витгиг):
(СН3)2С=О + (СбН5)3Р=СН2 — (СН3)2С=СН2 + (СбН5)3Р=О
илид фосфора трифенил-
фосфиноксид
4.3. Физические свойства, пространственное и электронное
строение, реакционная способность
Первые представители гомологического ряда алкенов (С2-С4) при обычных
температурах представляют собой газы, следующие члены ряда - бесцветные
жидкости или кристаллические вещества. Этилен и пропен имеют слабый за-
IIUX. При увеличении числа углеродных атомов и разветвлении цепи запах ста-
новится едким, раздражающим слизистую оболочку.
Н\ 1.33 А У11
с=с Л нб°
Н'говА
31
Молекула этилена плоская, углы между связями близки к 120° (тригональ-
ные углы); связь С=С (1.33 А) намного короче связи С-С (1.54 А).
Для объяснения строения и реакционной способности олефинов исполь-
зуют гипотезу о ^//-гибридизации атома углерода.
Углеродный атом в этилене образует три и-связи с использованием трех
$р1 2-гибридных орбиталей и одну л-связь за счет перекрывания р-орбиталей.
it
К” ч С+2> z
или
н fl__________fl н н \
Наличие л-связи объясняет новые свойства алкенов по сравнению с алка-
нами, так как л-связь, во-первых, менее прочна, чем а (65 и 88 ккал/моль, соот-
ветственно); во-вторых, пространственно более доступна для атаки и, в-
третьих, легко поляризуема, т.е. реакционноспособна.
Отрыв электрона от л-орбитали требует затраты меньшего количества
энергии, чем отрыв ота-орбитали примерно на 1-1,5 эВ.
Энергия отрыва электрона характеризуется энергией ионизации (ЭИ).
Энергия ионизации у этана равна 11,6 эВ. _______________________________
Алкен СН2=СН2 СН3СН=СН2 СН3 сПз— i=cH2 СН3 СН3 снз- i=i-CH3
ЭИ, эВ 10,5 9,7 9,2 8,3
То есть алкены имеют более сильно выраженные электронодонорные
свойства. Введение метильных групп также благоприятствует понижению ЭИ.
л-Связь можно представить в виде «сгустка электронной плотности». По-
этому основным типом реакций алкенов будут реакции электрофильного при-
соединения (AdE).
4.4. Химические свойства
4.4.1. Реакции электрофильного присоединения (AdE)
Алкены являются электронодонорными молекулами и проявляют , в пер-
вую очередь, слабые нуклеофильные свойства. Поэтому их реакция с электро-
фильными реагентами является основной.
1. Галогенирование
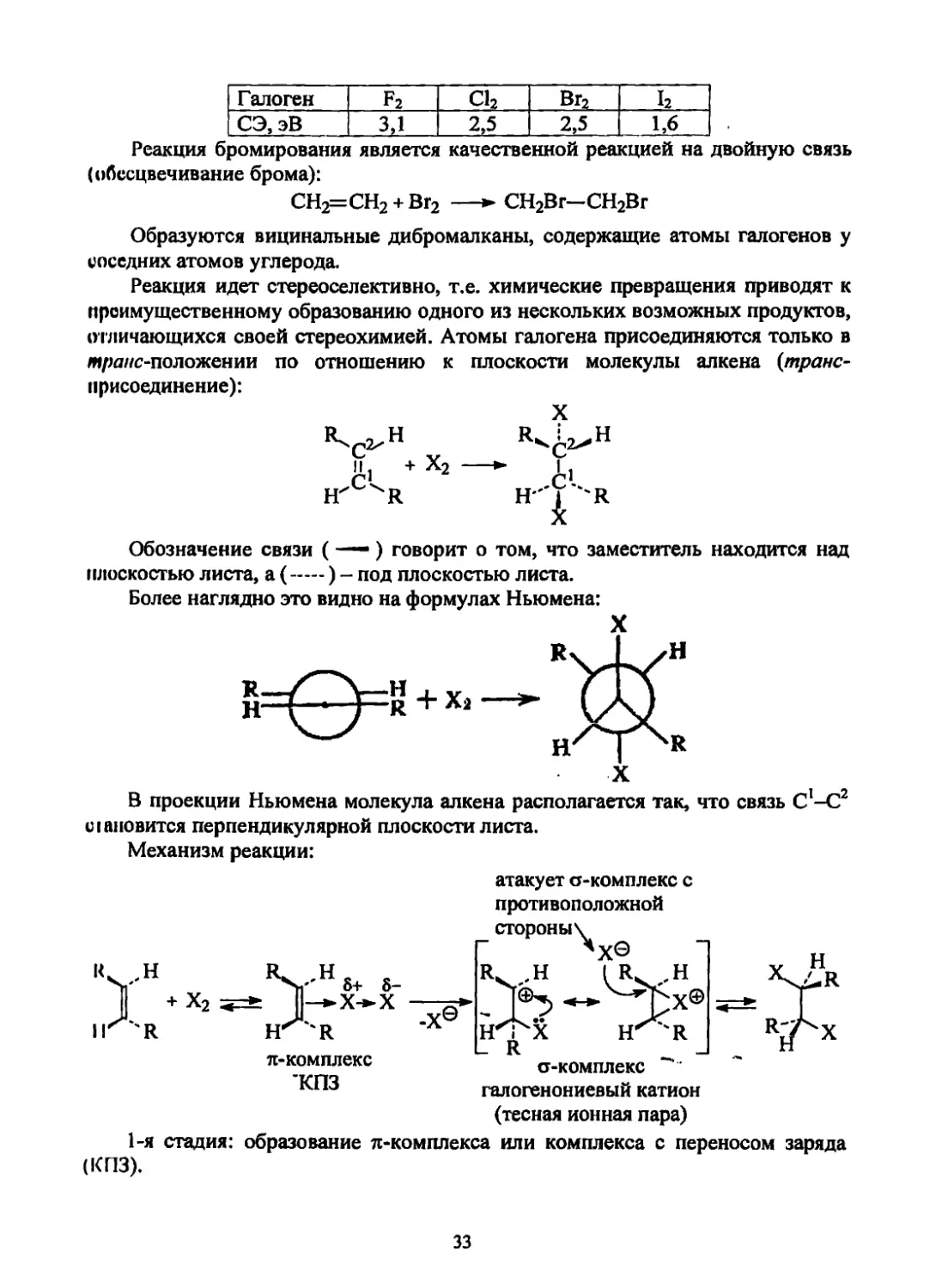
Алкены легко присоединяют молекулы галогенов Х2 (X = F, Ci, Br, I).
Очень энергично, даже со взрывом, реагирует фтор, медленно реагирует йод.
Это согласуется со сродством к электрону (СЭ) молекул галогенов:
32
Галоген f2 С12 Вг2 12
СЭ, эВ 3,1 2,5 2,5 1,6
Реакция бромирования является качественной реакцией на двойную связь
(обесцвечивание брома):
СН2=СН2 + Вг2 —► СН2Вг—СН2Вг
Образуются вицинальные дибромалканы, содержащие атомы галогенов у
соседних атомов углерода.
Реакция идет стереоселективно, т.е. химические превращения приводят к
преимущественному образованию одного из нескольких возможных продуктов,
отличающихся своей стереохимией. Атомы галогена присоединяются только в
мра//с-положении по отношению к плоскости молекулы алкена (транс-
присоединение):
X
It , Н R. :?.Н
п + х2 —I J
H"'^"'R
Обозначение связи (—) говорит о том, что заместитель находится над
плоскостью листа, а (-) - под плоскостью листа.
Более наглядно это видно на формулах Ньюмена:
В проекции Ньюмена молекула алкена располагается так, что связь С!-С2
наловится перпендикулярной плоскости листа.
Механизм реакции:
л-комплекс
КПЗ
атакует a-комплекс с
противоположной
а-комплекс
галогенониевый катион
(тесная ионная пара)
1-я стадия: образование л-комплекса или комплекса с переносом заряда
(КПЗ).
33
2-я стадия: образование a-комплекса карбокатиона (К), который тут же
преобразуется в циклический ион галогенония (ИГ).
3-я стадия: атака X® галогенониевого иона с противоположной стороны
(теранс-присоединение).
Образующийся на второй стадии ион может реагировать и с любым иным
нуклеофилом.
Например:
сн2=сн2 —
8-
Вг2, СН3ОН
CH2Br—CH2Br
СН2(ОСН3)—CH2Br
8- 3+
С12+Н2О [НО-С1]
хлорноватистая I ‘
кислота
СН2-СН2С1
хлоргидрины
гликолей
Это свидетельствует в пользу по-стадийного протекания реакции.
2. Присоединение Н-электрофилов (сильных кислот)
а) Присоединение галогеноводородов:
X
rch=ch2 r6h-ch3
Присоединение идет в соответствии с правилом Марковникова: протон
присоединяется к тому углеродному атому, у которого меньше углеводородных
заместителей (к «более гидрированному»).
Объяснение этому можно дать в рамках двух подходов.
1-й подход. Анализ основного состояния молекулы.
Алкильная группа как электронодонор поляризует молекулу, увеличивая
8- на атоме углерода группы СН2.
СнД»-СН:^СН2
В соответствии с механизмом реакции электрофильного присоединения
протон стремится присоединиться к атому углерода, несущему избыток элек-
тронной плотности.
2-й подход. Анализ переходного состояние молекулы:
© И
СН3-^СН-*-СН3 -п
Вг
сн3-£н-сн3
Н©
/ 1 НВг
СН3-СН=СН2
Вг0
г
© "1
II
34
Карбокатион I образуется легче (меньше Еах ), чем карбокатион II, т.к. он
более стабилен за счет лучшей делокализации положительного заряда (®) дву-
мя электронодонорными метильными группами
(11-эффект), чем карбокатион II, в котором (Ф)
делокализуется за счет одной этильной группы.
Кроме того, стабилизации карбокатиона I спо-
собствуют стерические (пространственные) эф-
фекты двух метильных групп. Естественно,
кпрбокатион - очень реакционная частица и ее
образование в ходе реакции в чистом виде ма-
ловероятно. Более вероятно образование ион-
ных пар, карбокатионов с анионами.
Скорость реакции уменьшается в ряду: HI > HBr > НС1.
В настоящее время присоединение кислот рассматривается как тримолеку-
лмрный процесс, в котором одна молекула кислоты поляризует л-связь алкена с
образованием л-комплекса (КПЗ), а вторая дает анион:
8+ 8- 8'+ 8'+ НХ
RCH=CHR + Н-Х RCH=CHR >
^Н-Х5"
8'+ Ч8'+ I Г
—*- RCH=CHR ---► R-C-C-R
S+^8- A A x®
б) Присоединение воды (в присутствии серной кислоты или другого ки-
слого катализатора):
RCH=CH2 НОН RCH-CH3
6н
Кислота способствует образованию карбокатиона.
3. Закономерности реакции Ade
1. Природа алкена._______________________
Алкен Скорость бромирования
СН2=СН2 3-10
Et-CH=CH2 2,9-103
(СНэ)2С=С(СН3)2 2,8-107
СН2=СН-Вг 1,1-10"3
cf2=cf2 не реагирует
Алкильные заместители увеличивают скорость реакции, а атомы галоге-
нов уменьшают.
35
Скорость реакции Ads уменьшается в ряду:
МеО^сО^ > Ме2С=СМе2 >^С=С^ » С1Хс=С^
/] 5+
НСГС’*-С=С''
т.е. электронодонорные заместители (+1, +М) увеличивают, а элекгроноакцеп-
торные заместители (-1, -М) уменьшают скорость реакции.
2. Природа реагента.
a) F2 > С12 > Вг2 > 12 (уменьшение сродства к электрону);
б) HI > HBr > НС1 (уменьшение силы кислот);
в) в присутствии кислот Льюиса скорость присоединения возрастает!
Х2 + А1С13 —► Х®[А1С13Х]0
Это объясняется возрастанием электрофильности атакующего агента при
переходе молекулы галогена в ионную пару с частицей X®.
3. Влияние природы растворителя.
С увеличением полярности среды растет скорость реакции, т.к. полярные
молекулы растворителя лучше стабилизируют карбокатионы и способствуют
их образованию.
4. Побочные процессы:
СН3. [сн^ © П С10
":С=СН2 + С12 —► "^С—СН2С1 -----► дихлорпроизводное
СН< [СН< J 20%
I—СН2=С-СН2С1
-Н® 6н3
80%
Промежуточные карбокатионы помимо взаимодействия с анионами гало-
генов способны к депротонированию с образованием алкенов.
5. Стереохимия присоединения.
Атомы галогена присоединяются только в транс-положение по отноше-
нию к плоскости молекулы алкена (транс-присоединение).
4.4.2. Реакции радикального присоединения (AdR)
Хараш установил, что если в реакционной среде присутствуют перекиси
или свободные радикалы, то реакция присоединения идет против правила Мар-
ковникова. Она идет по свободно-радикальному механизму.
Галогенирование
! -я стадия. Инициирование:
RO^OR RO (RCOO)
36
RO + Н^Вг ---------► Вг
гомоли- "ВОН бром-радикал
тический
разрыв
Радикалы RO, как и карбокатионы, являются слабо электронодефицитны-
ми частицами. Поэтому бром-радикал атакует л-электронную плотность моле-
кулы.
2-я стадия. Рост цепи:
СН3СН2СН2Вг Вг
СН3СН=СН2 + Вг -
^СН3СН-СН2вГ]
[сН3СНВг-СН^-^^*СН3СНВг~СН3 + Вг
II
Так как устойчивость радикалов падает в ряду: третичный > вторичный >
первичный, то энергетически выгодным является радикал I. Отсюда, в соответ-
ствии с правилом Хараша, конечным продуктом является первичный, а не вто-
ричный бромид - бромистый пропил.
3-я стадия. Обрыв цепи.
Димеризация радикалов.
4.4.3. Восстановление
1. Каталитическое гидрирование
Алкены присоединяют водород только в присутствии катализатора:
Ni Pt Pd
RCH=CH2 + H2 RCH2CH3
Взаимодействие атомов водорода с алкенами проходит как i/uc-присоеди-
псние, т.е. с одной стороны относительно плоскости молекулы алкена.
Н
Й
2. Гидроборирование
б(сн3)2с=сн2 + в2Нб - ВНз~* > 2[(СН3)2СН-СН7|3В
Н2О | | Н2О2
СН(СН3)3 (СН3)2СН-СН2ОН
Боран ВНз получается из диборана В2Нб.
37
M 1' 3. Оксосинтез (гидроформилирование)
Алкены реагируют с оксидом углерода и водородом под давлением в при-
сутствии кобальтовых катализаторов. В результате реакции образуются альде-
гиды, в отдельных случаях кетоны:
сн2=сн2 + со + н2 Со2.90~150°с>, сн3сн2с^°
10-30 МПа Н
Реакция имеет важное промышленное значение. »
4.4.4. Окисление
Алкены легко окисляются. В зависимости от окислителя и условий реак-
ции образуются различные продукты: двухатомные спирты (гликоли), эпокси-
ды, альдегиды, кетоны, карбоновые кислоты. Обычно окислители взаимодейст-
вуют с двойной связью. В определенных условиях окисление затрагивает ал-
лильное положение.
/. Окисление КМпО4 в слабощелочной среде (Вагнер)
Фиолетовый раствор КМпО4 при взаимодействии с алкеном обесцвечива-
ется, выделяется коричневый осадок (МпО2) - качественная реакция «на нена-
сыщенность».
Реакция проходит стереоспецифично, как i/пс-присоединение с разрывом
л-связи, и приводит к гликолям:
Кч~н о. о о
и + 'Mnz —► I Мп^
H,C..R о- ОТГ Н' о-к+
2Н2О
НО о
Мп*
нсГ 'сгкЛ
|н2о
МпО2 + кон
2. Окисление КМпО4 или СгОз в кислой среде
Окисление идет с разрывом о-связи С-С:
СН3хч
СН<
С=СН-СН3
б'
КМпО4, Н\
или СгО3
5с-снз
о^
нет
|[О]
с-сн3
38
3. Окисление пероксикислотами и кислородом
Алкены легко реагируют с пероксикислотами (Прилежаев). В результате
реакции образуются эпоксиды (оксираны):
RCH=CHR + СбН5С^^_о_н —► RCH-CHR + С6Н5СООН
пероксибензойная О
кислота
При взаимодействии алкенов с кислородом или воздухом в присутствии
NNi iunnaropoB тоже могут быть получены эпоксиды:
О2(катализатор)
RCH=CHR ——-------------RCH-CHR
У
4. Озонирование (Гарриес)
Алкены очень легко реагируют с озоном и образуют взрывчатые продукты
присоединения - мольозониды и озониды. Озониды при взаимодействии с во-
пий гидролизуются с образованием карбонильных соединений альдегидов и ке-
IDIIOD и перекиси водорода:
СГ'°'ЧО \О /
11(’П=СН2 —RCH—СН2 —► RCH ?CH2 Н2° > R-C?°+ СН2=О
мольозонид Ъ-Ю -Н2О2
озонид
Реакцию используют для определения положения двойной связи.
4.4.5. Полимеризация
Алкены в присутствии активных промежуточных частиц (карбокатионов,
кярПппионов, свободных радикалов) способны взаимодействовать друг с дру-
|<>м с образованием длинных цепей из углеродных атомов. Это процесс поли-
мер» шции, приводящий к димерам, тримерам, полимерам.
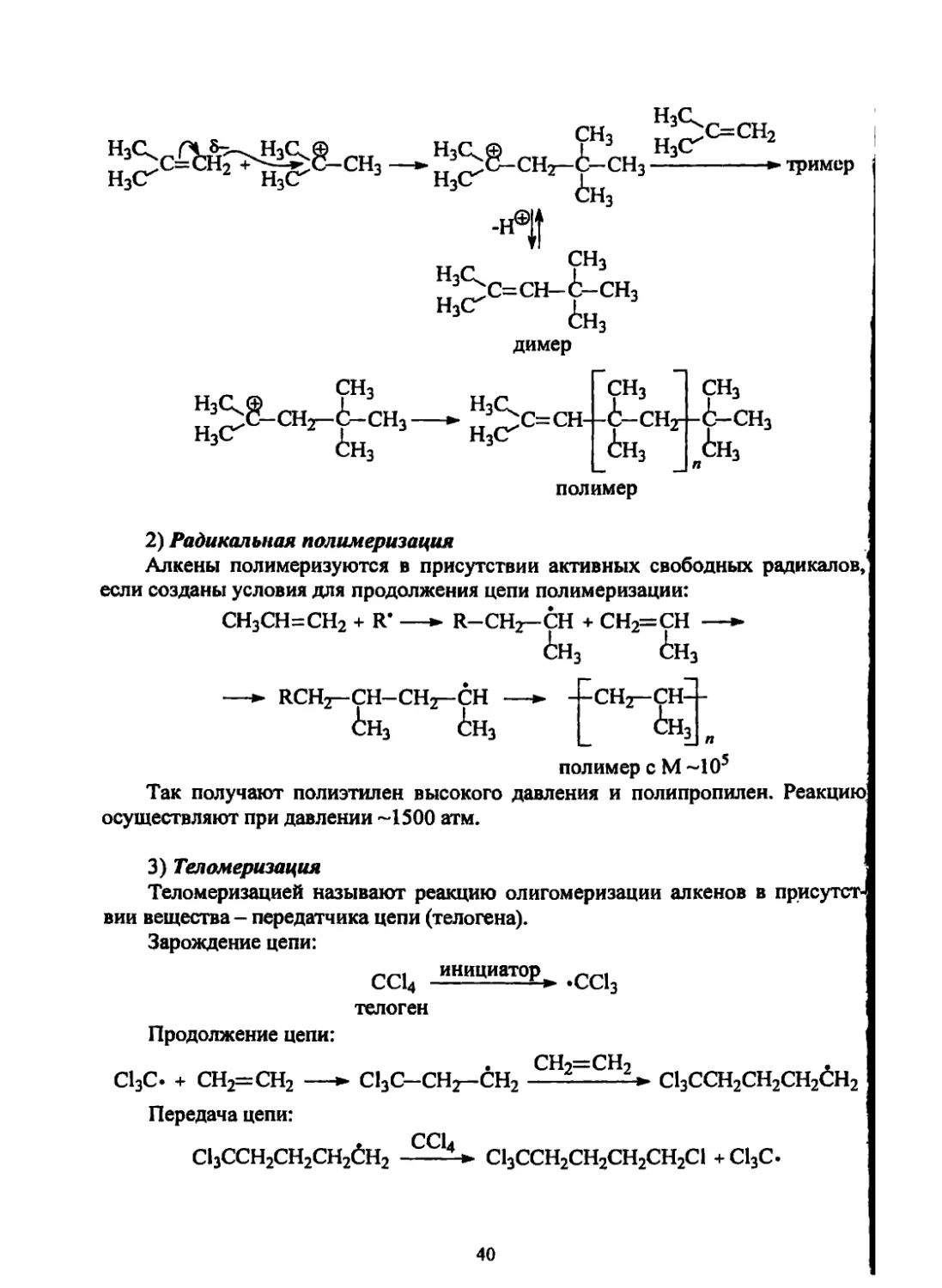
I) Катионная полимеризация
Й присутствии серной кислоты (используются также кислоты Льюиса BF3,
ЛК'h) способны полимеризоваться только алкены с несколькими электронодо-
нориыми алкильными группами (изобутилен). В зависимости от количества
серной кислоты получаются димер, тример или полимер:
39
H3cr 2 Н3СГ
H3C.S ?Нз НзС^™2
„ ^С-СНт-ё-СНз---------► тример
Нз<Г <Ьн3
-н®||
нс ?Нз
Зрс=сн- 6- СНз
НзСГ 6н3
димер
СН3
НзС.® сн. к сн г н3с_
и^С-снз-с-снз—► н3(г'
СН3
СН3 СН3
С= CH- -i-CHr - 6—сн3
£н3 (Ьн3
__ _____ я
полимер
(Ьн3
2) Радикальная полимеризация
Алкены полимеризуются в присутствии активных свободных радикалов,
если созданы условия для продолжения цепи полимеризации:
СН3СН=СН2 + R’ —► R-CHt-CH + СН2=СН —►
6н3 6н3
—► RCHz- СН-СНг- СН —► --СНг-СН--
6н3 6н3 _ <Ьнз я
полимер с М ~105
Так получают полиэтилен высокого давления и полипропилен. Реакцию
осуществляют при давлении ~1500 атм.
