/


























































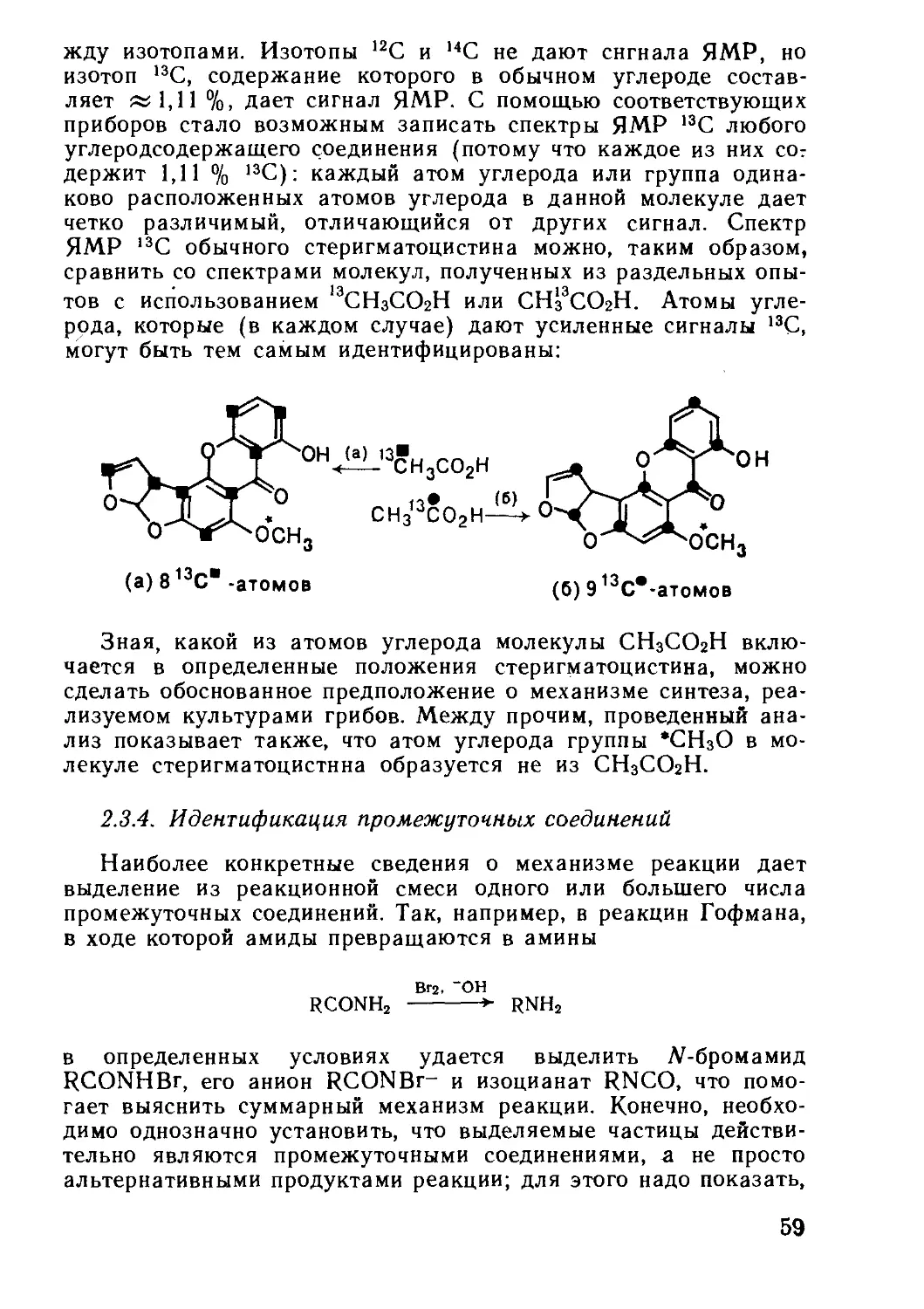

































































































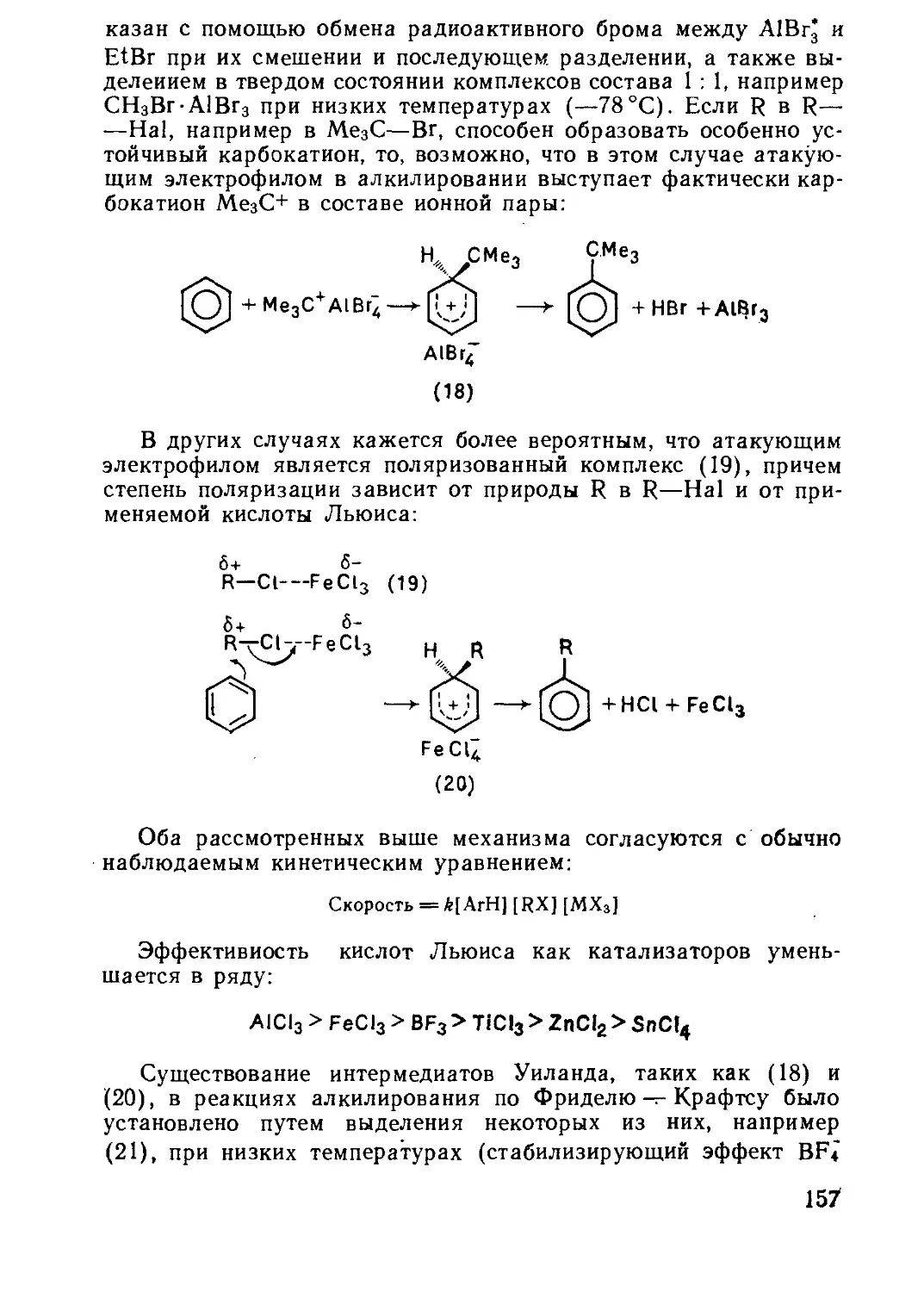



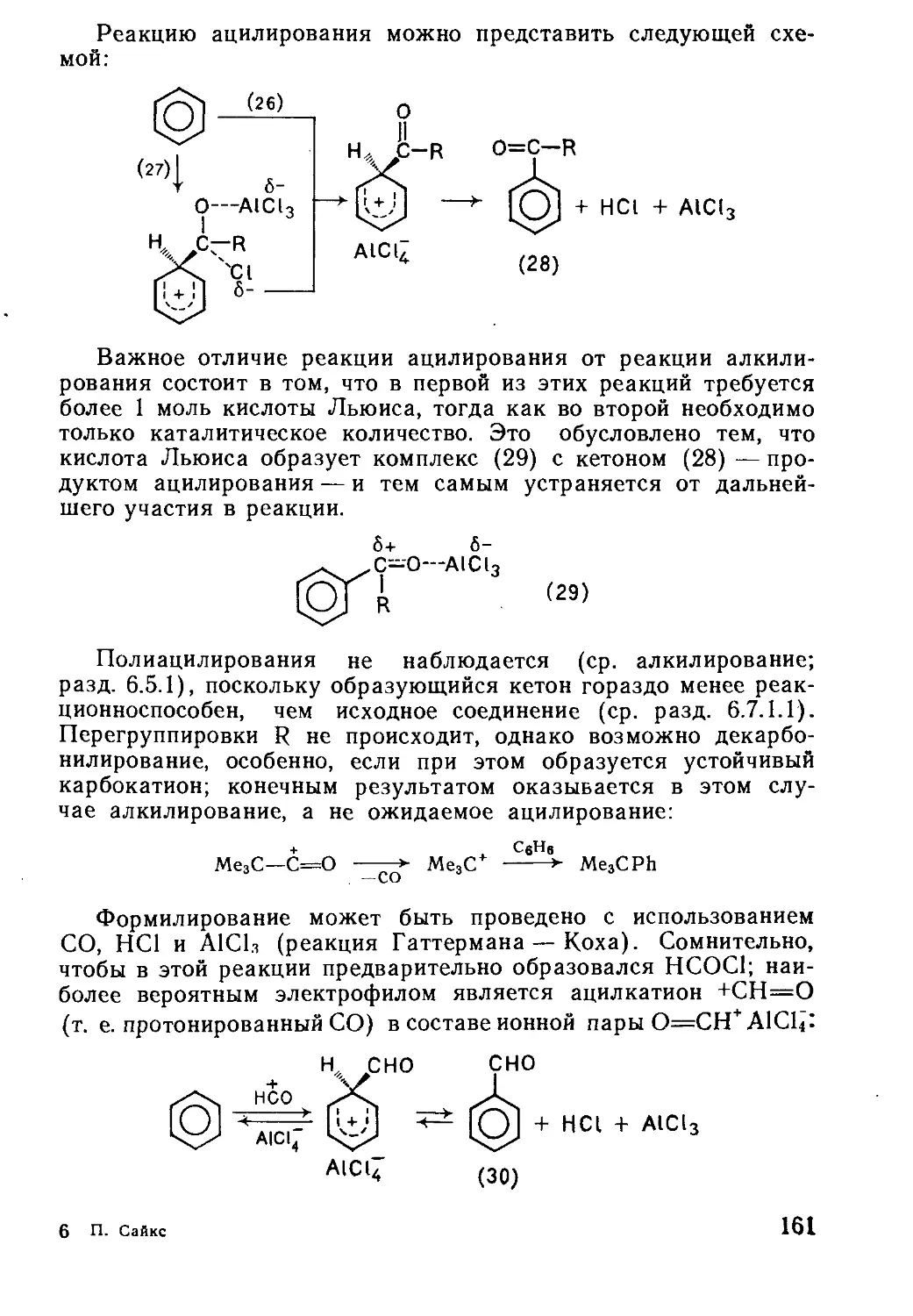





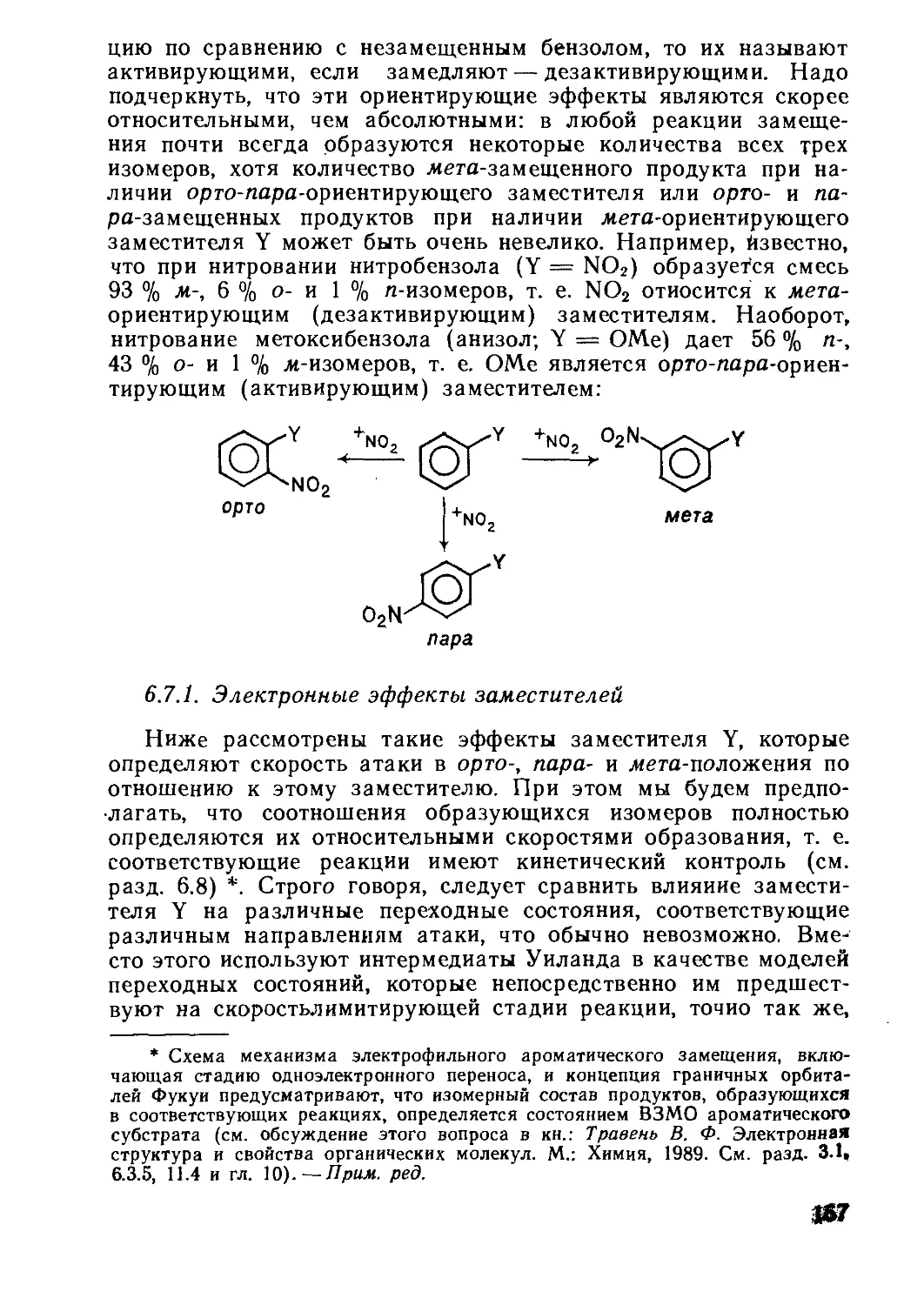











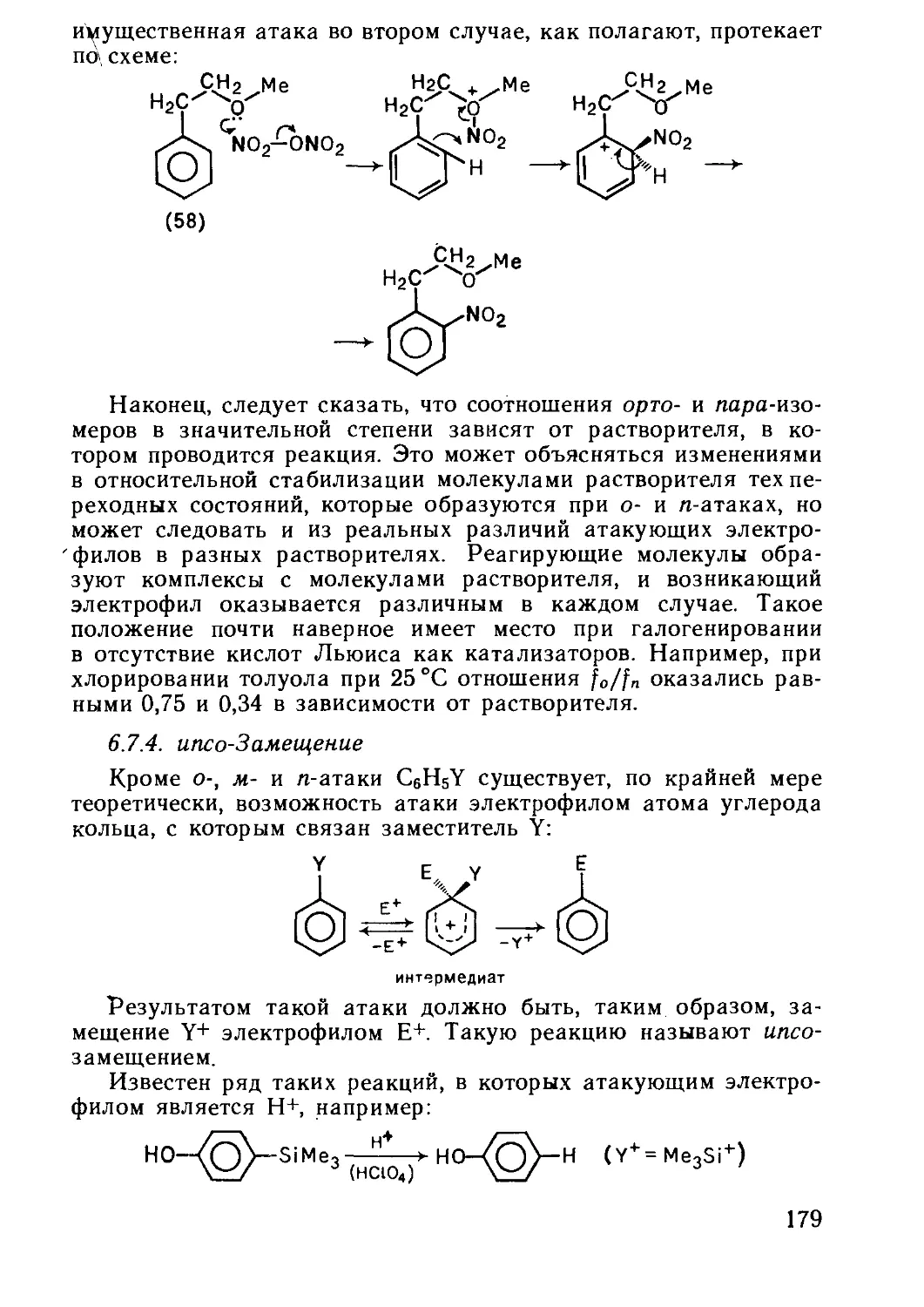







































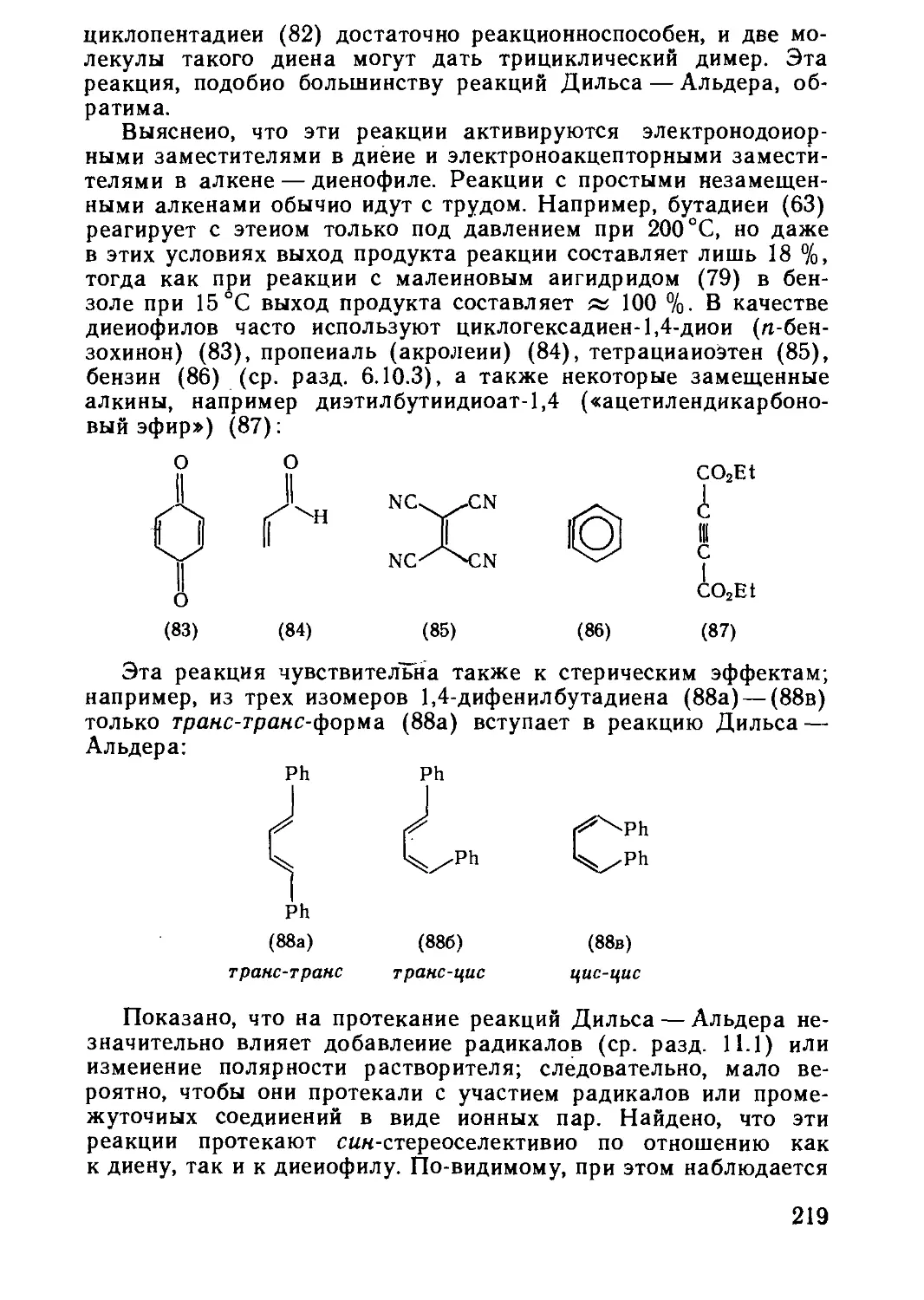

































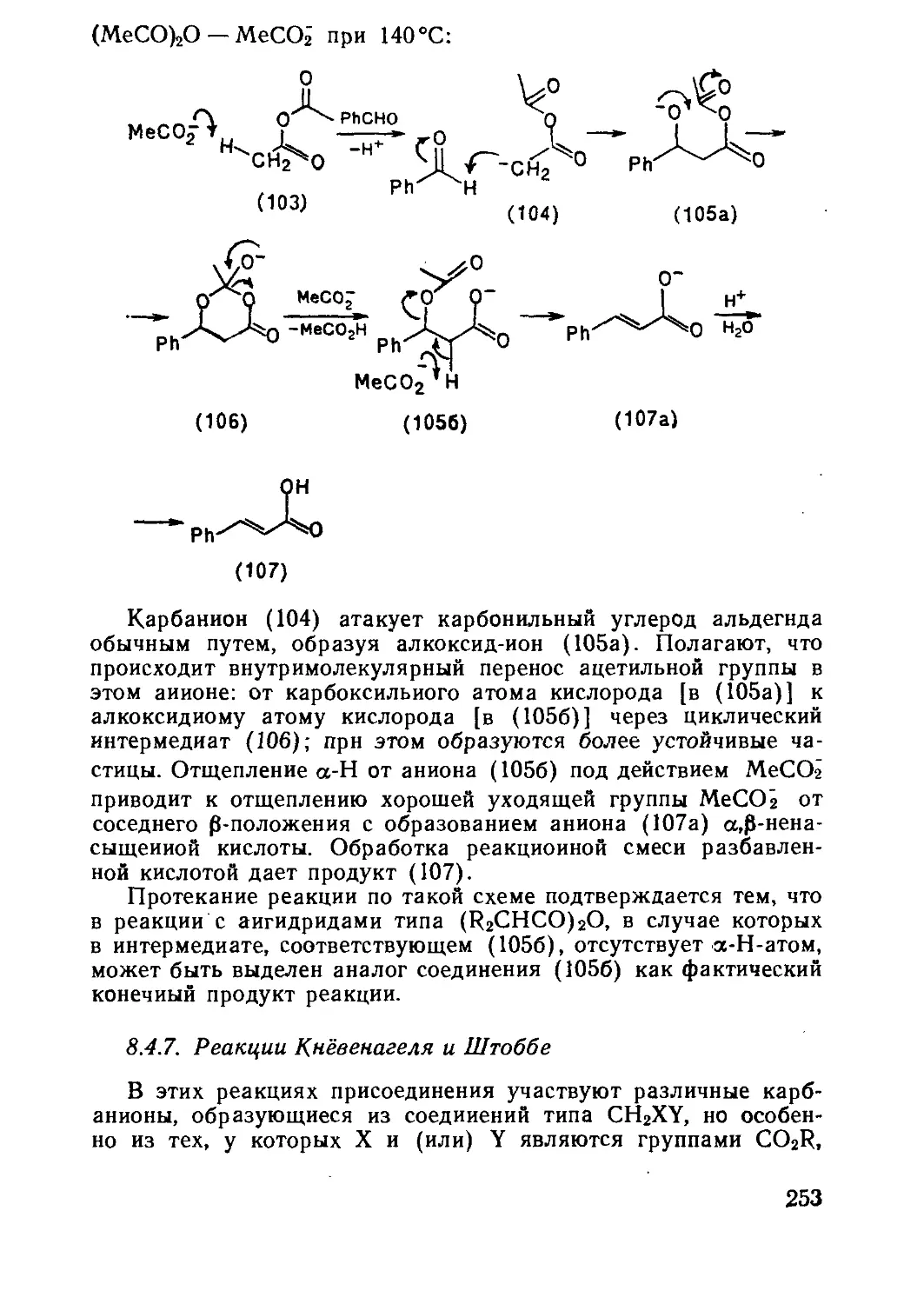








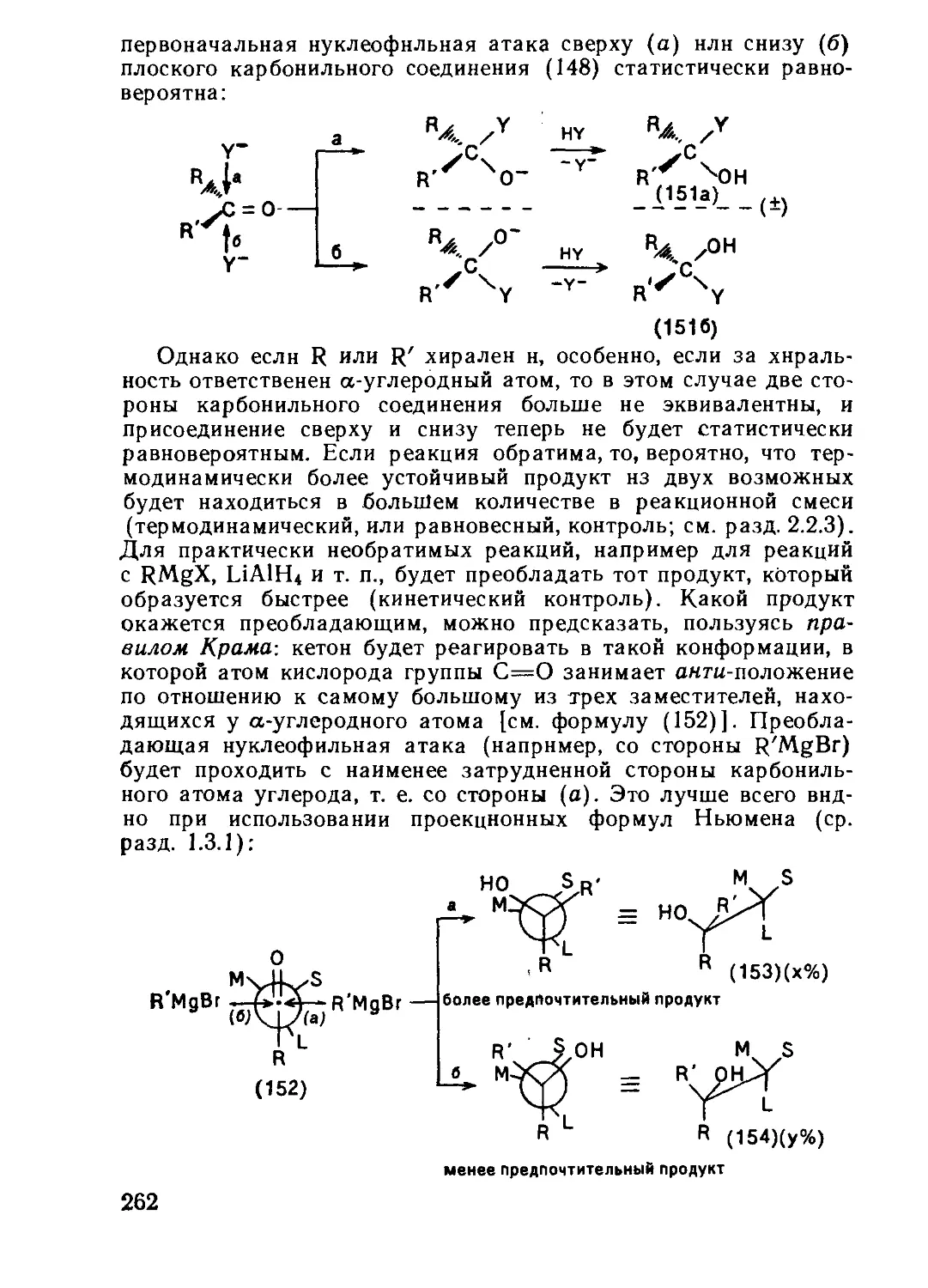





























































































































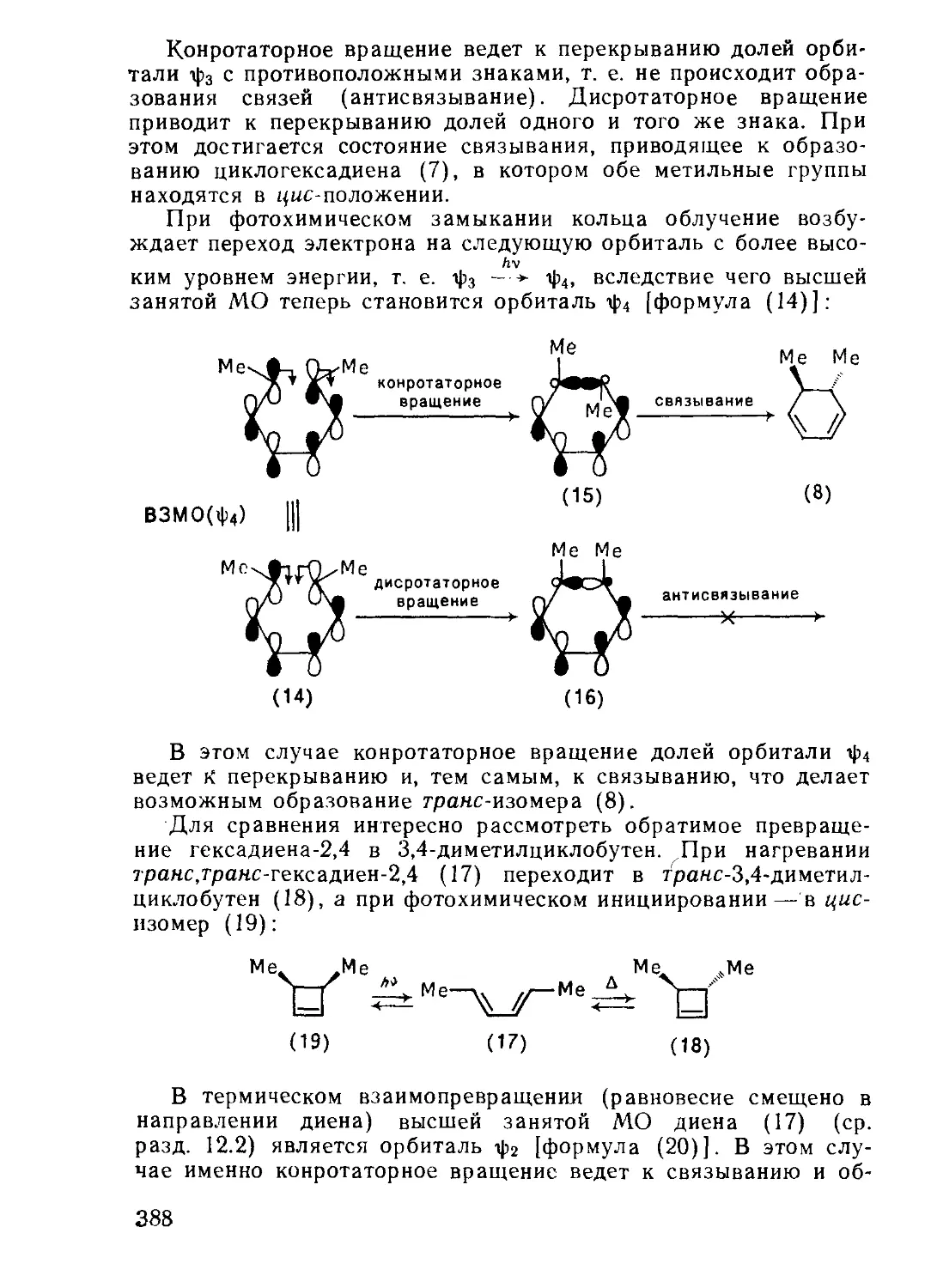









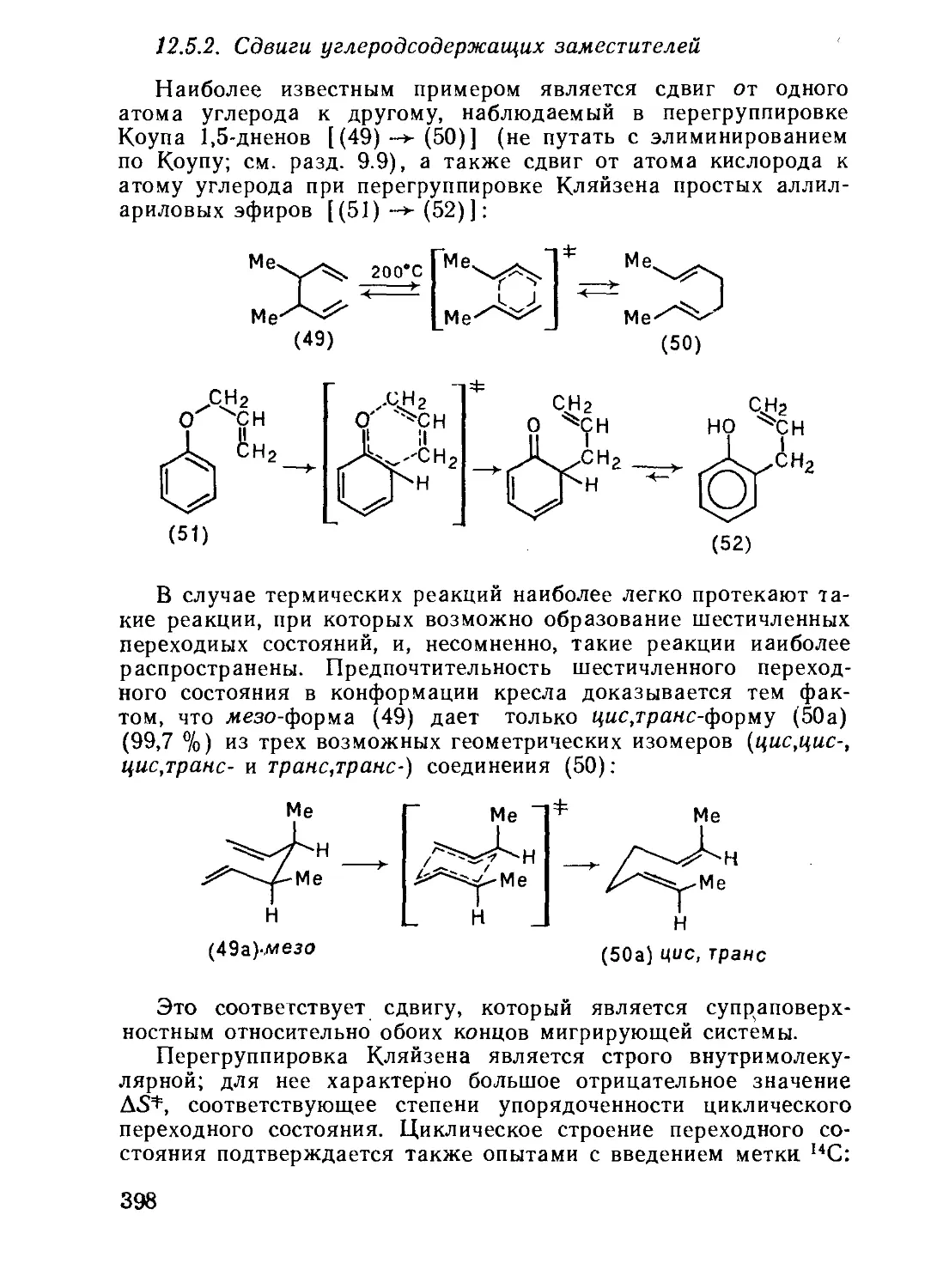





































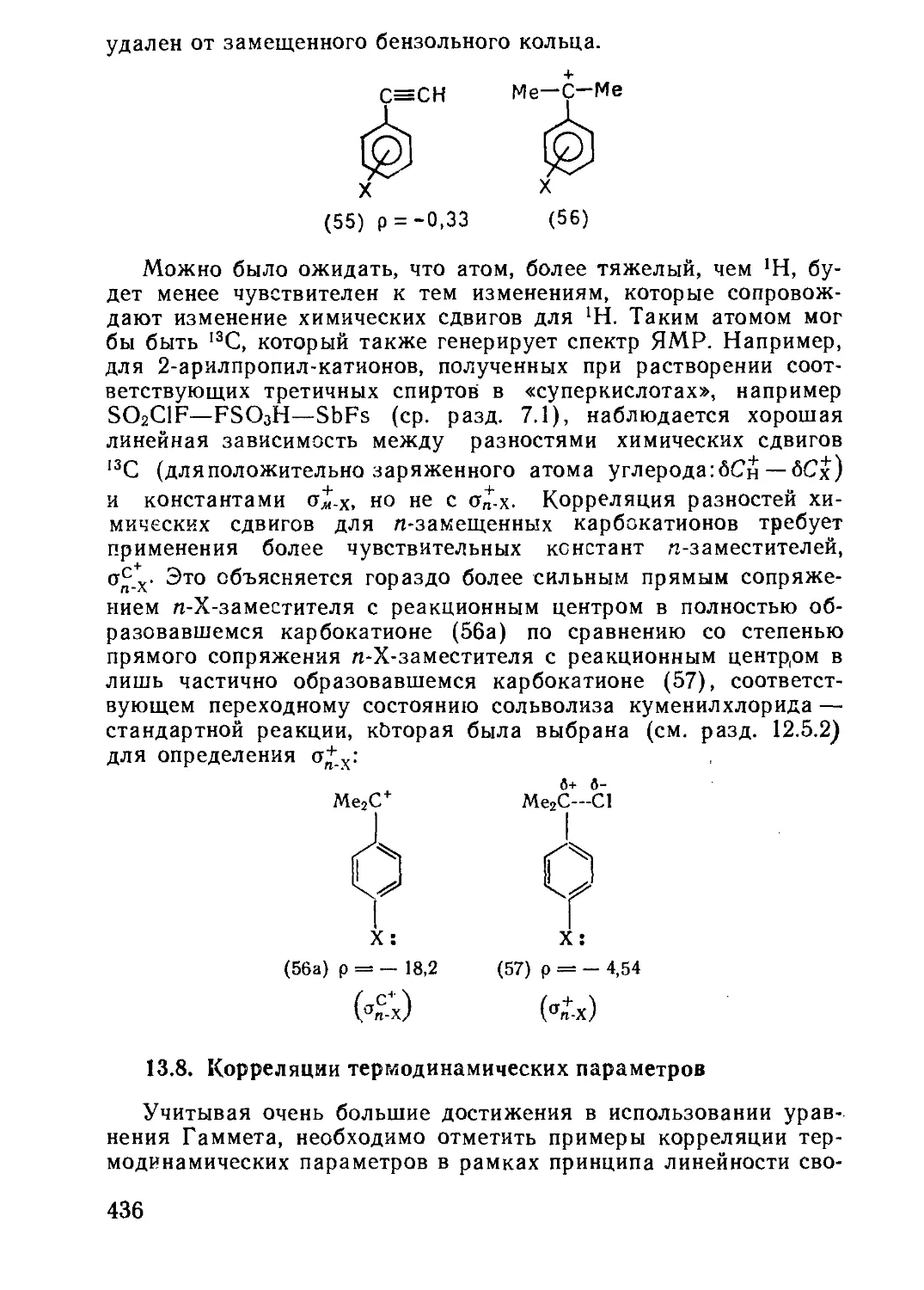

















Текст
A guidebook to mechanism in organic chemistry
Peter Sykes M.Sc., Ph.D., F.R.S.C., C.Chem.
Fellow and Vice-Master, Christ's College, Cambridge
Longman Scientific& S? Technical
Copublished in the United States with John Wiley & Sons, Inc., New York
П.Сайкс
РЕАКЦИЙ В ОРГАНИЧЕСКОЙ ХИМИИ
ИЗДАНИЕ 4-е
Перевод с английского канд. хим. наук Н. Г. ЛУЦЕНКО
Под редакцией докт. хим. наук В. Ф. ТРАВЕНЯ
МОСКВА «ХИМИЯ» 1991
Эта книга первоначально опубликована иа английском языке издательством Лонгман Троуп ЮК Лтд., Лондон
УДК 547:541.124
Механизмы реакций в органической химии/П. Сайкс.— 4-е изд. Пер. с англ./Под ред. В. Ф. Травеня. — М.: Химия, 1991. — Пер. изд.: Великобритания, 1986. — 448 с. ISBN 5—7245—0191—0
Книга представляет собой перевод 6-го английского издания. Приведены современные представления о строении органических соединений, о связи строения этих соединений с их реакционной способностью. Рассмотрены основные типы механизмов химических реакций. По сравнению с предыдущими изданиями (3-е изд. на русском языке— 1977 г.— являлось переводом 3-го английского издания) материал существенно обновлен и переработан, добавлены новые главы.
Для широкого круга химиков-органиков, преподавателей вузов, аспирантов и студентов.
Ил. 17. Библиогр.: 57 назв.
1705000000-052
050(01)-91 '
С
© Peter Sykes 1961, 1985
Third and subsequent editions © Longman Group Ltd 1970, 1975, 1981, 1986
ISBN 5—7245—0191—0 © Перевод на русский язык, Н. Г. Луценко © Предисловие, комментарии, В. Ф. Травень, 1991
ОГЛАВЛЕНИЕ*
Предисловие редактора перевода ................................... 6
Предисловие А. Р. Тодда............................................7
Предисловие автора к шестому английскому изданию...................8
Глава 1
Структура, реакционная способность и механизмы реакций.............9
Глава 2
Энергетика, кинетика и исследование механизмов реакций 42
Глава 3
Сила кислот и оснований......................................... 63
Глава 4
Нуклеофильное замещение у насыщенного атома углерода 89
Глава 5
Карбокатионы, электроиодефицитные атомы азота и кислорода и их реакции ................116
Глава 6
Электрофильное и нуклеофильное замещение в ароматических системах 145
Глава 7
Электрофильное и нуклеофильное присоединение по связи С=С . . . 197
Глава 8
Нуклеофильное присоединение по связи С=0.........................225
Глава 9
Реакции элиминирования ........................................ 273
Глава 10
Карбанионы и их реакции ........................................302
Глава 11
Радикалы и их реакции............................................335
Глава 12
Реакции, контролируемые симметрией ..............................382
Глава 13
Соотношения линейности свободных энергий.........................400
Рекомендательная литература .................................... 439
Предметный указатель.............................................441
* Подробное содержание приведено в начале каждой главы.
5
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Монография Питера Сайкса не нуждается в многословном представлении. Она уже выдержала шесть изданий и переведена на многие языки. Книга имеет широкую читательскую аудиторию, поскольку сложные понятия теоретической органической химии и схемы механизмов реакций большинства классов органических соединений автор излагает лишь на основе выдержавших испытание временем концепций. Оставаясь верным, как и в предыдущих изданиях, электронным представлениям, Питер Сайкс при интерпретации богатейшего экспериментального материала ограничивается понятиями, введенными Льюисом, Полингом, Робинсоном, Ингольдом, Хюккелем.
По сравнению с третьим изданием, перевод которого на русский язык был опубликован в 1977 г., настоящее издание содержит большой объем новых сведений. Практически в каждой главе появились новые разделы или переработаны прежние. Заново написаны две заключительные главы: глава 12 «Реакции, контролируемые симметрией» и глава 13 «Соотношения линейности свободных энергий». Эти главы имеют значительный объем и существенно обогащают химика в его стремлении осознать тонкие особенности механизмов органических реакций. В частности, глава 12 знакомит с применением орбитальных концепций Фукуи, Вудварда н Гофмана для объяснения протекания и результатов перициклических и родственных им реакций ненасыщенных соединений. Можно надеяться, что в последующих изданиях Питер Сайкс не ограничится экспериментальными данными главы 12 для популяризации орбитальных представлений.
Завершает монографию глава 13. И хотя к настоящему времени изложению результатов применения ор-метода посвящено огромное число монографий и обзоров и химики уже реже обращаются к его помощи, восхищает талант Питера Сайкса выбрать главное в интерпретации соотношений ЛСЭ и показать информативность даже простейших соотношений типа уравнения Гаммета для изучения механизмов органических реакций.
Новое издание получившей широкое признание монографии Питера Сайкса, несомненно, окажется полезным студентам старших курсов и аспирантам в качестве учебного пособия, представит интерес для всех химиков-органиков.
В. Травень
ПРЕДИСЛОВИЕ А. Р. ТОДДА
Пятьдесят лет тому назад студент, начавший увлекаться органической химией — а я говорю об этом из собственного опыта — был бы, наверное, отослан к тому или иному учебнику, обычно известному по именам его авторов, например, таких как Холлеман, Бернтсен, Шмидт, Каррер и Гаттерман. На этих учебниках воспитано не одно поколение химиков, и не в одной стране, поскольку они были переведены на несколько языков. Но эти хорошо знакомые учебники уже большей частью устарели. В те времена общее число доступных книг было достаточно мало по сравнению с настоящим потоком учебников по органической химии, появившихся за последние десятилетия. Такой рост числа учебников может быть объяснен отчасти увеличением числа студентов, но основной причиной является революционизирующее влияние изучения механизмов реакций (на элементарном уровне) на наш подход к органической химии. При таком изобилии учебников автору довольно трудно стать известным, независимо от предмета изучения. Однако это произошло с доктором Питером Сайксом и его книгой «Механизмы реакций в органической химии».
В Предисловии, которое я имел честь написать к первому изданию этой книги в 1961 г., описана моя точка зрения на происходящее в органической химии, а также на метод обучения, который предпочитает доктор Сайкс. Зная и наблюдая его в течение многих лет, сначала как студента, затем как коллегу и всегда как друга, я и тогда был уверен в том, что он написал прекрасную книгу, которая, по крайней мере на мой взгляд, должна способствовать повышению интереса к органической химии. Но ее успех намного превзошел все даже самые большие мон ожидания, и при последующих изданиях книга не потеряла своей прелести.
Настоящее издание продолжает эту традицию. В очередной раз автор «прочесал» появившуюся в последнее время литературу с целью поиска лучших примеров, иллюстрирующих принципы химических реакций. Особый интерес вызывает глава, в которой рассматриваются реакции, контролируемые параметрами орбитальной симметрии. До тех пор, пока я не прочел эту главу, я не был убежден в том, что эта очень важная новая разработка теории органических реакций может быть так просто и в то; же время с пользой рассказана студентам начального этапа обучения. То, что автор сумел это сделать, только еще раз подчеркивает дар учителя и писателя, присущий доктору Сайксу, и я уверен в том, что это новое издание будет иметь еще больший успех, чем предыдущие.
Кембридж Тодд
ПРЕДИСЛОВИЕ АВТОРА
К ШЕСТОМУ АНГЛИЙСКОМУ ИЗДАНИЮ
Прошло уже двадцать пять лет с тех пор, как вышло первое издание настоящей книги, и совсем неудивительно, что этот ее вариант очень сильно отличается от первого издания. В течение этих лет были приложены большие усилия по включению нового материала и удалению старого, целью которых было не стремление отразить текущие тенденции и популярные направления, а проиллюстрировать значительные изменения в наших фундаментальных представлениях в органической химии, и особенно необходимо было решить, как наилучшим образом донести эти изменения до аудитории, состоящей в основном из студентов младших курсов. В то же самое время особое внимание придавалось сохранению основной схемы построения и структуры книги, поскольку такой путь оказался очень полезным.
В настоящем издании не появилось новых глав, но введены некоторые новые вопросы, например unco-замещение в ароматических системах, выявление типов механизмов в нуклеофильном замещении, более широко использованы активационные параметры, особенно в гидролизе сложных эфиров, параметр Димрота Ет, корреляция данных спектроскопии с параметром Гам мета <ух, ЯМР 13С в биогенезе и др. Непопулярный теперь термин «карбониевый ион» заменен на «карбокатион», преимущество использования которого состоит в том, что он является естественной антитезой «карбаниону». Ограничено также применение достаточно двусмысленного альтернативного термина «кар-бениевый ион». Наряду с этими изменениями весь текст, строка за строкой, был перечитан с целью замены неясных мест более ясными и обоснованными объяснениями и более яркими примерами.
У меня всегда было чувство, что авторам многих учебников не удается раскрыть полностью свои возможности, поскольку автор никогда не решает до конца для себя, адресован ли данный учебник только студентам или, по крайней мере частично, преподавателям, в то время как требования в этих двух случаях в конечном счете разные. Новое издание, как и предшествующие ему, адресовано, вне всяких сомнений, студентам, так что я верю, что эта книга будет полезна всем студентам-химикам независимо от специальности, которую они изучают.
Я глубоко признателен многочисленным авторам писем, которые указали мне на ошибки, неточности и внесли полезные предложения; всегда, когда это было возможно, эти замечания были учтены в новом издании. Я буду очень благодарен за подобное содействие в будущем *.
Кембридж Сентябрь 1985 г. Питер Сайкс
* Заключительная часть Предисловия перенесена в качестве примечания в начало главы 13. — Прим. ред.
8
Глава 1
СТРУКТУРА, РЕАКЦИОННАЯ СПОСОБНОСТЬ И МЕХАНИЗМЫ РЕАКЦИЙ
1.1 Атомные орбитали.................................................9
1.2. Гибридизация...................................................12
1.3. Образование связей в соединениях углерода......................13
1.3.1. Простые углерод-углеродные связи........................15
1.3.2. Двойные углерод-углеродные связи........................17
1.3.3. Тройные углерод-углеродные связи........................18
1.3.4. Углерод-кислородные и углерод-азотные связи..............19
1.3.5. Сопряжение...............................................20
1.3.6. Бензол и ароматичность...................................23
1.3.7. Условия делокализации электронов.........................28
1.4. Разрыв и образование связей....................................30
1.5. Факторы, влияющие на доступность электронов....................31
1.5.1. Индуктивный эффект и эффект поля..................... . 31
1.5.2. Мезомерный эффект (эффект сопряжения)....................33
1.5.3. Динамические эффекты.................................... 34
1.5.4. Сверхсопряжение (гиперконъюгация)........................35
1.6. Стерические эффекты............................................37
1.7. Классификация реагентов........................................39
1.8. Классификация реакций..........................................41
Главное преимущество подхода к изучению органической химии с точки зрения механизмов реакций заключается в том, что существующие внешне совершенно различные факты могут быть систематизированы и объяснены на основе использования сравнительно небольшого числа основополагающих принципов. Более того, такой подход позволяет предвидеть, каким образом изменится протекание известных реакций в новых условиях и какие продукты будут получены в новых реакциях. В этой главе кратко изложены некоторые из этих основополагающих принципов и показано, как они действуют. Так как по существу речь дальше пойдет о соединениях углерода, необходимо обсудить способы образования связей между атомами углерода и другими атомами и, в частности, способы образования углеродуглеродной связи.
1.1. Атомные орбитали
Атом углерода имеет шесть электронов, которые, согласно теории Бора о строении атома, располагаются вокруг ядра на орбитах возрастающего диаметра. Эти орбиты соответствуют уровням с постепенно возрастающей энергией: уровень
9
с наименьшей энергией 1s содержит два электрона, следующий уровень 2s также содержит два электрона; остальные два электрона атома углерода находятся на уровне 2р, который может содержать всего шесть электронов.
Из рассмотрения поведения электрона с точки зрения законов волновой механики и, в частности, с учетом принципа неопределенности Гейзенберга следует, что орбиты как таковые не существуют. Вместо этого волноподобные электроны характеризуются в настоящее время волновыми функциями Y, а классические орбиты Бора заменены трехмерными атомными орбиталями, различающимися уровнями энергии. Размер, форма и ориентация этих атомных орбиталей — областей пространства, где с наибольшей вероятностью можно обнаружить электрон, соответствующий данному квантовому энергетическому уровню,— описываются одной из волновых функций: Ч'дЧ'в, Ч’с и т. д. Таким образом, орбитали представляют собой трехмерные электронные «контурные карты», в которых функция 'Г2 определяет относительную вероятность нахождения электрона в данной конкретной точке орбитали.
Относительные размеры атомных орбиталей увеличиваются по мере возрастания их энергий и определяются главным квантовым числом и, а их форма и ориентация в пространстве (по отношению к ядру и друг к другу)—квантовыми числами I и пг соответственно *. Электроны на орбиталях характеризуются также спиновым квантовым числом, которое может принимать значения от 1 /2 до —1/2. Ограничение, которое вводит теория в отношении таких орбиталей, состоит в том, что на каждой орбитали может находиться не более двух электронов, отличающихся друг от друга противоположными спинами (спаренные спины) **. Это ограничение следует из принципа Паули, согласно которому ни у какого атома не может быть двух электронов, характеризующихся одним и тем же набором квантовых чисел.
Квантовомеханическими расчетами можно показать, что 1s-орбиталь (определяемая квантовыми числами n=l, I = О, m = 0 и соответствующая К-оболочке) имеет сферическую симметрию относительно ядра атома; 2х-орбиталь (п — 2, 1 = 0, tn = 0) также имеет сферическую симметрию, но находится на большем расстоянии от ядра. Между этими орбиталями существует область, где вероятность нахождения электрона близка
* п может иметь значения 1, 2, 3, ...: I — значения 0, 1, 2, .... п. — 1; m — значения 0, ±1, ±2, ..., ±1. Мы будем обычно рассматривать только значения I, равные 0 и 1; соответствующие орбитали называют (по спектроскопической терминологии) s- и р-орбиталями соответственно; например, 1s-, 2s-, 2р-орбитали и т. д.
** Одни электрон имеет спиновое квантовое число +1/2, другой —1/2.
10
к нулю (сферическая узловая область) *:
Зр
3s------
М-оболочка
2р------
2s -----
L-оболочка
1s ------- К-оболочка
Сказанное выше о s-орбиталях не противоречит классическому представлению об орбитах, по которым движутся электроны, однако при рассмотрении 2р-уровня (продолжение L-оболочки) различие становится очевидным. Согласно теории, должны существовать три 2р-орбитали (и. = 2, I = 1, m = -f-l, О и —1), имеющие одинаковую форму и одинаковый уровень энергии (орбитали с одинаковыми уровнями энергии называют вырожденными), но различающиеся ориентацией в пространстве. Эти орбитали располагаются вдоль взаимно перпендикулярных осей х, у и z и обозначаются 2рх, 2ру и 2рг соответственно. Эти три орбитали не обладают сферической симметрией, подобно 1s- и 25-орбиталям, а напоминают гантели. Плоскости, в которых вероятность нахождения электрона равна нулю (узловые плоскости), проходят через ядро под прямыми углами соответственно к осям х, у и z и разделяют каждую из «гантелей» на две половинкй\
Узловая плоскость
плоскость
2рх 2ру
Шесть электронов атома углерода располагаются на атомных орбиталях, имеющих возрастающие уровни энергии, до тех пор, пока они не будут заполнены. Так, два электрона со
* Квантовомеханическое содержание понятия «атомная орбиталь» не может быть в полной мере передано графически; на эскизе показаны общепринятые условные изображения атомных орбиталей. — Прим. ред.
11
спаренными спинами займут ls-орбиталь, еще два электрона — 25-орбиталь, а оставшиеся два электрона займут 2р-уровень, причем будут располагаться или на одной и той же орбитали, например 2рх, или на различных, например 2рх и 2ру. Согласно правилу Гунда, два электрона избегают занимать одну и ту же орбиталь до тех пор, пока другие энергетические эквивалентные (т. е. вырожденные) орбитали свободны. Таким образом, электронную конфигурацию атома углерода можно представить как ls22s22p^2p^, причем орбиталь 2рг остается незанятой. Эта конфигурация характеризует основное состояние свободного атома углерода, в котором только два неспаренные электрона (на 2рх- и 2р!/-орбиталях) могут участвовать в образовании связей с другими атомами, т. е. на первый взгляд может показаться, что углерод может быть только двухвалентным.
Однако такой вывод противоречит экспериментальным данным, согласно которым соединения, содержащие атом углерода, связанный только с двумя другими атомами, например СС12 (см. разд. 9.8), очень неустойчивы; в подавляющем большинстве соединений углерод является четырехвалентным, как, например, в метане СН4. Это может достигаться расспариванием электронной пары 2s2 и переходом одного из этих электронов на вакантную 2рг-орбиталь. В результате атом углерода переходит в состояние с повышенной энергией (возбужденное состояние) ls22sl2p{x2p{y2p{z, имея в результате этого четыре неспаренных электрона вместо двух, способен образовать связи уже не с двумя, а с четырьмя другими атомами или группами. Большое количество энергии, выделяющееся при образовании этих двух дополнительных связей, значительно превышает энергию [«406 кДж/моль (97 ккал/моль)], необходимую для рас-спаривания двух 2в2-электронов и перехода 2s—>~2p.
1.2. Гибридизация
Очевидно, что при связывании атома углерода с четырьмя другими атомами не используются одна 25-орбиталь и три 2р-орбитали, так как это привело бы к образованию трех взаимно перпендикулярных связей (с тремя 2р-орбиталями) и одной связи, отличающейся от них и не имеющей направления (со сферически, симметричной 25-орбиталью). В действительности, четыре С—Н-связи, например в метане, как известно, равноценны и расположены симметрично (тетраэдрически) под углом 109°28' друг к другу. Это можно объяснить тем, что одна 2s- и 2р-орбитали объединяются так, чтобы образовать четыре новые (идентичные) орбитали, способные дать более прочные связи (ср. разд. 1.5). Эти новые орбитали известны под названием вр3-гибридных атомных орбиталей, а процесс их образования
12
называется гибридизацией:
Следует, однако, подчеркнуть, что в приведенной схеме речь идет о гибридизации не самих орбиталей, а математических функций, которые их описывают.
Сходную, хотя и несколько отличную перестройку можно представить в тех случаях, когда атом углерода связывается с тремя другими атомами, например в этене (этилен) (см. разд. 1.3.2); при этом три $р2-гибридные атомные орбитали располагаются в одной плоскости под углами 120° друг к другу (плоскостная, или тригональная, гибридизация). Наконец, когда атом углерода связывается с двумя другими атомами, например в этине (ацетилен) (см. разд. 1.3.3), две ъ’р'-гибридные атомные орбитали расположены под углом 180° (дигональная, или линейная, гибридизация). В каждом случае в гибридизации участвует s-орбиталь, поскольку она имеет самый низкий уровень энергии.
Указанные перестройки одной 2$-орбитали и трех 2р-орби-талей выгодны, так как в случае $р2-гибридизации становится доступной одна негибридизованная р-орбиталь (см. разд. 1.3.2), а в случае яр'-гибридизации — две р-орбитали (см. разд. 1.3.3). Выгодны и такие способы гибридизации, когда гибридные орбитали необязательно идентичны, например в СНгС12 (в отличие от орбиталей в СС14 и СН4). Реализуется тот способ гибридизации, при котором атом углерода может образовать максимально прочные связи, а другие связываемые с ним атомы (и электронные пары, образующие эти связи) возможно дальше отстоят друг от друга, так что общая внутренняя энергия полученного соединения оказывается минимальной.
1.3. Образование связей в соединениях углерода
Образование связи между двумя атомами можно рассматривать как результат перекрывания атомных орбиталей двух участвующих в ней атомов: чем сильнее такое перекрывание (интеграл перекрывания), тем прочнее образующаяся связь.
13
Согласно расчетным данным, относительные эффективности перекрывания атомных орбиталей SOTH возрастают следующим образом:
Орбиталь Орбиталь
s 1,00 sp2 1,99
р 1,72 sp3 2,00
sp1 1,93
Из этих данных ясно, почему использование гибридных орбиталей, например $р3-орбиталей в связывании одного атома углерода с четырьмя атомами водорода при образовании молекулы метана, приводит к возникновению более прочных связей.
Можно показать, что когда атомы оказываются на достаточно близком расстоянии друг от друга, обе атомные орбитали переходят в две молекулярные орбитали, одна из которых находится на более низком энергетическом уровне, а другая — на более высоком, чем- первоначальные орбитали:
антисвязывающая (<г*) молекулярная орбиталь
атомная орбиталь А _
атомная орбиталь В
связывающая (ст) молекулярная орбиталь
Эти две новые молекулярные орбитали распространяются на оба атома; на каждой из них могут находиться по два электрона. Молекулярную орбиталь с более низкой энергией называют связывающей орбиталью: заполнение ее электронами приводит к образованию стабильной связи между двумя атомами. В этом случае пара электронов, образующаяся связь, стремится сосредоточиться между двумя положительно заряженными атомными ядрами, что, можно полагать, и удерживает эти ядра друг около друга. Молекулярную орбиталь с более высокой энергией называют антисвязывающей орбиталью-, она соответствует такому состоянию, при котором межъядерное пространство в значительной мере свободно от электронов, что приводит к отталкиванию двух положительно заряженных атомных ядер. В основом состоянии молекулы антисвязывающая орбиталь не заполняется электронами и в дальнейшем при обсуждении образования стабильных связей между атомами рассматриваться не будет.
14
Если две атомные орбитали перекрываются вдоль их главных осей, то возникающую при этом связывающую орбиталь называют ст-орбиталью *, а образующуюся связь — ст-связью. Молекулярная ст-орбиталь и находящиеся на ней электроны локализованы симметрично относительно линии, соединяющей ядра атомов, участвующих в образовании связи. Так, при образовании связей с атомами водорода в метане четыре гибридные $р3-орбитали атома углерода перекрываются с ls-орбиталями четырех атомов водорода, образуя четыре идентичные прочные ст-связи, располагающиеся под углом 109°28z друг к другу (стандартный тетраэдрический угол). Сходная строго симметричная тетраэдрическая структура возникает также, например, при образовании ССЬ; если же атомы, образующие связи с углеродом, неодинаковы, например в случае СН2С12, пространственная структура будет несколько отличаться от полностью симметричной, хотя по существу она остается тетраэдрической (ср. разд. 1.2).
1.3.1. Простые углерод-углеродные связи
Связывание двух атомов углерода, например, в этане происходит в результате перекрывания двух атомных $р3-орбита-лей (по одной от каждого из атомов), которое приводит к образованию прочной ст-связи. Длина углерод-углеродной связи в насыщенных соединениях равна 0,154 нм. Это относится, однако, к простой углерод-углеродной связи между $р3-гибриди-зованными атомами. Было показано, что длина аналогичной простой связи между двумя $р2-гибридизованными атомами углерода =СН—СН= составляет «0,147 нм, а между двумя зр’-гибридизованными атомами углерода =С—Сг она равна «0,138 нм. Эти различия неудивительны, поскольку s-орбиталь и находящиеся на ней электроны расположены ближе к ядру, чем р-орбиталь и находящиеся на ней электроны. То же самое наблюдается и для других гибридных орбиталей — возрастание s-компонента приводит к тому, что при образовании связей между двумя атомами углерода межатомные расстояния сокращаются при переходах: sp3—sp3-»-sp2—sp2-»-sp1—sp'.
Однако для этана нельзя предложить какую-либо единственную структуру: ст-связь, соединяющая два углеродных атома, симметрична по отношению к линии, соединяющей эти атомы, и теоретически возможно неограниченное число структур с различным относительным расположением атомов водорода, связанных с атомами углерода. Два крайних случая возможных
* Соответствующую антисвязывающую молекулярную орбиталь обозначают как а*-орбиталь.
15
структур получилй названия форм:
заслоненной и заторможенной
Изображения указанных конформаций известны как проекции типа «козел* и проекции Ньюмена соответственно. Заслоненная и заторможенная формы и бесконечное множество структур, промежуточных между ними, называют конформациями молекулы этана; конформации определяют как различные расположения одной и той же группы атомов в пространстве, которые могут переходить друг в друга без разрыва связей.
Заторможенная конформация, вероятно, более стабильна, поскольку в ней атомы водорода максимально удалены друг от друга [0,310 им (3,1 А)] и любое так называемое несвязывающее взаимодействие между ними сведено к минимуму. В случае же заслоненной конформации они максимально сближены [до 0,230 нм (2,3 А)], что лишь немногим меньше суммы их ван-дер-ваальсовых радиусов. Давно известный принцип свободного вращения вокруг простой углерод-углеродной связи в случае этана, однако, не нарушается: как показывают расчеты, разница энергий заслоненной и заторможенной конформаций при 25 °C составляет «12 кДж/моль (3 ккал/моль); эта разница достаточно мала и не препятствует превращению одной формы в другую при комнатной температуре за счет энергии обычного теплового движения — вращательная частота при 25 °C составляет «1012 с-1. Тот факт, что очень тесное расположение атомов может привести к реальному ограничению вращения вокруг простой углерод-углеродной связи, был подтвержден выделением двух форм СНВг2СНВг2, однако только при низких температурах, когда энергия столкновений между молекулами еще недостаточна для того, чтобы обеспечить взаимные превращения конформеров.
16
1.3.2. Двойные углерод-углеродно связи
В этене каждый атом углерода связан с тремя другими атомами — с двумя атомами водорода и одним атомом углерода. Прочные a-связи с этими тремя атомами образуются с использованием трех орбиталей, возникающих в результате гибридизации 2$-орбитали и двух (из трех) 2р-орбиталей: число гибридных орбиталей, которое может образовать данный атом в основном состоянии, равно числу атомов или групп, с которыми он образует прочные ст-связи. Все три результирующие $р2-гибридные орбитали лежат в одной и той же плоскости и образуют друг с другом углы, равные 120° {плоскостные тригональные орбитали). При образовании молекулы этена две sp2-орбитали каждого атома углерода перекрываются с ls-орбита-лями двух атомов водорода, образуя прочные ст-связи С—Н, тогда как третьи $р2-орбитали каждого атома углерода перекрываются друг с другом аксиально с образованием прочной ст-связи С—С. Показано экспериментально, что углы Н—С—Н и Н—С—С равны 116,7 и 121,6° соответственно. Отклонение от 120° едва ли можно считать неожиданным, поскольку речь идет о разных триадах атомов, образующих связи.
Таким образом, у каждого из атомов углерода остается одна негибридизоваииая 2р-орбиталь, перпендикулярная плоскости, в которой находятся атомы углерода и водорода. Когда эти две орбитали становятся параллельными одна другой, они перекрываются с образованием связывающей молекулярной орбитали, охватывающей оба атома углерода. Молекулярная орбиталь располагается над и под плоскостью молекулы (в плоскости молекулы эта орбиталь имеет «узел»), в которой находятся оба атома углерода и четыре атома водорода (пунктирными клиньями показаны связи с атомами, расположенными за плоскостью чертежа, а зачерненными клиньями — связи с атомами, расположенными перед плоскостью чертежа:
нхА АЧн н
Эту новую связывающую молекулярную орбиталь называют л-орбиталью *, а находящиеся на ней электроны — л-электро-нами. Образованная таким образом новая связь, называемая л-связью, сближает атомы углерода: длина связи С=С в этене
* Аналогично образуется антисвязывающая (л*) молекулярная орбиталь (ср. разд. 1.3.5).
17
равна 0,133 нм, тогда как длина связи С—С в этане равна 0,154 нм. Боковое перекрывание атомных орбиталей, обусловливающее образование л-связи, менее эффективно, чем перекрывание вдоль главной оси, ведущее к образованию о-связи; именно поэтому л-связи менее прочны, чем ст-связи. Это проявляется, в частности, в том, что энергия двойной углерод-углеродной связи превышает энергию одинарной связи, однако менее чем в два раза. Так, энергия связи С—С в этане равна 347 кДж/ /моль (83 ккал/моль), тогда как энергия связи С—С в этене составляет только 598 кДж/моль (143 ккал/моль).
Степень бокового перекрывания двух атомных 2р-орбиталей, а следовательно, и прочность л-связи максимальна, если два атома углерода и четыре атома водорода расположены строго в одной плоскости, т. е. если они копланарны, поскольку только в этом случае атомные 2р-орбитали точно параллельны одна другой и поэтому способны к максимальному перекрыванию. Любое отклонение от копланарного состояния вследствие поворота вокруг ст-связи, соединяющей два атома углерода, приведет к уменьшению степени перекрывания и соответственно к снижению прочности л-связи; л-связь, таким образом, способствует сохранению плоскостности молекулы. В этом заключается теоретическое объяснение давно известного явления препятствия вращению вокруг двойной углерод-углеродной связи. Распределение л-электронов над и под плоскостью молекулы, т. е. за пределами оси углерод-углеродной связи, означает существование области отрицательного заряда, готовой к взаимодействию с любыми электронодефицитными реагентами (например, с окислителями). Неудивительно поэтому, что реакции с такого рода реагентами наиболее характерны для двойной углерод-углеродной связи (ср. разд. 7.1). Таким образом, классическая картина двойной связи заменяется моделью, согласно которой две связи, соединяющие атомы углерода, не только не идентичны, но и различны по природе, прочности и расположению в пространстве.
1.3.3. Тройные углерод-углеродные связи
В этине каждый атом углерода связан только с двумя другими атомами — с одним атомом водорода и одним атомом углерода. С этими двумя атомами образуются прочные ст-связи за счет двух гибридных орбиталей, образованных вследствие гибридизации 2«-орбитали, и только одной из атомных 2р-орби-талей углерода. Результирующие дигональные зр'-гибридные орбитали расположены на одной прямой. Так, при образовании молекулы этнна (ацетилен) эти гибридные орбитали используются для образования прочных о-связей между каждым из ато-тов углерода и одним атомом водорода и между атомами угле
18
рода, в результате чего образуется линейная молекула, в которой каждый из двух атомов углерода имеет по две негибриди-зованные атомные 2р-орбитали, расположенные под прямым углом друг к другу. Атомные орбитали одного атома углерода параллельны соответствующим орбиталям другого атома углерода и, следовательно, могут попарно перекрываться, что приводит к образованию двух л-связей во взаимно перпендикулярных плоскостях:
Таким образом, молекула этина как бы находится внутри цилиндра из отрицательного заряда. Энергия связи С = С составляет 812 кДж/моль (194 ккал/моль), так как вклад третьей связи еще меньше, чем вклад второй связи при переходе от простой связи к двойной. Длина связи С=С равна 0,120 нм: атомы углерода в этине расположены еще ближе друг к другу, чем в этене, но уменьшение длины связи при переходе С = С —>-С=С меньше, чем при переходе С—С —С=С.
1.3.4. Углерод-кислородные и углерод-азо,тные связи
Атом кислорода имеет электронную конфигурацию ls22s22p2x2p'y2p'z и при связывании с другими атомами может так же, как и углерод, давать гибридные орбитали, обеспечивающие образование наиболее прочных связей.
Так, при связывании с атомами углерода двух метильных групп в молекуле метоксиметана (диметиловый эфир) СН3—О—СНз атом кислорода мог бы использовать четыре s/Агибридные орбитали: две — для образования a-связей путем перекрывания с хр3-орбиталями каждого из двух атомов углерода, а две другие — для оставшихся свободными двух пар электронов. Показано, что угол С—О—С составляет 110°, длина связи С—О равна 0,142 нм (1,42 А), а энергия связи равна 360 кДж/моль (86 ккал/моль).
Атом кислорода может образовывать и двойные связи с атомом углерода; например, в пропаноне (ацетон) Ме2С=О: атом кислорода использует три 5р2-гибридные орбитали: одну для образования a-связи путем перекрывания с $р2-орбиталью
19
атома углерода, а две другие — для оставшихся двух свободу ных пар электронов. При этом как у атома кислорода, так м у атома углерода остается по одной негибридизованной р-орби-тали, которые могут давать боковое перекрывание (ср. для связи С=С, разд. 1.3.2) с образованием л-связи:
Найдено, что угол С—С—О составляет « 120°, длина связи С=О равна 0,122 нм (1,22 А), а ее энергия равна 750 кДж/ /моль (179 ккал/моль). Тот факт, что энергия связи С = О несколько больше удвоенной энергии связи С—О, в то время как энергия связи С=С заметно меньше удвоенной энергии связи С—С, объясняется, вероятно, тем, что в связи С=О свободные пары электронов атома кислорода более удалены друг от друга и поэтому более устойчивы, чем в связи С—О. Свойства атомов углерода и кислорода в образовании связей, таким образом, различны. Лишний раз об этом говорит и то, что связь углерод-кислород в отличие от связи углерод-углерод полярна.
Атом азота имеет электронную конфигурацию ls22s22p*2p^2p^ и также может давать гибридные орбитали при образовании простых С—N, двойных C=N и тройных C = N связей с углеродом. В каждом случае одна из орбиталей служит для локализации на ней свободной пары электронов атома азота; при образовании двойной и тройной связей соответственно одна и две л-связи также образуются в результате бокового перекрывания негибридизованных р-орбиталей атомов азота и углерода. Средние значения длин и энергий для простой азот-углеродиой связи равны соответственно 0,147 нм (1,47 А) и 305 кДж/моль (73 ккал/моль); для двойной связи они равны 0,129 нм (1,29 А) и 616 кДж/моль (147 ккал/моль) и для тройной связи равны 0,116 нм (1,16 А) и 893 кДж/моль (213 ккал/моль).
1.3.5. Сопряжение
При рассмотрении молекул, содержащих более одной кратной связи, например диенов с двумя С=С-связями, можно заметить, что соединения, в которых эти связи сопряжены (т. е. соединения, в которых кратные связи чередуются с простыми), например (1а) и (16), несколько более стабильны, чем соединения, в которых эти связи изолированы, например соедине
20
ния (2а) и (26):
(2а)
Большая термодинамическая устойчивость (более низкая энергия) сопряженных молекул проявляется в том, что соединения (1) характеризуются меньшей теплотой сгорания и меньшей теплотой гидрирования, чем соединения (2), и что изолированные двойные связи, как правило, могут легко мигрировать до тех пор, пока не станут сопряженными:
основание
МеСН=СН—СН2—С==О ------------МеСН2—СН=СН—С=О
। катализатор ।
Me Me
Безусловно, сопряжение не ограничивается кратными углерод-углеродными связями.
В соединениях (1а) и (2а) боковое перекрывание р-атом-ных орбиталей соседних атомов углерода могло бы привести к образованию двух локализованных л-связей и можно было бы ожидать, что эти соединения сходны с этеном, только как будто удвоенным! Такое сходство, действительно, было обнаружено для соединения (2а); соединение (1а) отличается, однако, несколько большей устойчивостью (о чем упоминалось выше), спектроскопическими свойствами (см. ниже) и способностью более легко вступать в реакции присоединения по сравнению с изолированными диенами (см. разд 7.5). При более внимательном рассмотрении выясняется, что в структуре (1а) боковое перекрывание может осуществляться между всеми четырьмя р-орбиталями соседних атомов углерода. Такое перекрывание приводит к образованию четырех молекулярных орбиталей —
21
двух связывающих (Ti и W2) и двух антисвязывающих (Чгз и Чг4) [перекрывание п атомных орбиталей (АО) всегда дает п молекулярных орбиталей (МО)]:
^4
«‘б
> антисвязывающие
V5J
Энергия л-орбитали
t 4 р-АО л-^
л-МО
1±_ _н_у t4 3
.
связывающие
атомы
этилен (хз)
МО бензол
t 4
у2
Как видно, при расположении четырех электронов в сопряженном диене (1а) на двух связывающих орбиталях суммарная энергия соединения ниже, чем (по аналогии с этеном) при расположении электронов на двух локализованных л-связях. Электроны в диене становятся делокализованными и принадлежат всей сопряженной системе, а не локализуются над двумя парами атомов углерода в л-связях, как в этене или структуре (16). Распределение четырех электронов на связывающих молекулярных орбиталях Ti и Ч^ ведет к образованию делокализованного заряженного электронного облака, как, например, в структуре (3):
При такой делокализации четыре атомные р-орбитали в структуре (1а) должны быть практически параллельными, и это неизбежно может вызвать серьезные ограничения вращения вокруг связи‘ С-2—С-3 в структуре (3), которая находится в наиболее предпочтительной конформации. Можно было бы также ожидать, что плотность л-электроиов между атомами С-2 и С-3 придаст этой связи характер частично двойной связи и она окажется короче, чем простая С—С-связь. Длина связи С-2—• —С-3 действительно несколько укорочена [0, 147 нм (1,47 А)], однако она не меньше длины одинарной связи между зр2-гиб-ридизованными атомами углерода (ср. разд. 1.3.2). Энергия стабилизации сопряженного диена относительно невелика [«17 кДж/моль (4 ккал/моль)], но даже этот выигрыш энер-
22
.гии нельзя приписать только делокализации электронов: необходимо принимать во внимание состояние гибридизации атомов углерода и различие в прочности связей между ними.
Делокализация гораздо более важна для стабилизации возбужденных состояний диенов и вообще полиенов, т. е. для снижения энергии этих состояний. Эффект делокализации при этом сводится к тому, что уменьшается щель между энергетическими уровнями основного и возбужденного состояний сопряженных молекул по сравнению с соответствующими характеристиками молекул с изолированными двойными связями. Действительно, по мере увеличения степени сопряжения указанная энергетическая щель постепенно уменьшается. Количество энергии, необходимое для перехода молекулы из основного состояния в возбужденное, снижается, вследствие чего длина волны излучения, поглощаемого сопряженной системой, возрастает. Простые диены поглощают в УФ-области спектра, однако по мере увеличения степени сопряжения поглощение постепенно смещается в видимую область, т. е. соединения приобретают окраску. Это можно проиллюстрировать на примере а, ю-дифеиилполиеиов:
CeH5(CH=CH)nCeH5
п Окраска
1 Нет
2—4 Желтая
п Окраска
5 Оранжевая
8 Красная
1.3.6. Бензол и ароматичность
Одна из главных проблем фундаментальной органической химии связана со структурой бензола. Известное плоскостное строение его молекулы предполагает х/Я-гибридизацию с р-ор-биталями на каждом из шести атомов углерода, направленными под прямыми углами к плоскости кольца [см. формулу (4)]:
Возможно, конечно, перекрывание орбиталей атомов 1, 2; 3, 4; 4г 6 или 1, 6; 5, 4; 3, 2, что привело бы к образованию соответствующих структур Секуле (4а) и j(46). Однако имеется и другой вариант: перекрывание всех шести смежных р-орби-талей, как в сопряженных диенах (см. разд. 1.3.5), что приводит к формированию шести молекулярных орбиталей — трех связывающих (Wi -> Ws) и трех аитисвязывающих (Чг4->-Чгб);
23
энергетические уровни этих орбиталей представлены ниже:
Энергия л-орбитали
21______
P-АО f | л-------
л-МО
этилен(х 2)
У4-
tit/3
антисвязывающие
р-АО ЦТ—LL
t 4
у,——
МО бутадиен
связывающие
Связывающая МО с самой низкой энергией (Т,) является циклической и охватывает все шесть атомов углерода, т. -е. делокализована. Эта МО имеет узловую плоскость, совпадающую с плоскостью молекулы, так что существуют два кольцевых электронных облака, располагающихся над и под плоскостью кольца [из которых только верхнее показано в структуре (5а)]: на этой МО распределены два электрона. Две другие связывающие МО (ХР2 и Тз) с равными энергиями (вырожденные МО) также охватывают все шесть атомов углерода, но кроме узловой плоскости, совпадающей с плоскостью кольца, каждая из них имеет еще и узловую плоскость, расположенную перпендикулярно плоскости молекулы. Таким образом, каждая из указанных МО имеет четыре доли, из которых только верхние пары показаны в структурах (56) и (5в):
Узловая плоскость I
(5а) (56) (5в)
На каждой МО ХР2 и также находятся по два электрона— всего в молекуле бензола оказывается шесть л-электро-нов. В результате формируется кольцевое электронное облако над и под плоскостью кольца (6):
24
Влияние этого облака отрицательного заряда на различные типы реагентов, которые могут атаковать молекулу бензола, рассмотрено в разд. 6.1.
Приведенная схема распределения электронов в молекуле бензола подтверждается тем, что все его углерод-углеродные связи имеют одну и ту же длину*, равную 0,140 им (1,40 А); молекула бензола представляет собой правильный шестиугольник, расстояния между атомами углерода в котором находятся между значениями длин простой [0,154 им (1,54А)] и двойной [0,133 им (1,33 А)] углерод-углеродиых связей. Чтобы подчеркнуть равиоцеииость всех связей в молекуле бензола, ее изображают ие формулами Кекуле, в которых связи неравноценны, а формулой вида:
Остается, одиако, выяснить вопрос о причинах сильно выраженной термодинамической устойчивости бензола. Эта устойчивость объясняется расположением трех ст-связей каждого из атомов углерода в одной плоскости под оптимальным углом 120° по отношению друг к другу и в большей степени тем, что все шесть л-электроиов размещены иа циклических, делокализованных молекулярных орбиталях. Такое распределение электронов значительно более устойчиво (соответствует более низкому уровню энергии), чем распределение иа трех локализованных л-ор-биталях (см. выше). Гораздо большая устойчивость бензола по сравнению, например, с сопряженными диенами (ср. разд. 1.3.5), вероятно, определяется тем, что молекула бензола является циклической (т. е. закрытой) симметричной системой.
Примерную оценку устойчивости бензола по сравнению с простыми циклическими ненасыщенными структурами можно получить, сравнивая теплоты гидрирования бензола, циклогексена (7) и циклогексадиеиа-1,3 (8):
ДЯ = — 120 кДж/моль (—28,6 ккал/моль)
ДЯ= —232 кДж/моль (—55,6 ккал/моль)
кН = — 208 кДж/моль (—49,8 ккал/моль)
* Связи С—Н в бензоле также имеют одинаковую длину, равную 0,108 нм (1,08 А).
25
Теплота гидрирования циклического диена (8) почти вдвое больше теплоты гидрирования циклогексена; следовало бы ожидать, что теплота гидрирования трех двойных связей в структуре Кекуле должна быть равна 3(—120) кДж/моль = = —360 кДж/моль (—85,8 ккал/моль). Однако в действительности при гидрировании бензола выделяется только —208 кДж/ /моль (—49,8 ккал/моль). «Реальная» молекула бензола по термодинамической устойчивости превосходит гипотетический «циклогексатриен» иа 151 кДж/моль (36 ккал/моль). Для сравнения следует отметить, что сопряженный диен стабильнее его аналога, в молекуле которого нет взаимодействия между электронами двойных связей, лишь на 17 кДж/моль (4 ккал/моль).
В отличие от бензола теплота гидрирования циклооктатетра-ена (9) до циклооктаиа (10) равна —410 кДж/моль (—98 ккал/моль), а теплота гидрирования циклооктена (11) равна —96 кДж/моль (—23 ккал/моль).
О) (10) (11)
АЯ = — 410 кДж/моль АЯ = —96 кДж/моль (—98 ккал/моль) (—23 ккал/моль)
Разница в значениях АД соединения (9) и 4АЯ соединения (11) составляет лишь —26 кДж/моль (—6 ккал/моль); цикло-октатетраеи, в отличие от бензола, не обнаруживает характерной стабилизации при сравнении с подходящим гипотетическим полиеном (в действительности он слегка стабилизирован), т. е. не является ароматическим. Отсутствие ароматических свойств у молекулы циклооктатетраена неудивительно, поскольку перекрывание циклических р-орбиталей, как и для бензола, возможно лишь в том случае, если соединение (9) имеет плоское строение и, следовательно, угол С—С—С равен 135°; это привело бы к очень большому напряжению в кольце, образованному sp2-гибридизованными атомами, для которых предпочтителен угол 120°. Такое напряжение может быть ослаблено деформацией кольца, но при этом становится невозможным перекрывание р-орбиталей. То, что такая деформация кольца действительно имеет место, подтверждено данными рентгеиоструктурного анализа, согласно которым циклооктатетраен имеет конформацию «ванны» (9а) с чередующимися двойными [0,133 нм (1,33 А)] и простыми [sp2 — sp2-, 0,146 нм (1,46 А); ср. разд. 1.3.1] углерод-углеродными связями:
од33 им (9а)
0,146 нм
Условия, необходимые для того, чтобы циклические полиены обладали ароматическими свойствами, рассмотрены ниже.
26
Энергию, на величину которой молекула бензола стабилизирована по сравнению с молекулой гипотетического циклогекса-триена, следовало бы называть энергией стабилизации, однако ее часто называют энергией делокализации, хотя вопрос о том, в какой мере стабилизация бензола действительно обусловлена делокализацией его шести л-электронов, еще не ясен. Широко используется также термин энергия резонанса, однако он совершенно неудовлетворителен с семантической точки зрения, поскольку слово резонанс обычно ассоциируется с быстрыми переходами (осцилляциями) между различными структурами (например, в случае бензола — между структурами Кекуле, которые на самом деле не существуют; ср. разд. 1.3.7).
Можно сформулировать следующие условия, необходимые для проявления стабилизации и ароматического характера полиенов: а) молекула должна быть плоской, для того чтобы было возможно циклическое перекрывание орбиталей; б) все связывающие орбитали должны быть полностью заполнены. Последнее условие выполняется в циклических системах с числом л-электронов, равным 4п-j- 2* (правило Хюккеля), чаще всего в ароматических соединениях при п = 1, т. е. для структур, содержащих шесть л-электронов. Десять л-электронов (п = 2) имеются в молекуле нафталина (12), энергия стабилизации которого равна 255 кДж/моль (61 ккал/моль), по 14л-электронов (п = = 3) —в молекулах антрацена (13) и фенантрена (14), энергии стабилизации которых равны 352 и 380 кДж/моль (84 и 91 ккал/моль) соответственно.
(14)
14л-электронов (п = 3)
(12)
Юл-электронов (п = 2)
(13)
14л-электронов (п = 3)
Хотя названные соединения ие являются моноциклическими, как бензол, и для них правило Хюккеля не может строго выполняться, наличие связи, соединяющей противолежащие атомы кольца и превращающей эти молекулы в би- и трициклические соответственно, приводит лишь к относительно небольшим нарушениям их ароматичности, поскольку сохраняет делокализацию л-электронов в циклических структурах, содержащих десять или четырнадцать атомов углерода.
* Важно отметить, что цнклооктатетраен, содержащий 8 л-электронов (4л; л = 2), как уже было показано, не является ароматическим соединением (см. разд. 1.3.6).
27
Известны также квазиароматические структуры, в которых стабилизированная циклическая форма является ионом. Примерами таких структур являются циклогептатриенил-катион (тро-пилий) (15) (см. разд. 5.2) и циклопентадиенил-анион (16) (ср. разд. 10.2), в которых имеется по бл-электронов (п — 1) и, что особенно удивительно, циклопропенил-катион (17) (ср. разд. 5.2), который имеет 2л-электрона (п = 0):
А
(15) (16) (17)
Кольцевая структура необязательно должна быть чисто карбоциклической: например, пиридин (18) (ср. разд. 6.9), содержащий атом азота в кольце и бл-электронов (п = 1), так же высоко стабилизирован, как бензол:
(18)
Ценный экспериментальный критерий ароматичности молекул, кроме выше упомянутых, дает метод ядерного магнитного резонанса (ЯМР) *. Положение сигнала протона в спектре ЯМР зависит от природы, а точнее, от локального окружения атома углерода (или другого атома), с которым связан протон. Так, например, сигнал протона циклооктатетраена наблюдается при 6 5,6 (это типично для протонов неароматического циклического полиена), тогда как сигнал протона в случае бензола наблюдается при 6 2,8, что характерно для типичных ароматических соединений. ( ,
1.3.7. Условия делокализации электронов
Трудность нахождения адекватного способа изображения связей между атомами углерода в бензоле ясно указывает, что обычно используемый химиками способ изображения связей между атомами в виде одной, двух или трех черточек, обозначающих участие в этих связях соответственно двух, четырех или шести электронов, совершенно неудовлетворителен. В действительности в образовании некоторых связей участвуют другие и даже дробные числа электронов. Это можно видеть на примере эта-ноат-аниона (19) (ацетат-анион), в котором, по данным рентгеноструктурного анализа, атомы кислорода неразличимы, а обе
* Полезный пример использования ЯМР (н других) спектров можно найти в работе: Williams D. Н., Fleming I. Spectroscopic Methods in Organic Chemistry. McGraw-Hill, 3rd Edition, 1980.
28
углерод-кислородные связи имеют одинаковые межатомные расстояния, т. е. содержат одинаковое число электронов.
Этн трудности привели к тому, что молекулы, структуры которых не могут быть описаны в виде одной классической формулы, стали изображать комбинацией двух или большего числа классических структур, так называемых канонических структур, связанных стрелками с двумя остриями (-*->-). Путь, по которому одна из этих структур может переходить в другую, часто указывают с помощью изогнутой стрелки, «хвост» которой показывает, откуда движется электронная пара, а ее острие — куда она движется *:
сн3с
0~
(19) (19а) (196) (19аб)
Нельзя, однако, слишком настаивать на том, что этаноат-анион имеет не две возможные альтернативные, быстро взаимо-превращающиеся структуры, а лишь одну, реальную структуру (19аб), иногда рассматриваемую в качестве гибридной, по отношению к которой классические канонические структуры (19а) и (196) могут рассматриваться как предельные приближения.
При рассмотрении делокализации и изображения ее посредством двух или более классических структур следует иметь в виду определенные ограничения. Вообще говоря, чем больше канонических структур может быть написано для данного соединения, тем, следовательно, выше степень делокализации электронов и тем стабильнее должно быть такое соединение. Эти структуры не должны слишком отличаться друг от друга по уровню энергии, в противном случае структура с более высокой энергией будет вносить слишком малый вклад в гибридную структуру и практически не будет участвовать в ее образовании. Эффект стабилизации выражен особенно сильно, если канонические структуры равноценны по энергии, как в приведенных выше структурах (19а) и (196). Структуры с разделенными зарядами (ср. разд. 1.5.2) также можно рассматривать в качестве канонических, однако при прочих равных условиях они обычно богаче энергией, чем структуры, в которых отсутствует разделение заряда, и, следовательно, вносят меньший вклад в образование гибридной структуры. Все канонические структуры
* В последующем изложении, одиако, канонические структуры, например, такие как (19а) и (196), связанные стрелкой с двумя остриями, даиы без изогнутых стрелок. Изогнутые стрелки применяются в дальнейшем для обозначения реального перемещения электронных пар, например при образовании и расщеплении связей в ходе реальной реакции.
29
должны содержать одинаковое число спаренных электронов, и взаимное расположение всех входящих в молекулу атомов должно быть одним и тем же. Для того чтобы делокализация была значительной, все атомы, связанные с ненасыщенными центрами, должны лежать в одной плоскости или вблизи нее. Ниже обсуждены примеры, когда делокализация, а следовательно, и стабилизация практически невозможны из-за пространственных затруднений (см. разд. 1.6).
1.4. Разрыв и образование связей
Разрыв ковалентной связи между двумя атомами может про
исходить по-разному:
R • + X • R:X
R:+X+
R+ + :X
В случае а каждый атом отделяется с одним электроном, что приводит к образованию частиц, называемых радикалами и обладающих высокой реакционной способностью вследствие наличия неспаренного электрона; такой разрыв называют гомолитическим расщеплением связи. В случаях бив один атом может удерживать оба электрона, оставляя другой атом без электронов, в результате чего возникают отрицательный и положительный ионы соответственно. Если атомы R и X неидентичны, расщепление может идти по одному из таких путей в зависимости от того, какой атом — R или X — удерживает пару электронов. Такого рода разрывы носят название гетеролитиче-ского расщепления и приводят к образованию ионной пары. Образование ковалентной связи может происходить путем обращения любого из этих процессов, а также, безусловно, путем атаки других частиц только что образовавшимися радикалами и ионами (см. разд. 5.3 и 11.5.2.1):
R • + Вг—Вг —► R—Вг + Вг •
R++ Н2О —► R—ОН + Н+
Как будет показано ниже, радикалы или ионные пары образуются как активные промежуточные соединения в очень большом числе органических реакций. Реакции, включающие образование радикалов, протекают преимущественно в газовой фазе и в растворах неполярных растворителей; онн катализируются светом или добавлением других радикалов (см. разд. 11.1). Реакции, включающие образование ионных интермедиатов, быстрее протекают в полярных растворителях из-за более легкого разделения заряда в таких растворителях и, очень часто, из-за стабилизации образующихся ионных пар вследствие сольвата-
30
ции. Считают, что многие из этих ионных интермедиатов несут заряд на каком-то атоме углерода, хотя такие ионы часто стабилизируются в результате делокализации заряда (в большей или меньшей степени) между другими атомами углерода или атомами иных элементов:
~сн2=сн—сн2"
•• н+ + —н2о .
СН2=СН—СН2—ОН ч --ь СН2=СН—СН2—ОН2 < » Т
_сн2—сн=сн2_
О Г ° О’ 1
II -он II - 1
СНз—С—СНз =г=* LCH3—С—СН2 -<-4- СНз—С=СН2 J + Н2О
Если атом углерода несет положительный заряд, соединение называют карбокатионом, а если отрицательный заряд — карбанионом. Хотя такие ионы могут образоваться только временно и присутствовать лишь в очень низких концентрациях, тем не менее они часто чрезвычайно важны для результата реакции, в которой они участвуют.
Кроме радикалов, карбокатионов и карбанионов известны и другие интермедиаты, в которых атом углерода является активным центром; к ним относятся интермедиаты, включающие электронодефицитные частицы: карбены R2C: (см. разд. 9.8), нитрены RN: (см. разд. 5.6), а также арины (см. разд. 6.10).
••
1.5. Факторы, влияющие на доступность электронов
Учитывая изложенное выше, можно ожидать, что любые факторы, которые влияют на относительную доступность электронов (на электронную плотность) в определенных связях или на определенных атомах данного соединения, могут очень сильно влиять на его реакционную способность при взаимодействии с определенным реагентом. Область с высокой электронной плотностью атакуется, например, анионом ~ОН, с трудом или вообще не атакуется, в то время как область с низкой электронной плотностью атакуется легко; обратная картина будет наблюдаться в случае положительно заряженного реагента. К настоящему времени уже выявлен ряд таких факторов.
1.5.1. Индуктивный эффект и эффект поля
В случае простой ковалентной связи между двумя неодинаковыми атомами пара электронов, образующая ст-связь, никогда не поделена абсолютно поровну между двумя атомами, она несколько смещена к более электроотрицательному атому. Так, в алкилхлориде (20) электронная плотность несколько выше
31
у атома хлора, чем у атома углерода, так как первый более электроотрицателен; в общем виде это представлено в структурах (20а) и (206):
\в+ 6- \
—С—С1 — С->С1
(20а) (206)
Если атом углерода, связанный с атомом хлора, соединен и с другими атомами углерода, то этот эффект может передаваться по цепи:
4 3 2 1
С—С—С->С->->С1
Частичное оттягивание атомом хлора электронов связи углерод — хлор приводит к тому, что атом С-1 становится электронодефицитным; это в свою очередь вызывает смещение электронов, образующих связь между атомами С-1 и С-2 и далее по цепи. Однако влияние атома С-1 на С-2 меньше, чем влияние атома С1 на С-1; передача этого влияния быстро затухает в насыщенной цепи и после С-2 становится настолько мала, что его трудно обнаружить. Подобное влияние на распределение электронов в о-связях известно как индуктивный эффект.
Кроме индуктивного эффекта, действующего в соединении через связи, практически аналогичный эффект может проявляться или через пространство, окружающее молекулу, или — в растворе— с участием молекул растворителя, которые окружают молекулу данного соединения. Во многих случаях, однако, невозможно различить действие индуктивного эффекта как такового и очень сходного с ним (и действующего в том же направлении) эффекта поля. Следовательно, говоря об индуктивном эффекте, необходимо учитывать, что индуктивный эффект включает и эффект поля *.
Большинство атомов и групп, связанных с атомом углерода, оказывают такое же индуктивное влияние и в том же направлении, что и атом хлора, т. е. они оттягивают электроны от атома углерода вследствие большей, по сравнению с ним, электроотрицательности; исключение составляют алкильные группы, являющиеся донорами электронов**. Хотя индуктивный эффект в количественном -отношении довольно невысок, он ответствен за
* Во многих случаях индуктивный эффект и эффект поля были разделены: при этом исходили из того, что индуктивный эффект зависит только от природы связей, а эффект поля определяется геометрией молекулы (см. обсуждение этих понятий в кн.: Марг Дж. Органическая химия: Пер. с англ./Под ред. И. П. Белецкой. М.: Мир, 1987. С. 32).— Прим. ред.
** Атомы металлов, например, в литийалкилах или реактивах Гриньяра (эти соединения содержат в основном ковалентные связи) также являются донорами электронов; это приводит к отрицательно поляризованному атому углерода в каждом случае: R-«-Li и R-«-MgHal (ср. разд. 8.4).
32
возрастание основности, которое наблюдается при замещении одного из атомов водорода в молекуле аммиака на алкильную группу (см. разд. 3.2.2), и отчасти объясняет более легкое замещение атомов водорода в ароматическом кольце метилбен-зола по сравнению с бензолом (см. разд. 6.7.1.2).
Индуктивные эффекты приводят к поляризации молекулы в ее основном состоянии и поэтому отражаются на ее физических характеристиках, например на дипольном моменте.
1.5.2. Мезомерный эффект (эффект сопряжения)
Мезомерный эффект также является результатом перераспределения электронов, которое происходит в ненасыщенных и, особенно, в сопряженных системах с участием их л-орбиталей. Например, свойства карбонильной группы (см. разд. 7.6.3) полностью не описываются ни классической формулой (21а), ии граничной структурой (216), получаемой путем сдвига л-элек-тронов:
(21а) (216) (21аб)
Действительная структура представляет собой нечто промежуточное, т. е. гибрид (21аб), для которого структура (21а) и (216) являются каноническими формами. Здесь также возможен индуктивный эффект, как это показано в структуре (21аб), но он гораздо меньше, чем мезомерный эффект, поскольку о-электроны намного меньше поляризуются и поэтому менее подвижны, чем л-электроны.
Если группа С=О сопряжена со связью С=С, поляризация может передаваться дальше посредством л-электронов, как, например, для соединения (22):
СН СН
АД
СН СН
МеХ \fi\r
(22а) (226)
Г б+
С(1 ZCH м/ xffV' (22аб)
При этом происходит делокализация заряда (ср. с 1,3-дие-нами; разд. 1.3.5), что приводит к нехватке электронов на атомах С-3 и С-1, как в простом карбонильном соединении. Различие между такой передачей через сопряженную систему и индуктивным эффектом в насыщенной системе состоит в том, что в данном случае при передаче по цепи эффект ослабляется в гораздо меньшей степени, а полярность соседних атомов углерода чередуется.
2 П. Сайкс
33
Стабилизация вследствие делокализации положительного или отрицательного заряда в ионе с участием его л-орбиталей может явиться основным фактором, обусловливающим образование соответствующего иона (ср. разд. 3.1.2). Например, стабилизация феноксид-иона (23) вследствие делокализации его заряда с участием делокализованных л-орбиталей ароматического кольца в значительной степени ответственна за кислотные свойства фенола (ср. разд. 3.1.3):
(23а) (236) (23в) (23г)
Внешне сходная делокализация рованном феноле (24) с участием нов атома кислорода:
имеет место и в недиссоции-неподеленной пары электро-
+ОН +0Н 1
(24а) (246) (24в) (24г)
Но в этом случае происходит разделение заряда, вследствие чего такая делокализация будет соответственно менее эффективной, чем при стабилизации феноксид-иона.
Мезомерные эффекты, подобно индуктивным, вызывают поляризацию молекул в их основных состояниях и поэтому сказываются на физических свойствах соединений. Существенное различие между индуктивными и мезомерными эффектами состоит в том, что если индуктивные эффекты могут действовать как в насыщенных, так и в ненасыщенных соединениях, то мезомерные эффекты могут действовать только в ненасыщенных и, особенно, в сопряженных соединениях. Индуктивные эффекты связаны с электронами ст-связей, а мезомерные — с электронами л-связей и орбиталей. Индуктивные эффекты передаются только на сравнительно короткие расстояния в насыщенной цепи, в то время как мезомерные эффекты могут передаваться от одного конца сравнительно больших молекул к другому при условии наличия сопряжения (т. е. делокализованных л-орбиталей).
1.5.3. Динамические эффекты
Некоторые исследователи пытались найти различия между такими эффектами, как два рассмотренных выше, которые обусловливают поляризацию молекул в основном состоянии, и
34
теми изменениями распределения электронов, которые происходят при приближении реагента или, в более частном случае, обусловлены образованием соответствующего переходного состояния после первоначальной атаки молекулы (см. разд. 2.2.2). Эти изменяющиеся во.времени эффекты, по аналогии с рассмотренными выше постоянно действующими, были названы соответственно индуктомерными и электромерными эффектами. Можно считать, что такие эффекты обусловливают скорее поляризуемость, чем поляризацию, поскольку распределение электронов возвращается к таковому в основном состоянии атакованной молекулы, если реагент удаляется до того, как реакция прошла, или если образовавшееся переходное состояние распадается с образованием исходных веществ.
Такие изменяющиеся во времени эффекты, будучи по существу временными, не должны, конечно, отражаться на физических свойствах соответствующих соединений. Часто невозможно экспериментально разграничить постоянные и изменяющиеся во времени эффекты, однако следует подчеркнуть, что приближение реагента может оказать глубокое влияние, увеличивая реакционную способность реагирующей молекулы и облегчая тем самым реакцию.
1.5.4. Сверхсопряжение (гиперконъюгация)
Индуктивный эффект алкильных групп, как и следовало ожидать, уменьшается в ряду.
Me. ..
\ Ме^.
Ме->С-> > С.СН-»- >Ме-<-СН2—► >СН3~►
м/ ме
Однако, если алкильные группы связаны с ненасыщенной системой, например с двойной связью или бензольным кольцом, этот порядок нарушается и в случае некоторых сопряженных систем изменяется на обратный. Таким образом, по-видимому, алкильные группы способны при некоторых обстоятельствах вызывать смещение электронов с помощью механизма, отличного от индуктивного эффекта. Это удалось объяснить, расширив понятие сопряжения или мезомерного эффекта и предположив, что делокализация электронов происходит с участием электронов. соседних ст-связей:
Н Н+
I
н—с—сн=сн2 -<-► н—с=сн—сн2 I I
н н
(25а) (256)
2*
3S
(26а) (266) (26в) (26г)
Этот эффект получил название сверхсопряжения (гиперконъюгации) ; с его помощью удалось объяснить ряд непонятных ранее явлений. Следует подчеркнуть, что в действительности не происходит отщепление протона в соединениях (25) или (26), поскольку, если он переместится из первоначального положения, то одно из условий, необходимых для делокализации, будет нарушено (см. разд. 1.3.7).
Обращение ожидаемого (индуктивного) ряда электронодонорных свойств (СНз > МеСНг > МегСН > Ме3С) можно объяснить тем, что эффект гиперконъюгации зависит от наличия атомов водорода, связанных с а-углеродным атомом ненасыщенной системы. Ясно, что эффект гиперконъюгации должен быть максимальным у СН3-группы [(25)] и отсутствовать у Ме3С-группы [(29)]:
Н Me
I I
н—с—сн=сн2 н—с—сн=сн2
I I
н н
(25) (27)
Me I Me—С— СН=СН2 I н
(28)
Me I Me—С— СН=СН2 1 Me
(29)
В гиперконъюгации могут участвовать как С—С-, так и С—Н-связи, а различия в относительной реакционной способности, наблюдаемые в каком-либо ряду соединений, в действительности могут быть результатом не только эффекта гипер-коиъюгации, ио и действия растворителя.
Эффект гиперконъюгации был использован для объяснения большей термодинамической устойчивости таких алкеиов, у которых двойная связь не является концевой, например в соединении (30), по сравнению с изомерными соединениями с концевой двойной связью, например ,(31); в соединении (30) имеется девять а-водородных атомов, способных участвовать в гиперконъюгации, а в соединении (31) — только пять:
CHS
I
СНз—с=сн—сн»
(30)
СНз I МеСН2—С=СН2
(31)
36
В результате этого преимущественно образуются алкены с неконцевой двойной связью в реакциях, в которых возможно также образование соответствующих изомеров с концевой двойной связью, а также наблюдается очень быстрая изомеризация менее стабильного соединения в более стабильное, например (31)->-(30).
1.6. Стерические эффекты
До сих пор обсуждались факторы, которые могут влиять на относительную доступность электронов в связях или на отдельных атомах соединения и, следовательно, определять реакционную способность этого соединения. Действие этих факторов, однако, может быть изменено или даже исключено под влиянием стерических факторов; например, эффективная делокализация с участием орбиталей может происходить только в том случае, если р- или л-орбитали на атомах, участвующих в делокализации, могут стать параллельными или почти параллельными. Если же это условие не выполняется, то не происходит перекрывания орбиталей и делокализация может быть нарушена. В качестве примера можно рассмотреть А\2У-диметиланилин (32) и его 2,6-диалкилпроизводные, например (33). Группа NMe2 соединения (32), будучи электронодонорной (из-за наличия непо-деленной пары электронов на атоме азота, взаимодействующей с делокализованными л-орбиталями ароматического кольца), облегчает атаку кольца катионом диазоиия PhN2> например, в реакции азосочетания; это приводит к замещению в пара-положение (ср. разд. 6.7.1.3):
(32)
PhN=N +
PhN^ ----->
Однако 2,6-диметильное производное (33) не подвергается азосочетанию в этих условиях, несмотря на то что введенные в молекулу метильные группы малы по объему и не могут мешать атаке в пара-положение. Отсутствие сочетания по этому положению обусловлено в действительности тем, что две метильные группы, находящиеся в орто-положениях по отношению к группе NMe2, создают пространственные затруднения двум метильным группам, связанным с атомом азота, для расположения их в плоскости бензольного кольца. Это означает, что р-орбитали атома азота и атома углерода кольца, с которым связан азот, не могут расположиться параллельно друг к другу и их перекрывание
37
ингибируется. Электронное взаимодействие с ароматическим кольцом поэтому сильно затрудняется, и перенос заряда, как это было в соединении (32), в этом случае не происходит (ср. разд. 3.2.3),
Наиболее обычным стерическим эффектом является классическое пространственное затруднение, при котором достаточно объемистые группы непосредственно влияют на реакционную способность того или иного участка молекулы, затрудняя подход реагента к реакционному центру или создавая напряжение в переходном состоянии (ср. разд. 2.2.1), и не влияют на доступность электронов. Это явление было тщательно исследовано в связи с проблемой устойчивости комплексов, образуемых триме-тилбором с различными аминами. Так, комплекс (34), образованный с триэтиламином, диссоциирует очень легко, тогда как комплекс (35) с хинуклидином очень стабилен, так как в нем три этильные группы, связанные с атомом азота, оттянуты «назад», что исключает возможность образования конформации, в которой могли бы возникнуть стерические препятствия атаке по атому азота:
(34) (35)
Тот факт, что это различие не обусловлено различной доступностью электронов атомов азота в этих соединениях, подтверждается тем, что оба эти амина очень мало отличаются по основности (ср. разд. 3.2.4) захват протона сопряжен с гораздо меньшими стерическими помехами, чем захват относительно громоздкой группы ВМе3. Среди других реакций особенно чувствительными к пространственным затруднениям можно назвать этерификацию и гидролиз сложных эфиров (ср. разд. 8.6.3).
Следует подчеркнуть, что такое стерическое ингибирование является крайним случаем; любые факторы, которые нарушают Или тормозят определенную взаимную ориентацию реагирующих
38
частиц, мешая им приблизиться друг к другу, также могут оказывать решающее влияние на скорость реакции; это часто встречается в реакциях биологических систем.
1.7. Классификация реагентов
Ранее уже были упомянуты электронодонорные и электроноакцепторные группы и их способность обогащать или обеднять определенные участки молекулы электронами. Такие группы, несомненно, влияют на тип реагента, с которым данное соединение будет наиболее легко взаимодействовать. Богатое электронами соединение, например феноксид-анион (36), наиболее легко атакуется положительно заряженными катионами, например катионом диазония CeHsN^ (см. разд. 6.6) или другими соединениями, которые хотя и не являются катионами, но содержат электронодефицитный атом или центр, например атом серы в триоксиде серы (37) (в реакции сульфирования; см. разд. 6.4):
(36а) (366)
б- 6+++/'° а
ч'Об
(37)
Такие реагенты, поскольку они стремятся атаковать субстрат в положение (или положения) с высокой электронной плотностью, называют электрофильными реагентами, или электрофилами.