3) Теломеризация
Теломеризацией называют реакцию олигомеризации алкенов в присутст-
вии вещества - передатчика цепи (телогена).
Зарождение цепи:
СС14 .сс13
телоген
Продолжение цепи:
СН2=СН2 ,
С13С. + СН2=СН2 —► С13С-СН2-СН2----------С13ССН2СН2СН2СН2
Передача цепи:
С13ССН2СН2СН26Н2 1д-> С13ССН2СН2СН2СН2С1 +С13С.
40
Реакция осуществляется в промышленном масштабе и в результате полу-
чаются тетрахлоралканы С1(СН2СН2)„СС13 с п = 3-5, которые являются исход-
ными для гидрокси- и аминокислот.
4) Полимеризация в присутствии металлоорганических соединений
Алкены взаимодействуют с такими металлоорганическими соединениями
(МОС), в которых атом металла имеет свободные орбитали. Наибольшее прак-
тическое значение в данной реакции нашли катализаторы Циглера и Натта (на-
пример, (С2Н5)зА1Т1С14). Они способны легко полимеризовать алкены до поли-
меров с высокой молекулярной массой. Например, полимеризация этилена
оиуществляется при умеренном давлении и дает полиэтилен низкого давления с
М -100000-1000000. Активными частицами в катализаторах Циглера и Натта
являются титанорганические соединения R-TiCI3:
СН2=СН2
R—TiCl3 +СН2=СН2 —► R-CHz-CHz-TiCh-------------
СН2=СН2 .
—► RCH2CH2CH2CH2TiCl3-----------* R(CH2CH2)/ICH2CH2 + TiCl3
I
диспропорционирование,
рекомбинация
4.4.6. Реакции алкенов в аллильное положение (SR)
В большинстве алкенов присутствуют алкильные группы, поэтому они,
1ВКЖС как и алканы, способны вступать в реакции Sr. При этом двойная связь
особенно активирует связи С-Н у атома углерода, который находится рядом с
ней, г.е. аллильное положение алкена. Энергия гомолитического разрыва связи
(' II в пропене 77 ккал/моль, что значительно ниже, чем в (СН3)3С-Н -
V0 ккал/моль:
СН2=СН-СНг$-Н —
сн2=сн-сн2
If —^Ich^c-chJ
CHz-CH=CH2 III
И алилльный
радикал
Сравнительная легкость образования аллильного радикала связана с боль-
iiioll его стабильностью по сравнению с трет.-бутильным радикалом. Это, в
смою очередь, обусловлено высокой делокализацией неспаренного электрона
вллплыюго радикала, что показано на «граничных» структурах I, II и делокали-
к»лонной структуре III.
С промежуточным образованием аллильного радикала проходят следую-
щие реакции Sr.
41
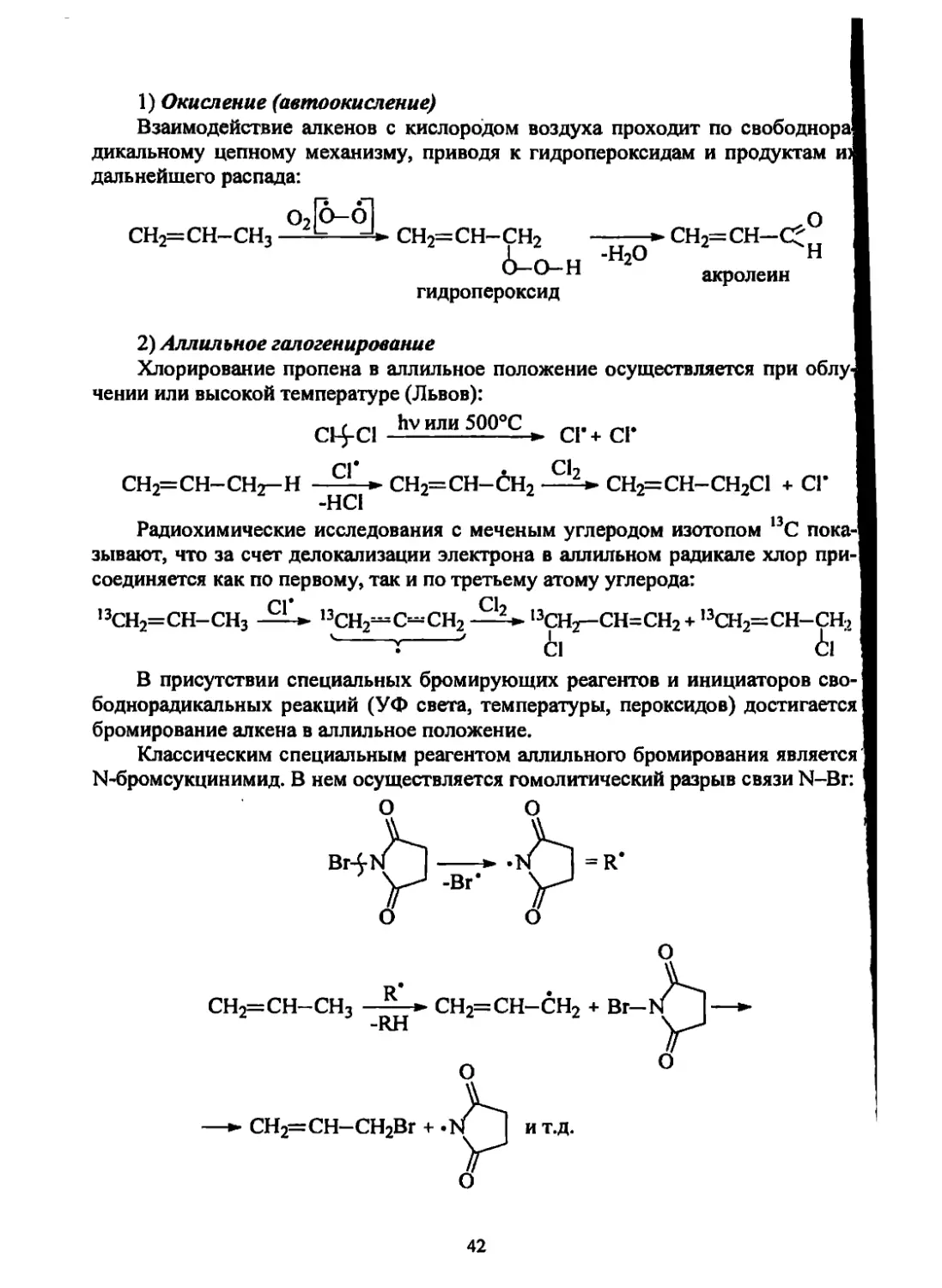
1) Окисление (автоокисление)
Взаимодействие алкенов с кислородом воздуха проходит по свободнора
дикальному цепному механизму, приводя к гидропероксидам и продуктам из
дальнейшего распада:
о21о— о] О
сн2=сн-сн3 —СН2=СН-СН2 -----------------► СН2=СН-С*н
О-Н 2 акролеин
гидропероксид
2) Аллильное галогенирование
Хлорирование пропена в аллильное положение осуществляется при облу-
чении или высокой температуре (Львов):
hv или 500°С * с|.+ ср
СН2=СН-СНг-Н - » СН2=СН-СН2 СН2=СН-СН2С1 + СГ
Радиохимические исследования с меченым углеродом изотопом 13С пока-
зывают, что за счет делокализации электрона в аллильном радикале хлор при-
соединяется как по первому, так и по третьему атому углерода:
,3СН2=СН-СН3 13СН^С~СН213СН2— СН=СН2 + ,3СН2=СН-СН2
' : ' ci ii
В присутствии специальных бромирующих реагентов и инициаторов сво-
боднорадикальных реакций (УФ света, температуры, пероксидов) достигается
бромирование алкена в аллильное положение.
Классическим специальным реагентом аллильного бромирования является
N-бромсукцинимид. В нем осуществляется гомолитический разрыв связи N-Br:
О О
R*
СН2=СН-СН3 ——
-RH
О
СН2=СН-СН2 + Вг-
о
о
СН2=СН-СН2Вг +.
ит.д.
42
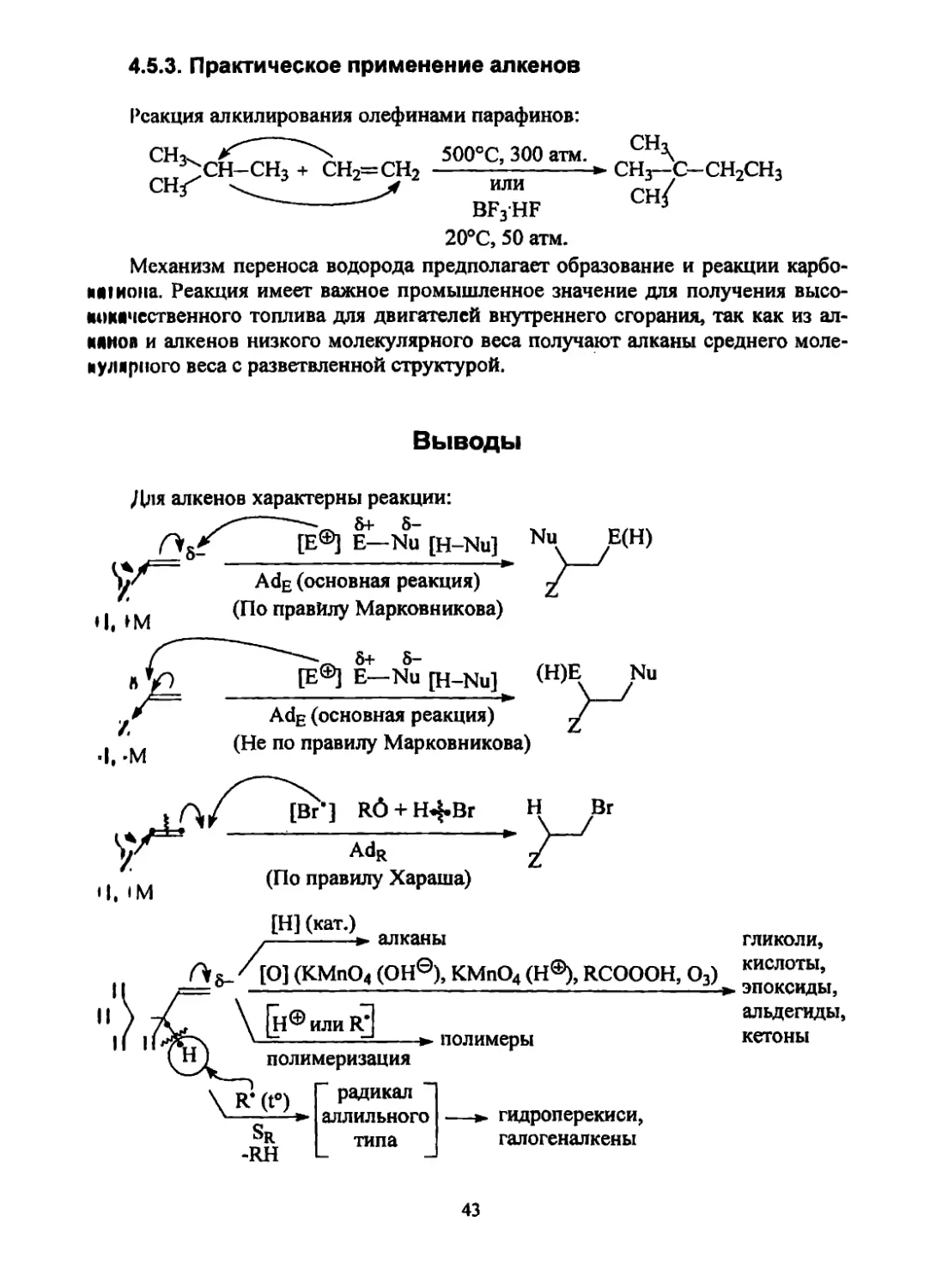
4.5.3. Практическое применение алкенов
Реакция алкилирования олефинами парафинов:
СНз^
СН<
СН-СНз + сн2=сн2
500°С, 300 атм.
или
BF3HF
20°С, 50 атм.
СН,
СН3—С-СН2СН3
сн/
Механизм переноса водорода предполагает образование и реакции карбо-
ми иона. Реакция имеет важное промышленное значение для получения высо-
кокачественного топлива для двигателей внутреннего сгорания, так как из ал-
кинов и алкенов низкого молекулярного веса получают алканы среднего моле-
кулярного веса с разветвленной структурой.
Выводы
Для алкенов характерны реакции:
5+ 5-
Е— Nu [H-Nu]
AdE (основная реакция)
(По правилу Марковникова)
5+ 5"
[Е®] Е—Nu [H-Nu]
AdE (основная реакция)
(Не по правилу Марковникова)
• I. IM
[Вг’ ] R6 + НЦ-Вг
AdR
(По правилу Хараша)
II
[Н] (кат.)
--------► алканы
[О] (КМпО4 (ОН®), KMnO4 (Н®), RCOOOH, О3)
Н® или R’
—--------=----полимеры
полимеризация
гликоли,
кислоты,
эпоксиды,
альдегиды,
кетоны
Sr
-RH
радикал
аллильного
типа
гидроперекиси,
галогеналкены
I, М
43
5. Алкадиены
Углеводороды с открытой цепью углеродных атомов, содержащие дв|
двойные связи, называются алкадиенами. Общая формула алкадиенов C2H2nJ
Этой общей формуле соответствуют также углеводороды о одной тройной свя|
зью ~ алкины. I
5.1. Номенклатура I
В молекулах алкадиенов может быть различное расположение двойным
связей: I
а) диены с соседним положением двойных связей называются алленовыми]
или кумулированными'. I
СН2=СН=СН2 - аллен или пропадиен-1,2
б) между двойными связями в молекуле алкадиена может находиться одна'
одинарная связь. Такие алкадиены называются сопряженными диенами',
СН2=СН-СН=СН2 - дивинил, бутадиен-1,3
СН2=С(СН3)-СН=СН2 - изопрен, 2-метилбутадиен-1,3
в) диены с изолированными двойными связями называются несопряжен-
ными диенами'.
СН2=СН-СН2-СН2-СН=СН2 - диаллил
Номенклатура ИЮПАК как у алкенов:
а) обе двойные связи должны входить в главную цепь,
б) положения двойных связей должны иметь наименьшие номера,
в) в названии наличие двух, трех двойных связей обозначают суффиксами
диен, триен и т.д.
6 5 4 3 2 1
CHj—СН—СН=СН—СН=СН2 - 5-метилгексадиен-1,3
6н3
5.2. Аллен (алкадиен-1,2) и его гомологи
5.2.1. Способы получения
а) из пропилена (Львов):
С12, 500°С , С12
СН2=СН-СН3 —-------► СН2=СН-СН2С1 —СН2С1СНС1СН2С1—►
КОН (сп.р-р) Zn
----iСН2=С(С1}-СН2С1 —► сн2=с=сн2
44
П) из глицерина:
________ ОСОСНз ОСОСН3ОСОСН3
СН3СООН I I 1
< 11,011-СНОН-СН2ОН------------► сн2---сн-----сн2
этерификация
‘ * СН2ВгСНВгСН2Вг ^Л0Н <СП-Р~Р)^ СН2=С=СН2
2) Zn
1.2.2. Пространственное и электронное строение, реакционная
алоообность аллена
Сиособразно пространственное строение молекулы аллена. Четыре атома
мн лоро да молекулы находятся не в одной плоскости, а в двух взаимно перпен-
дикулярных плоскостях. Все три углеродных атома аллена размещены на одной
прямой:
И , Н
\sp2 sp sp2/
Углеродные атомы в молекуле аллена находятся в различных состояниях
» иПридизации: sp2, sp, sp2. Средний углеродный атом образует две двойные (тс)
ннпи. Длина С-С связи 1,31 А:
Возможна стереоизомерия для двузамещенных алленов:
Н СН3
\ у
Н3С Н
1
И3С н
с=с=с
/ \
н сн3
II
Структура 1 является зеркальным изображением структуры II. Эти моле-
кулы хиральны.
6.2.3. Химические свойства аллена
За счет тс-связей аллены вступают в реакции электрофильного присоедине-
нии (АДе). Центром электрофильной атаки являются «боковые» углеродные
я । омы (зр2-гибридизированные).
45
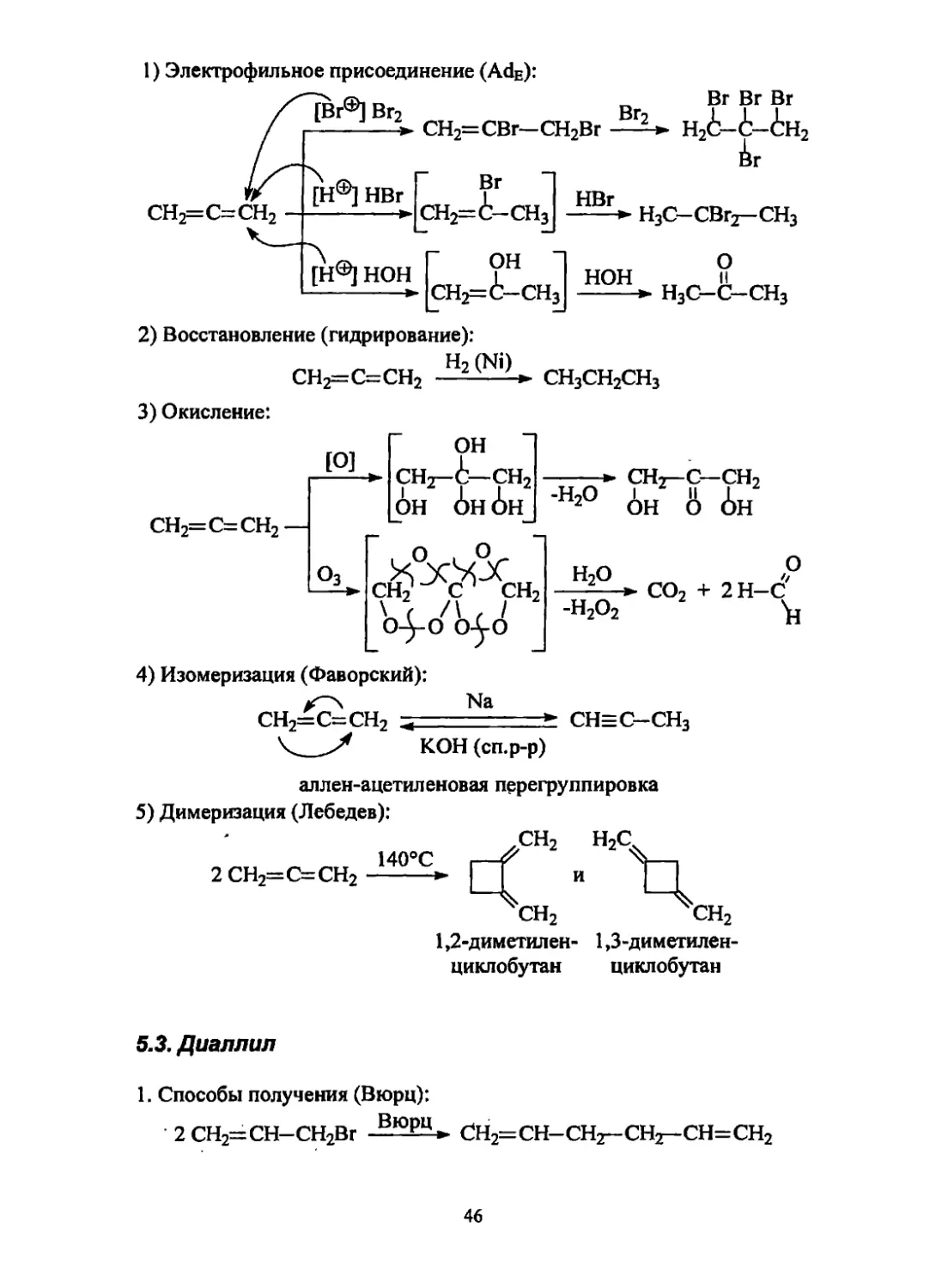
1) Электрофильное присоединение (AdE):
го о г, Вг Вг Вг
-Г? -Г» СН2=СВг—CH2Br H26-t-(!:H2
ir
сн2=с=сн2
[H®] HBr
Вг
СН2=6-СН3
НВг
---► Н3С-СВГ2-СН3
[Н®1 нон
он
СН2=(Ь—СН3
о
НОН II
----► H3C-C-CH3
2) Восстановление (гидрирование):
Н2 (Ni)
СН2=С=СН2 -» СН3СН2СН3
3) Окисление:
СН2=С=СН2 —
Оз
ОН
► снз-i—сн2
он 6н 6н_
сн2' с сн2
\ г ( 1
о-уо 04-0
СНз—с—сн2
6н о 6н
о
► СО2 + 2 Н-С
4) Изомеризация (Фаворский):
Na
СН2=С=СН2 < - СН=С-СН3
\___КОН (сп.р-р)
аллен-ацетиленовая перегруппировка
5) Димеризация (Лебедев):
2 СН2=С=СН2
140°С
1,2-диметилен- 1,3-диметилен-
циклобутан циклобутан
5.3. Диаллил
1. Способы получения (Вюрц):
2 СН2=СН-СН2Вг -ВюрЦ» СН2=СН-СН2-СН2-СН=СН2
46
1 Химические свойства похожи на свойства алкенов. Сначала вступает в
рмацию одна л-связь, затем другая.
9.4. Алкадиены-1,3
1.4.1. Получение
I) Получение бутадиена-1,3 (дивинила):
I) дегидрирование алканов - бутан-бутеновой фракции переработки нефти
I ЙЯМ1<ДИн, Богдан).