Наоборот, электронодефицитный центр, например атом углерода в хлорметане (38), наиболее легко атакуется анионами, такими как ~ОН, -CN и т. д., или другими частицами, которые, не являясь истинными анионами, содержат атом или центр, богатые электронами, например атом азота в аммиаке H3N: или аминах R3N:. Такие реагенты вследствие их тенденции атаковать субстрат в положение (или положения) с низкой электронной плотностью, т. е. в участке, где заряд атомного ядра не полностью компенсирован орбитальными электронами, называют нуклеофильными реагентами, или нуклеофилами.
в+ в-
Н3С->С1 (38)
Необходимо отметить, что для преимущественного направления реакции достаточно даже слегка асимметричного распределения электронов. Наличие полного заряда на реагирующей частице, конечно, помогает протеканию реакции, но ни в коей мере не является обязательным. Действительно, требующееся асимметричное распределение заряда может быть индуцировано
39
в результате взаимной поляризации реагента и субстрата при их сближении, например при взаимодействии брома с этеном (см. разд. 7.1).
Электрофилы и нуклеофилы можно рассматривать как частные случаи кислот и оснований. Классическое определение кислот и оснований основано на том, что кислоты являются донорами протонов, а основания — акцепторами протонов. Более общее определение было дано Льюисом, который определил кислоты как соединения, способные принимать электронные пары, а основания — как соединения, которые могут предоставлять такие пары. Под это определение подходит ряд соединений, которые ранее не считали кислотами или основаниями. Например, трифторид бора (39) действует как кислота, принимая пару электронов атома азота триметиламина с образованием комплекса (40), и поэтому его называют кислотой Льюиса.
F3B-|-:NMe3 F3B:NMe3
(39) (40)
Электрофилы и нуклеофилы в органических реакциях можно рассматривать как акцепторы и доноры электронов соответственно, принимающие их от одних атомов и отдающие их другим атомам, чаще всего атомам углерода. Электрофилы и нуклеофилы имеют, конечно, сходство с окисляющими и восстанавливающими агентами, поскольку первые рассматривают как акцепторы электронов, а вторые — как доноры электронов. Наиболее часто встречающиеся электрофилы и нуклеофилы приведены ниже (звездочкой отмечены атомы, которые принимают электроны от субстрата или передают их ему):
Электрофилы'. Н+, Н3О+, +NO2, +NO, PhNJ, R3C+,
SO3, CO2, BF3, A1C13, ICl, Br, O3.
Нуклеофилы: H-, BF^, HSOJ, HO~, RO”, RS , 'CN, RCOL RC=C~, ~CH(CO2Et)2 (41)
He всегда можно четко разграничить реагент и субстрат; так, +NO2, “ОН R т. д. обычно рассматриваются как атакующие реагенты, а карбанион (41) в зависимости от условий может быть атакующим реагентом или субстратом, когда взаимодействует, например, с алкилгалогенидом. Действие карбаниона на алкил-галогенид является нуклеофильной атакой, тогда как действие алкилгалогенида на карбанион можно рассматривать как электрофильную атаку; однако независимо от этого сущность этой реакции остается неизменной.
40
Следует напомнить, что существуют реакции, протекающие с участием радикалов. Эти реакции гораздо менее чувствительны к изменениям электронной плотности в субстрате по сравнению с реакциями, включающими промежуточные соединения полярной природы. Однако на эти реакции сильно влияет,добавление даже следовых количеств веществ, способных образовывать или связывать радикалы (подробнее см. разд. 11.5).
1.8. Классификация реакций
Можно назвать четыре основные типа реакций, в которых участвуют органические соединения: замещение (вытеснение), присоединение, элиминирование, перегруппировки.
В реакциях первого типа замещение обычно происходит у атома углерода, но замещаемый атом может быть атомом водорода или каким-либо другим атомом или группой атомов. Прн электрофильном замещении чаще всего замещается атом водорода; хорошим примером этого служит классическое ароматическое замещение (см. разд. 6.1):
no2 + н+
2
При нуклеофильном замещении чаще замещается не атом водорода, а другие атомы (см. разд. 4.1), например:
NC~ + R—Вг —► NC—R + Вг"
Однако известны реакции нуклеофильного замещения атома водорода (см. разд. 6.10), а также реакции замещения, индуцируемые радикалами, например галогенирование алканов (ср. разд. 11.5.2).
Реакции присоединения также могут быть электрофильными, нуклеофильными или радикальными в зависимости от типа частиц, инициирующих процесс. Присоединение к обычным двойным углерод-углеродным связям индуцируется, как правило, электрофилом или радикалом. Например, присоединение НВг
Н
\ / НВг \ | /
чс=с7 -----х—с;
/ \ / |\
Вг
может начинаться с атаки двойной связи протоном Н+ (см. разд. 7.3) или радикалом Вг • (см. разд. 11.5.1.2). В отличие от этого, реакции присоединения, идущие с участием двойной связи углерод — кислород в простых альдегидах и кетонах, являются обычными нуклеофильными реакциями (см. разд. 7.6.3).
41
Примером может служить катализируемое основанием образование циангидрина в жидком HCN:
"CN ч ,0“ — > ;сС
медленно '
нем ч ОН
^^/C\CN + "CN
Реакции элиминирования по существу обратны реакциям присоединения; наиболее обычный тип такой реакции — отщепление атома водорода и другого атома или группы от соседних атомов углерода с образованием алкенов (см. гл. 9):
Н Н
Перегруппировки также могут протекать через промежуточные соединения, представляющие собой катионы, анионы или радикалы; чаще всего эти реакции идут с образованием карбокатионов или других электронодефицитных частиц. Перегруппировки могут включать существенную перестройку углеродного скелета, как, например, в ходе превращения 2,3-диметилбутан-диола-2,3 (пинакол) (42) в 2,2-диметилбутанон-З (пинаколон) (43) (ср. разд. 5.4.2.3):
НО ОН О
I I н+ II
Ме2С—СМе2 ----->- Ме3С—СМе
(42) (43)
За-стадией собственно перегруппировки в таких реакциях часто следуют стадии замещения, присоединения или отщепления, приводящие к образованию стабильного конечного продукта. >
Глава 2
ЭНЕРГЕТИКА, КИНЕТИКА И ИССЛЕДОВАНИЕ МЕХАНИЗМОВ РЕАКЦИЙ
2.1. Энергетика реакции............................................43
2.2. Кинетика реакции..............................................46
2.2.1. Скорость реакции и свободная энергия активации..........47
2.2.2. Кинетика и скоростьлимитирующая стадия.................49
2.2.3. Кинетический и термодинамический контроль..............52
42
2.3. Исследование механизмов реакций...............................53
2.3.1. Идентификация продуктов реакции . ....................53
2.3.2. Кинетические доказательства.............................54
2.3.3. Изотопные эффекты...................................... 56
2.3.4. Идентификация промежуточных соединений..................59
2.3.5. Стереохимические доказательства.........................61
Выше были рассмотрены электронные и стерические факторы, которые могут влиять на реакционную способность каких-либо соединений в определенных условиях, а также типы реагентов, которые будут легко атаковать определенные центры таких соединений. Однако почти ничего не было сказано о том, каким образом электронные и стерические факторы влияют на энергетические и кинетические характеристики, определяющие направление и скорость реакции. Такие характеристики не в меньшей мере важны и потому, что они могли бы существенно облегчить понимание механизмов рассматриваемых реакций.
2.1. Энергетика реакции
Один из вопросов, возникающих при изучении превращения исходных веществ в продукты реакции, состоит в том, насколько «далеко» может пройти реакция в сторону образования конечных продуктов. Поскольку любая система стремится к наиболее устойчивому состоянию, можно ожидать, что чем более устойчивы продукты реакции по сравнению с исходными веществами, тем более равновесие между ними будет смещено в сторону продуктов, т. е. чем больше Аустойчивости (рис. 2.1), тем в большей степени исходные вещества должны превращаться в продукты.
Очевидно, однако, что просто изменение общей энергии при переходе от исходных веществ к продуктам реакции, которое можно легко определить путем измерения теплоты реакции А/7*, не может служить мерой различия их устойчивости, поскольку часто не наблюдается непосредственной корреляции между значениями АН и константой равновесия реакции К. Известны сильно экзотермические реакции с малыми константами равновесия (небольшая степень превращения исходных веществ в продукты) и некоторые реакции с большими константами равновесия, являющиеся фактически эндотермическими (энтальпия продуктов реакции выше, чем энтальпия исходных веществ); это ясно указывает на то, что при оценке относительной устойчивости химических соединений кроме энтальпии необходимо учитывать какой-то дополнительный фактор.
* Величина И является мерой теплосодержания, или энтальпии, соединения; ДЯ имеет знак минус, если продукты реакции имеют более низкое теплосодержание, чем исходные вещества: в этом случае, когда имеет место уменьшение энтальпии, реакция является экзотермической.
43
г
Исходные дешестда
в
*» Ё
Продукты
Этот вывод находится в соответствии со вторым законом термодинамики, согласно которому упорядоченные системы стремятся к разупорядоче-нию; мерой степени разупорядоченности системы является ее энтропия S. В поисках наи-Рис. 2.1. большей устойчивости систе-
мы стремятся к минимальному значению энергии (т. е. к минимуму энтальпии) и к максимуму энтропии (беспорядок или случайность). Таким образом, условия, отвечающие относительной устойчивости системы, должны определяться компромиссом между значениями Н и S, что выражается свободной энергией Гнббса G, определяемой уравнением:
G = Н - TS,
где Т — абсолютная температура.
Изменение свободной энергии в ходе реакции при данной температуре:
bG = Mi -TbS.
Показано, что стандартное изменение свободной энергии при переходе от исходных веществ к продуктам реакции, AG° (AG° характеризует изменение свободной энергии в стандартных условиях: при активности, равной единице; менее точно — в расчете на I моль при давлении 1 атм) связано с константой равновесия /С уравнением:
— ДО° = 2,303/?Г 1g К, т. е. чем сильнее уменьшается свободная энергия (поэтому минус AG°) при переходе от исходных веществ к продуктам реакции, тем больше К. и тем больше равновесие сдвинуто в сторону продуктов реакции. Очевидно, что минимальное значение свободной энергии соответствует равновесию между исходными веществами и продуктами реакции. В реакции, в которой свободная энергия не изменяется (AG° = 0), /С = I, что соответствует 50 %-му превращению исходных веществ в продукты реакции. Возрастанию положительных значений AG° соответствует быстрое уменьшение /С (поскольку эта величина стоит под знаком 1g), соответствующее чрезвычайно малому превращению исходных веществ в продукты, и наоборот, возрастанию отрицательных значений AG° соответствует быстрое увеличение /С. Так, изменению свободной энергии AG°, равному —42 кДж/моль (—10 ккал/моль), соответствует значение константы равновесия, равное ~107, т. е. исходные вещества практически пол-
44
иостью превращаются в продукты реакции. Знание стандартных свободных энергий исходных веществ и продуктов, которые определены для большого числа органических соединений, дает возможность предсказать ожидаемую степень превращения исходных веществ в продукты.
Вклад члена А/Лв изменение свободной энергии может быть оценен по разности энергии связей исходных веществ и продуктов; приблизительное значение А// для данной реакции часто может быть предсказано, исходя из табличных значений стандартных энергий связи. Вряд ли неожиданным является тот факт, что в первую очередь на основании данных А// были составлены таблицы средних значений энергий связи!
Энтропийный член не может быть объяснен так же просто, но его обоснованно связывают с числом возможных путей, с помощью которых суммарная энергия системы может быть распределена между составляющими ее молекулами, а также с числом возможных способов, при помощи которых энергия каждой молекулы может быть распределена на поступательную, вращательную и колебательную составляющие (из которых поступательная, вероятно, самая большая). Так, для реакции А В + С, в ходе которой происходит увеличение числа частиц при переходе от исходных веществ к продуктам реакции, должна, вероятно, заметно возрастать энтропия из-за увеличения поступательных степеней свободы. В таких случаях член —TAS может оказаться достаточным для того, чтобы уравновесить член -(-А// в эндотермической реакции и обеспечить общее уменьшение свободной энергии AG; в результате состоянию равновесия будет соответствовать высокая концентрация продуктов. Если же реакций экзотермична (член А// отрицателен), то AG будет, конечно, даже более отрицателен, а константа равновесия /С соответственно еще больше. Когда число реагирующих частиц уменьшается при переходе от исходных веществ к продуктам реакции, энтропия уменьшается (член AS отрицателен):
А + В С Дб = ДН — (—) Г ДЗ.
Поэтому до тех пор, пока реакция не будет достаточно экзотермична (член А// отрицателен и довольно велик), чтобы уравновесить это снижение AS, значение AG будет положительным и равновесие будет смещено в сторону исходных веществ.
Реакции циклизации, например циклизация алкенов, также могут сопровождаться уменьшением энтропии:
Н2С—СН2
СН3(СН2)3СН==СН2 +=> Н2С\ /СН2
Н2С—сн2
45
В этом случае поступательная составляющая энтропии может и не изменяться, однако вращательная составляющая уменьшается, так как в циклическом продукте, в отличие от линейного углеводорода, сильно ограничено вращение вокруг простой уг-лерод-углеродиой связи. Вращательный компонент энтропии, однако, меньше, чем поступательный компонент, что проявляется в реакциях, где число реагирующих частиц уменьшается при образовании продуктов. Это положение иллюстрируется преимущественным образованием внутримолекулярных водородных связей по сравнению с межмолекулярными водородными связями в случае 1,2-диолов:
внутримолекулярная
водородная связь
межмолекулярная
водородная связь
Не следует упускать из виду, что энтропийный член (TAS), в отличие от энтальпийного члена (А//), включает температуру, поэтому их относительные вклады в изменение свободной энергии для одной и той же реакции зависят от температуры ее проведения.
2.2. Кинетика реакции
Отрицательное значение AG° является необходимым условием для того, чтобы реакция вообще протекала в данных условиях. Однако этого условия недостаточно, так как оно ничего не говорит о том, насколько быстро исходные вещества превращаются в продукты реакции. Так, для реакции окисления целлюлозы:
(CeHioOs)^ Т" 6/zOg 6/zCOa Т- блНдО
значение AG° отрицательно, а абсолютное ее значение достаточно велико, в результате чего равновесие практически полностью смещено в сторону продуктов СОг и НгО, однако газету (в основном состоящую из целлюлозы) можно читать на воздухе (и даже в кислородной палатке!) в течение длительного времени без опасности превращения ее в газообразные продукты, так как скорость превращения целлюлозы в этих условиях чрезвычайно мала, хотя, конечно, увеличивается при более высоких температурах. Превращение исходных веществ в продукты реакции, несмотря на отрицательное значение AG°, почти никогда не протекает гладко (рис. 2.2); обычно на пути к продук-
46
там реакции молекулам исходных веществ приходится преодолевать энергетический барьер (рис. 2.3).
2.2.1. Скорость реакции и свободная энергия активации
Точка х на энергетической диаграмме (см. рис. 2.3), характеризующей изменение свободной энергии в ходе реакции, соответствует наименее устойчивой конфигурации, через которую молекулы исходных веществ проходят на пути их превращения в продукты; эта конфигурация носит название активированного комплекса, или, как его чаще называют, переходного состояния. Следует подчеркнуть, что переходное состояние — это очень неустойчивое состояние, через которое реагирующие молекулы проходят в процессе реакции, а не дискретная молекулярная форма, или промежуточное соединение, которое реально может быть определено и даже выделено (ср. разд. 2.3.4). Примером переходного состояния может служить состояние (1) при щелочном гидролизе бромметана, в ходе которого связь НО—С образуется еще до полного разрыва связи С—Вг, и три атома водорода, связанные с атомом углерода, проходят через конфигурацию, в которой все Они лежат в одной плоскости (под прямым углом к плоскости бумаги):
Н
но"+ н
' н Н 4
6- \ Z 5-НО—С—Вг —
Н
< + Вг~
Н
(D
Механизм этой реакции подробно рассмотрен ниже. Высоту барьера AG* (см. рис. 2.3) называют свободной энергией активации для данной реакции (чем выше этот барьер, тем медленнее идет реакция); можно считать, что она состоит из энтальпийного (А/7*) и энтропийного (Т AS*) членов:
AG* = \Н* - Т AS*.
47
Величина А/7* (энтальпия активации) соответствует энергии, необходимой для растяжения или даже разрыва связей, т. е. энергии, обеспечивающей возможность протекания реакции [например, растяжения связи С—Вт в переходном состоянии (1)J. Таким образом, реагирующие молекулы в момент столкновения должны обладать минимальным запасом энергии, для того чтобы реакция стала возможной (часто эту энергию называют просто энергией активации, ЕаКт, но связывают ее с величиной AW*- Хорошо известное увеличение скорости реакции при повышении температуры обусловлено по существу возрастанием доли молекул, запас энергии которых превышает требуемый минимум.
Значение Еакт для данной реакции может быть подсчитано на основе константы скорости реакции k (ср. разд. 2.2.1), определяемой экспериментально при двух различных температурах Ту и Т2, с использованием уравнения Аррениуса:
k = Ae~E/RT или lgA = — £акт/(2,303/?Г) + 1g Л,
где R — универсальная газовая постоянная, равная 8,32 Дж/(моль-град); А —константа реакции, не зависящая от температуры и пропорциональная той части столкновений от общего числа столкновений между реагирующими молекулами, которые приводят к успешному превращению в продукты реакции.
Энергия активации может быть получена графически в координатах lg k—1/Т или путем превращения приведенного выше уравнения в уравнение вида
1g (*i/M = — 2,303/? (у?-??)
и последующих расчетов.
Член AS* (энтропия активации) характеризует упорядоченность системы. Это мера изменения в степени организации, или упорядоченности, как самих молекул, так и распределения энергии между ними при переходе от исходных веществ к переходному состоянию; энтропия активации AS* определяет член А приведенного выше уравнения Аррениуса. Если для достижения переходного состояния необходима высокая степень организации в момент сближения реагирующих молекул друг с другом, а концентрация их энергии в отдельных связях может оказаться такой, что приведет к их окончательному разрыву, тогда достижение такого переходного состояния должно сопровождаться заметным уменьшением энтропии (неупорядоченности) и самой вероятности его образования.
48
2.2.2. Кинетика и скоростьлимитирующая стадия
Экспериментальное исследование кинетики реакции состоит в измерении скоростей исчезновения исходных веществ и (или) появления продуктов реакции при данной (постоянной) температуре и в установлении зависимости скорости реакции от концентрации одного (или всех) реагирующего вещества. Ход реакции можно контролировать различными методами, например непосредственно, отбирая аликвотные части реакционной смеси и проводя титриметрическое определение, или косвенно, наблюдая колориметрические, кондуктометрические, спектроскопические и другие изменения. В каждом случае наиболее важной частью исследования является поиск зависимости полученных кинетических данных от концентрации; это осуществляют графически или путем расчетов. Так, для реакции
СН3Вг + ‘ОН —► СН3ОН + ВГ
кинетическое уравнение имеет вид:
Скорость = &[СН3Вг] ['ОН].
Величину k называют константой скорости данной реакции. Эту реакцию в целом следует считать реакцией второго порядка, но по отношению отдельно к СН3Вг и ~ОН она является реакцией первого порядка.
Такое совпадение стехиометрии и кинетического уравнения бывает довольно редким; обычно кинетическое уравнение можно вывести только с помощью экспериментально полученных данных. Так, для катализируемой основаниями реакции бромирования пропанона (ацетон)
-он
СН3СОСН3 + Вг2 ---СН3СОСН2Вг + НВг
находим следующее уравнение скорости реакции:
Скорость = &[СН3СОСН3] [”ОН],
т. е. ее скорость не зависит от концентрации брома, но зависит от концентрации ионов ~ОН (ср. разд. 10.5.6). Ясно, что, поскольку бром входит в состав конечного продукта, он должен участвовать в какой-либо из стадий реакции; но, очевидно, концентрация брома не влияет на стадию, скорость которой мы реально измеряем. Таким образом, суммарная реакция должна включать по крайней мере две стадии: в одной из них, скорость которой мы измеряем, бром не участвует, а в другой — участвует. В действительности очень немногие органические реакции являются одностадийными процессами, как это показано на рис. 2.3. Это можно показать на примере образования гексаметилентетрамина:
6СН2О + 4NH3 —► C6H12N4 + 6Н2О
49
Вероятность одновременного столкновения шести молекул СН2О и четырех молекул NH3 с образованием комплекса из десяти молекул практически равна нулю. Но даже в более простых случаях реакции обычно проходят через ряд последовательных стадий (часто с участием двух частиц), из которых мы определяем самую медленную, т. е. скорость-лимитирующую стадию * реакции, являющуюся как бы кинетическим «бутылочным горлышком» иа пути превращения исходных веществ в продукты реакции.
Как показано на рис. 2.4, исходные вещества превращаются через переходное состояние Xi в промежуточное соединение, которое затем распадается иа продукты реакции через второе переходное состояние х2. Видно, что для образования промежуточного соединения через х{ необходимо больше энергии (AG^AG?), чем для второй стадии, поэтому первая стадия должна быть более медленной, т. е. это именно та стадия, скорость которой реально измеряют в кинетических экспериментах. За этой стадией следует быстрое (требующее меньше энергии), ие лимитирующее скорость всей реакции превращение промежуточного соединения в продукты. Рассмотренное выше бромирование пропанона при определенных условиях может протекать по идеализированному пути, показанному иа рис. 2.4, во время которого медленное (скоростьлимитирующее) удаление протона под действием основания приводит к образованию промежуточного карбаииоиа (2), который претерпевает быструю (ие лимитирующую скорость) атаку бромом с образованием бромпропа-нона:
НО^Н Вг-^Вг вг
CH2COCH3-_°Hq > 4)Н2СОСНз-£^Ц>- СН2СОСН3 + Вг~ медленно (2)
Следует подчеркнуть, что хотя это объяснение соответствует кинетическому уравнению, полученному иа основе эксперимен
* Эту стадию часто называют «лимитирующей стадией». Имея в виду, что протекание органической реакции может лимитировать как кинетический, так и термодинамический фактор (см. разд. 2.2.3), при подготовке перевода было решено использовать термин «скоростьлимитирующая стадия», точнее соответствующий применяемому автором термину «rate-limiting step». — Прим. ред.
50
тальных данных, тем не менее это уравнение нельзя рассматривать как однозначное доказательство правильности этого объяснения. Полученное экспериментальным путем кинетическое уравнение содержит информацию о частицах, участвующих в реакции на скоростьлимитирующей стадии; это уравнение в действительности определяет состав, а не структуру переходного состояния скоростьлимитирующей стадии. Это уравнение не несет непосредственной информации ни о промежуточных соединениях, ни о частицах, которые участвуют в быстрых процессах, не являющихся скоростьлимитирующими.
При рассмотрении возможного влияния изменения условий, например растворителя, или структуры исходных веществ на скорость реакции следует выяснить, какой эффект эти изменения будут оказывать на устойчивость (свободную энергию) переходного состояния: любые факторы, способствующие стабилизации, должны привести к более быстрому образованию переходного состояния, и наоборот. Детальную информацию о богатых энергией переходных состояниях удается получить очень редко; самое лучшее, что можно реально сделать, — это рассматривать соответствующие промежуточные соединения как модели переходных состояний и выяснить, какой эффект можно ожидать в результате упомянутых изменений при использовании этих моделей. Такой подход не является бессмысленным; образующееся промежуточное соединение (см. рис. 2.4) достаточно jopoino соответствует (в рамках уровней свободной энергии) предшествующему ему переходному состоянию и может иметь сходную с ним структуру. Несомненно, что такое промежуточное соединение является гораздо лучшей моделью переходного состояния, чем исходное вещество. Например, а-комп-лексы (интермедиаты Уиланда) в реакциях электрофильного замещения в ароматических соединениях используют как модели переходных состояний, являющихся их непосредственными предшественниками (см. разд. 6.7.1.1).
В присутствии катализатора скорость реакции увеличивается, так как при этом становится возможным протекание реакции по альтернативному пути/требующему меньше энергии, часто через образование нового, более устойчивого (с более низкой энергией) промежуточного соединения (рис. 2.5).
Так, гидратация алкена при непосредственном действии воды
ОН
Хчс=с// + н2о —>- ^С— / \ / |Х
н
протекает обычно крайне медленно, но она может быть
51
ПС для мекаталазируемой реакции
Исходные веществ
Продукты
€ для скорость-лимитирующей стадии катализируемой реакции
значительно ускорена добавлением кислотного катализатора, который на первой стадии протонирует алкен до промежуточного карбокатиона. За этой стадией следует быстрая атака положительно заряженного карбокатиона молекулой воды, выступающей в качестве нуклеофила; гидратация завершается отщеплением прото
на, способного опять функционировать в качестве катализатора (см. разд. 7.4.2):
Рис. 2.5.
Механизм кислотно-основного катализа рассмотрен в разд. 3.3.
2.2.3. Кинетический и термодинамический контроль
Если исходные вещества могут превращаться в два или большее число альтернативных продуктов, как, например, при электрофильной атаке ароматических соединений, уже содержащих какой-либо заместитель (см. разд. 6.7), соотношение между образующимися альтернативными продуктами часто определяется относительными скоростями их образования: чем быстрее образуется продукт, тем выше его содержание в смеси конечных продуктов. Поскольку в этом случае состав смеси продуктов определяется кинетическими параметрами, соответствующий контроль называют кинетическим. Однако этот тип контроля не всегда имеет место. Если одна или большее число альтернативных реакций обратимы или если образующиеся альтернативные продукты легко превращаются друг в друга непосредственно в условиях протекания реакции, то состав смеси конечных продуктов может определяться уже не относительными скоростями образования различных продуктов, а их относительными термодинамическими устойчивостями в реакционной системе; такой контроль носит название термодинамического, или равновесного. Так, показано, что нитрование толуола контролируется кинетически, тогда как алкилирование его по Фриделю — Крафтсу— термодинамически (см. разд. 6.8). Тип контроля, который проявляется в реакции, может зависеть в ряде случаев от условий проведения реакции; например, сульфирование нафталина
52
концентрированной H2SO4 при 80 °C контролируется в основном кинетически, в то время как при 160 °C — термодинамически (см. разд. 6.9).
2.3. Исследование механизмов реакций
Детальную информацию о структурных, термодинамических и стереохимических особенностях протекания той или иной реакции получить чрезвычайно трудно; никакой механизм реакции точно доказать невозможно. Тем не менее можно собрать достаточное количество данных, чтобы показать, что один (или более) из теоретически возможных механизмов не согласуется с экспериментальными результатами и (или) что один из нескольких оставшихся альтернативных механизмов более вероятен, чем другие.
2.3.1 Идентификация продуктов реакции
Возможно, что наиболее полную информацию о механизме реакции получают при установлении структуры образующихся продуктов и при сравнении их структуры со структурой исходных веществ. Если, как это часто бывает в случае органических реакций, образуются несколько продуктов, необходимо определить соотношение этих продуктов и определить, какому контролю — кинетическому или термодинамическому — подчиняется реакция. В прошлом на это затрачивали значительный труд, выделяя продукты вручную, сейчас это достигается более легко и точно с помощью весьма совершенных хроматографических методов или косвенным путем с помощью спектроскопических методов.
Установление точной структуры продуктов реакции имеет очень важное значение, однако это не всегда удается сделать. Например, трифенилметильный радикал (3) желтого цвета (ср. разд. 11.1), полученный в 1900 г. при действии серебра на три-фенилметилхлорид, легко образует бесцветный димер (мол. масса 486), который, как полагали, является гексафенилэтаном (4) с тридцатью «ароматическими» атомами водорода. Лишь спустя почти семьдесят лет (в 1968 г.) получили ЯМР-спектр (ср. разд. 1.3.6) этого димера (только с двадцатью пятью «ароматическими», четырьмя диеновыми и одним «насыщенным» атомами водорода) и доказали, что данный димер не может иметь строение гексафенилэтана, а фактически имеет строение (5):
^Х± РЬаС—СРЬа
(5) (3)
(4)
53
После установления правильной структуры многие частные детали поведения соединения (3) и его димера, которые ранее казались аномальными, сразу же стали понятны.
Сведения о строении продуктов реакции могут быть особенно ценными, если образуются совершенно неожиданные продукты. Например, показано, что реакция 4-метил-1-хлорбензола (п-хлортолуол) (6) с ионом ~NH2 в жидком аммиаке (см. разд. 6.10.3) приводит не только к ожидаемому 4-метилфенил-амину (п-толуидин) (7), но и к совершенно неожиданному 3-ме-тилфениламину (л-толуидин) (8), который фактически является основным продуктом:
С1
Me.
~nh2
NH3 (жидк.)
(6) (7) (8)
Очевидно, что л-толуидин не может быть получен из соединения (6) путем простого замещения и либо образуется по какому-то другому пути, чем тот, который ведет к продукту (7). либо, если оба продукта образуются через некоторый общий интермедиат, соединение (7) не может образоваться путем прямого замещения.
2.3.2. Кинетические доказательства
Хотя наибольший объем информации о механизмах реакции получен (и продолжает поступать) в результате кинетических исследований, тем не менее интерпретация кинетических данных (ср. разд. 2.2.2) не всегда столь проста, как может показаться с первого взгляда. Так, эффективно реагирующие частицы, концентрация. которых действительно определяет скорость реакции, могут отличаться от тех частиц, которые вводят в реакционную смесь и изменения концентрации которых пытаются измерить. Например, при ароматическом нитровании эффективно атакующей частицей является +МОг (ср. разд. 6.2), хотя в реакционную смесь вводят HNO3 и определяют изменение ее концентрации. Зависимость между концентрациями +NO2 и HNO3 может быть достаточно сложной и, следовательно, сложной окажется зависимость скорости реакции от концентрации HNO3. Таким образом, даже в тех случаях, когда механизм реакции сравнительно несложен, его выяснение на основании анализа наблюдаемых величин может оказаться не простой задачей.
54
Так, было найдено, что гидролиз алкилгалогенидов RHal в водном растворе подчиняется кинетическому уравнению:
Скорость = k i[ RHal ],
Однако неверно делать вывод, что в скоростьлимитирующей стадии не участвуют молекулы воды на том основании, что концентрация воды не входит в уравнение скорости. Вода используется как растворитель, она присутствует в очень большом избытке, и ее концентрация практически остается неизменной, независимо от того, участвует она реально в скоростьлимитирующей стадии или нет. Этот вопрос может быть, по-видимому, решен при проведении гидролиза в другом растворителе, например в НСО2Н, и при использовании гораздо меньших концентраций воды. Тогда может оказаться, что гидролиз подчиняется кинетическому уравнению:
Скорость = fc2[RHal] [Н2О].
Однако действительный механизм гидролиза мог существенно измениться при замене растворителя, так что можно так и остаться в неведении относительно того, что же происходит на самом деле при гидролизе алкилгалогенидов в водном растворе.
Огромное большинство органических реакций протекает в растворах, и сравнительно небольшие изменения в используемом растворителе могут оказывать глубочайшие влияния на скорости реакций и их механизмы. Особенно ярко это проявляется в тех случаях, когда промежуточные соединения имеют полярную природу, например при образовании входящих в ионные пары карбокатионов или карбанионов, поскольку такие частицы обычно создают вокруг себя окружение из молекул растворителя. Это окружение сильно влияет на устойчивость частиц (и легкость их образования) и зависит от состава и природы используемого растворителя, особенно от его полярности и способности сольватировать ионы. Реакции, протекающие с образованием радикалов (см. разд. 11.1), гораздо меньше зависят от природы растворителя (за исключением растворителей, способных реагировать с радикалами), но сильно зависят от добавления источников радикалов (например, пероксидов), веществ, поглощающих радикалы (например, хинонов), или от воздействия света, который может инициировать реакцию образования радикалов вследствие фотохимической активации, например:
hv
Вг2 ----► Вг • • Вг
Если на основе кинетического исследования показано, что реакция может протекать необычно быстрее или медленнее по сравнению с аналогичными реакциями в подобных условиях, то
55
можно предположить наличие другого или модифицированного механизма, отличающегося от общего механизма реакций такого типа. Так, на основе последовательности, найденной для наблюдаемых скоростей гидролиза хлорзамещенных метанов сильными основаниями в сравнимых условиях
СНаС1 » СН2С12 < СНС13 » СС14, можно предположить, что гидролиз трихлорметана протекает по иному механизму, чем гидролиз других хлорметанов (ср. разд. 9.9).
2.3.3. Изотопные эффекты
Во многих случаях важно узнать, разрывается ли данная связь или нет на скоростьлимитирующей стадии. Из простых кинетических данных нельзя получить такую информацию, и для решения этого вопроса необходимы дополнительные более совершенные подходы. Например, в случае разрыва связи С—Н ответ на этот вопрос может быть получен путем сравнения скоростей реакции исследуемого соединения (в одним и тех же условиях) и его точного аналога, в котором эта связь заменена связью С—D. Обе эти связи имеют одну и ту же химическую природу как связи, образованные изотопами одного и того же элемента, но их частоты колебаний, а следовательно, и их энергии диссоциации должны немного различаться, потому что массы атомов, образующих связь, различны: чем больше масса, тем прочнее связь. Это различие обусловливает, конечно, и различие в скоростях разрыва этих двух связей при сходных условиях: менее прочная связь С—Н должна расщепляться быстрее, чем более прочная связь С—D; согласно квантово-механическому расчету, максимальное различие скоростей йн/йо при 25 °C должно быть «7.
Например, найдено, что Ph2CHOH окисляется в 6,7 раз быстрее, чем Ph2CDOH, т. е. для этой реакции характерен первичный кинетический изотопный эффект, и разрыв связи С—Н должен, очевидно, происходить на скоростьлимитирующей стадии:
МпО", "он
Ph2CHOH --------> Ph2C=O
Однако в других случаях, например при нитровании бензола CgH6 (см. разд. 1.8) и гексадейтеробензола C6De, скорости реакций одинаковы; это означает, что разрыв связи С—Н должен произойти на такой стадии суммарного процесса, которая не является скоростьлимитирующей (ср. разд. 6.2).
Кроме пары водород — дейтерий первичные кинетические изотопные эффекты наблюдаются также и для других пар изотопов. Эти эффекты, однако, не столь выражены, поскольку различие относительных масс других пар изотопов не так ве
56
лико. В частности, получены следующие данные (при 25°C):
ЧМ) (|4) КИС!
,2сна1 + но~ —» 12СНаОН + Г Л =1,09 я ( с)
(37) (37) Х./35ГП
3SClCH2Ph + Н2О PhCH2OH + Н+ 35СГ = 1,0076
я(37С1)
Необходимо подчеркнуть, что первичные кинетические изотопные эффекты, получаемые экспериментально, имеют значения, промежуточные между максимальными расчетными и единицей (что соответствует отсутствию изотопного эффекта); данные об отсутствии изотопного эффекта также могут быть полезны, поскольку они дают важную информацию о расщеплении отдельных связей в переходном состоянии.
Изотопы могут быть использованы и для решения некоторых вопросов, относящихся к механизму реакции, но не связанных с кинетикой. Так, гидролиз сложных эфиров в водном растворе, приводящий к образованию кислоты и спирта, теоретически мог бы протекать путем разрыва связи алкил — кислород (а) или путем разрыва связи ацил — кислород (б):
О
О
О
б
RC—18ОН + Н—OR
II 1 I «
RC4-O-HR' + Н18ОН ------
RC—ОН + HI8O—R
б а
Если реакция протекает в воде, обогащенной тяжелым изотопом кислорода 18О, это приведет в первом случае к спирту, обогащенному 18О, и кислоте, не содержащей этот изотоп, тогда как во втором случае получают кислоту, обогащенную |8О, и обычный спирт. Показано, что при гидролизе большинства эфиров образуется кислота, обогащенная 18О, т. е. в этих условиях гидролиз протекает через разрыв связи ацил — кислород (см. разд. 8.6.1). Следует иметь в виду, что эти результаты могут иметь значение только в том случае, если ни спирт, ни кислота, образующиеся при гидролизе, не могут обменивать свои атомы кислорода на атом кислорода воды, обогащенной 18О, как было показано в приведенном выше случае. '
В подобных случаях для выяснения механизма реакции часто используют тяжелую воду D2O. Например, при изучении реакции Ка-нниццаро с участием бензальдегида (см. разд. 8.2.6)
О О О
II II -он II PhC—Н + PhC—Н -—* PhC—О' + PhCH2OH Н2О
(9)
возникает вопрос, является ли источником второго атома водорода, связанного с атомом углерода в молекуле образующегося
57
фенилметанола (бензиловый спирт) (9), растворитель (Н2О) или другая молекула бензальдегида. Оказалось, что если проводить реакцию в D2O, то PhCHDOH не образуется; это показывает, что источником второго атома водорода является не вода, а вторая молекула бензальдегида.
При изучении механизмов реакций используют также другие изотопные метки, например 3Н, 13С, 14С, 15N, 32Р, 35S, 37С1, 1311 и др. Основными трудностями при работе с изотопными метками являются: а) необходимость гарантии того, что метка включается только в требуемое положение (я) в исследуемом соединении; б) точное определение места вхождения метки в продукт(ы) после завершения исследуемой реакции.
Чрезвычайное увеличение селективности современных синтетических методов позволяет преодолеть первую трудность, но вторая долго оставалась главной, особенно при использовании изотопов углерода. Применение изотопов углерода имеет особое значение, потому что атом углерода присутствует во всех органических соединениях. В частности, изотоп 14С широко используют при исследовании механизмов биосинтеза — процессов, с помощью которых в живых организмах создаются чрезвычайно сложные молекулы.
Так, были все основания считать, что пентациклическое соединение стеригматоцистин (10), найденный в культурах некоторых грибов, строится постадийно из молекул этановой (уксусной) кислоты. Основное подтверждение этой гипотезы было получено при добавлении к подходящим культурам грибов иСНзСОгН или СНрСОгН соответственно (в раздельных опытах). По данным определения радиоактивности (14С — источник P-излучения) двух выделенных образцов стеригматоцистина С18Н12О6 было показано, что использование 14СН3СО2Н приводит к включению восьми атомов 14С, а использование СНз4СОгН —девяти атомов 14С. Остается, однако, открытым вопрос о том, где точно- в молекуле стеригматоцистина локализованы эти меченые атомы углерода:
14СН3СО2Н (а) или
СН‘4СО2Н (б)
(а) 814С-атомов
(б) 914С-атомов
Не так давно этот вопрос могли решать только с помощью чрезвычайно трудоемких и часто не дающих однозначных результатов опытов по селективному расщеплению, но с появлением спектроскопии ЯМР легко определяются все различия ме
58
жду изотопами. Изотопы 12С и 14С не дают сигнала ЯМР, но изотоп 13С, содержание которого в обычном углероде составляет ~ 1,11 %, дает сигнал ЯМР. С помощью соответствующих приборов стало возможным записать спектры ЯМР 13С любого углеродсодержащего соединения (потому что каждое из них сот держит 1,11 % 13С): каждый атом углерода или группа одинаково расположенных атомов углерода в данной молекуле дает четко различимый, отличающийся от других сигнал. Спектр ЯМР 13С обычного стеригматоцистина можно, таким образом, сравнить со спектрами молекул, полученных из раздельных опытов с использованием 13СН3СОгН или СНз3СОгН. Атомы углерода, которые (в каждом случае) дают усиленные сигналы 13С, могут быть тем самым идентифицированы:
Зная, какой из атомов углерода молекулы СН3СО2Н включается в определенные положения стеригматоцистина, можно сделать обоснованное предположение о механизме синтеза, реализуемом культурами грибов. Между прочим, проведенный анализ показывает также, что атом углерода группы *СН3О в молекуле стеригматоцистина образуется не из СН3СО2Н.
2.3.4. Идентификация промежуточных соединений
Наиболее конкретные сведения о механизме реакции дает выделение из реакционной смеси одного или большего числа промежуточных соединений. Так, например, в реакции Гофмана, в ходе которой амиды превращаются в амины
Вг2. _он
RCONH2 ------► RNH2
в определенных условиях удается выделить ЛГ-бромамид RCONHBr, его анион RCONBr- и изоцианат RNCO, что помогает выяснить суммарный механизм реакции. Конечно, необходимо однозначно установить, что выделяемые частицы действительно являются промежуточными соединениями, а не просто альтернативными продуктами реакции; для этого надо показать,
59
что эти частицы могут быть превращены в обычных условиях данной реакции в обычные продукты реакции со скоростью, по крайней мере, такой же, как скорость суммарной реакции в этих же условиях. Важно также установить, что эти реально выделяемые частицы непосредственно участвуют в реакции, а не просто находятся в равновесии с истинными промежуточными соединениями.
Часто совсем не удается выделить какие-либо интермедиаты, но это вовсе не означает, что они не образуются: просто они могут быть слишком неустойчивыми и короткоживущими, чтобы можно было их выделить. К заключению об их существовании часто приходят на основе физических, особенно спектроскопических, данных, полученных при исследовании реакции. Например, при образовании оксимов ряда карбонильных соединений в результате реакции с гидроксиламином (см. разд. 8.3.1)
R4 R\ ..
xC=O + NH2OH —► C=N, + H2O
r/ r/ xoh
характеристическая полоса группы C=O в ИК-спектре. исходного соединения быстро исчезает и может пропасть полностью до того, как начинает появляться характеристическая полоса группы C=N продукта реакции. Очевидно, образуется промежуточное соединение; на основании результатов последующих исследований считают, что это соединение—гидроксиаминоспирт (Н), который быстро образуется, а затем довольно медленно распадается с образованием продуктов реакции — оксима и воды.
R4 ^ОН
(И) Rz ^NHOH
Если можно предположить участие в реакции определенной частицы в качестве лабильного интермедиата, то эти предположения можно подтвердить, вводя в реакционную смесь реакционноспособные частицы, способные легко реагировать с предполагаемым промежуточным соединением. В этом случае лабильный интермедиат можно «поймать в ловушку» и выделить стабильную частицу, в которую он включен. Например, предполагали, что при гидролизе трихлорметана сильными основаниями (ср. разд. 2.3.2) лабильным интермедиатом является сильно электронодефицитный дихлоркарбен СС12 (см. разд. 9.9); ди-хлоркарбен был «пойман» путем введения в реакционную смесь электронодонорного цис-бутена-2 (12), а образовавшееся стабильное производное циклопропанона (13) затем было выде
60
лено; для его образования вряд ли можно представить себе какой-либо другой путь, чем тот, который приведен ниже:
С1
СС12 I
(12) (13)
Успешное исследование промежуточных соединений не только представляет несомненные свидетельства в пользу того или иного механизма реакции, но и может служить подтверждением предполагаемого строения соответствующих переходных состояний, для которых они часто оказываются хорошими моделями (ср. разд. 2.2.2) *.
2.3.5. Стереохимические доказательства
Информация о стереохимических изменениях, сопровождающих какую-либо реакцию, также может быть полезна для понимания ее механизма и может стать решающим критерием для выбора предполагаемого механизма реакции. Например, катализируемое основанием бромирование оптически активного стереоизомера кетона (14) ведет к оптически неактивному рацемическому продукту (см. разд. 10.5.6). Это свидетельствует о том, что реакция должна проходить через образование плоского промежуточного соединения, которое может подвергаться атаке с равной вероятностью с обеих сторон с образованием равных количеств двух зеркальных форм продукта.
. ВГ2. "ОН
PhCOCHMeEt --------► PhCOCBrMeEt
(14) (+) (±)
Аналогично, образование только транс-дибромида (16) при бромировании циклопентена (15) бромом в полярной среде свидетельствует о том, что реакция не является простым одностадийным присоединением молекулы брома к двойной связи, поскольку это должно было бы привести к цис-дибромиду (17). Присоединение должно быть, по крайней мере, двухстадийным
* Согласно постулату Хэммонда, геометрия переходного состояния похожа на геометрию тех веществ, к которым оно ближе по свободной энергии. Этот постулат особенно полезен при изучении реакций, протекающих с образованием промежуточных соединении. — Прим. ред.
61
процессом (ср. разд. 7.1).
Вг
(15) (16)
Вг Вг (17)
Такого рода реакции, которые протекают с образованием главным образбм (или полностью) одного стереоизомера из двух возможных, называют стереоселективными.
Как было показано, многие реакции элиминирования протекают намного легче с тем членом пары геометрических изомеров, в котором отщепляемые атомы или группы находятся в траке-положении по отношению друг к другу, чем с изомером, в котором эти группы находятся в цис-положении. Это можно видеть на примере различия в легкости реакций отщепления с участием ацетатов анти- и сик-альдоксимов, в ходе которых образуется один и тот же цианид *.
Ph^ /Н ph Ph^ /Н
С но" I С
II ------► С -«-------- ||
. легко ц| труднее .
МеСОС)/ N \эСОМе
анти син
Очевидно, это различие накладывает ограничения при решении вопроса о механизме реакции, поскольку ее результат противоречит основному принципу «химической ловушки», согласно которому отщепление двух групп происходит наиболее легко в том случае, когда они расположены ближе всего друг к другу:
Pl\
/C=N4
О? „ОСОЙej
Для того чтобы быть уверенным в том, что предполагаемый механизм описывает действительное течение данной реакции, недостаточно, чтобы он просто находился в соответствии с уже известными фактами; необходимо посмотреть, в какой мере этот механизм позволяет заранее предсказывать характер изменений скорости реакций или даже природы образующихся
* Речь идет о стереоэлектронных эффектах, подробно изученных на примере реакций элиминирования (см. разд. 9.4.1) и получивших надежное толкование в рамках орбитальных концепций (см. кн.: Deslongchamps Р. Stereo-electronic Effects in Organic Chemistry. Oxford: Pergamon Press, 1983. 375 p.). — Прим. ped.
62
продуктов при изменении условий протекания реакций или структуры исходных веществ. Таким образом можно выбрать механизм, который лучше других согласуется с имеющимися фактами. Обобщение результатов систематического исследования механизмов реакций вносит ясность в огромную массу разрозненной информации о равновесиях, скоростях реакций и относительной реакционной способности органических соединений. Далее мы рассмотрим ряд примеров, иллюстрирующих это общее положение.
Глава 3
СИЛА КИСЛОТ И ОСНОВАНИЙ
3.1. Кислоты.........................................................64
3.1.1. рКа -.................................................... 64
3.1.2. Кислотность органических соединений.......................65
3.1.3. Влияние растворителя . 67
3.1.4. Незамещенные алифатические кислоты........................68
3.1.5. Замещенные алифатические кислоты..........................70
3.1.6. Фенолы.................................................. 72
3.1.7. Ароматические карбеновые кислоты..........................73
3.1.8. Дикарбоновые кислоты......................................75
3.1.9. Зависимость рКа от температуры............................76
3.2. Основания . .............................................77
3.2.1. рКа, рК(ВН+) и рКа........................................77
3.2.2. Алифатические основания...................................78
3.2.3. Ароматические основания...................................81
3.2.4. Гетероциклические основания...............................84
3.3. Кислотно-основной катализ...................................... 86
3.3.1. Специфический и общий кислотный катализ...................86
3.3.2. Специфический и общий основный катализ....................88
Современные электронные теории органической химии оказались весьма полезными для многих корреляций свойств и структуры, в том числе и для объяснения относительной силы орга-4 нических кислот и оснований. Согласно определению Аррениуса, кислотами являются соединения, которые в растворе дают ионы водорода Н+, тогда как основания образуют гидроксид-ионы _ОН. Эти определения вполне приемлемы для реакций, идущих только в водных растворах, но поскольку кислотно-оснойные взаимодействия имеют чрезвычайно важное и практическое значение, то постепенно были сформулированы гораздо более общие концепции. Так, Бренстед определил кислоты как вещества, которые способны отдавать протоны, т. е. как доноры протонов,
63
а основания — как акцепторы протонов. Рассмотрим в качестве примера первую ступень ионизации серной кислоты в водном растворе:
h2so4 + н2о: =?=> н3о+ + hso;
кислота основание
сопря- сопряженное женная основание кислота
В этом случае вода действует как основание, принимая протон, и превращается в так называемую сопряженную кислоту Н3О+, тогда как H2SO4, отдавая протон, превращается в сопряженное основание HSOi-
Более обобщенная концепция кислот и оснований была предложена Льюисом, который определил кислоты как молекулы или ионы, способные к координации с неподелениыми парами электронов, а основания — как молекулы или ионы, которые имеют иеподеленные пары электронов, пригодные для такой координации (см. разд. 1.7). Кислоты Льюиса включают такие соединения, как трифторид бора (1), который реагирует с триметиламином с образованием твердой соли (т. пл. 128°C):
Ме3мГ> BF3^Me3N: BF3
(1)
Другие хорошо известные примеры кислот Льюиса — хлорид алюминия, хлорид олова(IV), хлорид цинка и т. д. Ниже мы остановимся на протонных кислотах, причем в первую очередь рассмотрим влияние структуры на силу органических кислот и оснований. Соединения, у которых ионизируется связь С—Н, будут рассмотрены позже (см. разд. 9.9).
3.1. Кислоты
3.1.1. рКа
Силу кислоты НА в воде, т. е. степень ее диссоциации, можно определить, рассматривая равновесие:
Н2О:+НА =<=> Н3О+ + А~
Константа такого равновесия выражается уравнением:
Аа = [Н3О+] Га"]/[НА]
Концентрация Н2О входит в это уравнение, так как вода присутствует в таком избытке, что ее концентрация заметно не изменяется. Необходимо иметь в виду, что Ка — константа кислотности данной кислоты в воде — только приблизительно выражается таким уравнением, поскольку в нем вместо активностей используются концентрации. Однако это допущение обосиован-
64
но, так как речь идет об очень разбавленном растворе. На константу кислотности влияют состав раствора, в котором находится кислота (см. ниже), а также другие факторы, но тем не менее она достаточно полно характеризует силу кислоты. Для удобства вместо Ка обычно используют величину рКа (рКа = = —1g Ка). Так, для этановой (уксусной) кислоты в воде при 25°C Ка = 1,79-10“5, а рКа = 4,76. Чем меньше численное значение рКа, тем сильнее соответствующая кислота.
Очень слабые кислоты, для которых рКа больше, чем ~ 16, в водной среде не проявляют себя как кислоты, поскольку концентрация Н3О+', которую они создают в воде, оказывается меньше, чем концентрация НзСН, создаваемая при автолизе самой воды:
Н2О + Н2О: Н3О+ + 'ОН
Вследствие этого относительные кислотности (рКа) слабых ’ кислот совсем не могут быть определены в воде. Если кислоты достаточно сильны (довольно низкие рКа), они практически полностью ионизируются в воде и будут, по-видимому, иметь одинаковую силу, например НС1, HNO3, НСЮ4 и т. д. Это явление называют выравнивающим эффектом воды.
Область сравнительного определения рКа может быть расширена при использовании в качестве растворителя как более сильных, так и более слабых оснований, чем Н2О. Проводя соответствующие измерения в ряде растворителей с увеличивающейся основностью (и используя в качестве эталона кислоту, чья кислотность находится почти у нижнего предела значений кислотности в одном растворителе и у почти верхнего предела — в другом растворителе), можно определить силу кислоты вплоть до таких слабых кислот, как метан (рКа ~ 43).
3.1.2. Кислотность органических соединений
Кислотность органического соединения НА зависит от прочности связи Н—А, от электроотрицательности А, от факторов, стабилизирующих А- по сравнению с НА, и от природы растворителя.
Первый из этих факторов обычно не является определяющим. Влияние электроотрицательности А отчетливо выражено, в частности, в том, что для метанола СН3ОН рКа ~ 16, а для метана рКа ~ 43, что обусловлено значительно большей электроотрицательностью кислорода по сравнению с углеродом. Для метановой (муравьиной) кислоты рКа = 3,77. Это отчасти обусловлено наличием электроноакцепторной карбонильной группы, усиливающей сродство к электрону того атома кислорода, к которому присоединен отщепляющийся протон; одиако гораздо важнее возможность стабилизации образующегося аниона
3 п. Сайкс
65
метаноата по сравнению с недиссопиированной молекулой метановой кислоты:
При этом наблюдается чрезвычайно эффективная делокализация заряда и, как следствие, —- стабилизация метаноат-анио-на, состоящего из двух одинаковых по энергии канонических структур. Делокализация возможна и в молекуле недиссоции-рованной метановой кислоты, однако она влечет за собой разделение заряда и, следовательно, гораздо менее эффективна. Это различие в стабилизации в определенной степени уменьшает тенденцию протона связываться с метаноат-анионом и смещает равновесие вправо так, что метановая кислота, по стандартам органической химии, является умеренно сильной кислотой.
В случае спиртов нет фактора, способствующего стабилизации алкоксид-аниона RO- по сравнению с молекулой самого спирта, поэтому спирты обладают значительно меньшей кислотностью, чем карбоновые кислоты. Однако у фенолов снова появляется возможность относительной стабилизации аниона (2) в результате делокализации его отрицательного заряда путем взаимодействия с л-орбиталями ароматического ядра:
Делокализация отрицательного заряда возможна и в недис-социированной молекуле фенола (см. разд. 1.5.2), однако, учитывая разделение заряда, она менее эффективна, чем в анионе (2), что ведет к некоторому снижению стремления аниона связываться с протоном. Действительно, фенолы являются более сильными кислотами, чем спирты (для самого фенола рКа = = 9,95), но значительно более слабыми, чем карбоновые кис
66
лоты. Это объясняется тем, что делокализация отрицательного заряда в карбоксилат-анионе осуществляется между одинаковыми по энергии структурами (см. выше), а два центра, участвующие в ней, являются сильно электроотрицательными атомами кислорода. В то же время в феноксид-анионе (2) структуры с отрицательным зарядом на атомах углерода ароматического кольца, вероятно, более богаты энергией, чем структура с отрицательным зарядом на атоме кислорода, и только один из центров, участвующих в делокализации, является сильно электроотрицательным атомом кислорода. Относительная стабилизация аниона по сравнению с недиссоциированной молекулой в случае фенола, вероятно, менее эффективна, чем в случае карбоновой кислоты, что и является причиной его меньшей относительной кислотности.
3.1.3. Влияние растворителя
На кислотность того или иного соединения кроме уже рассмотренных особенностей структуры большое влияние часто оказывает растворитель, особенно в том случае, когда, как это обычно бывает, растворителем является вода.
Недостатком воды как ионизирующего растворителя для органических соединений является низкая растворимость в ней неионизированных форм этих соединений. За исключением этого ограничения, вода — уникальный эффективный ионизирующий растворитель, что связано прежде всего с ее высокой диэлектрической проницаемостью (е = 80), а также со способностью сольватировать ионы. Чем выше диэлектрическая проницаемость (полярность) растворителя, тем ниже электростатическая энергия любой пары ионов, находящихся в нем, и, следовательно, тем легче возникают такие пары ионов, тем выше их стабильность в растворе и тем меньше склонность к взаимодействию друг с другом.
Ионы в растворе вызывают сильную поляризацию находящихся вблизи них молекул растворителя; вокруг ионов образуется, таким образом, сольватная оболочка. Чем выше степень такой сольватации, тем более эффективна стабилизация ионов вследствие распределения или делокализации их заряда. Молекулы воды чрезвычайно легко поляризуются, они относительно невелики по размеру; вследствие этого вода может эффективно сольватировать и таким путем стабилизировать как катионы, так и анионы. Стабилизация особенно заметна для анионов, поскольку их сольватация может сопровождаться образованием водородных связей (см. ниже). Аналогичная сольватация с образованием водородных связей невозможна для катионов, однако протон Н+, отщепляющийся от молекулы кислоты, также может сольватироваться, образуя водородные связи
3*
67
с молекулами воды:
Н—Y + пН2О
/И
но н—он
+ Н-У’Н но—н Ан
ч=± н—б
Поскольку молекулы спиртов, например МеОН, не слишком громоздки, то их способность к сольватации сходна со способностью воды. Показано, например, что НС1 является сильной кислотой и в метаноле. Не следует, однако, забывать, что основное требование к растворителю состоит в том, чтобы он мог функционировать как основание; чем слабее его основные свойства, тем ниже степень диссоциации кислоты. Так, легко убедиться, что, например, в метилбензоле (толуол) НС1 существует практически полностью в недиссоциированной форме.
3.1.4. Незамещенные алифатические кислоты
Как и следовало ожидать, замещение негидроксильного атома водорода в молекуле метановой (муравьиной) кислоты на алкильную группу приводит к образованию более слабой кислоты, поскольку электронодонорный индуктивный эффект алкильной группы должен понижать остаточное сродство к электрону атома кислорода, связанного с протонизируемым водородом, и, следовательно, уменьшать силу кислоты. В алкилзаме-щенном анионе повышенная электронная плотность на атоме кислорода способствует его рекомбинации с протоном по сравнению с системой метаноат-анион — метановая кислота:
/,0*1" Г У°" ~
Ме->С< Н—С?
X)J L ^0
Таким образом, следует ожидать, что равновесие в этой системе будет более смещено в сторону неионизированной формы, чем в системе метановая кислота — метаноат-анион. Действительно, для этановой кислоты рКа = 4,76, а для метановой кислоты рКа = 3,77. Однако степень структурного изменения, проявляющегося в такой небольшой молекуле, как метановая кислота, при замене атома водорода на метильную группу настолько велика, что приведенное выше простое объяснение может и не отражать основные причины наблюдаемого различия. Вполне возможно, что различная способность к сольватации в случае этих двух кислот объясняется заметным различием форм их молекул, а также различным распределением заряда.
Следует напомнить, что константа кислотности Ка связана с изменением стандартной свободной энергии ионизации AGP
68
уравнением
AG° = 2.303ЯГ 1g К.
и что ДО0 включает как энтальпийный, так и энтропийный члены:
AG° = ДЯ° - Т bS°.
Для ионизации этановой кислоты в воде при 25 °C (Ка = = I,79-I0-5) ДС° = 27,2 кДж (6,5 ккал), Д/У° =—0,5 кДж (—0,13 ккал) и Д3° = —92 Дж/град (—22 кал/град) [т. е. 7Д5 = —27,6 кДж (—6,6 ккал)], тогда как для метановой кислоты (Ка = 17,6-10-5) ДС° = 21 кДж (5,1 ккал), Д№ = — —0,3 кДж (—0,07 ккал), Д5° = —74 Дж/град (—18 кал/град) [т. е. ТДЗ =—21,3 кДж (—5,17 ккал)]. Столь малые значения Д№ почти полностью обусловлены тем фактом, что энергия, необходимая для диссоциации связи О—Н в не-диссоциированных карбоновых кислотах, компенсируется энергией, выделяемой при сольватации образующихся ионов.
Таким образом, различие в значениях ДС° для метановой и этановой кислот, обусловливающее различие их Ка, является результатом различия их энтропийных (Д£°) членов. Поскольку в каждом равновесии участвуют по два типа частиц с одной и другой стороны, различия в энтропии перехода при диссоциации будут невелики. Однако по одну сторону равновесия обе частицы являются нейтральными молекулами, а по другую — ионами. Поэтому основной вклад в величину Д3° вносит, судя по всему, большая степень упорядоченности сольватных оболочек молекул воды, окружающих ионы RCOj и Н3О+, по сравнению со степенью упорядоченности этих молекул в самой воде. Увеличение упорядоченности, однако, совсем не так велико, как можно было бы ожидать, поскольку в самой воде упорядоченность уже достаточно велика. Таким образом, различие в силе метановой и этановой кислот в действительности надо связывать с различной сольватацией анионов этих кислот, как это и предполагалось выше.
Дальнейшее введение алкильных групп в этановую кислоту оказывает гораздо меньший эффект. Этот эффект является по существу эффектом второго порядка, и его влияние на силу кислоты не всегда можно предсказать, поскольку определенную роль могут играть стерические и другие факторы. Значения рКа для ряда гомологов этановой кислоты приведены ниже:
Р*а Р*а
CH3CO2H 4,76 Ме3ССО2Н 5,05
МеСН2СО2Н 4,88 Ме(СН2)2СО2Н 4,82
Ме2СНСО2Н 4,86 Ме(СН2)3СО2Н 4,86
69
Если рядом с карбоксильной группой находится атом углерода, имеющий двойную связь, сила кислоты возрастает. Так, для пропеновой (акриловой) кислоты, СН2=СНСО2Н, рКа — = 4,25, тогда как для ее насыщенного аналога — пропановой кислоты — рКа = 4,88. Это связано с тем, что у ненасыщенного а-атома углерода, находящегося в $р2-гибридизованном состоянии, из-за относительно большего вклада s-орбиталей электроны оттянуты несколько ближе к ядру, чем у насыщенного атома углерода, находящегося в $р3-гибридизованном состоянии. Вследствие этого $р2-гибридизованные атомы углерода обладают меньшей способностью отдавать электроны, чем насыщенные $р3-гибридизоваиные атомы; именно поэтому пропеновая кислота, уступающая по силе метановой, все же несколько сильнее пропановой. Этот эффект еще больше выражен для sp'-гиб-ридизоваиного атома углерода тройной связи; так, значение рКа пропиновой (пропиоловой) кислоты CHsCCO2H равно 1,84. Аналогичная ситуация наблюдается и для атомов водорода в этене и этине; в этене атомы водорода имеют несколько более выраженные кислотные свойства, чем в этане, в то время как в этине атомы водорода являются настолько кислыми, что легко замещаются некоторыми металлами.
3.1.5. Замещенные алифатические кислоты
Еще более заметное влияние оказывает введение в молекулы алифатических кислот электроиоакцепториых заместителей. Так, введение галогена, индуктивный эффект которого действует в направлении, противоположном индуктивному эффекту алкильной группы, должно увеличивать силу галогензамещеиной кислоты, что действительно и наблюдается, как это видно из приведенных ниже значений рКЛ ряда кислот:
₽*а Рха
СН3н>-СО2Н 4,76 С1
F<-CH2<-CO2H 2,57
С1«-СН2«-СО2Н 2,86 С1«-СН<-СО2Н 1,25
Вг«-СН2«-СО2Н 2,90 С1 • t
1<-СН2<-СО2Н 3,16 С1ч-С«-СО2Н 0,65
4 С1
Относительное влияние различных галогенов соответствует ожидаемому: фтор является наиболее электроотрицательным (электроноакцепторным), и сила фторэтановой кислоты в 100 раз выше силы этановой кислоты. Индуктивный эффект атомов галогенов гораздо выше индуктивного эффекта, вызываемого (в противоположном направлении) введением алкильных групп, и дальнейшее введение атомов галогена в молекулу еще
70
более усиливает кислотность; так, трихлорэтановая кислота — уже очень сильная кислота.
Важно напомнить, что Ка (а следовательно, и р/Са) связана с AG° ионизации и что AG° включает как АД°, так и AS°. Было показано, что в ряду галогензамещенных этановых кислот АД° при переходе от одного соединения к другому изменяется незначительно. Наблюдаемое в этом ряду изменение AG° определяется главным образом различие в значениях Д5°, которое в свою очередь обусловлено влиянием атома галогена на делокализацию отрицательного заряда во всем анионе:
,0
F*-CH2*-C^
.О СНз^С: х'0_
В анионе галогензамещенной кислоты отрицательный заряд распределен более равномерно пр сравнению с незамещенным этаноат-анионом, заряд которого сконцентрирован в основном на группе СОг; это приводит к тому, что введение атома галогена облегчает возможность окружения иона молекулами воды, т. е. его сольватацию. Таким образом, ионизация галогензамещенных этановых кислот сопровождается меньшим снижением энтропии по сравнению с самой этановой кислотой. Этот эффект особенно сильно проявляется в случае CF3CO2H (рКа = 0,23), для которой AG° = 1,3 кДж (0,3 ккал), тогда как для СН3СО2Н AG° = = 27,2 кДж (6,5 ккал); в то же время значения ДД° для обеих кислот мало отличаются друг от друга.
Введение атома галогена в положение, более удаленное от карбоксильной группы, чем a-положение, оказывает гораздо меньшее влияние. Это объясняется тем, что индуктивный эффект быстро ослабевает при передаче вдоль насыщенной цепи, в результате чего отрицательный заряд становится все менее «размытым», оставаясь концентрированным в карбоксилат-ани-оне. В этом случае кислота все больше напоминает соответствующую незамещенную алифатическую кислоту, как это можно видеть из приведенных ниже значений рАа:
МеСН2СН2СО2Н 4,82 МеСНС1СН2СО2Н 4,06
С1СН2СН2СН2СО2Н 4,52 МеСН2СНС1СО2Н 2,84
Другие электроноакцепторные группы, например NR3, CN, NO2, SO2R, СО, CO2R, увеличивают силу простых алифатических кислот. Аналогичное влияние оказывают гидроксильные и метоксильные группы. Неподелеиные электроны атомов кислорода групп ОН и ОМе не способны проявлять мезомерный эффект в направлении, противоположном индуктивному эффекту, из-за наличия в молекуле насыщенного атома углерода.
71
Значения p/Ca соответствующих замещенных кислот приведены ниже:
O2N«-CH2«-CO2H 1,68 MeCO«-CH2«-CO2H 3,58
Me3N«-CH2<-CO2H 1,83 MeO«-CH2«-CO2H 3,53
NC<-CH2<-CO2H EtO2C^CH2<-CO2H 2,47 3,35 нд«-сн2<-со2н 3,83
3.1.6. Фенолы
Аналогичные эффекты можно наблюдать и для замещенных фенолов: наличие электроноакцепторных групп в ароматическом кольце повышает нх кислотность. При введении ннтрогруппы индуктивный эффект, как и предполагали, уменьшается с увеличением расстояния при переходе от орто- к мета- и пара-ни-трофенолам. Если ннтрогруппа находится в орто- или пара-по-ложенин (но не в лета-положении), то проявляется электроноакцепторный мезомерный .эффект, дополнительно усиливающий ионизацию из-за стабилизации (через делокализацию отрицательного заряда) образующегося аниона. Можно, таким образом, ожидать, что о- и «-нитрофенолы будут обладать большей кислотностью, чем лета-производные, что фактически и наблюдается:
Дальнейшее введение групп NO2 заметно усиливает кислотность; например, показано, что 2,4,6-тринитрофенол (пикриновая кислота) — очень сильная кислота. Значения рЛа соответствующих фенолов приведены ниже:
С6Н5ОН 9,95
o-O2NC6H4OH 7,23
jk-O2NC6H4OH 8,35
n-O2NCeH4OH 7,14
2,4-(O2N)2C6H3OH 4,01
2,4,6-(O2N)3CeH2OH 1,02
И в этих случаях значения ДН° для о-, м- и «-нитрофенолов очень близки, а различия AG° для этих фенолов обусловлены различиями в значениях TAS°, т. е. различиями в характере сольватации феноксид-анионов вследствие различного распределения в них отрицательного заряда.
72
Введение электронодонорных алкильных групп в ароматическое кольцо мало влияет на кислотность фенолов:
Р*а
Р*а
С6Н5ОН 9,95 л-МеС6Н4ОН 10,08
о-МеС6Н4ОН 10,28 п-МеС6Н4ОН 10,19
Алкилзамещенные фенолы являются несколько более слабыми кислотами по сравнению с фенолом, однако влияние алкильных групп выражено слабо и проявляется нерегулярно. Это свидетельствует о том, что при введении такого рода заместителей дестабилизация феноксид-иона (вследствие взаимодействия его отрицательного заряда с делокализованными л-орбита-лями ароматического кольца) меньше, чем можно было бы ожидать.
3.1.7. Ароматические карбоновые кислоты
Бензойная кислота (pAa = 4,20) является более сильной кислотой, чем ее насыщенный аналог — циклогексанкарбоновая кислота (рАа = 4,87). Это дает основание предположить, что фенильная группа, подобно двойной связи, является менее электронодонорной (по сравнению с насыщенной системой углеродных атомов) по отношению к карбоксильной группе из-за наличия $р2-гибридизованного атома углерода, присоединенного к карбоксильной группе (ср. разд. 3.1.5). Введение алкильных групп в ароматическое кольцо очень слабо влияет на силу образующейся кислоты (ср. с влиянием таких групп в фенолах, разд. 3.1.6):
Р*а
С6Н5СО2Н 4,20 л-МеС6Н4СО2Н 4,24 п-МеС6Н4СО2Н 4,34
Однако при введении электроноакцепторных групп, например группы NO2, кислотность увеличивается, причем этот эффект, как и в случае фенолов, наиболее сильно выражен при замещении в орто- и пара-положениях:
₽*а
СвН5СО2Н 4,20
o-O2NCeH4CO2H 2,17
л-О2ЫС6Н4СО2Н 3,45
n-O2NCeH4CO2H 3,43
3,5-(O2N)2CeH3CO2H 2,83
73
Особенно заметное влияние МО2-группы в орто-положении обусловлено, по-видимому, тем, что на очень коротком расстоянии индуктивный эффект действует сильнее всего, но нельзя исключать и прямое взаимодействие между соседними группами NO2 и СО2Н.
Наличие таких групп, как ОН, ОМе или галоген, обладающих электроноакцепторным индуктивным эффектом, но эл'ектро-нодонорным мезомерным эффектом в орто- или пара-положениях, может привести к тому, что n-замещенные кислоты окажутся слабее л-замещенных, а иногда и даже слабее исходной незамещенной кислоты, как, например, в случае п-гидроксибен-зойной кислоты [рХа незамещенной кислоты (X—Н) равно 4,20]:
рХа ХСвЩСОгН при X. равном
Н С1 Вг
о-Изомер 4,20 2,94 2,85
л-Изомер 4,20 3,83 3,81
п-Изомер 4,20 3,99 4,00
ОМе ОН
4,09 2,98
4,09 4,08
4,47 4,58
Следует отметить, что этот компенсирующий эффект становится более выраженным при переходе С1 ~ Вг->ОН, т. е. при возрастании способности атома, связанного с ароматическим кольцом, отделяться вместе со своей электронной парой.
Важно подчеркнуть, что здесь, как и в приведенных выше случаях, возможно, сказывается влияние различного распределения заряда в анионах на их сольватацию, т. е. на значение TAS0, связанное со степенью упорядоченности молекул растворителя в сольватной оболочке, что может стать причиной наблюдаемых различий в значениях рХа.
Поведение о-замещенных кислот, как отмечалось выше, часто аномально. Иногда они оказываются более сильными, чем можно было ожидать, исходя из предположения о прямом взаимодействии соседних групп. Так, внутримолекулярные водородные связи (ср. разд. 2.2) стабилизируют анион (4) о-гидрокси-бензойной (салициловой) кислоты (3) за счет делокализации его заряда, что нехарактерно ни для м- и n-изомеров, ни для
74
о-метоксибензойной кислоты.
Внутримолекулярные водородные связи могут, конечно, действовать в молекуле недиссоциированной кислоты так же, как и в ее анионе, но, вероятно, в последнем случае более эффективно, чем в первом (с соответствующей относительной стабилизацией), так как отрицательный заряд на атоме кислорода в анионе способствует образованию более сильной водородной связи. Этот эффект даже более выражен, когда водородные связи образуются с гидроксильными группами в обоих орто-положениях; так, для 2,6-дигидроксибеюзойной кислоты рЛа = 1,30.
3.1.8. Дикарбоновые кислоты.
Поскольку карбоксильная группа сама проявляет электроноакцепторный индуктивный эффект, можно ожидать, что присутствие второй такой группы в кислоте будет повышать силу кислоты. Это и наблюдается в действительности:
рка РКа
НСО2Н 3,77 НО2ССО2Н 1,23
СН3СО2Н 4,76 НО2ССН2СО2Н 2,83
сн3сн2со2н 4,88 НО2ССН2СН2СО2Н 4,19
С6Н5СО2Н 4,17 о-НО2СС6Н4СО2Н 2,98
jh-HO2CCsH4CO2H 3,46
п-НО2СС6Н4СО2Н 3,51
Данный эффект выражен весьма отчетливо, одиако он быстро уменьшается по мере того, как карбоксильные группы оказываются разделенными более чем одним насыщенным атомом углерода. цпс-Бутендиовая (малеиновая) кислота (5) (рЛа = = 1,92) гораздо сильнее, чем транс-бутендиовая (фумаровая кислота (6) (рЛа = 3,02); это объясняется стабилизацией ^«с-моиоаниона (малеат-иона) (7) путем образования внутримолекулярной водородной связи, что невозможно в случае
75
фумаровой кислоты (ср. с приведенной выше о-гидроксибензой-ной кислотой):
11 Нх^о
fll Н ц-с\ с=о 1*\ . о—н
(5) (6) (7)
В то же время вторая ступень диссоциации транс-бутендио-вой кислоты (рЛа — 4,38) протекает гораздо легче, чем в случае ^«с-кислоты (рДа — 6,23) вследствие большей трудности удаления протона из отрицательно заряженной циклической системы аниона (7), образующегося из малеиновой кислоты. Этандиовая (щавелевая), пропандиовая-1,3 (малоновая) и бутандиовая-1,4 (янтарная) кислоты уступают по второй ступени диссоциации метановой и пропановой кислотам соответственно. Это объясняется тем, что в случае дикарбоновых кислот второй протон приходится удалять из отрицательно заряженных частиц, содержащих электронодонорный заместитель, т. е. группу СОг, которая, как можно ожидать, дестабилизирует анион по сравнению с недиссоциированной кислотой в большей степени — эффект, который отсутствует при диссоциации незамещенной кислоты:
Z-0
О о С—* С Н о ’>“'> С i
/,0 СН3->С/
Ч)
3.1.9. Зависимость рДа от температуры
Как отмечалось выше (см. разд. 3.1.2), значение Да, а следовательно, и рДа для какой-либо кислоты не является отличительным свойством, присущим самой кислоте: оно зависит от используемого растворителя и от системы, составной частью которой является кислота. Значения рДа обычно приводят для водного раствора, если это специально не оговорено, поскольку большая часть данных получена для водных растворов, причем при температуре 25 °C. Температура должна быть точно установлена, поскольку константа равновесия изменяется в зависимости от температуры. Выше была рассмотрена относительная кислотность различных кислот и сделаны небезуспешные попытки установить ее зависимость от структуры кислоты. Однако сле
76
дует отметить, что в зависимости от температуры изменяются не только значения Ка кислот, но и относительная сила кислот. Так, этановая кислота—более слабая, чем а-этилбутановая кислота Е12СНСОгН при температуре < 30 °C, но более сильная при температуре > 30 °C. Такие обращения относительной кислотности в зависимости от изменения температуры довольно обычны, поэтому не стоит терять слишком много времени на корреляции между относительной кислотностью и структурой при 25 °C!
3.2. Основания
3.2.1. рКь, рК(ВН+) и рКа
Силу основания В: в воде можно оценить, рассматривая равновесие:
В : + НОН ^=fc ВН+ + 'ОН
Константа равновесия в воде Къ выражается уравнением:
Кь « [ВН+] ["ОН]/[В:].
Концентрация воды [Н2О] входит в Кь, поскольку вода является растворителем и ее концентрация заметно не изменяется; как уже говорилось ранее, концентрации можно использовать вместо более точных значений активностей при условии, что раствор достаточно разбавлен.
В настоящее время силу оснований также принято описывать в терминах Ка и рКа; тем самым устанавливается единая непрерывная шкала как для кислот, так и для оснований. Чтобы сделать это возможным, в качестве эталонной реакции для оснований используют равновесие
ВН+ + Н2О: 4=t В : + Н3О+,
для которого затем можно написать уравнение:
Ка«[В!][Н3О+]/[ВН+],
где Ка (и рКа) — мера кислотности сопряженной с основанием Вх кислоты ВН+. Эта мера легкости, с которой ВН+ будет отщеплять протон, является в то же время мерой затруднения, с которым основание Вх будет принимать протон; приходится принять, что чем сильнее ВН+ как кислота, тем слабее В: как основание. Таким образом, чем меньше численное значение рКа для ВН+, тем слабее В х как основание. Когда используют рК, чтобы указать силу основания В, соответствующие величины следует обозначать как рК(ВН+), но уже стало обычным, хотя это и некорректно, писать просто рКа.
77
Рассмотрим в качестве примера ион +NH4 (рЛа = 9,25): +NH4 + H2O: «=* NH3 + Н3О+
В этом случае ДО° = 52,7 кДж (12,6 ккал); Д№ — 51,9 кДж (12,4 ккал) и Д5° = —2,9 Дж/град (—0,7 кал/град) [т. е. TAS° =—0,8 кДж (—0,2 ккал)] при 25°C. Таким образом, положение приведенного выше равновесия эффективно определяется величиной ДД°, влиянием же величины Д5° можно пренебречь; этот результат заметно противоречит поведению многих кислот (см. разд. 3.1.4). Причина такого небольшого влияния Д59 состоит в том, что в этом случае в равновесии с каждой стороны участвуют по одной заряженной частице и по одному положительному иону, которые примерно одинаково влияют на степень упорядоченности окружающих их молекул воды, в результате чего энтропии их сольватации взаимно компенсируются.
3.2.2. Алифатические основания
Поскольку повышение силы азотистых оснований связано с возрастанием их готовности принимать протоны и, следовательно, с доступностью неподеленной пары электронов на азоте, можно было бы ожидать увеличения основности в ряду NH3-> RNH2 R2NH R3N в результате возрастания индуктивного эффекта при увеличении числа алкильных групп, что делает атом азота все более отрицательным. Однако в действительно-
сти это не так. Значения Ptfa некоторых аминов приведены
ниже: NH3 Me^NH2 9,25 10,64 Et->NH2 10,67
Me->NH«-Me 10,77 Et^-NH^Et 10,93
Me 4 Me-*N«-Me 9,80 Et 4 Et->N«-Et 10,88
Как и следовало ожидать, введение первой алкильной группы заметно увеличивает силу основания. Введение второй алкильной группы также увеличивает силу основания, но суммарный эффект от введения второй алкильной группы гораздо менее выражен, чем эффект от введения первой группы. Введение третьей алкильной группы приводит к третичному амину, ио фактически сила основания уменьшается. Это объясняется тем, что основность амина в воде определяется не только доступностью электронов иа атоме азота, но и тем, насколько катион, образовавшийся путем захвата протона, может подвергаться сольватации и таким образом стабилизироваться. Чем больше атомов водорода связано с атомом азота в катионе, тем больше возможностей для сильной сольватации в результате образова
78
ния водородных связей между атомами водорода и водой; стабилизация катионов уменьшается в ряду:
Н2О:-Н Н2О:-Н Н2О:-Н
1+ 1+ 1+
R—N—Н-:ОН2 > R—N—R > R—N—R
I ' I I
Н2О:-Н Н2О:--Н R
Таким образом, в ряду NH3-> RNH2-> R2NH-> R3N индуктивный эффект увеличивает основность, но последовательно снижающаяся стабилизация катиона в результате гидратации приводит к уменьшению основности. Суммарный эффект последовательного введения алкильных групп постепенно уменьшается, а при переходе от вторичного амина к третичному наблюдается фактическое обращение ряда основности. Если это объяснение верно, то подобного обращения не должно наблюдаться в тех случаях, когда основность измеряют в растворителе, в котором невозможно образование водородных связей. Действительно, в хлорбензоле был установлен следующий ряд основности бутил-аминов:
BuNH2 < Bu2NH < Bu3N
Значения рАа этих аминов в воде равны 10,61, 11,28 и 9,87 соответственно.
Известно, что при обработке четвертичных алкиламмониевых солей, например R4N+ 1~, влажным гидроксидом серебра AgOH получают растворы оснований, сравнимых по силе с минеральными щелочами. Это легко понять, поскольку образующееся при этом основание R4N+ -ОН должно быть полностью ионизированным: исключена возможность (имеющаяся у третичных и других аминов) перехода в неионизированную форму:
R3NH + “ОН —-+ R3N : + Н2О
Введение электроноакцепторных групп, например Cl, NO2, вблизи центра основности должно приводить к уменьшению основности вследствие электроноакцепторного индуктивного эффекта этих групп (ср. замещенные анилины, разд. 3.2.3). Так, амин (F3C<—)3N: практически обладает основными свойствами из-за наличия трех сильных электроноакцепторных СЕз-групп.
Изменение основности происходит при введении С=О, причем не только вследствие связывания атома азота, имеющего свободную пару электронов, с $р2-гибридизованным атомом углерода электроноакцепторной группы (ср. разд. 3.1.4), но и вследствие электроноакцепторного мезомерного эффекта:
О О'
II •• I +
R—C<-NH2 -м- R—С—-NH2
79
Известно, что амиды в воде являются очень слабыми основаниями [р/G этанамида (ацетамид) равно »0,5]. Если присутствуют две группы С=О, то соответствующие имиды, например бензолдикарбоксимид-1,2 (фталимид) (8) ие только "не имеют основных свойств, но часто обладают достаточно высокой кислотностью и образуют соли с щелочными металлами.
(8)
Влияние делокализации заряда на повышение основности амина можно видеть на примере гуанидина (9), который, за исключением гидроксидов алкиламмония, является одним из наиболее сильных органических азотистых оснований (рКа ~ 13,6). Как нейтральная молекула, так и катион (10), образующийся при ее протонировании, стабилизируются путем делокализации:
: NH2 +NH2 : NH2
I - II •• - I
HN=C—NH2 *—> HN—C—NH2 *—> HN—C=NH2 Нейтральная (9a) (96) (9в) молекула
: NH2 +NH2 : NH2 ^h+
H2N=C—NH2 -<-► H2N—NH2 -<-► H2N— <^=NH2 Катион
(10a) (106) (10b)
В катионе положительный заряд распределен симметрично вследствие вклада в гибридную структуру трех полностью эквивалентных по энергии структур. В нейтральной молекуле (где две структуры включают разделение заряда) делокализация не так эффективна. Таким образом, катион стабилизирован сильнее, чем нейтральная молекула, протонирование в целом оказывается «энергетически выгодным», вследствие чего гуанидин является чрезвычайно сильным основанием. Аналогичная ситуация наблюдается и в случае (11):
R R
I •• - I ♦
HN=C—NH2 -<—► HN—C=NH2 Нейтральная (11a) (116) молекула
+ I •• •• I +
H2N=C—NH2 H2N— C=NH2 Катион
(12a) (126)
80
Хотя нельзя ожидать, что стабилизация из-за делокализации в катионе (12) будет столь же эффективной, как и в катионе (10), однако этанамидин CH3C(=NH)NH2 (рКа — 12,4) является значительно более сильным основанием, чем этиламин МеС'НгМНг (рКа = 10,67).
3.2.3. Ароматические основания
Картина, обратная только что описанной, характерна для анилина (13), который является очень слабым основанием (рКа = 4,62) по сравнению с аммиаком (рКа = 9,25) или цик-логексиламином (рКа = 10,68). В анилине атом азота также связан с зр2-гибридизованным атомом углерода, однако, что более важно, неподеленная пара электронов атома азота может взаимодействовать с делокализованными л-орбиталями ароматического кольца:
(13а) (136) (13в) (13г)
В анилиний-катионе (14), образующемся при протонировании анилина любое такое взаимодействие, приводящее к стабилизации, оказывается запрещенным, поскольку электронная пара на атоме азота перестает быть свободной:
(14)
В результате молекула анилина более стабилизирована, чем анилиний-катион. Именно вследствие этого для анилина «энергетически невыгодно» присоединение протона. Эффект снижения основности, естественно, более выражен при замещении атомов водорода в NH2-rpynne анилина фенильными группами: дифениламин Ph2NH является крайне слабым основанием (рКа = = 0,8), а трифениламин Ph3N по обычным стандартам совсем не имеет основных свойств.
Введение алкильных, например метильных, групп в NH2-группу анилина приводит к небольшому увеличению рКа:
C6H5NH2 4,62
C6H5NHMe 4,84
C6H5NMe2 5,15
o-MeC6H4NH2 jK-MeC6H4NH2 n-MeC6H4NH2
4,38
4,67
5,10
81
В отличие от эффекта, наблюдаемого при введении алкиль-ных групп в алифатические амины (см. разд. 3.2.2), это увеличение основности постепенно нарастает. Предполагают, что стабилизация катиона посредством сольватации с образованием' водородных связей, которая была причиной необычного поведения алифатических аминов, в случае анилина имеет гораздо меньшее влияние на суммарный эффект. Основным фактором, определяющим основность алкилзамещенных анилинов, остается ме-зомерная стабилизация молекулы анилина (13), по сравнению с катионом (14), что подтверждается нерегулярным эффектом, оказываемым введением Me-групп в орто-, мета- и пара-положения анилина. Сходные нерегулярные эффекты были отмечены при введении Me-групп в орто-, мета- и лара-положения молекулы фенола (см. разд. 3.1.6).
Группа, обусловливающая более мощный (электроноакцепторный) индуктивный эффект, например NO2, оказывает гораздо более сильное влияние. Электроноакцепторность усиливается, если нитрогруппа находится в орто- или пара-положении, так как при этом взаимодействие неподеленной пары электронов аминного азота с делокализованной системой л-орбиталей ароматического кольца усиливается. В этом случае нейтральная молекула стабилизируется еще больше, чем катион, что ведет к дальнейшему ослаблению основности. Значения рКа нитроанилинов приведены ниже:
PhNH2 +4,62 jh-O2NC6H4NH2 +2,45
o-O2NC6H4NH2 —0,28 n-O2NC6H4NH2 +0,98
Дополнительный эффект, снижающий основность при введении заместителя в орто-положение, частично обусловлен малым расстоянием, на котором действует индуктивный эффект, а также прямым взаимодействием, как пространственным, так и путем образования водородных связей с МН2-группой (ср. случай о-замещенных бензойных кислот, разд. 3.1.7). о-Нитроанилин оказывается настолько слабым основанием, что его соли в значительной степени гидролизованы в водном растворе; 2,4-дини-троанилин нерастворим в водных кислотах, а 2,4,6-тринитроани-лин по свойствам напоминает амид. Его называют пикрамидом, и он легко гидролизуется до пикриновой кислоты (2,4,6-трини-трофенол).
82
Такие заместители, как ОН и ОМе, имеющие неподеленные пары электронов, могут проявлять электронодонорный, т. е. усиливающий основность, мезомерный эффект, если они находятся в орто- или пара-положении (но не в мета-положении):
Это приводит к тому, что n-замещенный анилин оказывается более сильным основанием, чем соответствующее м-замещен-ное. м-Замещенный анилин является более слабым основанием, чем сам анилин, вследствие электроноакцепторного индуктивного эффекта, проявляемого атомом кислорода заместителя. Довольно часто влияние о-заместителя оказывается несколько аномальным из-за прямого взаимодействия с МН2-группой с участием как пространственных, так и полярных эффектов. Замещенные анилины имеют следующие значения рКа:
PhNH2 4,62
o-HOC6H4NH2 4,72 o-MeOC6H4NH2 4,49
jh-HOC6H4NH2 4,17 jK-MeOC6H4NH2 4,20
n-HOC6H4NH2 5,20 n-MeOC6H4NH2 5,29
Интересно сопоставить основности 2,4,6-тринитро-.У,ЛЛдиме-тиланилина (15) и 2,4,6-тринитроанилина (16): основность первого почти в 40 000 раз (АрКа = 4,6) выше, чем второго (в отличие от МАЛдиметиланилина и анилина, основности которых близки). Это объясняется тем, что объем ЫМе2-группы достаточно велик и она может пространственно взаимодействовать с очень большими МО2-группами в обоих орто-положениях. Вращение вокруг связи атома углерода кольца с атомом азота группы NMe2 снижает стерические взаимодействия между атомами кислорода нитрогруппы и метильными группами фрагмента NMe2, однако при этом р-орбитали атомов азота становятся уже не параллельны р-орбиталям атомов углерода кольца. В результате этого мезомерный сдвиг неподеленной пары электронов от NMe2 к атомам кислорода NO2-rpynn с участием р-орбиталей атомов углерода кольца оказывается невозможным (см. разд. 3.2.3) и ожидаемого снижения основности (из-за мезомер-иого оттягивания электронов) не происходит (см. разд. 1.6)(. Тем самым снижающее основность влияние трех нитрогрупп
83
ограничивается по существу их индуктивными эффектами:
Напротив, в 2,4,6-трииитроанилине (16) NH2-rpynna сравнительно невелика по объему, что исключает вышеупомянутые пространственные ограничения: водородные связи между атомами кислорода o-NOz-rpynn и атомами водорода МН2-группы удерживают эти группы в требуемой плоской конформации. р-Орбитали могут принять параллельную ориентацию; тем самым становится возможным резкое снижение основности NH2-группы в результате очень сильного электроноакцепторного ме-зомерного эффекта трех NO2-rpynn:
(16а) (166)
3.2.4. Гетероциклические основания
Пиридин (17) является ароматическим соединением (см. разд. 1.3.6), в котором атом азота находится в $р2-гибридизо-ванном состоянии и вносит один электрон в систему из 6 л-элек-тронов (4n-|-.2; n= 1); при этом остается свободной пара электронов на атоме азота (расположенная на $р2-гибридной орбитали), вследствие чего пиридин и является основанием (рКа = ==5,21). Его основность, однако, гораздо ниже основности алифатических аминов (например, для Et3N: рКа = 10,75). Такая низкая основность характерна для оснований, в которых атом азота соединен кратными связями. Это объясняется тем, что по мере возрастания кратности связей у атома азота его неподе-ленная пара электронов располагается на орбитали, все больше приближающейся по характеру к s-орбитали. Такая электрон-
84
Ная пара смещается ближе к ядру атома азота, удерживается им более прочно, становится поэтому меиее доступной для образования связи с протоном, что и приводит к снижению основности соединения (см. разд. 3.1.4). Так, в соединениях, содержащих связи —Ns, /N: u=N:, например R3N:, C5H5N s,
RfeN s, неподеленные пары электронов занимают sp3-, sp2- и sp’-орбитали соответственно. Снижение основности иллюстрируется приведенными выше значениями рКа (для пиридина и триэтилалкина), а также очень низкой основностью алкилциа-нидов (для MeCN рКа = —4,3).
(17) (18) (19)
Однако в случае хинуклидина (18) неподеленная пара электронов также занимает $р3-орбиталь, а его рКа (10,95) очень мало отличается от рКа триэтиламина (10,75).
Пиррол (19), как известно, в некоторой степени обладает ароматичностью (хотя она выражена и не так сильно, как в случае бензола или пиридина) и не проявляет свойств сопряженного диена, как можно было бы ожидать при отсутствии ароматичности. Для достижения ароматичности 6 л-электронов (4/1 + 2; /1=1) атомов кольца должны заполнить три связывающие молекулярные орбитали (ср. разд. 1.3.6). Это делает необходимым вклад двух электронов атомов азота в общую 6 л-электронную систему. Образующееся электронное облако несколько деформировано в сторону атома азота из-за более высокой электроотрицательности этого атома по сравнению с атомами углерода, тем не менее электронная пара азота уже не настолько доступна, и атом азота не способен принимать протон:
(19а) (20)
В самом деле, протонирование пиррола в соответствующих условиях идет не по атому азота, а по а-углеродиому атому [см. формулу (20)]. Это объясняется включением свободной пары электронов атома азота в ароматическую систему 6 л-электронов, вследствие чего атом азота становится частично положительно заряженным. Он ие склонен присоединять протоны,
85
поэтому они и захватываются соседним а-углеродным атомом. Это напоминает уже рассмотренный случай с анилином (см. разд. 3.2.3) в том отношении, что катион (20) дестабилизирован по сравнению с нейтральной молекулой (19а)..В случае пиррола эта дестабилизация, однако, выражена гораздо сильнее; чтобы функционировать в качестве основания, пиррол должен полностью утратить ароматический характер, а следовательно, и стабилизацию; сравнение значений рХа пиррола (—0,27) и анилина (4,62) показывает, что пиррол является очень слабым основанием. Он может фактически функционировать как кислота, хотя и очень слабая, так как атом водорода группы NH может быть удален сильным основанием, например иоцом ~NH2; образующийся при этом анион (21), таким образом, сохраняет ароматический характер пиррола, в отличие от катиона (20) :
-nh2
(19а) Q
№ :NH
(21) (22)
Приведенные выше рассуждения не имеют, конечно, никакого отношения к полностью восстановленному пирролу — пирролидину (22), значение рХа для которого (11,27) очень близко к рКа для диэтиламина (11,04).
3.3. Кислотно-основной катализ
О катализе в гомогенном растворе уже упоминалось (см. разд. 2.2.2) как о процессе, делающем возможным альтернативный ход реакции с более низкой энергией активации, часто вследствие образования нового и более стабильного (с меньшим содержанием энергии) интермедиата. Несомненно, наиболее обычными и важными катализаторами в органической химии являются кислоты и основания.
3.3.1. Специфический и общий кислотный катализ
Простейшим случаем кислотного катализа является такой, когда скорость реакции пропорциональна концентрации ионов Н+ (например, Н3О+ в водной среде); при уменьшении pH скорость реакции увеличивается. Характерный пример — гидролиз простых ацеталей (см. разд. 8.2.2), например MeCH(OEt)2, скорость которого можно выразить уравнением:
Скорость = fe[H3O+] [MeCH(OEt)2].
Это случай специфического кислотного катализа, так как НзО4-—единственная кислотная частица, которая катализирует реакцию. Показано, что скорость реакции не зависит от добавления других возможных доноров протона (кислот), таких как
86
+NH4, при условии, что [НэО4-], т. е. pH, не изменяется при их добавлении. Полагают, что механизм гидролиза ацеталей можно представить схемой *, в которой быстрое необратимое протонирование субстрата предшествует медленной, скоростьлимитирующей стадии реакции:
OEt Со
Et / быстро / медленно; - EtOH
МеСН + Н30+ ч— * МеСН ----------------------------------
Х+ (скоростьлимитирующая
OEt X-OEt стадия)
Н --->. MeCH=OEt -------> MeCH = O + EtOH быстро
Известны также реакции, катализируемые не только Н3О+, но и другими кислотами, находящимися в реакционной системе. Примером такой реакции служит гидролиз ортоэфиров типа MeC(OEt)3 в присутствии кислоты НА:
Скорость = *НзО+[Н30+] [MeC(OEt)3] + 6НА [НА] [MeC(OEt)3],
Это пример так называемого общего кислотного катализа: катализ осуществляется донорами протонов вообще, а не только Н3О+. Общий кислотный катализ часто проявляется при более высоких pH. например при pH 7, когда [Н3О+] аг 10~7, в то время как [НА] может быть равна 1—2 моль/л. Общий кислотный катализ возможен и при более низких pH, однако в этом случае он может быть менее заметен из-за большого вклада Н3О+. Считают, что гидролиз ортоэфиров протекает по схеме (показана только НА, однако частицы Н3О+ ведут себя аналогично):
МеС—OEt
\х. Г*
О* Н—А
I
Et
медленно ------------->•
(скорость -лимитирующая стадия)
OEt
МеС^+
X. быстро OEt
+ EtOH + А-
Н2О
---МеС^
OEt
+ EtOH
Для общего кислотного катализа характерно, что протонирование субстрата протекает медленно (скоростьлимитирующая стадия), а превращение промежуточного соединения в продукты реакции идет быстро.
* Стрелку здесь и далее используют для того, чтобы показать суммарное превращение, которое протекает через несколько стадий.
87
3.3.2. Специфический и общий основный катализ
Как при кислотном катализе, в случае специфического Основного катализа скорость реакции пропорциональна pH; в данном случае она возрастает по мере увеличения pH, т. е. она пропорциональна концентрации ионов “ОН. Так, показано, что реакция, обратная альдольной конденсации (ср. разд. 8.4.4), подчиняется кинетическому уравнению:
Скорость = Л[ "ОН] [Ме2С(ОН)СН2СОМе],
которое позволяет выразить ход реакции следующим образом:
Ме2С С
СН2 Me
"ОН р?
: - L Ме2С С быстро \ Z \
СН2 Me
медленно
—-------------►
(скорость-лимитирующая стадия)
О
-->• Ме2С=О +
Н2С Me
По аналогии со схемой кислотного катализа, специфический основный катализ характерен для реакций, в которых стадия быстрого, обратимого удаления протона из субстрата предшествует медленной, скоростьлимитирующей стадии.
В случае общего основного катализа в реакции могут участвовать не только “ОН, но и другие основания. Например, при катализируемом основаниями бромировании ацетона (см. разд. 10.5.6) в ацетатном буферном растворе показано, что
Скорость = ^-он1~ОН] [МеСОМе] + ^меСоДМеСО'] [МеСОМе].
Предполагают, что реакция протекает по схеме:
Вг2;
•медленно -- ---------
(скорость—
лимитирующая
Вг + Вг~ I
стадия)
Вг—Вг
* х быстро
СН2—С—Me
В: = "ОН, МеСО ~
88
И опять же, по аналогии с кислотами, общий основный катализ характерен для реакций, в которых удаление протона из субстрата — медленная, скоростьлимитирующая стадия, за которой следует быстрое превращение промежуточного соединения в продукты.
Глава 4
НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ У НАСЫЩЕННОГО АТОМА УГЛЕРОДА
4.1. Кинетика и механизм реакции...................................90
4.2. Влияние растворителя..........................................93
4.3. Влияние структуры реагирующих соединений......................94
4.4. Стереохимические аспекты механизма реакций...................101
4.4.1. Механизм Sn2: обращение конфигурации . ..............101
4.4.2. Определение относительной конфигурации . ..............102
4.4.3. Механизм SnI: рацемизация?.............................104
4.4.4. Конкуренция механизмов 105
4.4.5. Механизм SNi: сохранение конфигурации..................106
4.4.6. Участие соседних групп: «сохранение» конфигурации .... 108
4.5. Влияние вступающих н уходящих групп..........................110
4.5.1. Вступающая группа......................................110
4.5.2. Уходящая группа........................................113
4.6. Другие реакции нуклеофильного замещения......................115
Нуклеофильное замещение у насыщенного атома углерода изучено более детально, чем реакции какого-либо другого типа, в основном благодаря фундаментальным работам Ингольда и его школы. Классическим примером реакции замещения является превращение алкнлгалогенида в спирт'под действием водного раствора щелочи:
HO’ + R—Hal —► НО—R + НаГ
Кинетические исследования реакций, в которых алкилгало-гениды атакуются различными нуклеофилами Nu:, показали, что имеются два типа таких реакций. Один из них следует кинетическому уравнению (4.1), а второй — кинетическому уравнению (4.2), в котором отсутствует член [Nu:], т. е. в этом случае скорость реакции не зависит от концентрации нуклеофила:
Скорость = k%[RHal] [Nu :] (4.1)
Скорость = &i[RHal] (4.2)
89
Найдено, что в некоторых случаях уравнение скорости относится к «смешанному» или осложненному механизму, но известны примеры, которые точно подчиняются приведенным выше простым уравнениям.
4.1. Кинетика и механизм реакции
Гидролиз первичного алкилгалогенида, например метилбро-мида (бромметан), в водном растворе щелочи, как было показано, протекает согласно уравнению (4.1), т. е. в стадии, лимитирующей скорость реакции (т. е. самой медленной), участвуют как алкилгалогенид, так и гидроксид-ион. Ингольд предположил существование переходного состояния, в котором частичное образование связи между атакующим гидроксид-ионом и реагирующим атомом углерода происходит до полного отщепления уходящего бромид-иона; при этом часть энергии, необходимой для разрыва связи С—Вг, возмещается за счет энергии, выделяющейся при образовании связи НО—С. С помощью квантовомеханического расчета показано, что наименьшая затрата энергии требуется в том случае, когда приближение гидроксидиона происходит вдоль линии, соединяющей центры атомов углерода и брома. Это можно представить следующей схемой:
и н°-с<,//Н н
НО“ +
Н
}с—Вг
Н“/ Н
Н
6- I 6-
НО с —-ВГ
У Н Н
+ Вг-
(П (2)
переходное состояние
В переходном состоянии отрицательный заряд распределяется, перемещаясь от гидроксид-иона к атому брома, а атомы водорода, соединенные с атакуемым атомом углерода, находятся в одной плоскости (под прямым углом к плоскости чертежа). $р3-Гибридизованный атом углерода становится в переходном состоянии $р2-гибридизованным, а НО и Вг оказываются связанными с двумя долями негибридизованной р-орбитали. Механизм такого типа Ингольд обозначил как Sn2 (Substitution Nucleophilic bimolecular — бимолекулярное нуклеофильное замещение).
Напротив, гидролиз третичного галогенида 2-метил-2-хлор-пропана (трет-бутилхлорид) (3) в щелочной среде, как было показано, описывается уравнением (4.2), и поскольку скорость не зависит от [~ОН], то гидроксид-ион не участвует в лимитирующей стадии реакции. Был сделан вывод, что скоростьлимитирующей стадией является медленная ионизация алкилгалогенида (на самом деле происходит завершение поляризации связи
90
R—>-На1, которая, как уже было показано, происходит в такого рода молекулах), что приводит к образованию ионной пары R+ С1~ (4); затем наступает быстрая стадия — атака ионом -ОН или, если возможно, растворителем, причем последний случай часто предпочтительнее из-за очень высокой концентрации растворителя:
Me
\ медленно
,..С—С1 -------->
1е' /
Me
(3)
Me
Me Me СГ (4)
Me
°- >но-с(
быстро p'Me
Me
Me н2о \
быстро
Me
х Me
-Ht \
+-ОН
мХ/
Me
С
Механизм реакции такого типа был обозначен SnI (Subslitu-tion Nucleophilic unimolecular — мономолекулярное нуклеофильное замещение). Энергия, необходимая для ионизации, компенсируется в основном за счет энергии, выделяемой при сольватации образующейся ионной пары. Значения энтропии активации AS* отражают предпочтительность такого процесса диссоциации (ср. разд. 2.2.2); например, AS* для гидролиза Ме3СС1 равна +51 Дж/(моль-К), тогда как для гидролиза СН3С1 она составляет —17 Дж/(моль-К). Катион ионной пары (4), в котором центральный атом углерода несет положительный заряд, является, конечно, промежуточным карбокатионом, и в ходе его образования первоначально $р3-гибридизованный атом углерода переходит в более стабильное плоское $р2-состояние, в котором три метильнце группы максимально удалены друг от друга. Атака ионом “ОН или молекулой растворителя (например, Н2О:) может происходить с обеих сторон этого плоского интермедиата. Если возникновение такого плоского интермедиата тормозится стерическими или какими-либо другими факторами (см. разд. 4.3), промежуточный карбокатион будет образовываться с трудом, если вообще сможет образоваться; в этом случае ионизация, а следовательно, и протекание реакции по механизму SnI не будут происходить.
Таким образом, наиболее характерное различие между реакциями, протекающими по механизмам Sn2 и SnI, состоит в том, что реакции типа Sn2 протекают только в одну стадию, через переходное состояние, в то время как реакции, протекающие по механизму SN1, — в две стадии, через реальное промежуточное соединение (карбокатион).
Некоторая путаница встречается как в учебниках, так и в другой литературе в отношении использования терминов
91
порядок и молекулярность в приложении к реакциям. Порядок— это экспериментально определяемая величина; общий порядок реакции равен сумме степеней членов (концентраций), входящих в кинетическое уравнение:
Скорость = £3[А] [В] [С] (третий общий порядок),
Скорость ==/г3[А]2[В] (третий общий порядок), Скорость = £2[А]2 (второй общий порядок).
В общем, однако, порядок относительно какого-либо реагента (или реагентов) более интересен и важен, чем общий порядок реакции; например, приведенные выше реакции имеют по отношению к А первый порядок. Известны также реакции нулевого и дробного порядков по отношению- к какому-либо реагирующему веществу.
Молекулярность относится к числу частиц (молекул, ионов и т. д.), которые участвуют в стадии расщепления и (или) об-, разования связей в одной и той же стадии реакции, обычно в скоростьлимитирующей стадии. Важно понять, что молекулярность нельзя определить экспериментально; она важна только в связи с определенным механизмом, выбранным для данной реакции, и при появлении дополнительных экспериментальных данных может быть пересмотрена, что ие должно иметь места с порядком реакции. Молекулярность реакции в целом важна только тогда, когда реакция протекает в одну стадию (элементарная реакция), как это предполагают, например, в случае гидролиза метилбромида (см. разд. 4.1). Порядок и молекулярность в этой реакции совпадают: реакция имеет второй общий порядок (первый порядок по отношению к каждому реагирующему веществу) и является бимолекулярной. Однако порядок и молекулярность не всегда или необязательно имеют одно и то же значение.
Однако простых кинетических измерений может быть недостаточно, чтобы установить, по какому нз двух механизмов, SnI или Sn2, действительно протекает, например, гидролиз галогенида. В частности, как было показано (см. разд. 2.3.2), если растворитель, например НгО, может действовать как нуклеофил (сольволиз), можно ожидать реакцию типа Sn2:
Скорость = A2[RHal] [Н2О],
но если концентрация воды остается фактически постоянной, реально наблюдаемое кинетическое уравнение будет иметь внд;
Скорость = /гнабл [RHalJ.
На основании простых кинетических измерений в водном растворе можно, таким образом, ошибочно предположить, что реакция протекает по механизму SnI.
92
Различие в кинетике между механизмами SN1 и SN2 часто может быть установлено при изучении влияния добавления конкурирующего нуклеофила, например азид-аниона Njj, на общую скорость реакции. Общая концентрация нуклеофила при этом увеличивается, и поэтому при механизме SN2, где концентрация нуклеофила входит в уравнение скорости, это приведет к повышению скорости реакции. Наоборот, при механизме SN1 величина [Nu s] не входит в уравнение скорости реакции, т. е. нуклеофил не участвует в скоростьлимитирующей стадии реакции, хотя, естественно, будет влиять на состав продуктов реакции.
4.2. Влияние растворителя
Растворитель, в котором протекает реакция, часто оказывает значительное влияние на ее скорость; замена растворителя может даже привести к изменению механизма реакции. Так, например, если гидролиз галогенида протекает по механизму SN1, то увеличение полярности растворителя (т. е. увеличение диэлектрической постоянной е) и (или) его сольватирующей способности приводит, как было показано, к очень заметному увеличению скорости реакции. Так, скорость сольволиза третичного галогенида Ме3СВг в 50 %-м водном этаноле в 3-104 раз больше, чем в чистом этаноле. Это происходит потому, что в случае механизма SnI в переходном состоянии образуется и концентрируется заряд, который отсутствует в исходном соединении:
б+ в-
R— Hal —► [R—На1]+ —► R+ НаГ
Энергия, необходимая для такого процесса, снижается при увеличении е; процесс облегчается также при повышении сольватации (и, следовательно, стабилизации) образующейся ионной пары по сравнению с исходным соединением, содержащим лишь ковалентные связи. То, что такие эффекты, в частности сольватация, имеют первостепенное значение, подтверждается тем фактом, что реакции типа SnI крайне редко идут в газовой фазе.
Для механизма Sn2, однако, увеличение полярности растворителя, как показано, имеет гораздо меньшее значение и приводит лишь к незначительному уменьшению скорости реакции. Это объясняется тем, что в данном случае не образуется новый заряд, а заряд, имеющийся в исходных реагентах, в переходном состоянии делокализован:
в- в-
Nu'+R—Hal —► [Nu - R - HalJ* —► Nu—R + Hal
В этом случае сольватация переходного состояния, вероятно, будет несколько менее эффективной, чем сольватация исходного нуклеофила, что приводит к некоторому уменьшению скорости.
93
Эти различия в скоростях реакции при изменении растворителя могут быть использованы для распознавания механизмов SN1 и Sn2.
Значительное влияние на скорость реакций Sn2 оказывает замена полярных гидроксилсодержащих растворителей на полярные, не содержащие гидроксильных групп растворители. Так, скорость реакции первичного галогенида Mel при добавлении N3' при О °C увеличивается в 4,5-104 раз при замене метанола (е=33) на А,А-диметилметанамид (диметилформамид, ДМФА) HCONMe2, имеющий практически такую же полярность (е = = 37). Такое большое различие в скорости возникает вследствие того, что атакующий нуклеофил N3 при проведении реакции в метаноле сильно сольватирован вследствие образования водородных связей (ср. разд. 3.1.3), в то время как в ДМФА он гораздо менее сильно сольватирован и не образует водородные связи. Несольватированный анион N3 (в ДМФА) является гораздо более сильным нуклеофилом, чем в окружении снижающей нуклеофильность сольватной оболочки (в МеОН), вследствие чего в первом случае скорость реакции возрастает. Увеличение скорости до 109 раз наблюдали при замене, например, МеОН в реакции SN2 на другой полярный апротонный растворитель— диметилсульфоксид (ДМСО) Me2SO (е = 46).
Таким образом, при замене растворителя происходит фактическое изменение механизма реакции. Увеличение полярности растворителя и способности сольватировать ионы может (ио необязательно), в частности, изменить тип реакции Sn2 на SnI. Замена гидроксилсодержащего растворителя на полярный, но апротонный растворитель (например, ДМСО) может изменить (и часто изменяет) тип реакции SnI на Sn2 вследствие сильного роста эффективности нуклеофила в этой системе.
4.3. Влияние структуры реагирующих соединений
Интересная закономерность наблюдается в реакции галогенидов (5) — (8) с основанием. Как известно, галогениды (5) и (8) легко гидролизуются, а соединения (6) и (7) более устойчивы.
СН3—Вг МеСН2—Вг Ме2СН—Вг Ме3С—Вг
(5) (6) (7) (8)
Кинетические кривые гидролиза этих галогенидов в разбавленном водно-спиртовом растворе гидроксида натрия приведены на рис. 4.1 *.
* По данным Ингольда; см.: Ingold С. К. Structure and Mechanism in Organic Chemistry (имеется русский перевод: Ингольд К. Механизм реакций и строение органических соединений: Пер. с англ./Под ред. И. Л. Кнунянца и Я. Ф. Комиссарова. М.: Мир, 1959. 673 с. — Прим, ред.)
94
Рис. 4.1.
С помощью кинетических исследований обнаружено, что порядок реакции и, возможно, механизм изменяются в точке пересечения кинетических кривых. Так, скорости гидролиза бромметана (5) * и бромэтана (6) описываются уравне- -нием реакции второго порядка. Скорость гидролиза 2-бромпропана (7) подчиняется уравнению реакции смешанного первого и второго порядков, причем относительная доля реакции второго порядка зависит от исходной концентрации ионов -ОН (чем выше эта концентрация,
тем больше доля реакции второго порядка); общая скорость реакции для этого соединения минимальна в рассматриваемом ряду. Гидролиз 2-бром-2-метилпропана (8), как показано, подчиняется кинетическому уравнению первого порядка.
Для того чтобы найти объяснение предполагаемому переходу от одного механизма к другому, необходимо рассмотреть в каждом случае влияние на переходное состояние как электронных, так и стерических факторов. В случае реакции Sn2 можно ожидать, что увеличение индуктивного эффекта с ростом числа метильных групп в рассматриваемом ряду соединений снижает положительный заряд на углеродном атоме, связанном с бромом, вследствие чего этот атом все труднее атакуется ионом -ОН. Это влияние, вероятно, невелико, и более важны стериче-ские факторы; так, _ОН все с большим трудом атакует связанный с бромом атом углерода при увеличении числа заместителей у этого атома. Еще более важно, что в переходном состоянии, отвечающем механизму Sn2, вокруг атома углерода должно находиться пять групп, тогда как в исходном галогениде — только четыре группы. Это ведет к увеличению «тесноты» при переходе от исходного галогенида к переходному состоянию, причем относительная «теснота» будет нарастать по мере увеличения размеров заместителей (Н->-Ме). Чем больше стерическая напряженность переходного состояния по сравнению с исходными соединениями, тем выше его энергия и тем медленнее оно образуется. Таким образом, следует ожидать, что скорость реакции, протекающей по чистому механизму Sn2, должна уменьшаться, когда приведенные выше кинетические кривые пересекаются. Фактически можно осуществить нуклеофильное замещение (Br~+RCl) в ряду галогенидов, аналогичных рассмотренным выше таким образом, чтобы реакции всех членов ряда строго подчинялись уравнению второго порядка (механизм Sn2). Относительные скорости 5ы2-реакций таких галогенидов приведены
95
ниже:
СН3С1 1 Ме2СНС1 4,9-10 4
МеСН2С1 27 • 10~2 Ме3СС1 2,2 10“5
В реакции, протекающей по механизму SnI, в переходном состоянии наблюдается значительное разделение заряда; образующуюся при этом промежуточную ионную пару часто принимают в качестве модели переходного состояния. При переходе от СН3Вг к МезСВг в приведенном выше ряду галогенидов увеличивается стабилизация карбокатиона ионной пары, т. е. увеличивается скорость образования переходного состояния. Это увеличение стабилизации обусловлено действием как индуктивного эффекта
Н—С—Н < Н—С—Н < Ме->С—Н < Ме.>Сч-Ме, | ф f
Н Me Me Me
так и эффекта сверхсопряжения (см. разд. 1.5.4) с участием атомов водорода, связанных с а-углеродиыми атомами, например:
Me—С—Me Me—С—Me
Н—СН2 Н+ СИ2
Карбокатионы приведенного выше ряда имеют 0, 3, 6 и 9 таких атомов водорода соответственно.
Подтверждением взаимодействия связей Н—С с атомом углерода, несущим положительный заряд, является тот факт, что замещение Н на D в исходном галогениде снижает скорость образования ионной пары на » 10 % в расчете на каждый внедренный атом дейтерия; этот результат сравним с примерами, когда в ионизацию вовлекаются только связи Н—С. Указанное явление называется вторичным кинетическим изотопным эффектом-, вторичным потому, что в данном случае разрывается другая связь — ие та, что содержит изотопную метку (см. разд. 2.3.3). Относительные вклады эффекта сверхсопряжения и индуктивного эффекта ие определены; важно то, что некоторые карбокатионы вообще могут образоваться только тогда, когда они могут принимать плоское строение, т. е. в состоянии, в котором сверхсопряжение может действовать наиболее эффективно (см. разд. 5.1.3).
Пространственные затруднения уменьшаются при переходе от исходного галогенида с тетраэдрическим расположением четырех заместителей вокруг $р3-гибридизоваииого атома углерода к карбокатиону с плоским расположением трех заместителей (ср. переходное состояние в случае механизма Sn2 с пятью заместителями) вокруг «р2-гибридизоваииого атома углерода. Эти три заместителя в плоском карбокатионе максимально удалены
96
друг от друга, н относительное снижение напряженности (при переходе от галогенида к карбокатиону) будет увеличиваться по мере увеличения размеров заместителей (Н -> Ме-> Ме3С). Можно было бы ожидать, что скорость реакции, протекающей по механизму SN1, будет заметно возрастать (на основании как электронных, так и стерических критериев) в приведенном выше ряду галогенидов в направлении слева направо. Оказалось, однако, невозможным подтвердить это экспериментально: не удается создать такие условия, чтобы каждый из галогенидов (5) — (8) реагировал в соответствии с механизмом SN1.
Таким образом, становится ясным, почему скорость реакции Sn2 уменьшается, а скорость реакции SN1 возрастает в ряду соединений, приведенных на рис. 4.1: причины изменения скоростей реакции заключаются в изменении механизма реакции.
Сходное, хотя и значительно более быстрое изменение механизма происходит и для соединений:
СН3—С1 СбН5СН2-С1 (С6Н5)2СН-С1 (С6Н5)3С-С1
(9) (Ю) (11) (12)
Так, скорость гидролиза фенилхлорметана (бензнлхлорид) (10) в 50 %-м водном ацетоне описывается уравнением смешанного второго и первого порядков, причем при использовании воды, не содержащей ацетон, реакция почти полностью идет по механизму SnI. Скорость гидролиза дифеннлхлорметана (11) описывается уравнением первого порядка, а общая скорость реакции очень сильно возрастает. Ионизация трифеннлхлорметана (тритилхлорид) (12) выражена настолько сильно, что раствор этого соединения в жидком SO2 обладает электрической проводимостью. Главная причина столь ярко выраженных примеров легкости ионизации и обусловленного ею перехода к механизму SnI в этом ряду состоит в значительной стабилизации карбокатиона путем делокализации его положительного заряда:
Это классический пример иона, стабилизированного путем делокализации заряда при содействии делокализованных л-орбиталей бензольного ядра (ср. отрицательно заряженный фено-ксид-ион; разд. 1.5.2). С увеличением числа фенильных групп у реакционного центра в галогенидах (11) и (12) этот эффект становится все более выраженным, возрастают возможности делокализации положительного заряда в соответствующих карбокатионах, вследствие чего протекание реакций по механизму SnI облегчается еще сильнее.
4 П. Сайкс
97
Показано, что группа СН2 в соединении (10) атакуется по механизму Sn2 практически с такой же скоростью, как и в соединении МеСН2С1. Это дает возможность предположить, что любое неблагоприятное стерическое затруднение в переходном состоянии, вызванное объемистой группой С6Н5 компенсируется небольшим электронным (индуктивным?) эффектом, ускоряющим реакцию.
Аналогичная стабилизация карбокатиона возможна и при гидролизе аллилгалогенидов, например 3-хлорпропена:
СН2=СН—СН2С1 —>- [СН2=СН—СН2 СН2—CH=CH2J СГ
Атака по механизму SnI в этом случае облегчается, и аллил-галогениды, подобно бензилгалогенидам, обычно более активны, чем, например, СН3СН2СН2С1 и С6Н5СН2СН2СН2С1, для которых подобная стабилизация карбокатиона невозможна. Атака по механизму Sn2 также облегчается по сравнению с СНзСН2СН2С1, возможно, из-за некоторого электронного эффекта двойной связи, ускоряющего реакцию, причем это ускорение не устраняется неблагоприятным стерическим эффектом, как и в случае объемистой С6Нз-группы в С6Н5СН2С1 (см. выше). Показано, что относительный вклад каждого из двух механизмов в суммарную реакцию зависит от определенных условий: для более сильных нуклеофилов предпочтительнее механизм Sn2 (см. разд. 4.5).
Наоборот, винилгалогениды, такие как хлорэтен СН2 = СНС1, и галогенбензолы, очень нереакционноспособны по отношению к нуклеофилам. Это следует из того факта, что атом галогена в этих соединениях связан с зр2-гибридизованным атомом углерода, в результате чего электронная пара связи С—С1 оттянута к углероду больше, чем в связях с участием зр3-гибридизован-ного атома углерода. Связь С—С1 в указанных соединениях более прочная и поэтому менее легко разрывается, чем в случае, например, СН3СН2С1; дипольный момент связи С—О, соседней с двойной связью, меньше, а следовательно, слабее тенденция к ионизации (SnI); положительный заряд на атоме углерода недостаточен, чтобы этот атом мог атаковаться ионом -ОН (Sn2); л-электроны двойной связи препятствуют близкому подходу атакующего нуклеофила. Двойная связь не может способствовать стабилизации ни переходного состояния, возникающего по механизму Sn2, ни карбокатиона, образующегося при механизме SnI. Очень многие из таких же соображений приложимы и к га-логеибензолам с их зр2-гибрндизованными атомами углерода и л-орбитальной системой ароматических колец; реакции с участием этих соединений, которые хотя часто и бимолекулярны, в действительности не являются просто реакциями Sn2 (см. разд. 5.2).
98
Влияние стерических факторов на механизм реакции особенно отчетливо наблюдается, когда замещение происходит в (3-положении. Ниже для ряда алкилбромидов приведены относительные скорости реакций (протекающих по механизму Sn2) с ЕЮ~ в EtOH при 55 °C:
СНз—СН2Вг МеСН2—СН2Вг Ме2СН—СН2Вг Ме3С— СН2Вг
(6) 1,0 (13) 2,8-10"1 (14) 3,0-10 2 (15) 4,2-10“®
Какие-либо различия в электронном эффекте групп Me через два насыщенных атома углерода должны быть очень небольшими. Причиной различия в скоростях может быть только сте-рический фактор: возросшая трудность подхода ЕЮ- «с тыла» атома углерода, связанного с Вг, и повышенная напряженность образующегося переходного состояния. Особенно значительное снижение скорости при переходе от 1-бром-2-метилпропана (14) к 1-бром-2,2-диметилпропану (неопентилбромид) (15) объясняется тем, что переходное состояние для первого, хотя и несколько напряженное, может путем вращения вокруг связи а-С—С-[3 принять конформацию (14а), в которой атакующему нуклеофилу EtO~ мешает только атом Н. В то же время снижение напряженности в переходном состоянии (15а) для соединения (15) невозможно (см. также разд. 5.4.2).
(14а)
Переходное состояние (15а) будет, следовательно, иметь большую энергию, его AG+ (см. разд. 2.2.1) будет больше, а скорость реакции соответственно ниже.
Влияние структуры на относительную реакционную способность особенно заметно, когда атом галогена расположен у «головного» атома углерода бициклической системы. Относительные скорости сольволиза ряда соединений в 80 %-м водном этаноле приведены ниже:
Вг Вг Вг
(8)1 (16) =И0~6 (17) -10~14
4*
99
Все эти соединения — третичные галогениды, так что атака по механизму SN2 для соединений (16) или (17) невозможна, как и для соединения (8) (ср. разд. 4.3).
Атака по механизму SN2 «с тыла» атома углерода, связанного с Вг, будет предотвращена как в соединении (16), так и в соединении (17), поскольку их структуры имеют форму клетки и являются очень жесткими, что исключает достижение переходного состояния с его обязательным плоским расположением связей по отношению к «головному» атому углерода (см. разд. 4.3). Сольволиз через скоростьлимитирующую стадию образования ионной пары (SN1), как в случае соединения (8), также исключается: образование карбокатионов из соединений (16) и (17) крайне затруднено тем, что жесткая структура не допускает перехода этих карбокатионов в плоское состояние, в котором возможна их стабилизация. Таким образом, эти промежуточные карбокатионы обладают гораздо большим запасом энергии, чем обычные интермедиаты, вследствие чего образуются крайне медленно. Резкое снижение скорости сольволиза соединения (17) по сравнению с соединением (16) отражает большую жесткость структуры относительно «головного» (катионного) атома углерода при наличии одноуглеродного мостика в случае соединения (17) по сравнению с двухуглеродным в случае соединения (16). Эта жесткость еще выше в 1-бромтрипти-цене (19), в котором, как было показано, атом брома фактически инертен к нуклеофилам.
(18)1
Найдено, что, несмотря на формальное сходство окружения атома брома в соединениях (19) и (18), различие в скоростях их реакций при соответствующих условиях достигает порядка 10~ . И Это происходит потому, что стабилизация карбокатиона, полученного из соединения (18), возможна путем делокализации его заряда с участием л-орбитальных систем трех бензольных колец, тогда как чрезвычайно жесткая структура (19) будет удерживать свободную орбиталь катиона (после потери Вт-) перпендикулярно л-орбитали всех колец, препятствуя, таким образом, делокализации положительного заряда.
100
4.4. Стереохимические аспекты механизма реакций
Гидролиз оптически активной формы хирального* галогенида имеет некоторые интересные стереохимические особенности и поэтому заслуживает отдельного рассмотрения.
4.4.1. Механизм Sn2: обращение конфигурации
Как будет показано ниже, пространственное расположение трех заместителей R, R' и R", связанных с атакуемым атомом углерода, таково, что при атаке они «выворачиваются» внутрь. Такой атом углерода, как говорят, подвергается обращению (инверсии) конфигурации (при этом имеют в виду изменение пространственного расположения заместителей, связанных с этим атомом) :
НО” +
+ Вг~
R'
(?)
Действительно, если бы продуктом реакции вместо соответствующего спирта оказался бромид, то было бы обнаружено, что он вращает плоскость поляризации плоско поляризованного света в противоположном направлении по сравнению с исходным веществом и является, следовательно, его зеркальным отражением (см. разд. 4.4.2). Однако продуктом этой реакции является спирт, и, к сожалению, нельзя сказать, просто наблюдая направление его оптического вращения, имеет ли он ту же конфигурацию, что и бромид, из которого образован, илн обратную. Для соединений, не являющихся зеркальными отражениями, но имеющих противоположные конфигурации, необязательно противоположное направление вращения плоскости поляризации, в то время как для веществ, имеющих одинаковые конфигурации, необязательно одинаковое направление вращения плоскости поляризации. Таким образом, для того чтобы подтвердить, что указанная выше реакция Sn2 действительно сопровождается обращением конфигурации, как этого требует теория, необходим независимый метод сопоставления конфигураций исходного вещества и продукта, в частности бромида и соответствующего спирта, как в приведенном выше примере.
* Хиральным называют соединение, не совместимое со своим зеркальным изображением.
101
4.4.2. Определение относительной конфигурации
Такое определение основано на том, что если хиральное соединение вступает в реакцию, в ходе которой разрывается связь, соединяющая одну из групп с хиральным центром, то этот центр может претерпевать обращение конфигурации, хотя это и необязательно; если же в ходе реакции такая связь не разрывается, то конфигурация хирального центра должна остаться неизменной.
Известно, например, что в серии реакции тозилирования оптически активного (+)-спирта (20) 4-метилбензолсульфонил-хлоридом (тозилхлорид) связь С—О спирта не разрывается *; следовательно, тозилат (21) должен иметь ту же конфигурацию, что и исходный спирт.
R R R
,\ ArSOjCl \ Вг" /
R,|r С—ОН------> R7" C-j-OSOgAr--Вг—С •"« r'
r"Z R"Z
(20) (+) (21) (24)
jMeCO“
F ZR R = PhCHp, R'= Me,
HO—C R' <---МеСО-ьО—C-„r R" = h, Ar = n-MeC6H4
R" r"
(23) (-) (22)
Известно также, что взаимодействие эфира (21) с МеСОг является реакцией замещения, во время которой группа Аг50з(Аг = п-МеС6Н4) вытесняется и на ее место вступает группа МеСО2; следовательно, в ходе этой реакции происходит разрыв связи С—О и при образовании ацетата (22) возможно обращение конфигурации. Щелочной гидролиз ацетата [(22)—>-(23)], как можно показать, не приводит к разрыву связи С—О**, так что спирт (23) должен иметь ту же конфигурацию, что и ацетат (22). Поскольку спирт (23) является зеркальным отражением исходного соединения (20) (имеет противоположное направление оптического вращения), это свидетельствует лищь о том, что вся серия реакций приводит к обращению конфигурации. Однако ясно, что это обращение может произойти только во время
* Это было показано с использованием спирта, меченного ,8О по ОН-группе; обнаружено, что этот атом не отщепляется при образовании тозилата, однако он удаляется при взаимодействии тознлата с МеСО“.
** Гидролиз ацетата, в котором атом кислорода помечен 18О, не приводит к замещению его кислородом воды; тем самым подтверждается, что связь алкил—кислород ацетата в ходе его гидролиза не разрывается (см. разд. 3.3).
102
реакции МеСОг с тозилатом (21). Показано, что при реакциях тозилата (21) с другими нуклеофилами также наблюдается обращение конфигурации. Следовательно, можно полагать, что подобное обращение происходит и при реакции с ионом Вг- с образованием бромида (24), т. е. что бромид (24), подобно ацетату (22), имеет конфигурацию, противоположную конфигурации исходного спирта (20).
Общее правило, согласно которому реакции бимолекулярного замещения (Sn2) сопровождаются обращением конфигурации, было установлено с помощью изящного и очень остроумного эксперимента, в котором галогенид-ион в оптически активном алкилгалогениде замещают таким же самым, но меченным га-логенид-ионом (нуклеофил), например, при действии радиоактивного 1281~ на (+)-2-иодоктан (25):
Ф
РбН13
’28Г+ 'С—I
Me”*/ н
(25) (+)
Cg Н и 128s” 1 б-
,28|---С---)
м/ чн
/С6Н]3
3|-с( + Г
""Me н
(25а)(-)
Замещение контролировали, наблюдая изменение распределения радиоактивного 1281 между неорганическим иодидом (Nal) и 2-иодоктаном. Было показано, что в этих условиях наблюдается суммарный второй порядок реакции (первый порядок по отношению к 1281~ и к 2-иодоктану); Аг = 3,00 ± 0,25-10-5 (при 30 °C).
Если происходит обращение конфигурации (согласно механизму Sn2), то оптическая активность раствора уменьшается до нуля, т. е. идет рацемизация. Это должно происходить из-за обращения конфигурации молекулы (+)-(25), что приведет к образованию ее зеркального отражения (—)-(25а). Молекула (—)-(25а) «спаривается» с молекулой (+)-(25) с образованием (±)-рацемата; таким образом, наблюдаемая скорость рацемизации будет вдвое больше скорости обращения конфигурации. Реакцию контролировали поляриметрически, измеряя скорость рацемизации. На основе этих данных была вычислена скорость обращения конфигурации; величина k оказалась равной 2,88 ± ±0,03-10~5 (при 30 °C).
Таким образом, скорость замещения и скорость обращения конфигурации в пределах ошибки опыта одинаковы, из чего следует, что каждый акт бимолекулярного замещения должен протекать с обращением конфигурации. Считают доказанным, что реакции типа Sn2 сопровождаются обращением конфигурации; если независимо показано, что данная реакция протекает по механизму Sn2, то этот факт часто используют для корреляции между конфигурациями исходного вещества и продукта реакции.
103
4.4.3. Механизм SN1: рацемизация?
Если карбокатион, образующийся на медленной, скоростьлимитирующей стадии реакции, является плоским, то можно ожидать, что последующая атака нуклеофилом, например ионом -ОН или молекулой растворителя (Н2О :), будет происходить с равной вероятностью с любой стороны этого плоского карбокатиона, что приведет к образованию смеси, где 50 % молекул имеют такую же конфигурацию, как исходное вещество, а 50 % — противоположную, т. е. будет происходить рацемизация, приводящая к оптически неактивному продукту
(+)
I? _
I он
С+ ------
Z ч
R' R"
ВГ
R / ?—СГ„ \ "R
R" (50%)
, ,.с—он
R ' / R"
(50)%
На практике, однако, ожидаемая рацемизация — и только рацемизация — наблюдается редко, она почти всегда сопровождается в некоторой степени инверсией. Показано, что относительные доли каждого из этих процессов зависят от структуры галогенида, в частности от стабильности образующегося из него карбокатиона, а также от растворителя, в частности от его способности выступать в качестве нуклеофила. Чем более стабилен карбокатион, тем больше степень рацемизации; чем более нуклеофилен растворитель, тем больше степень инверсии. Эти закономерности становятся понятными, если стадию ионизации, лимитирующую процесс SnI, представить схемой:
б+ б-
R—Вг
R+ В Г
R+
+ ВГ
(26)
(27) (28)
В этой схеме (26) представляет собой внутреннюю ионную сильно ассоциированы и между ними нет молекул растворителя, (27) —разделенную растворителем ионную пару, а (28) —диссоциированную и раздельно сольватированную пару ионов.
При сольволизе атака карбокатиона R+ молекулой растворителя, например Н2О :, в случае (26) ведет, вероятно, к обра-пару, в которой совместно сольватированные противоионы очень
104
щению конфигурации, поскольку атака может происходить (с участием сольватной оболочки) с «задней» стороны R+, но не с «передней», где нет молекул растворителя и которая прикрыта противоионами Вг- Атака в (27), возможно, идет с обеих сторон, приводя к рацемизации; атака в (28) с равной вероятностью может происходить с обеих сторон. Таким образом, чем больше время существования R+, т. е. чем дольше он избегает нуклеофильной атаки, тем больше доля рацемизации. Время существования R+, вероятно, тем больше, чем он стабильнее (см. выше), но тем меньше, чем более сильным нуклеофилом является растворитель (см. выше).
Например, сольволиз (+)-С6Н5СНМеС1, который может образовать стабилизированный карбокатион бензильного типа (см. разд. 4.3), приводит к 98 %-й рацемизации, в то время как сольволиз (4-)-СбН]3СНМеС1, в ходе которого нет аналогичной стабилизации, приводит только к 34 %-й рацемизации. Сольволиз (4-)-СбН5СНМеС1 в смеси ацетона (80%) и воды (20%) приводит, как и выше, к 98 %-й рацемизации, но в более нуклеофильном растворителе—чистой воде — только к 80 %-й рацемизации.
Такие же общие рассуждения применимы к реакциям нуклеофильного замещения нуклеофилами Nu:, однако в этом случае время существования R+ больше отчасти из-за того, что сольватная оболочка должна быть удалена перед атакой R+ нуклеофилом Nu:. Важно отметить, что рацемизация, конечно, предъявляет гораздо меньшее стереохимическое требование в реакциях SnI, чем обращение конфигурации в реакциях Sn2.
4.4.4. Конкуренция механизмов
Выше уже отмечалось (см. разд. 4.3), что реакции некоторых субстратов, например вторичных галогенидов, могут быть описаны уравнением смешанного первого и второго порядка. В этом случае возникает вопрос, протекает ли такая реакция по механизмам Sn2 и SnI одновременно (их относительные доли могут зависеть от растворителя и т. д.), или же она протекает по некоторому специфическому «промежуточному» механизму?
В реакциях сольволиза, подобных тем, что уже были рассмотрены, в которых растворитель сам является нуклеофилом, такая смешанная кинетика может не обнаруживаться, независимо от того, что происходит в действительности, потому что скорости реакций как SnI так и SN2 должны, очевидно, подчиняться следующему уравнению:
Скорость = &[RX).
ЭтО объясняется тем, что в реакции SN2 концентрация нуклеофила остается практически постоянной на протяжении всего
105
процесса, поскольку нуклеофил, а он является также и растворителем, присутствует в очень большом избытке. Возникает вопрос, обусловлено ли одновременное протекание рацемизации и обращения конфигурации, наблюдаемое в таких случаях, совместным действием механизмов SnI и Sn2 или же оно является следствием приведенной выше гипотезы, предусматривающей образование различных ионных пар.
По крайней мере, в некоторых случаях можно доказать, что «смешанный» SnI + Sn2 механизм не действует. Например, упомянутый выше сольволиз (+)-С6Н5СНМеС1 в МеСО2Н, как было показано, приводит к 88 %-й рацемизации и 12 %-му обращению конфигурации:
Me Me
I MeCOjH |
PhCH—Cl ------► PhCH—OCOMe
(+) (рацемизация 88%,
инверсия 12%)
При добавлении в реакционную смесь гораздо более сильного нуклеофила МеСО2 (в виде МеСОг Na+) было обнаружено, что общая скорость реакции и доля обращения конфигурации не увеличиваются. Имеются все основания полагать, что наблюдаемое в этом случае обращение конфигурации не обусловлено той частью суммарной реакции, которая протекает по механизму Sn2 одновременно с механизмом SnI (основной механизм). Если бы это было так, то можно было бы ожидать, что применение гораздо более сильного нуклеофила МеСОг (вместо МеСО2Н) приведет к заметному увеличению как общей скорости, так н доли обращения конфигурации.
Очень интересно (независимо от приведенных выше данных) обсудить вопрос: возможен ли непрерывный спектр механизмов, промежуточных между Sn2 и SnI, т. е. имеются ли механизмы, незаметно переходящие один в другой — через постепенно изменяющиеся переходные состояния — со стороны чистого механизма Sn2, и механизмы, проходящие через постепенно изменяющиеся комбинации ионная пара — растворитель со стороны чистого механизма SnI? Эта область теории незаметно переходит, однако, в семантику, если даже не в теологию!
4.4.5. Механизм S^i: сохранение конфигурации
Несмотря на то что было сказано выше о реакциях замещения, приводящих к обращению конфигурации или к рацемизации или к тому н другому, известен ряд реакций, протекающих с фактическим сохранением конфигурации, т. е. реакций, в которых исходное соединение и продукт обладают одинаковой конфигурацией. Одной из таких реакций, в которой это было обна
106
ружено, является замещение ОН на С1 с использованием тио-нилхлорида SOCl2:
Me Me
\ soci2 \ Ph"'"/C'"0H-----^Ph<C-Cl + S°2 + HCl
H H
(29) (30a)
Показано, что эта реакция подчиняется кинетическому уравнению второго порядка:
Скорость = &2[ROH[ [SOCIj].
Очевидно, она не может протекать по обычному 5м2-меха-низму, так как это должно было бы привести к обращению конфигурации продукта (см. разд. 4.4.1), что на самом деле не обнаружено.
При проведении этой реакции в более мягких условиях удается выделить алкилхлорсульфит ROSOC1 (31) и показать, что он является истинным промежуточным соединением. В соединении (31) сохраняется конфигурация исходного спирта, связь R—О не расщепляется во время реакции. Было показано, что скорость, с которой алкилхлорсульфит (31) расщепляется с образованием продукта реакции RC1 (30а), возрастает с увеличением полярности растворителя, а также с увеличением стабильности карбокатиона R+: почти наверняка при этом образуется ионная пара (32). Переход этой ионной пары в продукты реакции протекает быстро, т. е. во внутренней ионной паре (33) внутри сольватной оболочки (ср. разд. 4.4.3). Атака ионом Cl-происходит с той же стороны карбокатина R+, от которой отошел ион “OSOC1, т. е. с сохранением конфигурации:
Me О Me О Me Me
й Ph Н Ph \ й
(31) (32) (33) (30а)
Вопрос о том, расщепляются ли связи С—О и S—С1 одновременно или же сначала расщепляется связь С—О, все еще остается спорным.
Интересно, что при реакции SOC12 с ROH (29) в присутствии пиридина продукт RC1 претерпевает обращение конфигурации с образованием соединения (306). Это происходит потому, что НС1, выделяющийся при образовании хлорсульфита (31) из ROH и SOC12, превращается в пиридине в C5H5NH+ С1~ и,
107
являясь эффективным нуклеофилом, атакует соединение (31) «с тыла», как в обычной реакции SN2, т. е. с обращением конфигурации:
С1“ +
Ме О \ II ,,с—osct—►
Ph*'/ Н
(31)
Me О I* Ме
I 116- /
— с-—OSCI —>-ci-c +so2+ci
Р|/ чн r*ph
(ЗОб)
4.4.6. Участие соседних групп: «сохранение» конфигурации
Известны некоторые примеры сохранения конфигурации в реакциях нуклеофильного замещения, общей особенностью которых является то, что атом .или группа, расположенный рядом с атакуемым атомом углерода, имеет свободную пару электронов. Эта соседняя группа может использовать свою пару электронов, чтобы взаимодействовать «сзади» с атомом углерода, у которого происходит замещение, препятствуя тем самым атаке нуклеофильным агентом. Атака может происходить, следовательно, «спереди», что и приводит к сохранению конфигурации. Например, гидролиз 1,2-хлоргидрина (34) в присутствии оснований приводит к образованию 1,2-диола (35), имеющего ту же самую конфигурацию:
HOCEt2 CEt2
\» '“ОН _ / \* о
'С—С1-----(О / ,С—С1
Ме'"*/ ,
Н Ме Н CEt2
(34) (36) 0<Зс’J-°H
H°CEt2 CEt2 H №
(35) (38)
1, 2 - обращение конфигурации
Первоначальная атака соединения (34) основанием приводит к алкоксид-аниону (36), из которого в результате внутримолекулярной атаки образуется эпоксид (37); при этом происходит обращение конфигурации у асимметрического атома С (такие циклические интермедиаты во многих случаях могут
108
быть выделены). Этот атом углерода * в свою очередь подвергается обычной атаке нуклеофилом -ОН по механизму Sn2 с повторным обращением конфигурации. Образовавшийся алкоксид-анион (38) отрывает протон от молекулы растворителя с образованием продукта реакции—1,2-диола (35), имеющего ту же конфигурацию, что и исходное соединение (34). Таким образом, кажущееся сохранение конфигурации является результатом двух последовательных обращений конфигурации.
Другим примером участия кислорода как соседней группы является гидролиз аниона 2-бромпропаноата (39) при низких концентрациях -ОН; было показано, что эта реакция также протекает с сохранением конфигурации. Показано, что скорость этой реакции не зависит от концентрации ионов “ОН. Полагают, что реакция протекает по схеме:
/G\ .С° СО
V медленно _ / \ . ZS. быстро _ / \ °M’z?~Br----------0;-"С? 0Н ---------U О >-ОН
Me t .. / \ «.Xi
Н Me н Me
(ЗЭ) (40) (41)
Является ли интермедиат (40) цвиттер-ионом, как показано на схеме, или очень лабильным а-лактоном (40а), точно ие установлено. При увеличении концентрации нуклеофила (“ОН) увеличивается и доля обычной атаки «сзади» по механизму SN2 с обращением конфигурации.
/С\ .^Ме °— с-н
(40а)
Участие соседних групп наблюдалось не только в случае атома кислорода, но и со стороны других атомов, например атомов серы и азота. Неожиданное увеличение скорости позволяет предположить изменение механизма реакции. Например, показано, что соединение (42) гидролизуется в 104 раз быстрее, чем его кислородсодержащий аналог (44), по-видимому, вследствие участия атома S х, действующего как соседняя группа:
ГХ, медленно _ + ^2° ___
EtS\ /СН2 Cl ———>- EtS —-СН2 EtS^ СН2ОН
сн2 СН2 СН2
(42) (43)
EtOCH2CH2Cl (44)
* Атака преимущественно идет по этому, а не по другому атому углерода трехчленного кольца, поскольку он более положителен, будучи связан только с одной электроиодЪиориой алкильной группой.
109
Наоборот, атом О: в соединении (44), будучи весьма электроотрицательным, не отдает электронную пару (в отличие от О- в RO" и RCO2; см. выше), и гидролиз соединения {44) протекает, таким образом, через обычную атаку Sn2 внешним нуклеофилом, которая, очевидно, идет гораздо медленнее, чем внутримолекулярная нуклеофильная атака в реакции (42) ->
(43). Образование циклической сульфониевой соли (43) подтверждается гидролизом соединения (45), при котором образуются два спирта [причем неожидавшийся спирт (47) — с высоким выходом], что указывает на участие несимметричного интермедиата (46):
Me
медленно
Me
СН2
н2о ------- быстро
(45)
Me Me
I I
--->- EtSCH2-СИОН + EtS-CH-CH2OH
(47)
В соответствующих реакциях атом N: также может действовать как соседняя группа. Это наблюдается, например, при гидролизе Me2NCHCH2Cl, однако его скорость заметно меньше (в сравнимых условиях), чем скорость обсуждавшегося выше гидролиза соединения (42), вследствие большей стабильности промежуточного циклического иммониевого иона, аналогичного (43). Такие циклические формы образуются в ходе гидролиза горчичного газа [ди(хлорэтил)сульфид] S(CH2CH2C1)2 и его азотсодержащих аналогов, например MeN(CH2CH2Cl)2; циклические иммониевые соли, образующиеся из последнего, являются сильными нейротоксинами. л-Орбитальная система бензольного кольца также может действовать в качестве соседней группы (см. разд. 5.2 и 13.4.3.1).
4.5. Влияние вступающих и уходящих групп
4.5.1. Вступающая группа
Изменение в применяемом нуклеофильном реагенте, т. е. замена вступающей группы, не приводит непосредственно к изменению скорости реакции замещения по механизму SnI, потому что нуклеофильный реагент не участвует в скоростьлимитирую-щей стадии. При протекании реакции по механизму SN2 с увеличением нуклеофильности реагента скорость реакции должна
110
Увеличиваться. Казалось бы, можно ожидать, что нуклеофиль-ндрть реагента должна коррелировать с его основностью, по-скбдьку оба понятия включают доступность электронных пар и легкость, с которой они могут быть представлены. Эту параллель. однако, нельзя считать строгой; основность определяется способностью передавать электронную пару атому водорода, в то время как нуклеофильность — способностью передавать электронную пару другому атому, очень часто атому углерода; кроме того, основность является термодинамической характеристикой, тогда как нуклеофильность — кинетической; основность мало зависит от стерических факторов, тогда как нуклеофильность очень чувствительна к ним.
Эти различия вытекают, в некоторой степени, из введенного недавно различия между жесткими и мягкими основаниями. Жесткое основание — это основание, в котором атом — донор является очень электроотрицательным, мало поляризуемым и трудно окисляемым; к ним относятся, например, “ОН, ~OR, :NR3. Мягкое основание содержит атом — донор с низкой электроотрицательностью, высокой поляризуемостью и легкой окис-ляемостью; к ним относятся, например, RS~, I~, SCN-. В определенном интервале основности мягкость способствует нуклеофильности. Данные по основности гораздо более доступны и могут быть использованы как для предсказания ожидаемой нуклеофильности, так и для сравнения. Так, если атакующий атом один и тот же, то можно полагать, что основность и нуклеофильность изменяются параллельно: чем сильнее основание, тем более мощным нуклеофилом оно является:
ЕЮ' > PhO' > МеСО; > NO'
При замене нуклеофила возможно также изменение механизма; например, при использовании таких нуклеофилов, как Н2О:, НСОз, МеСО2, замещение идет по механизму SnI, а при использовании -ОН и ЕЮ- оио может пойти по механизму Sn2.
Найдено, что нуклеофильность очень сильно зависит от размера атакующего атома в нуклеофиле, по крайней мере при сравнении в пределах одной и той же группы или подгруппы в Периодической системе элементов, например:
Г > Вг' > СГ RS' > RO'
От размера атома, а также от его электроотрицательности зависит и поляризуемость (ср. упомянутые выше мягкие основания); когда размер атома увеличивается, действие его ядра иа периферические электроны уменьшается, в результате чего оии становятся более легко поляризуемыми и более склонными к связыванию. Кроме того, чем больше размер нуклеофильного иона или группы, тем меньше энергия его сольватации, т. е. тем
111
более легко он превращается в эффективный, преимущественно несольватированный, нуклеофил. Теплоты гидратации ионов I-и F- равны 284 и 490 кДж/моль соответственно. В результате сочетания выше обсужденных факторов большой, хорошо поляризуемый, слабо сольватированный иодид-ион I- является гораздо более эффективным нуклеофилом, чем небольшой, трудно поляризуемый, сильно сольватированный (вследствие образования водородных связей с гидроксилсодержащим растворителем) фторид-ион F-. На этом основании можно было бы ожидать, что скорость реакции при переходе от гидроксилсодержащего растворителя к полярному апротонному растворителю (см. разд. 4.2) увеличивается в гораздо меньшей степени для 1~, чем, например, для Вг~ или С1~, что и было на самом деле обнаружено (в Ме2СО Вг- является лучшим нуклеофилом, чем 1~).
Другой интересный вопрос возникает в отношении нуклеофилов, которые содержат обычно два соответствующих атома, посредством которых они могут атаковать субстрат, — амбидентных нуклеофилов:
[~X~Y *+ X—Y~]
Показано, что в очень полярных реакциях SnI атаку карбокатионного интермедиата R+ осуществляет атом нуклеофила, на котором выше электронная плотность. Например, в случае галогенидов, которые с трудом реагируют по механизму SnI, атака может быть облегчена использованием аниона соли серебра, например AgCN. Ион серебра Ag+ ускоряет образование R+ вследствие осаждения AgHal (ср. разд. 5.1.1):
[~C=N C=N-j медленно быстро +
R—Вг + Ag+ [CN]' R+ + [CN]' --► R-ffeC
В отсутствие иона серебра при использовании Na+ [CN]-, например, протекает реакция Sn2 с преимущественной атакой со стороны того атома в нуклеофиле, который более поляризуем:
в- в-
NC’ + R—Вг —> [NC--R—Вг]* —>- N=C—R + Вг
переходное состояние
Это и понятно, так как, в отличие от реакции SnI, образование связи в таком случае происходит в переходном состоянии в ходе скоростьлимитирующей стадии, для которой легкая поля
112
ризуемость атакующего атома нуклеофила несомненно важнее. Это различие в поведении AgCN и NaCN долгое время использовали в препаративных целях. Аналогично, было показано, что нитрит-ион NO2 образует алкилнитриты RON=O в условиях реакции SnI (атом О имеет более высокую электронную плот-ность) и нитроалканы RNO2— в условиях реакции Sn2 (атом N является более легко поляризуемым атомом).
4.5^2. Уходящая группа
Очевидно, что замена уходящей группы будет изменять скорость реакций как SnI, так и Sn2, поскольку в обоих случаях разрыв связи с уходящей группой осуществляется на медленной скоростьлимитирующей стадии. Можно ожидать, что относительная способность частицы Y в соединении R—Y быть уходящей группой должна определяться: а) прочностью связи R—Y; б) поляризуемостью этой связи; в) стабильностью Y-; г) степенью стабилизации посредством сольватации образующегося иона Y~ в переходных состояниях как механизма SnI, так и механизма Sn2.
Реакционная способность галогенидов (в реакциях SnI или Sn2) уменьшается в ряду:
R—I > R—Вг > R—Cl > R—F
Это дает возможность предположить, что упомянутые выше условия (а) и (б) более важны, чем (в) и (г). Для большого числа потенциальных уходящих групп условие (в) могло бы означать, что чем слабее Y- как основание (или чем сильнее Н—Y как кислота), тем лучше частица Y как уходящая группа. Это подтверждается в некоторой степени рядом уходящих групп, у которых атом, с помощью которого Y связан с R, один и тот же. Например, анионы сильных «кислородных кислот», таких как л-МеСбН48Оз (тозилат; см. разд. 4.4.2) и CF3SO3 (триф лат), являются хорошими уходящими группами (такими же, как анионы галогенидов); для таких О-уходящих групп условия (в) и (г) особенно важны. Относительная активность уходящей группы может, однако, изменяться при изменении растворителя [влияние условия (г)]. Это изменение относительной активности отмечено, в частности, при замене гидроксилсодержащего растворителя биполярным апротонным растворителем (например, Me2SO, HCONMe2 и т. д.); при этом большее значение приобретают не факторы (в) и (г), а факторы (а) и (б).
Высокая поляризуемость делает анион I- хорошей как вступающей, так и уходящей группой; иодид-ион поэтому часто может быть использован в качестве катализатора медленных
113
реакций замещения, например:
медленно
Н2О s + R—С1 -------НО—R + Н+ С1
быстро
F + R-C1 --------1
I-R4-CF
быстро I
Н+ I" + R—ОН -«------1
Это явление называют нуклеофильным катализом. Чем более сильным и жестким основанием является уходящая группа, тем труднее она замещается; в частности такие группы, как -ОН, “OR, -NH2, связанные с атомом углерода с помощью ие1 больших, очень электроотрицательных мало поляризованных атомов (названных выше жесткими основаниями), обычно не могут замещаться непосредственно другими нуклеофилами.
Реакции замещения, которые по каким-либо причинам затруднены или ддже невозможны, можно активировать путем модификации уходящей группы, например посредством протониро-вания, сделав ее, таким образом, более слабым и (или) более мягким основанием. Например, группа ~ОН не может быть непосредственно замещена бромид-ионом Вг- Замещение, однако, легко протекает, если ему предшествует протонирование:
Вг' + R—ОН —х-> Вг—R + 'ОН
• • н+||
Br' + R—ОН —► Вг—R + Н2О
Н
Это объясняют двумя основными причинами: а) Вг- после протонироваиия субстрата атакует положительно заряженную, а не нейтральную частицу; б) очень слабое основание НгО является гораздо лучшей уходящей группой, чем сильноосиовный гидроксид-иои -ОН. Хорошо известное использование HI для расщепления простых эфиров основывается на том, что I- является наиболее сильной нуклеофильной частицей, которая может образоваться в сильиокислом растворе, необходимом для первоначального протонироваиия:
н+ + г
R—OPh ч=* R—OPh ► R—I + PhOH
Н
114
4.6. Другие реакции нуклеофильного замещения
При обсуждении нуклеофильного замещения у насыщенного атфма углерода основное внимание было уделено атаке поляризованных нейтральных частиц, прежде всего алкилгалогенидов R—Hal, нуклеофильными ионами :Nu~, особенно -ОН. На самом же деле, кроме упомянутых выше реакций к реакциям такого типа часто относят реакции поляризованных нейтральных частиц с незаряженными нуклеофилами:
Me3N : + Et—Вг —> Me3NEt + ВГ
Et2S: + Me—Вг —► Et2SMe + ВГ
реакции положительно заряженных частиц с нуклеофильными анионами:
Г + С6Н13—ОН2 —> С6Н13—1+Н2О:
ВГ + Ме—NMe3 —► Me—Вг + : NMe3
а также реакции положительно заряженных частиц с незаряженными нуклеофилами (в этом случае, возможно, наилучшей уходящей группой является N2):
Н2О: + PhN2 —> PhOH + N2 + Н+
Кроме галогенид-ионов хорошими уходящими группами являются, например, тозилат-анион (ср. разд. 4.4.2) и «внутренние» уходящие группы (ср. разд. 4.4.6):
МеСО2 + ROSO2C6H4Me-n -* MeCO2R + n-MeCgl-HSOf
СН2- СН2->С1СН2СН2О“
Существуют также реакции нуклеофильного замещения, представляющие значительный интерес для синтеза, в которых атакующим атомом является атом углерода в составе карбаниона (см. разд. 10.5.3.2) или группы, являющейся источником отрицательно поляризованного углерода (см. разд. 8.4); в ходе этих реакций образуются новые углерод-углеродные связи:
_NH2 РгВг
НС=СН ------> НС=С~ ----> НС=С-Рг + ВГ
EtO“ - PhCH2Br
CH2(CO2Et)2 ч=ь (EtO2C)2CH -------(EtO2C)2CH—CH2Ph
- Br* d+ d-
BrMg— Ph + C6H13—Br -—► MgBr2 + Ph—C6H13
Необходимо помнить, что в приведенных выше примерах то, что является нуклеофильной атакой по отношению к одному
115
реагенту, является электрофильной атакой по отношению к дру/ тому реагенту. Поэтому любое обозначение такого процесса имеет в некоторой степени произвольный характер, как и в случае наших прежних представлений относительно того, что ^читать реагентом, а что субстратом (см. разд. 8.7). /
Вряд ли удивительно, что не все реакции нуклеофильного замещения протекают со 100 %-м выходом желаемых продуктов. Как и в других случаях, происходящие при этом побочные реакции дают неожиданные, а в препаративном смысле — и нежелательные продукты. Основной побочной реакцией является элиминирование, приводящее к ненасыщенным соединениям (подробнее см. разд. 8.7).
Глава 5
КАРБОКАТИОНЫ, ЭЛЕКТРОНОДЕФИЦИТНЫЕ АТОМЫ АЗОТА И КИСЛОРОДА И ИХ РЕАКЦИИ
5.1. Методы образования карбокатионов..............................117
5.1.1. Гетеролитический распад нейтральных частиц..............117
5.1.2. Присоединение катионов к нейтральным частицам...........118
5.1.3. Образование карбокатионов из других катионов............119
V 5.2. Стабильность и структура карбокатионов.........................119
5.3. Реакции с участием карбокатионов..............................122
>. 5.4. Перегруппировки карбокатионов.................................124
5.4.1. Перегруппировки без изменения углеродного скелета .... 125
5.4.1.1. Аллильные перегруппировки........................125
5.4.2. Перегруппировки с изменением углеродного скелета .... 126
5.4.2.1. Неопентильные перегруппировки....................126
5.4.2.2. Перегруппировки углеводородов....................127
5.4.2.3. Пинаколииовая перегруппировка....................128
5.4.2.4. Стереохимия перегруппировок......................131
5.4.2.5. Перегруппировка Вольфа...........................133
5.5. Катионы диазоиия..............................................134
5.6. Миграция к электронодефицятному атому азота.............. . . .137
5.6.1. Реакции Гофмана, Курциуса, Лоссеиа и Шмидта.............137
5.6.2. Перегруппировка Бекмана.................................139
5.7. Миграция к электроиодефицитиому атому кислорода..............142
5.7.1. Окисление кетонов по Байеру — Виллигеру.................142
5.7.2. Перегруппировка пероксидов..............................144
В предыдущей главе уже упоминалось об образовании карбокатионов (в ионных парах) в качестве интермедиатов некоторых реакций замещения при насыщенном атоме углерода, на
116
пример при сольволизе алкилгалогенидов по механизму SN1. Эуи ионы встречаются, однако, в гораздо большем числе реакций, и хотя их существование часто является только кратковременным, они играют важную роль в разнообразных химических превращениях.
5.1. Методы образования карбокатионов
5.1.1. Гетеролитический распад нейтральных частиц
Примером этого процесса является обычная ионизация, когда группа, связанная с атомом углерода, отщепляется со связывающими электронами; при этом образуется ионная пара R+-Y-, например:
Ме3С—Вг Ме3С+ Вг- Ph2CH—С1 Ph2CH+ СГ
МеОСН2—С1 МеОСН2 СГ
Для образования карбокатионов обычно необходима сильнополярная (высокое значение е), сильно сольватирующая ионы среда. Выше уже рассматривалось каталитическое действие ионов Ag+ (см. разд. 4.5.1), часто приводящее к изменению механизма реакции SN2 на SnI:
Ag++ R—Вг —► AgBr| + R+
Каталитическое действие Ag+ может осложняться, однако, тем, что выпадающий в осадок галогенид серебра сам может действовать как гетерогенный катализатор.
Ионизация может быть вызвана также кислотами Льюиса, например BF3; в этом случае образуется ацил-катион, причем на равновесие сильно влияет очень большая стабильность аниона BFi •
MeCOF + BF3 4=t MeCO BF;
Аналогичный процесс происходит и при действии А1С13. В этом случае относительно неустойчивый ацил-катион распадается с образованием очень стабильного карбокатиона Ме3С+, причем равновесие сдвинуто вправо вследствие выделения СО:
Ме3ССОС1 + А1С13 Ме3ССО A1CIJ —► Ме3С+ А1СЦ + COf
При использовании SbF5 в качестве кислоты Льюиса и жидкого SO2 (или избытка SbFs) в качестве растворителя были получены простые алкил-катионы в условиях, позволяющих проводить их детальное изучение как методом ЯМР-спектроскопии, так и другими методами.
R—F + SbFs R+ SbFj
117
Использование «суперкислот», например SbFs/FSChH, приводит К образованию алкил-катионов даже из алканов:
Ме3С—Н + SbF5/FSO3H —► Н2 + Ме3С+ (SbF6)FSO3
Стабильность Ме3С+ иллюстрируется тем фактом, что изомерный карбокатион +СНМеСН2Ме, полученный из МеСН2СН2Ме в этих условиях, практически мгновенно перегруппировывается в Ме3С+. Соотношение между структурой и устойчивостью карбокатионов, а также их перегруппировки рассмотрены ниже (см. разд. 5.1.3 и 5.4.1).
5.1.2. Присоединение катионов к нейтральным частицам
Наиболее обычным катионом является Н+, присоединяющийся к ненасыщенным связям, т. е. участвующий в протонировании, например в катализируемой кислотами гидратации алкенов (см. разд. 7.4.2):
Н Н +ОН2 Н ОН
н+ | + н2о I | —н+ | |
—СН=СН— —сн—СН— ч — * —СН—СН— ч=* —СН—сн—
Эта реакция обратима, причем обратная реакция больше известна как катализируемая кислотами дегидратация спиртов (см. разд. 9.1). Протонирование может проходить также и по атому кислорода связи С=О:
\в+ в- н+ г\ + \+ п н2о \ /ОН
V=O С=ОН ч->- С—ОН I ч=* +Н+
/ L/ / J / \qH
Это приводит к образованию положительно заряженного атома углерода, который и подвергается атаке нуклеофилом, в данном случае Н2О s, в катализируемой кислотой гидратации карбонильных соединений (см. разд. 8.2). То, что такое протонирование действительно происходит, подтверждается тем, что при растворении кетонов в концентрированной серной кислоте (в отсутствие воды) наблюдается депрессия температуры замерзания, вдвое большая, чем теоретическая; при этом идет следующая реакция:
C=O + H2SO4
с—он 4-н so; * 4
Карбокатионы могут образоваться также при протонировании атомов, содержащих неподеленную пару электронов, если протонируемый атом превращается вследствие этого в лучшую уходящую группу и ионизация таким образом ускоряется:
h2so4 + H2SO4
Ph3C—ОН ч=* HSO; + Ph3C—OH2 4=i Ph3C++ H3O++ 2HSO;
118
Эту реакцию можно сравнить с приведенным выше протонированием группы ОН, однако в этом случае отсутствует атом водорода, который мог бы (в виде Н+) отщепиться от соседнего атома углерода. Карбокатионы могут образоваться и при использовании кислот Льюиса
С=О: + А1С13 ч=*
С—ОА1С13
а также других катионов, например +NO2 при нитровании бензола (см. разд. 6.2):
Н NO2
В этом случае промежуточным соединением (1) является делокализованный карбокатион.
5.1.3. Образование карбокатионов из других катионов
Карбокатионы могут быть получены при разложении других катионов, например при разложении катионов диазония, образующихся при действии NaNO2 и НС1 на RNH2 (см. разд. 5.5):
[r— N=N*-*R-^-N=N] —R++N=Nt
Для получения карбокатионов используют также легко доступные карбокатионы; это позволяет получать менее доступные карбокатионы (см. разд. 5.2), например:
Ph3C+' +
5=±Ph3C—Н +
5.2. Стабильность и структура карбокатионов
Стабильность простых алкильных карбокатионов (см. разд. 4.3) уменьшается в ряду:
Ме3С+ > Ме2СН > МеСН2 > СН3
Это вызвано тем, что увеличение числа метильных групп у атома углерода карбокатиона приводит к увеличению делокализации положительного заряда (с последующей постепенно нарастающей стабилизацией) при участии как индуктивного
119
эффекта, так и эффекта сверхсопряжения (гиперконъюгации). Высокая стабильность иона Ме3С+ подтверждается тем, что этот катион часто возникает в жестких условиях в результате изомеризации других ранее образовавшихся катионов (ср. разд. 5.1.1), а также тем, что он остается неизменным после нагревания при 170 °C в SbF5/FSO3H в течение четырех недель!
Причиной такой стабилизации является плоское строение карбокатиона, поскольку только при плоской конфигурации делокализация может быть эффективной. Квантовомеханические расчеты простых алкил-катионов показывают, что плоская конфигурация (sp2) стабильнее пирамидальной (sp3) иа «84 кДж/моль (20 ккал/моль). Если плоская конфигурация нарушается (или же затрудняется ее достижение), стабильность катиона уменьшается, а следовательно, быстро увеличивается трудность его образования. Это было показано на примере 1-бромтриптицена (см. разд. 4.3), который крайне инертен по отношению к атаке по механизму SnI из-за неспособности «головного» атома углерода принять плоскую конфигурацию, что препятствует образованию карбокатиона. Ожидаемая плоская конфигурация простых катионов была подтверждена методами ЯМР- и ИК-спектроскопии для таких частиц, как Ме3С+ SbFe; они оказались структурно подобны изоэлектронным им триал-килборам R3B.
Основным фактором, определяющим стабильность более сложных катионов, является опять-таки возможность делокализации заряда, особенно если в этом могут участвовать л-орби-тали:
СН2=СН—СН2СН2—СН =СН2
+ 6+
Г: 6+!
Me—О —СН2Me—О=СН2
Об активности аллил- и бензилгалогенидов в реакциях SnI уже говорилось ранее. Из-за наличия свободной пары электронов на атоме кислорода сольволиз МеОСН2С1 протекает по крайней мере в 10м раз быстрее по сравнению с СН3С1.
Стабилизация может также осуществляться (по механизму делокализации) вследствие влияния соседней группы: это сопровождается образованием «мостикового» карбокатиона. Например, при действии SbFs в жидком SO2 на п-МеОСбН4СН2СН2С1 (2) образуется катион (3), а не ожидаемый катион (4), поскольку фенил действует как соседняя группа
120
(см. разд. 4.4.6 и 13.4.3.1):
Такие частицы с мостиковой фенильной группой называют феиоииевыми ионами. Эффект соседней группы еще более выражен при наличии группы ОН, а не ОМе в качестве заместителя в пара-положении. В сравнимых условиях сольволиз происходит в « 106 раз быстрее, что объясняется возможностью образования мостикового интермедиата (5), который уже ие является карбокатионом:
МеО^Н-гО л-о ~0
$ ч
Н2С-СН2-Вг Н2С—СН2’"ОМе СН2-СН2ОМе
(5)
Стабилизация по механизму делокализации может сопровождаться также ароматизацией. Было показано, что 1-бром-циклогептатриен-2,4,6 (тропилийбромид) (6), изомерный СбНзСНгВг, в отличие от последнего соединения, является твердым кристаллическим веществом (т. пл. 208 °C) и хорошо растворим в воде, т. е. он не обладает ковалентной структурой (6), а образует ионную пару (7). Причина такого поведения состоит в том, что циклический катион (7) имеет 6 л-электроиов, которые могут быть расположены иа трех молекулярных орбиталях, делокализованных с участием семи атомов углерода. Эта система -электронов, как и в молекуле бензола (см. разд. 1.3.6), подчиняется правилу Хюккеля (4п + 2; п= 1), а соединение в целом обнаруживает квазиароматические свойства:
121
В этом случае стабилизация плоского карбокатиона дости-гается переходом в ароматическое состояние. Существование такой делокализованной структуры подтверждается и тем, что в спектре ЯМР обнаруживается сигнал только одного протона, т. е. все семь атомов водорода эквивалентны. Эффективность такой ароматической стабилизации отражается в стабильности карбокатиона (7), превышающей в «10й раз стабильность сильно делокализованного катиона Ph3C+. Как уже отмечалось (см. разд. 5.1.3), катион (7) может быть генерирован действием Ph3C+ на циклогептатриен.
Особенно интересный случай стабилизации карбокатионов наблюдается в системах, в которых выполняется правило Хюк-келя (4л + 2; л = 0), т. е. в циклических системах с двумя л-электронами (см. разд. 1.3.6). Так, производные 1,2,3-трипропилциклопропена (8) очень легко образуют ионные пары, содер- / жащие соответствующий циклопропенил-катион (9).
Pr у Рг
Рг Рг Рг Рг (8) (9)
SbCle
Показано, что катион (9) более стабилен, чем катион (7) (в « 103 раза); даже в воде при pH 7 он примерно на 50 % все еще является карбокатионом. Совсем недавно удалось выделить в виде твердого белого кристаллического вещества ионную пару, содержащую циклопропенил-катион (10). Метод ЯМР 13С (ср. разд. 2.3.3) оказался полезным в этом случае, так как положение сигнала положительно заряженного углерода коррелирует с плотностью электронов на этом атоме (ср. разд. 13.7).
5.3. Реакции с участием карбокатионов
Карбокатионы могут участвовать в реакциях четырех основных типов: а) взаимодействие с нуклеофилом; б) отщепление протона; в) присоединение к ненасыщенной связи; г) перегруппировка.
Реакции первых двух типов приводят к образованию стабильных конечных продуктов, а реакции типов (в) и (г) — к образованию новых карбокатионов, которые могут участвовать в дальнейших превращениях. Это хорошо видно на примере реакции 1-аминопропана (11) с нитритом натрия и разбавленной хлороводородной кислотой [поведение катионов диазония, иа-
122
пример (12), рассмотрено в разд. 5.5]:
MeCH2CH2NH2 NHap,°'-» МеСН2СН2 г N = N —:т-> I4UI — 1’2
(11) (12)
+ н,о —МеСН2СН2 МеСН2СН2ОН (13) (14)
- Н + |(б) | (г) он
МеСН = СН2 MeCHCH3-^j->- МеСНСН3
(15) (16) (17)
Реакция 1-пропил-катиона (13) с водой [реакция типа (а)] дает пропанол-1 (14); отщепление протона от катиона (13) приведет к пропену (15) [реакция типа (б)]; перегруппировка катиона (13) [реакция типа (г) с миграцией Н_] приведет к 2-про-пйл-катиону (16). При отщеплении протона от катиона (16) образуется пропен (15), а при реакции с водой — пропанол-2 (17). В типичных условиях реакции в полученной смеси продуктов содержалось 7 % пропанола-1, 28 % пропена и 32 % пропанола-2. Более высокий выход пропанола-2 по сравнению с пропанолом-1 объясняется большей устойчивостью катиона (16) по сравнению с катионом (13).
Суммарный выход перечисленных выше продуктов составляет лишь 67 %, и, следовательно, при этом идут и другие реакции. Действительно, в системе присутствуют и другие нуклеофилы, например СГ и NO2, способные реагировать как с катионом (13), так и с катионом (16). При взаимодействии с NO? возможно образование как RNO2, так и RONO (нитриты могут образоваться и при прямой этерификации образующегося вначале ROH). Катионы (13) и (16) могут также реагировать с ранее образовавшимся ROH, давая эфиры ROR, или реагировать с исходным RNH2, образуя RNHR, который дальше может алкилироваться или нитрозироваться (ср. разд. 5.5). Наконец, любой из этих катионов может присоединиться к двойной связи ранее образовавшегося пропена (15) [реакция типа (в); ср. разд. 7.4.2] с образованием катионов +CHMeCH2R, которые сами могут участвовать в большом числе реакций. Состав смеси реально получаемых продуктов в значительной мере зависит от условий проведения реакции, поэтому неудивительно, что эту реакцию редко используют как удовлетворительный препаративный путь для превращения RNH2—>-ROH.
123
Реакция типа (г) осложняет также алкилирование бензола по Фриделю — Крафтсу (см. разд. 6.5) 1-бромпропаном МеСН2СН2Вг в присутствии бромида галлия GaBr3 — кислоты Льюиса, являющейся катализатором. В этом случае атакующий электрофил является сильнополяризованным комплексом R6++GaBr4--; большая стабильность этого комплекса, в котором Re++ несет положительный заряд в основном на вторичном, а не на первичном атоме углерода (т. е. Me2CHe++GaBr+~~, а не MeCH2CH2e++GaBrJ-~), и в этом случае приводит к гид-ридному сдвигу (см. выше), так что основным продуктом реакции является Me2CHC6Hs.
Такие перегруппировки не всегда протекают так просто, как кажется, например как простая миграция Н_. Это можно показать на примере взаимодействия 13СН3СН2СН3 с А1Вг3. В этом случае продукт реакции состоит из двух частей 13СН3СН2СН3 и одной части СН33СН2СН3, как определено масс-спектрометрическим анализом полученных фрагментов. Возможно, что перераспределение метки проходит при содействии протонированного циклопропанового интермедиата (18):
.С.Н2 ___ РН2
Н3 С Yh3 н3 С хСН2А1НВгз
СНз /С<?
/ + А1Вг-±=? ,, HAlBrZ
Н213С—СН3 Н213С—j—СН2 3
Н
(18)
Было показано, что метка находится (в течение нескольких часов) в исходном 2-пропил-катионе СН33СН+СН3, образующемся из СН33СНС1СН3 в присутствии SbF5 при —60 °C.
Реакции отщепления карбокатионов [реакции типа (б)] подробнее рассмотрены в разд. 9.2.
5.4. Перегруппировки карбокатионов
Перегруппировки с участием карбокатионов можно разделить на два типа: перегруппировки с изменением углеродного скелета и перегруппировки без изменения углеродного скелета. Более важны перегруппировки первого типа, однако сначала кратко остановимся на перегруппировках, протекающих без изменения углеродного скелета.
124
5.4.1. Перегруппировки, без изменения углеродного скелета
Ранее уже была рассмотрена перегруппировка такого типа (см. разд. 5.3): перегруппировка 1-пропил-катиона в 2-пропил-катион путем миграции атома водорода вместе с электронной парой (т. е. в виде Н_) от С-2 к С-1 карбокатиона, т. е. 1,2-гид-ридный сдвиг:
Н Н
сн3—сн—сн2 —► СНз—сн—<!:н2
Это свидетельствует о большей стабильности вторичного, а не первичного карбокатиона. Сдвиг в обратном направлении может, однако, происходить, если он предоставляет возможность для большей делокализации л-электронов бензольного кольца (т. е. при переходе от третичного карбокатиона к вторичному):
ОН Н Н
I FSo3H I * I
PhCH2—СМе2 —sbF--> PhCH—СМе2 —► PhCH—СМе2
Существуют и еще более интересные перегруппировки, присущие делокализованным катионам, например аллильные перегруппировки.
5.4.1.1. Аллильные перегруппировки
При сольволизе по механизму SnI З-хлорбутена-1 (19) в EtOH образуется не один эфир, а смесь двух изомерных простых эфиров; такая же смесь (т. е. такие же эфиры и приблизительно в тех же соотношениях) получается при подобном сольволизе 1-хлорбутена-2 (20):
OEt
МеСНСН=СН2
С1 /91)
I EtOH ' , ' EtOH
МеСНСН=СН2 -----► + ---- МеСН=СНСН2С1
(19) MeCH=CHCH2OEt }
(22)
Это можно обяснить тем, что в ходе обеих реакций образуется один и тот же промежуточный делокализованный аллильный катион (23) (ср. разд. 5.2), способный претерпевать последующую быструю нуклеофильную атаку молекулой EtOH как по атому С-1, так и по атому С-3:
[MeCH—СН=СН2 МеСН=СН—СН2] СГ (23)
Интересно, что если при реакции соединения (19) вместо этанола в качестве нуклеофила использовать ЕЮ- в очень высокой
125
концентрации, реакция протекает по механизму SN2 и образуется только эфир (21). Аллильные перегруппировки наблюдались и в ходе реакций замещения, которые протекают как бимолекулярный процесс. Такие реакции относят к реакциям типа SN2'; полагают, что они протекают следующим образом:
_ R R
О I р I
Nu”: *СН2 = CH - CH - С1 —► Nu - СН2- CH = СН + Cl-
Эти реакции легко протекают при наличии у а-углеродного атома объемистых заместителей, заметно уменьшающих скорость обычного замещения по механизму SN2 у этого атома. Адлильные перегруппировки распространены довольно широко, но установление детального механизма, по которому они протекают, является довольно сложным.
5.4.2. П ерегруппировки с изменением углеродного скелета
5.4.2.1. Неопентильные перегруппировки
Как уже отмечалось (см. разд. 4.3), гидролиз 1-бром-2,2-ди-метилпропана (неопентилбромид) (24) по механизму SN2 идет медленно из-за стерических затруднений. В условиях, благоприятствующих протеканию этой реакции по механизму SnI, скорость реакции увеличивается, однако продуктом реакции оказывается 2-метилбутанол-2 (28), а не ожидаемый 2,2-диме-тилпропанол (неопентиловый спирт) (26), т. е. происходит неопентильная перегруппировка:
sn> + н2о
Ме3С—СН2Вг -----► Ме3С—СН2 -Х->- Ме3С—СН2ОН
(24) (25) (26)
Me ОН
Ме\ /ме + | Н2О |
>С=С/ Ме2С—СН2 -----> Ме2С—СН2Ме
Ме/ \н
(29) (27) (28)
Большая стабильность третичного карбокатиона (27) по сравнению с исходным первичным карбокатионом (25) способствует расщеплению связи С—С, сопровождающему миграцию метильной группы вместе с ее электронной парой. Такие изменения углеродного скелета с участием карбокатионов называют перегруппировками Вагнера — Меервейна. Подтверждением участия катиона (27) является одновременное образование ал
126
кена — 2-метилбутена-2 (29) (вследствие потери протона), который не может образоваться из катиона (25).
Возможность протекания таких существенных перегруппировок углеродного скелета в ходе, казалось бы, однозначных реакций, очевидно, наиболее важна при интерпретации результатов экспериментов по установлению структуры, особенно в том случае, когда фактически получаемый продукт является изомером ожидаемого продукта. Некоторые перегруппировки этого типа очень сложны, например в случае образования природных продуктов, таких как терпены, и часто чрезвычайно трудно дать однозначную оценку механизма такой реакции. Для решения этого вопроса нельзя использовать данные о предполагаемом строении продуктов реакции; строение этих продуктов должно быть доказано обычным путем. Ценную информацию можно получить с помощью методов ЯМР ’Н и ЯМР 13С.
Интересно отметить, что в то время как бромид неопентильного типа (30) перегруппировывается в ходе гидролиза, протекающего по механизму SnI, его фенильный аналог не подвергается перегруппировке:
Вг Me
I SN> . + I
Ме3С—СНМе ------► Ме3С—СНМе ——► Ме2С—СНМе Продукты
(30)
Вг Me
I SN1 + + |
Ме3С—CHPh ------► Ме3С—CHPh — х-> Ме2С—CHPh
(31) (32) (33)
I
Продукты
Это свидетельствует о большей стабильности бензильного катиона (32), хотя он является вторичным, а не третичным, как катион (33), который мог бы образоваться, но в действительности не образуется, в ходе перегруппировки (ср. разд. 5.2).
5А.2.2. Перегруппировки углеводородов
Перегруппировки типа Вагнера — Меервейна происходят также в процессе крекинга углеводородов нефти, при котором в качестве катализаторов используют кислоты Льюиса. Такие катализаторы генерируют карбокатионы из неразветвленных алканов (ср. изомеризацию меченного 13С пропана; разд. 5.3). Получаемые карбокатионы стремятся перегруппироваться в продукты с разветвленной цепью. В процессе крекинга происходит также расщепление углеродного скелета, однако процессы изомеризации особенно важны, потому что образующиеся при
127
этом алканы вызывают меньшую детонацию в двигателях внутреннего сгорания по сравнению с их неразветвленными изомерами. Необходимо упомянуть, однако, что крекинг нефти можно проводить в присутствии катализаторов, которые способствуют протеканию радикальных реакций (см. разд. 11.2.2).
Перегруппировка алкенов легко протекает в присутствии кислот:
н+ +
Ме3С—СН=СН2 Ме3С—СН—СН3 —►
Ме + I -н+
—> Ме2С—СН—СН3 *=* Ме2С=СМе2
Эта относительно легко протекающая перегруппировка может оказаться досадной помехой при присоединении кислот, например галогеноводородов (см. разд. 7.3), к алкенам или при катализируемой кислотами гидратации алкенов (см. разд. 7.4.2); при этом могут получаться трудно разделяемые смеси продуктов или (в неблагоприятных условиях) целевой продукт вообще может быть не получен. Кроме того, может происходить присоединение карбокатионов к исходным алкенам или продуктам реакции (см. разд. 7.4.3).
Перегруппировки ди- и полиалкилбензолов также легко происходят в присутствии кислот Льюиса (см. разд. 6.7.4) и в ходе диенон-фенольной перегруппировки (см. разд. 5.4.2.3).
5.4.2.3. Пинаколиновая перегруппировка
Еще одним примером миграции группы, в простейшем случае Ме, к атому углерода карбокатиона является катализируемая кислотами перегруппировка 1,2-диолов в кетоны, например перегруппировка пинакона (34) (ср. разд. 8.2.7) в пинаколин (37):
НО ОН НО г ОН,
I I н+ 14. -HjO МегС — СМе2 ч Ме2С—СМе2ч- *
ОН
I + Ме2С—СМе2---*•
(34) (35)
НО: Ме НО Ме
II II I МеС—СМе2^-> МеС—СМег
О Ме
II । МеС—СМе^
(37)
(36)
1,2-Сдвиг метильной группы в катионе (35), который уже является третичным карбокатионом, обусловлен сверхстабилизацией, достигаемой в карбокатионе (36) в результате делокализации заряда с участием пары электронов атома кислорода;
128
катион (36) может также легко отдавать протон с образованием стабильного конечного продукта (37). Можно ожидать, что аналогичная реакция могла бы проходить с другими соединениями, способными образовывать ключевой карбокатион (35). Это в действительности и было обнаружено. Так, соответствующий 1,2-бромгидрин (38) и 1,2-аминоспирт (39) образуют пинаколин (35) при обработке их ионами Ag+ и смесью NaNO2—НС1 соответственно:
НО Вг
II Ад *
Ме2С—СМе2 --------*- (35) > (37)
(38) -АдВг 4 j
НО NH2 НО xN=N
I I NaN02 I Ч
Ме2С—СМе2------>- Ме2С—СМе2
(39) НС’
Был проведен ряд экспериментов, чтобы определить относительную миграционную способность различных групп в пинако-линовой перегруппировке; по относительной легкости миграции группы располагаются в ряд:
Ph > MejC > МеСН2 > Me
Необходимо представлять, что существуют значительные трудности в выборе подходящих моделей для таких экспериментов и интерпретации результатов, которые при этом получаются. Так, при перегруппировке диола (40) показано, что мигрирует именно Me, а не Ph, как можно было бы ожидать из приведенного выше ряда. Однако в этом случае реакция контролируется преимущественным протонированием той ОН-груп-пы, при котором первоначально образуется более стабильный карбокатион (41) [а не (42)], что и предопределяет предпочтительность миграции Me, а не Ph:
НО ОН Н2СК ОН ОН
I I Н+ Р I +1
Ph2C—СМе2----► Ph2C—СМе2—0» Ph2C—СМе2
(40) 2 (41)
^Н+
НО ^6н2 но
Ph2C—СМе ц > Ph2C—СМе,
О * *
(42)
5 П. Сайкс
129
Такого рода затруднений можно избежать, выбирая симметричные 1,2-диолы, например (43):
1. н+
2. -Н2О
(43)
Ph. zPh
>-CZ
Аг | ^Аг НО
(44)
4
(45)
В опытах с такими соединениями при определении относительных количеств образующихся кетонов (44) и (45) было установлено, что относительная миграционная способность различных групп уменьшается в ряду:
n-MeOC6H4 > n-MeC6H4 > С6Н5 > п-С1С6Н4 > о-МеОС6Н4
500 15,7 1,0 0,7 0,3
Эта последовательность может быть объяснена (кроме о-МеОСбН4), исходя из уменьшения электронодонорности мигрирующей группы, которая перемещается со своей электронной парой к положительному центру—атому углерода в карбокатионе. Электронодонорными свойствами мигрирующей группы можно также объяснить последовательность, полученную для алкильных групп (см. выше). Группа о-МеОС6Н4, несмотря на то что она является электронодонорной, мигрирует медленнее, чем C6Hs. Большое значение имеют также относительные напряженности переходных состояний. В приведенном выше случае с о-МеОС6Н4 они могут стать преобладающими над электронными эффектами.
Перегруппировкой, которая напоминает перегруппировку, обратную пицаколиновой (ретропинаколиновую перегруппировку), является диенон-фенольная перегруппировка:
(46) <47)
В ходе этой перегруппировки протонирование исходного дие-дона (46) позволяет полностью восстановить ароматическое со-
130
стояние (47) в результате 1,2-миграции алкильной группы:
5.4.2.4. Стереохимия перегруппировок
Для выяснения стереохимии карбокатионных перегруппировок важно получить ответы на следующие вопросы: что происходит с конфигурацией у атома углерода, от которого уходит мигрирующая группа (источник миграции); что происходит с конфигурацией у атома углерода, к которому присоединяется мигрирующая группа (катионный атом углерода, конечный пункт миграции); что происходит с конфигурацией мигрирующей группы, если она хиральна (например, PhMeCH)? Довольно интересно, что для одного и того же соединения на все эти вопросы никогда не были получены ответы, несмотря на огромное число работ, выполненных по исследованию карбокатионов.
Показано, что мигрирующая группа не становится свободной во время перегруппировки. В совместном эксперименте с двумя пинаконами (48) и (49), которые очень сходны по строению и перегруппировываются почти с одинаковой скоростью, но имеют различные мигрирующие группы (перекрестный эксперимент), перекрестная миграция не была обнаружена:
Et О “ 1 II РЬ2С—СМе или Ме О 1 II _ Ph2C—CEt _ <~Х-— - но он ~ 1 1 РЬ2С—СМе2 (48) + НО он 1 1 _ Ph2C—CEt2_
(49)
Ме ОН ~ I I Ph2C—СМе +
Et О
I II Ph2C—CEt _
Аналогично, при проведении перегруппировок, в которых происходит гидридный сдвиг (ср. разд. 5.4), в дейтерированном растворителе (например, D2O, MeOD и т. д.) дейтерий не обнаруживается в новой С—H(D)-связи конечного продукта перегруппировки. В обоих рассмотренных случаях, таким образом, перегруппировка протекает строго внутримолекулярно, т. е. мигрирующая группа не отделяется от остальной части молекулы,
5*
131
в отличие от межмолекулярной перегруппировки, в ходе которой это происходит.
Это дает возможность предположить, что мигрирующая группа R очень тесно связана с конечным пунктом миграции до полного разрыва ее связи с источником миграции; таким образом можно было бы рассчитывать, что изменение конфигурации невозможно, т. е. конфигурация мигрирующей хиральной группы R* должна сохраняться. Это подтверждается приведенной ниже реакцией (ср. разд. 5.4.2.4), при которой хиральная группа R* действительно мигрирует с сохранением ее конфигурации:
R‘ I н2о
• nariVn | + I при |
МеСН—СН2------->-МеСН—СН2——>-МеСН—СН2——>-МеСН—СН
• игч _» —К1„ I
| НС1
NH2
ОН
(+)
R* = Ме2СНСН2СНМе
I
Что касается первых двух вопросов (см. выше), то экспериментальные данные подтверждают преобладание обращения конфигурации как в источнике миграции (а), так и в ее конечном пункте (б):
Показано, что в циклических соединениях, где вращение во-Г связи С-1—С-2 сильно затруднено, наблюдается почти пол-обращение конфигурации; в значительной мере обращение конфигурации обнаруживается и в ациклических соединениях. Это можно объяснить, исходя из представления о мостиковом интермедиате (ср. бромониевый ион; разд. 7.1) или переходном состоянии:
Однако нельзя считать, что образование мостика во время перегруппировки происходит обязательно, даже в том случае, когда мигрирующей группой является СвНэ, л-орбитальная система которой, как можно было бы ожидать, способствует ста
132
билизации мостикового карбокатиона посредством делокализации (см. разд; 5.2).
Это отчетливо показано при пинаколиновом дезаминировании (разд. 5.4.2.3) оптически активной формы аминоспирта (50). Такие реакции происходят через антиперипланарную конформацию (50а) или (506), в которой мигрирующая (Ph) и уходящая (:NH2 в виде : N2; ср. разд. 5.4.2.3) группы находятся в транс-положении друг к другу. Перегруппировка через мостиковый карбокатион неизбежно должна привести к 100 %-му обращению конфигурации в конечном пункте миграции образующегося кетона (51а), независимо от того, какая исходная конформация — (50а) или (506) — участвовала в реакции:
Me Н Ph
(50а) (-) (51а) (+)
Me Н
Ph ОН
(506) (-)
Фактически найдено, однако, что, хотя обращение конфигурации и было преобладающим [выход кетона (51а) составил 88 %], в продукте реакции содержалось значительное количество (12%) зеркального изомера (516); таким образом, суммарная реакция не может протекать только через образование мостикового карбокатиона. Простейшее объяснение состоит в том, что хотя бы часть общей перегруппировки происходит через немостиковый карбокатион (52а), в котором возможно вращение вокруг связи С-1—С-2 [(52а) (526)], что ведет к образо-
ванию кетона (516), в котором сохранилась исходная конфигурация [ср. (50а) или (506) и (516)]:
Соотношение продуктов обращения (51а) и сохранения конфигурации (516) в таком случае могло бы быть определено относительной скоростью вращения вокруг связи Q-1—С-2 по сравнению со скоростью миграции группы Ph.
5.4.2.5. Перегруппировка Вольфа
Эта перегруппировка рассматривается отдельно от истинных карбокатионных перегруппировок, поскольку она включает миграцию к незаряженному, хотя и электроиодефицитному,
133
карбеноподобному атому углерода (ср. разд. 9.8), а не к положительно заряженному атому углерода. Реакция включает отщепление азота от а-диазокетонов (53) и перегруппировку с образованием очень реакционноспособных кетенов (55):
/R
>С—СН—N=N------/С—СН—> О=С=С'
// U. -n2 V \
(53) (54) (55)
Кетены затем могут легко реагировать с любым нуклеофилом, присутствующим в системе, например с Н2О.
Реакция может осуществляться в ходе фотолиза, термолиза или обработкой оксидом серебра. В первых двух случаях карбеновый интермедиат (54), возможно, образуется, как показано выше; в реакции, катализируемой серебром, выделение азота и миграция R могут происходить более или менее одновременно. Если группа R хиральна, например С4Н9С*МеРЬ, то она, как было показано, мигрирует с сохранением конфигурации (см. разд. 5.4.2.4).
Диазокетоны (53) могут быть получены путем действия диазометана CH2N2 на хлорангидрнды кислот; последующая перегруппировка Вольфа в присутствии воды приводит к образованию гомолога исходной кислоты (метод Арндта — Эйстерта):
О О о
II SOCI2 II ch2n2 1|
RC—ОН ------* RC—С1 -----*- RC—CHN2
(53)
Н ОН
н2о | | “
—► RCH=C=O -------RCH—С=О
Ag2O'
—N2
(55)
При проведении реакции не в воде, а в аммиаке или спирте присоединение также происходит по связи С=С кетена с образованием соответственно амида или сложного эфира гомолога исходной кислоты.
Перегруппировка Вольфа имеет формальное сходство с реакцией Гофмана и родственными ей реакциями (ср. разд. 5.6.1), в которых миграция происходит к электронодефицитному атому азота с образованием в качестве интермедиата изоцианата RN = C=O.
5.5. Катионы диазония
Нитрозирование первичных аминов RNH2, например, нитритом натрия и разбавленной кислотой (ср. разд. 5.3) ведет к об
134
разованию катионов диазония (56):
Н
I
RN—N=0 ~+±
•• 1 н^* Г •’ + + "1
—Ч± RN=N—ОН—;► I RN=N-*—->-R-rN=N---------------R+
2.-НгО L О J -n2|
(56)
Сама HNO2, вероятно, не является эффективным нитрози-рующим агентом; полагают, что при относительно низкой кислотности среды она существует в виде частицы N2O3 (X =* = ONO), образующейся в ходе превращения:
2HNO2 ONO—NO + H2O
При увеличении кислотности более эффективной нитрози-рующей частицей становится протонированная азотистая кислота Н2О+—NO (Х = Н2О), а затем ион нитрозония +NO (ср. разд. 6.1.1). Однако при проведении нитрозирования должен быть найден компромисс между возрастающей эффективностью нитрозирующего агента при увеличении кислотности раствора и снижением концентрации исходного амина вследствие его протонирования и снижения, тем самым, его реакционной способности.
В случае простых алифатических аминов катионы диазония (56) расщепляются чрезвычайно легко с образованием карбокатионов (ср. разд. 5.3), которые по не совсем ясным еще причинам гораздо 'более реакционноспособны, чем карбокатионы, образующиеся в других гетеролитических процессах, например RBr-*-R+ Вг-. В тех случаях, когда основной целью превращения является генерирование карбокатионов, нитрозированию лучше подвергать производные амина (чтобы избежать образования Н2О) в безводной среде:
coci2 +no sbFe-
RNH2-------RNCO -----------► R+ SbF6- + N2f + CO2f
Однако если R содержит сильную электроноакцепторную группу, то отщепляется Н+, а не молекула N2, с образованием замещенных диазоалканов. Примером такой реакции является превращение этиламиноацетата в этилдиазоацетат:
Н2МСНгСО2Е1
NaNO2 ------►
НС1
N=N-r-CHCO2Et —-> N=N=CHCO?Et
<| -н+ 2
H
135
Неустойчивость алифатических диазониевых катионов в отсутствие какого-либо стабилизирующего структуру фактора в очень большой степени обусловлена эффективностью N2 как уходящей группы. В ароматических диазониевых катионах стабилизирующим фактором является л-орбитальная система ароматического ядра:
. Поскольку первичные ароматические амины — более слабые основания (нуклеофилы), чем алифатические (вследствие взаимодействия электронной пары атома азота с л-электронами ароматического ядра), для их нитрозирования требуется довольно сильный нитрозирующий агент; поэтому реакцию проводят в условиях относительно высокой кислотности, при этом сохраняется достаточная равновесная концентрация непротонированного ароматического анилина (так как он является слабым основанием). В то же время эта концентрация достаточно низка, что препятствует еще недиазотированному амину вступить в реакцию сочетания с ранее образовавшимся ArN2 (ср. разд. 6.6).
Ароматические диазонийхлориды, -сульфаты, -нитраты и т. д. довольно стабильны в водном растворе при комнатной или более низкой температуре, но не могут быть легко выделены без разложения. Фторбораты ArN2 BFi более стабильны (ср. стабилизирующее влияние BFi на другие ионные пары; разд. 6.1.1) и могут быть выделены в твердом состоянии; термолиз сухих твердых фторборатов является важным препаративным методом получения фтораренов:
д
ArNj BF< ---► Аг— F + N2f + BF3^
Как и следовало ожидать, заместители в ароматическом ядре заметно влияют на устойчивость катиона ArN2, причем электронодонорные группы имеют заметный стабилизирующий эффект:
Me2N---------N=N Me2N=</ ^>=N=N
Нитрозированию могут подвергаться и вторичные амины, но в этом случае реакция останавливается на стадии образования устойчивого А-нитрозопроизводного R2N—N=O. Третичные алифатические амины сначала превращаются в нитрозотриалкил-аммоний-катион R3N+—N=O, после чего легко разрывается связь С—N с образованием относительно сложных продуктов реакции. В случае ароматических третичных аминов ArNR2 ни-
136
трозирование также происходит, но не по атому азота, а по активированному пара-положению ядра (ср. разд. 6.1.1) с образованием С-нитрозосоединения:
5.6. Миграция к электронодефицитному атому азота
Все рассмотренные выше перегруппировки имеют одну общую черту: миграция алкильной или арильной группы со своей электронной парой происходит к атому углерода, который, независимо от того, является ли он карбокатионным центром или нет, является электронодефицитным. Другим атомом, который может стать электронодефицитным, является атом азота, например, в составе частицы R2N+ или RN: (нитрен; ср. с карбеном). Можно ожидать, что и к таким центрам будет происходить миграция алкильных илн арильных групп, как это было в случае R3C+ и R2C :.
5.6.1. Реакции Гофмана, Курциуса, Лоссена и Шмидта
Типичным примером реакций этого типа является превращение амида (57) в амин (62), содержащий на один атом углерода меньше, под действием гипобромита в щелочной среде (реакция Гофмана):
(57)
ВгО-
>С=0-----*
H2Nz
“ОН R'
"N-rBr
(58) (59)
Н
t- + “1 НгО I -он
RN—С=о -<-> RN—С—О *-RN~C=O-----------RNH2
J | -НСО3
ОН
(60) (61) (62)
Интересным промежуточным соединением в этой реакции является изоцианат (60), очень напоминающий кетеновый интермедиат в реакции Вольфа (см. разд. 5.4.2.4). Он также присоединяет воду, но образующаяся при этом карбаминовая кислота (61) неустойчива н легко декарбоксилируется с образованием
137
аминй (62). В тщательно контролируемых условиях удаетйя выделить бромамид (58), его анион (59) и изоцианат (60) в качестве интермедиатов; предполагаемый механизм реакции, таким образом, необыкновенно хорошо обоснован. Скоростьлими-тирующей стадией реакции является, вероятно, отщепление иона Вг- от изоцианата (60). При этом возникает вопрос, протекает ли это отщепление согласованно с миграцией группы R, или же образуется карбонилнитреновый интермедиат RCON:, который затем перегруппировывается. Тот факт, что перегруппировка ArCONH2 ускоряется, когда Аг содержит электронодонорные заместители (ср. пинаколиновую перегруппировку; разд. 5.4.2.3), а также то, что образование гидроксамовой кислоты RCONHOH (которое можно было бы ожидать в результате атаки растворителя Н2О на RCON:) никогда не было обнаружено, подтверждают согласованный механизм. Перекрестные эксперименты не приводят к смешанным продуктам, что указывает на строго внутримолекулярный характер перегруппировки. Наконец, было показано, что если группа R хиральна, например С6Н5МеСН, то при миграции ее конфигурация остается неизменной.
Известны реакции (реакции Лоссена, Курциуса и Шмидта), родственные реакции Гофмана. Каждая из них включает образование изоцианата (60) в результате перегруппировки интермедиата, аналогичного соединению (59):
Rx -он R4
\с=О ?>С=О ------R—N=C=O
HN—OCOR' "N-^OCOR’
(63) (64) (6°)
।1 R4 NaNOz R\ HN3 R\
\c=o------->C=O -«------— ;C=O
/ HCt /ъ e /
HN—NH2 6 "n—N^N H0
(65) (67) (66)
a - реакция Лоссена; б - реакция Курциуса; а - реакция Шмидта
Реакция Лоссена протекает при действии основания на О-ацилпроизводное (63) гидроксамовой кислоты RCONHOH; группой, отщепляющейся от интермедиата (64), является группа R'CO2 [ср. отщепление Вг-от интермедиата (59)]. Реакция протекает (.также и с самой гидроксамовой кислотой, но не так легко, как с ее ацилпроизводными, поскольку R'COz — лучшая уходящая группа, чем -ОН. Согласованный механизм перегруп
138
пировки подтверждается тем фактом, что эта реакция облегчается не только при наличии электронодонорных заместителей в остатке R (ср. с реакцией Гофмана), но и при наличии электроноакцепторных заместителей в R'; таким образом, обе эти группы участвуют в скоростьлимитирующей стадии реакции. Как в реакции Курциуса, так и в реакции Шмидта изоцианат образуется в результате отщепления N2 от азидного интермедиата (67). В этих реакциях миграция R также протекает по согласованному механизму. Азид может быть получен при нитрози-ровании гидразида (65) (реакция Курциуса) или при взаимодействии азидоводородной кислоты HN3O с карбоновой кислотой (66) (реакция Шмидта).
5.6.2. Перегруппировка Бекмана
В ряду перегруппировок, при которых группа R переходит от атома углерода к атому азота, наиболее известной, безусловно, является перегруппировка, сопровождающая превращение ке-токсимов в А-замещенные амиды — перегруппировка Бекмана: RR'C=NOH —► R'CONHR или RCONHR'
Эта реакция катализируется различными кислотными реагентами, например H2SO4, SO3, SOC12, Р2Оэ, PCI5, BF3 и т. д., и протекает не только с самими кетоксимами, но и с их О-эфи-рами. В обычных условиях проведения этой реакции только немногие альдоксимы претерпевают перегруппировку, однако при использовании в качестве катализаторов полифосфорной кислоты их число увеличивается. По-видимому, наиболее интересной особенностью этой перегруппировки является то, что, в отличие от уже рассмотренных выше реакций, вопрос о том, какая из групп, R или R', будет мигрировать, определяется не природой этих групп (в частности, не их относительной электронодо-норностью), а их пространственным расположением. Было показано, что практически всегда к атому азота мигрирует группа R, находящаяся в снты-положении к группе ОН (т. е. образуется только R'CONHR):
R^ /R' НОХ ZR' /R'
С кислота С с
II ---------*- II I
: N N: N
\эн r/ , r/ Ni
Для подтверждения этого требуется достоверно установить конфигурации пары оксимов. Это было сделано следующим образом. Показано, что при обработке оксимов (68) ч (69) щелочью один из них циклизуется в бензизоксазол (70) на холоду, тогда как другой устойчив к циклизации даже в гораздо
139
более жестких условиях. Исходя из этого, легко циклизующемуся оксиму была приписана конфигурация (68), в которой атом углерода кольца, связанный с атомом брома, и атом кислорода группы ОН, которая атакует атом углерода, находятся в непосредственной близости:
В оксиме (69), устойчивом к циклизации, реагирующие атомы удалены друг от друга и могут оказаться на расстоянии, достаточном для взаимодействия, только при расщеплении связи C=N в этом оксиме. Конфигурации других пар оксимов могут быть установлены путем сравнения их физических констант с соответствующими константами тех пар оксимов, конфигурации которых уже установлены. Поскольку надежно показано, что в бекмановской перегруппировке мигрирует группа R, находящаяся в цнты-положении, по структуре образующегося амида можно судить о конфигурации оксима, из которого он образуется. Например, как и ожидалось, было найдено, что из соединения (68) образуется только М-метилзамещенный бензамид (71), тогда как из соединения (69) — только арилзамещенный ацетанилид (72):
О
II
АгС—NHMe
О
II MeC-NHAr
(68) (71) (69) (72)
Отсутствие прямого обмена групп R и ОН было показано путем проведения перегруппировки Ph?C = NOH оксима бензофенона в бензанилид PhCONHPh в среде Нг8О. Поскольку ни исходный оксим, ни образующийся анилид не обменивают свой кислород на 18О при растворении в Нг8О (как это было точно установлено), простой внутримолекулярный обмен между Ph и ОН не может привести к включению !8О в продукт перегруппировки. Оказалось, что образующийся бензанилид содержит 18О в той же пропорции, что и исходная вода. Это свидетельствует о том, что перегруппировка должна происходить с отщеплением ОН-группы оксима и последующим введением кислорода из молекулы воды. Основная функция кислотного катализатора фак
140
тически состоит в том, чтобы превратить ОН-группу оксима в лучшую уходящую группу с помощью цротонирования, этерификации и т. д.
Предполагают, что перегруппировка протекает по схеме:
О НО
)С— NHR^r~ pc=NR
FT R'
(77) (76)
(75)
В присутствии сильных кислот перегруппировка протекает путем О-протонирования оксима с образованием соединения (73а) с последующим отщеплением воды и образованием катиона (74). При действии на оксимы хлорангидридов, PCI5 и т. д. образуется промежуточный эфир (736), в котором анион -ХО~ является хорошей уходящей группой, что также способствует образованию промежуточного соединения (74). Был получен ряд эфиров (736), и было показано, что они легко перегруппировываются в ожидаемые амиды в отсутствие кислотного катализатора в нейтральных растворителях. Чем сильнее кислота ХОН, т. е. чем более способен анион к независимому существованию, тем лучшей уходящей группой должна быть ХО~ и тем быстрее, следовательно, будет происходить перегруппировка. Это наблюдается в следующем ряду ХО": СН3СО2 < <С1СНаСО2 < РИБОз- Утверждение, что указанная ионизация является скоростьлимитирующей стадией реакции, согласуется с тем наблюдением, что скорость реакции увеличивается с повышением полярности растворителя.
Как и в случае перегруппировок, рассмотренных выше, отщепление уходящей группы и миграция R, как полагают, происходят преимущественно одновременно в ходе превращения (73) в (74). Это подтверждается четким внутримолекулярным характером реакции (не обнаружены никакие перекрестные продукты; ср. разд. 5.4.2.4), уже упоминавшейся высокой стереоселективностью (мигрирует только R, но не R') и тем фактом, что
141
группа R, если она хиральна, например PhCHMe, сохраняет конфигурацию при миграции. Возможно, это отражает также большую стабильность катионного интермедиата (74) (который был обнаружен методом ЯМР-спектроскопии) по сравнению с интермедиатом RR'C=N+, который мог бы быть получен, если бы отщепление уходящей группы предшествовало миграции R. Перегруппировка завершается атакой воды (именно на этой стадии происходит включение |8О в рассмотренной выше перегруппировке оксима бензофенона в среде 18ОН2) катионного атома углерода интермедиата (74) с образованием соединения (75) с последующим отщеплением протона и образованием енольной формы (76) амида (77).
Об использовании бекмановской перегруппировки для выяснения вопросов стереохимии при установлении конфигурации кетоксимов уже упоминалось; эта реакция применяется также при промышленном синтезе полимера найлона-6, применяемого при изготовлении синтетических волокон, из оксима циклогексанона (78) через циклический амид (лактам) (79):
5.7. Миграция к электронодефицитному атому кислорода
Можно ожидать, что должны существовать аналогичные перегруппировки, в которых конечным пунктом миграции является электронодефицитный атом кислорода; действительно, такие перегруппировки известны.
5.7.1. Окисление кетонов по Байеру — Биллигеру
Окисление кетонов пероксидом водорода или пероксикисло-тамн RCO2OH (ср. разд. 11.5.2.2) приводит к сложным эфирам:
О О
II НгО2 ||
R—С—R ----->• R—С—OR
142
Циклические кетоны превращаются в лактоны (циклические эфиры):
н2о2
Реакции, вероятно, протекает по следующей схеме:
О
R—С—R
ОН I 1.RCO3H
R—С—R -----—
+ 2.-Н+
ОН
R—С—R —;---
-R COJ
R'COO-rO
(80)
(81)
О-т-Н
О
| ~н+
OR
(82)
R—С
OR (83)
Протоиирование кетона (80) с последующим присоединением пероксикислоты приводит к аддукту (81); отщепление хорошей уходящей группы RCO2 и миграция R к образующемуся электронодефицитному атому кислорода приводят к протонированной форме (82) эфира (83). Этот механизм подтверждается тем фактом, что окисление 18O=CPh2 приводит к образованию только PhC18OOPh, т. е. не наблюдается «разбрасывания» метки 18О в образующемся эфире. То, что отщепление R'CO2 и миграция R протекают согласованно, подтверждается ускорением реакции при наличии электроноакцепторных заместителей в уходящей группе R' и электронодонорных заместителей в мигрирующей группе R: согласованное превращение (81) в (82), таким образом, оказывается скоростьлимитирующей стадией реакции. Показано также, что хиральная группа R мигрирует с сохранением конфигурации. При окислении несимметричного кетона RCOR' могла бы мигрировать любая из двух групп, однако экспериментально показано, что мигрирует, обычно более нуклеофильная группа, способная лучше стабилизировать положительный заряд (ср. с пинаколиновой перегруппировкой; разд. 5.4.2.3). Однако, например в последней реакции, стерические эффекты также имеют значение и могут иногда заметно изменять порядок миграционной способности групп, ожидаемый на основе их элек-тронодонорности.
143
5.7.2. Перегруппировки пероксидов
Перегруппировка, очень сходная с описанной выше реакцией Байера — Виллигера, наблюдается при катализируемом кислотой разложении гидропероксидов ROOH, где остаток R — содержит вторичный или третичный атом углерода, связанный с алкильными или арильными группами. Хорошим примером является разложение гидропероксида (84), полученного при окислении кумола (изопропилбензол) кислородом воздуха; этот процесс используют в промышленности для получения фенола и ацетона:
О—ОН О-гОНо OPh
1 Н+ Л । и J 1-Н2°
Ph—СМе2 Ph—СМе2 -------------> СМе2-----►
(84) (85) "Н2° (86) 2’"н+
OPh
I н+, н2о
—► НО—СМе2---------► О=СМе2 + PhOH
(87)
И в этом случае отщепление уходящей группы (Н2О) и миграция Ph к образующемуся электронодефицитному атому кислорода в (85) очевидно согласованны. Присоединение воды к карбокатиону дает полуацеталь (87), который легко гидролизуется при данных условиях реакции до фенола и ацетона. Показано, что в (85) преимущественно мигрирует Ph, а не Me, как и можно было ожидать на основе предыдущего опыта. Показано, что наличие электронодонорных заместителей в мигрирующей группе увеличивает скорость реакции и усиливает способность такой группы к миграции по сравнению с ее незамещенным аналогом. Возможно, что превосходная способность к миграции фенильной группы объясняется образованием мостикового переходного состояния:
6t zCMe2
Phf | 6+
'О—он2 61-
Интермедиаты типа (86) были обнаружены в растворах суперкислот (ср. разд. 5.1.1); их строение подтверждено методом ЯМР-спектроскопии. • •
В приведенных примерах рассмотрено по существу гетеро-литическое расщепление пероксидных связей —О: О— —► —>—О+:О' в полярных растворителях. В подходящих условиях возможно и гомолитическое расщепление с образованием радикалов—О: О— —> —О- "О— (см. разд. 11.2.2).
144
Глава 6
ЭЛЕКТРОФИЛЬНОЕ И НУКЛЕОФИЛЬНОЕ
ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКИХ СИСТЕМАХ
6.1. Электрофильная атака бензола..............................145
6.1.1. л- н <т-Комплексы................................. 145
6.2. Нитрование................................................148
6.3. Галогенирование............................................153
6.4. Сульфирование.............................................155
6.5. Реакции Фриделя — Крафтса.................................156
6.5.1. Алкилирование.......................................156
6.5.2. Ацилирование........................................160
6.6. Азосочетание............................................. 162
6.7. Электрофильная атака монозамещенных бензолов..............166
6.7.1. Электронные эффекты заместителей....................167
6.7.1.1. У = +NR3, CCI3, NO2, СНО, СО2Н и др..........168
6.7.1.2. У = Aik, Ph..................................169
6.7.1.З. У = OCOR, NHCOR, OR, ОН, NH2, NR2............171
6.7.1.4. - У = Cl, Вг, I..............................173
6.7.2. Факторы парциальных скоростей и селективность.......174
6.7.3. Соотношение орто- и мра-нзомеров....................176
6.7.4. «псо-Замещение......................................179
6.8. Кинетический и термодинамический контроль.................181
6.9. Электрофильное замещение в других ароматических системах . . 182
6.10. Нуклеофильное замещение в ароматических системах.........186
6.10.1. Замещение атомов водорода................... . . . 186
6.10.2. Замещение неводородных атомов......................188
6.10.3. Замещение по ариновому механизму...................193
Ранее уже обсуждался вопрос о структуре бензола и, в частности, о его делокализованных л-орбиталях (см. разд. 1.3.6). Для бензола характерно расположение электронного облака над и под плоскостью кольца, образованного атомами углерода. Можно было бы ожидать, что это будет защищать кольцо атомов углерода от атаки нуклеофильными реагентами и, наоборот, облегчать атаку катионами Х+ или электронодефицитными, т. е. электрофильными, реагентами. Показано, что оба эти предположения действительно реализуются.
6.1. Электрофильная атака бензола
6.1.1. л- и а-Комплексы
Можно предположить, что на первой стадии реакции происходит взаимодействие между соответствующим электрофилом н делокализованными л-орбиталями. Действительно, показано,
145
что при этом образуются так называемые л-комплексы (1):
Так, метилбензол (толуол) при —78 °C образует с хлороводородом комплекс состава 1 : 1, причем реакция легко обратима. Тот факт, что не происходит образования связи между атомом углерода кольца и протоном из НС1, подтверждается при проведении этой реакции с DCL В этом случае также возникает л-Комплекс, но его образование и распад не приводят к обмену дейтерия на атом водорода кольца. Это означает, что связь С—D в комплексе не образуется. Ароматические углеводороды образуют л-комплексы также с галогенами и ионами Ag+; хорошо известны комплексы с пикриновой кислотой 2,4,6-(O2N)3C6H2OH, представляющие собой устойчивые окрашенные кристаллические соединения, температуры плавления которых можно использовать для характеристики углеводородов. Такие аддукты называют комплексами с переносом заряда. Было показано, что в комплексе, который бензол образует с бромом, молекула галогена ориентирована вдоль оси симметрии Сб перпендикулярно плоскости бензольного кольца.
В присутствии соединения, имеющего вакантную орбиталь, например кислоты Льюиса типа А1С13, образуется комплекс, отличающийся от описанного выше. Если в такой системе заменить НС1 на DC1, то атом водорода быстро обменивается на дейтерий. Такой обмен свидетельствует об образовании о-комплек-са * (2), который называют также интермедиатом Уиланда (см. разд. 2.2.2). В этом комплексе Н+ или D+ ковалентно связан с атомом углерода кольца. В этом карбокатионном интермедиате положительный заряд распределяется по остальным пяти атомам углерода с участием л-орбиталей, а атомы дейтерия и водорода находятся в плоскости, перпендикулярной плоскости кольца:
(2а) (26) (2в) (2)
Тот факт, что л- и о-комплексы, образованные, например, метилбензолом и HCI, действительно отличаются друг от друга, подтверждается различием в их свойствах. Так, при образовании л-комплекса получающийся раствор не обладает электри-
* Такие частицы называют арениевыми, или аренониевыми, ионами; более общий термин — карбокатионные интермедиаты.
146
\ческой проводимостью, цвет раствора не изменяется; обнаруживаются лишь незначительные изменения в УФ-спектре, что свидетельствует об отсутствии существенных нарушений в распределении электронов в исходном метилбензоле. В присутствии же А1С13 раствор приобретает зеленую окраску, электрическую проводимость, а УФ-спектр исходного метилбензола резко изменяется, что указывает на образование о-комплекса типа (2), так как нет оснований считать, что хлорид алюминия образует комплексы типа Н+ AICI4.
Реакция может быть завершена путем удаления протона из о-комплекса (2) под действием А1СЦ с образованием продукта замещения (4). Если в реакции участвует НС1, происходит только обмен атомов водорода; если же применяют DC1, то атом водорода замещается в некоторой степени дейтерием, т. е. весь процесс в целом является электрофильным замещением. Теоретически возможен альтернативный путь завершения реакции, так как комплекс (2) может реагировать с AIC^, отнимая от него ион С1~. Этот путь привел бы к суммарной реакции электрофильного присоединения [(2) -> (3)], как в случае обычной двойной углерод-углеродной связи (см. разд. 7.1). Такое превращение вызвало бы, однако, снижение степени стабилизации молекулы бензола, обусловленное наличием делокализованных л-орбиталей, которые охватывают все шесть атомов углерода ароматического кольца, поскольку продукт присоединения уже не был бы ароматическим соединением. При отщеплении Н+, т. е. в ходе замещения, а не присоединения, полностью заполненные делокализованные л-орбитали вновь формируются в продукте (4) и может снова наблюдаться характерная для ароматических соединений стабильность.
(3)
присоединение
AICI7 ------—(2) (+СГ)
AICI-4
(4) замещение
Повышение стабилизации при переходе (2) -> (4) помогает обеспечить реакцию энергией, необходимой для разрыва прочной связи С—Н, что требутся при отщеплении Н+. Например, при взаимодействии НС1 с алкенами (см. разд. 7.3) отсутствует такой фактор, ускоряющий замещение, и поэтому, как правило, происходят реакции присоединения.
Можно было бы ожидать, что для превращения бензола в о-комплекс (2), приводящего к утрате ароматической стабилизации, необходимо значительное количество энергии, т. е. энергия
147
активации этого процесса должна быть достаточно высокой, а его скорость, следовательно, небольшой. В действительности же многие реакции электрофильного замещения в ароматических соединениях протекают очень быстро при комнатной температуре. Это обусловлено наличием двух факторов, снижающих энергетический барьер, препятствующий образованию комплекса (2). Первый состоит в возможности использования энергии, освобождающейся при полном образовании новой связи с атакующим электрофилом, а второй — в стабилизации положительно заряженного сг-комплекса, т. е. снижения его энергетического уровня в результате делокализации заряда [см. формулу (2)]. Однако нельзя считать, что электронная плотность в этом ионе распределена равномерно, как показано в формуле (2); то, что это не так, ясно при написании для этого иона канонических структур (2а), (26) и (2в) (см. выше).
Если допущение о том, что электрофильное замещение ароматических соединений проходит через стадию образования про- межуточного сг-комплекса, правильно (об этом свидетельствует, в частности, выделение таких комплексов в ходе некоторых реакций замещения; см. разд. 6.2), то в таком случае следует принять, что реакции, которые мы обычно называем «ароматическим замещением», фактически представляют собой начальное присоединение с последующим отщеплением. Рассмотрим теперь, как эта основная теория подтверждается на примере типичных реакций электрофильного замещения бензола *.
6.2. Нитрование
Реакция нитрования является одной из наиболее широко изученных реакций ароматического замещения. На примере этой реакции можно, вероятно, получить наиболее детальное представление о механизме замещения. Для препаративных целей нитрование чаще всего проводят смесью концентрированных азотной и серной кислот, так называемой нитрующей смесью. «Классическое» объяснение необходимости присутствия серной кислоты состоит в том, что она поглощает воду, образующуюся при нитровании
С6Н6 + HNO3 —> CeH5NO2 + H2O
* При ознакомлении со схемой механизма, предлагаемой П. Сайксом, полезно иметь в виду многочисленные доказательства одноэлектронного переноса, сопровождаемого образованием катион-радикальных состояний ароматического субстрата—см. работы: Fukuzumi S., Kochi J. К.Hi. Amer. Chem. Soc. 1981. v. 103. N 24, P. 7240—7252; Fukuzumi S„ Kochi J. К.ЦЗ. Org. Chem. 1981. v. 46. N 21. P 4116—4126; Минкин В. И., Миняев Р. М., Юаилевич И. А., Клецкий М. Е.//УК. орг. химии. 1985. Т. 21. № 5. С. 926— 939; Морковник А. С.//Успехи химии. 1988. № 2. С. 254—280. — Прим. ред.
148
и препятствует тем самым протеканию обратной реакции. Такое объяснение неудовлетворительно во многих отношениях. В частности, остается непонятным, почему образовавшийся нитробензол в условиях реакции не атакуется водой! Несомненно, что в отсутствие серной кислоты нитрование проходит медленно, хотя сама серная кислота практически не взаимодействует с бензолом в обычных условиях реакции нитрования. Таким образом, можно предположить, что серная кислота взаимодействует в этой системе с азотной кислотой, а не с бензолом. Это подтверждается тем фактом, что растворы азотной кислоты в чистой серной кислоте имеют почти четырехкратную молярную депрессию температуры замерзания (фактически i а? 3,82), что интерпретируется как образование следующих четырех ионов:
НО - N02 + H2SO4 H2f- N02+ HSO7
н2(Р- no2 + h2so4 ч=± н3о+ + hso; + +no2
HNO3 + 2H2SO4^± +no2 + H30+ + 2HSO;
Небольшое снижение значения i (С 4), возможно, обусловлено неполным протонированием Н2О в этих условиях.
Наличие +NO2 (ион нитрония) как в этом растворе, так и в составе ряда солей (некоторые из которых, например +NO2C1O4, были выделены) подтверждено спектроскопически: для каждой из этих солей наблюдается только одна линия в спектре К.Р при 1400 см-1, которая может быть приписана только частицам, являющимся одновременно линейными и трехатомными. Сама азотная кислота в концентрированной серной кислоте практически полностью превращается в +NO2, и почти не вызывает сомнений в том, что в этих условиях ион нитрония является эффективным электрофилом. Если функция серной кислоты заключается просто в создании сильнокислой среды, в которой облегчено образование +NO2 из НО—NO2, то можно было ожидать, что и другие сильные кислоты, например НСЮ4, также будут способствовать нитрованию. Это предположение подтвердилось: система HF + BF3 также является очень эффективным катализатором. Незначительная эффективность самой азотной кислоты в реакции нитрова-ния бензола объясняется низким содержанием иона +NO2. Этот ион в небольшом количестве образуется в ходе двухстадийного процесса, в котором азотная кислота сначала быстро превращается в свою сопряженную кислоту, а затем
149
более медленно — в +NO2:
.. быстро +
НО - NO2 + HNO3 —-~t Н2О - NO2 + NOJ медленно
Н2О- NO2 + HNO3 ...— > Н3О++ NO3 + +no2
Найдено, что скорость нитрования ароматических соединений, более реакционноспособных, чем бензол, часто не зависит от их концентрации. Это означает, что именно образование +NO2 является медленной, скоростьлимитирующей стадией всего процесса. То, что образовавшийся +NO2 является высокоэффективным нитрующим агентом, подтверждается быстрым нитрованием относительно нереакционноспособных ароматических молекул при применении +NO2 BF4 при комнатной или даже более низкой температуре.
Многие реакции нитрования, протекающие с участием нитрующей смеси, подчиняются «идеализированному» уравнению скорости:
Скорость = &[АгН] [+NO2].
Однако реальную кинетику не всегда легко описать или объяснить. Это обусловлено различными причинами. Например, растворимость самого бензола в нитрующей смеси достаточно низка, поэтому скорость нитрования должна определяться скоростью, с которой этот несмешивающийся углеводород растворяется в кислотном слое. При использовании нитрующей смеси концентрация +NO2 прямо связана с концентрацией добавленной HNO3, поскольку HNO3 быстро и полностью превращается в +NO2. Однако при нитровании в других растворителях могут возникать другие осложнения. Зависимость концентрации эффективного электрофила (а это почти всегда +NO2) от концентрации HNO3 или другого потенциального нитрующего агента может оказаться весьма не простой.
Приведенное выше общее идеализированное уравнение скорости согласуется, по.крайней мере, с тремя различными возможными механизмами нитрования: одноступенчатым согласованным механизмом I [уравнение (6.1)], включающим одно переходное состояние (5), в котором образование связи С—NO2 и расщепление связи С—Н происходят одновременно; или с двухступенчатыми механизмами Па и Пб [уравнение (6.2)], включающими интермедиат Уиланда (о-комплекс) (6), в которых медленной, скоростьлимитирующей стадией может быть либо стадия (а) — образование связи С—NO2 (механизм Па), либо
150
стадия
(6)—расщепление связи С—Н (механизм Пб):
Г 6+ 6+ “|*
(6.1)
• (5)
(6)
(6.2)
Разрыв связи С—Н должен произойти при протекании реакции по любому из этих трех механизмов, однако на медленной, скоростьлимитирующей стадии это происходит только в случае механизмов I (всего одна стадия) и Пб. Если разрыв этой связи действительно протекает на скоростьлимитирующей стадии, то при замене СбНб на C6D6 должен проявляться первичный кинетический изотопный эффект (см. разд. 2.3.3). Для сравнения были использованы реакции нитрования C6H5NO2 и CeD5NO2 (это не важно для доказательства). Было показано, что при 25 °C Ah/Ad ~ 1,00; это соответствует отсутствию первичного кинетического изотопного эффекта. Таким образом доказано, что связь С—Н не расщепляется на стадии, лимитирующей скорость всей реакции, следовательно, механизмы I и Пб могут быть исключены из рассмотрения. Это, конечно, не означает, что нитрование протекает по механизму Па (медленная, скорость-лимитирующая стадия образования связи С—NO2 сопровождается быстрой стадией разрыва связи С—Н), однако это единственный механизм (из всех рассмотренных выше), который согласуется с экспериментальными данными.