СН3— СН2-СН2— СН3—
СН2=СН-СН2-СН3 —
СН3-СН=СН-СН3 —
(цис- и транс-)
б) из этанола (Лебедев):
Сг2О3/А12О3
600°С
СН2=СН-СН=СН2
2 С2Н5ОН
А12О3, ZnO
400°С
- О О Н2
2 СН3С^ -► CH3CHCH2(t? —
н 6н н
-► СН3СН-СН2СН2ОН
он
А12О3
сн2=сн-сн=сн2
-Н2О
в) на основе ацетилена:
СН^СН-СнСН H2(Fe)> СН2=СН-СН=СН2 (Ньюленд)
Г||а('П
Н2О О как в способе б)
----► 2 СИ3С? -----------------
Н восстановление
СН2= СН-СН= СН2 (Кучеров)
нс? о
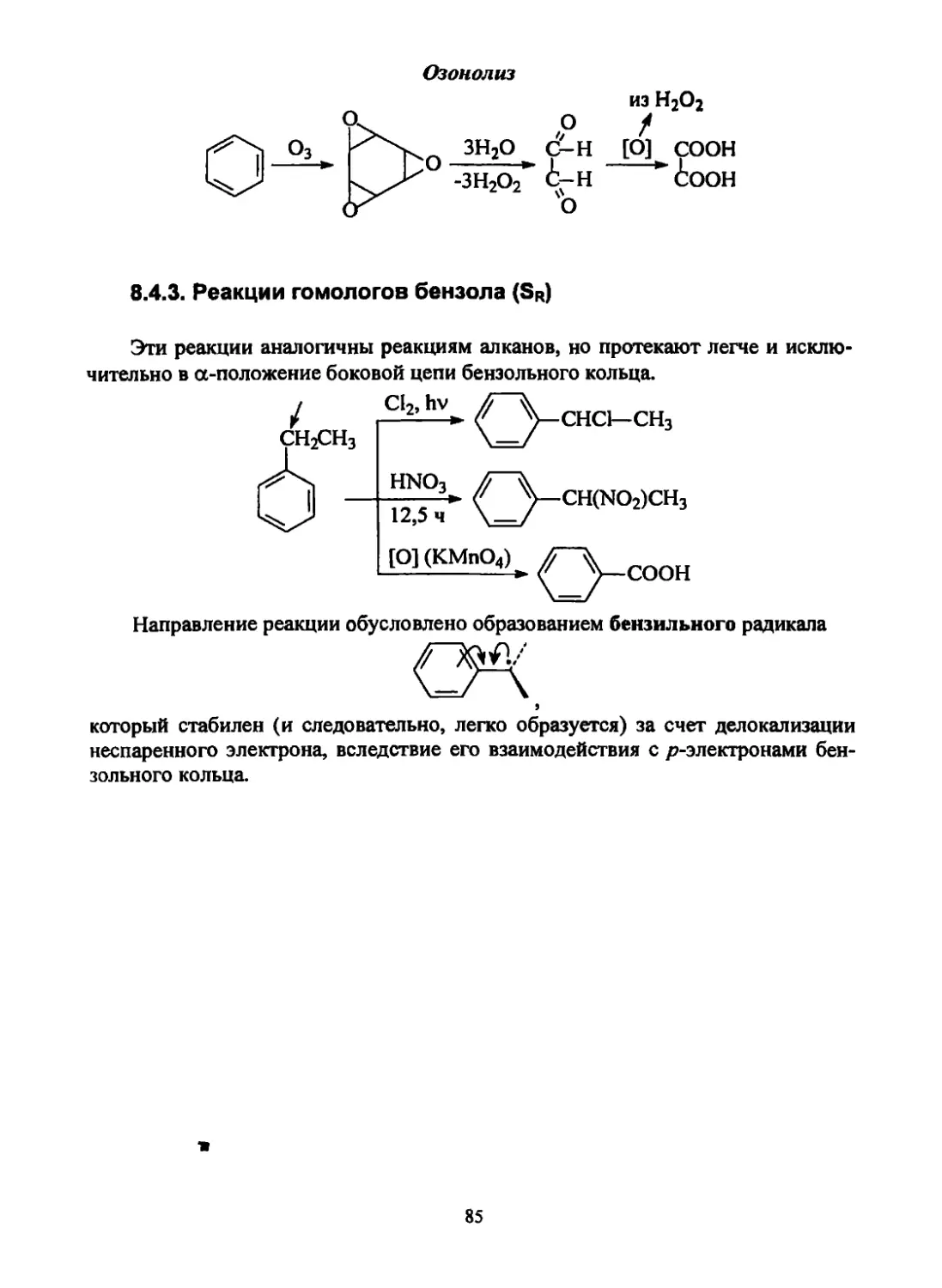
---НОН2С-С=С-СН2ОН Hz (pd)
—» НОН2С-СН2СН2-снрн ^^►сн2=сн-сн=сн2
2 (Фаворский, Реппе)
2) Получение изопрена:
й) аналогично дивинилу, только из изопентан-изопентеновой фракции пе-
реработки нефти;
47
б) из ацетилена и ацетона (Фаворский):
он
»3^С=О + CHECH И’^-СЕСН
H3C7 H3C7
он
—- !^i-CH=CH2 сн2=с-сн=сн2
н3<г -Н2О
в) из изобутилена и формальдегида (Принс):
СН3.
™^С=СН2 + 2^C=O CH
Цкат-д» сн с_сн=сн
~Н2° 6нз
-СН2О 3
5.4.2. Пространственное и электронное строение, реакционная
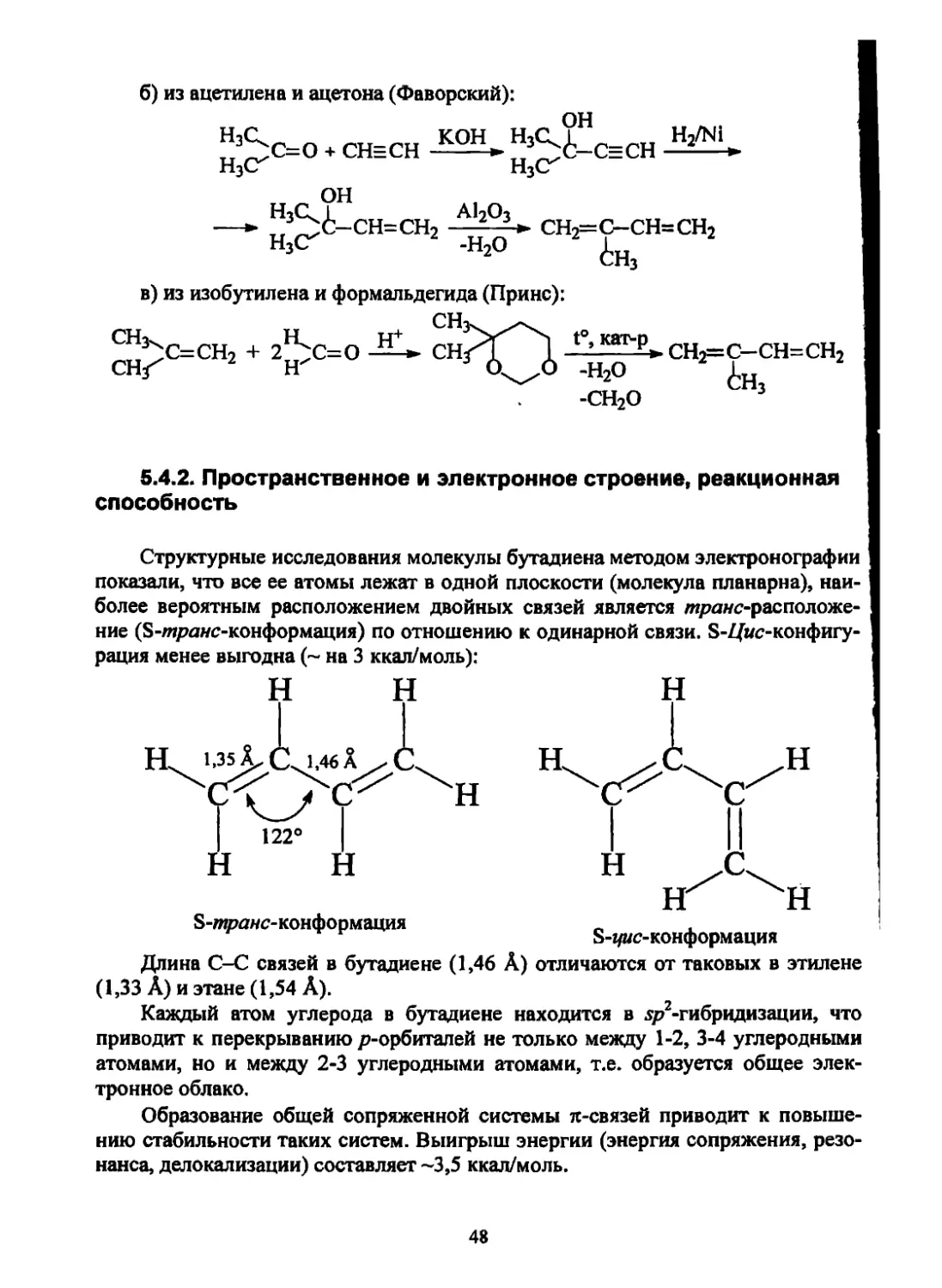
способность
Структурные исследования молекулы бутадиена методом электронографии
показали, что все ее атомы лежат в одной плоскости (молекула планарна), наи-
более вероятным расположением двойных связей является транс-расположе-
ние (S-транс-конформация) по отношению к одинарной связи. S-Z/uc-конфигу-
рация менее выгодна (~ на 3 ккал/моль):
S-г/ыс-конформация
S-транс-конформация
Длина С-С связей в бутадиене (1,46 А) отличаются от таковых в этилене
(1,33 А) и этане (1,54 А).
Каждый атом углерода в бутадиене находится в 5р2-гибридизации, что
приводит к перекрыванию р-орбиталей не только между 1-2, 3-4 углеродными
атомами, но и между 2-3 углеродными атомами, т.е. образуется общее элек-
тронное облако.
Образование общей сопряженной системы л-связей приводит к повыше-
нию стабильности таких систем. Выигрыш энергии (энергия сопряжения, резо-
нанса, делокализации) составляет ~3,5 ккал/моль.
48
11рисутствие л-электронного облака, как и в алкенах, приводит к тому, что
длп пл кадиенов-1,3 основным типом реакции будет реакция электрофильного
присоединения (AdE).
6.4.3. Химические свойства
5.4.3.1. Реакции электрофильного присоединения (AdE)
Особенности реакции AdE в случае алкадиенов-1,3 рассмотрим на примере
реакции галогенирования.
I) Галогенирование:
Вг2
сн2=сн-сн=сн2 —сн2=сн-сн=сн2--------------►
k V1*0
Вг—Вг
л-комплекс
—► СН2=СН-?Н-СН2 (?Н2-СН=СН-СН2
Ar Аг _
|+Br0 |+Bre
сн2=сн-сн-сн2 сн2-сн=сн-сн2
Ar Ar Ar Ar
1,2-присоединение 1,4-присоединение
На соотношение продуктов влияют температура реакции, полярность рас-
июрителя, характер реагента.
При -80°С: 80% продукта 1,2- и 20% продукта 1,4-присоединения.
При 40°С: 20% продукта 1,2- и 80% продукта 1,4-присоединения.
При низких температурах получается больше того продукта, скорость об-
Iлиования которого больше (кинетически контролируемый продукт реакции).
При высоких температурах образуется термодинамически более стабильный
продукт (термодинамически контролируемый продукт реакции).
При повышенных температурах процесс обратим и накапливается наибо-
лее стабильный продукт 1,4-присоединения:
а) при переходе от С12 к Вг2 и 12 возрастает доля продукта 1,4-присоедине-
ння,
б) 1,4-присоединение возрастает в ряду дивинил < изопрен < 2,3-диметил-
бутадиен.
2) Присоединение галогеноводородов:
НВг
СН2=СН-СН=СН2 ---------► СН3СН=СНСН2Вг
основной продукт
1,4-присоединения
49
5.4.3.2. Восстановление (гидрирование)
сн2=сн-сн=сн2 —
[H](Zn+HCQ
-----------► Urij— Сг1=Сп—СПз
1,4-присоединсние
И (Ni, Pd) Л [Н]
—-+ СН2=СН-СН2-СН3 —
1,2-присоединение
СП3СН2СН2СН3
5.4.3.3, Окисление гл ОН он он он — J* CHz-£h-£h-£h2 (Вагнер) КМпОд
СН2=СН-СН=СН2 — эритриты О3 О Ох ~ О \ —2 Н-С? + >С-С* (Гарриес) НН н
5.4.3.4. Присоединение диенофилов (диеновый синтез) (Дильс-Альдер)
Алкадиены-1,3 могут присоединяться к двойной (или тройной) связи с об-
разованием циклического продукта ((4+2]циклоприсоединение):
диен диенофил КПЗ согласованное
циклоприсоединение
(перициклическая
реакция)
Реакция Дильса-Альдера идет легче, если:
а) диен имеет электронодонорные заместители;
б) диенофил имеет электроноакцепторные заместители (например, это мо-
гут быть и-бензохинон, малеиновый ангидрид, акрилонитрил, акролеин);
в) скорость реакции увеличивается в присутствии кислот Льюиса.
5.4.3.5. Полимеризация
Алкадиены-1,3 легко полимеризуются в присутствии свободных радикалов
или металлорганических соединений. Полимеризация обычно происходит по
свободнорадикальному или по анионному механизму.
1. Радикальная полимеризация
Свободные радикалы генерируются из пероксидов.
50
R.'*+^CH2=^^-CH=^H2 —►(rCH2-CH= CH-CHj] ДИВИН”Л»
радикал делокализован
активная частица
-£сНг-СН= СН-СН^-
1,4-полимер
Получаются полимерыс нерегулярным строением.
Иногда наблюдается присоединение примеси или звеньев 1,2-полимера:
R. + СН2= ОТ
6н=сн2
дивинил
--СН—СН2—
сн=сн2 п
2. Анионная полимеризация
Происходит под действием металлорганических соединений, например
+’СН2=СН-СН=СН2'
RCH2—СН=СН-СН?
карбанион -*
активная частица
1,4-полимер
3. Катионная полимеризация
На катализаторах Циглера-Натта (алюминий- и титанорганические соеди-
нения) полимеризация идет очень легко. При этом в основном образуются сте-
рсорегулярные полимеры - г/ис-полиалкадиены:
5.5. Каучук
5.5.1. Натуральный каучук
Натуральный каучук представляет собой эластичную при низких темпера-
турах, пластичную при более высоких температурах массу, которую получают
51
из молочного сока латекса бразильской гевеи. В этом соке в виде эмульсии на-
ходится каучук. Из эмульсии при нагревании под действием кислот получают
эластичную массу - каучук-сырец. Каучук представляет собой ненасыщенный
ациклический полимер, молекула которого состоит из периодически повто-
ряющихся изопреновых групп 1,4-типа:
(С5Н8)Л
СН2-С=СН-СН
6н3
-I л
Молекулярная масса каучука 100000... 150000. Молекулы за счет вращения
вокруг одинарных связей принимают скрюченную конформацию. Отсюда эла-
стичность - при нагрузке распрямляется, в отсутствие нагрузки принимает ис-
ходное положение.
Натуральный каучук имеет строение г/нс-полиизопреиа. В природе встре-
чается также mptwc-полиизопрен, называемый гуттаперчей, который имеет
худшие механические свойства:
—CHz-CH, СНг-СН, CHj-CH,—
"cU 4c=d
сн/ Yi сн/ Yi
натуральный каучук (г/мс-строение)
—СН2-СН2 Н СН, СНг-СН
сн/ 4CHr-dH2 'й
гуттаперча (транс-строение)
5.5.2. Синтетический каучук
Первый промышленный метод получения синтетического каучука был
разработан Лебедевым в 1927 г. Бутадиен он получал из этилового спирта, а
полимеризация велась в присутствии натрия.
Начиная с 1950 г. исследовалась возможность получения стереорегулярно-
го каучука. Впервые стереорегулярную полимеризацию изопрена в присутст-
вии лития осуществил Коротков. В последующие годы при использовании ком-
плексных катализаторов Циглера-Натта было разработано промышленное про-
изводство стереорегулярного полиизопрена. По свойствам он не уступал нату-
ральному.
В 1956 г. (Долгоплоск) была разработана методика стереорегулярной по-
лимеризации бутадиена и получения z/wc-бутадиенового каучука. В настоящее
время не только производят изопреновый и бутадиеновый каучуки, но и полу-
чают хлоропреновый и фторопреновый каучук.
При обработке каучука серой получается материал с отличными механиче-
скими свойствами - резина (содержание серы 0,5-5%). Этот процесс был назван
52
иулкннизацией. В процессе вулканизации за счет двойных связей каучука обрв>
чуются мостиковые связи C-S-C или C-S-S-C между макромолекулами, кото-
рые соединяются таким образом в пространственные структуры.
Выводы
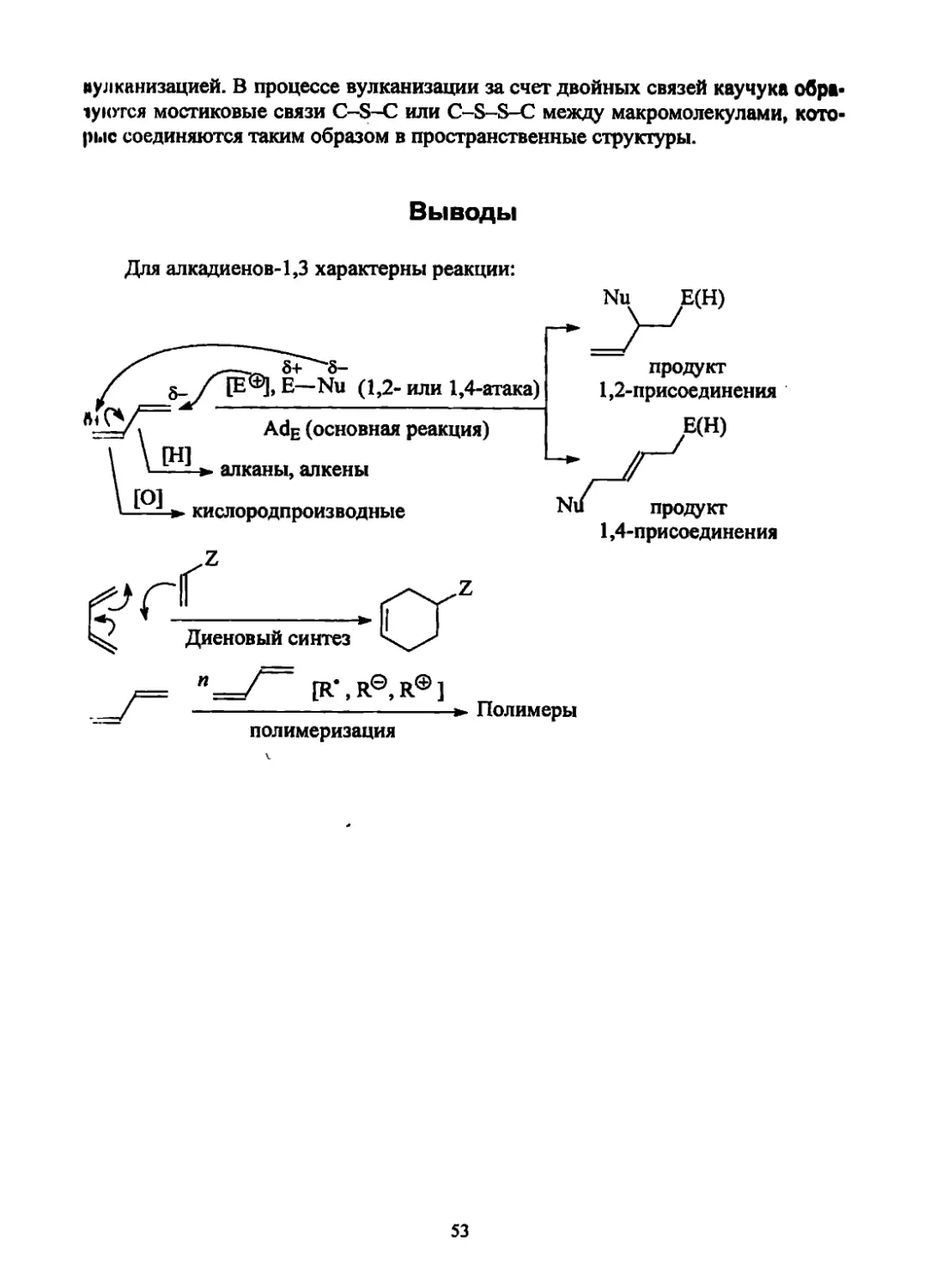
Для алкадиенов-1,3 характерны реакции: .—-.Г"з+ 8- / Ъ-/ [Е®1>Е—Nu (1,2-или 1,4-атака) Adg (основная реакция) \ \ И и- \ \- - - >. алканы, алкены \ [°] Ni/ * L J » кислородпроизводные .Qz f= n—/ [R*,R®, R®] / ► Полимеры полимеризация Nu^ /Е(Н) продукт 1,2-присоединения Е(Н) продукт 1,4-присоединения
53
в. Алкины
Углеводороды с тройной связью в открытой цепи углеродных атомов ru
зываются алкинами или ацетиленами. Их общая формула такая же, как дл
алкадиенов - СпН2п-2.
6.1. Номенклатура
Названия алкинов образуют, заменяя в названиях алканов суффикс -ан н;
-ин. Для первого представителя ряда алкинов С2Н2 сохраняется тривиальное на;
звание ацетилен, поэтому иногда гомологи ацетилена называют как замещен
ные ацетилены (рациональная номенклатура) и тройную связь - ацетиленовой
связью.
Углеродная цепь алкинов нумеруется таким образом, чтобы тройная связ!
получила наименьшую цифру (ИЮПАК).