То обстоятельство, что расщепление достаточно сильной связи С—Н должно быть быстрым, кажется менее удивительным, если вспомнить, что отщепление катиона Н+ от интермедиата (6) способно восстановить высоко стабилизированное ароматическое состояние в образующемся нитробензоле. Отрыв протона происходит вследствие атаки основанием, например HSO< в нитрующей смеси, но иногда — молекулами растворителя. Вероятность образования таких частиц, как (6), в качестве интермедиатов достаточно высока, так как аналогичные частицы, например (8), выделены при нитровании трифторметилбензола (7)
151
под действием NO2F—BF3:
(7) (8)
Комплекс (8) весьма устойчив при температуре ниже —50 °C, но при нагревании превращается в обычный продукт нитрования — соединение (7). Относительная устойчивость комплекса (8) отчасти обусловлена наличием аниона BF( в ионной паре (ср. разд. 5.1.1). Выделение этого комплекса не доказывает, что подобные интермедиаты обязательно образуются в реакциях нитрования с использованием нитрующей смеси, но в совокупности с кинетическими и другими данными участие таких частиц кажется чрезвычайно вероятным.
Скорости реакций замещения в ароматическом ряду лимитируются образованием переходного состояния (ПС1), непосредственно предшествующего образованию интермедиата (6) (рис. 6.1). Детальную информацию о таких частицах получить трудно; в качестве моделей используют интермедиаты, для которых переходные состояния являются непосредственными предшественниками, поскольку для них такая информация более доступна. Справедливость такого моделирования подтверждается принципом Хаммонда (см. примечание на с. 61). Несомненно, что интермедиат (6) в приведенной выше последовательности является лучшей моделью для ПСь чем исходное соединение. Известен и ряд других примеров аналогичного использования ст-комплексов в качестве моделей тех переходных состояний, которые нм предшествуют (см. разд. 6.7.1).
Было обнаружено, что высоко реакционноспособные ароматические соединения, например фенол, легко нитруются даже в разбавленной азотной кислоте, причем гораздо быстрее, чем
152
это можно было бы предположить, исходя из концентрации присутствующих в смеси ионов +NO2. Как было показано, это связано с наличием в системе азотистой кислоты, которая нитрози-рует реакционноспособное ароматическое кольцо ионом нитро-зония +NO (или другими частицами, способными осуществлять нитрозирование, см. разд. 5.5):
HNO2 + 2HNO3 ч=± нзО+ + 2NO3 + +NO
Нитрозофенол (10), который может быть выделен, очень быстро окисляется азотной кислотой, образуя п-нитрофенол (11) и азотистую кислоту; образование дополнительного количества азотистой кислоты приводит к ускорению процесса. Следует подчеркнуть, что азотистая кислота не должна обязательно содержаться в исходной азотной кислоте, так как небольшое количество ее образуется из азотной кислоты в ходе окисления фенола. Предполагают, что и в этом случае стадией, лимитирующей скорость всей реакции, является образование интермедиата (9). Одновременно происходит и прямое нитрование реакционноспособных ароматических соединений ионами +NO2, причем соотношение этих двух процессов зависит от условий проведения реакции.
Показано, что многие другие реакции ароматического электрофильного замещения также протекают по обсуждавшемуся выше общему механизму II, причем обычно по механизму 11а, хотя известен ряд реакций, протекающих по механизму Пб. Истинная природа электрофильной частицы, которая участвует в атаке ароматического ядра, еще не выяснена.
6.3. Галогенирование
В отличие от нитрования, при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены, например С12 и Вг2, могут легко атаковать активированное ароматическое ядро (см. разд. 6.7), например, фенола, но ие способны реагировать с бензолом (фотохимическая активация может привести к присоединению, однако при участии свободных атомов галогенов; см. разд. 11.5.1.2). Для поляризации атакующей молекулы галогена
153
необходимы катализаторы типа кислот Льюиса, такие как А1С13, с помощью которых в молекуле галогена появляется так называемый «электрофильный конец»: энергия, требующаяся для образования катиона С1+, очень велика. Уравнение скорости такой реакции часто имеет вид:
Скорость = Л[АгН] [На12] [Кислота Льюиса].
Бензол, вероятно, образует л-комплекс (12), например, с Вг2 (ср, разд, 6.1.1), с которым затем взаимодействует кислота Льюиса. Катализатор, вероятно, поляризует связь Вг—Вг, способствуя образованию ст-связи между теперь уже электрофильным концом молекулы брома и атомом углерода кольца, а на Последующей стадии — удалению возникающего бромид-иона с образованием комплекса (13):
Анион FeBri в свою очередь помогает удалению протона из ст-комплекса (13). Классический катализатор галогенирования — железные опилки — начинает действовать, конечно, только после превращения в кислоту Льюиса FeX3.
Кинетические изотопные эффекты не наблюдались при хлорировании и очень редко наблюдались при бромировании, т. е. эти реакции обычно следуют по механизму Па, подобно реакции нитрования. При иодировании, которое происходит только при взаимодействии самого иода с активированными субстратами, наблюдается кинетический изотопный эффект. Возможно, это происходит потому, что реакция обратима (в отличие от других реакций галогенирования); иод отщепляется из ст-комплекса (14) легче, чем Н, т. е. й-i йг:
Например, отношение йн/йо, измеренное для иодирования фенола и 2,4,6'-тридейтериофенола, равно « 4, т. е. реакция идет по механизму Нб. Иодирование часто облегчается в при
154
сутствии оснований или окисляющих агентов, которые удаляют HI и таким образом сдвигают равновесие вправо. При действии окисляющих агентов из молекулы 12 образуется катион 1+ или комплекс, содержащий положительно поляризованный код, т. е. более эффективный электрофил. Галогенирование может осуществляться также при применении соединений типа 6+Вг—С1*~, 6+1—С16- и т. д. При этом атака проходит с помощью менее электроотрицательного галогена, поскольку он является «электрофильным концом» молекулы. Показано, в частности, что при использовании ВгС1 и IC1 осуществляются соответственно бромирование и иодирование.
Галогенирование можно проводить также с применением 6- 64-
КИСЛОТ типа НО—Hal. Реакция в этом случае идет гораздо медленнее, чем с молекулярным галогеном, поскольку НО- в НОНа! — худшая уходящая группа, чем Hal- в молекуле галогена. Реакция тем не менее ускоряется в присутствии Hal, так как НО—Hal превращается при этом в более ракционноспособ-ный На12, например:
-ОС1 + СГ + 2Н+ —► С12 + Н2О
Однако в присутствии сильной кислоты НО—Hal становится очень сильным галогенирующим агентом вследствие образования сильно поляризованного комплекса (15):
НО - На! + Н+--► Н2Од На1-*-*-Н2О + На1 +
(15)
Существуют доказательства, что комплекс (15) является эффективным электрофилом в данных условиях и не превращается в На1+, т. е. ведет себя иначе, чем комплекс ЩО+—NO2 (см. разд. 6.2); однако НОС1 + кислота может быть еще более эффективным хлорирующим агентом, чем С12 + AICI3. Молекула фтора энергично реагирует с бензолом, но при этом разрываются связи С—С, и поэтому реакция не имеет препаративного значения (ср. разд. 6.10.2).
6.4. Сульфирование
Детали механизма сульфирования исследованы менее подробно по сравнению с нитрованием или галогенированием. Сам бензол сульфируется довольно медленно горячей концентриро; ванной серной кислотой, но быстро — олеумом (при этом скорость сульфирования зависит от содержания SO3) или SO3 в инертных растворителях. Природа электрофильной частицы зависит от условий реакции, ио, вероятно, это всегда SO3: или в свободном состоянии, или связанный с «носителем», например
155
в виде H2SO4-SO3 (H2S2O7) в серной кислоте. Небольшие количества SO3 образуются в H2SO4:
2h2so4 so3 + н3о+ + hso;
Атаку ароматического субстрата осуществляет атом серы, поскольку он сильно положительно поляризован, т. е. электронодефицитен:
б+э+^О®-*
Сульфирование, подобно иодированию, является обратимым процессом и, как полагают, в концентрированной серной кислоте происходит по схеме:
(16) (17)
> быстро
SO3H
Считают, что в олеуме ст-комплекс (16) протонируется по группе SO3 до разрыва связи С—Н с образованием 8О3Н-ана-лога соединения (17). Подобно иодированию, при сульфировании наблюдается кинетический изотопный эффект, т. е. разрыв связи С—Н происходит на скоростьлимитирующей стадии реакции k-i k2.
Практическое значение имеет обратимость реакции сульфирования: при обработке сульфокислот водяным паром происходит замещение группы SO3H на водород. Таким образом, можно ввести группу SO3H как заместитель, ориентирующий требуемым образом последующие реакции (ср. разд. 6.7), а затем ее отщепить. Некоторые интересные особенности имеет сульфирование нафталина (см. разд. 6.9).
6.5. Реакция Фриделя — Крафтса
Для удобства рассмотрим отдельно реакции алкилирования и ацилирования.
6.5.1. Алкилирование б+ б“
Атом углерода алкилгалогенидов R—Hal является электрофильным, однако часто этого недостаточно для осуществления замещения в ароматическом субстрате; необходимо наличие катализатора в виде кислоты Льюиса, например А1С13. Тот факт, что алкилгалогениды реагируют с кислотами Льюиса, был до-
156
казан с помощью обмена радиоактивного брома между AlBrj и EtBr при их смешении и последующем разделении, а также выделением в твердом состоянии комплексов состава 1 : 1, например СН3Вг-А1Вг3 при низких температурах (—78°С). Если R в R— —Hal, например в Ме3С—Вг, способен образовать особенно устойчивый карбокатион, то, возможно, что в этом случае атакующим электрофилом в алкилировании выступает фактически карбокатион Ме3С+ в составе ионной пары:
А1Вг4
(18)
В других случаях кажется более вероятным, что атакующим электрофилом является поляризованный комплекс (19), причем степень поляризации зависит от природы R в R—Hal и от применяемой кислоты Льюиса:
б+ 6-
R—Cl---FeCl3 (19)
(20)
Оба рассмотренных выше механизма согласуются с обычно наблюдаемым кинетическим уравнением:
Скорость = £[АгН] [RX] [МХ3]
Эффективность кислот Льюиса как катализаторов уменьшается в ряду:
А1С13 > FeCl3 > BF3> TiCI3 > ZnCI2> SnCI4
Существование интермедиатов Уиланда, таких как (18) и (20), в реакциях алкилирования по Фриделю — Крафтсу было установлено путем выделения некоторых из них, например (21), при низких температурах (стабилизирующий эффект BF4
157
в ионных парах рассмотрен в разд. 6.2):
(21)
Промежуточное соединение (21) представляет собой твердое кристаллическое вещество оранжевого цвета, которое плавится с разложением при —15 °C; из него образуется ожидаемый продукт алкилирования с практически количественным выходом (см. разд. 6.2).
В ходе некоторых реакций алкилирования по Фриделю — Крафтсу образуются продукты, которые содержат алкильную группу, претерпевшую перегруппировку. Например, при действии Ме3ССН2С1 + А1С1з на бензол образуется РЬСМе2СН2Ме. Этот результат можно объяснить, исходя из того, что первоначально образующийся электрофильный комплекс достаточно поляризован, чтобы произошла перегруппировка в более устойчивый комплекс (ср. относительную устойчивость карбокатионов; разд. 5.2):
в+ Л .
[Ме3ССН2]—Cl—AlClg —> [Ме2ССН2Ме]—Cl-AlClg'
Наоборот, при действии Ме3ССН2С1 + FeCl3 на бензол в основном образуется неперегруппированный продукт PhCH2CMe3. По-видимому, в комплексе с более слабой кислотой Льюиса, FeCl3, поляризация алкилгалогенида недостаточна, чтобы произошла изомеризация. Показано также, что и температура влияет на количество продукта перегруппировки: при данных галогениде и кислоте Льюиса его образуется тем меньше, чем ниже температура.
Найдено, что во многих случаях соотношения получаемых продуктов не всегда соответствуют относительной устойчивости образующихся карбокатионов (перегруппированного и непере-группированного). Это следует из того факта, что относительные скорости реакции этих карбокатионов с ароматическими субстратами не соответствуют их относительным устойчивостям, а могут быть даже диаметрально противоположными им. Атака ароматической молекулы образовавшимся поляризованным комплексом может проходить быстрее, чем его перегруппировка. Исследование этих перегруппировок осложняется тем фактом, что кислоты Льюиса способны катализировать перегруппировки как исходных галогенидов, так и конечных продуктов реакции
158
алкилирования:
СН2СНМе2 А1с,з
^х^СНСН2Ме
(Oj Me
Кроме алкилгалогенидов для алкилирования ароматических соединений могут быть использованы алкены и спирты. При этом необходимо присутствие протонной кислоты, чтобы протонировать алкен или спирт. В этих реакциях в качестве катализатора типа кислоты Льюиса часто используют BF3:
н+ + с,ня
МеСН = СН2^г=>МеСНСН3—Me2CHPh
Il BF3
II -нго и+
МеСНСН3^=>:МеСНСН,
I I 5
:он +он2
Катализаторы типа кислот Льюиса могут осуществлять также деалкилирование, т. е. реакция алкилирования обратима. Например, этилбензол (22) в присутствии BF3 и HF претерпевает диспропорционирование:
Et Et Et
(22) (45%) (10%) (45%)
Эта реакция должна быть, безусловно, межмолекулярной, однако перегруппировки, приводящие к изменению относительных положений заместителей в бензольном кольце, также хорошо изучены, и они являются внутримолекулярными. Например, нагревание n-диметилбензола (п-ксилол) (23) с А1С13 и НС1 ведет к превращению большей части этого соединения в более стабильный (см. разд. 6.8) м-диметилбензол (м-ксилол) (25). Полагают, что указанное превращение включает. миграцию метильной группы в первоначально протонированной частице (24):
Me Me Me Me
(23) (24) (25)
159
Помимо возможности перегруппировки основным недостатком при препаративном использовании реакции алкилирования по Фриделю — Крафтсу является полиалкилирование (ср. разд. 6.7.1.2). Наличие электроноакцепторных заместителей обычно ингибирует алкилирование по Фриделю— Крафтсу. Так, нитробензол часто используют как растворитель для этой реакции, поскольку А1С13 легко растворяется в нем, и таким образом удается избежать гетерогенности процесса.
6.5.2. Ацилирование
Ацилирование по Фриделю—Крафтсу в тех случаях, когда кинетика может быть легко прослежена, часто подчиняется тому же общему кинетическому закону, что и алкилирование:
Скорость = k[АгН] [RCOC1] [А1С13]
И в этом случае появляется сходная общая дилемма: выступает ли в качестве эффективного электрофила ацилкатион в составе ионной пары (26) или же поляризованный комплекс (27):
+ 6+- 5-
RC=Q AICI4 RC—O-AICI3
Cl (26) (27)
Ацилкатионы были обнаружены в ряде твердых комплексов, в жидком комплексе МеСОС! и А1С13 (с помощью ИК-спектро-скопии), в полярных растворителях и в ряде случаев, в которых R являлась очень объемистой группой. В менее полярных растворителях и в ряде других случаев ацилкатионы не обнаруживаются, однако при этом должен существовать поляризованный комплекс, который действует как электрофил.
Показано, что в зависимости от условий электрофилом является ацилкатион или комплекс (27). Так, при бензоилировании толуола получают одну н ту же смесь продуктов (1 % м-, 9 % о- и 90 % п-), независимо от того, какая кислота Льюиса является . катализатором, и проводилось ли бензоилирование бензоилхлоридом или бензоилбромидом, хотя скорости реакций оказываются, конечно, разными. По-видимому, во всех случаях атакующей частицей является +COPh. Однако во многих случаях количество образующегося орто-продукта очень мало по сравнению с его количеством в других реакциях электрофильного замещения, например в реакции нитрования. Это свидетельствует о том, что электрофильная частица очень громоздка; это, по-видимому, больше соответствует комплексу (27), чем иону +COR. Ясно, что в любом случае природа электрофила очень сильно зависит от условий.
160
Реакцию ацилирования можно представить следующей схемой:
Важное отличие реакции ацилирования от реакции алкилирования состоит в том, что в первой из этих реакций требуется более 1 моль кислоты Льюиса, тогда как во второй необходимо только каталитическое количество. Это обусловлено тем, что кислота Льюиса образует комплекс (29) с кетоном (28) — продуктом ацилирования — и тем самым устраняется от дальнейшего участия в реакции.
6+ 6-
С-О—А1С13 I
R
(29)
Полиацилирования не наблюдается (ср. алкилирование; разд. 6.5.1), поскольку образующийся кетон гораздо менее реакционноспособен, чем исходное соединение (ср. разд. 6.7.1.1). Перегруппировки R не происходит, однако возможно декарбонилирование, особенно, если при этом образуется устойчивый карбокатион; конечным результатом оказывается в этом случае алкилирование, а не ожидаемое ацилирование:
+ С6нв
Ме3С—С=О —Ме3С ----------► Me3CPh
Формилирование может быть проведено с использованием СО, НС1 и А1С1з (реакция Гаттермана — Коха). Сомнительно, чтобы в этой реакции предварительно образовался НСОС1; наиболее вероятным электрофилом является ацилкатион +СН=О (т. е. протонированный СО) в составе ионной пары О=СН+ AlCl^-
6 П. Сайкс
161
В этой реакции фактически устанавливается равновесие, неблагоприятное для образования продукта, однако оно смещается вправо в результате образования комплекса альдегида (30) с кислотой Льюиса (катализатор).
Ацилирование может осуществляться также ангидридами кислот (RCO)2O и кислотами Льюиса (эффективным электрофилом может быть +COR или, в некоторых случаях, RCOC1, который образуется при действии А1С13 на исходный ангидрид), а также самими кислотами. В последнем случае ацилирование ускоряется сильными кислотами, например H2SO4, HF, гтак же, как и кислотами Льюиса, и может идти через образование ацил-катионов путем протонирования:
О О
II .. || + + ,
RC - ОН + H2SO4 RC - OH25=±RC = О + Н2О
Это реализуется, в частности, в реакциях циклизации:
Поскольку полиацилирование не наблюдается (см. выше), часто алкилбензолы предпочитают получать путем ацилирования с последующим восстановлением по Клемменсену или другим методом, а не путем прямого алкилирования:
6.6. Азосочетание
Еще одним классическим примером электрофильного замещения в ароматических соединениях является реакция азосочетания, в которой эффективным электрофилом является катион диазония (ср. разд. 5.5):
4- 4-
PhN=N ч->- PhN=N
Однако этот катион — более слабый электрофил, чем, например, +NO2, и способен атаковать только очень реакционноспособные ароматические соединения, такие как фенолы и амины,
162
ие реагируя в то же время с анизолом PhOMe, достаточно реакционноспособным в других электрофильных реакциях. Введение электроноакцепторных групп в орто- или пара-положения диа-зониевого катиона усиливает его электрофильный характер, увеличивая положительный заряд на диазогруппе:
Так, катион 2,4-динитрофенилдиазония способен сочетаться с PhOMe, а 2,4,6-тринитрофенилдиазоний-катион— даже с углеводородом— 2,4,6-триметилбензолом (мезитилен). Катионы диазония существуют в кислой и слабощелочной средах (в силь-иощелочной среде они превращаются в диазогидраты PhN = = NOH, а затем в диазотат-анионы, PhN = NO_), поэтому реакции азосочетания проводят только в указанных условиях. Оптимальное значение pH зависит от свойств атакуемого соединения. В случае фенолов реакцию проводят в слабощелочной среде, так как именно PhO-, а не PhOH подвергается атаке электрофилом АгИг! скорость реакции подчиняется уравнению:
Скорость = &[АгГ<2 ] [PhO-].
Сочетание с феноксид-иоиом может протекать либо по атому кислорода, либо по атому углерода. В соответствии с относительной электронной плотностью можно было бы- ожидать предпочтительности реакции по атому кислорода, однако важную роль играет также прочность образующейся связи. Так, при реакции с фенолами, как и в других электрофильных реакциях фенолов, образуется лишь С-замещенный продукт (32):
Удалению протона из промежуточного соединения (31) (обычно ие на скоростьопределяющей стадии) способствует присутствующее в растворе основание. Сочетание обычно происходит в основном в пара-положение, а не в орго-положеиие (ср. разд. 6.7.1.3), при условии, что оно доступно, из-за значительного объема атакующего электрофила ArNa (ср. разд. 6.7.3).
Ароматические амины обычно реагируют несколько труднее, чем фенолы, и сочетание часто проводят в слабокислой среде, что обеспечивает высокую концентрацию катиона PhNj без
6*
163
заметного превращения амина ArNH2 в нереакционноспособный протонированный катион ArNHa' (ароматические амины представляют собой очень слабые основания, ср. разд. 3.2.3). Диазотирование первичных ароматических аминов проводят в сильно-кнслой среде, так как при этом еще непрореагировавший амин превращается в катион н таким образом предохраняется от сочетания с образующейся солью диазония.
В случае ароматических аминов возможна атака либо по атому азота, либо по атому углерода; в отличие от фенолов, атака первичных н вторичных аминов происходит в основном по ато1Лу азота, что приводит к диазоаминосоединениям (33):
HN—N=NAr
(33)
Для большинства первичных аминов такой продукт является фактически единственным; для вторичных аминов (т. е. А-ал-киланилинов) сочетание может идти также и по атому углерода бензольного ядра; в случае третичных аминов (т. е. А,А-диал-киланилинов) образуется только продукт сочетания по атому углерода — соединение (35):
Показано, что реакция сочетания подчиняется кинетическому уравнению:
Скорость = AfArNj] [PhNR2]-
В некоторых случаях она катализируется основаниями и сопровождается кинетическим изотопным эффектом, т. е. k_\
k2; расщепление связи С—Н в промежуточном соединении (35) происходит на скоростьлимитирующей стадии.
Интересным примером внутримолекулярной реакции сочетания является реакция, протекающая при диазотировании о-ди-аминобензола (36); при этом образуется бензотриазол (37)
164
с выходом 75 %:
о
(36)
NaNO2 HCI *
---►
-H+
(37)
Различие в местах преимущественной атаки первичных и вторичных ароматических аминов (по сравнению с фенолами) объясняется, по-видимому, различиями в относительной электронной плотности соответствующих участков молекулы. В отличие от ряда других реакций электрофильного замещения ароматических соединений, реакция азосочетания чувствительна к относительно небольшим различиям в электронной плотности (что отражает довольно низкую реакционную способность катиона PhNj как электрофила). Аналогичные различия в электронной плотности, естественно, имеют место и для фенолов, но для этих соединений, как уже отмечалось, выбор места атаки в большей мере зависит .от относительной прочности образующихся связей; в случае аминов это различие для двух альтернативных продуктов сочетания выражено в гораздо большей степени.
Образование диазоаминосоедииений при сочетании ArN£ с первичными аминами не препятствует получению препаративных количеств продуктов сочетания по бензольному кольцу, поскольку диазоамииосоедииения при нагревании в кислой среде могут перегруппировываться в аминоазосоединения (39):
Показано, что перегруппировка в этих условиях является межмолекулярным процессом, т. е. катион диазония освобождается в ходе реакции, после чего он может атаковать фенолы, ароматические амины и другие подходящие соединения, добавленные в раствор. Оказалось, что перегруппировка легче всего идет в присутствии кислотного катализатора при избытке амина, который первоначально подвергается сочетанию с образованием диазоамииосоедииения (33). Возможно, что этот амин непосредственно атакует протонированное диазоаминосоедине-
165
нне (38), вытесняя PhNH2, а затем теряет протон:
+NH3Ph
н2Й‘-Чк X N = NAr ———->H,N=C7' V"Н —(39)
* -PhNH2 2C\=/SN=NAr -н
(38)
В заключение следует упомянуть, что, хотя подавляющее большинство реакций электрофильного замещения ароматических соединений связано с замещением водорода, могут также замещаться другие атомы или группы. Так, мы уже рассматривали замещение SO3H в обратимой реакции сульфирования (см. разд. 6.4), замещение алкила при деалкилировании (см. разд. 6.5.1) и, наконец, менее распространенное замещение группы SiR3 в реакции протодесилилирования (ср. разд. 6.7.4):
ArSiR3 + H+ ► Ar—H + +SiR3
Для реакций такого типа характерны все особенности ароматического электрофильного замещения (влияние заместителей и т. д.; см. ниже), но они имеют гораздо меньшее препаративное значение, чем реакции, которые уже были рассмотрены выше.
Следует отметить, что известны также реакции гомолитического замещения ароматических соединений, т. е. реакции, протекающие с участием радикалов (см. разд. 11.5.2.3), а также реакции, включающие атаку нуклеофилами (см. разд. 6.10).
6.7. Электрофильная атака монозамещенных бензолов
Когда монозамещенные бензолы C6H5Y подвергаются дальнейшему электрофильному замещению, например нитрованию, то новый заместитель может вступать в орто-, мета- или параположение, а суммарная скорость замещения может быть больше или меньше скорости замещения самого бензола. Экспериментально установлено, что при наличии заместителя в кольце замещение проходит так, что в основном образуется либо мета-изомер, либо смесь орто- и пара-изомеров. В первом случае суммарная скорость реакции всегда меньше, чем скорость реакции с самим бензолом, а во втором — суммарная скорость обычно больше, чем скорость реакции с бензолом. Основное влияние, как было показано, оказывает уже находящийся в кольце заместитель Y. Влияние заместителя можно объяснить, исходя из его электронных эффектов. Этот заместитель может, конечно, обнаруживать и стерический эффект, но действие этого фактора ограничено по существу атакой в орто-положение (см. разд. 6.7.3).
Заместители Y, таким образом, подразделяют на мета- или орто-пара-ориентирующие; если они ускоряют суммарную реак
166
цию по сравнению с незамещенным бензолом, то их называют активирующими, если замедляют — дезактивирующими. Надо подчеркнуть, что эти ориентирующие эффекты являются скорее относительными, чем абсолютными: в любой реакции замещения почти всегда образуются некоторые количества всех трех изомеров, хотя количество .мета-замещенного продукта при наличии орто-пара-ориентирующего заместителя или орто- и пара-замещенных продуктов при наличии мета-ориентирующего заместителя Y может быть очень невелико. Например, йзвестно, что при нитровании нитробензола (Y = NO2) образуемся смесь 93 % М-, 6 % °- и 1 % n-изомеров, т. е. NO2 относится к метаориентирующим (дезактивирующим) заместителям. Наоборот, нитрование метоксибензола (анизол; Y = ОМ.е) дает 56 % п-, 43 % о- и 1 % м-изомеров, т. е. ОМ.е является орто-пара-ориен-тирующим (активирующим) заместителем:
6.7.1. Электронные эффекты заместителей
Ниже рассмотрены такие эффекты заместителя Y, которые определяют скорость атаки в орто-, пара- и мета-положения по отношению к этому заместителю. При этом мы будем предполагать, что соотношения образующихся изомеров полностью определяются их относительными скоростями образования, т. е. соответствующие реакции имеют кинетический контроль (см. разд. 6.8) *. Строго говоря, следует сравнить влияние заместителя Y на различные переходные состояния, соответствующие различным направлениям атаки, что обычно невозможно. Вместо этого используют интермедиаты Уиланда в качестве моделей переходных состояний, которые непосредственно им предшествуют на скоростьлимитирующей стадии реакции, точно так же,
* Схема механизма электрофильного ароматического замещения, включающая стадию одноэлектронного переноса, и концепция граничных орбита-лей Фукуи предусматривают, что изомерный состав продуктов, образующихся в соответствующих реакциях, определяется состоянием ВЗМО ароматического субстрата (см. обсуждение этого вопроса в кн.: Травень В. Ф. Электронная структура и свойства органических молекул. М.: Химия, 1989. См. разд. 3.1, 6.3.5, 11.4 и гл. 10). — Прим. ред.
Ж
как это было сделано при обсуждении отдельных реакций электрофильного замещения (ср. разд. 6.2). Обсудим несколько различных типов заместителей Y.
6.7.1.1. Y = +NR3, СС13, NOz, СНО, СОгН и др.
Для всех перечисленных групп, а также таких групп, как SO3H, CN, COR и др., характерно наличие положительно заряженного (положительно поляризованного) атома, смежного с атомом углерода бензольного кольца:
PhNR3
С1б 6+++/ t PhC—С1°~ ^Cl6-
Таким образом, все эти группы являются электроноакцепторными по отношению к бензольному кольцу, т. е. ароматическая частица, содержащая любую из этих групп, представляет собой диполь, положительный конец которого локализован на бензольном кольце. При Y=+NR3 о-комплексы, образующиеся при атаке электрофилом Е+ (например, +NO2), орто-, мета- и лара-положений по отношению к исходному заместителю, имеют вид:
Заместитель +NR3 будет проявлять суммарный сильный электроноакцепторный, т. е. дестабилизирующий, индуктивный (по-
168
лярный) эффект в каждом из трех положительно заряженных о-комплексов (40), (41) и (42) в отличие от о-комплекса, образующегося при аналогичной атаке бензола (ср. разд. 6.1.1). Так, при бромировании CeHaNRs атака любого положения бензольного кольца происходит' с меньшей скоростью, чем в .случае бензола; при Y — Me3N+~ ^(C6H5Y)[k(С6Н6) — 1,6-IO-5.
Группа +NR3 избирательно дестабилизирует структуру (40в) о-комплекса, образующегося» в случае о-атаки, и структуру (426), образующуюся в случае n-атаки, потому что в каждой из этих структур положительные заряды находятся на соседних атомах. Положительный заряд кольца будет, таким образом, делокализован в меньшей мере в комплексах (40) и (42), чем в комплексе (41). Переходное состояние, для которого в качестве модели взят комплекс (41), должно, следовательно, находиться на более низком энергетическом уровне, чем те переходные состояния, для которых моделями служат комплексы (40) и (42); свободная энергия активации AG* для комплекса (41) будет ниже, он образуется быстрее, что и ведет к преобладанию м-изомера среди продуктов реакции.
Показано, что в том случае, когда в заместителе на атоме, связанном с бензольным ядром, действительно имеется положительный заряд (например, когда Y =+NR3, а не NO2), влияние заместителя в о-комплексе; на бензольное кольцо проявляется через эффект поля (ср. разд. 1.5.1), действующий через пространство, помимо полярного (индуктивного) эффекта, действующего через связи. Дезактивирующее влияние заместителя Y на ядро уменьшается; общая скорость замещения увеличивается в ряду:
+NR3<NO2<CN <SO3H <С = О <СО2Н ,
Этот ряд, однако, является условным, поскольку он изменяется при переходе от одного процесса замещения к другому и зависит в определенной степени от природы атакующего электрофила. Неудивительно, что дезактивирующее влияние таких заместителей, как +NR3, особенно сильно проявляется в реакциях замещения, в которых атакующий электрофил положительно заряжен, например +NO2; для реакции нитрования при Y ~ +NMe3 £(C6H5Y)/£(C6H6) = 1,5-10~8.
6.7.1.2. Y = Aik, Ph
Алкильные группы являются электронодонорными по сравнению с атомом водорода и могут избирательно стабилизировать те канонические состояния для о- и n-атаки соответственно, в которых положительный заряд локализован на соседнем атоме
169
углерода бензольного ядра, — (43) н (446) [ср. структуры (40в) н (426), которые избирательно дестабилизированы]:
(43) (44)
Указанные факторы не действуют в комплексе, образующемся в случае м-атаки [ср. (41а)—(41в)], вследствие чего замещение в основном идет в орто- и пара-положения. Вследствие суммарного электронодонорного индуктивного (полярного) эффекта атака любого положения алкилбензола протекает быстрее, чем в самом бензоле [ (Л(СбН5Ме)Д(СбНб) = 3,4 • 102 в случае хлорирования].
Можно, таким образом, предполагать, что С6Н5СМе3 будет атаковываться быстрее, чем СеН5СНз, вследствие большего электронодонорного индуктивного эффекта группы Ме3С. Именно это и наблюдается при нитровании. Однако этот порядок становится обратным при хлорировании: эта менее полярная реакция контролируется подачей электронов по механизму гиперконъюгации, которая гораздо более эффективна в С6Н5СН3 [(45а) -«->-(456)], чем в С6Н5СМе3 [(46)]. Кроме того, имеют значение и относительные размеры этих алкильных групп (ср. разд. 6.7.3).
(45а) (456) (46)
Специфическая стабилизация канонических форм о-комплек-сов, образующихся при атаке орто- н пара-положений, осуществляется в случае фенильной группы, например, для структур (47а) и (476):
(47а) (476)
Показано, что суммарная скорость атаки для бифенила выше, чем для бензола [&(СбН5¥)/£(СбНб) =4,2-102 (для хлорирования)].
170
6.7.1.3. Y = OCOR, NHCOR, OR, OH, NH2, NR2
Для каждой из этих групп характерно, что атом, связанный с бензольным ядром, может проявлять электроноакцепторный (индуктивный, полярный) эффект (ср., например, N в NO2),.однако этот атом имеет также свободную электронную пару (например, :ОМе), которая может содействовать специфической стабилизации о-комплексов, образующихся при о- и п-атаке [ (48в) (48г) и (49в) (49г) соответственно], но не при
jH-атаке.[(50а) (50в)]:
о-атака:
(48а) (486) (48в)
м-атака:
(50а) (506) (50в)
Эта стабилизация примечательна не только тем, что в ней участвует еще одно (четвертое) каноническое состояние о-ком-плексов, образующееся при о- и n-атаках [(48г) и (49г) соответственно]. В этих структурах положительный заряд локализован на атоме кислорода, и они более устойчивы, чем три другие формы, в которых положительный заряд находится на атоме углерода [ср. (48а)-<—► (48в) и (49а)-<—► (49в) ]. Этот эффект намного превосходит индуктивный эффект, также действующий в этих двух о-комплексах. В результате замещение почти полностью идет в орто- и пара-положения (при нитровании PhOMe, образуется <С 1 % At-изомера) и гораздо быстрее, чем замещение в самом бензоле [А(С6Н5ОМе)/£(С6Н6) = 9,7-106 (для хлорирования)].
171
Действие электроноакцепторного индуктивного эффекта проявляется, однако, в том, что атака в мета-положение (для которой нет специфической стабилизации a-комплекса посредством делокализации) осуществляется медленнее, чем атака самого бензола (ср. разд. 6.7.2). В случае феноксид-иоиа (51) индуктивный эффект проявляется в обратном направлении; так как отрицательный заряд находится на атоме кислорода, феноксидион более реакционноспособен, чем фенол. Даже мета-положение в этом' случае атакуется легче, чем сам бензол (однако продукт лета-замещения образуется в небольших количествах или не образуется вообще). Многие реакции электрофильного замещения проходят в кислой среде, так что феноксид-ион не может в них участвовать; однако реакцию азосочетания (см. разд. 6.6) проводят с фенолами в слабощелочной среде (ср. разд. 6.6).
(51)
Активирующее влияние заместителя Y на бензольное кольцо возрастает (при этом общая скорость замещения увеличивается) в ряду:
OCOR<NHCOR<OR<OH <NH2<NR2
Группа NR2 является более сильной активирующей группой, чем NH2( из-за электронодонорного эффекта групп R. Не следует забывать, что в кислой среде, например при нитровании, обе эти группы превращаются в +NHR2 и +NH3 соответственно. В этом случае бензольное ядро будет дезактивироваться, и замещение будет происходить преимущественно в мета-положеиие (ср. +NR3; разд. 6.7.1.1). Группа ОН также является достаточно сильно активирующей, так что фенол бромируется мгновенно бромной водой при комнатной температуре с образованием 2,4,6-трибромпроизводного (атакуются пара- и оба орто-положения). Группы OCOR и NHCOR менее сильно активируют ядро, чем ОН и NH2, соответственно из-за снижения доступности электронов на атомах О и N вследствие делокализации заряда с участием электроноакцепторной карбонильной группы:
О О’ О О’
—О—С—R — 6=С—R —HN—С—R <-> — HN=C—R
Группа NHCOR не протонируется в кислой среде, поэтому нитрование анилина с целью получения о- и n-производных можно проводить, применяя СОМе в качестве защитной группы с последующим ее удалением.
172
6.7.1.4. Y == Cl, Br, I
У галогенбензолов атом, связанный с бензольным ядром, также имеет свободную электронную пару; поэтому и в этом случае происходит специфическая стабилизация о-комплексов, образующихся при о- ил-атаках [ (51 а)(51 б) и (52а) -<-► (526) соответственно]:
(51а) (516)
(52а) (526)
Таким образом, галогены также являются орто-пара-ориен~ тантами. Электроноакцепторные индуктивные эффекты галогенов, однако, таковы, что атака галогенбензолов проходит медленнее, чем самого бензола: атомы галогенов являются дезактивирующими заместителями [£(СбН5С1)/£(С6Нб) = 3-10~2 (для нитрования)]. Это суммарное электроноакцепторное действие галогенов видно при сравнении дипольных моментов хлорбензола (53) в основном состоянии, у которого положительный конец находится на ядре, и анизола (54), дипольный момент которого имеет противоположное направление:
|1 = 1,2Д
ц = 1,6Д (53)
Суммарный эффект любого заместителя, конечно, обусловлен вкладами индуктивного эффекта, эффекта поля и мезомер-ного эффекта. В случае группы ОМе (см. разд. 6.7.1.3) суммарный эффект таков, что стабилизация положительно заряженных интермедиатов (48) и (49), образующихся при о- и л-атаке соответственно, гораздо больше, чем стабилизация соответствующего интермедиата (2) (см. разд. 6.1.1), образующегося при атаке самого бензола; о- и л-атака CeHsOMe поэтому проходит гораздо быстрее, чем атака СвНв- В случае галогенов как заместителей (например, С1) суммарный эффект вследствие сильных электроноакцепторного эффекта и эффекта поля таков, что стабилизация интермедиатов (51) и (52), образующихся при о- и л-атаке соответственно, несколько меньше, чем стабилизация интермедиата, образующегося при атаке самого бензола; в результате о- и л-атака проходит немного медленнее, чем атака СвНв.
173
Очень сходная ситуация наблюдается при электрофильном присоединении несимметричных частиц (например, НВг) к ви-нилгалогенидам (например, СН2=СНВг); в этом случае индуктивный эффект галогена контролирует скорость присоединения, а относительная мезомерная стабилизация промежуточного карбокатиона— ориентацию присоединения (см. разд. 7.3).
6.7.2. Факторы парциальных скоростей и селективность
Более совершенные кинетические методы и возможность очень точного определения относительных количеств образующихся о-, м- и n-изомеров, например с помощью спектроскопических методов, а не путем их выделения, как это делали в прошлом, обеспечивают более строгий подход к вопросу о соотношении изомеров в реакциях ароматического замещения. Ценную информацию получают, определяя факторы парциальных скоростей, т. е. отношение скоростей атаки одного положения, например пара-положения, в C6H5Y к скорости атаки одного положения бензола (в данном случае fn).
Факторы парциальных скоростей могут быть определены с помощью кинетического измерения общих констант скоростей Л(С6Н5У) и А(С6Н6) в аналогичных условиях [или в эксперименте с конкурирующими реакциями, в котором эквимольные количества C6H5Y и С6Н6 конкурируют в реакциях с электрофилом, что отражается на отношении £(СбН5У)/£(С6Нб)], и на основе анализа относительных количеств о-, м- и п-продуктов, образующихся из C6H5Y (распределение изомеров-, обычно количество данного изомера дается в процентах от общего количества полученных продуктов замещения). С учетом того, что в молекуле СбН6 находится шесть положений, доступных для атаки, а в молекуле CeHsY — два орто-положения и одно пара-положение имеем:
ko feCeH5Y/2 выход о-изомера(%)
° feCeHe/6 100
(2 орто-положения; 6 Н-положеннй)
kM fec6HSY/2 выход л-изомера (%) f м== too
(2 лета-положения; 6 Н-положений)
kn fec6H5Y/i выход п-нзомера (%)
п kH feC#He/6 100
(1 пара-положение; 6 Н-положеннй)
Найдено, например, что для реакции нитрования толуола азотной кислотой в уксусном ангидриде при 0°С A(C6HsMe)/
174
/fe(C6H6)=27, распределение изомеров: 61,5 (о-), 1,5 (л-), 37,0 % (п-)- Рассчитанные на основе этих данных значения факторов парциальных скоростей приведены ниже:
(хлорирование)
Ме
2400
(бромирование)
Сравнение факторов парциальных скоростей для реакций нитрования, хлорирования и бромирования толуола показывает, что они различны в зависимости от атакующего электрофила. Другими словами, относительные ориентирующие эффекты в C6H5Y зависят как от Y, так и от Е+. Выше было отмечено, что абсолютные значения факторов парциальных скоростей, т. е. ky/kw, увеличиваются в ряду: нитрование < хлорирование < < бромирование, т. е. по мере уменьшения реакционной способности атакующего электрофила. Этот кажущийся парадокс оказывается при некотором размышлении вполне понятным: если бы Е+ был достаточно реакционноспособным, то каждое столкновение вело бы к замещению, атакующий реагент был бы в этом случае совершенно неселективным, а каждый фактор парциальной скорости был бы равен единице. Однако при уменьшении реакционной способности Е+ не каждое столкновение приводит к реакции, которая все больше зависит от относительной способности орто-, мета- и пара-положений в C6HsY и положений в С6Н6 поставлять электронную пару для образования связи с Е+. Атакующий реагент действует все более избирательно, его селективность возрастает, а абсолютные значения факторов парциальных скоростей увеличиваются, как и относительные различия между ними; именно это и отражается в цифрах, приведенных выше. Относительную избирательность лучше всего оценивать при сравнении только факторов fn и fM поскольку фактор fo зависит от стерических эффектов (размер Y и относительный размер атакующего реагента; ср. разд. 6.7.3), а не только от электронных эффектов, которые влияют на все три положения.
Использование факторов парциальных скоростей позволяет провести более точный анализ ориентирующих эффектов, чем это можно было сделать до сих пор. Тот факт, что факторы парциальных скоростей для толуола > 1, указывает на то, что СНз-группа (см. разд. 6.7.1.2) активирует все положения ядра по сравнению с бензолом. То же справедливо и для группы СМе3, но в этом случае для реакции нитрования С6НбСМез
175
fin = 3,0, тогда как для толуола он равен 1,3, т. е. группа СМе3 проявляет больший электронодонорный индуктивный (полярный) эффект, чем СН3-группа. Наоборот, в бифениле (Y = = С6Н5) (см. разд. 6.7.1.2) для хлорирования fM = 0,7, т. е. атака лета-положения идет медленнее, чем в бензоле [котя Л(С6Н5¥)/А(СвН6) = 4,2-102], поскольку $р2-гибридизованный атом углерода, с помощью которого СбН5-заместитель связывается с бензольным кольцом, проявляет электроноакцепторный индуктивный (полярный) эффект [см. формулу (55)].
Ph :OPh
(55) (56)
Сходный эффект наблюдается также и в случае орто-пара-ориентирующих, активирующих заместителей, когда в исследуемой реакции образуется достаточное для определения количество продукта лета-замещения, например при дейтерировании (дейтериевый обмен) C6H5OPh (56) под действием сильной кислоты CF3CO2D. Очень высокие значения факторов f0 и fn отражают способность электронной пары на атоме кислорода избирательно стабилизировать переходные состояния, образующиеся при о- и га-атаке (ср. разд. 6.7.1.3), в то время как значение fM < 1 отражает дестабилизацию (по сравнению с атакой бензола) переходного состояния в случае л-атаки вследствие электроноакцепторного индуктивного (полярного) эффекта атома кислорода.
Факторы парциальных скоростей, а следовательно, и распределение изомеров в каждой реакции замещения также зависят от температуры. Увеличение температуры оказывает наибольший относительный эффект на реакцию замещения, имеющую максимальное значение AG+ (из трех возможных альтернативных атак C6H5Y), т. е. на самую медленную реакцию. Влияние увеличения температуры направлено, таким образом, подобно эффекту увеличения реакционной способности Е+, на «выравнивание» различий между факторами парциальных скоростей и на то, чтобы сместить распределение изомеров в продукте несколько ближе к статистическому.
6.7.3. Соотношение орто- и пара-изомеров
После всего, что мы рассмотрели до сих пор, вряд ли покажется удивительным то обстоятельство, что отношение количеств орто- и тгара-изомеров, получаемых при замещении C6HSY,
176
В. котором Y является орто-пара-ориентантом, редко (если бывает вообще) равно статистическому отношению 2:1. Показано, чтр существует очень близкое соответствие между расчетными данными и данными ЯМР-спектроскопии относительно распределения положительного заряда (п- > о- Л-) на различных атомах циклогексадиенил-катиона (57), который представляет собой интермедиат Уиланда в реакции обмена протона в бензоле (ср. разд. 6.1.1):
(57) (57а) (576)
Электронодонорный заместитель Y должен облегчать атаку Н+ в пара-положение (57a; R = Н) больше, чем в орто-положение (576; R =Н) по отношению к заместителю Y, потому что в результате достигается несколько более эффективная делокализация положительного заряда. Цифры, приведенные в формуле (57), соответствуют ожидаемым отношениям логарифмов факторов парциальных скоростей, 1g fo/lg fn ~ 0,87; действительно, значения, очень близкие к приведенным, были получены при протонировании ряда соединений CeHsY.
Стерические требования Н+, конечно, очень невелики, но если CeH5Y атакуется любым другим электрофилом Е+, который неизбежно является более громоздким, взаимодействие между Е и Y в переходном состоянии, образующимся во время атаки орто-положения по отношению к Y (576; R = Е), усилится из-за увеличения размеров атакующего электрофила и заместителя. Такого увеличения взаимодействия не можт быть в переходном состоянии, образующемся во время атаки пара-положения (57a, R = E). Эти особенности отражаются в увеличении AG* для о-атаки; такая реакция будет, следовательно, более медленной, и относительная доля продукта орто-замещения будет уменьшаться по мере увеличения размеров Е и (или) Y. Это иллюстрируется снижением отношений fo/fn для реакции нитрования различных алкилбензолов в сравнимых условиях:
Увеличение размера Y
Содержание изомера, %
Y орто пара
СН3 58 37 0,78
СН2Ме 45 49 0,46
СНМе2 30 62 0,24
СМе3 16 73 0,11
177
и для реакций хлорбензола с различными электрофилами:
Увеличение размера Е +
Реакция
Содержание изомера, %
орто пара fo/f п
Хлорирование 39 55 0,35
Нитрование 30 70 0,21
Бромирование 11 87 0,06
Сульфирование 1 99 0,005
То, что стерический фактор не является единственным определяющим, видно из данных для нитрования галогеибензолов: галогены ведут себя как орто-пара-ориентанты, но общая скорость атаки галогеибензолов немного меньше, чем скорость , атаки бензола (см. разд. 6.7.1.4):
Увеличение размера Y
Содержание изомера, %
Y орто пара ^о^п
F 12 88 0,07
С1 30 69 0,22
Вг 37 62 0,30
I 38 60 0,32
Несмотря на увеличение размеров заместителя Y от F к I, доля орто-изомера (а следовательно, и отношение fo/fn) фактически возрастает. Увеличивающийся стерический эффект, как и в случае алкилбензолов, действует в направлении ингибирования о-атаки, ио этот эффект конкурирует с электроноакцепторным индуктивным эффектом и эффектом поля, проявляемыми атомом галогена Y. Индуктивный эффект уменьшается по мере удаления от атома галогена и проявляется несколько слабее в пара-положении по сравнению с соседним орто-положением. Оттягивание электронов будет особенно отражаться в орто-положении при наличии очень электроотрицательного атома фтора, и поэтому наблюдается. относительно медленная атака фтор-бензола в орто-положение, несмотря на малые размеры атома фтора. Электроноакцепторный эффект галогена заметно падает от F к I (наибольшее изменение наблюдается при переходе от F к С1), что и приводит к усилению атаки в орто-положение, несмотря на увеличение объема Y.
Известно несколько случаев, когда орто-замещение происходит при почти полном отсутствии атаки в пара-положение. Такие реакции объясняются взаимодействием (по механизму комплексообразования) заместителя, уже находящегося в кольце, и атакующего электрофила, так что последний «направляется» в соседнее орто-положение. Например, при нитровании простого эфира—1-метокси-2-фенилэтана (58)—нитрующей смесью получают 32 % орто-изомера и 59'% пара-изомера (вполне нормальное распределение), но при нитровании действием N2Os в MeCN образуется 69 % орто- и 28 % пара-изомеров. Эта пре-
178
имущественная атака во втором случае, как полагают, протекает по, схеме:
(58)
Наконец, следует сказать, что соотношения орто- и пара-изомеров в значительной степени зависят от растворителя, в котором проводится реакция. Это может объясняться изменениями в относительной стабилизации молекулами растворителя тех переходных состояний, которые образуются при о- и n-атаках, но может следовать и из реальных различий атакующих электрофилов в разных растворителях. Реагирующие молекулы образуют комплексы с молекулами растворителя, и возникающий электрофил оказывается различным в каждом случае. Такое положение почти наверное имеет место при галогенировании в отсутствие кислот Льюиса как катализаторов. Например, при хлорировании толуола при 25 °C отношения f0/fn оказались равными 0,75 и 0,34 в зависимости от растворителя.
6.7.4. ипсо-Замещение
Кроме о-, м- и n-атаки C6H5Y существует, по крайней мере теоретически, возможность атаки электрофилом атома углерода кольца, с которым связан заместитель Y:
интермедиат
Результатом такой атаки должно быть, таким образом, замещение Y+ электрофилом Е+. Такую реакцию называют ипсо-замещением.
Известен ряд таких реакций, в которых атакующим электрофилом является Н+, например:
(Y+= Me3sr)
179
В этом случае заместитель Me3Si может быть замещен особенно легко (протодесилилирование); аналогичное замещение происходит в обратимой реакции сульфирования (протодесу/tb-фирование) (см. разд. 6.4): /
Главным фактором, содействующим суммарной реакции unco-замещения, является легкость образования Y+. Можно надеяться поэтому, что реакции такого типа возможны при наличии вторичных и третичных алкильных заместителей вследствие относительной устойчивости соответствующих вторичных и третичных карбокатионов R+. Показано, что это случается в реакциях нитрования (нитродеалкилирование), приведенных ниже:
СНМе2 (56%)
(Y+ = +CMe3)
No+ не-
возможно замещение не только алкильных, но и других групп (например, нитродегалогенирование):
(Y = Br+,31%)
(Y = I , 40%)
Однако аналогичное нитродехлорироваиие не наблюдается, так как энергия образования С1+ выше, чем в случае Вг+ и I+. Хотя многие из наблюдавшихся реакций unco-замещения являются реакциями нитрования, оно может происходить также и во время атаки другими электрофилами (например, бромдеал-
180
килирование):
(Y+=+CMe3)
(71%)
Несомненно, что такой ыпсо-атаке, как и в некоторых других приведенных выше реакциях деалкилирования, благоприятствует ингибирование нормальной электрофильной атаки тех положений в кольце, которые окружены объемистыми алкильными группами. Важно отметить, что с возможностью протекания реакций ыпсо-замещения следует считаться, когда предполагается осуществить препаративное электрофильное замещение полиза-мещенных производных бензола.
6.8. Кинетический и термодинамический контроль
Во всех случаях, о которых шла речь выше, предполагалось, что соцтношение альтернативных продуктов, образующихся в ходе реакции, например о-, м- и n-изомеров, определяется соотношением скоростей их образования, т. е. регулируется кинетически (см. разд. 2.2.3). Однако это не всегда наблюдается на практике. Например, при алкилировании по Фриделю — Крафтсу толуола (Me — орто-па/щ-ориентант) бензилбромидом при 25 °C в присутствии GaBr3 (кислота Льюиса; катализатор) найдено следующее соотношение изомеров:
Время, с Содержание изомера, % орто мета пара
0,01 40 21 39
1,0 23 46 31
Даже через очень короткий промежуток времени с начала реакции (0,01 с) сомнительно, чтобы соотношение изомеров (в небольшом количестве уже успевшего образоваться продукта) контролировалось чисто кинетически — доля м-изомера уже относительно велика. Это подтверждается составом смеси, полученной через 10 с: м-бензилтолуол— термодинамически наиболее устойчивый изомер — преобладает, и контроль теперь уже, очевидно, определяется равновесными характеристиками, т. е. становится термодинамическим (см. разд. 2.2.3).
Такая ситуация должна иметь место в тех случаях, когда альтернативные продукты способны взаимно превращаться друг в друга в условиях реакции. Это возможно или путем прямой изомеризации, или вследствие обращения реакции с образованием исходных веществ, которые затем подвергаются новым
181
атакам и дают термодинамически более устойчивые изомеры. Важно подчеркнуть, что соотношения образующихся альтернативных продуктов будут определяться их относительной термодинамической устойчивостью в условиях данной реакции, которые могут отличаться от условий существования изолированных молекул. Например, при нагревании jw-диметилбензола (jw-кси-лол) при 82 °C с HF в присутствии каталитического количества BF3 содержание трех изомерных диметилбензолов в продукте реакции очень близко к содержанию, вычисленному на основе их термодинамических устойчивостей:
Содержание, %
эксперимент расчет
о-Изомер 19 18
л-Изомер 60 58
п-Изомер 21 24
Однако при использовании избытка BF3 продукт реакции содержит 97 % jw-диметилбензола; это связано с тем, что диметил-бензолы в этих условиях могут превращаться в соответствующие соли, например:
Равновесие вследствие этого будет смещаться в сторону наиболее основного изомера, а именно мета-изомера, который образует наиболее стабильный катион (59) в составе ионной пары с BFJ. Известны также примеры реакций, в которых тип контроля зависит от температуры (см. ниже).
6.9. Электрофильное замещение в других ароматических системах
В случае нафталина электрофильное замещение (например, нитрование) происходит преимущественно в положение 1(a), а не в положение 2(Р). Это можно объяснить более эффективной делокализацией, а следовательно, и стабилизацией, которая имеет место в интермедиате Уиланда, в случае атаки в положение 1 (60а) (606), чем при атаке в положение 2 (61):
(61)
182
В каждом случае можно записать еще несколько форм интермедиатов, в которых положительный заряд делокализован во втором кольце. Для замещения в положение 1 таких форм может быть семь, для замещения в положение 2 — шесть. Однако наиболее важны те формы, в которых второе кольцо сохраняется нетронутым и сохраняет полностью делокализованные л-орбитали; различие между а- и ^-замещением при этом становится особенно заметным, так как первому случаю соответствуют две такие формы, а второму — одна. Возможность делокализации заряда в ст-комплексе нафталина выше, чем в ст-комплексе бензола. Это позволяет предположить, что электрофильная атака в случае нафталина будет протекать легче, что и наблюдается в действительности *.
Было показано, что сульфирование нафталина концентрированной H2SO4 при 80 °C приводит почти полностью к а-замеще-нию, тогда как скорость образования альтернативной 2-сульфо-кислоты при этой температуре очень мала, т. е. имеет место кинетический контроль. В то же время сульфирование при 160 °C приводит к продукту, содержащему не менее 80 % 2-сульфокислоты и лишь 20 % 1-изомера. Тот факт, что в этих условиях действует термодинамический контроль, подтверждается наблюдением, что при нагревании чистой нафталин-1- или нафталин-2-сульфокислоты в концентрированной H2SO4 при 160 °C образуется равновесная смесь такого же состава: 80 % 2-сульфокислоты и 20 % 1-сульфокислоты. Большая устойчивость 2-сульфокислоты по сравнению с 1-сульфокислотой объясняется тем, что в 1-сульфокислоте наблюдается дестабилизирующий эффект из-за стерического взаимодействия между объемистой группой SO3H и атомом Н в соседнем положении (положение8), тогда как в 2-сульфокислоте атомы водорода в положениях 1 и 3 более удалены от группы SO3H.
Взаимное превращение 1- и 2-сульфокислот в H2SO4 при 160 °C может осуществляться или в результате прямой внутримолекулярной изомеризации, или вследствие обращения реакции сульфирования с образованием нафталина, который подвергается новой атаке уже в другом месте. Для того чтобы решить, по какому из этих путей идет взаимодействие, проводят реакцию в H25SC>4. В первом случае образующиеся сульфокислоты не должны содержать 35S, тогда как во втором случае включение 35S должно иметь место. Экспериментально показано, что включение 35S действительно происходит, но со скоростью более медленной, чем скорость взаимопревращения кислот. Это можно
* О применении концепции граничных орбиталей для объяснения соотношения изомеров в реакциях электрофильного ароматического замещения в нафталине и других полициклических ароматических углеводородах см. прим, ред. иа с. 167. — Прим. ред.
183
объяснить тем, что либо оба пути осуществляются одновременно, либо тем, что после десульфирования новая атака на образующиеся молекулы нафталина освобождающимися молекулами H2SO4 происходит быстрее, чем атака молекулами H^SCh- Вопрос о том, какой путь преобладает, остается открытым.
Пиридин (62), подобно бензолу, имеет шесть л-электронов (один из которых поставляется азотом) на делокализованных n-орбиталях, но, в отличие от бензола, л-орбитали пиридина деформированы из-за смещения электронов в сторону атома азота, так как азот более электроотрицателен, чем углерод. Это проявляется в наличии у пиридина дипольного момента; отрицательный конец диполя находится на атоме азота, а положительный — на ядре:
и = 2,ЗД(62)
Пиридин, следовательно, является л-дефицитным гетероциклом и, по аналогии с бензольным кольцом, содержащим электроноакцепторный заместитель, например NO2 (см. разд. 6.7.1.1), можно ожидать, что он будет дезактивирован в отношении электрофильной атаки. Замещение происходит с трудом в положение 3, поскольку при этом образуется наиболее устойчивый интермедиат Уиланда (63); каждый из интермедиатов, образующихся в результате атаки положений 2 и 4 [(64) и (65) соответственно], имеет каноническое состояние, в котором положительный заряд локализован на двухвалентном атоме азота; это состояние чрезвычайно неустойчиво, т. е. обладает высокой энергией:
Н. Я
(63)
(65)
Здесь имеется некоторая формальная аналогия с м-атакой нитробензола (ср. разд. 6.7.1.1), но пиридин замещается с большим трудом, чем нитробензол. Так, нитрование, хлорирование, бромирование и реакции Фриделя — Крафтса практически не могут быть проведены, а сульфирование происходит только при нагревании с олеумом в течение 24 ч при 230 °C в присутствии Hg2+ (стабилизатор). Такая затрудненность атаки обусловлена отчасти тем, что пиридин имеет свободную электронную пару на
184
атоме азота и может, следовательно, протонироваться или взаимодействовать с электрофилом [формулы (66) и (67) соответственно]. Возникающий при этом положительный заряд должен, безусловно, дестабилизировать любой из ст-комплексов, образующихся при электрофильном замещении, как при наличии заместителей типа +NR3 в бензольном ряду (см. разд. 6.7.1.1). Однако в случае пиридина дестабилизация будет гораздо более выражена, так как положительный заряд в протонированном пиридине находится на одном из атомов кольца, а не просто на заместителе.
(66) (67)
, Пиррол (68) также имеет шесть л-электронов на делокализованных л-орбиталях, причем его атом азота вносит в л-элек-тронную систему два электрона, чтобы довести их число до шести (поэтому пиррол становится практически неосновным), а диполь пиррола, как было найдено, имеет противоположное направление, т. е. положительный конец — на атоме азота, а отрицательный конец — на ядре.
(69)
Пиррол, таким образом, является л-избыточным гетероциклом. Пиррол ведет себя аналогично реакционноспособному производному бензола, например анилину (см. разд. 6.7.1.3), очень легко подвергаясь электрофильной атаке. Эта атака осложняется тем, что в сильнокислой среде пиррол очень легко протони-руется, несмотря на его низкую основность; протонирование происходит по атому углерода в положении 2 [(формула (69)], а не по атому азота (ср. разд. 3.2.4). Вследствие этого соединение утрачивает ароматический характер; катион ведет себя аналогично сопряженному диену, очень легко подвергаясь полимеризации.
Электрофильное замещение пиррола можно тем не менее осуществить в специфических условиях [например, ацилирование (МеСО)2О—BF3, сульфирование комплексом пиридина с SO3(C6H5N-SO3); ср. (67)], обеспечивающих преимущественную атаку в положение 2, а не 3. Это объясняется несколько большей устойчивостью интермедиата Уиланда (70), образующегося в первом случае, по сравнению с интермедиатом (71), который
185
образуется во втором случае:
(70а) (706) (70в)
Различие в стабильности этих двух структур не очень велико из-за высокоактивированного состояния ядра, и замещение легко идет в положение 3, если положение 2 занято. Действительно, довольно легко можно наблюдать замещение всех четырех атомов углерода, например', при бромировании бромом в уксусной (этановой) кислоте.
6.10. Нуклеофильное замещение в ароматических системах
6.10.1 Замещение атомов водорода
Можно ожидать, что нуклеофильная атака незамещенного бензольного ядра протекает с гораздо большим трудом, чем электрофильная. Это связано с тем, что л-электронное облако ядра (см. разд. 6.1), вероятно, отталкивает приближающийся нуклеофил, и с тем, что л-орбитальная система бензольного ядра гораздо менее способна к делокализации (а следовательно, и к стабилизации) двух лишних электронов в отрицательно заряженном комплексе (72), чем в положительно заряженном интермедиате Уиланда (73):
(72) (73)
Оба эти условия в некоторой степени преодолеваются, если присутствует достаточно сильный электроноакцепторный заместитель. Нуклеофильная атака в этом случае становится возможной (ср. присоединение нуклеофилов к алкенам, содержащим электроноакцепторные заместители; разд. 7.6). Так, показано, что при сплавлении нитробензола с КОН иа воздухе образуется
186
о-нитрофенол (75) н небольшое количество п-нитрофенола:
(74) (75)
Для анионных частиц (74) (ср. интермедиаты Уиланда) можно написать и другие канонические структуры, но наиболее важна структура, в которой отрицательный заряд накапливается (и стабилизируется) на атоме кислорода нитрогруппы. Это возможно только в том случае, если атакующая группа -ОН занимает орто- или пара-положения по отношению к NO2-rpynne (ср. специфическую стабилизацию ст-комплексов во время электрофильной атаки орто- и пара-положений к группе ОМе; разд. 6.7.1.3). Частица (74) может восстановить ароматические свойства при отщеплении из положения 2 группы -ОН или иона Н~; в первом случае образуется продукт восстановления исходного вещества (нитробензола), в последнем — продукт (75). Гид-рид-ион Н~—плохая уходящая группа (в отличие от гораздо лучшей уходящей группы Н+ при электрофильной атаке), так что равновесие стремится сместиться влево. Группа -ОН, являясь лучшей уходящей группой, отщепляется гораздо чаще, чем Н~, если только не присутствует какой-либо окисляющий агент, например кислород воздуха, KNO3 или КзРе(СМ)6, который способствует отщеплению гидрид-иона и связывает его, как только он образуется. Некоторые превращения происходят в отсутствие какого-либо окисляющего агента, потому что нитробензол сам может действовать как окисляющий агент (восстанавливаясь до азоксибензола), но в этом случае выход нитрофенола очень мал.
Как можно было бы ожидать, электроноакцепторный заместитель NO2, направляющий электрофильную атаку в мета-по-ложение, нуклеофильную атаку направляет в орто- и пара-положения.
В случае пиридина, атом азота которого обладает способностью оттягивать электроны, атака сильными нуклеофилами возможна и при отсутствии таких заместителей. Так, пиридин реагирует с ионом +NH2 (амид натрия NaNH2) в Л\#-диметил-анилине (растворитель) — реакция Чичибабина:
(76)
(77) (77а)
187
Уходящая группа Н~ отщепляет протон от вводимой группы NH2, высвобождая таким путем Н2 и превращая амин (77) в анион (77а). Этот анион может превратиться при обработке водой в целевой 2-аминопиридин (77), который является полезным исходным соединением для дальнейших синтезов.
6.10.2. Замещение неводородных атомов
Поскольку Н- (в противоположность Н+)—очень плохая уходящая группа, то при простом ароматическом нуклеофиль^ ном замещении ыпсо-атака (ср. разд. 6.7.4) является правилом, а не исключением. Такие группы, как СГ, Вг', N2, SO3, 'NR2 и т. д., входят в число наиболее эффективных уходящих групп. Поэтому для соответствующих ароматических субстратов могут наблюдаться процессы, аналогичные нуклеофильному замещению у насыщенного атома углерода (см. разд. 4.1).
Типичным примером является замещение N2 в реакциях с солями диазония ArN2, имеющее очень большое препаративное значение:
ArN£ + Y’ —> ArY + N2
Показано, что эта реакция подчиняется кинетическому уравнению
Скорость = fe[ArNj],
т. е. скорость реакции не зависит от концентрации Y- (аналогия с механизмом 5ы 1; см. разд. 4.1). Это соответствует медленному, скорость лимитирующему образованию арил-катиоиа, например (78), с последующей быстрой его реакцией с присутствующим нуклеофилом:
-N2 (78)
Сходство с механизмом 5ы1 усиливается тем фактом, что добавленные нуклеофилы (СН, МеОН и др.) влияют на состав продуктов реакции, но не на скорость реакции; это соответствует приведенному выше кинетическому уравнению.
Образование очень неустойчивого фенил-катиона (78), в котором положительный заряд не может быть делокализован с помощью л-орбитальной системы, на первый взгляд кажется несколько удивительным. Однако это возможно багодаря чрезвычайной эффективности N2 как уходящей группы [энергия связи N=N равна 946 кДж/моль (226 ккал/моль)]. Важно отметить,
188
что эта реакция, по-видимому, является единственной реакцией, в ходе которой в растворе могут генерироваться простые арил-катионы. Эти катионы очень реакционноспособны и поэтому мало избирательны по отношению к нуклеофилам. Так, для катиона СбН5+ &(С1-)/&(Н2О) = 3, тогда как для МезС+ это отношение равно 180. Очень высокая активность CgHs проявляется и в его способности рекомбинировать с N2; разложение катиона диазония является, таким образом, обратимый, что было обнаружено, в частности, при изучении захвата метки 15N:
+ 15 15 15
N=N N=N N=N +
(79a) (796)
Особенно важной реакцией солей диазония ArN2 является введение в бензольное ядро атома фтора (это невозможно путем прямого взаимодействия с F2; ср. разд. 6.4):
д
ArNj BF; ------>- Аг—F + Njf + BF3f
Фторбораты, в отличие от других солей диазония, относительно устойчивы. Их можно выделить, а затем нагреть в сухом виде, чтобы получить чистый ArF; другие продукты выделяются в виде газов. Некоторые реакции солей диазония, особенно в менее полярных растворителях, могут протекать также и через первоначальное образование радикалов (ср. разд. 11.5.2.3).
Наиболее типичной реакцией ароматического нуклеофильного замещения является замещение Hal- в галогенпроизвод-ном, активированном электроноакцепторными группами, например в соединении (80) :
Такие реакции обычно протекают в соответствии с кинетическим уравнением
Скорость — k [ArX] [ Y"),
так что для них можно отметить некоторое формальное сходство с механизмом SN2. Однако в приведенном выше механизме атака нуклеофилом Y- не может происходить «с тыла» атома
189
углерода, связанного с уходящей группой (ср. механизм SN2; разд. 4.1), а должна происходить сбоку. Такой механизм часто называют механизмом 5ы2аРомат. Кроме того, исходя из приведенного выше кинетического уравнения, эта реакция может быть согласованной (подобно Sn2) [в этом случае структура (81) является переходным состоянием] или протекать по ступенчатому механизму, в котором медленной, скоростьлимитирующей стадией является стадия 1 или стадия 2 [в этом случае (81) является промежуточным соединением]. Последняя гипотеза подтверждается тем, что можно выделить и охарактеризовать мето: дами ЯМР-спектроскопии и рентгеноструктурного анализа ряд частиц, аналогичных (81), например (82), а также так называемый комплекс Нейзенгеймера (84) — кристаллическое вещество красного цвета. Комплекс (84) образуется при действии ЕЮ- на метиловый эфир (83) или при действии МеО~ на этиловый эфир (85). Подкисление реакционной массы приводит к образованию из обоих субстратов смеси (83) + (85) одного и того же равновесного состава. Это, конечно, не доказывает, что обычные реакции замещения, например, ароматических галогенидов проходят через образование промежуточных соединений, но это делает их образование более вероятным.
(82) Y=H,MeO
(83)
(85)
(84)
Прямым подтверждением ступенчатого механизма является сравнение скоростей реакций ряда субстратов, имеющих различные уходящие группы, с одним и тем же нуклеофилом, например реакций 2,4-динитрогалогенбензолов (86) с пиперидином (87):
(86) (87)
190
Было найдено, что относительные скорости реакций при X = С1, Вт и I равны 4,3, 4,3 и 1,0 соответственно. Разрыв связи С—X, следовательно, не может происходить на скоростьлимити-рующей стадии реакции, так как в этом случае следовало бы ожидать значительно больших различий в скоростях в ряду I > Вт 2> С1. Эта реакция не может быть, следовательно, одноступенчатой, т. е. согласованной (ср. Sn2), а в двухступенчатом механизме, предложенном выше, стадия 1 — атака нуклеофилом — должна быть скоростьлимитирующей стадией. Интересно также отметить, что относительная скорость приведенной выше реакции в случае X = F равна 3300. Это является результатом очень сильного электроноакцепторного ускоряющего действия атома фтора на стадию Г. а) путем превращения атома углерода, с которым связан фтор, в более положительный и поэтому более легко атакуемый нуклеофилом; б) путем содействия стабилизации анионного интермедиата (88):
NO?
2,4-Динитрофторбензол (86; X — F) из-за его высокой реакционной способности часто используют для связывания МНг-группы концевой аминокислоты при анализе концевых групп белка. Присоединившуюся при этом 2,4-динитрофенильную группу очень трудно отщепить; она сохраняется даже при последующем гидролизе белка на составляющие его аминокислоты.
Различие в скоростях нуклеофильного ароматического замещения объясняется тем, что атака соединения (86) зависит от способности атома галогена X (благодаря оттягиванию электронов) влиять на относительную легкость атаки субстрата нуклеофилом. Образующийся при этом ряд обратен ряду относительной способности галогенид-ионов быть уходящими группами. Относительные скорости реакции 2,4-динитрогалогенбензолов с C6H5NHMe (в нитробензоле при 120 °C) при X = F, С1 и Вг соответственно равны 1, 15 и 46, что соответствует их способности быть уходящими группами. Возможно, что в этой реакции стадия 2 входит, по крайней мере отчасти, в скоростьлимитирую-щую стадию.
Таким образом, к известным ранее реакциям типа Sn2, в которых разрыв связи с уходящей группой и образование связи с нуклеофилом происходят одновременно, и реакциям типа SnI, где разрыв связи с уходящей группой происходит до образования связи с нуклеофилом, следует добавить реакции типа 5м2аромат, в которых разрыв связи с уходящей группой происходит после образования связи с нуклеофилом. Это нуклеофильное
191
ароматическое «замещение» является по существу процессом присоединения — отщепления, очень сходным с электрофильным ароматическим «замещением» и отличающимся от него лишь атакующими частицами. Другими важными реакциями нуклеофильного ароматического замещения, имеющими препаративное значение, являются замещения SO3’ в солях щелочных металлов сульфокислот, например ArSOs Na+, нуклеофилами ~ОН и _CN, и менее важное замещение ~NR2 в п-нитрозо-А,А-диалкил-анилинах группой 'ОН.
Способность электроноакцепторного заместителя стабилизировать анионный интермедиат, например (81), объясняется только мезомерным эффектом, т. е. если нитрогруппа, например, находится в орто- или пара-положении по отношению к уходящей группе. Реакционная способность галогенпроизводных увеличивается в ряду:
Аналогично, 2- и 4-галогенпиридины, но не 3-галогенпири-дины, легко подвергаются нуклеофильному замещению. Мезо-мерное взаимодействие с электроноакцепторным заместителем будет снижено или исключено вовсе, если р-орбиталь атома, связанного с ядром, например атома азота в NO2, не может стать параллельной р-орбитали атома углерода ядра, с которым этот атом связан (стерическое ингибирование делокализации; ср. разд. 3.2.3). Относительные скорости нуклеофильной атаки ряда галогенпроизводных приведены ниже:
Различия в скоростях для соединений (91) и (92) очень невелики, поскольку Me-группы не препятствуют мезомериому электроноакцепторному эффекту CN-группы, имеющей линейное строение. Однако это различие гораздо более выражено для со-
192
единений (89) и (90), так как Me-группы в соединении (90) мешают атому кислорода нитрогруппы находиться в той же плоскости, что и бензольное ядро: перекрывание р-орбиталей атомов азота и соседнего атома углерода, таким образом, значительно снижено.
Наконец, необходимо упомянуть, что ряд некоторых реакций нуклеофильного замещения активированных галогенаренов можно проводить в биполярных непротонных растворителях, таких как диметилсульфоксид (ДМСО) Me2S+—О-. В этом случае отсутствует сольватная оболочка, образованная (как, например, в МеОН) за счет водородных связей, которую необходимо удалить от Y-, прежде чем он начнет действовать как нуклеофил. Вследствие этого значения AG* в таких растворителях гораздо ниже, а скорость реакции соответственно выше. Различие в скоростях не менее чем в 109 раз наблюдали при замене растворителя МеОН на Me2SO. Хлорбензол в этих условиях легко реагирует с Ме3СО~:
дмсо
Ph—С1 + Ме3СО" ----► Ph—ОСМез + СГ
6.10.3. Замещение по ариновому механизму
Неактивированные галогенарены относительно инертны к действию нуклеофилов в обычных условиях, однако их реакционная способность значительно повышается, если нуклеофилы являются очень сильными основаниями. Так, хлорбензол легко превращается в анилин в ходе реакции с ~NH2 (NaNH2) в жидком аммиаке при —33 °C:
жидк. NH3
PhCl + "NH2 -------->- PhNH2 + СГ
—33 °C
Предполагают, что такое различие в реакционной способности объясняется возможностью протекания реакции по механизму иному, нежели 5м2аромат- Некоторая информация об этом может быть получена при исследовании аналогичной реакции /г-метилхлорбензола (93), в которой одинаково легко образуется не только ожидаемый /г-аминометидбензол (94), но и неожиданный л-аминометилбензол (95), причем с относительно большим выходом:
~nh2
жидк. ЫНз, -33 *С
(94) (38%) (95) (62%)
7 П. Сайкс
193
о-Изомер при этом не образуется, а соединения (94) и (95), как было показано, в условиях реакции ие превращаются друг в друга. Эти данные в сочетании с тем фактом, что _NH2, как известно, способен удалять протоны (дейтероны) из бензольного кольца [причем скорость удаления протона (дейтерона) из дейтерированного фторбензола в 108 раза выше, чем из самого дей-териобензола]
жидк. NH3, -33 °C
дают возможность предположить, что в данном случае _NH2 атакует как основание атом водорода, который находится в орто-положении по отношению к атому хлора, а не атакует как нуклеофил атом углерода связи С—С1:
Me
Н
(95)
Отщепление протона от соединения (93) может протекать согласованно с отщеплением атома хлора или предшествовать ему; в результате образуется ариновый интермедиат (97). Этот интермедиат имеет два альтернативных положения [ср. формулы (1016) и (101 в)] для атаки -NH2. Образование продукта завершается отщеплением протона из растворителя NH3. Суммарным результатом реакции оказывается формальное присоединение NH3 в два альтернативных положения кольца. Не следует ожидать, что относительные количества таких двух альтернативных продуктов будут одинаковыми, так как интермедиат (97) имеет несимметричное строение, т. е. два возможных направления атаки -NH2 неодинаковы.
В этом случае реакция идет по механизму отщепления — присоединения, в отличие от присоединения — отщепления в случае механизма £м2арОмат, и формально соответствует реакции элиминирования простых алкилгалогенидов (см. разд. 8.7). Подтверждением аринового механизма является тот факт, что галоге-
194
ниды (98), (99) н (100) реагируют с ~NH2 в условиях, более жестких, чем те, которые можно было бы объяснить стериче-скими эффектами о-Ме-групп в этих соединениях. Причина, видимо, заключается в том, что ни одно из этих соединений не содержит атом водорода в орто-положении к галогену; это требование, как показано выше, необходимо для начала реакции, протекающей через ариновый интермедиат.
Арины не могут быть отнесены к ацетиленам, так как потребовалась бы очень сильная деформация бензольного кольца, чтобы расположить связи под углом 180°, как это требуется для зр'-гибридизованных атомов в алкине (разд. 1.3.3). Более вероятно, что делокализованные л-орбитали ароматической системы в основном не затрагиваются (ароматическая устойчивость таким образом сохраняется), а два доступных электрона располагаются на исходных зр2-гибридных орбиталях (101):
(Ю1а) (Ю1б) (101в)
Перекрывание между этими орбиталями (с учетом пространственных требований) оказывается слабым, как и соответствующие связи; поэтому арины очень реакционноспособны по отношению к нуклеофилам (и к электрофилам), хотя некоторую селективность к их действию они все же проявляют.
Существование аринов доказано в экспериментах с их улавливанием и по данным спектроскопии. Сам бензин (дегидробензол) был выделен в твердом аргоне при 8 К. Генерирование бензина (101) в присутствии фурана (102) приводит к получению аддукта реакции Дильса — Альдера (см. разд. 7.5.2) (103), который подвергается быстрому катализируемому кислотой разрыву кольца с выделением нафтола-1 (104):
(101) (102) (ЮЗ)
О
7~
195
Если беизии образуется в таких условиях, в которых отсутствуют частицы, с которыми бы ои взаимодействовал, тогда он очень быстро димеризуется («самоулавливаиие») в устойчивый дифеиилеи (105):
(101) (101) (Ю5)
Очень убедительное доказательство существования бензина получено физическими методами. В нагретую трубку масс-спектрометра вводили цвиттер-ион (106)—соль диазотированной о-аминобензойной (антраниловой) кислоты. Полученный масс-спектр оказался очень простым и содержал пики с т/е 28, 44, 76 и 152:
+ N2
m/e 28
Пик с т/е 76 быстро исчезал, а пик с т/е 152 увеличивался во времени, что указывает иа постепенную димеризацию бензина в более устойчивый дифенилен (105).
Для препаративного получения аринов использовали такие методы, как пиролиз соли (106), что не требует сильНощелочиой среды, и окисление 1-амииобензотриазола (107) в присутствии тетраацетата свинца:
РЬ(ОАс)«
—* (Ю1)+ 2N,
Вполне возможно, что реакции неактивироваииых галоген-аренов с основаниями, более слабыми, чем ~ОН, которые идут в значительно более жестких условиях, протекают как с образованием ариновых интермедиатов, так и по механизму 5ы2аромат. Относительные вклады каждого из этих механизмов в суммарное превращение зависят от соотношения нуклеофила и субстрата, от структуры ароматического субстрата и от условий реакции.
196
Глава 7
ЭЛЕКТРОФИЛЬНОЕ И НУКЛЕОФИЛЬНОЕ ПРИСОЕДИНЕНИЕ ПО СВЯЗИ С=С
7.1. Присоединение галогенов......................................198
7.2. Влияние заместителей на скорость присоединения...............202
7.3. Ориентация присоединения.....................................203
7.4. Другие реакции присоединения.................................206
7.4.1. Присоединение других галогенпроизводных................206
7.4.2. Гидратация.............................................207
7.4.3. Присоединение карбокатионов............................208
7.4.4. Гидроксилирование......................................209
7.4.5. Гидрирование...........................................211
7.4.6. Озонолиз...............................................212
7.5. Присоединение к сопряженным диенам...........................215
7.5.1. Электрофильное присоединение...........................216
7.5.2. Реакция Дильса — Альдера............................. 218
7.6. Нуклеофильное присоединение..................................220
7.6.1. Цианэтилирование...................................... 221
7.6.2. Реакция Михаэля........................................221
7.6.3. Присоединение к системе связей С —С—С = О............ 222
Как уже говорилось (см. разд. 1.3.2), двойная углерод-углеродная связь состоит из сильной о-связи и более слабой л-связи.
Пара электронов на л-орбитали более подвижна, менее прочно удерживается ядрами атомов углерода и, следовательно, легче поляризуется, чем электроны о-связи, что ведет к характерной для таких ненасыщенных соединений реакционной способности. Поскольку в углерод-углеродной двойной связи л-электроны пространственно наиболее доступны, естественно ожидать, что они должны экранировать молекулу от- атаки нуклеофильными реагентами. Показано, что л-электроны действительно играют такую роль (ср., однако, разд. 7.6). Неудивительно, что наиболее характерными реакциями двойной углерод-углеродной связи оказались реакции, инициируемые электронодефицитиыми частицами, такими как Х+ и X • (радикалы можно рассматривать как электронодефицитиые частицы, поскольку они стремятся присоединить еще один электрон для образования связи), причем катионы индуцируют гетероциклическое, а радикалы — гомолитическое расщепление л-связи. Показано, что первое преобладает в полярных растворителях, а второе — в неполярных растворителях, особенно при действии света. Реакции присоединения, индуцируемые радикалами, обсуждаются ниже (см. разд. 11.5.1.1).
197
7.1. Присоединение галогенов
Одним из классических тестов на ненасыщенную связь и, возможно, наиболее известной реакцией присоединения к алкенам является обесцвечивание брома, особенно в растворе СС1«. Эта реакция обычно легко протекает в отсутствие катализаторов, и заманчиво допустить, что имеет место простой одноступенчатый механизм:
Однако имеющиеся экспериментальные данные опровергают это предположение.
Во-первых, если присоединение брома проводить в присутствии нуклеофила Y" или Y: (например, СГ, NO3 Н2О:), то кроме ожидаемого 1,2-дибромида (3) образуются и такие продукты (4), которые содержат один атом брома и один атом Y (или группу), присоединившиеся по двойной связи:
вг2 + у-
СН2=СН2 -------> ВгСН2-СН2Вг + ВгСН2—ch2y
(3) (4)
Это, очевидно, не согласуется с одноступенчатым механизмом, подобным приведенному выше, в котором нет возможности для атаки нуклеофилом Y". Важно, конечно, установить, что соединение (4) не образуется просто в результате последующей атаки нуклеофилом Y~ ранее образовавшегося соединения (3); экспериментально найдено, что образование продукта (4) в этих условиях происходит с гораздо большей скоростью, чем скорость реакции нуклеофильного замещения. Возможное объяснение образования продукта (4) —это конкуренция между Y- и Вг- (образовавшимся из Вг2) в ходе формирования обычного интермедиата (см. ниже).
Во-вторых, показано, что [в случае таких простых алкенов, в которых это можно обнаружить, например в транс-бутене-2 (5)] атомы брома присоединяются с противоположных сторон плоской молекулы алкена, т. е. происходит owru-присоединение:
(6) мезо (анти-присоединение)
198
Продуктом реакции является симметричный мезо-дибромид (6), тогда как если бы имело место cuw-присоединение (оба атома брома присоединились бы с одной и той же стороны), то получали бы несимметричный (±)-дибромид (7):
(7)(±)
(син-присоединение)
Экспериментально показано, что в случае соединения (5), как и в реакциях с другими простыми ациклическими алкенами, присоединение почти полностью протекает стереоселективно, т. е. на « 100 % является сити-присоединением. Этот результат также не согласуется с одноступенчатым механизмом, поскольку атомы в молекуле брома расположены слишком близко друг к другу и не могут одновременно участвовать в анти-при-соединении.
Приведенные выше факты объясняются наличием такого механизма, в котором один конец молекулы брома становится положительно поляризованным вследствие отталкивания л-элек-тронов алкена и поэтому образует с алкеном л-комплекс (8) (ср. Вг2 + бензол; разд. 6.3). Затем л-комплекс расщепляется с образованием циклического бромониевого иона (9) — альтернативной канонической формы карбокатиона (10). Присоединение завершается нуклеофильной атакой оставшимся Вг- (или добавленным Y") одного из двух атомов углерода первоначальной двойной связи со стороны, противоположной бромониевому иону Вг+ с образованием мезо-дибромида (6):
(6)
199
Значительная взаимная поляризация брома и алкена при их взаимодействии [см. формулу (8) ] должна привести к образованию бромониевого катиона (9). Поляризация молекулы брома может быть усилена путем добавления кислоты Льюиса, например А1Вг3 (ср. бромирование бензола; разд. 6.3), что ведет к увеличению скорости реакции. Образование катиона (9), невидимому, обычно является скоростьлимитирующей стадией реакции.
Предположение об образовании циклического бромониевого иона в качестве интермедиата для объяснения высокостереосе-лективного аяти-присоединения, часто наблюдаемого для простых ациклических алкенов, было сделано еще в 1938 г. Данные, подтверждающие существование такого интермедиата, получены из нескольких различных источников: его оказалось возможным определить только физическими методами, используя «суперкислоты» (см. разд. 6.1.1) и ЯМР-спектроскопию. Например, при реакции 1,2-дибромида (11) с SbF5 в жидком SO2 при —60 °C образуется ионная пара, но при этом в спектре ожидавшегося соединения (12) обнаружили не два сигнала (по одному от каждой из двух различных групп, имеющих по шесть эквивалентных протонов), а только один сигнал (б 2,9), что указывало на эквивалентность всех двенадцати протонов, т. е. очень вероятно, что образуется бромониевый ион (9а):
Вг
Вг
/\CH3CH3
Н3С СН3
(11) 62, О
СН3
SbF5
жидк. SO2, у\ СНз
-60°С Н3С СНз Н3С СНз
(12) (9а) 62, 9
Участие брома в качестве соседней группы (ср. разд. 4.4.6) не доказывает, конечно, -что присоединение к алкенам проходит через циклический бромониевый ион, но означает, что такие частицы могут быть весьма вероятными интермедиатами.
При попытках присоединить Вг2 к очень необычному алкену (13) было доказано, что действительно можно выделить циклический бромониевый ион (14):
200
Это возможно только потому, что дальнейшая атака анионом Вг- ранее образовавшегося интермедиата (14) полностью предотвращается чрезвычайно объемистой, похожей на клетку структурой на каждом конце исходной двойной связи; таким образом, обычное суммарное присоединение Вг2 не может иметь места.
Степень анти-стереоселективности, обнаруживаемой при присоединении галогенов к алкенам, зависит от относительной устойчивости (в данных условиях реакции) промежуточного циклического галогенониевого иона, например (9а), и соответствующего карбокатионного интермедиата, например (12). Так, из-за большей электроотрицательности хлора по сравнению с бромом и соответствующей меньшей склонности разделять свою пару электронов можно ожидать, что присоединение хлора к некоторым алкенам будет менее стереоселективным, чем присоединение брома, что в действительности и происходит. Можно было бы также ожидать, что особенности строения, приводящие к спецйфической стабилизации карбокатионов, могли бы также привести к меньшей анти-стереоселективности. Это наблюдается, например, в случае транс-1 -фенилпропена (15):
сии-присоединение; 30%
анти-присоединение; 70%
Из-за возможности образования делокализованного карбокатиона бензильного типа в этом случае анти-стереоселективность гораздо ниже (70 %), чем при реакции транс-бутена-2 (5) (анти-стереоселективность составляет « 100 %; см. разд. 7.1), для которого подобная делокализация невозможна. Показано, что возрастание полярности и сольватирующей способности растворителя также стабилизирует карбокатионный интермедиат по сравнению с бромониевым ионом; это также уменьшает анти-стереоселективность. Так, найдено, что присоединение брома к 1,2-дифенилэтену (стильбен) в растворителях с низкой диэлектрической проницаемостью проходит на 90—100 % как анти-присоединение, а в растворителе с е = 35 — только на ~ 50 %.
Обычно невозможно присоединить фтор непосредственно к алкенам, так как реакция настолько экзотермична, что происходит разрыв связи. Многие алкены не присоединяют также и иод, а если реакция происходит, то она обычно легко обратима. Выяснено также, что алкины, например бутиндиовая кислота (17), подвергаются преимущественному, хотя и не исключитель-
201
ному, анти-присоедииеиию галогенов:
СО2Н
Вг СО2Н
со2н
но2с
(17) (70%) (30%)
7.2. Влияние заместителей на скорость присоединения
Интермедиат, образующийся в реакции присоединения брома, является ли ои бромоииевым иоиом или карбокатионом, положительно заряжен. Поскольку он образуется на скоростьли-митирующей стадии реакции, следовало бы ожидать, по аналогии с электрофильным замещением ароматических соединений (см. разд. 6.7.1.2), что и интермедиат, и переходное состояние, которое предшествует его образованию, могут быть стабилизированы электроиодоиориыми заместителями, т. е. такими заместителями [(18)], которые могли бы увеличить скорость электрофильного присоединения, а противоположный эффект наблюдался бы при наличии электроиоакцепториых заместителей 1(19)]:
Вг Вг+
(19)
Относительные скорости реакций некоторых алкенов в аналогичных условиях приведены ниже:
СН2=СН->Вг « СН2=СН^СО2Н < СНг=СН2 < Et^>CH=CH2 <
3- 10~2
1 9,6-10
Me. ,Et ^C=CZ н/
Me, ,Ме
ХС=СХ н/ ^Et
Me, ,Ме
ХС=СХ
Me7* ^Ме
4,6-10’ 1,19-105 9,25-10s
Эти относительные скорости очень чувствительны к изменению условий реакции. Наблюдаемое увеличение скорости при
202
введении последующих электронодонорных алкильных групп оказывается меньше ожидаемого. Это обусловлено увеличением пространственных затруднений в переходном состоянии, вызванных этими алкильными группами. Фенильная группа также значительно (в 4-103 раза) увеличивает скорость электрофильного присоединения благодаря стабилизации, которая проявляется в интермедиате (20) и в переходном состоянии, которое ему предшествует:
СН-СН2Вг
(20)
СН—СН2Вг
7.3. Ориентация присоединения
Если присоединяющийся электрофил, в отличие от галогенов, имеет несимметричное строение, то в случае несимметричных алкенов, например пропена, возникает проблема ориентации присоединения, как, например, при присоединении галогеново-дородов. Легкость присоединения галогеноводородов к алкеиу уменьшается в ряду
HI > НВг > НС1 » HF
т. е. по мере уменьшения их кислотности. Это позволяет предполагать, что скоростьлимитирующей стадией реакции является присоединение протона к алкену; последующая быстрая нуклеофильная атака анионом На1~ завершает присоединение. В неполярных растворителях источником протона является HHal, а в полярных, особенна в гидроксилсодержащих, растворителях более вероятно, что протон поставляется сопряженной кислотой такого растворителя, например Н3О+ в Н2О.
Мостиковый интермедиат, аналогичный бромониевому иону, не может образоваться, поскольку атом водорода не имеет свободной электронной пары, однако возможно, что в некоторых случаях интермедиатом является л-комплекс (21). Как правило, интермедиат изображают в виде карбокатиона; относительная устойчивость возможных альтернативных карбокатионов, например (23) и (24), очевидно, и определяет суммарную ориентацию присоединения, например, при присоединении НВг к протону (22) в полярных растворителях:
Н+ t Me—СН=СН2 (21)
203
н
Me—СН->СН2
н+ (23)
Me—СН=СН2 -----
(22) + У
—► Ме->СН«-СН2
Н
Me—(^Н— СН2
Вг
Н
Вг- |
--->- Me—СН-СН2
Вг
(24) (25)
Как было показано ранее (см. разд. 5.2), вторичные карбокатионы более устойчивы, чем первичные, а поскольку это относится к переходным состояниям, которые им предшествуют, то преимущественно должен образовываться карбокатион (24). В действительности, по-видимому, образуется только этот карбокатион, так как единственным продуктом присоединения является 2-бромпропан (25). Протекающую таким путем реакцию, при которой галоген (или более отрицательная часть какого-либо другого несимметричного реагента) присоединяется к более замещенному атому углерода двойной связи, называют присоединением па Маркавникаву.
Предположение о том, что присоединение ННа! обычно проходит через промежуточный карбокатион, подтверждается образованием из некоторых алкенов аномальных (т. е. неожиданных) продуктов присоединения, которые могли бы возникнуть только в результате перегруппировки карбокатиона (ср. разд. 5.4.2.2), например при взаимодействии 3,3-диметилбутена (26) н HI:
Me Me Н Me Н
I н* I + I г | |
Me2C—CH=CH2 -------*- Me2C—CH—CH2 -----► Me2C—CH—CH2
I
(26) вторичный ожидаемый
карбокатион продукт
I
Me H Me H
Me2C—<^Н—СН2 —Ме2С— <^Н— СН2 + I
I
третичный неожиданный
карбокатион продукт
При присоединении ННа! к алкенам в водной среде или в других гндроксилсодержащих растворителях конкурирующей
204
реакцией может быть катализируемая кислотой гидратация (см. разд. 7.4.2) или сольватация. В менее полярных растворителях возможно образование радикалов; в этом случае присоединение идет против правила Марковникова с образованием 1-бром-пропана МеСН2СН2Вг через преимущественное образование радикального интермедиата МеСНСН2Вг (подробнее см. разд. 11.5.1.2).
Электрофильное присоединение к 1-галогеналкенам, например (27), имеет ряд сходных черт с электрофильным замещением- в галогенбензолах (см. разд. 6.7.1.3), Так, в стабилизации промежуточных карбокатионов также участвует электронная пара атома брома, которая контролирует ориентацию присоединения (ср. орто-пара-ориентацию в СеНзВг);
Н Н
Н+ + | Вг- J
СН2=СН— Вг —Х-> СН2—СН—Вг --------► СН2—СН—Вг
(27) (28) В г
|н-
СН2—СН—Вг I
н
СН2—СН=Вг н
(29а) (296)
Вг СН2—СН—Вг н (30)
Карбокатион (29), в отличие от карбокатиона (28), стабилизирован, поэтому он преимущественно и образуется; фактически единственным продуктом реакции является 1,1-дибромэтан (30). Скорость присоединения, однако, контролируется электроноакцепторным индуктивным эффектом атома галогена. Показано, что алкен (27) присоединяет НВг примерно в 30 раз медленнее, чем этен (ср.: бромбензол атакуется электрофилами медленнее, чем бензол), т. е. карбокатион (29) менее устойчив и образуется медленнее, чем карбокатион (31):
Н Н
I I
СН2->СН->Вг сн2->сн2
(29) (31)
Присоединение галогеноводородов к простым алкенам идет несколько менее стереоселективно, чем присоединение галогенов, так как гораздо больше зависит от природы алкена и от условий реакции.
205
7.4. Другие реакции присоединения
7.4.1. Присоединение других галогенпроизводных
Межгалоидные соединения присоединяются к алкенам гораздо легче, чем сами галогены; реакционная способность уменьшается в ряду:
BrCl > Вг2 > IC1 > IBr > 12
Присоединение инициируется положительно поляризованным концом (менее электроотрицательный атом галогена) несимметричной молекулы, причем, вероятно, образуется циклический галогенониевый интермедиат. Присоединение I—С1 особенно стереоселективно (анти) из-за легкости образования (и относительной устойчивости по сравнению с карбокатионами) циклических иодониевых ионов. В случае несимметричных алкенов, например 2-метилпропена (32), атом углерода с более объемистыми алкильными группами будет быстрее образовывать карбокатион [т. е. этот атом менее прочно связан с Вг в ионе (33)], а поэтому преимущественно будет атаковаться оставшимся нуклеофилом С1~. Суммарная ориентация присоединения будет, таким образом, подчиняться правилу Маркоцникова; в результате образуется соединение (34):
б+
Вг D
S+ S- х++ а\ х ?г
р. _ о / 0+ I
Ме2С = СН2 -—Ме2С~ СН2 — Ме2С - СН2 (►oct Cl
(32) (33) (34)
Полагали, что кислоты типа НО—Hal, например НО6-— Вгб+ (бромная вода), присоединяются точно таким же путем, однако имеются данные, что истинным электрофилом может быть и сам галоген, например Вг2, и что как 1,2-дибромид (35), так и 1,2-бромгидрин (36) могут получаться вследствие конкуренции Вг- и H20s за взаимодействие с промежуточным бромониевым ионом:
Вг I СН2-СН2
Вг (35)
Nu = Br
Н2С = СН2 |Вг2 <Вг+ У\
Н2С—СН2 Nu-)
МцгНгО
Вг I сн2-сн2 он
(36)
206
7.4.2. Гидратация
Катализируемая кислотой гидратация алкенов является процессом, обратным катализируемой кислотами дегидратации спиртов до алкенов (протекающей по механизму £1; см. разд. 9.2):
Н +ОН2 ОН
н+ + | Н2О I -н+ I
МеСН=СН2 МеСН—СН2 МеСН—СН3 МеСН—СН3
(37)
Образование карбокатионного интермедиата (37) (непосредственно или через л-комплекс), по-видимому, является скоростьлимитирующей стадией, а ориентация присоединения в целом подчиняется правилу Марковникова. Имеются данные о некоторой анги-стереоселективиости, но в этом случае она не очень выражена и зависит от природы алкена и условий реакции.
Кислоты, содержащие слабые нуклеофильные анионы, например HSO< в разбавленной водной H2SC>4, используют в качестве катализаторов, причем их анионы слабо конкурируют с Н2О; эфиры ROSO3H, если даже и образуются, в любом случае будут гидролизоваться до ROH в условиях реакции. Может происходить перегруппировка карбокатионного интермедиата, a также его электрофильное присоединение к еще непротониро-ванному алкену (см. разд. 7.3). Эту реакцию используют в промышленности для превращения алкенов, полученных из продуктов крекинга нефти, в спирты; гидратацию алкенов при этом проводят водяным паром над гетерогенным кислотным катализатором. В условиях кислотного катализа к алкенам могут присоединяться спирты с образованием простых эфиров, а также карбоновые кислоты RCO2H— с образованием сложных эфиров.
Гидратация против правила Марковникова может быть проведена косвенно, путем присоединения В2Н6 (гидроборирование) с последующим окислением полученного триалкилбора (38) щелочным раствором Н2О2:
BzHg Н2О2
МеСН=СН2 ------*- (МеСН2СН2)3В ---► МеСН2СН2ОН + В(ОН)3
(38) (39)
Диборан образуется (in situ или отдельно) из NaBH4 и Et2O+—BF3 и, возможно, образует комплекс, подобно мономерному ВНз, с растворителем (эфир), используемым для реакции. ВН3 является кислотой Льюиса и присоединяется к наименее замещенному атому углерода алкена. Суммарный процесс присоединения завершается переносом гидрид-иона к соседнему
207
положительно заряженному атому углерода:
Н—ВН2 н ВН2
ВН3 I II
МеСН=СН2 ----->- МеСН—СН2 —► МеСН—СН2
(40)
Возможно, что комплекс (40) имеет циклическое строение, так как суммарный процесс присоединения ВН3 в некоторых случаях протекает как сын-стереоселективное присоединение. Первоначально образовавшийся RBH2 реагирует с алкеном с образованием триалкилбора R3B (38). Последующее окисление действием Н2О2 ведет к разрыву связи С—В, в результате чего получается спирт (39). Общим результатом этой гидратации, идущей против правила Марковникова, часто оказывается син-стереоселективный процесс с очень хорошими выходами продуктов.
7.4.3. Присоединение карбокатионов
Как показано ранее, протонирование алкенов приводит к образованию карбокатионов; в отсутствие других эффективных нуклеофилов (например, Н2О; см. разд. 7.4.1) эти ионы могут действовать как электрофилы по отношению к еще не протонированному алкену (ср. разд. 5.3), например, в реакции с 2-ме-тилпропеном (41):
Ме2С = СН2 === МезС СН^= СМе2 —Ме3С - СН2 -СМе2
(41) (42)
(43)
СН2 = СМе2 I _н+
+
Ме3С - СН2- СМе2- СН2- СН2- СМе2 Ме3С - СН = СМе2
(45) (44)
ИТ. р.
Первоначально образовавшийся катион (42) может присоединиться ко второй молекуле 2-метилпропена (41) с образованием нового (димерного) катиона (43). Последний в свою очередь может потерять протон и превратиться в алкен (44) или присоединиться к третьей молекуле алкена и образовать (три-мериый) катион (45) и т. д.
208
Следует отметить, что как протонирование, так и последующее присоединение происходят с образованием наиболее устойчивого катиона.
д-Метилпропен может участвовать в процессе образования высб^ополимерных соединений — катионной полимеризации; однако^ большая часть простых алкенов образует лишь димеры или тримеры. В качестве мономеров в промышленности используют 2-метилпропен (в производстве бутилового каучука) и простые виниловые эфиры ROCH=CH2 (клеящие вещества). Катионная полимеризация часто инициируется катализаторами типа кислот Льюиса, например BF3; необходимо добавление источников протонов, так называемых сокатализаторов, например следов Н2О и т. д. В таких системах полимеризация происходит при низких температурах и обычно протекает очень быстро. Однако гораздо большее число алкенов полимеризуется по пути, индуцируемому радикалами (см. разд. 11.5.1.3).
7.4.4. Гидроксилирование
Существует ряд реагентов, с помощью которых возможно присоединение двух групп ОН к алкенам. Так, тетраоксид осмия OsO4 присоединяется к алкенам, образуя циклический эфир (47), в котором связи Os—О легко гидролизуются, что приводит к 1,2-диолу (48):
ОН
+ (HO)2OsO2
н
(48) цезо
Из цис-бутена-2 (46а) образуется мезо-1,2-диол (48), т. е. гидроксилирование в целом сын-стереоселективно, как и следовало ожидать в результате разрыва связи Os—О в циклическом эфире (47), являющемся, безусловно, цис-изомером. Недостатком этой реакции как препаративного метода является дороговизна и токсичность OsO4. Однако OsO4 можно использовать в каталитических количествах в сочетании с Н2О2, который окисляет образующуюся осмиевую кислоту (HO)2OsO2 до OsO4.
Щелочной раствор перманганата — реагент, используемый в классическом тесте на ненасыщенные связи, — также может участвовать в стереоселективном сын-присоединении. Полагают, что этот процесс, по аналогии с рассмотренным выше, протекает через циклические (цис) эфиры. Такие эфиры еще не удалось выделить (некоторые из иих обнаружены спектроскопически),
209
но было показано, что использование Мп18О4 приводит к 1,2-дио'-лу [например, (48)], в котором оба атома кислорода являются атомами 18О, т. е. их источником является ион Мп18О4, а не Й2О (растворитель). Это служит подтверждением, что интермедиа-том является марганцевый аналог (47) при условии отсутствия обмена атомов кислорода между Мп'8О4 и Н2О, как и показано в данном случае. Недостатком применения MnOi для гидроксилирования является то, что образующийся 1,2-диол (48) очень чувствителен к дальнейшему окислению под действием МпОО
Пероксикислоты RCOOOH также окисляют алкены, например транс-бутен-2 (466), присоединяя атом кислорода к двойной связи с образованием эпоксида (49):
Эпоксиды, хотя и не заряжены, имеют формальное сходство с циклическими бромониевыми интермедиатами (ср. разд. 7.1), но, в отличие от них, устойчивы и могут быть легко выделены. Однако они подвергаются нуклеофильной атаке в условиях кислотного или основного катализа, образуя 1,2-диолы. В обоих случаях атака атома углерода нуклеофилом будет происходить со стороны, противоположной кислородному мостику в эпоксиде (49). Такая атака будет проходить с обращением конфигурации (ср. разд. 4.4.6):
210
Выяснено, что атакуется только один из двух возможных атомов углерода, причем в эпоксиде (49) и катионе (50) эти атомы разные. Атака другого атома углерода в каждом случае приведет к одному и тому же продукту — мезо- 1,2-диолу (51). Сравнивая конфигурации соединений (51) и (466), можно видеть, что протекает стереоселективное анти-гидроксилирование.
Следовательно, подбирая подходящие реагенты, можно провести син- или анти-гидроксилирование алкенов.
7.4.5. Гидрирование
Присоединение водорода к ненасыщенным соединениям — одна из самых обычных и важных реакций присоединения. Именно поэтому она и рассматривается в этом разделе, а не среди радикальных реакций. Прямое присоединение водорода обычно проводят в условиях гетерогенного катализа тонкоиз-мельченными металлами, такими как Ni, Pt, Pd, Ru, Rh. Атомы металла, расположенные на поверхности кристалла, в отличие от атомов внутри кристалла, имеют «нескомпенсированные валентности», направленные в стороны от поверхности. В результате этого как алкены (например, этеи), так и водород реагируют с поверхностью металлического катализатора, например никеля, экзотермически и обратимо. Взаимодействие алкенов с металлом осуществляется с помощью л-электроиов, тогда как алканы не могут адсорбироваться подобным образом. В молекуле водорода нет л-электронов, и при его адсорбции должно происходить заметное ослабление его <т-связи, хотя и необязательно с полным разрывом связи и образованием Н •.
Большое значение, безусловно, имеет расположение атомов металла на поверхности кристалла, вследствие чего одна поверхность кристалла обладает каталитическими свойствами, а другая не обладает. Это зависит от того, насколько близко расстояния между атомами металла соответствуют длинам связей в молекулах алкена и водорода. Показано, что только относительно небольшая часть общей поверхности металла обладает каталитической активностью и образует так называемые активные центры. Эти центры адсорбируют алкен и немедленно десорбируют образовавшийся алкан, освобождаясь таким путем для дальнейшей адсорбции алкена.
В соответствии с тем, что молекулы алкена «выстраиваются» на поверхности катализатора и учитывая возможное их сближение с активированными молекулами водорода, адсорбированными на поверхности металла, можно предполагать, что гидрирование протекает син-стереоселективно. Это действительно так, и такое гидрирование используют как для установления
211
структуры, так и для целей синтеза, например:
Н Н Н СМез
Ме3СС ее ССМе3--------—- С = С V - с
° * катализатор / \ ь — и
Линдлара Ме3С' ЧМез Me3CZ \н
(52) (53)
Алкины часто могут быть селективно восстановлены до алкенов при использовании катализатора Линдлара [Pd на СаСОз, частично «отравленный» РЬ(ОАс)г]. В этом случае процесс также протекает син-стереоселективно, несмотря на то что образуется более пространственно затрудненный, термодинамически менее устойчивый цис-алкен (52), а не транс-изомер (53).
син-Стереоселективность не всегда достигает 100 %, поскольку она очень зависит от условий. Фактический механизм гидрирования был детально изучен с применением D2 и оказался очень сложным; наряду с другими фактами, показано, что в алкенах происходит водородный обмен. Установлено, что два атома водорода не присоединяются к алкену одновременно, вследствие чего становится понятной и причина отсутствия 100 %-й син-стереоселективности. Выяснено также, что цис-алкены, например цис-бутен-2, обычно гидрируются гораздо быстрее, чем транс-алкены, например транс-бутен-2. Скорость гидрирования уменьшается с увеличением степени замещения в алкене.
Предложены гомогенные катализаторы гидрирования, например ЁЬС1(РЬ3Р)з, растворимые в реакционной смеси. Полагают, что такие катализаторы переносят атом водорода к алкену с помощью промежуточного гидрида металла. Эти катализаторы также обеспечивают значительную син-сТереоселективность в процессе присоединения водорода.
7.4.6. Озонолиз
Присоединение озона к алкенам с образованием озонидов и последующее их расщепление с образованием карбонильных соединений хорошо изучено:
, Оз Г ^21 H2/Pt ,
R2C=CR2 —► 4- -—> r2c=o 4- o=cr2 + H2O
L Оз J
озонид
Однако структура озонида долгое время была предметом споров. Легко представить себе 1,3-диполярное присоединение озо
212
на, инициируемое электрофильным концом его молекулы. Действительно, из продуктов реакции озона с алкеном (54) при —70 °C был выделен кристаллический аддукт (55):
(57)
Структура этого интермедиата была подтверждена данными ЯМР-спектроскопии и восстановлением его натрием в жидком аммиаке до 1,2-диола (56). Трудно сказать, каким образом каталитическое восстановление интермедиата (55) может привести непосредственно к нормальному карбонильному конечному продукту, однако было найдено, что при повышении температуры он превращается в соединение (57), которое может давать (и действительно, это так) нормальные карбонильные продукты при каталитическом восстановлении.
Соединение (55) называют мольозонидом, а (57) — нормальным озонидом. Превращение (55) в (57), как полагают, проходит следующим путем:
(58)
Р —* ° 9 = —* R2C^cr2
R2C-CR2 я2с сд2 ^0-0 0-0
(55а) (58) (59) (59) (57а)
Предполагают, что озонид (55а) распадается на карбонильное соединение (58) и пероксицвиттер-нон (59), причем последний активно вступает в реакцию 1,3-диполярного присоединения с первым и образует озонид (57а). Альтернативные реакции цвиттер-иона (59), включающие полимеризацию, приводят к «аномальным» продуктам, которые иногда образуются в ходе присоединения к озониду. Если озонолиз проводится в МеОН (растворитель), то ион (59) «улавливается», как только он
213
образуется, благодаря его превращению в относительно устойчивый а-гидропероксиэфир (60). Тем самым предотвращается взаимодействие цвиттер-иона (59) с кетоном (58) с образованием озонида, и оба эти соединения могут быть выделены и идентифицированы. При проведении озонолиза с препаративными целями важно разрушить озонид (57а), используя подходящий восстановительный процесс, иначе образуется Н2Ог (как это и происходит, например, при разложении озонидов водой), который далее окисляет чувствительные к окислению карбонильные соединения, например альдегиды, в карбоновые кислоты.
МеО—CR2—О—ОН (60)
Предложенный выше путь удовлетворительно объясняет основной путь озонолиза, но этого недостаточно для объяснения наблюдаемой стереохимии этой реакции. Например, транс (или цис) -алкены часто образуют, как и следовало ожидать, смеси цис- и транс-озонидов, тогда как транс-алкен (54) дает только транс-озонид (57). Этот пример требует высокой степени стереоселективности как при разложении озонида (55) до альдегида и пероксицвиттер-иона, так и при их последующей рекомбинации в озонид (57). Это требование не согласуется безоговорочно с обсужденным выше механизмом.
Озонолиз ранее применяли для определения положения двойной связи (или связей) в ненасыщенных соединениях неизвестного строения (в основном из-за простоты идентификации образующихся карбонильных продуктов), но сейчас этот метод вытесняется физическими методами и прежде всего ЯМР-спек-троскопией. Бензол образует триозонид, который разлагается с образованием трех молекул глиоксаля ОНС—СНО; это единственная реакция бензола, которая позволяет предположить, что он может содержать три «реальные» двойные связи в структуре Кекуле! Алкины также подвергаются озонолизу, но с гораздо меньшей скоростью, чем алкены.
Для расщепления алкенов с препаративными целями лучше использовать следующую последовательность реакций:
МПО4 но он . или 1 | . NaIC>4
r2c=cr'2-^- r2c—cr'-------->- R2C=O + O^CR2
Данный процесс может быть проведен в одну стадию, если использовать метапериодат натрия, который разлагает 1,2-диол в количестве, достаточном для реокисления только каталитических количеств MnOi нли OsO^ необходимых на быстрой первой стадии.
1,3-Диполярное присоединение к алкенам происходит при использовании не только озона, но и других частиц; часто при этом образуются более устойчивые продукты, чем лабильные
214
мольозониды (55). Например, присоединение азидов (61) приводит к дигидротриазолам (62):
1,3-Диполярное присоединение к алкенам рассмотрено в разд. 12.4.2.
7.5. Присоединение к сопряженным диенам
Сопряженные диены, например бутадиен (63), несколько более устойчивы, чем аналогичные диены, в которых двойные связи не сопряжены (см. разд. 1.3.5). Это отражается на их тепло-тах гидрирования (см. разд. 1.3.7), хотя энергия делокализации, обусловленная образованием сопряженной системы л-орбита-лей, составляет только 17 кДж/моль (4 ккал/моль). Тем не менее сопряженные диены вступают в реакции присоединения несколько легче, чем несопряженные диены. Это объясняется тем, что промежуточные соединения (64) н (65) (и, что еще более важно, предшествующие нм переходные состояния), возникающие в результате первоначальной атаки электрофилами и радикалами соответственно, имеют аллильное строение (ср. разд. 5.2 н 11.4) и стабилизируются путем делокализации в гораздо большей степени, чем исходные диены. Эти интермедиаты более стабильны, чем соответствующие интермедиаты (66) и (67), полученные при аналогичном присоединении к простым алкенам:
Вг Вг
Вг2 I + I +
сн2=сн—сн=сн2 —сн2—сн—сн=сн2 сн2—сн=сн—сн2
(63) (64)
2|вГ2
Вг Вг
^н2—сн—сн=сн2 сн2—сн=сн—сн2
(65)
• ВГ2 ВГ2
СН2—СН2 -*—2— СН2=СН2 —Н2С—сн2
Вг Вг+
(67) (66)
1 — электрофильное присоединение; 2 — радикальное присоединение
215
7.5.1. Электрофильное присоединение
Во всех случаях на первой стадии происходит атака концевого атома углерода сопряженной системы, иначе не мог бы образоваться промежуточный карбокатион (64), стабилизированный путем делокализации заряда. Вследствие такой стабилизации образуется именно промежуточный карбокатион, а не циклический бромониевый ион [ср. соединение (66)]. Реакция завершается нуклеофильной атакой Вт- соединения (64) и может происходить либо по атому С-2 (1,2-присоединение) (а) с образованием продукта (68), либо по атому С-4 (1,4-присоединение) (б) с образованием продукта (69):
S+ 6+
(64) ВгСН2 - СН—СН—СН2 (а) Вт Вт (б)
а Л Л
ВгСН2 - СН - СН = сн2 Вг
(68)
ВгСН2 -СН® СН- СН2Вг (69)
2-1, 2-присоединение; б - 1, ^присоединение
Обычно получаются оба продукта, но их относительные количества очень сильно зависят от условий реакции, например от температуры. Так, при взаимодействии НС1 с бутадиеном (63) при —60 °C образуется только. 20—25 % 1,4-аддукта (остальное— 1,2-аддукт), тогда как при более высоких температурах образуется « 75 % 1,4-аддукта. Полагают, что бромирование при низких температурах контролируется кинетически (ср. разд. 2.2.3), и в этом случае 1,2-аддукт образуется быстрее, чем 1,4-аддукт. При более высокой температуре и (или) более продолжительном времени реакции действует равновесный, или тер* модинамический, контроль; основным продуктом в этом случае становится термодинамически более устойчивый 1,4-аддукт. Это следует из того факта, что при более высоких температурах как чистый 1,2-, так и чистый 1,4-аддукт могут быть превращены при данных условиях реакции в одну и ту же равновесную смесь 1,2-и 1,4-аддуктов. При этом 1,4-присоединению благоприятствует увеличение полярности растворителя.
Существует лишь возможность того, что присоединение брома к бутадиену с образованием 1,4-аддукта может происходить через ненапряженный пятичленный циклический бромониевый иои (70). Это могло бы привести при нуклеофильном его рас
216
щеплении под действием Вг- к Zfuc-дибромиду (71):
НС=СН СН2Вг
/ \ Вг- I
н2с/ 'сн2 ----► нс=сн нс=сн
\/ I I I
Вг+ ВгН2С СН2Вг СН2Вг
(70) (71) (72)
Однако фактически получается только транс- 1,4-дибромид (72). Таким образом, частицы, подобные (70), не могут быть получены, и, вероятно, обычным промежуточным соединением является ионная пара (73), включающая делокализованный карбокатион. Взаимопревращения 1,2- и 1,4-аддуктов (68) и (69) соответственно могут проходить также через интермедиат (73):
Вг 8г"
I 6+ 6+
сн2 - сн - сн = сн2 сн2-^сн—сн - сн2
Вг (68) Вг (73)
Вг I сн2- сн = сн- сн2
В случае несимметричных диенов (74а) и (746) и несимметричных аддуктов возникает проблема ориентации присоединения (ср. разд. 7.3). Первоначальная атака также происходит по концевому атому углерода сопряженной системы с образованием делокализованного аллильного интермедиата, однако преимущественно будет атаковаться тот концевой атом углерода, который будет давать более устойчивый из двух возможных катионов, например (75), а не (76), и (77), а не (78):
Н Н I
♦ I ♦ I
МеСН=СН—СН—СН2 -<—>• МеСН—СН=СН—СН2 J (75)
Н +
МеСН=СН—СН=СН2 (74а)
I + хн+
4 [Н Нт
I I +
МеСН— СН—СН=СН2 МеСН—СН=СН—CH2J (76)
217
H Me
I I
,CH2—C—CH=CH2 +
H Me
I I CH2—C=CH—CH2
(77)
Me
I
CH2=C— CH=CH2 (746)
I . xh+
Me H
CH2=C—CH—CH2
Me
♦ I
CH2—C=CH—
(78)
Для диенов известны также реакции каталитического гидрирования (1,2- и 1,4-), эпоксидирования (только 1,2- и более медленно, чем соответствующие простые алкены); очень редко диены подвергаются гидратации.
7.5.2. Реакция Цильса — Альдера
Эта реакция, классическим примером которой является взаимодействие бутадиена (63) и малеинового ангидрида (79)
О О
(63) (79)
включает 1,4-присоединение алкена к сопряженному диену. Реакция обычно протекает легко и быстро и приводит к образованию углерод-углеродной связи. Диен должен реагировать в цисоидной (80), а не в трансоидной (81) конформации:
(81) (80) (82)
Показано, что циклические диены, которые находятся в цисоидной конформации, например диен (82), реагируют гораздо быстрее, чем ациклические диены, в которых требуемая конформация должна быть достигнута путем вращения вокруг простой связи (трансоидная конформация обычно более устойчива). Так,
218
циклопентадиеи (82) достаточно реакционноспособен, и две молекулы такого диена могут дать трициклический димер. Эта реакция, подобно большинству реакций Дильса — Альдера, обратима.
Выяснено, что эти реакции активируются электронодоиор-ными заместителями в диене и электроноакцепторными заместителями в алкене — диенофиле. Реакции с простыми незамещенными алкенами обычно идут с трудом. Например, бутадиен (63) реагирует с этеном только под давлением при 200 °C, но даже в этих условиях выход продукта реакции составляет лишь 18 %, тогда как при реакции с малеиновым ангидридом (79) в бензоле при 15°C выход продукта составляет « 100 %. В качестве диенофилов часто используют циклогексадиен-1,4-диои (п-бен-зохинон) (83), пропеиаль (акролеин) (84), тетрациаиоэтен (85), бензин (86) (ср. разд. 6.10.3), а также некоторые замещенные алкины, например диэтилбутиидиоат-1,4 («ацетилендикарбоно-вый эфир») (87):
(83) (84) (85)
CO2Et
А С
CO2Et
(87)
Эта реакция чувствительна также к стерическим эффектам; например, из трех изомеров 1,4-дифенилбутадиена (88а) — (88в) только транс-транс-форма (88а) вступает в реакцию Дильса — Альдера:
С Ph
Ph
(88а) (886)
транс-транс транс-цис
(88в) цис-цис
Показано, что на протекание реакций Дильса — Альдера незначительно влияет добавление радикалов (ср. разд. 11.1) или изменение полярности растворителя; следовательно, мало вероятно, чтобы они протекали с участием радикалов или промежуточных соединений в виде ионных пар. Найдено, что эти реакции протекают син-стереоселективио по отношению как к диену, так и к диенофилу. По-видимому, при этом наблюдается
219
согласованный механизм, в котором образование связи и расщепление связи происходят более или менее одновременно, хотя и необязательно (до определенной степени) в одном и том же переходном состоянии. Это циклическое переходное состояние является плоским, имеет ароматический характер и стабилизируется вследствие циклического перекрывания шести р-орбита-лей диена и диенофила. Такие перициклические реакции подробно рассмотрены в разд. 12.1.
7.6. Нуклеофильное присоединение
Как уже отмечалось, введение электроиоакцепторных групп в ароматическое ядро приводит к ингибированию электрофильного замещения (см. разд. 6.7.1). То же самое справедливо и для реакций присоединения к алкенам. Так, уже было показано, что введение электроноакцепторных групп ингибирует присоединение, инициируемое электрофилами (см. разд. 7.2), однако те же самые группы активируют присоединение, инициируемое нуклеофилами. Эффективность обоих процессов уменьшается в следующем ряду заместителей:
СНО > COR > CO2R > CN > NO2
Аналогичное влияние оказывают такие заместители, как SOR, SO2R и F. Эти заместители действуют, уменьшая плотность л-электронов на атомах углерода алкена и облегчая тем самым подход нуклеофила Y~ и, что особенно важно, способствуя делокализации отрицательного заряда на образующемся карбанионном интермедиате, например (89) и (90). Эта делокализация, как правило, еще более эффективна, когда она включает мезомерную делокализацию, как в интермедиате (89), а не только индуктивный электроноакцепторный эффект, как в интермедиате (90):
Ориентация присоединения несимметричного реагента HY или XY к несимметричному замещенному алкену должна определяться образованием более стабилизированного карбаниона
220
(ср. предпочтительное образование более стабилизированного карбокатиона при электрофильном присоединении; разд. 7.3). Стереоселективность таких реакций нуклеофильного присоединения к ациклическим алкенам изучена мало. Нуклеофильное присоединение происходит также и с подходящими алкинами, причем более легко, чем с соответствующими алкенами.
Ряд таких реакций нуклеофильного присоединения представляет значительный интерес для синтеза.
7.6.1. Цианэтилирование
Одной из важных реакций является реакция с участием цианозамещенного этена (акрилонитрил) (91). Атака У- или У по незамещенному атому углерода акрилонитрила с последующим отрывом протона от молекулы растворителя приводит к присоединению 2-циаиоэтильной группы к исходному нуклеофилу:
PhOH ROH
PhOCH2CH2CN -«--- ----► ROCH2CH2CN
rnh2 ~ CH2=CHCN — H2S
RNHCH2CH2CN ► HSCH2CH2CN
(91)
Этот процесс называют цианэтилированием. Его часто проводят в присутствии основания, чтобы превратить НУ в более сильный нуклеофил У-. Эта реакция позволяет ввести в молекулу три атома углерода, причем концевая циаиогруппа может быть модифицирована восстановлением, гидролизом и т. д. для дальнейшего использования в синтезе. Присоединение У-, например -NHz, ведет к образованию карбаниона YCH2CHCN, который в отсутствие доноров протона может присоединиться к следующей молекуле акрилонитрила, что может привести в результате многократного повторения к анионной полимеризации (ср. разд. 8.4.5).
7.6.2. Реакция Михаэля
Реакцию, в которой нуклеофил, атакующий замещеииый ал-кеи, является карбанионом (ср. разд. 10.5), называют реакцией Михаэля. Она представляет собой общий метод образования углерод-углеродной связи, например:
EtO" EtOH
R2CH-CH0 5=* R2C-CHO^=£R2C-СНО +EtO~
*£й2 Генсы £н2сн2сы
(91)
221
Реакция ускоряется различными основаниями, используемыми только в каталитических количествах, под действием которых генерируется карбанион /92). Реакция обратима; ее скоростьлимитирующей стадией считают образование углерод-углеродной связи, т. е. реакцию .карбаниона (92) с замещенным алкеном (91). В ней может быть использовано большое число различных замещенных алкенов и карбанионов. Наиболее типичные карбанионы — это, возможно, те, что образуются из CH2(CO2Et)2 (см. ниже), МеСОСНгСОгЕ!, МССНгСОгЕЕ RCH2NO2 и т. д. Во многих реакциях Михаэля в качестве замещенного алкена используют соединения, содержащие группировку С=С—С=О.
7.6.3. Присоединение к системе связей
С=С—С=О
К числу наиболее типичных заместителей, «активирующих» алкен для нуклеофильной атаки, относится группа С=О в таких а,|3-ненасыщенных соединениях, как RCH=CHCHO, RCH= = CHCOR', RCH = CHCO2Et и т. д. Поскольку карбонильная группа в таких соединениях сама может подвергаться нуклеофильной атаке (ср. разд. 7.6.3), возникает вопрос, будет ли преобладать присоединение по связи С=С или С=О или 1,4-присоединение по системе С=С—С=О. Действительно, последний тип присоединения [см. формулу (93)] при применении, например, реактива Гриньяра обычно приводит к тому же продукту [95], который можно было получить при присоединении по связи С=С, из-за таутомерного превращения первоначально образующегося енола (94):
R2C=CH—С=О
Ph R II. н+
R2C—СН=С-О- MgBr > п 2'-*
(93)
Ph R
R2C—СН=С—ОН
Ph R
I I
R2C—СН—с=о
н
(94)
(95)
Между прочим, 1,4-электрофильное присоединение (например, НВг) по той же самой причине также дает С=С-аддукт (97) и может формально рассматриваться как катализируемое
222
кислотами присоединение нуклеофила Вг- к аддукту (96):
R R
I Н+ | Вг-
R2C=CH—С=О r2c—сн=с—он ------►
(96)
Вг R Вг R
—> R2i—СН=С—ОН R2C—СН—С=О
н
(97)
Менее сильные нуклеофилы, такие как ROH, также могут участвовать в 1,4-присоединении в условиях кислотного катализа.
Будет ли нуклеофильное присоединение протекать преимущественно как 1,4-присоединение или же по группе С=О, зависит от того, обратима ли данная реакция или нет. Если она обратима, тогда образование продукта контролируется термодинамически (равновесно; ср. разд. 2.2.3) и преобладающим будет 1,4-присоединение. Это связано с тем, что С=С-аддукт (98), получаемый в ходе 1,4-присоединения, термодинамически более устойчив, чем С=О-аддукт (99), поскольку первый содержит остаточную л-связь группы С=О, а она сильнее остаточной л-связн группы С=С:
(98)
Y
R2C=CH—С—ОН
R (99)
Стернческое затруднение иа одном участке молекулы может сильно активировать присоединение на другом ее участке. Например, было показано, что PhCH=CHCHO почти на 100 % присоединяет PhMgBr к группе С=О, тогда как в случае соединения PhCH=CHCOCMe3 тот же реагент почти на 100 % присоединяется к С=С-связи. Это также отражает снижение «карбонильной» активности С=О-группы в ряду альдегид > кетон > эфир (ср. разд. 8.1), приводящее к возрастанию доли С=С-присоединеиия.
Амины, тиолы, -ОН (см. разд. 8.4.5) и т. д. также присоединяются к p-углеродному атому а,Р-ненасыщенных соединений и сложных эфиров, ио наиболее важные реакции соединений, со-держащих группировку С=С—С=О, — это реакции Михаэля с карбанионами; в этих реакциях образуются углерод-углеродные
223
связи. Хорошим примером является синтез 1,1-диметилцикло-гександиона-3,5 (димедон) (104) из 2-метилпентен-2-она-4 (окись мезитила) (100) и карбаниона _СН (CO2Et)2:
О О
Ме^Лнз £21 Мез^'^Нз 2°S.
"CHCO2Et 4CHCO2Et
CO2Et CO2Et
О // H2C - с Ме2с( (^СН2 НС - С - OEt I ею2с о
(100) (101)
0 /9
H2/C-<f _____ Нгр-С
Ме2Сч СН ► Ме2С £н2
Н2С —С н2с—с
1.Гидролиз
2. Декарбоксилирование
(104а) (104)
(102) 1_-ои
’ О
//
н2с-с
Ме2С СН2
НС - С
1
EtO2C и
(ЮЗ)
Собственно реакция Михаэля завершается образованием аддукта (101), ио его обработка основанием (~OEt) приводит к карбаниону (102), который в свою очередь может атаковать карбонильный атом углерода одной из CO2Et-rpynn. Поскольку “OEt — хорошая уходящая группа, то она отщепляется и в результате циклизации образуется соединение (ЮЗ), что напоминает реакцию Дикмана (см. разд. 8.4.8). Гидролиз и декарбоксилирование оставшейся СО2Е(-группы приводят затем к желаемому конечному продукту (104), который на х 100 % существует в енольной форме (104а).
Димедон является ценным реагентом для избирательной идентификации и разделения альдегидов и кетонов, так как он легко образует производные (105) с альдегидами, но ие с кетонами. Эта избирательность, несомненно, вызвана главным образом стерическими причинами,
224
Глава 8
НУКЛЕОФИЛЬНОЕ ПРИСОЕДИНЕНИЕ ПО СВЯЗИ С=О
8.1. Строение и реакционная способность............................227
8.2. Простые реакции присоединения.................................230
8.2.1. Гидратация..............................................230
8.2.2. Присоединение спиртов...................................232
8.2.3. Присоединение тиолов....................................234
8.2.4. Присоединение циановодорода.............................235
8.2.5. Присоединение нона HSO^" и других анионов...............236
8.2.6. Реакции с участием гидрид-ионов.........................237
8.2.6.1. Ионы комплексных гидридов металлов...............238
8.2.6.2. Реакция Меервейна — Поиидорфа....................239
8.2.6.3. Реакция Канниццаро...............................240
8.2.7. Реакции с участием электронов...........................241
8.3. Реакции присоединения — отщепления............................243
8.3.1. Реакции с производными аммиака .........................244
8.4. Присоединение нуклеофилов с углеродным центром................246
8.4.1. Взаимодействие с реактивами Гриньяра и другими металлор-
ганическими соединениями.................................246
8.4.2. Присоединение ацетилид-ионов.......................... 248
8.4.3. Реакции с другими анионами..............................248
8.4.4. Альдольная конденсация................................ 249
8.4.5. Присоединение ннтроалканов..............................252
8.4.6. Реакция Перкина.........................................252
8.4.7. Реакции Кнёвенагеля и Штоббе ......................... 253
8.4.8. Сложноэфириая конденсация Кляйзена.................... 254
8.4.9. Бензоиновая конденсация ............................... 257
8.4.10. Бензиловая перегруппировка.............................259
8.4.11. Реакция Виттига.......................?................260
8.5. Стереоселективиость реакций присоединения к карбонильным соединениям ............................................................261
8.6. Реакции присоединения— отщепления производных карбоновых кислот ...............................................................263
8.6.1. Присоединение реактивов Гриньяра и других металлорганиче-ских соединений ...............................................265
8.6.2. Реакции с другими нуклеофильными реагентами............266
8.6.3. Реакции, катализируемые кислотами.......................268
8.7. Присоединение по связи C = N..................................272
Карбонильные соединения обладают дипольным моментом р, так как атом кислорода группы С=О является более электроотрицательным, чем атом углерода:
О
II zc^ Me ЧМе
Н = 2,ЗД
И = 2,8Д
8 П. Сайкс
225
С -> О-Индуктивный эффект, характерный для связи, соединяющей эти два атома, сказывается и на более легко поляризуемых л-электронах, так что карбонильную группу лучше всего представить гибридной структурой (1):
+ - 6+ в- Р
R2C = О *► R2C - О т. е. R2C^O = R2C-*>0 (1а) (16) (1аб)
По аналогии со связью С=С (см. разд. 7.1), можно ожидать, что связь С=О будет участвовать в реакциях присоединения, но если полярная атака связи С=С обычно начинается только электрофилами, то атака связи С=О из-за ее полярной природы может начинаться либо электрофильной атакой Х-+ или X по атому кислорода, либо нуклеофильной атакой У- или У : по атому углерода (радикальные реакции присоединения к карбонильным соединениям очень редки). Оказалось, что первичная электрофильная атака атома кислорода имеет небольшое значение, за исключением тех реакций, в которых электрофилом является кислота (или кислота Льюиса), когда быстрое обратимое протонирование может предшествовать медленной, скоростьлимитирующей атаке нуклеофилом атома углерода, что и завершает присоединение; присоединение в этом случае катализируется кислотой.
Протонирование усиливает положительный характер карбонильного атома углерода [(2)]
н+ + +
r2C=O: R2C=OH -*-* R2C—ОН
и таким путем облегчает нуклеофильную атаку по этому атому. Подобная активация, хотя и в меньшей степени, может быть следствием образования водородных связей кислоты (3) или даже молекул гидроксилсодержащего растворителя (4) с атомом кислорода карбонильной группы:
6 + 3 +
R2C = О r2 с = о-
. ’ ч 6-/R
Н-А®“ Н-0
(3) (4)
В отсутствие такой активации слабые нуклеофилы, например Н2О :, могут реагировать только очень медленно, а сильные нуклеофилы, например _С = N, не требуют активации вообще. Присоединение могут катализировать и основания, которые превращают слабый нуклеофил НУ в более сильный нуклеофил У-(например, HCN основание->-_CN). Наконец, хотя кислоты
226
HY
^R2C -0’2 Й2С - ОН + Y~
могут активировать карбонильный атом углерода для нуклеофильной атаки, одновременно оии могут уменьшать эффективную концентрацию нуклеофила, например:
"CN4-HA —► HCN4-A"
RNH2 + НА —► RNH3 + А'
Таким образом, для многих реакций присоединения к карбонильным соединениям существуют оптимальные значения pH; это важно учитывать в препаративной практике.
8.1. Строение и реакционная способность
В обычных реакциях нуклеофильного присоединения, в которых скоростьлимитирующей стадией является атака нуклеофила У-, положительный характер карбонильного атома углерода уменьшается при переходе от исходного соединения (5) к переходному состоянию (6) :
6+ 4-Y-f «- 6-
R2C =0 R2C—'О
(5) (6)
Следовательно, можно ожидать, что скорость присоединения должна уменьшаться при введении в молекулу карбонильного соединения электронодонориых групп и увеличиваться при введении электроиоакцепториых групп. Так, показано, что скорость присоединения уменьшается в следующем ряду*:
Н\ R. R.
\з=о > \з=О > ^С=О Н/ Н/ RZ
Соединения, у которых группа С=0 сопряжена со связью С=С (в этом случае конкурирующей реакцией может быть 1,4-присоедииение; ср. разд. 7.6.1) или с бензольным кольцом, также более медленно вступают в реакции присоединения, чем их насыщенные аналоги. Это связано с нарушением стабилизации (путем делокализации) при переходе от исходных
* Поскольку на первой скоростьлимитирующей стадии карбонильные соединения подвергаются атаке со стороны нуклеофильного агента, объективная оценка и$ реакционной способности требует учета доступности (по энергии) их низших свободных молекулярных орбиталей. Например, по данным ЭТС, доступность НСМО резко снижается в ряду: Н2С = О > СНзСН = О > > (СН3)гС=О; значения их электронного сродства соответственно равны: —0,86, —1,19, —1,51 эВ. (Подробное обсуждение этого вопроса и терминах граничных орбиталей см. и кн.: Травень В. Ф. Электронная структура и свойства органических молекул. М.: Химии, 1989, Разд. 8.5.3.)—Прим. ред.
8*
227
карбоксильных соединений (7) и (8) к аддуктам (9) и (10) и предшествующим им переходным состояниям:
R + R f
R2C = СН - С = О -R2C - СН = С - 0“ J -R2C = СН - С - 0"
Y
(7)
О)
Y (8) (10)
В приведенных выше примерах стерические эффекты, как и электронные, могли бы влиять на относительные скорости реакций. Влияние электронных эффектов заместителей видно иа примере замещенных бензальдегидов (11). Для них относительные скорости реакций уменьшаются в следующем ряду заместителей X:
Н
X —С=О (11) X = NO2, Н, ОМе NO2 > Н > ОМе
Что касается стерических эффектов, то минимум энергии обеспечивается при подходе нуклеофила к атому углерода карбонильной группы сверху или снизу плоскости, в которой расположена карбонильная группа, причем этот подход, вероятно, осуществляется немного «с тыла» [см. формулу (12)] из-за кулоновского отталкивания между подходящим нуклеофилом и высокой электронной плотностью на карбонильном атоме кислорода *:
(12)
* Обсуждаемая схема взаимодействия нуклеофила с фрагментом /С=О и роль орбитального контроля в этом взаимодействии блестяще доказаны иеэмпирическими квантовохимическими расчетами модельной реакции между гидрид-ионом и формальдегидом; см., например: Минкин В. И., Симкин Б. Я., Миняев Р. М. Квантовая химия органических соединений. Механизмы реакций. М.: «Химия», 1986. Разд. 4.1. — Прим. ред.
228
При увеличении объема групп R реакция замедляется, так как зр2-гибридизованиый атом углерода в исходном соединении (угол R—С—R « 120°) превращается в зр3-гибридизоваииый атом углерода в аддукте и в предшествующем ему переходном состоянии (угол R—С—R » 109°). Группы R по мере протекания реакции сближаются, переходное состояние становится пространственно более затрудненным, уровень его энергии увеличивается, а скорость реакции уменьшается. Наблюдаемое снижение скоростей реакции в ряду Н2С=О > RHC=O > > R2C=O определяется, следовательно, как электронными, так и стерическими эффектами. По аналогичной причине увеличение размера нуклеофила при взаимодействии с данным карбонильным соединением также может привести к уменьшению скорости реакции.
Кроме реакций с сильнейшими нуклеофилами, например А1Щ (см. разд. 8.2.6.1), RMgBr (см. разд. 8.4.1), многие реакции присоединения к карбонильным соединениям являются обратимыми. Факторы, от которых зависит скорость реакции, влияют таким же образом и на константу равновесия К. Это объясняется тем, что строение переходного состояния в простых реакциях присоединения гораздо ближе к строению аддукта, чем к строению исходного карбонильного соединения. Влияние стерических и электронных факторов на константы равновесия реакций различных карбонильных соединений с HCN (см. разд. 8.2.4) видно из приведенных ниже данных:
К К
GH3cHo Очень велика СН3СОСН2СН3 38
n-O2NCeH«CHO 1420 СвН5СОСН3 0,8
CeH5CHO 210 CeHsCOCeH5 Очень мала
n-MeOCeH4CHO 32
Сильно затрудненные кетоны, например МезССОСМез, могут вообще ие реагировать с нуклеофилами, за исключением, может быть, очень небольшого числа высоко реакционноспособных нуклеофилов.
Для данного карбонильного соединения значение К зависит от размеров нуклеофила; так, в реакции присоединения очень объемистого бисульфит-аииоиа SjOl" (см. разд. 8.2.5) к (МеСН2)2С = О К = 4-10-4, тогда как в реакции присоединения HCN (см. выше) к очень похожему кетону МеСН2СОМе К = 38. Значение К зависит также от природы того атома в нуклеофиле, который атакует карбонильный атом углерода, и от связи, которая при этом образуется. Так, значения К в реакциях приведенных ниже нуклеофилов с одним и тем же карбонильным соединением уменьшаются в ряду:
"CN > RNH2 > ROH
229
Ниже подробнее рассмотрены некоторые наиболее характерные реакции присоединения. Они разделены на три основные группы: простые реакции присоединения, реакции присоединения — отщепления и реакции присоединения углеродных нуклеофилов.
8.2. Простые реакции присоединения
8.2.1. Гидратация
Многие карбонильные соединения подвергаются в водном растворе обратимой гидратации:
R2C=O -f- Н2О ч *. R2C(OH)2
Значения К при 20 °C для Н2С=О, МеНС=О и Ме2С=О равны 2-Ю3, 1,4 и 2-Ю-3 соответственно; этот ряд отражает постепенно нарастающее влияние электронодонорных групп. Легкая обратимость гидратации проявляется в том, что Н2С = О может быть выделен путем перегонки из его водного раствора. Динамическое равновесие действительно устанавливается в случае Ме2С=О, хотя концентрация образующегося гидрата очень мала (его присутствие обнаружено, однако, в замороженных смесях Ме2СО—Н2О); это показано в опытах с Н28О:
О
II
Ме2С + Н]’О
,8ОН I Ме2С—ОН
i«O
МбгС Н2О
(13)
Включение 18О в кетоны в этих условиях, т. е. при pH 7, происходит с большим трудом, но в присутствии следов кислоты или основания оно идет [через гидрат (13)] очень быстро. Тот факт, что карбонильное соединение гидратировано, не влияет иа необратимые реакции нуклеофильного присоединения. Одиако гидратация может влиять на положение равновесия в обратимых реакциях присоединения, а также на скорость таких реакций, поскольку эффективная концентрация свободного карбонильного соединения при гидратации, естественно, уменьшается.
Показано, что гидратация протекает в условиях как общего кислотного, так и общего основного катализа (см. разд. 3.3), т. е. скоростьлимитирующая стадия включает или протонирование карбонильного соединения [общий кислотный катализ; см. переходное состояние (14)], или превращение Н2О в более силь-
230
ный нуклеофил ~ОН [общий основный катализ; см. переходное состояние (15)].
Нх хн О
R2C = O
медленно
Н- А
ГН\5уН О
i
r2c
^0
Н—А6-
(14)
/0Н2 r2c хон
/ОН r2c
\)Н
В-- Нч /Н О медленно
R2C = O *
б+
В—Н\ /Н О
r2cx ^'об-
ч20
/ОН
R2C х0"
(15)
В отличие от Ме2СО, Н2СО гидратируется очень быстро при pH 7, т. е. его более положительно заряженный карбонильный атом углерода атакуется Н2О: без предварительного протонирования карбонильного атома кислорода; тем не менее при pH 4 или 11 он гидратируется гораздо быстрее.
Электронодонорные заместители ингибируют образование гидрата, тогда как электроиоакцрпторные заместители активируют его образование. Так, для гидратации альдегида (16) К = = 2,7-104, и этот альдегид (трихлорэтаналь, хлораль) действительно образует кристаллический гидрат (17), который можно выделить. Сильно электроноакцепторные атомы хлора дестабилизируют исходное карбонильное соединение, но не гидрат, образование которого, таким образом, активируется:
(115)
С1
I
CI—C —с = о ~
1 I
С1 н
С1 он
I ♦
С1-С-*С-0Н
I I
С1 н
• Н\
С! ?
С1 - С - С - н
CI о ‘•н7
(17а)
Для превращения гидрата в исходное карбонильное соединение ои должен потерять _ОН или Н2О, что затруднено из-за наличия электроиоакцепторных групп. Гидрат, образованный
231
из хлораля, также стабилизируется (как показано методом ИК-спектроскопии) путем образования водородных связей [(17а)] между его ОН-группами и электроотрицательными атомами хлора. Карбонильные группы также могут участвовать в стабилизации гидратов, возможно, вследствие образования водородных связей или вследствие оттягивания электронов, как, например, в случае дифенилпропантриона (18), который кристаллизуется из воды в виде гидрата (19):
(19)
Еще одним примером легко выделяемого гидрата является гидрат (21) циклопропанона (20):
н,о ОН
О=о—
юн
(20) (21)
В этом случае гидратации способствует снижение степени напряжения связей при переходе от карбонильного соединения (угол С—С—С равен 60°, тогда как при хр2-гибридизации этот угол равен 120°) к гидрату (угол С—С—С равен 60°, а при spa-гибридизации он равен 109°).
8.2.2. Присоединение спиртов
Реакции карбонильных соединений со спиртами приводят к образованию полуацеталей (22) и протекают по той же схеме, что и образование гидрата:
OR'
I
R2C=O+R'OH R2C—он
(22)
Этот процесс также идет в условиях общего кислотного катализа, но для системы МеСНО—EtOH К — 0,5, а для системы МеСНО—Н2О К= 1,4. Устойчивые полуацетали могут быть, однако, выделены при взаимодействии карбонильных соединений, содержащих электроноакцепторные группы, например Вг3ССНО с EtOH. Превращение полуацеталя в ацеталь (24) идет в условиях специфического кислотного катализа (см. разд. 3.3), т. е. с отщеплением воды (SnI; см. разд. 4.2) от протонированной
232
формы (23) полуацеталя, причем эта стадия является медленной, скоростьлимитирующей стадией, с последующей быстрой атакой нуклеофилом R'OH:
ОН +ОН2
| Н+ | -Н2О + R'OH
RCH—OR' 7 ~ RCH—OR' -(-----------RCH—OR' 7-----------
быстро медленно быстро
(22) (23)
HOR' _н+ OR'
RCH—OR' ---------- RCH—OR'
быстро
(24)
Кетоны обычно не реагируют с простыми спиртами, однако с 1,2-диолами, например (25), они образуют циклические ацетали (26):
О
НО—СН2 Н+ / \СН2 r2c=o+ | r2c; i + н2о
НО—СН2 \ /СН2
о
(25) (26)
Протекание данной реакции с диолами, а не со спиртами R'OH обусловлено более благоприятным значением А5° для реакции диолов, чем для спиртов, так как в последнем случае число молекул при переходе от исходных соединений к продукту реакции уменьшается. Альдегиды и кетоны, которые трудно превратить в ацетали, часто образуют их с помощью ортоэфиров, например триэтоксиметана («этилортоформиат») НС (OEt) з в присутствии NH4CI в качестве катализатора.
Образование ацеталей является обратимым (К дляМеСНО— —EtOH равна 0,0125), но на положение равновесия влияют относительные количества спирта и НгО. Получение ацеталей в препаративных количествах обычно проводят в избытке спирта в присутствии безводного кислотного катализатора. Равновесие может быть смещено вправо либо азеотропной перегонкой (чтобы удалить НгО, которая образуется в ходе реакции), либо путем использования избытка кислотного катализатора, например непрерывно пропуская газообразный НС1 (для превращения Н2О: в нуклеофильную частицу Н3О+). Ацетали легко гидролизуются разбавленными кислотами до исходных карбонильных соединений, однако они устойчивы к гидролизу основаниями, так как при этом в системе нет протона, который может быть отщеплен от атома кислорода (ср. вызванный основанием гидролиз гидратов). Устойчивость ацеталей к действию оснований позволяет использовать реакцию их образования для защиты
233
группы С=0, очень чувствительной к атаке основаниями (см. разд. 8.4.4). Подобная защита позволяет проводить катализируемое основанием отщепление НВт от ацеталя (28); образующийся при этом ненасыщенный ацеталь (29) легко гидролизуется до ненасыщенного карбонильного соединения (30). Таким образом, удается осуществить именно ту реакцию, которая не могла быть проведена непосредственно с бромальдегидом (27) из-за его полимеризации под действием основания:
EtOH
ВгСН2—СН2СНО -------► ВгСН2—CH2CH(OEt)2
н+
(27) (28)
X "ОН
4 ’
Н2О
СН2=СНСНО -<-------- CH2=CHCH(OEt)2
н+ (30) (29)
Ацетали отвечают трем основным требованиям, предъявляемым к эффективным защитным группам: легко присоединяются, остаются прочно связанными до тех пор, пока они необходимы, н, наконец, легко отщепляются.
8.2.3. Присоединение тиолов
Карбонильные соединения реагируют с тиолами RSH с образованием полутиоацеталей и тиоацеталей гораздо легче, чем со спиртами. Это объясняется большей нуклеофильностью серы по сравнению с кислородом. Тиоацетали относительно устойчивы в разбавленных кислотах, однако они легко разлагаются смесью Н2О—HgCl2—CdCO3. Используя образования тиоацеталя, можно изменить на противоположную полярность карбонильного атома углерода в альдегиде; электрофильный центр карбонильного соединения превращается в нуклеофильный центр в анионе (31):
R\e+ в-
;с=о н/
CH2SH £h2sh
основание ---------->
D2O
тиоацеталь (31) анион
тиоацеталь
HgCl2. CdCOs
Н2О
234
Такое обращение полярности атома (umpolung) не может быть осуществлено непосредственно на самом RCHO. Из аниона (31) при обработке D2O и последующем гидролизе образуется дейтерированный аналог RCDO исходного альдегида RCHO, причем с хорошим выходом и селективно. Анион (31) может быть алкилирован (например, R'l); в этом случае исходный альдегид превращается в кетон RR'CO.
При применении никеля Ренея в качестве катализатора может быть проведена десульфуризация тиоацеталей; в результате происходит превращение группы С=О в группу СН2:
r'sh h2/ni
r2c=o -----► r2c(sr')2-----► r2ch2
Такое превращение обычно трудно непосредственно осуществить в препаративных целях.
8.2.4. Присоединение циановодорода
Хотя присоединение HCN следовало бы рассматривать как реакцию с участием карбаниона, обычно считают, что в этой реакции участвует простой анион. Эта реакция необыкновенно интересна в том отношении, что она, почти несомненно, является первой органической реакцией, для которой был установлен механизм (Лэпворт, 1903 г.). HCN — недостаточно сильный нуклеофил, чтобы атаковать С=О. Для превращения HCN в более сильный нуклеофил ~CN реакцию проводят в условиях основного катализа. В этом случае она подчиняется Кинетическому уравнению:
Скорость = й[р2С=О] [ “CN]
Присоединение _CN обратимо и в отсутствие донора протонов имеет тенденцию смещаться в сторону исходных веществ. В присутствии донора протонов равновесие смещается вправо, так как равновесие, включающее циангидрин, более благоприятно, чем равновесие, включающее промежуточный анион (32):
О' ОН
“CN | HY |
R2C=O " R2C—CN * R2C—CN + Y”
медленно быстро
(32) (33)
Атака под действием “CN является медленной, скоростьлимитирующей, тогда как перенос протона от HCN или от протонного растворителя, например Н2О, происходит быстро. Влияние строения карбонильного соединения на положение равновесия при образовании циангидрина уже было рассмотрено (см. разд. 8.1). С препаративными выходами эта реакция идет с альдегидами, алифатическими и циклическими кетонами; в случае
235
жирноароматических кетонов ArCOR выходы очень малы, а с ароматическими кетонами АгСОАг реакция не идет совсем. В случае АгСНО с образованием циангидрина может конкурировать бензоиновая конденсация, а при наличии системы связей С=С—С=О конкурирующей реакцией будет 1,4-присоединение (см. разд. 7.6.2).
Из карбонильных соединений, для которых равновесие с HCN не смещено в сторону образования циангидрина, производные циангидрина могут быть получены с удовлетворительными выходами реакцией cxM.e3SiCN:
OSiMe3 ОН
Me3SiCN | LIAIH4 |
R2C=O -------► R2C—CN --------► r2c—ch2nh2
Это возможно в результате выделения большого количества энергии при образовании очень прочной связи Si—О. Для препаративных целей получение Ме35ьпроизводных циангидринов обычно используют для осуществления дальнейших превращений группы CN (например, путем восстановления, гидролиза и т. п.). Такие превращения следует проводить в условиях, которые исключают возможность протекания обратной реакции, приводящей к образованию исходного карбонильного соединения. Дополнительным преимуществом Me3SiCN, по сравнению с самой HCN, является то, что взаимодействие его с системой С=С—С=О идет как реакция 1,2-присоединения, а при взаимодействии с АгСНО бензоиновая конденсация не идет (см. разд. 8.4.9).
8.2.5. Присоединение иона HSO$ и других анионов
Другой классической анионной реакцией является присоединение гидросульфит-аниона к карбонильным соединениям с образованием кристаллических аддуктов. Строение этих аддуктов долгое время было предметом споров. Установлено, что они являются солями сульфоновых кислот (34); это объясняется тем, что в атакующем анионе атом серы обладает большей нуклеофильностью, чем атом кислорода. Эффективным нуклеофилом является ион SO3”, а не ион HSO3, хотя концентрация иона HSO3 выше:
НО" + HSO3 Н20 + sof“
н2о
r2c = О R2C - О" R2C - ОН
♦> I 1
"0-§ = 0 SOf SOJ
236
Атакующий анион уже присутствует в растворе, так что реакция в целом не требует основного катализа. Анион SO3' является достаточно сильным нуклеофилом и не требует активации (протонирования) карбонильной группы, а также присутствия кислотного катализатора. Однако этот нуклеофил является весьма громоздким, так что значения К для этой реакции обычно значительно меньше, чем значения К для реакции образования циангидрина с тем же карбонильным соединением (см. разд. 8.1). С препаративными выходами гидросульфитные соединения образуются в случае альдегидов, метилкетонов и некоторых циклических кетонов. Такие карбонильные соединения могут быть выделены из смесей и (или) очищены путем получения, очистки и последующего разложения их гидросульфитных производных.
Галогенид-ионы также могут действовать как нуклеофилы по отношению к альдегидам в условиях кислотного катализа, но образующееся при этом, например, 1-гидрокси-1-хлорпроиз-водное (35) очень неустойчиво; равновесие этой реакции сильно смещено в сторону исходных веществ. При применении раствора НС1 в спирте ROH могут быть получены 1-алкокси-1-хлорсоеди-нения; например, из СН2О и МеОН получен 1-метокси-1-хлор-метан (а-хлордиметиловый эфир) (36) (ср. образование ацеталя; разд. 8.2.1); необходимым условием является нейтрализация реакционной смеси до выделения продукта реакции:
СН2=О
н+ +
* СН2—ОН
С1
I н+
сн2—он =<=
• •
(35)
Cl 1- МеОН С1
| + -Н2О + 2- -н+ |
+=> СН2—ОН2 -fe СН2—С1 \ СН2—ОМе
(36)
8.2.6. Реакции с участием гидрид-ионов
Карбонильные группы могут быть каталитически гидрированы, как и углерод-углеродные ненасыщенные связи (см. разд. 7.4.5). Однако обычно каталитическое гидрирование связи С = О труднее осуществить, чем гидрирование связей С — С, CsC, C=N или C = N, так что селективное восстановление группы С=О в присутствии любой из названных связей не может быть проведено каталитически. Тем не менее это возможно при использовании различных, обычно комплексных, гидридов металлов.
237
8.2.6.1. Ионы комплексных гидридов металлов
Наиболее эффективным восстановителем является литийалю-минийгидрид Li+ А1Щ, который может восстанавливать группу С=О в альдегидах, кетонах, кислотах, сложных эфирах и амидах до группы СН2, не затрагивая связи С=С или С=С, также присутствующие в данном соединении (иногда затрагиваются, однако, связи С=С, сопряженные с С=О). Эффективным восстановителем является ион А1Н4, который действует как сильный донор гидрид-ионов Н~. Восстановление в этом случае не может проводиться в протонных растворителях, например Н2О и ROH, так как они активно отщепляли бы протон. В качестве растворителей обычно используют простые эфиры; в некоторых из них LiAlH4 заметно растворим.
Нуклеофильная частица А1Щ необратимо отдает Н~ атому углерода карбонильной группы, а А1Н3 затем присоединяется к атому кислорода с образованием комплексного иона (37), который переносит затем гидрид-ион к трем другим молекулам R2C=O с образованием комплекса (38). При последующей обработке комплекса (38) протонным растворителем (R'OH) образуется спирт (39). Таким образом, источником одного атома водорода в этом спирте является ион А1Н4, а второго — R'OH:
А1Н> ЗНэС — О г й'пм
R2C = O—♦R2C-OAIH3——«► R2C-0 АГ —♦ R2C - ОН I I I
H-AIH3 н н J4 н
(37) (38) (39)
При восстановлении кислот возможно образование литиевой соли RCO2 Li+ и ее выделение из эфирного раствора, что приводит к прекращению восстановления. Этого можно избежать, превращая сначала кислоту в эфир, например в метиловый или этиловый. При восстановлении такого эфира первоначальная нуклеофильная атака частицей А1Н4 приводит к реакции присоединения— отщепления [OR' является хорошей уходящей группой в соединении (40)] с последующей обычной атакой, как показано выше, полученного карбонильного соединения (41) с образованием первичного спирта (42):
OR' fOR' _ Н
I /-* Ч кл 1. aih4 I
RC = О ------ RC - О- RC = О-—-2- R<? ~ ОН
| -OR | 2. R OH I
H-AIH3 н н н
(40) (41) (42)
Менее эффективным комплексным гидридом является Na+
ВН4, который может восстанавливать только альдегиды и ке-
238
трны и не восстанавливает производные карбоновых кислот. Он не атакует также, в отличие от Li+ А1Щ, группы NO2 или CsN, присутствующие в одном и том же соединении. Большое преимущество NaBH< состоит в том, что его можно применять в гидроксилсодержащих растворителях. Применяют также большое число других реагентов, таких как МЩ, MH3OR~, MH2(OR)2; их относительная эффективность зависит как от нуклеофильности, так и от размера иона.
8.2.6.2. Реакция Меервейна—Понндорфа
В данной реакции происходит обратимый гидридный перенос от атома углерода (см. схему) к атому углерода карбонильной группы. Классическим примером является восстановление кетонов, например (44), под действием А1(ОСНМе2)3 (43) в пропаноле-2:
СМе2
? н (2-PrO)2AI q>CR2 (43) (44)
СМе2 0^' "-н
(2-PrO)2Al >CR2
(45)
(46)
* СМе2
Н
^(2-PrO)2AI + cr2 х0
(47)
Поскольку пропанон (46) является самым низкокипящим компонентом данной системы, равновесие можно сместить практически полностью вправо, непрерывно отгоняя его из смеси. При использовании избытка пропанола-2 этот спирт взаимодействует со смешанным алкоксидом алюминия (47) с выделением продукта восстановления R2CHOH. В этом случае также один атом водорода поставляется за счет гидридного переноса, а другой — гидроксилсодержащим растворителем. Вследствие специфической природы этого равновесия и пути, по которому оно устанавливается, никакие другие группы, присутствующие в исходном кетоне, не восстанавливаются.
Наличие специфического гидридного переноса от углерода к углероду подтверждается образованием R2CDOH при использовании (Me2CDO)3Al. Возможно, реакция протекает через циклическое переходное состояние типа (45), хотя в некоторых случаях обнаружено, что в реакции участвуют две молекулы алкоксида: одна — в переносе гидрид-иона, другая присоединяется к карбонильному атому кислорода. Вместо этой реакции часто используют восстановление действием МЩ. Однако в ряде случаев эту реакцию применяют для осуществления обратного процесса — окисления; в качестве катализатора при этом используют А1(ОСМе3)3, а для смещения равновесия реакции влево
239
образующийся пропанон должен быть в большом избытке. Этсуг обратный процесс (окисление) обычно называют реакцией Qn-пенауэра.
8.2.6.3. Реакция Канниццаро
В этой реакции происходит перенос гидрид-иона от молекулы альдегида, не содержащей а-Н-атома, например НСНО, R3CCHO, АгСНО, ко второй молекуле того же альдегида или, иногда, к молекуле другого альдегида («перекрестная» реакция Канниццаро). Реакция идет в присутствии сильных оснований; в случае, например PhCHO, скорость ее подчиняется уравнению:
Скорость = £[ PhCHO)2 [ 'ОН].
Полагают, что реакция протекает по схеме:
ГОН _ ОН >0 ОН О 0“ НО
*• ОН | П PhCHO 1 II
PhC —Н PhC т Н. CPh -^-PhC + Н - CPh^PhC + Н - CPh
CU быст- | медлен- || | I I
Ч0 ро (_0 Н ОН ОН
(48) (49) (50) (51)
Быстрое обратимое присоединение ~ОН к PhCHO приводит к образованию потенциального донора гидрид-иона (48), после чего следует медленная, скоростьлимитирующая стадия переноса гидрид-иона к атому углерода карбонильной группы второй молекулы PhCHO и образование соединения (49). Реакция завершается быстрым обменом протона с образованием более устойчивой пары (50) и (51). Взаимное окисление — восстановление двух молекул альдегида происходит, следовательно, так, что образуются карбоксилат-ион (50) и первичный спирт (51).
При проведении реакции PhCHO в D2O атом D не включается в СН2-группу соединения (51). Это указывает на то, что перенос Н (D) должен происходить (согласно приведенному выше механизму) непосредственно от одной молекулы альдегида к другой, а не каким-то косвенным путем с участием растворителя.
В очень концентрированных основаниях уравнение скорости, например в случае НСНО, имеет вид:
Скорость = £[НСНО]2 [’ОН]2.
В этом случае отщепление протона из аниона (53), аналогичного иону (48), приводит к дианиону (54), который является
240
гораздо более
(48):
сильным гидридным
донором, чем ион (53) или
"ОН ОН
> "он | нс-н^=* нс-н
0-"он I НС-Н
(52)
О" (53)
О’
(54)
Некоторые диальдегиды также могут претерпевать внутримолекулярный гидридный перенос. Так, в реакции Канниццаро этандиаль-1,2 (глиоксаль) (55) превращается в гидроксиэтано-ат-анион (гликолят) (56):
Н
0=С—С=О
I Н
"ОН
Н
I 0=С—С—ОН
I I
о н
(55) (56)
Для этой реакции, как и ожидалось, найдено следующее кинетическое уравнение:
Скорость = £[ОНССНО] [ "ОН].
Альдегиды, которые содержат атомы водорода у атома углерода, соседнего с СНО-группой (а-углеродиый атом), не вступают в реакцию Канниццаро с основаниями, но гораздо быстрее вступают в реакцию альдольной конденсации (см. разд. 8.4.4).
8.2.7. Реакции с участием электронов
Атомы ряда сильно электроположительных металлов, например Na, Кит. д., могут в подходящих условиях в растворе давать сольватированные электроны:
жидк.
NH3
Na. =?==* Na+ + e-(NH3)„
Такие электроны могут действовать как нуклеофилы и присоединяться к атому углерода группы С=О с образованием ра-дикал-аниона (57) часто в виде ионной пары с катионом металла М+:
R2C=O + М+ + е R2C—О" М+
(57)
241
Так, при растворении натрия в отсутствие кислорода возу духа в эфирном растворе ароматических кетонов появляется голубая окраска в результате образования диарилкетила натрия (58) (в этом анион-радикале в делокализации принимает участие как ароматическая система, так и группа С=О):
[Аг2С—О 1
| 2Na+
Аг2С—OJ
(58)
(59)
Частица (58) находится в равновесии со своим димером (59)—динатриевой солью 1,2-диола (пинакона). В соответствующих условиях при добавлении донора протонов, например ROH, можно получить пинакон (в препаративных количествах). Лучшие результаты дает использование ароматических, а не алифатических кетонов, однако пропанон (46) при взаимодействии с магнием (и последующей обработке) легко превращается в 2,3-диметилбутандиол-2,3 (61) называемый пинаконом :
Ме2С=О Mg
: Mg =f=
Ме2С=О
(46)
О
Ме2С/ \
1 >g
Ме2С\ / О f
н+. н2о ,Ме2С— ОН
Ме2С—ОН
(60) (61)
Препаративное превращение кетонов (особенно ароматических} в пинаконы может проводиться также фотохимически под действием УФ-облучения в присутствии донора водорода, например Ме2СНОН.
Аналогичное нуклеофильное присоединение электронов может также идти к карбонильному атому углерода диэфиров, таких как (62), например, из растворов натрия в растворителях типа ксилола. Образующийся дианион (63), однако, отличается от аниона (59) тем, что содержит прекрасную уходящую группу (например, _OEt), так что общим результатом оказывается аци-
242
\доиновая конденсация:
OEt OEt
I I
•С = О Na- - O~ -~OEt /0 = 0 Na zC - 0"
C = o"* ^C-O"”* '0 = 0 " 'C-0-
I I
OEt OEt
(62) (63) (64)
zC-0" R'oH z-C - OH ________ <9 = 0
”— Q-0- ~ ^C-OH <CH-OH
(65) (66) (67)
Конечным продуктом этой реакции является 2-гидроксикетон (ацилоин) (67). Реакция протекает, вероятно, как показано выше, через 1,2-дикетон (64), который сам способен затем принимать электроны от натрия. При проведении реакции в ксилоле конечным продуктом является днанион (65) (в виде Na-соли), однако при последующем добавлении спирта R'OH он протони-руется и образуется ендиол-1,2 (66), более устойчивой таутомерной формой которого является ацилоин (67).
Эту реакцию используют для препаративного получения макроциклических гидроксикетонов путем циклизации длинноцепочечных диэфиров EtO2C(CH2)nCO2Et; при л = 8— 18 выходы соответствующих кетонов (с 10—20-членными кольцами) составляют 60—95 %.
8.3. Реакции присоединения — отщепления
Известен ряд реакций нуклеофильного присоединения к группе С=О, в которых присоединяющийся нуклеофил содержит кислый протон, поэтому становится возможным последующее отщепление Н2О от соединения (68), приводящее к продукту (69), т. е. происходит замещение атома кислорода:
ОН
NUH2 I —Н2О
R2C=O R2C—NuH < > R2C=Nu
(68) (69)
К наиболее известным реакциям такого типа относятся реакции с производными аммиака (HONH2, NH2CONHNH2, PhNHNH2 и др.). Эти реакции использовали для превращения жидких карбонильных соединений в твердые производные с целью их идентификации. Особенно полезным в этом отношении является 2,4-динитрофенилгидразин (О2М)2С6НзМНМН2.
243
8.3.1. Реакции с производными аммиака
Если, например, за ходом реакции между пируват-анионоуй (70) и гидроксиламином NH2OH при pH 7 следить, регистрируя ИК-спектр реакционной смеси, то характеристическое поглощение группы С=О (vMaKc 1710 см-1) в исходном веществе (70) исчезает полностью до того, как появится характеристическое поглощение группы C=N (vMaKc 1400 см-'1) образующегося оксима (72). Это свидетельствует об образовании промежуточного соединения, которым, по-видимому, является производное а-гидро-ксиа’мииоспирта (71). Такая частица действительно была обнаружена методом ЯМР-спектроскопии в реакции МеСНО с NH2OH:
Me. nh2oh Меч ОН V=O *==*
"O2CZ ~O2Cz 'NHOH
(70) <71)
^макс!7Ю CM 1
—H2O
Me.
XC=NOH "о2с/
(72) ^макс!400 CM 1
При увеличении кислотности реакционной смеси уменьшается скорость исчезиовеиия поглощения группы С=О; NH2OH при этом постепенно превращается в +NH3OH, который не является нуклеофилом, и заметно увеличивается скорость появления поглощения группы C=N, образующейся в результате дегидратации соединения (71) под действием кислоты. Реакция протекает по схеме:
_ „ .. Н+' f+ н2о
R2C = Os* R2C - О- si R2C - OH si R2C - OH2 -^*R2C = NY Н2ЙУ h2ny hny H-^Y
В случае сильных нуклеофилов, например NH2OH (Y=OH), ие требуется присутствие кислоты для их первоначального присоединения к С=О, но для более слабых нуклеофилов, таких как PhNHNH2 (Y — PhNH) и H2NCONHNH2 (Y = NHCONH2), присутствие, кислоты необходимо для активирования группы С=О (ср. разд. 8.1; фактически в этом случае имеет место общий кислотный катализ). В зависимости от pH раствора ско-ростьлимитирующей стадией может стать либо первоначальная стадия присоединения, либо стадия дегидратации. В нейтральной и щелочной средах такой стадией обычно является дегидратация, например (71) -* (72), т. е. этот переход является медленным и лимитирующим скорость реакции (см. выше). Однако в кислой среде обычно первоначальная стадия присоединения нуклеофила, например (70) -> (71), становится медленной и ско-ростьлимитирующей. Это имеет важное значение для препаративного получения этих соединений. В каждом случае реакцию
244
следует проводить при оптимальном значении pH, которое зависит от природы карбонильного соединения и используемого производного аммиака; например, для образования оксима из пропанона Ме2СО оптимальное значение pH равно « 4,5.
В случае альдегидов (и несимметричных кетонов RCOR') возможно, конечно, образование альтернативных син- и анти-геометрических изомеров:
ZC=N
XC—N н/ "
/Y
анти
Установлено, что син-изомер обычно преобладает; в случае RCOR' это изомер, в котором Y находится ближе всего к меньшей из групп, R или R'.
При реакции самого аммиака с карбонильными соединениями образуются имины R2C=NH, но они неустойчивы и могут полимеризоваться. Классические «альдегидоаммиаки», как показано, являются гидратированными циклическими тримерами. Однако из альдегидов, содержащих сильные электроноакцепторные заместители, можно получить простые аддукты (73) [ср. (71), гидраты (см. разд. 8.2.1) и полуацетали (см. разд. 8.2.2)].
ОН
С13С—СН—NH2 (73)
Продуктами взаимодействия аминов RNH2 с карбонильными соединениями также являются имины, они обычно также неустойчивы, за исключением соединений, у которых хотя бы один из заместителей у карбонильного атома углерода является ароматическим. Например, имины ArCH=NR являются устойчивыми продуктами; они известны как основания Шиффа. При взаимодействии с вторичными аминами R2NH сначала образуется аддукт (74), дегидратация которого обычным путем невозможна. Некоторые из таких частиц были выделены, но они оказались не особенно устойчивыми. Однако, если такой аддукт имеет «-Н-атомы, то дегидратация возможна; в этом случае образуется енамин (75):
Енамины представляют определенный интерес как промежуточные соединения для ряда синтезов.
245
8.4. Присоединение нуклеофилов с углеродным центром
При обсуждении этой группы реакций мы не будем проводить формального различия между реакциями, представляющими собой простое присоединение, и реакциями, протекающими как присоединение — отщепление. Они рассматриваются вместе, как отдельная группа, потому что приводят к образованию углерод-углеродных связей. Многие из них очень полезны и важны в синтетической органической химии. Рассмотрим сначала две специфические реакции нуклеофильного присоединения.
8.4.1. Взаимодействие с реактивами Гриньяра и другими металларганическими соединениями
Состав и строение реактивов Гриньяра RMgX до сих пор окончательно не установлены. Как известно, они зависят от природы R, а также от растворителя, в котором реактив находится или растворяется. Так, по данным спектра ЯМР, MeMgBr в Et2O представляет собой в основном MgMe2 + MgBr2. В то же время, по данным рентгеноструктурного анализа кристаллического PhMgBr, выделенного из эфирного раствора, это соединение имеет состав PhMgBr -2Et2O с четырьмя лигандами, тетраэдрически расположенными вокруг атома Mg. Независимо от строения реактивы Гриньяра можно рассматривать как источники отрицательно поляризованного углерода, т. е. как e~RMgXe+. Доказано, что атом магния реактива Гриньяра связан с атомом кислорода карбонильной группы [см. формулу (76)]. Показано, что в реакции присоединения участвуют две молекулы RMgX, причем, по крайней мере в некоторых случаях, реакция, возможно, идет через циклическое переходное состояние, например (77):
/Л- Г V в
6++ |б- — f R
Я'2СЧо,МдХ Я^М9Х R^C^gX л L J V
(76)
(77)
Вторую молекулу RMgX можно рассматривать как катализатор типа кислоты Льюиса, увеличивающий положительную поляризацию атома углерода карбонильной группы путем присоединения к атому кислорода. Действительно, показано, что добавление кислот Льюиса, например MgBr2, ускоряет присо-
246
единение реактивов Гриньяра. Достоверных сведений о механизме присоединения реактивов Гриньяра к группе С=О удивительно мало для такой хорошо известной реакции. Механизм, очень сходный с приведенными выше [т. е. через (77а) и (776)], можно использовать для объяснения двух важных обстоятельств: а) реактивы Гриньяра, имеющие атомы водорода у р-углерод-ных атомов, например RCH2CH2MgX (78), восстанавливают группу С=О до СНОН, превращаясь при этом в алкены (80) (происходит перенос атома водорода, а не группы RCH2CH2); б) стерически затрудненные кетоны, имеющие атомы водорода у а-углеродных атомов, например (81), превращаются в енолы (82). При этом реактив Гриньяра превращается в углеводород RH.
С HR
Н ^СН2
(78)
.CHR
Н" '^СН2
CHR <8°) Н ^СН2
R^o^MsX
(77а) (79)
4
„ /Н Г
RjC R R2C R
I I —* !
R’C^.MgX R’c^Mgx
(81) (776)
R2C % II
R'C MgX 0 (82)
Реактивы Гриньяра ведут себя как сильные нуклеофилы, и реакция их присоединения практически необратима. Конечными продуктами присоединения, получаемыми после гидролиза водой (например, после гидролиза R3COMgX), являются спирты (R3COH). Однако важно подчеркнуть, что реакции с реактивами Гриньяра, в частности реакции присоединения к группе С=О, представляют собой общий метод связывания различных групп с атомом углерода, поскольку спирты, первоначально получаемые как продукты реакции, далее могут быть модифицированы с помощью самых разнообразных реакций. В прошлом аналогично использовали цинкорганические соединения, которые затем все более вытеснялись реактивами Гриньяра; реактивы Гриньяра в свою очередь сейчас все больше вытесняются литий-алкилами RLi и литийарилами ArLi. Эти реактивы дают больше продуктов присоединения нормального строения при взаимодействии со стерически затрудненными кетонами, а также больше продуктов 1,2- и меньше продуктов 1,4-присоединения при взаимодействии с системой С=С—С=О, чем реактивы Гриньяра (ср. разд. 7.6.3).
247
8.4.2. Присоединение ацетилид-ионов
Ацетилен HCssCH и его гомологи RCssCH обладают заметными кислотными свойствами и могут быть превращены с помощью сильных оснований, например ~NH2 в жидком аммиаке, в соответствующие анионы (ср. разд. 10.2), которые несколько более нуклеофильны, чем -CN. Хотя такие частицы, например RC =С_, безусловно являются анионами, их рассматривают отдельно, так как для их стабилизации, в отличие от других анионов, не требуется наличия электроноакцепторных групп, таких как С=О, CsN, NO2 и т. д. Таким образом, к связи С=О может быть присоединена цепочка атомов углерода. Подобная реакция имеет большое значение, потому что связь С^С далее может быть модифицирована различными путями, например восстановлена до алкена (83) водородом на катализаторе Линдлара (ср. разд. 7.4.5):
R2C = О ”C = CR'
R2C - О" C = CR'
жидк. NH3
и
R2C - он -----r2c - он
I катали
C = CR' затор си — cud'
Линд- СН — CHR
8.4.3. Реакции с другими анионами
Эти реакции обычно катализируются основаниями, так как для получения карбаниона -CXYZ, являющегося эффективным нуклеофилом, необходимо удалить протон от HCXYZ. Одна из групп X, Y или Z является, как правило, электроноакцепторной, чтобы стабилизировать карбанион. Первоначальный аддукт (84) отщепляет протон от растворителя (часто от воды или спирта) с образованием простого продукта присоединения (85). Последующая дегидратация с образованием соединения (86) зависит от наличия атома водорода у а-углеродного атома, если X, Y или Z — Н; возможность дегидратации зависит и от того, будет ли вводимая С=С-связь сопряжена с другой С=С- или С=О-связью в продукте реакции:
R2C = О
(*
"CXYZ
R ОН
R2C ~ О" 5=±
I CXYZ
(84)
х = н
R2C-OH R2C = CYZ I CXYZ
(85) (86)
Первоначальное образование углерод-углеродной связи [->-(84)] часто обратимо, и последующая стадия, например дегидратация, может быть необходима, чтобы сместить равновесие. Известно большое число реакций карбанионов (в том числе
248
именных реакций), отличающихся друг от друга только природой карбонильного соединения (альдегид, кетон, сложный эфир и т. д.) и типом применяемого карбаниона.
8.4,4- Альдольная конденсация
В этих реакциях карбанион (88), полученный при действии основания (как правило, _ОН), на а-Н-атом одной молекулы карбонильного соединения (87), присоединяется к карбонильному углероду другой молекулы (87) с образованием р-гидро-ксикарбонильного соединения. Например, в случае этаналя СН3СНО продуктом реакции является 3-гидроксибутаналь (89) — альдоль:
Н 40 I “ОН \П / СНгСНОз^-МеС ♦ ’ I (87J Н (87) 0 0“ II 2 1 "СН2 - С - Н ^=*МеСН - СН2СНО 1 1|Нг° 0“ он 1 1 сн2 = с-н МеСН-СН2СНО (88) (89)
Показано,’ что в случае СН3СНО равновесие сдвинуто вправо, в сторону образования альдоля. Прямая реакция (2) и обратная реакция (/) практически конкурируют друг с другом. Однако при проведении реакции в D2O не удалось обнаружить включения дейтерия в СН3-группу еще не вступившего в реакцию этаналя, и, следовательно, скорость реакции (2) настолько выше скорости обратной реакции (/), что последняя становится фактически необратимой.
Найдено, что даже для простых кетонов, например пропанона (90), равновесие сильно смещено влево [выход соединения (92) равен «2 %], т. е. в случае кетонов атака карбонильного атома углерода идет с большим трудом, чем для альдегидов-
Н <0 О"
I “ОН 'II ГЛ 2 1
СН2СОМе^^Ме2С+ “CH2COMes=±Me2C - СН2С0Ме
н2о
(90) (90) (91)
ОН I Ме2ССН2СОМе
(92)
249
Было найдено, что в случае пропанона (90) проведение реакции в D2O приводит к включению дейтерия в СНз-группу еще не вступившего в реакцию пропанона, т. е. реакция (2) не идет быстрее, чем реакция (/).
Соединение (92) можно получить в препаративных количествах. С этой целью реакцию проводят путем повторных операций отгонки и сифонирования в аппарате Сокслета над твердым Ва(ОН)2 (в качестве основного катализатора). Равновесную смесь, содержащую ~2% гидроксикетона (92), отделяют си-фонированием. Затем эту смесь снова перегоняют над Ва(ОН)2; при этом отгоняется только пропанон (т. кип. 56°C), а 4-ги-дрокси-4-метилпентанон-2 («диацетоновый спирт»; т. кип. 164 °C) (92) остается («2%). Повторение указанных операций даст еще ~ 2 % соединения (92). Таким образом можно осуществить более или менее полное превращение (90)-*- (92).
Однако такие малоэффективные альдольные реакции с большей легкостью идут в условиях кислотного катализа. Кислота активирует образование достаточного количества енольной формы (94), например, пропанона (90), которая подвергается атаке протонированной формой второй молекулы карбонильного соединения — карбокатионом (93):
О н+ ОН___________ОН ОН OjH
Ме2С «± Ме2С+ *СН2 = СМе - Ме2С - СН2 - СМе^~(92) (90) (93) (94) +
В кислой среде третичный спирт (92) почти всегда претерпевает катализируемую кислотой, дегидратацию (ср. разд. 9.1) с образованием а,|3-ненасыщенного кетона — 2-метилпентен-З-она-2 (окись мезитила) (95):
НО Н г +н+
| ) 2. -н2о
Ме2С—СНСОМе Ме2С=СНСОМе
(92) (95)
Дегидратация альдолей может происходить также под действием оснований; так, альдоль (89) превращается в бутен-2-аль (кротоновый альдегид) (97):
ОН Н _ - н
МеСН - СНСНО ^=*МеСН - СНСНО МеСН = СНСНО
(89) (96) (97)
Катализируемые основаниями реакции дегидратации относительно необычны, и их протекание в этих условиях объясняет-
250
ся тем, что альдоль (89) содержит а-Н-атомы, которые могут отщепляться основанием с образованием карбаниона (96), имеющего хорошую уходящую группу ~ОН у соседнего (£) углеродного атома. Возможность такого отщепления может сместить равновесие вправо в ряде простых реакций альдольного присоединения, в которых иначе равновесие могло бы быть сдвинуто влево. Однако важно помнить, что суммарный процесс альдольного присоединения и дегидратации является обратимым, т. е. (89) (97), и что «.^-ненасыщенные карбонильные
соединения расщепляются основаниями при подходящих условиях. Следует отметить, что соединение (97) является альдегидом и может дальше присоединять карбанион с последующей дегидратацией и т. д. Именно так получают низкомолекулярные полимеры при нагревании простых алифатических альдегидов с водным раствором NaOH; чтобы остановить реакцию на стадии получения альдоля в качестве катализатора лучше использовать основную ионообменную смолу.
Перекрестные альдольные конденсации, которые протекают, когда оба альдегида (или другие подходящие карбонильные соединения) имеют а-Н-атомы, обычно не могут быть использованы в препаративных целях, так как при этом может образоваться смесь четырех различных продуктов. Однако такие реакции можно использовать в том случае, когда один альдегид не имеет а-Н-атома и может поэтому действовать только как акцептор карбаниона. Примером является конденсация ароматических альдегидов (98) с простыми алифатическими альдегидами или кетонами (обычно с метилкетонами) в присутствии 10 %-го водного раствора КОН (реакция Кляйзена — Шмидта) (дегидратация в этих условиях всегда происходит после присоединения карбаниона):
“СН2СНО “СН2СОМе
АгСН=СНСНО -<----АгСНО ► АгСН=СНСОМе
'ОН “ОН
(98)
Как и следовало ожидать, электронодонорные группы в арильном остатке замедляют реакцию; например, п-МеОСбН4СНО реагирует в 7 раз медленнее, чем C6HsCHO. В этих условиях важной конкурирующей реакцией является са-моконденсация алифатического альдегида, однако реакция Канниццаро идет настолько медленно, что ею можно пренебречь. Такая конденсация может происходить также в условиях кислотного катализа (см. выше).
Наконец, альдольная конденсация при наличии подходящих карбонильных соединений, например (99), может протекать
251
внутримолекулярно, т. е. происходит циклизация:
Ме—С=О
Н2с/ /СН2СОМе
Н2С—СН2
"ОН
Me—С—ОН
“ОН
С—Ме
Н2С^ ^ССОМе
Н2С—СН2
(99)
8.4.5. Присоединение нитроалканов
Другой широко используемой реакцией является присоединение к альдегидам и кетонам карбанионов (101), полученных из алифатических нитросоединений, например нитрометана (100):
ОН fO О
II I “ОН '‘II II
R2C+CH2NO2^=*R2C т "CH2-N-O“
+
о-I
R2c - CH2NO2
(100)
J J|h2o
0" OH
I I
CH2= N - 0“ R2C - CH2NO2
(101) (102)
Для получения карбанионов используют основания, такие как _ОН и ~OEt; возможность последующей дигидратации образовавшегося Р-гидроксинитросоедииения до R2C=CHNO2 зависит от условий реакции. Если карбонильное соединение является альдегидом, возможна его альдольная самоконденсация, однако делокализованный карбанион (101) обычно образуется значительно легче, чем карбанион -CHRCHO, и поэтому такая опасность относительно невелика Полученные нитросоединения могут быть восстановлены до аминов, а также модифицированы другими способами.
8.4.6. Реакция Перкина
В этой реакции карбанион (104) образуется путем отщепления а-Н-атома от молекулы ангидрида кислоты (103) под действием аниона соответствующей кислоты как основания. В качестве карбонильного акцептора могут выступать ароматические альдегиды. Продуктами реакции являются а,р-ненасы-щенные кислоты, например 3-фенилпропеновая (коричная) кислота (107), получаемая при взаимодействии PhCHO с избытком
252
(МеСО^О — МеСОг при 140 °C:
(106) (1056) (107а)
(Ю7)
Карбанион (104) атакует карбонильный углерод альдегида обычным путем, образуя алкоксид-ион (105а). Полагают, что происходит внутримолекулярный перенос ацетильной группы в этом анионе: от карбоксильного атома кислорода [в (105а)] к алкоксидному атому кислорода [в (1056)] через циклический интермедиат (106); при этом образуются более устойчивые частицы. Отщепление а-Н от аниона (1056) под действием МеСО-> приводит к отщеплению хорошей уходящей группы МеСО2 от соседнего p-положения с образованием аниона (107а) а,р-нена-сыщеииой кислоты. Обработка реакционной смеси разбавленной кислотой дает продукт (107).
Протекание реакции по такой схеме подтверждается тем, что в реакции с ангидридами типа (R2CHCO)2O, в случае которых в интермедиате, соответствующем (1056), отсутствует а-Н-атом, может быть выделен аналог соединения (1056) как фактический конечный продукт реакции.
8.4.7. Реакции Кнёвенагеля и Штоббе
В этих реакциях присоединения участвуют различные карбанионы, образующиеся из соединений типа CH2XY, но особенно из тех, у которых X и (или) Y являются группами CO2R,
253
например CH2(CO2Et)2. В качестве катализаторов в этих реакциях используют органические основания. В большинстве случаев промежуточный альдоль дегидратируется до а,р-ненасы-щенного продукта (сложного эфира). Интересным примером является реакция карбанионов (108), полученных из эфиров 1,4-бутандиовой (янтарной) кислоты, например (СНгСОгЕ^г, с альдегидами или кетонами с применением алкокси-ионов в качестве основных катализаторов — конденсация Штоббе. Эти эфиры реагируют намного легче, чем можно было бы ожидать, причем в ходе реакции одна из групп CO2R превращается в COg. Продуктом реакции является а,р-ненасыщенное производное (110); альдоль не образуется. Механизм, с помощью которого можно объяснить все эти акты, включает образование циклического интермедиата (109):
CO2Et R2c^CHCH2co2Et (108)
CHCO2Et
R2C СНо
OEt
СНСО2Е1 r2cx чсн2 6—c-?ro’-
CoEt
CO2Et
R2C = CCH2CO2
/"CCO2Et 2C XCH2 O*-C^
^0
~OEt
CHCO2Et r2cx 'ch2
O-Q ^0
R
(110) (109)
В некоторых случаях такие циклические интермедиаты удалось выделить.
8.4.8. Сложноэфирная конденсация Кляйзена
Это еще одна реакция, в которой участвуют карбанионы, полученные из сложных эфиров, например карбанион (111), но в этом случае карбанион присоединяется к карбонильному атому углерода другой молекулы сложного эфира. Основанием для рассмотрения этой реакции именно здесь, а не в ряду реакций производных карбоновых кислот (см. разд. 8.6) является то, что производных карбоновых кислот (см. разд. 8.6) является то, что дольной конденсации альдегидов (см. разд. 8.4.4). Например, для этилэтаноата (ацетат) (112) соответствующие превращения
254
могут быть представлены схемой:
СН2 = С - О"
OEt (111)
Н I СН2- CO2Et
гС?
szr MeC- CH2 CO2Et^f
-CoEt
(112) (112) (113)
он о о-
II I “OEt г II - I
S=t МеС - CHCO2Et - CHCO2Et *-* МеС = CHCO2Et I
(114) (115)
Однако в этом случае, в отличие от простой альдольной реакции, образующийся аддукт (ИЗ) содержит хорошую уходящую группу (OEt); поэтому вместо присоединения протона, как в собственно альдольной реакции (см. разд. 8.4.4), отщепляется ~OEt и образуется 0-оксоэфир — этил-3-оксобутаноат (этилацетоацетат) (114). Этот эфир на заключительной стадии превращается под действием основания (“OEt) в стабилизованный (делокализованный) карбанион (115).
В классическом варианте для получения небольших количеств этоксида натрия Na+ “OEt (основный катализатор) используют немного более 1 моль Na (в виде проволоки или в тонко-измельченном виде) и небольшое количество EtOH. Дополнительные количества EtOH образуются на стадии 1 и образуют с натрием Na+ “OEt; таким образом, концентрация иона “OEt поддерживается постоянной. Целый моль этого иона необходим для достаточно полного превращения Р-оксоэфира (114) в его анион (115) [кислотность эфира (114) выше кислотности EtOH] при условии, что вся последовательность равновесий будет смещена вправо.
Такое условие является необходимым, поскольку равновесие стадии (/) (образование карбаниона) смещено влево, причем в большей степени, чем, например, при реакции МеСНО, так как карбанион (111) менее стабилизирован посредством делокализации заряда, чем карбанион (116), образующийся из альдегида:
~СН2—С=О ч—► "СН2—С—О'
I I
: OEt OEt
(Ш)
~сн2—с=о I н
СН2=С—О' I н
(116)
255
Смещением равновесия (/) влево объясняется и тот факт, что реакция R2CHCO2Et в присутствии -OEt не идет, хотя в этом случае мог бы образоваться обычный р-оксоэфир R2CHCOCR2CO2Et. Однако в этом оксоэфире отсутствуют атомы водорода в a-положении, и, следовательно, ои ие может быть превращен в карбанион, соответствующий карбаниону (115), т. е. стадия (<3) не может иметь места! В присутствии сильного основания, например Ph3C~ Na+, стадия (/) практически полностью смещается вправо:
R2CHCO2Et + Ph3C' RCCO2Et + Ph3CH
В этом случае эфир R2CHCO2Et вступает в обычную реакцию Кляйзена, хотя протекание стадии (5) оказывается невозможным.
Важно подчеркнуть, что в подходящих условиях обычная реакция Кляйзена полностью обратима, например, при так называемом кислотном разложении [оба продукта — (118) и (119) — являются производными кислот] р-оксоэфира (117):
, я -
RC - CHR'CO2Et «=* RC - CHR CO2Et RC + R'CHCO2Et ^OEt OEt OEt
(117) (118) (119)
1,3-Дикетоны, например (120), также расщепляются в этих условиях с образованием производного кислоты (121) аниона кетона (122):
RC - CH2COR ("6Et
RC - CH2COR OEt
0 II _ i=iRC + CH2COR
OEt
(120) (121) (122)
«Перекрестные» реакции Кляйзена с двумя различными сложными эфирами, каждый из которых имеет а-Н-атомы, используют редко, поскольку при этом возможно образование четырех продуктов. Однако реакции такого типа находят применение в том случае, когда один из эфиров не имеет а-Н-атомов, например HCO2Et, ArCO2Et, (CO2Et)2 и др., и служит лишь акцептором карбаниона. Такие соединения являются действительно хорошими акцепторами, а побочная реакция самоконденса-
256
ции второго эфира, например RCH2CO2Et, не столь существенна. Если обе группы CO2Et входят в состав одной и той же молекулы [например, (123)], происходит внутримолекулярная конденсация, называемая реакцией Дикмана. Она протекает легче всего при обычных условиях, если возможно образование анио,-нов 5-, 6- или 7-членных циклических p-оксоэфиров [например, (124)], в частности с EtO2C (СН2) nCO2Et при п = 4—6:
Et0\ Ef<\ ею. zo;
с с
Н2С CH2CO2Et^* Н2С CHCO2Et **Н2С \нС02Е1
Н2С—(СН2)Х Н2С- (СН2)Х Н2С- (СН2)Х
(123) х=1-3
0 “OEt /с\~° ZC<k °
^H2q CHCO2Et =S=^H2C 9CO2Et*-*H2C XCCO2Et H2C— (CH2)x H2C—(CH2)x H2C—(CH2)x
(124)
Макроциклические кетоны (ср. ацилоиновую конденсацию; разд. 8.2.7) могут быть получены также при использовании очень разбавленных растворов; в этом случае углеродный атом карбаниона сложного эфира имеет больший шанс для взаимодействия с карбонильным атомом углерода на другом конце его собственной молекулы, чем с аналогичным атомом другой молекулы (межмолекулярная реакция).
8.4.9. Бензоиновая конденсация
Эта реакция ароматических альдегидов АгСНО с ионом ~CN сходна с реакцией Канниццаро в том, что начальная атака [быстрая и обратимая; стадия (/)] осуществляется по карбонильному атому углерода молекулы альдегида («донор») (125). Вместо гидридного переноса (ср. реакцию Канниццаро; разд. 8.2.6.3) карбанион (127) присоединяется к карбонильному атому углерода второй молекулы АгСНО («акцептор») (128). Эта реакция наряду с реакцией образования циангидрина (ем. разд. 8.2.4) была одной из первых реакций, для которых был установлен механизм (причем верно!) в 1903 г. Скорость реакции, как и можно было предполагать, выражается уравнением:
Скорость = £[АгСНО]2[ *CN].
9 П. Сайкс
257
Считают, что реакция протекает по схеме:
t°
Ч S' 1
ArC f “CN^
I Н
(125)
ArC-ОН
II
C = N“
(1276)
I 0“ он он о*
I I АгСНО Г гл ||7 ,
ArC -CN АгС" ----------» АгС"*' САг
I I 2 I I
Н C=N CN Н
(126) (127а) (127а) (128)
НО 0“
Arip- САг CN Н (129)
СГ)ОН АгС-САг ^CN Н
(130)
О ОН II I АгС-САг I Н
Если эту реакцию проводят в МеОН, то ни стадия (2) [образование карбаниона (127)], ни стадия (<?) [присоединение этого карбаниона к карбонильному атому углерода акцепторной молекулы (128)] не являются полностью скоростьлимитирующими. За этими стадиями следуют быстрый перенос протона (129) -> —>(130) и, наконец, быстрое отщепление ~CN — хорошей уходящей группы — с образованием 2-гидроксикетона (131) (реакция, обратная образованию циангидрина; см. разд. 8.2.4). Если Ar — Ph, образующийся продукт называется бензоином, а реакция полностью обратима.
Долгое время считали, что эту реакцию катализирует только ион ~CN. Полагали, что такая способность обусловлена тем, что эта частица способна быть нуклеофилом, способна быть уходящей группой и, что особенно важно, способна благодаря своим электроноакцепторным свойствам увеличивать кислотность связи С—Н в ионе (126) и стабилизировать карбанион [(127а)-<->( 1276)], что приводит к отщеплению протона.
Недавно было найдено, что илиды (132), образующиеся в небольших количествах при pH 7 в растворах солей М-алкил-тиазолия (не содержащих заместителя в положении 2), являются прекрасными катализаторами бензоиновой конденсации. На первый взгляд, илиды — частицы, имеющие заряды противоположного знака на соседних атомах, — сильно отличаются от ~CN, однако они, как и ожидалось, обладают перечисленными
258
выше свойствами группы -CN.
н
R-N^)s
>=< рк=7
Y"
(132)
8.4.10. Бензиловая перегруппировка
При окислении бензоина PhCH(OH)COPh (см. выше) образуется бензил PhCOCOPh (133), который, как и не способные к енолизации 1,2-дикетоны, претерпевает катализируемую основанием перегруппировку с образованием аниона а-гидр-оксикислоты—бензилат-аниона Ph2C(OH)CO2 (135). Эта перегруппировка, почти наверное, является первой изученной молекулярной перегруппировкой. Ее кинетическое уравнение имеет вид:
Скорость == /s[PhCOCOPh] [ "ОН].
Считают, что реакция идет по следующей схеме:
Г” рл „
о = с - С = 0^0 - с - с = о —*
-к I II
Н0"; Ph НО Ph
Ph Ph
। - I
0 = C - C - 0-^0 = C - c - OH II II
но Ph "0 Ph
(133) (134)
(135)
Медленной, скоростьлимитирующей стадией, почти несомненно, является миграция фенила в первоначальном “ОН-аддукте (134). Эта реакция в ряду 1,2-дикетонов аналогична внутримолекулярной реакции Канниццаро с 1,2-диальдегидом — глиоксалем ОНССНО (см. разд. 8.2.6.3). Однако в случае глиоксаля со своей электронной парой мигрирует атом водорода (т. е. гид-рид-ион) к соседней группе С=О, тогда как в случае бензила со своей электронной парой мигрирует фенильная группа (в виде карбаниона). По этой причйне эту реакцию рассматривают как присоединение (внутримолекулярное) карбаниона к группе С=О.
2-Оксоальдегид PhCOCHO также реагирует с ~ОН, превращаясь в PhCH(OH)CO2, Однако один и тот же продукт может образоваться при миграции .как атома водорода, так и фенильной группы. Эксперименты с PhCOCHO, меченной D или НС, показывают, что на самом деле мигрирует только водород. В случае кетонов межмолекулярные реакции Канниццаро, по-видймому, не происходят; эти реакции должны были бы
9*
259
включать миграцию R с электронной парой от одной молекулы к другой:
2r2co rco; + R3C0H
8.4.11. Реакция Виттига
Эта реакция находит широкое применение в синтезе алкенов. Она заключается в присоединении фосфониевого илида (139), называемого фосфораном, к карбонильной группе альдегида или кетона. Илид представляет собой карбанион, имеющий соседний гетероатом. Такие частицы образуются в ходе реакции алкил-галогенидов RR'CHX (137) с триалкил- или триарилфосфином (136), очень часто с Ph3P, с последующим отщеплением протона от фосфониевой соли (138) действием очень сильного основания, например PhLi:
X
+ PhLi 4- -
Ph3P + RR'CHX —> Ph3P—CHRR' --------► Ph3P—CRR'
(136) (137) (138) j
Ph3P=CRRz
(139)
Полагают, что присоединение реагента Виттига (139) к группе С=О протекает по следующей схеме:
(140)^
R'aC = О 1 R'aC-o-. 2 R2C-0
, * I I ~
RRC-PPh3 RRC-PPh3 RR'C-PPh3
Ra"C о и II RR С + PPh3
(139) (141) (142)
Однако не во всех случаях реакция протекает именно таким образом: стадия (/) может быть, а может и не быть равновесной, могут различаться также и скоростьлимитирующие стадии. В некоторых случаях удалось обнаружить (при —80 °C) окса-фосфетановый интермедиат (141), который при повышении температуры (до 0°С) разрушается до продуктов реакции. Движущей силой, лежащей в основе этой довольно необычной реакции, является выделение большого количества энергии при образовании очень прочной связи Р=О (535 кДж/моль). Если исходная фосфониевая соль (138) хиральна (у атома фосфора), например RR'R"P+CH2R, конфигурация при атоме фосфора, как показано, сохраняется в продукте реакции — фосфиноксиде RR'R"P=O.
Поскольку в данной реакции могут участвовать различные галогениды и различные карбонильные соединения, она служит
260
универсальным методом синтеза замещенных алкенов. Наличие связей С=С или С = С, даже если они сопряжены с группой С = О не мешает ходу реакции. Группа COzR, хотя и реагирует с илидом (139), но гораздо медленнее, чем с группой С=О, и, таким образом, также не мешает реакции. Эта реакция особенно полезна, например, для получения соединений с экзоцикли-ческой метиленовой группой [(143)] или р,у-ненасыщенных кислот (144), которые невозможно получить другими методами:
(ИЗ)
СН2
R2C=O + Ph3P—СНСН2СО2 —>
R2C=CHCH2CQ; + PhjP=O
(144)
При получении кислоты (144) другими методами происходит изомеризация в термодинамически более устойчивую, сопряженную (а,р-ненасыщенную) кислоту. Реакция Виттига была использована также для внутримолекулярного синтеза циклических алкенов, содержащих от 5 до 16 атомов углерода.
8.5. Стереоселективность реакций присоединения к карбонильным соединениям
При нуклеофильном присоединении HY по связи С=О независимо от того, протекает ли оно цис- или транс-стереоселектив-но, образуются одни и те же продукты [ (148)->( 149) или (150)], в отличие от электрофильного присоединения HY по связи С = С [(145)-*(146) или (147)]:
(147)
(150)
HY транс
(149)
(149) = (150)
В аддукте (151), образующемся при присоединении HY к RR/C=O, имеется хиральный центр. Однако в результате этой реакции образуется (±)-форма — рацемат, поскольку
261
первоначальная нуклеофильная атака сверху (а) или снизу (б) плоского карбонильного соединения (148) статистически равновероятна:
(±)
(1516)
Однако если R или R' хирален н, особенно, если за хиральность ответственен а-углеродный атом, то в этом случае две стороны карбонильного соединения больше не эквивалентны, и присоединение сверху и снизу теперь не будет статистически равновероятным. Если реакция обратима, то, вероятно, что термодинамически более устойчивый продукт из двух возможных будет находиться в большем количестве в реакционной смеси (термодинамический, или равновесный, контроль; см. разд. 2.2.3). Для практически необратимых реакций, например для реакций с RMgX, ЫА1Н4 и т. п., будет преобладать тот продукт, который образуется быстрее (кинетический контроль). Какой продукт окажется преобладающим, можно предсказать, пользуясь правилом Крама: кетон будет реагировать в такой конформации, в которой атом кислорода группы С=О занимает анти-положение по отношению к самому большому из трех заместителей, находящихся у а-углеродного атома [см. формулу (152)]. Преобладающая нуклеофильная атака (например, со стороны R'MgBr) будет проходить с наименее затрудненной стороны карбонильного атома углерода, т. е. со стороны (а). Это лучше всего видно при использовании проекционных формул Ньюмена (ср. разд. 1.3.1):
менее предпочтительный продукт
262
Другими словами, преобладающая реакция будет проходить через менее затрудненное (с более низкой энергией) переходное состояние. Таким образом, можно ожидать, что отношение выходов (х/у) соединений (153) и (154) будет тем выше, чем больше разница в размерах заместителей М и S и чем больше размер остатка R' в R'MgBr. Показано, что при взаимодействии MeMgl с PhCHMeCHO (152; L = Ph, М = Me, S = Н, R = Н) х : у = 2 : 1, тогда как при замене Me-группы более объемистой Et-группой х : у = 2,5 : 1. Аналогично, при использовании вместо MeMgl гораздо большего по объему PhMgBr х:у = 4:1.
Правило Крама было изучено в основном на реакциях присоединения реактивов Гриньяра и некоторых реакций присоединения гидридов к группе С=О. В общем, это правило позволяет достаточно точно предсказать, какой продукт будет преобладающим, но при этом наблюдается и ряд исключений. Это неудивительно, так как согласно этому правилу контроль за образованием продукта зависит только от стерических взаимодействий, тогда как важную роль играют также комплексообразование (между группами в субстрате, например образование водородных связей, или между субстратом и атакующим нуклеофилом, например RMgX и карбонильным атомом кислорода) и диполь-дипольное взаимодействие. В качестве примера последнего эффекта можно отметить, что а-хлоральдегиды и а-хлоркетоны реагируют (вследствие электростатического отталкивания) в конформации, например (155), в которой атом хлора и карбонильный атом кислорода находятся в анти-положении друг к другу, независимо от размера (по отношению к атому хлора) других групп, примыкающих к а-углеродному атому. Любое из указанных взаимодействий может преобладать над чисто сте-рическими причинами при определении геометрии преобладающего переходного состояния.
О
8.6. Реакции присоединения — отщепления производных карбоновых кислот
Реакции этого типа протекают по следующей общей схеме:
(О (“О О
о \!l v Y" -X- П
R - С - X s=r R - С - X <—» R - С + X "
Y Y Y
(156) (157)
263
Они представляют собой нуклеофильное присоединение — отщепление, протекающее через образование так называемого тетраэдрического интермедиата (157) и приводящее к образовав нию продукта замещения. Производные карбоновых кислот (156), в отличие от простых карбонильных соединений (альдегидов и кетонов), имеют у карбонильного атома углерода группу X, являющуюся хорошей потенциальной уходящей группой (в виде X-), тогда как в простых карбонильных соединениях потенциальная уходящая группа (R- или Н~) является очень плохой уходящей группой. Относительная реакционная способность производных карбоновых кислот (156) по отношению к данному нуклеофилу Y~ (например, -ОН) зависит от относительной электронодонорной или -акцепторной способности группы X по отношению к карбонильному атому углерода и от относительной способности этой группы выступать'в качестве уходящей группы. Относительная реакционная способность производных карбоновых кислот обычно уменьшается в ряду:
О О О О О
« II II’ 1 I
RC—Cl > RC—OCOR > RC—OR' > RC—NH2 > RC—NR2
Так, хлорангидриды и ангидриды легко реагируют с ROH и NH3 с образованием соответственно сложных эфиров и амидов, а сложные эфиры реагируют с NH3 или аминами с образованием амидов. Обратные реакции — образование сложных эфиров из амидов — хотя и возможны, но обычно их довольно трудно осуществить. Относительная реакционная способность может также зависеть как от электронного, так и, что более существенно, от стернческого влияния группы R. Несколько необычной уходящей группой является группа _СХ3 (например, -С13); в результате этой реакции образуется галоформ (158) (ср. разд. 10.5.6):
лО /"О О О
41 "он Ч .11 _ v (I
R—С—СХ3 ч—± R—С—СХ3—>-R—С+ 6X34=* R— С + НСХ3
z* I I _1
О'" ОН ОН ОН О
(158)
Кинетическое уравнение, которому подчиняются эти реакции, как правило, имеет обычный вид:
Скорость = fe[RCOX] [Y’].
Естественно, возникает вопрос: могут ли такие реакции протекать как прямое, одностадийное (ср. SN2) замещение у карбонильного атома углерода? Обычно невозможно выделить тетраэдрические интермедиаты, такие как (157). Однако известно об-
264
разованне подобного интермедиата в том случае, когда R содержит сильно электроноакцепторные атомы или группы (ср. CI3CCHO; разд. 8.2.1); например, интермедиат (160) образуется в результате присоединения -OEt к CF3CO2Et (159) (растворитель — днбутнловый эфир):
Г°
41 ~ОЕ\
CF3 —С—OEt .>
<JOEt (159)
О I CF3—С—OEt
OEt
(160)
Этот аддукт (выход «.100 %) может быть выделен и охарактеризован; более слабые нуклеофилы Н2О нлн EtOH к нему не присоединяются. При переходе от исходного производного карбоновой кислоты (156) к тетраэдрическому интермедиату (157) карбонильный атом углерода изменяет свою гибридизацию от sp2 до sp3, а поскольку переходное состояние, образующееся на скоростьлимитирующей стадии суммарной реакции, сходно с ннтермеднатом (157), можно считать, что этн реакции чувствительны к стернческим влияниям, как это н было обнаружено (см. ниже).
Ранее уже было рассмотрено присоединение карбанионов к RCO2Et (сложноэфирная конденсация Кляйзена; см. разд. 8.4.8) и восстановление карбонильных соединений под действием L1AIH4 (см. разд. 8.2.6). Другие реакции нуклеофильного присоединения рассмотрены ниже.
8.6.1. Присоединение реактивов Гриньяра и других металлорганических соединений
Атака сложных эфиров (161) реактивами Гриньяра протекает по общему механизму, приведенному выше, причем первоначальным продуктом присоединения — отщепления (~OR как уходящая группа) является кетон (162):
<5 13 ‘М9Х
R - С ’R" - MgX— R - с - R" -R'O *OR'
О 0~ +МдХ
о R R"MgX D । „
R - С ----2*- R - С - R
I I
R" R"
(161)
Карбоинльиый атом углерода в этом кетоне, одна ко, более реакционноспособен по отношению к нуклеофилу, чем карбонильный атом углерода исходного эфира (161), вследствие электронодоиорного мезойериого эффекта атома кислорода
265
в сложном эфире:
R—C-fQR" 6+-+<->'
(161)
(162)
Таким образом, образующийся кетон (162) с успехом конкурирует с еще йе прореагировавшим эфиром (161) в реакции с реактивом Гриньяра R"MgX, так что фактическим продуктом реакции является соль третичного спирта (163) (две алкильные группы в этой соли получены от реактива Гриньяра).
Ацилгалогениды, например RCOC1, образуют точно такие же продукты с реактивами Гриньяра; однако при использовании кадмийорганического соединения CdR" реакция может быть остановлена на стадии образования кетона. Реакция останавливается на стадии образования кетона и в случае сложных эфиров, если вместо R"MgX используют R"Li и ведут реакцию при более высоких температурах.
8.6.2. Реакции с другими нуклеофильными реагентами
Объектом большого числа исследований явилась реакция гидролиза сложных эфиров, например (164), действием водного раствора щелочи. Выяснено, что эта реакция подчиняется уравнению второго порядка; в экспериментах со сложным эфиром (164), меченным 18О, показано, что обычно разрывается связь ацилкислород (ср. разд. 2.3.3); метка 18О обнаружена только в EtOH. Это подтверждает, что реакция идет через тетраэдрический интермедиат (165):
4 О О“Ч О О
VII Х-ч II ,в_ И
R<pr “OH^ffcRC - ОН—*-RC - ОН + 18OEt ~3feRC - 0‘+ H18OEt
18OEt 18C?Et
(164) (165) (166) (167)
Скоростьлимитирующей стадией, почти несомненно, является атака исходного эфира (164) ионом -ОН. Это подтверждается параметрами активации для индуцируемого основанием гидролиза MeCOjEt: АД* = 112 кДж/моль, AS* = 109 Дж/(моль • К). Относительно большое отрицательное значение AS* указывает на уменьшение поступательной составляющей энтропии (см. разд. 2.1) для системы частиц (МеСОгЕ! + ~0Н), реагирующих (в ассоциативном процессе) с образованием соединения (165) на скоростьлимитирующей стадии реакции. В целом реакция
266
практически необратима, поскольку -OEt обычно отщепляет протон от кислоты (166), а не атакует ее карбонильный атом углерода, тогда как карбоксилат-анион (167) не чувствителен к атаке EtOH или ЕЮ-. Этот механизм обычно обозначают как Вас* (катализируемое основанием расщепление связи ацил — кислород, бимолекулярное). Если нуклеофилом является -OR, а не ~ОН, то протекает переэтерификация и образуется равновесная смесь двух эфиров— (164) и (168). Положение равновесия зависит от относительных концентраций и нуклеофильных способностей -OEt и -OR.
RC* ~0R^=- RC - OR
OEt CoEt
0
RC - OR + “OEt
(164) <168)
Атака группой ~OH амидов RCONH2 протекает аналогично реакции со сложными эфирами (см. выше), однако уходящей группой является в этом случае -NH2, а не -OEt. Она отрывает протон от соединения (166), образуя более устойчивую пару — карбоксилат-ион (167)4-NH3. Удаление аммиака из горячего щелочного раствора сдвигает равновесие этой реакции вправо. Взаимодействие сложных эфиров, например (164), с аминами приводит к амидам (169); эта реакция идет по той же общей схеме, что и в приведенных выше примерах (было показано, что RNH-, сопряженное основание RNH2, не участвует в нуклеофильной атаке сложного эфира):
у О 0“ _ 0“
I + RNHj | вн
RC* :NH2R'=^ RC - NH2R RC - NHR
OEt OEt OEt
(164) (170)
0
RC - NHR'+ EtOH+ B‘
(169)
По-видимому, медленной, скоростьлимитирующей стадией является отщепление уходящей группы от аниона (179); для этого обычна необходима содействие донора протона ВН, например Н2О.
Ацилхлориды RCOC1 легко атакуются более слабыми нуклеофилами, например Н2О и ROH. При этом1, однако, возникает вопрос; будет ли эта реакция (при наличии лучшей потенциальной
267
уходящей группы С1_) протекать в одну стадию, по механизму типа Sn2 (см. разд. 4.1) с участием переходного состояния, в котором атака Y- н отщепление СН по существу синхронны, нлн же по механизму типа SnI (см. разд. 4.1), при котором медленной стадией является диссоциация RCOC1->RCO+ С1~, за которой следует быстрая атака Y~ ацнл-катнона RCO+? В действительности большинство реакций ацилхлоридов, вероятно, протекает по уже знакомому механизму с образованием «тетраэдрического интермедиата».
Ангидриды кислот (RCO2)O также часто реагируют с более слабыми нуклеофилами, хотя н медленнее, чем хлорангндрнды, причем реакция обычно не протекает ин по механизму SnI, нн по механизму Sn2. Ангидриды по существу занимают промежуточное положение по реакционной способности между хлоран-гндрндамн н сложными эфирами, что соответствует ряду активности уходящих групп:
СГ > RCOj > RO’
8.6.3. Реакции, катализируемые кислотами
Атаку карбонильного атома углерода кислот RCO2H (171) нуклеофилами Y~ трудно осуществить, так как прн этом происходит отщепление протона, а образующийся RCO2 нуклеофилами не атакуется. Более слабые нуклеофилы YH, например ROH, хотя н не отщепляют протон, но их реакции с относительно мало реакционноспособным карбонильным атомом углерода в RCO2H протекают медленно. Однако реакционную способность карбонильной группы можно увеличить путем ее протоннрова-ння, например проводя реакцию этернфнкацнн [ (171)-> (176) ] в условиях кислотного катализа:
О ОН НО
II н+ I EtOH I медленно
RC-OH5=t RC - ОН RC+-ОН - —
+ <Т.
HOEt
Н?
RC-OHs=±=
I
HOEt +
(171) (172) (173)
н?->.
OEt
НО+ II
RC I OEt
О II
RC I OEt
(174) (175) (176)
Данные ЯМР-спектров подтверждают преимущественное протонированне карбонильного атома кислорода кислоты (172)
268
в прямой реакции (этерификация) и такого же атома кислорода в молекуле эфира (175) в обратной реакции (гидролиз). В присутствии кислотного катализатора облегчается отщепление уходящей группы, т. е. легче отщепить Н2О от иона (174) при этерификации или EtOH от иона (173) при гидролизе, чем, например, ЕЮ- от иона (165) (см. выше). Образование тетраэдрического интермедиата (процесс образования связи) иа скоростьлимитирующей стадии [(175)-«—»-(174) при гидролизе] подтверждается параметрами активации, полученными для катализируемого кислотой гидролиза эфира уксусной кислоты: ДЯ+=75 кДж/моль, AS+=—105 Дж/(моль-К) (ср. разд. 8.6.2). Равновесие обычно смещается в желаемо!м направлении применением избытка ROH (или Н2О при гидролизе). Этот механизм обычно обозначают как Адс2 (катализируемое кислотой расщепление связи ацил — кислород, бимолекулярное). Реакция R'OH с RCO2R" в этих условиях приводит к переэтерификации, причем положение равновесия определяется относительными количествами R'OH и R"OH. Ангидриды кислот и амиды подвергаются катализируемому кислотами гидролизу точно так же, как и сложные эфиры.
В случае сложных эфиров RCO2R', у которых алкильная группа R' может образовать относительно устойчивый карбокатион, например (178) из (177), как было показано в опытах с 18О, происходит расщепление связи алкил — кислород:
18ОСМе3 18О-СМе3 180
I Н+ медленно II
RC = О ^=RC-OH < — RC-OH + +CMe3
(177) + (178)
нго
180 180.
II -Н+ II +
RC - ОН + НО - СМе3.-=^ RC - ОН + Н2О - СМе3
(180) (179)
Были найдены следующие параметры активации для катализируемого кислотой гидролиза МеСО2СМе3 ДД+= 112 кДж/моль, AS+ = -]-55 Дж/(моль-К). В этом случае положительное значение ДД+ (указывающее на увеличение поступательной составляющей энтропии при образовании переходного состояния на лимитирующей стадии) дает возможность предположить, что эта стадия является процессом диссоциации; в качестве примера можно привести рассмотренный выше механизм реакции
269
расщепления протонированного сложного эфира на две отдельные частицы — карбоновую кислоту и карбокатион (178). Этот механизм обычно обозначают как AalI (катализируемое кислотой расщепление связи алкил — кислород, одномолекулярное). Аналогичный механизм реализуется также с эфирами типа RCO2CHPh2 и т. п. При попытках осуществить реакцию переэтерификации между эфиром (177) и R'OH в качестве продукта получили не ожидаемый сложный эфир RCO2R', а смесь кислоты RCO2H с простым эфиром R'OCMe3. Последний образуется при атаке R'OH карбокатионного интермедиата (178) [ср. приведенное выше превращение (178) -* (179) под действием Н2О]. Если алкильная группа R в сложном эфире RCO2R' имеет достаточно большие размеры, например R3C, то бимолекулярный гидролиз через тетраэдрический интермедиат ингибируется (из-за пространственных затруднений в переходном состоянии). Известны, хотя и наблюдаются относительно редко, реакции, протекающие по механизму ААс1 (катализируемое кислотой расщепление связи ацил — кислород, одномолекулярное); этот механизм реализуется только в сильноионизирующих растворителях:
Q0 О
П н+ II П -R'oh II,
R3CC - OR' R3CC - OR' -s==: НзСС+ + HOR |-| медленно
(181) (182) (183)
н2о
О + О
II -Н || +
R3CC - ОН + R'0№=s R3CC - 0Н2 + HOR
(185) (184)
Точно такие же соображения можно высказать и относительно этерификации пространственно-затрудненных кислот (185) (обратная реакция). Следует отметить, что при этом для образования ацилкарбокатионного интермедиата (183) необходимо протонирование атома кислорода гидроксильной группы (менее благоприятное; ср. разд. 8.6.3). Кроме кислот, содержащих группы R3C, хорошо известным примером пространственно-затрудненных кислот является 2,4,6-триметилбензойная кислота (186), которая не образует ^ложные эфиры в обычных условиях кислотного катализа, а ее эфиры (188) не гидролизуются. Растворение кислоты или её сложного эфира в концентрированной H2SO< и выливание этого раствора в холодный спирт или воду приводит соответственно к практически количественной этерификации или гидролизу; реакция протекает через
270
Образование йцил-катиона (187):
О=С—ОН О=С+ О=С—ОМе
Me Me Ме
(186) (187) (188)
Образование катиона (187) подтверждается тем, что растворение стерически незатрудненной бензойной кислоты (189) в концентрированной H2SO4 приводит к ожидаемой двукратной депрессии температуры замерзания, тогда как растворение затрудненной кислоты (186)—к четырехкратной депрессии температуры замерзания:
О ОН
II I
PhC—ОН + H3SO4 =<=> PhC—ОН + HSO4
(189) (190)
АгСО2Н + 2H2SO4 ArC=O + Н3О++ 2HSO;
(186) (187)
Более того, при растворении эфира 2,4,6-трифенилбензойной кислоты (188а) в концентрированной H2SO4 появляется яркая окраска вследствие образования 1,3-дифенилфлуоренона в результате замыкания цикла (внутримолекулярное ацилирование по Фриделю — Крафтсу) в ацил-катионе (187а):
Если бы кислота (186) была протонирована обычным образом (по карбонильному атому кислорода; ср. разд. 7.4.4), две объемистые о-Ме-группы могли бы заставить две соседние ОН-группы расположиться в плоскости, практически перпендикулярной к плоскости бензольного кольца:
271
Тем самым предотвращается нуклеофильная атака катионного атома углерода, например атака МеОН, с любой стороны. Наоборот, аномальное протонирование (ср. разд. 7.3) атома кислорода гидроксильной группы способствует образованию (вследствие отщепления НгО) плоского ацил-катиона (187). Легкая, незатрудненная атака катионного атома углерода молекулой МеОН может в этом случае проходить по двум направлениям под прямым углом к плоскости кольца. Протекание кислотного гидролиза сложных эфиров бензойной кислоты и 2,4,6-тризаме-щенных бензойных кислот по разным механизмам — ДАс2 и Лас1 соответственно—подтверждается соответствующими параметрами активации:
ДЯ+. кДж/моль ДЗФ, Дж/(моль-К)
PhCO2Me 79 - 110(Лдс2)
Эфир (188) 115 +57(ЛА(-1)
Основным фактором, ответственным за изменение механизма реакции, является стерический фактор. Это иллюстрируется тем, что кислоты (191) и (192) и их сложные эфиры легко вступают в реакции этерификации гидролиза по типу обычного механизма Лас2:
8.7. Присоединение по связи C = N
Связь C=N формально сходна со связью С=О + — 6+5-
RC=N-«->RC = N т.е. RC = N = RC=N
(193а) (1936) (193аб)
н можно было бы ожидать, что она вступает в ряд аналогичных реакций нуклеофильного присоединения. Так, соединения со связью C=N присоединяют реактивй Гриньяра с образованием солей иминов (194), гидролиз которых дает кетоны (195):
RC=N-----► RC = N’+MgX Н ’-2»RC = О
R'-i-MgX R' R’.
(194) (195)
272
Однако в случае соединений типа RCH2CN возможен отрыв ^ротона от. группы СН2 реактивами Гриньяра, приводящий к бо-лее сложным реакциям. Восстановление с использованием LiAlH4 (ср. разд. 8.2.6.1), дает RCH2NH2. При реакции с аммиаком в присутствии NH4 СГ образуются соли амидинов RC(NH2)=NH2Cr. Катализируемое кислотой присоединение спиртов, например EtOH, приводит к образованию солей иминоэфиров (196) (ср. полуацетали; разд. 8.22):
RC5N Rg = NH RC = NH «at RC = j$H2 HOEt HOEt OEt
(193) + (196)
Присоединение H2O (гидролиз) может катализироваться и кислотой, и основанием:
н+
RC=N «=*
(193)
RC=N~
I ОН
н2о
RC=NH
н2о
RC=NH I ОН
RC—NH3
(197)
Сначала образуется амид (197), но он также легко подвергается кислотному или щелочному гидролизу (см. выше), и фактическим продуктом реакции обычно является карбоновая кислота RCO2H или ее анион.
Глава 9
РЕАКЦИИ ЭЛИМИНИРОВАНИЯ
9.1. 1,2-Элиминирование (Р-элийинироваине) . .................275
9.2. Механизм £1.............................................. 276
9.3. Механизм £1сВ............................................. 278
9.4. Механизм £2................................................280
273
9.4.1. Стереоспецифичность элиминирования по механизму Е2 . . . 282
9.4.2. Ориентация элиминирования по механизму £2. Правила Зай- / цева и Гофмана..................................................285
9.5. Конкуренция реакций элиминирования и замещения.................290
9.6. Влияние активирующих групп.....................................293
9.7. Другие реакции 1,2-элиминирования............................ 295
9.8. 1,1-Элиминирование (а-элиминирование) 297
9.9. Пиролитическое син-элиминирование..............................300
В ходе реакций элиминирования происходит удаление из молекулы двух атомов или групп их замещения другими атомами или группами. В подавляющем большинстве случаев атомы или группы отщепляются от соседних углеродных атомов; одной из отщепляющихся частиц очень часто является протон, а другой— нуклеофил Y: или Y~. В результате такой реакции образуется кратная связь (1,2-элиминирование, или а,0-элиминирование):
Известны реакции элиминирования не только от атомов углерода, но и от других атомов, например
Агх /ОСОМе \z=NZ
--------►
—МеСО2Н
ArC=N
НН Н
I I I
R-c-° R-c=°
CN
а также реакции отщепления от одного и того же атома, т. е. 1,1-элиминирование (а-элиминирование) (ср. разд. 9.8), реакции 1,4-элиминирования, т. е. реакции, обратные 1,4-присоединению (ср. разд. 7.5.1), а также 1,5- и 1,6-элиминирования, приводящие к циклизации. Реакции 1,2-элиминироваиия наиболее распространены и важны, поэтому им уделено наибольшее внимание.
274
9.1. 1,2-Элиминирование (0-элиминирование)
В реакциях 1,2-элиминирования с участием атомов углерода (т. е. в большинстве реакций) атом, от которого отщепляется У, обычно называют а-углеродным атомом, а атом, от которого отщепляется (обычно) Н, — 0-углеродным атомом. В более старой а,^-терминологии букву а обычно опускают, и такие реакции называют реакциями 0-элиминирования. Наиболее известным» примерами таких реакций являются: отщепление галогеноводо-рода от алкилгалогенидов, особенно от алкилбромидов (1) в присутствии основания (наиболее типичная реакция элиминирования вообще); катализируемая кислотой дегидратация спиртов (2) и расщепление гидроксидов тетраалкиламмония (3) по Гофману:
"ОН
RCH2CH2Br -----► RCH=CH2 + Н2О + Вг-
(1)
н+
rch2cr2oh -----► RCH=CR2 + H3O+
(2)
RCH2CH2NMe3-OH —► RCH=CH2 + H20 + NMe3
(3)
Известны и другие отщепляющиеся группы: SR2, SO2R, OSO2Ar и др. Реакции 1,2-элиминирования являются основным способом получения алкенов.