Двойная связь старше тройной, поэтому нумеруют с двойной связи:
СН2=СН-СН=СН-С=СН гексадиен-1,3 -ин-5
6.2. Получение
1) Прямой синтез. Для получения ацетилена может быть использован
прямой синтез из элементов:
2С + Н2 Н—С=С—Н
2) Реакции пиролиза (промышленный метод). Ацетилен получается при
пиролизе метана или этана (этилена):
1500°С
2СН4 — > Н-С-С-Н + ЗН2
СНз— СНз СН2=СН2 + Н2 Н-С=С-Н + 2Н2.
3) Реакции карбидов (промышленный метод). В реакции карбидов метал-
лов с водой образуются алкины. Карбид кальция образует ацетилен:
СаС2 + 2Н2О -------► Н-С=С-Н
-Са(ОН)2
Карбид кальция можно рассматривать как металлорганическое соедине-
ние, содержащее сильно полярную связь углерод-металл:
8+ 8-
—Са—С=С—Са—С=С—
з- 5G+
но-н
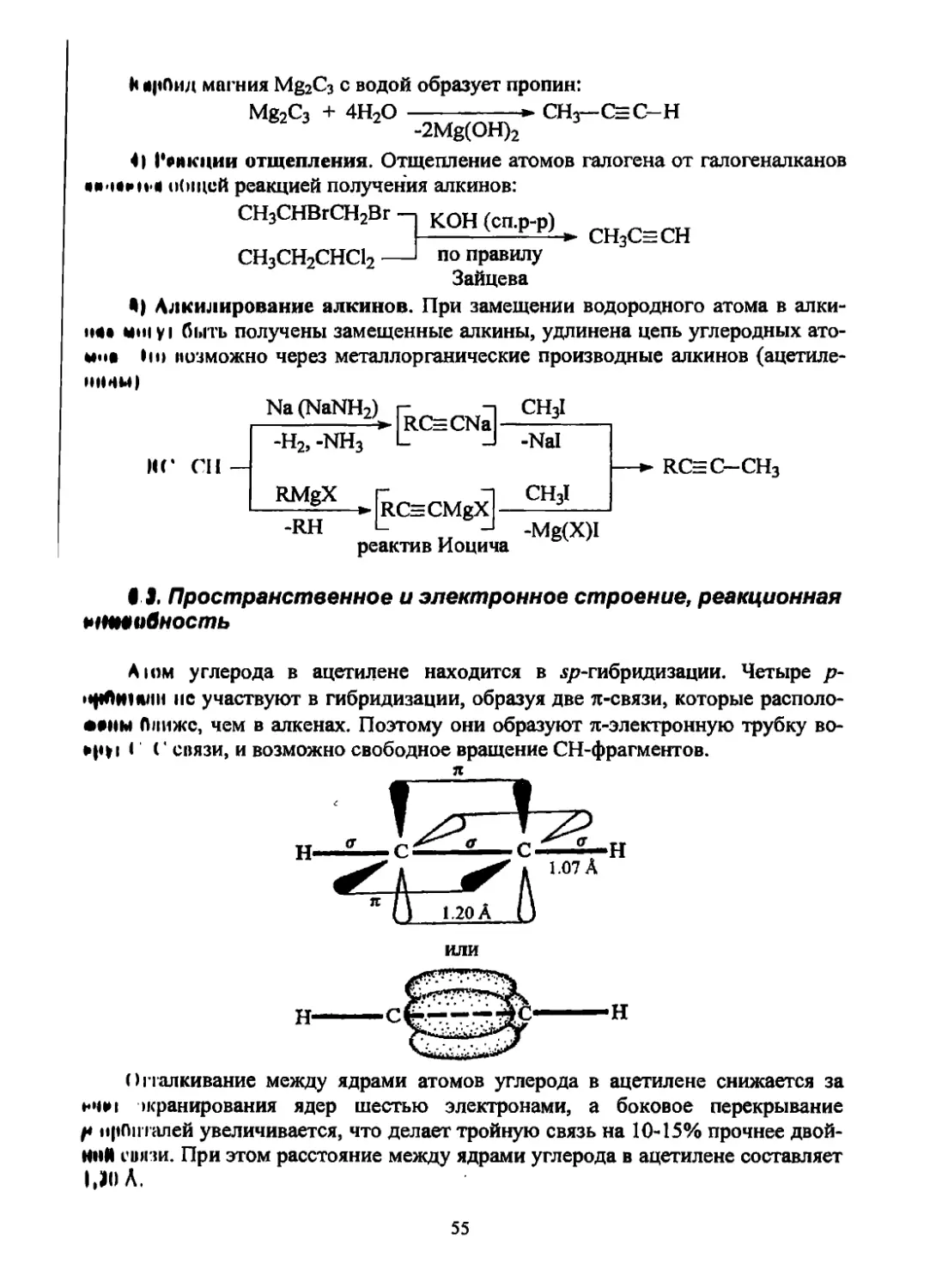
54
И ярПид магния Mg2C3 с водой образует пропин:
Mg2C3 + 4Н2О ———* СН3-С=С-Н
-2Mg(OH)2
4) 1'яйкцни отщепления. Отщепление атомов галогена от галогеналканов
«»'*•••»•>« о()|цей реакцией получения алкинов:
СН3СНВгСН2Вг —1 кОН(сп.р-р)
СН3СН2СНС12---1 по правилу
Зайцева
СН3С=СН
4) Алкилирование алкинов. При замещении водородного атома в алки-
не* мн| у । быть получены замещенные алкины, удлинена цепь углеродных ато-
м<'« ho возможно через металлорганические производные алкинов (ацетиле-
Н1ИЫ)
МГ СН —
-Н2, -NH3 L J -Nal
RC=C-CH3
RMgX Г “I СН31
--------- RC= CMgX---------
RH L J -Mg(X)I
реактив Иоцича
9 9. Пространственное и электронное строение, реакционная
тнюибность
Aiом углерода в ацетилене находится в ^-гибридизации. Четыре р-
’«pftWIiuiH не участвуют в гибридизации, образуя две л-связи, которые располо-
••им ближе, чем в алкенах. Поэтому они образуют л-электронную трубку во-
< (' связи, и возможно свободное вращение СН-фрагментов.
или
() ггалкивание между ядрами атомов углерода в ацетилене снижается за
ннн жранирования ядер шестью электронами, а боковое перекрывание
р орбиталей увеличивается, что делает тройную связь на 10-15% прочнее двой-
НнМ связи. При этом расстояние между ядрами углерода в ацетилене составляет
1,10 А.
55
Реакционная способность в реакциях электрофильного присоединения вы
сока, но меньше, чем в алкенах. тс-Электронная плотность сильнее втягивается i
пространство между атомами углерода, поэтому ядра атомов углерода доступ
нее для атаки нуклеофильных реагентов.
Энергия ионизации ацетилена больше, чем этилена, Это означает, чт<
электронодонорные свойства ацетилена меньше, чем у этилена. ЭИ снижаете:
при введении алкильных групп:_______________________________________
Углеводород СНз-СНз СН2=СН2 сн-сн СНз-CsCH 1 10,4
ЭИ, эВ 11,6 10,5 1L4
Таким образом, алкины способны участвовать как в реакциях AdE, так и в]
реакциях AdN. ]
Электроотрицательность углерода в 572-гибридизации выше (2,75), чем в'
sp2- (2,69) и ^-гибридизации (2,5). Эти особенности строения ацетиленов при-:
водят к поляризации связи =С<—Н5+. Поэтому более кислый водород в ацетиле-
не может замещаться металлами. Ацетилены проявляют СН-кислотность, кото-
рая намного больше, чем у алкенов и алканов.
6.4. Химические свойства
Алкины вступают в реакции присоединения как с электрофильными, так и
нуклеофильными реагентами. Важны реакции присоединения СО (карбонили-
рование). Они восстанавливаются и окисляются, а также участвуют в различ-
ных реакциях олиго- и полимеризации. В отличие от алкенов у алкинов с трой-
ной связью на конце молекулы (терминальные алкины) имеется реакционно-
способная группировка sC-H и возможны реакции с участием этого «ацетиле-
нового водорода».
6.4.1. Реакции электрофильного присоединения (AdE)
1. Присоединение галогенов
Реакция происходит медленнее, чем в случае алкенов, при этом могут об-
разоваться как транс-, так и гр/с-дигалогеналкены.
По аналогии с реакциями присоединения к алкенам сначала образуется
КПЗ, затем карбокатион. При этом происходит предпочтительно транс-присое-
динение нуклеофильного реагента:
Вг2 г и 1Вг®1 Г © “I Вг® Br. Н
СН3С=СН —[КПЗ] -------------*-|CH3C=CHBrj-------*СНГ %г
2. Присоединение галогеноводородов (Н-электрофилов) т
Взаимодействие с галогеноводородами (гидрогалогенирование) идет мед-
ленно, особенно для ацетилена. Алкилпроизводные ацетилена реагируют быст-
56
рсс и согласно правилу Марковникова. Как правило, происходит транс-при-
соединение:
X н
СН3С=СН + Н-Х —* сн^С==ССн
Реакцию присоединения галогеноводородов к тройной связи резко ускоря-
ет присутствие солей одновалентной меди. Здесь фактически происходит нук-
леофильное присоединение к тройной связи, активированной ионом металла.
11оэтому многие реакции присоединения к тройной связи, катализируемые ио-
нами металлов (CiT, Hg2+), могут рассматриваться как реакции нуклеофильного
присоединения.
6.4.2. Реакции нуклеофильного присоединения (AdN)
Ацетилен и его производные могут присоединять такие нуклеофильные
реагенты, как вода, спирты, карбоновые и другие кислоты, но в основном в
присутствии катализаторов - солей меди (I) или ртути (II).
1. Присоединение воды '
Алкины присоединяют воду в кислой среде в присутствии солей ртути
(Кучеров). Ацетилен в этой реакции дает уксусный альдегид, а другие алкины -
кетоны:
СН3С=СН + Н2О
СН3С=СН2 —► СНз-С-СНз
X и
он о
енол
Первичным продуктом присоединения является енол, который быстро пе-
регруппировывается в карбонильное соединение.
Вероятный механизм присоединения воды к ацетилену предполагает обра-
кшание л-комплекса между ацетиленом и ионом ртути:
Н Н „©
X Н 5+Л
ш + Hg2+X2" + Н®+ ;ОСН ^in-^Hf+X2------►
А Н-б: А
I
И л-комплекс
т_т
Н н® н-б"% н н®
Н - н®
57
2. Присоединение OUUpPUm I
Алкины присоединяют спирты как а приоуплаим аллаЙ ртути или Mej
так и в присутствии алкоксидов при нагриаиим. И атой раакции образуюч
простые алкениловые эфиры:
t° (КОН)
Hg2*
RC=CH
R’OH
II
Н
алмиилоам
«фирм
В случае ацетилена получаются виниловые эфиры.
3. Присоединение кислот |
Алкины присоединяют карбоновые кислоты, при ЭТОМ образуются ело:
ные алкениловые эфиры:
RteCH RlC°0H>
4. Присоединение цианистого водорода 1
В результате присоединения к ацетилену цианистого водорода образуете
нитрил акриловой кислоты (акрилонитрил):
HCN Н
----►
Ы ^C=N
НС=СН
6.4.3. Карбонилирование
В присутствии никелевых катализаторов алкины, в основном ацетилен
реагируют с оксидом углерода (Реппе). В этой реакции в случае ацетилена об
разуются акриловая кислота или ее производные (эфиры, амиды):
Ni(CO)4
НС=СН + СО + НХ —-—СН2=СН-С*
Л
х = он, ОС2Н5, nh2
Реакцию катализирует тетракарбонил никеля, который активирует трой
ную связь в результате образования тс-комплекса:
Н Н
A s+A
ш + Ni(CO)4^zfeg+^->Ni(CO)3 + CO
A A
58
Чатем следует присоединение СО. Предполагается, что в качестве промо*
логичного продукта образуется циклопропенон, который реагирует с водой,
ипиртом или аммиаком, присутствующими в реакционной среде:
н s+i Г <ГН” О—и НХ ^тт ^0 Г'Т-Т-.— Р14 Г*'
-Г _ III
5+С । -Ni(CO)3 % X
н
6.4.4. Гидрирование /
Алкины присоединяют водород в присутствии катализатора (Ni, Pt, Pd).
I идрирование осуществляется ступенчато. Обычно реакция происходит несте-
реосслективно и образуются как цис-, так и транс-алкены, а затем алканы:
RC=CH + Н2 Кат » RCH=CH2 —» RCH2-CH3
кат.
в.4.5. Окисление
Окисление алкинов проходит труднее алкенов. Окислителями являются
перманганат калия, дихромат в кислой среде, озон. В результате окисления ал-
кинов происходит расщепление молекулы по тройной связи и конечным про-
дуктом реакции являются карбоновые кислоты:
R—С=С—R1 » R-COOH + R1—СООН
6.4.6. Димеризация, циклоолигомеризация и полимеризация ал-
кинов
В присутствии катализаторов алкины могут образовывать димеры, цикли-
ческие тримеры и тетрамеры, линейные полимеры.
Димеризация
Ацетилен легко димеризуется в кислой среде в присутствии ионов Си'
(Ньюленд). Эта реакция рассматривается как присоединение молекулы ацети-
лена к активированной тройной связи:
Cu+, Н®,
Н
*СН
59
2. Димеризация
Циклотримеризация приводит к бензолу (Зминокий) и его производны!
Реакция осуществляется термически, в присутствии концентрированной серне
кислоты или лучше всего в присутствии металлическим катализаторов (соед)
нения Сг, Ni, Со):
tO’ H2S°4 (конц.)
j Г1 ———————
ЗНС=СН С(а—-‘А
R
3. Тетрамеризация :
При циклотетрамеризации ацетилена на катализаторе Ni(CN)2 образует»
циклооктатетраен (Реппе):
4НС=СН 1SaT-2_80L129°C>р
4. Полимеризация
В присутствии инициаторов (свободных радикалов) или в присутствш
специальных металлорганических катализаторов алкины полимеризуются. I
результате образуются сопряженные полиены:
RC=CR’
Такие системы в виде порошка или пленки обладают своеобразными элек-
трофизическими свойствами: повышенной электропроводностью, способно-*
стью повышать электропроводность при освещении (фотопроводимость). Такие
материалы называются органическими полупроводниками.
Своеобразная полимеризация возможна в случае ацетилена в присутствии’
ионов Си* и окислителей:
нс=сн • =С=С=С=С=-
карбин
Молекула полимера состоит практически только из углеродных атомов в
состоянии ^-гибридизации. Доказано, что полимерная цепь состоит только из
кумулированных двойных связей. Этот полимер, названный карбином, может
быть рассмотрен как новая аллотропная модификация углерода (наряду с алма-
зом и графитом).
60
6.4.7. Реакции по связи =С-Н
1. Образование ацетиленидов
Благодаря значительной полярности связи С-Н терминальных ацетиленов
й присутствии сильных оснований (:В) возможна ионизация ацетиленов с обра-
тит! ием ацетил енид-анионов:
8— 5+ iQ ©
R—С=С—Н +:В R—С=С: + НВ
В роли сильного основания могут выступать амид натрия NaNH2, металл-
органические соединения, концентрированные растворы щелочей:
R—С=С-Н
© ©
NaNH2 „ VT
——R—С=С—Na + NH3
8- 5+
CH3-MgX ______v
------—► R—C=C—MgX + CH4
© © реактив Иоцича
NaOH
!« - » R—C=C—Na + H2O
незначительное
количество
С ионами некоторых тяжелых металлов образуются малорастворимые аце-
тилениды:
7-1 ©
Н-С=С-Н ------—----► Си—С=С—Си|
Cu(NH3)2C1 красно-бурый’
осадок
R—С=С—Н ------►
Ag(NH3)2NO3
Ацетилениды меди и особенно серебра являются термически нестабиль-
ными соединениями. Производные самого ацетилена взрывчаты.
Малая растворимость ацетиленидов меди и серебра в воде объясняется
сильным межмолекулярным взаимодействием донорно-акцепторного типа:
f
R—С=С—Си
f
R—С=С—Си
t
R—С=С—Си
f
Ацетилениды используются в различных синтезах, в реакциях с электро-
фильными реагентами.
2. Алкилирование и арилирование
Ацетилениды реагируют с галогенпроизводными углеводородов как нук-
леофильные реагенты и в реакции образуются замещенные ацетилены:
61
[к—C=C-MgXl+ CH3I --------► RC=C—CH3
В реакции могут быть использованы также ацетилениды меди, особенно
при взаимодействии с алкилиодидами. Подарены вступают в реакцию архивиро-
вания алкинов только с ацетиленидами меди:
R—С=С—Си + С6Н51 -----► RC=C-C6H5
-Cui
3. Присоединение к альдегидам и кетонам
Ацетилен и монозамещенные ацетилены в присутствии КОН или NaOH
присоединяются к карбонильной группе кетонов с образованием ацетиленовых
спиртов (Фаворский). В этой реакции ацетиленцд-анион действует как сильный
нуклеофильный реагент:
©О л
R—С=С—Н + КОН R-C=C°K®
R-C=CeK© +^С=О —► RC=C-(pOK RC=C-i-OH
ацетиленовые
спирты
6.4.8. Изомеризация
Изомеризация алкинов проходит под действием металлов или щелочей
(Фаворский):
КОН (сп.), 170°С „ „тт
I - СН3— С=С-СН3
Na
СНз. КОН (сп.)
^сн-с=сн ..... \
снз Na
СН?с=с=сн2
аллены
Алкины с третичными радикалами не изомеризуются.
62
Выводы
СН3-
Для алкинов характерны реакции:
8+ 8— 5+ 8—
/ |_E®|,E-Nu (H-Nu)___________________________
8+ 8- Adj.
E-Nu (кат., RcP) (H-Nu)
СН
Е(Н)
AdN
CO, HX (кат.)
/ карбонилирование
[Н]
----► алкены, алканы
[О]
----► кислоты
п -=- (кат.)
------i----+ олиго-, полимеры
КОН
СНТ
СН2=»=СН2
изомеризация
В _А1
-ВН
©8-5+
[R°]R-MgX Гсп
-R*H L 7
О ©
<=>-MgX —
R—»>Х
СН3—&-R
5+ 5-
сн3-
i-он
Н
63
7. Циклоалканы
Алициклические соединения - углеводороды, в которых углеродные цепи
образуют циклы. Общая формула моноциклических циклоалканов такая же, как
для алкенов - СпН2п.
7.1. Изомерия и номенклатура
Изомерия
а) боковых цепей;
6) положения заместителей в цикле;
в) величины цикла;
г) геометрическая
СН3 СН3 СН3
цис-
транс-
1,2-диметилциклобутан
д) оптическая.
Зеркальные изомеры имеют z/wc-изомеры с двумя разными заместителями
и транс-изомеры с двумя одинаковыми заместителями.
транс-
Моноциклы разделяются на:
малые (3,4-членные): циклопропан , циклобутан ;
нормальные (5,6,7-членные): циклопентан Циклогексан
ЦИКЛО-
средние (8,9,10,11 -членные),
большие (макро-) (от 12 и более).
64
В названии цикла добавляется приставка цикло- к соответствующему на-
1ИП11ИЮ линейного углеводорода (ИЮПАК)
Me
1,2-диметилциклогексан
Me
7.2. Получение
Циклопропан, циклобутан
1. Действием диазометана на алкены:
CH2N2,t[CH2:]
-N2
Диазометан при нагревании образует карбен (метилен) - электрофильную
частицу с двумя неспаренными электронами, которая присоединяется по
в *связи алкена.
2. Димеризация алленов:
2 СН2=С=СН2
^СН2
сн3
^СНз
3. Внутримолекулярная реакция (Вюрц):
Na или Zn, спирт
-NaBr
Li (Hg)r
-LiBr
4. С помощью натриймапонового эфира:
Na2C(COOC2H5)2
-2NaBr
СООС2Н5
СООС2Н5
СООН
NaOH, t
-Na2CO3
H2O,H+,t^
-C2H5OH *
-CO2
Циклопентан, циклогексан
I. Внутримолекулярная реакция (Вюрц):
С Вт Li (Hg)^ Г~\
Вг -LiBr * \/
65
2. Пиролиз солей дикарбоновых кислот (Пириа):
циклопентанон
3. Циклизация алканов:
4. Диеновый синтез (Дильс-Альдер):
5. Тримеризация алкинов (Реппе, Зелинский):
6. Внутримолекулярная сложноэфирная конденсация (Дикман):
С0Р«Т СДОЧа,
СЦ!г/сООС2Н5 ‘С2Н5ОН
^^СООС2Н5
Н2О, NaOH, t
-С2Н5ОН
-Na2CO3
Средние и макроциклы
1. Тетрамеризация ацетиленов:
2. Разложение солей дикарбоновых кислот (Ружичка):
СНз—CQO'\
(Н2С)п Y*.
чсн2-:соо/
t
-СаСО3
66
.1. Внутримолекулярная сложноэфирная конденсация (Дикман):
zCH2COOC2H5 c2H5ONa
(H2C)n —--------
ЧСН2СООС2Н5 “С2Н5ОН
(СН2)п
Необходимо отметить, что «средние» и макроциклы получить трудно в
« вит с энергетической невыгодностью конформации этих колец.
7.3. Пространственное и электронное строение
/(ля объяснения уменьшения устойчивости (стабильности) моноциклов в
|«|ду
h«Mop (1885 г.) предложил теорию углового напряжения циклов. Она базиру-
vn ни двух положениях:
а) циклы имеют плоское строение;
П| отклонение валентных углов в цикле от тетраэдрического является мерой
напряженности (устойчивости) цикла.
Отклонение углов от тетраэдрического рассчитывается по формуле
109.28-х
2
। на v угол в цикле.
24044' 9044' 0°44' -5°16* -9°33'
11о Байеру 3- и 4-членные циклы малостабильны, а 5- и 6-членные наибо-
't** устойчивы.
11сдостатком теории Байера является постулат о плоском строении циклов.
В 1890 г. Сакс высказал предположение о неплоском строении циклогек-
сана, н Мор в 1918 г. предложил для него две формы («ванна» и «кресло»).