Для реакций 1,2-элиминирования могут быть предложены три различных простых механизма. Эти механизмы отличаются друг от друга последовательностью расщепления связей Н—С и С—Y. Механизм может быть согласованным, т. е. процесс является одноступенчатым и проходит через одно переходное состояние (4); его называют механизмом £2 (элиминирование бимолекулярное), причем он в какой-то мере напоминает механизм Sn2 (ср. разд. 4.1):
вЛн
R2C - СН2 —
4
В—н
ВН+ r2c = ch2 Y~
(4)
275
Связи Н—С и С—Y могут быть разорваны раздельно в двухступенчатом процессе. Если связь С—Y разрывается первой, то возникает карбокатионный интермедиат (5):
Н вГ*Н ВН+
। kl. ± кг
H2C-CR2S=t H2C-CR2—- H2C = CR2
4 ¥. ¥-
<5)
Такой механизм называют механизмом Е1 (элиминирование мономолекулярное). Он напоминает механизм SN1 (ср. разд. 4.1), а карбокатионные интермедиаты для механизмов 5n1 и fl, несомненно, идентичны.
Наконец, первой может быть разорвана связь Н—С, так что образуется карбанионный интермедиат (6):
ВЛН . ВН+ вн+
I ГЧ кг
Х2С - CH2 s=* Х2С-СН2 —* Х2С = СН2
У ’ (у Y-
(6)
Такой механизм называют механизмом Е 1сВ [элиминирование от сопряженного основания, т. е. (6)]. Известны примеры реакций, протекающих по каждому из этих трех механизмов. Наиболее распространены реакции, идущие по механизму Е2, наименее — по механизму flcB. Эти три механизма ниже рассмотрены по порядку; не следует представлять себе, что каждый из них является лишь предельным случаем (ср. 5N1/5N2), в действительности существует непрерывный спектр механизмов, определяющих последовательность расщепления двух связей.
9.2. Механизм Е1
Если, как обычно, карбокатион, например (5), образуется на медленной, скоростьлимитирующей стадии (т. е. #2>&i), кинетическое уравнение для реакции, например, с бромидом МеСН2СМе2Вг имеет вид:
Скорость =Л[МеСН2СМе2Вг].
Процесс элиминирования в этом случае завершается образованием алкена (8) путем быстрого нелимитирующего скорость отщепления протона от карбокатиона (7), обычно проте-
276
дающего с участием молекулы растворителя, в данном случае ЕЮН:
OEt
| EtOH + EtOH
МеСН2СМе2 ------ МеСН2СМе2 -----»- МеСН=СМе2
(9) (7) (8)
Можно было бы утверждать, что такое сольволитическое элиминирование (£1) должно быть кинетически неотличимо от бимолекулярного (Е2) элиминирования, в котором EtOH действует как основание, потому что член [EtOH] в кинетическом уравнении реакции Е2
Скорость = &[МеСН2СМе2Вг] [ЕЮН]
должен оставаться постоянным. Эти два механизма часто можно, однако, различить. Если при добавлении небольшого количества сопряженного основания растворителя, в данном случае _OEt, не наблюдается заметных изменений в скорости, механизм Е2 можно исключить, потому что если ~OEt не участвует в качестве основания, то гораздо более слабое основание EtOH тем более не может участвовать.
Карбокатион (7) идентичен карбокатиону, который образуется при сольволизе, протекающем по механизму SN1 (см. разд. 4.1). Сольволиз, таким образом, приводит к продукту замещения (9) и обычно конкурирует с элиминированием. Эти два процесса действительно имеют общий интермедиат, поскольку их соотношение (£1/Sn1) оказывается достаточно постоянным для данной алкильной группы и не зависит от уходящей группы Y". Тем не менее эти процессы протекают через различные переходные состояния; факторы, которые определяют соотношение процессов элиминирования и замещения, обсуждаются ниже (см. разд. 9.5).
Факторы, которые способствуют мономолекулярному [в отличие от бимолекулярного (£2)] элиминированию, точно такие же, как и факторы, которые делают предпочтительным замещение 5n1 по сравнению с SN2: а) алкильная группа в субстрате должна быть способна к образованию относительно устойчивого карбокатиона; б) реакция должна идти в хорошо ионизирующем, сольватирующем ионы растворителе. Фактор (а) проявляется в том, что в случае галогенидов элиминирование fl ускоряется в ряду: первичные < вторичные < третичные в соответствии с относительной устойчивостью образующихся карбокатионов. Первичные галогениды вряд ли вообще подвергаются El-элиминироНаиию. Наличие разветвления у 0-углеродиого атома благоприятствует El-элиминированию; так, МеСН2СМе2С1
277
дает только 34 % алкена, а в случае Ме2СНСМе2С1 выход алкена составляет 62 %. Возможно, это связано с тем, что во втором случае образуется более замещенный и, следовательно, термодинамически более устойчивый алкен (ср. разд. 1.5.4). Это обстоятельство в случае fl-реакций является основным контролирующим фактором (см. отщепление по Зайцеву; разд. 9.4.2) в ориентации элиминирования, когда возможно образование более одного алкена в результате отщепления различных 0-прото-нов от карбокатионного интермедиата (7):
(1)Н Н(2)
(2) 1+1 (I)
МеСН2—С=СН3 -«------- МеСН—С—СН3 -------► МеСН=С—Ме
I -н+ | -Н+ |
Me Me Ме
(Ю) (7) (8)
В частности, в приведенном выше случае было найдено, что продукт отщепления содержит 82 % алкена (8). Алкены неожиданного строения могут появляться вследствие перегруппировки первоначального карбокатионного интермедиата перед отщеплением протона. Показано, что реакции fl-элиминирования протекают с образованием карбокатиона. Однако (в зависимости от природы растворителя; ср. разд. 4.1) эти реакции часто могут сопровождаться образованием ионных пар, различающихся по степени разделения в них ионов.
9.3. Механизм Е1сВ
Если бы, как можно было бы ожидать для этого механизма, образование карбанионного интермедиата (6) было быстрым и необратимым, а отщепление уходящей группы Y~—медленным и скоростьлимитирующим, т. е. k-\ > kz, тогда эта реакция описывалась бы уравнением:
Скорость — /г[RY] [В],
и была бы кинетически неотличимой от согласованного (f2) механизма. Эти механизмы можно было бы, однако, различить, наблюдая обмен изотопной метки между еще не изменившимся субстратом и растворителем, ведущий к быстрому, обратимому образованию карбаниона (6); как раз это, безусловно, и не может происходить, в одноступенчатом, согласованном механизме Е2. Хорошим примером для проверки механизма является реакция отщепления с бромидом (11), в котором группа Ph у ₽-уг-леродного атома, как можно ожидать, усиливает кислотность Р-Н-атомов, а также стабилизирует образующийся карбанион
278
(12) путем делокализации:
Р^Н-СНг^ PhCH ’СН2 ^Х PhCH = СН2 I EtOH г1 2 Вг *Вг
(11) (12) (13)
EtOD||~OEt
D I PhCH-CH2 (14)
Вг
Реакцию проводят с _OEt в EtOD. Бромид (11) выделяют из реакционной смеси после превращения примерно половинного его количества в алкен (13). Показано, что выделенный бромид не содержит дейтерия, т. е. соединение (14) не образуется. В алкене (13) также не обнаружен дейтерий, что было бы возможно при элиминировании галогеноводорода из образовавшегося соединения (14). Следовательно, это потенциально благоприятное превращение протекает по механизму £1сВ в той форме, как описано выше, хотя нельзя исключить возможность того, что k2 что соответствует необратимому образованию карба-
ниона.
В действительности реакции, протекающие с промежуточным образованием карбаниона, крайне редки. Это неудивительно, так как, согласно расчетам, энергия активации для реакций, протекающих по механизму Е2, ниже, чем для реакций по механизму £1сВ, в большинстве случаев на «г 30—60 кДж/моль (7—14 ккал/моль) [обращение стадии 2 потребовало бы присоединения Y- к связи С=С, что, конечно, происходит совсем не легко]. Однако есть один пример, почти полностью соответствующий механизму flcB, — это отщепление HF от соединения (15; Х = Hal):
В: *Н
I а
Х2С - CF2
F
быстро _ » Медленно ,, _ __
у х2с - cf2 -----—* х2с = cf2
(15)
(16)
Соединение (15) обладает всеми требуемыми для этого свойствами: а) наличие электроотрицательных атомов галогена
279
у p-углерода, что делает 0-Н-атом более кислым; б) стабилизация карбаниона (16) в результате электроноакцепторного действия атомов галогена, связанных с карбанионным атомом углерода; в) наличие плохой уходящей группы — атома фтора. Относительная активность ряда различных уходящих групп Y в реакции, протекающей по механизму £1сВ, уменьшается в ряду:
В:%
I
PhSOoCH - СН2 PhSO2CH - СН2 -PhSO2CH = СН2 1 Г*
Y *Y
(18). . (19)
у; PhSe >PhO >PhS ~phso2> PhSO>MeO »CN
Оказалось, однако, что наблюдаемый ряд активности уходящих групп не коррелирует ни со значениями рКа групп YH, ни с прочностью связей С—Y, ни с полярным эффектом заместителя Y! Даже в этой простой реакции способность быть уходящей группой оказывается очень сложным фактором.
В качестве других примеров реакций, протекающих по механизму £1сВ, можно назвать образование дегидробензола из C6HsF (ср. разд. 6.10.3), обращение простого нуклеофильного присоединения к группе С=О, в частности индуцируемое основанием отщепление HCN от циангидринов (20) (ср. разд. 8.2.4)
вГ*н
I
O-CR2^± О - CR2 —^O = CR2+"CN CN ^CN
(20)
а также протекающая в присутствии основания дегидратация альдолей с образованием «.^-ненасыщенных карбонильных соединений (ср. разд. 8.4.4).
9.4. Механизм Е2
Наиболее типичным механизмом элиминирования является одностадийный согласованный механизм £2, в частности для индуцируемого основанием отщепления НВг от галогенида (21). Кинетическое уравнение имеет вид:
Скорость — £[RCH2CH2Br] [В].
Как правило, основание В может выступать в качестве как нуклеофила, так и основания, поэтому отщепление часто сопро-
280
вождается одностадийным согласованным (Sn2) нуклеофильным замещением (ср. разд. 4.1):
Е2:
ВЛ Н
RCH - СН2 —*
‘ЧЗг
(21)
(22)
5N2:
rch2ch2
<Вг
Вб+
rch2ch2
м-
ВН+
rch = ch2
Вг-
в+
—*rch2ch2
Вг~
(21) (23)
Факторы, которые способствуют отщеплению по сравнению с замещением, обсуждаются ниже (см. разд. 9.5). Разрыв связи С—Н на скоростьлимитирующей стадии реакции, как этого требует согласованный механизм, подтверждается наличием первичного кинетического изотопного эффекта (см. разд. 2.3.3), когда атом водорода у p-углеродиого атома замещается на дейтерий.
Одним из факторов, влияющих на скорость реакции £2, является, как и следовало ожидать, сила применяемого основания; она уменьшается в ряду:
’NH2 > ’OR > ’ОН
Такое исследование проводили для оснований типа АгО~, так как в этом случае возможно изменение силы основания (путем введения различных заместителей в пара-положение бензольного кольца) без существенного изменения его пространственного строения. Показано, что в случае любого основания замена гидроксилсодержащёго растворителя (Н2О или EtOH) биполярным апротонным растворителем, например HCONMe2 (ДМФА) или Me2S+—О- (ДМСО), очень сильно влияет на его силу; сила оснований _ОН, -OR, например, при этом чрезвычайно возрастает. Это объясняется тем, что основание в апротонном растворителе ие имеет оболочки из молекул растворителя, связанных водородными связями, которые должны быть удалены, прежде чем частица начнет действовать как основание (ср. влияние на нуклеофильность в реакциях Sn2; разд. 4.2). Для некоторых пар субстрат — основание такая замена растворителя может привести к изменению механизма £1 на механизм £2.
281
Для того чтобы объяснить зависимость скорости реакции соединения R—Y от природы группы Y (при одном и том же R), следует учитывать: а) влияние Y на скорость разрыва связи С—Н (реакция типа £2— согласованная реакция); б) прочность связи С—Y; в) стабильность иона Y~ (что находит отражение в значении p£a Н—Y). Таким образом, ясно, что предсказать относительную способность Y быть уходящей группой не так легко! Если в группе Y атом, непосредственно связанный с атомом углерода в RY и с атомом водорода в HY, один и тот же, например кислород, то наблюдается удовлетворительная обратная корреляция скорости реакции соединения RY со значениями рХа кислоты HY: чем сильнее кислота, тем лучшей уходящей группой является ее анион. Например, п-МеСбН480з (то-зилат Ts) является значительно более лучшей уходящей группой, чем “ОН, поскольку п-МеС6Н480зН — гораздо более сильная кислота (более низкое значение р£а), чем Н2О. Если атом, с помощью которого группа Y связана с атомом углерода в R—Y, не остается одним и тем же, такой корреляции часто не наблюдается. Важность прочности связи С—Y (а не р£а Н—Y) подтверждается приведенными ниже значениями относительных скоростей, полученными для реакций PhCH2CH2Hal в присутствии “OEt—EtOH:
PhCH2CH2F PhCH2CH2Cl
PhCH2CH2Br PhCH2CH2I
1 70 4,2-Ю3 2,7-Ю4
Начальная сольватация зарождающегося Y~ в переходном состоянии, например (22), путем образования водородных связей или другими способами также может влиять на устойчивость уходящей группы, и эта устойчивость может соответствовать или не соответствовать силе кислоты HY и (или) прочности связи С—Y. Замена растворителя, таким образом, может изменять (и действительно изменяет) устойчивость уходящих групп Y~.
Протеканию элиминирования по механизму £2 благоприятствуют такие изменения структуры субстрата, которые способствуют стабилизации образующегося аллена или, что важнее, предшествующего ему переходного состояния. К ним относятся: увеличение числа алкильных групп как у а-, так и у 0-углерод-ных ато'мов (что увеличивает термодинамическую стабильность алкенов) и введение фенильной группы, которая может оказаться сопряженной с образующейся двойной связью.
9.4.1. Стереоспецифичность элиминирования
по механизму Е2
В случае ациклических соединений можно представить себе, что элиминирование протекает в двух предельных конформациях этих молекул — антиперипланарной (24а) или синперипла-
282
нарной (246):
Очевидные преимущества имеет отщепление из конформации, в которой атомы Н, Ср, Са и Y находятся в одной и той же плоскости, поскольку р-орбитали, формируемые на атомах Ср и Са при отщеплении Н+ и Y~, становятся параллельными и. способны поэтому к максимальному перекрыванию с образованием л-связи. Для атакующего атома основания В также энергетически выгодно находиться в этой общей плоскости.
Определив предпочтительность элиминирования из плоской конформации, остается решить вопрос, какая из конформаций — (24а) или (246) — будет более предпочтительной.
Элиминированию от антиперипланарной конформации (24а) благоприятствуют три фактора: а) элиминирование в этом случае происходит от более бедной энергией «заторможенной» конформации по сравнению с более богатой энергией «заслоненной» конформацией (246) (ср. разд. 1.3.1) (это различие в энергии, вероятно, будет отражаться и на соответствующих переходных состояниях); б) атакующее основание В: и отщепляющаяся уходящая группа Y~ будут максимально возможно удалены друг от друга в переходном состоянии; в) электронная пара, образующаяся из исходной С—Н-связи, будет атаковать а-углерод-ный атом со стороны, противоположной той, от которой будет отщепляться электронная пара исходной С—Y-связи (ср. благоприятную атаку «с тыла» в механизме Sn2, разд. 4.1). Наиболее важным, по-видимому, является фактор (а). Таким образом, можно предсказать преобладание анти-элиминирования («с противоположных сторон») Н и Y от (24а), а не син-элимини-рования («с той же стороны») от (246).
Если, как в приведенном выше соединении (24), оба атома углерода хиральны, отщепление от этих двух конформаций приведет к разным продуктам — транс-алкену (25а) из (24а) и цис-алкену (256) из (246). Таким образом, зная конфигурацию исходного диастереоизомера [например, (24)] и определив конфигурацию образующихся изомеров, можно установить степень стереоспецифичности процесса элиминирования. В большинстве случаев отщепление от простых ациклических соединений по механизму анти-элиминирования является более предпочтительным; таким путем можно определить, например, какой из
283
конформеров— (26) или (27) — участвует в реакции.
(26)
~OEt EtOH
Me н
D Me
"OEt EtOH
Me
Me
(29)
При Y =.Br, Ts или +NMe3 элиминирование протекает иа 100 % анти-стереоспецифично: только (28) получен из (26) и только (29) — из (27). Однако для соединений с Y = +NR3, где R— алкильный остаток с более длинной цепью, возможно син-элимииироваиие 0-Н-атома через образование циклического переходного состояния (30), включающего ионную пару — четвертичный аммоний — гидроксид-аииои:
(30)
На степень стереоспецифичности в некоторой мере могут влиять также полярность и иои-сольватирующая способность растворителя:
В циклических соединениях конформация, из которой может происходить элиминирование, в значительной степени предопределена относительной жесткостью кольцевой структуры. Так, при отщеплении HY от соединений (CH2)nCHY выходы продуктов син-элимииироваиия составили:
п Выход. % п Выход, %
3 (циклобутил) 90 5 (циклогексил) 4
4 (циклопентил) 46 6 (циклогептил) 37
Некоторое снижение стереоспецифичности для производных циклопеитаиа (п = 4) отражается иа поведении его транс- и цис-изомеров (31) и (32). Если отщепление происходит только по механизму £2, каждый из них будет превращаться в одни и тот же алкен (33): изомер (31) — путем син-элимииироваиия, а изомер (32) — путем анти-элиминирования:
син-элими-иирование
----------->
транс-элиминирование
(31) (33) (32)
Выяснено, что анти-элиминирование (32) -> (33) протекает только в 14 раз быстрее, чем син-элимииироваиие (31) (33);
284
это подтверждает тот факт, что энергия, необходимая для трансформации кольца при переходе изомера (32) в примерно анти-периплаиарную конформацию, почти превосходит обычную предпочтительность в энергии заторможенной конформации по сравнению с синпериплаиарной, заслоненной конформацией, т. е. (31).
Заметная ант«-стереоселективиость, наблюдаемая в случае производных циклогексана (см. выше), обусловлена его способностью принимать так называемую транс-диаксиальиую конформацию (34), которая иключительно благоприятна для элимини
рования:
Например, один из геометрических изомеров гексахлорциклогексана С6Н6С16 как выяснено, отщепляет НС1 со скоростью, в (7—24) • 103 раза меньшей, чем другие изомеры. Установлено, что этот изомер является изомером (35), который не может принимать транс-диаксиальную конформацию.
9.4.2. Ориентация элиминирования по механизму Е2.
Правила Зайцева и Гофмана
Субстраты, которые имеют альтернативные доступные 0-атомы водорода, могут образовать при элиминировании более одного алкена. Например, в случае соединения (36) возможно образование двух алкенов:
(2)Н Н(1)
"OEt I I "OEt
МеСН2СН=СН2 -------- МеСН—СН—СН2 --------► МеСН=СНСН3
(о | (О
Y
(37) (36) (38)
У Выход, %
(37) (38)
Вг 19 81
+ SMe2 74 26
+NMe3 95 5
Для предсказания наиболее вероятного хода реакции долгое время использовали два противоположных эмпирических правила. Согласно правилу Гофмана (1851 г.; соединения типа RN+Me3, т. е. Y = +NMe3), преобладющим будет тот алкен,
285
который содержит наименьшее число алкильных заместителей у атомов углерода, образующих двойную связь, т. е. алкен (37). Согласно правилу Зайцева (1875 г.; соединения типа RBr, т. е. Y — Вг), преобладающим алкеном будет тот, который имеет наибольшее число алкильных заместителей у атомов углерода, образующих двойную связь, т. е. (38). Как видно из приведенных выше выходов продуктов отщепления, верны оба обобщения; состав смеси алкенов, получаемых при элиминировании, зависит от природы уходящей группы Y.
Элиминирование по Зайцеву, которое, по-видимому, имеет место, когда Y является нейтральной частицей (например, OTs и т. п., а также Вг), приводит к более устойчивому (т. е. более замещенному; ср. разд. 1.5.4) алкену. Поэтому можно предположить, что реакция в этом случае протекает через переходное состояние, в котором в значительной степени уже образовалась алкеновая структура; алкильные заместители поэтому становятся способными проявлять стабилизирующий (понижающий энергию) эффект на самой ранней стадии механизма £2, например, в структуре (38а):
EtO*"H
МеСН’—СНСНз
(38а)
О преобладании элиминирования по Зайцеву в механизме £1 уже говорилось выше (см. разд. 9.3).
Кажется совершенно логичным (это как раз то, что можно было бы ожидать от механизма £2) выяснить: почему положительно заряженный Y может способствовать отклонению от этого рчевидного пути? При Y = +NMe3 из-за сильного электроноакцепторного, индуктивного эффекта этой группы (и эффекта поля) увеличивается кислотность атомов водорода, связанных с p-углеродными атомами [формула, (36а)]. Вследствие этого эти атомы гораздо легче отщепляются основанием, чем в том случае, когда Y = Вг. Сильный электроноакцепторный эффект группы -+NMe3 будет также стабилизировать карбокатион, начинающий образовываться при отщеплении любого из этих атомов водорода. Этот эффект при отщеплении атома Н<2> несколько уменьшается из-за электронодонорного влияния Ме-группы у p-углеродного атома. Такой ослабляющий кислотность эффект отсутствует при отщеплении атома поэтому этот атом водорода является более кислым, чем атом Н<2\ а следовательно,
286
легче отщепляется основанием. Эффекта группы +NMe3, по-видимому, достаточно, чтобы определяющим фактором стала относительная кислотность протона, а не потенциальная стабильность алкена. Реакция, таким образом, проходит через переходное состояние (37а), обладающее в некоторой степени «карбанионным характером», но практически не обладающее «свойствами алкена».
(2)Н Н(1)
♦ ♦
Me— СН— |Н—СН2 +NMe3
6-H--OEt
МеСН2-СН-СН2 I 6 +NMe3
(36a) (37a)
Таким образом, реакции элиминирования по механизму Е2 могут включать различные переходные состояния, характер которых может в значительной степени зависеть от природы группы Y.
Интересно, что если Y = F, то, несмотря на то что в этом случае группа Y не заряжена положительно, также наблюдается тенденция к образованию продуктов расщепления по Гофману; например, отщепление от EtCH2CHFCH3 приводит к EtCH2CH= =СН2 с выходом не менее 85 %. Этот «неожиданный» результат является следствием чрезвычайно сильного электроноакцепторного эффекта фтора (ср. с +NMe3), а также того, что фтор является очень плохой уходящей группой, вследствие чего разрыв связи С—F проходит в переходном состоянии. Роль кислотности протона и возможность образования переходного состояния, обладающего «карбанионными свойствами», в ходе отщепления по Гофману подтверждаются тем фактом, что увеличение силы основания, атакующего RY (независимо от того, заряжена ли группа Y положительно или нет), также ведет к увеличению выхода продукта отщепления по Гофману. р-Заместители, которые могли бы помочь стабилизировать образующийся отрицательный заряд, способствуют образованию продукта по Гофману; однако такие заместители, как Ph, С=С и т. п., активируют образование любого алкена, двойная связь' которого сопряжена с системой связей в таких заместителях. Другая характерная особенность элиминирования по Гофмаиу состоит в том, что если, как в случае соединения (39), имеются альтернативные потенциальные уходящие группы RNMe2, всегда преимущественно образуется наименее замещенный алкен, т. е. (40),
287
а не (41):
(1) (t)
Н—СН2—СН2 —L,
\ “OEt 1
/NMe2 -------
Me >CII—СН2
I 2
Н(2)
(39)
1 — отщепление по Гофману,
СН2=СН2 + MeCH2CH2NMe2 (40)
МеСН=СН2 + CH3CH2NMe2 (41)
2 — отщепление по Зайцеву
Влияние группы Y на тип элиминирования может также включать и стерический фактор. Так, показано, что увеличение размера Y и, что более важно, наличие разветвлений в его структуре ведут к увеличению доли отщепления по Гофману. Например, в случае соединений (42) выходы продуктов отщепления по Гофману (при отщеплении Н(1) и Y) составляют:
(1)Н Н(2)
I I СН2—СН—СН—СН2Ме (42)
Y
Y Выход, %
Вг 31
+ SMe2 87
+ NMe3 98
Найдено также, что доля элиминирования по Гофману возрастает с увеличением степени разветвленности алкильной группы субстрата (при постоянных Y и основании), а также молекулы основания. Например, для бромида (43), в случае которого можно было бы ожидать преимущественного элиминирования по Зайцеву, при использовании более разветвленных оснований, чем ЕЮ-, увеличивается выход продукта элиминирования по Гофману:
Основание
ЕЮ'
Ме3СО~
(DH Н(2)
I I
СН2—С(Ме)— СНМе (43):
I
Вг
Выход, % Основание.
30 Me2EtCO~
72 Et3CO“
Выход,
77
78
Следует отметить, что для анализа образующихся смесей алкенов был использован метод газожидкостной хроматографии.
288
При наличии любого пространственного затруднения, независимо от его происхождения, переходное состояние (45), характерное для элиминирования по Зайцеву [удаление протона (2) из (44а)], будет более затрудненным, чем переходное состояние (46), характерное для элиминирования по Гофману [удаление протона (1) из (446)]. Это различие будет нарастать по мере увеличения затруднения (в R, Y или В), так что элиминирование по Гофману будет постепенно преобладать над элиминированием по Зайцеву:
<2)Н Н(1)
ВгС-^Ме-СНг (44) Y
1 - отщепление по Зайцеву; 2 - отщепление по Гофману
Во многих случаях невозможно различить действие электронных и стерических эффектов, поскольку они часто приводят к одному и тому же конечному результату. Тем не менее за исключением тех' случаев, когда пространственные помехи становятся крайне выраженными, вероятнее всего основную роль играют электронные эффекты.
Для циклических систем правила Зайцева илн Гофмана не применимы. В этих случаях действуют другие ограничения. Например, в производных циклогексана преобладает элиминирование от транс-аксиальиой конформации (ср, разд. 9.4.1). Кроме того, обычно невозможно провести элиминирование таким образом, чтобы получить двойную связь у узлового мостикового
10 П. Сайкс
289
углеродного атома в системе с конденсированными кольцами» (правило Бредта); например, превращение соединения (47) в соединение (48) осуществить не удается:
основание
*
Возможно, что в этом случае формирующиеся в реакции Е2 р-орбитали, будучи далеко не копланарными (ср. разд. 9.4.1), фактически находились бы под прямым углом одна к другой [см. формулу (49)] и поэтому не могли бы перекрываться в достаточной мере для образования двойной связи. Относительно небольшая кольцевая система является слишком жесткой, поэтому деформация, которая необходима для эффективного перекрывания р-орбиталей, энергетически недостижима. В этом случае, по-видимому, нет причин рассматривать, какой механизм— £1 или £1сВ— мог бы оказаться предпочтительным: бициклогептен (48) не был получен. В случае колец большего размера, например бициклононена (50), или в случае более гибкой системы (51), возможна деформация, позволяющая получить двойную связь в ходе реакции элиминирования:
(50)
(51)
9.5. Конкуренция реакций элиминирования и замешеиия
Реакции элиминирования, протекающие по механизму £1, как правило,' сопровождаются реакциями замещения типа SnI. поскольку обе реакции имеют общий (карбокатионный) интермедиат, хотя он и превращается либо в продукты элиминирования, либо в продукты замещения через различные переходные состояния в ходе быстрой нелимитирующей стадии. Аналогично, элиминирование по механизму £2 часто сопровождается замещением Sn2, хотя и в этом случае параллельные согласован-, ные процессы протекают по совершенно различным механизмам на всем протяжении реакции. Таким образом, при рассмотрении соотношения реакций элиминирования и замещения следует иметь в виду три основных аспекта: а) факторы, влияющие на
290
соотношение продуктов El и SnI; б) факторы, влияющие на соотношение продуктов Е2 и Sn2; в) факторы, влияющие на изменение механизма, т. е. E1/Sn1 ->E2/Sn2 (или наоборот), поскольку такое изменение часто влияет на соотношение элиминирования и замещения.
Последний из этих факторов, вполне возможно, наиболее важен. Так, сольволиз Ме3СВг и EtMe2CBr в EtOH (при 25 °C) по механизмам E1/Sn1 дает 19 и 36 % алкена соответственно, тогда как при добавлении 2 моль ЕЮ-, что сдвигает механизм, по крайней мере отчасти, в сторону E2/Sn2, выходы алкена увеличиваются до 93 и 99 % соответственно. Найдено, что для любого субстрата соотношение E2/SN2 всегда существенно выше, чем соотношение E1/Sn1. Следует иметь в виду, что для препаративных целей отщепление НВг от алкилбромидов следует проводить в менее полярном растворителе (процессу E1/Sn1 благоприятствует полярная, ионсольватирующая среда) — спиртовом, а не водном растворе К2СО3. Изменение механизма может быть также вызвано увеличением концентрации используемого основания, например -ОН, поэтому обычно используют концентрированные, а не разбавленные растворы К2СО3.
Что касается факторов (а) или (б), то следует отметить, что доля элиминирования зависит от строения углеродного скелета субстрата и увеличивается в ряду: первичный < вторичный < третичный. С точки зрения электронных эффектов, это является следствием увеличения относительной стабилизации переходного состояния для реакции элиминирования, так как число алкильных групп у атомов углерода, образующих двойную связь, возрастает (ср. разд. 9.4.2). Так, найдено, что при действии ЕЮ~ в EtOH на первичные алкилбромиды выход алкена составляет as 10 %, на вторичные— as 60 °/о и на третичные— > 90 %. Это объясняется не только увеличением скорости отщепления, но и уменьшением скорости замещения. Аналогично, такие заместители, как С=С и Аг, которые могут стабилизировать образующуюся двойную связь с помощью сопряжения (ср. разд. 9.4), также сильно благоприятствуют отщеплению: при сравнимых условиях СН3СН2Вг дает as 1 % алкена, тогда как РЬСН2СН2Вг— as 99 %.
При протекании реакций E1/SN1 увеличение разветвления в R—Y приводит к увеличению доли элиминирования. Это происходит в результате возрастания устойчивости образующегося более замещенного алкена и, что более важно, переходного состояния, предшествующего образованию алкена из карбокатионного интермедиата. Стерический фактор также может благоприятствовать отщеплению: др2-гибридизованный атом углерода в карбокатионе (52) остается др2-гибридизованным (углы связей as 120°) в продукте элиминирования (53), но становится
10*
291
«р3-гибридизованным (углы связей « 109°) в продукте замещения (54):
RCH RCH2 RCH2
Ё Ъ-Е
/ \ £1 / \ sn’ R “7
R R R R R
(53) (52) (54)
Стерическое напряжение в переходном состоянии при замещении увеличивается; в гораздо меньшей степени это происходит (если имеет место вообще) в переходном состоянии при элиминировании. При этом различие между переходными состояниями будет становиться все больше, благоприятствуя элиминированию, по мере увеличения размера и степени разветвленности групп R. Однако значительным это различие станет только тогда, когда группы R будут еще большими по объему или еще более разветвленными, чем в Me3CY. Периферийный атом водорода гораздо доступнее, чем относительно затрудненный атом углерода карбокатиона. Поэтому следует ожидать, что доля элиминирования должна возрастать по мере увеличения размеров атакующего основания (нуклеофила), как это и наблюдается в действительности: для осуществления реакций отщепления использование Ме3СО~ обычно дает лучшие результаты, чем в случае EtO-.
Аналогичные стерические эффекты следует учитывать при относительной оценке стабильностей переходного состояния для элиминирования Е2 и переходного состояния для замещения <Sn2.
Соотношение продуктов реакций Е1 и SN1, конечно, существенно не зависит от природы уходящей группы Y, но это не совсем так в случае механизма E2/SN2, когда расщепляющаяся связь С—Y включается в каждое альтернативное переходное состояние. Доля элиминирования увеличивается в следующем ряду заместителей Y:
Тозилат < Вг < SMe2 < NMe3
Природа атакующего основания (нуклеофила), очевидно, также важна. В идеале необходима частица, которая является сильным основанием, но слабым нуклеофилом. В препаративной работе часто используют третичные амины, например Et3N и пиридин, чтобы стимулировать элиминирование. Хотя эти амины не являются особенно сильно основными, они имеют довольно слабые нуклеофильные свойства из-за стерических влияний; например, разветвление в Et3N препятствует нуклеофильной атаке по атому углерода, но не препятствует атаке основания по пе
292
риферийному атому водорода. Хорошие результаты дает также использование оснований с относительно высокой температурой кипения (см. ниже).
Наконец, элиминирование, независимо от механизма £1 или £2, будет преобладать над замещением при повышении температуры. Возможно, это обусловлено тем, что отщепление приводит к увеличению числа частиц, тогда как при замещении этого не происходит. Отщепление, таким образом, имеет более положительный энтропийный член (см. разд. 8.6.3), и.поскольку его значение (AS*) умножается на Т в уравнении для свободной энергии активации (AG+ = А//+ —- Т AS+; см. разд. 2.2.1), этот член при увеличении температуры все больше перевешивает менее благоприятный член АН.
9.6. Влияние активирующих групп
До сих пор мы рассматривали влияние алкильных заместителей на предпочтительность реакций элиминирования в подходящих субстратах, а также, более кратко, влияние Аг и С=С-заместителей. В общем, элиминирование индуцируется наиболее сильными электроиоакцепторными заместителями, например CF3, NO2, ArSCh, CN, СО, COjEt и т. д. Их влияние может проявляться в повышении кислотности 0-Н-атомов [см. формулу (55)], что облегчает их отщепление, а также в стабилизации образующегося карбаниона вследствие оттягивания электронов [(56)] или, в некоторых случаях, в стабилизации образующейся двойной связи путем сопряжения [(57)]:
В:>Н
F3C-*CH-CHz I Y
(55)
б+
;n—сн -сн2-
'-</♦ {
(56)
о+
В---Н
О “--С—сн--=сн2
OEt Vs"
(57)
Чем более сильным является электроноакцепторный заместитель, тем больше вероятность того, что переходное состояние при отщеплении по механизму £2 будет напоминать карбанион (ср. разд. 9.4.2) или, даже, что механизм реакции будет смещаться в сторону механизма £1сВ (ср. разд. 9.3); это возможно, например, в случае NO2 или АгЗОг, особенно, если уходящая группа является плохой уходящей группой.
Наличие в альдоле (58) группы СНО облегчает элиминирование, поэтому возможна катализируемая основанием дегидратация с образованием «.^-ненасыщенного альдегида (60)
293
(см. разд. 8.4.4):
в-1н
О=С-СН-СНМе Н ОН
О^-с^сн—СНМе
Н 6"ОН
(59)
+ВН
О=С-СН=СНМе
I
н
(60)
Реакции дегидратации катализируются обычно кислотами; протонирование группы ОН превращает ее в +ОН2, которая является лучшей уходящей группой, чем -ОН. Катализируемое основанием элиминирование в рассматриваемом случае становится возможным из-за повышения кислотности атома водорода в p-положении под влиянием группы СНО и стабилизации образующегося карбаниона. В переходном состоянии (59) образующаяся двойная связь стабилизируется путем сопряжения, однако не совсем ясно, как велика роль этой стабилизации. Электроноакцепторные заместители обычно гораздо более эффективно содействуют элиминированию, когда они находятся у р-, а не у а-углеродного атома; они могут вступать в сопряжение с образующейся двойной связью одинаково хорошо в любом из этих двух положений, однако могут увеличивать кислотность только р-Н-атома и стабилизировать карбанионы только в р-по-ложении. Это ясно видно в случае индуцируемого основанием отщепления НВг от 1- и 2-бромкетонов (61) и (62) соответственно:
Н' ;В
Me
О = С-СН-СН2 Вг
(61)
медленно
6+ Н—В
Me Г -
I :
О = С-СН—сн2
Вг6"
В:% I О = С*-СН-СН2
Me Вг
(62)
быстро
б+В—Н
О— С—С—СН2 6"Вг
0 = С-СН = СН2
*/ Me / <63>
дают один и тот же а,Р-ненасыщенный (т. е.
Оба субстрата сопряженный) кетон (63), но в аналогичных условиях кетон (62) отщепляет НВг гораздо быстрее, чем кетон (61). Такие
294
p-заместители часто достаточно эффективны, чтобы активировать отрыв более необычных (и плохих) уходящих групп, таких как OR, NH2 и т. п. (о группе ОН см. выше).
9.7. Другие реакции 1,2-элиминирования
Выше были рассмотрены реакции элиминирования, в которых атом водорода отщепляется в виде протона от р-углерод-ного атома. Эти реакции являются наиболее важной группой реакций элиминирования. Однако известны реакции, в которых происходит отщепление других атомов или групп (кроме Н) от p-углеродного атома. Наиболее типичными из них являются реакции 1,2-дегалогенирования, в частности 1,2-дебромирование. Оно может быть индуцировано рядом различных частиц, включая иодид-ион I-, металлы (например, цинк) и некоторые ионы металлов (например, Fe2+). Показано, что реакция с F в ацетоне описывается приведенным ниже кинетическим уравнением (после допущения, что I- образует комплекс с 12, выделяющимся в этой реакции):
Скорость = k[ 1,2-Дибромид] [I-],
которое согласуется с механизмом £2.
Это подтверждается высокой анти-стереоселективностью, которая наблюдается в случае ациклических соединений (см. разд. 9.4.1), когда один или оба атома брома связаны с вторичным или третичным атомами углерода, например:
|Вг + Г- 12 + ВГ
Из соединения (64) образуется только транс-алкен (65). Однако, если один или оба атома брома связаны с первичными атомами углерода, например в соединении (66), суммарная реакция протекает син-стереоселективно, т. е. единственным продуктом является цис-алкен (68) . Полагают, что этот несколько удивительный результат не отражает Стереохимического изменения в самом отщеплении, но вытекает из сложного взаимоотношения механизмов SN2/£2. Замещение Вг на I при действии иодид-иона по механизму Sn2 с обращением конфигурации [(67)] сопровождается анти-стереоселективным элиминированием от 1-иод-2-бромида (67) с образованием алкена (68); в целом
295
суммарная реакция является кажущимся сик-элиминированием [(66)->(68)]:
(68)
Подтверждением того, что в каждом случае стадия отщепления действительно протекает по механизму Е2, служит тот факт, что введение алкильных заместителей у а- и 0-углеродных атомов приводит, как правило, к увеличению скоростей реакций из-за увеличения относительной термодинамической устойчивости образующегося алкена.
Бромид- и хлорид-ионы гораздо менее эффективны в содействии дегалогенированию, чем иодид-ион, но металлы, особенно Zn, используются для этой цели уже давно. Реакция в этом случае протекает гетерогенно на поверхности металла. Растворитель «восстанавливает» активную поверхность металла путем удаления образующегося галогенида. В качестве примера рассмотрим реакцию дибромида (69), проходящую с высокой степенью а«ти-стереоселективности. Эта реакция, вероятно, протекает по механизму Е2, хотя, безусловно, поверхность металла участвует в реакции:
- Me Н Zn^Br /^4 г Н Ме
ZnBr+ + Вг -► ZnBr2
(69)
Однако в случае 1,2-дибромидов с более длинными цепями, т. е. >С4, строгая а«ти-стереоселективность не наблюдается. В этом случае реакция может быть индуцирована Mg, поэтому невозможно получить реактивы Гриньяра из простых 1,2-дибромидов. Для дегалогенирования использовали также ионы металлов; в этом случае реакция протекает в гомогенной среде. Дебромирование редко используют для препаративных целей, так как исходные 1,2-дибромиды обычно получают путем добавления брома к алкену! Бромирование — дебромирование, однако, иногда используют для «защиты» двойных связей, например при окислении (70)->(71), которое нельзя осуществить непосред
296
ственно из-за одновременного окисления двойной связи:
Вг
Вг2 I HNO3
rch=chch2oh -------> rch—снсн2он —
(70) Вг
Вг
—> RCH—снсо2н -z->- rch=chco2h I
Вг (71)
Реакции элиминирования протекают также с рядом соединений HalCH2CH2Y, где Y = ОН, OR, OCOR, NH2 н т. п. Такие реакции обычно идут в более жестких условиях, чем в случае 1,2-дигалогенидов. Лучшие результаты при этом дает использование металлов или катионов металлов, чем иодид-иона. Показано, что стереохимия этих реакций элиминирования может быть различной. Отщепление СО2 н Вг~ от диастереомера (72) 2,3-дибром-3-фенилпропаноата в Ме2СО достигает, однако, 100 %-й анти-стерерселективности, причем при чрезвычайно мягких условиях:
Вг
9.8. 1,1-Элиминирование (а-элиминирование)
Известно относительно небольшое число примеров реакций 1,1-элиминирования (а-элиминирование), в которых как атом водорода, так и уходящая группа Y отщепляются от одного и того же углеродного атома, например:
В.^Н Н и
I» О I—* 7
МеСН2СН2СН - С1 —> МеСН2СН ^СН —> МеСН2СН = СН (73) (74) (75)
Этим реакциям благоприятствуют: а) наличие сильных электроноакцепторных групп, так как они увеличивают кислотность а-Н-атомов и стабилизируют отрицательный заряд на а-угле-родном атоме; б) использование очень сильных оснований В; в) отсутствие р-Н-атомов (это необязательно) (ср. разд. 3.2.4).
Полагают, что в некоторых (необязательно во всех) случаях отщепление Н+ и С1~ протекает согласованно и приводит
непосредственно к карбенному интермедиату (74) (ср. разд. 2.3.5). Для образования алкена необходима последующая миграция атома водорода с его электронной парой от 0-углерод-ного атома. Реакция 1,1-элиминирования (£а) кинетически неотличима от 1,2-элиминирования (£2). Возможность его протекания доказана в опытах с изотопными метками и косвенными данными об образовании карбенов, например (74).
Так, показано, что при введении двух атомов дейтерия в a-положение соединения (73) один из них теряется при переходе к алкену (75), тогда как в случае механизма £2 должны были бы сохраниться оба атома. В то же время при введении двух атомов дейтерия в 0-положен^е соединения (73) оба они обнаружены в алкене (75), хотя один из них теперь находится у концевого [а-С в (73) ] атома углерода; он должен был отщепиться в случае £2. С помощью меченых атомов можно определить, в какой степени элиминирование протекает как 1,1-элиминирование и в какой — как 1,2-элиминирование. При действии раствора С6Н5О“ Na+ (чрезвычайно сильное основание) в декане на соединение (73) выход продукта 1,1-элиминирования достигает 94 %, тогда как при использовании Na+ NH2 он гораздо меньше, а при действии Na+ ОМе~ отщепление вообще вряд ли можно осуществить, т. е. здесь проявляется действие фактора (б) (см. выше). Найдено также, что при действии одного и того же основания алкилбромиды и -иодиды подвергались 1,1-элими-нированию в гораздо меньшей степени, чем соответствующие хлориды, в соответствии с действием фактора (а). Предположение об образовании карбенного интермедиата (74) подтверждается выделением циклопропана (76) из реакционной смеси:
/СН2 /СН2
Ме—Н2С | —> Ме—НС^ |
: СН ^СН2
(74) (76)
Такие внутримолекулярные «внедрения» с образованием циклопропанов обычны для реакций соответствующих карбенов. Их можно рассматривать как примеры «внутренней ловушки» (ср. разд. 2.3.4). Выход соединения (76) при реакции хлорида (73) составил всего 4 %, однако в результате 1,1-элиминирова-ния изомерного хлорида МеСНОСНгСНз он составил не менее 32 %.
Наиболее известными и наиболее изученными реакциями 1,1-элиминирования являются реакции соединений, у которых нет доступных-р-Н-атомов, например гидролиз галоформов. Так, гидролиз СНС13 (77) сильными основаниями включает первоначальное 1,1-элиминирование, протекающее, возможно, по двух
298
стадийному, т. е. £1сВ, механизму, с образованием дихлоркар-бенного интермедиата (78):
Н HgO ~он
| / медлен-
СС13 СС12 СС12-Р— СО + Не-
быстро быстро с-
(77) С1 (78)
Эта реакция, как и ожидалось, подчиняется следующему кинетическому уравнению:
Скорость = &[СНС13] [ ~ОН].
Наличие быстрой обратимой первой стадии подтверждается тем фактом, что дейтерированный хлороформ CDC13 вступает в катализируемый основанием обмен с Н2О (потеря D) гораздо быстрее, чем протекает его гидролиз.
Дальнейшим подтверждением приведенного выше механизма являются данные о том, что СНС13 относительно инертен к действию PhS-, однако при добавлении -ОН реагирует с ним очень быстро с образованием CH(SPh)3, т. е. PhS-, являясь недостаточно нуклеофильным, чтобы атаковать СНС13, будет атаковать очень реакционноспособный дихлоркарбен (78). Дихлоркарбен является очень электронодефицитной частицей и (если генерируется в. апротонном растворителе) присоединяется к двойной связи богатых электронами алкенов, например ^ис-бутену-2 (79) с образованием циклопропанов, например (80), — реакция «улавливания» (ср. разд. 2.3.4):
СНС13
(79)
Ме3СО”
С6Не
(78) (80)
При подходящих условиях эту реакцию можно использовать для препаративного получения циклопропанов; другой препаративной. реакцией «улавливания» с участием СС12 является его электрофильная атака фенолов в реакции Раймера — Тимана (см. разд. 10.5.3.3).
Следует, однако, отметить, что в протонных растворителях с обычными основаниями и с субстратами, содержащими 0-Н-атомы, 1,1-элиминирование проходит в очень незначительной степени, если происходит вообще.
299
9.9. Пиролитическое син-элиминирование
Ряд органических соединений, в том числе сложные эфиры [в частности, ацетаты, ксантаты (дитиокарбонаты)], оксиды аминов и галогениды, претерпевают пиролитическое элиминирование HY без добавления реагента, в инертных растворителях или в отсутствие растворителя, в некоторых случаях — в газовой фазе. Такие реакции обычно подчиняются следующему кинетическому уравнению:
Скорость = ^[Субстрат].
Однако они отличаются от реакций элиминирования по механизму Е1 (которые следуют тому же самому уравнению) тем, что для них характерно преобладание снн-стереоселективности. Иногда эти реакции обозначают как Е;-элимииирование (элиминирование внутримолекулярное). Степень их стереоселективности выражает, в какой мере эти реакции протекают через циклические переходные состояния типа (82), которые и предопределяют протекание син-элиминирования.
Наибольшее применение в синтезе имеет, по-видимому, реакция Коупа, так как протекает при относительно низких температурах; в ней участвуют третичные оксиды аминов, например
(81):
Н (83)
Н -Н NMe2
Уходящие группы, Н и ONMej, должны занять синперипла-нарную ориентацию по отношению друг к другу, чтобы стало возможным их сближение и образование связи О”Н в переходном состоянии (82). Продуктами реакции оказываются алкен (83) и М,АЕдиметилгидроксиламин. Реакция Коупа, протекая через напряженное плоское пятичленное переходное состояние, обнаруживает высочайшую степень син-стереоселективности по сравнению с любой другой реакцией этого типа.
Пиролиз дитиокарбонатов (84) (реакция Чугаева) и эфиров карбоновых кислот (86) отличается от приведенной выше реакции тем, что протекает через шестичленные циклические пере-
300
ходные состояния, например, (85) и (87) соответственно:
(84)
HSCOSMe-» COS + MeSH
(86)
А *500°
Шестичленные кольца в этих переходных состояниях более гибки, чем пятичленные переходные состояния (82), и необязательно должны быть плоскими (ср. циклогексаны и циклопентаны). Таким образом, отщепление может происходить, по крайней мере отчасти, из других конформаций, а не из синперипла-нарной, в результате чего степень син-стереоселективности в этих реакциях элиминирования может быть иногда ниже, чем это наблюдается в реакции Коупа. Обе рассматриваемые реакции идут при более высоких температурах, чем реакция Коупа, особенно в случае эфиров карбоновых кислот.
Одним из основных преимуществ этой группы реакций элиминирования как препаративного метода получения алкенов является то, что условия их проведения относительно мягкие: прежде всего, низкие значения кислотности или основности. Это означает, что можно синтезировать алкены, которые лабильны, т. е. изомеризуются в ходе альтернативных методов синтеза, например, вследствие миграции связи (в направлении сопряжения с другими связями) или молекулярной перегруппировки. Так, пиролиз дитиокарбоната (89), полученного из спирта (88), приводит к образованию перегруппированного алкена (90), в то время как более обычная катализируемая кислотой дегидратация соединения (88) приводит к перегруппировке в карбокатионный интермедиат (91) (ср. разд. 5.4.2.1) и, следовательно,
301
к образованию термодинамически более стабильного алкена (92): MeS- С = S
А <
ОН О ) U
I , CS2, “ОН V Д
Ме3С-СН-СН3 - Ме3С-СН-СН2 ------•>Ме3ССН = СН2
(88) (89)
1 .+Н+ “
2 . -Н2О,,
I + +1
Ме2С- CHMe — Ме2С - СМе-*
V| -н+
Н
Ме2С = СМе2
(91а) (916) (92)
Пиролиз алкилхлоридов и -бромидов (алкилфториды слишком устойчивы, а в случае алкилиодидов образуется некоторое количество алкана вследствие восстановления образующегося алкена отщепляющимся HI) приводит к образованию алкенов, но идет при температурах до 600 °C. Такие реакции редко используют в препаративных целях. Как ни удивительно, но именно такие реакции наиболее детально изучены. Полностью согласованное 1,2-элиминирование галогеноводорода должно было бы проходить через очень напряженное четырехчленное переходное состояние. Поэтому вполне возможно, что разрыв связи С—Hal в основном происходит до разрыва связи С—Н: высокая степень «карбокатионного характера», таким образом, возникает у атома углерода связи С—Hal. Поэтому не приходится удивляться, что реакции элиминирования галогеноводорода обнаруживают меньшую си«-стереоселективность, чем другие реакции элиминирования. Далее будут обсуждены согласованные реакции элиминирования а также другие реакции с участием циклических переходных состояний (см. разд. 12.1).
Глава 10
КАРБАНИОНЫ И ИХ РЕАКЦИИ
10.1. Образование карбанионов..................................303
10.2. Стабилизация карбанионов..................................305
10.3. Конфигурация карбанионов..................................309
10.4. Карбанионы и таутомерия...................................311
302
10.4.1. Механизмы таутомерных превращений......................311
10.4.2. Скорость таутомеризации................................314
10.4.3. Положение равновесия и строение таутомеров.............315
10.5. Реакции карбанионов...........................................318
10.5.1. Реакции присоединения..................................318
10.5.1.1. Карбоксилирование..............................318
10.5.2. Реакции элиминирования.................................319
10.5.2.1. Декарбоксилирование............................320
10.5.3. Реакции замещения......................................322
10.5.3.1. Дейтеро-водородный обмен.......................322
10.5.3.2. Реакции карбанионных нуклеофилов...............323
10.5.3.3. Реакция Раймера — Тимана.......................325
10.5.4. Перегруппировки........................................327
10.5.5. Реакции окисления......................................330
10.5.6. Галогенирование кетонов................................330
Теоретически любое органическое соединение типа (1), которое содержит С—Н-связь, т. е. почти все органические соединения, может вести себя как кислота в классическом смысле слова, отдавая протон подходящему основанию и образуя сопряженную кислоту (2), являющуюся карбанионом (ср. разд. 1.4):
R3C-Н + В: R3C’4-BH+
(1) (2)
При рассмотрении вопроса об относительной кислотности, как правило, интересуются только термодинамическим аспектом перехода протона; значение рКа кислоты (ср. разд. 3.1.1) может быть измерено, исходя из приведенного выше равновесия. Кинетические характеристики таких реакций обычно не имеют существенного значения, поскольку отрыв протона от таких атомов, как атомы кислорода, азота и т. п., происходит в растворе чрезвычайно быстро. Однако в случае С—Н-кислот типа (1) скорость переноса протона к основанию может быть достаточно мала, чтобы стать лимитирующим фактором. Кислотность соединения (1) в таком случае контролируется кинетически, а не термодинамически (ср. разд. 10.4.3).
Карбанионы могут образовываться не только путем отрыва протона (см. ниже). Карбанионы представляют большой интерес не только сами по себе; они участвуют в большом числе различных реакций. Многие из этих реакций особенно ценны тем, что приводят к образованию связей С—С (ср. разд. 8.4).
10.1. Образование карбанионов
Наиболее общим методом образования карбанионов является отщепление атома или группы X от атома углерода, причем электронная пара остается на атоме углерода:
R3C—X + Y R3C' + XY+
303
Поскольку, как было упомянуто выше, обычно уходящей группой X является атом водорода, то именно протон удаляется при переходе (1)->-(2). Однако известны и другие уходящие группы, например СО2 [при декарбоксилировании (см. разд. 10.5.2.1) RCO2 (3)] или С1~ [при отщеплении хлора от Ph3CCl образуется окрашеииая в кроваво-красный цвет растворимая в эфире соль (4)]:
[Г- С ^О~ —R" + СО2
О
(3)
РЬ3С - Cl Ph3C’ Na+
(4)
Тенденция алканов отщеплять протон и образовывать карбанионы проявляется мало, так как они не содержат заместителей, которые бы усиливали кислотность атомов водорода или могли бы стабилизировать карбанионный центр (ср. карбоновые кислоты; разд. 3.1.2). Так, для СН4 рУа ~ 43, тогда как для МеСО2Н р/Са = 4,76. Обычные методы определения рКа для алканов нельзя испольарвать; соответствующие значения были получены при изучении равновесий в системах иодид—металл-оргаиическое соединение:
RM + R'I =<=> RI + R'M
Предполагают, что чем сильнее кислота RH, тем большая часть ее будет в форме RM (например, М = Li), а не в форме RI. Определение константы равновесия А позволяет измерить относительную кислотность RH и R'H и путем подбора подходящих пар поднять шкалу р/Са так, чтобы сделать возможным прямое сравнение с соединением R—Н, значение р/Са которого измерено другими способами. Так, было найдено, что для Ph3CH (5) рК.а = 33, т. е. это более сильная кислота, чем СН4, карбанион (4) может быть препаративно получен из соединения (5) при действии амида натрия, т. е. ~NH2, в жидком аммиаке:
жидк.
NH3
Ph3C—Н + Na+ ~NH2 ” Ph3C~ Na++ NH3
(5) (4)
Анион (4) может быть получен также при действии натрия на Ph3CCl в инертном растворителе; образующийся раствор трифенилметилнатрия используют как очень сильное органическое основание (ср. разд. 8.4.8) вследствие способности его
304
карбаниона (4) присоединять протон. Алкены являются немного более сильными кислотами, чем алканы: для этена (этилен) СН2=СН2 рКа = 37; зато алкины — гораздо более сильные кислоты: рАа этина (ацетилен) НС=СН равно 25. Карбанион НС=С~ (или RC=C-) может быть получен из соответствующего углеводорода действием ~NH2 в жидком аммиаке; такие анионы находят применение в синтезе (ср. разд. 8.4.2).
При введении электроноакцепторных заместителей увеличивается кислотность атомов водорода, связанных с атомом углерода. Так, при действии сильных оснований на хлороформ образуется неустойчивый карбанион ~СС13 (ср. разд. 9.8). К этому можно добавить, что значения рКа HCF3 и HC(CF3)3 равны « 28 и 11 соответственно. Еще большее влияние оказывают заместители, которые могут способствовать делокализации отрицательного заряда и обладают, кроме того, электроноакцепторным индуктивным эффектом. Так, значения рКа MeCN, МеСОМе и MeNO2 равны 25, 20 и 10,2 соответственно. В случае CH3NO2 соответствующий карбанион ~CH2NO2 может быть получен при действии _OEt в EtOH или даже ~ОН в Н2О (ср. разд. 8.4.6); в небольших количествах он образуется в водных растворах даже из менее кислых карбонильных соединений, способных к альдольной конденсации (ср. разд. 8,4.4).
Значения рКа некоторых углеродных кислот приведены
ниже: Соединение Р*а Соединение
сн4 43 CH2(CO2Et)2 13,3
сн2=сн2 37 CH2(CN)2 12
С6Н6 37 CH(CF3)3 11
РЬСНз 37 MeCOCH2CO2Et 10,7
Ph3CH 33 MeNO2 10,2
CF3H 28 (MeCO)2CH2 8,8
СН=СН 25 (MeCO)3CH 6
MeCN 25 C H2(NO2)2 4
МеСОМе 20 CH(NO2)3 0
PhCOMe 19 CH(CN)3 0
10.2. Стабилизация карбанионов
Известен ряд структурных факторов, увеличивающих кислотность атомов водорода в соединениях R—Н и облегчающих тем самым отрыв атома водорода под действием оснований. Кроме того, существуют некоторые факторы, способствующие стабилизации образующегося карбаниона R-; в некоторых случаях оба эффекта вызываются одним и тем же структурным фактором. Основными структурными факторами, способствующими стабилизации образующегося карбаниона R-, являются
305
следующие (ср. с факторами, способствующими стабилизации карбокатионов; разд. 5.2): а) увеличение s-характера карбанионного углерода; б) электроноакцепториые индуктивные эффекты; в) сопряжение свободной электронной пары карбаниона с поляризованной кратной связью; г) ароматизация.
Действие фактора (а) проявляется в увеличении кислотности атомов водорода:
СНз—СНз < СН2=СН2 < СН^СН
Как 'уже отмечалось, увеличение кислотности особенно заметно при переходе от алкенов к алкинам. Это объясняется увеличением s-характера гибридной орбитали, участвующей в образовании о-связи с атомом водорода, т. е. sp3 < sp2 < sp1. s-Орбитали находятся ближе к ядру, чем соответствующие р-ор-битали, и имеют более низкий энергетический уровень. Электронная пара на вр'-орбитали находится ближе к ядру атома углерода, чем электронная пара на sp2- или вр3-орбитали (кажущаяся электроотрицательность- атома углерода увеличивается). Это не только способствует более легкому отрыву атома водорода без электронной пары, т. е. повышению его кислотности, но и стабилизирует образующийся карбанион.
Действие фактора (б) проявляется, например, в случае соединений HCF3 (рКа = 28) и HC(CF3)3 (рКа — 11), изменение значений рКа которых по сравнению с СН4 (рКа ~ 43) достигается за счет сильного электроноакцепторного индуктивного эффекта атомов фтора (именно этот эффект делает атом водорода более кислым), стабилизирующего образующиеся карбанионы ~CF3 и ~C(CF3)3 путем оттягивания электронов. Этот эффект, естественно, более выражен в случае HC(CF3)3, в котором имеются девять атомов фтора (тогда как в CF3 их только три), даже несмотря на тот факт, что эти девять атомов фтора не действуют непосредственно на атом углерода карбаниона. Аналогичный электроноакцепторный индуктивный эффект наблюдается при образовании ~СС13 из НСС13 (ср. разд. 9.9). Он, вероятно, менее эффективен в случае хлора, чем в случае более электроотрицательного фтора, но это может в определенной степени компенсироваться за счет делокализации электронной пары карбаниона с участием вакантных d-орбиталей хлора, что, конечно, невозможно в случае фтора.
При введении алкильных групп из-за дестабилизирующего влияния электронодонорного индуктивного эффекта стабильность карбанионов уменьшается в ряду:
СНз > RCH2 > R2CH > R3C
Приведенный ряд противоположен ряду, полученному при оценке устойчивости карбокатионов (см. разд. 4.3).
306
Фактор (в) является наиболее типичным стабилизирующим эффектом; в качестве примера показана стабилизация при наличии групп CN [(6)], С=О [(7)], NO2 [(8)], CO2Et [(9)]:
В:\Н
СН2 &+=N [’СН2 - C=N СН2 = С = N"] + ВН+
(6) (10а) (106)
рКа = 25
ВЛН Me Me Me
’ll 1 1
CH2 » > С = О л- ** СН2 - С — О г » СН2 — с - о
(7) (11а) (116)
+ ВН+
рКа = 20
ВЛН
I +
CH2^-»N = O
6"
(8)
"СН2- N = О I О
(12а)
СН2 = Й-О" 0“ (126)
+ ВН +
рКа=10,2
ВЛН I СН
б+ 6-1-*С =0 -Г1 .OEt (9) рКа=24
"СН2- С = Оч->сн2 = С - О-’•OEt (136)
.OEt (13а)
В каждом случае действует электроноакцепторный индуктивный эффект, увеличивающий кислотность атомов водорода у будущего карбанионного центра, однако стабилизация образующегося карбаниона путем делокализации имеет, вероятно, гораздо большее значение. Интересно, что группа NO2 проявляет намного более сильное стабилизирующее действие, чем это можно было ожидать. Заметный эффект при увеличении числа стабилизирующих групп у атома углерода можно видеть из приведенных выше (см. разд. 10.1) значений p^a; так, CH(CN)3 и CH(NO2)3 являются такими же сильными кислотами в воде, как НС1, HNO3 и др. Естественно, возникает вопрос: можно ли рассматривать структуры (Юаб), (Наб) и (12аб) как карбанионы?
307
Атомы кислорода и азота более электроотрицательны, чем атом углерода, и структуры (106), (116) и (126), вероятно, вносят более заметный вклад в гибридную анионную структуру, чем (10а), (11а) и (12а) соответственно.
Сложноэфирная группа, например COzEt в соединении (9), менее эффективна в стабилизации карбанионного центра, чем группа С—О в простых альдегидах и кетонах. Это можно видеть при сравнении приведенных ниже значений рКа:
CH2(CO2Et)2 MeCOCH2CO2Et СН2(СОМе)2
13,3 10,7 8,8
Причиной указанного эффекта является электронодонорная способность свободной пары электронов у атома кислорода группы OEt.
Как отмечено выше, в случае элементов третьего периода любой индуктивный эффект, который они способны проявлять, может дополняться делокализацией пары электронов атома углерода карбаниона с участием свободных d-орбиталей атома элемента; это может иметь место, например, для атома серы в заместителе ArSOz, а также для атома фосфора в заместителе R3P+.
Действие фактора (г) наблюдается в циклопентадиене (14), у которого рКа = 16, тогда в случае простого алкена рКа = 37. Образующийся карбанион — циклопентадиенид-анион (15) представляет собой делокализованную систему из шести л-электро-нов, отвечающую правилам Хюккеля (4п + 2; п=1) (ср. разд. 1.3.6). Эти шесть л-электронов могут быть расположены на трех стабилизированных л-молекулярных орбиталях, подобных орбиталям бензола. Таким образом, анион обнаруживает квазиароматическую стабилизацию, т. е. стабилизируется путем ароматизации:
Н Н
(14) (15)
Его ароматичность не может быть, конечно, проверена опытами по электрофильному замещению, поскольку атака электрофилом Х+ приведет к простой комбинации с анионом. Тем не менее истинно ароматический характер (например, участие в реакции Фриделя — Крафтса) обнаруживается в интересном ряду чрезвычайно стабильных, нейтральных соединений, получаемых из аниона (15) и называемых металлоценами. В частности, ферроцен (16), в котором атом металла удерживается л-связями в центре молекулярного «сэндвича» между двумя циклопентадие-
308
нильными структурами, реагирует как типичный ароматический субстрат:
(16)
При обработке калием неплоского неароматического (ср. разд. 1.3.6) циклооктатетраена (17) возможно присоединение двух электронов; в результате образуется устойчивая кристаллическая соль дианиона (18):
(17) (18)
Для дианиона (18) также выполняется правило Хюккеля (4п + 2; п = 2). Следует особо подчеркнуть, что в этом случае квазиароматическая устойчивость (т. е. стабилизация путем ароматизации) происходит в системе .имеющей двойной отрицательный заряд.
10.3. Конфигурация карбанионов
Теоретически для простого карбаниона типа R3C~ может быть принята пирамидальная (sp3) или планарная (sp2) конфигурация или же промежуточная между ними (в зависимости от природы R). Пирамидальная конфигурация могла быть предпочтительнее по энергетическим соображениям, поскольку не-поделенная электронная пара в таком случае располагалась бы на вр3-орбитали [(19)], а не на более энергетически богатой не-гибридизованной р-орбитали в планарной конфигурации карбаниона. Пирамидальную конфигурацию, безусловно, имеют третичные амины R3N:, изоэлектронные простым карбанионам R3C_. Как и амины, карбанионы способны к легкому обращению конфигурации, например (19а) (196):
R"
(19а) (1эб)
Предпочтительность вр3-конфигурации карбанионов подтверждается, в частности, тем, что реакции, включающие
309
образование карбанионных интермедиатов в мостиковых системах, протекают достаточно легко; это не наблюдается для реакций, протекающих через образование соответствующих карбокатионных интермедиатов (ср. разд. 4.3).
В металлорганических соединениях типа RR'R"C—М связь С—М может иметь различную природу; она может быть полярно-ковалентной RR'R"C6~—М6+ и практически ионной RR'R"C“— —М+. В реакциях этих соединений наблюдали как преимущественное сохранение конфигурации, так и рацемизацию и обращение конфигурации; стереохимический результат в каждом случае зависит не только от алкильного остатка, но и от металла, а также, в значительной мере, от растворителя. Даже в случае наиболее ионизируемых субстратов, по-видимому, мало вероятно, что мы имеем дело с простым карбанионом. Так, при взаимодействии EtI с (PhCOCHMe)- М+ (М = Li, Na, К) относительные скорости реакции в аналогичных условиях отличаются не менее чем в « 104 раз.
Карбанионы, которые имеют заместители, способные к делокализации электронной пары по механизму сопряжения, должны иметь планарную конфигурацию (sp2), в которой возможно максимальное перекрывание р-орбиталей карбанионного центра с соответствующими орбиталями заместителей, как, иапример, в анионах (4) и (10):
Если такой конфигурации препятствуют структурные или стерические особенности, ожидаемой стабилизации не наблюдается. Так, пентадион-2,4 (20) (рКа = 8,8) и циклогександион-1,3 (21) легко растворяются в водном NaOH (но не в воде) и дают красное окрашивание с раствором FeCl3 (ср. фенол), тогда как в случае 1,3-дикетона (22), формально сходного с соединениями (20) и (21), этого не происходит.
(20)
(22)
В соединении (22) атом водорода, защищенный двумя С=О-группами, вряд ли проявляет более кислые свойства, чем анало-
310
гичный атом в соответствующем углеводороде. Иное поведение дикетона (22) объясняется тем, что после отщепления протона неподеленная электронная пара карбаниона должна была бы оказаться на 5р3-орбитали под более или менее прямым углом к р-орбиталям каждого из соседних карбонильных атомов углерода (ср. разд. 9А2). В этом случае невозможно перекрывание sp3- и р-орбиталей и, следовательно, не будет и стабилизации отрицательного заряда вследствие делокализации, и поэтому карбанион не образуется.
10,4. Карбанионы и таутомерия
Таутомерия, строго говоря, может быть использована для описания любых обратимых взаимопревращений изомеров при любых условиях. На практике применение этого термина ограничено главным образом изомерами, которые легко подвергаются взаимным превращениям и отличаются друг от друга только распределением электронов и положением относительно подвижного атома или группы атомов. Подвижным атомом в подавляющем большинстве случаев является атом водорода; в этом случае соответствующее явление называют прототропией. Хорошо известными примерами прототропии могут служить р-оксоэфи-ры, например этил-2-оксобутаноат (этилацетоацетат) (23), и алифатические нитросоединения, например нитрометан (24):
О
МеС—CHCO2Et
Н
(23а) оксоформа
ОН
I
MeC=CHCO2Et
(236) енольная форма
Н
СН2—N=O
I
О'
ch2=n—oh I О'
(24а) псевдокислота
(246) а^и-форма
Такие взаимопревращения катализируются как кислотами, так и основаниями.
10.4.1. Механизмы таутомерных превращений
Прототропные взаимопревращения изучены очень подробно; в этом случае хорошие результаты дает использование дейтериевой метки, вводимой как в молекулу растворителя, так и в
311
молекулу субстрата, а также изучение стереохимии превращения оптически активных субстратов, у хирального центра которых имеется способный к отщеплению атом водорода. Возможные граничные механизмы (ср. SnI и Sn2) таковы: а) межмолекулярный механизм, в ходе которого отрыв протона от субстрата и присоединение протона (из растворителя) происходят раздельно, — в этом случае образуется карбанионный интермедиат; б) внутримолекулярный механизм, в ходе которого протон переносится внутри молекулы субстрата:
В:>Н
1 в-
(a) R2C - СН = Y t r'oh
r2c-ch = y
t
r2c = ch - Y"
(25)
H
R2C = CH- Y
карбанионный интермедиат
(6)
H
I B:
R2C ~ CH = Y=^
В Ф
A
R< > -в:
CH
H
r2c = ch - y
(26) переходное состояние
Многие соединения, которые легко подвергаются кето-еноль-ным прототропным превращениям, катализируемым основаниями; p-оксоэфиры, 1,3-дикетоны (fj-дикетоны), алифатические ии-тросоединения и др. образуют относительно стабильные карбанионы, например (25), которые часто могут быть выделены. В частности, можно получить карбанионы из оксоформ р-оксо-эфиров (23а) и нитрометана (24а) и в подходящих условиях протонировать их для получения енольных форм (236) и (246) соответственно. Поэтому весьма вероятно, что эти взаимопревращения следуют внутримолекулярному механизму (а). Чем более кислыми свойствами обладает субстрат, т. е. чем более устойчивый карбанион из него образуется, тем больше возможность прототропного взаимопревращения с участием карбанионного интермедиата.
На примере реакции, протекающей по межмолекулярному механизму, можно показать различие между таутомерией и мезомерией, которые часто путают. Так, в случае этил-2-оксобута-
312
ноата (23)
О
О
II
МеС—CHCO2Et
Н
(23а) оксоформа
В :
ROH
МеС—CHCO2Et
R'OH
СГ
_ MeC=CHCO2Et.
(27) карбанионный интермедиат
ОН Mei=CHCO2Et
(276) енольная форма
соединения (23а) и (236) являются таутомерами; это совершенно разные частицы, которые можно различить химическими методами; они легко превращаются друг в друга, однако в данном случае их можно выделить в чистом виде. Две структуры, которые написаны для карбанионного интермедиата (27), являются мезомерами; они в действительности не существуют. Это просто не совсем точная попытка изобразить распределение электронов в карбанионе, который является единственной индивидуальной частицей. Карбанион (27) лучше изображать формулой (27а):
О6-
I; б-
МеС—CHCO2Et (27а)
Однако и она не отражает тот важный факт, что больший отрицательный заряд в- анионе располагается на более электроотрицательном атоме кислорода, а не на атоме углерода. Действительно, хотя мы назвали такие частицы, как (27) (и будем называть далее), карбанионами, их называют также (и, возможно, это более точно) енолят-анионами. Очень часто оказывается, что той или иной паре таутомеров, например (23а) и (236), соответствует один и тот же стабилизированный карбанион енолят-анион (27).
Другой граничный механизм — полностью внутримолекулярный перенос протона — наблюдается в случае Et3N :, катализирующего превращение оптически активного субстрата (28) в соединение (30):
(30)
Me
При изучении этой оптической активности
реакции найдено, что скорость потери и скорость изомеризации идентичны, а
313
если реакция проводится в присутствии D2O (5 моль на 1 моль субстрата), дейтерий не включается в продукт реакции. Реакция, таким образом, в этих условиях является полностью внутримолекулярной, идет без участия карбаниона; полагают, что она протекает через мостиковое переходное состояние типа (29). Для ряда субстратов наблюдается как меж-, так и внутримолекулярный механизм, причем относительная доля каждого из них зависит не только от природы субстрата, но и от применяемых основания и растворителя.
10.4.2. Скорость таутомеризации
Фактически во всех тех случаях, о которых мы говорили, медленной скоростьлимитирующей стадией является разрыв (или образование) связи С—Н; в этом главное отличне реакций С—Н-кислот от реакций тех кислот, в которых потенциальный протон связан с атомом кислорода или азота (и т. п.): Скорость разрыва связи С—Н часто измеряют путем определения скорости дейтеро-водородного обмена с подходящим донором протонов (дейтеронов), таким как D2O, EtOD и др. Интересно, что кинетическая кислотность (определяемая с помощью k) непосредственно не коррелирует с термодинамической кислотностью (определяемой с помощью К), т. е. со шкалой p^a:
*1
R3C—Н 4-В : R3C“-|-BH (K = fe1/fe_1)
Это объясняется тем, что кинетическая кислотность основывается на значении AG*, а термодинамическая — на значении Дб°. В таком случае связь между соответствующими шкалами не обязательна. В то же время хорошо известно, что структурные изменения в субстрате, приводящие к повышению термодинамической кислотности, ведут также и к более быстрому превращению его в карбанион. Однако это справедливо не для всех соединений, как можно видеть из приведенных ниже значений р^Са и ki:
рха fe,. с 1
CH2(NO2)2 4 8,3-10“’
СН2(СОМе)2 8,8 1,7 • 10“2
MeNOg 10,2 4,3 10“’
MeC0CH2CO2Et 10,7 1,2 -10“® .
CH2(CN)2 12 1,5- 10“2
CH2(CO2Et)2 13,3 2,5 • 10~5
MeCOMe 20 4,7-10“'°
Особенно мала скорость ионизации простых нитросоедине-
ний, несмотря на их относительно высокую кислотность. Так, 314
MeNO2 и MeCOCH2CO2Et имеют очень близкие значения рКа, но первое соединение ионизируется медленнее почти в 105 раз. Возможно, это отражает большую степень делокализации заряда в карбанионе, образовавшемся из CH3NO2, по сравнению с карбанионом, образовавшимся из CH3COCH2CO2Et. В таких случаях как отрыв протона, так и его присоединение протекают медленнее, чем в случае таких углеродных кислот, в карбанионах которых заряд более сконцентрирован на атоме углерода. Так, при введении цианогрупп можно было бы ожидать, что отрицательный заряд в карбанионе будет делокализован меньше, чем при введении группы С=О. Показано, например, что значения ki для CH2(CN)2 и СН2(СОМе)2 почти одинаковы, хотя рКа первого больше (т. е. кислотность ниже). На соотношение между рКа и kt может сильно влиять и растворитель.
10.4.3. Положение равновесия и строение таутомеров
Зависимость положения равновесия от строения таутомеров наиболее полно изучена для оксо-енольных систем. Относительная доля каждого из таких таутомеров обычно определяется химическими методами, например титрованием енольной формы бромом в таких условиях, когда скорость кето-енольного взаимопревращения очень низка. Однако более точно и более удобно проводить соответствующий анализ спектроскопически, например, методом ИК-спектроскопии. Соответствующие значения vMaKc для этил-3-оксобутаноата приведены ниже:
О 11(1) (2)
МеС—СН2—С=О
I OEt
(23а)
СВЯЗЬ VMaKc, см-1
(1) 1718
(2) 1742
ОН I (3)
МеС=СН С=О
OEt (236) Связь Умакс, СМ~
(3) (1650)
В простых карбонильных соединениях, например МеСОМе, содержание енольной формы в равновесной смеси чрезвычайно мало. Основные структурные факторы, которые способствуют увеличению доли этого таутомера, видны из приведенных ниже
данных:
Содержание енола, %
МеСОМе 1,5- io~4
CH2(CO2Et)2 7,7- 10~3
NCCH2CO2Et 2,5- 10"*
Циклогексанон 1,2
Содержание енола» %
MeCOCH2CO2Et 8,0
MeCOCHPhCO2Et 30,0
МеСОСН2СОМе 76,4
PhCOCH2COMe 89,2
315
Главный фактор — это наличие кратной связи нли л-орби-тальной системы (например, как у Ph), которые могут образовать сопряженную систему с двойной С=С-связью в енольной форме. В этом отношении С=О-группа является, несомненно, эффективной, причем обычная карбонильная С = О-группа значительно более эффективна, чем группа С = О в составе сложно-эфирной группировки [ср. содержание енольной формы для MeCOCH2CO2Et (8%) и CH2(QO2Et)2 7,7-Ю"3 %)]. Дополнительное влияние Ph-группы видно прн сравнении MeCOCH2CO2Et (8%) с MeCOCHPhCO2Et (30 %) и МеСОСН2СОМе (76,4%) с PhCOCH2COMe (89,2%).
Другим фактором, который может способствовать стабилизации енольной формы, является возможность образования внутримолекулярных водородных связей, как, например, в случае МеСОСН2СОМе (31) и MeCOCH2CO2Et (23):
Н О' '•О I II Me—С^. /С—Me
СН
(31)
Н
О/ '••О
I II
Me—/С—OEt
СН
(23)
Помимо повышения стабилизации образование внутримолекулярных водородных связей в молекуле енола уменьшает его полярность и повышает степень компактности его молекулы («свернутая» конформация) по сравнению с более вытянутой конформацией оксоформы. В результате этого енольная форма обычно имеет более низкую температуру плавления, чем оксоформа (это обнаружено в тех случаях, когда обе формы удается разделить), несмотря на наличие гидроксильной группы. Влияние внутримолекулярных водородных связей на преимущественную стабилизацию енольной формы видно также при сравнении содержания енольной формы соединения (31) в различных растворителях, а также в его жидкой и газовой фазах:
Содержание енола, % Содержание енола, %
Газовая фаза 92 MeCN 58
Гексан 92 Н2О 15
Жидкая фаза 76
Так, содержание енола в неполярном растворителе — гексане— такое же, как и в газовой фазе, и выше, чем в чистом жидком еноле (последний действует как несколько полярный растворитель); содержание енола уменьшается в более полярном растворителе MeCN и еще более резко в воде. Наблюдается, таким образом, увеличение относительной устойчивости оксоформы из-за сольватации. Это особенно заметно в воде, где оксоформа может образовывать межмолекулярные водородные
316
связи с молекулами растворителя. Аналогично ведет себя соединение (23); жидкий эфир на 8% находится в виде енола; содержание енола увеличивается до 46 % в гексане и до 50 % в газовой фазе, но уменьшается до 0,4 % в разбавленном водном растворе. Содержание енола зависит также и от температуры.
Особенно интересно сравнение МеСОСОМе (32) и циклического 1,2-дикетона — циклопентандиона-1,2 (33):
(32а) (326) (33а) (336)
(5,6-10“3%) (*100%)
В случае соединения (32), несмотря на образование внутримолекулярных водородных связей в его енольной форме (326), равновесие по существу полностью смещено в сторону оксоформы (32а), поскольку она может принимать амти-конформацию, в которой оба электроотрицательных атома кислорода максимально удалены один от другого, а карбонильные диполи ориентированы противоположным образом. В случае дикетона (33) С=О-группы закреплены в сим-конформации, как в оксо- (33а), так и в енольной (336) формах; образование же внутримолекулярных водородных связей возможно лишь в енольной форме (336).
В приведенных выше примерах состав равновесной смеси при данных условиях определяется относительной термодинамической устойчивостью оксо- и енольной форм. Интересная ситуация возникает, однако, в случае алифатических нитросоединений, например фенилнитрометана (34). В этом случае нитроформа (34а) (масло желтого цвета) более стабильна, чем аци-форма (346), и преобладает в равновесной смеси, тогда как аци-форма почти полностью отсутствует:
но~>н
• + “он б- + б- Н+
PhCH - N = Оз=* PhCH^N—О ч=Ь
О" Н “О”
I PhCH = N- О I 0“
(34а) (35) (346)
нитро-форма аци-форма
При подкислении выделенной натриевой соли этого нитросоединения [карбанионный интермедиат имеет строение (35)] образуется только менее устойчивая аци-форма (346)—бесцветное твердое соединение. Это происходит потому, что имеет место более быстрое протонирование в положение с более высокой
317
электронной плотностью, т. е. образование продукта в этих условиях контролируется кинетически. Энергетический профиль для этой системы имеет форму, показанную на рис. 10.1.
Переходное состояние (ПС2) между (35) и (346) находится на более низком энергетическом уровне, чем переходное состояние (ПС1) между (35)
и (34а), т. е. связь О—Н разрывается легче, чем связь С—Н. Непосредственным результатом подкисления интермедиата (35) является образование формы (346), однако она претерпевает спонтанную реионизацию: ацы-форма («кислородная» кислота) отщепляет протон быстрее, чем нитроформа («углеродная» кислота). Таким образом, постепенно устанавливается равновесие, ведущее к медленному, но непременному образованию более устойчивой формы (34а). Таким образом, окончательный состав продукта контролируется термодинамически.
10.5. Реакции карбанионов
Карбанионы могут участвовать в большом числе реакций, например, в реакциях присоединения, элиминирования, замещения, перегруппировках и т. д., а также в некоторых реакциях окисления. Кроме того, они являются интермедиатами в ряде других процессов. Многие реакции карбанионов находят применение в синтезе, потому что они приводят к образованию углерод-углеродной связи.
10.5.1. Реакции присоединения
Выше уже была рассмотрена большая группа реакций, в которых карбанионы присоединяются к группе С=О (см. разд. 8.4), в том числе реакции внутримолекулярного присоединения карбанионов, например альдольная конденсация (см. разд. 8.4.4), реакция Дикмана (см. разд. 8.4.8) и бензиловая перегруппировка (см. разд. 8.4.10); было рассмотрено также присоединение к системе С=С—С=О (см. разд. 7.6.2) —реакция Михаэля.
10.5.1.1. Карбоксилирование
Интересной реакцией карбанионов, а также металлорганиче-ских соединений (источник карбанионов) является присоединение к очень слабому электрофилу СО2 с образованием соответ-
318
ствующего карбоксилат-аниона (36) — карбоксилирование:
R^)M+ О = С = 0 -
R I 0 = С-0“ м+
(36)
Эта реакция находит применение в синтезе. Она протекает с алкильными и арильными производными металлов, с ацетиленидами металлов (более электроположительных, чем магний), а также с реактивами Гриньяра. Обычно ее проводят, добавляя раствор металлорганического соединения в инертном растворителе к взятому в большом избытке твердому мелкоизмельчен-ному СО2. Особенно широко эту реакцию применяют для получения кислот ацетиленового ряда. Другим примером реакции такого типа является реакция Кольбе —Шмидта (см. разд. 10.5.3.3).
Реакцию карбоксилирования применяют при изучении карбанионов для обнаружения их образования путем превращения в устойчивые продукты, которые можно идентифицировать. Например, так было обнаружено значительное сохранение конфигурации в алкенильном карбанионе (38) при реакции соединения (37) с литием и последующем взаимодействии с СО2:
Mev Вг
\ = сх — н ЧМе
Me Li+ Me СОг Li+
с = Сх —с = с
п Me » '''ме
(37) (38) (39)
Выход соли (39) составил ~ 75. %, в то геометрического изомера не превысил 5 %.
время как выход ее
10.5.2. Реакции элиминирования
Ранее уже были рассмотрены реакции элиминирования, в которых карбанионы участвовали как интермедиаты, например карбанион (40), т. е. реакции, которые протекают по механизму £1сВ (см. разд. 9.4), например:
В^Н
I
PhSO2CH - СН2 * PhSO2CI-p- СН2 - PhSO2CH = сн2 Y *Y
(40)
К реакциям такого типа относится также декарбоксилирование.
319
10.5.2.1. Декарбоксилирование
Полагают, что отщепление СО2 от карбоксилат-анионов (41) проходит с образованием карбанионного интермедиата (42), который затем захватывает протон из растворителя или другого источника:
-Л о. н+
О — С R-------*- С 02 + R R — Н
ц медленно * быстро 11 О
(41) (42)
Отщепление СО2 является обычно скоростьлимитирующей стадией, т. е. кинетическое уравнение имеет вид
Скорость = fe[RCOj],
а последующее присоединение протона протекает быстро. Таким образом, декарбоксилирование должно ускоряться электроноакцепторными заместителями, которые могут стабилизировать карбанионный интермедиат (42) путем делокализации его отрицательного заряда. Это подтверждается гораздо более легким декарбоксилированием нитрозамещенного карбоксилат-иона (43) по сравнению с самим МегСНСОь:
О- С - CMe2NO2 --*
II -сог
О
Ме2С - N = О । О~
Iu+
—Me2CHNO2
Ме2С = N - О"
0"
(43) (44)
Декарбоксилирование легко протекает с Hal3CCH2CO2, 2,4,6-(O2N)3C6H2COi и др. Обычно эту реакцию не используют в препаративных целях для анионов простых алифатических кислот, кроме МеСО2. Участие карбанионных интермедиатов, например (44), подтверждается данными, полученными при декарбоксилировании в присутствии брома. Показано, что бром не влияет на суммарную скорость реакции, однако конечным продуктом реакции является МезСВгЬЮг, а не Me2CHNO2, хотя известно, что в условиях реакции ни Me2C(NO2)CO2, ни Me2CHNO2 не бромируются. Бромированный продукт (45) возникает при быстрой атаке бромом карбанионного интермедиата (44), ко-
320
торый таким образом «улавливается» (ср. катализируемое основанием бромирование кетонов; разд. 10.5.6):
Вг - Вг
Вг
I
Me2CNO2 - Me2CNO2 + Br"
(44) (45)
Группа С=О действует аналогично группе NO2; анионы
Р-оксокислот (46) декарбоксилируются очень легко:
- ♦ о
О - С - СН2СОМе --*
II -сог
о
(46)
СН3СОМе-
Найдено, однако, что суммарная скорость декарбоксилирования зависит от концентрации оксокислоты (47), а также от концентрации ее аниона. Быстрое декарбоксилирование р-оксо-кислоты (47), возможно, обусловлено переносом образующегося протона к группе С=О путем образования водородной связи:
Н
'"'О
^СМе
СН2
(47) (48)
± СО2 + СН3СОМе
Подтверждением такого механизма декарбоксилирования свободной кислоты может служить идентификация (путем «улавливания») енольного интермедиата (48). Возможно, что аналогично протекает декарбоксилирование р,у-ненасыщенных кислот (49)
xC<yCR
О С<2
(49)
° iiR2
.(X ^CR 0 "сн2
Н
XCR2
-CR
CH2
ЦП. Сайкс
321
а также a,p-ненасыщенных кислот R2CHCR=CHCO2H, так как было показано, что они изомеризуются в соответствующие р,у-ненасыщенные кислоты до декарбоксилирования.
Другим примером легкого декарбоксилирования свободной кислоты является декарбоксилирование пиридинкарбоновой-2 кислоты (50), которое идет гораздо легче, чем декарбоксилирование соответствующих 3- и 4-кислот, и проходит с образованием карбанионного интермедиата (51), который фактически является илидом:
Интермедиат (51) может быть «пойман» при проведении декарбоксилирования в присутствии карбонильных соединений, например PhCOMe; при этом образуется соответствующий продукт присоединения карбаниона, например (52). Этот процесс может быть использован в препаративных целях. Значительно более легкое декарбоксилирование соединения (50) по сравнению с его 3- и 4-изомерами объясняется тем, что в этом случае наблюдается стабилизация, обусловленная влиянием положительно заряженного атома азота на соседний атом углерода карбаниона в илидном интермедиате (51).
10.5.3. Реакции замещения
Карбанионы (или родственные частицы) участвуют в разнообразных реакциях замещения в качестве интермедиатов или атакующих нуклеофилов.
10.5.3.1. Цейтеро-водородный обмен
Показано, что кетон (53) обменивает свой а-водородный атом на дейтерий при обработке основанием (_OD) в растворе D2O. Если реакцию проводят с оптически активной формой этого соединения, происходит потеря оптической активности (ра
322
цемизация) с той же скоростью, с какой 'происходит дейтеро-водородный обмен. Когда аналогичное соединение, содержащее дейтерий вместо водорода, подвергали обмену в Н2О, то было найдено, что при сравнении скоростей обмена этих двух соединений обнаруживается кинетический изотопный эффект (йн/£о).
О -
О MeEtC—CPh О
II ~ОР d2o ||
> MeElL—СРп
। медленно быстро |
н О' D
_MeEtC=CPh_
(53) (+)
(54)
Все это позволяет предположить, что медленной, скоростьлимитирующей стадией является разрыв связи С—Н с образованием стабилизированного карбанионного интермедиата (54); затем происходит быстрый захват D+ нз растворителя D2O. Потеря оптической активности происходит при каждом акте разрыва связи С—Н, поскольку связи карбанионного атома углерода должны принять плоскую конфигурацию, если имеет место стабилизация вследствие делокализации с соседней группой С=О. Последующее присоединение D+ может происходить, следовательно, равновероятно с обеих сторон. Такое медленное, ско-ростьлимитирующее образование карбанионного интермедиата, сопровождаемое быстрой электрофильной атакой для завершения общего замещения, формально сходно со скоростьлимити-рующим образованием карбокатиона в механизме SnI, поэтому его обозначают как SeI-
10.5.3.2. Реакции карбанионных нуклеофилов
Как сами карбанионы, так и металлорганические соединения, например реактивы Гриньяра, являются сильными нуклеофилами, как это было показано в реакциях их присоединения к группе С=О (см. разд. 8.4), поэтому реакции замещения с участием этих реагентов идут по механизму Sn2. В препаративных целях особенно широко применяют карбанионы, полученные из так называемых активных метиленовых соединений — CH2(CO2Et)2, p-оксоэфиров, р-дикетонов, например (55), а-циа-ноэфиров, нитроалканов и др.:
"OEt - RBr
(МеСО)2СН2 <- (МеСО)2СН -------->- (МеСО)2СН—R + Вг~
(55) ‘ (56)
П* 323
5м2-Характер этого процесса подтвержден кинетически; в соответствующих случаях обнаружено обращение конфигурации у углеродного атома, атакуемого RBr. Алкилированный продукт (56) также содержит «кислый» атом водорода, и процесс может повториться с образованием диалкилированного продукта (MeCOJzCRR'. Широко применяется алкилирование ацетилид-иона (57):
“NH2 RBr
НС=СН НС=С“ -------------► НС==С—R + Вг'
(57)
В этом случае также возможно второе алкилирование с образованием RC = CR или R'C = CR. Следует, однако, отметить, что приведенные выше карбанионы, особенно ацетилид-ион (57), являются анионами очень слабых кислот, а следовательно, сами являются сильными основаниями, а также сильными нуклеофилами. Вследствие этого они могут способствовать как элиминированию (см. разд. 9.5), так и замещению. Реакция с третичными галогенидами поэтому часто приводит к образованию алкена, а не продукта алкилирования.
Реактивы Гриньяра также могут действовать как источники отрицательно заряженного атома углерода в реакциях замещения, например, в важной реакции с триэтоксиметаном (этилортоформиат) (58) с образованием ацеталей (59), а затем альдегидов (60):
6- н+, н2о „
R / CH(OEt)2 —* RCH(OEt)2--------*RCHO
„ I-7 rl
Brl^g 'OEt
(58) (59) (60)
В подходящих условиях можно получить алкильные производные более электроположительных металлов, например натрия, и затем провести их взаимодействие с алкилгалогенидами (реакция Вюрца):
2Na- R'Br
RGH2CH2—Cl ----► RCH2CH2 Na+ -----> RCH2CH2R'
(61)
Подтверждением такого механизма с участием карбанионов (в определенных условиях, однако, возможно участие радикалов) является тот факт, что в некоторых случаях при использовании оптически активных галогенидов можно обнаружить обращение конфигурации у атома углерода, подвергающегося нуклеофильной атаке. Карбанион, например (61), может дей
324
ствовать и как основа-ние и вызывать элиминирование:
R Н
I ГЧ I
RCH2CH2 Н-^СН^-СН2-Cl Na+-RCH2CH2+RCH = CH2+NaCI (61) (62) (63)
Это приводит к диспропорционированию с образованием алкана (62) и алкена (63). Такая реакция часто является побочной реакцией при проведении реакции Вюрца.
Интересное внутримолекулярное замещение происходит в реакции Дарзана, в которой карбанионы, полученные из а-гало-генэфиров, реагируют с карбонильными соединениями с образованием а-эпоксиэфиров:
а-хлорэфир карбанион
R. С? Д'
Eto2c Cl R'
И, ₽-эпоксиэфир
енолят-анион
Иногда, например в случае а-хлорэфиров, удается выделить енолят-анионный интермедиат.
10.5.3.3. Реакция Раймера— Тимана
В этой реакции участвуют ионы (64), а также ион ~СС13, получаемый при действии сильных оснований на НСС13 (см. разд. 9.8); ион _СС13 существует только в переходном состоянии, распадаясь затем до дихлоркарбена СС12 — высокоэлектронодефицитного электрофила, который атакует ароматическое ядро:
(64а) (646) (65)
325
Конечным продуктом реакции, образующимся после подкисления, является о-гидроксиальдегид (66) (салициловый альдегид); в небольшом количестве образуется его пара-изомер. Однако, если оба орто-положения в исходном феноксид-ионе замещены, образуется п-гидроксиальдегид.
Справедливость приведенного выше механизма подтверждается данными, полученными в аналогичной реакции с анионом n-гидрокситолуола (п-крезол) (67):
В этом случае кроме ожидаемого о-гидроксиальдегида (68) удается выделить негидролизованное дихлорпроизводное (70). Атака дихлоркарбеном пара-положения иона (67в) приводит к интермедиату (69); этот интермедиат, в отличие от интермедиата, образующегося при атаке в орто-положение, не имеет атома водорода, который можно было бы оторвать в виде Н+ для восстановления ароматичности. При подкислении ион (69) присоединяет протон с образованием соединения (70). Дихлорпроизводное (70) устойчиво к гидролизу отчасти из-за его нерастворимости в водно-щелочной среде, а также из-за стерически затрудненных атомов хлора — их окружение в соединении (70) подобно структуре неопентила (ср. разд. 4.3).
В несколько аналогичной реакции Кольбе — Шмидта СОг действует как электрофил, атакуя феноксид натрия (646):
326
Продуктом является в основном о-гидроксибензоат (салицилат) натрия (71), образуются лишь следы лара-изомера. Однако если реакцию проводят с феноксидом калия, то основным продуктом является л-гидроксибензоат. Предполагают, что преимущественное образование о-гидроксибензоата при использовании феноксида натрия связано с тем, что образующееся в результате атаки орто-положения переходное состояние (72) стабилизировано в результате образования хелатного соединения в виде ионной пары:
(72)
Размеры катиона К+ больше, и, вероятно, он менее эффективен в качестве хелатирующего агента, так что атака лара-положения становится поэтому более предпочтительной.
10.5.4. Перегруппировки
Перегруппировки с участием карбанионов, как показано, гораздо менее распространены, чем формально аналогичные им перегруппировки с участием карбокатионов (см. разд. 5.4). Это становится понятнее при сравнении переходных состояний для 1,2-сдвига алкильной группы в карбокатионе и карбанионе:
Д+ I*
/С'—С\ J карбокатионное ПС (2е)
карбанионное ПС
(4е)
Первое включает делокализацию двух электронов (электронов, принадлежавших исходной связи R—С), в то время как второе — делокализацию четырех электронов. Два электрона могут быть размещены на доступных связывающих молекулярных орбиталях, а дополнительные два электрона в карбанионном переходном состоянии могут быть размещены только на разрыхляющей молекулярной орбитали с более высокой энергией. Известны, однако, также 1,2-сдвнги арильных групп, например, в реакции хлорида (73) с натрием; однако в этом случае возможна некоторая стабилизация карбанионного переходного состояния в результате делокализации дополнительных
327
электронов с участием мигрирующей фенильной группы:
Ph Na+ Ph
Na I _ _ I
Ph3C—CH2C1 -----> Ph2C—CH2 Na+ —► Ph2C—CH2
(73) (74) (75)
Na* ~O2C H
Ph2C— CH2Ph Ph2i—CH2Ph
(77) (76)
Как и следовало ожидать, продуктом является алкилнатрий, однако при последующих протонировании и карбоксилировании образуются перегруппированные продукты (76) и (77) соответственно. Не выяснено,образуется ли сначала неперегруппировав-шийся алкилнатрий (74), который затем перегруппировывается, или же отщепление атома хлора и миграция фенильной группы происходят в значительной степени согласованно, так что непосредственно образуется перегруппированный алкилнатрий (75). Однако если вместо натрия использовать литий, то можно получить неперегруппированный алкиллитий, соответствующий (74), что подтверждается образованием соответствующих продуктов протонирования и карбоксилирования; последующая перегруппировка возможна при повышении температуры. Найдено, что способность соединения (73) к перегруппировке при взаимодействии с металлами или их производными уменьшается в ряду: К ~ Na > Li > Mg, т. е. в порядке уменьшения ионного характера связи углерод—металл. Эти данные в сочетании с данными, полученными при изучении относительной способности к миграции лара-замещенных арильных групп (предполагают, что мигрирует Аг+, а не Аг»), подтверждает предположение о том, что 1,2-сдвиг происходит с образованием карбанионов, а не радикалов.
Простые 1,2-сдвиги алкильной группы от углерода к углероду, которые бы имели карбанионный характер, практически неизвестны. Тем не менее известны примеры, когда 1,2-сдвиг алкильной группы происходит от других атомов, например от атома азота или серы, к атому углерода карбаниона — перегруппировка Стивенса-.
Н Li+
+ I PhLi +
Me2N—CHPh -------> Me3N—CHPh —> Me2N—CHPh
I I I
Me Me Me
(78)
328
н
+ I “ОН + -
MeS—CHCOPh MeS — CHCOPh •—> MeS—CHCOPh
I 1 1
PhCH2 PhCH2 PhCH2
(79)
Имеются, однако, данные, на основании которых можно предположить, что в некоторых реакциях могут участвовать радикальные, а не карбанионные интермедиаты. Для отрыва протона от положительно заряженной частицы (78), не содержащей электроноакцепторного заместителя типа С=О-группы, как, например, в соединении (79) необходимо использовать очень сильные основания, например PhLi. Выяснено, что миграция группы PhCH2, например, в соединении (79) предпочтительнее миграции метильной группы; поскольку группа PhCH2 более стабильна (ср. разд. 5.2). Аллиловые и бензиловые простые эфиры, например (80), претерпевают аналогичную перегруппировку Виттита (не путать с реакцией Виттига в синтезе алкенов; см. разд. 8.4.1.1):
Н Li+
| PhLi - Н+, Н2О
О—CHPh -► О—CHPh —> Li+ “О—CHPh -► НО—CHPh
II II
Me Me Me Me
(80)
Наконец, существуют индуцируемые основанием перегруппировки с участием карбанионов, которые протекают путем 1,3-элиминиров.ания с образованием циклопропаноновых интермедиатов, например (82), — перегруппировка Фаворского для а-галогенкетонов, например (81):
/С<° -он ^С = О HoQ,c^O PhHC CH2CI»=*phHC CH2-CI—*PhHC—^СН2 —*•
но^н
(81) (82)
НО^С-СГ НО-С = О
—- PhHC——-СН2—»- PhCH - СН2 —-~PhCH2CH2COf
(83) (84)
Образовавшийся интермедиат (82) присоединяет -ОН, после чего происходит раскрытие цикла с образованием более
329
устойчивого (из двух возможных) карбаниона (83) (бензил >• > Р-фенилэтил). Затем следует миграция протона и образование карбоксилат-иона (84).
10.5.5. Реакции окисления
Карбанионы могут быть окислены в соответствующих условиях; например, трифенилметил-анион (85) очень медленно окисляется кислородом воздуха:
Ph3C’ Na+ 3=* Ph3C • + NaO2. Na/Hg
(85) (86) (87)
Образующийся радикал (86) в свою очередь может быть восстановлен обратно до карбаниона путем встряхивания с амальгамой натрия. В некоторых случаях окисление карбанионов, например (88), одноэлектронными окисляющими агентами, обычно иодом, может быть использовано для образования углерод-углеродной связи, так как образующийся радикал (89) затем димеризуется:
(МеСО)2СН (МеСО)2СН (МеСО)2СН
- — : — I
(МеСО)2СН (МеСО)2СН (МеСО)2СН
(88) (89) (90)
Окислительная димеризация алкинов RC = CH под действием солей меди (II), например ацетатов, в растворе пиридина также находит применение в синтезе:
2RO=C~
Cu2+
--->• 2RC==C • —► RC==C—C=CR
Вероятно, что ацетиленид-анион, образующийся в щелочном растворе,-окисляется под действием меди(II) (другой одноэлектронный окислительный агент) до соответствующего радикала, который затем димеризуется.
10.5.6. Галогенирование кетонов
Одно из наиболее ранних наблюдений, позволивших высказать предположение о возникновении карбанионов в качестве интермедиатов реакций, состояло в том, что бро\Й1рование ацетона в присутствии водного раствора щелочи подчиняется кине
330
тическому уравнению
Скорость = /г[МеСОМе] [ ОН]
и не зависит от концентрации брома. Позднее было показано, что в аналогичных условиях иодирование происходит с такой же скоростью, как и бромирование, что также согласуется с приведенным выше кинетическим уравнением. Как уже отмечалось (см. разд. 10.5.3.1), индуцируемый основанием дейтеро-водородный обмен (в D2O) и рацемизация оптически активного кетона (53) происходят с одной и той же скоростью и обнаруживают кинетический изотопный эффект (&н>&о), когда а-Н-атом замещается дейтерием, т. е. расщепление связи С—Н является медленной, скоростьлимитирующей стадией. Все это свидетельствует об образовании карбанионного интермедиата, например (91):
О О6- О
« И "ОН 6- ;i Hal, II
MeEtC - CPh <----MeEtC—CPh ----------- MeEtC - CPh
медлен- быстро I
но Hal
(91) (92)(±)
быстро|d20
MeEtC - COPh (93)(±)
I D
Этот интермедиат затем быстро атакуется каким-либо электрофилом — С12, Вг2, I2, Н2О, D2O и т. д. — с образованием конечных продуктов реакции, таких, как (92), (93) и др., скорости образования которых одинаковы. Этот процесс формально напоминает медленную, скоростьлимитирующую стадию образования карбокатионного интермедиата с последующей быстрой нуклеофильной атакой в механизме SnI; поэтому этот процесс обозначается как SeI-
В случае кетонов типа (94), которые имеют альтернативные группы а-Н-атомов, возникают два вопроса: а) какая из групп— СН2 или СНз—атакуется преимущественно; б) если один из атомов водорода замещается галогеном, будет ли второй галоген связываться с тем же самым а-углеродным атомом или с Другим? Было показано, что при бромировании, например, МеСН2СОСН3 I- и 3-бромбутаноны образуются практически в равных количествах (оба эти бромкетона затем очень быстро подвергаются дальнейшей реакции; ср. разд. 10.5.6). Индуктивный эффект, проявляемый простой алкильной группой R таким образом, по-видимому, оказывает относительно небольшое влияние на кислотность атома Н(2) или на устойчивость
331
образующегося аниона (96):
(1)
(2)Н Н(1)
I 4
R-СН—С—СН2
О
~он
медленно
rch2c - сн2— rch2c = сн2
0‘ 6“
(95)
(2) Г "]
R-CH-CCH3-R-CH = CCH3 О 6“
(96)
Найдено также, что введение в молекулу кетона атома галогена, например Вг, как в случае соединения (97), очень заметно влияет на направление дальнейшего галогенирования:
(2)Н н(1)
* 4
R-CH-»C—СН-Вг II О
(97)
-он
медленно
RCH2C - СН—Вг - RCH2C = СН—Вг
О 0-
(98)
R—СН - CCH2Br — R—СН = ССН2Вг
О 0“
(99)
Сильный электроноакцепторный индуктивный эффект и эффект поля, проявляемый бромом, увеличивают кислотность а-Н-атомов группы СН2Вг по сравнению с а-Н-атомами группы RCH2 и могут способствовать также стабилизации образующегося карбаниона (98) по сравнению с (99). Таким образом, образование карбаниона (98) будет более предпочтительным, и дальнейшее бромирование, по-видимому, будет идти по группе СН2Вг, а не по группе RCH2. Кроме того, из-за электроноакцепторного эффекта, проявляемого атомом брома, анион (98) образуется быстрее, чем, например, анион (95), т. е. второе бромирование будет идти быстрее первого, а третье бромирование группы СН3 будет идти еще быстрее. Следовательно, можно ожидать, что конечным продуктом эт^го катализируемого основанием галогенирования будет RCH2COCX3 (100). Возможно, однако, обратимое присоединение -ОН к группе С=О этого кетона; группа СХз в этом случае может оказаться хорошей уходящей группой, в результате чего происходит разрыв связи
332
С—С (ср. разд. 8.6):
- G? ft В
RCH2C**CX3-₽*RCH2C ^СХ3—RCH2C +CX3^RCH2C + НСХ3
Г*" I | I
он он ОН 0-
(100) (101) (102) (103)
Группа СХ3 является хорошей уходящей группой из-за электроноакцепторного индуктивного эффекта трех атомов галогена; это активирует атом углерода карбонильной группы в соединении (ЮО) для нуклеофильной атаки, а также стабилизирует отщепляющийся анион (101). Конечным продуктом кроме карбо-ксилат-иона (102) является галоформ (103). Суммарный процесс
RCHjCOCrii —> RCH2COj + НСХз
известен как галоформная реакция. Ее используют для обнаружения метнлкетонов с применением нода и водного раствора щелочи; образующийся при этом СН31 (йодоформ) представляет собой нерастворимый в реакционной среде продукт желтого цвета с характерным запахом.
Галогенирование кетонов катализируется также кислотами (общий кислотный катализ; ср. разд. 3.3); эта реакция подчиняется кинетическому уравнению
Скорость = й[Кетон] [Кислота].
Как и в случае катализируемой основанием реакции, скорости бромирования, иодирования, дейтеро-водородного обмена н рацемизации идентичны. Промежуточным соединением, образующимся на медленной, скоростьлимитирующей стадии, является обычно енол (104)
О 4 Н - А А~ Н О-Н
IK I
СН3 - СМе Ч=е СН-?- СМе
медлен но
О,-Н<А
СН^СМе Вг
(Ю4)
А
СН2-С
О Н-А Me Br~ Вг
который подвергается затем быстрой атаке бромом или другим нуклеофилом.
333
Чтобы решить, а-Н-атомы какой из групп будут преимущественно замещаться в RCH2COCH3, необходимо сравнить возможности образования соответствующих енолов (105) и (107):
он
I ВГ2
RCH=CMe ----->
(Ю5)
О
II
RCH—СМе I
Вг
(Ю6)
ОН о
I ВГ2 II
rch2c=ch2 -—>- RCH2C—сн2
I
Вг
(107) (108)
Енол (105), вероятно, более устойчив, чем енол (107), так как он содержит сильно замещенную двойную связь (ср. разд. 1.5.4). Поэтому можно полагать, что преимущественным продуктом бромирования будет соединение (106). В самом деле, при катализируемом кислотой бромировании М.еСН2СОСН3 выход 3-бромбутанона (106; R — Me) в три раза выше выхода 1-бромбутанона (108; R = Me).
Обнаружено также, что, в отличне от бромирования, проводимого в условиях основного катализа, дальнейшее введение брома в монобромкетон идет с большим трудом, чем введение первого атома брома. Таким образом, в кислой среде бромирование можно остановить на стадии образования монобромпроиз-водного, например (106), тогда как в условиях основного катализа дальнейшее бромирование не может быть предотвращено, что в ряде случаев может привести к отщеплению галоформа (см. выше).
Причиной затруднения дальнейшей атаки галогенкетонов в условиях кислотного катализа является то, что промежуточное соединение (и переходное состояние), участвующее в образовании енола, например в образовании (104) из СН3СОМе, несет положительный заряд. Соответствующий положительно заряженный интермедиат, участвующий в образовании енола из ВгСН2СОМе, будет, таким образом, дестабилизирован (по сравнению с енолом из СН3СОМе) вследствие электроноакцепторного индуктивного эффекта и эффекта поля, проявляемого атомом брома. Исходный СН3СОМе будет, следовательно, подвергаться енолизации и последующему (быстрому) бромированию предпочтительнее, чем ВгСН2СОМ.е. Показано, что при дальнейшем бромировании основным продуктом является 1,1-ди-бромкетон Вг2СНСОМе, однако в условиях этой реакции 1,1-дибромкетон частично изомеризуется в 1,3-дибромкетон ВгСН2СОСН2Вг.
334
Глава 11
РАДИКАЛЫ И ИХ РЕАКЦИИ
11.1. Введение.................................................335
11.2. Образование радикалов....................................339
11.2.1. Фотолиз...........................................339
11.2.2. Термолиз..........................................341
11.2.3. Окислительно-восстановительные реакции............342
11.3. Методы обнаружения радикалов.............................343
11.4. Пространственное строение и стабилизация радикалов.......346
11.5. Реакции радикалов........................................350
11.5.1. Реакции присоединения.............................351
11.5.1.1. Присоединение галогенов...................351
11.5.1.2. Присоединение бромоводорода...............354
11.5.1.3. Другие реакции присоединения ........ 358
11.5.1.4. Винильная полимеризация ..................359
11.5.2. Реакции замещения.................................363
11.5.2.1. Галогенирование...........................363
11.5.2.2. Автоокисление.............................368
11.5.2.3. Ароматическое замещение................. 372
11.5.3. Перегруппировки...................................377
11.6. Бирадикалы...............................................379
11.1. Введение
Большая часть рассмотренных ранее реакций протекает с участием полярных реагентов и интермедиатов, т. е. карбокатионов и карбанионов или родственных высокополяризованных частиц; в ходе этих реакций происходит гетеролитический разрыв и образование ковалентных связей, например:
R3C: Х+ RaC—X R3C+ : X’
Однако возможен и гомолитический разрыв ковалентной связи, в результате которого образуются частицы, обладающие неспаренным электроном, — радикалы, например (1) и (2):
RsC—X < -A- R3C • ~Г • X
(1) (2)
Энергия гомолитического разрыва связи R3C—X в газовой фазе всегда меньше, чем энергия гетеролитического разрыва. Одиако при проведении реакции в полярных растворителях эта разница в энергиях уменьшается, поскольку при сольватации образующихся в ходе гетеролитического разрыва ионов выделяется энергия.
335
Реакции с участием радикалов часто протекают в газовой фазе; горение любого органического соединения — почти всегда радикальная реакция, а окислительное расщепление алканов в двигателях внутреннего сгорания является наиболее широко распространенной химической реакцией. Радикальные реакции протекают также и в растворе, особенно, если они проводятся в неполярных растворителях и если они катализируются светом или веществами, способными разлагаться с образованием радикалов, например органическими пероксидами. Радикалы, образовавшиеся в растворе, обычно менее избирательны при атаке других частиц или альтернативных положений в одной и той же частице по сравнению с карбокатионами или карбанионами.
Радикальные реакции, однажды начавшись, часто протекают с большой скоростью вследствие развития цепных процессов с низким расходом энергии, как, например, в случае галогенирования алканов (3) (ср. разд. 11.5.2.1):
Вг—Вг
|Av
R—Н + • Вг —> R • + Н—Вг
(3) f I
Вг2
• Вг + R—Вг
В этом случае радикал Вг •, полученный фотохимически, при взаимодействии с нейтральным субстратом R—Н (3), генерирует другой радикал R •, который в свою очередь реагирует с другой молекулой Вг2, снова давая Вг •; цикл, таким образом, протекает как цепная реакция, не требующая в дальнейшем фотохимического образования Вг • т. е. развивающаяся самостоятельно.