(' точки зрения современных представлений строение и стабильность цик-
ана будут зависеть от трех видов напряжений:
I) уI лопое (Байер);
>1 lopuiioiiHoe (Питцер) - напряжение, которое возникает за счет отталкивания
ннсидних (вицинальных) атомов, находящихся в заслоненных положениях;
I) непнлентное взаимодействие (Прелог, Ван-дер-Ваальс), возникающее за счет
Сниженных в пространстве атомов.
Молекулы циклоалканов принимают такие конформации, в которых вес
•I* андп напряжений (взаимодействий) минимальны.
67
Циклопропан
К энергии углового напряжения добавляется анергия отталкивания трех
пар атомов водорода:
Н
Ешцф 27.6 ккал/моль
116-118°
По современным понятиям стремление молекулы циклопропана к умень-
шению углового напряжения приводит к изменению характера a-связей цик-
ла.
Это выражается в том, что максимумы перекрывания электронных облаков
С-С связи цикла располагаются не на прямых, соединяющих атомы углерода, а
вне линий связи, как у тс-связей в алкенах.
Угловое напряжение уменьшается, но связи при этом приобретают нена-
сыщенный характер (банановые связи), а угол между осями орбиталей С-С свя-
зей увеличивается от 60 до 106°. ;
О значимом вкладе р-орбитали в образование связей циклопропана свиде-
тельствует сравнительно низкая энергия ионизации.
Пропан - 11,5 эВ, циклопропан - 10,5 эВ, пропен - 9,7 эВ.
Циклобутан
Важной особенностью циклобутана является его непланярность. Угловое
напряжение меньше, чем в циклопропане.
11.57 А, I
Чтобы уменьшить торсионное напряжение, один из атомов углерода вы-
деляется над плоскостью молекулы. Однако это приводит к тому, что появляет-
ся дополнительное невалентное взаимодействие между первым и третьим ато-
мами углерода (/ = 2,2 А, £иапР. = 26,2 ккал/моль).
Циклопентан
Углового напряжения почти нет. Торсионное - незначительно: 5 пар ато-
мов водорода находятся в «скошенных» положениях.
68
Благодаря силам отталкивания между атомами водорода два атома углеро-
да выходят из плоскости цикла. В таком положении оказываются все атомы уг-
лерода, т.е. кольцо как бы находится в постоянном волнообразном движении
псевдо вращении.
Енапр. = 6,5 ккал/моль
Циклогексан
Угловое напряжение отсутствует в трех конформациях:
"кресло" "ванна" "твист"
Конформация «кресло» наиболее выгодна, т.к. в ней отсутствует и торси-
онное напряжение (все атомы водорода в заторможенных положениях), по
сравнению с конформацией «ванна» и конформацией «твист». Поэтому равно*
веская смесь конформаций циклогексана при 20°С содержит 99,99% формы
«кресло».
Циклогексан имеет в форме «кресло» 2 типа атомов водорода, находящих-
ся в экваториальных и аксиальных положениях. Шесть экваториальных атомов
водорода располагаются в плоскости молекулы, а шесть аксиальных связей С-
11 направлены перпендикулярно плоскости молекулы.
Объемные заместители предпочитают занимать наиболее выгодные эква-
юриальные положения.
Конформация «кресло» находится в состоянии конверсии, т.е. взаимопре-
вращений (10s раз в 1 сек), при которой аксиальные атомы и группы становятся
экваториальными и наоборот.
акс-метил-
циклогексан
экв-метил-
циклогексан
95%
Конформация «ванна» на 8 ккал/моль менее выгодна, чем «кресло».
69
Это связано с тем, что в этой конформации 8 связей С-Н находятся в за-
слоненном положении.
Конформация «твист» приблизительно на 2 ккал/моль стабильнее конфор-
мации «ванна».
Средние циклы
В средних циклах встречаются оба эффекта: а) питцерное напряжение;
б) трансанулярное взаимодействие групп, находящихся в противоположных
сторонах цикла, но сближенных в пространстве.
Например, для циклодекана в самой выгодной конформации шесть атомов
водорода направлены внутрь цикла и вызывают появление Ван-дер-ваальсовых
сил отталкивания.
\_/ ' I
/_/ /2
з
Атомы углеродов в положениях 1 и 5 сближены на 1,8 А.
7.4. Химические свойства
Малые циклы
Циклопропан проявляет двойственную химическую природу - реагирует
как алкен - с присоединением (раскрытием) цикла (основная реакция типа AdE)
и как алкан (по реакции Sr) с замещением атомов водорода.
ВГ2 > ВгСН2СН2СН2Вг
Adg
НВг
——► СН3СН2СН2Вг
ме-/^ -^р^сн3)Дсн2сн3]
Me Е
(СН3)2С-СН2СН3
ir
Циклопропан напоминает соединение с л-связями, поэтому винилциклоп-
ран реагирует как сопряженная система - 1,5-присоединение.
Д -Вг » СН3СНг-СН=СН-СН2СНВг
^^СН=СН2 AdE
70
[Н], Н2 (Ni, 80°С)
СН3СН2СН3
н2/°он
'соон
—1 (Pt) Pd)—► СН3СН=СН2
изомеризация
Хлорирование циклопропана при обучении УФ светом приводит к реали-
зации реакции Sr.
CI2 (hv)
Sr
С1
Хотя в циклобутане о-связи также менее прочны, чем в алканах, но эти от-
клонения меньше, и он по свойствам ближе к алканам (реакции типа AdE и рас-
крытия цикла идут труднее).
НВг
СН3СН2СН2СН2Вг
— Н2, Ni, 120°С
------—-------► СН3СН2СН2СН3
[О] СНг-СООН
(Ьнг— СООН
С другой стороны, реакции SR идут также как в алканах.
-------------------------
Вг
Циклопентан и циклогексан в реакциях с галогенами, азотной и серной ки-
слотами ведут себя как алканы (по механизму SR).
HiC Cl
Cl2 (hv)
SO2+C12 (hv)
H3Q^SO2C1
71
Раскрытие кольца в циклопентане возможно только при высоких темпере
турах (300°С) и в присутствии катализатора (Pt).
Реакции сужения и расширения циклов
Наблюдаются при реакциях галоген*, окси*, амино*, оксосоединений.
Основные закономерности перегруппировок алициклических функцис
нальных соединений установлены Демьяновым.
а) Если функциональная группа находится у атома углерода, соседнего
циклом, то идет расширение цикла.
б) Если функциональная группа находится у атома углерода цикла, то иде?
сужение цикла.
ОН®
-Н2О*
Средние циклы
Средние циклы кроме реакции Sr вступают в транс-анулярные реакции
что связано с их конформациями.
Например, нуклеофильное замещение у циклодекана.
Вследствие того, что в наиболее выгодной конформации циклодекана ато
мы углерода в положениях 1 и 5 сближены в пространстве, реакция замещение
72
«тома хлора на гидроксильную группу проходит с переносом катионного рма-
। (ионного центра из положения 1 в положение 5.
Выводы
Для циклоалканов характерны реакции:
5+ 5-
[Е® ] (E-Nu)
аналогичен Adp
(основная реакция)
[Xе], Х2, (hv,t°)
U
Sr
X
кат.
изомеризация
[Н], кат.
СООН
'соон
® б+ б"
[Е®] (E-Nu)
аналогичен AdE
Sr
X
73
74
8. Арены
Бензол был получен в 1825 г. Фарадеем из продуктов перегонки каменно-
угольной смолы. Позже Либих назвал это вещество бензолом. Бензол и его
производные имели своеобразный запах и свойства и были названы аромати-
ческими углеводородами. Их общая формула CnHjn.6«
Бензол, его гомологи и производные являются моноциклическими арена-
ми.
8.1. Изомерия и номенклатура
Изомерия:
•) боковых цепей;
I) положения боковых цепей.
1I оменклатура тривиальная и ИЮПАК.
Монозамещенные:
цимол
СН(СН3)2
о
кумол
I ри замещенные:
I
о-ксилол
мезитилен
1,3,5-триметилбензол
75
Тетразамещенные:
дурол
Арены рассматриваются также как продукты замещения атомов водорода-
бензоле. 1
Радикалы:
фенил бензил о-толил 2,3-ксилил 2-этилфенил
о-фенилен л<-фенилен «-фенилен
8.2. Способы получения
'.1) Сухая перегонка каменного угля (1000°С, без доступа воздуха). Полу-
чают сухой остаток (кокс), каменноугольную смолу и газы. В газе и смоле со-
держатся бензол, толуол, ..., ксилол (фракция 80-170°С при разгонке каменно-
угольной смолы).
2) Из нефти, а также при каталитическом крекинге нефтепродуктов.
3) Из углеводородов (ароматизация парафинов).
300°С
------►
Pt
н-гептан
I + ЗН2
450-500°С
Сг20з/А120з
4) Из ацетилена (Зелинский, Реппе; катализатор - активированный уголь
или катализатор Циглера-Натта).
ЗНС=СН kat’ 600°С>
76
5) Щелочная плавка солей кислот (Дюма).
C6H5COONa + NaOH--------►
-Na2CO3
6) Гомологи бензола из бромпроизводных (Вюрц-Фиттиг).
СбН5Вг + СН3Вг СУсНз + + С2Нб
7) Алкилирование (Фридель-Крафтс).
А1С13
СбНб + С2Н5Вг----+ У т—с2н5
8) Восстановление по Клеменсену.
ZA_rft_rH znfflfeH® /ГАг н
< )—СО—Сг13 ----------► V у—С2П5
9) Конденсация кетонов.
СН3СОСН3
10) Из сульфокислот.
н И Н2°>НзРО4
Н3С—Г у— a(J3n ------------► и у—СН3
\—/ -H2SO4 \ — /
11) Через диазосоединения.
Xе С2Н5ОН
-N2
-СН3СНО
8.3, Строение бензола. Ароматичность. Правило Хюккеля
Формула бензола СбН6. Исходя из этой формулы можно считать бензол
очень ненасыщенным соединением. Однако факты говорят об обратном:
Бензол:
1) не склонен к реакциям присоединения;
2) не окисляется под действием большинства окислителей;
3) не полимеризуется;
4) термически стоек;
5) вступает в реакции замещения в присутствии кислот Льюиса.
77
Эти свойства бензола называют комплексом ароматичности.
Строение бензола
а) при восстановлении превращается в циклогексан;
б) получается тримеризацией ацетилена.
Следовательно, в бензоле есть цикл из шести атомов углерода.
Для бензола были предложены следующие структурные формулы:
Кекуле Клаус Байер Дьюар Ладенбург
1865 г.
Осцилляция связей
В настоящее время для изображения бензола принята формула Кекуле.
Современные представления о структуре бензола
1. Все связи углерод-углерод в бензоле одинаковы и их длина составляет
1,4 А (для сравнения двойная связь углерод-углерод - 1,33 А, простая связь уг-
лерод-углерод - 1,54 А).
2. Углеродные атомы в системе бензола находятся в $р1 2-гибридизации, ка-
ждый образует три о-связи и предоставляет одну р-орбиталь для образования
сопряженной системы из шести тс-электронов, т.е. образуется общее электрон-
ное облако р-электронов.
Такое строение придает особую устойчивость молекуле бензола. Энерге-
тической мерой устойчивости (ароматичности) является энергия сопряжения
трех двойных связей в цикле (энергия резонанса, сопряжения, делокализации),
т.е. энергия, которую нужно затратить на нарушение системы сопряжения (ко-
торая выделяется при образовании системы сопряжения).
Следовало бы ожидать, что ДН гидрирования трех двойных связей должна
быть 3-28,6 = 85,8 ккал/моль. Однако в действительности при гидрировании
бензола выделяется только 49,8 ккал/моль. «Реальная» молекула бензола по
термодинамической устойчивости превосходит гипотетический «циклогекса-
триен» на 36 ккал/моль. Эту энергию и называют энергией делокализации (со-
пряжения). Существует целый класс подобных соединений.
!
i
78
Правило Хюккеля
На основании квантово-химических расчетов Хюккель сформулировал
правило согласно которому ароматическими (стабильными) являются цикли*
чсские системы, содержащие
а) (4и+2)р-электронов (и = О,1,2, ...);
б) имеющие непрерывную цепь сопряжения (т.е. каждый атом углерода
должен находиться в состоянии др2-гибридизации);
в) такие системы являются плоскими.
л = 0; 2 р-электрона
Примеры
циклопропенилия
л = 1; 6 р-электронов
бензол
пиридин
пиррол
79
8.4. Реакционная способность и химические свойства бензола
и еао аомолоаое
/
Для бензола и его гомологов характерны три типа реакций:
1) реакция электрофильного замещения (Sn) - основная реакция;
2) присоединение к системе циклогексатриона проходит трудно: при ин<
тенсивном облучении или с активными реагентами и катализаторами;
3) реакция в боковую цепь - радикального замещения (Sr).
8.4.1. Реакции электрофильного замещения (8е)
+ ЕХ
©е
электро-
фильный
л-комлекс
п-комплекс
реагент (КПЗ)
1. Электрофил - катион или положительно заряженная часть (катиноид-
ная частица) диполя сначала притягивается к л-элекгронам кольца, образу^
л-комплекс.
2. Затем вырывается пара электронов ароматического кольца и образуете?
a-комплекс (интермедиат Уэланда или аренониевый катион).
3. ст-Комплекс - высокореакционное соединение, которое стремится к ста?,
билизации путем выброса протона. Протон Н ® связывается с анионной части*
цейХ®.
Этот механизм доказан - выделены как л-, так и о-комплеке.
Нитрование
HNO3(k.),H2SO4(k.)[NO2®] а х1л
АгН -----------------------► Ar—NO2
Сульфирование
АгН
H2SO4(k.) [SO3]
Ar—SO3H
Алкилирование
ArH R~C1’A1C13 [R^AlCtP]
Фридель-Крафтс
Ar-R
80
Ацилирование
RCOC1, AICI3 [R?=OAlClp]
АгН ------------------------
О
R
Фридель-Крафтс
"н 5+ "
сгс=0
HCI + СО
АгН ---------
Гатгерман-Кох
Галогенирование
АгН С12 нлИВг2(Ре,А1С!з)^Аг_с|(Вг)
X = AI, Вг
Хлорметилирование
Н2С=О + НС1 ^8-ОН С1°
АгН -----------------------► Аг—СН2С1
Влияние электронодонорных и электроноакцепторных заместителей
на скорость и направление реакции
По ориентирующему действию заместители в кольце делятся на 2 группы.
Ориентанты I рода - в о- и «-положение.
Ориентанты II рода - в ^/-положение.
I рода обладают +/- или +Л/-эффектами:
-^О- ——NR2 > -—OR > <— СН3 (алкил)> —кЙсОСНз > -*На1
II рода обладают-/- или -М-эффектами:
NH3 > -NO2 > -SO3H > -ON > -CCI3 > — > — С* > — С<_ >
п. К Url
,^о
("OR
Ориентанты I рода - ускоряют реакцию (кроме галогенов).
Ориентанты II рода - замедляют реакцию.
Направление реакции определяется прежде всего Л/-эффектом, а затем
/ (ффектом; скорость реакции - суммой /- и Л/-эффектов.
Анализ направления атаки электрофильным реагентом бензольного кольца
»• позиций статического эффекта (в основном состоянии молекулы) и динамиче-
ского эффекта (в переходном состоянии молекулы).
81
Ориентанты I рода
Статический эффект
+Л/-Эффект группы ОН создает избыток электронной плотности в о-
л-положениях кольца, что и определяет атаку Е ®.
Динамический эффект
I II III
При атаке в о- и «-положения по отношению к ориентанту I рода осущест-
вляется эффективная делокализация положительного заряда за счет +Л/-эффек-
та группы ОН, что показано на граничной структуре III.
ОН
При атаке в ^-положение делокализация за счет +Л/-эффекта группы ОН
осуществляется неэффективно и +Л/-эффект группы ОН не достигает атомов '
углерода кольца, несущих 8+.
Ориентанты IIрода
В случае ориентантов II рода все наоборот.
Статический эффект
Н^Об“
5+ \
-M-Эффект карбонильной группы создает недостаток электронной плот-,
ности, особенно в о- и «-положениях кольца. В jw-положении этот недостаток
электронов меньше, что и определяет атаку Е® в лг-положение. .
82
Динамический эффект
н _о
При атаке электрофила в о-положение электроноакцепторная группа
СН=О, связанная непосредственно с атомом углерода и несущая положитель-
ный заряд, еще более ухудшает делокализацию карбокатионного состояния мо-
лекулы.
н _о
5+г<^|8+
k Ее
5+
В случае атаки в ^-положение неблагоприятные -I- -А/-эффекты группы
СН~О будут сказываться меньше.
Согласованная и несогласованная ориентация
При наличии в молекуле двух заместителей возможна их согласованная и
несогласованная ориентация атаки электрофильных реагентов.
Согласованная ориентация
2 изомера 2 изомера 1 изомер
Несогласованная ориентация
а) если ориентанты I рода находятся в «ара-положении, то ориентировать
Л у дет наиболее сильная группа
О® > NR2 > NHR > NH2 > OR> ОН > Aik > Hal
83
б) если имеются ориентанты I и II рода, то ориентировать будет ориентан
I рода
в) если имеются ориентанты II рода, то ориентировать будет наиболее
сильная группа
R3N © > NO2 > SO3H > ON > CF3, CCIj > —
ri vri
8.4.2. Реакции присоединения
Нехарактерные для бензола реакции присоединения по формально двой
ным связям кольца осуществляются с активными реагентами в «жестких» уело
виях при облучении УФ светом, высокой температуре и в присутствии катали
заторов.
гексахлоран
ЗН2, Pt, Pd, 300°С
Ni Ренея
О
[Q3
V2O5
малеиновый
ангидрид
| О + 2СО2 + Н2О
84
Озонолиз
ЗН2О
-ЗН2О2'
из Н2О2
[О] соон
*"(!:оон
8.4.3. Реакции гомологов бензола (SR)
Эти реакции аналогичны реакциям алканов, но протекают легче и исклю-
чительно в a-положение боковой цепи бензольного кольца.
HNO3
12,5 ч
Cl2, hv
СНС1—СН3
CH(NO2)CH3
[О] (КМпО4)
СООН
Направление реакции обусловлено образованием бензильного радикала
который стабилен (и следовательно, легко образуется) за счет делокализации
неспаренного электрона, вследствие его взаимодействия с р-электронами бен-
зольного кольца.
85
Выводы
Для аренов характерны реакции:
86
9. Многоядерные ароматические соединения
9.1. Номенклатура
Полициклические арены с изолированными циклами:
1) ароматические системы, в которых бензольные кольца связаны непо*
средственно друг с другом.
3 2 2' 3*
Бифенил Терфенил
Кватерфенил
2) ароматические кольца соединяются через 1 или несколько атомов угле-
рода.
3 2 2‘ 3' ] \ 3 2 2* 3’
Дифенилметан Трйфенилметан 1,2-Дифен ил этан
3) если ароматические кольца имеют 2 или более общих атома углерода, то
они образуют группы конденсированных углеводородов.
мезо
Нафталин Антрацен Фенантрен
9.2. Дифенил (бифенил)
9.2.1. Получение
1) Пиролиз бензола (термическое дегидрирование) (Бертло)
t = 800°С
2С6Н6 ---------1
-н2
87
2) Реакцией арилгалогенидов с металлами (Na, Си, Mg) (Вюрц)
Na
С$Н5Вг ------
-NaBr
(Ульман)
Си (порошок)
C6H5I -----—------>
-Cui
3) Арилирование замещенных бензолов солями диазония (Гомберг)
NH2
NH-NH—/ 4 H2S°4» H2N
бензидин
гидразобензол
О t,C2H5OH
-2СН3СНО
Ч
9.2.2. Строение, атропизомерия
Молекула бифенила является планарной, т.е. оба цикла находятся в одной
плоскости.
Если в положениях 2, 2' и 6, 6' имеются заместители, то за счет простран- j
огненных препятствий одно кольцо развернуто по отношению к другому, а по- j
порог вокруг связи С-С' затруднен. В случае разных заместителей возможно 1
существование двух зеркальных изомеров (энантиомеров): j
88
Такой вид оптической изомерии в ряду дифенила, когда при отс/пивии
асимметрического атома углерода молекула в целом хиральна, нмываггиа
атропэнантиомерией (атропизомерией).
Например:
о,о'-замещенные дифеновые кислоты
9.2.3. Реакционная способность и химические свойства
Молекула дифенила имеет более сопряженную систему, чем бензол, по-
этому электронодонорные свойства у него выражены более ярко (энергия иони-
зации (8,2 эВ). Это значительно облегчает наиболее характерные для него реак-
ции электрофильного замещения (SE).
Дифенил легко галогенируется, нитруется. Реакции идут в положения
4 и 4'.
4-бромбифенил 4,4'-дибромбифенил
Особенность! Вторая молекула электрофильного реагента встает в пара-
цоложение второго кольца. Даже, например, в реакции нитрования дифенила:
HN°3, o2n-^2^—ии03»
4-нитробифенил
4,4'-динитробифенил
Это значит, что кольца обладают определенной самостоятельностью.
89
9.3. Полиарилметаны
9.3.1. Получение (на примере трифениметана)
1) Реакция Фриделя-Крафтса
ЗСбН6
СНС13, АICI3, t
5ici *
(С6Н5)3СН
2) Восстановление карбинола
СН3СООН i-Zn, ГН]
(СбН5)3СОН ----3--------(СбН5)3СН
3) Конденсация с карбонильными соединениями
с6н5с;° + гСбНе (с6н5)3сн
Н -HjO
9.3.2. Реакционная способность и химические свойства трифе-J
нилметана ]
Трифенилметан содержит 3 изолированных кольца. Для него характер»
реакции электрофильного замещения и реакции у центрального атома углероде
I. Реакции электрофильного замещения идут в «qpa-положение кольца.
(С6Н5)3СН Вгг>.А1С1з> ^вг—0)СН
II. Реакции у центрального атома углерода связаны с достаточно легкий
образованием весьма стабильных промежуточных частиц: карбаниона, свобод
ного радикала и карбокатиона.
1. Образование карбаниона
Повышенная кислотность трифенилметана обусловлена влиянием трех фе
нильных радикалов на центральный атом углерода, что облегчает депротониза
цию молекулы (рКа» 32). (Для сравнения рКа > 40 - метан, 25 - ацетилен). По
этому при действии сильного основания амида натрия в жидком аммиаке воз
можны отрыв протона и образование трифенилметанид-иона (карбанион крас
ного цвета).
90
Легкость образования трифенилметанид-иона обусловлена эффективной
делокализацией отрицательного заряда по всем кольцам, несмотря на некото-
рую непланарность аниона, который имеет структуру воздушного винта с уг-
лом поворота колец относительно друг к другу на угол -30-40°. Электроноак-
цепторные группы еще более увеличивают устойчивость аниона.
Трифенилметилнатрий — ионно построенное соединение, высоко реакци-
онный нуклеофильный реагент.
(С6Н5)3С^-^^=Я> (С6Н5)3С-COONa (СбН5)3С-СООН
2. Образование радикала, свободнорадикальные реакции (реакции S^).
Трифенилметан легко бромируется при нагревании и окисляется.
Вг2 [Вг]
> (С6Н5)3С—Вг
(С6Н5)3СН —
[О] г (С н ' с он
-----* (,СбН5)3С—ин
Причина легкого протекания реакции Sr связана с легким образованием
радикала трифенилметила за счет эффективной делокализации неспаренного
электрона, несмотря также на некоторую непланарность колец (30-40°).
Уб)
91
Квантово-химические расчеты показывают, что спиновая плотность сосре-
доточена как на центральном атоме (0,5-0,7), так и в орто- и иара-поолженияХ
колец, 0,115 и 0,127 соответственно. 4
Впервые образование этого радикала наблюдал Гомберг в 1900 г. при изу-
чении реакции
Zn
(СбН5)3СС1 - » 2(С6Н5)зС.^* (СбН5)3С-С(СбН5)3
, -ZnCb
трифенил- предположительное
хлорметан соединение
Он обнаружил, что бензольные растворы предположительно гексафенилэ-
тана ведут себя странно: раствор окрашен в желтый цвет, при нагревании окра-
ска углубляется, при отгонке растворителя исчезает. Гомберг высказал мысль*
что в растворе существуют свободные радикалы. ,
Действительно трифениметил-радикал желтого цвета. В растворе он суще?
ствует в равновесии со своим димером. В растворе его -3-5%, он легко реагин
рует с кислородом и образует пероксид, реагирует с йодом и образует трифени
л подметан.
г—(СбН5)3С—О—О—С(СбН5)3
(С6Н5)3С. -
L-2— (c6H5)3ci
Долгое время считали, что при димеризации трифенилметила образуете!
гексафенил этан. Однако, по-видимому, из-за пространственных затруднени!
образование последнего маловероятно и димеризация осуществляется с участий
ем одного атома углерода бензольного цикла в иара-положении.
3-диф енил метилиден-6-трифенил-
метилциклогексадиен-1,4
Нужно отметить, что стерические эффекты очень важны как факторы, ста^
билизирующие свободные радикалы.
Например, орто-заместители стабилизируют три(о-толил)радикал, поэтому
доля его в растворе резко возрастает по сравнению с трифенилметильным ра-
дикалом:
92
I
87%
3. Образование карбокатиона
Производные трифенилметана, имеющие электроноакцепторную группи-
ровку при «метановом» атоме углерода, легко образуют трифенилмегил-катион
(красно-оранжевого цвета).
(C6H5)3C-*Ci + А1С13 (С6Н5)3?А1С14
(СбН5)3С-*ОН + H2SO44=*= (C6H5)3CHS§4
Легкость гетеролиза связи С->Х обусловлена легкостью образования кар-
бокатиона, стабилизированного эффективной делокализацией положительного
заряда тремя бензольными кольцами.
54-
Введение электронодонорных заместителей, особенно в пара-положение,
еще более стабилизирует карбокатион.
Относительная стабильность карбокатионов:
ТФМ 0%
п-СНз 7%
П-СН3О 20%
n-(CH3)2N 70%
93
9.3.3. Трифенил метановые красители
Малахитовый зеленый
Получается конденсацией бензальдегида с NjN-диметиланилином.
CsHs-Cj® + C6H5N(CH3h
п
лейко-основание
(4,4-тетраметилдиамино-
трифенилметан)
HSO®
94
Получается конденсацией кетона Михлера с диметиланилином:
Аминогруппы играют решающую роль в устойчивости катиона и его ово*
топоглощении и называются ауксохромами.
Образование стабильного катиона приводит к понижению его энергетичв-
ского уровня в возбужденном состоянии и, следовательно, увеличению длины
волны падающего света.
Выводы
9.4. Конденсированные ароматические углеводороды
9.4.1. Нафталин
9.4.1.1. Способы получения
1) Содержится в каменноугольной смоле (~6%)
2) Дегидроциклизация
СН3
300°С
-Н2
СН3
а-метилнафталин
95
3) Конденсация ацетилена (Зелинский, Казанский)
5 СН=СН
С,400°С
4) Конденсация бензола с ангидридом янтарной кислоты (Хеуорс)
9.4.1.2. Строение и реакционная способность
Структура нафталина была доказана следующим образом:
96
Длины связей в нафталине различны.
Энергия делокализации (61 ккал/моль), что ниже (Д = И), чем для двух
молекул бензола (36x2 = 72). Это означает, что образование конденсированной
системы из двух циклов дает меньший выигрыш энергии и в растворах будет
легче идти присоединение и превращение нафталина в систему дизамещенного
бензола.
Электронная плотность сконцентрирована в a-положении, поэтому реак-
ции электрофильных реагентов будут идти в а-п сложение.
В молекуле нафталина связи более локализованы, поэтому оправдано при-
менение обычных формул Кекуле:
9.4.1.3. Химические свойства
Реакция электрофильного замещения (Se) (основная реакция)
Реакции SE идут легче, чем для бензола, и протекают аналогичным обра-
юм. Одно кольцо в нафталине является формально +М-донором по отношению
к другому.
а-комплекс
(Р-атака)
о-Комплекс с ^-гибридизированным углеродным атомом в а-положении
более стабилен, чем в 3-положении. Поэтому реакции в a-положение идут бы-
97
стрее, но в некоторых случаях, когда реакция обратима, при повышенных тем-
пературах образуется термодинамически более стабильный 0-изомер.
Примеры:
(продукт (продукт
кинетического термодинамического
контроля) контроля)
Это примеры кинетически и термодинамически контролируемой реакции.
0-Изомер термодинамически более стабилен, чем а-изомер, поэтому в об-
ратимой реакции сульфирования при высоких температурах, где достигается
термодинамическое равновесие, больше образуется 0-изомера (термодинамиче-
ски контролируемая реакция). При низких температурах термодинамическое
равновесие не достигается, поэтому получается а-изомер, скорость образования
которого выше.
Для нафталина также характерны реакции присоединения, которые проте-
кают значительно легче, чем для бензола. При этом одно из колец нафталина
проявляет свойства диеновой системы и дает продукты типа 1,4-присоеди-
нения.
98
H Cl
Окисление
9.4.2. Антрацен
9.4.2.1. Способы получения
1) Выделение из каменноугольной смолы (Дюма, 1832 г.)
99
2) Циклизация 2-метилбензофенона (Эльбе)
О
3) Ацилирование бензола фталевым ангидридом (Байер)
4) Диеновый синтез (через бензохинон)
5) Димеризация хлористого бензила
9,10-дигидроантрацен
6) Сшивание по Вюрцу-Фитгигу
СН^Вг Вг^
l)Na
2)S
Гвг'ЙСН/
100
9.4.2.2. Строение и реакционная способность
Структура антрацена доказана следующим образом:
1) при гидрировании присоединяется 14 атомов водорода, что соответству-
ет циклу с 7 двойными связями;
2) при окислении образуется антрахинон, синтез которого свидетельствует
о наличии в молекуле трех конденсированных колец.
О
1. Длины связей в антрацене различны.
2. Энергия делокализации 86 ккал/моль, что ниже на 22, чем у трех моле-
кул бензола (36x3 = 108). Меньше выигрыш энергии при образовании конден-
сированной системы из трех циклов. Следовательно, более чем характерны ре-
акции присоединения.
3. Энергия ВЗМО значительно ниже, чем для бензола и нафталина (ЭИ =
7,4 эВ).
4. Наибольшая концентрация электронной плотности на ВЗМО в положе-
ниях 9 и 10 (мезо-углеродные атомы).
5. Поэтому взаимодействие антрацена с электрофильными реагентами про-
ходит легко и во многих случаях сначала образуются продукты присоединения
и положениях 9 и 10 (это важная особенность его реакционной способности).
При нагревании продукты присоединения превращаются в замещенные антра-
цены.
9.4.2.3. Химические свойства
Реакции присоединения (по крайним атомам углерода формально тетрае-
ноной системы антрацена).
101
В этих реакциях антрацен ведет себя на первой стадии как тетраеновая
система.
Реакции Se
Эти реакции менее характерны для антрацена и порой неселективны.
102
9.4.3. Фенантрен
9.4.З.1. Получение
1. Пиролиз стильбена.
Также можно получить пиролизом дибензила и о,о'-дитолила.
2. Взаимодействие нафталина с янтарным ангидридом (Хеуорс).
3. Конденсация о-нитробензальдегида с фенилуксусной кислотой (Пшорр).
103
9.4.3.2. Строение, реакционная способность и химические свойства
9 10
1) молекула фенантрена является более стабильной, чем антрацена, так как
два ядра обладают ароматическим характером;
2) энергия делокализации 99 ккал/моль;
3) донорные свойства выражены слабее, чем у антрацена (ЭИ = 7,9);
4) наиболее активными положениями являются атомы углерода в положе-
ниях 9 и 10, а связь между ними напоминает двойную;
5) многие реакции идут в положения 9 и 10, однако во многих случаях эти
продукты нестабильны и являются промежуточными;
Реакции электрофильного замещения (Se).
CH3COCI
—► 3-ацетилфенантрен
смесь 1-, 2-, 3-, 4-, 9-нитрофенантренов
смесь 1-, 2-, 3-, 9-сульфофенантренов
Реакции нитрования и сульфирования фенантрена неселекгивны.
104
Выводы
Для многоядерных углеводородов характерны реакции:
(основная реакция)
105
10. Галогенпроизводные углеводородов
Наиболее простыми производными углеводородов являются галогенпро-
изводные, в которых один или несколько атомов водорода замещены атомами
галогенов (F, Cl, Вг, I).
В основу классификации может быть положена принадлежность углеводо-
рода к тому или иному классу, например галогенпроизводные алифатическо-
го или ароматического ряда.
Однако с точки зрения общности химических свойств важен тип атома
углерода, с которым связан галоген.
В основу этой классификации положена гибридизация атома углерода в
связи С-Х:
1) галогенпроизводные со связью С($р3)-На1 общей формулы CnH2n+iX, где
X = F, С1, Вг, I. Наиболее важный класс - моногалогеналканы.
2) галогенпроизводные со связью C(sp2)-Hal. Наиболее важный класс - га-
логенарены.
10.1. Галогенпроизводные со связью C(sp3)-Hal
10.1.1. Изомерия и номенклатура
Изомерия
1) структурная (углеродного скелета);
2) положение галогена.
Номенклатура
1) тривиальная:
а) из названий радикалов алканов - алкилов с добавлением слова хлори-
стый и т.д.:
СНзО - хлористый метил Н —Вг - бромистый циклогексил
б) для вицинальных дигалогенпроизводных добавляется приставка —ен к
алкилу:
СНз-СН(С1)Ч2Н2С1 - хлористый пропилен
в) для геминальных дигалогенпроизводных добавляется приставка -идея к
алкилу:
СН3СН2СНВг2 - бромистый пропил идеи
г) с концевыми атомами галогена указывается количество метиленовых
групп:
С1-СН2СН2СН2~С1 - хлористый триметилен
106
д) у полностью замещенных добавляется приставка пер:
CF3CF3 - перфторэтан
2) ИЮПАК
Те же правила.
CH3F - фторметан
СН2=СН-СН2С1 - 3-хлорпропен
10.1.2. Способы получения
1. Галогенирование алканов (SR):
Х2 (hv)
R-H - > R-X
Х2
Ph-СН2СН3 ---Ph-CH-CH3
X = F, Cl, Br
Cl2, 500°C
CH2=CH-CH3 —-----------► CH2=CH-CH2CI (Львов)
Фторирование. Выделяется много теплоты вследствие большой энергии
образования связи C-F, поэтому реакция идет очень энергично, иногда со взры-
вом. Используют специальные условия: разбавление фтора азотом; переносчи-
ки фтора - фториды металлов: CoF2 + F2 -> [C0F3], которые фторируют алканы.
2. Присоединение галогенов и галогеноводородов к алкенам и алкинам
(AdE):
Х2
r-ch=ch2 —
нх
Jc
X
R— (*ZH- СН2Х
(виц.)
--- > R-CX2-CHX2
R—С=СН —
----► R-CX2-CH3
(гем.)
3. Реакции замещения (SN):
а) гидроксильной группы в спиртах
НВг (гапогеноводороды)
ROH < =fe R— В г
Выделяющаяся вода связывается избытком галогеноводорода.
107
РС15 (или РС13, РВг3, РВг5, Р13)
ROH -
SOC12 (SF4)
R—Cl
-HC1, -SO2 (-2HF, - SO2)
Скорость реакции зависит от природы спирта и галогеноводорода:
третичные > вторичные > первичные (только с ZnCl2)
Н1>НВг>НС1
б) карбонильного кислорода в альдегидах и кетонах
РС15 \
----/СС12
C=0 -
SF4 \
—/CF2
в) гидроксильной группы в карбоновых кислотах
.О РС15 .О PCL,t,p
R-c?OH -^R-^c|^JUR-ccl3
г) взаимное замещение атомов галогена в галогенпроизводных
© © © ©
RCHr-X + NaX'-------► RCHj-X’ + NaX
Практически используется для получения йодпроизводных.
4. Галогенметилирование аренов:
М ZnX2, t
Ar-H + Т1^С=О + НХ-------Аг—СН2Х
Н" -Н2О
X = CI, Вг
Реакция галогенметилирования является примером электрофильного
мещения у углеродного атома арена. Электрофильным реагентом является п
роксиметил-катион:
Н 5+ 5- 5+ 8-
_JC=O + Н-Х
н
H^S+OH
н ©
"С-°н
н X©
10.1.3. Строение, реакционная способность, химические свой-
ства
Атомы галогена более электроотрицательны, чем атом углерода, поэто
связь
5+ 5—
R-CH2-^X
полярна (дипольный момент 2.0-2.3 D). Электроны сдвигаются в сторону ато
ranoi’ena и на атомах углерода и галогена появляются дробные заряды.
108
Полярности связей (судя по дипольному моменту) не сильно отличаются
друг от друга и меняются в последовательности:
C-F (2,3)« С-С1 (2,3) > С-Вг (2,2) > С-1 (2,0),
т.к. на дипольный момент связей влияют не только величины электроотрица-
тельностей атомов, но и наличие неподеленных пар электронов и сравнитель-
ное ковалентное число.
Еще более важным фактором, определяющим легкость протекания реак-
ции, является поляризуемость, т.е. способность молекулы (связи) перераспре-
делять свою электронную плотность под действием атакующего реагента,
Наименее электроотрицательные атомы в ряду галогенов являются наибо-
лее поляризуемыми.
Поляризуемость связей (судя по рефракции связей R) увеличивается в ряду
C-F (1,7) « С-С1 (6,5) < С-Вг (9,6) < С-1 (14,6).
и различается почти в 10 раз.
Наиболее прочной является связь С-F, наименее - С-1.
В соответствии с этим в реакциях нуклеофильного замещения галогенов
(Sn) йодпроизводные максимально активны, фторпроизводные - мало реакци-
онноспособны.
Механизмы реакций нуклеофильного замещения
Механизм Sn2 (нуклеофильное замещение 2 порядка), бимолекулярное за-
мещение.
~ Н. S+ S-
Y© + Н-^С-Х
ну к- Н
лео-
фил
переходное
Y—С--Н + X®
Н нук-
лео-
фуг
состояние
активированный
комплекс
1) Скорость реакции зависит от концентрации двух веществ: реагента
(Y ® ) и субстрата (СН3-Х).
2) Y ® - атакует субстрат со стороны противоположной уходящей группы
X. В активированном комплексе связи Y-С и С-Х выравниваются, а атомы во-
дорода находятся в плоскости, перпендикулярной оси Y-C-X. X ® - нуклеофуг
(уходящая группа) выталкивается Y ® - нуклеофилом.
3) Происходит обращение конфигурации: S-конфигурация переходит в R-
и наоборот, т.е. реакция S^2 принадлежит к стереоспецифическим реакциям.
В соответствии с этим механизмом на скорость реакции оказывают влия-
ние: а) внутримолекулярные факторы - электронные и пространственные эф-
фекты реагента и субстрата; б) внешние факторы, т.е. условия реакции (раство-
ритель, температура и т.д.).
109
Реакции нуклеофильного замещения благоприятствуют:
I. В молекуле субстрата:
I) электроотрицательные заместители с -I- и -М-эффектами (увеличивают
б-l на карбонильном атоме углерода);
2) заместители с малыми объемами (например атом водорода не препятст-
вует подходу Y0), т.е. с малыми пространственными эффектами. Реализация
этого механизма характерна для первичных и частично вторичных алкилгало-
генидов;
3) наиболее поляризуемые атомы галогена, т.е. в ряду R-F < R-C1 <
R-Br < R-I. Наилучшие уходящие группы - слабые основания. В ряду галоге-
нид-ионов слабое основание Г.
II. В молекуле реагента:
Увеличение нуклеофильности нуклеофильной частицы.
Нуклеофильность возрастает при увеличении концентрации электронной
плотности на частице и способности ее отдавать электронодефицитному центру
С8+.
1) Чем правее в периоде стоит элемент, тем менее реакционноспособны его
неподеленные пары:
NH3 > Н2О > HF
СН30 >NH20 >ОН0 >F0
2) В группе нуклеофильноть возрастает сверху вниз:
I© > Br0 >CI0 >F0
3) Соседние группы также оказывают влияние на нуклеофильность реаген-
та:
а) электронодонорные повышают:
NH3 < CH3NH2 < (CH3)2NH ,
б) электроноакцепторные понижают:
Н2О> СНэ-С^и
4) Иногда в нуклеофильной частице есть несколько нуклеофильных цен*
трое (например два), связанных системой сопряжения. Например, енолят-ионы
/ ^.С— СТ . В этом случае оба центра могут вступать в ре*»
акцию Sn2.
Влияние растворителя
Протонные растворители замедляют реакции Sn2, т.к. сольватируют нук*
леофильные реагенты.
Апротонные растворители, особенно биполярные апротонные, ускоряю?
реакцию, в частности, за счет сольватации катиона они продуцируют высоко*!
нуклеофильные анионы. I
110
Механизм SN1
Для третичных алкил галогенидов подход Y® к субстрату затруднен объ-
емными радикалами, поэтому процесс идет по ионному механизму.
Этот механизм включает в себя 2 стадии.
6ыстр% >C-Y Л
Г/
На первой стадии под действием растворителя, который сольватирует за-
ряженные частицы (медленная стадия), идет ионизация и образуются ионные
пары (тесные или сольватно-разделенные) или даже диссоциация (до свобод-
ных ионов). В данном случае показан плоский тре/и-бутил-катион.
На второй стадии идет быстрая реакция с Y ®.
1. Скорость реакции зависит только от первой стадии (ионизации или дис-
социации /лре/л-галогеналкила) и следовательно от концентрации галогенпро-
изводного, т.е. реакция 1-го порядка.
2. Атака катиона возможна с двух сторон, поэтому стереохимический ре-
зультат - рацемизация.
Реакции SN1 способствуют
в галогенпроизводном:
1. Электронодонорные заместители и пространственные объемные замес-
тители.
Это связано с устойчивостью карбокатиона, чем он устойчивее, тем легче
должна идти реакция SN1. Например скорость сольволиза возрастает в ряду:
(СНз)зСС! < С6Н5(СН3)СНС1 < (СбН5)2СНС1 < (СбН5)зСС1
Объяснение: эффективная делокализация положительного заряда карбока-
тиона фенильными кольцами.
Также и в ряду:
СН3
СН2=СН-СН2С1 < СН3-СН=СНСН2С1 < CH2=CH-i-Cl
6н3
Объемные заместители дестабилизируют исходные молекулы и стабили-
шруют карбокатион, способствуя переходу атома углерода из состояния sp3 в
v> •
2. Уходящая группа.
Например, в ряду галогенов способность к диссоциации растет в ряду
I' « С1 < Вг < I. Это связано с уменьшением энергии полной диссоциации.
В протонных растворителях этот ряд может изменяться за счет специфиче-
ской сольватации (водородные связи X—HSoI); атомами водорода, несущими
мистичные или полные положительные заряды уходящих групп. Способность к
сольватации падает в ряду:
RO>F>Cl>Br>I
111
3. Влияние растворителя.
С ростом полярности растворителя растет и скорость диссоциации.
В протонных растворителях скорость возрастает за счет специфической,
сольватации, которая тем больше, чем выше кислотность молекул растворите-
ля.
4. Ионизирующая сила растворителя.
Ионизация галогенпроизводных растет в ряду кислот: ,
уксусная кислота < муравьиная кислота < трифторуксусная кислота
Реакции замещения сопровождаются побочными реакциями. >
Реакция элиминирования
Возможность этой реакции связана с тем, что любой нуклеофильный реа-
гент одновременно является основанием, т.е. способен атаковать не только
атом углерода, но и атом водорода.