Радикальные реакции могут ингибироваться путем введения веществ, которые сами особенно легко реагируют с радикалами (ингибиторы, или «улавливатели:», радикалов), например фенолов, хиионов, дифениламина, иода и др. Эти и родственные нм вещества можно использовать для того, чтобы остановить уже начавшуюся радикальную реакцию.
Первыми радикалами, которые были изучены, оказались менее реакционноспособные радикалы, способные к длительному существованию. Первым таким радикалом был Ph3C *(4), полученный в 1900 г. при взаимодействии Ph3CCl с тонкоизмель-чеиным серебром (ср. разд. 2.3.1). Этот радикал реагировал с галогенами, образуя трифенилметилгалогенид (5), или с кислородом воздуха, образуя пероксид (6) (все радикалы легко реа-
336
Гцруют с кислородом воздуха):
Ph3C-
Ph3C • + X—X —► Ph3C—X + X • ----2Ph3C—X
(4) (5) (5)
Ph3C.
Ph3C • + O2 —► Ph3COO • -->• Ph3COOCPh3
(4) (6)
Радикал (4), окрашенный в желтый цвет, находился в равновесии в растворе инертного растворителя с бесцветным димером, причем доля радикала увеличивается при разведении и при повышении температуры. Так, разбавленный раствор димера в бензоле содержит « 2 % Ph3C ° при 20 °C и « 10 % при 80 °C; при удалении растворителя получается только димер. Естественно, предполагали, что димер представляет собой гексафенилэтан Ph3C—CPh3, и, как уже упоминалось (см. разд. 2.3), только спустя 70 лет методом спектроскопии ЯМ.Р ‘Н было показано, что этот димер имеет строение (7):
Ph3C\/=\
H)Q=CPb, <7)
Гексафенилэтан даже не был получен и вряд ли мог бы существовать в обычных условиях вследствие чрезвычайно высоких стерических затруднений. Причины относительно высокой устойчивости Ph3C • обсуждаются ниже (см. разд. 11.4).
Простые алкильные радикалы гораздо более реакционноспособны и впервые были систематически изучены только в 1929 г. Радикалы были получены термическим разложением металлор-ганических соединений типа РЬМе4 при пропускании их через стеклянную трубку в струе инертного газа-носителя, например азота:
PbMe, Pb + 4Ме .
Было обнаружено, что тонкое свинцовое зеркало, отложенное на различных расстояниях на внутренней стенке трубки, атакуется радикалами и исчезает. Измеряя расстояние, на которое распространилась атака радикалов и зная скорость пропускания газа-носителя, можно точно определить полупериод жизни алкильных радикалов; для радикалов Ме , в частности, он составлял 8-10-3 с. При отсутствии металлического зеркала, которое можно атаковать, такие алкильные радикалы обычно димеризуются:
СН3.+ .СН3 —>- СН3—СН3
Позднее было обнаружено, что радикалы в качестве интермедиатов участвуют в целом ряде реакций (см. ниже).
337
Радикалы различной степени устойчивости, содержащие кроме атома углерода и другие атомы, — гетерорадикалы — также были выявлены. Так, в 1911 г. было обнаружено, что нагревание Л/',Л/',Л/",./У/-тетраарилгидразинов, например (8), в неполярном растворителе приводит к образованию окрашенного в зеленый цвет радикала (9):
мпо;
2Ph2NH --------► Ph2N—NPh2 Ph2N* + «NPh2
(8) (9)
Другим важным азотсодержащим радикалом является 1,1-ди-фенил-2-пикрилгидразил (11), полученный окислением триарил-гидразина (10) PbOj:
NHNPh2 NNPh2
no2 no2
(10) (11)
Этот радикал достаточно устойчив (о причинах его устойчивости см. разд. 11.4), и его можно подвергнуть перекристаллизации из различных растворителей; он получается в виде фиолетовых призм, которые могут храниться более или менее долгое время. Радикал (11) относительно нереакционноспособен по отношению к нейтральным молекулам, но легко реагирует с другими радикалами; его используют в качестве «ловушки», так как он образует устойчивые продукты, например (12), почти с любым другим радикалом Ra ;
Ph2N—NAr + Ra .
/Ra
Ph2N—
\Ar
(H)
(12)
Растворы радикала интенсивно окрашены, а при его взаимодействии с другими радикалами образуются бесцветные продукты, что можно контролировать колориметрически.
Было найдено, что растворы дифенилдисульфида (13) при нагревании окрашиваются в желтый цвет, а при охлаждении окраска снова исчезает:
А
PhS—SPh PhS. + «SPh
(13) (14)
338
Образующиеся при этом радикалы (14) могут быть «пойманы», например, радикалом (11). Простые алкилтиильные радикалы типа MeS • были обнаружены в качестве промежуточных соединений ряда реакций; они очень реакционноспособны. Известны также относительно устойчивые кислородсодержащие радикалы, например феноксильный радикал (16):
Этот радикал существует в мономерной форме (а не в виде димера) как в растворе, так и в твердом состоянии и представляет собой твердое вещество синего цвета (т. пл. 97°C). Его относительная устойчивость объясняется, почти наверное, пространственными затруднениями, создаваемыми объемистыми группами СМ.е3 в обоих орто-положениях для подхода другой молекулы (16) или какой-либо другой частицы к атому кислорода радикала.
11.2. Образование радикалов
Существует ряд путей образования радикалов из нейтральных молекул. Наиболее важными из них являются: фотолиз, термолиз и окислительно-восстановительные реакции с переносом одного электрона при участии неорганических ионов, металлов или в ходе электролиза.
11.2.1. Фотолиз
Этот метод основан на способности молекулы, подвергаемой фотолизу, поглощать излучение в ультрафиолетовой или видимой области спектра. Так, ацетон в газовой фазе разлагается под действием света с длиной волны « 320 нм (ftv = — 375 кДж/моль) :
О
II Av
Me—С—Me -----
О
Me • + • С—Me
СО+ 2. Me
(17) (18) (17)
Разложение происходит потому, что карбонильные соединения поглощают излучение именно в этой области. При фотохимическом разложении ацетона сначала образуются радикалы
339
(17) и (18); последний затем самопроизвольно разлагается с об/ разованием метильного радикала и устойчивой частицы СО. Фотолизу легко подвергаются также алкилгипохлориты (19) и Нитриты (21); оба эти процесса можно использовать для генерирования алкоксильных радикалов (20):
RO—С1 — > RO. + .C1 RO—NO —RO • + • NO
(19) (20) (21) (20)
Другим важным примером фотолиза является превращение молекулярного галогена в атомарный
hv hv
Cl—Cl ----► C1- + .C1 Вг—Вг ----► Br*+*Br
являющееся первой стадией, например, галогенирования алканов (см. разд. 11.5.2) илн присоединения галогенов к алкенам (см. разд. 11.5.1.f).
Основные преимущества фотолиза перед термолизом (см. ниже) при получении радикалов состоят в следующем: а) возможность расщепления прочных связей (которые расщепляются с трудом или не расщепляются совсем) при умеренных температурах, например, в азоалканах:
hv
R—N=N—R ------► R • + N=N + • R
б) поглощение молекулой энергии только одного определенного уровня, так что фотолиз является более специфическим методом проведения гомолиза, чем пиролиз. Так, при фотолизе диацилпероксидов, например (22), в отлнчие от термолиза, не протекают побочные реакции:
О О О
II II -hv ||
RCO—OCR ------► 2R—С—О. —*- 2R • + 2СО2
(22)
Эффективным способом генерирования радикалов является флеш-фотолиз, при котором используют очень сильный импульс излучения (видимого илн УФ) очень короткой длительности. Это приводит к высокой мгновенной концентрации радикалов, которые могут быть обнаружены (как и продукты их превращений) спектроскопическим путем посредством нх облучения одним или большим числом последующих импульсов света меньшей интенсивности при соответствующей длине волны. Этот способ используют для изучения радикалов, а не для их препаративного получения. В некоторых случаях радикалы могут быть получены также путем облучения нейтральных молекул рентгеновскими лучами или у-лучами, т. е. путем радиолиза.
340
11.2.2. Термолиз
В ранних работах короткоживущие радикалы (см. разд. 11.1) получали в газовой фазе путем разложения алкильных производных металлов, например (23):
PbR4 Pb-|-4R.
(23)
Связи Pb—R лабильны, т. е. легко подвергаются термическому разрыву, и радикалы могут быть генерированы как в растворе инертного растворителя, так и в газовой фазе; энергия диссоциации этих связей ниже « 165 кДж/моль (40 ккал/моль). В образовании таких связей часто участвуют не только атомы углерода, но и атомы других элементов; главным источником радикалов в растворе является термолиз подходящих пероксидов (связи О—О) и азосоединений (связи С—N). Однако, если субстрат не содержит заместителей, способных стабилизировать образующийся радикал или вызывающих первоначальное разложение пероксида, для разрыва таких связей необходимы относительно жесткие условия. Так, (Ме3ССОО)2 имеет полупериод жизни «200 ч при 100°C, тогда как (PhCOO)2 при той же температуре имеет полупериод жизни только « 0,5 ч. Как уже упоминалось выше, простые алкилазосоединения слишком устойчивы и не могут подвергаться термолизу при умеренных температурах, но при введении подходящих заместителей, как, например, в соединении (24), могут стать источниками радикалов:
CN CN
I I А .
Ме2С—N=N—СМе2 -----► 2[Ме2С—C=N ч—► Me2C=C=N] + N=N
(24)
В частности, MeN = NMe, несмотря на то, что гоуппа N = N, является наилучшей из всех уходящих групп, устойчив до « 200 °C, в то время как (24) имеет полупериод жизни только «5 мин при 100°C.
В отсутствие других частиц, с которыми радикал может реагировать (например, отрыв Н из подходящего растворителя), радикалы в основном димеризуются
СН3СН2-+ • СН2СН3--> СН3СН2—СН2СН3
или подвергаются диспропорционированию:
СН3СН2. + н—сн2сн2. —> СН3СН3 + сн2=сн2
Использование PbEt4 как антидетонирующего агента в бензине зависит отчасти от способности этильных радикалов, образующихся при его термическом разложении, соединяться
341
с радикалами, возникающими при сверхбыстром сгорании углеводородов бензина. Таким образом, эффективно обрываются цепные реакции, ведущие к взрыву (детонации). Полностью механизм действия PbEt4 не выяснен, но имеются некоторые данные о том, что мельчайшие частицы РЬО, образующиеся из PbEt4, также могут ингибировать цепной процесс.
Образование радикалов путем разрыва углерод-углеродной связи наблюдается в индуцируемом радикалами крекинге длинноцепочечных алканов при » 600 °C. Радикалы, введенные в систему в начальный период, возможно, действуют, отрывая атом водорода от СН2-группы цепи; образующийся неконцевой радикал с длинной цепью (25) затем расщепляется в p-положении по отношению к углеродному атому радикала. Это приводит к образованию более низкомолекулярного алкена (26) и другого радикала (27), который поддерживает цепную реакцию:
Ra • Н
RCH—CH2R' „ ,> RCH—CH2R' ------> RCH=CH2 + .R'
—RaH
(25) (26) (27)
Прекращение реакции вследствие радикал-радикального взаимодействия вряд ли имеет место в заметной степени до тех пор, пока концентрация алкана с длинной цепью не станет очень мала.
11.2.3. Окислительно-восстановительные реакции
Генерирование радикалов при таких реакциях происходит путем одноэлектронного переноса; в них могут принимать участие такие ионы, как Fe2+ и Fe3+, а также Си+ и Си2+. Так, найдено, что ионы Си+ сильно ускоряют разложение ацилпероксидов, например (28):
[О 1 О
II + II
АгСО _|2 + Си —> АгС—О • -|- АгСО2 + Си2
(28) (29)
Эту реакцию можно использовать для генерирования радикалов АгСО2*, так как при термолизе соединения (28) возможно разложение радикала (29) до Аг»4-СО2. Ионы Си+ участвуют также в разложении солей диазония ArN2 СГ до АгС1 + + N2 (реакция Зандмейера), при котором весьма вероятно образование Аг- в качестве промежуточного соединения:
ArNj -)- Cu+ —> Ar • -)- N2 -)- Cu2+
Обе эти реакции являются реакциями восстановления. В реакциях окисления водными растворами пероксида водорода 342
в качестве катализатора используют ион Fe2+:
Н2О2 + Fe2+ —► НО • + “ОН + Fe3+
Эту смесь называют реагентом Фентона; действующим окисляющим агентом этой системы является радикал НО-. Этот радикал особенно хорошо отщепляет атом водорода и может быть использован либо для получения необходимого радикала, например (30), для дальнейшего изучения, либо в некоторых случаях для препаративного получения продукта димеризации (31) радикала (30):
(30)
НО • + Н—СН2СМе2ОН ——• СН2СМе2ОН ---------►
—Н2О
(30)
—► НОСМе2СН2СН2СМе2ОН
(31)
Прямое восстановление карбокатионов обычно не происходит, однако его можно осуществить при использовании, например, хлорида ванадия(П):
Ph3C+ + V2+ —> Ph3C. + V3+
Генерирование радикала в ходе окислительного процесса, возможно, происходит при инициировании автоокисления бензальдегида (см. разд. 11.5.1.3), которое катализируется рядом ионов тяжелых металлов, способных к одноэлектронному переносу, например Fe3+:
О О
II , II
PhC—H + Fe3+ —> PhC • + Н + Fe2
Ранее уже упоминалось (см. разд. 11.1) о получении устойчивого феноксильного радикала (16) путем одноэлектронного процесса окисления с участием Fe(CN)e
(l5) + Fe(CN)3- —> (16) + Fe(CN)46-
а также об окислении карбанионов, например (32), иодом (см. разд. 10.5.5) с образованием продукта димеризации радикала (33):
i2
2(МеСО)2СН ----► 2(МеСО)2СН --->- (МеСО)2СН—СН(СОМе)2
(32) (33)
Радикалы (34), которые впоследствии димеризуются, могут быть получены путем анодного окисления карбоксилат-ионов
343
RCO2 при электролитическом синтезе углеводородов по Кольбе:
2RCOj —2RCO2 > 2R- —> R—R анод —СО2
(34)
Электролиз кетонов (35) приводит к их катодному восстановлению в анион-радикалы (36), которые затем димеризуются, образуя дианионы (37):
-Ге • R2C—О н+ R2C—ОН
2R2C=O ----> 2R2C—О —► I -------► |
катод R2C—О’ R2C—ОН
(35) (36) (37)
Аналогичные анион-радикалы образуются при получении пинаконов восстановлением кетонов натрием или магнием (см. разд. 8.2.7), а также при восстановлении сложных эфиров натрием в ходе ацилоиновой конденсации (см. разд. 8.2.7).
Следует, однако, подчеркнуть, что все рассмотренные выше методы образования радикалов включают первоначальное генерирование радикалов из нейтральных молекул или из ионов. В действительности же радикалы часто образуются в ходе атаки подходящей частицы радикалами Ra •, заранее полученными специально для этой цели из предшественников, таких как пероксиды или азоалканы:
R—Н + Ra • —> R • + Н—Ra
СХ2=СХ2 + Ra • —► . СХ2—СХ2— Ra
11.3. Методы обнаружения радикалов
Мы уже видели, как высокая химическая активность короткоживущих радикалов может быть использована для их обнаружения по исчезновению металлического зеркала на внутренней стенке стеклянной трубки (см. разд. 11.1). Переход неспаренного электрона в радикале с одного энергетического уровня на другой требует меньше энергии, чем переход спаренных электронов в стабильной исходной молекуле; это означает, что радикал имеет тенденцию поглощать свет при более длинных волнах. Поэтому многие радикалы окрашены (тогда как их предшественники не окрашены) и могут быть легко обнаружены таким путем, например радикалы (11) и (16) (см. разд. 11.1). Радикалы могут быть также обнаружены по обесцвечиванию растворов, содержащих, например, такие частицы, как 1,1-дифенил-2-пикрилгидразил (11).
Другой химический метод обнаружения радикалов основан на их способности инициировать полимеризацию (см. разд. 11.5.1.4). Полимеризация может быть инициирована катио
344
нами, анионами, а также радикалами, однако действие этих частиц может быть различным. Так, при использовании в качестве субстрата смеси (50:50) фенилэтена (стирол) PhCH= = СНг и метил-2-метилпропеноата (метилметакрилат) СН2 = = С(Ме)СО2Ме было найдено, что под действием катионных инициаторов образуется только полистирол, под действием анионных — только полиметилметакрилат, в то время как радикалы вызывают образование сополимера, включающего равные количества этих двух мономеров.
Однако наиболее распространенным методом обнаружения радикалов является метод электронного парамагнитного резонанса (ЭПР). Этот метод использует постоянный магнитный момент, присущий радикалу за счет спина его неспаренного электрона (радикалы являются парамагнетиками). Спин электрона может быть равен -р/г или —’/г (ср. разд. 1.1), что в присутствии внешнего магнитного поля соответствует наличию у радикалов двух различных уровней энергии. Между этими уровнями возможны переходы, реализация которых делает возможной регистрацию характеристических спектров поглощения. Спектроскопия ЭПР неспаренных электронов является, таким образом, аналогом ЯМР-спектроскопии ядер (например, 'Н, 13С и др.), которые также имеют постоянный магнитный момент. Неудивительно поэтому, что эти переходы наблюдаются в различных областях спектра: неспаренный электрон имеет гораздо больший магнитный момент, чем протон, и для реализации соответствующего перехода требуется большая энергия.
Спектроскопия ЭПР регистрирует взаимодействие («расщепление») между неспаренным электроном и соседними магнитными ядрами, особенно 'Н, что приводит к очень сложному набору линий. Их анализ может дать много ценной информации о пространственной и электронной структуре радикала. Так, отщепление водорода от циклогептатриена (38) с помощью -ОН приводит к радикалу, имеющему очень простой ЭПР-спектр: восемь расположенных на одинаковом расстоянии друг от друга линий, указывающих на взаимодействие неспаренного электрона с семью эквивалентными ядрами 'Н. Образующийся радикал, следовательно, не может иметь структуру (39), которая должна была бы иметь гораздо более сложный спектр ЭПР; он должен быть делокализованной частицей в соответствии со структурой (40) (ср. разд. 5.2):
Н Н (38) (39) (40)
345
Радикалы методом спектроскопии ЭПР регистрируются, как правило, при низких концентрациях (« 10-8 моль/л). Исследуемые радикалы могут быть генерированы (например, облучением) непосредственно в кювете спектрометра. Если это не удается, то их можно генерировать и вне кюветы, применив в этом случае «метод остановленной струи» для поддержания постоянной их концентрации в кювете спектрометра. Недостатком этого метода является, однако, то, что в таком случае требуются относительно большие объемы и количества исходного вещества. Чем больше срок жизни радикала, тем больше возможность получения его спектра; например, такие частицы, как Ph3C-, легко наблюдать, а частицы, подобные Ph-, PhCH2-, С2Н5- и др., наблюдать намного труднее. Для «продления» жизни короткоживущих частиц вводят подходящее диамагнитное вещество, например (41), которое взаимодействует с промежуточно образовавшимся радикалом и превращает его в долгоживущий радикал (42), который может быть легко обнаружен:
Ra I Ra • + Ме3С—N=O —> Ме3С—N—О •
(41)
Такие соединения называют «спиновыми ловушками». Другой метод, который применяют для исследования короткоживущих радикалов, заключается в том, что их получают фотолитическим путем из предшественников в твердой инертной матрице, например в замороженном аргоне. Срок жизни радикалов тем самым искусственно удлиняется, поскольку исключается возможность их столкновения друг с другом и с другими частицами, что могло бы привести к прекращению их существования.
Необходимо подчеркнуть, что помимо этих специфических физических методов обнаружения радикалов существуют и более обычные методы, основанные на высокой чувствительности реакций, в которых возможно образование радикалов, к добавлению инициаторов или ингибиторов радикальных реакций (ср. разд. 11.5 и 11.1) и относительной нечувствительности (по сравнению с полярными реакциями) к изменению растворителя.
11.4. Пространственное строение и стабилизация радикалов
Как в случае карбокатионов (см. разд. 5.2) и карбанионов (см. разд. 10.3), возникает вопрос, располагается ли электрон в простых радикалах типа R3C • на р-орбитали [плоская форма (43)], или на sp-гибридной орбитали [пирамидальная форма
346
(44)] или на орбитали
(43)
промежуточной гибридизации.
(446)
r*""7 xr
R
(44а)
Прямое доказательство плоского строения радикала • СН3 было получено с помощью спектра ЭПР радикала 13СН3' Анализ линий, образованных при взаимодействии между неспаренным электроном и парамагнитным ядром |3С, дает информацию о степени s-характера орбитали, на которой располагается неспаренный электрон. Найдено, что в |3СН3’ вклад s-орбитали очень мал или отсутствует вовсе, следовательно, этот радикал является практически (отклонение составляет « 5 %) плоским, т. е. имеет строение (43). Этот вывод подкрепляется данными УФ- и ИК-спектров. Показано, что s-характер полузаполненной орбитали увеличивается в ряду:
•СН3 < -CH2F < .CHF2 < • CF3
В радикале • CF3 неспаренный электрон практически находится на sp-орбитали, а сам радикал, следовательно, имеет пирамидальную форму (44; R = F). Радикалы ‘СН2ОН и • СМе2ОН также заметно «изогнуты».
Сравнение легкости образования и реакционной способности мостиковых радикалов (45) и (46) с их ациклическими аналогами дает возможность предположить, что для алкильных радикалов действительно предпочтительнее плоское строение. Это нельзя сравнивать с предпочтительностью плоской конформации в случае карбокатионов; в отличие от карбокатионов (см. разд. 4.3), генерирование радикальных центров в мостиковых положениях не составляет особого труда.
(45) (46)
Относительная устойчивость простых алкильных радикалов уменьшается в ряду:
• CR3 > -CHR2 > -CH2r > -СН3
Это связано с относительной легкостью гомолитического разрыва связи С—Н в алкане — предшественнике и, что более важно, со снижением стабилизации вследствие гиперконъюгации или других факторов. В том же ряду уменьшается влияние
347
пространственного напряжения при переходе от хр3-гибридизо-ванного предшественника к хр2-гибридизованному радикалу. Однако относительное различие в устойчивости этих радикалов гораздо меньше, чем различие в устойчивости соответствующих карбокатионов.
Радикалы аллильного RCH=CHCH2 (47) и бензильного PhCHR (48) типов более устойчивы и менее реакционноспособны, чем простые алкильные радикалы, вследствие возможности делокализации неспаренного электрона в каждой из названных структур по всей л-орбитальной системе:
[RCH—СН—СН2]
(47) (48)
Оба радикала по существу являются плоскими, т. е. радикальный атом углерода хр2-гибридизован, поскольку только в этой конфигурации возможно максимальное перекрывание р-и л-орбиталей, что и определяет наблюдаемую стабилизацию. Устойчивость радикала увеличивается еще более при возрастании степени возможной делокализации; так, Ph2CH> более устойчив, чем PhCH2-, a Ph3C • (ср. разд. 11.1) — чрезвычайно устойчив.
Геометрия радикала Ph3C« (49) представляет особый интерес, так как она прекрасно иллюстрирует стабилизацию благодаря эффективной делокализации неспаренного электрона. Центральный углеродный атом радикала (49), безусловно, является £/?2-гибридизованным, т. е. связи, соединяющие его с тремя бензольными кольцами, лежат в одной плоскости. Максимальная стабилизация могла бы быть, однако, достигнута лишь тогда, когда все три бензольных кольца могли бы располагаться в одной плоскости [(49а)], так как только в этой конформации р-орбиталь центрального атома углерода может взаимодействовать одинаково и в максимальной степени с л-орбитальной системой каждого из трех ядер. В действительности триарилметильные радикалы, как было показано спектроскопическими и рентгеноструктурными методами, имеют форму пропеллера (496), причем бензольные кольца находятся под углом около 30° к общей плоскости.
(49а)
(496)
348
Таким образом, хотя делокализация в радикале (49) и имеет место (как определено с помощью спектров ЭПР), она не является максимальной и ненамного выше степени делокализации в радикале Ph2CH2*. Главная причина большей «стабильности» РЬ3С", которая выражается, в частности, в его большей устойчивости к димеризации, должна быть, следовательно, в основном стерической: затруднения возникают, когда два чрезвычайно объемистых радикала Ph3C • пытаются соединиться друг с другом. Из-за наличия таких пространственных затруднений димер, когда он образуется, имеет строение не ожидавшегося гексафенилэтана Ph3C—CPh3 (ср. разд. 11.1), а строение (7); реакция объемистого радикала Ph3C • с другим радикалом Ph3C • направляется в более легко доступное, а следовательно, реакционноспособное (вследствие делокализации электрона) положение:
OPh3C, /==\ =CPh2 V )=CPh2
(7)
Бензольные кольца выталкиваются из копланарной конформации вследствие пространственного взаимодействия о-Н-ато-мов соседних ядер друг с другом. Как и следовало ожидать, было найдено, что о-заместители, более объемистые, чем атом водорода, увеличивают диэдральный угол (поворот кольца по отношению к положению в копланарной конформации) ароматического кольца до 50° и более. В этом случае делокализация должна снижаться еще более, однако радикалы с объемистыми о-заместителями тем не менее более устойчивы, т. е. еще более неохотно образуют димеры, чем Ph3C-. Это, конечно, должно быть обусловлено стерическим эффектом: о-заместители очень близко расположены к радикальному атому углерода и поэтому мешают его приближению к другой частице или мешают другим частицам приближаться к нему [ср. (16); разд. 11.1]. В свете сказанного выше важно, что эффективность о-заместителей в экранировании радикального атома углерода увеличивается по мере того, как бензольные кольца отклоняются от копланарной конформации, т. е. с увеличением диэдрального угла.
Если каждое ароматическое ядро в радикале имеет объемистый заместитель в пара-положении, как, например, в радикале (50), то независимо от размера заместителя в орто-положении димеризация будет сильно ингибироваться или даже будет предотвращена [ср. (7); разд. 11.1]:
(fi-RC6H4)3C • + •/ \=C(C6H4R-fi)2 —Х-> Димер r/\=/
(50а) (506)
349
Гетерорадикалы (9), (11), (14), (16) (см. разд. 11.1) относительно устойчивы [по сравнению с димерами, кроме 1,1-ди-фенил-2-пикрилгидразила (И)]- Это связано с относительно малой прочностью связей N—N, S—S и О—О, с делокализацией с участием ароматических ядер и со стерическим ингибированием приближения к атомам, содержащим неспаренный электрон, или к лара-положению ароматического ядра [ср. (50)]. Этот последний фактор вносит большой вклад (в дополнение к непрочности связи О—О) в устойчивость радикала (16) (ср. разд. 11.1). В стабилизации радикала (51), который полностью диссоциирован в растворе, участвуют все перечисленные выше факторы:
В этом радикале, как было показано на основе анализа спектров ЭПР, n-фенильная группа копланарна с центральной феноксигруппой, но обе о-фенильные группы расположены к нему под углом 46°. Таким образом, л-фенильная группа может способствовать максимальной делокализации, а также может действовать как объемистая группа, ингибирующая димеризацию [ср. радикал (50)], тогда как два расположенных под углом о-заме-стителя препятствуют подходу к атому кислорода и, следовательно, образованию О—О-димера [димеризация происходит в твердом состоянии, однако через одно из лара-положений; ср. образование димера (7), разд. 11.1].
11.5. Реакции радикалов
Логично было бы классифицировать разнообразные реакции радикалов по отношению к самому радикалу: а) мономолеку-ляриые реакции, например фрагментация, перегруппировки; б) бимолекулярные реакции между радикалами, например димеризация, диспропорционирование; в) бимолекулярные реакции между радикалами и молекулами, например присоединение, замещение, отрыв атома (часто Н). Такая классификация, однако, не отличается от традиционной классификации органических реакций. Поэтому мы будем рассматривать реакции, в которых участвуют радикалы, независимо от того, являются ли они исходными реагирующими соединениями или интермедиатами в
350
типичных процессах присоединения, замещения или перегруппировки.
Важно отметить, что в любой реакции радикала с нейтральной молекулой продуктом реакции также является радикал (ср. разд. 11.3); тем самым развивается цепная реакция, для поддержания которой потом уже не нужно участия инициатора радикалов. В относительно редких случаях такая цепная реакция может оборваться путем взаимодействия двух радикалов друг с другом (концентрации радикалов обычно очень малы). Такое взаимодействие ведет к димеризации или диспропорционированию (ср. разд. 11.2.2), новый радикал при этом не образуется.
11.5.1. Реакции присоединения
Реакции присоединения к связи С=С образуют наиболее важную группу реакций с участием радикалов. Наибольшее внимание привлекает винильная полимеризация (см. разд. 11.5.1.4); важное значение имеют также реакции присоединения галоиенов и галогеноводородов.
11.5.1.1. Присоединение галогенов
Присоединение галогенов к алкенам может протекать не только по ионному механизму (см. разд. 7.1), но и через радикальные интермедиаты. Ионному механизму реакции благоприятствуют полярные растворители и присутствие катализаторов типа кислот Льюиса, а образованию радикальных интермедиатов— неполярные растворители (илн газовая фаза), действие солнечного света или УФ-облучения, а также добавление радикальных предшественников (инициаторов). Примером радикальной реакции является катализируемое фотохимическим путем присоединение хлора к тетрахлорэтану (52), представляющее собой цепную реакцию (ср. разд. 11.1):
Cl—С1
С12С=СС12 + .С1 —► С12С—СС13
(52) (53)
С1-С1
• С1 + С1ЭС—СС1з
(54)
Каждая молекула хлора при фотохимическом расщеплении дает два атома хлора, т. е. два радикала, каждый из которых
351
способен инициировать цепную реакцию. То, что каждый поглощаемый квант энергии действительно приводит к инициированию двух цепных реакций, подтверждается тем, что скорость реакции пропорциональна квадратному корню интенсивности поглощенного света.
Атомы хлора являются электрофилами (так как хлор — электроотрицательный элемент, и радикал С1 • легко захватывает электрон, чтобы дополнить свой октет) и поэтому легко присоединяются к двойной связи соединения (52), образуя радикал (53). Этот радикал в свою очередь может оторвать атом хлора от второй молекулы (этот процесс можно также рассматривать как реакцию радикального замещения в молекуле С1—С1) с образованием конечного продукта присоединения (54) и еще одного атома хлора, который продолжает цепную реакцию, т. е. очень быстрая, продолжающаяся реакция возникает под действием каждого атома хлора — инициатора, образованного фотохимическим путем. Найдено, что каждый квант поглощенной энергии приводит к превращению нескольких тысяч молекул (52) в (54); цепные реакции в этом случае, как говорят, имеют высокий квантовый выход, являются «длинными». Вплоть до более поздних стадий реакции, когда почти весь алкен (52) и хлор израсходуются, концентрации радикалов (53) и С1 • очень малы по сравнению с концентрациями исходных веществ; столкновение радикала с молекулой будет поэтому происходить гораздо чаще, чем столкновение радикала с другим радикалом. Тем не менее цепная реакция прекращается после столкновения двух радикалов, например:
С13С—СС12 + СС12—СС13 —> С13С—СС12—СС12—СС13 (53) (53) (55)
Реакции радикального хлорирования ингибируются в присутствии кислорода, поскольку молекула кислорода имеет два неспаренных электрона и ведет себя как бирадикал «О—О- (ср. разд. 11.6), хотя и не очень реакционноспособный. В рассмотренных выше реакциях присоединения он может соединяться с очень реакционноспособными радикальными интермедиатами, превращая их в гораздо менее реакционноспособные пероксиль-ные радикалы RO—О • , не способные продолжать цепную реакцию, т. е. ведет себя как эффективный ингибитор. Взаимодействие кислорода главным образом с пентахлорэтил-радикалами (53) доказано, например, образованием соединения (56) при ингибировании реакции присоединения кислородом:
. .о-о.
С13С—СС1 ► С13С—С=0 I I
С1 С1
(53) (56)
352
Реакционная способность галогенов при гомолитическом присоединении к алкенам уменьшается в том же ряду, что и при электрофильном присоединении, т. е. F2 > С12 > Вг2 > 12. Присоединение фтора, не требующее фотохимической или какой-либо другой активации, идет слишком бурно и сопровождается побочными реакциями; поэтому эта реакция не нашла широкого применения. Присоединение хлора обычно протекает быстро (как «длинная» цепная реакция) и необратимо, за исключением реакций при температурах > 200 °C. При повышении температуры, однако, растет тенденция к отрыву атома водорода, что приводит в соответствующих случаях к полному замещению хлором, а не к присоединению (ср. разд. 11.5.2.1). Бромирование проходит легко (как более «короткая» цепная реакция) и обычно является обратимым, в то время как иодирование проходит с трудом (или не идет совсем) и очень легко становится обратимым. Оказалось, что увеличение числа алкильных групп у атомов углерода, образующих двойную связь, относительно мало влияет на скорость присоединения галогена, во всяком случае, гораздо меньше, чем в реакциях присоединения по полярному механизму (ср. разд. 7.2). При накоплении атомов галогенов, например атомов хлора, атомов углерода, связанных двойной связью,' скорость реакции уменьшается; например, С12С=СС12 присоединяет хлор гораздо медленнее, чем СН2=СН2.
Обратимость присоединения брома и иода, особенно иода, использовали для изомеризации (менее стабильного в более стабильный) пары геометрических изомеров, содержащих двойную связь: в простейших случаях для превращения цис-изомера в транс-изомер, например (57) -+ (58). Такую изомеризацию можно провести с помощью УФ-облучения в присутствии каталитических количеств брома или иода:
(57) (58)
В полученной равновесной смеси обычно преобладает более устойчивая форма. Протекание изомеризации путем присоединения и отщепления Вг • было показано путем применения радиоактивного Вг2 в качестве катализатора: в конечной равновесной смеси были обнаружены оба изомера [(57) и (58)], причем оба содержали радиоактивный бром.
Было показано, что присоединение хлора и брома к бензолу— одна из немногих реакций присоединения к незамещенному бензольному ядру — также протекает по радикальному
12 П. Сайкс 353
механизму, т. е. катализируется светом или добавлением пероксидов и замедляется или полностью тормозится обычными ингибиторами. Хлорирование, вероятно, протекает по схеме:
С1-,С12
-----i С6Н6С16
(59)
В результате образуется смесь нескольких (из восьми возможных) геометрических изомеров гексахлорциклогексана (59). В отсутствие света или пероксидов реакция не идет; в присутствии кислот Льюиса происходит электрофильное замещение по схеме присоединение — отщепление (см. разд. 6.3). В случае других радикалов, например Ph •, суммарный процесс гомолитического замещения в бензоле также может происходить по механизму присоединения — отщепления (см. разд. 11.5.2.3).
При хлорировании метилбензола (толуол) (60) радикальная атака приводит к преимущественному отрыву атома водорода под действием С1 •, в результате чего происходит замещение в СНз-группе, а не присоединение к ядру. Это объясняется большей устойчивостью первоначально образующегося (делокализованного) бензильного радикала PhCH2* (61), а не гексадиениль-ного радикала (62), в котором нарушена ароматическая стабилизация исходного соединения:
11.5.1.2. Присоединение бромоводорода
Присоединение НВг к пропену (63) в полярных условиях приводит к 2-бромпропану (см. разд. 7.3). В присутствии пероксидов (или в других условиях, способствующих образованию радикалов) происходит быстрая цепная реакция и образуется 1-бромпропан (65), т. е. присоединение идет против правила
354
Марковникова (пероксидный эффект). Различная ориентация присоединения НВг обусловлена тем фактом, что в первом случае (полярная среда) присоединение инициируется Н+ и протекает через более устойчивый (вторичный) карбокатион, в то время как во втором случае (радикальный процесс) присоединение инициируется Вг • и протекает через более устойчивый (вторичный) радикал (64):
RO— Н + Вг • *— RO • + Н—Вг
МеСН=+Н2 + Вг- —► МеСН—СН2Вг
(63)
(64)
НВг
Вг • + МеСН2—СН2Вг
(65)
МеСНВг—СН2 (66)
Инициирование присоединения действием Вг •, как и отрыв водорода под действием RO • ют НВг, энергетически более благоприятно, чем альтернативное отщепление брома с образованием ROBr + Н Альтернативное присоединение Вг • к алкену (63) с образованием радикала (66) не происходит, поскольку вторичные радикалы, например (65), более устойчивы (ср. разд. 11.4), чем первичные, например (66).
Из всех галогеноводородов только НВг легко присоединяется к алкенам по радикальному механизму. Причина этого видна при сравнении значений ДЯ, приведенных ниже для двух стадий цепной реакции присоединения НХ к СН2=СН2: (1) X • + + СН2=СН2 (2) ХСН2СН2 + НХ s
ДЯ (1), кДж/моль • ДЯ (2), кДж/моль
Н—F — 188 (—45) + 155 (+37)
Н—С1 — 109 (-26) +21 (+5)
Н-Вг -21 (-5) -46 (-11)
H-I +29 (+7) -113 (-27)
• В скобках приведены значения ДЯ в ккал/моль.
12*
355
Только в случае присоединения НВг обе стадии цепной реакции экзотермичны; при присоединении HF вторая стадия очень эндотермична, что объясняется прочностью связи Н—F и трудностью ее разрыва; в случае НС1 вторая стадия также эндотермична, хотя и не в такой степени; при присоединении Ш первая стадия эндотермична, т. е. энергия, выделяющаяся при образовании слабой связи I—С, меньше энергии, которая затрачивается при расщеплении связи С=С. Известно лишь несколько радикальных реакций присоединения НО, но при обычных температурах скорости их невелики, а длины реакционных цепей малы.
Даже в случае присоединения НВг длины реакционных цепей весьма малы, гораздо меньше, чем в случае присоединения галогена; для получения достаточного количества радикалов обычное количество добавляемого пероксида должно быть выше, чем следовое; так, для препаративных целей требуется 0,01 моль пероксида на 1 моль алкена. Однако на практике того количества пероксида, которое образуется в реакционной среде вследствие автоокисления алкена кислородом воздуха (см. разд. 11.5.2.2), может быть достаточно для автоинициирования радикального присоединения НВг, независимо от того, желательно оно или нет. Начавшись, реакция по этому механизму протекает гораздо быстрее, чем конкурирующее присоединение по полярному механизму, поэтому в реакционной смеси будет преобладать продукт присоединения против правила Марков-никова, например (65). Если необходимо получить продукт присоединения по Марковникову, например 2-бромпропан из пропена, необходима тщательная очистка алкена перед применением или добавление ингибиторов (хорошие акцепторы радикалов, такие как фенолы, хиноны и др.), чтобы удалить любые присутствующие в алкене радикалы или потенциальные источники радикалов. Указанный продукт гораздо легче получить препаративно. Практически полный контроль ориентации присоединения НВг в любом направлении может быть достигнут в препаративных условиях путем введения в реакционную смесь либо пероксидов (инициаторы радикалов), либо ингибиторов. Введение инициаторов или ингибиторов радикалов для контроля ориентации используется не только в случае незамещенных алкенов; 3-бромпропен, например, может быть превращен в 1,2-или 1,3-дибромпропан.
При любом рассмотрении стереоселективности радикального присоединения к ациклическим субстратам при интерпретации результатов необходимо учитывать возможность превращения алкенов, по крайней мере частично, в их геометрические изомеры в присутствии следов брома (или НВг, т. е. радикалом Вг-; ср. разд. 11.5.1.1). Это превращение, однако, можно свести к ми-
356
нимуму, проводя реакцию при низких температурах и используя высокие концентрации НВг. Так, было найдено, что при прибавлении жидкого НВг при —80 °C к цис-2-бромбутену-2 (67) присоединение идет с высокой степенью транс-стереоселективности и приводит почти исключительно к соединению (69):
Me Н
+ Вг-
(68) (69)
В аналогичных условиях присоединение НВг к транс-2-бром-бутену (71) происходит практически транс-стереоселективно. Предположили, что в этом случае присоединение протекает через циклический бромониевый радикал (70), аналогичный циклическому бромониевому катиону, образующемуся при полярном присоединении брома к алкенам (см. разд. 7.1):
(70)
+ Вг*
(69)
Присоединение завершается атакой НВг с менее затрудненной стороны (вдали от мостикового атома брома) с образованием продукта транс-присоединения (69). Однако присоединение НВг к алкену (67) при комнатной температуре и более низкой концентрации НВг приводит к образованию смеси продуктов транс- и цис-стереоселективного присоединения (69) и (73) соответственно в соотношении 78:22. Такая же смесь продуктов и такого же состава получается в этих условиях из транс-изомера (71). Это позволяет предполагать, что в этих условиях вращение вокруг центральной углерод-углеродной связи является достаточно быстрым для установления конформационного равновесия между радикальными интермедиатами (68) и (72), прежде чем они смогут оторвать атом водорода от НВг, чтобы
357
завершить присоединение:
Имеются причины полагать, что образование одной и той же смеси продуктов из каждого, из двух алкенов— (67) и (71) — не обусловлено установлением равновесия между исходными веществами перед соответствующим присоединением. Вполне возможно, что наблюдаемая более высокая степень трамс-стерео-селективности (см. выше) для присоединения при более низкой температуре и более высокой концентрации НВг является результатом не образования циклического бромониевого радикала (70), а более медленного вращения вокруг центральной углеродуглеродной связи. Относительно быстрый перенос атома водорода (при более высокой концентрации НВг) может в этом случае происходить с менее затрудненной стороны в интермедиате (68) или (72), приводя к преимущественному суммарному трамс-присоединению.
В циклических алкенах, где такое равновесие радикальных интермедиатов невозможно, наблюдается преимущественное, но не исключительное (кроме циклогексенов) суммарное транс-присоединение.
11.5.1.3. Другие реакции присоединения
Тиильныё радикалы RS • могут быть получены путем отрыва атома водорода от тиолов RSH и способны легко присоединяться к алкенам по цепной реакции, аналогичной реакции присоединения НВг. Эту реакцию используют для препаративного получения диалкилсульфидов, однако она обратима:
RCH=CH2 4- R'SH RCH2CH2SR'
358
Сульфенилхлориды, например CI3CSCI, также могут служить источниками тиильных радикалов, но в этом случае присоединение инициируется радикалом С1 •, и поэтому R'S будет связываться с другим атомом углерода двойной связи:
SCC13 Cl. . CIsCSCI I
RCH=CH2 ------► RCH—СН2С1 ---------► RCHCH2C1 + C1«
Углерод-углеродные связи наряду с другими частицами могут образоваться путем присоединения галогенметильных радикалов к алкенам. Соответствующие радикалы «СХ3 (X = Br, С1) могут быть получены при действии пероксидов на СХ4 или при фотолизе СХ4:
С1
• СС13 ссц |
rch=ch2 --------->- RCHCH2CC13 -------► RCHCH2CC13 + . СС13
(74) (75)
То, что относительно инертный СС14 присоединяется таким образом, кажется немного удивительным, однако обе стадии этой реакции являются экзотермическими; АЯ =—75 и — 17 кДж/моль (—18 и —4 ккал/моль) соответственно. Первоначально образующийся радикал (74) может, однако, конкурировать с -СС13 за присоединение к RCH=CH2; в результате этого кроме обычного продукта присоединения (75) в некоторых случаях возможно образование низкомолекулярных полимеров.
11.5.1.4. Ванильная полимеризация
Эта реакция была предметом многих теоретических исследований и изучения механизма главным образом вследствие технического значения тех полимеров, которые образуются в ходе полимеризации. Подобно другим рассмотренным выше радикальным реакциям, она включает три стадии: инициирование, рост цепи и прекращение реакции (обрыв цепи).
Инициирование:
(1) Образование инициатора Ra«, например, из пероксидов или азосоеди-ирний
(2) Ra • + СН2=СН2 —> RaCH2CH2
Рост цепи:
. (ге-1)СН2=СН2
RaCH2CH2 Ra(CH2)2rt.
Обрыв цепи:
(1) Ra(CH2)2rt-+ • Ra —> Ra(CH2)2nRa
(2) Ra(CH2)2n. + •(CH2)2rtRa —► Ra(CH2)4nRa
(3) Ra(CH2)xCH2. + • CH2CH2(CH2)1,Ra —► Ra(CH2)xCH3+CH2=CH(CH2)yRa
359
Рост цепи обычно идет очень быстро. Мономерные алканы могут реагировать с кислородом воздуха с образованием пероксидов, которые легко распадаются до соответствующих радикалов, вызывающих автоинициирование и называемых инициаторами. Поэтому для стабилизации мономера при хранении к нему добавляют небольшое количество ингибитора, например хинона. Для последующей полимеризации необходимо добавить достаточное количество радикального инициатора, чтобы «насытить» ингибитор, перед тем как начнется полимеризация. Именно этим объясняется наличие индукционного периода, часто наблюдаемого при проведении процесса полимеризации.
Радикальные инициаторы не являются, строго говоря, катализаторами (хотя их часто именно так и называют), поскольку каждый радикал, инициирующий рост полимерной цепи, необратимо связывается с ней, и в ряде случаев его удается обнаружить в полученном полимере. Эффективность некоторых инициаторов настолько велика, что после определенного индукционного периода каждый радикал вызывает образование полимерной цепи.
Обрыв растущей цепи может произойти вследствие ее столкновения как с инициатором, так и с другой растущей цепью (см. выше), причем вторая реакция более важна; большая часть инициирующих радикалов расходуется для зарождения цепей. Обрыв цепи может происходить также в результате реакции диспропорционирования (ср. разд. 11.2.1) между растущими цепями (см. выше). Отрыв атома водорода возможен при атаке растущей цепью уже сформированного полимера, что приводит к появлению новой точки роста, а следовательно, к образованию разветвленного продукта (76):
Н
| Ra(CH2)2fI. . nCHz=CHz
Ra(CH2)JtCH(CH2)vRa Ra(CH2)xCH(CH2)„Ra ---------->
Xa(Ci П2/2 д_। С- 413
(СН2)2„.
—► Ra(CH2)xCH(CH2)j,Ra
(76)
Степень разветвления оказывает глубокое влияние на физические и механические свойства образующегося полимера.
Свойства полимера в значительной степени зависят также от его средней молекулярной массы, т. е. от средней длины полимерных молекул, которая может варьировать от нескольких мономерных единиц до многих тысяч. Кроме того, важное значение имеет фактический диапазон отклонений длин молекул полимеров от среднего значения. Например, свойства двух полимеров с примерно равной средней молекулярной массой различны,
360
если один из них состоит из молекул, большая часть которых имеет одну и ту же длину, в то время как другой содержит как очень длинные, так и очень короткие молекулы. Длину цепи образующегося полимера можно контролировать несколькими путями. Так, увеличение концентрации инициатора по отношению к концентрации алкена будет приводить к образованию более коротких цепей, так как число растущих цепей увеличится и, следовательно, их обрыв станет более возможным, чем рост цепи. Обрыв цепи может быть вызван добавлением терминаторов или агентов переноса цепи. Они представляют собой соединения типа ХН, которые могут реагировать с растущей полимерной цепью, теряя атом водорода и тем самым обрывая цепь. При этом образуется новый радикал Х-, который способен инициировать образование новой цепи (77) из мономера. В качестве агентов переноса цепи часто применяются тиолы RSH:
Ra(CH2)rtCH2 пСН2=СН2
RSH ^а(Сн7гаСЩ RS--------------" RS(CH2)2„.
(77)
Новая растущая цепь получается, таким образом, без замедления суммарного процесса превращения мономера. Если добавленное соединение ХН должно играть роль терминатора, то его подбирают с таким расчетом, чтобы образующийся радикал X • был достаточно нереакционноспособным и не мог инициировать образование новой цепи из мономера.
Инициируемая радикалами полимеризация простейших алкенов, например этена и пропена, идет только в жестких условиях, в том числе при очень высоком давлении, однако многие замещенные алкены мономера полимеризуются легко. К ним относятся, например, СН2=СНС1 [полимер — поливинилхлорид (ПВХ) — используют для изготовления водопроводных труб и др.], СН2 = СМеСО2Ме (полимер — полиметилметакрилат, органическое стекло); PhCH=CH2 (полимер — полистирол — используют для изготовления изоляционных материалов), CF2= =CF2 (полимер — тефлон, обладающий чрезвычайно низким коэффициентом трения, высокой химической инертностью и высокой температурой плавления). Свойства полимера можно изменять в чрезвычайно широких пределах путем сополимеризации двух различных мономеров, в ходе которой оба мономера включаются в молекулы полимера поровну или в иных пропорциях; например, многие синтетические каучуки являются сополимерами стирола и бутадиена. Как уже отмечалось (см. разд. 11.3), сополимеризацию PhCH2=CH2 и СН2=СМеСО2Ме (50 : 50) используют в аналитических целях, чтобы отличить вызываемую радикалами полимеризацию от полимеризации, вызываемой анионами или катионами (ср. разд. 7.4.3).
361
Индуцируемая радикалами полимеризация имеет, однако, некоторые недостатки. Так, в ряде случаев образуются разветвленные полимеры вследствие отрыва атомов водорода от растущей цепи (см. выше). При использовании мономеров типа СН2=СНХ (т. е. обычных мономеров кроме СН2=СН2 и CF2= =CF2) возможна неупорядоченная ориентация заместителей X относительно атомов углерода в цепи твердого полимера. Такие полимеры, называемые атактическими, например атактический полипропилен, не получаются в кристаллической форме, обладают низкой плотностью, низкой температурой плавления, малой механической прочностью. Однако при использовании в качестве катализатора TiCl3-AlEt3 полимеризация, например, пропена идет в очень мягких условиях и приводит к полимеру с одинаковой ориентацией всех метильных групп. Показано, что такой полимер — изотактический полипропилен — является кристаллическим, обладает высокой плотностью, высокой температурой плавления, высокой механической прочностью. Полагают, что протекание такой регулярной, координационной полимеризации объясняется наличием на поверхности гетерогенного катализатора групп атомов, действующих как матрица таким образом, что каждая следующая молекула мономера может присоединиться к растущей полимерной цепи только благодаря «координации», т. е. лишь при определенной ориентации на поверхности катализатора.
Когда мономерами являются сопряженные диены, например бутадиен-1,3 СН2=СН—СН=СН2 или 2-метилбутадиен-1,3 (изопрен) СН2=СМеСН=СН2, полимерная цепь, образующаяся путем обычного 1,4-присоединения (см. разд. 7.5.1), все же содержит по одной углерод-углеродной связи в каждой мономерной единице, т. е. сохраняет реакционную способность, что позволяет осуществить «сшивание» двух полимерных цепей. Так, при реакции такого полимера с серой (вулканизация каучука) образуются S—S-мостики между полимерными цепями. Найдено, что при относительно небольшом числе поперечных связей полимер обладает эластичностью, тогда как увеличение степени связывания придает ему жесткую трехмерную структуру. Части полимерных цепей могу! быть по-разному ориентированы относительно двойной связи в цепи; они могут быть в цис- или транс-положении по отношению друг к другу, как, например, в случае полиизопрена:
---СН2х /СН2 -СН2\ /Н /С=с^--------------------------------V=cz
Ме/ Ж Ме/ \СН2----
цис транс
Как и следовало ожидать, различия в стереохимии заметно влияют на свойства полимера. Так, природные полиизопрены —
362
натуральный каучук и гуттаперча — обладают разными свойствами. Натуральный каучук (до вулканизации мягкий и липкий) содержит в цепи quc-звенья, тогда как гуттаперча (твердая и хрупкая) содержит все звенья в транс-положении.
11.5.2. Реакции замещения
Хотя большинство реакций такого типа в целом являются реакциями замещения, однако не всегда такой результат достигается непосредственно (ср. Sn2). В некоторых случаях радикал получают из субстрата путем отщепления (как правило, атома водорода), после чего этот радикал участвует в реакции замещения в другой частице или присоединяется к ней. Однако в ряде случаев суммарное замещение достигается путем последовательной реализации процессов присоединения и отщепления.
11.5.2.1. Галогенирование
Алканы чрезвычайно легко атакуются галогенами в условиях, которые способствуют образованию радикалов. Это находится в заметном противоречии с их крайней устойчивостью к атаке электрофилами или нуклеофилами, которое объясняется очень низкой полярностью связи С—Н в алканах. Суммарная реакция замещения у атома углерода алканов, например при хлорировании, включает (после первоначального образования С1 •) отщепление атома водорода от R—Н радикалом С1 • и отщепление атома хлора от С1—С1 радикалом R • (эту стадию можно рассматривать как прямое замещение); эти две стадии чередуются в быстро развивающейся цепной реакции:
С1—С1
R—НЦ--С1 —> R.+H-C1
I |С|-С'
-Cl + R—С1
Для метана СН4 число превращений RH—>-RCl на один радикал С1 •, образующийся при фотолизе, равно да 106, вследствие чего при солнечном освещении реакция может протекать со взрывом. Хлорирование можно инициировать и термическим способом, однако для осуществления процесса С12 —► 2С1-требуется значительное повышение температуры. Хлорирование С2Н6 в темноте при 120 °C практически не идет, одиако при
363
введении небольших добавок PbEt4 скорость реакции достаточно высока, поскольку тетраэтилсвинец при указанной температуре распадается с образованием этильных радикалов Et-, которые могут действовать как инициаторы:
Et • + Cl—С1 -► Et—С1 + С1.
Хлорирование простых алканов, однако, редко используют для получения монохлорпроизводных, так как первоначально образовавшийся продукт подвергается дальнейшей атаке очень реакционноспособным хлором, что приводит к образованию сложной смеси продуктов.
Найдено, что легкость атаки атомов водорода в алкане увеличивается в ряду (под формулами приведены относительные скорости отщепления атомов водорода под действием С1 • при 25 °C)
И\ Н Н\ х
Н—С—Н < Н—С—Н < —С—Н < —С—н
и/ / / /
первичный вторичный третичный
1 4,4 6,7
что соответствует ослаблению связи С—Н и увеличению стабильности образующегося радикала (ср. разд. 11.3). Эту последовательность часто может нарушать статистический эффект, т, е. относительные количества различных типов атомов водорода. Например, в (СН3)3СН имеется девять первичных атомов водорода и лишь один третичный атом водорода; при хлорировании (СН3)3СН образуются 65 % (СН3)2СНСН2С1 и 35 % (СН3)3СС1, что согласуется с приведенными выше отношениями скоростей после введения статистической поправки. Показано, что если хлорирование проводится в растворе, распределение изомеров зависит от природы растворителя, особенно от его способности образовывать комплекс с С1-. Стабилизация таким путем радикала хлора увеличивает селективность по сравнению с селективностью реакции хлора в газовой фазе. Показано, что селективность галогенирования снижается с повышением температуры.
Галогенирование, особенно хлорирование, в отличие от большинства радикальных реакций, заметно зависит от присутствия в субстрате полярных заместителей, так как радикал С1- из-за электроотрицательности хлора является электрофильным реагентом и может преимущественно атаковать участки с более высокой электронной плотностью (см. разд. 11.5.1.1). Хлорирование, таким образом, замедляется в присутствии электроноакцепторных групп, как это видно из относительных количеств продуктов замещения у четырех различных атомов углерода
364
в 1-хлорбутане (78) при инициируемом фотохимическом хлорировании при 35°C:
СН3—СН2—СН2—СН2—С1 (78)
(25%) (50%) (17%) (3%)
Это различие свидетельствует о затухании электроноакцепторного влияния атома хлора по мере удаления от него: СН2-группа в -у-положении ведет себя аналогично СН2-группам в СН3СН2СН2СН3. Уменьшение эффективности атаки группы СН3 отражает большую трудность разрыва связи С—Н в СН3, чем в СН2 (см. выше).
В случае пропена (79) возможно как присоединение хлора к двойной связи, так и атака группы СН3. Найдено, что при повышенных температурах, например при да 450 °C (С1 • в этом случае образуется путем термолиза С12), образуется только продукт замещения. Это объясняется тем, что аллильный радикал (80), полученный путем отрыва атома водорода, стабилизирован в результате делокализации, тогда как в радикале (81), полученном при присоединении С1», такая стабилизация невозможна; образование радикала (81) при повышенных температурах является обратимым процессом, равновесие которого сдвинуто влево:
Н Н
I Ct- | .
СН2—СН=СН2 ^=±-СН2—СН—СН2С1
(79) (81)
сф-нО
[сн2==сн—-сн2]'+ НС1
(80)
Циклогексен подвергается аналогичному «аллильному» хлорированию по тем же причинам.
Ниже приведены значения ДЯ для двух стадий цепных реакций галогенирования (см. разд. 11.5.2.1) метана:
(1)Х. + Н—СН3 (2) СНз» + Х2
ДЯ (1), кДж/моль * ДЯ (2), кДж/моль *
F2 -134 (-32) -292 (-70)
С12 —4( —1) -96 (-23)
Вг2 4-63(4-15) -88 (-21)
12 4-138(4-33) -75 (-18)
• В скобках даны значения ДЯ в ккал/моль.
Данные, приведенные для реакций фторирования, отражают слабость связи F—F [150 кДж/моль (36 ккал/моль)]. и прочность связи Н—F [560 кДж/моль (134 ккал/моль)]. Обычно
365
фторирование не требует специального инициирования (см. разд. 11.5.2.1) и протекает со взрывом, если не применяют сильно разбавленные растворы. Протекание фторирования по радикальному механизму, несмотря на отсутствие специфического инициирования, подтверждается тем, что хлорирование можно инициировать в темноте при комнатной температуре путем введения в смесь следов фтора. В сравнимых условиях бромирование протекает намного медленнее хлорирования, так как стадия (1) — отрыв атома водорода под действием Вг •—обычно является эндотермической. Эта стадия настолько эндотермична для иодирования действием I •, что прямое иодирование алканов, как правило, не происходит.
Заметно сниженная реакционная способность Вг- по сравнению с С1 • на стадии отрыва атома водорода означает, что бромирование является гораздо более селективным, чем хлорирование. Легкость отщепления атомов водорода при первичном, вторичном и третичном атомах углерода увеличивается в ряду (под формулами приведены относительные скорости отщепления водорода под действием Вг • при 25 °C):
И\ Н\
Н-С—Н < — С—Н < — С—Н
1 80 1600
Меньшая реакционная способность Вг • по сравнению с С1 • может быть использована в препаративных целях. Так, бромирование (СНз)зСН приводит к образованию только (СН3)3СВг (ср. хлорирование). Соответствующий эффект еще более выражен при наличии заместителей, стабилизирующих первоначально образующийся радикал; так, в ряду СН4, PhCH3, Ph2CH2 и Ph3CH относительные скорости бромирования различаются примерно в 109 раз, тогда как для хлорирования — только в 103раз. Селективность уменьшается с ростом температуры.
Галогенирование оптически активной формы хирального алкана RR'R"CH обычно приводит к рацемическому (±)-галогениду. Этот результат ничего не говорит о преимущественной конформации промежуточного радикала RR'R"C •, поскольку рацемизация может .наблюдаться как для плоской, так и для быстро превращающейся пирамидальной структуры (ср. разд. 11.4). Однако бромирование (+)-1-бром-2-метилбутана (82) дает оптически активный бромид (—) -1,2-дибром-2-метил-бутан (84), что соответствует суммарному замещению с сохранением конфигурации. Полагают, что это является результатом экранирования атомом брома, находящимся в положении 1, той стороны промежуточного радикала (83), которая противополож-
366
на стороне отщепления атомд водорода, что и приводит к сохранению конфигурации:
Ме, .СН2Вг
Ус
El i
(82) (+)
К "
мч ]/СН2
Вг—Й
Ф
-НВг *
мч
Etx
(83)
Ме„ ,СН2Вг "'Л X Л E.-V Вг
+ Вг •
(84) (-)
Бромирование оптически активной формы соответствующего хлоралкана — 2-метил-1-хлорбутана — также приводит к оптически активному продукту и сохранению конфигурации. Возможно, что мостиковый радикал образуется и при хлорировании, однако соответствующее экранирование, по-видимому, не столь эффективно, поскольку найдено, что галогенирование более реакционноспособным радикалом хлора ведет к полной рацемизации.
Радикальное галогенирование (особенно хлорирование), осуществленное не самими галогенами, а другими галогенсодержащими реагентами, находит широкое применение в синтезе вследствие большей селективности этой реакции. Так, хлорирование может быть проведено при взаимодействии с алкилгипо-хлоритами ROC1 (например, R—Ме3С) в присутствии инициаторов, которые отрывают атом хлора от ROC1 с образованием радикала RO •. Этот радикал, как было показано, на следующей стадии отрывает атом водорода от RH. Этот реагент применяется особенно часто для аллильного хлорирования. Другим полезным реагентом для препаративного хлорирования является SO2C12; в этом случае инициатор отрывает атом хлора, образуя • SO2C1, который, как и радикал С1 •, образующийся после отщепления SO2 от • SO2Cl, может отрывать атом водорода от RH.
Широкое применение в качестве бромирующего реагента получил JV-бромсукцинимид (NBS) (88), который с высокой степенью избирательности атакует только слабые С—Н-связи, т. е. аллильные, бензильные и аналогичные положения. При использовании этого реагента необходимо присутствие инициаторов. Было показано, что при проведении бромирования необходимо поддерживать постоянную, но очень низкую концентрацию Вг2. Это достигается путем взаимодействия НВг, образующегося в ходе реакции, с Af-бромсукцинимидом (см. ниже). Как правило,
367
в А-бромсукцинимиде присутствуют следы Вг2 или НВг, при реакции которых с инициатором образуется радикал Вг-, который и начинает реакцию:
Вг2 (НВг) + Инициатор —>- Вг •
Вг
(85)
(86) (87)
(88)
Концентрация брома поддерживается за счет реакции N-бромсукцинимида с НВг [которая является быстрой (хотя и ионной)], образующимся в цепной реакции. Альтернативная реакция присоединения Вг • к двойной связи с образованием радикала (89) является обратимой
(85)
^Вг (89)
тогда как образование радикала (86) — необратимая реакция. Суммарное замещение, таким образом, преобладает над присоединением до тех пор, пока концентрация брома мала. Радикал (86) также стабилизирован делокализацией, тогда как радикал (89) не стабилизирован (ср. разд. 11.4). Подтверждением приведенного выше объяснения механизма действия А-бромсук-цинимида являются следующие факты: 1) действие А-бромсукци-нимида так же селективно, как и действие брома; 2) циклогексен (85) при высоких концентрациях брома вступает в основном в реакцию присоединения, а при низких концентрациях — в реакцию замещения (при этом необходимо удалять образующийся НВг, что и происходит при применении А-бромсукцинимида).
11.5.2.2. Автоокисление
Автоокисление — это окисление органических соединений кислородом при низкой температуре по механизму радикальной ценной реакции. На начальной стадии, как правило, образуются гндропероксиды (RH—>-ROOH). Суммарный процесс является замещением, хотя он включает отрыв атома водорода и присо
368
единение 02 (см. ниже). Образующиеся на начальной стадии гидропероксиды часто вступают в дальнейшие реакции. Авто-окисЛение играет важную роль в процессе затвердевания масляных красок; ненасыщенные эфиры масел обычно образуют гидропероксиды, распад которых до RO- инициирует полимеризацию, в результате чего образуется защитная полимерная пленка. С процессами автоокисления связаны также нежелательные изменения, особенно в веществах, содержащих ненасыщенные связи, например прогоркание жиров и старение резины. Постепенный распад большинства органических соединений под действием воздуха и солнечного света обусловлен фотосенсибилизи-рованным автоокислением. Автоокисление может инициироваться следами ионов металлов (см. ниже), светом и обычными инициаторами.
Реакция протекает в две стадии с отрывом атома водорода на одной из стадий:
.0-0.
Ra. + H-R R. -------> RO—О
f (90)
R • + RO—ОН
(91)
В определенных условиях гидропероксид (91) сам распадается на радикалы RO- и -ОН, которые действуют как инициаторы; в этом случае реакция становится автокаталитической. Присоединение О2 к R- протекает очень быстро и часто контролируется диффузией. Образующиеся пероксидные радикалы (90) обычно обладают сравнительно низкой реакционной способностью (см. радикал -О—О-; разд. 11.5.1.1), и отщепление атома водорода из различных положений идет высокоизбирательно. Так, аллильные и бензильные связи С—Н сравнительно легко атакуются, потому что прочность этих связей несколько меньше, а образующиеся радикалы стабилизируются делокализацией. В частности, при атаке циклопеитеиа затрагивается аллильное положение и образуется гидропероксид (92). В простых алканах, как правило, атакуется только третичный атом углерода. Так, в случае декалина образуется гидропероксид (93):
(92)
ООН
ООН
(93)
369
Относительные скорости отщепления атомов водорода /йод действием RO2> при 30 °C увеличиваются в ряду (отщепляющиеся атомы водорода подчеркнуты):
PhCH3 < PhCH2 < PhCH2CH=CH2
1
30 63
Автоокисление алкенов может включать присоединение RO2-к двойной связи и (или вместо него) отщепление атома водорода, особенно если в алкене отсутствуют доступные аллильные, бензильные или третичные С—Н-группы. Влияние присутствия пероксидов на ориентацию присоединения НВг к алкенам рассмотрено выше (см. разд. 11.5.1.2). Простые эфиры особенно склонны к автоокислению, причем первоначальная атака происходит по а-С—Н-связи по отношению к атому кислорода с образованием стабильного радикала. Образовавшийся пероксид реагирует далее с образованием диалкилпероксидов, которые взрываются при нагревании; об этом следует помнить при упаривании эфирных растворов досуха! Накапливающиеся при хранении эфиров пероксиды могут быть безопасно разложены перед использованием эфира путем промывания раствором восстанавливающего агента, например FeSO4.
Автоокисление в некоторых случаях может иметь препаративное значение. Так, при промышленном производстве фенола и ацетона протекает катализируемая кислотой перегруппировка гидропероксида, образующегося из 2-фенилпропана (кумол) (см. разд. 5.7.2). Другим примером является реакция, включающая образование гидропероксида (94) при окислении тетрагидронафталина (тетралин) кислородом воздуха при 70 °C; при последующем действии основания образуется кетон (а-тетралон) (95), а при восстановлении происходят разрыв связи О—О и образование спирта (а-тетралол) (96):
(96) (94) (95)
Альдегиды, особенно ароматические, очень чувствительны к автоокислению. Так, бензальдегид (97) быстро превращается в бензойную кислоту (101) на воздухе при комнатной температуре. Эта реакция катализируется светом и обычными инициаторами, а также следами ионов металлов, которые могут действовать как одноэлектронные окисляющие агенты (ср. разд. 11.2.3), на
370
пример Fe3+, Со3+ и др;:
О О О
II Fe3+ (а) || .О2. ||
PhC— Н --------*• PhC • --► PhC—О—О*
(97) ~Fe ’ ~Н (98) (99)
(б) (97)
О о
II II
PhC • + PhC—О—ОН
(98) (100)
ООО
II II Н+ II
PhC—О—ОН + PhC—Н -► 2PhC—ОН
(в)
(100) (97) (101)
При инициировании окисления ионами Fe3+ образуется бензоильный радикал (98), который затем присоединяет молекулу кислорода, образуя радикал (99). При реакции этого радикала с бензальдегидом (97) образуются пербензойная кислота (100) и бензоильный радикал (98); эти две стадии и формируют цепную реакцию. Однако конечным продуктом оказывается не пербензойная кислота (100), так как она подвергается быстрой, катализируемой кислотой нерадикальной реакции с бензальдегидом (97) с образованием бензойной кислоты (101). Эта реакция, катализируемая кислотой, ускоряется при повышении концентрации образующейся бензойной кислоты, являясь, таким образом, автокаталитической. Участие бензоильного радикала (98) в процессе следует из того, что ври проведении реакции при более высоких температурах (» 100 °C) и при низкой концентрации кислорода выделяется СО, образующийся в результате реакции:
PhCO —► Ph • + СО
Автоокисление альдегидов и других органических соединений может быть значительно уменьшено очень тщательной очисткой для удаления присутствующих пероксидов, следов ионов металлов и т. д.; гораздо больший эффект дает добавление подходящих ингибиторов, являющихся в данном случае антиоксидантами. Лучшими из них являются фенолы и ароматические амины, которые содержат легко отщепляемый атом водорода; образующийся в результате такого отщепления радикал обладает относительно низкой реакционной способностью, является плохим инициатором (взаимодействие с новой молекулой субстрата), но способен хорошо обрывать цепь путем взаимодействия с другим радикалом.
371
Интересным примером автоокисления является фотоокислё-ние углеводородов типа 9,10-днфенилантрацена (102) в таких растворителях, как CS2. Поглощаемый свет превращает углеводород в стабилизированный бирадикал (103) (ср. разд. 11.6) или подобный ему. Стабилизация этого бирадикала основана на делокализации его неспаренных электронов, а также на превращении частично ароматического состояния (102) в полностью ароматическое состояние (103). Бирадикал затем присоединяет молекулу кислорода с образованием трансаннулярного пероксида (104):
Легкость подобного фотоокисления увеличивается при увеличении числа бензольных колец в полициклических ароматических углеводородах линейного строения, т. е. при уменьшении степени ароматичности. Это происходит легко, например, с углеводородом гексаценом (105) (окрашен в очень темно-зеленый цвет), что не позволяет работать с ним на солнечном свету и на воздухе (ср. разд. 11.6).
11.5.2.3. Ароматическое замещение
Ароматические молекулы могут атаковаться радикалами, а также электрофилами и нуклеофилами (см. разд. 6.10.1). Как и при атаке этими полярными частицами, гомолитическое ароматическое замещение протекает по механизму присоединения — отщепления:
(106) (107)
Отрыв атома водорода от делокализованного циклогексадие-нильного радикального интермедиата (106) с образованием продукта замещения (107) ие протекает самопроизвольно, а требует участия еще одного радикала Ra- для отщепления атома водорода. Реакция между радикалом (106) и радикалом, отщеп
372
ляющим атом водорода, вероятно, идет быстро и не лимитирует скорость реакций; заметного кинетического изотопного эффекта не наблюдается. Атака Ra- исходного ароматического субстрата, таким образом, является скоростьлимитирующей стадией. Изучены реакции замещения, в которых Ra - являются Аг-(особенно, Ph-), PhCO2- (и некоторые RCO2), R- и НО-. Атака под действием НО-—гидроксилирование — особенно важна в биологических системах как первая стадия обезвреживания «чужих» ароматических молекул. Известно также небольшое число реакций, в которых кроме атома водорода замещаются иные атомы или группы, например галоген, МеО. Однако наиболее детально исследовано замещение атома водорода на арильную группу — арилирование.
Показано, что атака, например, радикалом Ph- ароматического соединения, например бензола, приводит к продуктам, отличающимся от тех, которые образуются в результате простого замещения [(107; Ra = Ph)]. Это объясняется тем, что промежуточный радикал (106) способен не только превращаться в соединение (107) (после отрыва атома водорода), но может и димеризоваться, образуя соединение (108), и(или) диспропор-ционировать, образуя смесь соединений (107) и (109):
(Ю9)
Для простоты выше показаны только продукты взаимодействия в пара-положение радикала (106); взаимодействие по орто-положению может приводить к о-дигидроизомеру соединения (109) и к продуктам, как о,о-, так и о,п-сочетания, изомерным соединению (108). Таким образом, при арилировании ароматических соединений образуются довольно сложные смеси продуктов.
Что касается суммарной реакции замещения [с образованием соединения (107)], то заметные отличия от электрофильной и нуклеофильной атак можно увидеть, сравнивая поведение различных замещенных бензолов C6H5Y. Так, найдено, что гомолитическая атака CeH5Y протекает быстрее, чем атака С6Н6, независимо от того, является ли Y электронодонорной или электроноакцепторной группой. Это следует из сравнения
373
относительных скоростей атаки различных соединений под действием радикала Ph<
C6H5Y
Y
&отн
н 1,0 CN 3,7
ОМе 1,2 no2 4,0
Вг 1,8 Ph 4,0
Ме 1,9
В этом случае при изменении заместителя Y относительные скорости изменяются незначительно, в отличие от электрофильного замещения, например нитрования того же субстрата, при котором относительные скорости могут увеличиваться в «s 108 раз. При этом необходимо помнить, что фенилирование включает атаку частицей с низкой полярностью.
При сравнении факторов парциальных скоростей (ср. разд. 6.7.2) реакций различных защищенных бензолов CeHsY видно, что независимо от природы заместителя Y в C6H5Y предпочтительность атаки орто-, мета- и пара-положений радикалом Ph • уменьшается в ряду: о- > п- > м- (за исключением случая, когда Y = Ме3С; при этом стерический эффект заместителя
Y может препятствовать о атаке):
fo In
PhOMe 5,60 1,23 2,31
PhNO2 5,50 0,86 4,90
PhMe 4,70 1,24 3,55
PhCl 3,90 1,65 2,12
PhBr 3,05 1,70 1,92
PhCMe3 0,70 1,64 1,81
Предпочтительность о- и n-атак МОЖНО объяснить тем, что
электрон, переданный атакующим радикалом Ph • в интермедиат (106), может быть делокализован (что приводит к стабилизации интермедиата) при наличии электроноакцепторного [в соединении (110)] или электронодонорного [в соединении (111)] заместителя, как это показано ниже для случая атаки параположения:
Н Ph Н Ph 0-^ O=N—О" "О—N—О + *
(110a) (1106)
Н Ph Н Ph
(111a) (1116)
Однако нет вполне удовлетворительного объяснения тому, почему аналогичная м-атака CeHsY протекает быстрее, чем атака СбНв, или почему о-атака C6HsY обычно протекает быстрее, чем n-атака. Относительно небольшие различия в значениях
374
факторов парциальных скоростей для каждого соединения C6H5Y означают, что гомолитическое ароматическое замещение обычно приводит к более сложной смеси продуктов, чем электрофильная атака той же молекулы.
Все приведенные выше данные относятся к фенилированию радикалом Ph«, полученным из (PhCO2)2. Этот и другие диацилпероксиды часто используют для этой цели, но поскольку образование радикала Ph • включает стадию
PhCO2« —> Ph • + СО2
обычно невозможно предотвратить образование некоторого количества сложных эфиров либо вследствие взаимодействия Ph • с PhCO2’, либо, что более обычно, вследствие атаки радикалом PhCO2- ароматического субстрата (ацилоксилирование). Это затруднение можно преодолеть, получая Аг. при термическом разложении А-нитрозопроизводных ацетилированных ароматических аминов ArN (NO) СОМе или солей диазония ArN2 в слабощелочных средах. Последнее превращение представляет собой реакцию Гомберга для синтеза несимметричных биарилов Аг— —Аг'. В каждом случае предшественник радикала Аг разлагается в присутствии избытка ароматического субстрата, который часто фактически используется как растворитель. Выход продукта классической реакции Гомберга можно намного увеличить путем диазотирования свободного амина ArNH2 действием CoHjiONO в растворе ароматического субстрата. Никакой кислоты при этом не применяют; реакция в этом случае полностью гомогенная. Некоторые синтезы Аг—Аг' проходят вполне удачно, но в целом реакция не нашла широкого применения.
Однако внутримолекулярное радикальное арилирование проходит вполне надежно, в частности реакции Пшорра. Она включает термическое разложение диазониевых солей, например (112), в присутствии порошка меди (катализатор) и используется для синтеза фенантренов типа (113):
375
фторирование не требует специального инициирования (см, разд. 11.5.2.1) и протекает со взрывом, если не применяют сильно разбавленные растворы. Протекание фторирования по радикальному механизму, несмотря на отсутствие специфического инициирования, подтверждается тем, что хлорирование можно инициировать в темноте при комнатной температуре путем введения в смесь следов фтора. В сравнимых условиях бромирование протекает намного медленнее хлорирования, так как стадия (1) — отрыв атома водорода под действием Вг — обычно является эндотермической. Эта стадия настолько эндотермична для иодирования действием I •, что прямое иодирование алканов, как правило, не происходит.
Заметно сниженная реакционная способность Вг- по сравнению с С1 • на стадии отрыва атома водорода означает, что бромирование является гораздо более селективным, чем хлорирование. Легкость отщепления атомов водорода при первичном, вторичном и третичном атомах углерода увеличивается в ряду (под формулами приведены относительные скорости отщепления водорода под действием Вг • при 25 °C):
Н\ Н\ \ н-с—н < —с—н < — с—н
1 80 1600
Меньшая реакционная способность Вг • по сравнению с С1 • может быть использована в препаративных целях. Так, бромирование (СНз)зСН приводит к образованию только (СН3)3СВг (ср. хлорирование). Соответствующий эффект еще более выражен при наличии заместителей, стабилизирующих первоначально образующийся радикал; так, в ряду СН4, PhCH3, Ph2CH2 и Ph3CH относительные скорости бромирования различаются примерно в 109 раз, тогда как для хлорирования — только в 103раз. Селективность уменьшается с ростом температуры.
Галогенирование оптически активной формы хирального алкана RR'R"CH обычно приводит к рацемическому (±)-галогениду. Этот результат ничего не говорит о преимущественной конформации промежуточного радикала RR'R"C •, поскольку рацемизация может .наблюдаться как для плоской, так и для быстро превращающейся пирамидальной структуры (ср. разд. 11.4). Однако бромирование (+)-1-бром-2-метилбутана (82) дает оптически активный бромид (—)-1,2-дибром-2-метил-бутан (84), что соответствует суммарному замещению с сохранением конфигурации. Полагают, что это является результатом экранирования атомом брома, находящимся в положении 1, той стороны промежуточного радикала (83), которая противополож-
366
на стороне отщепления атомд водорода, что и приводит к сохранению конфигурации:
Ме„ _СН2Вг хс
Е' Л (82)(+)
'К ’
Ме//у, | J>CH2 Et-f
Вг—Н
Ф
----
-НВг
мч
Et*'
(83)
Вг2 2 *
Me, ХН2Вг
Et-V Вг
+ Вг •
(84) (-)
Бромирование оптически активной формы соответствующего хлоралкана — 2-метил-1-хлорбутана — также приводит к оптически активному продукту и сохранению конфигурации. Возможно, что мостиковый радикал образуется и при хлорировании, однако соответствующее экранирование, по-видимому, не столь эффективно, поскольку найдено, что галогенирование более реакционноспособным радикалом хлора ведет к полной рацемизации.
Радикальное галогенирование (особенно хлорирование), осуществленное не самими галогенами, а другими галогенсодержащими реагентами, находит широкое применение в синтезе вследствие большей селективности этой реакции. Так, хлорирование может быть проведено при взаимодействии с алкилгипо-хлоритами ROC1 (например, R=Me3C) в присутствии инициаторов, которые отрывают атом хлора от ROC1 с образованием радикала RO •. Этот радикал, как было показано, на следующей стадии отрывает атом водорода от RH. Этот реагент применяется особенно часто для аллильного хлорирования. Другим полезным реагентом для препаративного хлорирования является SO2CI2; в этом случае инициатор отрывает атом хлора, образуя • SO2C1, который, как и радикал С1 •, образующийся после отщепления SO2 от • SO2C1, может отрывать атом водорода от RH.
Широкое применение в качестве бромирующего реагента получил jV-бромсукцинимид (NBS) (88), который с высокой степенью избирательности атакует только слабые С—Н-связи, т. е. аллильные, бензильные и аналогичные положения. При использовании этого реагента необходимо присутствие инициаторов. Было показано, что при проведении бромирования необходимо поддерживать постоянную, но очень низкую концентрацию Вг2. Это достигается путем взаимодействия НВг, образующегося в ходе реакции, с jV-бромсукцинимидом (см. ниже). Как правило,
367
иеспареиного электрона с участием л-орбитальной системы бензольного ядра. Важно, что в радикале (116) мигрирует только Ph-группа, хотя миграция Me-группы могла бы привести к еще более стабилизированному радикалу Ph2CCH2Me; это еще раз подчеркивает энергетическую предпочтительность миграции через мостиковое делокализованное переходное состояние типа (117). При отсутствии Ph-группы, например, в EtM.e2CCH2-, образованном из EtM^CCHjCHO, миграция не происходит совсем, и конечным продуктом является EtMe2CCH3.
Арильные миграции не ограничиваются углерод-углеродными перегруппировками, например (ср. разд. 11.1):
Ph Ph Ph2C—OPh
д I .1 I
PhjCO—OCPhj ---► 2Ph2C—O. —► 2Ph2C—О —> Ph2C—OPh
(120) (121) (122)
Реакция в этом случае протекает через мостиковое переходное состояние; движущей силой перегруппировки является гораздо большая устойчивость радикала (122) по сравнению с радикалом (121). Наряду с 1,2-арильными сдвигами известны аналогичные миграции винильной, ацильной и ацилоксигрупп, также протекающие через мостиковые переходные состояния или интермедиаты, а также 1,2-сдвнги хлора, при которых делокализация неспаренного электрона в мостиковом интермедиате, например (126), осуществляется с участием вакантной d-орбитали атома галогена. Так, катализируемое светом присоединение НВг к алкену (123) приводит не к ожидавшемуся соединению (125), а только к соединению (127):
Н
С13ССН = СН2-^С13ССНСНгВг-^С13ССНСН2Вг + Вг
(123) (124а) (125)
/.\ • I цог I I
Cl2C—СНСН2Вг—э-С12С - СНСН2Вг ^-ci2c - СН - CH2Bf
(126) (1246) (127)
Движущей силой этой реакции является образование более устойчивого радикала, т. е. неспаренный электрон более эффективно делокализуется с участием атома хлора в радикале (1246), чем с участием атома водорода в радикале (124а). Ми
378
грация фтора не наблюдается, поскольку его d-орбитали недоступны, а миграция Вг происходит лишь изредка, так как в этом случае радикальные интермедиаты подвергаются элиминированию (с превращением в алкен) легче, чем перегруппировке.
1,2-Алкильные сдвиги не наблюдаются в растворе, однако окисление Ме3СН в режиме «холодного пламени» (в газовой фазе при 480 °C) приводит к образованию значительных количеств кетона (129):
. 1 02 I I I
Me— С—НMe— С—ОО--------->• Me—С—О—ОН 4^ С=О
Me Сн2—Н 'ч-*-СН2 Me— СН2
(128) (129)
Полагают, что образование этого кетона протекает путем межмолекулярного отщепления образовавшегося первоначально пероксильного радикала (128) (ср. разд. 11.5.2.2) с последующей миграцией Ме». Возможно, что используемые при этом жесткие условия облегчают 1,2-алкильный сдвиг или что сдвиг метильной группы может включать фрагментацию с последующим присоединением, а не прямую миграцию.
Известна также радикальная миграция атома водорода, хотя и на более длинные расстояния по сравнению с 1,2-сдвигами, например 1,5-сдвиг к кислороду через шестичленное циклическое переходное состояние при фотолизе нитрита (130) в реакции Бартона:
Н .о
Ziv / \
Ph(CH2)5ONO ----> PhCH2—Н(/ \:Н2 —►
(130) н^с—сн2
PhCH2—НС\ /СН2
Н2С—сн2
Н—О 1 +
—> PhCH2CH(CH2)3OH
11.6. Бирадикалы
Как уже упоминалось, молекулу кислорода — парамагнитную частицу с неспаренным электроном на каждом атоме — можно рассматривать как бирадикал, хотя и нереакционноспособный. Фотохимическое возбуждение антрацена с образованием бирадикала или подобной ему частицы также уже упоминалось (см. разд. 11.5.2.2). Если такое возбуждение проводится в отсутствие кислорода воздуха, вместо трансаннулярного
379
пероксида (104) получается фотодимер (131):
Н Н I
Бирадикалы образуются также в качестве интермедиатов при восстановлении магнием кетонов до пинаконов (см. разд. 8.2.7) и как анион-радикалы в ацилоиновой конденсации сложных эфиров (см. разд. 8.2.7). Полагают, что термолиз циклопропана (132) до пропена (135) при яа 500 °C также идет с образованием бирадикальных интермедиатов, например (133) и (134):
СН2 • СН2 н—СН2 СНз
/ \ а I I I
н2с—сн2 —> н—нс—сн2 —► сн—сн2 —> сн=сн2
(132) (133) (134) (135)
При образовании бирадикала (133) молекула циклопропана сначала претерпевает колебательное возбуждение за счет столкновения с другой молекулой; связь С—С вследствие этого может разрываться при условии, что избыточная энергия не теряется слишком быстро из-за последующих столкновений *. При этом имеется возможность для 1,2-сдвига водорода, в отличие от монорадикалов (см. разд. 11.5.3), вследствие спаривания электронов в бирадикале (134) с образованием л-связи (с выделением энергии). Имеются данные, что скоростьлимитирующей стадией реакции обычно и является миграция атома водорода.
Приведенные выше бирадикалы, за исключением молекулы кислорода, очень неустойчивы; однако известны гораздо более устойчивые частицы, которые также обнаруживают свойства бирадикалов. Так, в частности, углеводород (136) в растворе ведет себя как бирадикал:
CPh2
Ph2C
* Кинетическая неустойчивость бирадикалов очень осложняет экспериментальное изучение реакций их образования. Квантовохнмнческнй анализ механизма раскрытия циклопропанового кольца с образованием 1,3-бирадикала см. в кн.: Минкин В. И., Симкин Б. Я., Миняев Р. М. Квантовая химия органических соединений. Механизмы реакций. М.: Химия, 1986. Разд. 8.3. — Прим. ред.
380
Как ни удивительно, ио он ведет себя подобно радикалу Ph3C- (см. разд. 11.1). Вне раствора он существует в виде бесцветного твердого вещества, однако возможно, что он является при этом полимером, а не димером, как в случае Ph3C-. Это твердое вещество диссоциирует в растворе в такой же степени, как и димер радикала Ph3C-. Неспаренные электроны в бирадикальной форме (136) не могут взаимодействовать друг с другом, чтобы образовать диамагнитную частицу, поскольку такое взаимодействие с участием центральных бензольных ядер привело бы к возникновению jw-хиноидных форм, которые не могут существовать. Электроны, таким образом, «внутренне изолированы» друг от друга. Такая внутренняя разобщенность электронов в бирадикалах может быть вызвана стерическими, а не электронными причинами. Так, частица (137) существует в растворе как бирадикал (до ~ 17 %), находясь в равновесии с полимером, подобно бирадикалу (136):
С1 С!
С1 С1
Ph2C—(О)—(О/ CPh2^Ph2C
CPh2
Cl Cl (138)
В этом случае нет формального препятствия для взаимодействия электронов, т. е. их спаривания с образованием диамагнитной частицы (138). Однако этого в действительности не происходит из-за наличия объемистых атомов хлора в орто-положениях, мешающих бензольным кольцам принять конформацию, достаточно близкую к плоской, в которой было бы возможно перекрывание р-орбиталей, необходимое для спаривания электронов.
Довольно интересно, что некоторыми свойствами бирадикалов обладают системы, аналогичные (137), даже если они не содержат громоздких атомов хлора, способных стерически ингибировать делокализацию. Например, в случае системы (139)
(140) частицы как с п = 3, так и с п = 4 являются парамагнитными в твердом состоянии, находясь на 8 % в виде бирадикала при л = 3 и на 15 % — при п = 4 при 20 °C.
381
Глава 12
РЕАКЦИИ, КОНТРОЛИРУЕМЫЕ СИММЕТРИЕЙ
12.1. Введение.......................................................382
12.2. Симметрия орбиталей............................................384
12.3. Электроциклические реакции.....................................386
12.4. Реакции циклоприсоединения.....................................390
12.4.1. Реакция Дильса — Альдера................................391
12.4.2. Реакции 1,3-биполярного присоединения...................394
12.5. Сигматропные перегруппировки...................................395
12.5.1. Сдвигр атомов водорода..................................395
12.5.2. Сдвиги углеродсодержащих заместителей...................398
12.1. Введение
Известна небольшая группа реакций, которые, как оказалось, не протекают ни По полярному, ни по радикальному механизмам. В этих реакциях не участвуют полярные реагенты и, следовательно, на них не влияют изменение полярности растворителя, наличие инициаторов (или ингибиторов) или других катализаторов. Все попытки выделить, обнаружить или «поймать в ловушку» интермедиаты оказались безуспешными. Некоторые из таких реакций были рассмотрены ранее, например реакция Дильса — Альдера (см. разд. 7.5.2), заключающаяся в 1,4-присоединении (обычно) замещенных алкенов к сопряженным диенам
пиролитическое расщепление эфиров карбоновых кислот (1) и ксантатов (дитиокарбонатов) (3) (см. разд. 9.8) с образованием алкенов (2):
RHC — СН2 RHC=CH2 (3)
Н /°. \ /?,
O=CR О—CR
RHC—СН2 RHC=CH2 О) н \ Н о
S=CSMe S—CSMe
(2)
Такие реакции являются, по-видимому, согласованными, т. е. электронные перегруппировки, включающие как образование, так и расщепление связей, протекают одновременно в одностадийном процессе. Тем не менее совсем необязательно, чтобы каждая имеющаяся связь возникла или разорвалась в одинако
382
вой мере к тому моменту, когда достигается переходное состояние. Переходные состояния являются циклическими, содержащими преимущественно (хотя и необязательно) шесть р-элек-тронов (ср. ароматичность; разд. 1.3.6), а реакции, как правило, достигают высокой степени стереоселективности (ср. разд. 9.8). Многие из этих реакций обратимы, например реакция Дильса — Альдера, хотя равновесие часто сдвинуто в ту или другую сторону.
Для согласованных реакций, которые протекают через циклические переходные состояния, был предложен термин пери-циклические. На перициклические реакции не влияют полярные реагенты, изменение растворителей, радикальные инициаторы и т. д. На них влияют лишь термические и фотохимические воздействия, однако результаты этих воздействий могут быть различными как в отношении легкости (или вообще возможности) протекания реакции, так и в отношении их стереохимии. Так, реакция Дильса — Альдера (пример процесса циклоприсоединения) обычно может быть индуцирована термически, но не фотохимически, тогда как циклоприсоединение двух молекул алкена, например (4), с образованием циклобутана (5) может быть индуцировано фотохимически, но не термически.
Ph Ph
(4) (4) (5)
Различный стереохимический эффект отчетливо виден в случае тра«с,цис,тра«с-октатриена-2,4,6 (6). Найдено, что он циклизуется при нагревании, образуя только цис-1,2-диметилцикло-гексадиен-3,5 (7), тогда как при фотохимическом облучении он циклизуется с образованием только транс-1,2-диметилциклогек-садиена-3,5 (8):
< (8) (6)
д
130 ®с
(7)
Реакции этого типа, независимо от того, включают ли они циклизацию полиена, как в рассматриваемом случае, или же раскрытие кольца циклического соединения с образованием полиена, называют электроциклическими реакциями.
Возможность протекания такой реакции в одну стадию, по согласованному механизму, или через большее число стадий, через образование радикального или биполярного интермедиата,
383
определяется относительными значениями AG* (см. разд. 2.2) для первого механизма н для скоростьлимитирующей стадии последнего. Как известно, AG* зависит как от AS*, так и от А//*. Найдено, что для согласованных реакций характерны большие отрицательные значения AS* и небольшие положительные значения А//*; первое отражает степень упорядоченности участвующих молекул или групп, что необходимо для образования циклического переходного состояния, а последнее — степень, с которой энергия, полученная при образовании связи в переходном состоянии, может способствовать обязательному разрыву связи. Необходимо, однако, подчеркнуть, что высокие отрицательные значения AS* и низкие положительные значения А//* для какой-либо конкретной реакции не всегда свидетельствуют о протекании реакции по согласованному механизму.
Что касается величины А//*, можно предположить, что более благоприятным механизмом протекания какой-либо реакции должен быть тот, при котором в переходном состоянии сохраняется максимальное число связей, что означает сохранение перекрывания орбиталей. Для выявления условий, обеспечивающих сохранение такого перекрывания, необходимо рассмотреть свойства атомных и молекулярных орбиталей с точки зрения их симметрии.
12.2. Симметрия орбиталей
Как уже говорилось (см. разд. 1.1), отдельные электроны атома могут быть выражены волновыми функциями ф; поведение такого «волноподобного» электрона можно сравнить со стоячими волнами, которые могут возникать в струне, закрепленной у обоих концов, — модель «электрона в одномерном ящике». Первые три вида возможных колебаний показаны на рис. 12.1.
В первом случае (ф1) амплитуда волны увеличивается от нуля до максимума, а затем снова уменьшается до нуля. Во втором случае (ф2) амплитуда увеличивается до максимума, затем уменьшается, проходя через нуль (узел, отмеченный точкой), до минимума, а затем снова возвращается к нулю, т. е. знак функции изменяется один раз. В третьем случае (ф3) амплитуда изменяется от нуля до максимума, проходит через нуль до минимума, снова через нуль до максимума и, наконец, возвращается к нулю, т. е. в этом случае имеются два узла (^меченные точкой), а знак функции изменяется дважды. Перемещения выше узловой плоскости для удобства обозначены знаком «+», а перемещения ниже плоскости — знаком «—». Доли, например, 2р-атомцой орбитали, которая имеет одну узловую плоскость, таким образом, отличаются по знаку и для удобства обозначены знаками «+» и «—», как, например в формуле (9).
384
Однако это может привести к недоразумению из-за привычной ассоциации с положительным и отрицательным зарядами *, поэтому различия в знаках, которые являются чисто относительными, могут быть обозначены также в виде темных и светлых долей орбитали, т, е. так, как в (10):
узловая _w_ плоскость Q
69) (Ю)
Молекулярные орбитали (МО) получаются путем линейной комбинации атомных орбиталей, и для них также следует рассмотреть вопрос о симметрии. Так, имеются две МО (л и л*; ср. разд. 1.3.5), возникающие из двух атомных р-орбиталей (АО) в этене, и четыре МО (”ф1, трг. фз и ф<), возникающие из четырех р-М) в бутадиене (ср. рис. 1.2, разд. 1.3.5) в цисоидной конформации (см. разд. 7.5.2):
антисвязывающая
связывающая
* Важно отметить, что функция ф2, которая отражает вероятность нахождения электрона в какой-либо части пространства, всегда будет положительной, независимо от того, является ли величина ф положительной иля отрицательной.
13 П. Сайкс
385
Важно отметить, что перекрываются только те орбитали, которые имеют одинаковую симметрию (находятся в одной и той же фазе) — такое перекрывание приводит к образованию связи; орбитали, которые имеют различную симметрию, находятся в разных фазах (разрыхление).
Рассматривая относительные фазы, а следовательно, и общую симметрию участвующих орбиталей, Вудвард и Гофман смогли сформулировать в 1965 г. ряд правил. Они не только объяснили протекание перициклических реакций, которые были к тому времени известны, но и точно предсказали направление многих предполагаемых реакций. Эти предсказания были связаны с возможностью термического или фотохимического индуцирования реакции и подробной стереохимией, которая должна в этом случае наблюдаться. Их заслуга тем более велика, что некоторые предсказания (после оказавшиеся верными) были сделаны в то время, когда они казались совершенно невероятными. Чтобы сделать такие предсказания, надо было рассмотреть относительные фазы, т. е. симметрию, всех орбиталей, участвующих в процессе превращения реагирующих веществ в продукты. Вместе с тем, оказалось возможным получить достаточное представление о направлении реакций и гораздо более просто, путем применения концепции граничных орбиталей. В рамках этого подхода принимают, что электроны высшей занятой молекулярной орбитали (ВЗМО) реагирующего вещества аналогичны внешним (валентным) электронам атома. Реакция в этом случае включает перекрывание ВЗМО одного реагента (потенциальный донор электронов) с низшей свободной молекулярной орбиталью (НСМО) другого реагента (потенциальный акцептор электронов). В тех случаях, как, например, в электроциклических реакциях, когда в реакции участвует только одна частица, с использованием этого подхода должна быть рассмотрена только НСМО. Ниже анализируется ряд перициклических реакций.
12.3. Электроциклические реакции
Мы уже видели, что циклизация транс,цис,транс-октатриена-2,4,6 (6) при нагревании протекает с образованйем только цис-1,2-диметилциклогексадиена-3,5 (7), а при фотохимическом инициировании образуется только соответствующий транс-изомер (8). В любом случае равновесие почти полностью сдвинуто в сторону образования циклического продукта. Стереоселективность фактически так велика, что при термической циклизации образуется менее 0,1 % транс-изомера (8), несмотря на то что он термодинамически более устойчив, чем цнс-форма (7). Шесть МО соединения (6) —фь ф2, фз. ф<> Фз и фв, — формируемые из шести p-АО, можно изобразить следующим образом (ср. ор-
386
Шесть л-электронов распределяются по два на орбиталь, так что высшей занятой МО становится орбиталь ф3 [формула (11)]. Чтобы образовать ст-связь С—С при циклизации, каждая из долей орбитали г|?3, локализованных на концевых атомах углерода сопряженной системы (атомы С-2 и С-7, связанные с метильными группами), должна повернуться на 90° до взаимного перекрывания (p/sp2sp3; регибридизация). При этом должно произойти либо вращение обеих долей в одном и том же направлении— конротаторное вращение [формула (12)], либо вращение каждой из них в противоположном направлении — дис-ротаторное вращение [формула (13)]:
Ме
ВЗМО (¥3): И]
Me Me
(11) (13) (7)
13* 387
Конротаторное вращение ведет к перекрыванию долей орбитали ф3 с противоположными знаками, т. е. не происходит образования связей (антисвязывание). Дисротаторное вращение приводит к перекрыванию долей одного и того же знака. При этом достигается состояние связывания, приводящее к образованию циклогексадиена (7), в котором обе метильные группы находятся в цис-положении.
При фотохимическом замыкании кольца облучение возбуждает переход электрона на следующую орбиталь с более высо-hv
ким уровнем энергии, т. е. ф3 ф4, вследствие чего высшей занятой МО теперь становится орбиталь ф4 [формула (14)]:
ВЗМО(Ф4) III
Me Me
(14)
(8)
В этом случае конротаторное вращение долей орбитали ф4 ведет К перекрыванию и, тем самым, к связыванию, что делает возможным образование транс-изомера (8).
Для сравнения интересно рассмотреть обратимое превращение гексадиена-2,4 в 3,4-диметилциклобутен. При нагревании транс,транс-гексадиен-2,4 (17) переходит в транс-3,4-диметил-циклобутен (18), а при фотохимическом инициировании — в цисизомер (19):
(19) (17) (18)
В термическом взаимопревращении (равновесие смещено в направлении диена) высшей занятой МО диена (17) (ср. разд. 12.2) является орбиталь ф2 [формула (20)]. В этом случае именно конротаторное вращение ведет к связыванию и об-
388
разованию транс-диметилциклобутена (18).
связывание
(21)
Me Ме
(18)
ВЗМО(Ъ): HI
дисротаторное вращение
антисвязывание
------X-------
(20) (22)
В фотохимическом взаимопревращении (равновесие смещено в сторону циклобутана) облучение диена возбуждает переход электрона на орбиталь с более высоким уровнем энергии, т. е.
Av
ф2 —* ф3; высшей занятой МО теперь становится орбиталь ф3 [формула (23)]. Дисротаторное вращение ведет к связыванию и образованию цис-диметилциклобутена (19).
Ме
конротаторное вращение
антисвязывание
(24)
Таким образом, различия в стереохимическом результате этих реакций определяются относительными знаками (у концевых атомов углерода) молекулярных орбиталей этих (и аналогичных им) пя-электронных систем, т. е. симметрией их орбиталей. Как отмечалось ранее, доли орбиталей концевых атомов углерода имеют одинаковые знаки в ВЗМО (ф3) триена (6л-электронов) и в ВЗМО (ф3) диена (4я-электрона) после облучения. Эти же доли имеют противоположные знаки в ВЗМО (ф2) диена и в ВЗМО (фД триена после облучения. Две концевые доли МО, имеющие одинаковые знаки, должны совершить дисротаторное вращение для образования — расщепления связи.
389
Напротив, доли МО концевых атомов, имеющих противоположные знаки, должны совершить конротаторное вращение перед образованием — расщеплением связи. Это различие в результатах термического и фотохимического воздействий можно суммировать следующим образом:
Число л-электроиов Условия реакции Тип вращения, необходимый для связывания
4п Термические Конротаторный
4п Фотохимические Дисротаторный
4п + 2 Термические Дисротаторный
4п + 2 Фотохимические Конротаторный
Электроциклические реакции имеют важное значение для образования углерод-углеродной связи, так как их стереоспецифичность гораздо выше, чем в большинстве других, несогласованных реакций, протекающих через бирадикальные или биполярные интермедиаты.
12.4. Реакции циклоприсоединения
В реакциях циклоприсоединения обычно участвуют два компонента, и возможность согласованного процесса будет определяться возможностью перекрывания ВЗМО одного компонента и НСМО другого. Так, в системе диен — моноен
ВЗМО (Y2)
НСМО (п*)
НСМО (Т3)
ВЗМО (л)
взаимодействие оказывается связывающим: наблюдают согласованное присоединение независимо от того, какой компонент представляет ВЗМО, а какой — НСМО. В этом случае говорят, что циклоприсоединение разрешено по симметрии. Наоборот, если оба компонента являются моноенами
ВЗМО
<п> н
НСМО (л*)
взаимодействие оказывается несвязывающим, согласованное присоединение становится невозможным. В этом случае говорят о циклоприсоединении, запрещенном по симметрии.
390
Существует общее положение для термических, согласованных реакций присоединения: реакции, в которых участвуют 4л-и 2л-электронные системы, протекают легко, например реакция Дильса — Альдера, тогда как реакции, в которых участвуют две 2л-электронные системы, например циклодимеризация алкенов, не протекают. Однако можно ожидать, что фотохимическая циклодимеризация алкенов будет реакцией, разрешенной по симметрии, так как облучение возбуждает электрон одного из компонентов для перехода на орбиталь с более высоким уровнем энергии, т. е. л —-> л’, и интересующая нас ВЗМО становится л*-орбиталью:
hv
ВЗМО(-» л*)
НСМО (л*)
Многие реакции такого типа можно осуществить в препаративных целях в фотохимических условиях, хотя по причинам, которые мы здесь не будем рассматривать (речь идет о деталях механизма фотохимических изменений), они часто не являются согласованными, однако протекают через бирадикальные интермедиаты. Одна из реакций фотохимического (2л + 2л)-циклоприсоединения, на которую мы уже ссылались, действительно протекает как согласованный процесс:
Ph Ph
(4) (4)
Ph Ph (5)
Важность же (4л + 2л)-термического согласованного циклоприсоединения настолько велика, что оно заслуживает отдельного рассмотрения.
12.4.1. Реакция Дильса — Альдера
Реакция Дильса—Альдера является одной из наиболее известных реакций (4л + 2л)-циклоприсоединения. Она уже обсуждалась в некоторой степени (см. разд. 7.5.2) в связи с тем фактом, что протекает строго сим-стереоспецифично как по
391
отношению к диену (26), так и по отношению к диенофилу (27):
(26) (27)
Это является убедительным доказательством согласованного механизма, предусматривающего одновременное образование обеих новых о-связей в переходном состоянии. Однако, поскольку на реакцию заметно влияет электронный эффект заместителя, можно предположить, что в переходном состоянии обе новые связи необязательно образуются в одинаковой степени. Найдено, в частности, что реакция активируется электронодонор-ными заместителями в диене и электроноакцепторными заместителями в диенофиле, причем в отсутствие последних она идет очень медленно, или не идет совсем. Такие заместители понижают уровень энергии НСМО в диенофиле и повышают уровень энергии ВЗМО в диене, тем самым повышая степень взаимодействия между ними. Наличие заместителей и даже гетероатомов в системе, по-видимому, не влияет на симметрию участвующих орбиталей.
Заместители в диене могут также влиять на циклоприсоединение стерически, определяя положение равновесия между цис-и т/щнс-формами: для взаимодействия с диенофилом необходима цисоидная конформация диена. Так, объемистый 1-цис-за-меститель в соединении (28) замедляет, реакцию, тогда как объемистый заместитель в положении 2 диена (29) ускоряет ее:
(29)
Со стереохимической точки зрения, важно также, что в некоторых реакциях Дильса — Альдера существуют альтернативные пути присоединения, приводящие к образованию экзо- (32) и эндо- (33) аддуктов, например при реакции циклопентадиена
392
(30) с малеиновым ангидридом (31) (диенофил):
Несмотря на тот факт, что экзо-аддукт, вероятно, термодинамически более устойчив, часто (хотя и не всегда) в реакциях Дильса — Альдера эндо-аддукт является основным или даже единственным продуктом. Чтобы объяснить это, предположили, что при эндо-присоединении возможна стабилизация переходного состояния (скорость реакции поэтому возрастает) вследствие вторичного взаимодействия долей ВЗМО, например, в диене (30) и НСМО в диенофиле (31), которые сами по себе не участвуют непосредственно в образовании связи (при условии, что они имеют одни и те же знаки). Подобные взаимодействия были бы невозможны, конечно, в переходном состоянии экзо-присоединения, так как соответствующие фрагменты в соединениях (30) и (31) отстоят слишком далеко один от другого. Таким образом, образование эндо-аддукта контролируется кинетически. В этой связи важно, что относительное содержание экзо-аддуктов может иногда увеличиваться при большой продолжительности реакции; образовавшийся на начальном этапе эндо-продукт (кинетический) затем превращается в более устойчивый экзо-продукт (термодинамический) путем обращения реакции и последующего экзо-реприсоединения (ср. разд. 10.4.3).
Большим преимуществом реакции Дильса — Альдера (как
процесса, приводящего к образованию углерод-углеродной связи) является ее универсальность. Число различных диенофилов, которые могут быть использованы в препаративных целях, очень велико. Возможны вариации и в ряду диенов, но не столь широкие. В соответствующих условиях реакции циклоприсоединения обычно идут с удовлетворительными выходами. Подобно другим реакциям циклоприсоединения, реакции Дильса — Альдера потенциально обратимы: в некоторых случаях обратный процесс — ретрореакция Дильса — Альдера — также может использоваться в препаративных целях. Например, циклопентадиен (30) легко вступает в автореакцию Дильса — Альдера с образованием трициклического димера; он обычно сохраняется в этой относительно стабильной форме и вновь превращается (если потребуется) в (30) при нагревании, например при перегонке. Термическое расщепление циклогексена (аддукт
393
реакции Дильса — Альдера из бутадиена и этена, хотя и не получаемый этим путем!) было применено как удобный метод получения бутадиена в лабораторных условиях. Известно несколько реакций Дильса — Альдера, особенно для соединений, содержащих гетероатомы и (или) высокополярные заместители, которые протекают через несогласованный, двухстадийный механизм, включающий цвиттер-ионные интермедиаты. Однако до сих пор не удавалось наблюдать реакции, протекающие по двухстадийному механизму с промежуточным образованием бирадикала. Пиролитическое сия-элиминирование сложных эфиров карбоновых кислот и дитиокарбонатов (см. разд. 9.9) также можно рассматривать как аналог ретрореакции (4л + 2л)-циклоприсоединения.
12.4.2. Реакции 1,3-биполярного присоединения
В реакции (4л + 2л)-циклоприсоединения 4л-электронный компонент не обязательно должен быть четырехатомной системой (как в случае 1,3-диенов) и состоять только из атомов углерода (при условии выполнения требований симметрии ВЗМО и НСМО компонентов для согласованного механизма). Чаще всего такие недиеновые 4л-электронные системы содержат три атома и имеют одну или несколько биполярных канонических структур, например (34а). Этим определяется и название этих реакций— 1,3-биполярное присоединение. Вместе с тем такие реакции не нуждаются в большом постоянном диполе [ср. диазометан (34а)-«-► (346)]:
_ N n
-«-> h2C^>N
34а) (346)
Первоначальное присоединение озона к алкенам с образованием мольозонидов (см. разд. 7.4.6) можно рассматривать как 1,3-биполярное присоединение; многие подобные реакции имеют большое значение для получения пятичленных гетероциклических систем. Так, уже было рассмотрено получение 1,2,3-три-азола из PhN~—N=N+ (см. разд. 7.4.6); аналогично получают дигидропиразол (35) из диазометана (34):
V2
EtO2C—НС_ ,N
--->- EtO2C
(34) (35)
394
12.5. Сигм атропные перегруппировки
Известна большая группа перициклических реакций, протекающих с миграцией о-связи (отсюда и название этих реакций) в пределах л-электронной структуры. Простейшие примеры включают миграцию о-связи с атомом водорода.
12.5.1. Сдвиги атомов водорода
Такие реакции в ациклических в обобщенной форме:
полиенах можно представить
Н Н
I , । ,
R2C(CH=CH)xCH=CR2 ----► R2C=CH(CH=CH)xCR2
(36) (37)
При обсуждении возможности протекания этих сдвигов как согласованных процессов, т. е. через циклические переходные состояния, необходимо рассмотреть симметрию участвующих орбиталей. Модель переходного состояния может быть построена на допущении, что мигрирующая о-связь С—Н может быть расщеплена с образованием водорода с ls-орбиталью и углерода с 2р-орбиталью. Для соединения (36; х= 1) переходное состояние можно рассматривать как пентадиенильный радикал (38) с атомом водорода (один электрон на ls-орбитали), мигрирующим между концевыми атомами углерода 5л-электронной системы (всего в системе участвует шесть электронов):
По аналогии с рассмотренными ранее перициклическими реакциями, возможность такой миграции может быть решена на основе анализа знаков долей высшей занятой МО на концевых атомах углерода системы, т. е. на основе анализа симметрии ВЗМО пентадиенильного радикала (38). Он является 5л-элек-тронной системой, и его электронная конфигурация должна быть записана как Его ВЗМО является, следовательно,
орбиталь ф3. Можно показать, что эта МО имеет концевые атомные орбитали одинакового знака, так что перекрывание между ls-орбиталью атома водорода и обеими концевыми долями МО радикала (38) оказывается возможным в переходном состоянии (39).
395
Термические 1,5-сдвиги атомов водорода являются, таким образом, разрешенными и вследствие симметрии переходного состояния (39), поскольку атом водорода в продукте (37; х= 1) оказывается на той же стороне общей плоскости полиена, на которой он находился в исходном веществе (36; х=1); такой сдвиг называют супраповерхностным. Это утверждение ие могло быть проверено экспериментально в приведенном выше примере. Однако то, что термические 1,5-сдвиги (которые достаточно широко распространены) действительно включают строго супраповерхностную миграцию, обнаружено на примере соединения (40). Было показано, что при нагревании этого соединения получается смесь диенов (41) и (42), образующихся вследствие супраповерхностных сдвигов в альтернативных конформациях (40а) и (406) соответственно:
(40а) (41)
(406) (42)
Концевые доли ВЗМО имеют одни и те же знаки в нонате-траенильном радикале (36; х = 3); 1,9-сдвиги (система содержит 10 электронов) должны быть также разрешены и являться супраповерхностными. Однако образование требуемого 10-членного переходного состояния должно быть стерически затрудненным, и сомнительно, чтобы реально можно было наблюдать такие согласованные 1,9-сдвиги. Супраповерхностные термические сдвиги не наблюдаются и в других «разрешенных», т. е. (4п + + 2)-электронных системах, например (36; х = 3, 5, ит. д.).