В периоде нуклеофильность-основность меняется параллельно:
-NR2>-OR>-F
В группе - в противоположных направлениях (основность падает, нуклед-
фильность растет):
F > С1 > Вг > I (основность)
F < С1 < Вг < I (нуклеофильность)
Для реакции элиминирования предложено 2 механизма. j
Механизм Е2 (реакция идет по правилу Гофмана)
CH3
(Ьн
сн2
Ч ГЧб-
сн24-с!
t__f
-ci©
-HY
По этому механизму легко протекают реакции при действии реагенто]
амидов металлов, алкоголятов металлов, щелочей в спиртовых растворах.
Реакция Е2 легче протекает в рядах:
иодид < бромид < хлорид
жреш-углеродный атом > ewop-углеродный атом > первичный углероднь!
атом.
Механизм Е1 (реакция идет по правилу Зайцева) (
Медленная стадия - ионизация, затем быстрый распад на алкен и выбрш
протона Н ©, который связывается нуклеофилом. *
Н3С СН3 Н3С СН3 Н3С СН3 1
V-CI sol 0 V© V I
I — О -----► и 1
СН2 НС-Н -HCI с 1
(Ьн3 (Ьн3 rf ЧСН3 1
Перегруппировки характерны для реакций, протекающих по механизм®
SN1. Это обусловлено тем, что образующийся карбокатион может перегрупцш
роваться в более стабильный. W
112
СНх
СНРс-СНСНз < >- СНз—С-СНСН3 --►
СН/ (kj -Cl® CH3Z
вторичный
Y СН3
с1М-6н -
сн^ 6н,
третичный
СН3
сн^ 6н,
____СНз^ СНз
_Н®* СНГ ^сн3
Например, реакция с неопентилхлоридом (под действием Ag®) идет о перо*
группировкой через трет-карбокатион.
Примеры реакций нуклеофильного замещения.
Н2О
» ROH
2^Urnh2
Na—R- » ROR'
AgOCOR>> RCOOR'
RNO2(RONO)
NaCN
----► RCN
Nal
----► RI
NaSH
—s---► R.SH
R-X —
R-X
Восстановление
[H] (C2HSOH+Na)
-NaCl
Отношение к металлам:
а) Вюрц
R-Cl + Na +C1—R’ -► R-R + R-R* + R —R'
б)Зайцев
CH3
CH2=CHCH2I + Zn + CH3COCH3 CH2=CHCH2—6-OH
' v J CH3
[CH2=CHCH2Znf]
113
п) Гриньяр
Rd Mg.M>HP> R MgC[
10.2. Галогенпроизводные co связью C(sp2)-Hal
10.2.1. Получение галогенаренов
1. Прямое галогенирование:
а) хлорирование идет легко с катализатором Fe+FeCh (механизм Sg
(С1+[А1С14Г);
б) бромирование идет труднее (Fe+FeBrj);
в) йодирование требует специальных условий.
2. Фторирование только обменной реакций с использованием KF
(300°С, р).
3. Через арендиазониевые соли.
ArF (Шиман)
CuCl
(Зандмейер)
10.2.2. Строение, реакционная способность, химические свой-
ства
Сравнение дипольных моментов галогеналканов и галогенапкенов, а также
галогенаренов показывает, что полярность связи C(sp2)-X меньше, чем
C(sp3)-X.
Соединение СНзСНг-Cl СН2=СН-С1 С1
Дипольный момент (D) 2,0 1,44 1,58
Причиной уменьшения полярности связи является изменение состояния
| нОриди шции углеродного атома sp3 на sp2, что вызывает увеличение электро-
отрицательности этого атома.
114
Неподеленная пара электронов вступает также в сопряжение и это вызыва-
ет смещение тс-электронной плотности от атома галогена в сторону углеродных
атомов. Другими словами, атом галогена проявляет +М-эффект, особенно в пе-
реходном состоянии.
5->~л8+ Iх
СНг— Cl 72 А
Н
Вследствие этого изменяется характер связи С-Х. Она становится короче и
прочнее с 1,76 А (С С!) до 1,72 А (=С С1).
Галогеналкены и галогенарены малоактивны в реакциях нуклеофиль-
ного замещения.
Реакции проходят трудно, необходимы жесткие условия и при этом проте-
кают побочные реакции.
а) Реакции Sn галогеналкенов - при взаимодействии с нуклеофильными
реагентами в большинстве случаев не удается осуществить. Доминируют по-
бочные реакции - отщепление, присоединение, полимеризация.
б) Галогенарены взаимодействуют с нуклеофильными реагентами при
температуре выше 200°С. Сама система бензола при этих температурах ста-
бильна.
, , NaOH, 300°С, Си или Cu(I)
сбн5с1---------------:----------—
с6н5он
Легче реагируют йодарены (Си* - Ульман).
Механизм Sn?
карб- j
анион
1. Чем устойчивей карбанион, тем легче будет идти реакция Sn2. Устойчи-
вость повышается при введении в кольцо электроноакцепторных заместителей
в орто- и пара-положения (NO2, CN, SO3H, СООН).
Группы NO2 стабилизируют переходное состояние типа I и в этом случае
реакция, например, с AgOH протекает в обычных условиях.
115
2. Скорость реакции зависит также от уходящей группы в галогенарене.
Оказалось, что она возрастает в ряду:
I < Br < Cl « F
Это связано с тем, что более электроотрицательный F лучше стабилизиру-
ет образующийся карбанион.
3. Скорость реакции увеличивается с увеличением нуклеофильности реа-
гента.
Кроме механизма Sn2 может реализовываться и другой тип замещения
элиминирование-присоединение.
Если на арилгалогенид действовать сильным основанием (NaNH2 в NH3),
то реакция идет следующим образом.
Механизм элиминирования-присоединения
"Дегидробензол"
На первой стадии вырывается протон из орто-положения. На второй ста-
дии образуется «дегидробензол». На третей стадии происходит присоединение.
Элиминирование - первая и вторая стадии, присоединение - третья стадия.
Структура «дегидробензола» очень активна. Это структура бирадикала:
Структура с тройной связью маловероятна, т.к. перекрытие р-орбиталей в
радикале небольшое. Уловить «дегидробензол» можно диенофилами при по-
мощи диенового синтеза.
Доказательство образования «дегидробензола»:
NH3 48% 52%
«Дегидробензол» легко реагирует с нуклеофильными реагентами и сам с
собой.
116
Реакция с металлами
Галогеналкены реагируют с металлами по-разному:
а) активные металлы могут вызвать полимеризацию;
б) магний реагирует только в ТГФ.
ТГФ
CH2=CHCI + Mg » CH2=CH-MgCl
Галогенарены взаимодействуют как обычно.
Аг—Аг
Ar—Li
Ar-MgCl
в обычных условиях не реагирует
Li
Ar—С1 — Mg r
Zn
Реакции замещения
Реакции замещения типа Se характерны для галогенаренов (нитрование,
сульфирование, галогенирование).
Скорости реакции ниже, чем скорости подобных реакций, например для
толуола. Атомы галогена в этих реакциях выявляют свои электронодонорные
свойства на л-электронном уровне и ориентируют вступающие группы в орто-
и лара-положения.
5— 5—
V толуола » ^галогснбензола
117
Выводы
1--------------► [карбокатионы] —*•
перегруппировки
—► продукты замещения
и элиминирования
Z (-1, -М) - электроноакцепторный заместитель
ВН - сильное основание
118
11. Гидроксилсодержащие производные углеводоро-
дов. Спирты. Алканолы
11.1. Гидроксилпроизводные углеводородов со связью
C(sp3)-OH
В эту группу входят:
а) гидроксилпроизводные алканов - насыщенные одноатомные спирты,
или алканолы CnH2n+lOH;
б) гидроксилпроизводные алкенов или алкинов - ненасыщенные одно-
атомные спирты, или алкенолы или алкинолът.
C=C-(C)n-OH (CnHin-iOH) C=C-(C)n-OH (СпН2п.3ОН)
в) гидроксилпроизводные циклоалканов или циклоалкенов - циклоалкано-
лы и циклоалкенолы:
а ОН
(QH^OH) |f J (СпН2п.3ОН)
г) гидроксилпроизводные циклоаренов с гидроксильной группой в боковой
цепи - арилалканолы:
Аг-<С)п-ОН (СпН2п.7ОН)
д) дигидроксилпроизводные — двухатомные спирты, или диолы
С„Н2п(ОН)2;
е) тригидроксилпроизводные и полигидроксилпроизводные — трехатомные
и многоатомные спирты, или триолы и полиолы:
CnH2n.!(OH)3 CnH2n+2.m(OH)m
11 .1.1. Алканолы
Наиболее важный тип спиртов - алканолы. Гидроксильная группа может
быть связана с различными углеродными атомами, отсюда различают первич-
ные, вторичные и третичные спирты.
11.1.1.1. Номенклатура
1. Тривиальная (по названию радикалов) - радикально-функциональная.
СН3ОН - метиловый
(СН3)2СНОН - изопропиловый
(СН3)3СОН - трет, -бутиловый
СН2=СНСН2ОН - аллиловый
119
СНиС-CHjOH - пропаргиловый
СЛ11j-Cl IjOH - бензиловый ,
CHjOH-CHjOH - этиленгликоль
Cl laOH CHOH-CH2OH - глицерин
2. Рациональная. Спирты рассматриваются как производные СН3ОН, или
карбинола.
(СбН5)2СНОН - дифенилкарбинол
3. ИЮПАК:
а) выбирается наиболее длинная цепь углеродных атомов с группой ОН;
б) группа ОН должна иметь наименьший номер;
в) к названию углеводорода добавляется окончание -ал;
г) если есть старшая группа, то гидроксильную группы обозначают пре-
фиксом -окси (-гидрокси).
С1СН2СН=С(С2Н5)СН2ОН - 4-хлор-2-этилбутен-2-ол-1
НОСН2СН2СОСНз - 4-гидроксибутанон-2
Глицерин - пропантриол-1,2,3
и
11.1.1.2. Способы получения
1. Ферментативный гидролиз углеводов
С6Н]2Об ДР0ЖЖИ>. С2Н5ОН(14%)
глюкоза а также С3Н7ОН, /-С4Н9ОН, /-С5Нj jOH
2. Окисление алканов (через стадию перекисей)
3. Гидратация алкенов
НОН [Н®] I
RCH=CH2 ------------—*- RCH-СН3
(H2SO4, ZnCl2) 1
4. Гидролиз галогенпроизводных (используются щелочи для смещения
равновесия)
НОН (ОН0)
RX <------ROH
НХ
СН2=СНСН2С1 ------► аллиловый спирт 1 ГИДролизует
СбН5СН2С1-----► бензиловый спирт J очень легко
5. Восстановление альдегидов, кетонов, производных кислот
H2(Ni)
RCOR1
LiAIH4
RCH(OH)R1
А1(ОС3Н7-0з
-(СН3)2СО
120
Механизмы восстановления
б) восстановление по Меервейну-Пондорфу-Верлею
i V»
. Na+ROH [Н] .
RCOOR1---------—RCH2OH + R’OH
метод
б) Буво-Блана
LiAlH4
в) RCOHal RCH2OH
6. Металлоорганический синтез
В 1900 г. В. Гриньяр широко развил реакцию с использованием металлоор-
ганических соединений. Сущность ее состоит в следующем:
I стадия
RX + Mg----► RMgX (ДЭЭ, ТГФ и т.д.)
II стадия
О ® Ч8+Ах I НОН I
R MgX + ^С=О5- —► R-C-OMgX---------------► R-C-OH
I -Mg(OH)Cl |
СН2=О -> первичный спирт
R-CH=O -> вторичный спирт
RCOR1 -> третичный спирт
7. Гидрирование оксида углерода (промышленный способ)
Н2, ZnO, Сг2О3, 500°С, р л
СО — --------------------——--------СН3ОН
Со, Rh, t°
СО --------—
синтол
(смесь спиртов)
11 .1.1.3. Физические свойства
Особенность - высокая температура кипения спиртов. Она, например, вы-
ше, чем для соответствующих йодалканов, хотя молекулярная масса спиртов
121
значительно ниже. Это вызвано значительным межмолекулярным взаимодейст-
вием - ассоциацией молекул.
Температура кипения возрастает в ряду:
третичный < вторичный < первичный
Трем.-бутиловый спирт - кристаллический (т.пл. 25°С).
Температура кипения резко возрастает в ряду:
одноатомный < двухатомный < трехатомный < многоатомный
11 .1.1.4. Строение, реакционная способность, химические свойства
Алканолы являются полярными соединениями. Они содержат две поляр-
ные связи:
5+ S— 5— б+
С—»-0 и О<-Н
p = 0.9D p=1.5D
Суммарный эффект с учетом влияния НЭП = 1.6-1.8 D. Например, у
СН3ОН (газовое состояние) М = 1.69 D.
Полярность связи О<—Н больше, чем С->0.
По сравнению с водой алканолы (их НЭП) проявляют большие электроно-
донорные свойства.________________________________________
Н2О СН3ОН С2Н5ОН
эИ (эВ) 12.6 10.8 10.6
Полярная связь О-Н и НЭП кислорода определяют возможность межмо-
лекулярной ассоциации.
5— 5+ 5— 5+
О*-Н ... О-«-Н...
Ассоциация в основном осуществляется за счет электростатического при-
тяжения разноименно заряженных частей молекул. Такое взаимодействие на-
зывают водородной связью (энергия невелика 5-7 ккал/моль).
У алканолов сохраняются обычные длины связей С-С и С-Н, атомы угле-
рода и кислорода находятся в состоянии $р3-гибридизации, угол 106°.
Имеются два подхода для объяснения электронного состояния спиртов: I -
состояние с полной £р3-гибридизацией атома кислорода (обе электронные пары
находятся на одинаковых орбиталях; угол HOR равен 109°28'); II - состояние
без гибридизации (одна электронная пара находится на р-орбитали, другая на
5-орбитали; угол HOR равен 90°):
122
oqc)
H R
I состояние
др3-гибридизации
И состояние
без гибридизации
Первый подход более адекватно отражает величину реального угла HOR,
равного 106°.
1. Кислотность спиртов
Алканолы являются очень слабыми ОН-кислотами, менее кислыми, чем
НОН.
R-O-H «Д-» RCP+ HSol®
Анион алканола, называемый алканолят-ионом (алкоксид- или алкоголят-
ионом), представляет собой сильное основание.
Кислотность алканолов
Алканол Z-BuOH z-PrOH EtOH МеОН Н2О СС13СН2ОН СН3СООН PhOH
рКа для водных растворов (“1g ^дис) 19 17 16 15 15.7 12 5 10
Такое изменение кислотности можно объяснить +1-эффектом алкильной
группы.
Отсюда, например, анионы третичных спиртов наиболее сильные основа-
ния.
В водных растворах кислые свойства спиртов проявляются слабо. Только в
концентрированных водных растворах щелочей частично происходит иониза-
ция алканолов.
Обычно для получения алканолятов используют реакцию безводного
спирта и активного металла
2ROH + 2Na----> 2RONa + Н2
Алканоляты являются не только сильными основаниями, но и сильными
нуклеофилами, использующимися в реакциях конденсации (как основания), ал-
килирования и ацилирования (как нуклеофилы).
2. Основные свойства спиртов
Неподеленная пара электронов (НЭП) кислорода может взаимодействовать
с электрофильными (Е) реагентами с образованием донорно-акцепторной (се-
миполярной) связи:
R-O-H + Е R-SCg©
Е - кислоты Льюиса (BF3, AIC13, TiCl4, Н®)
123
ROH + H®
Основные свойства спиртов выражены сильнее, чем у воды.
11ри взаимодействии спиртов с Н® образуется катион оксония:
® Н
-%
катион
оксония
В сильных кислотах (20%-ная серная кислота, pH = -1) метанол на 10% J
превращается в катион метил-оксония.
В слабых кислотах (pH > 0) концентрация катионов алкил-оксония состав- 1
ляет доли процента. Большинство молекул в этих условиях образуют водород-
ные связи.
Во многих случаях продукты реакции алканолов с Е-реагентами образуют-
ся через промежуточные карбокатионы:
5+ ® Н д, Н к
R-ОС , » R®+(T
н н
8+ ® Н ~ Н
r-o;e ^R®+o;Ee Я
Реакции с разрывом связи О-Н
1. Кислотность и образование алканолятов.
2. Образование сложных эфиров кислот (реакция этерификации).
[н®| Л)
ROH + R'COOH < > R— С?
Н20 OR
Механизм реакции этерификации (в условиях кислотного катализа):
R"°^H
®z ОН^нуклеофил
СНН;
,0 Н
ОН
ОН
О
-Н2О U“K
-Н®
Скорость реакции этерификации уменьшается в ряду:
первичный > вторичный » третичный
Реакции с отщеплением группы ОН
1. 11уклеофильное замещение группы ОН.
Y® + ROH R-Y + ОН®
124
ОН значительно труднее замещается в реакция Sn, чем атомы 1'алоганов (I,
Br, С1). Это обусловлено невыгодностью образования богатого энергией гидро-
ксид-аниона. Поэтому эта реакция осуществляется с ограниченным числом
нуклеофильных реагентов или требует специальных условий.
a) ROH R—X
-Н2О
X = CI, Br, I
HF не дает желаемых продуктов, т.к. имеет меньшую кислотность:
HI HBr HCI HF
J>Ka_ -10 -9.5 -7.4 -3.2
Эту реакцию можно рассматривать как нуклеофильное замещение прото-
нированной гидроксильной группы.
В образующемся ионе оксония дробный положительный заряд (8+) на ато-
ме углерода увеличивается
является хорошей уходящей группой) и ста-
новится возможной реакция Sn2.
Наиболее легко реакция идет с третичными спиртами по механизму SnI,
т.к. в этом случае образуется стабильный трет.-карбокатион.
Для замены группы ОН на галоген используют такие неорганические гало-
гениды, как РС1Э, РС15, SOCI2.
Например,
СгНзбН^РС^
С2Н5ОРС14----►
-РОС13
С2Н5С1
< 4°
ROH + S^Cl
Cl
О
-НС1
Sfti
- RC1
-SO2
сложный эфир
хлоран гидрида
сернистой кислоты
б) Образование простых эфиров
Алканолы в присутствии сильных кислот образуют простые эфиры:
Н®
--------------------------------► R-O-R
2R-OH
-Н2О
125
Эфиры образуются, если кислота взята в недостатке, а температура реак-
ции достаточно низка.
Механизм заключается в нуклеофильной атаке одной молекулы спирта
протонированного алканола (иона оксония) или образующегося из него карбо-
катиона.
Н® 8+ © н
R—ОН ► R-O<
н
r®+h2o
R—ОН
Реакция идет с хорошим выходом с первичными спиртами. В случае вто-
ричных и третичных спиртов в продуктах реакции преобладают алкены.
в) Дегидратация спиртов (образование алкенов)
Отщепление воды происходит в присутствии сильных кислот или над ка-
тализатором при повышенной температуре.
[Н®] (H2SO4, изб.), А12О3 (300°С)
RCH2CH2OH -------------------------------► RCH=CH2
-H2O
F IJ
сн3сн2бн —
-H2O
сн2=сн2
н
Реакция идет по механизму Е1 (по правилу Зайцева). При избытке спирта
образуется простой эфир.
При дегидратации спиртов, содержащих третичный радикал рядом с угле-
родом, связанным с гидроксильной группой, наблюдается ретропинаколино-
вая перегруппировка.
НЭС Н
Н3С-С-С-СН3
н3г бн
ИзС Н
Н3С—С—С—СН3
Н3с Н
н,с-с-с-сн,
(йА/®
н®
-н20
Н" "Н
cSc^chj
Н3С сн3
Н3С сн3
сн3
126
Происходит отнятие воды как бы от одного атома углерода с одновремен-
ным смещением к нему алкильного радикала.
Окисление и дегидрогенизация
Окислителями являются KM11O4+H2SO4, К2СГ2О7+H2SO4. Кислородом воз-
можно в присутствии катализаторов Си, СиО:
а) первичные
ГО] (СгО3) [О]
С2Н5ОН —-------СН3СН=О -^►CHjCOOH
б) вторичные
[О]
(СН3)2СНОН (СН3)2С=О
в) третичные окисляются только в кислой среде в жестких условиях, через
алкен.
Считается, что окисление является гомолитическим процессом, в ходе ко*
торого образуются свободные радикалы.
Близка по результату к реакции окисления реакция каталитической де-
гидрогенизации под действием оксидов тяжелых металлов (Fe, Zn, Си) ИЛИ в
присутствии Си, Pt, Pd.
r-ch2oh t; KaT--> R-C*° + H2
H
11.2. Многоатомные спирты
К ним относятся спирты, которые содержат несколько гидроксильных
групп. Две — диолы или гликоли, три — глицерины, четыре - эритриты.
Гем-диолы неустойчивы (исключение СС1зСН(ОН)г). Однако их производ-
ные ацетали и кетали типа >C(OR)2 достаточно стабильны.
11.2.1. Способы получения
1) Гидролиз вмг/-дигалогенпроизводных (гидролиз сложных эфиров).
2) Окисление кратной связи по Вагнеру.
3) Восстановление карбонильных производных.
4) С помощью магнийорганических соединений.
Примеры
1Ч СН2С1 Н2О
1) I —-—► этиленгликоль
CH2C1
127
[О], Н2О
2) Н2С=СН2 --------;— ---------► этиленгликоль
С12, НОН , Н2О, OHet
I—-----► СН2ОН-СН2С1 ——--------1
.. НЭС<„ „ Na(Hg),H2O НЭС<„ СН3
’ ГнСН’
пинакон
.О Н3С СН3
pOEt 4CH3MgI Yf-OMgl НОН
4) _ ------ - -> -------► пинакон
^OEt C-OMgl
*0 H3cf хсн3
ЭТИЛЕНГЛИКОЛЬ
Химические свойства
Реакции аналогичны реакциям алканолов (спиртов). Реагируют одна или
обе гидроксильные группы - образуются 2 ряда производных: полные и не-
полные.
Кислотность (рКа = 15.1). Этиленгликоль - более кислый, чем этанол (-/-
эффект второй группы ОН).