Возможно, что сферически симметричная ls-орбиталь атома водорода могла бы перекрываться через плоскость атомов углерода полиена в тех случаях, когда концевые доли его ВЗМО имели бы противоположные знаки; при этом наблюдалось бы антараповерхностное перекрывание. Концевые доли ВЗМО имеют противоположные знаки для соединения (36) при х = 0, 2, 4 и т. д., что могло бы привести к переходному состоянию
396
типа (43), например, для х = 0 (система из четырех электронов) :
(43)
Такое переходное состояние является, однако, весьма напряженным; никаких 1,3-антараповерхностных сдвигов в действительности не наблюдали. В то же время на примере соединений ряда витаминов D наблюдали 1,7-термический антараповерх-ностный сдвиг в системе (36; х = 2); в этом случае переходное состояние, по-видимому, является гораздо менее напряженным, т. е. способно принимать требуемую спиральную конфигурацию.
Вместе с тем 1,3-фотохимические сдвиги разрешены и являются супраповерхностными [(44)->-(46)], так как ВЗМО переходного состояния (45) (ф3; благодаря переходу —► -—► Ф[Фз) в этом случае имеет концевые доли одинакового знака:
R2C— ch=cr2—*
Н
(44)
R*W^R-~-R2C=CH~CR2 н н
(45) (46)
Действительно, оказалось, что такие 1,3-сдвиги довольно обычны. 1,5-Фотохимические сдвиги в системах (36; х=1) должны были бы быть антараповерхностными, однако соответствующее переходное состояние должно быть весьма напряженным, так что примеры 1,5-сдвигов не известны. 1,7-Фотохимиче-ские сдвиги в системах (36; х = 2) должны быть разрешены и являться супраповерхностными; действительно, наблюдали переход (47) -► (48):
(47)
Реализация 1,7-фотохимического сдвига атома водорода в этом соединении сама по себе не означает, что этот сдвиг протекает по супраповерхностному механизму, однако относительно жесткая циклическая структура (47) должна исключать возможность антараповерхностного сдвига.
397
12.5.2. Сдвиги углеродсодержащих заместителей
Наиболее известным примером является сдвиг от одного атома углерода к другому, наблюдаемый в перегруппировке Коупа 1,5-дненов [(49)-► (50)] (не путать с элиминированием по Коупу; см. разд. 9.9), а также сдвиг от атома кислорода к атому углерода при перегруппировке Кляйзена простых аллилариловых эфиров [(51) (52)]:
В случае термических реакций наиболее легко протекают такие реакции, при которых возможно образование шестичленных переходных состояний, и, несомненно, такие реакции наиболее распространены. Предпочтительность шестичленного переходного состояния в конформации кресла доказывается тем фактом, что мезо-форма (49) дает только цис,транс-форму (50а) (99,7 %) из трех возможных геометрических изомеров (цис,цис-, цис,транс- и транс,транс-) соединения (50):
Это соответствует сдвигу, который является супраповерх-ностным относительно обоих концов мигрирующей системы.
Перегруппировка Кляйзена является строго внутримолекулярной; для нее характерно большое отрицательное значение AS+, соответствующее степени упорядоченности циклического переходного состояния. Циклическое строение переходного состояния подтверждается также опытами с введением метки 14С:
398
\
положение атома |4С в аллильной группе «обращается» во время миграции [ (51а)(52а) ]:
(51а) (53а) (52а)
Диеновый интермедиат (53а), как и его енольная форма (52а), тоже способен подвергаться перегруппировке Коупа с образованием второго диенона [ср. (56а)], енольная форма которого является пара-замещенным фенолом [ср. (57а)]. Еиоли-зация, как правило, преобладает; однако, если в соединении (51) имеются заместители в орто-положениях, как, например, в (54а), «о-енолизация» невозможна и образуется только п-за-мещенный фенол (57а). Найдено, что этот продукт действительно образуется не путем прямой миграции аллильной группы, а путем двух последовательных сдвигов, включающих «двойную инверсию» положения 14С-метки в аллильной группе:
(54а) (55а)
(56а) (57а)
Другим подтверждением двукратного сдвига и двойной инверсии положения метки 14С является «улавливание» (ср. разд. 2.3.4) первого диенового интермедиата (55а) с помощью малеинового ангидрида в реакции Дильса — Альдера. Аналогичная
399
перегруппировка происходит в аллильных эфирах типа (58/:
(58)
(59)
Эта реакция также является согласованной и протекает через шестичленное переходное состояние, но в этом случае частица (59), соответствующая еноновому интермедиату (53а) в ароматической перегруппировке Кляйзена, фактически является конечным продуктом. Это объясняется отсутствием в случае (59) того энергетического фактора, который был бы сравним с реароматизацией в ходе превращения (53а) -> (52а).
Наконец, следует подчеркнуть, что в тех случаях, когда какая-либо электроциклическая реакция (циклоприсоединение или сигматропные перегруппировки, рассмотренные выше) описывается как реакция, запрещенная по симметрии, это относится только к согласованному механизму. Вполне может оказаться энергетически осуществимым несогласованный механизм, включающий цвиттер-ионные или бирадикальные интермедиаты. Равным образом, утверждение, что реакция является разрешенной по симметрии, вовсе не является гарантией того, что она будет легко протекать в действительности; достижение переходного состояния, имеющего требуемую геометрию, может быть ограничено размером соответствующего кольца, наличием тех или иных заместителей или другими факторами.
Глава 13
СООТНОШЕНИЯ ЛИНЕЙНОСТИ СВОБОДНЫХ ЭНЕРГИЙ
13.1. Введение..............'.......................................401
13.2. Первые зависимости Гаммета....................................401
13.3. Уравнение Гаммета.............................................404
13.3.1. Вывод уравнения Гаммета .............................. 404
13.3.2. Константа заместителя ох..........................404
13.3.3. Константа реакции р....................................405
13.3.4. Физическое содержание константы ох.....................406
13.3.5. Физическое содержание константы р.....................409
400
13.3.6. Прямое полярное сопряжение. Константы и . .411
13.3.7. Уравнение Юкава — Цуно............................... 415
Г3.4. Применение уравнения Гаммета................................ 416
13.4.1 . Расчеты значений k и К................................417
13.4.2 . Отклонения от линейных зависимостей...................417
13.4.3 . Зависимости, отклоняющиеся вверх.................... 418
13.4.3.1 . Ацетолиз З-арил-2-бутил-п-бромбензолсульфонатов 418
13.4.3.2 . Гидролиз ArCOzR в 99%-й H2SO4................421
13.4.4 . Зависимости, отклоняющиеся вниз.......................423
13.4.4.1 . Циклодегидратация диарил (бифенилил-2)метанолов 423
13.5. Стерические эффекты...........................................426
13.5.1. Уравнение Тафта........................................426
13.5.2. Стерические константы Е, и б......................... 428
13.6. Эффекты растворителя . .......................................431
13.6.1. Зависимость константы р от природы растворителя .... 431
13.6.2. Уравнение Грюнвальда—Уинстейна . ......................432
13.6.3. Параметр Димрота (параметр £т) 434
13.7. Корреляции спектроскопических параметров......................435
13.8. Корреляции термодинамических параметров.......................436
13.1. Введение
В предыдущих главах была рассмотрена относительная реакционная способность различных соединений в специфических реакциях, например при нуклеофильном замещении действием ЕЮ" в ряду бромалканов (ср. разд. 4.3):
СН3СН2Вг > МеСН2СН2Вг > Ме2СНСН2Вг > Ме3ССН2Вг
Наблюдаемые ряды активности пытались объяснить действием электронных и стерических эффектов. Этот путь является и полезным, и благодарным, однако существенным недостатком таких исследований и объяснений является то, что они остаются качественными; необходим метод для сопоставления структуры и реакционной способности на количественной основе.
13.2. Первые зависимости Гаммета
Гаммет еще в 1933 г. показал, что при взаимодействии метиловых эфиров ряда карбоновых кислот (1) с NMe3 скорости реакций прямо связаны с константами диссоциации (в воде) соответствующих карбоновых кислот (2):
k
RCO2Me + NMe3 ----> RCOJ + +NMe<
(1)
К
RCO2H + H2O RCO2 + H3O+
(2)
Так, откладывая значение lg£ для реакции сложных эфиров, например (1) против 1g К. для диссоциации кислот (2) (факти-
401
чески откладывали значения —1g), получили удовлетворительную линейную зависимость (рис. 13.1).
Константы равновесия К и константы скорости k связаны с изменениями свободной энергии (см. разд. 2.1 и 2.2.2) в соответствующих реакциях следующим образом:
1g к = - Д0о/(2,303ЯГ),
lg k = - AG+/(2,303RT) + lg[k'T/h],
где k' — константа Больцмана; h — константа Планка.
Линейная зависимость (см. рис. 13.1) между —lg k реакции эфиров (1) и —1g К ионизации в воде соответствующих карбоновых кислот (2) означает, что существует также линейная зависимость между AG+ (свободная энергия активации) реакции эфиров и AG° (стандартная свободная энергия) диссоциации кислот в воде. Линейные графики, подобные графику, приведенному на рис. 13.1, обычно называют соотношениями линейности свободных энергий *.
Другим примером первых обобщений Гаммета является график (рис. 13.2), который представляет собой зависимость lg k катализируемого основанием гидролиза этиловых эфиров (3) некоторых карбоновых кислот от 1g К диссоциации в воде соответствующих карбоновых кислот (2):
к
RCO2Et + ‘ОН ----► RCO2 + ЕЮН
(3)
Однако в этом случае линейная зависимость сохраняется лишь для бензойной кислоты и ее n-Ме- и п-МОг-производных, тогда как точки, соответствующие o-NO2- и о-С1-бензойным кислотам, сильно отклоняются по одну сторону от этой прямой ли-
* Теоретическое обоснование этих соотношений получило название принципа линейности свободных энергий (ЛСЭ). Подробнее об этом см. в кн.: Пальм В. А. Основы количественной теории органических реакций. Л.: Химия, 1967.--Прим. ред.
402
нии, а точки, соответствующие ' алифатическим карбоновым кислотам — этановой и 2-гид- в роксипропановой,— сильно от- s клоняются по другую сторону -? от той же прямой линии. Гам-мет обнаружил, что линейная зависимость часто не получается, если в соответствующие зависимость' включают данные для реакций о-замещенных производных бензола или али
фатических соединений. Вместе с тем он показал, что если при построении такой зависимости ограничиться реакциями м- и n-замещенных производных бензола, то получается превосходная линейная зависимость, как, например, в случае гидролиза сложных эфиров (рис. 13.3), причем это справедливо для большого числа различных реакций таких производных.
Причину такого несоответствия о-замещенных бензола и алифатических производных найти нетрудно. Так, в сложном эфире (За) заместитель в мета- или пара-положении удален от реакционного центра и в такой жесткой молекуле не может оказывать на него никакого стерического влияния (щелочной гидролиз; см. разд. 8.6.1). В эфире (36) заместитель в орто-положении
(За) - м- или n-замещенные ароматические сложные эфиры; (36) - о-замещен-ный ароматический сложный эфир; (Зв) - алифатический сложный эфир; (Зг) -(Зе) - тетраэдрические интермедиаты; ; - скоростьлимитирующая стадия
403
расположен вблизи реакционного центра (ср. разд. 8.6.3), что приводит к повышению напряженности переходного состоя-/ ния при образовании тетраэдрического интермедиата во времй медленной, скоростьлимитирующей атаки эфира (36) ионом ~ОН. Практически то же самое справедливо и для гибких Молекул алифатических эфиров типа (Зв). Подобные стерические влияния будут гораздо меньше (или не будут проявляться вообще) при удалении атома водорода от группы СО2Н под действием Н2О (процесс ионизации карбоновых кислот в воде).
13.3. Уравнение Гаммета
Несмотря на установление линейных зависимостей для большого числа реакций м- и n-замещенных производных бензола, все еще отсутствовало какое-либо четкое количественное соотношение, которое можно было бы использовать для изучения и предсказания новых фактов; в этом отношении Гаммет был первым, кто смог решить эту проблему.
13.3.1. Вывод уравнения Гаммета
Общее уравнение прямой линии у = тх + с для прямой линии, приведенной на рис. 13.3, приобретает вид:
lgfex = P1gy<x + c- (13-1)
где р— тангенс угла наклона прямой линии; с — отрезок, отсекаемый на оси ординат; X — заместитель в мета- или пара-положении бензольного кольца рассматриваемого соединения.
Аналогичное уравнение справедливо в случае незащищенных эфира и кислоты, т. е. если X = Н:
lgfeH = P18y<H + f- О3-2)
Вычитая уравнение (13.2) из уравнения (13.1), получаем уравнение (13.3), которое может быть записано также в виде уравнения (13.4):
MMhWOsKx - ’еКн)’ <13-3)
>g[^H] = p’gHx/W (13-4)
13.3.2. Константа заместителя <Тх
Гаммет рассматривал диссоциацию м- и n-замещенных бензойных кислот в воде при 25 °C в качестве стандартной реакционной серии. Он выбрал эту реакцию потому, что для большого
404
числа различных м- и n-замещенных бензойных кислот уже были определены с достаточной точностью значения констант Диссоциации в воде Кх- Зная Кн и Кх, можно определить стх:
ax = lg[Ax/KH],
(13.5)
где <Ух является константой заместителя, которая постоянна для конкретного заместителя в конкретном положении (м- или п-) независимо от природы реакции, в которой участвует производное бензола, содержащее данный заместитель. Подставляя значение 1£(Хх/Хн) из уравнения (13.5) в уравнение (13.4), получим уравнение (13.6), которое обычно называют уравнением Гаммета **:
’В [*х/*н] = Р°х- (13-6)
Используя известные значения Кх (или рХа) для диссоциации в воде м- и n-замещенных бензойных кислот [или измеряя Кх (или рХа), если они еще не известны для данного м- или «-заместителя], можно рассчитать ох. Значения таких констант для некоторых заместителей X приведены ниже:
X ам-Х ап-Х X ам-Х °п-Х
Ме3С —0,10 -0,20 С1 +0,37 +0,23
Me -0,07 -0,17 МеСО +0,38 +0,50
Н 0 0 Вг +0,39 +0,23
МеО + 0,12 —0,27 CN + 0,56 +0,66
НО +0,12 —0,37 no2 +0,71 +0,78
F +0,34 +0,06
Вряд ли удивительно, что значение ах для данного заместителя зависит от его положения и имеет разные значения для мета- и пара-положений.
13.3.3. Константа реакции р
Зная значения констант ох для ряда заместителей, можно рассчитать значение константы реакции р в уравнении (13.6); часто это делают графически. Так, чтобы оценить р, например, для щелочного гидролиза м- и п-замещенных этил-2-арилэтаноа-
* Уравнение (13.5), конечно, можно записать в форме ах = рКа(Н)— —рАа(Х), так что численное значение ах для конкретного заместителя получается путем простого вычитания рАа замещенной кислоты (если она известна) из рКа самой бензойной кислоты.
** Атом водорода в уравнениях Гаммета рассматривается в качестве стандартного заместителя; его константа — значение ан — принята равной нулю. — Прим. ред.
405
тов (4)
(4)
можно было бы, исходя из кинетических измерений (или по литературным данным, если они имеются), получить kH для незамещенного эфира и kx по крайней мере для трех различных замещенных эфиров. Зная значение ох для каждого из трех этих заместителей, можно построить график зависимости lg(£x/£H) от ох. Константа р [см. уравнение 13.6] представляет собой тангенс угла наклона прямой линии; в частности, в данном случае она равна +0,82 при гидролизе в водном растворе этанола при 30 °C. Значения р для различных реакций м- и п-замещенных производных бензола приведены ниже:
Реакция Константа р
(1) ArNH2 + 2,4-(O2N)2C6H3Cl(EtOH, 25 °C) k -3,19
(2) ArNH2 + PhCOCl (C6H6, 25 °C) k -2,69
(3) Сольволиз ArCH2Cl (води. Me2CO, k —1,88
69,8 °C)
(4) ArO~ + EtI (EtOH, 25 °C) k -0,99
(5) ArCO2H + MeOH (кисл. катализ, 25 °C) k —0,09
(6) Кислотный гидролиз ArCO2Me (води. k +0,03
MeOH, 25 °C)
(7) Диссоциация АгСН2СО2Н (H2O, 25 °C) К +0,47
(8) АгСН2С1 + Г(Ме2СО, 20°C) k +0,79
(9) Щелочной гидролиз ArCH2CO2Et (води. k +0,82
EtOH, 30 °C)
(10) Диссоциация АгСО2Н (Н2О, 25°C)* К +1,00
(11) Диссоциация АгОН (Н2О, 25°С) К +2,01
(12) ArCN+H2S (щел. среда, EtOH, 60,6 °C) k +2,14
(13) Щелочной гидролиз ArCO2Et k +2,51
(води. EtOH, 25°C)
(14) Диссоциация ArNH* (Н2О, 25 °C) К +2,73
* Стандартная реакционная серия-
Для стандартной реакционной серии, т. е. диссоциации в воде м- или «’-замещенных бензойных кислот при 25°С, р = 1,0, что является обязательным условием определения ох в уравнении (13.5) и его использования в уравнении (13.6). Значение константы реакции р для данной серии в определенных условиях постоянно, независимо от положения заместителей (мета- или пара) в реагирующем соединении.
13.3.4. Физическое содержание константы ох
Прежде чем продолжить рассмотрение применения уравнения Гаммета, необходимо обсудить физическое содержание кон
406
стант (Ух и р в терминах уже знакомых нам факторов, влияющих на скорости и равновесия реакций.
Если рассмотреть сначала константу заместителя <ух и сравнить значения <тм.х (см. разд. 13.3.2), то можно увидеть, что константы для л-Ме3С- и л-Ме-групп имеют небольшие отрицательные значения. При X = Н значение пх = 0, тогда как для других ^-заместителей константы пх имеют возрастающие положительные значения. Изменение знака констант [от (—) к (+)] соответствует изменению направления (электронодонорный -> электроноакцепторный) индуктивного эффекта, проявляемого этими заместителями. Заместители могут также проявлять эффект поля (ср. разд. 1.5.1), действующий через среду, но он имеет то же направление, что и индуктивный эффект. Таким образом, можно сказать, что (УЛ х как по направлению, так и по численному значению представляют собой меру общего полярного эффекта, оказываемого заместителем X на реакционный центр.
Это видно при сравнении скоростей щелочного гидролиза (ср. разд. 8.6) замещенных этилбензоатов (5) и (6), содержащих соответственно заместители л-NOz и л-Ме, и незамещенного эфира. В этой реакции медленной, скоростьлимитирующей стадией является атака эфира ионом -ОН (см. разд. 8.6.1):
(5) переходное тетраэдрический
состояние
интермедиат
°м-МОг _ +0171 ^m-NOz^h - 63,5
(6)
переходное тетраэдрический
состояние
интермедиат
О^-МеО ~~0»07 ^м-Ме/^н — 0,66
407
л-Нитроэфир (5), для которого o\<no2 = + 0,71, гидролизуется в 63,5 раза быстрее, чем незамещенный эфир (сильное оттягивание электронов заместителем способствует атаке карбонильного углеродного атома ионом ~ОН и стабилизации переходного состояния на пути к Образованию отрицательно заряженного тетраэдрического интермедиата). В то же время л-Ме-эфир (6) (сг^-ме = — 0,07) гидролизуется лишь в 0,66 раза быстрее, чем незамещенный эфир (очень слабое электронодонорное действие заместителя ингибирует атаку ионом "ОН и т. д.).
При сравнении значений констант оп-х (см. разд. 13.3.2) видно, что ол-х для каждого конкретного заместителя X отличается не только по численному значению от ом-х, но может отличаться и по знаку, как это имеет место, например, в случае м- и п-МеО. Сравнение эффектов заместителя л-МеО и п-МеО для эфиров (7) и (8) соответственно в одной и той же реакции (щелочной гидролиз) делает понятной причину этого измене-
ния знака:
(7)
переходное тетраэдрический
состояние
интермедиат
°м-МеО = + °>12 ^м-МеО>*н
(8) переходное тетраэдрический
состояние
интермедиат
°л-МеО ~ ” °.07 ^Н^л-МеО
При наличии группы МеО в лета-положении электроотрицательный атом кислорода метоксигруппы проявляет электроно-
408
акцепторный индуктивный эффект (Ол-мео = + 0,12), поэтому гидролиз протекает быстрее, чем в случае незамещенного эфира [ср. .м-МОг-эфир (5)]. В пара-положении МеО-группа также проявляет электроноакцепторный индуктивный эффект, однако, кроме того, благодаря своей паре электронов эта группа может проявлять и электронодонорный мезомерный эффект по отношению к атому углерода кольца, с которым связана группа CO2Et. Мезомерный эффект, вследствие того что он проявляется в более легко поляризуемой системе л-электронов, является преобладающим среди этих двух эффектов, поэтому результатом оказывается суммарный электронодонорный эффект (сг„.мео = =—0,27). В соответствии с этим п-МеО-эфир (87) гидролизуется заметно медленнее, чем незамещенное соединение (ср. разд. 6.7.1.3).
Таким образом, константу сгх можно рассматривать как меру общего полярного влияния (общий полярный эффект) заместителя X на реакционный центр. Знак константы сгх определяет характер эффекта [(—)—электронодонорное, (+)—электроноакцепторное), а ее значение — количественную оценку влияния, которое оказывает заместитель X, по сравнению с тем эффектом, который имеет атом водорода. Таким образом, предполагаемое постоянство константы сгх в большом числе самых разнообразных реакций не обязательно означает, что абсолютный полярный эффект заместителя X всегда остается постоянным, а означает, что этот эффект постоянен лишь относительно эффекта атома водорода.
13.3.5. Физическое содержание константы р
Рассмотрим теперь константу реакции р. Сравнивая значения констант р (см. разд. 13.3.3), выберем для начала реакцию с отрицательным значением р, например реакцию (2) — бензоилирование м- и п-замещенных анилинов (9) (р = —2,69), и обсудим эту реакцию более подробно:
Показано, что ее медленной, скоростьлимитирующей стадией является первоначальная атака парой электронов атома азота замещенного анилина (9) по карбонильному углеродному атому хлорангидрида. Это ведет к появлению положительного заряда
409
на реакционном центре — атоме азота, непосредственно связанном с замещенным бензольным кольцом в формирующемся интермедиате (10). Вследствие этого реакция ускоряется электронодонорными заместителями, которые помогают делокализовать положительный заряд, наводимый в переходном состоянии, ведущем к интермедиату (10), и соответственно замедляется электроноакцепторными заместителями. Найдено, что это положение справедливо для всех реакций с отрицательными значениями р.
Ранее уже была рассмотрена реакция с положительным значением р — реакция (13) (см. разд. 13.3.3), т. е. щелочной гидролиз м- и n-замещенных этилбензоатов (11), для которой р = = +2,51:
В медленной, скоростьлимитирующей стадии этой реакции в переходном состоянии, приводящем к интермедиату (12), вблизи реакционного центра наводится отрицательный заряд. Суммарная реакция, как мы уже видели (см. разд. 13.3.4), ускоряется электроноакцепторными и замедляется электронодонорными заместителями.
Константу р, таким образом, можно рассматривать как меру чувствительности реакции к электронодонорному или электроноакцепторному эффекту, проявляемому заместителем X по сравнению, конечно, с чувствительностью (к такому же заместителю) стандартной реакции — диссоциации в воде м- и n-замещенных бензойных кислот при 25 °C, для которой значение р принято равным +1,00. Отрицательное значение константы р означает наведение положительного заряда (или, очевидно, снижение отрицательного заряда) в реакционном центре переходного состояния на скоростьлимитирующей стадии общей реакции, и, наоборот, положительное значение означает наведение отрицательного заряда (или снижение положительного заряда) в реакционном центре. Константу р можно рассматривать, следовательно, как меру изменения плотности заряда в реакционном центре переходного состояния или при изменении положения равновесия.
На этом основании можно ожидать, что константа р для ряда аналогичных реакций будет уменьшаться при удалении реак
410
ционного центра от заместителя, который оказывает на этот центр полярный электронный эффект. Это видно при сравнении констант р для диссоциации ряда кислот (13) — (16) в воде.
Кислота р Кислота р
ХС6Н4СО2Н (13) 1,00* ХС6Н4СН2СН2СО2Н (15) 0,21
ХС6Н4СН2СО2Н (14) 0,49 ХС6Н4СН=СНСО2Н (16) 0,47
* Стандартная реакционная серия
Введение сначала одной, а затем двух СН2-групп между бензольным кольцом и группой СО2Н постепенно снижает чувствительность процесса диссоциации кислоты к полярному эффекту заместителя X в бензольном кольце. Однако эта чувствительность, оцениваемая значением р, возрастает для кислоты (16), поскольку группа СН=СН заметно лучше проводит электронные влияния, чем группа СН2—СН2.
13.3.6. Прямое полярное сопряжение.
Константы а~ и о+
Прежде чем продолжить анализ соотношений линейности свободных энергий, необходимо более внимательно посмотреть, насколько постоянным является значение константы <тх для данного заместителя. Если построить график, откладывая значения lg(Kx/tfH) для диссоциации в воде м- и л-замещенных бензойных кислот (13) и lg(Xx/An) для диссоциации соответствующих замещенных фенолов (17), то для большого числа различных заместителей получается весьма удовлетворительная линейная зависимость (рис. 13.4).
(1) ХС6Н4СО2Н + Н2О ХС6Н4СО2 + Н3О+
(13)
(2) ХС6Н4ОН + Н2О ХС6Н4О” 4- Н3О+
(17)
Однако два заместителя — сильные электроноакцепторные группы n-NO2 и n-CN — лежат выше прямой линии; это означает, что л-нитро- и л-циано-фенолы являются фактически более сильными кислотами, чем можно было бы ожидать. При- * чина этого становится ясной, если написать структурные формулы частиц, участвующих в обоих равновесиях, на примере соединений (18) и (19) и оценить полярные электронные эффекты, которые могут дей-
411
ствовать в этих структурах:
(18а) (186)
(19а)
(196)
Для каждой частицы не учитывали индуктивный эффект «-МО2-заместителя, который по существу одинаков в каждом ряду частиц; учитывали лишь мезомерный эффект, или эффект сопряжения. В стандартной реакции (18а) ^ (186), которая была использована для оценки <тга хо2> эффект сопряжения п-МО2-заместителя передается на реакционный центр в основном через индуктивное влияние на СО2Н- или СО2-группу от углеродного атома кольца, с которым эта группа связана. При переходе (19а) (196) эффект сопряжения передается, однако, от
п-МО2-заместителя непосредственно электронной паре атома кислорода, который и является в этой системе реакционным центром. Этот эффект особенно заметен в (196), где анион стабилизирован в основном делокализацией его отрицательного заряда и где равновесие диссоциации для п-МО2-фенола тем самым смещено вправо, в сторону аниона, вследствие чего и возрастает кислотность фенола.
Значение <тга-мо2 > полученное с помощью стандартной реакции (18а) (186), очевидно, не учитывает повышенный эффект
этого прямого сопряжения, из-за чего точки на графике для n-NO2 и для n-CN находятся вне прямой линии (см. рис. 13.4). Такое прямое полярное сопряжение, однако, позволяет использовать диссоциацию фенолов в воде для определения ряда новых, альтернативных значений констант а для n-NO2 и других сходных электроноакцепторных заместителей; эти новые значения могут быть затем использованы для рассмотрения реакций, в которых может иметь место прямое сопряжение.
412
Это достигается следующим образом: строят график lg(Ax/AH)-ax только для jn-замещенных фенолов (кото-рые не могут быть вовлечены в прямое сопряжение); тан- S гене угла наклона полученной прямой дает значение р для этой реакции. Используя это значение в обычном уравнении Гаммета (13.6), рассчитывают
новое, уточненное значение константы <тх для n-NO2 и аналогичных заместителей, способных к прямому сопряжению. Эти уточненные значения обычно обозначают о~. Некоторые из таких констант приведены ниже (для сравнения приведены также значения обычных констант <тга.х):
О Y V
П‘Л П-Л
CO2Et 0,68 0,45
СОМе 0,84 0.50
CN 0,88 0,66
X °П-Х %-Х
СНО 1,03 0,43
no2 1,27 0,78
Аналогичная ситуация возникает в том случае, когда появляется возможность прямого сопряжения между подходящими электронодонорными заместителями в пара-положении и реакционным центром, в котором наводится положительный заряд. Хорошим примером является сольволиз (SnI) третичных галогенидов— 2-арил-2-хлорпропанов (20) (рис. 13.5).
Показано, что сольволиз п-МеО- и п-Ме-замещенных хлоридов протекает быстрее (для п-МеО в « 800 раз), чем можно было предсказать, исходя из значений соответствующих констант ап- Это является следствием проявления, прямого сопряжения в карбокатионных интермедиатах (21а) й (216), которые образуются во время медленной, скоростьлимитирующей стадии
413
всей реакции, т. е. Следствием их стабилизации:
(21а)
Тот факт, что наведение положительного заряда в переходном состоянии этой медленной стадии является существенным, следует из большого отрицательного значения константы р (—4,54) для этой реакции. Используя сольволиз как новую стандартную реакционную серию, можно так же, как и для о~, аналогично получить константы а+, что позволит оценить прямое сопряжение с участием сильных электронодонорных п-замести-телей; несколько таких констант приведены ниже (для сравнения даны соответствующие значения а):
X ап-Х ап-Х X G* Y П-Х an-X
Ph —0,18 —0,01 nh2 — 1,30 -0,66
Me —0,31 -0,17 NMe2 -1,70 -0,83
МеО —0,78 —0,27
Таким образом, для каждого n-заместителя мы имеем теперь две альтернативные константы: а~х и ага Х для электроноакцепторных заместителей и а+х и агаХ для электронодонорных заместителей. Их применение зависит от того, имеет ли место прямое сопряжение в конкретной реакции между п-заместите-лем и реакционным центром или нет. Хотелось бы думать, что эти двойные значения констант заместителей учитывают все возможности. Исследовано не менее восьмидесяти различных реакций, чтобы увидеть, приводит ли использование констант о~ или о„ „ и о+ или ст Y к линейной зависимости в каж-Л-Х П-Х- П-Х П~Х
дом случае. Было показано, что требуемые значения, например, для n-NO2 не группируются вокруг значений 0,78 (а) или. 1,27 (о-), а распределяются более или менее равномерно между этими двумя предельными значениями; то же самое наблюдается и для констант n-MeO-группы: они распределяются между значениями —0,27 (а) и —0,78 (а+).
Это не удивительно. Степень изменения электронной плотности на реакционном центре (т. е. на атоме, связанном непо
414
средственно с бензольным кольцом) в этих реакциях во время медленной, скоростьлимитирующей стадии будет, очевидно, различной для разных реакций. Поэтому будет отличаться также и мера эффекта (благодаря прямому сопряжению), проявляемого одним и тем же «-заместителем в различных реакциях. Исходя из этого, становится очевидной неизбежность ряда различных значений а„.х для каждого «-заместителя, отражающих разные степени прямого сопряжения в различных реакциях.
13.3.7. Уравнение Юкава — Цуно
Было предпринято несколько попыток путем введения дополнительных параметров в уравнение Гаммета количественно выразить этот дифференцированный отклик (обусловленный прямым сопряжением) со стороны «-заместителя. Среди наиболее известных попыток можно назвать уравнение Юкава — Цуно (13.7):
1g(fex/feH) = P[ox + ''(oJ-°x)]- (13-7)
которое применимо для электронодонорных «-заместителей; для электроноакцепторных заместителей константы должны быть заменены константами ах. Новый параметр г, который представляет собой меру прямого сопряжения, действующего в данной реакции, был принят равным 1,00 для сольволиза третичных галогенидов — 2-арил-2-хлорпропанов (20). Для такой реакции уравнение (13.7), очевидно, можно упростить до уравнения (13.8):
lg (fex/feH) = Р°х • (13.8)
которое кажется достаточно объективным, поскольку именно указанная реакция была применена для определения констант а+ (см. разд. 13.3.6) электронодонорных заместителей, способных к значительному прямому сопряжению. Аналогично, для реакции, в которой нет прямого сопряжения между заместителем и реакционным центром, г = 0, и уравнение (13.7) преобразуется в обычное уравнение Гаммета [см. уравнение (13.6)].
Чтобы оценить значение г для других реакций, можно получить р для данной реакции, измеряя ky только для соединений с .«-заместителями, а затем определить kx для соединений с «-заместителями, для которых значения агаХ и о+х или ап Х уже известны. Используя уравнение (13.7), можно затем определить г путем вычисления или графическими методами. Так, было определено, что для щелочного гидролиза «-замещенных
415
фенокситриэтилсиланов (22) г = 0,50.
(24)
Высокая степень прямого сопряжения с участием такого заместителя, как n-NO2, предполагает наведение значительного отрицательного заряда (р = +3,52) в переходном состоянии (23) скоростьлимитирующей стадии. Вместе с тем обсуждаемый заряд не столь выражен, как в переходном состоянии (24) стандартной реакции (сольволиз галогенидов, где г= 1,00), в котором наводится положительный заряд (р = —4,54). Так как в каждом случае накопление заряда в переходном состоянии идет одновременно >с расщеплением связи между реакционным центром и уходящей группой, то значение г может восприниматься как некоторая мера степени этого разрыва связи к моменту достижения переходного состояния.
Не следует, однако, вводить новые параметры в уравнение Гаммета только ради того, чтобы достичь лучшего соответствия ему экспериментальных данных. Это особенно верно тогда, когда, как в’некоторых случаях, отсутствует реальный физический смысл того или иного нового параметра. В действительности возможно, как мы увидим, получить достоверную информацию о путях реакции, используя только обычное уравнение Гаммета *.
13.4. Применение уравнения Гаммета
Константы ох и р с успехом используют для изучения реакций и. механизмов, по которым они протекают.
* Призыв автора не увлекаться варьированием различных констант в соотношениях ЛСЭ оказался запоздалым. К настоящему времени известно свыше 40 различных типов констант заместителей. Широкое распространение получили и так называемые множественные корреляционные уравнения. Подробнее об этом см.: Shorter J. Correlation analyses of organic reactivity with particular reference to multiple regression. Chichester: Res. Stud. Press, 1982. 235 p. — Прим. ped.
416
13.4.1. Расчеты значений k и К
Простейшее применение уравнения Гаммета — расчет k и К для конкретной реакции какого-либо конкретного соединения, если такая информация отсутствует или если это соединение даже еще не получено. Так, известно, что щелочной гидролиз этил-м-нитробензоата протекает в 63,5 раза быстрее, чем гидролиз соответствующего незамещенного эфира в этих же условиях. Какова же будет в этих условиях скорость щелочного гидролиза этил-п-метоксибензоата? Из приведенных ранее значений ах (см. разд. 13.3.2) следует, что ал-мо2 = 0,71 тогда как о = —0,27. Тогда уравнение Гаммета (см. разд. 13.3.2) примет вид:
*8 (^л-МОг/^н) — Рстл-МО2> (13.6а)
т. е. 1g (63,5/1) = р • 0,71, откуда р = 2,54. 'в (^n-Meo/^н) =° РСТп-МеО (13.66)
т. е. ’g (fen-Meo/feH) = 2>54 (—°.27), откуда ^n-Мео/^Н = °.21-
Когда kn-мео впоследствии определили экспериментально, отношение kn-Meo/ku оказалось равным 0,21, что точно совпало с расчетным значением. На практике аир редко используют с такой целью; гораздо чаще их используют для получения соответствующих данных о механизмах реакций.
13.4.2. Отклонения от линейных зависимостей
Мы уже видели (см. разд. 13.3.5), как знак и численное значение константы реакции р могут дать полезную информацию о возникновении (или исчезновении) заряда (положительного или отрицательного) при переходе от исходных веществ к переходному состоянию на скоростьлимитирующей стадии реакции. Мы также видели (см. разд. 13.3.6), как отклонения от линейных зависимостей при использовании обычных констант заместителей ах позволяют определить значения а+ или оценивающие прямое сопряжение между /i-заместителями и реакционным центром. Необходимость применения других констант, кроме обычных ах, указывает на существование такого прямого сопряжения в данной реакции, а параметр Юкава — Цуно г указывает на степень такого сопряжения.
Может показаться парадоксальным, но зависимости Гаммета, как правило, наиболее информативны в тех случаях, когда
14 П. Сайкс
417
они отклоняются от линейности; основной вывод, который может быть сделан на основе этого отклонения, зависит от того, направлено ли такое отклонение «вверх» или «вниз».
13.4.3. Зависимости, отклоняющиеся вверх
13.4.3.1. Ацетолиз З-арил-2-бутил-п-бромбензолсульфонатов
Для реакции ацетолиза З-арил-2-бутил-п-бромбензолсульфо-натов (З-арил-2-бутилброзилатов) (25) зависимость Гаммета приведена на рис. 13.6. Нижняя правая часть графика (сильные электроноакцепторные заместители) является прямой линией, тангенс угла наклона которой определяет константу р для данной реакции, равную —1,46. В левой части этого графика (менее электроноакцепторные заместители) кривая начинает отклоняться вверх, т. е. скорость ацетолиза этих соединений выше, чем можно было бы ожидать на основе значений стх для этих заместителей.
*х
АсО~, АсОН
(25)
МеСН—СНМе
(26)
Bs = п -ВгСсНдЗОг (броэилат)
Можно предполагать, что эта 5м2-замещением (см. разд. 4.5.1) (брозилат-анион) ацетат-ионом:
реакция является обычным хорошей уходящей группы
X
X
*
МеСН—СНМе des
(25)
Ю 6-ХГХ ОАс
МеСН—QHMe
6-OBs
(27)
-"OBs
(26)
Небольшое отрицательное значение р (—1,46) согласуется с этим механизмом и означает, что в переходном состоянии (26) связь С—OBs «расщепляется несколько раньше», чем образуется связь АсО—С. Это приводит в результате к появлению небольшого положительного заряда на реакционном центре. Такая трактовка плохо согласуется с двумя обстоятельствами: а) реакционный центр находится на вторичном атоме углерода; б) на-
418
Рис. 13.6
личие хорошей уходящей группы. Указанному механизму должно было бы способствовать, хотя и в небольшой степени, ослабление электроноакцепторных свойств заместителей X, т. е. скорость ацетолиза должна была бы увеличиваться постепенно и линейно при переходе от правой части графика к левой, как показано пунктиром на рис. 13.6.
Чтобы объяснить наблюдаемое отклонение от линейности, когда X становится все более электронодонорным, следует предположить, что замещенное бензольное кольцо в некоторой степени начинает проявлять прямое влияние на реакционный центр в эфире (25), по сравнению с тем влиянием, какое обычно имеет место для механизма SN2. При этом важно, что при увеличении электронодонорности заместителя X будет увеличиваться нуклеофильность замещенного бензольного кольца и повышаться его способность функционировать (конкурируя с -ОАс) в качестве соседней группы (см. разд. 4.4.5) или «внутреннего» нуклеофила, например, при Х = МеО [см. формулу (28)]. Этот альтернативный механизм реакции должен включать, таким образом, в качестве медленной, скоростьлимитирующей стадии образование циклического фенониевого иона (29) как промежуточного соединения (ср. разд. 5.2). Затем происходит его быстрое раскрытие под действием ~ОАс с образованием обычного продукта ацетолиза (30):
МеСН—СНМе
ОАс (30)
ОМе
Предположение о том, что показанная на рис. 13.6 зависимость действительно отражает изменение механизма реакции, подтверждается результатами ацетолиза трео-диастереомера (31) брозилата. Ацетолиз приводит к двум различным диастереомерам, относительные количества которых зависят от того, в
14*
419
какой степени общая реакция протекает путем внешней нуклеофильной атаки по механизму Sn2 [образование эрытро-продукта (32)] и в какой — путем внутримолекулярной нуклеофильной атаки через образование интермедиата в виде фенониевого циклического иона [образование трсо-продукта (33)]:
Два альтернативных продукта ацетолиза—(32) и (33),— являющиеся диастереомерами, а не зеркальными отражениями, могут затем быть разделены (или их относительные количества могут быть определены спектроскопическими методами). Оказалось, что выход трсо-продукта (33) зависит от природы заместителя X в бензольном кольце:
X Выход соединения (33)*, % X Выход соединения (33)* %
п-МеО 100 л-С1 39
п-Ме 88 л-С1 12
л-Ме 68 л-СК3 6
Н 59 n-NO2 1
* Выход соединения (33) соответствует доле реакции, протекающей путем внутримолекулярной нуклеофильной атаки.
Когда X = n-МеО (самый сильный электронодонорный заместитель; см. рис. 13.6), ацетолиз полностью проходит путем внутримолекулярной нуклеофильной атаки п-МеОСбН4. Когда X = jh-C1, то только 12 % общей реакции проходит благодаря внутримолекулярной нуклеофильной атаке. Наконец, при X — = n-NO2 (самый сильный электроноакцепторный заместитель), только 1 % всей реакции протекает путем такой атаки.
Если на обычном графике Гаммета наблюдается отклонение вверх (см. рис. 13.6), то это можно считать доказательством
420
изменения механизма общей реакции при изменении природы заместителя. То, что изменение механизма реакции приводит к отклонению вверх, вполне понятно: на рис. 13.6 именно в той точке, где начинается отклонение от линейности, ничто не мешает действию первоначального механизма Sn2 (что показано пунктирной линией экстраполяции). Любое изменение в сторону какого-либо нового механизма должно обеспечивать менее затрудненный и поэтому более быстрый альтернативный путь (что, без
условно, ведет к кривой, отклоняющейся вверх). Конечно, и исходный механизм реакции мог бы продолжать оставаться преобладающим; никакого отклонения от исходной прямой линии в этом случае не наблюдалось бы.
13.4.3.2. Гидролиз ArCOiR. в 99 %-й H2SOi
Иногда отклонение от прямой линии является намного более резким, чем на рис. 13.6. Особенно хорошим примером этого является гидролиз в 99,9 %-й H2SO4 замещенных метил- (34а) и этилбензоатов (346) (рис. 13.7):
Рассмотрим сначала более простой случай — прямолинейный график для гидролиза метиловых эфиров (34а), для которых р = —3,25. Ясно, что в этом случае реакция не может протекать по обычному ЛАс2-механизму (см. разд. 8.6.3), характерному для кислотного гидролиза эфиров, для которого, как известно [см. разд. 13.3.3; реакция (6)], р — +0,03. Однако такое значение р относится к гидролизу, осуществляемому разбавленной серной кислотой, тогда как в приведенном выше примере используется 99,9 %-я серная кислота, т. е. концентрация воды, доступной для гидролиза, в этом случае очень мала.
Мы уже рассматривали альтернативный кислотный гидролиз (ЛАс1; см. разд. 8.6.3), в котором молекулы воды не участвуют в медленной, скоростьлимитирующей стадии. В этой стадии
421
возникает значительный положительный заряд на реакционном центре протонированного эфира (35а), превращающегося в ацилкатионный интермедиат (36а); это является обязательным требованием для реакции с большим отрицательным значением р (—3,25):
Н20л Н2(У НО
медленно + I I
Аг—С=О < 1 ± Аг—С—О ^=±Аг—С=О Аг—С=О
.1> +
+ОМе
Н
(35а) (36а)
Именно этот механизм ЛАс1 должен действовать первоначально и в случае гидролиза этиловых эфиров (346) (см. рис. 13.7); значение р (—3,25) для этой реакции является точно таким же, как и для метиловых эфиров (34а). Как только заместитель в бензольном кольце становится более электроноакцепторным, наблюдается резкое изменение хода зависимости при гидролизе этиловых эфиров — переход в новую прямую линию с р, равной +2,0. При таком значении р необходима медленная, скоростьлимитирующая стадия для гидролиза, в которой положительный заряд в реакционном центре уменьшается — общая реакция все больше ускоряется, по мере того как заместитель в бензольном кольце становится более электроноакцепторным.
Это соответствует еще одному механизму кислотного гидролиза эфиров (Даь1; см. разд. 8.6.3):
Аг-С = О
Fid—СН2Ме
медленно.
Аг - С = О + + СН2Ме НО
(356)
(376)
(376) НО - СН2Ме + Н*
Отщепление катиона (376) приводит к заметному уменьшению положительного заряда вблизи реакционного центра (если бы это могло быть на самом реакционном центре, положительное значение р было бы гораздо больше). Карбокатионный интермедиат (376) затем быстро реагирует с любой доступной молекулой воды с образованием этанола.
Возникает вопрос: почему подобное изменение механизма реакции не происходит в случае метиловых эфиров (34а)? Такое изменение могло бы привести к неизбежному образованию катиона +СН3, а не катиона (376), в ходе медленной, скорость-
422
лимитирующей стадии. Однако катион +СН3, как известно, получить гораздо труднее, чем катион +СН2Ме. Это различие, по-видимому, достаточно велико, чтобы исключить на основании энергетических соображений изменение механизма Aac1->Aal1 в случае метиловых эфиров, несмотря на сильное содействие механизму Aal 1 со стороны электроноакцепторных заместителей.
13.4.4. Зависимости, отклоняющиеся вниз
Существуют, однако, примеры отклонений от простых зависимостей Гаммета, в которых кривизна имеет противоположное направление, — зависимости, отклоняющиеся вниз, причем эти отклонения имеют весьма различное толкование.
13.4.4.1. Циклодегидратация диарил(бифенилил-2)метанолов
Хорошим примером реакции с отклонением вниз от зависимости Гаммета (рис. 13.8) является циклодегидратация некоторых замещенных диарнл(бифеннлнл-2)метанолов (38) в 80 %-м водном растворе этановой кислоты, содержащей 4 % H2SO4, при 25°C с образованием соответствующих тетраарилметанов (39):
Два бензольных кольца в соединении (38) содержат в пара-положении заместители (X и Z соответственно), и значение ст, отложенное на графике (см. рис. 13.8), фактически является Ест, т. е. суммой констант ст для заместителей X и Z. На этом графике имеются две прямые линии, для одной из которых (слева) р= +2,67, а для другой (справа) р=—2,51.
Не вызывает сомнения, что 5 суммарная реакция протекает * по четырехстадийному меха-низму; при этом первые две стадии представляют собой £1-элиминирование (см. разд. 9.1) воды с образованием
423
карбокатионного интермедиата (40), который затем в ходе оставшихся двух стадий подвергается внутримолекулярному электрофильному замещению с образованием соединения (39):
Тогда возникает вопрос: какая из этих стадий суммарной реакции будет медленной, а следовательно, скоростьлимитирующей? Вряд ли это стадия 1 (первоначальное протонирование при кислотной дегидратации обычно протекает быстро) или стадия 4 (отщепление протона при ароматическом электрофильном замещении также, как правило, протекает быстро). На стадии 2 положительный заряд на реакционном центре (атом углерода, связанный с двумя замещенными Аг-группами) увеличивается, тогда как на стадии 3 положительный заряд на реакционном центре уменьшается. Как это соответствует зависимости, показанной на рис. 13.8?
Отрицательное значение константы р (—2,51) (правая часть графика на рис. 13.8) указывает на возникновение значительного положительного заряда на реакционном центре в ходе скоростьлимитирующей стадии. Это может, конечно, соответствовать стадии 2, но не стадии 3. Для левой части графика справедливо совершенно обратное: положительное значение р (4-2,67) указывает на существенное уменьшение положительного заряда на реакционном центре, что должно быть совместимым со стадией 3 как скоростьлимитирующей, но не со стадией 2.
Важно, что заместители, приведенные в левой части графика [(38; X, Z = МеО)], являются сильноэлектронодонорными и поэтому способны стабилизировать карбокатион (41а)-«-► (416), образующийся на стадии 2, путем делокализации его положительного заряда. Действительно, показано, что зависимость 1g£набл — S<r+ в левой части графика (см. рис. 13.8) имеет более линейный характер, чем зависимость ^Анабл — So вследствие прямого сопряжения [ (41а) ► (416) ] между л-заместите-
424
лями и реакционным центром:
В соединении (38; X, Z = МеО) эта стабилизация путем сопряжения приводит к легкому образованию карбокатиона (41), т. е. к быстрой стадии 2, однако в результате последующей делокализации положительного заряда [заряд удаляется от реакционного центра; (41а)-*->-.(416)] этот катион становится менее эффективным электрофилом, т. е. стадия 3 — электрофильная атака бензольного ядра — идет поэтому медленно. Таким образом, именно стадия 3 является медленной, а следовательно, скоростьлимитирующей в случае соединения (38; X, Z = OMe).
При уменьшении электронодонорной способности заместителей делокализация положительного заряда уменьшается, а реакционный центр становится более электрофильным. Ско-ростьлимитирующая стадия 3 в этих условиях, следовательно, ускоряется, скорость суммарной реакции поэтому возрастает, наклон графика поднимается вверх при переходе слева направо (р является положительной). При переходе слева направо уменьшается, кроме того, и прямое сопряжение, так как заместители становятся менее электронодонорными, что делает образование карбокатиона более трудным. Стадия 2, таким образом, замедляется (в то время как стадия 3 ускоряется), и наступает ситуация, при которой скорости стадий 2 и 3 выравниваются. При дальнейшем уменьшении электронодонорности заместителей стадия 2 станет более медленной (чем стадия 3), т. е. скоростьлимитирующей стадией всей реакции. Такое изменение скоростьлимитирующей стадии и наблюдается для соединения (38; X, Z = Ме) (см. рис. 13.8).
Дальнейшее еще большее уменьшение электронодонорности заместителей ведет к еще большему замедлению стадии 2 — теперь уже скоростьлимитирующей стадии, а следовательно, и к замедлению общей реакции: правая часть графика отклоняется вниз (р является отрицательной). Для реакции, в которой наблюдается такое изменение скоростьлимитирующей стадии (при изменении электронодонорной или электроноакцепторной способности заместителей), должен быть один заместитель или ограниченное число заместителей, для которых соотношение между
425
скоростями стадий 2 и 3 таково, что скорость суммарной реакции оказывается максимальной.
Именно это и происходит (см. рис. 13.8) для соединения (38; X, Z=Me). С каждой стороны этого максимума соответствующая скоростьлимитирующая стадия постепенно замедляется и, следовательно, уменьшается скорость суммарной реакции. Таким образом, путем анализа направленных вниз отклонений от линейности на графике Гаммета удается различить изменения в скоростьлимитирующей стадии в пределах одного и того же механизма, в отличие от отклонений, направленных вверх, которые, как это было показано (см. разд. 13.3.3), указывают на изменение всего механизма реакции.
13.5. Стерические эффекты
С самого начала обсуждение соотношений линейности свободных энергий (см. разд.. 13.2) было ограничено реакциями в боковых цепях м- и n-замещенных производных бензола. Реакции о-замещенных производных бензола и реакции алифатических соединений были исключены из-за стерических и других эффектов, которые приводили к нелинейным зависимостям или даже часто к отсутствию каких-либо зависимостей.
Полезность уравнений Гаммета (в том числе то, что они часто даже более ценны при отклонении от линейности) побудила ряд исследователей искать, вводя подходящие модификации, возможности применения зависимостей Гаммета для более широкого круга соединений. Наиболее общие и успешные из этих работ были проведены Тафтом.
13.5.1. Уравнение Тафта
По предложению Ингольда Тафт сравнил относительную чувствительность к полярным эффектам заместителей (константы р) реакций кислотного (ЛАс2; см. разд. 8.6.3) и щелочного гидролиза (ВАС2; см. разд. 8.6.2) ряда м- и n-замещенных эфиров бензойной кислоты (42).
ЛО О') О
А "он г г И <?
Ar—С—OEt - > Ar—С д-OEt----->-Ar—С +' OEt~
“ОН ОН ОН
(42)
О
____II
~4=tAr-C—О + EtOH
Вас; р = 2,51
426
О:
II н+
Аг—С—OEt4= 1
(42)
ОН Н
Аде2; Р = 0,03
Ar—С—OEt
<7
+ОН О
-EtOH II ~Н+ ||
' > Ar—С Ar—С 4 | 5 |
он он
Значение р для щелочного гидролиза является положительным и достаточно большим (+2,51), что соответствует наведению не такого уж незначительного отрицательного заряда на реакционном центре в скоростьлимитирующей стадии — атаке этого центра ионом ~ОН (стадия 1 в механизме Вдс2). Напротив, значение р для кислотного гидролиза близко к нулю (+0,03), т. е. скорость такого гидролиза заметно не изменяется от одного эфира к другому, независимо от присутствия м- или л-замести-теля. Такое небольшое значение константы р, несмотря на делокализацию значительного положительного заряда в медленной стадии (стадия 2), связано с тем, что скорость реакции, т. е. £набл (которую используют для графического определения р) зависит не только от скорости fe2 этой медленной стадии, но и от константы равновесия Ki предшествующей обратимой стадии 1. Эти два эффекта почти компенсируют друг друга (имея в виду чувствительность этих двух стадий к электронодонорности илн электроноакцепторности полярных заместителей), а суммарная константа р для этой реакции, таким образом, оказывается равной нулю.
При рассмотрении щелочного (Вас2) и кислотного (Ддс2) гидролиза любых эфиров, включая алифатическое RCO2Et, можно увидеть большое сходство между переходными состояниями (426) и (42а) для скоростьлимитирующей стадии в каждом из двух механизмов: переходные состояния являются тетраэдрическими и отличаются только тем, что во втором из них на два протона больше, чем в первом:
II R— С—OEt
6н
(426)
ПС для Вдс2
(42а)
ПС для Адс2
ОН
С—OEt
ОН2
427
Протоны, будучи очень малы по размеру, оказывают сравнительно небольшое пространственное влияние. Поэтому можно допустить, что любой стерический эффект группы R практически одинаков как при кислотном, так и при щелочном гидролизе вследствие равенства объемов, занимаемых в пространстве обоими переходными состояниями *. При таком допущении уравнение Гаммета можно написать в виде уравнения (13.9), что соответствует действию только полярного эффекта заместителя R при гидролизе эфира:
(Шс. - М*к/*о)кисл = РЧ- (’3.9)
Поскольку стерический эффект, проявляемый заместителем R, практически одинаков в обоих типах гидролиза, две соответствующие величины аннулируют одна другую и не входят в это уравнение.
Тафт затем определил значение р* (2,48) путем вычитания значения р для кислотного гидролиза бензоатов (0,03) из значения р для щелочного гидролиза тех же эфиров (2,51). Он использовал в качестве стандартного заместителя R = Me, а не R = Н; поэтому k0 в уравнении (13.9) относится к MeCO2Et, а не к HCO2Et. Определив скорости кислотного и щелочного гидролиза ряда эфиров, у которых группы R не являются метильными группами, можно с помощью уравнения (13.9) оценить ст* каждого из соответствующих заместителей R по отношению к Ме-группе, для которой как для стандартного заместителя аМе~0 (сР-: ан~0 для ионизации бензойной кислоты; см. разд. 13.3.2). Принимая значение р*, равным 2,48, можно видеть, что получающееся значение ст^, которое является мерой только полярного эффекта, проявляемого R, не отличается очень сильно от уже известных значений стх, ст^ и стх (см. разд. 13.3.2).
Затем, подставляя значения ст^ и kR и kMe в более общее уравнение (13.10), можно определить константы р* и для других реакций ряда алифатических соединений, кроме сложных эфиров:
,В(йкЛме) = р’°'к- (1310)
13.5.2. Стерические константы Es и б
Получение прямолинейной зависимости Гаммета может, на первый взгляд, показаться довольно удивительным результатом, особенно при том, что в соотношении (13.10) учитывается толь
* Такое допущение, однако, не исключает возможность того, что степень сольватации положительно и отрицательно заряженных переходных состояний может заметно отличаться н сильно влиять тем самым на относительные скорости этих двух реакций гидролиза.
428
ко полярный эффект, проявляемый заместителями R. Однако это вовсе не означает, что в реакции не проявляется никаких стерических эффектов. Это означает только, что не происходит существенных изменений в действии таких эффектов при переходе от исходных веществ к переходному состоянию скоростьлимитирующей стадии (или при переходе от исходных веществ* к продуктам для равновесного процесса).
Нетрудно обнаружить реакции алифатических соединений, отклоняющиеся от линейных зависимостей типа (13.10); эти отклонения, как и отмеченные выше (см. разд. 13.4.2), дают значительно больше информации о механизме соответствующих реакций, нежели сами прямые линии. Установлено, что в тех случаях, когда наблюдаются отклонения от линейных зависимостей (отражающих только полярные эффекты), можно предполагать действие значительных (и неодинаковых) стерических эффектов и вводить стерический параметр заместителя, Es, для оценки таких эффектов. Ниже иллюстрируется способ определения параметров £s. Как уже было показано (см. разд. 13.5.1), кислотный гидролиз м- и л-замещенных эфиров бензойной кислоты (42) (р = 0,03) по существу не зависит от какого-либо полярного эффекта, проявляемого заместителем X. Заместитель, кроме того, достаточно далеко отстоит от реакционного центра и, очевидно, не способен проявлять какое-либо стерическое влияние на этот центр. Вследствие этого скорости кислотного гидролиза таких эфиров практически одинаковы.
CO2Et
^) + Н2О X
(42)
Нет оснований полагать, что кислотный гидролиз эфиров алифатических карбоновых кислот RCO2Et будет более чувствительным к полярным эффектам, чем соответствующий гидролиз эфиров бензойной кислоты, поэтому наблюдаемые различия в скоростях кислотного гидролиза эфиров алифатических карбоновых кислот при изменении R должны быть обусловлены различными стерическими эффектами, проявляемыми этими группами R. Принимая метильную группу в качестве стандартного заместителя и используя уравнение (13.11):
te (feRCO2Et/feMeCO2Et)Knca ~ ЕS (13-Н)
можно оценить £s — стерический параметр заместителя R.
429
Значения £s, полученные таким путем для различных заместителей R в эфирах RCO2Et, приведены ниже:
R R
н + 1,24 Ме(СН2)3 —0,39
Me • 0 МеСНСН2 — 1,13
Et —0,07 Ме3С — 1,54
С1СН2 —0,24 Ме3ССН2 — 1,74
1СН2 —0,37 Ph2CH — 1,76
PhCH2 —0,38 Et3C —3,81
* Стандартный заместитель.
Исходя из вида уравнения (13.11), Es для Me как стандартного заместителя будет, конечно, равна нулю. Все другие заместители, кроме атома водорода, имеют отрицательные значения Es, так как размеры В >> Me-группы. Скорость гидролиза любого эфира RCO2Et(R¥=H) будет, следовательно, меньше, чем скорость гидролиза MeCO2Et в ходе реакции, скорость которой определяется исключительно стерическим эффектом R.
На практике оказалось, что стерический параметр Es для конкретной группы R несколько изменяется при переходе от одной реакции к другой. Это неудивительно, так как при этом изменяются и локальное окружение группы R, и размеры атакующего реагента. Таким образом, оказалось, что при включении Es в уравнение Гаммета типа (3.12) необходимо ввести еще один параметр, б, как меру чувствительности данной реакции к стерическим эффектам:
lg (*R/feMe) = p’aR + <13-12)
В этом смысле константа б аналогична константе р*, которая характеризует чувствительность реакции к полярным эффектам. Параметр б принимается равным 1,00 для кислотного гидролиза эфиров (стандартная реакция); его значение для других реакций может быть определено экспериментально обычным путем.
Теперь, когда стерические параметры введены в соотношения ЛСЭ, можно рассмотреть реакции о-замещенных производных бензола. Найдено, что для кислотного гидролиза о-замещенных бензамидов (43) константа б = 0,81. Эта реакция, по-видимому, несколько менее чувствительна к стерическому эффекту заместителей, чем стандартная реакция — кислотный гидролиз о-замещенных эфиров. В целом, однако, попытки количественно оценить эффекты, о-заместителей были не очень успешными. Здесь химики снова сталкиваются с той же дилеммой, что и в случае уравнения Юкава — Цуно (см. разд. 13.4): насколько полученная дополнительная информация оправдывает те очень большие усилия, которые необходимы для экспериментальной оценки со-
430
ответствующих дополнительных параметров?
(43)
н+
н2о
13.6. Эффекты растворителя
До сих пор при обсуждении соотношений линейности свободных энергий не учитывалась роль, которую играет в реакциях растворитель. И это несмотря на то, что подавляющее большинство органических реакций на самом деле протекает в растворе.
13.6.1. Зависимость константы р
от природы растворителя
Показано, что значение константы р для данной реакции, как правило, изменяется при изменении растворителя, в котором протекает реакция. Так, для реакций (13.13) и (13.14) в различных растворителях значения р составляют:
ArCO2H + H2O АгСО;+Н3О+ (13.13)
(44) (45)
ArCO2Et + 'ОН —> АгСО; + ЕЮН (13.14)
Реакция Растворитель Р
(13.13) Н2О 1,00
50 %-й водн. EtOH 1,60
EtOH 1,96
(13.14) 70 %-й водн. диоксан 1,83
85% -й водн. EtOH 2,54
* Стандартная реакция.
В случае диссоциации м- н п-замещенных бензойных кислот (44) гидроксилсодержащий растворитель способен сольватировать как недиссоциированную кислоту (44), так и образующийся карбоксилат-ион (45). Различие в эффективности сольватации отрицательно заряженного аниона (45) и нейтральной не-диссоциированной кислоты (44) является главным фактором, определяющим положение равновесия, т. е. значение К*. При переходе от воды (е = 79) к этанолу (е = 24) преимущественная сольватация аниона (45) по сравнению с нейтральной молекулой кислоты (44) снижается. Относительный вклад полярного эффекта, проявляемого электроноакцепторнымн заместителями, в общую стабилизацию карбоксилат-иона (т. е. в усиление кислотности: возрастание Кх) будет, следовательно, увеличиваться, по мере того как диэлектрическая проницаемость растворителя уменьшается. Значение константы р, характеризующей
431
чувствительность реакции к полярному эффекту заместителя, будет также, следовательно, увеличиваться при переходе от воды к этанолу.
13.6.2. Уравнение Грюнвальда — Уинстейна
Попытки коррелировать изменения скорости какой-либо реакции, когда она протекает в различных растворителях, с диэлектрическими проницаемостями этих растворителей оказались безуспешными. Поэтому были предприняты попытки установить эмпирические зависимости реакционной способности от природы растворителя в рамках уравнения Гаммета. Одна из наиболее важных таких попыток была предпринята Грюнвальдом и Уин-стейном для реакции сольволиза галогенидов. Они попытались установить параметр растворителя, обозначенный Y, который можно было бы коррелировать с различными койстантами скорости, определенными для сольволиза одного и того же галогенида в различных растворителях.
В качестве стандартной была избрана реакция сольволиза третичного галогенида — 2-метил-2-хлорпропана (46), протекающая по механизму SnI; в качестве стандартного растворителя был избран 80%-й водный этанол (80 % EtOH и 20 % Н2О):
Sol
Ме3С—Cl -------» Ме3С+ Cl----ёыстпГ Me3C-Sol
медленно оыстро
(46) (47)
интермедиат
Sol — растворитель
Для этой реакции оказалось возможным записать соотношение, подобное уравнению Гаммета:
lg*A-lgk0 = rA-Y0. (13.15)
В уравнении (13.15) &А и k0—константы скорости сольволиза третичного галогенида (46) в растворителе Айв стандартном растворителе (80 %-й водный EtOH) соответственно; параметры Ya и Yo являются эмпирическими параметрами растворителя А и стандартного растворителя. Подставляя значение Yo, равное нулю, и измеряя Аа для сольволиза галогенида (46) в различных растворителях, можно, используя уравнение (13.15), определить значение Ya для каждого из них:
Растворитель А ГА 8 Растворитель А ГА г
Н2О +3,49 78,5 МеОН — 1,09 32,7
50 %-й води. МеОН + 1,97 — МеСО2Н — 1,64 6,2
hconh2 +0,60 109,5 EtOH —2,03 24,3
70 %-й водн. EtOH +0,59 — Ме2СНОН —2,73 18,3
80 %-й водн. EtOH* 0 — Ме3СОН -3,26 12,2
80 %-й водн. Ме2СО —0,67 —
• Этот растворитель рассматривается как стандартный.
432
Оказалось, что значения УА прямо не коррелируют со значениями диэлектрической проницаемости для рассматриваемых растворителей. Вместе с тем, диэлектрическая проницаемость связана, по-видимому, определенным образом с Ya, поскольку разделение противоположных зарядов является решающим признакам скоростьлимитирующей стадии реакции, протекающей по механизму SN1: реакция идет через переходное состояние, приводящее к интермедиату типа ионной пары (47). Кроме того, параметр Уд должен включать и способность растворителя к специфической сольватации разделяющихся зарядов, а возможно, и другие свойства растворителя. Как правило, величину Уд интерпретируют как меру ионизирующей способности растворителя А.
Теперь можно сделать следующий шаг и написать знакомое соотношение (13.16), которое описывает сольволиз галогенидов вообще, а не одного стандартного галогенида (46):
1g(feA/M = mrA (13.16)
В этом уравнении k\ и ko — константы скорости сольволиза данного галогенида в растворителе Айв стандартном растворителе соответственно; Уд — параметр растворителя, характеризующий ионизирующую способность растворителя А; т — параметр соединения, характеризующий данный галогенид [для стандартного галогенида (46) он принимается равным 1,00]. Параметр т может быть определен как мера чувствительности сольволиза данного галогенида к ионизирующей способности растворителя УА. Значения т для некоторых галогенидов приведены ниже:
Галогенид т
РЬСН(Ме)Вг (48) 1,20
Ме3СС1 (46) 1,00
Ме3СВг 0,94
Е1Ме2СВг 0,90
Галогенид т
СН2=СНСН(Ме)С1 0,89
EtBr (49) 0,34
Ме(СН2)3Вг (50) 0,33
Альтернативным толкованием параметра т является то, что его рассматривают как оценку степени ионности пары, образующейся в переходном состоянии скоростьлимитирующей стадии реакции сольволиза; в этом смысле он может быть использован для прогнозирования механизма реакции. Так, известно, что ионная пара хорошо образуется в переходном состоянии сольволиза 2-метил-2-хлорпропана (46) по механизму SnI; этот галогенид служит в качестве стандартного галогенида, для которого т = 1,00; неудивительно, что для 1-бром-1-фенилэтана (48), в случае которого образующийся катион бензильного типа [PhCHMe]+ стабилизируется делокализацией его положительного заряда по всей л-системе соседнего бензольного кольца (ср. разд. 4.3), значение т оказывается даже большим (1,20).
433
Наоборот, для первичных галогенидов — бромэтана (49) и 1/ бромбутана (50)—значения т гораздо ниже (0,34 и 0,33 соответственно). Эти значения, указывающие на низкую чувствительность к ионизирующей способности растворителя, характерны для галогенидов, сольволиз которых, как известно, проге/ает по механизму Sn2. В общем, значение т, равное 0,5, можно/счи-тать пограничным между механизмами SnI и Sn2 в реакциях сольволиза этого типа.
Главным недостатком уравнения Грюнвальда — Уинстейна является ограниченность области его применения. Это уравнение можно использовать не только в случае реакций сольволиза галогенидов, но, в общем, его применение ограничено теми реакциями, в которых скоростьлимитирующая стадия записывается следующим образом:
k
А—В --------► А+ В'
медленно
13.6.3. Параметр Димрота (параметр Ет)
Были предприняты попытки определить параметры полярности растворителя. Наилучшие результаты дает использование сольватохромных сдвигов, т. е. сдвигов по оси — длина волны (или частота) полосы поглощения в спектре соответствующей частицы в результате ее взаимодействия с молекулами различных растворителей. Особенно большие сдвиги наблюдались для цвиттер-иона (51):
Максимум поглощения этого соединения смещается от 450 до 1000 нм в зависимости от растворителя: соединение (51) имеет желтый цвет в МеОН, красный — в Ме2СНОН и голубой— в СНС13. Димрот принял за меру полярности растворителя величину £т — энергию возбуждения (основное состояние -* возбужденное состояние), соответствующую максимуму поглощения в данном растворителе. Физический смысл значений Ет определяется тем, что основное состояние соединения (51) (намного более полярное, чем возбужденное состояние) должно быть стабилизировано полярным растворителем в несравненно большей степени. Полагая, что влияние изменения растворителя на уровень энергии возбужденного состояния очень мало, изменение значения Ет можно считать мерой относительной стабилизации основного состояния; Ет возрастает по
434
мере того, как стабилизация, а следовательно, полярность растворителя увеличиваются:
Растворитель £т ГА Растворитель £т ГА
Н2О 63,1 + 3,49 ЕЮН 51,9 —2,03
hconh2 56,6 + 0,60 Ме2СНОН 48,6 — 2,73
80 %-й водн. EtOH 53,7 0 Ме3СОН 43,9 —3,26
80 %-й водн. Ме2СО 52,2 —0,67 СНС13 39,1 —•
МеОН 55,5 — 1,09
Для сравнения приведены и значения Тд (ср. разд. 13.6.2) для этих растворителей. Из этих двух параметров параметр £т представляется более удачным и имеет несколько более широкое применение.
13.7. Корреляции спектроскопических параметров
Мы уже обсуждали зависимость между химическими свойствами Х-замещенных молекул и значениями полярных констант ох заместителей X. Неизбежно возникает вопрос: могут ли быть установлены аналогичные корреляции между константами Ох и физическими свойствами молекул? Для этой цели были использованы, например, спектроскопические параметры.
Известно много попыток найти взаимосвязь между константами ох и частотой и (или) интенсивностью полосы поглощения в ИК-спектре соответствующего замещенного ароматического соединения. Среди наиболее успешных попыток надо назвать корреляции частот колебаний группы С=О в рядах соединений (52) и (53) и корреляцию интенсивности колебаний кольца при 1600 см-1 в ряду соединений (54):
Можно было бы ожидать наличия корреляции между константами ох и значениями химических сдвигов 6 (ср. разд. 1.3.6) по данным ЯМР, которые отражают степень экранирования или дезэкранирования электронов у соответствующих атомов. В действительности корреляции значений 6 для ’Н со значениями ох оказались не очень хорошими. Исключение составляет ряд соединений (55), в которых соответствующий протон весьма
435
удален от замещенного бензольного кольца.
(55) р = -0,33
Можно было ожидать, что атом, более тяжелый, чем ’Н, будет менее чувствителен к тем изменениям, которые сопровождают изменение химических сдвигов для ‘Н. Таким атомом мог бы быть 13С, который также генерирует спектр ЯМР. Например, для 2-арилпропил-катионов, полученных при растворении соответствующих третичных спиртов в «суперкислотах», например SO2C1F—FSO3H—SbFs (ср. разд. 7.1), наблюдается хорошая линейная зависимость между разностями химических сдвигов 13С (дляположительно заряженного атома углерода:бСн — бСх) и константами оХх, но не с о„-х. Корреляция разностей химических сдвигов для n-замещенных карбокатионов требует применения более чувствительных констант и-заместителей, цС*. Это объясняется гораздо более сильным прямым сопряжением n-X-заместителя с реакционным центром в полностью образовавшемся карбокатионе (56а) по сравнению со степенью прямого сопряжения n-X-заместителя с реакционным центром в лишь частично образовавшемся карбокатионе (57), соответствующем переходному состоянию сольволиза куменилхлорида — стандартной реакции, которая была выбрана (см. разд. 12.5.2) для определения о+_х:
в+ 6-
МегС+ МегС—С1
Ф 9
X: X:
(56а) р = — 18,2 (57) р = - 4,54
(ап-х) (ап-х)
13.8. Корреляции термодинамических параметров
Учитывая очень большие достижения в использовании уравнения Гаммета, необходимо отметить примеры корреляции термодинамических параметров в рамках принципа линейности сво-
436
брдных энергий. Выше рассматривалась (см. разд. 13.1) зависимость между изменением свободной энергии AG и 1g k или 1g Л и отмечалось, что AG состоит из энтальпийного ЛЯ и энтропийного AS компонентов:
AG° = — 2.303RT 1g Л; AG° = Л/Г - Т bS°.
&G^ = - 2,303Rf 1g [k (h/k'T)]; bG + = Д//+ — T bS*,
где k' — константа Больцмана; h — константа Планка.
Возвращаясь к одному из рассмотренных выше примеров (см. рис. 13.3; разд. 13.2) — зависимости между 1g К (для диссоциации АгСОгН) и lg R (для щелочного гидролиза ArCChEt), следует отметить, что прямая линия означает линейную зависимость между значениями AG° для первой реакции и значениями AG+ — для второй. Такая зависимость может наблюдаться только в том случае, если для каждой реакционной серии выполняется одно из следующих условий: 1) АН линейно связана с AS; 2) АЯ постоянна; 3) AS постоянна.
Любое из этих условий представляет собой жесткое ограничение, и всегда остается некоторое сомнение, в какой мере каждое из них действительно выполняется в реакциях, которые обнаруживают вполне удовлетворительные прямолинейные зависимости Гаммета. Именно поэтому получение прямолинейных зависимостей всегда остается загадочным. Тем не менее известны примеры, для которых действительно показано, что они удовлетворяют одному из названных выше условий. Так, для щелочного гидролиза сложных эфиров (58)
CO2Et
СО2 + EtOH
"OH
---------------►
56% водн. Me2C0,
25°C
AS* * const.
AG*~AH*~OX
(58)
найдено, что значение AS+ практически постоянно [условие (3)] и значения AG+ или АЯ+, таким образом, пропорциональны Ох- Совсем неудивительно отсутствие примеров, в которых было бы выполнено условие (2). Напротив, условие (1), по-ви-димому, является одним из тех, которое выполняется чаще всего. Интересно, что ранее выражалось сомнение в отношении того, удовлетворяет ли даже стандартная реакционная серия — диссоциация в воде м- и n-замещенных бензойных кислот при 25 °C — этому условию.
Основным препятствием для решения вопроса о том, насколько справедливым является указанное сомнение, стали
437
экспериментальные трудности, с которыми приходится ветре? чаться при проведении измерений диссоциации бензойных кислот. Растворимость этих кислот в воде очень низка, а А//° оч^йь невелики, так что получаемые результаты оказывались недостаточно достоверными и точными. Однако относительно недавно AG°, Д//° и Д3° были определены с большой точностью для серии из десяти м- и n-замещенных бензойных кислот. Используя эти данные, получили строго линейные зависимости между Д//° и Д3°, Дб° и Д//° и между Дб° и Д3°. Вероятно, что Гаммет в самом деле не ошибся в выборе стандартной реакционной серии.
Важно, однако, помнить, что теоретическая интерпретация, которая дана уравнению Гаммета, имела истоком случайные совпадения, а не строгое рассмотрение. Это уравнение остается эмпирическим, и по этой причине нет никакой необходимости оценивать константы ох и р с точностью до десятых долей. Даже информация на уровне «порядка величины» часто оказывается достаточной для выяснения механизма реакции: является ли константа р положительной или отрицательной, велико или мало ее численное значение, наблюдаются ли заметные отклонения от линейности в графиках ах — 1g kx и т. д. Это относится и к многоп ар а метровым уравнениям. Признавая их чрезвычайный интерес для химиков-специалистов по физической органической химии, приходится сомневаться (если речь идет в целом о химиках-экспериментаторах), действительно ли труднейшая работа по оценке всех этих дополнительных параметров вознаграждается качеством получаемой при этом информации.
Важно помнить, что число и разнообразие полезных корреляций, которые основаны на уравнении Гаммета, поистине поразительны, особенно имея в виду простоту и удобство этого метода. В действительности экспериментально устанавливаемые соотношения линейности свободной энергии в целом представляют собой контрольный тест для теоретического применения концепций, которые по своему происхождению являются чисто эмпирическими.
РЕКОМЕНДАТЕЛЬНАЯ ЛИТЕРАТУРА
Теория строения и спектроскопия
Dewar М. J. S., Dougherty R. С. The РМО Theory of Organic Chemistry (Plenum, 1975) [Дьюар M., Догерти Р. Теория возмущений молекулярных орбиталей в органической химии: Пер. с англ./Под ред. Л. А. Яновской. М.: Мир, 1977. 696 с.].
Fleming /. Frontier Orbitals and Organic Chemical Reactions (Wiley, 1976). McWeeny R. Coulson’s Valence (OUP, 3rd Edition, 1979).
Tedder J. M., Nechvatal A. Pictorial Orbital Theory (Pitman, 1985).
Williams D. H., Fleming I. Spectroscopic Methods in Organic Chemistry (McGraw-Hill, 3rd Edition, 1980).
438
Строение и механизм реакции
Alder R. W., Baker R., Brown J. M. Mechanism in Organic Chemistry (Wiley-Interscience, 1971).
Amis E. S. Solvent Effects on Reaction Rates and Mechanisms (Academic Press, 1966) [Амис Э. Влияние растворителя на скорость и механизм химических реакций: Пер. с англ./Под ред. М. И. Кабачника. М.: Мир, 1968. 328 с.].
Buncel Е. Carbanions: Mechanistic and Isotopic Aspects (Elsevier, 1975).
Buncel E., Durst T. (Eds). Comprehensive Carbanion Chemistry. Parts A and В (Elsevier, 1980, 1984).
Capon B., McManus S. P. Neighbouring Group Participation (Plenum, 1976). Carpenter В. K. Determination of Organic Reaction Mechanisms (Wiley, 1984). Collins C. J., Bowman N. S. (Eds). Isotope Effects in Chemical Reactions (Van Nostrand Reinhold, 1970).
De La Mare P. B. D. Electrophilic Halogenation (CUP, 1976).
De La Mare P. B. D., Bolton J. Electrophilic Addition to Unsaturated Systems (Elsevier, 2nd Edition, 1982) [Де ла Map П., Болтон P. Электрофильное присоединение к ненасыщенным системам: Пер. с англ./Под ред. В. И. Соколова. М.: Мир, 1968. 320 с].
De Mayo Р. (Ed.) Rearrangements in Ground and Excited States (Academic Press, Vols. I—III, 1980).
Eliel E. L. Stereochemistry of Carbon Compounds (McGraw-Hill, 1962) [Илиэл Э. Основы стереохимии: Пер. с англ./Под ред. В. М. Потапова. М.: Мир, 1971. 200 с.).
Forrester A. R., Hay J. М., Thomson R. Н. Organic Chemistry of Stable Free Radicals (Academic Press, 1968).
Garratt P. J. Aromaticity (McGraw-Hill, 1971).
Gilchrist T. L., Rees C. W. Carbenes, Nitrenes and Arynes (Nelson, 1969).
Gilchrist T. L., Storr R. C. Organic Reactions and Orbital Symmetry (CUP, 2nd Edition, 1979) [Джилкрист T., Сторр P. Органические реакции и орбитальная симметрия: Пер. с англ./Под ред. М. Е. Дяткиной. М.: Мир, 1975. 352 с.].
Gilliom R. D. Introduction to Phisical Organic Chemistry (Addison-Wesley, 1970).
Hartshorn S. R. Aliphatic Nucleophilic Substitution (CUP, 1973).
Hine J. Structural Effects on Equilibria in Organic Chemistry (Wiley, 1975).
Hoffmann R. W. Dehydrobenzene and Cycloalkynes (Academic Press, 1967).
Hoggett J. G., Moodie R. B., Penton J. R., Schofield K. Nitration and Aromatic Reactivity (CUP, 1971).
Ingold С. K- Structure and Mechanism in Organic Chemistry (Bell, 2nd Edition, 1969) [Ингольд К- Теоретические основы органической химии: Пер. с англ./Под ред. И. П. Белецкой. М.: Мир, 1973. 1055 с.].
Isaacs N. S. Reactive Intermediates in Organic Chemistry (Wiley, 1974).
Jencks W. P. Catalysis in Chemistry and Enzymology (McGraw-Hill, 1969) [Дженкс В. Катализ в химии и энзимологии: Пер. с аигл./Под ред. И. В. Березина. М.: Мир, 1972. 467 с.|.
Johnson С. D. The Hammett Equation (CUP, 1973) [Джонсон К. Уравнение Гаммета: Пер. с англ./Под ред. И. П. Белецкой. М.: Мир, 1977. 240 с.].
Jones R. A. Y. Physical and Mechanistic Organic Chemistry (CUP, 1979).
Kirmse W. Carbene Chemistry (Academic Press, 2nd Edition, 1971) [Кирмсе В. Химия карбенов: Пер. с англ./Под ред. Д. Н. Курганова. М.: Мир, 1966. 324 с.].
Klumpp G. W. Reactivity in Organic Chemistry (Wiley, 1982).
Kochi J. K. (Ed.). Free Radicals (Wiley-Interscience, Vols. I, II, 1973).
Lowry T. H., Richardson K. S. Mechanism and Theory in Organic Chemistry (Harper and Row, 1976).
Lwowski W. (Ed.). Nitrenes (Interscience, 1970).
439
Marchand A. P., Lehr R. E. (Eds.) Pericyclic Reactions (Academic Press, Vols. I. II, 1977).
Miller J. Aromatic Nucleophilic Substitution (Elsevier, 1968).
Nonhebel D. C., Walton J. C. Free-radical Chemistry (CUP, 1974).
Norman R. О. C., Taylor R. Electrophilic Substitution in Benzenoid Compounds (Elsevier, 1965).
Olah G. A. Schleyer P. von R. (Eds). Carbonium Ions (Interscience, Vols. I— V, 1968—1976).
Ritchie C. D. Physical Organic Chemistry (Dekker, 1975).
Saunders W. H., Cockerill A. F. Mechanisms of Elimination Reactions (Wiley-Interscience, 1973).
Shorter J. Correlation Analysis in Organic Chemistry (OUP, 1973).
Stevens T. S., Watts N. E. Selected Molecular Rearrangements (Van Nostrand Reinhold, 1973).
Testa B. Principles of Organic Stereochemistry (Dekker, 1979).
Thyagarajan B. S. (Ed.). Mechanisms of Molecular Migrations (Interscience, Vol. I, 1968; Vol. II, 1969; Vol. JU, 1971).
Vogel P. Carbocation Chemistry (Elsevier, 1985).
Wentrup C. Reactive Molecules: The Neutral Reactive Intermediate in Organic Chemistry (Wiley, 1984).
Обзоры
Advances in Physical Organic Chemistry. Gold. V. and Bethell D. (Eds) (Academic Press, Vol. I, 1963).
Organic Reaction Mechanisms. Knipe A. C. and Watts W. E. (Eds) (Interscience, Vol. I, 1966).
Progress in Physical Organic Chemistry. Taft R. W. (Ed.) (Interscience, Vol. I, 1963).
Reactive Intermediates—A Serial Publication, Jones, M. and Moss, R. A. (Eds) (Wiley-Interscience, Vol. I, 1978).
Видеокассеты
The following titles in the «Chemistry Cassette» series are published by, and are available from, The Chemical Society, Blackhorse Road, Letchworth, Herts., SG6 1HN, England:
CC3 Sykes P. Some Organic Reaction Pathways: (A). Elimination: (B). Aromatic Substitution (1975).
CC6 Sykes P. Some Reaction Pathways of Double Bonds: (A). C=C; (B). C=O(1977).
CC7 Sykes P. Some Reaction Pathways of Carboxylic Acid Derivatives (1979).
CC9 Sykes P. Radicals and their Reaction Pathways (1979).
CC11 Sykes P. Linear Free Energy Relationships (1980).
Книги советских авторов *
Дашевский В. Г. Конформация органических молекул. М.: Химия, 1974. 432 с.
Днепровский А. С., Темникова Т. И. Теоретические основы органической химии. Л.: Химия, 1979. 515 с.
Жданов Ю. А. Теория строения органических соединений. М.: Высшая школа, 1971. 288 с.
Коптюг В. А. Аренониевые иоиы. Новосибирск: Наука, 1983. 269 с.
Минкин В. И., Симкин Б. Я., Миняев Р. М. Квантовая химия органических соединений. Механизмы реакций. М.: Химия, 1986. 246 с.
Несмеянов А. Н„ Несмеянов Н. А. Начала органической химии. В 2-х т. М.: Химия, 1974. Т. 1. 623 с.; Т. 2. 724 с.
* Дополнение редактора.
440
Пальм В. А. Введение в теоретическую органическую химию. М.: Высшая школа, 1974. 455 с.
Реутов О. А. Теоретические основы органической химии. М.: Изд. МГУ, 1964. 700 с.
Современные проблемы органической химии. Вып. 1—5. Л.: Изд. ЛГУ, 1969— 1976.
Степаненко Б. Н. Курс органической химии. М.: Высшая школа, 1976. Ч. 1. 448 с.; Ч. 2. 303 с.
Травень В. Ф. Электронная структура и свойства органических молекул М.: Химия, 1989. 384 с.
Яновская Л. А. Современные теоретические основы органической химии. М.: Химия, 1978. 358 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоокислеиие 368 сл.
Азосочетаиие 37, 162 сл.
Активированный комплекс 47 сл., 327, 377, 395
Алкаиы 304, 342, 363 сл., 369
Алкеиы 36, 260, 301, 305 автоокисление 370 галогенирование 61, 198 сл.,
363 сл.
гидратация 51 сл., 118, 128,
207 сл.
озонолиз 212 сл., 394
реакции 128, 159, 197 сл., 207, 358 циклизация 45 циклодимеризация 391
Алкилгалогеииды 89, 156 сл., 275, 366 сл.
реакции 47, 55, 90 сл., 92 сл., 94 сл., 98, 126, 302
Алкилирование 52, 124, 156 сл., 181, 324
Алкил-катиоиы 117 сл., 119 сл., 123 Алкииы 201 сл., 214, 305 сл., 330 Аллилгалогеииды 98 Аллил-катиои 125
Аллильные перегруппировки 125 сл.
Альдегиды 224, 370 сл.
Амиды 80, 137 сл., 139, 264, 267 о-Амииобеизойиая кислота 196 Амииометилбеизолы 193 сл. 1-Амииопропаи 122 сл.
Амины 137 сл., 375
основность 78 сл., 81 сл.
реакции 134 сл., 162 сл.
Анизол 163, 167
Анилин (ы) 81 сл., 83, 409 сл.
Аииои-радикалы 344
Аитараповерхиостиые сдвиги 396 сл.
Антраниловая кислота 196
Антрацен 27, 379
Арилироваиие 373 сл.
Арил-катиоиы 188 сл.
Арины 31, 194 сл.
Арндта — Эйстерта метод 134 Ароматичность 23 сл.
Аррениуса уравнение 48 Ацетали 232 сл.
Ацетилен 18 сл.
Ацетилид-иоиы 248, 324
Ацетолиз 418 сл.
Ацетон см. Пропанон
Ацилгалогеииды 264, 266 сл.
Ацилирование 160 сл.
Ацил-катиоиы 117, 160
Ацилоксилироваиие 375
Ацилпероксиды 342
Байера — Виллигера реакция 142 сл.
Бартона реакция 379
Бекмана перегруппировка 139 сл.
Бензальдегид 57 сл., 370 сл.
Беизилгалогеииды 97, 98, 181
Бензин 195 сл., 219
Бензоилирование 160, 409 сл.
Бензойные кислоты 271, 371, 402 сл., 404 диссоциация 431, 438 кислотность 73
Бензол 23 сл.
реакпии 56, 124 сл., 214, 353 сл., 374, 403
п-Бензохиион 219
Бирадикалы 379 сл.
Бифенил 176
Бромбутаиы 366, 434 2-Бромбутеиы 357 Бромдеалкилирование 180 сл. 1-Бром-2,2-диметилпропан 99, 126 Бромирование 61, 169, 172, 366 алкенов 61, 198 сл. бутадиена 216 сл. галогеиалкаиов 366 сл.
кетонов 49 сл., 61, 88, 330 сл., 334 радикальное 353, 366 сл.
толуола 175
441
Бромирование хлорбензола 178
Бромметан 47 сл., 90, 95
Бромонневый катион 200
Бромпропаны 95, 99, 124, 354, 356
JV-Бромсукцинимид 367 сл.
1-Бром-1-фенилэтан 433 1-Бромциклогептатриен-2,4,6 121 Бромэтан 95, 434
Бутадиен 215 сл., 218 сл., 362, 385
Бутендновые кислоты 75 сл.
Бутены-2 60, 198 сл., 201, 299 трет-БутилхлориД 90 сл., 432 сл.
Бутиндиовая кислота 201 сл.
Вагнера — Меервейна перегруппировка 126, 127 сл.
Вннилгалогеннды 98
Виттига перегруппировка 329
Виттига реакция 260
Вольфа перегруппировка 133 сл.
Восстановление 162, 238 сл., 342 сл.
Вудварда — Гофмана правила 386
Вюрца реакция 324
Галогенарены 98, 178, 193 сл.
Галогенирование 153 сл., 201 сл., 330 сл., 351 сл., 363 сл.
Г алогенпирнднны 192
Галоформная реакция 333
Гаммета зависимости 401 сл.
Гаммета уравнение 404 сл., 416 сл, Гаттермана — Коха реакция 161 сл. Гексадиен-2,4 388 сл.
Гексаметилентетрамин 49 сл.
Гексафеннлэтан 53, 337
Гексахлорциклогексан 354
Гетероциклы 184 сл.
Гидразины 338
Гидратации
алкенов 51 сл., 118, 128, 207 сл.
карбонильных соединений 230 сл., 232
Гидриды металлов комплексные 238 Гидридиый сдвиг 131, 239, 240 сл.
Гидрирование 25 сл., 211 сл., 237 сл.
Гидроборирование алкенов 207
Гидроксамовая кислота 138
Гидрокснбеязойные кислоты 74 Гидроксилирование 209 сл., 373 4-Гидрокси-4.;Метилпентанон-2 250 2-Гндрокснпропановая кислота 403 п-Гидрокситолуол 326 Гидролиз 109 сл., 417 алкилгалогенидов 47 сл., 55, 90 сл., 92 сл., 94 сл., 98, 126 амидов 267 беизилхлорнда 97
Г идролиз бензоатов 272, 407 сл., 410, 417, 421 сл., 426, 429 2-бромпропаноат-аниона 109 галоформов 60, 298 нитрилов 273 ортоэфиров 87 сложных эфиров 38, 57, 266, 269 сл., 405 сл., 427, 429, 437 фенокситрнэтилсилаиов 416 1,1-хлоргидрина 108
Гндроперокснды 144, 368 сл.
Гнперконъюгация (Сверхсопряжение) 35 сл., 170
Глиоксаль 241
Гомберга реакция 375
Гофмана правило 285 сл., 287 сл.
Гофмана реакция 59, 137 сл., 286 сл.
Гриньяра реактивы 246 сл., 265, 324
Г рюнвальда — Уинстейна уравнение
432
Гуанидин 80
Дарзана реакция 325
Деалкилирование 159
Дегалогенирование 295 сл.
Дегидратация
альдолей 250, 254, 293 сл.
спиртов 118, 275
Дегидробензол 195 сл., 219
Дезаминирование 133
Дейтерирование 176
Дейтеро-водородный обмен 322 сл.
Декарбоксилирование 320 сл.
Делокализация
заряда 66, 80, 97
электронов 22 сл., 28 сл., 35, 37
Десульфуризация тноацеталей 235 Диазоаминосоединения 164 сл.
Диазоалканы 134, 135, 394
Диазогндраты 163
Диазокетоны 134
Диазоннй-катион 39, 134 сл., 162 сл. реакции 119, 188 сл.
Диазоннн солн 136, 188 сл., 375
Диазотат-ионы 163
Диалкилбензолы 128, 159, 182
о-Диаминобензол 164
Днацетоновый спирт 250
Диацилпероксиды 375 1,2-Днбром-2-метнлбутан 366 Дигидропиразол 394
Днены 20 сл., 215 сл., 362
Дикмана реакция 257
Дильса — Альдера реакции 195, 218 сл., 382 сл., 391 сл., 393
Днмеризацнн 330, 341, 349, 376
ЛГ,ЛГДиметиланилииы 37 п-Диметилбензол 159
442
2,3-Диметилбутаидиолы 42, 242
2,2-Диметнлбутанои-З 42
3,3-Диметилбутен 204
N, АГ-Диметилметанамид 94
Диметиловый эфир 19 2,2-Диметилпропанол 126 Диметнлсульфоксид 94, 193
Диметилформамид 94
3,4-Днметилцнклобутен 388 сл.
1,2-Диметнлциклогексадиены 383,
386, 388
Димрота параметр 434
2,4-Динитроаиилин 82
2,4-Динитрогалогенбензолы 190 сл.
2,4-Динитрофенилгидразин 243
2,4-Динитрофеннлдиазоний-катион 163
1,2-Диолы 128 сл., 242
Дипольные моменты 184 сл., 225
Дисротаторное вращение 387 сл.
Диссоциация
кислот 401 сл., 431, 437 сл.
фенолов 412
Дитиокарбонаты 300, 382
9,10-Дифенилантрацен 372
1,4-Дифенилбутадиен 219
Дифенилен 196
1,1 -Дифеиил-2-пикрилгидразил 338, 350
1,3-Дифенилфлуоренон 271
Дихлоркарбен 60, 299, 325
Енамины 245
Енолы 315 сл.
Енолят-анионы 313
Зайцева правило 286 сл.
Зайцева реакция 286 сл.
Замещение 114
ароматическое 372 сл.
механизмы 290 сл.
нуклеофильное см. Нуклеофильное замещение
радикальное 41, 363 сл.
с участием карбанионов 322 сл.
электрофильное см. Электрофильное замещение
ипсо-Замещение 179 сл., 188
Зандмейера реакция 342
Изопрен 362
Изопропилбензол 144, 370
Илиды 258, 260, 322
Имины 245
Инверсия (Обращение) конфигурации 101 сл., 132 сл.
Ингибирование стерическое 38 Ингибиторы 352, 360, 371
Индуктивный эффект 31 сл., 34 сл., 96, 171
электроиоакцепториый 168 сл., 173, 176, 306 сл., 407, 408 сл., 410 электронодонорный 170 сл., 407, 409 сл.
Иодирование 353
(+)-2-Иодоктан 103
Канниццаро реакция 57 сл., 240 сл. Карбанионы 31, 305 сл., 308 сл., 311 образование 252 сл., 303 сл. реакции 252 сл., 318 сл.
Карбены 31, 298 сл.
Карбокатионы 31, 97 сл., 119 сл. образование 117 сл., 119 сл. реакции 122 сл., 124 сл., 343
Карбоксилат-аниои 67 Карбоксилирование 318 сл. Карбонильная группа 33, 226, 234 сл. Карбонильные соединения 225 сл., 252 сл., 315 сл.
Карбоновые кислоты 401 сл. реакции 238, 263, 268 сл., 270 сл., 320 сл.
Катализ гетерогенный 211 кислотный 86 сл., 230 сл., 232 сл., 268, 333 нуклеофильный 114 основный 88 сл., 230 сл.
Квазиароматические структуры 28 Кето-енольная таутомерия 312 Кетены 134
Кетоны 143, 224 реакции 142 сл., 162, 239, 330, 344
Кинетика реакций 46 сл., 49 сл. гидролиза 55 нитрования 54 нуклеофильного замещения 90 сл. смешанная 105 сл.
Кислотность органических соединений 65 сл., 304 сл., 314
Кислоты 40
Льюиса 40, 117, 119, 127, 154, 157, 161
протонные 64 сл. сила 63 сл., 68 сл. сопряженные 64, 77 С—Н-кислоты 314 углеродные 304 сл.
Кляйзена конденсация ?г4 сл. Кляйэена перегруппировка 398 сл. Кляйзена — Шмидта реакция 251 Кнёвенагеля реакция 253 сл.
Кольбе синтез 344
Кольбе — Шмидта реакция 319, 326
443
Комплекс (ы) активированный 47 сл., 327, 377, 395
Мейзеигеймера 190 с переносом заряда 146 я-Комплексы 146 сл., 154 о-Комплексы (Уиланда интермедиаты) 51, 146 сл., 150 сл., 168 сл., 170 сл., 173 Конденсация
альдольная 249 сл., 251 ацилоиновая 380 бензоиновая 257 сл. Кляйзена 254 сл. Штоббе 254 Конротаторное вращение 387 сл. Константы
диссоциации 401
заместителей 404 сл., 406 сл., 413, 414
кислотности 64 сл.
равновесия 229 сл., 402, 417 реакции 405 сл., 409 сл., 428, 431 скорости 402, 417 стерические 428 сл.
Конфигурация 102 карбанионов 309 сл. обращение 101 сл., 132 сохранение 106 сл., 108 сл.
Конформации молекул 16, 133 Коупа перегруппировка 398 сл. Коупа реакция 300 Крама правило 262 сл.
п-Крезол 326
Крекинг 127 сл., 342
п-Ксилол 159 Кумол 144, 370 КурЦиуса реакция 138 сл.
Лактоны 143
Лоссена реакция 138 сл.
Малеиновая кислота 75 ангидрид 218 сл., 393
Меервейна — Понндорфа реакция 239 сл.
Мезомерия З12.сл.
Металлоргаиические соединения 310, 323, 328, 337, 341
Металлоцены 308 сл.
Метан 65, 363
Метаиол 65, 68, 94 Метилбеизол см. Толуол 4-Метилбензолсульфоиилхлорид 102 Метнлбромид 47 сл., 90, 95 2-Метнлпентен-2-он-4 224, 250 2-Метилпропеи 208 сл. 4-Метил-1-хлорбензол 54, 193
444
2-Метил-2-хлорпропаи 90 сл., 432 сл. 1-Метокси-2-фенилэтаи 178 сл.
1-Метокси-1-хлорметаи 237 Механизм
Ядс 1 270, 421 сл.
ЯАС 2 269
Яде 1 270, 422
Вас2 267
Е 1 276 сл., 290 сл.
Е 1сВ 276, 278 сл., 319
Е 2 275, 280 сл., 290 сл.
Зц 1 290 сл.
SN 2 290 сл.
Sn 2аромат. 190, 191 сл.
Михаэля реакция 221 сл., 223 сл.
Молекулярность реакции 92
Нафталин 27, 52 сл., 182 сл.
Неопеитилбромид 99, 126
Нитрены 31
Нитроалкаиы 252, 311 сл.
Нитроанилииы 82
Нитробензол 151, 167, 186 сл.
Нитрование 170, 172, 180
анизола 167
ареиов 54, 150 сл., 182 сл.
бензолов 56, 177, 178 1-метокси-2-фенилэтаиа 178 сл. нитробензола 167 толуола 52, 174 сл.
Нитродеалкилироваиие 180 Нитродегалогеиирование 180 Нитрозирование аминов 134 сл. Нитрозофеиол 153 Нитрофенолы 72, 153, 187 Нуклеофильное замещение 41, 101 сл., 186 сл.
ариновый механизм 193 сл. атомов водорода 186 сл.
бимолекулярное 90 сл., 101 сл.
в бензоле 186
в галогенпиридинах 192
в нитробензоле 186 сл.
в пиридине 187 сл.
в солях диазония 188 сл.
в хлорбензоле 193
галогеннд-ионов 189 сл., 192 сл.
механизм SN 2,роиат. 190, 191 сл. моиомолекулярное 91, 104 сл. неводородиых атомов 188 сл. у атома углерода 89 сл.
Нуклеофильные реагенты 39 сл., 110 сл., 323 сл.
Обращение
конфигурации 101 сл., 132 сл.
полярности атома 235
Озонолиз 212 сл., 394
Окисление 56, 342 сл.
алкенов 210
бензальдегида 343
бензоина 252
карбанионов 330
карбоксилат-ионов 343 сл.
кетонов 142 сл.
тетралина 370
фенолов 376
Окись мезитила 224, 250
Окснмы 60, 62, 139 сл.
Октатриен-2,4,6 383, 386, 387
Орбитали
антисвязывающие 14, 22, 385, 387
атомные 9 сл., 13 сл., 385
вырожденные 11
гибридные 12, 15, 17 сл., 19 сл.
молекулярные 14 сл., 17 сл., 23 сл., 385
перекрывание 13 сл., 15, 18, 20сл., 23 сл.
плоскостные (тригональные) 17 связывающие 14 сл., 22, 385, 387 симметрия 384 сл.
о- н я-орбитали 17
Ориентация
замещения 166 сл., 175 сл., 187
присоединения 203 сл., 217, 220, 355 сл.
элиминирования 285 сл.
Основания 40, 63
алифатические 78 сл.
ароматические 81 сл.
гетероциклические 84 сл.
жесткие н мягкие 111
сила 77 сл., 281
Параметр (ы)
Димрота 434
растворителя 432 сл.
спектроскопические 435 сл.
термодинамические 436 сл.
стерический заместителя 429 сл.
Пербензойная кислота 371
Перегруппировка (и) 42
алкенов 128
алкилбензолов 128
аллильные 125 сл.
без изменения углеродного скелета 125 сл.
Бекмана 139 сл.
бензиловая 259 сл.
Вагнера — Меервейна 126, 127 сл.
Виттига 329
Вольфа 133 сл.
диенон-фенольная 128, 130 сл.
1,2-диолов 128 сл.
карбокатионов 123, 124 сл.
Перегруппировка(и) Кляйзена 398 сл. Коупа 398 сл. неопентильные 126 сл. оксимов 139 сл. пероксидов 144 пинакола 42 пинаколиновая 128 сл., 133 ретропинаколиновая 130 сигматропные 395 сл. с изменением углеродного скелета 126 сл.
с миграцией групп 124 сл., 137 сл., 142 сл.
стереохимия 131 сл.
Стивенса 328 сл.
с участием карбанионов 327 ----- радикалов 377 сл.
Фаворского 329 сл.
Переходное состояние 47 сл. 327, 377, 395
Перициклические реакции 383 Пероксид(ы) 142, 144, 341 Пероксикислоты 142 Пикриновая кислота 72, 82 Пинаконы 128 сл., 242
Пиридин 28, 84 сл., 184 сл., 187
Пиридинкарбоновая кислота 322 Пиролиз 300 сл., 302, 382 Пиррол 85 сл., 185 сл.
Полиены 27
Полимеризация 209, 344 сл., 359 сл.
Порядок реакции 92 Присоединение 41, 202 ел.
1,3-диполярное 214, 394 катионов 118 сл.
к производным карбоновых кислот 265
к системе связей С = С—С = О 222 сл.
к сопряженным диенам 215 сл. нуклеофильное 41, 220 сл., 225 сл. по Марковникову 204 по связи С —С 128, 197 сл., 214, 351 сл., 378
по связи CsN 272 сл. радикальное 41, 351 сл. с участием гидрид-нонов 237 сл. ----- электронов 241 сл.
электрофильное см. Электрофильное присоединение
Присоединения — отщепления реакции 243 сл., 263 Пропанолы 123, 249
Пропанон (Ацетон) 19 сл., 144, 239, 242, 370
бромирование 49 сл., 88, 330 сл. конденсация 249 сл.
оксим 245
445
Пропанон фотолиз 339 сл. Пропен 123, 380
реакции 354 сл., 356, 361, 365
Простые эфиры 114, 125, 370 Протодесилилирование 180 Протодесульфонирование 180 Протонирование 118 сл., 185, 226, 268-, 270 сл.
Прототропия 311 сл.
Пшорра реакция 375
Радикалы 41, 335 сл., 344 сл., 347 сл. азотсодержащие 338 алкильные 336 сл., 347, 359 кислородсодержащие 339 сл., 376 образование 30, 337, 339 сл., 342 сл.
реакции 41, 336 сл., 341, 349, 350 сл, 363 сл, 376
структура 345 сл.
тиильные 339, 358 сл.
Разрыв связи 30, 117 сл, 335
металл — углерод 341
углерод—водород 56, 314
углерод — углерод 342
Раймера — Тимана реакция 299 сл, 325 сл.
Рацемизация 104
Салициловая кислота 74 Салициловый альдегид 326 Сверхсопряжение (Гиперконъюгация) 35 сл, 170
Связь (и)
водородные 46, 74, 75 сл, 79, 316 двойные 17 сл, 20 сл.
кислород — кислород 341 ковалентные 30, 335
простые (а-связи) 15 сл, 20 сл.
прочность 65
разрыв 30 сл, 117 сл, 314, 335, 341 сл.
сопряженные 20 сл.
тройные 18 сл, 20
углерод — азот 272, 341
углерод — кислород 226
углерод — углерод 15 сл, 221 сл, 342
Сложные эфиры 264, 270, 401 конденсация 380 гидролиз 38, 57, 266, 269 сл, 405, 427, 429, 437
пиролиз 300 сл, 382 Сольватация 67, 74, 78 сл. Сольватохромиые сдвиги 434 Сольволиз 104 сл, 125, 291, 413, 415, 432 сл.
Сопряжение прямое полярное 411 сл, 415
Спирты 66, 68, 118, 275 Стабилизация
енолов 316
ионов 67, 97 сл, 120 сл, 305 сл.
карбонильных соединений 231 сл. радикалов 347 сл.
Стереохимия
бромирования 61, 366 дегалогенирования 295 сл. нуклеофильного замещения 101 сл. перегруппировок 131 сл.
присоединения к карбонильным соединениям 261 сл.
циклизации 389
циклоприсоединения 392 элиминирования 282 сл, 291 сл. Стивенса перегруппировка 328 сл. Сульфирование '39, 52 сл, 155 сл, 178, 183
Суперкислоты 118
Супраповерхностные сдвиги 396
Таутомерия 311 сл, 315 сл.
Тафта уравнение 426 сл.
Термолиз 340, 341 сл, 380
Тозилирование 102
Толуол (Метилбензол) 68, 174 сл.
реакции 52, 146, 160, 175, 179, 181, 354
2,4,6-Триметилбензойная кислота 270
2,4,6-Триннтрофенилдиазоний-катион 163
1,2,3-Трипропилциклопропеи 122 Тритилхлорид 53, 97
2,4,6-Трифенилбензойная кислота 271 Трифеиилметилнатрий 304 Трифторметилбензол 151 сл.
Трихлорметан 60
Трихлорэтановая кислота 71
Триэтиламин 38, 85
Тропилий 28
Тропилийбромид 121
Уиланда интермедиаты (п-Комплек-сы) 51, 146, 157 сл, 184, 186
Уксусная кислота 65, 68 сл, 77, 403
Факторы парциальных скоростей 175 сл, 374
Фаворского перегруппировка 329 сл.
Фенантрены 27, 375
Фенил-катион 188 сл.
2-Фенилпропан 144, 370
транс- 1-Фенилпропен 201
Фенилхлорметан 97
Феноксид-анион 34, 66 сл.
реакции 39, 163, 172
Фенол(ы) 34, 144, 370, 412 кислотность 66, 72 сл.
446
Фенол(ы) реакции 152 сл., 162, 376
Фенониевые ионы 121
Фентона реагент 343
Формилирование 161
Фриделя — Крафтса реакция 124, 156 сл., 181
Фторирование 365 сл.
Хаммонда принцип 152
Хинуклидин 38, 85
Хлорбензол 178, 193
Хлорбутеиы 125 а-Хлордиметиловый эфир 237 Хлорирование 175 сл., 178 сл., 352 сл., 363 сл.
Хэммонда постулат 61 Хюккеля правило 27
Циангидрины 42, 235 Цианэтилирование 221 Циклизация 45, 162, 252, 383, 386, 388 сл.
Циклогексадиеинл-катион 177 Циклогексаидион-1,3 310 Циклогексен 25, 365 Циклогептатриенил-катиои 28 Циклодегидратация 423 сл. Циклодимеризация 391 Циклооктатетраеи 26, 309 Циклопеитадиеи 308, 392 сл.
Циклопентадиенид-аиион 28, 308
Циклопеитадиенильный радикал 395 Циклопентен 61, 369 Циклоприсоединение 390 сл.
Циклопропан (ы) 298 сл., 380 Циклопропанон 60 сл., 232 Циклопропенил-катиои 28, 122
Чичибабина реакция 187 Чугаева реакция 300 сл.
Шиффа основания 245 Шмидта реакция 138 сл. Штоббе реакции 253 сл.
Экзотермические реакции 43
Электроиоакцепториые заместители 39, 71, 73, 168, 410 сл., 418
Электронодонорные заместители 39, 73 сл., 169 сл., 410, 411 сл., 413 сл., 424 сл.
Электрофильное замещение 41 в бензолах 145 сл., 166 сл. в нафталине 182 сл. в пиридине 184 сл. в пирроле 185 сл. соотношение изомеров 176 сл.
Электрофильное присоединение 41, 222 сл.
к 1-галогеналкенам 204 к сопряженным диеиам 216 сл. по связи С = С 197 сл.
Электрофильные реагенты (Электрофилы) 39 сл.
Электроциклические реакции 383,
386 сл.
Элиминирование 42, 282 сл., 291 сл., 304
1,1- (а) 297 сл.
1,2- (Р) 273 сл., 275 сл., 295 сл., 302
пиролитическое син 300 сл.
по Гофману 286 сл.
по Зайцеву 286 сл. по механизму Е1 276 сл.,1 290 сл. -----Е 1 сВ 276, 278 сл., 319 -----Е 2 275, 280 сл., 290 сл.
с участием карбанионов 319 сл.
Этандиаль-1,2 241
Этановая кислота 65, 68 сл., 77, 403 Этен 17, 22, 24, 361, 385 Этерификация 38, 268 а-Этилбутановая кислота 77 Этилен 17, 22, 24, 361, 385 Этин 18 сл.
Эффект(ы) выравнивающий воды 65 динамические 34 сл. изотопные 56 сл., 96 индуктивный см. Индуктивный эффект иидуктомерный 35 мезомерный (сопряжения) 33 сл., 35, 192, 409, 412 поля 31 сл. полярный 407, 431 сл.
стерические 37 сл., 228 сл., 426 сл. электро мерный 35 электронные 167 сл.
Юкава — Цуно параметр 415, 417 уравнение 415 сл.
Научное издание
Сайкс П.
МЕХАНИЗМЫ РЕАКЦИЙ В ОРГАНИЧЕСКОЙ ХИМИИ
Редактор М. Н. Пастушенко
Художник И. К- Капралова
Художественный редактор К К Федоров
Технический редактор В. В. Лебедева
Корректоры М. А. Ивлиева, Л. В. Лазуткина
ИБ № 2531
Сдано в набор 24.07.90. Подписано в печать 26.06.91. Формат бумаги 60x88^/16- Бум. офс. №2. Гарнитура литературная. Печать офсетная. Усл. печ. л. 27,44. Усл. кр.-отг. 27,44. Уч.-изд. л. 28,0. Тираж 11500 экз. Заказ. 613. Цена 3 р. 50 к.
Ордена «Знак Почета» издательство «Химия». 107076, Москва, Стромынка, 21, корп. 2
Набрано в Ленинградской типографии № 2 головного предприятия ордена Трудового Красного Знамени Ленинградского объединения «Техническая книга» им. Евгении Соколовой Государственного комитета СССР по печати. 198052, г. Ленинград, Л-52, Измайловский проспект, 29.
Отпечатано в Ленинградской типографии № 4 Государственного комитета СССР по печати. 191126, Ленинград, Социалистическая ул., 14.
Отсканировал Семенсченко Владимир chem_vova@mail.univ.kiev.ua; vova2002@mail.ru
П.Сайкс
МЕХАНИЗМЫ
РЕАКЦИИ
л
е