а) Образование алконолятов (гликолятов):
СН2ОН Na CH2OH-CH2ONa
I z > или
CH2OH CH2ONa-CH2ONa
б) Легко идет замещение на тяжелые металлы:
Н
СН2ОН Си(ОН)2~ СНг—Оч Д)-СН2
6н2он ^Н2-о-,Си"о-(5н2
ъ
Происходит образование хелатных ярко окрашенных соединений (качест-
венная реакция на а-гликоли).
в) Образование простых эфиров:
Н®
этиленгликоль + СН3ОН--► НОСН2СНОСН3 или СН3ОСН2СН2ОСН3
Метилцеллозольв и диметоксиэтан (моноглим) применяются как раствори-
тели.
г) Образование сложных эфиров:
НС1 (сухой) RCO-OCH2CH2OH
этиленгликоль + RCOOH-----или
RCO-OCH2CH2O-COR
128
д) Гидроксильные группы могут быть замещены атомами галогена:
НС1 РС15
этиленгликоль----► НОСН?—СН2С1-------CICH2—СН2С1
I
этиленхлоргидрин
Соединение I имеет большое значение для синтеза новокаина, индиго, ип-
рита, аминоспиртов, окиси этилена.
е) При нагревании с кислотами образуется циклический эфир - диоксан:
ж) Дегидратация:
СН2ОН ZnCl2 СН2
I 2 -----+ и 2 ----►СН3СНО
сн2он -н2о снон 3
Дегидратация пинаконов сопровождается пинаколиновой перегруппи-
ровкой:
н3с< сн3 н®
,СН3
H3Q
н3с<8
Н3СГ
з) Окисление.
этиленгликоль —
_О
СН2ОН-С?
н
гликолевый
альдегид
СН2ОН-СООН
гликолевая
кислота
*->. нсо-сно * нсо-соон —► ноос-соон
глиоксаль глиоксалевая щавелевая
кислота кислота
Окисление иодной кислотой (метод Малапраде), тетрацетатом свинца яв-
ляется способом установления расположения гидроксильных групп в гликолях,
сахарах и других сложных оксисодержащих органических молекулах.
129
HIO4
CHj—CH—С2Нз------
6н 6н
Н С2Н5
сн-сн
о^он
-----► НСНО + С2Н5СНО
-НЮ3
ГЛИЦЕРИН
Глицерин - бесцветная сиропообразная жидкость со сладким вкусом.
Получение
1) Впервые был получен Шееле при обработке жиров щелочью в присутст-
вии оксида свинца.
2) Из аллилхлорида (или аллилового спирта):
НОС1 ?н НОН (ОН°)
СН2=СНСН2С1------» С1СН2СНСН2С1-----------+ глицерин
3) Из акролеина:
О [Н] [О] НОН
СН2=СН-С? —СН2=СН-СН2ОН —---------------► глицерин
н
Химические свойства
Глицерину присущи все характерные реакции алканолов. Дает три ряда
производных: моно-, ди- и три-.
Кислотность (рКа ~ 14). По кислотности превышает гликоли (влияние -/
групп ОН). Предполагают, что ионизируется средняя гидроксильная группа.
Очень легко образует глицераты тяжелых металлов, например глицерат
меди (II), в котором ион металла связан в виде внутреннего комплекса, поэтому
гидроксид металла щелочью не осаждается (качественная реакция):
Действие кислот
а) Азотная кислота:
\ ONO2ONO2ONO2
\ глицерин ----U (Ьнг—(Ьн—6н2
тринитроглицерин
Тринитроглицерин с добавками - динамит (Нобель).
130
б) Дикарбоновые кислоты - образуются полиэфиры (алкидные смолы), ко-
торые применяются в лакокрасочной промышленности.
в) Щавелевая кислота:
(СООНЪ t°
глицерин 2----СНт-СН-СН2ОН------------► СН2=СН-СН2ОН
-2Н2О J О "2С°2
Ьо-со
г) Соляная кислота:
НС! ?H2Cl НС! глицерин ► СН2ОН i СН2С1 ►
6н2он 6н2он I
а-монохлор- дихлор- эпихлор-
гидрин гидрин гидрин
глицерина глицерина глицерина
Дегидратация
глицерин
t°,H+(KHSO4)
-2Н2О
Н Н
xc=c=d
И он
о
—► СН2=СН-С^
акролеин
Окисление
Образуется целый ряд продуктов.
Мягкое окисление:
глицерин
СН2ОН-СН2ОН-С*°
глицериновый
альдегид
СН2ОН-СО-СН2ОН
диоксиацетон
Окисление по Малапраде:
НЮ4
глицерин------► 2 СНг=О + НСООН
11.3. Гидроксилпроизводные углеводородов со связью
C(sp2)-OH
1) Енолы
2) Фенолы (нафтолы, арендиолы - двухатомные фенолы, арентриолы -
। рсхатомные фенолы).
131
11.3.1. Енолы (непредельные спирты)
Представитель - виниловый спирт или этенол, СН2=СН-ОН. Эти вещества
нестабильны, существуют как промежуточные соединения.
11.3.1.1. Получение и строение
1) Реакции алкинов с водой (Кучеров):
R—С=С—R
НОН (Hg2+)
2) Гидролиз сложных эфиров енолов:
НОН [Н®] г 1
R— соосн=сн2-----------► носн=сн2
-RCOOH L -1
Методом микроволновой спектроскопии удалось оценить длины связей в
молекуле винилового спирта:
Все связи имеют обычную длину, связь С-0 укорочена.
11.3.1.2. Химические свойства
Енолы не стабильны. Их, как правило, не удается выделить из реакцион-
ной смеси, т.к. они перегруппировываются в карбонильные соединения (пере-
группировка Эльтекова-Эрленмейера), причем перегруппировка носит межмо-
лекулярный характер:
Время жизни енолов колеблется от 10 с до 30 мин. Получено несколько
стабильных енолов, например:
ОН
I
F 3С— С= CF 2 пентафторпропен-1 -ол-2
132
11.3.1.3. Таутомерия сполов и карбонильных соединений
Принципиально возможен нс только переход енола в карбонильное совди-
некие, но и наоборот. Следовательно, образуется два (или более) изомера, пе-
реходящих один в другой при миграции протона. Такое явление называют ди-
намической изомерией или таутомерией.
Таутомерную систему карбонильное соединение енол (К Е)
характеризуют константой таутомерного равновесия Кт или иногда процентным
содержанием енола.
Обычно содержание енола от 10~2 до 10"^%, т.к. положение равновесия оп-
ределяется сравнительной термодинамической стабильностью К и Е, т.е. тепло-
той образования. Карбонильные соединения на 15-18 ккал/моль более термоди-
намически стабильнее, чем енолы.
рКа енолов ~11-12 (алканолы 15-19)
рКа карбонильных соединений «16-20
Ph(Me)C=CHOH - 8.7%
(СР3)гС=СНОН-100%
Таким образом, система Е К является равновесной системой, со-
стоящей из сравнительно сильной ОН-кислоты и слабой СН-кислоты.
Обе кислоты при ионизации образуют один и тот же анион - амбидентный
енолят-анион.
анион имеет 2 реакционных центра
Енолят-анион является сопряженным ионом, заряд в нем делокализован
между О- и С-атомами.
Производные енолов - простые и сложные эфиры - не содержат подвиж-
ного атома водорода и в обычных условиях стабильны, однако легко гидроли-
зуются. Используются для синтеза полимеров.
11.3.2. Фенолы
11.3.2.1. Номенклатура
Тривиальные названия:
фенол крезолы резорцин
пирогаллол
133
По И ЮНАК названия фенолов образуют от названий соответствующих
аренов, а наличие гидроксильной группы обозначают суффиксом -ол.
11.3.2.2. Получение
1) Из легкого масла каменноугольной смолы.
2) Кумольный метод (Удрис и Сергеев).
СН3
СН(СН3)2 0 65°С)
----------► 1| О-О-Н----------------1
КОН, 130°С
кумилпероксид
*
3) Сплавление аренсульфоновых кислот с щелочами.
NaOH, 300°С Н® .
C6H5SO3Na -------------► C6H5ONa ------► фенол
-Na2SO3
4) Из хлорбензола (реакция Ульмана).
NaOH, Си, ЗОО°С ,
СбН5С1 -------------------------------► фенол
5) Через соли диазония.
НОН
----► АгОН
6) Окисление реактива Гриньяра.
О2 C6H5MgX Н® .
C6H5MgX —C6H5OOMgX - » CeH5OMgX----► фенол
7) Прямое гидроксилирование (катализаторы соли металлов — Fe
Си (И), Ti (III)).
(П),
НООН, кат-р
----------► фенол
134
113.2.3. CipoeiiHo, роякциопняя способность, химические свойстве
Фенол и сп» гомологи являются полярными соединениями, их дипольные
моменты 1.5-1.6 D. При этом вектор дипольного момента в отличие от алкано-
лов и циклоалканолов направлен в сторону бензольного кольца.
5-
р = 1.5Д
Это связано с взаимодействием НЭП с тс-электронной системой кольца.
+М-Эффект кислородного атома преобладает над его -1-эффектом, переда-
ваемом по системе a-связей. Образование такой сопряженной системы увели-
чивает электронодонорные свойства. Энергия ионизации фенола 8.5-8.6 эВ, чю
ниже ЭИ бензола, толуола, алканолов. Реакции определяются присутствием
ОН-группы ароматического кольца.
Кислотность и реакции с участием атома кислорода
Фенол на ~8 порядков более кислый, чем алифатические спирты. Его рКа *
10. Поэтому фенол называют карболовой кислотой.
Это определяется двумя факторами:
1) большой полярностью связи О<—Н;
2) образованием сопряженного фенолят-иона, в котором отрицательный
заряд значительно делокализован.
__ 5“
О-@+^о1 5=^ 8-<^~yO)Q + HSol
5-
Введение элпектроноакцепторных групп повышает, а электронодонорных
групп понижает кислотность:
рКа
7.2
10.2
0.2
135
Фенол реагирует не только с щелочными металлами как алканолы, но бла-
годаря высокой кислотности взаимодействует и со щелочами с образованием
фенолятов.
NaOH
-Н2О
О-Na
Фенолят натрия более сильный нуклеофил, чем фенол.
Фенол образует также с солями тяжелых металлов (например с FeCI3) ком-
плексные феноляты с ионом Fe (III). Это качественная реакция на фенол -
красно-фиолетовое окрашивание.
Феноляты - активные нуклеофилы, поэтому легко алкилируются и ацили-
руются.
Образование простых эфиров
СН31
C6H5ONa -
—^СбНзОСНз
(CH3hso4
Для аллилового эфира фенола известна перегруппировка Кляйзена (сигма-
тропная перегруппировка).
Перегруппировка Кляйзена - строго внутримолекулярная. Для нее ха-
рактерно большое отрицательное значение AS*, соответствующее высокой сте-
пени упорядоченности циклического переходного состояния.
Доказательством такого механизма служит метка 14С.
Образование сложных эфиров
RCOC1
PhONa-
(RCO)2O
О
R-C
Ъ- Ph
Ацилированию может подвергаться и сам фенол, но только сильными аци-
лирующими реагентами (не кислотой), например ангидридами кислот.
(RCO)2O
PhOH -----RCOOPh
Фениловые эфиры кислот подвергаются перегруппировке Фриса.
136
Суть перегруппировки заключается в том, что фенолят-ион является поли-
дентным нуклеофилом, имеющим 4 реакционных центра: атом кислорода и
атомы углерода в орто- и пара-положениях бензольного кольца. Именно в эти
положения проходит атака хлорацила, образующегося в ходе реакции.
Нуклеофильное замещение гидроксильной группы на нуклеофил идет с
‘большим трудом (практически не идет).
g Реакции ароматического кольца (Se)
облегчает прохождение реакций с
О
ВГ Вг2 ВГ
Гидроксильная группа значительно
юктрофильными реагентами.
1) Галогенирование
•г
2,4,4,6-тетрабром-
циклогексадиенон
Чтобы получить монопродукт, нужно проводить реакцию при 0°С в инерт-
пара-
ОМ
NO2
2,4,6-тринитрофенол
(пикриновая кислота)
137
4) Ацилирование по Фриделю-Крафтсу
5) Алкилирование
В случае б) и в) кислоты с алканолами и алкенами генерируют карбока-
тионы R ® — сильные электрофильные частицы.
6) Нитрозирование (нитрозофенол - хинон оксимная перегруппировка)
HNO2^
N—ОН
п-нитрозофенол монооксим
п-бензохинона
7) Азосочетание
НО—/"А А-Г^ Ar—N=N—rV0H
138
8) Карбонилирование:
а) Вильсмейер
" ОРОС12 “
нс^й(сн3)2
® t
HCON(CH3)2 + POCI3
он о
б) Реймер-Тиман
139
В этой реакции электрофильным реагентом является гидроксиметилкатион
Н
ХС=О + НХ
X©
Соединение I в присутствии кислот может выступать алкилирующим реа-
гентом:
В более жестких условиях фенол с формальдегидом образует высокомоле-
кулярные продукты поликонденсации - фенолформальдегидные смолы.
Возможна также поликонденсация с образованием СН2-мостиков между
отдельными цепями. Так образуется сетчатая структура полимера.
11) Реакция с кетонами
ОН
11
13) Окисление
Такие окислители, как FeCl3, окисляют фенолы до арилоксирадикалов (фе-
ноксильный радикал).
ОН О» О®
5»
делокализация
в орто- и ияра-положения
140
Радикал может димеризоваться:
4,4'-диоксидифенил
Стабильность радикала увеличивается при введении пространственно объ-
емных заместителей. Например,
Ионол образует устойчивый феноксильный радикал (кристаллический).
Выводы
[карбокатионы]
141
он
продукты окисления
[О]
легко
sol (В0)
- *----------- » [фенолят-анион]
кислотность „©
рКа~10
> 5+ 5- ОН
Е®(Е—X)
-Н®
Sp(легко)
О* “
L_ _
феноксильный
радикал
Библиографический список
1. Агрономов А.Е. Избранные главы органической химии. М.: Химия,
1990. 559 с.
2. Робертс Д., Касерио М. Основы органической химии. М.: Мир, 1978. Кн.
1.842 с.
3. Моррисон Р., Бойд Р. Органическая химия. М.: Мир, 1974. 1132 с.
4. Гауптман 3., Грефе Ю., Ремане X. Органическая химия. М.: Химия,
1979. 832 с.
5. Шабаров Ю.С. Органическая химия. М.: Химия, 1994. Ч. 1. 494 с.
6. Нейланд О.А. Органическая химия. М.: Высшая школа. 1990. 750 с.
142
Оглавление
1. Валентное состояние атома углерода, о- и я-связи.................3
1.1. Орбитали.....................................................3
1.2. Основное и возбужденное электронное состояние атома углерода.4
1.3. Гибридизация.................................................4
1.4. сг-Связи и л-связи......................................... 7
2. Электронные эффекты (механизмы) влияния атома в молекулах,
классификация реакций...............................................8
2.1. Электронные смещения в насыщенных молекулах (индуктивный
эффект, I).......................................................8
2.2. Электронные смещения в ненасыщенных молекулах (мезомерный
эффект или эффект сопряжения, М)................................9
2.3. Классификация реакций.......................................12
3. Алканы..........................................................15
3.1. Гомологический ряд, изомерия и номенклатура.................15
3.2. Получение...................................................18
3.3. Физические свойства, пространственное и электронное строение,
реакционная способность........................................19
3.4. Химические свойства.........................................21
3.4.1. Реакция радикального замещения (SR).....................21
3.4.2. Термическое расщепление алканов.........................25
3.4.3. Изомеризация и перегруппировки алканов..................26
Выводы.............................................................27
4. Алкены..........................................................28
4.1. Изомерия и номенклатура................................... 28
4.2. Получение...................................................30
4.3. Физические свойства, пространственное и электронное строение,
реакционная способность........................................31
4.4. Химические свойства.........................................32
4.4.1. Реакции электрофильного присоединения (AdE)............ 32
4.4.2. Реакции радикального присоединения (AdR)................36
4.4.3. Восстановление..........................................37
4.4.4. Окисление...............................................38
4.4.5. Полимеризация...........................................39
4.4.6. Реакции алкенов в аллильное положение (SR)..............41
4.5.3. Практическое применение алкенов.........................43
Выводы.............................................................43
5. Алкадиены.......................................................44
5.1. Номенклатура................................................44
5.2. Аллен (алкадиен-1,2) и его гомологи.........................44
5.2.1. Способы получения.......................................44
143
5.2.2. Пространственное и электронное строение, реакционная способность
аллена.......................................................45
5.2.3. Химические свойства аллена............................45
5.3. Диаллил...................................................46
5.4. Алкадиены-1,3.............................................47
5.4.1. Получение.............................................47
5.4.2. Пространственное и электронное строение, реакционная
способность..................................................48
5.4.3. Химические свойства...................................49
5.4.3.1. Реакции электрофильного присоединения (AdE).......49
5.4.3.2. Восстановление (гидрирование).....................50
5.4.3.3. Окисление.........................................50
5.4.3.4. Присоединение диенофилов (диеновый синтез)
(Дильс-Альдер).............................................50
5.4.3.5. Полимеризация........................................50
5.5. Каучук....................................................51
5.5.1. Натуральный каучук.........:..........................51
5.5.2. Синтетический каучук..................................52
Выводы......................................................... 53
6. Алкины........................................................54
6.1. Номенклатура..............................................54
6.2. Получение.................................................54
6.3. Пространственное и электронное строение, реакционная способность.. 55
6.4. Химические свойства.......................................56
6.4.1. Реакции электрофильного присоединения (AdE)...........56
6.4.2. Реакции нуклеофильного присоединения (Adhi)...........57
6.4.3. Карбонилирование......................................58
6.4.4. Гидрирование..........................................59
6.4.5. Окисление.............................................59
6.4.6. Димеризация, циклоолигомеризация и полимеризация алкинов.59
6.4.7. Реакции по связи =С-Н.................................61
6.4.8. Изомеризация........................................ 62
Выводы......................................................... 63
7. Циклоалканы...................................................64
7.1. Изомерия и номенклатура...................................64
7.2. Получение.................................................65
7.3. Пространственное и электронное строение...................67
7.4. Химические свойства.......................................70
Выводы...........................................................73
8. Арены.........................................................75
8.1. Изомерия и номенклатура...................................75
8.2. Способы получения.........................................76
8.3. Строение бензола. Ароматичность. Правило Хюккеля..........77
8.4. Реакционная способность и химические свойства бензола и его
гомологов.....................................................80
144
{ 8.4.1 Рмации М»й1рофипы1о1о мм«|ц»нии (Ян)........................80
: 8.4.2 Ртнии 11|1н*н1«лин»ния......................................84
j- 8.4.1 Ртиии |0М(»жнпв Офикмй (Nr)................................85
выводы . 86
^Мнтоядермм» йрими1мч»имие оовдипвния...............................87
i 9.1. НпмйИклвгуря.................................................87
f 9.2. Днфвпил (Оифвннл)............................................87
; 9,2.1 Получение...................................................87
4 9.2.2. (Чроение. атропизомврия....................................88
9.2.3, Рммционни способность и химические свойства..............89
9.3. Полиарилмгганы...............................................90
9.3.1. Получение (на примере трифениметана).....................90
9.3.2. Реакционная способность и химические свойства
трифенилметана................................................90
9.3.3. Трифенилметановые красители..............................94
Выводы..............................................................95
9.4. Конденсированные ароматические углеводороды..................95
9.4.1. Нафталин.................................................95
9.4.1.1. Способы получения....................................95
9.4.1.2. Строение и реакционная способность...................96
9.4.1.3. Химические свойства..................................97
9.4.2. Антрацен.................................................99
9.4.2.1. Способы получения....................................99
9.4.2.2. Строение и реакционная способность..................101
9.4.2.3. Химические свойства.................................101
9.4.3. Фенантрен...............................................103
9.4.3.1. Получение...........................................103
9.4.3.2. Строение, реакционная способность и химические свойства... 104
Выводы..........................,..................................105
10. Галогенпроизводные углеводородов...............................106
10.1. Галогенпроизводные со связью C(sp3)-Hal....................106
10.1.1. Изомерия и номенклатура................................106
10.1.2. Способы получения......................................107
10.1.3. Строение, реакционная способность, химические свойства.108
10.2. Галогенпроизводные со связью C(sp2)-Hal....................114
10.2.1. Получение галогенаренов................................114
10.2.2. Строение, реакционная способность, химические свойства.114
Выводы.............................................................118
11. Гидроксилсодержащие производные углеводородов. Спирты. Алканолы 119
11.1. Гидроксилпроизводные углеводородов со связью C(sp3)-OH.....119
11.1.1. Алканолы...............................................119
11.1.1.1. Номенклатура.......................................119
11.1.1.2. Способы получения..................................120
11.1.1.3. Физические свойства................................121
11.1.1.4. Строение, реакционная способность, химические свойства... 122
145
11.2. Многоатомные спирты........................................127
11.2.1. Способы получения......................................127
11.3. Гидроксилпроизводные углеводородов со связью C(sp2)-OH.....131
11.3.1. Енолы (непредельные спирты)............................132
11.3.1.1. Получение и строение...............................132
11.3.1.2. Химические свойства................................132
11.3.1.3. Таутомерия енолов и карбонильных соединений........133
11.3.2. Фенолы.................................................133
11.3.2.1. Номенклатура.......................................133
11.3.2.2. Получение..........................................134
11.3.2.3. Строение, реакционная способность, химические свойства... 135
Выводы.............................................................141
Библиографический список...........................................142
146
Учебное издание
Щепин Василий Викторович
ОРГАНИЧЕСКАЯ ХИМИЯ
ЧАСТЬ 1
Редактор Н.П. Стрекаловская
Корректор Л. И. Перова
Компьютерный набор и верстка Ловриков С.Б.
Подписано в печать 20.04.2006,
Формат 60х80*/1б. Бум. ВХИ. Печать офсетная. Усл.печ.л. 8,6.
Уч.-изд.л. 5,25. Тираж 250 экз. Заказ 21Ф.
Редакционно-издательский отдел Пермского университета
614990, Пермь, ул. Букирева, 15
Типография Пермского университета
614990, Пермь, ул. Букирева, 15